Hereditäre Bindegewebserkrankungen - Derma-Net
Werbung

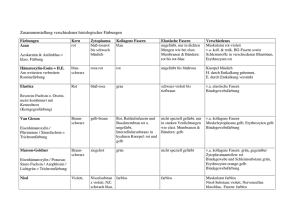
7. Hereditäre Erkrankungen 1 7.2 Hereditäre Bindegewebserkrankungen von <Ingrid Haußer> Das Bindegewebe oder die extrazelluläre Matrix ist das Füll- und Stützgewebe des Körpers. Es erfüllt vielfältige Aufgaben, z.B. als Stützgewebe für die Gefäße, als Füllgewebe zwischen und um Organe oder als Bestandteil des Bewegungsapparats in Form von Sehnen und Bändern. In der Haut gehören Dermis und Subkutis zum Bindegewebe. Hauptbestandteile der extrazellulären Matrix sind: • Ein Grundgerüst aus Kollagenfasern, die dem Gewebe Zugfestigkeit verleihen. • Elastische Fasern, die die Verformbarkeit und Elastizität des Gewebes ermöglichen. • Eine Grundsubstanz von gelartiger Konsistanz aus Glykosaminoglykanen, Proteoglykanen und anderen nichtkollagenen Glykoproteinen, die beide Faserarten einbettet. Kollagene umfassen eine große Familie verwandter Strukturproteine; elastische Fasern bestehen aus Elastin und einer komplexen Mischung assoziierter mikrofibrillärer Proteine. Insgesamt besteht Bindegewebe aus mehr als 200 verschiedenen chemischen Molekülarten, deren Zusammensetzung in Bindegeweben unterschiedlicher Lokalisation verschieden ist (Royce 2002). Beeinflusst wird die extrazelluläre Matrix von der strukturellen Umgebung, von altersbedingten Veränderungen, mechanischem Stress, Umwelteinflüssen und genetischen Einflüssen. Unter diesen Einflüssen stehen damit naturbedingt viele Organsysteme wie Skelett, Intestinaltrakt, Augen, kardiovaskuläres System, Lunge, Haut und zentrales Nervensystem. Im Folgenden werden die wichtigsten erblichen Bindegewebserkrankungen vorgestellt, die auch die Haut betreffen. Eine besondere Schwierigkeit stellen bei diesen seltenen Konditionen recht häufige und deutliche klinische Überlappungen dar. Zunehmend werden in der letzten Zeit Kandidatengene für definierte Krankheitsbilder identifiziert, wodurch in spezialisierten Einrichtungen die diagnostische Einordnung zwar möglich, aber auch immer komplizierter wird. Aus der Sicht der Patienten ist, mit Blick auf Diagnostik und Therapie, eine gute interdisziplinäre Zusammenarbeit (z.B. Pädiater, Orthopäden, Genetiker, Dermatologen) von grundlegender Bedeutung. Für eine Reihe von erblichen Bindegewebserkrankungen wurden ultrastukturelle Veränderungen an Komponenten des dermalen Bindegewebes als wichtige diagnostische und pathogenetische Wegweiser etabliert. Dadurch wurde z.B. die Suche nach eventuell zu sequenzierenden Kandidatengenen möglich oder vereinfacht. Gute aktuelle Informationsmöglichkeiten zum Stand der Pathogeneseforschung, aber auch zu den diagnostischen und therapeutischen Möglichkeiten und zu internationalen Ansprechpartnern finden sich im Internet: Allgemein: www.orphanet.de; www.geneskin.idi.it Patienten-Selbsthilfegruppen: http://www.ehlers-danlos-initiative.de/; http://www.rheuma-liga.de; www.pxe-groenblad.de; http://www.pxe-network.de. 2 7.2 Hereditäre Bindegewebserkrankungen 7.2.1 Ehlers-Danlos-Syndrom (Synonym: Cutis hyperelastica) Das Ehlers-Danlos-Syndrom (EDS) umfasst eine Gruppe von erblichen Erkrankungen, die