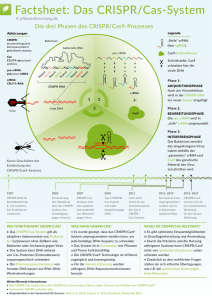
Nutzen und Risiken des Genomic Editings
Werbung

Diplomarbeit Nutzen und Risiken des Genomic Editings eingereicht von Roland Mühlhauser zur Erlangung des akademischen Grades Doktor der gesamten Heilkunde (Dr. med. univ.) an der Medizinischen Universität Graz ausgeführt am Institut für Humangenetik unter Anleitung von Priv.-Doz. Mag. Dr.scient.med. Thomas Schwarzbraun Graz, am 20.05.2016 Eidesstattliche Erklärung Ich erkläre ehrenwörtlich, dass ich die vorliegende Arbeit selbstständig und ohne fremde Hilfe verfasst habe, andere als die angegebenen Quellen nicht verwendet habe und die den benutzten Quellen wörtlich oder inhaltlich entnommenen Stellen als solche kenntlich gemacht habe. Graz, am 20.05.2016 Roland Mühlhauser, eh. Vorwort und Danksagung Die vorliegende Diplomarbeit entstand im Wintersemester 2015/16 im Rahmen meines Studiums der Humanmedizin an der Medizinischen Universität Graz. Die Möglichkeit, das menschliche Genom gezielt zu verändern und zu manipulieren, existiert schon seit einigen Jahrzehnten. Jedoch machen neue Verfahren die genetische Manipulation so einfach, effizient und leicht zugänglich wie nie zuvor. Die Einsatzmöglichkeiten dieser Technik in der Wissenschaft und insbesondere in der Medizin sind enorm. Mit der Leichtigkeit dieser Methoden steigt auch das Risiko negativer Auswirkungen durch die Manipulation am menschlichen Erbgut. Sind die Risiken und Nebenwirkungen, die solche Verfahren mit sich bringen überhaupt noch vertretbar? Aufgrund dieser Fragestellung und der Aktualität dieser Thematik entstand in der Absprache mit Priv.-Doz. Mag. Dr.scient.med. Thomas Schwarzbraun das Thema „Nutzen und Risiken des Genomic Editings“. An dieser Stelle geht zu allererst mein Dank an meinen Betreuer Priv.-Doz. Mag. Dr.scient.med. Thomas Schwarzbraun, der mir während des gesamten Zeitraums der Erstellung dieser Arbeit zur Seite stand und mir eine sehr kompetente, unkomplizierte und freundliche Beratung und Betreuung bot. Ich danke weiters meiner gesamten Familie, insbesondere meinen Eltern, die mich nicht nur während dieses Schreibprozesses, sondern während meines gesamten Studiums in jeglicher Hinsicht unterstützt haben und mir alle Chancen eröffnet haben. Ohne ihren Rückhalt und ihre Unterstützung wäre mein Werdegang nicht möglich gewesen. Roland Mühlhauser Graz, 20.05.2016 Zusammenfassung Einleitung: In den letzten Jahren wurden zunehmend schnellere und effizientere Verfahren geschaffen, die ein gezieltes Eingreifen in das Genom verschiedener Spezies ermöglichen. Das Genomic Targeting wird durch neuere Durchführungsmöglichkeiten sowohl praktikabler, als auch erschwinglicher. Somit wird die Veränderung bzw. der Austausch von DNA leichter zugänglich und durchführbar. Welche Risiken sind mit diesem Trend verbunden? Überwiegen weiterhin die positiven Aspekte der Methode? Neben den Vorteilen, die verschiedene Verfahren des Genomic Editings bedeuten, werden auch die möglichen Gefahren und Risiken aufgezeigt, die bei Eingriffen in das Genom von Organismen entstehen. Die Anfänge und der wissenschaftliche Fortschritt des Genomic Editings werden beschrieben und verschiedene Verfahren erläutert. Material und Methodik: Die Thematik dieser Diplomarbeit wurde als Literaturrecherche behandelt. Für die Bearbeitung der Themen wurden diverse Online-Datenbanken für wissenschaftliche Literatur und Suchdienste herangezogen. Gesucht wurden Artikel, Journale, Studien und Fachzeitschriften in deutscher und englischer Sprache. Ergebnisse: Die Anwendungsgebiete des Genomic Targetings erstrecken sich von der genetischen Manipulation an Mäusen für die Arzneimittelforschung, über den Einsatz der Methoden in der Pflanzenzucht und Landwirtschaft, bis hin zur genetischen Manipulation von menschlichen Embryos. Durch die Entwicklung dieser Verfahren soll zukünftig ermöglicht werden, Ursachen für Krankheiten auf genetischer Ebene auszurotten. Ebenfalls soll der lebenswichtige Einsatz von tierischen Organen in der Transplantationschirurgie ohne Abstoßungsreaktionen ermöglicht werden. Abstract Introduction: In the last few years people have been finding ways to interfere with the genome of different species which are faster and even more efficient. Due to these new possibilities of influencing the genome, Genomic Targeting has become more practicable as well as more affordable. Therefore the transformation or exchange of DNA is both easier to access and to perform. Which risks are associated with this trend? Do the positive aspects still outweigh the negative ones? Aside from the advantages which come with the different methods of Genomic Editing this thesis is concerned with possible hazards, which can occur when interfering with an organism’s genome, as well. The beginnings and the scientific progress of Genomic Editing are being pictured and different methods explained. Methods and process: The topic of this thesis has been discussed using literature research. Various online databases containing numerous papers and other search engines have been used in order to work with the issue. Articles, journals, studies and other papers have been browsed whereas English and German literature has been included. Results: The application range of Genomic Targeting reaches from genetic manipulation in mice in favour of pharmaceutic research and usage in plant breeding and agriculture to the manipulation of even the human genome in embryos. Via the construction of these methods extirpating the causes of diseases on the genetic level should be made possible. The usage of animal organs, which is vital in graft surgery, is to be made more reliable due to fewer organ rejections. Inhaltsverzeichnis Eidesstattliche Erklärung Vorwort und Danksagung Zusammenfassung Abstract Inhaltsverzeichnis Abbildungsverzeichnis/Tabellenverzeichnis Abkürzungen 1 Einleitung s.1 1.1 Genome Editing s.1 1.2 Restriktionsendonukleasen s.3 1.3 Zinkfingernuklease-Verfahren s.5 1.3.1 Gene Targeting mit dem Zinkfingernuklease-Verfahren s.6 1.3.2 Genetische Aspekte des Genomic Editings durch das Zinkfingernuklease-Verfahren s.7 1.3.3 Spezifität des Zinkfingernuklease-Verfahren s.9 1.3.4 Zukunft der Zinkfingernuklease-Technik s.10 1.4 Meganuklease-Verfahren 1.4.1 Ablauf des Meganuklease-Verfahrens 1.5 Transcription Activator-like Effector Nuclease 1.5.1 Aufbau s.11 s.12 s.13 s.13 1.5.2 Verwendung der TALEN-Methode s.15 1.5.3 Zukunft des TALEN-Verfahrens s.16 1.6 Das CRISPR/Cas-System s.17 1.6.1 Das mikrobielle Immunsystem CRISPR/Cas s.17 1.6.2 Genomic Editing mit dem CRISPR/Cas9 System s.21 1.6.3 Spezifität des CRISPR/Cas9-Verfahrens s.24 2 Material und Methodik s.25 3 Ergebnisse s.27 3.1 Das Mausmodell s.27 3.1.1 Simultanes Genomic Targeting von bis zu fünf Genen 3.1.2 Transfektion durch direkte Mikroinjektion in Zygoten s.27 s.29 3.1.3 Einschränkungen und negative Aspekte des Verfahrens 3.2 Die genetische Modifikation menschlicher Embryos 3.2.1 Die genetische Veränderung des HBB Gens s.33 s.34 s.36 3.2.2 Genomic Editing in menschlichen Tripronuklearen Zygoten durch das CRISPR/Cas9 Verfahren s.40 3.2.3 Die negativen Aspekte dieses Verfahrens s.41 3.2.4 Die Reparatur der Doppelstrangbrüche s.43 3.3 Der Einsatz von Gentechnikverfahren in der Pflanzenzucht s.44 3.4 Die Nutzung tierischer Organe zur Transplantation in Menschen s.45 4 Diskussion s.46 Literaturverzeichnis s.50 Abbildungsverzeichnis s.53 Abbildungsverzeichnis/Tabellenverzeichnis Tabelle 1 Tabelle der vier verschieden Gruppen von Endonukleasen Abbildung 1 Abbildung des “Synthesis-dependant strand s.4 -annealing mechanism of homologous recombination“ s.9 Abbildung 2 Ablauf der TALEN-Methode s.15 Abbildung 3 1. Adaptation: Integration der neuen Spacer in CRISPR. 2. Expression: Transkription des CRISPR-Bereiches. 3. Interferenz: Erkennen und Zerstören der viralen Abschnitte s.18 Abbildung 4 Abbildung der Prozessierung und Interferenz von crRNA s.20 Abbildung 5 Abbildung des CRISPR/Cas9-Ribonukleinkomplexes s.21 Abbildung 6 Abbildung eines Cas9 induzierten Doppelstrangbruches s.23 Abbildung 7 Abbildung des Genomic Editings in embryonalen Stammzellen und die genetische Veränderung an Mäusen s.31 Abbildung 8 Abbildung einer polyspermatischen Zygote s.35 Abbildung 9 Graphik von drei gRNA Seiten des Genomic Targetings s.37 Abbildung 10 Abbildung eines T7E1 Assays s.38 Abbildung 11 Abbildung einer Transfektion des G1gRNA-Cas9 Vektors in ansteigender Konzentration in den 293T Zellen Abbildung 12 Schematischer Ablauf der ersten 48h nach s.39 der Transfektion s.40 Abbildung 13 Abbildung der Off-Target Spaltung in menschlichen Tripronuklearen Zygoten Abbildung 14 Abbildung zweier Reparaturmechanismen Doppelstrangbrüchen in menschlichen Zellen s.42 von s.43 Abkürzungen 3PN Tripronukleare Zygote ATP Adenosintriphosphat CRISPR Clustered regularly interspaced short palindromic repeats DNA Desoxyribonukleinsäure DSBR Double-strand break Fok Flavobacterium okeanokoites GFP Grün fluoreszierendes Protein gRNA Guide RNA GTP Guanosintriphosphat HBD Hämoglobin delta HBE Exkretorisches Hämoglobin HBG1 Hämoglobin gamma-1 HBG2 Hämoglobin gamma-2 HR, HDR Homologe Rekombination mRNA Messenger RNA NGG, PAM Sequenzen NHEJ Non-homologous end-joining PCR Polymerase chain reaction PERV Porcine edogenous retrovirus pre-crRNA Precursor CRISPR RNA RFLP Restriction fragment length polymorphism RVD Repeat variable diresidue SDSA Synthesis-dependent strand annealing sgRNA Eine chimäre nicht-kodierende RNA Sry Sex determining region of Y ssDNA Single-stranded DNA TALEN Transcription activator-like effector nuclease Tet Ten-eleven translocation methylcytosine dioxygenase tracrRNA Trans-activating crRNA Uty Ubiquitously transcribed tetratricopeptide repeat containing ZFN Zinkfingernukleasen 1 Einleitung 1.1 Genome Editing Schon früh kamen erste Ideen auf, das menschliche Genom nach den eigenen Wünschen zu verändern und zu gestalten. Erste Versuche, ein maßgeschneidertes Genom zu kreieren, gab es bereits 1972, als zum ersten Mal ein DNA-Molekül eines bakteriellen Genoms generiert wurde. Unter den Begriffen „Genome Editing“ oder auch „Genome Targeting“ versteht man heute speziell entwickelte Verfahren und Techniken, die eine gezielte Veränderung, beziehungsweise einen Austausch von DNA ermöglichen (1). Diese speziellen Eingriffe ermöglichen sowohl das Ausschneiden, sowie das Einfügen neuer DNAAbschnitte in das Genom (2). 1972 handelte es sich bei den erstmaligen Eingriffen in das Erbgut um das Genom des Bakteriums Escherichia coli und das Genom der Bakteriophage Lambda. Über Jahrzehnte hinweg wurden die Hefebakterien (Saccharomyces cerevisiae) bevorzugt als Anwendungsgebiet verwendet (3). Zu diesem Zeitpunkt war die Erfolgsrate dieser Methode von mehreren Faktoren abhängig. Die Schwierigkeit bestand einerseits darin, unter Laborbedingungen DNA-Segmente der Hefe gezielt zu entfernen, und andererseits veränderte beziehungsweise fremde DNA erfolgreich am gewünschten Ort einsetzen zu können. Auch die Qualität des Zusammenspiels zwischen der Spender-DNA und der Ziel-DNA spielte eine entscheidende Rolle. Das Genom der Hefebakterien und auch anderen Bakterienarten und Pilzen erfüllten zum größten Teil diese Anforderungen und waren somit beliebte Forschungsobjekte. Zu dieser Zeit stellten das menschliche Genom, sowie jenes anderer Eukaryoten, eine unüberwindbare Hürde für die Wissenschaft dar. Bei diesen Zellen war vorerst die Frequenz der Rekombination zu niedrig oder es mangelte, aus rechtlichen oder auch technischen Gründen, an der Verfügbarkeit embryonaler Stammzellen. An linearer Spender-DNA konnten durchaus bessere 1 Werte erzielt werden (1). Im Vergleich zu heutigen Verfahren war jedoch auch in Hefezellen die Rate der Rekombinationen zwischen den Zellen des Spenders und den Zellen des Ziels relativ niedrig (1). Das erste genetisch veränderte Tiermodell wurde bereits 1974 hergestellt und präsentiert (4). Dafür werden präzise und effiziente Technologien benötigt, um ein möglichst zielgerichtetes Ausschneiden der DNA-Sequenzen zu garantieren. Es stellt eine große Herausforderung dar, ein einzelnes Gen in einem größeren Abschnitt überhaupt genau lokalisieren zu können (1). Weiterhin wird nach neuen und effizienteren Möglichkeiten gesucht. Es wird größtenteils versucht, durch neuere Methoden den Schnitt in der Sequenz so genau wie möglich zu tätigen und größere Änderungen im genetischen Material zu vermeiden. Die