ZIP - FreiDok plus
Werbung

Quasiklassischer Ansatz
für
volldifferentielle
Ionisationsquerschnitte
INAUGURAL-DISSERTATION
zur
Erlangung des Doktorgrades
der
Fakultät für Physik
der
Albert-Ludwigs-Universität
Freiburg im Breisgau
vorgelegt von
Tihamér Geyer
aus
Aachen
2000
Dekan:
Leiter der Arbeit:
Referent:
Korreferent:
Prof. Dr. K. Königsmann
Prof. Dr. J.-M. Rost
Prof. Dr. J.-M. Rost
Prof. Dr. A. Blumen
Tag der Verkündigung des Prüfungsergebnisses: 18. Juli 2000
Übrigens , sagte IIa schließlich.
Was bedeutet Quantum? IIb zuckte mit den Achseln.
Es
bedeutet, daß man eine weitere Null
hinzuf ügen muß. Oh , murmelte IIa. Das ist alles? Terry Pratchett
Inhaltsverzeichnis
1 Warum klassisch?
1
2 Bisherige klassische Rechnungen
2.1 Wie alles begann . . . . . . . . . . . . . . . . .
2.2 Verbesserungen/-änderungen . . . . . . . . . .
2.2.1 Energieabhängige Anfangsverteilungen
2.2.2 Die Wignerverteilung in der CTMC . .
2.2.3 Zusatzpotentiale . . . . . . . . . . . .
2.2.4 Modifikationen der Propagation . . . .
2.3 Zusammengefasst . . . . . . . . . . . . . . . .
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
5
5
7
7
7
8
9
10
3 Der quasiklassische Ansatz
3.1 Das System
. . . . . . . . . . . . . . . . . .
3.2 Dreiteilung des Streuvorgangs . . . . . . . . . . . . . .
3.3 Wiederholung der Standard–CTMC . . . . . . . . . . .
3.4 Die Anfangsverteilung . . . . . . . . . . . . . . . . . .
3.4.1 Korrespondenz und Phasenraumverteilungen . .
3.4.2 Das Wigner–Weyl–Bild der Quantenmechanik .
3.4.3 Die klassisch genäherte Anfangsverteilung . . .
3.4.4 Vorteile und Probleme . . . . . . . . . . . . . .
3.4.5 Anders motivierte Anfangsverteilungen . . . . .
3.4.6 Das Projektil . . . . . . . . . . . . . . . . . . .
3.4.7 Die komplette Anfangsverteilung . . . . . . . .
3.5 Die Propagation . . . . . . . . . . . . . . . . . . . . . .
3.5.1 Der Ansatz nach Møller . . . . . . . . . . . . .
3.5.2 Die Propagation im Wignerbild . . . . . . . . .
3.5.3 Zur Gültigkeit des Møllerformalismus . . . . . .
3.6 Die Auswertung . . . . . . . . . . . . . . . . . . . . . .
3.6.1 Der Endzustand . . . . . . . . . . . . . . . . . .
3.6.2 Wirkungsquerschnitte . . . . . . . . . . . . . .
3.7 Vergleich mit bisherigen Ansätzen . . . . . . . . . . . .
3.7.1 Die mikrokanonische Anfangsverteilung . . . .
3.7.2 Die Propagation in der CTMC . . . . . . . . . .
3.7.3
Cut–off–Wigner“ und Cohens Energieverteilung
”
3.7.4 Zur numerischen Implementation des Ansatzes .
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
12
12
13
13
14
15
18
20
21
22
24
25
25
25
27
29
29
29
32
36
36
38
38
39
i
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
INHALTSVERZEICHNIS
ii
3.8
4
5
6
7
Abschließende Bemerkungen . . . . . . . . . . . . . . . . . . . . . . . . .
3.8.1 Grenzen der Phasenraum–Darstellung . . . . . . . . . . . . . . . .
3.8.2 Anregung in der klassischen Beschreibung . . . . . . . . . . . . .
Erweiterung auf Mehrelektronentargets
4.1 Das (klassische) Heliumatom . . . .
4.2 Erweiterungen des Formalimus . . .
4.2.1 Anfangsverteilung . . . . .
4.2.2 Propagation . . . . . . . . .
4.2.3 Auswertung . . . . . . . . .
4.3 Anmerkungen . . . . . . . . . . . .
39
39
41
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
43
43
43
44
45
45
47
Rechnungen an Wasserstoff
5.1 Die verwendeten Phasenraumverteilungen . . . .
5.1.1 Bindungsenergie und Energiebreite . . .
5.1.2 Radiale Orts- und Impulsverteilungen . .
5.2 Betrachtete Querschnitte . . . . . . . . . . . . .
5.2.1 Ein technischer Einschub zur Auswertung
5.3 Verlauf des totalen Querschnitts . . . . . . . . .
5.4 Einschußenergie 250eV . . . . . . . . . . . . . .
5.4.1 Totaler Ionisationsquerschnitt . . . . . .
5.4.2 Einfach differentiell . . . . . . . . . . .
5.4.3 Zweifach differentiell . . . . . . . . . . .
5.4.4 Voll differentiell . . . . . . . . . . . . .
5.4.5 Zusammenfassung . . . . . . . . . . . .
5.5 Einschußenergie 54,4eV . . . . . . . . . . . . .
5.5.1 Totaler Querschnitt . . . . . . . . . . . .
5.5.2 Einfach differentiell . . . . . . . . . . .
5.5.3 Zweifach differentiell . . . . . . . . . . .
5.5.4 Voll differentiell . . . . . . . . . . . . .
5.5.5 Zusammenfassung . . . . . . . . . . . .
5.6 Einschußenergie 17,6eV . . . . . . . . . . . . .
5.6.1 Totaler Querschnitt . . . . . . . . . . . .
5.6.2 Einfach differentiell . . . . . . . . . . .
5.6.3 Zweifach differentiell . . . . . . . . . . .
5.6.4 Voll differentiell . . . . . . . . . . . . .
5.6.5 Zusammenfassung . . . . . . . . . . . .
5.7 Die Ergebnisse im Überblick . . . . . . . . . . .
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
49
49
50
52
52
54
55
56
57
58
59
61
65
66
66
66
67
69
71
72
72
73
73
74
76
78
Erste Ergebnisse an Helium
6.1 Die Anfangsverteilung . . . . .
6.2 Der totale Ionisationsquerschnitt
6.3 Differentielle Querschnitte . . .
6.4 Anmerkungen . . . . . . . . . .
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
80
80
81
83
84
Zusammenfassung
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
86
INHALTSVERZEICHNIS
iii
A Zur Liouville–Gleichung
A.1 Klassische Stationarit ät . . . . . . . . . . . . . . . . . . . . . . . . . . . .
A.2 Die Hamiltonschen Bewegungsgleichungen . . . . . . . . . . . . . . . . .
89
89
90
B Diskretisierung der Phasenraumverteilungen
B.1 Mikrokanonische Verteilung . . . . . . .
B.2 Produktverteilung . . . . . . . . . . . . .
B.3 Wignerverteilung . . . . . . . . . . . . .
B.4 Energieverteilung nach Cohen . . . . . .
.
.
.
.
92
92
93
93
94
C Numerische Integration der Trajektorien
C.1 Symplektische Integration . . . . . . . . . . . . . . . . . . . . . . . . . .
C.2 Regularisierung des Coulombpotentials . . . . . . . . . . . . . . . . . . .
95
95
96
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
iv
Kapitel 1
Warum klassisch?
Erstaunlich oft werden in der Atomphysik, einem eigentlich typisch quantenmechanischen
Arbeitsgebiet, klassische Näherungen und Modelle verwendet. Dafür gibt es natürlich verschiedene Gründe:
Zum einen ist unsere gesamte Alltagserfahrung von der klassischen Mechanik geprägt, es
ist daher gar nicht so einfach, nicht in diesen dort gewonnenen Erfahrungen und Bildern zu
denken, selbst wenn es um das Verständnis atomarer Vorgänge geht. Diese klassischen Bilder helfen im Gegenteil sogar erstaunlich oft, atomare Prozesse zu verstehen“ — nicht im
”
physikalischen“ Wortgebrauch, daß ein Rechenweg gefunden sei, sondern in dem Sinne,
”
daß ein Vorgang durch Bilder des Alltags“ dargestellt, und damit der Intuition zugänglich
”
gemacht wird.
Bereits bevor die Quantenmechanik entwickelt war, wurden die ersten klassischen Atommodelle aufgestellt, so z.B. von Thomson oder Bohr. Das Bohrsche Planentensysteme“–
”
Modell war, als die Sommerfeldsche Wirkungsquantisierung hinzugefügt wurde, in der Lage das Linienspektrum des Wasserstoffatoms zu erklären. Doch dieses intuitive und vertraute Bild eines Elektrons, das wie ein Planet um die Sonne“ Proton umläuft, hat mit ernst”
haften Problemen zu kämpfen: das klassische, auf seiner Kreisbahn beschleunigte Elektron
sollte seine Energie innerhalb kürzester Zeit durch elektromagnetische Strahlung abgegeben
haben und es konnte nie ein stabiles klassisches Mehrelektronenatom konstruiert werden.
An dieser Stelle zeigt sich, daß das vertraute Bild unseres Sonnensystems, das nun bereits
seit Menschengedenken“ existiert, nicht ohne weiteres auf ein Atom, das bei korrekter Be”
handlung sogar deutlich langlebiger ist als das Sonnensystem, übertragen werden kann. Und
doch ist das Bohrsche Modell eines Atoms aus unserer Vorstellung nicht mehr zu wegzudenken.
Ein weiterer wichtiger Grund ist rein praktischer Natur: Der numerische Aufwand, der
erforderlich ist, voll quantenmechanische Rechnungen in mehreren Dimensionen, d.h. mit
mehreren beteiligten Teilchen, durchzuführen, ist deutlich höher als für das entsprechende
klassische Modell. So konnte erst kürzlich das (sechsdimensionale) Drei–Körper–Coulomb–
Problem mit immensem Aufwand numerisch exakt gelöst werden [RBIM99]. Das betrachtete System, die Elektronenstoßionisation an atomarem Wasserstoff, ist, da es das theoretisch einfachste Dreiteilchensystem ist, bereits seit langem ein Standardproblem, an dem
bisher viele quantenmechanische Näherungen getestet wurden — es wird auch in dieser
1
2
KAPITEL 1. WARUM KLASSISCH?
Arbeit betrachtet werden. Höherdimensionale Probleme, d.h. mit mehr als drei Teilchen,
werden sicher auch in näherer Zukunft nicht voll quantenmechanisch exakt zugänglich sein.
Klassische Modelle und Näherungen sind daher oft der einzige mögliche Zugang zu komplexeren Systemen. Es ist sogar zu erwarten, daß für komplexere Systeme die klassische
Näherung eine bessere Beschreibung liefern kann, als bei dem sehr einfachen Wasserstoffatom, da dann eine größere Zahl klassischer Reaktionswege zur Verfügung steht und quantenmechanische Effekte wie Tunneln für die Dynamik immer weniger bestimmend werden.
Dahinter steht die Beobachtung, daß, wenn es einen direkten klassischen“ Reaktionsweg
”
gibt, dieser gegenüber z.B. quantenmechanischem Tunneln vorgezogen wird. Die klassischen Reaktionspfade werden also immer bedeutender, je mehr Freiheitsgrade ein System
hat, das System daher immer klassischer“.
”
Aus diesen Gründen, d.h. wegen ihrer intuitiven Verständlichkeit und des geringeren numerischen Aufwands, werden klassische Modelle und Näherungen häufig verwendet. Dabei
treten aber sofort die beiden Fragen auf, ob eine solche, so grob genäherte Beschreibung
einem quantenmechanischen Problem angemessen sein kann und, bezogen auf die Anwendung, wie gut z.B. gemessene Querschnitte wiedergegeben werden können. Diese beiden
Fragen sollen auch durch die weitere Darstellung dieser Arbeit leiten.
In dieser Arbeit wird in erster Linie das bereits erwähnte Testproblem“ der Streuphy”
sik, die Elektronenstoßionisation an atomarem Wasserstoff betrachtet. Dieses System kann
quantenmechanisch korrekt beschrieben werden, da die unabhängigen Wellenfunktionen
der beteiligten Teilchen jeweils analytisch bekannt sind und der Streuoperator im Møllerbild
(zumindest als Grenzwert) exakt beschrieben wird. Genauer wird dies weiter unten im Rahmen der Herleitung beschrieben werden. Diese quantenmechanische Formulierung wird
dann über das Wignerbild in eine Phasenraumdarstellung übertragen und aus dieser heraus klassisch genähert.
Elektronenstoßionisation an Wasserstoff hat darüber hinaus den Vorteil, daß eine Vielzahl gemessener (und gerechneter) Querschnitte vorliegen, also ein sehr breiter Bereich an
Energie– und Winkelverteilungen in verschiedenen Geometrien getestet werden kann.
Die Näherung wird mit dem rein klassischen Ansatz des Problems verglichen: die Elektronen und das Proton sind darin geladene Punktteilchen, die den klassischen Bewegungsgleichungen gehorchen — das Wasserstoffatom wird im Bohrschen Atommodell beschrieben. Diese Methode ist als CTMC“ bekannt, als classical trajectory Monte–Carlo“–Rech”
”
nung. Der Name kommt daher, daß die Querschnitte analog einer Monte–Carlo–Integration
bestimmt werden, wobei der Integrand“ die klassische Zeitentwicklung der ausgewürfel”
ten Phasenraumpunkte beschreibt. Dieses rein klassische Modell ist eine in sich konsistente
Beschreibung des Systems, sie kann allerdings keine quantenmechanischen Effekte berücksichtigen — diese sind nicht darin enthalten.
Es stellt sich natürlich sofort die Frage, ob diese CTMC“ auch der korrekte klassische
”
Limes der genäherten quantenmechanischen Beschreibung ist, oder aber nur ein klassisches
Modell, das auf dieses System angewandt wird.
Neben dieser mehr theoretisch motivierten Betrachtung soll auch die Anwendung“ der
”
hier vorgestellten klassischen Näherung betrachtet werden: wie gut werden Querschnitte
wiedergegeben, bzw., stellt diese Näherung eine Verbesserung gegenüber dem klassischen
Modell dar? Liefert sie entweder bei vergleichbarem Aufwand verbesserte Querschnitte
3
oder kann evtl. bereits mit geringerem Einsatz an Rechenzeit die gleiche Aussage getroffen
werden? Oder ist der Unterschied gar vernachlässigbar?
Zusätzlich wurden in dieser Arbeit zum ersten Mal in einer klassischen Rechnung voll
differentielle Ionisationsquerschnitte an Wasserstoff bestimmt. Das ist unseres Wissens nach
bisher zumindest nicht veröffentlicht worden. Ein Grund mag darin liegen, daß die Standard–
CTMC diese Querschnitte praktisch nicht beschreiben kann. Diese voll differentiellen Querschnitte aber erlauben eine sehr differenzierte Aussage über die Qualität verschiedener
Näherungen und ermöglichen es, die Fehler zu lokalisieren; sie sollen in dieser Untersuchung nicht in erster Linie zur Vorhersage möglicher Messergebnisse dienen.
Aus der skizzierten Vorgehensweise ergibt sich die Gliederung der vorliegenden Untersuchung:
Zuerst wird, um die Problematiken einer klassischen Betrachtung zu verdeutlichen, in
Kapitel 2 eine Bestandsaufnahme bisheriger klassischer Rechnungen zur Stoßionisation
durchgeführt. Dazu wird kurz das Modell der Standard–CTMC vorgestellt sowie die wichtigsten Versuche, dessen Einschränkungen zu beseitigen.
Der folgende, etwas umfangreichere Abschnitt in Kapitel 3 beschreibt die Herleitung des
quasiklassischen Ansatzes“: es wird gezeigt, wie die zeitabhängige quantenmechanische
”
Streuung über das Wignerbild klassisch genähert wird. Die für die Herleitung wichtigen
Grundlagen werden anfangs (kurz) wiederholt. Dieses Kapitel beschreibt auch, wie weit
die Näherung durchgeführt wird und welche Veränderungen zur Standard–CTMC bestehen
bleiben. Dabei wird gezeigt, daß das Modell der CTMC im Rahmen der hier gezeigten
Näherung der korrekte klassische Limes ist.
Ein wichtiger Aspekt dieses Abschnittes wird es sein, die Interpretation der beiden klassischen Rechnungen — CTMC und quasiklassischer Ansatz — gegeneinander abzugrenzen,
da hierin die wesentlichen Unterschiede bestehen, nicht so sehr in den Formeln.
Im Gegensatz zur CTMC kann der hier vorgestellte quasiklassische Ansatz auf Mehr–
Elektronen–Targets erweitert werden, ohne daß zusätzliche Annahmen gemacht werden
müssten. Dies ist eine der wesentlichen Stärken dieser Näherung. Die Erweiterungen werden in Kapitel 4 am Beispiel des Heliumatoms beschrieben.
Größeren Platz nimmt auch die Darstellung der an Wasserstoff gerechneten Querschnitte
ein, da sie darüber Aufschluß geben, wie gut die Näherung, auch im Vergleich zur Standard–
CTMC, tatsächlich ist und wo ihre Grenzen liegen (Kapitel 5). Im Rahmen der Näherung
ergibt sich eine gewisse Wahlfreiheit in der Phasenraumbeschreibung des Targets, neben
dem bisherigen mikrokanonischen Modell der CTMC werden drei weitere Beschreibungen
auf ihre Eignung bei verschiedenen Gesamtenergien hin untersucht.
Ausführliche Rechnungen an Mehrelektronentargets sind sehr aufwendig, daher kann
in Kapitel 6 lediglich gezeigt werden, daß mit dem vorgestellten Ansatz das klassische
Heliumatom mit allen Wechselwirkungen konsistent behandelt werden kann. Dies wird an
ersten einfachen Querschnitten der Doppelionisation gezeigt.
In Kapitel 7 wird eine Zusammenfassung der Ergebnisse sowie ein Ausblick auf die
nächsten Schritte gegeben.
4
KAPITEL 1. WARUM KLASSISCH?
In den Anhang wurden noch drei mehr technische Aspekte aufgenommen: die Entkopplung der Stützstellen einer Phasenverteilung, die auf die klassischen Trajektorien“ führt,
”
die konkret verwendete numerische Rechentechnik sowie eine detailiertere Auflistung, wie
die verglichenen Phasenraumverteilungen für die Rechnung präpariert wurden.
Im weiteren werden, wenn nicht anders gekennzeichnet, atomare Einheiten verwendet,
also und . Die wichtigsten Umrechnungen sind damit die Energie, für sie gilt
1a.u. = 27,2eV und als Längeneinheit der Bohrsche Radius = 0,53 10 cm = 1a.u. Ein
Elektron auf der klassischen Bahn des Wasserstoffgrundzustands hat in diesen Einheiten
eine Geschwindigkeit von einer atomaren Einheit.
Quantenmechanische Wellenfunktionen und Dichtematrizen sind normiert, d.h. "!$#%!'&() .
Die symbolische Größe *$+ ist in der Praxis eine ausreichend“ große Zahl, bei der die
”
Rechnung im Rahmen der numerischen Genauigkeit konvergiert ist; für analytische Rechnungen ist, wenn nicht explizit angegeben, der Grenzübergang nach Unendlich gemeint.
Zur Beschreibung von Reaktionen und Stoßsystemen wird zum Teil eine in der Hochenergiephysik übliche Notation verwendet: Nach einem Kürzel für das Target stehen in
,-
.
Klammern das Projektil und die Ejektile, dahinter das Target nach der Reaktion.
bedeutet also, daß ein Elektron ( ) auf atomaren Wasserstoff ( ) geschossen wird, die aus
laufenden Teilchen sind zwei Elektronen ( ), ein Proton bleibt zurück. Anregung von mo0/1(
"235/ 4
.
lekularem Wasserstoff mit Protonen wäre dagegen
Kapitel 2
Bisherige klassische Rechnungen
2.1
Wie alles begann . . .
Bereits 1912 berechnete Sir Thomson klassisch den totalen Ionisationsquerschnitt für die
-
Elektronenstoßionisation an atomarem Wasserstoff (
) [Tho12], indem er die Energie betrachtete, die in Abhängigeit vom Stoßparameter bei der Streuung des Projektilelektrons auf das (ruhende) Targetelektron übertragen wird. Aus dieser Rechnung ergibt sich
das klassische Hochenergieverhalten des totalen Ionisationsquerschnittes ( 6798:7; <= ).
Das Maximum des totalen Querschnitts liegt dabei bei einer Einschußenergie, die der doppelten Bindungsenergie entspricht und damit deutlich zu niedrig (siehe Abb. 2.1). Mit den
damals verfügbaren Messungen jedoch war diese Berechnung verträglich.
Die ersten Rechnungen, die später unter dem Namen CTMC“ bekannt werden sollten,
”
wurden 1966 von Abrines und Percival durchgeführt [AP66a, AP66b]. Sie berechneten
1(
"2
den totalen Ionisationsquerschnitt für
. Das Wasserstoffatom wird dabei klassisch
im Bohrschen Modell (die Elektronen laufen auf Keplerbahnen um den Kern) beschrieben. Die Menge der zulässigen Anfangsbedingungen im Phasenraum (PR) wird in Winkel–
Wirkungsvariablen auf der Energieschale zur Bindungsenergie (–0,5a.u.) gleichmäßig gefüllt 1 . Diese mikrokanonische“ Verteilung hat die gleiche (radiale) Impulsverteilung wie die
quantenmechanische Wellenfunktion des Wasserstoffgrundzustands, die Ortsverteilung ist
allerdings deutlich anders, da — wegen der festen Energie — der Bereich außerhalb von
2a.u. für die klassischen Elektronen unerreichbar ist; dort befinden sich jedoch etwa 24%
der quantenmechanischen Aufenthaltswahrscheinlichkeit. Diese Verteilung wird diskretisiert und jeder dieser Phasenraumpunkte als Startwert für eine Bahn des Targetelektrons
verwendet. Dieses Atom wird mit dem Projektil beschossen“ und die klassischen Bewe”
gungsgleichungen des System integriert (eine Trajektorie“). Der Anteil der Schüsse“, bei
”
”
denen das Targetelektron am Ende eine positive Energie besitzt, definiert den Ionisationsquerschnitt.
1
Die kartesischen Phasenraumvariablen Ort und Impuls werden dazu durch eine kanonische Transformation
auf Winkel und Wirkungen (Energie > , Quadrat des Drehimpulses ?@ und dessen Projektion auf eine Achse
?BA ) abgebildet; diese sind im Keplerproblem Konstanten der Bewegung. Die Bezeichnung mikrokanonisch
entstammt der Thermodynamik, da diese Verteilung eine feste Energie hat und in den restlichen Freiheitsgraden,
hier ? @ und ?2A , gleichverteilt ist.
Für eine konkrete Vorschrift zur Auswahl der Startwerte siehe z.B. [AP66b] oder [RT85].
5
KAPITEL 2. BISHERIGE KLASSISCHE RECHNUNGEN
6
Bereits kurze Zeit später wurden mit dem selben Modell die ersten Querschnitte zur Elektronenstoßionisation an Wasserstoff gerechnet [APV66]. Dieses aus dem klassischen Grenzfall hergeleitete Modell führt zu brauchbaren“ absoluten totalen und energieabhängigen
”
Querschnitten (vgl. Abb. 2.1), Winkelabhängigkeiten sind aber — v.a. bei kleinen Elektronenenergien — z.T. einfach falsch (vgl. Abb. 2.2). Nun ist nicht klar, welcher Teil dieses
klassischen Modells, die klassische Propagation oder die unzureichende Anfangsverteilung,
zu diesem Fehler führt.
σ [10-17cm2]
Shah
10
1
Shah
CTMC
Thomson
10
100
Eein [eV]
1000
Abbildung 2.1: totaler Elektronenstoßionisationsquerschnitt an atomarem Wasserstoff: Vergleich einer Standard–CTMC–Rechnung mit der Messung von Shah [SEG87] und dem von
Thomson berechneten Querschnitt [Tho12].
TDCS [10-18 cm2 sr-2 ev-1]
15
mikrokanonisch
Messung
10
5
0
-180
-90
0
θb
90
180
.C
Abbildung 2.2: volldifferentieller
–Querschnitt in Ehrhardt–Geometrie ( =D-E1F =
8
250eV, GCH = 8 und =DI = 5eV): Vergleich einer Standard–CTMC–Rechnung mit einer Messung von Ehrhardt [EKSJ85].
Das Bohrsche Modellatom ist stabil, solange nur ein einzelnes Elektron betrachtet wird2 ,
2
Die elektromagnetische Abstrahlung, die zu einem Energieverlust des kreisenden Elektrons führt, soll in
dieser Betrachtung vernachlässig werden — die experimentelle Beobachtung zeigt, daß die Orbits der Elektronen im Atom bei quantisierter Wirkung stabil sind.
2.2. VERBESSERUNGEN/-ÄNDERUNGEN
7
bei mehreren Elektronen kommt es zur Autoionisation: eines der Elektronen fällt in den
Kern, die anderen werden dadurch aus dem Atom herausgehoben. Weiterhin ist es auch
nicht eindeutig, wie die mikrokanonische Verteilung auf mehrere Elektronen erweitert werden müsste: jedes Elektron mit der gleichen Bindungsenergie oder mit unterschiedlichen
Energien bei korrekter Gesamtenergie?
Um zu besseren winkelabh ängigen Querschnitten zu gelangen und um die Probleme mit
Mehr–Elektronen–Atomen in den Griff zu bekommen, wurden bisher eine Reihe von Veränderungen vorgeschlagen, z.T. an der Anfangsverteilung, z.T. an der Propagation. Hier
sollen allerdings nur Modifikationen erwähnt werden, die im Rahmen der CTMC“ bleiben,
”
d.h. Rechnungen mit einzelnen, unabhängigen, klassisch propagierten Trajektorien, über die
am Ende summiert wird.
2.2
2.2.1
Verbesserungen/- änderungen
Energieabhängige Anfangsverteilungen
Einer der naheliegenden Ansätze, die im Vergleich zur Quantenmechanik falsche Ortsraumdichte der mikrokanonischen Verteilung zu verbessern, ist es, mikrokanonische Verteilungen mit unterschiedlicher Bindungsenergie so zu überlagern, daß in der Summe die quantenmechanische Ortsverteilung möglichst gut nachgebildet wird, ohne dadurch die Impulsverteilung deutlich zu verändern. Hardie und Olson [HO83] verwendeten acht mikrokanonische Verteilungen mit Bindungsenergien von –2 bis –0,25a.u., deren Gewichte durch einen
Fit der überlagerten Ortsraumdichte an die quantenmechanische Dichte bestimmt wurden.
Damit wurden Ionisations- und Ladungstransferquerschnitte mehrfach geladener (nackter)
Ionen an atomarem Wasserstoff berechnet.
Ein anderer Weg wurde von Cohen beschritten [Coh85]: Er betrachtet, wie aus einer
angesetzten energieabhängigen Anfangsverteilung Orts– und Impulsraumdichten bestimmt
werden und löst diese Gleichungen nach der quantenmechanischen Ortsraumdichte auf.
Startwerte zu freien Elektronen, bei denen das Elektron bereits ionisiert starten würde, werden dabei durch die Konstruktion (d.h. die Integrationsgrenzen) verhindert. Die so erhaltene
analytische Anfangsverteilung reproduziert die Impulsverteilung ebenfalls recht gut. Auch
hier wurden wieder Ionisations– und Transferquerschnitte der Protonenstoßionisation an
Wasserstoff bestimmt.
2.2.2
Die Wignerverteilung in der CTMC
Die Wignerverteilung ist eine Phasenraumdarstellung der quantenmechanischen Dichtema
trix [Wig32], ihre Zeitentwicklung wird im klassischen =0–Limes von der Liouvillegleichung, der Bewegungsgleichung klassischer Phasenraumverteilungen, beschrieben. Dies
legt es nahe, die Wignerverteilung eines Wasserstoffgrundzustands als Anfangsverteilung
in der CTMC zu verwenden. Eichenauer etal [EGS81] mussten allerdings die Wignerverteilung in der Energie begrenzen ( cut–off wigner“), da sie Anteile enthält, die zu ungebunde”
nen Elektronen gehören (etwa 17%). Die obere und untere Energiegrenze =KJ H3L und =MJ E1F
wurden durch einen Fit der Wirkungsquerschnitte bestimmt; = J HNL,O 0,25a.u. lieferte
die besten mit dem Experiment konsistenten Querschnitte. = J EPF wird so bestimmt, daß die
KAPITEL 2. BISHERIGE KLASSISCHE RECHNUNGEN
8
mittlere Bindungsenergie dem Wasserstoffgrundzustand entspricht. Diese begrenzte Verteilung reproduziert Orts- und Impulsverteilung zwar nicht korrekt, aber beide recht gut. Die
Verteilung ist nicht station är, ihre Phasenraumdichte ändert sich zeitlich (siehe dazu An1(
"2
hang A). Auch hier wurden Rechnungen wieder zu
durchgeführt. Ein Vergleich
mit Ergebnissen mit der mikrokanonischen Verteilung ist in [EGS82] zu finden.
2.2.2.1
Schwierigkeiten bei verbreiterter Energieverteilung
Dieses Abschneiden der Energieverteilung ist schwierig zu rechtfertigen, denn diese Einschränkung schafft eine unphysikalische“, eckige“ Verteilungsfunktion und es gibt dafür
”
”
keine theoretisch fundierte Begründung.
Ein weiteres gravierendes Problem tritt bei der Bestimmung energiedifferentieller Querschnitte auf, bzw. aller Querschnitte, bei denen die Energie der ionisierten Elektronen betrachtet wird: die Gesamtenergie ist kein fester Wert, für jede Trajektorie gilt eine andere
Energieskala, auf der der energiedifferentielle Querschnitt aufzutragen ist; bei Elektronenstoßionisation z.B. reicht es nicht mehr aus, die Energie eines Elektrons anzugeben, die des
anderen ist wegen der unscharfen Verteilung der Bindungsenergie nicht mehr automatisch
bekannt. Dieses Problem betrifft alle Verteilungen, auch die, die energetisch begrenzt sind
oder nach Konstruktion keine freien Startwerte enthalten.
Bei der abgeschnittenen Wignerverteilung sind zudem die Querschnitte noch von den
Energiegrenzen abhängig, für unterschiedliche Wirkungsquerschnitte kann das auf unterschiedliche Breiten der Bindungsenergie führen, für die der Querschnitt jeweils am besten
wiedergeben wird. Die Vorhersagekraft solcher Anpassungen ist natürlich begrenzt.
2.2.3
Zusatzpotentiale
Um die autoionisierenden klassischen Mehrelektronenatome zu stabilisieren, wird in der
Regel entweder auf hochsymmetrische Anfangskonfigurationen zurückgegriffen, die sich
zumindest in einem labilen Gleichgewicht befinden, oder die Wechselwirkung der Targetelektronen untereinander wird ganz vernachläßigt, bzw. erst nach/zum Zeitpunkt des Stoßes
eingeschaltet“, um PCI–Effekte3 zu berücksichtigen ( independent electron approximati”
”
on“). Hierfür existiert allerdings keine schlüssige Herleitung oder Begründung, zudem führt
dieses Einschalten“ zu Schwierigkeiten bei der Erhaltung der Gesamtenergie.
”
Kurz sollen noch zwei Erweiterungen des Anfangszustandes erwähnt werden, die über
den Hamiltonoperator RQ / @ O)T S des Wasserstoffatoms hinausgehen:
Wenn der Spin des Elektrons nicht vernachlässigt wird, verhindert dieser, daß ein radial
auf den Kern zulaufendes Elektron mit diesem kollidiert: es wird kurz davor in dessen elektromagnetischem Feld abgelenkt. Das ist die Grundidee von Gryzińskis erweitertem free–
”
fall–modell“ (siehe [Gry72], für Anwendungen und Erweiterungen z.B. auch [Gry95])
Einen anderen Ansatz verfolgen Kirschbaum und Wilets [KW80], die geschwindigkeitsabhängige Zusatzpotentiale einführen, um die Heisenbergsche Unschärferelation und das
3
PCI = post collision interaction“: langreichweitige Wechselwirkungen zwischen den auslaufenden Teil”
chen verändern die Energie- und Winkelverteilung der Endprodukte, nachdem der eigentliche Stoß bereits stattgefunden hatte.
2.2. VERBESSERUNGEN/-ÄNDERUNGEN
9
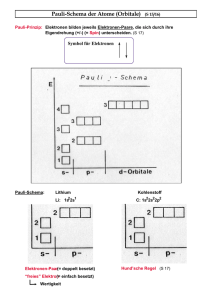
Pauliprinzip zu modellieren. Mit diesen Potentialen können klassische Atome aufgebaut
werden, in denen die Elektronen stabil ruhen“, indem Position und Impuls der Elektro”
nen solange variiert werden, bis die Gesamtenergie minimal ist; es entsteht dabei eine der
Realität ähnliche Schalenstruktur.
Durch diese geschwindigkeitsabhängigen Zusatzpotentiale sind für die klassischen Elektronen Impuls und Geschwindigkeit nicht mehr proportional und in dynamischen Problemen bleibt die Energie nicht erhalten, trotzdem wurden in [ZM86] Ionisationsquerschnitte
für und He UVU auf He berechnet.
Eine Variante (und kritische Anmerkungen zu diesem Ansatz) wird von Cohen vorgeschlagen [Coh96]: Zusatzpotentiale, die für jedes Elektron einzeln eine untere Schranke
für dessen Energie setzen. Dieses Potential greift nicht ganz so drastisch in die Dynamik
ein, aber auch hier bleibt die Energie nicht erhalten, sodaß auch mit diesem Ansatz keine
sinnvollen energieabhängigen Querschnitte bestimmt werden können.
2.2.4
Modifikationen der Propagation
Neben den Ansätzen, die Anfangsverteilung zu verändern, gab es auch Versuche, bei denen
die klassische Propagation modifiziert wurde, um höhere Ordnungen der –Entwicklung
der Quantenmechanik zu berücksichtigen.
Von Keller et al wurde vorgeschlagen [KAD93], die korrekte Propagation durch einen
stochastischen Algorithmus zu ersetzen, der die quantenmechanische Unschärfe modelliert.
Dazu wird die Zeitentwicklung der Wignerfunktion in einen klassischen und einen (da
gegen sehr kleinen) quantenmechanischen Liouvilleoperator, der die Terme mit enthält,
aufgeteilt. Der quantenmechanische Anteil läßt sich als Markovscher stochastischer Prozess
darstellen [SCU 84]. In diesem Modell wird die klassische Propagation immer wieder durch
eine (quantenmechanische) statistische Störung unterbrochen“. Mit dem Modell wurden
”
an Wasserstoff totale Ionisations- und Transferquerschnitte durch Protonenstoß berechnet
und mit der Standard–CTMC verglichen. Die Unterschiede sind sichtbar, jedoch gering.
Ein weiterer Ansatz, der allerdings nicht mehr direkt CTMC“ ist, wird von Eichenauer et
”
al vorgeschlagen [EGS82]: Hier wird die Zeitentwicklung vieler Phasenraumpunkte gleichzeitig durchgeführt, das Projektil spürt die gesamte Verteilung, nicht nur einen Punkt davon.
Auf diese Weise soll der Effekt nachgebildet werden, daß in der Quantenmechanik nur die
komplette Wellenfunktion die vollst ändige Information enthält (Stichwort Nichtlokalit ät“).
”
Als Anfangsverteilung wurden dabei die mikrokanonische und die abgeschnittene Wignerverteilung benutzt. Die gezeigten Querschnitte sind allerdings nicht aussagekräftig genug,
um zu erkennen, ob dieses Vorgehen Vorteile gegenüber einer Standard–CTMC–Rechnung
bietet.
Einen ähnlichen Ansatz zeigen Reinhold und Falcón, die über eine fluid–dynamische
Interpretation der Schrödingergleichung ein zusätzliches (quantenmechanisches) Potential
einführen, das von der Phasenraumdichte abhängig ist [RF88b] (Querschnitte dazu sind in
[RF88a] für nackte Ionen auf wasserstoffartige Targets berechnet). In diesem Modell hat der
Anfangszustand durch das quantenmechanische Zusatzpotential die richtige Ortsverteilung
und die (korrekte) scharfe Bindungsenergie, es muß allerdings die komplette Phasenraumverteilung propagiert werden, um das Zusatzpotential mitführen zu können. An den berechneten totalen Ionsations– und Transferquerschnitte ist allerdings kein großer Unterschied
10
KAPITEL 2. BISHERIGE KLASSISCHE RECHNUNGEN
zur Standard–CTMC zu erkennen.
2.3
Zusammengefasst
Betrachtet man die verschiedenen Ansätze, so ergibt sich folgendes Bild: Abrines und Percival begannen mit dem rein klassischen Bohrschen Modell des Wasserstoffatoms, d.h. die
Startwerte und die Propagation entsprechen der klassischen Mechanik, ohne jegliche Quanteneinflüße. Der einzige Parameter ist die experimentell zugängliche Bindungsenergie. Dieses Modell wurde explizit klassisch angesetzt, wie die folgende Stelle aus [AP66a] zeigt:
Die mit diesem makroskopischen“ Modell berechneten Querschnitte werden gewisser”
maßen erst nach der Rechnung auf das Wasserstofftarget skaliert. Für totale, bzw. nur energieabhängige Querschnitte scheint dieses Modell die wesentlichen Prozesse zu beschreiben.
Es ist in sich schlüssig und komplett, ohne daß weitere Annahmen gemacht werden müssten.
Quantenmechanische Effekte wie z.B. Tunneln sind in diesem Modell nicht enthalten, über
sie liefert es daher auch keine Informationen. Unklar ist lediglich, ob die angesetzte Verteilung des Drehimpulses der Keplerellipsen korrekt ist, da quantenmechanisch WYX gilt.
