In vitro-Untersuchungen zum Tat
Werbung

In vitro-Untersuchungen zum
Tat-abhängigen Transport des Fusionsproteins
TorA-PhoA
INAUGURAL-DISSERTATION
zur Erlangung der Doktorwürde
der Fakultät für Biologie
der Albert-Ludwigs-Universität, Freiburg i.Br.
Vorgelegt im März 2008 von
Sascha Panahandeh
geb. in Teheran, Iran
Diese Arbeit wurde angefertigt am Institut für Biochemie und Molekularbiologie, der
medizinischen Fakultät an der Albert-Ludwigs-Universität, Freiburg i.Br.
Betreuer der Arbeit:
Prof. Dr. Matthias Müller
Dekan der Fakultät:
Prof. Dr. Ralf Reski
Promotionsvorsitzender:
Prof. Dr. Samuel Rossel
1. Koreferent:
JunProf. Dr. Michael Schroda
2. Koreferent:
Dr. Katja Arndt
Tag der Verkündigung des Ergebnisses:
19.06.2008
Wesentliche Teile dieser Arbeit wurden zur Publikation eingereicht:
Panahandeh S., Moser, M., Maurer C., DeLisa M.P., and Müller, M. (2008)
Following the path of a twin-arginine precursor along the TatABC translocase of Escherichia
coli. EMBO Journal
Im Rahmen dieser Arbeit wurde veröffentlicht:
Moser, M., Panahandeh, S., Holzapfel, E., and Müller, M. (2007). In vitro analysis of the
bacterial twin-arginine-dependent protein export. Methods in Molecular Biology, 390, 63-80
Panahandeh, S., Holzapfel, E., and Müller, M. (2008)
The twin-arginine translocation pathway. Horizon Book Chapter, Review (in press)
Inhaltsverzeichnis
Inhaltsverzeichnis
I. Zusammenfassung
1
II. Einleitung
3
II.1. Das sekretorische (Sec)-System in E. coli
II.1.1. Das Sec-Translokon
II.1.2. Zielsteuerung und Erkennung
II.1.3. SecA/SecB-abhängiger posttranslationaler
Transport sekretorischer Proteine
II.1.4. Signal recognition particle (SRP)–abhängige
cotranslationale Integration von Innenmembranproteinen
4
4
5
6
7
II.2. Das twin arginine translocation (Tat)–System
II.2.1. Tat-Substrate
II.2.2. Die Komponenten der Tat-Translokase
II.2.2.1. TatA
II.2.2.2. TatB
II.2.2.3. TatC
II.2.3. Tat-Komplexe
II.2.3.1. Der TatBC-Rezeptor-Komplex
II.2.3.2. Der TatA-Komplex
II.2.4. Tat-spezische Signalsequenz und Zielsteuerung
II.2.5. Transportmechanismus
II.2.5.1. Kontakt des Tat-Substrates mit dem
TatBC–Rezeptor–Komplex
II.2.5.2. Translokationsmodelle
II.2.6. Energetisierung der Tat-abhängigen Translokation
II.2.7. Qualitätskontrolle des Tat-Systems
II.2.8. Spezifische Chaperone der Tat-Substrate
8
8
9
10
11
12
13
13
14
15
16
II.3. Zielsetzung
23
III. Material und Methoden
III.1. Material
III.1.1.
III.1.2.
III.1.3.
III.1.4.
III.1.5.
Escherichia coli-Stämme
Plasmide
Oligonukleotide
Antibiotika
Chemikalien
17
18
19
20
21
24
24
24
24
25
26
26
i
Inhaltsverzeichnis
III.2. Methoden
III.2.1. Molekularbiologische Methoden
III.2.1.1. Anzucht und Lagerung von Escherichia coli
III.2.1.2. Herstellung kompetenter E. coli-Zellen und Transformation
nach der TSB-Methode
III.2.1.3. Plasmidpräparation im großen Maßstab (Maxiprep)
III.2.1.4. Plasmidpräparation im kleinen Maßstab (Miniprep)
III.2.1.5. Agarose-Gelelektrophorese
III.2.1.6. Mutagenisierende PCR (polymerase chain reaction)
27
27
III.2.2. Biochemische Methoden
III.2.2.1. Denaturierende SDS-Polyacrylamidgelelektrophorese
III.2.2.2. Visualisierung von Proteinen in Polyacrylamidgelen
III.2.2.2.1. Autoradiographie
III.2.2.2.2. Coomassie-Blau-Färbung von
Polyacrylamidgelen
III.2.2.2.3. Western-Blotting
III.2.2.3. Bestimmung der Proteinkonzentration mit Amidoschwarz
III.2.2.4. Überexpression und affinitätschromatographische
Aufreinigung von TorD
III.2.2.5. Präparation der Komponenten des zellfreien Systems
III.2.2.5.1. Präparation des Zellextraktes S135
III.2.2.5.2. Präparation von inside-out
Innenmembranvesikeln (INV)
III.2.2.5.3. Präparation von T7-RNA-Polymerase
III.2.2.6. Versuche im zellfreien System aus E. coli
III.2.2.6.1. Gekoppelte in vitro-Transkription/Translation
III.2.2.6.2. In vitro-Translokation und
Protease-Resistenz-Test
III.2.2.6.3. Ortsgerichtete Quervernetzung mit
para-Benzoyl-L-phenylalanin (pBPA)
III.2.2.7. Immunpräzipitation
III.2.2.8. Kovalente Modifikation freier Sulfhydrylgruppen von
in vitro-synthetisiertem Protein
III.2.2.9. Größenchromatographische Analyse eine
in vitro-Transport-Ansatzes
III.2.2.10. Affinitätschromatographische Aufreinigung von in vitrosynthetisiertem Protein
31
31
33
33
IV. Ergebnisse
IV.1. Etablierung des Tat-abhängigen in vitro-Transports von TorA-PhoA
IV.1.1. In vitro-Transport von TorA-PhoA findet nur unter
oxidierenden Synthesebedingungen statt
IV.1.2. Durch oxidierende Synthesebedingungen wird vollständig
oxidiertes TorA-PhoA synthetisiert
IV.1.3. Oxidiertes TorA-PhoA wird TatABC-abhängig transportiert
ii
27
27
28
28
29
29
33
33
35
35
36
36
38
40
41
41
43
44
45
47
48
48
50
51
53
55
58
Inhaltsverzeichnis
IV.1.4. In vitro-Transport des oxidierten pTorA-PhoA ist abhängig
von der TorA-Signalsequenz
IV.1.4.1. TorD-Bindung an die TorA-Signalsequenz
verhindert den Transport von pTorA-PhoA
IV.1.4.2. Das Doppel-Arginin des Konsensusmotifs ist
essentiell für den Transport von pTorA-PhoA
IV.2. Charakterisierung der Membranbindung von TorA-PhoA
IV.2.1. Etablierung eines Nachweises für die Bindung von
TorA-PhoA an die INV-Membran
IV.2.2. Transportkompetentes, oxidiertes TorA-PhoA assoziiert
Tat-spezifisch mit der Membran
IV.2.3. Auch transportinkompetentes, reduziertes TorA-PhoA
assoziiert Tat-spezifisch mit der Membran
IV.3. Charakterisierung molekularer Interaktionen von TorA-PhoA
mit den Tat-Proteinen
IV.3.1. Zielgerichteter Einbau des photoaktivierbaren Quervernetzers
pBPA in die Signalsequenz von TorA-PhoA
IV.3.2. Reduziertes TorA-PhoA zeigt eine stark abgeschwächte
Interaktion der Signalsequenz mit TatC
IV.3.3. Reduziertes TorA-PhoA zeigt eine abgeschwächte
Interaktion der Signalsequenz mit TatB
IV.3.4. Die Signalsequenz von TorA-PhoA interagiert mit
TorD und FkpA
IV.4. Charakterisierung eines potentiellen Transportintermediates
von TorA-PhoA
IV.4.1. Triton-Resistenz von iTorA-PhoA wird nur für die
oxidierte Form beobachtet
IV.4.2. Die Bildung von iTorA-PhoA ist abhängig vom Doppel-Arginin-Motif
IV.4.3. Translokationsarrest findet vor dem ∆pH-abhängigen Schritt
der Translokation statt
IV.4.4. Translokationsarrest findet vor der Abspaltung der Signalsequenz statt
IV.4.5. Der C-Terminus von iTorA-PhoA ist nicht proteasegeschützt
IV.4.6. Überlastung der Tat-Translokase könnte die Ursache für den
Translokationsarrest sein
IV.4.7. Translokationsarrest findet in der unmittelbaren Nähe von TatA statt
61
61
62
64
64
67
68
71
71
73
75
78
80
80
81
83
86
87
89
90
V. Diskussion
95
V.1. Etablierung des Tat-abhängigen in vitro-Transportes von TorA-PhoA
95
V.1.1. Faltungszustand von in vitro-synthetisiertem TorA-PhoA
V.1.2. Der in vitro-Transport des oxidierten TorA-PhoAs ist Tat-abhängig
96
98
iii
Inhaltsverzeichnis
V.2. TorA-PhoA bindet faltungsunabhängig und TatABC-spezifisch an die
Membran
100
V.3. Erkennung der Faltungszustände von TorA-PhoA durch das Tat-System
104
V.4. Charakterisierung eines potentiellen Transportintermediates
106
V.5. Translokationsarrest findet in der unmittelbaren Nähe von TatA statt
109
VI. Literaturverzeichnis
113
VII. Anhang
125
VII.1. TorA-PhoA-Sequenz
125
VII.2. Abkürzungsverzeichnis
126
VII.3. Danksagung
129
iv
I. Zusammenfassung
I. Zusammenfassung
Das Cytoplasma Gram-negativer Bakterien ist von einer Zellhülle umgeben, die sich aus der
Innenmembran (Cytoplasmamembran), der Außenmembran und dem dazwischen liegenden
Periplasma zusammensetzt. Proteine, die in einem dieser Kompartimente lokalisiert sind,
müssen nach ihrer Synthese im Cytoplasma spezifisch an ihren Wirkungsort transportiert
werden. Der posttranslationale Transport einiger dieser Proteine wird durch das twin arginine
translocation (Tat)-System vermittelt, das aus den drei Innenmembranproteinen TatA, TatB
und TatC aufgebaut ist. Substrate des Tat-Systems besitzen in ihrer N-terminalen
Signalsequenz das Konsensusmotif S-R-R-x-F-L-K, dessen Arginin-Paar Namensgeber
dieses Transportsystems ist. Eine Besonderheit des Tat-Systems stellt dessen Fähigkeit dar,
Proteine im vollständig gefalteten Zustand zu transportieren. Tat-Substrate falten sich bereits
vor ihrem Transport, da sie entweder Kofaktoren benötigen, die im Cytoplasma eingebaut
werden, als Oligomere transportiert werden, oder vermutlich eine sehr schnellen
Faltungskinetik aufweisen.
Im Rahmen dieser Arbeit sollte Einblick in den Mechanismus des Tat-abhängigen
Transportes in Escherichia coli gewonnen werden. Dazu kam das Tat-abhängige
Fusionsprotein TorA-PhoA zum Einsatz, das sich aus der Signalsequenz des Tat-Substrates
Trimethylamin-N-Oxid-Reduktase (TorA) und dem reifen Teil der alkalischen Phosphatase
(PhoA) aus Escherichia coli zusammensetzt. In einer früheren Studie konnte gezeigt werden,
dass die Bildung der beiden intramolekularen Disulfidbrücken im reifen Teil von TorA-PhoA,
und somit das Erlangen der nativen Faltung, Voraussetzung für den Tat-abhängigen
Transport in Escherichia coli-Zellen war. In der vorliegenden Arbeit ist es gelungen, diese
Ergebnisse
reproduzieren.
in
einem
gekoppelten
TorA-PhoA
wurde
in vitro-Transkriptions/Translations-System
in vitro
synthetisiert
und
Tat-abhängig
zu
in
Innenmembranvesikel aus Escherichia coli transportiert. Dabei konnte gezeigt werden, dass
im Gegensatz zu der reduzierten Form, nur die nachweislich vollständig oxidierte Form des
TorA-PhoA-Proteins transportkompetent war.
Membranbindungsstudien ergaben, dass die TatABC-spezifische Zielsteuerung des TorAPhoA-Proteins an die Membran unabhängig vom Faltungszustand des Fusionsproteins, und
sogar bei Fehlen des
konservierten Arginin-Paars erfolgt. Die Inhibition dieser Bindung
durch Dicyclohexylcarbodiimid (DCCD) deutet auf eine Beteiligung von membranintegralen,
sauren Aminosäureresten des TatBC-Rezeptor-Komplexes an dieser Interaktion hin.
Um die molekulare Interaktion des TorA-PhoA-Proteins mit den Tat-Proteinen zu
charakterisieren, wurde der photoaktivierbare Quervernetzer pBPA ortsspezifisch in die
1
I. Zusammenfassung
Signalsequenz von TorA-PhoA eingebaut. Es konnte gezeigt werden, dass die Region um
das Konsensusmotif der Signalsequenz mit TatC interagiert, während die h-Region der
Signalsequenz mit TatB interagiert. Im Gegensatz zur Membranbindung, waren diese
Interaktionen abhängig vom Redoxzustand des TorA-PhoA-Proteins, da die reduzierte,
transportinkompetente Form des Proteins eine deutlich schwächere Interaktion mit TatB und
insbesondere mit TatC aufwies. Auf Grund dieser Beobachtung muss der TatBC-RezeptorKomplex die Fähigkeit haben, den Faltungszustand des TorA-PhoA-Proteins zu detektieren.
Weiterhin wurde ein Transportintermediat charakterisiert, das nur im Fall des oxidierten,
transportkompetenten TorA-PhoA entstand. Es konnte gezeigt werden, dass die Bildung des
Intermediates die Anwesenheit der Tat-Proteine sowie eines intakten Doppel-Arginin-Motifs
voraussetzt. Die Bildung des Intermediates erfolgt nach der initialen Bindung an den TatBCRezeptor-Komplex, jedoch vor dem ∆pH-abhängigen Schritt der Translokation, sowie vor der
Signalpeptidase-spezifischen
Prozessierung
des
TorA-PhoA-Proteins.
Eine
partielle
Proteaseresistenz des Intermediates auch in Gegenwart verschiedener Detergenzien
deutete darauf hin, dass sich das Intermediat in einer schützenden Membranstruktur
befindet. Schließlich konnte demonstriert werden, dass die Bildung des Intermediates sowohl
in Abhängigkeit von TatA als auch in dessen unmittelbarer Umgebung stattfindet. Diese
Beobachtung, zusammen mit der Vorstellung einer durch den TatA-Komplex gebildeten Pore
des Tat-Translokons, läßt den Schluss zu, dass sich das Intermediat noch in eben solch
einer Pore befindet. Somit konnte erstmals ein direkter Kontakt eines Tat-abhängigen
Transportintermediates mit TatA nachgewiesen werden, wodurch Translokationsmodelle mit
einem porenbildenden TatA-Komplex unterstützt werden.
2
II. Einleitung
II. Einleitung
Das Prinzip der Kompartimentierung eukaryontischer sowie prokaryontischer Zellen durch
Biomembranen erlaubt eine Unterteilung der Zelle in separate Reaktionsräume. Im
Gegensatz zu Eukaryonten ist die Anzahl der Kompartimente bei Prokaryonten gering. So
besitzt das Gram-negative Bakterium Escherichia coli (E. coli) nur vier Zellkompartimente.
Das Cytoplasma ist von der Innenmembran, der so genannten Cytoplasmamembran
umgeben, an die sich nach außen das Periplasma und schließlich die äußere Membran
anschließen. Da Proteinsynthese ausschließlich im Cytoplasma stattfindet, stellt die
Kompartimentierung der Zelle allerdings ein Problem für die Proteine dar, deren Funktionen
nicht im Cytoplasma liegen. Etwa ein Drittel des im Cytoplasma synthetisierten bakteriellen
Proteoms besteht aus Proteinen, die entweder in die innere Membran eingebaut
(Membranproteine) oder durch diese transportiert werden müssen (sekretorische Proteine),
da sich ihr finaler Wirkungsort im Periplasma, in der äußeren Membran oder sogar im
zellumgebenden Medium befindet. Dazu gehören beispielsweise hydrolytische Enzyme,
Toxine, Strukturproteine sowie membranintegrale Proteine, die an selektivem Ionen- und
Metabolitentransport, Energiekonvertierung, Zellteilung, Signalleitung
und Membran-,
beziehungsweise Zellwandbiogenese beteiligt sind. Proteine, die für den Transport bestimmt
sind, besitzen eine intrinsische Signalsequenz, wodurch sie von cytoplasmatischen
Proteinen unterschieden werden können. Die Signalsequenz beinhaltet darüber hinaus die
Information durch welches der Transportsysteme das Protein exportiert oder in die Membran
integriert werden soll. In Bakterien haben sich zu diesem Zweck komplexe Transportwege
entwickelt.
Der Haupttransportweg in oder durch die Cytoplasmamembran ist das sekretorische (Sec-)
Transportsystem, welches ausschließlich Proteine im entfalteten Zustand post- oder
cotranslational
transportiert.
Einige
Proteine
allerdings
erlagen
auf
Grund
von
Kofaktorinsertion, Assemblierung zu oligomeren Strukturen oder schnellen Faltungskinetiken
bereits im Cytoplasma ihre native Form. Diese Gruppe von Proteinen wird posttranslational
durch
das
twin arginine translocation (Tat)-System
in
und
vor
allem
über
die
Cytoplasmamembran transportiert. Dabei zeigt das System die bemerkenswerte Fähigkeit,
den Faltungszustand der Tat-Substrate zu erkennen, um nur Proteine mit korrekten
Konformationen zu transportieren.
In Gram-negativen Bakterien sind auch Transportsysteme in und über die äußere Membran
der Zellhülle nötig. Zu diesen gehören beispielsweise die Sekretionssysteme des Typs I-VI,
3
II. Einleitung
die entweder Proteine, die durch das Sec- oder Tat- System ins Periplasma gelangt sind
weitertransportieren, oder sogar eigenständig den gesamten Transport über die innere und
äußere Membran vermitteln.
II.1. Das sekretorische (Sec)-System in E. coli
Das Sec-System ist außer in der Plasmamembran von Bakterien auch in der
Plasmamembran der Archaebakterien sowie in eukaryontischen Organellen wie dem
Endoplasmatischen Reticulum und Chloroplasten zu finden. In E. coli basiert dieses System
im Wesentlichen auf der Funktion des heterotrimeren Membrankomplexes SecYEG
(bestehend aus den Membranproteinen SecY, SecE und SecG), der die porenbildende
Komponente darstellt, durch die Proteine in oder durch die Plasmamembran transportiert
werden. Da die Pore an sich passiv ist, benötigt sie Partner, welche Energie für den
Proteintransport durch die Pore zur Verfügung stellen und sich zu diesem Zweck peripher an
den SecYEG-Komplex lagern. Dabei handelt es sich entweder um das translatierende
Ribosom selbst oder um das ATP-abhängige Motorprotein SecA. Weitere Proteine des SecSystems (SecD, SecF, YaiC) erhöhen durch ihre Interaktion mit dem SecYEG-Komplex die
Effizienz des Translokationsprozesses.
II.1.1. Das Sec-Translokon
Den Kern des SecYEG-Komplexes bildet das essentielle SecY (SecY in Bakterien und
Archaebakterien, cpSecY in Chloroplasten, Sec61α in Säugern und Sec61p in Hefe), ein 48
kDa großes Protein mit zehn membrandurchspannenden Domänen, wobei der N- und der CTerminus cytoplasmatisch lokalisiert sind [Akiyama und Ito, 1987]. Das ebenfalls essentielle
SecE-Protein (SecE in Bakterien und Archaebakterien, cpSecE in Chloroplasten, Sec61γ in
Säugern und Sss1p in Hefe) ist 14 kDa groß und durchspannt die Membran dreimal, wobei
nur der N-Terminus cytoplasmatisch lokalisiert ist [Schatz et al., 1989]. SecG (SecG in
Bakterien, Secβ in Archaebakterien, Sec61β in Säugern und Sbh in Hefe) ist ein 12 kDa
großes Protein, das die Plasmamembran zweimal durchspannt. N- und C-Terminus sind
periplasmatisch lokalisiert [Nishiyama et al., 1996]. Anders als SecY und SecE ist SecG nicht
essentiell für die Zelle [Bost und Belin, 1995]. Der SecYEG-Komplex bildet sich als Resultat
der Interaktion von SecY mit SecE. In Abwesenheit von SecE ist SecY instabil und wird von
der membrangebundenen Protease FtsH abgebaut [Kihara et al., 1995]. Für die Bildung
eines SecYE-Komplexes ist SecG nicht nötig [Akimaru et al., 1991], aber durch seine
4
II. Einleitung
Interaktionen scheint der SecYE-Komplex stabilisiert zu werden [Homma et al., 1997].
In vitro-Studien zeigten, dass SecG auch eine Bedeutung als stimulierender Faktor der
Translokation zukommt [Nishiyama et al., 1994; Hanada et al., 1996]. Dabei scheint eine
Topologieinversion des SecG-Proteins von Bedeutung zu sein [Nishiyama et al., 1996; Sugai
et al., 2007].
Neueste Daten zur Struktur des Komplexes aus einem Archaebakterium [Van den Berg et
al., 2004], sowie aus E. coli [Breyton et al., 2002; Bostina et al. 2005] zeigen eine Sanduhrähnliche Struktur des Komplexes mit jeweils einer trichterförmigen Öffnung des Kanals zur
cytoplasmatischen und periplasmatischen Seite der Membran. Die Spitzen der beiden
Trichter treffen sich in der Mitte der Membran, wo sich die engste Stelle des Kanals befindet.
Diese wird aus einem Ring von sechs hydrophoben Aminosäuren gebildet, wodurch die
selektive Permeabilität der Membran erhalten bleibt. Unterstützt wird dies in einem
nichttranslozierenden SecYEG-Kanal durch eine kurze α-Helix („plug“), die den Trichter zur
periplasmatischen Seite hin verschließt und erst während einer Proteintranslokation den
Weg durch den Kanal freigibt [Saparov et al., 2007]. Unklar ist bislang, ob während der
Proteintranslokation ein einzelner SecYEG-Komplex oder mehrere, zu Oligomeren
zusammengelagerte Komplexe, nötig sind [Rapoport, 2007].
II.1.2. Zielsteuerung und Erkennung
Substrate des Sec-Systems sind sekretorische Proteine, die durch die Membran transportiert
werden oder Membranproteine, die in die Membran inseriert werden müssen. Sec-Substrate
werden als solche erkannt und zur Sec-Translokase geleitet. Sekretorische Proteine werden
von Proteinen, die nicht für den Export bestimmt sind, durch eine N-terminale Signalsequenz
unterschieden. Diese besteht aus durchschnittlich 20 Aminosäuren und besitzt eine
dreigeteilte Struktur, die sich zusammensetzt aus einer meist positiv geladenen N-terminalen
Region (n-Region), einem hydrophoben Kern aus etwa 10–15 Aminosäuren (h-Region),
sowie einer polaren C-terminalen Region (c-Region) [von Heijne, 1990]. Die Signalsequenz
wird postranslokational durch eine Signalpeptidase vom reifen Teil des Proteins abgespalten.
Membranproteine dagegen besitzen in der Regel keine derartige Signalsequenz. Die
Funktion eines intrinsischen Signals für die Zielsteuerung des Proteins zum SecYEGKomplex wird von einer N-terminalen hydrophoben Transmembrandomäne, der SignalAnker-Sequenz, übernommen, die nach der Insertion des Proteins nicht abgespalten wird.
In E. coli existieren für Sec-abhängige Proteine zwei unterschiedliche Zielsteuerungswege.
In der Regel werden sekretorische Proteine posttranslational durch das molekulare
Chaperon SecB, Membranproteine dagegen cotranslational durch das sog. signal
5
II. Einleitung
recognition particle (SRP) zum SecYEG-Komplex gelenkt. Welcher der beiden Wege
eingeschlagen wird, entscheidet sich zu einem frühen Zeitpunkt der Translation, wenn das
naszierende Protein aus dem Ribosom austritt [Koch et al., 1999] und eine Interaktion
entweder mit SRP oder der Peptidyl-Prolyl-cis/trans-Isomerase trigger factor (TF) eingeht.
Die Hydrophobizität der Kette ist ausschlaggebend für die Wahl des Interaktionspartners. Je
hydrophober die naszierenden Kette (bspw. Transmembrandomäne), desto wahrscheinlicher
ist eine Interaktion mit SRP und somit eine cotranslationale Zielsteuerung [Valent et al.,
1995]. In Konkurrenz dazu steht die Interaktion mit TF, der eine Interaktion einer wenig
hydrophoben Kette mit SRP verhindert und somit eine Bindung von SecB an die wachsende
Kette zu einem späteren Zeitpunkt ermöglicht, wodurch das Protein posttranslational zum
SecYEG-Komplex geleitet wird [Beck et al., 2000]. TF bindet in vitro unspezifisch an
cytoplasmatische und sekretorische Proteine [Valent et al., 1995; Hesterkamp et al., 1996],
jedoch nicht an Membranproteine [Beck et al., 2000].
II.1.3. SecA/SecB-abhängiger, posttranslationaler Transport sekretorischer
Proteine
In der Regel werden sekretorische Proteine, die durch das SecYEG-Translokon transportiert
werden, durch den SecA/SecB-abhängigen Weg zum Translokon geleitet. Da die SecSubstrate ausschließlich in einer ungefalteten Konformation transportiert werden und
ungefaltete Proteine im Cytosol instabil sind, wird nach der Vollendung der Translation die
ungefaltete Konformation sekretorischer Proteine durch die Bindung des cytosolischen,
tetrameren Chaperons SecB stabilisiert und somit in einer transportkompetenten Form
gehalten [Reyes et al., 2007]. SecB übergibt die Polypetidkette an ein an den SecYEGKomplex gebundenes SecA-Protein, das eine hohe Affinität zu diesem Komplex besitzt [Hartl
et al., 1990]. SecA ist ein ATP-abhängiges Motorprotein, das mit SecB interagiert und sich
sowohl an die Signalsequenz als auch an den reifen Teil des sekretorischen Proteins binden
kann [Walter und Johnson, 1994; Valent et al., 1998; Prinz et al., 2000]. Die Translokation
beginnt mit der Bindung von ATP an SecA, wodurch sich SecA über SecY in die Membran
inseriert und dabei die Signalsequenz des Polypeptids in einer Haarnadel-ählichen Form in
die Translokationspore einführt [Ramamurthy und Oliver, 1997]. Dieser Schritt kann durch
die protonenmotorische Kraft stimuliert werden, die wahrscheinlich die Orientierung der
Signalsequenz in der Pore beeinflusst [Nouwen et al., 1994; van Dalen et al., 1999].
Hydrolyse des gebundenen ATPs induziert die Deinsertion des SecA aus der Membran
sowie dessen Dissoziation von der inserierten Polypetidkette [Schiebel et al., 1991], um
anschließend erneut an die Polypeptidkette zu binden, wodurch diese weiter durch die Pore
6
II. Einleitung
geschoben wird. Durch erneute ATP-Bindung wird SecA erneut inseriert und die
Polypeptidkette nochmals weiter durch die Pore geschoben [Schiebel et al., 1991; van der
Wolk et al., 1997]. Der beschriebene Zyklus aus ATP-Bindung und -Hydrolyse, also
wiederholter SecA-Insertion und -Deinsertion schiebt in jedem Zyklus etwa 5 kDa der Kette
weiter durch die Pore, bis schließlich die Translokation vollständig ist [van der Wolk et al.,
1997]. Der Protonengradient unterstützt die SecA-Deinsertion [Nishiyama et al., 1999] und
kann sogar zu einem späten Zeitpunkt die Translokation ohne SecA vorantreiben [Schiebel
et
al.,
1991].
Außerdem
bestimmt
der
Protonengradient
wahrscheinlich
die
Transportrichtung, indem ein Zurückgleiten des Proteins in das Cytoplasma verhindert wird.
Nach vollendeter Translokation wird die Signalsequenz des sekretorischen Proteins durch
eine membranständige Signalpeptidase abgespalten und anschließend abgebaut.
II.1.4. Signal recognition particle (SRP)–abhängige cotranslationale Integration
von Innenmembranproteinen
In der Regel werden Innenmembranproteine, die durch den SecYEG-Komplex in die
Cytoplasmamembran integriert werden, durch den signal recognition particle (SRP)–
abhängigen Weg zum Translokon geleitet. Das SRP in E. coli besteht aus einer 4,5 S RNA
und der 48 kDa großen GTPase Ffh (fifthy-four homolog) [Poritz et al., 1990]. Sobald die
hydrophobe Signal-Anker-Sequenz translatiert wurde und sich außerhalb des Ribosoms
befindet, wird diese durch SRP erkannt und gebunden [Valent et al., 1997]. Der Komplex aus
Ribosom, naszierender Kette und dem SRP bindet an den membranständigen SRPRezeptor FtsY [Bernstein et al., 1989]. FtsY ist ebenfalls eine GTPase, die entweder an
anionische Phospholipide der Membran [de Leeuw et al., 2000] oder direkt an den SecYEGKomplex binden kann [Angelini et al., 2005]. Durch Bindung des Komplexes aus Ribosom,
naszierender Kette und SRP an FtsY bindet GTP an SRP sowie an FtsY [Shan und Walter,
2005; Reyes et al., 2007]. SRP und FtsY sind beide GTP-abhängige GTPase-aktivierende
Proteine (GAPs) für das jeweils andere Protein, wodurch eine gegenseitig Regulation erfolgt
[Keenan et al., 2001]. Eine anschließende GTP-Hydrolyse an FtsY als auch an SRP führt zur
Übergabe des Ribosoms mit der naszierenden Kette an den SecYEG-Komplex [Valent et al.,
1998]. Dabei geht das Translokon einen engen Kontakt mit dem Ribosom ein, wodurch die
Pore des Translokons zum Cytoplasma hin abgedichtet wird [Prinz et al., 2000]. Durch die
fortlaufende Translation bewegt sich nun die naszierende Kette direkt aus dem Austrittskanal
des Ribosoms in die Pore des SecYEG-Komplexes. Während das Ribosom für die Synthese
der Polypeptidkette GTP benötigt, ist die Bewegung der naszierenden Kette durch den
SecYEG-Komplex unabhängig von Nukleotidhydrolysen [Connolly und Gilmore, 1986].
7
II. Einleitung
Transmembrandomänen der naszierenden Kette werden durch eine laterale Öffnung des
SecYEG-Komplexes in die Membran inseriert, ein Vorgang, der unter Mithilfe des
Membranproteins YidC erfolgt [Beck et al., 2001].
Eine Besonderheit stellt die Mithilfe von SecA bei der Integration von Membranproteinen mit
großen periplasmatischen Domänen dar. SecA fungiert auch in diesem Fall als Motorprotein,
das schrittweise die große periplasmatische Domäne durch das Translokon befördert [Scotti
el al., 1999; Neumann-Haefelin et al., 2000].
II.2. Das twin arginine translocation (Tat)–System
Das twin arginine translocation (Tat)-System ist ein Proteintransportsystem in Bakterien,
Archaebakterien und Chloroplasten, das in der Lage ist, vollständig gefaltete Proteine über
die Cytoplasmamembran bzw. Thylakoidmembran zu transportieren. Der Name ist auf zwei
hochkonservierte Arginine (Doppel-Arginin-Motif) in der n-Region der Signalsequenz Tatabhängiger Proteine zurückzuführen. Prätranslokationale Faltung der Tat-Substrate findet
auf Grund von Kofaktorinsertion, Assemblierung zu oligomeren Strukturen oder schnellen
Faltungskinetiken statt.
Das Tat-System unterscheidet sich in wesentlichen Punkten vom Sec-System. Der
bemerkenswerteste Unterschied liegt in der Fähigkeit des Tat-Systems, vollständig gefaltete
Proteine zu transportieren, während die Vorraussetzung für den Sec-abhängigen Transport
eine entfaltete Form des Substrates ist. Ein weiterer Unterschied besteht im Aufbau der
Signalsequenz, die in Tat-Substraten länger ist und ein N-terminales Konsensusmotif besitzt,
das die zwei konservierten Arginine beinhaltet. Substrate, denen das Doppel-Arginin-Motif
fehlt oder unvollständige Faltung besitzen, werden in der Regel nicht transportiert. Darüber
hinaus arbeitet das Tat-Translokon unabhängig von Nukleotidhydrolysen und wird exklusiv
durch die protonenmotorische Kraft (PMF) an der Membran energetisiert, während das SecSystem ATP (für die ATPase SecA), GTP (für die GTPasen SRP und FtsY) als auch die
PMF nutzt.
II.2.1. Tat-Substrate
Proteintranslokation durch das Tat-System wird in Bakterien in unterschiedlicher Intensität
genutzt. In E. coli werden wahrscheinlich nur 26 Proteine durch das Tat-System transportiert,
während beispielsweise im Gram-positiven Actinomyceten Streptomyces coelicolor 250 TatSubstrate vorausgesagt wurden [Widdick et al., 2006]. Halophile Archaebakterien nutzen für
8
II. Einleitung
den Export sekretorischer Proteine sogar hauptsächlich das Tat-Sytem. Dies ist vermutlich
auf die hohe Salzkonzentration im Cytoplasma und der damit verbundenen schnellen
Faltung neu synthetisierter Proteine zurückzuführen [Rose et al., 2002]. In Chloroplasten hält
sich die Anzahl der Tat- bzw Sec-abhängigen Proteine die Waage.
Ein Transportsystem für gefaltete Proteine kann aus unterschiedlichen Gründen nötig sein.
So sind die meisten Tat-Substrate in E. coli kofaktortragende Redoxproteine, deren
Kofaktoren, wie Moybdopterin, FAD, NADP+, [FeS] - und [NiFe] - Cluster und Kupferionen,
bereits im Cytoplasma eingebaut werden müssen [Berks et al., 2003]. Diese Proteine
übernehmen in E. coli Funktionen in der anaeroben Atmung als Hydrogenase, FormatDehydrogenase,
Trimethylamin-N-Oxid (TMAO)–Reduktase,
Dimethylsulfoxid
(DMSO)–
Reduktase und Nitrat-Reduktase, sowie als Enzym der Biogenese der Zellhülle und als
Virulenzfaktor [Lee et al., 2006]. Darüber hinaus existieren in E. coli kofaktorlose TatSubstrate wie beispielsweise periplasmatische Amidasen oder auch das SufI-Protein
(suppressor of FtsI), das Zellteilungsdefekte von ftsI-Mutanten subprimieren kann. Diese
Proteine besitzen wahrscheinlich eine schnelle Faltungskinetik, wodurch sie inkompatibel für
das Sec-System sind.
Einige Tat-abhängige Proteine werden durch den sog. Hitchhiker-Mechanismus transportiert.
Sie selbst besitzen keine eigene Tat-Signalsequenz, werden aber in einem Komplex mit
Proteinen transportiert, die eine solche besitzen. So weist nur die DmsA-Untereinheit der
dimeren DMSO-Reduktase eine Tat-Signalsequenz auf. Gleiches gilt auch für eine der
Untereinheiten der Hydrogenase-1 oder -2 in E. coli [Rodrigue et al., 1999].
Das Tat-System spielt auch in der Pathogenese eine Rolle. So ist das Tat-Translokon
essentiel für die Virulenz sowohl von Yersinia pseudotuberculosis [Lavander et al., 2006] als
auch von Agrobacterium tumefaciens [Ding and Christie, 2003]. Zwei Phospholipasen aus
Pseudomonas aeruginosa wurden als Tat-abhängige Virulenzfaktoren identifiziert [Voulhoux
et al., 2001; Ochsner et al., 2002].
Darüber hinaus sind auch Membranproteine als Tat-Substrate bekannt. So sind in E. coli
mindestens fünf der Tat-Substrate vorausgesagte Innenmembranproteine, die durch eine Cterminale Transmembranhelix verankert sind [Hatzixanthis et al., 2003]. Auch das RieskeFeS-Protein in Paracoccus denitrificans [Bachmann et al., 2006] und in Legionella
pneumophila [DeBuck et al., 2007] sind Tat-abhängige Membranproteine.
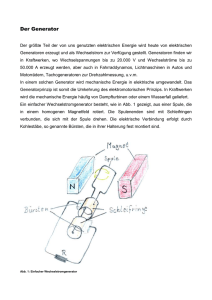
II.2.2. Die Komponenten der Tat-Translokase
Das Tat-System in E. coli besteht aus den drei Innenmebranproteinen TatA (TatE), TatB und
TatC. Die orthologen Proteine in der Thylakoidmembran des Chloroplasten werden als Tha4,
9
II. Einleitung
Hcf106 und cpTatC bezeichnet. Sowohl in E. coli als auch im Chloroplasten sind diese
Proteine essentiell für die Tat-abhängige Proteintranslokation [Sargent et al., 1998; Bogsch
et al., 1998; Sargent et al., 1999; Mori et al., 1999 und 2001]. TatE aus E. coli ist zu 50%
sequenzhomolog zu TatA und kann auch funktionell TatA ersetzen, weshalb nur eine
Deletion beider Proteine zu einer Inhibition des Tat-abhängigen Transportes führt [Sargent et
al., 1998]. TatA liegt allerdings in der 50–200fach höheren Menge als TatE in der Zelle vor,
wodurch eine TatA-Deletion wesentlich schwerwiegendere Folgen hat als die einer TatEDeletion [Jack et al., 2001]. TatD, dessen Gen in einem Operon mit den Genen von TatA,
TatB und TatC organisiert ist, ist eine Nuklease, die nicht am Tat-System beteiligt ist [Wexler
et al., 2000].
Periplasma
TatA
TatB
TatC
N
N
C
C
N
Cytoplasma
C
Abb. II.1 Topologie der Membranproteine TatA, TatB und TatC
II.2.2.1. TatA
TatA ist die kleinste und mit einem 25–50fach höheren Expressionsniveau als TatB oder
TatC auch die häufigste der drei Komponenten der Tat-Translokase in E. coli [Jack et al.,
2001].
Da
TatA
dazu
neigt,
homooligomere,
ringförmige
Komplexe
in
der
Cytoplasmamembran zu bilden, wird angenommen, dass TatA die porenbildende
Komponente der Translokase repräsentiert. TatA besteht aus 89 Aminosäuren und zeigt
20 % Sequenzhomologie zu TatB. Zudem wurde für beide Proteine eine ähnliche Toplogie
vorgeschlagen, mit einer N-terminalen, transmembranen α-Helix die über eine „Gelenk“Region mit einer amphiphatischen Helix verbunden ist, an die sich ein unstrukturierter CTerminus anschließt, der im Fall von TatA wesentlich kürzer ist [Porcelli et al., 2002] (Abb.
II.1). Die tatsächliche Orientierung des TatA-Proteins ist noch ungeklärt. Untersuchungen zur
Topologie zeigten eine cytoplasmatische Lokalisierung der amphiphatischen Helix und des
unstrukturierten C-Terminus, wodurch der N-Terminus zum Periplasma hin orientiert ist
[Porcelli et al., 2002]. Neuere Studien deuten jedoch auf eine umgekehrte Orientierung mit
einer dualen Topologie des periplasmatisch lokalisierten C-terminalen Teils des Proteins, der
10
II. Einleitung
von einer membranassoziierten Position zu einer in die Membran eingebetteten Position
wechseln kann [Gouffi et al., 2004; Chan et al., 2007]. Es konnte sogar eine Abhängigkeit
der Topolgieänderung des C-terminalen Teils vom Membranpotential gezeigt werden.
Basierend auf dieser Annahme wurde ein Model für einen TatA-Komplex mit einem 50 Å
breiten Kanal vorgeschlagen, dessen periplasmatische Öffnung durch die amphiphatischen
Helices aller TatA-Untereinheiten in Gegenwart eines Membranpotentials verschlossen wird.
Dieses Model einer geschlossen TatA-Pore würde allerdings eine aus energetischen
Gesichtspunkten ungünstige Konformation besitzen [Chan et al., 2007].
Eine Vielzahl von Mutationsstudien identifizierten für die Funktion von TatA essentielle
Aminosäuren in der Transmembranhelix, der „Gelenk“-Region sowie der amphiphatischen
Helix.
So
führten
Transmembranhelix),
Alanin-Substitutionen
Phe20
und
Gly21
der
Aminosäuren
(„Gelenk“-Region),
Glu8
(N-Terminus
Phe39
und
der
geladene
Aminosäuren an Position 41 (C-Terminus der amphipathischen Helix) teilweise zur
vollkommenen Inhibition des Tat-abhängigen Transportes von TorA in E. coli [Hicks et al.,
2003 und 2005, Green et al., 2007]. Der unstrukturierte C-Terminus des Proteins scheint für
die Funktion von TatA bedeutungslos zu sein, da die Aktivität von TatA auch ohne seine 40
C-terminalen Aminosäuren voll erhalten bleibt [Lee et al., 2002].
In den gram-positiven Bakterien Streptomyces lividans [De Keersmaeker et al., 2005] und
Bacilus subtilis [Westermann et al., 2006] konnten lösliche Formen von TatA im Cytoplasma
nachgewiesen werden. Das TatAd-Protein aus Bacillus subtilis ist spezifisch für den
Transport des Tat-Substrates PhoD. Der löslichen Form des TatAd muss möglicherweise
eine Funktion als Zielsteuerungs-Faktor eingeräumt werden, da es sowohl an die
Signalsequenz PhoD bindet als auch mit TatCd interagiert [Schreiber et al., 2006].
II.2.2.2. TatB
Für TatB wurde eine ähnliche Topologie wie für TatA vorgeschlagen (Abb. II.1). Mit 171
Aminosäuren ist es größer als TatA und besitzt dadurch einen deutlich längeren
unstrukturierten C-Terminus als TatA [De Leeuw et al., 2001]. Trotz der Homologien
zwischen den beiden Proteinen erfüllen sie verschiedene Aufgaben und können funktionell
einander nicht ersetzen
[Sargent et al. 1999]. TatB bildet zusammen mit TatC den
Erkennungskomplex für die Tat-Signalsequenz. TatB fungiert wahrscheinlich als Vermittler
zwischen TatC und TatA [Cline und Mori, 2001; Alami et al., 2003].
Einige konservierte Aminosäuren wie das Glu8 (N-Terminus der Transmembranhelix), Gly21
und Pro22 („Gelenk“-Region), sowie Leu25, Pro26 und drei Glutamate an Position 49, 53
und 58 (C-Terminus der amphipathischen Helix) wurden Mutationsstudien unterzogen. Die
stärksten Inhibitionen wurden durch Substitution der Aminosäuren in der „Gelenk“-Region,
sowie in der amphiphatischen Helix beobachtet. Dennoch scheint keine der konservierten
11
II. Einleitung
Aminosäuren essentiell für den Tat-abhängigen Transport zu sein [Barrett et al., 2003; Hicks
et al., 2003]. Außerdem kann der unstrukturierte C-Terminus genau wie bei TatA ohne
Aktivitätsverlust entfernt werden [Lee et al., 2002].
Während TatB essentiell für den Transport natürlicher Tat-Substrate in E. coli ist [Sargent et
al., 1999], konnte für Fusionsproteine (TorA–maltose binding protein, TorA-Colicin V) auch in
einem TatB-Deletionsstamm Tat-abhängiger Transport beobachtet werden [Ize et al., 2002;
Blaudeck et al., 2005]. Transport des TorA–maltose binding protein in einem TatBDeletionshintergrund konnte durch Mutationen im extremen N-Terminus von TatA, einer
Region wo das TatB-Protein ein konserviertes Aspartat an Position 3 aufweißt, deutlich
gesteigert werden. Wahrscheinlich ist diese Region entscheidend für die spezifische
Funktion von TatB [Blaudeck et al., 2005]. Die erwähnten TatA-Mutanten sind sogar in der
Lage in der Abwesenheit von TatB das natürliche Tat-Substrat TorA effektiv zu
transportieren, selbst wenn sie zusätzlich an TatC fusioniert sind [Barrett et al., 2007].
Tatsächlich existieren in Archaebakterien und Gram-positiven Bakterien auch natürliche
Beispiele für TatAC-Typ Tat-Systeme, in denen TatA nicht nur seine vermutliche Funktion als
porenbildende Komponente erfüllt, sondern auch die spezifische Funktion von TatB als
Mediator zwischen TatC und TatA übernimmt [Dilks et al., 2003, Jongbloed et al., 2004].
II.2.2.3. TatC
TatC ist mit 258 Aminosäuren das größte der Tat-Proteine. Eine Toplogie mit sechs
transmembranen Helices mit beiden Termini auf der cytoplasmatischen Seite wurde
vorgeschlagen [Mori et al., 2001; Behrendt et al., 2004; Ki et al., 2004]. Gouffi et al. schlugen
eine TatC-Toplogie mit nur vier Transmembrandomänen vor, wobei eine große
periplasmatische Schleife die transmembranen Helices vier und fünf des anderen Modells
beinhaltet [Gouffi et al., 2002]. Neueste Studien unterstützen allerdings eher die 6Transmembrandomänen-Topologie [Punginelli et al., 2007] (Abb. II.1).
TatC bildet zusammen mit TatB den Erkennungskomplex für die Tat-Signalsequenz [Cline
und Mori, 2001; Alami et al., 2003]. Trotz ausgedehnter Mutationsstudien konnte bislang
keine definierte Region identifiziert werden, die in Kontakt zur Signalsequenz kommt.
Vielmehr scheint die gesamte N-terminale Hälfte des Proteins bei der Erkennung der TatSignalsequenz beteiligt zu sein [Holzapfel et al., 2007]. Lediglich die Identifizierung von
Suppressor-Mutanten in TatC, die eine erhöhte Toleranz gegenüber mutierten TatSignalsequenzen aufweisen, deuten auf einen Einfluss der ersten cytoplasmatischen
Schleife von TatC auf die Signalsequenz-Bindungsregion des TatBC-Rezeptor-Komplexes
hin [Kreutzenbeck et al., 2007]. Darüber hinaus führen Mutationen in dieser Schleife zur
Reduktion der Tat-Transport-Effizienz [Strauch und Georgiou, 2007].
12
II. Einleitung
II.2.3. Die Tat-Komplexe
Die drei E. coli Tat-Komponenten TatA, TatB und TatC bilden Komplexe in der inneren
Membran. Die Zusammensetzung isolierter Tat-Komplexe scheint von der verwendeten
Aufreinigungsmethode abhängig zu sein. Nichtsdestotrotz zeigten mehrere Studien eine
Organisation der Tat-Komponenten in hauptsächlich zwei Typen von Komplexen. Zum einen
der Rezeptor-Komplex oder Tat(A)BC-Komplex, der vorwiegend durch TatB und TatC
gebildet wird und, wenn aus Membranen aus E. coli gereinigt, Spuren von TatA enthält
[Bohluis et al, 2001; McDevitt et al., 2005]. Der zweite Komplex ist der TatA-Komplex, der
hauptsächlich durch TatA gebildet wird und, wenn aus Membranen aus E. coli gereinigt,
Spuren von TatB enthält [Sargent et al., 2001; Oates et al., 2003; McDevitt et al., 2005 und
2006]. Trotz geringer Sequenzhomologien der Tat-Komponenten verschiedener Organismen
scheint die strukturelle Organisation der Tat-Proteine unter Bakterien konserviert zu sein.
Dies zeigten elektronenmikroskopische Studien aufgereinigter Tat-Komplexe aus E. coli,
Agrobacterium tumefaciencs und Salmonella thyphimorium [Oates et al., 2003].
II.2.3.1. Der TatBC-Rezeptor-Komplex
Die Bezeichnung als Rezeptor-Komplex leitet sich von dessen Funktion als Rezeptor der
Tat-spezifischen Signalsequenz ab. Die Bindung Tat-abhängiger Proteine an den RezeptorKomplex wurde in in vitro-Studien mit einem ortsspezifischen photoaktiven Quervernetzer für
das natürlich Tat-Substrat SufI aus E. coli gezeigt [Alami et al., 2003], sowie für pflanzliche
Tat-Substrate, die spezifisch an den thylakoidalen Rezeptor-Komplex, dem Hcf106–cpTatCKomplex assozierten [Cline und Mori, 2001]. In diesen Studien war das Vorhandensein des
Doppel-Arginin-Motifs Voraussetzung für die spezifische Bindung des Tat-Substrates an den
Rezeptor-Komplex. Dies war in einer neueren in vivo-Studie nicht der Fall, in der SufI
überexprimiert, die Tat-Komponenten dagegen auf Wildtyp-Niveau belassen wurden. Das
SufI-Protein war unter diesen Bedingungen an den aufgereinigten TatBC-Rezeptor-Komplex
auch dann gebunden, wenn das Doppel-Arginin-Motif durch zwei Lysinreste substituiert
wurde [McDevitt et al., 2006].
Die
molekulare
Masse
des
Rezeptor-Komplexes
variiert
in
Abhängigkeit
der
Aufreinigungsmethode zwischen 360-700 kDa [Oates et al., 2005; McDevitt et al., 2005].
Unter Berücksichtung der molekularen Massen von TatB (18,4 kDa) und von TatC (28,9
kDa) muss der TatBC-Rezeptor-Komplex eine hochmolekulare Struktur besitzen, dessen
stöchiometrische Verhältnisse von TatB zu TatC mit 1:1 bestimmt wurden [Bolhius et al.,
2001; McDevitt et al., 2005]. In E. coli bildet sich der Rezeptor-Komplex unabhängig von
13
II. Einleitung
TatA [Behrendt et al., 2007; Orriss et al., 2007]. Auf Grund von Quervernetzungs-Studien
geht man bislang davon aus, dass der TatBC-Rezeptor-Komplex einer internen Organisation
unterliegt, in der die TatB-Protomere im Zentrum lokalisiert sind, während die TatC-Proteine
sich peripher anordnen [Lee et al., 2006; Punginelli et al., 2007]. Da translationale Fusionen
aus einem TatB- und einem TatC-Protein separate TatB- und TatC-Proteine funktionell
ersetzen können, muss von einer strukturellen und funktionellen Einheit aus je einem
Protomer von TatB und TatC innerhalb des Rezeptor-Komplexes ausgegangen werden
[Bohlius et al., 2001].
Elektronenmikroskopische Analysen des TatBC-Rezeptor-Komplexes zeigen eine ovale
Struktur mit einem maximalen Durchmesser von 13 Å, wodurch die Funktion eines Kanals
ausgeschlossen werden kann, da dieser in der Lage sein sollte, vollständig gefaltete
Proteine bis zu einer Größe von 100 kDa zu transportieren [Oates et al., 2003]. Diese Daten
unterstützen somit das Modell von einer aus TatA-Proteinen gebildeten Pore.
Neuste Blaue Nativ Gelelektrophorese-Analysen zeigen, dass überproduziertes TatB bzw.
TatC in Abwesenheit des jeweilig anderen, stabile homooligomere Komplexe bilden kann
[Orriss et al., 2007; Behrendt et al., 2007]. TatB assembliert in Oligomere von 100 kDa
[Orriss et al., 2007; Behrendt et al., 2007], die wiederum Multimere von bis zu 880 kDa bilden
können [Behrendt et al., 2007]. TatC dagegen bildet distinkte Oligomere von 220–250 kDa
[Orriss et al., 2007; Behrendt et al., 2007]. Diese homomultimeren Komplexe könnten
Vorstufen zur Bildung des TatBC-Rezeptor-Komplexes darstellen.
II.2.3.2. Der TatA-Komplex
In der bakteriellen Cytoplasmamembran und in der Thylakoidmembran ist TatA bzw. Tha4
das mengenmäßig häufigste der drei Tat-Komponenten. TatA bildet in der bakteriellen
Membran ringförmige homooligomere Komplexe mit einer molekularen Masse von 100–
700 kDa [Barrett et al., 2005; McDevitt et al., 2006]. Es wird angenommen, dass der TatAKomplex den proteinschleusenden Kanal bildet, der sich durch modularen Aufbau seiner
Komplexe den unterschiedlich großen Tat-Substraten anpassen kann. Diese Anpassung
muss sehr präzise erfolgen, um die Membranpermeabilitätsbarriere während der
Translokation zu erhalten.
Elektronenmikroskopische Analysen in Kombination mit Strukturrekonstruktionen von
gereinigten TatA-Komplexen aus E. coli zeigten eine Poren-ähnliche Struktur von TatAKomplexen unterschiedlicher Größe, die sich in vier Klassen einteilen ließen. Darunter waren
Komplexe zu finden mit molekularen Größen von 130–390 kDa, Protomer-Anzahlen von 12–
35 und Kanal-Durchmessern von 30–70 Å [Gohlke et al., 2005]. Diese Kanal-Durchmesser
würden zu den Größen der E. coli Tat-Substrate passen, wobei das größte Substrat der
FdnGH-Subkomplex der Format-Dehydrogenase mit 70 Å im Durchmesser und das kleinste
14
II. Einleitung
Substrat das NrfC-Protein mit 20–30 Å im Durchmesser wäre [Berks et al., 2003]. Darüber
hinaus wurde eine Deckel-ähnliche Struktur auf der cytoplasmatischen Seite des TatAKomplexes entdeckt, die vermutlich durch die amphiphatische Helix aller TatA-Protomere
gebildet wird. Solch eine Struktur könnte den Zugang zur Pore des TatA-Komplexes
kontrollieren, indem es zum einen den Eintritt des Tat-Substrates reguliert und zum anderen
den unspezifischen Fluss von Ionen durch den Kanal verhindert [Gohlke et al., 2005].