fortschrittlicheren Methoden sollten sowohl erschwinglicher als auch praktikabler und schneller als die bisher bewährten Systeme sein (5). Viele der bekannten Systeme leiden an der zu geringen Effizienz oder der zu hohen Laborkosten (6). Der Durchbruch in der gezielten Veränderung des Genoms gelang mit der erstmaligen Durchführung des Genome Editings. Ab diesem Zeitpunkt war ein gezieltes und verschiedene effizientes Eingreifen in das Erbgut durchführbar. Es gibt Verfahren des Genome Editings, die für die genetischen Veränderungen ortsspezifische Nukleasen verwenden (2). Diese Nukleasen ermöglichen einen ortsspezifischen Schnitt an einer gewünschten Sequenz (2). Es gibt verschiedene Ausführungen dieser Endonukleasen. Die bisher bekannten und gebräuchlichsten Methoden sind die Zinkfingernukleasen, die TALENs (Transcription Activator-like Effector Nucleases) und die Meganukleasen (2, 6). Eine neue Methode in diesem Bereich ist beispielsweise das Genome Editing durch das CRISPR/Cas System (7). Diese Arbeit soll neben den Vorteilen, die verschiedene Verfahren des Genomic Editings bedeuten, auch die möglichen Gefahren und Risiken aufzeigen, die bei Eingriffen in das Genom von Organismen entstehen. Die Anfänge und der wissenschaftliche Fortschritt des Genomic Editings werden beschrieben und verschiedene Verfahren genau erläutert. Welche Herausforderungen bringt die Durchführung der unterschiedlichen Verfahren mit sich? Neben den seit Jahren 2 bewährten Verfahren, werden auch neu entwickelte Verfahren des Genomic Targetings beschrieben. Immer schnellere und effizientere Methoden werden entwickelt, die ein gezieltes Eingreifen in das Genom ermöglichen. Das Genomic Targeting wird sowohl praktikabler als auch erschwinglicher. Somit wird die Veränderung bzw. der Austausch von DNA leichter zugänglich. Welche Risiken sind aber mit diesem Trend verbunden? Und überwiegen weiterhin die positiven Aspekte der Methode? 1.2 Restriktionsendonukleasen Enzyme, welche die spezifische DNA-Sequenz erkennen und lokalisieren, werden als Restriktionsendonukleasen bezeichnet. Diese Enzyme sind sowohl für das Erkennen, als auch das Schneiden der DNA-Abschnitte verantwortlich. Bisher sind 4 verschiedene Klassen von Restriktionsendonukleasen bekannt (8). Je nach der Erkennungssequenz der Endonukleasen werden die Gruppen I, II, III und IV unterschieden. (8). Typ I Enzyme besitzen drei verschiedene Subunits, welche ein multifunktionales Enzym bilden. Sie sind sowohl für die Restriktion als auch die Modifikation der DNA verantwortlich. Ihre Enzymaktivität reicht von der Endonuklease über die Methyltransferase bis hin zur ATPase. Diese Gruppe ist nur in der Lage, eine Sequenz an einer zufällig ausgewählten Stelle zu finden. Außerdem ist es dieser Gruppe nicht möglich, die DNA in der Nähe der Erkennungssequenz zu schneiden (8). Typ II Enzyme besitzen hingegen zwei idente Subunits. Bei der Enzymaktivität dieser Gruppe handelt es sich um die Endonuklease oder die Methyltransferase. Im Vergleich zur Typ I Endonuklease ist die Typ II Endonuklease in der Lage, die Sequenz direkt bei oder zumindest in der Nähe der Erkennungssequenz zu schneiden (8). 3 Typ III Enzyme besitzen zwei verschiedene Subunits. Vergleichbar mit Typ I fungieren sie als Endonuklease, Methyltransferase und ATPase. Dieser Subtyp ist in der Lage die DNA-Sequenz ungefähr 25-27 Basenpaare entfernt von der Erkennungssequenz zu schneiden (8). Enzyme vom Typ IV besitzen ebenfalls zwei verschieden Subunits. Die Enzymaktivität reicht von Endonuklease bis hin zu GTPase. Im Vergleich zu den ersten drei Typen ist die Typ IV Endonuklease nur in der Lage methylierte DNA zu schneiden (8). Typ I Enzyme Typ II Enzyme Typ III Enzyme Typ IV Enzyme Aufbau/ Multifokales 2 idente Subunits 2 verschieden 2 verschieden Struktur Enzym Subunits Subunits 3 verschiedene Subunits Endonuklease Endonuklease Endonuklease Endonuklease Enzym- Methyltransferase Methyltransferase Methyltransferase GTPase aktivität ATPase ATP- ATP Notwendigkeit ATPase Kein ATP ATP Tabelle 1 Es gibt vier verschieden Gruppen von Endonukleasen. Je nach der Erkennungssequenz der Endonukleasen werden die Typen I, II, III und IV unterschieden. Jede von ihnen weist individuelle Werte bezüglich Aufbau, Enzymaktiviät und Versorgung durch ATP auf. 4 1.3 Zinkfingernuklease-Verfahren Die effektivste und bewährteste Möglichkeit des Genomic Targetings war in den letzten Jahren die Methode der Zinkfingernukleasen. Erstmals wurden Zinkfingernukleasen 1996 präsentiert. Von da an fand das System Anwendung in verschiedensten Organismen und Zelltypen (1). Die Methode beschränkte sich zu ihren Anfängen lediglich auf die prokaryotische Spaltungsdomäne (9). Damals war noch nicht eindeutig geklärt, dass diese Spaltungsdomäne auch an der DNA Anwendung finden würde. Zinkfingernukleasen sind keine von Natur aus vorkommenden Restriktionsenzyme, sondern werden gentechnisch hergestellt. Diese haben separate funktionelle Domänen. Eine Domäne ist in der Lage, DNA zu binden (Zinkfingerdomäne), die andere, DNA zu spalten (Nukleasedomäne) (1). Diese Domänen wurden künstlich zusammengefügt. Die Nukleasedomäne gehört zum Typ IIS der Restriktionsendonukleasen, welcher auch FokI genannt wird (8). Dieses FokI wurde von Natur aus vorkommend aus dem Flavobakterium Okeanoloites gebildet (10). Durch ihre Konstruktion werden genaue und genspezifische Doppelstrangbrüche ermöglicht (11). Die wirkungsvollste Domäne zum Erkennen und Binden der DNA-Struktur ist ein Set von Cys2His2 Zinkfingern (zwei Cystein-und zwei Histidinreste). Diese Einheit von 30 Aminosäuren ist in der Lage, ein einzelnes Zinkatom zu binden (1). Bei den ersten Versuchen wurde noch nicht festgestellt, dass die FokI Spaltungsdomäne im Arbeitsprozess ein Dimer bilden muss, um überhaupt in der Lage zu sein, DNA zu spalten (12). Somit ist der beste Weg für eine erfolgreiche Spaltung, zwei Zinkfingersets zu konstruieren, die nahe aneinander angebracht werden. Jedes Set wählt damit eine monomere Spaltungsdomäne. Sobald jede der zwei Zinkfingersets an ihre für die Bindung zuständige Domäne bindet, wird die Spaltung deutlich erleichtert (1). Die Erkennungssequenz von einzelnen Zinkfingernukleasen, ohne Verbindung zu anderen, entspricht ungefähr einer Größe von 9 bis 12 Basenpaaren (bp). Im Vergleich dazu kann die Erkennungssequenz durch Zinkfingersets auf ungefähr 18 bis 24 Basenpaare (bp) erhöht werden. Für die optimale Verbindung zwischen der Domäne des Proteins und der bindenden Seite wird ein Abstand von 5-6 Basenpaaren beschrieben. Auch ein Abstand von 7 Basenpaaren gilt als möglich 5 für eine erfolgreiche Spaltung (1, 13). Zinkfingernukleasen arbeiten mit der Unterstützung von vom Zelltyp abhängigen Promotoren, die den Ablauf der Expression regeln, und mit Vektoren, die durch Transduktion mit Hilfe eines Virus, die DNA-haltigen Zellen einbringen können (1). 1.3.1 Gene Targeting mit dem Zinkfingernuklease-Verfahren In den frühen Entwicklungsstadien des Verfahrens wurden Zinkfingernukleasen lediglich als chimäre Restriktionsendonukleasen erzeugt, mit der Aufgabe, in vitro Aktivität aufzuzeigen und nachzuweisen (9). Zu diesem Zeitpunkt wurde daran gezweifelt, ob die prokaryote Spaltungsdomäne auch in eukaryoter DNA verwendet werden könnte. Erste Experimente zeigten jedoch eine hohe Spaltungs- und Rekombinationsrate in Froscheizellen mit extrachromosomalen Substraten und Zinkfingernukleasen mit bekannter Spezifität. Der erste Erfolg mit neu produzierten Zinkfingernukleasen zeigte sich beim Gene Targeting in der Fruchtfliege. Die Veränderung fand am „yellow locus“ im Zellkörper und in den Keimzellen statt. Der „yellow locus“ ist jener genetische Abschnitt der Fruchtfliege, der für die Ausprägung der Pigmentierung und somit für das Muster und die Farbe verantwortlich ist. Von diesem Zeitpunkt an wurde das Verfahren in individuellen Genen und verschiedensten Organismen angewandt. Wichtig für den Erfolg des Verfahrens war die Übertragung der Zinkfingernukleasen. Expressionsfaktoren für Zinkfingernukleasen benutzen vom Zelltyp abhängige DNA-Abschnitte, die eine regulierte Expression einzelner Gene ermöglichen und Vektoren, welche bei der Transfektion von DNA unter Mithilfe von Viren eingesetzt werden. Dieselben Methoden werden auch dazu verwendet die DNA des Spenders einzusetzen. Frühere Experimente an Fruchtfliegen beschäftigten sich mit der gentechnischen Integration von Spender-DNA und Zinkfingernukleasen-codierten Sequenzen über eine P-Element-vermittelte Transformation. Als P-Element bezeichnet man eine 6 spezielle DNA-Sequenz, die häufig bei Fruchtfliegen zu finden ist. Eine weitere Anwendung ist das Einbringen von Zinkfingernukleasen-mRNA und Spender-DNA in Embryos. Diese Methode des Einbringens von mRNA für die Expression der Zinkfingernukleasen wurde in diversen Organismen ausgetestet (1). Diese Organismen sind beispielsweise die Fruchtfliege (Drosophila melanogaster), der Seidenspinner (Bombyx mori), der Seeigel (Hemicentrotus pulcherrimus) und der Krallenfrosch (Xenopus tropicalis). Die homologe Rekombination mit Hilfe der Spender-DNA fand bei Mäusen und Ratten Anwendung. Diverse Studien beschreiben die Herausforderungen, die beim Übertragen des Materials auftreten. Zum Beispiel gab es anfangs hohe Raten von Mutationen sowohl im Genom als auch in extrachromosomalen Arealen durch die Anwendung des Hitzeschock-Promotors bei der Zinkfingernukleasenexpression. Obwohl Genveränderungen in vielen Fällen beim Zinkfingernuklease-Verfahren festgestellt werden, erfolgt jedoch nicht bei allen ein Genaustausch. Auch bei Zebrafischen wird ein hohes Maß an Spaltung erzielt, wobei bis jetzt noch kein non-homologer Genaustausch stattgefunden hat. Es gibt kein Problem mit dem Einfügen der Spender-DNA, jedoch stellt die Rekombination mit der Zielsequenz ein Hindernis dar (1). 1.3.2 Genetische Aspekte des Genomic Editings durch das Zinkfingernuklease-Verfahren Grundsätzlich kann man sagen, dass das Standardverfahren der homologen Rekombination und des „non-homologen end-joinings“ in den meisten Fällen völlig ausreichend ist. In gewissen Situationen ist es jedoch unbedingt nötig, wichtige Variationen durchzuführen oder andere speziellere Komponenten zu verwenden (1). Beispielsweise wird bei der Fruchtfliege in den meisten Fällen die DNAbindenden Proteine Rad51 und Rad54 zur DNA-Reparatur von Doppelstrangbrüchen eingesetzt. In manchen Fällen kann es jedoch notwendig 7 sein, Rad51-unabhängige Mechanismen zu verwenden. Der größte Anteil des „non-homologen end joinings“ benutzt eine spezielle DNA-Ligase, Lig 4, um Doppelstrangbrüche wieder zu verbinden (1). In den meisten Zellen und Organismen werden bei der homologen Rekombination die Enden an der Zielsequenz von 5´ nach 3´ geschnitten (14). Daraufhin wird das übriggebliebene 3´-Ende durch die DNA-Polymerase verlängert. Nach der folgenden Synthese kapselt sich das verlängerte Ende ab und legt sich an das andere Ende des Strangbruchs. Die Liganden verbinden die beiden Bruchstücke um die Kontinuität des Strangs wieder einwandfrei herzustellen. Die Länge der bearbeiteten Sequenz variiert je nach Verwendungszweck und Organismus. Beim Genomic Editing an der Fruchtfliege weist die entnommene Sequenz der SpenderDNA eine Länge von mehreren Kilobasen auf. Bei Säugetierzellen werden jedoch kürzere Sequenzen, im Bereich von ungefähr 100 bis 200 Basenpaaren (bp) bearbeitet (1). 