in unterschiedlichem Ausmaß eine Hyperelastizität der Haut, aberrante Narbenbildung, Hypermobilität der Gelenke und Fragilität von Gefäßen und Geweben aufweisen (Abb. 1-5). Sie beruhen auf genetisch bedingten Veränderungen von Kollagenfibrillen bzw. der Kollagensynthese (Steinmann 2002). Bisher wurden Mutationen in Strukturgenen für Kollagenketten identifiziert, aber auch Mutationen in Genen von Enzymen, die für die Synthese (Colige 2004), den Zusammenbau oder für nicht-fibrilläre Bestandteile der Grundsubstanz verantwortlich sind. Diese Komponenten sind notwendig für die Funktion des Gesamtgefüges der extrazellulären Matrix und damit mutmaßlich indirekt an der Synthese bzw. am Arrangement von Kollagenfibrillen beteiligt. (Byers 2002, Uitto 2003). Entsprechend der Nosology of Villefranche (Beighton 1998) unterscheidet man sechs EDS-Haupttypen (Tab. 1) sowie weitere, sehr seltene Nebentypen, die teilweise auf Einzelbeschreibungen beruhen (Typ V, Typ VIII). Letztere sind nur unzureichend charakterisiert; das Typ IX/Menkes-Syndrom/occipital horn syndrome wurde aus der EDS-Gruppe herausgenommen (siehe 7.2.2). Abb. 1: Ehlers-Danlos-Syndrom, Hypermobilität der kleinen Gelenke. Abb. 2: Ehlers-Danlos-Syndrom, Hyperleastizität der Haut. Die Kombination aus sorgfältiger klinischer Untersuchung und elektronenmikroskopischer Analyse des dermalen Bindegewebes erlaubt in vielen Fällen eine diagnostische Zuordnung. Damit ist dann auch der Weg geebnet für eine weitergehende Gensequenzierung und die spezifische genetische Beratung (Proske 2006). 7. Hereditäre Erkrankungen 3 Abb. 4: Ehlers-Danlos-Syndrom Klassischer Typ, Wundheilungsstörungen; aberrante Narbenbildung an der Stirn. Abb. 3: Ehlers-Danlos-Syndrom klassischer Typ, Wundheilungsstörungen; narbige und indurierte Bereiche an den Schienbeinen (besonders auffällig bei Kleinkindern) Pat. 4 J. Abb. 5: Ehlers-Danlos-Syndrom, vaskukärer Typ: Durchscheinende Haut mit deutlicher Gefäßzeichnung. Bisher werden leider noch nicht alle Patienten systematisch und vollständig durchuntersucht, was Vergleichsstudien und therapeutische Ansätze erschwert. Klinische Überschneidungen bestehen mit anderen, noch wenig definierten Hypermobilitätssyndromen, insbesondere zwischen dem hypermobilem EDS und dem familiären Hypermobilitätssyndrom. Gelegentlich werden Fälle provisorisch dem EDS zugeordnet, auch wenn es an einem Kandidatengen fehlt, aber regelmäßige ultrastrukturelle Veränderungen am Kollagen im dermalen Bindegewebe zu verifizieren sind. Ein Großteil von Patienten mit spontanen zervikozerebralen Dissektio- 4 7.2 Hereditäre Bindegewebserkrankungen nen, aber ohne weitere Symptomatik eines EDS oder einer anderen erblichen Bindgewebeerkrankung, weisen ebenfalls ultrastrukturelle, EDS-ähnliche Veränderungen am Kollagen auf. Nur zu einem geringen Prozentsatz liegen jedoch Mutationen in EDS-verursachenden Genen vor (Hausser 2004). Zunehmend wurden weitere Entitäten definiert, die Überlappungen von Syndromen darstellen: Osteogenesis imperfecta + EDS (Makareeva 2006) wird von Kollagen-1 Mutationen verursacht, die mit dem N-Propetid-Processing interferieren. Das