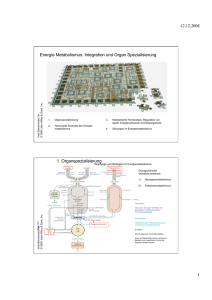
Beeinflussung körpereigener Regulationsmechanismen zur
Werbung

Beeinflussung körpereigener Regulationsmechanismen zur Therapie von Typ-2-Diabetes Bei der Entwicklung neuer oraler Medikamente zur Behandlung des Typ-2-Diabetes konzentrieren sich die Forscher auf die Aktivierung oder Hemmung körpereigener Enzyme und Rezeptoren, die eine Rolle im Glukosestoffwechsel spielen. Bevor sich ein Typ-2-Diabetes (T2D) mit allen den auf S. 14 aufgeführten Symptomen manifest ausprägt, vergehen oft bis zu 40 Jahre mit «diabetogenem» Lebensstil, gekennzeichnet durch kalorienreiche Ernährung, Übergewicht und mangelnde körperliche Bewegung. Meist sind Jahre vergangen, in denen die Betroffenen bereits eine gestörte Glukosetoleranz und Symptome eines sog. metabolischen Syndroms, aber noch keine Anzeichen eines klinisch manifesten T2D aufwiesen. Was passiert, wenn sich ein Typ-2-Diabetes entwickelt Vorstadium metabolisches Syndrom Warum und wie entwickelt sich ein metabolisches Syndrom? Unter einem metabolischen Syndrom versteht man ein ganzes Bündel verschiedener Symptome, deren wichtigste Ursache eine zunehmende Insulinresistenz der Gewebe ist. Was passiert dabei auf zellulärer Ebene? Bisher weiss man, dass bei der Insulinresistenz eine normale Bindung des Insulins (siehe Kasten) an den Insulinrezeptor auf den Zelloberflächen erfolgt. Der Signalreiz, der dadurch ausgelöst werden sollte, bleibt jedoch aus. Mit anderen Worten: Die nach dem Schlüssel-Schloss-Prinzip erfolgende Bindung von Insulin an den Insulinrezeptor setzt nicht mehr die normalerweise ablaufenden intrazellulären Signalkaskaden in Gang. Die Glykolyse, d. h. der Abbau von Glukose unter Bildung des Energiespeichers und -lieferanten Adenosintriphosphat (ATP), sowie die Synthese des Reservekohlenhydrates Glykogen aus Glukose sind gehemmt. Diese Herunterregulierung der durch Insulin-Insulinrezeptor-Bindung eingeleiteten Signalwege versucht der Körper durch ein vermehrtes Ausschütten des Botenstoffes Insulin (Hyperinsulinämie) auszugleichen. Die Insulinresistenz ist aber nur ein Kennzeichen des metabolischen Syndroms. Es ist weiterhin gekennzeichnet durch Übergewicht, Fettstoffwechselstörungen und Bluthochdruck. Klinisch-manifester Typ-2-Diabetes Die ständige Überbeanspruchung der insulinproduzierenden -Zellen in den Langerhans’schen Inseln des Pankreas führt letztendlich zu ihrer Erschöpfung und zum Funktionsverlust. Diese -Zelldysfunktion auf der einen Seite und eine Insulinresistenz der wichtigsten Zielgewebe der Insulinwirkung, ❚ der Skelettmuskeln, ❚ der Leber und ❚ des Fettgewebes, 48 Insulin Insulin vermittelt seine kurzzeitige Stoffwechselaktivität und seine langfristige, wachstumsfördernde Wirkung durch Bindung an den Insulinrezeptor an der Zelloberfläche. Es stimuliert die Aufnahme von Einfachzuckern (Monosacchariden), Aminosäuren und Fettsäuren in die Zellen. Es fördert die Synthese und Einlagerung von Kohlenhydraten, Fetten und Proteinen, während es deren enzymatischen Abbau und Freisetzung in den Blutkreislauf hemmt. Insulin erhöht den Transport von Glukose in Fettund Muskelzellen durch Stimulierung der Verlagerung des Glukosetransporters GLUT4 vom Zellinneren zur Plasmazellmembran [4]. Die Insulinsynthese wird schon durch Glukosekonzentrationen von über 2–4 mmol/l im Blut angeregt. Zur Insulinausschüttung sind allerdings Glukosekonzentrationen