Durch die Symmetrie des Wasserstoffatomes ist allerdings die Summe aller Drehimpulse
gleich Null. Dies ist übrigens die einzige Inkonsistenz des Modells, denn in dem klassischen Bild der tatsächlichen Flugbahnen der Elektronen sollte jedes einzelne Targetelektron,
d.h. jede Trajektorie, bereits verschwindenen Drehimpuls besitzen. Bei der Bestimmung der
Drehimpulsverteilung haben also die ansonsten echten Trajektorien lediglich den Status von
Stützstellen einer Phasenraumverteilung.
Die Probleme beginnen dann, wenn versucht wird, Teile dieses Modells zu erweitern bzw.
auszutauschen, indem nur einer davon aus der Quantenmechanik hergeleitet/-gen ähert wird:
alle bisherigen Erweiterungen betrachten jeweils nur einen Aspekt des CTMC–Modells
(Anfangszustand, bzw. Propagation), ohne die anderen zu modifizieren. Das ist zumindest
inkonsequent und kann zu Inkonsitenzen führen, wenn in verschiedenen Teilen einer Rechnung in unterschiedlichen Ordnungen genähert wird.
Das zeigt sich deutlich bei der Anfangsverteilung: wird hier versucht, die falsche Ortsverteilung zu verbessern, so wird die anfangs scharfe Energieverteilung aufgeweicht. Eine
Verteilung in der Energie bedeutet aber, daß ein (kleiner) Anteil ungebundener Startwerte
vorhanden sein kann, wenn die Energieverteilung physikalisch sinnvoll“, d.h. glatt, sein
”
soll. Im Rahmen der CTMC bedeuten freie Elektronen, daß Ionisation stattgefunden hat;
es kommt also ohne Targeteinwirkung bei Wasserstoff zur teilweisen (Auto–)Ionisation.
2.3. ZUSAMMENGEFASST
11
Natürlich liegt es dann nahe, diese bereits ionisierten Startwerte wegzudiskutieren“, indem
”
die Energie willk ürlich begrenzt wird. Dabei bleibt allerdings das Problem erhalten, die
Endenergie der Elektronen festzulegen (siehe Abschnitt 2.2.2.1).
Das Problem der Autoionisation tritt bei Mehr–Elektronen–Targets bereits im mikrokanonischen Modell4 auf, auch hier wurde ad hoc die Elektron–Elektron–Wechselwirkung
modifiziert, ohne daß dafür eine Begründung gegeben werden kann5 .
Auch wenn bei den bisherigen Ansätzen die Propagation modifiziert wurde, blieb der
Rest des Modells erhalten. Es existiert bisher keine zusammenhängende Herleitung der
CTMC aus der Quantenmechanik, anhand der sich nachvollziehen läßt, welche Näherung
an welcher Stelle stattfindet und welche Bedeutung diese dabei hat.
Diese Herleitung wird im folgenden Kapitel an der Elektronenstoßionisation aufgezeigt:
ausgehend von der (korrekten) quantenmechanischen Beschreibung werden die einzelnen
Teile der Rechnung in gleicher Ordnung ZX genähert. Das ergibt ein schlüssiges Bild,
in dem auch die bisherige CTMC ihren Platz findet.
4
. . . wobei eine mikrokanonische“ Verteilung für ein Mehrelektronenatom bisher klassisch nicht hergeleitet
”
werden konnte.
5
Die Rechtfertigung dafür lautet, das die Rechnung ohne dieses Vorgehen nicht durchzuf ühren sei — was
für das klassisch angesetzte Mehrelektronenmodell so natürlich korrekt ist.
Kapitel 3
Der quasiklassische Ansatz
3.1
Das System
[]\_^a`cbd^Ve3f
Die konkreten Rechnungen werden an der Elektronenstoßionisation von atomarem Wasserstoff getestet, die Herleitung ist natürlich unabhängig von diesem speziellen Streusystem.
Da aber immer wieder Beispiele eingefügt werden, soll dieses kurz vorgestellt werden. Die
Adaption auf andere Projektile sollte eindeutig sein, die Erweiterung auf andere Targets
wird im folgenden Kapitel am Beispiel des Heliumatoms aufgezeigt werden.
Das komplette System besteht aus drei Teilchen: den beiden Elektronen und dem Kern.
Dessen Masse g wird als unendlich angenommen, sodaß durch den Kern der Ursprung des
Koordinatensystems definiert wird.
Die volle Hamiltonfunktion
/
/
/
/
k
h " .h / " / ij
.k$
S l Yl Y
(3.1)
l h
h/ O h
i O h/
i
S S
g
# O
#
# O
#
# O
#
S
S
i
i
reduziert sich damit wegen m op n YX und YX zu:
/
/
/
S O h l O h /
(3.2)
l
h
h /
# O
#
S
S
Im Eingangskanal befindet sich ein Elektron beim Kern, das andere ist weit“ weg: das
”
Gesamtsystem besteht aus einem neutralen Atom und einem freien Elektron, also:
/
/
/
E
E
/E
(3.3)
S l
)O h /
l
S
/E
ist bekannt, für den GrundDie quantenmechanische Lösung des Wasserstoffproblems
zustand gilt
-h / T HNvw
!
q rts @.u
Das freie Projektil
E
wird im einfachsten Fall durch eine ebene Welle beschrieben:
S
Ezy3{ TN|
x h E
- s ~} /
S
u
12
3.2. DREITEILUNG DES STREUVORGANGS
3.2
13
Dreiteilung des Streuvorgangs
Eine zeitabhängige Beschreibung der Streuung kann in drei Schritte aufgeteilt werden:
die Präparation des Anfangszustandes,
dessen Propagation und
die Bestimmung von Querschnitten aus dem propagierten Anfangszustand.
Diese Gliederung entspricht der Durchführung eines Experiments mit Präparation der
Apparatur, Durchführung der eigentlichen Messung und (späterer) Auswertung.
Jeder dieser drei Schritte wird im Folgenden zuerst quantenmechanisch formuliert und
dann klassisch genähert. Der genaue Weg dieser Näherung über eine Phasenraumdarstellung der Quantenmechanik wird an der Ableitung des klasischen Anfangszustands erläutert
werden. Auf diese Weise wird ersichtlich, welche Näherungen gemacht werden und welchen Einfluß sie an dieser Stelle, bzw. auf die gesamte Herleitung haben.
Doch damit die z.T. nur geringen Unterschiede zwischen diesem Ansatz und der Standard–
CTMC deutlich werden, soll diese zunächst wiederholt werden, bevor die eigentliche Herleitung des quasiklassischen Ansatzes vorgestellt wird.
3.3
Wiederholung der Standard–CTMC
Eine ausführliche Beschreibung der CTMC, wie sie von Abrines und Percival eingeführt
.C
-Reaktionen
wurde, ist z.B. in [RT85] zu finden, sodaß hier nur ein kurzer Abriß der für
wichtigen Formeln folgt (siehe auch Abschnitt 2.1).
Diese Methode basiert auf der Zeitentwicklung klassischer Phasenraumverteilungen. Diese gehorchen der Liouville–Gleichung:
7"
h
"(
N
T
Q
O
O
l
T
Q
mit Poissonklammer
Für die Berechnung wird die PR–Verteilung diskretisiert ( test particle ansatz“):
”
h
"(
N
h
h N
3
'Ft
O F
O F
(3.4)
(3.5)
(3.6)
bestimmt dabei die Normierung, die Gewichte 'F sind oft konstant auf eins gesetzt,
h
"2
h " dann liegen an Stellen, wo
groß ist, viele PR–Punkte F F .
Setzt man diese diskretisierte Verteilung in die Liouvillegleichung ein, so entkoppelt die
Zeitentwicklung jedes einzelnen PR–Punktes von den anderen und wird durch die klassischen Bewegungsgleichungen bestimmt. Jeder (evtl. hochdimensionale) PR–Punkt beschreibt also eine von den Anfangswerten abhängige Trajektorie“ im Phasenraum. Dieser
”
Schritt ist in Anhang A.2 detailliert beschrieben:
h
7 F und
7 F)O h
F
F
KAPITEL 3. DER QUASIKLASSISCHE ANSATZ
14
Die Erwartungswerte einer Phasenraumverteilung werden klassisch aus dem Phasenraumintegral bestimmt:
~
"
&
h
Y
h
"2
h
~
(3.7)
Ist der Erwartungswert ein volldifferentieller Querschnitt, so testet“ , ob die Endwerte
hc
"2
”
passen:
der Trajektorien zum gewählten Endzustand
h
"2
1
1B
bzw. wegen
t=tK =
J =YO=
O
Q
Durch die Diskretisierung Gl. (3.6) wird das Integral in Gl. (3.7) zur Summe über die
Endwerte der berechneten Trajektorien:
O
"
&
~
F
h
F
~_
"
F
N
(3.8)
Die Normierung der einlaufenden Welle“ hat die Einheit eines Flußes, d.h. wenn J H3L
”
der maximale betrachtete Stoßparameter ist:
F
s /
J HNL
Damit erhält man den voll differentiellen Ionisationsquerschnitt (ohne Symmetrisierung)
zu:
s /
}
6
J HNL
/
Ft = F O=
H F$OKH FOKI
(3.9)
S
S
tMH_tKI~c=DH
Endwerte
mit
F , d.h. normiert auf ein Targetelektron und einen einfallenden Fluß von
s /
< J HNL . Die Energie des zweiten Elektrons ist durch die Erhaltung der Gesamtenergie
festgelegt, sie braucht also nicht mehr explizit abgefragt werden.
¡ " Die Verteilung der Startwerte F F (t= OK+ ) erfolgt über Torusquantisierung in Winkel–
Wirkungsvariablen, d.h. Gleichverteilung in den kanonischen Winkelvariablen. Die so erhaltene mikrokanonische“ Anfangsverteilung für ein Wasserstoffatom im Grundzustand
”
wird durch feste Energie und gleichverteiltes Drehimpulsquadrat beschrieben (siehe auch
Abschnitt 2.1):
/
/
w¤£
wwCw
a.u.
(3.10)
=Z¢OKX
W )¥¤X
W J H3L§¦ gleichmäßig.
3.4
Die Anfangsverteilung
Die Präparation einer Anfangsverteilung ist der erste Schritt in einer zeitabhängigen Rechnung. In der Standard–CTMC werden dafür Messgrößen des betrachteten Targets/Systems
zugrunde gelegt: die Bindungsenergie und der Drehimpuls (und implizit die Kugelsymmetrie des Problems).
Da wir einen Zugang suchen, der in der Quantenmechanik beginnt, soll in unserer Herleitung die Wellenfunktion der Ausgangspunkt sein.
3.4. DIE ANFANGSVERTEILUNG
3.4.1
15
Korrespondenz und Phasenraumverteilungen
Die wesentliche Verbindung zwischen Quantenmechanik und klassischer Physik sind die
Korrespondenzregeln, die beschreiben, wie klassische Observablen in quantenmechanische
Operatoren übersetzt werden.
©
Dazu wird der Erwartungswert eines quantenmechanischen Operators ¨ betrachtet:
©
&ª
t«d!
4 © :B « ¨ «
! «
(3.11)
Dieser Erwartungswert soll nun klassisch formuliert werden. Damit wird eine Phasen "B
raumverteilung ¬ «
definiert und an Stelle des Operators tritt die klassische Funktion
­ « :B :
©
"2 ­ B
(3.12)
&ªYt«d ¬ «
«
:B
¬ «
wird auch als quasi probability distribution“ bezeichnet [Coh66], da sie ähnliche
”
Eigenschaften wie eine klassische Wahrscheinlichkeitsverteilung besitzt.
Die gesuchte Verteilung ¬ soll dabei als Randbedingungen“ die korrekten Dichten der
”
Wellenfunktion reproduzieren:
/
:B
(3.13)
#%! « #
¬ «
/
"2
12
#! ®
#
(3.14)
t«d¬ «
Um daraus die zugrunde liegende Korrespondenzregel zu bestimmen, wird durch Fou .¯
:B
der PR–Verteilung ¬ «
beriertransformation die charakteristische Funktion“ g G
”
stimmt (siehe z.B. [Coh66]). Vergleich mit Gl. (3.12) zeigt, daß diese dem Erwartungswert
Ez°± E1²
U Q entspricht.
von
¯
G
g
Ez°~± 1E ²
"B
U Q ¬ «
E°~± E1²
U Q &
´ F ´"¯ J
G
F JZ¸
µd¶ ¶
· «
F³ J
t«d
(3.15)
(3.16)
In der letzten Zeile ist der Erwartungswert der Exponentialfunktion als Taylorreihe geschrieben. Damit wird das Problem, die Korrespondenz zu bestimmen, darauf reduziert,
F die Erwartungswerte der Momente « J klassisch und quantenmechanisch zu formulieren,
¹ F J F J
bzw. den zu «
gehörigen Operator ¨ « ¨ ¨
festzulegen, denn es gilt:
"«
F J
&º
t«d!
4 ¹ F J « ¨ «
! « ¹
F J
Bei gegebener Korrespondenzregel, die «
in ¨
:B
verteilung ¬ «
über die Rücktransformation der
der Wellenfunktion verknüpft:
¬
:
B
«
rts /
cGª
c«d
¬
"
B F J
«
«
(3.17)
F J «¨ ¨
übersetzt, ist die PhasenraumGln. (3.15) bzw. (3.16) eindeutig mit
¯ Ez°~± 1E ² E°~± 1E ²
Q
U Q & (3.18)
KAPITEL 3. DER QUASIKLASSISCHE ANSATZ
16
Die Korrespondenzregel beschreibt also, wie Gl. (3.11) in Gl. (3.12) zu übersetzten ist,
©
d.h. den Zusammenhang zwischen ! und ¬ bzw. ¨ und ­ .
Von Cohen stammt die folgende Verallgemeinerung, die die Bestimmung fast beliebiger
Phasenraumverteilungen und der zugehörigen Korrespondenzregeln erlaubt [Coh66]. Dafür
wird Gl. (3.17) in Gl. (3.18) eingesetzt und etwas umgeschrieben:
¬
"
(»¼V
«
rcs /
cGa
¯
c½
E°~± 1E ² E°:¾ ¼ª ¯ 4 Q U
G
!
½ O
0
/° ! ½ l
°/ (3.19)
¼ª ¯
¼ª X
G X ¢
¼ 4 ¯
¼¿
¯
Wenn für die gutmütige“ Hilfsfunktion
gilt, dann ist ¬ real und
G
OKG O
©
”
der zugehörige Operator ¨ ist hermitesch. Diese Hilfsfunktion darf sogar eine Funktion der
Wellenfunktion ! sein.
¼
Nach einigen Zeilen elementarer Rechnung zeigt sich, wie die Korrespondenz be
"
2
der klassischen Variablen « und wird nach der folgenden
schreibt: die Funktion ­ «
Vorschrift abgeleitet, das Ergebnis normal geordnet (alle Produkte von « stehen vor den
Produkten von ) und dann « und durch die Operatoren « ¨ und ¨ ersetzt:
/
© ¼5À EÂzà @ ñ~à ­ "B
¨ «¨ ¨
(3.20)
´
´ ªÁ S u
OBÅ
Q_Ä «
u
«
mit
3.4.1.1
verschiedene Korrespondenzregeln
Bisher wurden in der Literatur fünf Korrespondenzregeln vorgeschlagen (siehe z.B. [She59]
und [Meh64]):
(a) Dirac schlug vor, Kommutatoren von Operatoren über Poissonklammern der Funktionen
zu bestimmen [Dir26]:
©
©
´ ¹ © ¨ ¨ ¬
¬ ¨ ¨ O ¨ ¬Y
(b) Von Neumanns Regel(n) [VN27]:
¹ ¹ -¼¿ 3
¼ª
¨
1 Wenn ¨ ¨ , dann gilt ¨
¹ ÆÇ
¹ È
¹ Æ
Æ
È
2 Wenn ¨
¨ und ¨
¨ , dann gilt ¨
Æ
È
tauschbarkeit von ¨ und ¨ zu fordern
l
È
Æ
¨ l
È
¨ , ohne die Ver-
Diese Korrespondenzregel führt, wie die Diracsche, auf Widersprüche, da damit z.T. die
gleiche klassische Funktion durch verschiedene quantenmechanische Operatoren dargestellt
werden kann, siehe z.B. [She59].
(c) die wohl bekannteste Korrespondenz ist die Weylsche:
É
É
J
µ Ì_Í
F J
F J
«
Å F ɤÊ
«¨ ¨ «¨
Ë
(3.21)
Von Moyal wurde 1949 gezeigt, daß aus dieser Korrespondenzregel die Wignerverteilung
folgt [Moy49]. In Cohens Ansatz, Gl. (3.20), eingesetzt, folgt daraus
¼
w
)
3.4. DIE ANFANGSVERTEILUNG
17
(d) die symmetrische oder Riviersche Regel [Riv57]:
J F F J
F J
«
Å «¨ ¨ l ¨ «¨
Sie liefert
¼
(3.22)
¯ w
ÏÎ_ÐÑ G <
Daraus folgt eine Phasenraumverteilung der Form (siehe dazu auch [MH61] und [Meh64])
4 i$jÒ "B
¯' E1²
¯Ó w
Q !
(3.23)
¬ «
rts
! «
«DO
(e) die Korrespondenz nach Born und Jordan [BJ25]
É
É
J
J
F J
F «
Å
ÉÔÊ ¨ « ¨ ¨
l (3.24)
¯ G <
w
¯ G <
¼
Auch aus dieser Korrespondenz folgt durch Einsetzen von in Gl. (3.19) eine Phasenraumverteilung.
führt auf
¼
ÑNÕÖ
Die in der CTMC bisher übliche mikrokanonische Verteilung ist ein rein klassischer Ansatz, sie ist auf diese Weise nicht ableitbar. Das zeigt sich bereits daran, daß die Ortsverteilung nicht mit der quantenmechanischen übereinstimmt, also nicht der Randbedingung“
”
Gl. (3.13) gehorcht1.
3.4.1.2
Allgemeine Bewegungsgleichungen
Cohen gibt in [Coh66] auch die Zeitentwicklung einer Phasenraumverteilung nach Gl. (3.19)
an ( . . . a direct but somewhat lenghty calculation. . .“):
”
"(
N
¼
´ 2Ø ´ 2Ø ¼
´ 2Û ´ 2Û ¼ ´ 2Ø
´ 2Û ´ 2Ø
´ 2Û
S× ±
× O
× ± l
7"¬ «
l
± O
±
QÚÙ
QÙ
Q
QÙ
2Û 2Ø :B "
3
2Ø 2Û
Ñ3ÕzÖ Ü
«
¬ «
l
± O
±ºÝ
Q
Q
¼
´ Ø ´ Ø
m× O
± O
:(
3~
Q Ù
(3.25)
l ¼
´ Ø ´ Ø ¬ «
× O
± O
Q Ù
Ø
Ø
Û
Û
Die Differentiationen
und ± wirken auf ¬ allein,
und ± nur auf . Dieser
Q
Q
Ausdruck reduziert sich auf die klassische Liouvillegleichung (Gl. (3.4), siehe auch Anhang
¼
A), wenn für als Funktion von gilt:
Þ
Þ
¼
¼
)
und àá_Õzâ ß m YX
àá_Õzâ ß ¼
Alle im vorigen Abschnitt aufgeführten erfüllen diese Bedingung, d.h. alle diese Kor
respondenzen führen im =0–Limes zur klassischen Liouvillegleichung.
1
Es existiert keine klassische Phasenraumverteilung, die ohne zusätzliches Potential eine scharfe Energie
besitzt und die Ortsraumdichte des Wasserstoffatoms korrekt reproduzieren kann — mindestens der Bereich für
ãªäå a.u. ist hierbei klassisch nicht zugänglich, kann also überhaupt nicht nachgebildet werden.
KAPITEL 3. DER QUASIKLASSISCHE ANSATZ
18
3.4.2
Das Wigner–Weyl–Bild der Quantenmechanik
Die prominenteste der oben vorgestellten Phasenraumverteilungen ist die Wignerverteilung,
die auf der Weylschen Korrespondenz aufbaut. Ursprünglich wurde sie von Wigner aus
der quantenmechanischen Dichtematrix abgeleitet [Wig32]. Da sie die gebräuchlichste ist,
soll an ihr gezeigt werden, wie die klassische Rechnung aus dem quantenmechanischen
Streuproblem als Näherung hervorgeht, die Herleitung läßt sich genauso mit den anderen
vorgestellten Korrespondenzen durchführen.
Vorher wird aber noch ein Überblick über die benötigten Aspekte des Wignerformalismus
gegeben. Eine ausführliche Darstellung, speziell für die Anwendung auf Streuprobleme, ist
z.B. in [CZ83] gegeben.
3.4.2.1
Definition der Wignerverteilung
Die quantenmechanische Dichtematrix æ für einen reinen Zustand ! ist
4 ¡ 3~ ¡ / N w
æçè#%!'&_"!$#Y!
!
S
Nun werden Relativ- und Schwerpunktskoordinaten
¡
¡ /
h
¡
¡ /
l
S und éD
O
S
eingeführt und und die Relativkoordinate fouriertransformiert. Das ist die Definition der
Wignerverteilung 2 [Wig32]:
~êëh
:(»3~
E àá 4 h
3 h
3~
!
s cé Qíì u !
!
O /ì
(3.26)
l /ì
h
Die Größen und sind Phasenraumvariablen, keine Operatoren; die Wignertransformierte einer beliebigen Wellenfunktion ist reell, allerdings nicht notwendig überall positiv.
Durch Integration über den Orts- bzw. Impulsanteil erhält man die korrekten (quantenmechanischen) Aufenthaltswahrscheinlichkeiten:
/
/
~ê
h
h' ~ê
12
!
î#%!
#
!
è#! ¨
#
Die Norm der Wellenfunktion bleibt erhalten:
¡ 0¼CêÚ(ï Vï¡ 3~
"!$#%!'&(
-¡ .
¡ / Analog zu Gl. (3.26) wird ein beliebiger Operator
ins Wignerbild übersetzt:
S
ðêñ-h
"(»N
E àá
h
.h
ò
cé Q_ì u
Oé<
(3.27)
l éC<
h
Für einen Hamiltonoperator der Form
/ óQ J @ lõô
ist dessen Wignertransformierte
h
"2
h
gleich der Hamiltonfunktion
der Variablen und , im folgenden wird also statt
5~ê
geschrieben.
x x
Ein anderer wichtiger Operator ist der Projektionsoperator ö÷$ø# &_ # auf den Zustand
x
# &s . Aus den obigen Definitionen (Gln. (3.26) und (3.27)) folgt, daß er bis auf den Faktor
x
der Wignertransformierten des betrachteten Zustands # & entspricht.
2
Die Wignerverteilung wird hier nur eindimensional dargestellt, die Erweiterung auf mehrere Dimensionen
ist eindeutig [Wig32].
3.4. DIE ANFANGSVERTEILUNG
3.4.2.2
19
Erwartungswerte
Erwartungswerte eines Operators werden aus der Dichtematrix über die Spur bestimmt,
daraus folgt, daß im Wignerbild über den Phasenraum integriert werden muss3 :
ù.ú K
æ
h j ~êç ~ê
!
"D&
(3.28)
~ê
Diese Form entspricht der Bestimmung klassischer Mittelwerte: !
übernimmt die
ê
Rolle der Verteilung, während
die betrachtete Meßgröße beschreibt.
Die Wignertransformation ist keine lineare Operation, denn:
sondern:
æ
¨¨
~ê'h
"Bû
ðêÚh
"2
æ¨ ¨
â
â
~ ê'h
"B' ðêÚh
"2
æ¨
¨
ð êÚh
"2
ýü K ðêÚh
"2
æ¨
Ñ3ÕzÖ
¨
(3.29)
üý
Oîþ T wirkt nach rechts und nach links. Der Ausdruck
Der Differentialoperator ÿþ T
Q
Q
:üý üý
wird auch, in Anlehnung an die Poissonklammer, die durch
beschrieben wird
Ñ3ÕzÖ
(siehe Gln. (3.4) und (3.5)), Moyalklammer genannt.
Die Wignertransformierte einer Produktwelle ist das Produkt der Wignertransformierten4 :
h x h / 3 ê
-h 3ðêÚ x h / 3~ê
(3.30)
!
!
S
S
3.4.2.3
Zeitentwicklung
Aus der Schrödingergleichung folgt für die Zeitentwicklung einer Dichtematrix, bzw. eines
Operators
´
7æ O ¥
æ ¦
´
¥
7
¦
¼
Werden beide Seiten wignertransformiert, so ergibt sich (siehe Gl. (3.25) mit ) ):
/
àá / ~ê
ðê
7 !
O àá
Ñ3ÕzÖ ×
!
(3.31)
ðÙê
y
!
h
O
l
y
y ~ê
/ àá |
|
|
S
U U
U U
U U
!
ô
E
h | wwCw h | wwCw ¶ wCww ¶
l
{ ungerade
F
S
àá /
~ê
~ê
àá
7
Ñ3ÕzÖ × /
(3.32)
Ù
3
Beweis durch Einsetzen der Definitionen.
Beweis durch einsetzten: die Fourierintegrale sind unabhängig, da die Variablen ã | und ã unabhängig sind
@
— in Gl. (3.29) sind und Funktionen der gleichen Variablen.
4
KAPITEL 3. DER QUASIKLASSISCHE ANSATZ
20
â
Oèþ T
Q
Q
Die ersten Terme der Gl. (3.31) sehen ausgeschrieben so aus:
wieder mit
~ê
7 !
þ T
ýü
â
ð ê
~ ê
T !
T
!
O
l
àá/ Q } } ê
áà / Q ê
!
O @ T
!
l
l T
Q
Q
S O
(3.33)
Die beiden Terme ohne in Gl. (3.33) entsprechen der klassischen Liouvillegleichung
h
"2
Gl. (3.4) für Phasenraumverteilungen
. Daraus wird ein klassischer Liouville–Operator definiert, der, analog zur Schrödingergleichung, die Zeitentwicklung klassischer Phasenraumverteilungen beschreibt:
7"
O
Q
O
´
T
Q
(mit Poissonklammer)
T l
3
(mit Liouvilleoperator)
(3.34)
Analog kann aus Gl. (3.31) ein quantenmechanischer“ Liouvilleoperator '± für die kom”
plette Zeitentwicklung einer Wignerfunktion definiert werden:
/
àá / ê
´
ê
E
á
à
7 !
± ZO
(3.35)
)O ± !
!
Ñ3ÕzÖ ×
Ù
Diese Definitionsgleichungen lassen sich formal integrieren; damit können — analog zur
EÛ 7
Quantenmechanik, wo "
gilt — Zeitentwicklungsoperatoren definiert werden:
"ª±
"'
3.4.3
E$#&%~7
E$#&(:7
Die klassisch genäherte Anfangsverteilung
Oben wurde gezeigt, wie über die Korrespondenzregeln Wellenfunktionen/Dichtematrizen
eindeutig mit Phasenraumverteilungen verknüpft sind. Diese Verteilungen sind also nur eine
andere Darstellung der Quantenmechanik.
Diese Verteilungen werden nun klassisch genähert, d.h. èX gesetzt, und für die Integration der Zeitentwicklung diskretisiert:
¬
-h
"B
Å
'FÚ
h
O
h
F
1
O
F
(3.36)
Der klassische Limes X hat zur Folge, daß alle Anteile einer Wignertransformier
ten (oder einer anders bestimmten Verteilung), die explizit von abhängen, entweder verschwinden oder so schnell oszillieren, daß sich die Beiträge infinitesimal voneinander entfernter Phasenraumpunkte, die zu praktisch den selben Endwerten führen, gegenseitig aufheben. Sie können daher von Anfang an aus der Verteilung herausgelassen werden5 . Bei den
5
Bei einem chaotischen System, bei dem auch sehr nahe benachbarte Phasenraumpunkte nicht mehr nebeneinander im Querschnitt beitragen, muß im Einzelfall überprüft werden, ob diese Argumentation der stationären
Phase zutrifft. Die hier betrachtete Elektronenstoßionisation an Wasserstoff ist nur schwach chaotisch (es handelt sich um ein Dreikörperproblem), d.h. die chaotischen Anteile des Phasenraums sind für diese Anwendung
vernachlässigbar klein.
3.4. DIE ANFANGSVERTEILUNG
21
bisher vorgestellten Verteilungen treten beim Wasserstoffatom keine expliziten –Abhängigkeiten auf, sie können daher direkt als klassische Anfangsverteilung verwendet werden.
Die Dichte der Stützstellen wird entsprechend des Betrags der Verteilung ausgewählt, Ort
und Impuls werden dazu gleichmäßig im Phasenraum verteilt ausgewürfelt“. Die Wahrk0h " ”
scheinlichkeit
F
F , einen Phasenraumpunkt als Startwert auszuwählen, ist mit der
h
"B
:
normierten Verteilung ¬
k0h " h : F
F î#%¬
F F #
(3.37)
h : Ist die Verteilung bei F F negativ, so bekommt diese Trajektorie das Gewicht O$ 6 :
)
-h : ¸
F
F
X
für ¬
l -h : 'F
(3.38)
F
F · X
für ¬
O$
Im Gegensatz zu diesem importance sampling“ kann auch der Phasenraum auf einem
”
Gitter diskretisiert werden, jede Trajektorie bekommt dann das Gewicht der Phasenraumh " F
F .
verteilung am Stützpunkt, d.h. 'FjY¬
Der Normierungsfaktor dieser diskretisierten Verteilung ist gegeben durch:
h
3.4.4
¬
h
"B
F
Vorteile und Probleme
Die Vorteile einer solchen Herleitung liegen auf der Hand: die einzige Wahlmöglichkeit“,
”
die noch bleibt, ist die Entscheidung für eine bestimmte Korrespondenzregel.
Mit vergleichbaren Ansätzen wurde bisher bereits mehrfach versucht, die Standard–CTMC zu verbessern“. Doch wenn eine wie hier aus der Wellenfunktion abgeleitete Pha”
senraumverteilung naiv“ eingesetzt wird, so ergeben sich daraus eine Reihe von Schwie”
rigkeiten (vgl. Abschnitt 2.2.2.1):
Die Verteilungen sind nicht positiv definit. Das ist leicht ersichtlich, wenn man das
Skalarprodukt zweier orthogonaler (Wellen–)Funktionen im Wignerbild betrachtet:
Damit das Phasenraumintegral verschwindet, müssen zumindest Teile einer der beiden Funktionen negatives Vorzeichen haben. In der Standard–CTMC werden diese
PR–Verteilungen direkt als Wahrscheinlichkeiten interpretiert, negative Wahrscheinlichkeiten stellen aber ein konzeptionelles Problem dar.
Diese Verteilungen sind unter klassischer Propagation nicht station är, die Poissonklammer mit der Hamiltonfunktion verschwindet nicht (siehe Anhang A.1):
û
¬
X
(3.39)
¸
Das ist in erster Linie auf den Anteil freier“ Startwerte, für die Q / @ O T S
X ist,
”
zurückzuführen, gilt aber auch für alle Verteilungen, die nicht aus vollst ändigen mikrokanonischen Beiträgen aufgebaut werden können (z.B. die von Eichenauer etal.
Der Fall *,+,- braucht nicht explizit beachtet zu werden, da dann die Wahrscheinlichkeit, eine Trajektorie
zu starten, ebenfalls verschwindet.
6
KAPITEL 3. DER QUASIKLASSISCHE ANSATZ
22
verwendete cut–off Wigner“–Verteilung [EGS81]). Hier besteht das Problem darin,
”
daß das Target zerläuft“, während das Projektil, in einem großen Abstand gestartet,
”
herankommt. Die Rechnung sollte aber unabhängig davon sein, bei welchem Abstand
(in Raum, bzw. Zeit) das Projektil präpariert wurde.
Ionisation ist in der CTMC durch positive Endenergie der beteiligten Teilchen definiert. Wenn eine Trajektorie nun bereits mit positiver Energie gestartet wurde, ist
diese Definition unbrauchbar.
Wie bereits in Abschnitt 2.2.2.1 dargestellt, führt die Breite in der Energieverteilung
der Startwerte dazu, daß energiedifferentielle Querschnitte nicht mehr eindeutig definiert werden können.
Diese Probleme zeigen, daß es nicht ausreicht, nur einen Teil des klassischen Modells
durch quantenmechanische“ Größen zu ersetzen.
”
Im weiteren Verlauf zeigt sich, daß diese Schwierigkeiten erst gar nicht auftreten, wenn
die Herleitung der klassischen Näherung aus der Quantenmechanik konsequent durchgeführt
wird. Das ist bereits am ersten Punkt zu erkennen: Startwerte, bei denen die Verteilung negativ ist, werden in der Auswertung ebenfalls negativ gezählt, im Wirkungsquerschnitt also
abgezogen. Das Problem der Standard–CTMC ist der Versuch, dies als negative klassische
Wahrscheinlichkeit zu interpretieren. Man darf die Wignerverteilung aber nicht auf eine
Stufe mit klassischen Wahrscheinlichkeitsdichten stellen, sie ist eine Phasenraumverteilung,
aus der erst in der Auswertung Wahrscheinlichkeiten bestimmt werden.
Der wesentliche Unterschied zur bisherigen CTMC liegt also in der Interpretation der
propagierten Verteilung: dort wird mit echten Wahrscheinlichkeiten gearbeitet, die gesuchte Größe (die Wahrscheinlichkeit) ist von Anfang an vorhanden, hier sind es Phasenraumverteilungen, aus denen Wahrscheinlichkeiten erst abgeleitet werden müssen. Daher kann
in der CTMC eine Trajektorie als eine mögliche Realisierung des Experiments angesehen
werden, hier jedoch ist eine einzelne Trajektorie nur eine Stützstelle (siehe Gl. (3.36)) und
allein quasi wertlos“ — erst die gesamte Verteilung beschreibt das System mit allen seinen
”
Realisierungen.
3.4.5
Anders motivierte Anfangsverteilungen
Neben der Möglichkeit, über die Korrespondenzregeln Phasenraumverteilungen zu bestimmen, existieren noch andere Ansätze für klassische Verteilungen, die Verbesserungen gegenüber der mikrokanonischen darstellen, was Orts- und Impulsverteilung betrifft (siehe
Gln. (3.13) und (3.14)). Da Cohen zeigte, wie das Konzept der Korrespondenz verallgemeinert werden kann, um beliebige PR–Verteilungen zu konstruieren, kann für die nachfolgend
vorgestellten Ansätze angenommen werden, es existiere eine jeweils passende Korrespondenzregel (Abschnitt 3.4.1). Die folgenden Argumente wären dann eine alternative Herleitung der Verteilung (vgl. Abschnitt 3.4.2.1).
3.4.5.1
Produktverteilung
Die Heisenbergsche Unschärferelation verknüpft den Kommutator zweier Operatoren ¨
Æ
und ¨ mit der minimalen Unschärfe, mit der die zugehörigen Observablen gleichzeitig
3.4. DIE ANFANGSVERTEILUNG
.
bestimmt werden können:
23
.
Æ0/21 11 ´ 3¥2 ¨ Æ ¨ ¦ & 11
1
1
1
Der Wasserstoffgrundzustand ist ein Zustand minimaler Unschärfe, es kann daher nur
entweder die Orts- oder die Impulsverteilung angegeben werden. In der klassischen Nähe
rung wird nun X gesetzt, damit verschwindet der Kommutator und Ort und Impuls
werden unabhängig voneinander. Das führt auf den Ansatz einer Phasenraumverteilung aus
dem Produkt der Dichten:
/
/
h
"B
h
12
(3.40)
¬
î#%!
# #! ®
#
Diese Produktverteilung“ liefert die korrekten Erwartungswerte für Ort, Impuls und
”
mittlere Bindungsenergie und besitzt ebenfalls einen Anteil ungebundener Startwerte, so
wie die Verteilungen in Abschnitt 3.4.1.1.
Hier wird also zuerst die klassische Näherung Å X durchgeführt, danach erst die
Wellenfunktion in eine Phasenraumverteilung übersetzt; die Reihenfolge ist gegenüber der
Wignerverteilung (und deren Näherung) umgedreht.
Diese Phasenraumverteilung läßt sich auch mit dem Ansatz nach Cohen (Gl. 3.19) mit
der Hilfsfunktion
/
/
D Ez°~± EP²
1B
t
d
«
%
#
!
«
#
#
!
#
U
®
Q
¼ª ¯
G
¯ ª Ez°:¾
¯ 4 ½0O
<
! ½ l
<
t½
!
herleiten.
3.4.5.2
Energieverteilung nach Cohen
In [Coh85] wird von Cohen7 ein anderer Weg beschritten: Da bei fester Energie der Startwerte die Ortsverteilung des Wasserstoffgrundzustands nicht nachzubilden ist, läßt er eine
Energieverteilung zu und modelliert die Dichten durch Überlagern mikrokanonischer Verteilungen verschiedener Energie (vgl. Abschnitt 2.2.1):
h
:B
¬
Y¬
=
h
:B3
Es soll also gelten:
rtq s
æ
h
12
6
rcs
S
u
T
c=Y¬
t=Y¬
=
=
æ J zE y T 8
h
6 J Ezy T 8
1(
=
3
=
/
s
3 s /
T
(3.41)
l
6 / (3.42)
54
1
æ J Ezy T 8
= und 6 J Ezy T 8
=
sind die Dichten in Ort und Impuls einer mikrokanonischen Verteilung zur Bindungsenergie = .
Gl. (3.42) kann, wie bekannt, durch eine Delta–Funktion
der Energie gelöst werden,
s } w¤£ = l X
durch die mikrokanonische Verteilung: ¬ = ¢<
h
7
Diesmal ein anderer Cohen. . .
KAPITEL 3. DER QUASIKLASSISCHE ANSATZ
24
Es existiert keine Lösung, die beide Gleichungen gleichzeitig erfüllt — das System ist
durch die Festlegung der Drehimpulsverteilung der überlagerten mikrokanonischen Verteilungen überbestimmt, da auf eine der Variablen“ verzichtet wird —, aber die Energie”
verteilung, bei der die Ortsverteilung korrekt ist, gibt die auch Impulsverteilung recht gut
wieder.8
Der Ansatz schließt freien Trajektorien explizit aus, die obere Integrationsgrenze der
Energie ist Null. Die Verteilung ist daher station är. Die Unsicherheit in der Bestimmung
energieabhängiger Querschnitte bleibt allerdings weiter bestehen.
