Infoblatt 10 Augenbeteiligung - Tuberöse Sklerose Deutschland eV
Werbung

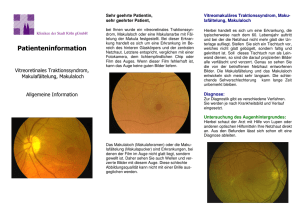
Tuberöse Sklerose Deutschland e. V. www.tsdev.org I N F O R M AT I O N S B L AT T 1 0 Augenbeteiligung bei Tuberöse Sklerose Complex (TSC) 1. Einleitung Campbell, ein Schüler des französischen Erstbeschreibers der Erkrankung Bourneville, beschrieb 1905 erstmals eine Augenbeteiligung bei TSC. 15 Jahre später wurden durch den Augenarzt van der Hoeve die Veränderungen am Auge genauer untersucht und diskutiert. Er stellte fest, dass die Auswirkungen der Erkrankung vor allem im Bereich der Retina (Netzhaut) und des Nervus opticus (Sehnerv) zu finden sind. Man weiß heute, dass nahezu die Hälfte der TSC-Patienten eine Augenbeteiligung aufweist. Papille 2. Einführung in die Anatomie des Auges Die Netzhaut ist die innerste der drei zwiebelschalenartig den Augapfel umschließenden Hüllen. Durch das Sehloch, die sogenannte Pupille, fällt das Licht auf die Netzhaut. Der Lichtstrahl passiert dabei Hornhaut, Linse und Glaskörper, also die brechenden Elemente des Auges. Die Netzhaut enthält Nerven- und Sehsinneszellen. Zu den Sehsinneszellen zählen Stäbchen und Zapfen. Die Stäbchen werden für das Sehen bei Dämmerung, die Zapfen für das Farbensehen verwendet. Durch den Lichteinfall zerfällt ein bestimmter Sehfarbstoff in den Sinneszellen. Durch den Zerfall dieses Sehpurpurs wird Energie freigesetzt. Die Nervenzellen verarbeiten diese energetischen Potenziale und leiten sie über den Sehnerv, der rückseitig am Auge austritt, an die Sehbahn zum Sehzentrum des Großhirns weiter. Hier befindet sich die zentrale Datenverarbeitung, wo die Einzeleindrücke gesammelt und zu einem sinnvollen Bild zusammengesetzt werden. Anschrift Tuberöse Sklerose Deutschland e. V. Vereinsbüro Im Brückfeld 15 65207 Wiesbaden Kontakt Tel. 0611/469-2707 Fax 0611/469-2708 eMail [email protected] www.tsdev.org Das Schema wurde den Wikimedia Commons entnommen. Als Autor ist dort TALOS verzeichnet. 3. Krankhafte Veränderungen am Auge bei Patienten mit TSC 3.1 Angiofibrome der Augenlider Die TSC-typischen Angiofibrome des Gesichts sind gutartige Tumoren, die aus Gefäßen und Bindegewebe bestehen. Bei TSC-Patienten zeigt sich meist eine charakteristische Schmetterlingsverteilung dieser Tumoren. Auch die Augenlider können mit betroffen sein. 2001 zeigte eine britische Studie, dass 39 % der untersuchten TSC-Patienten Angiofibrome an den Augenlidern aufwiesen (Rowley et al.). Aufgrund der Spendenkonten Sparkasse Ettlingen BLZ 660 512 20 , Konto 123 54 64 Commerzbank Frankfurt BLZ 500 400 00, Konto 33 90 33 300 1 Mitgliedschaften des TSD e.V. Kindernetzwerk e.V. Allianz Chronischer Seltener Erkrankungen (ACHSE) Tuberous Sclerosis International (TSI) Tuberous Sclerosis Europe (TSE) Gutartigkeit der Tumoren ist eine Entfernung jedoch nicht notwendig. 3.2 Pigmentstörungen der Netzhaut Die bereits genannte britische Studie und eine Studie in den USA konnten darstellen, dass ebenfalls 39 % der TSC–Patienten Pigmentstörungen der Netzhaut im Sinne einer Minderpigmentierung haben (Franz 2004). Durch die vermindert pigmentierten Netzhautareale entsteht der Eindruck, die Netzhaut sei von ausgestanzten Löchern durchsetzt. Es besteht eine Ähnlichkeit zu den weißen Flecken der Haut, den sogenannten „white spots“ bei TSC-Patienten. Punktuell vermehrt pigmentierte Netzhautareale können ebenfalls vorkommen. Allerdings ist unklar, ob diese Veränderungen für TSC spezifisch sind. Es handelt sich dabei wahrscheinlich um eine angeborene Hypertrophie (Vergrößerung) des Pigmentepithels der Netzhaut. Beide Veränderungen sind harmlos und müssen nicht therapiert werden. 