Interessant in diesem Zusammenhang ist ein Modell in dem sich die amphiphatischen
Helices der TatA-Protomere eines TatA-Komplexes von einer cytoplasmatisch lokalisierten
Position in die Membran hinein bewegen und dadurch in ihrer Gesamtheit einen hydrophilen
Kanal ausbilden [Gouffi et al., 2004]. Diese Modelle wären unabhängig von der Orientierung
des TatA-Proteins in der Membran.
II.2.4. Tat-spezische Signalsequenz und Zielsteuerung
Wie die Signalsequenz Sec-abhängiger Proteine besitzt auch die Tat-spezifische
Signalsequenz eine dreigeteilte Struktur aus einer kurzen, basischen, aminoterminalen nRegion, gefolgt von einer längeren hydrophoben h-Region an die sich die carboxyterminale
c-Region anschließt, die meist ein Erkennungsmotif für eine Signalpeptidase besitzt.
Charakteristisch für die Tat-Signalsequenz ist das Konsensusmotif S-R-R-x-F-L-K in der nRegion, wobei x eine polare Aminosäure ist. Die beiden hochkonservierten Arginine in
diesem Konsensusmotif sind der Namensgeber des Tat-Systems. Die Aminosäuren um
dieses Arginin-Paar sind weniger stark konserviert und sind in pflanzlichen TatSignalsequenzen variabler als in Bakterien.
Bakterielle Tat-Signalsequenzen sind in der Regel länger und besitzen eine weniger
hydrohobe h-Region als Sec-Signalsequenzen [Cristobal et al., 1999]. Außerdem besitzt die
c-Region der Tat-Signalsequenz häufig basische Aminosäuren, während die c-Regionen von
Sec-Signalsequenzen praktisch nie geladen sind [Berks et al., 2003]. Obwohl nicht alle TatSignalsequenzen dieses Merkmal aufweisen, geht man von einer sog. Sec-avoidanceFunktion dieser Region aus [Blaudeck et al., 2003]. Kürzlich wurde berichtet, dass zur TatSpezifität einer Signalsequenz die Ladung der c-Region als auch die Ladung der Nterminalen Region des reifen Proteins beitragen [Tullmann-Ercek et al., 2007].
Es gibt Hinweise auf eine RR-unspezifische Zielsteuerung von Tat-Substraten zur Membran,
die einhergeht mit der Insertion der Signalsequenz in die Membran noch bevor ein Kontakt
zur Tat-Translokase besteht [Hou et al., 2006; Shanmugham et al., 2006]. Nichtsdestotrotz
gilt es als gesichert, dass die initiale Kontaktstelle der Tat-Signalsequenz an die TatTranslokase durch den TatBC-Rezeptor-Komplex gebildet wird [Cline and Mori, 2001; Alami
15
II. Einleitung
et al., 2003; McDevitt et al., 2006]. Obwohl das Doppel-Arginin-Motif praktisch unerlässlich
für den Tat-abhängigen Transport in vivo und in vitro ist, ist dessen Bedeutung für die
Bindung des Tat-Substrates an den TatBC-Rezeptor-Komplex weniger klar. So zeigten
in vitro-Quervernetzungsversuche mit dem natürlichen Tat-Substrat SufI aus E. coli [Alami et
al.,
2003]
sowie
Blaue Nativ Gelelektrophorese-Analysen
des
Tat-abhängigen
Fusionsproteins HiPIP-PhoA in vivo [Richter und Brüser, 2005], dass die Substitution des
Arginin-Paares durch zwei Lysine zu einem Verlust der Interaktion zwischen dem TatSubstrat und dem TatBC-Rezeptor-Komplex führen. Dies war nicht der Fall in vivo, wenn
SufI überexprimiert, die Tat-Proteine allerdings auf Wildtyp-Niveau belassen wurden
[McDevitt et al., 2006]. Unter diesen Bedingungen fand Bindung zwischen SufI und dem
Rezeptor-Komplex auch dann statt, wenn das Arginin-Paar durch zwei Lysine bzw. zwei
Alanine ersetzt wurde. Auch im pflanzlichen Tat-System konnte beobachtet werden, dass
transport-inkompetente Mutanten des natürlichen Tat-Substrates OE17 auch dann geringe
Interaktionen zum Tat-Translokon aufwiesen, wenn das Arginin-Paar durch zwei Lysine oder
zwei Glutamine ersetzt wurde [Alder und Theg, 2003].
In diesem Zusammenhang ist es interessant zu erwähnen, dass auch einige wenige
natürliche Tat-Substrate existieren, die von dem hochkonservierten Doppel-Arginin-Motif
abweichen. Beispiele sind die TtrB-Untereinheit der Tetrathionat-Reduktase aus Salmonella
enterica (KR) [Hinsley et al., 2001], die Präpropenizillinamidase aus E. coli (RNR) [Ignatova
et al., 2002], sowie das Rieske-Protein des Cytochrom b6f-Komplexes in Chloroplasten (KR)
[Molik et al., 2001]. Darüber hinaus zeigt die Doppel-Lysin-Mutante des Tat-abhängigen
Reporterproteins TorA-ColV eine geringe Transportkompetenz in E. coli-Zellen [Ize et al.,
2002]. Diese Beobachtungen lassen den Schluss zu, dass das Doppel-Arginin-Motif nicht die
einzige Determinante für die Tat-abhängige Zielsteuerung des Tat-Substrates zum TatBCRezeptor-Komplex sein kann. Diese Annahme wird bestätigt durch die Identifizierung von
Tat-Translokasen mit Mutationen in der ersten cytosolischen Domäne von TatC, die in der
Lage sind, die Transportinkompetenz des Reporterproteins TorA-PhoA, dessen DoppelArginin-Motif durch ein Lysin und ein Glutamin ersetzt wurde, zu unterdrücken [Kreutzenbeck
et al., 2007].
II.2.5. Transportmechanismus
Die Größe der Tat-Substrate die durch die Cytoplasmamembran ins Periplasma (Bakterien
und Archaebakterien), bzw. durch die Thylakoidmembran ins Thylakoidlumen transportiert
werden (Chloroplast), reicht von 10-100 kDa [Müller, 2005], mit Durchmessern von 20–70 Å
[Berks et al., 2000]. Um Substrate solch unterschiedlicher Größe in einer vollständig
16
II. Einleitung
gefalteten Konformation durch eine Membran zu transportieren, ohne deren Funktion als
Permeabilitätsbarriere zu stören, bedarf es eines Transportmechanismus, der sich
grundlegend von den bekannten Proteintranslokations-Systemen unterscheidet.
Der Translokationsprozess lässt sich in zwei Schritte unterteilen. Zum einen die Bindung des
Tat-Substrates an den TatBC-Rezeptor-Komplex und zum anderen die PMF-abhängige
Rekrutierung
eines
TatA-Komplexes
an
den
Substrat-TatBC-Rezeptorkomplex
zur
Assemblierung der vollständig aktiven Translokase, wodurch die eigentliche Translokation
stattfindet. Nach der Translokation zerfällt die Translokase in ihre Subkomplexe.
II.2.5.1. Kontakt des Tat-Substrates mit dem TatBC–Rezeptor–Komplex
In in vitro-Experimenten mit einem photoaktiven ortsgerichteten Quervernetzer in der
Signalsequenz des natürlichen Tat-Substrates SufI aus E. coli wurde eine Hierarchie in der
Interaktion der Signalsequenz zu den Tat-Komponenten beobachtet [Alami et al., 2003]. So
findet der erste Kontakt der Signalsequenz zu TatC statt. Anschließend bindet an die
Signalsequenz TatB, das vermutlich den darauf folgenden Kontakt der Signalsequenz zu
TatA vermittelt. Der Kontakt zu TatA fand nur statt, wenn die in dieser Studie benutzten,
invertierten Membranvesikel durch eine intakte PMF energetisiert waren. Durch die
Veränderung der Position des Quervernetzers in der Signalsequenz konnte gezeigt werden,
dass TatC mit der Region um das Konsensus-Motif interagiert, während TatB mit der
gesamten Signalsequenz sowie mit dem N-terminalen Teil des reifen Proteins in Kontakt
steht [Alami et al., 2003].
Im Prinzip wurden die gleichen Ergebnisse für das Tat-System des Chloroplasten erhalten,
einzig mit der Ausnahme, dass kein Kontakt zu TatA nachgewiesen wurde [Gerald und Cline,
2006]. Dieser Unterschied kann mit der Beobachtung erklärt werden, dass die thylakoidalen
Tha4-Komplexe relativ instabil und von transienter Natur sind. In der gleichen Studie wurde
auch dann noch Transport beobachtet, wenn die Signalsequenz kovalent an das cpTatCProtein gebunden war, was darauf hindeutet, dass der Transfer der Signalsequenz von
cpTatC auf andere Tat-Komponenten keine Voraussetzung für die Translokation des reifen
Teils des Substrates ist [Gerald und Cline, 2006]. An der Erkennung der Signalsequenz
durch TatC ist dessen gesamte N-terminale Hälfte beteiligt [Holzapfel et al., 2007], wobei die
erste cytoplasmatische Schleife von TatC wahrscheinlich von besonderer Bedeutung ist
[Kreutzenbeck et al., 2007].
Der TatBC-Rezeptor-Komplex erkennt nicht nur die Signalsequenz, sondern scheint diese
auch in die Membran zu inserieren noch bevor die Translokation des reifen Proteins
stattfindet. Die Insertion der Signalsequenz ermöglicht es der an der periplasmatischen Seite
der Membran assoziierten Signalpeptidase die spezifische Schnittstelle am C-Terminus der
17
II. Einleitung
Signalsequenz zu erkennen, noch bevor die Translokation des reifen Proteins stattgefunden
hat [DiCola und Robinson, 2005].
II.2.5.2. Translokationsmodelle
Nach der Bindung des Tat-Substrates an den TatBC-Rezeptor-Komplex erfolgt in einem
zweiten Schritt die eigentliche Translokation des reifen Teils des Proteins durch die
Membran. Um diesen Prozess zu beschreiben wurden verschiedene Modelle vorgeschlagen.
In allen Modellen gilt TatA als die Tat-Komponente, die die Passage des Proteins durch die
Membran ermöglicht. Nach der Bindung des Tat-Substrates an den TatBC-RezeptorKomplex wird TatA PMF-abhängig rekrutiert, wodurch sich die vollständige Translokase
assembliert [Mori und Cline, 2002; Alami et al., 2003; Dabney-Smith et al., 2006]. In
Bakterien wird überwiegend davon ausgegangen, dass dazu ein bereits assemblierter TatAKomplex an den Substrat-Rezeptor-Komplex bindet [Sargent et al., 2001; Alami et al., 2003],
während im Chloroplasten die Tha4-Oligomerisierung durch die Bildung des SubstratRezeptor-Komplexes ausgelöst wird [Dabney-Smith et al., 2006]. In beiden Fällen bildet der
TatA-(Tha4)-Komplex einen hydrophilen Kanal, durch den der reife Teil des Proteins
transportiert wird. Diese Annahme fußt im Wesentlichen auf elektronenmikroskopischen
Analysen
von
TatA-Komplexen
der
die
E. coli-Membran,
ringförmige
Strukturen
unterschiedlicher Größen aufweisen. Man geht davon aus, dass die Größe des TatSubstrates
durch
das
Tat-System
detektiert
wird
und
dadurch
entweder
der
Kanaldurchmesser sich dem Substrat anpasst oder alternativ dazu, einfach ein TatAKomplex passender Größe rekrutiert wird [Gohlke et al., 2005].
In einer neuen Studie wurde ein Tat-abhängiges Fusionsprotein in Chloroplasten benutzt,
dessen Transport unvollständig verläuft und dadurch ein Transportintermediat generiert, das
in der Thylakoidmembran stecken bleibt. Dieses Intermediat weist keinen Kontakt zu den
Tat-Komponenten auf [Cline and McCaffery, 2007]. Dies bedeutet entweder das Substrat
befand sich zu keinem Zeitpunkt in einer Poren-ähnlichen Struktur oder die Translokase hat
das Substrat lateral in die Membran entlassen als dessen Transportinkompetenz detektiert
wurde.
Nicht-Poren-Modelle bevorzugen die Idee einer Membran-schwächenden Funktion von TatA.
In diesen Modellen wird die Translokation durch die Membran durch eine Destabilisierung
der Membran erreicht. Dies wird entweder durch die Ansammlung der Transmembranhelices
der TatA-Protomere in direkter Nähe zum Substrat-Rezeptor-Komplex [Brüser und Sanders,
2003], oder durch die Insertion der amphiphatischen Helices der TatA-Protomere in die
Membran erreicht, wodurch ein passiver, transienter Kanal gebildet wird [Dabney-Smith et
al., 2006]. Letzteres könnte durch einen Falltür-Mechanismus funktionieren, in dem auf
Grund einer mechanischen Kraft, die das Substrat durch die Membran bewegt, die
18
II. Einleitung
amphipathischen Helices in die Membran inserieren. Die hydrophilen Seiten der inserierten
amphiphatischen Helices blieben dabei in engem Kontakt zum reifen Teil des Substrates
[Dabney-Smith et al., 2006]. Völlig unklar ist bislang, wie die PMF der Membran in
mechanische Arbeit konvertiert wird. Die spekulative Annahme TatC könne durch eine PMFabhängige Topologieänderung die PMF in mechanische Energie umwandeln [Brüser und
Sanders, 2003] basiert auf Ergebnissen einer TatC-Topologie-Studie, die eine 4- statt 6Transmembran-Topologie für TatC vorschlägt [Gouffi et al., 2002].
II.2.6. Energetisierung der Tat-abhängigen Translokation
Der Tat-abhängige Transport wird exklusiv durch die PMF energetisiert, die sich aus dem
elektrischen Potential (∆ψ) und dem pH-Gradienten (∆pH) der Membran zusammensetzt.
Ein
wesentlicher
Unterschied
zu
anderen
Protein-Translokationssystemen
ist
die
Unabhängigkeit des Tat-Systems von Nukleotidtriphosphat-Hydrolysen [Cline et al., 1992;
Mould und Robinson, 1991]. Die ursprüngliche Bezeichnung des Tat-Systems als ∆pHSystem rührt von der exklusiven Abhängigkeit der Tat-abhängigen Translokation in
Chloroplasten vom pH-Gradienten der Thylakoidmembran. Die Abhängigkeit vom pHGradienten sowie die Unabhängigkeit von Nukleotidtriphosphat-Hydrolysen konnte in einem
zellfreien System aus E. coli bestätigt werden, da Tat-Substrate effizient in Abwesenheit von
ATP transportiert wurden, der Transport aber inhibiert war, wenn der pH-Gradient zerstört
wurde [Yahr und Wickner et al., 2001; Alami et al., 2002]. Die Bindung des Tat-Substrates an
den TatBC-Rezeptor-Komplex erfolgt PMF-unabhängig [Ma und Cline, 2000; Alami et al.,
2002].
Die meisten Studien zur Energetisierung des Tat-Systems wurden im pflanzlichen TatSystem durchgeführt, da die Bildung der PMF an der Thylakoidmembran über die
photosynthetische
Transportkette
durch
Bestrahlung
mit
Licht
erfolgt.
Die
Proteintranslokation durch die cpTat-Translokase benötigt einen minimalen pH-Gradienten,
der vom jeweiligen Substrat abhängt [Alder und Theg, 2003]. Alder und Theg waren in der
Lage, den energetischen Aufwand für die Translokation eines Tat-Substrates mit 7,9 x 104
Protonen zu beziffern. Dieser Protonenfluss ist äquivalent zu der Energie, die aus der
Hydrolyse von 104 ATP-Molekülen gewonnen werden kann [Alder und Theg, 2003].
Neuste Ergebnisse deuten darauf hin, dass der ∆pH durch den ∆ψ der Membran als
treibende Kraft des cpTat-abhängigen Transportes ersetzt werden kann [Braun et al., 2007].
Die Austauschbarkeit der beiden Gradienten deutet auf die Existenz eines proton well
innerhalb der Tat-Translokase hin [Alder und Theg, 2003; Theg et al., 2005]. Dieses Prinzip
der Energiekonvertierung ist bereits von der Funktionsweise der ATP-Synthase bekannt
19
II. Einleitung
[Mitchell, 1968]. Kürzlich wurde sogar ∆ψ als einzige Energiequelle für den Tat-abhängigen
Transport in E. coli vorgeschlagen. Die Autoren postulierten, daß der Transport in
Abwesenheit eines pH-Gradienten in zwei distinkten, ∆ψ-abhängige Schritte abläuft
[Bageshwar und Musser, 2007].
Einen indirekten Hinweis auf die Bedeutung der PMF für das Tat-System liefert das phage
shock protein (PspA), das an der Stabilisierung der PMF unter Stressbedingungen beteiligt
ist [Kleerebezem et al, 1996] und dessen Überproduktion den Tat-Transport in E. coli
[DeLisa et al., 2004] als auch in Streptomyces lividans verstärkt [Vrancken et al., 2007].
II.2.7. Qualitätskontrolle des Tat-Systems
Die Tatsache, dass das Tat-System vollständig gefaltete Proteine transportiert, wirft die
Frage nach einer intrisischen Qualitätskontrolle oder eines Korrektur-Mechanismus auf. Dies
würde den Transport nicht-reifer und somit inaktiver Tat-Substrate ins Periplasma oder eine
Konkurrenz falsch gefalteter mit korrekt gefalteten Substraten am Tat-Translokon verhindern.
Unklar ist bislang, wie dieser Mechanismus funktionieren könnte und ob das Tat-Translokon
selbst in der Lage ist, den Faltungszustand des Substrates zu erkennen um es entweder
abzuweisen
oder
zu
transportieren.
Starke
Indikationen
für
eine
intrinsische
Qualitätskontrolle durch die Tat-Tranlokase selbst liefern Experimente mit Disulfid-haltigen
Tat-abhängigen Substraten, die im reduzierten Zustand nicht transportiert werden. So konnte
gezeigt werden, dass PhoA (alkalische Phosphatase) aus E. coli sowie andere Disulfidhaltige Proteine, die über eine Tat-spezifische Signalsequenz verfügen, nur in einem E. coliStamm mit einem oxidierenden Cytoplasma Tat-abhängig transportiert werden [DeLisa et al,
2003]. Dies konnte für verschiedene an PhoA-fusionierte Tat-Signalsequenzen gezeigt
werden, wodurch die Beteiligung Signalsequenz-spezifischer Chaperone ausgeschlossen
werden kann, so dass nur die Tat-Translokase selbst als Detektor des Redox- bzw.
Faltungszustandes der Substrate in Frage kommt [DeLisa et al., 2003]. Unterstützt wird
diese Annahme durch die Beobachtung des Tat-abhängigen Transportes von Cytochrom c,
der nur dann stattfindet, wenn die Reifung und Faltung des Proteins im Cytoplasma
ermöglicht ist [Sanders et al., 2001]. Musser und Theg postulierten eine Qualitätskontrolle,
die nach der Erkennung der Signalsequenz, aber vor dem Transport stattfindet. Diese
Annahme basiert auf der Beobachtung, dass die Tat-spezifische Signalsequenz einer großen
Avidineinheit vom Rezeptor-Komplex erkannt wird, aber Translokation nicht stattfindet
[Musser und Theg, 2000].
Die Idee der Qualitätskontrolle durch die Tat-Translokase selbst wurde angezweifelt durch
die Beobachtung, dass auch reduziertes und somit ungefaltetes PhoA-Protein an die Tat-
20
II. Einleitung
Translokase binden kann, wodurch angenommen wurde, dass weder TatC noch TatB die
Funktion einer intrinsischen Qualitätskontrolle ausüben [Richter und Brüser, 2005]. Wurde
das transportinkompetente PhoA unter diesen Bedingungen überexprimiert, wiesen die E.
coli-Zellen einen defekten Protonengradienten auf, was damit erklärt wurde, dass das
ungefaltete PhoA fehlerhaft transportiert wurde. Demnach würde auch nach der Erkennung
durch den TatBC-Rezeptor-Komplex keine Qualitätskontrolle, beispielsweise durch TatA,
stattfinden [Richter und Brüser, 2005].
In einer anderen Studie wurde für das E. coli Tat-System gezeigt, dass Tat-abhängiger
Transport von ungefalteten Proteinen von der Hydrophobizität ihrer Oberfläche abhängt, da
erst die Insertion einer hydrophoben Sequenz zur Transportinkompetenz der ungefalteten
Proteine führte. Somit wäre eine Vorraussetzung für den Tat-abhängigen Transport eine
hydrophobe Oberfläche des Proteins, wie es bei allen vollständig gefalteten Proteinen der
Fall ist [Richter et al., 2007]. Die Transport-Effizienz der unstrukturierten Proteine sank dabei
mit
steigender
Größe.
Andererseits
konnte
ein
unvollständiger
Transport
langer,
unstrukturierter Tat-Substrate bestehend aus 120 Aminosäuren im cpTat-System beobachtet
werden [Cline und McCaffery, 2007]. Unvollständig deshalb, weil sie während des
Translokationsprozesses
in
der
Membran
stecken
blieben.
Ein
Kontakt
dieses
Transportintermediats zu einem der Tat-Komponenten konnte allerdings nicht nachgewiesen
werden. Dies könnte bedeuten, dass Transport-inkompetente Substrate lateral in die
Membran entlassen werden. Diese Vorstellung unterstützt die Idee, dass Qualitätskontrolle
nicht über die Zurückweisung des Substrates durch die Translokase geschieht, sondern
durch den Abbau transportinkompetenter Substrate durch Membranproteasen erfolgt [Brüser
und Sanders, 2003].
Somit herrscht bislang keine Einigkeit darüber, ob das Tat-System auch ungefaltete Proteine
transportiert und falls nicht, ob die Tat-Translokase selbst an der Detektion des
Faltungszustandes beteiligt ist.
II.2.8. Spezifische Chaperone der Tat-Substrate
Reifungsprozesse der natürlichen Tat-Substate wie Faltung, Kofaktorinsertion oder sogar
Assemblierung zu oligomeren Komplexen finden prätranslokational im Cytoplasma statt und
verleihen dadurch den Substraten Transportkompetenz. Da am Reifungsprozess einiger TatSubstrate spezifische Chaperone beteiligt sind, könnten diese auch eine KorrekturleseFunktion ausüben [Turner et al., 2004; Sargent, 2007].
Eines der am besten charakterisierten Chaperone ist TorD, das spezifisch mit der
Signalsequenz der Trimethyl-N-Oxidoreduktase (TorA) interagiert. Beide Proteine sind im
21
II. Einleitung
torCAD- Operon codiert. TorA bildet zusammen mit TorC das Trimethylamin-N-Oxid
(TMAO)-reduzierende
System,
ein
respiratorisches
System,
das
unter
anaeroben
Bedingungen zur Reduktion von TMAO als alternativer Elektronenakzeptor genutzt wird.
TorD erkennt spezifisch die TorA-Signalsequenz [Jack et al, 2004] und scheint diese
dadurch vor proteolytischem Abbau zu schützen [Genest et al., 2006]. Da TorA bereits vor
der Insertion des Molybdän-Kofaktors in TorA an dessen Signalsequenz bindet, wurde
vermutet TorD assistiert in einer frühen Phase der Reifung von TorA, indem es durch seine
Bindung TorA in einer Kofaktor-Insertion-kompetenten Form hält [Ilbert et al., 2003].
TorD konnte aus Shewanella massilia als Mono- und als Dimer aufgereinigt werden. Beide
Formen binden an TatA, wobei das Dimer stärker interagiert [Tranier et al., 2002]. Das TorAProtein weist zwei Bindestellen für TorD auf: zum einen die Signalsequenz und zum anderen
eine im reifen Teil von TorA [Jack et al, 2004].
Ein anderes Tat-Signalsequenz-bindendes
Chaperon in E. coli ist DmsD, dem eine
essentielle Funktion in der Biogenese der Dimethylsulfoxid (DMSO)–Reduktase zukommt,
und das an die Signalsequenz der DMSO-Reduktase-Untereinheit DmsA bindet [Oresnik et
al., 2001]. DmsD interagiert auch mit der Signalsequenz von TorA [Oresnik et al., 2001], was
auf die Sequenzhomologie von TorD und DsmD zurückzuführen ist [Sargent et al., 2002].
Unter anaeroben Bedingungen bindet DmsD an die Cytoplasmamembran. Diese Assoziation
setzt die Präsenz von TatB und TatC aber nicht die von DmsA voraus [Papish et al., 2003],
weswegen spekuliert wurde, dass DmsD als Zielsteuerungsprotein fungieren könnte. Dabei
kann es sich aber nicht um eine essentielle Funktion handeln, da GFP mit der Signalsequenz
von TorA oder DmsA auch in Abwesenheit von DmsD Tat-abhängig transportiert wird [Ray et
al., 2003].
Weitere bekannte Tat-spezifische E. coli-Chaperone sind HyaE und HybE, die spezifisch mit
den Tat-Signalsequenz-tragenden Untereinheiten der Hydrogenase-1, HyaA, und der
Hydrogenase-2, HybO interagieren [Dubini and Sargent, 2003]. Den beiden Chaperonen
wird wiederum eine Funktion bei der Reifung der Untereinheiten, sowie der Assemblierung
des Hydrogenase-Komplexes zugesprochen [Dubini and Sargent, 2003]. So konnte gezeigt
werden, dass HybE die Translokation von HybO in Abwesenheit seines Komplexpartners
HybC verhindert [Jack et al., 2004].
Ein erst kürzlich beschriebenes Chaperon aus E. coli ist NapD, ein Signalsequenz-bindendes
Chaperon des Tat-Substrates NapA, das durch seine Bindung die Interaktion unreifen NapAProteins an die Tat-Translokase verhindert. NapA ist eine katalytische Untereinheit der
periplasmatischen Nitrat-Reduktase, die als Kofaktoren sowohl einen [4Fe-4S]-Cluster als
auch ein bis-Molybdopterin-Guanin-Dinukleotid (bis–MGD) besitzt [Maillard et al., 2007].
Diese Ergebnisse deuten darauf hin, dass spezifischen Chaperonen eine KorrekturleseFunktion beim Reifungsprozess Kofaktor-tragender Tat-Substrate zukommen könnte.
22
II. Einleitung
II.3. Zielsetzung
In der vorliegenden Arbeit sollten Einblicke in den Transport-Mechanismus des Tat-Systems
gewonnen werden. Dabei war das Tat-abhängige Fusionsprotein TorA-PhoA von
besonderem Interesse. Für dieses Protein konnte bereits gezeigt werden, dass Tatabhängiger Transport nur in einem E. coli-Stamm mit einem oxidierenden Cytoplasma
möglich ist, wodurch die Bildung der beiden intramolekularen Disulfidbrücken im PhoA-Teil
des Proteins und somit das Erlangen einer nativen Faltung erlaubt war [DeLisa et al., 2003].
Da nur diese Form des TorA-PhoA-Proteins Tat-abhängig transportiert wurde, konnte diese
Beobachtung als Indiz für eine intrinsische Qualitätskontrolle bzw. Erkennung von
Faltungszuständen durch das Tat-System gewertet werden. Ziel der vorliegenden Arbeit war
es zunächst, den Tat-abhängigen in vitro-Transport von TorA-PhoA in Abhängigkeit seines
Faltungszustandes zu etablieren. Anschließend sollten die molekularen Interaktionen des
gefalteten, transportkompetenten Proteins und des ungefalteten, transportinkompetenten
Proteins mit den Tat-Proteinen durch verschiedene Methoden untersucht werden.
Unterschiede in den Interaktionen mit den Komponenten der Tat-Translokase würden
Rückschlüsse auf den Mechanismus zur Erkennung von Faltungszuständen der TatSubstrate durch das Tat-System erlauben.
23
III. Material und Methoden
III. Material und Methoden
III.1. Material
III.1.1. Escherichia coli-Stämme
Tabelle III.1.1 In der vorliegenden Arbeit verwendete Bakterienstämme
Stamm
DH5α
Resistenz
BL21(DE3)
pLysS
Cam
Beschreibung
supE44 ∆lacU169 Φ80lacZ∆M15
hsdR17 recA1 endA1 gyrA96
thi-1 relA1
F ompT hsdSB(rBmB) gal dcm (DE3)
pLysS
MC4100
F- araD139 ∆(argF-lac) U169
rpsL150 relA1 flbB3501 deoC1
ptsF25 rbsR
DEAD
MC4100 ∆tatABCDE
Top10
Str
M15
F- mcrA ∆(mrr-hsdRMS-mcrBC)
φ80lacZ∆M15 ∆lacX74 recA1
araD139 ∆(araleu)7697 galU galK
rpsL (StrR) endA1 nupG
NalS, StrS, RifS, Thi-, Lac-, Ara+,
Gal+, Mtl-, F-, RecA+, Uvr+, Lon+
(Phänotyp)
Verwendung
Plasmid DNAIsolierung
Referenz
Sambrook
et al., 1984
Überexpression
von TatA-D
und Tat+-INVPräparation
Überexpression
von TatB-D und
∆TatA-INVPräparation
∆Tat-INVPräparation
Präparation von
S135 und
Expression des
orthogonalen
tRNASynthetase/tRNA
CUA-Paares
Überexpression
von TorD
Novagen
Silhavy et
al., 1984
Sargent et
al., 1999
Invitrogen
Quiagen
III.1.2. Plasmide
Tabelle III.1.2 In der vorliegenden Arbeit verwendete Plasmide
Plasmid
pET28a-TorA-PhoA
Resistenz
Kan
pET28a-TorA-PhoAF15
Kan
pET28a-TorA-PhoAV24
Kan
24
Beschreibung und Verwendung
Expressionsvektor für T7-abhängige in
vitro Synthese
Derivat von pET28a-TorA-PhoA zur
Einführung von pBPA an Position 15
von TorA-PhoA
Derivat von pET28a-TorA-PhoA zur
Einführung von pBPA an Position 24
von TorA-PhoA
Referenz
DeLisa M.P.,
unveröffentlicht
Die vorliegende
Arbeit
Die vorliegende
Arbeit
III. Material und Methoden
pET28a-TorA-PhoAKR
Kan
pET28a-TorA-PhoAKK
Kan
pET28a-TorA-PhoA∆SP1
Kan
pET28a-TorA-PhoA∆SP2
Kan
pKSM717-SufI
Amp
p8737
Amp
pDULE
Tet
pQE60-TorD
Amp
pFat222
Amp
pRep4
Kan
Derivat von pET28a-TorA-PhoA zur in
vitro Synthese von TorA-PhoA, dessen
erstes Arginin im Doppel-Arginin-Motif
durch Lysin substituiert ist (R12K)
Derivat von pET28a-TorA-PhoA zur in
vitro Synthese von TorA-PhoA, dessen
beide Arginine im Doppel-Arginin-Motif
durch Lysine substituiert sind (R12K,
R13K)
Derivat von pET28a-TorA-PhoA zur in
vitro Synthese von TorA-PhoA, mit der
Mutation A40L. Zwischenprodukt bei
der Herstellung von pET28a-TorAPhoA∆SP2
Derivat von pET28a-TorA-PhoA zur in
vitro Synthese von nichtprozessierbarem TorA-PhoA, mit den
Mutationen A40L und A41L
Expressionsvektor für T7-abhängige in
vitro Synthese
Expressionsvektor zur in vivo Synthese
von TatA-D (Tat+-INV). tatABCD in
pET22b
Expressionsvektor zur in vivo Synthese
des orthogonalen tRNA- SynthetasetRNACUA-Paares
Expressionsvektor zur in vivo Synthese
von TorD
Expressionsvektor zur in vivo Synthese
von TatB-D (∆TatA-INV)
Expressionsvektor zur in vivo Synthese
des Lac-Repressor-Proteins
Die vorliegende
Arbeit
Die vorliegende
Arbeit
Die vorliegende
Arbeit
Die vorliegende
Arbeit
Alami et al.,
2003
Alami et al.,
2002
Farrell et al.,
2005
Hatzixanthis et
al., 2005
Sargent et al.,
1999
Qiagen
III.1.3. Oligonukleotide
Tabelle III.1.3 In der vorliegenden Arbeit verwendete Oligonukleotide
Oligonukleotid
TorA-PhoAF15for
TorA-PhoAF15rev
TorA-PhoAV24for
TorA-PhoAV24rev
TorA-PhoAR12KRfor
TorA-PhoAR12KRrev
TorA-PhoAKR13Kfor
Sequenz
5´-CA TCA CGT CGG CGT
TAG CTG GCA CAA CTC GGC
GGC-3´
5´-GCC GCC GAG TTG TGC
CAG CTA ACG CCG ACG TGA
TG-3´
5´-CAA CTC GGC GGC TTA
ACC TAG GCC GGG ATG CTG
G-3´
5´- C CAG CAT CCC GGC CTA
GGT TAA GCC GCC GAG
TTG-3´
5´-CTC TTT CAG GCA TCA
AAG CGG CGT TTT CTG GCA
CAA CTC GGC-3´
5´-GCC GAG TTG TGC CAG
AAA ACG CCG CTT TGA TGC
CTG AAA GAG-3´
5´-CTC TTT CAG GCA TCA
Verwendung
zur Einfügung von TAG-Codon in
TorA-PhoA anstelle des Codons
für F15
zur Einfügung von TAG-Codon in
TorA-PhoA anstelle des Codons
für F15
zur Einfügung von TAG-Codon in
TorA-PhoA anstelle des Codons
für V24
zur Einfügung von TAG-Codon in
TorA-PhoA anstelle des Codons
für V24
zur Einfügung der Mutation R12K
in TorA-PhoA
zur Einfügung der Mutation R12K
in TorA-PhoA
zur Einfügung der Mutation R13K
25
III. Material und Methoden
TorA-PhoAKR13Krev
TorA-PhoA40LAfor
TorA-PhoA40LArev
TorA-PhoLA41Lfor
TorA-PhoLA41Lrev
AAG AAG CGT TTT CTG GCA
CAA CTC GGC-3´
5´-GCC GAG TTG TGC CAG
AAA ACG CTT CTT TGA TGC
CTG AAA GAG-3´
5´-CCG CGA CGT GCG ACT
CTG GCG CAA GCG GCG-3´
5´- CGC CGC TTG CGC CAG
AGT CGC ACG TCG CGG-3´
5´-CCG CGA CGT GCG ACT
CTG CTG CAA GCG GCG-3´
5´-CGC CGC TTG CAG CAG
AGT CGC ACG TCG CGG-3´
in TorA-PhoAKR
zur Einfügung der Mutation R13K
in TorA-PhoAKR
zur Einfügung der Mutation
in TorA-PhoA
zur Einfügung der Mutation
in TorA-PhoA
zur Einfügung der Mutation
in TorA-PhoAA40L
zur Einfügung der Mutation
in TorA-PhoAA40L
A40L
A40L
A41L
A41L
III.1.4. Antibiotika
Tabelle III.1.4 In der vorliegenden Arbeit verwendete Antibiotika
Antibiotikum
Ampicillin
Chloramphenicol
Kanamycin
Streptomycin
Tetracyclin
Konzentration der
Stammlösung (mg/ml)
50
35
50
10
5
Lösungsmittel
H2O
EtOH
H2O
H2O
70 % (v/v) EtOH
Endkonzentration
(µg/ml)
100
35
50
50
2,5 oder 25
Alle wässrigen Antibiotika-Stammlösungen wurden mit einem 0,22 µm Spritzenfilter steril
filtriert (Rotilabo-Spritzenfilter steril, Roth) und bei -20°C gelagert.
III.1.5. Chemikalien
Im Rahmen der vorliegenden Arbeit wurden Chemikalien der Firmen ICN, Pharmacia,
Roche, Roth, Serva, Bachem und Sigma verwendet. Enzyme wurden von den Firmen NEB,
und Stratagene, Plasmidpräparations-Kits von Qiagen bezogen. Von Amersham stammte
der
35
S-Methionin/35S-Cystein-Mix, zur radioaktiven Markierung von Proteinen. Die Hersteller
verwendeter Spezialchemikalien werden in den beschrieben Methoden explizit genannt.
26
III. Material und Methoden
III.2. Methoden
III.2.1. Molekularbiologische Methoden
III.2.1.1. Anzucht und Lagerung von Escherichia coli
E. coli wurde, wenn nicht anders angegeben, in LB-Medium (pro Liter dest. Wasser: 10 g
pankreatisch verdautes Casein, 10 g NaCl, 5 g Hefeextrakt) unter aeroben Bedingungen
(Schüttler: 180 rpm) bei 37°C angezogen. Das Medium wurde vor der Anzucht der Bakterien
autoklaviert. Nach dem Abkühlen wurden je nach Bedarf Antibiotika zugeben. Die
Bakterienzellen aus diesen Anzuchten dienten zur Aufreinigung überexprimierter Protein,
oder als Ausgangsmaterial für die Präparation von Zellextrakten, Membranen und
Plasmiden.
Verdünnungsausstriche zur Isolierung von Einzelzellkolonien, sowie zur Kultivierung und
Selektion von transformierten Zellen erfolgten auf 2 % Agar-LB-Platten (pro Liter dest.
Wasser: 10 g pankreatisch verdautes Casein, 10 g NaCl und 5 g Hefeextrakt, 20 g AgarAgar) über Nacht bei 37°C im Brutschrank. Je nach Bedarf wurden Antibiotika zur Selektion
der Bakterien dem handwarmen Agar-Medium vor dem Gießen der Platten zugeben. Bei 4°C
wurden Kulturen auf Agar-LB-Platten ca. 4 Wochen gelagert.
Für eine längere Lagerung wurden Glycerinkulturen hergestellt. Dazu wurde ein Volumen
Bakterien-Kultur mit einem Volumen autoklaviertem LB-Medium, das 30 % (w/v) Glycerin
enthielt, versetzt, in flüssigem Stickstoff schockgefroren und bei -70°C gelagert.
III.2.1.2. Herstellung kompetenter E. coli-Zellen und Transformation nach der
TSB-Methode
Zur Herstellung kompetenter Zellen wurde der gewünschte E. coli-Stamm in 3 ml LBMedium, bei Bedarf in Anwesenheit des entsprechenden Antibiotikums, über Nacht
angezogen. Für jeden Transformationsansatz wurden die Über-Nacht-Kultur im Verhältnis
1:100 in 3 ml LB-Medium überimpft und ca. zwei weitere Stunden bis zu einer OD600nm von
0,3 im Schüttler (180 rpm) inkubiert. Danach wurden je 1,5 ml Zellkultur zentrifugiert
(Tischzentrifuge: 14000 rpm, 3 min, RT). Das Zellpellet wurde in 150 µl TSB resuspendiert
(TSB: 10 % (w/v) Polyethylenglykol 6000, 5 % (v/v) DMSO, 10 mM MgCl2, 10 mM MgSO4, in
LB-Medium gelöst und steril filtriert) und 10 min auf Eis inkubiert. Den nun kompetenten
Zellen wurde die DNA zugegeben (ca. 100-300 ng/Transformationsansatz), vorsichtig
vermengt und weitere 90 min auf Eis inkubiert. Anschließend erfolgte die Zugabe von 900 µl
TSB sowie von 12,5 µl 20 %iger (w/v) Glukose-Lösung und eine weitere Inkubation der
Proben für 1 h bei 37°C im Thermomixer (200-300 rpm). Die Zellen wurden daraufhin erneut
27
III. Material und Methoden
abzentrifugiert (Tischzentrifuge: 14000 rpm, 3 min, RT) und der Überstand vollständig
abgenommen. Das Zellpellet wurde in 100 µl LB-Medium resuspendiert, zur Selektion
erfolgreich transformierter Zellen auf LB-Agarplatten mit Antibiotika ausplattiert und über
Nacht bei 37°C inkubiert.
III.2.1.3. Plasmidpräparation im großen Maßstab (Maxiprep)
Plasmid-DNA wurde insbesondere für die in vitro-Synthese von Proteinen benötigt. Für die
Präparation von Plasmid-DNA wurden Zellen in 400 ml LB-Medium in einem 2 lErlenmeyerkolben, eventuell mit den ensprechenden Antibiotika, bei 37°C über Nacht im
Schüttler (180 rpm) angezogen und in der Regel nach ca. 15 h durch Zentrifugation geerntet
(SCL 3000 Rotor (Sorvall), 7000 rpm (8650 xg), 4°C, 10 min). Zur Aufreinigung der PlasmidDNA aus den Zellen wurde das Qiagen Maxiprep-Kit verwendet und dabei den Anweisungen
des Herstellers gefolgt. Die Funktionsweise des Kits basiert auf dem Prinzip der alkalischen
Lyse
von
Zellen
und
der
anschließenden
Aufreinigung
der
Plasmid-DNA
durch
Ionenaustauscher-Chromatographie. Abweichungen vom Hersteller-Protokoll lauten wie
folgt: Nach der Isopropanol-Fällung der Plasmid-DNA und anschließender Zentrifugation
wurde
das
DNA-Pellet
in
700
µl
TE-Puffer
(10 mM Tris-HCl, pH 8, 1 mM EDTA)
resuspendiert, in ein 2 ml-Reaktionsgefäß (Eppendorf) überführt, mit 70 µl QiagenNeutralisationspuffer P3 (3 M Kaliumacetat, pH 5,5) sowie 1,2 ml EtOH p.a. versetzt und
entweder für 20 min bei -70°C oder über Nacht bei -20°C inkubiert. Anschließend wurde
durch
Zentrifugation
(Kühltischzentrifuge: 16000 g, 15 min, 4°C)
das
DNA-Präzipitat
sedimentiert. Das daraus resultierende Pellet wurde einmal mit 1 ml 70 % EtOH p.a.
gewaschen und anschließend an der Luft getrocknet. Das getrocknete DNA-Pellet wurde in
50 µl TE-Puffer oder 50 µl autoklaviertem, dest. Wasser aufgenommen und bei 4°C gelagert.
III.2.1.4. Plasmidpräparation im kleinen Maßstab (Miniprep)
Plasmidpräparationen aus E. coli wurden im kleinen Maßstab zur Überprüfung von
Mutagenesen durchgeführt. Für die Präparation von Plasmid-DNA wurden E. coli-Zellen in
5 ml LB-Medium in einem verschließbaren 50 ml-Reaktionsgefäß (Falcon), eventuell mit den
entsprechenden Antibiotika, bei 37°C über Nacht im Schüttler (180 rpm) angezogen und in
der Regel nach ca. 15 h durch Zentrifugation geerntet (5000 xg, 10 min, 4°C). Das Zellpellet
wurde mit 5 ml dest. H2O gewaschen und anschließend nach den Angaben des Herstellers
unter Verwendung eines Quiagen Miniprep-Kits aufgereinigt. Die Funktionsweise des Kits
basiert, wie die des Maxiprep-Kits, auf dem Prinzip der alkalischen Lyse und der
anschließenden Aufreinigung der Plasmid-DNA durch Ionenaustauscher-Chromatographie.
Die so präparierte DNA wurde entweder gelchromatographisch analysiert und diente als
Material für Sequenzierungen (Seqlab, Göttingen) und Transformationen.
28
III. Material und Methoden
III.2.1.5. Agarose-Gelelektrophorese
Diese Methode wurde zur Auftrennung von DNA-Fragmenten nach ihrer Größe benutzt.
Dazu wurde die DNA-haltige Probe (beispielsweise aus Miniprep oder PCR) zunächst mit
1/10 Volumen 10x Ladepuffer (30 % (v/v) Glycerin, 0,25 % (w/v) Bromphenolblau, 0,25 %
(w/v) Xylecyanol) versetzt. Je nach potentieller Größe des Molekulargewichts der zu
analysierenden DNA-Moleküle betrug die Konzentration der Agarose im Gel zwischen 0,8 %
(z.B. Plasmide: zwischen ca. 3 kbp und 10 kbp) und 2 % (z.B. PCR-Produkte: 100 bp bis
3 kbp). Zum Gießen des Agarosegels wurde die Agarose zunächst in 70 ml 1 x TAE-Puffer
(50 x TAE-Puffer: 242 g Tris Base, 57,1 ml Essigsäure p.a., 100 ml 0,5 M EDTA pH 8, ad.
1 l dest. H2O) aufgekocht, unter Rühren auf ca. 50°C wieder abgekühlt, mit 7 µl
Ethidiumbromid (10 mg/ml) versetzt und vor dem Erkalten in die Flachbettapparatur
gegossen. Der Lauf erfolgte mit 3 V/cm Elektrodenabstand in 1 x TAE Laufpuffer. Die DNA
Banden konnten im Gel mittels UV-Licht auf einem UV-Tisch sichtbar gemacht werden.
III.2.1.6. Mutagenisierende PCR (polymerase chain reaction)
Ortsgerichtete Mutation einzelner Codons der Signalsequenz-codierenden Region des TorAPhoA-Gens wurde unter Verwendung der Turbo-Pfu-DNA-Polymerase (2,5 U/µl, Stratagene)
durchgeführt. Durch diese Methode wurden sämtliche Mutanten des TorA-PhoA-Gens
generiert (Tabelle III.1.2). Dazu wurden zwei komplementäre mutagene Primer benutzt die
jeweils etwa in der Mitte ihrer Sequenz das mutierte Codon enthielten (Tabelle III.1.3). Die
Synthese der in den PCRs verwendeten Primer wurde bei der MWG-Biotech AG in Auftrag
gegeben.
Ein 50 µl PCR-Ansatz wurde wie folgt angesetzt:
Komponente
Menge/Volumen
Matrize (Plasmid)
10-20 ng
Primer 1 (for.)
125 ng
Primer 2 (rev.)
125 ng
Turbo-Pfu-DNA-Polymerase
1 µl
10x Polymerase-Reaktionspuffer
5 µl
dNTP-Mix*
1 µl
H2O autokl., bidest.
ad 50 µl
→ Turbo-Pfu-Polymerase, Reaktionspuffer, dNTP-Mix und H2O wurden häufig
zusammen als Mix angesetzt, während Matrize und Primer im PCRReaktionsgefäß vorgelegt wurden.
→ Als DpnI-Kontrolle wurde immer auch ein Ansatz ohne Matrize angesetzt.
29
III. Material und Methoden
* dNTP-Mix wurde wie folgt vorbereitet: die einzelnen dNTP-Stammlösungen (je 100 mM,
Desoxynucleoside Triphosphate Set, Roche) wurden auf Eis aufgetaut und 10 µl aus jeder
Stammlösung mit autokl., bidest. H2O in 100 µl Endvolumen verdünnt.
PCR wurde in einem Thermocyler (Mastercycler, Eppendorf) und mit dem folgenden
Programm durchgeführt:
Zyklus
Funktion
Temperatur
Dauer
Wiederholungen
1
Denaturierung
95°C
30 sec
1
2
Denaturierung
Annealing
Elongation
95°C
55°C
68°C
30 sec
1 min
10 min
16
3
Elongation
68°C
20 min
1
Nach Beendigung der PCR wurde jeder Ansatz mit dem Restriktionsenzym DpnI behandelt.
Dazu wurde jedem Ansatz 1 µl DpnI-Lösung (20000 U/ml, New England Biolabs) zugegeben
und 3 h bei 37°C im Thermomixer bei 500 rpm inkubiert. Durch diese Behandlung wurde auf
Grund seiner Methylierung selektiv nur das als Matrize eingesetzte Plasmid verdaut. Das in
der PCR synthetisierte, mutierte Plasmid war dagegen nicht methyliert und blieb deshalb
erhalten.
25 µl eines DpnI-behandelten PCR-Ansatzes wurde zur Transformation des E. coliBakterienstamm DH5α eingesetzt. Nach erfolgreicher Transformation wurde das Plasmid
präpariert und sequenziert (SeqLab, Göttingen) um die Mutation im Gen von TorA-PhoA zu
bestätigen.
30
III. Material und Methoden
III.2.2. Biochemische Methoden
III.2.2.1. Denaturierende SDS-Polyacrylamidgelelektrophorese
Die Auftrennung denaturierter Proteine in Abhängigkeit des Molekulargewichtes erfolgte
durch SDS-Polyacrylamid-Gelelektrophorese (SDS-PAGE) nach Laemmli (1970). Zum
Gießen von vertikalen Gelen wurden zunächst Glasplatten mit 10 % Acrylamid (AA), 0,26 %
Bisacrylamid, 0,15 % TEMED und 0,075 % APS abgedichtet. Je nach Größe der zu
trennenden Proteine variierte die Konzentration des Acrylamids zwischen 8 % und 15 %. Die
angegebenen Volumina waren ausreichend für ein Gel mit ca. 100ml Volumen:
Trenngel:
15 %ig: 30% Acrylamid, 0.8% Bisacrylamid
50 ml
oder
10 %ig: 30% Acrylamid, 0.8% Bisacrylamid
33,3 ml
oder
8 %ig: 30% Acrylamid, 0.8% Bisacrylamid
Unmittelbar
vor
26,7 ml
2 M Tris-HCL, pH 8,8
20 ml
25 % (w/v) SDS
400 µl
TEMED
40 µl
dest. H2O
ad 100 ml
dem
Eingießen
zwischen
die
Glasplatten
wurden
10%
Ammoniumperoxodisulfat (1 ml/100 ml) als Starter der Vernetzungsreaktion hinzugegeben.
Das Gel wurde mit Isobutanol überschichtet. Nach vollständiger Polymerisation des
Trenngels wurde das Isobutanol entfernt und das Sammelgel eingegossen wobei sofort ein
passender Kamm für die Bildung der Probentaschen eingeschoben wurde:
Sammelgel (pro Gel):
30 % Acrylamid, 0,8% Bisacrylamid
5 ml
0,5 M Tris-HCl, pH 6,8
3,6 ml
25 % (w/v) SDS
120 µl
TEMED
12 µl
10 % APS
0,3 ml
dest. H2O
ad 30 ml
31
III. Material und Methoden
Nach der vollständigen Polymerisation des Sammelgels wurde der Kamm entfernt, das Gel
in die Gelelektrophoresevorrichtung (Eigenbau) eingespannt und die Kammern mit
Laufpuffer befüllt:
Tris-Glycin-Puffer (5 x):
Tris
150 g
Glycin
720 g
dest. H2O
ad 5 l
SDS-PAGE-Laufpuffer:
Tris-Glycin-Puffer (5 x)
400 ml
25 % (w/v) SDS
8 ml
dest. H2O
ad 2 l
Die Proteinproben konnten anschließend in die Probentaschen geladen werden. Dazu wurde
die zu analysierende Proteinlösung mit dem gleichen Volumen oder, im Falle eines Pellets
aus einer Proteinfällung, mit 30 µl SDS-PAGE-Probenpuffer versetzt und im Thermomixer
bei 95°C und 1400 rpm für 5 min inkubiert.
Lösung 1 :
Tris Base
0,2 M
EDTA, pH 8
20 mM
Lösung 2:
Tris Base
83,3 mM
Glycerin
29,2 % (w/v)
SDS
8,3 %
Bromphenolblau
0,03 %
SDS-PAGE-Probenpuffer (jeweils frisch angesetzt):
Lösung 1
0,5 ml
Lösung 2
0,4 ml
1 M DTT
0,1 ml
Als Molekulargewichtstandard wurde vorgefärbte Proteinmarkerlösung (Prestained Molecular
Weight Marker, Bio Rad) verwendet. Die Gele liefen entweder über Nacht mit 25 mA oder
über Tag mit 50 mA.
32
III. Material und Methoden
III.2.2.2. Visualisierung von Proteinen in Polyacrylamidgelen
III.2.2.2.1. Autoradiographie
Radioaktiv
markierte
Proteine
aus
einer
in vitro-Proteinsynthese
wurden
mittels
Autoradiographie sichtbar gemacht. Dazu wurde zunächst nach der Beendigung der
Gelelektrophorese das Gel durch eine 15 min Inkubation in Fixierer-Lösung (10 % (v/v)
Essigsäure p.a., 35 % (v/v) EtOH) behandelt und anschließend zweimal für 10 min
gewässert. Anschließend wurde das Gel auf Filterpapier (Whatman) bei 65°C für 90-120 min
Vakuum-getrocknet und das getrocknete Gel unter einem Phosphoimiger-Screen (Molecular
Dynamics) exponiert, dessen Beschichtung empfindlich gegenüber radioaktiver Strahlung ist.
Zur
Detektion
der
radioaktiven
Anregung
der
Screen-Beschichtung
diente
ein
Phosphoimager (Molecular Dynamics). Die Quantifizierung der radioaktiven Signale erfolgte
mittels des Programms ImageQuant 5.2 (Molecular Dynamics).
