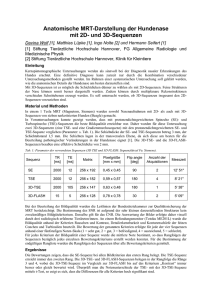
Vorhersage von RNA-Sekundärstrukturen
Werbung

Aktuelle Themen der Bioinformatik
Vorhersage von RNASekundärstrukturen
-drei verschiedene
Methoden zur Vorhersage
von Pseudoknoten der RNA
Natalie Jäger
Johann-Wolfgang-Goethe Universität, Frankfurt am Main
1/89
Vorhersage von
Pseudoknoten
• Grundlagen – Aufbau der RNA
3 Methoden für RNA Secondary Structure
Prediction:
• Stochastisches Modellieren durch parallele
Grammatiken
• Graph-theoretischer Ansatz
• „Iterated Loop Matching“ Algorithmus
Johann-Wolfgang-Goethe Universität, Frankfurt am Main
2/89
Vorhersage von
Pseudoknoten
• Stochastic modeling of RNA pseudoknotted structures:
a grammatical approach; Cai, Russell, Wu; 2003
• A graph theoretical approach to predict common RNA
secondary sructure motifs including pseudoknots in
unaligned sequences; Yongmei, Stormo, Xing; 2004
• An iterated loop matching approach to the prediction of
RNA secondary structures with pseudoknots; Ruan,
Stormo, Zhang; 2004
Johann-Wolfgang-Goethe Universität, Frankfurt am Main
3/89
Biologische Aspekte der RNA
-besteht ZuckerphosphatRückgrat, sowie einer Abfolge
von 4 möglichen Basen (A, U, G,
C)
-Unterschied zur DNA: Zucker ist
die Ribose, und eine der vier
Basen, nämlich T (Thymin) ist
ersetzt durch U (Uracil)
-Jeweils drei Nukleotide bilden
ein Codon, mit dessen Hilfe sich
eine spezifische Aminosäure,
eindeutig bestimmen lässt
Johann-Wolfgang-Goethe Universität, Frankfurt am Main
4/89
Funktion der RNA
- im Gegensatz zur doppelsträngigen DNA einsträngige Polynukleotide
- dieser Unterschied erhöht die katalytische Funktion
der RNA und erlaubt ihr chemische Reaktionen, die
der DNA nicht möglich sind
- mRNA, Boten-RNA: kopiert die in einem Gen auf der
DNA liegende Information und trägt sie zum
Ribosom, wo mit Hilfe dieser Information die
Proteinbiosynthese stattfinden kann
Johann-Wolfgang-Goethe Universität, Frankfurt am Main
5/89
•tRNA, Transfer-RNA:
kodiert keine genetische
Information, sondern dient
als Hilfsmolekül bei der
Proteinbiosynthese, indem
sie eine einzelne
Aminosäure aus dem
Cytoplasma aufnimmt und
zum Ribosom transportiert
•Paarungen konjugierender
Basen über Wasserstoffbrücken kleeblattartige
Struktur
Johann-Wolfgang-Goethe Universität, Frankfurt am Main
6/89
• rRNA, ribosomale RNA: trägt ähnlich wie die tRNA
keine genetische Information, sondern ist am Aufbau
des Ribosoms beteiligt und erfüllt dort auch
Stoffwechselfunktion
• snRNA, small nuclear-RNA: im Zellkern von
Eukaryoten, verantwortlich für die enzymatische
Spaltung der RNA (Splicing)
Johann-Wolfgang-Goethe Universität, Frankfurt am Main
7/89
RNA-Sekundärstrukturen
• werden durch Interaktionen zwischen
komplementären Nucleotid-Paaren festgelegt (über
H-Brücken), die nah oder weit voneinander im
Molekül entfernt sind
• genau diese Interaktionen falten die RNA in solche
Formen wie Stem Loops oder die komplizierteren
Pseudoknoten
• Sekundärstruktur hängt mit der Funktion der RNA
zusammen daher versucht man Sekundärstruktur
der RNA vorherzusagen
Johann-Wolfgang-Goethe Universität, Frankfurt am Main
8/89
Stem Loops (Haarnadelstruktur)
- Doppelhelixbereich, der durch Basenpaarung zwischen
benachbarten, komplementären Sequenzen innerhalb eines
RNA-Stranges entsteht
Johann-Wolfgang-Goethe Universität, Frankfurt am Main
9/89
Pseudoknoten
Pseudoknoten wegen c und c`, die zusammen
eine Base-Paired-Region sind, also eine
Doppelhelix bilden
Johann-Wolfgang-Goethe Universität, Frankfurt am Main
10/89
Definition Pseudoknoten:
1.
2.