3.3 Hamartome der Netzhaut und des Sehnerven Häufiger sind die Hamartome der Netzhaut oder des Sehnerven, die sogenannten retinalen Hamartome. Sie kommen bei 30 bis 50 % der TSC-Patienten vor und ähneln sehr den Tuberomen des Gehirns. Es gibt drei Arten von Hamartomen: Nichtverkalkte Hamartome, verkalkte Hamartome und Mischtypen. Bei vielen TSC- Patienten ist mehr als ein Typ nachweisbar. Werden retinale Hamartome sehr früh im Baby- oder Kleinkindesalter entdeckt und sind noch keine klassischen Symptome der Tuberösen Sklerose auffällig, können sie bereits ein wichtiger Hinweis auf TSC sein. Retinale Hamartome können schon vor der Geburt auftreten. Retinaler Maulbeertumor, Foto: GE Lang, Augenklinik Erlangen 2 Hamartome sind ebenso wie die Angiofibrome gutartige Tumoren. Sie wachsen nicht zerstörerisch und bilden auch keine Metastasen (Tochtergeschwülste). Im Gegensatz zu den Angiofibromen der Lider kann man die Hamartome von außen aber nicht erkennen. Nur der Augenarzt kann über eine weit getropfte Pupille in das Innere des Auges schauen und dort den Fundus (Augenhintergrund) und somit die Netzhaut, den Sehnerv und die durch die Netzhaut schimmernde Aderhaut beurteilen. Man nennt diese Untersuchung auch Augenspiegelung oder Ophthalmoskopie. Da der Patient bei der Untersuchung bestimmten Aufforderungen nachkommen muss, kann die Spiegelung des Augenhintergrunds bei geistig behinderten oder unkooperativen Patienten schwierig werden. Bei einem guten, einfühlsamen Augenarzt ist aber nur selten eine Sedierung, also eine medikamentöse Ruhigstellung des Patienten, notwendig. Bei 34 – 50 % der TSC-Patienten kommen die Hamartome bilateral, also in beiden Augen, vor. Glücklicherweise verursachen Hamartome kaum einen Verlust der Sehkraft. Somit ist eine Behandlung der Hamartome nur selten angezeigt. • Nicht verkalkte Hamartome In der Augenspiegelung stellen sich nicht verkalkte Hamartome als ovale, leicht erhabene, transparente und gut umschriebene Strukturen dar. Sie sind reich an Gefäßen und kommen einzeln oder auch gehäuft vor. TSC-Patienten mit Hamartomen weisen zu 70 % nicht verkalkte Hamartome auf. Eine 2007 veröffentlichte Studie stellte einen Zusammenhang der Hamartome zum männlichen Geschlecht und zu einer TSC2Mutation her (Au et al.). • Verkalkte Maulbeerhamartome Diese Veränderungen werden bei 50 % der TSCPatienten mit Hamartomen nachgewiesen. Sie sind durch den Augenarzt relativ leicht am Augenhintergrund zu erkennen. Aufgrund der erhabenen, bläschenartigen oder knötchenhaften Erscheinung ähneln diese Hamartome Kaviar oder Maulbeeren. Sie können mehrere Millimeter dick werden und sind reich an Calcium. So können sie entsprechend röntgenologisch wie auch sonografisch, also mittels Ultraschall gut identifiziert werden. Maulbeerhamartome treten gewöhnlich in der Peripherie (Umfeld) des Augenhintergrunds auf, sind aber auch zentral zu finden. Gelegentlich können Gefäße die knotigen Strukturen durchziehen. Es gibt Beobachtungen darüber, dass sich die kleinen Bläschen der Maulbeerhamartome in den Glaskörper des Auges entleeren und sich auch wieder füllen können. Der