8 Abbildung 1 Abbildung des “Synthesis-dependant strand annealing mechanism of homologous recombination“. Die Zellen werden an der Zielsequenz von 5´ nach 3´ geschnitten. Das 3´ Ende wird durch die DNA-Polymerase verlängert. Nach der Synthese kapselt sich das verlängerte Ende ab und legt sich an das andere Ende des Strangbruchs. Die Liganden verbinden die beiden Bruchstücke (15). (Abbildung modifiziert nach Cox MM, Battista JR. Deinococcus radiodurans - the consummate survivor. Nature reviews. Microbiology 2005; 3(11):882–92) 1.3.3 Spezifität der Zinkfingernuklease-Verfahren Für das Genome Targeting eines einzelnen Gens werden mehrere Paare benötigt und ausgetestet. Teilweise ist es von Vorteil, neue Kombinationen von Zinkfingern zu erzeugen und somit neue Arrays zu schaffen (1, 16). Als besonders bewährt gilt die Methode, in der Sets durch drei Zinkfinger gebildet werden. Teilweise wird 9 jedoch auch die Methode unter Verwendung von Zweifingersets angewandt (1, 17). Die Spezifität ist davon abhängig, woran die Zinkfinger besser, beziehungsweise leichter binden (1). Grundsätzlich lässt sich jedoch sagen, dass jegliche neue Kombination von Zinkfingern, sowohl die Sets mit drei Zinkfingern als auch die Sets mit zwei Zinkfingern, nachweislich sowohl die Spezifität des Verfahrens verbessert als auch die Bindungsaffinität deutlich erleichtert. Somit steigt die Rate der erfolgreichen Rekombinationen deutlich an (16). Kommt es zu einer unplanmäßigen Bindung, spricht man von einer Off-target Reaktion. In diesem Fall binden die Zinkfinger an eine weitere, nicht gewünschte, Stelle des Genoms und es kann zu einer Off-target Spaltung kommen (1). Diese zusätzlichen Spaltungen können zu schwerwiegenden Veränderungen im genetischen Material der Zelle und im schlimmsten Fall auch zum Zelltod durch fehlende Stabilität im führen. Meist Genom führen kleineste Fehler im Zinkfingerpaar zu solchen Veränderungen. Diese Effekte werden in einem weiteren Kapitel dieser Arbeit noch genauer erläutert. Bei Organismen, die rein der Forschung oder der genetischen Analyse dienen, ist eine niedrige Frequenz von Off-target Reaktionen noch vertretbar und akzeptabel. Wird das Zinkfingernuklease-Verfahren jedoch in der menschlichen Gentherapie eingesetzt, ist jegliche Rate von Fehlmutationen inakzeptabel (1). Optimal wäre ein Zinkfingerpaar, das genau die gewünschte Sequenz erkennt, die gespalten werden soll und an diese bindet. Danach sollte genau ein einzelner Doppelstrangbruch getätigt werden. 1.3.4 Zukunft der Zinkfingernukleasentechnik Prinzipiell kann jede Zelle aus nahezu allen Organismen durch diese Technik verändert und neu gestaltet werden. Doch wie weit kann der wissenschaftliche Fortschritt noch gehen? 10 Bisher galt die Ratte als bevorzugter Organismus, um diese Technik zu erforschen und weiterzuentwickeln. Schon seit Jahren ist es möglich ZinkfingernukleasenmRNA einzubringen und somit ein Genomic Targeting durchzuführen (1, 18). Auch die Mutagenese und Genveränderung in Mäusen ist durch Einbringen von Zinkfingernukleasen schon längst keine Herausforderung mehr für die Wissenschaft. Der Einsatz dieser Technik in der Pflanzenzucht ermöglicht ungeahnte Möglichkeiten. Erwähnenswert ist jedoch auch, dass immer neuere und effizientere Techniken entwickelt werden. Diese Techniken ermöglichen nahezu jedem Forscher, Genome von verschiedensten Organismen ohne großen Aufwand nach seinen Wünschen neu zu gestalten. Somit wird die Zinkfingernuklease-Technik mehr und mehr in den Hintergrund gedrängt, da sie ein gewisses Maß an finanziellen Mitteln und Know-how erfordert. In diversen Studien werden Möglichkeiten beschrieben, die das Risiko von ungewollten Mutationen deutlich senken können. Beispielsweise besteht die Möglichkeit Zinkfingernukleasen so zu nutzen, dass sie statt eines Doppelstrangbruches nur einen Einzelstrangbruch durchführen. Somit wird der DNA erleichtert, den Bruch auszugleichen (1, 19). 1.4 Meganuklease-Verfahren Eine weitere Methode um eine doppelsträngige DNA an einer gewissen Erkennungssequenz zu lokalisieren und zu schneiden ist die MeganukleaseTechnik. Dieses Verfahren ist vergleichbar mit dem der Restriktionsendonukleasen. Im Vergleich dazu arbeitet dieses Verfahren jedoch effizienter und sequenzspezifischer. Die gentechnisch veränderten Enzyme sind sogenannte Homing Endonukleasen. Diese speziellen Endonukleasen sind dazu in der Lage mittellange bis lange DNA Abschnitte zu erkennen und zu spalten. Man spricht dabei von ungefähr 20-30 aneinander liegenden Basenpaaren, die auf diese Weise lokalisiert und gespalten werden können (20). Homing Endonukleasen zählen zu den spezifischen Enzymen, die an einer gewissen 11 Sequenz Doppelstrangbrüche generieren können (20). Hauptsächlich kommen sie in mikrobiellen selbstspleißenden Sequenzen vor. In diesen selbstspleißenden Sequenzen wirken sie entweder in Gruppe 1 oder in Gruppe 2 als Introns. Andererseits können sie auch in speziellen Aminosäuresequenzen, sogenannten Inteinen wirken. In diesen Bestandteilen von Proteinen bewirken sie das Einbringen ihrer DNA Sequenz (21). Ihr Einbringen in eine fremde Zelle dient nur ihrer eigenen Lebenserhaltung und somit in keiner Weise dem befallenen Wirtsgen, schädigt dessen Funktion jedoch nicht. Somit können sie ungestört mikrobiellen Genomen von Wirten besetzten und in dort auch weitere Kopien von Sequenzen einfügen (20). 1.4.1 Ablauf des Meganuklease-Verfahrens Der Ablauf der Herstellung einer genetisch veränderten eukaryotischen Zelle durch Meganukleasen wurde bereits 2005 publiziert und 2012 zum Patent angemeldet (22). Nach der Auswahl des zu infizierenden Chromosoms wird die gewünschte Nukleinsäure in die Zielzelle eingesetzt. Die erste eingesetzte Nukleinsäuresequenz kodiert für eine Meganuklease. Danach wird eine weitere Sequenz eingesetzt, die im Vergleich zur ersten die exogene Sequenz enthält (22). Nach der Infizierung durch die erste Nukleinsäuresequenz wird durch die Meganuklease eine Spaltstelle in der eukaryoten Zelle erzeugt. In dieser Spaltstelle kann somit die exogene Sequenz, die beim Infizieren der zweiten Nukleinsäuresequenz eingefügt wird, ihren Platz finden und sich einbringen. Bei dieser Methode ist die verwendete eukaryote Zelle keine menschliche Stammzelle (22). Die für dieses Verfahren verwendeten Materialien sind ein MeganukleaseMonomer und ein Meganuklease Heterodimer. Diese bestehen aus einem Peptid, das eine Ähnlichkeit von mindestens 85% zu den Sequenzen der eukaryoten Zelle aufweist. Die Affinität der Dimerbildung wird ausgetauscht (22). 12 1.5 Transcription Activator-like Effector Nuclease Die transkriptionsaktivatorartige Effektornuklease, kurz TALEN genannt, ist eine Restriktionsendonuklease, mit deren Hilfe es möglich ist ein sequenzspezifisches Genomic Targeting durchzuführen. Wie bei den bereits erwähnten Verfahren, erfolgt das Genomic Doppelstrangbrüchen. Targeting Erstmals durch wurden die diese Erzeugung von DNA- transkriptionsaktivatorartigen Effektoren in den Bakterien Xanthomonas oryzae und Xanthomonas oryzae pv.oryzicola entdeckt, wo sie als Virulenzfaktoren wirken. Die Bakterien können das Genom einer Pflanze gezielt nach ihren Bedürfnissen verändern. Diese Effektorproteine sind durch eine Genfamilie namens avrBs3/PthA kodiert. Das TALEN-Verfahren findet in diversen Organismen Anwendung. Das Anwendungsgebiet reicht von einfachsten prokaryoten Organismen bis hin zu menschlichen Stammzellen (20). 1.5.1 Aufbau Ähnlich wie bei den Zinkfingernukleasen, ist die eigene DNA-Bindungsdomäne mit der FokI Nukleasedomäne fusioniert. Der Unterschied zwischen den TALENs und den Zinkfingernukleasen ist jedoch ihre DNA-Bindungsdomäne. Anstatt der Zinkfingerdomäne Wiederholungseinheit, besitzt wobei die TALEN eine eine Domäne in 33-35-Aminosäureetwa 12 bis 27 Wiederholungseinheiten aufweist. Diese Einheit ist dazu in der Lage ein einzelnes Basenpaar zu erkennen. Somit ermöglicht die Technologie der TALENs, mit Hilfe eines simplen DNA-Proteincodes an eine individuelle Base zu binden. Dies geschieht über Aminosäurereste, über die sich die Wiederholungseinheit an die Base heften kann (23). Durch diese Möglichkeit an Basen zu binden, ist die TALEN-Methode leichter und effizienter zu handhaben als das Verfahren mit 13 Zinkfingernukleasen. Sowohl bei den Kosten, als auch bei der Spaltungsrate schneidet die TALEN-Methode besser ab, als das Zinkfingernuklease-Verfahren. Die schon vorhin erwähnten Xantomonas-Bakterien sind in der Lage, durch ihr eigenes Sekretionssystem, Effektorproteine zu produzieren. Diese Effektorproteine werden Typ III-Effektoren genannt und stammen direkt aus dem Typ III-Sekretionssystem der Bakterien (23). Daraufhin können sie in den Wirtszellen die Transkription regulieren. Das Typ III-Sekretionssystem ist durch einen speziellen Gen-Cluster namens hrp (hypersensitive response and pathogenicity) codiert. Dieser Gen-Cluster ermöglicht den Transport von Proteinen von Bakterien zu eukaryoten Zellen. Dies geschieht durch die Produktion von Genen (24). An Position 12 und 13 der Domäne sitzen individuelle Aminosäuren, sogenannte Repeat Variable Diresidue (RVD), welche als Erkennungscode der DNA und somit der DNA- Bindungsspezifität der TALEN-Proteine dienen können (19). Es gibt vier verschieden RVDs (NN, NI, HD, NG), die mit einer der vier Basen (Adenin, Guanin, Cytosin und Thymin) korrespondieren. NN erkennt die Base Guanin NI erkennt die Base Adenin HD erkennt die Base Cytosin NG erkennt die Base Thymin Somit sind sie in der Lage lange DNA-Sequenzen mit einer Länge von mehr als 30 Basenpaaren zu erkennen (19, 25). Um diese Effekt zu verdeutlichen kann man beispielsweise sagen, dass ein gewisses RVD NK spezifischer für Guanin ist, als NN, da NN zusätzlich noch die Base Adenin erkennen kann. Jedoch läuft die Reaktion bei ihnen um einiges langsamer und mit geringerer Aktivität ab, als mit NN (19). 14 Abbildung 2 Ablauf der TALEN-Methode. Herstellung eines TAL-Codes als spezifisches Bindemolekül. An Position 12 und 13 der Domäne sitzen individuelle Aminosäuren, sogenannte Repeat Variable Diresidue (RVD), welche als Erkennungscode der DNA und somit der DNA-Bindungsspezifität der TALEN-Proteine dienen können (23). (Abbildung modifiziert nach Zhang M, Wang F, Li S, Wang Y, Bai Y, Xu X. TALE: a tale of genome editing. Progress in biophysics and molecular biology 2014; 114(1):25–32) 1.5.2 Verwendung der TALEN-Methode Wie auch die anderen Verfahren des Genomic Editings wurde die TALENMethode in den letzten Jahren in verschiedenste Organismen angewandt. Beginnend bei einfachsten Prokaryoten wurden fortlaufend sowohl die Techniken als auch die Organismen immer komplexer. Organismen, die früher als ungeeignet für die Veränderung des Erbmaterials galten, wurden zu beliebten Modellen für die TALEN-Methode. Die Liste an Versuchstieren reichte von Fruchtfliegen, 15 Zebrafischen, Fröschen, Mäusen Ratten, Hasen, Hamster und Schweinen, bis hin zu Kühen und sogar menschlichen pluripotenten Stammzellen. Größtenteils wird ein einzelnes TALEN-Paar für eine NHEJ (non-homologous endjoining)-induzierte Mutation eingesetzt. Beim NHEJ erfolgt die Verknüpfung bei den, beim Doppelstrangbruch erzeugten, Enden über spezielle Enzyme(19). Diverse Studien berichten jedoch auch von zwei TALEN-Paaren, die für Deletionen, beziehungsweise Inversionen eingesetzt wurden (26). Weitere Anwendungsgebiete der TALEN-Methode sind die Pflanzenzucht und auch die Landwirtschaft. 1.5.3 Zukunft des TALEN-Verfahrens Obwohl seit einigen Jahren weiterhin exzessiv an der Verbesserung der Verfahren gearbeitet wird, gibt es noch immer Risiken bei der Verwendung der TALENMethode. Es besteht ein erhebliches Risiko für das Auftreten von unvorhergesehenen Folgen. Die durch NHEJ-induzierte Mutation neigt weiterhin zu ungewollten Veränderungen am Genom. Die große Herausforderung besteht darin, eine Balance zwischen Effizienz, Spezifität und den Risiken einer Off-target Mutation zu finden. Zukünftig wird es besonders wichtig sein, diese Aspekte in Einklang zu bringen. Off-target Mutationen können zu schwersten chromosomalen Veränderungen oder sogar zum Zelltod führen. Die Methode steht jedoch auch anderen Domänen außer der Nukleaseaktivität zu Verfügung. Beispielsweise wird an Möglichkeiten geforscht, Proteinfusionen mit TALE-basierten Aktivatoren oder Repressoren durchzuführen. Das Ziel ist es, das TALEN-Verfahren sowohl leichter, als auch für mehr verschiedene Organismen und Zelltypen, zugänglich zu machen (19). Obwohl noch einige ungeklärte Fragen bezüglich der Anwendbarkeit und der Effizienz der Methode bestehen, ist der Umfang an Möglichkeiten dieser Technologie enorm. Das TALEN-Verfahren bleibt für die Forschung ein aufregendes und wichtiges Untersuchungsobjekt. 