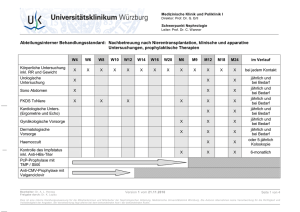
ZIP - FreiDok plus
Werbung

Aus der Chirurgischen Universitätsklinik Abteilung Allgemein- und Viszeralchirurgie der Albert-Ludwigs-Universität Freiburg im Breisgau Differentielle Analyse der Genexpression in der Niere der Ratte nach Hirntodinduktion INAUGURAL DISSERTATION zur Erlangung des Medizinischen Doktorgrades der Medizinischen Fakultät der Albert-Ludwigs-Universität Freiburg im Breisgau Vorgelegt 2008 von Carolin Horn geboren in Frankfurt am Main Dekan: Prof. Dr. med. Christoph Peters 1.Gutachter: Priv. Doz. Dr. med. Robert Obermaier 2.Gutachter: Priv. Doz. Dr. med. Jan Kaminsky Jahr der Promotion: 2008 I Inhaltsverzeichnis 1. EINLEITUNG............................................................................................................................. 1 1.1 Nierentransplantation – Indikation und Problematik............................................................ 1 1.1.1 Indikation und Nutzen........................................................................................................... 1 1.1.2 Anzahl der Spenderorgane .................................................................................................... 2 1.1.3 Art der Spenderorgane .......................................................................................................... 2 1.1.4 Komplikation und Gründe für den Transplantatverlust ........................................................ 2 1.1.5 Antigenabhängige - antigenunabhängige Faktoren für die chronische Abstoßung .............. 4 1.1.6 Der Hirntod als antigenunabhängiger Faktor........................................................................ 5 1.2 Hirntod ........................................................................................................................................ 6 1.2.1 Entwicklung der Definition „Hirntod“.................................................................................. 6 1.2.2 Der Einfluss des Hirntodes auf das Transplantat.................................................................. 7 1.3 Hämodynamik – Makrozirkulation – Mikrozirkulation........................................................ 8 1.4 Endokrines Systems ................................................................................................................... 9 1.5 Morphologische Unterschiede................................................................................................. 10 1.6 Apoptose .................................................................................................................................... 11 1.7 Inflammatorisches System....................................................................................................... 12 Einzelne Komponenten der Entzündungsreaktion .......................................................................... 13 1.7.1 Zytokine .............................................................................................................................. 13 1.7.1.1 Interleukin – 6 .............................................................................................................. 14 1.7.1.2 Chemokine ................................................................................................................... 14 1.7.1.3 Interleukin-1 und Interleukin-1-rezeptor-Antagonist................................................... 15 1.7.2 Selektine – Vertreter der Adhäsionsmoleküle .................................................................... 15 1.7.3 Makrophagen....................................................................................................................... 16 1.7.4 Die Heat-Shock-Proteine - Hämoxygenase-1 .................................................................... 17 1.8 Ansatzpunkt dieser Arbeit....................................................................................................... 18 2. MATERIAL UND METHODEN............................................................................................ 20 2.1 Tiermodell ................................................................................................................................. 20 2.2 Anästhesie und Monitoring ..................................................................................................... 21 2.3 Parameter zur Überwachung der Kreislaufstabilität........................................................... 22 2.4 Schrittweise Hirntodinduktion ............................................................................................... 22 2.5 Multiorganentnahme ............................................................................................................... 23 2.6 RNA-Extraktion ....................................................................................................................... 24 2.6.1 RNA-Miniprep-Protokoll .................................................................................................... 25 2.6.2 Quantitative Bestimmung.................................................................................................... 26 2.6.3 Qualitative Bestimmung...................................................................................................... 26 II 2.6.4 RNA- Pooling...................................................................................................................... 27 2.7 Genchip-Hybridisierung.......................................................................................................... 28 2.7.1 DNA-Microarray................................................................................................................. 28 2.7.2 Affymetrix-Standardprotokoll der cDNA-Synthese ........................................................... 29 2.7.3 Aufbau und Durchführung der DNA-Microarrays ............................................................. 32 2.7.4 Chiphybridisierung.............................................................................................................. 33 2.7.5 Waschen, Färben und Antikörperamplifikation.................................................................. 34 2.7.6 Scannen ............................................................................................................................... 36 2.7.7 Datenanalyse ....................................................................................................................... 36 2.8 Quantitative PCR ..................................................................................................................... 38 2.8.1 Durchführung der RT-PCR ................................................................................................. 38 2.8.2 Durchführung der PCR ....................................................................................................... 39 2.9 Statistik...................................................................................................................................... 41 3. ERGEBNISSE .......................................................................................................................... 43 3.1 Makrohämodynamik ............................................................................................................... 43 3.2 PH-Wert .................................................................................................................................... 46 3.3 pCO2 und pO2 ........................................................................................................................... 46 3.4 Analytisches Agarosegel der Gesamt-RNA............................................................................ 48 3.5 RNA-Extraktion und Poolung................................................................................................. 48 3.6 Ergebnisse der Chiphybridisierung........................................................................................ 49 3.7 Bestätigung der Genchip-Daten mit Real – Time - PCR...................................................... 55 4. DISKUSSION ........................................................................................................................... 57 4.1 Studienaufbau........................................................................................................................... 57 4.1.1 Relevanz der Studie ............................................................................................................ 57 4.1.2 Versuchsmodell des Hirntods ............................................................................................. 57 4.1.3 Zeitlicher Rahmen des Versuches....................................................................................... 58 4.1.4 Auschluß von Einflußfaktoren auf das Ausmaß des Hirntods............................................ 58 4.2 Diskussion der einzelnen Komponenten ................................................................................ 59 4.2.1 Zytokine .............................................................................................................................. 60 Il-6.................................................................................................................................................. 60 Il-1RA ............................................................................................................................................ 61 Calgranuline................................................................................................................................... 61 4.2.2 Chemokine .......................................................................................................................... 63 4.2.3 Adhäsionsmoleküle ............................................................................................................. 63 4.2.4 Transkriptionsfaktoren........................................................................................................ 65 4.2.5 Transportergene .................................................................................................................. 66 4.2.6 Apoptose ............................................................................................................................. 67 4.2.7 Gerinnung............................................................................................................................ 68 4.2.8 Heat-Shock-Proteine und Regenerationsproteine ............................................................... 69 4.3 Limitation und Ausblick.......................................................................................................... 71 III 5. ZUSAMMENFASSUNG.......................................................................................................... 73 6. LITERATURVERZEICHNIS UND QUELLENANGABEN .............................................. 74 7. ABKÜRZUNGSVERZEICHNIS ............................................................................................ 96 8. SELBSTSTÄNDIGKEITSERKLÄRUNG............................................................................. 98 9. DANKSAGUNG ....................................................................................................................... 99 10. LEBENSLAUF ....................................................................................................................... 100 Abbildungsverzeichnis IV Abbildungsverzeichnis Abbildung 1: Versuchsablauf in der Hirntod- und in der Kontrollgruppe ........................................ 21 Abbildung 2: Ratte nach gradueller Hirntodinduktion ...................................................................... 23 Abbildung 3: Agarosegel der Gesamt-RNA ...................................................................................... 27 Abbildung 4: Schema der Repräsentation eines Gens auf Mikroarrays von Affymetrix .................. 33 Abbildung 5 Prinzip der Chipanalyse nach Affymetrix®Gene Expression Assay ........................... 37 Abbildung 6: Mittlerer arterieller Druck (Mittelwerte ± SEM) während und kurz nach der Hirntodinduktion mit initialem Druckanstieg der HT-Gruppe.................................... 44 Abbildung 7: Vergleich der Herzfrequenz (Mittelwerte ± SEM) von Hirntodgruppe und Kontrollgruppe ............................................................................................................ 45 Abbildung 8: Vergleich des mittleren pH-Werte (Mittelwerte ± SEM) der Kontrollgruppe sowie der Hirntodgruppe. ............................................................................................ 46 Abbildung 10: Vergleich des mittleren pCO2-Wertes (Mittelwerte ± SEM) der Kontrollgruppe mit der Hirntodgruppe, Messabstand eine Stunde............................. 47 Abbildung 11: Elektrophorese des RNA-Pools .................................................................................. 49 Abbildung 12: Boxplots der mRNA Expression der drei Einzelgene HO-1, ELAM und CCL2 im Vergleich zwischen Hirntod- und Kontrollgruppe ...................................... 56 Tabellenverzeichnis V Tabellenverzeichnis Tabelle 1: Posttransplantative Organüberlebensraten (DSO) ................................................................. 6 Tabelle 2: Genspezifische RT-PCR Primer .......................................................................................... 40 Tabelle 3: RNA-Konzentration nach Poolung ...................................................................................... 48 Tabelle 4: Ergebnisse der Chiphybridisierung...................................................................................... 55 1 Einleitung 1. Einleitung 1.1 1.1.1 Nierentransplantation – Indikation und Problematik Indikation und Nutzen Die häufigsten Ursachen für Nierenerkrankungen im Endstadium sind die diabetische Nephropathie und verschiedene primäre und andere sekundäre Glomerulopathien (109). Im terminalen Stadium der Niereninsuffizienz ist zur Korrektur des Elektrolyt- und Säure-Base-Haushaltes, zur Elimination von Wasser und harnpflichtigen Substanzen, sowie zur Vermeidung weiterer Komplikationen die Nierenersatzbehandlung indiziert (56). Bezüglich der Patientenanzahl, die sich in einer chronischen Nierenersatztherapie befindet, ist von 1997 bis 2005 im Durchschnitt ein jährlicher Zuwachs der Prävalenz von 4,8 % und der Inzidenz von 5,2 % zu verzeichnen (Eurotransplant, Jahresbericht 2006). Nach Angaben von Eurotransplant befinden sich in Deutschland aktuell ca. 87.000 Personen in chronischer Nierenersatztherapie. 72,8% werden mit Dialyseverfahren behandelt, von denen 3% eine Peritonealdialyse erhalten und sich in etwa 27,2% Personen in der Transplantationsnachsorge befinden. Im Jahre 2005 wurden in Deutschland 2712 Patienten nierentransplantiert. Eine primäre Nierentransplantation ohne vorherige Dialysebehandlung (präemptive Transplantation) wurde für ca. 0,6% aller Transplantationen verzeichnet. Die Nierentransplantation wird standardmässig auch im Rahmen einer simultanen Pankreas-Nierentranplantation (SPKT) zur Behandlung niereninsuffizienter Diabetiker angewandt (50), mit guten Langzeiterfolgen hinsichtlich Überlebensdauer und Lebensqualität der Patienten (45). Für Diabetiker ist die Nierentransplantation bzgl. Mortalität und Morbidität günstiger als die Dialyse. Die Überlebensrate nach Nierentransplantation liegt bei ca. 60 % für Typ-2Diabetiker nach 5 Jahren. Dagegen beträgt die 5-Jahres-Überlebensrate unter Hämodialyse und Peritonealdialyse bei dieser Patientengruppe weniger als 25 % (90). Die Patientenüberlebensrate von Typ1-Diabetikern liegt nach alleiniger Nierentransplantation noch höher als bei Typ-2-Diabetikern. Bei Patienten mit Typ-1-Diabetes kann mit einer kombinierten Nieren-Pankreas-Transplantation sogar eine weitere Reduktion der Mortalität im Vergleich zur alleinigen Nierentransplantation beobachtet werden (180). Einleitung 1.1.2 2 Anzahl der Spenderorgane Auf der Warteliste des Jahres 2005 für Nierentransplantationen konnte die dreifache Zahl der tatsächlich durchgeführten Transplantationen, nämlich 8850 Patienten, verzeichnet werden. Die mittlere Wartezeit für eine Nierentransplantation beträgt ca. 40 Monate. Obwohl die Nierentransplantation als Mittel der Wahl und erfolgreichste kausal rehabilitierende und resozialisierende Therapieform der terminalen chronischen Niereninsuffizienz bezeichnet werden kann, ist nicht jedem Betroffenen eine solche Behandlung zu ermöglichen (81). 1.1.3 Art der Spenderorgane Ein wichtiger weiterer Punkt in der Beurteilung der Nierentransplantation ist die Differenzierung in Lebendspenderorgane und postmortale Spenderorganen. Im Jahre 2005 belief sich die Quelle der 2712 Nierentransplantationen in Deutschland zu über 80% auf postmortale Spender, der Anteil der Nierenlebendspender lag bei 19,2% (143). 1.1.4 Komplikation und Gründe für den Transplantatverlust Eine der Hauptkomplikationen der Transplantation ist die Abstoßung des Organs. Wenn die Behandlung einer Abstoßung erfolglos ist, muss der Empfänger zur Dialyse zurückkehren und auf eine erneute Transplantationschance warten. Weitere spätere Komplikationen sind die Nierentoxizität der Immunsuppression, Infektionen, Nierenerkrankungen und das Auftreten von Tumoren. Das Risiko eines Karzinoms oder Lymphoms ist 30-mal höher als in der Normalbevölkerung (142, 206). Die Abstoßungsreaktionen Die 5-Jahres-Funktionsraten der transplantierten Nieren unterscheiden sich je nach ihrer Spenderquelle. Durch Fortschritte in der Transplantationsmedizin kann aktuell eine 5- Jahresfunktionsrate von 71% nach postmortalen Nierenspenden, bzw. von 84% nach Lebendspenden erreicht werden. Dies ist neben verbesserten chirurgischen Techniken und postoperativen Nachbehandlungen vor allem der Entwicklung hochpotenter Immunsuppressiva zu verdanken (143). Einleitung 3 Hyperakute Abstoßung Eine Transplantatabstoßung lässt sich in verschiedene Kategorien einteilen. Die seltene hyperakute Abstoßung, auch Typ-II-Immunreaktion genannt, tritt bei allogenen Transplantationen innerhalb von Minuten bis Stunden nach der erfolgten Transplantation und der Wiederherstellung des Blutflusses auf. Sie wird durch präformierte, allospezifische Antikörper oder durch blutgruppenspezifische Antikörper, die zum Zeitpunkt der Transplantation bereits vorhanden sind (zytotoxische Antikörper gegen AB0- oder HLA-Antigene des Transplantats), verursacht. Nach der Komplementaktivierung kommt es durch intravasale Gerinnungsvorgänge zur vollkommenen Thrombosierung der Gefäße des Transplantats und somit zur irreversiblen Schädigung des Organs (178). Akute Abstoßung Von der akuten Abstoßung spricht man bei spezifisch gegen die Transplantationsantigene gerichteten Immunreaktionen, die zwischen Tagen bis einigen Monaten die Transplantatvitalität bedrohen. Eine Abstoßung zwischen dem 2. und 5. Tag nach der Operation bezeichnet man auch als akzelerierte Abstoßung. Neben dem ABO-Blutgruppensystem sind die HLA-Komplexe (HLA= humanes Leukozytenantigen) (engl. Major histocompatibility complex, MHC) mit ihren Histokompatibilitätsantigenen von entscheidender Bedeutung für die akute Immunantwort. Als Transplantationsantigene spielen sie sowohl eine entscheidende Rolle bei der immunologischen Erkennung und Abwehr, als auch bei der Antigenpräsentation (178). HLA-I-Antigene sind als Glykoproteine auf der Zelloberfläche kernhaltiger Zellen in die Zytoplasmamembran integriert und exprimiert und sind wichtiger Bestandteil für die Erkennung körpereigener und fremder Antigene. Endothelzellen werden zur vermehrten Expression von MHC-Antigenen angeregt, welche wiederum die Immunogenität des Spenderorgans erhöhen, und somit das fortlaufende Zugrundegehen des Organs weiter triggern. HLA-Klasse-II-Antigene sind hauptsächlich auf BLymphozyten, dendritischen Zellen, sowie stimulierten Makrophagen zu finden. Wichtige Ursache für die zelluläre Immunreaktion und nachfolgende akute Abstoßung des Organs sind die aktivierten Lymphozyten, die aus Kapillaren in das Interstitium wandern und dort Nekrosen hervorrufen (177, 195, 196). Chronische Abstoßung Die wichtigsten Gründe für einen langfristigen Transplantatverlust stellt die chronische allogene Transplantatnephropathie (CAN) dar (78). Die chronische Abstoßung eines Transplantats ist hauptsächlich durch eine über Monate und Jahre fortschreitende, langsame und kaum beeinflussbare Funktionsverschlechterung des Transplantates gekennzeichnet (62). Meist beruht diese chronische Abstossungsreaktion vorwiegend auf einer obliterierenden Arteriopathie mit Intimafibrose. Sie ist vermutlich eine überschießende Antwort auf eine unentwegte Schädigung der Gefäßwandzellen durch die Kom- Einleitung 4 plexe von Transplantationsantigenen mit den Wirtsantikörpern (190). Chronische Aktivierung zellulärer und humoraler Abstoßungsmechanismen führen zu einer chronischen Tranplantatvaskulopathie, die vor allem die Rindenarterien betrifft und zu renaler Ischämie mit tubulärer Atrophie, Nephronzerstörung und interstitieller Nephrose führt, bis hin zur Schrumpfung des Nierenparenchyms (207). Einige Vermutungen werden dahingehend angestellt, dass die chronische Abstoßung ein Ergebnis ständiger kleinerer Verletzungen des Endothels darzustellen scheint, das dadurch in einer ständigen Aktivierung gehalten wird (70, 153). Dies kann in eine Expressionsstimulation von zum Beispiel Adhäsionsmolekülen münden, gefolgt von der Aufrechterhaltung einer Entzündungsreaktion (73). Die zuvor durchgemachte akute Abstoßung oder eine Entzündung des Organs sind dabei Risikofaktoren bei der Niere, die eine spätere chronische Abstoßung oder Dysfunktion wahrscheinlich machen (178). Es besteht eine hohe Korrelation zwischen der Anzahl akuter Abstoßungsreaktionen und der Wahrscheinlichkeit eine chronischen Abstoßung zu erfahren (6). 1.1.5 Antigenabhängige - antigenunabhängige Faktoren für die chronische Abstoßung Pathophysiologisch definiert man antigenabhängige, aber auch antigenunabhängige Risikofaktoren, die die progrediente Entwicklung einer chronischen Abstoßung entscheidend beeinflussen können (199). Antigenabhängige Ursachen Immunogene Gründe für eine Transplantatabstoßung sind seit langem bekannt. So ist eine schlechtere HLA-Kompatibilität mit einer höheren Anzahl an Abstoßungsreaktionen assoziiert. Antikörper gegen HLA-klasse 1 (A, B, C) oder Klasse 2 (DR, DQ) können vermehrt in Gruppen gefunden werden, die vorher gegen diese Glykoproteine sensibilisiert wurden (33), z.B. durch Schwangerschaft, Bluttransfusionen oder eine vorhergehende HLA-unpassende Transplantation (84). Vorhergehend stark immunisierte Patienten haben eine statistisch höhere Abstoßungsrate, als nicht-sensibilisierte Personen, was die antigenabhängige Beeinflussung im Outcome aufweist (163, 214). Antigenunabhängige Ursachen Obwohl sich frühere Erklärungen für die progrediente Entwicklung einer chronischen Abstoßung von Spenderorganen vornehmlich auf antigenabhängige Unterschiede zwischen Spendern und Empfängern stützten, rückte in den letzten Jahren die Erkenntnis über antigenunabhängige Beeinflussung durch nicht-immunologische Faktoren stark in den Vordergrund (154). Ihre Einflussnahme auf renale Transplantatabstoßungen gilt inzwischen als bekannt (49). Klinische Datenbanken und wissenschaftliche Studien konnten beweisen, dass nicht-immunologischen, spenderspezifischenden Risikofaktoren wie Spenderalter (39), Vorerkrankungen (144, 169), Rasse (86), die Möglichkeiten der intensivmedi- Einleitung 5 zinische Betreuung bei Organentnahmen (24, 35, 91), aber auch der Hirntod (170, 201, 212) und der Ischämie- und Reperfusionsschadens (93, 146) einen Einfluss auf die Qualität des Organs haben. Pathophysiologische Veränderungen sorgen für eine immunologische Grundaktivierung des zu transplantierenden Organs, und somit zu erhöhten Entzündungsreaktionen im Körper des Empfängers und des Spenders (212). Der Ischämie-/Reperfusionsschaden, als ebenfalls anerkannter antigen-unabhängiger Faktor, folgt dem Hirntod im Prozess der Transplantation, beginnend mit der Minderperfusion und Ischämie nach der Organentnahme des Spenders. Forschungsarbeiten konnten aufweisen, dass diese zu einer Verschlechterung der mikrovaskulären Durchblutung, einer Leukozytenendothelreaktion, dem Austritt von Entzündungszellen und Mediatoren aus den Gefäßen in das Gewebe, sowie der Freisetzung von freien Sauerstoffradikalen und proteolytischen Enzymen führt (132). Durch Präkonditionierungsversuche konnten in einem nächsten Schritt die klinischen Auswirkungen dieser Organschäden beeinflusst werden. Es zeigte sich zum Beispiel, daß durch die Vorbehandlung mit Endotoxin exprerimentell eine signifikant geringere Ausprägung des Ischämie/Reperfusionsschadens auftrat. (133, 134). 1.1.6 Der Hirntod als antigenunabhängiger Faktor Parallel zu den Studienfortschritten des Ischämie/Reperfusionsschadens, wurde in den letzten Jahren klar, dass die schlechteren Langzeiterfolge hirntoter Spenderorgane betreffend Funktion, Abstoßungsrate und Transplantatüberlebenszeit ihren grundlegenden Ursprung in pathophysiologischen Veränderungen während des Hirntodprozesses haben (28). Vertiefungen der Forschung in diesem Bereich erscheinen auch deswegen so sinnvoll, da die postmortalen Spendernieren gut 80% aller Organtransplantate ausmachen. Erwiesenermaßen liegt die Halbwertszeit der Funktion von hirntoten Spendernieren aber signifikant unter den Kurz– und Langzeitergebnissen von verwandten, aber auch unverwandten Lebendspendern (32, 189). In der Vergangenheit galten immunogene Faktoren, also möglichst identische Histokompatibilitätskomplexe, als primäre Prognosefaktoren für das Transplantatüberleben. HLA-identische Zwillinge zeigen in der Tat statistisch beste Funktionsergebnisse bei Nierentransplantationen. Weitere Studien konnten jedoch zeigen, dass HLA-angepasste, hirntote Spendernieren in Kurz- und Langzeitresultaten trotzdem weit unter den Ergebnissen HLA-unangepasster, lebendgespendeter Nieren liegen (202). Beispielsweise zeigen gesammelte Daten über die Ergebnisse von Nierentransplantationen zwischen Ehegatten oder anderen unverwandten und HLA-unangepassten Lebendspendern im Vergleich zu postmortalen Spendern bessere Transplantat- und Patientenüberlebensraten (United Network of Organ Sharing; UNOS, Sollaborative Transplant Study, DSO) (63, 143, 194). 