Lösungen Seite 105
Werbung

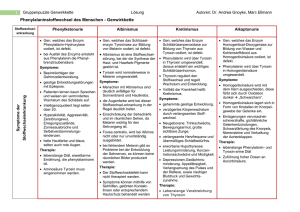
Lösungen zur Kompetenzen-Seite Ein Gen ist molekularbiologisch betrachtet ein Abschnitt auf einem der beiden Stränge der DNA (codogener Strang) mit einer bestimmten Basen- bzw. Nucleotidsequenz, in der die Reihenfolge der Aminosäuren der Polypeptidkette, aus der das Enzym besteht, in Form von Tripletts codiert ist. Aufgrund einer Mutation im Gen für Phenylalaninhydroxylase wird ein Enzym hergestellt, dessen aktives Zentrum stark verändert ist, sodass kein Phenylalanin gebunden und in Tyrosin umgewandelt wird. In der Folge kommt es zu einem Überschuss an Phenylalanin und zu einem Mangel an Tyrosin. © 2010 Cornelsen Verlag, Berlin. Alle Rechte vorbehalten. Bei Missense-Mutationen bewirkt ein verändertes Codon den Einbau einer falschen Aminosäure. Dieser bleibt ohne Folgen, wenn die neue Aminosäure ähnliche Eigenschaften hat oder wenn der Austausch in einem funktionell unwichtigen Teil des Proteins stattfindet; erfolgt er dagegen in funktionell wichtigen Bereichen, kommt es zu erheblichen Veränderungen der Konformation, die in der Regel zum Funktionsverlust führen. Eine Deletion bedeutet den Verlust eines oder mehrerer Nucleotidpaare, wodurch das Leseraster verschoben wird; die Auswirkungen hängen von der Lage und der Zahl der Nucleotide ab. Bei Nonsense-Mutationen wird ein Aminosäure-Codon in ein Stopp-Codon umgewandelt; das führt zu einem vorzeitigen Abbruch der Translation. Das Enzym wird überhaupt nicht hergestellt und fehlt. Stumme Mutationen zeichnen sich dadurch aus, dass die veränderten Codons wegen der Redundanz des genetischen Codes zum Einbau von identischen Aminosäuren führen. Bei der Proteinbiosynthese wird zunächst die gesamte Basensequenz eines Gens mit allen Exons und Introns in eine komplementäre RNA-Basensequenz transkribiert. Danach schneiden spezielle Enzyme aus dieser Prä-mRNA die Introns heraus und verbinden die Exons zu einem kontinuierlichen RNA-Molekül. Eine Mutation an einer Schnittstelle bewirkt, dass ein Intron mit einem Exon verbunden bleibt. Das führt an den Ribosomen zum Einbau zusätzlicher Aminosäuren, die die Konformation des Proteins völlig verändern und daher dessen Funktionsverlust zur Folge haben. Arginin wird in der mRNA entweder durch die Tripletts CGG, CGA, CGC, CGU oder durch AGG und AGA codiert. Tryptophan wird in der mRNA ausschließlich durch das Triplett UGG codiert. Die dazu komplementäre Basensequenz der DNA ist ACC. Als DNA-Tripletts für Arginin kommen daher nur GCC und TCC in Frage. Demnach sind zwei verschiedene Mutationen möglich: A statt G oder A statt T. In Leberzellen ist das Gen zugänglich und kann jederzeit transkribiert werden. In allen anderen Körperzellen ist das Gen entweder gar nicht zugänglich ist, weil es durch Histone oder andere chromosomale Proteine blockiert wird, oder es wird durch Silencer-Sequenzen kontrolliert, die über Regulatorproteine eine Transkription verhindern. Grundlagen der Genetik Molekulargenetik Seite 1 von 2 Lösungen zur Kompetenzen-Seite © 2010 Cornelsen Verlag, Berlin. Alle Rechte vorbehalten. In der Regel werden bei der Proteinbiosynthese beide Gene transkribiert. Ist nur eines der beiden Gene für Phenylalaninhydroxylase mutiert, werden funktionsfähige Enzyme hergestellt, allerdings in geringerer Menge. Diese Menge reicht bei normaler Aufnahme von Phenylalanin mit der Nahrung aus, um dessen Konzentration im Blut auf Werte zu senken, die keine physiologischen und damit keine phänotypischen Auswirkungen haben. Um den 5. Lebenstag (bei Frühgeburten um den 10. Lebenstag) wird aus der Ferse des Neugeborenen Blut auf eine Filterpapierkarte getropft und nach dem Trocknen in ein Labor eingesandt. Dort werden Scheiben von definierter Größe ausgestanzt und auf einen Nährboden aufgetragen, der mit dem Bakterium Bacillus subtilis beimpft ist. Dem Nährboden wird außerdem ein Wachstums-Hemmstoff (2-Thienylalanin) zugefügt, dessen Wirkung durch die Anwesenheit von Phenylalanin aufgehoben wird. Ein Wachstum der Bakterienkultur um die Blutprobe herum lässt auf erhöhte Phenylalaninwerte und die Größe des Bakterienhofs auf die ungefähre Konzentration der Aminosäure schließen. Die Methode ist allerdings mit Fehlerquellen behaftet. So hängt die Konzentration von Phenylalanin stark von der Menge der aufgenommenen Nahrung in den ersten Lebenstagen ab. War diese zu gering, kann auch bei Vorliegen der gesuchten Erkrankung der Phenylalaninspiegel in der untersuchten Probe unterhalb der Nachweisgrenze liegen. Eine Behandlung mit einem Antibiotikum kann das Bakterienwachstum allgemein hemmen und somit zu falsch-negativen Ergebnissen führen, obwohl eine Störung des Phenylalaninstoffwechsels vorliegt. Da der Fetus über den mütterlichen Blutkreislauf versorgt wird, führen hohe Phenylalaninkonzentrationen im Blut von Schwangeren zu Schädigungen des Fetus, die sich nach der Geburt in Form von geistiger Behinderung bemerkbar machen. Die Grafik zeigt, dass das Ausmaß der Behinderung mit steigender Phenylalaninkonzentration zunimmt, allerdings nicht linear. Die kritische Konzentration liegt zwischen 11 und 16 mg/dl. Schwangere Frauen mit PKU sollten deshalb durch Diät ihre Phenylalaninkonzentration unter 10 mg/dl halten. (Anmerkung: In der Praxis werden 2–6 mg/dl angestrebt). Die frühere Empfehlung beruhte auf der Annahme, dass das Gehirn im Erwachsenenalter „ausgereift“ sei und keinen Schaden mehr nehmen könne. Vermutlich hat sich diese Annahme nicht bestätigt, zumal inzwischen belegt ist, dass auch im erwachsenen Gehirn massive Umbauprozesse stattfinden können, vor allem bei Lernprozessen („Neuroplastizität“). Grundlagen der Genetik Molekulargenetik Seite 2 von 2