3.4.6
Das Projektil
Die Wahl der PR–Verteilung des Projektils ist eindeutiger: Im Eingangskanal läuft das Projektilelektron auf ein neutrales Atom zu, es ist — weit weg vom Atom — frei.
q Die quanten=MEPF :
mechanische Lösung des Problems ist eine ebene Welle mit Impuls 7E1F
Ezy3{ T
x
E( s } /
u
Deren Wignertransformierte ist eine Deltafunktion des Impulses — die anderen oben
vorgestellten Ansätze liefern bis auf Vorfaktoren9 die gleiche Abhängigkeit:
} 1
x ðê
O 7EPF
8
s
E
(3.43)
}
Für die klassische Propagation wird auch diese Verteilung diskretisiert (vgl. Abschn. 3.4.3),
die Startwerte werden gleichmäßig auf der Ebene senkrecht zu 7E1F verteilt. Dabei reicht es
aus, die beschossene“ Fläche zu begrenzen, da ab einem bestimmten Stoßparameter keine
”
Reaktionen mehr stattfinden; üblich ist eine Kreisscheibe mit Radius J H3L , einem maximalen Stoßparameter. Das so bestimmte Wellenpaket“ wird durch die folgende Phasenraum
: 9
”
verteilung beschrieben, E1F
¨ :
Q
h
:B
s
/
J HNL
E1F;
'Fc
}
O
=<
E1F
(3.44)
Mits / E1;F F wird die Verteilung auf die Flächendichte — und damit auf den Fluß —
<
J HNL normiert. Der Stoßparameter kann, da das Target bereits rotationssymmetrisch
um die Einschußrichtung ist, in einer Richtung senkrecht dazu ausgewählt werden. Hier
¡
wurde die -Achse verwendet. Die Verteilung im Stoßparameter erfolgt somit nach
/
/
wwCw
w
gleichmäßig in ¥¤X
J H3LC¦
Das Target ist quantenmechanisch ein Eigenzustand, also station är; die Übersetzung in
eine klassische Verteilung kann daher zu jedem beliebigen Zeitpunkt erfolgen. Aus praktischen Gründen, die leider erst im nächsten Abschnitt klar werden, wurde der Zeitpunkt
8
Vergleiche mit dem Vorgehen von Eichenauer etal, die die Abschneideparameter des Energiebereichs der
Wignerverteilung durch einen Fit an gemessene Querschnitte bestimmten [EGS81]. Cohens Ansatz ist dagegen
parameterfrei“.
” 9
Die Vorfaktoren betreffen nur die Normierung und sind in jeder der Darstellungen konsistent miteinander.
Lediglich bei einem Wechsel zu einer anderen Korrespondenzregel innerhalb der Rechnung muß dies explizit
in Betracht gezogen werden.
3.5. DIE PROPAGATION
25
der Übersetzung für jeden Startwert auf den Nulldurchgang des Projektils durch die Ebene senkrecht zum Targetmittelpunkt (dem Kern) gesetzt, daher die Delta–Funktion in z in
Gl. (3.44):
3.4.7
Die komplette Anfangsverteilung
Die gesamte Anfangsverteilung für Target und Projektil ist ein Produktansatz der beiden
Verteilungen, da das Problem separiert (Gl. (3.3)), die Diskretisierung (Abschnitt 3.4.3)
erfolgt für Target und Projektil gemeinsam: die dreidimensionalen Orte und Impulse in den
Gln. (3.37) und (3.38) werden zu sechsdimensionalen Phasenraumpunkten, die Verteilung
ist eine Funktion aller Koordinaten und Impulse.
-h : h " A
@
h " ¬
Y?
¬ >
> >
¬
n
n
n
k0h
:
F
F
'F
Es gilt wieder
E1F;ª
h
Q
F
"
#¬
)
h
F
l O$
"
F
#
(3.45)
-h " ¸
für ¬
F
- h "
F für ¬
F
F ·
X
(3.46)
X
F , die komplette Anfangsverteilung lautet also:
F
s
J 3H L E1FB
/
FÚ
-h
O
h
F
1
O
F
(3.47)
Das ist die gleiche Form wie in der Standard–CTMC. Der Unterschied besteht tatsächlich
h
nur in der Auswahl der Startwerte 'F , F und F , sowie in der Interpretation: in der CTMC
h " C
ist 'F die Wahrscheinlichkeit, daß echte Elektronen die durch F F
beschriebene
Bahn durchlaufen. Im hier vorgestellten quasiklassischen Ansatz handelt es sich nur um
einen Phasenraumpunkt, der z.T. wegen der Unschärferelation nicht zu einer echten Bahn
gehören darf.
3.5
Die Propagation
Als zweiter der drei Teile soll nun die Propagation betrachtet werden. Sie beschreibt die
Dynamik des Stoßes: sie überführt die Anfangsverteilung in den gestreuten Zustand“, d.h.
”
in die Phasenraumverteilung des Endzustandes. Dieser wird dann im dritten und letzten
Schritt ausgewertet.
3.5.1
Der Ansatz nach Møller
Das wesentliche Element in der quantenmechanischen Streuung ist die Übergangsamplitude
x x
2C
E vom asymptotischen Anfangszustand E in den
bzw. –wahrscheinlichkeit !
asymptotischen Endzustand ! ; der Streuoperator D beschreibt die Reaktionsdynamik:
/
x
E
C x
!
E î#1"! # D
# E9&#
(3.48)
Der Ansatz von Møller geht davon aus, daß asymptotisch, d.h. für sehr große Abstände,
das Wechselwirkungspotential vernachlässigt werden kann, ein echter Streuzustand ! —
26
KAPITEL 3. DER QUASIKLASSISCHE ANSATZ
x
genauer: ein Wellenpaket10 — von einem freien Wellenpaket H J nicht zu unterschei
ì=F
OK+ , kann das formal
den ist (siehe z.B. [Tay72]). Für den einlaufenden Ast, also Å
geschrieben werden als:
Þ
11 !dE O x EH J ~ 11 YX
(3.49)
7 â Õß
ì=F
1
4 1
G
Die zeitliche Entwicklung der Wellenfunktionen wird durch Zeitentwicklungsoperato~
beschrieben, wobei alle Wechselwirkungen enthält und das asymptotische
ren "
Problem beschreibt, in dem die Wechselwirkung, die zur Streuung führt, abgeschaltet ist:
EÛ 7
~
E Û v~7
und "a
(3.50)
"
~
!
"
!
x
~ x
H J
"a
H J
Hì F
Hì F
Gl. (3.49) erhält damit die Form
Þ
11 " ~ !dEBOI"a x EH J 11 YX
(3.51)
7 â Õzß
ì=F 1
4 1
G
Die analoge Gleichung gilt für den Endkanal. Durch Umstellen ergeben sich die folgenden Ausdrücke für die echten Streuzustände des Eingangs–, bzw. des Ausgangskanals !dE
und ! :
Þ
~ E ~ x E
(3.52)
! E "KJ
" H J
7 â Õzß
=ì F
5
4
Þ
~
x
!
" J
" (3.53)
H J
7 â Õzß
=ì F
U 4
Das führt zur Definition der Mølleroperatoren M
L . Sie stellen die Verbindung zwischen
einem asymptotischen Zustand, d.h. ohne die Wechselwirkung, die zur Streuung/Reaktion
führt, und dem echten Streuzustand, der eine — oft analytisch unbestimmbare — Lösung
des vollen Hamiltonians ist, her:
Þ
~ E ~
"KJ
" (3.54)
7 â Õzß
U
Þ G4
Pz P1
J
Õzß
" J
"
(3.55)
7ON â
U 4
Man beachte, daß Anfangs- und Endkanal durch den gleichen vollen
Hamiltonoperator
E
beschrieben werden, während die asymptotischen Äste ( und ) nicht identisch sein
E
müssen. Durch das Møllerschema existiert eine kontinuierliche Abbildung von auf .
E
E
u
JL
M
5
L
(3.56)
u
Der (Teilchen–)Strom bleibt erhalten und es können absolute Querschnitte berechnet werden11 . Mit diesen Definitionen und den Gln. (3.52) und (3.53) in Gl. (3.48) erhält man für
den Streuoperator
DY J U
(3.57)
10
Nur für Wellenpakete ist der Begriff Abstand“ sinnvoll definierbar, eine kontinuierliche Lösung (ebene
”
Welle, Coulombwellen, . . . ) ist unendlich ausgedehnt. Die Wechselwirkung wird nie vernachlässigbar.
11
Das ist ein typisches Problem quantenmechanischer Querschnitte, die in Bornscher Näherung berechnet
werden. Hier sind Anfangs– und Endzustand Lösungen verschiedener Probleme (Hamiltonoperatoren), die
Stromerhaltung ist verletzt.
3.5. DIE PROPAGATION
27
Der so definierte Streuoperator erhält die Gesamtenergie12 . , denn nach Gl. (3.56) wird
E
auch ein Eigenzustand von auf einen Eigenzustand zu abgebildet.
xQ
x
Im folgenden wird
der gestreute Anfangszustand mit # & RD
# & bezeichnet, das Max Q
trixelement mit "! # & .
3.5.2
Die Propagation im Wignerbild
Die Zeitentwicklung der Wignerfunktion wurde bereits in Abschnitt 3.4.2.3 beschrieben.
Wie dort gezeigt, können für quantenmechanische Wellenfunktionen, für Wignerfunktionen
und für klassische Phasenraumverteilungen jeweils vergleichbare Zeitentwicklungsoperatoren definiert werden, Gln. (3.50), (3.34) und (3.35):
Wellenfunktion:
"
Wignerfunktion:
"ª±
" klassische Phasenraumverteilung:
~
EÛ 7
ES# % 7
ES#T(:7
Für die Anwendung des Møllerformalismus im Wignerbild, bzw. in dessen klassischer
=0–Näherung, brauchen also nur die Wellenfunktionen durch Phasenraumverteilungen ersetzt zu werden; dabei sind natürlich die Propagatoren und Skalarprodukte entsprechend
auszutauschen; das Propagationsschema bleibt erhalten.
Ê
àá ê
"B
!
OBÅ
!
OBÊ Å
¬ «
àá ~
"
OBÅ
" ±
OBÊ Å
" àá x
ðê¿ x ~ê
"!$# &
OBÅ
!
OBÅ
¬ U ¬d÷
c«a
c«a Da die Notation mit Wellenfunktionen am übersichtlichsten ist, soll sie noch für die
nächsten Formeln beibehalten werden, die Aussagen gelten aber, mit obigen Übersetzungen, auch für (klassische) Phasenraumverteilugen.
Das (quantenmechanische) Streumatrixelement (Gl. (3.48)) hat mit ausgeschriebenen
Mølleroperatoren die folgende Form:
Þ
Þ
~
P E P :! V# DD#%!dE9&ª 7 â Õzß
"! W# " J
"
" J
" #%!dE&
(3.58)
â Õzß
O
7
N
4
4
5
Dieses Streumatrixelement besitzt einen ausgezeichneten Zeitpunkt, an dem es berechnet
wird: X ist der Bezugspunkt, auf den der asymptotische Anfangs- und Endzustand
bezogen sind.
klassischen Rahmen erweist es sich als praktischer, die letzte
Für die Anwendung im
3
freie Propagation d.h. " J
auf den Endzustand anzuwenden (vgl. Abb. 3.1):
Þ
Þ
P E P "! # DD#%!dE&ª 7 â Õß
Õâ ß
"
! W# "
K
"
J
" !dE &
(3.59)
4 7N G
4
Auf diese Weise kann in der Auswertung die korrekte asymptotische Form des Endzustandes — ebene Wellen — verwendet werden (siehe z.B. [BBK89]). Das Matrixelement
12
siehe z.B. [Tay72], S. 342ff
KAPITEL 3. DER QUASIKLASSISCHE ANSATZ
28
φiasym (t=0)
U0i(t)
ψi(t=–∞)
<φf |ψs >(t=+∞)
U(t)
U0f(t)
φfasym (t=0)
t
Abbildung 3.1: Bei der hier benutzten Propagation nach Møller (Gl. (3.59) wird der An
E
fangszustand zuerst ohne Wechselwirkung (unter " ) nach OK+ propagiert, dann mit
Wechselwirkung nach l + . Hier wird der Überlapp mit dem ebenfalls nach l +
propagierten Endzustand gebildet.
ist immer noch auf X bezogen, auch wenn die Auswertung“ (das Skalarprodukt der
”
beiden Wellenfunktionen) zu + berechnet wird, es ist lediglich die Reihenfolge von
Propagation und Projektion vertauscht.
Zu den drei verschiedenen Propagationsschritten gehören die entsprechenden Zeitentwicklungsoperatoren (hier nur für Wellenfunktionen gezeigt):
E Û v{ 7
" E
EÛ 7
E
ê2
ê2ê
E ê
"
l
l
Û vX 7
EY
" Für die hier betrachtete Elektronenstoßionisation an Wasserstoff haben die einzelnen Anteile die folgende Form:
/
/
/
E
ê2
E ê
(3.60)
)O h l ¡
¡ /
S l ZO h /
#
O
#
/
/
S
S
/
ê2ê
(3.61)
¡
¡ / O h O h /
S l #
O
#
S
S
Mit diesem Propagationsschema ist die Nichtstationarit ät der klassischen Anfangsverteilungen, der zweite Punkt in Abschnitt 3.4.4, kein Problem mehr, denn die Verteilung wird
auf den Zeitpunkt des Stosses präpariert; beim Zurückpropagieren zerläuft sie zwar, wird
aber, während das Projektil herankommt, wieder auf etwa die Form zurückfokussiert, mit
der sie erstellt wurde. Lediglich eine Polarisation“ durch das herankommende Projektil
”
findet statt, das aber ist ein Vorgang, der auch im Experiment auftritt. Es können also beliebige klassische Anfangsverteilungen ohne weitere Annahmen oder Hilfspotentiale in der
nun klassischen Rechnung verwendet werden — ein länger bekanntes klassisch nicht stabiles System ist das Heliumatom, bei dem Autoionisation stattfindet. Auch dafür eignet sich
das vorgestellte Propagationsschema, wie weiter hinten gezeigt wird.
Der Vergleich mit der Propagation der Standard–CTMC verdeutlicht den Unterschied in
der Behandlung des Targets: in der CTMC wird die Anfangsverteilung von einem Zeitpunkt
3.6. DIE AUSWERTUNG
29
auf die gestreute Verteilung an einem späteren Zeitpunkt abgebildet. Ohne die Einwirkung
des Projektils darf keine Ionisation auftreten, das Target muß in diesem Fall vom Beginn
der Rechnung an bis zum Ende der Propagation stabil bleiben. Hier, im quasiklassischen
Ansatz, sind Anfangsverteilung und gestreute Verteilung zum gleichen Zeitpunkt definiert
(die Auswertung bei î+ ist dabei als technischer Trick“ zu werten), die Phasenraum”
verteilung wird zum jeweils gleichen Zeitpunkt betrachtet: dafür muss sie nicht station är
sein. Die CTMC ist also explizit zeitabhängig, im quasiklassischen Ansatz ist die Zeit eine
Hilfsvariable“, die nur für die spezielle Darstellung des Streuoperators benötigt wird.
”
3.5.3
Zur Gültigkeit des Møllerformalismus
Wenn die Wechselwirkungen bei der Streuung Coulombpotentiale sind, ist der Møller–
Ansatz wegen deren unendlicher Reichweite nicht definiert: für das Abschalten“ der Wech
h
”
selwirkung
bei *$+ , d.h. bei Y+ , ist es wichtig, daß diese Wechselwirkung schneller
/
h
als <
abfällt.
Im betrachteten Streusystem hat man es im Eingangskanal mit einem neutralen Target
mit endlicher Ausdehnung zu tun, dessen Potential ist auf der Skala einiger Atomradien exponentiell abgeklungen. Im Ausgangskanal werden Zustände mit der korrekten Asymptotik
h
bei )+ eingesetzt, das Abschalten“ der Wechselwirkung wird vom Endzustand über”
”
nommen“ und damit korrekt beschrieben; es muß nicht explizit in der Rechnung stattfinden
(Gl. (3.59)). Der Møller–Ansatz ist für die Elektronenstoßionisation also gerechtfertigt.
3.6
Die Auswertung
In den vorherigen Abschnitten wurde beschrieben, wie die Anfangsverteilung präpariert und
diese dann propagiert wird. Hier wird nun, als dritter und letzter Teil, die klassische Näherung der Auswertung besprochen. Es wird gezeigt, wie aus der propagierten Phasenraumverteilung Ionisationsquerschnitte bestimmt werden, welche Probleme sich dabei ergeben
und wie sie umgangen werden.
3.6.1
Der Endzustand
Wie in Abschnitt 3.5.2 beschrieben, wird die letzte freie Propagation des Streuoperators auf
b d f `ce bb ,
[ ^a`ceg
den Endzustand angewendet (Gl. (3.59)). Der Endzustand ist die Lösung zu Z\]_
d.h. ohne alle Potentiale. Dessen korrekte asymptotische Beschreibung ist ein Produktansatz ebener Wellen für die beiden Elektronen [BBK89]. In Abschnitt 3.5.1 wurde gesagt,
daß für den Møller–Ansatz Wellenpakete nötig sind, um Abstände definieren zu können.
Im asymptotischen Endzustand allerdings sind die ionisierten Elektronen und der Kern unabhängig voneinander, der Hamiltonian enthält keine Ortsinformation — Lokalisierung ist
nicht nötig.
hjilkm no
d=tvuw tvx
^qp:rSs =y z|{?}r$~ s b
m
m
(3.62)
Dieser Endzustand beschreibt die Messung, daß an den (weit entfernten) Detektoren
Elektronen mit den Impulsen und e gemessen werden; eine Aussage über den Zeitpunkt des Eintreffens wird nicht gemacht. Der Endzustand muß also translationsinvariant
KAPITEL 3. DER QUASIKLASSISCHE ANSATZ
30
in der Zeit (und damit in Richtung des Impulses) sein. Ein Wellenpaket als Endzustand
würde dazu führen, daß zusätzlich durch die Korrelation zwischen gestreutem Wellenpaket
und Endzustandswellenpaket die Zeitdauer der Reaktion abgefragt wird, während im Experiment nur die Wahrscheinlichkeit gemessen wird, daß die Elektronen bestimmte Impulse
haben.13
Die der Wellenfunktion des Endzustands entsprechende Größe im klassischen, bzw. im
Wignerbild ist der Projektor auf den Endzustand (siehe Abschnitt 3.4.2.1).
Größen mit Zahlen sollen im weiteren den gesuchten Endzustand kennzeichnen, die
Buchstaben–Indizes bezeichnen die propagierten Endwerte der Trajektorie(n).
3.6.1.1
Wiederholung: Der klassische Endzustand
Bei einem klassischen Ansatz interessiert man sich dafür, wieviele Teilchen mit einer bestimmten Energie in eine bestimmte Richtung auslaufen. Das wird formal mit einem Projektor auf den Endzustand beschrieben; dessen Erwartungswert (vgl. Gl. (3.28)) ergibt den
y z
[
Anteil der Trajektorien mit passenden Endwerten. Also (für zwei Elektronen, d.h. p| p ))
in unserem Fall (siehe auch Gl. (3.9)):
y=Yl8 e } e =y M8
^R |z
e|
[
} e =y l8 e }
Über die Erhaltung der Gesamtenergie ist mit der Energie eines Elektrons die andere
festgelegt, so daß diese nicht mehr explizit abgefragt werden muß (siehe z.B. [Ros98]).
Da beide Elektronen ununterscheidbar sind, muß dieser Querschnitt noch symmetrisiert,
d.h. die beiden Elektronen auch vertauscht abgefragt werden. Somit lautet der komplette
klassische Projektor:
3.6.1.2
[
^
8 e } e =y } e y= 8 e }
=
y
z
f
&e
y=!A8 } e y=M8 } e =y M }
e
f z
(3.63)
Herleitung im Wignerbild
Auch quantenmechanisch sind die beiden Elektronen ununterscheidbar, die Endzustandwellenfunktion muß, entsprechend der beiden Spins, (anti)symmetrisiert werden, w für den
[
Singlett–, für den Triplettendzustand.
[
^d
[
b
y=
:} e y=}?
y=} e y=:}
(3.64)
Um den Projektor auf den Endzustand zu bestimmen, berechnen
z|}
yHwir
die
Wignertransw ^
f
R
formierte dieser Wellenfunktion. Mit den Abkürzungen
, ^
13
Das ist kein Widerspruch zu time of flight“–Messungen: Dabei wird der Impuls der Elektronen aus der
”
makroskopischen Flugstrecke zwischen Target und Detektor bestimmt. Ein Wellenpaket, das auf dieser Skala
lokalisiert ist, kann, gemessen an den atomaren Dimensionen, für den eigentlichen Stoßvorgang immer noch
unendlich ausgedehnt“ sein.
”
3.6. DIE AUSWERTUNG
y
yH
z}
}
[
^
^
31
z|}
=y
f
, usw. ergibt sich:
e
y } yH
}
y [ :} y ¡ e } ¡ y } y e :}£
f ¢d
¢d
~
~
²
&
§
S
®
¯
¯
¢¤ d §¨ª©«!¬ ¬ pl¦ ­ u ` ucw x ` x°'± w e w e f w e w e
b¥c¦
, e w ^
(3.65)
(3.66)
Zeile (3.65) beschreibt den klassischen“ Anteil, Zeile (3.66) die quantenmechanische“
”
”
Interferenz.
kann also, je für Singlett und Triplett, geschrieben werden:
[
y=z|{A´³ }µ|y
^
[
[
}
^
s
·¶
(3.67)
Für die Wellenfunktionen der auslaufenden Elektronen werden nun ebene Wellen eingesetzt, Gl. (3.62):
m5
y=z|{?} ~ y
Der klassische Anteil
s
:}
^
~ y
¡
}
und entsprechend
erhält damit die Form:
m5
~ y
^¹º ¸ ± ¡
s
} ~ y A
¡
m
~ y M
e } f
¡
m
~
e } y ?
¡
m
}²
(3.68)
Um den Interferenzterm (3.66) zu bestimmen, wird zuerst das Integral mit dem ersten
Summanden ausgerechnet, beim zweiten Summanden
sind lediglich die Indizes vertauscht:
»
~
«2¬ ¬
¿
§d
p ­ÁÀ ¦
^
^
^
~
§&®S¯ u uw ¯ x ` x° e¼;¾½ ©
® °
p'¦ ­ `
® t u wM u ° § b ® t x w  x ° § d ® t x  x ° § b ® t u  u °ÄÃ
b p ­WÀ ¦
b p ­ÁÀ ¦
b p ­ÁÀ ¦
b
¾
½
® d t u
tvx=w dtxw b t u °ÆÅ
rSs
p Sr s rSs y=b z|{Ç´³ rS}s µ
~ ~
« ¬ ¬
® d
°
p r s sb
® ` u ¯ u  u ® dvw °Æ°OÅ ½¾
­b s sb
p r
® t u tvx°OÅ ½¾ ~ y dvw e
¡
s
® ` x ¯ x  x ® d w °Æ°ÆÅ ½¾
­b s s b
p r
} ~ y
}
dw e b
bs ¡
s s
Entsprechend erhält man
» e ^
¾½ ~ y
} ~ y
}
® d
° ® t u tx°ÆÅ
vd w e
d w e
¡
s sb ¡
s sb
p r s sb
^ » e , ihre Summe ergibt für den Interferenzterm:
~ y
} ~ y l
}
± ® d b °½¾ ® t u tvx° ²
dw e b
dw e b
(3.69)
¶ ^qd
s
s
s s
s s
¡
¡
bÈ:É|Ê
f ¶ , im Triplett–Fall gilt ^
¶.
Der Singlett–Querschnitt ist w ^
[
[
s
Um den korrekten Wirkungsquerschnitt
zus erhalten, müssen diese Terme entsprechend
der
Es gilt dabei
»
Spinstatistik addiert werden, also einmal Singlett und dreimal Triplett:
[
^
^
f Ë
w j
yº[ 8
z [ }
¶
s
KAPITEL 3. DER QUASIKLASSISCHE ANSATZ
32
Damit erhält man schließlich als Projektor auf den korrekt symmetrisierten Endzustand
aus zwei ebenen Wellen:
3.6.1.3
m }
º y
m
m
m
¶
~ y s
~ y
} ~ y e}
}e ~ y
}Ì
f
¡
¡
~ y M
¡
} ~ y ¡ A
}
± ® d b °½¾ ® t u t x ° ²
d
v
w
dw e b
e
b ¡
s
s
s
s
s
s
¡
È:É|Ê
^
[
^
(3.70)
Die klassische Näherung
Der Projektor auf den Endzustand wird klassisch genähert, d.h. wie bei der PR–Verteilung
´³
des Anfangszustandes ^ÎÍ gesetzt (siehe Abschnitt 3.4.3). Somit entfällt der Interferenz–Term oszilliert dann sehr schnell, die Beiträge verschiedener Trajektorien
term (der
ÉÊ Mittel auf), übrig bleibt die klassische Symmetrisierung:
heben sichÈ:im
m
m
m
m
[
^ º
s
^
~ y
¡
} ~ y
¡
~ y
e} f
¡
~
e } y
¡
}
(3.71)
Für die anderen vorgestellten Phasenraumverteilungen erhält man das gleiche Ergebnis für den klassischen Anteil des Endzustandsprojektors. Dabei kann, entsprechend der
gewählten Korrespondenzregel, ein zusätzlicher Faktor auftreten, siehe dazu auch Abschnitt
3.4.6.
3.6.2
Wirkungsquerschnitte
Ð
bestimmEin Wirkungsquerschnitt
Ï in einem
y=
} ist definiert als Verhältnis der Reaktionsrate
ten Endkanal [ [ , hier bestimmt durch die Winkel [ und Energien [ der Endprodukte, zum einfallenden Fluß Ñ .
Ò
Anstatt aus den momentanen rªWerten
kann der Wirkungsquerschnitt
y= auch
} aus den Zeitintegralen bestimmt werden, d.h. aus der Anzahl der Ereignisse Ó ¯Ô [ [ und der gesam^ÎÖ ¬|×Ñ
ten Menge an Projektilen Õ
in der gleichen Zeit:
rªÒ
=
y
}
Ø
[ [ ^
rªÒ
}
Ð =y
Ï [ [ ^
Ñ
rÙÒ
Ó ¯Ô
y=
Õ
[
[
}
(3.72)
rÙÒ
Der einfallende Fluß wird durch Dichte und Geschwindigkeit der Projektile beschrieben:
Ñ
rªÒ
^ÛÚ Ü rÙÒ Ô Ý
Þ
Dabei ist
Ú rªÒ Ô die gesammte Menge des einlaufenden Zustands im ”IntegrationsvoluÜ
men“ , der Impuls
liege, wie üblich, parallel zur z–Achse.
Mit der in Abschnitt 3.4.6 angegebenen Verteilung für das Projektil (Gl. (3.44)) stellt
á Anzahl der Trajektorien dar, das Volumen wird durch den maximalen
Ú rªÒ Ô die (gewichtete)
ßà
Stoßparameter
und in Einschußrichtung durch die Länge ÝÞlâäã beschrieben (vergleiche dazu [Ros98]: Energieerhaltung bedingt Translationsinvarianz in der Zeit).
Somit ergibt sich für den Fluß
Ñ
rªÒ
^
{ e Ú å rÙá Ò Ô
ßà
âäãçæ
3.6. DIE AUSWERTUNG
33
Es mag überraschen, daß der Fluß nun von einer Zeit abhängen soll, aber
Ú rÙÒ Ô ist der
Anteil der PR–Verteilung innerhalb der Länge ÝÞlâäã , so daß Ñ
konstant ist.
Über die Zeit aufintegriert, wird innerhalb âäã die Menge rªÒ
Õ
^
rªÒ
«|è?éS¬×?Ñ
{ Ú e ªr Ò Ô á
ßà
^
rªÒ
(3.73)
an Projektilen auf das Target geschossen.
Für die Anzahl der Reaktionen Ó ¯Ô gilt mit der Wahrscheinlichkeit ê , den gewünschten
Endzustand zu erreichen:
y=
}
(3.74)
Ó ¯Ô ^
m n
m5 m
Ô ê
[ [
Ú rªÒ
y
}
e aus dem AnDie Wahrscheinlichkeit, einen bestimmten Impulsendzustand ^
fangszustand ë zu erreichen,m nist
gegeben:
ì quantenmechanisch
kªím nÄkîYk k durch
kªím ndas
k Überlappintegral
k
r
ê
y
}
ë
r
^
e
ë
rOï
^
ë ¯
e
(3.75)
ï
Für das Wignerbild wird der Projektionsoperator auf den Endzustand benötigt. Dessen
[
Wignertransformierte in der klassischen Näherung, hier ohne Symmetrisierung angegeben,
dazu siehe Gl. (3.70), lautet:
m n
[
^
^
y
µ [ y ¡ ð
¡
m }n
}
y } ¯
ist diskretisiert (Abschnitt 3.4):
µ yH y }} µ y
y } ¯ y=
}
y } }
^ ñ d
Ñ
ê
× ¡
×
¡ ×
¡
Ò
Ò
Ò
­Áòólô
Die Übergangswahrscheinlichkeit wird damit zu:
kªím n k
k
m n
e
µ
µ
y
}
=
y
}
y
}
^
Ñ ¯
«õ¬ ¬ ¡ Ñ ¯
× ¡ ð
¡
m n
[
ï
y }} µ y
}
µ
µ y
^
ê ñ d ¡ m n¡ × ¡
« ¬ ¡
Ò µ|y y }'
}Ò
­Áòó
ô ê
^
ñ d
¡ ×
Ò
Ò
­Wòcó ô
(3.76)
Die gestreute Wignerfunktion Ñ
n
(3.77)
(3.78)
Für den Wirkungsquerschnitt muß noch über das Phasenraumvolumen â des Endzustands integriert werden. Um den Querschnitt in Energie und Winkeln anzugeben, wird auf
Kugelkoordinaten gewechselt, dabei gilt14:
~ y }
y } e y=M}
^
¸
e
¡
¡
n
¡
yH 8!} y= M:} yH e jy=Î8Y
}å} yH e 8v}
â ^
(3.79)
m
m5 y jö z
=y Y:}
!}
^Û
14
Das folgt aus Ö÷:ø=ù?ú:ûcTüù ýAþIÿ in kartesischen bzw. Kugelkoordinaten aus den Volumenelementen ÷:øÆù þ
ù b ÷ù?÷ .
KAPITEL 3. DER QUASIKLASSISCHE ANSATZ
34
Alles eingesetzt:
y
ê
n ì
}
Ñ ¯
â
«|è¬ ^
m n kªím n k
µ
m m
e
«|è ¬ ¬
m m
e
|« è ¬ ¬
m n
e
«|
è ¬ ¬
^
^
^
k
e
Ñ ¯
ï m
e
e
m
e
e
ô
m
kªím n k
k
e
¯
Ñ
m n
ï
µ y y }'
}
ê ¡ ×
mÒ
e y y ô }' } y= Ò y }Ì8 }
ê ¡ ×
×
[
Ò
Ò
Ò
¬ e¬ e
m
¬ e¬ e
(3.80)
Mit Gl. (3.79) kann das Integral ausgeführt werden, die Übergangswahrscheinlichkeit
lautet dann:
ì
ê
y=
[
}
[
^
^
ê
y=! M
Y
M
}
Ñ
y= 8
Y:} e =y ?M
} Hy e ?!} e =y e ?M}
ê
Ò
Ò
Ò
Ò
Ò
(3.81)
ô
Eingesetzt in Gl. (3.72)
erhält man mit den Gln. (3.73) und (3.74) den volldifferentiellen,
nicht symmetrisierten Querschnitt zu:
Ø =y [
^
[
}
3.6.2.1
8 } e y=
? } yH e ? } e yH e ? }
(3.82)
ê
¥ ñ x b u
Ò
Ò
Ò
Ò
Ò
òóôFormel für den klassischen volldifferentiellen CTMC–Wirkungsquerschnitt,
Das entspricht ­Áder
man erhält — bis auf die Deltafunktion für e — Gl. (3.9).
y=
Energieübertrag als Maß
Hier sollte sich Einspruch regen, denn zum einen filtert Gl. (3.82) die Trajektorien her
^
aus,
deren
Gesamtenergie
der
mittleren
Gesamtenergie
des
Experiments
entspricht
(
f
), zum anderen wurde weiter oben eine Anfangsverteilung
vorgeschlagen, die aus
e
^
f
Trajektorien zu unterschiedlicher Energie
besteht. An dieser Stelle wird
Ò
Ò zurückgeworfen,
Ò
man (anscheinend) wieder auf die Standard–CTMC
da auf diese Weise
nur die Trajektorien zum Querschnitt beitragen können, die von Anfang an die korrekte
Gesamtenergie — und damit die korrekte Bindungsenergie des Targetelektrons — hatten:
die Anfangsverteilungen aus Abschnitt 3.4 werden auf eine mikrokanonische Verteilung
reduziert.15
Dieses Problem läßt sich umgehen, denn die absoluten Endenergien können auf Energiedifferenzen umgeschrieben werden:
Für jede einzelne Trajektorie gilt Energieerhaltung (ohne
den Zählindex Ó geschrieben):
Umstellen liefert
15
^
r f
^
[ ^
[ f
r
[ ^
r
f
y= 8
}
[ ^ â
r
æ
e
Bei früheren Ansätzen, in der CTMC eine in der Energie unscharfe Anfangsverteilung zu verwenden,
wurde dieser Punkt nicht beachtet. Die zweite Deltafunktion der Energie in Gl. (3.82) kann in Gl. (3.9) nur
vernachlässigt werden, wenn die Gesamtenergie fest, bzw. bekannt ist.
3.6. DIE AUSWERTUNG
35
â
beschreibt die Energie, die vom Projektil auf das Target übertragen wird. Für den
â
zum Anfangszustand definiert:
Endzustand wird ebenfalls eine Energiedifferenz
^
rÙÒ
â
Damit gilt (vgl. Gl. (3.82)):
y= }
=y
j
y=
}å}
y Î
}
^ f â
â ^ â
â [
r
y= e }
y= 8
y=Î
8rÙ Ò }}
y
}
^
^
^ â â
[
[
æ ææ
Auf diese Weise tragen zum Wirkungsquerschnitt all die Trajektorien bei, bei denen der
Energieübertrag vom Projektil auf das Target kompatibel zum gewählten Endzustand ist.
Diese Vorgehen entspricht einem Franck–Condon–Ansatz, bei dem die Target–Energie–
Verteilung ohne Änderung um â
angehoben wird. Der Anteil der Anfangsverteilung, der
diese Energieübertrag aufweist, bestimmt den energiedifferentiellen Querschnitt.
Vertauschen der Endwerte — die klassische Symmetrisierung, wie in Abschnitt 3.6.1.3
beschrieben — führt auf:
y= e }
y=
jy=Î
} }
yyHz|
82
}
'
ä}
^
f â
^
â
â
[
r â
ªr Ò
y= 8 }
y=
jy=
Î
}}
y jrªy=Ò z|
8Î
}}
^
^
f
[
â
â
â
â
r
rªÒ
rªÒ
Die Richtungen (Winkel) der Endwerte bleiben von dieser Betrachtung unberührt, sie
werden weiterhin aus der Richtung der Trajektorien gewonnen.
Einschließlich der klassischen Symmetrisierung schlagen wir somit zur Auswertung voll
y z }
differentieller p| p –Querschnitte vor:
ø u
x
u
^
¼
b u
­Áòó
ê
y
â
Endwerte
f ê
ô
Ò
y
} =y
â
Î
â
Ò
y=z|
rªÒ
M:} Hy e M}
}} yH 8v} yH e 8
}
â
(3.83)
Diese Umrechnung von den absoluten Energien auf den Energieübertrag muß nach der klassischen Näherung des Querschnitts (Gl. (3.82)) erfolgen, denn nur dann kann die Energie
als Parameter/Variable betrachtet werden — quantenmechanisch ist sie ein Erwartungswert,
der aus dem tatsächlichen Impuls bestimmt wird.
Mit dieser Betrachtung, (relativer) Energiedifferenzen ist auch der dritte und letzte Teil
der Rechnung aus der Quantenmechanik hergeleitet. Jetzt werden auch die restlichen in
Abschnitt 3.4.4 genannten Einwände gegenstandslos:
Ionisation bedeutet nun, daß der Energieübertrag auf das Target größer ist als die
Bindungsenergie, unabhängig von der Gesamtenergie dieser Trajektorie.
Wenn alle Trajektorien bei der korrekten Bindungsenergie gestartet werden, d.h. aus
einer mikrokanonischen Verteilung, so ist die Abfrage der positiven Gesamtenergie
äquivalent zur hier vorgeschlagenen Abfrage des Energieübertrags. Ein Unterschied
besteht nur, wenn die Anfangsenergien verschmiert waren.
KAPITEL 3. DER QUASIKLASSISCHE ANSATZ
36
Für alle Trajektorien ist damit eine konsistente Basis gelegt, nach der energiedifferentielle Querschnitte zu berechnen sind, die Schwierigkeit der Zuordnung existiert
nicht mehr. Da in praktisch jedem Querschnitt die Energie betrachtet wird, betraf
diese Einschränkung so gut wie alle gemessenen (nichttotalen) Querschnitte.
Im weiteren Verlauf ist es wichtig, zwischen der tatsächlichen Energie“ der Elektronen,
”
die sich aus dem Impuls berechnet, und der nominellen Energie“, die die Differenz zur
”
anfänglich ausgewürfelten Energie beschreibt, zu unterscheiden.