6 Einleitung Niere 1-Jahres-Überleben 5-Jahres-Überleben Totspender-Organ 85.0% 71,0% Lebendspender-Organ 95.1% 84,0% Tabelle 1: Deutsche Stifung für Organtransplantation; Collaborative Transplant Study (CTS), 2005 Die These, dass der Hirntod als antigenunabhängiger Faktor per se die Donororgane vor der Explantation beeinflusst, sensibler für Preservations- und Ischämie/Reperfusionsschäden macht und zu einer gesteigerten immunogenen Aktivität des Spenderorgans führt, welche bei der späteren Transplantation die Immunantwort des Empfängers beschleunigt, konnte bereits in verschiedenen Tierexperimenten und in klinischen Studien bewiesen werden (24, 91, 92, 172, 174, 204). Hierauf soll im Folgenden etwas genauer eingegangen werden. 1.2 1.2.1 Hirntod Entwicklung der Definition „Hirntod“ Mit Einführung der mechanischen Ventilation, suffizienter Oxygenierung peripheren Gewebes und verbessertem Management kritisch kranker Patienten geriet die traditionelle Definition des Todes als Herz-Kreislauf-Stillstand und Aussetzen des Herzschlages in Diskussion (44). Auf der Basis der in Frankreich publizierten Definition des „irreversiblen Komas“ von 1950, transplantierten französische und belgische Chirurgen einige Jahre später erstmals Nieren von herzkreislaufstabilen, hirntoten Spendern (16, 124, 183). Dadurch konnte die Ischämiezeit um ein Vielfaches gesenkt und die effiziente Wiederaufnahme der renalen Funktion erhöht werden (153). Die Benutzung solcher hirntoter Spender initiierte weltweit rechtliche und moralische Debatten (191). Eine legale Basis wurde erst im August 1968 in Form der Harvard Kriterien des Hirntods geschaffen, die diesen Zustand als irreversiblen Endpunkt des Daseins definierten und in seinen Kriterien genau differenzierten (1). Der Hirntod lässt sich als vollständiger und irreversibler Ausfall des menschlichen Bewusstseins definieren, einhergehend mit dem Verlust aller zerebralen Funktionen (Großhirn, Kleinhirn, Hirnstamm) bei künstlich aufrechterhaltener Herz-Kreislauf-Funktion und kontrollierter Beatmung (191). Einleitung 1.2.2 7 Der Einfluss des Hirntodes auf das Transplantat Die Annahme, der Hirntod sei eine gegebene statische Kondition hat sich anhand verschiedener experimenteller Forschungsarbeiten als falsch bewiesen (153). Tierexperimentelle Studien mit gleichen Bedingungen betreffend Explantation, Ischämiezeit und Transplantationsprozess zwischen Hirntodmodellen und Kontrollgruppen zeigten, dass das Kadaverorgan zum Zeitpunkt der Transplantation neben systemischen auch schon organspezifische, komplexe funktionelle und morphologische Veränderungen durchlaufen hat und dementsprechend, unabhängig von direkten antigenabhängigen Einflüssen, eine schlechtere Funktion aufweist (158, 174, 189, 203, 204, 215). So wurde in den letzten Jahren klar, dass die Ursache der unterschiedlichen Erfolgsraten zwischen lebendgespendeten Nieren und denen hirntoter Spender besonders auch auf dem Hirntod basieren, und nicht nur, wie vielfach angenommen, in den Unterschieden der kalten Ischämiezeit (24, 91, 136). Die Ätiologie, also Tempo und Ausdehnung des Hirntods, spielt ebenfalls eine große Rolle (176). Man unterscheidet einen explosionsartigen Typ des Hirntodes, der die peripheren Organe auf ein vielfaches stärker beeinflusst und eine graduierte Form, bei der der Hirntod des Individuums schrittweise hervorgerufen wird (157). 8 Einleitung 1.3 Hämodynamik – Makrozirkulation – Mikrozirkulation Der Effekt des Hirntods zeigt sich in rapiden Schwankungen der Hämodynamik (Mittlerer arterieller Blutdruck und Herzfrequenz) und daraus resultierenden pathologischen Folgeereignissen. (153) Die Auswirkungen auf das kardiopulmonale System und die hämodynamische Stabilität des Donors kann generell in zwei Phasen unterteilt werden. Hypertensive Phase Nach der sehr frühen parasympathischen Episode mit Bradykardie folgt eine intensive sympathische Stimulation als Folge der zerebralen Ischämie und Einklemmung des Hirnstamms, die für einige Minuten andauert (8). Diese Dynamik wird entweder durch direkte neuronale Aktivität oder durch die Ausschüttung endogener Katecholamine ausgelöst (23, 34, 77, 149, 157). Es besteht eine Korrelation zur Art der Hirntodinduktion, entsprechend auch zu Anstiegskurve des intrakraniellen Druckes (76, 176). Es konnten in diesem Zusammenhang weit über das Ausgangsniveau reichende Level an Adrenalin, Norepinephrin und Dopamin nachgewiesen werden Dabei konnten teilweise Steigerungen des Dopaminlevels bis 800% und eine 700%ige Erhöhung des Norepinephrins aufgezeigt werden (65). Diese initiale Phase nach Hirntodinduktion wird mit dem Begriff des „Autonomic storm“ zusammengefasst, der Alterationen betreffend den Blutdruck, der Gerinnung und den Elektrolyt- und Hormonhaushalt mit sich zieht (157). Der massive Anstieg des systemischen Katecholaminlevels zieht bei gleichzeitiger Aktivitätssteigerung des Herz-Kreislaufsystem mit positiv chronotroper, inotroper und dromotroper Wirkung einen veränderten vaskulären Widerstand mit sich, der in der Niere auf das Vierfache ansteigt, in der Milz sogar auf das Neunfache (77). Diese gravierende Vasokonstriktion und der erhöhte Perfusionsdruck führen zu einer signifikant erniedrigten Organperfusion (76). Eine andere Ursache des veränderten Organwiderstandes sind Veränderungen im mikrovaskulären Strombett. Intravitalmikroskopische Untersuchungen an der Leber haben gezeigt, dass es bereits drei Stunden nach Hirntod zu einer reduzierten sinusoidalen Perfusion und zu einer gesteigerten Leukozyten-Endothelinteraktion in der mikrovaskulären Strombahn kommt (130, 138, 139). Es konnte ein vergleichbarer Effekt für die pankreatische Mikrozirkulation gezeigt werden (133). Hypotensive Phase Auf die hypertensive folgt eine spätere normo- bis hypotensive Phase, die nach abgeschlossener Hirntodinduktion durch die irreversible Entkopplung des Rückenmarks von höheren Hirnarealen erklärt wird. 9 Einleitung Sie wird durch eine Reduktion hämodynamischen Parameter definiert (68). Inotropie und Chronotropie, sowie der kardiale Ausstrom werden erniedrigt (68). Der reduzierte autonome Tonus führt zu einer veränderten vaskulären Autoregulation mit erniedrigter Blut- und Sauerstoffversorgung der Organe und Gewebe. Sowohl anfängliche als auch spätere Zirkulationsveränderungen können also schon vor der Organentnahme zu schweren ischämischen Schäden und zu schlechter Funktion im Empfänger führen (148, 153). Die Hypotension kann durch gutes klinisches Management, Volumenaufrechterhaltung, adäquater Sauerstoffversorgung und Gewebeperfusion kontrolliert werden (36, 150). 1.4 Endokrines Systems Desweiteren besteht ein Zusammenhang zwischen Hirntod und der Störung der HypothalamusHypophysen-Achse (53, 110, 112, 119). In verschiedenen Tiermodellen konnte eine Dysfunktion der Hypothalamus-Hypophysenachse in Form einer Verminderung der Hypophysenhomone Vasopressin und ACTH dargestellt werden. ACTH sinkt 45 Minuten nach Hirntodinduktion sogar auf Werte nahe dem Nullpunkt und auch Vasopressin (Antidiuretisches Hormon, ADH) ist zum Ende der Versuchsreihe nicht mehr messbar. Es entsteht ein neurogener Diabetes insipidus. Stark erhöhte, hypotone Urinausscheidungen konnten beobachtet werden (181). Messungen des Plasmaspiegels von ACTH und Vasopressin könnten daher möglicherweise als diagnostische Hilfsinstrumente für den Hirntod dienen. Die Erklärung für die Abnahme liefert dabei die Histologie der Hypophyse, bei der Veränderungen vom Ödem bis hin zur Nekrose gefunden werden (10). Hier scheint sich die zugrunde liegende Störung des Regelkreises zu begründen. Auch der signifikante Abfall von Aldosteron weist neben Cortisol auf eine Einbeziehung der Nebeniere, als eine der Endstrecken der Hypothalamus-Hypophysenachse, hin (52). Adrenerge Insuffizienz ist in verschiedenen Studien ebenfalls als Folge des Hirntodes zu registrieren. Markanter Abfall von Schilddrüsenhormonen (Triiodthyronin und Thyroxin) (168), Glucagon und des Cortisols konnte in einer retrospektiven Studie bei 76% der Patienten mit Kriterien des Hirntods dargestellt werden (43, 67, 82, 131). Andere Studien, z.B. von Powner und Lopau (107, 151), zeigten wiederum keine signifikanten Veränderungen der hormonellen Konzentrationen nach Hirntod, bzw. nicht zwangsweise endokrines Versagen. Leichte Schwankungen in Hormonkonzentrationen (TSH, Insulin, Laktat) konnten zwar fast überall aufgezeigt werden, es bestand aber keine Korrelation zu metabolischen und hämodynamischen Fehlfunktionen, sowie der Organfunktion nach Transplantation (67, 107). 10 Einleitung Hormonelle Substitutionstherapien führten in verschiedenen Studien zu einer Verbesserung des kardiovaskulären Status, verminderten perioperativen inotropen Medikation und signifikant weniger Bikarbonatgabe durch Verminderung des Serumlaktats (131). Progressiver Energieverlust und Stoffwechseleinschränkungen infolge mitochondrialer Schädigung als Hauptfolge hormoneller Schwankungen, konnten ebenfalls erfolgreich mit einer Kombination von T3, Cortisol und Insulin unterbunden werden (131). 1.5 Morphologische Unterschiede Studien über genaue morphologische und funktionelle Veränderungen als Effekte des Hirntods sind an verschiedenen Organen erläutert worden. Am Herzen am besten untersucht, werden diese als katecholamininduzierte Toxizität beschrieben und entsprechen mit petechialen, subendothelialen Hämorrhagien und Myozytennekrosen Veränderungen, die auch bei hoher experimentell-exogener Katecholaminzufuhr beobachtet werden (119, 197, 218). Dieselben Schäden ließen sich bei Patienten aufweisen, die an akuten zerebralen Veletzungen starben (17). Diese myozytotoxischen Mechanismen führen zu ausgeweiteten Herzmuskelnekrosen sogar im gut durchbluteten Herzen (118). Eine Studie von Yeh et al. (217) erweiterte diese Bilanzen mit der Hypothese, dass neben der Toxizität auch eine veränderte myokardiale Genexpression zur Dysfunktion beiträgt. 2002 untersuchten sie in einer Studie die Auswirkung einer zentralen sympathomimetischen Blockade, um die Effekte der hohen Katecholaminausschüttung durch den „Autonomic storm“ auf die myokardiale Genexpression zu unterbinden. Sie konnten zeigen, dass durch die erniedrigten Level an Katecholaminen sowohl die hämodynamische Instabilität, die elektrokardiographischen Veränderungen, als auch die myozellulären Schäden, die durch den Hirntod aufzuweisen waren, signifikant verringert waren (152, 161, 217). Bei Lungentransplantaten zeigt sich eine chronische Abstoßung morphologisch meist als obliterierende Bronchiolitis. Histologisch zeigt sich ein fibroproliferativer Prozess, der durch eine Entzündungsreaktion sowie eine Fibrose gekennzeichnet ist (15). Doch auch der direkte und akute Einfluss des „Autonomic storm“ auf die Lunge bewirkt eine Schädigung, sofern er nicht medikamentös zu kontrollieren ist. Die Entwicklung einer pulmonalen Dysfunktion beispielsweise im Sinne eines neurogenen Ödems, ist als Komplikation bei zentralnervösen Traumata beschrieben. Studien zeigen, dass eine Behandlung während der hypertensiven Phase mit α-Rezeptor Antagonisten die Infiltration des pulmonalen Gewebes mit Entzündungszellen vermindert, während in der hypotensiven Phase Noradrenalin eine verbesserte pulmonale Oxygenierung gewährleisten kann (12). 11 Einleitung Eine Studie von van der Hoeven et al. über die Funktion der von hirntoten Ratten transplantierten Leber, zeigte signifikante perizentrale Nekrosen und profunde Vakuolisierungen überall am Transplantat, die sich bei lebendtransplantierten Kontrolllebern nicht auffinden ließen (201, 202). Kardiovaskuläre Instabilität, Hypotension und nachfolgende Ischämie stellen Hauptgründe für Nierenschäden dar (198). Eine hohe Inzidenz tubulärer Nekrosen wurde posttransplantativ an Nieren von hirntoten Spendern mit instabilem hämodynamischen Status beobachtet (99). Die histopathologischen Veränderungen in der Niere nach Hirntod liegt in einer glomerulären Hyperämie mit nachfolgender Entwicklung einer Glomerulitis, endotheliale Proliferation und Periglomerulitis (126). Innerhalb von 3 Tagen präsentieren sich Degeneration und Atrophie tubulärer Zellen. In Studien von Pratschke et al. (154, 158) über kurzfristige und langfristige Auswirkungen des Hirntods auf die Abstoßung von Nierentransplantaten, ließen sich jeweils signifikante Unterschiede zugunsten der Lebendspender demonstrieren. Eine progrediente Funktionsverschlechterung ging parallel mit morphologischen Veränderungen einher: Alle Nierenbiopsien zeigten zwei Wochen nach Transplantation histologische Merkmale von Gewebeschaden. Allerdings präsentierte sich das Gewebe der hirntoten Spenderorgane, im Vergleich zu Lebendspendern, kortikointerstitiell zwei- bis dreifach stärker mit Monozyten infiltriert, zeigte einen zweifach höhergradigen tubulären Schaden und früheres Einsetzen von interstitieller Fibrose. Nach acht Wochen war das Interstitium markant mit wirtseigenen mononukleären Zellen infiltriert. Im histologischen Bild ließ sich verstärkt eine glomeruläre Hypertrophie und interstitielle Fibrose mit parallel zunehmender tubulärer Atrophie demonstrieren. Nach 48 Wochen zeigte sich das Organ im finalen Stadium und morphologisch ausgebrannt (158). Eine Studie am hirntoten Schwein zeigt eine signifikante Erniedrigung von intrazellulärem Natrium und Kalium im Vergleich zu lediglich anästhestetisierten Tieren (211). Diese Elektrolytparameter der Nierenfunktionstüchtigkeit korellierten mit der funktionellen und morphologischen Qualität der Kadaverorgane. 1.6 Apoptose Die Apoptose führt zum programmierten Zelltod infolge von Verletzungen, Ischämie, metabolischem Ungleichgewicht oder anderen Stressfaktoren (60, 69). Sie wird hauptsächlich durch eine Reihe von Proteinen kontrolliert, die man als Kaspase–Kaskade bezeichnet. Die Beteiligung von TNF-alpha durch Bindung an seinen Oberflächenrezeptor ist in diesem Zusammenhang bekannt und seine Erhöhung nach Hirntodinduktion gilt als bewiesen (2, 113). Eine andere Auslösung des programmierten Zelltods stellen Zytokine dar, die direkt durch die Zellmembran diffundieren, und die Kaspase Kaskade in Gang setzen. Die nachfolgenden Schritte führen zu Veränderungen in der Zellmembran, Kon- 12 Einleitung densation von nuklearen und zytoplasmatischen Strukturen und zur Spaltung der DNA-Helix (104, 184). Die Zelle zerfällt schließlich und wird von Gewebsmakrophagen abgeräumt und absorbiert. Neuere Studien zeigen eine deutliche Verknüpfung zwischen Hirntod, inflammatorischer Antwort und erhöhter Apoptoserate in der Leber. Es wurde bereits beschrieben, dass durch den Hirntod proapoptotische Proteine verstärkt exprimiert werden und die apoptotische Kaskade im Donororgan vermehrt induziert wird (38, 145). Erhöhte Apoptoseinduktion übt dementsprechend einen negativen Einfluss auf die Organvitalität posttransplantativ aus. Das Resultat ist wiederum eine schlechte Kurz-und Langzeitfunktion des Spenderorgans im Empfänger (22, 203). Kalte Konservierung und KaspaseInhibitoren konnten den Zelltod in Studien am Pankreas bereits erfolgreich minimieren (105). 1.7 Inflammatorisches System Stressoren, wie in diesem Fall der Hirntod, aber auch Schock, oxidativer Stress, oder Hyperthermie, rufen eine Stress- und Entzündungsantwort des Körpers hervor, die auf komplexen ineinander greifenden Kaskaden endogener Mediatoren basiert (155, 176). Die inflammatorische Reaktionsabfolge beginnt mit der Immunaktivierung, es folgen die zelluläre Rekrutierung, das Rolling, die Adhäsion, Diapedese und schließlich die Infiltration durch inflammatorische Zellen. Im Mittelpunkt steht die These, dass das Organ eines hirntoten Spenders schon im Vorfeld für erhöhte Entzündungsreaktionen sensibilisiert wurde, das heisst, dass Organe von postmortalen Spendern zum Zeitpunkt der Einpflanzung schon molekulargenetische Veränderungen durchgemacht haben und dazu programmiert sind, spätere Entzündungsreaktionen im Empfänger zu triggern und Tempo und Intensität immunologischer Antworten in großem Ausmaß zu verstärken (35, 64, 177). Erhöhtes Tempo und Intensität der immunologischen Aktivierung durch hirntote Spenderorgane im Vergleich zu lebendgespendeten Transplantaten, lässt sich anhand der Expressionslevel der inflammatorischen Proteine beweisen (153). Die Aktivierung des inflammatorischen Systems besteht aus verschiedenen kaskadenartigen Reaktionen auf molekularer und zellulärer Ebene, durch erhöhte Expression inflammatorischer Gene, aber auch durch Reaktionsabfolgen in der Peripherie. Eine Schädigung des zentralen Nervensystems, in unserem Fall die Induktion des Hirntodes, ruft eine direkte und massive Expression inflammatorischer Induktoren, z.B. den Histokompatibilitätsantigenen (HLA I und II), aber auch von Adhäsionsmolekülen, Zytokinen und anderer Akut-PhasenProteine in den peripheren Organen hervor. Die erhöhte Expression von Haupthistokompatibilitätsantigenen HLA I und HLA II wird als direkte Verstärkung der Immunogenität gesehen (71, 125). Die Einleitung 13 kortikale Nekrose führt zu einer Aktivierung entzündungsspezifischer Zelltypen mit Freisetzung unterschiedlicher proinflammatorischer Zytokine (PIC). Das zentrale Ereignis triggert also prompte, unspezifische Entzündungsvorgänge, die später wiederum die Intensität, sowie das Eintreten spezifischer immunologischer Reaktion, im Sinne einer Abstoßungsreaktion, beschleunigen können (199). Tierstudien zeigen ein frisch transplantiertes und perfundiertes Transplantat eines hirntoten Spenders immunhistologisch schon nach drei Stunden von neutrophilen Abwehrzellen infiltriert, kurz gefolgt von größeren Zahlen an Makrophagen und TZellen (96, 154), signifikant stärker als im Vergleich zu lebendgespendeten Transplantaten. Im Folgenden soll auf einzelne Komponenten des Entzündungssystems und ihre Auswirkungen auf die Transplantatniere während der unterschiedlichen Reaktionsschritte eingegangen werden. Einzelne Komponenten der Entzündungsreaktion 1.7.1 Zytokine Die Familie der Zytokine, als primärer Überbegriff für eine große, in den sechziger Jahren entdeckte Gruppe endogener Proteine, ist in ihren zahlreichen Funktionen bei entzündlichen, angiogenetischen, antitumorösen und wachstumsbeeinflussenden Prozessen schwer zu überschauen und zu klassifizieren. Zusammenfassungen von Xing (216) und Raeburn (159) zeigen die Funktion der Zytokine als Mediator zwischen somatischen und myeoloischen Zellen. Neben essentiellen positiven Effekten der Zytokine auf Zellen und Organismus, sowie ihrer Funktion als iatrogen eingesetzte Therapeutika, sind auch krankhafte Zustände durch zu hohe oder zu niedrige Zytokinkonzentrationen im Körper bekannt (104, 149). Eine veränderte Zytokinproduktion nach Hirntod wurde bereits intensiv untersucht und beschrieben (66, 102, 107). Lopau et al. (107) konnten zum Beispiel nach Hirntodinduktion einen Anstieg von IL6, TNF-α, als auch von IL-2 als Marker für die immunogene Aktivierung aufzeigen. Amado et al (7) zeigten Il-6 im Vergleich zu der Zytokin-Konzentration von Kontrollgruppen signifikant erhöht. Die erhöhte Konzentration von IL-6 läßt sich durch die im Hirn entstehende Ischämie, bzw. durch die exzessive Katecholaminausschüttung erklären (158, 197, 217). 14 Einleitung 1.7.1.1 Interleukin – 6 Interleukin-6 stellt den bekanntesten Vertreter der proinflammatorischen Zytokine dar. Es ist Initiator in der Immunreaktion des Körpers und ist entscheidend bei der Umwandlung von BLymphozyten in Plasmazellen (108), sowie an der vermehrten Synthese von inflammatorischen Proteinen in der Leber beteiligt. Il-6 ist einer der stärksten Vermittler der Akuten-Phase-Proteins-Synthese und induziert eine grosse Reihe systemischer Folgeeffekte, endokriner und metabolischer Veränderungen. Im Gegenzug zu seinen proinflammatorischen Eigenschaften inhibiert es jedoch die Aktivität von Il-1, was seine Funktion als dualer Effektor kennzeichnet (140). Il-6 ist bei Patienten mit akutem Gehirnschaden sowohl in der zerebrospinaler Flüssigkeit - durch intrathekale Ausschüttung - , als auch im Serum und im geschädigten Gewebe selber stark erhöht (13, 102). 1.7.1.2 Chemokine In den letzten Jahren konnten verschiedene chemotaktische Zytokine, die Chemokine, als spezifische Lockboten für die Infiltration von Entzündungszellen während einer akuten Abstoßung verantwortlich gemacht werden (94, 165). Der Prozess der Leukozytendiapedese von der zirkulatorischen Seite zu der des betroffenen Gewebes beinhaltet eine grosse Anzahl von Interaktionen zwischen löslichen Faktoren und Oberflächenmolekülen (25). Die meisten der heutzutage bekannten Chemokine sind Mitglieder zweier Subfamilien, den CC-Chemokinen und den CxC-Chemokinen, unterteilt durch die Präsenz oder Absenz einer Aminosäure zwischen den ersten zwei Cysteinmolekülen. CC-Chemokine sind für die Aktivierung von TZellen und Makrophagen/ Monozyten zuständig, CxC-Chemokine wirken eher auf Granulozyten (95). Chemokine werden von unterschiedlichen Zellarten, einschließlich der Leukozyten produziert. Histomorphologisch korrelierend, kommt es es im Zusammenhang mit dem Hirntod zu einer Entzündungsreaktion. Vielfach wurde gezeigt, dass der Hirntod selbst durch die entstehende kortikale Nekrose mit Aktivierung einer Vielzahl von Zelltypen zu einer Freisetzung proinflammatorischer Cytokine, so auch den Chemokinen, führt. Ihre Konzentration findet sich bei Abstoßungsreaktionen transplantierter Nieren erhöht. So scheinen endotheliale Nierenzellen, tubulointerstitielle Epithelien, mesangiale Zellen und Fibroblasten Chemokine konstitutiv oder als Antwort auf zerebralen Schaden zu exprimieren (175). Interessant erscheint ihre signifikant unterschiedlich erhöhte Konzentration in Vergleich zwischen Lebend- und Totspendernieren, was die These einer hintodinduktiv erhöhten Genexpression nahe legt. 15 Einleitung 1.7.1.3 Interleukin-1 und Interleukin-1-rezeptor-Antagonist Eines der potentesten proinflammatorischen Vermittler ist das früh agierende Zytokin Il-1, ein Schlüsselenzym bei der Mediation interzellulärer Kaskaden. Die IL-1 Familie besteht aus drei Polypeptiden, den Proteinen Il-1alpha und Il-1beta, welche eine große Reihe biologischer transduktorischer Signale übermitteln und dem Il-1-rezeptor-Antagonisten (114). Il-1RA ist in der Lage, die von Il-1 induzierte T-Zell-Proliferation und die Aktivierung von B-Zellen zu unterbinden. Il-1-RA reguliert also die Aktivität von Il-1-alpha und beta als gegenläufiger Ligand (192). Il-1 wird von aktivierten MonozytenMakrophagen produziert und bindet an Zell-Oberflächen –Rezeptoren. Interessanterweise zeigt sich nach Hirntod diese Aktivierungsachse im Vergleich zu Kontrollgruppen stark angeregt. In den Nieren hirntoter Spender werden deutlich erhöhte Konzentrationen an Il-1 gemessen. Dies erklärt sich durch die funktionelle Beteiligung von Il-1 an B- und T-Zell-Aktivierung, Fieber-Reaktionen und der Initiierung an der Akute-phase-Protein-Synthese, alles Prozesse, die durch den Hirntod aktiviert werden (37). Vorrangegangene Studien zeigen an postmortal gespendeten Nieren eine ebenfalls erhöhte Konzentration von Il-1-RA, was eine gegenläufige Kompensation des Körpers der erhöhten immunogenen Aktivierung präsentiert. Fraglich scheint eine Korrelation der Abstoßungshäufigkeit mit den Expressionsleveln von Il-1-RA. 1.7.2 Selektine – Vertreter der Adhäsionsmoleküle Die zentrale Schädigung bei Hirntod führt einerseits zur prompten Katecholaminsekretion mit resultierender peripherer Vasokonstriktion und Gewebsischämie, andererseits zu einer Ausschüttung von Hormonen und entzündlichen Mediatoren, die die Organfunktion direkt beeinflussen können. Eine Gruppe dieser hochregulierten Mediatoren sind die adhäsiven Akute-Phase-Moleküle. Wichtige Vertreter dieser Gruppe sind die Selektine (61). Im März 1989 wurde die Entdeckung der primären Sequenzen von drei unabhängig studierten Zelloberflächen-Glykoproteinen publiziert, die man auf Endothel, Thrombozyten und Leukozyten gefunden hatte (20). Jedes Molekül beinhaltet eine NH2-terminale Lektin-ähnliche Domäne, einen EGFrepeat und eine diskrete Anzahl von Modulen, ähnlich zu solchen, die man in Komplement-bindenden Proteinen gefunden hatte. Die Kollektion dieser Domänen wurde zum Markenzeichen einer neuen Molekülfamilie, die heute als Selektine bekannt sind (115). Die Bezeichnung Selektin wurde ursprünglich vorgeschlagen um die Präsenz der Lektin-domäne hervorzuheben, aber auch, um die selektive Expression und Funktion der Moleküle hervorzuheben. Jedes Familienmitglied wurde entspre- Einleitung 16 chend seines erst-detektierten Zelltyps klassifiziert: E-selektin (Endothel), P-Selektin (Platelets) und L-Selektin (Lymphozyten). Alle drei Selektinarten agieren in Zusammenarbeit mit anderen Zelladhäsions-Molekülen, wie z.B. ICAM-1 (Intercellular Cell Adhesion Molecule), VCAM-1 (Vascular) oder PECAM (Platelet-Endothelial), die aufgrund ihrer molekularen Struktur der ImmunglobulinSuperfamilie zugehörig sind. Sie alle bewerkstelligen Interaktionen zwischen Leukozyten, Plättchen und endothelialen Zellen (98). Durch die Selektine wird die Infiltration von Entzündungszellen in das Gewebe ermöglicht. Ischämie, Hypoxie und Azidose führen zu einer progressiven Verlangsamung des Leukozytenverkehrs entlang der Gefäßwände (117, 138, 139). Selektine reagieren dann mit Liganden zirkulierender polymorphonukleärer Leukozyten (PMN) und bewirken den „Rolling-Effekt“ derselben entlang der Gefässwände, einem Prozess, der der festen Haftung und Diapedese in das entzündliche Gewebe vorangeht (205). Eine erhöhte Induktion der De-Novo-Synthese dieser Adhäsionsmoleküle bewirken unterschiedliche Zytokine, wie z.B. Il-1, INF gamma oder TNF-alpha, welche wiederum, wie oben genannt, nach Eintritt des Hirntodes massiv ausgeschüttet werden. Vergleiche zwischen hirntoten und lebendverwandten Spendern zeigen einen signifikanten Unterschied in der Selektinexpression- und konzentration (72). Studien zeigen, dass die Selektinexpression in hirntoten Spendern schon vor der Organentnahme deutlich erhöht ist. Sie erreicht ihr Maximum vier bis sechs Stunden nach Hirntodinduktion (61, 92). Im Gegensatz dazu, zeigt sich die Selektinexpression und korrelierbare Leukozyteninfiltration in lebendgespendeten Nieren nach Reperfusion gleich null (93). 