III.2.2.2.2. Coomassie-Blau-Färbung von Polyacrylamidgelen
Um alle Proteine eines Polyacrylamidgeles sichtbar zu machen konnten die Proteinbanden
mit Coomassie-Blau angefärbt werden. Dazu wurde das Gel nach Beendigung der
Elektrophorese in Coomassie-Blue-Lösung (40 % (v/v) EtOH tech., 10 % (v/v) Essigsäure,
1 % (w/v) Coomassie Brilliant Blau R250) für mind. 2 h inkubiert und anschließend wieder mit
10 % (v/v) Essigsäure entfärbt. Dabei wurde der Gelhintergrund schneller entfärbt als die
angefärbten Proteinbanden. Zur Dokumentation wurde das gefärbte Gel entweder
fotografiert oder zwischen Cellophan-Folien bei 65°C für 90-120 min Vakuum-getrocknet.
III.2.2.2.3. Western-Blotting
Durch Western-Blotting konnten gelelektrophoretisch getrennte Proteine durch spezifische
Antikörper immunologisch nachgewiesen werden. In der vorliegenden Arbeit wurde diese
Methode eingesetzt um den Gehalt der Tat-Proteine (TatA, TatB und TatC) in präparierten
Membranvesikeln zu bestimmen.
Dazu wurden nach der Auftrennung durch SDS-PAGE die Proteinproben auf eine PVDFMembran (Millipore) transferiert (geblottet), die kurz in Methanol inkubiert, und dann in
folgender Anordnung in die Nassblottapparatur eingesetzt wurde. Sämtliche Komponenten
des Transfers wurden 5 min in Transferpuffer inkubiert und anschließend, bei der Kathode
beginnend, in der folgenden Reihenfolge angeordnet: Saugschwamm, 3 Lagen Filterpapier
(Whatman), SDS-PAGE-Gel, PVDF-Membran, 3 Lagen Filterpapier (Whatman) und
Saugschwamm:
33
III. Material und Methoden
5 x Tris-Glycin-Puffer:
Tris
150 g
Glycin
720 g
dest. H2O
ad 5 l
Transferpuffer:
5x Tris-Glycin-Puffer
600 ml
MeOH tech.
600 ml
25 % (w/v) SDS
2 ml
dest. H2O
ad 3 l
Der Transfer erfolgte entweder bei 400 mA für 2 h oder bei 20 mA über Nacht. Anschließend
konnte zur Kontrolle die Membran einige Minuten in Ponceau-S-Lösung (0,2 % (w/v)
Poinceau S, 3 % (v/v) TCA) inkubiert werden, wodurch die transferierten Proteine sichtbar
gemacht werden konnten. Nach dem Entfärben der Membran mit dest. H2O wurde der Blot
unter leichtem Schwenken 2 h in Blockierungslösung (5 % (w/v) Milchpulver in TTBS)
inkubiert und daraufhin dreimal 5 min in 50 ml TTBS gewaschen:
10x TBS:
Tris-HCl, pH 7,5
0,2 M
NaCl
1,49 M
TTBS:
Tween-20 in 1x TBS
0,1 %
Um die Antikörperadsorption zu ermöglichen, wurde das gewünschte Antiserum in
entsprechender Verdünnung in TTBS für 1 h unter leichtem Schwenken mit dem Blot
inkubiert.
Antiserum (Kaninchen)
αTatA
αTatB
αTatC
Verdünnung
1 : 2500
1 : 2500
1 : 2500
Herkunft
AG M. Müller
AG M. Müller
AG M. Müller
Anschließend wurde der Blot zunächst dreimal 5 min in 50 ml TTBS gewaschen und dann
mit dem sekundären Antikörper (anti-Kaninchen-Antikörper von der Ziege, MeerrettichPeroxidase-gekoppelt, Amersham Bioscience) in einer Verdünnung von 1:5000 in TTBS für 1
h unter leichtem Schwenken inkubiert. Nach einer erneuten Waschung (dreimal 5 min in 50
34
III. Material und Methoden
ml TTBS) und nach dem Trocknen des Blots durch kurzes Abstreifen auf Filterpapier wurde
zur Detektion der antikörpermarkierten Proteinbanden das „Enhanced Chemiluminescence
ECL Westernblotting System“ (Amersham Bioscience) verwendet. In diesem Verfahren
erkennt der sekundäre Antikörper den primären Antikörper und kann diesen durch eine
chemoluminiszierende Reaktion der gekoppelten Peroxidase über Belichtung eines
Rötgenfilms sichtbar machen. Dazu wurden Lösung 1 und 2 des ECL Detektionskits 1:1
frisch vermischt und durch vorsichtiges Pipettieren gleichmäßig auf der abgetrockneten
Membran verteilt und 1 min inkubiert, die Lösung entfernt, der Blot abgetrocknet und in eine
Blotkassette gelegt. Durch Auflegen eines Röntgenfilms (Hyperfilm, Amersham Bioscience)
für ca. 10 sec und anschließende Filmentwicklung wurden die durch den ersten Antikörper
erkannten Proteine als schwarze Banden sichtbar. Um die Membran nach der Entwicklung
mit einem weiteren Antikörper dekorieren zu können, mußten die vorher verwendeten
Antikörper von der Membran abgelöst werden. Hierfür wurde die Membran für 30 min in
5 % (v/v) Essigsäure tech. und 0,5 M NaCl unter leichtem Schwenken inkubiert. Nach 3
Waschschritten mit TTBS begann das Verfahren bei der Inkubation mit der Blockierlösung
wieder von neuem.
III.2.2.3. Bestimmung der Proteinkonzentration mit Amidoschwarz
Die zu bestimmende Proteinlösung wurde mit dest. H2O auf 200 µl aufgefüllt und mit 800 µl
Färbelösung (10 % (v/v) Essigsäure, 90 % (v/v) Methanol p.a., Spatelspitze Amidoschwarz)
versetzt und für 10 min bei RT inkubiert. Nach einem Zentrifugationsschritt (Tischzentrifuge,
14000 rpm, 20 min, RT) wurde der Überstand abgenommen und das Proteinpellet mit 1 ml
Waschlösung (10 % (v/v) Essigsäure, 90 % (v/v) Ethanol p.a.) gewaschen und erneut
zentrifugiert. Diese Prozedur wurde so lange wiederholt bis der Überstand farblos war. Der
farblose Überstand wurde abgenommen und das Pellet in 1 ml 0,2 M NaOH gelöst. Diese
Lösung wurde bei 615 nm im Photometer vermessen. Anhand einer BSA-Eichkurve wurde
die Proteinkonzentration bestimmt.
III.2.2.4. Überexpression und affinitätschromatographische Aufreinigung von TorD
Zur Überexpression des TorD-Proteins wurde der E. coli-Stamm M15pRep4, der mit dem
Plasmid pQE60-TorD transformiert wurde (siehe III.2.1.2.), welches für eine His-getaggte
Version des TorD-Proteins codierte [Hatzixanthis et al., 2005] (siehe Tabellen III.1.1 und
III.1.2) verwendet. Der transformierte Stamm wurde über Nacht in 10 ml autoklaviertem
Medium in einem 100 ml-Erlenmeyerkolben unter aeroben Bedingungen (Inkubator: 180
rpm) und in Gegenwart von Kanamycin (50 µg/ml) sowie Ampicillin (100 µg/ml) bei 37°C
angezogen. Für eine Über-Tag-Kultur wurden 100 ml autoklaviertes LB-Medium in einem
1000 ml-Erlenmeyerkolben im Verhälnis 1:100 mit der Über-Nacht-Kultur in Gegenwart der
35
III. Material und Methoden
oben genannten Antibiotika überimpft und unter den gleichen Bedingungen angezogen. Bei
einer optischen Dichte (OD600) der Kultur von 0,6 wurde die Expression des TorD-Proteins
durch die Zugabe von 1 mM IPTG induziert und die Zellen nach 4 h Induktion durch
Zentrifugation geerntet (SCL 3000 Rotor (Sorvall): 7000 rpm (8650 xg), 10 min, 4°C). Das
Zellpellet wurde in Pufferlösung (1,5 ml Puffer /g Zellen, Puffer: 10 mM Tris-HCl, pH 7, 10 %
(w/v) Glycerin) resuspendiert und mit einem Proteaseinhibitormix (“complete, EDTA-free“,
Roche) versetzt. Die Zellsuspension wurden im French-Press-Verfahren (French Press von
SCM Aminico) in einer gekühlten 40k-Zelle (Spectronic Unicam) mit einem Druck von 8000
psi in zwei Durchgängen aufgeschlossen. Anschließend wurde die Suspension zentrifugiert
(30000 g, 30 min, 4°C). Im Folgenden wurde versucht das überexprimierte His-getaggte
TorD-Protein im batch-Verfahren affinitätschromatographisch aufzureinigen. Dazu wurde der
Überstand der Zentrifugation mit gewaschener Talon-Sepharose (0,15 ml SepharoseSuspension/ml Überstand, Talon Polyhistidine-Tag Purification Resins von Clontech) auf Eis
inkubiert und im Abstand von 15 min kurz gevortext. Nach 1,5 h wurde durch Zentrifugation
(Kühltischzentrifuge: 2000 rpm, 2 min, 4°C) das Säulenmaterial sedimentiert und
anschließend mit Puffer A gewaschen (Puffer A: 10 mM Tris-HCl, 7,0; 300 mM NaCl; Vol. an
Puffer A = 10 x Vol. an Talon). Nach zwei weiteren Waschungen mit Puffer A wurde einmal
mit Puffer B gewaschen (Puffer B: 10 mM Tris-HCl, pH 7, 5 mM Imidazol; Vol. an Puffer B =
10 x Vol. an Talon). Anschließend wurde die Suspension in eine Einweg-Säule überführt
(Polypropylene column 1 ml, Qiagen) wodurch Puffer B weitgehend entfernt wurde. Die
Elution des an das Säulenmaterial gebundene TorD-Proteins erfolgte durch Applikation von
Elutionspuffer (10 mM Tris-HCl, pH 7, 50 mM Imidazol; Vol. an Eluationspuffer = 4 x
Volumen an Talon). Das Eluat wurde in 1 ml-Fraktion aufgefangen und die Reinheit der
Fraktionen durch SDS-PAGE und anschließender Coomassie-Blue-Färbung des Gels
analysiert. TorD-haltige Fraktionen wurden mit Glycerin in einer Endkonzentration von 20 %
(v/v) versetzt und bei -20°C gelagert.
III.2.2.5. Präparation der Komponenten des zellfreien Systems
III.2.2.5.1. Präparation des Zellextraktes S135
Um Proteine in vitro zu synthetisieren wurde ein Zellextrakt aus E. coli benötigt, der
sämtliche cytosolische Komponenten enthält, die zur Transkription und Translation
gebraucht wurden [Müller und Blobel, 1984]. Ein solcher Extrakt (S135) wurde in der
vorliegenden Arbeit aus Zellen der E. coli-Stämme BL12(DE3)pLys des Top10pDULE (siehe
Tabellen III.1.1 und III.1.2) gewonnen. Zellextrakt aus der Präparation des Top10pDULEStammes enthielt neben den nötigen Komponenten der Transkription und Translation auch
das orthogonale Paar aus einer mutierten Aminoacyl-tRNA-Synthetase und einer amber-
36
III. Material und Methoden
Suppressor-tRNA (tRNACUA), deren beider Gene auf dem pDULE-Plasmid codiert vorlagen
[Farrell et al., 2005]. Die Anwesenheit des orthogonalen Paares im Zellextrakt war
Voraussetzung für die ortsgerichtete Integration des photoaktiven Aminosäurenderivates
pBPA in das TorA-PhoA-Protein.
Zur Herstellung des Zellextraktes S135 wurden zunächst folgende Lösungen hergestellt:
S30-Medium:
pankreatisch verdautes Casein
9 g/l
Hefeextrakt
0,8 g/l
NaCl
5,6 g/l
1 M NaOH
1 ml/l
→ Nach dem Autoklavieren des Mediums wurde zugesetzt:
20% (w/v) Glukose (autoklaviert)
4 ml/l
S30-Puffer:
Triethanolaminacetat (TeaOAc), pH 7,5
10 mM
Mg(OAc)2
14 mM
KOAc, pH 7,5
60 mM
DTT
1 mM
Zur Anzucht der E. coli-Zellen wurde eine Über-Nacht-Kultur des jeweiligen Stammes
(100 ml S30-Medium in einem 500 ml-Erlenmeyerkolben) in Gegenwart des entsprechenden
Antibiotikums (siehe Tabelle III.1.4) bei 37°C und unter Schütteln bei 180 rpm angezogen.
Für eine Über-Tag-Kultur wurden 4x 1 l S30-Medium in einem 5 l-Erlenmeyerkolben im
Verhälnis 1:100 mit der Über-Nacht-Kultur in Gegenwart von Antibiotika überimpft und unter
den gleichen Bedingungen angezogen. Um die Expression des orthogonalen Paares zu
optimieren wurde im Fall des Top10pDULE-Stamms die Konzentration des Tetracyclins in
der Über-Tag-Kultur auf 2,5 µg/ml gesenkt. Nach Erreichen einer optischen Dichte (OD600)
von 1,0-1,2 wurde die Kultur in einem Wasser/Eis-Gemisch kaltgestellt und anschließend
durch Zentrifugation geerntet (SCL 3000 Rotor (Sorvall): 7000 rpm (8650 xg), 10 min, 4°C).
Das Zellpellet wurde mit S30-Puffer gewaschen und erneut zentrifugiert. Anschließend
wurde das Zellpellet in 1 ml S30-Puffer pro 1 g Zellen resuspendiert und die Zellen im
French-Press-Verfahren (Presse von SCM Aminico, 40k-Zelle von Spectronic Unicam) mit
einem Druck von 8000 psi in 2-3 Durchgängen aufgeschlossen. Durch eine weitere
Zentrifugation (SS 34 Rotor (Sorvall): 15500 rpm (30000 xg), 30 min, 4°C) wurden
Zellbruchstücke und nicht-aufgeschlossene Zellen sedimentiert. Der S30-Überstand wurde
abgenommen und durch eine anschließende „read out“-Behandlung die darin enthaltenen
37
III. Material und Methoden
endogenen mRNAs vollständig translatiert. Dies war eine Voraussetzung für die spezifische
in vitro-Proteinsynthese. Dazu wurden pro ml S30-Überstand folgende Komponenten
zugesetzt:
1 M TeaOAc, pH 7,5
60 µl
1 M DTT
0,6 µl
1 M Mg(OAc)2
1,6 µl
1 mM 18 Aminosäuren-Mix
6 µl
1 mM Cystein
6 µl
1 mM Methionin
6 µl
0,25 M ATP (neutralisiert)
2 µl
0,2 M PEP
27 µl
2 mg/ml Pyruvat-Kinase
2,4 µl
Die „read out“-Behandlung wurde bei 37°C für 1 h durchgeführt. Um die zugegebenen
niedermolekularen Komponenten, insbesondere nicht radioaktives Methionin und Cystein,
aus dem S30-Überstand zu entfernen, wurde anschließend zweimal für je 1 h und ein drittes
Mal über Nacht bei jeweils 4°C gegen je 1 l S30-Puffer dialysiert. Dazu wurde ein
Dialyseschlauch mit einer Ausschlußgröße von 14 kDa benutzt (Visking, Roth).
Der so gewonnene S30 konnte nun in Aliquots in flüssigem Stickstoff eingefroren und dann
bei –80°C aufbewahrt oder direkt zu S135 weiterverarbeitet werden. Dazu wurde je 1 ml
S30-Überstand zentrifugiert (Ultrazentrifuge Beckman, Rotor: TLA 100.2, 90000 rpm, 13 min,
4°C), wodurch endogene Membranen abgetrennt wurden. Je 750 µl des Überstands wurden
als S135 abgenommen. Der S135-Zellextrakt wurde aliquotiert, in flüssigem Stickstoff
schockgefroren und bei −70°C gelagert.
III.2.2.5.2. Präparation von inside-out Innenmembranvesikeln (INV)
Um den Transport von sekretorischen Proteinen über die Innenmembran von E. coli in vitro
untersuchen zu können, wurden sogenannte inside-out Vesikel (INV), Vesikel der
Innenmembran mit umgedrehter Orientierung, im Wesentlichen nach dem Protokoll von
Moser et al. (2007) hergestellt. Zur Präparation der INVs wurden die in Tabelle III.1.1 und
III.1.2 beschriebenen Stämme und Plasmide verwendet. Zunächst wurden folgende
Lösungen angesetzt:
38
III. Material und Methoden
INV-Medium:
dest. H2O mit 10g pankreatisch verdautem Casein
und 10 g Hefeextrakt
753 ml
1 M KH2PO4
41 ml
1 M K2HPO4
166 ml
25 % (w/v) Glukose
40 ml
→ Die Lösungen wurden getrennt von einander autoklaviert und
erst kurz vor Beginn der Anzucht in den angegebenen Volumina
zu 1 l INV-Medium vereint.
Puffer A:
Triethanolaminacetat (TeaOAc), pH 7,5
50 mM
Saccharose
0,25 M
EDTA
1 mM
DTT
1 mM
PMSF
0,5 mM
→ PMSF wurde erst kurz vor dem Aufschluß der Zellen aus
einer frisch zubereiteten 0,1 M-Lösung in EtOH p.a. zugegeben
INV-Puffer:
Triethanolaminacetat (TeaOAc), pH 7,5
50 mM
Saccharose
250 mM
DTT
1 mM
Zur Präparation der INVs wurde zunächst eine Über-Nacht-Kultur (100 ml INV-Medium in 1 lErlenmeyerkolben, entsprechendes Antibiotikum) angezogen. Für eine Über-Tag-Kultur
wurden 4x 1 l INV-Medium in je einem 5 l-Erlenmeyerkolben im Verhälnis 1:50 mit der ÜberNacht-Kultur in Gegenwart von Antibiotika überimpft und bei 37°C unter Schütteln bei
180 rpm angezogen. Gegebenfalls wurde die Expression plasmidkodierter Proteine durch die
Zugabe von 1 mM IPTG bei einer OD600 von 0,6 induziert (Tat+-INVs, ∆TatA-INVs). Die Ernte
der Zellen erfolgte bei einer OD600 von 1,5-1,8. Dazu wurde die Kultur in einem Wasser/EisGemisch kalt gestellt und anschließend durch Zentrifugation geerntet (SCL 3000 Rotor
(Sorvall): 7000 rpm (8650 xg), 10 min, 4°C). Alle folgenden Schritte wurden auf Eis bzw. bei
4°C durchgeführt. Nach einem Waschschritt mit Puffer A wurde das Zellpellet in 1 ml
Puffer A pro 1 g Zellen resuspendiert und die Zellen im French-Press-Verfahren (Presse von
SCM Aminico, 40k-Zelle von Spectronic Unicam) mit einem Druck von 8000 psi in 2-3
Durchgängen aufgeschlossen. Zelltrümmer und nicht aufgeschlossene Zellen wurden durch
39
III. Material und Methoden
einen niedertourigen ersten Zentrifugationsschritt sedimentiert (Rotor: SS-34 (Sorvall),
5000 rpm, 5 min). Durch Ultrazentrifugation des Überstandes (Rotor: 50.2 Ti (Beckmann),
40000 rpm, 2 h) wurde das Rohmembranen-Pellet sedimentiert, das aus Innenmembranen,
Außenmembranen sowie Ribosomen bestand. Dieses Pellet wurde anschließend mit Hilfe
eines Dounce Homogenisators (“loose fit“) in 8 ml Puffer A resuspendiert. Auf Grund der
unterschiedlichen Dichten konnten im folgenden Präparationsschritt die Außenmembranen
und Ribosomen des Rohmembranen-Pellets von den Innenmembranvesikeln weitgehend
getrennt werden. Dazu wurden 0,77 M, 1,44 M und 2,02 M Saccharoselösungen in
Saccharose-freiem Puffer A (50 mM TeaOAc, pH 7,5, 1 mM EDTA, 1 mM DTT, 0,5 mM
PMSF (frisch)) hergestellt. Durch Unterschichtung von 12 ml der 0,77 M Saccharoselösung
mit 12 ml der 1,44 M Lösung und schließlich mit 10 ml der 2,02 M Lösung entstand ein
Saccharosegradient, der für 1 h bei 4°C äquilibriert wurde. Nach Überschichtung von 2–2,5
ml der Rohmembranen pro Gradient erfolgte ein weiterer Ultrazentrifugationsschritt
(Ausschwingrotor SW28 (Beckmann), 25000 rpm, mindestens 16 h). Auf Grund ihrer
spezifischen Dichte bewegten sich die Innenmembranvesikel zu der oberen Interphase
zwischen der 0,77 M und 1,44 M Saccharoselösung und waren hier als gelbliche Bande zu
erkennen. Diese wurde mittels einer Kanüle und einer Spritze abgezogen. Um die
Saccharosekonzentration der Innenmembranvesikellösung zu verringern, wurde diese 1:4
mit
50 mM
Triethanolaminacetat,
pH 7,5
verdünnt.
Durch
einen
weiteren
Ultrazentrifugationsschritt (Rotor: 50.2 Ti (Beckmann), 40000 rpm, 2 h) wurden die INVs
pelletiert und anschließend mit Hilfe eines Homogenisators in insgesamt 1 ml INV-Puffer
resuspendiert, um eine ausreichend hohe Vesikel-Konzentration zu erhalten. Zur
Absorptionsmessung (OD280) wurden die Vesikel 1:100 in 2 % SDS verdünnt.
Präparierte INVs wurden durch Western-Blotting (siehe III.2.2.2.3.) mit Antikörpern gegen die
Tat-Proteine TatA, TatB und TatC, sowie durch Bestimmung ihrer in vitro-Transportaktivität
charakterisiert (siehe III.3.2.).
III.2.2.5.3. Präparation von T7-RNA-Polymerase
Die in der vorliegenden Arbeit zur in vitro-Synthese verwendeten Plasmide codierten für
Proteine, deren Gene unter der Kontrolle des T7-Promotors standen. Zur Optimierung der
Synthese
wurde
daher
dem
Transkriptions/Translations-System
exogene
T7-RNA-
Polymerase zugegeben. Diese wurde aus einem überproduzierenden E. coli-Stamm von D.
Trescher nach der in Beck et al., (2000) beschriebenen Methode aufgereinigt.
40
III. Material und Methoden
III.2.2.6. Versuche im zellfreien System aus E. coli
III.2.2.6.1. Gekoppelte in vitro-Transkription/Translation
In vitro-Proteinsynthese erfolgte in der vorliegenden Arbeit im Wesentlichen nach dem
Protokoll von Moser et al., 2007. Ein Ansatz zur Synthese eines radioaktiv-markierten
Proteins enthielt neben einem aus E. coli präpariertem S135-Extrakt folgende Komponenten:
Ein Plasmid, das für das T7-Promotor-kontrollierte Gen des zu synthetisierenden Proteins
codiert, T7-RNA-Polymerase und für die Transkription notwendige Nukleotide; ein ATPregenerierendes System aus Kreatinphosphat (CP), Kreatinphosphatkinase (CPK) und
Phosphoenolpyruvat (PEP); 20 Aminosäuren, darunter mit
35
S radioaktiv-markiertes Cystein
und Methionin; Polyethylenglycol zur Simulierung des „molecular crowding“; Spermidin, das
als Polyamid die Ribosomen stabilisiert, sowie DTT, das die im Cytoplasma herrschenden
reduzierenden Bedingungen etabliert. Dieses System wurde je nach Fragestellung durch den
Zusatz weiterer Komponenten erweitert.
Entscheidend für die Effizienz der in vitro-Proteinsynthese war die Etablierung stabiler
Ionenverhältnisse. Gleichbleibende Konzentrationen an Tea+- (Triethanolamin), K+- und
insbesondere Mg2+-Ionen wurden durch die Verwendung eines Kompensationspuffers
gewährleistet, dessen ionische Zusammensetzung sich nach der Art und Menge der zu dem
in vitro-System zugesetzten Komponenten richtete. So wurde beispielsweise durch den
Einsatz von 1 µl S135 10 nmol Tea+-, 60 nmol K+- und 14 nmol Mg2+-Ionen in das in vitroSystem eingetragen. Um das Konzept des Kompensationspuffers zu verdeutlichen wird im
Folgenden die Beispielrechnung eines 5x Kompensationspuffers für einen Syntheseansatz
(25 µl) dargestellt, in der 5 µl S135 und 1µl Plasmid-DNA verwendet wurden:
10
60
14
5µl S135 (T K Mg )
1 µl Plasmid-DNA (T10)
Gewünschte Endkonzentration
Dies entspricht in 25µl
∆ nmol
Konzentration der
Stammlösungen
Volumen für 500µl 5x
Kompensationspuffer
Tea+
50 nmol
10 nmol
40 mM
1000 nmol
940
1M
K+
300 nmol
140 mM
3500 nmol
3200
4M
Mg+
70 nmol
11 mM
275nmol
205
1M
Spermidin
0,8 mM
20 nmol
20
0,1M
H20
94µl
80 µl
20,5 µl
20µl
ad
500µl
Nach einer S135-Präparation wurde die optimale Menge an Zellextrakt/Syntheseansatz
durch Titration neu ermittelt. Ein weiteres Mittel zur Optimierung der Syntheseleistung war
außerdem die Titration der Mg+-Konzentration.
41
III. Material und Methoden
Der eigentliche Syntheseansatz wurde wie folgt zusammenpipettiert:
Kompensationspuffer
Polyethylenglykol (PEG)
18 Aminosäuren
DTT
NTP-Mix: ATP, GTP, CTP, UTP
Phosphoenolpyruvat (PEP)
Kreatinphosphat (CP)
Kreatinphosphatkinase (CPK)
S135
DNA
T7- RNA- Polymerase
35
S- Met/Cys
sonstige Komponenten
dest. H2O
[Stammlösung]
Endkonzentration
5x
40%
je 1mM
200mM
50, 10, 10 ,10 mM
0,2M
0,5M
10mg/ml
1x
3,2%
je 0,04mM
2mM
2,5 ; 0,5; 0,5; 0,5 mM
12mM
8mM
40µg/ml
1µg/µl
40µg/ml
15µCi/µl
0,3µCi/µl
µl/ 25µl
Ansatz
5
2
1
0,25
1,25
1,5
0,4
0,1
5
1
0,1
0,5
x
ad 25
Die Komponenten wurden entweder einzeln, oder zur Vereinfachung, in Mix-Lösungen aus
mehreren Komponenten pipettiert. So wurden PEG, die 18 Aminosäuren und der NTP-Mix
als sog. Master-Mix angesetzt, während alle anderen Komponenten einzeln pipettiert
wurden. Die Komponenten, die in gleicher Menge in allen Syntheseansätzen vorkamen,
wurden erst in einem so genannten Reaktions-Mix zusammenpipettiert. Dabei wurde das
Volumen des Reaktions-Mixes für n+1 Ansätze berechnet. Dieser konnte dann auf die
einzelnen Reaktiongefäße verteilt werden. Komponenten, die auf Grund der experimentellen
Fragestellung nicht in allen Ansätzen vorhanden war, wurden für die entsprechenden
Ansätze einzeln in die Reaktionsgefäße vorgelegt oder zu definierten Zeitpunkten
zugegeben. Nahmen die zusätzlichen Komponenten Einfluss auf die ionischen Verhältnisse
des
in vitro-Systems,
so
wurde
dies
im
Vorhinein
in
die
Berechnung
des
Kompensationspuffers einbezogen. Ansätze denen diese Komponenten fehlten, wurden
stattdessen nur mit dem Puffer der zusätzlichen Komponente versetzt. Die zugesetzten
Komponenten mussten auch bei der Berechnung des Endvolumens des Reaktionsansatzes
(25 µl) berücksichtigt werden. Die radioaktiven Aminosäuren wurden zuletzt zugegeben. Die
Transkriptions-/Translationsreaktion erfolgte bei 37°C für 30-35 min. Die Synthese wurde
entweder durch eine Fällung mit 5 % TCA und einer anschließenden Inkubation von 30 min
auf Eis gestoppt, oder durch die Zugabe des Antibiotikums Puromycin in einer
Endkonzentration von 0,8 mM und einer Inkubation von 10 min bei 37°C beendet
(Stammlösung Puromycin: 11 mM, pH 6,5). Durch Puromycin wird die naszierende
Polypeptidkette vom Ribosom abgelößt. Die Ursache hierfür liegt in der Struktur des
Antibiotikums, die der eines Aminoacylterminuses einer Aminoacyl-tRNA ähnelt. Dadurch
bindet Puromycin an die leere A-Stelle im Ribosom, die Peptidyltransferaseaktivität des
Ribosoms katalysiert eine Peptidbindung zwischen der αAminogruppe des Puromycins und
42
III. Material und Methoden
der Carboxylgruppe der letzten Aminosäure der naszierenden Kette, wodurch das PeptidylPuromycin-Molekül vom Ribosom abdissoziiert.
III.2.2.6.2. In vitro-Translokation und Protease-Resistenz-Test
Das in der vorliegenden Arbeit benutzte in vitro-System konnte auch zur Untersuchung von
Translokationsprozessen des in vitro-synthetisierten Proteins über die Cytoplasmamembran
von E. coli benutzt werden. Dazu wurden dem in vitro-Syntheseansatz 5-10 min nach dem
Start der Synthese invertierte Innenmembranvesikel (INV) aus E. coli zugesetzt und für
weitere 25 min inkubiert. Die Zugabe der INVs wurde im Kompensationspuffer für die
Synthese berücksichtigt (1 µl INV: (T50)). Ein Spezialfall stellte der Tat-abhängige in vitroTransport des TorA-PhoA-Proteins da, der von „oxidierenden“ Synthesebedingungen
abhängig war. Oxidierende Bedingungen wurden durch die Zugabe von 5 mM oxidiertem
Glutathion (GSSG) etabliert. Dieses wurde den entsprechenden Ansätzen zu Beginn der
in vitro-Synthese zugegeben. Dazu wurden einem bereits fertiggestellten Reaktionsmix
0,625 µl/25 µl-Ansatz einer 200 mM GSSG-Lösung zugesetzt und erst dann auf die
Reaktionsgefäße verteilt. Da GSSG in dest. H2O gelöst war hatte dieser Zusatz keinen
Einfluss auf die ionischen Verhältnisse des in vitro-Systems, allerdings mußte das
zugegebene Volumen der GSSG-Lösung mit in die Kalkulation einbezogen werden.
Zur Inhibition des PMF-abhängigen in vitro-Tat-Transportes konnten dem in vitro-Ansatz
Entkoppler-Substanzen
Protonengradienten
zugesetzt
wurde
werden.
das
Zur
Protonophor
Zerstörung
CCCP
eines
bestehenden
(Carbonylcyanid-m-
chlorophenylhydrazon, Sigma) in einer Endkonzentration von 0,1 mM unmittelbar vor der
Zugabe der INVs zugesetzt. Die CCCP-Arbeitslösung wurde immer frisch in einer
Konzentration von 10 mM in DMSO angesetzt. Zur Inhibition der ATPase der zugesetzten
INVs und somit zur Inhibition der Formation eines Protonengradienten wurde DCCD
(Dicyclohexylcarbodiimid, Fluka) in einer Endkonzentration von 0,5 mM unmittelbar vor der
Zugabe der INVs dem in vitro-Transportansatz zugesetzt. Die DCCD-Arbeitslösung wurde in
einer Konzentration von 50 mM in DMSO aus einer 1 M Stammlösung in Tetrahydrofuran
(THF) angesetzt. Die Stammlösung wurde immer frisch angesetzt. Als Kontrolle wurde
Ansätzen ohne Entkoppler das entsprechende Volumen DMSO zugesetzt.
Transportaktivität wurde nach Proteinsynthese und Inkubation des Ansatzes mit den INV
mittels eines Protease-Resistenz-Tests überprüft. Dazu wurde 25 min nach der Zugabe der
INVs 15µl eines Doppelansatzes (50 µl) direkt mit TCA (Endkonzentration 5 %) gefällt,
während 30 µl mit 30 µl Proteinase K (1 mg/ml) für 25 min bei 25°C inkubiert wurden.
Während des 25-minutigen Proteaseverdaus wurden Proteine, die nicht durch die
Membranvesikel geschützt wurden, vollständig verdaut. Proteine, die dagegen durch
Transport ins Lumen des Membranvesikel gelangten, waren dem proteolytischem Verdau
43
III. Material und Methoden
durch Proteinase K nicht ausgesetzt. Proteinas K-Behandlung konnte in Gegenwart von
Detergenzien (bspw. 1 % Triton X100) durchgeführt werden, die zusammen mit der
Proteinase K zugesetzt wurden. Durch Detergenzien wurde die Integrität der Membran
zerstört, wodurch auch das Vesikellumen der Proteinase K zugänglich wurde. TCA-Pellets
von Ansätzen denen Detergenzien zugesetzt worden waren, wurden zunächst mit 1 ml
Aceton p.a. gewaschen und erst dann in SDS-PAGE-Probenpuffer gelöst.
PK-behandelte Proben wurden mit TCA gefällt und, um eine vollständige Inaktivierung der
Proteinase K zu erreichen, für 10 min bei 56°C inkubiert und erst danach mind. 30 min bei
4°C inkubiert. Die Proben wurden dann wie beschrieben mittels SDS-PAGE analysiert.
III.2.2.6.3. Ortsgerichtete Quervernetzung mit para-Benzoyl-L-Phenylalanin (pBPA)
Um molekulare Interaktion des in vitro-synthetisierten Proteins zu analysieren wurden in der
vorliegenden Arbeit eine in vitro-Quervernetzungsmethode mit dem photoaktivierbaren
Quervernetzer para-Benzoyl-L-Phenylalanin (pBPA, Bachem) etabliert. pBPA wurde dabei
ortsspezifisch in das in vitro-synthetisierte Protein eingebaut. Vorbild dieser Methode bildete
das von Farrell et al., 2005 etablierte in vivo-System zum ortsspezifischen Einbau von pBPA
in Proteine.
In der vorliegenden Arbeit wurde zunächst an einer gewünschten Position der DNA-Sequenz
des plasmidcodierten Proteins ein Aminosäurecodon zu einem amber-Stop-Codon (TAG)
mutiert, wodurch es zu einem Abbruch der in vitro-Translation an dieser Stelle kam. Durch
den Einbau von pBPA konnte der Translationsstopp supprimiert werden, wodurch ein Protein
synthetisiert wurde, das einen photoaktivierbaren Quervernetzer an einer definierten Position
der Aminosäuresequenz enthielt. Die Grundlage für den Einbau von pBPA an dieser Position
bildet ein orthogonales Paar aus einer amber-Suppressor-tRNA (tRNAUAG) und einer
mutierten Tyrosyl-tRNA-Synthetase. Beide Komponenten waren bereits in dem für diese
Methode verwendeten Zellextrakt vorhanden, da der E. coli-Stamm aus dem der Extrakt
präpariert wurde das pDULE-Plasmid (siehe Tabelle III.1.2) trug, auf dem die Gene dieser
Komponenten codiert waren. Beide Komponenten wurden in vivo unter der Kontrolle eines
endogenen llp-Promotors aus E. coli exprimiert [Farrell et al., 2005]. Durch die Zugabe von
pBPA zum in vitro-Syntheseansatz wurde die amber-Suppressor-tRNA in einer durch die
mutierten Tyrosyl-tRNA-Synthetase-katalysierten Reaktion mit pBPA aminoacyliert, wodurch
eine pBPA-amber-Suppressor-tRNA (pBPA-tRNAUAG) entstand. Diese supprimierte das
amber-Stop-Codon in der transkribierten mRNA des mutierten Proteins, wodurch das
photoaktivierbare pBPA an dieser Stelle während der Translation in die Polypeptidkette
eingebaut und das Protein bis zum regulären Stop-Codon am 3’ Ende fertig synthetisiert
wurde (siehe Abb. IV.11 in IV. Ergebnisse).
44
III. Material und Methoden
Im Einzelnen wurde wie folgt verfahren: Da pBPA lichtempfindlich ist wurden alle folgenden
Schritte im Dunkeln durchgeführt, d.h. alle pBPA-haltigen Lösungen wurde mit Alufolie vor
Licht geschützt. Zunächst wurde eine 1 M pBPA-Stammlösung in 1 N NaOH angesetzt.
Daraus wurde anschließend durch eine 1:500 Verdünnung mit dest. H2O eine 2 mM pBPAArbeitslösung hergestellt. Um die Stop-Codon-Mutation zu supprimieren wurden 0,5 µl dieser
Arbeitslösung/25 µl Syntheseansatz (Endkonzentration pBPA: 40 µM) entweder einzeln in
die Reaktionsgefäße vorgelegt oder direkt in den Reaktionsmix pipettiert und wie
beschrieben bei 37°C für 30 min inkubiert. Das Volumen der zugegebenen pBPA-Lösung
wurde bei der Berechnung des Reaktionsmixes einkalkuliert.
Für die Quervernetzungsexperimente mußten die Synthesebedingungen der Stop-CodonMutanten optimiert werden. Die Suppression des amber-Stop-Codons hing sehr stark von
der Mg2+-Konzentration im Syntheseansatz ab. Bei zu hoher Mg2+-Konzentration wurde das
amber-Stop-Codon, auch ohne Zugabe von pBPA, von den Ribosomen zum Teil überlesen
(readthrough). Bei zu geringen Mg2+-Konzentrationen war die Syntheserate deutlich
reduziert. In Vorversuchen wurde die optimale Mg2+ Konzentration ermittelt, die meist bei 89 mM lag.
Der eigentliche Quervernetzungsversuch fand nach dem Stopp der in vitro-Synthese durch
Puromycin statt. Dazu wurde der Syntheseansatz (Doppelter Syntheseansatz: 50 µl)
gegebenenfalls mit Membranvesikeln bei 37°C für 20 min inkubiert, um anschließend zu
gleichen Teilen aufgeteilt zu werden, wobei eine Hälfte durch die Zugabe von TCA in einer
Endkonzentration von 5 % gefällt, und die zweite Hälfte durch eine Bestrahlung mit UV-Licht
(365 nm, Handlampe VL-6.L von Vilbert Lourmat) für 20 min auf Eis behandelt wurde. Nach
der
Bestrahlung
wurde
auch
diese
Probe
mit
TCA
gefällt.
Eventuelle
Quervernetzungsprodukte konnten nach SDS-PAGE über Autoradiographie sichtbar
gemacht werden.
III.2.2.7. Immunpräzipitation
Durch
Immunpräzipitation
(IP)
können
Proteine
in
Lösungen
(bspw.
in
vitro-
Syntheseansätze) mit Hilfe von Antikörpern nachgewiesen werden. Auf diese Weise konnten
in der vorliegenden Arbeit Interaktionspartner des in vitro-synthetisierten Proteins identifiziert
werden. Zur Immobilisierung der Antikörper wurde Protein A-Sepharose (CL-4B, GE
Healthcare) verwendet, deren Protein A den Fc-Teil von IgG-Antikörpern bindet. Je nach
Fragestellung wurde eine native oder eine denaturierende Immunpräzipitation angewandt. Im
Einzelnen wurde wie folgt verfahren:
Für Immunpräzipitationen (IP) mit Antikörpern gegen TatC wurden 20 mg, für alle anderen
IPs 10 mg ProteinA-Sepharose eingesetzt. In jedem Fall wurde die ProteinA-Sepharose
zunächst in 1 ml kaltem Triton-Puffer (25 mM Tris-HCl, pH 6,8, 0,9 % (w/v) NaCl, 1 % (v/v)
45
III. Material und Methoden
Triton X100, 5 mM EDTA, pH 8) für 5 min auf Eis gequollen und anschließend durch
Zentrifugation (Tischzentrifuge, 14000 rpm, 1 min, RT) pelletiert. Die Protein A-Sepharose
wurde in 1 ml kaltem Triton-Puffer gewaschen um schließlich in demselben Volumen TritonPuffer wieder aufgenommen und mit dem spezifischen Antiserum versetzt zu werden:
Antiserum (vom Kaninchen)
µl pro Immunpräzipitation
α TatA
α TatB
α TatC
α FkpA
α TorD
α PhoA
30
30
80
20
30
10
Herkunft
AG M. Müller
AG M. Müller
AG M. Müller
J.M. Betton
F. Sargent
Abcam (ab354)
Der Antikörper-haltige Ansatz wurde ca. 90 min bei 4°C in einem Überkopfschüttler inkubiert.
In dieser Zeit bindet der Antikörper an die ProteinA-Sepharose. Nach zwei Waschschritten
mit jeweils 1 ml kaltem Tritonpuffer zur Abtrennung nicht gebundener Antikörper, wurden die
Antikörper-Protein-A-Sepharose-Komplexe
wiederum
in
0,5 ml
kaltem
Tritonpuffer
aufgenommen. An dieser Stelle konnte der zu untersuchende in vitro-Ansatz zugesetzt
werden. Dieser wurde im Fall einer denaturierenden IP mit 1 % SDS versetzt, bei 56°C für
10 min in einem Thermomixer inkubiert, auf RT abgekühlt und anschließend zu der
präparierten Antikörper-Protein-A-Sepharose-Suspension pipettiert. Entsprechend dem
Volumen des zugesetzten Ansatzes wurde die Suspension mit Triton-Puffer bis zu einer
SDS-Konzentration von 0,1 % verdünnt. Im Fall einer nativen IP wurde auf die Inkubation mit
SDS
verzichtet
und
der
in vitro-Ansatz
direkt
zur
Antikörper-Protein-A-Sepharose-
Suspension pipettiert und mit Triton-Puffer auf 1 ml aufgefüllt. Handelte es sich um einen mit
Proteinase K-behandelten Ansatz wurde dieser nach der 25 minütigen Inkubation mit
Proteinase K (Endkonzentration 0,5 mg/ml) bei 25°C mit 0,5 mM PMSF (Stammlsg: 0,1 M
PMSF in EtOH p.a.) sowie 3 µl „Complete“-Lösung (“Complete“, EDTA-frei, Roche: 1
Tablette in 1 ml dest. H2O) versetzt, bei 25°C für 20 min inkubiert und erst dann zu der
Antikörper-Protein A-Sepharose-Suspension pipettiert. In jedem Fall wurden die IP-Ansätze
für ca. 3 h oder über Nacht bei 4°C in einem Überkopfschüttler inkubiert. Schließlich wurden
die Proben zweimal mit 1 ml Triton-Puffer gewaschen, um nicht gebundene Proteine zu
entfernen, und einmal mit 1 ml Äquilibrierungspuffer (25 mM Tris-HCl, pH 6,8) zur Entfernung
des Tritons. Das Sepharose-Pellet wurde in 40 µl SDS-PAGE-Probenpuffer aufgenommen,
für 10 min bei 56°C im Thermomixer (14000 rpm) inkubiert und, zur Sedimentierung der
Sepharose, für 1 min zentrifugiert. Der SDS-PAGE-Probenpuffer wurde mit einer HamiltonSpritze (50 µl-Hamiltonspritze) abgenommen und auf ein SDS-PAGE-Gel aufgetragen.
46
III. Material und Methoden
III.2.2.8. Kovalente Modifikation freier Sulfhydrylgruppen von in vitro-synthetisiertem
Protein
Um den Oxidationszustand von Sulfhydrylgruppen eines Proteins zu bestimmen wurden
reduzierte und somit freie Sulfhydrylgruppen kovalent modifiziert. Durch die Alkylierung freier
Sulfhydrylgruppen mit Iodacetamid wird eine Oxidation des Proteins während der Analyse
durch SDS-PAGE verhindert. Dazu wurde ein in vitro-Syntheseansatz (25 µl) nach einer 30
minütigen Synthese mit 5 % TCA gefällt und 30 min auf Eis inkubiert. Durch anschließende
Zentrifugation (Tischzentrifuge: 14000 rpm, 10 min, RT) wurde das Präzipitat sedimentiert,
mit 1 ml 100 % Aceton gewaschen und in gleicher Weise erneut zentrifugiert. Das
gewaschene Pellet wurde in 25 µl SDS-PAGE-Probenpuffer (siehe III.2.2.1.) aufgenommen,
der 100 mM Iodacetamid aber kein DTT (nicht-reduzierend) enthielt. Die Probe wurde
daraufhin 30 min bei 25°C in einem Thermomixer bei 1400 rpm inkubiert und anschließend
über eine SDS-PAGE analysiert.
Eine weitere Methode zur kovalenten Modifikation freier Sulfhydrylgruppen stellt die
Behandlung
mit
AMS
(4´-Acetamido-4´-maleimidylstilbene-2,2´-Disulfonsäure, Molecular
Probes) dar. Freie Sulfhydrylgruppen reagieren mit AMS, wodurch dem Molekulargewicht
des Proteins ca. 500 Da/AMS-Molekül zugefügt werden und dadurch ein verändertes
Laufverhalten des Proteins in der anschließenden SDS-PAGE-Analyse auftritt. Dazu wurde
erneut ein in vitro-Syntheseansatz (25 µl) nach einer 30 minütigen Synthese mit 5 % TCA
gefällt, 30 min auf Eis inkubiert und anschließend zentrifugiert (16000 g, 30 min, 4°C). Die
AMS-Behandlung wurde nach Hoffmann et al., 2004 durchgeführt. Hierfür wurde das Pellet
der TCA-Präzipitation in 20 µl ASB-A-Puffer aufgenommen und in einem Thermomixer bei
25°C und 900 rpm für 1 h im Dunkeln inkubiert:
ASB-A-Puffer:
Harnstoff
6M
Tris-HCl, pH 8,5
200 mM
EDTA
10 mM
SDS
0,5% (w/v)
→ zu dieser Pufferlösung wird frisch zugegeben:
AMS
15 mM
Nach Beendigung der Inkubation wurde die gesamte Probe in 10 µl SDS-PAGEProbenpuffer aufgenommen, bei 95°C für 3 min gekocht und durch SDS-PAGE analysiert.
47
III. Material und Methoden
III.2.2.9. Größenchromatographische Analyse eines in vitro-Transport-Ansatzes
Um die Bindung des in vitro-synthetisierten Proteins an zugegebene Membranen zu
analysieren wurden nach dem Gelfiltrationsprinzip die Membranen vom restlichen TransportAnsatz nach Beendigung des in vitro-Transportes getrennt.
Dazu wurde zunächst ca. 1,5 ml Säulenmaterial (Sepharose CL-6B, Sigma-Aldrich), das
einen Trennbereich von 10 kDa –4 MDa aufweist, unter Berücksichtung der HerstellerAngaben in eine Einweg-Säule (Polypropylene column 1 ml, Qiagen) gepackt und mit fünf
Säulenvolumen dest. Wasser gespült. Anschließend wurde die Säule mit 5 Säulenvolumen
Puffer I bei RT äquilibriert:
Puffer I
HEPES, pH 7,5
50 mM
KAc
140 mM
MgAc
10 mM
DTT
2,2 mM
→ wurden Transportansätze analysiert, die oxidiertes Glutathion (GSSG)
enthielten:
GSSG
5 mM
Für die größenchromatographischen Analysen eines Transportansatzes wurde dieser nach
dem Stopp der Synthese durch die Zugabe von Puromycin (siehe III.3.1.) 20 min mit 16000 g
bei 4°C zentrifugiert um eventuelle Aggregate des synthetisierten Proteins zu sedimentieren.
100 µl des Überstandes wurden bei RT auf die Säule appliziert. Eluation fand mit 2 ml Puffer
I statt. Dabei wurden 20 Fraktionen à 100 µl aufgefangen, mit TCA in einer Endkonzentration
von 5 % versetzt und mind. 30 min auf Eis inkubiert. Durch anschließende Zentrifugation
(Tischzentrifuge: 14000 rpm, 10 min, RT)
wurde
das
Präzipitat
sedimentiert.
Zur
anschließenden SDS-PAGE- Analyse wurde das Pellet der Zentrifugation in 30 µl SDSPAGE-Probenpuffer aufgenommen (siehe III.2.2.1.).
III.2.2.10. Affinitätschromatographische Aufreinigung von in vitro-synthetisiertem
Protein
In vitro-synthetisiertes,
His-getaggtes
TorA-PhoA
wurde
im
batch-Verfahren
affinitätschromatographisch aufgereinigt. Dazu wurde zunächst 50 µl einer Ni2+-SepharoseSuspension (Ni-Sepharose 6 fast flow, GE healthcare) mit 1 ml dest. H2O versetzt und
anschließend zentrifugiert (500 xg, 2 min, RT). Der Überstand wurde entfernt und die
48
III. Material und Methoden
Sepharose in 1 ml Äquilibrierungspuffer (25 mM Tris-HCl, pH 8,0, 150 mM NaCl, 1 % (v/v)
Triton X100) aufgenommen, erneut zentrifugiert (500 xg, 2 min, RT) und der Überstand
entfernt. Dieser Schritt wurde ein weiteres Mal wiederholt und schließlich der Überstand mit
einer Spritze vollständig abgenommen (Hamilton, 50 µl). Auf die so äquilibrierte Ni2+Sepharose wurde 50 µl eines in vitro-Syntheseansatzes gegeben, der mit Puromycin
gestoppt und zusätzlich mit Triton X100 in einer Endkonzentration von 1 % (v/v) versetzt
wurde. Nach einer Inkubationszeit von 15 min bei RT wurde erneut zentrifugiert und der
Überstand als ungebundenes Material vollständig mit der Spritze abgenommen. Die Ni2+Sepharose wurde mit 50 µl Äquilibrierungspuffer versetzt, 15 min bei RT inkubiert um
anschließend die Probe zu zentrifugieren und den Überstand mit der Spritze abzunehmen.
Dieser Schritt wurde einmal wiederholt und die Überstände aus beiden Schritten als
Waschfraktionen 1 und 2 gesammelt. Elution des an die Sepharose gebundenen Proteins
erfolgte durch Inkubation der Ni2+-Sepharose mit 50 µl Elutionspuffers (25 mM Tris-HCl, pH
8,0, 150 mM NaCl, 1 % (v/v) Triton X100, 50 mM EDTA) für 15 min bei RT. Nach erneuter
Zentrifugation wurde der Überstand mit der Spritze abgenommen und als Eluat-Fraktion
gesammelt. Sämtliche Fraktionen wurden mit TCA in einer Endkonzentration von 5 %
versetzt und mind. 30 min auf Eis inkubiert. Durch anschließende Zentrifugation
(Tischzentrifuge: 14000 rpm, 10 min, RT)
wurde
das
Präzipitat
sedimentiert.
Zur
anschließenden SDS-PAGE-Analyse wurde das Pellet der Zentrifugation in 30 µl SDSPAGE-Probenpuffer aufgenommen (siehe III.2.2.1.).
49
IV. Ergebnisse
IV. Ergebnisse
Ziel dieser Arbeit war es, detaillierte Einblicke in den Mechanismus des twin-argininetranslocation (Tat)-Systems, einem Proteintranslokations-Systems aus E. coli zu erhalten. In
E. coli wird der Tat-abhängige Transport von den drei essentiellen Membranproteinen TatA,
TatB und TatC vermittelt. Tat-abhängig transportierte Proteine tragen zwei konservierte
Arginine (twin arginine) in ihren N-terminalen Signalsequenzen. Die Bedeutung des TatSystems liegt in der Translokation von Proteinen, bei denen die Ausbildung von tertiären und
quartären Strukturen im Cytoplasma auf Grund von Kofaktor-Insertion, Assemblierung mit
Untereinheiten eines Komplexes oder schneller Faltungskinetik dem Transport voraus
gehen. Dadurch werden diese Proteine inkompatibel für den sekretorischen (Sec)
Transportweg, der nur vollständig ungefaltete Proteine befördern kann. Das Tat-System
muss in der Lage sein den Faltungszustand der Tat-Substrate zu erkennen, da ungefaltete
Proteine nicht transportiert werden. Dies wurde bereits durch Untersuchungen mit Tatabhängigen Proteinen definierter Faltung in vivo gezeigt. Unklar ist bislang, welche
Komponente des Tat-Systems und in welchem Schritt des Translokationsprozesses die
Detektion des Faltungszustandes stattfindet.
Im Fokus der Untersuchungen der vorliegenden Arbeit stand die Fähigkeit des Tat-Systems,
Faltungszustände des Translokations-Substrates zu unterscheiden. Hierfür wurde das Tatabhängige Fusionsprotein TorA-PhoA in einem zellfreien, gekoppelten TranskriptionsTranslations-System aus E. coli synthetisiert und die Interaktionen von TorA-PhoA mit den
Komponenten des Tat-Systems charakterisiert. Mit dem Tat-abhängigen Fusionsprotein
TorA-PhoA
war
in
vivo
der
Zusammenhang
zwischen
Faltungszustand
und
Transportkompetenz des Substrates untersucht und dabei nachgewiesen worden, dass nur
die vollständig gefaltete Form des Proteins Tat-abhängig transportiert wird [DeLisa et al.,
2003]. In dieser Studie wurde die vollständige Faltung des Proteins wegen zweier
intramolekularer Disulfidbrücken im reifen Teil des Proteins nur in einem E. coli-Stamm mit
einem „oxidierenden“ Zytoplasma erreicht. In der vorliegenden Arbeit wurde versucht, einen
faltungsabhängigen Transport von TorA-PhoA in vitro durch das Tat-System in einem
zellfreien gekoppelten Transkriptions-Translations-System aus E. coli zu etablieren.