In der RNA-Sequenz s beinhaltet die Teilsequenz t
eine potential region, wenn eine Base-Region zu
einer Helix beiträgt in s, aber nicht zu einer Helix in
t c und c´ sind potential regions
Die Teilsequenz t ist eine P-Structure, wenn sie
eine potential region enthält. t ist nicht-triviale PStructure, wenn die potential region zwischen zwei
base-paired regions liegt
Johann-Wolfgang-Goethe Universität, Frankfurt am Main
11/89
3. s ist eine RNA-Sequenz. s ist eine pseudogeknotete Struktur, wenn sie zwei nichtüberlappende P-Strukturen enthält, wobei eine davon
nicht-trivial ist (hier: t1), und beide potential regions
bilden eine Doppelhelix
so können alle RNA-Pseudoknoten definiert werden
Johann-Wolfgang-Goethe Universität, Frankfurt am Main
12/89
Vorhersage von
RNA-Sekundärstrukturen
• Grammatiken nach Chomsky sind ideal um zum
Modellieren von Interaktionen zwischen Nucleotiden
( Stems sind palindromartig)
• Stem Loops kann man mit stochastischen
kontextfreien Grammatiken (SCFG) modellieren
• Pseudoknoten sind aber komplexer als Stem Loops
und würden formal eine kontextsensitive Grammatik
erfordern, was aber Komplexität stark erhöht
Johann-Wolfgang-Goethe Universität, Frankfurt am Main
13/89
Parallele Grammatiken
• zum Vorhersagen von Pseudoknoten werden hier
parallel communicating grammar systems
(PCGS) benutzt
• PCGS besteht aus einer Anzahl an Chomsky
Grammatiken - den Components Gi
• eine Component kann Sequenzen anfragen, die von
anderen Grammatiken erzeugt wurden
• mehrere Components können gleichzeitig anfragen
Johann-Wolfgang-Goethe Universität, Frankfurt am Main
14/89
Parallele Grammatiken
• so kann eine kontextsensitive Struktur, wie ein
Pseudoknoten, durch eine kontextfreie Grammatik
synchronisiert mit einer Vielzahl an regulären
Grammatiken generiert werden
• Stochastische Version von PCGS wird dadurch so
einfach wie bei SCFG
• die (eine) CFG beinhaltet spezielle query symbols
als Nichtterminale für potentielle base-pairing
regions, welche die für Pseudoknoten typische
Doppelhelix formen (einziger Unterschied zu SCFG)
Johann-Wolfgang-Goethe Universität, Frankfurt am Main
15/89
PCGS
• ein PCGS G besteht aus mehr als einer Chomsky
Grammatik G0, G1,..., Gk – den Components
• Grammatik G0 wird Master genannt
• Grammatiken teilen sich Alphabet (Terminale; hier: a,
c, g, u) und Variablen (Nonterminale)
• es gibt zusätzlich spezielle Nonterminale: Query
Symbols – diese sorgen für die Kommunikation
zwischen den Grammatiken
Johann-Wolfgang-Goethe Universität, Frankfurt am Main
16/89
PCGS
• die Ableitung des Systems ist das Umschreiben jeder
Grammatik (Components)
• Synchronisierung zwischen dem Umschreiben der
Components erhält man durch die Query Symbols Qi
• die Sprache, die durch das PCGS schließlich erzeugt
wird, ist eine Menge von Strings, welche die MasterGrammatik G0 erzeugt
Johann-Wolfgang-Goethe Universität, Frankfurt am Main
17/89
PCGS Beispiel
-zeigt die 3 regulären (Hilfs-)Grammatiken G1, G2 , G3
-Synchronisierung zwischen G1 und G2 erhält man
durch die Produktion S1 Q2 , weil dadurch in G2
zuerst abgeleitet wird
Johann-Wolfgang-Goethe Universität, Frankfurt am Main
18/89
PCGS Beispiel
-Beispiel für das parallele Ableiten von den zwei basepaired Regionen acg und cgu
Johann-Wolfgang-Goethe Universität, Frankfurt am Main
19/89
PCGS Beispiel
- zeigt die kontextfreie Master-Grammatik G0
-G0 beschreibt zwei nicht-überlappende P-Structures;
eine davon ist nicht-trivial
Johann-Wolfgang-Goethe Universität, Frankfurt am Main
20/89
PCGS Beispiel
-Ableitungsbaum des PCGS, so dass eine pseudogeknotete Struktur entsteht
Johann-Wolfgang-Goethe Universität, Frankfurt am Main
21/89
PCGS: Stochastische Version
• Wahrscheinlichkeiten mit den Produktionsregeln
jeder Component (Grammatik) der PCGS assoziieren
• am einfachsten durch Definieren einer
Wahrscheinlichkeits-Verteilung für jede Component
als unabhängige SCFG
• die Wahrscheinlichkeit für einen parallen
Ableitungsschritt muss aber die bedingten
Wahrscheinlichkeiten berücksichtigen, die durch die
Kommunikation zwischen Grammatiken entstehen
Johann-Wolfgang-Goethe Universität, Frankfurt am Main
22/89
PCGS: Stochastische Version
• die Wahrscheinlichkeit für einen Pseudoknoten lässt
sich durch die Master-CFG G0 berechnen, wenn die
Wahrscheinlichkeiten für Crossing Helices (Q1, Q2),
die durch Hilfsgrammatiken generiert werden,
bekannt sind:
• Sei S = {a, u, c, g}, G eine PCGS mit m Components.