1.7.3 Makrophagen Die Makrophagen sind eine sehr komplexe und heterogene Zellpopulation. Makrophagen können in großer Konzentration bei der akuten und chronischen Abstoßung nach Nierentransplantation detektiert werden, sowohl bei der Antikörper-vermittelten und vaskulären Abstossung, als auch bei der akuten zellulären Abstossungsreaktion, alles Ereignisse, die bei Nieren von hirntoten Spendern signifikant häufiger vorkommen, als bei lebendgespendeten Nieren. Monozyten-Makrophagen sind antigen-präsentierende Zellen, die Interleukine produzieren, T-ZellAktivierung durch Kostimulationen bewerkstelligen und die Entwicklung von Th1-CD4+ Zellen fördern, welche eine dominante Rolle im Abstoßungsprozess spielen (220). Makrophagen verhalten sich als phagozytierende Zellen, die nekrotische Zellkomponenten bei tubulärer Nekrose nach einer Transplantation abbauen. Im aktivierten Zustand schütten sie selber Chemokine aus, z.B. Il-8, MCP-1 oder MIP1a, zudem sind sie für ungünstige Prozesse der Postreperfusionsperiode verantwortlich, indem sie Sauerstoffradikale und Lipidperoxidation induzieren (47). Zu nennen Einleitung 17 ist trotzdem auch die von Monozyten-Makrophagen gegenzügliche Exprimierung antiinflammatorischer Moleküle. Dazu gehören Il-1RA, Il-10, TGF-b und Lipoxine (220). Die Anzahl der Makrophagen zeigt sich nach Hirntodinduktion im peripheren Gewebe stark erhöht. Ihre Rekrutierung und Aktivierung wird mitunter durch C-C-Chemokine hervorgerufen. Nach Rolling und Adhäsion präsentieren die in das Transplantat eingewanderten Leukozyten-MakrophagenPopulationen andere Klassen von adhäsiven Molekülen (87) und aktivieren proinflammatorische Lymphokine (155), sowie das Komplementsystem (189, 210). Somit wirken sie als Trigger der inflammatorischen Reaktionskaskade nach Hirntodinduktion in der Peripherie. Nach Eintritt des Hirntodes infiltrieren sie entzündetes Gewebe und verursachen dadurch Gewebsschaden (140). Dies zeigt sich in der Niere mit einer halbmondförmigen Glomerulonephritis und progredienter tubulärer Atrophie (158). Diese ausgeprägte Makrophageninfiltration zeigt sich im Vergleich, bei lebendgespendeten Nieren, nicht (156). 1.7.4 Die Heat-Shock-Proteine - Hämoxygenase-1 Stressoren führen zur Denaturierung von Proteinen und zu Fehlfaltung in der Proteinstrukur (40). Die Zellantwort auf Stress ist vielfältig und beinhaltet unter anderem die Aktivierung der Transkription und die Induktion von unterschiedlichen Genen (188). Ein ziemlich genau bekannter Mechanismus (4) der Stressantwort der Zelle ist die Induktion der Heat-shock-Familie, der Heat-Shock-Proteine (HSP) (18, 193, 208). Heat-Shock-Proteine gehören zu einer Multigenfamilie und bestehen aus verschiedenen, strukturell nicht miteinander verwandten Gruppen von Proteinen. Neben dem Hitzeschock gehören zellschädigende Faktoren, wie schwere intrazelluläre Azidose (pH< 6.7), Entleerung der ATP-Reserven (<65%) (18, 171), Schwermetalle, Aminosäureananloga, Entzündungen (216), ischämiescher und oxidativer Stress (89) zu den Stressoren, die die Expression von HSP´s triggern. In unserer Arbeit bezieht sich die HSP-Expression als Antwort auf den Hirntod als systemischer „Stressfaktor“. Charakteristischerweise wird nach einem starken Stressreiz durch die Phosphorylierung von Initiationsfaktoren die allgemeine Proteinsynthese inhibiert (219), im Gegensatz dazu aber die HSP-Gene besonders stark exprimiert und die zugehörigen Proteine translatiert. Auf diese Weise können innerhalb von Minuten nach einem Stressereignis die Heat-Shock-Proteine bis zu 25% aller intrazellulären Proteine ausmachen und in den wichtigen Zellkompartimenten Proteine vor Denaturierung schützen (18). Einige cytosolische HSP´s translozieren sogar in den Nukleolus und verhindern dort die Auslösung von stressinduzierten Apoptosemechanismen (116). 18 Einleitung Von den 3 Isoformen HO-1, HO-2 und HO-3 ist HO-1 die am weitesten verbreitete Form. HO-1, die induktive Isoform entspricht HSP32 und wird durch diverse Stressoren induziert (18). HSP32 mediiert genau wie die NO-Synthase guanylatcyclaseabhängig eine Thrombozyteninhibition und eine Vasodilatation. Endogenes oder exogen zugeführtes NO führt zudem zu einem bis zu 6-fachen Anstieg der HSP32-Genexpression, was auf eine synergistische Wirkungsweise schließen lässt. Für die Induktion der HSP32-Genexpression sind vor allem Heat-Shock-Faktor 1 (HSF1) und der durch Hypoxie induzierte TF-1 (Transkriptionsfaktor 1) von Bedeutung. HSP32-Konzentrationen zeigen sich bereits in Studienergebnissen vorangegangener Arbeiten nach Nierentransplantation hirntoter Spender signifikant erhöht (172). Der Hirntod scheint als Stressinduktion eine deutliche Expressionserhöhung zu bewirken. Die HO-1 ist ein Heat-Shock-Protein, das antioxidative, antiinflammatorische und antiapoptotische Funktionen aufweist und die Regulation des Gefäßtonus beeinflussen kann. Diese klaren zytoprotektiven Fähigkeiten konnten in verschiedenen experimentellen Modellen der Organtransplantationsforschung dargestellt werden (18, 101). 1.8 Ansatzpunkt dieser Arbeit Der hirntote Spender stellt die größte Quelle für Nierentransplantationen dar (63). Dies begründet die intensive Forschung an diesem Spenderkollektiv, denn trotz vielfältiger Verbesserungen der Immunsuppression bleibt die Erfolgsrate der Kurz- und Langzeitorganfunktion signifikant unter der von sowohl lebend-verwandten, als auch lebend- unverwandten Spendern (194). Basierend auf bisherigen Forschungsarbeiten scheint es erwiesen, dass die komplexe Reaktionenskaskade des gesamten Organismus als Antwort auf den Hirntod bereits vor der Explantation, durch verzweigte pathophysiologische Prozesse, eingeleitet wird. Die erhöhte Frequenz schwerer akuter und chronischer Abstoßungsreaktionen scheint ihren Ursprung auf molekularer Ebene zu haben. Die Erkennung, Beeinflussung und Kontrolle der frühen Faktoren, die den Verlauf nach Organtransplantation beeinflussen können, ist ein vordringliches Ziel in der Weiterentwicklung der Transplantationsmedizin. Das übergeordnete Bestreben dieses Projektes besteht daher in der weiteren Aufklärung der Auswirkung des Hirntodes auf die Niere, da die komplexen Mechanismen, die nach dem massiven Hirnschaden ausgelöst werden, noch immer ungenau verstanden sind und somit inadäqut behandelt werden können. Der Ansatz der Überexpression/ bzw. Abschaltung zahlreicher Gene und Proteine nach Hirntodinduktion wurde in spezifischen Bereichen schon erforscht und beschrieben. Die Resultate weisen 19 Einleitung bereits auf den relevanten Zusammenhang zwischen molekularer und funktioneller Ebene hin (19, 217, 218). Die von uns benutzte standardisierte DNA-Microarray-Technologie bietet den Vorteil der simultanen Analyse von zahlreichen Genen mit dem Potential eine Vielzahl von Veränderungen der Genexpression in der Niere zu detektieren. Frühere Forschungsmethoden basierten hauptsächlich auf Zuhilfenahme von PCR oder Gelelektrophorese bei einzelnen selektierten Genen. Durch hochdichte Genchips ist die Untersuchung der Genexpression für einen Großteil des Gesamtgenoms gleichzeitig durchzuführen und diese im selben Schritt quantitativ zu erfassen. Das Wissen über den Hirntod als elementares Ereignis und als antigenunabhängiger Risikofaktor in der Transplantationsmedizin, lässt sich anhand dieser Methoden auf seine molekulare Ebene ausweiten. Bereits dargestellte Erkenntnisse über morphologische oder zelluläre Veränderungen peritransplantativ lassen sich nun auf Genebene erklären und verifizieren. Aufschluss über Gene und Proteine unterschiedlicher Funktionsgruppen, die bisher nicht im Zusammenhang mit dem Hirntod diskutiert wurden, könnte gewonnen werden. Nach Beweis ihrer veränderten Expression könnte dann eine entsprechende iatrogene Stimulation oder Inhibition Folgeschäden reduzieren. Letztlich können so neuartige therapeutische Ansatzpunkte für die Transplantationsmedizin und die Organerhaltung von hirntoten Spendern geschaffen werden. In der aktuellen Tranplantationsmedizin sind peritransplantative Abläufe und Transplantationsresultate schon lange durch Intensivmangagement und moderne Pharmaka verbessert worden. Zukunftsgerichtete Arbeiten zeigen jedoch bereits die Möglichkeiten, auf genetischer Ebene anzugreifen, um so prophylaktisch bestimmte pathologische Reaktionen im Ursprung zu stoppen, oder protektive körpereigene Mechanismen zu fördern (152, 217) Der experimentelle Teil dieser Arbeits lässt sich wie folgt untergliedern: 1. Tier-Modell des Hirntodes und Organentnahme (Niere) an der Ratte 2. RNA-Extraktion aus dem renalen Gewebe 3. Genchip-Hybridisierung und Auswertung der Genexpression 4. Quantitative PCR zur Verifizierung der Gensexpression Material und Methoden 2. 20 Material und Methoden Die Durchführung der Tierexperimente wurde den Vorschriften des Deutschen Tierschutzgesetzes und der „European Convention on Animal Care“ untergeordnet und durch die Veterinärbehörde des Regierungspräsidiums Freiburg mit der Registriernummer G-01/73 genehmigt 2.1 Tiermodell Als Versuchstierte dienten männliche Wistar Ratten mit einem Körpergewicht von 300-400g. Die Tiere wurden paarweise in Plexiglaskäfigen gehalten, die Fütterung erfolgte mit handelsüblicher Trockennahrung, sowie ständigem Zugang zu Frischwasser. Für die Studie wurde ein von Pratschke et al. (135, 157) etabliertes Tiermodell angewandt. Dies ist ein kontrollierter Versuchsaufbau, auf dessen Basis die Herz-Kreislauffunktion der Ratten bei Isothermie über mehrere Stunden aufrechterhalten werden kann, nachdem ein graduierter, normotoner Hirntod induziert wurde. Nachfolgend konnten entsprechende Organe zur weiteren Untersuchung entnommen werden. Versuchsgruppen Um die Ergebnisse als alleinige Folgen des Hirntods definieren zu können, benötigt man den Vergleich zu einer Kontrollgruppe. Die Ratten wurden den zwei Versuchsgruppen randomisiert zu je 5 Tieren zugeteilt. Material, Zeitablauf und Operationsvorgang wurden exakt gleichgestellt. Kontroll-Gruppe (n=5) Multiorganentnahme nach Narkoseinduktion und einer Beobachtungsperiode von sechs Stunden ohne Hirntodinduktion. Hirntod-Gruppe (n=5) Multiorganentnahme nach Narkoseinduktion und Auslösen des Hirntodes bei einer Beobachtungsperiode von sechs Stunden nach Hirntodinduktion. 21 Material und Methoden HIRNTOD-GRUPPE Narkoseinleitung + Präparation < ▲ ca.10 min Hirntodinduktion >< Messung MAD HF und BG nach Katheterimplantation 5min >< 360min >< ▲ ▲ ▲ ▲ ▲▲ ▲ ▲ ▲ ▲ minütliche Messung alle 60min Messung MAD, HF, pH, pO2, CO2 von MAD und HF KONTROLL-GRUPPE Narloseinleitung + Präparation < ▲ ca.10 min >< ▲ ca.10min ▲ > MultiorganEntnahme 6 h Liegezeit 360min ▲ ▲ Messung MAD HF und BG nach Katheterimplantation MultiorganEntnahme 6 h Liegezeit ▲ >< ▲ ca.10min ▲ > Alle 60min Messung MAD, HF, pH, pO2, CO2 Abbildung 1: Darstellung des Versuchsablaufes bei der Kontroll- und Hirntodgruppe in der Zeitachse. Die dargestellten Pfeile (▲) markieren die Messzeitpunkte von mittlerem arteriellen Druck (MAD), Herzfrequenz (HF) und Blutgasanalyse (BG). 2.2 Anästhesie und Monitoring Zur Narkoseeinleitung werden die Ratten in einer Kleintierbeatmungskammer ( Klaus Thoma GmbH, Freiburg ) mit 4.0 Vol% Isofluran und einem Flow von 4 l Sauerstoff (O2) /min bis zum Verlust des Stellreflexes narkotisiert. Anschließend wird die Narkose mit einer Kleintierbeatmungsmaske mit 1.31.8 Vol% Isofluran, 30 Vol% Lachgas und 60 Vol% Sauerstoff fortgesetzt. Die Tiere werden in Rückenlage auf einer Wärmeplatte (Temperaturmesser von KE medizinisch technischer Gerätebau Typ OP-T, Pfaffing) platziert und die Körpertemperatur mittels eines rektalen Temperaturfühlers zwischen 35º C und 37º C automatisch konstant gehalten und protokolliert. Zur Prämedikation werden 5 µg Atropin (Atropinsulfat, Braun, Melsungen) subkutan injiziert. Nach Rasur der Bauchdecke und des Halses wird ein querer Kragenschnitt durchgeführt, die Halsmuskulatur stumpf lateralisiert und die Trachea freigelegt. Diese wird in der Vorderwand halbseitig inzidiert und ein Tubus zur kontrollierten Beatmung in die Luftröhre eingeführt und fixiert. Die Beatmungsmaschine (Kleintierbeat- Material und Methoden 22 mungspumpe Typ 50ml, RHG Rhemalabortechnik, Hofheim) hält das Atemzugvolumen um 4.5 - 5ml und die Atemfrequenz auf 35 - 40 min-1 volumenkontrolliert konstant. Der inspiratorische Sauerstoffgehalt wird dabei im Bereich von 65 Vol% gehalten (Oxygen Monitor 810, Kontron medical). Die genaue Justierung der Beatmungspumpe, also die adäquate Einstellung von Atemzugvolumen und Atemfrequenz, erfolgt in Adaption an stündlich durchgeführte Blutgasanalysen (Radiometer GmbH, Willich, Copenhagen, ABL 725). Angestrebte Zielparameter sind dabei in allen Versuchsgruppen ein pH-Wert von 7.4 ± 0.1, ein pO2 von 110 – 250 mmHg und ein pCO2 von 25 - 50 mmHg. Diese kontinuierliche Überwachung der Kreislaufparameter kann nach erfolgter Kanülierung der rechten Arteria carotis communis und der linken Vena jugularis mit einem Polyethylenkatheter (Fine-Bore Polyethylen Tubing, ID 0.5mm; 1,0mm OD Sims Portex Ltd, Uk) erfolgen. Der venöse Katheter wurde mithilfe eines DruckSpülsystem (DPS Monitoring-Set, Medex Medical Ltd, Lancshire, Great Britain) mit isotoner Kochsalzlösung (ca. 0.5ml/h, 0.9% NaCl, Braun Schiwa, Glandorf) kontinuierlich gespült und somit offengehalten. Die Infusion dient ebenfalls als Volumensubstitution, ihre Menge pro Zeit wird mit Hilfe des Perfusors an den MAD adaptiert. 2.3 Parameter zur Überwachung der Kreislaufstabilität Zur Bestimmung der Makrozirkulation dienen die Parameter mittlerer arterieller Druck (MAD) und die Herzfrequenz (HF). Beide werden kontinuierlich gemessen (Sirecust 403 P, Siemens), zum Zeitpunkt der Katheterimplantation und nachfolgend stündlich protokolliert. Als Ausschlusskriterien gelten Tiere, bei denen der MAD im Verlauf des Versuchs länger als fünf Minuten unter 50mmHg liegt, sowie der biologische Tod der Tiere vor Ablauf des jeweiligen Untersuchungszeitraumes. 2.4 Schrittweise Hirntodinduktion Die tracheotomierten und intubierten Tiere der Hirntodgruppe werden in Bauchlage umgelagert und mit Klebestreifen fixiert, um Lageveränderungen durch tonisch-klonische Krämpfe während der Hirntodinduktion zu vermeiden. Am dorsoparietalen Skalpgebiet werden Fell, Haut und Muskeln entfernt. Mit Hilfe einer Kanülenspitze werden Periost und Schädeldecke manuell vorgebohrt. An diesem Ansatz wird mit einem Dremel (Dremel, Multipro Cordless) 0.2 – 0.5 mm lateral der sagittalen Naht ein 2 mm großes Loch gebohrt. Ein Forgarty-Katheter (Edward Lifescience, Größe 4F, Länge 80 cm) Material und Methoden 23 wird dann durch das Loch intrakraniell eingeführt und fixiert. Zur Erhöhung des Hirndrucks erfolgt nun die graduelle Inflation des Ballons mit Kochsalzlösung. In dieser Phase werden die Kreislaufparameter minütlich protokolliert. Das Erreichen der Hirntodinduktion und der Einklemmung des Hirnstamms wird durch den Ausfall der Hirnstammreflexe, Apnoe über eine Minute nach Ausschalten der Beatmungsmaschine und bilateral maximal dilatierte und fixierte Mydriasis definiert. Das durchschnittliche Ballonvolumen zur Auslösung des Hirntods liegt bei 0.3 - 0.4 ml NaCl über einen Zeitraum von fünf Minuten, analog des von Pratschke et al. beschriebenen Modells (157). Der Ballon bleibt während der nun folgenden sechsstündigen Adaptionszeit expandiert. In Fällen, in denen die Tiere hypotensiv werden und ein zirkulatorischer Kollaps droht, wird das intrakranielle Ballonvolumen um 10 ul NaCl pro Minute reduziert und die venöse Volumenzufuhr erhöht. Als Folge der Dekompression erreichen die Tiere wieder Normotension. Abbildung 2: Beatmete Ratte nach Hirntodinduktion mit platziertem Forgarty-Katheter und maximal dilatierten Pupillen. 2.5 Multiorganentnahme Die Multiorganentnahme erfolgt bei der Kontroll- und Hirntodgruppe nach sechsstündiger Beobachtung. Dazu werden die Tiere wieder in Rückenlage positioniert. Es folgt ein Oberbauchquer -und Medianschnitt mit medianer Sternotomie. Die Schnittränder werden mit Hilfe von Fixierklemmen nach außen Material und Methoden 24 offengehalten. Nach Präparation der infrarenalen Aorta abdominalis wird diese nach kaudal und kranial ligiert, temporär kranial mit einem Gefäßclip abgeklemmt und dazwischen quer eröffnet. Nach Einführung eines Katheters infrarenal in die abdominelle Aorta (Vasofix Braunüle, Venenverweilkanüle, 14G, 2.1 x 50mm, Fa.Braun) werden 2 ml Blut entnommen. Nach der Durchtrennung der Vena cava inferior werden die Organe durch Infusion einer gekühlten isotonischen NaCl Lösung aortal blutleer gespült. Danach wurden nacheinander erst das Pankreas, dann beide Nieren, Herz und Teile der Leber entfernt, steril in verschlossenen Eppendorfröhren aserviert und sofort in flüssigem Stickstoff schockgefroren. Die Lagerung erfolgt in -80° C Kühlschränken. 2.6 RNA-Extraktion Die Isolierung von Gesamt-RNA aus kleinen Mengen von Nierengewebe beinhaltet verschiedene Reaktionsschritte, die mithilfe von RNeasy Kits, Qiagen (Hilden, Deutschland) erfolgen, und sich an das vorgegebene Aufarbeitungsprotokoll (Trizol/Qiagen, RNA Miniprep) halten. RNA kann unter nativen Bedingungen leicht enzymatisch degradiert oder katalytisch hydrolisiert werden. Die RNA-Stabilisierung in biologischen Ausgangsmaterialien ist eine essentielle Voraussetzung, um die Genexpression zuverlässig zu analysieren, da es sonst unmittelbar nach Probenentnahme durch spezifische und unspezifische RNA-Abbauprozesse und durch die Induktion der Transkription zu Veränderungen im Genexpressionsmuster kommt. Die Qualität der von uns gewonnen GesamtRNA, und damit auch unseres Analyseergebnisses, wird dabei bereits durch die ersten Schritte bei der Probenentnahme mitbeeinflusst. Da es bei den Versuchen mit anschließender Genchipanalyse und PCR gerade darum geht, zuverlässige quantitative Genexpressions-Analysedaten zu gewinnen, werden die Organproben bis zur Durchmischung mit dem Trizolreagenz ständig mit Flüssigstickstoff gekühlt und damit in dem schon direkt nach der Organentnahme schockgefrosteten Zustand belassen. Die RNeasy Methode isoliert RNA-Moleküle mit einer Länge von über 200 Nukleotiden. Das Verfahren trennt RNA-Spezies mit weniger als 200 Nukleotiden Länge (z.B. 5.8SrRNA, 5srRNA und tRNAs, die zusammen 15 - 20% der Gesamt-RNA ausmachen) selektiv ab, so dass es zu einer Anreicherung der mRNA kommt. Das spezielle Hochsalz-Puffersystem ermöglicht die Bindung von bis zu 100µg RNA an die RNeasy Silicagel-Membran, während die übrigen Substanzen effizient ausgewaschen und somit abgetrennt werden. Die isolierte RNA wird dann mit RNase-freiem Wasser eluiert. Material und Methoden 2.6.1 25 RNA-Miniprep-Protokoll Das bei -80° C gefrorene Nierengewebe wird als erstes unter ständiger Kühlung mit flüssigem Stickstoff in einem Metallmörser pulverisiert. Ein Auftauen des Materials muss unbedingt vermieden werden. Bis zu 40 mg werden in einer Kugelmühle (Kugelmühle MM2000, Retsch, Haan, Germany) von 8 mm Mahlkugeln zweimal eine Minute lang zermahlen und homogenisiert. Es folgt die zweimalige Extraktion mit einem TRIZOL® Reagent (GibcoBRL manufactured for LIFE Technologies), Chloroform (Isomayl-Alkohol 24:1, Amresco®, Solon, Ohio, USA) und 2Mercaptoethanol (Merck, Darmstadt). Das benutzte TRIZOL® Reagent ist ein gebrauchsfertiges Reagenz zur Isolierung von Gesamt-RNA aus Zellen und Geweben. Es setzt sich zusammen aus Phenol und Guanidin. Das ebenso hinzu gegebene Chloroform-Isomaylalkoholgemisch hilft bei der Denaturierung und Phasierung und teilt die nach Zentrifugierung entstandene Lösung in eine wässrige und organische Phase auf, wobei die RNA in der wässrigen Phase zurückbleibt. 2-Mercaptoethanol fällt die RNA aus und verfügt über eine antioxidante Wirkung. Nach Mischung des Homogenats mit den Reagenzien erhalten wir nach der Zentrifugation einen farblosen Überstand. Dieser farblose Überstand wird in ein frisches Röhrchen pipettiert und ein gleiches Volumen von Ethanol (70%, J.T. Baker, Deventer, Holland, 8006) hinzugefügt. Ethanol stellt die Bindebedingungen für die Säule ein, an der die RNA später haften soll. Das Reagenz wird gemischt und sofort auf RNeasy Säulchen (Qiagen, RNeasy –kit, Hilden, Deutschland, Cat.nr. 74104) pipettiert und für 15 sec mit 10.000 rpm zentrifugiert. Der Durchlauf wird verworfen, die RNA haftet jetzt an der Säule. Falls das vorherige Volumen zu groß ist, wird die Säule ein zweites Mal geladen. Im nächsten Schritt werden 350 µl RW1 (Waschpuffer, Qiagen, RNeasy-kit, Hilden) aufgetragen, um die übrigen Substanzen auszuwaschen. Nach 15 Sekunden Zentrifugation bei 10.000 rpm wird der Durchlauf verworfen. Zur Verdauung von Resten organischer DNA werden danach 10 µl DNase1 (RNase-frei) mit 70 µl RDD gemischt und auf die Säulenmembran pipettiert. Die Röhrchen werden jetzt 15 min bei Raumtemperatur (20 - 30° C) inkubiert. Danach wird die Säulenmatrix erneut mit 350 µl RW1 gewaschen, zentrifugiert und die Säulchen danach in frische Sammelröhrchen gesteckt. Im weiteren Schritt werden 500 µl RPE (Waschpuffer, Qiagen, RNeasy-kit, Hilden) aufpipettiert, zentrifugiert und der Durchlauf verworfen. Dieser Waschschritt wird wiederholt und die Säulenmembran danach noch mal 1min bei 10.000 rpm zentrifugiert und somit getrocknet. Es folgt das Herauslösen der RNA in destilliertes RNase-freies Wasser. Material und Methoden 2.6.2 26 Quantitative Bestimmung Um die Konzentration im Eluat zu bestimmen, benötigte man eine 1:100 Verdünnung. Dafür wurde 1 µl RNA-wasser mit 99 µl destilliertem H2O gemischt. Die einzelnen Konzentrationen konnten jetzt quantitativ im Photometer (Pharmacia Biotech, Ultrospec 2000, Modell 80-2106-06, Cambridge, UK) bei 260 nm bestimmt werden Lagen die Konzentrationen unter 0.2 µg / µl musste das Prozedere wiederholt werden, da die Menge an RNA ungenügend war. 2.6.3 Qualitative Bestimmung Zur Qualitäts- und Intaktheitsüberprüfung der extrahierten RNA erfolgte eine AgaroseGelelektrophorese mit anschließender Ethidiumbromidimprägnation. Hierbei sollte sich zeigen, ob die Gesamt-RNA frei von Kontamination mit genomischer DNA war, und ferner, ob die RNA intakt war. Ethidiumbromid wird zum Anfärben von Nukleinsäure bei der Gelelektrophorese verwendet. Einzelne Ethidiumbromidmoleküle interkalieren dabei zwischen die Basen der RNA. Dadurch verändert sich das Anregungsspektrum von Ethidiumbromid und die Fluoreszenz der Substanz wird so durch ultraviolettes Licht stark erhöht. Die Lichtintensität ist dabei proportional zur vorliegenden RNAKonzentration sowie zur Länge der Nukleinsäure. Entscheidend für die Integrität der RNA waren dabei die Fragmente, die auf die 18S und 28S RNA zurückzuführen sind. Die Intensitäten der 18S-Banden und 28S RNA-Banden sollten dabei ein Verhältnis von 1:1.5 - 2.5 zeigen (siehe Abbildung 3). Zur Vorbereitung des Gels wurde TBE (Trisborat-EDTA, pH 7.5, 10x) aus Tris, EDTA und Bovicacid als Ladepuffer hergestellt. Zur Herstellung eines 10x10 cm 1% igen Agarosegels benötigte man 80ml TBE und 0.8 g Agarose, (Invitrogen™, Life Technologie). Das Gemisch wurde in der Mikrowelle (Siemens) einige Minuten erhitzt und aufgekocht, bis sich das Pulver vollständig im TBE gelöst hatte. Es folgte eine Abkühlung auf 60° C bevor man dem Gel 4 µl Ethidiumbromid (EtBr, Amresco) zur Anfärbung der Banden zufügte. Das Gel wurde auf die vorbereiteten Platten gegossen und ein Kamm mit 40 µl Taschenvolumen eingesteckt. Das erkaltete Gel wird in die mit 1-fach TAE-Puffer gefüllte Laufkammer gelegt und mit der gewonnen RNA befüllt. Die elektrophoretische Auftrennung der Nukleinsäuren erfolgt bei einer Spannung von 120 V und einer Stromstärke von 100 mA (P25 Powerpack, Biometra, Göttingen). Die Wanderung der RNA entsprechend ihrer Ladung von Minus nach Plus dauert ungefähr 60-70 Minuten. Als Kontrolle dient ein 1kb DNA- Ladepuffer (MBI Fermentas, 0.1 mgDNA/ ml, Gene Ruler, SM0313), der im Gel mitläuft. Danach wurde das Gel in die UV- Kammer (UV-System, Intas) gelegt und die Banden im Fotogramm angeschaut und dokumentiert. Das Verhältnis der Färbeintensitäten von 28S-rRNA zu 18S-rRNA sollte hierbei ungefähr 2:1 betragen (Abb. 3). 27 Material und Methoden Ein abweichendes Verhältnis, oder eine unscharfe und undeutliche RNA-Bande, wies auf eine RNADegradation während der Präparation hin. Dieser Versuchsdurchlauf wurde dann wiederholt. 28S-rRNA Bande 18S-rRNA Bande 1kb DNA Ladder Abbildung 3: Agarosegel der Gesamt-RNA, Betrachtung unter UV-Licht nach elektrophoretischer Auftrennung. Die Intensitäten der 18S-Banden und 28S RNA-Banden zeigen ein Verhältnis von 1: 2. 2.6.4 RNA- Pooling Um jetzt die Genexpressionsänderungen als Querschnitt der einzelnen Gruppen auf den Genchips identifizieren zu können, wird die RNA pro Gruppe gepoolt. Hierzu wird von jeder der fünf extrahierten RNAs jeder Gruppe sechs µg RNA entnommen und mit den anderen RNA´s der Gruppe vermengt. Das entstandene Volumen wird dann mit RNase-freiem Wasser auf 100 µl aufgefüllt. Nach Zugabe von 350 µl Buffer RLT und 250 µl 96%igem Ethanol wird das Gemisch auf die RNeasy Mini Säule aufgetragen. Nach zweimaligem Waschen mit 500 µl Buffer RPE und einem Durchgang der Trockenzentrifugation wird die RNA wieder in RNase freiem Wasser eluiert und mittels Photometer und Gelelektrophorese quantitativ und qualitativ kontrolliert. Material und Methoden 2.7 28 Genchip-Hybridisierung Die Arbeiten erfolgen in Kooperation mit der Arbeitsgruppe von Dr. Koczan (Universitätsklinikum Rostock, Immunologisches Institut Prof. Dr. Thiessen). 