Unterschiede in den Interaktionen der gefalteten zur ungefalteten Form von TorA-PhoA mit
den Komponenten des Tat-Systems würden Rückschlüsse auf die intrinsische Detektion des
Tat-Substrat-Faltungszustands zulassen.
50
IV. Ergebnisse
IV.1. Etablierung des Tat-abhängigen in vitro-Transports von TorA-PhoA
Für die in vitro-Synthese eines radioaktiv-markierten Proteins wurden die Komponenten des
Transkriptions- und Translationsapparates eines E. coli-Zellextrakts (S135) verwendet.
Weitere zugesetzte Komponenten waren: Plasmid-DNA mit dem Gen des zu exprimierenden
Proteins unter der Kontrolle des T7-Phagen-Promotors, gereinigte T7-RNA-Polymerase,
Nukleotide, sämtliche proteinogenen Aminosäuren, darunter
35
S-markiertes Methionin und
Cystein, Ribosomen-stabilisierende Polyamine wie Spermidin und Putrescin [Kakegawa et
al., 1986], Polyethylenglykol, ein ATP-regenerierendes System aus Kreatinphosphat,
Kreatinphosphatkinase und Phosphoenolpyruvat, definierte Konzentrationen an Mg2+ und K+,
sowie DTT als Reduktionsmittel.
Für den Tat-abhängigen in vitro-Transport wurden invertierte Innenmembranvesikel (INV)
von E. coli verwendet. In diesen Vesikeln zeigt die cytoplasmatische Seite der Membran
nach außen und die periplasmatische Seite nach innen. Daher entspricht der Transport von
Proteinen über die Membran ins Innere der Vesikel dem Proteintransport in einer intakten
Zelle aus dem Cytoplasma in das Periplasma. Dies bildet die Grundlage für den Nachweis
des in vitro-Proteintransportes durch Proteinase K (PK). Sämtliches radioaktiv-markiertes
Protein, das nicht transportiert wurde und sich deshalb nicht im Inneren der Vesikel befindet,
wird durch Proteinase K verdaut, während transportiertes Protein Proteinase K-geschützt ist.
Die für das in vitro-System verwendeten INVs wurden aus einem E. coli-Stamm präpariert, in
dem die Tat-Proteine Plasmid-codiert unter der Kontrolle eines lac-Promotors vorlagen und
überexprimiert wurden. Der Grund für die Überexpression liegt in der Beobachtung früherer
Studien, wonach ein Tat-abhängiger in vitro-Transport nicht mit Wildtypmengen an TatProteinen möglich ist. [Yahr und Wickner, 2001; Alami et al., 2002].
Das zellfreie gekoppelte Transkriptions-Translations-System aus E. coli wurde bereits
erfolgreich für die Synthese und den Transport des natürlichen Tat-Substrates SufI
eingesetzt [Alami et al., 2002; Moser et al., 2007]. SufI (supressor of FtsI, Abb. IV.1) ist ein
Tat-abhängiges, periplasmatisches E. coli-Protein, das im Gegensatz zu den meisten
natürlichen E. coli-Tat-Substraten keinen Kofaktor trägt und sich daher sehr gut für das in
vitro-System eignet. SufI, dessen in vitro-Synthese und Transport in diversen Studien
durchgeführt und charakterisiert wurde [Alami et al., 2002 und 2003; Bageshwar und Musser,
2007], diente in der vorliegenden Arbeit als etabliertes Kontrollprotein für den Tatabhängigen Transport.
TorA-PhoA (Abb. IV.1) ist ein Tat-abhängiges Fusionsprotein, bestehend aus der
Signalsequenz des E. coli Tat-Substrates TorA (Trimethylamin-N-Oxid-Reduktase) und dem
reifen Teil des E. coli Sec-Substrates PhoA (alkalische Phosphatase). TorA ist ein
51
IV. Ergebnisse
periplasmatisches Protein, dessen Signalsequenz bereits in mehreren Studien über das TatSystem zum Einsatz kam.
MSLSRRQFIQASGIALCAGAVPLKASA
Signalsequenz
SufI
SP
Signalsequenz TorA
mTorA
MANNNDLFQASRRRFLAQLGGLTVAGMLGPSLLTPRRATAAQAATDA
TorA-PhoA
TorA ◄ ► reifes PhoA
Abb. IV.1: Schematische Darstellung der Tat-abhängigen Proteine.
SufI: Signalsequenz mit blau hervorgehobenem Konsensusmotif. TorA-PhoA: TorA-Anteil am Tat-abhängigen
Fusionsprotein bestehend aus der TorA-Signalsequenz mit blau hervorgehobenem Konsensusmotif sowie den
ersten 7 Aminosäuren des reifen TorA (mTorA). SP markiert die potentielle Schnittstelle der Signalpeptidase I.
Wie aus Abb. IV.1 deutlich wird, ist das TorA-Signalpeptid in TorA-PhoA über die ersten
sieben Aminosäuren des reifen Teils von TorA mit dem reifen Teil von PhoA verknüpft. Dies
war vermutlich aus klonierungsstrategischen Gründen nötig und ist sogar von Vorteil, da die
positiven Ladungen am N-Terminus des reifen Teils Tat-abhängiger Substrate im
Allgemeinen dazu beitragen Sec-abhängigen Transport zu verhindern [Tullman-Ercek et al.,
2007]. Die alkalische Phosphatase (PhoA) wird normalerweise Sec-abhängig ins Periplasma
transportiert, wo sich durch die dort herrschenden oxidierenden Bedingungen zwei
intramolekulare Disulfidbrücken ausbilden. Die Disulfidbrücken sind kritisch für die Stabilität
und für die Formation eines katalytisch aktiven Homodimers. Die Ausbildung von
Disulfidbrücken und somit die Faltung von PhoA wird durch die reduzierenden Bedingungen
des Cytoplasmas verhindert, wodurch Sec-abhängiger Transport erst möglich wird. Nur in
einem mutierten E. coli-Stamm, der durch die Deletion der Gene der GlutathionOxidoreduktase und der Thioredoxin-Reduktase ein oxidierendes Cytoplasma besitzt, wird
die Bildung von Disulfidbrücken im reifen PhoA vor dem Membrantransport ermöglicht
[Bessette et al., 1999]. Für TorA-PhoA konnte in solch einem Stamm Tat-abhängiger
Transport beobachtet werden [DeLisa et al., 2003], während in einem Stamm mit
reduzierendem Cytoplasma weder Tat- noch Sec-abhängiger Transport zu beobachten war
[DeLisa et al., 2003; Tullman-Ercek et al., 2007]. Diese in vivo-Untersuchungen mit TorAPhoA zeigten eindeutig den Zusammenhang zwischen Faltungszustand des Substrats und
dessen
Transportkompetenz.
Das
Tat-System
scheint
demnach
eine
intrinsische
Qualitätskontrolle zu besitzen, wodurch es in der Lage ist, ungefaltetes, reduziertes TorAPhoA oder
52
transportinkompetentes Substrat von gefaltetem, oxidierten TorA-PhoA oder
IV. Ergebnisse
transportkompetentem Substrat zu unterscheiden. Ziel dieser Arbeit war es, Unterschiede in
den molekularen Interaktionen der beiden Faltungszustände von TorA-PhoA mit dem TatSystem zu charakterisieren. Voraussetzung hierfür war es, einen Tat-abhängigen in vitroTransport von TorA-PhoA zu etablieren.
IV.1.1. In vitro-Transport von TorA-PhoA findet nur unter oxidierenden
Synthesebedingungen statt
Von der Entwicklung eines Tat-abhängigen in vitro-Transportes war bekannt [Yahr und
Wickner, 2001; Alami et al., 2002], dass dieser mit E. coli-Komponenten nur dann möglich
ist, wenn Vesikel mit hohem TatABC-Gehalt verwendet wurden. In dieser Arbeit wurden
invertierte Innenmembranvesikel (INV) von einem BL21 (DE3) pLys
E. coli-Stamm
verwendet, der mit dem p8737-Plasmid (siehe Tabelle III.1.2) transformiert worden war,
wodurch die Überexpression von TatABC durch Induktion mit IPTG möglich wurde. Die
Aktivität des Tat-Transportes neu präparierter INVs wurde in vitro mit SufI, die Aktivität des
Sec-Transportes mit OmpA (outer membrane protein A) getestet. Die optimale Menge aktiver
INVs für den Tat-abhängigen in vitro-Transport ist durch Titration ermittelt worden.
Abb. IV.2A zeigt exemplarisch einen Tat-abhängigen in vitro-Transport von SufI in
Abwesenheit oder Anwesenheit von INVs eines TatABC-Überproduzenten (Tat+-INV). SufI
wurde in einem gekoppelten Transkriptions-Translations-System synthetisiert und dabei
radioaktiv
markiert.
Um
die
Syntheseleistung,
sowie
eventuelle
Transportaktivität
nachzuweisen wurde jeder Ansatz zu einem Drittel mit Trichloressigsäure (TCA) gefällt,
während zwei Drittel mit Proteinase K (PK) behandelt und erst anschließend mit TCA gefällt
wurden. Sämtliches Protein, das durch Translokation in die Vesikel gelangt, ist vor dem
proteolytischen Verdau durch die Proteinase K geschützt. Die PK-unbehandelte Probe in
Spur 1 zeigt das synthetisierte SufI-Vorläuferprotein bei ca. 50 kDa. Durch Proteinase KBehandlung wurde das Protein komplett proteolytisch verdaut (Abb. IV.2A, Spur 2). Da die
Spuren 1 und 2 keine INVs enthielten und somit kein Proteintransport erfolgen konnte, war,
wie zu erwarten, auch keine Proteaseresistenz zu beobachten. In Spur 3 und 4 dagegen
wurde fünf Minuten nach Synthesestart Tat+-INVs zugesetzt. In diesen Spuren war
Translokation zu beobachten. Es war ein um die Länge des Signalpeptides kleineres
Fragment unterhalb des Vorläuferproteins (pSufI) zu beobachten, bei dem es sich um das
reife, prozessierte Protein (mSufI) handelte (Abb. IV.2A, Spur 3). Dieses entstand nach dem
erfolgten Transport in die Vesikel und der anschließenden Abspaltung des Signalpeptids
durch
die Signalpeptidase
der Membranvesikel.
Bestätigt
wurde
dies
durch
die
anschließende Proteinase K-Behandlung (Abb. IV.2A, Spur 4), wodurch sämtliches Protein,
53
IV. Ergebnisse
das nicht transportiert und daher nicht durch die Vesikel geschützt wurde, durch Proteinase
K verdaut worden war. Neben der reifen Form von SufI war eine gewisse Menge an pSufI in
der Proteinase K-behandelten Probe zu finden, da nicht das gesamte transportierte Protein
prozessiert wurde.
A
B
Tat+-INV
-
PK
Tat+-INV
+
+
+
GSSG [mM]
+
-
PK
50 kDa
p
m
1
2
3
2
+
3
+
5
+
+
SufI
p
TorAm
PhoA
i
50 kDa
4
1
2
3
4
5
6
7
8
Abb. IV.2: Die Transporteffizienz von TorA-PhoA ist abhängig von der GSSG-Konzentration.
A: SufI wurde in zwei 50 µl Ansätzen bei 37°C in einem zellfreien E. coli-Transkriptions/Translations-System
35
35
synthetisiert und durch Verwendung von S-Met/ S-Cys radioaktiv markiert. 5 min nach Synthesestart wurden
+
Vesikel der inneren Membran eines TatABC-überproduzierenden E. coli-Stammes (Tat -INV), beziehungsweise
Membranpuffer zugesetzt. Nach weiteren 25 min wurden 15 µl jedes Ansatzes mit 5 % TCA gefällt (Spur 1 und 3),
während 30 µl für weitere 25 min bei 25°C mit Proteinase K (PK; 0,5 mg/ml) inkubiert und anschließend mit 5 %
TCA gefällt wurden (Spur 2 und 4). Proteine, die durch Transport in die Vesikel gelangen sind der PK nicht
zugänglich und erlangen PK-Resistenz (Vorläuferprotein: p, reifes Protein: m).
B: TorA-PhoA wurde in vier 50 µl Ansätzen bei 37°C in einem zellfreien E. coli-Transkriptions/TranslationsSystem synthetisiert und durch Verwendung von 35S-Met/35S-Cys radioaktiv markiert. Die Ansätze in den Spuren
3-8 enthielten oxidiertes Glutathion (GSSG) in ansteigender Konzentration. Allen Ansätzen wurden 5 min nach
Synthesestart Tat+-INVs zugesetzt. Nach weiteren 25 min wurden 15 µl jedes Ansatzes mit 5 % TCA gefällt
(Spuren 1, 3, 5, 7), während 30 µl für weitere 25 min bei 25°C mit Proteinase K (PK; 0,5 mg/ml) inkubiert und
anschließend mit 5 % TCA gefällt wurden (Spuren 2, 4, 6, 8). Proteine, die durch Transport in die Vesikel
gelangen sind der PK nicht zugänglich und erlangen PK-Resistenz (Vorläuferprotein: p, reifes Protein: m,
Intermediat: i). Sämtliche Proben aus A und B wurden mittels SDS-PAGE (10%iges Gel) aufgetrennt und die
radioaktiv markierten Proteine mittels Autoradiographie sichtbar gemacht.
In Abb. IV.2B wurde in gleicher Weise TorA-PhoA synthetisiert und dabei radioaktiv markiert.
Allen Ansätzen wurde 5 Minuten nach Synthesestart Tat+-INV zugesetzt. Die Spuren 1 und 2
sind direkt mit den Bedingungen in Abb. IV.2A in Spuren 3 und 4 vergleichbar. Dennoch ist
kein Transport von TorA-PhoA zu beobachten, da in der Proteinase K-behandelten Probe
(Abb. IV.2B, Spur 2) keine Protease-geschützen Fragmente sichtbar wurden. Auch das
Fragment unterhalb des Vorläuferproteins (pTorA-PhoA) in Spur 1 bei ca. 50 kDa ist kein
Hinweis
auf
Signalpeptidase-spezifische
Prozessierung,
sondern
rührt
von
einer
proteolytischen Aktivität des in vitro-Systems her, auf die im Abschnitt IV.1.3. ausführlich
eingegangen wird. Um oxidierende Bedingungen zu etablieren, welche Vorraussetzung für
die Ausbildung von Disulfidbrücken im reifen Teil von TorA-PhoA sind, wurden den Ansätzen
in den Spuren 3–8 in Abb. IV.2B zu Beginn der Synthese steigende Konzentrationen an
oxidiertem Glutathion (GSSG) zugesetzt. Tatsächlich treten mit steigender Konzentration an
GSSG in den Proteinase K-behandelten Proben (Abb. IV.2B, Spuren 4, 6 und 8) drei
Protease-geschützte Proteine in ansteigenden Mengen auf. Das größte Protein läuft bei ca.
54
IV. Ergebnisse
55 kDa auf der Höhe des Vorläuferproteins. Dabei handelt es sich um transportiertes TorAPhoA-Protein, das nicht durch die Signalpeptidase prozessiert wurde (pTorA-PhoA).
Darunter, bei ca. 50 kDa, läuft das reife, prozessierte TorA-PhoA-Protein (mTorA-PhoA), das
nach dem Transport durch die Signalpeptidase um die Größe des Signalpeptids verkleinert
wurde. Diese Zuordnung wurde auch in späteren Experimenten mit einer TorA-PhoAMutante, deren Signalpeptidase-Spaltstelle fehlt, bestätigt. Darüber hinaus ist ein drittes
Protease-geschütztes Fragment - kleiner als das mTorA-PhoA-Protein - bei ca. 46 kDa zu
finden, das hier als Intermediat oder iTorA-PhoA bezeichnet wird und auf das ausführlich im
letzten Teil dieser Arbeit eingegangen wird.
Das Auftreten der beschriebenen Protease-geschützten TorA-PhoA-Spezies in Abhängigkeit
der GSSG-Konzentration lässt auf eine Translokation des Proteins in Abhängigkeit seines
Redoxzustandes
schließen.
Mit
ansteigenden
oxidierenden
Bedingungen
werden
ansteigende Mengen an pTorA-PhoA transportkompetent. Der Redoxzustand von TorAPhoA wurde im Folgenden näher untersucht.
IV.1.2. Durch oxidierende Synthesebedingungen wird vollständig oxidiertes
TorA-PhoA synthetisiert
TorA-PhoA besitzt insgesamt vier Cysteinreste die im vollständig oxidierten TorA-PhoA zwei
Disulfidbrücken ausbilden (Cys-168-Cys-178 und Cys-286-Cys-336). Um den Einfluss von
oxidiertem Glutathion auf die Bildung der Disulfidbrücken von in vitro-synthetisiertem TorAPhoA zu analysieren, wurde im Folgenden die Mobilität des Proteins in der SDSgelelektrophoretischen Analyse untersucht. So ist von der reduzierten Form des PhoAProteins bekannt, eine geringere Mobilität im Vergleich zur oxidierten Form des PhoAProteins zu besitzen (Akiyama et al., 1992). In Abb. IV.3A wurde TorA-PhoA in vitro
synthetisiert und dabei radioaktiv markiert. Synthese fand entweder in Gegenwart von 5 mM
GSSG (Abb. IV.3A, Spuren 2 und 4, oxidierende Bedingungen) oder in dessen Abwesenheit
statt
(Abb. IV.3A, Spuren 1 und 3,
reduzierende
Bedingungen).
30
Minuten
nach
Synthesestart wurde jeder Reaktionsansatz mit 5 % Trichloressigsäure gefällt und für 30
Minuten auf Eis inkubiert. Das Pellet dieser Fällung wurde in einem DTT-freien SDSProbenpuffer
aufgenommen,
der
entweder
100
mM
Iodacetamid
(Abb.
IV.3A, Spuren 3 und 4) oder kein Iodacetamid enthielt (Abb. IV.3A, Spuren 1 und 2). Die
Proben wurden dann für 30 Minuten bei 25°C inkubiert um anschließend durch SDSGelelektrophorese analysiert zu werden. Unabhängig von den Synthesebedingungen zeigte
pTorA-PhoA
gleiches
Laufverhalten
in
Abwesenheit
von
Iodacetamid
(Abb. IV.3A, Spuren 1 und 2). Erst durch die Zugabe von Iodacetamid in den SDS-
55
IV. Ergebnisse
Probenpuffer verringerte sich die Mobilität des pTorA-PhoA-Proteins, welches unter
reduzierenden Bedingungen synthetisiert wurde, um ca. 1-2 kDa (Abb. IV.3A, Spur 3)
gegenüber
dem,
welches
unter
oxidierenden
Bedingungen
synthetisiert
wurde
(Abb. IV.3A, Spur 4). Die verringerte Mobilität des unter reduzierenden Bedingungen
synthetisierten pTorA-PhoAs deutet auf
eine erfolgreiche Alkylierung reduzierter
Sulfhydrylgruppen durch Iodacetamid hin, wodurch Oxidation und somit Faltung in eine
kompaktere, mobilere Form des Proteins während der nicht-reduzierenden SDSGelelektrophorese verhindert wurde. Dies war nur möglich da das Protein nach der in vitroSynthese nicht vollständig oxidiert vorlag. pTorA-PhoA, das unter oxidierenden Bedingungen
synthetisiert wurde, konnte nicht durch Iodacetamid alkyliert werden, was darauf deutete,
dass das Protein zumindest partiell oxidiert vorlag. Da Sone et al. zeigen konnte, dass allein
die Ausbildung der C-terminalen Disulfidbrücke (Cys-286-Cys-336) ausreichen kann um den
beobachteten Mobilitätsunterschied durch die Iodacetamid-Behandlung zu bewirken, [Sone
et al., 1997] konnte mit diesem Experiment keine Aussage über den Zustand der Nterminalen Disulfidbrücke (Cys-168-Cys-178) gemacht werden. Um dies zu klären wurde ein
weiteres, in Abb. IV.3B dargestelltes Experiment durchgeführt.
A
Red.
Probenpuffer
Ox.
- Iodacetamid
Red.
Ox.
+Iodacetamid
pTorA-PhoA
50 kDa
1
2
3
4
B
Red.
AMS
Ox.
+
+
pTorA-PhoA
50 kDa
1
2
3
4
Abb. IV.3: TorA-PhoA ist unter oxidierenden Synthesebedingungen vollständig oxidiert.
A: TorA-PhoA wurde in zwei 50 µl Ansätzen bei 37°C in vitro synthetisiert und dabei radioaktiv markiert.
Proteinsynthese fand entweder in Gegenwart von 5 mM oxidiertem Glutathion (Ox.) oder in dessen Abwesenheit
(Red.) statt. Nach 30 min Synthese wurde jeder Ansatz in gleiche Teile geteilt und mit 5 % TCA für 30 min auf Eis
gefällt. Das Pellet der Proteinfällung wurde in 30 µl DTT-freiem SDS-PAGE-Probenpuffer aufgenommen, der im
Falle der Proben 3 und 4 zusätzlich mit 100 mM Iodacetamid versetzt war und anschließend für 30 min bei 25°C
und 14000 rpm in einem Thermomixer inkubiert wurde.
B: TorA-PhoA wurde in zwei 50 µl Ansätzen bei 37°C in vitro synthetisiert und dabei radioaktiv markiert.
Proteinsynthese fand entweder in Gegenwart von 5 mM oxidiertem Glutathion (Ox.) oder in dessen Abwesenheit
(Red.) statt. Nach 30 min Synthese wurde jeder Ansatz in gleiche Teile geteilt und mit 5 % TCA 30 min auf Eis
gefällt. Das Pellet der Proteinfällung wurde in 20 µl ASB-A-Puffer aufgenommen (6M Harnstoff; 0,2M Tris-HCL,
pH 8,5; 10 mM EDTA; 0.5% (w/v) SDS), der im Fall der Proben 2 und 4 zusätzlich 15 mM AMS enthielt. Sämtliche
Proben wurden für 1 h bei 25°C und 900 rpm in einem Thermomixer inkubiert und anschließend in 30 µl SDSPAGE-Probenpuffer aufgenommen und 5 min bei 95°C gekocht. Sämtliche Proben aus A und B wurden mittels
SDS-PAGE (10%iges Gel) aufgetrennt und die radioaktiv markierten Proteine mittels Autoradiographie sichtbar
gemacht.
56
IV. Ergebnisse
Grundlage dieses Versuchsansatzes ist wiederum die Alkylierung nicht-reduzierter
Sulfhydrylgruppen,
diesmal
jedoch
durch
4-Acetamido-4’-maleimidylstilbenen-2,2’-
disulfonsäure oder kurz AMS. Durch die Alkylierung des pTorA-PhoA-Proteins mit einem
AMS-Molekül wird das Molekulargewicht des Proteins um 490 Da erhöht, wodurch eine
Verringerung der Mobilität allein durch die vergrößerte Masse des Proteins in der SDSGelelektrophorese sichtbar werden sollte. In Abb. IV.3B wurde TorA-PhoA in vitro
synthetisiert und dabei radioaktiv markiert. Synthese fand entweder in Gegenwart von 5 mM
GSSG (Abb. IV.3B, Spuren 3 und 4, oxidierende Bedingungen) oder in dessen Abwesenheit
statt
(Abb. IV.3B, Spuren 1 und 2,
reduzierende
Bedingungen).
30
Minuten
nach
Synthesestart wurde jeder Reaktionsansatz mit 5 % Trichloressigsäure gefällt und für 30
Minuten auf Eis inkubiert. Das Pellet der Proteinfällung wurde im Puffer aufgenommen, der
im Fall der Proben 2 und 4 zusätzlich 15 mM AMS enthielt. Sämtliche Proben wurden für 1 h
bei 25°C inkubiert und anschließend in DTT-haltigem SDS-Probenpuffer aufgenommen und
SDS-gelelektrophoretisch analysiert. Aus Abb. IV.3B wird deutlich, dass pTorA-PhoA,
welches in Gegenwart von 5 mM GSSG synthetisiert wurde, durch die Behandlung mit AMS
nicht in seiner Mobilität beeinträchtigt wurde (Abb. IV.3B, Spuren 3 und 4). Daraus konnte
eindeutig auf die vollständige Oxidierung des pTorA-PhoA-Proteins während der in vitroSynthese in Gegenwart von 5 mM GSSG geschlossen werden. pTorA-PhoA, welches in
Abwesenheit von GSSG synthetisiert wurde, zeigt dagegen eine verringerte Mobilität, wenn
es mit AMS behandelt wurde (Abb. IV.3B, Spuren 1 und 2). Durch die Alkylierung von
reduzierten Sulfhydrylgruppen des Proteins durch AMS wurde das Molekulargewicht des
Proteins um ca. 1-2 kDa vergrößert, woraus geschlossen werden konnte, dass sich 2-4
AMS-Moleküle an das Protein gebunden haben. Dies war nur möglich, weil es sich hierbei
um eine partiell oder vollständig reduzierte Form des pTorA-PhoA-Proteins handelte.
Aus den gezeigten Experimenten in Abb. IV.3 geht eindeutig hervor, dass durch die
Gegenwart von GSSG vollständig oxidiertes
pTorA-PhoA in vitro synthetisiert wurde,
während in Abwesenheit von GSSG dies nicht der Fall ist. Demnach muss vollständig
oxidiertes pTorA-PhoA zwei Disulfidbrücken ausgebildet haben, was gleichgesetzt werden
kann mit der vollständigen Faltung des Proteins. Über die Qualität der Disulfidbrücken lässt
sich allerdings keine Aussage machen, wodurch nicht-native Disulfidbrücken nicht
ausgeschlossen werden konnten. Ungeachtet dessen wurde in dieser Arbeit von nun an das
TorA-PhoA-Protein in Abhängigkeit der Synthesebedingungen als reduziert bzw. oxidiert
bezeichnet.
57
IV. Ergebnisse
IV.1.3. Oxidiertes TorA-PhoA wird TatABC-abhängig transportiert
In den bisherigen Experimenten konnte gezeigt werden, dass in Gegenwart von 5 mM GSSG
ein vollständig oxidiertes, gefaltetes pTorA-PhoA-Protein in vitro synthetisiert wurde
(Abb. IV.3). Darüber hinaus wurde deutlich, dass die oxidierte, gefaltete Form des Proteins
Voraussetzung für die erfolgreiche in vitro-Translokation in Gegenwart von Tat+-INVs war
(Abb. IV.2). In den folgenden Experimenten sollte geklärt werden, ob diese Translokation
Tat-abhängig erfolgte. Hierfür wurden Translokationsexperimente von reduziertem und
oxidiertem pTorA-PhoA in Abhängigkeit der TatABC-Proteine durchgeführt. Abb. IV.4A zeigt
ein solches Experiment, in dem pTorA-PhoA unter reduzierenden Bedingungen oder unter
oxidierenden Bedingungen synthetisiert wurde. Fünf Minuten nach Synthesestart wurden
entweder Tat+-INVs eines TatABC-überproduzierenden E. coli-Stammes, ∆Tat-INVs eines
TatABC-defizienten E. coli-Stammes (DEAD-Stamm) oder keine INVs zugesetzt.
In den Proben ohne INVs (Abb. IV.4A, Spuren 1-4) war unter reduzierenden als auch
oxidierenden
Bedingungen
nur
das
Vorläuferprotein
pTorA-PhoA
zu
erkennen
(Abb. IV.4A, Spuren 1 und 3). Darüber hinaus traten wie erwartet in den membranfreien
Ansätzen keine Protease K-resistenten Proteine und somit keine Transportaktivität auf
(Abb. IV.4A, Spuren 2 und 4). Ähnlich verhielt es sich für die ∆Tat-INV-haltigen Ansätze
(Abb. IV.4A, Spuren 5-8), denn auch hier konnten keine Proteinase K-resistenten Proteine
und somit keine Transportaktivität beobachtet werden, unabhängig davon, ob es sich um das
reduzierte (Abb. IV.4A, Spur 6) oder das vollständig oxidierte pTorA-PhoA handelte
(Abb. IV.4A, Spur 8). Auch das kleinere TorA-PhoA-Fragment unterhalb des pTorA-PhoA
(Abb. IV.4A, Spuren 5 und 7) bei ca. 50 kDa ist kein Indiz für Prozessierung durch die
Signalpeptidase nach dem Transport von pTorA-PhoA, sondern rührt von einer Tatunabhängigen proteolytischen Aktivität der ∆Tat-INVs her, die in Abb. IV.4B charakterisiert
ist.
Signifikante Mengen an Proteinase K-resistentem pTorA-PhoA (Vorläuferprotein), mTorAPhoA (reifes TorA-PhoA) und iTorA-PhoA (intermediäres TorA-PhoA) und somit TransportAktivität traten nur im Fall des oxidierten, gefalteten pTorA-PhoA-Proteins in Gegenwart von
Tat+-INVs auf
Proteins
(Abb. IV.4A, Spur 12). Im Fall des reduzierten, entfalteten pTorA-PhoA-
waren
nur
sehr
geringe
Mengen
an
mTorA-PhoA
zu
beobachten
(Abb. IV.4A, Spur 10). Diese geringen Mengen an Proteinase K-resistentem mTorA-PhoA
waren wahrscheinlich nicht das Ergebnis des Transports von reduziertem, entfaltetem
pTorA-PhoA, sondern vielmehr auf nicht vollständig etablierte reduzierende Bedingungen in
diesem Ansatz zurückzuführen.
Als Kontrolle wurde einem Transportansatz mit oxidiertem, gefaltetem pTorA-PhoA, der Tat+INVs enthielt, das Detergenz Triton X100 (1%) während der Proteinase K-Behandlung
58
IV. Ergebnisse
zugesetzt (Abb. IV.4A, Spur 14), wodurch die Integrität der Vesikel zerstört wurde und
transportiertes Protein im Inneren der Vesikel nicht mehr vor Proteinase K-Verdau geschützt
war. Die transportierten Proteine pTorA-PhoA und mTorA-PhoA wurden praktisch vollständig
verdaut, wodurch die Validität des Proteinase K-Tests für diese Spezies bestätigt wurde. Das
iTorA-PhoA dagegen war auch in Gegenwart von Triton X100 proteaseresistent
(Abb. IV.4A, Spur 14). Auf diesen Umstand wird im letzten Teil dieser Arbeit ausführlich
eingegangen. Jedoch sei schon an dieser Stelle betont, dass auch die Formation dieser
Spezies (iTorA-PhoA) abhängig war von der oxidierten, gefalteten Form des pTorA-PhoA
und nur in Gegenwart der TatABC-Proteine in der Membran auftrat.
A
Triton X100 1%
+
GSSG (5mM)
-
+
INV
-
-
Proteinase K
+
-
+
Tat+
∆Tat
+
+
+
+
+
+
+
p
m TorA-PhoA
i
50 kDa
1
2
3
4
5
6
7
8
9
10
11
12
13
14
B
INV
∆TatA
-
+DFP
+Cu2+
-
pTorA-PhoA
Fragment aus Pseudoprozessierung
50 kDa
1
2
3
4
Abb. IV.4: TorA-PhoA wird nur in Anwesenheit der Tat-Proteine transportiert.
A: TorA-PhoA wurde in 50 µl Ansätzen bei 37°C in vitro synthetisiert und dabei radioaktiv markiert. Die Ansätze der
Spuren 3, 4, 7, 8 und 11-14 enthalten oxidiertes Glutathion (GSSG). Den Ansätzen in den Spuren 5-8 wurden 5 min
nach Synthesestart Vesikel der inneren Membran eines TatABCDE-deletierten E. coli-Stammes (∆Tat-INV)
zugesetzt, während den Ansätzen in Spuren 9-14 Tat+-INVs zugesetzt wurden. Nach weiteren 25 min wurden 15 µl
jedes Ansatzes mit 5 % TCA gefällt, während 30 µl für weitere 25 min bei 25°C mit Proteinase K (PK, 0,5 mg/ml)
inkubiert und anschließend mit 5 % TCA gefällt wurden. Proteine, die durch Transport in die Vesikel gelangen sind
der PK nicht zugänglich und erlangen PK-Resistenz (Vorläuferprotein: p, reifes Protein: m, Intermediat: i). Zur
Kontrolle der PK-Aktivität wurde der PK-Verdau in Spur 14 in Gegenwart von 1 % Triton X100 durchgeführt, wodurch
die Integrität der Vesikel zerstört wird und Proteine innerhalb des Vesikel nicht länger PK-geschützt sind.
B: TorA-PhoA wurde in vier 25 µl Ansätzen bei 37°C in vitro synthetisiert und dabei radioaktiv markiert.
Proteinsynthese fand in Gegenwart von 5 mM oxidiertem Glutathion (GSSG) statt. Allen Ansätzen wurde 5 min nach
Synthesestart Vesikel der inneren Membran eines ausschließlich TatBC-überproduzierenden E. coli-Stammes
(∆TatA-INV) zugesetzt. Zum selben Zeitpunkt wurde dem Ansatz in Spur 2 50 mM DFP (Diisopropylflurphosphat)
und dem Ansatz in Spur 4 5mM CuCl2 zugegeben. Nach weiteren 25 min wurde jeder Ansatz mit 5% TCA gefällt.
Sämtliche Proben aus A und B wurden durch SDS-PAGE (10%iges Gel) aufgetrennt und die radioaktiv markierten
Proteine mittels Autoradiographie sichtbar gemacht.
59
IV. Ergebnisse
Zusammenfassend zeigen die Transport-Ansätze in Abb. IV.4A, Spur 1–12 eindeutig, dass
Transport von pTorA-PhoA sowohl die oxidierte, gefaltete Form des Proteins als auch die
Gegenwart der TatABC-Proteine in den zugesetzten Membranen voraussetzt. Dies impliziert
zum einen, dass der Transport des oxidierten, gefalteten pTorA-PhoA TatABC-abhängig
erfolgt und zum anderen, dass das Tat-System auch in diesem in vitro-System in der Lage
ist, den Faltungszustand des Tat-Substrates zu erkennen um selektiv nur das oxidierte,
gefaltete pTorA-PhoA zu transportieren. Diese Ergebnisse decken sich in dieser Hinsicht mit
dem Tat-abhängigen in vivo-Transport von TorA-PhoA in E. coli [DeLisa et al., 2003].
Wie bereits erwähnt trat in Gegenwart von ∆Tat-INVs (Abb. IV.4A, Spuren 5 und 7) ein TorAPhoA-Fragment bei ca. 50 kDa auf, das eine minimal geringere Mobilität als pTorA-PhoA
aufwies (Abb. IV.4A, Spuren 7 und 9). Da die ∆Tat-INVs aber nicht zu Tat-abhängiger
Translokation von TorA-PhoA in der Lage waren (Abb. IV.4A, Spuren 6 und 8), war dieses
Fragment höchstwahrscheinlich nicht das Ergebnis einer Prozessierung durch die
Signalpeptidase, sondern musste von einer anderen proteolytischen Aktivität stammen.
Tatsächlich weist die TorA-Signalsequenz eine spezifische Spaltstelle für OmpT (outer
membran protease T) zwischen den Argininen an Positionen 36 und 37 auf (Abb. IV.1)
[Dekker at al., 2001], unmittelbar vor der Spaltstelle der Signalpeptidase. Das daraus
resultierende Fragment hätte damit die zu erwartende Größe, da es nur vier Aminosäuren
länger und daher praktisch gleich groß wäre wie das reife TorA-PhoA-Protein. Um OmpTAktivität als Ursache dieser Pseudoprozessierung nachzuweisen, wurde pTorA-PhoA in vitro
synthetisiert und fünf Minuten nach Synthesestart mit ∆TatA-INVs versetzt. ∆TatA-INVs
wurden aus einem ausschließlich TatBC-überproduzierenden E. coli-Stamm präpariert und
sind daher nicht zu einem Tat-abhängigen Transport und somit zu keiner Prozessierung
durch die Signalpeptidase nach einem Tat-abhängigen Transport fähig. Die hier verwendete
Präparation wies eine besonders hohe Pseudoprozessierungs-Aktivität auf. Durch die
Zugabe von Diisopropylfluorphosphat (DFP) (Abb. IV.4B, Spur 2) sowie von Kupferionen
(Cu2+) (Abb. IV.4B, Spur 4) wurde, im Vergleich zu den entsprechenden Syntheseansätzen
ohne
Zusätze
(Abb. IV.4B, Spuren 1 und 3),
die
Menge
des
Produktes
der
Pseudoprozessierung bei ca. 50 kDa in beiden Fällen deutlich reduziert. DFP, als auch Cu2+
sind als OmpT-Inhibitoren bekannt [Sugimura und Nishihara, 1988; Baneyx und Georgiou,
1990]. Ein Nebeneffekt der Zugabe von Kupferionen zum Syntheseansatz ist die Reduktion
der Syntheseleitung (Abb. IV.4B, Spur 4). Dies ist jedoch ohne Bedeutung für die Aussage
des
Experiments,
da
nur
das
Verhältnis
Pseudoprozessierung entscheidend ist.
60
von
pTorA-PhoA
zum
Produkt
der
IV. Ergebnisse
Damit konnte gezeigt werden, dass die Pseudoprozessierung des pTorA-PhoA-Proteins das
Resultat
einer
proteolytischen
OmpT-Aktivität
war,
die
als
Verunreinigung
präparationsbedingt in den INVs vorliegen kann. Die OmpT-Spaltstelle in der TorASignalsequenz ist wahrscheinlich auch Ziel nicht näher definierter, cytoplasmatischer
Protease-Aktivitäten [Li et al., 2006; Genest et al., 2006], wodurch die geringe
Pseudoprozessierungs-Aktivität auch in Abwesenheit von INVs zu erklären ist, die in vielen
der Versuchen dieser Arbeit zu finden ist (Abb. IV.2B, 18B, 19B). Auch eine Kontamination
des für die in vitro-Synthese verwendeten Zellextraktes (S135) mit Bestandteilen der
Außenmembran und somit mit OmpT kann nicht ausgeschlossen werden. Als eindeutigen
Nachweis für den in vitro-Transport von TorA-PhoA wurde in der vorliegenden Arbeit daher
nur die Resistenz gegenüber der Behandlung mit Proteinase K gewertet.
IV.1.4. In vitro-Transport des oxidierten pTorA-PhoA ist abhängig von der
TorA-Signalsequenz
IV.1.4.1. TorD-Bindung an die TorA-Signalsequenz verhindert den Transport von
pTorA-PhoA
Nachdem die Abhängigkeit des pTorA-PhoA-Transportes von den TatABC-Proteinen
demonstriert wurde, sollte im Folgenden die Abhängigkeit des Transportes von der Tatspezifischen TorA-Signalsequenz untersucht werden.
Dazu wurde zunächst eine His-tag-Version des TorA-spezifischen Chaperons TorD in E. coli
überexprimiert
und
affinitätschromatographisch
bis
zur
Homogenität
aufgereinigt
(Abb. IV.5A1 und A2). TorD ist ein spezifisches TorA-Signalsequenz-bindendes Chaperon,
dessen Funktion als Anti-Zielsteuerungs-Chaperon vermutet wird, das so lange an der TorASignalsequenz bindet, bis Kofaktor-Insertion und somit Faltung des reifen TorA-Proteins
stattgefunden hat [Dubini und Sargent, 2003; Jack et al., 2001 und 2004]. In Abb. IV.5B
wurde pTorA-PhoA unter oxidierenden Bedingungen synthetisiert und in Anwesenheit von
Tat+-INVs transportiert. Durch die Zugabe von aufgereinigtem TorD-Protein in steigenden
Konzentrationen nimmt die Menge an Proteinase K-resistentem Material proportional ab
(Abb. IV.5B, Spuren 5–10). Die Interpretation dieser Beobachtung ist, dass TorD durch seine
Bindung an die Signalsequenz diese gegen Interaktionen mit den Tat-Proteinen abschirmte,
wodurch der TatABC-abhängige Transport verhindert wurde. Demnach ist die Tat-abhängige
Signalsequenz essentiell für den in vitro-Transport von oxidiertem TorA-PhoA.
61
IV. Ergebnisse
rk
er
IP
TG
Ma
[kDa]
[kDa]
75
75
50
50
37
37
25
W1-W4
E1-E3
+
TG
A2
IP
-
+
M
ar
ke
r
A1
25
TorD
TorD
20
20
15
15
1
2
1
2
3
4
5
6
7
8
B
Ox.
TorD
-
-
1µg
4µg
10µg
TorD-Puffer
-
+
-
-
-
PK
+
+
+
+
+
p
TorA-PhoA m
i
50 kDa
1
2
3
4
5
6
7
8
9
10
Abb. IV.5: Der TorA-PhoA-Transport wird durch die Bindung von TorD an die Signalsequenz inhibiert.
A1: Eine His-tag-Version von TorD wurde in einer E.coli–Kultur durch Induktion mit 1 mM IPTG überexprimiert
(Spur 2). Im Vergleich dazu eine nicht induzierte Kultur gleicher optischer Dichte (Spur 1).
A2: 2 l Zellsuspension wurden bei einer optischen Dichte (600 nm) von 0,6 mit 1mM IPTG induziert und die
Zellen nach 4 h Wachstum bei 37°C durch das French-press-Verfahren aufgeschlossen. Der Überstand einer 30
minütigen Zentrifigation mit 30000 g diente als Ausgangsmaterial einer affinitätschromatographischen
Aufreinigung des TorD-Proteins mittels „Talon“ im batch-Verfahren (1). Spuren 2-5 zeigen die Fraktionen der
Waschschritte W1-W4, wobei der letzten Waschung dem Waschpuffer 5 mM Imidazol zugefügt wurden. Spuren
6-8 zeigen die Elution des TorD-Proteins durch die Elutionsschritte E1-E3 mit Waschpuffern mit steigenden
Imidazol-Konzentrationen von 50 mM, 100 mM und 200 mM Imidazol. Sämtliche Proben in A1 und A2 wurden
mittels SDS-PAGE (15%iges Gel) aufgetrennt und das Gel mit Coomassie-Blau gefärbt.
B: TorA-PhoA wurde in 50 µl Ansätzen in Gegenwart von 5 mM oxidiertem Glutathion (Ox.) in vitro synthetisiert
und dabei radioaktiv markiert. Den Ansätzen in den Spuren 5-10 wurde zusätzlich aufgereinigtes TorD-Protein
mit ansteigenden Mengen zugesetzt. Allen Ansätzen wurden Tat+-INVs zugesetzt. Nach 30 min wurden 15 µl
jedes Ansatzes mit TCA gefällt, während 30 µl zuerst mit Proteinase K verdaut wurden (Vorläuferprotein: p,
reifes Protein: m, Intermediat: i). Sämtliche Proben wurden durch SDS-PAGE (10%iges Gel) aufgetrennt und die
radioaktiv markierten Proteine mittels Autoradiographie sichtbar gemacht.
IV.1.4.2. Das Doppel-Arginin des Konsensusmotifs ist essentiell für den Transport
von pTorA-PhoA
Des Weiteren wurden die hochkonservierten Doppel-Arginine des Tat-Konsensusmotifs
gegen zwei Lysine ersetzt (Abb. IV.6A). In Tat-Substraten führt diese Mutation im
Allgemeinen zur kompletten Inhibition des Tat-abhängigen Transports in vivo als auch in
vitro.
RR-TorA-PhoA sowie KK-TorA-PhoA wurden unter oxidierenden Bedingungen in vitro
synthetisiert. Während das RR-TorA-PhoA Proteinase K-resistentes Material und somit
62
IV. Ergebnisse
Transportaktivität aufwies (Abb. IV.6B, Spur 2) war im Fall des KK-TorA-PhoAs der
Transport vollkommen blockiert, da kein Proteinase K-resistentes Protein detektierbar war
(Abb. IV.6B, Spur 4). Somit wurde ein weiteres spezifisches Merkmal, nämlich die
Notwendigkeit des Doppel-Arginins, für den Tat-abhängigen Transport bestätigt.
Die Inhibition des TorA-PhoA-Transports durch die Abschirmung der Signalsequenz auf
Grund der Bindung des TorA-spezifischen Chaperons TorD an die Signalsequenz oder durch
die Mutation der Doppel-Arginine des Konsensusmotifs demonstriert eindeutig die
Abhängigkeit des Transports von TorA-PhoA von dessen Tat-spezifischer Signalsequenz.
Zusammen mit der Abhängigkeit des Transports von den TatABC-Proteinen in der Membran
konnte somit zweifellos die Tat-Abhängigkeit des in vitro-Transports von TorA-PhoA
demonstriert werden.
A
Signalsequenz TorA
mTorA
MANNNDLFQASRRRFLAQLGGLTVAGMLGPSLLTPRRATAAQAATDA
TorA-PhoA
TorA ◄ ► reifes PhoA
Signalsequenz TorA
mTorA
MANNNDLFQASKKRFLAQLGGLTVAGMLGPSLLTPRRATAAQAATDA
KK-TorA-PhoA
TorA ◄ ► reifes PhoA
B
Ox.
TorA-PhoA
RR
Proteinase K
KK
+
+
p
TorA-PhoA m
i
50 kDa
1
2
3
4
Abb. IV.6: Das Doppel-Arginin des Konsensusmotifs ist essentiell für den Transport von pTorA-PhoA
A: RR-TorA-PhoA: Signalsequenz mit blau hervorgehobenem Konsensusmotif sowie den ersten 7
Aminosäuren des reifen TorA (mTorA). KK-TorA-PhoA: Signalsequenz mit blau hervorgehobenem
Konsensusmotif in dem beide konservierten Arginine gegen Lysine (rot) ausgetauscht sind.
B: Die TorA-PhoA-Varianten RR- und KK-TorA-PhoA wurden in je einem 50 µl Ansatz in Gegenwart von 5mM
+
oxidiertem Glutathion (Ox.) in vitro synthetisiert und dabei radioaktiv markiert. Allen Ansätzen wurden Tat -INVs
zugesetzt. Nach 30 min wurden 15 µl jedes Ansatzes mit TCA gefällt, während 30 µl zuerst mit Proteinase K
verdaut wurden (Vorläuferprotein: p, reifes Protein: m, Intermediat: i). Sämtliche Proben wurden durch SDSPAGE (10%iges Gel) aufgetrennt und die radioaktiv markierten Proteine mittels Autoradiographie sichtbar
gemacht.
Zusammenfassend lässt sich über die Etablierung des in vitro Transportes von TorA-PhoA
sagen, dass in Gegenwart von 5 mM GSSG ein vollständig oxidiertes, gefaltetes TorA-PhoAProtein synthetisiert und in Gegenwart von Tat+-INVs Tat-abhängig transportiert wurde. Im
Transportnachweis durch Proteinase K erschien für das oxidierte TorA-PhoA neben dem
63
IV. Ergebnisse
pTorA-PhoA und dem mTorA-PhoA eine weitere Tat-abhängige Proteinase K-resistente
Spezies, iTorA-PhoA, die auch in Gegenwart von Triton X100 proteaseresistent blieb.
Reduziertes TorA-PhoA dagegen wurde nicht transportiert, was den Schluss zuließ, dass
das Tat-System zwischen den beiden Redoxzuständen des TorA-PhoA unterscheiden
konnte. Somit konnten die Daten von DeLisa et al. zum selektiven Tat-abhängigen in vivoTransport der oxidierten Form von TorA-PhoA auch in vitro bestätigt werden.
IV.2. Charakterisierung der Membranbindung von TorA-PhoA
Nach der erfolgreichen Etablierung des Tat-abhängigen in vitro-Transports von TorA-PhoA
sollten in den folgenden Experimenten Interaktionen des reduzierten, ungefalteten und des
oxidierten, gefalteten TorA-PhoA-Proteins mit den TatABC-Proteinen näher charakterisiert
werden. Hierzu wurde zum einen die Bindung des Proteins an die Membran untersucht und
zum anderen durch ortsgerichtete Quervernetzungsversuche molekulare Interaktionen des
TorA-PhoA-Proteins zu den Tat-Proteinen analysiert.
IV.2.1. Etablierung eines Nachweises für die Bindung von TorA-PhoA an die
INV-Membran
Um die Bindung von TorA-PhoA an die Membranen invertierter Innenmembranvesikel (INV)
zu analysieren, wurde ein Versuchsansatz etabliert, in dem die eingesetzten INVs
größenausschlusschromatographisch
von
den
restlichen
Komponenten
eines
Transportversuchs getrennt wurden. Neben dem transportkompetenten oxidiertem TorAPhoA (Ox. TorA-PhoA) wurde wiederum als Kontrolle das natürliche Tat-Substrat SufI
verwendet.
SufI bzw. oxidiertes TorA-PhoA wurden in vitro synthetisiert und dabei radioaktiv markiert.
Fünf Minuten nach Synthesestart wurden Tat+-INVs bzw. INV-Puffer zugesetzt. Nach
weiteren 25 Minuten wurde die Synthese durch das Antibiotikum Puromycin gestoppt und
wie in den vorhergehenden Versuchen ein Teil des Transportansatzes mit 5 %
Trichloressigsäure gefällt (TCA), während ein weiterer Teil mit Proteinase K (PK) behandelt
wurde, um Transportaktivität nachzuweisen. Der größte Teil eines solchen Ansatzes jedoch
wurde zunächst mit 16000 xg, 20 min bei 4°C zentrifugiert, um mögliche Aggregate des in
vitro-synthetisierten Proteins zu sedimentieren. Ein Teil des Überstandes (10 µl) wurde mit
5 % Trichloressigsäure gefällt (10%), während das restliche Volumen (100 µl) auf eine
64
IV. Ergebnisse
äquilibrierte Gelfiltrationssäule appliziert wurde. Die anschließende Elution erfolgte in 100 µlFraktionen und das Elutionsprofil wurde durch SDS-Gelelektrophorese analysiert.
A
SufI
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
10% TCA PK
p
-INV
B
p
m
p
m
+Tat+-INV
Ox. TorA-PhoA
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
10% TCA PK
p
-INV
p
m
+Tat+-INV
C
Ox. TorA-PhoA
4
+PK
p
m
i
5
m
i
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
TCA
PK
p
m
i
Abb. IV.7: Größenchromatographische Isolation der INVs eines in vitro-Transportansatzes zeigt die
Assoziation des oxidierten, transportkompetenten TorA-PhoA an Tat+-INVs.
A und B: SufI (A) bzw. TorA-PhoA (B) wurden in je einem 175 µl Ansatz in vitro synthetisiert und dabei
radioaktiv markiert. Proteinsynthese fand entweder in Gegenwart von 5 mM oxidiertem Glutathion (Ox. TorA+
+
PhoA) oder in dessen Abwesenheit (SufI) statt. Den Ansätzen Tat -INVs (+Tat -INV) oder entsprechendes
Volumen INV-Puffer (-INV) zugesetzt. Nach 30 min wurde die Proteinsynthese durch Puromycin gestoppt und
anschließend 15 µl jedes Ansatzes mit TCA gefällt (TCA), während 30 µl zuerst mit Proteinase K (PK) verdaut
wurden (Vorläuferprotein: p, reifes Protein: m, Intermediat: i). Der restliche Ansatz wurde mit 16000 xg, 20 min
bei 4°C zentrifugiert. 10 µl des Überstandes wurden mit TCA gefällt (10%) während 100 µl auf eine
Gelfiltrationssäule (Sepharose CL-6b) aufgetragen wurden. Die Elution der Proteine erfolgte in 100 µl Fraktionen
(4-20), die mit 5 % TCA gefällt wurden. Da die INVs in den Fraktion 6-8 (rot) eluierten, sind sowohl
Vorläuferprotein (p) als auch reifes Protein (m) in diesen Fraktionen zu finden.
C: Die Fraktionen 4-20 (je 100 µl) der Gelfiltration eines Synthese-Ansatzes von oxidiertem TorA-PhoA (wie in A
beschrieben) wurden 25 min bei 25°C mit Proteinase K (PK, 0,5 mg/ml) inkubiert und anschließend mit 5 % TCA
gefällt. Proteine, die durch Transport in die Vesikel gelangen sind der PK nicht zugänglich und erlangen PKResistenz (reifes Protein: m, Intermediat: i). Sämtliche Proben aus A-C wurden durch SDS-PAGE (15%iges Gel)
aufgetrennt und die radioaktiv markierten Proteine mittels Autoradiographie sichtbar gemacht.
Abb. IV.7A zeigt das Elutionsprofil eines SufI-Transportansatzes in Abwesenheit von INVs (INV) mit den Fraktionen 4-20. Aus dem Profil geht hervor, dass der Großteil des radioaktiv
markierten SufI-Proteins in den Fraktionen 10-15 eluierte. Transportaktivität wurde wie
erwartet nicht beobachtet (PK). Erst in Gegenwart von Tat+-INV konnte proteaseresistentes
Vorläuferprotein (p) als auch reifes, prozessiertes SufI (m) in der Proteinase K-behandelten
Probe und somit Transport nachgewiesen werden (Abb. IV.7A, +Tat+-INV, PK). Außerdem ist
durch die Gegenwart der Tat+-INVs das Elutionsprofil deutlich verändert, da signifikante
Mengen des radioaktiven pSufI in den Fraktionen 6-8 eluierten (Abb. IV.7A, +Tat+-INV, 4–
20). Ausschließlich diese Fraktionen enthielten darüber hinaus das reife, prozessierte
65
IV. Ergebnisse
Protein, das sich durch Tat-abhängigen Transport im Innern der INVs befinden musste. Aus
diesem Grund konnten die Fraktionen 6-8 als INV-haltige Fraktionen identifiziert werden (rotmarkierte Fraktionen). Die Tatsache, dass in einem Tat+-INV-haltigen Transport eine
signifikante Menge des Vorläufers von SufI mit den INVs coeluierte ließ den Schluss zu,
dass dieses Material zwar nicht transportiert wurde, aber anscheinend an die Membran
assoziiert vorlag.