16 1.6 Das CRISPR/Cas-System 1.6.1 Das mikrobielle Immunsystem CRISPR/Cas Viren sind bedeutende Faktoren in der Ökologie und in der Entwicklung des Lebens. Sie dienen als Vermittler und Prädikatoren für den genetischen Austausch in Organismen und Zellen. Aus diesem Grund wurde eine Vielzahl an Mechanismen entwickelt, um einer Virusinfektion entgegen wirken zu können. Ein großer Vorteil in der Abwehr von Viren kann es sein, das Abwehrsystem adaptieren zu können. Somit schufen immer mehr Organismen Mechanismen, die es ermöglichten, die speziellen Fähigkeiten eines Pathogens zu erkennen und zu bekämpfen. Im menschlichen Abwehrsystem existieren B- und T-Zellen, die Proteine und andere Strukturen erkennen und somit Pathogene oder infizierte Zellen zerstören können (27, 28). Über Jahrzehnte hinweg war man davon überzeugt, dass die Möglichkeit, ein adaptives Immunsystem zu besitzen lediglich den Wirbeltieren vorbehalten war. Auf Grund der Komplexität des Systems war es für die Wissenschaft sehr überraschend, dass diese Art des Abwehrsystems auch in Prokaryoten zu finden war. Das System der Prokaryoten wird nach einer Region der DNA “Clustered Regularly Interspaced Short Palindromic Repeats“, kurz CRISPR, benannt (29). Diese sich wiederholenden Abschnitte der DNA oder RNA dienen dem Genom als Schutz gegen Viren oder verschiedenste andere Pathogene (29). Erstmals wurde dieser Abschnitt der DNA in Escherichia coli Bakterien im Jahr 1987 nachgewiesen, doch mit fortlaufender Forschung auf diesem Gebiet wurde die Region in immer mehr Prokaryoten lokalisiert (27, 30). Heutzutage ist die CRISPR-Region ist in 84% aller Archaeen und in 45% aller bekannten Bakterien nachweisbar. Jedoch muss man an dieser Statistik berücksichtigen, dass bisher schätzungsweise 20mal mehr Bakterienstämme untersucht wurden als Archaeen. CRISPR sind kurze repetitive Sequenzen von DNA-Abschnitten, die Repeats genannt werden. Die Bereiche werden dazu verwendet, Virus-Genome zu erkennen, zu lokalisieren und zu zerstören. Diese Aktivität funktioniert durch die 17 Anwesenheit des „CRISPR-associated“ Proteinkomplexes Cas, der die fremde DNA erkennt. Neue Spacer werden üblicherweise in die CRISPR eingefügt. Dieser Immunisierungsprozess wird in drei Stadien aufgeteilt. Im ersten Stadium, der Adaptation, werden neue Spacer in die Abschnitte der CRISPR integriert. Im Vergleich dazu werden im zweiten Stadium, der Expression, die Cas-Gene hergestellt und die CRISPR in lange pre-crRNA („precursor CRISPR RNA“) transkribiert (27). Diese Vorläufer-crRNA bildet mit der crRNA eine gemeinsame Struktur. Der dritte Abschnitt wird als Interferenz bezeichnet. In diesem wird die virale Sequenz durch den gemeinsamen Einsatz von crRNA und Cas-Proteinen erkannt und zerstört. Abbildung 3 1. Adaptation: Integration der neuen Spacer in CRISPR. 2. Expression: Transkription des CRISPR-Bereiches. 3. Interferenz: Erkennen und Zerstören der viralen Abschnitte. (27) (Abbildung modifiziert nach Rath D, Amlinger L, Rath A, Lundgren M. The CRISPR-Cas immune system: Biology, mechanisms and applications. Biochimie 2015; 117:119–28.) 18 Die Größe der CRISPR variiert zwischen 23 und 47 Basenpaaren (bp) und die der Spacer zwischen 21 zu 72 Basenpaaren (bp). In diversen Organismen kann sich die DNA-Abfolge der CRISPR stark unterscheiden (28). CRISPR können nach den Cas-Genen in drei verschiedene Typen aufgeteilt werden. Es können auch mehrere dieser Typen in einem einzelnen Organismus auftreten. Im Typ I-System ist das Signalprotein Cas3 verantwortlich für die Prozessierung beziehungsweise das Binden der crRNA an der Ziel-DNA. Derzeit sind noch weitere sechs Subtypen des Typ I-Systems bekannt (27). Die Signalproteine, die in dem TYP II-System wirken, sind Cas1, Cas2 und Cas9. Die Anwendungsgebiete von Cas9 sind die Anpassung, die crRNA-Prozessierung und die Spaltung der Ziel-DNA durch crRNA und tracrRNA. Im Vergleich zu Typ I sind bei diesem System nur drei weitere Subtypen bekannt. Typ II ist besonders erwähnenswert, da es die Grundlage für das Genomic Targeting darstellt. Typ III hingegen besitzt nur die Cas10-Nukleasen, die jedoch keine bisher bekannte Funktion aufweisen. Das System ist in der Lage sowohl fremde RNA als auch DNA zu schneiden. Typ II Systeme wurden bisher nur in Bakterien nachgewiesen, im Vergleich dazu fand man Systeme von Typ I und III sowohl in Bakterien als auch in Archaeen (27). 19 Abbildung 4 Abbildung der Prozessierung und Interferenz von crRNA in den verschiedenen CRISPR Systemen. Im Typ I-System wird die pre-crRNA durch Cas5 und Cas6 bearbeitet. Das Typ II-System verwendet RNase III und tracrRNA um reife crRNA herzustellen. Daraufhin erfolgt die Weiterleitung zur Ziel-DNA. Typ III benutzt ebenfalls Cas6 zur Bearbeitung von crRNA. Zusätzlich kommt ein unbekannter Faktor zum Einsatz, der eine 3´-End-Kürzung durchführt (27). (Abbildung modifiziert nach Rath D, Amlinger L, Rath A, Lundgren M. The CRISPR-Cas immune system: Biology, mechanisms and applications. Biochimie 2015; 117:119–28.) Cas9 ist ein Protein, das die zwei katalytisch wirkenden Untereinheiten RuvC und HNH, besitzt. Diese können Doppelstrangbrüche durchführen. RuvC schneidet die-DNA am Spacer und HNH spaltet auf der gegenüberliegenden Seite die ZielDNA. PAM, welches in der Nähe des C-terminalen Endes am Photospacer sitzt, beinhaltet die Basenkombination NGG. 20 Abbildung 5 Der CRISPR/Cas9-Ribonukleinkomplex mit den beiden Untereinheiten RuvC und HNH. RuvC wirkt an der Nicht-Ziel-DNA, wohingegen HNH an der gegenüberliegenden Seite, an der Ziel-DNA, wirkt. Sie sind dazu in der Lage DNAStränge zu schneiden. PAM beinhaltet die Basenkombination NGG und sitzt in der Nähe des C-terminalen Endes am Photospacer. (31) (Abbildung verändert nach Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE et al. RNA-guided human genome engineering via Cas9. Science (New York, N.Y.) 2013; 339(6121):823–6) 1.6.2 Genomic Editing mit dem CRISPR/Cas9 System Bereits 2012 wurden erste bedeutende Ergebnisse in Bezug auf eine neue Methode veröffentlicht, die es ermöglichte das Genomic Editing noch leichter und effizienter durchzuführen als bisher. Wie bereits erwähnt, können bei den bisherigen Methoden einige unerwünschte Begleiterscheinungen auftreten. Doch welche Vorteile bringt das CRISPR/Cas9 System? Noch nie war es so leicht, schnell und billig möglich, das Genom, egal ob pflanzlich, tierisch oder menschlich, nach eigenen Wünschen zu bearbeiten. Doch welche sicherheitstechnischen Bedenken sind mit dem Verfahren verbunden? Nahezu jedes Labor bzw. sogar jede Privatperson könnte es ohne großen finanziellen 21 Aufwand betreiben. Können somit potenziell riskante Genmanipulationen durchgeführt werden? Die Anwendungsgebiete des Systems reichen von genetischer Manipulation in Bakterien, Mäusen, Fruchtfliegen und Zebrafischen bis hin zu menschlichen Embryonen (5, 32, 33). Ein weiteres Forschungsgebiet ist die genetische Ausrottung von Krankheiten. In diversen Studien wird die Möglichkeit beschrieben, das CRISPR/Cas9 Verfahren dazu zu verwenden, die Erkrankung Tyrosinämie auf genetischer Ebene zu behandeln (34). Andere Studien wiederum beschäftigen sich mit der Anwendung des Verfahrens auf die Manipulation von Blutstammzellen (35). Auch für die Landwirtschaft könnte die Methode in Zukunft von großer Bedeutung sein. Wie bereits erwähnt ist das Typ II System der CRISPR dazu in der Lage gezielt Doppelstrangbrüche durchzuführen. Die erste gezielte Veränderung an eukaryoten Mauszellen fand 2013 statt. Ausgangspunkt dieser Studie war das Bakterium Streptococcus pyogenes (SpCas9) (33). Verschiedenste Vektoren, zum Beispiel rcRNA, tracrRNA und Cas9 werden daraus entnommen und für die Herstellung der Doppelstrangbrüche verwendet. Jahrelang beschränkte sich das Genomic Targeting beispielsweise auf die Verwendung von Homologer Rekombination in Hefezellen oder Mäusen. Oftmals überwogen auch die negativen Effekte gegenüber dem eigentlichen Ziel in der Durchführung. Nuklease-induzierte Doppelstrangbrüche können in nahezu allen Organismen und Zelltypen auf zwei verschiedene Wege repariert werden. Einerseits durch die des „non-homologous end joining“ (NHEJ) oder durch den komplexeren und somit präziseren Reparaturmechanismus „homologous recombination“ (HR). NHEJ kann durch seinen ungenaueren Mechanismus zu Mutationen, zum Beispiel Insertionen und Deletionen, führen (19). Der HR Reparaturmechanismus führt hingegen zu kleineren Punktmutationen oder Insertionen und ist abhängig von sogenannten DNA-Templates. Die Rate dieser Veränderungen bei der Erzeugung von Nuklease-induzierten Doppelstrangbrüchen kann von ungefähr 1% bis 50% aufweisen. Weitere Studien werden notwendig sein, um die Unterschiede in der Effizienz von HDR und Indel Mutationen aufzuzeigen (19). 22 Abbildung 6 Diese Abbildung zeigt den durch Cas9 induzierten Doppelstrangbruch. Daraufhin erfolgt auf der linken Seite der Graphik die NHEJ-Reparatur, die wie abgebildet zu einer Indel Mutation führen kann. Es kommt jedoch nicht notwendigerweise zu einem Gen-Knockout. Auf der rechten Seite wird im Vergleich dazu die HR-Reparatur dargestellt. Wie hier zu sehen ist, ist diese Methode unter Mithilfe der blau dargestellten DNA-Templates der präzisere Reparaturmechanismus (36). (Abbildung modifiziert nach Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. Genome engineering using the CRISPR-Cas9 system. Nature protocols 2013; 8(11):2281–308) Im Vergleich zu dieser Methode basieren herkömmliche Systeme des Genomic Targetings, wie Meganukleasen, bereits beschrieben, Zinkfingernukleasen auf oder DNA-Bindungsspezifitäten wie transkriptionsaktivatorartige Effektornukleasen (TALENs). Diese sind für die Anwenderin oder den Anwender jedoch mit größerem Aufwand, beziehungsweise Schwierigkeiten verbunden. Das Zinkfingernuklease-Verfahren und die TALEN-Methode nutzt die Protein-DNA Interaktion, wobei sich das CRISPR/Cas9-System auf die Regeln der einfachen Basenpaarung zwischen der zu bearbeiteten RNA und der Ziel-DNA zu Nutzen macht (19). Die Einfachheit von CRISPR/Cas9 Genomic Targeting findet auch in großen gRNA Libraries Anwendung. Diese besitzen unzählige Gensequenzen von verschiedensten Organismen und garantieren somit die weltweite Erfassung von DNA und RNA. Sie benutzen die Cas9 Nuklease um Knockout-Mutation zu 23 verursachen. Die enorme Anzahl von 64.000 bis 87.000 verschiedenen gesammelten gRNAs demonstrieren die große Kapazität der Libraries (19). 1.6.3 Spezifität des CRISPR/Cas9-Verfahrens Obwohl die Nukleasen größtenteils die gewünschten Spaltungen durchführen, gibt es noch immer eine gewisse Rate an Off-Target Effekten und unerwünschten Indel Mutationen. Um die Spezifität bestimmen zu können, kreierten Forscher geänderte gRNAs. Es stellte sich heraus, dass eine Mutation am 5´-Ende der 20 Nukleotide (nt) Region an der das Genomic Targeting durchgeführt wird, besser toleriert wird, als am 3´-Ende. Diese Erkenntnis stimmt mit den vorigen Forschungsergebnissen an Bakterien bzw. bei in vitro Versuchen überein. Anhand dieser lässt sich sagen, dass die 8-12 Basenpaare (bp) am 3´-Ende für die Zielerkennung ausschlaggebend sind. Jedoch lässt die Lokalisation der Mutation keinen bedingungslosen Rückschluss auf das Ausmaß dieser zu. Manche OffTarget Effekte an 5´-Enden können gravierende Folgen für den Organismus haben, wohingegen diverse Mutationen an 3´-Enden nahezu keine relevante Auswirkungen haben. Es lässt sich nicht einwandfrei feststellen, dass alle Substitutionen von Nukleotiden an einer gewissen Position überhaupt irgendeine Aktivität mit sich führen (19). Als besonders relevant gilt die Sequenz NAG als Alternative zu PAM. Die Frequenz der Indel Mutationen an dieser Off-Target Seite kann hoch genug sein, um die T7 Endonuklease I einzusetzen, die Mismatch-DNA erkennt und spaltet. Man muss jedoch erwähnen, dass die Effizienz dieser Methode nicht zufriedenstellend ist (19). Ein weiteres Anwendungsbeispiel sind die Cas9 Nickasen im Gene Editing. Ziel ist es, die Rate an Off-Target Mutationen so niedrig wie möglich zu halten. Bei D10A und H840A Mutationen in der RuvC1- oder HNH-Domäne werden Cas9 Nickasen dazu verwendet, komplementäre und nicht-komplementäre Zielstränge zu 24 inaktivieren. Somit wird es der Cas9-Nuklease ermöglicht, einen einzelnen Strang zu schneiden. Kommt es bei der Verwendung von Nickasen beim Genomic Editing mit dem CRISPR/Cas9 System zu Off-Target Effekten, so erfolgt dabei lediglich ein Einzelstrangbruch (31). Der nächste Teil dieser Arbeit beschäftigt sich mit speziellen Anwendungsbeispielen der bisher beschriebenen Methoden. Es werden diverse Publikationen beschrieben, deren Inhalt von der Anwendung des CRISPR/Cas9 Systems in Mäusen bis hin zu menschlichen Tripronuklearen Zygoten handelt (25, 33, 36). Abschließend wird noch die Frage behandelt, welche Methoden angewandt werden, um die Off-Target Effekts vom CRISPR/Cas9 System zu reduzieren. 