von über 4–6 mmol/l erforderlich. Normalerweise beträgt beim Menschen die Konzentration von Insulin im Blut 1 ng/ml. Dieses einzige blutzuckersenkende Hormon wird schnell aus dem Blutkreislauf entfernt und durch spezifische insulinabbauende Enzyme in den Zellen der Leber und anderer Gewebe inaktiviert [5]. ABBILDUNG 1: Dreidimensionale Darstellung des humanen Insulinmoleküls. Das Peptidhormon ist aus einer 30 Aminosäuren enthaltenden A-Kette (rot dargestellt) und aus einer 21 Aminosäurenbausteinen bestehenden B-Kette (grün markiert) aufgebaut. Zwei Disulfidbrücken (gelb) halten die beiden Ketten zusammen. Die Abbildung wurde von Dr. Bernd Kuhn, Molecular Design, F. Hoffmann-La Roche AG, nach den in [6] angegeben Daten erstellt. auf der anderen Seite sind die Ursachen für den erhöhten Blutzuckergehalt (Hyperglykämie) bei T2D ohne Behandlung (siehe Abb. 2). Durch die Insulinresistenz sinkt die Glukoseaufnahme in Muskel- und Fettzellen. In der Leber bewirkt die Insulinresistenz eine Steigerung der körpereigenen Glukoseproduktion, denn Insulin hemmt dort normalerweise die Glukoneogenese. Insulinsekretionsstörung und Insulinresistenz sind bei verschiedenen Patienten unterschiedlich stark ausgeprägt. Beide Faktoren können sich gegenseitig bedingen. Zum Zeitpunkt der Diagnose T2D sind 40–50 % der Betroffenen übergewichtig [1]. Mit zunehmender Erkrankungsdauer nimmt bei T2D-Patienten leider oft auch das Körpergewicht und der Anteil von Fettgewebe zu. Fettgewebe wirkt wie ein endokrines Organ und fördert durch Freisetzung von Botenstoffen die Lipolyse (Fettspaltung). Dadurch steigt die Konzentration an freien Fettsäuren1 im Blut. Eine erhöhte Konzentration an freien 1 Freie Fettsäuren sind nicht veresterte Fettsäuren. Fette sind Mono-, Di- und Triester des Glyzerins mit Fettsäureresten. Triglyzeride sind Triester des Glyzerins. Typ-2-Diabetes: Entwicklung oraler medikamentöser Therapien 49 -Zelldysfunktion Insulinresistenz erhöhte Konzentration freier Fettsäuren Bauchspeicheldrüse erhöhte Fettspaltung Leber + -Zellen schütten weniger Insulin aus erhöhte Adipozytokinwerte Muskel niedriger Insulinplasmaspiegel Fettgewebe erhöhte Freisetzung von Glukose Hyperglykämie ABBILDUNG 2: Wie Insulinresistenz und -Zelldysfunktion zu Hyperglykämie bei Typ-2-Diabetes führen: Insulin senkt nicht nur den Blutzuckerspiegel, es reduziert auch die Glukoseproduktion in der Leber (Glukoneogenese) und erhöht die Glukoseeaufnahme, -verwertung und -speicherung in Muskel- und Fettgewebe. Steht weniger Insulin zur Verfügung, so kommt es zur Erhöhung der Glukoneogenese. Fettzellen spielen eine bedeutende Rolle im Stoffwechsel. Sie setzen freie Fettsäuren (FFA) und verschiedene erniedrigter Glukosetransport, erniedrigte Aktivität des Glukosetransporters GLUT4 Adipozytokine, wie z. B. Tumornekrosefaktor- (TNF) oder Leptin, frei. Adipozytokine regulieren die Nahrungsaufnahme, Energieverbrauch und Insulinsensitivität. Ist mehr Fettgewebe vorhanden, so wird vermehrt TNF- freigesetzt. Dadurch wird die Fettspaltung (Lipolyse) gesteigert, der FFA-Spiegel steigt. Die FFA aber reduzieren die Glukoseaufnahme durch Muskelzellen, die Insulinfreisetzung durch die -Zellen und steigern die Glukoneogenese. (Abbildungsquelle: J. Mizrahi, F. Hoffmann-La Roche AG) Fettsäuren verstärkt nicht nur Hyperinsulinämie und Insulinresistenz. Sie hat auch noch andere unangenehme Folgeerscheinungen: Sie stimuliert die Synthese von triglyzeridreichen Lipoproteinen sehr geringer Dichte, VLDL (von engl.: very lowdensity lipoprotein)2. Auch Cholesterin wird von diesen freien Fettsäuren verestert und an Lipoproteine hoher Dichte (HDLCholesterin, von engl.: high-densitiy lipoprotein) oder Lipoproteine niedriger Dichte (LDL-Cholesterin, von engl.: low-densitiy lipoprotein) gebunden. Man hat erniedrigte HDL-Cholesterin-Werte und erhöhte Konzentration der LDL-Cholesterin2 50 Die Fettsäuren werden zu Fetten verestert und im Körper zusätzlich an sog. Apolipoproteine gebunden, denn nur so wird ihr Transport in wässriger Umgebung (Blutplasma) möglich. Partikel gefunden. Die LDL-Partikel tragen wesentlich zur Bildung atherosklerotischer Plaques bei. Lösen sich solche Plaques und gehen mit dem Blutstrom auf Wanderschaft, so können sie Verstopfungen in kleinen Blutgefäßen verursachen. Herzinfarkt und Schlaganfall drohen. Die Hyperinsulinämie sorgt darüber hinaus auch für eine erhöhte Rückresorption von Na+-Ionen in der Niere. Diese wiederum bewirkt über eine Volumenzunahme eine Blutdruckerhöhung. In Industrieländern weisen Typ-2-Diabetiker mit 53 % sehr viel häufiger erhöhte Blutdruckwerte auf als die altersgleiche Allgemeinbevölkerung mit 17,3 % [3]. 1998 wurden die Ergebnisse der bislang größten medizinischen Langzeitstudie überhaupt, der UKPDS (United Kingdom Prospective Diabetes Study) veröffentlicht. 5102 neu diagnostizierte Menschen mit T2D wurden im Mittel über einen Zeitraum von 11,1 Jahren beobachtet. Dabei zeigte sich deutlich: Wenn durch geeignete Maßnahmen Blutzucker und Blutdruck der Betroffenen im physiologischen Bereich gehalten werden konnten, nahm die Wahrscheinlichkeit der Entwicklung von Spätschäden signifikant ab [7]. Nach den Empfehlungen der International Diabetes Foundation sollten der HbA1c-Wert3 und der Blutzuckerspiegel, nüchtern sowie nach Mahlzeiten, in den in Tab. 1 gezeigten Bereichen Frühe Therapie kann Folgeschäden verhindern TABELLE 1: Kontrollparameter zur Verhinderung von Diabetes-Folgeschäden [7]. Parameter Risiko von Makroangiopathien, Risiko von Mikroangiopathien (Herzinfarkt, Schlaganfall) (Nephropathie, Retinopathie, Neuropathien) HbA1c unter 7,5 % Nüchternplasma- unter 7,0 mmol/l unter 6,5 % unter 6,0 mmol/l glukosegehalt (bzw. 126 mg/dl) (bzw. 108 mg/dl) Blutzuckergehalt unter 9,0 mmol/l unter 7,5 mmol/l nach Mahlzeiten (bzw. 162 mg/dl) (bzw. 135 mg/dl) 3 Zur Definition des HbA1c-Wertes siehe Erläuterung auf S. 13. Typ-2-Diabetes: Entwicklung oraler medikamentöser Therapien 51 TABELLE 2: Bisherige Medikamente zur Senkung des Blutzuckergehaltes. Medikamenten- chem. Substanzklassen gruppe (Beispielpräparate) Insuline biotechnologisch her- wie körpereigenes Insulinhormon Dosierung ohne gestellte Humaninsuline mit verzögertem Eintreten der kontinuierliches mit Modifikationen Wirkung bei Retardpräparaten Monitoring schwierig insulin- Sulfonylharnstoffe regen die Insulinausschüttung in Gefahr der Unterzuckerung sekretions- (z. B. Glibenclamide), den pankreatischen -Zellen an anregende neuere Generation der schöpfung der -Zellen durch Substanzen Meglitinide Überstimulierung Insulin- Thiazolidindione Herabsetzung der bei 85 % aller Zunahme des Sensitizer (Glitazone) Typ-2-Diabetiker vorliegenden Körpergewichtes (z. B. Rosiglitazon, peripheren Insulinresistenz durch Pioglitazon, Troglitazon) Wirkungsweise Probleme