LoeysDietz-Syndrom II wird definiert durch TGFBR-Mutationen und Symptome eines vaskulären EDS (Loeys 2006). ). Tabelle 1: Klassifikation des Ehlers-Danlos-Syndroms: Haupttypen EDS-Typ Klinische (frühere BeSymptome zeichnung) Klassisch Überdehnbare Haut (I, II) Hypermobile Gelenke Aberrante Narbenbildung Fragilität von Geweben und Gefäßen Hypermobil (III) Vaskulär (IV) Kyphoskoliose (okulärer Typ, VI) Arthrochalasis (VIIA, B) Dermatosparaxis (VIIC) Hypermobil Erbgang Verantwortliches Gen/ Protein Ultrastrukturelle Merkmale AD* COL5A1, COL5A2/a1und a2-Kette von Kollagen V (ca. 40% der untersuchten Patienten) ? Regemäßig zahlreiche veränderte Kollageneinzelfibrillen in der gesamten Dermis mit unregelmäßig ausgefransten Umrissen der Querschnitte (Abb. 6,7) Hypermobile Gelenke Dislokationen, Gelenkschmerzen und frühzeitige Arthritis Samtartige Haut Dünne durchscheinende Haut Überstreckbare kleine Gelenke in der Kindheit, später eher steife Gelenke und straffe Haut Ausgeprägte Gefäß- und Gewebefragilität, bes. Arterien, Darm, Uterus Hypotonie, hypermobile Gelenke, kongenitale Skoliose, Augenbeteiligung AD Ausgeprägte Hypermobilität der Gelenke, kongenitale Dislokationen und Luxationen, milde Hautbeteiligung, Skoliose, Verletzlichkeit Ausgeprägte Verletzlichkeit und Überdehnbarkeit der Haut und von Geweben, Hernien, blaue Skleren Hypermobile Gelenke Milde Hautverletzlichkeit Regelmäßig einzelne oder mehrere veränderte Kollagenfibrillen in Bündeln der retikulären Dermis AD COL3A1/a1Kette von Kollagen III AR* PLOD/Lysylh Lockere Dermis mit ydroxylase wenigen veränderten Kollagenfibrillen AD COL1A1, COL1A2/a1und a2Ketten von Kollagen I ADAMTS-2/ ProkollagenN-Peptidase Meist regelmäßiges Auftreten von großkalibrigen, aberranten Kollageneinzelfibrillen TNX/ Tenascin X Geringfügig verminderte Dichte der Kollagenfibrillen im Bündel AR AR (*AD Autosomal-dominant, AR Autosomal-rezessiv) Sehr kurze, locker aufgebaute Dermis, Rarefizierung von Kollagen/Vermehrung von elastischen Fasern, kleinkalibrige, variable Kollagenfibrillen Spezifische hieroglyphenartige Kollagenfibrillen (Abb. 8) 7. Hereditäre Erkrankungen 5 Manche Patienten haben einen marfanoiden Habitus, das Marfan-Syndrom an sich ist jedoch eindeutig durch Mutationen im Fibrillin-Gen charakterisiert (Kielty 2006). Im Vordergrund der Patientenversorgung stehen die individuelle Aufklärung über das Krankheitsbild und die diagnostische Abklärung eventuell betroffener Familienmitglieder. Die vielfältigen Symptome sind zwar meist nicht heilbar, aber behandelbar durch Schmerztherapie, Muskelaufbautraining (Entlastung des bindegewebigen Halteapparats, Ausgleich der Dysfunktion) und andere sportliche Aktivitäten wie Schwimmen und isometrisches Kraftraining. Zur Früherkennung lebensbedrohlicher Aneurysmen werden regelmäßige Ultraschalluntersuchungen der großen Gefäße empfohlen. Abhängig vom Typ der Erkrankung sind auch ophthalmologische Untersuchung und eine orthopädischchirurgische Versorgung erforderlich. Die oft entstellenden, ausgedehnten, teilweise hyperpigmentierten Narben mancher EDS-Patienten (typabhängig) können wegen der Neigung zu Wundheilungsstörungen