gallertige Glaskörper besteht nahezu nur aus Wasser (Hydrogel) und erhält die Form des Augapfels. Er hat außerdem die Aufgabe, die lichtempfindliche Netzhaut zu schützen. Gelangt nun blutiges Sekret in den Glaskörper, kann es vorübergehend zu einer Sehstörung kommen. Sinneszellen durchsetzte Netzhaut geschädigt wird. Dieser Schaden kann zusätzlich zu einem partiellen Sehverlust führen. Die Behandlungsmethode muss also genau gegen den Begleitschaden an der Netzhaut abgewogen werden. Hamartomatöses Riesenzellenastrozytom der Netzhaut, Foto: U. Mayer, Augenklinik Erlangen • Mischtyp Dieser Typ weist charakteristische Merkmale beider bereits genannten Hamartomarten auf. Zum einen ist die Peripherie des Tumors flach und durchscheinend, zum anderen ist sein Zentrum verkalkt und knötchenhaft verändert. Der Mischtyp wird bei 10 % der TSC-Patienten mit Hamartomen nachgewiesen. Wie bereits erwähnt, führen Hamartome kaum zu Beeinträchtigungen des Sehens. In seltenen Fällen kann es bei den Hamartomen aber auch zu Komplikationen kommen. Die gutartigen Tumoren weisen eine gute Durchblutung auf, sodass sie bluten, sehr stark anwachsen oder auch absterben können. Folge dieser Komplikationen kann eine Sehbeeinträchtigung bis zur Erblindung sein. Hamartome, die zentral in der Nähe des Sehnervs gelegen sind, haben eine höhere Wachstumstendenz als Hamartome, die in der Peripherie zu finden sind. Die meisten Komplikationen oder Symptome treten bei den nicht verkalkten Hamartomen auf. Werden Komplikationen bemerkt, ist eine engmaschige Verlaufskontrolle notwendig. Eine Sehverschlechterung im Rahmen der Komplikation, hervorgerufen durch Flüssigkeitsansammlungen unter der Netzhaut, Abhebung der Netzhaut von der ernährenden Aderhaut oder Vergrößerung des Hamartoms im Bereich des Gefäß-Nervenaustritts, kann sich nach ein paar Wochen spontan auflösen. Wenn die Sehbeeinträchtigung jedoch anhält muss eine Behandlung in Betracht gezogen werden. Dazu ist eine genaue Diagnostik notwendig. In diesem Fall ist eine Gefäßdarstellung mit Fluorescein hilfreich. Wird ein „Leck“ in einem Gefäß des Tumors entdeckt, kann der Defekt mit Laserphotokoagulation verschlossen werden. Die Blutung wird damit gestoppt. Handelt es sich um ein sehr großes, wachsendes Hamartom, können die den Tumor ernährenden Gefäße ebenfalls mit Hilfe der Laserphotokoagulation verschlossen werden. Der Tumor wird dadurch nicht mehr ernährt und stirbt ab. Bei dieser Therapiemöglichkeit besteht allerdings die Gefahr, dass auch die benachbarte, mit Nerven und Eine andere Methode „bedrohliche“ Hamartome zu verkleinern oder ganz zu eliminieren ist die photodynamische Therapie (PDT). Dabei handelt es sich um ein Verfahren, bei dem Licht in Kombination mit einer lichtempfindlichen Substanz (Photosensibilisator) zum Einsatz kommt. Der Sensibilisator wird dem Patienten verabreicht und reichert sich selektiv im Tumorgewebe an. Nach einer gewissen Zeit wird der Tumor mit Licht einer bestimmten Wellenlänge bestrahlt. Durch photophysikalische Prozesse entstehen giftige Substanzen, die den mit dem Sensibilisator angereicherten Tumor gezielt zerstören. Vorteil des Verfahrens ist die relativ geringe Laserleistung. Dadurch können die Netzhaut und die darin befindlichen Nerven- und Sehsinneszellen geschont werden. Die bisherigen Ergebnisse zeigen, dass dieses Verfahren das Mittel der ersten Wahl werden könnte. 