Dann ist L G
die Menge aller pseudogeknoteten Strukturen die G
generiert
Es gilt:
Johann-Wolfgang-Goethe Universität, Frankfurt am Main
23/89
PCGS: Stochastische Version
• die Wahrscheinlichkeit für die Ableitung S0 * s1r1s2r2s3
• weil die Generierung von r1 und r2 (potential regions)
synchron ist
Johann-Wolfgang-Goethe Universität, Frankfurt am Main
24/89
PCGS: Stochastische Version
• der Produktterm ist die Wahrscheinlichkeit für das
komplementäre Alignment zwischen r1 und r2
• die stochastische Version des PCGS ist somit nur die
stochastische Version der kontextfreien MasterGrammatik G0
• einziger Unterschied zu sonstigen SCFG: die Query
Symbols, die als Nonterminale dazu dienen
Pseudoknoten (Crossing Helices) zu spezifizieren
Johann-Wolfgang-Goethe Universität, Frankfurt am Main
25/89
Automatisierter Algorithmus für
Pseudoknoten-Vorhersage
• Resultat aus PCGS: ein System, das automatisch
einen „Pseudoknoten-Vorhersage-Algorithmus“ für
jede pseudogeknotete Struktur generiert
• zum Modellieren von Crossing Helices,
repräsentiert durch die Query Symbols, benötigt man
eine 5x5 probabilistische Matrix
• diese Matrix beschreibt die WahrscheinlichkeitsVerteilung (der 4 Basen + gap für bulges) in den
Crossing Helices
Johann-Wolfgang-Goethe Universität, Frankfurt am Main
26/89
Automatisierter Algorithmus für
Pseudoknoten-Vorhersage
• basiert auf Dynamischem Programmieren, ähnlich
dem CYK-Algorithmus
• für die Eingabe-Sequenz x[1..n] (eine SCFG in CNF)
berechnet der Algorithmus für jedes Nonterminal X
die maximale Wahrscheinlichkeit für jede Teilsequenz
x[i..j]
• der Algorithmus unterscheidet 3 Kategorien von
Teilsequenzen: stem-loops, Pseudoknoten und PStructures
Johann-Wolfgang-Goethe Universität, Frankfurt am Main
27/89
Automatisierter Algorithmus für
Pseudoknoten-Vorhersage
1.Berechnung für stem-loops folgt dem CYKAlgorithmus
2. Berechnung von Pseudoknoten erfolgt über eine
Hilfsfunktion H , welche für jedes Paar an
Teilsequenzen die maximale Wahrscheinlichkeit
angibt, eine Crossing Helix zu bilden
Johann-Wolfgang-Goethe Universität, Frankfurt am Main
28/89
Exkurs: CYK-Algorithmus
Der Cocke-Younger-Kasami-Algorithmus (CYKAlgorithmus) ist ein Algorithmus, der das
Wortproblem für gegebene kontextfreie Sprachen
effizient löst. Die Sprache muss dazu in Form einer
Grammatik in CNF vorliegen. Laufzeit O(n³)
• Anstatt sofort zu berechnen, ob sich das Wort w der
Länge m aus dem Startsymbol ableiten lässt, wird
zuerst ermittelt, aus welchen Variablen sich
einstellige Teilworte von w ableiten lassen. Danach
wird für alle zweistelligen Teilworte berechnet, aus
welchen Variablen sie sich ableiten lassen.
Johann-Wolfgang-Goethe Universität, Frankfurt am Main
29/89
Automatisierter Algorithmus für
Pseudoknoten-Vorhersage
• für das Nonterminal X wird die maximale
Wahrscheinlichkeit, aus X einen Pseudoknoten x[i..j]
abzuleiten, so berechnet:
- wobei Y und Z Teilsequenzen sind, die potentielle basepairing regions x[h..l] und x[u..v] enthalten
Johann-Wolfgang-Goethe Universität, Frankfurt am Main
30/89
Automatisierter Algorithmus für
Pseudoknoten-Vorhersage
Johann-Wolfgang-Goethe Universität, Frankfurt am Main
31/89
Automatisierter Algorithmus für
Pseudoknoten-Vorhersage
3. die maximale Wahrscheinlichkeit, für das Nonterminal
X eine P-Structure x[k..l] aus der Teilsequenz x[i..j]
abzuleiten, ist so definiert:
- bzw. rekursiv:
Johann-Wolfgang-Goethe Universität, Frankfurt am Main
32/89
Automatisierter Algorithmus für
Pseudoknoten-Vorhersage
Johann-Wolfgang-Goethe Universität, Frankfurt am Main
33/89
Implementierung und Tests
• Laufzeit im worst case: O(n6) für CPU-Zeit / O(n4) für
Speicher (RAM)
• die Eingabe besteht aus der SCFG G0(inklusive
query symbols), die in CNF vorliegen muss, und
einer 5x5 probabilistischen Matrix, welche die
Wahrscheinlichkeiten für das Base-Pairing enthält
• getestet wurden 36 tmRNA Sequenzen (alle
Pseudoknoten vorab bekannt): in 34 Sequenzen
wurde ein Pseudoknoten vorausgesagt, wenn auch
nur in 7 Sequenzen absolut korrekt
(7+18)/36=69%
Johann-Wolfgang-Goethe Universität, Frankfurt am Main
34/89
Zusammenfassung
• stochastisches Modellieren von RNA-Pseudoknoten
durch Parallel Communicating Grammar Systems
(PCGS)
• eine kontextfreie Grammatik synchronisiert mit einer
Anzahl an regulären Grammatiken – kontextsensitive
Regeln vermieden
• dieses Modell erlaubt die automatische Generierung
eines Pseudoknoten-Vorhersage-Algorithmus für jede
spezifische pseudogeknotete Struktur
Johann-Wolfgang-Goethe Universität, Frankfurt am Main
35/89
Zusammenfassung
Algorithmus (ähnlich CYK):
via PCGS
RNAPrimärsequenz
SCFG
G0 in
CNF
und 5x5
Matrix
Für jedes X der SCFG wird
maximale Ws. für
1. stem loop
2. Pseudoknoten
3. P-Structure
berechnet
Ausgabe: RNA-Sekundärstruktur mit
maximaler Wahrscheinlichkeit
Johann-Wolfgang-Goethe Universität, Frankfurt am Main
36/89
Graph-theoretischer Ansatz
• um RNA-Sekundärstrukturen in einer Menge von
funktionell oder evolutionär verwandten Sequenzen
vorherzusagen
• Methode basiert auf dem Vergleich von Stem-Loops
zwischen Sequenzen
• Algorithmus findet Menge von stabilen Stem-Loops,
die in mehreren Sequenzen konserviert vorliegen –
daraus lässt sich Konsensus-Sekundärstruktur
formen
Johann-Wolfgang-Goethe Universität, Frankfurt am Main
37/89
Graph-theoretischer Ansatz
Das generelle Schema dieser Methode:
1.