chirurgisch nur selten kosmetisch verbessert werden. Abb. 6: Ehlers-Danlos-Syndrom, Klassischer Typ, Elektronenmikroskopie: Regelmäßige und ausgeprägte Veränderungen am Kollagen des dermalen Bindegewebes. Zahlreiche Einzelfibrillen zeigen keine normalen kreisrunden Querschnitte, sondern Formen mit unregelmäßig ausgefranste Umrissen. Abb. 7: Ehlers-Danlos-Syndrom, Klassischer Typ, Elektronenmikroskopie: Längsschnitte von aberranten verdrillten und deutlich verbreiterten Einzelfibrillen. 6 7.2 Hereditäre Bindegewebserkrankungen Abb. 8: Ehlers-Danlos-Syndrom, Dermatosparaxis, Elektronenmikroskopie: Sehr spezifische ultrastrukturell hieroglyphenartig geformte Kollageneinzelfibrillen. 7.2.2. Cutis laxa und Progeriesyndrome (Synonyme der Cutis laxa: Dermatochalasis, Elastolysis, Elastorrhexis) Die Cutis laxa (CL) ist eine seltene, klinisch und genetisch variable Erkrankung (Uitto 2002, Davidson 2002). Kardinalsymptom ist eine Haut von stark verringerter Elastizität. Dies manifestiert sich durch schlaffe oder stehende Hautfalten, die im Gesicht den Eindruck einer starken Voralterung („Trauergesicht“) hervorrufen (Abb. 9, 10). Die Hautsymptomatik besteht meist kongenital und verstärkt sich lebenslang. Es gibt aber auch erbliche und erworbene Cutis laxa–Formen, die sich erst später (2.-3. Lebensdekade) manifestieren (late-onset). Abb. 9: Cutis laxa, Klinische Symptome: Vorgealtertes Aussehen des Rumpfes mit hängenden Falten (Patientin 18 Monate alt). Abb. 10: Cutis laxa, Neugeborenes mit stehenden Hautfalten am Bauch. 7. Hereditäre Erkrankungen 7 Die klinischen Symptome können auf die Haut beschränkt sein und lediglich ein kosmetisches Problem darstellen. In anderen Fällen sind sie jedoch assoziiert mit teilweise schweren, systemischen Symptomen wie Lungenemphysemen, Darmdivertikeln, Pylorusstenosen und Hernien. Die Gelenke sind nicht hypermobil, die Haut ist nicht fragil, es bestehen keine Wundheilungsstörungen. Selten ist eine lokalisierte Beteiligung der Haut um die Augen (Blepharochalasis), der Palmoplantarhaut oder der Haut am Rumpf, den Händen und Füßen (wrinkly skin-Syndrom) (Gupta 2006). Die histopathologische Untersuchung des dermalen Bindegewebes ergibt meist eine ausgeprägte Rarefizierung, Disorganisation und Fragmentierung von elastischen Fasern. Neben quantitativen Abweichungen, können auch qualitative Veränderungen der elastischen Fasern bestehen, die sich durch eine elektronenmikroskopische Untersuchung erschließen (Ledoux-Corbusier 1985; Jung 1996). Die Fasern können: • Sehr klein, aber in sich regelrecht aufgebaut sein. • Innerhalb des dermalen Bindegewebe von oben nach unten immer mehr degenerieren oder: • Eine gestörte Ablagerung und Aggregation ihrer Hauptkomponenten Elastin und elastische Mikrofbrillen zeigen (Abb. 11) Abb. 11 Cutis laxa; Elektronenmikroskopie: Getrennte Ablagerung von Elastin und elastischen Mikrofibrilen in sehr kleinen und seltenen elastischen Fasern der retikulären Dermis. Der Erbgang ist je nach Form autosomal-dominant, autosomal-rezessiv oder Xchromosomal. Es existieren mindestens zwei Kandidatengene für dominante Formen (Fibulin-5, Elastin) (Urban 