4. Weitere Ursachen für Sehbeeinträchtigung oder Erblindung bei TSC Eine Sehbeeinträchtigung oder Erblindung im Zusammenhang mit TSC ist selten. Neben den bereits beschriebenen Komplikationen der Hamartome gibt es noch einige andere ursächliche Möglichkeiten, die eine Beeinträchtigung des Sehens zu Folge haben können: • Ein Tumor im Gehirn drückt auf das Sehzentrum des Großhirns, also den Ort der Datenverarbeitung der visuellen Sinneseindrücke. • Tumorwachstum im Gehirn führt zu einem erhöhten Hirndruck mit der Folge der Kompression und Schädigung des Nervus opticus (Sehnerv). • Es kommt zu einer Einblutung in den Glaskörper. Die Sehminderung ist aber nur vorübergehend, da das Blut mit der Zeit wieder resorbiert wird. Eine Abnahme der Sehkraft oder Gesichtsfeldausfälle können aber auch infolge einer ärztlichen Maßnahme hervorgerufen werden. Bei schwersten Fällen von Epilepsie kann beispielsweise ein neurochirurgisches Verfahren zur Anwendung kommen, das man als Hemisphärektomie bezeichnet. Dabei wird eine Hirnhälfte entfernt. Folge sind Lähmungen und Gesichtsfeldeinschränkungen. Unter dem Gesichtsfeld versteht man alle zentralen und peripheren Punkte des Außenraums, die bei ruhiger Kopfhaltung und geradeaus gerichtetem Blick visuell wahrgenommen werden können. Beim Erwachsenen beträgt beispielsweise die horizontale Ausdehnung des Gesichtsfelds beider Augen 180°. Mit Hilfe der Perimetrie kann das Gesichtsfeld genau vermessen werden. 3 Ebenso führen einige Medikamente zu Gesichtsfeldausfällen. Vigabatrin (Sabril ®) wird gerade bei TSC-Patienten u. a. zur Behandlung der BNS-Anfälle bevorzugt eingesetzt. Vigabatrin verursacht jedoch signifikant bei 1/3 – 1/4 der Patienten (mit oder ohne TSC) Gesichtsfeldeinschränkungen, die sich nicht wieder zurückbilden. Der Gesichtsfelddefekt stellt sich meist als konzentrische Einengung oder als ringförmiger, der Nase zugewandter Defekt dar. Es gibt Hinweise dafür, dass das Auftreten von Gesichtsfeldeinschränkungen von der Gesamtdauer der Behandlung und der kumulativen Dosis von Vigabatrin (also der Anhäufung des Wirkstoffs) abhängig ist. Vor Beginn einer Vigabatrinbehandlung und im Verlauf müssen deshalb regelmäßige augenärztliche Kontrollen erfolgen! 5. Ähnliche Krankheitsbilder (Differentialdiagnosen) Es gibt TSC-Patienten, die als einziges klinisches Zeichen Hamartome der Netzhaut aufweisen. Zu dieser Gruppe gehören vor allem Neugeborene und Kinder, bei denen sich andere Symptome evtl. erst nach Jahren zeigen. Der Nachweis einer Vielzahl von knotig veränderten Hamartomen der Netzhaut ist charakteristisch für TSC und gehört zu den diagnostischen Hauptkriterien der Erkrankung. Allerdings ist zu beachten, dass die Hamartome der Netzhaut bei TSC-Patienten den Veränderungen der Patienten mit Neurofibromatose gleichen. Das heißt also, dass die Diagnose TSC dann vorsichtig zu stellen ist, wenn Hamartome zunächst die einzigen klinischen Veränderungen darstellen. Eine entsprechende Diagnostik mit Untersuchung der Familienangehörigen ist dann anzuschließen. Maulbeerhamartome können auch mit Einlagerungen in der Netzhaut verwechselt werden, den sogenannten Drusen, die keinen Krankheitswert haben. Maulbeerhamartome erkennt man im Vergleich zu Drusen daran, dass sie die Gefäße der durchscheinenden Aderhaut gerne verdecken. Ebenso können diese Hamartome, vor allem wenn sie peripher in der Netzhaut gelegen