3.6.2.2
gebundene“ Ionisationen
”
h
o
Die vorgestellte Auswertung nach Energiedifferenzen
kann nun dazu führen, daß ein Elek
»
Í .
tron als ionisiert zu zählen wäre (d.h. â
), aber trotzdem nicht frei ist: ` be
t
í
Die zugehörige Bahn ist eine Keplerellipse, dieser läßt sich nicht ohne weiteres
eine Richy }
tung des Endimpulses zuordnen, auch ist der mittlere Impuls ¡ ^ Ö ¬× ¡ × Null.
ï
Um im realen Experiment als ionisiert zu zählen, muß ein Elektron
den Detektr erreichen.
Der Projektor auf den Endzustand (Gl. (3.70)) muß also um eine Abfrage erweitert werden,
ob das Elektron den Abstand zwischen Reaktionsvolumen und Detektor überwinden wird.
Das ist gleichbedeutend
mit der Frage, ob der mittlere Impuls größer oder gleich Null ist.
Aus diesem Grund werden für den Querschnitt nur die Ereignisse gezählt, deren tatsächliche
Energie `cbe
größer Nul ist.
t
Es soll nicht verschwiegen werden, daß diese Art, mit gebundenen Ionisationen“ umzu”
gehen, nicht die einzig denkbare ist, sie ist jedoch die eindeutigste Variante.
3.7
Vergleich mit bisherigen Ansätzen
Der hier vorgestellte quasiklassische Ansatz beruht also darauf, den zeitabhängigen quantenmechanischen Streuformalismus über das Wignerbild klassisch zu nähern. Auf diese
Weise ist die Anfangsverteilung eindeutig über eine Korrespondenzregel definiert, es sind
keine weiteren Annahmen nötig, lediglich die Wellenfunktion von Anfangs– und Endzustand muß bekannt sein. Das ist — wenigstens genähert — für beliebige Probleme möglich.
Durch die Propagation entsprechend des Møllerschemas muß die so ausgewählte Anfangsverteilung nicht station är sein, da sie nur an dem einen Zeitpunkt des Stoßes das System korrekt beschreiben muß.
Die so propagierte Verteilung trägt dann als Ganzes in der Auswertung bei, da nur der
Energieübertrag als Maß genommen wird, nicht die tatsächliche Energie der auslaufenden
Elektronen. Damit ist auch die Unsicherheit in der Beschreibung energieabhängiger Querschnitte eliminiert.
Es ist interessant, die bisher unternommenen Anstrengungen, die CTMC zu verbessern,
noch einmal vor diesem Hintergrund zu betrachten: an welcher Stelle wurden dabei Änderungen vorgenommen und wie fügen sie sich in die hier vorgestellte Herleitung ein?
3.7.1
Die mikrokanonische Anfangsverteilung
In Abschnitt 3.4 konnte die mikrokanonische Verteilung nicht aus einer Korrespondenzregel
hergeleitet werden — sie ist ein rein klassisches Modell. Sie kann aber in die Rechnung
3.7. VERGLEICH MIT BISHERIGEN ANSÄTZEN
37
eingeführt werden, wenn die Auswertung der Endwerte nicht über den Energieübertrag,
sondern mit den tatsächlichen Energien (Gl. 3.82) erfolgt. Dann tragen nur Trajektorien zum
Querschnitt bei, bei denen das Targetelektron 16 bei der echten Bindungsenergie gestartet
ist. Alle anderen Trajektorien
können ignoriert werden, aus der Phasenraumverteilung wird
^ –0,5a.u. herausgeschnitten“. Die so erhaltene Verteilung muß
die Energieschale zu
”
lediglich neu auf die Anzahl der verbleibenden Trajektorien normiert werden.
rel. Wahrscheinlichkeit
0.1
mikro
Wigner
Produkt
0.05
0
0
0.25
0.5
L2
0.75
1
Abbildung 3.2: Verteilung des Drehimpulsquadrats verschiedener PR–Verteilungen des
µ
Wasserstoffgrundzustands für feste Energie von -0,5a.u. (je Verteilung etwa 1,5 Í Start¸
werte, verteilt in 20 Intervalle). Die mikrokanonische Verteilung ist nach Konstruktion
gleichverteilt.
Bei fester Bindungsenergie reicht als zweiter Parameter die Verteilung des Drehimpulsquadrats aus, um eine Phasenraumverteilung zu charakterisieren, wenn die Richtungen des
17
Drehimpulses (die restlichen
Y Freiheitsgrade) isotrop im Raum verteilt sind . Wie Bild
3.2 zeigt, führt eine um =–0,5a.u. herum begrenzte Wignerverteilung des Wasserstoffgrundzustands zur gleichen Verteilung des Drehimpulsquadrates wie die mikrokanonische,
während die Produktverteilung (siehe Abschnitt 3.4.5.1) kleinere Drehimpulse bevorzugt.
Die Energieverteilung, die von Cohen hergeleitet wurde [Coh85] (vgl. Abschnitt 3.4.5),
ist aus mikrokanonischen Beiträgen verschiedener Energie zusammengesetzt, sie reduziert
sich für feste Energie also aus ihrer Konstruktion heraus ebenfalls auf die mikrokanonische
Verteilung. Die mikrokanonische Verteilung besitzt — bei gleichmäßig verteiltem Quadrat
des Drehimpulses — die korrekte (radiale) Impulsverteilung des Wasserstoffatoms.
Auf diese Weise läßt sich konsistent die von Abrines und Percival aus der (semi)klassischen Torusquantisierung abgeleitete Anfangsverteilung bestätigen, sie ist eine Folge der
klassischen Auswertung des quantenmechanischen Problems. Diese klassische Quantisie16
Auch wenn hier von einem Elektron die Rede ist, so handelt es sich doch nur um eine Stützstelle“ der
”
Phasenraumverteilung.
17
Generell hat ein Phasenraumpunkt in drei Dimensionen sechs Koordinaten, durch die Randbedingung fester
Energie reduziert sich deren Anzahl um eins. Hier sind diese fünf Koordinaten das Quadrat des Drehimpulses,
dessen Projektion auf die z–Achse und die drei Euler–Winkel für die Orientierung im Raum
38
KAPITEL 3. DER QUASIKLASSISCHE ANSATZ
rung konnte allerdings bis heute z.B. für Helium nicht durchgeführt werden, hier wird also
ein anderer, nichtklassischer Zugang möglich.
3.7.2
Die Propagation in der CTMC
Im klassischenBild
der von Abrines und Percival eingeführten CTMC wird^ der
Anfangszu^
präpariert und unter der vollen Wechselwirkung nach ×
stand bei ×
propagiert.
Die beiden Äste des Møllerschemas, bei der die Verteilung von × ^ÎÍ nach × ^
und von
× ^
nach × ^ Í frei zurückpropagiert wird, fehlen. Wenn aber in der Standard–CTMC
eine station äre Phasenraumverteilungen (im Sinne von Anhang A.1) zum Einsatz kommt,
bedeutet die freie Rückwärtspropagation lediglich eine Verschiebung der Phase, d.h. der
Position des Elektrons auf der geschlossenen Keplerellipse. Diese Phasenverschiebung ist,
da alle Startwerte die gleiche Energie, und damit die gleiche Umlaufzeit haben, für alle
ausgewürfelten Startwerte die gleiche. Es verschiebt sich nur die absolute Phase, nicht aber
die relative Position der Startwerte zueinander. Bei einer station ären Verteilung ist der gesamte zur Verfügung stehende Phasenraum gleichmäßig gefüllt, durch diese gleichmäßige
Verschiebung der (zyklischen) Phase bleibt die Phasenraumdichte unverändert.
Es macht also technisch keinen Unterschied, ob bei einer station ären Verteilung der
anfängliche freie Ast des Møllerschemas explizit beachtet wird oder nicht. Dieser Unterschied tritt erst zu Tage, wenn in die Standard–CTMC (ohne die freien Propagationen)
nichtstation äre Verteilungen eingesetzt werden sollen (z.B. dieWignerverteilung).
Im Ausgangskanal werden hier ähnliche Formeln verwendet wie in der Standard–CTMC,
allerdings ist die Interpretation anders: hier beschreibt der Endzustandsprojektor eine frei
nach × ^ f
propagierte ebene Welle, deren Bezugspunkt bei × ^0Í liegt, dort ein freies
existiert. Da diese ebene Welle durch eine (klassisch)
Teilchen, das zum Zeitpunkt × ^ f
station äre Verteilung beschrieben wird, führt die freie Propagation wieder nur auf eine Phasenverschiebung, die in der CTMC ignoriert wird. Hier wird sie zwar beachtet, führt aber
zu keinem anderen Ergebnis.
Für station äre Anfangs– und Endzustandsverteilungen kann die hier vorgestellte Møller”
propagation“ also durch die analytische (Nicht)Beachtung der jeweils konstanten Phasenverschiebung auf die Standard–CTMC, d.h. auf den korrekten klassischen Limes zurückgeführt werden.
3.7.3
Cut–off–Wigner“ und Cohens Energieverteilung
”
Die bisherigen Versuche, andere Anfangsverteilungen als die klassische mikrokanonische
in die Standard–CTMC einzubauen, hatten zu Schwierigkeiten oder gar Inkonsistenzen
geführt.
Meist wurde das Augenmerk auf die ungebundenen Startwerte gelegt, nicht aber auf die
unklare Definition energieabhängiger Querschnitte (vgl. Abschnitt 2.2.2.1). Nach den vorangegangegen Ausführungen erkennt man, daß der Versuch, eine mehr quantenmechani”
sche“ Anfangsverteilung in die klassische Rechnung zu implementieren, bedeutet, daß die
einzelnen Teile der quantenmechanischen Rechnung unterschiedlich weit genähert wurden:
Die Anfangsverteilung bringt Beiträge in die Rechnung ein, die — bei dieser Näherung
— gar nicht behandelt werden können: in der Auswertung, wenn diese (klassisch) korrekt
3.8. ABSCHLIESSENDE BEMERKUNGEN
39
durchgeführt wird, trägt nur der Anteil der Anfangsverteilung bei, der auf der Energieschale liegt, so wie weiter oben die mikrokanonische Verteilung eingeführt wurde (Abschnitt 3.7.1). Allerdings hatte keiner der Autoren, die in die Standard–CTMC eine andere
als die mikrokanonische Verteilung eingebaut hatten, diese Diskrepanz des Modells auch
nur erwähnt — diese Versuche mögen zwar zu besseren (totalen) Querschnitten führen,
wenn die Fitparameter korrekt gewählt werden, sind aber vom Ansatz her falsch.
3.7.4
Zur numerischen Implementation des Ansatzes
Das vorangegangene Kapitel hatte gezeigt, das der quasiklassische Ansatz letzten Endes
der Standard–CTMC sehr ähnlich sieht, was die verwendeten Formeln betrifft. Die größten
Unterschiede liegen in der Auswahl der Startwerte und der Auswertung, sowie im Propagationsschema, nicht aber in den Bewegungsgleichungen, die in der numerischen Anwendung
die meiste Rechenzeit beanspruchen.
Ein bestehender CTMC–Code kann also ohne große Schwierigkeiten an den quasiklassischen Ansatz angepaßt werden:
Die Auswahl der Startwerte ist die umfangreichste Änderung, da hier eventuell komplizierte Phasenraumverteilungen bestimmt werden müssen (siehe Abschnitt 3.4 und
Anhang B).
An der eigentlichen Propagationsroutine braucht keine Änderung vorgenommen zu
werden, für die erste Rückwärtspropagation unter ] r ist lediglich ein Teil der Wechselwirkungen abzuschalten (Abschnitt 3.5.2).
Die Bestimmung der Wirkungsquerschnitte wird so modifiziert, daß die Endenergien nicht mehr aus der absolute Energie sondern über die Energiedifferenz festgelegt
werden (siehe Abschnitt 3.6.2.1).
Was aus der CTMC jedoch nicht übernommen werden darf, ist die Interpretation der
Trajektorien als echte Bahnen der Elektronen.
Diese Modifikationen zeigen, daß mit dem quasiklassischen Modell in der praktischen
Anwendung pro Trajektorie nur unwesentlich mehr Rechenzeit benötigt wird. Da aber das
Target nun deutlich ausgedehnter sein kann, muß dann evtl. mit einem größeren Bereich des
Stoßparameters gearbeitet werden, was letztlich die Anzahl der zu berechnenden Trajektorien gegenüber der Standard–CTMC erhöht, bis vergleichbar viele Ionisationsereignisse zur
Verfügung stehen. Kapitel 5 vermittelt einen Eindruck, welcher Mehraufwand im Vergleich
zur CTMC nötig ist, zeigt aber auch deutlich die Vorteile dieses Vorgehens.
3.8
3.8.1
Abschließende Bemerkungen
Grenzen der Phasenraum–Darstellung
An dieser Stelle sollen noch einmal zwei Probleme der Phasenraumdarstellung, sei es im
Wignerbild oder einer anderen Darstellung, erwähnt werden. Die beiden Schwierigkeiten
sind an sich seit längerem bekannt, werden aber immer wieder in der Anwendung übersehen:
KAPITEL 3. DER QUASIKLASSISCHE ANSATZ
40
Die erste Schwierigkeit wurde von Heller als dangerous cross terms“ bezeichnet [Hel76]:
”
wird eine Wellenfunktion in einzelne lokalisierte Anteile zerlegt und wignertransformiert,
so entstehen dabei — analog zur Symmetrisierung (Abschnitt 3.6.1.2, speziell Gln. (3.65)
und (3.66)) — Kreuzterme zwischen den einzelnen, an verschiedenen Orten zentrierten Anteilen. Diese Kreuzterme haben ihren Schwerpunkt um den mittleren Ort und den mittleren
Impuls der beiden Anteile. Bei z.B. einem Doppelspaltexperiment (siehe Abb. 3.3) laufen
die beiden Anteile und e einer Wignerfunktion durch je einen der beiden Spalte und
erzeugen auf dem Schirm die klassische Verteilung ohne Interferenzstreifen. Die Mischterme e ^ e hingegen, zwischen
den beiden anderen zentriert, würden in der klassisch
´³
genäherten Propagation mit ^aÍ (Gl. (3.34)) an der Wand zwischen den Spalten reflektiert. Die vollst ändige Propagation nach Gl. (3.31) jedoch erkennt“, woher die Kreuzterme
”
stammen und propagiert sie korrekt auf den Schirm, wo sie dann die quantenmechanischen
Interferenzen erzeugen.
Γ1
Γ12
Γ2
Abbildung
3.3: Bei einem Doppelspaltexperiment im Wignerbild werden in der klassischen
´³
=0–Näherung der Propagation die Kreuzterme e ^ e an der Wand zwischen den
beiden Spalten reflektiert, die Interferenzstreifen verschwinden (genauer wird dies im Text
erläutert).
Dies kann auch anders formuliert werden: In ³ der klassisch genäherten Propagation in
´
Gl. (3.34) fehlen die Terme aus Gl. (3.33), die enthalten. Diese Terme aber bestehen
aus höheren Ableitungen des Potentials und der Phasenraumverteilung. Um diese Ableitungen bestimmen zu können, muß also die Phasenraumverteilung auch in der um den jeweils
betrachteten Phasenraumpunkt herum liegenden Umgebung bekannt sein. Auf diese Weise werden auch weiter auseinander liegende Phasenraumpunkte miteinander verbunden“,
”
der nichtlokale Charakter einer quantenmechanischen Wellenfunktion kommt darin zum
Ausdruck18. In der klassischen Näherung entkoppeln die einzelnen Stützstellen zu klassi”
schen Trajektorien“, die keine Information über ihre Umgebung mehr beinhalten. Das ist
der Grund, warum die Trajektorien, wie weiter hinten in Abb. 5.11 gezeigt, aus der korrekten Richtung heraus abgelenkt werden (können). In der vollst ändigen Propagation findet
eine Art gegenseitige Stabilisierung“ und/oder Interferenz der miteinander verknüpften
”
Trajektorien statt.
18
Um in der Schrödingergleichung die zweite Ableitung der Wellenfunktion an einem Ort bestimmen zu
können, muß der Verlauf in dessen Umgebung bekannt sein, für die Zeitentwicklung dieser Nachbarpunkte wird
aber wieder deren Umgebung benötigt, sodaß die Entwicklung eines Punktes von der ganzen Wellenfunktion
abhängt; die Schrödingergleichung ist in diesem Sinne nichtlokal“: eine Wellenfunktion ist immer im ganzen
zu finden (sie kann allerdings stellenweise verschwindend
”
klein sein).
ò
3.8. ABSCHLIESSENDE BEMERKUNGEN
41
Neben dieser in der klassischen Näherung vernachlässigten Nichtlokalit ät der Wellenfunktion existiert noch eine wesentlich Einschränkung der Phasenraumdarstellung der Quantenmechanik: Shewell zeigte, daß es mit den bisher vorgeschlagenen Korrespondenzregeln
(siehe Abschnitt 3.4.1) und eventuellen Verallgemeinerungen davon nicht immer möglich
ist, höhere Potenzen von Operatoren eindeutig zu definieren [She59]. Er zeigt dies exemplarisch an einer verallgemeinerten Weylschen Korrespondenz (vgl. Gln. (3.20) und (3.21)).
Diese läßt sich verallgemeinern zu:
%'&
" ! y$#ä} ^
e
# y ! +! }
_
! s!
+
)] ( s * s ¡ s-, ?¡
ôs
* *
"! y }
Ê
Wenn nun z.B. für die höheren Potenzen des Hamiltonoperators
"! y
Z Ò
}
^
" ! y }
Z
Ò
mit
Z
z¡
^
e
f
Z
(3.84)
gefordert wird, daß
Ü y+ }
(3.85)
gilt, so
werden.
e Ó ^ ¸ und
z können daraus die Entwicklungskoeffizienten
bestimmt
³
´³ Für
^ ( s e/. ´ und e ^
ist die Gleichung erfüllt, wenn ] ^
,
gesetzt wird.
Ó ^
¸ (
(
(
~
Wenn diese Koeffizienten festgelegt sind, gibt es keine Möglichkeit mehr,
und so zu
(
(
bestimmen, daß Gl. (3.85) für Ó ^ Ë erfüllt wäre. Es ist also auch mit einer Verallgemeinerung der Weylschen Korrespondenz nicht möglich, alle Ordnungen des Hamiltonoperators
eindeutig darzustellen. Das bedeutet z.B., daß für Phasenraumverteilungen von Energieeigenfunktionen die höheren Momente der Energie (Breite der Verteilung, etc.) nicht alle
verschwinden können, im Gegensatz zur Quantenmechanik.
Eine Phasenraumdarstellung kann also gar nicht alle Aspekte der Quantenmechanik reproduzieren, auch wenn oft gesagt wird, die Wignerdarstellung sei eine korrekte, eindeutige
und vollst ändige Darstellung der Quantenmechanik.
3.8.2
Anregung in der klassischen Beschreibung
Bisher war im Rahmen der Herleitung des quasiklassischen Ansatzes nur von Ionisationsquerschnitten die Rede. Das liegt daran, daß hierbei die Auswertung eindeutig ist: der Raum
der freien Eigenzustande ist quantenmechanisch und klassisch kontiniuierlich. Zu jedem
Impuls existiert eine quantenmechanische Eigenfunktion und ihre entsprechende Wignertransformierte (bzw. der Projektor auf diesen Endzustand). Für die gebundenen Zustände
ist das nicht der Fall, deren Hilbertraum ist in der Natur (und in der quantenmechanischen
Beschreibung) diskret, in der klassischen Näherung dagegen kontinuierlich. Das entspricht
dem klassischen Grenzfall der zur Schwelle immer dichter liegenden Rydbergzustände für
sehr hohe Quantenzahlen, wo diese Näherung keine wesentliche Einschränkung bedeutet:
zu praktisch jeder Bindungsenergie und jedem Drehimpuls existiert an der Schwelle ein gebundener Zustand. Es gibt aber für die untersten gebundenen Zustände keine Abbildung, die
eindeutig und umkehrbar den diskreten quantenmechanischen Funktionenraum der gebundenen Eigenzust ände auf den klassischen kontinuierlichen abbildet. Es treten in der klassischen Rechnung also nicht nur die zu den diskreten Niveaux gehörenden Energieüberträge
auf, sondern beliebige, es kann auch passieren, daß nach dem Stoß das Targetelektron fester
gebunden ist und das Projektil dadurch Energie gewinnt.
42
KAPITEL 3. DER QUASIKLASSISCHE ANSATZ
Um im Rahmen des vorgestellten quasiklassischen Ansatzes Anregung bestimmen zu
können, müsste der
asymptotische Target–Endzustand unter dem vollen Wasserstoffhamiltonian nach × ^
propagiert werden — oder die gestreute Verteilung für eines der ElektroÎ
^
Í
nen nach ×
zurück — und der Überlapp mit der Wignertransformierten eines angeregten
Zustandes bestimmt werden. Dieser Projektor ist nun zusätzlich ortsabhängig.
Anregung wurde in der vorliegenden Arbeit nicht untersucht. In diesem Ansatz kann aber,
im Gegensatz zur Standard–CTMC, eine eindeutige Definition der Anregungswahrscheinlichkeit gegeben werden. Wie gut nun die Querschnitte tatsächlich wiedergegeben werden,
wagt der Autor jedoch nicht vorherzusagen.
Kapitel 4
Erweiterung auf
Mehrelektronentargets
Der große Vorteil, den der hier vorgestellte quasiklassische Ansatz bietet, ist die Freiheit
in der Wahl der Anfangsverteilung, die das System beschreibt. Es wurde gezeigt, welche
verschiedenen Möglichkeiten bestehen, ein Wasserstoffatom durch eine klassische Phasenraumverteilung darzustellen. Diese Verteilungen müssen dabei nicht station är sein, wie bisher in der CTMC nötig, auch darf die Energie der diskretisierten Startwerte von der Schale
der korrekten Bindungsenergie abweichen.
Hier wird nun gezeigt, wie der Formalismus auf Mehrelektronentargets, die ja klassisch
prinzipiell instabil sind, zu erweitern ist. Diese Erweiterung wird am Beispiel des Heliumatoms dargestellt, für andere Systeme ist das Vorgehen analog.
4.1
Das (klassische) Heliumatom
z
Das Heliumatom besteht aus einem Kern mit Ladung 0 ^
und zwei Elektronen; die
Spins sollen hier außer acht gelassen werden, sie können aber durch die entsprechenden
î
Anteile an Ortho- und Parahelium im Target berücksichtigt werden — hier wird also nur
der ^ÎÍ –Singlett–Fall betrachtet.
Das gesamte Streusystem wird durch den folgenden Hamiltonoperator beschrieben (in
atomaren Einheiten und mit
unendlicher Kernmasse, vgl. Gln. (3.1) und (3.2)):
^
Z
^
1
¡z
e
e
e
0 f ¡ z e 0 f ¸ f ¡ z ` 0 f ¸ f ¸
e
e 5 1624345
e
243
1 `
24` 3
` 5
]
]
G
f Z
f
Z 7
Z
`
Das Problem zerfällt
dabei in einen Teil, der das ungestörte Target beschreibt, Z
]
G
freie Projektil, Z , und die zur Reaktion führenden Wechselwirkungen, Z
.
7
]
(4.1)
, das
`
4.2
Erweiterungen des Formalimus
In Kapitel 3 wurde die Herleitung des quasiklassischen Ansatzes in drei Teile zerlegt: Präparation des Anfangszustandes, Propagation und Auswertung. Diese Aufteilung ist natürlich
43
44
KAPITEL 4. ERWEITERUNG AUF MEHRELEKTRONENTARGETS
auch für Mehrelektronentargets sinnvoll. Im Folgenden wird beschrieben, welcher Schritt
auf welche Weise erweitert werden muß, wenn anstatt des Wasserstoffatoms z.B. ein Heliumatom als Target verwendet wird.
4.2.1
Anfangsverteilung
Im ersten der drei Schritte wird die Phasenraumverteilung des Anfangszustandes aus der
Wellenfunktion bestimmt. Diese ist für das Wasserstoffatom analytisch bekannt, für Helium muß eine genäherte Wellenfunktion verwendet werden. Das kann
z.B. eine korree
lierte Hylleraas–Wellenfunktion sein oder auch eine unkorrelierte “–Lösung aus zwei
”¸
Wasserstofforbitalen im abgeschirmten Coulombpotential. Diese Wellenfunktion wird dann
nach einem der in Abschnitt 3.4 vorgestellten Verfahren in eine Phasenraumverteilung des
Targets übertragen. Ein Beispiel dazu wird bei den zu Helium durchgeführten Rechnungen gezeigt. Da das Target nun zwei Elektronen besitzt, muß die Phasenraumverteilung,
wie bereits die Wellenfunktion, symmetrisiert werden. Nach der Argumentation in Abschnitt 3.6.1.2 (dort auf den Endzustand bezogen) bedeutet dies in der klassischen Näherung, daß die beiden Elektronen vertauscht werden. Dies kann aber auch bei der Auswertung
durchgeführt werden, es sind dann entsprechend alle Permutationen der drei Elektronen explizit zu berücksichtigen.
Das Projektil wird analog zu Abschnitt 3.4.6 modelliert.
Die Schwierigkeit dieses Schrittes besteht hier also in der Bestimmung der (genäherten)
Wellenfunktion (deren Wahl kann die Übersetzung in eine Phasenraumverteilung durchaus
vereinfachen oder erschweren).
Die Gesamtenergie
des Targets setzt sich aus drei Beiträgen zusammen: den unabhängi”
`cb 98
^ e
gen“ Energien
der beiden Elektronen und der Elektron–Elektron–Wechselt
;::
­ beiden
^ r . Die
Elektronen sind gleichwertig, sollten also mit der gleiwirkung
­
t=<><
»
chen Energie gebunden sein. Das bedeutet, daß sie beide mit dem Ionisationspotential e
des He w –Ions gebunden sind, die Elektron–Elektron–Abstoßung bedingt die Differenz zwi»?
»?
schen e und dem Ionisationspotentials des Atoms,
. Die Gesamtenergie ist die Sum
me dieser beiden:
é
z »? e
::
^ » f » e ^
f
(4.2)
Quantenmechanisch berechnet
alle@diese
sind
:: Energien scharf, in der klassischen Phasene
raumverteilung können nun ,
und
jeweils eine Verteilung aufweisen, die bereits
beim Wasserstoff angesprochene Problematik unscharfer Energien (siehe Abschnitt 2.2.2.1)
führt hier zu noch größeren Unsicherheiten in der Bestimmung der absoluten Energien.
Das soll an den folgenden Beispielen erläutert werden: setzt man eine klassische motivierte
Anfangsverteilung aus zwei unabhängigen mikrokanonischen Verteilungen ( –Ansatz“)
”¸
an, so werden die beiden
@:: Elektronen zwar jeweils für sich eine feste Energie haben, aber ihr
Abstand, und damit
, ist variabel, man erhält eine Breite der Gesamtenergieverteilung.
Um mit einem klassischen Modell eine scharfe Gesamtenergie zu erhalten, müsste also der
Elektron–Elektron–Abstand festgehalten werden.
Wird nun z.B. eine korrelierte Wellenfunktion wignertransformiert, so sind alle drei Energien unscharf verteilt. Eine Phasenraumverteilung für das Heliumatom ist also — abgesehen
von pathologischen“, auf feste Gesamtenergie hin konstruierten Verteilungen — unscharf
”
in den Energien, wenn sie wie hier beschrieben aus einer Wellenfunktion bestimmt wird.
4.2. ERWEITERUNGEN DES FORMALIMUS
45
In bisherigen Ansätzen wurden, um die beiden Schwierigkeiten zu umgehen, im wesentlichen drei Ansätze verwendet: im einfachsten Fall wird das Heliumatom durch ein Elektron,
das sich mit der ersten Ionisierungsenergie gebunden in einem abgeschirmten Potential bewegt, dargestellt. Das zweite Elektron wird in Form der Abschirmung berücksichtigt. Mit
diesem Modell ist es natürlich unmöglich, Zweifachionisation zu beschreiben, Einfachionisation dagegen wird mit der für die CTMC üblichen Genauigkeit wiedergegeben.
Der zweite Weg wurde von Olson vorgeschlagen: in der nCTMC“ sind beide Elektronen
”
mit der ersten Ionisierungsenergie gebunden. Um Autoionisation zu vermeiden, wird ihre
gegenseitige Abstoßung vollst ändig vernachlässigt. In diesem Modell kann Zweifachionisation nur durch zwei (hintereinander erfolgende) Einfachstöße des Projektils mit den beiden
Targetelektronen erfolgen. In [SMO92] werden diese beiden Ansätze anhand einfach und
zweifach differentieller Querschnitte miteinander vergleichen.
In der dritten Variante wird zwar die Elektron–Elektron–Abstoßung voll berücksichtigt,
die Startwerte werden aber auf hochsymmetrische Elektronenorbits beschränkt. Auf ihnen befinden sich die beiden Elektronen in einem labilen Gleichgewicht, die Verteilung
ist zumindest eine gewissen Zeit lang stabil und kann als CTMC–Target verwendet wer
den. Dabei kann durch die Größe des Bahnradius
die Gesamtenergie oder die
entweder
Bindungsenergie der einzelnen Elektronen (d.h. Å e ) festgelegt werden, aber nicht beides
unabhängig. Von diesem Modell ist daher zu erwarten, daß auch energieabhängige Querschnitte das Experiment lediglich qualitativ beschreiben können1 .
4.2.2
Propagation
Hier ist das Verfahren das gleiche wie für Wasserstoff, das in Abschnitt 3.5 dargestellt wurde; lediglich die zu den einzelnen Ästen gehörenden Hamiltonoperatoren sind dem Heliumatom angepaßt (vgl. Gln. (3.60) und (3.61)). Für den Propagationsast vor dem Stoß gilt
nun:
e
e
e
Z ] r ^R¡ z
0 f ¡ z e 0 f ¸ f ¡ z `
e
e
Z r ^
0
`
f
¸
`
f
¸e
`
(4.3)
Hier muß die Rückpropagation am Anfang auf alle Fälle explizit durchgeführt werden,
denn bei den instabilen Mehrelektronentargets läßt sich die Zeitentwicklung nicht mehr, wie
beim mikrokanonischen Modell des Wasserstoffs, als reine Phasenverschiebung darstellen.
4.2.3
Auswertung
Für die Auswertung wird der Endzustand nach × ^
propagiert und dort der Überlapp mit
der gestreuten Verteilung bestimmt, wie am Wasserstofftarget beschrieben. Diesmal müssen
aber, im Gegensatz zum Wasserstoff, mehrere Fälle berücksichtigt werden:
a) es tritt Zweifachionisation auf: dann wird der Endzustand durch drei freie Teilchen
beschrieben, oder
1
Aber gerade das qualitative, klassisch intuitive Bild, das eine CTMC–Rechnung liefert, kann helfen, Reaktionsmechanismen zu verstehen — dies ist die Stärke des klassischen Ansatzes, nicht die quantitativ korrekte
Beschreibung. Das zeigt gerade dieses Modell: die Wechselwirkungen werden korrekt behandelt, aber die Parameter (Bindungsenergien) haben von Anfang an die falschen Zahlenwerte.
KAPITEL 4. ERWEITERUNG AUF MEHRELEKTRONENTARGETS
46
b) nur eines der Elektronen wird ionisiert, dann besteht der Endkanal aus zwei freien
Elektronen und dem (angeregten) He w –Ion, oder aber
c) beide Elektronen werden angeregt. Dieser Fall führt bei realem Helium immer über
einen Augerprozeß zur Einfachionisation, könnte klassisch aber wegen der kontinuierlichen gebundenen Zustände existieren (dieser Fall führt bekannterweise auch
klassisch zur Autoionisation).
4.2.3.1
Zweifachionisation
Im Falle der Zweifachionisation besteht der Endzustand aus drei freien Elektronen, der entsprechende Projektor des Endzustands fragt die drei Impulsvektoren ab. Da die Elektronen
ununterscheidbar sind, muß der Endzustand symmetrisiert werden. In der klassischen Näherung bedeutet das, die Indizes zu permutieren (siehe Abschnitt 3.6.1.3):
m5
~ y M
^ ¡
[
m
} ~ y A
¡
~
e} y Ô
¡
m
~ } f
Permutationen
(4.4)
Um wieder, wie in Abschnitt 3.6.2.1, die absoluten Endenergien auf Energiedifferenzen
umschreiben zu können, werden zuerst die einzelnen Beiträge benannt: Die unabhängige
Energie eines der Targetelektronen ist im Anfangszustand
BA
^
r
`bue C x
8
, im Endkanal
t Du C x
BA A
c
`
b
x
D
u
C
C
E
^
e
. Der Index a, bzw. b bezeichnet die Targetsind alle Elektronen frei, d.h. [
`
elektronen, der Index p das Projektil. Die drei Energien, die den Endzustand beschreiben,
werden mit , e und ~ bezeichnet. Die Gesamtenergie setzt sich daher zusammen als:
GF :
r f
`
[` f
~
f
^
¯
^
^
f f ::
r
r
r
[ f [
e
f
Die Elektron–Elektron–Wechselwirkung des Anfangszustandes sorgt, im Vergleich zum
Wasserstoff, für einen zusätzlichen Term in der Energiebilanz, in einer klassischen Beschreibung müssen daher immer noch, trotz@Energieerhaltung,
alle drei Endenergien ab::
gefragt werden. Quantenmechanisch gilt für
:
@::
z »? e 8 F :
» e »
^
¯ ^
(4.5)
Aber auch hier können die absoluten Energien auf Energieüberträge umgeschrieben werden, diese sind
für die Elektronen
â
^
` r 8
^
â
[
Y
^
â
[
Y`
`[
r
r
für den Endzustand
8 ~
â
` ^
rªÒ »? e
f
â
^
e f ?» e
â
e ^
Das schnellste Elektron wird dabei dem Endzustand mit der Nummer drei zugeordnet,
meist ist es das (ehemalige) Projektil.
4.3. ANMERKUNGEN
47
~ ^
Mit diesen
;:: Definitionen gilt, wenn â
rekten“
aus Gl. (4.5):
â
`
f
@::
Y
^ â
â
f â
e beachtet wird und mit dem korf â
Y
”
^ â
f â
e
(4.6)
Zweifachionisation bedeutet also, daß das Projektil mindestens die Gesamtenergie des
Heliumgrundzustandes an das Target abgegeben hat und die Targetelektronen jeweils min»?
destens die Ionisierungsenergie e aufgenommen haben. Hinzu kommt, daß alle Elektro
nen am Ende frei sind, also [ , [ und [ größer Null sein müssen. Der Energieanteil des
`
damit die folgende Form:
Wirkungsquerschnitts (vgl. Gl. (3.83)) bekommt
~ } y â â
} y â â
e }H =y }DH y= }DH y= }
â
â
`
`
f Permutationen
y
4.2.3.2
(4.7)
Einfachionisation
Bei der Einfachionisation bleibt eines der Elektronen gebunden, das andere wird ionisiert.
Im Bild der Energieüberträge hat das Projektil mindestens die erste Ionisierungsenergie
»
»?
an das Target abgegeben, eines der Targetelektronen hat mindestens e aufgenommen
»? e
und ist frei, der Übertrag auf das andere ist kleiner als
. Für das gebunden gebliebene
I8
`
b
x
^
e
Elektron, hier mit Index ß , gilt nun [
. Analog zur Zweifachionisation erhält
tvx
man mit den Energieüberträgen
â
â
e
^
^
f
»? e
e
»? e
f
mit e
h
Í
für den Energieanteil des Wirkungsquerschnitts
nun:
y
â
4.2.3.3
`
} y Y
}H y »? e
Y}H =y }H yH!:}
f
â
~ â
â
â
`
Perm.
(4.8)
Zweifachanregung
Bei der Zweifachanregung treten wieder die gleichen systematischen Probleme auf, wie
bereits beim Wasserstofftarget (vgl. bschnitt 3.8.2): in der klassischen Näherung existiert
keine diskrete Schalenstruktur der gebundenen Zustände mehr, jede beliebige Anregung ist
möglich.
Auch hier bei Helium müsste der Endzustand wieder ohne die Wechselwirkung mit dem
ungebundenen Elektron nach × ^¹Í zurückpropagiert und auf die Phasenraumdarstellung
der verschiedenen angeregten Zustände projiziert werden. Wie auch am Wasserstoff wird
die Anregung im Rahmen dieser Arbeit nicht betrachtet.
4.3
Anmerkungen
Bei der Einfachionisation kann zusätzlich Anregung auftreten. In der Realität sind die gebundenen Zustände diskret in der Energie, klassisch kann aber das Elektron auf jede beliebige Energie angeregt werden, sogar auf Energien, die tiefer liegen als der Grundzustand. Das
48
KAPITEL 4. ERWEITERUNG AUF MEHRELEKTRONENTARGETS
ist ein Problem des klassischen Modells, das den diskreten Funktionenraum der gebundenen Zustände auf einen kontinuierlichen abbildet, nicht ein Problem des hier vorgestellten
Formalismus (vgl. Abschnitt 3.8.2).
Zur Berechnung von Einfachionisation ohne Anregung empfiehlt es sich daher, mit einem
Ein–Elektronen–Modell zu rechnen und das zweite Elektron durch ein effektives Potential
zu beschreiben, während für die Zweifachionisation der vorgestellte quasiklassische Ansatz
in der Lage ist, alle Energien und Wechselwirkungen korrekt zu beschreiben.
Weiter oben wurde angemerkt, daß kein mikrokanonisches“ Modell des Heliumatoms
”
existiert. Analog zum Vorgehen am Wasserstoff in Abschnitt 3.7.1 könnte aber eine Phasenraumverteilung mit scharfer Gesamtenergie konstriuert werden, indem aus der Wignertransformierten einer Heliumwellenfunktion nur die Startwerte mit der korrekten Gesamtenergie ausgewählt werden. Diese Verteilung ist natürlich nicht station är, im Gegensatz zur
mikrokanonischen Verteilung des Wasserstoffs. Diese Idee wird hier jedoch nicht weiter
verfolgt werden, denn die Ergebnisse an Wasserstoff werden zeigen, daß eine solche energetisch gestutzte“ Verteilung eine schlechtere Abbildung des Atoms darstellt, als z.B. die
”
volle Wignertransformierte.
