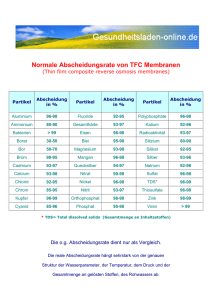
Nichtwässrige Nano-Kolloide : Halbleiter-, Metall
Werbung

Nichtwässrige Nano-Kolloide Halbleiter-, Metall- und Kompositpartikel aus Metallorganischen Precursoren Inaugural-Dissertation Vorgelegt von Diplom-Chemikerin Julia Hambrock 2003 Dissertation zur Erlangung der Doktorwürde der Fakultät für Chemie der Ruhr-Universität Bochum vorgelegt von Diplom-Chemikerin Julia Hambrock aus Hannover Mündliche Prüfung: April 2003 Gutachter: Prof. Dr. R. A. Fischer Prof. Dr. M. Epple Dem Mann meiner Träume Die vorliegende Arbeit entstand unter der Leitung von Prof. Dr. R. A. Fischer in der Zeit von Mai 1999 bis April 2003 am Institut für Anorganische Chemie II, Organometallics & Materials Chemistry, der Ruhr-Universität Bochum. Meinem hochverehrten Lehrer und Mentor Herrn Professor Dr. Roland Augustinus Fischer danke ich von ganzem Herzen für die außergewöhnliche Aufgabenstellung und die im gleichen Zuge zu ihrer Durchführung bereitgestellten wissenschaftlichen und finanziellen Freiheiten. Sein Interesse und seine immerwährende Ansprechbarkeit, die fruchtbaren Diskussionen und Denkanstöße und nicht zuletzt die Möglichkeit, an bedeutenden Konferenzen teilzunehmen, waren für mich von unschätzbarem Wert. Ich danke ihm insbesondere für das entgegengebrachte Vertrauen und die Atmosphäre knisternder Motivation, die dieser Arbeit ihren Rahmen gegeben und sie über weite Strecken zu einem Vergnügen haben werden lassen. Mein Dank gilt ferner... ... den Kolleginnen Sabine Bendix, Uschi Bossek, Dr. Anjana Devi, Ursula Fischer, Cornelia Gregorec, Silvia Grum, Heike Kampschulte, Eva Maile, Sabine Masukowitz, Dr. Nicola Oberbeckmann, Urmila Patil, Helga Rudack, Dr. Ulrike Weckenmann, Dr. Barabara Wehner, Dr. Dana Weiß, Dr. Pia Wennek, Eliza Gemel und den Kollegen Arne Baunemann, Ralf Becker, Rhagunandan Bhakta, Dr. Qingmin Cheng, Mirza Cokoja, Rolf Deibert, Dr. Holger Fölsing, Dr. Christian Gemel, Stephan Hermes, Frank Hipler, Dr. Holger Hoffmann, Thorsten Johann, Jayaprakash Khanderi, Heinz-Jürgen Klußmann, Dr. Lianhai Lu, Dr. André Manz, Dr. Jens Müller, Harish Parala, Dr. Wolfram Rogge, Matthias Ruttert, Rainer Schäffer, Dr. Oliver Segnitz, Stephan Spöllmann, Dr. Oliver Stark, Frank Stowasser, Tobias Steinke, Dr. Jurij Weiß, Dr. Holger Winkler, Dr. Carl Winter und Andreas Wohlfart. Die überaus angenehme Arbeitsatmosphäre und nahezu uneingeschränkte Hilfsbereitschaft haben maßgeblich zum Gelingen dieser Arbeit beigetragen. ... Marie Katrin Schröter und Christian Schirrmacher, die sich unermüdlich den Schwierigkeiten der Kolloidchemie gestellt und durch ihre praktische Arbeit einen außerordentlich wertvollen Beitrag geleistet haben. Nana wünsche ich viel Glück und Erfolg bei der Fortsetzung dieses hochinteressanten und (fast) nie langweiligen Themas! .... Dr. Alexander Birkner nicht nur für die Einführung in die Transmissionselektronenmikroskopie, ohne die diese Arbeit schlichtweg nicht möglich gewesen wäre, sondern darüber hinaus für zahlreiche und fruchtbare Diskussionen zur Interpretation von TEM-Egebnissen. ... Prof. Drieß, Dr. Klaus Merz und Robert Schönen (Anorganische Chemie I) für die Zusammenarbeit auf dem Gebiet der ZnO-Kolloide. ... Prof. Winter, Dr. Gerd Gebauer und Thomas Galka (Institut für Experimentalphysik) für die Zusammenarbeit auf dem Gebiet der Mie-Ellipsoemtrie. ... Prof. Muhler, Lamy Khodeir, Dr. Wilma Busser und Dr. Elke Löffler (Technische Chemie) für die Zusammenarbeit auf dem Gebiet der CO-Adsorption von Kupfer-Kolloiden. ... dem Graduiertenkolleg „Dynamische Prozesse an Festkörperoberflächen“ für die zweijährige finanzielle Unterstützung während dieser Arbeit und all seinen Mitgliedern für ihre Offenheit, die schönen gemeinsamen Stunden und (nicht zu vergessen) die Musik. ... den ungezählten Mitarbeitern der Institute und Werkstätten der Ruhr-Universität Bochum, die mich bei dieser Arbeit unterstützt haben. I ... und nicht zuletzt meinen Eltern, Rosi Simon und meinem wundervollen Mann für ihre Geduld mit meinen Zweifeln und ihre uneingeschränkte Unterstützung in den weniger leichten Momenten. Inhaltsverzeichnis Abkürzungsverzeichnis ......................................................................................................... IV 1 Einleitung ..........................................................................................................................1 2 Theoretische Aspekte .......................................................................................................5 2.1 Grundlegende Prinzipien in der Synthese von Nanopartikeln........................................ 5 2.1.1 Metallische Nanopartikel.................................................................................... 6 2.1.2 Halbleiter-Nanopartikel...................................................................................... 8 2.2 Optische und physikalische Eigenschaften von Nanopartikeln...................................... 9 2.2.1 Grundlagen ......................................................................................................... 9 2.2.2 Halbleiter .......................................................................................................... 11 2.2.3 Metalle .............................................................................................................. 13 2.3 Methoden der Charakterisierung .................................................................................. 14 2.3.1 UV/VIS Spektroskopie ....................................................................................... 15 2.3.2 Photolumineszenz (PL) ..................................................................................... 16 2.3.3 Transmissionselektronenmikroskopie (TEM) ................................................... 18 3 CdSe-Nanopartikel .........................................................................................................20 3.1 Einleitung und Aufgabenstellung ................................................................................. 20 3.2 Etablierung einer Syntheseroute für CdSe-Nanopartikel.............................................. 22 3.2.1 Stand der synthetischen Forschung .................................................................. 22 3.2.2 Synthese der Edukte und Vorbereitung der Thermolyse................................... 25 3.2.3 Thermolytische Synthese von CdSe-Nanopartikeln .......................................... 26 3.3 Alternative Precursoren für die Synthese von CdSe-Nanopartikeln durch Thermolyse von organometallischen Verbindungen .................................................... 28 3.3.1 Einleitung.......................................................................................................... 28 3.3.2 Beschreibung und Synthese der Precursoren und ihre Thermolyse in Trioctylphosphinoxid ........................................................................................ 29 3.3.3 Charakterisierung der CdSe-Nanopartikel....................................................... 31 3.4 Oberflächenmodifizierung von TOPO-gecappten CdSe-Nanopartikeln ...................... 34 3.4.1 Einleitung.......................................................................................................... 34 3.4.2 Austausch von oberflächengebundenem Trioctylphosphinoxid gegen 2Naphtylthiol....................................................................................................... 36 3.5 Zusammenfassung ........................................................................................................ 40 4 Zinkoxid-Nanopartikel...................................................................................................42 4.1 Stand der Forschung und Aufgabenstellung................................................................. 42 I 4.2 Synthese von ZnO-Nanopartikeln durch Thermolyse von organometallischen Precursoren ................................................................................................................... 44 4.2.1 Auswahl der Precursor ..................................................................................... 44 4.2.2 Thermolyse von [MeZnOSiMe3] 4 bei niedrigen Temperaturen ........................ 46 4.2.3 Thermolyse von [MeZnOSiMe3] 4 bei hohen Temperaturen.............................. 50 4.2.4 Thermolyse von [Zn(OCH(Me)CH2NMe2)2]..................................................... 51 4.3 Zusammenfassung ........................................................................................................ 53 5 Kupfer-Nanopartikel......................................................................................................55 5.1 Metallische Nanopartikel: ein Überblick...................................................................... 55 5.2 Stand der Forschung und Aufgabenstellung................................................................. 58 5.3 Synthese von Kupfer-Nanopartikeln durch Thermolyse des organometallischen Precursors [Cu(OCH(Me)CH2NMe2)2] ........................................................................ 60 5.3.1 Vorstellung des Precursors............................................................................... 60 5.3.2 Thermolyse........................................................................................................ 62 5.3.3 Analytische Charakterisierung ......................................................................... 63 5.3.4 Variation der Reaktionsparameter ................................................................... 67 5.4 Oberflächenmodifizierung von HDA-gecappten Cu-Nanopartikeln............................ 70 5.4.1 Einleitung.......................................................................................................... 70 5.4.2 Verhalten gegenüber Wasser und Sauerstoff.................................................... 71 5.4.3 Verhalten gegenüber Thiolen............................................................................ 73 5.4.4 Verhalten gegenüber Carbonsäuren................................................................. 75 5.4.5 Verhalten gegenüber Kohlenmonoxid............................................................... 76 5.5 Untersuchungen zum Wachstumsmechanismus: Mie-Ellipsometrie ........................... 78 5.6 Zusammenfassung ........................................................................................................ 81 6 Palladium-Nanopartikel.................................................................................................85 6.1 Stand der Forschung und Aufgabenstellung................................................................. 85 6.2 Synthese von Palladium-Nanopartikeln unter Verwendung des organometallischen Precursors [(Cp)(Allyl)Pd] ........................................................................................... 88 6.2.1 Precursor .......................................................................................................... 88 6.2.2 Thermolyse von (η5-Cyclopentadienyl)(η3-Allyl)Pd......................................... 89 6.2.3 Oberflächenmodifizierung von Palladium-Nanopartikeln ............................... 96 6.3 Synthese von Palladium-Nanopartikeln unter Verwendung des organometallischen Precursors [Pd(OCH(Me)CH2NMe2)2]......................................................................... 97 6.3.1 Vorstellung des Precursors............................................................................... 97 6.3.2 Thermolyse von [Pd(OCH(Me)CH2NMe2)2] .................................................. 101 6.4 Zusammenfassung ...................................................................................................... 102 II 7 Nickel-Nanopartikel .....................................................................................................104 7.1 Stand der Forschung und Aufgabenstellung............................................................... 104 7.2 Synthese von Nickel-Nanopartikeln unter Verwendung des organometallischen Precursors [Ni(OCH(Me)CH2NMe2)2]....................................................................... 106 7.2.1 Vorstellung des Precursors............................................................................. 106 7.2.2 Pyrolyse........................................................................................................... 109 7.3 Zusammenfassung ...................................................................................................... 111 8 Modellsysteme für den Cu/ZnO Katalysator in der Methanolsynthese..................112 8.1 Einleitung und Aufgabenstellung ............................................................................... 112 8.2 Cu und ZnO in räumlicher Trennung: Imprägnierung von ZnO mit KupferNanopartikeln.............................................................................................................. 