sind, leicht mit dem bösartigen und im Kindesalter vorkommenden Retinoblastom verwechselt werden. Bei einem zweifelhaften Befund ist deshalb eine wöchentliche Verlaufskontrolle notwendig. Das Retinoblastom wächst immens schnell, da es bösartig ist. Das Maulbeerhamartom ist gutartig und weist keine oder nur eine geringe Wachstumstendenz auf. 6. Therapie In den meisten Fällen ist bei den TSC-typischen Läsionen keine Therapie notwendig, da eine Zunahme oder Änderung des Befundes selten zu beobachten ist. Bei Patienten mit sehr großen Netzhauthamartomen, fortschreitendem Tumorwachstum oder Veränderungen, die sekundär in den Glaskörper oder die Netzhaut einbluten, sind allerdings sorgfältige und regelmäßige Kontrollen notwendig. Insbesondere sollten neue oder sich verändernde Läsionen mittels Angiografie (Gefäßdarstellung mit einem fluoreszierenden Farbstoff) untersucht werden. Erst dann ist eine entsprechende Therapie, wie bereits oben erwähnt, einzuleiten. Bei der Diagnose eines Papillenödems ist sofort der Neurologe zu informieren, da unbedingt ein erhöhter Hirndruck ausgeschlossen werden muss. Die Papille ist der Kopf des Sehnervs. Bei erhöhtem Hirndruck kommt es zu Wassereinlagerungen um den Sehnerv. Dadurch wird die Papille in das Augeninnere vorgewölbt. Diese Veränderung kann mittels Spiegelung des Augenhintergrundes festgestellt und gemessen werden. Dabei bemerkt der Patient anfangs noch keine Sehverschlechterung. Weitere Hirndruckzeichen sind Erbrechen, Mattigkeit, Kopfschmerzen, verlangsamter Herzschlag, hoher Blutdruck, aktuelle Auffälligkeiten im Verhalten und eventuell eine erhöhte Anfallsfrequenz. Ein erhöhter Hirndruck kann durch eine Hirnblutung, einen stetig wachsenden Hirntumor oder durch eine Abflussbehinderung des Hirnwassers, z. B. durch ein Riesenzellastrozytom, einer Komplikation der Tuberösen Sklerose, verursacht werden. Damit es nicht zu einer Hirnschädigung durch Kompression des Hirngewebes kommt, ist eine rasche operative Entlastung notwendig. Weitere Informationen über Riesenzellastrozytome können in dem Informationsblatt „Subependymale Riesenzellastrozytome (SEGA) bei Tuberöse Sklerose (TSC)“ nachgelesen werden. Bemerkung: Ein erhöhter Hirndruck, hat nichts mit einem erhöhten Augeninnendruck (Glaukom/Grüner Star) zu tun, der durch einen mangelnden Abfluss des Kammerwassers im Auge bedingt ist. Beide Erkrankungen können aber zur Erblindung führen und machen anfänglich keine Sehverschlechterung. 7. Empfehlung für TSC-Patienten Nach Diagnosestellung der Tuberösen Sklerose sollte relativ zeitnah eine augenärztliche Untersuchung erfolgen. Danach sind regelmäßige augenärztliche Kontrollen erforderlich. Im Säuglings- und Kleinkindalter wird eine halbjährliche Untersuchung empfohlen, da das Auge in dieser Zeit einer raschen Entwicklung unterworfen ist. Danach sind jährliche Kontrollen angemessen. Betroffene und ihre Eltern sollten daher einen Augenarzt ihres Vertrauens suchen. Bei behinderten Kindern ist ein Arzt mit Erfahrung in der Kinderophthalmologie zu empfehlen. Diese Ärzte sind in der Regel Augenärzte, die mit einer/m Orthoptistin/ en zusammenarbeiten. Dabei handelt es sich um eine 4 Fachkraft, die geduldig Sehvermögen und Funktionen der Augen überprüfen kann. So sind auch bei behinderten Kindern die notwendigen Untersuchungen in der Regel erstaunlich gut durchführbar. 