In gleicher Weise wie mit SufI wurde mit dem transportkompetenten, oxidierten TorA-PhoA
(Ox. TorA-PhoA) verfahren (Abb. IV.7B). In Abwesenheit von Membranen (Abb. IV.7B, -INV)
fand sich kein proteaseresistentes Material (Abb. IV.7B, -INV, PK). Das Elutionsprofil des
radioaktiv markierten TorA-PhoA-Vorläuferproteins (p) war praktisch identisch mit dem von
pSufI im vergleichbaren Versuchsansatz, da wiederum der Großteil des pTorA-PhoAs in den
Die Anwesenheit von Tat+-INVs
Fraktionen 10-15 eluierte (Abb. IV.7B, -INV, 4-20).
ermöglichte den Transport von TorA-PhoA. Sichtbar wurde dies durch die Detektion des
prozessierten, reifen mTorA-PhoA und des intermediären iTorA-PhoA in der Proteinase Kbehandelten
Probe
(Abb. IV.7B, +Tat+-INV, PK).
Außerdem
veränderte
sich
das
Elutionsprofil dahingehend, dass nun signifikante Mengen des pTorA-PhoAs in den
Fraktionen 6–8 eluierten (Abb. IV.7B, +Tat+-INV, 4-20). Diese Fraktionen wurden in den oben
beschriebenen Elutionsprofilen mit pSufI als die INV-haltigen Fraktionen definiert, da sie das
prozessierte, reife mSufI enthielten. Im Fall von TorA-PhoA ist ebenfalls das prozessierte,
reife mTorA-PhoA in diesen Fraktionen zu finden, welches Tat-abhängig in die INVs
transportiert wurde. Allerdings ist radioaktiv markiertes Protein derselben Größe auch in den
mittleren, INV-freien Fraktionen zu finden. Dies ist auf die im ersten Teil dieser Arbeit
erläuterte Pseudoprozessierung zurückzuführen (siehe Erläuterung zu Abb. IV.4B). Protein,
das aus einer Prozessierung des pTorA-PhoA durch die Signalpeptidase stammt, sollte sich
von dem Protein, das aus einer Pseudoprozessierung stammt durch seine Resistenz
gegenüber einer Proteinase K-Behandlung unterscheiden lassen. Aus diesem Grund wurden
sämtliche Fraktionen der Elution einer Gelfiltration eines Transportansatzes mit oxidiertem
TorA-PhoA in Anwesenheit von Tat+-INVs mit Proteinase K behandelt (Abb. IV.7C, +PK), mit
dem Resultat, dass tatsächlich nur in den Fraktionen 6-8 Proteinase K-resistentes mTorAPhoA und iTorA-PhoA zu finden war (Abb. IV.7C, +PK, 4-20). Nur diese Fraktionen
enthielten INVs, die als Folge des Transports und anschließender Prozessierung durch die
Signalpeptidase mTorA-PhoA in ihrem Inneren enthielten.
Somit ist es gelungen eine Methode zu etablieren, um die Assoziation von TorA-PhoA an die
Membran unter Transportbedingungen nachzuweisen. Wie bereits bei SufI beobachtet
coeluiert eine signifikante Menge des oxidierten TorA-PhoAs mit den INVs in den Fraktionen
6-8 (Abb. IV.7A und B, +Tat+-INV). Oxidiertes pTorA-PhoA ist wie pSufI an die INVs
assoziiert.
66
IV. Ergebnisse
IV.2.2. Transportkompetentes, oxidiertes TorA-PhoA assoziiert Tat-spezifisch
mit der Membran
In den folgenden Experimenten sollte der Frage nachgegangen werden, ob die Assoziation
von oxidiertem TorA-PhoA mit der Membran Tat-abhängig erfolgte, oder ob es sich hierbei
um unspezifische Bindung handelte. Diese Frage ist von besonderem Interesse, da es
Hinweise für unspezifische Insertion der Tat-Signalsequenz von Tat-Substraten in die LipidDoppelschicht der Membran gibt, die dem Kontakt der Signalsequenz mit dem TatBCRezeptor-Komplex vorausgehen könnte (Hou et al., 2006; Shanmugham et al., 2006).
A
Ox. TorA-PhoA
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
10 % TCA PK
p
-INV
+Tat+- INV
20
p
m
i
p
m
p
+∆Tat- INV
B
25
Tat+-INV
20
delta Tat-INV
-INV
%
15
10
5
0
0
1
2
3
4
5
6
7
8
9 10 11 12 13 14 15 16 17 18 19 20 21
Fraktion
membranhaltige Fraktionen
Abb. IV.8: Oxidiertes, transportkompetentes TorA-PhoA bindet Tat-spezifisch an die Membran.
A: TorA-PhoA wurde in einem 175 µl Ansatz in vitro synthetisiert und dabei radioaktiv markiert. Proteinsynthese
+
fand in Gegenwart von 5 mM oxidiertem Glutathion (Ox. TorA-PhoA) statt. Den Ansätzen wurden Tat -INVs
+
(+Tat -INV), INV-Puffer (-INV) oder ∆Tat-INVs (+∆Tat-INV) zugesetzt. Nach 30 min wurde die Proteinsynthese
durch Puromycin gestoppt und anschließend 15 µl jedes Ansatzes mit TCA gefällt (TCA), während 30 µl zuerst
mit Proteinase K (PK) verdaut wurden (Vorläuferprotein: p, reifes Protein: m, Intermediat: i). Der restliche Ansatz
wurde mit 16000 xg, 20 min bei 4°C zentrifugiert. 10 µl des Überstandes wurden mit 5 % TCA gefällt (10%)
während 100 µl auf eine Gelfiltrationssäule (Sepharose CL-6b) aufgetragen wurden. Die Elution der Proteine
erfolgte in 100 µl Fraktionen (4-20), die mit 5 % TCA gefällt wurden. Fraktionen 6-8 (rot) sind die
membranhaltigen Fraktionen. Sämtliche Proben wurden durch SDS-PAGE (15%iges Gel) aufgetrennt und das
radioaktiv markierte Protein mittels Autoradiographie sichtbar gemacht.
B: Graphische Darstellung des TorA-PhoA-Elutionsprofils der in A gezeigten Versuchsansätze. Das Rechteck
markiert die membranhaltigen Fraktionen 6-8. 100 % entsprechen der Summe des quantifizierten Proteins aus
allen Fraktionen. Die Quantifizierung erfolgte mit dem Programm ImageQuant 5.2
Hierzu wurde oxidiertes TorA-PhoA in in vitro-Ansätzen synthetisiert, denen entweder keine
INVs (-INV), Tat+-INVs (+Tat+-INV) oder ∆Tat-INVs (+∆Tat-INV) zugesetzt wurden
(Abb. IV.8). Diese Ansätze dienten, wie oben beschrieben, als Ausgangsmaterial für eine
67
IV. Ergebnisse
Auftrennung nach Größe durch Gelfiltration. Ansätze mit oxidiertem TorA-PhoA, die keine
INVs bzw. Tat+-INVs enthielten, zeigten die Elutionsprofile wie in Abb. IV.7B bereits
beschrieben: Oxidiertes TorA-PhoA aus dem Ansatz ohne INVs eluiert überwiegend in den
Fraktionen 10-15 (Abb. IV.8A, -INV, 4-20). Nur in Anwesenheit von Tat+-INVs eluiert eine
signifikante Menge des Proteins in den INV-haltigen Fraktionen 6-8 (Abb. IV.8A, +Tat+INVs, 4-20). Wurde dagegen ein Ansatz der ∆Tat-INVs enthielt analysiert, war kein oxidiertes
TorA-PhoA-Protein in den INV-haltigen Fraktionen 6–8 zu finden. Vielmehr eluierte der
größte Teil des Proteins in den Fraktionen 10–15 (Abb. IV.8A, +∆Tat-INV, 4–20). Der Ansatz
ohne INVs und der Ansatz mit ∆Tat-INVs wiesen somit praktisch identische Elutionsprofile
auf. Transportaktivität in Gegenwart der ∆Tat-INVs war wie erwartet nicht zu erkennen
(Abb. IV.8A, +∆Tat-INV, PK).
Die hier beschriebenen Experimente wurden zwei- bis dreimal unabhängig voneinander
wiederholt und quantitativ mit dem Programm ImageQuant 5.2 ausgewertet, um die
Elutionsprofile graphisch darzustellen (Abb. IV.8B). Daraus wurde klar ersichtlich, dass nur in
Gegenwart der TatABC-Proteine ca. 38 % des auf die Gelfiltrationssäule aufgetragenen,
oxidierten TorA-PhoA in den INV-haltigen Fraktionen 6–8 eluierten (Abb. IV.8B, blaue Linie).
In Abwesenheit von INVs (Abb. IV.8B, schwarze Linie) oder in Anwesenheit von INVs eines
TatABC-deletierten E. coli-Stamms (Abb. IV.8B, grüne Linie) eluierte weniger als 1 % des
TorA-PhoA in den Fraktionen 6–8.
Somit stellt die Koelution des oxidierten TorA-PhoA mit den Tat+-INVs eine Tat-spezifische
Assoziation des Proteins mit der Membran dar.
IV.2.3. Auch transportinkompetentes, reduziertes TorA-PhoA assoziiert
Tat-spezifisch mit der Membran
Das reduzierte TorA-PhoA wurde, im Gegensatz zur oxidierten Form, nicht in vitrotransportiert,
erlangte
also
keine
Transportkompetenz
für
das
Tat-System
(Abb. IV.2B und 4A). Da bislang die Tat-Spezifität der Membranassoziation von oxidiertem
TorA-PhoA
bewiesen
wurde
(Abb. IV.8)
sollte
im
Folgenden
einer
möglichen
Membranassoziation des reduzierten TorA-PhoA nachgegangen werden. Ist auch das
transportinkompetente, reduzierte TorA-PhoA in der Lage Tat-spezifisch zu binden?
Dazu wurde reduziertes TorA-PhoA in in vitro-Ansätzen synthetisiert, denen entweder keine
INVs (-INV), Tat+-INVs (+Tat+-INV) oder ∆Tat-INVs (+∆Tat-INV) zugesetzt wurden. Diese
Ansätze dienten wiederum als Ausgangsmaterial für eine Auftrennung nach Größe durch
Gelfiltration. Abb. IV.9A zeigt zum Vergleich zunächst das Elutionsprofil des oxidierten TorAPhoAs in Gegenwart von Tat+-INVs (Abb. IV.9A, +Tat+-INV, 4-20).
68
IV. Ergebnisse
A
Ox. TorA-PhoA
4
5
7
8
9
10
11
12
13
14
15
16
17
18
19
20
TCA
PK
p
m
+Tat+-INV
B
6
p
m
i
Red. TorA-PhoA
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
10% TCA PK
p
+Tat+-INV
p
- INV
p
+∆Tat INV
C
Membranbindung TorA-PhoA
25
ox. TorA-PhoA,Tat+-INV
red.TorA-PhoA, Tat+-INV
20
red. TorA-PhoA, deltaTat-INV
red TorA-PhoA, -INV
%
15
10
5
0
0
1
2
3
4
5
6
7
8
9 10 11 12 13 14 15 16 17 18 19 20 21
Fraktion
membranhaltige Fraktionen
D
Red. KK-TorA-PhoA
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
10% TCA PK
p
+Tat+-INV
Abb. IV.9: Reduziertes, transportinkompetentes TorA-PhoA bindet im selben Maße wie das oxidierte
transportkompetente TorA-PhoA Tat-spezifisch an die Membran. Die Bindung erfordert aber kein intaktes
RR-Motif
A und B: TorA-PhoA wurde in einem 175 µl Ansatz in vitro synthetisiert und dabei radioaktiv markiert.
Proteinsynthese fand in Gegenwart von 5 mM oxidiertem Glutathion (Ox. TorA-PhoA) oder in dessen
Abwesenheit (Red. TorA-PhoA) statt. Den Ansätzen wurden Tat+-INVs (+Tat+-INV), INV-Puffer (-INV) oder ∆TatINVs (+∆Tat-INV) zugesetzt. Nach 30 min wurde die Proteinsynthese durch Puromycin gestoppt und
anschließend 15 µl jedes Ansatzes mit TCA gefällt (TCA), während 30 µl zuerst mit Proteinase K (PK) verdaut
wurden (Vorläuferprotein: p, reifes Protein: m, Intermediat: i). Der restliche Ansatz wurde mit 16000 g, 20 min bei
4°C zentrifugiert. 10 µl des Überstandes wurden mit TCA gefällt (10%) während 100 µl auf eine
Gelfiltrationssäule (Sepharose CL-6B) aufgetragen wurden. Die Elution der Proteine erfolgte in 100 µl Fraktionen
(4-20), die mit TCA gefällt wurden. Fraktionen 6-8 (rot) sind die membranhaltigen Fraktionen. Sämtliche Proben
wurden durch SDS-PAGE (15%iges Gel) aufgetrennt und das radioaktiv markierte Protein mittels
Autoradiographie sichtbar gemacht.
C: Graphische Darstellung des TorA-PhoA-Elutionsprofils der in A und B gezeigten Versuchsansätze. Das
Rechteck markiert die membranhaltigen Fraktionen 6-8. 100 % entsprechen der Summe des quantifizierten
Proteins aus allen Fraktionen. Die Quantifizierung erfolgte mit dem Programm ImageQuant 5.2.
D: Red. KK-TorA-PhoA wurde wie in B beschrieben synthetisiert und durch anschließende Gelfiltration analysiert.
Sämtliche Proben wurden durch SDS-PAGE (15%iges Gel) aufgetrennt und das radioaktiv markierte Protein
mittels Autoradiographie sichtbar gemacht.
69
IV. Ergebnisse
In Gegenwart von Tat+-INVs entspricht das Elutionsprofil des reduzierten TorA-PhoA im
Prinzip dem des oxidierten Proteins, da auch in diesem Fall eine bedeutende Menge von
TorA-PhoA in den INV-haltigen Fraktionen 6-8 eluierte (Abb. IV.9B, +Tat+-INV, 4–20). Da
kein Transport stattfand (Abb. IV.9B, +Tat+-INV, PK), war auch in den INV-haltigen
Fraktionen 6-8 kein mTorA-PhoA zu finden (Abb. IV.9B, +Tat+-INV, 4-20). Signifikante
Mengen des reduzierten TorA-PhoA eluierten nicht in Fraktionen 6–8, wenn keine INVs
zugesetzt (Abb. IV.9B, -INV, 4–20), oder wenn INVs aus einem TatABC-deletierten E. coliStamm zugesetzt worden waren (Abb. IV.9B, +∆Tat-INV, 4–20). In beiden Fällen eluierte der
Großteil des reduzierten Proteins in den Fraktionen 10–15. Proteinase K-resistentes Material
war in keinem der beiden Fälle zu beobachten (Abb. IV.9B, -INV, PK oder +∆Tat-INV, PK).
Sämtliche Versuche mit dem reduzierten TorA-PhoA wurden wieder drei- bis fünfmal
unabhängig voneinander wiederholt und quantitativ mit dem Programm ImageQuant 5.2
ausgewertet, um die Elutionsprofile graphisch darzustellen (Abb. IV.9C). Daraus wurde
deutlich, dass das reduzierte TorA-PhoA im vergleichbaren Maße (ca. 45 %, grüne Linie) wie
das oxidierte TorA-PhoA (ca. 38%, blaue Linie) zusammen mit den INVs in den Fraktionen
6–8 eluierte, während in Abwesenheit von INVs (schwarze Linie) oder in Abwesenheit der
TatABC-Proteine in der Membran der INVs (rote Linie) nur 9 % bzw. 12 % in den INVhaltigen Fraktionen 6–8 zu finden war. Dies bedeutet, dass das reduzierte TorA-PhoA im
selben Maße an die Membran assozierte wie das oxidierte Protein, obwohl es sich dabei um
die transportinkompetente Form handelt. Diese Assoziation war auch im Fall des reduzierten
TorA-PhoAs Tat-spezifisch. Somit besteht hinsichtlich der Tat-spezifischen Membranbindung
kein
erkennbarer
Unterschied
zwischen
der
transportkompetenten
und
transportinkompetenten Form des Proteins.
In einem abschließenden Versuch wurde die Membranassoziation von KK-TorA-PhoA
untersucht (Abb. IV.9D). Bei dieser Version des Proteins handelte es sich um eine Mutante,
deren Doppel-Arginin-Motif durch zwei Lysine ersetzt wurde, wodurch der Transport auch für
das oxidierte Protein vollständig inhibiert war (Abb. IV.6).
Obwohl der Tat-abhängige
Transport durch die Mutation in der Signalsequenz des Proteins zu einer Inhibition der
Transports führt, ist die Bindung von TorA-PhoA in seiner reduzierten Form an die Membran
überraschenderweise nicht beeinträchtigt, da wiederum eine signifikante Menge des Proteins
mit den Tat+-INVs in den Fraktionen 6–8 eluierte (Abb. IV.9D).
70
IV. Ergebnisse
IV.3. Charakterisierung molekularer Interaktionen von TorA-PhoA mit den
Tat-Proteinen
Bislang wurde eine Tat-spezifische Assoziation des TorA-PhoA-Proteins an eine TatABChaltige Membran bewiesen. Dabei war diese Bindung unabhängig von dem Redox- bzw.
Faltungszustand des Proteins. Da das Tat-System selektiv nur das oxidierte, gefaltete TorAPhoA transportierte, sollte nach Unterschieden der beiden Faltungszustände des Proteins in
deren Interaktion mit den Tat-Proteinen gesucht werden. In Quervernetzungsexperimenten
mit dem natürlichen Tat-Substrate SufI konnten definierte Kontakte zwischen der
Signalsequenz und den Tat-Proteinen in vitro nachgewiesen werden (Alami et al. 2003). Aus
diesem Grund wurde auch in dieser Arbeit versucht, Kontakte dieser Art nachzuweisen.
IV.3.1. Zielgerichteter Einbau des photoaktivierbaren Quervernetzers pBPA
in die Signalsequenz von TorA-PhoA
Um Interaktionen des TorA-PhoA-Proteins mit den Tat-Proteinen zu untersuchen, wurde eine
neue Methode etabliert, durch die der photoaktivierbare Quervernetzer p-Benzoyl-LPhenylalanin (pBPA) ortspezifisch in die Signalsequenz eingebaut werden konnte. pBPA ist
ein Phenylalaninderivat, das in
OH
O
OH
O
seiner Hydrophobizität und der
NH 2
Größe seiner Seitenkette dem
OH
O
NH 2
80-120µs
NH 2
Tryptophan
O
Durch
Bestrahlung mit UV-Licht findet
hν
~ 350 nm
O
ähnelt.
C
eine
O
R3
R2
der
Carbonylgruppe der Seitenkette
OH
O
H
Umwandlung
zu einem hochreaktiven Biradikal
NH 2
R1
statt, das bevorzugt mit C-HHO
R1
R2
R3
Abb. IV.10: Reaktionsmechanismus des Quervernetzers pBPA
Bindungen reagiert und dabei
kovalente
Bindungen
benachbarten
mit
Molekülen
ausbildet. Findet sich kein geeigneter Reaktionspartner in der unmittelbaren Nähe des
Biradikals, fällt es zurück in seinen Grundzustand (Abb. IV.10) [Kauer et al., 1986].
Abb. IV.11 illustriert den ortspezifischen Einbau des Quervernetzers in das in vitrosynthetisierte Protein. Um den Einbau von pBPA zu ermöglichen, war es zunächst nötig, das
Codon einer Aminosäure bestimmter Position mittels mutierender PCR in ein amber-StopCodon (TAG) umzuwandeln.
71
IV. Ergebnisse
A
C
C
amberSuppressortRNA
pBPA
pBPA
TetR
AUC
Mutierte
Tyrosyl-tRNASynthetase
pDULE
3´
Aminosäure-Codon
ACG
A
C
C
amber-Stop-Codon
TAG
Plasmid
AUC
TAG
5´
mutiertes Protein
mit ortsspezifisch
inseriertem pBPA
mRNA
Plasmid
Abb. IV.11: Ortsspezifische Insertion von pBPA in Proteine
Das Stop-Codon würde bei einer in vitro-Synthese zum vorzeitigen Abbruch der Translation
führen. Doch durch die Anwesenheit einer pBPA-amber-Suppressor-tRNA während der in
vitro-Synthese des mutierten Proteins wird ein Abbruch verhindert und ein Protein
synthetisiert, dem ortsgerichtet ein pBPA-Molekül inkorporiert wurde. Die pBPA-amberSuppressor-tRNA wird erst nach Zugabe von pBPA zum in vitro-Proteinsynthese-Ansatz
durch eine mutierte Tyrosyl-tRNA-Synthetase aus Methanococcus Jannaschii beladen.
Dabei handelt es sich um eine hochspezifische Reaktion, da die Synthetase hierfür keine der
endogenen E. coli-tRNAs und keine der 20 proteinogenen Aminosäuren, sondern nur pBPA
und die amber-Suppressor-tRNA als Substrat akzeptiert [Chin et al., 2002]. Sowohl die
amber-Suppressor-tRNA als auch die mutierte Tyrosyl-tRNA-Synthetase sind auf einem
Plasmid (pDULE) codiert, das in den E. coli-Stamm transformiert worden war, aus dem der
für die in vitro-Synthese verwendeten Zellextrakt (S135) präpariert wurde [Farrell et al.,
2005]. Da die Tyrosyl-tRNA-Synthetase und die amber-Suppressor-tRNA unter der Kontrolle
eines endogenen E. coli-Promotors in vivo exprimiert wurden, sind diese somit bereits im
Zellextrakt vorhanden, so dass für eine Suppression des amber-Stop-Codons in vitro nur die
Zugabe von pBPA notwendig ist.
Die Positionen für den Einbau von pBPA in die Signalsequenz von TorA-PhoA ergaben sich
daraus, dass möglichst hydrophobe Aminosäuren gegen den Quervernetzer ausgetauscht
werden sollten. Es wurden ein Phenylalanin auf Position 8 direkt vor dem Konsensusmotif,
ein weiteres Phenylalanin an Position 15 in direkter Nachbarschaft zum Doppel-Arginin, ein
Valin an Position 24 in der hydrophoben Region und ein Leucin an Position 32 im Cterminalen Bereich der Signalsequenz ausgetauscht (Abb. IV.12).
72
IV. Ergebnisse
Signalsequenz TorA
mTorA
MANNNDLFQASRRRFLAQLGGLTVAGMLGPSLLTPRRATAAQAATDA
TorA-PhoA
TorA ◄ ► reifes PhoA
Abb. IV.12: Schematische Darstellung der Stop-Codon-Mutanten
Der TorA-Anteil am Tat-abhängigen Fusionsprotein TorA-PhoA bestehend aus der TorA-Signalsequenz mit blau
hervorgehobenem Konsensusmotif sowie den ersten 7 Aminosäuren des reifen TorA (mTorA). Rot sind die
Aminosäurereste markiert, deren Codon durch ein amber-Stop-Codon ersetzt wurde, um den Einbau von pBPA
an diesen Positionen zu ermöglichen.
Alle Stop-Codon-Mutanten zeigten vergleichbare Transportaktivitäten (bezogen auf mTorAPhoA-Mengen) wie das nicht mutierte TorA-PhoA (nicht gezeigt). Membranabhängige
Quervernetzungsprodukte allerdings wurden nur für zwei der vier Stop-Codon-Mutanten
beobachtet, auf die im Folgenden näher eingegangen wird (F15-TorA-PhoA und V24-TorAPhoA).
IV.3.2. Reduziertes TorA-PhoA zeigt eine stark abgeschwächte Interaktion der
Signalsequenz mit TatC
Aus
früheren
in
vitro-Experimenten
mit
einem
vergleichbaren,
photoaktivierbaren
Quervernetzer (Tmd-Phe: Trifluoromethyldiazerin-Phenylalanin) und dem natürlichen TatSubstrat SufI aus E. coli war bekannt, dass die Region um das Konsensusmotif mit TatC
interagiert [Alami et al., 2003].
Um zu überprüfen, ob Interaktion auch zwischen dem Konsensusmotif der Signalsequenz
des reduzierten bzw. oxidierten TorA-PhoAs mit den Tat-Proteinen stattfindet, wurde unter
Verwendung der Stop-Codon-Mutante F15-TorA-PhoA im folgenden Versuch pBPA anstelle
des Phenylalanins an Position 15 der Signalsequenz innerhalb des Konsensusmotifs
eingebaut (Abb. IV.13A).
Reduziertes bzw. oxidiertes F15-TorA-PhoA wurde in vitro in Gegenwart von pBPA
synthetisiert (Abb. IV.13B). Zur Aktivierung des Quervernetzers wurde mit UV-Licht bestrahlt,
wobei
immer
eine
Probe
als
Negativkontrolle
unbestrahlt
blieb
(Abb. IV.13B, Spuren 1 und 6). Durch Bestrahlung traten mehrere hochmolekulare Banden
auf. In Proben, denen keine Tat+-INVs zugesetzt worden waren, erschien nach UVBestrahlung eine Gruppe von Quervernetzungsprodukten mit molekularen Größen von
ca. 75–85 kDa. Diese Produkte traten unabhängig vom Redoxzustand des TorA-PhoAs auf
(Abb. IV.13B, Spuren 2 und 7). Unter Berücksichtigung des Molekulargewichts von TorAPhoA mit ca. 55 kDa resultiert daraus ein theoretisches Molekulargewicht der
Quervernetzungspartner von ca. 20–30 kDa. Da diesen Ansätzen keine INVs zugesetzt
73
IV. Ergebnisse
waren,
handelte
es
sich
um
lösliche,
nicht-membranabhängige
Faktoren.
Diese
+
Quervernetzungsprodukte waren nach Zugabe von Tat -INVs sowohl im Fall des reduzierten
als auch des oxidierten TorA-PhoAs deutlich reduziert (Abb.IV.13B, Spuren 3 und 8).
Zugleich
erschien
im
Fall
des
oxidierten
Proteins
Quervernetzungsprodukt bei ca. 70 kDa (Abb. IV.13B, Spur 8).
produkt
war
in
der
vergleichbaren
Probe
des
ein
neues
Dieses
reduzierten
intensives
QuervernetzungsTorA-PhoAs
nicht
zu erkennen (Abb.IV.13B, Spur 3).
A
Signalsequenz TorA
mTorA
MANNNDLFQASRRRFLAQLGGLTVAGMLGPSLLTPRRATAAQAATDA
TorA-PhoA
TorA ◄ ► reifes PhoA
B
Red. F15-TorA-PhoA
Ox. F15-TorA-PhoA
INV
-
-
+
-
-
+
UV
-
+
+
-
+
+
IP
α
TatB
α
TatC
α
TatB
α
TatC
100 kDa
75 kDa
*
pF15-TorA-PhoA ►
25 µl Ansatz
1
2
3
4
5
6
7
8
9
10
2x
2x
2x
8x
8x
2x
2x
2x
8x
8x
Abb. IV.13: Die Signalsequenz des oxidierten TorA-PhoA interagiert mit TatC.
A: Der TorA-Anteil am Tat-abhängigen Fusionsprotein TorA-PhoA bestehend aus der TorA-Signalsequenz mit blau
hervorgehobenem Konsensusmotif sowie den ersten 7 Aminosäuren des reifen TorA (mTorA). Rot markiert an
Position 15 befindet sich das Phenylalanin, dessen Codon durch ein amber-Stop-Codon ersetzt wurde, um den
Einbau von pBPA an diesen Positionen zu ermöglichen (F15-TorA-PhoA).
B: F15-TorA-PhoA wurde in einem 25 x Ansatz in Gegenwart von pBPA synthetisiert und dabei radioaktiv markiert.
25 µl entsprechen einem Ansatz. Der verwendete Zellextrakt stammt von einem mit dem pDULE-Plasmid
transformierten E. coli-Stamm und enthält daher die für die Suppression nötigen Komponenten. Proteinsynthese
fand in Gegenwart von 5 mM oxidiertem Glutathion (Ox. F15-TorA-PhoA) oder in dessen Abwesenheit statt (Red.
F15-TorA-PhoA). Nach 30 min wurde die Synthese durch Puromycin gestoppt und der 25 x Ansatz aufgeteilt auf je
zwei 2x Ansätze (1,2,6,7) und je ein 18 x Ansatz (3,8). Letzteren wurden Tat+-INVs zugesetzt, allen anderen
Ansätzen das gleiche Volumen INV-Puffer. Nach einer Inkubation von 15 min bei 37°C wurden Ansätze 1 und 6 mit
TCA gefällt, während die restlichen Ansätze 20 min auf Eis mit UV-Licht bestrahlt wurden um den eingebauten
Quervernetzer zu aktivieren. Anschließend wurden Ansätze 2 und 7 mit TCA gefällt. Ansätze 3 und 8 wurden
aufgeteilt um je 50 µl mit TCA zu fällen, während der restliche Ansatz in gleichen Teilen (je 8 x) für
Immunpräzipitationen verwendet wurde. Alle TCA-Pellets, sowie die für die Immunpräzipitation verwendete Protein
A-Sepharose wurden in SDS-PAGE-Probenpuffer bei 56°C und 1400 rpm für 15 min in einem Thermomixer
inkubiert. Sämtliche Proben wurden durch SDS-PAGE (8%iges Gel) aufgetrennt und die radioaktiv markierten
Proteine mittels Autoradiographie sichtbar gemacht. Rote Klammern oder Sterne markieren die
Quervernetzungsprodukte. Rote Pfeile markieren präzipitiertes Material.
74
IV. Ergebnisse
Durch Immunpräzipitationen mit Antikörpern gegen TatC konnte die Bande bei 70 kDa als
ein Quervernetzungsprodukt zwischen der Signalsequenz des oxidierten TorA-PhoA und
TatC identifiziert werden (Abb. IV.13B, Spur 10). Immunpräzipitation mit dem Antikörper
gegen TatB ergab eine Bande auf gleicher Höhe wie die mit TatC-Antikörpern
(Abb. IV.13B, Spur 9). Dabei handelte es sich höchstwahrscheinlich nicht um ein echtes
Signal, sondern viel mehr um eine Verunreinigung dieser Spur durch die benachbarte Probe
der αTatC-Immunpräzipitation, da aus anderen TatB-Quervernetzungsstudien, sowie aus
Ergebnissen dieser Arbeit bekannt ist, dass TatB-Quervernetzungsprodukte ca. 5 kDa höher
laufen als die von TatC.
Obwohl im Falle des reduzierten TorA-PhoAs kein vergleichbares Quervernetzungsprodukt
erkennbar war, konnte mit Antikörpern gegen TatC eine geringe Menge an radioaktivem
Material immunpräzipitiert werden, das auf derselben Höhe lief wie das Signal der αTatCImmunpräzipitation des oxidierten TorA-PhoAs (Abb. IV.13B, Spur 5). Dies könnte auf eine
sehr schwache Interaktion der Signalsequenz des reduzierten TorA-PhoAs und TatC
hindeuten, die nur in der Immunpräzipitation zu erkennen war, da im Vergleich zur Probe in
Spur 3 die vierfache Menge an Material für die Immunpräzipitation benutzt wurde
(Abb. IV.13B, Spur 5).
Die Ergebnisse der Quervernetzungsexperimente zeigen, dass eine direkte Interaktion der
Region um das Konsensusmotif mit TatC besteht, und dass die Stärke dieser Interaktion sich
zwischen den beiden Redoxzuständen bzw. Faltungszuständen des Proteins deutlich
unterscheidet, wobei die reduzierte Form eine stark abgeschwächte Interaktion aufweist.
Demnach ist die Bindung dieser Region der Signalsequenz abhängig von der Konformation
bzw. vom Redoxzustand des TorA-PhoA-Proteins. Die Tatsache, dass das oxidierte sowie
das reduzierte Protein in gleicher Stärke Tat-spezifisch an die Membran binden (Abb. IV.9C),
lässt den Schluss zu, dass die stark abgeschwächte Interaktion der Region um das
Konsensusmotif des reduzierten TorA-PhoAs mit TatC nicht den primären Kontakt an den
TatBC-Rezeptor-Komplex widerspiegelt.
IV.3.3. Reduziertes TorA-PhoA zeigt eine abgeschwächte Interaktion der
Signalsequenz mit TatB
Aus früheren in vitro-Experimenten mit dem photoaktivierbaren Quervernetzer Tmd-Phe und
dem natürlichen Tat-Substrates SufI aus E. coli war bekannt, dass die hydrophobe Region
(h-Region) der Signalsequenz mit TatC und TatB interagiert [Alami et al., 2003].
Um zu überprüfen, ob Interaktion auch zwischen der h-Region der Signalsequenz des
reduzierten bzw. oxidierten TorA-PhoAs mit den Tat-Proteinen stattfindet, wurde unter
75
IV. Ergebnisse
Verwendung der Stop-Codon-Mutante V24-TorA-PhoA im folgenden Versuch pBPA anstelle
des Valins an Position 24 der Signalsequenz innerhalb des Konsensusmotifs eingebaut
(Abb. IV.14A).
Signalsequenz TorA
A
mTorA
MANNNDLFQASRRRFLAQLGGLTVAGMLGPSLLTPRRATAAQAATDA
TorA-PhoA
TorA ◄ ► reifes PhoA
B
Red. V24-TorA-PhoA
Ox. V24-TorA-PhoA
INV
-
-
+
-
-
+
UV
-
+
+
-
+
+
IP
α
TatB
α
TatC
α
TatB
α
TatC
100 kDa
75 kDa
*
*
pV24-TorA-PhoA ►
25 µl Ansatz
1
2
3
4
5
6
7
8
9
10
2x
2x
2x
8x
8x
2x
2x
2x
8x
8x
Abb. IV.14: Die Signalsequenz des reduzierten V24-TorA-PhoA zeigt eine abgeschwächte Interaktion mit TatB.
A: Der TorA-Anteil am Tat-abhängigen Fusionsprotein TorA-PhoA bestehend aus der TorA-Signalsequenz mit blau
hervorgehobenem Konsensusmotif sowie den ersten 7 Aminosäuren des reifen TorA (mTorA). Rot makiert an Position
24 befindet sich das Valin, dessen Codon durch ein amber-Stop-Codon ersetzt wurde, um den Einbau von pBPA an
diesen Positionen zu ermöglichen (V24-TorA-PhoA).
B: V24-TorA-PhoA wurde in einem 25 x Ansatz in Gegenwart von pBPA synthetisiert und dabei radioaktiv markiert. 25
µl entsprechen einem Ansatz. Der dafür verwendete Zellextrakt stammt von einem mit dem pDULE-Plasmid
transformierten E. coli-Stamm und enthält daher die für die Suppression nötigen Komponenten. Proteinsynthese fand
in Gegenwart von 5 mM oxidiertem Glutathion (Ox. V24-TorA-PhoA) oder in dessen Abwesenheit statt (Red. V24TorA-PhoA). Nach 30 min wurde die Synthese durch Puromycin gestoppt und der 25x Ansatz aufgeteilt auf je zwei 2 x
Ansätze (1,2,6,7) und je ein 18 x Ansatz (3,8). Letzteren wurden Tat+-INVs zugesetzt, allen anderen Ansätzen das
gleiche Volumen INV-Puffer. Nach einer Inkubation von 15 min bei 37°C wurden Ansätze 1 und 6 mit TCA gefällt,
während die restlichen Ansätze 20 min auf Eis mit UV-Licht bestrahlt wurden um den eingebauten Quervernetzer zu
aktivieren. Anschließend wurden Ansätze 2 und 7 mit TCA gefällt. Ansätze 3 und 8 wurden aufgeteilt um je 50 µl mit
TCA zu fällen während der restliche Ansatz in gleichen Teilen (je 8 x) für Immunpräzipitationen verwendet wurde. Alle
TCA-Pellets, sowie die für die Immunpräzipitation verwendete Protein A-Sepharose wurden in SDS-PAGEProbenpuffer bei 56°C und 1400 rpm für 15 min in einem Thermomixer inkubiert. Sämtliche Proben wurden durch
SDS-PAGE (8%iges Gel) aufgetrennt und die radioaktiv markierten Proteine mittels Autoradiographie sichtbar
gemacht. Rote Klammern oder Sterne markieren die Quervernetzungsprodukte. Rote Pfeile markieren präzipitiertes
Material.
76
IV. Ergebnisse
Reduziertes bzw. oxidiertes V24-TorA-PhoA wurde in vitro in Gegenwart von pBPA
synthetisiert, wodurch der Quervernetzer an Position 24 in der hydrophoben Region der
Signalsequenz eingebaut wurde (Abb. IV.14B). Wie im vorherigen Experiment beschrieben,
wurde pBPA durch Bestrahlung mit UV-Licht photoaktiviert (Abb. IV.14B). Dadurch traten im
Vergleich zu unbestrahlten Proben (Abb. IV.14B, Spuren 1 und 6) mehrere hochmolekulare
Banden auf (Abb. IV.14B, Spuren 2 und 7). In Proben, denen keine Tat+-INVs zugesetzt
waren erschien nach UV-Bestrahlung erneut eine Gruppe von Quervernetzungsprodukten
mit molekularen Größen von ca. 75–85 kDa. Diese Produkte traten unabhängig vom
Redoxzustand des TorA-PhoAs auf und wurden bereits für die oben beschriebene StopCodon-Mutante
F15-TorA-PhoA
beobachtet
(Abb. IV.14B,
Spuren 2 und 7).
Diese
Quervernetzungsprodukte waren nach Zugabe von Tat+-INVs sowohl im Fall des reduzierten
als auch des oxidierten TorA-PhoAs deutlich reduziert (Abb. IV.14B, Spuren 3 und 8).
Zugleich
erschien
unabhängig
Quervernetzungsprodukt
bei
75
von
kDa
Redoxzustand
des
TorA-PhoAs
(Abb. IV.14B, Spuren 3 und 8),
das
ein
mit
neues
TatB
Antikörpern immunpräzipitiert werden konnte und daher auf die Interaktion zwischen der
Signalsequenz und TatB zurückzuführen war. (Abb. IV.14B, Spuren 4 und 9). Die durch
TatB-Antikörper immunpräzipitierte Bande des reduzierten TorA-PhoAs war deutlich
schwächer als die des oxidierten TorA-PhoA. Demnach interagiert die Signalsequenz des
reduzierten TorA-PhoA im Vergleich zum oxidierten Protein in einer abgeschwächten Weise
mit TatB.
Dieses Quervernetzungsexperiment verdeutlichte, dass die h-Region der Signalsequenz von
TorA-PhoA mit TatB interagiert, und dass auch diese Interaktion vom Redox- bzw.
Faltungszustand des Proteins beeinflusst war, wobei die reduzierte Form eine weniger starke
Interaktion aufwies. Der Unterschied in den Intensitäten der Interaktion des reduzierten und
oxidierten TorA-PhoAs war jedoch schwächer ausgeprägt als es für die Interaktion der
Region um das Konsensusmotifs (F15-TorA-PhoA) mit TatC zu beobachten war
(Abb. IV.13B).
Einen Kontakt der h-Region zu TatC, wie es für SufI gezeigt wurde [Alami et al., 2003],
konnte nicht nachgewiesen werden. Interaktionen mit TatA konnten weder für die F15-, noch
für die V24-Stop-Codon-Mutante beobachtet werden. Darüber hinaus führte der Einbau von
pBPA an die bereits erwähnten Positionen F8 (vor dem Konsensusmotif) und L31 (Cterminale Region der Signalsequenz) zu keinen Interaktionen mit den Tat-Proteinen (nicht
gezeigt).
77
IV. Ergebnisse
IV.3.4. Die Signalsequenz von TorA-PhoA interagiert mit TorD und FkpA
In den Experimenten in Abb. IV.13B und Abb. IV.14B trat in Proben, denen keine Tat+-INVs
zugesetzt worden waren, nach UV-Bestrahlung eine Gruppe von Quervernetzungsprodukten
mit molekularen Größen von ca. 75–85 kDa auf (jeweils Spuren 2 und 7). Um diese zu
identifizieren, wurde exemplarisch reduziertes F15-TorA-PhoA in Gegenwart von pBPA
synthetisiert und mit UV-Licht bestrahlt, wobei eine Probe als Negativkontrolle unbestrahlt
blieb (Abb. IV.15, Spuren 1 und 2).
Red. F15-TorA-PhoA
INV
UV
-
+
IP
αFkp
A
αTorD
100 kDa
75 kDa
pF15-TorA-PhoA ►
25 µl Ansatz
1
2x
2
2x
3
4x
4
4x
Abb. IV.15: FkpA und TorD interagieren
mit der Signalsequenz von TorA-PhoA.
F15-TorA-PhoA wurden in einem 12 x
Ansatz
in
Gegenwart
von
pBPA
synthetisiert und dabei radioaktiv markiert.
25 µl entsprechen einem Ansatz. Der dafür
verwendete Zellextrakt stammt von einem
mit dem pDULE-Plasmid transformierten
E. coli-Stamm und enthält daher die für die
Suppression nötige amber-SuppressortRNA, sowie die pBPA-spezifische TyrosyltRNA-Synthetase. Nach 30 min wurde die
Synthese durch Puromycin gestoppt und
der 12 x Ansatz in einen 2 x Ansatz sowie
einen 10 x Ansatz aufgeteilt. Der 2 x
Ansatz wurde mit TCA gefällt (1), während
der 10 x Ansatz 20 min mit UV-Licht auf
Eis bestrahlt wurde um den eingebauten
Quervernetzer zu aktivieren. Nach der
Bestrahlung wurden 50 µl des bestrahlten
Ansatzes mit TCA gefällt (2), während der
restliche Ansatz zu gleichen Teilen für die
Immunpräzipitationen verwendet wurde
(3,4). Alle TCA-Pellets, sowie die für die
Immunpräzipitation verwendete Protein ASepharose
wurden
in
SDS-PAGEProbenpuffer bei 56°C und 1400 rpm für
15 min in einem Thermomixer inkubiert.
Sämtliche Proben wurden durch SDSPAGE (8%iges Gel) aufgetrennt und die
radioaktiv markierten Proteine mittels
Autoradiographie sichtbar gemacht. Die
rote
Klammer
markiert
die
Quervernetzungsprodukte. Rote Pfeile
markieren präzipitiertes Material.
Wie erwartet trat diese Gruppe von Quervernetzungsprodukten bei ca. 75–85 kDa auf
(Abb. IV.15, Spur 2).
Durch
Immunpräzipitationen
konnte
diese
Gruppe
von
Quervernetzungsprodukten als Interaktionen der Signalsequenz mit FkpA (27 kDa)
(Abb. IV.15, Spur 3) und mit TorD (22,5 kDa) (Abb. IV.15, Spur 4) identifiziert werden.
Angesichts der Größe des Signals der Quervernetzungsprodukte werden jedoch noch
weitere Interaktionen mit nicht-identifizierten Proteinen vermutet. FkpA ist in dem für die in
vitro-Synthese benutzen Zellextrakt (S135) enthalten. Dabei handelt es sich um eine
periplasmatische PPIase mit Chaperon-Funktion. Als periplasmatisches Protein hat FkpA
78
IV. Ergebnisse
wahrscheinlich keine physiologische Bedeutung für den Tat-abhängigen Transport, dennoch
interagiert es mit der Signalsequenz von TorA-PhoA (Abb. IV.15) als auch mit der von TorA23K, einem anderen Tat-abhängigen Fusionsprotein, sowie mit der Signalsequenz des
natürlichen Tat-Substrates SufI. Diese Interaktion beeinflusst allerdings nicht den Tatabhängigen in vitro-Transport [E. Holzapfel, Dissertation].
TorD ist ebenfalls in dem für die in vitro-Synthese benutzen Zellextrakt enthalten und ist ein
spezifisches cytoplasmatisches Chaperon des TorA-Proteins [Jack et al., 2001 und 2004]. In
Abb. IV.5B wurde gezeigt, dass TorD in hohen Mengen durch seine Bindung an die
Signalsequenz den Transport von TorA-PhoA inhibiert. Dieser Effekt ist für die
Konzentrationen an TorD im Zellextrakt nicht zu erwarten. Dafür spricht auch die
Beobachtung, dass die Interaktion von TorD als auch von FkpA in Anwesenheit der TatProteine nach Zugabe der Tat+-INVs deutlich reduziert war (Abb. IV.13B und 14B) und
deshalb wahrscheinlich keine Konkurrenz für die Bindung der Signalsequenz an den BCRezeptor-Komplex darstellten.
Zusammenfassend lässt sich über die ortsgerichteten Quervernetzungsexperimente sagen,
dass es gelang einen Kontakt der Region um das Konsensusmotif der Signalsequenz mit
TatC, als auch einen Kontakt der hydrophoben Region der Signalsequenz mit TatB
nachzuweisen. Die Intensitäten der Interaktionen waren dabei abhängig vom Redoxzustand
des Proteins. Während die Interaktion des reduzierten TorA-PhoA-Proteins zu TatC im
Gegensatz zu der des oxidierten Proteins kaum erkennbar war, zeigte das reduzierte TorAPhoA im Vergleich zum oxidierten Protein eine abgeschwächte Intensität seiner Interaktion
zu TatB. Diese Unterschiede sind bislang, neben der Transportkompetenz, die einzigen
Hinweise auf eine faltungsabhängige Erkennung des TorA-PhoA-Proteins durch das TatSystem.
79
IV. Ergebnisse
IV.4. Charakterisierung eines potentiellen Transportintermediates von
TorA-PhoA
Der letzte Teil der vorliegenden Arbeit befasst sich mit der Spezies, die in den ersten Teilen
dieser Arbeit als Intermediat oder iTorA-PhoA bezeichnet aber noch nicht näher beschrieben
wurde. Es konnte bereits gezeigt werden, dass iTorA-PhoA zusammen mit mTorA-PhoA
(reifes TorA-PhoA) nur dann als Proteinase K-resistente Proteine auftraten, wenn die für den
Transportversuch verwendeten invertierten Vesikel der Innenmembran aus einem TatABCüberproduzierenden Stamm präpariert wurden (Abb. IV.4A). Dies spricht für die TatAbhängigkeit beider Spezies. Anders als mTorA-PhoA blieb iTorA-PhoA auch dann noch
Proteinase K-resistent, wenn der Verdau in Gegenwart von 1 % Triton X100 stattfand
(Abb. IV.4A), wodurch die Integrität der INVs zerstört wird und das ins Innere der Vesikel
transportierte Material nicht länger vor dem Verdau durch Proteinase K geschützt ist. Dieses
Phänomen wurde in den folgenden Experimenten näher untersucht.
IV.4.1. Triton-Resistenz von iTorA-PhoA wird nur für die oxidierte
Form beobachtet
Um die Abhängigkeit der Triton-Resistenz des iTorA-PhoA vom Redoxzustand zu
untersuchen wurden die reduzierte als auch die oxidierte Form des Proteins in Gegenwart
von Tat+-INVs in vitro synthetisiert (Abb. IV.16).
TorA-PhoA
Red.
Ox.
Tat+
INV
Triton 1%
+
-
Proteinase
K
+
-
+
+
p
m
i
50 kd
1
2
3
4
5
6
Abb. IV.16: Formation des potentiellen Intermediats ist von dem Redoxzustand des TorA-PhoA-Proteins
abhängig.
TorA-PhoA wurde bei 37°C in vitro synthetisiert und dabei radioaktiv markiert. Proteinsynthese fand in
Gegenwart von 5 mM oxidiertem Glutathion (4-6, Ox.) oder in dessen Abwesenheit statt (1-3, Red.). Den
Ansätzen 1 und 4 ( je 75 µl) wurden Tat+-INVs zugesetzt. Nach 30 min wurden 15 µl jedes Ansatzes mit TCA
gefällt (1 und 4), während je zweimal 30 µl (2,3 und 5,6) zuerst mit Proteinase K verdaut wurden
(Vorläuferprotein: p, reifes Protein: m, Intermediat: i). Die Ansätze 3 und 6 enthielten während der Proteinase
K–Behandlung zusätzlich 1% Triton X100. Sämtliche Proben wurden durch SDS-PAGE (10%iges Gel)
aufgetrennt und die radioaktiv markierten Proteine mittels Autoradiographie sichtbar gemacht.
80
IV. Ergebnisse
Transportaktivität wurde nur für das oxidierte TorA-PhoA festgestellt, da Protease-resistentes
pTorA-PhoA, mTorA-PhoA und iTorA-PhoA detektierbar war (Abb. IV.16, Spur 5), während
das reduzierte Protein vollständig verdaut wurde (Abb. IV.16, Spur 2). Proteinase Kbehandelte Proben, denen zusätzlich 1 % Triton X100 zugesetzt wurden, wiesen nur für das
oxidierte TorA-PhoA proteaseresistentes iTorA-PhoA auf (Abb. IV.16, Spur 6), während das
reduzierte TorA-PhoA keine Resistenzen aufwies (Abb. IV.16, Spur 3).
Demnach ist die Bildung des Triton-resistenten iTorA-PhoAs an die oxidierte Form des
Proteins gekoppelt. Bemerkenswert allerdings ist die Tatsache, dass trotz der Zerstörung der
INV-Integrität durch Triton X100 nur das Vorläuferprotein und das reife Protein (pTorA-PhoA
und mTorA-PhoA) verdaut wurden. Dies ließ die Vermutung aufkommen, dass iTorA-PhoA
nicht im Inneren der Vesikel zu finden war, sondern noch von Membranstrukturen umgeben
war, die das Protein vor proteolytischem Verdau schützten. Allein auf Grund der Menge an
detektierbaren iTorA-PhoA kann dieses Protein nur das Produkt eines partiellen Verdaus des
pTorA-PhoAs sein. Da diese partielle Proteaseresistenz des oxidierten pTorA-PhoAs nur in
Gegenwart von Tat+-INVs auftrat (Abb. IV.4A) ist die Formation des iTorA-PhoA
höchstwahrscheinlich abhängig vom Tat-spezifischen Transportapparat.
IV.4.2. Die Bildung von iTorA-PhoA ist abhängig vom Doppel-Arginin-Motif
Um die Annahme, die Bildung von iTorA-PhoA sei abhängig vom Tat-spezischen Transport
zu untermauern, wurde die Bedeutung des Doppel-Arginin-Motifs für die Bildung des iTorAPhoAs untersucht, indem das erste (KR-TorA-PhoA) oder beide hochkonservierten Arginine
(KK-TorA-PhoA) im Konsensusmotiv der Signalsequenz durch Lysin ersetzt wurden
(Abb. IV.17A). In Tat-Substraten führt diese Mutation im Allgemeinen zur kompletten
Inhibition des Tat-abhängigen Transports in vivo als auch in vitro.
RR-TorA-PhoA, KR-TorA-PhoA und KK-TorA-PhoA wurden unter oxidierenden Bedingungen
in Gegenwart von Tat+-INVs in vitro synthetisiert (Abb. IV.17B). Transportaktivität wurde
durch die Behandlung mit Proteinase K überprüft. Transport war wie erwartet für das RRTorA-PhoA
zu
beobachten,
da
proteaseresistentes
mTorA-PhoA
und
iTorA-PhoA
detektierbar waren, wobei in diesem Versuch verhältnismäßig viel iTorA-PhoA auftrat
(Abb. IV.17B, Spur 2). Die Menge an iTorA-PhoA wurde durch den Austausch des ersten
Arginins durch Lysin im Doppel-Arginin-Motif stark reduziert, während die Menge an mTorAPhoA unbeeinflusst blieb (Abb. IV.17B, Spur 4). Kein Transport war im Fall des KK-TorAPhoA zu beobachten, da keine Proteaseresistenz zu detektieren war (Abb. IV.17B, Spur 6).
Diese Ergebnisse verdeutlichten die Bedeutung des Doppel-Arginin-Motifs für den Tatabhängigen Transport von TorA-PhoA.
81
IV. Ergebnisse
Signalsequenz TorA
A
mTorA
MANNNDLFQASRRRFLAQLGGLTVAGMLGPSLLTPRRATAAQAATDA
TorA-PhoA
TorA ◄ ► reifes PhoA
Signalsequenz TorA
mTorA
MANNNDLFQASKRRFLAQLGGLTVAGMLGPSLLTPRRATAAQAATDA
KR-TorA-PhoA
TorA ◄ ► reifes PhoA
Signalsequenz TorA
mTorA
MANNNDLFQASKKRFLAQLGGLTVAGMLGPSLLTPRRATAAQAATDA
KK-TorA-PhoA
TorA ◄ ► reifes PhoA
B
Ox.