2 Material und Methodik Die Thematik dieser Diplomarbeit wurde als Literaturrecherche behandelt. Es werden im einleitenden Teil verschiedene Verfahren gegenübergestellt und hinsichtlich ihrer Vor- und Nachteile beschrieben. Es sollten im Verlauf der Arbeit sowohl medizinischer Fortschritt in diesem Bereich, als auch aktuelle Anwendungsgebiete, sowie moralische und ethische Aspekte der Anwendung beleuchtet werden. Die Vor- und Nachteile der jeweiligen Verfahren werden in der abschließenden Diskussion durch eigene Analyse behandelt. Für die Bearbeitung der Themen wurden diverse Online-Datenbanken für wissenschaftliche Literatur und Suchdienste, wie Pubmed, Google Scholar und Oxford Medicine Online, herangezogen. Aufgrund der Aktualität des Themas wurden ebenfalls verschiedene elektronische Fachzeitschriften, medizinische Journale und auch Artikel in Tagespressen zur Auseinandersetzung mit der Thematik verwendet. Um die Grundlagen der Genetik zu wiederholen, 25 beziehungsweise zu vertiefen, wurde in diversen Sach- und Lehrbüchern recherchiert. Für das bessere Verständnis einzelner Begriffe besser, wurde über Standard Internetsuchmaschinen wissenschaftlich glaubwürdige Seiten gesucht und gelesen. Die Suchbegriffe enthielten dabei sowohl die Namen der jeweiligen Verfahren, als auch die der Autoren. Weitere Suchkriterien waren auch die Aktualität der Literatur sowie der öffentliche Zugang mit Hilfe der Zugriffsmöglichkeiten durch die Medizinische Universität Graz. Die verwendeten Bilddateien, Abbildungen und Tabellen wurden entweder aus Studien entnommen, modifiziert und daraufhin zitiert oder teilweise selbst kreiert. Gesucht wurden Artikel, Journale und Fachzeitschiften in deutscher und englischer Sprache. Bevorzugt wurde Erstliteratur und bedeutende aktuelle Quellen verwendet. Die Anzahl an Publikationen in den vergangenen Jahren ist enorm umfangreich, was eine sehr ausgiebige und zeitaufwendige Recherche nach passender Literatur für die Arbeit mit sich brachte. Der Zeitraum für die Erhebung der für die Diplomarbeit relevanten Daten war von September 2015 bis Dezember 2015. Zusätzlich wurde während des Schreibprozesses immer wieder nach relevanter und aktueller Literatur recherchiert und diese dann umgehend behandelt. Um die Quellen sowohl des Textes als auch der Abbildung besser wiedergeben zu können wurde Citavi als Zitierprogramm verwendet. 26 3 Ergebnisse 3.1 Das Mausmodell 3.1.1 Simultanes Genomic Targeting von bis zu fünf Genen Die Grundlagenforschung an Tiermodellen ist für die Weiterentwicklung von Genomic Editing Systemen von großer Bedeutung. Neben einfachsten Organismen sind die Versuche an Mäusen besonders relevant. Stammzellen von Mäusen weisen im Vergleich zu anderen Säugetieren große Ähnlichkeit mit jenen des Menschen auf. Somit sind sie als Modelle besonders gut dazu geeignet, neue Therapiemöglichkeiten im Bereich der Gentherapie auszutesten und für den Einsatz an menschlichen Zellen zu perfektionieren. Das Mausmodell dient dem Verständnis der Entwicklung von Krankheiten und deren Bekämpfung auf genetischer Ebene. Einerseits werden sie dazu verwendet, sequenzielle Rekombination in embryonalen Stammzellen durchzuführen oder andererseits Kreuzungen zwischen Mäusen mit einer einzelnen Mutation durchzuführen. Letzteres ist jedoch deutlich zeitaufwendiger. Das folgende Modell beschreibt die Auswirkungen des CRISPR/Cas Systems auf den simultanen Bruch von fünf Genen (Tet 1, 2, 3, Sry, Uty-8) in embryonalen Mauszellen (33). Bei der Verwendung von konventionellen Gene Targeting Systemen werden Mutationen durch homologe Rekombination in den Stammzellen erzeugt. Die dadurch veränderten Stammzellen werden in Wildtyp-Blastozysten injiziert. Somit können sie zur Veränderung der Keimbahn chimärer Tiere und zur dadurch entstehenden genetischen Modifikation führen. Diese Methode, die bereits 2005 erstmals publiziert wurde, ist im Vergleich zu neueren Methoden deutlich teuer und auch zeitintensiver. Wenn man bedenkt, dass bereits ein einzelner GeneKnockout sehr zeitaufwendig ist, kann man daraus schließen, welcher Aufwand für eine Doppelmutation notwendig wäre. Alternative Methoden arbeiteten bisher durch direktes Einbringen von DNA und mRNA von seitenspezifischen Nukleasen um dadurch Doppelstrangbrüche an einer ortspezifischen Stelle zu generieren. 27 Wie bereits erwähnt kann eine Reparatur dieser entweder durch NHEJ (nonhomologous end jouning) oder durch homologe Rekombination erfolgen. Jedoch kann durch die Verwendung von Zinkfingernukleasen oder TALENs die Effizienz des Genomic Editings stark variieren und genaue Ergebnisse sind so nur schwer voraussehbar (33). Um die Möglichkeit zu haben, funktionelle redundante Gene derselben Familie genetisch zu verändern, werden sogenannte sgRNAs designt. Diese sind in der Lage, Genomic Targeting an Mitgliedern der Ten-eleven-Translokation-Familie, Tet1, Tet2 und Tet3, durchzuführen. Tet Proteine verändern 5-Methylcytosin zu 5Hydroxymethylcytosin in verschiedenen embryonalen und adulten Zellen. Dadurch entstehen gentechnisch veränderte Mäuse in jeder dieser drei Gruppen durch Homologe Rekombination in embryonalen Stammzellen. Daraufhin werden ein optimiertes Cas9 und eine sgRNA dazu verwendet, in embryonalen Stammzellen von Mäusen Gene Targeting durchzuführen und die Effizienz des Verfahrens bzw. der Spaltung darzustellen. Bei der anschließenden Analyse zeigt sich, dass alle drei Cas9 sgRNA Transfektionen Spaltungen am Zielort erzeugen. Die durchaus hohe Effizienz beträgt bei Tet1 36%, bei Tet2 48% und bei Tet3 36%. Weil jeder Zielort eine Restriktionsenzym-Erkennungsseite besitzt, wird durch Polymerasenkettenreaktion (PCR) ein 500 bp großes Fragment in jede TargetSeite eingebracht. Somit verbinden sich die PCR Produkte mit den jeweiligen Enzymen. Ein korrekt verändertes Allel würde die Restriktionsseite verlieren. Unter der Verwendung des RFLP („restriction fragment lenght polymorphism“) Assay, werden pro Durchgang 48 Klone von embryonalen Stammzellen erkannt. Eine hohe Anzahl der Klone haben Mutationen in beiden Allelen. Zwischen 65% und 81% der getesteten Klone tragen Mutationen in Tet Genen und bis zu 77% zeigen Veränderungen in beiden Allelen (33). Durch die hohe Effizienz der „single-gene“ Modifikationen wurden die Wissenschaftler dazu bewegt, die Möglichkeit zu testen, Gene Targeting an allen drei Genen simultan durchzuführen. Aus diesem Grund werden die embryonalen Stammzellen mit Cas9 und drei sgRNAs, von Tet1, Tet2 und Tet3, zusammen transferiert. Von den 96 untersuchten Klonen, zeigen 20 Mutationen in allen sechs Allelen der drei Gene. Um diese Ergebnisse zu bestätigen, werden verschiedenste Verfahren wie Southern Blot verwendet um falsch positive Resultate 28 auszuschließen. Bei den Mutationen der Allele handelt es sich hauptsächlich um Deletionen und kleine Insertionen. Um herauszufinden, ob diese Modifikationen die Funktion der Tet Gene beeinflussen oder sogar gänzlich verhindern, vergleicht man das Level von 5-Hydroxymethylcytosin der veränderten Klone mit den embryonalen Stammzellen der Wildtypen. In nahezu allen durchgeführten Untersuchungen wird eine Reduktion des Levels von 5-Hydroymethylcytosin an den Klonen, welche Mutationen an beiden Allelen trugen, festgestellt (33). Ein weiterer Test des Potentials des CRISPR/Cas Systems beim Gene Targeting multiplexer Gene beschäftigt sich mit der Herstellung von zwei Y-linked Genen, Sry und Uty. In diesem Fall werden die embryonalen Stammzellen mit Cas9 und drei sgRNAs und den beiden Y-linked Genen zusammen transferiert. Von wiederum diesen 96 hergestellten Klonen, kann bei 10% Mutationen nachgewiesen werden, die sich auf alle acht Allele der fünf Gene erstreckt. Somit wird die Kapazität dieser Methode eindrucksvoll nachgewiesen (33). 3.1.2 Transfektion durch direkte Mikroinjektion in Zygoten Ein weiterer Ansatzpunkt um genetisch veränderte Mäuse herzustellen, ist die Möglichkeit einer direkten Manipulation der Embryonen in vivo. Bei dieser Methode wird Cas9 mRNA durch in vitro Transkription erzeugt und zusammen mit der schon vorhin erwähnten sgRNA injiziert. Eine besondere Schwierigkeit liegt in der Berechnung der optimalen Menge von Cas9 mRNA. Es werden unterschiedliche Mengen davon mit einer konstanten Konzentration von sgRNA in das pronukleäre Stadium der Einzell-Maus injiziert und die dadurch entstandenen Mutationen erfasst und berechnet. Wie erwartet führt eine höhere Konzentration von Cas9 mRNA zu einer erheblich höheren Rate an genetischen Veränderungen (33). Man muss jedoch erwähnen, dass ab einem gewissen Wert die Rate an Mutationen wieder absinkt. Somit ist bei der am höchsten gemessenen 29 Konzentration an Cas9 mRNA eine normale Blastozystenentwicklung zu beobachten. Um die postnatalen Auswirkungen zu generieren werden Tet1 und Tet2 zusammen mit verschiedenen Konzentrationen von Cas9 mRNA verabreicht. Southern Blot und Sequenzanalysen zeigen, dass ungefähr 50% bis 90% Mutationen an beiden Allelen tragen. Zusätzlich wird auch die Kombination von Tet3 sgRNA mit Cas9 mRNA in Blastozysten getestet. Von acht untersuchten Embryos weisen drei homozygote und drei heterozygote Mutationen auf. Einige Mäuse zeigen postnatal eine deutlich verringerte Größe oder manche sterben kurz nach der Geburt. Bei Mäusen, die eine Tet3 Mutation an beiden Allelen tragen, ist ein Überleben über die Neonatalphase hinaus nicht möglich (37). Die bisher erwähnten Versuche beschreiben die Veränderungen von EinzelGenen an Mäusen. Jedoch werden auch Versuche durchgeführt, die eine DoppelGen- Mutation oder auch multiple Gen-Mutation bewirken (33). Ein weiterer Teil derselben Versuchsreihe beschäftigt sich mit der Möglichkeit Doppel-Gen-Mutationen aus einem einzelnen Embryo zu generieren. Hierbei werden Tet1 und Tet2 sgRNAs zusammen mit Cas9 mRNA in die Zygote injiziert. Anhand dieser Methode können aus 144 transferierten Embryos 28 lebensfähige Mäuse erzeugt werden. Das ergibt eine Überlebensrate von 21%. Von diesen weisen wiederum 22 eine nachweisbare Mutation auf zwei Allelen in zwei verschiedenen Genen auf. Verschiedenste Versuche anhand dieser Methode zeigen, dass die Überlebensrate der Mäuse stark von der Konzentration der Cas9 mRNA abhängig ist. Erwähnenswert hierbei ist, dass die Überlebensrate zuerst proportional mit der Konzentration auf ungefähr 25% steigt, sie bei deutlich erhöhter Konzentration von Cas9 mRNA allerdings auf 21% absinkt. Somit wird bewiesen, dass es möglich ist, mit Hilfe der CRISPR/Cas9 Methode innerhalb kürzester Zeit biallelische Mutationen auf zwei unabhängigen Genen zu erzeugen. Ein weiterer Vorteil dieser Methode ist die vergleichsweise hohe 30 Effizienz, die bei einer Versuchsdauer von ungefähr einem Monat erreicht werden kann. Abbildung 7 Diese Abbildung zeigt einerseits das Genomic Targeting in embryonalen Stammzellen und andererseits die genetische Veränderungen an Mäusen durch multiple Mutationen. Die mittlere Abbildung stellt multiple Mutationen (Indels), induziert durch NHEJ, dar. Der im unteren Bildteil abgebildete Vorgang beschreibt Mutationen, induziert durch HDR-mediierte Reparatur (33). (Abbildung modifiziert nach Wang H, Yang H, Shivalila CS, Dawlaty MM, Cheng AW, Zhang F et al. One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell 2013; 153(4):910–8.) Die Herstellung von multiplen Mutationen ist eine weitere Möglichkeit, dieses Verfahren anzuwenden. Hierbei werden, dem ohnehin schon sehr präzisen HDRmediierten Genomic Editing, Cas9 mRNA, sgRNAs und Einzelstrang-DNA coinjiziert. Zusätzlich werden Tet1 und Tet2 verwendet. Tet1 verändert zwei Basenpaare der Restriktionsseite und kreiert somit eine EcoRI Seite. Tet2 konvertiert die schon vorhandene EcoRV Seite in eine EcoRI Seite. Durch diese 31 Veränderungen kann das HDR-mediierte Genomic Editing leichter erfolgen. Als EcoRI bezeichnet man eine spezielle Endonuklease, gebildet aus dem Stamm des Bakterium Escherichia coli (38). Wenn Cas9 mRNA, sgRNAs, Tet1 sowie Tet2 in Zygoten zusammen injiziert werden, weisen von vierzehn Embryos vier eine Veränderung am Tet1 Locus und sieben eine am Tet2 Locus auf. Ein Embryo weist ein Allel von beiden Genen auf. Die Rate an lebendig geborenen Mäusen beträgt durch diese Methode 21%. Diese Resultate zeigen, dass Mäuse durch HDR-mediierte Mutationen in multiplen Genen in einem Schritt durch CRISPR/Cas mediiertes Genomic Editing erzeugt werden können (33). Zusammenzufassend lässt sich sagen, dass die genetische Manipulation an Mäusen unerlässlich für die Entwicklung neuer Arzneistoffe sowie die Erforschung von Krankheiten ist. Jedoch muss man erwähnen, dass die homologe Rekombination an embryonalen Stammzellen und die Produktion von chimären Mäusen intensiver Laborarbeit bedarf und einen Zeitaufwand von ungefähr sechs bis zwölf Monaten in Anspruch nimmt. Die Studie beschreibt drei verschieden Anwendungen genetischer Veränderungen an Mäusen. Es wird gezeigt, dass Genome Editing durch das CRISPR/Cas Verfahren in embryonalen Stammzellen simultane Mutationen von mehreren Genen mit hoher Effizienz ermöglicht. Der erste Teil beschreibt die Möglichkeit, in einem einzigen Schritt Zellen zu produzieren, die Mutationen in fünf verschiedenen Genen aufweisen. Ebenfalls wird bestätigt, dass in Embryos von Mäusen direkt durch die Injektion von Cas9 mRNA und sgRNA biallelische Mutationen in einem bestimmten Gen erzeugt werden können. Durch die Coinjektion Cas9 mit Tet1 und Tet2 sgRNAs in Zygoten können Mutationen in beiden Genen hergestellt werden. Die Versuche zeigen, dass bei einer Injektion einer einzigen sgRNA, die Neugeborenen eine biallelische Mutationsrate von 95% aufweisen und bei einer Injektion mit zwei verschiedenen sgRNAs bis zu 80% biallelische Mutationen in beiden Genen tragen (33). 