bei Überdosierung, Er- Senkung der freien Fettsäuren im Blut, dadurch Ankurbelung des Glukosestoffwechsels und Senkung der körpereigenen Glukoseproduktion in der Leber (Glukoneogenese) und Senkung des Blutzuckergehaltes Biguanide Metformin Drosselung der Glukoneogenese, gastro-intestinale Erhöhung der Effektivität der Beschwerden und sehr selten AMP-Kinase, die die Glukoseauf- Milchsäureazidose nahme in den Muskel stimuliert und z. B. die Neubildung von Cholesterol, Lipiden und Triglyzeriden in der Leber hemmt Kohlenhydrat- Acarbose, Hemmung der -Glukosidase, gestörte Verdauung, aufnahme- Miglitol die aus Stärke Glukose freisetzt, Blähungen hemmer Freisetzung der Glukose aus Nahrungskohlenhydraten wird verzögert, postprandiale Hyperglykämien werden reduziert Fettaufnahmehemmer Xenical ® Reduktion der Fettspaltung gestörte Verdauung, im Magen-Darm-Trakt und Blähungen damit der Fettaufnahme um 30 % gehalten werden [8]. Dies gelingt oft schon durch eine fettarme Diät mit einem hohen Anteil komplexer Kohlenhydrate und mehr körperliche Aktivität, denn durch Muskelarbeit wird nicht nur eine Insulinresistenz verbessert [1], sondern auch eine insulinunabhängige Glukoseaufnahme in den Muskel möglich. Reichen Ernährungs- und Bewegungstherapie nicht aus, um die in Tab. 1 aufgeführten Werte zu realisieren, stehen gegenwärtig 52 neben Insulin andere medikamentöse Möglichkeiten zur Verfügung. Meist kommt eine Kombination mehrerer oraler Antidiabetika zum Einsatz. Der Vorteil solcher Kombinationstherapien liegt in den im Vergleich zu Monotherapien geringeren einzusetzenden Dosen der Präparate. Da die verschiedenen Klassen oraler Antidiabetika unterschiedliche Wirkmechanismen haben, gelingt dadurch ein maximaler Effekt bei Minimierung der Nebenwirkungen [9]. Die bisherigen oralen Antidiabetika haben die in Tab. 2 aufgeführten Nebenwirkungen, wie Erhöhung des Körpergewichtes, Gefahr der Unterzuckerung und makrovaskuläre Komplikationen oder Durchfall. Sie wirken überdies auch nur in frühen Stadien eines T2D, wenn der Blutzuckergehalt im nüchternen Zustand unbehandelt nicht höher als 180 mg/dl liegt. Bei 40 % der Menschen mit T2D kann derzeit mit oralen Antidiabetika keine ausreichende Kontrolle des Blutzuckers erzielt werden, sie müssen zusätzlich Insulin erhalten. Ein Bedarf an neuen, wirksameren medikamentösen Behandlungsansätzen besteht. Die Forschung konzentriert sich dabei auf eine bessere Früherkennung und eine «aggressivere» Therapie. Dies bedeutet im Einzelnen: ❚ Früherkennung der Erkrankung, bevor sich ein klinisch manifester T2D sowie kardiovaskuläre Komplikationen und andere Schäden z. B. an Nieren und Augen entwickelt haben, ❚ Vorbeugung makrovaskulärer Schäden durch intensive Behandlung der Risikofaktoren des metabolischen Syndroms, ❚ Verbesserung der Dyslipoproteinämie, gekennzeichnet durch erhöhte Triglyzeridwerte und erniedrigte HDL-CholesterinSpiegel sowie ❚ Kontrolle des Blutdrucks [10]. Ein orales Antidiabetikum sollte als Monotherapie oder in Kombination mit anderen Medikamenten nahezu alle bei T2D aus den Fugen geratenen Mechanismen beeinflussen können. Es sollte ❚ die Insulinsensitivität wieder herstellen, ❚ die Funktion der insulinproduzierenden -Zellen der Bauchspeicheldrüse verbessern, ❚ die Menge freier Fettsäuren im Blut senken, ❚ vorteilhafte Wirkungen auf die Lipide und andere Komponenten des metabolischen Syndroms haben, ❚ vorteilhafte Effekte auf die Gefäßendothelfunktionen ausüben, Ein ideales orales Antidiabetikum Typ-2-Diabetes: Entwicklung oraler medikamentöser Therapien 53 gestörte Insulinausschüttung GLP-1, DPP-IV Bauchspeicheldrüse Glukose Fettgewebe freie Fettsäuren Darm Glukoseaufnahme Glukosefreisetzung aus der Leber Hyperglykämie AMPK Glukokinase Glukoseaufnahme Muskel Leber Glykogenphosphorylase Insulin-Sensitizer PPAR-Coagonisten ABBILDUNG 3: Zielorte bzw. Angriffspunkte, um mittels medikamentöser Ansätze (orange Pfeile) die physiologischen Mechanismen zu beeinflussen, die bei Typ-2-Diabetikern gestört bzw. außer Kontrolle gera- ten sind. Erläuterungen der Abkürzungen und detaillierte Erklärung im Text. (Abbildungsquelle: J. Mizrahi, F. Hoffmann-La Roche AG) ❚ ❚ die Glukoseproduktion in der Leber unterdrücken sowie eine Langzeitkontrolle des Blutzuckers gewährleisten. Daneben sollte es selbstverständlich gut verträglich sein [10]. Wie lässt sich dies erreichen? Die Forscher konzentrieren sich auf die Beeinflussung körpereigener Mechanismen, Rezeptoren und Enzyme, die eine Rolle im Glukosestoffwechsel spielen und von denen bisher bekannt ist, dass sie bei T2D eine gestörte Funktion aufweisen. Abbildung 3 zeigt eine Übersicht der verschiedenen Möglichkeiten, die im Folgenden näher erläutert werden sollen. Glukokinasemodulatoren: den Glukoseabbau fördern Das Enzym Glukokinase gehört zu den Transferasen. Die Aufgabe des Enzyms im Organismus besteht darin, eine Phosphatgruppe von ATP auf eine -OH-Gruppe des Glukosemoleküls zu übertragen, zu «transferieren». Diese Phosphorylierung ist der erste Schritt bei der Verstoffwechselung der Glukose. Glukokinasen befinden sich in der Leber und in der Bauchspeicheldrüse. In der Leber leitet das Enzym den Abbau Beeinflussung wichtiger Stoffwechselenzyme 54 der Glukose und die Speicherung von Glukose in Form des Reservekohlenhydrats Glykogen ein. In der Bauchspeicheldrüse hat die Glukokinase eine Funktion als Glukosesensor und ist an der Regulation der Insulinausschüttung beteiligt. Mit der Aktivierung dieses Enzyms kann man also eine Senkung des Blutzuckergehaltes auf zwei Wegen erreichen: ❚ durch Verbesserung der durch Glukose stimulierten Insulinausschüttung und ❚ durch erhöhte Glukoseaufnahme der Leber. Forscher der F. Hoffmann-La Roche AG fanden niedermolekulare Substanzen, sog. kleine Moleküle, welche die Fähigkeiten der Glukokinase zurVerstoffwechselung von Glukose verbessern konnten. Dafür identifizierten sie die Arzneimittelbindungsstelle im Glukokinasemolekül, die für eine Enzymaktivierung verantwortlich ist. In verschiedenen Diabetes-Tiermodellen konnten Glukosemodulatoren den Blutzuckerspiegel senken [13]. Hemmung der Glykogenphosphorylase: den Abbau von Reservekohlenhydraten hemmen Das Enzym Glykogenphosphorylase spaltet die Bindung zwischen zwei Glukosebausteinen im Glykogen phosphorolytisch unter Bildung von Glukose-1-Phosphatbausteinen4. Glykogenphosphorylasen kommen im Muskelgewebe und in der Leber vor. In der Leber sorgt das Enzym für den Abbau von Glykogen, um so normalerweise bei niedrigem Blutglukosegehalt Glukose zum Transport in andere Organe zur Verfügung zu stellen. Hohe Blutglukosegehalte können es hemmen. Eine medikamentöse Hemmung dieses Enzyms könnte helfen, die bei Typ-2-Diabetikern erhöhte körpereigene Glukosefreisetzung aus der Leber zu drosseln. Aktivierung der AMP-Kinase: körperliche Aktivität vorgaukeln Muskelarbeit hat einen insulinähnlichen Effekt: Sie fördert die Aufnahme von Glukose in die Zellen. Andauernde Muskelarbeit z. B. durch ein Ausdauertraining hat außerdem positive Auswirkungen auf die Insulinsensitivität. Dies wird z. T. auf die durch andauernde Muskelarbeit erhöhte Konzentration des Glukosetransporters GLUT4 im Muskelgewebe zurückgeführt. 