TorA-PhoA
RR
Proteinase K
KR
+
KK
+
+
p
TorA-PhoA m
i
50 kDa
1
2
3
4
5
6
Abb. IV.17: Der TorA-PhoA-Transport, sowie die Bildung des potentiellen Intermediates sind abhängig
von einem intakten RR-Motif der Signalsequenz.
A: RR-TorA-PhoA: Signalsequenz mit blau hervorgehobenem Konsensusmotif sowie den ersten 7
Aminosäuren des reifen TorA (mTorA). KR-TorA-PhoA: Signalsequenz mit blau hervorgehobenem
Konsensusmotif in dem das erste der beiden konservierten Arginine gegen Lysin (rot) ausgetauscht ist. KKTorA-PhoA: Signalsequenz mit blau hervorgehobenem Konsensusmotif in dem beide konservierten Arginine
gegen Lysine (rot) ausgetauscht sind.
B: Die TorA-PhoA-Varianten RR-,KR- und KK-TorA-PhoA wurden in Gegenwart von 5mM oxidiertem
+
Glutathion (Ox.) in vitro synthetisiert und dabei radioaktiv markiert. Allen Ansätzen wurden Tat -INVs
zugesetzt. Nach 30 min wurden 15 µl jedes Ansatzes mit TCA gefällt, während 30 µl zuerst mit Proteinase K
verdaut wurden (Vorläuferprotein: p, reifes Protein: m, Intermediat: i). Sämtliche Proben wurden durch SDSPAGE (10%iges Gel) aufgetrennt und die radioaktiv markierten Proteine mittels Autoradiographie sichtbar
gemacht.
Überraschend war allerdings die drastische Auswirkung der KR-Mutation auf die Bildung des
iTorA-PhoAs. Offensichtlich ist eine abgeschwächte Interaktion der Doppel-Arginin-Motifs
zum TatBC-Rezeptor-Komplex, hervorgerufen durch den Austausch des ersten der beiden
Arginine, von besonderer Bedeutung für die Bildung von iTorA-PhoA.
Klar ist jedoch, dass für die Bildung von iTorA-PhoA eine intakte TorA-Signalsequenz für die
Interaktion mit dem TatBC-Rezeptor-Komplex benötigt wird. Da das Auftreten von iTorAPhoA alle Erfordernisse für den Tat-abhängigen Transport erfüllt, stellt dessen Bildung
vermutlich ein translokationsarretierte Form der TorA-PhoA-Proteins dar, die erst nach der
produktiven Erkennung durch den TatBC-Rezeptor-Komplex entsteht.
82
IV. Ergebnisse
IV.4.3. Translokationsarrest findet vor dem ∆pH-abhängigen Schritt der
Translokation statt
Um die translokationsarretierte Form des Proteins weiter zu charakterisieren, sollte im
folgenden Versuch der Frage nachgegangen werden, ob für die Bildung von iTorA-PhoA
Energie in Form der protonenmotorischen Kraft (PMF) benötigt wird. Im bakteriellen
zellfreien System wurde gezeigt, dass Tat-Substrate ohne ATP transportiert werden, aber
der Transport inhibiert werden kann durch Zerstörung des transmembranen pH-Gradienten
(Yahr und Wickner, 2001; Alami et al. 2002). Der transmembrane pH-Gradient wurde in
diesem Versuch durch den Einsatz von DCCD (Dicyclohexylcarbodiimid) oder CCCP
(Carbonylcyanid-m-chlorophenylhydrazon)
manipuliert. DCCD ist ein lipophiles Reagenz,
das im Allgemeinen mit protonierten Carboxylresten in einer hydrophoben Umgebung
reagiert. Bekannt ist es insbesondere als Inhibitor der F1FO-ATPase, in der DCCD mit dem
protonierten Carboxylrest eines konservierten Aspartatrestes der c-Untereinheiten des F0Teils der F1FO-ATPase reagiert, und durch kovalente Modifikation dieses Restes den
Protonenfluss über die Membran durch den FO-Teil verhindert [Sebald et al., 1980; Hermolin
und Fillingame, 1989]. Da sich im zellfreien in vitro-Proteinsynthese-System ein pH-Gradient
an der Membran der INVs durch die Hydrolyse von ATP über die FOF1-ATPase aufbaut,
verhindert
DCCD
die
Bildung
des
pH-Gradienten.
CCCP
dagegen
ist
ein
protonenspezifisches Ionophor, das einen bereits bestehenden Protonengradienten zerstört
indem es selbst Protonen durch die Membran transportiert.
Zunächst wurde die Wirkung von DCCD und CCCP auf den Tat-abhängigen SufI-Transport
analysiert. SufI wurde in vitro synthetisiert und mit Tat+-INV versetzt, wobei einem Ansatz als
Kontrolle nur INV-Puffer zugesetzt wurde (Abb. IV.18A). Durch Proteinase K-Behandlung
wurde
Translokationsaktivität
nur
in
Gegenwart
von
Tat+-INVs
nachgewiesen
(Abb. IV.18A, Spur 4), während in dem Ansatz ohne Vesikel keine Transportaktivität
detektierbar war (Abb. IV.18A, Spur 2). Durch den Einsatz von 0,5 mM DCCD während des
Transports wurde dieser praktisch vollständig inhibiert, da kein Proteinase K-resistentes
Material mehr detektierbar war (Abb. IV.18A, Spur 6). Gleiches gilt für den Einsatz von 0,1
mM CCCP (Abb. IV.18A, Spur 8). Demnach waren beide Substanzen in der Lage den Tatabhängigen Transport von SufI vollkommenen zu inhibieren. Dieser Versuch wurde in
gleicher Weise mit dem oxidierten TorA-PhoA durchgeführt (Abb. IV.18B). Auch hier wurde
wie erwartet Translokationsaktivität nur in Gegenwart von Tat+-INVs nachgewiesen
(Abb. IV.18B, Spur 4), während in dem Ansatz ohne Vesikel keine Transportaktivität
detektierbar war (Abb. IV.18B, Spur 2). Wie bei SufI zu beobachten war, wurde auch hier
durch den Einsatz von 0,5 mM DCCD der Transport praktisch vollständig inhibiert, da kein
Proteinase K-resistentes Material mehr detektierbar war (Abb. IV.18A, Spur 6).
83
IV. Ergebnisse
A
DCCD
0,5 mM
INV
CCCP
0,1 mM
Tat+
-
Proteinase K
+
+
+
+
p
50 kd
SufI
m
1
2
3
4
5
B
6
DCCD
0,5 mM
INV
7
8
CCCP
0,1 mM
Tat+
-
Proteinase K
+
+
+
+
p
m
i
50 kd
1
C
3
4
5
6
7
8
2
9
10
3
11
4
12
13
5
14
6
15
16
7
17
TorA-PhoA
8
18
19
10% TCA
PK
-
p
m
i
+CCCP
i
+DCCD
Abb. IV.18: Die Bildung des potentiellen Intermediats ist nicht ∆pH-abhängig, wird aber durch DCCD
inhibiert.
A: SufI wurde in vitro synthetisiert und dabei radioaktiv markiert. 5 min nach Synthesestart wurden den
Ansätzen Tat+-INVs (Spuren 3-8) bzw. INV-Puffer (Spuren 1 und 2) zugesetzt. Zusammen mit den INVs wurden
den Ansätzen in den Spuren 5 und 6 0,5 mM DCCD und den Ansätzen in den Spuren 7 und 8 0,1 mM CCCP
zugesetzt. Nach weiteren 25 min wurden 15 µl jedes Ansatzes mit TCA gefällt, während 30 µl zuerst mit
Proteinase K verdaut wurden (Vorläuferprotein: p, reifes Protein: m).
B: TorA-PhoA wurde in vitro synthetisiert und dabei radioaktiv markiert. Proteinsynthese fand in Gegenwart von
5 mM oxidiertem Glutathion statt. Der restliche Versuchsverlauf wurde wie in A beschrieben durchgeführt.
Sämtliche Proben aus A und B wurden mittels SDS-PAGE (10%iges Gel) aufgetrennt und die radioaktiv
markierten Proteine mittels Autoradiographie sichtbar gemacht.
C: TorA-PhoA wurde in drei 175 µl Ansätzen in vitro in Gegenwart von 5 mM oxidiertem Glutathion synthetisiert
+
und dabei radioaktiv markiert. 5 min nach Synthesestart wurden den Ansätzen Tat -INVs zugesetzt. Zusätzlich
wurde 0,1 mM CCCP (+CCCP) oder 0,5 mM DCCD (+DCCD) zugesetzt. Nach weiteren 25 min wurde die
Proteinsynthese durch Puromycin gestoppt. 15 µl jedes Ansatzes wurden mit TCA gefällt (TCA), während 30 µl
zuerst mit Proteinase K verdaut wurden (Vorläuferprotein: p, reifes Protein: m, Intermediat: i). Der restliche
Ansatz wurde mit 16000 xg, 20 min bei 4°C zentrifugiert. 10 µl des Überstandes wurden mit TCA gefällt (10%)
während 100 µl auf eine Gelfiltrationssäule (Sepharose CL-6b) aufgetragen wurden. Die Elution der Proteine
erfolgte in 100 µl Fraktionen (3-19), die mit TCA gefällt wurden. Fraktionen 6-8 (rot) sind die membranhaltigen
Fraktionen. Sämtliche Proben wurden mittels SDS-PAGE (15%iges Gel) aufgetrennt und die radioaktiv
markierten Proteine mittels Autoradiographie sichtbar gemacht.
Durch den Einsatz von 0,1 mM CCCP jedoch blieb selektiv iTorA-PhoA proteaseresistent,
während pTorA-PhoA und mTorA-PhoA durch Proteinase K verdaut wurden.
Demnach sind DCCD als auch CCCP in der Lage den Tat-abhängigen Transport zu
inhibieren, zeigen allerdings unterschiedliche Effekte auf die Bildung von iTorA-PhoA. Da die
84
IV. Ergebnisse
Bildung von iTorA-PhoA durch CCCP nicht beeinflusst wurde, muß dessen Bildung vor dem
∆pH-abhängigen Schritt des Translokationsprozesses stattfinden. Demnach muss das
Verschwinden von iTorA-PhoA durch DCCD unabhängig von dessen inhibitorischen Wirkung
auf die FOF1-ATPase sein, und kann nur durch eine unbekannte, direkte Hemmung des TatTransportes erklärt werden.
Um die Tat-spezifische Bindung von TorA-PhoA an die Membran in Gegenwart von DCCD
zu untersuchen, wurden im folgenden Experiment größenchromatographische Analysen
betrieben, wie sie bereits in einem vorhergehenden Teil der vorliegenden Arbeit zur
Anwendung kamen (Abb. IV.7-9).
Dazu wurden die Elutionsprofile der Gelfiltration einer in vitro-Synthese von oxidiertem TorAPhoA analysiert (Abb. IV.18C). Alle Ansätze enthielten Tat+-INVs, während zwei der drei
Ansätze CCCP oder DCCD zugesetzt wurde. Der Ansatz ohne Zusatz von CCCP bzw.
DCCD zeigte Transportaktivität (Abb. IV.18C, -, PK). Darüber hinaus wies das Elutionsprofil
dieses Ansatzes signifikante Mengen an pTorA-PhoA in den INV-haltigen Fraktionen 6–8
auf. Da die Transportaktivität in diesem Versuchsansatz relativ gering war, ist im Vergleich
zu Abb. IV.7 kein mTorA-PhoA in den Fraktionen 6–8 zu erkennen (Abb. IV.18C, -, 4-20).
Das Elutionsprofil des CCCP-haltigen Ansatzes war praktisch identisch mit dem des
Ansatzes ohne Zusatz (Abb. IV.18C, +CCCP, 4-20). Demnach wurde die Bindung des
Proteins an die Membran nicht durch CCCP beeinflusst. Wie erwartet, zeigte in Gegenwart
von
CCCP
nur
das
iTorA-PhoA
Resistenz
gegenüber
Proteinase
K
(Abb. IV. 18C, +CCCP, PK). Das Elutionsprofil des DCCD-haltigen Ansatzes unterschied
sich von den zwei vorhergehenden dahingehend, dass deutlich geringere Mengen an pTorAPhoA in den INV-haltigen Fraktionen 6–8 eluierten (Abb. IV.18C, +DCCD, 4-20). Die
Proteinase K-Behandlung dieses Ansatzes wies keine proteaseresistenten Proteine auf
(Abb. IV.18C, +DCCD, PK). DCCD verhinderte offensichtlich die Tat-spezifische Bindung des
pTorA-PhoAs an die Membran.
Basierend auf diesen Beobachtungen kann davon ausgegangen werden, dass die Bindung
von pTorA-PhoA an die Membran Voraussetzung für die Bildung des iTorA-PhoAs nach
Behandlung mit Proteinase K ist. Zusätzlich zeigen diese Experimente, dass DCCD neben
seiner inhibitorischen Wirkung auf den Tat-abhängigen Transport durch die Inhibition der
F1FO-ATPase noch eine direkte Wirkung auf das Tat-System besitzt. Vorstellbar wäre eine
Interaktion von DCCD mit einem für die Bindung des Tat-Substrates bedeutende Region des
TatBC-Rezeptor-Komplexes, über den nachweislich der initiale Kontakt des Substrats zum
Tat-System hergestellt wird [Cline und Mori, 2001; Alami et al., 2003].
85
IV. Ergebnisse
IV.4.4. Translokationsarrest findet vor der Abspaltung der Signalsequenz statt
Im folgenden Versuch wurde der Frage nachgegangen, ob dem Translokationsarrest der zur
der Bildung von iTorA-PhoA führt eine Prozessierung durch die Signalpeptidase vorausgeht.
Dazu wurden die beiden möglichen Spaltstellen (Ala-X-Ala, Abb. IV.19A) am Übergang der
Signalsequenz zum reifen Teil des Proteins dadurch inaktiviert, dass das C-terminale Alanin
der Signalsequenz, sowie das N-terminale Alanin des reifen Proteins durch Lysine ersetzt
wurde (Abb. IV.19A). Die daraus resultierte Spaltmutante wies dadurch kein Ala-X-Ala-Motif
mehr auf und wurde als ∆SP-TorA-PhoA bezeichnet.
A
SP
Signalsequenz TorA
mTorA
MANNNDLFQASRRRFLAQLGGLTVAGMLGPSLLTPRRATAAQAATDA
TorA-PhoA
TorA ◄ ► reifes PhoA
∆SP
Signalsequenz TorA
mTorA
MANNNDLFQASRRRFLAQLGGLTVAGMLGPSLLTPRRATLLQAATDA
∆SP-TorA-PhoA
TorA ◄ ► reifes PhoA
B
TorA-PhoA
INV
∆SPTorA-PhoA
Tat+
-
Proteinase K
+
+
+
p
m TorA-PhoA
i
1
2
3
4
5
6
Abb. IV.19: Translokationsarrest findet vor der Abspaltung des Signalpeptids statt.
A: TorA-PhoA: Signalsequenz mit blau hervorgehobenem Konsensusmotif sowie den ersten 7 Aminosäuren des
reifen TorA (mTorA). SP markiert die potentielle Schnittstelle der Signalpeptidase.
∆SP-TorA-PhoA:
Signalsequenz mit blau hervorgehobenem Konsensusmotif sowie den ersten 7 Aminosäuren des reifen TorA
(mTorA). Die beiden Alanine unmittelbar neben der potentiellen Signalpeptidase-Schnittstelle sind durch Leucine
ersetzt.
B: TorA-PhoA und ∆SP-TorA-PhoA wurden in Gegenwart von 5mM oxidiertem Glutathion (Ox.) in vitro
synthetisiert und dabei radioaktiv markiert. Allen Ansätzen wurden Tat+-INVs zugesetzt. Nach 30 min wurden 15
µl jedes Ansatzes mit TCA gefällt, während 30 µl zuerst mit Proteinase K verdaut wurden (Vorläuferprotein: p,
reifes Protein: m, Intermediat: i). Sämtliche Proben wurden mittels SDS-PAGE (10%iges Gel) aufgetrennt und die
radioaktiv markierten Proteine mittels Autoradiographie sichtbar gemacht.
TorA-PhoA und ∆SP-TorA-PhoA wurden unter oxidierenden Bedingungen in Gegenwart
oder Abwesenheit von Tat+-INVs in vitro synthetisiert (Abb. IV.19B). Durch Proteinase KBehandlung wurde Translokationsaktivität nur in Gegenwart von Tat+-INVs nachgewiesen
86
IV. Ergebnisse
(Abb. IV.19B, Spur 4), während in dem Ansatz ohne Vesikel keine Transportaktivität
detektierbar war (Abb. IV.19B, Spur 2). Transport war auch für
∆SP-TorA-PhoA
zu
beobachten, da proteaseresistentes pTorA-PhoA und iTorA-PhoA detektierbar waren
(Abb. IV.19B, Spur 6). Wie erwartet, trat in dieser Spur praktisch kein mTorA-PhoA auf, da
Signalpeptidase-spezifische Prozessierung durch die Mutation in der Spaltmutante nicht
möglich war. Aus demselben Grund trat pTorA-PhoA stärker auf. Die Menge an iTorA-PhoA
der Spaltmutante unterschied sich nicht von der des spaltbaren TorA-PhoA-Proteins, woraus
geschlossen werden kann, dass der Translokationsarrest vor der Abspaltung der
Signalsequenz stattfindet. Außerdem wird damit klar, dass die Bildung von iTorA-PhoA das
Resultat der proteolytischen Verdaus des pTorA-PhoA-Proteins und nicht der reifen Form
des Proteins sein muss.
IV.4.5. Der C-Terminus von iTorA-PhoA ist nicht proteasegeschützt
Die bisherigen Ergebnisse zur Charakterisierung von iTorA-PhoA zeigen, dass die Tatspezifische Bindung von pTorA-PhoA an die Membran zu einer Interaktion des Proteins mit
den Tat-Proteinen führt, die dem pTorA-PhoA-Protein partielle Proteinase K-Resistenz
verleiht. Durch Proteinase K-Behandlung wurde das Protein gemäß Analyse durch SDSGelelektrophorese um ca. 9 kDa verkleinert. Unklar ist, welcher Teil des Proteins verdaut
wurde. Um dies zu klären, wurde der Umstand ausgenutzt, dass TorA-PhoA einen His-tag
am C-Terminus trägt.
Oxidiertes TorA-PhoA wurde in vitro in Gegenwart von Tat+-INVs synthetisiert (Abb. IV.20).
Nach
Synthesestopp
durch
Puromycin
konnte
durch
Proteinase
K-Behandlung
Transportaktivität nachgewiesen werden (Abb. IV.20, Spur 2). Entgegen den meisten
Experimenten tritt in diesem Versuch relativ wenig iTorA-PhoA auf. Fand die Proteinase KBehandlung in Gegenwart von 1 % Triton X100 statt, blieb, wie oben bereits mehrfach
gezeigt, nur noch iTorA-PhoA proteaseresistent (Abb. IV.20, Spur 4, (IV)). Dieses Material
wurde für eine affinitätschromatographische Analyse zum Bindungsverhalten von iTorAPhoA verwendet. Zum Vergleich diente eine mit 1 % Triton X100 versetzte Probe, die nicht
mit Proteinase K behandelt wurde, wodurch kein iTorA-PhoA generiert, aber pTorA-PhoA
und mTorA-PhoA aus den Vesikeln freigesetzt wurde (Abb. IV.20, Spur 3, (III)). Beide
Proben ((III) und (IV)) wurden mit Ni2+-Sepharose inkubiert (Abb. IV.20, Spuren 5–9, (III) und
Spuren 10-15, (IV)), während letztere Probe ((III)) als Negativ-Kontrolle auch mit Ni2+-freier
Sepharose inkubiert wurde (Abb. IV.20, Spuren 15–19, (III)).
87
IV. Ergebnisse
Ni2+- freie
Sepharose
Ni2+-Sepharose
Triton 1%
PK
-
-
+
+
-
+
-
+
1
2
U
W1 W2 E1 E2
U
W1 W2 E1
E2
U
W1 W2 E1 E2
10
11
14
15
16
p
m
i
Ansatz
Vol.:15µl
3
4
5
6
7
8
9
12 13
17
I
II
III
IV
III
IV
III
1x
2x
1x
2x
3x
3x
3x
18
19
Abb. IV.20: Der C-Terminus des potentiellen Intermediats ist ungeschützt.
TorA-PhoA wurde in einem 15x Ansatz in vitro in Gegenwart von 5 mM oxidiertem Glutathion synthetisiert und
dabei radioaktiv markiert. 15 µl entsprechen einem Ansatz. Dem 15x Ansatz wurden 5 min nach Synthesestart
Tat+-INVs zugesetzt. Nach 25 min wurde die Proteinsynthese durch Puromycin gestoppt. Anschließend
wurden 15 µl (1x) mit 5 % TCA gefällt (I) während 30 µl (2x) für weitere 25 min bei 25°C mit Proteinase K (PK,
0,5 mg/ml) inkubiert und anschließend mit 5 % TCA gefällt wurden (II). Darüber hinaus wurden 105 µl (7x, III)
mit 1 % Triton X100, sowie 75 µl (5x, IV) mit 1 % Triton X100 und Proteinase K (PK, 0,5 mg/ml) versetzt.
Beide Ansätze wurden 25 min bei 25°C inkubiert. Zuletzt wurden 15 µl aus Ansatz III (1x) und 30 µl aus
Ansatz IV (2x,) mit 5 % TCA gefällt. Nach der Inaktivierung der Proteinase K durch 0,5 mM PMSF dienten die
restliche Volumina der Ansätze III und IV als Ausgangsmaterial für eine His-tag-Aufreinigung mit Ni2+Sepharose im batch-Verfahren. Um unspezifische Bindung des TorA-PhoA-Proteins auszuschließen, wurde
als Kontrolle parallel auch Ni2+-freie Sepharose verwendet. Ungebundenes Protein (U), Waschschritte 1 und 2
(W1, W2) sowie Eluate aus zweimaliger Elution mit 50 mM EDTA (E1, E2) wurden mit 5 % TCA gefällt.
Sämtliche Proben wurden mittels SDS-PAGE (10%iges Gel) aufgetrennt und die radioaktiv markierten
Proteine mittels Autoradiographie sichtbar gemacht.
Dabei zeigte sich, dass der überwiegende Teil des pTorA-PhoA, sowie das gesamte mTorAPhoA an die Ni2+-Sepharose gebunden (Abb. IV.20, Spur 5) und erst durch den EDTAhaltigen Puffer wieder eluiert wurde (Abb. IV.20, Spuren 8 und 9). Dabei muss es sich um
eine spezifische Bindung des His-tags an die Ni2+-Ionen handeln, da praktisch kein Protein
an die Ni2+-freie Sepharose gebunden wurde (Abb. IV.20, Spuren 15–19). iTorA-PhoA
dagegen wurde offensichtlich nicht durch die Ni2+-Sepharose gebunden, da das gesamte
Material im Durchlauf zu finden war (Abb. IV.20, Spur 10). Da iTorA-PhoA nicht an die Ni2+Sepharose binden konnte, kann davon ausgegangen werden, dass der mit dem His-tag
versehene C-Terminus des Proteins durch die Proteinase K abgespalten wurde. Demnach
befindet sich ein vermutlich ca. 9 kDa großes Stück des C-Terminus nicht in einer
schützenden Umgebung, welche durch Interaktion des pTorA-PhoAs mit den Tat-Proteinen
vermittelt wird. Statistisch gesehen befindet sich nach fast jedem zweiten Aminosäurerest
eine Proteinase K-spezifische Schnittstelle in der TorA-PhoA-Aminosäuresequenz (233
Schnittstellen; pTorA-PhoA: 517 Aminosäuren).
88
IV. Ergebnisse
IV.4.6. Überlastung des Tat-Apparates könnte die Ursache für den
Translokationsarrest sein
Unklar ist bislang, wieso ein Teil des TorA-PhoA-Proteins vollständig transportiert wird,
während eine signifikante Menge des Proteins nur teilweise transportiert wird. Um einen
Einblick in die Entstehung der transportarretierten Form zu bekommen, wurde eine Kinetik
des TorA-PhoA-Transportes durchgeführt.
A
Transport [Min]
0
Proteinase K
1
+
2
+
3
+
5
+
15
+
25
+
+
p
m TorA-PhoA
i
B
100
100
100
91,7
90
80
70
%
60
Transportiertes TorA-PhoA
50
40
30
Transport-Intermediat
41,4
+Tat+-INV
20
10
0
0
10,4
5,6
0
00,1
1
29,2
27,9
0
2
7,3
0
3
5
15
25
min Transport
Abb. IV.21: Die Formation des potentiellen Intermediats findet zu einem späten Zeitpunkt des
Transportes statt.
A: TorA-PhoA wurde in einem 350 µl Ansatz in vitro in Gegenwart von 5 mM oxidiertem Glutathion
synthetisiert und dabei radioaktiv markiert. Dem Ansatz wurden 5 min nach Synthesestart Tat+-INVs
zugesetzt. Um eine Transportkinetik zu erstellen wurden 0, 1, 2, 3, 5, 15 und 25 min nach der Zugabe der
Tat+-INVs (t = 0 min) 45 µl entnommen. Davon wurden 15 µl mit TCA gefällt während 30 µl zuerst mit
Proteinase K verdaut wurden (Vorläuferprotein: p, reifes Protein: m, Intermediat: i). Sämtliche Proben wurden
mittels SDS-PAGE (10%iges Gel) aufgetrennt und die radioaktiv markierten Proteine mittels Autoradiographie
sichtbar gemacht.
B: Graphische Darstellung der Transportkinetik von TorA-PhoA. Zur Quantifizierung wurde das Programm
ImageQuant 5.2 benutzt. Quantifizierte Signale des Vorläuferproteins (p) und des reifen Proteins (m) jedes
Proteinase K-behandelten Ansatzes wurden summiert (Transportiertes TorA-PhoA). Das Signal des
potentiellen Transport-Intermediates wurde separat quantifiziert (Transport-Intermediat). Die Prozentangaben
beziehen sich auf den höchsten gemessenen Wert (100 %) beider Spezies.
Oxidiertes TorA-PhoA wurde in einem Ansatz in vitro synthetisiert, dem fünf Minuten nach
Synthesestart Tat+-INVs zugesetzt wurden. Die Zugabe der INVs wurde als Zeitpunkt Null
definiert. Zu diesem Zeitpunkt wurde zum ersten Mal eine Probe aus diesem Ansatz
entnommen. Weitere Proben wurden nach 1, 2, 3, 5, 15 und 25 Minuten entnommen. Alle
entnommenen Proben wurden durch SDS-Gelelektrophorese analysiert (Abb. IV.21A) und
89
IV. Ergebnisse
die Signale von pTorA-PhoA, mTorA-PhoA und iTorA-PhoA mit ImageQuant 5.2 quantifiziert,
um diese graphisch darzustellen (Abb. IV.21B). Aus dieser Graphik ist die kontinuierliche
Zunahme an vollständig transportiertem Protein (p + mTorA-PhoA) zu erkennen, die nach 15
Minuten bereits ihr Maximum erreichte (Abb. IV.21B, blaue Linie). Somit war nach 15
Minuten der vollständige Transport des Proteins bereits abgeschlossen. Zu diesem Zeitpunkt
lagen dagegen weniger als 30 % der maximal gebildeten Menge an iTorA-PhoAs vor. Erst in
den letzten 10 Minuten der Reaktion nahm dessen Menge drastisch zu und erreichte den
höchsten aufgenommenen Wert nach 25 Minuten (Abb. IV.21B, grüne Linie). Kinetiken,
aufgenommen über längere Zeiträume als 25 Minuten, führten zu keiner weiteren Zunahme
von iTorA-PhoA (nicht gezeigt). Aus der Kinetik in Abb. IV.21B geht hervor, dass die
überwiegende Menge an iTorA-PhoA in einer späten Phase des Transportversuchs entsteht.
Seine stärkste Zunahme erfährt es zu einem Zeitpunkt, zu dem der vollständige Transport
des Proteins bereits sein Maximum erreicht hat. Möglicherweise stehen diese beiden
Beobachtungen in kausalem Zusammenhang zueinander, in einer Weise, dass, wenn der
vollständige Transport des TorA-PhoA-Proteins vollständig zum Erliegen kommt, dies die
Bildung von iTorA-PhoA zur Folge hat. Eine mögliche Erklärung könnte eine Überlastung der
Tat-Translokase der Membranvesikel sein. Dieser Effekt ist bereits bei Überproduktion von
Tat-abhängigen Substraten in vivo beobachtet worden (Yahr und Wickner, 2001).
IV.4.7. Translokationsarrest findet in der unmittelbaren Nähe von TatA statt
Interaktion des pTorA-PhoAs mit den Tat-Proteinen führte zur einer partiellen Resistenz
gegenüber Proteinase K, die auch nach Solubilisierung der Vesikel mit Triton X100 erhalten
blieb. Diese Beobachtung könnte für eine Solubilisierung von Membranstrukturen sprechen,
die das TorA-PhoA-Protein enthalten und in einer schützenden Weise umgeben.
In den meisten Modellen des Tat-Transports wird von einer hydrophilen Pore als
Komponente der Tat-Translokase ausgegangen, durch die die Passage des Substrates
ermöglicht wird. Nach dieser Vorstellung könnte sich die in seinem Transport arretierte Form
des TorA-PhoA-Proteins in oder in der Umgebung einer potentiellen Pore befinden. Als
Poren-bildende Komponente hat sich das TatA-Protein als wahrscheinlichster Kandidat für
diese Funktion herausgestellt, da TatA in homooligomeren, wahrscheinlich ringförmigen
Komplexen in der bakteriellen Membran vorliegt. In diversen Studien konnte der TatAKomplex durch Solubilisierung aus der inneren Membran gelöst werden.
Dazu kamen
verschiedene Detergenzien zum Einsatz, die auch im folgenden Versuch verwendet wurden,
um deren Wirkung auf die Protease-Resistenz des iTorA-PhoAs zu testen.
90
IV. Ergebnisse
Ox. TorA-PhoA
Tat+
INV
Proteinase K
-
+
Detergenz
-
-
Triton
1%
1
2
3
C12E9
1%
Chaps
50 mM
OG
50 mM
Triton
2%
p
TorA-PhoA m
i
4
5
6
7
Abb. IV.22: Das potentielle Intermediat tritt in Gegenwart verschiedener Detergenzien auf.
TorA-PhoA wurde in einem 200 µl Ansatz in vitro synthetisiert und dabei radioaktiv markiert. Proteinsynthese
fand in Gegenwart von 5 mM oxidiertem Glutathion (Ox. TorA-PhoA) statt. Dem Ansatz wurden Tat+-INVs
zugesetzt. Nach 30 min wurden 15 µl des Ansatzes mit 5 % TCA gefällt (1), während 6x 30 µl (2-7) zuerst mit
Proteinase K verdaut wurden (Vorläuferprotein: p, reifes Protein: m, Intermediat: i). Die Ansätze 3 bis 7
enthielten während der Proteinase K–Behandlung zusätzlich Detergenzien in den angegebenen
Konzentrationen (OG: Octylglucosid). Sämtliche Proben wurden durch SDS-Gelelektrophorese (10%iges Gel)
aufgetrennt und die radioaktiv markierten Proteine mittels Autoradiographie sichtbar gemacht.
Oxidiertes TorA-PhoA wurde in vitro in Gegenwart von Tat+-INVs synthetisiert (Abb. IV.22).
Transport wurde durch das Auftreten von Proteinase K-resistenten pTorA-PhoA, mTorAPhoA und iTorA-PhoA nachgewiesen (Abb. IV.22, Spur 2). Durch gleichzeitige Zugabe von
1 % oder 2 % Triton X100 wurden wie erwartet pTorA-PhoA und mTorA-PhoA verdaut,
während iTorA-PhoA resistent blieb (Abb. IV.22, Spuren 3 und 7). Dies war auch dann der
Fall, wenn statt Triton andere Detergenzien während der Proteinase K-Behandlung zum
Einsatz kamen: 1 % Nonaethylenglykol-monododecylether oder C12E9 (Abb. IV.22, Spur 4),
60 mM Chaps (Abb. IV.22, Spur 5) und 60 mM Octylglucosid oder OG (Abb. IV.22, Spur 6).
Die verwendeten Konzentrationen entsprachen denen, die für die Solubilisierung von TatAKomplexen aus bakteriellen Rohmembranen verwendet wurden [de Leeuw et al., 2002; Ida
Porcelli, Dissertation; McDevitt et al., 2006]. Die Beobachtung, dass auch diese
Detergenzien
nicht
die
Protease-Resistenz
von
iTorA-PhoA
aufhoben,
kann
als
Solubilisierung des iTorA-PhoAs aus der Membran gewertet werden, die der von TatAKomplexen ähnelt.
Falls iTorA-PhoA tatsächlich von einer durch TatA gebildeten Struktur vor proteolytischem
Verdau geschützt wird, sollte in Abwesenheit von TatA dieser Effekt nicht auftreten. Um
dieser Annahme nachzugehen wurden im folgenden Versuch INVs benutzt, die aus einem
ausschließlich TatBC-überproduzierenden Stamm präpariert wurden, wodurch die Formation
eines TatBC-Rezeptors in der Membran gewährleistet ist, die potentielle TatA-Pore
allerdings nicht vorhanden ist. Oxidiertes TorA-PhoA wurde in vitro in Gegenwart von Tat+INVs, ∆TatA-INVs oder ∆Tat-INVs synthetisiert (Abb. IV.23A). ∆TatA-INVs wurden aus
einem ausschließlich TatBC-überproduzierenden E. coli-Stamm präpariert. Transportaktivität
wurde durch Proteinase K in Gegenwart von Tat+-INVs nachgewiesen (Abb. IV.23A, Spur 2).
91
IV. Ergebnisse
A
Ox- TorA-PhoA
INV
Tat+
1 % Triton X100
∆Tat
∆TatA
-
+
Proteinase K
+
+
+
+
p
TorA-PhoA m
i
1
B
2
Red. TorA-PhoA
-
Triton 1%
4
5
6
7
8
Ox. TorA-PhoA
+
-
Proteinase K
3
-
+
+
-
+
Native IP
αTat
A
αTat
B
αTatC
αPho
A
p
m
i
15 µl Ansatz
50 kDa
1
2
3
4
5
6
7
8
9
10
1x
2x
2x
1x
2x
2x
4x
4x
4x
4x
C
Red. TorA-PhoA
-
Triton 1%
Proteinase K
Ox. TorA-PhoA
+
-
-
+
+
-
+
αTatA
Native IP
αTatB
αTatC
p
m
i
50 kDa
1
15 µl Ansatz
1x
2
3
4
5
6
2x
2x
1x
2x
2x
6
+ αTatA
2x
7
8
9
4x
4x
4x
Abb. IV.23: Translokationsarrest findet in der unmittelbaren Nähe von TatA statt.
A: TorA-PhoA wurde in vitro in Gegenwart von 5 mM oxidiertem Glutathion synthetisiert und dabei radioaktiv markiert.
Den Ansätzen in den Spuren 1-4 wurden Tat+-INVs, den Ansätzen in den Spuren 5 und 6 Vesikel der Innenmembran
eines ausschließlich TatBC-überproduzierenden E. coli-Stammes (∆TatA-INV) und den Ansätzen in den Spuren 7-8
∆Tat-INVs zugesetzt. Nach 30 min wurden 15 µl jedes Ansatzes mit TCA gefällt, während 30 µl zuerst mit Proteinase
K verdaut wurden (Vorläuferprotein: p, reifes Protein: m, Intermediat: i). PK-Verdau der Ansätze in Spuren 3-8 fand in
Gegenwart von 1% Triton X100 statt. Sämtliche Proben wurden mittels SDS-PAGE (10%iges Gel) aufgetrennt.
B: TorA-PhoA wurde in einem 5x Ansatz (Red. TorA-PhoA) und in einem 21x Ansatz (Ox. TorA-PhoA) in vitro
synthetisiert und dabei radioaktiv markiert. Proteinsynthese fand in Gegenwart von 5 mM oxidiertem Glutathion (Ox.
TorA-PhoA) oder in dessen Abwesenheit (Red. TorA-PhoA) statt. Jedem Ansatz wurden Tat+-INVs zugesetzt. Nach
30 min wurden 15 µl (1x) des 5x Ansatz (Red. TorA-PhoA) mit TCA gefällt (1), während zweimal 30 µl ( 2 und 3, je 2x)
für weitere 25 min bei 25°C mit Proteinase K (PK, 0,5 mg/ml) inkubiert wurden. Aus dem 21x Ansatz (Ox. TorA-PhoA)
wurden ebenfalls 15 µl (1x) mit TCA gefällt (4), während einmal 30 µl (5, 2x) und einmal 270 µl (6, 18x) für weitere 25
min bei 25°C mit Proteinase K (PK, 0,5 mg/ml) inkubiert wurden. Die Ansätze in den Spuren 3 und 6 enthielten
während der Proteinase K–Behandlung 1 % Triton X100. Nach der PK-Behandlung wurden die Ansätze in den
Spuren 2, 3, 5 und 30µl aus Ansatz 6 (6, 2x) mit TCA gefällt. Der restliche Ansatz 6 wurde mit 0,5 mM PMSF versetzt
um die Proteinase K zu inhibieren. Anschließend wurde der Ansatz in gleichen Teilen à 60µl aufgeteilt und für native
Immunpräzipitationen (6-9) mit den angegebenen Antikörpern verwendet. Alle TCA-Pellets sowie die für die
Immunpräzipitation verwendete Protein A-Sepharose wurden in SDS-PAGE-Probenpuffer bei 56°C und 1400 rpm für
15 min in einem Thermomixer inkubiert. Sämtliche Proben wurden durch SDS-PAGE (8%iges Gel) aufgetrennt und
die radioaktiv markierten Proteine mittels Autoradiographie sichtbar gemacht.
C: Wiederholung des Versuchs wie in B beschrieben, mit dem Unterschied, dass die doppelte Menge an Ansatz 6
verwendet wurde um diesen in gleiche Teile à 30µl zu teilen: in Ansatz 6 und Ansatz 6+αTatA. Um die Verschiebung
des Signals des radioaktiven Proteins im SDS-Polyacrylamidgel durch den Antikörper zu demonstrieren, wurde dem
SDS-PAGE-Probenpuffer des 6+αTatA-Ansatzes die gleiche Menge an TatA-Antikörper zugegeben, wie sie in der
Immunpräzipitation mit TatA-Antikörpern im Ansatz 7 enthalten war.
92
IV. Ergebnisse
In Gegenwart von 1% Triton X100 wurde Proteinase K-resistentes iTorA-PhoA nur im Fall
der Tat+-INVs nachgewiesen (Abb. IV.23A, Spur 4), während in Gegenwart von ∆TatA-INVs
oder
∆Tat-INVs
die
Bildung
von
iTorA-PhoA
nicht
zu
beobachten
war
(Abb. IV.23A, Spuren 6 und 8). Die Menge an pTorA-PhoA in den ∆TatA-INVs- oder ∆TatINVs-haltigen Ansätzen wurde durch OmpT-abhängige Pseudoprozessierung deutlich
reduziert. Da aus Transportversuchen mit Tat+-INVs bekannt ist, dass bei einer OmpTabhängigen Reduktion des Vorläuferproteins pTorA-PhoA um bis zu 70 % die Bildung von
iTorA-PhoA immer noch detektierbar ist (nicht gezeigt), ist auch die verbliebene Menge an
pTorA-PhoA in dem hier beschriebenen Versuch für die Bildung von iTorA-PhoA
ausreichend. Die Tatsache, dass iTorA-PhoA dennoch nicht erkennbar war spricht für die
Abhängigkeit der Formation von iTorA-PhoA von überproduzierten Mengen an TatA.
Ob iTorA-PhoA tatsächlich zu TatA oder möglicherweise einem anderen Tat-Protein in
Kontakt steht, sollte im folgenden Versuch durch native Immunpräzipitationen untersucht
werden. Dazu wurden reduziertes sowie oxidiertes TorA-PhoA in vitro in Gegenwart von
Tat+-INVs synthetisiert. Transportaktivität wurde wiederum durch Behandlung mit Proteinase
K kontrolliert, mit dem Ergebnis, dass wie erwartet keine proteaseresistenten Proteine für
das reduzierte TorA-PhoA zu detektieren waren (Abb. IV.23B, Spur 2). Gleiches gilt für einen
mit 1 % Triton X100 versetzten Proteinase K-Verdau (Abb. IV.23B, Spur 3). Wie erwartet,
war Transportaktivität nur für das oxidierte TorA-PhoA zu beobachten (Abb. IV.23B, Spur 5).
Um eine Probe zu erhalten, die nur iTorA-PhoA enthielt, wurde in Gegenwart von 1 % Triton
X100
pTorA-PhoA
und
mTorA-PhoA
durch
Proteinase
K-Behandlung
verdaut
(Abb. IV.23B, Spur 6). Diese Probe wurde zu gleichen Teilen aufgeteilt und für native
Immunpäzipitationen mit Antikörpern gegen TatA, TatB, TatC und PhoA benutzt. Dabei
konnte radioaktives Material durch TatA-Antikörper (Abb. IV.23B, Spur 7) sowie durch PhoAAntikörper (Abb. IV.23B, Spur 10) präzipitiert werden. Dies war nicht der Fall mit Antikörpern
gegen
TatB
und
TatC
(Abb. IV.23B, Spuren 8 und 9).
Die
Bande
der
TatA-
Immunpräzipitation lief niedriger, als die des iTorA-PhoA in Spur 6. Diese Verschiebung war
die Folge einer Überladung des Gels in diesem Bereich durch die große Menge des
verwendeten Antikörper (Antikörper-Shift). Darauf deuteten zum einen die Form des Signals
in der TatA-Immunpräzipitation hin (Abb. IV.23B, Spur 7), und zum anderen die Tatsache,
denselben Effekt auch bei der Positivkontrolle mit Antikörpern gegen PhoA beobachtet zu
haben (Abb. IV.23B, Spur 10). Um diese Annahme zu bestätigen, wurde im Prinzip derselbe
Versuch wiederholt (Abb. IV.23C). Ausgangsmaterial für die Immunpräzipitationen war
wiederum eine Probe, die nur iTorA-PhoA enthielt, da sie in Gegenwart von 1 % Triton X100
durch Proteinase K verdaut wurde (Abb. IV.23C, Spur 6). Doch diesmal wurden dieser
Probe, um den Antikörper-Shift zu simulieren, Antikörper zugesetzt, (Abb. IV.23C, Spur 7),
wodurch die Bande des iTorA-PhoAs auf die gleiche Höhe gedrückt wurde wie die Bande in
93
IV. Ergebnisse
der Immunpräzipitation mit Antikörpern gegen TatA (Abb. IV.23C, Spur 8).
Daraus wird
deutlich, dass TatA-Antikörper in der Lage sind, iTorA-PhoA zu präzipitieren. Dies bedeutet
wiederum iTorA-PhoA muss sich in direktem Kontakt zu TatA befinden.
Zusammengefasst lassen die Ergebnisse dieser letzten Experimente den Schluss zu, iTorAPhoA stellt eine transportarretierte Form des TorA-PhoA dar, die in Kontakt zu TatA steht.
Erklärt werden könnte dies durch die Vorstellung von einer Protein-schleusenden Pore, die
durch TatA gebildet wird und in der sich iTorA-PhoA befindet. Somit wäre iTorA-PhoA das
erste
Transportintermediat
dem
nachgewiesen werden konnte.
94
ein
direkter
Kontakt
zu
einem
der
Tat-Proteine
V. Diskussion
V. Diskussion
Die wohl herausragendste Eigenschaft des twin arginine translocation (Tat)–Systems ist die
Fähigkeit, Proteine über die Membran zu transportieren, die bereits eine tertiäre bzw. sogar
quartäre Struktur ausgebildet haben. Dabei scheint das Tat-System auch in der Lage zu
sein, den Faltungszustand des zu transportierenden Proteins zu erkennen und dieses als
transportinkompetent abzulehnen, falls das Protein ungefaltet vorliegt. Neben Studien mit
natürlichen Tat-Substraten wurde insbesondere mit Tat-abhängigen Fusionsproteinen der
Zusammenhang von Faltungszustand und Transportkompetenz in E. coli in vivo untersucht.
Überzeugende Ergebnisse lieferten Untersuchungen mit Fusionsproteinen, für deren Tatabhängigen Transport native Faltung durch die Bildung von Disulfid-Bindung im reifen Teil
des Proteins Voraussetzung war [DeLisa et al., 2003; Masip et al., 2004; Ribnicky et al.,
2007].
Der Mechanismus, der der Erkennung von Faltungszuständen zu Grunde liegt, ist
vollkommen unbekannt. Zwar wurden beim prätranslokationalen Reifungsprozess einiger
Kofaktor-tragender Tat-Substrate in E. coli spezifische Chaperone identifiziert, die eine
Korrekturlese-Funktion ausüben könnten (siehe II. Einleitung), aber nicht essentiell für den
Transport dieser Proteine sind. Für den Transport von Proteinen, die eine Tat-spezifische
Sequenz tragen, sind in E. coli die Tat-Proteine TatA, B und C nötig und auch ausreichend,
weshalb spekuliert wurde, dass eine intrinsische Kontrolle des Substrat-Faltungszustandes
(Qualitätskontrolle) durch diese Membranproteine vermittelt werden könnte.
Ziel der vorliegenden Arbeit war es, grundlegende Einblicke in die Interaktionen eines
faltungsabhängig-transportierten
Tat-Substrates
mit
den
Tat-Proteinen
zu
erhalten.
Grundlage für diese Untersuchungen waren die Ergebnisse einer Studie mit dem Tatabhängigen Fusionsprotein TorA-PhoA, das in E. coli nur dann transportiert wurde, wenn die
Bildung der zwei intramolekularen Disulfidbrücken vor der Translokation über die
Cytoplasmamembran ermöglicht war [DeLisa et al., 2003].
V.1. Etablierung des Tat-abhängigen in vitro-Transportes von TorA-PhoA
TorA-PhoA
besteht
aus
der
Signalsequenz
des
natürlichen
Tat-Substrates
TorA
(Trimethylamin-N-Oxid (TMAO)-Reduktase) sowie dem reifen Teil des Sec-Substrates PhoA
(alkalische Phosphatase) aus E. coli. Das reife PhoA besteht aus 450 Aminosäuren und
95
V. Diskussion
enthält vier Cyteinreste, aus denen zwei intramolekulare, konsekutive Disulfidbrücken
zwischen den Cysteinresten Cys-168 und Cys-178 sowie zwischen Cys-286 und Cys-336
gebildet werden [Bradshaw et al., 1981]. Die Bildung dieser Disulfidbrücken erfolgt nach dem
Sec-abhängigen Transport von PhoA im oxidierenden Milieu des Periplasmas [Derman und
Beckwith, 1991]. Nach der oxidierenden Proteinfaltung lagern sich zwei PhoA-Einheiten zu
einem katalytisch aktiven, Zn2+-haltigen Homodimer zusammen [Wyckoff, 1987]. Im
Allgemeinen ist die Bildung von Disulfidbrücken in Proteinen im reduzierenden Cytoplasma
nicht möglich. Die reduzierenden Bedingungen werden durch das Thioredoxin- und
Glutaredoxin-System aufrechterhalten [Prinz et al., 1997]. Es konnte gezeigt werden, dass
durch die Deletion der Gene der Thioredoxin- sowie der Glutathion-Reduktase diese beiden
Systeme ausgeschaltet werden und ein E. coli-Stamm mit einem „oxidierenden“ Cytoplasma
entsteht, in dem die Formation der Disulfidbrücken des reifen PhoAs erlaubt ist [Bessette et
al., 1999]. In solch einem Stamm wird PhoA, versehen mit der Tat-abhängigen
Signalsequenz von TorA, Tat-abhängig ins Periplasma transportiert [DeLisa et al., 2003].
Dies ist nicht der Fall in einem Wildtypstamm mit einem reduzierenden Cytoplasma [Delisa et
al., 2003; Tullmann-Ercek et al., 2007]. Bildung der Disulfidbrücken und somit Formation
einer nativen Faltung ist die Voraussetzung für den Tat-abhängigen Transport von TorAPhoA in vivo [DeLisa et al., 2003].
V.1.1. Faltungszustand von in vitro-synthetisiertem TorA-PhoA
In der vorliegenden Arbeit ist es erstmals gelungen ein in vitro-System zu etablieren, in dem
ein Tat-abhängiges Protein in Abhängigkeit von seinem Redox- bzw. Faltungszustand
transportiert wird.
Es konnte gezeigt werden, dass ein in vitro-Transport des Tat-abhängigen Fusionsproteins
TorA-PhoA nur unter oxidierenden Bedingungen stattfindet (Abb. IV.2B und 4A). Oxidierende
Bedingungen während in vitro-Synthese und -Transport von TorA-PhoA wurden durch die
Zugabe von oxidiertem Glutathion (GSSG) etabliert. Die Menge an transportiertem Protein
nahm dabei proportional bis zu einer GSSG-Konzentration von 5 mM zu (Abb. IV.2B). Durch
Unterschiede in der elektrophoretischen Mobilität des TorA-PhoAs konnte gezeigt werden,
dass nur in Gegenwart von 5 mM GSSG das in vitro-synthetisierte TorA-PhoA-Protein
vollständig oxidiert vorlag, während in dessen Abwesenheit das Protein teilweise oder
vollständig reduziert war (Abb. IV.3A und B). Durch die Anwesenheit von GSSG in dem
verwendeten in vitro-System werden Redox-Bedingungen etabliert, die die Bildung von
Disulfidbrücken im reifen Teil von TorA-PhoA ermöglichen (oxidiertes TorA-PhoA), wodurch
das Protein eine Konformation annimmt, die dem Protein Transportkompetenz verleiht. So
wurde bereits in früheren Studien in einem vergleichbaren in vitro-System gezeigt, dass
durch GSSG in vitro-synthetisiertes reifes PhoA oxidiert wird, wodurch eine enzymatisch
96
V. Diskussion
aktive Form des Proteins erhalten wurde [Akiyama et al., 1992; Akiyama und Ito, 1993].
Dabei erfährt das in vitro-synthetisierte reife PhoA nach der Bildung der beiden
intramolekularen Disulfidbrücken durch GSSG eine Faltung zu einem teilweise Proteaseresistenten Monomer. In Gegenwart von Zn2+ assemblieren zwei Monomere zu einem
enzymatisch aktiven Dimer, das vollständig Protease-resistent ist [Akiyama und Ito, 1993].
Sone et al. konnten darüber hinaus zeigen, dass für das Erlangen der enzymatischen
Aktivität und der damit verbundenen Dimerisierung nur die Bildung der C-terminalen
Disulfidbrücke (Cys-286-Cys-336) nötig ist, Proteaseresistenz dagegen setzt die Bildung
beider intramolekularer Disulfide voraus [Sone et al., 1997]. Da die enzymatische Aktivität
des in vitro-synthetisierten TorA-PhoAs nicht bestimmt wurde, bleibt unklar, ob die oxidierte
Form des Proteins zur Dimerisierung in der Lage ist, was grundsätzlich möglich wäre, da das
im in vitro-System enthaltene Mg2+ das für die Dimerisierung benötigte Zn2+ ersetzen kann
[Schlesinger und Barrett, 1965].
Obwohl das in der vorliegenden Arbeit in vitro-synthetisierte TorA-PhoA-Protein nachweislich
vollständig oxidiert war, konnte dennoch keine Protease-Resistenz, beispielsweise in
Abwesenheit von Membranen oder unter Transport-verhinderten Bedingungen, beobachtet
werden (Abb. IV.4A, 18B, 19B). Dies weist auf eine nicht-native, aber vollständig oxidierte
Konformation des Proteins hin. Eine mögliche Erklärung für die Bildung solch einer
Konformation wäre die Formation nicht-nativer Disulfide im reifen Teil von TorA-PhoA.
Obwohl diese Möglichkeit nicht ausgeschlossen werden kann, neigen im Allgemeinen
hauptsächlich
Proteine
mit
zahlreichen,
und
insbesondere
mit
nicht-konsekutiven
Disulfidbrücken zur Ausbildung von nicht-nativen Disulfidbrücken [Messens und Collet,
2006]. TorA-PhoA dagegen weist nur zwei konsekutive Disulfide auf. Wahrscheinlicher ist,
dass der reife Teil von TorA-PhoA native Disulfide enthält, eine native Faltung aber
verhindert wird durch die 47 Aminosäuren von TorA am N-Terminus (Abb. IV.1) und/oder
den His-Tag am C-Terminus von PhoA, so dass eine Protease-resistente Konformation des
oxidierten TorA-PhoA nicht erreicht werden kann. Für diese Annahme sprechen
insbesondere die Beobachtungen aus den oben genannten Studien zur Faltung von in vitrosynthetisiertem PhoA, die zeigten, dass das Vorläuferprotein im Gegensatz zu dem reifen
PhoA ein schlechtes Substrat der in vitro-Faltung des Proteins darstellt und deshalb in
diesen Studien ausschließlich nur der reife Teil von PhoA synthetisiert wurde [Akiyama et al.,
1992; Akiyama und Ito, 1993]. Es ist anzunehmen, dass die TorA-Signalsequenz einen
ähnlichen Effekt auf die in vitro-Faltung des reifen PhoAs hat wie dessen natürliche, 20
Aminosäuren lange Signalsequenz. Die physiologische Bedeutung der Faltungs-Inhibition
durch das Signalpeptid liegt vermutlich in der Aufrechterhaltung der Transportkompetenz
Sec-abhängiger Proteine [Park et al., 1988], insbesondere solcher, die wie PhoA SecB–
unabhängig exportiert werden.
