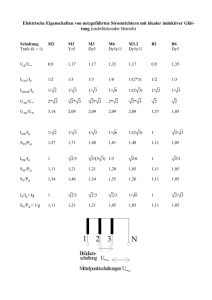
Charakterisierung und in vitro Expression einer Protein Disulfid
Werbung

Aus dem Institut für Parasitologie der Tierärztlichen Hochschule Hannover Charakterisierung und in vitro Expression einer Protein Disulfid Isomerase (PDI) zur Immunisierung gegen Dictyocaulus viviparus INAUGURAL-DISSERTATION zur Erlangung des Grades einer Doktorin der Veterinärmedizin (Dr. med. vet.) durch die Tierärztliche Hochschule Hannover Vorgelegt von Sonja Wolken aus Sande Hannover 2006 Wissenschaftliche Betreuung: Univ.-Prof. Dr. T. Schnieder 1. Gutachter: Univ.-Prof. Dr. T. Schnieder 2. Gutachter: Apl.-Prof. Dr. L. Haas Tag der mündlichen Prüfung: 16. November 2006 Teile dieser Arbeit wurden bereits auf folgenden Tagungen vorgestellt: Sonja Wolken, Georg von Samson-Himmelstjerna, Thomas Schnieder (2005): „Herstellung eines rekombinanten Vakzinekandidaten für Dictyocaulus viviparus“ Tagung der DVG-Fachgruppe „Parasitologie und Parasitäre Erkrankungen“ Potsdam, 22. – 24.06.2005 Sonja Wolken, Georg von Samson-Himmelstjerna, Thomas Schnieder (2006): „Characterisation of Dictyocaulus viviparus protein disulfid isomerase and testing of its immunological potential” 22. Jahrestagung der Deutschen Gesellschaft für Parasitologie (DGP) Wien, 22. – 25.02.2006 Inhaltsverzeichnis 1 2 Einleitung.........................................................................11 Literaturübersicht.............................................................13 2.1 Die Dictyocaulose des Rindes.....................................................................13 2.1.1 Der Erreger ......................................................................................... 13 2.1.2 Entwicklungszyklus und Pathogenese ................................................ 14 2.1.3 Epidemiologie und wirtschaftliche Bedeutung ..................................... 17 2.1.4 Immunität ............................................................................................ 20 2.1.5 Diagnose ............................................................................................. 23 2.1.6 Bekämpfung ........................................................................................ 26 2.1.6.1 Weidetechnische Maßnahmen..................................................... 27 2.1.6.2 Anthelminthika ............................................................................. 27 2.1.6.3 Vakzinierung ................................................................................ 28 2.1.6.4 Biologische Bekämpfung.............................................................. 30 2.2 Antigene und Immunisierungsversuche bei D. viviparus .............................30 2.3 Die Protein Disulfid Isomerase (PDI)...........................................................36 2.3.1 Struktur der PDI und PDI-ähnlicher Enzyme ....................................... 37 2.3.2 Funktionen der PDI ............................................................................. 41 2.3.2.1 Redox-Isomerase-Funktion der PDI............................................. 41 2.3.2.2 Die PDI als Untereinheit der Kollagen-Prolyl-4-Hydroxylase ....... 42 2.3.2.3 Chaperon-Funktion und Anti-Chaperon-Funktion der PDI ........... 43 2.3.2.4 Die PDI als Untereinheit des mikrosomalen .................................... Triglycerid-Transfer-Proteins (MTP)............................................. 45 2.3.2.5 Funktionen des ERp57 ................................................................ 45 2.3.2.6 Weitere Funktionen der PDI......................................................... 46 2.3.3 Lokalisation der PDI außerhalb des ER .............................................. 46 2.3.4 PDIs bei Helminthen ........................................................................... 47 3 Material und Methoden....................................................54 3.1 Materialien...................................................................................................54 3.1.1 Puffer und Lösungen........................................................................... 54 3.1.2 Medien und Platten ............................................................................. 56 3.1.3 Bakterien und Vektoren....................................................................... 56 3.1.4 Reagenzien, Reaktionskits und Geräte............................................... 57 3.1.5 Chemikalien und Verbrauchsmaterialien............................................. 60 3.2 Gewinnung und Aufbereitung des parasitologischen Probenmaterials .......61 3.2.1 Isolierung von Lungenwurmlarven aus dem Kot ................................. 61 3.2.2 Isolierung von adulten Lungenwürmern aus der Lunge ...................... 62 3.2.3 Herstellung von D. viviparus Rohantigen ............................................ 62 3.3 Molekularbiologische Methoden ..................................................................63 3.3.1 mRNA Isolierung ................................................................................. 63 3.3.2 Isolierung genomischer DNA............................................................... 64 3.3.3 cDNA-Synthese................................................................................... 64 3.3.3.1 cDNA-Synthese für die konventionelle PCR ................................ 64 3.3.3.2 cDNA-Synthese für die RACE-PCR ............................................. 65 3.3.4 Polymerasekettenreaktion (polymerase chain reaction, PCR) ............ 66 3.3.4.1 Allgemeines zur Methode............................................................. 66 3.3.4.2 PCR zur Erstamplifikation der PDI von D. viviparus..................... 67 3.3.4.3 Rapid amplification of cDNA ends (RACE PCR).......................... 69 3.3.4.4 PCR zur Selektion von Kolonien .................................................. 70 3.3.4.5 PCR zur Ermittlung der genomischen Sequenz........................... 71 3.3.4.6 PCR zur Klonierung des ORF (open reading frame) der PDI....... 72 3.3.5 Darstellung von Nucleinsäuren mittels Agarosegelelektrophorese ..... 73 3.3.6 Isolierung von PCR-Amplifikaten aus Agarosegelen........................... 74 3.3.7 Klonierung von PCR-Amplifikaten ....................................................... 74 3.3.7.1 Klonierung in pCR®4-TOPO® ..................................................... 74 3.3.7.2 Klonierung in pGEX4T-3 .............................................................. 75 3.3.8 Transformation kompetenter Zellen .................................................... 76 3.3.8.1 E. coli TOP 10.............................................................................. 76 3.3.8.2 E. coli BL 21 (DE3) ...................................................................... 77 3.3.8.3 E. coli BL 21-Codon Plus® .......................................................... 77 3.3.9 Plasmidpräparation ............................................................................. 78 3.3.9.1 Miniprep ....................................................................................... 78 3.3.9.2 Midiprep ....................................................................................... 79 3.3.10 Restriktionsenzymverdau .................................................................... 79 3.3.10.1 Restriktionsfragment-Längenpolymorphismus (RFLP) ................ 79 3.3.10.2 Darstellung des Inserts ................................................................ 80 3.3.11 Sequenzierung und Sequenzbearbeitung ........................................... 80 3.4 Proteinbiochemische Methoden ..................................................................81 3.4.1 Proteinexpression ............................................................................... 81 3.4.2 Gewinnung des Zelllysats ................................................................... 82 3.4.2.1 Zelllyse mittels MagneGST™ Protein Purification System............... Lysispuffer (PROMEGA) ............................................................. 82 3.4.2.2 Zelllyse mittels Lysisprotokoll nach SAMBROCK (1989) ............. 82 3.4.2.3 Zelllyse mittels Lysispuffer und Ultraschall................................... 82 3.4.3 Proteinaufreinigung ............................................................................. 83 3.4.3.1 Magne GST™ Protein Purification System .................................. 83 3.4.3.2 Glutathione Sepharose™ High Performance ............................... 84 3.4.3.3 FPLC mit GSTrap™ HP Affinitätssäulen...................................... 85 3.4.4 Proteinproduktion für die Immunisierungen......................................... 86 3.4.5 Darstellung von Proteinen ................................................................... 87 3.4.5.1 SDS-Polyacrylamid-Gelelektrophorese........................................ 87 3.4.5.2 Coomassie-Brilliant-Blue-Färbung von Proteinen ........................ 88 3.4.5.3 Transfer Blot ................................................................................ 88 3.4.5.4 Western-Blot ................................................................................ 89 3.4.6 Bestimmung der Proteinkonzentration ................................................ 90 3.4.6.1 Bradford ....................................................................................... 90 3.4.6.2 Agilent 2100 Bioanalyzer ............................................................. 91 3.4.7 Aufkonzentrieren der Proteinlösung .................................................... 92 3.4.8 Identifizierung der PDI mittels MALDI-TOF-MS................................... 92 3.5 Tierexperimentelle Untersuchungen............................................................92 3.5.1 Versuchsplan ...................................................................................... 92 3.5.2 Genehmigung...................................................................................... 94 3.5.3 Tiermaterial ......................................................................................... 95 3.5.4 Tierhaltung .......................................................................................... 95 3.5.5 Verwendete Adjuvantien und Zubereitung des Impfstoffs ................... 95 3.5.6 Applikation des Impfstoffs ................................................................... 96 3.5.7 Experimentelle Infektion...................................................................... 96 3.5.8 Überwachung der Tiergesundheit ....................................................... 96 3.5.9 Medikamentöse Behandlung erkrankter Tiere .................................... 97 3.5.10 Gewinnung von Probenmaterial und dessen Aufbereitung ................. 97 3.5.10.1 Blutproben.................................................................................... 97 3.5.10.2 Kotproben .................................................................................... 98 3.5.11 Sektion ................................................................................................ 98 3.5.12 Größenbestimmung der adulten Stadien ............................................ 98 3.6 Serologische Untersuchungen ....................................................................99 3.6.1 Enzyme linked immunosorbant assay (ELISA) ................................... 99 3.6.2 Überprüfung der Serokonversion mittels Western-Blot ..................... 101 3.7 Quantitative real-time PCR (QRT-PCR) ....................................................101 3.7.1 Allgemeines zur Methode.................................................................. 101 3.7.2 Konstruktion der Primer und Sonden ................................................ 104 3.7.3 Gesamt-RNA Isolierung .................................................................... 104 3.7.4 DNAse-Verdau der Gesamt-RNA...................................................... 105 3.7.5 cDNA-Synthese für die quantitative real-time PCR ........................... 105 3.7.6 Absolute Quantifizierung ................................................................... 106 3.7.7 Relative Quantifizierung .................................................................... 107 3.7.8 Durchführung der QRT-PCR ............................................................. 107 4 Ergebnisse.....................................................................109 4.1 Molekularbiologische Ergebnisse ..............................................................109 4.1.1 Amplifikation der Protein Disulfid Isomerase von D. viviparus........... 109 4.1.2 Vergleich positiver Klone mittels RFLP ............................................. 110 4.1.3 RACE-PCR ....................................................................................... 110 4.1.4 Identitätsvergleiche der PDI1 und PDI2 von D. viviparus mit PDIs ......... anderer Spezies ................................................................................ 112 4.1.5 Identifizierung konservierter Domänen.............................................. 115 4.1.6 Ermittlung der genomischen Sequenz der PDI2 von D. viviparus ..... 116 4.2 Proteinbiochemische Ergebnisse ..............................................................120 4.2.1 Optimierung der Proteinaufreinigung ................................................ 120 4.2.1.1 Proteinexpression ...................................................................... 121 4.2.1.2 Vergleich unterschiedlicher Lysebedingungen........................... 122 4.2.1.3 Unterschiedliche Aufreinigungbedingungen im Magne ................... GST™-Protokoll und mittels Sepharose™-Medium................... 123 4.2.1.4 Aufreinigung mittels FPLC ......................................................... 123 4.2.2 Proteinproduktion für die Immunisierungen....................................... 124 4.2.3 Ergebnisse der MALDI-TOF-MS-Analyse ......................................... 127 4.3 Tierexperimentelle Ergebnisse ..................................................................128 4.3.1 Tiergesundheit .................................................................................. 128 4.3.1.1 Allgemeinuntersuchung.............................................................. 128 4.3.1.2 Gewichtsentwicklung ................................................................. 131 4.3.1.3 Lokale Impfreaktionen................................................................ 132 4.3.1.4 Behandlung erkrankter Tiere...................................................... 134 4.3.1.5 Differentialblutbild ...................................................................... 134 4.3.2 Larvenausscheidung ......................................................................... 135 4.3.3 makroskopische Beurteilung der Lungen .......................................... 137 4.3.4 Bestimmung der Anzahl adulter D. viviparus..................................... 139 4.3.5 Bestimmung der Wurmgröße ............................................................ 141 4.4 Ergebnisse der serologischen Untersuchungen ........................................142 4.4.1 ELISA................................................................................................ 142 4.4.2 Überprüfung der Serokonversion mittels Westernblot....................... 144 4.5 Ergebnisse der quantitativen real-time PCR .............................................147 4.5.1 Absolute Quantifizierung ................................................................... 147 4.5.2 Relative Quantifizierung .................................................................... 147 5 Diskussion .....................................................................150 5.1 Identifizierung der PDI von D. viviparus ....................................................150 5.2 Expression und Aufreinigung der rekombinanten PDI...............................152 5.3 Durchführung der Immunisierungsversuche..............................................157 5.3.1 Wahl des Tiermaterials ..................................................................... 157 5.3.2 Immunisierung und experimentelle Infektion ..................................... 158 5.4 Ergebnisse der Immunisierungsversuche .................................................159 5.4.1 Befunde der täglichen Beobachtung und klinischen Untersuchung .. 159 5.4.2 Etablierung der Infektion in den Kontrollgruppen .............................. 160 5.4.3 Larvenausscheidung und Wurmbürden ............................................ 161 5.4.4 Ergebnisse der serologischen Untersuchungen................................ 164 5.4.5 Quantitative real-time PCR................................................................ 166 5.5 Zusammenfassende Beurteilung und Ausblick .........................................168 6 7 8 9 Zusammenfassung ........................................................170 Summary .......................................................................172 Literaturverzeichnis .......................................................174 Anhang ..........................................................................199 9.1 9.2 9.3 9.4 9.5 9.6 9.7 9.8 9.9 9.10 Vollständige cDNA-Sequenz und abgeleitete ................................................. Aminosäurensequenz der PDI1 von D. viviparus ......................................199 Vollständige cDNA-Sequenz und Aminosäurensequenz................................. der PDI2 von D. viviparus..........................................................................200 Eosinophile Granulozyten .........................................................................203 Larvenausscheidung im I. u. II. Immunisierungsversuch...........................204 Längen und Breiten der adulten Würmer ..................................................205 Indices für verschiedene Serumantikörpertiter ..........................................206 Verzeichnis der verwendeten Abkürzungen ..............................................208 Symbole der Aminosäuren ........................................................................211 Abbildungsverzeichnis...............................................................................212 Tabellenverzeichnis...................................................................................213 Einleitung 11 1 Einleitung Parasitosen stellen weltweit die häufigsten und verlustreichsten Infektionskrankheiten bei Mensch und Tier dar. Zu dieser Situation trägt wesentlich die Tatsache bei, dass sich die Möglichkeiten zur Bekämpfung parasitärer Erkrankungen fast ausschließlich auf den Einsatz von chemotherapeutischer Anthelminthika Substanzen birgt beschränken. jedoch das Der intensive ständige Risiko Einsatz einer Resistenzentwicklung bei den Erregern. Aus vielen Ländern existieren bereits Berichte über das Vorliegen von Resistenzen bei veterinärmedizinisch relevanten Parasiten, wobei sich diese zum Teil gegen alle verfügbaren Substanzklassen richten. Aus diesen Gründen beschäftigen sich weltweit Forschergruppen mit der Entwicklung von Vakzinen gegen Parasiten, wobei im Vergleich zu viralen und bakteriellen Erregern bislang erst wenige Erfolge erzielt wurden. Gründe hierfür sind unter anderem die komplexe Struktur und hohe genetische Organisation der Parasiten. Die vorliegende Arbeit befasst sich mit einem Vakzinekandidaten gegen Dictyocaulus viviparus, dem großen Lungenwurm des Rindes. Die von D. viviparus verursachte parasitäre Bronchitis ist eine der wichtigsten und verlustreichsten Weideparasitosen in gemäßigten Klimazonen, wobei Rinder im ersten Weidejahr besonders gefährdet sind. Eine in den 1950er Jahren entwickelte Lebendvakzine kann für einige Monate eine belastbare Immunität induzieren, die jedoch durch erneute Infektion aufgefrischt werden muss. Lebendvakzinen haben den Nachteil, dass ihre Produktion sehr aufwendig und teuer ist und die Handhabung aufgrund der begrenzten Haltbarkeit und Notwendigkeit zur kühlen Lagerung eingeschränkt ist. Die aktuelle Vakzineentwicklung hat daher zum Ziel, Impfstoffe in rekombinanter Form herzustellen. Hierdurch ist die Produktion unabhängig von der Kultivierung des Erregers und die rekombinanten Antigene können in großem Umfang bei gleichbleibender Reinheit und Qualität hergestellt werden. Gegenstand dieser Arbeit ist die Protein Disulfid Isomerase (PDI). Dieses Enzym wurde bereits bei verschiedenen Helminthen beschrieben und stellt durch 12 Einleitung seine vielfältigen Funktionen und nachgewiesene Immunogenität einen potentiellen Vakzinekandidaten dar. Unter anderem wird der PDI eine essentielle Bedeutung bei der Kutikula-Synthese der Nematoden zugesprochen. Ziel dieser Arbeit ist die Identifizierung und Charakterisierung der PDI bei D. viviparus. Weiterhin erfolgt in Immunisierungsversuchen eine Überprüfung der Immunogenität und Protektivität des Enzyms. Literaturübersicht 13 2 Literaturübersicht 2.1 Die Dictyocaulose des Rindes 2.1.1 Der Erreger Bereits 1727 wurde von RUYSCH über das Vorkommen von Lungenwürmern beim Rind berichtet. Die Erstbeschreibung erfolgte im Jahre 1782 durch BLOCH, der diesem Parasiten den Namen Gordius viviparus gab. Seinen endgültigen Namen Dictyocaulus viviparus erhielt der Rinderlungenwurm 1907 durch RAILLIET und HENRY. Dictyocaulus viviparus, der große Lungenwurm des Rindes, gehört taxonomisch der Ordnung Rhabditida und der Familie der Trichostrongylidae an. Die geschlechtsreifen Stadien leben in den Bronchioli, Bronchien und der Trachea und verursachen eine parasitäre Bronchitis. Bis heute beinhaltet die Gattung Dictyocaulus sieben Arten, die zum Teil mehrere Wirtstierspezies befallen. Hierbei zeigen die Parasiten eine unterschiedliche Anpassung an ihre Wirte, was sich unter anderem in der Ausbildung einer patenten Infektion äußert. Rinder sind für D. viviparus empfänglicher als Wildwiederkäuer (BIENIOSCHEK 1994). D. arnfieldi parasitiert in Equiden. Esel sind häufiger befallen als Pferde, zeigen jedoch seltener Symptome (GOTHE 1983; HASSLINGER 1989). D. filaria ist bei kleinen Haus- und Wildwiederkäuern zu finden (ENIGK u. HILDEBRANDT 1969; GOTHE u. KANDELS 1984). Eine erstmals bei Rentieren beschriebene Art, D. eckerti (SKRJABIN 1931), wurde aufgrund morphologischer Unstimmigkeiten lange Zeit nicht als solche anerkannt. Molekulargenetische Untersuchungen konnten jedoch die Eigenständigkeit dieser Spezies beweisen (EPE et al. 1995; EPE et al. 1997; SCHNIEDER et al. 1996b). Neben Rentieren konnten als weitere Wirte für D. eckerti Rotwild, Elche und Moschusochsen identifiziert werden (DIVINA et al. 2002; HÖGLUND et al. 2003). Eine experimentelle Übertragung von D. eckerti auf das Rind ist möglich, es besteht jedoch nur eine geringe Empfänglichkeit (BIENIOSCHEK 1994). 14 Literaturübersicht Eine neue Spezies, D. capreolus, wurde kürzlich bei Rehwild und Elchen in Schweden beschrieben (GIBBONS u. HÖGLUND 2002; HÖGLUND et al. 1999). Eine experimentelle Übertragung dieser Spezies aufs Rind war nicht erfolgreich (DIVINA u. HÖGLUND 2002). Bei afrikanischen Paarhufern existieren als weitere Arten D. cameli und D. africanus (GIBBONS u. KHALIL 1988). Des Weiteren entdeckten DIVINA et al. (2002) ein Dictyocaulus-Isolat in Damwild, das molekulargenetisch keiner bekannten Spezies zugeordnet werden konnte. 2.1.2 Entwicklungszyklus und Pathogenese Mit dem Wirtstierkot werden die 390 bis 450 µm langen, unbescheideten Erstlarven ausgeschieden, die sich in der Außenwelt unter günstigen klimatischen Bedingungen über ein einfach bescheidetes zweites Larvenstadium zur doppelt bescheideten, infektiösen dritten Larve entwickeln. Während dieser Entwicklungsphase nehmen die Larven keine Nahrung auf, sondern zehren von in Granula gespeicherten Reservestoffen (PFEIFFER u. SUPPERER 1980; TAYLOR 1951). Nach URQUHART et al. (1973) lässt sich der Krankheitsverlauf in die vier Phasen Penetrationsphase, Präpatenz, Patenz und Postpatenz gliedern, die jede für sich eine eigene Symptomatik aufweisen. Der Schweregrad der klinischen Erscheinungen wird von der initial aufgenommenen Larvenmenge bestimmt. Darüber hinaus sind geringes Alter, ein schlechter Ernährungszustand und ungünstige Witterungsbedingungen prädisponierend für schwere Erkrankungen (TAYLOR 1951). Auch ein zusätzlich vorhandener Befall mit Magen-Darm-Würmern kann zu einer erhöhten Anfälligkeit führen (PFEIFFER 1971). Die Penetrationsphase umfasst die Aufnahme der Larven bis zu ihrer Anwesenheit in der Lunge. Nach oraler Aufnahme werden die infektiösen Larven im Dünndarm durch den Kontakt mit Gallenflüssigkeit aktiviert (JØRGENSEN 1973) und durchdringen unter Verlust ihrer Scheiden die Dünndarmschleimhaut des Wirtes. Die hierdurch hervorgerufenen Läsionen führen zu einer subklinischen Enteritis. Über das Lymphgefäßsystem erreichen die Larven die Mesenteriallymphknoten, wo die Häutung zur vierten Larve erfolgt (JARRETT et al. 1957). Diese gelangen über den Ductus thoracicus und die Vena cava cranialis in die rechte Herzkammer, von wo aus sie über die Arteria pulmonalis das Kapillargebiet der Lunge erreichen. Hier Literaturübersicht 15 überwinden die Larven die arterio-alveoläre Schranke, häuten sich in den Alveolen zum geschlechtlich differenzierten, etwa 1 mm großen, fünften Larvenstadium und ereichen etwa eine Woche post infectionem die Bronchioli und Bronchien (PFEIFFER u. SUPPERER 1980). Klinisch ist in dieser Phase lediglich ein leichter Hustenreiz zu beobachten (PFEIFFER 1971). Die Präpatenzphase, auch alveolarbronchiale Phase genannt, umfasst den Zeitraum vom 8. bis etwa zum 21. Tag p.i. . Die fünften Larven zeigen ein intensives Größenwachstum und verursachen eine Entzündung der luftführenden Wege. In der zweiten Woche p.i. sind gelegentlicher Husten und ein Anstieg der Atemfrequenz feststellbar (PFEIFFER u. SUPPERER 1980). Diese Symptome können wieder abklingen oder sich bei schwerem Krankheitsverlauf verstärken, sodass Atemfrequenzen von über 70 Atemzüge pro Minute, frequenter Husten und Dyspnoe zu beobachten sind. In Abhängigkeit von der Schwere der Erkrankung kommt es zu einer mehr oder weniger weitreichenden Zerstörung des Zilienbesatzes der Bronchialepithelien mit daraus resultierender Verminderung der mukoziliären Clearence. Histologisch zeigt sich ein Ersatz des zerstörten Bronchialepithels durch hyperplastische Basalzellen, das flache Alveolarepithel wird durch kubische Typ II-Pneumozyten ersetzt. Der vermehrt gebildete Schleim obstruiert mit Zelltrümmern und Entzündungszellen die Bronchiolen, wodurch nachgeschaltete Alveolen atelektatisch werden. Klinisch tritt Nasenausfluss in Erscheinung (SCHNIEDER et al. 1989). Drei bis vier Wochen p.i. liegen die ersten geschlechtsreifen Lungenwürmer vor. Männliche Adulte sind 3 bis 4 cm, weibliche 3 bis 8 cm lang (SCHNIEDER 2006). Die Eier werden embryoniert abgelegt, gelangen über das Flimmerepithel in den Larynx und werden vom Wirtstier abgeschluckt. Ein geringer Teil wird mit dem Sputum ausgehustet (TAYLOR 1951). Während der Magen-Darm-Passage schlüpfen die ersten Larven und gelangen mit dem Kot in die Außenwelt. Die Phase der Patenz ist durch eine Verschlechterung des klinischen Bildes gezeichnet (JARRETT et al. 1957). Das Allgemeinbefinden der Tiere ist gestört. Sie zeigen Fieber, Inappetenz und Dyspnoe. Aufgrund der fortschreitend erschwerten Atmung nehmen die Tiere zur Entlastung eine sägebockartige Haltung mit nach vorne gestrecktem Kopf ein (MICHEL u. SHAND 1955). Es kommt zu Hustenanfällen und der Nasenausfluss nimmt mukösen 16 Literaturübersicht Charakter an (PFEIFFER u. SUPPERER 1980). Das histologische Bild zeigt Massen von in die Alveolen eingewanderten Entzündungszellen. Makrophagen umschließen Würmer und aspirierte Eier und fließen zu Fremdkörperriesenzellen zusammen (JARRETT et al. 1957; SCHNIEDER et al. 1989). Aufgrund der erschwerten Atmung werden die terminalen Bronchioli gedehnt und können reißen. Hierbei kommt es zur Ausbildung eines interstitiellen Lungenemphysems, das sich weiter ausdehnen kann und in schweren Fällen als subkutanes Knistern palpierbar ist (JARRETT et al. 1957). Neben echten Sekundärinfektionen können sich im geschädigten Lungengewebe auch fakultativ pathogene Keime etablieren, die bereits vor der Infektion mit Lungenwurmlarven katarrhalisch-eitrige vorhandenen Bronchopneumonie, waren. wodurch Die das Folge ist eine Infektionsgeschehen verkompliziert wird (SCHNIEDER et al. 1989). Bei laktierenden Tieren sinkt die Milchleistung und in der späten Phase der Trächtigkeit kann es zu Aborten kommen (MICHEL u. SHAND 1955). Acht bis zehn Wochen nach der Infektion sind die Parasiten eliminiert und die Krankheitssymptome klingen ab. In der Phase der Postpatenz nehmen Ausgangsgewicht die zurück Tiere wieder (JARRETT et Futter al. auf 1957). und Je kehren nach zu ihrem Ausmaß der vorangegangen Schädigung können jedoch respiratorische Symptome in Form von Husten und erhöhter Atemfrequenz auch noch mehrere Monate anhalten. Bei massiver Larvenaufnahme durch hochempfängliche Tiere kann es zu einem so schweren Verlauf kommen, dass die Tiere nach wenigen Tagen sterben (MICHEL u. SHAND 1955). Wenn Kühe plötzlich einem hohen Infektionsdruck ausgesetzt werden, z.B. durch Grasen auf stark kontaminierten Kälberweiden, kann es zum so genannten Reinfektionssyndrom kommen. Bei unzureichender Immunität gelangen viele Larven in die Lunge und führen zu einer akuten, schweren Erkrankung. Werden Larven in der Außenwelt Temperaturen unter 8°C ausgesetzt, tritt nach Aufnahme durch den Wirt eine Entwicklungshemmung im frühen präadulten Stadium ein (Hypobiose). Im nächsten Frühjahr wird die Entwicklung fortgesetzt und die Tiere scheiden erneut Larven aus (PFEIFFER u. SUPPERER 1980). Bezüglich der Literaturübersicht 17 Mechanismen der Hypobiose soll an dieser Stelle auf die Arbeit von STRUBE (2004) verwiesen werden. 2.1.3 Epidemiologie und wirtschaftliche Bedeutung Dictyocaulus viviparus ist weltweit in Gebieten verbreitet, wo es Rinder gibt und zumindest zeitweise gemäßigte Temperaturen von 15-20°C herrschen. Die Larven auf den Weiden sind empfindlich gegen Hitze und Trockenheit (JØRGENSEN 1980). Feuchte Weiden und langes Gras haben einen positiven Einfluss auf die Überlebenszeit (PFEIFFER u. SUPPERER 1980; TAYLOR 1951). Höhere Temperaturen beschleunigen die Entwicklung, sodass es bei warmer, feuchter Witterung zu einer schnellen Anhäufung infektiöser Stadien kommt. Da Rinder es vermeiden, in unmittelbarer Nähe von Kothaufen zu grasen, ist zur Aufrechterhaltung der Infektionskette eine Translokation der Larven nötig. Die Larven selbst zeigen kaum ein gerichtetes Wandervermögen, sodass dieser Prozess über belebte Vektoren wie Käfer und Vögel oder mechanisch durch Zertreten der Kotfladen erfolgt. Des Weiteren haben koprophile Pilzen der Gattung Pilobolus eine Bedeutung bei der Verbreitung der Larven. Die Sporen dieser Pilze wachsen im Rinderkot zu Hyphen aus und bilden Fruchtträger (Sporangien). Die Larven wandern auf die Sporangien oder bohren sich basal in diese ein. Bei steigenden Temperaturen und Lichteinfall explodieren die Sporangien und die Sporen werden mitsamt den Larven bis zu einem Meter vom Kotfladen weggeschleudert (DONCASTER 1981). JØRGENSEN et al. (1982) zeigten, dass bei Kälbern, die auf einer Pilobolus-freien Weide gehalten wurden, die Larvenausscheidung reduziert war und weniger klinische Symptome auftraten. Wildwiederkäuer können mit D. viviparus infiziert sein und als Erregerreservoir dienen (BIENIOSCHEK 1994). In der Regel liegen jedoch Infektionen mit D. eckerti oder D. capreolus vor. Da Rinder für D. eckerti nur eine sehr geringe und für D. capreolus keine Empfänglichkeit zeigen, besteht hierin nur eine geringe Infektionsgefahr (BIENIOSCHEK 1994; DIVINA u. HÖGLUND 2002). Berichte von Ausbrüchen und Prävalenzstudien stammen überwiegend aus den Ländern Deutschland, Niederlande, England und Schweden. Sporadisch wird über Ausbrüche in Nordamerika und Kanada berichtet, wobei dies nach Meinung von 18 Literaturübersicht JOHNSTONE (1998) nicht die tatsächlichen Verhältnisse wiederspiegelt, da der Erreger aus Unwissenheit häufig nicht diagnostiziert werde. Grundsätzlich können alle Rinder ohne belastbare Immunität erkranken. Dies sind in erster Linie erstsömmrige Kälber. Jedoch können auch ältere Tiere betroffen sein, da eine erlangte Immunität nach etwa einem halben bis einem Jahr wieder verloren geht, wenn keine Reinfektion stattfindet (DÜWEL 1971; MICHEL u. SHAND 1955). Prävalenzstudien in Norddeutschland (SCHNIEDER et al. 1993), den Niederlanden (PLOEGER et al. 2000) und Schweden (HÖGLUND et al. 2004b) zeigten, dass die Seroprävalenz bei Jährlingsherden bei etwa 40% liegt. Verschiedene Autoren berichteten in den letzten Jahren von einem vermehrten Vorkommen von Dictyocauloseausbrüchen, wobei ungewöhnlich häufig Milchkühe betroffen waren. So verzeichnete VIDA (Veterinary Investigation Diagnosis Analysis) im Jahr 1993 135 Dictyocauloseausbrüche in England und Wales, wobei es sich in 60% der Fälle um erwachsene Tiere handelte (DAVID 1993). In den Folgejahren verzeichnete VIDA einen kontinuierlichen Anstieg mit einem Peak von 543 Ausbrüchen Im Jahr 1997. Bis 2004 schwankte die Zahl der jährlichen Ausbrüche zwischen 250 und 480 (VETERINARY LABORATORIES AGENCY 2006). Diese steigende Prävalenz wird auf den exzessiven Einsatz von Anthelminthika statt der Vakzinierung und einer damit verbundenen mangelhaften Immunitätsbildung zurückgeführt (PLOEGER 2002). In Schweden wird vermutet, dass die Zunahme an ökologisch arbeitenden Betrieben mit dem damit verbundenen Verbot der prophylaktischen Behandlung zur Erhöhung der Prävalenz in den letzten Jahren geführt hat. HÖGLUND et al. (2001) fanden bei einer Prävalenzstudie auf Ökobetrieben in Schweden 80% seropositive Betriebe. In einer späteren Studie konnte dies jedoch nicht bestätigt werden. Bei der Untersuchung von zehn Herden fand sich nicht ein seropositives Tier (HÖGLUND et al. 2004b). Für die Aufrechterhaltung der Infektionskette sind vor allem zwei Faktoren von Bedeutung. Zum einen können Lungenwurmlarven unter geeigneten Bedingungen auf den Weiden überwintern (ENIGK u. DÜWEL 1962; JARRETT et al. 1955; OAKLEY 1979) und im nächsten Frühjahr von empfänglichen Tieren aufgenommen werden, zum anderen können ältere Tiere den Erreger im hypobiotischen Stadium Literaturübersicht 19 über den Winter beherbergen und so als symptomlose Ausscheider fungieren, die im nächsten Frühling die Weiden kontaminieren (MICHEL u. SHAND 1955). EYSKER et al. (1994) fanden bei einer Pävalenzstudie in den Niederlanden im Frühling bei sechs von 40 Kuhherden mindestens ein patentes Tier. Die Anzahl positiver Herden nahm im Laufe der Weidesaison ab, jedoch waren auch im August noch Ausscheider vorhanden. Die Rolle von auf der Infektionsgeschehen wird klimabedingt Weide regional überwinterten von Larven verschiedenen im Autoren unterschiedlich bewertet (POUPLARD 1968). SCHNIEDER et al. (1989) stellten in Norddeutschland fest, dass Herden, die vor dem 15.5. ausgetrieben wurden, signifikant häufiger mit Lungenwürmern infiziert waren als die später ausgetriebenen, was die Bedeutung der überwinterten Larven unterstreicht. Ebenso wurde aus England, Schottland und Belgien von überwinterten Larven als Infektionsquelle berichtet, während diese in Österreich, der Schweiz, in Dänemark, in den Niederlanden und in Schweden kaum auftreten bzw. für das Infektionsgeschehen keine große Bedeutung haben (EYSKER et al. 1994; HU et al. 2002; JARRETT et al. 1955; JØRGENSEN 1980; VERCRUYSSE et al. 1998). Typischerweise entwickeln sich im Laufe einer Weideperiode drei bis vier Lungenwurmgenerationen. Besteht anfangs ein niedriger Infektionsdruck, werden nur wenige Tiere schwach befallen und klinische Symptome bleiben aus. Diese Tiere tragen jedoch zu einer fortschreitenden Kontamination der Weiden bei, sodass es in den folgenden Generationen zu massiven Infektionen und schweren klinischen Erscheinungen bei empfänglichen Tiere kommt (DÜWEL 1971; MICHEL u. SHAND 1955). Eine hohe Besatzdichte erhöht weiterhin den Infektionsdruck (TAYLOR 1951). Aufgrund der Generationsfolge werden die meisten Dictyocauloseausbrüche im Herbst verzeichnet. Neuere Erkenntnisse zur Epidemiologie der Dictyocaulose liefern populationsdynamische Studien. Die Bestimmung populationsgenetischer Marker in Dictyocaulus-Isolaten von verschiedenen Farmen in Schweden zeigte, dass innerhalb eines Isolats eine geringe genetische Vielfalt vorhanden ist, die Populationen sich untereinander jedoch deutlich unterscheiden (HÖGLUND et al. 2004a; HÖGLUND et al. 2006; HU et al. 2002). Es konnte keine größere Ähnlichkeit 20 Literaturübersicht zwischen Isolaten von benachbarten Farmen als zwischen Isolaten von weit entfernten Farmen festgestellt werden. Aufgrund dieses geringen genetischen Flusses halten die Autoren es für wahrscheinlich, dass Dictyocauloseausbrüche im Zusammenhang mit Tiertransporten eher so interpretiert werden können, dass empfängliche Tiere in die Herde eingebracht werden, als dass der Erreger eingeschleppt wird. Populationsdynamische Studien haben auch Bedeutung für die Verbreitung von Resistenzen. Aufgrund der gefundenen populationsgenetischen Strukturen in Schweden vermuten die Autoren, dass im Falle des Auftretens einer Anthelminthikaresistenz bei Dictyocaulus, die Ausbreitung der Resistenz verzögert erfolgen würde (HÖGLUND et al. 2004a). Für andere Regionen liegen noch keine populationsgenetischen Untersuchungen vor, sodass keine Aussagen über die Allgemeingültigkeit der gefundenen Daten getroffen werden können. Es wird vermutet, dass die geringe genetische Vielfalt innerhalb der Isolate auch mit der schlechten Überwinterungsrate der Larven in Schweden zusammenhängt, da hieraus ein starker Selektionsdruck resultiert (HÖGLUND et al. 2006). Wirtschaftliche Verluste entstehen durch Todesfälle, Lebendmasseverluste, verringerte Milchleistung und allgemeine Beeinträchtigung der Leistungsfähigkeit durch chronische Lungenschäden (SCHNIEDER et al. 1989). WOOLEY (1997) kalkulierte für einen mittelschweren Ausbruch in einer Herde mit 100 Milchkühen unter der Annahme, dass alle Tiere behandelt werden, einen Verlust von 20.000£. Hierin einbezogen waren die Kosten für Milchverluste, reduzierte Fertilität, Behandlung und Totalverluste von erkrankten Tieren und durch Aborte. Allerdings erfolgte diese Kalkulation unter der Annahme, dass aufgrund der Wartezeit ein dreitägiger Milchverlust während der Behandlung erfolgt. Da mit der Substanz Eprinomectin (GIBB et al. 2005) inzwischen ein Präparat ohne Wartezeit zur Verfügung steht, würde eine Kalkulation zum jetzigen Zeitpunkt moderater ausfallen. 2.1.4 Immunität Die Ausbildung einer Immunität ist im Infektionsgeschehen der Dictyocaulose von großer Bedeutung. Nach einer Infektion wird eine belastbare Immunität entwickelt, deren Grad allerdings schon nach 6 Monaten wieder abnimmt, wenn es zwischenzeitlich nicht zu einer erneuten Infektion kommt. Dies führt dazu, dass Tiere, Literaturübersicht 21 die länger als zwölf Monate keinen Kontakt mit dem Erreger hatten, erneut an einer Infektion erkranken. Dies geschieht unabhängig vom Alter; eine Altersresistenz besteht nicht. MICHEL (1958) zeigte, dass nach einem Jahr reinfizierte Tiere zwar nicht patent wurden, sich in der Schwere der klinischen Symptome aber nicht von den erstmals exponierten Tieren unterschieden. Die Ausbildung der Immunität verläuft phasenweise. Schon im Stadium der Larvenwanderung, 8 bis 10 Tage nach Infektion, entwickelt sich eine frühe Teilimmunität, die durch die Anwesenheit der adulten Stadien verstärkt wird. PLOEGER u. EYSKER (2002) zeigten, dass die von Larven vermittelte Immunität abhängig von der aufgenommenen Larvendosis ist, jedoch die spätere Gesamtimmunität unabhängig von der Infektionsdosis ist. In einem früheren Infektionsversuch konnten EYSKER et al. (2001) zeigen, dass bereits die Applikation von 30 Larven 35 Tage vor einer Belastungsinfektion zu einer um 80% verringerten Larvenausscheidung und einer um mehr als 70% verringerten Wurmbürde führte. Die genauen Mechanismen der Immunitätsbildung und Elimination der Parasitenstadien aus der Lunge sind nicht bekannt. Eine Beteiligung humoraler Komponenten an der Immunantwort wurde schon 1957 von JARRETT et al. demonstriert. Ihnen gelang die passive Immunisierung von Rindern mit dem Serum experimentell infizierter Tiere. Auch MCKEAND et al. (1995b) zeigten, dass die Wurmbürden passiv immunisierter Meerschweinchen signifikant geringer waren als die einer Kontrollgruppe. Die Bestimmung der lokalen und peripheren Antikörperspiegel für IgG, IgG1, IgG2, IgM und IgA in einer Langzeitstudie mit drei Infektionen (Tag 0, 65, 112) zeigte einen Anstieg aller Antikörperklassen nach jeder Infektion in der bronchoalveolären Lavageflüssigkeit (BALF), wobei höchste Titer nach der dritten Infektion erreicht wurden. Im Gegensatz dazu zeigte sich, außer bei IgA, kein Anstieg der Serumantikörper nach der dritten Infektion, sondern ein kontinuierlicher Abfall. Der bei allen Infektionen vorhandene frühe Anstieg der schleimhautschützenden IgAAntikörper wird mit der Larvenmigration in Zusammenhang gebracht. Weiterhin zeigte sich eine Korrelation zwischen Larvenausscheidung und IgG2a(1). Tiere mit hohen Ausscheidungsraten hatten auch hohe IgG2a(1)-Titer, sowohl im Serum als 22 Literaturübersicht auch in der BALF. Da die IgG2-Produktion von IFNγ und IL-2 stimuliert wird, wurde vermutet, dass bei diesen Tieren eine Th1-Antwort vorherrscht (SCOTT et al. 1996). Die Bedeutung von IgE bei der Immunantwort wurde von KOOYMAN et al. (2002) demonstriert. Die Autoren zeigten, dass die gesamt IgE-Antikörperspiegel im Serum nach Reinfektion positiv mit der Protektion gegen eine Belastungsinfektion korreliert sind, wobei bereits zehn Tage nach Reinfektion ein Anstieg zu verzeichnen war. Weiterhin war die Ausbildung einer Eosinophilie positiv mit dem IgE-Spiegel, jedoch nicht mit der Protektion korreliert. Hohe IgE-Spiegel und Eosinophilie sind Ausdruck einer Th2-Immunantwort, sodass davon auszugehen ist, dass dieser Reaktionstyp bei der Infektion mit D. viviparus vorherrscht. Eine besondere Bedeutung wird in diesem Zusammenhang einem Glykanantigen mit Lewisx-Epitop auf der Oberfläche von D. viviparus beigemessen. Lewisx-Epitope sind eine Hauptkomponente der Glykokonjugate bei Schistosomen, jedoch ist D. viviparus der einzige Nematode, bei dem diese Struktur bislang nachgewiesen wurde. Lewisx-Strukturen fungieren als Immunmodulatoren, indem sie die Th2-Immunantwort fördern und so möglicherweise die zelluläre Immunantwort des Wirtes auf den Parasiten einschränken (HASLAM et al. 2000). Die lokale Cytokinexpression im Lungengewebe, in der Trachea und in den Bronchiallymphknoten nach experimenteller Infektion wurde von JOHNSON et al. (2005) untersucht. Die Autoren fanden ein gemischtes Expressionsmuster, da sowohl die Th2-Cytokine IL-4, IL-5 und IL13, als auch die Th1-Cytokine IL-p35 und IFNγ am Tag 15 nach der Infektion hochreguliert waren. Dies korrespondiert mit dem Zeitpunkt der Larvenmigration durch die Lunge. Gegen Ende des Versuchs fielen die Cytokinspiegel wieder ab und am Tag 42 zeigte sich in der Sektion, dass nur noch wenige Lungenwürmer in der Lunge vorhanden waren, also eine Erregerelimination stattgefunden haben musste. Die Serum-IgG1-Spiegel waren hingegen am Ende des Versuchs am höchsten, also negativ mit der Cytokinantwort korreliert. Ob die Th1Cytokine essentiell für die Immunität sind oder die Th2-Antwort modulieren, ist noch nicht geklärt. HAGBERG et al. (2005) untersuchten mononukleäre Zellen in der BALF. Sowohl nach einmaliger Infektion als auch nach Reinfektion waren γ/δ-TCR-Zellen signifikant Literaturübersicht 23 erhöht. Dieser Zelltyp erfüllt wichtige Funktionen bei der Regulierung der T- und BZellaktivität sowie bei der Rekrutierung eosinophiler Granulozyten und der Produktion von IgE. Eosinophile Granulozyten waren sowohl in der BALF, als auch im Blut erhöht, wobei nach Reinfektion höhere Spiegel erreicht wurden, als nach Erstinfektion. Für CD4-, CD8-, CD14- und Ig-Zellen konnten in der BALF keine signifikanten Unterschiede zwischen infizierten und nicht infizierten Tieren festgestellt werden. Des Weiteren spielen individuelle Unterschiede des Wirtes bei der Antigenerkennung eine Rolle bei der Immunantwort. BRITTON et al. (1992a) lieferten Hinweise, dass diese Individualität bei der Antigenerkennung genetischen Ursprungs ist. Die Seren vakzinierter oder infizierter Rinder wiesen eine deutliche Heterogenität im Antigenerkennungsprofil auf. Dies war auch für einen Meerschweinchen- Auszuchtstamm der Fall. Der Vergleich zweier Inzuchtstämme zeigte eine deutliche Heterogenität zwischen den Stämmen, jedoch wenig individuelle Unterschiede innerhalb der Stämme. Ähnliche Beobachtungen konnten auch MCKEAND et al. (1994a) bei Immunisierungsversuchen mit stämmen machen. Neben qualitativen Antikörperrepertoire waren die verschiedenen Meerschweinchen- und quantitativen Unterschieden im Stämme nach der Immunisierung auch unterschiedlich gut gegen eine Belastungsinfektion geschützt. Da die Inzuchtstämme identisch bezüglich des MHC Klasse I Locus waren, wurde vermutet, dass die Unterschiede auf Gene des MHC II Locus zurückzuführen sind. 2.1.5 Diagnose Die Symptome der Dictyocaulose sind nicht pathognomonisch. Leichte respiratorische Symptome sechs bis acht Wochen nach Weideaustrieb geben zwar einen Verdacht, erfordern aber weitere Nachweisverfahren. Dies sind zum einen direkte Verfahren, die den Erreger selbst nachweisen, zum anderen serologische Verfahren zum Nachweis der Immunantwort auf den Parasiten. Ein häufig angewandtes Verfahren zum Nachweis der Dictyocaulus-Larven im Kot ist das Trichterauswanderverfahren nach BAERMANN (1917), bei dem ein mit Kot beladenes Sieb in einen wassergefüllten, unten verschlossenen Trichter gelegt wird. Die aktiv aus dem Kot auswandernden Larven sammeln sich am unteren Ende des 24 Literaturübersicht Trichterhalses und können abgelassen werden. Sensitivität und Spezifität des Verfahrens hängen von der Art der Probennahme und der Durchführung der Methode ab. Negativen Einfluss auf die Sensitivität haben lange Lagerung des Kotes, feste Kotkonsistenz, hohe Temperaturen und kurze Auswanderzeiten. RODE u. JØRGENSEN (1989) zeigten, dass nach 24stündiger Lagerung bei 20°C die Auswanderrate im Vergleich zur sofortigen Probenverarbeitung um 60% reduziert ist. Des Weiteren war nach zehnstündigem Ansatz erst ein sehr geringer Teil der Larven (<10%) ausgewandert. EYSKER et al. (1994; 1997) zeigten, dass die Methode für jüngere Tiere eine höhere Sensitivität aufweist als für ältere, da bei letzteren durch die größere Kotmenge ein Verdünnungseffekt der Larven auftritt. Darüber hinaus erfolgt die Larvenausscheidung intervallweise und quantitativ unterschiedlich (BUCHWALDER 1963; DÜWEL 1971). Daher sollte zur Absicherung eines negativen Ergebnisses die Probennahme an mehreren aufeinander folgenden Tagen durchgeführt werden Ein erster Nachweis von Larven im Kot kann drei Wochen p.i. erfolgen (DÜWEL 1971). Erste serologische Nachweisverfahren wurden in den 50er Jahren entwickelt. Auf Grundlage eines aus adulten Parasiten gewonnenen Gesamtantigens war ein Titeranstieg komplementbindender Antikörper vier bis fünf Wochen nach Infektion zu verzeichnen (CORNWELL 1960; JARRETT et al. 1959). Nach zweimaliger Applikation einer Vakzine aus röntgenattenuierten Larven konnten keine oder nur sehr geringe Titer nachgewiesen werden. Patente Infektionen lassen sich ebenfalls im Agargelpräzipitationstest nachweisen. Allerdings zeigten PARFITT u. SINCLAIR (1967) Kreuzreaktionen mit D. filaria. BOKHOUT et al. konnten mittels indirektem Hämaglutinationstest einen Titeranstieg sowohl nach einer Infektion als auch nach einer Vakzinierung zeigen (BOKHOUT et al. 1979). Die ersten Enzyme Linked Immunosorbent Assays (ELISA) für D. viviparus erwiesen sich als sehr sensitiv. Mit dem von MARIUS et al. (1979) vorgestellten indirekten ELISA auf Grundlage von Adultenantigen war ein Titeranstieg drei (BOON et al. 1982) bis vier (MARIUS et al. 1979) Wochen nach Infektion nachweisbar. Unter Einsatz von somatischem Antigen aus in vitro kultivierten vierten Larven konnten BOS u. BEEKMAN ( 1985 ) bereits zwei Wochen p.i. einen signifikanten Titeranstieg Literaturübersicht 25 nachweisen. DE LEEUW und CORNELISSEN (1991) fanden für den RohantigenELISA Kreuzreaktionen mit Cooperia oncophora, Ostertagia ostertagi, Ascaris suum und Fasciola hepatica. Den Autoren gelang jedoch die Isolierung eines 17 kDaAntigens aus adulten Würmern, das im ELISA und im Western-Blot nicht mit Antiseren der genannten Spezies kreuzreagierte. Ein Titeranstieg war mit dem 17 kDa-Antigen vier Wochen p.i. nachweisbar. Der Vergleich eines RohantigenELISAs mit dem 17 kDa-ELISA und einem kompetitiven ELISA, bei dem ein gegen das 17 kDa-Antigen gerichteter monoklonaler Antikörper eingesetzt wurde, zeigte eine Sensitivität des Rohantigen-ELISAs von 99%, des 17 kDa-ELISAs von 97% und der kompetitive ELISA hatte eine 73%ige Sensitivität. Die Spezifität war beim kompetitiven und beim 17 kDa-ELISA in etwa gleich (97%), im Rohantigen-ELISA mit 67% jedoch wesentlich geringer. Die Seren vakzinierter Tiere reagierten nicht (DE LEEUW u. CORNELISSEN 1993). Der Vergleich des 17 kDa-ELISAs mit dem indirekten Hämagglutinationstest (IHA) zeigte für beide eine hohe Spezifität (99,2% und 99,6%). Die Sensitivität des ELISAs war jedoch mit 100% signifikant höher als die des IHA (78,1%). Darüber hinaus konnten mit dem ELISA bereits 28-42 Tage p.i. Antikörper nachgewiesen werden, was mit dem IHA erst nach 42-70 Tagen möglich war. Mit dem IHA konnten jedoch bis zum Ende des Experiments (Tag 196), mit dem ELISA nur bis Tag 168 Antikörper nachgewiesen werden. Weiterhin wurde der ELISA in einem Ringversuch auf seine Reproduzierbarkeit getestet. Zwischen fünf Labors konnte kein signifikanter Unterschied festgestellt werden (CORNELISSEN et al. 1997). Die erhaltenen Ergebnisse führten dazu, dass der ELISA 1995 den bis dahin eingesetzten IHA als Routinetest für die Tiergesundheitsdienste in den Niederlanden ablöste. Dieser ELISA ist mittlerweile als kommerzieller Kit unter der Bezeichnung CEDITEST erhältlich. TENTER et al. (1993) etablierten einen ELISA auf Grundlage von Adultenantigen, der sich als sehr geeignet für den Einsatz bei epidemiologischen Fragestellungen erwies. Die Kalkulation von Sensitivität, Spezifität, positivem und negativem prädiktivem Wert lieferte die sichersten Ergebnisse für einen Untersuchungszeitraum von 13 bis 17 Wochen nach Weideauftrieb. 