2.
3.
Finden aller möglichen stabilen Stems in jeder
Sequenz und diese vergleichen mit denen aller
anderen Sequenzen
Finden aller potentiell konservierten Stems, die in
Teilmengen der Sequenzen gemeinsam vorliegen
Zusammenfügen der besten Mengen von
konservierten Stems um eine KonsensusSekundärstruktur zu konstruieren
Johann-Wolfgang-Goethe Universität, Frankfurt am Main
38/89
Graph-theoretischer Ansatz
Johann-Wolfgang-Goethe Universität, Frankfurt am Main
39/89
a) Finden aller stabilen Stems
• Definition stem: palindromische Helix in einer Sequenz,
welche die Basenpaare AU oder GC (oder wobble:GU)
umfasst; mit minimaler Länge von L Basenpaaren
• um Suchraum zu reduzieren werden nur stabile stems
betrachtet
• Evaluieren der Stabilität eines stems durch seine
Stacking-Energie (nach Turner) nur stems mit StackingEnergie niedriger als cutoff E (Default:-5kcal) gelten als
stabil
• Auflisten aller mögliche stems durch einen branch-andbound Algorithmus (Programm dotplot)
Johann-Wolfgang-Goethe Universität, Frankfurt am Main
40/89
b) Vergleichen von Stems über
mehrere Sequenzen
• globales Alignieren von 2 Sequenzen nach
Needleman-Wunsch-Algorithmus, um große
Sequenzähnlichkeit auszunutzen
• im Alignment sucht man nun highly conserved
regions Region ist 10 nt oder länger, mit
mindestens 80% Sequenzidentität
• highly conserved regions dienen als Anker für stemVergleiche
Johann-Wolfgang-Goethe Universität, Frankfurt am Main
41/89
Vergleichen von Stems über
mehrere Sequenzen
• zwei stems von 2 Sequenzen können nur verglichen
werden, wenn die dazu gehörenden 5‘ oder 3‘ halfstems in der gleichen Anker oder Nicht-Anker Region
liegen
Johann-Wolfgang-Goethe Universität, Frankfurt am Main
42/89
Vergleichen von Stems über mehrere
Sequenzen
• zudem dürfen die 5‘ oder 3‘ half-stems in der Anker
Region um maximal 10 nt versetzt sein (in NichtAnker Region keine Constraints)
• wenn nach Alignieren die Ähnlichkeit zwischen 2
Sequenzen nicht groß ist (keine highly conserved
regions ), gilt die ganze Sequenz als Nicht-Anker
Region und somit wird jeder stem der beiden
Sequenzen miteinander verglichen ( erhöht
Laufzeit)
Johann-Wolfgang-Goethe Universität, Frankfurt am Main
43/89
Vergleichen von Stems über mehrere
Sequenzen
• die Funktion S(ix, jy) misst die Ähnlichkeit zwischen zwei
stems i und j aus den Sequenzen x und y
• Ähnlichkeit zwischen zwei Stems anhand von 5
Eigenschaften messbar:
1. Helix-Länge
2. Helix-Sequenz
3. Loop-Sequenz (abgeschlossen durch stem)
4. Stem-Stabilität
5. Relative Positionen des Starts und Ende des stems
Johann-Wolfgang-Goethe Universität, Frankfurt am Main
44/89
Vergleichen von Stems über mehrere
Sequenzen
• S(ix, jy) ist definiert als die gewichtete Summe dieser
5 Ähnlichkeits-Scores, geteilt durch die Summe des
Stabilitäts-Scores der beiden stems (skaliert wurde
mit stability adjusting factor f):
Johann-Wolfgang-Goethe Universität, Frankfurt am Main
45/89
Vergleichen von Stems über mehrere
Sequenzen
• wobei sl(ix, jy) der Ähnlichkeits-Score zwischen den
stems ix, jy ist, bezogen auf eine (aus den 5 möglichen)
spezielle Eigenschaft l
• so berechnet man sl(ix, jy) (außer für Helix oder Loop
Sequenz):
sl(ix, jy) = min{sl(ix), sl(jy)}/max{sl(ix), sl(jy)}
• wl ist das Gewicht für jede Eigenschaft l und liegt
zwischen 0 und 1 (alle 5 Gewichte aufsummiert ergibt
1)
Johann-Wolfgang-Goethe Universität, Frankfurt am Main
46/89
Vergleichen von Stems über mehrere
Sequenzen
• Werte von r liegen zwischen 0 und 1 – je stabiler ein
stem, desto niedriger der r-Wertrx(i)= (ei-e`)/(e``-e`)
• der Wert von S(ix, jy) liegt zwischen 0 und 1 – je
höher der Wert um so wahrscheinlicher, dass 2
stems Instanzen eines konservierten stems sind
• nur die Paare an stems werden als potentiell
eingestuft, für die gilt S(ix, jy) >= S (für einen
Schwellwert S)
Johann-Wolfgang-Goethe Universität, Frankfurt am Main
47/89
conserved stems
• Setzen eines Signifikanz-Levels p (0< p <=1),
welches der minimale prozentuale Anteil aller n