113 8.3 Cu und ZnO in räumlicher Nähe: Mischungen von Cu- und ZnO-Kolloiden ............ 114 8.4 Inniger Kontakt zwischen Kupfer und Zink: Coating von Kupfer-Nanopartikeln mit Zink 115 8.4.1 Precursor und Synthese .................................................................................. 116 8.4.2 Analytische Charakterisierung ....................................................................... 118 8.4.3 Elektronenmikroskopie (TEM)........................................................................ 119 8.4.4 Elektronenbeugung (SAED und XRD) ............................................................ 120 8.5 Zusammenfassung ...................................................................................................... 122 9 Zusammenfassung ........................................................................................................124 10 Experimenteller Teil.....................................................................................................128 10.1 Allgemeine Arbeitsvorschriften.................................................................................. 128 10.2 Charakterisierung........................................................................................................ 128 10.3 Chemikalien ................................................................................................................ 130 10.4 Synthesen .................................................................................................................... 131 10.4.1 Vorschriften zum Kapitel Cadmiumselenid .................................................... 131 10.4.2 Vorschriften zum Kapitel ZnO ........................................................................ 134 10.4.3 Vorschriften zum Kapitel Kupfer .................................................................... 134 10.4.4 Vorschriften zum Kapitel Palladium............................................................... 135 10.4.5 Vorschriften zum Kapitel Nickel ..................................................................... 136 10.4.6 Vorschriften zum Kapitel Cu/ZnO-Systeme .................................................... 136 11 Literaturverzeichnis .....................................................................................................138 12 Anhang.............................................................................................................................. V 12.1 Publikationen, Vorträge, Posterbeiträge ........................................................................ V 12.2 Lebenslauf....................................................................................................................VII III Abkürzungsverzeichnis AOT AAO Bipy CVD DLS DMAP EDX en ES FAB FK HDA IR MS NAC OPR PL PVP SAED SAM Schmp. SSP TBP TEM THF TOAB TOPO UV/VIS vdW XRD (2-Ethylhexyl)sulfosuccinat Anodisches Aluminiumoxid 2,2’-Bipyridin Chemical Vapour Deposition (chemische Dampfabscheidung) Dynamische Lichtstreuung Dimetyhlaminopyridin Energy-dispersive X-Rays Ethylendiamin Electrospray Fast atom bombardement Festkörper n-Hexadecylamin Infrarot Mass spectrometry Non-aqueous colloid Oberflächen-Plasmonen-Resonanz Photolumineszenz Polyvinylpyrrolidon Selected Area Electron Diffraction Self Assembled Monolayer Schmelzpunkt Single-source Precursor Tributylphosphin Transmission electron microscopy Tetrahydrofuran Tetraoctylammoniumbromid Tri-n-octylphosphinoxid Ultraviolett/Visible van-der-Waals X-Ray diffraction Einige englische Begriffe wurden (teilweise zum besseren Verständnis) unübersetzt übernommen, da sie meist auch im deutschen Sprachraum so verwendet werden: bandgap, biological labelling, bottom-up, coating, core-shell, dangling bond, deep und shallow traps, grid, nanowire, onset, precursor, quantum confinement, shift, top-down. Sie werden im Text kursiv hervorgehoben. IV 1 Einleitung “I imagine experimental physicists…”, and I might add, chemists as well, “…must often look with envy at men like Kamerlingh Onnes, who discovered a field like low temperature, which seems to be bottomless and in which one can go down and down. Such a man is then a leader and has some temporary monopoly in a scientific adventure. Percy Bridgman, in designing a way to obtain higher pressures, opened up another new field and was able to move into it and to lead us all along. The development of ever higher vacuum was a continuing development of the same kind. I would like to describe a field, in which little has been done, but in which an enormous amount can be done in principle. This field is not quite the same as the others in that it will not tell us much of fundamental physics (in the sense of, ``What are the strange particles?'') but it is more like solid-state physics in the sense that it might tell us much of great interest about the strange phenomena that occur in complex situations. Furthermore, a point that is most important is that it would have an enormous number of technical applications. What I want to talk about is the problem of manipulating and controlling things on a small scale…”[1] Diese visionären Worte sind der Beginn eines Vortrags, den der Physiker Richard Feynman vor mehr als 40 Jahren bei dem Jahrestreffen der Amerikanischen Physikalischen Gesellschaft gehalten hat. Der Vortrag trug den Titel „There`s Plenty of Room at the Bottom“, nicht etwa nur „There`s Room at the Bottom”, denn der spätere Nobelpreisträger rechnete seinen Zuhörern vor, dass die Gesamtheit der Encyclopedia Brittanica in einen Stecknadelkopf passt, wenn man nur einen Weg findet, die Buchstaben um das 25.000-fache zu verkleinern. Und er führte aus, dass das im Grunde nur noch eine Frage der Zeit sein sollte. Auch wenn dieser Vortrag fast ein halbes Jahrhundert zurückliegt – Nanopartikel und Nanomaschinen begleiten die Menschheit, seit sie angefangen hat zu existieren: Zellen lesen und schreiben Informationen im Nanometermaßstab, sie erzeugen Energie, Wärme und neues Material. Daher kann zu Recht behauptet werden, dass Nanotechnologie schon seit 3.5 Milliarden Jahren existiert. Aber auch anorganisches, nanoskaliges Material wurde von Menschen verwendet, ohne dass es ihnen bewusst war. Beispielsweise in Autoreifen. Beispielsweise in der heterogenen Katalyse. Dass es ihnen nicht bewusst war, lag aber lediglich daran, dass sie es einfach nicht sehen konnten. Erst im 20. Jahrhundert zündete eine entscheidende Idee den Boom, der mit dem Wort nano verknüpft ist. Louis de Broglie postulierte, dass Elektronen als Wellen aufgefasst werden können, wobei ihre Wellenlänge um ein Vielfaches kürzer ist als die des Lichts. Damit sollte es möglich sein, wesentlich kleinere Abstände aufzulösen und es dauerte nicht lange, bis die Anwendung von Elektronen in einem ElektronenMikroskop vorgeschlagen wurde. Der kritische Punkt zur Realisierung des Elektronenmikroskops war die Erfindung eines Analogons zu der im Lichtmikroskop zur Fokussierung Einleitung / 1 und Defokussierung verwendeten Linse, und die Entwicklung der elektromagnetischen Linse wurde später (1986) mit dem Nobelpreis an Ruska belohnt. Seitdem weiß der Mensch von der Nanowelt und seitdem träumt er von ihren unbegrenzt erscheinenden Anwendungsmöglichkeiten. Der größte Traum der Nanotechnologen, von vielen eher als Utopie belächelt, ist es, einzelne Atome willentlich und mit System anzuordnen. Natürlich nur in sinnvoller Art und Weise, nicht so, dass sie chemisch instabil sind. Eine Vielfalt neuer Materialien könnte dadurch entstehen oder auch nur eine Vielzahl neuer Eigenschaften von bereits bekannten Materialien. Selbst wenn man Materialien, die wir bereits kennen, Atom für Atom zusammenbauen würde, wäre der Gewinn enorm: es entstünde kein Abfall, weil 100% der „Edukte“ genutzt würden, demzufolge wäre die Umweltbelastung minimal bzw. nicht vorhanden und die Herstellung wäre billig, denn es gäbe keine Nebenprodukte, die abgetrennt und entsorgt werden müssten. Zum heutigen Zeitpunkt existiert zwar ein fundiertes Wissen über Selbstorganisation von Systemen, also die Anordnung von großen Anzahlen an Atomen, wir können einzelne Atome manipulieren und es ist sogar möglich, im Nanometer-Maßstab zu „schreiben“, doch der gezielte Aufbau von makroskopischen, dreidimensionalen Gegenständen liegt in weiter Ferne. Ganz zu schweigen von den Nanomaschinen, die diese Aufgabe erledigen sollten... Etwas höher auf der Realitäts-Skala lassen sich die Versuche ansiedeln, die Bauteile eines Computers zu verkleinern. Einige Vorschläge sind dazu gemacht worden, z. B. die Verwendung von Nanopartikeln als SETs (single electron transistor), wobei die Zustände 1 und 0 durch die An- oder Abwesenheit eines Elektrons auf diesem Partikel realisiert werden können. Oder die Anordnung von Nanoröhrchen in Gittern, wobei jeder Knotenpunkt in zwei Konfigurationen vorliegen und damit 1 und 0 darstellen kann. Die Funktionsweise beider Vorschläge ist bereits bewiesen worden, aber die Frage nach der Adressierung der einzelnen Funktionselemente und ihrer Verknüpfung zu geordneten Strukturen ist ein nach wie vor ungelöstes Problem. Gelänge es, dafür eine Lösung zu finden, wäre der Gewinn zugegebenermaßen gigantisch. Nanopartikel sind jedoch nicht nur für die Realisierung von SETs interessant, sie haben auch andere außergewöhnliche Eigenschaften, die bereits zu realen Anwendungen in verschiedensten Bereichen des täglichen Lebens geführt haben. • Beispielsweise können Schichten aus Nanopartikeln extrem hart und kratzfest sein, da die Haftkraft und Bindung zu anderen Partikeln direkt mit der Größe der Oberfläche zusammenhängt und das Verhältnis von Oberfläche zu Volumen für Nanopartikel extrem groß ist. Dies ermöglicht die Herstellung kratzfester Lacke. • Sonnencremes schützen die Haut vor der schädlichen Einwirkung von UV-Licht durch den Zusatz von organischen Molekülen als UV-Filter. Diese schützen die Haut jedoch nicht gleichmäßig, da sie beim Auftragen in die Hautfalten abfließen und sie rufen teilweise Allergien hervor. ZnO oder TiO2 filtern das UV-Licht ebenso, aber leiden nicht 2 / Einleitung • • • • • unter den oben genannten Nachteilen. Mikrometer große Partikel erscheinen jedoch auf der Haut weiß und waren daher zu Beginn ein sozusagen kosmetisches Problem. Verringert man jedoch die Größe in den Nanometerbereich, werden die Partikel für sichtbares Licht durchlässig und können als kosmetisch unbedenklicher UV-Schutz verwendet werden. Schon lange ist bekannt, dass Tumore absterben, wenn sie erhitzt werden. Bislang war es jedoch nicht möglich, tiefer sitzende Geschwülste zu behandeln, da die Wärme nicht weit genug in den Körper eindringt bzw. nicht präzise steuerbar ist. An der Charité in Berlin werden nun (bereits in der ersten klinischen Studie!) Eisenpartikel benutzt, die direkt in einen Tumor eingespritzt und dann über ein elektrisches Feld erhitzt werden können. Darüber hinaus stellte sich heraus, dass je nach Krebsart ein jeweils anderes Nanopartikel mit einer speziellen Oberflächenstabilisierung am besten wirkte. Schering verwendet Nanopartikel als Kontrastmittel, da ihre Oberfläche so modifiziert werden kann, dass sie sich an ganz bestimmten Orten im menschlichen Körper sammeln. Roche Diagnostics verwenden magnetische Nanopartikel für die Untersuchung von Erbmaterial in Blutproben. Die Quantum Dot Corporation benutzt in ähnlicher Weise halbleitende Nanopartikel für verschiedenste Untersuchungen von biologischen Proben. Gilead Sciences benutzt Nanopartikel als Träger für Arzneimittel. Darüber hinaus führt allein die Formulierung von Medikamenten in nanopartikulärer Form zur Löslichkeit im Blutstrom, was insbesondere für die Pharmazie ungeahnte Möglichkeiten bietet, da über 50 % der neuen Medikamente aufgrund von Löslichkeitsproblemen nie auf den Markt gebracht werden. Degussa beschichtet Papier mit nanopartikulären Oxiden und ermöglicht somit den Druck mit strahlenden Farben. Dies ist nur eine relativ kleine Auswahl an Beispielen, die aber deutlich macht, dass Nanopartikel ein breites Anwendungsspektrum und darüber hinaus enorme ökonomische Perspektiven aufweisen. Es dürfte auch klar sein, dass nanostrukturierte Materialien größtenteils synthetische Chemikalien sind und dass ihre Synthese allem anderen, auch ihrer Anwendung, vorausgeht. Begonnen hat die Synthese von Nanopartikeln mit bottom-up Methoden, d. h. zunächst mit Studien an kleinen Aggregaten von Metallatomen, die durch Verdampfung von Metallen erzeugt wurden, bevor dann die chemischen Methoden so weit fortgeschritten waren, dass die Synthese von kleinen Partikeln durch die Reduktion von Metallsalzen und später dann die Synthese von Nanopartikeln nahezu jeden Materials möglich wurde. Aufgabe der Chemiker, insbesondere der anorganischen (und physikalischen) Chemiker, ist es also, synthetische Wege für die Herstellung von nanostrukturiertem Material zu eröffnen. Dieser Zugang sollte so beschaffen sein, dass er eine Kontrolle über die Eigenschaften der Partikel ermöglicht und Partikel mit genau definierter Größe, Form und einheitlichen Eigenschaften liefert. Dies ist nicht nur Voraussetzung für ihre Anwendung, sondern auch für die Einleitung / 3 Klärung von grundlegenden physikalischen Fragen. Die Organometallchemie bietet (eine der) Möglichkeiten, genau diese Anforderungen zu erfüllen, da sie es ermöglicht, Precursoren mit Hilfe ihrer Liganden maßzuschneidern, und die vorliegende Arbeit hatte zur Aufgabe, genau dieses Wissen der Chemie in die Synthese von Nanopartikeln einzubringen. Die einzelnen Kapitel dieser Arbeit beschäftigen sich mit jeweils einem Materialssystem, zunächst mit halbleitenden Materialien (CdSe und ZnO), nachfolgend mit metallischen Partikeln (Kupfer, Nickel, Palladium) und schließlich mit ihrer Koexistenz (Cu/ZnO). Jedes dieser Systeme bietet einen besonderen Anlass für die Verwendung von organometallischen Precursoren, z. B. den Ersatz von toxischen und zusätzlich sicherheits-gefährdenden Precursoren durch einfach handhabbare Substanzen oder das Design von Precursoren, die keiner weiteren Zusätze bedürfen oder die Etablierung von organometallischen Verbindungen, die die Synthese von nicht-wässrigen Kolloiden, sogenannten NACs (non-aqueous colloids) ermöglichen. Einleitend zu jedem Kapitel finden sich daher eine materialspezifische Motivation und ein Überblick über bereits veröffentlichte Arbeiten. Die wichtigsten theoretischen Aspekte werden in Kapitel 2 angesprochen, d. h. prinzipielle Aspekte der Synthese, mit der Größe der Partikel verbundene spezielle optische und physikalische Eigenschaften sowie relevante Einzelheiten der Analytik. Eine spezielle Einleitung zu metallischen Nanopartikeln wird stellvertretend für Kupfer, Nickel und Palladium in Kapitel 5 gegeben. 