26 Literaturübersicht Den ersten Einsatz eines rekombinanten Antigens in einem Dictyocaulus-ELISA beschrieb SCHNIEDER (1992). Hierbei handelte es sich um ein adultenspezifisches 18 kDa Antigen, das als GST-Fusionsprotein exprimiert wurde. Nach experimenteller Infektion hatte der ELISA sowohl mit Fusionsprotein (DvGST3-14) als auch mit Thrombin-geschnittenem Protein (Dv3-14) eine Spezifität von >99%. Die Sensitivität lag bei 93% (DvGST3-14) und 91% (Dv3-14). Bei Feldseren lagen Spezifität und Sensitivität bei 90 und 85% (DvGST3-14) bzw. >99 und 70% (Dv3-14). Eine Serokonversion wurde 30 Tage p.i. festgestellt. Auf Basis des rekombinanten Antigens wurde ein Diagnostik-Schnelltest entwickelt. Grundlage dieses DipstickAssays ist ein optimierter, verkürzter Immunoblot, der bereits nach 90 Minuten ein Ergebnis liefert. Das Dv3-14-Antigen ist hierbei an Filterpapierstreifen gebunden, die mit Serum oder auch mit Vollblut beprobt werden. Nach experimenteller Infektion zeigte der Test eine Spezifität und Sensitivität von mehr als 99% zwischen Tag 30 und 85 p.i.. Die mit Antigen beladenen Filterpapierstreifen zeigten nach zwölfmonatiger Lagerung bei –20°C bzw. sechswöchiger Lagerung bei 4°C keinen Verlust der Sensitivität (SCHNIEDER 1993a). Der dem Dipstick-Assay zugrundeliegende Immunoblot wird in der Routinediagnostik des Instituts für Parasitologie der Stiftung Tierärztliche Hochschule Hannover eingesetzt. In der Bestimmung der akute Phase Proteine (APP) Haptoglobin, Serum Amyloid A und Fibrinogen sehen GANHEIM et al. (2004) ein mögliches diagnostisches Instrument. In Infektionsversuchen wurde gezeigt, dass der durch die Parasiten verursachte Gewebsschaden in der Lunge zu einem signifikanten Anstieg der APP führt. Bei gleichzeitigem Vorliegen einer Eosinophilie sehen die Autoren hierin einen Indikator für das Vorliegen einer Infektion mit Lungenwürmern. Allerdings ist der Anstieg der APP dosisabhängig und niedrige Spiegel bei gleichzeitiger Eosinophilie schließen die Infektion nicht aus. 2.1.6 Bekämpfung Die Bekämpfung der Dictyocaulose kann durch weidetechnische Maßnahmen, Vakzinierung und den Einsatz von Anthelminthika erfolgen (ROMMEL u. SCHNIEDER 1989). Da eine vollständige Unterbindung der Infektion kaum möglich ist (SELMAN 1984), besteht das Ziel in der Vermeidung wirtschaftlicher Verluste. Literaturübersicht 27 Grundsätzlich und besonders in enzootischen Gebieten sollte die Ausbildung einer protektiven Immunität bei erstsömmrigen Rindern angestrebt werden. 2.1.6.1 Weidetechnische Maßnahmen Ein später Weideauftrieb kann das Infektionsrisiko signifikant senken, da überwinterte Larven absterben. Durch Mähen des ersten Aufwuchses zur Heu- oder Silageverarbeitung kann die Anzahl vorhandener Larven reduziert werden (SCHNIEDER u. BELLMER 1993). Da Feuchtigkeit für das Überleben der Larven notwendig ist, sollten Jungrinderweiden möglichst trocken sein. Die Tiere sollten über künstliche Tränken versorgt werden, da eine Verbreitung der Larven über Grabenwasser möglich ist (PFEIFFER 1971). Eine zu hohe Besatzdichte fördert das Grasen in der Nähe von Kothaufen. Durch Zufütterung kann das Risiko gesenkt werden (BELLMER 1990). In Gebieten mit geringer Inzidenz kann versucht werden, die Infektion durch Austrieb auf lungenwurmfreie Weiden zu verhindern. Ein gemeinsames Weiden von Kälbern und älteren Tieren, die als Ausscheider fungieren können, sollte grundsätzlich vermieden werden. Auch sollten Kälber nicht auf Weiden getrieben werden, die vorher von Ausscheidern beweidet wurden. Die in der Literatur angegebenen Wartezeiten reichen von fünf Wochen bis zu einem Jahr und sind stark klimaabhängig. Da die ersten Larven mindestens vier Tage brauchen, um Infektionsreife zu erlangen, kann ein Weidewechsel im Abstand von vier Tagen massiven Infektionen empfängliche Tiere vorbeugen täglich (POUPLARD kontrolliert werden, 1968). um Grundsätzlich beim ersten sollten Auftreten respiratorischer Symptome eingreifen zu können. Bei gesicherter Diagnose sollte die ganze Herde behandelt werden (SCHNIEDER et al. 1993). 2.1.6.2 Anthelminthika Für den pro- und metaphylaktischen Einsatz bei der Bekämpfung der Dictyocaulose stehen verschiedene Breitbandanthelminthika zur Verfügung. Größte Bedeutung hat hierbei die Substanzklasse der makrozyklischen Laktone mit den Vertretern Ivermectin, Doramectin, Moxidectin und Eprinomectin. Injektions- und Pour-onPräparate bewirken eine mindestens 99%ige Reduktion der Wurmbürde für etwa vier bis sechs Wochen. (EDDI et al. 1993; EPE et al. 1999; GOUDIE et al. 1993; 28 Literaturübersicht HUBERT et al. 1997; STROMBERG et al. 1999; VERCRUYSSE et al. 1997; WEATHERLEY et al. 1993; WHELAN et al. 1995). Intraruminale Boli geben den Wirkstoff kontinuierlich oder intervallweise ab, wodurch die Wirkungsdauer auf etwa drei bis vier Monate verlängert wird. (SCHNIEDER et al. 1996a). Ein intensiver Anthelminthikaeinsatz wird von verschiedenen Autoren als Ursache für ein seit den 1990er Jahren vermehrtes Auftreten von Dictyocaulosefällen bei älteren Tieren genannt (CONNAN 1993; DAVID 1993). Studien zur Immunitätsentwicklung unter Anthelminthikaeinsatz führten zu gegensätzlichen Ergebnissen, wobei eine Abhängigkeit vom herrschenden Infektionsdruck vorhanden zu sein scheint. Werden große Larvenmengen aufgenommen, erhöht sich die Chance, dass einzelne Larven die Darmwand durchdringen und die Immunitätsbildung stimulieren bevor sie abgetötet werden (PLOEGER 2002). Bei geringem Antigenstimulus unter Langzeitbehandlung wird jedoch möglicherweise keine ausreichende Immunität ausgebildet, sodass starke Infektionen zu einem späten Zeitpunkt zu massiven Erkrankungen führen können (SCHNIEDER et al. 1998). 2.1.6.3 Vakzinierung Die Grundlagen der Immunisierung von Kälbern gegen den Befall mit Lungenwürmern wurden von JARRETT et al. (1957) erarbeitet. Hierauf basierend wurde eine Lebendvakzine entwickelt, die 1959 auf den Markt kam. Dieser Impfstoff besteht aus dritten Larven, deren Virulenz durch Röntgenbestrahlung mit 400 Gy abgeschwächt ist. Das antigene Potential ist erhalten, sodass für einige Monate eine belastbare Immunität induziert wird (ECKERT 1972). Vor allem in endemischen Gebieten wird die Impfung erstsömmriger Kälber und älterer Tiere, die länger keinen Lungenwurmkontakt hatten empfohlen (PFEIFFER u. SUPPERER 1980). Eine Impfdosis enthält etwa 1000 Larven. Die Verabreichung erfolgt oral zweimal im Abstand von vier Wochen. Alle Tiere einer Weidegruppe sollten immunisiert werden. Zum Zeitpunkt der ersten Vakzinierung sollten die Kälber mindestens acht Wochen alt und gesund sein. Zwischen den Impfungen und drei bis vier Wochen danach sollten die Tiere aufgestallt bleiben (DÜWEL 1963). Die Vakzine ist für die Tiere weitgehend unschädlich. Da allerdings ein großer Teil der Larven in die Lunge einwandert und sich zum Teil zum fünften Stadium entwickelt bevor sie absterben, Literaturübersicht 29 können sieben bis zwölf Tage nach der Vakzinierung leichter Husten und eine Erhöhung der Atemfrequenz auftreten (ECKERT 1972). Die Impfung schützt die Tiere bei einer Reinfektion vor dem Auftreten klinischer Symptome, erzielt aber keine sterile Immunität. Ein Teil der Tiere scheidet in geringem Maße Larven aus, wodurch eine gewisse Weidekontamination stattfindet. Dies ist notwendig, da die durch die Impfung erlangte Immunität durch natürliche Infektionen aufgefrischt werden muss. Ohne diesen Boostereffekt würde die Immunität lediglich drei bis fünf Monate anhalten (MICHEL u. MACKENZIE 1965). Die Vakzine ist bei 2 bis 4°C zehn bis vierzehn Tage haltbar. Fehlerhafte Aufbewahrung und Verwendung überalteter Vakzine, aber auch die Impfung zu junger Tiere und ein zu langer Abstand zwischen Vakzinierung und Austrieb können zum Versagen der Impfung führen. Erkrankungen wie infektiöse Pneumonien anderer Genese oder ein starker Befall mit Magen-DarmStrongyliden können zu Störungen der Immunitätsbildung oder zu einem Impfdurchbruch führen. Deshalb sollte eine Bekämpfung der Magen-Darm-Parasiten immer mit einbezogen werden (ECKERT 1972). Im Zuge der Problematik des vermehrten Auftretens von Dictyocaulosefällen bei erwachsenen Tieren (siehe Punkt 2.1.6.2) wurde der Einsatz der Vakzine bei Rindern während der ersten Trächtigkeit und Laktation untersucht. HOLZHAUER et al. (2005) konnten keinen negativen Einfluss der Vakzine auf die Trächtigkeit und Milchproduktion feststellen. Die Larvenvakzine kann also auch bei Jungrindern sicher angewendet werden, wenn deren Immunitätsstatus fragwürdig ist und in einem Problembestand Erkrankungsfälle bei adulten Tieren aufgetreten sind. Eine Studie zum Einsatz der Larvenvakzine bei Rotwild in Neuseeland zeigte, dass bei dieser Spezies keine zufriedenstellende Effektivität gegeben ist. Weder bei den mit D. viviparus, noch den mit D. eckerti infizierte Tieren zeigten die vakzinierten Tiere eine signifikant niedrigere Larvenausscheidung. Eine gewisse Protektivität war gegeben, da die vakzinierten Tiere signifikant weniger Würmer in den Lungen aufwiesen (JOHNSON et al. 2003). Die Lungenwurmvakzine wird von der Firma Schering Plough Animal Health unter dem Handelsnamen Dictol® und von Intervet unter der Bezeichnung Bovilis® vertrieben. Zur Zeit hat die Vakzine keine Zulassung in Deutschland. 30 Literaturübersicht 2.1.6.4 Biologische Bekämpfung Ein mögliches Instrument zur biologischen Bekämpfung stellt der nematophage Pilz Duddingtonia flagrans dar. Dieser Pilz wurde aus Rinderkot isoliert und ist in der Lage, Nematodenlarven in einem Hyphennetz zu immobilisieren und zu zerstören (LARSEN 1999). HENRIKSEN et al. (1997) fanden nach Zusatz von 50.000 Chlamydosporen/g Kot eine Reduktion der D. viviparus-Larven um durchschnittlich 86%. Nach LARSEN (1999) könnte der Pilz als Futterzusatzstoff oder in Form von Lecksteinen eingesetzt werden und durch Reduktion der infektiösen Larven auf der Weide klinische Erkrankungen vermeiden. Auf Neuseeländischen Wildfarmen wurde die Beobachtung gemacht, dass Tiere, denen Chicoree gefüttert wurde, weniger wurmbelastet waren als Tiere auf normalem Grasland. Als hierfür verantwortliche Substanzen wurden kondensierte Tannine und Sesquiterpen Lakton identifiziert. Diese Stoffe haben durch einen noch nicht bekannten Mechanismus hemmenden Einfluss auf die Motilität der larvalen Stadien. Im Larven-Migrations-Inhibitions-Test führten Konzentrationen, wie sie auch bei der Fütterung erreicht werden, bis zu einer 73%igen Inhibition (MOLAN et al. 2003). 2.2 Antigene und Immunisierungsversuche bei D. viviparus Die ersten näher bei D. viviparus untersuchten Enzyme waren Proteasen. REGE et al. (1989) identifizierten in Extrakten von adulten D. viviparus eine Cysteinprotease mit der in vitro Fähigkeit Typ I-Kollagen und Hämoglobin zu lysieren. Es wurde vermutet, dass das Enzym eine Rolle bei der Nahrungsaufnahme des Parasiten spielt und eventuell am Pathomechanismus der Infektion beteiligt ist. In von BOS et al. (1989) durchgeführten Immunisierungsversuchen wurden Meerschweinchen zweimal im Abstand von vier Wochen mit je 100 µg aufgereinigter Cysteinprotease bzw. Wurmextrakt und Freund’s inkompletem Adjuvans (FIA) intramuskulär immunisiert. Zwei Wochen nach der Boosterimpfung erfolgte die Belastungsinfektion. Die Cysteinprotease-immunisierten Tiere zeigten eine signifikante Reduktion der Wurmbürde um 42%. Bei den Wurmextrakt-immunisierten Tieren war eine Reduktion um 66% vorhanden. In einem Rinderimmunisierungsversuch war dieser Effekt nicht Literaturübersicht 31 konstant reproduzierbar. BRITTON et al. (1992b) untersuchten Extrakte von dritten Larven und Adulten und lösliche Komponenten, die vom Parasiten an die Umgebung abgegeben werden (excretory-secretory-products, ES-Produkte) auf proteolytische Aktivitäten. Mit Hilfe spezifischer Inhibitoren identifizierten sie Serin-, Cystein- und Metalloproteasen, wobei die ES-Produkte eine höhere proteolytische Aktivität aufwiesen. In den Adulten-ES-Produkten wurden die Enzyme von Serum IgGAntikörpern vakzinierter oder infizierter Rinder erkannt und inhibiert, jedoch nicht in den Wurmhomogenaten. In den Larvenextrakten reagierten nur Serum IgGAntikörper vakzinierter Tiere. Dies spricht dafür, dass die somatischen Proteasen normalerweise nicht für das Immunsystem zugänglich sind, jedoch bei der Immunisierung frei werden, da attenuierte Larven vermehrt absterben. MCKEAND et al. (1995b) führten Meerschweinchenimmunisierungsversuche mit Extrakten aus adulten D. viviparus und dritten Larven sowie mit ES-Produkten von Adulten durch. Es zeigte sich, dass nur die mit Adulten-ES-Produkten immunisierten Tiere signifikant besser geschützt waren als die Kontrollgruppe. Die Meerschweinchen wurden zweimal im Abstand von vier Wochen subkutan immunisiert. In der ersten Immunisierung wurden 60 µg Protein mit einem equivalenten Volumen Freund’s komplettem Adjuvans (FCA) eingesetzt. Die zweite Vakzinedosis bestand aus 100 µg Protein mit FIA. Vier Wochen nach der zweiten Impfung erfolgte die Belastungsinfektion mit 3000 L3. Sechs Tage später wurden die Tiere seziert. In der mit ES-Produkten immunisierten Gruppe war die Anzahl fünfter Larven um 85,5% reduziert. In einer zweiten Versuchsreihe wurden Meerschweinchen passiv immunisiert, um die protektive Rolle von Antikörpern bei der Immunantwort zu untersuchen. Hierzu wurde von mit ES-Produkten immunisierten oder mit dritten Larven infizierten Meerschweinchen Serum gewonnen und anderen Meerschweinchen intraperitoneal appliziert. Eine Stunde nach der Seruminjektion wurden die Tiere mit 6000 L3 infiziert. Bei der Sektion war sowohl in der Gruppe, die das Anti-L3-Serum erhalten hatte, als auch in der Anti-ES-Gruppe die Wurmbürde signifikant reduziert. Im Vergleich zu einer Kontrollgruppe, die Serum nicht immunisierter und nicht infizierter Tiere erhalten hatte, war eine Reduktion von 79,7% bzw. 64,6% vorhanden. 32 Literaturübersicht Ein Enzym, das bereits in den ES-Produkten diverser parasitischer Nematoden nachgewiesen wurde und dem große Bedeutung bei der Immunabwehr des Wirtes beigemessen wird, ist die Acetylcholinesterase (AChE). Es konnte gezeigt werden, dass AChEs von Nematoden sezerniert werden, jedoch konnte die biologische Bedeutung der sezernierten Formen noch nicht vollständig geklärt werden. Es wird davon ausgegangen, dass durch die Hydrolyse des wirtseigenen Acetycholins Funktionen blockiert werden, die zur Parasitenabwehr beitragen. Hierzu zählen Darmperistaltik und Schleimsekretion, aber auch Effekte des Acetylcholins auf Zellen des Immunsystems wie Neutrophilenchemotaxis, Mastzelldegranulation und Lymphozytenproliferation (MCKEAND et al. 1994b; MCKEAND et al. 1995a). MCKEAND et al. (1994b) konnten in Extrakten und ES-Produkten adulter D. viviparus Acetylcholinesteraseaktivität nachweisen, wobei in ES-Produkten eine 200fach höhere Aktivität vorhanden war als in den Extrakten. Hieran zeigte sich, dass es sich bei der Acetylcholinesterase (AChE) um ein von D. viviparus sezerniertes Enzym handelt. Vergleiche mit dritten und vierten Larven wiesen darauf hin, dass das Protein erst in einem späten Entwicklungsstadium produziert wird. Bei dritten Larven konnte keine, bei vierten Larven nur eine geringe AChE-Aktivität nachgewiesen werden. Nach gelelektrophoretischer Trennung und Färbung der Extrakte und ES-Produkte wurden fünf Isoformen des Enzyms identifiziert, die alle mit Serum IgG von natürlich und experimentell infizierten Rindern reagierten, wodurch ihre Antigenität demonstriert wurde. Das Enzym reagierte jedoch nicht mit dem Serum von mit röntgenattenuierten Larven vakzinierten Kälbern, was die Autoren darauf zurückführten, dass nicht genügend Larven ein Entwicklungsstadium erreichen, in dem ausreichend AChE produziert wird, um eine Immunantwort hervorzurufen. Dass es sich bei den fünf Isoformen um Produkte verschiedener Gene handelt, konnten Sequenzanalysen und Aktivitätsessays für zwei Formen bestätigen (LAZARI et al. 2003; SELKIRK et al. 2005). Weiterhin wurde eine gewebsständige Form der AChE identifiziert (LAZARI et al. 2004). Das protektive Potential der AChE wurde in einem Immunisierungsversuch überprüft (MCKEAND et al. 1995a). Meerschweinchen wurden mit einer AChE-angereicherten Fraktion der adulten ES-Produkte immunisiert. Eine weitere Gruppe erhielt nicht angereichertes Literaturübersicht 33 ES-Produkt. Die Impfdosen enthielten 80 µg Protein beim nicht angereicherten ESProdukt und 20 µg bei der angereicherten Fraktion sowie FIA bei der ersten und FCA bei der zweiten Immunisierung. Acht Wochen nach der zweiten Impfung wurden die Tiere mit 6000 L3 infiziert. Die mit der AChE-angereicherten Fraktion immunisierte Gruppe wies eine signifikante Reduktion der Wurmbürde im Vergleich zur AdjuvansKontrollgruppe auf (-64,8%). Im Gegensatz zum vorangegangen Immunisierungsversuch mit ES-Produkten (MCKEAND et al. 1995b) zeigte jedoch die mit nichtangereicherten ES-Produkten immunisierte Gruppe keine signifikante Reduktion der Wurmbürde (-43,4%). Die Autoren führten dies auf das veränderte Versuchsprotokoll zurück. Da nicht ausreichend Larven zur Verfügung standen, erfolgte die Infektion acht statt vier Wochen nach der zweiten Immunisierung. In einem Rinderimmunisierungsversuch mit rekombinanter AChE und Adulten-ES-Produkten konnte das protektive Potential der AChE nicht bestätigt werden (MATTHEWS et al. 2001). Für diesen Versuch wurde eine 10% kürzere Teilsequenz der AChE in einen Expressionsvektor kloniert und als His-getagtes Fusionsprotein in E. coli exprimiert. Die Immunogenität des rekombinanten Proteins wurde durch die Reaktion mit dem Serum Dictyocaulus-infizierter Rinder und mit dem Serum von mit ES-Produkten immunisierten Meerschweinchen bestätigt. Die rekombinante AChE wies jedoch keine AChE-Aktivität auf. Im Immunisierungsversuch erhielt eine Gruppe zweimal im Abstand von vier Wochen 100 µg des aufgereinigten Fusionsproteins mit FIA. Eine weitere Gruppe Kälber wurde mit ES-Produkten immunisiert. Die Challengeinfektion erfolgte acht Wochen nach der zweiten Impfung. Obwohl die Tiere AChE-spezifische Antikörper bildeten, zeigte keine der beiden Immunisierungsgruppen eine Reduktion der Wurmbürde im Vergleich zur Kontrollgruppe. Die Fusionsprotein-immunisierte Gruppe hatte mit 97 Würmern sogar eine höhere mittlere Wurmbürde als die Kontrolltiere, bei denen im Durchschnitt 80 Würmer vorhanden waren. Mögliche Ursachen für den fehlgeschlagenen Versuch sehen die Autoren in der Versuchsdurchführung, aber auch im Enzym selbst. Rechtliche Grundlagen machten es notwendig, statt FCA wie in den vorhergegangen Versuchen, FIA als Adjuvans einzusetzten. Andere Autoren hatten bei einem Wechsel von FCA zu FIA ebenfalls einen Verlust des protektiven Potentials beobachtet. Weiterhin führten die Autoren 34 Literaturübersicht auf, dass es sich bei der rekombinant hergestellten AChE nicht um ein vollständiges Transkript handelte und dass die Expression in E. coli aufgrund inkorrekter Faltung zu einem Verlust wichtiger immunogener Epitope geführt haben könnte. Insgesamt halten die Autoren die AChE nicht für einen geeigneten Vakzinekandidaten. Das Vorhandensein stadienspezifischer Oberflächenantigene wurde von BRITTON et al. (1993) demonstriert. Mit dem Serum vakzinierter Rinder konnte die Oberfläche bescheideter L3 erkannt werden, jedoch nicht die von Adulten, Eiern und ersten Larven. Letztere Stadien wurden jedoch von Serumantikörpern patent infizierter Rinder detektiert. Dies zeigt, dass Adulte, Eier und erste Larven Oberflächenantigene besitzen, für die vakzinierte Tiere keine Antikörper bilden, da es hier nicht zur Ausbildung einer patenten Infektion kommt. Weiterhin demonstriert das Vorhandensein von Antikörpern gegen Obeflächenantigene von bescheideten L3, dass die infektiösen Stadien bei der Penetration der Darmwand ihre Scheide nicht abstreifen bzw. dass zumindest ein ausreichender Anteil die Scheide behält und eine Immunantwort im Wirt hervorruft. Mittels monoklonaler Antikörper identifizierten GILLEARD et al. (1995b) ein immunodominantes Antigen auf der Oberfläche der Scheide von dritten Larven. Dieses 29-40 kDa große Protein erwies sich als innerhalb strongylider Nematoden konserviert, da der monoklonale Antikörper auch Oberflächenproteine der Scheide diverser anderer Spezies detektierte. Jedoch detektierten Seren von mit gastrointestinalen Nematoden (H. contortus, C. oncophora, O. ostertagi) infizierten Tieren dieses Antigen nicht, was von den Autoren darauf zurückgeführt wird, dass diese Parasiten während ihres Lebenszyklus im Verdauungstrakt verbleiben (GILLEARD et al. 1995a). Ob dieses Antigen ein effektives Ziel der Immunabwehr des Wirtes ist oder, da die Scheide frühzeitig während der Migration abgestreift wird, Teil der Strategie des Parasiten ist, den Immunmechanismen auszuweichen, konnte nicht geklärt werden. Ein weiteres Antigen, dem eine wichtige Funktion im Infektionsgeschehen beigemessen wird, wurde von BRITTON et al. (1994) identifiziert. Die Autoren wiesen in ES-Produkten und Extrakten von Adulten und dritten Larven SuperoxidDismutase-Aktivität Umwandlung von nach. zwei Superoxid-Dismutasen Superoxidanionen (O2-) (SOD) in katalysieren die Wasserstoffperoxid und Literaturübersicht 35 molekularen Sauerstoff. Superoxidanionen werden während des „oxidatory burst“ von Phagozyten freigesetzt und schädigen Krankheiterreger durch die Oxidation von Membranen, Proteinen und Nukleinsäuren. Es wird davon ausgegangen, dass Parasiten SOD synthetisieren, um diese toxischen Substanzen zu neutralisieren. Bei D. viviparus identifizierten BRITTON et al. (1994) mehrere Isoformen des Enzyms, die alle ein Kupfer-Zink-Abhängigkeit aufwiesen. Nur eine Isoform war sowohl in den ES-Produkten als auch in den Adulten- und Larven-Extrakten vorhanden. Eine weitere, nur in ES-Produkten vorhandene Isoform wies darauf hin, dass die SOD tatsächlich von D. viviparus sezerniert wird. Die SOD-Aktivität konnte durch SerumIgG von infizierten und vakzinierten Rindern inhibiert werden. Ein immunodominantes Protein mit möglicher Rolle bei der Lungenpathologie im Verlauf der D. viviparus Infektion wurde von BRITTON et al. (1995) in somatischen Extrakten und ES-Produkten identifiziert und charakterisiert. Das DvA-1 (D. viviparus Antigen-1) weist Ähnlichkeiten zum ABA-1 von A. suum auf. Von diesem ist bekannt, dass es zu den Hauptallergenen des Parasiten gehört und eine IgE-Antwort induziert. Ferner konnte bei Brugia malayi infizierten Patienten eine Korrelation zwischen IgE-Reaktion auf das Allergen und der Entwicklung pathologischer Veränderungen gezeigt werden. In einem Schafmodel zur Untersuchung der lokalen Immunmechanismen in der Lunge nach Antigenapplikation zeigten COLLIE et al. (2001), dass Schafe, die vorher durch systemische Applikation des rekombinanten DvA-1 sensibilisiert worden waren, auf die lokale Applikation des Antigens mit einem starken Anstieg an neutrophilen und eosinophilen Granulozyten in der BALF sowie mit einer lokalen Antikörperproduktion reagierten (siehe auch Punkt 2.1.4). Dies bestätigt die Rolle des DvA-1 im Pathomechanismus der Lungenwurminfektion. Als Vakzinekandidat ist dieses Antigen wahrscheinlich nicht geeignet, da mit einer Sensibilisierung gerechnet werden muss. BRITTON u. MURRAY (2002) identifizierten bei D. viviparus eine innerhalb der Nematoden konservierte Cathepsin L Protease. In C. elegans konnte gezeigt werden, dass dieses Enzym essentiell für die Embryonalentwicklung ist. RNAInterferenz (RNAi) des Gens führte bei 98% der Embryonen zum Stillstand der 36 Literaturübersicht Embryonalentwicklung in einer frühen Phase (HASHMI et al. 2002). Ob die Cathepsin L Protease immunogenes Potential hat, wurde nicht gezeigt. KÖHRMANN (2002) immunisierte Rinder mit in E. coli exprimiertem Major Sperm Protein (MSP) (SCHNIEDER 1993b). Die Wurmbürde war in der immunisierten Gruppe um 71% niedriger. Dieses Ergebnis war jedoch aufgrund der geringen Tierzahl in der Impfgruppe (n = 3) statistisch nicht signifikant. Um neue Vakzinekandidaten zu identifizieren, trennten MATTHEWS et al. (2004) Adulten-ES-Produkte mittels zweidimensionaler Gelelektrophorese auf und testeten diese im Immunoblot. Hierbei wurden Seren von drei verschiedenen Meerschweinchenstämmen eingesetzt, die nach Immunisierung mit ES-Produkten eine Unterschiedliche Empfänglichkeit für Belastungsinfektionen gezeigt hatten. Durch Vergleich der von den Seren im Immunoblot erkannten Spots wurden sieben Spots identifiziert, die nur mit den Seren von Meerschweinchen reagierten, die nach einer Immunisierung mit ES-Produkten signifikant geschützt waren. Bei der MALDITOF-Analyse dieser Spots wurde ein Protein als Major Sperm Protein, MSP; (SCHNIEDER 1993b) identifiziert. Die Fingerprints der übrigen Proteine zeigten keine signifikanten Ähnlichkeiten mit Sequenzen anderer Nematoden. Die Autoren beabsichtigen Proteinsequenzierungen dieser Spots durchzuführen, um degenerierte Primer für die Amplifikation der Kandidaten konstruieren zu können (MATTHEWS et al. 2004). 2.3 Die Protein Disulfid Isomerase (PDI) Die Protein Disulfid Isomerase wurde erstmals von GOLDBERGER et al. (1963) in Rattenleberlysaten identifiziert. Nachdem die PDI auch aus Humangeweben und diversen weiteren eukaryotischen Spezies isoliert werden konnte, wird heute davon ausgegangen, dass es sich um ein ubiquitär vorhandenes Protein handelt. Die PDI ist ein multifunktionales Enzym mit Hauptfunktionen bei der Ausbildung und Modifikation von Disulfidbrückenbindungen sowie als molekulares Chaperon und Untereinheit zweier Enzymkomplexe. Literaturübersicht 37 2.3.1 Struktur der PDI und PDI-ähnlicher Enzyme Neben der PDI existieren eine Reihe strukturell und funktionell ähnlicher Enzyme, die in der Familie der Protein Disulfid Isomerasen zusammengefasst werden. Die Enzymfamilie der humanen PDIs zählt bis heute 14 Mitglieder und drei weitere, nicht publizierte Sequenzen sind in Datenbanken zu finden (ELLGAARD u. RUDDOCK 2005). Aufgrund der Sequenzidentität der in dieser Arbeit identifizierten PDIs soll im folgenden Überblick in erster Linie auf die PDI selbst und auf das nahe verwandte Enzym Erp57 intensiver eingegangen werden. Allen Protein Disulfid Isomerasen gemeinsam ist das Vorkommen mindestens einer Thioredoxin-ähnlichen Domäne, womit sie zur Thioredoxin-Superfamilie gehören. Thioredoxin ist ein ubiquitär vorkommendes Protein mit einer Größe von etwa 100 Aminosäuren. Das Enzym besteht aus einer einzelnen Domäne mit einer zentralen fünfsträngigen β-Faltblattstruktur, die von vier α-Helices umgeben ist. Die beiden Cysteine des aktiven -Cys-Gly-Pro-Cys-Zentrums können eine intramolekulare Disulfidbrücke ausbilden, womit das Molekül an diversen Redoxreaktionen beteiligt ist (MARTIN 1995). Bereits die erste Sequenzierung der PDI aus Rattenleber-cDNA durch Edman et al. (1985) führte zur Aufstellung eines Vier-Domänen-Modells. Es wurden zwei Sequenzbereiche mit hoher Ähnlichkeit zueinander und zu Thioredoxin identifiziert. Diese als a und a’ bezeichneten Bereiche wiesen beide die Aminosäurenabfolge -Trp-Cys-Gly-His-Cys-Lys- auf, und es wurde vermutet, dass es sich um die aktiven Zentren des Enzyms handelt. Des Weiteren fanden sich im mittleren Teil des Proteins zwei Sequenzbereiche b und b’, die gewisse Ähnlichkeit zueinander aufwiesen, für die jedoch zuerst keine Identität zu anderen Proteinen identifiziert werden konnte. Die Autoren konnten mit ihrer Strukturanalyse nicht das gesamte Protein abdecken. Weitere Studien führten zur Aufstellung eines SechsDomänen-Modells mit der Struktur a-e-b-b’-a’-c (FREEDMAN et al. 1994). Aufgrund der Identität zu einer Östrogen-bindenden Domäne des humanen Östrogenrezeptors wurde zwischen der a- und der b-Domäne eine e-Domäne vermutet. Dieses Konzept wurde jedoch wieder verworfen und zur ursprünglichen vier-Domänen-Struktur zurückgekehrt, wobei die Domänengrenzen allerdings anders als ursprünglich definiert wurden (KEMMINK et al. 1997). Zusätzlich zu diesen vier Domänen findet 38 Literaturübersicht sich am C-Terminus ein Sequenzbereich überwiegend saurer Aminosäurereste (PIHLAJANIEMI et al. 1987). Die biologische Bedeutung dieser als c-Domäne bezeichneten Struktur ist nicht bekannt. Am C-Terminus befindet sich weiterhin die Sequenz –KDEL, (HAUGEJORDEN bei et der al. es sich 1991). um ein Klonierung ER-Retentionssignal der einzelnen handelt Domänen und Aktivitätsstudien konnten schließlich bestätigen, dass es sich bei den schon von Edman et al. 1985 beschriebenen aktiven Zentren um eben solche handelt (HAWKINS u. FREEDMAN 1991). Kernresonanzspektroskopie (NMR, nuclear magnetic resonance) brachte weitere Einblicke in die Struktur der Domänen. KEMMINK et al. lieferten 1996 ein dreidimensionales Bild der a-Domäne, 1997 folgte die Darstellung der b-Domäne (KEMMINK et al. 1996; KEMMINK et al. 1997). Die a-Domäne zeigt eine typische Thioredoxin-ähnliche β-Faltblatt-α-Helix-Struktur: β-α-β-α-β-α-β-β-α. Obwohl die bDomäne keinerlei Sequenzidentitäten zu Thioredoxin aufweist, zeigt auch sie einen diesem Molekül ähnlichen dreidimensionalen Aufbau. Aufgrund der Sequenzidentität der a- und a’-Domänen sowie der b- und b’-Domänen ist es wahrscheinlich, dass die PDI aus vier Thioredoxin-ähnlichen Strukturen besteht, von denen zwei katalytisch aktive Zentren besitzen (KEMMINK et al. 1997). Die Bedeutung des Vorkommens katalytisch inaktiver Strukturen konnte noch nicht vollständig geklärt werden, es konnte jedoch gezeigt werden, dass sich in der b’-Domäne eine Peptidbindungsstelle befindet. Phylogenetische Arbeiten zeigten, dass die molekulare Evolution der PDIs von einem Enzym mit zwei Thioredoxin-Domänen ausging (KANAI et al. 1998; MCARTHUR et al. 2001; PEDONE et al. 2004). Die vier-Domänen-Struktur entwickelte sich möglicherweise aus internen Genverdoppelungen, die im Laufe der Zeit ihre Homologie verloren, aber die Faltung beibehielten. Bis heute existiert kein vollständiges dreidimensionales Bild der PDI. Die Analyse der genomischen Sequenz zeigte, dass die humane PDI aus elf Exons und zehn Introns besteht (TASANEN et al. 1988). Die anderen Mitglieder der PDI-Familie unterscheiden sich von der PDI im Vorkommen und der Anzahl katalytisch aktiver und inaktiver Domänen. Literaturübersicht 39 Die folgende Abbildung zeigt einen Überblick aller bis heute identifizierten Mitglieder der humanen PDI-Familie. a CGHC b b‘ a‘ CGHC CGHC KDEL PDI CGHC CGHC QEDL ERp57 CGHC CTHC KEEL PDIp CGHC CGHC KEEL ERp72 SKQS SKKG KEEL PDILT KVEL ERp27 KEEL PDIr KEEL ERp28 KDEL ERdj5 KDEL P5 EDEL ERp18 RDEL ERp44 KDEL ERp46 ? TMX KKDK TMX2 KKKD TMX3 ? TMX4 CSMC CSHC CPPC CGPC CGHC CHPC CPHC CGPC CGPC CGPC CRFS CGHC CGHC CPAC SNDC CGHC CPAC Abb. 1: CGHC Schematische Darstellung der PDI-Familie. (Modifiziert nach ELLGAARD u. RUDDOCK 2005). 40 Literaturübersicht ERp57 (in älterer Literatur und bei anderen Spezies auch als ERp60 bezeichnet) weist die größte Ähnlichkeit zur PDI auf. Die Domänenorganisation ist gleichartig gestaltet (FERRARI u. SOLING 1999). Dabei weisen die a und a’-Domänen ebenfalls die aktiven Zentren -CGHC- auf, ERp57 fehlt jedoch die C-terminale saure Sequenz und die ER-Retentionssequenz weist die Variante –QEDL auf. PDIp ist eine im Pankreas vorkommende Isoform der PDI mit 66%iger Ähnlichkeit zur PDI auf Aminosäurenebene (DESILVA et al. 1996) und gleicher Domänenstruktur (FERRARI u. SOLING 1999), wobei jedoch ein aktives Zentrum zu -CTHCmodifiziert ist. Weitere Unterschiede bestehen im ER-Retentionssignal, welches bei PDIp die Sequenz –KEEL aufweist (DESILVA et al. 1996). Während in älterer Literatur davon ausgegangen wird, dass es sich bei der PDIp um eine pankreasspezifische Isoform handelt, konnten CONN et al. (2004) die PDIp in dopaminergen Neuronen im menschlichen Gehirn nachweisen. Eine erhöhte PDIpExpression wird hier mit dem Auftreten von Lewy-Körperchen bei der Parkinson Krankheit in Verbindung gebracht. ERp72 weist am 5’-Ende eine zusätzliche a-Domäne mit gleichem -CGHC-Motiv auf. Am N-Terminus befindet sich ein Bereich aus 42 vorwiegend sauren Aminosäuren (FERRARI u. SOLING 1999). ERp27 und ERp28 weisen keine a-Domänen-ähnlichen Bereiche auf. NMRStrukturanalysen zeigten für das ERp28-Homolog in der Ratte (ERp29) eine bDomänen-ähnliche Faltung am 5’-Ende und einen C-terminalen Bereich mit rein helicaler Struktur. Im Menschen hat das ERp28 ein Molekulargewicht von 28 kDa (LIEPINSH et al. 2001). Die von HAYANO u. KIKUCHI 1995 identifizierte PDIr weist drei a-Domänenähnliche Strukturen auf, von denen lediglich eine die -CGHC-Sequenz der PDI besitzt. ERjd5 (CUNNEA et al. 2003) weist vier der a-Domäne ähnliche Bereiche auf. Zusätzlich befindet sich N-terminal ein Sequenzbereich mit Homologie zu DnaJ. DnaJs sind Proteine, die funktionell mit Chaperonen kooperieren. Die übrigen Mitglieder der PDI-Familie weisen eine bis drei a-Domänen auf, wobei die aktiven Zentren zum Teil Modifikationen aufweisen. Im ERp44 ist das C- Literaturübersicht 41 terminale, im TMX2 das N-terminale Cystidin gegen Serin getauscht. Die zentralen Aminosäuren werden bei ERp44 durch Arginin (R) und Phenylalanin (F), bei TMX durch Prolin (P) und Alanin (A), bei TMX2 durch Asparagin (N) und Asparaginsäure (D) und bei TMX4 durch Prolin (P) und Serin (S) verkörpert. 2.3.2 Funktionen der PDI Die Erstbeschreibung durch GOLDBERGER et al. (1963) identifizierte die PDI als den Katalysator der Disulfidbrückenbildung bei der Faltung von RNAse A. Inzwischen wurden diverse weitere Funktionen dieses ubiquitär vorhandenen Enzyms beschrieben. Neben Oxidation, Reduktion und Isomerisation erfüllt die PDI auch redoxunabhängige Funktionen bei der Faltung von Proteinen. Des Weiteren fungiert sie als Untereinheit zweier Enzymkomplexe. 2.3.2.1 Redox-Isomerase-Funktion der PDI Die Grundlage der Redoxaktivität der PDI bilden die aktiven Zentren der a- und a’Domäne. Die beiden Cysteine des -CGHC-Motivs können entweder eine intramolekulare Disulfidbindung ausbilden (oxidierte PDI), oder als Dithiol vorliegen (reduzierte PDI). Oxidierte PDI vermag Reduktionsäquivalente von einem Dithiol aufzunehmen und kann somit Disulfidbrücken in ein Substratprotein einfügen. Reduzierte PDI kann umgekehrt eine Disulfidbindung angreifen und entweder das Disulfidsubstrat zu einem Dithiol reduzieren oder in einer Isomerisationsreaktion zu einer intramolekularen Verschiebung der Disulfidbindung führen (HAWKINS u. FREEDMAN 1991). Welcher Reaktionstyp wann in vivo stattfindet, konnte noch nicht abschließend geklärt werden (ELLGAARD u. RUDDOCK 2005), fest steht jedoch, dass diverse Faktoren hierauf Einfluss nehmen. Zum einen spielt das Redoxpotential der Umgebung und damit das zur Verfügungstehen von Reduktionsequivalenten eine Rolle. Andererseits nimmt die Struktur der aktiven Seite selbst, vor allem die Identität der beiden Aminosäuren zwischen den Cysteinen und Aminosäuren in der unmittelbaren Umgebung der aktiven Seite, Einfluss auf das Redoxpotential des aktiven Zentrums. Studien zeigten, dass eine Mutation der aktiven -CGPC-Seite des Thioredoxins, das seinerseits eher als Reduktant wirkt, zur aktiven -CGHC-Seite der 42 Literaturübersicht PDI die oxidativen Fähigkeiten des Thioredoxins zehnfach erhöht (LUNDSTROM et al. 1992). Mutationsstudien der a und a’ aktiven Seiten zeigten, dass jedes Aktivitätszentrum 50% der Gesamtaktivität der PDI ausmacht, was für ein unabhängiges Agieren der beiden Zentren spricht (VUORI et al. 1992a). Es zeigte sich jedoch, dass ein Vorhandensein aktiver Domänen nicht für das volle Reaktionsspektrum ausreicht. Indem sie die katalytischen Aktivitäten der Einzeldomänen und von verschiedenen Domänenkombinationen untersuchten, zeigten DARBY et al. (1998), dass einfache Oxidationsreaktionen nur der a- und a’-Domäne bedürfen, für einfache Isomerisationen zusätzlich die b’-Domäne benötigt wird, und Isomerisationen, die einen ausgeprägten Konformationswechsel im Substrat erfordern, nur bei Vorhandensein aller vier Domänen vonstatten gehen. 2.3.2.2 Die PDI als Untereinheit der Kollagen-Prolyl-4-Hydroxylase (C-P4H) Das Enzym Kollagen-Prolyl-4-Hydroxylase katalysiert die für die Ausbildung der Tripelhelixstruktur von Kollagenen essentielle Hydroxylierung von Prolinen (MYLLYHARJU 2003). In Vertebraten stellt das Enzym ein Tetramer aus zwei α- und zwei β-Untereinheiten dar. Das Gesamtmolekulargewicht des Komplexes beträgt ca. 240 kDa, wobei auf die α-Untereinheiten je 63 kDa, auf die β-Untereinheiten je 55 kDa entfallen. In Vertebraten existieren drei verschiedene Isoformen der αUntereinheiten: α(I) , α(II) (ANNUNEN et al. 1997; HELAAKOSKI et al. 1995) und α(III) (VAN DEN DIEPSTRATEN et al. 2003), die jeweils mit der gleichen βUntereinheit Tetramere bilden. Als Hauptform tritt in den meisten Geweben das [α(I)]2β2-Tetramer, auch als Typ I C-P4H bezeichnet, auf, während die Typ II C-P4H, ein [α(II)]2β2-Tetramer, nur etwa 30% ausmacht (ANNUNEN et al. 1997). 1987 wurde herausgefunden, dass es sich bei der β-Untereinheit um PDI handelt (KOIVU et al. 1987; PIHLAJANIEMI et al. 1987). Die isolierten Untereinheiten hatten keine C-P4HAktivität, und es konnte durch Inaktivierung der aktiven Seiten der PDI gezeigt werden, dass für eine vollständige C-P4H-Aktivität keine Redox-Isomerase-Aktivität der PDI notwendig ist. Jedoch entfaltet die intakte PDI im C-P4H-Komplex 50% der Wildtyp-Aktivität in der Reaktivierung von „scrambled“ RNAse (reduzierte und denaturierte RNAse) (KOIVU et al. 1987). Dies konnte durch Inaktivierung der C- Literaturübersicht 43 terminalen aktiven Seite der PDI verhindert werden, nicht jedoch wenn die Nterminale aktive Seite inaktiviert wurde. Dies spricht dafür, dass das N-terminale aktive Zentrum der PDI in P4H-Komplex nicht zugänglich ist. Diese Fakten und die Tatsache, dass die isolierten α-Untereinheiten unlösliche Aggregate bilden und inaktiv sind (HELAAKOSKI et al. 1995; VEIJOLA et al. 1994; VUORI et al. 1992b; VUORI et al. 1992c), sprechen dafür, dass die PDI im C-P4H-Komplex in erster Linie die Funktion erfüllt, die α-Untereinheiten in löslicher Form zu halten. Darüber hinaus wird vermutet, dass die PDI für das Zurückhalten der C-P4H im Endoplasmatischen Retikulum verantwortlich ist, da die α-Untereinheiten selbst kein ER-Retentionssignal besitzen (VUORI et al. 1992c). VEIJOLA et al. (1996b) vermuten jedoch, dass die PDI noch weitere Aufgaben bezüglich der Struktur und Funktion der C-P4H haben muss. Ihnen gelang mit der Koexpression der α-Untereinheiten zusammen mit dem molekularen Chaperon BiP die Formung eines löslichen Komplexes, der aber keine Prolyl-4-Hydroxylase-Aktivität besaß. In diesem Zusammenhang zeigte PIRNESKOSKI (2003), dass für die Bildung eines aktiven Tetramers nur die b’- und a’-Domäne notwendig sind, eine volle Aktivität aber nur mit Konstrukten erreicht wird, die alle Domänen beinhalten. Die essentielle Bedeutung der PDI bei der Kollagensynthese konnte im Nematoden Caenorhabditis elegans gezeigt werden (siehe Kapitel 2.3.4). 2.3.2.3 Chaperon-Funktion und Anti-Chaperon-Funktion der PDI WANG u. TSOU stellten 1993 erstmals die Hypothese auf, dass die PDI neben der Redox-Isomerase-Funktion auch ungefalteten als Proteinen als Chaperon Faltungshelfer, fungiert. wobei Chaperone nur dienen nicht-kovalente Wechselwirkungen beeinflusst werden. Durch das spezifische Interagieren mit aggregationsanfälligen Proteinen wird deren Aggregation verhindert. Die Arbeitsgruppe lieferte weitere Beweise für die Chaperon-Funktion der PDI, indem sie den Einfluss der PDI auf die Reaktivierung zweier Proteine, die keine Disulfide enthalten, zeigten: GAPDH (CAI et al. 1994) und Rhodanese (SONG u. WANG 1995) zeigen beide nach Denaturierung nur eine langsame, spontane Reaktivierung. Im Beisein von PDI wird die Ausbeute an renaturiertem Protein stark erhöht. Weiterhin zeigte die Arbeitsgruppe, dass dieser Effekt unabhängig vom aktiven 44 Literaturübersicht Zentrum ist, indem sie die Cysteine der -CGHC-Sequenz durch Alkylierung inaktivierten (QUAN et al. 1995). Darüber hinaus zeigten sie, dass die Chaperonaktivität der PDI auch bei der Faltung von Proteinen, die Disulfidbindungen enthalten, von Bedeutung ist (YAO et al. 1997). Um die Funktion als Faltungshelfer ausführen zu können, muss die PDI in der Lage sein, Peptide zu binden oder mit ihnen zu interagieren. Erste Berichte über eine mögliche Peptidbindungsstelle finden sich bei NOIVA et al. (1993). Diese sollte die letzten drei Aminosäuren der a’-Domäne und den größten Teil der c-Domäne beinhalten. In diesem Zusammenhang demonstrierten DAI u. WANG (1997), dass die Deletion der c-terminalen 51 Aminosäuren der PDI die Rückfaltung denaturierten Lysozyms verhindert. Zu einem anderen Ergebnis kamen PIRNESKOSKI et al. (2004). Hier wurde in Deletionsstudien gezeigt, dass die c-Domäne keine Rolle bei den Funktionen der PDI als Chaperon spielt, sondern das Ende der a’-Domäne essentiell ist. Eine zweite Peptidbindungsstelle wurde 1998 von KLAPPA et al. identifiziert. Sie konnten zeigen, dass die b’-Domäne ausreicht, um kurze Peptide zu binden, die Bindung langer Substrate jedoch das Vorhandensein der anderen Domänen erfordert. Ein 28-Aminosäurenfragment konnte von einem b’-a’-c- oder ab-b’-Konstrukt , „scrambled“ RNAse jedoch nur von einem a-b-b’-a’-Konstrukt effektiv gebunden werden. Die Chaperon-Funktion der PDI verhindert eine Aggregation von Proteinen. Allerdings scheint die PDI unter bestimmten Bedingungen einen gegenteiligen Effekt zu haben. PUIG u. GILBERT (1994) demonstrierten, dass reduziertes, denaturiertes Lysozym in der Gegenwart niedriger Konzentrationen PDI stark aggregiert, hohe PDI-Konzentration jedoch zu einer effektiven Faltung führen. Es wird vermutet, dass bei Proteinen, die eine Eigentendenz zur Selbstassoziation besitzen, niedrige PDIKonzentrationen die Quervernetzung fördern. Die Autoren sehen hierin eine Analogie zur klassischen Immunopräzipitation. Bei hohen PDI-Konzentrationen - analog zum Antikörperexzess - erhöht sich die Wahrscheinlichkeit, dass ein ungefaltetes Protein an ein noch freies PDI-Molekül bindet und eine Quervernetzung verhindert wird. Literaturübersicht 45 2.3.2.4 Die PDI als Untereinheit des mikrosomalen Triglycerid-TransferProteins (MTP) WETTERAU et al. (1990) identifizierten die kleinere Untereinheit des mikrosomalen Triglycerid-Transfer-Proteins (MTP) als PDI. Das MTP ist ein αβ-Heterodimer mit Funktionen im Triglyceridtransport zwischen Membranen. Wie bei der C-P4H scheint die PDI auch hier den Komplex in Lösung zu halten, denn Studien mit den isolierten Untereinheiten zeigten, dass die PDI keine Lipidtransferaktivität besitzt und die α-Untereinheit allein funktionslos ist und unlösliche Aggregate bildet (WETTERAU et al. 1991). LAMBERG et al. (1996) zeigten fernerhin, dass die Aktivität des MTP-Dimers unabhängig von der RedoxFunktion der PDI ist, indem sie die aktiven Seiten der PDI zu -SGHC- modifizierten. Die Koexpression dieser redoxinaktivierten PDI und der α-Untereinheit resultierte in einem aktiven Dimer. Bezüglich der Rolle der PDI bei der Assoziation der beiden Untereinheiten zeigten RICCI et al. (1995), dass die C-terminalen 30 Aminosäuren essentiell für die Interaktion sind. Zusätzlich fanden WETTERAU et al. (1990) heraus, dass die PDI im MTP nur sehr wenig Redox-Isomerase-Aktivität bei der Reaktivierung reduzierter, denaturierter RNAse entfaltet. Dies könnte darauf hindeuten, dass die aktiven Zentren der PDI im MTP-Komplex nicht zugänglich sind. 2.3.2.5 Funktionen des ERp57 Trotz hoher Ähnlichkeiten zur PDI, konnte gezeigt werden, dass ERp57 nicht in der Lage ist, die PDI im C-P4H-Komplex strukturell oder funktionell zu ersetzen (KOIVUNEN et al. 1996). OLIVER et al. zeigten, dass ERp57 eine Rolle bei der Faltung von Glycoproteinen spielt. (OLIVER et al. 1997). Ein in den Nematoden C. elegans (NATSUKA et al. 2001) und Dirofilaria immitis (CHANDRASHEKAR et al. 1998) identifiziertes ERp57-Homolog weist neben der PDI-Aktivität noch eine Transglutaminaseaktivität auf. Transglutaminasen katalysieren die Quervernetzung von Glutamin- und Lysinresten, (ESCHENLAUER u. PAGE 2003). was zur Gewebestabilisierung beiträgt 46 Literaturübersicht 2.3.2.6 Weitere Funktionen der PDI YAMAUCHI et al. (1987) fanden heraus, dass ein von CHENG et al. (1986) identifiziertes Trijodthyronin-bindendes Protein (THBP) identisch mit der PDI ist. Die physiologische Bedeutung dieser Entdeckung ist unklar. Weiterhin zeigt die PDI ein hohes Kalziumbindungsvermögen (MACER u. KOCH 1988). KRISHNA RAO u. HAUSMAN (1993) zeigten, dass die PDI identisch mit Retina-Cognin ist, einem Protein, das in die Zellerkennung und neuronale Differenzierung der embryonalen Retina involviert ist. 2.3.3 Lokalisation der PDI außerhalb des ER Trotz des vorhandenen ER-Retentionssignals wurden die PDIs PDI, ERp57 und ERp72 auch in nicht-ER-Lokalisationen identifiziert. Für die PDI wurde eine Sekretion aus Hepatozyten (TERADA et al. 1995), exokrinen Pankreaszellen (YOSHIMORI et al. 1990), Endothellzellen (HOTCHKISS et al. 1998) und Blutplättchen (CHEN et al. 1992), für ERp72 und ERp57 von Zellkulturen bei Überexpression beschrieben (DORNER et al. 1990; HIRANO et al. 1995). Wie die PDIs trotz des Retentionssignals aus dem ER entweichen können, ist nicht bekannt. Verschiedene Mechanismen werden diskutiert, wie z.B. Übersättigung des Rückhalteapparates, Entfernung der KDEL-Sequenz oder Sekretion durch Komplexbildung mit anderen Molekülen (JOHNSON et al. 2001). Des Weiteren wurden PDIs bei verschiedenen Zellen auf der Zelloberfläche detektiert. TERADA et al. (1995) vermuten, dass die PDI nach Sekretion durch elektrostatische Interaktionen an die Zelloberfläche bindet. Grundsätzlich scheinen die Thioredoxindomänen auf der Zelloberfläche reduzierende Aktivität zu entfalten, so dass Disulfidbrücken in Molekülen, die auf der Zelloberfläche vorhanden sind oder mit dieser in Kontakt kommen, reduziert oder isomerisiert werden (MANDEL et al. 1993). Obwohl die Rolle des daraus resultierenden vermehrten Vorkommens an Thiolen an der Zelloberfläche nicht geklärt ist, konnte ihre Bedeutung bei verschiedenen physiologischen und pathologischen Mechanismen beschrieben werden. So ist z.B. die Aktivierung von Blutplättchen von einem Auftreten von Thiolgruppen abhängig (BURGESS et al. 2000). Eine pathologische Bedeutung konnte beim Zellinvasionsmechanismus des Literaturübersicht 47 HIV-Virus gezeigt werden. Dieser ist von der Reduktion von Disulfidbrücken eines viralen Hüllglycoproteins abhängig und die PDI konnte bei dieser Reaktion als reduzierendes Agens identifiziert werden (FENOUILLET et al. 2001). Des Weiteren konnte ERp72 auf der Neutrophilenoberfläche (WEISBART 1992) und ERp57 auf der Spermienmembran (BOHRING et al. 2001) lokalisiert werden. Außerdem wurden für die PDI und ERp57 Lokalisationen im Cytosol und im Nucleus beschrieben (TURANO et al. 2002). 