Sequenzen ist, die eine gemeinsame Struktur
besitzen
•
es gilt, die konservierten stems zu finden, die in
mindestens k Sequenzen vorkommen (k = [p * n] )
• das wird erreicht durch n-partite ungerichtete
gewichtete Graphen
Johann-Wolfgang-Goethe Universität, Frankfurt am Main
48/89
n-partite Graphen
• jeder Koten des Graphen repräsentiert einen stem
• der Graph ist unterteilt in n Teile; jeder Teil umfasst
die Anzahl an stems einer Sequenz
• nur Knoten von verschieden Teilen können
verbunden werden
Johann-Wolfgang-Goethe Universität, Frankfurt am Main
49/89
c) n-partite Graphen
• potentiell gleiche stems aus verschiedenen
Sequenzen, die einen Ähnlichkeits-Score größer S
aufweisen, werden verbunden und gewichtet
• in der Graphen-Theorie repräsentiert eine Clique
einen vollständigen Teilgraphen, in dem jeder Knoten
mit allen anderen verbunden ist
• eine Clique ist maximal, wenn sie nicht in einer
größeren Clique enthalten ist ist
Maximierungsproblem und ist NP-vollständig
Johann-Wolfgang-Goethe Universität, Frankfurt am Main
50/89
n-partite Graphen
• das Finden aller potentiell konservierten stems in
mindestens k Sequenzen, entspricht dem Finden
aller maximaler Cliquen der Größe >= k im n-partiten
Graphen (ist NP-hartes Problem)
Johann-Wolfgang-Goethe Universität, Frankfurt am Main
51/89
n-partite Graphen
• besser: enumerativer Algorithmus basierend auf
Depth-First Suche im Graphen
• Input ist der n-partite Graph; Output ist eine Array mit
maximalen Cliquen größer/gleich k
• je größer die Clique, desto ähnlicher sind die stems
zueinander, also wahrscheinlich Instanzen
konservierter stems
Johann-Wolfgang-Goethe Universität, Frankfurt am Main
52/89
n-partite Graphen
• worst-case Laufzeit: O(mn), wobei m die maximale
Anzahl an stems in einer Sequenz ist, und n die
Anzahl an Sequenzen
• aber average case in diesem Algorithmus erzielt
bessere Laufzeit, weil die Eingabe-Graphen meist
geringe Dichte aufweisen (wegen Definition der
Anker-Regionen)
• Cliquen werden nach absteigendem Score
angeordnet – haben zwei Cliquen mehr als 70%
gleiche stems, wird die Clique mit niedrigerem Score
entfernt
Johann-Wolfgang-Goethe Universität, Frankfurt am Main
53/89
d) Zusammenfügen von
konservierten Stems
• jede erhaltene maximale Clique entspricht einer
Menge ähnlicher stems aus verschiedenen
Sequenzen; die Größe der Clique liegt zwischen k
und n
• ein stem block repräsentiert die Menge an stems,
die der maximalen Clique entspricht
• Ziel ist, die bestmögliche Zusammenstellung an stem
blocks zu finden, welche so die Konsensus-Struktur
repräsentieren
Johann-Wolfgang-Goethe Universität, Frankfurt am Main
54/89
Zusammenfügen von konservierten
Stems
Johann-Wolfgang-Goethe Universität, Frankfurt am Main
55/89
e) Zusammenfügen von
konservierten Stems
• man konstruiert einen gerichteten Graphen, in dem
jeder Knoten einem stem block, also einer maximalen
Clique entspricht
• innerhalb einer Sequenz liegt stem s1 vor stem s2 ,
wenn der Helixanfang von s1 vor dem von s2 liegt
• stem s1 und stem s2 sind kompatibel wenn sie in
ihren Helix-Regionen nicht überlappen
Johann-Wolfgang-Goethe Universität, Frankfurt am Main
56/89
Zusammenfügen von konservierten
Stems
- bezogen auf die relative Start- und Endposition, kann
die Anordnung beider stems nur in 3 Mustern erfolgen:
Johann-Wolfgang-Goethe Universität, Frankfurt am Main
57/89
Zusammenfügen von konservierten
Stems
• eine Kante zwischen den stem blocks b1 und b2 im
gerichteten Graphen ist nur möglich, wenn b1 vor b2
und kompatibel ist, sowie eines der 3 möglichen
Muster formt
• diese Kriterien müssen in mindestens einer
kritischen Anzahl c von Sequenzen erfüllt sein
• c ist k oder Hälfte der Anzahl an Sequenzen, die
stems aus einem der blocks besitzen (je nach dem
was größer ist)
Johann-Wolfgang-Goethe Universität, Frankfurt am Main
58/89
f) Maximale Pfade an stem blocks
Johann-Wolfgang-Goethe Universität, Frankfurt am Main
59/89
Maximale Pfade an stem blocks