4 / Einleitung 2 Theoretische Aspekte 2.1 Grundlegende Prinzipien in der Synthese von Nanopartikeln Im Prinzip lassen sich Nanopartikel als sehr kleine Ausschnitte aus dem Gitter eines Festkörpers verstehen. Sie können deshalb auf dieselbe Weise hergestellt werden wie der entsprechende Festkörper auch. Der Haken dabei ist jedoch, dass Atome bzw. Ionen, die sich an der Oberfläche befinden, eine ungesättigte Koordinationssphäre haben und deshalb dangling bonds aufweisen (siehe die auf zwei Dimensionen projizierte Darstellung eines Kristallgitters in Abb. 1a). Die Konsequenz ist die Reaktion mit einem weiteren Atom oder Ion, das die Bindungen absättigt und nach weiterem Wachstum schließlich zur Ausbildung eines Festköpers führt. Für die Herstellung von Nanopartikeln muss also das Wachstum eines Festkörpers bei einer Größe von wenigen Nanometern gestoppt werden, indem die Oberflächenatome elektronisch abgesättigt oder aber sterisch vor einer weiteren Anlagerung geschützt werden. In der Praxis sind diese beiden Strategien jedoch oft nicht unterscheidbar, da sie sich meistens gegenseitig bedingen. (a) (b) L L K e rn H ü lle Abb. 1: Schematische Darstellung der (a) unterschiedlichen Koordinationssphären von Volumen- und Oberflächenatomen und (b) der Situation in Nanopartikeln. Synthetisch arbeitende Naturwissenschaftler haben dafür im Laufe der letzten Jahrzehnte eine Reihe von Lösungsansätzen gefunden und Nanopartikel hergestellt, die wie in Abb. 1b dargestellt ganz allgemein als ein System aus Kern und Hülle verstanden werden können. Der Kern besteht aus einem nanometergroßen Ausschnitt eines Festkörpers – das kann ein Metall sein, aber auch ein Halbleiter oder ein Isolator – und enthält zwischen wenigen hundert bis mehreren tausend Atome oder Ionen. Um den Kern herum befindet sich eine Hülle, die die notwendige elektronische oder sterische Abschirmung des Kerns gegenüber einem weiteren Wachstum gewährleistet. Die Abkürzung L steht für organische Liganden und ist die am häufigsten verwendete „Hülle“. Es handelt sich dabei um Moleküle mit einer Kopfgruppe, die über freie Elektronenpaare an Atome auf der Oberfläche der Partikel koordinieren kann, und einem Rest, der entweder eine zusätzliche sterische Abschirmung bewirkt (lange Alkylketten) Theorie / 5 oder weitere Funktionalitäten aufweist. Typische Beispiele sind langkettige Amine und Phosphine, Lösungsmittelmoleküle wie Tetrahydrofuran, Polymere oder Tenside. Dabei führen lipophile Schutzmoleküle zu Partikeln, die in organischen Medien löslich sind, während hydrophile Agentien wasserlösliche Partikel ergeben. Eine andere Möglichkeit der Abschirmung ist die elektrostatische Stabilisierung. Sie basiert auf der Coulombschen Abstoßung zwischen zwei benachbarten Partikeln, die durch die durch adsorbierte Ionen und ihre Gegenionen gebildete elektrische Doppelschicht hervorgerufen wird. Diese Methode wird häufig bei metallischen Partikeln angewandt und arbeitet typischerweise mit Citraten oder quartären Ammoniumionen. Eine ganz andere Art von Hülle bietet sich bei der Verwendung von strukturierten Materialien als „Nanoreaktoren“ an. Das Wachstum des Kerns dauert gerade so lange an bis die Hohlräume des Gastgebers, z. B. sphärische Poren oder längliche Kanäle, angefüllt sind und die Wände der Kavität das Wachstum stoppen. Dieses Syntheseprinzip wird auch als bottom-up Methode bezeichnet, da die Materialien aus einzelnen Atomen bzw. Ionen bis zur gewünschten Größe aufgebaut werden. Genauso lassen sich aber auch top-down Methoden anwenden, bei denen die Größe eines ausgedehnten Festkörpers durch z. B. mechanische Einwirkung bis zu nanoskaligen Dimensionen verkleinert wird. Da diese Methoden jedoch meist aufwändige Apparaturen erfordern, sind die aufbauenden Synthesen wesentlich weiter verbreitet. Für die Stabilisierung dieser Nanopartikel gelten indes dieselben Prinzipien wie für die bottom-up Methoden. Beide Methoden können zu nanometergroßen Dimensionen in verschiedenem Ausmaß führen und werden folgendermaßen benannt: • Festkörper sind in alle drei Raumrichtungen ausgedehnt und werden daher als dreidimensionale 3-D Objekte bezeichnet • Liegt die Ausdehnung einer der Raumrichtungen im Bereich von Nanometern, spricht man von 2-D Objekten oder quantum wells • Reduktion der Größe in zwei Dimensionen führt zu Objekten, die nur noch in eine Raumrichtung ausgedehnt sind. Sie werden mit der Bezeichnung 1-D versehen und heißen quantum wires oder Nanodrähte • Dreidimensional auf den Nanometerbereich eingeschränkte Objekte werden mit 0-D bezeichnet und heißen quantum dots oder Quantenpunkte 2.1.1 Metallische Nanopartikel Metallische Nanopartikel lassen sich ganz allgemein durch die chemische oder elektrochemische Reduktion von Metallionen in Gegenwart von Stabilisatoren herstellen. Diese Methode wurde bereits 1857 von Faraday publiziert[2] und ist seitdem die am weitesten verbreitete und für viele Bereiche leistungsfähigste Methode. Die ersten reproduzierbaren synthetischen Protokolle wurden von Turkevich für die Herstellung von Goldkolloiden durch Reduktion von HAuCl4 mit Natriumcitrat veröffentlicht.[3-5] In seinen Veröffentlichungen hat 6 / Theorie Turkevich postuliert, dass die relativen Raten von zwei entscheidenden Vorgängen die Größe der (Metall-)Partikel beeinflussen: Nukleation und Wachstum (als dritter Schritt ist die Terminierung des Wachstums zu ergänzen). Dieses Postulat ist bis heute weitgehend akzeptiert und wird zur Erklärung der Effekte von Reaktionsparametern herangezogen. Nukleation und Wachstum sind jedes für sich genommen extrem komplizierte Prozesse, die nur für wenige Fälle genauer beleuchtet worden sind.[6-9] Demnach sind geladene Dimere die ersten Zwischenprodukte, die gebildet werden. Nach der Bildung eines stabilen Nucleus, dessen Größe unter einem Nanometer, abhängig von der Stärke der Metall-Metall-Bindung, vermutet wird, sind verschiedene Mechanismen für das weitere Wachstum denkbar: Zum Beispiel eine Art oberflächenkatalysiertes Wachstum, bei dem Atom für Atom an den Nukleationskeim angelagert wird, oder aber eine Agglomeration durch Kollision von instabilen Anhäufungen von Metallatomen (siehe Schema 1). Schema 1: Bildung von Metallkolloiden nach der Reduktion des Metallkations.[10] In der Theorie wird das Wachstum homogen verteilter Nukleationskeime von zwei gegenläufigen energetischen Faktoren bestimmt: auf der einen Seite nimmt die Festköperenergie ab und favorisiert somit ein Wachstum der Nukleationskeime, auf der anderen Seite nimmt jedoch die Oberflächenenergie zu, was ein Schrumpfen der Kristallite zur Folge hat: û* = 4ŒU 2 s −l − 4Œ 3 r û 3 s −l ⋅n Gleichung 1: Zusammenhang zwischen der freien Energie und dem Radius von sphärischen Kristalliten ∆G ist die freie Energie für einen sphärischen Kristallit, r der Radius, γ die freie Energie der Kristall-Flüssigkeit interface pro Flächeneinheit, ∆µ die Differenz zwischen flüssigem und festem chemischen Potential und n die Zahl der Partikel im Kristallit. Wachstum ist demnach energetisch erst dann möglich, wenn eine kritische Größe rc erreicht ist. Das Maximum der obigen Gleichung gibt den Radius dieses kritischen Nukleus an: rc = 2γ /(∆µn) (Gleichung 2). Trotz seiner Wichtigkeit ist seine Größe jedoch weder experimentell gemessen noch Theorie / 7 theoretisch berechnet worden. In der klassischen Nukleationstheorie wird angenommen, dass die Form der Nukleationskeime wegen ihrer Oberflächenspannung sphärisch ist, eine experimentelle Bestätigung dieser Annahme existiert jedoch nicht. Lediglich bei der Bildung von kolloidalen Kristallen ist bekannt, dass die Oberfläche rauh und die Oberflächenspannung demzufolge geringer ist. Zusätzlich wurden während der Kristallisation nicht-sphärische Kristallformen beobachtet, deren Verhältnis zwischen kürzester und längster Achse selbst bei größeren Kristalliten 0.65 ± 0.15 nicht überstieg.[11] 2.1.2 Halbleiter-Nanopartikel Auch für Nanopartikel, die aus halbleitendem Material bestehen, haben sich die ersten veröffentlichten Methoden als die bis heute gebräuchlisten herausgestellt. Die schwer löslichen Halbleiter wie CdSe, CdS u. a. lassen sich sehr einfach durch Fällungsreaktionen in Gegenwart von Stabilisatoren herstellen, während Oxide durch Dehydratisierung der kolloidalen Hydroxide zugänglich sind. Im englischen Sprachgebrauch bezeichnet man diese Methoden als arrested precipitation oder kinetic trapping. Im Falle der halbleitenden CdSe-Nanopartikel sind eine Reihe von Fakten über den Wachstumsprozess bekannt. Die Gruppe um Alivisatos hat sich mit der zeitlichen Entwicklung der Größenverteilung beschäftigt. Abb. 2 zeigt, dass die Größenverteilung der Partikel zu Beginn bei einer Standardabweichung von etwa 20 % liegt. Nach der Injektion der Precursor (Me2Cd und Tributylphosphinselenid) folgen zwei verschiedene kinetische Bereiche: Während der ersten 22 Minuten steigt der Partikeldurchmesser rasch von 2.1 auf 3.3 nm an und die Standardabweichung wird von 20 % auf 8 % fokussiert. Anschließend wachsen die Partikel wesentlich langsamer von 3.3 auf 3.9 nm und die Größenverteilung verbreitert sich wieder. Eine weitere Injektion steigert die Wachstumsrate und fokussiert die Größenverteilung erneut. Während der Fokussierung und Defokussierung ändert sich die Anzahl der Partikel nicht, während die Monomerkonzentration durch die Fokussierung abnimmt und während der Defokussierung konstant bleibt. Abb. 2: Die mittlere Größe (oben) und die Größenverteilung (unten) von CdSe-Nanopartikeln als Funktion der Zeit. Pfeile indizieren Injektionen der Precursoren Me2Cd und TBPSe.[12] Daraus ergibt sich das folgende Bild: Die Nukleation von CdSe erfolgt sehr kurz nach der Injektion und endet, sobald die Temperatur und die Monomerkonzentration unter einen 8 / Theorie kritischen Wert fallen. Wie oben beschrieben gibt es für jede gegebene Monomerkonzentration einen kritischen Radius. Nanokristallite, deren Größe darunter liegt, haben negative Wachstumsraten, d. h. sie lösen sich auf, während Partikel mit r > rc größer werden. Eine Fokussierung der Größenverteilung tritt dann ein, wenn die vorhandenen Nanokristallite alle eine Größe haben, die etwas über dem kritischen Radius liegt. Dann nämlich wachsen die kleineren Kristallite schneller als die größeren. Wenn allerdings die Monomerkonzentration aufgrund des Wachstums erschöpft ist, übersteigt die kritische Größe die mittlere Partikelgröße und es beginnt eine Defokussierung, weil sich Partikel mit r < rc beginnen aufzulösen, während Partikel mit r > rc wachsen. Dieser Bereich der Defokussierung wird auch Ostwald ripening genannt. Die Verteilung wird erneut fokussiert, wenn die Monomerkonzentration durch eine weitere Injektion ansteigt, weil dann die kritische Größe wieder auf einen kleineren Wert fällt. Dass Fokussierung und Defokussierung empfindlich von den Reaktionsparametern abhängen, zeigt die Beobachtung, dass in dem System Trioctylphosphinoxid (TOPO) – Hexadecylamin (HDA) – Trioctylphosphin (TOP), also durch Zugabe von Aminen, eine wesentlich schnellere Fokussierung und nahezu keine Defokussierung erfolgt.