32 3.1.3 Einschränkungen und negative Aspekte des Verfahrens Es gibt natürlich wie bei jedem Verfahren, beziehungsweise jeder Technologie, einige Hindernisse und Einschränkungen, die die Durchführung des Versuchs erschweren können. Beispielsweise limitiert die Notwendigkeit der NGG PAM Sequenz des Streptococcus pyogenes Cas9 im Mausgenom. Es stellt sich heraus, dass das Streptococcus thermophilus LMD-9 Cas9 im Vergleich dazu eine andere PAM Sequenz verwendet, die eine Spaltung in Säugetierzellen durchführen kann (5). Somit kann es notwendig sein, durch zeitaufwendige Vergleiche die richtigen Cas9 Proteine auszutesten und auszuwählen. Auch für die speziell kreierten sgRNAs ist eine aufwendige Entwicklungs- und Forschungsarbeit notwendig, um sie so zu gestalten, dass sie die höchste Effektivität erzielen. Genau diese ist notwendig um das Genomic Targeting überhaupt zu ermöglichen. Ein weiterer Punkt ist die erhebliche Rate an Off-target Mutationen. Immer wieder treten genetische Veränderungen an Stellen des Genoms auf, die zum größten Teil negative Auswirkungen haben. Um diese zu verhindern, ist, wie bereits erwähnt, eine genaueste Analyse der eingesetzten sgRNAs und der Cas9 Proteine notwendig, um die Folgen des Verfahrens für den Embryo bestmöglich voraussehen zu können (33). Außerdem bringt auch der Reparaturmechanismus NHEJ diverse Negativaspekte mit sich. Einerseits ist dadurch zwar ein präzises Genomic Editing möglich, andererseits kann das andere (nicht behandelte) Allel massive Mutationen aufweisen. Mehrere Untersuchungen beweisen, dass eine niedrigere Konzentration an Cas9 mRNA, Mäuse mit heterozygoten Mutationen hervorbringt. Der Einsatz von Cas9 Nickasen würde höchstwahrscheinlich die Rate dieser Komplikationen reduzieren oder die auch verhindern. Cas9 Nickasen würden Einzelstrangbrüche verursachen, die normalerweise durch HDR repariert werden könnten (5, 31). 33 3.2 Die genetische Modifikation menschlicher Embryos Der weltweit erste dokumentierte Versuch Genomic Editing bei menschlichen Embryos durchzuführen, wurde 2015 publiziert. Diese Studie löste eine weitreichende mediale Debatte über die Ethik solcher Verfahren aus. Die Forscher rechtfertigten ihre Versuchsreihe mit dem Standpunkt, nicht-lebensfähige Embryos für ihre Untersuchungen verwendet zu haben. Der Grundgedanke der Studie war es, das Gen, welches für die Ausbildung der Beta-Thalassämie verantwortlich ist, zu verändern (39). Die Forscher verwendeten für ihre Untersuchungen tripronukleare Zygoten (3PN) um die CRISPR/Cas9 Technologie an menschlichen Zellen zu untersuchen. Die Entscheidung fiel auf Grund der hohen Effektivität, hohen Spezifität und der geringen Off-Target Effekte auf diese Technologie. Da die Verwendung von lebensfähigen Embryos aus ethischen Gründen nicht vertretbar war, wurden tripronukleare Zygoten mit einem Oozyten Nukleus und zwei Spermien Nuklei ausgewählt. Diverse Studien zeigen, dass solche polyspermatischen Zygoten eine gute Alternative zur Verwendung von normalen Embryos darstellen (40). Polyspermatische Zygoten, welche in 2-5% aller Zygoten im Rahmen der In-VitroFertilisation entstehen, können in vitro Blastozysten generieren. Jedoch versagt bei diesen unweigerlich die in vivo Entwicklung. Somit sind sie ideal geeignet für Untersuchungen der positiven und negativen Effekte und Auswirkungen der genetischen Manipulation während der frühen menschlichen Entwicklung (40). 34 Abbildung 8 Diese Abbildung zeigt eine polyspermatische Zygote. Diese tripronukleare Zygote (3PN) mit einem Oozyten Nukleus und zwei Spermien Nuklei kann zu genetischen Untersuchungen eingesetzt werden, da sie in diesem Zustand nahezu nicht lebensfähig ist. (Abbildung nach http://de.dexeus.com/reproduktionsmedizin/in-vitro- fertilisation/beurteilung-der-befruchtung) Das CRISPR/Cas9 System ist hierbei in der Lage, die endogenen Gene in tripronuklearen Zygoten effizient zu spalten und Doppelstrangbrüche zu verursachen. Diese können wie bereits beschrieben entweder durch NHEJ oder HDR repariert werden. Die Reparaturvorlage der HDR kann sowohl dem endogenen homologen Gen, als auch der exogenen DNA Sequenz entsprechen. Dieser Konkurrenzkampf zwischen endogener und exogener Sequenz kann die Analyse der möglichen Genome Editing Produkte deutlich komplizierter gestalten. Auch mögliche Fehlmutationen können die Interpretation erschweren. All diese Faktoren müssen zur besseren Verständlichkeit des Verfahrens erkannt und berücksichtigt werden, um es für den klinischen Einsatz zu verbessern (40). 35 3.2.1 Die genetische Veränderung des HBB Gens Das menschliche Beta-globin Gen kodiert eine Untereinheit des adulten Hämoglobins und ist im Rahmen der Beta-Thalassämie verändert. Beispielsweise sind in China CD14/15, CD17 und CD41/42, welche Frameshifts des Beta-Globins sind, drei der meist verbreitetsten Beta-Thalassämie Mutationen. Lokalisiert am Chromosom 11, befindet sich HBB innerhalb des Beta-Globin-Cluster, welcher noch vier weitere Globin Gene beinhaltet. In 5´ bis 3´ Richtung sind dies HBE, HBG2, HBG1, HBD und HBB (41). Da HBD und HBB eine sehr ähnliche Struktur aufweisen, kann HBD auch als Reparaturvorlage für HBB verwendet werden. Der von HBD hinterlassene Abdruck am reparierten HBB Locus ermöglicht die Untersuchung und Abschätzung, ob und wie endogene homologe Sequenzen als HDR Templates verwendet werden können. Diese Information kann zukünftig jenen dienen, die sich mit der Möglichkeit beschäftigen, das CRISPR/Cas9 System dazu verwenden, Genorte mit sich wiederholenden Sequenzen zu reparieren (41). Es werden zu Beginn drei gRNAs (G1, G2 und G39) designt und entwickelt, die ein Genomic Targeting an den verschiedenen Regionen des HBB Gens durchführen können. 36 Abbildung 9 Diese Graphik zeigt drei gRNA Seiten des Genomic Targetings, welche für den HBB Locus ausgewählt werden. Die Sequenz für jede gRNA (G1, G2 und G3) wird mit der PAM Sequenz in Grün dargestellt. In Rot sind die Mutationen der BetaThalassämie abgebildet (40). (Abbildung modifiziert nach Liang P, Xu Y, Zhang X, Ding C, Huang R, Zhang Z et al. CRISPR/Cas9-mediated gene editing in human tripronuclear zygotes. Protein & cell 2015 [cited 2015 Oct 3]; 6(5):363–72.) Daraufhin werden die gRNA Expressionsvektoren in menschliche 293T Zellen transferiert. Zusammen mit dem GFP Pseudovektor zeigen die G1 und G2 gRNAs eine deutliche Spaltungsaktivität. Diese werden durch das T7E1 Assay deutlich nachgewiesen. Sequenzanalysen der zwei durch Genomic Editing veränderten Regionen zeigen unterschiedliche Präferenzen bezüglich der Art der NHEJ Reparatur (40). 37 Abbildung 10 Die 293T Zellen werden individuell mit den drei gRNA-Cas9 Expressionsvektoren transfiziert. 48h nach der Transfektion werden sie für die genomische DNA Isolation gesammelt. Ein GFP Expressionsvektor wird als Kontrollorgan der Transfektion eingesetzt. Die Regionen der gRNA, die am Genomic Editing beteiligt sind, werden für das T7E1 Assay durch PCR vergrößert. Die blaue Markierung zeigt die erwartete Größe der „no mismatch“ PCR Produkte (40). (Abbildung modifiziert nach Liang P, Xu Y, Zhang X, Ding C, Huang R, Zhang Z et al. CRISPR/Cas9-mediated gene editing in human tripronuclear zygotes. Protein & cell 2015 [cited 2015 Oct 3]; 6(5):363–72.) Von bisherigen Studien war stets bekannt, dass das Genomic Targeting durch das CRISPR/Cas9 Verfahren am Beta-globin Locus eine deutlich erhöhte Rate an OffTarget Mutationen in menschlichen Zellen aufwies. Aus diesem Grund wurden vor Beginn des Verfahrens spezifische PCR-Primer designt. Diese sind für die sieben bevorzugten Off-Target Seiten des Genoms sowohl für die G1 als auch die G2 gRNAs, zusammen mit jener der G1 gRNA der HBD Gene bestimmt (40). Im folgenden Schritt werden die Off-Target Effekte der G1 und G2 gRNAs in menschlichen 293T Zellen durch die Verwendung des T7E1 Assays beurteilt. Die Unterschiede bezüglich dieser Raten sind erheblich. Während an den G1 gRNA Seiten nahezu keine Fehler erkennbar beziehungsweise nachweisbar sind, weisen die G2 gRNAs eine sehr niedrige Rate an Fehlmutationen in den Bereichen zwischen den Genen auf. 38 Abbildung 11 Diese Abbildung zeigt die Transfektion des G1gRNA-Cas9 Vektors in ansteigender Konzentration Expressionsvektor zur in die 293T Zellen. Wiederum wird ein GFP Kontrolle verwendet. Die Off-Target Effekte der sieben bevorzugten Regionen werden durch die T7E1 Assays beurteilt. HBB stellt das erfolgreiche Genomic Editing am HBB Genlocus dar (40). (Abbildung modifiziert nach Liang P, Xu Y, Zhang X, Ding C, Huang R, Zhang Z et al. CRISPR/Cas9-mediated gene editing in human tripronuclear zygotes. Protein & cell 2015 [cited 2015 Oct 3]; 6(5):363– 72.) Nach der Analyse dieser Daten wird die G1 gRNA zu weiteren Untersuchungen verwendet, da im Vergleich zur G2 gRNA die Nebenwirkungsrate reduziert ist. Im nächsten Schritt wird ssDNA Oligonukleotid Template synthetisiert, welches sechs stille Mutationen codiert. Diese ist auch dazu in der Lage, das Oligonukleotid alleine oder auch zusammen mit der G1 gRNA in die 293T Zellen zu transportieren (40). Nach 48 Stunden kann die DNA aus den Zellen für die PCR Amplifikation entnommen werden. Eine Analyse der unabhängigen Klone bestätigt die vorhergehende Annahme, dass eine vergleichsweise hohe Effizient beim Gene Editing am HBB Locus in Zellen erzielt werden kann. Von 29 untersuchten unabhängigen Klonen können bei vierzehn (48,3%) davon perfekt übereinstimmende Oligonukleotid Templates beobachtet werden (40). 39 Abbildung 12 48 Stunden nach der Transfektion kann die DNA aus den Zellen für die PCR Amplifikation entnommen werden. Daraufhin folgt die PCR Amplifikation. Die daraus entstandenen PCR Produkte werden für die Sequenzanalyse in Vektoren subkloniert (40). (Abbildung modfiziert nach Liang P, Xu Y, Zhang X, Ding C, Huang R, Zhang Z et al. CRISPR/Cas9-mediated gene editing in human tripronuclear zygotes. Protein & cell 2015 [cited 2015 Oct 3]; 6(5):363–72.) 3.2.2 Genomic Editing in menschlichen Tripronuklearen Zygoten durch das CRISPR/Cas9 Verfahren Um die Spezifität und Effektivität des Genomic Targetings in Tripronuklearen Zygoten zu untersuchen, werden G1 dRNA, Cas9 mRNA, GFP mRNA und ssDNA Oligonukleotide zusammen in das Zytoplasma dieser eingebracht. Um die Auswirkungen zu verdeutlichen, werden sie in verschiedenen Konzentrationen injiziert. Wie schon im Mausmodell beschrieben, sind nach 48 Stunden, basierend auf deren Morphologie, ungefähr 80% der Embryos lebensfähig (33). Daraufhin werden alle GFP-positiven Embryos gesammelt, um das gesamte Genom amplifizieren zu können. Dies geschieht durch eine Multidisplacement Amplifikation mit darauf folgender PCR Amplifikation der G1 gRNA Region (40). Von den 54 PCR-amplifizierten Embryos werden 28, das entspricht einer Effizienz von 52%, durch Cas9 gespalten. Von diesen 28 benutzen vier das ssDNA Template zur Reparatur. Es ist zu beobachten, dass in sieben Embryos HDR das 40 HBD Gen als Reparaturtemplate verwendet, da an gewissen Stellen Punktmutationen auftreten. Diese Beobachtung deutet auf eine Rekombination des HBB Gens mit HBD in sieben Embryos hin. Dies geschieht auch in Anwesenheit des exogenen ssDNA Templates. Ähnliches geschieht auch bei Mäusen, wo endogene homologe Templates gefunden werden, die mit den ssDNA Oligonukleotiden konkurrieren (33, 40). Auf Grund der Präferenz des fehlerbehafteten NHEJ Reparaturmechanismus, zeigt die HBB Sequenz am Sequenzchromatographen Spitzen. Dieses Phänomen ist jedoch nur in der Nähe der PAM Seite und nur bei Cas9 induzierter Spaltung zu beobachten (40). 