• Rekursive Depht-First-like-Suche wird auf den
gerichteten Graphen angewendet
• so findet man die maximalen Pfade an stem blocks,
die in mindestens k Sequenzen vorliegen (hier k=4)
• Algorithmus ist ähnlich dem zum Finden maximaler
Cliquen – erster stem block wird inkrementell
erweitert um neuen stromabwärts liegenden stem
block, der mit allen anderen blocks verbunden sein
muss
Johann-Wolfgang-Goethe Universität, Frankfurt am Main
60/89
Maximale Pfade an stem blocks
• jeder maximale Pfad bekommt eine Score, der die
Summer aller Scores der stem blocks im Pfad ist
• diese potentiellen Konsensus-Strukturen werden
entsprechend ihres Scores gelistet und die n-besten
(Default 10) gelten als Kandidaten für KonsensusStruktur
• jede dieser Strukturen wird noch verfeinert: refolding/
internal loops und bulges nun erlaubt
Johann-Wolfgang-Goethe Universität, Frankfurt am Main
61/89
Testergebnisse
Johann-Wolfgang-Goethe Universität, Frankfurt am Main
62/89
Testergebnisse
• Statistische Signifikanz von COMRNA: Vergleichen
von Struktur-Score Verteilung zwischen echten und
random Strukturen
Johann-Wolfgang-Goethe Universität, Frankfurt am Main
63/89
Zusammenfassung
• Graph-theoretischer Ansatz für Vorhersage häufiger
RNA Sekundärstruktur-Muster (KonsensusStrukturen) in einer Menge von Sequenzen
• basierend auf Suchen und Zusammenfügen
konservierter stems ( stems dienen dem
Algorithmus als Vergleichseinheit)
• gute Ergebnisse für bis zu 20 RNA-Sequenzen mit
einer Länge < 300 nt
Johann-Wolfgang-Goethe Universität, Frankfurt am Main
64/89
Zusammenfassung
• der Algorithmus kann große Sequenzähnlichkeit
ausnutzen, indem im Alignment Anker-Regionen
definiert werden – Suchraum für stems wird dadurch
kleiner
• weiterer Vorteil: diese Methode gibt eine Menge an
möglichen Konsensus-Strukturen wieder – weil beste
Struktur nicht der realen entsprechen muss, und es
ist hilfreich um mögliche alternative Strukturen zu
entdecken
• Nachteil: die worst case Laufzeit für maximale
Clique/Path-Algorithmus liegt in NP
Johann-Wolfgang-Goethe Universität, Frankfurt am Main
65/89
Iterated Loop Matching
• eine Sekundärstruktur ist eine Liste von Basenpaaren
• die Basenpaare (i,j) und (k,l) sind kompatibel, wenn
sie juxtaposed (i<j<k<l) oder nested (i<k<l<j) sind
• sonst sind sie inkompatibel, also i<k<j<l
• eine inkompatible Struktur ist eine Pseudoknoten (C,
D)
Johann-Wolfgang-Goethe Universität, Frankfurt am Main
66/89
Iterated Loop Matching
• ein dynamic-programming-Algorithmus, der RNASekundärstruktur inklusive Pseudoknoten
vorhersagen kann
• Algorithmus nutzt thermodynamische und
vergleichende (Covarianz) Information aus, und kann
jeden Typ von Pseudoknoten vorhersagen, in
alignierten und einzelnen Sequenzen
• basiert auf „Loop-Matching-Algorithmus“ nach
Nussinov et al. (1978) ohne Pseudoknoten
Johann-Wolfgang-Goethe Universität, Frankfurt am Main
67/89
Loop-Matching-Algorithmus
• der LM-Algorithmus findet die best-score
Sekundärstruktur ohne Pseudoknoten
• gegeben ist Basepair-Score Matrix B, wobei B(i,j) der
Score für Basenpaarung zwischen der Base i und
Base j ist
• da Sekundärstruktur hier kompatibel sein muss, kann
man sie in kleinere Strukturen unterteilen
Johann-Wolfgang-Goethe Universität, Frankfurt am Main
68/89
Loop-Matching-Algorithmus
• für jede Teilsequenz S[i,j] mit i+1<j gibt es nur 3
Möglichkeiten für Struktur: 1) i ist single-stranded
2) i und j gepaart 3) i und k sind gepaart (i<k<j)
• somit lässt sich der Score einer optimalen
Teilsequenz wie folgt rekursiv berechnen in der
NxN-Matrix Z:
Johann-Wolfgang-Goethe Universität, Frankfurt am Main
69/89
Loop-Matching-Algorithmus
• am Ende des LM-Algorithmus ist Z(1,N) der Score
der optimalen Struktur für Sequenz S[1..N]; durch
traceback in Z erhalten diese Berechnung und
traceback in O(n³) möglich
• im einfachsten Fall ist B(i,j)=1 wenn Base i mit Base j
ein Watson-Crick oder G-U Basenpaar bildet, sonst 0
• LM-Algorithmus findet Sekundärstruktur mit