Kapitel 5
Rechnungen an Wasserstoff
Obwohl der hier vorgestellte quasiklassische Ansatz in der Lage ist, auf konsistente Weise
Mehrelektronentargets zu beschreiben, wurden doch zuerst ausführliche Rechnungen und
Vergleiche an atomarem Wasserstoff durchgeführt, denn hier kann direkt mit der bisherigen
Standard–CTMC verglichen werden, ohne daß bei ihr zusätzliche Stabilisierungsmaßnahmen ergriffen werden müssten. Zum anderen sind die quantenmechanischen analytischen
Wellenfunktionen bekannt, die Phasenraumverteilung des Targets wird also aus der exakten
Wellenfunktion bestimmt. Ein dritter, nicht zu vernachlässigender Punkt ist die Statistik“:
”
ein volldifferentieller Querschnitt an Helium ist fünffach differentiell, an Wasserstoff nur
dreifach, d.h. es müssen deutlich weniger Trajektorien propagiert werden, um daraus volldifferentielle Querschnitte bestimmen zu können. Deutlich weniger“ bedeutet aber immer
”
noch, daß mehrere Dutzend bis Hundert Millionen Trajektorien gerechnet wurden, der Aufwand betrug immer noch“ mehrere Wochen CPU–Zeit pro Einschußenergie.
”
Vor der Darstellung der gerechneten Querschnitte werden noch die verwendeten Phasenraumverteilungen vorgestellt. Das numerische Vorgehen ist in Anhang C dargestellt, hier
soll nur erwähnt werden, daß mittels eines symplektischen Integrators der Hamiltonian in
die unabhängige Keplerbewegung der beiden Elektronen und ihre gegenseitige Wechselwirkung aufgeteilt wird. Die Keplerbewegungen werden regularisiert und können durch
analytische Bahngleichungen ohne die Coulombsingularit ät dargestellt werden. Auf diese
Weise wird eine sehr gute Energiestabilit ät der Trajektorien bei gleichzeitig hoher Rechengeschwindigkeit erreicht. Die Zeitintegration ist so stabil, daß nur sehr wenige (eine unter
mehr als Í-J ) Trajektorien insgesamt wegen numerischer Fehler abgebrochen werden mus¸
sten.
5.1
Die verwendeten Phasenraumverteilungen
Der hier vorgestellte Ansatz ist parameterfrei“, es besteht
allerdings die Wahl zwischen
´³
”
verschiedenen Korrespondenzregeln, anhand derer die ^ Í –Näherung durchgeführt wird
(siehe Abschnitt 3.4). Jede dieser Korrespondenzregeln führt auf eine andere Phasenraumverteilung zur Beschreibung des Wasserstoffatoms. Dem gegenüber steht in der Standard–
CTMC die klassisch hergeleitete mikrokanonische Anfangsverteilung. Aus den verschiedenen Konzepten und Wahlmöglichkeiten wurden die folgenden vier Verteilungen ausgewählt
49
KAPITEL 5. RECHNUNGEN AN WASSERSTOFF
50
und anhand verschiedener Querschnitte bei drei unterschiedlichen Einschußenergien miteinander verglichen (siehe auch Abschnitt 3.4.5):
a) die voll klassische mikrokanonische Verteilung (siehe Abschnitte 2.1 und 3.7.1), wie
sie ursprünglich von Abrines und Percival eingeführt wurde [AP66a],
b) die Wignerverteilung als prominentester Vertreter einer aus einer Korrespondenzregel
abgeleiteten Phasenraumverteilung [Moy49] (Abschnitt 3.4.1.1),
c) der Produktansatz [Coh66], der dem naiven“
”
und
´³ ^ÎÍ
-Zugang entspricht (Abschnitt 3.4.5.1)
d) die erweitert klassische“ Energieverteilung, mit der Cohen allein mit gebundenen
”
Orbits die Ortsraumdichte korrekt nachbildete [Coh85] (Abschnitt 3.4.5.2).
Die Verteilungen a)–c) besitzen dabei die korrekte radiale Impulsverteilung, b)–d) die
richtige Ortsraumdichte.
Die Wignerverteilung und der Produktansatz sind Lösungen der gewünschten Randbedingungen — Orts- und Impulsverteilung sollen korrekt wiedergegeben werden — ohne
daß weitere Vorgaben an z.B. Energie oder Drehimpuls gestellt werden, während bei der
mikrokanonischen und Cohens Energieverteilung die Bindungsenergie und die Drehimpulsverteilung vorgegeben bzw. angepaßt wurden.
Das konkrete Vorgehen, wie diese Verteilungen für die Rechnung präpariert werden, ist
in Anhang B dargestellt.
Mit der mikrokanonischen Verteilung reduziert sich der vorgestellte quasiklassische Ansatz auf die bisherige Standard–CTMC (siehe Abschnitt 3.7). Die Ergebnisse mit dieser Verteilung zeigen auf, was dieses voll klassische Modell zu beschreiben vermag und wo seine
Grenzen liegen. Hier wurde die mikrokanonische Verteilung jedoch den anderen Verteilungen gleichwertig gegenübergestellt: alle Rechnungen und Auswertungen wurden — jeweils
bis auf die Auswahl der Startwerte — mit dem gleichen Programmcode durchgeführt.
Die anderen Verteilungen zeigen, welche Verbesserungen durch die quantenmechanisch
motivierten Anfangsverteilungen erreicht werden können.
5.1.1
Bindungsenergie und Energiebreite
Alle untersuchten Verteilungen besitzen die korrekte mittlere Bindungsenergie des Wasserstoffgrundzustands 1. Für die nach Abschnitt 3.4.7 diskretisierten Verteilungen bedeutet
í
das:
y=
}
ï
K ê Ò Z Ò ¡ Ò
K ê Ò
^
^
z ¸ a.u.
Die Breite der jeweiligen Energieverteilungen ist unterschiedlich: nur die mikrokanonische Verteilung hat, wie die quantenmechanische Lösung, die Breite Null. Für die diskretisierten Verteilungen berechnet sich die Breite der Energieverteilung
wie folgt:
í
y
1
â
ä} e
^
K
ê
Ò
y
Z
Wenn nicht, so wäre das ein Ausschlußkriterium. . .
yH
¡
Ò Ò
K ê Ò
}'
}e
ï
(5.1)
5.1. DIE VERWENDETEN PHASENRAUMVERTEILUNGEN
Verteilung
mikrokanonisch
Wigner
Produkt
Cohen
ö y
â
}e
Í
Í
51
[a.u.]
Mæ zL-zL
z
͸ æ º
æ
Tabelle 5.1: Breite der Energieverteilung der veschiedenen Phasenraumverteilungen des
Wasserstoffgrundzustandes.
In Tabelle 5.1 sind die (z.T. numerisch bestimmten) Breiten der verwendeten Phasenraumdarstellungen des Wasserstoffatoms aufgeführt. Die beiden klassischen“ Verteilungen
”
haben eine geringe Breite, während die rein aus der quantenmechanischen Wellefunktion
abgeleiteten recht breit in der Energie sind (vgl. auch Abb. 5.1). Hier zeigt sich wieder
die Problematik der klassischen Näherung: nicht alle Eigenschaften einer Wellenfunktion
können gleichzeitig klassisch dargestellt werden2 .
Hardie & Olson
Cohen
Wigner
Produkt
rel. Wahrsch.
0.2
0.1
0
-2
-1.6
-1.2
-0.8
-0.4
0
Eb 0.4
Abbildung 5.1: Vergleich der Energieverteilungen verschiedener Phasenraumverteilungen
(siehe Text).
In Abb. 5.1 sind die relativen Energieverteilungen des Ansatzes von Cohen und die Fitparameter von Hardie und Olson der Wignerverteilung und der Produktverteilung“ ge”
genübergestellt.
2
Im Grunde ist das nur eine Schwierigkeit, wenn versucht wird, die klassischen Trajektorien als echte Bahnen zu interpretieren: wenn eine Wellenfunktion in eine beliebige Basis zerlegt wird, muß auch nicht jede
dieser Basisfunktionen die korrekte Energie“ des ursprünglichen Zustands aufweisen. So kann z.B. der Was”
serstoffgrundzustand durch ebene Wellen dargestellt werden (d.h. im Impulsraum), diese haben aber nicht die
Bindungsenergie“ NPORQ S a.u. Die in diesen klassischen Rechnungen verwendeten Startwerte sind quasi die Ba”
”
sisfunktionen“ der klassischen Hamiltonschen Bewegungsgleichungen.
KAPITEL 5. RECHNUNGEN AN WASSERSTOFF
52
5.1.2
Radiale Orts- und Impulsverteilungen
Die radialen Orts– und Impulsverteilungen des Wasserstoffgrundzustands sind:
e
T y=B} ^
Ø y }
¡
^
e
p t
º
z e
Ë
{y ¡ e } f
¸ ¡
(5.2)
(5.3)
Die mikrokanonische Verteilung besitzt zwar die korrekte radiale Impulsverteilung, die
Ortsverteilung jedoch ist deutlich anders als die korrekte Lösung (siehe Abb. 5.2):
T à
r$s
tU
yH;}
^
z| e z
{
¸
für
Vjz
(5.4)
1
quantenmechanisch
mikrokanonisch
radiale Dichte
0.8
0.6
0.4
0.2
0
0
1
2
r [a.u.] 3
4
Abbildung 5.2: Vergleich der quantenmechanischen und der mikrokanonischen radialen
Ortsaufenthaltswahrscheinlichkeiten nach Gln. (5.2) und (5.4).
z
Der Bereich außerhalb ^
a.u. ist bei dieser Bindungsenergie klassisch nicht erreichbar, außerdem haben die Elektronen am äußeren Umkehrpunkt der Bahn die größte Verweildauer. Die mikrokanonische Ortsraumdichte hat daher
ihr z Maximum weiter außen als die
a.u. Die anderen Verteilungen
quantenmechanische und einen scharfen Rand“ bei ^
”
sind so konstruiert, daß die Ortsverteilung stimmt.
Wie in Abschnitt 3.4.5.2 bereits erwähnt, kann Cohens Energieverteilung die Impulsverteilung nicht richtig wiedergeben, da durch die Vorgaben — keine ungebundenen Anteile und Drehimpulsverteilung wie bei der mikrokanonischen Verteilung — die Lösung des
kompletten Gleichungssystems Gln. (3.41) und (3.42) nicht mehr möglich ist.
5.2
Betrachtete Querschnitte
Numerische Rechnungen wurden für die drei Einschußenergien 17,6eV, 54,4eV und 250eV
durchgeführt, die Impulse des Projektils sind dabei 1,14a.u., 2a.u. bzw. 4,29a.u. Sie liegen
jeweils im Bereich knapp oberhalb der Ionisationsschwelle, bei mittleren Energien und am
5.2. BETRACHTETE QUERSCHNITTE
53
radiale Impulsdichte
1.2
quantenmechanisch
Energieverteilung
0.9
0.6
0.3
0
0
0.5
1
1.5
2
p [a.u.]
2.5
Abbildung 5.3: Vergleich der quantenmechanischen radialen Impulsaufenthaltswahrscheinlichkeit mit der der Energieverteilung nach Cohen.
Anfang des Hochenergiebereichs. Die gewählten Energien sind die, für die im jeweiligen
Bereich die meisten experimentellen Daten verfügbar sind.
Bei jeder Energie wurden viele Millionen von Trajektorien berechnet und die Endwerte
der Trajektorien, die nach Abschnitt 3.6.2 als ionisiert zu zählen sind, gespeichert. Aus dieser Datenbank“ können dann — zeitlich unabhängig von der eigentlichen Rechnung — die
”
jeweils zu einem Querschnitt passenden Ereignisse herausgefiltert werden, ein Vorgehen,
das der offline“–Auswertung im Experiment entspricht. Die Berechnung der Trajektorien
”
muß also für eine bestimmte Energie nur ein einzges Mal erfolgen, daraus können dann alle gewünschten Querschnitte nachträglich bestimmt werden, egal ob voll differentiell oder
integriert.
Da die einzelnen Trajektorien voneinander unabhängig sind, kann auch im Nachhinein
noch die Statistik verbessert werden, wenn es sich zeigt, daß für einen gewünschten Querschnitt zu wenig Ereignisse vorhanden sind.
Bei jeder Energie wurden nun aus den Endwerten der gespeicherten Trajektorien bestimmt:
der totale Ionisationsquerschnitt Ø
der energieabhängige einfach differentielle Querschnitt
7 7,
U
,
die in Energie und Winkel zweifach differentiellen Querschnitte
und schnelle Elektronen sowie
die voll differentiellen Querschnitte
Y ø x
u
x
Wb X
je für langsame
, für die Messungen vorliegen.
Neben dieser konventionellen“ Methode, die Daten als einzelne Querschnitte darzustel”
len, können, wie bei experimentellen Auswertungen häufig verwendet, beliebige Schnitte
durch den Raum der Endzustände gelegt werden und als Scatterplots“ dargestellt werden.
”
Diese Art der Auftragung ist quantitativ weniger anspruchsvoll“ als konventionelle Quer”
schnitte, bietet aber oft einen qualitativ besseren Überblick, vor allem, wenn relativ wenig
54
KAPITEL 5. RECHNUNGEN AN WASSERSTOFF
Ereignisse zur Verfügung stehen. Bei dieser Auftragung spielt die von der Berechnung getrennte Auswertung ihre Stärken aus, denn um solche Verteilungen quantenmechanisch zu
bestimmen, ist es nötig, für jeden Punkt der Ebene getrennt den (z.T. integrierten) Querschnitt zu berechnen, während hier bereits alle Daten vorhanden sind und nur aus einer
großen Datei herausgesucht werden müssen.
Als quantenmechanischer Vergleich — neben den verfügbaren experimentellen Querschnitten — dienen die kürzlich veröffentlichen CCC–Rechnungen von Bray etal. [Bra99],
die sehr konsistent den gesamten betrachteten Energiebereich abdecken und daher auch
dort, wo keine Messungen verfügbar sind, als Vergleich benutzt werden.
Die Ergebnissen werden jeweils nach Einschußenergie getrennt vorgestellt, angefangen
bei der höchsten. Für jede Energie werden dann die Querschnitte, die mit den verschiedenen
Anfangsverteilungen erhalten werden, aufgeführt. Propagation und Auswertung werden immer auf die gleiche Weise durchgeführt, wie dies in Kapitel 3 erläutert wurde; das generelle
Vorgehen ist parameterfrei“, der einzige Freiheitsgrad“ ist die verwendete Anfangsvertei”
”
lung. Dabei zeigt es sich, daß Unterschiede bei den totalen und den wenig differentiellen
Querschnitten am geringsten sind, der strengste Test sind jeweils die voll differentiellen
Querschnitte.
Bei dieser z.T. recht tabellarischen Auflistung ist daran, wie gut oder schlecht die jeweiligen Querschnitte sind, zu erkennen, welche Prozesse in der klassischen Näherung bei den
verschiedenen Einschußenergien jeweils dominant sind. Daraus wird ersichtlich, welche
Näherungen im jeweiligen Energiebereich ausschlaggebend sind.
Da die Begriffe im folgenden Teil recht häufig verwendet werden, werden für die verschiedenen Querschnitte die folgenden (englischen) Abkürzungen benutzt: der einfach differentielle Querschnitt wird mit SDCS bezeichnet ( single differential cross section“), der
”
zweifach differentielle mit DDCS ( doubly differential cross section“) und der dreifach
”
differentielle mit TDCS ( triply . . .“, natürlich); für den totalen Querschnitt wird z.T. die
”
Abkürzung Ø 7 U 7 verwendet.
5.2.1
Ein technischer Einschub zur Auswertung
In der Berechnung der Querschnitte in Gl. 3.83 werden die Endwerte durch Delta–Funktionen
gefiltert. In numerischen Rechnungen muß dabei natürlich eine der asymptotischen Darstellungen verwendet werden, die für CTMC–Rechnungen
praktischste Form ist die Rechteck
V
k
l
i
k
funktion:
i
V è á
è á
y }
e
für
Á^[Z Í
(5.5)
sonst
Eine andere, hier auch verwendete
ist eine normierte Gaußfunktion mit verschwin Form
i
i
i
dender Breite:
y }
y e z Ø }
^ \ e¼
mit Ø^] Í
(5.6)
p ¡
Diese Funktionen simulieren einen Detektor mit endlicher Auflösung, bzw. endlichem
Akzeptanzwinkel. Die Breite hat Einfluß auf die Qualität der Wirkungsquerschnitte, denn,
5.3. VERLAUF DES TOTALEN QUERSCHNITTS
55
ist sie zu groß, führt das zu einer Verschmierung, bei der Signaturen des Querschnitts verloren gehen können, ist sie aber zu schmal, wird wegen der endlichen Anzahl an Ereignissen, aus denen dann immer weniger ausgewählt werden, die Statistik des Querschnitts zu
schlecht.
Ein technischer Trick“, die begrenzte Anzahl an Ereignissen, die für voll differentiel”
le Querschnitte zur Verfrügung steht, besser auszunutzen, ist es, Ereignisse mehrfach zu
zählen: Gl. 3.83 bestimmt den Querschnitt an einem Punkt“ mit bestimmten Parametern.
”
Um einen kompletten Verlauf des Querschnitts zu erhalten, muß diese Auswahl für jeden
gewünschten Winkel durchgeführt werden, üblicherweise in gleichmäßigen Winkelschritten.
Diese beiden Intervalle — die Detektorakzeptanz“ und der Abstand der Datenpunk”
te des Querschnitts — sind unabhängig, der Detektor“ kann auch um kleinere Intervalle
”
verschoben werden, als seine Auflösung beträgt. Das bedeutet, daß ein gerechnetes Ereignis in der Auswertung in zwei oder mehreren nebeneinanderliegenden Datenpunkten eines
Querschnitts beitragen kann. Auf diese Weise wird die Statistik für jeden einzelnen Punkt
des Querschnitts verbessert, ohne die Form durch zu wenige Stützstellen unkenntlich zu
machen. Auf diese Weise sind nebeneinander liegende Punkte des Querschnitts aber nicht
mehr statistisch unabhängig.
Die in den voll differentiellen Querschnitten angegeben Fehlerbalken sind aus dem statistischen Fehler bestimmt:
â Ø
5.3
r
^
Ø
_ r
Ó
(5.7)
r
Verlauf des totalen Querschnitts
Die historisch erste Anwendung einer CTMC–Rechnung in der Atomphysik diente der Bestimmung des totalen Ionisationsquerschnittes [AP66b], daher soll dieser auch hier am Anfang der vorgestellten Rechnungen stehen. Er hat für alle hier untersuchten Anfangsverteilungen im wesentlichen den gleichen Verlauf (siehe Abb. 5.4): das Maximum liegt bei einer
Einschußenergie von etwa 40eV, knapp einen Faktor zwei niedriger als im Experiment,
:
und das Hochenergieverhalten läuft jeweils gegen den klassischen Limes `
. Die
¸
Wigner– und die Produktverteilung führen etwa zur gleichen Höhe am Maximum, sierªÒ liegen
um etwa 10% über dem zur mikrokanonischen bzw. Cohens Energieverteilung gehörenden.
Der totale Querschnitt zeigt, daß die Wahl der Anfangsverteilung für integrierte Querschnitte kaum einen Einfluß hat, wenn man beachtet, daß diese doch recht unterschiedlichen
Ansätze untereinander eine kleinere Streuung aufweisen, als die jeweilige Abweichung zum
Experiment — speziell der Verlauf, d.h. Position des Maximums und der Hochenergielimes,
wird durch die Anfangsverteilung nur minimal beeinflußt. Der Grund dafür ist die bei allen
Rechnungen gleiche klassische Näherung, die das Gesamtproblem in einzelne unabhängige
Trajektorien auflöst“: Für den totalen Querschnitt ist nur wichtig, mit welcher Wahrschein”
lichkeit ein gewisser minimaler Energieübertrag vom Projektil auf das Target stattfindet,
weniger, an welcher Stelle (im Phasenraum) dies stattfindet (vgl. [Tho12]).
KAPITEL 5. RECHNUNGEN AN WASSERSTOFF
56
10
σtot [10 -17cm2]
Shah etal.
mikrokan.
Produkt
1
10
σtot [10 -17cm2]
Shah etal.
Cohen
Wigner
1
10
100
E in [eV]
1000
Abbildung 5.4: Verlauf des totalen Ionisationsquerschnitts, gerechnet für verschiedene Anfangsverteilungen des Wasserstoffatoms: oben mikrokanonisch und Produktverteilung, unten Wignerverteilung und Energieverteilung nach Cohen. Die dargestellte absolute Messung
wurde von Shah etal. durchgeführt [SEG87].
5.4
Einschußenergie 250eV
Die Einschußenergie von 250eV ist ein häufig untersuchter Bereich, da hier bereits der
Hochenergielimes der Bornschen Reihe3 zu brauchbaren Ergebnissen führt, der totale Querschnitt aber noch so hoch ist, so daß im Experiment in vernünftigen Zeiten“ die nötige
”
Statistik erreichbar ist.
Bei dieser Energie wurden auch von uns die ersten Rechnungen durchgeführt, da hier zu
erwarten ist, daß die Elektronen bereits so schnell sind, daß eine klassische Billardkugel–
”
Beschreibung“ die Schwerpunktdynamik der Wellenpakete beschreiben kann: der Stoß ist
dann so schnell, daß im Vergleich dazu das Zerlaufen der quantenmechanischen Wellenpakete deutlich langsamer erfolgt. Auf der anderen Seite aber ist bekannt, daß das klassische Hochenergieverhalten, das bereits von 1912 von Thomson bestimmt wurde [Tho12],
3
Hochenergielimes bedeutet hier zum einen, daß die Elektronen — klassisch gedacht(!) — so schnell sind,
daß das Projektil und das schnellere der auslaufenden Elektronen in einer quantenmechanischen Rechnung als
ebene Wellen dargestellt werden können. Andererseits verändert die post collision interaction“ die Querschnit”
te nicht mehr in ihrer Form, lediglich die Lage der Maxima wird um ein paar Grad verschoben. Das langsamere
Elektron wird dabei meist durch eine Coulombwelle beschrieben.
5.4. EINSCHUSSENERGIE 250EV
57
vorhersagt und damit vom korrekeinen Abfall des totalen Querschnitts proportional <
Ø
ten quantenmechanischen Verlauf mit 7 U 7 `badc abweicht:
bei sehr hohen Energien tragen
­Wò
also quantenmechanische Prozesse, vor allem Tunneln, signifikant zum Querschnitt bei —
diese werden aber von einer voll klassischen Theorie überhaupt nicht erfaßt; entgegen der
Intuition ist also zu erwarten, daß bei sehr hohen Energien die klassische Beschreibung
zunehmend versagt.
In Tabelle 5.2 sind die berechneten
Ereignisse aufgelistet: für jede Verteilung ist jeweils
åá
der maximale Stoßparameter ß à
, die Anzahl der gerechneten sowie die Anzahl der als
ionisiert gespeicherten Trajektorien angegeben.
Verteilung
mikrokan.
Produkt
Wigner
Cohen
ßà
åá
[a.u.]
3,5
12
15
7,5
Anz.(Traj.)
6,1 Mio.
186 Mio.
104 Mio.
17,75 Mio.
Anz.(ionis.)
155’484
395’691
339’242
83’492
á
Tabelle 5.2: Übersicht über maximalen Stoßparamter ( ß à “), Anzahl der für die Ein”
schußenergie von 250eV gerechneten ( Anz.(Traj.)“) und der als ionisiert gespeicherten
”
( Anz.(ionis.)“) Trajektorien mit den betrachteten Anfangsverteilungen des Wasserstof”
fatoms (siehe Abschnitt 5.1): mikrokanonische, Produkt-, Wigner- und Energieverteilung
nach Cohen. Bei der Wignerverteilung sind die reinen Anzahlen angeben, bei der Berechnung der Querschnitte müssen die einzelnen Trajektorien noch mit dem jeweiligen Vorzeichen gewichtet werden.
5.4.1
Totaler Ionisationsquerschnitt
Aus den (effektiven) Anzahlen der gerechneten Trajektorien erhält man die in Tabelle 5.3
angegebenen totalen Ionisationsquerschnitte.
Verteilung
mikrokan.
Produkt
Wigner
Cohen
Messung von Shah etal. [SEG87]
Messung von Shyn [Shy92])
Ø 7 7 [ Í 6J e e ]
U ¸
2,76
2,71
2,52
2,33
3,43
(4,9)
Tabelle 5.3: totaler Ionisationquerschnitt für die verwendeten Anfangsverteilungen bei
;:
^ 250eV. Zum Experiment von Shyn siehe die Anmerkung im Text.
rªÒ
Die berechneten Querschnitte sind alle um etwa 20% kleiner als der gemessene Wert.
Bei dieser Energie weist der klassische totale Ionisationsquerschnitt bereits einen steileren
Verlauf auf als das Experiment, quantenmechanische Beiträge scheinen bereits eine Rolle
zu spielen.
Der totale Querschnitt beschreibt einen effektiven geometrischen Querschnitt des Targets. Da, bis auf die mikrokanonische Verteilung, die Dichte im Ortsraum korrekt beschrie-
KAPITEL 5. RECHNUNGEN AN WASSERSTOFF
58
ben wird, muß der zu kleine Querschnitt so interpretiert werden, daß die Verteilung zu
durchlässig“ ist. Das ist eine Folge der klassischen Näherung der Propagation, die die ein”
zelnen Stützstellen der Phasenraumverteilung entkoppelt, die Verteilung in gewisser Weise
aufweicht“.
”
Der von Shyn gemessene Querschnitt [Shy92] wurde in Klammern gesetzt, da dieser Wert
nicht zuverlässig ist. Bei diesem Experiment wurden zweifach differentielle Querschnitte
gemessen und diese zu einfach differentiellen und totalen aufsummiert. Die DDCS aber,
wie noch gezeigt wird, sind bei kleinen Winkeln systematisch zu groß, somit auch der totale
Querschnitt.
5.4.2
Einfach differentiell
Um eine Stufe detailliertere
Informationen liefert der einfach differentielle energieabhängi
ge Querschnitt Y . Hier sind zwischen den verschiedenen Phasenraumverteilungen praktisch keine Unterschiede zu erkennen, ihr gemeinsamer Verlauf ist allerdings etwas flacher
als im Experiment (Abb. 5.5).
10
SDCS [10-18 cm2/eV]
E0 = 250eV
Shyn * 0.7
mikrokan.
Produkt
Wigner
Cohen
1
0.1
0.01
0
80
E [eV]
160
;:
z/f
^
Í eV: zwischen
Abbildung 5.5: Energiedifferentieller Wirkungsquerschnitt bei
ª
r
Ò
den verschiedenen Anfangsverteilungen sind praktisch keine Unterschiede zu erkennen. Die
Messung von Shyn wurde entsprechend Tab. 5.3 auf den korrekten totalen Querschnitt skaliert.
Der gleiche Verlauf für die verschiedenen Phasenraumverteilungen läßt sich mit der hier
eingeführten Definition der Ionisation erklären: Bei 250eV Einschußenergie ist das Projektil
bereits so schnell, daß der Ort, an dem der Stoß stattfindet, praktisch keinen Einfluß auf den
Energieübertrag auf das Targetelektron hat — ob die Ortsverteilung korrekt ist, spielt also
keine Rolle, wenn eine Wechselwirkung eintritt.
Der flachere Verlauf des SDCS bedeutet, daß bei der klassischen Rechnung der Energieübertrag auf das Targetelektron im Mittel etwas höher ist als im Experiment, schnellere
ionisierte Elektronen sind wahrscheinlicher. Das wird nachvollziehbar, wenn man die beim
totalen Querschnitt gegebene Interpretation berücksichtigt: die einzelnen Phasenraumpunkte sind in der klassischen Rechnung entkoppelt, das Projektil stößt also nicht auf eine ver”
schmierte“, nachgiebige“ Ladungsverteilung, sondern auf eine Punktladung. Der Stoß ist
”
härter“: es wird mehr Energie übertragen.
”
5.4. EINSCHUSSENERGIE 250EV
59
Bereits dieser klassisch einfach zu bestimmende Querschnitt zeigt die Vorteile, die der
hier vorgestellte quasiklassische Ansatz durch die Betrachtung des Energieübertrags hat: es
spielt keine Rolle, wie die Energie der Startwerte verteilt ist, jedesmal läßt sich ein konsistenter energieabhängiger Querschnitt bestimmen. Das wäre nicht möglich, wenn Ionisation
wie bisher über die absolute Energie der Elektronen definiert würde, da dann jede Trajektorie eine andere Gesamtenergie hätte, für jede Trajektorie also eine andere Skalierung der
Achsen gälte.
5.4.3
Zweifach differentiell
Der zweifach differentielle Querschnitt Wb wird hier als Winkelverteilung der Elektronen bei fester Energie aufgetragen, für die beiden zusammengehörigen Energien 5eV und
231,4eV.
Um den Verlauf des Querschnitts besser vergleichen zu können, wurden die DDCS jeweils auf den korrekten totalen Querschnitt skaliert; diese Skalierung zeigt besser, wie die
Winkelverteilung durch die Anfangsverteilungen beeinflußt wird. Dabei wird die Annahme gemacht, daß sich die Fehler in den Querschnitten gleichmäßig auf alle Konfigurationen
verteilen, daß also in der klassischen Näherung keine zusätzlichen Reaktionskanäle geöffnet
oder geschlossen sind, verglichen mit der Quantenmechanik.
Für die schnellen Elektronen (Abb. 5.6) existieren keine Messungen, sodaß hier mit den
CCC–Rechnungen von Bray verglichen wird. Man erkennt, daß die Winkelverteilung etwas
breiter ist als die Rechnung, aber im wesentlichen bei allen Phasenraumverteilungen das
gleiche Verhalten zeigt.
DDCS [10-19 cm2/sr eV]
1000
CCC (15,5)
Born
mikrok. * 1,24
Produkt * 1,26
Wigner * 1,36
Cohen * 1,48
E0 = 250eV
Eb = 231eV
100
10
1
0.1
0.01
0
30
ϑ [°]
60
Abbildung 5.6: Winkelverteilung der Elektronen mit 231,4eV bei
90
@:
rªÒ
^ 250eV.
Anders sieht das Bild bei den langsamen Elektronen aus (Abb. 5.7): Die Wigner- und die
Produktverteilung haben in etwa den gleichen Verlauf wie die CCC–Rechnungen: bei etwa
gÍU
hat der Querschnitt ein Maximum, den sogenannten binary peak“, der durch einen
”
Elektron–Elektron–Stoß ohne Beteiligung des Kerns erklärt wird.
Die Messungen von Shyn jedoch zeigen einen anderen Verlauf, hier steigt der Querschnitt
zu kleinen Winkeln hin stark an. Diese Abweichung wurde bereits von Berakdar und Klar
[BK93] und Bray [Bra99] festgestellt. Es handelt sich hierbei um einen systematischen
Fehler im Experiment, denn für diese Elektronenenergien ist im (koplanaren) TDCS die
KAPITEL 5. RECHNUNGEN AN WASSERSTOFF
60
CCC (15,5)
Born
mikrok. * 1,24
Produkt * 1,26
Shyn
DDCS [10-19 cm2/sr eV]
E0 = 250eV
Eb = 5eV
6
3
DDCS [10-19 cm2/sr eV]
0
CCC (15,5)
Born
Wigner * 1,36
Cohen * 1,48
Shyn
6
3
0
0
45
90
ϑ [°]
135
Abbildung 5.7: Winkelverteilung der Elektronen mit 5eV bei h@i>jlknm
gen sind auf den korrekten totalen Querschnitt skaliert.
180
250eV. Die Rechnun-
bestimmende Struktur der binary peak“, der auch nach der Integration über den Winkel
”
zwischen den Streuebenen noch zu erkennen sein sollte; es ist auch nicht nachvollziehbar,
bei welchen Geometrien dieser starke Beitrag in Vorwärtsrichtung zu finden sein sollte,
wenn er in der (und um die) koplanaren Konfiguration nicht auftritt (zur Form der TDCS
bei 5eV Elektronenenergie siehe weiter unten). Diese Abweichungen führen dann dazu, daß
der aus diesem Messungen aufsummierte totale Querschnitt zu groß ist.
Aus diesem Grund kann bei kleinen Streuwinkeln nicht mit den Messungen verglichen
werden, diese scheinen erst bei größeren Winkeln verläßlich zu sein.
Mit der mikrokanonischen Verteilung läßt sich bereits der DDCS für die langsamen Elektronen nicht mehr korrekt bestimmen, hier ist die Verteilung zu großen Winkeln hin verschoben, auch ist das Maximum des binary peaks“ nicht vorhanden. Die Form der Ortsvertei”
lung scheint also einen Einfluß auf die Winkelverteilung der Elektronen zu haben: andersherum spielt nun der Ort, an dem der Stoß stattfindet, eine Rolle, indem er den weiteren
Weg der Elektronen beeinflußt. Das geschieht bei langsamen Elektronen natürlich stärker
als bei schnellen.
Cohens Energieverteilung liegt zwischen den anderen Verteilungen: Das Maximum zwar
ist vorhanden, aber bei zu großen Winkeln, während der recoil“–Bereich wie bei der mi”
krokanonischen Verteilung stark ausgeprägt ist.
Die recht gute Übereinstimmung des skalierten DDCS ist so zu interpretieren, daß lediglich die Wahrscheinlichkeit für eine Ionisation zu klein ist (siehe Abschnitt 5.4.1), der
5.4. EINSCHUSSENERGIE 250EV
61
Vorgang als solcher aber im wesentlichen richtig beschrieben wird.
5.4.4
Voll differentiell
Der volldifferentielle Ionisationsquerschnitt, bei dem die Winkel und Energien der beiden
Elektronen in Koinzidenz gemessen werden, ist der härteste Test für eine theoretische Rechnung, denn diese Messung gibt Aufschluß über die korrellierte Dynamik der beiden Elektronen, wobei auch kleinere Strukturen des Querschnitts nicht hinter einer Mittelung verborgen
bleiben.
Auch an unsere klassische Rechnung stellt der TDCS die höchsten Anforderungen bezüglich der Statistik der Rechung: Aus dem Raum der Endwerte der Trajektorien wird in jeder
Dimension“ ein kleiner Teil ausgewählt, letzten Endes bleiben nur sehr wenige passende
”
Ereignisse übrig, die zu einem TDCS beitragen. In die nachfolgend vorgestellten Querschnitte gehen jeweils nur etwa hundert (oder wenige hundert) der vielen Millionen gerechneter Trajektorien ein4 .
Alle verfügbaren Experimente und Rechnungen sind bei dieser Energie in koplanarer
asymmetrischer Geometrie durchgeführt: eines der Elektronen läuft schnell unter einem
festgehaltenen kleinen Streuwinkel aus, die Winkelverteilung des anderen Elektrons in der
durch das schnelle definierten Streuebene wird gemessen (Abb. 5.8). Diese Anordnung ist
als Ehrhardt–Geometrie“ bekannt, da die ersten und bekanntesten absoluten Messungen
”
von Ehrhardt etal. durchgeführt wurden [EKSJ85].
Dieser TDCS wird durch zwei Maxima bestimmt: den sogenannten binary peak“, bei
”
dem das langsame Elektron in Richtung des Impulsübertrags ausläuft, verbreitert durch die
Verteilung des Anfangsimpulses im gebundenen Zustand, und den recoil peak“, bei dem
”
das langsame Elektron nach der Streuung noch einmal am Kern reflektiert wird und in entgegengesetzter Richtung zum binary peak“ das Atom verläßt. Mit steigender Energie sinkt
”
die Wahrscheinlichkeit für diese Rückstreuung, dieser Peak geht zurück, während er für
vernachlässigbar kleine Impulsüberträge, im sog. Photoionisationslimit gleich groß wie das
binäre Maximum ist. Die Position dieser beiden Maxima wird noch durch die Abstoßung
der beiden auslaufenden Elektronen etwas zu größeren Streuwinkeln hin verschoben, diese
Wechselwirkung wird oft als PCI“, als post collision interaction“ bezeichnet.
”
”
Für die folgenden Winkel und Energien wurden die TDCS bestimmt, es sind dies die
Parameter, für die absolute Messungen vorliegen. Die jeweiligen Breiten der ausgewählten
Bereiche wurden so weit verkleinert, bis gerade noch genügend Ereignisse zum Querschnitt
beitrugen, sie erreichen z.T. die experimentelle Auflösung. Aus Platzgründen wird jedoch
nur eine (repräsentative) Auswahl der Querschnitte gezeigt, an denen sich die Charakteristika der Anfangsverteilungen darstellen lassen.
Auch die TDCS wurden wieder auf den korrekten totalen Querschnitt skaliert, wie bereits
beim DDCS durchgeführt.
4
Eine einfache Abschätzung demonstriert die Größenordnung: es wird in vier Dimensionen ausgewählt,
in den zwei Streuwinkeln, im Winkel zwischen den Streuebenen und in der Energie. Mit den in Tab. 5.4 angegebenen Breiten schätzt man ab, daß eine von etwa 30 Millionen Ionisationen zum Querschnitt beitrüge,
wenn die Endwerte isotrop in allen Winkeln und Energien verteilt wären. Zum Glück weisen die Endwerte eine
ausgepr ägte Struktur auf. . .