3.2.3 Die negativen Aspekte dieses Verfahrens Um die Auswirkungen des Verfahrens genau voraussagen zu können und den klinischen Einsatz überhaupt zu ermöglichen, müssen alle Aspekte und Auswirkungen bekannt sein. Das beinhaltet somit auch alle negativen Mutationen und Nebenwirkungen, die während oder durch den Untersuchungsablauf auftreten können. Um diese zu determinieren werden wiederum die sieben bevorzugten Seiten von Off-Target Effekten und zusätzlich die der HBD Gene untersucht. Um die Fehler erkennen zu können wird das T7E1 Assay verwendet. Dieses zeigt auch sofort, dass Off-Target Spaltung im OPCML Intron sowie im TULP Intron stattfindet, obwohl scheinbar keines dieser Seiten in menschlichen 293T Zellen gespalten wird (40). Im folgenden Schritt werden sechs HBB gespaltene Embryos durch Zufall ausgewählt und zur Sequenzierung des gesamten Genoms vorbereitet. Indels sind hier in allen Beispielen vertreten. An zwei weiteren Seiten sind fehlerhafte Exons zu finden. An genau diesen wurde jedoch eine geringere Konzentration von Cas9 mRNA und gRNA verwendet und sie liegen in den Exons der C1QC und Transtyretin Gene. 41 Diese Ergebnisse beweisen die möglichen negativen Auswirkungen, die durch das Genomic Targeting in menschliche 3PN Embryos entstehen können. Auch die geringsten Fehlmutationen können erhebliche Folgen für die Entwicklung des späteren Säuglings haben. Jegliche Art von Off-Target Effekten muss vor der klinischen Reife behoben werden (40). Abbildung 13 Diese Abbildung zeigt die Off-Target Spaltung in menschlichen Tripronuklearen Zygoten. Die PAM Sequenz wird in Grün dargestellt. HBB symbolisiert die regelrechte Spaltung am HBB Locus. G1-OT1 bis G1-OT2 entsprechen den sieben bevorzugten Stellen für Fehlmutationen. HBD, hier in der unteren Zeile abgebildet, demonstriert die bevorzugte Seite für Off-Target Effekte (40). (Abbildung modifiziert nach Liang P, Xu Y, Zhang X, Ding C, Huang R, Zhang Z et al. CRISPR/Cas9-mediated gene editing in human tripronuclear zygotes. Protein & cell 2015 [cited 2015 Oct 3]; 6(5):363– 72) 42 3.2.4 Die Reparatur der Doppelstrangbrüche Wie im bisherigen Text schon umfassend geschildert, können durch CRISPR/Cas9 induzierte Doppelstrangbrüche auf zwei unterschiedliche Wege repariert werden. Einerseits existieren fehlerhafte Reparaturmechanismen wie das NHEJ, anderseits gibt es die deutlich effizientere Methode der HDR. Die drei unterschiedlichen Wege des letzteren Mechanismus ist das SDSA (synthesisdependent strand annealing), die DSBR (non-crossover double-strand break repair) und die Crossover DSBR (40, 42). Abbildung 14 Die Abbildung zeigt zwei unterschiedliche Reparaturmechanismen von Doppelstrangbrüchen in menschlichen Zellen. Links wird der DSBR (non-crossover double-strand break repair) Mechanismus dargestellt, rechts das SDSA (synthesisdependent strand annealing). DSBR kann sowohl Crossover als auch Non-Crossover auftreten (42). (Abbildung modifiziert nach Bugreev DV, Pezza RJ, Mazina OM, Voloshin ON, Camerini-Otero RD, Mazin AV. The resistance of DMC1 D-loops to dissociation may account for the DMC1 requirement in meiosis. Nat Struct Mol Biol 2011; 18(1):56–60.) 43 Bidirektionaler Sequenzaustausch zwischen den rekombinierten Genen wird mittels Crossover repariert, während unidirektionaler Sequenzaustausch ohne Cossover erfolgt. Von den 3PN Embryos wird der Großteil mit Hilfe des endogenen HBD Gens rekombiniert. Einige können durch Verwendung des ssDNA Oligonukleotid Templates repariert werden. Zusammenfassend kann man sagen, dass Homologe Rekombination in der frühen menschlichen Entwicklung vorzugsweise durch den Non-Crossover HDR Mechanismus erfolgt (40). 3.3 Der Einsatz von Gentechnikverfahren in der Pflanzenzucht Einsatzmöglichkeiten der bisher beschriebenen Verfahren haben in den letzten Jahren auch zunehmend in anderen Bereichen Anklang gefunden. Beispielsweise profitiert die Landwirtschaft aus den neuen, immer effizienteren und leichter handhabbaren Methoden, um in die Genetik der Pflanzen eingreifen zu können und sie somit widerstandsfähiger gegen Schädlinge zu machen oder deutlich schneller wachsen zu lassen. Doch welche Verfahren sind für diese genetischen Veränderungen am besten geeignet? Grundsätzlich kann man sagen, dass jeder Effekt, der durch das neu entwickelte CRISPR/Cas9 System bewirkt wird, auch durch die ZFNs oder TALENs erreicht werden kann. Die Vorteile dieses Verfahrens liegen jedoch in der Einfachheit, der Verfügbarkeit und Kosten. Somit wird die genetische Veränderung auch in anderen Gebieten außerhalb der Medizin immer attraktiver (43). Die Nutzungsmöglichkeiten von Pflanzen sind enorm. Sie dienen der Herstellung von verschiedensten Materialien, Treibstoff, Chemikalien und letztendlich der Nahrung des Menschen. Deren Zucht ist jedoch mit einigen Hindernissen und Problemen verbunden. Die konventionelle Pflanzenzucht beruht auf dem Vorhandensein natürlicher genetischer Variationen sowie teuren Verfahren, die nötig sind um diese zu verändern. Die Vorteile des CRISPR/Cas9 Verfahrens liegt in der Möglichkeit multiple Eigenschaften simultan gentechnisch zu verändern, beziehungsweise zu verbessern. 44 NHEJ induzierte Genveränderung ist die einfachste Form des Genomic Editings. Es wird beispielsweise dazu verwendet Gene, die einen negativen Effekt in der Qualität der Nahrung hervorrufen oder leicht durch Pathogene angreifbar sind, zu eliminieren. Dies geschieht durch Modifikation der Allele, die für die Krankheit der Pflanze verantwortlich sind. Ein Beispiel für diese Art der genetischen Manipulation ist das Knockout aller drei Allele, die für die Ausprägung des Schimmelpilzbefalls einer Pflanze verantwortlich sind (43). Die Insertion von großen Sequenzen durch NHEJ oder HDR erlaubt auch das Einbringen von Transgenen an bestimmten Orten, die daraufhin eine „high-level“ Transkription durchführen und nicht mit der Aktivität der endogenen Gene interferieren. 3.4 Die Nutzung tierischer Organe zur Transplantation in Menschen Ein weiteres Anwendungsgebiet des CRISPR/Cas9 Verfahrens ist die Möglichkeit der Verwendung von tierischen Organen in der Transplantationschirurgie. Schon seit Jahrzehnten träumen Forscherinnen/Forscher und Ärztinnen/Ärzte von der Möglichkeit, Organe in Schweinen wachsen zu lassen und sie daraufhin Menschen zu transplantieren. In einer 2015 veröffentlichten Studie wird die Verwendung des CRISPR/Cas9 Verfahrens in Schweinezellen zur Zerstörung von DNA Sequenzen im Genom erstmals beschrieben (44). Bisher scheiterte die Verwendung tierischer Organe durchwegs an der enorm starken Immunantwort der Schweinezellen. Deren DNA ist gespickt mit einer großen Anzahl an Kopien von DNA-Sequenzen, die Reste von Viren beinhalten, die noch immer in der Lage sind bedrohliche Viruspartikel zu produzieren (44, 45). Diese Zellen werden PERV (porcine endogenous retrovirus) genannt. Genau diese Sequenzen sind die Ansatzpunkte des Experiments mit dem CRISPR/Cas9 Verfahren (44). 45 Wie bereits im einleitenden Teil der Arbeit beschrieben ist die Basis der Methode der Verteidigungsmechanismus von Bakterien gegen die virale DNA. In dieser Studie zielt eine speziell designte gRNA auf die Veränderung von 62 Genen in Schweinembryos. In diesen ist es durch das Verfahren möglich die PERVs in den Nierenzellen der Schweine zu inaktivieren (45). Eine besondere Schwierigkeit stellt das Überleben der Zellen dar, obwohl ihre DNA an 62 Stellen verändert wurde. Es wird vermutet, dass das Experiment durch die Hilfe eines Phänomens, genannt „Gene Conversion“, funktioniert, in welchem bereits inaktivierte DNASeiten helfen, dass andere Seiten nicht wieder korrekt durch die Zelle repariert werden können. Diese Methode kann zukünftig enorme Auswirkungen auf die Beschaffung und die Verfügbarkeit von Organen haben. Allein in den Vereinigten Staaten von Amerika warten rund 120.000 Menschen derzeit auf ein lebensrettendes Organ. Und die Zahl der benötigten Transplantate steigt Jahr für Jahr weiter an. Auch die Übertragung von Pathogenen im Rahmen der Transplantation kann durch die Anwendung von CRISPR/Cas9 verhindert werden (46). 4 Diskussion Im Verlauf dieser Arbeit wurden nur einige der unzähligen Anwendungsgebiete des Genomic Editings beschrieben. Die hier erwähnten Artikel, Studien oder sonstige Publikationen zeigen nur die aktuellsten und wissenschaftlich relevantesten Möglichkeiten, die das jeweilige Verfahren mit sich bringt. 2014 wurden allein über die CRISPR/Cas9-Methode hunderte Publikationen in den namhaften Online-Datenbanken für wissenschaftliche Literatur, in diversen Journalen und in Fachzeitschriften veröffentlicht. Unzählige Patente wurden ausgestellt und der finanzielle Aufwand zur Weiterentwicklung dieser Verfahren ist weiterhin immens. Forscherinnen und Forscher die jahrelang Methoden, wie das 46 TALEN-Verfahren, das Zinkfingernuklease-Verfahren sowie das MeganukleaseVerfahren, bearbeitet und perfektioniert haben, wurden durch die deutlich billigere, effizientere und einfachere CRISPR/Cas9-Methode in den Hintergrund gedrängt. Verfahren, die früher nur mit enormen, finanziellen und zeitlichen Aufwand möglich waren, können nun durch kleinste Labore und mit geringster wissenschaftlicher Kenntnis durchgeführt werden. Die jahrzehntelange Ausbildung und der Aufwand zur Weiterentwicklung herkömmlicher Verfahren wurden auf einen Schlag, durch den Einsatz der CRISPR/Cas9-Methode, überholt. Labore mussten ihre langwierige Arbeit beenden, da sie ohne den Einsatz von CRISPR nicht mehr konkurrenzfähig waren. Kurz zusammengefasst wäre heutzutage jede Studentin und jeder Student dazu in der Lage das CRISPR/Cas9-Verfahren mit geringstem finanziellen Aufwand und wenig Erfahrung in der wissenschaftlichen Arbeit, mit allen Risiken und Nebenwirkungen, die die Manipulation am Erbgut mit sich führt, durchzuführen. Die auch in dieser Arbeit erläuterte Grundlagenforschung an Tiermodellen ist für die Weiterentwicklung von Genomic Editing Systemen von großer Bedeutung. Das Mausmodell dient beispielsweise dem Verständnis der Entwicklung von Krankheit und soll zukünftig dazu dienen, Krankheiten bereits vor ihrem Ausbruch auf genetischer Ebene zu bekämpfen. Doch muss man auch verdeutlichen, dass jeglicher Eingriff in das Erbmaterial mit erheblichen Risiken und unvorhergesehen Nebenwirkungen verbunden ist. Hierbei stellt sich die Frage, ob der Einsatz dieser Technik bei der immensen Rate an Off-target Mutationen überhaupt vertretbar ist. Durch den raschen Fortschritt dieser Technik ist das Wissen über die möglichen negativen Konsequenzen solcher Verfahren begrenzt. Die kleinsten Fehler bei der Durchführung können ungeahnte Auswirkungen auf die Organismen und deren Nachkommen haben. Durch den Versuch, Krankheiten genetisch zu manipulieren, können auf der Ebene des Erbguts wiederum neue Krankheiten entstehen (33). Noch beunruhigender ist die Vorstellung von unvorhergesehenen Off-target Mutationen, durch das Genomic Editing, in menschlichen 3PN Embryos. Bereits kleinste Fehler in der Durchführung und somit geringste Fehlmutationen können erhebliche Folgen für die Entwicklung des späteren Lebewesens und zukünftiger Generationen mit sich führen. Viele Expertinnen und Experten halten diesen Umstand für gefährlich und ethisch inakzeptabel (47). In der aktuell frühen Phase 47 der Entwicklung dieser Technik sollte ein Eingriff in das menschliche Genom nicht durchgeführt werden. Es ist nicht vorhersehbar, inwiefern sich das Verfahren auf andere Zellen auswirkt. Wie viele Zellen werden modifiziert? Wie kann man die Mutationen kontrollieren? Welche Argumente sprechen gegen den Einsatz von Maßnahmen zur genetischen Manipulation von menschlichen Embryos? All diese Fragen sollten sowohl medizinisch, ethisch als auch gesetzlich geklärt werden, bevor nahezu jede Forscherin und jeder Forscher diese gefährlichen Verfahren anwenden kann (40, 47). Obwohl das Genomic Targeting ein enorm starkes Verfahren mit nahezu grenzenlos wissenschaftlichen und medizinischen Potenzial ist, muss überlegt werden, ob die Grenze des Akzeptierbaren nicht bereits überschritten ist. Auch der soziale und ethische Standpunkt muss hierbei betrachtet werden. Ist es ethisch vertretbar das menschliche Erbgut gezielt zu manipulieren und zu verändern? Die gesetzliche Regelung in Bezug auf den Einsatz des Genomic Editings an menschlichen Embryos und die menschliche Stammzellenforschung ist größtenteils noch unklar (48). Immer mehr Studien beschäftigen sich hierbei mit der Frage über die Legalisierung von Verfahren wie dem Genomic Editing, dem reproduktivem Klonen, der Stammzellenforschung, der Gentherapie und der Präimplantationsdiagnostik. Die Forscherinnen und Forscher, die eine Manipulation an menschlichen 3PN Embryos durchführten, argumentieren auf Vorwürfe, ethisch und moralisch nicht einwandfrei gehandelt zu haben, mit der Rechtfertigung, dass die Embryos ihrer Versuchsreihe schon im Vorhinein nicht lebensfähig waren. Doch hierbei stellt sich die Frage, wo die Grenze des Vertretbaren zu ziehen ist. Im Falle der Legalisierung dieser Verfahren, wenn ein einwandfreier therapeutischer Nutzen für das spätere Leben des Embryos nachgewiesen werden kann, stellt sich wiederum die Frage nach den schon mehrmals genannten Nebenwirkungen. Diese sind bei jedem Verfahren möglich. Solange sie nicht vollständig auszuschließen sind, müssen die Verfahren vor dem Einsatz am Menschen weiterentwickelt und verbessert werden. Die heutige Gesetzeslage hat auf Grund verschiedener Definitionen noch Grauzonen bezüglich der Durchführbarkeit diverser Untersuchungen und Verfahren. Solange diese Ungereimtheiten nicht beseitigt werden, kann hierbei auch keine klare Regelung getroffen werden (48). 