maximaler Anzahl an Basenpaaren (in diesem Fall)
Johann-Wolfgang-Goethe Universität, Frankfurt am Main
70/89
Iterated-Loop-Matching-Algorithmus
• Erweiterung des LM-Algorithmus, der auch
Pseudoknoten in Vorhersage der Sekundärstruktur
einbezieht
• da Pseudoknoten der Interaktion zwischen 2 LoopRegionen entspricht, kann mal LM–Algorithmus
zweimal darauf anwenden (mehr Iterationen für
kompliziertere Pseudoknoten)
• dieses Vorgehen versagt aber oft in Praxis
Johann-Wolfgang-Goethe Universität, Frankfurt am Main
71/89
Iterated-Loop-Matching-Algorithmus
• die Basen, die für Pseudoknoten vorgesehen waren,
können im ersten Durchlauf des LM-Algorithmus in
falsch-positive Basenpaarungen geraten
• lässt sich so vermeiden: LM-Algorithmus mehrmals
anwenden, und es werden nur Basenpaare mit
höchstem Score akzeptiert
Johann-Wolfgang-Goethe Universität, Frankfurt am Main
72/89
Iterated-Loop-Matching-Algorithmus
1.
Erstellen der Basepair Scorematrix B[1..n][1..n] aus
einer Sequenz oder Sequenzalignment
2.
Aufruf des (basic) LM-Algorithmus; Matrix B wird
dazu benutzt um Matrix Z zu erzeugen; Traceback
in Z, so dass man Basepair-Liste L erhält
3.
Identifiziere alle Helices in L in kombiniere Helices,
die durch internal Loops oder Bulges getrennt sind;
existieren keine Helices, gehe zu Schritt 7.
Johann-Wolfgang-Goethe Universität, Frankfurt am Main
73/89
Iterated-Loop-Matching-Algorithmus
4. jede Helix wird mit Score versehen (ergibt sich durch
Summieren der Scores von Basenpaaren in Helix);
Helix H mit höchstem Score wird in Basenpaar-Liste
S gemischt
5. die Positionen von H werden aus Initialsequenz
genommen; Update von Scorematrix B
6. Schritte 2-5 wiederholen bis keine Basen mehr übrig
sind
7. Ausgabe ist Basenpaar-Liste S; Termination
Johann-Wolfgang-Goethe Universität, Frankfurt am Main
74/89
Iterated-Loop-Matching-Algorithmus
• nicht alle Elemente in Matrix Z müssen in jeder
Iteration neu berechnet werden
• folgende 3 Dreiecke der Matrix müssen nicht neu
berechnet werden: A,B,C (und obere und untere
Dreiecksmatrix symmetrisch hier), nur Teil D neu
In A: i< j< p
In B: p< i< j< q
In C: q< i <j
In D: i< p <j oder i< q <j
Johann-Wolfgang-Goethe Universität, Frankfurt am Main
75/89
Iterated-Loop-Matching-Algorithmus
• die Rekursion um Z neu zu berechnen:
• im Array M sind die Indizes der restlichen
ungepaarten Basenpaare gespeichert
Johann-Wolfgang-Goethe Universität, Frankfurt am Main
76/89
Iterated-Loop-Matching-Algorithmus
• VLOOP_LENGTH beschreibt die minimale virtuelle
Distanz, die zwischen Basenpaaren eingehalten
werden muss nach der ersten Iteration (Default:3)
• M[i] ist die i-te übrige ungepaarte Base, und p und q
(mit p<q) sind die Endpunkte einer Helix aus
vorheriger Iteration
• in der ersten Iteration gilt M[i] = i und p und q sind
noch nicht definiert Rekursion wie in LM
Johann-Wolfgang-Goethe Universität, Frankfurt am Main
77/89
Iterated-Loop-Matching-Algorithmus
• worst case Laufzeit: der LM-Algorithmus nach
Nussinov braucht O(n³); dieser wird m (= Anzahl an
Helices, die Algorithmus vorhersagt) mal wiederholt
• da gilt m <= n/2k, wobei k die minimale Helixlänge ist,
kann der worst case an Laufzeit im ILM-Algorithmus
O(n4) sein
• m ist aber typischerweise klein und Matrix Z muss
nur teilweise neu berechnet werden in jeder Iteration,
daher im average case O(n³)
Johann-Wolfgang-Goethe Universität, Frankfurt am Main
78/89
Base-Pairing Score-Matrix B
• wird hier als Kombination aus mutual Information und
Helix Plot berechnet
• mutual information ist ähnlich der relativen Entropie
(Kullback-Leibler-Distanz)
• gibt an, wie viel Information eine zufällige Variable
(hier Base) über eine andere enthält
• relative Entropie zwischen der gemeinsamen
Verteilung fij(XY) und dem Produkt der einzelnen
Verteilungen
Johann-Wolfgang-Goethe Universität, Frankfurt am Main
79/89
Base-Pairing Score-Matrix B
•
mutual Information: multiples Sequenz-Alignment
von n Sequenzen liegt vor
- fij(XY) gibt an wie oft man Base X an alignierter
