link zum Infoblatt
Werbung

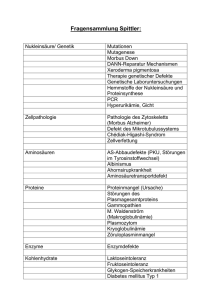
Medizinisches Versorgungszentrum der UMG Bereich Humangenetik Heinrich-Düker-Weg 12 37073 Göttingen Informationsblatt: Morbus Osler Stand: 30.09.2015 Der Morbus Osler (auch unter den Bezeichnungen Osler-Weber-Rendu-Syndrom und Hereditäre Hämorrhagische Teleangiektasie (HHT) bekannt) ist eine seltene, autosomal dominant vererbte Erkrankung, die sich hauptsächlich durch Gefäßfehlbildungen auszeichnet. Zwar sind die meisten Blutgefäße von Morbus Osler Patienten normal ausgebildet, doch vereinzelt kommt es zu Blutgefäßerweiterungen (Teleangiektasien), die infolge ihrer hohen Verletzlichkeit z. B. an den Schleimhäuten zu Blutungen neigen. Die Penetranz ist altersabhängig und im Alter von 40 bis 45 Jahren nahezu vollständig. Erste Erkrankungsanzeichen, wie z. B. spontanes Nasenbluten treten aber häufig schon im Alter von 10 bis 20 Jahren auf. Über die Verbreitung von Morbus Osler gibt es unterschiedliche Angaben, sie wird auf 1-10 auf 100000 geschätzt. Kürzlich wurden folgende 4 Kriterien festgelegt, von denen 3 vorliegen müssen, um die Diagnose Morbus Osler zu stellen: 1. Epistaxis 2. Teleangiektasien 3. viszerale Beteiligung 4. familiäre Häufung Die Epistaxis (spontanes, häufiges Nasenbluten) tritt bei mehr als 90% aller Patienten mit Morbus Osler auf. Dieses Nasenbluten kann sehr stark und anhaltend sein, so dass manche Patienten an Anämie (Blutarmut) leiden. Viele Morbus Osler-Patienten bilden Teleangiektasien aus, diese sind als kleine, etwa stecknadelkopfgroße rote Punkte in der Haut zu erkennen. Betrachtet man diese Punkte genauer, so erkennt man, dass von ihnen feine Verästelungen ausgehen. Diese Punkte findet man vor allem an den Fingerspitzen, an der Nase und im Bereich der Lippen, der Zunge und in der Mundhöhle. Ihre Ursache liegt in der Erweiterung kleiner Blutgefäße, wobei die Zahl der Kapillaren zwischen Arteriolen und Venolen reduziert und einzelne Kapillaren derart erweitert sind, dass das Blut direkt von den Arteriolen zu den Venolen fließen kann ("Kurzschluss"). Solche Teleangiektasien findet man jedoch nicht nur in der Haut, sondern es kann auch eine viszerale Beteiligung, d. h. eine Beteiligung der inneren Organe, vorliegen. Besonders betroffen sind hier die Lunge, das Gehirn und der Verdauungstrakt. Die Gefäßveränderungen werden hier als arteriovenöse Malformationen (AVM) bezeichnet. Das Risiko für die Patienten besteht nicht nur aufgrund der Gefahr innerer Blutungen. Durch den Kurzschluss zwischen Arteriolen und Venolen in der Lunge können auch Gerinnsel und Erreger passieren und so zu Schlaganfällen und Gehirnabszessen führen. Von einer familiären Häufung spricht man, wenn mindestens zwei Verwandte ersten Grades von der Erkrankung betroffen sind. Genetik Beim Morbus Osler wurden bislang Mutationen in drei verschiedenen Genen gefunden. Bei dem einen Gen handelt es sich um Endoglin (ENG), das auf Chromosom 9 lokalisiert ist, bei dem anderen um das Gen für die Aktivin Rezeptor-ähnliche Kinase (ACVRL-1), das auf Chromosom 12 liegt. Beide Gene werden in Endothelzellen exprimiert. Sie kodieren für Proteine, die in der Zellmembran lokalisiert und Bestandteil des Rezeptorkomplexes für den Transforming Growth Factor β (TGF-β) sind. Des weiteren spielt das dritte Gen (das SMAD4-Gen auf Chromosom 18) eine wichtige Rolle in der Transduktion der Signale der TGF-β−Ligandensuperfamilie von der Zellmembran in den Zellkern. Da TGF-β Seite 1 von 2 Version: 1.0-0915 Die Humangenetik des MVZ der UMG ist im Bereich der Medizinischen Laboratoriumsdiagnostik in den Untersuchungsgebieten Molekulare Humangenetik und Zytogenetik akkreditiert nach DIN EN ISO 15189:2014 Medizinisches Versorgungszentrum der UMG Bereich Humangenetik Heinrich-Düker-Weg 12 37073 Göttingen Informationsblatt: Morbus Osler Stand: 30.09.2015 an verschiedenen Prozessen der Endothelzellentwicklung beteiligt ist (Zellteilungen, Wanderung der Zellen, Zelladhäsion), liegt dem Morbus Osler vermutlich eine Störung der Gefäßentwicklung oder der Reparaturmechnismen der Gefäße zugrunde. In der klinischen Ausprägung unterscheiden sich Patienten mit Endoglin-Mutationen (HHT Typ 1) von Patienten mit ACVRL-1 Mutationen (HHT Typ 2). Patienten des Typ 1 haben ein Risiko von etwa 30 %, pulmonale AVM auszubilden. Dieses Risiko liegt bei Typ 2 Patienten nur bei 3 %, auch der gesamte Krankheitsverlauf ist milder als bei Typ 1 Patienten. Das SMAD4 Gen ist bei Patienten mit juveniler Polyposis (JP) häufig mutiert. Mutationen im SMAD4 Gen sind ebenfalls ursächlich für das kombinierte Syndrombild (JP + HHT). Kürzlich haben Gallione et al. (2006) Mutationen im SMAD 4 Gen in HHT Patienten ohne JP gefunden. Es ist davon auszugehen, dass SMAD4 das dritte Gen für Morbus Osler ist. Allerdings müssen Morbus Osler-Patienten mit Mutationen im SMAD4 Gen auf das Vorliegen von Magendarmtraktpolypen untersucht werden. Diagnostik Im Rahmen der Diagnostik werden die Gene ENG, ACVRL-1 und SMAD4 mittels PCR amplifiziert und direkt sequenziert. Bei einem unauffälligen Ergebnis kann eine MLPA Analyse (Multiplex Ligation dependent Probe Amplification) zur Detektion von großen Rearrangements (Deletionen/Duplikationen) Gens angeboten werden. Material Für die Untersuchung werden 10 - 20 ml EDTA-Blut, bei Kindern mindestens 2 ml, benötigt. Der Versand kann auf dem normalen Postweg erfolgen. Kontakt Dr. rer. nat. Silke Kaulfuß Tel.: 0551 / 39-9019 E-Mail: [email protected] Seite 2 von 2 Version: 1.0-0915 Die Humangenetik des MVZ der UMG ist im Bereich der Medizinischen Laboratoriumsdiagnostik in den Untersuchungsgebieten Molekulare Humangenetik und Zytogenetik akkreditiert nach DIN EN ISO 15189:2014