8. Allgemeine augenärztliche Untersuchung bei Kindern mit Behinderung Grundsätzlich sollte im Kindesalter die Funktionstüchtigkeit der Augen untersucht werden. Viele Störungen wie z. B. Weit- oder Kurzsichtigkeit, Hornhautverkrümmung und Strabismus (Schielen) werden nicht bemerkt. Schwächen der Augenmuskeln im Zusammenspiel der Augen und der fließenden Augenbewegungen können ständig gestört und damit sehr anstrengend sein. Raum für eigene Notizen: 5 Beim Kinderarzt fallen solche leichten Sehstörungen zunächst nicht auf, haben aber für ein ungestörtes Blick- und Fixierverhalten hohes Störpotenzial. Die Kinder suchen wenig Blickkontakt zu Personen und können sich schlecht auf etwas konzentrieren (Blickhalteschwäche). Für das Lernen und Begreifen sind das sehr ungünstige Störfaktoren, die gerade Kindern mit Behinderung erspart werden sollten. Feinmotorik und Zielgenauigkeit sind schon bei geringer Sehbeeinträchtigung auffällig. Auch wenn Sie keinen unmittelbaren Verdacht auf eine Sehbeeinträchtigung Ihres Kindes haben, scheuen Sie sich nicht zu einem Augenarzt zu gehen, der mit Kindern Erfahrung hat. Sehr hilfreich ist auch die Vorstellung bei einer/m Orthoptistin/en (Sehschule). Dort werden teilweise noch weiterreichendere Untersuchungen durchgeführt. Weiterführende Literatur Autorin Franz, D.N. (2004) Non-neurologic manifestations of tuberous sclerosis complex. J.Child Neurol., 19, 690698 Dr. med. Carmen Gallitzendorfer Bundesvorstandsmitglied des TSD e. V. Rowley, S.A., O`Callaghan, F.J. and Osborne, J.P. (2001) Ophthalmic manifestations of tuberous sclerosis: a population based study. Br. J. Ophthalmol. 85, 420423 Au, K.S. et al (2007), Genotype/phenotype correlation in 325 individuals referred for a diagnosis of tuberous sclerosis complex in the Unitet States. Genet. Med., 9, 88-100 Lektorat Sandra Hoffmann Grafik & Layout Sandra Welz Kwiatkowski D.J., Whittemore V.H. Thiele E.A. (Hrsg.) (2010) Tuberous Sclerosis Complex. Genes, Clinical features, and Therapeutics. Wiley-Blackwell Weinheim, Deutschland www.neuropaediatrie.com Danksagung TSD e. V. dankt Frau Christine Paul, Orthoptistin in Ravensburg, für ihre Anregungen und ihre Unterstützung bei der Aktualisierung dieses Informationsblattes. Mit freundlicher Unterstützung der 6 Rechtlicher Hinweis: Mit den Infoblättern des Tuberöse Sklerose Deutschland e.V. werden Basisinformationen für Betroffene, deren Angehörige und weitere Kontaktpersonen bereitgestellt. Sie sollen Hilfestellung im Umgang mit der Erkrankung geben und zur weiteren Aufklärung hierüber beitragen. Die Informationen berücksichtigen den jeweils aktuellen Stand der Wissenschaft und werden regelmäßig aktualisiert. Ungeachtet dessen sind sie kein Ersatz diagnostischer und / oder therapeutischer Maßnahmen durch den Facharzt und sollten keinesfalls Anlass für eine eigenmächtige Veränderung oder den Abbruch ärztlicher Verordnungen sein. Dies kann zu lebensbedrohlichen Situationen führen! Die Informationsblätter wollen auch nicht für einzelne Personen und / oder Institutionen werben oder Ratschläge erteilen. Eine Weitergabe des Informationsblattes an den behandelnden Arzt ist sinnvoll und erwünscht. Soweit in einzelnen Informationsblättern auf Links verwiesen wird, welche nicht vom Verfasser stammen, distanziert sich dieser ausdrücklich und erklärt, dass ein rechtsgeschäftlicher Wille mit der Bereitstellung solcher Verweise nicht verbunden ist. Stand: 08.08.2010