2.3.4 PDIs bei Helminthen Die erste Beschreibung einer PDI bei Nematoden findet sich bei WILSON et al. (1994). Die Autoren isolierten beim Durchmustern einer Onchocerca volvulus cDNABank mit L3-Antiserum aus Kaninchen einen Klon mit hoher Ähnlichkeit zur PDI anderer Spezies. Der offene Leserahmen hat eine Länge von 1491 bp und es konnten die bei anderen PDIs beschriebenen aktiven Regionen mit der Sequenz -CGHC- identifiziert werden. Die Aminosäurensequenz zeigt eine hydrophobe Signalsequenz, jedoch kein KDEL-ER-Retentionssignal. Auf Aminosäureebene hat die O. volvulus-PDI eine 50%ige Identität zur humanen PDI. Aufgrund der bei anderen Spezies beschriebenen Rolle der PDI bei der Kollagensynthese vermuten die Autoren, dass die PDI bei O. volvulus eine Hauptfunktion in der Biosynthese der Kutikula besitzt. Die Kutikula der Nematoden besteht aus einem Netzwerk von Kollagenmolekülen, das über intramolekulare Disulfidbrücken stabilisiert wird (COX 1992). Passend zur aufgestellten Hypothese konnten WILSON et al. (1994) mittels In situ Hybridisierung hohe mRNA-Spiegel in sich entwickelnden Mikrofilarien detektieren, wobei das Signal mit zunehmender Reifung der Mikrofilarien schwächer wurde. In adulten Weibchen konnten Signale nur in der Hypodermis, dem Syntheseort der Kutikulabestandteile, detektiert werden. Der Nachweis der PDIExpression mittels Immunhistochemie korrespondierte mit den Ergebnissen der In situ Hybridisierung. Weiterhin konnten die Autoren bei 16% von 108 patent O. volvulus infizierten Patienten Antikörper gegen PDI detektieren. Aufgrund des fehlenden ER-Retentionssignals vermuten die Autoren, dass eine weitere ERständige PDI bei O. volvulus vorkommt, bzw. räumen ein, dass möglicherweise ein anderer ER-Retentionsmechanismus vorliegt. 48 Literaturübersicht EPE et al. (1998) identifizierten mittels Immunoscreening ein Fragment der PDI aus A. caninum. Das rekombinant hergestellte Protein hatte ein Molekulargewicht von etwa 38 kDa und reagierte im Western-Blot mit dem Antiserum experimentell mit A. caninum L3 infizierter Kaninchen und mit Antikörpern gegen hydrophile Proteine des Darms, der Zephal- und der Zervikaldrüsen adulter Würmer. Antiserum von mit 1000 L3 perkutan infizierten Hunden zeigte keine Reaktion im Western Blot, jedoch gelang eine Detektion mit Antiserum von mit 100.000 L3 infizierten Hunden. Die Aminosäuresequenz des identifizierten Klones zeigte eine 82%ige Identität mit einem 334 Aminosäuren-Segment der PDI aus C. elegans und eine 73%ige Identität mit einem 334 Aminosäuren großen Abschnitt der PDI aus O. volvulus. Die Autoren vermuten, dass die PDI während der Hautpenetration und der Gewebsinvasion freigesetzt wird. Die von EPE et al. (1998) identifizierte Teilsequenz wurde von POHL (2006) vervollständigt. Dabei ergab sich eine 1784 bp lange Sequenz. Der offene Leserahmen codiert für ein Protein aus 493 Aminosäuren, das zwei hochkonservierte Thioredoxin-Domänen aufweist und ein Molekulargewicht von etwa 55 kDa besitzt. Auf Aminosäuren-Ebene zeigt die PDI eine 92%ige Identität zur PDI2 von Ostertagia ostertagi, eine 82%ige Identität zur PDI2 von C. elegans und eine 74%ige Identität zur PDI von Brugia malayi. Die genomische Sequenz beinhaltet vier Introns der Längen 431 bp 104 bp, 425 bp und 170 bp. Das C-terminale Ende der PDI von A. caninum weist eine HTEL-Sequenz auf, wobei es sich um eine Modifikation des Standard-ER-Retentionssignals KDEL handelt, welches als ebenso effektiv bei der Zurückhaltung eines Proteins im ER gilt (ANDRES et al. 1990). Aufgrund der hohen Identität der PDI von A. caninum zur PDI2 von O. ostertagi und diverser außerhalb des ER nachgewiesener Proteine mit KDEL-Signal, hält POHL es jedoch für wahrscheinlich, dass die PDI von A. caninum ebenfalls sezerniert wird (POHL 2006). Eine von CHANDRASHEKAR et al. (1998) bei D. immitis identfizierte Transglutaminase (TGase) zeigte keine Homologie zu anderen TGasen, aber zeigte eine 43-52%ige Identität mit ERp60 (=ERp57) von verschiedenen Spezies. Mit Serum von mit rekombinanter Transglutaminase immunisierten Kaninchen konnten die Autoren das Protein in Extrakten von dritten und vierten Larven, von Adulten und in ES-Produkten von Larven und Adulten mittels Immunoblot nachweisen. Darüber Literaturübersicht 49 hinaus konnte die Bedeutung des Enzyms für die Kutikulasynthese demonstriert werden (CHANDRASHEKAR et al. 2002). Durch den Einsatz von TGase-Inhibitoren wurde der Schlupf von dritten zu vierten Larven verhindert. Weiterhin zeigten die Autoren mittels Immunhistochemie die Lokalisation der TGase in der Hypodermis, im Darmepithel, in Muskelzellen und in Embryonen. VERCAUTEREN et al. (2003) identifizierten eine Protein Disulfid Isomerase beim Durchmustern einer cDNA-Bank des Trichostrongyliden Ostertagia ostertagi. Diese zeigt auf Aminosäurenebene eine 88%ige Identität zur PDI von S. mansoni. Eine weitere PDI bei O. ostertagi, als PDI2 bezeichnet, wurde von GELDHOF (2003) identifiziert. Die Sequenz zeigt ebenfalls zwei Thioredoxin-Domänen mit der aktiven Seite –WCGHCK-. Am N-Terminus konnte eine Signalsequenz identifiziert werden, und der C-Terminus weist das modifizierte ER-Retentionssignal -HTEL auf. Auf Aminosäureebene zeigt sich eine höchste Identität zur PDI2 von C. elegans (78%). Im Western Blot konnte das Protein mit Kaninchen-Antiserum gegen L4- und Adultenantigen im Extrakt von L3, L4 und Adulten nachgewiesen werden. Darüber hinaus konnte die PDI im exkretorisch-sekretorischen (ES) Material adulter Würmer und vierter Larven identifiziert werden. Ebenso gelang die Detektion mit dem Serum O. ostertagi infizierter Kälber. Mittels Immunhistochemie konnte die PDI in Teilen der Hypodermis, des Darms und des Pharynx adulter Parasiten und vierter Larven nachgewiesen werden. Bei dritten Larven konzentrierte sich die Färbung auf die Darmzellen. Darüber hinaus testeten die Autoren affinitätsaufgereinigte O. ostertagi PDI2-Antikörper auf Kreuzreaktion an Extrakten und ES-Material von adulten Cooperia oncophora Würmern. Im Western-Blot zeigte sich eine etwa 56 kDa große Bande im Extrakt, jedoch nicht im ES-Material dieses nahe verwandten Parasiten. In der Immunhistochemie wurden ähnliche Lokalisationen identifiziert wie bei O. ostertagi. Aufgrund der hypodermalen Lokalisation der PDI in Ostertagia vermuten die Autoren eine ähnliche Bedeutung bei der Kutikulasynthese wie diese bei C. elegans demonstriert werden konnte (WINTER u. PAGE 2000). Die Bedeutung der PDI im Darm konnte nicht geklärt werden. Es wird vermutet, dass auch Nematoden ein dem MTP ähnliches Lipidtransportsystem im Darm besitzen, bei dem die PDI eine Rolle spielt. Unterstützt wird diese Hypothese durch die Identifikation eines 50 Literaturübersicht Proteins in C. elegans mit Homologie zum humanen MTP (GELDHOF 2003). Aufgrund der Detektion der PDI in ES-Material spekuliert der Autor über eine mögliche Sekretion der PDI trotz des vorhandenen ER-Retentionssignals und räumt ein, dass die PDI möglicherweise für eine immunologische Kontrolle der Ostertagiose geeignet sei. Allerdings gelang es ihm nicht, ausreichend PDI für einen Immunisierungsversuch aufzureinigen. VEIJOLA et al. (1996a) exprimierten zwei PDIs in C. elegans und zeigten deren funktionelle Unterschiede. Die erste PDI (als PDI/β bezeichnet) wies eine 61%ige, die zweite (als PDI Isoform bezeichnet) eine 44%ige Identität mit der humanen PDI (PDI/β) auf. Die beiden identifizierten PDIs untereinander waren zu 48% identisch. Hauptunterschiede waren in der c-Domäne zu finden, sowohl im Vergleich mit der humanen PDI als auch zwischen den beiden C. elegans PDIs. In Koexpressionsstudien der humanen und der C. elegans PDI/β mit der humanen bzw. C. elegans α(I)-Untereinheit der Prolyl-4-Hydroxylase zeigte sich, dass sowohl die humane PDI/β als auch die C. elegans PDI/ β mit der humanen α(I)-Untereinheit ein aktives α2β2-Tetramer, mit der C. elegans α-Untereinheit jedoch ein aktives αβ-Dimer bildet. Der Grund für Tetramer- oder Dimerbildung wurde daher in der C. elegans α-Untereinheit vermutet. Das Entfernen eines nur bei C. elegans vorhandenen C-terminalen Extensionsbereiches der α-Untereinheit führte zur vollständigen Aufhebung der Dimerbildung, allerdings wurde mit dieser verkürzten αUntereinheit auch kein Tetramer gebildet. Dies spricht dafür, dass der C-terminale Bereich zwar in die Dimerbildung involviert ist, aber noch andere strukturelle Unterschiede in der α-Untereinheit bestehen müssen, die über eine Dimer- oder Tetramerbildung entscheiden. Außerdem wurde gezeigt, dass die Prolyl-4Hydroxylasebildung mit der humanen PDI/β sehr viel katalytisch effektiver ist als mit der C. elegans PDI/β. Der Grund hierfür wurde in den unterschiedlichen c-Domänen vermutet. Die Konstruktion eines PDI/β-Hybrids, bestehend aus dem vorderen C. elegans Abschnitt und der humanen c-Domäne, führte zu einer Steigerung der Prolyl-4-Hydroxylasebildung im Vergleich zur reinen C. elegans PDI/β. Umgekehrt führte der Austausch der c-Domäne der humanen PDI/β gegen die C. elegans c-Domäne zu einer verringerten Prolyl-4-Hydroxylaseformation. Allerdings konnte die Literaturübersicht 51 Anwesenheit der humanen c-Domäne in der C. elegans PDI/β die C-P4H-Formation nicht zu einem Maß steigern, wie es mit der unmodifizierten humanen PDI/β beobachtet wurde. Bezüglich der zweiten identifizierten PDI (PDI Isoform) zeigten die Autoren, dass diese zwar gleiche Redox-Isomerase-Aktivität wie die humane PDI/β besitzt, jedoch weder mit der humanen, noch mit der C. elegans α-Untereinheit CP4H bildet. Darüber hinaus besitzt diese PDI in der aktiven Seite der a-Domäne die unübliche Sequenz -CVHC-. Die als PDI/β bezeichnete Form wird in GenBank und späterer Literatur als PDI2, die hier als Isoform bezeichnete PDI als PDI1 weitergeführt. Nach der Identifikation eines zweiten für die C-P4H-α-Untereinheit kodierenden Gens (phy-2) konnte gezeigt werden, dass die C-P4H auch bei C. elegans überwiegend als Tetramer vorkommt. Allerdings handelt es sich hierbei um ein, im Gegensatz zu Vertebraten, gemischtes Tetramer der Form PHY-1/PHY2/(PDI2)2. Daneben existieren Dimere der PDI2 mit beiden α-Untereinheiten (MYLLYHARJU et al. 2002). Die essentielle Bedeutung der PDI2 im C-P4H-Komplex für die Viabilität von C. elegans konnte von WINTER u. PAGE (2000) demonstriert werden. Mittels RNA-Interferenz zeigten die Autoren, dass das Ausschalten des PDI2-Gens zu embryonaler Mortalität führt. Die Embryonen entwickelten sich normal bis zur Ausbildung der ersten Kutikula. Hierbei verloren sie ihre normale Morphologie und starben. Die Autoren zeigten weiterhin, dass die Expression der PDI2 in den hypodermalen Zellen wellenförmig verläuft, wobei die vier Expressionsspitzen während der Entwicklung mit der Ausbildung der vier Scheiden korrespondierten. Ein weiterer Expressionspeak war bei Adulten vorhanden, was möglicherweise für eine zusätzliche Funktion der PDI2 bei Adulten spricht, da dieser Expressionspeak nicht bei der α-Untereinheit der C-P4H vorhanden war und somit wahrscheinlich nicht mit der Kutikulasynthese in Verbindung gebracht werden kann. Ein weiteres von NATSUKA et al. (2001) identifiziertes PDI-ähnliches Enzym mit Homologie zu ERp57 zeigte neben der Redox-Isomeraseaktivität ebenfalls Transglutaminaseaktivität. ESCHENLAUER u. PAGE (2003) zeigten für das von ihnen als PDI3 bezeichnete ERp57-Homolog eine vergleichbare Lokalisation wie bei D. immitis. RNAi hatte auf den C. elegans Wildtyp keinen Effekt, bei Mutanten mit instabiler Kutikula konnten jedoch massive morphologische Veränderungen 52 Literaturübersicht hervorgerufen werden. Dies lässt vermuten, dass die Transglutaminaseaktivität der PDI3 zur Stabilisierung der Kutikula beiträgt. Weiterhin konnten die Autoren auch für die beiden anderen C. elegans PDIs und für die humane PDI eine Transglutaminaseaktivität feststellen. Es wird vermutet, dass es sich um eine allgemeine Funktion der PDIs handelt. Im humanpathogenen Trematoden S. mansoni identifizierten FINKEN et al. (1994) eine PDI, die das Produkt eines single copy Gens zu sein scheint. Der offene Leserahmen der identifizierten Sequenz hat eine Länge von 482 Aminosäuren und weist die zwei charakteristischen Thioredoxin-ähnlichen Sequenzbereiche mit den aktiven Seiten -CGHC- sowie eine C-terminale KDEL-Sequenz auf. Die Schistosomen-PDI zeigte eine 48%ige Identität mit der humanen PDI. Die evolutionäre Konservierung dieses Gens zeigt sich auch in der Struktur der genomischen Sequenz. Die sieben identifizierten Introns befinden sich an exakt den gleichen Positionen wie im humanen PDI-Gen. Mittels Immunhistochemie und In situ Hybridisierung identifizierten die Autoren die Gastrodermis, die Wandzellen der Protonephridia und die Sustentaculumzellen im Hoden als Orte höchster Genexpression. Es wird vermutet, dass das konzentrierte Auftreten der PDI in diesen Geweben mit deren sekretorischer Aktivität korreliert, wie dies für die PDI der Vertebraten (FREEDMAN 1989) gezeigt werden konnte. SALAZAR-CALDERON et al. (2003) identifizierten eine PDI des Trematoden F. hepatica beim Durchmustern einer adulten cDNA-Expressionsbank mit KaninchenAntiserum gegen exkretorisch-sekretorisches Material. Der offene Leserahmen der cDNA codiert für ein 489 AS großes Protein mit einem Molekulargewicht von 55 kDa. Im Immunoblot mit Antiserum gegen das rekombinante Protein zeigte sich eine Bande ähnlicher Größe im exkretorisch-sekretorischen Material adulter Parasiten. Im ELISA mit rekombinanter PDI als Antigen zeigten experimentell mit F. hepatica infizierte Kaninchen PDI-spezifische Antikörper zwischen Tag 21 und Tag 28 p.i.. Maximale Antikörperkonzentrationen wurden bis Tag 35 erreicht. Da die PDI von F. hepatica im ES-Material identifiziert wurde, vermuten die Autoren eine extrazelluläre Lokalisation des Proteins trotz des vorhandenen ER-Retentionssignals HEEL. Es Literaturübersicht 53 wird darüber spekuliert, dass die PDI eine Rolle im Verteidigungskonzept gegen den oxidativen Stress, den dieser Parasit in seinem Lebensraum Wirtsleber erfährt, spielt. 54 Material und Methoden 3 Material und Methoden 3.1 Materialien 3.1.1 Puffer und Lösungen AP-Puffer: 100 mM NaCl, 5 mM MgCl2 x 6 H20, 100 mM Tris-HCl, pH 9,5 in A. bidest. BCIP-Stocklösung: 50 mg/ml 5-Brom-4-Chloro-3-Indolylphosphat in 100%igem Dimethylformamid Blotpuffer für Westernblot: 24 mM Tris, 192 mM Glycin, 20% Methanol in A. bidest. Coomassie-Blue Stammlösung: 0,1% Brilliant blue R 250 Coomassie-Fixier-Färbelösung: 40% Methanol, 10% Eisessig, 10% 0,01%-ige Coomassie-Blue Stammlösung, 40% A. bidest. Coomassie-Entfärbelösung: 10% Eisessig, 90% A. bidest. DEPC-Bidest: 0,1% Diethylpyrocarbonat in A. bidest. ELISA-Substrat: 12,5 ml A. bidest., 6,075 ml Zitratpuffer, 6,425 ml Phosphatpuffer, 10 mg OPD, 10 µl H2O2 (30%ig) Elutionspuffer, FPLC: 50 mM Tris-HCl, 10 mM red. Glutathion, pH 8,0 in A. bidest. Geltrocknungslösung: 40% Methanol, 10% Glycerol, 7,5% Eisessig, 42,5% A. bidest. GIT-Puffer: 4 M Guanidinium, 0,1 M Tris (pH 7,5), 1% β-Mercaptoethanol in A. bidest. Glycerol für Stocks: 65% Glycerol, 100 mM MgSO4, 25 mM Tris-HCl (pH 8,0) Laufpuffer für SDS-PAGE, 10X: 25mM Tris, 192 mM Glycin, 0,1% SDS Material und Methoden 55 (pH 8,5) in A. bidest. Ladungspuffer, 6x für Agarosegele: 0,25% Bromphenolblau, 40% Saccharose in A. bidest. NBT-Stocklösung: 50 mg/ml Nitro-Tetrazolium-Chlorid in 70%igem Dimethylformamid PBS, 1x: 150 mM NaCl, 16 mM Na2HPO4 x 7 H2O, 4 mM NaH2PO4 x 2 H2O in A. bidest. PBS-Tween: 1x PBS, 0,05% Tween 20 Phosphatpuffer für ELISA: 0,2 M Na2HPO4 in A. bidest. SDS-Sammelgel, 5%ig (4 ml): 2,7 ml A. bidest., 0,67 ml Acrylamidmix (30%ig), 0,5 ml 1 M Tris (pH 6,8), 40 µl SDS (10%ig), 40 µl APS (10%ig), 4 µl TEMED SDS-Probenpuffer (9 ml) 5,4 ml A. bidest., 2 ml SDS (10%ig), 1 ml Glycerol, 0,5 ml Tris (1 M, pH 6,8) 0,1 ml Bromphenolblau (Stocklösung 5 mg/100 µl), kurz vor Gebrauch 10% 1 M DTT zugeben SDS-Trenngel, 10%ig (10 ml): 4 ml A. bidest., 3,3 ml Acrylamidmix (30%ig), 2,5 ml 1,4 M Tris (pH 8,8), 100 µl SDS (10%ig), 100 µl Ammoniumpersulfat (10%ig), 4 µl TEMED Stoplösung für ELISA: 2,5 M H2SO4 in A. bidest. TAE-Puffer, 1x: 40 mM Tris, 1 mM EDTA, pH 8,0 TBS, 1x: 140 mM NaCl, 10 mM Tris-Base (pH 7,3) TBS-Tween: 1x TBS, 0,05% Tween 20 Westernblot-Färbelösung: 50 ml AP-Puffer, 660 µl NBT-Stocklösung, 330 µl BCIP-Stocklösung Zitratpuffer: 0,1 M C6H6O7 in A. bidest. 56 Material und Methoden Alle Puffer und Lösungen wurden, soweit möglich, autoklaviert oder aus autoklavierten Stammlösungen hergestellt. 3.1.2 Medien und Platten LB-Medium (ROTH): 25 g/l (entspricht 10 g/l Trypton, 5 g/l Hefeextrakt, 10 g/l NaCl; (pH 7,0 ± 0,2) LB-Agarplatten: LB-Medium mit 1,5% Agar (w/v) Alle Nährmedien wurden autoklaviert. Vor Verwendung wurden 100 µg Ampicillin pro ml Medium bzw. Agar zugesetzt. 3.1.3 Bakterien und Vektoren Escherichia coli One Shot ™ TOP 10 F-mcrA ∆(mrr-hsdRMS-mcrBC) Φ80lacX (INVITROGEN) Z∆M15 ∆lacX74 recA1 araD139 ∆(araleu) 7697 galU galK rpsL (StrR) end A1 nupG Escherichia coli One Shot™ BL21 (DE3) F- ompT hsdSB (rB-mB-) gal dcm (DE3) (INVITROGEN) Escherichia coli BL 21-Codon Plus® E. coli B F- ompT hsdSB (rB-mB-) dcm+ Tetr (DE3)-RIPL gal λ(DE3) endA Hte [argU proL Camr] (STRATAGENE) [argU ileY leuW Strep/Specr] pCR®4-TOPO® TA-Klonierungsvektor, lac Promoter, LacZα- (INVITROGEN) ccdB Genfusion, Topoisomerase I kovalent gebunden, Kanamycin- und Ampicillin- Resistenz pGEX4T Expressionsvektor, Proteinexpression als (AMERSHAM BIOSCIENCES) Fusionsprotein mit GST aus Schistosoma japonicum, tac Promoter (IPTG-induziert), Ampicillin-Resistenz Material und Methoden 57 3.1.4 Reagenzien, Reaktionskits und Geräte AGILENT TECHNOLOGIES, Waldbronn 2100 Bioanalyzer, Protein 200 PLUS Chip AMERSHAM BIOSCIENCES, Freiburg Spektralphotometer Gene Quant pro, Ultraspec 2000, QuickPrep micro mRNA Purification Kit; Glutathione Sepharose™ High Performance; GSTrap™ HP columns, 1 ml; ÄKTA FPLC BD CLONTECH, Heidelberg SMART™ PCR cDNA Synthesis Kit; SMART™ RACE cDNA Amplification Kit, Advantage™ 2 Polymerase Mix BECKMAN COULTER GMBH, Krefeld J2-21 MIE Centrifuge BIOMETRA, Göttingen Standard Power Pack P25, UNO Thermoblock BIOMOL GMBH, Hamburg Maus-anti-PDI, monoklonal BIO-RAD, München Quick Start™ Bradford Protein Assay, Mini Protean II™, Power Supply Modell 1000/500 BIO TEK, Bad Friedrichshall ELISA Reader Elx800™ BIOZYM, Hessisch Oldendorf MJ Research Multicycler PTC 200, Gel Star® 58 Material und Methoden COLORA MESSTECHNIK GMBH, Lorch Magnetrührer Magnetomix® EFLAB, Helsinki, Finnland Mehrkanalpipette Titertek® EPPENDORF, Hamburg Centrifuge 5415, Pipetten Eppendorf Research (10 µl, 100 µl, 1000 µl, 5000 µl), elektrische Pipetten Eppendorf Research Pro (10 µl, 100 µl, 1000 µl) FRESENIUS, Bad Homburg Ampuwa FRÖBEL LABORTECHNIK, Lindau Schütteltisch Rocky® GERBU BIOTECHNIK GMBH, Gaiberg Alhydrogel Adjuvant (Aluminiumhydroxid Gel 2,0% Suspension), Saponin QuilA GFL, Burgwedel Inkubationsschrank 3033, Wasserbad 1083, Überkopfschüttler 3025 HERAEUS, Osterode MinifugeT, Omnifuge 2.0 RS, Biofuge pico, Inkubationsschrank B 5042 E INTAS, Göttingen UV-Tisch TF-M 20 x 40 cm, 312 nm; Video-Geldokumentations-Anlage Material und Methoden 59 INVITROGEN, Karlsruhe TRIZOL® Reagent; TOPO TA Cloning® Kit for Sequencing; 250 bp DNA Ladder; Low DNA MASS™ Ladder; SeeBlue® Prestained Standard, SuperScript ™ III Reverse Transkriptase, Primer JOUAN, Unterhaching Zentrifuge BR4i MACKEREY-NAGEL, Düren NucleoSpin® Plasmid; Nucleobond® AX 100, NucleoSpin™ Tissue Kit, porablot NCL, Blotting Papier MN 440B MSE SCIENTIFIC INSTRUMENTS, Sussex, Großbritannien Ultraschallgerät PK 1653 NUNC, Wiesbaden Nunc Immobilizer™ Amino, nunc-Immuno™ wash 12 OLYMPUS, Hamburg Stereolupe 1x4x VMT, Digitalkamera C5050 mit Software PHARMACIA LKB, Bromma, Schweden Multidrive XL 2303 PROMEGA, Mannheim MagneGST™ Protein Purification System, RNAsin™ Ribonuclease Inhibitor, DNAseI (RQ1 Rnase-Free Dnase), Stop-Lösung (20 mM EGTA), Gel Drying Film QIAGEN, Hilden Taq DNA Polymerase 60 Material und Methoden RENNER GMBH, Dannstadt Sterilfilter 0,2 µm Celluloseacetate Membrane ROCHE, Grenzach-Wylen Complete mini, RsaI ROTH, Karlsruhe Agarose NEEO Ultra Qualität Rotiagarose™, Roti®-Block, Roti®-Mark Prestained, Rotiphorese® NF-Acrylamid/BIS Lösung 30% SARTORIUS, Göttingen Elektronische Präzisionswaage L610D, Vivaspin 4 Konzentratoren (VIVASCIENCE) SEROTEC, Düsseldorf Schaf-anti-Rind IgG:HRP, IgG1:HRP, IgG2:HRP, IgA:HRP, IgM:HRP SIGMA-ALDRICH, Deisenhofen Maus-anti-GST, Ziege-anti-Maus:AP, Maus-anti-Rind:AP STRATAGENE, Amsterdam, Niederlande MX 4000® Multiplex Quantitative PCR System, Brilliant® QPCR Master Mix ZEISS, Jena Mikroskop 473011-9901 3.1.5 Chemikalien und Verbrauchsmaterialien Alle nicht im einzelnen aufgeführten Chemikalien und Verbrauchsmaterialien wurden von den Firmen Merck (Darmstadt), Sigma-Aldrich (Deisenhofen) und Roth (Karlsruhe) erworben. Als Reaktionsgefäße wurden 0,5 ml Safe-Lock-Gefäße der Firma Eppendorf (Hamburg), 1,5 ml und 2 ml Gefäße sowie 1,5 ml und 2 ml DEPCGefäße der Firma Biozym (Hessisch Oldendorf) verwendet. Des Weiteren kamen 11 Material und Methoden 61 ml und 50 ml Gefäße der Firma Sarstedt (Nürnberg) zum Einsatz. In der quantitativen real-time PCR dienten 8x strip tubes (0,2 ml) mit optischen Deckeln (8x strip optical cap) der Firma Stratagene (Amsterdam/Niederlande) als Reaktionsgefäße. Beim Umgang mit RNA und in der PCR-Vorbereitung wurden gestopfte Pipettenspitzen (Biosphere® Filter tips) der Firma Sarstedt eingesetzt. Das Beladen von SDS-Gelen erfolgte mit ausgezogenen Pipettenspitzen (Multiflex®) der Firma Roth. In allen anderen Fällen wurden 10 µl und 100 µl Pipettenspitzen der Firma Sarstedt und 1000 µl Pipettenspitzen der Firma Ratiolab GmbH (Dreieich) eingesetzt. Die Blutentnahme erfolgte mittels Serum- und EDTA-Monovette® (Sarstedt). 3.2 Gewinnung und Aufbereitung des parasitologischen Probenmaterials 3.2.1 Isolierung von Lungenwurmlarven aus dem Kot Die für RNA-Isolierungen und experimentelle Infektionen benötigten D. viviparus Larven wurden von Stammhaltungsrindern des Instituts für Parasitologie gewonnen. Hierzu wurde der rektal entnommene Kot entsprechend des Trichter- Auswanderverfahrens (BAERMANN 1917) auf Siebe verteilt und diese wiederum auf wassergefüllte, unten mit einem Quetschhahn nach MOHR verschlossene Trichter gegeben. Nach Übernachtinkubation konnten die aktiv aus dem Kot ausgewanderten ersten Larven aus dem Trichter abgelassen werden. Um die Larven weiter aufzureinigen, wurde die entnommene Larvensuspension in 500 ml Standzylindern über Nacht sedimentiert, der Überstand abgenommen und die Larven einem zweiten Auswanderverfahren unterzogen. Hierbei wurden Siebe mit einer Maschenweite von 50 µm eingesetzt und diese noch zusätzlich mit drei Lagen Zellstoff ausgeschlagen. Die so gewonnenen Larven wurden in mit Leitungswasser gefüllten Erlenmeyerkolben bei Raumtemperatur inkubiert. Nach frühestens sechs Tagen hatten sie sich zum doppelt bescheideten, infektiösen dritten Larvenstadium (L3) entwickelt und konnten für experimentelle Infektionen verwendet, bzw. für weitergehende Untersuchungen behandelt werden. 62 Material und Methoden Zur Lagerung für spätere RNA-Isolierungen wurden die Larven zunächst entscheidet. Hierzu wurden Portionen von etwa 100.000 Larven in 5 ml DEPC-Bidest unter Zugabe von 250 µl Natriumhypochlorid (NaOCl) für 15 Minuten bei 37°C unter ständigem Schütteln (GFL) inkubiert. Mit einem Mikroskop (ZEISS) wurde die Entscheidung kontrolliert. Zur Entfernung des Natriumhypochlorids schlossen sich drei Waschschritte mit 10 ml DEPC-Bidest an. Zwischen den Waschschritten wurden die Larven bei 600 g (HERAEUS) für zehn Minuten pelletiert. Zum Schluss wurde das Larvenpellet in 500 µl GIT-Puffer resuspendiert und in einem 1,5 ml DEPCGefäß bei –80°C gelagert. 3.2.2 Isolierung von adulten Lungenwürmern aus der Lunge Nach der Sektion der experimentell infizierten Tiere (siehe Abschnitt 3.5.11) wurden die adulten Lungenwürmer aus den exenterierten Lungen gewonnen. Hierzu wurden ausgehend von der Trachea alle Bronchen bis in die kleinen Brochioli mit einer Schere eröffnet und die Lungenwürmer mit einer Pinzette gesammelt. Bis zur Weiterverarbeitung wurden die Lungenwürmer in Bechergläsern in eiskalter, 0,9%iger NaCl-Lösung verwahrt. Die Geschlechtsdifferenzierung erfolgte mithilfe eines Stereomikroskops (ZEISS). Die für die quantitative real-time PCR bestimmten Würmer wurden zur Reinigung mehrfach in DEPC-Bidest geschwenkt und anschließend einzeln in DEPC-Gefäßen mit 500 µl GIT-Puffer bei –80°C gelagert. 3.2.3 Herstellung von D. viviparus Rohantigen Zur Herstellung von Rohantigen wurden ca. 50 adulte D. viviparus (Männchen und Weibchen gemischt) in einen Glashomogenisator gefüllt und durch drehende Bewegung des geschliffenen Kolbens manuell zerkleinert. Nach etwa zwei Minuten wies der Wurmbrei eine homogene Konsistenz auf. Zum vollständigen Gewebeaufschluss wurde das Wurmhomogenisat mit 0,5 ml PBS in einen 13 mlFalkon überführt und einer Ultraschallbehandlung (MSE) ausgesetzt. Hierbei wurden die Ultraschalimpulse über 3 min verabreicht (15 sec an/aus). Während der Behandlung erfolgte eine Kühlung im Eiswasserbad. Anschließend erfolgte eine Überführung in 1,5 ml-Reaktionsgefäße und eine 30minütige Zentrifugation bei Material und Methoden 63 13000 g und 4°C (EPPENDORF). Der proteinhaltige Überstand wurde abgenommen und in 1,5 ml-Reaktionsgefäßen bei –80°C gelagert. 3.3 Molekularbiologische Methoden 3.3.1 mRNA Isolierung Zum Einsatz in der cDNA-Synthese für die konventionelle und die RACE-PCR wurde messenger RNA aus entscheideten 3. D. viviparus-Larven gewonnen. Zunächst wurden ca. 400.000 Larven unter flüssigem Stickstoff gründlich zermörsert, um einen möglichst vollständigen Aufschluss des Gewebes zu erreichen. Das zerkleinerte Larvenmaterial wurde in ein 1,5 ml DEPC-Gefäß überführt und die mRNA mithilfe des Quick Prep™ Micro mRNA Purification Kits (AMERSHAM BIOSCIENCES) nach Angaben des Herstellers aufgereinigt. Bei diesem Kit erfolgt die Isolierung der mRNA über die den meisten eukaryotischen mRNAs eigene Poly-Adenin-Sequenz am 3’Ende. Diese hybridisiert mit einer kovalent an Zellulose gebundenen Poly-ThymidinKette. Zunächst erfolgt Extraktionspuffers durch ein Zugabe weiterer des Guanidium-Thiocyanat-(GTC)-haltigen Aufschluss des Larvenmaterials und eine Inaktivierung endogener RNAsen. Das anschließende Zufügen von Elutionspuffer bewirkt ein erstes Ausfällen von Proteinen und reduziert die GTC-Konzentration, um die Anlagerung der Poly-A-Sequenzen an die Oligo(dT)-Zellulose zu ermöglichen. Nach Anlagerung der mRNA an die Zellulose werden in mehreren Waschschritten mit Hoch- und Niedrigsalzpuffern DNA und andere Zellbestandteile entfernt. Abschließend erfolgt die Gewinnung der mRNA mittels eines Elutionspuffers. Die Präzipitation der mRNA erfolgte über Nacht bei –80°C unter Zugabe von Glycogen, Kaliumacetat und 95%igem Ethanol. Am Folgetag wurde die mRNA durch einstündige Zentrifugation bei 13000 g und 4°C (JOUAN) pelletiert. Der Überstand wurde verworfen und das Pellet mit 1 ml 70%igem Ethanol vorsichtig gewaschen, ohne es vollständig zu resuspendieren. Nach erneuter Zentrifugation für 30 min bei 13000 g und 4°C wurde der Überstand verworfen und das Pellet einige Minuten bei Raumtemperatur getrocknet. Eventuell vorhandene Flüssigkeitsreste wurden mit 64 Material und Methoden einer ausgezogenen Pasteurpipette entfernt. Anschließend wurde das mRNA-Pellet in 11 µl DEPC-Bidest resuspendiert. 3.3.2 Isolierung genomischer DNA Die Präparation von genomischer DNA erfolgte mit dem NucleoSpin™ Tissue Kit (MACKEREY & NAGEL) nach Angaben des Herstellers. Hierbei fand eine Aufreinigung von DNA durch selektive Bindung an eine Silica-Membran statt. Ein adulter D. viviparus wurde mittels einer Proteinase K/SDS Lösung lysiert und auf eine Schleudersäule verbracht. Mittels eines Guanidium-Hydrochlorid-haltigen Puffers wurden Proteine und sonstige Kontaminanten ausgewaschen. Anschließend wurde die genomische DNA mit auf 70°C vorgewärmtem A. bidest. eluiert. Die Lagerung erfolgte bei –20°C. 3.3.3 cDNA-Synthese Um das Ausgangsmaterial für die PCR zu erhalten, wurde die isolierte RNA in cDNA (complementary DNA) umgeschrieben. Hierbei wird die Fähigkeit der Reversen Transkriptase (RT), RNA in einzelsträngige DNA umzuschreiben genutzt. Abhängig von der Weiterverwendung der cDNA wurden unterschiedliche Verfahren angewendet. 3.3.3.1 cDNA-Synthese für die konventionelle PCR Der Vorgang der reversen Transkription ist unter normalen Umständen oftmals nicht vollständig. Bei besonders langen mRNAs oder vorhandenen Sekundärstrukturen erreicht die Reverse Transkriptase häufig nicht das 5’-Ende, sodass dieser Genbereich in einer cDNA-Population unterrepräsentiert sein kann. Um für die Suche nach der PDI-Gensequenz einen möglichst vollständigen cDNA-Pool zu erhalten, wurde das SMART™ PCR cDNA Synthesis Kit der Firma BD CLONTECH eingesetzt. Die SMART™-Methode (Switching Mechanism At 5’-End of RNA Template) nutzt eine besondere Fähigkeit der MMLV (moloney murine leukemia virus) Reversen Transkriptase: Sobald die Erststrangsynthese das 5’-Ende der mRNA erreicht hat, sorgt die terminale Transferase-Aktivität der RT dafür, dass Material und Methoden 65 einige zusätzliche Nukleotide an das 3’-Ende der cDNA gehängt werden. Dies sind vor allem Desoxycytidine, sodass hierbei vom „d(C)-tailing“ gesprochen wird. Mit dieser Oligo(dC)-Struktur hybridisiert die am 3’-Ende des zugefügten SMART™ II Oligonukleotids lokalisierte Oligo(dG)-Sequenz. Die Reverse Transkriptase wechselt das Template und verlängert die cDNA komplementär zum SMART™ II Oligonukleotid. Die terminale Transferaseaktivität ist nur am Ende des Templates optimal, sodass der template switch hauptsächlich nur bei vollständigen cDNA-RNAHybriden stattfindet. In der sich anschließenden long distance PCR zur Zweitstrangsynthese werden Primer eingesetzt, die Sequenzen komplementär zum SMART™ II Oligonukleotid und zum CDS-Primer aufweisen (Ankersequenz). Dies hat zur Folge, dass nur cDNAs amplifiziert und angereichert werden, die in voller Länge vorliegen. Der gesamte Prozess der cDNA-Synthese erfolgte entsprechend den Herstellerangaben. 3.3.3.2 cDNA-Synthese für die RACE-PCR Die Herstellung von cDNA zum Einsatz in der RACE-PCR (Rapid Amplification of cDNA Ends) erfolgte mit dem BD SMART™RACE cDNA Amplification Kit der Firma BD CLONTECH. Das Grundprinzip der cDNA-Synthese mittels reverser Transkription entspricht dem unter 3.3.3.1 beschriebenen. Allerdings werden für die RACE zwei verschiedene cDNA Populationen generiert, und es erfolgt keine Zweitstrangsynthese mittels long distance PCR, sondern das RNA/DNA-Hybrid wird direkt in der RACE-PCR eingesetzt. Für die 5’-RACE-cDNA-Synthese wurde ein Oligo(dT)-Primer (5’-CDS-Primer) und das BD SMART II™ A Oligonukleotid eingesetzt. Wie unter 3.3.3.1 beschrieben, erfolgte am 5’-Ende der cDNA ein „d(C)-tailing“, Anlagerung des SMART II™ A Oligonukleotids, template switch der MMLV RT und Verlängerung des 5’-Endes um die Sequenz des Oligonukleotids. Für die Synthese der 3’-RACE-cDNA wird nur ein Oligo(dT)-Primer (3’-CDS-Primer) eingesetzt. Allerdings enthält dieser neben der Poly-Thymidinsequenz zusätzlich einen Sequenzbereich des SMART II™ A Oligonukleotids am 5’-Ende. Dies hat den Vorteil, dass sowohl in der 5’-RACE-PCR als auch in der 3’-RACE-PCR der gleiche Universalprimer eingesetzt werden kann. 66 Material und Methoden Die gesamte RACE-cDNA-Synthese erfolgte entsprechend den Herstellerangaben. Es wurden jeweils etwa 0,7 µg mRNA eingesetzt. 3.3.4 Polymerasekettenreaktion (polymerase chain reaction, PCR) 3.3.4.1 Allgemeines zur Methode Mithilfe der Polymerasekettenreaktion lässt sich ein beliebiger Abschnitt eines DNAMoleküls (template) selektiv vervielfältigen. Voraussetzung hierfür ist, dass Sequenzen bekannt sind, die den gewünschten Bereich flankieren. Anhand dieser Sequenzen werden kurze Oligonukleotide (Primer) konstruiert, die während der DNA-Synthesereaktion als Startpunkt für die DNA-Polymerase dienen. Im Allgemeinen besteht eine PCR aus den drei Reaktionsschritten Denaturierung, Annealing (Anlagerung) und Elongation (Verlängerung). Im Denaturierungsschritt führen hohe Temperaturen zu einem Auseinanderweichen der beiden TemplateDNA-Stränge. Das anschließende Absenken der Temperatur in der Annealing-Phase ermöglicht den Primern, an den ihnen komplementären Bereichen zu hybridisieren. Im Elongationsschritt wird die Temperatur auf das Arbeitsoptimum der Polymerase erhöht. Das Enzym synthetisiert ausgehend von den Primern den Zweitstrang und damit den gewünschten DNA-Bereich. Diese drei Schritte werden in mehreren Zyklen wiederholt. Da von einem Zyklus zum nächsten die DNA-Menge annähernd verdoppelt wird, kommt es zu einer selektiven Anreicherung des gewünschten DNAAbschnitts. Für eine erfolgreiche Amplifikation ist die Primerauswahl von entscheidender Bedeutung. Hierbei sind gewisse Grundregeln zu beachten. Primer sollten eine Länge von 18-30 Basen und einen Guanidin- und Cytosinanteil von 40 – 60% aufweisen. Mit der Plazierung von ein bis zwei G oder C am 3’-Ende kann eine bessere Bindung und Elongation erreicht werden. Um Fehlhybridisierungen und Leserasterverschiebungen zu vermeiden, sollten nicht mehr als vier gleiche Basen nacheinander und nicht mehr als drei G oder C am 3’-Ende vorhanden sein. Durch hohe Annealingtemperaturen wird die Spezifität erhöht, daher sollte die Schmelztemperatur der Primer auch entsprechend hoch sein. Idealerweise sollten Vorwärts- und Rückwärtsprimer den gleichen Schmelzpunkt aufweisen. Des Material und Methoden 67 Weiteren sollten die Primer keine internen Sekundärstrukturen bilden und nicht miteinander hybridisieren. Mit Ausnahme der degenerierten Primer zur Erstamplifikation der PDI, deren Erstellung manuell erfolgte, wurden alle PCR-Primer mithilfe des Programms Primer Select (DNA STAR Version 5.06) konstruiert. 3.3.4.2 PCR zur Erstamplifikation der PDI von D. viviparus Zur Erstamplifikation der Protein Disulfid Isomerase von D. viviparus wurden anhand bekannter PDI-Sequenzen anderer Nematodenspezies Primer konstruiert. Mithilfe des DNA Star Alignment Programms MegAlign™ konnten zwei hochkonservierte Bereiche identifiziert werden, die hierfür geeignet erschienen. Neben der Beachtung der allgemeinen Regeln zum Primerdesign wurde miteinbezogen, dass die Sequenzen der verschiedenen Spezies an einigen Positionen unterschiedliche Basen aufweisen. Durch die Angabe der Platzhalter Y-W-D-R erfolgt bei der Primersynthese die Generierung einer Primerpopulation, die an den entsprechenden Stellen verschiedene Basen aufwiesen (degenerierte Primer). Hierdurch erhöht sich die Wahrscheinlichkeit des Vorhandenseins Sequenzähnlichkeit mit der unbekannten entsprechenden Bereiche zu hybridisieren. von Sequenz Primern, die aufweisen, ausreichend um an die 68 Material und Methoden O.v. PDI A.c. PDI C.e. PDI2 C.e. PDI1 C.e.PDI3 O.o. PDI1 O.o. PDI2 5-TTYTAYGCWCCDTGGTG-3 degenerierter Vorwärtsprimer DVPDID F O.v. PDI A.c. PDI C.e. PDI2 C.e. PDI1 C.e.PDI3 O.o. PDI1 O.o. PDI2 3’-TGGTGRGGDCARTGRAA-5’ degenerierter Rückwärtsprimer DVPDID R Abb. 2: Y = C oder T W = A oder T D = A, T oder G R = A oder T Konstruktion degenerierter Primer. (O.v.: Onchocerca volvulus, A.c.: Ancylostoma caninum, C.e.: Caenorhabditis elegans, O.o. Ostertagia ostertagi) Für eine erfolgreiche Amplifikation war es erforderlich, die PCR zu optimieren. Folgende Parameter wurden getestet (jeweils im 25 µl-Ansatz): • Annealing-Temperatur (Gradienten-PCR mit 48°C bis 56°C) • MgCl2-Konzentration (1,5 mM, 2,5 mM, 3,5 mM, 4,5 mM, 5,5 mM) • cDNA (18 Zyklen, 21 Zyklen, 24 Zyklen der Zweitstrangsynthese) Material und Methoden 69 • eingesetzte cDNA-Menge (1 – 8 µl; 1:10, 1:100 Verdünnungen) • Primermenge (25-500 pmol) • Polymerase (Taq Polymerase, Amplitaq Gold Polymerase) Eine erfolgreiche Amplifikation gelang mit folgendem Ansatz: 2,5 µl 10X PCR-Puffer (QIAGEN) 1,0 µl dNTP Mix (10 mM, PROMEGA) 2,0 µl MgCl2 (25 mM, QIAGEN) 0,5 µl DVPDID R (50 µM) 0,5 µl DVPDID F (50 µM) 0,2 µl Taq DNA Polymerase (QIAGEN) 4,0 µl Template (21 Zyklen cDNA) 14,3 µl Temperaturprofil 95°C 1 min 40 Zyklen 95°C 30 sec 50°C 30 sec 72°C 1 min 72°C 6 min AMPUWA 3.3.4.3 Rapid amplification of cDNA ends (RACE PCR) Zur Ermittlung der 5’- und 3’-Gensequenzen wurden anhand des bei der Erstamplifikation der PDI erhaltenen Fragments genspezifische Primer (PDIDV2RC5 und PDIDV2RC3) entworfen, die zusammen mit dem Universalprimer aus dem BD SMART™ RACE cDNA Amplification Kit in der PCR eingesetzt wurden. Um im Falle einer problematischen Amplifikation eine nested PCR durchführen zu können, wurden des Weiteren die Primer PDIDV2RCN5 und PDIDV2RCN3 konstruiert. Für die Ermittlung der 5’- und 3’-Gensequenzen eines zweiten PDI-Gens wurden ferner die Primer PDIDV1RC5, PDIDV1RC3, PDIDV1RCN5 und PDIDV1RCN3 konstruiert. RACE Primer PDIDV2RC5 PDIDV2RCN5 PDIDV2RC3 PDIDV2RCN3 PDIDV1RC5 PDIDV1RCN5 PDIDV1RC3 PDIDV1RCN3 Tab. 1: RACE Primer Sequenz 5’-3’ TAGCAACATCGCCATGAACGGTAG CTGTGAGGCAGCTTTGGCATATTCC CCTGAAGATTGGGATAAAGCACCTGT GAACTTCGACCAAGTAGCACTGGAC CCAGCTTGCCCACGCATATACTTGACG AGGTCCCTCATATGCCTGAGCTTCCG CCGTCGGAATGGCTGGGATTCGCAC CGTTGCAGCTGTGACAAATGAAGGC 70 Material und Methoden 5’-RACE 3’-RACE RC3 RCN3 5’ 3’ - A A A A A - 3’ T T T T T - 5’ RCN5 RC5 Mittels degenerierter Primer amplifiziertes Fragment Unbekannter Genbereich Abb. 3: Schema der RACE-PCR Die 50 µl-Ansätze enthielten folgende Komponenten: 5’-RACE 3’-RACE 5 µl 10X BD Advantage 2 PCR-Puffer 1 µl dNTP Mix (je 10 mM) 2,5 µl 5’-RACE-Ready cDNA 2,5 µl 3’-RACE-Ready cDNA 5 µl 10X Universal Primer A Mix 1 µl PDIDV2RC5 bzw. 1 µl PDIDV2RC3 bzw. 1 µl PDIDV2RCN5 1 µl PDIDV2RCN3 1 µl 50X BD Advantage 2 Polymerase Mix 41,5 µl AMPUWA 3.3.4.4 PCR zur Selektion von Kolonien Um eine große Anzahl von Kolonien auf das Vorhandensein von Inserts der richtigen Größe zu überprüfen, wurden Einzelkolonien in 25 µl LB-Medium für drei bis vier Stunden bei 37°C inkubiert (GFL) und dann direkt in der PCR als Template eingesetzt. Als Primer dienten die M13 Vorwärts- und Rückwärts-Primer aus dem TOPO TA Cloning® Kit (INVITROGEN). Material und Methoden 71 Reaktionskomponenten der 25 µl-Ansätze: 2,5 µl 10X PCR-Puffer (QIAGEN) 1,0 µl dNTPs (10 mM) (PROMEGA) 1,0 µl MgCl2 (25 mM) (QIAGEN) 0,5 µl M13 Forward (50 µM) 0,5 µl M13 Reverse (50 µM) 0,2 µl Taq DNA Polymerase (QIAGEN) 5,0 µl Zellen 14,3 µl Temperaturprofil 95°C 5 min 35 Zyklen 95°C 30 sec 55°C 30 sec 72°C 1 min 72°C 6 min AMPUWA 3.3.4.5 PCR zur Ermittlung der genomischen Sequenz Um die genomische Sequenz der PDI von D. viviparus zu ermitteln, wurde die erhaltene D. viviparus cDNA-Sequenz mit der bereits bekannten genomischen Sequenz der PDI von Ancylostoma caninum (GenBank AY582129) verglichen. Da die Sequenzen schon auf cDNA Ebene hohe Ähnlichkeiten aufwiesen, wurde vermutet, dass auch die Lage der Introns ähnlich sein könnte. Die Primerpaare wurden so gewählt, dass die amplifizierten Sequenzbereiche bei der PDI von A. caninum Introns überspannt hätten. Intron Vorwärtsprimer: PDI2G1: 5’-CGGTGTGTCGGAGTGCTCTCTT-3’ 1 u. 2 Rückwärtsprimer: PDI2G2: 5’-CATCTTCAGAGGTGATTCCAAATGG-3’ Intron Vorwärtsprimer: 3 Rückwärtsprimer: PDI2G4: 5’-CTGAAGATTGGGATAAAGCACTGTA-3’ Intron Vorwärtsprimer: 4 Tab. 2: PDI2G3: 5’-CTGTATTCACTCGGCTTCCACTG-3’ PDI2G5: 5’-AATATGCCAAAGCTGCCACACA-3’ Rückwärtsprimer: PDI2G6: 5’-TCACCATCACTTATCAACGATCACAAT-3’ Sequenzen der Primer zur Amplifikation der genomischen DNA 72 Material und Methoden Reaktionskomponenten der 25 µl-Ansätze: Temperaturprofil 2,5 µl 10x PCR-Puffer (QIAGEN) 1,0 µl dNTPs (10 mM, PROMEGA) 2,0 µl MgCl2 (25 mM, QIAGEN)) 0,5 µl Vorwärtsprimer (50 µM) 0,5 µl Rückwärtsprimer (50 µM) 0,2 µl Taq DNA Polymerase (QIAGEN) 16,3 µl 94°C 3 min 25 Zyklen 94°C 1 min 65°C 30 sec 72°C 1 min 72°C 6 min AMPUWA 3.3.4.6 PCR zur Klonierung des ORF (open reading frame) der PDI Um für die Genexpression den offenen Leserahmen der Protein Disulfid Isomerase zu erhalten, wurden Primer anhand der Start- und Stopsequenzen konstruiert. Zusätzlich wurden an die Primer Zielsequenzen für Restriktionsenzyme angefügt. Dies ermöglichte eine direkte Klonierung des PCR-Produkts ohne Verschiebung des Leserahmens. Um geeignete Zielsequenzen zu finden, wurden die Schnittstellen der cloning site des Expressionsvektors pGEX4T mit der PDI-Sequenz verglichen. Für das 5’-Ende wurde eine Sal I-Schnittstelle, für das 3’-Ende eine Not I-Schnittstelle gewählt. Da die PDI im pGEX4T-Vektor als Fusionsprotein mit GST exprimiert werden sollte, wurde bei der Primerkonstruktion zusätzlich ein Basentausch am Startcodon vorgenommen. A wurde gegen C getauscht, um an dieser Stelle keinen Transkriptionsstart zu ermöglichen. Material und Methoden Vorwärtsprimer PDIDV2FLFOR: 73 SalISchnittstelle PDI-Sequenz 5’-ATAGTCGACCTGTTCAAACTCGCCTGC-3’ Modifiziertes Startcodon Rückwärtsprimer PDIDV2FLREV: NotISchnittstelle PDI-Sequenz 5’ATAGCGGCCGCTTACAGCTCAGTATGACC-3’ Stopcodon Abb. 4: Konstruktion der Full length-Primer 3.3.5 Darstellung von Nucleinsäuren mittels Agarosegelelektrophorese Mittels Agarosegelelektrophorese können DNA-Stränge ihrer Größe nach getrennt werden. Durch Anlegen eines elektrischen Feldes werden die negativ geladenen DNA-Moleküle durch das Gel gezogen, das hierbei wie ein Sieb wirkt: Kleinere Moleküle wandern schneller, wobei die Trennschärfe von der Agarosekonzentration abhängig ist. Für die Herstellung wurde Agarose (Agarose NEEO Ultra Qualität Rotiagarose™, ROTH) in einer Konzentration von 0,5 bis 2% in 1x TAE-Puffer durch Aufkochen gelöst und anschließend auf ca. 52°C abgekühlt. Die noch flüssige Agarose wurde mit dem Farbstoff GelStar® (BIOZYM) versetzt (0,01% (v/v)) und in Flachbettformen gegossen. Durch den Einsatz von Kämmen wurden während des Polymerisierens Geltaschen zur Aufnahme der Proben erzeugt. Nach einer Polymerisationszeit von 30 bis 60 min wurde das Gel in eine mit TAE-Puffer gefüllte Elektrophoresekammer (HOEFER) verbracht und mit den zu untersuchenden Proben sowie einem Größenstandard beladen. Zum Beschweren und um die Lauffront kontrollieren zu können, wurden die Proben vorher mit einer entsprechenden Menge 6x 74 Material und Methoden Ladungspuffer versetzt. Dieser enthält Sacharose, um ein Absinken der Proben in den Geltaschen zu ermöglichen und den Farbstoff Bromphenolblau. Mit einer Spannungsquelle (BIOMETRA) wurde je nach Größe und Konzentration der Gele für 45 bis 90 min eine Spannung von 85 bis 120 Volt angelegt. Während des Laufs interkalierte der Farbstoff Gelstar® mit der DNA und ermöglichte deren Sichtbarmachung durch Fluoreszenz auf einem UV-Leuchttisch (312 nm, INTAS). Die Dokumentation der Gele erfolgte über eine Kamera (Video-Dokumentationsanlage, INTAS). 3.3.6 Isolierung von PCR-Amplifikaten aus Agarosegelen Die zu klonierenden Banden wurden unter UV-Kontrolle mit einer sterilen Skalpellklinge aus dem Agarosegel herausgeschnitten. Eine gestopfte 100 µl Pipettenspitze wurde zu einem Filter umfunktioniert. Hierzu wurde das untere Drittel abgeschnitten und diese dann in ein 1,5 ml Reaktionsgefäß verbracht. Das ausgeschnittene Gelstück wurde in den Konus der Pipettenspitze gegeben und das Reaktionsgefäß anschließend für 1 min bei Raumtemperatur und 13000 U/min zentrifugiert (HEREAUS). Die so eluierten PCR-Produkte wurden für Klonierungen eingesetzt. 