2.7.1 DNA-Microarray Die Firma Affymetrix ist Produzent von DNA-Microarrays, mit deren Hilfe wir die RNA-Profile unterschiedlicher Organe erstellen konnten. In unserer Arbeit verwendeten wir den GeneChip® Rat Genome 230 2.0 Array. Das Prinzip der Mikroarray-Technologie ist die Miniaturisierung eines bekannten analytischen Verfahrens, das auf der Hybridisierung und Detektion eines freien, markierten Nukleinsäurestrangs mit einem immobilisierten, komplementären Nukleinsäurestrang beruht. Dieses Prinzip findet in zahlreichen Anwendungen bereits Verwendung (Southern Blot, Northern Blot, Dot Blot). Das Besondere an Mikroarrays ist die sehr hohe Dichte, mit der Tausende von unterschiedlichen Sequenzen durch neuartige Technologien nebeneinander auf einen Träger aufgebracht werden, und innerhalb eines Testansatzes analysiert werden können. Der Einsatz von Mikroarrays ermöglicht die schnelle und parallele Untersuchung der Genexpression von bekannten Genen (probe genes) oder Gensequenzen und deren Veränderung zwischen zwei Experimentgruppen. Für den synthetischen Oligonukleotid-Mikroarray von Affymetrix ist im RoutineEinsatz eine Ausgangsmenge von 5 µg Gesamt-RNA ausreichend. Die folgenden Hauptschritte werden dabei bei der Expressionsanalyse der Genchips durchlaufen: 1. Probengewinnung (siehe RNA Phenolextraktion) sowie Weiterbearbeitung der gepoolten RNA vor Hybridisierung 2. Chiphybridisierung 3. Waschen, Färben und Antikörperamplifikation 4. Scannen 5. Datenanalyse 29 Material und Methoden 2.7.2 Affymetrix-Standardprotokoll der cDNA-Synthese Bei der vorherigen Phenolextraktionsmethode gewonnenen RNA handelt es sich um messenger-RNA (mRNA), also eine transkribierte Arbeitskopie eines Gens der DNA, welches im normalen Stoffwechsel der Zelle aus dem Zellkern herauswandert und dann mit Hilfe der Ribosomen in Proteine translatiert wird. Durch Zuhilfenahme von Reverser Transkriptase (RNA-abhängige DNA-Polymerase) wird im nächsten Schritt aus dieser mRNA zuerst eine doppelsträngige copyDNA (cDNA) gewonnen, aus welcher danach durch in vitro-Transkription eine cRNA gewonnen werden kann. In diesem Schritt findet auch schon durch Einbau eines bestimmten mit Fluorochrom markierten Nukleotids die Vorbereitung zur späteren Erkennung statt. Die Herstellung der copyDNA (cDNA) erfolgt in zwei Schritten, beginnend mit der Erststrangsynthese. Hierzu werden 10 µg des Gesamt-RNA-Eluats in ein frisches 1.5 ml Safe Lock Reaktionsgefäß (Eppendorf Biopur) überführt und 2 µl einer Glykogenstammlösung (Ambion, P/N 9510) hinzugesetzt. Es folgt die Ethanolpräzipation, bei der 50 % einer 7.5M Ammoniumacetatlösung (Sigma, P/N A2706), sowie das Dreifache des jetzt entstandenen Volumens an 96%igen Ethanol hinzugemischt werden. Die Mixtur wird danach in flüssigem Stickstoff schockgefroren, bevor sie 30 Minuten lang bei -3° C zentrifugiert wird. Der Überstand wird dekantiert, die übrige Lösung kurzzeitig zentrifugiert, und der adhärenten Rest des nun entstandenen Eluates abpipettiert. 600 µl 80%igen Ethanols werden hinzugegeben, danach kurz mit dem Vortex aufgerüttelt und erneut drei Minuten zentrifugiert. Das neue Eluat wird bis auf ca. 50 µl mit einer Pipette abgenommen und erneut kurz anzentrifugiert. Bei offenem Reaktionsgefäß und Zimmertemperatur erfolgt nun die Trocknung. Ein HPLC-gereinigter T7-(dT)24 Primer (5`-GGCCAGTGAATTGTAATACGACTTCACTATAGGG AGGCGG-(dT)24-3` wird nun zum Priming der Erststrangsynthese in einer Konzentration zu 10 pmol/µl mit RNase freiem Wasser (Ambion) vorverdünnt. 10 µl dieser Primerlösung werden zum vorher getrockneten Inhalt des Reaktionsgefäßese hinzugegeben. Es erfolgt eine dreiminütige Inkubation bei 70° C. Danach wird die Lösung auf Eis heruntergekühlt und eine Minute lang in die Zentrifuge gestellt. Es erfolgt die Beisetzung eines Mastermixes. Die Rezeptur setzt sich wie folgt zusammen: 1 µl dNTP Stammlösung ( je 10mM dATP, dCTP, dGTP, dTTP) 2 µl Dithiothreitol 4 µl Erststrang 5x Puffer (GibcoBRL, Invitrogen) _________________________________________ ∑ 7 µl Mastermix 30 Material und Methoden Danach werden noch 3.5 µl SuperScript™ II RNase H Reverse Transcriptase (GibcoBRL, Invitrogen) hinzugegeben und anschließend bei 42° C für ein bis zwei Stunden inkubiert. Es folgt ein mindestens zweiminütiges Abkühlen auf Eis. Anschließend wird für einige Sekunden zentrifugiert, der am Boden des Reaktionsgefäßes befindliche Reaktionsansatz abpippetiert und zur Zweitstrangsynthese weiterverwendet. Die Zweitstrangsynthese wird mit einem anderen Mastermix durchgeführt. Die Rezeptur setzt sich wie folgt zusammen: 91 µl RNase freies Wasser (DEPC-treated water, Ambion) 30 µl 5x Second Strand Buffer (GibcoBRL, Invitrogen) 3 µl dNTP Stammlösung (je 10mM dATP, dCTP, dGTP, dTTP) 4 µl DNA Polymerase I von E.coli (5-10 Units/µl; GibcoBRL, Invitrogen) 1 µl E.coli DNA Ligase (10 Units/µl; GibcoBRL, Invitrogen) 1 µl Ribonulease H (1-4 Units/µl; GibcoBRL, Invitrogen) _________________________________________ ∑ 130 µl Mastermix Der Mastermix wird mit dem Reaktionsgemisch der Erststrangsynthese vermischt und zwei Stunden bei 16° C inkubiert. Es folgt die „End-polishing“- Auffüllreaktion, bei der 2 µl T4 DNA Polymerase (1-8 units/µl; GibcoBRL, Invitrogen) hinzugegeben werden und die Mischung für fünf Minuten bei 16° C inkubiert wird. Die Reaktion wird durch Zugabe von 10 µl 0,5M EDTA Lösung (Sigma) beendet. Anschließend erfolgt die Aufreinigung durch eine Phenolextraktion. Es werden hierzu 160 µl Phenol/Chloroform (Phenol-Chloroform-Isoamylalkohol, Ambion) zur zuvor gewonnen Lösung hinzugegeben und kräftig im Vortex vermischt. Es folgt eine fünfminütige Abkühlung auf Eis. Danach wird zehn Minuten bei Raumtemperatur zentrifugiert und die wässrige Phase in ein 1.5 ml Safe Lock Reaktionsgefäß (Eppendorf Biopur) pipettiert. Der Mixtur werden dann 2 µl Glykogenstammlösung zugesetzt (Ambion, P/N 9510). Hiernach erfolgt erneut eine Präzipitation mit Ethanol und darauf ein Waschgang mit 600 µl 80% igen Ethanol. Affymetrix-Standardprotokoll der cRNA-Transkription und Markierung mit Biotin Als Ergebnis erhält man eine cDNA, die wesentlich stabiler ist, als die RNA, dem Transkriptom eines Zellpools in einem bestimmten Zustand jedoch identisch ist. Sie bietet sich daher als Ausgangspunkt für Untersuchungen der Genexpression an. Nach Anleitung des von Affymetrix empfohlenen Stan- 31 Material und Methoden dardprotokolles folgt die In-Vitro-Transkription dieser cDNA mit gleichzeitiger Markierung durch biotinylierter Cytidintriphosphate (CTP) und Uridintriphosphate (UTP). Hierzu wird das BioArrayTM High YieldTM RNA TranscriptLabeling Kit (P/N 900182, Enzo) verwendet. Es wird ein anderer Ansatz eines neuen Mastermixes benötigt. Die Rezeptur setzt sich wie folgt zusammen: 22 µl RNase freies Wasser (DEPC treated water, Ambion) 4 µl 10x HY Reaction Buffer 4 µl 10x Biotin Labeled Ribonucleotides 4 µl 10x DTT 4 µl 10xRNase Inhibitor Mix 2 µl 20x T7 RNA Polymerase _____________________________________ ∑ 40 µl Mastermix Nach Hestellung des Mastermix wird dieser in das getrocknete Hütchen pipettiert. Es folgt eine Inkubation für fünf bis 15 Stunden, abhängig von der eingesetzten Gesamt-RNA. Anschließend wird der Reaktionsansatz aufgereinigt: Hierzu wird der komplette Reaktionsansatz auf eine RNeasy Minisäule (Qiagen, Hilden) pipettiert und bei 10000 G für 15 Minuten zentrifugiert. Der Durchsatz wird ein zweites Mal auf die Säule geben und zentrifugiert. Insgesamt folgt eine dreimalige Waschung der Säule. Dabei wird im erstenWaschschritt die Waschung mit 700 µl RW (RNeasy Kit) im zweiten und dritten Waschschritt mit 500 µl RPE (RNeasy Kit) durchgeführt, sowie jeweils 15 Sekunden bei 10000 G zentrifugiert. Eine Trockenzentrifugation der Säule für 2 Minuten bei 19940 G erfolgt im letzten Schritt. Anschließend wird die Säule in ein frisches RNase freies 1.5 ml Safe Lock Reaktionsgefäß (Eppendorf Biopur) überführt, um die RNA zu eluieren. Es folgt eine zweimalige Waschung mit je 30 µl RNase freien Wasser (DEPC-treated water, Ambion). Nach einminütiger Inkubation wird für eine weitere Minute bei 19940 G zentrifugiert. Danach werden die gewonnenen 60 µl in ein frisches, RNase freies 1.5 ml Safe Lock Reaktionsgefäß (Eppendorf Biopur) umpipettiert. Nach Herstellung einer 1:50 Verdünnung der eluierten cRNA (1.4 µl + 68.6 µl Wasser) folgt zur Quantifizierung (1 OD = 40 µg/ml) eine photometrische Messung bei 260 nm. Um RNA-Fragmente gleicher Länge und Hybridisierungskinetik, sowie gleicher Anzahl inkorporierter Biotinmoleküle herzustellen, die alle eine gleich hohe Spezifität zu dem entsprechenden Oligonukleotid auf dem Chip haben, efolgt nun die Fragmentierung, die auch als saure Hydrolyse bezeich- Material und Methoden 32 net wird. Hierzu gibt man 12.5 µl fünffach Fragmentierungspuffer (Trizma Base, Sigma P/NT 1503) pro Reaktionsansatz hinzu und inkubiert dies bei 94° C für 35 Minuten. Anschließend wird mit Eis Heruntergekühlt und kurzzeitig zentrifugiert. Zur anschließenden Qualitätskontrolle und Volumenbestimmung des nachfolgenden Hybridisierungsgels werden zunächst die Gelkammer, der Schlitten und der Kamm mindestens 30 min mit 3 %igem Wasserstoffperoxid inkubiert und anschließend abgewaschen. Danach wird ein 1.6 %iges TBE-Agarosegel hergestellt, in das 1 µl der zuvor fragmentierten cRNA auftragen wird (Elektrophorese: 10 V/cm, max. fünf Zentimeter Lauflänge). Mittels einer Geldokumentationsanlage über 254 nm UV-Licht erfolgt die Fotodokumentation und Datenspeicherung. 2.7.3 Aufbau und Durchführung der DNA-Microarrays Bei dem cDNA-Microarray handelt es sich meist um einen Siliziumträger, auf den durch Photolithographie spezifische DNA-Sequenzen punktförmig und in systematischer Reihenfolge auf 11x11 µ m große, so genannte Sondenzellen, fixiert werden. Jede Sondenzelle ist also mit einem einzigartigen Oligonukleotid bestückt, das eine Länge von 25 Basenpaaren hat und in ca. 107 Kopien pro Sondenzelle vorkommt. Jede mRNA-Referenzsequenz wird durch 20 verschiedene Sondenpaare (= ein Sondensatz) repräsentiert. So ist es möglich auf einer 1.28 cm2 großen Fläche 1.3 Millionen Oligonukleotide für die Analyse von bis zu 54000 Zielsequenzen zu erstellen. Jede Sondenzelle mit einem so genannten Perfect-Match (PM) enthält ein zur Referenzsequenz genau komplementäres Oligonukleotid. Ihr gegenübergestellt liegt eine Miss-Match-Sondenzelle (MM) mit einem Oligonukleotid, welches bis auf einen homomeren Austausch an Position 13 mit dem Oligonukleotid der gegenüberliegenden PM-Sondenzelle identisch ist. Sie bilden gemeinsam ein Sondenpaar. Bei Anwesenheit des Transkripts in der Probe des Experiments, wird das Oligonukleotid der PM-Sondenzelle besser mit dem Transkript hybridisieren, als das Oligonukleotid der MMSondenzelle. Dadurch wird das gemessene Fluoreszenzsignal der PM-Sondenzelle (heller dargestellt) über dem Signal der MM-Sondenzelle (dunkler dargestellt) liegen. Die Software von Affymetrix ermittelt dann die durchschnittliche Differenz zwischen PM- und MMSondenzellen über die Gesamtheit des Sondensatzes und trifft eine Aussage über die Anwesenheit und die Signalstärke des Transkripts. Die Signalsstärke wird verwendet, um den Faktor der Expressionsveränderung zwischen den zwei Gruppen zu bestimmen. Beim Auftreten von unspezifischen Hybridisierungen betreffen diese PM- und MM-Sondenzellen annähernd gleichermaßen, da nur eine Base zwischen den Oligonukleotiden unterschiedlich ist. Dadurch ergibt sich für die durchschnittliche Differenz der Fluoreszenzsignale ein Wert bei Null. Dies führt zu einer deutlichen Reduktion falschpositiver Signale und Fehlinterpretationen der Software. 33 Material und Methoden Abbildung 4: Schema der Repräsentation eines Gens auf Mikroarrays von Affymetrix Jede Referenzsequenz wird in bis zu 20 verschiedene Sondenpaare eingeteilt. Das Sondenpaar besteht aus Oligonukleotiden, die identisch mit der Referenzsequenz sind, Perfect Match (PM), oder einen einzigen Basenaustausch besitzen, Miss Match (MM). Nach der Hybridisierung wird die Fluoreszenzintensität der markierten Probe aller Sondenzellen gemessen und berechnet, ob und in welcher Signalstärke ein Transkript anwesend war. Bei Anwesenheit ist die Fluoreszenz der PM-Sondenzelle in der Regel höher als die der MM-Sondenzelle (103). 2.7.4 Chiphybridisierung Affymetrix-Standardprotokoll der Chip-Hybridisierung Die Herstellung eines zweifachen Hybridisierungspuffers, muß vor der Durchführung der eigentlichen Genhybridisierung erfolgen. Die Rezeptur setzt sich wie folgt zusammen: ∑ 8.3 ml der 12x MES-Stammlösung (70,4 g 2-(N-Morholino)Ethansulfonsäure Natriumsalz (Sigma, M 3058) plus 800 ml Wasser (Molecular Biology Grade, BioWhittaker, P/N 16-001 Y)) 17.7 ml 5 M NaCl (RNase frei, Ambion, P/N 9760) 4.0 ml 0.5 M EDTA (Sigma, P/N E 7889) 0.1 ml 10 % Tween 20 (Pierce, P/N 28230) 19.9 ml RNase freies Wasser (DEPC treated water,Ambion) 50 ml 2x Hybridisierungspuffer 34 Material und Methoden Danach wird ein Hybridisierungsmix synthetisiert. Die Rezeptur setzt sich wie folgt zusammen: ∑ 5 µg cRNA Fragmentierungsansatz 50 µl 2x Hybridisierungspuffer 1 µl Bovines Serumalbumin (50mg/ml, GibcoBRL, P/N 15561-020) 1 µl Heringsspermien DNA (10 mg/ml, Promega P/N D 1811) 1.7 µl B2-Oligo Bio-GTC GTC AAG ATG CTA CCG TTC AGG A, HPLC gereinigt, Sigma) 5 µl 20x Kontrollspikes (BioB, 150 pM; BioC, 500 pM; BioD 2,5 nM; Cre,10 nM) x µl RNase freies Wasser (DEPC treated water, Ambion) bis zu einem Gesamtvolumen von 100µl 100µl Hybridisierungsmix Es folgt die Hybridisierung, für die die die Arrays mit 200 µl einfach Hybridisierungspuffer aufpipettiert werden, um danach bei 45° C und 60 rpm im Hybridisierungsofen (Hybridization Oven 640, Affymetrix) vorhybridisiert zu werden. Schließlich wird für den Schritt der eigentlichen Hybridisierung der zuvor gemischte Hybridisierungsmix für fünf Minuten auf 99° C erhitzt und anschließend für fünf Minuten bei 45°C abgekühlt. Nach Zentrifugation wird die Vorhybridisierungslösung vom Chip abpipettiert und mit dem eigentlichen Hybridisierungsmix befüllt. Die Durchführung der Hybridisierung dauert 16 Stunden bei 45° C und 60 rpm im Hybridisierungsofen (Hybridization Oven 640, Affymetrix). 2.7.5 Waschen, Färben und Antikörperamplifikation Nach dem Protokoll von Affymetrix erfolgt anschließend der Wasch- und Anfärbevorgang in der halbautomatischen Waschstation (Fluidics Station 400). Es erfolgt die Herstellung von Waschpuffern. Die Rezeptur setzt sich wie folgt zusammen: Stringenter Waschpuffer (1000 ml): 83.3 ml 12x MES-Stammlösung 5.2 ml 5 M NaCl (Ambion, P/N 9760) 1.0 ml 10 % Tween 20 (Pierce, P/N 28320) 35 Material und Methoden ∑ 910.5 ml Wasser (Molecular Biology Grade, BioWhittaker) 1000 ml Stringenter Waschpuffer Nicht-stringenter Waschpuffer (1000ml): ∑ 300 ml 20x SSPE (3 M NaCl; 0,2M NaH2PO4;0,02M EDTA, BioWhittaker, P/N 16-010Y) 1.0 ml 10 % Tween 20 (Pierce, P/N 28320) 699 ml Wasser (Molecular Biology Grade, BioWhittaker) 1000 ml Nicht-stringenter Waschpuffer Zweifach Stain Buffer (250ml) ∑ 41.7 ml 12x MES Stammlösung 92.5 ml 5 M NaCl (RNase frei, Ambion, P/N 9760) 2.5 10 % Tween 20 (Pierce, P/N 28320) ml 112.8 ml RNase freies Wasser (DEPC treated water, Ambion) 250 Stain Buffer ml Färbelösung ∑ 300 µl 2x Stain Buffer 270 µl RNase freies Wasser 24 µl Acetyliertes BSA (50 mg/ml, GibcoBRL P/N 15561-020) 6 R-Phycoerythrin Streptavidin (Molecular Probes, P/N S-866) µl 600 µl Färbelösung 36 Material und Methoden Antikörperlösung 300 ∑ µl Zweifach Stain Buffer 266.4 µl RNase freies Wasser 24 µl Acetyliertes BSA 6 µl 10 mg/ml Ziegen-IgG (Sigma, P/N I5256; gelöst in PBS GibcoBRL ohne Ca/Mg) 3.6 µl 0.5 mg/ml biotinylierter Anti-streptavidin Antikörper (Ziege) (Vector Laboratories, P/N BA-0500) 600 µl Antikörperlösung Durch zweimaliges Anhängen an die halbautomatische Waschstation werden die Arrays mit der beschriebenen Färbelösung gefärbt. Dazwischen erfolgt die Antikörperamplifikation durch das Anhängen der Antikörperlösung. 2.7.6 Scannen Durch Substrahierung der unspezifischen Hybridisierungsmerkmale von den Signalen der PerfectMatch-Proben werden die Ergebnisse ausgewertet. Ebenso werden die gescannten Resultate aller aufgetragenen Oligonukleotiden für ein Gen mit einem Scanner (GeneChip Scanner 3000, Hewlett Packard, Affymetrix) untereinander verrechnet. Der Scanner kann bei einer Auflösung von 1.56 micron eine Laseranregung der Farbteilchen erzeugen und die Fluoreszenz in den verschiedenen Fragmenten quantitativ erfassen. Die Ergebnisse repräsentieren hinterher die quantitative Expression des Gens: Das Ausmaß der Fluoreszenz gibt demnach die Menge der zuvor gewonnenen mRNA wieder und lässt weiterhin gute Rückschlüsse auf die Proteinexpression zu. 2.7.7 Datenanalyse Die Datenanalyse und Koordinierung der Affymetrixgeräte wie zum Beispiel des Scanners, inkl. Färbung und Auswertung der gescannten Daten erfolgt mit GCOS 1.2 (GeneChip Operating Software) und dem Data Mining Tool 3.1. Dazu werden zwei zu analysierende Proben, die auf zwei DNAMicroarrays aufgebracht werden, miteinander verglichen, um Veränderungen zu bestimmen und zu quantifizieren. Die Software dient der Analyse folgender Daten: - Spezifikation der experimentellen Daten (Experimentname, Array-Typ usw.) 37 Material und Methoden - Scannen der gefärbten Chips - Umwandlung der Rohbilddaten in Intensitätsdaten - Umwandeln der Intensitätsdaten in Maßzahlen, wie beispielsweise Signal, Detection Call - Report über Qualitätskriterien wie Background und und 3'/5' Ratio der Housekeeping-Gene - Exportieren der Maßzahlen nach Excel oder ins txt-Format Eine Exceltabelle liefert die Ergebnisse als Vergleichswerte zweier gescannter Gruppen. Durch eine Vernetzung des Programms mit unterschiedlichen Datenbanken können bei der Auswertung des betreffenden Gens Name, Funktion und Similaritätswahrscheinlichkeiten aufgezeigt werden. Extraktion der Poly-A - RNA Zellpool aus Gewebeproben oder Zellkulturen Amplifikation und Markierung der RNA Chip-hybridisierung Oligonukleotid Auslesen des Fluoreszenzsignals Chipzelle Abbildung 5 Prinzip der Chipanalyse nach Affymetrix®Gene Expression Assay im vereinfachten Schema Gepoolte, markierte und fragmentierte cRNA hybridisiert mit Oligonukleotidsonden auf dem Chip. Entfernen nicht hybridisierter Nukleotide im Waschvorgang und Auswertung der quantitativen Bindung durch Markieren und Scannen der gebundenene cRNA (Darstellung in Anlehnung an Affymetrix®Gene Expression Assay) 38 Material und Methoden 2.8 Quantitative PCR Die Genchip-Hybridisierung wird mit dem Querschnitt der Gene, dem RNA-Pool, durchgeführt. Für die quantitative PCR wird ein spezifisches Gen eines jeden Tieres vorher ausgewählt und einzeln untersucht, um zu verifizieren, dass die beobachteten Expressionsveränderungen, die im Pool der Genchiphybridisierung aufgezeigt wurden, auch auf ein einzelnes Tier zutreffen. Außreißer können so dargestellt werden. Das genetische Screening durch die Chiphybridisierung fördert eine große Bandbreite hoch- und runterregulierter Gene zutage. Die 2-Step-Real-Time-PCR (Taqman, core facility) wird in unserer Arbeit für exemplarisch selektierte Gene durchgeführt. Das Prinzip der PCR-Reaktion beruht auf der enzymatischen Amplifizierung eines bestimmten DNA-Abschnittes, der zwischen zwei Startersequenzen, den so genannten Primern, liegt. Diese markieren den zu vervielfältigenden Genabschnitt. Da wir nach der Phenolextraktion RNA produziert haben und diese mittels PCR-Reaktion nicht amplifiziert werden kann, muss diese durch reverse Transkriptase in einem ersten Schritt in cDNA (copy DNA) umgeschrieben werden. Da RT und PCR separat in zwei Reagenzgefäßen durchgeführt werden, spricht man von zweistufiger oder 2-Step-RT-PCR. 2.8.1 Durchführung der RT-PCR Erstellung von cDNA Zur Erstellung von cDNA benötigt man zuerst folgende Substratzusammensetzung: ∑ x µl RNA (1µg RNA) 1 µl Random Hexamers 1 µl dNTPs 8-x µl H2O 10 µL RNA-Mix Zu diesem RNA-Mix werden 10 µl eines ReverseTranskriptase (RT) -Mixes folgender Zusammensetzung hinzugegeben und eine Stunde bei 50 °C inkubiert: 39 Material und Methoden ∑ 2 µl 10x RT-Puffer 4 µl MgCl2 (25mM) 2 µl DTT (10mM) 1 µl RNase out (40U/µL) 1 µl Superscript III (200 U/µL) 10 µl RT-Mix Bei 85° C erfolgt die Denaturierung der RT bei fünfminütiger Inkubationszeit. Anschließend folgt die Denaturierung der RT durch fünfminütige Inkubation. Zur Zerstörung der zurückgebliebenen RNA wird im Anschluss ein µl RNaseH hinzugegeben und 20 Minuten bei 37 ° C inkubiert. 2.8.2 Durchführung der PCR Mit der nun vorhandenen DNA erfolgt die eigentliche PCR, welche im Wesentlichen aus der zyklischen Wiederholung von drei Reaktionsschritten besteht: Bei 95° C erfolgt eine Hitzedenaturierung des DNA-Doppelstrangs, um die beiden DNA-Stränge voneinander zu trennen (melting) Es folgt bei 52° C ein Hybridisierungsschritt, mit Bindung der beiden Primer an den jeweils komplementären Strang (annealing). Im letzten Schritt wird der zwischen den Primern liegende DNA-Abschnitt mithilfe der hitzestabilen DNA-Polymerase und der im Reaktionsmix vorhandenen Desoxyribonucleotide bei ca. 72° C selektiv synthetisiert (extension). Zu Reaktionsbeginn nimmt die Zahl der amplifizierten DNA-Fragmente exponentiell mit der Zykluszahl zu. Eine Abweichung von dem exponentiellen Wachstum tritt nach ca. 17 Zyklen auf und erreicht nach ca. 35 Zyklen ein Maximum. Ausschlaggebend hierfür ist unter anderem die begrenzte Verfügbarkeit der beteiligten Komponenten. Diese Amplifizierunsschritte werden mit einem PCR-Cycler durchgeführt. Durch die Auswahl bestimmter Primer (Tabelle 2) und 35-40-faches Durchlaufen dieser Schritte kommt es zur Amplifizierung der Ausgangs-DNA. Material und Methoden Gen Il-1-RA CCL2 / MCP-1 Hmox-1 ELAM Il-6 40 Primersequenzen 5´-GAATGTGTTCTTGGGCATCC-3´ 5´-GATTCGAAGCTGGTGGTAGG-3` 5´-GGTCTCTGTCACGCTTCTGG-3´ 5´-CTGGACCCATTCCTTATTGG-3´ 5´-CAGAGTTTCTTCGCCAGAGG-3´ 5´-ATGGCATAAATTCCCACTGC-3´ 5´-GTACCTCATCCGGTGTCTGG-3´ 5´-CTTGTGCACTGCATTCAACC-3´ 5´-GATGGATGCTTCCAAACTGG-3´ 5´-AGGAGAGCATTGGAAGTTGG-3´ Fragmentlänge (bp) 177 211 169 201 233 Tabelle 2: Genspezifische RT-PCR Primer und deren Länge Früher wurde zur Detektierung hauptsächlich Ethidiumbromid (EtBr) verwendet (79), heutzutage scheinen andere interkalierende Farbstoffe ein zum Teil besseres Signal-Hintergrund-Verhältnis aufzuzeigen und werden bevorzugt benutzt. Wir verwenden das SYBR®-Green I (Molecular Probes, Portland, Oregon). Dies ist ein Fluoreszenzfarbstoff, der sich – wie EtBr – unspezifisch in Doppelstrang-DNA einlagert, wodurch es mit fortschreitender PCR-Reaktion zu einem Fluoreszenzanstieg kommt. Dieser so genannte fluoreszierende Reporterfarbstoff macht es möglich, die Reaktion darzustellen und einen mit der Produktmenge proportionalen Anstieg anzuzeigen. Die Messung erfolgt mit einem optischen Detektionsmodul, das die ansteigenden Fluoreszenzwerte messen kann. Der CT-Wert oder Threshold Cycle basiert auf einem bestimmten Fluoreszenz Schwellenwert und dient der Quantifizierung der PCR. Der CT-Wert ist jener PCR-Zyklus, bei dem die Reporterfluoreszenz die Hintergrundfluoreszenz signifikant übersteigt. Am Anfang der PCR-Reaktion wird nur die Basis- oder Hintergrundfluoreszenz gemessen, da die Reporterfluoreszenz aufgrund der geringen Templatekonzentration im Reaktionsgefäß während der ersten PCR-Zyklen normalerweise nicht detektierbar ist. Die Kinetik der PCR-Reaktion, nicht die absolute Menge des PCR-Produktes ist für die Quantifizierung der DNA-Menge relevant. Der CT-Wert gilt als richtungsweisend, da die ansteigende Produktmenge zu diesem Zeitpunkt exponentiell ist und es in dieser Phase der PCR-Reaktion keine limitierenden Faktoren, wie Primer- oder Nukleotidmangel, sowie nachlassende Enzymaktivität gibt. Gleichzeitig werden in jedem PCR-Lauf bekannte Templatemengen von18s rRNA amplifiziert, sodass man Templatemenge und CT-Wert korrelieren kann. Ein niedriger CT in der abschließenden Messung signalisiert, dass der exponentielle Anstieg in einer Gruppe, z.B. unserer Hirntodgruppe schnel- Material und Methoden 41 ler erreicht wurde, als in der Kontrollgruppe. Dies erklärt sich durch eine höhere Ausgangsmenge an DNA in der Hirntodgruppe, als in der Kontrollgruppe. Die Gleichung für dieser relativen Messung bildet die ∆∆C(t)- Methode. C (t) ist der schon oben beschriebene CT-Wert, entspricht also der Zyklusnummer, bei der das Thresholdlevel erreicht wird. Die Formel, die hierbei Verwendung findet, lautet: ∆∆C(t) = ∆C(t) Hirntodgruppe - ∆C(t) Kontrollgruppe ∆C(t) Hirntodgruppe und ∆C(t) Kontrollgruppe lassen sich wie folgt berechnen: ∆C(t) Hirntodgruppe = C(t) Hirntodgruppe - C(t) 18s bzw. ∆C(t) Kontrollgruppe = C(t) Kontrollgruppe - C(t) 18s Zum Beweis oder Ausschluss eines signifikanten Expressionsunterschiedes zwischen beiden Gruppen, kann mit folgender Formel, nach Standardisierung neu gewonnener Werte der Versuchsgruppen durch Referenzgene, weitergerechnet werden: 2-∆∆C(t) Dieser Zahlenwert entspicht der relativen Expressionsveränderung der untersuchten Gruppe zur Referenzgruppe. Als Normwert gilt ein sogenanntes House-keeping-gene, auch Hausgen genannt. Im Vergleich mit den Ergebnissen der Genchipanalyse können sich die Werte zu diesem quantitativ unterscheiden, qualitativ sind sie immer übereinstimmend. Mit einer Gel-Elektrophorese wird die PCR überprüft. Die PCR-Reaktionsergebnisse werden mit je 4 µl 6x Gel-Ladepuffer vermischt und in die Geltaschen pipettiert, daneben 4 µl des Molekulargewichtsmarkers. Nach einer Stunde der Wanderung bei einer Spannung von 120 V und einer Stromstärke von 100 mA werden die Nukleinsäuren im Gel elektrophoretisch aufgetrennt. Die Dokumkentation der Ergebnisse wird photographisch digitalisiert und gespeichert. 2.9 Statistik Die Darstellung der Daten erfolgt mit Mittelwerten ± SEM, in Normalverteilung. Die Makrohämodynamik wird anhand der gemessenen Kreislaufparamter mit zwei verschiedenen Tests pro Parameter präsentiert. Die Nullhypothese wird einerseits anhand des Vergleiches gleicher Mittelwerte der Hirntod- und Kontrllgruppe gegen die Alternative signifikant verschiedener Mittelwerte durch den t-Test kontrolliert. Zum anderen werden Hirntod- und Kontrollgruppe mittels eines einfaktoriellen ANOVATests separat darauf überprüft, ob die Mittelwerte zu den jeweilig aufgenommenen Zeitpunkten mit- Material und Methoden 42 einander verträglich sind. Als Nullhypthese wird hier ebenfalls angenommen, dass alle Mittelwerte innerhalb einer Gruppe miteinander verträglich sind. Alle Tests werden bei einem a priori gewählten Signifikanzniveau von p < 0.05 durchgeführt. Analog dazu werden die Daten der PCR ausgewertet. Zur optischen Vereinfachung werden diese als Boxplots dargestellt. Zur Berechnung der Statistik aller Daten, sowie zur Darstellung der Boxplots wird die Statistik Toolbox von MATLAB® The MathWorks sowie das Program R und SPSS verwendet. Die Darstellung der makrohämodynamischen Parameter erfolgt mit Sigmaplot 9.0. 