Position i findet, und Y an Position j
- mutual-Information-Score zwischen Position i
und j, also Mij wird so berechnet:
Johann-Wolfgang-Goethe Universität, Frankfurt am Main
80/89
Base-Pairing Score-Matrix B
• Helix Plot Score: Helix Plots sind Mittelwerte von
Basepair-Scores – sie kombinieren phylogenetische
und thermodynamische Information
• für jede Sequenz im multiplen Alignment wird eine
Score-Matrix erstellt
• indem Watson-Crick- oder Wobble-Basenpaare good-pair
Scores bekommen (=1) und andere Basenpaare bad-pair
Scores (=2)
• Penalty Scores für gaps (=3)
• für lange Helices gibt es Bonus Scores/ für zu kurze auch badpair Score
• alle Score-Matrizen werden addiert zu einer ScoreMatrix
Johann-Wolfgang-Goethe Universität, Frankfurt am Main
81/89
Base-Pairing Score-Matrix B
• mutual Information und Helix Plot Scores werden
addiert um die Score-Matrix B zu generieren, die ILM
schließlich nutzt:
a und b sind relative Gewichtungen für mutual
Information und Helix Plot Scores
• HPij ist Helix Plot Score eines potentiellen
Basenpaares; N ist Anzahl der Sequenzen im
Alignment
Johann-Wolfgang-Goethe Universität, Frankfurt am Main
82/89
Base-Pairing Score-Matrix B
• Extended Helix Plot Scores: wenn nur eine einzige
Sequenz statt eines Alignments vorliegt, und somit
keine Covarianz
• dann kann es natürlich keine mutual Information (also
wechselseitige Beziehung zwischen Sequenzen)
geben und nur der Helix Plot Score ist nicht
ausreichend
• somit wird hier noch die Faltungs-Thermodynamik
der RNA miteinbezogen
Johann-Wolfgang-Goethe Universität, Frankfurt am Main
83/89
Base-Pairing Score-Matrix B
• hier hängt der good-pair Score GPij vom Typ des
Basenpaares ab (G-C 80, A-U 50, G-U 30)
• der Helix-Bonus BONUSij ist proportional zur
Stacking-Energie der Helix:
Johann-Wolfgang-Goethe Universität, Frankfurt am Main
84/89
Ergebnisse und Tests
• 2 Testreihen wurden durchgeführt: a) ILM und MWMAlgorithmus testen mit alignierten homologen
Sequenzen, mit Kombination aus Helix Plot und
mutual Information als Score
• b) die drei Algorithmen ILM, PKNOTS und MWM
testen mit Menge von einzelnen Sequenzen; als
Score Extended Helix Plot
• Sensitivität: TP/EP
Genauigkeit: TP/(TP+FP)
Johann-Wolfgang-Goethe Universität, Frankfurt am Main
85/89
Ergebnisse und Tests
• in 8 – 12 homologen Sequenzen identifiziert ILM
mehr als 90% der Basenpaare in kurzen Sequenzen
(<300nt), und in mittleren Sequenzen ca. 80%
• ILM sagte alle Pseudoknoten in alignierten
Sequenzen korrekt voraus (nur ein weitreichender
Pseudoknoten in rRNA verfehlt)
Johann-Wolfgang-Goethe Universität, Frankfurt am Main
86/89
Ergebnisse und Tests
• ILM hat in der Menge von einzelnen Sequenzen alle
Basenpaare (außer für TMV-3´-end) korrekt
vorhergesagt; einen Pseudoknoten des TMV sowohl
up- als auch down-stream nicht erkannt
Johann-Wolfgang-Goethe Universität, Frankfurt am Main
87/89
Zusammenfassung
• der Iterated-Loop-Matching Algorithmus ist
Erweiterung des LM-Algorithmus nach Nussinov
• basiert auf folgendem Prinzip: iteratives Vorhersagen
einer nicht-pseudogeknoteten Struktur (Initialschritt),
daraus Auswählen der wahrscheinlichsten Helix,
diese aus Sequenz entfernen
• LM-Algorithmus wieder auf diese verkürzte Sequenz
anwenden bis keine Basen mehr vorhanden sind
oder keine Helices mehr gefunden werden
Johann-Wolfgang-Goethe Universität, Frankfurt am Main
88/89
Vergleich der 3 Methoden
• Stochastisches Modellieren durch parallele
Grammatiken : Vorhersage der Sekundärstruktur nur
für eine Sequenz; für alle Typen an Pseudoknoten;
Laufzeit O(n6) im worst case
• Graph-theoretischer Ansatz: Vorhersage der
Sekundärstruktur nur für Alignment von Sequenzen;
die worst case Laufzeit für maximale Clique/PathAlgorithmus liegt in NP
• Iterated Loop Matching: Vorhersage der
Sekundärstruktur für Alignment von Sequenzen und
für einzelne Sequenz; für alle Typen an
Pseudoknoten; Laufzeit O(n4) im worst case
Johann-Wolfgang-Goethe Universität, Frankfurt am Main
89/89