3.3.7 Klonierung von PCR-Amplifikaten 3.3.7.1 Klonierung in pCR®4-TOPO® Alle zu klonierenden Amplifikate wurden im ersten Schritt in den pCR®4-TOPO® Vektor aus dem TOPO TA Cloning® Kit for Sequencing (INVITROGEN) kloniert. Der linearisierte Vektor besitzt an den 3´-Enden einen überhängenden Desoxythymidinrest. Durch die nicht templateabhängige Terminale TransferaseAktivität der bei der PCR eingesetzen Taq DNA-Polymerase, wird an den 3´-Enden des Amplifikationsprodukts ein Desoxyadenosinrest angehängt. Dieser kann mit dem Desoxythymidinrest des Vektors hybridisieren. Die Religation des Vektors wird von der Topoisomerase I des Vaccinia Virus kathalysiert. Dieses Enzym bindet doppelsträngige DNA und spaltet in einem Strang das Phosphodiester-Rückgrat nach der Sequenz 5´-CCCTT. Die dabei freiwerdende Energie wird in einer Material und Methoden 75 kovalenten Bindung zwischen einem Tyrosyl-Rest der Topoisomerase I und dem 3´Phosphat des geschnittenen Stranges gespeichert. Diese Bindung kann wiederum von der 5´-OH-Gruppe des geschnittenen Stranges angegriffen werden. Dadurch wird die Spaltungsreaktion rückgängig gemacht und die Topoisomerase I freigesetzt. Im pCR®4-TOPO®-Vektor wird diese Reaktion zur Insertion eines PCR-Produkts genutzt, indem sich die 5´-CCCTT-Sequenz komplementär an den 3´-Enden mit dem angehängten A-Rest befindet. Des Weiteren enthält der Vektor Gene für Ampicillin- und Kanamycinresistenz. Hierdurch wird eine selektive Anzucht der Wirtszellen ermöglicht. Damit nur Zellen wachsen, die Vektoren enthalten, welche das Insert tragen, befindet sich auf dem Vektor das letale E. coli control of cell death B (ccdB)-Gen. Dieses ist mit dem CTerminus des LacZα-Fragments fusioniert. Da sich die Insertionsstelle innerhalb des LacZα-Fragments befindet, unterbricht die Ligation eines Inserts die Expression des LacZα-ccdB-Fusionsgens und ermöglicht so ein Wachstum der Wirtszelle. Der Ligationsansatz enthielt 2 µl PCR-Produkt, 1 µl Salzlösung, 2 µl A. bidest. und 1 µl des pCR®4-TOPO®-Vektors. Dieser Ansatz wurde 5 min bei Raumtemperatur inkubiert und anschließend auf Eis gekühlt. Die Transformation erfolgte in E. coli TOP 10 Zellen (siehe Abschnitt 3.3.8.1) 3.3.7.2 Klonierung in pGEX4T-3 Für die Proteinexpression wurde die Sequenz des open reading frame der PDI in den Expressionsvektor pGEX4T-3 kloniert. In diesem Vektor wird das Zielprotein als Fusionsprotein mit GST (Glutathion-S-Transferase) aus Schistosoma japonicum exprimiert. Die Expression steht unter der Kontrolle des tac Promoters, welcher durch IPTG (Isopropyl-β-D-Thiogalactosid) induziert wird. Der GST-Anhang (tag) ermöglicht die Aufreinigung des Fusionsproteins über ein Affinitätsmedium. Nach erfolgreicher T/A-Klonierung in den pCR®4-TOPO®-Vektor, Transformation in TOP 10 Zellen und Anzucht erfolgte eine Plasmidpräparation mittels Miniprep (siehe Abschnitt 3.3.9.1). Anschließend wurden sowohl die Plasmide als auch der pGEX 4T-3-Vektor einem Restriktionsverdau mit den Enzymen Sal I und Not I unterzogen: 76 Material und Methoden PCR®4-TOPO®-Verdau 8 µl Plasmid-Miniprep 8 µl autokl. A. bidest. PGEX4T-Verdau 2 µl pGEX4T-3 14 µl autokl. A. bidest. 2 µl Puffer O (FERMENTAS) 2 µl Puffer O 1 µl Sal I (10 U, FERMENTAS) 1 µl Sal I 1 µl Not I (10 U, FERMENTAS) 1 µl Not I Beide Ansätze wurden für 4 h bei 37°C inkubiert und dann auf ein 1%iges Agarosegel geladen. Anschließend wurde die Bande des geschnittenen pGEXVektors und die Insertbande des TOPO-Vektors mit einer sterilen Skalpellklinge aus dem Gel ausgeschnitten und die DNA, wie unter Abschnitt 3.3.6 beschrieben, eluiert. Daraufhin erfolgte die Ligation des PDI-Inserts mit dem pGEX4T-Vektor. Der Ligationsansatz enthielt folgende Komponenten: 3 µl pGEX4T, geschnitten 6 µl PDI2 Insert 7 µl autoklav. A. bidest. 2 µl Ligase Puffer (FERMENTAS) 2 µl T4-Ligase(1 U/µl, FERMENTAS) Inkubation bei 14°C über Nacht Die Transformation des pGEX4T-PDI-Expressionsplasmids erfolgte in die E. coliStämme TOP 10, BL 21 (DE 3) und BL 21-Codon Plus®(DE 3)-RIPL. 3.3.8 Transformation kompetenter Zellen 3.3.8.1 E. coli TOP 10 Die Vermehrung des pCR®4-TOPO®-Vektors für Sequenzierungen und die Langzeitlagerung des pGEX4T-PDI-Expressionsplasmids erfolgte in E. coli TOP 10Zellen (One Shot® TOP 10, INVITROGEN). Für die Transformation wurden 2 µl des Ligationsansatzes aus 3.3.7.1 oder 2 µl einer Plasmidpräparation (3.3.9) zu den auf Eis aufgetauten, chemisch kompetenten TOP 10-Zellen gegeben und diese für 30 min auf Eis inkubiert. Anschließend erfolgte ein Hitzeschock im 42°C Wasserbad Material und Methoden 77 für 30 sec und darauffolgend wieder eine Kühlung auf Eis. Durch starke Temperaturschwankungen sollte die Permeabilität der Bakterienzellmembran erhöht werden, um das Einschleusen des Vektors zu erleichtern. Nach Zugabe von 250 µl S.O.C.-Medium (INVITROGEN) wurde die Bakteriensuspension für eine erste Zellvermehrung und Expression der Resistenzgene bei 37°C und 200 rpm für eine Stunde inkubiert (GFL). Die Anzucht erfolgte auf mit Ampicillin (100 µg/ml Agar) versetzten LB-Agar-Nährböden. Hierzu wurden zwei verschiedene Volumina der Bakteriensuspension (i.A. 50 und 100 µl) mit einem sterilen Drigalskispatel ausgestrichen und über Nacht bei 37°C inkubiert (HEREAUS). 3.3.8.2 E. coli BL 21 (DE3) Zur Expression wurde der E. coli BL 21 (DE3)-Stamm eingesetzt. Dieser enthält das λDE3 Lysogen für die T7 RNA Polymerase unter der Kontrolle des lacUV5 Promoters. Die Expression der T7 RNA Polymerase wird durch IPTG induziert. Der Stamm ist lon Protease und outer membrane Protease (OmpT) defizient, wodurch die Degradation heterologer Proteine reduziert wird. Für die Transformation wurden 5 µl des Ligationsansatzes aus 3.3.7.2 zu 50 µl One Shot® BL 21 (DE3)-Zellen (INVITROGEN) gegeben. Das weitere Vorgehen entsprach dem Transformationsprotokoll für One Shot ®TOP 10-Zellen (3.3.8.1). 3.3.8.3 E. coli BL 21-Codon Plus® Für Optimierungsversuche der Proteinexpression erfolgte eine Transformation des pGEX4T-PDI-Expressionsplasmids in den Wirtszellstamm E. coli BL 21- CodonPlus®(DE3)-RIPL. CodonPlus®-Zellen enthalten zusätzliche Genkopien für tRNAs, die in E. coli selten vorkommen, aber oftmals bei der Expression heterologer Proteine in größerer Anzahl benötigt werden. Der RIPL-Stamm enthält die tRNAGene argU (AGA, AGG), ileY (AUA), proL (CCC) und leuW (CUA) und kann dadurch die Expression heterologer Proteine von Organismen mit AT- oder GC-reichen Genomen verbessern. Der Transformationsprozess entsprach im Wesentlichen der Transformation in den E. coli TOP 10-Stamm. 2 µl einer Miniprep wurden zu 75 µl Zellen gegeben und 30 78 Material und Methoden min auf Eis inkubiert. Der Hitzeschock bei 42°C war auf 20 sec verkürzt. Das weitere Vorgehen entsprach dem unter 3.3.8.1 beschriebenen. 3.3.9 Plasmidpräparation 3.3.9.1 Miniprep Von den LB-Agarplatten (3.3.8.1) wurden gut separiert gewachsene Klone nach dem Zufallsprinzip ausgewählt. Diese wurden mit einer Pipettenspitze abgenommen und in 5 ml LB-Ampicillin-Medium überführt. Die Anzucht der Flüssigkultur erfolgte bei 37°C und ständigem Schütteln (200 rpm, GFL) über Nacht. Vor der Plasmidpräparation wurden 600 µl der Flüssigkultur abgenommen, mit 600 µl Glycerol für Glycerolstocks versetzt und bei –20°C gelagert. Die restliche Flüssigkultur wurde für eine Plamidpräparation im kleinen Maßstab (Miniprep) im NucleoSpin® Plasmid-Kit (MACHEREY & NAGEL) eingesetzt. Hierzu wurde die Bakteriensuspension für 20 min bei 4000 g (HERAEUS) zentrifugiert und der Überstand sorgfältig entfernt. Das zurückbleibende Zellpellet wurde in einem RNAse A-haltigen Puffer vollständig resuspendiert. Anschließend erfolgte mittels eines NaOH/SDS-haltigen Puffers eine Denaturierung der Proteine. Durch Zugabe eines Neutralisationspuffers wurden die erforderlichen Bedingungen für die Bindung der Plasmid-DNA an eine Silicamembran geschaffen und die denaturierten Proteine gefällt. Zelltrümmer und SDS-Präzipitate wurden anschließend mittels Zentrifugation entfernt und der geklärte Überstand auf eine Silica-Schleudersäule verbracht. Nach einem Waschschritt mit ethanolhaltigem Puffer erfolgte die Elution der Plasmid-DNA von der Silica-Membran. Bis auf den Elutionsschritt, der mit autoklaviertem A. bidest. statt des im Kit enthaltenen Elutionspuffers durchgeführt wurde, fanden alle Arbeitsschritte dem Herstellerprotokoll entsprechend statt. Die gewonnene Plasmid-DNA wurde bei ausreichender Menge für Sequenzierungen eingesetzt oder als Reserve für Transformationen bei –20°C gelagert. Material und Methoden 79 3.3.9.2 Midiprep Wurde für Sequenzierungen oder Klonierungen eine größere Menge Plasmid-DNA benötigt, erfolgte die Isolierung mit dem NucleoBond® AX 100 Midi-Kit (MACKEREY & NAGEL) aus einer 50 ml Flüssigkultur. 100 µl Glycerolstock wurden in 50 ml LB-Medium über Nacht bei 37°C unter ständigem Schütteln (200 rpm, GFL) angezüchtet. Durch dreißigminütige Zentrifugation wurden die Zellen pelletiert und entsprechend des Herstellerprotokolls weiterverarbeitet. Hierbei erfolgte die Zelllyse und erste Reinigung des Zelllysats analog zur Miniprep. Im Weiteren jedoch erfolgte die Separation der Plasmid-DNA über eine Anionenaustauschersäule. Das negativ geladene Phosphat-Rückgrad der Plasmid-DNA bindet hierbei an eine funktionelle Gruppe des Silica-Betts. Durch schrittweise Erhöhung der Salzkonzentration werden Verunreinigungen und kleinere Nucleinsäuremoleküle ausgewaschen. Die eluierte Plasmid-DNA wurde durch Zugabe von 100% Isopropanol präzipitiert und das Pellet mit 70%igem Ethanol gewaschen. Mittels ausgezogener Glaspipette erfolgte eine weitestgehende Entfernung der Alkoholverunreinigungen. Anschließend wurde das Pellet in 50 µl A. bidest. resuspendiert und bei –20°C gelagert. 3.3.10 Restriktionsenzymverdau 3.3.10.1 Restriktionsfragment-Längenpolymorphismus (RFLP) Um Klone der Erstamplifikation der PDI ohne Sequenzierung zu vergleichen, wurden diese nach der PCR zur Selektion von Kolonien einem Verdau mit RsaI unterzogen. RsaI ist ein 4-base cutter, d.h. das Enzym schneidet statistisch alle 44 = 256 Basenpaare die Zielsequenz GT↓AC. Für sichtbare Unterschiede in den Laufeigenschaften der Fragmente müssen die Sequenzen allerdings relativ große Unterschiede aufweisen. Daher erhält man mit dieser Methode nur einen groben Vergleich der Sequenzen. Für den Verdau wurden 15 µl PCR-Produkt mit 2 µl Puffer L (ROCHE), 0,3 µl RsaI (10 U/µl, ROCHE) und 2,7 µl AMPUWA versetzt und für 2 h bei 37°C inkubiert. Anschließend wurden 15 µl auf einem 1%igen Agarosegel überprüft. 80 Material und Methoden 3.3.10.2 Darstellung des Inserts In wenigen Fällen führt beim TOPO TA Cloning® Kit eine Leserahmenverschiebung zur Unterbrechung des LacZα-ccdB-Leserahmens und ermöglicht damit transformierten Kolonien ein Wachstum, die Vektoren ohne Inserts tragen. Um einen schnellen Überblick zu erhalten, ob der bearbeitete Klon ein Insert der erwarteten Größe enthält, kann dieses mittels Restriktionsenzymverdau dargestellt werden. Im pCR®4-TOPO®-Vektor wird das Insert von zwei EcoRI-Schnittstellen flankiert. In einem 20 µl-Ansatz wurden 8 µl einer Miniprep bzw. 2 µl einer Midiprep mit 2 µl 10XReaktionspuffer und 1 µl EcoRI (1 U/µl, LIFE TECHNOLOGIES) versetzt und eine Stunde bei 37°C inkubiert. Anschließend wurde der gesamte Ansatz zur Überprüfung auf ein 1%iges Agarosegel geladen. 3.3.11 Sequenzierung und Sequenzbearbeitung Die Sequenzierung zu untersuchender Proben wurde von der Firma SEQUENCE LABORATORIES, Göttingen durchgeführt. Nach Erhalt der Sequenzen auf elektronischem Weg wurden diese mit dem Datenverarbeitungsprogramm Align™ Plus (Sequence Alignment Program Version 4.0, S & E SOFTWARE) bearbeitet. Mit diesem Programm erfolgte das Auffinden von Primersequenzen, Übersetzung in die entsprechenden Aminosäuresequenzen und der Identitätsvergleich zwischen verschiedenen Klonen bzw. der Identitätsvergleich mit Sequenzen anderer Spezies. Alignments der PDI-Sequenzen verschiedener Spezies wurden ferner mit dem Programm MegAlign™(DNA Star) durchgeführt. Erhaltene Sequenzen wurden im Internet mit bereits publizierten Sequenzen anderer Organismen verglichen. Hierzu wurde das „basic local alignment search tool“ (BLAST) (NCBI, USA) genutzt. Im BLASTN werden eingespeiste Sequenzen auf Nukleinsäureebene, mit dem BLASTP auf Proteinebene mit bereits in GenBank (NCBI, USA) vorhandenen Daten verglichen. Mit dem RPSBLAST erfolgte die Suche nach konservierten Domänen in der Aminosäuresequenz. Bei Unstimmigkeiten zwischen Vorwärts- und Rückwärtssequenzen oder nicht eindeutig detektierten Basen, wurden die mit den Sequenzen erhaltenen Material und Methoden Elektropherogramme 81 mithilfe des Programms CHROMAS (Version 2.31, Technelysium Pty Ltd.) überprüft. 3.4 Proteinbiochemische Methoden 3.4.1 Proteinexpression Die Expression des GST-PDI-Fusionsproteins erfolgte in E. coli BL 21(DE3) und BL 21(DE3) Codon Plus® Zellen. 100 µl Glycerolstock wurden in 50 ml LB/Ampicillin-Medium bei 37°C und 200 rpm (GFL) über Nacht zu einer Vorkultur herangezüchtet. Mit dieser Vorkultur wurde frisches LB/Ampicillin-Medium auf eine OD600 von 0,1 bis 0,15 (AMERSHAM BIOSCIENCES) eingestellt. Hierzu war ca. 1/10 Volumen Vorkultur nötig. Diese Hauptkultur wurde bis zu einer OD600 von 0,5 bis 0,6 weitergezüchtet. Dies war nach ca. 2 Stunden erreicht. Um zu überprüfen, ob eine basale Expression stattfindet, wurde ein 1 ml Aliquot von der Kultur entnommen, welches später parallel mit der induzierten Kultur lysiert und mittels SDS-PAGE (3.4.5.1) untersucht wurde. Die Hauptkultur wurde mit IPTG induziert und weiter bei 37°C und 200 rpm inkubiert. Zur Optimierung wurde mit 50 ml Kulturen gearbeitet. Folgende Parameter wurden bei der Expression in E. coli BL 21 (DE3) getestet: • Anzucht bei 37°C, 30°C, Raumtemperatur • Induktion bei OD600 ca. 0,25 und 0,5 • Induktion mit 0,2 mM IPTG und 1,0 mM IPTG • Expression für 1 h, 2 h, 3 h, 4 h, 5 h und über Nacht Die Anzucht des E. coli BL 21 (DE3) Codon Plus® Stammes erfolgte bei 37°C. Die Induktion erfolgte mit 1 mM IPTG bei einer OD600 von 0,5. Die Kultur wurde für 5 h inkubiert. Im Anschluss an die Expression wurden die Zellen für 20 Minuten bei 4000 g (HEREAUS) abzentrifugiert, der Mediumüberstand entfernt und die Zellpellets bei -80°C eingefroren. 82 Material und Methoden 3.4.2 Gewinnung des Zelllysats 3.4.2.1 Zelllyse mittels MagneGST™ Protein Purification System Lysispuffer (PROMEGA) Ein Zellpellet aus einer 50 ml-Kultur wurde in 2 ml MagneGST™ Lysispuffer (PROMEGA) resuspendiert und in einem 10 ml Gefäß für 30 Minuten rotierend inkubiert (GFL). Der unlösliche Zelldebris wurde durch Zentrifugation bei 13000 g und 4°C für 30 Minuten (EPPENDORF) entfernt. Das geklärte Zelllysat wurde in der Proteinaufreinigung mittels des MagneGST™ Protein Purification System-Kits (PROMEGA) eingesetzt. 3.4.2.2 Zelllyse mittels Lysisprotokoll nach SAMBROCK (1989) Zellpellets wurden mit 3 ml Lysispuffer (SAMBROCK 1989) pro Gramm Zellen resuspendiert. Es erfolgte die Zugabe von 25 µl 40X Complete-Lösung (ROCHE), 25 µl DNAse (1 mg/ml) und 200 µl Lysozym (10 mg/ml) pro Milliliter Lysispuffer. Nach einer zwanzigminütigen Inkubation auf Eis auf einem Schwenktisch erfolgte die Zugabe von 4 mg Desoxycholsäure pro Milliliter Lysispuffer und eine Inkubation bei 37°C für zehn Minuten (GFL). Anschließend wurden 25 µl DNAse (1 mg/ml) pro Milliliter Lysispuffer hinzupipettiert und der Ansatz für 15 min bei Raumtemperatur inkubiert. Abschließend wurden unlösliche Zellbestandteile durch dreißigminütige Zentrifugation bei 13000 g und 4°C abgetrennt. Das geklärte Lysat wurde im MagneGST™ Protein Purification System-Kit eingesetzt. 3.4.2.3 Zelllyse mittels Lysispuffer und Ultraschall Zellpellets wurden in 2 ml Lysispuffer (SAMBROCK oder MagneGST™-Kit) pro 50 ml ursprünglicher Zellkultur resuspendiert und mittels Ultraschallsonde (MSE) lysiert. Die Behandlung erfolgte im Eiswasserbad und die Ultraschallimpulse wurden stoßweise verabreicht (15 sec an/15 sec aus). Die Intensität wurde nur so hoch geregelt, dass es zu keiner Schaumbildung kam. Zwischenzeitlich wurde die Zellsuspension aufgeschüttelt. Die Impulse wurden so lange verabreicht, bis eine deutliche Klärung der Zellsuspension und Abnahme der Viskosität zu erkennen war. Dies war in der Regel bei einem 50 ml-Pellet nach etwa 3 min der Fall. Das Zelllysat Material und Methoden 83 wurde durch dreißigminütige Zentrifugation bei 13000 g und 4°C geklärt und mittels MagneGST™-Kit, Glutathione Sepharose™ High Performance (AMERSHAM BIOSCIENCES) und FPLC aufgereinigt. 3.4.3 Proteinaufreinigung 3.4.3.1 Magne GST™ Protein Purification System Das Magne GST™ Protein Purification System (PROMEGA) ermöglicht die Aufreinigung von GST-Fusionsproteinen über magnetische Partikel. An die Oberfläche der magnetischen Partikel ist reduziertes Glutathion kovalent gebunden. Hieran kann die GST des Fusionsproteins binden. Die Aufreinigung erfolgt in drei Schritten. Im ersten Schritt werden die magnetischen Partikel zum Zelllysat gegeben. Nach einer Inkubationszeit werden die Partikel mittels eines von außen an das Gefäß gehaltenen Magneten fixiert und der Überstand abgenommen. Im zweiten Schritt erfolgt die Zugabe eines Waschpuffers, um nicht gebundene Verunreinigungen von den Partikeln zu waschen. Anschließend werden die Partikel wieder fixiert, um den Waschpuffer zu entfernen. Im letzen Schritt wird das Fusionsprotein mittels eines Glutathion-haltigen Elutionspuffers von den Partikeln eluiert. Pro 2 ml Zelllysat wurden ca. 400 µl der Partikelsuspension eingesetzt. Die Elution erfolgte mit 400 µl Elutionspuffer für 15 min. Zur Optimierung der Aufreinigung wurden folgende Parameter variiert: Anlagerung: • Inkubationszeit: 15 min, 30 min, 1 h • Temperatur: Raumtemperatur, 4°C 84 Material und Methoden Waschen • Anzahl der Waschschritte: 3, 5, 10 • Volumen des Waschpuffers: 1 ml, 2 ml, 5 ml, 10 ml • Dauer der Waschschritte: 5 min, 20 min, 30 min • Zusatz von NaCl zum Waschpuffer (500 mM) • Zusatz von - 50% Glycerol - 2% Triton - 2% Tween 20 - 20 mM β-Mercaptoethanol - 6 M Guanidium HCl - 8 M Urea - 3 M DTT Elution • Original-Elutionspuffer mit 50 mM Glutathion und 50 mM Tris-HCl • Glutathion-Konzentration: 100 mM, 50 mM, 25 mM, 12,5 mM, 6,25 mM 3.4.3.2 Glutathione Sepharose™ High Performance Bei der Proteinaufreinigung mittels Glutathione Sepharose™-Medium (AMERSHAM BIOSCIENCES) erfolgt die Isolierung eines GST-Fusionsproteins aus einem Zelllysat ebenfalls durch Bindung der GST an Glutathion. Das Glutathion ist hierbei an quervernetzte Agarosepartikel gebunden. Analog zu dem unter 3.4.3.1 beschriebenen Prozess besteht auch hier der Aufreinigungsvorgang aus den drei Schritten Anlagerung des Fusionsproteins, Auswaschen von Verunreinigungen und Elution des gebundenen Proteins. Um die Sepharose™-Partikel von den zugefügten Flüssigkeiten zu trennen, werden die Partikel mittels Zentrifugation sedimentiert, und der flüssige Überstand wird abpipettiert. 500 µl der 60%igen Glutathione Sepharose™ Suspension wurden entsprechend den Herstellerangaben equilibriert und mit 200 µl Zelllysat versetzt. Es folgte eine dreißigminütige Inkubation auf einem Überkopf-Schüttelinkubator (GFL). Durch Material und Methoden 85 Zentrifugation für fünf min bei 500 g und Raumtemperatur (HERAEUS) wurde die Sepharose™ sedimentiert und der Zelllysatüberstand wurde abpipettiert. Anschließend wurden die Partikel durch Zugabe von 5 ml PBS und Inkubation für 10 min auf einem Schüttelinkubator gewaschen und darauffolgend wieder sedimentiert. Die Waschlösung wurde abpipettiert und der Waschvorgang zweimal wiederholt. Abschließend erfolgte eine zweimalige Elution für 30 min durch Zugabe von je 250 µl Elutionspuffer. Die Eluate wurden bei –80°C gelagert. 3.4.3.3 FPLC mit GSTrap™ HP Affinitätssäulen Mittels FPLC (Fast pressure liquid chromatography) lässt sich der unter 3.4.3.2 beschriebene Aufreinigungsprozess automatisieren. Hierzu wurde das Gerät ÄKTA FPLC (AMERSHAM BIOSCIENCES) eingesetzt. Die Anlage wird vom Computer mittels der zugehörigen Unicorn Software gesteuert. Das Affinitätsmedium Glutathione Sepharose™ befindet sich dicht gepackt in einer Säule, die einen Zuund Abfluss besitzt. Über einen zuführenden Schlauch wird das Zelllysat mittels einer Pumpe automatisch durch die Säule gepumpt. Anschließend führt die Pumpe den Waschpuffer zu. Für die Elution tritt eine zweite Pumpe in Kraft, die den Elutionspuffer über die Säule befördert. Der Säulendurchfluss passiert eine UVLampe, deren Signale an die Steuereinheit weitergegeben und auf einem Bildschirm als UV-Kurve ausgegeben werden. Durch Kontrolle der OD lässt sich der Aufreinigungsprozess verfolgen und z.B. feststellen, wann der Waschdurchfluss keine Verunreinigungen mehr enthält oder nach welchem Elutionsvolumen das Protein vollständig eluiert wurde. Mittels des zum System gehörenden Fraktionssammlers kann das eluierte Protein in beliebig großen Fraktionen automatisch gesammelt werden. Es wurden GSTrap™ HP- Säulen (AMERSHAM BIOSCIENCES) mit einem Volumen von 1 ml eingesetzt. Das verwendete Zelllysat wurde durch einen 0,2 µm Filter (RENNER) sterilfiltriert. Die eingesetzten Puffer wurden, soweit möglich, autoklaviert und ansonsten mittels Magnetrührer (COLORA MESSTECHNIK) entgast. Mit der Unicorn Software wurden unterschiedliche Programme kreiert, Aufreinigungsprozess zu optimieren. Folgende Parameter wurden getestet: um den 86 Material und Methoden 1. Beladen der Säule mit Zelllysat • unterschiedliche Volumina reinen Zelllysats • unterschiedliche Volumina einer 1:5 Verdünnung des Zelllysats mit PBS • Flussgeschwindigkeit beim Beladen: 1 ml/min, 0,5 ml/min, 0,2 ml/min 2. Waschen der Säule • Flussgeschwindigkeit: 1 ml/min, 0,2 ml/min • Waschvolumen: Es wurde so lange gewaschen, bis die UV-Kurve wieder konstant auf der Basislinie war 3. Elution • Elution mit Standardelutionspuffer (10 mM Glutathion) • Elution mittels Puffergradienten (0-100% Elutionspuffer, 10 mM Glutathion) Das Eluat wurde in Fraktionen von 1 ml gesammelt und bei –80°C gelagert. 3.4.4 Proteinproduktion für die Immunisierungen Aus den oben beschriebenen Vorversuchen zur Expression und Aufreinigung ergab sich das Protokoll für die Herstellung großer Fusionsproteinmengen für die Immunisierungen. Es wurde der BL 21(DE3)-Stamm verwendet. 500 ml Kulturen wurden bei einer OD600 von ca. 0,5 mit 500 µl IPTG (1 M) für 5 h induziert. Die Zellsuspension wurde für 1 h bei 4000 g und 4° C (BECKMAN COULTER GMBH) zentrifugiert. Das Zellpellet wurde in 15 ml Sambrock-Lysispuffer resuspendiert und mit 300 µl 40x Complete-Lösung versetzt. Hiervon wurden 5 ml-Fraktionen über 5 min mittels Ultraschall lysiert (30 sec an/15 sec aus). Das Zelllysat wurde durch dreißigminütige Zentrifugation bei 13000 g und 4°C geklärt und durch einen 0,2 µm Filter sterilfiltriert. Das so vorbereitete Zelllysat wurde 1:5 mit PBS verdünnt und in der Aufreinigung mittels FPLC eingesetzt. 10 ml des verdünnten Zelllysats wurden mit einer Flussgeschwindigkeit von 0,2 ml/min auf die Säule geladen. Anschließend wurde die Säule mit 30 ml Waschpuffer bei einer Flussgeschwindigkeit von 1 ml/min gewaschen. Die darauffolgende Elution fand mit 10 ml Elutionspuffer (10 mM Glutathion) bei einer Flussrate von 0,5 ml/min statt. Das eluierte Protein wurde in 1 ml-Fraktionen gesammelt und bei –80°C gelagert. Material und Methoden 87 3.4.5 Darstellung von Proteinen 3.4.5.1 SDS-Polyacrylamid-Gelelektrophorese Die SDS-Page nach LÄMMLI (1970) ermöglicht die Auftrennung von Proteinen nach ihrem Molekulargewicht. Natriumdodecylsulfat (sodiumdodecylsulfate, SDS) ist ein anionisches, amphipatisches Detergenz. Durch Bindung an die kationischen Reste von Proteinen erhalten diese eine negative Gesamtladung und können so im elektrischen Feld wandern. Des Weiteren wird die zu untersuchende Probe durch die Zugabe von DTT (Dithiothreitol) und Erhitzen denaturiert, wodurch die native Oberflächenstruktur keinen Einfluss mehr auf die Wanderungseigenschaften der Proteine hat. Unter diesen Bedingungen ist die elektrophoretische Beweglichkeit der meisten Proteine Trägermedium dem dienen Logarithmus ihrer Polyacrylamidgele. Masse Bei der direkt proportional. Polymerisation Als entstehen abhängig von den Konzentrationen an Acrylamid und dem quervernetzenden Reagenz Bisacrylamid unterschiedlich große Poren, die wie ein Sieb wirken. Kleinere Moleküle vermögen die Netzstruktur schneller zu durchwandern als große, wobei eine hohe Trennschärfe erreicht wird. Auf das eigentliche Trenngel wird ein großporiges Sammelgel gegossen. Dieses wird von den Proteinen schnell durchwandert und die Proben sammeln sich in einem engen Bereich an der Grenze zum Trenngel. Hierdurch erhalten alle die gleiche Ausgansposition, wodurch die Trennschärfe nochmals erhöht wird. Es wurde die vertikale Elektrophoresekammer Mini Protean II™ der Firma BIO-RAD eingesetzt. Alle selbstgegossenen Gele hatten eine Konzentration von 10% Acrylamid und eine Dicke von 1 mm. Das Trenngel wurde mit A. bidest. überschichtet, um eine gerade Oberkante zu erhalten. Nach einer Polymerisationszeit von einer Stunde wurde das Wasser entfernt und das Trenngel mit einem 5%igen Sammelgel überschichtet. In dieses wurden 10- oder 15-wellKämme zu Erzeugung von Probentaschen eingesetzt. Nach einer halbstündigen Polymerisationszeit wurden die Kämme entfernt, zwei Gelkassetten in den Elektrophoreseständer eingespannt und die innere Pufferkammer mit 1x Elektrophoresepuffer gefüllt. Die Proben wurden 1:2 mit SDS-Probenpuffer versetzt und für fünf Minuten bei 99°C erhitzt. Dem SDS-Probenpuffer wurde vorher 10% 88 Material und Methoden frisch aufgetautes DTT (1 M) zugesetzt. Die Proben wurden mit ausgezogenen Pipettenspitzen (ROTH) in die Probentaschen verbracht. Zur Ermittlung der Molekulargewichte wurde in einer Spur ein Proteingrößenstandard aufgetragen. Zum Einsatz kam entweder der Marker Roti®-Mark Prestained (ROTH) oder der SeeBlue® Prestained Standard (INVITROGEN). Die Elektrophoresekammer wurde in den mit 1x Elektrophoresepuffer gefüllten Puffertank eingesetzt. Für das Passieren des Sammelgels wurde anfangs für 20 Minuten eine Spannung von 70 V angelegt, die anschließend auf 120 V erhöht wurde. Nach 70 – 90 Minuten hatte die Lauffront die untere Gelkante erreicht, und die Elektrophorese wurde beendet. 3.4.5.2 Coomassie-Brilliant-Blue-Färbung von Proteinen Zur direkten Sichtbarmachung der Proteine wurde das Gel im Anschluss an die Elektrophorese für eine Stunde in Coomassie-Brilliant-Blue-Färbelösung eingelegt und auf einem Wipptisch (FRÖBEL) geschwenkt. Der Farbstoff bindet unspezifisch an Proteine, wobei die Detektionsgrenze ca. 0,1 µg pro Bande beträgt. Um überschüssige Hintergrundfärbung zu entfernen, wurde das Gel unter ständigem Schwenken in Entfärbelösung für mehrere Stunden bis zur deutlichen Sichtbarwerdung der Proteinbanden entfärbt. Für die Langzeitlagerung wurden die Gele zwischen zwei mit Geltrocknungslösung befeuchteten Membranen (Gel Drying Film, PROMEGA) in einen Rahmen gespannt und über mehrere Tage getrocknet. 3.4.5.3 Transfer Blot Statt einer direkten Färbung im Gel können Proteine im Anschluss an die Gelelektrophorese auf eine Nitrocellulosemembran übertragen und mittels WesternBlot spezifisch nachgewiesen werden. Bei der Übertragung macht man sich wiederum die negative Ladung der SDS-beladenen Proteine zunutze. Das Gel wird nach der Elektrophorese auf eine Nitrocellulosemembran verbracht. Durch Anlegen einer Spannung senkrecht zur ursprünglichen Laufrichtung verlassen die Proteine das Gel und werden an die Membran gebunden. Die Übertragung erfolgte in einer Semi-Phor™-Blotkammer (HOEFER) nach dem Semi-Dry-Verfahren. Drei mit Blotpuffer getränkte Filterpapiere (MACKEREY- Material und Methoden 89 NAGEL) wurden auf den Boden (Anode) der Blotkammer gelegt. Auf die Filterpapiere wurde luftblasenfrei die zuvor einige Minuten in Blotpuffer equilibrierte Nitrocellulosemembran (porablot NCL, MACKEREY-NAGEL) verbracht. Auf die Membran wurde das Gel gelegt und mit drei blotpuffergetränkten Filterpapieren abgedeckt. Der Proteintransfer erfolgte bei einer konstanten Stromstärke von 100 mA für 90 min (BIO-RAD). 3.4.5.4 Western-Blot Im Western-Blot können Proteine mithilfe einer Antigen-Antikörper-Reaktion spezifisch detektiert werden. Hierfür wird die Membran mit den daran gebundenen Proteinen in einer Lösung inkubiert, die für das nachzuweisende Protein spezifische Antikörper (Primärantikörper) enthält. Diese binden an das entsprechende Protein und fungieren im zweiten Schritt selbst als Antigen. Durch Zugabe eines zweiten, an Epitope des Primärantikörpers bindenden Antikörpers (Sekundärantikörper oder Konjugat), erfolgt die Detektion. Der Sekundärantikörper ist mit einem Enzym gekoppelt, das eine Farbreaktion katalysiert. Hierdurch werden Proteinbanden spezifisch sichtbar gemacht. Bei den hier eingesetzten Konjugaten handelt es sich um Alkalische Phosphatase gekoppelte Antikörper. Als Substrat dienen die beiden Farbstoffe NBT (Nitroblau Tetrazolium) und BCIP (5-Brom-4-Chlor-3- Indolylphosphat). Die Alkalische Phosphatase (AP) spaltet die Phospatgruppe des BCIP ab und reduziert NBT. Dephosporyliertes BCIP erscheint blau und reduziertes NBT bildet einen violetten, schwerlöslichen Niederschlag. So entsteht in den Bereichen, in denen AP-gekoppelte Antikörper gebunden wurden, aus beiden Farbstoffen ein blauviolettes Präzipitat und die Proteinbanden werden sichtbar. Alle Schritte des Western-Blots wurden auf einem Schwenktisch (FRÖBEL) durchgeführt. Nach erfolgtem Proteintransfer wurde die Membran für fünf Minuten in TBS-Puffer geschwenkt. Daraufhin erfolgte die Blockierung freier Bindungsstellen durch eine einstündige Inkubation in Roti®-Block (ROTH). Für eine 5,5 cm x 9 cm große Membran wurden 50 ml einer 1:10 Verdünnung des Reagenz eingesetzt. Anschließend wurde die Membran dreimal für fünf Minuten mit ca. 50 ml TBSTween-Puffer gewaschen. Im nächsten Schritt erfolgte eine einstündige Inkubation mit dem Primärantikörper und darauffolgend erneut drei Waschschritte mit TBS- 90 Material und Methoden Tween-Puffer. Nach Zugabe des Konjugats und einstündiger Inkubation wurde die Membran einmal für fünf Minuten mit TBS-Tween und zweimal mit TBS gewaschen. Anschließend wurde die Westernblot-Substratlösung zugegeben und bis zur deutlichen Sichtbarwerdung der Proteinbanden inkubiert. Die Reaktion wurde mit A. bidest. abgestoppt. Folgende Primärantikörper und Konjugate wurden eingesetzt: Primärantikörper Konjugat Maus-anti-PDI (BIOMOL) Ziege-anti-Maus IgG (SIGMA) Maus-anti-GST IgG (SIGMA) Ziege-anti-Maus IgG (SIGMA) 3.4.6 Bestimmung der Proteinkonzentration 3.4.6.1 Bradford Der Bradford Assay nutzt die Eigenschaft des Farbstoffs Coomassie Brilliant Blue G250 abhängig von den Reaktionsbedingungen, in verschiedenen Formen vorzuliegen (BRADFORD 1976). Unter sauren Bedingungen liegt der Farbstoff überwiegend als protoniertes Kation mit braunroter Farbe und einem Absorptionsmaximum bei 470 nm vor. Durch Bindung an Proteine nimmt der Farbstoff eine stabile, unprotonierte Form ein. Diese Reaktion führt zu einem blauen Farbumschlag und damit zu einer Verschiebung des Absorptionsmaximums zu 595 nm. Die Absorption bei 595 nm korreliert dabei direkt mit der Proteinmenge und ermöglicht so eine Quantifizierung. Es wurde der Quick Start™ Bradford Protein Assay (BIORAD) entsprechend den Herstellerangaben für den 1 ml-Küvettenansatz durchgeführt. Mittels des zum Kit gehörigen BSA-Standards wurde eine Standardverdünnungsreihe erstellt. Die Einstellung „Protein 595 nm Assay“ des Photometers (AMERSHAM BIOSCIENCES) berechnete aus den Absorptionswerten der eingegebenen Verdünnungsstufen eine Eichgrade, anhand derer die Proteinkonzentration der unbekannten Lösung automatisch ermittelt wurde. Da sich mit der Methode nach Bradford nur der Gesamtproteingehalt einer Lösung bestimmen läßt, die Proteinaufreinigung aber kein 100%ig reines Fusionsprotein Material und Methoden 91 lieferte, diente dieser Assay nur als grobe Richtlinie zur Abschätzung der bei den Aufreinigungen gewonnenen Proteinmenge. Um genauere Aussagen über den Anteil des Fusionsproteins an der Gesamtproteinlösung zu erhalten, wurde der Agilent 2100 Bioanalyzer eingesetzt. 3.4.6.2 Agilent 2100 Bioanalyzer Die „Lab on a chip“-Technologie des Agilent 2100 Bioanalyzers (AGILENT TECHNOLOGIES) ermöglicht es, unterschiedliche Fraktionen einer Proteinlösung quantitativ zu bestimmen. Das Grundprinzip des Bioanalyzers beruht auf der klassischen Gelelektrophorese, die auf ein Chipformat verkleinert wurde. Ermöglicht wird dies durch die Technologie der Mikrofluidik, der Steuerung winziger Flüssigkeitsmengen in miniaturisierten Systemen. Hierdurch wird die Trennzeit und die Menge des einzusetzenden Probenmaterials stark reduziert. Im Inneren eines Glaschips befinden sich winzige, eingeätzte Mikrokapillaren, die auf der Oberseite des Chips in größeren Öffnungen zum Einfüllen des Probenmaterials münden. Die Trennmatrix besteht wie bei der klassischen Gelelektrophorese aus einem gelartigen Polymer. Dieses wird zunächst zusammen mit einem Fluoreszenzfarbstoff in die Mikrokapillaren gefüllt. Anschließend werden die zu untersuchenden Proben und der Größenstandard in die entsprechenden Öffnungen pipettiert. Der so vorbereitete Chip wird in das Messgerät eingelegt. Von diesem Punkt an ist der ganze Prozess vollkommen automatisiert. Die Proben werden automatisch nacheinander aus den Probenkapillaren in den Trennkanal injiziert und durch den Siebeffekt des Polymers elektrophoretisch aufgetrennt. Durch laserinduzierte Fluoreszenz werden die Proteine detektiert, und die erhaltenen Daten als Elektropherogramme (Peaks) und Gelbanden-ähnliche Bilder ausgegeben. Die Größenbestimmung erfolgt mittels einer Standardkurve, die das Programm anhand der gemessenen Daten für die Proteine des Größenstandards berechnet. Die Probenbehandlung erfolgte entsprechend den Angaben des Herstellers für den PROTEIN 200 PLUS Chip (AGILENT TECHNOLOGIES). Um möglichst genaue Messwerte zu erhalten, wurden mehrere Messungen auf mehreren Chips vorgenommen und die erhaltenen Konzentrationen gemittelt. 92 Material und Methoden 3.4.7 Aufkonzentrieren der Proteinlösung Um ein applikables Injektionsvolumen für die Immunisierung zu erhalten, war es notwendig, das aufgereinigte Protein zu konzentrieren. Hierzu wurden VivaSpin 4Säulen (SARTORIUS) mit einem MWCO (molecular weight cut off) von 50 kDa eingesetzt. Die Säulen wurden so lange bei 4000 g zentrifugiert, bis das Volumen auf ca. 1/10 reduziert war. 3.4.8 Identifizierung der PDI mittels MALDI-TOF-MS Aus einem Coomassie-gefärbten SDS-Gel wurde die Fusionsproteinbande mit einer sterilen Skalpellklinge ausgeschnitten und an die Firma TOPLAB, Martinsried, zur Analyse geschickt. Bei der MALDI-TOF-MS (Matrix-Assisted-Laser- Desorption/Ionisation-Time-Of Flight-Mass-Spectrometry / Matrix-unterstützte Laserdesorptions/Ionisations-Flugzeit-Massenspektrometrie) werden Proteine anhand charakteristischer Peptidmassen identifiziert. Hierzu wird eine Gelbande mit Trypsin verdaut und das entstehende Peptidgemisch auf einem Probenteller mit einer Matrix kokristallisiert. Durch Laserbeschuss werden Protonen von der Matrix auf die Peptide übertragen. Die protonierten Peptide desorbieren von der Oberfläche und werden in einem elektrischen Feld beschleunigt. Dann erfolgt die Messung der Flugzeit auf einer Driftstrecke im feldfreien Raum. Anhand der Flugzeit der Teilchen lässt sich ihre Masse berechnen. Da bei dem Verdau eines Proteins charakteristische Peptide entstehen, ist es möglich, durch den Vergleich der gemessenen Peptidmassen einer Probe mit den theoretisch verdauten Massen einer Proteindatenbank ein Protein zu identifizieren. 3.5 Tierexperimentelle Untersuchungen 3.5.1 Versuchsplan Es fanden zwei Immunisierungsversuche nach identischem Schema statt. Der erste Immunisierungsversuch wurde vom 05.04.2005 bis zum 29.07.2005, der zweite vom 01.09.2005 bis zum 16.12.2005 durchgeführt. Am Tag 0 erhielten die Rinder die erste Impfung. Drei und sechs Wochen später erfolgten die erste und zweite Material und Methoden 93 Boosterimpfung. Wiederum drei Wochen später fand die experimentelle Infektion der Rinder mit Lungenwurmlarven statt. Am Tag 98 bzw.99 wurden die Tiere seziert. Kotproben Beobachtung Untersuchung 0 21 42 I. II. III. Impfung Abb. 5: 63 70 84 98/99 Tage Infektion Sektion Schematische Darstellung des Versuchsablauf Parallel zur Protein Disulfid Isomerase und in Kombination mit dieser wurden weitere Vakzinekandidaten getestet. Immunisierungsversuch zwei Darüber verschiedene Gruppeneinteilung gestaltete sich wie folgt: hinaus wurden Adjuvantien im eingesetzt. ersten Die 94 Material und Methoden I. Immunisierungsversuch II. Immunisierungsversuch Tierzahl Protein(e) Adjuvans Tierzahl Protein(e) Adjuvans 4 - (Kontrolle) Al(OH)3 6 - (Kontrolle) Quil A 4 PDI Al(OH)3 4 ACE Quil A 4 PDI Quil A 4 PARA Quil A 4 PNMT Al(OH)3 4 PNMT Quil A 4 PNMT Quil A 4 4 Tab. 3: PDI + PNMT + MSP PDI + PNMT + MSP Al(OH)3 PDI + ACE + 6 PARA + PNMT + Quil A MSP Quil A Gruppeneinteilung der Immunisierungsversuche Die vorliegende Arbeit stellt nur die die Protein Disulfid Isomerase betreffenden Ergebnisse dar. Bezüglich der anderen Proteine sei auf folgende Arbeiten verwiesen: MSP (Major Sperm Protein): (VON HOLTUM 2006) PNMT (Phenylethanolamin-N- Methyltransferase): (STRUBE et al. 2006) PARA (Paramyosin): (STRUBE et al. 2006) ACE (Acetylcholinesterase): (nicht veröffentlicht) 3.5.2 Genehmigung Der für die hier beschriebenen tierexperimentellen Untersuchungen erforderliche Antrag auf Genehmigung eines Tierversuchsvorhabens wurde von Herrn Prof. Dr. T. Schnieder bei der Bezirksregierung Hannover gestellt und am 13.09.2004 unter dem Aktenzeichen 04/853 genehmigt. Material und Methoden 95 3.5.3 Tiermaterial Es wurden männliche Kälber der Rasse Holstein-Frisian eingesetzt. Diese hatten zu Versuchsbeginn ein Alter von vier bis fünf Monaten und wogen zwischen 99 und 178 kg. Die Tiere wurden von der Dücker GbR, Misselwarden bezogen. Bei der Einstallung wurden die Tiere gewogen und in vier Gewichtsklassen eingeteilt. Anschließend erfolgte die Randomisierung in die verschiedenen Gruppen. 3.5.4 Tierhaltung Die Rinder wurden im Tierhaus des Instituts für Parasitologie gehalten. Hierbei handelt es sich um Einstreuställe mit Zugang zu betonierten Ausläufen. Die Ställe wurden täglich gemistet. Um Einschleppungen zu verhindern, war jeder Stall mit einer Desinfektionswanne am Eingang und stalleigenen Reinigungsgerätschaften ausgestattet. Die Fütterung erfolgte mit Heu und Kraftfutter. Leitungswasser stand den Tieren aus Selbsttränken ad libitum zur Verfügung. Die Versorgung der Tiere wurde vom Tierpflegerpersonal durchgeführt. 3.5.5 Verwendete Adjuvantien und Zubereitung des Impfstoffs Im ersten Immunisierungsversuch wurden Aluminiumhydroxid (Al(OH)3) (GERBU) und Quil A (GERBU) als Adjuvantien eingesetzt. Al(OH)3 wurde in einer Dosierung von 400 µl (entsprechen 4 mg Aluminium) eingesetzt. Vom Quil A wurden 75 µl einer 1%igen Lösung pro Impfdosis eingesetzt (entsprechen 750 µg). Pro Impfdosis wurden 200 µg Protein eingesetzt. Im zweiten Immunisierungsversuch wurde ausschließlich Quil A als Adjuvans eingesetzt. Die Proteindosis betrug bei diesem Versuchsdurchgang 100 µg pro Impfung. Die Impfdosen wurden mit Trispuffer auf ein Volumen von 1,5 ml (Al(OH)3) bzw 1,0 ml (Quil A) aufgefüllt. Die Injektionen für die Kontrollgruppen enthielten den Elutionspuffer aus der Proteinaufreinigung (50 mM Tris-HCl, 10 mM Glutathion, pH 8,0). Das hiervon eingesetzte Volumen berechnete sich aus dem Mittelwert der Volumina aller eingesetzten Impfproteinlösungen. Die Impfdosen wurden jeweils frisch vor der Applikation angemischt und für 90 min auf einem Schwenktisch inkubiert. 96 Material und Methoden I. Immunisierungsversuch 610 µl Elutionspuffer Al(OH)3 Kontrollgruppe Quil A 400 µl Al(OH)3 Kontrollgruppe 490 µl Trispuffer 604 µl PDI Al(OH)3 Quil A 400 µl Al(OH)3 Impfgruppe Impfgruppe 496 µl Trispuffer 422 µl Elutionspuffer 75 µl Quil A 503 µl Trispuffer 258 µl PDI (387,9 µg/ml) 75 µl Quil A 667 µl Trispuffer 604 µl PDI (331,5 µg/ml) Quil A 75 µl Quil A Impfgruppe Tab. 4: II. Immunisierungsversuch 321 µl Trispuffer Zusammensetzung der Impfdosen 3.5.6 Applikation des Impfstoffs Die Applikation des Impfstoffs bzw. der Kontrollsubstanz erfolgte intramuskulär in die seitliche Halsmuskulatur. Um eventuell auftretende lokale Impfreaktionen besser beurteilen zu können, wurde an der Applikationsstelle ein ca. 6 cm x 6 cm großes Hautareal frei geschoren. Bei den Kombinationsgruppen erfolgte die Applikation der verschiedenen Proteine einzeln. Um lokale Impfreaktionen voneinander abgrenzen zu können, wurde zwischen den freigeschorenen Hautarealen ein Abstand von mindestens fünf Zentimetern eingehalten. 3.5.7 Experimentelle Infektion An den Tagen 63, 64 und 65 erfolgte die experimentelle Infektion mit jeweils 1100 infektiösen D. viviparus L3, die, wie unter 3.2.1 beschrieben, von Stammhaltungsrindern gewonnen worden waren. Hierzu wurde die Larvendosis in Leitungswasser mit einer 50 ml Spritze aufgenommen und über den Zungengrund oral appliziert. 3.5.8 Überwachung der Tiergesundheit Während des gesamten Versuchs erfolgte eine tägliche Beobachtung der Tiere, die ab Tag 70 zu einer täglichen Untersuchung erweitert wurde. Die tägliche Beobachtung beinhaltete die Beurteilung des Allgemeinbefindens, des Auftretens von Husten und Nasenausfluss sowie lokaler Reaktionen an der Injektionsstelle. Ab Material und Methoden 97 Tag 70 wurden zusätzlich täglich Atem- und Herzfrequenz, Atemcharakter und Körpertemperatur bestimmt. Darüber hinaus wurden die Rinder am Tag der Einstallung (-7), an den drei Impfterminen (0, 21, 42), bei der ersten Infektion (63) und am Tag der Schlachtung (98/99) einer ausführlichen Allgemeinuntersuchung unterzogen und gewogen. 3.5.9 Medikamentöse Behandlung erkrankter Tiere Tiere, die im Versuchsverlauf deutliche Störungen des Allgemeinbefindens mit oder ohne Auftreten von Fieber zeigten, wurden medikamentös behandelt. Folgende Präparate kamen zum Einsatz: • Baytril® 10%, BAYER, Leverkusen Wirkstoff: Enrofloxacin 100 mg/ml; Dosierung: 5 ml/100 kg/p.d., i.v., 5 Tage • Finadyne® RPS, ESSEX TIERARZNEI, München Wirkstoff: Flunixin-Meglumin 50 mg/ml; Dosierung: 2,2 ml/50 kg/p.d., i.v. • Draxxin®, PFIZER, Karlsruhe Wirkstoff: Tulathromycin 100 mg/ml; Dosierung: einmalig 1 ml/40 kg, s.c. 3.5.10 Gewinnung von Probenmaterial und dessen Aufbereitung 3.5.10.1 Blutproben An den Tagen –7 und 0 und darauffolgend zweimal wöchentlich (Tag 4, 7, 11, 14 etc.) erfolgte die Entnahme von Blutproben. Nach Punktion der Vena jugularis mittels Strauss Kanüle (DISPOMED) wurden ca. 8 ml Blut in Serum-Röhrchen (Monovette® Serum, SARSTEDT) entnommen. Nach Inkubation über Nacht im Kühlschrank wurden die Blutproben für 20 Minuten bei 3000 g (HERAEUS) zentrifugiert, der Serumüberstand mit einer Pasteurpipette in ein 12 ml Gefäß überführt und bei –20°C gelagert. An den Tagen 0 (erste Immunisierung), 63 (Infektion), 84 (21 dpi), 91 (28 dpi) und 98/99 (Sektion) wurden zusätzlich EDTA-Blutproben (Monovette® EDTA, SARSTEDT) gewonnen. Diese wurden umgehend an die Klinik für kleine Klauentiere 98 Material und Methoden der Tierärztlichen Hochschule Hannover zur Bestimmung des Differentialblutbildes übergeben. 3.5.10.2 Kotproben An drei Tagen vor der ersten Immunisierung sowie an den drei Immunisierungstagen und am ersten Tag der experimentellen Infektion wurden zur Verifizierung der Lungenwurmfreiheit der Versuchsrinder Kotproben untersucht. Ab Tag 84 erfolgte eine tägliche Überprüfung der Larvenausscheidung. Der rektal entnommene Kot wurde gründlich vermengt und zwei mal zehn Gramm (Doppelansatz) von jedem Tier dem Trichterauswanderverfahren nach BAERMANN unterzogen. Hierbei wurden die eingesetzten Siebe noch mit einer einzelnen Lage Zellstoff ausgekleidet, um möglichst wenig Kotpartikel im abgelassenen Larvenmaterial zu erhalten, da diese das Auszählen der Larven erheblich erschweren. Am darauffolgenden Tag wurden die Larven in Zählkammern abgelassen und unter dem Mikroskop (ZEISS) ausgezählt. 3.5.11 Sektion Am Tag 98 bzw 99 (35/36 dpi) wurden die Rinder im Institut für Pathologie der Tierärztlichen Hochschule Tierschutzschlachtverordnung Hannover erfolgte eine getötet. Betäubung Entsprechend der Tiere der mittels Bolzenschuß, gefolgt von einem Ausbluten durch Eröffnen beider Vv. jugularia. Die Lungen wurden einschließlich der Trachea entnommen und einer makroskopischen Beurteilung unterzogen. Die pathologischen Veränderungen wurden für jeden Lungenlappen nach Art und Ausdehnung beschrieben. 3.5.12 Größenbestimmung der adulten Stadien Aus den exenterierten Lungen wurden die adulten D. viviparus-Stadien wie unter 3.2.2 beschrieben gewonnen. Pro Versuchstier wurden 25 männliche und 25 weibliche Würmer mit einer Digitalkamera (OLYMPUS) fotografiert. Mithilfe der Olympus Software 5050 wurden die Länge und Breite der Würmer bestimmt. Material und Methoden 99 3.6 Serologische Untersuchungen 3.6.1 Enzyme linked immunosorbant assay (ELISA) Mittels ELISA wurden die Seren der Versuchtiere auf PDI-spezifische Antikörper untersucht. Das Prinzip des hier eingesetzten ELISAs ist eine Antigen-AntikörperBindung, die mit einem enzymgekoppelten Sekundärantikörper (Konjugat) nachgewiesen wird. Der enzymatische Umsatz eines zugegebenen Substrats führt zu einer Farbreaktion, die photometrisch quantifiziert werden kann. Bei den hier eingesetzten Konjugaten handelte es sich um Horseradish Peroxidase (HRP)gekoppelte Anti-Rind-Antikörper. HRP setzt das Substrat ortho-Phenylendiamin (OPD) zu einem orangefarbenen Produkt um, dessen Absorption bei 490 nm gemessen wird. Als Antigen wurde das für den zweiten Immunisierungdurchgang aufgereinigte GSTPDI-Fusionsprotein eingesetzt. In Vorversuchen wurden durch Schachbretttitrationen geeignete Verdünnungsstufen für Antigen, Serum und Konjugat ermittelt. Hierbei wurden folgende Verdünnungen überprüft: • PDI: zweifach Verdünnungen von 10 µg/well bis 0,039 µg/well • Serum: 1:5, 1:10, 1:20, 1:40, 1:80 • Konjugat: 1:5000, 1.10000, 1:20000 Als Mikrotiterplatten wurden Nunc Immobilizer™ Amino (NUNC) 96-well-Platten eingesetzt. Die Platten wurden mit 100 µl der Protein-PBS-Verdünnung (entsprechen 0,31 µg Protein) pro Vertiefung beschichtet, mit einer selbstklebenden Folie abgedeckt und über Nacht (mind. 16 h) bei 4°C inkubiert. Am darauffolgenden Tag wurde die Proteinlösung abgeschüttet und die Wells mit PBS-Tween dreimal für fünf Minuten mit einem Mehrkanal-ELISA-Washer (nunc-Immuno™ wash 12; NUNC) gewaschen. Durch den Einsatz von Tween in der Waschlösung konnte bei diesen Platten auf einen Blockierungsschritt verzichtet werden. Nach dem Waschen wurden die Mikrotiterplatten durch Ausschlagen auf einem sauberen Handtuch getrocknet. Anschließend wurden die zu untersuchenden Seren 1:10 mit PBS-Tween verdünnt 100 Material und Methoden und hiervon 100 µl/Well im Doppelansatz aufgetragen. Auf jeder Platte wurden ein Plattenleerwert, eine Positivkontrolle und eine Negativkontrolle mitgeführt. Der Plattenleerwert wurde hierbei über Wells definiert, die zwar mit Protein, jedoch nicht mit Serum und Konjugat beschichtet wurden. Die Negativkontrolle lieferten Wells, die mit Serum von D. viviparus-Stammhaltungsrindern vor der Infektion beschichtet wurden. Da kein geeignetes externes Positivserum zur Verfügung stand, wurde die Positivkontrolle vom gepoolten Serum der PDI-immunisierten Rinder am Tag 21 verkörpert. Die mit Folie abgedeckten Platten wurden für eine Stunde bei 37°C (HEREAUS) im Glasperlenwasserbad inkubiert. Nach Abschütten der Serumverdünnung und dreimaligem Waschen mit PBS-Tween wurden die Platten mit einer 1:5000 Verdünnung der Konjugate (100 µl/Well) beschichtet und wiederum für eine Stunde bei 37°C inkubiert. Eingesetzte Konjugate IgG1:HRP IgG2:HRP IgG:HRP IgA:HRP IgM:HRP Schaf-anti-Rind (SEROTEC) Nach Entfernen der Konjugatlösung und erneutem Waschen mit PBS-Tween, wurden 50 µl der Substratlösung in jede Vertiefung pipettiert und die Mikrotiterplatte für 10 min unter Lichtabschluss bei Raumtemperatur inkubiert. Nach exakt 10 min wurde die Reaktion mit 50 µl 2,5 M Schwefelsäure pro Vertiefung abgestoppt, und die Platten unmittelbar im ELISA-Reader (BIO TEK) bei einer Wellenlänge von 490 nm eingelesen. Zur Auswertung der ELISAs wurden, um tages- und materialabhängige Schwankungen zu verringern, aus den Rohdaten Indexwerte gebildet, die nach folgender Formel berechnet wurden: INDEX = Mittelwert Probe – Mittelwert Leerwert Mittelwert Positivkontrolle – Mittelwert Leerwert Material und Methoden 101 3.6.2 Überprüfung der Serokonversion mittels Western-Blot Da es nicht gelang, rekombinante PDI ohne GST und ohne E. coli- Proteinverunreinigungen zu gewinnen, wurde die Serokonversion zusätzlich im Western-Blot überprüft, um auszuschließen, dass die im ELISA ermittelten Anstiege der OD ausschließlich auf einer Reaktion auf GST bzw. E. coli-Kontaminanten beruhten. Hierzu wurde eine SDS-Page mit D. viviparus-Rohantigen wie unter 3.4.5.1 durchgeführt. Es wurde ein Probenkamm eingesetzt, der eine Probentasche über die gesamte Breite des Gels erzeugte. Nach erfolgter Gelelektrophorese wurden die Proteine wie unter 3.4.5.3 beschrieben auf eine Nitrocellulosemembran geblottet. Anschließend wurde die Membran in 4 mm breite Streifen geschnitten. Im WesternBlot wurden die Streifen mit den Seren der PDI-immunisierten Tiere und der Kontrollgruppe aus dem I. Immunisierungsversuch vom Tag 0, Tag 63 und Tag 98 sowie mit einem Positiv- und einem Negativserumpool geprüft. Die Seren wurden in einer Verdünnung von 1:20 mit TBS-Tween eingesetzt und als Konjugat diente ein AP-gekoppelter Maus-Anti-Rind-Antikörper (SIGMA) in einer Verdünnung von 1:30000. 