43 Ergebnisse 3. Ergebnisse 3.1 Makrohämodynamik Zum Zeitpunkt 0 (Baseline, vor Hirntodinduktion) besteht zwischen den beiden Versuchsgruppen kein signifikanter Unterschied des mittleren arteriellen Blutdruckes (K-Gruppe 113.2 ± 10.5 mmHg versus HT-Gruppe 98 ± 2.3 mmHg). Nach Hirntodinduktion präsentiert sich ein signifikanter Anstieg des mittleren arteriellen Blutdruckes mit einem Maximalwert von 176 ± 14.6 mmHg (* p < 0.05) im Vergleich zu der Baseline der Kontrollgruppe. Nach 60 Minuten war der Unterschied noch signifikant mit Werten von 120 ± 13.5 mmHg zu 90 ± 7.5 mmHg (* p < 0.05), danach zeigten sich im Verlauf vergleichbare Werte des mittleren arteriellen Blutdruckes zwischen den Gruppen (siehe Abbildung 6). Auch die Herzfrequenz zeigt zwischen beidne Gruppen ausser bei dem letzten Messpunkt, nach 360 Minuten (p < 0.05), keine Signifikanz. 44 Ergebnisse * 200 * * * 180 HT-Gruppe MADMAP [mmHg] [mmHg] 160 * K-Gruppe * * 140 120 100 80 60 Ka 0 36 0 30 0 24 0 18 0 12 60 th Fo ete ga ri m H rtyk pl. ir n a to th. di nd . 1 2 3 4 5 0 Zeitverlauf [min] Abbildung 6: MAD (MW ± SEM) im Verlauf. Signifikante Unterschiede zeigen sich nur während der Hirntodinduktion durch Inflation des Forgathykatheters (* p < 0.05). 45 Ergebnisse 400 * 380 Herzfrequenz [1/min] 360 340 320 300 280 HT-Gruppe K - Gruppe 260 240 220 200 36 0 30 0 24 0 18 0 12 0 60 Ka th et e Fo rim ga pl. rty k Hi ath rn . to di nd . 0 Zeitverlauf min] Abbildung 7: Herzfrequenz (MW ± SEM) im Versuchsverlauf. Nur am Versuchsende (6 h nach Hirntodinduktion) bestand ein signifikanter Unterschied (* p < 0.05) zwischen Hirntod- und Kontrollgruppe. 46 Ergebnisse 3.2 PH-Wert Die Untersuchung des pH-Wertes, die beginnend mit der arteriellen Katheterisierung stündlich bestimmt wird, zeigt weder signifikanten Unterschiede in und zwischen den Gruppen. Der Zeitpunkt Null bezeichnet dabei sowohl bei der Hirntod-, wie auch bei der Kontrollgruppe den Zeitpunkt der ersten Blutentnahme direkt nach Positionierung des Katheters in die A. carotis. 7,55 K-Gruppe HT-Gruppe 7,50 pH [-log c(H+)] 7,45 7,40 7,35 7,30 7,25 7,20 0 60 120 180 240 300 360 Zeitverlauf [min] Abbildung 8: pH (MW ± SEM) im Versuchsverlauf 3.3 pCO2 und pO2 Die Messung von pO2 (Abb.9) und pCO2 (Abb.10) zeigt keine signifikanten Unterschiede (p< 0.05) in den jeweiligen Gruppen. 47 Ergebnisse H T -G ruppe K -G ruppe 300 250 PO2 [mmHg] 200 150 100 50 0 0 60 120 180 240 300 360 Z eitverla uf [m in ] Abbildung 9: Mittlerer pO2-Wert (MW ± SEM) der Kontrollgruppe und der Hirntodgruppe. Keine signifikanten Unterschiede. 60 H T-G ruppe K - G ruppe pC02 [mmHg] 50 40 30 20 10 0 0 60 120 180 240 300 360 Zeitverlauf [m in] Abbildung 10: Mittlerer pCO2-Wert (MW ± SEM) der Kontrollgruppe und der Hirntodgruppe. Keine signifikanten Unterschiede 48 Ergebnisse 3.4 Analytisches Agarosegel der Gesamt-RNA Die Gesamt-RNAs werden nach der DNaseI-Verdauung mittels 1 % iger Agarosegele überprüft. Die analytischen Agarosegele zeigen die beiden erwarteten, charakteristischen, diskreten RNABanden bei 1.9 und 4.5 kb, die die eukaryontische ribosomale 18S und 28S RNA repräsentieren. Das Auftreten dieser diskreten Banden bestätigt, dass die isolierte RNA intakt ist. Auch das Verhältnis der Bandenintensitäten 18S zu 28S liegt annähernd im Bereich von 1:1.5-2.5. (Abb. 11) Zusätzliche Banden, die bei kleineren kb-Werten auftreten, sind auf ribosomale 5S RNA und tRNA zurückzuführen. Die Effektivität des DNaseI-Verdaus wird bestätigt, da bei höheren kb-Werten keine Banden zu sehen sind, die auf eine noch vorhandene Kontamination mit genomischer DNA hingewiesen hätte. 3.5 RNA-Extraktion und Poolung Nach dem im Abschnitt Material und Methoden beschriebenen Poolen wird die eluierte RNA photometrisch quantitativ bestimmt. Für die Niere ergibt sich dabei in den verschiedenen Versuchsgruppen die in Tabelle 3 angegeben Konzentration. Pool-Nr. Gruppe Konzentrationen in µg/µl 1 Hirntodgruppe 0.41 2 Kontrollgruppe 0.47 Tabelle 3: RNA-Konzentration nach Poolung 49 Ergebnisse Kontrollpuffer Hirntodgruppe Shamgruppe Kontrollgruppe Abbildung 11: Elektrophorese des RNA-Pools: Zwei Banden zeigen die Intaktheit der gepoolten RNA an 3.6 Ergebnisse der Chiphybridisierung Die folgende Tabelle gliedert die im Gruppenvergleich unterschiedlich exprimierten Gene nach ihrer vermuteten oder gesicherten Funktion auf. Das Expressionsprofil der Nieren hirntoter Ratten zeigt im Microarray signifikante Unterschiede zu der Kontrollgruppe. Es werden die Gene präsentiert, die GCOS 1.2 und das Data Mining Tool 3.1 eindeutig zuordnen konnten und die als cut-off point, also als Mindestwert, ab welchem die Gene als unterschiedlich exprimiert gelten, eine mindestens zweifache Expressionsveränderung der Hirntodgruppe im Vergleich zur Kontrollgruppe aufweisen. Mit diesem Filter können wir nach 6 Stunden Hirntod im Verhältnis zur Kontrollgruppe 151 Gene hoch- und herunterreguliert darstellen. Aus dieser Gesamtmenge sind 88 relevante Gene nachfolgend in ihrer Funktion aufgegliedert dargestellt. Hauptsächliche Gruppen dieser identifizierten Transkripte stellen die der Chemokine/Cytokine, der Zelladhäsion, Ionenkanäle und Proteasen dar, aber auch Gene der Transkription und des Stoffwechsels zeigen bemerkenswerte Veränderungen. Interessanterweise werden auch protektive Gene der Abwehr und Reparatur in ihrer Expression hochreguliert, was den induzierten Zellschutz des Körpers kennzeichnet. Die Kategorisierung der Gene nach bekannter oder mutmaßlicher biologischer Funktion in verschiedene Gruppen erfolgt mit Hilfe von einer Recherche in Gen- und Proteindatenbanken wie Pubmed oder Expasy (Expert Protein Analysis System). 50 Ergebnisse Gruppe Abkürzung Cyp1b1 PDK4 PsPLA1 HMGCS2 Stoffwechsel Cytokine, Chemokine Beschreibung/Name Cytochrome P450, family 1, subfamily b, polypeptide 1 Pyruvate dehydrogenase kinase Isoenzyme 4 Phosphatidylserine-specificPhospholipase A 1 3-Hydroxy-3-Methylglutaryl-Coenzym A-Synthase 2 Expressionsveränderung +3.69 +3.38 +3.49 +2.88 CES1 Carboxylesterase 1 +2.64 BHMT Betaine-homocysteine methyltransferase +2.24 PPM2c Protein phosphatase 2C, magnesiumdependent, catalytic subunit +2.24 CA3 Carbonic anhydrase 3 -3.24 A4GALT Alpha 1,4-galactosyltransferase -3.31 IL-6 Interleukin 6 +22.45 IL-1R2 Interleukin 1 receptor, type II +135.5 S100a8 S100 calcium binding protein A8 (Calgranulin A) +48.98 CCL2 / MCP-1 Chemokine (C-C motif ) ligand 2 +5.75 S100a9 Calcium binding protein A9 (Calgranulin B) +25.57 CXCL-1 Chemokine (C-X-C motif ) ligand 1 +25.55 IL-1b Interleukin 1 beta +10.91 CCL20 Chemokine (C-C motif) ligand 20 +12.68 IL-1RA Interleukin 1 receptor antagonist +4.52 CCL4 Small inducible cytokine A4 +4.72 CCL3 Chemokine (C-C motif) ligand 3 +5.79 CXCL2 Chemokine (C-X-C motif) ligand 2 +7.29 MIP2 Macrophage inflammatory protein-2 +2.1 IL-6st Interleukin 6 signal transducer +4.02 51 Ergebnisse Gruppe Abkürzung Beschreibung/Name Expressionsveränderung Immediate early response Gene CSRP3 Cysteine-rich protein 3 +19.88 IER5 Immediate early response 5 +2.41 KIM-1 Kidney injury molecule 1 +9.45 ICAM1 Intercellular adhesion molecule 1 +4.4 ELAM-1 Selectin, endothelial cell +188.48 VCAM-11 Vascular cell adhesion molecule 1 +3.37 GLYCAM-1 Glycosylation dependent cell adhesion molecule 1 +4.68 SPIN2c Serine protease inhibitor +23.56 TIMP1 Tissue inhibitor of metalloproteinase 1 +7.4 ADAMTS-1 Adisintegrin-like and metalloprotease +4.68 SLPI Secretory leukocyte peptidase inhibitor +3.16 Zelladhäsion Proteasen/ -inhibitoren 52 Ergebnisse Gruppe Transkriptionsfaktoren Hormone / Wachtsumsfaktoren Abkürzung Beschreibung/Name Expressionsveränderung NFIL3 Nuclear factor, interleukin 3 regulated +3.09 ATF3 Activating transcription factor 3 +2.95 IRF1 Interferon regulatory factor 1 +2.55 IFRD1 Interferon-related developmental regulator 1 +2.44 STAT3 Signal transducer and activator of transcription 3 +2.27 CEBPD CCAAT/enhancer binding protein delta +2.18 DbP D site albumin promoter binding protein -3.96 NR4A1 Nuclear receptor subfamily 4 group A, member 1 -2.65 NR1D1 Nuclear receptor subfamily 1, group D, member 1 -3.19 Egr3 Early growth response gene 3 +2.8 Znf354a Zinc finger protein 354A -3.36 TGFb1i4 Transforming growth factor beta stimulated clone 22 +3.4 GDF15 Growth differentiation factor 15 +2.59 Nrg1 Neuroregulin 1n +4.84 DTR Diphtheria toxin receptor heparinbinding EGF-like growth factor +3.24 IGFBP1 Insulin-like growth factor binding protein 1 +5.4 53 Ergebnisse Gruppe Wachstums / Zellzyklus Abwehr / Reparatur Apoptose Abkürzung Beschreibung/Name Expressionsveränderung PRL 1 Tyrosine phosphatase +2.2 BTG 2 B-cell translocation gene 2, antiproliferive +3.9 BRFl Butyrate response factor 1 +2.1 GADD45g GADD45gamma +2.3 SOCS-3 Suppressor of cytokine signaling-3 +2.5 Clu Clusterin +4.81 HSP70 Heat shock protein 70 +2.9 PAIl Plasminogen activator inhibitor +2.8 HO-1 Hemeoxygenase 1 +30.5 GSTA2 Glutathione-S-transferase, alpha type2 -2.15 Bcl2a1 B-cell leukemia/lymphoma 2 related protein A1 +4.43 Birc3 Inhibitor of apoptosis protein 1 +3.85 Tnfrsf1a Tumor necrosis factor receptor superfamily member 1a +2.86 Aqp2 Aquaporin2 -2.0 Ttr Tranthyretin -4.28 Alb Albumin -24.48 Lcn2 Lipocalin 2 +8.67 Transporter 54 Ergebnisse Gruppe Blut / Gerinnung Abkürzung Expressionsveränderung Fgb Fibrinogen, B beta polypeptide +15.68 TFPI2 Tissue factor pathway inhibitor 2 +11.72 Fgg Fgg -fibrinogen, gamma polypeptide +7.07 Fgl2 Fibrinogen-like 2 +6.86 Kng1 Kininogen 1 +6.46 F3 Coagulation factor 3 +3.08 F2RL1 Coagulation factor II (thrombin receptor-like 1) +2.58 HRG Histidine-rich glycoprotein +2.06 Fxyd4 FXYD domain-containing ion transport regulator 4 -2.09 Fxyd6 FXYD domain-containing ion transport regulator 6 -2.15 SNFlk SNF-like-Kinase PIM3 Serin/Threonin-protein Kinase PAK1 P21 (CDKN1A)-aktivierte Kinase 1 NEK 6 Pik 3r1 NIMA (never in mitosis gene)-related expressed kinase 6 Mitogen-aktivierte ProteinKinase 1 Epidermal growth factor Rezeptor Phosphatidylinositol 3-Kinase Sphk 1 Sphingosinkinase 1 EFNA 1 Ephrin A1 Ionenkanäle Kinasen Beschreibung/Name MAP 3kl EGFR -2.2 -2.21 +2.12 +2.12 +2.57 +2.91 +3.18 +3.6 +3.79 55 Ergebnisse Gruppe Abkürzung Beschreibung/Name Expressionsveränderung Andere LOX Lysiloxydase +2.54 CaSR Calcium-sensing-Rezeptor -2.53 Tabelle 4: Ergebnisse der Chiphybridisierung, gruppenspezifische Gliederung der differentiell regulierten Gene 3.7 Bestätigung der Genchip-Daten mit Real – Time - PCR Zur Bestätigung der durch die Chip-Hybridisierung gewonnen Daten mit der extrahierten RNA wird eine quantitative 2-step RT-PCR durchgeführt. Exemplarisch werden drei Gene aus verschiedenen Funktionsgruppen ausgewählt, die im Einzelnen und im Vergleich zur Kontrollgruppe in der Genchipanalyse hochreguliert sind. Aus der Gruppe der Zytokine/ Chemokine wählen wir das proinflammatorische Gen MCP-1 / CCL2. Aus der Gruppe der protektiven Gene mit Funktionen in der Abwehr und Reparatur wird die Hämoxygenase (HO-1) ausgewählt. Die Gruppe der Zelladhäsion wird durch das Selektin (ELAM-1) repräsentiert. Die gemessenen Werte der mRNA-Expression der Einzelgene wurden in Relation zu den Werten des Housekeeping-Gens gesetzt. Die mediane mRNA Expression der untersuchten Gene (HO-1, ELAM und CCL2) ist in der Hirntodgruppe im Vergleich zur Kontrollgruppe signifikant erhöht. Im Ergebnis zeigt sich für alle drei Einzelgene eine statistisch signifikante mRNA Expressionserhöhung (p<0.05, Wilcoxon Test) der Hirntodgruppe gegenüber der Kontrollgruppe (Abbildung 12). Somit kann qualitativ mit zwei unterschiedlichen Techniken, Chipanalyse und RT-PCR, eine Hochregulierung dieser Gene nachgewiesen werden. 56 Ergebnisse P < 0.05 P = 0.043 P < 0.05 P = 0.043 P < 0.05 P = 0.042 Abbildung 12: Boxplots der mRNA Expression der drei Einzelgene HO-1, ELAM und CCL2 im Vergleich zwischen Hirntod- (HT) und Kontrollgruppe (K) 57 Diskussion 4. Diskussion 4.1 4.1.1 Studienaufbau Relevanz der Studie Die Gruppe der hirntoten Spender stellt weltweit die Hauptquelle für Spendernieren in der Transplantationsmedizin dar. Im Fokus unserer Untersuchung stehen die Auswirkungen des Hirntodes auf den Organspender als primär antigenunabhängiger Faktor und seine Einflüsse auf die spätere Transplantatfunktion. Transplantatüberlebensraten von lebendgespendeten Nieren standen im Langzeitoutcome immer über denen totgespendeter Organe, egal wie hoch die Übereinstimmung der HLA waren. Die von Ehepartnern gespendeten Nieren konnten in Studienresultaten als sehr ähnlich zu denen eingeordnet werden, die von Familienmitgliedern mit nur einem Haplotyp-Mismatch aufgefunden werden (31, 84, 194). Dies zeigt, dass die Transplantatprognose keiner rein antigenabhängigen Beeinflussung unterliegt. Es ist also von Nöten, auch die nicht-antigenabhängige Ebene, in diesem Fall die komplexen Auswirkungen des Hirntods, zu analysieren und zu verstehen, um in der Zukunft die Ergebnisse der Transplantatfunktion von postmortalen Spendern weiter zu verbessern. 4.1.2 Versuchsmodell des Hirntods In unserer Arbeit benutzten wir das von Pratschke et al. etablierte Rattenmodell, das die Durchführung einer graduellen, normotensiven Hirntodinduktion erlaubt (157). Dieses Vorgehen vermeidet im Gegensatz zu explosiven Hirntodmodellen ausgedehnte hypotensive Phasen und schließt somit pathologische Reaktionskaskaden basierend auf systemischer Hypotension weitestgehend aus. Tiermodelle mit explosiver Hirntodinduktion induzieren derart starke Kreislaufschwankungen mit ausgeprägten hypotensiven Phasen, dass die intensive Behandlung mit kreislaufstabilisierenden Medikamenten, wie z.B. Katecholaminen, benötigt wird, die die Ergebnisse der hirntodinduzierten Veränderungen mitbeeinflussen könnten (204). Studien zeigen, dass posttransplantative Folgeerscheinungen, wie z.B. die Morphologie des Transplantats, schon je nach Ätiologie des Hirntodes deutliche Unterschiede zeigen (176). In unserem Ansatz ist eine Berücksichtigung demographischer Faktoren nicht möglich. In der Praxis sind Resultate von älteren Spendern, die an intrazerebraler Blutung oder thrombotischem Schlaganfall gestorben sind, sicher schlechter, als Organe von jungen Spendern, die an akutem Schädel-Hirn-Trauma gestorben sind (154). Unser Set-up repräsentiert also eher den optimalen, gesunden Spender. Diskussion 4.1.3 58 Zeitlicher Rahmen des Versuches Um dem Körper eine ausreichende Zeit zur Einstellung auf die Narkose und den Hirntodstatus zu gewährleisten, musste eine Aufrechterhaltung des Kreislaufs und der Atmung über mehrere Stunden gewährleistet sein. Nur so kann die Adaption auf Ebene der Gene sichergestellt werden. Wir wählten in unserer Arbeit mit sechs Stunden Hirntod einen Zeitraum, der sich in vorhergehenden Studien als sinnvoll herausgestellt hatte, und auch von anderen Autoren als adäquat angesehen wird (76, 157, 172, 202, 218). 4.1.4 Auschluß von Einflußfaktoren auf das Ausmaß des Hirntods Indem die Organe vor der Explantation in etwa analog zur humanen Multiorganentnahme mit NaClLösung gespült wurden, konnte die klinische Situation der Nierentransplantation simuliert werden, da hier Transplantate vor der Ischämie mit einer Konservierungslösung perfundiert werden. Gruppen Mithilfe der Kontrollgruppe konnten wir den Anteil an Genexpressionsveränderungen herausfiltern, der durch die Faktoren Anästhesie, Präparation, Operationsdauer und Explantation entstanden ist. Sichtbar wird die differentielle Genexpression, die nicht durch die Induktion des Hirntods ausgelöst wurde, anhand der Daten der Kontrollgruppe. Änderungen auf Geneebene dieser Gruppe basieren primär auf dem Stress, dem die Nieren durch die Operation, die Versuchsdauer und die Präparation am Organ selbst ohne den Faktor „Hirntod“ ausgesetzt sind. Untersuchung der Genexpression Zur Untersuchung der Genexpression auf RNA-Ebene versucht man heutzutage, Alternativen zu der „Ein Gen-Ein Experiment“-Basis zu finden. Um die differentielle Genexpression in unserem Fall in der Niere von hirntoten Ratten versus Kontrollen zu analysieren, wurde im Rahmen dieser Arbeit die Affymetrix-Microarray-Technologie eingesetzt. Der Affymetrix-Chip ermöglicht die Untersuchung von bis zu 12000 verschiedenen Genen mit Kontrollen auf einer ca. 1.5 cm2 großen Fläche und somit die Erfassung der mRNAs tausender Gene eines Organismus für das ausgewählte Gewebe. Es können eine Vielzahl an Transkripten einer experimentellen oder einer klinischen Probe nachgewiesen werden, und damit auch Gentranskripte, die der Experimentator a priori noch nicht kennt, bzw. erwartet. Die Signifikanz unserer Arbeit liegt damit in einem relativ kompletten Screening der veränderten Genexpression nach Hirntodinduktion und der Aufdeckung herauf- oder herunterregulierter Gene, bzw. bisher unerkannter Gengruppen in der Spenderniere. 59 Diskussion 4.2 Diskussion der einzelnen Komponenten Hirntodgruppe Der massive Hirnschaden ist mit multiplen hämodynamischen, neurohormonalen und morphologischen Abweichungen assoziiert (96, 187, 189). Die initiale hypertensive Krise und der „autonomic storm“ kann über einen sekundären Anstieg des Sauerstoffverbrauchs und durch stärkste periphere Vasokonstriktion zu Ischämie, Endothelschaden und verringerter Mikrozirkulation führen. Vorangehende Studien begründen den Ursprung dieser Reaktionsabfolgen auf molekulargenetischer Ebene. Immunaktivierung Der Status des Hirntodes eines Spenders beeinflusst die posttransplantive Organfunktion und das Organüberleben bereits vor der Explantation und stellt somit einen wesentlichen primär nichtimmunogenen Risikofaktor dar. Klinische und experimentelle Studien weisen immer wieder auf die Bedeutung von proinflammatorischen Cytokinen, Chemokinen und Adhäsionsmolekülen in Zusammenhang mit einer Verschlechterung der Organqualität in Totspendern als sogenannte „Entzündungsantwort“ hin. Sie sind demnach mitentscheidend im Ablauf dieser inflammatorischen Kaskade. Diese Auswirkungen werden durch die Beobachtung unterstützt, dass im Transplantationsexperiment am Schwein selbst kurzzeitige hohe iatrogene Verabreichungen von Katecholaminen, entsprechend dem „autonomic storm“ des Hirntodes, zu erhöhten Zytokinkonzentrationen, schlechteren initialen Organfunktionen und reduzierten Überlebensraten führen. (147, 153) Zytokine werden im Prozeß des Hirntodes unter anderem von Astrozyten und Mikroglia des verletzten Hirngewebes sezerniert und gelangen dann über die im Hirntod geschädigte Blut-Hirn-Schranke in das periphere Blut, können aber auch von immunkompetenten Zellen des zirkulierenden Blutes selber, als auch von geschädigtem peripheren Gewebe freigesetzt werden (213). In den vergangenen Jahren stand der inflammatorische Status des Transplantats im Fokus zahlreicher Forschungen. Adhäsionsmoleküle und Zytokine finden sich in Spendernieren von Ratten (96) und Menschen (58, 92) immens hochreguliert. Unsere Ergebnisse bestätigen diese Thesen und zeigen das starke Ausmaß dieser Aktivierung anhand einer großen Gruppe von Genen, wie den Adhäsionsmolekülen (E-Selektine x 188, GLYCAM-1 x 4, u.a.), Chemokinen (CXCL-1 x 26, CCL20 x 13, MCP-1 x 6 u.a.), Zytokinen (IL-6 x 22, IL-1beta x 11, IL1-R2 x 135 u.a.) und anderen neutrophilen oder monozytären Lockstoffen, welche in der akuten Phase der inflammatorischen Antwort und im endothelialen Schaden eine Rolle spielen (97, 158). Unsere Daten zeigen eine signifikante Hochregulation von Genen unterschiedlicher Funktionsgruppen, die im Folgenden näher beschrieben werden sollen. Diskussion 4.2.1 60 Zytokine In Studien über Zytokinexpressionen nach postmortaler Nierentransplantation kann neben der deutlichen Steigerung von Serumkonzentrationen der makrophagen-assoziierten Produkte (Il-1, IL-6 und TNF – alpha) (21), auch ein Anstieg der von Th1-Zellen ausgeschütteten Faktoren (IL-2 und IFNgamma) und der mRNA von Makrophagen-Inhibitor-Protein-1 in der Niere nachgewiesen werden. Die Erhöhung zeigt sich im Vergleich mit Kontrollgruppen signifikant verstärkt (156, 172, 212), was die erhöhte inflammatorische Kaskade durch den Status des Hirntodes kennzeichnet. Korrelierend mit der quantitativen Steigerung dieser Zytokinkonzentrationen kann in NierenTransplantationsmodellen eine deutliche funktionelle und morphologische Verschlechterung der Organe gezeigt werden (13). In anderen Studien zeigen die lebendgespendeten Nieren der Kontrollgruppe eine deutlich niedrigere Erhöhung der Zytokin-mRNA-Expression im Vergleich zu den totgespendeten Nieren (96, 189). Il-6 In unserer Arbeit zeigte sich Interleukin-6 im Vergleich zur Kontrollgruppe deutlich hochreguliert (x 22.4) und in der qualititativen PCR bestätigt. Dies korreliert mit vorhergehenden Arbeiten (155, 172). Der Hirntod geht mit ausgedehnter kortikaler Nekrose einher. In diesen geschädigten Arealen stimulieren Zelllyse und weitere Entzündungsprozesse die eingewanderten Monozyten und Makrophagen zur Ausschüttung verschiedener Zytokine. Interleukin-6 stellt einen der wichtigsten Vertreter proinflammatorischer Zytokine dar. Es wird von praktisch jedem verletzten Gewebe gebildet, z.B. von Endothelium, Keratinozyten, Skelettmuskelzellen, Neuronen, Mikroglia und Astrozyten (7). Das Ausmaß der Gehirnläsion korreliert mit dem Anstieg dieser Produkte, deren Konzentration auch im peripheren Blut ansteigt und wiederum periphere Organschäden durch Verstärkung der endogenen Entzündungskaskade verursacht (7, 51, 210, 220). Man kann die zirkulierenden Blutkonzentration von Il-6 insofern als Marker für Hirnnekrose und potentiellen prognostischen Parameter für die spätere Transplantatfunktion anerkennen (120). Hancock et al. zeigten, dass in Nieren hirntoter Spender eine beständige intraglomeruläre Expression von Il-6 vorherrschte, dessen mitogener Effekt für mesangiale Zellen in vitro nachgewiesen ist. Il-6 gilt somit als Mediator für mesangiale Expansion, fortschreitenden glomerulären Untergang und Glomerulosklerose bei chronischer Abstoßung einzuschätzen ist (71). Die Hochregulierung von Il-6 kann somit eine schlechtere Nierentransplantatfunktion im Empfänger nach Hirntodspende bedeuten, aufgrund einer erhöhten bereits präexplantativen Entzündungsantwort im Transplantat. Il-6 führt zu einer vermehrten Einwanderung immunaktiver Zellen in die peripheren Organe, in unserer Arbeit die Niere, und somit zu chronischen Entzündungszuständen mit strukturellen Umwandlungen und verschlechterter Funktion. Diskussion 61 Il-1RA Il-1RA zeigte in unserer Arbeit in den Nieren hirntoter Ratten im Vergleich zu der Kontrollgruppe vierfach erhöht. Il-1 aktiviert B- und T-Zellen, amplifiziert Fieberreaktionen und triggert akute-Phase-Reaktionen peripherer Organe. Diese Reaktionen werden von Il-1-RA antagonisiert. Die Organe werden von endogener Seite vor diesen proinflammatorischen Kaskaden geschützt. Vorangegangene Studien zeigen, dass Il-1RA, der gegenläufige Ligand zu den immunogenen Aktivatoren IL1 alpha und beta, in wesentlich größeren Mengen bei posttransplantativ abstoßungsfreien Nieren gefunden wurde, als bei geschädigten, was seine anti-inflammatorische Wirkung demonstriert (111). Es existieren unterschiedliche Anzahlen an Tandem-repeat –Polymorphismen (VNTR) am Il-1-RA-gen. Bei manchen ist eine erhöhte IL-1-RA-Expression die Folge. Hier fallen akute Graft-versus-host-diseases (GvHD) häufig milder aus, als bei Kontrollen (114). Eine Studie von Suzuki et al bewies eine IL1-β induzierte erhöhte Apoptose am Herzen mit vermehrten Nekrosen von Kardiomyozyten und akuter Entzündungsreaktion. Eine induzierte Il-1-RA Überexpression konnte dieses Ausmaß verringern und erbrachte bessere Resultate (186). Eine Behandlung des Donors nach Hirntod mit rekombinantem Il-1RA, oder die Perfusion des Donororgans sofort nach Entnahme mit Il-1-RA blockiert die Aktivität von Il-1. Diese Vorbehandlung war in der Lage, bestimmte inflammatorische Il-1 getriggerte Kaskaden und die Hochregulation von MHC-Komplexen im Spender-gewebe zu reduzieren und später verringerte Abstoßungskomplikationen zu verringern. (186). Eine Studie von Endo zeigte im Tierexperiment, dass bei Lebertransplantationen von hirntoten Ratten versus lebenden Ratten anfangs deutliche erhöhte Il-1RA-Konzentrationen in der Gruppe des Hirntodes bestanden, die nach 24 Stunden unter die Konzentration der Lebendspender-Ratten fielen. Eine Erklärung ist die primäre Induktion des protektiven Genes durch den „Stressfaktor“ Hirntod mit nachfolgender Erschöpfung der Proteintranslation und somit unzureichenden Serumleveln nach circa 24 Stunden (48). Diese Ergebnisse bestätigen, dass einem besseren posttransplantativen Resultat eine schwächere immunologische Antwort vorausgeht. Calgranuline Eine weitere Bestätigung vorangegangener Arbeiten ist die Hochregulation von Genen in der Niere, die für Calgranulin A (S100A8) und Calgranulin B (S100A9) kodieren (97, 172). Beide Calgranuline, auch bekannt als Myeloid-related-Proteins (MRP8, MRP14) gehören zu der Familie kalziumbindender Proteine, einer neuen Gruppe proinflammatorischer Zytokine, die von Phagozyten ausgeschüttet wird (75). Diskussion 62 In unserer Arbeit zeigen sich hochregulierte Expressionswerte von x 49 für Calgranulin A (MRP8) und x 26 für Calgranulin B (MRP14). Calgranuline wirken proinflammatorisch, indem sie an Zelloberflächen kummulieren und durch Interaktionen mit karboxylierten Glykanen die Bindung von Phagozyten auf dem Gefäßendothel vereinfachen (14, 26). Ryckmann et al. sehen die vordergründige Bedeutung der Calgranuline in der Neutrophilen-Migration und der L-Selektin-Ausschüttung (166). Immunhistochemische Analysen menschlicher Nierenbiopsien mit unterschiedlichen Formen von Nephritiden weisen auf die Korrelation zwischen der Expression von MRP8/14-Komplexen und der Schwere des inflammatorischen Prozesses hin (57). Burkhardt et al. bestimmten Serumkonzentrationen von MRP8/14-Komplexen nach Nierentransplantation. Sie setzten die Werte in Korrelation zu Serumkreatinin-Leveln und Nierenbiopsien. Es zeigten sich stark erhöhte MRP8/14-Konzentrationen in Fällen der akuten Nierenabstoßung und dies im Durchschnitt 5 Tage vor Beginn der klinischen Abstoßungssymptomatik mit hoher Spezifität und Sensitivität. Dies beweist MRP8 und 14 als funktionstüchtige frühe Parameter im Monitoring dieses unwünschenswerten Prozesses (27). Die Stellung von Calgranulinen in Fällen vaskulärer oder traumatischer Hirnschädigung wurde ebenfalls vielfach untersucht. Studien zeigten eine Korrelation des Konzentrationsanstieges von Calgranulinen im Serum zu der Intensität der Hirnschädigung (42, 200). Weiter ansteigende Serumwerte konnten in den Patienten nachgewiesen werden, die später Hirntod erleiden sollten. Dies prädestiniert Calgranuline als Prädiktor und Parameter für das Ausmaß der Schädigung und die frühe Diagnose des Hirntodes (160). Diskussion 4.2.2 63 Chemokine Vertreter der Gruppe der Chemokine, z.B. CxCl1- (x 26)- und CxCl-2 (x 7), Ccl3 (x 6) und MCP1 (x 6), zeigten sich in unseren Daten gegenüber der Kontrollgruppe unterschiedlich stark erhöht. Funktionellen und morphologischen Veränderungen in der Niere liegen unter anderem einer Aktivierung der Makrophagen zugrunde. Ihre Rekrutierung und Einwanderung in entzündliches Gewebe scheinen vor allem von C-C-Chemokinen abhängig zu sein. C-C-Chemokine sind in gesunden Kontrollnieren nicht detektierbar, zeigen aber im Falle einer Glomerulonephritis eine starke Präsenz (100). Der entzündliche Prozess im Falle einer Abstoßung präsentiert sich mit einer Makrophagen- und Leukozyteninfiltration, Mesangiumproliferation und Gewebsfibrose, was unter anderem durch Chemokine gesteuert wird. Die Arbeitsgruppe um Liu untersuchte die glomeruläre Expression von C-C-Chemokinen in verschiedenen Formen von Glomerulonephritiden. Hochexprimiertes MCP-1, ein wichtiger Vertreter der C-C-Chemokine, das an vier Chemokinrezeptoren gleichzeitig koppeln kann, korreliert demnach mit einem verstärkten Entzündungsprozess und demzufolge mit einer schlechteren Transplantatfunktion und –prognose (106). Die Arbeitsgruppe von Notohamiprodjo zeigte, daß die systemische Behandlung mit dem ChemokinAntagonisten Met-RANTES eine direkte Suppression des akuten Schadens transplantierter Nieren durch die Blockade der Entzündungszellinfiltration erreicht werden kann. Durch präventive Suppression der MCP-1/ CCL 2-Funktion konnte eine Verbesserung der Posttransplantergebnisse erzielt werden (129). 