97
V. Diskussion
Bei der Betrachtung des Faltungszustandes des in vitro-synthetisierten TorA-PhoAs ist von
Bedeutung, dass zum einen in Gegenwart von GSSG das Protein vollständig oxidiert vorliegt
und zum anderen, dass diese Form eine kompaktere Faltung aufweist als die reduzierte
Form des Proteins, weshalb eine größere elektrophoretische Mobilität des oxidierten TorAPhoAs zu beobachten ist (Abb. IV.3A). Auf Grund der Proteasesensibilität stellt die
kompakte, oxidierte Form des Proteins zwar keine native aber vermutlich eine, der nativen
Konformation ähnelnde Konformation dar, die sich von der reduzierten Form des Proteins
derart
unterscheidet,
dass
das
Tat-System
die
oxidierte
Konformation
als
transportkompetentes Tat-abhängiges Protein erkennt, während das reduzierte Protein nicht
transportiert wird.
V.1.2. Der in vitro-Transport des oxidierten TorA-PhoAs ist Tat-abhängig
In vitro-Transport von TorA-PhoA wurde nur für das oxidierte Protein in Gegenwart von
invertierten Innenmembran-Vesikeln (INVs) eines TatABC-überproduzierenden E. coliStammes nachgewiesen (Abb. IV.4A, 8A, 19B). Resistenz von TorA-PhoA gegenüber
Proteinase K auf Grund der Lokalisation im Lumen der Vesikel wurde als Nachweis für
Transportaktivität gewertet. Signalpeptidase-spezifische Prozessierung des Vorläuferproteins
konnte nicht als Hinweis auf Transport angesehen werden, da eine unspezifische
Proteaseaktivität
(Pseudoprozessierung)
des
verwendeten
Zellextraktes
auftrat
und
insbesondere in Gegenwart einiger INVs verstärkt wurde. Bezogen auf das Molekulargewicht
unterschied sich das Produkt der Pseudoprozessierung in gelelektrophoretischen Analysen
praktisch nicht von dem der Signalpeptidase-spezifischen Prozessierung (Abb. IV.4A).
Demnach müssen, im Falle des N-terminalen Teils des Proteins, die Spaltstellen beider
proteolytischer
Aktivitäten
nahe
beieinander
liegen.
Tatsächlich
weist
die
TorA-
Signalsequenz eine spezifische Spaltstelle für OmpT (outer membrane protease T)
unmittelbar vor der Spaltstelle der Signalpeptidase zwischen den Argininen an Positionen 36
und 37 auf (Abb. IV.1) [Dekker at al., 2001]. Da die Pseudoprozessierungs-Aktivität, die
durch INVs verursacht wurde, sich durch den Einsatz von Cu2+-Ionen als auch den von
Diisopropylfluorphosphat (DPF) signifikant reduzieren ließ (Abb. IV.4B), und beide
Substanzen als OmpT-Inhibitoren gelten [Sugimura und Nishihara, 1988; Baneyx und
Georgiou,
1990],
ist die
Pseudoprozessierungs-Aktivität
höchstwahrscheinlich
einer
Kontamination der INVs durch OmpT zuzuordnen. Die Pseudoprozessierungs-Aktivität, die
in geringer Intensität auch in Abwesenheit von INVs auftrat (Abb. IV.7B), ergab ein
Degradationsprodukt gleicher Größe wie das der potentiellen OmpT-Aktivität. Dies könnte
zum einen bedeuten, dass geringe OmpT-Aktivität durch Kontamination des Zellextraktes mit
Bestandteilen der äußeren Membran vorlag, oder zum anderen, dass die vermeintliche
OmpT-Schnittstelle auch Ziel undefinierter cytoplasmatischer Proteaseaktivität war. Letztere
98
V. Diskussion
Vermutung wird unterstützt durch Studien mit anderen TorA-Signalsequenz-haltigen
Fusionsproteinen,
die
von
cytoplasmatischem,
proteolytischem
Verdau
der
TorA-
Signalsequenz berichten [Li et al., 2006; Genest et al., 2006, Sanders et al., 2001].
Auf Grund der beobachteten Pseudoprozessierungsaktivität wurde ausschließlich die
Resistenz gegenüber Proteinase K als Nachweis für Translokationsaktivität betrachtet. Auf
diesem Wege wurden drei Protease-resistente Spezies beobachtet, die als pTorA-PhoA
(Vorläuferprotein), mTorA-PhoA (reifes Protein) und iTorA-PhoA (Intermediat) bezeichnet
wurden (Abb. IV.2B). Zwei dieser Spezies repräsentieren transportiertes TorA-PhoA-Protein,
das nicht durch die Signalpeptidase prozessiert wurde (pTorA-PhoA), sowie reifes,
prozessiertes TorA-PhoA-Protein (mTorA-PhoA), das nach dem Transport durch die
Signalpeptidase um die Größe des Signalpeptids verkleinert wurde. Diese Zuordnung wurde
mit einer TorA-PhoA-Mutante, deren Signalpeptidase-Spaltstelle fehlt, bestätigt (Abb. IV.19).
Darüber hinaus lag ein potentielles Transportintermediat vor (iTorA-PhoA), auf das in
Abschnitt V.4. ausführlich eingegangen wird.
Es konnte gezeigt werden, dass die Bildung von Protease-resistentem mTorA-PhoA, pTorAPhoA und iTorA-PhoA -
und somit der in vitro-Transport von oxidiertem TorA-PhoA -
absolut Tat-abhängig erfolgte. Ein Indiz für einen Tat-abhängigen Transport war die
Tatsache, dass Transport nur in Anwesenheit von Membranen stattfindet, die aus einem E.
coli-Stamm präpariert wurden, in dem TatA, TatB und TatC überproduziert wurden
(Abb. IV.4A). Von der Entwicklung eines Tat-abhängigen in vitro-Transport-Systems ist
bekannt, dass dieser nur dann erfolgt, wenn Vesikel mit hohem TatABC-Gehalt verwendet
werden [Yahr und Wickner, 2001; Alami et al., 2002]. Darüber hinaus fand in Gegenwart von
INVs aus einem Tat-Deletionsstamm kein Transport von TorA-PhoA statt, was einen Secabhängigen Transport ausschließt (Abb. IV.4A), da diese Membranen grundsätzlich in der
Lage sind, Sec-Substrate zu transportieren (dies wurde standardmäßig mit dem Secabhängigen Substrat OmpA für jede INV-Präparation überprüft).
Ein weiteres sehr starkes Indiz für die Tat-Abhängigkeit des TorA-PhoA-Transportes ist die
vollkommene Inhibition des Transportes durch den Austausch des hoch-konservierten
Arginin-Paares in dem Konsensusmotif der TorA-Signalsequenz durch zwei Lysinreste (KKTorA-PhoA) (Abb. IV.6B und 17B). Aus einer Vielzahl von Studien ist bekannt, dass eine
derartige Mutation zur vollkommenen Blockade des Tat-abhängigen Transportes in E. coli in
vivo als auch in vitro führt [Cristobal et al., 1999; Stanley et al. 2000; Alami et al., 2002].
Außerdem wurde durch die Zugabe des TorA-Signalsequenz-bindenden Proteins TorD aus
E. coli der in vitro-Transport von oxidiertem TorA-PhoA proportional zur verwendeten TorDMenge inhibiert (Abb. IV.5B). TorD ist ein spezifisches TorA-Signalsequenz-bindendes
Chaperon, dessen Funktion als Anti-Zielsteuerungs-Chaperon vermutet wird, das so lange
an der TorA-Signalsequenz bindet, bis Kofaktor-Insertion und somit Faltung des reifen TorA-
99
V. Diskussion
Proteins stattgefunden hat [Dubini und Sargent, 2003; Jack et al., 2001 und 2004]. Der
inhibitorische Effekt des TorD kann nur dahingehend interpretiert werden, dass die im
Überschuss zugesetzten Mengen an TorD die TorA-Sequenz maskieren, wodurch diese
nicht mit dem TatBC-Rezeptor-Komplex in Interaktion treten kann und Transport
unterbunden wird.
Ein weiteres Charakteristikum des Tat-abhängigen in vitro-Transportes ist die exklusive
Energetisierung der Transportes durch den pH-Gradienten an der Membran [Yahr und
Wickner, 2001; Alami et al., 2002], wodurch es sich von allen anderen ProteinTransportwegen unterscheidet. Dies gilt auch für den in vitro-Transport des oxidierten TorAPhoAs, da durch den Einsatz von 0,1 mM CCCP der vollständige Transport des Proteins
(pTorA-PhoA + mTorA-PhoA) inhibiert werden konnte (Abb. IV.18B und 18C). CCCP ist ein
Protonophor, das einen bestehenden Protongradienten zerstört und dadurch den Tatabhängigen Transport von TorA-PhoA inhibiert.
Somit deutete die Abhängigkeit eines erfolgreichen Transportes von TorA-PhoA in INVs von
1.: überproduzierten Mengen an Tat-Proteinen in den verwendeten INVs, 2.: der Tatspezifischen Signalsequenz insbesondere deren Doppel-Arginin-Motifes und 3.: einem
intakten Protonengradienten, zweifelsfrei auf einen Tat-spezifischen Prozess hin. Die
Tatsache, dass nur die vollständig oxidierte Form des Proteins durch das Tat-System
transportiert wird, demonstriert die Fähigkeit des in vitro-Tat-Systems zur Erkennung der
beiden Redox- bzw. Faltungszustände von TorA-PhoA und bestätigt somit die mit diesem
Fusionsprotein gewonnenen in vivo-Daten [DeLisa et al., 2003]. Die erfolgreiche Etablierung
des Redox- bzw. Faltungs-abhängigen in vitro-Transportes in der vorliegenden Arbeit
ermöglichte die Charakterisierung der Interaktionen beider Konformationen mit dem TatSystem.
V.2. TorA-PhoA bindet faltungsunabhängig und TatABC-spezifisch an die
Membran
Die Etablierung des Tat-abhängigen und Faltungs-abhängigen in vitro-Transportes von TorAPhoA ermöglichte die Charakterisierung der Interaktionen beider konformationeller Zustände
des Proteins mit den Tat-Proteinen. Zunächst wurde die Zielsteuerung des Proteins durch
Untersuchungen zur Membranbindung von TorA-PhoA charakterisiert.
Dabei konnte gezeigt werde, dass das Protein unabhängig von seinem Faltungszustand mit
der Membran der INVs assoziierte (Abb. IV.9). Die Tat-abhängige Bindung von reduziertem
und somit ungefaltetem PhoA an den TatBC-Rezeptor-Komplex konnte bereits in einer in
100
V. Diskussion
vivo-Studie mit dem Tat-abhängigen Fusionsprotein HiPIP-PhoA durch Blaue NativeGelelektrophorese nachgewiesen werden [Richter und Brüser, 2005].
Außerdem konnte gezeigt werden, dass die Membranbindung des Tat-abhängigen Proteins
strikt TatABC-abhängig erfolgte, da mit Membranen, die keine Tat-Proteine enthielten eine
Assoziation nicht beobachtet wurde (Abb. IV.7 bis 9). Die Tat-spezifische Membranbindung
von
TorA-PhoA
ist
nicht
nur
hinsichtlich
des
Mechanismus
der
intrinsischen
Qualitätskontrolle von Bedeutung, sondern gibt auch Aufschluss über die Zielsteuerung der
Signalsequenz an die Membran.
So wurde eine unspezifische Zielsteuerung und Bindung der Signalsequenz von TatSubstraten an die Membran vorgeschlagen, die der initialen Interaktion mit der TatTranslokase vorausgeht. Dieses Modell basiert zum einen auf in vitro-Ergebnissen im
bakteriellen
als
auch
im
chloroplastidären
Tat-System,
in
denen
Tat-abängige
Fusionsproteine TatABC-unabhängig und auch unabhängig vom Doppel-Arginin-Motif
Bindung mit der Membran eingingen [Brüser et al., 2003; Hou et al., 2006]. Zum anderen
wurde Bindung von Tat-abhängigen GFP-Konstrukten an eine proteinfreie Modellmembran,
bestehend aus einer unilamellaren Schicht aus Phospholipiden durch Oberflächen-PlasmonResonanz-Spektroskopie beobachtet [Schanmugham et al., 2006]. Die Autoren dieser Studie
schlugen ein Modell für die Insertion der Signalsequenz vor, in dem die positiv geladenen
Reste der n-Region des Signalpeptids mit den negativen Ladung der MembranlipidKopfgruppen an der Oberfläche der Membran wechselwirken und anschließend die
hydrophobe h-Region des Signalpeptids in die Membran inseriert. Das so verankerte Protein
könnte durch laterale Diffusion in Kontakt zum TatBC-Rezeptor-Komplex gelangen
[Schanmugham et al., 2006], der nachweislich den initialen Kontakt des Tat-Substrates zum
Tat-System vermittelt [Cline und Mori, 2001; Alami et al., 2003]. Fraglich bleibt jedoch, ob die
Interaktion des Signalpeptids mit einer unilamellaren Membran mögliche Interaktionen mit
der Cytoplasmamembran in vivo widerspiegelt. Auf Grund der in der vorliegenden Arbeit
gewonnenen Ergebnisse, kann das Modell einer unspezifische Zielsteuerung des
Signalpeptids an die Membran nicht bestätigt werden, da eindeutig gezeigt wurde, dass die
Membranbindung von TorA-PhoA ausschließlich TatABC-abhängig erfolgte (Abb. IV.7 bis 9).
Eine Erklärung für die Diskrepanz der hier vorliegenden Ergebnisse zu denen in den oben
beschriebenen Studien liegt möglicherweise in den unterschiedlichen Versuchsbedingungen.
Denkbar wären beispielsweise Unterschiede in der Ionenstärke der verschiedenen
Versuchsansätze, wodurch eine mögliche elektrostatische Interaktion der Signalsequenz mit
der Membranoberfläche beeinflusst wäre.
Interessant ist in diesem Zusammenhang auch die Tatsache, dass der Austausch des
Doppel-Arginin-Motifs gegen zwei Lysinreste in der TorA-Signalsequenz von TorA-PhoA
zwar zu einer vollkommenen Inhibition des in vitro-Transportes führte (Abb. IV.6B und 17B),
101
V. Diskussion
die Bindung an die Membran dadurch aber nicht beeinträchtigt wurde, sondern im
vergleichbaren Maße zu der der Doppel-Arginin-Version des Proteins stattfand (Abb. IV.9D).
Interaktion von Tat-abhängigen Proteinen mit dem TatBC-Rezeptor-Komplex, deren DoppelArginin-Motif gegen zwei Lysinreste ausgetauscht wurde, konnte bereits in früheren Studien
beobachtet werden [Alder und Theg, 2003; McDevitt et al., 2006]. Allerdings wurden auch
gegenteilige Ergebnisse mit solchen Mutanten in vivo als auch in vitro beobachtet [Alami et
al., 2003; Richter und Brüser, 2005]. Es wird angenommen, dass das Doppel-Arginin-Motif
nicht die einzige Determinante der Tat-spezifischen Zielsteuerung darstellt [Kreutzenbeck et
al., 2007]. Die Ergebnisse zur Membranbindung der KK-Version von TorA-PhoA bestätigen
diese Annahme.
Die Ergebnisse dieser Arbeit zeigen, dass die TatABC-spezifische Membranbindung des
TorA-PhoA-Proteins weder ein Doppel-Arginin-Motif noch eine quasi-native Konformation
des oxidierten Proteins voraussetzt, so dass davon ausgegangen werden kann, dass die
primäre Zielsteuerung von Tat-Substraten unabhängig von dessen Transportkompetenz
erfolgt. Dies könnte bedeuten, dass sich der primären Zielsteuerung von Tat-Substraten ein
späterer, spezifischerer Erkennungsschritt anschließt, der über die Transportkompetenz des
Substrates entscheidet.
Interessanterweise kann die Bindung des oxidierten TorA-PhoA-Proteins an die Membran
durch den Einsatz von DCCD verhindert werden (Abb. IV.18C), einem Inhibitor der F1FOATPase,
der
mit
der
Carboxylgruppe
eines
konservierten
Aspartatrestes
der
c-
Untereinheiten des F0-Teils der F1FO-ATPase reagiert, und durch kovalente Modifikation
dieses Restes den Protonenfluss über die Membran durch den FO-Teil verhindert [Sebald et
al., 1980; Hermolin und Fillingame, 1989]. Ursprünglich wurde DCCD in der vorliegenden
Arbeit in vitro eingesetzt, um die Hydrolyse von ATP durch die in den INVs vorhandene F1FOATPase und der damit verbundenen Bildung eines für den Tat-abhängigen Proteintransport
benötigten Protonengradienten zu verhindern (Abb. IV.18A und 18B). Aus demselben Grund
wurde auch das Protonophor CCCP verwendet (Abb. IV.18A und 18B), von dem bereits
gezeigt wurde, den Tat-abhängigen Transport zu inhibieren [Alami et al., 2002; Blaudeck et
al., 2005]. CCCP war, wie auch DCCD, in der Lage den Tat-abhängigen in vitro-Transport
von TorA-PhoA zu inhibieren, zeigte allerdings im Gegensatz zu DCCD keine Hemmung der
Membranbindung von oxidiertem TorA-PhoA (Abb. IV.18C). Dies bedeutet, dass die
inhibitorische Wirkung von DCCD auf die Membranbindung des Proteins nicht auf der
Hemmung der F1FO-ATPase beruhte, was in Einklang zu anderen Befunden steht nach
denen die Bindung des Vorläuferproteins an den TatBC-Rezeptor-Komplex pH-Gradient
unabhängig verläuft [Cline und Mori, 2001; Alami et al., 2003, Dabney-Smith et al., 2006].
Über die Ursache der verhinderten Membranbindung durch DCCD kann nur spekuliert
werden. DCCD ist ein lipophiles Carbodiimid, das nur mit protonierten Carboxylresten in
102
V. Diskussion
einer hydrophoben Umgebung eine kovalente Bindung eingeht [von Ballmoos und Dimroth,
2007]. Neben der c-Untereinheit der F1FO-ATPase konnten inhibitorische Effekte von DCCD
auch für einen neuronalen Amintransporter [Suchi et al., 1991], einen Na+/H+-Austauscher
[Igarashi und Aronson, 1987] sowie einen H+/organischen Kationen-Antiporter der Niere [Kim
et al., 1993], als auch für den detoxifizierenden multidrug resistance efflux transporter EmrE
aus E. coli [Yerushalmi und Schuldiner, 2000] nachgewiesen werden. All diese Proteine sind
pH-Gradient-abhängige Transporter und Membranproteine, in denen eine für den
Mechanismus entscheidende und meist in der Membran eingebettete und konservierte
Carboxylgruppe durch DCCD kovalent modifiziert und dadurch inaktiviert wird. Als
potentielles Ziel einer DCCD-Modifikation im Tat-System, die die Bindung eines Substrates
beeinflussen könnte, kommen nur die Komponenten des TatBC-Rezeptor-Komplexes in
Frage, und von diesen insbesondere das TatC-Protein, von dem gezeigt wurde, die primäre
Erkennungsstelle des Signalpeptids zu besitzen und darüber hinaus diese Funktion auch in
Abwesenheit von TatB auszuüben [Alami et al., 2003]. TatC ist mit 248 Aminosäuren das
größte
der
drei
Tat-Proteine
und
besitzt
wahrscheinlich
eine
Topologie
mit
6
transmembranen Helices [Mori et al, 2001; Behrendt et al., 2004; Ki et al., 2004].
Vergleichende Sequenzanalysen ergaben, dass sich unter den etwa 120 Aminosäuren, die
die 6 Transmembrandomänen bilden, 3 saure Aminosäuren befinden (E170, D211, E227),
die ausnahmslos
hoch-konserviert sind [Buchanan et al., 2002]. Verschiedene
Mutationsstudien lieferten allerdings gegensätzliche Aussagen darüber, ob einer dieser
Reste essentiell für die Funktion von TatC ist [Buchanan et al., 2002; Barrett et al., 2005;
Holzapfel et al., 2007]. Dennoch stellen diese Aminosäuren in TatC potentielle
Reaktionspartner für DCCD dar. Da nicht ausgeschlossen werden kann, dass DCCD auch
mit sauren Aminosäuren außerhalb der vorhergesagten Transmembrandomänen reagiert,
soll hier noch die erste cytoplasmatische Schleife von TatC hervorgehoben werden. Diese
weist als potentiellen DCCD-Reaktionspartner einen absolut konservierten und auch sehr
wahrscheinlich essentiellen Glutamatrest an Position 103 auf [Buchanan et al., 2002;
McDevitt et al., 2005; Holzapfel et al., 2007]. Zwar ist bekannt, dass die gesamte N-terminale
Hälfte von TatC an der Bindung der Signalsequenz beteiligt ist [Holzapfel et al., 2007], doch
spielt diese cytoplasmatische Schleife wahrscheinlich eine Rolle in der Erkennung des
Doppel-Arginin-Motifes des Signalpeptides [Kreutzenbeck et al., 2007].
103
V. Diskussion
V.3. Erkennung der Faltungszustände von TorA-PhoA durch das Tat-System
In der vorliegenden Arbeit konnte gezeigt werden, dass das in vitro-synthetisierte TorAPhoA-Protein Tat-abhängig transportiert wird. Dabei wurde selektiv nur die oxidierte,
gefaltete Konformation des Proteins transportiert, wodurch die Fähigkeit des Tat-Systems
zur Erkennung von Faltungszuständen und somit von transportkompetenten Konformationen
demonstriert wurde. Es konnte außerdem gezeigt werden, dass das TorA-PhoA TatABCabhängig an die Membran bindet. Diese Interaktion war allerdings unabhängig von der
Transportkompetenz des TorA-PhoA-Proteins, was darauf hindeutete, dass die Erkennung
von transportkompetenten Konformationen nach dem initialen Kontakt des Tat-abhängigen
Proteins an den TatBC-Rezeptor-Komplex erfolgen musste.
In der vorliegenden Arbeit wurde versucht, Unterschiede in den molekularen Interaktionen
der Signalsequenz beider konformationeller Zustände des TorA-PhoA-Proteins zu den
Komponenten des Tat-Systems zu analysieren. Dazu wurde das photoaktivierbare
Phenylalaninderivat pBPA (para-Benzoyl-L-Phenylalanin) ortsspezifisch in die Signalsequenz
eingebaut. Grundlage für diese Herangehensweise waren die Ergebnisse aus in vitroQuervernetzungsstudien
mit
dem
photoaktivierbaren
Quervernetzer
Tmd-Phe
(Trifluoromethyldiazirino-Phenylalanin) und dem natürlichen Tat-Substrat SufI aus E. coli.
Darin konnte gezeigt werden, dass die gesamte Signalsequenz in Kontakt zu TatB steht,
während sich Interaktionen von TatC auf den Bereich um das Konsensusmotif beschränken
[Alami et al., 2003].
Durch die Inkorporation von pBPA konnten, in Abwesenheit von Membranen, Interaktionen
des Konsensusmotifs, als auch der hydrophoben Region der Signalsequenz mit den
löslichen Chaperonen FkpA und TorD nachgewiesen werden (Abb. IV.15), wodurch die freie
Zugänglichkeit der Signalsequenz demonstriert wurde, wenn diese nicht durch Tat-Proteine
erkannt wird. TorD ist ein TorA-Signalsequenz-spezifisches, cytoplasmatisches Chaperon
[Dubini und Sargent, 2003; Jack et al., 2001 und 2004], während FkpA ein periplasmatisches
Chaperon
ist,
das
in
dem
für
die
in
vitro-Synthese
verwendeten
Zellextrakt
präparationsbedingt enthalten ist und nachweislich keinen Einfluss auf den in vitro-Transport
Tat-abhängiger Proteine hat [E. Holzapfel, Dissertation].
In Anwesenheit von TatABC-haltigen Membranen (Tat+-INV) wurde die Interaktion zu den
löslichen Chaperonen verdrängt. Stattdessen konnte für die transportkompetente, oxidierte
Konformation des TorA-PhoA-Proteins ein Kontakt der Region um das Konsensus-Motif mit
TatC nachgewiesen werden (Abb. IV.13), wodurch die Ergebnisse der oben erwähnten
Studie mit dem natürlichen Tat-Substrat SufI bestätigt werden konnten. Kontakt zu TatB
allerdings konnte nur für die hydrophobe Region der Signalsequenz nachgewiesen werden
(Abb. IV.14), während die Region um das Konsensusmotif nicht in Kontakt zu TatB stand
104
V. Diskussion
(Abb. IV.13). Letzteres unterscheidet sich von den erwähnten Ergebnissen mit dem SufIProtein, dessen Region um das Konsensusmotif auch mit TatB interagiert. Die Ursache für
diese Diskrepanz ist nicht klar, könnte aber in der unterschiedlichen Reichweite, bedingt
durch unterschiedliche molekulare Struktur, der verwendeten Quervernetzer liegen.
Das reduzierte, transportinkompetente TorA-PhoA-Protein interagiert mit TatB, im Vergleich
zu dem transportkompetenten, oxidierten Protein, in abgeschwächter Stärke (Abb. IV.14).
Unterschiede zwischen den beiden konformationellen Formen des TorA-PhoA-Proteins
konnten insbesondere in der Interaktion der Region um das Konsensusmotif zu TatC
beobachtet werden, da das reduzierte, ungefaltete Protein praktisch keine Interaktion zu
TatC aufwies (Abb. IV.13).
Dies bedeutet, dass, obwohl das reduzierte, transportinkompetente Protein TatABCabhängig an die Membran bindet, es sich in seiner molekularen Interaktion zum TatBCRezeptor-Komplex von der des oxidierten transportkompetenten Proteins in der Stärke
seiner TatB-, aber vor allem TatC-Interaktion unterscheidet. Dies impliziert wiederum, dass
der Rezeptor-Komplex in der Lage zu sein scheint, Faltungsunterschiede des Substrates zu
erkennen, was sich in einer abgeschwächten Bindung des Signalpeptides an TatB und TatC
äußert. Die Beobachtung, dass TatB auch mit der transportinkompetenten, reduzierten Form
des TorA-PhoA-Proteins interagiert, zeugt womöglich, im Vergleich zu TatC, von einer
geringeren Spezifität der TatB-Interaktionen zu der Signalsequenz des Tat-Substrates.
Unterstützt wird diese Annahme von der Beobachtung, das eine TatB-Interaktion auch zu
einer transportinkompetenten KK-Mutante von SufI beobachtet wurde, während der Kontakt
zu TatC durch den Austausch des Doppel-Arginin-Paares gegen zwei Lysine vollständig
inhibiert war [Alami et al., 2003].
Die Ergebnisse der vorliegenden Arbeit unterstützen die Vorstellung von einer intrinsischen
Qualitätskontrolle des zu transportierenden Substrates durch das Tat-System. Ihnen zufolge
muss die Fähigkeit zur Erkennung von Faltungszuständen der Tat-Translokase selbst
zugesprochen werden, im speziellen wahrscheinlich dem TatC-Protein. Unklar dabei bleibt
allerdings
der
molekulare
Mechanismus
der
Detektion.
Interessant
in
diesem
Zusammenhang sind die Ergebnisse einer Studie, in der gezeigt werden konnte, dass kurze
hydrophile, ungefaltete Proteine Tat-abhängig in E. coli-Zellen transportiert werden, solange
keine hydrophoben Sequenzen in dem Protein enthalten waren [Richter et al., 2007]. Diese
Effekte wurden dahingehend interpretiert, dass Tat-Substrate grundsätzlich unabhängig von
ihrem Faltungszustand transportiert werden und erst durch die Interaktion exponierter
hydrophober Sequenzen mit Lipiden der Membran der Transport verhindert wird. Die
Autoren schlossen dabei die Existenz einer hydrophilen Pore aus, sondern postulierten eine
durch TatA verursachte strukturelle Schwächung der Membran, durch die das Tat-Substrat
105
V. Diskussion
hindurch gezogen würde [Richter et al., 2007, Sanders und Brüser, 2003]. Vor dem
Hintergrund der in dieser Arbeit präsentierten Ergebnisse ist eher ein Mechanismus zur
Verhinderung des Transportes eines nicht-nativen Substrates vorstellbar, der durch die
Erkennung hydrophober Seitenketten des ungefalteten Substrates durch den TatBCRezeptor-Komplex zustande kommt. Weiterhin wäre denkbar, dass durch diese Art von
Interaktion die Rekrutierung von TatA verhindert würde, wodurch die Assemblierung der
vollständigen Translokase und somit der Transport verhindert würde. TatA gilt auf Grund
seiner homooligomeren Organisation in TatA-Komplexen der E. coli-Membran, sowie auf
Grund der ringförmigen, porenähnlichen Struktur der TatA-Komplexe als potentielle Pore
[Porcelli et al., 2002; Oates et al., 2005; Gohlke et al., 2005], die nach der Bildung des
Substrat-Rezeptor-Komplexes
zur
vollständigen
Assemblierung
der
Tat-Translokase
rekrutiert wird [Mori und Cline, 2002; Alami et al., 2003; Dabney-Smith et al., 2006]. Diese
Vorstellung von einer Substrat-Transportkompetenz-abhängigen Assemblierung wäre
darüber hinaus eine mögliche Erklärung dafür, warum die Translokase bei jedem
Translokationsvorgang von neuem assembliert und nach vollendetem Transport wieder in
die zwei Subkomplexe TatBC-Rezeptor-Komplex und TatA-Komplex dissoziiert.
V.4. Charakterisierung eines potentiellen Transportintermediates
Wie bereits erwähnt, trat in Anwesenheit von Tat+-INVs während des Tat-abhängigen
Transportes von oxidiertem TorA-PhoA neben dem unprozessierten, transportierten pTorAPhoA und prozessierten, transportierten mTorA-PhoA eine weitere Protease-geschützte
Spezies mit einer geringeren elektophoretischen Mobilität als mTorA-PhoA auf, die als iTorAPhoA (intermediäres TorA-PhoA) bezeichnet wurde (Abb. IV.2B, 4A, 16). Die Bildung von
iTorA-PhoA war dabei abhängig zum einen, von der oxidierten Form des Proteins
(Abb. IV.16) und zum anderen, von der Gegenwart der Tat-Proteine der verwendeten
Membranen (Abb. IV.4A). Diese Abhängigkeiten ließen vermuten, es handle sich bei iTorAPhoA
um
ein
unvollständig
transportiertes
TorA-PhoA-Protein,
das
eine
partielle
Proteaseresistenz erlangt hatte.
Eine kinetische Analyse des in vitro-Transportes ließ die Formation von iTorA-PhoA zu
einem Zeitpunkt erkennen, zu dem der vollständige Transport des Proteins bereits sein
Maximum erreicht hat (Abb. IV.21). Möglicherweise stehen diese beiden Beobachtungen in
kausalem Zusammenhang zueinander, in einer Weise, dass, wenn der vollständige
Transport des TorA-PhoA-Proteins zum Erliegen kommt, dies die Bildung von iTorA-PhoA
zur Folge hat. Eine mögliche Erklärung für diese Beobachtung liefern die Ergebnisse einer
Studie, die eine Überlastung bzw. Sättigung der Tat-Translokase durch die Überproduktion
106
V. Diskussion
von Tat-abhängigem Substrat in vivo nachweisen konnte [Yahr und Wickner, 2001]. Der
gleiche Effekt könnte auch die Ursache für die Formation des iTorA-PhoAs sein. Ein weitere
Erklärung liegt möglicherweise in der generellen Begrenzung der Transportkapazität des
verwendeten in vitro-Systems, das wie Eingangs erwähnt, nur durch eine Überproduktion der
Tat-Proteine zu einem Tat-abhängigen Transport befähigt ist [Yahr und Wickner, 2001; Alami
et al., 2002], der beispielsweise für das natürliche Tat-Substrat SufI bei einer Effizienz von
ca. 20% liegt [Alami et al., 2002].
Durch Mutation des Doppel-Arginin-Motifs konnte gezeigt werden, dass für die Bildung von
iTorA-PhoA neben der Gegenwart der Tat-Proteine, auch ein intaktes Doppel-Arginin-Motif
nötig war, da durch die Substitution beider Arginine mit zwei Lysinen die Bildung aller
proteaseresistenter
Proteine
unterbunden
wurde
(Abb. IV.17B).
Auffällig
war
die
Beobachtung, dass die Substitution des ersten der beiden Arginine des Doppel-ArgininMotifs durch Lysin den vollständigen Transport nicht beeinflusste, während die Bildung von
iTorA-PhoA signifikant reduziert war (Abb. IV.17B). Offensichtlich ist eine abgeschwächte
Interaktion des Doppel-Arginin-Motifs zum TatBC-Rezeptor-Komplex, hervorgerufen durch
den Austausch des ersten der beiden Arginine, von besonderer Bedeutung für die Bildung
von iTorA-PhoA. Die abgeschwächte Interaktion führt möglicherweise zu einer geringeren
Überlastung der Tat-Translokase und somit zu einer verminderten Bildung des iTorA-PhoA.
Die Tatsache, dass ohne das Doppel-Arginin-Motif kein iTorA-PhoA gebildet wurde zeigt,
dass für die Bildung des iTorA-PhoAs eine produktive Bindung und Erkennung durch den
TatBC-Rezeptor-Komplex absolut notwendig ist. Dieses Verhalten würde man nur von einem
echten Transportintermediat erwarten. Außerdem wurde dadurch deutlich, dass im Fall des
iTorA-PhoAs der Transport an einem Punkt des Translokationsprozesses gestoppt wird, der
nach der produktiven Erkennung des Vorläuferproteins durch den Rezeptor-Komplex
stattfindet.
Um den Moment des Transportarrestes während des Transportvorganges genauer zu
definieren, wurde die Abhängigkeit der iTorA-PhoA-Bildung von dem für den Transport
notwendigen pH-Gradienten an der Membran untersucht. Als pH-abhängiger Schritt des
Translokationsmechanismus gilt die vollständige Assemblierung der Tat-Translokase aus
dem Substrat-Rezeptor-Komplex und dem TatA-Komplex und findet somit nach der
Erkennung des Tat-Substrates statt [Mori und Cline, 2002; Alami et al., 2003; Dabney-Smith
et al., 2006]. Im bakteriellen System wird ein bereits assemblierter TatA-Komplex rekrutiert
[Sargent et al., 2001; Alami et al., 2003], während im pflanzlichen Tat-System die
Assemblierung
des
TatA-Komplexes
durch
die
Bildung
Substrat-Rezeptor-Komplex
ausgelöst wird [Dabney-Smith et al., 2006].
107
V. Diskussion
Durch den Einsatz des Protonophors CCCP oder des F1FO-ATPase-Inhibitors DCCD
während eines in vitro-Transportes von TorA-PhoA, wurde der vorhandene Gradient zerstört,
bzw. der Aufbau eines Gradienten verhindert. Beide Inhibitoren waren in der Lage, den
Transport von TorA-PhoA als auch den des Kontrollproteins SufI zu blockieren (Abb. IV.18A
und 18B). Da die Bildung des Transportintermediates nicht durch CCCP beeinflusst wurde,
kann davon ausgegangen werden, dass dessen Formation noch vor dem pH-abhängigen
Schritt der Translokation stattfand.
Anders als CCCP hatte DCCD die Inhibition der Bildung von iTorA-PhoA zur Folge
(Abb. IV.18B). Wie bereits ausführlich erläutert, ist dieser Effekt sehr wahrscheinlich auf eine
Modifikation der TatBC-Komponenten durch DCCD zurückzuführen, die zu einer Hemmung
der Tat-spezifischen Bindung von oxidiertem TorA-PhoA an die Membran führt.
Durch Mutation der Signalpeptidase-spezifischen Schnittstellen (AXA) konnte eine TorAPhoA-Version konstruiert werden, die nach erfolgtem Transport nicht prozessiert wurde
(Abb. IV.19). Während durch diese Mutation kein reifes TorA-PhoA entstand aber im
gleichen Maße Vorläuferprotein Proteaseresistenz erlangte, wurde die Bildung des
Intermediates iTorA-PhoA nicht beeinflusst. Demzufolge stammt iTorA-PhoA nicht von der
reifen, prozessierten Form des Proteins ab, sondern entstand durch den partiellen
proteolytischen Verdau des Vorläuferproteins pTorA-PhoAs, das sich nicht im schützenden
Lumen der Membranvesikel befindet. Darüber hinaus wird deutlich, dass die Bildung von
iTorA-PhoA vor der proteolytischen Abspaltung des Signalpeptides passieren muss. Die
Signalpeptidase-spezifische Prozessierung findet höchstwahrscheinlich zu einem späteren
Zeitpunkt des Tat-abhängigen Translokationsprozesses statt. So wurde beispielsweise der
Transport des reifen Teils des Tat-abhängigen Proteins sogar dann beobachtet, wenn das
Signalpeptid während des Transportes kovalent an TatC gebunden war [Gérard und Cline,
2006], wodurch auch verdeutlicht wurde, dass Prozessierung keine Voraussetzung für
Transport ist und diese sich deshalb wahrscheinlich erst zu einem sehr späten Zeitpunkt der
Translokation ereignet.
Die iTorA-PhoA Form ist demnach das Produkt von oxidiertem Vorläuferprotein, das in eine
produktive Interaktion mit der Tat-Translokase getreten ist, die über die initiale Bindung
durch den TatBC-Rezeptor-Komplex hinaus ging, so dass partielle Proteaseresistenz erlangt
wurde. Durch affinitätschromatographische Analyse konnte bewiesen werden, dass im Fall
des iTorA-PhoA der His-Tag tragende C-Terminus durch den Verdau durch Proteinase K
verloren gegangen ist (Abb. IV.20). Demnach befindet sich der C-Terminus des Proteins
nicht
in
einer
Protease-geschützten
Umgebung.
Dennoch
kann
nicht
gänzlich
ausgeschlossen werden, dass iTorA-PhoA das Produkt eines Verdaus des C-Terminus als
108
V. Diskussion
auch N-Terminus ist. Gegen diese Möglichkeit spricht aber die aus unterschiedlichen Studien
resultierenden Annahme, die Signalsequenz sei während der Translokation durch den
TatBC-Rezeptor-Komplex tief in die Membran inseriert [Gerald und Cline, 2007;
Kreutzenbeck et al., 2007], wo diese vor dem proteolytischen Angriff durch eine Protease
geschützt wäre. Außerdem kann davon ausgegangen werden, dass sich der ungeschützte
C-Terminus auf der cytoplasmatischen Seite der Membran befindet, da nach der Zerstörung
der Integrität der Vesikel durch 1% Triton X100 die molekulare Größe von iTorA-PhoA
unverändert bleibt (Abb. IV.4A, 16, 20).
Zusammenfassend konnte in der vorliegenden Arbeit gezeigt werden, dass iTorA-PhoA das
Produkt eines partiellen proteolytischen Abbaus durch Proteinase K darstellt, das von einer
Population an oxidiertem TorA-PhoA-Protein stammt, welches sich nur in Gegenwart der
Tat-Proteine und in Abhängigkeit von dem Doppel-Arginin-Motif der TorA-Signalsequenz
bildet. Der Bildung von iTorA-PhoA liegt eine Erkennung des Proteins durch den TatBCRezeptor-Komplex zu Grunde, die nur dann produktiv ist, wenn das erkannte Protein im
gefalteten, oxidierten und somit transportkompetenten Zustand vorliegt. Die initiale Bindung
des Proteins an den TatBC-Rezeptor-Komplex kann durch DCCD inhibiert werden. Auf
Grund der Überlastung bzw. Sättigung der Tat-Translokase wird der Transport eines
signifikanten Teils des erkannten, oxidierten Proteins nicht vollständig transportiert. Der
Translokationsarrest erfolgt nach der initialen Bindung an den TatBC-Rezeptor-Komplex,
jedoch vor dem pH-abhängigen Schritt der Translokation und unabhängig von der
Prozessierung durch die Signalpeptidase. Dabei gelangt das Transportintermediat in eine
partiell Protease-geschütze Umgebung, die auch nach der Zerstörung der MembranIntegrität durch Triton X 100 erhalten bleibt. Wahrscheinlich ist lediglich der C-Terminus des
partiell
transportierten
Proteins
auf
der
cytoplasmatischen
Seite
der
Membran
proteolytischem Verdau ausgesetzt.
V.5. Translokationsarrest findet in der unmittelbaren Nähe von TatA statt
Interaktion des Vorläuferproteins pTorA-PhoA mit den Tat-Proteinen führte zur Bildung des
Transportintermediates iTorA-PhoA, die sich in einer partiellen Resistenz gegenüber
Proteinase K widerspiegelte (Abb. IV.4A, 16, 20). Die Protease-Resistenz von iTorA-PhoA
wurde auch nach der Zerstörung der Membranintegrität mit dem Detergenz Triton X100 nicht
beeinträchtigt (Abb. IV.4A, 16, 22). Auf Grund dieser Beobachtung wurde von einer
Solubilisierung von Membranstrukturen durch Triton X100 ausgegangen, die das TorAPhoA-Protein enthalten und in einer schützenden Weise umgeben. Es konnte gezeigt
109
V. Diskussion
werden, dass die Protease-Resistenz von iTorA-PhoA nicht auf Triton X100 beschränkt war,
sondern auch in Gegenwart von anderen Detergenzien, wie C12E9, CHAPS und
Octylglycosid zu beobachten war (Abb. IV.22). Dabei kamen Konzentrationen zum Einsatz,
von denen berichtet wurde, in der Lage zu sein, intakte TatA-Komplexe aus der
Cytoplasmamembran von E. coli zu solubilisieren [de Leeuw et al., 2002; McDevitt et al.,
2006]. Die Resistenz des Transportintermediates gegenüber diesen Detergenzien ähnelte
somit der von E. coli TatA-Komplexen. Tatsächlich konnte außerdem gezeigt werden, dass
die Bildung von iTorA-PhoA in Gegenwart von Membranen aus einem Stamm, der nur TatB
und TatC überproduziert, nicht zu beobachten war (Abb. IV.23A). Überproduzierte Mengen
an TatA waren demnach unbedingt für die Formation eines partiell Protease-resistenten
Transportintermediates
nötig.
Darüber
hinaus
konnte
ein
direkter
Kontakt
des
Transportintermediates zu TatA durch Präzipitation von iTorA-PhoA mit TatA-Antikörpern
nachgewiesen werden (Abb. IV. 23B und 23C).
Diese Ergebnisse weisen somit eine TatA-abhängige Formation des Transportintermediates
auf, wobei TatA in direktem Kontakt zu iTorA-PhoA steht. Zusammen mit der beobachteten
Resistenz von iTorA-PhoA gegenüber proteolytischem Verdau in Gegenwart der
verwendeten Detergenzien und unter der Annahme, TatA bildet TatA-Komplexe, die im E.
coli-Tat-System die Pore der Tat-Translokase repräsentieren, kann davon ausgegangen
werden, dass das Vorläuferprotein in dieser Pore und somit in der Membran während des
Transportes stecken bleibt. Das Vorläuferprotein ist dabei vom TatA-Komplex in einer Weise
umgeben, dass bis auf den C-Terminus auf der cytoplasmatischen Seite der Membran, das
TorA-PhoA-Protein vor proteolytischem Verdau geschützt wird. Da auch der TatBC-Komplex
geringe Mengen an TatA-Protomeren aufweist [Bolhuis et al., 2001; McDevitt et al., 2005],
kann zwar nicht ausgeschlossen werden, dass der TatA-Kontakt von TatA-Protomeren des
Rezeptor-Komplexes stammt. Da jedoch Kontakt von iTorA-PhoA zu TatB oder TatC nicht
nachgewiesen werden konnte, wird diese Möglichkeit als unwahrscheinlich betrachtet
(Abb. IV.23B und 23C). Diese Ergebnisse sind in mehrerer Hinsicht von Bedeutung.
Alle Modelle zur Tat-abhängigen Translokation schlagen TatA als die Komponente vor, die
die Translokation durch die Membran ermöglicht. Strittig ist allerdings, in welcher Weise dies
geschieht. Das gängigste Modell schlägt in Bakterien als auch in Chloroplasten den TatAKomplex als hydrophile Pore der Tat-Translokase vor, durch die das Tat-Substrat
hindurchgeschleust
wird
und
sich
dem
Substratmolekül
so
anpasst,
dass
die
Membranintegrität erhalten bleibt und ungewollter Ionenfluss dadurch verhindert wird. Diese
Annahme basiert im Wesentlichen auf der homooligomeren Organisation von TatA in der
Membran
[de
Leeuw
et
al.,
2001;
Porcelli
et
al.,
2002]
als
auch
auf
elektronenmikroskopischen Analysen von TatA-Komplexen der E. coli- Membran, die
ringförmige Strukturen unterschiedlicher Größen aufweisen [Gohlke et al., 2005].
110
V. Diskussion
Dem gegenüber stehen Nicht-Poren-Modelle eines Transmembrantransportes durch
Schwächung der Membranintegrität [Brüser und Sanders, 2003; Dabney-Smith et al., 2006].
Nach der Vorstellung von Brüser und Sanders geht dabei dem eigentlichen Transport eine
Insertion des Signalpeptids voraus, dem dann eine Erkennung durch den TatBC-RezeptorKomplex
folgt
und
schließlich
durch
die
punktuelle
Ansammlung
von
TatA-
Transmembranhelices um das Substrat eine Schwächung der Membranintegrität es
ermöglicht, das Protein durch die Membran zu transportieren, wobei die Membranlipide die
Membran ionisch dicht halten [Brüser und Sanders, 2003]. Unterstützt wird die Vorstellung
eines Transports durch einen destabilisierten Bereich der Membran von Studien zu einem
Transportintermediat des pflanzlichen Tat-Systems, das während des Transportes in der
Membran stecken blieb, aber keinen Kontakt zu einem der Tat-Proteine aufwies [Cline und
McCaffery, 2007]. Diese Ergebnisse wurden dahingehend interpretiert, dass eine stabile
Protein-schleusende
Pore
wahrscheinlich
nicht
existiert.
Dennoch
konnte
eine
hochdynamische, transiente Pore, die im Fall eines Translokationsarrestes dissoziiert, nicht
ausgeschlossen werden.
Die Ergebnisse der vorliegenden Arbeit unterstützen eher die Vorstellung von einer durch
den TatA-Komplex gebildeten Pore, die im direkten Kontakt zu dem Substrat-Protein steht
und dieses während des Transportes umgibt. Darüber hinaus findet dieser Kontakt in einer
Weise statt, die einen stabilen Komplex aus einem Transportintermediat wie iTorA-PhoA mit
TatA im Fall eines Translokationsarrestes erlaubt. Tatsächlich wurde ein vergleichbares
Transportintermediat
bereits
im
chloroplastidären
System
beobachtet.
Partielle
Proteaseresistenz eines Tat-abhängigen, nicht-pozessierbaren Proteins wurde in vitro als
direkte Folge einer Interaktion mit dem pflanzlichen Tat-System nachgewiesen [Di Cola und
Robinson, 2005]. Allerdings war, anders als bei iTorA-PhoA, die Proteaseresistenz nicht in
Anwesenheit von Triton X100 gegeben. Dieser Unterschied könnte sich aus der transienten
und instabilen Natur des Tha4-Komplexes ableiten [Dabney-Smith et al., 2006], wodurch ein
potentieller Komplex aus Substrat und Tha4-Komplex sensitiver gegenüber der Behandlung
mit Detergenzien sein könnte. Darüber hinaus konnte auch im pflanzlichen Tat-System mit
einem Fusionsprotein aus dem natürlichen Tat-Substrat 23kDa-Protein und Avidin erreicht
werden, dass dieses Konstrukt in der Thylakoidmembran stecken blieb. Die Tatsache, dass
dadurch der Transport von 23kDa-Protein ohne Avidin blockiert wurde, spricht für die
Blockierung der Tat-spezifischen Pore [Asai et al., 1999]. Ein vergleichbarer Effekt liegt
wahrscheinlich auch im Fall der Bildung von iTorA-PhoA vor.
Der nachgewiesen Kontakt des Transportintermediates iTorA-PhoA zu TatA ist auch
hinsichtlich der Assemblierung der Tat-Translokase aus deren Subkomplexen TatBCRezeptor-Komplex und TatA-Komplex interessant. In vitro-Studien im pflanzlichen als auch
im bakteriellen System haben gezeigt, dass die Rekrutierung von TatA zur Assemblierung
111
V. Diskussion
der vollständigen, aktiven Translokase abhängig ist von der Bildung des Substrat-TatBCRezeptor-Komplexes sowie eines intakten pH-Gradienten [Mori und Cline, 2002; Alami et al.,
2003]. Durch den Einsatz des Protonophors CCCP konnte für den Tat-abhägigen in vitroTransport von TorA-PhoA gezeigt werden, dass der Translokationsarrest vor dem pHabhängigen Schritt des Transportes stattfinden muss (Abb. IV.18B und 18C). Somit sind der
Kontakt des Transportintermediates iTorA-PhoA zu TatA und demzufolge auch die
Rekrutierung von TatA durch den Substrat-TatBC-Rezeptor-Komplex nicht von einem
intakten pH-Gradienten abhängig. Anders als die Bildung des Transportintermediates ist der
vollständige Transport des TorA-PhoA-Proteins allerdings pH-Gradient-abhängig. Eine
ähnliche Beobachtung wurde in pflanzlichen Tat-System in in vivo-Studien gemacht. Dabei
wurde eine nicht-prozessierbare Mutante eines Tat-abhängigen GFP-Proteins verwendet,
deren Transport zu 50% unvollständig verlief und dadurch ein durch die Thylakoidmembran
Protease-geschütztes Intermediat generierte. Durch den Einsatz des Entkopplers Nigericin
konnte gezeigt werden, dass für die Bildung des Intermediates kein pH-Gradient benötigt
wurde [Di Cola et al., 2005]. Diese Daten decken sich demnach mit den in der vorliegenden
Arbeit erhaltenen Ergebnissen, wonach ein pH-Gradient weder für eine funktionelle
Assemblierung des Tat-Translokase noch für die frühe Phase der eigentlichen Translokation
des Substrat-Proteins benötigt wird. Lediglich die vollständige Translokation ist pH-abhängig.
Demnach ist Energetisierung durch den pH-Gradienten erst zu einem späten Zeitpunkt der
Translokation von Bedeutung.
112
VI. Literaturverzeichnis
VI. Literaturverzeichnis
Akimaru, J., Matsuyama, S., Tokuda, H., and Mizushima, S. (1991). Reconstitution of a protein
translocation system containing purified SecY, SecE, and SecA from Escherichia coli. Proceedings of the
National Academy of Sciences of the United States of America 88, 6545-6549.
Akiyama, Y., and Ito, K. (1987). Topology analysis of the SecY protein, an integral membrane protein
involved in protein export in Escherichia coli. Embo Journal 6, 3465-3470.
Akiyama, Y., Kamitani, S., Kusukawa, N., and Ito, K. (1992). In vitro catalysis of oxidative folding of
disulfide-bonded proteins by the Escherichia coli dsbA (ppfA) gene product. The Journal of biological
chemistry 267, 22440-22445.
Akiyama, Y., and Ito, K. (1993). Folding and assembly of bacterial alkaline phosphatase in vitro and in
vivo. The Journal of biological chemistry 268, 8146-8150.
Alami, M., Trescher, D., Wu, L.-F., and Müller, M. (2002). Separate Analysis of Twin-arginine
Translocation (Tat)-specific Membrane Binding and Translocation in Escherichia coli. The Journal of
biological chemistry 277, 20499-20503.
Alami, M., Luke, I., Deitermann, S., Eisner, G., Koch, H.-G., Brunner, J., and Müller, M. (2003).
Differential Interactions between a Twin-Arginine Signal Peptide and Its Translocase in Escherichia coli.
Molecular Cell 12, 937-946.
Alder, N.N., and Theg, S.M. (2003). Protein transport via the cpTat pathway displays cooperativity and is
stimulated by transport-incompetent substrate. FEBS Letters 540, 96-100.
Angelini, S., Deitermann, S., and Koch, H.G. (2005). FtsY, the bacterial signal-recognition particle
receptor, interacts functionally and physically with the SecYEG translocon. EMBO Reports 6, 476-481.
Asai, T., Shinoda, Y., Nohara, T., Yoshihisa, T., and Endo, T. (1999). Sec-dependent pathway and
DeltapH-dependent pathway do not share a common translocation pore in thylakoidal protein transport.
The Journal of biological chemistry 274, 20075-20078.