3.7 Quantitative real-time PCR (QRT-PCR) Die quantitative real-time PCR (QRT-PCR) wurde eingesetzt, um zu überprüfen, ob die Immunisierung der Rinder mit PDI Einfluss auf die Expression der PDI in den Würmern hatte. Hierzu wurden je fünf männliche und weibliche Würmer der sechs Kontroll- und Kombitiere des zweiten Immunisierungsversuchs untersucht. 3.7.1 Allgemeines zur Methode Mithilfe der quantitativen real-time PCR lassen sich PCR-Produkte in „Echtzeit“ (realtime) während der laufenden PCR detektieren und anschließend quantifizieren. Dies ermöglicht einen Rückschluss auf die Menge an Ausgangs-DNA und damit auch auf die Transkriptionsrate eines bestimmten Gens. In der QRT-PCR werden neben Primern fluoreszenzmarkierte Sonden eingesetzt. Hierbei handelt es sich um DNASonden, die zu einem internen Bereich des Amplifikats komplementär sind. Die 102 Material und Methoden Sonden sind an beiden Enden mit Fluoreszenzfarbstoffen markiert. Der sogenannte Reporterfarbstoff am 5’-Ende besitzt eine Emissionswellenlänge, die der Anregungswellenlänge des als Quencher bezeichneten Farbstoffs am 3’-Ende entspricht. Bei intakter Sonde befinden sich die beiden Farbstoffe in enger räumlicher Nähe und es kommt zu einem als Fluoreszenz-Resonanz-Energie-Transfer (FRET) bezeichneten Mechanismus. Der Quencherfarbstoff nimmt die Emissionsenergie des Reporterfarbstoffs auf, wodurch dessen Lichtemission unterdrückt wird. Die in der QRT-PCR eingesetzte Thermus aquaticus (Taq)-DNA Polymerase besitzt eine 3’→ 5’-Exonukleaseaktivität. Hierdurch werden während der Extensionsphase vom 5’Ende doppelsträngiger DNA Nukleotide abgespalten. Erreicht die Taq-Polymerase die Sonde, wird diese hydrolysiert und die räumliche Nähe zwischen Reporter und Quencher aufgehoben. Hierdurch kann der Quencher den Reporter nicht mehr unterdrücken und dieser sendet Lichtenergie aus, die detektiert werden kann. Die Höhe des Fluoreszenzsignals ist hierbei direkt proportional zur Anzahl hydrolysierter Sondenmoleküle, wodurch auf die in der Probe vorhandene DNA-Menge des untersuchten Gens geschlossen werden kann. Da es einerseits in der frühen Phase der Reaktion einige Zyklen braucht, bis ein sich deutlich von der Hintergrundfluoreszenz abhebendes Signal entsteht und es andererseits gegen Ende der Reaktion durch Abnahme der Reaktionskomponenten oder Funktionsverlust der Polymerase zu einer Verlangsamung der Reaktion kommt, hat der Fluoreszenzanstieg nur dazwischen exponentiellen Charakter und nur dieser Zeitpunkt der Reaktion kann zur Quantifizierung herangezogen werden. Als Bezugsgröße für die Quantifizierung dient ein Reaktionszyklus an dem das Fluoreszenzsignal den Schwellenwert (threshold) der Hintergrundfluoreszenz konstant überschreitet. An diesem als Ct (cycle-threshold) bezeichneten Zyklus befindet sich in allen Reaktionsgefäßen die gleiche Menge an synthetisierter DNA und über die Höhe des Ct kann auf die Ausgangsmenge an DNA geschlossen werden. Befindet sich in einer Probe viel Ausgangsmaterial, wird der Schwellenwert bei einer niedrigen Zyklenzahl überschritten, also ein niedriger Ct erreicht und umgekehrt. Da die Hintergrundfluoreszenz in verschiedenen Proben unterschiedlich Material und Methoden 103 sein kann, wird den Proben ein Referenzfarbstoff zugesetzt und dessen Fluoreszenz zur Ermittlung des Schwellenwertes herangezogen. Reporter Quencher hν Sonde Vorwärtsprimer Denaturierung hν Annealing hν Extension Abb. 6: Taq Prinzip der QRT-PCR mit TaqMan™-Sonden Um verschiedene Proben vergleichen zu können, wird die Expression eines Zielgens in Relation zu einem zweiten Gen dargestellt. Dieses als „Housekeeping Gen“ (HKG) bezeichnete Referenzgen unterliegt nicht der Regulation, sondern wird homogen exprimiert. Als HKG wurde die 18S-rRNA gewählt. 104 Material und Methoden 3.7.2 Konstruktion der Primer und Sonden Das Design der Sonde und der zugehörigen Primer erfolgte mit der Primer Express™-Software der Firma APPLIED BIOSYSTEMS. Hierbei wurde der Sequenzbereich zur Positionierung von Primern und Sonde auf einen Intronüberspannenden Bereich eingeengt. Dies sollte verhindern, dass eine Amplifikation an eventuell vorhandenen Verunreinigungen mit genomischer DNA stattfindet. Für die PDI-Sonde konnte eine passende Primer-Sonden-Kombination im Bereich des ersten Introns identifiziert werden. Hierbei lagen der Vorwärtsprimer und die Sonde in 5’-3’-Richtung vor dem Intron und der Rückwärtsprimer über der Introngrenze. Als Reporterfarbstoff wurde FAM (6-Carboxy-Fluorescein), als Quencherfarbstoff TAMRA (6-Carboxy-Tetramethylrhodamin) gewählt. Die Primersynthese erfolgte bei der Firma INVITROGEN. Die Synthese der Sonden wurde bei der Firma APPLIED BIOSYSTEMS in Auftrag gegeben. Bezeichnung Sequenz 5’→ 3’ Tm PDI QPCR for CTTGCCGAATTTTATGCTCCTT 58°C PDI QPCR rev GTGTGGCAGCTTTGGCATATT 59°C PDI-Sonde: 6-FAM-CGGTCACTGTAAGGCACTTGCTCCG-TAMRA 68°C 18S QPCR for CGTCATTGCTGCGGTTAAAA 58°C 18S QPCR rev CGCCAAAGGCGAATCG 58°C 18S-Sonde: 6-FAM-CGTAGTTGGATCTGAGTTACATGCAA-TAMRA 68°C Tab. 5: In der QRT-PCR verwendete Primer und Sonden 3.7.3 Gesamt-RNA Isolierung Zum Einsatz in der cDNA-Synthese für die quantitative real-time PCR wurde RNA von den aus den Lungen der Versuchsrinder isolierten adulten Lungenwürmern gewonnen. Da ribosomale RNA keine Poly-A-Sequenz am 3’-Ende aufweist, über die eine Aufreinigung wie unter 3.3.1 möglich wäre, war es erforderlich, Gesamt-RNA zu isolieren. Dies erfolgte mit dem Trizol®-Reagenz (INVITROGEN) nach Angaben des Material und Methoden Herstellers. Es handelt 105 sich hierbei um eine Modifikation der sauren Guanidinisothiozianat-Phenol-Chloroform-Extraktion (CHOMCZYNSKI u. SACCHI 1987). Das Reagenz enthält Guanidinisothiozianat und Phenol in einer monophasischen Lösung. Die in 500 µl GIT-Puffer gelagerten Würmer wurden bei Raumtemperatur aufgetaut und mehrfach homogenisiert. Nach Zugabe von 1 ml Trizol®, 200 µl Chloroform und dreiminütiger Inkubation bei Raumtemperatur folgte ein fünfzehnminütiger Zentrifugationsschritt bei 12000 g und 4°C (EPPENDORF). Hierdurch erfolgte eine Phasenseparation in eine obere wässrige, RNA-haltige Phase, eine DNA-haltige Interphase und eine untere organische, proteinhaltige Phase. Die wässrige Phase wurde in ein neues 2 ml DEPC-Gefäß überführt. Durch Zugabe von 500 µl Isopropylalkohol erfolgte die Fällung der RNA. Nach zehnminütiger Inkubation bei Raumtemperatur wurde die RNA durch Zentrifugieren bei 12000 g und 4°C über zehn Minuten pelletiert. Der Überstand wurde entfernt und das RNA-Pellet mit 1 ml 75%igem Ethanol/DEPC-Bidest gewaschen. Nach fünfminütiger Zentrifugation bei 7500 g und 4°C und Entfernen des Alkohols, wurde das RNA-Pellet in 9 µl DEPCBidest resuspendiert und zum Schutz vor Abbau durch RNAsen mit 1 µl (40 U) RNAsin™ Ribonuclease Inhibitor (PROMEGA) versetzt. 3.7.4 DNAse-Verdau der Gesamt-RNA Um eventuell vorhandene DNA-Verunreinigungen der Gesamt-RNA zu entfernen, schloss sich an die RNA-Isolierung ein DNAse-Verdau an. Hierzu wurde die resuspendierte Gesamt-RNA mit 1 µl DNAse I (RQ1 Rnase-Free Dnase, PROMEGA) und 1 µl des zugehörigen 10x-Puffers versetzt und für 30 min bei 37°C inkubiert. Anschließend erfolgte die Zugabe von 1 µl Stop-Lösung (20 mM EGTA, PROMEGA) und eine zehnminütige Inkubation bei 65°C (BIOMETRA), um die DNAse zu inaktivieren. Von diesem Ansatz wurden 2 µl für die Konzentrationsbestimmung entnommen. 3.7.5 cDNA-Synthese für die quantitative real-time PCR Die hergestellte cDNA sollte für die Quantifizierung der PDI und eines weiteren, in einer anderen Arbeit untersuchten Gens, dem Major Sperm Protein (MSP) eingesetzt 106 Material und Methoden werden. Da ribosomale RNA keine Poly-A-Sequenz am 3’-Ende aufweist, war es nicht möglich, in der reversen Transkription ausschließlich einen Oligo(dT)-Primer einzusetzten. Deshalb wurden neben einem Oligo(dT)-Primer zusätzlich genspezifische Rückwärtsprimer eingesetzt, die später auch in der quantitativen realtime PCR Anwendung fanden. In einem 0,2 µl PCR-Gefäß wurden 8 µl Gesamt-RNA mit je 1 µl Oligo(dT)-Primer (50 µM, INVITROGEN), PDI QPCRrev-Primer (50 µM), MSP QPCRrev-Primer (50 µM), 18S QPCRrev-Primer (50 µM) und 1 µl dNTPs (10 mM) versetzt und für 7 Minuten bei 70°C inkubiert (Uno Thermoblock, BIOMETRA). Anschließend wurde der Ansatz sofort für 2 min auf Eis gekühlt und für einige Sekunden herunter zentrifugiert. Daraufhin erfolgte die Zugabe von 4 µl 5x Erststrang-Puffer, 1 µl DTT (10 mM), 1 µl SuperScript™ III Reverse Transkriptase und 1 µl DEPC-Bidest. Dieser 20 µl-Ansatz wurde für 60 Minuten bei 55°C inkubiert. Zur Inaktivierung der Reversen Transkriptase erfolgte ein abschließender Inkubationsschritt bei 72°C für 10 Minuten. Die cDNA wurde bei –20°C gelagert. 3.7.6 Absolute Quantifizierung Bei der absoluten Quantifizierung werden die am Ct vorliegenden Kopienzahlen des Zielgens anhand einer Standardreihe bestimmt. Hierzu wurden die in der QRT-PCR als Template dienenden Sequenzbereiche der PDI und der 18S-rRNA in einer konventionellen PCR amplifiziert: 50 µl Ansatz: 5,0 µl 10X PCR-Puffer (QIAGEN) 2,0 µl dNTPs (2 mM) (PROMEGA) 3,0 µl MgCl2 (25 mM) (QIAGEN) 1,0 µl Vorwärtsprimer (50µM) 1,0 µl Rückwärtsprimer (50µM) 0,1 µl Taq DNA Polymerase (QIAGEN) 36,9 µl 1,0 µl AMPUWA cDNA Temperaturprofil 95°C 15 min 35 Zyklen 95°C 1 min 61°C 1 min 72°C 1 min 72°C 10 min Material und Methoden 107 Nach Klonierung in den pCR®4-TOPO®-Vektor (3.3.7.1), Transformation in E. coli TOP 10-Zellen (3.3.8.1), Anzucht und Midiprep (3.3.9.2) wurde die Konzentration der Plasmid-DNA photometrisch bestimmt und eine Verdünnungsreihe mit 100 bis 107 Kopien/µl erstellt. Die Verdünnungsreihe wurde in der QRT-PCR eingesetzt. Aus den Cts der Verdünnungsstufen wurde eine Kalibrierungskurve erstellt, anhand derer die Kopienzahl einer Probe über den Ct der Probe bestimmt werden konnte. 3.7.7 Relative Quantifizierung Bei der relativen Quantifizierung werden keine Kopienzahlen bestimmt, sondern die Expression des Zielgens in einer Probe wird auf eine Standardprobe (Kalibrator) bezogen. Der in der untersuchten Probe ermittelte Ct für das Zielgen wird über den Ct des Referenzgen in der Probe normalisiert und unter Einbeziehung der PCREffizienz der beiden Gene auf den normalisierten Ct des Kalibrators bezogen. Als Kalibrator wurde ein männlicher Wurm aus der Kontrollgruppe willkürlich gewählt. Die relative Quantifizierung wurde mit dem „Comparative Quantitation“ Programm des Mx4000®, sowie mit der Effizienz-korrigierten Formel nach PFAFFL (2001) durchgeführt. Für die Ermittlung der QRT-PCR-Effizienz wurde von zehn Einzel-cDNAs der Würmer je 1 µl entnommen und zusammengeführt. Von diesem cDNA-Mix wurde eine 5fach-2fach-Verdünnungsreihe von 1:100 bis 1:100.000 erstellt. Eine Konzentrationsbestimmung war hierfür nicht nötig, da für die Berechnung der Effizienz die Ct-Werte herangezogen werden. 3.7.8 Durchführung der QRT-PCR Die quantitative real-time PCR wurde im Mx® 4000 Multiplex Quantitative PCR System der Firma STRATAGENE durchgeführt. Für die Durchführung der absoluten Quantifizierung wurde die Einstellung „Quantitative PCR“ gewählt. Die relative Quantifizierung erfolgte mit dem Programm „Comparative Quantitation“. Für die Reaktionsansätze wurde der Brilliant® QPCR MasterMix der Firma STRATAGENE eingesetzt. Dieser enthält bereits Polymerase, Nukleotide und MgCl2 in optimierter Konzentration. In Vorversuchen wurden verschiedene Primer- und 108 Sondenkonzentrationen Material und Methoden getestet. Dies führte zu folgendem optimierten Reaktionsansatz: 25 µl Ansatz: 12,50 µl Brilliant® MasterMix 0,30 µl Vorwärtsprimer (50 µM) 0,30 µl Rückwärtsprimer (50 µM) 0,50 µl Sonde (10 µM) 0,38 µl Referenzfarbstoff ROX (1:500) Temperaturprofil 95°C 10 min 40 Zyklen 95°C 30 sec 55°C 1 min 72°C 30 sec 10,02 µl AMPUWA 1,00 µl cDNA (1:10000) Für die Verdünnungsreihen der Plasmid-DNA und cDNA wurden Duplikate der 25 µl Reaktionen eingesetzt. In den QRT-PCR-Läufen für die absolute Quantifizierung wurden die Reaktionen für die Einzel-cDNAs der Würmer als Triplikate und für die relative Quantifizierung als Duplikate eingesetzt. Für alle Reaktionen wurden 8x strip tubes (0,2 ml) mit optischen Deckeln (8x strip optical cap) der Firma STRATAGENE verwendet. Ergebnisse 109 4 Ergebnisse 4.1 Molekularbiologische Ergebnisse 4.1.1 Amplifikation der Protein Disulfid Isomerase von D. viviparus Um die Sequenz der Protein Disulfid Isomerase bei D. viviparus zu bestimmen, wurden degenerierte Primer in einer PCR mit cDNA als Template eingesetzt. Nach diversen Optimierungsläufen gelang mit dem unter 3.3.4.2 beschriebenen PCRProtokoll die Amplifikation eines Produktes mit einer erwarteten Größe von ca. 1000 bp. 2000 bp 1200 bp 800 bp 1000 bp 400 bp Abb. 7: M K 1 2 3 4 5 Mit degenerierten Primern erhaltenes Amplifikat. 1%iges Agarosegel. M: Low DNA Mass Ladder, K: Negativkontrolle, 1: 4 µl Template, 5 µl Q-Solution, 2: 8 µl Template, 3: 6 µl Template, 4: 4 µl Template, 5: 2 µl Template Die Klonierung und Sequenzierung der etwa 1000 bp großen Bande lieferte eine Sequenz mit einer Länge von 1018 bp. Im Sequenzvergleich auf Nukleinsäureebene mittels BLASTN zeigte diese Sequenz höchste Identitäten mit der PDI2 von Ostertagia ostertagi (Genbank AJ419174) und der PDI von Ancylostoma caninum (GenBank AY582130). 110 Ergebnisse 4.1.2 Vergleich positiver Klone mittels RFLP Klone, die in der PCR Inserts der erwarteten Größe von ca. 1000 bp aufwiesen, wurden mittels RsaI-Verdau verglichen. Der überwiegende Anteil der Klone zeigte hierbei ein identisches Bandenmuster. Allerdings konnte ein Klon identifiziert werden, dessen Bandenmuster sich deutlich von dem der anderen unterschied. 2000 bp 1200 bp 800 bp 400 bp M Abb. 8: M Vergleich positiver Klone mittels RsaI-Verdau. Der Pfeil zeigt einen Klon mit abweichendem Bandenmuster. M: Low DNA Mass Ladder Die Sequenzierung dieses Klons lieferte eine 1024 bp große Sequenz, die im BLASTN höchste Identitäten mit der Protein Disulfid Isomerase 3 von Caenorhabditis elegans (GenBank AAF45013) aufwies. 4.1.3 RACE-PCR Durch den Einsatz genspezifischer Vorwärts- und Rückwärtsprimer sollte mittels RACE-PCR die mit degenerierten Primern erhaltene Teilsequenz der PDI vervollständigt werden. Sowohl in der 5’-RACE als auch in der 3’-RACE konnte mit beiden eingesetzten genspezifischen Primern ein Produkt amplifiziert werden, das sich im Agarosegel als klare Bande darstellte. Ergebnisse 111 2000 bp 1200 bp 800 bp 400 bp M Abb. 9: 1 2 3 4 5 6 PCR-Produkte der RACE-PCR. 1%iges Agarosegel. M: Low DNA Mass Ladder, 1: 3’-RACE (GSP PDIDV2RC3), 2: 3’-RACE (GSP PDIDV2RCN3), 3 und 4: Negativkontrollen, 5: 5’-RACE (GSP PDIDV2RC5), 6: 5’-RACE (GSP PDIDV2RCN5) Im Alignment mit der bereits bekannten Teilsequenz der PDI zeigte sich eine vollständige Übereinstimmung der Sequenzen im Überlappungsbereich. Darüber hinaus war am 5’-Ende ein 308 bp großer Sequenzbereich vorhanden, auf den in 5’Richtung folgend die Sequenz des SMART™-Olygonukleotids identifiziert werden konnte. Das 3’-RACE-Produkt lieferte auf den Überlappungsbereich folgend eine 386 bp große Teilsequenz mit darauffolgendem Poly-A-Schwanz. Die erhaltenen Sequenzen wurden mit der bereits vorhandenen Teilsequenz im Programm Align™Plus zusammengesetzt. Nach Übersetzung der 1796 bp großen Gesamtnukleotidsequenz in die Aminosäuresequenz konnte an der Basenposition 62 ein Startcodon identifiziert werden, auf das ein 493 Aminosäuren großer ORF folgte. Der offene Leserahmen endete an einem TAA-Stopcodon. Aufgrund der Identitäten zur PDI2 von O. ostertagi und C. elegans wurde die vollständige Sequenz als PDI2 von D. viviparus in der NCBI-Datenbank unter der Nummer DQ 011044 veröffentlicht. Das zweite identifizierte Gen konnte ebenfalls mittels 5’- und 3’-RACE vervollständigt werden und wurde aufgrund der Identitäten zur PDI1 von O. ostertagi als PDI1 von 112 Ergebnisse D. viviparus in der NCBI-Datenbank unter der Nummer DQ 011045 veröffentlicht. Die PDI1-Sequenz besteht aus 1743 bp und weist einen offenen Leserahmen über 484 Aminosäuren auf. Die vollständigen cDNA-Sequenzen und abgeleiteten Aminosäurensequenzen beider PDIs von D. viviparus finden sich in Anhang 9.1 und 9.2. 4.1.4 Identitätsvergleiche der PDI1 und PDI2 von D. viviparus mit PDIs anderer Spezies Das Alignment der kodierenden Sequenz der PDI2 mit in der NCBI-Datenbank vorhandenen Sequenzen anderer Nematodenspezies und der Homo sapiens Sequenz zeigte folgende Identitäten: D. A. T. O. C. H. v. c. c. o. e. s. PDI2 PDI PDI PDI2 PDI2 PDI 1 1 1 1 1 1 mfklaclcvfalsaf--aatveeeenvivltkdnfdevinghefvlaefyapwcghcka mfklaclclfalsaf--aatveeeenvlvltkdnfdevinghefvlaefyapwcghcka mfklacvcllalsaf--aatveeeknvivltkdnfdevinghefvlaefyapwcghcka mfklacvcllalsaf--aatveeeknvivltkdnfdevinghefvlaefyapwcghcka mfrlvglfflvlgas--aavieeeenvivltkdnfdevingnefilvefyapwcghcks mlrrallcl-avaalvradapeeedhvlvlrksnfaealaahkyllvefyapwcghcka D. A. T. O. C. H. v. c. c. o. e. s. PDI2 PDI PDI PDI2 PDI2 PDI 58 58 58 58 58 59 lapeyakaatqlkeegstiklakldatvhgdvaskfevrgyptlklfrsg---kpseys lapeyakaatqlkdegsaiklakldatvhgdvaskfevrgyptlklfrng---kpseyt lapeyekaatqlkeegseiklakldatvhgdvaskfevrgyptlklfrng---kpseyt lapeyektatqlkeegseiklakldatvhgdvaskfevrgyptlklfrng---kpseyt lapeyakaatqlkeegsdiklgkldatvhgevsskfevrgyptlklfrng---kpqeyn lapeyakaagklkaegseirlakvdateesdlaqqygvrgyptikffrngdtaspkeyt D. A. T. O. C. H. v. c. c. o. e. s. PDI2 PDI PDI PDI2 PDI2 PDI 114 114 114 114 114 118 ggrdaasiiawlkkktgpvaktlktaddvkslqeeadvvvvgyfksvegekakvfleva ggrdaasiiawlkkktgpvaktlktaddvkslqeeadvvvvgyyknvdgekakifleva ggrdaasivawlkkktgpvaktlktaddvkslqeeadvvvvgyfkkadgdkakvfleva ggrdaasivawlkkktgpvaktlktaddvkslqeeadvvvvgyfkkadgdkakvfleva ggrdhdsiiawlkkktgpvakpladadavkelqesadvvvigyfkdttsddaktfleva agreaddivnwlkkrtgpaattlpdgaaaeslvessevavigffkdvesdsakqflqaa D. A. T. O. C. H. v. c. c. o. e. s. PDI2 PDI PDI PDI2 PDI2 PDI 173 173 173 173 173 177 sgvdnipfgitsedaakkqlelkeegivllkkfddgravfdekltadnlktwiqanrla ggiddipfgitsedaakkqlelkdegivllkkfddgravfdekltadalktwiqanrlp agiddipfgistedaakkqlelkeegivllkkfdegrdvfdekltadnlktwiqanrla agiddipfgistedaakkqlelkeegivllkkfdegrdvfdekltadnlktwiqanrla agiddvpfgistedavkseielkgegivlfkkfddgrvafdekltqdglktwiqanrla eaiddipfgitsnsdvfskyqldkdgvvlfkkfdegrnnfegevtkenlldfikhnqlp D. A. T. O. C. H. v. c. c. o. e. s. PDI2 PDI PDI PDI2 PDI2 PDI 232 232 232 232 232 236 lvseftqetasvifggeikshnllfvskessefekletefknaarqfkgkvlfvyintd lvseftqetasvifggeikshnllfvskessefeklesefknaakqfkgkvlfvyintd lvseftqetasvifggeikshnllfvskessefeklekefknaakqfkgkvlfvyintd lvseftqetasvifggeikshnllfvskessefeklekefknaakqfkgkvlfvyintd lvseftqetasvifggeikshnllfvskessefakleqefknaakqfkgkvlfvyintd lviefteqtapkifggeikthillflpksvsdydgklsnfktaaesfkgkilfifidsd Ergebnisse 113 D. A. T. O. C. H. v. c. c. o. e. s. PDI2 PDI PDI PDI2 PDI2 PDI 291 291 291 291 291 295 vednvrimeffglkntdlpavrlisleedmtkfkpdfveintesivkftqayldgtlka vednarimeffglkkddlpavrlisleedmtkykpdfaeintenivkftqsyldgalkp vednarimeffglkkddlpavrlisleedmtkfkpdfaeintenivkftqsyldgalkp vednarimeffglkkddlpavrlisleedmtkfkpdfaeintenivkftqsyldgalkp veenarimeffglkkdelpairlisleedmtkfkpdfeeitteniskftqnyldgsvkp htdnqrileffglkkeecpavrlitleeemtkykpeseeltaeritefchrflegkikp D. A. T. O. C. H. v. c. c. o. e. s. PDI2 PDI PDI PDI2 PDI2 PDI 350 350 350 350 350 354 hlmseeipedwdkapvkvlvgknfdqvardntknvlvefyapwcghckqlaptwdklge hlmseeipedwdkapvkvlvgknfdqvardntknvlvefyapwcghckqlaptwdklge hlmseeipedwdkapvkvlvgknfeqvardntknvlvefyapwcghckqlaptwdklge hlmseeipedwdkapvkvlvgknfeqvardntknvlvefyapwcghckqlaptwdklge hlmsedipedwdknpvkilvgknfeqvardntknvlvefyapwcghckqlaptwdklge hlmsqelpedwdkqpvkvlvgknfedvafdekknvfvefyapwcghckqlapiwdklge D. A. T. O. C. H. v. c. c. o. e. s. PDI2 PDI PDI PDI2 PDI2 PDI 409 409 409 409 409 413 kyadheniiiakmdatanevedvkvqsfptikffpagsnk-iidytgdrtlegftkfle kyadheniiiakmdatanevedvkvqsfptikffpagsnk-vidytgdrtlegftkfle kyadheniiiakmdatanevedvkvqsfptikffpagsnk-vidytgdrtlegftkfle kyadheniiiakmdatanevedvkvqsfptikffpagsnk-vidytgdrtlegftkfle kfaddesiviakmdstlnevedvkiqsfptikffpagsnk-vvdytgdrtiegftkfle tykdheniviakmdstaneveavkvhsfptlkffpasadrtvidyngertldgfkkfle D. A. T. O. C. H. v. c. c. o. e. s. PDI2 PDI PDI PDI2 PDI2 PDI 467 467 467 467 467 472 sggkegagpsddekaaeeaedeghtel---------sggkegagpsddekaaeeaeeeghtel---------sggkegagpsddekaaeeaeeevhtel---------sggkegagpsddekaaeeaeeevhtel---------tngkegagaseeekaeeeadeeghtel---------sggqdgagddddledleeaeepdmeedddqkavkdel Abb. 10: Alignment der Aminosäurensequenz der PDI2 von D. viviparus mit PDIs anderer Spezies Unterschiede in den Sequenzen sind gelb unterlegt. Die aktiven Zentren sind rot, das ER-Retentionssignal ist grün umrandet. D. v.: D. viviparus, A. c.: Ancylostoma caninum, T. c.: Teladorsagia circumcincta, O. o.: Ostertagia ostertagi, C. e. Caenorhabditis elegans, H. s.: Homo sapiens. A. caninum PDI2 T. circumcincta PDI2 O. ostertagi PDI2 C. elegans PDI2 H. sapiens PDI2 Tab. 6: Nukleotide 80% 80% 80% 72% 59% Aminosäuren 94% 93% 92% 81% 56% Identitäten der PDI2 von D. viviparus mit PDIs anderer Spezies Die Sequenz der PDI1 von D. viviparus zeigte höchste Identitäten zur PDI1 von O. ostertagi und zur PDI3 von C. elegans: 114 Ergebnisse D. v. PDI1 O. o. PDI1 C. e. PDI3 1 ---mtfayvfavlflcasas-dvleytdaifedsikfheialvkfyapwcghckkmape 1 ---mrivcvlaalvlgisasgdvleytdsnfdeliasheva*vkfy*pwcghckklape 1 miwvqaalvasflafasagg-avleytdgnfddliqthdialvkfyapwcghckkiape D. v. PDI1 O. o. PDI1 C. e. PDI3 56 fdkastklksndppvalikvdctvekstcdkygvkgfptlkifrfgseaqayegprdad 57 fdkaatklkandppitlikvdctvekatcdkfgvkgfptlkifrngleaqsydgpread 59 yeraapklasndppvalvkvdcttektvcdkfgvkgfptlkifrngvpaqdydgprdad D. v. PDI1 O. o. PDI1 C. e. PDI3 115 givkymrgqagpsareiksinefkkaisgdenivigffenesklkdsflkvadterdrf 116 givkymrgqagpsakelktveefkkfvgg*enavvgffenesklkdsflkvadterdrf 118 givkfmrgqsgpsskelktvaefekftggdenvvigffesesklkdsylkvadterdrf D. v. PDI1 O. o. PDI1 C. e. PDI3 174 qfaytsdrsvlketgynddivvftpkqlhnkfdpnefkydgnydtdkiknflihetvgm 175 qfgyssna**l*eagytddivvytpkklhnkfdpnefkydgnydtdkiksflihdtvgm 177 sfahtsnkdiikkagysddvvvfvpkklhnkfdtnefkydgnydtdkiknflvhetvgf D. v. PDI1 O. o. PDI1 C. e. PDI3 233 agirthgnlfqfeqkplvivyynvdylkdpkgsnywrnrvlkvakdykrkvhfavsnke 234 agirtmgnlfqfe*kpl*i*yynvdylkdpkgsnyw*nrvlkvaqdykrkvhfavsnke 236 agirtqgnlfqfeqkpivivyynvdyvkdpkgsnywrnrvlkvaqnykrkvqfavsnke D. v. PDI1 O. o. PDI1 C. e. PDI3 292 efssevdqnglslrkdsdkpivaavtnegkfpmdnefsvdnlktfvedliagrlepymk 293 eftteidqnglaerkdsdkpivaavtndgkfpmddefsvenlkafvedvlagnldpymk 295 efsseietnglgerkdsdkpivailtnegkypmdqefsvdnlqqfvdevlagnaepymk D. v. PDI1 O. o. PDI1 C. e. PDI3 351 sepipentga--lkvavaknfkelvlnakkdvlvefyapwcghckalapkyeelaeklv 352 sepipenneseplkvavgrnfkelvmeadkdvlvefyapwcghckalapkye*laktar 354 sepipdeqgd--vkvavgknfkelimdadkdvliefyapwcghckslapkyeelaekln D. v. PDI1 O. o. PDI1 C. e. PDI3 408 ded-vlivkmdatandvpplfevngfptiywlpknkkgspvpysggrevddfisfiakh 411 rkk*vlivkmdatandvpplfevrgfptlywlpkktk-epvplqrgre*ndfinfiakh 411 ked-viiakmdatandvppmfevrgfptlfwlpknaksnpipynggrevkdfvsfiskh D. v. PDI1 O. o. PDI1 C. e. PDI3 466 stdglkqysrdgkkk-kael 469 stdglkgytrdgkkk-ksel 469 stdglkgfsrdgkkkkktel Abb. 11: Alignment der Aminosäuresequenz der PDI1 von D. viviparus mit PDIs anderer Spezies. Unterschiede in der Sequenz sind gelb unterlegt. Die aktiven Zentren sind rot, das ER-Retentionssignal ist grün umrandet. D. v.: D. viviparus, O. o.: Ostertagia ostertagi, C. e. Caenorhabditis elegans; *nicht eindeutig definiert O. ostertagi PDI1 C. elegans PDI3 Tab. 7: Nukleotide 72% 69% Aminosäuren 77% 73% Identitäten der PDI1 von D. viviparus mit PDIs anderer Spezies Ergebnisse 115 Ein Alignment mittels MegAlign™ (DNA Star) nach der Clustal W-Methode zeigte folgende phylogenetische Verwandtschaften der PDI1 und PDI2 von D. viviparus zu PDIs anderer Spezies: Abb. 12: Phylogenetischer Stammbaum 4.1.5 Identifizierung konservierter Domänen Bei der Suche nach konservierten Domänen mittels RPSBLAST in der conserved domain database (NCBI) zeigte sowohl die PDI1 als auch die PDI2 von D. viviparus einen Aufbau aus zwei redoxaktiven (a und a’) und zwei redoxinaktiven (b und b’) Thioredoxin-Domänen. Während die b-Domänen der PDI2 von D. viviparus höchste Ähnlichkeiten zu den entsprechenden Domänen der humanen PDI aufweisen, zeigt die b-Domäne der PDI1 von D. viviparus höhere Ähnlichkeiten zur b-Domäne von ERp57 und die b’-Domäne höhere Ähnlichkeiten zur b’-Domäne von ERp72 und ERp57. Die Domänen erstrecken sich über folgende Sequenzbereiche: 116 Ergebnisse PDI1 von D. viviparus 20 - 120 127 - 229 233 - 341 361 - 462 AS 364 - 462 AS PDI2 von D. viviparus 26 - 125 Abb. 13: 133 - 228 240 - 343 Domänenstruktur der PDI1 und PDI2 von D. viviparus. AS: Aminosäuren 4.1.6 Ermittlung der genomischen Sequenz der PDI2 von D. viviparus Die genomische Sequenz wurde nur für die PDI2 ermittelt. Alle Primerkombinationen zur Amplifikation der genomischen Sequenz lieferten mit genomischer DNA als Template ein größeres Produkt als in der PCR mit cDNA. Dies wies auf das Vorhandensein von Introns in den amplifizierten Bereichen hin. Abb. 8 zeigt die mit genomischer DNA erhaltenen Amplifikationsprodukte im Vergleich zum cDNA-Template. Ergebnisse 117 B 2000 bp A 2000 bp 1200 bp 800 bp 1200 bp 800 bp 400 bp 400 bp M Abb. 14: 1 2 3 4 5 6 M K PCR zur Ermittlung der genomischen Sequenz. 1%iges Agarosegel. A: M: Low DNA Mass Ladder, K: Negativkontrolle, 1, 3, 5: Template genomische DNA; 2, 4, 6: Template cDNA; 1, 2: Primerpaar PDI2G1/PDI2G2; 3, 4: Primerpaar PDI2G3/PDI2G4; 5, 6: Primerpaar PDI2G5/PDI2G6; Die Amplifikation der genomischen DNA mit dem Primerpaar PDI2G5/PDI2G6 (Spur 5) wurde wiederholt und ist in Abbildung B dargestellt. M: Low DNA Mass Ladder. Das Alignment der erhaltenen Sequenzen mit der cDNA-Sequenz der PDI2 zeigte eine vollständige Übereinstimmung der Sequenzen im Überlappungsbereich und lieferte vier Intronsequenzen. Insgesamt weist die genomische DNA der PDI2 von D. viviparus eine Länge von 2649 bp auf. 78 bp 151 bp 335 bp Abb. 15: 92 bp 306 bp 805 bp 361 bp 454 bp 67 bp Exon-Intron-Organisation der PDI2 von D. viviparus. Die vier Introns sind weiß, die Exons schwarz dargestellt. 118 1 61 121 181 241 301 361 421 481 541 601 661 721 781 841 901 961 1021 1081 1141 1201 1261 1321 1381 1441 1501 1561 1621 1681 1741 1801 1861 1921 1981 2041 2101 2161 2221 2281 2341 2401 2461 2521 2581 2641 Abb. 16: Ergebnisse ACGGTGTGTC AATGTTCAAA AGAAGAGGAG ACCTGACTGA ACTTACTCAT AAATGTTAGC CACAGTAGTG CTAAGCTTAT TTTTAGGACA GCTCCTTGGT TTTTTCCATC TTGCTCCGGA TAGCAAAACT ACCCAACACT CGGCATCAAT CTGCCGATGA AGGTATATAG TAGGCTTGAT TTACAAAAAC CGAATTATAC CTCGGAACAA TCACATAAGT GAGTGTCGAA TCCATTTGGA TATCGTGCTA TGACAATCTC GGAAACGGCA CAAAGAGTCC TAAGGGCAAA GGAATTCTTT GGATATGACT CACCCAAGCG AGATTGGGAT GGACAATACA ACTTGCTCCG TGCCAAAATG CCGAACGTTC TCCCGACAAT GGACTCTTGA CAGATGATGA TTGCTGCTAT TTTGCTCTAC TTTTCTCTTC AGAACTACAC AAAAAAAAA GGAGTGCTCT CTCGCCTGCC AATGTGATCG TCTGTCCGGG CTGAATCTGA AAAATTGCAA CGTTTTATAA TTACATTATT ACTTTGACGA GCGGTCACTG AGCTAGCTAT ATATGCCAAA AGATGCTACC GAAACTGTTC CATTGCATGG CGTCAAATCA TGTCGTTTAC AGTTATTTTT CAATCCCTTA ATTTCCCCAG CGGTGTTCCA AGCTATGTAT GGGGAGAAGG ATCACCTCTG TTGAAGAAGT AAAACATGGA TCTGTCATTT AGCGAGTTCG GTTCTCTTTG GGTCTTAAGA AAATTCAAAC TATTTGGATG AAAGCACCTG AAAAATGTTC ACTTGGGACA GACGCAACTG AATATTTTAA CAAGTTCTTC GGGATTCACT GAAGGCAGCT CTTACTGGGG TAATCACCCA TTCAATTGTG CACTGTGTTG CTTCACGGTG TTTGCGTCTT TGCTTACAAA TTTTCCGTGA CATTATATCG GGTTTTCGAA CATCTAACAG CTTTGGTCTT AGTTATTAAC TAAGGTTGGT TTCATCGTTT GCTGCCACAC GTTCATGGCG CGCAGTGGAA TTGAAGAAGA CTTCAAGAAG GTTAAGTTAC GGCCTTCAGC AATTCTTAAT CAGAAACTTA CTATGTTAGC TTTTTTTTCG CGAAAGTCTT AAGATGCTGC TCGACGATGG TCCAAGCCAA TTGGTGGAGA AAAAGTTAGA TATACATCAA ACACTGACTT CTGACTTTGT GAACTCTCAA TAAAAGTGCT TTGTTGAGTT AGCTTGGAGA CTAATGAGGT TTTGTCGCTT CCCGCAGGAT AAATTCCTTG GAAGAGGCTG TCGTACGGTC TGTACGAGTG ATCGTTGATA GAGAACTGGG TTCTAGACAA TGCTCTGTCG GGTAATTGAT AAGTTTCAAA CATCTTTGTT TTTGATCTAT ATTTCTATAA AATGTACGGT GGTCATGAGT CTACAAGTGC TAATATCGAC AGCTGAAGGA ATGTTGCTAG AGCCGAGTGA AGACTGGTCC AGGCTGATGT GATATCGAGA GTTATTTCTA GTTTATGTTG ATAAGTTTTG TTGATGGGGT CCATATAGTT TTTGGAAGTT AAAGAAGCAG ACGCGCAGTG CCGACTAGCC GATTAAAAGC AACTGAATTC TACGGATGTT GCCAGCAGTA CGAGATTAAC GGCTCACCTC TGTAGGAAAG CTATGCTCCA AAAGTATGCC GTGAAAATTG TGAAGGTCGA CAAATAAAAT AAAGCGGTGG AAGACGAGGG TGACGACCTG ATCTAAACAT AGTGATGGTG TTTAAATAAA ATTGTCATTC GCATTCGCCG TGTTTATGTA AACAATGTAC ATATTGTACA AATATTTGCA ATAATTCGAA CAATTCCTAT TTGTGCTTGC TTGATGTGCC TTTTGATTGA AGAGGGTTCA CAAGTTCGAG ATACAGCGGT GGTCGCGAAG TGTTGTCGTT ATTTTAAGTA ACGTACTTAG TCTAAAAATG TATTCATGCT AATAATTTCC CAATAATTTT GCAAGTGGAG CTAGAACTTA TTTGATGAGA CTTGTTTCGG CATAACCTTC AAAAATGCTG GAAGACAACG CGACTAATCT ACAGAGAGCA ATGTCCGAAG AACTTCGACC TGGTGTGGTC GACCACGAGA TTCATAGCTT GGACGTAAAA AATCGACTAC TAAGGAAGGG TCATACTGAG TTGTTGTTAC TGCCTTCCTG AAATTTATTT TATAACATTT ATTGGGGCGC CAACTGTTGA ATTATCGCGG TTTCCACAAC TGTCTCTAGT CATTTGGTTG AATTGTTTAA TTTGACAAAC CGAATTTTAT CAGTCGTTGT TTCCAGGCAC ACGATCAAAT GTTCGCGGGT GGACGCGATG ACATTAAAAA GGATACTTCA GAATATTATG TTGATCATAT TACAGGCAAA TGATGTATTC CTATTGATTC CTTTTCTTCA TTGATAATAT AGGAAGAAGG AGCTCACTGC AATTCACTCA TTTTTGTCTC CTAGACAATT TTCGTATAAT CATTAGAAGA TTGTTAAATT AGATTCCTGA AAGTAGCACG ACTGCAAGCA ACATCATAAT CTTCATTACG GTGCAATCAT ACTGGTGACA GCTGGACCAT CTGTAAGCAT AGTTAATTTA TATATGCGTT ATTTTTGTTC TGCCAAAAAA Vollständige genomische DNA-Sequenz der PDI2 von D. viviparus. Die vier Introns sind in rot dargestellt. Ein Vergleich der genomischen Sequenz der PDI2 mit der genomischen Sequenz der PDI von A. caninum zeigte, dass die Introns sich an den gleichen Positionen befinden, jedoch bei der PDI2 von D. viviparus wesentlich kleiner sind. A. caninum D. viviparus Ergebnisse 119 Exon 1 Intron 1 Exon 2 Intron 2 Exon 3 Intron 3 Exon 4 Intron 4 Exon 5 Tab. 8: 102 bp 431 bp 78 bp 104 bp 306 bp 425 bp 807 bp 179 bp 491 bp 151 bp 335 bp 78 bp 92 bp 306 bp 361 bp 805 bp 67 bp 454 bp Vergleich der Exon- u. Introngrößen von A. caninum und D. viviparus 120 Ergebnisse 4.2 Proteinbiochemische Ergebnisse 4.2.1 Optimierung der Proteinaufreinigung Die ersten Versuche zur Aufreinigung der PDI wurden mittels des Magne GST™ Purification Systems durchgeführt. Im Coomassie-gefärbten Gel zeigten sich neben der Fusionsproteinbande mit einer Größe von etwa 81 kDa noch diverse weitere Banden. Die Überprüfung im Western-Blot mit GST- und PDI-Antikörpern zeigte, dass es sich bei den Begleitbande um PDI-Bruchstücke und um Kontaminanten handelte. M kDa M M kDa 250 250 250 98 64 98 64 98 64 50 50 36 36 30 A Abb. 17: 30 B M kDa kDa 250 98 64 50 50 36 30 36 C 30 D Darstellung des aufgereinigten Fusionsproteins. M: Marker Seeblue® A: Coomassie-gefärbtes SDS Gel; B: WesternBlot mit Anti-GST-Ak; C: Western-Blot mit Anti-PDI-Ak; Western-Blot mit Serumpool von D. viviparus infizierten Rindern In den darauffolgenden Optimierungsversuchen gelang es nicht, die Begleitbanden vollständig zu eliminieren. Im Folgenden sind beispielhaft einige Ergebnisse der verschiedenen Versuchsbedingungen aufgeführt. Ergebnisse 121 4.2.1.1 Proteinexpression Um den Einfluss der Proteinexpression auf das Aufreinigungsergebnis zu überprüfen, wurden sowohl unterschiedliche Expressionsbedingungen im E. coli BL 21 (DE3)-Stamm getestet, als auch eine Expression im E. coli BL 21 (DE3) Codon Plus® Stamm durchgeführt. Es konnte gezeigt werden, dass weder mildere Expressionsbedingungen (Verkürzung der Inkubationszeit, geringere IPTG- Konzentration, niedrigere Inkubationstemperatur), noch der Einsatz des BL 21 (DE3) Codon Plus® Stammes das Aufreinigungsergebnis verbessern konnten. kDa M 250 98 64 50 36 30 1 Abb. 18: 2 3 4 5 6 7 8 Optimierung der Expression. Western Blot, Anti-GST-Antikörper. Aufreinigungsergebnis nach unterschiedlichen Expressionsbedingungen. M: Marker Seeblue®; 1, 2, 3, 4: 3 h Inkubation; 5, 6, 7, 8: 6 h Inkubation; 1, 2, 5, 6: Inkubation bei 25°C; 3, 4, 7, 8: Inkubation bei 30°C; 1, 3, 5, 7: Induktion mit 0,2 mM IPTG; 2, 4, 6, 8: Induktion mit 1,0 mM IPTG. Da keine Unterschiede zwischen den Stämmen vorhanden waren, wurde für nachfolgende Expressionen der BL 21 (DE3) Stamm verwendet. Mit einer IPTGKonzentration von 1,0 mM und einer Inkubation für 5 h bei 37°C war die Ausbeute an 122 Ergebnisse Fusionsprotein sehr hoch, sodass die Herstellung des Fusionsproteins für die Immunisierungen unter diesen Bedingungen erfolgte. 4.2.1.2 Vergleich unterschiedlicher Lysebedingungen Um die Auswirkungen unterschiedlicher Lysebedingungen zu überprüfen, wurde eine Zellsuspension in zwei Fraktionen aufgeteilt und pelletiert. Ein Pellet wurde ausschließlich für 45 min mittels Magne GST™-Lysepuffer lysiert, das andere zusätzlich mit Ultraschalimpulsen behandelt. Die anschließende Aufreinigung erfolgte bei beiden Proben mittels Magne GST-Partikel nach identischem Protokoll (1 h Anlagerung, 5 x Waschen für 10 min mit 2 ml Waschpuffer). kDa kDa B A 42 123 77 42 30 30 123 77 1 Abb. 19: 2 3 4 5 6 7 8 9 1 2 3 4 5 6 7 8 9 Western-Blot zum Vergleich der Aufreinigung nach unterschiedlichen Lysebedingungen. Antikörper: Anti-GST; A: Lysepuffer; B: Lysepuffer + Ultraschall; 1: Marker Roth; 2: Zelllysat; 3: Zelllysat nach Anlagerung; 4, 5, 6, 7: 1., 2., 3., 4. Waschschritt; 8: 1. Eluat; 9: 2. Eluat. Es konnte gezeigt werde, dass die Zelllyse mittels Ultraschall wesentlich effektiver ist, das Ergebnis der Aufreinigung jedoch nicht beeinflusst wird. Die Ultraschallbehandlung führte also nicht zu einer vermehrten Koaufreinigung bakterieller Kontaminanten bzw. konnte hierin nicht die Ursache für das Auftreten degradierten rekombinanten Proteins gesehen werden. Ergebnisse 123 4.2.1.3 Unterschiedliche Aufreinigungbedingungen im Magne GST™-Protokoll und mittels Sepharose™-Medium Bei der Aufreinigung mittels des Magne GST™ Purification Systems wurde versucht, durch Erhöhung der Anzahl und Länge der Waschzyklen und durch ein größeres Waschvolumen eine Verbesserung des Aufreinigungsergebnisses zu erzielen. Dies konnte jedoch nicht erreicht werden. Ebenso wenig führte eine Erhöhung der NaClKonzentration im Waschpuffer oder ein Zusatz der unter 3.4.3.1 aufgeführten Substanzen zu einer Verringerung der bakteriellen Kontaminanten. Auch der Einsatz von Glutathione Sepharose™-Medium konnte keine Verbesserung des Aufreinigungsergebnisses erzielen. 4.2.1.4 Aufreinigung mittels FPLC In der Aufreinigung mittels FPLC zeigten sich die gleichen Begleitbanden in vergleichbarer Intensität wie bei den anderen Aufreinigungsmethoden. Aufgrund der Automatisierung des Prozesses wurde dann letztendlich dieses Verfahren gewählt, um große Mengen Fusionsprotein für die Immunisierungen aufzureinigen. Abb. 14 zeigt fünf Peak-Fraktionen eines Laufes. 1a 1b 2a 2b Abb. 20: 3a 3b 4a 4b 5a 5b Coomassie-gefärbtes SDS-Gel der fünf Peak Fraktionen eines FPLC-Laufes. a: nicht aufkonzentriert, b: mit Vivaspin aufkonzentriert. 124 Ergebnisse 4.2.2 Proteinproduktion für die Immunisierungen Für die Immunisierungen wurde die PDI über Affinitätssäulen mittels FPLC aufgereinigt. Aus den einzelnen FPLC-Läufen wurden die Peak-Fraktionen zusammengeführt und mit Vivaspin-Säulen aufkonzentriert. Da es nicht möglich war, vor dem ersten Immunisierungsdurchgang ausreichend Protein für beide Immunisierungsversuche herzustellen, wurden die beiden Versuche mit zwei unterschiedlichen Proteinpools durchgeführt. Die Konzentrationsmessungen im Agilent 2100 Bioanalyzer lieferten folgende Werte: Abs. Protein- Rel. Konz. MW SD Konzentration Fusions-Protein (µg/ml) (µg/ml) (µg/ml) Fusions-Protein MW (%) (%) SD Chip 1: 499,3 453,1 502,5 342,2 316,6 350,2 68,5 69,9 336,3 17,6 69,7 69,4 0,8 Chip 2: 482,9 528,9 485,7 482,7 329,8 365,5 347,9 342,1 68,3 69,1 76,1 346,3 14,8 70,9 71,1 3,5 Chip 3: 470,6 445,5 425,8 482,6 406,4 MW 472,2 312,8 323,8 298,0 337,1 287,3 66,5 72,7 70,0 69,9 311,8 19,8 70,7 331,5 17,8 70,0 2,2 70,1 0,9 Tab. 9: Agilent-Messungen des I. Proteinpools. MW: arithmetischer Mittelwert, SD: Standardabweichung Ergebnisse 125 Rel. Konz. Abs. ProteinMW Mittelwert Fusions-Protein Konzentration (µg/ml) (µg/ml) (µg/ml) (µg/ml) 433,0 564,7 408,4 521,3 493,7 539,1 536,9 464,5 516,4 Tab. 10: 497,6 339,8 441,8 314,9 397,8 379,9 405,5 419,2 366,0 425,9 SD Fusions-Protein MW (%) (%) 387,9 41,7 78,5 78,2 77,1 76,3 77,0 75,2 78,1 78,8 82,5 SD 78,0 2,1 Agilent-Messungen des II. Proteinpools. MW: arithmetischer Mittelwert, SD: Standardabweichung Die folgende Abbildung zeigt beispielhaft die graphische Darstellung der bei der Messung des zweiten Proteinpools erhaltenen Ergebnisse. 126 Ergebnisse A Fusionprotein B Fusionsprotein C P eak 2 3 4 5 6 7 8 9 M ig.Tim e(secs) Corr.Area S ize(kDa) Rel.Conc.(µg/m l) % Total Observations 19,3 169,95 9,4 System Peak 22 2,62 18 9,4 2,2 23,15 3,34 23,7 11,9 2,8 26,35 3,41 45,2 12,2 2,8 27,1 2,84 50,5 10,1 2,3 28,85 6,58 65,4 23,5 5,4 29,8 95,07 77,6 339,8 78,5 36,85 7,31 166,3 26,1 6 Total Rel Conc: 433 µg/m l Abb. 21: Messung des II. Proteinpools. A: generiertes Gelbild der 9 Messungen, B: Elektropherogramm der 1. Messung, C: ermittelte Werte für die erste Messung. Ergebnisse 127 4.2.3 Ergebnisse der MALDI-TOF-MS-Analyse Die von der Firma TOPLAB durchgeführte Datenbanksuche mit den erhaltenen Peptidmassen identifizierte sowohl die GST aus S. japonicum als auch die Protein Disulfid Isomerase aus A. caninum und aus O. ostertagi. Da die beiden letzteren weitestgehend identische Peptidmassen aufwiesen, ist es (laut Mitteilung des Labors) wahrscheinlich, dass es sich bei der eingesandten Probe um das entsprechende Protein aus D. viviparus handelt. Die PDI aus D. viviparus konnte nicht direkt identifiziert werden, da sich die Sequenz zum Untersuchungszeitpunkt erst kurzfristig in der Datenbank befand. Für die GST aus S. japonicum ergab sich eine 70%ige Übereinstimmung der Fingerprints, für die Protein Disulfid Isomerase stimmten bei A. caninum 30% und bei O. ostertagi 32% der Fingerprints überein. 128 Ergebnisse 4.3 Tierexperimentelle Ergebnisse 4.3.1 Tiergesundheit 4.3.1.1 Allgemeinuntersuchung Mit Ausnahme eines Tieres (Nr. 544), das aufgrund respiratorischer Symptome und Fieber in der dritten Versuchswoche medikamentös behandelt werden musste, wiesen alle Tiere bis zur experimentellen Infektion ein ungestörtes Allgemeinbefinden auf. Außer einer bei den meisten Tieren vorhandenen Trichophytie konnten keine Anzeichen einer Erkrankung beobachtet werden. Die Trichophytie war gering bis mittelgradig ausgeprägt und führte nicht zu einer Beeinträchtigung der Tiere. Im Verlauf der experimentellen Infektion zeigten alle Tiere mehr oder weniger ausgeprägt Zeichen einer klinischen Dictyocaulose. Der überwiegende Anteil der Tiere zeigte keine oder nur eine geringgradige Störung des Allgemeinbefindens in Form von Mattigkeit. Bei einer deutlichen Störung des Allgemeinbefindens war in den meisten Fällen auch Fieber vorhanden, sodass ein medikamentöses Eingreifen notwendig war (siehe Abschnitt 4.3.1.4). Das Auftreten von Nasenausfluss erwies sich als nicht konstant zu beobachtender Parameter, da sich die Tiere den Ausfluss spontan und bei der Nahrungsaufnahme entfernten. Am Tag der Schlachtung wurden die Tiere früh morgens vor der Fütterung untersucht, sodass hier bei einem Großteil der Tiere mucopurulenter Nasenausfluss beobachtet werden konnte. Alle Tiere zeigten respiratorische Symptome in Form von Husten, Erhöhung der Atemfrequenz und Veränderung der Atmungsqualität. Z.T. waren bei der Auskultation pathologische Atemgeräusche vernehmbar. Ergebnisse 129 II. Immunisierungsversuch I. Immunisierungsversuch PDI Quil A PDI Al(OH)3 Tab. 11: Tier- HF KT 110 + 57 109 ↑ 795 ++ 58 112 ↑ 802 + 62 110 ↑ 805 + 60 117 ↑ 808 - 51 101 ↑ - 814 - 68 113 - 100 ↑ 754 + 44 98 ↑ 67 106 ↑ 762 + 53 105 ↑ - 71 93 ↑ 790 + 54 103 - 586 - 47 99 - 807 + 49 104 ↑ 597 - 59 99 - 810 + 51 104 ↑ 952 - 68 109 - 816 ++ 57 99 ↑ 540 - 60 111 ↑ 545 - 64 114 - 581 - 61 105 ↑ 752 - 64 110 ↑ 579 - 61 96 - 600 - 68 103 - 753 + 62 106 ↑ 948 - 59 105 - HF KT 588 - 55 99 - 596 - 60 103 - 598 - 54 86 - 950 + 72 108 ↑ 611 + 77 118 ↑ 749 - 66 101 757 - 62 914 - 544 Nr. Kontrolle Quil A AF AF Gruppe Tier- AB AB Kombi Quil A Kombi Quil A Kombi Al(OH)3 Kontrolle Al(OH)3 Gruppe Nr. Befunde der täglichen Untersuchung ab Tag 70. Bei den angegebenen Atem- und Herzfrequenzen handelt es sich um arithmetische Mittelwerte der Tage 70-98 (PDI Quil A, PDI Al(OH)3), 70-99 (Kontrolle Al(OH)3, Kombi Al(OH)3, Kombi Quil A) bzw. 70-97 (II. Immunisierungsversuch); AB = Allgemeinbefinden: - nicht gestört, + ggr. gestört, ++ deutliche Störung des Allgemeinbefindens; KT = Körpertemperatur: ↑: Erhöhung konnte beobachtet werden, - :konstant physiologisch (<40°C) Ergebnisse Atemzüge/min 130 90 80 70 60 50 40 30 20 I. Immunisierungsversuch -7 0 21 42 63 70 72 74 76 78 80 82 84 86 88 90 92 94 96 98 Versuchstage Atemzüge/min Kontrolle KontrolleAl(OH)3 Al(OH)3 90 80 70 60 50 40 30 20 Kombi Kombi Al(OH)3 Al(OH)3 Kombi Kombi Quil QuilAA I. Immunisierungsversuch -7 0 21 42 63 70 72 74 76 78 80 82 84 86 88 90 92 94 96 98 Versuchstage Atemzüge/min Kontrolle KontrolleAl(OH)3 Al(OH)3 90 80 70 60 50 40 30 20 PDI PDIAl(OH)3 Al(OH)3 PDI Quil QuilAA II. Immunisierungsversuch -7 0 21 42 63 70 72 74 76 78 80 82 84 86 88 90 92 94 96 Versuchstage Kontrolle KontrolleQuil QuilA A Abb. 22: Kombi Kombi Quil QuilAA Veränderung der Atemfrequenzen während des Versuchsverlaufs. Darstellung der Tagesmittelwerte je Gruppe. Ergebnisse 131 Abb. 22 zeigt die Gruppenmittel der Atemfrequenzen über den gesamten Versuchsverlauf. Die bei der Einstallung durch Beunruhigung der Tiere noch erhöhten Herzfrequenzen erreichten bis zum Tag 70 physiologische Werte. Dies ist auf eine Gewöhnung der Tiere an die Umgebung und an die Manipulationen bei der Untersuchung zurückzuführen. Ab Tag 70 zeigte sich in allen Gruppen ein kontinuierlicher Anstieg der mittleren Atemfrequenzen. 4.3.1.2 Gewichtsentwicklung An den Versuchstagen 0, 21, 42, 63 und vor der Sektion wurden die Tiere gewogen. Beim I. Immunisierungsversuch erfolgte die Wägung am Tag der Sektion (Tag 98 bzw. 99). Die Tiere waren hierbei nüchtern. Beim II. Immunisierungsversuch wurden die Rinder am Spätnachmittag vor der Sektion (Tag 97) gewogen, sodass es sich hierbei nicht um Nüchterngewichte handelt. Eine Vergleichbarkeit der beiden Versuche ist daher nicht gegeben. Abbildung 25 zeigt die Gruppenmittel der prozentualen Gewichtszunahmen von einer zur nächsten Wägung. I. Immunisierungsversuch II. Immunisierungsversuch 20 Tag 0 bis 21 Abb. 23: Tag 21 bis 42 * Tag 42 bis 63 * Kombi Quil A * Kontrolle Quil A -10 * PDI Quil A -5 Kombi Al(OH)3 * 0 PDI Al(OH)3 5 Kombi Quil A 10 Kontrolle Al(OH)3 Gewichtzunahme (%) 15 Tag 63 bis Sektion Gruppenmittel der prozentualen Gewichtszunahme. Die mit * gekennzeichneten Werte wurden auf Grundlage von Nüchterngewichten vor der Sektion bestimmt. 132 Ergebnisse Die Bestimmung des Nüchterngewichtes beim I. Immunisierungsversuch führte zu einem scheinbar erhöhten Gewichtsverlust post infectionem. 4.3.1.3 Lokale Impfreaktionen An den Injektionsstellen kam es bei einem Teil der Tiere zu lokalen Reaktionen in Form von nicht schmerzhaften Umfangsvermehrungen. Diese hatten bei fünf Tieren an den ersten Tagen des Auftretens einen Durchmesser von 5 bis 10 cm, in den übrigen Fällen blieben die Schwellungen kleiner als 5 cm im Durchmesser. Spätestens 12 Tage post injectionem hatten sich die Umfangsvermehrungen vollständig zurückgebildet. Ergebnisse 133 Tage nach 1. Impfung Tage nach 2. Impfung Tage nach 3. Impfung 1 2 3 4 5 6 7 8 9 10 11 12 13 1 2 3 4 5 6 7 8 9 10 1 2 3 4 5 6 7 8 9 1. Immunisierungsversuch 588 596 598 950 611 749 757 914 544 586 597 952 540 545 581 752 579 600 753 948 O O O O O O O O O O O O O O O O O O O O O O O O O O O O O O O O O O O O O O O O O O O O O O O O O O O O O O O O O O O O O O O O O O O O O O O O O O O O O O O O O O O O O O O O O O O O O O 2. Immunisierungsversuch 110 795 802 805 808 814 754 762 O 790 O 807 810 O O 816 O Tab. 12: O O O O O O O O O O O O O O O O O O O O O O O O O O O O O O O O O O O O Auftreten lokaler Impfreaktionen Ø > 5 cm O Ø < 5 cm 134 Ergebnisse 4.3.1.4 Behandlung erkrankter Tiere Bei folgenden Tieren war eine medikamentöse Behandlung notwendig: Tier-Nummer 544 540 581 Tab. 13: Gruppe Versuchstag I. Immunisierungsversuch 15–19 Kombi Quil A 88 88-92 90 PDI A 90-94 94 PDI A 94-98 II. Immunisierungsversuch 814 Kontrolle Quil A 89 805 Kontrolle Quil A 92 795 Kombi Quil A 807 Kombi Quil A 816 Kombi Quil A 810 Kombi Quil A 95-96 95-97 95 95-97 95 95-97 96 96-97 Medikamente Baytril 2,5% oral Finadyne, i.v. Baytril 10%, i.v. Finadyne, i.v. Baytril 10%, i.v. Finadyne, i.v. Baytril 10%, i.v. Finadyne, i.v. Draxxin, s.c. Finadyne, i.v. Draxxin, s.c. Finadyne, i.v. Baytril 10%, i.v. Finadyne, i.v. Baytril 10%, i.v. Finadyne, i.v. Baytril 10%, i.v. Finadyne, i.v. Baytril 10%, i.v. Behandlung erkrankter Tiere 4.3.1.5 Differentialblutbild Die Bestimmung des Differentialblutbildes erfolgte an den Versuchstagen 0, 63, 84, 91 und bei der Sektion. Folgende Tiere wiesen eine erhöhte Leukozytenzahl auf: 950: Tag 63 (12,9 G/l) Tag 99 (16,5 G/l) 581: Tag 63 (12,2 G/l) Tag 84 (12,5 G/l) 752: Tag 63 (13,4 G/l) 814: Tag 0 (12,1 G/l) 754: Tag 0 (14,1 G/l) Tag 91 (13,2 G/l) Ergebnisse 762: Tag 0 (12,7 G/l) 790: Tag 0 (12,3 G/l) 135 Tag 63 (12,1 G/l) Eine absolute Eosinophilie (> 1,5 G/l) war bei folgenden Tieren vorhanden: 544: Tag 91 (2,12 G/l) 952 Tag 91 (1,71 G/l) 805 Tag 91 (1,55 G/l) 810 Tag 84 (1,56 G/l) Eine vollständige Darstellung der Ergebnisse der Eosinophilenzählung nach der experimentellen Infektion findet sich im Anhang, Abschnitt 9.3, Tab. 18. Es konnten keine weiteren Veränderungen des Differentialblutbildes festgestellt werden (Normalwerte nach PLONAIT 1980). 4.3.2 Larvenausscheidung Ab Versuchstag 84 (21 dpi) erfolgte eine tägliche Kontrolle der Larvenausscheidung mittels des Trichterauswanderverfahrens nach BAERMANN (1917). Aus den Larvenzahlen des 2 mal 10 g Doppelansatzes wurde der arithmetische Mittelwert gebildet. Die folgenden Abbildungen zeigen den Verlauf der Larvenausscheidung in den Versuchsgruppen sowie die absolute Larvenausscheidung der Einzeltiere. 