4 Das sind Glukosemoleküle, die am Kohlenstoffatom 1 im Molekül eine Phosphatgruppe tragen. Typ-2-Diabetes: Entwicklung oraler medikamentöser Therapien 55 Die Erhöhung der Glukosetransporterfunktion wiederum hängt von der Aktivierung einer Protein-Kinase im Muskel ab, der durch Adenosinmonophosphat (AMP) aktivierten ProteinKinase (AMPK). Mit fortschreitender Dauer der Muskelkontraktion wird zur Deckung des dafür benötigten Energiebedarfs energiereiches ATP in Adenosindiphosphat (ADP) und AMP gespalten. Gleichzeitig wird zur Bereitstellung von genügend Nachschub an ATP ein Phosphatrest von Kreatinphosphat auf ADP übertragen. Wenn also der Muskel kontrahiert, nimmt daher in Abhängigkeit von der Intensität der Arbeitsleistung die Konzentration an AMP zu und die Konzentration von Kreatinphosphat ab. Durch erhöhte AMP-Konzentrationen während der Muskelarbeit wird die AMPK, wie der Name schon sagt, aktiviert und durch erhöhte Kreatinphosphatkonzentrationen inaktiviert [14]. Im Allgemeinen sind Protein-Kinasen Enzyme, die Phosphatgruppen übertragen und so die Aktivität bestimmter Proteine regulieren. Man nimmt an, dass die aktivierte AMPK Proteine, die Prozesse zur Energiegewinnung (also zur Bildung von ATP) einleiten, phosphoryliert und damit aktiviert. Dazu zählen z. B. die Verbrennung von Fettsäuren und die Glukoseaufnahme. Gleichzeitig sorgt sie dafür, dass solche energieverbrauchenden Prozesse wie die Synthese von Fettsäuren, Triglyzeriden und Cholesterin vorübergehend eingestellt werden. Sie wacht also über den Energiezustand der Zelle. Das Ausmaß ihrer Aktivierung hängt dabei von der Arbeitsleistung des Muskels und von der Konzentration des Langzeitglukosespeichers Glykogen ab. Eine therapeutische Beeinflussung dieses Enzyms eignet sich daher hervorragend zur T2D-Behandlung. Durch Aktivierung der AMPK sollten sich die physiologischen Prozesse in den Muskelzellen in Gang setzen lassen, wie sie normalerweise durch körperliche Anstrengung hervorgerufen werden. Körperliche Aktivität wird vorgegaukelt. PPAR: eine Wirkung am Zellkern Geht man noch einen Schritt weiter, so versucht man nicht nur, auf wichtige Stoffwechselenzyme direkt Einfluss zu nehmen, sondern über die Aktivierung bestimmter Rezeptoren z. B. das Ausmaß der körpereigenen Produktion dieser Enzyme zu beeinflussen. In Leber-, Muskel- und Fettgewebe sowie in Makrophagen kommen beispielsweise sog. Peroxisomen-Proliferation-aktivierte Rezeptoren (PPAR) vor. Es handelt sich um Zellkernrezeptoren, Noch einen Schritt weiter: die Aktivierung von Rezeptoren 56 die nach Aktivierung als Transkriptionsfaktoren wirken. Sie heften sich im Zellkern an Abschnitte der Erbinformation, welche die Bauanleitung für Proteine tragen, die beim Transport und Stoffwechsel von Glukose, Lipiden und Lipoproteinen eine Rolle spielen. Auf diese Art und Weise regulieren sie, wieviel dieser Proteine gebildet werden, und haben letztlich dadurch einen Einfluss auf den Stoffwechsel. Aktiviert man die -PPARezeptoren in Leber- und