4.2.3 Adhäsionsmoleküle Die zentrale Schädigung des Hirntodes führt neben einer massiven Veränderung der Makrohämodynamik und Ausschüttung von Hormonen und verschiedenen Mediatoren zu einer reaktiven inflammatorischen Kaskade, die unter anderem die erhöhte De-Novo-Synthese der Adhäsionsmoleküle beinhaltet. Die Adhäsionsmoleküle spielen eine zentrale Rolle in der Modulation des Entzündungsstatus des Transplantats (182). Sie sind oberflächliche Glykoproteine und interagieren mit inflammatorischen und proinflammatorischen Zellen, um deren Rekrutierung und Invasion in das Spenderorgan zu fördern. Studien zeigten bereits, dass die Expression von Selektinen, wichtige Vertreter der Adhäsionsmoleküle, bei hirntoten Spendernieren, im Vergleich zu lebendgespendeten Nieren, die eine deutlich geringere Adhäsionsmolekülexpression und Leukozyteninfiltration aufweisen, bereits vor der Explantatation deutlich erhöht ist (92, 93). Die Ergebnisse unserer Arbeit zeigen eine vierfach erhöhte Expression sowohl von V-CAM, I-CAM1, als auch von E-Selektin (ELAM-1, x 188). Da wir ein Gesamthomogenat der Niere verwendet ha- Diskussion 64 ben, können wir in unserer Studie nicht die Expression in unterschiedlichen anatomischen und funktionellen Kompartimente der Niere unterscheiden. Bestätigt wurde unsere Untersuchung mittels RTPCR von E-Selektin (ELAM-1), einem Repräsentanten der Gruppe der Adhäsionsmoleküle. Eine signifikante Hochregulation in der Hirntodgruppe konnte nachgewiesen werden (p<0.05). Vorausgehend untersuchten bereits unterschiedliche Arbeitsgruppen die Expression von ICAM-1, VCAM-1 oder E-Selektin sowohl in Kadavernieren von Ratten, als auch an menschlichen Spenderbiopsien. Im Tierversuch zeigten Skrabal et al eine signifikante Hochregulation von ICAM-1-mRNA in den Nieren hirntoter Ratten bereits vier bis sechs Stunden nach Hirntodinduktion (179). Yoshida et al. bestätigten im Tierversuch bei postmortal transplantierter Nieren von Ratten eine in erster Linie vermehrte VCAM-1-Expression auf den Endothelien der Glomeruli, wobei ICAM-1 auch in den Vasae afferentes und efferentes gefunden werden konnte (204). Die De-novo-Expression der adhäsiven Moleküle in renalen Gefässen und den Glomeruli erweist sich als diagnostischer Parameter für die Intensität inflammatorischer Reaktionen nach Transplantationen von Kadavernieren. Sie reflektieren die Schwere des strukturellen Schadens durch die entzündlichen Vorgänge in der Niere, die akute tubuläre Nekrose und chronische Nephropathien (72). Ähnliche Untersuchungen wurden auch in menschlichen Spenderbiopsien bestätigt: Koo et al. demonstrierten in prätransplantativen renalen Proben von hirntoten menschlichen Spendern eine Hochregulation von ICAM-1 und VCAM-1 auf den proximalen Tubuli, bzw. von E-Selektin in den intertubulären Kapillaren (92, 127). In einer immunhistochemischen Analyse von renalen Glomeruli und Interstitium vor der Explantation konnte in den Nieren hirntoter Patienten ein erhöhtes Level an E-Selektin, sowie eine entsprechend starke Infiltration inflammatorischer Zellen aufgewiesen werden. Diese Resultate wurden mit denen lebendgespendeter Nierentransplantationen verglichen. In den Ergebnissen zeigten sich 53% der Kadavernieren mit Leukozyten infiltriert bei hohen Leveln an ESelektin, während sich die lebendgespendeten Nieren nur zu 9% entzündlich infiltriert und mit hohen E-Selektin-Leveln zeigten. Die Werte korrelierten mit den späteren Funktionsraten der Nieren, die bei den lebendgespendeten Nieren deutlich über denen der totgespendeten lagen (92, 93). Gasser et al. untersuchten den negativen Einfluß der Selektine auf spätere Transplantatfunktionen in der Niere, indem die frühen entzündlich-adhäsiven Vorgänge durch einen löslichen rekombinanten Pselektin Glykoliganden unterdrückt wurden. Das Resultat bot deutlich bessere funktionelle und morphologische Ergebnisse, als unbehandelte hirntote Spenderorgane von Versuchsratten. Eine Behandlung des Spenders mit dem Liganden noch vor der Organentnahme bewirkte eine kompetitive Bindung der Selektinrezeptoren und somit eine Unterbrechung der negativen Kaskade posttransplantativ (61). Diskussion 65 Bemerkenswert im Zusammenhang der Adhäsionsmoleküle scheint ebenfalls die Hochregulation eines noch nicht vollständig erforschten Genes, dem Kidney-injury-molecule-1 (KIM-1), das für das Typ-1 transmembranale Glykoprotein kodiert (172). KIM-1 ist in der gesunden Niere nicht detektierbar, wird aber auf dedifferenzierten proximalen Tubuluszellen, die eine Regeneration nach toxischem oder ischämischem Schaden durchlaufen, stark erhöht exprimiert (14, 83, 85). Die Expression von KIM-1 korreliert mit der Ausscheidung von KIM-1 im Urin (uKIM-1) und zeigt sich mit progredienter renaler Fibrose und Entzündung des Organes assoziiert. In unserer Arbeit zeigte sich eine Hochregulation des Genes der hirntoten Gruppe im Vergleich zur Kontrollgruppe (x 9.5). Van Timmeren et al. erklären KIM-1 als möglichen non-invasiven Biomarker nephrologischer Erkrankungen und signifikanter Parameter zur Einschätzung von Transplantatverlust nach Nierentransplantation. 4.2.4 Transkriptionsfaktoren Regulatorische Vorgänge in Zellen müssen teilweise sehr schnell geschehen. Hierzu zählen zum Beispiel Vorgänge der Feinregulation des Zellstoffwechsels, welche durch die prompte Aktivierung oder Deaktivierung von Proteinen oder Enzymen gesteuert wird. Diesbezüglich wurde in letzter Zeit vermehrt die wichtige Rolle von Transkriptionsfaktoren in den Vordergrund gerückt. Ein Transkriptionsfaktor ist molekularbiologisch ein Protein, das für die Initiierung der RNA-Polymerase bei der Transkription von Bedeutung ist. Spezifische Transkriptionsfaktoren werden durch Rezeptor-vermittelte Proteinkinasen aktiviert und binden an den DNA-Abschnitt, dessen Gen transkribiert werden soll. Schuurs et al. zeigten diese Transkriptionsfaktoren als wesentliche Weichensteller für die nachfolgenden unterschiedlichen Expressionsmuster anderer Effektoren. Ein Großteil dieser Transkriptionsfaktoren gehört zu den sogenannten frühen Transkriptionsfaktoren, oder „immediate early genes“ (IEGs). IEGs antworten via Signaltranduktionswege auf extrazelluläre Stimuli, wie z.B. Hormone oder Streß, nach denen ihre Expression rapide induziert wird (172). Unsere Ergebnisse bestätigen diese Daten. Wir können in unserer Arbeit ein verändertes Expressionsmuster der Transkriptionsfaktoren zeigen (x 3 Atf3, x 3 Nfil3, x 2 Stat3, x 3 Irf1). Der aktivierende Transkriptionsfaktor-3 (ATF-3), zeigte in vorangegangenen Studien eine Korrelation zu Hirnischämie. Das Expressionsprofil von ATF-3 nach exogen induzierter Hirnischämie am Rattenmodell zeigte sich sofort, aber auch nachfolgend signifikant zur Kontrollgruppe erhöht. Betroffene Diskussion 66 Areale des Gehirns zeigten nach Schädigung vermehrte Expression von ATF-3, was seine Funktion als Marker für Neuronenschädigung aufzeigen kann (137, 172). Ebenso zeigt sich ATF-3 als Induktor der erhöhten Expression von Hämoxygenase-1, welche, wie unten näher beschrieben, eine wichtige Rolle in der Regeneration und Protektion der Niere spielt (173). Andere Arbeiten demonstrieren die erhöhte Expression von Transkriptionsfaktoren als profunden Trigger der inflammatorischen Kaskade, z.B. Signal-Transducer-and-Activator-of-Transcription-3, (STAT-3), das an der Genexpressionsinduktion der Akuten-Phase-Antwort beteiligt ist (162, 204). Interferon-Regulatory-Factor-1 (IRF-1), ein anderer Transkriptionsfaktor, spielt eine wichtige Rolle in der Induktion von Klasse1 und Klasse2 -MHC-Genen in der Niere und ist zudem in den zellulären Widerstand bei Nekrosevorgängen involviert (3, 80). Die erhöhte Expression ist mitunter ursächlich für die expansive Entzündungsreaktion in Nieren hirntoter Spender. 4.2.5 Transportergene Bekannt ist die biochemische Analyse von Serumparametern, die eine fortschreitende Nierenfunktionsverschlechterung anzeigen. Erhöhte Kaliumwerte im Serum lassen sich z.B. durch eine Dysregulation von Natrium- und Kaliumtransportern erklären. Eine andere Beobachtung ist die Herunterregulation des Aquaporin-2-Transporters in hirntoten Spendernieren. Unsere Ergebnisse zeigen eine Minderexpression von -2,0 der Hirntodgruppe im Vergleich zu der Kontrollgruppe. Aqp-2 ist ein Wasserkanal, der hauptsächlich in den Sammelrohrzellen der Niere platziert ist. Er spielt eine signifikante Rolle in der Rückresorption von Wasser und ist somit stark in die Rolle der Urinkonzentration integriert. Interessanterweise zeigt sich als eine Konsequenz des Hirntods die markant erhöhte Urinproduktion niedriger Konzentration, besser bekannt als Diabetes insipidus (29, 59). Unter diesem Aspekt scheint fraglich, ob die verminderte Aqp-2-Expression, diese Folgewirkung wenigstens teilweise erklären kann. Eine andere mögliche Ursache scheint die inadäquate Produktion von ADH durch den posterioren Anteil der Hypophyse zu sein, was Forschungsergebnisse einer Studie von Szabo et al. aufweisen (187). Desweiteren zeigen unsere Daten die Hochregulation des Lipocalin 2-Genes (x 8). Neutrophile-Gelatinase-bindende Protein (NGAL/ Lipocalin 2) ist ein wichtiger Vertreter der Lipocaline, die generell als extrazelluläre Transporterproteine klassifiziert werden. Sie haben eine große strukturelle und funktionelle Varianz und binden Retinoide, Fettsäuren und steroidale Proteine im Plasma. Sie weisen biologische Funktionen z.B. in der Zellhomeostase, der Immunmodulation, der Gustation und Olfaktion und des Pheromonhaushaltes auf. Zahlreiche Vertreter werden bislang be- Diskussion 67 schrieben, unter anderem das Rentinol-bindende Protein (RBA), Complement c8-Gamma-Lipocalin, Alpha-1-Mikroglobulin (A1M) und das Lipocalin 2/ NGAL (5, 55). Lipocalin 2/ NGAL wurde ursprünglich aus neutrophilen Granulozyten isoliert, ohne seine Funktion näher zu kennen. Es wirkt als Modulator der Immunantwort und spielt, wie c8-Gamma, eine wichtige Rolle in der Akuten-Phase-Antwort (54). Eine andere Arbeit zeigte, dass Lipocalin 2/NGAL protektive Komplexe mit Gelatinase B mit Auswirkungen auf regulatorische Prozesse der physiologischen und pathologischen Erneuerung von Gewebe, vor allem in der Angiogenese, bilden kann. Ebenso besitzt es bakterostatische Eigenschaften durch die kompetitive Bindung von Eisen zwischen Bakterien und Lipocalin (122). Die Arbeitsgruppe von Anwaar zeigte in Patienten mit hirnischämischem Schaden eine signifikante Erhöhung von NGAL im Vergleich zur Kontrollgruppe ohne Hirnschädigung. Dies kennzeichnet die chronische entzündliche Aktivität und endotheliale, sowie leukozytäre Aktivierung bei zerebrovaskulärem Schaden (9) In neueren Arbeiten konnte NGAL als Aktivator für Nephronenbildung in der embryonalen Niere identifiziert werden. Eine komplementäre DNA-Microarry-Analyse zeigte, dass Neutrophiles Gelatinase-assoziiertes Lipocalin 2 in Nieren nach ischämischem Schaden markant erhöht ist (185). Mishra et al. beobachteten in NGAL-behandelten Nieren eine verminderte apoptotische Antwort, was NGAL nierenprotektive Eigenschaften zuweisen lässt (121). Die schnelle Diagnose eines akuten Nierenschadens durch lokale oder systemische Schädigung ist häufig problematisch aufgrund der fehlenden frühen Biomarker für renale Dysfunktion. NGAL zeigte sich in verschiedenen Studien als günstiger früher Parameter, der bei Nierenschädigung bereits im frühen Stadium signifikant ansteigt. Dies geschieht 24 - 48 Stunden vor einem merklichen Kreatininanstieg. NGAL zeigt sich signifikant erhöht, sowohl im Serum als auch im Urin. Seine Verwendung als Marker für frühen Nierenschaden nach Transplantation ist daher sinnvoll und bereits in einigen Klassifikationen berücksichtigt (41, 164). 4.2.6 Apoptose Programmierter Zelltod wird als wichtiger Faktor in der Ursache des Transplantatschadens hirntoter Nierenspender akzeptiert (30). In der jetzigen Studie konnten wir eine Hochregulation der Expression unterschiedlicher Gene bestätigen, die sowohl proapoptotische als auch antiapoptotische Eigenschaften besitzen. (Bcl2al x 4, Birc3 x 4). Drognitz et al. konnten in der Freiburger Arbeitsgruppe einen Zusammenhang zwischen der Apoptose im azinären Pankreas einerseits und der verschlechterten Mikrozirkulation mit Zunahme der Leukozytenadhäsion auf der anderen Seite aufzeigen (46). Ebenfalls ist die Verknüpfung zum inflammatorischen Regelkreis zu beachten, denn hier zeigt IL-1β eine Diskussion 68 Mitbeteiligung bei der Aktivierung des Apoptoseweges. Der erhöhte Apoptoseprozess scheint bei der Verschlechterung der Organqualität von hirntoten Spendern im Vergleich zum Lebendspender eine signifikante Rolle zu spielen. Antiapoptotische Stimulierung zeigt im Umkehrschluß verbesserte Ergebnisse der Nierenfunktion von hirntoten Spendern. Eine Studie am Rattenmodell von Olszaneki et al. zeigte, dass eine Induktion der Expression protektiver und antiapoptotischer Gene, z.B. der Hämoxygenase-1, ein deutlich verbessertes hämodynamisches Gleichgewicht, verminderte Caspase-3-Aktivität, zudem aber erhöhte Konzentrationen von antiapoptotischen Molekülen, z.B. Bcl-2 und Bcl-Xl, mit sich führte (141). 4.2.7 Gerinnung Als wichtigster Trigger des plasmatischen Gerinnungssystems ist der „extrinsic pathway“ oder „tissue factor pathway“ anzusehen. Er wird initiiert, wenn Gewebethromboplastin, Faktor III, (tissue factor, TF) mit dem plasmatischen Faktor VII (FVII) in Kontakt kommt und einen Komplex bildet, wobei der Faktor VII zu Faktor VIIa (FVIIa) aktiviert wird. Gewebethromboplastin ist ein Phospholipoprotein, das aus verletzten Gewebezellen freigesetzt wird, ebenso auf Makrophagen erscheint und die weiterführende Gerinnungskaskade stimuliert. Auf einem unbeschädigten Endothel ist es dagegen gering oder gar nicht präsent. Die systemische Inflammation während des Hirntodes stimuliert die Gerinnung. Durch die veminderte Mikrozirkulation, durch den „autonomic storm“, kommt es zur Hochregulation von Prothrombin (158). Ebenso induzieren proinflammatorische Zytokine, z.B. Il-1, Il-6 und TNF, die Expression des Faktor III auf Endothelien und Monozyten. Durch Verminderung der antithrombotischen Eigenschaften dieser Zellen entsteht eine erhöhte Koagulationsneigung. In unserer Arbeit finden sich hochexprimierte Gene, die für Fibrinogen kodieren (Fgb x 16, Fgg x 7, Fgl2 x 7), aber auch für Prothrombin (x 3) und Gerinnungsfaktor III (x 3). Fibrinogen wird durch Thrombin in Fibrin umgewandelt, welches eines der wichtigsten Komponenten der Hämostase darstellt. Des Weiteren zeigen unterschiedliche Spaltformen von Fibrinogen chemotaktische Aktvität und regulieren Teile der Zelladhäsion (123). Der Zusammenhang zwischen Thrombose und Entzündung ist bereits durch Krankheitsbilder der Sepsis oder der Arteriosklerose bekannt und zeigt eine gegenseitige Beeinflussung der beiden Systeme auf. Studien zeigen bereits, dass PIC die Gerinnungsaktivität beeinflussen und im Umkehrschluss auch Elemente des Gerinnungssystems auf die Entzündungssituation einwirken (88) Vor mehr als zehn Jahren bewiesen Herfy et al. bereits profunde Störungen im Gerinnungssystem hirntoter Spender (74). Jüngste Forschungsergebnisse postulieren, dass die Induktion von Fgl2 auf Diskussion 69 vaskulärem Endothel eine Rolle in der Pathogenese akuter vaskulärer abstoßungsassoziierter Thrombosen spielen könnte (128). Tissue factor pathway inhibitor 2 ist ein Serinproteinase-Inhibitor, der als zentraler Inhibitor der exogenen Blutgerinnung bekannt ist, aber auch Funktionen im Bereich der Immunmodulation, Tumorsuppression und Arteriosklerose aufweist. Dieser Faktor zeigt sich in unserer Arbeit x 11.2 hochreguliert, was einerseits eine Gegenregulation des Körpers auf eine prokoagulatorische Situation darstellen könnte, oder unverstandene Prozesse im Bereich der Immunmodulation. Zusammenfassend lassen sich die Veränderungen im Gerinnungssystem hinsichtlich des Hirntodes als spenderspezifischer Faktor beschreiben, der die Qualität des Spenderorgans verschlechtern kann. Dabei bestätigen sich die Erkenntnisse, dass eine Hyperkoagobilität zu einer Entwicklung einer Thrombose in der Spenderniere führen könnte, die dann eine Mitursache für einen frühzeitigen Organverlust darstellt (172). 4.2.8 Heat-Shock-Proteine und Regenerationsproteine Die endothelialen Nahtstellen sind die ersten Barrieren in einem vaskularisierten Organ, die sich der Attacke des Immunsystems stellen. Das Endothel ist in der Lage, seinen Phänotyp drastisch zu verändern und Adhäsionsmoleküle zu exprimieren (z.B. Selektine, VCAMs, ICAMS, Chemokine, MCP-1 und Gewebsfaktoren). Es stellt somit eine proinflammatorische Umgebung her, die die zelluläre Invasion und Destruktion des Transplantats durch entzündliche Effektoren erleichtert und zur Apoptoseinduktion führen kann. Die Erhaltung eines integren Endotheliums sollte ein vaskularisiertes Tranplantat beschützen und sich positiv auf sein Überleben auswirken. Dieses Vermögen wird durch zytoprotektive Proteine gefördert. Die Definition eines zytoprotektiven Proteins beinhaltet sein Vermögen, das Überleben und die physiologischen Funktionen der Zellen aufrechtzuerhalten, also in Verteidigung und Reperatur der Organe involviert ist (11). Das Ausmaß des Transplantatschadens reflektiert die Balance zwischen schädigenden inflammatorischen Prozessen und protektiven Faktoren. Wie in der Einleitung beschrieben, besitzen Heat-Shock-Proteine zytoprotektive Eigenschaften und werden durch unterschiedliche Arten des Stresses induziert, z.B. Ischämie oder Toxineinwirkung. In der Niere wurde ihre Expression in den Glomeruli, tubulären epithelialen Zellen und den glatten Muskelschichten renaler Arterien detektiert. Diese zellulären Kompartimente werden bei der akuten und chronischen Transplantatabstoßung durch proinflammatorische Stimuli zur erhöhten Expression protektiver Gene angeregt (11, 101, 167). Die Hämoxygenase-1 zeigt sowohl antioxidative und antiinflammatorische Eigenschaften, ebenso moduliert sie den vaskulären Tonus. HO-1 induziert Bcl2, ein antiapoptotisches Gen und übt somit ebenfalls eine Hemmung der Apopotose aus (101). Diskussion 70 Trotzdem ist die Antwort dieser zellschützenden Gene oft nicht ausreichend, eine irreversible Abstoßung zu verhindern, obwohl sie vielleicht wirksam und nötig ist, das Leben von renalen Zellkomponenten aufrechtzuerhalten, bis die Immunattacke durch externe medikamentöse Immunsuppression zu kontrollieren ist (101). Hirntodinduzierte protektive Genantwort scheint in der Klinik nur insuffizienten Schutz für die Nierenzellen zu bieten, da die schädlichen Prozesse durch den zerebralen Schaden nicht aufzuwiegen sind. Die mRNA-Levels von HO-1 präsentieren sich in verschiedenen tierexperimentellen Forschungsarbeiten über Nierentransplantationen mit und ohne Hirntodinduktion mit unterschiedlichen Ergebnissen. Lemos et al. zeigten in hirntoten Spendernieren signifikant niedrigere mRNA-Level von HO-1, als in Nieren von Lebendspendern (101). Erklärt wird dies durch eine verlängerte kalte Ischämiezeit bei Nierentransplantationen hirntoter Spender und die defekte Anpassung des hirntoten Spenders an den Ischämie-Reperfusionsschaden (101, 167). In Arbeiten von Njiboer und Schuurs et al. zeigt sich eine signifikante Hochregulation der Expression von HO-1 im Vergleich zu Lebenspendern, was wir in unserer Arbeit bestätigen können (+31). Die Hochexpression zeigt die Reaktion des Körpers auf den Hirntod als systemischen Stressfaktor und den Versuch Gewebsdestruktion durch inflammatorische Invasion und Apoptoseinduktion zu reduzieren. In Nierentransplantationsmodellen ließ sich eine künstliche Überexpression von HO-1 durch pharmakologische Interventionen oder Hitze-Präkonditionierung erzeugen. Die so manipulierten Gruppen zeigten ein deutlich verbessertes Transplantatüberleben und signifikant erhöhte Langzeitfunktionen, bei gleichzeitig verminderter Apoptose und erniedrigten proinflammatorischen Zytokinkonzentrationen (162, 209). Limitationen und Ausblick 4.3 71 Limitation und Ausblick Unsere Arbeit untersucht den Hirntod und seine Auswirkungen auf genetischer Ebene durch die Chipanalyse einer großen Anzahl an Genen, die in ihrer Expression beeinflusst werden. Diese sind an einer Vielzahl pathophysiologischer Kaskaden beteiligt. Der Hirntod des Spenders und die unter dem Begriff des „Autonomic Storms“ zusammengefassten Veränderungen führen im Ergebnis zu einer Verschlechterung der Organqualität mit erhöhter Inflammation, Immunogenität und Thrombogenität des Transplantates. Dies kann als Konsequenz mit Funktionsverschlechterungen und einer erhöhten Frequenz von Transplantatabstoßungen korrelieren. Durch das elementare Ereignis des Hirntodes wird neben dem schädlichen Einfluss auf die peripheren Organe jedoch auch die Bildung protektiver Faktoren initiiert, was in unseren Ergebnissen dargestellt werden kann. Nicht berücksichtigt wird in unserer Untersuchung ein Nachweis für Veränderungen auf Proteinebene, die Limitation erfolgt hier auf die Genexpressionsveränderungen im Rahmen des Hirntodereignisses. Durch Verwendung eines Nierengesamthomogenats ist in unserer Studie eine Zuordnung der Expressionsveränderung auf bestimmten Kompartimenten der Niere, z.B. Endothel, Bindegewebe, Glomeruli, Tubuluszellen, oder auch adhärenten oder infiltrierten Leukozyten, nicht möglich. Mit einer Liegezeit von sechs Stunden wird weiterhin nur die Frühphase nach Hirntod dargestellt und somit lediglich die Gesamtveränderung der Genexpression innerhalb dieses Zeitfensters analysiert. Zusammenfassend lässt sich mit den von uns durchgeführten Untersuchungen unter Nutzung der DNA Microarrys eine Vielzahl von differentiell exprimierten Genen nach Hirntodinduktion identifizieren. Größtenteils korrelieren unsere Ergebnisse mit den Ergebnissen anderer Arbeitsgruppen und bestätigen mit den gefundenen Expressionsveränderungen die Aussage, dass der Hirntod als elementares Ereignis die erhöhte Expression schädlicher Fakoren schon vor Transplantation bewirkt. Die Ergebnisse ermöglichen die Weiterentwicklung non-invasiver peritransplantativer Serum- oder Urin-Biomarker, die ein Abschätzen der zukünftigen Langzeitfunktion von Spendernieren oder das Risiko einer akuten Abstoßung, ermöglichen. Ergebnisse unserer und anderer Arbeiten sollen die Basis weiterer Forschungsinterventionen bilden, beispielsweise die Analyse der Veränderungen einzelner Zellkompartimente in der Niere im Rahmen des Hirntodes und die Entwicklung weiterer Therapieoptionen. Anhand der erworbenen Erkenntnisse scheint es möglich, zukünftig gezielte Interventionen am hirntoten Spender zur Stabilisierung, bzw. zur Verbesserung der Transplantatqualität vor der Explantation und Konservierung durchzuführen. Durch iatrogene Stimulation oder Inhibition auf molekulargeneti- Limitationen und Ausblick 72 scher Ebene könnten schon prophylaktisch pathologische Kaskaden unterbunden, oder protektive, körpereigene Mechanismen gefördert. Zusammenfassung 5. 