KAPITEL 5. RECHNUNGEN AN WASSERSTOFF
62
Ein
ϑa
ϑb
Eb
Abbildung 5.8: Schema der TDCS–Messungen in Ehrhardt–Geometrie: das schnelle Elektron wird um den Winkel oqp gestreut, das langsame mit der Energie h@r detektiert, die Verteilung dieses Elektrons im Winkel oRr ist die Meßgröße.
h r
[eV]
5 s 0,5
10 s 1
14 s 1,5
50 s 2
op
tvuqw6s 0,3 x , y?x=w , z-x , {vuqw
z x , { u s 0,5 x
z/x , {|us 0,5 x
}qz x , ~/ x , ~/z x , / x , /z x sW~ x
w
Messung von
: Ehrhardt [EKSJ85, EJKS86]
Lohmann [LMSW84]
Weigold [WNHF79]
Tabelle 5.4: Übersicht über die Parameter, zu denen volldifferentielle Querschnitte in
Ehrhardt–Geometrie gemessen wurden. Für die fett markierten Parameter sind hier die berechneten Querschnitte gezeigt. Die Streuebene wurde auf s}q/x genau bestimmt, dieser
Wert hatte im Bereich zwischen sWz x und sW~/ x kaum Einfluß auf die Form des TDCS, nur
auf die Anzahl der gezählten Ereignisse, ist daher unproblematisch.
Als generelle Tendenz bei allen gerechneten Anfangsverteilungen stellt man fest (Abb. 5.9
und 5.10), daß praktisch alle TDCS im Vergleich zu den Messungen zu klein sind; das ist
konsistent mit den weniger differentiellen Querschnitten: bei den gerechneten TDCS sind
die Maxima i.a. breiter als im Experiment und dadurch in der Höhe niedriger.
Im einzelnen zeigen die Verteilungen das folgende Verhalten: bei der mikrokanonischen
Verteilung liegt der binary–peak entweder bei zu großen Winkeln ( h;rm 50eV) oder wird
völlig falsch wiedergegeben ( h@rm 5 . . . 14eV). Der Rückstreupeak ist im wesentlichen
vorhanden, aber da er, bezüglich der Vorwärtsrichtung, etwa symmetrisch zu den Resten
des binären Maximums ist, muß man ihn als Artefakt der Rechnung interpretieren.
Bei der Produktverteilung liegt der binary peak“ tendenziell bei etwas zu kleinen Win”
keln, die Breite ist meist etwas zu groß, das ist gut bei h r m 50eV zu sehen (hier nicht
gezeigt). Der recoil peak“ hingegen ist deutlich zu klein.
”
Auch bei der Wignerverteilung ist der recoil peak“ zu wenig ausgeprägt, hier stimmen
”
aber Position und Breite des binären Maximums recht gut. Zum Teil weist hier der Querschnitt negative Werte auf, das ist, neben den starken Oszillationen, ein Zeichen dafür, daß
immer noch nicht genug Ereignisse für den Querschnitt zur Verfügung stehen.
Cohens Energieverteilung zeigt, wie erwartet, ein Verhalten, das zwischen dem der klassisch motivierten mikrokanonischen Verteilung und dem der aus der Quantenmechanik be-
5.4. EINSCHUSSENERGIE 250EV
Eb = 5eV
ϑa = 3°
63
Ehrhardt
mikrokan. * 1,24
Ehrhardt
Eb = 5eV
ϑa = 8°
10
Ehrhardt
mikrokan. * 1,24
2.5
0
0
Ehrhardt
2.5
0
0
Ehrhardt
Wigner * 1,36
Ehrhardt
Wigner * 1,36
Ehrhardt
TDCS [10-17 cm2/eV sr2]
10
5
10
5
2.5
0
TDCS [10-17 cm2/eV sr2]
Ehrhardt
Produkt * 1,26
Ehrhardt
Produkt * 1,26
0
Ehrhardt
Cohen * 1,48
Ehrhardt
Ehrhardt
Produkt * 1,48
10
0
-180
5
-90
0
ϑ b [°]
90
5
2.5
-180
-90
0
ϑb
90
0
180
Abbildung 5.9: TDCS in Ehrhardt–Geometrie für hjkm 250eV, h@rm 5eV und oqpm-x und
x für die verschiedenen Anfangsverteilungen. Die Messungen sind absolut, die (absoluten)
Rechnungen nachträglich entsprechend des totalen Querschnitts skaliert.
stimmten Wignerverteilung liegt.
Das binäre Maximum wird mit einen Billardstoß der Elektronen erklärt, ein Bild das in
dieser Rechung recht gut wiedergegeben werden kann. Dabei spielt die Form der Ortsverteilung eine Rolle, d.h. die Position, an der sich das Targetelektron befindet, wenn es vom
Projektil gestoßen wird, beeinflußt die weitere Entwicklung der Trajektorie. Das ist schön
in Abb. 5.11 zu erkennen: hier findet der Stoß nahe beim Kern statt, das Targetelektron läuft
zuerst in Richtung des Impulsübertrags, wird aber dann, da es die Anziehung des Kerns
spürt, deutlich aus dieser Richtung abgelenkt. Die Abstände, auf denen der Energieübertrag,
also der Stoß der beiden Elektronen, erfolgt, ist dabei klein im Verhältnis zu einer typischen
Halbachse der gebundenen Bahn bzw., zeitlich betrachtet, zur typischen Umlaufzeit (dies
ist in Abb. 5.11 zu erkennen, obwohl nicht explizit gekennzeichnet).
Die mikrokanonische Verteilung nun hat das Maximum ihrer radialen Verteilung nahe
beim Kern, hier findet diese starke Ablenkung bei fast allen ionisierten Elektronen statt,
während die anderen Verteilungen auch einen wesentlichen Anteil an Elektronendichte weiter weg vom Kern haben, wo diese Störung deutlich schwächer ist: der binäre Stoß wird
kaum verfälscht. Am deutlichsten ausgeprägt ist dies bei der Wignerverteilung, ihre negativen Anteile befinden sich im wesentlichen bei großen Radien und Impulsen. Dort ist die
KAPITEL 5. RECHNUNGEN AN WASSERSTOFF
Eb = 10eV
ϑa = 8°
Eb = 14eV
ϑa = 8°
Lohmann
mikrokan. * 1,24
Lohmann
Lohmann
3
1.5
3
Lohmann
mikrokan. * 1,24
1.5
0
3
Lohmann
Produkt * 1,26
Lohmann
Produkt * 1,26
Lohmann
3
1.5
1.5
0
0
Lohmann
Wigner * 1,36
3
3
Lohmann
Wigner * 1,36
Lohmann
TDCS [10-17 cm2/eV sr2]
Lohmann
Lohmann
0
1.5
1.5
Lohmann
Cohen * 1,48
3
1.5
0
-180
0
3
Lohmann
Cohen * 1,48
Lohmann
Lohmann
0
-90
0
ϑ b [°]
90
TDCS [10-17 cm2/eV sr2]
64
1.5
-180
-90
0
ϑb
90
180
0
Abbildung 5.10: TDCS in Ehrhardt–Geometrie für hjlkm 250eV, oqpWm
x und h;r'm 10eV
bzw. 14eV für die verschiedenen Anfangsverteilungen. Die Messungen sind absolut, die
(absoluten) Rechnungen nachtrräglich entsprechend des totalen Querschnitts skaliert.
aufsummierte Dichte zwar gering, da die unterschiedlichen Vorzeichen in der Mittelwertbildung zu einer Auslöschung führen, es werden aber deutlich mehr Startwerte bei großen
Radien ausgewählt, als z.B. bei der Produktverteilung oder Cohens Energieverteilung. Das
stimmt mit der gängigen Interpretation überein, in der der binäre Stoß ohne Beteiligung des
Kerns stattfindet — in einer klassischen Rechnung muß diese Ionisation also möglichst weit
vom Kern entfernt ablaufen.
Bei der Rückstreuung wird das bereits ionisierte Elektron zum Teil am Kern reflektiert.
Dieses Bild setzt voraus, daß das Elektron als ausgedehnte Wellenfunktion realisiert ist,
denn bei einem klassischen Punktteilchen findet Rutherfordstreuung statt, die Rückstreuung ist also extrem unwahrscheinlich, während die kohärente Streuung einer (ebenen) Welle die Rückwärtsrichtung bevorzugt. Das Fehlen des Rückstreupeaks ist also die Folge der
klassischen Entkoppelung der einzelnen Phasenraumpunkte, die sich nun wie Punktteilchen
bewegen, der quantenmechanische Wellencharakter ist verloren gegangen. Oder anders formuliert: von einer klassischen Rechnung kann der quantenmechanische Rückstreupeak gar
nicht wiedergegeben werden, da die Rückstreuung in Gegenrichtung auf der Interferenz
zwischen ein– und auslaufender Wellen beruht. Vor diesem Hintergrund muß der bei der
mikrokanonischen Verteilung vorhandene recoil peak“ als zufällige Struktur interpretiert
”
5.4. EINSCHUSSENERGIE 250EV
65
4
0
∆p
∆p + ∂V/∂r
-4
0
4
Abbildung 5.11: Wenn die Ionisation des gebundenen Elektrons nahe beim Kern stattfindet,
so läuft dieses zwar zuerst in Richtung des Impulsübertrags, wird aber danach durch das
anziehende Feld des Kerns aus dieser Richtung abgelenkt. Auf diese Weise strägt es an
einer falschen Stelle zum Wirkungsquerschnitt bei.
werden, allenfalls noch als Ergebnis der Elektron–Elektron–Abstoßung.
5.4.5
Zusammenfassung
Für die Einschußenergie von 250eV läßt sich damit zusammenfassend folgendes feststellen:
Auf den einfach differentiellen Querschnitt, die Energieverteilung, hat die verwendete
Anfangsverteilung keinen Einfluß, der Stoß findet bei dieser Energie so schnell statt,
daß die anfängliche Energie des Targetelektrons keine Rolle spielt, die Ionisation
ist lokal“. Wesentlich ist hierbei nur die Wahrscheinlichkeit für einen bestimmten
”
Energieübertrag.
Die Winkelverteilung vor allem der langsamen Elektronen wird (indirekt) durch den
Ort beeinflußt, an dem die Wechselwirkung mit dem Projektil stattfindet, denn das
Targetelektron spürt“ auf seinem Weg nach draußen das Kernpotential, dieses führt
”
zu einer Ablenkung. Die Energie des Elektrons wird dadurch jedoch nicht mehr beeinflußt, da es sich um eine ungestörte Keplerbahn handelt.
Bei 250eV Einschußenergie findet die Reaktion i.w. an einer Momentaufnahme“ des
”
Targets statt, das Bild zweier die meiste Zeit unabhängiger Elektronen ist recht gut
erfüllt. Wichtig ist also, daß das Target die korrekte Form“ hat, die Energieverteilung
”
ist zweitrangig.
Aus diesen Gründen zeigt die klassisch hergeleitete mikrokanonische Verteilung die größten
Abweichungen bei den Streuwinkeln. Bessere Ergebnisse liefert die Energieverteilung, in
die Cohen zusätzlich noch die quantenmechanische Ortsraumdichte einfließen ließ. Die beiden rein aus der quantenmechanischen Wellenfunktion bestimmten Anfangsverteilungen,
die Produkt– und die Wignerverteilung, sind bei dieser Einschußenergie zur Berechnung
von Querschnitten im wesentlichen gleichwertig: die Wignerverteilung reproduziert die
Querschnitte etwas besser, benötigt dafür aber eine deutlich höhere Anzahl an gerechneten Trajektorien.
KAPITEL 5. RECHNUNGEN AN WASSERSTOFF
66
5.5
Einschußenergie 54,4eV
Die Einschußenergie von 54,4eV liegt im mittleren Enegiebereich in der Nähe des Maximums des totalen Ionisationsquerschnitts.
Die Darstellung folgt der für 250eV Einschußenergie, sodaß der folgende Teil eher knapp
gehalten ist.
Die bei dieser Energie gerechneten Mengen an Trajektorien sind in Tab. 5.5 aufgeführt.
Verteilung
mikrokan.
Produkt
Wigner
Cohen
4p
[a.u.]
6
22
22
9
Anz.(Traj.)
20 Mio.
80 Mio.
181 Mio.
25,67 Mio
Anz.(ionis.)
455’781
162’752
537’997
222’888
Tabelle 5.5: Übersicht über die Anzahlen der bei 54,4eV Einschußenergie gerechneten Trajektorien für die jeweiligen Anfangsverteilungen.
5.5.1
Totaler Querschnitt
Für den totalen Ionsationsquerschnitt erhält man die in Tabelle 5.6 gezeigten Werte:
Verteilung
mikrokan.
Produkt
Wigner
Cohen
Messung von Shah etal. [SEG87]
| x [ R} ?= ]
v
6~y
6/
v6|}
v6~/
vq}q
Tabelle 5.6: totaler Ionisationquerschnitt für die betrachteten Anfangsverteilungen bei
h@i>jlknmzyy eV.
5.5.2
Einfach differentiell
Für diese Energie existiert keine Messung des einfach differentiellen Querschnitts, daher
wurde, wie bereits von Bray [Bra99], die Messung von Shyn [Shy92] in der Energieskala
und, entsprechend des totalen Querschnitts, in der Höhe umskaliert.
Hier (Abb. 5.12) sind nun Unterschiede im Querschnitt zu sehen, nicht nur zwischen
Messung und Rechnung wie schon bei 250eV sondern auch zwischen den verschiedenen
Anfangsverteilungen. Der Verlauf ist bei der mikrokanonischen Verteilung am flachsten,
bei der Produktverteilung am steilsten, jedoch auch diese zeigt ein weniger ausgeprägtes
Minimum als die Messung. Der Grund ist der gleiche wie bei 250eV Einschußenergie: durch
die Diskretisierung ist jeder einzelne Stoß im Mittel etwas härter“, der Energieübertrag
”
daher zu groß.
5.5. EINSCHUSSENERGIE 54,4EV
67
1
CCC (15,5)
Shyn * 0.7
mikrokan.
Produkt
Wigner
Cohen
E0 = 54,4eV
SDCS [10-17 cm2/eV]
0.8
0.6
0.4
0.2
0
0
10
20
E [eV]
30
40
Abbildung 5.12: Einfach differentieller energieabhängiger Querschnitt: Verteilung der Energie der ionisierten Elektronen bei hjlknm 54,4eV.
5.5.3
Zweifach differentiell
Der zweifach differentielle Querschnitt wird wieder für zwei zusammengehörige Energien
betrachtet: 5eV und 35,8eV. Für die schnelleren Elektronen existiert wieder keine Messung,
für die langsameren wird die Messung, die Shyn bei 60eV durchgeführt hatte, als Vergleich
benutzt, diesmal jedoch nicht entsprechend des totalen Querschnitts skaliert.
1000
DDCS [10-19 cm2/sr eV]
E0 = 250eV
Eb = 231eV
CCC (15,5)
mikrokan.
Produkt
Wigner
Cohen
100
10
1
0.1
0
45
90
ϑ [°]
135
180
Abbildung 5.13: Winkelverteilung der Elektronen mit 35,8eV Energie bei hjlknm 54,4eV.
Für die schnelleren Elektronen (Abb. 5.13), in der klassischen Rechnung zum größten
Teil das gestreute Projektil, findet man das gleiche Verhalten wie bereits bei 250eV (vgl.
Abb. 5.6): die Winkelverteilungen sind etwas breiter als Brays CCC–Rechnung, die Unterschiede zwischen den Anfangsverteilungen wiederum sehr gering. Obwohl hier die Energie
des Projektils geringer ist, ist dieses immer noch so schnell, daß der Ort, an dem die Wechselwirkung stattfindet, wenig Einfluß auf den eigentlichen Ionisationsprozeß hat: ähnlich
wie in Abb. 5.11 ist es immer noch ein lokaler“ Prozeß.
”
Bei den langsamen Elektronen mit 5eV zeigt der gemessene Querschnitt von Shyn wieder
die gleiche systematische Überhöhung bei kleinen Winkeln wie bei 250eV Einschußener-
KAPITEL 5. RECHNUNGEN AN WASSERSTOFF
68
E0 = 54,4eV
Eb = 5eV
DDCS [10-19 cm2/sr eV]
9
CCC (15,5)
Shyn
mikrokan. * 0,85
Produkt * 0,71
Wigner * 0,78
Cohen * 1,0
6
3
0
0
45
90
ϑ [°]
135
180
Abbildung 5.14: Winkelverteilung der Elektronen mit 5eV Energie bei hjlkm 54,4eV. Die
Querschnitte sind auf den korrekten totalen Querschnitt skaliert, um die Winkelabhängigkeit
besser vergleichen zu können.
gie (siehe Abschnitt 5.4.3), auch hier wird wieder mit Brays CCC–Rechnung verglichen.
Die mit den verschiedenen Anfangsverteilungen gerechneten DDCS sind auf den korrekten
(gemessenen) totalen Querschnitt skaliert, um die Winkelabhängigkeit besser zu erkennen.
Betrachtet man die Winkelverteilung der langsamen Elektronen, so sind deutliche Unterschiede zwischen den verschiedenen Anfangsverteilungen zu erkennen: am besten wird
die Struktur des Querschnitts — der binary peak“ liegt bei etwa //x , der recoil peak“ bei
”
”
großen Winkeln — von der Wignerverteilung wiedergegeben, es fehlt aber aus den in Abschnitt 5.4.4 diskutierten Gründen das Rückstreumaximum; das binäre Maximum stimmt
dagegen in Position und Breite recht gut.
Die mikrokanonischen Verteilung beschreibt den Querschnitt völlig verkehrt, die Gründe
sind die gleichen wie bei ~/z- eV Einschußenergie: die Verteilung befindet sich zu nahe um
den Kern, die langsamen Elektronen werden nach dem Stoß deutlich abgelenkt.
Cohens halbklassische“ Energieverteilung liegt wieder in der Mitte zwischen mikroka”
nonischer und Wignerverteilung: das binäre Maximum ist zu größeren Winkeln verschoben
und der Querschnitt bei großen Winklen deutlich überhöht, jedoch nicht so ausgeprägt wie
bei der mikrokanonischen Verteilung.
Interessanterweise stimmt hier das Ergebnis der Produktverteilung in der Betonung der
kleinen Winkel mit der Messungen überein. Doch dieses Verhalten läßt sich auch mit der
Wignerverteilung reproduzieren, wenn hierbei die Ereignisse nicht entsprechend ihres Vorzeichens gewichtet werden, sondern nur die Anzahl der Endwerte je Winkelbereich betrachtet wird, siehe Abb. 5.15.
Die Beiträge zu kleinen Streuwinkeln kommen also aus den Bereichen, in denen die Wignerverteilung negative Anteile hat: das ist bei großen Radien und Impulsen, d.h. im Bereich
der ungebundenen Startwerten. Die mikrokanonische Verteilung hat keine Startwerte außerhalb des Radius von 2a.u., hier ist der Querschnitt bei kleinen Winkeln zu klein. Daraus
ergibt sich folgende Zuordnung: zu den ganz kleinen Streuwinkeln tragen Targetstartwerte
bei großen Radien bei, die sich aber bei der Wignerverteilung z.T. gegenseitig auslöschen
— man könnte von klassischer Interferenz“ sprechen (das ist auch an starken Fluktuation
”
der Datenpunkte des DDCS der Wignerverteilung zu erkennen). Die Produktverteilung ist
5.5. EINSCHUSSENERGIE 54,4EV
69
12
DDCS [10-19 cm2/sr eV]
CCC (15,5)
Shyn * 0,7
Produkt * 0,71
Wigner * 0,78
Anz.(Wigner) * 0,4
E0 = 54,4eV
Eb = 5eV
10
8
6
4
2
0
0
45
90
ϑ [°]
135
180
Abbildung 5.15: Wird bei der Wignerverteilung nur die Anzahl der Endwerte betrachtet, die
zu einem bestimmten Winkelbereich beitragen, so zeigt sich ein Verlauf, der ähnlich dem
des DDCS mit aus Produktverteilung ist (siehe auch Abb. 5.14): die Beiträge zu kleinen
Winkeln gehören also zu Startwerten mit großen Radien (siehe Text).
bei großen Radien zu dicht“, bzw. zu klassisch“, während die mikrokanonische Verteilung
”
”
durch ihre falsche Ortsraumdichte nur Beiträge zu großen Streuwinkeln liefern kann.
5.5.4
Voll differentiell
Für den TDCS liegen Messungen in koplanarer asymmetrischer (Ehrhardt–) Geometrie vor,
entsprechend wurden die Querschnitte für die folgenden Parameter bestimmt:
h@r
[eV]
5 s 0,5
4x
s
oqp
1 x , 10 x , 16 x , 23 x je s 2 x
Messung von
Röder etal. [R 96]
Tabelle 5.7: Übersicht über die Parameter, zu denen bei der Einschußenergie von 54,4eV
volldifferentielle Querschnitte in Ehrhardt–Geometrie berechnet wurden. Die Streuebene
wurde auf s 12 x genau bestimmt.
Neben der Skalierung auf den totalen Querschnitt wurde unabhängig von der Anfangsverteilung anhand des DDCS für die schnellen Elektronen (35,8eV) der TDCS für oRpWm 4 x
mit dem Faktor 0,25 in der Höhe skaliert, der TDCS für oqp@m 10 x mit 0,5 (vgl. den Verlauf
des DDCS bei kleinen Streuwinkeln in Abb. 5.13).
Die Unterschiede zwischen den einzelnen Anfangsverteilungen sind recht deutlich zu
erkennen (Abb. 5.16 und 5.17): bei der mikrokanonischen Verteilung wird das binäre Maximum überhaupt nicht wiedergegeben, dafür stimmt das Rückstreumaximum erstaunlich
gut, unabhängig vom Winkel des schnelleren Elektrons.
Die etwas mehr quantenmechanische“ Energieverteilung von Cohen zeigt die gleiche
”
gute Übereinstimmung beim recoil peak“, der binary peak“ wird besser wiedergegeben,
”
”
wenn oqp größer wird, liegt aber bei diesen Querschnitten immer bei zu großen Winkeln.
Ein völlig anderes Verhalten zeigt die Produktverteilung: hier wird der Querschnitt mit
steigendem Winkel des schnellen Elektrons zwar auch immer besser wiedergegeben, allerdings liegen hier deutlich zu viele Endwerte in Vorwärtsrichtung, d.h. bei zu kleinen
KAPITEL 5. RECHNUNGEN AN WASSERSTOFF
70
Röder
mikrokan. * 0,85 * 0,25
ϑa = 4°
Röder
mikrokan. * 0,85 * 0,5
ϑa = 10°
2
Röder
Röder
3
2
1
1
0
0
Röder
2
2
1
1
0
0
Röder
Wigner * 0,78 * 0,25
3
Röder
Wigner * 0,78 * 0,5
2
Röder
TDCS [10-17 cm2/eV sr2]
Röder
Röder
3
Röder
Produkt * 0,71 * 0,5
2
1
1
0
0
Röder
Cohen * 1,0 * 0,25
Röder
Cohen * 1,0 * 0,5
2
Röder
3
Röder
TDCS [10-17 cm2/eV sr2]
Röder
Produkt * 0,71 * 0,25
2
1
1
0
-180
-90
0
ϑ b [°]
90
180
-180
-90
0
ϑ b [°]
90
180
0
Abbildung 5.16: TDCS in Ehrhardt–Geometrie für hjlkm 54,4eV, h;rm 5eV und oqpm 4 x
und 10 x für die verschiedenen Anfangsverteilungen. Die Rechnungen sind entsprechend des
totalen und des zweifach differentiellen Querschnitts für die schnellen Elektronen skaliert
(siehe Text).
Ablenkwinkeln. Besonders deutlich ist dies bei o p m 4 x zu erkennen (Abb. 5.16 links): hier
laufen beide Elektronen in etwa die gleiche Richtung nach vorne aus. Dies sollte aber durch
die Elektron–Elektron–Abstoßung stark unterdrückt sein. Daran zeigt sich ein Problem der
hier eingeführten Auswertung: durch die Betrachtung der Energieüberträge stimmen nun
die nominelle Energie“ und der tatsächliche Impuls nicht mehr überein. Es wird weiter
”
unten bei der Einschußenergie von }q|6 eV noch deutlicher werden, welche Konsequenzen
dies hat, an dieser Stelle sei dieses Problem erst einmal festgestellt.
Die Verteilung, die am nächsten an der Quantenmechanik ist, die Wignerverteilung, ist
hier in der Lage, alle Querschnitte recht gut wiederzugeben, speziell Position und Höhe des
binären Maximums. Das Problem, daß beide Elektronen in die gleiche Richtung, nach vorn,
auslaufen können, tritt auch hier bei o p m 4 x auf, jedoch nicht so ausgeprägt, wie bei der
Produktverteilung.
5.5. EINSCHUSSENERGIE 54,4EV
Röder
mikrokan. * 0,85
ϑa = 23°
Röder
mikrokan. * 0,85
ϑa = 16°
Röder
Röder
1.5
71
1
0.5
0.5
0
0
Röder
1
0.5
0.5
0
0
Röder
Röder
Röder
Wigner * 0,78
Röder
Wigner * 0,78
1.5
1
0.5
0.5
0
0
Röder
Cohen * 1.0
Röder
Cohen * 1,0
Röder
1.5
Röder
TDCS [10-17 cm2/eV sr2]
TDCS [10-17 cm2/eV sr2]
Röder
Röder
Produkt * 0,71
Röder
Produkt * 0,71
1.5
1
0.5
0.5
0
-180
-90
0
ϑ b [°]
90
180 -180
-90
0
ϑ b [°]
90
180
0
Abbildung 5.17: TDCS in Ehrhardt–Geometrie für hjkm 54,4eV, h@rm 5eV und oqpm 16 x
und 23 x für die verschiedenen Anfangsverteilungen. Die Rechnungen sind entsprechend des
totalen Querschnitts skaliert (siehe Text).
5.5.5
Zusammenfassung
Bei dieser Energie ist beim Übergang von den klassischen“ Verteilungen (mikrokanonische
”
und Cohens Energieverteilung), die nur entweder die Impuls– oder die Ortsraumdichte korrekt wiedergeben, zu den mehr quantenmechanischen“ eine deutliche Verbesserung der
”
Querschnitte zu erkennen. Die Produktverteilung hat die korrekte Orts– und Impulsdichte, hier ist der binary peak“ bereits vorhanden, die Wignerverteilung hat zusätzlich durch
”
ihre negativen Anteile eine Art quantenmechanischer Struktur“ 5 , die dazu führt, daß klas”
sisch erlaubte, aber in der Realität nicht auftretende Beiträge/Trajektorien sich gegenseitig
auslöschen.
Bei der Einschußenergie von 54,4eV hat das Projektil einen Impuls von 2a.u., es ist etwas
schneller als ein (klassisches) Elektron im Grundzustand. Die Geschwindigkeit ist noch
ausreichend hoch, daß, wie bei 250eV, die geometrische Form des Targets im Phasenraum
5
Hier sei nur an die in der Quantenoptik verbreitete Interpretation erinnert, wonach der Anteil negativer
Beiträge der Wignertransformierten eines Systems als Maß genommen wird, wie stark quantenmechanische Eigenschaften beitragen; ein kleiner Anteil dieser Beiträge bedeutet, daß das System klassischer“ ist, ein größe”
rer, daß es sich quantenmechanischer“ verhält. Oder anders formuliert: ein klassisches System hat eine rein
”
positive Wignertransformierte, negative Anteile gehören zu Quanteneffekten“.
”
KAPITEL 5. RECHNUNGEN AN WASSERSTOFF
72
abgetastet wird, aber bereits niedrig genug, um eine innere Struktur“ des Targets auflösen
”
zu können.
5.6
Einschußenergie 17,6eV
Bei der Einschußenergie von 17,6eV ist das Projektil kaum schneller als das Targetelektron
auf der klassischen Bahn mit dem Bohrschen Radius, die Kollision verläuft recht langsam.
Es ist also zu erwarten, daß hier andere Eigenschaften der Anfangsverteilungen zum Tragen
kommen, als bei den höheren Einschußenergien, da nun die Dynamik mehr Zeit hat“, sich
”
zu entwickeln.
Angemerkt soll noch werden, daß diese Einschußenergie gar nicht so unklassisch“ ist,
”
wie die Erfahrung erwarten ließe: Die Gesamtenergie liegt nur um 4eV über der Ionisationsschwelle; das Verhalten des totalen Querschnits an der Schwelle wurde zuerst von
Wannier [Wan53] (siehe für Verallgemeinerungen auf mehr als zwei Teilchen z.B. [PR98])
aus rein klassischen Phasenraumüberlegungen bestimmt — an der Ionsationsschwelle kann
also ein aufbrechendes Mehrteilchensystem recht gut klassisch beschrieben werden. Der
Hauptgrund ist der, daß zum einen die Dynamik vom (klassisch beschreibbaren) Langzeitverhalten bestimmt wird, zum anderen quantenmechanische Effekte wie z.B. Tunneln bei
diesen Energien gegenüber den direkten Reaktionswegen vernachlässigbar sind.
Die Langzeitdynamik der auslaufenden Elektronen kann klassisch beschrieben werden.
Das bedeutet, das die Bewegung der quantenmechanischen Wellenpakete durch das Verhalten des Schwerpunkts beschrieben wird, der dabei den klassischen Bewegungsgleichungen
gehorcht. Das aber ist das Bild der Standard–CTMC, in dem eine Trajektorie die Bahn
(des Schwerpunkts) eines Elektrons beschreibt. Im quasiklassischen Ansatz aber bedeutet
das, daß alle Trajektorien, die zu diesem Wellenpaket gehören, praktisch die gleiche Bahn
beschreiben müssen. Das geht aber nur, wenn beim Auslaufen deren Energie sehr nahe beieinander liegt. Dazu darf aber die Anfangsverteilung keine breite Energieverteilung haben,
sonst würden — wegen der Auswertung über den Energieübertrag — Trajektorien mit zu
unterschiedlichem Impuls zum gleichen Wellenpaket gezählt werden.
Bei dieser Energie wurden die folgenden Mengen an Trajektorien berechnet:
Verteilung
mikrokan.
Produkt
Wigner
Cohen
4p
[a.u.]
4,5
9
22
8
Anz.(Traj.)
17,5 Mio.
16,5 Mio.
50,5 Mio.
14,9 Mio.
Anz.(ionis.)
320’244
79’146
47’077
80’780
Tabelle 5.8: Übersicht über die Anzahlen der bei 17,6eV Einschußenergie gerechneten Trajektorien für die jeweiligen Anfangsverteilungen.
5.6.1
Totaler Querschnitt
Für den totalen Ionsationsquerschnitt erhält man mit den oben aufgeführten Rechnungen die
in Tabelle 5.9 gezeigten Werte. In diesem Bereich ist der totale Querschnitt bei allen Verteilungen deutlich größer als im Experiment, wie schon der Verlauf des totalen Querschnitts
5.6. EINSCHUSSENERGIE 17,6EV
73
in Abschnitt 5.3 gezeigt hatte. Der Grund ist die klassische Näherung, die die Trajektorien entkoppelt und den Ionisationsvorgang dem Thomsonschen Bild entsprechen läßt, vgl.
Abb. 2.1.
Verteilung
mikrokan.
Produkt
Wigner
Cohen
Messung von Shah etal. [SEG87]
v x [ q} )
]
3,27
3,55
3,98
3,06
2,1
Tabelle 5.9: totaler Ionisationquerschnitt für die berechnten Anfangsverteilungen bei
h;ijlknm 17,6eV.
5.6.2
Einfach differentiell
Für den SDCS bei 17,6eV liegen keine Mesungen vor, hier werden die klassischen Rechnungen wieder mit Brays CCC–Rechnungen verglichen.
2
CCC (∞, 5)
mikrokan. * 0,64
Produkt * 0,59
Wigner * 0,53
Cohen * 0,69
SDCS [10-17 cm2/eV]
E0 = 17,6eV
1.5
1
0.5
0
0
1
2
3
4
E [eV]
Abbildung 5.18: SDCS bei h@i>jlkm 17.6eV: die Rechnungen sind auf den korrekten totalen
Querschnitt skaliert.
Bei dem auf den totalen Querschnitt skalierten einfach differentiellen energieabhängigen Querschnitt sind wieder kaum Unterschiede zwischen den Anfangsverteilungen zu erkennen, der Querschnitt ist praktisch flach. Lediglich bei der Produktverteilung bricht der
Querschnitt für ganz asymmetrische Energieaufteilungen ein. Deutlich schwächer ist dieser
Effekt auch bei der mikrokanonischen Verteilung zu erkennen. Die Tendenz, daß der SDCS
bei allen Verteilungen etwa den gleichen zu flachen Verlauf hat, setzt sich hier weiter fort.
5.6.3
Zweifach differentiell
Auch für den DDCS existieren bei dieser Energie keine Messungen, so daß wieder die
CCC–Rechnungen von Bray als Vergleich dienen. Dieser Querschnitt wurde zum einen
wieder auf den totalen Querschnitt skaliert, zum anderen zusätzlich auf den (skalierten)
KAPITEL 5. RECHNUNGEN AN WASSERSTOFF
74
SDCS: in Abb. 5.18 finden bei symmetrischer Energieaufteilung etwa um die Hälfte zuviele Ereignisse statt, die DDCS wurden zusätzlich mit den Faktor 0,66 skaliert, um die
Winkelverteilung besser vergleichen zu können.
E0 = 17,6eV
Eb = 2eV
DDCS [10-18 cm2/sr eV]
3
CCC (20,5)
mikro * 0,42
Produkt * 0,39
2
1
0
CCC (20,5)
Wigner * 0,35
Cohen * 0.46
DDCS [10-18 cm2/sr eV]
3
2
1
0
0
45
90
ϑ [°]
135
180
Abbildung 5.19: Winkelverteilung der Elektronen mit 2eV bei h i>jlk m 17.6eV, skaliert auf
den totalen Querschnitt und den SDCS bei 2eV (siehe Text und Abb. 5.18).
Bei dieser niedrigen Energie hat sich beim DDCS der Trend umgekehrt (Abb. 5.19): hier
liefern die beiden klassischen“ Verteilungen eine deutlich bessere Übereinstimmung mit
”
der quantenmechanischen Rechnung: die mikrokanonische Verteilung führt zur korrekten
Winkelverteilung, lediglich in einem Bereich von etwa 30 x um die Rückwärtsrichtung ist
der Querschnitt deutlich zu groß. Cohens Energieverteilung ist insgesamt etwas zu flach,
während bei den quantenmechanischeren“ Verteilungen, Wigner– und Produktverteilung,
”
zusätzlich das Minimum zu zu großen Winkeln hin verschoben ist.
Wie bei 54,4eV Einschußenergie hat auch hier die Produktverteilung im Bereich um 40 x
herum eine Überhöhung.
5.6.4
Voll differentiell
Bei dieser Energie liegen für verschiedene koplanare Geometrien Messungen vor: für den
vollsymmetrischen Querschnitt, bei dem oRp;m¡ oqr und h;pmh;r gilt, in koplanar asymmetrischer Ehrhardtgeometrie und für konstanten Winkel zwischen den Elektronen. Bei diesem
Querschnitt mit oqp4r@m9¢£¤¥ ist die Elektron–Elektron–Wechselwirkung unabhängig vom
Streuwinkel immer die gleiche, dieser Querschnitt ist also besonders auf die Wellenfunktion
5.6. EINSCHUSSENERGIE 17,6EV
75
bzw. Phasenraumverteilung des Targets sensitiv. Die vorliegenden Messungen und die hier
gezeigten Querschnitte sind in Tabelle 5.10 dargestellt.
Geometrie
symmetrisch
asymmetrisch
konst. oRp6r
Parameter
¦doRp §oRrq¦?¨ x ,
¦dh@p ©h@r6¦?¨ 0,3eV
oRp;m~-/x , ª¬«|u;s­/x , }y?/x ,
h@rm®~ eV s 0,2eV
oRp6r'm¯¬« u , }q- x , }q~- x , }qz- x , °|{« u s±z x ,
h@rm®~ eV s 0,2eV
Tabelle 5.10: Geometrien und Parameter, für die bei 17,6eV Einschußenergie Messungen
durchgeführt wurden. Die zu den fett markierten Winkeln gehörenden Querschnitte sind
hier gezeigt. Für alle Querschitte ist die Streuebene auf s 12 x festgelegt. Alle Messungen
wurden von Röder etal durchgeführt [RJE93, R 96]
Bei diesen Querschnitten ist bei Wigner– und Produktverteilung wieder das gleiche Problem zu erkennen, das bereits bei zyy eV Einschußenergie auftrat: beide Elektronen laufen
bei gleicher Energie in fast die gleiche Richtung, obwohl diese Beiträge durch die Elektron–
Elektron–Abstoßung unterdrückt sein sollten. Das ist besonders gut am vollsymmetrischen
Querschnitt und in Ehrhardtgeometrie zu erkennen (Abb. 5.20); beim symmetrischen Querschnitt sind die Beiträge bei kleinen Winkeln deutlich überhöht, in der Ehrhardtgeometrie
mit o p m 60 x ist bei o r'² 45 x v.a. bei der Produktverteilung ein ausgeprägtes Maximum zu
erkennen; das ist etwa die Richtung, in der das andere Elektron festgehalten ist. Durch die
Auswertung über Energiedifferenzen stimmen die nomienelle Energie und der Impuls nicht
mehr überein, wenn das Projektil also 15,6eV (die Bindungsenergie von 13,6eV plus 2eV)
abgegeben hat, hat das Targetelektron nicht die Energie von 2eV, wenn es nicht anfangs
mit der korrekten Bindungsenergie (d.h. –0,5a.u.) gebunden war. Dieser Effekt macht sich
besonders bei Querschnitten bemerkbar, bei denen die Energien der Elektronen gleich sind
oder nahe beieinander liegen, bei stark asymmetrischen Energieaufteilungen spielt er nur
eine sehr kleine Rolle, wie die Querschnitte bei höheren Energien gezeigt hatten.
Abb. 5.20 zeigt, daß die mikrokanonische Verteilung auch bei dieser Energie knapp
öberhalb der Ionisationsschwelle nicht in der Lage ist, das binäre Maximum wiederzugeben, während das Rückstreumaximum eine gewisse Übereinstimmung mit den Messungen
zeigt. Für konstanten Winkel zwischen den Elektronen wird der Querschnitt immer besser, je größer dieser Winkel ist (siehe Abb. 5.21). Das wird aber hauptsächlich durch die
Abstoßung der Elektronen verursacht, die bei dieser niedrigen Energie lange Zeit haben,
aufeinander einzuwirken.