Muskelgewebe, lassen sich ❚ eine erhöhte Fettverbrennung, ❚ eine Absenkung der Triglyzeridkonzentrationen im Blut sowie ❚ eine Verbesserung der HDL-Cholesterin-Werte feststellen. Die -PPA-Rezeptoren in Makrophagen und Fettzellen sind u. a. an der Regulierung des Glukosestoffwechsels und der Speicherung von Energie in Fettzellen beteiligt. Ihre Aktivierung kann eine Insulinresistenz senken. Das in Tab. 2 aufgeführte Troglitazon kann z. B. die Glukoseaufnahme der Zellen durch eine erhöhte Bildung der Glukosetransporter GLUT1 und GLUT4 steigern [9]. Forscher der F. Hoffmann-La Roche AG suchen derzeit nach weiteren Substanzen, welche die -PPA-Rezeptoren oder beide PPA-Rezeptortypen (PPARCoagonisten) aktivieren können. PPAR-Coagonisten sollten einen noch stärkeren Einfluss auf die Senkung der Insulinresistenz und die Beeinflussung des metabolischen Syndroms haben. Angriff auf die Insulinresistenz über den Eingriff in intrazelluläre Signalwege Ein weiterer interessanter Ansatzpunkt zur Bekämpfung einer Insulinresistenz ist sicherlich der, den intrazellulären Bereich des Insulinrezeptors zu aktivieren. Dies ist jener Bereich, der nach erfolgter Insulin-Insulinrezeptor-Bindung auf der Zelloberfläche, intrazellulär über eine Abfolge von Reaktionen verschiedene Signalwege aktiviert. Störungen dieser Prozesse sind, wie man annimmt (s. o.), für die Ausprägung einer Insulinresistenz bei T2D bedeutsam. GLP-1: ein Botenstoff mit vielen Funktionen Woher bekommen die insulinproduzierenden Zellen des Pankreas die Information, wann und wie viel Insulin sie nach einer Mahlzeit ausschütten müssen? Ein Schlüsselbotenstoff heißt Glukagon ähnliches Peptid GLP-1 Beeinflussung von Enzymen und Rezeptoren Typ-2-Diabetes: Entwicklung oraler medikamentöser Therapien 57 (von engl.: glucagon-like peptide). Es handelt sich um ein nur aus wenigen Aminosäurebausteinen bestehendes Peptidhormon. Dieses Peptidhormon wird im Darm, neben anderen Peptiden, aus dem in den -Zellen der Langerhans’schen Inseln der Bauchspeicheldrüse produzierten Proglukagon gebildet und zwar abhängig von der zugeführten Nahrung! GLP-1 kann nicht nur die Magenentleerung und damit die Ankunft der Nahrung im Darm verzögern. Abhängig von der Glukosekonzentration im Darm hemmt GLP-1 in der Bauchspeicheldrüse die Freisetzung von Glukagon5 und fördert gemeinsam mit Glukose die Freisetzung von Insulin, indem es GLP-1 an spezifische Rezeptoren auf der Oberfläche von insulinproduzierenden -Zellen in den Langerhans’schen Inseln der Bauchspeicheldrüse andockt. Dadurch werden intrazelluläre Signalkaskaden in Gang gesetzt, die zur Insulinfreisetzung führen [5]. Diese Steuerung der Insulinfreisetzung durch GLP-1 und Glukose stellt sicher, dass nicht zu viel Insulin freigesetzt wird. Es kann keine Unterzuckerung entstehen. GLP-1 wird schnell durch das Enzym Dipeptidyl-peptidase-IV (DPP-IV) inaktiviert. DPP-IV ist eine Protease, die im Blutkreislauf zirkulierendes Protein oder Peptid spaltet und somit inaktiviert. Dieser interessante physiologische Regelmechanismus bietet gleich mehrere Ansatzpunkte einer therapeutischen Beeinflussung, sofern noch eine Insulinproduktion bei Typ-2-Diabetikern vorhanden ist: ❚ Durch Gabe von GLP-1 lässt sich der Glukoseanstieg nach Nahrungsaufnahme verzögern [1] und die Freisetzung von Insulin steigern. ❚ Erhöhte GLP-1-Konzentrationen im Blut lassen sich z. B. aber auch dadurch