[13] 2.2 Optische und physikalische Eigenschaften von Nanopartikeln 2.2.1 Grundlagen Mie-Theorie Die Zusammenhänge zwischen Teilchengröße und Streulichtintensität sind sehr komplex und von sehr vielen Faktoren abhängig. Zu diesen Faktoren gehören die Wellenlänge des einfallenden Lichts, der Winkel zwischen einfallendem und gestreutem Licht, die Form der Teilchen und die physikalischen Eigenschaften des dispergierten Stoffs. Insgesamt werden drei Abhängigkeitsbereiche zwischen Streulichtintensität und Teilchengröße unterschieden. Zur Abgrenzung der verschiedenen Bereiche wird der sogenannte Mie-Parameter α (siehe Gleichung 3) herangezogen. α= 2πr λ Gleichung 3: Definition des Mie-Paraemters α mit r = Teilchenradius und λ = Wellenlänge des einfallenden Lichts. Das Streulichtverhalten kleiner, sphärischer Teilchen wurde 1871 von Rayleigh beschrieben, wobei der entsprechende Rayleigh-Bereich bis zu einem Mie-Parameter von α § gilt. In diesem Größenbereich ist die Streulichtintensität proportional zur zweiten Potenz des Teilchenvolumens (bzw. sechsten Potenz des Teilchendurchmessers), weswegen Streulichtmessungen zur Bestimmung des Molargewichts hochverdünnter makromolekularer Lösungen Theorie / 9 herangezogen werden. Bei größeren Teilchen kommt es hingegen zur Abschwächung des Streulichts durch Interferenzerscheinungen bis hin zur destruktiven Interferenz. Die führt dazu, dass die Intensitäten in Vorwärtsrichtung sehr viel höher sind als in Rückwärtsrichtung, was häufig als Mie-Effekt bezeichnet wird. Mit größer werdendem Teilchendurchmesser werden die Unterschiede immer größer, was messtechnisch in der Mehrwinkelanalyse der Photonenkorrelationsspektroskopie ausgenutzt wird. Mie entwickelte 1908 eine für sphärische Teilchen allgemeingültige Streulichttheorie (die später von Gans und Möglich auf dreiachsige Ellipsoide erweitert wurde). Die Streulichtintensität ist für Teilchen mit einem Mie-Parameter > 0.5 proportional zur zweiten Potenz des Teilchendurchmessers. Dieser Bereich wird auch Mie-Bereich genannt und ist zugleich der Gültigkeitsbereich der geometrischen Optik. Zwischen den beiden beschriebenen Bereichen gibt es einen nicht monotonen Übergangsbereich, der messtechnisch schwerer zugänglich ist. Die grundsätzlichen Vorstellungen der Interferenz- oder Mie-Theorie bestehen darin, dass die streuenden Teilchen durch eine Anzahl von elektrischen und magnetischen Oszillatoren steigender Ordnung ersetzt werden, die sich im Mittelpunkt des Teilchens befinden. Die Interferenzerscheinungen bei der Lichtstreuung an großen Partikeln werden durch eine Streufunktion P(θ) berücksichtigt. Das Streulicht wird mit wachsendem Beobachtungswinkel q geschwächt. Infolge der Interferenzwirkung ist P(θ) für θ > 0° kleiner als 1 und wird für θ = 0° gleich 1. Weiterhin besteht eine Abhängigkeit der Streufunktionen von den Winkeln und von der lokalen Anordnung bzw. molekularen Zusammensetzung der als selbständige Dipole wirkenden Volumenelemente. Letztlich steckt dahinter die Geometrie und der chemische Aufbau der Teilchen. Der gesamte Extinktionskoeffizient von kleinen Metallpartikeln ist in der Mie-Theorie als Summe über alle elektrischen und magnetischen Mulitpol-Oszillationen gegeben, die zur Absorption und Streuung beitragen. Bei Nanopartikeln, deren Größe sehr viel kleiner als die Wellenlänge des eingestrahlten Lichts ist, wird angenommen, dass nur der Dipolterm zur Absorption beiträgt (dipole approximation). Der Extinktionskoeffizient κ für N Partikel mit dem Volumen V ist dann durch die folgende Gleichung gegeben: κ= 18πNVε m 3/ 2 ⋅ ε2 (ε1 + 2ε m )2 + 2ε 2 2 Gleichung 4: Extinktionskoeffizient κ für kleine (< 25 nm) Partikel. λ = Wellenlänge des absorbierten Lichts, εm = Dielektrizitätskonstante des umgebenden Mediums, ε1 und ε2 = realer bzw. imaginärer Teil der Dielektrizitätskonstante des Materials. In Übereinstimmung mit Gleichung 4 ist die Plasmonen-Absorption innerhalb der DipolAnnäherung also unabhängig von der Teilchengröße. Lediglich experimentell konnte festgestellt werden, dass eine gewisse Abhängigkeit zwischen der Teilchengröße und der 10 / Theorie Bandbreite besteht: je kleiner der Radius r, desto größer die Bandbreite der entsprechenden Absorptionsbande. Für größere Partikel beobachtet man einen Shift des Absorptionsmaximums zu größeren Wellenlängen bzw. kleineren Energien, da die höheren Multipol-Terme nicht mehr vernachlässigt werden können. Dieser Zusammenhang ist jedoch theoretisch sehr schwer zu beschreiben und die Ansätze führen sogar zu widersprüchlichen Ergebnissen für die Abhängigkeit des Absorptionsmaximums von der Teilchengröße.[14] Plasmonen Plasmonen sind kollektive Anregungen der quantisierten Schwingungen von Elektronen in einem Metall. Man unterscheidet zwischen Volumen- und Oberflächenplasmonen. Polaritonen Polaritonen entstehen, wenn des Feld der elektromagnetischen Strahlung und das des Excitons überlappen. Wenn die Energien ähnlich sind, beeinflussen sich Licht und Exciton und es kommt durch quantenmechanische Überlagerung zu neuen, unterschiedlichen Objekten. Diese „Objekte“ werden Polaritonen genannt. 2.2.2 Halbleiter In einem halbleitenden Festkörper führt die große Anzahl von Atomen zur Ausbildung von Bändern, dem Valenzband und dem Leitungsband, die durch eine energetische Lücke (Bandlücke) voneinander getrennt sind. Bei 0 K ist das Valenzband mit Elektronen gefüllt, während das Leitungsband unbesetzt ist. Bei höheren Temperaturen können die Elektronen des Valenzbandes durch thermische Energie über die Bandlücke hinweg in das Leitungsband hinein angeregt werden. Das angeregte Elektron im Leitungsband bildet mit dem zurückgebliebenen, positiv geladenen Loch im Valenzband ein sogenanntes „Elektron-Loch-Paar“. Bei ausreichender räumlicher Nähe können diese Ladungsträger einen gebundenen Zustand ausbilden. Ein gebundenes Elektron-Loch-Paar wird Wannier-Exciton genannt und ist über den Festkörper delokalisiert. Der Bohrsche Radius des Festkörper-Excitons ist durch Gleichung 5 gegeben: aB = h2ε e2 1 1 + me mh Gleichung 5: Bohrscher Radius des Excitons im Festkörper. ε = optischer Dielektrizitätskoeffizient des Festkörpers, e = Elementarladung, me bzw. mh = effektive Massen des Elektrons bzw. Lochs. Halbleiter-Nanokristallite unterscheiden sich in zwei Punkten von Festkörpern: (1) das Verhältnis von Oberfläche zu Volumen ist größer und (2) die Größe der Partikel ist so klein, dass der Bohrsche Radius der Excitonen unterschritten wird und die Ladungsträger auf die drei Dimensionen des Partikels beschränkt werden: quantenmechanisch entspricht diese Situation Theorie / 11 der des „Teilchens-im-Kasten“, auch quantum confinement genannt. Die resultierenden Effekte nennt man quantum size effects. In Nanopartikeln sind also Elektron und Loch näher beieinander als im Festkörper. Deshalb kann die Coulomb-Wechselwirkung zwischen ihnen nicht mehr vernachlässigt werden und sie haben eine höhere kinetische Energie. Mit Hilfe der EMA (effective mass approximation) kann die größenabhängige Energie des ersten excitonischen Übergangs unter Verwendung von folgender Gleichung berechnet werden: h2π 2 1 1 1.8e 2 − ∆E ≅ + 2 R 2 me mh εR Gleichung 6: Energieunterschied zwischen Grundzustand und erstem angeregten Zustand. Aus Gleichung 6 kann man erkennen, dass der Coulomb Term den ersten angeregten Zustand mit R-1 zu niedrigeren Energien, während der erste Term ihn mit R-2 zu höheren Energien verschiebt. Das heißt, der erste excitonische Übergang bzw. die Bandlücke wächst mit abnehmendem Partikelradius. Diese Vorhersage ist in vielen Experimenten bestätigt worden und optisch besonders eindrucksvoll an fluoreszierenden Lösungen von CdSe-Partikeln zu sehen (Abb. 3). Die links abgebildete blaue Lösung enthält kleine Partikel mit großer Bandlücke, während die aufgrund ihres größeren Durchmessers und einer dadurch kleineren Bandlücke rot emittierenden Partikel am rechten Ende der Skala zu finden sind. Abb. 3: Fluoreszierende Lösungen von CdSe-Nanopartikeln mit von links nach rechts zunehmender Partikelgröße bzw. abnehmender Bandlücke (Felice Frankel, MIT, 1998). Die Größe der Bandlücke ist jedoch nicht die einzige Eigenschaft, die sich ändert, wenn die Größe der Halbleiter-Partikel verringert wird. Je kleiner die Partikel werden, desto größer wird auch das Verhältnis von Oberfläche zu Volumen. Dieser sogenannte Volumeneffekt ist neben dem quantum confinement der zweite wichtige Effekt, der bei der Reduktion der Größe auftritt. Die Folge ist, dass sich ein größerer Teil der Gesamtzahl an Atomen an der Oberfläche befindet und dort aufgrund der Oberflächenspannung eine höhere potentielle Energie besitzt. So befinden sich beispielsweise in 50 nm großen Partikel etwa 6 % der Atome an der Oberfläche, für 2-3 nm große Partikel sind es bereits 50 % und bei einem Durchmesser von 1.5 nm schließlich 75 % aller Atome. Oberflächenatome bzw. -ionen mit höherer Energie lassen sich wesentlich leichter bewegen oder gar aus dem Verbund entfernen. Die makro12 / Theorie skopische Konsequenz dieses Sachverhalts ist ein Absinken der Schmelztemperatur mit sinkendem Partikeldurchmesser. Beispielsweise hat der Halbleiter CdS im Festkörper einen Schmelzpunkt von TSchmp. = 1678 K. Für ein Partikel mit 4 nm Durchmesser liegt er bei TSchmp. (4 nm) = 1200 K und für 1.5 nm große Partikel ist der Übergang von fester zu flüssiger Phase bereits mehr als 1000 K niedriger als beim Festkörper (TSchmp. (1.5 nm) = 600 K).[15] Der Volumeneffekt wirkt sich außerdem auf die chemischen Eigenschaften der Partikel aus. Dadurch, dass sich ein Großteil der Atome an der Oberfläche befindet, befinden sich auch eine hohe Anzahl von Liganden in der Hülle um den Halbleiterkern. Da diese Liganden die chemischen Eigenschaften der Partikel im Verhältnis zur Umgebung bestimmen, kann ihre Auswahl gezielt zur Variierung des chemischen Verhaltens ausgenutzt werden, z. B. für die Löslichkeit in einem bestimmten Medium oder um sie durch Knüpfen einer Bindung an einer bestimmten Stelle zu verankern. 2.2.3 Metalle Diese beiden Effekte, Volumen- und Größenquantisierungseffekt, gelten nicht nur für Halbleiter, sondern auch für Metalle. Auch hier kommt es mit abnehmender Größe zu einer Vergrößerung der Bandlücke, wobei jedoch der Effekt nicht so ausgeprägt ist, da Valenz- und Leitungsband im makrokristallinen Material noch überlappen. Erst bei sehr kleinen Partikeln ist das Anwachsen der Bandlücke so weit fortgeschritten, dass halbleitende Eigenschaften beobachtet werden können. Die auffälligsten Merkmale von metallischen Nanopartikeln seien im Folgenden kurz genannt. Aus denselben Gründen wie bei halbleitenden Partikeln sinkt der Schmelzpunkt ebenfalls mit abnehmendem Partikeldurchmesser. Für 2-nm-Goldpartikel ist ein Schmelzpunkt von ca. 500 °C berechnet worden, der damit fast 600 °C unter dem für einen makrokristallinen Verbund liegt.[16] Sehr auffällig sind die intensiven Farben einiger metallischer Nanopartikel. Sie beruhen jedoch nicht auf der Absorption durch Anregung von Elektronen vom Valenz- in das Leitungsband, sondern auf der Anregung des Elektronengases. Diese sogenannte Oberflächenplasmonenresonanz tritt prinzipiell immer auf, ist jedoch in einem Festkörper nicht zu sehen, da der Anteil der Oberfläche im Verhältnis zum Volumen sehr gering ist. Eine weitere typische Eigenschaft von metallischen Festkörpern ist ihre elektrische Leitfähigkeit, die auf der freien Beweglichkeit der Elektronen im Leitungsband beruht. Bei einer Verkleinerung der Partikel ändert sich aber die Bänderstruktur, und aufgrund des Ausbildens von diskreten energetischen Niveaux ist auch das Ohmsche Gesetz nicht mehr gültig. Viele Forschungsgruppen haben deshalb kleine Partikel zwischen zwei Elektroden platziert und ihre Strom-Spannungskurven untersucht. Normalerweise, d. h. in einem Festkörper, würde das Tunneln eines Elektrons von der Elektrode auf das Partikel unbemerkt vonstatten gehen. Im Falle eines Nanopartikels ist damit jedoch eine beträchtliche Energieänderung verbunden, nämlich EC = e2 / 2C (C ist die Kapazität des Partikels; durch ihre geringe Größe, die im Bereich von aF liegt, wird der Nenner sehr klein und damit EC groß). Im Falle eines neuTheorie / 13 tralen Partikels ist eine externe Spannung von e/2C erforderlich, bevor Strom, d. h. ein erstes Elektron, fließen kann. Für jedes weitere Elektron muss die Spannung um weitere e/C erhöht werden. Das Ergebnis ist eine stufenartige Strom-Spannungs-Kurve. In der Realität wird meist nur die erste Stufe beobachtet, da (1) der Widerstand und die Kapazität für die zwei Verbindungen zu und von dem Partikel gleich sind, d. h. das Elektron tunnelt mit derselben Geschwindigkeit durch die Verbindungen und die Rückkopplung mit der Spannung geht verloren. Außerdem wird (2) durch thermische Effekte ebenfalls das Tunneln von Elektronen angeregt und damit werden mit steigender Temperatur die einzelnen Stromstufen herausgewaschen bzw. ein Ohmscher Verlauf in der I-V-Kurve beobachtet. Das Tunneln von einem Elektron durch ein nanometergroßes metallisches (oder auch halbleitendes) Nanopartikel ist die logische Operation eines single electron transistors (SET), eine der am vielversprechendsten Möglichkeiten, die Größe von elektronischen Bauteilen weiter zu verkleinern. Mit den heutigen Transistoren ist dies noch nicht möglich, da ein typischer Transistor eine Kapazität von etwa 10-12 F hat. Wenn man einen solchen Transistor mit einem einzigen Elektron beladen wollte, müsste man ein Potential von V = e/2C, also etwa 10-8 Volt anlegen. Darüber hinaus müsste der Transistor, um thermische Effekte zu vermeiden, bei einer Temperatur von 0.0005 K arbeiten, damit 10-8 > kT erfüllt ist. Normale Spannungen liegen aber bei etwa 100 mV, d. h. es würde nicht eines, sondern q = CV = 106 Elektronen auf den Transistor übergehen. 