Bei der mikrokanonischen Verteilung starten alle Targettrajektorien bei der richtigen Bindungsenergie, der tatsächliche Impuls dieser Elektronen stimmt mit der Energie überein, die
aus dem Energieübertrag berechnet wird: die Langzeitdynamik wird korrekt beschrieben.
Über das Verhalten an der Ionisationsschwelle ist nun bekannt, daß für ganz langsame
Elektronen eine Wannier–Konfiguration“ bevorzugt wird, d.h. die Elektronen sind so lang”
sam, daß sie sich letzten Endes immer auf einen maximalen Abstand untereinander abstoßen, was zu einem Winkel zwischen ihren Impulsen von 180 x führt, zur back–to–back“–
”
KAPITEL 5. RECHNUNGEN AN WASSERSTOFF
76
ϑ a = -ϑ b
ϑ a = 60°
Röder
mikrokan. * 0,42
Röder
mikrokan. * 0,42
0.5
Röder
Röder
0.09
0.06
0.25
0.03
0
0.5
0.25
2
0.03
Röder
Wigner * 0,35
0.06
0.5
0.25
0.03
0
0.09
0
Röder
Cohen * 0,46
Röder
Cohen * 0,46
0.5
Röder
Röder
Röder
Wigner * 0,35
Röder
TDCS [10
-17
Röder
0
0
0.09
TDCS [10-17 cm2/eV sr2]
0.06
cm /eV sr ]
Röder
Produkt * 0,35
Röder
Röder
Produkt * 0,39
2
Röder
0
0.09
0.06
0.25
0.03
0
0
45
90
135
ϑ a = -ϑ b [°]
180 -180
-90
0
ϑb
90
180
0
Abbildung 5.20: links der vollsymmetrischer koplanare Querschnitt, rechts der Querschnitt
in Ehrhardtgeometrie für h;rm 2eV und oqp@m 60 x .
Konfiguration.
Aus diesem Grund werden die Querschnitte, die stark von dieser linearen Anordnung geprägt sind, von der mikrokanonischen Verteilung recht gut wiedergegeben (z.B. o p4r m 180 x ),
kleinere Winkel dagegen eher schlecht (siehe die anderen gezeigten Querschnitte). Da die
Querschnitte zu konstantem oRp6r unabhängig von der Langzeitdynamik der auslaufenden
Elektronen sind, zeigen sie, daß die mikrokanonische Verteilung auch bei dieser Energie
nicht in der Lage ist, den eigentlichen Ionisationsvorgang korrekt zu beschreiben.
Im Gesamtbild liefert diesmal Cohens Energieverteilung die besten“ Querschnittte. Sie
”
ist die Verteilung, die, nach der mikrokanonischen, die geringste Breite in der Energieverteilung aufweist (siehe Tabelle 5.1). Sie kombiniert aber auf der einen Seite die richtige
Form“ im Phasenraum (mit der korrekten Ortsraum– und der fast korrekten Impulsver”
teilung), die wesentlichen Einfluß auf den eigentlichen Ionisationsvorgang hat, mit einer
geringen Energieunschärfe, die für die Langzeitdynamik bestimmend ist.
5.6.5
Zusammenfassung
Bei der Einschußenergie von 17,6eV zeigt sich also ein etwas anderes Bild, als bei den
höheren Energien: Hier zählt nicht nur, daß die Phasenraumbeschreibung möglichst direkt
5.6. EINSCHUSSENERGIE 17,6EV
Röder
mikrokan. * 0,42
77
ϑ a = 90°
ϑ ab = 180°
Röder
mikrokan. * 0,42
0.4
Röder
0.05
0.2
0
0
Röder
Produkt * 0,39
Röder
Produkt * 0,39
0.4
Röder
cm /eV sr ]
2
0.05
0.2
2
Röder
0.1
0
Röder
Wigner * 0,35
Röder
Wigner * 0,35
0.4
0.1
Röder
TDCS [10
Röder
-17
0
0.05
0.2
0
0
Röder
Cohen * 0,46
Röder
Cohen * 0,46
0.4
Röder
Röder
0.1
0.05
0
-180
TDCS [10-17 cm2/eV sr2]
Röder
0.1
-90
0
ϑ b [°]
90
0.2
180 -180
-90
0
90
Abbildung 5.21: TDCS für konstanten Elektron–Elektron–Winkel, links für
rechts für 180 x .
180
oRp6r§m
0
90 x ,
aus der Wellenfunktion bestimmt wird, sondern nun findet die Ionisation so langsam statt,
daß die Energie der Elektronen eine wichtige Rolle spielt. Das zeigt sich entsprechend darin,
daß diesmal Cohens Energieverteilung die besten Querschnitte liefert. Das ist die Verteilung
mit der geringsten Energiebreite, die gleichzeitig die Dichten recht gut beschreibt.
Die anderen Verteilungen haben entweder Schwierigkeiten wegen der falschen“ Ener”
gien, d.h. zu breiter Energieverteilung (Wigner– und Produktverteilung), oder wegen der
falschen Phasenraumdichte, der falschen Form“ des Targets (mikrokanonische Verteilung).
”
Erst für noch geringere Gesamtenergien, die noch näher an der Schwelle liegen, ist zu
erwarten, daß die mikrokanonische Verteilung korrekte Querschnitte liefert. Dies liegt aber
nicht daran, daß das Wasserstoffatom dann korrekt beschrieben würde, sondern daran, daß
dann die Langzeitdynamik noch dominanter wird; aber um diese richtig zu beschreiben, ist
es essentiell, die echten Energien in der Auswertung zu benutzen.
Der Vergleich mit der Messung zeigt, daß das Wanniersche Potenzgesetz für das Schwellenverhalten des totalen Querschnitts nur bis etwa 2eV Gesamtenergie gilt (siehe z.B. [Ros98],
besonders die Rechnungen am kolinearen Modell), die hier berechnete Energie liegt bereits
deutlich höher. Da üblicherweise der totale Querschnitt am unempfindlichsten gegenüber
Feinheiten einer Rechnung ist, ist zu erwarten, daß das Schwellenverhalten für voll differentielle Querschnitte bei bereits noch geringeren Energien nicht mehr gut reproduziert wird.
78
KAPITEL 5. RECHNUNGEN AN WASSERSTOFF
Die mikrokanonische Verteilung sollte also erst kurz oberhalb der Ionisationsschwelle gute volldifferentielle Querschnitte reproduzieren können, d.h. nur in einem extrem kleinen
Energiebereich.
5.7
Die Ergebnisse im Überblick
Faßt man die Ergebnisse dieses Kapitels zusammen, so ergibt sich das folgende Gesamtbild:
Eine Anfangsverteilung, die das Wasserstofftarget beschreibt, kann auf verschiedene Weisen hergeleitet werden: einerseits wird zuerst der klassische Limes des zeitabhängigen quantenmechanischen Formalismus bestimmt, dieser bereits klassische Rahmen führt zu einer
klassischen“ Anfangsverteilung. Das ist der Weg zur mikrokanonischen Verteilung. Auf
”
der anderen Seite wird die Wellenfunktion des Targets benutzt; auf diese Weise wird die
Wignerverteilung in die Rechnung eingeführt. Zwischen diesen beiden Extremen liegen Cohens Energieverteilung, die eine klassisch begründete Verteilung ist, in die Informationen
aus der Wellenfunktion einfließen, und die Produktverteilung, die nur aus den quantenmechanischen Dichten konstruiert ist.
Von den betrachteten vier Verteilungen führt die mikrokanonische über den gesamten
Energiebereich zu den schlechtesten Ergebnissen. Das zeigt, daß die korrekte Dichte in
Orts– und Impulsraum wichtiger ist, als eine scharfe Bindungsenergie, der Phasenraumpunkt, an dem der Stoß zwischen Target und Projektil stattfindet, also eine wesentliche Rolle spielt. Vor allem bei den höheren Energien wird das Target vom schnellen Projektil im
Phasenraum quasi abgebildet“. Bei 250eV Einschußenergie sind Wigner- und Produktver”
teilung in etwa gleichwertig und besser als Cohens Energieverteilung und die mikrokanonische Verteilung. Bei der Targetenergie von 54,4eV zeigt sich im wesentlichen das gleiche
Verhalten, hier sind jedoch bereits Unterschiede zwischen Wigner– und Produktverteilung
zu erkennen, die darauf zurückzuführen sind, daß die Wignerverteilung der Quantenmechanik näher steht. Bei niedrigen Energien, hier 4eV Gesamtenergie, kommt das Langzeitverhalten der auslaufenden Elektronen ins Spiel. Dafür ist die tatsächliche Energie wichtig,
sodaß nun die Verteilungen mit geringerer Energiebreite zu besseren Querschnitten führen.
Cohens Energieverteilung hat unter den betrachteten Verteilungen für diese Energie das
beste Verhältnis zwischen richtiger Beschreibung im Phasenraum und schmaler Energieverteilung. Oder anders herum formuliert: die Langzeitdynamik wird durch die Energien der
Elektronen bestimmt, die Kurzzeitdynamik (der Stoß“) durch die Geometrie des Problems
”
im Phasenraums. Für schnelle Stöße, d.h hohe bis mittlere Energien, muß die Anfangsverteilung die Form des Targets möglichst gut abbilden, während für (sehr) langsame Stöße,
d.h. für Energien knapp oberhalb der Schwelle, die Energie korrekt beschrieben werden
muß. Auf diese Weise läßt sich in Abhängigkeit der betrachteten Energieverhältnisse ein
Auswahlkriterium für eine Phasenraumverteilung zur Beschreibung des Targets formulieren.
Insgesamt zeigen die Ergebnisse, daß es möglich ist, mit einer klassischen Rechnung die
dominanten Strukturen und Reaktionsmechanismen auch atomarer Systeme zu beschreiben.
Wichtig ist dabei, daß die Beschreibung konsistent und in allen Teilen in gleicher Ordnung
genähert aus der Quantenmechanik abgeleitet wird.
Eine solche klasssische Beschreibung hat natürlich ihre Grenzen: die Näherung versagt
z.B. bei der Beschreibung des Tunnelns oder von Interferenzeffekten, die auf einer kohären-
5.7. DIE ERGEBNISSE IM ÜBERBLICK
79
ten Überlagerung der Wellenfunktionen beruhen. Diese Einschränkungen müssen aber im
richtigen Licht gesehen werden: der vorgestellte Ansatz ist eine recht drastische Näherung
der (korrekten) quantenmechanischen Beschreibung und von daher garnicht dazu gedacht,
hochpräzise Querschnitte zu erzielen. Das Ziel ist vielmehr, einen qualitativen Überblick
über den gesamten Bereich der Endwerte zu erhalten, ohne daß vor Beginn der Rechnung
bereits bekannt sein muß, an welcher Stelle überhaupt Ereignisse zu erwarten sind. Die
klassische Rechnung kann also vorhersagen, wo in etwa die wesentlichen Strukturen der
Querschnitte liegen werden und welche absolute Höhe in etwa zu erwarten ist.
Drastisch ist allerdings der Unterschied zur Standard–CTMC, die bereits bei einfachen
Winkelabhängigkeiten das falsche Verhalten vorhersagt und voll differentielle Querschnitte
gar nicht beschreiben kann.
Explizit betont werden soll noch einmal, daß die guten Ergebnisse nicht bedeuten, daß
ein klassisches Atommodell zur Beschreibung geeignet sei, wie es der Standard–CTMC
mit ihrer Identifikation der Trajektorien als tatsächliche Bahnen der Elektronen zugrunde
liegt, sondern daß ein klassische Näherung des quantenmechanischen Modells einen Großteil der Dynamik beschreiben kann. es handelt sich bei dem quasiklassischen Ansatz um
eine klassische Näherung, nicht um ein klassisches Modell.
Kapitel 6
Erste Ergebnisse an Helium
In Kapitel 4 wurde gezeigt, wie der quasiklassische Ansatz zu erweitern ist, um Zweifachionisation am Heliumtarget zu beschreiben. Passend dazu wurden Rechnungen für eine Einschußenergie von 2keV durchgeführt, die qualitativ mit Messungen von Dorn et al verglichen werden [D 99]. Diese wurden allerdings bei einer Einschußenergie von 3keV gemessen.
Die Schwierigkeiten bei Helium bestehen darin, daß für dieses hochkorrelierte Zweielektronensystem keine exakte Wellenfunktion verfügbar ist und daß der Querschnitt für
die Zweifachionisation sehr klein ist, die Rechnungen werden sehr langwierig. Wie bereits
in Abschnitt 3.8.2 erläutert, wird hier wegen der kontinuierlichen Struktur der klassischen
gebundenen Zustände — abgesehen vom totalen Ionisationsquerschnitt — nur die Zweifachionisation betrachtet.
6.1
Die Anfangsverteilung
Für das Heliumatom existiert kein klassisches Modell, es ist daher kein Vergleich mit einer
klassisch korrekten“ Anfangsverteilung möglich 1 .
”
Quantenmechanisch können lediglich genäherte Wellenfunktionen angegeben werden,
die bekanntesten sind die von Hylleraas eingeführten [Hyl29]. Der einfachste Ansatz vernachlässigt die Elektron–Elektron–Korrelation und berücksichtigt den Effekt des jeweils
anderen Elektrons durch ein effektives abgeschirmtes Potential:
³
m´¶µ ·¸¹lº¸ º»¼
(6.1)
Die Abschirmumg ist bei dieser Wellenfunktion unabhängig von der Kernladung, der
Parameter hat den Wert ½± §z/¾v}q , damit folgt ´¿m4À ¾-Á .
Für die hier durchgeführten ersten Rechnungen wurde diese einfachste Wellenfunktion,
die einen Produktansatz für die beiden Elektronen darstellt, durch eine Produktverteilung
analog zum Wasserstoff für jedes der Elektronen modelliert (vgl. Abschnitt 3.4.5.1). Diese
Verteilung hat die korrekte (mittlere) Gesamtenergie von 79eV, die Bindungsenergie eines
1
Wintgen etal. zeigten, daß die Dynamik eines (semi)klassisch beschriebenen Heliumatoms gemischt regulär und chaotisch ist [WRT92], solch eine Beschreibung eignet sich sicher nicht als klassischer Vergleich
zum hier verwendeten quasiklassischen Ansatz.
80
6.2. DER TOTALE IONISATIONSQUERSCHNITT
81
einzelnen Elektrons ist dabei keine sinnvolle Größe, denn es handelt sich, wie in der Quantenmechanik, um ein hochkorreliertes Zweielektronensystem, das korrekterweise nicht in
zwei unabhängige Teile zerlegt werden sollte. Wird diese Aufteilung versucht, so führt sie
zu Inkonsistenzen des Modells oder Schwierigkeiten der Interpretation.
Aus spektroskopischen Messungen ist bekannt, daß zum Herauslösen eines Elektrons aus
diesem Verbund“ eine Energie von etwa 24,6eV nötig ist, wenn sich danach das verblei”
bende Elektron im tiefstmöglichen Zustand befindet. Dort ist es mit 54,4eV, dem Vierfachen
der Bindungsenergie des Wasserstoffgrundzustands, gebunden. Werden beide Elektronen
gleichzeitig ionisiert, so müssen insgesamt 79eV an Energie aufgebracht werden.
An dieser Stelle zeigt es sich wieder, daß im quasiklassischen Ansatz das Heliumatom konsistent beschrieben wird: beide Elektronen werden gleich behandelt, es ist nicht
nötig, künstlich einzelne Bindungsenergien einzuführen, wie das bei bisherigen klassischen
Ansätzen nötig war, um die Autoionisation zu verhindern.
Mit dieser Verteilung wurden nun sowohl der totale Ionisationsquerschnitt für Einfach–
und Zweifachionisation bestimmt sowie bei einer Einschußenergie von 2keV Endwerte zur
Zweifachionisation gesammelt.
6.2
Der totale Ionisationsquerschnitt
Der totale Ionisationsquerschnitt an Helium wurde klassisch bisher nur für Einfachionisation bestimmt. Da es mit dem hier vorgestellten quasiklassischen Modell möglich ist,
konsistent die Zweifachionisation zu beschreiben, wurde auch dieser Querschnitt berechnet und mit Messungen verglichen. Dabei wird das (einfacher zu messende) Verhältnis von
Zweifach– zu Einfachionisation angegeben.
Exp.
IEM
nCTMC
Produkt
σtot [10-17cm2]
Exp.
10
1
100
E in [eV]
1000
Abbildung 6.1: Totaler Ionisationsquerschnitt für Einfachionisation an Helium: Vergleich
des quasiklassischen Ansatzes mit dem independent electron model“ und einer nCTMC”
”
-Rechnung, beide von Schultz etal. [SMO92]. Die Messung wurde von Montague etal.
durchgeführt [MHS84].
Vergleicht man die Einfachionisation im quasiklassischen Ansatz mit zwei anderen klassischen Rechnungen, einer im independent electron modell“ (IEM), einer Einteilchenbe”
KAPITEL 6. ERSTE ERGEBNISSE AN HELIUM
82
Andersen
Produkt * 0,33
0.6
++
Andersen
σ /σ+ [%]
0.9
0.3
0
100
E in [eV]
1000
Abbildung 6.2: Das Verhältnis von Zweifach– zu Einfachionisation für Helium: Wird das
Ergebnis des quasiklassischen Ansatzes um einen Faktor 0,33 skaliert, so stimmt der Verlauf
recht gut mit der Messung überein. Die aus mehreren Veröffentlichungen zusammengetragenen Messungen sind nach Andersen etal zitiert [A 87].
schreibung in einem effektiven Potential, und einer nCTMC“–Rechnung, in der die Wech”
selwirkung der Targetelektronen abgeschaltet ist, so sind diese beiden Querschnitte deutlich höher [SMO92]. Interessanterweise stimmt die Form unseres Querschnitts gut mit der
nCTMC“–Rechnung überein, wenn einer der Querschnitte in der Höhe mit den Faktor
”
1,6 skaliert wird. Ein Grund mag darin liegen, daß es in diesen beiden CTMC–Modellen
nicht möglich ist, die Bindungsenergien korrekt zu berücksichtigen und die Elektronen im
Mittel zu schwach gebunden sind: das Ein–Elektronen–Modell zeigt, daß im Heliumatom
die Korrellationen zwischen den beiden Elektronen sehr wichtig sind, besonders, wenn die
Ionisation langsam“ abläuft. Im nCTMC“–Ansatz hingegen, der die Elektron–Elektron–
”
”
Wechselwirkung vernachlässigt, ist die Gesamtenergie nicht korrekt darstellbar. Sie ist um
etwa 30eV zu klein, da jedes der beiden Elektronen nur mit der einfachen Ionisierungsenergie gebunden ist.
Für niedrige Einschußenergien kann der quasiklassische Ansatz die Messungen wiedergeben, er ist bei höheren Energien jedoch systematisch zu klein, wie auch die beiden anderen Rechnungen. Das Hochenergieverhalten ist bekanntermaßen klassisch nicht korrekt,
da Tunnelbeitr äge in diesem Modell nicht beschrieben werden können. Die Abweichung
von der Messung läßt sich nun so interpretieren, daß diese quantenmechanischen Beiträge
bereits ab dem Maximum des totalen Querschnitts einen deutlichen Beitrag zur gesamten
Ionisationswahrscheinlichkeit liefern.
Interessanterweise hat das Verhältnis von Zweifach– zu Einfachionisation ) ¾/ den
gleichen Verlauf wie die Messungen, es liegt lediglich um den Faktor Drei zu hoch: die
Ionisationsschwelle, das Maximum, der Anstieg dazwischen sowie der Anfang des Hochenergieverhaltens zeigen, daß zwar die Energieabhängigkeit der klassisch beschriebenen
Ionisation nicht stimmt, aber die Mechanismen der Ionisationsdynamik im wesentlichen
korrekt beschrieben werden. Die Größe der Skalierung paßt recht gut den Abweichungen
der absoluten Höhen der Querschnitte, wie sie bereits am Wasserstoff auftraten. Dort war es
ein Einelektronenprozeß, hier sind zwei Elektronen beteiligt — die absolute Abweichung
6.3. DIFFERENTIELLE QUERSCHNITTE
83
ist entsprechend größer. In Abschnitt 4.3 wurde empfohlen, die Einfachionisation in einem
Einelektronenmodell zu berechnen. Werden also in P¾/Â für die Einfachionisation die
Werte der nCTMC–Rechnung verwendet, so beträgt die Abweichung nur noch“ etwa einen
”
Faktor zwei.
6.3
Differentielle Querschnitte
Insgesamt wurden, um differentielle Querschnitte zu bestimmen, etwa 30 Millionen Trajektorien bei verschiedenen maximalen Stoßparametern gerechnet, letzlich standen knapp
7000 Zweifachionisationen zur Verfügung. Aus diesem Grund konnten keine hochdifferentiellen Wirkungsquerschnitte bestimmt werden, lediglich ein– und zweifach differentielle
Auftragungen waren möglich.
Wahrsch. [willk.]
E23 > 8a.u.
E23 < 2a.u.
0
0.5
1
E2/(E2+E3)
Abbildung 6.3: Relative Energieaufteilung der beiden langsamen Elektronen, einmal für
Ã
à der beiden Elektronen, einmal für höheres à .
niedrige Gesamtenergie Ã
ÀÄ
'Å
À
À
Unter den vielen möglichen Parametern, die einzeln und gegeneinander aufgetragen betrachtet werden können, sollen zwei Beispiele herausgegriffen werden, die trotz der geringen Anzahl an Doppelionisationen einen Vergleich mit dem Experiment zulassen.
Eine bisher häufig betrachtete Größe ist die relative Energieaufteilung der beiden langsamen Elektronen, siehe Abb. 6.3. Für niedrige Gesamtenergie Ã;Æ À
Ä Ã@Æ Å Ã À der beiden
Elektronen sind alle Verhältnisse etwa gleich häufig, bei höherem Energieübertrag ist die
Aufteilung asymmetrischer [PS93]. Diese Verhalten ist auch aus der Photodoppelionisation bekannt. So, wie an Wasserstoff die energieabhängigen Querschnitte zu flach waren,
zeigt sich auch hier dieses Verhalten: die Form der Energieaufteilung wird erst bei höheren
Energien erreicht als im Experiment.
Von Dorn et al wurde kürzlich eine kinematisch vollst ändige Messung bei einer Einschußenergie von 3keV durchgeführt [D Ç 99]. Dort wurde vorgeschlagen, zusätzlich zur relativen Energieaufteilung den Winkel zwischen den beiden langsamen Elektronen zu betrachten (siehe Abb. 6.4 rechts). Der gemessenen Querschnitt ist dabei für kleine Impulsübertrage des Projektils praktisch unabhängig von der Energieverteilung. Durch die Elektron–
Elektron–Wechselwirkung werden kleine Winkel zwischen ihnen stark unterdrückt, das
KAPITEL 6. ERSTE ERGEBNISSE AN HELIUM
84
Maximum der Winkelverteilung liegt bei etwa 130 È . Für gleiche Energie und einen Winkel von 180 È hat der Querschnitt ebenfalls ein Minimum. Die unterschiedliche Symmetrie
von Anfangs– und Endzustand verhindert diese back–to–back“-Konfiguration.
”
ÒdÓ ÔÒ Õ|Ö Ï×ØÙ
Ì ÐÑÉ
â ßãÌMÊ Ë Õ Ê áÊ
Ú Ö Ïàß Í Õ Ê áBÊ
Ì Ï/Ë
ÎÉ
ÍË
É
É)Ê É-É
É)Ê ËÉ
ÌMÊ ÉÉ
Ú ÖDÛ4Ü Ú ÖDÝ Ú Ï¬Þ
Abbildung 6.4: Zusätzlich zur relativen Energieaufteilung ist hier der Relativwinkel zwischen den beiden langsamen Elektronen dargestellt (siehe Text). Links die hier berechnete
Verteilung, rechts die Messung aus [D Ç 99], die bei 3keV Einschußenergie durchgeführt
wurde. Ausgewählt wurden dabei alle Ereignisse mit einem Impulsübertrag ädå|ä'æ 0,7a.u.
und einer Energie Ã Æ æ 20eV. In der klassischen Rechnung mussten wegen der gerigen
À
Statistik, und da die Energieverteilung flacher verläuft als im Experiment, diese Parameter
auf ädå|ä@æ 1,5a.u. und Ã Æ æ 4a.u. erhöht werden. Die Form der Verteilung wurde aber
À
dadurch kaum beeinflußt.
Vergleicht man die Messung mit der quasiklassischen Rechnung, Abb. 6.4 links, so wird
das Minimum bei kleinen Winkeln ebenfalls wiedergegeben, da die Abstoßung der Elektronen wird in der klassischen Rechnung voll berücksichtigt wird.
Wie häufig bei Wasserstoff auch, ist die Winkelverteilung insgesamt zu breit, speziell zu
kleinen Winkeln hin. Dieses Verhalten ist ebenfalls von demn Rechnungen an Wasserstoff
her bekannt und konnte dort einer zu breiten Energieverteilung der Startwerte zugeordnet
Æ
werden. Das ist auch hier der Fall: die angesetzte Produktverteilung der unkorrelierten 1s –
Wellenfunktion ist, bedingt durch die höhere Kernladung, noch breiter als beim Wasserstoff.
Hinzu kommt, daß nun zwei Elektronen betrachtet werden, die vergleichbare Energie haben,
ähnlich den Wasserstoffquerschnitten bei mittleren oder niedrigen Energien.
Erstaunlicherweise ist auch in der klassischen Rechnung das Minimum bei 180 È und gleicher Energie zu sehen, obwohl im klassischen Bild eine Argumentation über die Erhaltung
der Parität an der kontinuierlichen Drehimpulsverteilung scheitert — quantenmechanisch
betrachtet, hat der klassische Anfangszustand nur noch im Mittel eine çYèêé –Symmetrie.
6.4
Anmerkungen
Für die Elektronenstoßionisation an Helium konnten hiermit zum ersten Mal Querschnitte
unter der vollen Wechselwirkung aller vier Teilchen berechnet werden, ohne auf zusätzliche Stabilisierungspotentiale zurückgreifen zu müssen. Die Ergebnisse stimmen dabei in
6.4. ANMERKUNGEN
85
der Form im groben Verhalten mit dem Experiment überein, Verteilungen sind jedoch meist
zu breit verschmiert. Wie bereits bei den Rechnungen am Wasserstofftarget hat die klassische Näherung Probleme, den totalen Querschnitt korrekt zu beschreiben, der doch am
unempfindlichsten gegenüber Näherungen sein sollte.
Die Erklärung ist wahrscheinlich im Denken in klassischen Bildern zu suchen: für die
wesentlichen Strukturen eines Querschnitts existieren fast immer klassische, anschauliche
Erklärungen, die in einer klassischen Rechnung natürlich leichter beschreibbar sind. Zu
diesen Geometrien existieren die meisten Messungen, während in weniger strukturierten“
”
Geometrien kaum Querschnitte gemessen wurden — dies sind aber die Bereiche, in denen
quantenmechanische Interferenzen, Tunneln, etc. den Querschnitt bestimmen. Die klassischen Anteile werden nun in der klassischen Rechnung gut wiedergegeben, während die
quantenmechanischen Beiträge vollst ändig fehlen. Auf diese Weise können z.B. die hochdifferentiellen Querschnitte an Wasserstoff gut wiedergegeben werden, während der totale
Querschnitt bei höheren Energien zu klein ist, da all die quantenmechanischen Bereiche
nicht berücksichtigt werden können.
Kapitel 7
Zusammenfassung
In der vorliegenden Arbeit wurde aus der zeitabhängigen quantenmechanischen Streutheorie eine klassische Näherung abgeleitet: der quasiklassische Ansatz. Die Streuung wurde
dazu quantenmechanisch im Møllerbild formuliert und die einzelnen Teile dieses Ansatzes
— die Wellenfunktion des Anfangszustands, die Propagation in der Zeit und die Auswertung — über die Wignerdarstellung klassisch genähert. Der so hergeleitete quasiklassische
Ansatz hat in großen Teilen die gleiche Form wie das von Anfang an klassisch angesetzte
Modell, die CTMC, doch es bestehen wesentliche Unterschiede zwischen diesen beiden.
Im quasiklassischen Ansatz beginnt die Beschreibung der beteiligten Teilchen mit deren
Wellenfunktion. Das erlaubt es, für beliebige Systeme eine Phasenraumverteilung aufzustellen, auch, oder gerade dann, wenn kein klassisches Modell dafür existiert. Da die Festlegung
einer Korrespondenzregel, die diese Übersetzung definiert, nicht eindeutig ist, besteht hier
eine gewisse Wahlfreiheit.
Wird der quantenmechanische Streuoperator im Møllerbild formuliert, so ist dies ein
zeitabhängiges Bild und bietet sich daher für die klassische Näherung an. Der große Vorteil
dieser Formulierung liegt darin, daß die Präparation des Anfangszustands und die Auswertung am gleichen Zeitpunkt erfolgen, ebenso wie die eigentliche Reaktion; die Zeit ist
dabei nur eine Hilfsvariable“ zur Konstruktion der korrekten Streuzustände. Auf diese Wei”
se wird eines der größten Probleme bisheriger klassischer Modelle umgangen: die aus der
Wellenfunktion übersetzte Phasenraumverteilung muß nicht mehr station är sein, da sie quasi nur an einem Zeitpunkt benötigt wird. Das öffnet den Weg, auch komplexere Targets als
das Wasserstoffatom, wie z.B. das autoionisierende Helium, in einer klassischen Rechnung
zu benutzen.
Auch in der Auswertung kann die Freiheit in der Wahl einer Phasenraumverteilung berücksichtigt werden, wenn nicht, wie bisher üblich, mit den absoluten Energien der Trajektorien
gerechnet wird: durch die in einer klassischen Rechnung äquivalente Betrachtung des Energieübertrags vom Projektil auf das Target ist es möglich, Trajektorien mit beliebiger Anfangsenergie konsistent auszuwerten. Damit wird einerseits eine eindeutige Definition von
Ionisation gegeben, andererseits ist für energiedifferentielle Querschnitte eine konsistente
Basis vorhanden.
Dieser Ansatz wurde an einem der Standardprobleme der Atomphysik, der Elektronenstoßionisation von atomarem Wasserstoff bei drei Einschußenergien und mit verschiedenen
Anfangsverteilungen getestet und mit der Standard–CTMC verglichen. In der Standard–
86
87
CTMC wird das Wasserstoffatom durch eine Verteilung mit fester Energie beschrieben,
die jedoch die falsche Dichte im Ortsraum besitzt. Demgegenüber stellen die aus der Wellenfunktion bestimmten Anfangsverteilungen die Form des Atoms im Orts– und im Impulsraum korrekt dar, besitzen dafür aber keine scharfe Energie der Startwerte mehr. Es
zeigt sich, daß für den allergrößten Bereich der Einschußenergie die Form des Targets eine
deutlich wichtigere Rolle spielt als die Energieverteilung: die Standard–CTMC ist z.T. bei
zweifach differentiellen Querschnitte nicht mehr in der Lage, auch nur die grobe Struktur
korrekt zu beschreiben, während die anderen getesten Verteilungen, allen voran die Wignerverteilung, auch volldifferentielle Querschnitte beschreiben können. Erst bei recht niedriger
Gesamtenergie, hier bei 4eV über der Schwelle, zeigt sich der Einfluß der Breite der Energieverteilung.
Damit kann eine der Fragen aus der Einleitung beantwortet werden: der quasiklassische
Ansatz ist in der Lage, dieses quantenmechanische System in seinen wesentlichen Eigenschaften zu beschreiben. Der Aufwand ist höher als bei der Standard–CTMC, doch vergleicht man die Ergebnisse, so ist die Entscheidung eindeutig.
Eine so grob genäherte klassische Beschreibung hat natürlich ihre Grenzen, die Näherung der Propagation ist dabei die wesentliche Fehlerquelle: in der genäherten Zeitentwicklung entkoppeln die einzelnen Phasenraumpunkte zu klassischen Trajektorien. Das bedeutet, daß alle quantenmechanischen Phänomene, die z.B. auf Interferenz verschiedener Teile
der Wellenfunktion beruhen oder für die die Kohärenz innerhalb des Systems wesentlich ist,
nicht beschrieben werden können. Auch Tunneln ist in diesem Modell nicht enthalten. In
der Anwendung an Wasserstoff zeigte es sich allerdings, daß vor allem im mittleren Energiebereich die klassischen Reaktionspfade den größten Teil der Dynamik bestimmen, die
klassische Näherung kann die wesentlichen Strukturen der Querschnitte wiedergeben. Bei
komplizierteren Targets werden Auswahlregeln eine Rolle spielen, auch diese können hier
nicht wiedergegeben werden, ebensowenig wie z.B. Austauscheffekte.
Diese Einschränkungen müssen beachtet werden, wenn mit diesem klassischen Ansatz
gearbeitet wird: er war jedoch von Anfang an nicht als neue Rechenmethode gedacht, mit
der in kürzester Zeit hochpräzise Querschnitte bestimmt werden können, sondern um die
wesentlichen Strukturen aufzuzeigen und die Möglichkeit zu geben, diese in anschaulichen
Bildern zu verstehen. Dazu trägt bei, daß wie im Experiment alle Endwerte gleichzeitig bestimmt werden. Im Raum dieser gesammelten Ereignissen können dann beliebige Schnitte
gelegt werden, ohne jedesmal die Rechnung neu zu starten — der Ansatz ist sehr am Experiment orientiert.
Diese Nähe zum Experiment darf aber nicht dazu verleiten, die berechneten Trajektorien als tatsächliche Teilchenbahnen zu interpretieren. Dies wird allein schon durch die
Unschärferelation verboten. In diesem Ansatz haben die einzelnen Trajektorien ihren Ursprung in einer Diskretisierung der Phasenraumverteilung, d.h. sie sind lediglich Stützstellen, nicht mehr. Für eine korrekte Interpretation muß also über jeweils eine Klasse ähnlicher Trajektorien gemittelt werden, der Schwerpunkt dieser Gruppe kann dann wieder als
Teilchenbahn betrachtet werden. Dies ist der wesentliche Unterschied zum rein klassischen
Modell, der CTMC.
Das ist auch die Antwort auf die zweite große Frage der Einleitung: wird eine klassische Beschreibung einem atomaren System gerecht? Wenn sie als klassische Näherung der
88
KAPITEL 7. ZUSAMMENFASSUNG
Quantenmechanik interpretiert wird, dann ja. Der quasiklassische Ansatz ist kein klassisches Modell, sondern eine klassische Näherung.
Der quasiklassische Ansatz ist zwar von der Herleitung und vom Konzept her sehr anders
als die CTMC, doch kann diese, wenn die Näherung weitergeführt wird, schließlich auch
daraus abgeleitet werden. Von der Form der verwendeten Formeln allerdings sind sich die
beiden Zugänge so ähnlich, daß bisher bestehende CTMC–Codes nur geringfügig angepasst
werden müssen, um numerische Rechnungen mit dem quasiklassischen Ansatz durchführen
zu können. Auf diese Weise kann die gesamte Erfahrung, die bereits mit der CTMC gesammelt wurde, fast ohne Veränderung weiter genutzt werden — wenn die unterschiedliche
Interpretation beachtet wird.
Das größte Potential des hier vorgestellten Ansatzes ist zweifellos die Möglichkeit, mit
nichtstation ären Targets zu arbeiten. Das öffnet den weiteren Weg, klassische Rechnungen
über das Dreikörperproblem hinaus durchzuführen, ohne zusätzliche Annahmen machen
zu müssen, die weder aus einem klassischen noch aus einem quantenmechanischen Ansatz
heraus zu begründen sind.
Daß dieser Weg funktioniert, konnte hier an Helium gezeigt werden, für genauere Rechnungen ist es nötig, dafür eine bessere Phasenraumbeschreibung zu finden, als dieser einfache Ansatz, mit dem die ersten Tests durchgeführt wurden. Da die Elektronenstoßionisation
an Helium wegen der drei ununterscheidbaren Elektronen ein recht komplexes Verhalten
zeigt, liegt es nahe, diese Beschreibung zunächst an einem einfacheren System zu testen:
dazu bietet sich der Schwerionenstoß an, denn dabei kann die Dynamik von Ion und Heliumkern von der Bewegung der Elektronen abgetrennt werden, übrig bleibt die Bewegung
zweier Elektronen im Feld der beiden schweren Teilchen. Das ist nicht nur numerisch einfacher, dabei tritt auch die Darstellung des Targets in den Vordergrund, es ist quasi isoliert“
”
im Vergleich zum Elektronenstoß.
Bei höheren Teilchenzahlen besteht die Erwartung, daß dann eine klassische Beschreibung zunehmend besser wird, da dann immer mehr direkte, klassische Reaktionspfade zur
Verfügung stehen, die dann die Dynamik immer mehr kontrollieren. Außerdem werden
Kohärenzeffekte immer weniger wichtig, je größer ein System ist.
Hier wurde also gezeigt, daß eine konsequente, in allen Teilen gleich weit genäherte,
klassische Beschreibung sehr wohl in der Lage ist, auch typisch quantenmechanische Systeme zu modellieren, wenn vermieden wird, diese wie ein klassisch hergeleitetes Modell zu
interpretieren. Der Ansatz kann dann auch auf kompliziertere Systeme angewandt werden.
Diese Arbeit ist daher erst der Anfang, weitere Untersuchungen müssen noch folgen.
Anhang A
Zur Liouville–Gleichung
A.1
Klassische Stationarität
Die Liouvillegleichung ist die zentrale Zeitentwicklungsgleichung klassischer Phasenraumverteilungen.