2005, Hu 2006) und für rezessive Formen (Fibulin 5, Fibulin-4) (Hu 2006; Huctagowder 2006). Viele Fälle ohne bisher identifizierte Mutation bzw. Kopplung wie z.B. die autosomal-dominante late-onset-Form und viele autosomal-rezessive Fälle sprechen für weitere verursachende Gene. 8 7.2 Hereditäre Bindegewebserkrankungen Zusätzlich kompliziert wird die diagnostische und pathogenetische Zuordnung dadurch, dass z.B. Fibulin-5 Mutationen dominante und rezessive CL verursachen können, dass Veränderungen im Elastin-Gen auch für das Willams-Beuren-Syndrom und SVAS veantwortlich sind (Szabo 2006) und dass auch Homocysteinurie und aAntitrypsinmangel zu CL-ähnlichen Phänotypen führen können (Royce 2002). Tabelle 2: Erkrankungen assoziiert mit Veränderungen der elastischen Fasern Rarefizierung und/ oder Strukturveränderungen Erbliche/erworbene Cutis laxa Menkes-Syndrom Laminopathien, z.B. Restriktive Dermopathie Anetodermie Elastinveränderungen Proliferation elastischer Fasern Verwandte Konditionen Rezessive/dominante Cutis laxa Williams-Beuren Syndrom Supravalvuläre Aortenstenose SVAS Pseudoxanthoma elasticum Aktinische Elastose Progeriesyndrome Marfan-Syndrom Altershaut Erbliches Emphysem Buschke-OllendorfSyndrom Mieschers-Elastom Erbliche Divertikulose α-Antitrypsinmangel Homocysteinurie Die X-chromosomale Form, das Menkes-kinky-hair-Syndrom oder occipital horn syndrome (früher auch als EDS Typ IX bezeichnet), wird duch Mutationen im Gen für ATPA7 verursacht und beruht auf Störungen des Kupfertransports (Moller 2004). Diagnostisch sind ausgeprägte Pili torti, Rarefizierung von elastischen Fasern in der Hautbiopsie sowie verringerte Mengen an Kupfer und Ceruloplasmin im Serum bzw. erhöhte Kupferaufnahme in kultivierten Fibroblasten. Ein Verlust an Hautelastizität kann bedingt sein durch: • Normale Alterungsprozesse. • Beträchtlichen Verlust an Körpergewicht. • Erbliche Bindegewebserkrankungen wie EDS. Beim EDS ist die Haut jedoch hyperelastisch und es bestehen primär Veränderungen am Kollagennetzwerk. Der Begriff Cutis laxa sollte beschränkt sein auf Konditionen mit Veränderungen der Struktur oder der Menge der elastischen Fasern im ultrastukturellen Befund. In Fällen von Pseudoxanthoma elasticum unterscheiden sich bereits die lokalisierten klinischen Hautläsionen eindeutig von Cutis laxa. Die mid-dermal Elastolysis stellt eine erworbene CL-Form dar. Sie ist charakterisiert durch den Verlust elastischer Fasern in der mittleren Dermis. Klinisch beginnt sie bei jungen Erwachsenen mit einer feinen Fältelung der Haut an sonnenexponierten Arealen der Arme und des Oberkörpers. Eine Reihe von Progeriesyndromen ist ebenfalls mit CL oder rarefizierten elastischen Fasern assoziiert: Das De Barsy-Syndrom, die Cutis laxa Typ Debré (mit Hypermobilität der Gelenke), das Costello-Syndrom, das Wiedemann-RautenstrauchSyndrom usw (Tab. 2¸ Moulson 2005). 7. Hereditäre Erkrankungen 9 Therapeutisch ist bei der CL eine plastisch-chirurgische Behandlung von entstellenden Falten, auch mehrmals, erfolgreich möglich (im Gegensatz zum EDS!). Die internistische Symptomatik muss interdisziplinär behandelt werden. 