2.3 Methoden der Charakterisierung Grundsätzlich sind Nanopartikel zunächst allen Methoden der Festkörperanalyse zugänglich, wie z. B. der Elektronenmikroskopie oder der Röntgenbeugung, die die Untersuchung des Partikelkerns zum Ziel haben. Darüber hinaus sind Partikel, die mit organischen Liganden funktionalisiert sind, auch in organischen Solventien löslich, d. h. sie sind auch den traditionellen Methoden der organischen Chemie zugänglich. Je nach Art der Untersuchungsmethode werden so entweder Informationen über den anorganischen Kern oder über die meist organische Hülle erhalten. Im Rahmen dieser Arbeit wurde der Schwerpunkt auf Transmissionselektronenmikroskopie (TEM), Elektronenbeugung (SAED), Dynamische Lichtstreuung (DLS), Absorptionsspektroskopie (UV/VIS) und Fluoreszens (PL) gelegt. Einige dieser Methoden sollen daher in den folgenden Unterkapiteln kurz vorgestellt werden, wobei der Fokus auf die für Nanopartikel relevanten Einzelheiten gerichtet ist. 14 / Theorie 2.3.1 UV/VIS Spektroskopie Aufgrund der optischen Eigenschaften von halbleitenden Nanopartikeln, die in Kapitel 2.2 beschrieben worden sind, liefert die Absorptionsspektroskopie wichtige Informationen über die untersuchten Kolloide, die am Beispiel von CdSe erläutert seien. (1) Die Lage der lokalen Maxima entspricht jeweils einem bestimmten Übergang von einem Loch- in einen Elektronenzustand. Die beiden deutlich sichtbaren Banden des UV/VIS-Spektrums von CdSeNanopartikeln (Abb. 4) können Übergängen in den niedrigsten elektronischen Zustand 1Se aus zwei verschiedene Lochzuständen 1S3/2 und 3S3/2 heraus zugeordnet werden.[17] Der Übergang mit der geringeren Energiedifferenz, 1S3/2 – 1Se, entspricht dabei der langwelligeren Bande. Die Lage dieser Übergänge gibt (2) Aufschluss über die Dimension des entstandenen Materials. Material mit Abmessungen über dem Bohrschen Excitonradius weist dieselbe Absorption auf wie der Festkörper, während Partikel, die auf Größen unterhalb dieses Radius beschränkt sind, dem quantum confinement unterliegen, d. h. eine Aufweitung der Bandlücke erfahren, und daher eine Blauverschiebung in der Absorption zeigen. Aus der Extrapolation des steilen Anstiegs (siehe Abb. 4) erhält man (3) die genaue Lage der Bandkantenabsorption und damit die Größe der Bandlücke. Der Zusammenhang zwischen der Bandlücke und dem Radius der Partikel erlaubt es (4) auch, aus der UV/VIS-Absorption die Größe der Partikel zu berechnen. Die entsprechende Formel und ihre Anwendung ist in den Kapiteln über CdSe und ZnO zu finden. 1 ( Absorption [a. u.] 1P e E le ktronisc he Z ustä nd e 1S e 1S 3 /2 Absorption der Bandlücke im Festkörper 400 500 600 2S 3 /2 3S 3 /2 1P 3 /2 L oc h zu stän de 700 W ellenlänge [nm] Abb. 4: Optische Absorptionskante von nanodispergiertem CdSe in Toluol, extrapoliert an einen direkten Übergang. Aus der Halbwertsbreite der Absorptionsbande lässt sich (5) abschätzen, ob die Größenverteilung der in der Probe enthaltenen Partikel schmal oder breit ist. Angenommen, eine Probe besteht aus Partikeln, die einen kontinuierlichen Größenbereich abdecken: Jedes Partikel wird dann aufgrund der Abhängigkeit der Größe der Bandlücke vom Radius ein etwas anderes Absorptionsmaximum aufweisen. Da diese Absorption genau wie die Größenverteilung kontinuierlich ist, wird sie nicht aufgelöst, und die Folge ist eine Verbreiterung der entsprechenden Bande im Vergleich zur monodispersen Probe. Theorie / 15 Die UV/VIS-Spektroskopie an metallischen Nanopartikeln erlaubt nicht ganz so viele Rückschlüsse auf die Eigenschaften der Partikel. Zunächst einmal wird die Absorption von Licht nicht durch Anregung von Elektronen über die Bandlücke verursacht. Der physikalische Ursprung der Lichtabsorption liegt vielmehr in der durch Wechselwirkung mit dem elektromagnetischen Feld angeregten kohärenten Oszillation der Elektronen im Leitungsband. Diese Resonanzen sind als Oberflächen-Plasmonen bekannt und treten vor allem bei Kupfer, Silber und Gold auf, nicht jedoch bei Metallen mit gefüllten Schalen. Die energetische Lage, Breite und Amplitude der OPR (Oberflächenplasmonenresonanz) wird von der Dielektrizitätskonstante des konstituierenden Materials und der Umgebung bestimmt. Wenn die Partikel sphärisch sind und klein im Vergleich zur Wellenlänge des Lichts, d. h. etwa 3 bis 20 nm, und zudem getrennt voneinander in Lösung vorliegen, gibt es keine starke Abhängigkeit des Absorptionsspektrums von der Größe, da die höheren Terme der Mie-Theorie nicht mit eingehen, sondern nur der sogenannte Dipol-Term, der nicht von der Partikelgröße abhängt. Ist das Verhältnis der Achsen größer als 1 (wie z. B. bei nanorods), treten zwei verschiedene Resonanzen auf, die in der Regel als (1,0) Mode (Anregung entlang der kurzen Achse) und (1,1) Mode (Anregung in Richtung der längeren Achse) bezeichnet werden. Das Auftreten der OPR ist jedoch in jedem Fall ein Effekt kleiner Partikel, da sie weder in einzelnen Atomen noch im Festkörper beobachtbar ist. Damit erlaubt das UV/VIS-Spektrum eines Nanometalls folgende Informationen: (1) ist eine OPR sichtbar, liegt nanopartikuläres Material mit einem Radius < 25 nm vor. (2) Die Anzahl der Banden erlaubt Rückschlüsse auf die Form der Partikel: besteht das Spektrum aus einer einzigen Bande, liegen sphärische Partikel vor. Sind mehrere Banden sichtbar, ist die Morphologie anisotrop, d. h. es könnten sich z. B. Stäbchen gebildet haben. 2.3.2 Photolumineszenz (PL) Die Absorption von elektromagnetischer Strahlung durch Nanokristallite ist relativ geradlinig und umkompliziert. Ihr Lumineszenzverhalten ist dagegen wesentlich komplizierter und noch lange nicht vollständig verstanden. Im Rahmen dieser Arbeit sollen die Vorgänge jedoch zumindest qualitativ skizziert werden. Folgende Prozesse laufen während der PL in einem Halbleiter ab (siehe Schema 2): Zunächst wird durch Absorption von Licht der geeigneten Wellenlänge ein Elektron aus dem Valenz- in das Leitungsband angeregt. Das entstehende Elektronen-Loch-Paar bildet ein - + Wannier-Exciton aus, ein gebundenes e -h -Paar. Nach einer bestimmten Zeit, die durch die Lebensdauer des angeregten Zustands bestimmt wird, relaxiert das Elektron zurück in das Valenzband und emittiert dabei ein Photon. Da die Energie des emittierten Photons der Energielücke zwischen den Bändern entspricht, wird diese Emission Bandkantenlumineszenz genannt. Zwischen den Bandkanten existieren aber weitere Energieniveaux, sowohl nahe an den Bändern (shallow traps) als auch weiter weg (deep traps). Man nimmt an, dass shallow traps durch Oberflächendefekte entstehen, z. B. durch dangling bonds, aber auch durch die im 16 / Theorie Vergleich zum Inneren des Kristalls stark gestörten Bindungsverhältnisse, während deep traps von Kristalldefekten stammen. / Ein Elektron kann strahlungslos oder unter Abgabe eines Lichtquants in eines dieser Energieniveaux und von dort aus zurück in das Valenzband relaxieren, wobei auch hier der Übergang strahlend oder strahlungslos erfolgen kann (dies gilt sinngemäß natürlich auch für Löcher). L eitu n g sb and B an d lü ck e e - sh a llo w trap d eep tra p 1 2 h e 3 4 5 + - Valen zb an d Schema 2: Schematische Darstellung der während der Photolumineszenz ablaufenden Prozesse. Im Wesentlichen charakterisieren vier Merkmale die Eigenschaften der Partikelemission: Helligkeit, Farbe, Reinheit der Farbe und Stabilität der Emission. Da die Bandlücke aufgrund des quantum confinements vergrößert ist, wird auch die Emission kontinuierlich von rot nach blau verschoben. In der Regel ist die Emission von Nanopartikeln im Vergleich zur Absorption zu höheren Wellenlängen verschoben. Dieser Shift ist das Ergebnis von zwei Effekten, nämlich der Relaxation in shallow traps und der Größenverteilung, und wird StokesShift genannt. Die Größenverteilung beeinflusst darüber hinaus die Linienbreite der Emission: Die typische Halbwertsbreite der Emission von CdSe-Nanopartikeln beträgt 27-40 nm und ist damit aufgrund von inhomogener spektraler Verbreiterung beträchtlich breiter als die eines einzelnen Partikels (< 20 nm). Die Reinheit der Farbe ist aufgrund der starken Abhängigkeit der Emission von der Größe sehr von der Größenverteilung der Partikel abhängig: je schmaler die Größenverteilung, desto reiner die Emissionsfarbe. Die Helligkeit geht einher mit der Quantenausbeute. Es wird angenommen, dass eine hohe Quantenausbeute ein Merkmal von Kristalliten mit wenigen elektronischen Defekten ist, da die Quantenausbeute um ein Vielfaches gesteigert wird, wenn statt koordinierender organischer Liganden eine anorganische Hülle verwendet wird (core-shell Partikel), die eine bessere Passivierung der Oberflächenzustände gewährleistet, so dass Verluste aufgrund von strahlungsloser Emission verhindert werden. Insgesamt gibt ein Emissionsspektrum also Auskunft über: • • • die Lage der Bandlücke bzw. Farbe der Partikel die Größenverteilung der Partikel die Qualität der Partikel in Bezug auf elektronische und strukturelle Defekte. Theorie / 17 2.3.3 Transmissionselektronenmikroskopie (TEM) Die Prinzipien der Transmissionselektronenmikroskopie sind in guten Lehrbüchern (siehe z. B. [18]) bereits eingehend beschrieben worden und werden hier nicht weiter behandelt. Stattdessen wird die Auswertung der Real- und Beugungsbilder kurz erläutert. Die Realbilder sind eine direkte Abbildung des Leuchtschirms, auf dem die Elektronen die Struktur der Probe abgebildet haben, auf einen Photofilm. Die belichteten Negative werden im Photolabor entwickelt und ihr Positiv dient zur Auswertung. Dadurch, dass es sich um eine direkte Abbildung handelt, sind die auf dem Positiv gemessenen Distanzen direkt proportional zu den Distanzen der Probe. Um den realen Wert zu erhalten, wird der auf dem Positiv gemessene Wert zunächst um die Nachvergrößerung (Negativ zu Positiv) korrigiert und dann durch den geeichten Wert für die Vergrößerung dividiert. Dieser berechnet sich aus dem Produkt der aus der Messung bekannten, nominellen Vergrößerung und des Eichfaktors, der anhand von Asbest und dem Kreuzgitter von Plano berechnet worden ist. Die Auswertung der Beugungsbilder erfolgt anhand von einfachen geometrischen Überlegungen, die in Schema 3 dargestellt sind. Auf das rechtwinklige Dreieck, das durch die Kameralänge L, den Abstand R zwischen dem ungebeugten Strahl und dem Reflex, und den Winkel 2Θ aufgespannt wird, lässt sich der tangens-Satz (Quotient aus Gegen- und Ankathete entspricht dem Winkel zwischen diesen Seiten des Dreiecks) anwenden. Damit erhält man: R/L = 2Θ. Da der Winkel Θ mit etwa 0.5° sehr klein ist, wird dies angenähert zu R/L § 2Θ. Über die Bragg-Bedingung lässt sich nun eine Beziehung zu den Netzebenenabständen herstellen: λ/d = 2 sin Θ §2Θ, wobei auch hier aufgrund des kleinen Winkels angenähert werden kann. Kombiniert man nun diese beiden Gleichungen für 2Θ miteinander, so erhält man die Beziehung λL = Rd. K a m eralän g e L d 2Θ B ild sch irm R M ittelp u n k t R e flex Schema 3: Schematische Darstellung zur Berechnung der Ebenenabstände aus dem Beugungsbild einer Probe im Elektronenmikroskop. R entspricht dem Reflexabstand vom Mittelpunkt des Beugungsbildes (im Negativ, nicht im Positiv!). Die Kamerakonstante L ist aus der Messung bekannt und wird zusätzlich durch einen Eichfaktor korrigiert. Die Wellenlänge λ von Elektronen mit einer Beschleunigungs18 / Theorie spannung von 200 kV beträgt 2.507 pm. Damit lassen sich die Netzebenenabstände dhkl des Materials relativ einfach berechnen. Zusammenfassend erhält man aus den elektronenmikroskopischen Messungen folgende Informationen: • Größe der Partikel • Form der Partikel • Größenverteilung • Ordnung • Existenz von kristallinen Phasen • Material (aus Gitterabständen) • Zusammensetzung des Partikelkerns (aus EDX und Auswertung SAED) Theorie / 19 3 CdSe-Nanopartikel 3.1 Einleitung und Aufgabenstellung Zu Beginn unserer Arbeiten auf dem Gebiet der Nanopartikel wurde das System CdSe ausgewählt. Es bietet für „Neulinge“ auf diesem Gebiet eine Reihe von Vorteilen: (1) seine physikalischen und chemischen Eigenschaften sind sorgfältig untersucht worden und können mit den eigenen Ergebnissen verglichen werden. Im Vergleich dazu ist die Synthese von CdTeoder CdS-Partikeln lange nicht so fortgeschritten, z. B. gibt es keine Methode, die es erlaubt, die Form dieser Partikel gezielt zu variieren. (2) Es ist analytisch gut zugänglich, da CdSe als II-VI Halbleiter mit einer Bandlücke von 716 nm und vor allem CdSe-Nanopartikel mit einer Bandlücke <716 nm UV/VIS-aktiv sind und es wegen seiner elektronenreichen atomaren Bestandteile (34Se und 48Cd) einen guten Kontrast im Elektronenmikroskop zeigt. Darüber hinaus sind die Partikel nur geringfügig luftempfindlich. Die erste Aufgabe war daher, eine geeignete Syntheseroute für CdSe-Nanopartikel zu etablieren. Dieser Teil der Arbeit wird in Kapitel 3.2.1 beschrieben. Eine Reihe von möglichen, aber auch bereits realisierten Anwendungen haben CdSe-Nanopartikel zudem industriell interessant gemacht. Allen voran das biological labeling, das 1998 von Alivisatos[19] und Nie[20] publiziert wurde und heute von der Quantum Dot Corporation (homepage: www.qdots.com) kommerziell genutzt wird. Bei der biologischen Markierung wird der Oberflächeneffekt und die Modifizierbarkeit der Partikeloberfläche ausgenutzt, um die Partikel an chemisch definierte Orte in einer Zelle zu dirigieren. Die anschließende Lokalisierung erfolgt über laserangeregte, ortsaufgelöste Detektion der (sichtbaren) Lichtemission der Partikel. Abb. 5: Durchschnitt durch eine zweifach markierte Probe von 3T3 Fibroplasten einer Maus. Bildbreite: 84 µm. Grün fluoreszierende Partikel haben einen Durchmesser von 2nm und sind mit Acetatgruppen modifiziert (aufgrund der Hydrophilie Affinität zum Zellkern). Die größeren (d = 4 nm) Partikel fluoreszieren rot und haben eine Biotin modifizierte Oberfläche (Affinität zu den mit Streptavidin markierten Filamenten). 