erreichen, dass man das Enzym DPP-IV, welches diesen Botenstoff abbaut, hemmt. ❚ Eine Substanz, die wie GLP-1 an den GLP-1-Rezeptor auf der Oberfläche der -Zellen binden könnte, ein sog. GLP-1-Rezeptor-Agonist, könnte außerdem beispielsweise für eine Anregung der Insulinfreisetzung sorgen, ohne dass es langfristig zu einer Erschöpfung der insulinproduzierenden Zellen kommt, da nur im Zusammenspiel mit Glukose eine 5 58 Glukagon ist der natürliche Gegenspieler, ein Antagonist, des Insulins. Glukagon stimuliert also die Glukosesynthese und den Abbau des Glykogens und sorgt so für eine Erhöhung des Blutzuckerspiegels. Insulinfreisetzung erfolgt. Die bisher eingesetzten insulinsekretionsanregenden Substanzen, wie z. B. die Sulfonylharnstoffe (siehe Tab. 2) können längerfristig zu einer Erschöpfung der -Zellen führen, da sie die Freisetzung von Insulin unabhängig von Glukose oder anderen insulinstimulierenden Nahrungsbestandteilen vermitteln [1]. Wie gezeigt, gibt es viele hoffnungsvolle Ansätze, die bei T2D außer Kontrolle geratenen physiologischen Prozesse so zu beeinflussen, dass es zu einer Senkung des erhöhten Blutzuckergehaltes kommt. Der beste Weg im Kampf gegen T2D ist aber sicher der, ihn erst gar nicht entstehen zu lassen. Quellen 1. Mehnert, H., Standl, E., Usadel, K.-H.: Diabetologie in Klinik und Praxis. Thieme Verlag, Stuttgart, 1999 2. Fiedler, H.: Das metabolische Syndrom, Pathogenese, Diagnostik, Therapie. mta 12 (5): 326-327, 1997 3. Simon, A., Giral, P., Lebenson, J.: Extracoronary atherosclerotic plaque at multiple sites and total coronary calcification deposit in asymptomatic men: Association with coronary risk profile. Circulation 92: 1414–1421, 1995 4. Saltiel, A.R., Kahn, C.R.: Insulin signalling and the regulation of glucose and lipid metabolism. Nature 414: 799–806, 2001 5. Lexikon der Biochemie, Spektrum Akademischer Verlag, Heidelberg, Berlin, 2000 6. Smith, G.D., Pangborn, W.A., Blessing, 7. R.H.: Phase changes in T3R3f human insulin: temperature or pressure induced? Acta Crystallographica, Section D: Biological Crystallography D57 (8): 1091–1100, 2001 Turner, R.C., Cull, C.A., Frighi, V., Holmann, R.: Glycemic control with diet, sulfonylurea, metformin, or insulin in patients with type 2 diabetes mellitus: progressive requirement for multiple 8. 9. 10. 11. 12. 13. 14. therapies (UKPDS 49). UK Prospective Diabetes Study (UKPDS) Group. J Am Med Assoc 281: 2005-2012, 1999 Ganz, M.: Präsentation Roche Media Roundtable «New Approaches in Diabetes Care». Mannheim, 19.–20.09. 2002 Mehnert, H.: Typ-2-Diabetes. Pathogenese, Diagnostik, Therapie, Folgeschäden. Medikon Verlag, München, 2000 Mizrahi, J.: Präsentation Roche Media Roundtable «New Approaches in Diabetes Care», Mannheim 19.–20.09.2002 Mizrahi, J.: What's new in the field of oral antidiabetics: Insulin sensitizers as novel agents for the treatment of type 2 diabetes. Abstraktband Roche Media Roundtable «New Approaches in Diabetes Care», Mannheim 19.–20.09.2002 Wagman, A.S., Nuss, J.M.: Current therapies and emerging targets for the treatment of diabetes. Current Pharmaceutical Design 7: 417–450, 2001 Die erstaunlichen Fähigkeiten der Glukokinase. Roche Nachrichten 7: 8, 2001 Winder, W. W.: Energy-sensing and signalling by AMP-activated protein kinase in skeletal muscle. J Appl Physiol 91: 1017–1028, 2001 Typ-2-Diabetes: Entwicklung oraler medikamentöser Therapien 59