73 Zusammenfassung Einleitung: Die Nierentransplantation als Ersatztherapie bei fortgeschrittener Niereninsuffizienz ist Therapie der Wahl. Die Hauptquelle der Transplantate stellen postmortale Spenderorgane dar. Dennoch liegen diese in den Langzeitergebnissen signifikant unter den Nierenfunktionen von Lebendspendern. Dies ist unabhängig von bekannten antigenabhängigen Faktoren. Der Hirntod als primär antigenunabhängiger Faktor übt einen entscheidenden Einfluß auf posttransplantative Resultate aus. Im Fokus der Forschung stehen die weitreichenden systemmischen Veränderungen, die schon auf molekulargenetischer Ebene durch den Hirntod per se beeinflusst werden. Ziel unserer Arbeit war die Analyse und Identifikation von differentiell exprimierten Genen nach Hirntodinduktion im Vergleich zu einer Kontrollgruppe. Methoden: In unserer Arbeit wird ein Tiermodell des graduellen normotensiven Hirntodes verwendet. Nach Induktion des Hirntodes werden nach sechsstündiger Beobachtungszeit die Nieren und anderen Organe entnommen und im Folgenden einer RNA-Extraktion unterzogen. Die RNA wird nach Pooling durch die Affymetrix-Genchiphybridisierung weiterverarbeitet. Die Ergebnisse der unterschiedlich exprimierten Gene werden in unterschiedliche Funktionsgruppen gegliedert. Einzelne Gene unterschiedlicher Funktionsgruppen werden exemplarisch durch eine quantitative Two-step-RT-PCR verifiziert. Als Vergleich dient eine Kontrollgruppe mit gleichfalls sechsstündiger Beobachtungszeit (Narkose und Beatmung) ohne Hirntodinduktion. Ergebnisse: Neben der Darstellung des zeitlichen Verlaufes von Kreislauf- und Blutparametern zwischen Hirntod- und Kontrollgruppe, können durch die Chiphybridisierung 150 unterschiedlich exprimierte Gene aufgeschlüsselt und 88 relevante Gene in unterschiedliche Funktionsbereiche eingeteilt werden. Große Veränderungen zeigen die Genexpressionen der Zyto- und Chemokine, aber auch der Adhäsionsmoleküle, der Gerinnung, Transkription, der Apoptose, aber auch der Protektion und Reparatur. Die RT-PCR kann bei drei ausgewählten Genen diese Expressionsunterschiede auf qualitativer Ebene signifikant verifizieren. Schlussfolgerung: Die induzierten Expressionsveränderungen von Genen unterschiedlichster Funktionsgruppen, zeigen die Auswirkung des Hirntodes als primär antigenunabhängiger Faktor prätransplantativ. Der Hirntod per se induziert eine Herauf-/ oder Herabregulation von Genen, die z.B. eine verstärkte Inflammation, Immunogenität, Thrombosierung oder Apoptose schon vor der Explantation kodieren. Bemerkenswert ist der Versuch des Körpers, durch vermehrte Expression zytoprotektiver Gene (z.B. die Hämoxygenase-1) körpereigene Schutzmechanismen zu aktivieren. Unsere Ergebnisse bestätigen und ergänzen Ergebnisse vorhergehender wissenschaftlicher Arbeiten und sollen das Verständnis der Auswirkungen des Hirntodes in der Transplantationsmedizin erweitern. Zukunftsgerichtet liegen die Ziele dieses Forschungsschwerpunktes in den medizinischpharmazeutischen Möglichkeiten, in pathologische Mechanismen einzugreifen und die Resultate der Transplantationen bei postmortalen Spendern, Hauptquelle der Nierentransplantate, zu verbessern. Literaturverzeichnis und Quellenangaben 6. 74 Literaturverzeichnis und Quellenangaben 1. A definition of irreversible coma. Report of the Ad Hoc Committee of the Harvard Medical School to Examine the Definition of Brain Death (1968) Jama, 205: 337-40 2. S. Afford and S. Randhawa (2000) Apoptosis Mol Pathol, 53: 55-63 3. M. Afrouzian, V. Ramassar, J. Urmson, L. F. Zhu and P. F. Halloran (2002) Transcription factor IRF-1 in kidney transplants mediates resistance to graft necrosis during rejection J Am Soc Nephrol, 13: 1199-209 4. Z. Akcetin, R. Pregla, D. Darmer, H. J. Bromme and J. Holtz (2000) During ischemia-reperfusion in rat kidneys, heat shock response is not regulated by expressional changes of heat shock factor 1 Transpl Int, 13: 297-302 5. B. Akerstrom, D. R. Flower and J. P. Salier (2000) Lipocalins: unity in diversity Biochim Biophys Acta, 1482: 1-8 6. P. S. Almond, A. Matas, K. Gillingham, D. L. Dunn, W. D. Payne, P. Gores, R. Gruessner and J. S. Najarian (1993) Risk factors for chronic rejection in renal allograft recipients Transplantation, 55: 752-6; discussion 756-7 7. J. A. Amado, F. Lopez-Espadas, A. Vazquez-Barquero, E. Salas, J. A. Riancho, J. J. Lopez-Cordovilla and M. T. Garcia-Unzueta (1995) Blood levels of cytokines in brain-dead patients: relationship with circulating hormones and acute-phase reactants Metabolism, 44: 812-6 8. R. E. Anderson and J. L. Atkinson (2003) Intracranial pressure response to severe head injury induced apnea and catecholamine surge J Trauma, 54: 550-4 9. I. Anwaar, A. Gottsater, K. Ohlsson, I. Mattiasson and F. Lindgarde (1998) Increasing levels of leukocyte-derived inflammatory mediators in plasma and cAMP in platelets during follow-up after acute cerebral ischemia Cerebrovasc Dis, 8: 310-7 10. K. Arita, T. Uozumi, S. Oki, K. Kurisu, M. Ohtani and T. Mikami (1993) The function of the hypothalamo-pituitary axis in brain dead patients Acta Neurochir (Wien), 123: 64-75 Literaturverzeichnis und Quellenangaben 75 11. Y. Avihingsanon, N. Ma, E. Csizmadia, C. Wang, M. Pavlakis, M. Giraldo, T. B. Strom, M. P. Soares and C. Ferran (2002) Expression of protective genes in human renal allografts: a regulatory response to injury associated with graft rejection Transplantation, 73: 1079-85 12. V. S. Avlonitis, C. H. Wigfield, J. A. Kirby and J. H. Dark (2005) The hemodynamic mechanisms of lung injury and systemic inflammatory response following brain death in the transplant donor Am J Transplant, 5: 684-93 13. H. Azuma, K. Nadeau, M. Takada, H. S. Mackenzie and N. L. Tilney (1997) Cellular and molecular predictors of chronic renal dysfunction after initial ischemia/reperfusion injury of a single kidney Transplantation, 64: 190-7 14. V. Bailly, Z. Zhang, W. Meier, R. Cate, M. Sanicola and J. V. Bonventre (2002) Shedding of kidney injury molecule-1, a putative adhesion protein involved in renal regeneration J Biol Chem, 277: 39739-48 15. K. Bando, I. L. Paradis, S. Similo, H. Konishi, K. Komatsu, T. G. Zullo, S. A. Yousem, J. M. Close, A. Zeevi, R. J. Duquesnoy and et al. (1995) Obliterative bronchiolitis after lung and heart-lung transplantation. An analysis of risk factors and management J Thorac Cardiovasc Surg, 110: 4-13; discussion 13-4 16. C. N. Barnard (1968) Human cardiac transplantation. An evaluation of the first two operations performed at the Groote Schuur Hospital, Cape Town Am J Cardiol, 22: 584-96 17. G. Baroldi, G. Di Pasquale, M. D. Silver, G. Pinelli, A. M. Lusa and V. Fineschi (1997) Type and extent of myocardial injury related to brain damage and its significance in heart transplantation: a morphometric study J Heart Lung Transplant, 16: 994-1000 18. I. J. Benjamin and D. R. McMillan (1998) Stress (heat shock) proteins: molecular chaperones in cardiovascular biology and disease Circ Res, 83: 117-32 19. S. Benz, M. Lobler, R. Obermaier, B. Kortmann, F. Pfeffer, D. Koczan, H. J. Thiesen and U. T. Hopt (2002) New possible target genes in pancreatic ischemia/reperfusion-injury identified by microarray analysis Transplant Proc, 34: 2369-71 Literaturverzeichnis und Quellenangaben 76 20. M. P. Bevilacqua and R. M. Nelson (1993) Selectins J Clin Invest, 91: 379-87 21. E. J. Birks, P. B. Burton, V. J. Owen, N. Latif, B. Nyawo and M. H. Yacoub (2001) Molecular and cellular mechanisms of donor heart dysfunction Transplant Proc, 33: 2749-51 22. E. J. Birks, M. H. Yacoub, P. S. Burton, V. Owen, A. Pomerance, A. O'Halloran, N. R. Banner, A. Khaghani and N. Latif (2000) Activation of apoptotic and inflammatory pathways in dysfunctional donor hearts Transplantation, 70: 1498-506 23. H. B. Bittner, S. W. Kendall, K. A. Campbell, T. J. Montine and P. Van Trigt (1995) A valid experimental brain death organ donor model J Heart Lung Transplant, 14: 308-17 24. H. Boom, M. J. Mallat, J. W. de Fijter, A. H. Zwinderman and L. C. Paul (2000) Delayed graft function influences renal function, but not survival Kidney Int, 58: 859-66 25. M. Boratynska (1998) [The role of monocyte chemotactic peptide (MCP-1) in chronic renal allograft rejection] Pol Arch Med Wewn, 99: 272-80 26. G. Bouma, W. K. Lam-Tse, A. F. Wierenga-Wolf, H. A. Drexhage and M. A. Versnel (2004) Increased serum levels of MRP-8/14 in type 1 diabetes induce an increased expression of CD11b and an enhanced adhesion of circulating monocytes to fibronectin Diabetes, 53: 1979-86 27. K. Burkhardt, M. Radespiel-Troger, H. D. Rupprecht, M. Goppelt-Struebe, R. Riess, L. Renders, I. A. Hauser and U. Kunzendorf (2001) An increase in myeloid-related protein serum levels precedes acute renal allograft rejection J Am Soc Nephrol, 12: 1947-57 28. M. Busson, P. N'Doye, G. Benoit, L. Hannoun, R. Adam, A. Pavie, P. Romano and J. Hors (1995) Donor factors influencing organ transplant prognosis Transplant Proc, 27: 1662-4 29. M. A. Cadnapaphornchai, Y. W. Kim, A. K. Gurevich, S. N. Summer, S. Falk, J. M. Thurman and R. W. Schrier (2003) Urinary concentrating defect in hypothyroid rats: role of sodium, potassium, 2-chloride co-transporter, and aquaporins J Am Soc Nephrol, 14: 566-74 Literaturverzeichnis und Quellenangaben 77 30. M. P. Castaneda, A. Swiatecka-Urban, M. M. Mitsnefes, D. Feuerstein, F. J. Kaskel, V. Tellis and P. Devarajan (2003) Activation of mitochondrial apoptotic pathways in human renal allografts after ischemiareperfusion injury Transplantation, 76: 50-4 31. J. M. Cecka (1995) Living donor transplants Clin Transpl: 363-77 32. J. M. Cecka (2003) The OPTN/UNOS Renal Transplant Registry 2003 Clin Transpl: 1-12 33. J. M. Cecka and L. Cho (1988) Sensitization Clin Transpl: 365-73 34. E. P. Chen, H. B. Bittner, S. W. Kendall and P. Van Trigt (1996) Hormonal and hemodynamic changes in a validated animal model of brain death Crit Care Med, 24: 1352-9 35. G. M. Chertow, E. L. Milford, H. S. Mackenzie and B. M. Brenner (1996) Antigen-independent determinants of cadaveric kidney transplant failure Jama, 276: 1732-6 36. G. L. Clifton, C. S. Robertson, K. Kyper, A. A. Taylor, R. D. Dhekne and R. G. Grossman (1983) Cardiovascular response to severe head injury J Neurosurg, 59: 447-54 37. H. Cullup, A. M. Dickinson, G. H. Jackson, P. R. Taylor, J. Cavet and P. G. Middleton (2001) Donor interleukin 1 receptor antagonist genotype associated with acute graft-versushost disease in human leucocyte antigen-matched sibling allogeneic transplants Br J Haematol, 113: 807-13 38. M. A. Daemen, C. van 't Veer, G. Denecker, V. H. Heemskerk, T. G. Wolfs, M. Clauss, P. Vandenabeele and W. A. Buurman (1999) Inhibition of apoptosis induced by ischemia-reperfusion prevents inflammation J Clin Invest, 104: 541-9 39. J. W. de Fijter, M. J. Mallat, Doxiadis, II, J. Ringers, F. R. Rosendaal, F. H. Claas and L. C. Paul (2001) Increased immunogenicity and cause of graft loss of old donor kidneys J Am Soc Nephrol, 12: 1538-46 40. A. De Maio (1999) Heat shock proteins: facts, thoughts, and dreams Shock, 11: 1-12 Literaturverzeichnis und Quellenangaben 78 41. C. L. Dent, Q. Ma, S. Dastrala, M. Bennett, M. M. Mitsnefes, J. Barasch and P. Devarajan (2007) Plasma NGAL predicts acute kidney injury, morbidity and mortality after pediatric cardiac surgery: a prospective uncontrolled cohort study Crit Care, 11: R127 42. I. Dimopoulou, S. Korfias, U. Dafni, A. Anthi, C. Psachoulia, G. Jullien, D. E. Sakas and C. Roussos (2003) Protein S-100b serum levels in trauma-induced brain death Neurology, 60: 947-51 43. I. Dimopoulou, S. Tsagarakis, A. Anthi, E. Milou, I. Ilias, K. Stavrakaki, C. Charalambidis, M. Tzanela, S. Orfanos, K. Mandragos, N. Thalassinos and C. Roussos (2003) High prevalence of decreased cortisol reserve in brain-dead potential organ donors Crit Care Med, 31: 1113-7 44. L. Dosemeci, M. Yilmaz, M. Cengiz, B. Dora and A. Ramazanoglu (2004) Brain death and donor management in the intensive care unit: experiences over the last 3 years Transplant Proc, 36: 20-1 45. O. Drognitz, S. Benz, F. Pfeffer, C. Fischer, F. Makowiec, W. Schareck and U. T. Hopt (2004) Long-term follow-up of 78 simultaneous pancreas-kidney transplants at a single-center institution in Europe Transplantation, 78: 1802-8 46. O. Drognitz, R. Obermaier, X. Liu, H. Neeff, E. von Dobschuetz, U. T. Hopt and S. Benz (2004) Effects of organ preservation, ischemia time and caspase inhibition on apoptosis and microcirculation in rat pancreas transplantation Am J Transplant, 4: 1042-50 47. S. P. Eisenberg, M. T. Brewer, E. Verderber, P. Heimdal, B. J. Brandhuber and R. C. Thompson (1991) Interleukin 1 receptor antagonist is a member of the interleukin 1 gene family: evolution of a cytokine control mechanism Proc Natl Acad Sci U S A, 88: 5232-6 48. A. Endo, T. Yagi, A. Nakao, H. Matsukawa, Y. Okada, K. Fujisawa, H. Matsuda, M. Oishi, T. Matsuno, H. Iwagaki and N. Tanaka (1999) Cytokine analysis of cerebrohepatic organ interaction in porcine liver transplantation from brain-dead donors Transplant Proc, 31: 2698 49. X. Escofet, H. Osman, D. F. Griffiths, S. Woydag and W. Adam Jurewicz (2003) The presence of glomerular sclerosis at time zero has a significant impact on function after cadaveric renal transplantation Transplantation, 75: 344-6 Literaturverzeichnis und Quellenangaben 79 50. A. C. Farney, E. Cho, E. J. Schweitzer, B. Dunkin, B. Philosophe, J. Colonna, S. Jacobs, B. Jarrell, J. L. Flowers and S. T. Bartlett (2000) Simultaneous cadaver pancreas living-donor kidney transplantation: a new approach for the type 1 diabetic uremic patient Ann Surg, 232: 696-703 51. K. Fassbender, S. Rossol, T. Kammer, M. Daffertshofer, S. Wirth, M. Dollman and M. Hennerici (1994) Proinflammatory cytokines in serum of patients with acute cerebral ischemia: kinetics of secretion and relation to the extent of brain damage and outcome of disease J Neurol Sci, 122: 135-9 52. R. Ferrera, M. Ovize, B. Claustrat and G. Hadour (2005) Stable myocardial function and endocrine dysfunction during experimental brain death J Heart Lung Transplant, 24: 921-7 53. I. Finkelstein, L. H. Toledo-Pereyra and J. Castellanos (1987) Physiologic and hormonal changes in experimentally induced brain dead dogs Transplant Proc, 19: 4156-8 54. T. H. Flo, K. D. Smith, S. Sato, D. J. Rodriguez, M. A. Holmes, R. K. Strong, S. Akira and A. Aderem (2004) Lipocalin 2 mediates an innate immune response to bacterial infection by sequestrating iron Nature, 432: 917-21 55. D. R. Flower (1996) The lipocalin protein family: structure and function Biochem J, 318 ( Pt 1): 1-14 56. U. Frei, Schober-Halstenberg, H.-J. (2005) Renal Replacement Therapy in Germany QuaSi-Niere Annual Report, Berlin, Germany: 57. M. Frosch, T. Vogl, R. Waldherr, C. Sorg, C. Sunderkotter and J. Roth (2004) Expression of MRP8 and MRP14 by macrophages is a marker for severe forms of glomerulonephritis J Leukoc Biol, 75: 198-206 58. S. V. Fuggle, J. B. Sanderson, D. W. Gray, A. Richardson and P. J. Morris (1993) Variation in expression of endothelial adhesion molecules in pretransplant and transplanted kidneys--correlation with intragraft events Transplantation, 55: 117-23 59. K. Fushimi, S. Uchida, Y. Hara, Y. Hirata, F. Marumo and S. Sasaki (1993) Cloning and expression of apical membrane water channel of rat kidney collecting tubule Nature, 361: 549-52 Literaturverzeichnis und Quellenangaben 80 60. V. Gahtan, E. T. Olson and B. E. Sumpio (2002) Molecular biology: a brief overview J Vasc Surg, 35: 563-8 61. M. Gasser, A. M. Waaga, J. E. Kist-Van Holthe, S. M. Lenhard, I. Laskowski, G. D. Shaw, W. W. Hancock and N. L. Tilney (2002) Normalization of brain death-induced injury to rat renal allografts by recombinant soluble P-selectin glycoprotein ligand J Am Soc Nephrol, 13: 1937-45 62. M. Gasser, A. M. Waaga, I. A. Laskowski and N. L. Tilney (2000) The influence of donor brain death on short and long-term outcome of solid organ allografts Ann Transplant, 5: 61-7 63. D. W. Gjertson and J. M. Cecka (2000) Living unrelated donor kidney transplantation Kidney Int, 58: 491-9 64. N. Goes, J. Urmson, V. Ramassar and P. F. Halloran (1995) Ischemic acute tubular necrosis induces an extensive local cytokine response. Evidence for induction of interferon-gamma, transforming growth factor-beta 1, granulocyte-macrophage colony-stimulating factor, interleukin-2, and interleukin-10 Transplantation, 59: 565-72 65. C. J. Graf and N. P. Rossi (1978) Catecholamine response to intracranial hypertension J Neurosurg, 49: 862-8 66. D. I. Graham, T. K. McIntosh, W. L. Maxwell and J. A. Nicoll (2000) Recent advances in neurotrauma J Neuropathol Exp Neurol, 59: 641-51 67. H. J. Gramm, H. Meinhold, U. Bickel, J. Zimmermann, B. von Hammerstein, F. Keller, R. Dennhardt and K. Voigt (1992) Acute endocrine failure after brain death? Transplantation, 54: 851-7 68. H. J. Gramm, J. Zimmermann, H. Meinhold, R. Dennhardt and K. Voigt (1992) Hemodynamic responses to noxious stimuli in brain-dead organ donors Intensive Care Med, 18: 493-5 69. S. Gupta (2000) Molecular steps of cell suicide: an insight into immune senescence J Clin Immunol, 20: 229-39 70. R. D. Guttmann, R. D. Forbes and S. X. Zheng (1997) The pathophysiology of chronic rejection Am J Med Sci, 313: 302-4 Literaturverzeichnis und Quellenangaben 81 71. W. H. Hancock, W. D. Whitley, S. G. Tullius, U. W. Heemann, B. Wasowska, W. M. Baldwin, 3rd and N. L. Tilney (1993) Cytokines, adhesion molecules, and the pathogenesis of chronic rejection of rat renal allografts Transplantation, 56: 643-50 72. I. A. Hauser, R. Riess, B. Hausknecht, H. Thuringer and R. B. Sterzel (1997) Expression of cell adhesion molecules in primary renal disease and renal allograft rejection Nephrol Dial Transplant, 12: 1122-31 73. P. Hayry, A. Mennander, S. Yilmaz, J. Ustinov, A. Raisanen, A. Miettinen, I. Lautenschlager, K. Lemstrom, C. A. Bruggeman and T. Paavonen (1992) Towards understanding the pathophysiology of chronic rejection Clin Investig, 70: 780-90 74. T. R. Hefty, L. W. Cotterell, S. C. Fraser, S. H. Goodnight and T. R. Hatch (1993) Disseminated intravascular coagulation in cadaveric organ donors. Incidence and effect on renal transplantation Transplantation, 55: 442-3 75. C. W. Heizmann (2002) The multifunctional S100 protein family Methods Mol Biol, 172: 69-80 76. P. Herijgers and W. Flameng (1998) The effect of brain death on cardiovascular function in rats. Part II. The cause of the in vivo haemodynamic changes Cardiovasc Res, 38: 107-15 77. P. Herijgers, V. Leunens, T. B. Tjandra-Maga, K. Mubagwa and W. Flameng (1996) Changes in organ perfusion after brain death in the rat and its relation to circulating catecholamines Transplantation, 62: 330-5 78. I. Herrero-Fresneda, J. Torras, A. Vidal, N. Lloberas, J. M. Cruzado and J. M. Grinyo (2005) Reduction of postischemic immune inflammatory response: an effective strategy for attenuating chronic allograft nephropathy Transplantation, 79: 165-73 79. R. Higuchi, G. Dollinger, P. S. Walsh and R. Griffith (1992) Simultaneous amplification and detection of specific DNA sequences Biotechnology (N Y), 10: 413-7 80. M. Hobart, V. Ramassar, N. Goes, J. Urmson and P. F. Halloran (1997) IFN regulatory factor-1 plays a central role in the regulation of the expression of class I and II MHC genes in vivo J Immunol, 158: 4260-9 Literaturverzeichnis und Quellenangaben 82 81. U. T. Hopt (2003) [Transplant surgery] Zentralbl Chir, 128: 785 82. T. A. Howlett, A. M. Keogh, L. Perry, R. Touzel and L. H. Rees (1989) Anterior and posterior pituitary function in brain-stem-dead donors. A possible role for hormonal replacement therapy Transplantation, 47: 828-34 83. E. C. Hsiao, L. G. Koniaris, T. Zimmers-Koniaris, S. M. Sebald, T. V. Huynh and S. J. Lee (2000) Characterization of growth-differentiation factor 15, a transforming growth factor beta superfamily member induced following liver injury Mol Cell Biol, 20: 3742-51 84. A. Humar, B. Durand, K. Gillingham, W. D. Payne, D. E. Sutherland and A. J. Matas (2000) Living unrelated donors in kidney transplants: better long-term results than with nonHLA-identical living related donors? Transplantation, 69: 1942-5 85. T. Ichimura, J. V. Bonventre, V. Bailly, H. Wei, C. A. Hession, R. L. Cate and M. Sanicola (1998) Kidney injury molecule-1 (KIM-1), a putative epithelial cell adhesion molecule containing a novel immunoglobulin domain, is up-regulated in renal cells after injury J Biol Chem, 273: 4135-42 86. R. B. Isaacs, S. L. Nock, C. E. Spencer, A. F. Connors, Jr., X. Q. Wang, R. Sawyer and P. I. Lobo (1999) Racial disparities in renal transplant outcomes Am J Kidney Dis, 34: 706-12 87. W. Jassem, D. D. Koo, L. Cerundolo, M. Rela, N. D. Heaton and S. V. Fuggle (2003) Leukocyte infiltration and inflammatory antigen expression in cadaveric and livingdonor livers before transplant Transplantation, 75: 2001-7 88. K. Johnson, Y. Choi, E. DeGroot, I. Samuels, A. Creasey and L. Aarden (1998) Potential mechanisms for a proinflammatory vascular cytokine response to coagulation activation J Immunol, 160: 5130-5 89. C. Jolly and R. I. Morimoto (2000) Role of the heat shock response and molecular chaperones in oncogenesis and cell death J Natl Cancer Inst, 92: 1564-72 90. H. Kim and J. S. Cheigh (2001) Kidney transplantation in patients with type 1 diabetes mellitus: long-term prognosis for patients and grafts Korean J Intern Med, 16: 98-104 Literaturverzeichnis und Quellenangaben 83 91. O. H. Koning, R. J. Ploeg, J. H. van Bockel, M. Groenewegen, F. J. van der Woude, G. G. Persijn and J. Hermans (1997) Risk factors for delayed graft function in cadaveric kidney transplantation: a prospective study of renal function and graft survival after preservation with University of Wisconsin solution in multi-organ donors. European Multicenter Study Group Transplantation, 63: 1620-8 92. D. D. Koo, K. I. Welsh, A. J. McLaren, J. A. Roake, P. J. Morris and S. V. Fuggle (1999) Cadaver versus living donor kidneys: impact of donor factors on antigen induction before transplantation Kidney Int, 56: 1551-9 93. D. D. Koo, K. I. Welsh, J. A. Roake, P. J. Morris and S. V. Fuggle (1998) Ischemia/reperfusion injury in human kidney transplantation: an immunohistochemical analysis of changes after reperfusion Am J Pathol, 153: 557-66 94. B. Kruger, B. Schroppel, R. Ashkan, B. Marder, C. Zulke, B. Murphy, B. K. Kramer and M. Fischereder (2002) A Monocyte chemoattractant protein-1 (MCP-1) polymorphism and outcome after renal transplantation J Am Soc Nephrol, 13: 2585-9 95. T. Kurihara and R. Bravo (1996) Cloning and functional expression of mCCR2, a murine receptor for the C-C chemokines JE and FIC J Biol Chem, 271: 11603-7 96. M. Kusaka, J. Pratschke, M. J. Wilhelm, F. Ziai, K. Zandi-Nejad, H. S. Mackenzie, W. W. Hancock and N. L. Tilney (2000) Activation of inflammatory mediators in rat renal isografts by donor brain death Transplantation, 69: 405-10 97. M. Kusaka, K. Yamada, Y. Kuroyanagi, A. Terauchi, H. Kowa, H. Kurahashi and K. Hoshinaga (2005) Gene expression profile in rat renal isografts from brain dead donors Transplant Proc, 37: 364-6 98. M. Kusaka, K. Zandi-Nejad, S. Kato, F. Beato, H. Nagano, G. D. Shaw and N. L. Tilney (1999) Exploitation of the continuum between early ischemia/reperfusion injury and host alloresponsiveness: indefinite kidney allograft survival by treatment with a soluble Pselectin ligand and low-dose cyclosporine in combination Transplantation, 67: 1255-61 99. B. Lagiewska, M. Pacholczyk, M. Szostek, J. Walaszewski and W. Rowinski (1996) Hemodynamic and metabolic disturbances observed in brain-dead organ donors Transplant Proc, 28: 165-6 Literaturverzeichnis und Quellenangaben 84 100. E. Lazzeri, L. Lasagni, M. Serio, S. Romagnani and P. Romagnani (2002) [Cytokines and chemokines in nephropathies and renal transplant] G Ital Nefrol, 19: 641-9 101. F. B. Lemos, J. N. Ijzermans, P. E. Zondervan, A. M. Peeters, S. van den Engel, W. M. Mol, W. Weimar and C. C. Baan (2003) Differential expression of heme oxygenase-1 and vascular endothelial growth factor in cadaveric and living donor kidneys after ischemia-reperfusion J Am Soc Nephrol, 14: 3278-87 102. P. M. Lenzlinger, V. H. Hans, H. I. Joller-Jemelka, O. Trentz, M. C. MorgantiKossmann and T. Kossmann (2001) Markers for cell-mediated immune response are elevated in cerebrospinal fluid and serum after severe traumatic brain injury in humans J Neurotrauma, 18: 479-89 103. R. J. Lipshutz, S. P. Fodor, T. R. Gingeras and D. J. Lockhart (1999) High density synthetic oligonucleotide arrays Nat Genet, 21: 20-4 104. P. Lipton and T. S. Whittingham (1979) The effect of hypoxia on evoked potentials in the in vitro hippocampus J Physiol, 287: 427-38 105. X. Liu, O. Drognitz, H. Neeff, S. Benz and U. T. Hopt (2004) Apoptosis is caused by prolonged organ preservation and blocked by apoptosis inhibitor in experimental rat pancreatic grafts Transplant Proc, 36: 1209-10 106. Z. H. Liu, S. F. Chen, H. Zhou, H. P. Chen and L. S. Li (2003) Glomerular expression of C-C chemokines in different types of human crescentic glomerulonephritis Nephrol Dial Transplant, 18: 1526-34 107. K. Lopau, J. Mark, L. Schramm, E. Heidbreder and C. Wanner (2000) Hormonal changes in brain death and immune activation in the donor Transpl Int, 13 Suppl 1: S282-5 108. M. Lotz (1995) Interleukin-6: a comprehensive review Cancer Treat Res, 80: 209-33 109. J. Lutz, M. Stangl and U. Heemann (2003) [Kidney transplantation in Germany - 2002] Zentralbl Chir, 128: 816-20 110. J. A. Macoviak, I. R. McDougall, M. F. Bayer, M. Brown, H. Tazelaar and E. B. Stinson (1987) Significance of thyroid dysfunction in human cardiac allograft procurement Transplantation, 43: 824-6 Literaturverzeichnis und Quellenangaben 85 111. T. Mandrup-Poulsen, L. D. Wogensen, M. Jensen, P. Svensson, P. Nilsson, T. Emdal, J. Molvig, C. A. Dinarello and J. Nerup (1995) Circulating interleukin-1 receptor antagonist concentrations are increased in adult patients with thermal injury Crit Care Med, 23: 26-33 112. F. Masson, M. Thicoipe, M. J. Latapie and P. Maurette (1990) Thyroid function in brain-dead donors Transpl Int, 3: 226-33 113. M. P. Mattson, C. Culmsee and Z. F. Yu (2000) Apoptotic and antiapoptotic mechanisms in stroke Cell Tissue Res, 301: 173-87 114. P. L. McCarthy, Jr., S. Abhyankar, S. Neben, G. Newman, C. Sieff, R. C. Thompson, S. J. Burakoff and J. L. Ferrara (1991) Inhibition of interleukin-1 by an interleukin-1 receptor antagonist prevents graftversus-host disease Blood, 78: 1915-8 115. A. J. McLaren, S. E. Marshall, N. A. Haldar, C. G. Mullighan, S. V. Fuggle, P. J. Morris and K. I. Welsh (1999) Adhesion molecule polymorphisms in chronic renal allograft failure Kidney Int, 55: 1977-82 116. D. R. McMillan, X. Xiao, L. Shao, K. Graves and I. J. Benjamin (1998) Targeted disruption of heat shock transcription factor 1 abolishes thermotolerance and protection against heat-inducible apoptosis J Biol Chem, 273: 7523-8 117. M. D. Menger (1995) Microcirculatory disturbances secondary to ischemia-reperfusion Transplant Proc, 27: 2863-5 118. P. M. Mertes, P. Burtin, J. P. Carteaux, Y. Jaboin, C. Dopff, G. Pinelli, J. P. Villemot, C. Burlet and M. Boulange (1994) Brain death and myocardial injury: role of cardiac sympathetic innervation evaluated by in vivo interstitial microdialysis Transplant Proc, 26: 231-2 119. P. M. Mertes, K. el