Die Dynamik einer Phasenraumverteilung ëì>å?íîï wird durch eine Kontinuit ätsgleichung
beschrieben: die totale zeitliche Änderung der Dichte ë an der Stelle ì>å?íîï setzt sich zusammen aus der Bewegung der Phasenraumpunkte um ì>å?íîï herum, die durch die Hamiltonfunktion ð beschrieben wird, und einer expliziten Zeitabhängigkeit am Ort ì>å?íîï :
ñ
ë
ë
ñ-ò Äôó ëí6ðöõ Å[÷ ò
÷ ù
Dabei ist die Poissonklammer zweier Funktionen ø und
ø ù
ø ù
ó øÂí6ùvõ Ä ÷ å ÷ îöú ÷ î ÷ å
÷ ÷
÷ ÷
(A.1)
definiert als:
(A.2)
Die infinitesimale Bewegung eines PR–Punktes wird durch eine Kontakttransformation
beschrieben, diese Transformation ändert das Volumen eines Gebiets im Phasenraum nicht
(eine der Poincareschen Integralinvarianten). Ein Punkt, der sich anfangs in diesem Volumen befand, kann es auch nicht verlassen, da er sonst die gleiche Position im Phasenraum
einnehmen müsste, wie ein Punkt, der den Rand des Gebiets definiert — und ab da der
gleichen Dynmaik gehorchen. Die Dichte in diesem Volumen bleibt also konstant, daher
gilt
ñ
ë
ñ-ò Äûvü
(A.3)
Als Bewegungsgleichung einer Phasenraumverteilung erhält man die als Liouvillegleichung bekannte Beziehung
ë
(A.4)
÷ ò Ä ú ó ëí4ðöõ ü
Eine Phasenraumverteilung ist also÷ station är, wenn die Poissonklammer mit der Hamil-
tonfunktion verschwindet. Dann befindet sich das System im Gleichgewicht, die Dichte
ändert sich nicht zeitlich.
ò
ëì>å?íîPí ï Ä ëì>å?íîï
ýþ
89
ó ëí4ðöõ Äû
(A.5)
ANHANG A. ZUR LIOUVILLE–GLEICHUNG
90
Die Phasenraumverteilung muß dann eine Funktion der Konstanten der Bewegung sein.
Für das radialsymmetrische Wasserstoffproblem sind die Konstanten der Bewegung die
Æ
Energie à , das Drehimpulsquadrat ÿ und die Projektion des Drehimpulses auf eine Achse,
meist ÿ .
Die übliche mikrokanonische Verteilung des Wasserstoffproblems ist durch eine feste
Energie, die Bindungsenergie, gekennzeichnet für die anderen beiden Konstanten der Bewegung existiert keine Einschränkung. Es muß nur deren ganzer Bereich gleichmäßig in
der Verteilung enthalten sein, denn wenn das nicht der Fall ist, wenn z.B. immer die gleiche
Phase auf der Bahn als Startwert gewählt wird, dann wird der Phasenraumpunkt nicht dort
bleiben, sondern den ganzen Bereich der Phase durchlaufen — die anfangs ausgewählte Verteilung ist nicht station är. Bei gleichmäßiger Belegung des ganzen zur Verfügung stehenden
Volumens“ für die Phase ändern zwar die einzelnen Phasenraumpunkte ihre Position, aber
”
die Dichte bleibt konstant.
Für die Anwendung auf die hier betrachteten Phasenraumverteilungen erhält man die
folgenden Kriterien, ob eine Verteilung station är ist:
die mikrokanonische Verteilung ist station är, da die Winkel“ zu den Konstanten der
”
Æ
Bewegung, den Wirkungen“ Ã , ÿ und ÿ , im ganzen zugänglichen Bereich1 gleich”
verteilt sind.
auch Verteilungen mit scharfer Energie, die einen festen Drehimpuls ( ÿ
feste Orientierung der Bahnen im Raum ( ÿ ) haben, sind station är.
Æ
) oder eine
Wenn sich eine Verteilung aus mikrokanonischen Verteilungen (eventuell mit eingeschränktem Drehimpuls) zusammensetzen läßt, sind die einzelnen Beiträge station är,
damit auch die gesamte Verteilung. Ein Beispiel ist die Energieverteilung nach Cohen.
Eine Anfangsverteilung, die nicht den ganzen Bereich der Winkelvariablen abdeckt,
wie z.B. die abgeschnittene Wignerverteilung von Eichenauer etal. [EGS81], ändert
sich; bei der cut–off wigner“ kommen die Beiträge verschiedener Energien außer
”
Phase, so daß die Verteilung nach einer kurzen Zeit thermalisiert“, d.h. station är
”
aussieht.
A.2
Verteilungen, die ungebundene Anteile enthalten, wären station är, wenn der komplette Winkelbereich abgedeckt wäre — die zugehörige Dichteverteilung aber ist nicht
begrenzt, sondern erstreckt sich unendlich weit im Raum. Für lokalisierte Objekte (wie Atome) können aber nicht alle Bereiche beitragen. Wegen dieses Begrenzens einer kontinuierlichen Dichteverteilung ist eine PR–Verteilung, die freie Anteile
enthält, nicht station är — es liegt also nicht primär an der positiven Energie, sondern
an der Einschränkung des Variablenbereichs.
Die Hamiltonschen Bewegungsgleichungen
Eine Möglichkeit, die Zeitentwicklung einer Phasenraumverteilung entsprechend der Liouvillegleichung zu integrieren, ist der folgende Ansatz:
1
Der zugängliche Bereich“ der Energie ist hier die feste Bindungsenergie.
”
A.2. DIE HAMILTONSCHEN BEWEGUNGSGLEICHUNGEN
91
In die Liouvillegleichung (Gl. A.4)
÷
ë
Ä
ð
÷
ë
÷
ú ÷
ð
ë
÷
(A.6)
wird eine diskretisierte Phasenraumverteilung eingesetzt ( test particle ansatz“)
”
ò
ëì>å?íîÂí ï Ä
ò å ì ïï ìlî
ìå
ú
ò
î ì ïï ü
ú
(A.7)
Mit einer Hamiltonfunktion, die nur kinetische und potentielle Anteile enthält,
î Æ
ð­ì>å?íîï Ä >ì åï
ú
(A.8)
erhält man für die beiden Ableitungen
ð
Ä î
÷
und
ð
Ä
÷
ü
÷ (A.9)
Die direkte Zeitableitung der diskretisierten Phasenraumverteilung Gl. (A.7) ist
ë
÷
å
ú
Å ì>å ú
Ä
>ì å å ì ò Dï ï lì î î ì ò ïï
ò ú ú ò
å ÷ ì ïDï î ìlî î ì ïï ü
ú
÷
Für die Ableitungen nach Ort und Impuls gilt:
ë
÷ ë
÷
Ä
>ì å å ì ò ïï
ú ò
÷
ì>å å ì ïï ú
÷
Ä
Die einzelnen Teile zusammen ergeben:
î ïå
ú ú Å î ìlî ú î ï
÷
ìlî
>ì å å ï
ì>å å
ú ú
ú
÷ ì>å å ï
>ì å
ú
ú ÷ ú
ò
î ì ïï
ú ò
ì î î ì ïï
ú
ìî
ïî ìî î
ú
å ï÷ ìî î
ú
÷
(A.10)
(A.11)
(A.12)
ï
Å
ï Ä®û
(A.13)
Da diese Gleichung für beliebige Verteilungen gelten muß, muß jeder Term der Summe
einzeln verschwinden, die Zeitentwicklung jedes einzelnen Phasenraumpunktes wird unabhängig von den anderen durch die Hamiltonschen Bewegungsgleichungen beschrieben:
å Ä î
î
und
Ä ú ÷
(A.14)
Die Erweiterung auf mehrere Dimensionen ist einfach. Die Phasenraumverteilung ist
dann eine Funktion der Vektoren å und î und die Poissonklammer lautet (vgl. Gl. (A.2)):
ó øPí6ùvõ Ä
÷
֌
ø
÷
֔
ù
ø
ú ÷î ÷å
÷ ÷
ù (A.15)
In der CTMC wird die gesamte zwölfdimensionale Phasenraumverteilung von Projektil
und Target gemeinsam diskretisiert, während Eichenauer etal. Target und Projektil getrennt
diskretisierten [EGS82], siehe Abschnitt 2.2.4. In diesem Ansatz kann nicht mehr Trajektorie für Trajektorie berechnet werden, es muß die gesamte Verteilung für Target und Projektil
betrachtet werden.
Anhang B
Diskretisierung der
Phasenraumverteilungen
Im folgenden soll die Notation
ûüqüRü anzeigen, daß eine (Zufalls–)Zahl im Bereich zwischen und gleichverteilt ausgewählt wird, d.h. alle Werte in diesem Bereich gleich wahrû
scheinlich auftreten.
B.1
Mikrokanonische Verteilung
Diese Verteilung (siehe Abschnitte 2.1 und 3.7.1) wird durch feste Bindungsenergie und
gleichverteiltes Drehimpulsquadrat beschrieben. Ist er Winkel zwischen und î , so gilt
für den Drehimpuls:
(B.1)
ÿ Æ Æ î Æ "!$#|Æ&%
Ä
Gleichverteilung
bedeutet daher — für festes und î — Gleichverteilung von
der Zufallszahl , die im Intervall
ûüRüqü)( aufgewürfelt wird, wird der Winkel
chend
%
"!$#0/ mit Ä+*-,).
ûüRüqü"(
Ä
"!$#Æ'% . Mit
% entspre(B.2)
verteilt. Alle Keplerellipsen zur Bindungsenergie –0,5a.u. gehen durch den Punkt Ä1( ,
dort ist î
Ä2( . Als Anfangspunkte werden o.B.d.A. gewählt:
ì í í ï
î Ä înì û í "!$#% í .3 % ï
(B.3)
Ä
û
û
(
ò
Weiterhin ist die Phase des Elektrons auf der Keplerellipse gleichmäßig verteilt, die ge65
:
sammte Umlaufzeit beträgt 4
Ä
ò -5 (B.4)
Ä ûüqüRü
ò
Die oben ausgewählten Punkte und î werden nun um auf der Ellipse verschoben“.
”
Das Wasserstoffatom ist rotationssymmetrisch, Richtung und Orientierung der eben bestimmten Ellipse werden über eine Drehung mit den Eulerwinkeln 7 , 8 und 9 gleichmäßig
verteilt.
8
7
9
Ä
Ä
Ä
-5
û üqüRü *6 ,:..3 -ì 5 ú ( ï
ûüqüRü 92
mit
Ä
(B.5)
ûüqüqü;( (B.6)
(B.7)
B.2. PRODUKTVERTEILUNG
93
Die so erhaltenen Werte für Ort und Impuls werden als Startwerte der Rechnung verwendet, sie haben die korrekte Bidungsenergie und Verteilung des Drehimpulses und sind
isotrop im Raum orientiert.
B.2
Produktverteilung
In der Produktverteilung (siehe Abschnitt 3.4.5.1) sind Ort und Impuls unabhängig voneinander, sie werden beide nach dem gleichen Verfahren ausgewählt.
Dazu wird nach der radialen Dichte die Wahrscheinlichkeit für einen bestimmten Anfangsradius/%
impuls bestimmt und der Raumwinkel ì=<?í ï gleichmäßig auf der Einheitskugel verteilt.
Die radialen Orts- und Impulsverteilungen des Wasserstoffgrundzustands sind:
> ì ï
Ä
D ìlî¬ï
Ä
Die Winkel werden nach
Æ@BAÆ)C
î Æ
5 E
ì ( Å î Æ ï)F
%
65 ûüqüRü *6,:..3 ì
Ä
<
?
Ä
ï
ú (
mit
(B.8)
Ä
(B.9)
(B.10)
ûüqüRü"( (B.11)
ausgewählt. Die Startwerte sind damit für den Ort (für den Impuls analog mit unabhängig
bestimmten Winkeln):
N)O
B.3
'%K;!L# <
HGIJ . " 3!$#0%M"!$# < P
Ä
.3 <
(B.12)
Wignerverteilung
Die Verteilung der Startwerte nach der Wignerverteilung folgt dem Vorgehen von Eichenauer etal [EGS81]: Für den Wasserstoffgrundzustand
@ ARC
Q ì ï Ä / 5
(B.13)
"!$#
7 íî .3 7ï die Wignertransformawird für den Spezialfall Ä ì û í û í ï und î Ä ì û íî
tion aufgestellt. Das erste Integral läßt sich analytisch lösen, die beiden restlichen werden
numerisch integriert und die Wignerfunktion für Ort, Impuls und den Winkel dazwischen
tabelliert. Dieser Schritt erfolgt unabhängig von
der Berechnung der Trajektorien. Dabei
wurde der Bereich a.u. und î
Ä ûüRüqüS
Ä ûüRüqü=?¬üUT a.u. abgedeckt; außerhalb ist die
Wignerverteilung vernachlässigbar klein.
Um Startwerte für Trajektorien zu erhalten, wurden , î und 7 ausgewählt
7
î
Ä
Ä
Ä
ûüRüqüVXWY"Z VXWY"ZWÄ+S a.u.
ûüRüqü[ W\ Y;Z [ WY"Z Ä]? üUT a.u.
*6,)..3 ì ú ( ï mit
Ä ûüRüqü"( (B.14)
(B.15)
(B.16)
94
ANHANG B. DISKRETISIERUNG DER PHASENRAUMVERTEILUNGEN
Für diese Werte wird die Wignerverteilung aus den vorher tabellierten Werten interpoliert
und nach Abschnitt 3.4.3 die Wahrscheinlichkeit berechnet, daß diese Werte ein Startpunkt
sind. Dieser muß noch, wie im mikrokanonischen Fall, mit den Eulerwinkeln im Raum
gedreht werden.
B.4
Energieverteilung nach Cohen
Diese Verteilung (vgl. Abschnitt 3.4.5.2) ist aus mikrokanonischen Verteilungen veschiedener Energie zusammengesetzt“, das Verfahren, mikrokanonische Startwerte zu bestimmen,
”
wird also übernommen und für beliebige Bindungsenergien erweitert. Es wird zuerst nach
der Energieverteilung aus [Coh85] eine Bindungsenergie bestimmt, danach das Verfahren
nach Abschnitt B.1 durchgeführt mit:
4
î
^
/
^
^
ú
6( _
6_
ú 65
/ 6_a`
ú
(B.17)
(B.18)
(B.19)
Anhang C
Numerische Integration der
Trajektorien
Für die Elektronenstoßionisation an atomarem Wasserstoff kann der volle Hamiltonian in
zwei Anteile aufgeteilt werden. Der eine, ð Æ , beschreibt die jeweils ungestörte Keplerbeç
wegung der beiden Elektronen und ðcbb deren gegenseitige Wechselwirkung (vgl. Gl. 3.2):
^
ð
^
î Æç
(
e ú çhf d g
ð
î ÆÆ
ú (Æ
i
çÆ
d
d
( e ä ç jf ú g Æi ä
ðcbb
(C.1)
Ziel ist es nun, die Zeitentwicklung gemäß der beiden Teile getrennt jeweils mit einem
angepassten Integrationsschema zu behandeln. Dazu sind zwei Techniken nötig:
Die Wirkung der beiden nicht kommutierenden Anteile
ander verwoben“ werden und
”
ð çÆ
und
ðcbb
muss mitein-
die Singularit ät des anziehenden Coulombpotentials des Kerns durch eine Regularisierung in geeigneter Weise abgefangen“ werden.
”
Die Elektron–Elektron–Wechselwirkung ðcbb ist zwar auch singul är, verhindert aber, da
sie abstoßend wirkt, daß die beiden Elektronen sich (für die numerische Berechnung) zu
nahe kommen. Er kann ohne weitere Regularisierung berechnet werden.
Im Folgenden werden die beiden benötigten Techniken, die symplektische Integration,
die die einzelnen Operatoren miteinander verwebt, und die Regularisierung des Coulombpotentials genauer dargestellt.
C.1
Symplektische Integration
Für eine Hamiltonfunktion, deren kinetischer Term nur von den Impulsen abhängt und die
einen geschwindigkeitsunabhängigen Potentialterm besitzt, kann eine Integrationsvorschrift
ñ
ñ
für die Zeitentwicklung aufgestellt werden, die das Phasenraumvolumen îlk å explizit
erhält. Solch ein Integrator wird symplektisch genannt.
95
ANHANG C. NUMERISCHE INTEGRATION DER TRAJEKTORIEN
96
Dieses Konzept läßt sich aber auf beliebige nicht vertauschende Operatoren anwenden,
in unserem Fall die freie Kepler–Bewegung der Elektronen unter ð Æ und ihre Wechselwirç
kung unter ðmbb . Die grundlegende Gleichung ist die Baker–Campbell–Hausdorff–Formel:
Sie beschreibt für zwei nicht kommutierende Operatoren n und n , wie das Produkt der
@o und @p zu einer Exponentialfunktion zusammengefasst werden
Exponentialfunktionen
kann:
@ o @ p ^ @rq
(C.2)
Dabei ist
s
^
n
w ní
dKw tM
ì ndví u ní d w=u u x s ní í t
d u
t t
t $ d í jí n$ ï
d
íjnd $$ t üqt üRü
d
(C.3)
Im betrachteten System entspricht dem vollen Hamiltonian ð , n und den beiden
t
Teilen ð Æ bzw. ðcbb .
ç
Gl. C.3 wird nun für die praktische Anwendung in ein Produkt der einzelnen Exponenti
alfunktionen aufgeteilt:
@yBz o Ç p { ^ | r@ =y o @"y p
ì= Ç ç ï
}$~
d
ç
(C.4)
Die Zahl wird Ordnung des Integrators“ genannt. Aus dem Vergleich der letzten beiñ
”
den Gleichungen können die Parameter } und } bestimmt werden. Auf eine leicht modifizierte Art bestimmte Yoshida daraus Integratoren bis zur Ordnung ^ [Yos90]. Hier
wurde im wesentlichen der Integrator sechster Ordnung, Typ A“ aus [Yos90] verwendet.
”
Bei höherer ( ^ ) oder niedrigerer ( ^
? ) Ordnung war bei gleicher Genauigkeit die
Propagation langsamer.
Zur Schrittweitensteuerung des Integrationszeitschrittes genügte die einfache Abfrage
der Gesamtenergie: war diese nach einem Schritt nicht mehr innerhalb der Toleranzgrenze,
so wurde der Schritt mit halber Schrittweite wiederholt, andernfalls der nächste Schritt mit
berechnet und überprüft. Bei den betrachteten Einschußenergien war eine relative GeA bis A ausreichend, erst bei relativen Grenzen von
nauigkeit der Gesamtenergie auf
R
(
û
(qû
A F zeigten sich Abweichungen in den
etwa
Endwerten der Trajektorien.
(qû
Mittels dieses Integrators wurde abwechselnd die im nächsten Abschnitt vorgestellte analytische Bahngleichung der ungestörten Elektronen und die Änderung der Impulse durch
ð bb berechnet.
C.2
Regularisierung des Coulombpotentials
Ein klassisches Elektron bewegt sich Feld des Kerns auf Keplerbahnen. Dabei sind auch
sehr exzentrische Bahnen möglich, bei denen das Elektron dem Kern sehr nahe kommt oder
sogar direkt auf ihn zuläuft (d.h. der Drehimpuls ist Null). Für die numerische Integration der Bahn stellen diese Annäherungen ein großes Problem dar, da hier mit extrem kleinen Schrittweiten gerechnet werden muss, bzw. die Divergenzen zu numerischem Überlauf
C.2. REGULARISIERUNG DES COULOMBPOTENTIALS
97
führen können. Aus diesem Grund wird die freie Elektronenbewegung durch eine Verallgemeinerung der aus der Himmelsmechanik bekannten Levi–Civita–Transformation regularisiert. Kustaanheimo und Stiefel stellen eine Abbildung des ` auf den F vor, für den,
V
V
analog zur Levi–Civita–Transformation, gilt [KS65]:
`
Æ ^ ~
Æ ^ F ø }Æ
}$~
ç
Rç
Æ
(C.5)
Die Abbildung ist definiert über:
Die Matrix
V ` ø V F
^+ ø û
(C.6)
NO
ø F OO
ø
úø Æ` P
ø ç
(C.7)
hat die Gestalt
ø ç
G
I
v^ IIJ ø¬Æ
ø `
ø F
úø
ø
ú
ø Æ
ç
F
ø `
ø
ú ø `
úø F
ç
ø Æ
ú
Für die Differentiale gilt;
ñ
ñ
ñ
ñ
ø F ø ç ø ` ø Æ ø Æ ø ` ø ç ø F ^ û
ú
ú
d
`
Die Bewegungsgleichung im
Masse lautet mit der Kraft ë
V für ein Teilchen der
ò
} ì ï ^ ë
(C.8)
:
(C.9)
Unter der Bedingung (C.8) wird diese Bewegungsgleichung über den Lagrange–Formalismus
übersetzt, nun sind die ø } die kanonischen Koordinaten. Dazu werden die transformierten
ñ¡ Kräfte } benötigt. Sie werden aus der geleisteten Arbeit
bestimmt, die in beiden Koordinatensystemen gleich sein muss:
ñ¡ ^ ë }ñ } ^ }ñ ø }
}
(C.10)
Daraus folgt, daß sich die Kraft gemäß
ë
^+£¢¤
û
(C.11)
übersetzt. Für die kinetische Energie in beiden Systemen
gilt:
Æ ^ }Æ
4 ^ w
ù Æ ^ w
ø
}
u
u
ò
ì ï:
Eingesetzt in Gl. C.9 ergibt sich die Bewegungsgleichung für ø
ñ ? 1¥ ñ/ò ì ø } ï ø } } ø }hÆ ¦ ^ }
ú
(C.12)
(C.13)
98
ANHANG C. NUMERISCHE INTEGRATION DER TRAJEKTORIEN
Diese Gleichung ist immer noch singul är im Ursprung. Daher wird noch einmal die von
den Kräften geleistete Arbeit betrachtet: sie entspricht der Zunahme an kinetischer Energie.
Æ
Aus Gl. (C.12) folgt mit der anfänglichen kinetischen Energie Æç ù
é
( ù é Æ ü
d
}Æ ^
( ø
}
(C.14)
Des weiteren wird eine regularisierte Zeit § eingeführt:
ñ-ò
§ ^©¨
ñ
ñ
ñ/ò ^ ( ñ
§
þ
(C.15)
Mit diesen beiden Definitionen bekommt die Bewegungsgleichung der
folgende Form:
ñ Æ
? ñ Æ ø } ì ú
§
ù é Æ ï ø } ^+ }
d
Für den Fall des Coulombpotentials gilt
ì ï ^
} ^
þ
úlª und
^
(
ú
÷
(
÷ø } ú
(C.16)
ø }
ú ªÆ
(C.17)
ü
é ò
und ù beziehen sich auf den gleichen Zeitpunkt ^
û
é
é
ª
^
ø } , Gl. (C.13), die
(C.18)
. Eingesetzt in die Bewegungs-
gleichung Gl. (C.16) erhält man:
ñ Æ
ù éÆ
}
ú ª ø } ^ û
? ñ §Æ ø ú
é
(C.19)
Den Ausdruck in Klammern identifiziert man als die Gesamtenergie:
_
ù éÆ
^
«
é
ú ª é
(C.20)
Damit bekommt die Bewegungsgleichung eine Form analog zum Harmonischen Oszillator, jedoch sind hier auch negative Energien zugelassen:
ñ Æ
_
}
ñ Æ ø
é ø } ^ û
§
ú
(C.21)
Diese Gleichung weist nun keine Singularit ät am Ursprung mehr auf, damit sind Bahnen
beliebiger Exzentrizität ohne numerische Schwierigkeiten bestimmbar.
Mit der Definition ¬ Æ ^1Æ ­® gilt für die — nun analytischen — Bahngleichungen in der
W
regularisierten Zeit:
j¯
° "!$#R¯
ø ì=§qï ^ . 3 ¬\§ ; !L# ¬\§
.3
d
_2±
_ ² û
2
û
(C.22)
C.2. REGULARISIERUNG DES COULOMBPOTENTIALS
99
ò
Um für einen von außen vorgegebenen Zeitschritt die regularisierte Zeit § zu bestimmen,
muss Gl. (C.15) integriert werden:
¨
Daraus ergibt sich
ñ/ò ^ ¨´³ ñ
§?ìø 쵧qïï Æ
é
é
°Æ
ò ^ Æ
"!$#R¯ ¬\§ j ¯ \
d
.3 ¬ §
¬
(C.23)
°
_2±
" !$#R¯ Æ ¬\§ 6§ _
(C.24)
û
ú
d ¬
und
°Æ
°Æ
°
Æ "!$#
ò ^ Æ
_¶²
¬\§ .3 ¬\§ "$! # Æ ¬\§
(C.25)
d §
ú û
¬
¬
d
d
Diese Gleichungen müssen nun nach § aufgelöst werden. Da keine analytische Lösung
existiert, wurde hier ein numerisches Newton–Verfahren zur Nullstellensuche eingesetzt.
Auf diese Weise wird das singul äre Coulombpotential auf den harmonischen Oszillator
abgebildet und ist damit für numerische Berechnungen wesentlich einfacher zu handhaben.
Mit dem oben vorgestellten symplektischen Integrator werden nun in jedem Schritt unter
auf die hier vorgestellte Weise die analytischen Bahnen der Elektronen benutzt und in
den Schritten unter ðmbb ihre Dynamik wieder aufeinander abgeglichen“. Diese Zerlegung
”
bietet vor allem bei großen Abständen der Elektronen, bzw. wenn ðcbb klein gegenüber ð Æ
ç
ist, also auch, wenn ein Elektron dem Kern sehr nahe kommt, große numerische Vorteile:
Es kann immer mit recht großen Zeitschritten gerechnet werden, sodaß der deutlich höhere Aufwand pro Schritt gegenüber z.B. einer Runge–Kutta–Integration durch die deutlich
kleinere Anzahl an Schritten mehr als kompensiert wird.
Neben diesen Geschwindigkeitsargumenten bietet das hier verwendete Integrationsschema auch deutliche Vorteile in der Energiestabilit ät — mit den analytischen Bahngleichungen lassen sich in einem Schritt selbst mehrere Umläufe mit beliebiger Exzentrizität exakt
propagieren und auch der symplektische Integrator selbst erhält wegen seiner symmetrischen Form die Energie sehr gut.
Dieses Integrationsschema ist extrem zuverlässig: obwohl, auch durch die Streuung bedingt, alle möglichen Keplerbahnen im Kernpotential zugelassen sind, musste im Schnitt
nur eine unter mehr als zehn Millionen gerechneten Trajektorien wegen zu großer Energieabweichung abgebrochen werden. Dies ist vor allem im Hinblick auf die voll differentiellen
Querschnitte von Bedeutung: wenn aus 100 Millionen Trajektorien nur etwa einhundert
zum Querschnitt beitragen, ist es wichtig, daß die Anzahl der Fehlertrajektorien“ deutlich
”
kleiner ist, damit nicht etwa gerade die Trajektorien, die für einen bestimmten Querschnitt
wichtig sind, alle abgebrochen wurden. Dies sollte nicht unterschätzt werden, da zu erwarten ist, daß zu einem voll differentiellen Querschnitt hauptsächlich Trajektorien mit der
gleichen Struktur“ beitragen, die z.B. alle eine sehr nahe Annäherung an den Kern bein”
halten und damit bevorzugt zu numerischen Fehlern führen würden.
ð çÆ
Literaturverzeichnis
[A Ç 87]
L.H. Andersen et al., Multiple ionization of He, Ne and Ar by fast protons and
antiprotons, Phys. Rev. A 36 (1987), 3612–29.
[AP66a]
R. Abrines and I. C. Percival, Classical theory of charge transfer and ionization
of hydrogen atoms by protons, Proc. Phys. Soc. 88 (1966), 861–72.
[AP66b]
R. Abrines and I. C. Percival, A generalized correspondence principle and
proton–hydrogen collisions, Proc. Phys. Soc. 88 (1966), 873–83.
[APV66]
R. Abrines, I. C. Percival, and N. E. Valentine, Proc. Phys. Soc. 89 (1966), 515.
[BBK89]
M. Brauner, J. S. Briggs, and H. Klar, Triply-differential cross sections for
ionization of hydrogen atoms by electrons and positrons, J. Phys. B 22 (1989),
2265–87.
[BJ25]
M. Born and P. Jordan, Zur Quantenmechanik, Z. Phys. 34 (1925), 858–88.
[BK93]
J. Berakdar and H. Klar, Structures in triply and doubly differential ionization
cross sections of atomic hydrogen, J. Phys. B 26 (1993), 3891–913.
[Bra99]
I Bray, Electron impact ionization of atomic hydrogen from near threshold to
high energies, arXiv:physics9906008 v3, 1999.
[Coh66]
L. Cohen, Generalized phase space distribution functions, J. Math. Phys. 7
(1966), 781–6.
[Coh85]
J. S. Cohen, Classical phase–space distributions for atomic collisions, J. Phys.
B 18 (1985), 1759–69.
[Coh96]
James S. Cohen, Quasiclassical–trajectory Monte Carlo methods for collisions
with two-electron atoms, Phys. Rev. A 54 (1996), 573–86.
[CZ83]
P. Carruthers and F. Zachariasen, Quantum collision theory with phase-space
distributions, Rev. Mod. Phys. 55 (1983), 245–285.
[D Ç 99]
[Dir26]
A. Dorn et al., Double ionization of helium by fast electron impact, J. Phys. IV
9 (1999), PR6–89.
P. A. M. Dirac, Proc. Roy. Soc. (London) A110 (1926), 561.
100
LITERATURVERZEICHNIS
101
[EGS81]
D. Eichenauer, N. Grün, and W. Scheid, Classical trajectory calculations for
ð Ç – ð collisions with the Wigner function as initial phase space distribution,
J. Phys. B 14 (1981), 3929–41.
[EGS82]
D. Eichenauer, N. Grün, and W. Scheid, Classical–trajectory calculations of
ð collisions, J. Phys.
the differential cross section for charge transfer in ð¶Ç
ú
B 15 (1982), L17–20.
[EJKS86]
H. Ehrhardt, K. Jung, G. Knoth, and P. Schlemmer, Differential cross sections
of direkt single electron impact ionization, Z. Phys. D 1 (1986), 3–32.
[EKSJ85]
@ @
H. Ehrhardt, G. Knoth, P. Schlemmer, and K. Jung, Absolute ð ì í ï>î cross
section measurements: comparison with first and second order theory, Phys.
Lett. 110A (1985), 92–4.
[Gry72]
M. Gryziński, Free fall“ solution of the Kepler problem in the presence of the
”
magnetic moment, Phys. Lett. 41A (1972), 69–70.
[Gry95]
M. Gryziński, Alignment of hydrogen atoms in
Phys. Lett. A 200 (1995), 360–4.
[Hel76]
E. J. Heller, Wigner phase space method: Analysis for semiclassical applications, J. Chem. Phys. 65 (1976), 1289–98.
[HO83]
D. J. W. Hardie and R. E. Olson, Charge transfer and ionization processes
involving multiply charged ions in collision with atomic hydrogen, J. Phys. B
16 (1983), 1983–96.
[Hyl29]
E. A. Hylleraas, Neue Berechnung der Energie des Heliums im Grundzustande,
sowie des tiefsten Terms von Ortho–Helium, Z. Physik 54 (1929), 347–66.
[KAD93]
S. Keller, H. Ast, and R. M. Dreizler, Quantum effects in CTMC modells, J.
Phys. B 26 (1993), L737–42.
[KS65]
P. Kustaanheimo and E. Stiefel, Perturbation theory of Kepler motion based on
spinor regularization, J. Reine Angew. Math. 218 (1965), 204–19.
[KW80]
C. L. Kirschbaum and L. Wilets, Classical many-body modell for atomic collisions incorporating the Heisenberg and Pauli principles, Phys. Rev. A 21
(1980), 834–41.
@r·
and
@A
ionising collisions,
[LMSW84] B. Lohmann, I. E. McCarthy, A. T. Stelbovics, and E. Weigold, Electron–
@ @
impact ionization of atomic hydrogen: Comparison of asymmetric ì í ï measurements with theories, Phys. Rev. A 30 (1984), 758–67.
[Meh64]
C. L. Mehta, Phase-space formulation of the dynamics of canonical variables,
J. Math. Phys. 5 (1964), 677–86.
[MH61]
H. Margenau and R. N. Hill, Correlation between measurements in quantum
theory, Progr. Theor. Phys. (Kyoto) 26 (1961), 722–38.
LITERATURVERZEICHNIS
102
[MHS84]
R. G. Montague, M. F. A. Harrison, and A. C. H. Smith, A measurement of the
cross section for ionization of helium by electron impact using a fast crossed
beam technique, J. Phys. B 17 (1984), 3295–3310.
[Moy49]
J. E. Moyal, Quantum mechanics as a statistical theory, Proc. Cambridge Phil.
Soc. 45 (1949), 99–124.
[PR98]
T. Pattard and J.-M. Rost, Degenerate Wannier theory for multiple ionization,
Phys. Rev. Lett. 80 (1998), 5081.
[PS93]
D. Proulx and R. Shakeshaft, Double ionization of helium by a single photon
with energy 89–140ev, Phys. Rev. A (1993), R975–8.
·
[R 96]
J. Röder et al., Coulomb three–body effects in low–energy impact ionization,
Phys. Rev. A 53 (1996), 225–33.
[RBIM99] T. N. Rescigno, M. Baertschy, W. A. Isaacs, and C. W. McCurdy, Collisional
breakup in a quantum system of three charged particles, Science 286 (1999),
2474–79.
[RF88a]
C. O. Reinhold and C. A. Falcón, Classical charge transfer and ionisation
cross sections for one– and three–dimensional collision processes, J. Phys. B
21 (1988), 2473–83.
[RF88b]
C. O. Reinhold and C. A. Falcón, On the quantum foundations of some classical approximations for ion–atom collisions, J. Phys. B 21 (1988), 1829–43.
[Riv57]
D.C. Rivier, On a one–to–one correspondence between infinitesimal canonical transformations and infinitesimal unitary transformations, Phys. Rev. 83
(1957), 862(L).
[RJE93]
J. Röder, K. Jung, and H. Ehrhardt, Recent progress in low energy atomic hy@ @
drogen ì í ï measurements, Journal de Physique IV 3 (1993), 29–38.
[Ros98]
J.-M. Rost, Semiclassical S–matrix theory for atomic fragmentation, Phys.
Rep. 297 (1998), 271–344.
[RT85]
Lionel M. Raff and Donald L. Thompson, Theory of chemical reaction dynamics, vol iii, ch. 1, The classical trajectory approach to reactive scattering, CRC
Press, 1985, Hrsg.: M. Baer.
·
[SC 84]
M. Sirigue-Collin et al., Quantum dynamical time evolutions as stochastic
flows on phase space, Physica A 124 (1984), 561–74.
[SEG87]
M. B. Shah, D. S. Elliot, and H. B. Gilbody, Pulsed crossed–beam study of
the ionisation of atomic hydrogen by electron impact, J. Phys. B 20 (1987),
3501–14.
[She59]
J. R. Shewell, On the formation of quantum–mechanical operators, Am. J.
Phys. 27 (1959), 16–21.
LITERATURVERZEICHNIS
103
[Shy92]
T. W. Shyn, Doubly differential cross sections of secondary electrons ejected
from atomic hydrogen by electron impact, Phys. Rev. A 45 (1992), 2951–6.
[SMO92]
D. R. Schultz, L. Meng, and R. E. Olson, Classical description and calculation
of ionization in collisions of 100eV electrons and positrons with He and ðl¸ , J.
Phys. B 25 (1992), 4601–18.
[Tay72]
J. R. Taylor, Scattering theory, John Wiley & Sons, Inc., 1972.
[Tho12]
Sir J. J. Thomson, Ionization by moving electrified particles, Phil. Mag. S. 6
23 (1912), 449–57.
[VN27]
J. Von Neumann, Nachr. Akad. Wiss. Göttingen, Math.-phys. Kl. 252 (1927).
[Wan53]
G. H. Wannier, The threshold law for single ionization of atoms or ions by
electrons, Phys. Rev. 90 (1953), 817.
[Wig32]
E. Wigner, On the quantum correction for thermodynamic equilibriuim, Phys.
Rev. 40 (1932), 749–59.
[WNHF79] E. Weigold, C. J. Noble, S. T. Hood, and I. Fuss, Electron impact ionization of
@ @
atomic hydrogen: experimental and theoretical ì í ï differential cross sections, J. Phys. B 12 (1979), 291–313.
[WRT92]
D. Wintgen, K. Richter, and G. Tanner, The semicalssical helium atom, CHAOS 2 (1992), 19–32.
[Yos90]
H. Yoshida, Construction of higher order symplectic integrators, Phys. Lett. A
150 (1990), 262–8.
[ZM86]
D. Zajfman and D. Maor, Heisenberg core in classical–trajectory Monte Carlo
calculations of ionization and charge exchange, Phys. Rev. Lett. 56 (86), 320–
3.
104
105
Mein Dank gilt allen, die am Entstehen dieser Arbeit beteiligt waren:
In erster Linie meinem Betreuer Jan–Michael Rost für das interessante Thema, all die anregenden Diskussionen und dafür, daß die Betreuung trotz der großen räumlichen Entfernungen nicht abbrach.
Herrn Prof. Briggs für die Aufnahme in seine Arbeitsgruppe, deren gute Atmosphäre wesentlich durch ihn geprägt ist.
Michael und Massimo, meinen Zimmergenossen für die interessanten physikalischen Diskussionen, aber noch mehr für die persönliche Anteilnahme: es war eine schöne Zeit in
NB 04 047.
Allen jetzigen und früheren Mitgliedern des RISC–Teams für’s Rechner zähmen“ (oder
”
zum Laufen bringen, je nachdem. . . ) und für alles, was sie mir darüber beibrachten.
Der ganzen Arbeitsgruppe, in den wechselnden Besetzungen, die sehr wesentlich dazu beigetragen hat, daß es eine sehr interessante und gute Zeit war.
Bettina: vielen Dank!!!