136 Ergebnisse 1800 Mittelwert Larven 1600 I. Immunisierungsversuch 1400 1200 1000 800 600 400 200 0 84 85 86 87 88 89 90 91 92 93 94 95 96 97 98 Versuchstage I: Kombi Al(OH)3 Al(OH)3 I: Kontrolle KontrolleAl(OH)3 Al(OH)3 I:I: PDI PDI Al(OH)3 Al(OH)3 I: Kombi Quil A I: PDI Quil A 1800 Mittelwert Larven 1600 II. Immunisierungsversuch 1400 1200 1000 800 600 400 200 0 84 85 86 87 88 89 90 91 92 93 94 95 Versuchstage 97 98 II: Kombi Quil A II: Kontrolle Quil A Abb. 24: 96 Larvenausscheidung im I. und II. Immunisierungsversuch. Gruppenmittelwerte Übersichtlichkeit an wurde den auf Versuchstagen. Angabe der Aus Gründen der Standardabweichung verzichtet. Diese befinden sich im Anhang, Abschnitt 9.4, Tab. 19. Ergebnisse 137 I. Immunisierungsversuch II. Immunisierungsversuch 20 Larven ( x1000) 16 12 8 4 Kombi Al(OH)33 Al(OH) Kombi Quil A PDI Al(OH)3 Al(OH)3 PDI Quil A 611 749 754 914 544 586 597 952 540 545 581 752 579 600 753 948 Kontrolle Quil A Kombi Quil A 110 795 802 805 808 814 754 762 790 807 810 816 Kontrolle Al(OH)3 Al(OH) 3 588 596 598 950 0 Tiernummern Abb. 25: Larvenausscheidung im I. und II. Immunisierungsversuch. Gesamtlarvenausscheidung der Einzeltiere (Summe der Mittelwerte des 2 x 10 g Doppelansatzes). Zwischen den immunisierten und nicht-immunisierten Gruppen konnten keine statistisch signifikanten Unterschiede in der Larvenausscheidung festgestellt werden (p>0,05, Zweifaktorielle Varianzanalyse für abhängige Stichproben). Die absoluten Zahlen der Gesamtlarvenausscheidung sind in Abschnitt 4.3.4, Tab. 11, vorzufinden. 4.3.3 makroskopische Beurteilung der Lungen Bei der Sektion zeigten sich pathologische Veränderungen an den Lungen in Form von multifokal vorhandenen dunkelroten, atelektatischen Bezirken unterschiedlicher Ausdehnung. Einige Tiere zeigten eine herdförmige fibroblastische Pleuritis. Der Charakter der entzündlichen Veränderungen führte bei allen Tieren zur 138 Ergebnisse Verdachtsdiagnose katarrhalisch-eitrige Bronchopneumonie. Im Folgenden sind die an den Lungen erhobenen pathologisch-anatomischen Befunde aufgeführt: I. Immunisierungsversuch Verdachtsdiagnose Befunde Tier- PDI Quil A PDI Al(OH)3 Kombi Quil A Kombi Al(OH)3 Kontrolle Al(OH)3 Nr. Anteil der entzündlichen Hyperplasie Veränderungen der multifokale Pleuritis katarrhalischeitrige linke rechte Bronchial- Lunge Lunge lymphknoten 588 bis zu 35% 5% mgr. - ggr. 596 5-15% 5-10% ggr.-mgr. - ggr. 598 20-25% 5-10% mgr.-hgr. - ggr. 950 5-10% bis zu 40% mgr. ggr. ggr.-mgr. 611 bis zu 90% 10-15% mgr.-hgr. - ggr.-mgr. 749 10-15% 5-10% mgr. - ggr. 757 5-15% bis zu 60% mgr.-hgr. - ggr.-mgr. 914 bis zu 40% bis zu 90% mgr.-hgr. - mgr. 544 5-15% bis zu 60% mgr.-hgr. ggr.-mgr. ggr.-mgr. 586 5-10% 5% ggr.-mgr. - ggr. 597 5% 5% ggr.-mgr. ggr. ggr. 952 15% 5-10% mgr.-hgr. - ggr. 540 20-35% 5% mgr. ggr. ggr.-mgr. 545 10-50% 5-15% mgr.-hgr ggr. ggr.-mgr. 581 0-15% 5-10% mgr. - ggr.-mgr. 752 5-40% 5% mgr. - ggr.-mgr. 579 10-30% 5% mgr.-hgr. - ggr.-mgr. 600 10-25% 5% mgr.-hgr. ggr. ggr.-mgr. 753 5-70% 10-70% mgr.-hgr. - mgr. 948 5-15% 5-10% mgr. - ggr.-mgr. Tab. 14: Bronchopneumonie Pathologisch-anatomische Befunde des I. Immunisierungsversuchs Ergebnisse 139 II. Immunisierungsversuch Befunde Anteil der entzündlichen Tier- Kombi Quil A Kontrolle Quil A Nr. Veränderungen Verdachtsdiagnose multifokale Hyperplasie der Pleuritis katarrhalischeitrige linke rechte Bronchial- Lunge Lunge lymphknoten 110 30% 10% mgr. - mgr. 795 20% 20% hgr. ggr. mgr. 802 bis zu 60% 30-40% hgr. - mgr.-hgr. 805 5% bis zu 80% mgr. - mgr. 808 20-30% 20% mgr.-hgr. - mgr. 814 5-10% 5% ggr. - ggr. 754 5-10% 10% ggr. hgr. ggr. 762 20-30% 10% mgr. - ggr.-mgr. 790 15-20% 5-10% mgr. - ggr.-mgr 807 20-25% 15-20% mgr. - mgr. 810 20-30% 15% mgr. - mgr. 816 10% 15% mgr. ggr. ggr.-mgr. Tab. 15: Bronchopneumonie Pathologisch-anatomische Befunde des II. Immunisierungsversuchs 4.3.4 Bestimmung der Anzahl adulter D. viviparus Aus den exenterierten Rinderlungen wurden die adulten Lungenwürmer manuell gewonnen und anschließend mikroskopisch geschlechtsdifferenziert. Die folgende Tabelle zeigt die Anzahl männlicher und weiblicher Würmer pro Versuchstier sowie die Durchschnittswerte der Versuchsgruppen. Zur Berechnung der Fekundität, definiert als Larven pro weiblichem Wurm (Larven/w Wurm) ist die Gesamtzahl der während des Versuchs ausgeschiedenen Larven angegeben. Diese berechnete sich aus der Summe der Mittelwerte der 2 x 10 g Doppelansätze. 140 Ergebnisse Gruppe Tier AlOH3 AlOH3 Quil A AlOH3 Quil A Quil A Quil A Kontrolle Kombi Kombi PDI Kombi Kontrolle PDI Nr. Tab. 16: Larven total Adulte m w 588 596 598 950 611 749 757 914 544 586 597 952 540 545 581 752 579 600 753 948 187 105 276 349 323 44 133 80 141 189 89 90 138 319 172 344 70 231 356 107 282 159 395 474 513 82 175 152 269 261 156 217 300 471 195 490 168 387 582 178 110 795 802 805 808 447 324 77 159 142 689 474 136 294 291 814 754 762 790 807 810 816 260 179 289 199 283 192 84 391 291 476 337 489 325 145 total (Tier) MW (Gruppe) Tier Gruppe I. Immunisierungsversuch 469 5194 557 264 2816 32716 SD=243,1 671 5966 823 18740 836 14638 376 126 2078 25446 SD=315,9 308 5828 232 2902 410 2838 450 4855 353 11537 SD=93,9 245 1234 307 2610 438 5128 607 790 5884 25943 367 8015 SD=238,9 834 6916 238 817 520 618 3029 12500 SD=326,1 938 6755 285 1899 II. Immunisierungsversuch 1136 10490 798 5552 213 529 614 24310 453 2990 SD=324,7 433 3221 651 1529 470 4254 765 2096 548 536 1929 13299,5 SD=203,1 772 3769 517 961 229 292 Larven/w Wurm Tier 18,4 17,7 15,1 39,5 28,5 25,3 33,3 19,1 10,6 18,6 7,9 12 17,1 12,5 41,1 14,1 4,9 7,8 11,6 10,7 15,2 11,7 3,9 10,2 11,1 3,9 14,6 4,4 5,7 7,7 3,0 2,0 MW (Gruppe) 22,7 SD=11,3 26,6 SD=6,0 12,3 SD=4,5 21,2 SD=13,4 8,8 SD=3,0 9,3 SD=4,5 6,2 SD=4,6 Anzahl adulter Lungenwürmer und Larvenausscheidung. Die Unterschiede in den Wurmzahlen zwischen den Gruppen waren statistisch nicht signifikant (p = 0,58, Kruskal-Wallis Test). Ergebnisse 141 4.3.5 Bestimmung der Wurmgröße Von jedem Versuchstier wurden die Länge und Breite von 25 männlichen und 25 weiblichen Würmern mithilfe der OLYMPUS-Software 5050 bestimmt. II: Kombi Quil A 42,3 32,1 II: Kontrolle Quil A 47,3 35,3 I: PDI Quil A 44,1 35,2 I: PDI AI(OH)3 46,9 37,6 I: Kombi Quil A 46,3 34,0 I: Kombi AI(OH)3 45,3 33,2 I: Kontrolle AI(OH)3 40,5 0 10 20 30 40 50 52,7 60 70 Länge (mm) Abb. 26: Darstellung der mittleren Wurmlängen und Standardabweichungen in den Versuchsgruppen. grau: Länge der Weibchen; schraffiert: Länge der Männchen Im Anhang, Abschnitt 9.5, Tab. 20, sind je Tier Mittelwerte und Standabweichungen von Längen und Breiten der adulten Würmer dargestellt. Weibliche Würmer der vakzinierten Tiere im I. Immunisierungsversuch waren 11-16,3% kürzer als die der Kontrolltiere. Bei den männlichen Würmern der Vakzinegruppen war eine Längenreduktion von 7,2-18% vorhanden. Im II. Vakzineversuch waren die weiblichen Würmer der Kombigruppe 10,6%, die 142 Ergebnisse männlichen Würmer 9,1% kleiner als die der Kontrollgruppe. Diese Ergebnisse waren statistisch nicht signifikant (Ryan-Einot-Gabriel-Welsch Multiple Range Test, Kruskal-Wallis Test, p = 0,09 für weibliche Würmer; p = 0,12 für männliche Würmer). 4.4 Ergebnisse der serologischen Untersuchungen 4.4.1 ELISA Während des Versuchsdurchgangs wurde die Bildung von Anti-PDI Antikörpern der Tiere überprüft. Alle immunisierten Tiere zeigten einen Titeranstieg zwischen dem vierten und elften Versuchstag. Dieser Anstieg war bei allen untersuchten Antikörperklassen vorhanden, wobei die höchsten ODs bei IgG2 zu verzeichnen waren. Die Kontrollgruppen zeigten nach der experimentellen Infektion einen Titeranstieg bei IgG und IgG1. Bis zum Ende des Versuchs wurden jedoch nicht die Werte der Immunisierungsgruppen erreicht. Index Gruppenmittel Ergebnisse 143 IgG 1,6 1,2 0,8 0,4 0,0 Index Gruppenmittel 0 1,6 4 7 11 14 21 28 35 42 49 56 63 70 77 84 91 Tage S 7 11 14 21 28 35 42 49 56 63 70 77 84 91 Tage S 7 11 14 21 28 35 42 49 56 63 70 77 84 91 Tage S IgG1 1,2 0,8 0,4 0,0 Index Gruppenmittel 0 1,6 4 IgG2 1,2 0,8 0,4 0,0 0 4 I: Kontrolle Al(OH)3 3 I: Kombi Al(OH)33 II: Kombi Quil A I: PDI Al(OH)3 3 I: Kombi Quil A I: PDI Quil A II: Kontrolle Quil A Index Gruppenmittel 144 Ergebnisse IgA 1,6 1,2 0,8 0,4 0,0 Index Gruppenmittel 0 4 7 11 14 21 28 35 42 49 56 63 70 77 84 91 Tage S 7 11 14 21 28 35 42 49 56 63 70 77 84 91 Tage S IgM 1,6 1,2 0,8 0,4 0,0 0 4 I: Kontrolle Al(OH)3 3 I: Kombi Al(OH)33 II: Kombi Quil A Abb. 27: I: PDI Al(OH)3 3 I: Kombi Quil A I: PDI Quil A II: Kontrolle Quil A Gruppenmittelwerte der Indices für verschiedene Serumantikörpertiter Detaildaten incl. Standardabweichung finden sich aus Gründen der Übersichtlichkeit im Anhang, Abschnitt 9.6, Tab. 21. 4.4.2 Überprüfung der Serokonversion mittels Westernblot Da es nicht möglich war, rekombinante PDI ohne GST und ohne bakterielle Kontaminanten zu gewinnen, wurde die im ELISA beobachtete Serokonversion zusätzlich im Western-Blot überprüft. Hiermit sollte ausgeschlossen werden, dass Ergebnisse 145 der Anstieg der OD ausschließlich auf einer Reaktion auf GST oder bakteriellen Kontaminanten beruhte. Hierzu wurden die Seren der Kontrollgruppe und der PDIimmunisierten Tiere des I. Immunisierungsversuchs anhand von D. viviparus Rohantigen überprüft. Nach der dritten Immunisierung (Tag 63) zeigte sich bei den PDI-immunisierten Tieren eine Bande auf einer Höhe von etwa 55 kDa. Dies entspricht dem Molekulargewicht der PDI. Nach der Infektion zeigte sich auch mit den Seren der Kontrolltiere eine Bande auf gleicher Höhe, jedoch stellte sich diese schwächer dar als bei den immunisierten Tieren. Mit dem Serumpool D. viviparus-infizierter Stammhaltungrinder (Positivpool) konnte ebenfalls eine Bande auf gleicher Höhe detektiert werden, jedoch war auch hier die Reaktion schwach ausgeprägt. 146 Ergebnisse kDa 250 98 64 50 36 30 Tag 0 Tag 63 250 98 64 50 36 30 Tag 98 Abb. 28: 588 596 598 950 540 545 581 752 579 600 753 948 Positivpool Negativpool 250 98 64 50 36 30 Tiernummer Kontrolle Al(OH)3 Gruppe PDI Al(OH)3 PDI Quil A Überprüfung der Serokonversion im Western-Blot Ergebnisse 147 4.5 Ergebnisse der quantitativen real-time PCR 4.5.1 Absolute Quantifizierung In den ersten Versuchen zur Quantifizierung der PDI-Expression in den bei der Sektion gewonnenen Würmern wurde versucht, eine absolute Quantifizierung durchzuführen, wobei die Ergebnisse auf das Housekeeping-Gen 18S-rRNA bezogen wurden. Hierbei ließen sich keine verwertbaren Ergebnisse erzielen. Anhand der Plasmid-DNA-Verdünnungsreihen zeigte sich eine schlechte und von Lauf zu Lauf schwankende Sensitivität der Sonden. Die PDI-Sonde detektierte nur in einem von vier Läufen alle Verdünnungsstufen. In den übrigen Läufen konnten lediglich 103 Kopien sicher detektiert werden. Die 18S-rRNA-Sonde erwies sich als noch weniger sensitiv. Es konnte maximal die 103-Verdünnungsstufe detektiert werden. Die anhand der Plasmid-DNA-Verdünnungsreihe errechnete PCR-Effizienz lag bei der PDI-Sonde bei 76% und bei der 18S-rRNA-Sonde bei 55%. Trotz neuer Sondencharge konnte keine besserte PCR-Effizienz erzielt werden. Daher wurden Versuche zur Effizienz-korrigierten relativen Quantifizierung angeschlossen. 4.5.2 Relative Quantifizierung Im ersten Schritt wurde versucht, eine Effizienz-korrigierte relative Quantifizierung mithilfe des „Relative Quantitation“-Programms des Mx 4000® durchzuführen. Hierbei wird in jedem Lauf eine als Kalibrator bezeichnete Probe mitgeführt, für die die Cts von Zielgen (PDI) und Referenzgen (18S-rRNA) bestimmt wird. Die Expression des Zielgens in der untersuchten Probe wird auf den Kalibrators bezogen. Der Kalibrator wurde von einem willkürlich gewählten, männlichen Wurm der Kontrollgruppe (Tier Nr.: 795) verkörpert. Die PCR-Effizienz wurde in vorher durchgeführten Versuchen anhand von cDNA-Verdünnungsreihen bestimmt. Hierbei errechnete sich für die PDI-Sonde eine Effizienz von 1,704 (70,4%) und für die 18SrRNA-Sonde eine Effizienz von 1,479 (47,9%). Die extrem schlechte PCR-Effizienz der 18S-Amplifikation führte zu hohen Schwankungen der Cts. Dies zeigte sich beim Vergleich der für den Kalibrator in allen Läufen ermittelten Cts : 148 Ergebnisse PCR-Lauf 1 2 3 4 5 6 7 8 9 MW Tab. 17: MW Ct 18S 27,20 33,67 37,08 34,05 31,94 32,28 36,07 33,65 33,09 33,23 SD 0,22 # 0,13 0,05 # 0,37 0,11 0,00 0,16 MW Ct PDI 30,10 30,00 30,10 31,08 32,05 31,12 31,10 30,48 32,10 30,90 SD 0,12 0,01 0,12 0,04 0,06 0,14 0,04 0,12 0,09 Cts des Kalibrators in verschiedenen PCR-Läufen. MW: Mittelwert; SD: Standardabweichung; #: ein Duplikat ausgeschlossen Während bei der PDI-Amplifikation maximale Schwankungen von etwa zwei Cts vorlagen, schwankten die Cts der 18S-rRNA-Amplifikation um fast zehn Cts. Eine sinnvolle Auswertung war mit diesen Ergebnissen nicht möglich. Versuchsweise wurden die Mittelwerte der in den neun Läufen erhaltenen Cts für den Kalibrator in die Formel zur Effizienz-korrigierten relativen Quantifizierung eingesetzt und die Ratios der 120 Würmer bestimmt. Ratio = Abb. 29: (EZielgen)∆Ct Zielgen (Kontrolle – Behandlung) (EReferenzgen)∆Ct Referenzgen (Kontrolle – Behandlung) Ratioberechnung nach PFAFFL E: Effizienz Ergebnisse 149 Ratio = Abb. 30: (EPDI)∆Ct PDI (MW Kalibrator – Wurm X) (E18S)∆Ct 18S (MW Kalibrator – Wurm X) Anwendung der Formel nach PFAFFL. (MW = arithmetischer Mittelwert; X = Wurm Nr. 1 bis 120) Im Folgenden sind die Berechnungen der Ratios für 120 Würmer grafisch dargestellt. 2,0 Ratio 1,5 1,0 0,5 Tier-Nr. Abb. 31: Grafische Darstellung der Ratios für 120 Würmer grau: männliche Würmer; weiß: weibliche Würmer Diese Auswertung führte zu keinen sinnvollen Ergebnissen. 81 6 81 0 80 7 79 0 76 2 75 4 81 4 80 8 80 5 80 2 79 5 11 0 0,0 150 Diskussion 5 Diskussion Die vorliegende Arbeit hatte zum Ziel, die Protein Disulfid Isomerase (PDI) von D. viviparus zu identifizieren und auf DNA-Sequenzebene zu charakterisieren. Nach rekombinanter Herstellung und Aufreinigung des Proteins erfolgte in Immunisierungsversuchen die Überprüfung der Immunogenität und Protektivität der PDI. 5.1 Identifizierung der PDI von D. viviparus Durch eine PCR mit degenerierten Primern und anschließender 5’- und 3’-RACEPCR konnte anhand von cDNA eine Sequenz von 1796 bp Länge identifiziert werden. Der Sequenzidentitätsvergleich zeigte eine 80%ige Identität der gefundenen Sequenz zur PDI von A. caninum und T. circumcincta und zur PDI2 von O. ostertagi, sowie eine 72%ige Identität zur PDI2 von C. elegans. Diese Übereinstimmungen auf Nukleotidebene und eine spätere Analyse des exprimierten Proteins mittels MALDITOF-MS bestätigten, dass eine PDI von D. viviparus identifiziert worden war und führten zur Veröffentlichung der Sequenz als PDI2 von D. viviparus in der NCBIDatenbank unter der Nummer DQ011044. Die Überprüfung der AS-Sequenz auf das Vorliegen konservierter Domänen mittels RPSBLAST zeigte eine PDI-typische VierDomänen-Struktur mit zwei aktiven und zwei inaktiven Thioredoxin-Domänen. Die aktiven Thioredoxin-Domänen enthalten jeweils die Sequenz -WCGHCK-, hierbei handelt es sich um die aktiven Zentren des Enzyms. Die beiden Cysteine bilden die Grundlage der Redoxaktivität der PDI, indem diese zwischen dem Disulfid- und Dithiolzustand wechseln (HAWKINS u. FREEDMAN 1991). Des Weiteren erfüllt die PDI als Untereinheit der Prolyl-4-Hydroxylase eine essentielle Funktion bei der Synthese von Kollagen (MYLLYHARJU 2003). Bei C. elegans konnte demonstriert werden, dass diese Funktionen der PDI eine maßgebliche Bedeutung bei der Synthese der Kutikula haben, wobei hier die PDI2 eine essentielle Rolle besitzt, da die PDI1 nicht als Untereinheit der Prolyl-4-Hydroxylase fungiert (WINTER u. PAGE 2000). Die hohen Identitäten der PDI2 von D. viviparus zur PDI2 von C. elegans lassen für dieses Enzym ähnliche Funktionen bei der Synthese der Kutikula von D. viviparus vermuten. Diskussion 151 Am C-terminalen Ende der PDI2 konnte eine HTEL-Sequenz identifiziert werden, die sich in gleicher Form bei der PDI von A. caninum und T. circumcincta sowie bei der PDI2 von O. ostertagi und C. elegans findet. Hierbei handelt es sich um eine modifizierte Form des Standard-ER-Retentionssignal KDEL, das unter anderem bei der humanen PDI zu finden ist. Nach ANDRES et al. (1990) ist die HTEL-Sequenz ebenso effektiv bei der Zurückhaltung eines Proteins im Endoplasmatischen Reticulum wie das KDEL-Signal. Dies würde bedeuten, dass die PDI von D. viviparus im Lumen des ER lokalisiert ist. Allerdings wurden bereits diverse, eine KDELSequenz aufweisende Proteine, außerhalb des ER lokalisiert. Dies trifft auch für die PDI zu. So wird für die humane PDI eine Sekretion durch bestimmte Zellen und auch eine Lokalisation auf der Zelloberfläche beschrieben. Bei Helminthen konnte die PDI in ES-Produkten von O. ostertagi, D. immitis und F. hepatica beschrieben werden. Diese Beobachtungen zeigen, dass das Retentionssignal allein nicht ausreichend für eine Zurückhaltung des Proteins im ER ist und lassen eine extrazelluläre Lokalisation der PDI2 von D. viviparus möglich erscheinen. Die Bestimmung der genomischen Sequenz der PDI2 lieferte vier Intronsequenzen, die alle der GT/AG-Regel entsprachen. Ein Vergleich mit der genomischen Sequenz der PDI von A. caninum zeigte, dass sich die Introns an den gleichen Positionen befinden. Dies macht deutlich, dass die Organisation des Gens zwischen diesen nahe verwandten Spezies evolutionär konserviert ist. Nach der Klonierung des mit den degenerierten Primern amplifizierten PCR-Produkts wurden mehrere Klone verglichen. Hierbei konnte ein Klon mit von der PDI2 abweichender Sequenz identifiziert werden. Datenbankvergleiche zeigten, dass diese Sequenz höchste Ähnlichkeit zur PDI3 von C. elegans und zur PDI1 von O. ostertagi aufweist. Die Vervollständigung der Sequenz führte zur Beschreibung eines zweiten PDI-Gens von D. viviparus, welches unter der Nummer DQ011045 in der NCBI-Datenbank veröffentlicht wurde. Diese PDI weist ebenfalls die typische Vier-Domänen-Struktur auf, wobei die b- und b’-Domäne jedoch höhere Ähnlichkeiten zum humanen ERp57 aufweisen. Darüber hinaus liefert das Fehlen eines C-terminalen Bereichs saurer Aminosäuren bei der PDI1 von D. viviparus einen weiteren Hinweis auf die nähere Verwandtschaft dieser PDI zum ERp57. Die 152 Diskussion aktiven Thioredoxin-Domänen weisen ebenfalls die Sequenz -WCGHCK- auf und das ER-Retentionssignal hat die Form KAEL. Ob diese PDI von D. viviparus der PDI3 von C. elegans und dem humanen Analogon ERp57 auch funktionell ähnelt und nicht als Untereinheit der Prolyl-4-Hydroxylase fungiert, konnte aus den erhaltenen Informationen nicht ermittelt werden. Auch bei anderen Nematoden wurden verschiedene PDI-Gene beschrieben. So konnten bei O. ostertagi bislang zwei PDIs identifiziert werden, und bei C. elegans wurden drei PDIs näher charakterisiert. Ob wie bei C. elegans auch bei D. viviparus weitere PDIs existieren, konnte im Rahmen dieser Arbeit nicht geklärt werden. 5.2 Expression und Aufreinigung der rekombinanten PDI Nachdem die vollständige Sequenz der PDI ermittelt worden war, wurde die Sequenz des ORF in den pGEX-4T-Expressionsvektor kloniert und als GST-Fusionsprotein in E. coli exprimiert. Die Wahl des pGEX-Vektors bietet verschiedene Vorteile. In diesem Vektor erfolgt die Expression des Zielproteins in Fusion mit der Glutathion-STransferase aus S. japonicum. Über die GST lässt sich das Fusionsprotein leicht mit verschiedenen Systemen aufreinigen. Grundprinzip der Aufreinigung ist die reversible Bindung der GST an Glutathion. In der vorliegenden Arbeit wurden dazu sowohl Glutathion-gekoppelte Sepharosekügelchen als auch magnetische Partikel genutzt. Mit beiden Systemen gelang eine effektive Aufreinigung der rekombinant hergestellten PDI. Neben diesem technischen Vorteil erfolgte die Wahl des Expressionsvektors vor allem auch im Hinblick auf die geplanten Immunisierungsversuche. Aus der Literatur ist bekannt, dass die GST selbst immunmodulatorische Eigenschaften besitzt. GST-28 aus S. japonicum ist ein vielversprechender Vakzinekandidat gegen Schistosomatose beim Menschen und auch bei anderen Spezies konnte das protektive Potential dieses Enzyms nachgewiesen werden. Beim Einsatz des Fusionsproteins als Impfprotein könnte also die GST zur Immunogenität beitragen. Für die Herstellung rekombinanter Proteine stehen verschiedene pro- und eukaryotische Expressionssysteme zur Verfügung. Die Expression heterologer Proteine in E. coli hat den Vorteil, dass dieses System technisch einfach zu Diskussion 153 handhaben ist und das gewünschte Protein kostengünstig und in großen Mengen hergestellt werden kann. Hauptnachteil ist die Tatsache, dass Proteine in E. coli nach der Translation nicht in gleicher Art und Weise weiterverarbeitet werden wie in Eukaryoten. Bakterien führen keine posttranslationalen Modifikationen wie etwa die Abspaltung von Signalsequenzen oder die Anheftung von Zuckergruppen (Glykosilierung) an Proteine durch. Diese Abwandlungen sind allerdings häufig notwendig, damit beispielsweise Enzyme ihre katalytische Aktivität entfalten können. Weiterhin zeigen neuere Untersuchungen, dass gerade Kohlenhydratkomponenten ein hohes immunogenes Potential haben und in den Prozess der Immunantwort gegen parasitische Helminthen involviert sind (NYAME et al. 2004). Weiterhin wird das in E. coli exprimierte Protein nicht gefaltet. Wenn Proteine nicht die korrekte Tertiärstruktur annehmen, bleiben sie in der Regel unlöslich und bilden in den Bakterien Einschlusskörperchen (inclusion bodies). Die Umwandlung von Proteinen aus Einschlusskörperchen in die richtig gefaltete Form gestaltet sich in vitro äußerst schwierig und gelingt häufig nicht. Aus der Literatur ist bekannt, dass in E. coli exprimierte Proteine mit sehr unterschiedlichem Erfolg in Vakzinierungsversuchen eingesetzt wurden. Der einzige zur Zeit erhältliche rekombinante Impfstoff gegen Parasiten, Tickgard® - gegen den Schildzeckenbefall bei Rindern, wird in E. coli produziert. Des Weiteren waren Immunisierungsversuche gegen Cestoden mit in E. coli als GST-Fusionsproteine exprimierten Onkosphärenantigenen sehr erfolgreich. Bei Taenia ovis konnte durch eine zweimalige Immunisierung mit 100 µg des To 45W-GST unter Einsatz des Adjuvans Saponin eine Reduktion der Cysticerci um mindestens 97% erreicht werden. Das hohe protektive Potential des Antigens bestätigte sich auch in Feldversuchen (LAWRENCE et al. 1996). Zwei weitere Onkosphärenantigene, To 16.17 und To 18 konnten ebenfalls eine fast vollständige Immunität gegen experimentelle Infektionen vermitteln (HARRISON et al. 1996). Bei Taenia saginata führte die Kombination der Proteine Tsa 9 und Tsa 18 zu einer Protektion. Eine dreimalige Applikation von 200 µg GST-Fusionsprotein mit Quil A resultierte in einer 94%igen Reduktion der Cysticerci (LIGHTOWLERS et al. 1996). Auch gegen Echinococcus granulosus konnte ein Onkosphärenantigen mit hohem protektiven Potential identifiziert werden. Immunisierungen mit dem EG 95-GST- 154 Diskussion Fusionsprotein führten zu einer mindestens 96%igen Reduktion der Cysticerci (LIGHTOWLERS 1996; LIGHTOWLERS et al. 1999). Gegenüber diesen positiven Ergebnissen konnten bei Nematoden und Trematoden bislang keine Erfolge mit rekombinanten Proteinen Darmoberflächenproteine erzielt H11, werden. H-gal-GP Beispielsweise (Haemonchus zeigten die galactose-containing glycoprotein complex) und TSBP von H. contortus in nativer Form ein hohes protektives Potential. So konnten etwa in Immunisierungsversuchen mit H11 bis zu 99%ige Reduktionen der Eiausscheidung und bis zu 95%ige Reduktionen der Wurmbürden erreicht werden (NEWTON et al. 1995; SMITH et al. 1993; TAVERNOR et al. 1992). Die dreimalige Applikation von 100 µg H-gal-GP mit Quil A resultierte in einer 93%igen Reduktion der Eiausscheidung und einer 60%igen Reduktion der Wurmbürde (SMITH et al. 2000). Mit TSBP konnten bei dreimaliger Applikation von 200 µg 82%ige Reduktionen der Eiausscheidung und Wurmbürde erreicht werden (KNOX et al. 1999). Alle Antigene erwiesen sich jedoch als in E. coli exprimierte Proteine wirkungslos. Als eukaryotische Expressionssysteme haben sich Hefen, wie Saccharomyces cerevisae und Pichia pastoris, Insekten- und Säugerzelllinien etabliert. Diese Systeme haben den Vorteil, dass das exprimierte Protein posttranslational modifiziert wird und so dem Protein im Ursprungsorganismus möglicherweise stärker ähnelt. Allerdings ist zu beachten, dass jeder Organismus ein eigenes Repertoire an Enzymen besitzt, die posttranslationale Modifikationen ausführen, und die exprimierten Proteine nicht notwendigerweise die identischen Strukturen erhalten, wie sie im Ursprungsorganismus vorliegen würden. Beispielsweise finden bei S. cerevisiae Hyperglykosilierungen mit Oligosaccharidketten statt, die aus 50 bis 150 Resten bestehen. In P. pastoris werden bei N-Glykosilierungen hingegen 8 bis 14 Reste angefügt. Die in eukaryotischen Expressionssystemen durchgeführten posttranslationalen Modifikationen führen zwar möglicherweise dazu, dass das gewünschte Protein in katalytisch aktiver Form erhalten wird, allerdings besteht aufgrund der Unterschiede in Glykosilierungen und anderen posttranslationalen Diskussion 155 Modifikationen keine Garantie, dass das rekombinante Protein die gleiche Immunogenität besitzt. In den in dieser Arbeit durchgeführten Immunisierungsversuchen wurde in E. coli exprimierte PDI eingesetzt, da die Überprüfung der Immunogenität des rekombinanten Proteins im Western-Blot zeigte, dass die PDI mit dem Serum experimentell infizierter Rinder detektiert werden konnte, also antigene Determinanten zumindest teilweise vorhanden sein mussten. Aus der Literatur war weiterhin bekannt, dass in E. coli exprimierte PDI in der Regel katalytisch aktiv ist. Dies bestätigen auch verschiedene Versuche, in denen demonstriert werden konnte, dass die Koexpression von PDI die Expression heterologer Proteine in E. coli verbessern kann. Durch ihre Funktion als Chaperon trägt die PDI dazu bei, dass die Proteine ihre richtige Konformation erhalten und nicht unlöslich in Einschlusskörperchen vorliegen. Auch wenn es im Rahmen dieser Arbeit nicht gelang, einen Aktivitätstest für die PDI zu etablieren, ließ das Verhalten des Proteins bei der Aufreinigung die Vermutung zu, dass die PDI in katalytisch aktiver Form exprimiert wurde. Für die Aufreinigung des GST-PDI-Fusionsproteins wurden Glutathion-gekoppelte magnetische Partikel Optimierungsversuche und Glutathion-Sepharose gelang es nicht, die eingesetzt. im Trotz Western-Blot diverser sichtbaren Begleitbanden vollständig zu entfernen. Die Proteinmessung im Agilent 2100 Bioanalyzer zeigte für den in den Immunisierungsversuchen eingesetzten Proteinpool eine Reinheit des Fusionsproteins von etwa 70% im ersten Immunisierungsversuch bzw. 78% im zweiten Immunisierungsversuch. Laut Herstellerangaben ist es mit beiden Medien möglich, hochreines GST-Fusionsprotein zu gewinnen. Das Auftreten von Begleitbanden kann zum einen die Ursache haben, dass das Fusionsprotein durch Proteasen degradiert wird, zum anderen besteht die Möglichkeit der Koaufreinigung bakterieller Proteine. Im Western-Blot mit PDIAntikörpern zeigte sich, dass es sich bei den Begleitbanden nur zu einem geringen Teil um PDI-Bruchstücke handelte. Durch den Einsatz des Protease-defizienten 156 Diskussion E. coli Stammes BL21 und durch die Zugabe von Proteaseinhibitoren während der Aufreinigung konnte eine Proteindegradation weitgehend verhindert werden. Eine Koaufreinigung bakterieller Proteine kann verschiedene Gründe haben. So führt eine exzessive Ultraschallbehandlung der Zellsuspension zur Aggregation von Proteinen, die dann gemeinsam aufgereinigt werden. Dieser Grund kann im vorliegenden Fall ausgeschlossen werden, da nach der Zelllyse mittels des Lysisreagenz aus dem MagneGST™-Kit die gleichen Begleitbanden in der Aufreinigung identifiziert werden konnten, wie nach Ultraschallbehandlung. Weiterhin ist bekannt, dass es bei den verwendeten Aufreinigungmedien zu einer Koaufreinigung der bakteriellen Chaperonproteine DnaK, DnaJ, GrpE, GroEL und GroES kommen kann. Möglicherweise liegt bei der Aufreinigung der PDI ein ähnlicher Mechanismus vor. Unter der Vorraussetzung, dass die in dieser Arbeit rekombinant hergestellte PDI katalytisch aktiv ist, wäre es möglich, dass das Enzym während der Expression und Aufreinigung seine Funktion als Chaperon erfüllt und bakterielle Proteine gebunden hat, die hierdurch mit aufgereinigt wurden und erst unter den denaturierenden Bedingungen der SDS-Page wieder frei wurden. Für das Vorliegen eines derartigen Mechanismus spricht, dass unabhängig von den gewählten Aufreinigungsbedingungen immer exakt die gleichen Begleitbanden vorhanden waren. Für die Tatsache, dass die PDI in katalytisch aktiver Form exprimiert wurde, spricht weiterhin das konstante Vorhandensein einer Bande mit einem Molekulargewicht von etwas weniger als 250 kDa, die mit PDI-Antikörpern nachgewiesen werden konnte. Diese Bande war nach Zusatz des Disulfidbrückenspaltenden Agens β-Mercaptoethanol im Probenpuffer für die SDS-Page nicht mehr vorhanden (Ergebnisse nicht gezeigt). Dies könnte bedeuten, dass die PDI einen stabilen Komplex mit E. coli Proteinen gebildet hatte. Da es nicht möglich war, das Aufreinigungsergebnis über eine Optimierung der Affinitäts-Chromatographie zu verbessern, wurde das Fusionsprotein mit einer Reinheit von 70 bis 78% in den Immunisierungversuchen eingesetzt. Es wurde darauf verzichtet, weitere Aufreinigungsschritte wie etwa über Ionenaustauschersäulen oder Gelfiltrationssäulen anzuschließen, da hierdurch möglicherweise ein Verlust der für die PDI vermuteten katalytischen Aktivität Diskussion 157 stattgefunden hätte. Für die protektive Wirkung eines Vakzinekandidaten hat die Reinheit des Proteins primär keine ausschlaggebende Bedeutung. DEMPSTER et al. (1996) entwickelten ein Aufreinigungsprotokoll für die schnelle und kostengünstige Herstellung großer Mengen an GST-45W für die Immunisierung gegen T. ovis und erreichten mit einer Reinheit von 30-50% eine vollständige Protektion. Für die Immunisierung ist es wichtig, dass die Vakzine keine schädlichen Bestandteile enthält. Um die Sicherheit der in dieser Arbeit verwendeten Vakzine zu erhöhen, wurde das aufgereinigte Protein einer Sterilfiltration unterzogen. Bei der Entwicklung einer kommerziellen Vakzine muss ein Aufreinigungergebnis erzielt werden, dass eine gleichbleibende Qualität und Sicherheit garantieren kann. Für die in dieser Arbeit durchgeführten Immunisierungsversuche konnte davon ausgegangen werden, dass die erhaltene Reinheit ausreicht, um das protektive Potential der PDI zu überprüfen. 5.3 Durchführung der Immunisierungsversuche 5.3.1 Wahl des Tiermaterials Die Immunisierungsversuche wurden im natürlichen Wirt von D. viviparus durchgeführt. Das einzige Tiermodell, das bisher für D. viviparus beschrieben wurde, ist das Meerschweinchen. Bei dieser Spezies gelangen die Larven nach experimenteller Infektion in die Lunge, jedoch stoppt die Entwicklung im präadulten Stadium. Bei Immunisierungsversuchen lassen sich also keine Aussagen über einen möglichen Einfluss des Impfstoffs auf die adulten Stadien oder auf die Geschlechtsprodukte treffen. Weiterhin ist eine Übertragbarkeit der Ergebnisse vom Meerschweinchen auf das Rind vorsichtig zu beurteilen. Da das Meerschweinchen kein natürlicher Wirt von D. viviparus ist, greifen bei der Infektion möglicherweise nicht die gleichen Anpassungsmechanismen wie im Rind. Dies ist insbesondere in Bezug auf die Immunabwehr des Wirtes von Bedeutung. Für die Immunisierungsversuche wurden männliche Holstein Frisian Rinder mit einem Alter von 4-5 Monaten eingesetzt. Außer der Tatsache, dass die Tiere keinen Lungenwurm-Kontakt gehabt haben durften, war die Wahl des Tiermaterials unter anderem von praktischen Gesichtspunkten beeinflusst. Männliche Rinder sind 158 Diskussion einfacher zu beschaffen und kostengünstiger als weibliche Tiere. Weiterhin konnten die Bullkälber von einem einzigen Mäster bezogen werden, der die Tiere nach dem Absetzen aufgestallt und bis zu dem entsprechenden Alter aufgezogen hatte. Hierdurch wurde die Variabilität im Tiermaterial herabgesetzt, da alle Tiere durch die gemeinsame Aufzucht einen ähnlichen Immunstatus hatten. Wären weibliche Tiere eingesetzt worden, hätten diese aus mehreren Quellen bezogen werden müssen. Hierin hätte eine erhöhte Gefahr der Übertragung von Infektionserregern bei der gemeinsamen Aufstallung bestanden. Für Immunisierungen ist es jedoch von essentieller Bedeutung, dass die Tiere gesund sind. In der Literatur finden sich keine Hinweise darauf, dass männliche und weibliche Rinder eine unterschiedliche Empfänglichkeit für D. viviparus zeigen. Weiterhin finden sich keine Angaben bezüglich einer Rassedisposition. Es war also davon auszugehen, dass mit dem gewählten Tiermaterial erste Hinweise bezüglich der Wirksamkeit des Impfproteins erhalten werden können. Aufgrund der hohen Kosten der Versuchstiere musste im Hinblick auf die eingesetzte Tierzahl ein Kompromiss eingegangen werden. Eine Zahl von vier Tieren pro Versuchsgruppe wie im ersten Immunisierungsversuch kann im Zweifelsfall keine statistische Sicherheit bieten, ist jedoch geeignet, um Tendenzen aufzuzeigen. Immunisierungsversuchen Da um es den sich bei erstmaligen den hier Einsatz durchgeführten der PDI als Vakzinekandidaten handelt, bestand hierin das Hauptziel der Versuche. 5.3.2 Immunisierung und experimentelle Infektion Im ersten Immunisierungsversuch wurden die Rinder dreimal im Abstand von vier Wochen mit 200 µg PDI immunisiert. Die eingesetzte Proteinmenge orientierte sich hierbei an den Angaben in der Literatur. LIGHTOWLERS et al. (1996) testeten Proteinmengen von 20, 200, 500 und 1000 µg bei der Immunisierung von Rindern gegen T. saginata mit Tsa 9 + 18 und erreichten die höchste Protektion mit einer Proteinmenge von 200 µg. Neben anderen Faktoren hat die Wahl des Adjuvans einen entscheidenden Einfluss auf die Ausbildung einer protektiven Immunantwort. Adjuvantien können einerseits dem Transport des Antigens dienen, so dass eine schnellere und bessere Verarbeitung durch Antigen-präsentierende Zellen erfolgt, andererseits haben einige Diskussion 159 Adjuvantien selbst einen immunstimulierenden Effekt. Darüber hinaus können Adjuvantien die Immunantwort in eine bestimmte Richtung lenken. In den in dieser Arbeit durchgeführten Immunisierungsversuchen wurden Al(OH)3 und Quil A eingesetzt. Während Al(OH)3 fast ausschließlich eine Th2-Immunantwort fördert, gilt Quil A als starkes Stimulans sowohl einer Th1- als auch einer Th2-Antwort. Vier Wochen nach der zweiten Boosterimpfung wurden die Impf- und Kontrolltiere mit insgesamt 3300 dritten Larven oral infiziert. Die Larvendosis wurde aufgrund von Erfahrungswerten gewählt, die während der Haltung von Stammhaltungsrindern für D. viviparus im Institut für Parasitologie gesammelt worden waren. Es hatte sich gezeigt, dass die Verabreichung von 3300 Larven bei einem Rind mit einem Körpergewicht von etwa 100 kg zur Ausbildung einer klinisch manifesten Dictyocaulose mit deutlichen Krankheitssymptomen führt, aber in der Regel keine Gefahr für das Tier darstellt. Mit der gewählten Larvendosis konnte also davon ausgegangen werden, dass beurteilt werden konnte, ob die immunisierten Tiere Unterschiede zur Kontrollgruppe bezüglich der Ausprägung klinischer Symptome zeigten. 5.4 Ergebnisse der Immunisierungsversuche 5.4.1 Befunde der täglichen Beobachtung und klinischen Untersuchung Bis zur Belastungsinfektion blieben alle Tiere bis auf die Nummer 544 klinisch gesund. Dieses Tier musste aufgrund respiratorischer Symptome in der dritten Versuchswoche medikamentös behandelt werden. An den Injektionsstellen traten bei den Kontrolltieren des ersten Immunisierungsversuchs keine Schwellungen auf. Im zweiten Immunisierungsversuch zeigten zwei Tiere jeweils nach einer Impfung eine geringgradige Schwellung, die nach einem bzw. nach drei Tagen wieder abgeklungen war. In den immunisierten Gruppen wiesen 17 von 22 Tieren nach mindestens einer Impfung eine lokale Umfangsvermehrung auf, die z.T. Durchmesser von mehr als fünf Zentimetern erreichte. Die Schwellungen waren nicht schmerzhaft und spätestens 12 Tage p.i. wieder abgeklungen. Diese lokalen Reaktionen hatten ein zumindest für Nutztiere tolerierbares Ausmaß. Da die eingesetzten Adjuvantien ihrerseits eher selten zum Auftreten lokaler 160 Diskussion Entzündungsreaktionen führen, wie dies etwa vom Freunds Adjuvans bekannt ist, können die aufgetretenen Schwellungen auf die Proteinbestandteile der Vakzine zurückgeführt werden. Es konnte nicht beurteilt werden, ob die applizierte Vakzine noch pyrogenes Material enthielt, da im Anschluss an die Impfungen keine Temperaturmessung erfolgte. Da die Tiere jedoch keine Störung des Allgemeinbefindens zeigten, ist davon auszugehen, dass die Impfungen keinen negativen Einfluss hatten. Zwei Wochen p.i. zeigten die Tiere erste Symptome einer Lungenwurminfektion. Diese Beobachtung deckt sich mit Angaben aus der Literatur (PFEIFFER 1971). Nachdem die Larven die Dünndarmschleimhaut des Wirtes durchdrungen und über Ductus thoracicus, Vena cava cranialis, rechte Herzkammer und Arteria pulmonalis die Lunge erreicht haben, finden sich etwa eine Woche p.i. die fünften Larvenstadien in den Bronchioli und Bronchen. Die durch sie verursachte Entzündung der luftführenden Wege äußert sich ab der zweiten Woche p.i. in klinischen Symptomen. Bei den in dieser Arbeit durchgeführten Belastungsinfektionen konnten eine Erhöhung der Atemfrequenz und Veränderung des Atemtyps zu einer vermehrt flachen Atmung, gelegentlicher Husten, muköser bis purulenter Nasenausfluss und eine Erhöhung der Herzfrequenz festgestellt werden. Bei einem Großteil der Tiere waren Lungengeräusche wahrnehmbar. Die Symptome waren sowohl in den Kontrollgruppen, als auch in den vakzinierten Gruppen gleichermaßen vorhanden. Es konnten keine Unterschiede bezüglich der Ausprägung der Symptome zwischen den Gruppen festgestellt werden. Das Auftreten von Nasenausfluss erwies sich als nicht konstant zu beobachtender Parameter, da die Tiere sich den Ausfluss spontan und bei der Futteraufnahme entfernten. Z.T. zeigten die Tiere Fieber und waren in ihrem Allgemeinbefinden soweit gestört, dass eine medikamentöse Behandlung notwendig war. Hiervon waren ebenfalls sowohl Kontrolltiere als auch immunisierte Tiere betroffen. Bezüglich der klinischen Symptomatik konnte also keine Wirksamkeit der PDI bei dieser Infektionsdosis festgestellt werden. 5.4.2 Etablierung der Infektion in den Kontrollgruppen An den Versuchstagen 63, 64 und 65 wurden die Rinder mit je 1100 dritten Larven infiziert. Bei der Sektion zeigte sich, dass sich die Larven in sehr unterschiedlicher Diskussion 161 Anzahl als Adulte in den Lungen etabliert hatten. Bei den Kontrolltieren schwankte der Anteil reisolierter Würmer im ersten Immunisierungsversuch zwischen 8 und 24,9%; im zweiten Immunisierungsversuch konnten zwischen 6,6 und 34,3% der 3300 applizierten Larven als Adulte wiedergewonnen werden. Diese Beobachtung deckt sich mit Angaben aus der Literatur. MATTHEWS et al. (2001) fanden bei sechs Kontrolltieren eine Etablierung zwischen 3,1 und 24% nach einer Belastungsinfektion mit 700 L3. Auch PFEIFFER (1971) gab an, dass die Befallsstärken von Tier zu Tier stark schwankten. Bei sechs Tieren, die mit 40 Larven/kg infiziert worden waren, fanden sich zwischen 2,9 und 29,9% als Adulte in den Lungen wieder. Diese Beobachtungen machen zum einen deutlich, mit welchen großen individuellen Schwankungen bei einem Immunisierungsversuch gerechnet werden muss, zum anderen bestätigen die in dieser Arbeit bei den Kontrolltieren gefundenen Befallsstärken, dass sich die Infektion in einem zu erwartenden Maß etabliert hatte. Dies zeigt, dass die Infektiösität und Viabilität der Larven nicht herabgesetzt war. 5.4.3 Larvenausscheidung und Wurmbürden Am Tag 84 (= 21 dpi) war bei jeweils einem Tier der Al(OH)3-Kombi-Gruppe und der PDI-Quil-A-Gruppe jeweils eine Larve im Trichteransatz vorhanden. Am 23. Tag nach der Infektion waren alle Tiere patent. Lediglich bei einem Rind der PDI-Quil-AGruppe setzte die Larvenausscheidung erst am 24. Tag p.i. ein. Diese Beobachtung deckt sich mit den Angaben anderer Autoren. PFEIFFER (1971) ermittelte ein Einsetzten der Larvenausscheidung zwischen dem 20. und 25. Tag p.i., wobei in den meisten Fällen am Tag 23 die ersten Larven im Kot zu finden waren. In den in dieser Arbeit durchgeführten Immunisierungsversuchen konnten keine Unterschiede zwischen den Kontroll- und Impfgruppen bezüglich des Einsetzens der Patenz festgestellt werden. Sowohl die tägliche als auch die Gesamtlarvenausscheidung der Einzeltiere wies sehr hohe Standardabweichungen auf. Es konnten keine signifikanten Unterschiede zwischen den Gruppen festgestellt werden. Eine interessante Beobachtung konnte allerdings bei der Gesamtlarvenausscheidung der Gruppen im ersten Immunisierungsversuch gemacht werden. Die Impfgruppen, in denen jeweils das gleiche Adjuvans eingesetzt wurde, wiesen annähernd gleiche Gesamtlarvenzahlen auf. Hierbei wurden in den Al(OH)3-Impfgruppen mit 25446 und 162 Diskussion 25943 etwa doppelt so viele Larven ausgeschieden, wie in den Quil A-Gruppen, bei denen insgesamt 11537 bzw. 12500 Larven vorhanden waren. Die Kontrollgruppe wies mit 32716 eine Gesamtlarvenausscheidung im Bereich der Al(OH)3Impfgruppen auf. Ob es sich bei diesen Beobachtungen um zufällige Ergebnisse oder um den Ausdruck einer Adjuvans-abhängigen Stimulation der Immunantwort mit unterschiedlichen Effekten auf die Fekundität der Würmer handelt, kann nicht beurteilt werden. Möglich wäre, dass der beobachtete Effekt auf einer unspezifischen Stimulierung des Immunsystems beruht. Diese Eigenschaft ist bei Quil A, jedoch nicht bei Al(OH)3 vorhanden. Dass die ermittelten Unterschiede in den Gesamtlarvenzahlen nicht auf eine unterschiedlich starke Etablierung der Würmer in den Lungen zurückzuführen waren, zeigten die nach der Sektion ermittelten Wurmzahlen. Hierbei zeigte sich, dass sowohl die Kombi-Gruppen, als auch die PDIGruppen untereinander ähnliche mittlere Befallsstärken aufwiesen. In den KombiGruppen waren im Mittel 376 (Al(OH)3) bzw. 353 (Quil A), in den PDI-Gruppen 607 (Al(OH)3) bzw. 520 (Quil A) Würmer vorhanden. Insgesamt betrachtet konnten bei den Befallsstärken keine statistisch signifikanten Unterschiede zwischen den Gruppen festgestellt werden. Die beiden Kombi-Gruppen des ersten Immunisierungsversuchs zeigten mit mittleren Befallsstärken von 376 und 353 Würmern tendenziell eine geringere Wurmbürde als die Kontrollgruppe, deren Tiere eine mittlere Wurmbürde von 557 aufwiesen. Sollte es sich hierbei um einen Effekt der Immunisierung handeln, so ist dieser nicht auf die PDI-Komponente im Kombi-Impfstoff zurückzuführen, da die immunisierten Gruppen, die ausschließlich die PDI-Vakzine erhielten, mit 607 (PDI Al(OH)3) und 520 (PDI Quil A) Würmern pro Tier Befallsstärken im Rahmen der Kontrollgruppe aufwiesen. Betrachtet man jedoch die mittleren Wurmbürden aus Sicht der Etablierung der Infektion, so befinden sich auch die Kombi-Gruppen mit einer Etablierungsrate von 3,8% bis 25,3% in einem Bereich der standardmäßig bei experimentellen Infektionen erreicht wird. Weiterhin war im zweiten Immunisierungsversuch der Unterschied in der mittleren Befallsstärke zwischen der Kombi-Impfgruppe und der Kontrollgruppe wesentlich geringer als im ersten Versuch. Hier waren bei der Kontrollgruppe im Mittel 614 und bei den immunisierten Tieren 548 Würmer vorhanden. Dies zeigt, dass die im ersten Versuch Diskussion 163 beobachteten geringeren Befallsstärken der Kombi-Impfgruppen vorsichtig zu beurteilen sind. Eine Beobachtung, die konstant bei allen Versuchstieren gemacht werden konnte, war, dass grundsätzlich mehr weibliche als männliche Würmer vorhanden waren. Das Verhältnis weiblich zu männlich schwankte hierbei zwischen 1,3:1 und 2,0:1 bei den Kontrolltieren und zwischen 1,1:1 und 2,4:1 bei den immunisierten Tieren. Dieser Sachverhalt wurde auch von PFEIFFER (1971) beschrieben. Hier fanden sich bei sechs experimentell infizierten Rindern Verhältnisse von weiblichen zu männlichen Würmern zwischen 1,1:1 und 1,4:1. Hieran zeigt sich, dass die Immunisierung keinen unterschiedlichen Effekt auf weibliche und männliche Würmer hatte, wie dies etwa für Darmoberflächenantigene bei verschiedenen blutsaugenden Nematoden beschrieben wurde. Ein derartiger Effekt war auch aufgrund der anderen Biologie von D. viviparus und vermuteten Lokalisation des Impfproteins nicht zu erwarten. Die Bestimmung der Wurmgröße zeigte, dass in allen immunisierten Gruppen des ersten Versuchs sowohl weibliche als auch männliche Würmer eine geringere Körperlänge aufwiesen als die der Kontrollgruppe. Diese Ergebnisse waren jedoch nicht signifikant. Im zweiten Immunisierungsversuch waren die Würmer der KombiImpfgruppe ebenfalls kürzer als die der Kontrollgruppe, jedoch zeigte sich in diesem Versuch, dass die Würmer grundsätzlich kleiner waren, als im ersten Versuch. So hatten etwa die männlichen Würmer der Kontrollgruppe im zweiten Versuch etwa die gleiche Länge wie die männlichen Würmer der PDI-Quil-A-Impfgruppe im ersten Versuch. Insgesamt konnte beobachtet werden, dass die Wurmgrößen innerhalb eines Versuchstieres wenig schwankten, jedoch zwischen den Rindern deutliche Unterschiede vorhanden waren. So existierte beispielsweise in der Kontrollgruppe des zweiten Versuchs eine Differenz der mittleren Wurmlängen von annähernd 15 mm zwischen dem Rind mit den kürzesten und dem mit den längsten weiblichen Würmern. Es war in beiden Versuchen eine positive Korrelation zwischen der Wurmlänge und der Wurmbürde vorhanden. Allerdings konnte kein konstanter Zusammenhang zwischen Wurmlänge/Wurmbürde und Körpergewicht des Versuchstieres festgestellt werden. Während diese Parameter im ersten Versuch negativ korreliert waren, zeigte sich im zweiten Versuch, dass die schwersten Tiere 164 Diskussion die meisten und längsten Würmer beherbergten. Die Beobachtung einer vom Körpergewicht unabhängigen Etablierung der Lungenwürmer wurde bereits von PFEIFFER (1971) gemacht und zeigt auf, welche starken individuellen Unterschiede bei einer Infektion vorhanden sind. Weiterhin konnte kein konstanter Zusammenhang zwischen Befallsstärke und Schwere der klinischen Symptome festgestellt werden. Sowohl Tiere mit einer hohen Wurmbürde als auch Tiere, die geringere Befallsraten aufwiesen, zeigten ein klinisches Bild, dass einer medikamentösen Behandlung bedurfte. Dies ist wahrscheinlich auf eine individuell unterschiedliche Empfänglichkeit für Sekundärinfektionen, die zu einer Verkomplizierung des Krankheitsbildes führten, zurückzuführen und spiegelte sich auch in den pathologisch-anatomischen Befunden an den Lungen wieder. Zum Teil wiesen auch Tiere mit einer geringeren Wurmbürde ausgedehnte Lungenveränderungen auf. Die Bestimmung des Differentialblutbildes erwies sich als wenig aussagekräftig. Eine absolute Eosinophilie konnte nur bei vier Tieren beobachtet werden. Da allerdings erst drei Wochen nach der Infektion eine Bestimmung des Differentialblutbildes erfolgte, ist möglicherweise ein vorher vorhandener Anstieg unentdeckt geblieben. PFEIFFER (1971) beobachtete einen ersten Anstieg der eosinophilen Granulozyten in der zweiten Woche p.i.. Danach sanken die Werte teilweise wieder ab und stiegen erst fünf bis sechs Wochen später wieder an. Weiterhin wird von einem Fehlen der Eosinophilie bei wenig abwehrkräftigen Tieren und von einem Absinken oder Verschwinden der Zellen in schweren Krankheitsphasen berichtet. Welche Ursache für eine fehlende Eosinophilie in den hier durchgeführten Versuchen zugrunde liegt, kann aufgrund der wenigen Zeitpunkte der Bestimmung des Blutbildes nicht ermittelt werden. Die übrigen Parameter des weißen Blutbildes zeigten keine Veränderungen, die konstant mit der Infektion in Zusammenhang gebracht werden könnten. Eine von PFEIFFER (1971) beobachtete Neutropenie konnte nur relativ und unregelmäßig beobachtet werden. 