20 / CdSe Durch Verwendung von unterschiedlich großen Partikeln, die wegen ihrer größenabhängigen, schmalen Emissionsbanden voneinander unterschieden werden können, können simultan mehrere Umgebungen durch Anregung mit nur einer Wellenlänge abgefragt werden wie in Abb. 5 am Beispiel der oben zitierten Arbeit gezeigt ist. Großes Interesse besteht an der Anwendbarkeit von CdSe-Nanopartikeln als Single Electron Transistoren (SET). SETs sind eine mögliche Antwort auf die Frage nach einer Fortführung des empirisch von Moore gefundenen Gesetzes: die Größe von Transistoren verringert sich alle 12-18 Monate um den Faktor zwei. 1997 konnte von der Gruppe um McEuen erstmals ein solcher Einzelelektronen-Transistor mit nasschemisch synthetisierten, ca. 5 nm großen CdSe-Nanokristalliten verwirklicht werden[21, 22] (siehe Abb. 6). Abb. 6: Schematischer Aufbau eines Einzelelektronen-Transistors. Ein CdSe-Nanoteilchen befindet sich zwischen zwei Golddrähten (entnommen aus [22]). Auf eine elektrisch leitende Silicium-Oberfläche, die mit einer isolierenden Schicht SiO2 bedeckt ist, werden zwei Golddrähte so aufgebracht, dass ihre Enden nur etwa 10 nm voneinander entfernt sind. Auf diese Oberfläche wird eine Lösung aus CdSe-Partikeln getropft, deren Oberfläche mit Dithiolen modifiziert wurde. Durch die freie Thiolfunktion binden die Nanokristallite an die Golddrähte. Natürlich wird dadurch die Gesamtheit der Goldoberfläche mit CdSe-Partikeln bedeckt, doch in etwa 1 von 20 Fällen befinden sich einige von ihnen direkt in der Lücke zwischen den Golddrähten, und es ist möglich, den Strom zu messen, der in Abhängigkeit von der Spannung durch die Halbleiter-Nanopartikel fließt. Aufgrund der Coulomb-Blockade und des quantum confinements wird wie erwartet eine treppenartige Strom-Spannungs-Chrarakteristik erhalten, wobei allerdings aus physikalischen Gründen nur die erste Stufe zu erkennen ist. Aufgrund des Anwendungspotentials dieser Halbleiterpartikel besteht seitens der Industrie ein großer Bedarf an sicheren synthetischen Zugängen. Die in Kapitel 3.2 eingeführte Syntheseroute arbeitet jedoch mit Dimethylcadmium, das diesem Anspruch in keinster Weise genügt wie später ausgeführt wird. Die sich an die Etablierung der Synthese anschließende Aufgabe war es daher, mit Hilfe der organometallischen Chemie alternative Precursoren zu finden, die ein sichereres Arbeiten ermöglichen. Die Ansätze und ihre Ergebnisse für die Synthese von CdSe-Nanopartikeln werden in Kapitel 3.3 vorgestellt. Nach der Etablierung und Verbesserung des synthetischen Zugangs zu CdSe-Nanopartikeln sollte der Fokus konsequenterweise auf den zweiten Bereich gerichtet werden, der für die meisten der oben beCdSe / 21 schriebenen Anwendungsfelder relevant ist und in dem die Kenntnisse der Chemie gefordert sind: die Modifizierung der Partikel-Oberfläche. Das Konzept und die dafür ausgewählten Liganden werden zusammen mit den Ergebnissen dieser Arbeit in Kapitel 3.4 beschrieben. 3.2 Etablierung einer Syntheseroute für CdSe-Nanopartikel 3.2.1 Stand der synthetischen Forschung Über einen Forschungszeitraum von inzwischen über einem Jahrzehnt sind eine Vielzahl von Synthesen für CdSe- und CdS-Halbleiterpartikel vorgestellt worden (siehe Tabelle 1). Nanopartikel-Synthesen müssen einer Reihe von Kriterien genügen: Die Formulierung „high quality colloids oder quantum dots“ könnte etwa wie folgt definiert sein: • • • • • Einheitliche, möglichst durch Reaktionsparameter steuerbare und vor allem reproduzierbare Partikelgröße und –form über einen möglichst weiten Bereich Schmale Größenverteilung (Monodispersität) Kristalline Materialien mit wenigen Defekten und Verunreinigungen (insb. für Quantenausbeute bei der Anwendung als optische Bauteile wichtig) Stabilisierung der Nanokristallite gegen Agglomeration Löslichkeit in gebräuchlichen Lösungsmitteln („Kolloide“), d. h. eine Oberfläche mit den gewünschten Eigenschaften • Verwendung von möglichst einfach handhabbaren und ungiftigen Edukten • Unkomplizierte Reaktionsbedingungen und Verfahren Allen Methoden ist gemeinsam, dass das Wachstum der Halbleiter-Partikel auf der Stufe von nanopartikulärem Material beendet wird. Wässrige Methoden gehören zu den am weitesten verbreiteten synthetischen Zugängen, da das Wachstum durch die Nukleationskinetik oder durch Stabilisatoren kontrolliert werden kann. Es handelt sich bei diesen Methoden um eine arrested precipitation (gehinderte Fällung) von Cd2+ mit Se2- (bzw. S2-) in der Gegenwart von Stabilisatoren wie Thiolen, Aminen, Phophinoxiden oder geeigneten Lösungsmittelmolekülen. Dabei kann das Anion sehr elegant in-situ erzeugt werden, indem z. B. Thioharnstoff mit Mikrowellen bestrahlt wird (Bildung von S2-) oder Selen in Tetralin gelöst wird (Bildung von Se2-). Bei der Verwendung von elementarem Selen bzw. Schwefel muss der Lösung zusätzlich ein Reduktionsmittel beigefügt werden. Andere Methoden verwenden begrenzte Reaktionsräume wie sie in reversen Mizellen, Zeolithen oder porösem Aluminiumoxid vorliegen. Reverse Mizellen sind Wasser-in-Öl Tropfen, deren Größe durch das Verhältnis von Wasser zu Tensid bestimmt wird. Das am häufigsten benutzte Tensid ist AOT (Natriumdiethylsulfosuccinat oder Aerosol OT). Innerhalb des polaren Kerns kann Wasser gelöst werden, so dass im Prinzip dieselben Reaktionen wie oben beschrieben ablaufen können. Bei Matrices mit strukturell vorgegebenen Poren wird das Wachstum durch die Porenwände 22 / CdSe gestoppt, wobei die begrenzte Auswahl an Porengrößen die Zugänglichkeit der möglichen Partikelgrößen limitiert. Tabelle 1: Auswahl an Synthesemethoden für II-VI-Halbleiter. Methode Verwendete Precursoren Ligand Literatur [23] Elektrochemische Abscheidung Cd(ClO4)2 2H2O, Se in DMSO Gasphasensynthese Me2Cd, H2Se Pyridin [24] Metathese [Cd(N(SiMe3)2)2], Se=C=NR TOPO [25] Mikrowellenunterstütze Fällung Cd(OAc)2, Thioharnstoff DMF [26] Photoetching Technique CdS Thiophenol [27] Pyrolyse Me2Cd, TBPSe TOPO [28] Pyrolyse Me2Cd, TOPSe TOPO [29] Pyrolyse CdO, Phosphonsäuren, TBPSe TOPO [30] Pyrolyse [Cd(CH2CMe3)2] TOPO [31] Pyrolyse Me2Cd· Bipy TOPO [31] Pyrolyse [Cd(CH2CH2CH2NEt2)2] TOPO [31] Single-Source-Precursor Cd(SCNHNH2)2Cl2 TOPO [32] Single-Source-Precursor Cd[Se2CN-Me(C6H13)]2 TOPO [33] Single-Source-Precursor [Cd(Se2CN(C2H5)2)2]2 4-Ethylpyridin [34] Solventothermal CdC2O4, Se en [35] Solvothermal Cd-Stearat, Se/Tetralin Dodecanthiol [36] CdCl2, Se, KBH4 en [37] Cd(ClO4)2, Se(TMS)2 thf [38] Organosilicat Sol-Gel Matrix Cd-Ethoxyacetat, (Me3Si)2Se Polymer [39] Reverse Mizellen Cd2+, Se(TMS)2 AOT oder Ph [38] Polymer Matrix 2-Pyridylpolybutadien, Me2Cd, H2Se Polymer [40] MCM-41 Matrix Cd2+-Ionenaustausch, H2S [41] MCM-41 Matrix Me2Cd, TBPSe [42] Al2O3 Matrix CdSO4, SeO2, H2SO4 [43] SiO2 Sol-Gel Matrix Cd(OAc)2, SeO2 [44] Matrices Eine der am weitesten verbreiteten Methoden und die Methode der Wahl für die vorliegende Arbeit benutzt die Thermolyse von Metallalkylen in heißen, koordinierenden Lösungsmitteln, im Falle von CdSe die Injektion von Lösungen von Me2Cd und TBPSe in heißes TOPO. Diese Methode wurde ursprünglich von Bawendi[29] und Alivisatos[28] für CdSe entwickelt und führt zu qualitativ hochwertigen Nanopartikeln im Sinne der oben genannten Kriterien, d. h. nahezu einkristallinen Partikel mit einer Größenverteilung < 5 %, die durch die Koordination von TOPO sowohl geschützt sind als auch die Löslichkeit in Solventien wie CdSe / 23 Toluol ermöglichen. Außergewöhnlich hochwertige Partikel entstehen, wenn das koordinierende Lösungsmittel aus einer Mischung von HDA und TOPO besteht.[13] State of the art ist die synthetische Kontrolle über die Partikelmorphologie, bei der die Anisotropie der WurtzitStruktur von CdSe ausgenutzt wird. Bei langsamem Wachstum, d. h. bei einer niedrigen Konzentration an Monomer, ist das Wachstum auf allen Flächen gleich und es entstehen nahezu sphärische, aber immer noch facettierte Partikel. Wird die Wachstumsrate jedoch erhöht, d. h. das System mit einer hohen Monomerkonzentration übersteuert, dann ist das Wachstum entlang der c-Achse höher und das Ergebnis sind stäbchenartige Partikel.[45, 46] Eine Variation der Monomerkonzentration in Kombination mit zusätzlichen Injektionen an Monomer ermöglicht auch die Synthese von Partikeln in Form eines Pfeils oder einer Träne oder von vierarmigen CdSe-Nanokristalliten (tetrapods), von denen einige in Abb. 7 gezeigt sind.[47] Abb. 7: TEM Bilder von CdSe-Nanopartikeln, deren Form gezielt durch die Synthese beeinflusst wurde. Links: Tetrapods, Mitte: Stäbchen; Rechts: pfeilförmige Partikel.[47] Diese Prinzipien können ohne weiteres auf andere Systeme angewendet werden wie z. B. in einer kürzlich veröffentlichen Studie von Jinwoo Cheon[48] an Bleisulfid PbS, das in einer völlig symmetrischen Kochsalz-Struktur kristallisiert. Reaktionsparameter wie Temperatur und Monomerrate bestimmen das Wachstum von zwei unterschiedlichen Oberflächen, der (100)-Oberfläche mit hoher Oberflächenenergie und der (111)-Fläche mit niedriger Energie in Gegenwart von starken Adsorbatmolekülen, so dass sogar die Ausbildung von sternförmigen Partikeln oder isotropen Kuben möglich wird. Andere Beispiele sind Nickel[49] Cobalt[50] oder Indium[51]. Die Synthese von elongierten oder anders geformten CdSe-Nanopartikeln ist jedoch nicht nur von akademischem Interesse: werden 7x60 nm große Stäbchen in ein konjugiertes Polymer, Polyhexylthiophen, eingebettet, entsteht so etwas wie eine Solarzelle aus Plastik.[52] Normalerweise ist die Beweglichkeit von Elektronen in einem Polymer sehr gering. Durch die Kombination des Polymers mit anorganischen Halbleitern wird jedoch die Ladungstransferrate sehr hoch und damit der Ladungsverlust durch Rekombination reduziert. CdSe ist ein solcher Halbleiter, der sich aufgrund seiner (steuerbaren) Bandlücke im sichtbaren Bereich eignet. Aufgrund der nanoskaligen Natur der Lichtabsorption werden Nanopartikel von CdSe verwendet, wobei elongierte Partikel gegenüber sphärischen bevorzugt sind: sie haben einen natürlichen, gerichteten Weg für den elektrischen Transport. Ihre Länge kann an die Dicke 24 / CdSe des Bauteils angepasst werden, die letztendlich die optimale Absorption des einfallenden Lichts bestimmt. Die beiden Komponenten, die zusammen etwa 200 Nanometer dick sind, können wie Farbe auf fast jede beliebige Unterlage aufgetragen werden. Zwar ist der Wirkungsgrad dieser Solarzelle bisher noch sehr gering (2 %), doch sie lässt sich wesentlich einfacher herstellen als herkömmliche anorganische Solarzellen: ohne Hochvakuum und ohne Reinraumbedingungen. 