7.2.3 Pseudoxanthoma elasticum (Synonym: Grönblad-Strandberg-Syndrom, PXE) Das Pseudoxanthoma elasticum ist eine Systemerkankung des elastischen Gewebes. Elastische Fasern degenerieren zunehmend und lagern Kalziumapatit und andere anorganische Substanzen ein. Klinische Kardinalsymptome dieses Syndroms: • Haut: Gelbliche Papeln/Pseudoxanthome (Abb. 12,13) mit Prädilektionsstellen im Nacken, in den großen Beugen und der Inguinalregion sowie um den Nabel, die in der Kindheit oder später auftreten. • Augen: Sogenannte „angoid streaks“ aufgrund brüchiger Bruch’s Membran und andere chorioretinale Veränderungen. Gefahr der Erblindung! • Gefäßsystem: Durchblutungsstörungen, Hypertonie, gastrointestinale Blutungen, Herzinfarkt, Schlaganfall (Uitto 2003). Das Vollbild des PXE wird autosomal-rezessiv vererbt; das verantwortliche Gen ABCC6 codiert einen ATP-abhängigen ABC-Kassetten-Transporter, MPP6, der vor allem in Leber und Niere exprimiert wird (Efflux-Pumpe) und vermutlich an zellulären Entgiftungsprozessen beteiligt ist (Ladewig 2006). Pathogenetische Hypothesen vermuten, dass bei dieser sekundären erblichen Bindegewebserkrankung Mutationen in ABCC6 zu einer Akkumulation von Substanzen mit einer Affinität zu elastischen Fasern führen. Da regelmäßig auch Kollagenfibrillen in unmittelbarer Umgebung von degenerierten elastischen Fasern verändert sind, scheint der Gesamtaufbau der extrazellulären Matrix beeinträchtigt zu sein (Hendig 2006). Das PXE ist klinisch außerordentlich variabel, auch intrafamiliär besteht hohe Variabilität (Sherer 2001). Einige kardiovaskuläre Symptome scheinen dominant weitergegeben zu werden mit nur einem mutierten Allel. Die geschätzte Prävalenz von 1:100.000–1:150.000 lässt vermuten, dass das PXE unterdiagnostiziert ist (Schroder in press). Abb. 12: Pseudoxanthoma elasticum, Pseudoxanthome am Hals (9 j. Patient) Abb. 13: Pseudoxanthome und stehende Hautfalten in der Achselregion, (55 j. Patientin) 10 7.2 Hereditäre Bindegewebserkrankungen Die Diagnose erfolgt durch histopathologische (Abb. 14) und elektronenmikroskopische Untersuchung (Abb. 15) einer Hautbiopsie. In der retikulären Dermis fallen charakteristische korkenzieherartig gewundene, verklumpte und stark gefärbte elastische Fasern auf. In diesen finden sich elektronendichte Einschlüsse, umgeben von Kollagenbündeln mit Fibrillen (mit unregelmäßig ausgefransten Umrissen der Querschnitte) und Ablagerungen von bisher undefiniertem Matrixmaterial. Differenzialdiagnostisch kommen ähnliche Veränderungen nach D-PenicillinaminBehandlung, lokaler Einwirkung von Salpeter, im Endstadium schwerer Nierenerkrankungen und beim L-Tryptophan-induzierten Eosinophilie-Myalgie-Syndrom vor. Es fehlen dann jedoch die charakteristischen ophthalmologischen und kardiovaskulären Symptome und die Hautläsionen verschwinden nach entsprechender Behandlung. Unerklärt ist bisher das Auftreten des PXE bei bis zu 20% der Patientnen mit ßThalassämie und Sichelzellanämie in Griechenland. Ähnliche Hautläsionen zeigen sich auch bei der Amyloidose, jedoch mit deutlich verschiedener Morphologie (Amyloidablagerungen). Abb. 14: Pseudoxanthoma elasticum (Lichtmikroskopie,Semidünnschnitt, MethylenblauFärbung): Korkenzieherartig gewundene, verklumpte und stark angefärbte elastische Fasern in der retikulären Dermis. Abb. 15: Pseudoxanthoma elasticum, Elektronenmikroskopie: Elastische Fasern mit deutlichen, elektronendichten Einlagerungen. In der Nachbarschaft zahlreiche Kollageneinzelfibrillen mit unregelmäßig ausgefransten Umrissen der Querschnitte. 