5.4.4 Ergebnisse der serologischen Untersuchungen Die Rolle der humoralen Immunität bei der Immunantwort auf eine Infektion mit D. viviparus wurde noch nicht vollständig geklärt. Während nachgewiesen werden konnte, dass eine passive Immunisierung mit dem Serum infizierter Rinder möglich Diskussion 165 ist (JARRETT et al. 1957), konnten KOOYMAN et al. (2002) keinen Zusammenhang zwischen Protektion und der Höhe an IgA, IgG1, IgG2 und IgM ermitteln. Die Autoren fanden jedoch eine Korrelation zwischen dem Gesamt-IgE-Titer und der Protektion bei Reinfektion. Mittels ELISA wurde die Bildung von Anti-PDI-Antikörpern der Klassen IgG, IgG1, IgG2, IgA und IgM untersucht. Eine Untersuchung auf IgE-Antikörper konnte nicht durchgeführt werden, da ein Rinder Anti-IgE-Konjugat nicht kommerziell erhältlich war. Alle Tiere zeigten zwischen dem vierten und elften Tag nach der ersten Immunisierung einen Titeranstieg bei allen untersuchten Antikörperklassen. Die Titer blieben während des gesamten Versuchs gleichbleibend hoch. Da es nicht gelang, reine PDI ohne GST und bakterielle Kontaminanten zu gewinnen, wurde im ELISA das Impfprotein eingesetzt. Es ist also nicht auszuschließen, dass ein Teil des Titeranstiegs auf einer Serokonversion auf GST oder bakterielle Kontaminanten beruhte. Dass der Titeranstieg nicht ausschließlich auf nicht-PDI-Proteine zurückzuführen war, wurde im Western-Blot überprüft. Anhand von D. viviparus Rohantigen zeigte sich, dass die Seren vakzinierter Rinder spezifisch eine Bande auf Höhe der PDI erkannten. Auf einen ELISA mit reinem GST wurde verzichtet, da ein Vergleich mit dem Impfprotein-ELISA und eine quantitative Aussage bezüglich des GST-Anteils am Titeranstieg nicht möglich gewesen wäre. Ein Vorhandensein GSTspezifischer Antikörper bei den Tieren der Kombigruppe wurde von VON HOLTUM (persönliche Mitteilung) nachgewiesen. Der Nachweis PDI-spezifischer Antikörper zeigte, dass die in E. coli rekombinant hergestellte PDI zumindest teilweise in ihrer Konformation dem nativen Protein entsprechen muss und antigene Determinanten vorhanden sind. Bezüglich der Höhe der Antikörperspiegel konnten keine Unterschiede zwischen den Gruppen, die Al(OH)3 als Adjuvans und denen, die Quil A erhalten hatten, festgestellt werden. Dies und der in allen Gruppen ähnlich frühe Anstieg der Antikörper sprechen dafür, dass beide Adjuvantien die Immunabwehr in gleichem Maße positiv beeinflusst haben. Ein Antikörperanstieg in den Kontrollgruppen als Reaktion auf das natürliche Antigen nach der Infektion war wesentlich geringer und nur bei IgG und IgG1 vorhanden. Bei IgG1 zeigten beide Kontrollgruppen, bei IgG die Kontrollgruppe des ersten Versuchs ab Tag 91 (28 dpi) 166 Diskussion eine Immunantwort. Bei der Kontrollgruppe des zweiten Versuchs waren bereits sieben Tage nach der Infektion IgG-Antikörper vorhanden. Die geringen Titer an PDIspezifischen Antikörpern in den Kontrollgruppe nach der experimentellen Infektion können unterschiedlich interpretiert werden. Möglich wäre, dass die native PDI doch weitere Epitope besitzt, die immunogen wirken und die hierauf gebildeten Antikörper nicht anhand des rekombinanten Proteins detektiert werden können. Dies hieße, dass ein prokaryotisches Expressionssystem für die PDI nicht geeignet ist. Eine andere Erklärung wäre, dass das Protein während der Infektion nicht zugänglich ist und damit dem Immunsystem nicht präsentiert werden kann. Der Anstieg der Antikörper in den Kontrollgruppen könnte hierbei auf das Freiwerden des Proteins aus toten Larven oder Adulten zurückgeführt werden. Hauptgrundlage des Einsatzes der PDI als Vakzinekandidat war die von verschiedenen Autoren bei anderen Helminthen nachgewiesene Immunogenität und extrazelluläre Lokalisation des Proteins. So konnte die PDI bei O. ostertagi in ES-Produkten von vierten Larven und Adulten detektiert werden. Das Protein wurde ferner vom Serum Ostertagia-infizierter Rinder erkannt (GELDHOF et al. 2003). Eine Lokalisation im ES-Material von Larven und Adulten wurde des Weiteren für Dirofilaria immitis beschrieben (CHANDRASHEKAR et al. 1998) und auch in ES-Produkten des Trematoden F. hepatica konnte die PDI nachgewiesen werden (SALAZAR-CALDERON et al. 2003). Da keine Möglichkeit zur Gewinnung von ES-Produkten bestand, erfolgte im Vorfeld keine Überprüfung, ob die PDI hierin vorhanden ist, sondern es wurde lediglich überprüft, ob das rekombinante Protein vom Serum D. viviparus-infizierter Rinder erkannt wird. Es kann also nicht ausgeschlossen werden, dass die PDI von D. viviparus nicht sezerniert wird. 5.4.5 Quantitative real-time PCR Mithilfe der QRT-PCR sollte untersucht werden, ob eine Immunantwort auf die PDI unter der Belastungsinfektion Einfluss auf die PDI-Transkription hatte. Dies wäre unter der Voraussetzung, dass Immunmechanismen zur Inaktivierung des Enzyms geführt hätten, denkbar gewesen. Möglicherweise hätte dies einen erhöhten „Verbrauch“ des Enzyms vorgetäuscht und zu einer gesteigerten Transkriptionsrate in den Würmern immunisierter Tiere geführt. Diskussion 167 Als Housekeeping Gen für die Normalisierung der PDI-Transkription wurde die 18SrRNA gewählt. Grundvoraussetzung für die Funktion als Referenzgen ist, dass ein Gen in konstanter Höhe exprimiert wird. Diese Eigenschaft wird ribosomaler RNA zugesprochen. Verschiedene andere Gene, die häufig als Housekeeping Gen eingesetzt werden, wie etwa Glycerinaldehydphosphat-Dehydrogenase (GAPDH) oder β-Aktin, können unter bestimmten Voraussetzungen unterschiedliche Transkriptionsraten aufweisen. 18S-rRNA wird daher allgemein als sicherer Faktor angesehen (BUSTIN 2000; BUSTIN 2002; THELLIN et al. 1999). Bei der cDNA-Synthese erfolgte die Reverse Transkription der PDI und der 18SrRNA im gleichen Reaktionsansatz (ZHU u. ALTMANN 2005). Hierdurch wurde die Varianz reduziert, da Gewebeeffekte wie unterschiedliche RNA- Extraktionseffizienzen und Fehler bei der RT in einer Probe gleichermaßen das Zielgen und das HKG betreffen. Bei der Durchführung der QRT-PCR zeigte sich anhand der Plasmid- und cDNA-Verdünnungsreihen eine mangelhafte PCREffizienz, die bei der Amplifikation der 18S-rRNA besonders ausgeprägt war. Die Effizienz der PCR hat entscheidenden Einfluss auf das Ergebnis einer Quantifizierung. Aufgrund des exponentiellen Charakters der Reaktion haben geringste Unterschiede in der PCR-Effizienz extreme Auswirkungen auf die Kopienzahl. Zielgen und Referenzgen sollten daher die gleiche PCR-Effizienz aufweisen, oder es sollten Effizienz-korrigierte Modelle angewendet werden (PFAFFL 2004). In der hier durchgeführten Arbeit wurde versucht, eine relative Quantifizierung mithilfe der Effizienz-korrigierten Formel nach PFAFFL (2001) durchzuführen. Es gelang jedoch nicht, verwertbare Ergebnisse zu erzielen. Die Ursache hierfür wurde bei der Betrachtung der Cts des Kalibrators deutlich. Theoretisch hätte der Kalibrator in allen Läufen gleiche oder zumindest sehr ähnliche Cts aufweisen müssen. Während die Cts der PDI noch in einem vergleichsweise engen Rahmen von zwei Cts schwankten, waren bei den Cts der 18S-rRNA Unterschiede von bis zu zehn Cts vorhanden. Hierdurch war keine konstante Vergleichsgröße gegeben und es konnten keine auswertbaren Ergebnisse erzielt werden. 168 Diskussion Eine mangelhafte PCR-Effizienz kann diverse Gründe haben. Hierzu gehören u. a. die Länge des Amplifikats, Spezifität der Primer, GC-Gehalt des Templates, Sekundärstrukturen, Qualität der Sonde und PCR-Inhibitoren (BUSTIN 2004). Da die Konstruktion der Primer und Sonden mit dem Primer Express Programm erfolgte, erscheint ein suboptimales Primer/Sonden-Design als Ursache für die unzureichende PCR-Effizienz eher unwahrscheinlich. Weiterhin wurde die Spezifität der PCR durch Darstellung der PCR-Produkte im Agarosegel überprüft. Da sowohl für die PDI- als auch für die 18S-rRNA-Amplifikationen der gleiche cDNA-Pool und Mastermix verwendet wurde, scheint auch ein Vorhandensein von PCR-Inhibitoren nicht Hauptursache zu sein. Am problematischsten erscheint bei der Versuchsdurchführung die Wahl der Primer für die Reverse Transkription. Da 18SrRNA nicht über eine Poly-A-Sequenz am 3’-Ende verfügt, war es notwendig, einen genspezifischen Primer einzusetzten. Hierbei wurde der gleiche Rückwärtsprimer verwendet wie für die Amplifikation in der real-time PCR. Das heißt, dass die zu amplifizierende Sequenz sich am äußersten 3’-Ende der cDNA befand. Dies stellt eine ungünstige Position für eine korrekte und effektive Primerbindung dar und könnte die Ursache für die mangelhafte PCR-Effizienz der 18S-rRNA-Amlifikation sein. 5.5 Zusammenfassende Beurteilung und Ausblick Im Rahmen dieser Arbeit konnten die cDNA-Sequenzen zweier PDI Gene von D. viviparus identifiziert werden. Für das als PDI2 bezeichnete Gen erfolgte ferner die Bestimmung der genomischen Sequenz sowie eine Expression als GSTFusionsprotein in E. coli. Sequenzidentitätsvergleiche und eine MALDI-TOF-MSAnalyse des exprimierten Proteins konnten die Identität der PDI bestätigen. Die hohen Ähnlichkeiten der hier identifizierten PDI2 zur PDI2 von C. elegans lassen für dieses Enzym bei D. viviparus eine ähnlich essentielle Bedeutung bei der Synthese der Kutikula vermuten. Im Western-Blot konnte die rekombinante PDI mit dem Serum D. viviparus infizierter Rinder detektiert werden, wodurch die Immunogenität des Proteins bestätigt wurde. In darauffolgenden Immunisierungsversuchen an Rindern zeigte das GST- Diskussion 169 Fusionsprotein kein protektives Potential. Obwohl bei den immunisierten Tieren PDIspezifische Antikörper detektiert werden konnten, waren diese nicht in der Lage, einen Schutz gegen eine experimentelle Infektion zu vermitteln. Es konnte weder eine statistisch signifikante verminderte Larvenausscheidung noch eine Reduktion der Wurmbürde im Vergleich zur Kontrollgruppe beobachtet werden. Eine konstant zu beobachtende Reduktion der Wurmlänge bei den immunisierten Tieren erreichte aufgrund großer Schwankungen zwischen den Versuchstieren keine statistische Signifikanz. Die geringe serologische Reaktion der Kontrolltiere auf das GSTFusionsprotein nach der experimentellen Infektion ließen die extrazelluläre Lokalisation des Proteins bei D. viviparus fraglich erscheinen. Möglicherweise war die Detektion der PDI jedoch auch aufgrund fehlender Epitope in Folge der prokaryotischen Expression eingeschränkt. Bevor die PDI als Vakzinekandidat aufgegeben wird, sollten diese beiden Punkte überprüft werden. Sofern die PDI sich in ES-Material von D. viviparus nachweisen lässt und das native Protein eine höhere Immunogenität als die in E. coli exprimierte PDI besitzt, könnten Immunisierungen mit in eukaryotischen Expressionssystemen hergestellter PDI versucht werden. Weiterhin wäre es aufschlussreich, die Lokalisation der PDI im Wurm, beispielsweise mittels Immunhistochemie, zu bestimmen. 170 Zusammenfassung 6 Zusammenfassung Sonja Wolken (2006): Charakterisierung und in vitro Expression einer Protein Disulfid Isomerase (PDI) zur Immunisierung gegen Dictyocaulus viviparus Die Dictyocaulose ist eine der wirtschaftlich bedeutsamsten parasitären Erkrankungen der Weiderinder. Die im natürlichen Infektionsgeschehen vorhandene Ausbildung einer protektiven Immunität und Erfolge mit einer Larvenvakzine lassen die Entwicklung eines rekombinanten Impfstoffs möglich erscheinen. Als Vakzinekandidat wurde das Enzym Protein Disulfid Isomerase gewählt, dessen immunogene Eigenschaften bereits bei anderen Helminthen beschrieben worden waren. Mittels degenerierter Primer konnte ein zentraler Abschnitt der PDI-Sequenz bei Dictyocaulus viviparus identifiziert werden, welcher durch 5’- und 3’-Rapid Amplifikation of cDNA Ends (RACE) vervollständigt wurde. Die hieraus resultierende Sequenz weist einen offenen Leserahmen über 1482 bp auf und zeigt eine 80%ige Identität zur PDI von Ancylostoma caninum und Teladorsagia circumcincta sowie zur PDI2 von Ostertagia ostertagi und eine 72%ige Identität zur PDI2 von Caenorhabditis elegans. Diese Identitäten resultierten in der Beschreibung der identifizierten PDI als PDI2 von D. viviparus. Die Aminosäurensequenz weist zwei Thioredoxin-Domänen auf, wobei es sich um hochkonservierte Bereiche handelt, in denen die katalytisch aktiven Zentren des Enzyms lokalisiert sind. Die genomische Sequenz der PDI2 hat eine Größe von 2649 bp und besteht aus fünf Exons und vier Introns. Die Positionen der Introns im Gen stellen sich im Vergleich mit der A. caninum-Sequenz als vollständig konserviert dar. Die degenerierten Primer identifizierten eine zweite PDI bei D. viviparus mit höchsten Identitäten zur PDI1 von O. ostertagi und zur PDI3 von C. elegans. Die PDI2 wurde als GST-Fusionsprotein in E. coli exprimiert und mittels AffinitätsChromatographie aufgereinigt. Die Immunogenität des rekombinanten Proteins konnte im Western-Blot anhand von Serum experimentell mit D. viviparus infizierter Zusammenfassung 171 Rinder nachgewiesen werden. In zwei verschiedenen Immunisierungsversuchen erfolgte an insgesamt 22 Rindern die Überprüfung des protektiven Potentials des GST-PDI-Fusionsproteins in Kombination mit den Adjuvantien Al(OH)3 und Quil A. Trotz des Vorhandenseins PDI-spezifischer Antikörper bei allen immunisierten Tieren konnte durch die Impfung kein Schutz gegen eine experimentelle Infektion vermittelt werden. Weder in der Larvenausscheidung noch in Bezug auf die Wurmbürde konnte eine statistisch signifikante Reduktion festgestellt werden. Tendenziell zeigten Tiere, die das Adjuvans Quil A erhalten hatten, eine geringere Larvenausscheidung, wobei unklar ist, ob es sich hierbei um eine spezifische Immunreaktion handelte. Weiterhin war bei allen immunisierten Tieren eine Reduktion der Wurmgröße zwischen 7,2 und 18% im Vergleich zur Kontrollgruppe zu beobachten, die jedoch ebenfalls keine statistische Signifikanz erreichte. 172 Summary 7 Summary Sonja Wolken (2006): Characterisation and in vitro expression of a protein disulfide isomerase (PDI) for the immunisation against Dictyocaulus viviparus Dictyocaulosis is one of the most important parasitic diseases in grazing cattle with high economic impact. Since natural infections stimulate a protective immunity and good results were optained with an attenuated live vaccine, development of a recombinant vaccine seems to be feasible. Protein disulfide isomerase, an enzyme whose immunogenicity has been discribed in other helminths, was choosen as a vaccine candidate. Using degenerate primers, a central part of the PDI sequence in Dictyocaulus viviparus was identified. To complete the sequence, 5’- and 3’-rapid amplification of cDNA ends (RACE) was used. The full length sequence contains an open reading frame of 1482 bp and presents 80% identity to PDI of Ancylostoma caninum and Teladorsagia circumcincta as well as to PDI2 of Ostertagia ostertagi and 72% identity to PDI2 of Caenorhabditis elegans. Therefore the identified PDI was described as PDI2 of D. viviparus. The amino acid sequence shows two thioredoxin domains. These highly conserved regions contain the active sites for the enzyme activity. The genomic sequence of the PDI2 has a length of 2649 bp and consists of five exons and four introns, the introns being located at exactly the same positions as in the A. caninum sequence. This demonstrates the conserved organization of this gene. When using degenerate primers, a second PDI sequence of D. viviparus was identified. This PDI shows highest identities to PDI1 of O. ostertagi and to PDI3 of C. elegans. The PDI2 was expressed as GST fusion protein in E. coli and purified via affinity chromatography. The immunogenicity of the recombinant protein was demonstrated in western-blots using sera of cattle experimentally infected with D. viviparus. In two Summary 173 immunization trials the protective potential of the fusion protein in combination with the adjuvants Al(OH)3 and Quil A was tested in a total of 22 cattle. Despite the existence of PDI specific antibodies in all vaccinated cattle, the recombinant protein failed to induce protection against challenge. Neither in larvae counts nor in worm burdens a statistical significance in reduction was seen. Calves that had received Quil A showed a tendency to have lower total larvae counts, but it was not clear whether this effect was due to a specific immunresponse. Furthermore all immunized calves harboured worms that were 7,2% to 18% shorter than those of the control group. But again this was not statistical significant. 174 Literaturverzeichnis 8 Literaturverzeichnis ANDRES, D. A., I. M. DICKERSON u. J. E. DIXON (1990): Variants of the carboxyl-terminal KDEL sequence direct intracellular retention J. Biol. Chem. 265, 5952-5955 ANNUNEN, P., T. HELAAKOSKI, J. MYLLYHARJU, J. VEIJOLA, T. PIHLAJANIEMI u. K. I. KIVIRIKKO (1997): Cloning of the human prolyl 4-hydroxylase alpha subunit isoform alpha(II) and characterization of the type II enzyme tetramer. The alpha(I) and alpha(II) subunits do not form a mixed alpha(I)alpha(II)beta2 tetramer J. Biol. Chem. 272, 17342-17348 BAERMANN, G. (1917): Eine einfache Methode zur Auffindung von Ankylostomum-(Nematoden)-Larven in Erdproben Tijdschr. Diergeneesk. 57, 131-137 BELLMER, A. (1990): Seroepidemiologische Untersuchungen über das Vorkommen von DictyocaulusInfektionen bei Rindern in Niedersachsen mit Hilfe des ELISA Hannover, Tierärztl. Hochsch., Diss. BIENIOSCHEK, S. (1994): Experimentelle Wechselinfektionen zwischen Zerviden und Hauswiederkäuern mit großen Lungenwürmern (Dictyocaulus spp.) Leipzig, Univ., Veterinärmed. Fak., Diss. BOHRING, C., E. KRAUSE, B. HABERMANN u. W. KRAUSE (2001): Isolation and identification of sperm membrane antigens recognized by antisperm antibodies, and their possible role in immunological infertility disease Mol. Hum. Reprod. 7, 113-118 BOKHOUT, B. A., J. H. BOON u. J. HENDRIKS (1979): Operational diagnostics of lungworm infections in cattle. Preliminary investigation into the usefulness of the indirect haemagglutination Tijdschr. Diergeneesk. 104, suppl-203 BOON, J. H., A. KLOOSTERMAN u. R. VAN DEN BRINK (1982): The incidence of Dictyocaulus viviparus infections in cattle in the Netherlands. I. The Enzyme Linked Immunosorbent Assay as a diagnostic tool Vet. Q. 4, 155-160 Literaturverzeichnis 175 BOS, H. J., BEEKMAN J, J. R. BATTISON u. M. H. DRESDEN (1989): Isolation of a cysteinyl proteinase from adult Dictyocaulus viviparus and its potential as a vaccine Proc.13. Conf. WAAVP, Berlin, P 2-25 BOS, H. J. u. J. BEEKMAN (1985): Serodiagnosis Of lungworm Infection In calves using ELISA Develop. biol. Standard 62, 45-52 BRADFORD, M. (1976): A rapid and sensitive method for the quantification of microgram quantities of proteins utilizing the principle of protein-dye binding Anal. Biochem. 72, 248-252 BRITTON, C., G. J. CANTO, G. M. URQUHART u. M. W. KENNEDY (1992a): The antibody repertoire in infection and vaccination with Dictyocaulus viviparus: heterogeneity in infected cattle and genetic control in guinea pigs Vet. Immunol. Immunopathol. 31, 313-322 BRITTON, C., G. J. CANTO, G. M. URQUHART u. M. W. KENNEDY (1993): Stage-specific surface antigens of the cattle lungworm Dictyocaulus viviparus Parasite Immunol. 15, 625-634 BRITTON, C., D. P. KNOX, G. J. CANTO, G. M. URQUHART u. M. W. KENNEDY (1992b): The secreted and somatic proteinases of the bovine lungworm Dictyocaulus viviparus and their inhibition by antibody from infected and vaccinated animals Parasitology 105 (Pt 2), 325-333 BRITTON, C., D. P. KNOX u. M. W. KENNEDY (1994): Superoxide dismutase (SOD) activity of Dictyocaulus viviparus and its inhibition by antibody from infected and vaccinated bovine hosts Parasitology 109 (Pt 2), 257-263 BRITTON, C., J. MOORE, J. S. GILLEARD u. M. W. KENNEDY (1995): Extensive diversity in repeat unit sequences of the cDNA encoding the polyprotein antigen/allergen from the bovine lungworm Dictyocaulus viviparus Mol. Biochem. Parasitol. 72, 77-88 BRITTON, C. u. L. MURRAY (2002): A cathepsin L protease essential for Caenorhabditis elegans embryogenesis is functionally conserved in parasitic nematodes Mol. Biochem. Parasitol. 122, 21-33 BUCHWALDER, R. (1963): Untersuchungen zur Technik des Lungenwurmnachweises bei Rind und Schaf Monatsh. Veterinärmed. 18, 457-460 176 Literaturverzeichnis BURGESS, J. K., K. A. HOTCHKISS, C. SUTER, N. P. DUDMAN, J. SZOLLOSI, C. N. CHESTERMAN, B. H. CHONG u. P. J. HOGG (2000): Physical proximity and functional association of glycoprotein 1b alpha and protein disulfide isomerase on the platelet plasma membrane J. Biol. Chem. 275, 9758-9766 BUSTIN, S. A. (2000): Absolute quantification of mRNA using real-time reverse transcription polymerase chain reaction assays J. Mol. Endocrinol. 25, 169-193 BUSTIN, S. A. (2002): Quantification of mRNA using real-time reverse transcription PCR (RT-PCR): trends and problems J. Mol. Endocrinol. 29, 23-39 BUSTIN, S. A. (2004): A-Z of quantitative PCR International University Line, La Jolla, Kalifornien, USA CAI, H., C. C. WANG u. C. L. TSOU (1994): Chaperone-like activity of protein disulfide isomerase in the refolding of a protein with no disulfide bonds J. Biol. Chem. 269, 24550-24552 CHANDRASHEKAR, R., E. DEVARAJAN u. K. MEHTA (2002): Dirofilaria immitis: further characterisation of the transglutaminase enzyme and its role in larval molting Parasitol. Res. 88, 185-191 CHANDRASHEKAR, R., N. TSUJI, T. MORALES, V. OZOLS u. K. MEHTA (1998): An ERp60-like protein from the filarial parasite Dirofilaria immitis has both transglutaminase and protein disulfide isomerase activity Proc. Natl. Acad. Sci. U.S.A. 95, 531-536 CHEN, K., Y. LIN u. T. C. DETWILER (1992): Protein disulfide isomerase activity is released by activated platelets Blood 79, 2226-2228 CHENG, S. Y., S. HASUMURA, M. C. WILLINGHAM u. I. PASTAN (1986): Purification and characterization of a membrane-associated 3,3',5-triiodo-L-thyronine binding protein from a human carcinoma cell line Proc. Natl. Acad. Sci. U.S.A. 83, 947-951 Literaturverzeichnis 177 CHOMCYNSKI, P. u. N. SACCHI (1987): Single step method of RNA isolation by acid guanidinium thiocyanate-phenolchloroform extraction Anal. Biochem. 162, 156-159 COLLIE, D. D., C. N. MACALDOWIE, A. D. PEMBERTON, C. J. WOODALL, N. MCLEAN, C. HODGSON, M. W. KENNEDY u. H. R. MILLER (2001): Local lung responses following local lung challenge with recombinant lungworm antigen in systemically sensitized sheep Clin.Exp.Allergy 31, 1636-1647 CONN, K. J., W. GAO, A. MCKEE, M. S. LAN, M. D. ULLMAN, P. B. EISENHAUER, R. E. FINE u. J. M. WELLS (2004): Identification of the protein disulfide isomerase family member PDIp in experimental Parkinson's disease and Lewy body pathology Brain Res. 1022, 164-172 CONNAN, R. M. (1993): Calfhood vaccination for dictyocaulosis Vet. Rec. 133, 554-555 CORNELISSEN, J. B., F. H. BORGSTEEDE u. F. J. VAN MILLIGEN (1997): Evaluation of an ELISA for the routine diagnosis of Dictyocaulus viviparus infections in cattle Vet. Parasitol. 70, 153-164 CORNWELL, R. L. (1960): The complement fixing antibody response of calves to Dictyocaulus viviparus III. Vaccinated calves exposed to challenge J. Comp Pathol. 70, 499-513 COX, G. N. (1992): Molecular and biochemical aspects of nematode collagens J. Parasitol. 78, 1-15 CUNNEA, P. M., A. MIRANDA-VIZUETE, G. BERTOLI, T. SIMMEN, A. E. DAMDIMOPOULOS, S. HERMANN, S. LEINONEN, M. P. HUIKKO, J. A. GUSTAFSSON, R. SITIA u. G. SPYROU (2003): ERdj5, an endoplasmic reticulum (ER)-resident protein containing DnaJ and thioredoxin domains, is expressed in secretory cells or following ER stress J. Biol. Chem. 278, 1059-1066 DAI, Y. u. C. WANG (1997): A mutant truncated protein disulfide isomerase with no chaperone activity J. Biol. Chem. 272, 27572-27576 178 Literaturverzeichnis DARBY, N. J., E. PENKA u. R. VINCENTELLI (1998): The multi-domain structure of protein disulfide isomerase is essential for high catalytic efficiency J. Mol. Biol. 276, 239-247 DAVID, G. P. (1993): Increased prevalence of husk Vet. Rec. 133, 627 DE LEEUW, W. A. u. J. B. CORNELISSEN (1991): Identification and isolation of a specific antigen with diagnostic potential from Dictyocaulus viviparus Vet. Parasitol. 39, 137-147 DE LEEUW, W. A. u. J. B. CORNELISSEN (1993): Comparison of three enzyme immunoassays for diagnosis of Dictyocaulus viviparus infection Vet. Parasitol. 49, 229-241 DESILVA, M. G., J. LU, G. DONADEL, W. S. MODI, H. XIE, A. L. NOTKINS u. M. S. LAN (1996): Characterization and chromosomal localization of a new protein disulfide isomerase, PDIp, highly expressed in human pancreas DNA Cell Biol. 15, 9-16 DIVINA, B. P. u. J. HÖGLUND (2002): Heterologous transmission with Dictyocaulus capreolus from roe deer (Capreolus capreolus) to cattle (Bos taurus) J. Helminthol. 76, 125-131 DIVINA, B. P., E. WILHELMSSON, T. MORNER, J. G. MATTSSON u. J. HÖGLUND (2002): Molecular identification and prevalence of Dictyocaulus spp. (Trichostrongyloidea: Dictyocaulidae) in Swedish semi-domestic and free-living cervids J Wildl. Dis. 38, 769-775 DONCASTER, C. C. (1981): Observations on relationships between infective juveniles of bovine lungworm, Dictyocaulus viviparus (Nematoda: Strongylida) and the fungi, Pilobolus kleinii and P. crystallinus (Zygomycotina: Zygomycetes) Parasitology 82, 421-428 DORNER, A. J., L. C. WASLEY, P. RANEY, S. HAUGEJORDEN, M. GREEN u. R. J. KAUFMAN (1990): The stress response in Chinese hamster ovary cells. Regulation of ERp72 and protein disulfide isomerase expression and secretion J. Biol. Chem. 265, 22029-22034 Literaturverzeichnis 179 DÜWEL, D. (1963): Die künstliche Immunisierung beim Lungenwurmbefall des Rindes Monatsh. Tierheilk. 15, 334-345 DÜWEL, D. (1971): Die Dictyocaulose des Rindes Tierärztl. Umsch. 26, 152-158 ECKERT, J. (1972): Bekämpfung der Dictyocaulose und der Trichostrongylidosen des Rindes Schweiz. Arch. Tierheilk. 114, 652-667 EDDI, C., I. BIANCHIN, M. R. HONER, R. A. MUNIZ, J. CARACOSTANTOGOLO u. Y. A. DO NASCIMENTO (1993): Efficacy of doramectin against field nematode infections of cattle in Latin America Vet. Parasitol. 49, 39-44 EDMAN, J. C., L. ELLIS, R. W. BLACHER, R. A. ROTH u. W. J. RUTTER (1985): Sequence of protein disulphide isomerase and implications of its relationship to thioredoxin Nature 317, 267-270 ELLGAARD, L. u. L. W. RUDDOCK (2005): The human protein disulphide isomerase family: substrate interactions and functional properties EMBO Rep. 6, 28-32 ENIGK, K. u. J. HILDEBRANDT (1969): Zur Empfänglichkeit der Wiederkäuer für Dictyocaulus viviparus und Dictycaulus filaria (Strongyloidea, Nematoda) Zentralbl. Veterinärmed. B 16, 67-77 ENIGK, K. u. D. DÜWEL (1962): Beitrag zur Epizootiologie der Dictyocaulose des Rindes Dtsch. tierärztl. Wochenschrift 69, 72-78 EPE, C., S. BIENIOSCHEK, S. REHBEIN u. T. SCHNIEDER (1995): Comparative RAPD-PCR analysis of lungworms (Dictyocaulidae) from fallow deer, cattle, sheep, and horses Zentralbl. Veterinärmed. B 42, 187-191 EPE, C., C. KOHLMETZ u. T. SCHNIEDER (1998): A recombinant protein disulfide isomerase homologue from Ancylostoma caninum Parasitol. Res. 84, 763-766 180 Literaturverzeichnis EPE, C., G. V. SAMSON-HIMMELSTJERNA u. T. SCHNIEDER (1997): Differences in a ribosomal DNA sequence of lungworm species (Nematoda:Dictyocaulidae) from fallow deer, cattle, sheep and donkeys Res. Vet. Sci. 62, 17-21 EPE, C., S. WOIDTKE, M. PAPE, M. HEISE, F. KRAEMER, C. KOHLMETZ u. T. SCHNIEDER (1999): Strategic control of gastrointestinal nematode and lungworm infections with eprinomectin at turnout and eight weeks later Vet. Rec. 144, 380-382 ESCHENLAUER, S. C. u. A. P. PAGE (2003): The Caenorhabditis elegans ERp60 homolog protein disulfide isomerase-3 has disulfide isomerase and transglutaminase-like cross-linking activity and is involved in the maintenance of body morphology J. Biol. Chem. 278, 4227-4237 EYSKER, M. (1997): The sensitivity of the Baermann method for the diagnosis of primary Dictyocaulus viviparus infections in calves Vet. Parasitol. 69, 89-93 EYSKER, M., E. W. CLAESSENS, T. J. LAM, M. J. MOONS u. A. PIJPERS (1994): The prevalence of patent lungworm infections in herds of dairy cows in The Netherlands Vet. Parasitol. 53, 263-267 EYSKER, M., F. N. KOOYMAN u. H. W. PLOEGER (2001): Immunity in calves against Dictyocaulus viviparus following a low primary infection Parasitology 123, 591-597 FENOUILLET, E., R. BARBOUCHE, J. COURAGEOT u. R. MIQUELIS (2001): The catalytic activity of protein disulfide isomerase is involved in human immunodeficiency virus envelope-mediated membrane fusion after CD4 cell binding J. Infect. Dis. 183, 744-752 FERRARI, D. M. u. H. D. SOLING (1999): The protein disulphide-isomerase family: unravelling a string of folds Biochem. J. 339 (Pt 1), 1-10 FINKEN, M., A. SOBEK, P. SYMMONS u. W. KUNZ (1994): Characterization of the complete protein disulfide isomerase gene of Schistosoma mansoni and identification of the tissues of its expression Mol. Biochem. Parasitol. 64, 135-144 Literaturverzeichnis 181 FREEDMAN, R. B. (1989): Protein disulfide isomerase: multiple roles in the modification of nascent secretory proteins Cell 57, 1069-1072 FREEDMAN, R. B., T. R. HIRST u. M. F. TUITE (1994): Protein disulphide isomerase: building bridges in protein folding Trends Biochem. Sci. 19, 331-336 GANHEIM, C., J. HÖGLUND u. K. P. WALLER (2004): Acute phase proteins in response to Dictyocaulus viviparus infection in calves Acta vet. scand. 45, 79-86 GELDHOF, P. (2003): Vaccination of calves against the abomasal nematode Ostertagia ostertagi with parasite excretory-secretory antigens Gent, Univ., Fac. Vet. Med., Diss. GIBB, M. J., C. A. HUCKLE u. A. B. FORBES (2005): Effects of sequential treatments with eprinomectin on performance and grazing behaviour in dairy cattle under daily paddock stocking management Vet. Parasitol. 133, 79-90 GIBBONS, L. M. u. J. HÖGLUND (2002): Dictyocaulus capreolus n. sp. (Nematoda: Trichostrongyloidea) from roe deer, Capreolus capreolus and moose, Alces alces in Sweden J. Helminthol. 76, 1-7 GIBBONS, L. M. u. L. F. KHALIL (1988): A revision of the species Dictyocaulus Raillet & Henry, 1907 (Nematoda: Trichostrongyloidea) with the description of D. africanus n. sp. from African artiodactylids J. Afr. Zool. 102, 151-175 GILLEARD, J. S., J. L. DUNCAN u. A. TAIT (1995a): An immunodominant antigen on the Dictyocaulus viviparus L3 sheath surface coat and a related molecule in other strongylid nematodes Parasitology 111 ( Pt 2), 193-200 GILLEARD, J. S., J. L. DUNCAN u. A. TAIT (1995b): Dictyocaulus viviparus: surface antigens of the L3 cuticle and sheath Exp. Parasitol. 80, 441-453 GOLDBERGER, R. F., C. J. EPSTEIN u. C. B. ANFINSEN (1963): Acceleration of reactivation of reduced bovine pancreatic ribonuclease by a microsomal system from rat liver J. Biol. Chem. 238, 628-635 182 Literaturverzeichnis GOTHE, R. (1983): Zur Dictyocaulus arnfieldi Infektion der Equiden Berl. Münch. tierärztl. Wochenschr. 96, 364-368 GOTHE, R. u. R. KANDELS (1984): Lungenwürmer bei Schafen in Hessen: Untersuchungen zur Befallshäufigkeit von Dictyocaulus filaria und Protostrongyliden sowie zur Saisondynamik der Larvenausscheidung bei unterschiedlichen Haltungsformen Berl. Münch. tierärztl. Wochenschr. 97, 359-366 GOUDIE, A. C., N. A. EVANS, K. A. GRATION, B. F. BISHOP, S. P. GIBSON, K. S. HOLDOM, B. KAYE, S. R. WICKS, D. LEWIS, A. J. WEATHERLEY, C. I. BRUCE, A. HERBERT u. D. J. SEYMOUR (1993): Doramectin - a potent novel endectocide Vet. Parasitol. 49, 5-15 HAGBERG, M., E. WATTRANG, R. NISKANEN, M. TRAVEN, J. HÖGLUND u. A. LUNDEN (2005): Mononuclear cell subsets in bronchoalveolar lavage fluid during Dictyocaulus viviparus infection of calves: a potential role for gamma/delta TCR-expressing cells in airway immune responses? Parasite Immunol. 27, 151-161 HARRISON, G. B., D. D. HEATH, R. P. DEMPSTER, C. GAUCI, S. E. NEWTON, W. G. CAMERON, C. M. ROBINSON, S. B. LAWRENCE, M. W. LIGHTOWLERS u. M. D. RICKARD (1996): Identification and cDNA cloning of two novel low molecular weight host-protective antigens from Taenia ovis oncospheres Int. J. Parasitol. 26, 195-204 HASHMI, S., C. BRITTON, J. LIU, D. B. GUILIANO, Y. OKSOV u. S. LUSTIGMAN (2002): Cathepsin L is essential for embryogenesis and development of Caenorhabditis elegans J. Biol. Chem. 277, 3477-3486 HASLAM, S. M., G. C. COLES, H. R. MORRIS u. A. DELL (2000): Structural characterisation of the N-glycans of Dictyocaulus viviparus: Discovery of the Lewisx structure in a nematode Glycobiology 10, 223-229 HASSLINGER, M. A. (1989): Dictyocaulus arnfieldi in equines, present situation and future aspects J. Egypt. Vet. Med. Assoc. 49, 445-455 Literaturverzeichnis 183 HAUGEJORDEN, S. M., M. SRINIVASAN u. M. GREEN (1991): Analysis of the retention signals of two resident luminal endoplasmic reticulum proteins by in vitro mutagenesis J. Biol. Chem. 266, 6015-6018 HAWKINS, H. C. u. R. B. FREEDMAN (1991): The reactivities and ionization properties of the active-site dithiol groups of mammalian protein disulphide-isomerase Biochem. J. 275 ( Pt 2), 335-339 HAYANO, T. u. M. KIKUCHI (1995): Molecular cloning of the cDNA encoding a novel protein disulfide isomerase-related protein (PDIR) FEBS Lett. 372, 210-214 HELAAKOSKI, T., P. ANNUNEN, K. VUORI, I. A. MACNEIL, T. PIHLAJANIEMI u. K. I. KIVIRIKKO (1995): Cloning, baculovirus expression, and characterization of a second mouse prolyl 4hydroxylase alpha-subunit isoform: formation of an alpha 2 beta 2 tetramer with the protein disulfide-isomerase/beta subunit Proc. Natl. Acad. Sci. U.S.A. 92, 4427-4431 HENRIKSEN, S. A., M. LARSEN, J. GRONVOLD, P. NANSEN u. J. WOLSTRUP (1997): Nematode-trapping fungi in biological control of Dictyocaulus viviparus Acta vet. scand. 38, 175-179 HIRANO, N., F. SHIBASAKI, R. SAKAI, T. TANAKA, J. NISHIDA, Y. YAZAKI, T. TAKENAWA u. H. HIRAI (1995): Molecular cloning of the human glucose-regulated protein ERp57/GRP58, a thioldependent reductase. Identification of its secretory form and inducible expression by the oncogenic transformation Eur. J. Biochem. 234, 336-342 HÖGLUND, J., A. ENGSTROM, D. A. MORRISON u. J. G. MATTSSON (2004a): Genetic diversity assessed by amplified fragment length polymorphism analysis of the parasitic nematode Dictyocaulus viviparus the lungworm of cattle Int. J. Parasitol. 34, 475-484 HÖGLUND, J., D. A. MORRISON, B. P. DIVINA, E. WILHELMSSON u. J. G. MATTSSON (2003): Phylogeny of Dictyocaulus (lungworms) from eight species of ruminants based on analyses of ribosomal RNA data Parasitology 127, 179-187 184 Literaturverzeichnis HÖGLUND, J., D. A. MORRISON, J. G. MATTSSON u. A. ENGSTROM (2006): Population genetics of the bovine/cattle lungworm (Dictyocaulus viviparus) based on mtDNA and AFLP marker techniques Parasitology 133, 89-99 HÖGLUND, J., C. SVENSSON u. A. HESSLE (2001): A field survey on the status of internal parasites in calves on organic dairy farms in southwestern Sweden Vet.Parasitol. 99, 113-128 HÖGLUND, J., S. VIRING u. M. TORNQVIST (2004b): Seroprevalence of Dictyocaulus viviparus in first grazing season calves in Sweden Vet. Parasitol. 125, 343-352 HÖGLUND, J., E. WILHELMSSON, D. CHRISTENSSON, T. MÖRNER, P. WALLER u. J. G. MATTSSON (1999): ITS2 sequences of Dictyocaulus species from cattle, roe deer and moose in Sweden: molecular evidence for a new species Int. J. Parasitol. 29, 607-611 HOLZHAUER, M., W. G. HOLLAND u. H. W. PLOEGER (2005): Preventive vaccination of lactating and pregnant heifers against lungworm: safety and protection in three dairy herds Tijdschr. Diergeneesk. 130, 74-77 HOTCHKISS, K. A., L. J. MATTHIAS u. P. J. HOGG (1998): Exposure of the cryptic Arg-Gly-Asp sequence in thrombospondin-1 by protein disulfide isomerase Biochim. biophys. Acta 1388, 478-488 HU, M., J. HÖGLUND, N. B. CHILTON, X. ZHU u. R. B. GASSER (2002): Mutation scanning analysis of mitochondrial cytochrome c oxidase subunit 1 reveals limited gene flow among bovine lungworm subpopulations in Sweden Electrophoresis 23, 3357-3363 HUBERT, J., D. KERBOEUF, B. CARDINAUD, F. BLOND-RIOU u. R. FOURNIER (1997): Persistent efficacy of topical moxidectin against Dictyocaulus viviparus and Ostertagia ostertagi Vet. Parasitol. 68, 187-190 JARRETT, W. F. H., W. F. M. MCINTYRE, F. W. JENNINGS u. W. MULLIGAN (1957): The natural history of parasitic bronchitis with notes on prophylaxis and treatment Vet. Rec. 69, 1329-1340 Literaturverzeichnis 185 JARRETT, W. F. H., F. W. JENNINGS, W. I. M. MCINTYRE, W. MULLIGAN, B. A. C. THOMAS u. G. M. URQUHART (1959): Immunological studies on Dictyocaulus viviparus infection. The immunity resulting from experimental infection Immunol. 2, 252-267 JARRETT, W. F. H., W. I. M. MCINTYRE u. G. M. URQUHART (1955): Some factors in the epidemiology of parasitic bronchitis in cattle Vet. Rec. 67, 820-824 JOHNSON, D. R., J. SALES u. J. B. MATTHEWS (2005): Local cytokine responses in Dictyocaulus viviparus infection Vet. Parasitol. 128, 309-318 JOHNSON, M., R. E. LABES, M. J. TAYLOR u. C. G. MACKINTOSH (2003): Efficacy trial of an irradiated cattle lungworm vaccine in red deer (Cervus elaphus) Vet. Parasitol. 117, 131-137 JOHNSON, S., M. MICHALAK, M. OPAS u. P. EGGLETON (2001): The ins and outs of calreticulin: from the ER lumen to the extracellular space Trends Cell Biol. 11, 122-129 JOHNSTONE, C. (1998): Parasites and parasitic diseases of domestic animals [Internet: URL: http://cal.vet.upenn.edu/merial/Trichos/trich_10.htm] JØRGENSEN, R. J. (1973): In vitro effect of bile on the motility of Dictyocaulus viviparus third stage larvae Acta vet. scand. 14, 341-343 JØRGENSEN, R. J. (1980): Epidemiology of bovine dictyocaulosis in Denmark Vet. Parasitol. 7, 153-167 JØRGENSEN, R. J., H. RONNE, C. HELSTED u. A. R. ISKANDER (1982): Spread of infective Dictyocaulus viviparus larvae in pasture and to grazing cattle: experimental evidence of the role of Pilobolus fungi Vet. Parasitol. 10, 331-339 KANAI, S., H. TOH, T. HAYANO u. M. KIKUCHI (1998): Molecular evolution of the domain structures of protein disulfide isomerases J. Mol. Evol. 47, 200-210 KEMMINK, J., N. J. DARBY, K. DIJKSTRA, M. NILGES u. T. E. CREIGHTON (1996): Structure determination of the N-terminal thioredoxin-like domain of protein disulfide isomerase using multidimensional heteronuclear 13C/15N NMR spectroscopy Biochemistry 35, 7684-7691 186 Literaturverzeichnis KEMMINK, J., N. J. DARBY, K. DIJKSTRA, M. NILGES u. T. E. CREIGHTON (1997): The folding catalyst protein disulfide isomerase is constructed of active and inactive thioredoxin modules Curr. Biol. 7, 239-245 KLAPPA, P., L. W. RUDDOCK, N. J. DARBY u. R. B. FREEDMAN (1998): The b' domain provides the principal peptide-binding site of protein disulfide isomerase but all domains contribute to binding of misfolded proteins EMBO J. 17, 927-935 KNOX, D. P., S. K. SMITH u. W. D. SMITH (1999): Immunization with an affinity purified protein extract from the adult parasite protects lambs against infection with Haemonchus contortus Parasite Immunol. 21, 201-210 KÖHRMANN, A. (2002): Immunisierungsversuche mit rekombinantem Major Sperm Protein von Dictyocaulus viviparus Hannover, Tierärztl. Hochsch., Diss. KOIVU, J., R. MYLLYLA, T. HELAAKOSKI, T. PIHLAJANIEMI, K. TASANEN u. K. I. KIVIRIKKO (1987): A single polypeptide acts both as the beta subunit of prolyl 4-hydroxylase and as a protein disulfide-isomerase J. Biol. Chem. 262, 6447-6449 KOIVUNEN, P., T. HELAAKOSKI, P. ANNUNEN, J. VEIJOLA, S. RAISANEN, T. PIHLAJANIEMI u. K. I. KIVIRIKKO (1996): ERp60 does not substitute for protein disulphide isomerase as the beta-subunit of prolyl 4-hydroxylase Biochem. J. 316 ( Pt 2), 599-605 KOOYMAN, F. N., A. P. YATSUDA, H. W. PLOEGER u. M. EYSKER (2002): Serum immunoglobulin E response in calves infected with the lungworm Dictyocaulus viviparus and its correlation with protection Parasite Immunol. 24, 47-56 KRISHNA RAO, A. S. u. R. E. HAUSMAN (1993): cDNA for R-cognin: homology with a multifunctional protein Proc. Natl. Acad. Sci. U.S.A. 90, 2950-2954 LAMBERG, A., M. JAUHIAINEN, J. METSO, C. EHNHOLM, C. SHOULDERS, J. SCOTT, T. PIHLAJANIEMI u. K. I. KIVIRIKKO (1996): The role of protein disulphide isomerase in the microsomal triacylglycerol transfer protein does not reside in its isomerase activity Biochem. J. 315 (Pt 2), 533-536 Literaturverzeichnis 187 LARSEN, M. (1999): Biological control of helminths Int. J. Parasitol. 29, 139-146 LAWRENCE, S. B., D. D. HEATH, G. B. HARRISON, C. M. ROBINSON, R. P. DEMPSTER, T. K. GATEHOUSE, M. W. LIGHTOWLERS u. M. D. RICKARD (1996): Pilot field trial of a recombinant Taenia ovis vaccine in lambs exposed to natural infection N. Z. Vet. J. 44, 155-157 LAZARI, O., A. S. HUSSEIN, M. E. SELKIRK, A. J. DAVIDSON, F. J. THOMPSON u. J. B. MATTHEWS (2003): Cloning and expression of two secretory acetylcholinesterases from the bovine lungworm Dictyocaulus viviparus Mol. Biochem. Parasitol. 132, 83-92 LAZARI, O., M. E. SELKIRK, H. W. PLOEGER u. J. B. MATTHEWS (2004): A putative neuromuscular acetylcholinesterase gene from Dictyocaulus viviparus Mol. Biochem. Parasitol. 136, 313-317 LIEPINSH, E., M. BARYSHEV, A. SHARIPO, M. INGELMAN-SUNDBERG, G. OTTING u. S. MKRTCHIAN (2001): Thioredoxin fold as homodimerization module in the putative chaperone ERp29: NMR structures of the domains and experimental model of the 51 kDa dimer Structure 9, 457-471 LIGHTOWLERS, M. W. (1996): Vaccination against cestode parasites Int. J. Parasitol. 26, 819-824 LIGHTOWLERS, M. W., O. JENSEN, E. FERNANDEZ, J. A. IRIARTE, D. J. WOOLLARD, C. G. GAUCI, D. J. JENKINS u. D. D. HEATH (1999): Vaccination trials in Australia and Argentina confirm the effectiveness of the EG95 hydatid vaccine in sheep Int. J. Parasitol. 29, 531-534 LIGHTOWLERS, M. W., R. ROLFE u. C. G. GAUCI (1996): Taenia saginata: vaccination against cysticercosis in cattle with recombinant oncosphere antigens Exp. Parasitol. 84, 330-338 LUNDSTROM, J., G. KRAUSE u. A. HOLMGREN (1992): A Pro to His mutation in active site of thioredoxin increases its disulfide-isomerase activity 10-fold. New refolding systems for reduced or randomly oxidized ribonuclease J. Biol. Chem. 267, 9047-9052 188 Literaturverzeichnis MACER, D. R. u. G. L. KOCH (1988): Identification of a set of calcium-binding proteins in reticuloplasm, the luminal content of the endoplasmic reticulum J. Cell Sci. 91 (Pt 1), 61-70 MANDEL, R., H. J. RYSER, F. GHANI, M. WU u. D. PEAK (1993): Inhibition of a reductive function of the plasma membrane by bacitracin and antibodies against protein disulfide-isomerase Proc. Natl. Acad. Sci. U.S.A. 90, 4112-4116 MARIUS, V., S. BERNARD, J. P. RAYNAUD, P. PERY u. G. LUFFAU (1979): Dictyocaulus viviparus in calves: quantitation of antibody activities in sera and respiratory secretions by immuno-enzymatic analysis Ann. Rech. vet. 10, 55-63 MARTIN, J. L. (1995): Thioredoxin - a fold for all reasons Structure. 3, 245-250 MATTHEWS, J. B., A. J. DAVIDSON u. R. J. BEYNON (2004): The application of mass spectrometry to identify immunogenic components of excretory/secretory products from adult Dictyocaulus viviparus Parasitology 128 Suppl, S43-S47 MATTHEWS, J. B., A. J. DAVIDSON, K. L. FREEMAN u. N. P. FRENCH (2001): Immunisation of cattle with recombinant acetylcholinesterase from Dictyocaulus viviparus and with adult worm ES products Int. J. Parasitol. 31, 307-317 MCARTHUR, A. G., L. A. KNODLER, J. D. SILBERMAN, B. J. DAVIDS, F. D. GILLIN u. M. L. SOGIN (2001): The evolutionary origins of eukaryotic protein disulfide isomerase domains: new evidence from the Amitochondriate protist Giardia lamblia Mol. Biol. Evol. 18, 1455-1463 MCKEAND, J. B., D. P. KNOX, J. L. DUNCAN u. M. W. KENNEDY (1994a): Genetic control of the antibody repertoire against excretory/secretory products and acetylcholinesterases of Dictyocaulus viviparus Parasite Immunol. 16, 251-260 MCKEAND, J. B., D. P. KNOX, J. L. DUNCAN u. M. W. KENNEDY (1994b): The immunogenicity of the acetylcholinesterases of the cattle lungworm Dictyocaulus viviparus Int. J. Parasitol. 24, 501-510 Literaturverzeichnis 189 MCKEAND, J. B., D. P. KNOX, J. L. DUNCAN u. M. W. KENNEDY (1995a): Immunisation of guinea pigs against Dictyocaulus viviparus using adult ES products enriched for acetylcholinesterases Int. J. Parasitol. 25, 829-837 MCKEAND, J. B., D. P. KNOX, J. L. DUNCAN u. M. W. KENNEDY (1995b): Protective immunisation of guinea pigs against Dictyocaulus viviparus using excretory/secretory products of adult parasites Int. J. Parasitol. 25, 95-104 MICHEL, J. F. (1958): An experimental outbreak of husk among previously parasitised cattle Vet. Rec. 70, 554-556 MICHEL, J. F. u. A. MACKENZIE (1965): Duration of the aquired resistance of calves to infection with Dictyocaulus viviparus Res. Vet. Sci. 6, 344-395 MICHEL, J. F. u. A. SHAND (1955): A field study of the epidemiology and manifestations of parasitic bronchitis in adult cattle Vet. Rec. 67, 249-266 MOLAN, A. L., A. J. DUNCAN, T. N. BARRY u. W. C. MCNABB (2003): Effects of condensed tannins and crude sesquiterpene lactones extracted from chicory on the motility of larvae of deer lungworm and gastrointestinal nematodes Parasitol. Int. 52, 209-218 MYLLYHARJU, J. (2003): Prolyl 4-hydroxylases, the key enzymes of collagen biosynthesis Matrix Biol. 22, 15-24 MYLLYHARJU, J., L. KUKKOLA, A. D. WINTER u. A. P. PAGE (2002): The exoskeleton collagens in Caenorhabditis elegans are modified by prolyl 4hydroxylases with unique combinations of subunits J. Biol. Chem. 277, 29187-29196 NATSUKA, S., R. TAKUBO, R. SEKI u. K. IKURA (2001): Molecular cloning and expression of Caenorhabditis elegans ERp57-homologue with transglutaminase activity J. Biochem. (Tokyo) 130, 731-735 NEWTON, S. E., L. E. MORRISH, P. J. MARTIN, P. E. MONTAGUE u. T. P. ROLPH (1995): Protection against multiply drug-resistant and geographically distant strains of Haemonchus contortus by vaccination with H11, a gut membrane-derived protective antigen Int. J. Parasitol. 25, 511-521 190 Literaturverzeichnis NOIVA, R., R. B. FREEDMAN u. W. J. LENNARZ (1993): Peptide binding to protein disulfide isomerase occurs at a site distinct from the active sites J. Biol. Chem. 268, 19210-19217 OAKLEY, G. A. (1979): Survival of Dictyocaulus viviparus infection on pasture Vet. Rec. 104, 530-531 OLIVER, J. D., F. J. VAN DER WAL, N. J. BULLEID u. S. HIGH (1997): Interaction of the thiol-dependent reductase ERp57 with nascent glycoproteins Science 275, 86-88 PARFITT, J. W. u. I. J. SINCLAIR (1967): Cross resistance to Dictyocaulus viviparus produced by Dictyocaulus filaria infections in calves. Res. Vet. Sci. 8, 6-13 PEDONE, E., B. REN, R. LADENSTEIN, M. ROSSI u. S. BARTOLUCCI (2004): Functional properties of the protein disulfide oxidoreductase from the archaeon Pyrococcus furiosus: a member of a novel protein family related to protein disulfideisomerase Eur. J. Biochem. 