48 Die Abwägung von Nutzen und Risiken spielt in dieser Debatte eine bedeutende Rolle. Es gilt zu entscheiden, ob der Effekt einer Behandlung oder eines Verfahrens gegenüber möglicher negativer Auswirkungen überwiegt. Erst durch langwierige Tests und Versuche, sowie Erfahrung, kann daraufhin auf gesetzlicher Ebene entschieden werden. Das Ziel muss sein, die Rate an Off-target Mutationen zu senken und die Effizienz der Verfahren deutlich zu erhöhen um somit zu ermöglichen, Genomic Editing als anerkanntes Therapieverfahren gegen diverse Krankheiten gewinnen zu können (48). 49 1 Literaturverzeichnis 1. Carroll D. Genome engineering with zinc-finger nucleases. Genetics 2011; 188(4):773–82. 2. Wood AJ, Lo T, Zeitler B, Pickle CS, Ralston EJ, Lee AH et al. Targeted genome editing across species using ZFNs and TALENs. Science (New York, N.Y.) 2011; 333(6040):307. 3. Scherer S, Davis RW. Replacement of chromosome segments with altered DNA sequences constructed in vitro. Proceedings of the National Academy of Sciences of the United States of America 1979; 76(10):4951–5. 4. Bednarski C, Cathomen T. Maßgeschneidertes Genom — Designer-Nukleasen im Einsatz. Biospektrum 2015; 21(1):22–4. 5. Le Cong, Ran FA, Cox D, Lin S, Barretto R, Habib N et al. Multiplex genome engineering using CRISPR/Cas systems. Science (New York, N.Y.) 2013; 339(6121):819–23. 6. Esvelt KM, Wang HH. Genome-scale engineering for systems and synthetic biology. Molecular systems biology 2013; 9:641. 7. Cho SW, Kim S, Kim Y, Kweon J, Kim HS, Bae S et al. Analysis of off-target effects of CRISPR/Casderived RNA-guided endonucleases and nickases. Genome research 2014; 24(1):132–41. 8. Sistla S, Rao DN, Bitinaite J, Wah DA, Aggarwal AK, Schildkraut I. S-Adenosyl-L-methioninedependent restriction enzymes // FokI dimerization is required for DNA cleavage. Critical reviews in biochemistry and molecular biology 2004 // 1998; 39 // 95(1 // 18):1–19. 9. Jensen B, Unger KK, Uebe J, Gey M, Kim YM, Flecker P. Proteolytic cleavage of soybean Bowman-Birk inhibitor monitored by means of high-performance capillary electrophoresis. Implications for the mechanism of proteinase inhibitors. Journal of biochemical and biophysical methods 1996; 33(3):171–85. 10. Sugisaki H, Kanazawa S. New restriction endonucleases from Flavobacterium okeanokoites (FokI) and Micrococcus luteus (MluI). Gene 1981; 16(1-3):73–8. 11. Santiago Y, Chan E, Liu P, Orlando S, Zhang L, Urnov FD et al. Targeted gene knockout in mammalian cells by using engineered zinc-finger nucleases. Proceedings of the National Academy of Sciences of the United States of America 2008; 105(15):5809–14. 12. Bitinaite J, Wah DA, Aggarwal AK, Schildkraut I. FokI dimerization is required for DNA cleavage. Proceedings of the National Academy of Sciences of the United States of America 1998; 95(18):10570–5. 13. Costanzo V, Robertson K, Bibikova M, Kim E, Grieco D, Gottesman M et al. Mre11 protein complex prevents double-strand break accumulation during chromosomal DNA replication. Molecular cell 2001; 8(1):137–47. 14. Song SU, Gerasimova T, Kurkulos M, Boeke JD, Corces VG. An env-like protein encoded by a Drosophila retroelement: evidence that gypsy is an infectious retrovirus. Genes & Development 1994; 8(17):2046–57. 50 15. Cox MM, Battista JR. Deinococcus radiodurans - the consummate survivor. Nature reviews. Microbiology 2005; 3(11):882–92. 16. Carroll D, Morton JJ, Beumer KJ, Segal DJ. Design, construction and in vitro testing of zinc finger nucleases. Nature protocols 2006; 1(3):1329–41. 17. Maeder ML, Thibodeau-Beganny S, Osiak A, Wright DA, Anthony RM, Eichtinger M et al. Rapid "open-source" engineering of customized zinc-finger nucleases for highly efficient gene modification. Molecular cell 2008; 31(2):294–301. 18. Geurts AM, Cost GJ, Freyvert Y, Zeitler B, Miller JC, Choi VM et al. Knockout rats via embryo microinjection of zinc-finger nucleases. Science (New York, N.Y.) 2009; 325(5939):433. 19. Joung JK, Sander JD. TALENs: a widely applicable technology for targeted genome editing. Nature reviews. Molecular cell biology 2013; 14(1):49–55. 20. Stoddard BL. Homing endonucleases: from microbial genetic invaders to reagents for targeted DNA modification. Structure (London, England : 1993) 2011; 19(1):7–15. 21. Belfort M, Perlman PS. Mechanisms of intron mobility. The Journal of biological chemistry 1995; 270(51):30237–40. 22. Hellinga HW, Smith JJ, Jantz D. Rationally-designed meganucleases with altered sequence specificity and DNA-binding affinity: Google Patents; 2012. Available from: URL:http://www.google.com/patents/EP2357226B1?cl=en. 23. Zhang M, Wang F, Li S, Wang Y, Bai Y, Xu X. TALE: a tale of genome editing. Progress in biophysics and molecular biology 2014; 114(1):25–32. 24. Cornelis GR, van Gijsegem F. Assembly and function of type III secretory systems. Annual review of microbiology 2000; 54:735–74. 25. Cho SW, Kim S, Kim JM, Kim J. Targeted genome engineering in human cells with the Cas9 RNA-guided endonuclease. Nature biotechnology 2013; 31(3):230–2. 26. Carlson DF, Tan W, Lillico SG, Stverakova D, Proudfoot C, Christian M et al. Efficient TALENmediated gene knockout in livestock. Proceedings of the National Academy of Sciences of the United States of America 2012; 109(43):17382–7. 27. Rath D, Amlinger L, Rath A, Lundgren M. The CRISPR-Cas immune system: Biology, mechanisms and applications. Biochimie 2015; 117:119–28. 28. Horvath P, Barrangou R. CRISPR/Cas, the immune system of bacteria and archaea. Science (New York, N.Y.) 2010; 327(5962):167–70. 29. Barrangou R, Fremaux C, Deveau H, Richards M, Boyaval P, Moineau S et al. CRISPR provides acquired resistance against viruses in prokaryotes. Science (New York, N.Y.) 2007; 315(5819):1709–12. 30. Ishino Y, Shinagawa H, Makino K, Amemura M, Nakata A. Nucleotide sequence of the iap gene, responsible for alkaline phosphatase isozyme conversion in Escherichia coli, and identification of the gene product. Journal of bacteriology 1987; 169(12):5429–33. 31. Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE et al. RNA-guided human genome engineering via Cas9. Science (New York, N.Y.) 2013; 339(6121):823–6. 51 32. Hwang WY, Fu Y, Reyon D, Maeder ML, Tsai SQ, Sander JD et al. Efficient genome editing in zebrafish using a CRISPR-Cas system. Nat Biotech 2013; 31(3):227–9. 33. Wang H, Yang H, Shivalila CS, Dawlaty MM, Cheng AW, Zhang F et al. One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell 2013; 153(4):910–8. 34. Yin H, Xue W, Chen S, Bogorad RL, Benedetti E, Grompe M et al. Genome editing with Cas9 in adult mice corrects a disease mutation and phenotype. Nat Biotech 2014; 32(6):551–3. 35. Xu P, Tong Y, Liu X, Wang T, Cheng L, Wang B et al. Both TALENs and CRISPR/Cas9 directly target the HBB IVS2-654 (C T) mutation in β-thalassemia-derived iPSCs. Scientific reports 2015; 5:12065. 36. Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. Genome engineering using the CRISPR-Cas9 system. Nature protocols 2013; 8(11):2281–308. 37. Gu T, Guo F, Yang H, Wu H, Xu G, Liu W et al. The role of Tet3 DNA dioxygenase in epigenetic reprogramming by oocytes. Nature 2011; 477(7366):606–10. 38. Pingoud A. Structure and function of type II restriction endonucleases. Nucleic acids research 2001; 29(18):3705–27. 39. Cyranoski D, Reardon S. Chinese scientists genetically modify human embryos. Nature 2015. 40. Liang P, Xu Y, Zhang X, Ding C, Huang R, Zhang Z et al. CRISPR/Cas9-mediated gene editing in human tripronuclear zygotes. Protein & cell 2015 [cited 2015 Oct 3]; 6(5):363–72. 41. Schechter AN. Hemoglobin research and the origins of molecular medicine. Blood 2008; 112(10):3927–38. 42. Bugreev DV, Pezza RJ, Mazina OM, Voloshin ON, Camerini-Otero RD, Mazin AV. The resistance of DMC1 D-loops to dissociation may account for the DMC1 requirement in meiosis. Nat Struct Mol Biol 2011; 18(1):56–60. 43. Bortesi L, Fischer R. The CRISPR/Cas9 system for plant genome editing and beyond. Biotechnology advances 2015; 33(1):41–52. 44. Servick K. Gene-editing method revives hopes for transplanting pig organs into people. Science 2015. 45. Reardon S. Gene-editing record smashed in pigs. Nature 2015. 46. Yang L, Güell M, Niu D, George H, Lesha E, Grishin D et al. Genome-wide inactivation of porcine endogenous retroviruses (PERVs). Science (New York, N.Y.) 2015; 350(6264):1101–4. 47. Lanphier E, Urnov F, Haecker SE, Werner M, Smolenski J. Don't edit the human germ line; 2015. 48. Isasi R, Kleiderman E, Knoppers BM. GENETIC TECHNOLOGY REGULATION. Editing policy to fit the genome? Science (New York, N.Y.) 2016; 351(6271):337–9. 52 2 Abbildungsverzeichnis Abbildung 1 Cox MM, Battista JR. Deinococcus radiodurans - the consummate survivor. Nature reviews. Microbiology 2005; 3(11):882–92. Abbildung 2 Zhang M, Wang F, Li S, Wang Y, Bai Y, Xu X. TALE: a tale of genome editing. Progress in biophysics and molecular biology 2014; 114(1):25–32. Abbildung 3 Rath D, Amlinger L, Rath A, Lundgren M. The CRISPR-Cas immune system: Biology, mechanisms and applications. Biochimie 2015; 117:119–28. Abbildung 4 Rath D, Amlinger L, Rath A, Lundgren M. The CRISPR-Cas immune system: Biology, mechanisms and applications. Biochimie 2015; 117:119–28. Abbildung 5 Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE et al. RNA-guided human genome engineering via Cas9. Science (New York, N.Y.) 2013; 339(6121):823–6 Abbildung 6 Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. Genome engineering using the CRISPR-Cas9 system. Nature protocols 2013; 8(11):2281–308 Abbildung 7 Wang H, Yang H, Shivalila CS, Dawlaty MM, Cheng AW, Zhang F et al. One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell 2013; 153(4):910–8. Abbildung 8 http://de.dexeus.com/reproduktionsmedizin/in-vitro-fertilisation/beurteilung-derbefruchtung (28.12.2015) Abbildung 9 Liang P, Xu Y, Zhang X, Ding C, Huang R, Zhang Z et al. CRISPR/Cas9-mediated gene editing in human tripronuclear zygotes. Protein & cell 2015 [cited 2015 Oct 3]; 6(5):363–72. Abbildung 10 Liang P, Xu Y, Zhang X, Ding C, Huang R, Zhang Z et al. CRISPR/Cas9-mediated gene editing in human tripronuclear zygotes. Protein & cell 2015 [cited 2015 Oct 3]; 6(5):363–72. Abbildung 11 Liang P, Xu Y, Zhang X, Ding C, Huang R, Zhang Z et al. CRISPR/Cas9-mediated gene editing in human tripronuclear zygotes. Protein & cell 2015 [cited 2015 Oct 3]; 6(5):363–72. Abbildung 12 Liang P, Xu Y, Zhang X, Ding C, Huang R, Zhang Z et al. CRISPR/Cas9-mediated gene editing in human tripronuclear zygotes. Protein & cell 2015 [cited 2015 Oct 3]; 6(5):363–72. 53 Abbildung 13 Liang P, Xu Y, Zhang X, Ding C, Huang R, Zhang Z et al. CRISPR/Cas9-mediated gene editing in human tripronuclear zygotes. Protein & cell 2015 [cited 2015 Oct 3]; 6(5):363–72 Abbildung 14 Bugreev DV, Pezza RJ, Mazina OM, Voloshin ON, Camerini-Otero RD, Mazin AV. The resistance of DMC1 D-loops to dissociation may account for the DMC1 requirement in meiosis. Nat Struct Mol Biol 2011; 18(1):56–60. 54