7. Hereditäre Erkrankungen 11 Die Diagnostik und Therapie dieser Multisystemerkrankung erfordern den Einsatz vieler Disziplinen: Nach Bestätigung der Diagnose duch eine Hautbiopsie sollten ophthalmologische und kardiovaskuläre Untersuchungen erfolgen. Die Hautveränderungen stellen lediglich ein kosmetisches Problem dar, für das es bisher keine adäquate Therapie gibt. Für die ophthalmologische und vaskuläre Symptomatik können bisher nur präventive Maßnahmen wie jährliche Funduskopien, das Tragen von Sonnenbrillen, gesunder Lebensstil (keinNikotin, kein Alkoholmissbrauch) und Ernährung mit Antioxidanzien-reicher Kost, Vermeidung von Übergewicht und regelmäßige Bewegung empfohlen werden. Eine Therapie mit EDTA, das Kalzium und andere Minerale bindet, konnte bisher keine eindeutige klinische Verbesserung zeigen und ist mit starken Nebenwirkungen belastet. Eine Therapie mit niedrig dosierter Acetylsalicylsäure (100mg/Tag) kann, trotz des erhöhten Risikos für gastrointestinale Blutungen, zur Vermeidung von schweren kardiovaskulären und neurovaskulären Ereignissen indiziert sein. Literatur Beighton P, De Paepe A, Steinmann B, Tsipouras P, Wenstrup RJ. Ehlers-Danlos syndromes: revised nosology, Villefranche, 1997. Ehlers-Danlos National Foundation (USA) and EhlersDanlos Support Group (UK). Am J Med Genet 1998; 77:31-7. Byers PH, Schwarze U. Ehlers-Danlos Syndrome. In: Emery and Rimoin's Principles and Practice of Medical Genetics. 4th ed. Rimoin DL, Connor JM, Pyeritz RE, Korf BR eds. Edinburgh: Churchill-Livinigstone 2002; 4021-43. Colige A, Nuytinck I, Hausser I et al. Novel types of mutation responsible for the dermatosparaxis type of Ehlers-Danlos syndrome (Type VIIC) and common polymorphisms in the ADAMTS2 gene. J Invest Dermatol 2004; 123: 656-63. Davidson JM, Giro M, Cutis laxa and premature aging syndromes. In : Royce PM, Steinmann B (eds) Connective tissue and its heritable disorders. New York: Wiley-Liss 2002; 525-60. Gupta N, Phadke SR. Cutis laxa type II and wrinkly skin syndrome: distinct phenotypes: Pediatr Dermatol 2006; 23:225-30. Hausser I, Muller U, Engelter S et al. Different types of connective tissue alterations associated with cervical artery dissections. Acta Neuropathol 2004; 107:509-14. Hendig D, Schulz V, Arndt M et al. Role of fetuin-A: a major inhibitor of systemic calcification in pseudoxanthoma elasticum. Clin Chem 2006; 52:227-34. Hu Q, Loeys BL, Coucke PJ et al. Fibulin-5 mutations: mechanisms of impaired elastic fibre formation in recessive cutis laxa. Hum Mol Genet 2006: 11 (Epub ahead of print). Hu Q, Reymond JL, Pinel N et al. Inflammatory destruction of elastic fibres in acquired cutis laxa is associated with missense alleles in the elastin and fibulin-5 genes. J Invest