271, 3437-3448 PFAFFL, M. W. (2001): A new mathematical model for relative quantification in real-time RT-PCR Nucleic Acids Res. 29, e45 PFAFFL, M. W. (2004): Real-time RT-PCR: Neue Ansätze zur exakten mRNA Quantifizierung Biospektrum 10, 92-95 PFEIFFER, H. (1971): Zur Kenntnis der Dictyocaulose des Rindes Wien. tierärztl. Monatsschr. 58, 54-63 PFEIFFER, H. u. R. SUPPERER (1980): Die Dictyocaulose des Rindes Berl. Münch. tierärztl. Wochenschr. 93, 365-370 PIHLAJANIEMI, T., T. HELAAKOSKI, K. TASANEN, R. MYLLYLA, M. L. HUHTALA, J. KOIVU u. K. I. KIVIRIKKO (1987): Molecular cloning of the beta-subunit of human prolyl 4-hydroxylase. This subunit and protein disulphide isomerase are products of the same gene EMBO J. 6, 643-649 Literaturverzeichnis 191 PIRNESKOSKI, A. (2003): The significance of the domains of Protein disulfide isomerase for the different functions of the protein Oulu, Faculty of Medicine, University of Oulu, Diss. PIRNESKOSKI, A., P. KLAPPA, M. LOBELL, R. A. WILLIAMSON, L. BYRNE, H. I. ALANEN, K. E. SALO, K. I. KIVIRIKKO, R. B. FREEDMAN u. L. W. RUDDOCK (2004): Molecular characterization of the principal substrate binding site of the ubiquitous folding catalyst protein disulfide isomerase J. Biol. Chem. 279, 10374-10381 PLOEGER, H. W. (2002): Dictyocaulus viviparus: re-emerging or never been away? Trends Parasitol. 18, 329-332 PLOEGER, H. W., F. H. BORGSTEEDE, J. SOL, M. H. MIRCK, M. W. HUYBEN, F. N. KOOYMAN u. M. EYSKER (2000): Cross-sectional serological survey on gastrointestinal and lung nematode infections in first and second year replacement stock in the Netherlands: relation with management practices and use of anthelmintics Vet. Parasitol. 90, 285-304 PLOEGER, H. W. u. M. EYSKER (2002): Protection against and establishment of Dictyocaulus viviparus following primary infection at different dose levels Vet. Parasitol. 106, 213-223 PLONAIT, H. (1980): Labordiagnostik für die tierärztliche Praxis Verlag Paul Parey, Berlin, Hamburg POHL, S. (2006): Identifizierung, Expression und Charakterisierung der Protein Disulfid Isomerase von Ancylostoma caninum Hannover, Tierärztl. Hochsch., Diss. POUPLARD, L. (1968): Eine rationelle Bekämpfung des Lungenwurmbefalls Veterinärmed. Nachr. 1, 3-19 PUIG, A. u. H. F. GILBERT (1994): Protein disulfide isomerase exhibits chaperone and anti-chaperone activity in the oxidative refolding of lysozyme J. Biol. Chem. 269, 7764-7771 192 Literaturverzeichnis QUAN, H., G. FAN u. C. C. WANG (1995): Independence of the chaperone activity of protein disulfide isomerase from its thioredoxin-like active site J. Biol. Chem. 270, 17078-17080 REGE, A. A., C. Y. SONG, H. J. BOS u. M. H. DRESDEN (1989): Isolation and partial characterisation of a potentially pathogenic cysteine proteinase from adult Dictyocaulus viviparus Vet. Parasitol. 34, 95-102 RICCI, B., D. SHARP, E. O'ROURKE, B. KIENZLE, L. BLINDERMAN, D. GORDON, C. SMITH-MONROY, G. ROBINSON, R. E. GREGG, D. J. RADER u. J.R. WETTERAU: (1995): A 30-amino acid truncation of the microsomal triglyceride transfer protein large subunit disrupts its interaction with protein disulfide-isomerase and causes abetalipoproteinemia J. Biol. Chem. 270, 14281-14285 RODE, B. u. R. J. JØRGENSEN (1989): Baermannization of Dictyocaulus spp. from faeces of cattle, sheep and donkeys Vet. Parasitol. 30, 205-211 ROMMEL, M. u. T. SCHNIEDER (1989): Neue Anthelminthika und neue Behandlungssysteme zur Bekämpfung der Weideparasitosen der Rinder Angew. Parasitol. 30, 101-109 SALAZAR-CALDERON, M., J. M. MARTIN-ALONSO, A. M. CASTRO u. F. PARRA (2003): Cloning, heterologous expression in Escherichia coli and characterization of a protein disulfide isomerase from Fasciola hepatica Mol. Biochem. Parasitol. 126, 15-23 SAMBROCK, J., E. F. FRITSCH u. T. MANIATIS (1989): Molecular cloning: A laboratoy manual 2. Aufl. Cold Spring Harbour Laboratory Press, New York, USA SCHNIEDER T. (Hrsg.) (2006): Veterinärmedizinische Parasitologie 6. Aufl. Verlag Paul Parey, Berlin, S. 217 SCHNIEDER T., EPE C, SAMSON-HIMMELSTJERNA G VON, KOHLMETZ C, HEISE M u. S. WOIDTKE (1998): Immunitätsbildung gegen Lungenwürmer nach Einsatz des Ivomec SR Bolus oder des Panacur SR Bolus Prakt. Tierarzt 79, 446-457 Literaturverzeichnis 193 SCHNIEDER, T. (1992): Use of a recombinant Dictyocaulus viviparus antigen in an enzyme-linked immunosorbent assay for immunodiagnosis of bovine dictyocaulosis Parasitol. Res. 78, 298-302 SCHNIEDER, T. (1993a): A dipstick immunoassay using a recombinant antigen for the rapid diagnosis of bovine dictyocaulosis Res. Vet. Sci. 54, 278-282 SCHNIEDER, T. (1993b): The diagnostic antigen encoded by gene fragment Dv3-14: a major sperm protein of Dictyocaulus viviparus Int. J. Parasitol. 23, 383-389 SCHNIEDER, T. u. A. BELLMER (1993): Seroepidemiologische Untersuchungen zur Ermittlung von Risikofaktoren bei der Dictyocaulose des Rindes Tierärztl. Umsch. 48, 11-15 SCHNIEDER, T., A. BELLMER u. F. J. KAUP (1989): Neuere Erkenntnisse zur Ätiologie, Pathogenese und Bekämpfung der Dictyocaulose Wien. tierärztl. Monatsschr. 76, 372-376 SCHNIEDER, T., A. BELLMER u. A. M. TENTER (1993): Seroepidemiological study on Dictyocaulus viviparus infections in first year grazing cattle in northern Germany Vet. Parasitol. 47, 289-300 SCHNIEDER, T., C. EPE, G. SAMSON-HIMMELSTJERNA u. C. KOHLMETZ (1996a): The development of protective immunity against gastrointestinal nematode and lungworm infections after use of an ivermectin bolus in first-year grazing calves Vet. Parasitol. 64, 239-250 SCHNIEDER, T., C. EPE u. G. VON SAMSON-HIMMELSTJERNA (1996b): Species differentiation of lungworms (Dictyocaulidae) by polymerase chain reaction/restriction-fragment-length polymorphism of second internal transcribed spacers of ribosomal DNA Parasitol. Res. 82, 392-394 SCOTT, C. A., J. B. MCKEAND u. E. DEVANEY (1996): A longitudinal study of local and peripheral isotype/subclass antibodies in Dictyocaulus viviparus-infected calves Vet. Immunol. Immunopathol. 53, 235-247 194 Literaturverzeichnis SELKIRK, M. E., O. LAZARI, A. S. HUSSEIN u. J. B. MATTHEWS (2005): Nematode acetylcholinesterases are encoded by multiple genes and perform nonoverlapping functions Chem. Biol. Interact. 157-158, 263-268 SELMAN, I. E. (1984): Control measures for parasitic bronchitis in cattle Vet. annu. 4, 80-84 SKRJABIN, K. I. (1931): Dictyocauliosis of cattle in the USSR Veterinarnyi Spetsialist na Sotsialistischeskoj Stroike 19, 23-32 SONG, J. L. u. C. C. WANG (1995): Chaperone-like activity of protein disulfide-isomerase in the refolding of rhodanese Eur. J. Biochem. 231, 312-316 STROMBERG, B. E., G. A. AVERBECK, J. F. ANDERSON, B. W. WOODWARD, J. CUNNINGHAM, A. BRAKE u. T. SKOGERBOE (1999): Comparison of the persistent efficacy of the injectable and pour-on formulations of doramectin against artificially-induced infection with Dictyocaulus viviparus in cattle Vet. Parasitol. 87, 45-50 STRUBE, C., G. VON SAMSON-HIMMELSTJERNA u. T. SCHNIEDER (2006): Immunisierungsversuche gegen den bovinen Lungenwurm mittels rekombinanter Proteine Tagung der DVG-Fachgruppe „Parasitologie und parasitäre Erkrankungen“ Wetzlar, 7.-9.6.2006, Abstract S. 51 STRUBE, C. (2004): Differentielle Gentranskription bei Hypobiose induzierten und nicht induzierten dritten Larven von Dictyocaulus viviparus Hannover, Tierärztl. Hochsch., Diss. SMITH, T. S., E. A. MUNN, M. GRAHAM, A. S. TAVERNOR u. C. A. GREENWOOD (1993): Purification and evaluation of the integral membrane protein H11 as a protective antigen against Haemonchus contortus Int. J. Parasitol. 23, 271-280 SMITH, W. D., S. K. SMITH, D. PETTIT, G. F. NEWLANDS u. P. J. SKUCE (2000): Relative protective properties of three membrane glycoprotein fractions from Haemonchus contortus Parasite Immunol. 22, 63-71 Literaturverzeichnis 195 TASANEN, K., T. PARKKONEN, L. T. CHOW, K. I. KIVIRIKKO u. T. PIHLAJANIEMI (1988): Characterization of the human gene for a polypeptide that acts both as the beta subunit of prolyl 4-hydroxylase and as protein disulfide isomerase J. Biol. Chem. 263, 16218-16224 TAVERNOR, A. S., T. S. SMITH, C. F. LANGFORD, M. GRAHAM u. E. A. MUNN (1992): Immune response of Clun Forest sheep to vaccination with membrane glycoproteins from Haemonchus contortus Parasite Immunol. 14, 671-675 TAYLOR, E. I. (1951): Parasitic bronchitis In cattle Vet. Rec. 63, 859-867 TENTER, A. M., A. BELLMER u. T. SCHNIEDER (1993): Evaluation of an ELISA for Dictyocaulus viviparus-specific antibodies in cattle Vet. Parasitol. 47, 301-314 TERADA, K., P. MANCHIKALAPUDI, R. NOIVA, H. O. JAUREGUI, R. J. STOCKERT u. M. L. SCHILSKY (1995): Secretion, surface localization, turnover, and steady state expression of protein disulfide isomerase in rat hepatocytes J. Biol. Chem. 270, 20410-20416 THELLIN, O., W. ZORZI, B. LAKAYE, B. DE BORMAN, B. COUMANS, G. HENNEN, T. GRISAR, A. IGOUT u. E. HEINEN (1999): Housekeeping genes as internal standards: use and limits J. Biotechnol. 75, 291-295 TURANO, C., S. COPPARI, F. ALTIERI u. A. FERRARO (2002): Proteins of the PDI family: unpredicted non-ER locations and functions J. Cell Physiol. 193, 154-163 URQUHART, G. M., W. F. H. JARRETT u. W. I. M. MCINTYRE (1973): Pathology, clinical signs, epidemiology, treatment and control Helminth diseases of cattle, sheep and horses in Europe,Glasgow, 23-29 VAN DEN DIEPSTRATEN, C., K. PAPAY, Z. BOLENDER, A. BROWN u. J. G. PICKERING (2003): Cloning of a novel prolyl 4-hydroxylase subunit expressed in the fibrous cap of human atherosclerotic plaque Circulation 108, 508-511 196 Literaturverzeichnis VEIJOLA, J., P. ANNUNEN, P. KOIVUNEN, A. P. PAGE, T. PIHLAJANIEMI u. K. I. KIVIRIKKO (1996a): Baculovirus expression of two protein disulphide isomerase isoforms from Caenorhabditis elegans and characterization of prolyl 4-hydroxylases containing one of these polypeptides as their beta subunit Biochem. J. 317 (Pt 3), 721-729 VEIJOLA, J., P. KOIVUNEN, P. ANNUNEN, T. PIHLAJANIEMI u. K. I. KIVIRIKKO (1994): Cloning, baculovirus expression, and characterization of the alpha subunit of prolyl 4hydroxylase from the nematode Caenorhabditis elegans. This alpha subunit forms an active alpha beta dimer with the human protein disulfide isomerase/beta subunit J. Biol. Chem. 269, 26746-26753 VEIJOLA, J., T. PIHLAJANIEMI u. K. I. KIVIRIKKO (1996b): Co-expression of the alpha subunit of human prolyl 4-hydroxylase with BiP polypeptide in insect cells leads to the formation of soluble and insoluble complexes. Soluble alpha-subunit-BiP complexes have no prolyl 4-hydroxylase activity Biochem. J. 315 ( Pt 2), 613-618 VERCAUTEREN, I., P. GELDHOF, I. PEELAERS, E. CLAEREBOUT, G. BERX u. J. VERCRUYSSE (2003): Identification of excretory-secretory products of larval and adult Ostertagia ostertagi by immunoscreening of cDNA libraries Mol. Biochem. Parasitol. 126, 201-208 VERCRUYSSE, J., E. CLAEREBOUT, P. DORNY, D. DEMEULENAERE u. E. DEROOVER (1997): Persistence of the efficacy of pour-on and injectable moxidectin against Ostertagia ostertagi and Dictyocaulus viviparus in experimentally infected cattle Vet. Rec. 140, 64-66 VERCRUYSSE, J., P. DORNY, E. CLAEREBOUT u. A. WEATHERLEY (1998): Field evaluation of a topical doramectin formulation for the chemoprophylaxis of parasitic bronchitis in calves Vet. Parasitol. 75, 169-179 VETERINARY LABORATORIES AGENCY (2006): Veterinary Surveillance Report-VIDA (http://www.defra.gov.uk/corporate/vla/science/science-vida-intro.htm) VON HOLTUM, C. (2006): Immunreaktion im Kalb auf das rekombinante Major Sperm Protein von Dictyocaulus viviparus als Glutathion-S-Transferase Fusionsprotein Hannover, Tierärztl. Hochsch., Diss. Literaturverzeichnis 197 VUORI, K., R. MYLLYLA, T. PIHLAJANIEMI u. K. I. KIVIRIKKO (1992a): Expression and site-directed mutagenesis of human protein disulfide isomerase in Escherichia coli. This multifunctional polypeptide has two independently acting catalytic sites for the isomerase activity J. Biol. Chem. 267, 7211-7214 VUORI, K., T. PIHLAJANIEMI, M. MARTTILA u. K. I. KIVIRIKKO (1992b): Characterization of the human prolyl 4-hydroxylase tetramer and its multifunctional protein disulfide-isomerase subunit synthesized in a baculovirus expression system Proc. Natl. Acad. Sci. U.S.A. 89, 7467-7470 VUORI, K., T. PIHLAJANIEMI, R. MYLLYLA u. K. I. KIVIRIKKO (1992c): Site-directed mutagenesis of human protein disulphide isomerase: effect on the assembly, activity and endoplasmic reticulum retention of human prolyl 4hydroxylase in Spodoptera frugiperda insect cells EMBO J. 11, 4213-4217 WANG, C. C. u. C. L. TSOU (1993): Protein disulfide isomerase is both an enzyme and a chaperone FASEB J. 7, 1515-1517 WEATHERLEY, A. J., C. HONG, T. J. HARRIS, D. G. SMITH u. N. C. HAMMET (1993): Persistent efficacy of doramectin against experimental nematode infections in calves Vet. Parasitol. 49, 45-50 WEISBART, R. H. (1992): An antibody that binds a neutrophil membrane protein, ERp72, primes human neutrophils for enhanced oxidative metabolism in response to formyl-methionylleucyl-phenylalanine. Implications for ERp72 in the signal transduction pathway for neutrophil priming J. Immunol. 148, 3958-3963 WETTERAU, J. R., K. A. COMBS, L. R. MCLEAN, S. N. SPINNER u. L. P. AGGERBECK (1991): Protein disulfide isomerase appears necessary to maintain the catalytically active structure of the microsomal triglyceride transfer protein Biochemistry 30, 9728-9735 WETTERAU, J. R., K. A. COMBS, S. N. SPINNER u. B. J. JOINER (1990): Protein disulfide isomerase is a component of the microsomal triglyceride transfer protein complex J. Biol. Chem. 265, 9801-9807 198 Literaturverzeichnis WHELAN, N. C., W. A. CHARLESTON, W. E. POMROY u. A. M. ALEXANDER (1995): Evaluation of the efficacy of doramectin against an artificial infection of Dictyocaulus viviparus in calves N. Z. Vet. J. 43, 21-22 WILSON, W. R., R. S. TUAN, K. J. SHEPLEY, D. O. FREEDMAN, B. M. GREENE, K. AWADZI u. T. R. UNNASCH (1994): The Onchocerca volvulus homologue of the multifunctional polypeptide protein disulfide isomerase Mol. Biochem. Parasitol. 68, 103-117 WINTER, A. D. u. A. P. PAGE (2000): Prolyl 4-hydroxylase is an essential procollagen-modifying enzyme required for exoskeleton formation and the maintenance of body shape in the nematode Caenorhabditis elegans Mol. Cell Biol. 20, 4084-4093 WOOLLEY, H. (1997): The economic impact of "husk" in dairy cattle Cattle Practice 5, 315-318 YAMAUCHI, K., T. YAMAMOTO, H. HAYASHI, S. KOYA, H. TAKIKAWA, K. TOYOSHIMA u. R. HORIUCHI (1987): Sequence of membrane-associated thyroid hormone binding protein from bovine liver: its identity with protein disulphide isomerase Biochem. Biophys. Res. Commun. 146, 1485-1492 YAO, Y., Y. ZHOU u. C. WANG (1997): Both the isomerase and chaperone activities of protein disulfide isomerase are required for the reactivation of reduced and denatured acidic phospholipase A2 EMBO J. 16, 651-658 YOSHIMORI, T., T. SEMBA, H. TAKEMOTO, S. AKAGI, A. YAMAMOTO u. Y. TASHIRO (1990): Protein disulfide-isomerase in rat exocrine pancreatic cells is exported from the endoplasmic reticulum despite possessing the retention signal J. Biol. Chem. 265, 15984-15990 ZHU, L. u. S. W. ALTMANN (2005): mRNA and 18S-RNA coapplication-reverse transkription for quantitative gene expression analysis Anal. Biochem. 345, 102-105 Anhang 199 9 Anhang 9.1 Vollständige cDNA-Sequenz und abgeleitete Aminosäurensequenz der PDI1 von D. viviparus 1 gtttaattac ccaagtttga gggtgcaccg 1 46 tat gtt ttt gct gtg tta ttt ctg 5 y v f a v l f l 85 gat gtg tta gag tac acc gat gct 18 d v l e y t d a 124 atc aag ttc cat gaa atc gct ttg 31 i k f h e i a l 163 ccg tgg tgt ggc cac tgt aag aaa 44 p w c g h c k k 202 gac aaa gct tca act aag ctg aaa 57 d k a s t k l k 241 gtc gct tta att aag gtt gat tgc 70 v a l i k v d c 280 aca tgt gac aaa tat ggc gtc aaa 83 t c d k y g v k 319 aaa atc ttt cgc ttt gga tcg gaa 96 k i f r f g s e 358 gga cct cgt gat gcg gac ggt atc 109 g p r d a d g i 397 ggg caa gct ggt cct tct gcc aga 122 g q a g p s a r 436 aac gag ttc aag aaa gct atc agt 135 n e f k k a i s 475 gtt att ggc ttc ttc gag aat gag 148 v i g f f e n e 514 tca ttc ctc aaa gtt gct gac act 161 s f l k v a d t 553 cag ttt gca tac act tct gat cgt 174 q f a y t s d r 592 acg gga tat aac gat gat atc gtt 187 t g y n d d i v 631 cag ctc cat aat aag ttt gat cct 200 q l h n k f d p 670 gat ggc aac tat gat acg gac aaa 213 d g n y d t d k 709 att cat gaa acc gtc gga atg gct 226 i h e t v g m a 748 gga aat ttg ttc caa ttt gaa cag 239 g n l f q f e q 787 gta tat tac aat gtc gac tat ttg 252 v y y n v d y l 826 tcg aat tat tgg cgt aat cgt gta 265 s n y w r n r v 865 gat tac aag cgc aag gtt cat ttt 278 d y k r k v h f cgg atg m tgt gct c a atc ttt i f gtc aaa v k atg gct m a agc aac s n act gtc t v ggt ttc g f gct cag a q gtc aag v k gag ata e i gga gat g d agc aag s k gaa aga e r tct gta s v gtg ttc v f aat gaa n e atc aag i k ggg att g i aag cct k p aaa gat k d ttg aag l k gct gta a v aca t tcg s gag e ttc f cca p gat d gaa e cca p gca a tat y aaa k gaa e ttg l gac d ctc l acg t ttc f aat n cgc r ctt l cct p gtg v tca s ttt f gca a gac d tac y gaa e cct p aaa k acg t tat y atg m tct s aat n aaa k cgt r aaa k ccg p aaa k ttc f act t gtc v aaa k gcc a aat n gct a agc s tct s gct a ttt f ccc p tcc s tta l gag e cgt r atc i att i gat d ttc f gaa e aag k tat y ctc l cat h att i ggt g aaa k aaa k 200 Anhang 904 291 943 304 982 317 1021 330 1060 343 1099 356 1138 369 1177 382 1216 395 1255 408 1294 421 1333 434 1372 447 1411 460 1450 473 1489 1529 1569 1609 1649 1689 1729 gaa gaa ttt tca tca gaa gtt gat caa aat gga e e f s s e v d q n g ttg cgt aaa gac tca gat aaa cct atc gtt gca l r k d s d k p i v a aca aat gaa ggc aaa ttt cct atg gat aac gaa t n e g k f p m d n e gtt gac aat ctt aag acc ttc gtt gaa gat ttg v d n l k t f v e d l ggt aga ctt gaa cct tac atg aaa tct gaa cca g r l e p y m k s e p gag aat aca gga gca tta aag gtt gca gtt gcc e n t g a l k v a v a ttc aaa gag tta gta ctg aac gcc aaa aaa gat f k e l v l n a k k d gta gaa ttt tac gct ccg tgg tgc gga cac tgc v e f y a p w c g h c ttg gct ccg aaa tac gaa gaa ctt gcc gag aag l a p k y e e l a e k gat gaa gat gtt ctc att gta aaa atg gat gcc d e d v l i v k m d a aat gat gta cct ccc ttg ttt gaa gtc aat gga n d v p p l f e v n g aca ata tac tgg ttg cca aag aac aag aag ggg t i y w l p k n k k g gtt cct tac agt ggt ggc cga gaa gtg gac gac v p y s g g r e v d d tcc ttc ata gct aag cac tca acc gat ggc ctc s f i a k h s t d g l tat tca cgt gat gga aag aag aag aag gca gag y s r d g k k k k a e ttttgtaagc gttcattgtt gaagaagctt ttgcatggtg tttacaagaa catcattaat atgatacaat gcaacatttg cacttttttc cactgttatt tattatttcg aagggtggtc ttcacggttg tttgttgtta cataggctta tgatgtgcct tttatggttt tcacataatt ttattgtagc aagtatttat cagaagtcaa taaatcgttg tttctgttaa aaaaaaaaaa aaaaaaaaaa aaaaa Abb. 32: ctt l gct a ttc f att i att i aag k gtt v aag k tta l acc t ttt f tca s ttt f aag k ttg l tcc s gtg v agc s gct a cct p aat n ctt l gcg a gta v gct a cct p cca p atc i caa q taa - cDNA-Sequenz und abgeleitete Aminosäurensequenz der PDI1. Start- und Stopcodon sind fett gedruckt. 9.2 Vollständige cDNA-Sequenz und Aminosäurensequenz der PDI2 von D. viviparus 1 41 1 83 8 122 21 161 acggtgtgtc ggagtgctct cttcacggtg ttctagacaa attgtcattc attggggcgc a atg ttc aaa ctc gcc m f k l a tgc gtc ttt gct ctg tcg gca ttc gcc gca act c v f a l s a f a a t gaa gag gag aat gtg atc gtg ctt aca aag gac e e e n v i v l t k d gac gaa gtt att aac ggt cat gag ttt gtg ctt tgc c gtt v aac n gcc ctt l gaa e ttt f gaa Anhang 34 200 47 239 60 278 73 317 86 356 99 395 112 434 125 473 138 512 151 551 164 590 177 629 190 668 203 707 216 746 229 785 242 824 255 863 268 902 281 941 294 980 307 1019 320 1058 333 1097 346 1136 359 1175 372 1214 201 d ttt f ccg p ggt g cat h cca p tac y ttg l act t gtt v aag k aat n aag k aag k ctc l cga r tct s ctt l gaa e aaa k aac n gac d atg m gag e act t gat d aac n ctt e tat y gaa e tca s ggc g aca t agc s aag k gcc a gtt v gcg a att i cag q aag k act t cta l gtc v ttt f act t gtt v gtt v ttg l act t agc s ctc l tgg w ttc f gtt v gct a tat y acg t gat d ctg l ggt g aag k gat d gtc v aaa k cca p cta l ttc f gct a gcc a att i gtc v gaa e ctc l cgt r cca p aaa k att i aag k gat d gac d gag i cct p gcc a atc i gtt v aaa k gga g aag k gac d gtt v gtc v ttt f gaa e gac d gac d ctt l ttt f tcc s ttc f ttt f ata i gca a ttc f gtt v gct a aaa k caa q ttc n tgg w aaa k aaa k gct a ctg l cgc r act t gtc v gga g ttt f gga g ctt l gat d aat n gtt v ggt g aaa k aaa k gta v atg m gta v aaa k aaa k cac h gca a gta v tat g tgc c gct a tta l agc s ttc f gat d ggt g aaa k tac y ttg l atc i aag k gga g ctc l tcg s gga g gag e aat n tac y gaa e cga r cct p ttc f ctc l cct p gca a gct h ggt g gcc a gca a aag k cgc r gcg a ccg p tca s ttc f gaa e acc t gaa e cgc r aaa k gaa e gag e tcc s gct a atc i ttc f cta l gac d acc t atg m gta v cgg r cca e cac h aca t aaa k ttc f agt s gca a gtc v ctt l aag k gtt v tct s gaa e gca a aca t ttc f att i agc s gct a aat n ttt f atc i ttt f caa q tcc s aaa k gac d tgg f tgt c cag q cta l gag e gga g tca s gcg a caa q agt s gca a gaa e ggt g gtg v tgg w act t aaa k gag e aga r acg t ggt g tca s gtc v gcg a gaa e gtg v aat n tgt v aag k ctg l gat d gtt v aag k atc i aag k gaa e gtc v agt s gat d atc i ttt f atc i cag q agc s ttc f caa q gat d ctt l tta l gag e tat y gag e ctt l aca t ggt l gca a aag k gct a cgc r ccg p att i aca t gag e gaa e gga g gct a gtg v gat d caa q gaa e cat h gaa e ttt f gtt v aag k gaa e att i ttg l att i gta v aaa k cac a ctt l gaa e acc t ggg g agt s gca a tta l gct a ggg g gtt v gca a cta l gag e gcc a acg t aac n aag k aag k gaa e aac n gag e aac n gat d cct p gga g aat n tgc e gct a gag e gtt v tac y gaa e tgg w aaa k gat d gag e gat d aag k ttg l aag k aac n gca a ctt l tta l ggc g gac d act t gat d aca t gga g gaa e aag k gtt v aag 202 385 1253 398 1292 411 1331 424 1370 437 1409 450 1448 463 1487 476 1526 489 1564 1604 1644 1684 1724 1764 Anhang l v e f y a p w c g h c caa ctt gct ccg act tgg gac aag ctt gga gaa aag q l a p t w d k l g e k gcc gac cac gag aac atc ata att gcc aaa atg gac a d h e n i i i a k m d act gct aat gag gtc gag gac gta aaa gtg caa tca t a n e v e d v k v q s ccg aca atc aag ttc ttc ccc gca gga tca aat aaa p t i k f f p a g s n k atc gac tac act ggt gac agg act ctt gag gga ttc i d y t g d r t l e g f aaa ttc ctt gaa agc ggt ggt aag gaa ggg gct gga k f l e s g g k e g a g tca gat gat gag aag gca gct gaa gag gct gaa gac s d d e k a a e e a e d ggt cat act gag ctg taa gcatttgctg ctatcttact g h t e l ggggtcgtac ggtctgacga cctgttgttg ttacagttaa tttatttgct ctactaatca cccatgtacg agtgatctaa acattgcctt cctgtatatg cgttttttct cttcttcaat tgtgatcgtt gataagtgat ggtgaaattt atttattttt gttcagaact acaccactgt gttggagaac tgggtttaaa taaatataac attttgccaa aaaaaaaaaa aaa Abb. 33: k tat y gca a ttc f ata i act t cca p gag e cDNA-Sequenz und abgeleitete Aminosäurensequenz der PDI2. Start- und Stopcodon sind fett gedruckt. Anhang 203 9.3 Eosinophile Granulozyten Kombi Quil A Kontrolle Quil A PDI Quil A PDI Al(OH)3 Kombi Quil A Kombi Al(OH)3 Kontrolle Al(OH)3 Gruppe Tab. 18: Tier-Nr. 588 596 598 950 611 749 757 914 544 586 597 952 540 545 581 752 579 600 753 948 110 795 802 805 808 814 754 762 790 807 810 816 Eosinophile Granulozyten % G/l % G/l % G/l I. Immunisierungsversuch Tag 84 Tag 91 Sektion 11,5 1,36 4,5 0,41 5,5 0,51 2,0 0,13 4,0 0,30 3,0 0,21 8,0 0,86 3,0 0,25 6,0 0,58 4,5 0,41 2,5 0,15 4,0 0,66 10,0 0,82 11,0 1,06 5,0 0,50 5,5 0,49 2,0 0,17 8,0 0,61 4,5 0,48 3,0 0,33 4,0 0,33 11,5 1,12 10,5 0,72 17,5 1,30 1,5 0,13 18,0 2,12 2,0 0,18 10,5 0,58 5,0 0,27 3,0 0,14 4,0 0,35 5,0 0,43 4,0 0,32 10,5 1,00 16,0 1,71 5,5 0,43 1,0 0,07 1,5 0,11 2,5 0,15 3,0 0,31 3,5 0,38 2,5 0,23 4,0 0,50 5,5 0,73 6,5 0,76 10,0 0,80 5,5 0,48 5,0 0,40 8,0 0,82 3,5 0,31 4,5 0,38 7,0 0,53 1,0 0,08 1,5 0,09 10,0 0,86 9,5 0,81 4,5 0,35 21,5 2,06 11,5 0,84 6,0 0,43 II. Immunisierungsversuch Tag 84 Tag 91 Sektion 10,5 0,83 14,0 0,99 2,5 0,15 13,5 1,15 8,0 0,57 7,5 0,52 12,5 1,13 13,0 1,24 9,0 0,70 7,5 0,71 15,5 1,55 8,0 0,93 8,0 0,96 6,0 0,59 2,0 0,20 5,0 0,44 9,0 0,95 8,0 0,85 4,0 0,29 11,0 0,87 3,5 0,24 4,0 0,34 3,5 0,34 2,0 0,16 15,5 1,13 11,0 0,96 5,0 0,49 6,0 0,54 10,0 1,08 2,0 0,17 17,5 1,56 5,0 0,39 6,5 0,35 14,0 0,99 12,5 0,91 2,0 0,11 Eosinophile Granulozyten 204 Anhang 9.4 Larvenausscheidung im I. u. II. Immunisierungsversuch Kontrolle Al(OH)3 Tag 84 85 86 87 88 89 90 91 92 93 94 95 96 97 98 ∅ SD ∅ SD ∅ SD ∅ SD ∅ SD ∅ SD ∅ SD ∅ SD ∅ SD ∅ SD ∅ SD ∅ SD ∅ SD ∅ SD ∅ SD Tab. 19: I. Immunisierungsversuch Kombi Kombi PDI Quil A Al(OH)3 Al(OH)3 PDI Quil A II. Immunisierungsversuch Kontrolle Kombi Quil A Quil A Mittelwert Anzahl Larven und Standardabweichung SD 0,0 0,3 0,0 0,0 0,3 0,0 0,0 0,4 0,0 0,0 0,4 0,0 5,3 11,5 5,0 2,3 1,8 4,2 4,9 10,7 7,5 1,8 1,8 6,5 35,8 38,3 9,8 11,5 7,5 12,3 42,0 40,3 14,1 5,0 4,7 9,4 93,0 63,0 31,5 37,8 26,0 27,7 93,3 55,6 44,2 14,5 23,3 33,7 169,3 183,8 64,3 185,0 27,8 101,3 249,0 113,7 65,8 61,9 17,5 75,5 211,8 137,3 82,0 242,8 39,8 90,5 270,5 67,2 93,5 97,2 16,8 65,6 365,3 296,8 26,8 288,3 112,3 325,5 456,2 254,4 10,8 154,0 67,4 156,2 312,8 572,0 166,8 329,3 95,8 390,6 191,8 393,5 136,2 119,9 47,5 325,9 505,0 599,8 300,8 438,5 183,3 456,8 325,4 499,4 190,5 250,8 126,5 384,0 802,0 751,0 175,0 365,0 425,5 576,4 644,1 627,5 49,3 126,1 389,5 419,2 1233,0 355,0 401,3 968,5 415,5 437,3 545,7 140,7 240,3 237,3 314,8 535,0 992,0 848,0 720,8 915,0 632,3 396,4 604,6 615,2 395,9 505,6 383,0 317,7 1708,0 1069,0 425,0 1647,0 594,0 592,4 1313,6 1186,9 135,8 627,3 471,0 756,1 687,0 824,0 191,0 546,0 167,0 640,3 445,1 769,3 111,8 144,7 86,2 965,6 1059,0 612,0 284,5 509,0 396,5 1301,7 511,9 148,6 179,9 395,8 0,0 0,0 1,0 1,0 16,2 17,9 47,4 40,9 66,3 32,4 90,1 59,6 198,4 155,1 211,2 161,1 257,8 286,5 244,3 222,6 181,8 150,5 353,1 242,4 325,7 220,3 223,5 164,8 Larvenausscheidung im I. u. II. Immunisierungsversuch. Arithmetischer Mittelwerte (∅) der Gruppen mit Standardabweichungen an den Versuchstagen. Anhang 205 9.5 Längen und Breiten der adulten Würmer Tier Nr. 38,4 36,7 40,2 46,8 40,5 40,4 29,1 34,7 28,6 33,2 32,8 37,7 32,7 32,8 34,0 34,2 39,2 38,0 39,0 37,6 27,7 38,5 42,9 31,9 35,2 110 795 802 805 808 814 ∅ (Gruppe) 754 762 790 807 810 816 ∅ (Gruppe) 40,3 36,7 29,9 34,0 35,4 35,4 35,3 33,7 31,7 30,6 34,2 34,6 27,6 32,1 Al(OH)3 Kombi ∅ Kombi Quil A ∅ Al(OH)3 PDI ∅ PDI Quil A ∅ Kombi Quil A Kontrolle Quil A ∅ Tab. 20: m 588 596 598 950 (Gruppe) 611 749 757 914 (Gruppe) 544 586 597 952 (Gruppe) 540 545 581 752 (Gruppe) 579 600 753 948 (Gruppe) Al(OH)3 Kontrolle Gruppe Länge (m m ) SD w SD m I. Immunisie rungsv e rsuch 3,5 51,5 4,1 0,42 4,5 48,6 7,0 0,40 3,9 52,0 4,2 0,40 3,3 58,7 3,6 0,44 5,4 52,7 6,2 0,42 4,0 56,9 5,3 0,42 3,2 40,1 4,2 0,34 3,6 46,4 4,5 0,37 2,2 37,8 3,2 0,29 5,9 45,3 8,6 0,36 5,1 47,6 3,6 0,31 3,9 49,0 4,1 0,36 5,3 47,5 5,0 0,29 3,8 41,1 4,7 0,31 5,1 46,3 5,4 0,32 4,6 42,3 5,1 0,33 3,9 47,7 4,2 0,36 3,5 49,1 5,4 0,34 3,6 48,5 4,6 0,36 4,4 46,9 5,5 0,35 3,3 37,0 4,8 0,31 2,8 45,7 4,4 0,36 4,5 52,8 5,0 0,40 3,1 40,7 4,4 0,31 6,8 44,1 7,5 0,35 II. Immunisie rungsv e rsuch 4,7 55,5 5,0 0,43 2,8 47,2 4,8 0,41 2,6 40,6 5,3 0,30 3,2 48,0 4,4 0,36 3,6 46,1 4,7 0,38 4,9 46,5 6,8 0,35 3,2 47,3 5,1 0,37 3,6 42,8 4,3 0,34 3,4 44,3 4,8 0,33 3,1 42,4 4,0 0,32 3,1 42,6 5,0 0,34 3,5 44,9 5,0 0,38 3,5 37,2 3,5 0,28 4,2 42,3 5,3 0,33 B reite (m m ) SD w SD 0,05 0,06 0,05 0,05 0,06 0,05 0,06 0,05 0,03 0,07 0,05 0,05 0,05 0,05 0,05 0,04 0,05 0,06 0,05 0,05 0,05 0,06 0,04 0,05 0,06 0,42 0,39 0,43 0,45 0,42 0,47 0,36 0,42 0,35 0,40 0,41 0,41 0,42 0,36 0,40 0,45 0,47 0,44 0,47 0,46 0,42 0,48 0,49 0,43 0,45 0,05 0,05 0,05 0,04 0,05 0,06 0,05 0,06 0,05 0,07 0,05 0,06 0,05 0,04 0,05 0,04 0,08 0,07 0,05 0,06 0,07 0,05 0,06 0,06 0,07 0,06 0,04 0,05 0,05 0,04 0,06 0,04 0,05 0,04 0,04 0,05 0,06 0,04 0,05 0,47 0,43 0,34 0,44 0,41 0,41 0,42 0,36 0,36 0,34 0,33 0,37 0,29 0,34 0,06 0,06 0,06 0,04 0,05 0,07 0,04 0,04 0,06 0,04 0,05 0,05 0,04 0,06 Länge und Breite der adulten Würmer. Mittelwerte von 25 weiblichen (w) und 25 männlichen (m) Würmern pro Versuchstier. ∅: arithmetischer Mittelwert der Gruppe, SD: Standardabweichung. 206 Anhang 9.6 Indices für verschiedene Serumantikörpertiter Gruppe 0 4 7 11 PDI Quil A ∅ SD ∅ SD ∅ SD ∅ SD ∅ SD 0,28 0,06 0,28 0,05 0,35 0,09 0,25 0,04 0,30 0,03 0,27 0,04 0,38 0,13 0,49 0,11 0,43 0,09 0,44 0,05 0,25 0,03 0,41 0,16 0,82 0,09 0,32 0,10 0,77 0,16 0,33 0,04 0,97 0,24 0,95 0,10 1,17 0,04 0,95 0,07 Kontrolle Quil A Kombi Quil A ∅ SD ∅ SD 0,47 0,11 0,41 0,09 0,43 0,09 0,48 0,07 0,39 0,06 0,88 0,07 0,46 0,09 1,10 0,08 Kontrolle Al(OH)3 Kombi Al(OH)3 PDI Quil A ∅ SD ∅ SD ∅ SD ∅ SD ∅ SD 0,37 0,10 0,28 0,07 0,35 0,06 0,34 0,06 0,37 0,10 0,31 0,08 0,29 0,06 0,36 0,06 0,29 0,04 0,32 0,08 0,25 0,06 0,33 0,08 0,64 0,11 0,30 0,08 0,58 0,12 0,35 0,09 0,80 0,16 0,77 0,08 0,97 0,05 0,81 0,07 Kontrolle Quil A Kombi Quil A ∅ SD ∅ SD 0,34 0,07 0,40 0,07 0,35 0,07 0,38 0,07 0,36 0,06 0,70 0,10 0,34 0,07 0,91 0,06 Kontrolle Al(OH)3 Kombi Al(OH)3 PDI Quil A ∅ SD ∅ SD ∅ SD ∅ SD ∅ SD 0,12 0,04 0,14 0,11 0,16 0,10 0,40 0,24 0,18 0,08 0,10 0,03 0,23 0,23 0,21 0,12 0,39 0,21 0,18 0,06 0,08 0,02 0,14 0,08 0,39 0,16 0,26 0,09 0,36 0,19 0,11 0,02 1,04 0,35 0,88 0,23 1,47 0,18 0,91 0,08 Kontrolle Quil A Kombi Quil A ∅ SD ∅ SD 0,25 0,19 0,21 0,04 0,25 0,19 0,21 0,04 0,26 0,18 0,46 0,23 0,25 0,18 1,07 0,26 IgG Kontrolle Al(OH)3 Kombi Al(OH)3 Kombi Quil A PDI Al(OH)3 Tag 14 21 28 35 42 49 98 91 99 56 63 70 77 84 0,38 0,07 1,18 0,05 1,15 0,02 1,22 0,06 1,10 0,06 0,37 0,04 1,22 0,04 1,18 0,02 1,14 0,03 1,03 0,06 0,34 0,04 1,21 0,05 1,18 0,02 1,30 0,07 1,17 0,03 0,36 0,05 1,21 0,03 1,29 0,08 1,20 0,04 1,08 0,06 0,38 0,04 1,21 0,03 1,15 0,02 1,31 0,05 1,18 0,03 0,47 0,13 1,23 0,03 1,18 0,03 1,17 0,02 1,08 0,04 0,83 0,25 1,25 0,04 1,20 0,04 1,31 0,03 1,13 0,06 0,38 0,06 1,08 0,03 0,40 0,06 1,13 0,03 0,54 0,07 1,27 0,05 0,58 0,16 1,28 0,04 0,60 0,13 1,30 0,04 0,71 0,21 1,25 0,04 0,74 0,22 1,15 0,03 0,37 0,06 1,05 0,02 1,02 0,01 1,10 0,02 1,07 0,02 0,34 0,05 1,01 0,02 1,01 0,00 1,06 0,02 1,04 0,02 0,37 0,06 1,03 0,02 1,00 0,02 1,07 0,01 1,03 0,02 0,33 0,06 1,02 0,03 1,03 0,03 1,06 0,01 1,04 0,02 0,39 0,05 1,05 0,03 1,03 0,02 1,06 0,02 1,05 0,02 0,52 0,17 1,03 0,03 0,99 0,01 1,03 0,03 1,02 0,02 0,78 0,20 1,04 0,03 1,00 0,02 1,08 0,02 1,03 0,01 0,32 0,04 1,04 0,01 0,37 0,05 1,03 0,02 0,46 0,04 1,02 0,01 0,41 0,03 1,01 0,02 0,43 0,04 1,03 0,02 0,58 0,19 1,04 0,01 0,61 0,22 1,06 0,02 0,11 0,02 1,08 0,33 1,33 0,10 1,32 0,31 1,38 0,10 0,11 0,03 1,09 0,33 1,38 0,11 1,17 0,26 1,17 0,16 0,12 0,03 1,08 0,35 1,40 0,12 1,55 0,32 1,50 0,15 0,10 0,03 1,17 0,35 1,50 0,12 1,26 0,25 1,26 0,12 0,13 0,03 1,28 0,35 1,64 0,11 1,48 0,29 1,52 0,16 0,16 0,08 1,28 0,31 1,56 0,14 1,23 0,26 1,33 0,14 0,22 0,08 1,49 0,14 1,66 0,15 1,58 0,22 1,45 0,19 0,34 0,20 1,29 0,08 0,36 0,20 1,30 0,08 0,48 0,17 1,36 0,13 0,43 0,20 1,37 0,11 0,44 0,18 1,39 0,08 0,45 0,18 1,39 0,06 0,37 0,17 1,32 0,09 I. Immunisierungsversuch 0,41 0,07 1,01 0,09 1,08 0,11 1,20 0,04 1,13 0,07 0,19 0,02 1,00 0,09 1,06 0,10 1,04 0,06 1,04 0,08 0,39 0,03 1,10 0,03 1,13 0,04 1,12 0,04 0,99 0,06 0,22 0,06 1,10 0,05 1,14 0,04 1,05 0,03 0,96 0,05 0,43 0,05 1,13 0,03 1,15 0,04 1,20 0,04 1,01 0,04 0,22 0,03 1,22 0,11 1,09 0,03 1,01 0,05 0,95 0,03 IgG1 II. Immunisierungsversuch Kombi Quil A PDI Al(OH)3 0,43 0,10 1,10 0,09 0,43 0,09 1,12 0,10 0,41 0,09 1,05 0,02 0,46 0,11 1,07 0,03 0,39 0,07 1,02 0,02 0,35 0,06 1,04 0,04 I. Immunisierungsversuch 0,46 0,14 0,92 0,06 0,90 0,05 1,06 0,02 1,04 0,02 0,30 0,06 0,93 0,06 0,91 0,04 0,96 0,02 0,96 0,03 0,44 0,05 1,01 0,03 1,04 0,02 1,07 0,01 1,02 0,03 0,29 0,03 1,01 0,03 1,01 0,02 0,98 0,03 0,97 0,03 0,42 0,03 1,00 0,02 1,03 0,02 1,07 0,01 1,01 0,03 0,26 0,03 1,04 0,03 1,01 0,02 0,99 0,02 0,98 0,01 IgG2 II. Immunisierungsversuch Kombi Quil A PDI Al(OH)3 0,37 0,07 0,96 0,06 0,36 0,08 0,95 0,04 0,36 0,07 1,02 0,02 0,40 0,10 1,03 0,01 0,35 0,07 1,03 0,01 0,31 0,06 0,98 0,03 I. Immunisierungsversuch 0,14 0,03 1,32 0,19 1,37 0,32 1,68 0,15 1,47 0,04 0,08 0,02 1,38 0,23 1,45 0,29 1,39 0,15 1,22 0,08 0,13 0,03 0,98 0,37 1,24 0,06 1,29 0,24 0,87 0,13 0,08 0,01 1,08 0,48 1,24 0,08 1,07 0,18 0,85 0,10 0,14 0,01 1,06 0,40 1,26 0,07 1,43 0,26 0,96 0,04 0,08 0,02 1,06 0,35 1,25 0,11 0,99 0,29 0,95 0,03 II. Immunisierungsversuch 0,26 0,19 1,31 0,34 0,27 0,19 1,33 0,31 0,23 0,14 0,98 0,23 0,25 0,13 1,02 0,14 0,36 0,21 1,22 0,19 0,34 0,20 1,19 0,10 Anhang Gruppe 0 4 7 11 PDI Quil A ∅ SD ∅ SD ∅ SD ∅ SD ∅ SD 0,27 0,04 0,24 0,06 0,26 0,14 0,33 0,11 0,28 0,07 0,21 0,02 0,28 0,07 0,35 0,13 0,30 0,08 0,35 0,06 0,16 0,02 0,38 0,19 0,90 0,17 0,23 0,05 0,72 0,28 0,27 0,03 0,87 0,19 1,00 0,24 1,10 0,28 1,03 0,20 Kontrolle Quil A Kombi Quil A ∅ SD ∅ SD 0,39 0,15 0,43 0,15 0,34 0,08 0,40 0,11 0,34 0,07 0,99 0,16 0,33 0,06 1,14 0,12 PDI Quil A ∅ SD ∅ SD ∅ SD ∅ SD ∅ SD 0,53 0,11 0,38 0,10 0,31 0,06 0,54 0,11 0,49 0,04 0,48 0,17 0,42 0,11 0,48 0,08 0,47 0,09 0,59 0,14 0,44 0,24 0,61 0,26 1,08 0,09 0,41 0,06 1,11 0,17 0,52 0,20 0,94 0,16 1,07 0,14 1,04 0,18 1,16 0,14 Kontrolle Quil A Kombi Quil A ∅ SD ∅ SD 0,66 0,13 0,64 0,15 0,59 0,08 0,65 0,11 0,62 0,07 1,12 0,07 0,63 0,08 1,12 0,10 Kontrolle Al(OH)3 Kombi Al(OH)3 IgA 207 Kombi Quil A PDI Al(OH)3 Tag 14 21 28 35 42 49 98 91 99 56 63 70 77 84 0,52 0,42 1,19 0,11 1,29 0,05 0,97 0,15 1,02 0,06 0,47 0,26 1,17 0,10 1,26 0,03 1,07 0,18 1,13 0,10 0,58 0,34 1,18 0,10 1,24 0,02 1,14 0,17 1,12 0,10 0,41 0,22 1,20 0,12 1,37 0,05 1,10 0,21 1,14 0,11 0,46 0,22 1,18 0,11 1,28 0,06 1,15 0,21 1,17 0,10 0,40 0,14 1,17 0,13 1,28 0,04 1,08 0,28 1,19 0,07 0,54 0,17 1,27 0,15 1,32 0,06 1,35 0,16 1,26 0,03 0,28 0,14 1,10 0,08 0,28 0,11 1,12 0,06 0,33 0,09 1,25 0,16 0,38 0,22 1,24 0,13 0,45 0,28 1,26 0,12 0,51 0,27 1,26 0,09 0,43 0,18 1,20 0,10 0,61 0,14 0,98 0,21 1,21 0,03 0,85 0,12 1,05 0,04 0,48 0,03 0,99 0,19 1,20 0,03 0,92 0,13 1,09 0,04 0,58 0,04 0,96 0,17 1,15 0,04 1,03 0,14 1,16 0,08 0,43 0,05 0,99 0,18 1,27 0,03 0,98 0,17 1,10 0,05 0,59 0,07 0,99 0,27 1,30 0,04 1,10 0,16 1,18 0,06 0,48 0,03 0,96 0,29 1,29 0,04 1,00 0,19 1,14 0,03 0,65 0,07 1,07 0,23 1,31 0,05 1,22 0,11 1,21 0,04 0,50 0,08 1,09 0,07 0,52 0,08 1,11 0,02 0,51 0,06 1,24 0,12 0,57 0,23 1,23 0,10 0,58 0,24 1,22 0,09 0,61 0,22 1,23 0,06 0,62 0,13 1,23 0,08 I. Immunisierungsversuch 0,39 0,07 1,01 0,15 1,11 0,30 1,38 0,20 1,34 0,15 0,16 0,01 0,96 0,13 1,03 0,28 1,01 0,21 0,98 0,21 0,29 0,03 1,17 0,09 1,26 0,11 1,07 0,15 1,24 0,13 0,17 0,06 1,19 0,16 1,19 0,07 0,90 0,12 0,98 0,09 0,35 0,17 1,17 0,14 1,24 0,08 1,14 0,15 0,99 0,02 0,31 0,26 1,21 0,15 1,18 0,06 0,92 0,14 0,97 0,07 II. Immunisierungsversuch IgM Kontrolle Al(OH)3 Kombi Al(OH)3 Kombi Quil A PDI Al(OH)3 0,33 0,05 1,12 0,14 0,27 0,07 0,96 0,18 0,27 0,07 1,05 0,04 0,33 0,09 1,08 0,05 0,29 0,10 1,18 0,14 0,30 0,15 1,12 0,09 I. Immunisierungsversuch 0,60 0,16 0,97 0,12 1,06 0,19 1,20 0,14 1,22 0,12 0,46 0,08 0,85 0,17 0,95 0,22 1,10 0,16 1,06 0,13 0,51 0,05 1,03 0,10 1,19 0,10 0,98 0,12 1,24 0,09 0,34 0,05 0,97 0,26 1,17 0,06 0,78 0,16 0,98 0,11 0,51 0,05 0,94 0,27 1,14 0,04 1,01 0,13 0,95 0,12 0,35 0,05 0,96 0,26 1,13 0,04 0,77 0,13 0,94 0,12 II. Immunisierungsversuch Tab. 21: 0,60 0,04 1,11 0,09 0,55 0,06 0,97 0,13 0,51 0,05 0,94 0,11 0,49 0,05 0,93 0,11 0,54 0,06 1,11 0,08 0,51 0,10 1,09 0,07 Indices für verschiedene Serumantikörpertiter. ∅: arithmetischer Mittelwert der Gruppe, SD: Standardabweichung. 208 Anhang 9.7 Verzeichnis der verwendeten Abkürzungen A Adenin Abb. Abbildung A. bidest zweifach destilliertes Wasser (lat.: aqua bidestillata) AP Alkalische Phosphatase APS Ammoniumpersulfat BCIP 5-Brom-4-Chloro-3-Indolylphosphat BLASTN/P basic local alignment search tool nucleotide/protein bp Basenpaare bzw. beziehungsweise C Cytosin Ct Cycle threshold °C Grad Celsius ca. circa cDNA komplementäre DNA (engl.: complementary DNA, copy DNA) cm Zentimeter DEPC Diethylpyrocarbonat DNA Desoxyribonukleinsäure (engl.: deoxyribonucleic asid) DNTP Desoxynucleosidtriphosphat dpi Tage nach der Infektion (engl.: days post infectionem) DTT Dithiothreitol E Effizienz EDTA Ethylendiamintetraessigsäure ELISA Enzym gekoppelter Immunabsorptionsassay (engl.: enzyme-linked immunosorbent assay) ER Endoplasmatisches Retikulum ES exkretorisch-sekretorisch et al. lat.: et alii FPLC Fast Pressure Liquid Chromatography g Gravitationskonstante Anhang 209 g Gramm G Guanin GIT Guanidin-Isothiocanat GST Glutathion-S-Transferase HRP Meerrettich-Peroxidase (engl.: horseradish peroxidase) i. A. im Allgemeinen i. m. intramuskulär i. v. intravenös IgA Immunglobulin A IgG Immunglobulin G IgM Immunglobulin M IPTG Isopropylthiogalactosid kDa Kilodalton kg Kilogramm l Liter LB Luria-Bertani L3 dritte Larven M molar (Mol/Liter) mA Milliampere mg Milligramm µg Mikrogramm min Minute ml Milliliter µl Mikroliter mm Millimeter mM millimolar µM mikromolar MW Mittelwert mRNA Boten-RNA (engl.: messenger RNA) NaCl Natriumchlorid NCBI National Centre for Biotechnology Information 210 Anhang NBT Nitroblau-Tetrazoliumchlorid nm Nanometer OD optische Dichte OPD ortho-Phenylendiamin-Dihydrochlorid ORF Offener Leserahmen (engl.: open reading frame) PCR Polymerase Kettenreaktion (engl.: polymerase chain reaction) p.d. pro Tag (engl.: per day) PDI Protein Disulfid Isomerase pH negativer dekadischer Logarithmus der H+-Ionen-Konzentration RACE Rapid Amplification of cDNA Ends RNA Ribonukleinsäure (engl.: ribonucleic acid) ROX 5-Carboxy-X-Rhodamin rpm Umdrehung pro Minute (engl. rounds per minute) rRNA ribosomale RNA RT Reverse Transkriptase s.c. subcutan SD Standardabweichung SDS Natriumdodecylsulfat (engl.: sodiumdodecylsulfate) sec. Sekunde T Thymin Tab. Tabelle TAE Tris-Acetat-EDTA-Puffer Taq Thermus aquaticus TBS Tris-gepufferte Natriumchlorid-Lösung (engl.: tris-buffered saline) TEMED N,N,N’,N’-Tetramethylendiamin Tm Schmelztemperatur U Enzymeinheiten (engl.: unit) UV ultraviolett V Volt v/v Volumen pro Volumen w/v Gewicht pro Volumen (engl.: weight per volumen) Anhang 211 9.8 Symbole der Aminosäuren a Alanin b Asparagin oder Asparaginsäure c Cystein d Asparaginsäure e Glutaminsäure f Phenylalanin g Glycin h Histidin i Isoleucin k Lysin l Leucin m Methionin n Asparagin p Prolin q Glutamin r Arginin s Serin t Threonin v Valin w Tryptophan y Tyrosin z Glutamin oder Glutaminsäure 212 Anhang 9.9 Abbildungsverzeichnis Abb. 1: Abb. 2: Abb. 3: Abb. 4: Abb. 5: Abb. 6: Abb. 7: Abb. 8: Abb. 9: Abb. 10: Abb. 11: Abb. 12: Abb. 13: Abb. 14: Abb. 15: Abb. 16: Abb. 17: Abb. 18: Abb. 19: Abb. 20: Abb. 21: Abb. 22: Abb. 23: Abb. 24: Abb. 25: Abb. 26: Abb. 27: Abb. 28: Abb. 29: Abb. 30: Abb. 31: Abb. 32: Abb. 33: Schematische Darstellung der PDI-Familie ............................................ 39 Konstruktion degenerierter Primer. ......................................................... 68 Schema der RACE-PCR......................................................................... 70 Konstruktion der Full length-Primer ........................................................ 73 Schematische Darstellung des Versuchsablauf...................................... 93 Prinzip der QRT-PCR mit TaqMan™-Sonden ...................................... 103 Mit degenerierten Primern erhaltenes Amplifikat. ................................. 109 Vergleich positiver Klone mittels RsaI-Verdau. ..................................... 110 PCR-Produkte der RACE-PCR............................................................. 111 Alignment der Aminosäurensequenz der PDI2 von D. viviparus mit PDIs anderer Spezies ................................................. 112 Alignment der Aminosäuresequenz der PDI1 von D. viviparus mit PDIs anderer Spezies. ................................................ 114 Phylogenetischer Stammbaum ............................................................. 115 Domänenstruktur der PDI1 und PDI2 von D. viviparus. ........................ 116 PCR zur Ermittlung der genomischen Sequenz.................................... 117 Exon-Intron-Organisation der PDI2 von D. viviparus. ........................... 117 Vollständige genomische DNA-Sequenz der PDI2 von D. viviparus..... 118 Darstellung des aufgereinigten Fusionsproteins. .................................. 120 Optimierung der Expression. ................................................................ 121 Western-Blot zum Vergleich der Aufreinigung nach unterschiedlichen Lysebedingungen. ................................................................................ 122 Coomassie-gefärbtes SDS-Gel der fünf Peak Fraktionen eines FPLC-Laufes......................................................................................... 123 Messung des II. Proteinpools................................................................ 126 Veränderung der Atemfrequenzen während des Versuchsverlaufs...... 130 Gruppenmittel der prozentualen Gewichtszunahme. ............................ 131 Larvenausscheidung im I. und II. Immunisierungsversuch. .................. 136 Larvenausscheidung im I. und II. Immunisierungsversuch. .................. 137 Darstellung der mittleren Wurmlängen ................................................. 141 Gruppenmittelwerte der Indices für verschiedene Serumantikörpertiter 144 Überprüfung der Serokonversion im Western-Blot ............................... 146 Ratioberechnung nach PFAFFL ........................................................... 148 Anwendung der Formel nach PFAFFL. ................................................ 149 Grafische Darstellung der Ratios für 120 Würmer ................................ 149 cDNA-Sequenz und abgeleitete Aminosäurensequenz der PDI1. ........ 200 cDNA-Sequenz und abgeleitete Aminosäurensequenz der PDI2. ........ 202 Anhang 213 9.10 Tabellenverzeichnis Tab. 1: Tab. 2: Tab. 3: Tab. 4: Tab. 5: Tab. 6: Tab. 7: Tab. 8: Tab. 9: Tab. 10: Tab. 11: Tab. 12: Tab. 13: Tab. 14: Tab. 15: Tab. 16: Tab. 17: Tab. 18: Tab. 19: Tab. 20: Tab. 21: RACE Primer .......................................................................................... 69 Sequenzen der Primer zur Amplifikation der genomischen DNA............ 71 Gruppeneinteilung der Immunisierungsversuche.................................... 94 Zusammensetzung der Impfdosen ......................................................... 96 In der QRT-PCR verwendete Primer und Sonden ................................ 104 Identitäten der PDI2 von D. viviparus mit PDIs anderer Spezies .......... 113 Identitäten der PDI1 von D. viviparus mit PDIs anderer Soezies .......... 114 Vergleich der Exon- u. Introngrößen von A. caninum und D. viviparus. 119 Agilent-Messungen des I. Proteinpools. ............................................... 124 Agilent-Messungen des II. Proteinpools. .............................................. 125 Befunde der täglichen Untersuchung ab Tag 70................................... 129 Auftreten lokaler Impfreaktionen ........................................................... 133 Behandlung erkrankter Tiere ................................................................ 134 Pathologisch-anatomische Befunde des I. Immunisierungs-versuchs .. 138 Pathologisch-anatomische Befunde des II. Immunisierungs-versuchs . 139 Anzahl adulter Lungenwürmer und Larvenausscheidung. .................... 140 Cts des Kalibrators in verschiedenen PCR-Läufen............................... 148 Eosinophile Granulozyten..................................................................... 203 Larvenausscheidung im I. u. II. Immunisierungsversuch. ..................... 204 Länge und Breite der adulten Würmer.................................................. 205 Indices für verschiedene Serumantikörpertiter...................................... 207 Erklärung nach § 3 Abs. 2 Nr. 7, Promotionsordnung vom 28.08.2000 Hiermit erkläre ich, dass ich die Dissertation mit dem Titel „Charakterisierung und in vitro Expression einer Protein Disulfid Isomerase (PDI) zur Immunisierung gegen Dictyocaulus viviparus“ selbstständig verfasst habe. Bei der Anfertigung wurden folgende Hilfen Dritter in Anspruch genommen: Es wurden Materialien und Geräte des Instituts für Parasitologie sowie des Instituts für Pathologie der Tierärztlichen Hochschule Hannover in Anspruch genommen. Die Bestimmung von Differentialblutbildern erfolgte durch die Klinik für kleine Klauentiere und forensische Medizin der Tierärztlichen Hochschule Hannover. Ich habe keine entgeltliche Hilfe von Vermittlungs- bzw. Beratungsstellen (Promotionsberater oder andere Personen) in Anspruch genommen. Niemand hat von mir unmittelbar oder mittelbar entgeltliche Leistungen für Arbeiten erhalten, die im Zusammenhang mit dem Inhalt der vorgelegten Dissertation stehen. Ich habe die Dissertation an folgender Institution angefertigt: Institut für Parasitologie der Tierärztlichen Hochschule Hannover Die Dissertation wurde bisher nicht für eine Prüfung oder Promotion oder für einen ähnlichen Zweck zur Beurteilung eingereicht. Ich versichere, dass ich die vorstehenden Angaben nach bestem Wissen vollständig und der Wahrheit entsprechend gemacht habe. Sonja Wolken DANKSAGUNG Herrn Prof. Dr. Thomas Schnieder danke ich für die Überlassung des interessanten und abwechslungsreichen Themas, die Bereitstellung des Arbeitsplatzes und die freundliche Beratung während der Anfertigung dieser Dissertation. Weiterhin möchte ich mich dafür bedanken, dass mir die Teilnahme an verschiedenen Kongressen ermöglicht wurde. Dies war eine sehr große fachliche und persönliche Bereicherung für mich. Herrn Prof. Dr. Georg von Samson-Himmelstjerna sei vielmals für alle fachlichen Diskussionen und Hilfestellungen gedankt. I thank Mr. Ph.D. M.Sc.William Blackhall for showing me all his little tricks when dealing with DNA. Frau Ph.D. Christina Strube danke ich für viele persönliche und fachliche Gespräche. Meinem Mitdoktoranden Christian von Holtum danke ich ganz herzlich für die freundschaftliche und konstruktive Zusammenarbeit. Danke CvH! Allen jetzigen und ehemaligen Mitarbeitern, Doktoranden und Praktikanten, die beim „Picken“ der Lungenwürmer geholfen haben und mir ansonsten mit Rat und Tat zur Seite standen, sei ganz herzlich gedankt. Ohne die vielen fleißigen Hände wäre diese Arbeit nicht möglich gewesen. Ulla Küttler sei hierbei vielmals für die Unterstützung beim Larvenzählen gedankt. Ganz besonderer Dank geht an Sandra Buschbaum, die mit ihrem guten Geist alles zusammenhält, Ricarda Hüsken, Claudia Welz und Nina Fischer. Eure Freundschaft und euer Vertrauen haben meine Doktorandenzeit zu etwas ganz Besonderem gemacht. Den Tierpflegern Norbert Schulze, Adam Rosalski, Ivonne Strickrodt, Sandra Horchler und Eileen Winkler danke ich für die gute Zusammenarbeit im Stall und diverse lebensrettende Sofortmassnahmen in Form von Kaffee und Tee. Norbert und Adam sei besonders für ihre Hilfe bei der Sektion gedankt. Den Laboranten der Klinik für kleine Klauentiere danke ich für die Erstellung der Differentialblutbilder. Frau Ph.D. Ilse Jacobsen danke ich ganz besonders für ihre Freundschaft und die fachlichen Diskussionen in diversen Lokalitäten. Meiner Familie gilt besonderer Dank für die Unterstützung während meines Studiums und der Anfertigung dieser Dissertation. Der größte Dank geht an meinen Partner und Seelengefährten Jörg Giere. Danke Jörg, für die viele Hilfe und all das, was nicht in Worte zu fassen ist.