3.2.2 Synthese der Edukte und Vorbereitung der Thermolyse Dimethylcadmium, eine extrem giftige Substanz mit einem hohen Dampfdruck (78 Torr bei 52 °C) dient sowohl bei der Herstellung von Nanopartikeln als auch bei CVD-Prozessen als organometallische Cd-Quelle. Die farblose Flüssigkeit (Schmp. –4 °C) ist thermisch instabil, bildet innerhalb von Minuten elementares Cadmium, wenn sie Tageslicht ausgesetzt wird, raucht bei Luftkontakt und zersetzt sich in Wasser. Auch wenn Me2Cd nicht pyrophor ist, detoniert es doch spontan bei Temperaturen über 90 °C. Es ist für einen Preis von etwa 500 pro 25 g käuflich erwerblich, aber auch leicht synthetisch zugänglich durch die GrignardReaktion von wasserfreiem CdCl2 mit MeMgI in Diethylether. Aufgrund seiner Lewisacidität bildet es mit dem Lösungsmittel das Addukt Me2Cd 2 Et2O, das wegen des Freisetzens von 2 Äquivalenten Ether nicht in die bei hohen Temperaturen durchgeführte Pyrolyse eingesetzt werden kann. Lösemittelfreies bzw. Lösemittel-reduziertes Me2Cd lässt sich auf zwei alternativen Wegen erhalten: durch Abdestillieren des Lösungsmittels bei Temperaturen über der Zersetzung des Addukts (120 °C) oder durch Bildung des kristallinen 2,2’-Bipyridin-Addukts, das bei Erwärmen im Vakuum Dimethylcadmium freisetzt. Der Ethergehalt nach Destillation beträgt etwa 4 % (1H NMR), das nach der Pyrolyse des Bipy-Addukts aufgefangene Me2Cd ist lösemittelfrei. Für die Herstellung der Cd-Stammlösung werden 7 mmol Me2Cd in 10 g trockenem Tri-nbutylphosphin (TBP) gelöst. Das 31P-Signal des Tributylphosphins in d6-Benzol wird bei Zugabe von 0.5 Äq. Me2Cd nicht verschoben. TBP bildet also im Gegensatz zu Diethylether kein Lewis-Säure-Base Addukt mit dem Metallkation aus, so dass Dimethylcadmium in der Vorratslösung unverändert voliegt. Als Selenquelle dient eine Lösung von 5 mmol elementarem Selen in 10 g trockenem TBP, wobei durch Reduktion des Selens der organometallische Precursor Tributylphosphinselenid (TBPSe) entsteht. Im Gegensatz zur Literaturvorschrift wurden die Lösungen getrennt voneinander unter Argon bei 0 °C im Dunklen aufbewahrt und erst kurz vor der Reaktion miteinander vermischt, da die Mischungen wegen der Niederschlagsbildung nicht über einen längeren Zeitraum gelagert werden können. Mittels 31P NMR wurde überprüft, ob sich in Lösung ein Säure-BaseAddukt ausbildet, das dann als Single-Source-Precursor verwendet werden könnte. Die Lösung aus 1 Äq. Me2Cd, 2 Äq. Selen und 3 Äq. TBP zeigte jedoch keine Verschiebung des 31 P-Signals im Vergleich zu TPB-Selenid, so dass man von Me2Cd und TBP-Selenid als organometallischen Precursoren in der Pyrolyse ausgehen kann. CdSe / 25 Als koordinierendes Lösungsmittel diente Tri-n-octylphosphinoxid (TOPO), eine bei Raumtemperatur wachsartige, weiße Substanz mit einem Schmelzpunkt von 50-54 °C. Die Koordination des TOPOs erfolgt über die freien Elektronenpaare des Sauerstoffs an die CdIonen an der Oberfläche der Nanokristallite. Dadurch sind sowohl die Metallkationen elektronisch stabilisiert als auch die Partikel durch die langen Alkylketten des Phosphinoxids sterisch gegen Agglomeration geschützt. Reines TOPO (•97 %) koordiniert relativ schwach an Cd2+. Diese Tatsache limitiert die Konzentration an Precursor in der Pyrolyse auf 0.4 mL Stammlösung pro 6 g TOPO und macht damit eine Synthese von größeren Mengen an CdSeNanopartikeln unhandlich. 3.2.3 Thermolytische Synthese von CdSe-Nanopartikeln Absorption [a. u.] Für die Synthese von hoch kristallinen Partikeln einer bestimmten Größe und Größenverteilung sind definierte Reaktionsparameter ausschlaggebend (siehe Kapitel 2.1). Einer der wichtigsten Parameter sind die exakten hohen Temperaturen bei der Injektion der organometallischen Precursor und während des Wachstums der Partikel. Um reproduzierbare Reaktionen zu ermöglichen, wurde daher mit einem Heizpilz gearbeitet, der durch Temperaturkontrolle mit einem 1/10-DIN Platin-Temperaturfühler über einen taktenden Temperaturregler gesteuert wurde. Die Pyrolyse erfolgt durch Injektion der organometallischen Precursor in TOPO bei 350.0 °C. Dabei ist darauf zu achten, dass die Injektion sehr schnell erfolgt (< 1s) 1 und durch schnelles Rühren eine homogene Vermischung der Reaktionspartner gewährleistet ist. Beendet wird die Reaktion durch Entfernen des Heizpilzes und Abkühlen auf JH-12a λ =524nm JH-12b λ =502nm Raumtemperatur. Abb. 8 zeigt JH-12c λ =495nm JH-12d λ =514nm die UV/VIS-Spektren einer λ =531nm JH-12f Reaktionsreihe, bei der die 0 Reaktionsparameter exakt 400 500 600 700 W ellenlänge [nm] eingehalten wurden. Abb. 8: UV/VIS-Spektren einer Reihe von CdSe-Nanopartikel-Lösungen, hergestellt durch Pyrolyse von 0.4 mL Stammlösung in 6 g TOPO (97 %). Reaktionsparameter: TInjektion = 350.0 °C, t = 1.0 Min. Das Maximum der Absorptionsbande ist wegen seiner Abhängigkeit von der Größe und der Größenverteilung (siehe Kapitel 2.1) ein empfindliches Maß für einen Vergleich der synthetisierten CdSe-Partikel. Trotzdem dieselben Reaktionsparameter eingehalten wurden, wiechen die Maxima der Absoptionskurven voneinander ab. Die Abweichung beträgt 5 %. (Weitere Charakterisierung von CdSe-Nanopartikeln siehe Kapitel 3.3). 26 / CdSe Die so hergestellten Partikel haben ein mittleres Absorptionsmaximum von 520 nm, was einem Durchmesser von etwa 2.8 nm entspricht. Die Größe der Partikel lässt sich durch eine längere Wachstumszeit einfach variieren. Um dies zu testen, wurden über einen Zeitraum von 1 h 20 Proben aus der Reaktionsmischung (Parameter siehe unten) entnommen und im UV/VIS-Spektrometer vermessen (siehe Abb. 9). Nach einem anfänglich starkem Anstieg des UV/VIS-Maximums und damit einem starken Größenwachstum, flacht die Kurve merklich ab und es ist abzusehen, dass sie einem Maximalwert zustrebt. Nach 60 Minuten ist das Absorptionsmaximum von anfänglich 520 nm auf 540 nm gestiegen, was einem Partikelwachstum von 2.8 nm auf etwa 3.2 nm entspricht. Um die Größe merklich zu steigern, wurde der Reaktionsmischung durch Injektion von kleinen Mengen Stammlösung weitere Cd- und SePrecursoren zugeführt (markiert durch Pfeile). Jede Injektion hat einen merklichen Anstieg des Absorptionsmaximums zur Folge und führt nach weiteren 1.5 h (entsprechend 8 0.2 mL = 1.6 mL Stammlösung) zu Partikeln mit λmax = 600 nm. Damit konnte der Durchmesser der Partikel insgesamt von 2.8 auf 4.6 nm erhöht werden. 620 Absorption [nm ] 600 580 560 540 520 0 20 40 60 80 100 120 140 160 Zeit [M in.] Abb. 9: Entwicklung des UV/VIS-Absorptionsmaximums von CdSe-Nanopartikeln während der Synthese. Reaktionsparameter: 4 g TOPO (technisch), 0.7 mL Stammlösung, TInjektion = 350 °C, TWachstum (Soll) = 332 °C. Die Pfeile indizieren eine weitere Injektion von jeweils 0.2 mL Stammlösung in die Reaktionsmischung. In den ersten, oben beschriebenen Synthesen sind pro 6 g TOPO (97 %) 0.2-0.4 mL Stammlösung in die Reaktion eingesetzt worden. Das entspricht, wenn man von einer vollständigen Umsetzung der Precursor in Nanopartikel ausgeht, einer Masse an CdSe in den gecappten Nanopartikeln von etwa 11 mg (für 0.2 mL). Der Reaktionsansatz lässt sich zwar problemlos verdoppeln, doch für eine Synthese von einer (industriell gesehen immer noch kleinen) Menge von 1 g CdSe müssten 20 mL Stammlösung in die 100-fache Menge an TOPO, also 600 g, injiziert werden. Neben dem apparativen Aufwand stellt dies ein erhöhtes Gefahrenpotential und einen übermäßig hohen Anfall von Nebenprodukten (überschüssiges TOPO) dar. Deshalb sollte nach der erfolgreichen Implementierung der CdSe-Synthese versucht werden, die Menge an Nanomaterial pro Reaktion zu erhöhen. Wie in Abb. 10 zu erkennen ist, lässt sich dies nicht durch simples Erhöhen der Precursorkonzentration erreichen. Das UV/VIS-Spektrum der Pyrolyse von 2.4 mL Stammlösung in 4 g TOPO CdSe / 27 (untere Kurve) zeigt nur eine leichte Schulter im Bereich von etwa 650 nm. Dies deutet auf das Entstehen von größeren Nanopartikeln (Lage der Absorptionsbande) mit einer breiten Größenverteilung (Breite der Absorption) hin und lässt sich durch die unzureichende Koordination des Trioctylphosphinoxids an die Oberfläche der Nanopartikel erklären. Erhöht man die Menge an TOPO bei gleichbleibender Menge an Stammlösung (mittlere Kurve), ist eine gute Koordination wieder gewährleistet und man erhält ein UV/VISSpektrum mit einer ausgeprägten und schmalen Bande bei 562 nm. Auf diese Weise lässt sich also keine signifikante Erhöhung der Precursorkonzentration erreichen. Nach mündlichen Informationen durch Prof. A. P. Alivisatos (später publiziert im Zusammenhang mit der Formkontrolle von CdSe Nanokristalliten[45]) kann die Koordination des Lösungsmittels an die Partikel verbessert werden, indem TOPO von technischer Qualität verwendet wird. Der Unterschied zu reinem (97 %) TOPO besteht darin, dass es Verunreinigungen von Alkylphosphonsäuren und Phosphinsäuren enthält, die eine stärkere Koordination zu Cd2+ eingehen. Trägt man dieser Tatsache Rechnung, lässt sich die Menge an verwendetem TOPO erheblich verringern: die Injektion von 20 mL Stammlösung in 40 g TOPO (techn.) ergibt CdSe-Nanopartikel, die eine ähnlich ausgeprägte und schmale Bande im UV/VIS-Spektrum zeigen wie die in 97 % TOPO synthetisierten Proben (Abb. 10, obere Kurve). Absorption [a. u.] 4 3 2 1 JH-29a JH-19f 0 JH-19c 500 600 700 800 W ellenlänge [nm] Abb. 10: UV/VIS-Spektren von CdSe-Nanopartikeln. Unten: 4 g TOPO (97 %), 2.4 mL Stammlösung (JH-19c); Mitte: 25 g TOPO (97 %), 2.4 mL Stammlösung (JH-19f); Oben: 40 g TOPO (technisch), 20 mL Stammlösung (JH-29a). 3.3 Alternative Precursoren für die Synthese von CdSe-Nanopartikeln durch Thermolyse von organometallischen Verbindungen 3.3.1 Einleitung Die Synthese von CdSe-Nanopartikeln durch die Injektion von organometallischen Precursoren in heiße Lösungsmittel muss wegen der Verwendung von Dimethylcadmium, einer flüchtigen und hoch giftigen Verbindung, unter erhöhten Sicherheitsvorkehrungen durch28 / CdSe geführt werden. Zwar lässt sich dies umgehen, wenn man Einkomponeten (Single-Source) Precursoren verwendet, doch diese sind nicht käuflich erwerblich und erfordern den Umgang mit ebenfalls hochgiftigen und flüchtigen Selenverbindungen wie CSe2 während ihrer Synthese. Systematische Studien über die Verwendung von alternativen Cadmium-Quellen sind in der Literatur allerdings rar und es gibt nach wie vor Bedarf an bequemeren präparativen Methoden für die Synthese von CdSe-Partikeln unter Verwendung von einfach zugänglichen Precursoren. Daher wurden eine Reihe von Sauerstoff-freien organometallischen Cd-Verbindungen auf ihre Eignung als alternative und sicherere Precursoren für die Synthese von CdSeNanopartikeln getestet: Dineopentylcadmium (Np2Cd), Bis(3-diethylaminopropyl)cadmium (RN2Cd) und (2,2’-Bipyridin)dimethylcadmium (BipyCd) (s. Schema 4). Me Cd Me Cd Me Et2N Cd Me Cd NEt2 N N Schema 4: Alternative organometallische Cadmium-Quellen für Me2Cd in der Synthese von CdSe-Nanopartikeln durch Pyrolyse in koordinierenden Lösungsmitteln. 3.3.2 Beschreibung und Synthese der Precursoren und ihre Thermolyse in Trioctylphosphinoxid Dineopentylcadmium ist eine Dimethylcadmium vergleichbare Alkyl-Cadmium-Verbindung, die jedoch einige Vorteile gegenüber letzterem aufweist. Der Neopentylrest wird wegen seines sterischen Anspruchs und des tertiären β-C-Atoms, das eine β-Eliminierung verhindert, häufig zur Stabilisierung von Metall-Alkyl-Verbindungen eingesetzt. Dineopentylcadmium hat konsequenterweise einen wesentlich höheren Schmelzpunkt als Me2Cd (40 °C im Vergleich zu –4 °C), kann ohne Zersetzung destilliert werden und ist wesentlich weniger flüchtig (3 Torr bei 78 °C im Vergleich zu 78 Torr). Die erhöhte thermische und photolytische Stabilität vereinfacht sowohl die Lagerung der Verbindung als auch deren Handhabung und die Sicherheitsvorkehrungen. Eine andere Möglichkeit, die Cadmium-Quelle chemisch zu stabilisieren, eröffnet sich durch intramolekulare Addukt-Stabilisierung mit N,N-Dialkylaminopropyl-Resten. Dieser Typ Ligand wird häufig für organometallische Precursoren verwendet, die für die Abscheidung von dünnen Metall- oder Halbleiter-Filmen und -Schichten in der MOCVD (metalorganic chemical vapour deposition) geeignet sind, da sie zu weniger emCdSe / 29 pfindlichen, gegenüber Hydrolyse stabileren und einfacher handhabbaren Verbindungen mit einer guten Abgangsgruppe führen. Bis(3-diethylaminopropyl)cadmium ist im Gegensatz zu Me2Cd ein farbloses Öl mit einer geringen Flüchtigkeit. Es kann ebenfalls ohne Zersetzung destilliert werden, ist aber instabiler gegenüber Licht und Wärme als Dineopentylcadmium. Als dritter Precursor wurde (2,2’-Bipyridin)dimethylcadmium ausgewählt. Das Lewis-Base Addukt ist ein gelber, kristalliner Feststoff (Schmp. 