Abassi, Y. Jaboin, P. Burtin, G. Pinelli, J. P. Carteaux, C. Burlet, M. Boulange and J. P. Villemot (1994) Changes in hemodynamic and metabolic parameters following induced brain death in the pig Transplantation, 58: 414-8 120. E. Minambres, A. Cemborain, P. Sanchez-Velasco, M. Gandarillas, G. Diaz-Reganon, U. Sanchez-Gonzalez and F. Leyva-Cobian (2003) Correlation between transcranial interleukin-6 gradient and outcome in patients with acute brain injury Crit Care Med, 31: 933-8 Literaturverzeichnis und Quellenangaben 86 121. J. Mishra, K. Mori, Q. Ma, C. Kelly, J. Yang, M. Mitsnefes, J. Barasch and P. Devarajan (2004) Amelioration of ischemic acute renal injury by neutrophil gelatinase-associated lipocalin J Am Soc Nephrol, 15: 3073-82 122. K. Mori and K. Nakao (2007) Neutrophil gelatinase-associated lipocalin as the real-time indicator of active kidney damage Kidney Int, 71: 967-70 123. M. W. Mosesson, K. R. Siebenlist and D. A. Meh (2001) The structure and biological features of fibrinogen and fibrin Ann N Y Acad Sci, 936: 11-30 124. J. E. Murray, N. L. Tilney and R. E. Wilson (1976) Renal transplantation: a twenty-five year experience Ann Surg, 184: 565-73 125. K. C. Nadeau, H. Azuma and N. L. Tilney (1995) Sequential cytokine dynamics in chronic rejection of rat renal allografts: roles for cytokines RANTES and MCP-1 Proc Natl Acad Sci U S A, 92: 8729-33 126. T. Nagareda, Y. Kinoshita, A. Tanaka, M. Takeda, T. Sakano, K. Yawata, T. Sugimoto, Y. Nishizawa and N. Terada (1993) Clinicopathology of kidneys from brain-dead patients treated with vasopressin and epinephrine Kidney Int, 43: 1363-70 127. W. N. Nijboer, T. A. Schuurs, J. A. van der Hoeven, S. Fekken, J. Wiersema-Buist, H. G. Leuvenink, S. Hofker, J. J. Homan van der Heide, W. J. van Son and R. J. Ploeg (2004) Effect of brain death on gene expression and tissue activation in human donor kidneys Transplantation, 78: 978-86 128. Q. Ning, Y. Sun, M. Han, L. Zhang, C. Zhu, W. Zhang, H. Guo, J. Li, W. Yan, F. Gong, Z. Chen, W. He, C. Koscik, R. Smith, R. Gorczynski, G. Levy and X. Luo (2005) Role of fibrinogen-like protein 2 prothrombinase/fibroleukin in experimental and human allograft rejection J Immunol, 174: 7403-11 129. M. Notohamiprodjo, R. Djafarzadeh, A. Mojaat, I. von Luttichau, H. J. Grone and P. J. Nelson (2006) Generation of GPI-linked CCL5 based chemokine receptor antagonists for the suppression of acute vascular damage during allograft transplantation Protein Eng Des Sel, 19: 27-35 Literaturverzeichnis und Quellenangaben 87 130. D. Novitzky (1997) Detrimental effects of brain death on the potential organ donor Transplant Proc, 29: 3770-2 131. D. Novitzky, D. K. Cooper and B. Reichart (1987) Hemodynamic and metabolic responses to hormonal therapy in brain-dead potential organ donors Transplantation, 43: 852-4 132. R. Obermaier, S. Benz, E. Von Dobschuetz, O. Drognitz, W. Schareck, L. Jonas, K. Messmer and U. T. Hopt (2002) Characterization of microcirculatory disturbance in a novel model of pancreatic ischemia-reperfusion using intravital fluorescence-microscopy Pancreas, 25: 142-8 133. R. Obermaier, O. Drognitz, A. Grub, E. von Dobschuetz, W. Schareck, U. T. Hopt and S. Benz (2003) Endotoxin preconditioning in pancreatic ischemia/reperfusion injury Pancreas, 27: e51-6 134. R. Obermaier, P. Michel, E. von Dobschuetz, M. Lobler, H. J. Thiessen, D. Koczan, U. T. Hopt and S. Benz (2005) Protective effect of endotoxin preconditioning in pancreas: analysis with DNA chip arrays Transplant Proc, 37: 1632-4 135. R. Obermaier, E. von Dobschuetz, T. Keck, H. H. Hopp, O. Drognitz, W. Schareck, U. T. Hopt and S. Benz (2004) Brain death impairs pancreatic microcirculation Am J Transplant, 4: 210-5 136. G. Offermann (1998) What is a reasonably short cold ischemia time in kidney transplantation? Transplant Proc, 30: 4291-3 137. N. Ohba, M. Maeda, S. Nakagomi, M. Muraoka and H. Kiyama (2003) Biphasic expression of activating transcription factor-3 in neurons after cerebral infarction Brain Res Mol Brain Res, 115: 147-56 138. S. Okamoto, C. O. Corso, T. Kondo, R. Leiderer, W. Rascher, Y. Yamamoto, Y. Yamaoka and K. Messmer (1999) Changes in hepatic microcirculation and histomorphology in brain-dead organ donors: an experimental study in rats Eur J Surg, 165: 759-66 139. S. Okamoto, C. O. Corso, D. Nolte, W. Rascher, J. Thiery, Y. Yamaoka and K. Messmer (1998) Impact of brain death on hormonal homeostasis and hepatic microcirculation of transplant organ donors Transpl Int, 11 Suppl 1: S404-7 Literaturverzeichnis und Quellenangaben 88 140. J. G. Oliveira, P. Xavier, S. Neto, A. A. Mendes and L. E. Guerra (1997) Monocytes-macrophages and cytokines/chemokines in fine-needle aspiration biopsy cultures: enhanced interleukin-1 receptor antagonist synthesis in rejection-free kidney transplant patients Transplantation, 63: 1751-6 141. R. Olszanecki, R. Rezzani, S. Omura, D. E. Stec, L. Rodella, F. T. Botros, A. I. Goodman, G. Drummond and N. G. Abraham (2007) Genetic suppression of HO-1 exacerbates renal damage: reversed by an increase in the antiapoptotic signaling pathway Am J Physiol Renal Physiol, 292: F148-57 142. D. Ondrus, V. Pribylincova, J. Breza, P. Bujdak, M. Miklosi, J. Reznicek and V. Zvara (1999) The incidence of tumours in renal transplant recipients with long-term immunosuppressive therapy Int Urol Nephrol, 31: 417-22 143. D. S. Organtransplantation and www.dso.de (2006): 144. J. Parameshwar, J. Foote, L. Sharples, J. Wallwork, S. Large and P. Schofield (1996) Lipids, lipoprotein (a) and coronary artery disease in patients following cardiac transplantation Transpl Int, 9: 481-5 145. T. Patel and G. J. Gores (1995) Apoptosis and hepatobiliary disease Hepatology, 21: 1725-41 146. T. G. Peters, T. R. Shaver, J. E. t. Ames, E. A. Santiago-Delpin, K. W. Jones and J. W. Blanton (1995) Cold ischemia and outcome in 17,937 cadaveric kidney transplants Transplantation, 59: 191-6 147. H. Pienaar, I. Schwartz, A. Roncone, Z. Lotz and R. Hickman (1990) Function of kidney grafts from brain-dead donor pigs. The influence of dopamine and triiodothyronine Transplantation, 50: 580-2 148. B. M. Power and P. V. Van Heerden (1995) The physiological changes associated with brain death--current concepts and implications for treatment of the brain dead organ donor Anaesth Intensive Care, 23: 26-36 149. D. J. Powner (2003) Effects of gene induction and cytokine production in donor care Prog Transplant, 13: 9-14; quiz 15-6 150. D. J. Powner and J. M. Darby (2000) Management of variations in blood pressure during care of organ donors Prog Transplant, 10: 25-30; quiz 31-2 Literaturverzeichnis und Quellenangaben 89 151. D. J. Powner, A. Hendrich, R. G. Lagler, R. H. Ng and R. L. Madden (1990) Hormonal changes in brain dead patients Crit Care Med, 18: 702-8 152. J. Pratschke, G. Kofla, M. J. Wilhelm, A. Vergopoulos, I. Laskowski, G. D. Shaw, S. G. Tullius, H. D. Volk, P. Neuhaus and N. L. Tilney (2001) Improvements in early behavior of rat kidney allografts after treatment of the braindead donor Ann Surg, 234: 732-40 153. J. Pratschke, M. J. Wilhelm, M. Kusaka, M. Basker, D. K. Cooper, W. W. Hancock and N. L. Tilney (1999) Brain death and its influence on donor organ quality and outcome after transplantation Transplantation, 67: 343-8 154. J. Pratschke, M. J. Wilhelm, M. Kusaka, F. Beato, E. L. Milford, W. W. Hancock and N. L. Tilney (2000) Accelerated rejection of renal allografts from brain-dead donors Ann Surg, 232: 263-71 155. J. Pratschke, M. J. Wilhelm, M. Kusaka, W. W. Hancock and N. L. Tilney (1999) Activation of proinflammatory genes in somatic organs as a consequence of brain death Transplant Proc, 31: 1003-5 156. J. Pratschke, M. J. Wilhelm, M. Kusaka, W. W. Hancock and N. L. Tilney (1999) Acute rejection of rat renal allografts is accelerated by donor brain death Transplant Proc, 31: 874-5 157. J. Pratschke, M. J. Wilhelm, M. Kusaka, I. Laskowski and N. L. Tilney (2000) A model of gradual onset brain death for transplant-associated studies in rats Transplantation, 69: 427-30 158. J. Pratschke, M. J. Wilhelm, I. Laskowski, M. Kusaka, F. Beato, S. G. Tullius, P. Neuhaus, W. W. Hancock and N. L. Tilney (2001) Influence of donor brain death on chronic rejection of renal transplants in rats J Am Soc Nephrol, 12: 2474-81 159. C. D. Raeburn, F. Sheppard, K. A. Barsness, J. Arya and A. H. Harken (2002) Cytokines for surgeons Am J Surg, 183: 268-73 160. A. Regner, M. Kaufman, G. Friedman and I. Chemale (2001) Increased serum S100beta protein concentrations following severe head injury in humans: a biochemical marker of brain death? Neuroreport, 12: 691-4 161. A. Reutzel-Selke, S. G. Tullius, T. Zschockelt, M. Nieminen-Kelha, U. Bachmann, S. Jonas, J. Pratschke, G. Schmidbauer, W. O. Bechstein, H. Volk and P. Neuhaus (2001) Donor pretreatment of grafts from marginal donors improves long-term graft outcome Transplant Proc, 33: 970-1 Literaturverzeichnis und Quellenangaben 90 162. G. A. Ricchetti, L. M. Williams and B. M. Foxwell (2004) Heme oxygenase 1 expression induced by IL-10 requires STAT-3 and phosphoinositol-3 kinase and is inhibited by lipopolysaccharide J Leukoc Biol, 76: 719-26 163. S. Rigden, O. Mehls and R. Gellert (1999) Factors influencing second renal allograft survival. Scientific Advisory Board of the ERA-EDTA Registry. European Renal Association-European Dialysis and Transplant Association Nephrol Dial Transplant, 14: 566-9 164. C. Ronco (2007) N-GAL: Diagnosing AKI as soon as possible Crit Care, 11: 173 165. M. Ruster, H. Sperschneider, R. Funfstuck, G. Stein and H. J. Grone (2004) Differential expression of beta-chemokines MCP-1 and RANTES and their receptors CCR1, CCR2, CCR5 in acute rejection and chronic allograft nephropathy of human renal allografts Clin Nephrol, 61: 30-9 166. C. Ryckman, K. Vandal, P. Rouleau, M. Talbot and P. A. Tessier (2003) Proinflammatory activities of S100: proteins S100A8, S100A9, and S100A8/A9 induce neutrophil chemotaxis and adhesion J Immunol, 170: 3233-42 167. A. A. Salahudeen, J. K. Jenkins, H. Huang, K. Ndebele and A. K. Salahudeen (2001) Overexpression of heme oxygenase protects renal tubular cells against cold storage injury: studies using hemin induction and HO-1 gene transfer Transplantation, 72: 1498-504 168. A. Salim, P. Vassiliu, G. C. Velmahos, J. Sava, J. A. Murray, H. Belzberg, J. A. Asensio and D. Demetriades (2001) The role of thyroid hormone administration in potential organ donors Arch Surg, 136: 1377-80 169. C. E. Sanders, Jr. and J. J. Curtis (1995) Role of hypertension in chronic renal allograft dysfunction Kidney Int Suppl, 52: S43-7 170. F. W. Schildberg, E. Pratschke and H. Berger (1992) [Intra-abdominal infections: the importance of imaging procedures for therapeutic planning] Chirurg, 63: 153-61 171. E. Schutz, E. Wieland, L. Heine, A. Hensel, A. Schmiedl, V. W. Armstrong, J. Richter, P. Schuff-Werner, E. Gunther and M. Oellerich (1997) Acceleration of hepatocellular energy by idebenone during early reperfusion after cold preservation ameliorates heat shock protein 70 gene expression in a pig liver model Transplantation, 64: 901-7 Literaturverzeichnis und Quellenangaben 91 172. T. A. Schuurs, F. Gerbens, J. A. van der Hoeven, P. J. Ottens, K. A. Kooi, H. G. Leuvenink, R. M. Hofstra and R. J. Ploeg (2004) Distinct transcriptional changes in donor kidneys upon brain death induction in rats: insights in the processes of brain death Am J Transplant, 4: 1972-81 173. T. A. Schuurs, A. M. Morariu, P. J. Ottens, N. A. t Hart, S. H. Popma, H. G. Leuvenink and R. J. Ploeg (2006) Time-dependent changes in donor brain death related processes Am J Transplant, 6: 2903-11 174. L. D. Segel, D. W. vonHaag, J. Zhang and D. M. Follette (2002) Selective overexpression of inflammatory molecules in hearts from brain-dead rats J Heart Lung Transplant, 21: 804-11 175. S. Segerer, Y. Cui, F. Eitner, T. Goodpaster, K. L. Hudkins, M. Mack, J. P. Cartron, Y. Colin, D. Schlondorff and C. E. Alpers (2001) Expression of chemokines and chemokine receptors during human renal transplant rejection Am J Kidney Dis, 37: 518-31 176. B. Shivalkar, J. Van Loon, W. Wieland, T. B. Tjandra-Maga, M. Borgers, C. Plets and W. Flameng (1993) Variable effects of explosive or gradual increase of intracranial pressure on myocardial structure and function Circulation, 87: 230-9 177. D. A. Shoskes, N. A. Parfrey and P. F. Halloran (1990) Increased major histocompatibility complex antigen expression in unilateral ischemic acute tubular necrosis in the mouse Transplantation, 49: 201-7 178. Y. W. Sijpkens, Doxiadis, II, M. J. Mallat, J. W. de Fijter, J. A. Bruijn, F. H. Claas and L. C. Paul (2003) Early versus late acute rejection episodes in renal transplantation Transplantation, 75: 204-8 179. C. A. Skrabal, L. O. Thompson, E. V. Potapov, R. E. Southard, D. L. Joyce, K. A. Youker, G. P. Noon and M. Loebe (2005) Organ-specific regulation of pro-inflammatory molecules in heart, lung, and kidney following brain death J Surg Res, 123: 118-25 180. Y. F. Smets, R. G. Westendorp, J. W. van der Pijl, F. T. de Charro, J. Ringers, J. W. de Fijter and H. H. Lemkes (1999) Effect of simultaneous pancreas-kidney transplantation on mortality of patients with type-1 diabetes mellitus and end-stage renal failure Lancet, 353: 1915-9 Literaturverzeichnis und Quellenangaben 92 181. M. Smith (2004) Physiologic changes during brain stem death--lessons for management of the organ donor J Heart Lung Transplant, 23: S217-22 182. K. Solez, L. C. Racusen, F. Abdulkareem, E. Kemeny, E. von Willebrand and L. D. Truong (1997) Adhesion molecules and rejection of renal allografts Kidney Int, 51: 1476-80 183. T. E. Starzl, L. J. Koep, C. G. Halgrimson, J. Hood, G. P. Schroter, K. A. Porter and R. Weil, 3rd (1979) Fifteen years of clinical liver transplantation Gastroenterology, 77: 375-88 184. K. Strle, J. H. Zhou, W. H. Shen, S. R. Broussard, R. W. Johnson, G. G. Freund, R. Dantzer and K. W. Kelley (2001) Interleukin-10 in the brain Crit Rev Immunol, 21: 427-49 185. S. Supavekin, W. Zhang, R. Kucherlapati, F. J. Kaskel, L. C. Moore and P. Devarajan (2003) Differential gene expression following early renal ischemia/reperfusion Kidney Int, 63: 1714-24 186. K. Suzuki, B. Murtuza, R. T. Smolenski, I. A. Sammut, N. Suzuki, Y. Kaneda and M. H. Yacoub (2001) Overexpression of interleukin-1 receptor antagonist provides cardioprotection against ischemia-reperfusion injury associated with reduction in apoptosis Circulation, 104: I308-I3 187. G. Szabo (2004) Physiologic changes after brain death J Heart Lung Transplant, 23: S223-6 188. L. Tacchini, L. Radice and A. Bernelli-Zazzera (1999) Differential activation of some transcription factors during rat liver ischemia, reperfusion, and heat shock J Cell Physiol, 180: 255-62 189. M. Takada, K. C. Nadeau, W. W. Hancock, H. S. Mackenzie, G. D. Shaw, A. M. Waaga, A. Chandraker, M. H. Sayegh and N. L. Tilney (1998) Effects of explosive brain death on cytokine activation of peripheral organs in the rat Transplantation, 65: 1533-42 190. R. Tardanico and S. Sandrini (2004) [Clinical anatomical features of chronic dysfunction in the transplanted kidney] G Ital Nefrol, 21 Suppl 26: S34-8 Literaturverzeichnis und Quellenangaben 93 191. R. M. Taylor (1997) Reexamining the definition and criteria of death Semin Neurol, 17: 265-70 192. A. M. Teppo, E. Honkanen, J. Ahonen and C. Gronhagen-Riska (1998) Does increased urinary interleukin-1 receptor antagonist/interleukin-1beta ratio indicate good prognosis in renal transplant recipients? Transplantation, 66: 1009-14 193. H. Terajima, G. Enders, A. Thiaener, C. Hammer, T. Kondo, J. Thiery, Y. Yamamoto, Y. Yamaoka and K. Messmer (2000) Impact of hyperthermic preconditioning on postischemic hepatic microcirculatory disturbances in an isolated perfusion model of the rat liver Hepatology, 31: 407-15 194. P. I. Terasaki, J. M. Cecka, D. W. Gjertson and S. Takemoto (1995) High survival rates of kidney transplants from spousal and living unrelated donors N Engl J Med, 333: 333-6 195. P. I. Terasaki, S. Takemoto, M. S. Park, J. M. Cecka and M. R. Mickey (1991) Molecular HLA matching Transplant Proc, 23: 365-7 196. R. J. Tesi, E. A. Elkhammas, M. L. Henry, E. A. Davies, A. Salazar and R. M. Ferguson (1993) Acute rejection episodes: best predictor of long-term primary cadaveric renal transplant survival Transplant Proc, 25: 901-2 197. G. L. Todd, G. Baroldi, G. M. Pieper, F. C. Clayton and R. S. Eliot (1985) Experimental catecholamine-induced myocardial necrosis. II. Temporal development of isoproterenol-induced contraction band lesions correlated with ECG, hemodynamic and biochemical changes J Mol Cell Cardiol, 17: 647-56 198. C. Troppmann, K. J. Gillingham, E. Benedetti, P. S. Almond, R. W. Gruessner, J. S. Najarian and A. J. Matas (1995) Delayed graft function, acute rejection, and outcome after cadaver renal transplantation. The multivariate analysis Transplantation, 59: 962-8 199. S. G. Tullius, M. Nieminen, W. O. Bechstein, S. Jonas, T. Steinmuller, Y. Qun, J. Pratschke, E. Graser, P. Sinha, H. D. Volk, P. Neuhaus and N. L. Tilney (1998) Contribution of early acute rejection episodes to chronic rejection in a rat kidney retransplantation model Kidney Int, 53: 465-72 200. J. Unden, J. Bellner, P. Reinstrup and B. Romner (2004) Serial S100B levels before, during and after cerebral herniation Br J Neurosurg, 18: 277-80 Literaturverzeichnis und Quellenangaben 94 201. J. A. Van der Hoeven, S. Lindell, R. van Schilfgaarde, G. Molema, G. J. Ter Horst, J. H. Southard and R. J. Ploeg (2001) Donor brain death reduces survival after transplantation in rat livers preserved for 20 hr Transplantation, 72: 1632-6 202. J. A. van der Hoeven, G. Molema, G. J. Ter Horst, R. L. Freund, J. Wiersema, R. van Schilfgaarde, H. G. Leuvenink and R. J. Ploeg (2003) Relationship between duration of brain death and hemodynamic (in)stability on progressive dysfunction and increased immunologic activation of donor kidneys Kidney Int, 64: 1874-82 203. J. A. Van Der Hoeven, H. Moshage, T. Schuurs, M. Nijboer, R. Van Schilfgaarde and R. J. Ploeg (2003) Brain death induces apoptosis in donor liver of the rat Transplantation, 76: 1150-4 204. J. A. van der Hoeven, R. J. Ploeg, F. Postema, I. Molema, P. de Vos, A. R. Girbes, P. T. van Suylichem, R. van Schilfgaarde and G. J. Ter Horst (1999) Induction of organ dysfunction and up-regulation of inflammatory markers in the liver and kidneys of hypotensive brain dead rats: a model to study marginal organ donors Transplantation, 68: 1884-90 205. A. Varki (1994) Selectin ligands Proc Natl Acad Sci U S A, 91: 7390-7 206. J. L. Veale and H. A. Gritsch (2006) Lymphoma after kidney transplantation: risk factors and timing of development Nat Clin Pract Nephrol, 2: 422-3 207. A. M. Waaga, M. Gasser, I. Laskowski and N. L. Tilney (2000) Mechanisms of chronic rejection Curr Opin Immunol, 12: 517-21 208. A. C. Wagner, H. Weber, L. Jonas, H. Nizze, M. Strowski, F. Fiedler, H. Printz, H. Steffen and B. Goke (1996) Hyperthermia induces heat shock protein expression and protection against ceruleininduced pancreatitis in rats Gastroenterology, 111: 1333-42 209. M. Wagner, P. Cadetg, R. Ruf, L. Mazzucchelli, P. Ferrari and C. A. Redaelli (2003) Heme oxygenase-1 attenuates ischemia/reperfusion-induced apoptosis and improves survival in rat renal allografts Kidney Int, 63: 1564-73 210. X. Wang, T. L. Yue, F. C. Barone, R. F. White, R. C. Gagnon and G. Z. Feuerstein (1994) Concomitant cortical expression of TNF-alpha and IL-1 beta mRNAs follows early response gene expression in transient focal ischemia Mol Chem Neuropathol, 23: 103-14 Literaturverzeichnis und Quellenangaben 95 211. W. N. Wicomb, D. K. Cooper and D. Novitzky (1986) Impairment of renal slice function following brain death, with reversibility of injury by hormonal therapy Transplantation, 41: 29-33 212. M. J. Wilhelm, J. Pratschke, M. Kusaka, W. W. Hancock and N. L. Tilney (1999) Donor brain death affects tempo and intensity of acute rejection of rat chronic allografts Transplant Proc, 31: 1008-9 213. C. Woiciechowsky, B. Schoning, W. R. Lanksch, H. D. Volk and W. D. Docke (1999) Mechanisms of brain-mediated systemic anti-inflammatory syndrome causing immunodepression J Mol Med, 77: 769-80 214. K. L. Womer, J. P. Vella and M. H. Sayegh (2000) Chronic allograft dysfunction: mechanisms and new approaches to therapy Semin Nephrol, 20: 126-47 215. K. E. Wood, B. N. Becker, J. G. McCartney, A. M. D'Alessandro and D. B. Coursin (2004) Care of the potential organ donor N Engl J Med, 351: 2730-9 216. Z. Xing and J. Wang (2000) Consideration of cytokines as therapeutics agents or targets Curr Pharm Des, 6: 599-611 217. T. Yeh, Jr., A. S. Wechsler, L. Graham, K. E. Loesser, D. A. Sica, L. Wolfe and E. R. Jakoi (2002) Central sympathetic blockade ameliorates brain death-induced cardiotoxicity and associated changes in myocardial gene expression J Thorac Cardiovasc Surg, 124: 1087-98 218. T. Yeh, Jr., A. S. Wechsler, L. J. Graham, K. E. Loesser, D. A. Sica, L. Wolfe and E. R. Jakoi (1999) Acute brain death alters left ventricular myocardial gene expression J Thorac Cardiovasc Surg, 117: 365-74 219. J. M. Zapata, F. G. Maroto and J. M. Sierra (1991) Inactivation of mRNA cap-binding protein complex in Drosophila melanogaster embryos under heat shock J Biol Chem, 266: 16007-14 220. Q. H. Zhai, N. Futrell and F. J. Chen (1997) Gene expression of IL-10 in relationship to TNF-alpha, IL-1beta and IL-2 in the rat brain following middle cerebral artery occlusion J Neurol Sci, 152: 119-24 Abkürzungsverzeichnis 7. 96 Abkürzungsverzeichnis A. = Arteria A = arteriell Alb = Albumin Aqp2 = Aquaporin 2 ATP = Adenosin-tri-phosphat CAN = Chronische allograft Nephropathie (Abstoßungs-) Ccl2 = (=MCP-1), Chemokin – ligand 2 CO2 = Kohlendioxid DNA = Desoxyribucleic-acid, deutsch DNS (Desoxyribonukleinsäure) DNES = Diffuses neuroendokrines System EtBr = Ethidiumbromid Fgb = Fibrinogen GAG = Glykosaminoglykane GvHD = Graft versus host disease H = Stunde HF = Herzfrequenz HLA = Humanes Leukozytenantigen HO = Hämoxgenase HSP = Heat-Shock-Protein HT-Gruppe = Hirntod-Gruppe ICAM-1 = Intercellular adhesion molecule - 1 Il = Interleukin Il-1RA = Interleukin Rezeptor Antagonist i.a. = Intraarteriell i.v. = Intravenös I/R = Ischämie/Reperfusion µg = Mikrogramm K-Gruppe = Kontrollgruppe KIM = Kidney Injury Molecole Lcn 2 = Lipocalin 2 MAP = Mittlerer arterieller Druck MCP = Makrophagen-chemotaktisches-Protein MHC = master histocompatibility complex Min = Minute Abkürzungsverzeichnis 97 NaCl = Natriumchlorid NH2 = Ammoniak NO = Stickstoff NTX = Nierentransplantation O2 = Sauerstoff PIC = Pro-inflammatorische Cytokine PMN = Polymorphonukleäre Leukozyten PCR = Polymerase Chain Reaction RNA = Ribunucleic acid RR = Blutdruck RT = Reverse Transkriptase S100a8 = Calcium bindendes Protein A8/ Calgranulin A S100a9 = Calcium bindendes Protein A9/ Calgranulin B SPKT = Simultaneous pancreas and (cadaver) kidney transplantation STAT 3 = Signal transcducer and activator of transcription factor 3 TGF-β = Transforming Growth Factor beta TNF-α = Tumor Nekrose Faktor α TPFI-2 = Tissue factor pathway inhibitor 2 TX = Transplantation VCAM-1 = Vascular cell adhesion molecule – 1 V. = Vena VEGF = Vasoactive endothelial growth factor ZNS = Zentrales Nervensystem Vol% = Volumenprozent Selbstständigkeitserklärung 8. 98 Selbstständigkeitserklärung Hiermit versichere ich, dass ich die vorliegende Arbeit ohne unzulässige Hilfe Dritter und ohne Benutzung anderer, als der angegebenen, Hilfsmittel angefertigt habe. Die aus anderen Quellen direkt oder indirekt übernommenen Daten und Konzepte sind unter Angabe der Quelle gekennzeichnet. Insbesondere abe ich hierfür nicht die entgeltliche Hilfe von Vermittlungs- beziehungsweise Beratungsdiensten (Promotionsberater oder anderer Personen) in Anspruch genommen. Niemand hat von mir unmittelbar oder mittelbar geldwerte Leistungen für Arbeiten erhalten, die im Zusammenhang mit dem Inhalt der vorgelegten Dissertation stehen. Die Arbeit wurde bisher weder im In- noch im Ausland in gleicher oder ähnlicher Form einer anderen Prüfungsbehörde vorgelegt. Danksagung 9. 99 Danksagung Ich bedanke mich bei meinem Doktorvater PD Dr. med. Robert Obermaier für die Inspiration und Überlassung des Themas, die freundliche Unterstützung und Förderung des Forschungsprojektes. Ich danke PD Dr. Jan Kaminsky für die freundliche Übernahme des Zweitgutachtens. Mein besonderer Dank für die hilfreiche Unterstützung gilt allen Mitarbeitern des Forschungslabores der chirurgischen Abteilung. Allerbesten Dank vorallem an Silke Hempel, Jessica Pfannstiel und Kolleginnen, die mich in die Materie eingearbeitet haben und mir immer mit freundlichen Ratschlägen zur Seite standen. Herrn PD Dr. Löbler danke ich für die Hilfe bei der Auswertung der gewonnen Daten. Bedanken möchte ich mich auch bei Dr. Kozcan und seinem Team in Rostock für die Kooperation bezüglich der Chiphybridisierung und Datengewinnung. Herrn Dr. Pilarsky danke ich für die Hilfe bei der Auswertung der gewonnen Chip-Daten. Ich möchte mich herzlich bei Herrn Dr. med. Frank Hengl für seine langjährige Freundschaft und Mithilfe als Mitarbeiter unserer Forschungsgruppe bedanken. Am meisten danke ich meiner Familie, die mir das Studium in dieser Form ermöglicht hat, meinem Bruder Michael Horn und meinem Freund Peter Grimminger für die stetige Unterstützung. 100 Lebenslauf 10. Lebenslauf Personalien Name: Carolin M. Y. Horn Geburtsdatum: 13.06.1980 Geburtsort: Frankfurt am Main Adresse: Eifelstrasse 33 50677 Köln Schulischer Werdegang 1986-1990 Grundschule Hofheim 1990-1999 Main-Taunus-Gymnasium Hofheim 1999 Abitur Universitärer Werdegang WS 1999/2000 Beginn des vorklinischen Studiums der Humanmedizin an der Semmelweisuniversität in Budapest, Ungarn WS 2001-2006 Fortsetzung des klinischen Studiums der Medizin an der Albert-Ludwigs-Universität in Freiburg am Breisgau April 2006 3. Medizinisches Staatsexamen und Erlangung der Approbation (Gesamtnote 1,6) Beruflicher Werdegang Seit August 2006 Assistenzärztin der Hals-Nasen-Ohrenheilkunde ders Universitätsklinikums Köln unter der Leitung von Univ.-Prof. Dr. med. Dr. h.c. K.-B. Hüttenbrink