Bageshwar, U.K., and Musser, S.M. (2007). Two electrical potential-dependent steps are required for
transport by the Escherichia coli Tat machinery. The Journal of cell biology 179, 87-99.
Baneyx, F., and Georgiou, G. (1990). In vivo degradation of secreted fusion proteins by the Escherichia
coli outer membrane protease OmpT. Journal of bacteriology 172, 491-494.
Barrett, C.M.L., Mathers, J.E., and Robinson, C. (2003). Identification of key regions within the
Escherichia coli TatAB subunits. FEBS Letters 537, 42-46.
Barrett, C.M.L., Mangels, D., and Robinson, C. (2005). Mutations in Subunits of theEscherichia coli
Twin-arginine Translocase Block Function via Differing Effects on Translocation Activity or Tat Complex
Structure. Journal of molecular biology 347, 453-463.
Barrett, C.M., Freudl, R., and Robinson, C. (2007). Twin arginine translocation (Tat)-dependent export
in the apparent absence of TatABC or TatA complexes using modified Escherichia coli TatA subunits that
substitute for TatB. The Journal of biological chemistry 282, 36206-36213.
113
VI. Literaturverzeichnis
Beck, K., Wu, L.F., Brunner, J., and Müller, M. (2000). Discrimination between SRP- and SecA/SecBdependent substrates involves selective recognition of nascent chains by SRP and trigger factor. Embo
Journal 19, 134-143.
Beck, K., Eisner, G., Trescher, D., Dalbey, R.E., Brunner, J., and Müller, M. (2001). YidC, an
assembly site for polytopic Escherichia coli membrane proteins located in immediate proximity to the
SecYE translocon and lipids. EMBO Reports 2, 709-714.
Behrendt, J., Standar, K., Lindenstrauss, U., and Brüser, T. (2004). Topological studies on the twinarginine translocase component TatC. FEMS microbiology letters 234, 303-308.
Behrendt, J., Lindenstrauss, U., and Brüser, T. (2007). The TatBC complex formation suppresses a
modular TatB-multimerization in Escherichia coli. FEBS Letters 581, 4085-4090.
Berg, B.v.d., Clemons, W.M., Collinson, I., Modis, Y., Hartmann, E., Harrison, S.C., and Rapoport,
T.A. (2004). X-ray structure of a protein-conducting channel. Nature 427, 36-44.
Berks, B.C., Sargent, F., and Palmer, T. (2000). The Tat protein export pathway. Molecular microbiology
35, 260-274.
Berks, B.C., Palmer, T., and Sargent, F. (2003). The Tat protein translocation pathway and its role in
microbial physiology. Advances in microbial physiology 47, 187-254.
Bernstein, H.D., Poritz, M.A., Strub, K., Hoben, P.J., Brenner, S., and Walter, P. (1989). Model for
signal sequence recognition from amino-acid sequence of 54K subunit of signal recognition particle.
Nature 340, 482-486.
Bessette, P.H., Aslund, F., Beckwith, J., and Georgiou, G. (1999). Efficient folding of proteins with
multiple disulfide bonds in the Escherichia coli cytoplasm. Proceedings of the National Academy of
Sciences of the United States of America 96, 13703-13708.
Blaudeck, N., Kreutzenbeck, P., Müller, M., Sprenger, G.A., and Freudl, R. (2005). Isolation and
Characterization of Bifunctional Escherichia coli TatA Mutant Proteins That Allow Efficient Tat-dependent
Protein Translocation in the Absence of TatB. The Journal of biological chemistry 280, 3426-3432.
Blaudeck, N., Kreutzenbeck, P., Freudl, R., and Sprenger, G.A. (2003). Genetic analysis of pathway
specificity during posttranslational protein translocation across the Escherichia coli plasma membrane.
Journal of bacteriology 185, 2811-2819.
Bogsch, E.G., Sargent, F., Stanley, N.R., Berks, B.C., Robinson, C., and Palmer, T. (1998). An
Essential Component of a Novel Bacterial Protein Export System with Homologues in Plastids and
Mitochondria. The Journal of biological chemistry 273, 18003-18006.
Bolhuis, A., Mathers, J.E., Thomas, J.D., Barrett, C.M.L., and Robinson, C. (2001). TatB and TatC
Form a Functional and Structural Unit of the Twin-arginine Translocase from Escherichia coli. The Journal
of biological chemistry 276, 20213-20219.
Bost, S., and Belin, D. (1995). A new genetic selection identifies essential residues in SecG, a
component of the Escherichia coli protein export machinery. EMBO Journal 14, 4412-4421.
Bostina, M., Mohsin, B., Kuhlbrandt, W., and Collinson, I. (2005). Atomic Model of the E. coli
Membrane-bound Protein Translocation Complex SecYEG. Journal of molecular biology 352, 1035-1043.
Bradshaw, R.A., Cancedda, F., Ericsson, L.H., Neumann, P.A., Piccoli, S.P., Schlesinger, M.J.,
Shriefer, K., and Walsh, K.A. (1981). Amino acid sequence of Escherichia coli alkaline phosphatase.
Proceedings of the National Academy of Sciences of the United States of America 78, 3473-3477.
114
VI. Literaturverzeichnis
Braun, N.A., Davis, A.W., and Theg, S.M. (2007). The chloroplast Tat pathway utilizes the
transmembrane electric potential as an energy source. Biophysical journal 93, 1993-1998.
Breyton, C., Haase, W., Rapoport, T.A., Kuhlbrandt, W., and Collinson, I. (2002). Three-dimensional
structure of the bacterial protein-translocation complex SecYEG. Nature 418, 662-665.
Brüser T., Sanders C. (2003). An alternative model of the twin arginine translocation system.
Microbiological research 158, 7-17.
Buchanan, G., de Leeuw, E., Stanley, N.R., Wexler, M., Berks, B.C., Sargent, F., and Palmer, T.
(2002). Functional complexity of the twin-arginine translocase TatC component revealed by site-directed
mutagenesis. Molecular microbiology 43, 1457-1470.
Chan, C.S., Zlomislic, M.R., Tieleman, D.P., and Turner, R.J. (2007). The TatA Subunit of Escherichia
coli Twin-Arginine Translocase Has an N-in Topology. Biochemistry 46, 7396-7404.
Chin, J.W., Martin, A.B., King D.S., Wang L. and Schultz P.G. (2002). Addition of a photocrosslinking
amino acid to the genetic code of Escherichia coli. Proceedings of the National Academy of Sciences of
the United States of America 99, 11020-11024
Cline, K., Ettinger, W.F., and Theg, S.M. (1992). Protein-specific energy requirements for protein
transport across or into thylakoid membranes. Two lumenal proteins are transported in the absence of
ATP. The Journal of biological chemistry 267, 2688-2696.
Cline, K., and McCaffery, M. (2007). Evidence for a dynamic and transient pathway through the TAT
protein transport machinery. EMBO Journal 26, 3039-3049.
Cline, K., and Mori, H. (2001). Thylakoid {Delta}pH-dependent precursor proteins bind to a cpTatCHcf106 complex before Tha4-dependent transport. The Journal of cell biology 154, 719-730.
Connolly, T., and Gilmore, R. (1986). Formation of a functional ribosome-membrane junction during
translocation requires the participation of a GTP-binding protein. The Journal of cell biology 103, 22532261.
Cristobal, S., de Gier, J.-W., Nielsen, H., von Heijne, G. (1999). Competition between Sec- and Tatdependent protein translocation in Escherichia coli. EMBO Journal 18, 2982-2990.
Dabney-Smith, C., Mori, H., and Cline, K. (2006). Oligomers of Tha4 Organize at the Thylakoid Tat
Translocase during Protein Transport. The Journal of biological chemistry 281, 5476-5483.
de Leeuw, E., te Kaat, K., Moser, C., Menestrina, G., Demel, R., de Kruijff, B., Oudega, B., Luirink,
J., and Sinning, I. (2000). Anionic phospholipids are involved in membrane association of FtsY and
stimulate its GTPase activity. EMBO Journal 19, 531-541.
de Leeuw, E., Porcelli, I., Sargent, F., Palmer, T., and Berks, B.C. (2001). Membrane interactions and
self-association of the TatA and TatB components of the twin-arginine translocation pathway. FEBS
Letters 506, 143-148.
de Leeuw, E., Granjon, T., Porcelli, I., Alami, M., Carr, S.B., Müller, M., Sargent, F., Palmer, T., and
Berks, B.C. (2002). Oligomeric properties and signal peptide binding by Escherichia coli Tat protein
transport complexes. Journal of molecular biology 322, 1135-1146.
Dekker, N., Cox, R.C., Kramer, R.A., and Egmond, M.R. (2001). Substrate specificity of the integral
membrane protease OmpT determined by spatially addressed peptide libraries. Biochemistry 40, 16941701.
115
VI. Literaturverzeichnis
DeLisa, M.P., Tullman, D., and Georgiou, G. (2003). Folding quality control in the export of proteins by
the bacterial twin-arginine translocation pathway. Proceedings of the National Academy of Sciences of
the United States of America 100, 6115-6120.
DeLisa, M.P., Lee, P., Palmer, T., and Georgiou, G. (2004). Phage shock protein PspA of Escherichia
coli relieves saturation of protein export via the Tat pathway. Journal of bacteriology 186, 366-373.
Derman, A.I., and Beckwith, J. (1991). Escherichia coli alkaline phosphatase fails to acquire disulfide
bonds when retained in the cytoplasm. Journal of bacteriology 173, 7719-7722.
Di Cola, A., Bailey, S., and Robinson, C. (2005). The Thylakoid {Delta}pH/{Delta}{Psi} Are Not Required
for the Initial Stages of Tat-dependent Protein Transport in Tobacco Protoplasts. The Journal of biological
chemistry 280, 41165-41170.
Di Cola, A., and Robinson, C. (2005). Large-scale translocation reversal within the thylakoid Tat system
in vivo. The Journal of cell biology 171, 281-289.
Dilks, K., Rose, R.W., Hartmann, E., and Pohlschroder, M. (2003). Prokaryotic Utilization of the TwinArginine Translocation Pathway: a Genomic Survey. Journal of bacteriology 185, 1478-1483.
Ding, Z., and Christie, P.J. (2003). Agrobacterium tumefaciens twin-arginine-dependent translocation is
important for virulence, flagellation, and chemotaxis but not type IV secretion. Journal of bacteriology 185,
760-771.
Dubini, A., and Sargent, F. (2003). Assembly of Tat-dependent [NiFe] hydrogenases: identification of
precursor-binding accessory proteins. FEBS Letters 549, 141-146.
Farrell, I.S., Toroney, R., Hazen, J.L., Mehl, R.A., and Chin, J.W. (2005). Photo-cross-linking
interacting proteins with a genetically encoded benzophenone. Nature methods 2, 377-384.
Genest, O., Seduk, F., Ilbert, M., Mejean, V., and Iobbi-Nivol, C. (2006). Signal peptide protection by
specific chaperone. Biochemical and biophysical research communications 339, 991-995.
Gerard, F., and Cline, K. (2007). The thylakoid proton gradient promotes an advanced stage of signal
peptide binding deep within the Tat pathway receptor complex. The Journal of biological chemistry 282,
5263-5272.
Gerard, F., and Cline, K. (2006). Efficient Twin Arginine Translocation (Tat) Pathway Transport of a
Precursor Protein Covalently Anchored to Its Initial cpTatC Binding Site. The Journal of biological
chemistry 281, 6130-6135.
Gohlke, U., Pullan, L., McDevitt, C.A., Porcelli, I., de Leeuw, E., Palmer, T., Saibil, H.R., and Berks,
B.C. (2005). The TatA component of the twin-arginine protein transport system forms channel complexes
of variable diameter. Proceedings of the National Academy of Sciences of the United States of America
102, 10482-10486.
Gouffi, K., Santini, C.-L., and Wu, L.-F. (2002). Topology determination and functional analysis of the
Escherichia coli TatC protein. FEBS Letters 525, 65-70.
Gouffi, K., Gerard, F., Santini, C.-L., and Wu, L.-F. (2004). Dual Topology of the Escherichia coli TatA
Protein. The Journal of biological chemistry 279, 11608-11615.
Greene, N.P., Porcelli, I., Buchanan, G., Hicks, M.G., Schermann, S.M., Palmer, T., and Berks, B.C.
(2007). Cysteine scanning mutagenesis and disulfide mapping studies of the TatA component of the
bacterial twin-arginine translocase. The Journal of biological chemistry 282, 23937-23945.
116
VI. Literaturverzeichnis
Hanada, M., Nishiyama, K., and Tokuda, H. (1996). SecG plays a critical role in protein translocation in
the absence of the proton motive force as well as at low temperature. FEBS Letters 381, 25-28.
Hatzixanthis, K., Palmer, T., and Sargent, F. (2003). A subset of bacterial inner membrane proteins
integrated by the twin-arginine translocase. Molecular microbiology 49, 1377-1390.
Hatzixanthis, K., Clarke, T.A., Oubrie, A., Richardson, D.J., Turner, R.J., and Sargent, F. (2005).
Signal peptide-chaperone interactions on the twin-arginine protein transport pathway. Proceedings of the
National Academy of Sciences of the United States of America 102, 8460-8465.
Hermolin, J., and Fillingame, R.H. (1989). H+-ATPase activity of Escherichia coli F1F0 is blocked after
reaction of dicyclohexylcarbodiimide with a single proteolipid (subunit c) of the F0 complex. The Journal
of biological chemistry 264, 3896-3903.
Hesterkamp, T., Hauser, S., Lutcke, H., and Bukau, B. (1996). Escherichia coli trigger factor is a prolyl
isomerase that associates with nascent polypeptide chains. Proceedings of the National Academy of
Sciences of the United States of America 93, 4437-4441.
Hicks, M.G., Lee, P.A., Georgiou, G., Berks, B.C., and Palmer, T. (2005). Positive selection for loss-offunction tat mutations identifies critical residues required for TatA activity. Journal of bacteriology 187,
2920-2925.
Hicks, M.G., de Leeuw, E., Porcelli, I., Buchanan, G., Berks, B.C., and Palmer, T. (2003). The
Escherichia coli twin-arginine translocase: conserved residues of TatA and TatB family components
involved in protein transport. FEBS Letters 539, 61-67.
Hinsley, A.P., Stanley, N.R., Palmer, T., and Berks, B.C. (2001). A naturally occurring bacterial Tat
signal peptide lacking one of the `invariant' arginine residues of the consensus targeting motif. FEBS
Letters 497, 45-49.
Hoffmann, J.H., Linke, K., Graf, P.C., Lilie, H., and Jakob, U. (2004). Identification of a redox-regulated
chaperone network. EMBO Journal 23, 160-168.
Holzapfel, E., Eisner, G., Alami, M., Barrett, C.M., Buchanan, G., Luke, I., Betton, J.M., Robinson,
C., Palmer, T., Moser, M., et al. (2007). The entire N-terminal half of TatC is involved in twin-arginine
precursor binding. Biochemistry 46, 2892-2898.
Homma, T., Yoshihisa, T., and Ito, K. (1997). Subunit interactions in the Escherichia coli protein
translocase: SecE and SecG associate independently with SecY. FEBS Letters 408, 11-15.
Hou, B., Frielingsdorf, S., and Klosgen, R.B. (2006). Unassisted membrane insertion as the initial step
in DeltapH/Tat-dependent protein transport. Journal of molecular biology 355, 957-967.
Igarashi, P., and Aronson, P.S. (1987). Covalent modification of the renal Na+/H+ exchanger by N,N'dicyclohexylcarbodiimide. The Journal of biological chemistry 262, 860-868.
Ignatova, Z., Hornle, C., Nurk, A., and Kasche, V. (2002). Unusual Signal Peptide Directs Penicillin
Amidase from Escherichia coli to the Tat Translocation Machinery. Biochemical and Biophysical
Research Communications 291, 146-149.
Ilbert, M., Mejean, V., Giudici-Orticoni, M.T., Samama, J.P., and Iobbi-Nivol, C. (2003). Involvement of
a mate chaperone (TorD) in the maturation pathway of molybdoenzyme TorA. The Journal of biological
chemistry 278, 28787-28792.
117
VI. Literaturverzeichnis
Ize, B., Gérard, F., and Wu, L.-F. (2002). In vivo assessment of the Tat signal peptide specificity in
Escherichia coli. Archives of Microbiology 178, 548-553.
Jack, R.L., Sargent, F., Berks, B.C., Sawers, G., and Palmer, T. (2001). Constitutive Expression of
Escherichia coli tat Genes Indicates an Important Role for the Twin-Arginine Translocase during Aerobic
and Anaerobic Growth. Journal of bacteriology 183, 1801-1804.
Jack, R.L., Buchanan, G., Dubini, A., Hatzixanthis, K., Palmer, T., and Sargent, F. (2004).
Coordinating assembly and export of complex bacterial proteins. EMBO Journal 23, 3962-3972.
Jongbloed, J.D.H., Grieger, U., Antelmann, H., Hecker, M., Nijland, R., Bron, S., and van Dijl, J.M.
(2004). Two minimal Tat translocases in Bacillus. Molecular microbiology 54, 1319-1325.
Kakegawa, T., Sato, E., Hirose, S., and Igarashi, K. (1986). Polyamine binding sites on Escherichia coli
ribosomes. Archives of biochemistry and biophysics 251, 413-420.
Kauer, J.C., Erickson-Viitanen, S., Wolfe, H.R., Jr., and DeGrado, W.F. (1986). p-Benzoyl-Lphenylalanine, a new photoreactive amino acid. Photolabeling of calmodulin with a synthetic calmodulinbinding peptide. The Journal of biological chemistry 261, 10695-10700.
Keenan, R.J., Freymann, D.M., Stroud, R.M., and Walter, P. (2001). The signal recognition particle.
Annual review of biochemistry 70, 755-775.
Keersmaeker, S.D., Mellaert, L.V., Schaerlaekens, K., Dessel, W.V., Vrancken, K., Lammertyn, E.,
Anne, J., and Geukens, N. (2005). Structural organization of the twin-arginine translocation system in
Streptomyces lividans. FEBS Letters 579, 797-802.
Ki, J.J., Kawarasaki, Y., Gam, J., Harvey, B.R., Iverson, B.L., and Georgiou, G. (2004). A periplasmic
fluorescent reporter protein and its application in high-throughput membrane protein topology analysis.
Journal of molecular biology 341, 901-909.
Kihara, A., Akiyama, Y., and Ito, K. (1995). FtsH is required for proteolytic elimination of uncomplexed
forms of SecY, an essential protein translocase subunit. Proceedings of the National Academy of
Sciences of the United States of America 92, 4532-4536.
Kim, Y.K., Kim, T.I., Jung, D.K., Jung, J.S., and Lee, S.H. (1993). Inhibition of H+/organic cation
antiport by carboxyl reagents in rabbit renal brush-border membrane vesicles. The Journal of
pharmacology and experimental therapeutics 266, 500-505.
Kleerebezem, M., Crielaard, W., and Tommassen, J. (1996). Involvement of stress protein PspA
(phage shock protein A) of Escherichia coli in maintenance of the protonmotive force under stress
conditions. EMBO Journal 15, 162-171.
Koch, H.G., Hengelage, T., Neumann-Haefelin, C., MacFarlane, J., Hoffschulte, H.K., Schimz, K.L.,
Mechler, B., and Müller, M. (1999). In vitro studies with purified components reveal signal recognition
particle (SRP) and SecA/SecB as constituents of two independent protein-targeting pathways of
Escherichia coli. Molecular biology of the cell 10, 2163-2173.
Kreutzenbeck, P., Kroger, C., Lausberg, F., Blaudeck, N., Sprenger, G.A., and Freudl, R. (2007).
Escherichia coli Twin Arginine (Tat) Mutant Translocases Possessing Relaxed Signal Peptide
Recognition Specificities. The Journal of biological chemistry 282, 7903-7911.
Laemmli, U.K. (1970). Cleavage of structural proteins during the assembly of the head of bacteriophage
T4. Nature 227, 680-685
118
VI. Literaturverzeichnis
Lavander, M., Ericsson, S.K., Broms, J.E., and Forsberg, A. (2006). The twin arginine translocation
system is essential for virulence of Yersinia pseudotuberculosis. Infection and immunity 74, 1768-1776.
Lee, P.A., Buchanan, G., Stanley, N.R., Berks, B.C., and Palmer, T. (2002). Truncation Analysis of
TatA and TatB Defines the Minimal Functional Units Required for Protein Translocation. Journal of
Bacteriology 184, 5871-5879.
Lee, P.A., Orriss, G.L., Buchanan, G., Greene, N.P., Bond, P.J., Punginelli, C., Jack, R.L., Sansom,
M.S.P., Berks, B.C., and Palmer, T. (2006). Cysteine-scanning Mutagenesis and Disulfide Mapping
Studies of the Conserved Domain of the Twin-arginine Translocase TatB Component. The Journal of
biological chemistry 281, 34072-34085.
Li, S.Y., Chang, B.Y., and Lin, S.C. (2006). Coexpression of TorD enhances the transport of GFP via the
TAT pathway. Journal of biotechnology 122, 412-421.
Ma, X., and Cline, K. (2000). Precursors bind to specific sites on thylakoid membranes prior to transport
on the delta pH protein translocation system. The Journal of biological chemistry 275, 10016-10022.
Maillard, J., Spronk, C.A., Buchanan, G., Lyall, V., Richardson, D.J., Palmer, T., Vuister, G.W., and
Sargent, F. (2007). Structural diversity in twin-arginine signal peptide-binding proteins. Proceedings of
the National Academy of Sciences of the United States of America 104, 15641-15646.
Masip, L., Pan, J.L., Haldar, S., Penner-Hahn, J.E., DeLisa, M.P., Georgiou, G., Bardwell, J.C., and
Collet, J.F. (2004). An engineered pathway for the formation of protein disulfide bonds. Science (New
York, NY 303, 1185-1189.
McDevitt, C.A., Hicks, M.G., Palmer, T., and Berks, B.C. (2005). Characterisation of Tat protein
transport complexes carrying inactivating mutations. Biochemical and Biophysical Research
Communications 329, 693-698.
McDevitt, C.A., Buchanan, G., Sargent, F., Palmer, T., and Berks, B.C. (2006). Subunit composition
and in vivo substrate-binding characteristics of Escherichia coli Tat protein complexes expressed at
native levels. FEBS Journal 273, 5656-5668.
Messens, J., and Collet, J.F. (2006). Pathways of disulfide bond formation in Escherichia coli. The
international journal of biochemistry & cell biology 38, 1050-1062.
Mitchell, P. (1968). Chemiosmotic coupling and energy transduction. Glynn Research, Bodmin, UK
Molik, S., Karnauchov, I., Weidlich, C., Herrmann, R.G., and Klosgen, R.B. (2001). The Rieske Fe/S
Protein of the Cytochrome b6/f Complex in Chloroplasts. Missing link in the evolution of protein transport
pathways in chloroplasts? The Journal of biological chemistry 276, 42761-42766.
Mori, H., Summer, E.J., Ma, X., and Cline, K. (1999). Component specificity for the thylakoidal Sec and
Delta pH-dependent protein transport pathways. The Journal of biological chemistry 146, 45-56.
Mori, H., Summer, E.J., and Cline, K. (2001). Chloroplast TatC plays a direct role in thylakoid (Delta)pHdependent protein transport. FEBS Letters 501, 65-68.
Moser, M., Panahandeh, S., Holzapfel, E., and Müller, M. (2007). In vitro analysis of the bacterial twinarginine-dependent protein export. Methods in Molecular Biology, 390, 63-80.
Mould, R.M., and Robinson, C. (1991). A proton gradient is required for the transport of two lumenal
oxygen-evolving proteins across the thylakoid membrane. The Journal of biological chemistry 266,
12189-12193.
119
VI. Literaturverzeichnis
Müller, M. (2005). Twin-arginine-specific protein export in Escherichia coli. Research in microbiology 156,
131-136.
Müller, M., and Blobel, G. (1984). In vitro translocation of bacterial proteins across the plasma
membrane of Escherichia coli. Proceedings of the National Academy of Sciences of the United States of
America 81, 7421-7425.
Musser, S.M., and Theg, S.M. (2000). Characterization of the early steps of OE17 precursor transport by
the thylakoid DeltapH/Tat machinery. European journal of biochemistry 267, 2588-2598.
Neumann-Haefelin, C., Schafer, U., Müller, M., and Koch, H.G. (2000). SRP-dependent cotranslational targeting and SecA-dependent translocation analyzed as individual steps in the export of a
bacterial protein. EMBO Journal 19, 6419-6426.
Nishiyama, K., Hanada, M., and Tokuda, H. (1994). Disruption of the gene encoding p12 (SecG)
reveals the direct involvement and important function of SecG in the protein translocation of Escherichia
coli at low temperature. EMBO Journal 13, 3272-3277.
Nishiyama, K., Suzuki, T., and Tokuda, H. (1996). Inversion of the membrane topology of SecG
coupled with SecA-dependent preprotein translocation. Cell 85, 71-81.
Nishiyama, K., Fukuda, A., Morita, K., and Tokuda, H. (1999). Membrane deinsertion of SecA
underlying proton motive force-dependent stimulation of protein translocation. EMBO Journal 18, 10491058.
Nouwen, N., Tommassen, J., and de Kruijff, B. (1994). Requirement for conformational flexibility in the
signal sequence of precursor protein. The Journal of biological chemistry 269, 16029-16033.
Oates, J., Mathers, J., Mangels, D., Kuhlbrandt, W., Robinson, C., and Model, K. (2003). Consensus
Structural Features of Purified Bacterial TatABC Complexes. Journal of molecular biology 330, 277-286.
Oates, J., Barrett, C.M.L., Barnett, J.P., Byrne, K.G., Bolhuis, A., and Robinson, C. (2005). The
Escherichia coli Twin-arginine Translocation Apparatus Incorporates a Distinct Form of TatABC Complex,
Spectrum of Modular TatA Complexes and Minor TatAB Complex. Journal of molecular biology 346, 295305.
Ochsner, U.A., Snyder, A., Vasil, A.I., and Vasil, M.L. (2002). Effects of the twin-arginine translocase
on secretion of virulence factors, stress response, and pathogenesis. Proceedings of the National
Academy of Sciences of the United States of America 99, 8312-8317.
Oresnik, I.J., Ladner, C.L., and Turner, R.J. (2001). Identification of a twin-arginine leader-binding
protein. Molecular microbiology 40, 323-331.
Orriss, G.L., Tarry, M.J., Ize, B., Sargent, F., Lea, S.M., Palmer, T., and Berks, B.C. (2007). TatBC,
TatB, and TatC form structurally autonomous units within the twin arginine protein transport system of
Escherichia coli. FEBS Letters 581, 4091-4097.
Papish, A.L., Ladner, C.L., and Turner, R.J. (2003). The twin-arginine leader-binding protein, DmsD,
interacts with the TatB and TatC subunits of the Escherichia coli twin-arginine translocase. The Journal of
biological chemistry 278, 32501-32506.
Park, S., Liu, G., Topping, T.B., Cover, W.H., and Randall, L.L. (1988). Modulation of folding pathways
of exported proteins by the leader sequence. Science 239, 1033-1035.
120
VI. Literaturverzeichnis
Porcelli, I., de Leeuw, E., Wallis, R., van den Brink-van der Laan, E., de Kruijff, B., Wallace, B.A.,
Palmer, T., and Berks, B.C. (2002). Characterization and membrane assembly of the TatA component of
the Escherichia coli twin-arginine protein transport system. Biochemistry 41, 13690-13697.
Poritz, M.A., Bernstein, H.D., Strub, K., Zopf, D., Wilhelm, H., and Walter, P. (1990). An E. coli
ribonucleoprotein containing 4.5S RNA resembles mammalian signal recognition particle. Science 250,
1111-1117.
Prinz, W.A., Aslund, F., Holmgren, A., and Beckwith, J. (1997). The role of the thioredoxin and
glutaredoxin pathways in reducing protein disulfide bonds in the Escherichia coli cytoplasm. The Journal
of biological chemistry 272, 15661-15667.
Prinz, A., Behrens, C., Rapoport, T.A., Hartmann, E., and Kalies, K.U. (2000). Evolutionarily
conserved binding of ribosomes to the translocation channel via the large ribosomal RNA. EMBO Journal
19, 1900-1906.
Punginelli, C., Maldonado, B., Grahl, S., Jack, R., Alami, M., Schroder, J., Berks, B.C., and Palmer,
T. (2007). Cysteine-scanning mutagenesis and topological mapping of the Escherichia coli twin-arginine
translocase TatC component. Journal of Bacteriology 189, 5482-5494
Ramamurthy, V., and Oliver, D. (1997). Topology of the integral membrane form of Escherichia coli
SecA protein reveals multiple periplasmically exposed regions and modulation by ATP binding. The
Journal of biological chemistry 272, 23239-23246.
Rapoport, T.A. (2007). Protein translocation across the eukaryotic endoplasmic reticulum and bacterial
plasma membranes. Nature 450, 663-669.
Ray, N., Oates, J., Turner, R.J., and Robinson, C. (2003). DmsD is required for the biogenesis of
DMSO reductase in Escherichia coli but not for the interaction of the DmsA signal peptide with the Tat
apparatus. FEBS Letters 534, 156-160.
Reyes, C.L., Rutenber, E., Walter, P., and Stroud, R.M. (2007). X-ray structures of the signal
recognition particle receptor reveal targeting cycle intermediates. PLoS ONE 2, e607.
Ribnicky, B., Van Blarcom, T., and Georgiou, G. (2007). A scFv antibody mutant isolated in a genetic
screen for improved export via the twin arginine transporter pathway exhibits faster folding. Journal of
molecular biology 369, 631-639.
Richter, S., Lindenstrauss, U., Lucke, C., Bayliss, R., and Brüser, T. (2007). Functional Tat transport
of unstructured, small, hydrophilic proteins. The Journal of biological chemistry 282, 33257-33264.
Richter, S., and Brüser, T. (2005). Targeting of Unfolded PhoA to the TAT Translocon of Escherichia
coli. Journal of biological chemistry 280, 42723-42730.
Rodrigue, A., Chanal, A., Beck, K., Müller, M., and Wu, L.F. (1999). Co-translocation of a periplasmic
enzyme complex by a hitchhiker mechanism through the bacterial tat pathway. The Journal of biological
chemistry 274, 13223-13228.
Rose, R.W., Brüser, T., Kissinger, J.C., and Pohlschroder, M. (2002). Adaptation of protein secretion
to extremely high-salt conditions by extensive use of the twin-arginine translocation pathway. Molecular
microbiology 45, 943-950.
Sambrook, J., Fritsch, E.F. and Maniatis, T. (1989). Molecular cloning – a laboratory manual. Cold
Spring Harbor Laboratory Press, Cold Spring Harbor, New York, USA.
121
VI. Literaturverzeichnis
Sanders, C., Wethkamp, N., and Lill, H. (2001). Transport of cytochrome c derivatives by the bacterial
Tat protein translocation system. Molecular microbiology 41, 241-246.
Saparov, S.M., Erlandson, K., Cannon, K., Schaletzky, J., Schulman, S., Rapoport, T.A., and Pohl,
P. (2007). Determining the Conductance of the SecY Protein Translocation Channel for Small Molecules.
Molecular Cell 26, 501-509.
Sargent, F. (2007). Constructing the wonders of the bacterial world: biosynthesis of complex enzymes.
Microbiology 153, 633-651.
Sargent, F., Bogsch, E.G., Stanley, N.R., Wexler, M., Robinson, C., Berks, B.C., and Palmer, T.
(1998). Overlapping functions of components of a bacterial Sec-independent protein export pathway.
EMBO Journal 17, 3640-3650.
Sargent, F., Stanley, N.R., Berks, B.C., and Palmer, T. (1999). Sec-independent Protein Translocation
in Escherichia coli. A DISTINCT AND PIVOTAL ROLE FOR THE TatB PROTEIN. Journal of biological
chemistry 274, 36073-36082.
Sargent, F., Gohlke, U., de Leeuw, E., Stanley, N.R., Palmer, T., Saibil, H.R., and Berks, B.C. (2001).
Purified components of the Escherichia coli Tat protein transport system form a double-layered ring
structure. European Journal of Biochemistry 268, 3361-3367.
Schatz, P.J., Riggs, P.D., Jacq, A., Fath, M.J., and Beckwith, J. (1989). The secE gene encodes an
integral membrane protein required for protein export in Escherichia coli. Genes & development 3, 10351044.
Schatz, P.J., Bieker, K.L., Ottemann, K.M., Silhavy, T.J., and Beckwith, J. (1991). One of three
transmembrane stretches is sufficient for the functioning of the SecE protein, a membrane component of
the E. coli secretion machinery. EMBO Journal 10, 1749-1757.
Schiebel, E., Driessen, A.J., Hartl, F.U., and Wickner, W. (1991). Delta mu H+ and ATP function at
different steps of the catalytic cycle of preprotein translocase. Cell 64, 927-939.
Schlesinger, M.J., and Barrett, K. (1965). The reversible dissociation of the alkaline phosphatase of
Escherichia coli. I. Formation and reactivation of subunits. The Journal of biological chemistry 240, 42844292.
Schreiber, S., Stengel, R., Westermann, M., Volkmer-Engert, R., Pop, O.I., and Müller, J.P. (2006).
Affinity of TatCd for TatAd Elucidates Its Receptor Function in the Bacillus subtilis Twin Arginine
Translocation (Tat) Translocase System. The Journal of biological chemistry 281, 19977-19984.
Scotti, P.A., Valent, Q.A., Manting, E.H., Urbanus, M.L., Driessen, A.J., Oudega, B., and Luirink, J.
(1999). SecA is not required for signal recognition particle-mediated targeting and initial membrane
insertion of a nascent inner membrane protein. The Journal of biological chemistry 274, 29883-29888.
Sebald, W., Machleidt, W., and Wachter, E. (1980). N,N'-dicyclohexylcarbodiimide binds specifically to
a single glutamyl residue of the proteolipid subunit of the mitochondrial adenosinetriphosphatases from
Neurospora crassa and Saccharomyces cerevisiae. Proceedings of the National Academy of Sciences of
the United States of America 77, 785-789.
Shan, S.O., and Walter, P. (2005). Molecular crosstalk between the nucleotide specificity determinant of
the SRP GTPase and the SRP receptor. Biochemistry 44, 6214-6222.
Shanmugham A., W.F.S.H.W., Bollen Y.J.M.,Lill H. (2006). Membrane binding of twin arginine
preproteins as an early step in translocation. Biochemistry 45, 2243-2249.
122
VI. Literaturverzeichnis
Silhavy, T.J., and Beckwith, J. (1983). Isolation and characterization of mutants of Escherichia coli K12
affected in protein localization. Methods in enzymology 97, 11-40.
Sone, M., Kishigami, S., Yoshihisa, T., and Ito, K. (1997). Roles of disulfide bonds in bacterial alkaline
phosphatase. The Journal of biological chemistry 272, 6174-6178.
Stanley, N.R., Palmer, T., and Berks, B.C. (2000). The twin arginine consensus motif of Tat signal
peptides is involved in Sec-independent protein targeting in Escherichia coli. The Journal of biological
chemistry 275, 11591-11596.
Strauch, E.M., and Georgiou, G. (2007). Escherichia coli tatC mutations that suppress defective twinarginine transporter signal peptides. Journal of molecular biology 374, 283-291.
Suchi, R., Stern-Bach, Y., Gabay, T., and Schuldiner, S. (1991). Covalent modification of the amine
transporter with N,N'-dicyclohexylcarbodiimide. Biochemistry 30, 6490-6494.
Sugai, R., Takemae, K., Tokuda, H., and Nishiyama, K. (2007). Topology inversion of SecG is essential
for cytosolic SecA-dependent stimulation of protein translocation. The Journal of biological chemistry 282,
29540-29548.
Sugimura, K., and Nishihara, T. (1988). Purification, characterization, and primary structure of
Escherichia coli protease VII with specificity for paired basic residues: identity of protease VII and OmpT.
Journal of bacteriology 170, 5625-5632.
Theg, S.M., Cline, K., Finazzi, G., and Wollman, F.A. (2005). The energetics of the chloroplast Tat
protein transport pathway revisited. Trends in plant science 10, 153-154.
Tranier, S., Mortier-Barriere, I., Ilbert, M., Birck, C., Iobbi-Nivol, C., Mejean, V., and Samama, J.P.
(2002). Characterization and multiple molecular forms of TorD from Shewanella massilia, the putative
chaperone of the molybdoenzyme TorA. Protein science 11, 2148-2157.
Tullman-Ercek, D., DeLisa, M.P., Kawarasaki, Y., Iranpour, P., Ribnicky, B., Palmer, T., and
Georgiou, G. (2007). Export Pathway Selectivity of Escherichia coli Twin Arginine Translocation Signal
Peptides. The Journal of biological chemistry 282, 8309-8316.
Turner, R.J., Papish, A.L., and Sargent, F. (2004). Sequence analysis of bacterial redox enzyme
maturation proteins (REMPs). Canadian journal of microbiology 50, 225-238.
Valent, Q.A., Kendall, D.A., High, S., Kusters, R., Oudega, B., and Luirink, J. (1995). Early events in
preprotein recognition in E. coli: interaction of SRP and trigger factor with nascent polypeptides. EMBO
Journal 14, 5494-5505.
Valent, Q.A., de Gier, J.W., von Heijne, G., Kendall, D.A., ten Hagen-Jongman, C.M., Oudega, B.,
and Luirink, J. (1997). Nascent membrane and presecretory proteins synthesized in Escherichia coli
associate with signal recognition particle and trigger factor. Molecular microbiology 25, 53-64.
Valent, Q.A., Scotti, P.A., High, S., de Gier, J.W., von Heijne, G., Lentzen, G., Wintermeyer, W.,
Oudega, B., and Luirink, J. (1998). The Escherichia coli SRP and SecB targeting pathways converge at
the translocon. EMBO Journal 17, 2504-2512.
von Ballmoos, C., and Dimroth, P. (2007). Two distinct proton binding sites in the ATP synthase family.
Biochemistry 46, 11800-11809.
van Dalen, A., Killian, A., and de Kruijff, B. (1999). Delta psi stimulates membrane translocation of the
C-terminal part of a signal sequence. The Journal of biological chemistry 274, 19913-19918.
123
VI. Literaturverzeichnis
van der Wolk, J.P., de Wit, J.G., and Driessen, A.J. (1997). The catalytic cycle of the Escherichia coli
SecA ATPase comprises two distinct preprotein translocation events. EMBO Journal 16, 7297-7304.
von Heijne, G. (1990). Protein targeting signals. Current opinion in cell biology 2, 604-608.
Voulhoux, R., Ball, G., Ize, B., Vasil, M.L., Lazdunski, A., Wu, L.F., and Filloux, A. (2001).
Involvement of the twin-arginine translocation system in protein secretion via the type II pathway. EMBO
Journal 20, 6735-6741.
Vrancken, K., De Keersmaeker, S., Geukens, N., Lammertyn, E., Anne, J., and Van Mellaert, L.
(2007). PspA overexpression in Streptomyces lividans improves both Sec- and Tat-dependent protein
secretion. Applied microbiology and biotechnology 73, 1150-1157.
Walter, P., and Johnson, A.E. (1994). Signal sequence recognition and protein targeting to the
endoplasmic reticulum membrane. Annual review of cell biology 10, 87-119.
Westermann, M., Pop, O.I., Gerlach, R., Appel, T.R., Schlormann, W., Schreiber, S., and Müller, J.P.
(2006). The TatAd component of the Bacillus subtilis twin-arginine protein transport system forms homomultimeric complexes in its cytosolic and membrane embedded localisation. Biochimica et Biophysica
Acta (BBA) - Biomembranes 1758, 443-451.
Wexler, M., Sargent, F., Jack, R.L., Stanley, N.R., Bogsch, E.G., Robinson, C., Berks, B.C., and
Palmer, T. (2000). TatD is a cytoplasmic protein with DNase activity. No requirement for TatD family
proteins in sec-independent protein export. The Journal of biological chemistry 275, 16717-16722.
Widdick, D.A., Dilks, K., Chandra, G., Bottrill, A., Naldrett, M., Pohlschroder, M., and Palmer, T.
(2006). The twin-arginine translocation pathway is a major route of protein export in Streptomyces
coelicolor. Proceedings of the National Academy of Sciences of the United States of America 103, 1792717932.
Wyckoff, H.W. (1987). Phosphate metabolism and cellular regulation in microorganism. American
Society of Microbiology Press, Washington, D.C., USA.
Yahr, T.L., and Wickner, W.T. (2001). Functional reconstitution of bacterial Tat translocation in vitro.
EMBO Journal 20, 2472-2479.
Yerushalmi, H., and Schuldiner, S. (2000). An essential glutamyl residue in EmrE, a multidrug antiporter
from Escherichia coli. The Journal of biological chemistry 275, 5264-5269.
124
VII. Anhang
VII. Anhang
VII.1. Sequenz des Fusionsproteins TorA-PhoA (in pET28a)
1
61
121
181
241
301
361
421
481
541
601
661
721
781
841
901
961
1021
1081
1141
1201
1261
1321
1381
1441
1501
ATGGCAAACAATAACGATCTCTTTCAGGCATCACGTCGGCGTTTTCTGGCACAACTCGGC
M A N N N D L F Q A S R R R F L A Q L G
GGCTTAACCGTCGCCGGGATGCTGGGGCCGTCATTGTTAACGCCGCGACGTGCGACTGCG
G L T V A G M L G P S L L T P R R A T A
GCGCAAGCGGCGACTGACGCTTCTAGACGGACACCAGAAATGCCTGTTCTGGAAAACCGG
A Q A A T D A S R R T P E M P V L E N R
GCTGCTCAGGGCGATATTACTGCACCCGGCGGTGCTCGCCGCTTAACGGGTGATCAGACC
A A Q G D I T A P G G A R R L T G D Q T
GCCGCTCTGCGTGATTCTCTTAGCGATAAACCTGCAAAAAATATTATTTTGCTGATTGGC
A A L R D S L S D K P A K N I I L L I G
GATGGGATGGGGGATTCGGAAATTACTGCCGCACGTAATTATGCCGAAGGTGCGGGCGGC
D G M G D S E I T A A R N Y A E G A G G
TTTTTTAAAGGTATCGATGCCTTACCGCTTACCGGGCAATACACTCACTATGCGCTGAAT
F F K G I D A L P L T G Q Y T H Y A L N
AAAAAAACCGGCAAACCGGACTACGTCACCGACTCGGCTGCATCAGCAACCGCCTGGTCA
K K T G K P D Y V T D S A A S A T A W S
ACCGGTGTCAAAACCTATAACGGCGCGCTGGGCGTCGATATTCACGAAAAAGATCACCCA
T G V K T Y N G A L G V D I H E K D H P
ACGATTCTGGAAATGGCAAAAGCCGCAGGTCTGGCGACCGGTAACGTTTCTACCGCAGAG
T I L E M A K A A G L A T G N V S T A E
TTGCAGGATGCCACGCCCGCTGCGCTGGTGGCACATGTGACCTCGCGCAAATGCTACGGT
L Q D A T P A A L V A H V T S R K C Y G
CCGAGCGCGACCAGTGAAAAATGTCCGGGTAACGCGCTAGAAAAAGGCGGGAGAGGATCG
P S A T S E K C P G N A L E K G G R G S
ATTACCGAACAGCTGCTTAACGCTCGTGCCGATGTTACGCTTGGCGGCGGCGCAAAAACC
I T E Q L L N A R A D V T L G G G A K T
TTTGCTGAAACGGCAACCGCCGGTGAATGGCAGGGAAAAACGCTGCGTGAACAGGCACAG
F A E T A T A G E W Q G K T L R E Q A Q
GCGCGTGGTTATCAGTTGGTGAGCGATGCTGCCTCACTGAATGCGGTGACGGAAGCGAAC
A R G Y Q L V S D A A S L N A V T E A N
CAGCAAAAACCCCTGCTAGGACTGTTTGCTGACGGCAATATGCCAGTGCGCTGGCAAGGA
Q Q K P L L G L F A D G N M P V R W Q G
CCGAAAGCAACGTACCACGGCAATATCGACAAGCCCGCAGTTACCTGTACGCCTAATCCG
P K A T Y H G N I D K P A V T C T P N P
CAACGTAATGACAGCGTACCGACCCTGGCGCAGATGACTGATAAAGCCATTGAATTGTTG
Q R N D S V P T L A Q M T D K A I E L L
AGTAAAAATGAGAAAGGCTTTTTCCTGCAAGTTGAAGGTGCATCAATCGATAAACAGGAT
S K N E K G F F L Q V E G A S I D K Q D
CACGCTGCGAATCCTTGTGGGCAAATTGGCGAGACGGTCGATCTCGACGAAGCCGTACAA
H A A N P C G Q I G E T V D L D E A V Q
CGGGCGCTGGAATTCGCTAAAAAGGATGGCAACACGCTGGTCATAGTCACCGCTGATCAC
R A L E F A K K D G N T L V I V T A D H
GCCCACGCCAGCCAGATTGTTGCGCCGGACACCAAAGCGCCGGGCCTCACCCAGGCGCTA
A H A S Q I V A P D T K A P G L T Q A L
AATACCAAAGATGGCGCAGTGATGGTGATGAGTTACGGGAACTCCGAAGAGGATTCACAA
N T K D G A V M V M S Y G N S E E D S Q
GAACATACCGGTAGTCAGCTGCGTATTGCGGCGTATGGCCCACATGCCGCCAATGTCGTT
E H T G S Q L R I A A Y G P H A A N V V
GGACTGACCGACCAGACCGATCTCTTCTACACCATGAAAGCCGCTCTGGGGCTGAAAGTC
G L T D Q T D L F Y T M K A A L G L K V
GACAAGCTTGCGGCCGCACTCGAGCACCACCACCACCACCACTGA
D K L A A A L E H H H H H H *
TorA-Signalpeptid
reifes TorA
reifes PhoA
Stop-Codon-Position
Cysteine in TorA-PhoA
klonierungsbedingt
eingefügte Aminosäuren
MCS des pET28aPlasmids
His-tag
125
VII. Anhang
VII.2. Abkürzungsverzeichnis
Å
A
Abb.
Ac
ATP
AK
Ala
Amp
AMS
APS
Arg
Asn
Asp
ATP
bis–MGD
BSA
bzw.
bspw.
C
°C
ca.
CCCP
Ci
CTP
Cys
D
Da
dATP
DCCD
dCTP
dest.
dGTP
d.h.
DMSO
DNA
dNTP
DPF
DTT
dTTP
dUTP
E
E. coli
EDTA
EtOH
F
FAD
G
g
Gln
Glu
Gly
GSSG
126
Ångstöm
Alanin
Abbildung
Acetat
Adenosin-5´-Diphosphat
Antikörper
Alanin
Ampicillin
4´-Acetamido-4´-maleimidylstilbene-2,2´-Disulfonsäure
Ammoniumperoxodisulfat
Arginin
Asparagin
Aspartat
Adenosin-5´-Triphosphat
bis-Molybdopterin-Guanin-Dinukleotid
Rinderserumalbumin
beziehungsweise
beispielsweise
Cystein
Grad Celsius
Circa
Carbonylcyanid-m-chlorophenylhydrazon
Curie
Cytidin-5´-Triphosphat
Cystein
Asparaginsäure
Dalton
3´-Desoxy-Adenosin-5´-Triphosphat
Dicyclohexylcarbodiimid
3´-Desoxy-Cytidin-5´-Triphosphat
destilliert
3´-Desoxy-Guanosin-5´-Triphosphat
das heißt
Dimethylsulfoxid
Desoxyribonukleinsäure
3´-Desoxy-Nucleosid-5´-Triphosphat
Diisopropylfluorphosphat
Dithiotreitol
3´-Desoxy-Thymidin-5´-Triphosphat
3´-Desoxy-Uridin-5´-Triphosphat
Glutaminsäure
Escherichia coli
Ethylendiamintetraacetat
Ethanol
Phenylalanin
Flavinadenindinukleotid
Glycin
Gramm, Milligramm, Mikrogramm
Glutamin
Glutamat
Glycin
oxidiertes Glutathion
VII. Anhang
GTP
H
h
HEPES
His
i
I
Ile
INV
IPTG
K
Kan
kDa
l
L
LB
Leu
Lys
µg
µl
µM
m
M
M, Met
mA
MCS
MDa
mg
min
ml
mM
MOPS
mRNA
N
NADP(H)
ng
nm
NTP
OD
OmpA
OmpT
p
P
p.a.
PAGE
pBPA
PEG
PEP
Phe
PhoA
PK
PMF
PMSF
PPIase
Pro
PVDF
Guanosin-5´-Triphosphat
Histidin
Stunde
N-(2-Hydoxyethyl)piperazin-N‘-(2-Ethan-Sulfonsäure)
Histidin
Intermediat
Isoleucin
Isoleucin
Inside-out Innenmembranvesikel
Isopropyl-β-D-thiogalactosid
Lysin
Kanamycin
Kilodalton
Liter
Leucin
Luria Broth-Medium
Leucin
Lysin
Mikrogramm
Mikroliter
mikromolar
reif
molar
Methionin
Miliampere
multiple cloning site
Megadalton
Milligram
Minute
Milliliter
millimolar
( N-Morpholino )-Propansulfonsäure
messenger RNA
Asparagin
Nicotinamidadenindinucleotidphosphate (reduziert)
Nanogramm
Nanometer
Nukleosidtriphosphat
Optische Dichte
outer membrane protein A
outer membrane protease T
Vorläufer
Prolin
pro analysi
Polyacrylamid-Gelelektrophorese
para-Benzoyl-L-Phenylalanin
Polyethylenglycol
Phosphoenolpyruvat
Phenylalanin
alkalische Phosphatase
Proteinase K
proton-motive force
Phenylmethylsulfonylfluorid
Peptidyl-Prolyl-cis/trans-Isomerasen
Prolin
Polyvinylidenfluorid
127
VII. Anhang
Q
R
RNA
rpm
RT
S
s
SDS
Ser
sog.
SRP
T
Tab.
Tat
TCA
Tea
tech.
TEMED
Tet
TF
THF
Thr
TMAO
Tmd-Phe
TorA
Tris
tRNA
tRNACUA
Trp
Tyr
u
UTP
UV
UZ
v/v
V
V, Val
w/w
W
xg
Y
z.B.
128
Glutamin
Arginin
Ribonukleinsäure
rotation per minute
Raumtemperatur
Schwefel, Serin,
Sekunde
Natriumdodecylsulfat
Serin
sogenannt
signal recognition particle
Threonin
Tabelle
twin arginine translocation
Trichloressigsäure
Triethanolamin
technisch
N, N, N‘, N‘-Tetramethylethylendiamin
Tetracylin
Trigger Faktor
Tetrahydrofuran
Threonin
Trimethylamin-N-Oxid
L-4’-(3-[trifluoromethyl]-3H-diazirin-3yl)phenylalanine
Trimethylamin-N-Oxid (TMAO)–Reduktase
Trihydroxymethylaminomethan
transfer-Ribonukleinsäure
amber-Suppressor-transfer-Ribonukleinsäure
Tryptophan
Tyrosin
unit(s)
Uridin-5´-Triphosphat
ultraviolett
Ultrazentrifuge
volume per volume
Volumen
Valin
weight per weight (Gewicht pro Gewicht)
Tryptophan
x-fache Erdbeschleunigung
Tyrosin
zum Beispiel
VII. Anhang
VII.3. Danksagung
Mein besonderer Dank gilt Prof. Dr. Matthias Müller, für die herzliche Aufnahme in
seine Arbeitsgruppe, sowie für die Bereitstellung eines spannenden und
anspruchsvollen Themas. Sein Ideenreichtum, seine ständige und uneingeschränkte
Diskussionsbereitschaft sowie sein motivierender Optimismus waren unerlässlich für
die Anfertigung dieser Arbeit.
Ebenfalls besonderer Dank gilt auch Dr. Michael Moser und Dr. Evchen Holzapfel,
für die erstklassige Einstiegshilfe im Labor und der immer fortwährenden
Unterstützung bei Planung, Durchführungen und Interpretation von Experimenten,
sowie für eine wunderbare Atmosphäre und eine schöne Zeit im 5.OG.
Für eine gute Zusammenarbeit und einem angenehmen, freundlichen Arbeitsklima
möchte ich mich außerdem bedanken bei der gesamten AG Müller: Carlo, Diana,
Jörg, Michi, Dr. Thuy-Anh Tran-Thi, Silke und ganz speziell auch bei unseren
ehemaligen Doktorandinen Carmen und Raluca.
Dorothea Trescher, Inge Friese und insbesondere Cornelia Gribben-Pfirrmann danke
ich außerdem für die ständige Hilfsbereitschaft in technischen und administrativen
Angelegenheiten.
Besonders herzlich danke ich meinen lieben Eltern, für die ständige und
bedingungslose Unterstützung in der Zeit des Studiums und Dissertation.
Mein uneingeschränkter Dank gilt meiner geliebten Frau Sandra, die mir eine nie
versiegende Energiequelle und ein Ruhepol zu allen Zeiten dieser Arbeit war.
129