Dermatol 2006; 126:283-90. Huctagowder V, Sausgruber N, Kim KH et al. Fibulin-4: a novel gene for an autosomalrecessive cutis laxa syndrome. Am J Hum Genet 2006; 78:1075-80. Jung K, Ueberham U, Hausser I et al. Autosomal recessive cutis laxa syndrome. Acta Derm Venereol 1996; 76:298-301. Kielty CM Elastic fibres in health and disese. Expert Rev Mol Med 2006; 8:1-23. Ladewig MS, Götting C, Szliska C et al. Pseudoxanthoma elasticum: genetische Grundlagen, Manifestationen und therpeutische Ansätze. Der Opthalmologe 2006; 6:537-51. Ledoux-Corbusier M. Les elastolyses hereditaires. Dermatologica 1985; 171:407-18. Loeys BL, Schwarze U, Holm T et al. Aneurysm syndrome caused by mutation in the TGF-beta receptor. N Engl J Med 2006; 355:788-98. Makareeva E, Cabral WA, Marini JC, Leikin S. Molecular mechanisms of alpha 1(I)osteogenesis imperfecta/Ehlers-Danlos syndrome: unfolding of an N-ancor domain at the Nterminal end of the type I collagen triple helix. J Biol Chem 2006; 281:6463-70. 12 7.2 Hereditäre Bindegewebserkrankungen Moller LB, Hausser I, Emeis M et al. Variable clinical expression of an identical mutation in the ATPA7 gene for Menkes disease/occipital horn syndrome in three affected males in a single family. J Pediatr 2004; 145:119-21. Moulson CL, Go G, Gardner JM, et al. Homozygous and compound heterozygous mutations in ZMPSTEE24 cause the laminopathy restrictive dermopathy. J Invest Dermatol 2005; 125:913-9. Proske S, Hartschuh W, Enk A et al. Ehlers-Danlos-Syndrom – 20 Jahre Erfahrungen in der Diagnostik und Klassifikation an der Universitäts-Hautklinik Heidelberg. JDDG 2006; 4:308-18. Royce PM, Steinmann B (eds) Connective tissue and its heritable disorders. New York: WileyLiss, 2002. Schröder F, Hausser I, Szliska C et al. Pseudoxanthoma elasticum: under-recognized cause of early onset peripheral arterial disease? Vasc Med 2006, in press. Sherer DW, Bercovitsch L, Lebwohl M. Pseudoxanthoma elasticum: significance of limited phenotypic expression on parents of affected offsprings. J Am acad Dermatol 2001; 44:534-37. Steinmann B, Royce PM, Superti-Furga A. The Ehlers-Danlos syndrome. In. Royce PM, Steinmann B (eds) Connective tissue and its heritable disorders. New York: Wiley-Liss, 2002:351407. Szabo Z, Crepeau MW, Mitchell AL et al. Aortic aneurysmal disease and cutis laxa caused by defects in the elastin gene. J Med Genet 2006; 43:255-8. Uitto J, Pulkkinen L. Heritable disorders affecting the elastic tissues: cutis laxa, pseudoxanthoma elasticum and related disorders. In: Principles and Practice of Medical Genetics; 4th ed. Rimoin DL, Connor JM, Pyeritz RE, Korf BR eds. Edinburgh: Churchill-Livinigstone 2002. Uitto J, Ringpfeil F, Pulkkinen L. Heritable disorders of connective tissue - Ehlers-Danlos syndrome, Pseudoxanthoma elsticum and cutis laxa. In: Dermatology. Bologna JL, Jorizzo JL, Rapini RP eds. Harcourt Publishing, London, 2003; 1519-36. Urban Z, Gao J, Pope FM et al. Autosomal dominant Cutis laxa with severe lung disease: synthesis and matrix deposisition of mutant tropoelastin. J Invest Dermatol 2005; 124:1193-9. Zurück zum Inhaltsverzeichnis: DNO © BBS-Verlag Wiesbaden