75-78 °C) und immer noch sehr luftempfindlich wie die Farbveränderung von gelb nach weiß (freie Base) innerhalb von Minuten bei Luftkontakt zeigt. Im Vergleich zu den anderen Cadmium-Quellen zeigt es die geringste Flüchtigkeit und ist in Bezug auf Handhabung und Sicherheit der am besten geeignete Precursor. Aus Studien an CdTe OMVPE (organometallic vapor-phase epitaxy) unter Verwendung von Dimethylcadmium und Dimethyltellurid weiß man, dass die Abscheidung des Films über einen radikalischen Mechanismus abläuft. Dabei laufen Reaktionen an der Oberfläche ab, die zu Alkylradikalen in der Gasphase führen. Wenn keine Oberfläche vorhanden ist, wird davon ausgegangen, dass der thermische Zerfall des Dampfes sowohl heterolytisch als auch homolytisch abläuft und verschiedene Produkte wie elementares Cadmium, Alkane (Methan, Ethan) und Alkene (Ethylen) gebildet werden. Da die Cd-C Bindungsdissoziationsenergie D0 nur 281 kJ/mol beträgt, ist es sehr wahrscheinlich, dass auch der erste Schritt nach Injektion des Cd-Precursors bei hohen Temperaturen in Trioctylphosphinoxid der Cd-C-Bindungsbruch ist. Die vorgestellten Precursoren haben alle eine Cd-C-Bindung, manche mit weiteren Liganden, die aber über eine schwächere koordinative Bindung gebunden sind. Dies führte zu der Annahme, dass die Reaktion unabhängig von der Wahl des Precursors ist und eine Vermeidung des Dimethylcadmiums ermöglicht. Für den Vergleich der Precursoren wurde besonderer Wert auf die Konstanz der Reaktionsparameter gelegt, da bereits kleine Abweichungen die Eigenschaften des Produkts verändern (s. oben). In allen Fällen wurden 7 mmol des Precursors in 10 g trockenem TBP gelöst und das Verhältnis Cadmium:Selen konstant gehalten. Die Injektion der Precursor (0.4 mL) erfolgte in 350.0 °C heißes TOPO (4 g, technische Reinheit), das Wachstum erfolgte über 10.0 Minuten bei einer Temperatur von 315 °C. Nach der Aufarbeitung wurden die strukturellen und optischen Eigenschaften der CdSe-Nanopartikel mit Hilfe von UV/VIS, PL, IR, MS und TEM miteinander verglichen. Die Bezeichnungen wurden entsprechend der verwendeten Precursoren gewählt, d. h. Me2Cd-CdSe, Np2Cd-CdSe, (RN)2Cd-CdSe und BipyCdCdSe. 30 / CdSe 3.3.3 Charakterisierung der CdSe-Nanopartikel UV/VIS-Spektroskopie Die Absorptionsspektren der aus der Pyrolyse der vier verschiedenen Cd-Precursor erhaltenen CdSe-Nanopartikel sind in Abb. 11 gezeigt. Alle Absorptionsmaxima sind deutlich blauverschoben im Vergleich zur Absorption des Festkörpers (1.74 eV bzw. 713 nm) und zeigen dadurch eindeutig den Effekt des quantum confinements. Dies ist bereits der erste Hinweis darauf, dass alle Precursor zu Nanopartikeln führen. Da im Falle von Halbleiter-Nanoparti- Absorption [a. u.] keln die Absorption von sichtbarem bzw. ultraviolettem Licht von der Breite der Bandlücke und wegen des quantum confinements demzufolge auch von der Größe der Partikel abhängt, kann man aus der Lage des Absorptionsmaximums sowohl die Bandkante als auch die Größe der Partikel berechnen. 0,5 a b c 0,0 d 400 500 600 W ellenlänge [nm] Abb. 11: UV/VIS-Spektren der verschiedenen Lösungen der Nanopartikel: (a) Me2Cd-CdSe, (b) Np2Cd-CdSe, (c) (RN)2Cd-CdSe und (d) BipyCd-CdSe. Die optische Bandlücke Eg erhält man durch Anpassen der Absorptionsdaten an einen direkten Übergang durch lineare Regression und anschließende Extrapolation (siehe Kapitel 2.3.1). Daraus lässt sich dann mit Hilfe von Gleichung 7[53] die Partikelgröße berechnen. In Tabelle 2 sind die so erhaltenen Daten für die einzelnen Proben zusammengestellt. Man kann erkennen, dass die Lage des Absorptionsmaximums für die verschiedenen Proben leicht variiert. Dies liegt daran, dass es unter den gegebenen apparativen Bedingungen sehr schwierig ist, alle relevanten Reaktionsparameter exakt konstant zu halten: Tabelle 2: Lage des Absorptionsmaximums und der berechneten Werte für die Bandkante und den Durchmesser der verschiedenen CdSe-Nanopartikel-Proben. λmax [nm] Bandkante [nm] Bandkante [eV] Durchmesser [nm] Me2Cd-CdSe 560 617 ± 1 nm 2.01 ± 0.005 3.72 ± 0.03 Np2Cd-CdSe 568 624 1.99 3.87 (RN)2Cd-CdSe 545 602 2.06 3.42 BipyCd-CdSe 567 621 2.00 3.80 CdSe / 31 û( g = h2 8a 2 1 1 + me mh Gleichung 7: Beziehung zwischen ∆Eg (Verschiebung der Bandkante im Vergleich zum Festkörper) und dem Partikeldurchmesser a. me bzw. mh sind die effektiven Massen der Elektronen bzw. Löcher; mh = 0.13 m0, mh = 0.44 m0 (Daten des Festkörpers CdSe) mit m0 = 9.1095 10-31 kg als Elektronenruhemasse. Die ersten Arbeiten mit Me2Cd als Precursor, die in Kapitel 3.2.3 vorgestellt wurden, haben bereits gezeigt, dass die Lage der Absorptionsmaxima trotz gleicher Reaktionsparameter leicht voneinander abweichen. Im Vergleich zu der dort beobachteten Abweichung von 5 % ist die Abweichung der hier vorgestellten Absorptionsmaxima mit 2.5 % wesentlich geringer. Trägt man dieser Tatsache Rechnung, ist es ganz offensichtlich so, dass die Wahl des CdPrecursors keinen Einfluss auf die Größe der CdSe-Nanokristallite hat. Darüberhinaus ist die Halbwertsbreite der Banden für alle vier Spektren vergleichbar, was darauf hindeutet, dass die Größenverteilung der Partikel ähnlich ist. Partikel-Oberfläche Wie in Kapitel 2.1 bereits beschrieben worden ist, wird die Bildung von Nanopartikeln erst durch die Koordination von Liganden an die Atome bzw. Ionen an der Oberfläche der Partikel möglich. Die Koordination eines Liganden wird sich vor allem in der Bindungsordnung der beteiligten Bindung niederschlagen, weswegen (neben NMR, MS u. a.) meist die IRSpektroskopie zur Charakterisierung der Oberfläche herangezogen wird. 90 Transmission [%] a b d 80 c 70 P-O-Streckschwingung -1 (1093 cm ) 60 3000 2000 1000 -1 Frequenz [cm ] Abb. 12: IR-Spektren der verschiedenen Proben, aufgenommen als KBr-Presslinge: (a) Me2Cd-CdSe, (b) Np2Cd-CdSe, (c) (RN)2Cd-CdSe, (d) BipyCd-CdSe Alle IR-Spektren (Abb. 12) zeigen dieselben Absorptionen in Bezug auf Wellenzahl und Intensität und weisen damit darauf hin, dass auch die Oberflächenbedeckung der Partikel unabhängig vom verwendeten Cd-Precursor ist. Die P-O-Streckschwingung des Trioctylphosphinoxids (ν = 1147 cm-1) ist deutlich zu kleinern Wellenzahlen (ν = 1093 cm-1) verschoben. Die Ursache für diese Verschiebung liegt in der Koordination des Sauerstoffs an die Oberfläche der Nanopartikel, was eine Verringerung der Elektronendichte in der benachbarten 32 / CdSe Bindung zwischen Phosphor und Sauerstoff zur Folge hat. Da ν proportional zur Bindungsordnung ist, nimmt somit auch die Absoprtionsfrequenz ab. Im Bereich von 2800 bis 3000 cm-1 sind die C-H-Streckschwingungen der Octylreste des TOPOs zu erkennen, jedoch können keine aromatischen C-H-Streckschwingungen (> 3000 cm-1) detektiert werden, d. h., bei Verwendung von Me2Cd Bipy als Precursor findet keine Koordination durch Bipyridin statt. Demnach besteht die Ligandenhülle der Nanopartikel im Wesentlichen aus TOPO, was auch durch Massenspektrometrie der Proben bestätigt werden konnte: Die FAB-Spektren aller Nanopartikel zeigen ein Signal für TOPO, jedoch keine Signale für 2,2’-Bipyridin oder Diethylpropylamin. Transmissionselektronenmikroskopie (TEM) Die TEM-Aufnahmen der Nanopartikel (Abb. 13) illustrieren ebenfalls die Ähnlichkeit der vier Proben. Die gebildeten Nanopartikel sind einheitlich in Größe und Größenverteilung, sie haben eine sphärische Form und einen mittleren Durchmesser von etwa 4 nm. Dies stimmt mit den Werten überein, die aus der UV/VIS-Absorptionsspektroskopie mit Hilfe von Gleichung 7 erhalten wurden. Trotz des einfachen Modells ist die Übereinstimmung mit den experimentellen Daten erstaunlich gut und legt diese Methode als eine einfache Alternative für die Abschätzung der Partikelgröße nahe. Abb. 13: TEM- und EDX-Aufnahmen der verschiedenen Proben: (a) Me2Cd-CdSe (inset 8.2 nm2), (b) Np2CdCdSe (inset 14.8 nm2), (c) (RN)2Cd-CdSe (inset 16.6 nm2), (d) BipyCd-CdSe (inset 12.1 nm2). Skalierung = 56 nm Die TEM-Aufnahmen in höherer Vergrößerung (siehe Insets von Abb. 13) zeigen für jede Probe eindeutig die (100)- oder (002)-Netzebenen von hexagonalem CdSe und bestätigen damit nicht nur die kristalline Struktur, sondern auch das Vorliegen von CdSe. Letzteres wird durch die EDX-Analyse untermauert (beispielhaft für Np2Cd-CdSe im Inset von Abb. 13(b) CdSe / 33 gezeigt). Die zusätzlichen Signale für P, O, Cu und C stammen von koordiniertem TOPO, dem verwendeten Kupfer-Grid bzw. dem Kohlenstofffilm, mit dem das Grid überzogen ist. 3.4 Oberflächenmodifizierung von TOPO-gecappten CdSeNanopartikeln 3.4.1 Einleitung Neben dem rein wissenschaftlichen Interesse gibt es im Wesentlichen zwei Gründe, die Oberfläche von Nanopartikeln zu modifizieren. Erstens, um eine Affinität zu bestimmten Moleküle zu erreichen: bei der Verwendung von Nanopartikeln als biologische Marker kann die Oberfläche beispielsweise mit Biotin-Molekülen modifiziert werden, damit eine Bindung an Streptavidin-reiche Bereiche einer Zelle erfolgt. Ist ein Nanosensor das Ziel, muss die Oberfläche chemisch so modifiziert sein, dass sie die Zielmoleküle, z. B. DNA, bindet. Der zweite Grund für eine Oberflächenmodifizierung ist die Anordnung von Nanopartikeln in geordnete Strukturen, die für viele technische Anwendungen erforderlich ist. Dafür sind sowohl bifunktionelle Liganden verwendet worden, die an zwei verschiedene Partikel binden können (z. B. Dithiole[54]), als auch Liganden, die in einer nachfolgenden Reaktion mit dem Liganden eines zweiten Partikels reagieren[55]. Das Ergebnis sind Di- und Oligomere oder ein zweibzw. dreidimensionales Netzwerk. Daneben sind Oberflächenmodifizierungen vorgenommen worden, um die chemischen Eigenschaften zu steuern (Löslichkeit in hydro- bzw. lipophilen Lösungsmitteln[56]) oder um die Oberfläche besser zu passivieren (anorganische Hüllen, vor allem für die Steigerung der Quantenausbeute bei Halbleitern[19, 20, 57, 58]). Im Prinzip hängt also die Verwendung von Nanopartikeln, egal in welchem Bereich der Nanotechnologie, ganz entscheidend von der Möglichkeit ab, erstens die chemischen Eigenschaften ihrer Oberfläche gezielt zu modellieren und sie dann zweitens mittels dieser Eigenschaften an spezifische Stellen zu binden. Angenommen, das Ziel sei ein Partikel, dessen Oberfläche mit der Funktionalität B bedeckt ist, dann kann die Modifizierung dieser Oberfläche grundsätzlich auf drei Wegen erfolgen (siehe Schema 5). Da die Liganden erstmals während der Synthese eingeführt werden, kann schon dort durch Wahl eines anderen Liganden, HY-B anstatt HY-A, Einfluss auf die Oberflächenbedeckung genommen werden (Schema 5A). Eine weitere Möglichkeit besteht darin, den primär gebundenen Liganden zu modifizieren. Dies ist in der Regel dann möglich, wenn er eine zweite funktionelle Gruppe aufweist (hier mit A bezeichnet), die den gängigen organischen Reaktionen zugänglich ist (Schema 5B). Die dritte Möglichkeit ist der postsynthetische Austausch. Dies setzt voraus, dass der Ligand, der das Partikelwachstum in der Synthese terminiert, keine starke Bindung zu den Oberflächenatomen ausbildet, sondern möglichst koordinativ oder ionisch gebunden ist. Dann nämlich kann er in einem nachfolgenden, also 34 / CdSe postsynthetischen, Reaktionsschritt durch einen stärker koordinierenden Liganden ausgetauscht werden, typischerweise durch Ausbildung einer kovalenten Bindung (Schema 5C). P re cu rsor + Z u sä tze HX A HY A B A A A A B A A A B B A A A B B A P re cu rsor + Z u sä tze + HX A B B A A P re cu rsor + Z u sä tze + HX A A A A A B A A Z B HY B B B A* A* B B B B A* B B B B B A* A* A* B B A* A* A A A B A B B $ % & Schema 5: Prinzipien der Oberflächenmodifizierung. (A) in der Synthese, (B) Modifizierung des Liganden, (C) Postsynthetischer Ligandaustausch. Die von uns verwendete synthetische Methode liefert bereits hochwertige, kristalline Nanopartikel mit einer schmalen Größenverteilung, die koordinativ mit Trioctylphosphinoxid bedeckt sind. Da bei der Wahl eines neuen Liganden für die Synthese (Weg A) die Reaktionsparameter immer wieder neu optimiert werden müssen und sich die Alkylketten, die durch Verwendung von TOPO die Oberfläche terminieren, nicht für nachfolgende Reaktionen eignen (Weg B), wurde Weg C für eine Oberflächenmodifizierung der CdSe-Nanopartikel ausgewählt. Dieser Weg eignet sich auch deshalb gut, weil TOPO koordinativ an die Oberfläche der Partikel bindet. Dadurch sollte es möglich sein, das Phosphinoxid durch einen Liganden zu ersetzen, der eine kovalente Bindung zu den Cadmiumionen ausbildet, z. B. durch ein Thiol. Für den Austausch des gebundenen TOPOs wurde der neue Ligand so ausgewählt, dass der Austausch analytisch einfach zu verfolgen ist: Bei einem Austausch von Trioctylphosphinoxid gegen 2-Thionaphtol sollte im IR-Spektrum eine Abnahme bzw. ein kompletter Verlust der Alkyl-C-H-Schwingungen des TOPOs (2960, 2926 und 2856 cm-1) CdSe / 35 und eine Zunahme der aromatischen C-H-Schwingungen des Thiols (3054 cm-1) zu beobachten sein. 3.4.2 Austausch von oberflächengebundenem Trioctylphosphinoxid gegen 2Naphtylthiol Die für die Reaktionen (Übersicht siehe Tabelle 3) verwendeten TOPO-gecappten CdSeNanopartikel wurden nach ihrer Synthese standardmäßig gewaschen, um überschüssiges, nicht gebundenes Trioctylphosphinoxid zu entfernen. Analog wurde mit den nach der Austauschreaktion erhaltenen Lösungen bzw. Niederschlägen verfahren, um freigewordenes TOPO bzw. ungebundenes Naphtylthiol abzutrennen. Werden die CdSe-TOPO-Nanopartikel nach dem Waschen in trockenem Pentan aufgenommen und mit einem Überschuss Naphtylthiol versetzt, so lässt sich eine allmähliche Entfärbung der roten Lösung und schließlich die Bildung eines Niederschlags beobachten (Reaktion A). Das IR-Spektrum dieser Partikel (siehe Abb. 14) zeigt in den Bereichen der C-H-Schwingungen und der P-OStreckschwingung Banden bei 2960, 2927 und 1138 cm-1. Die ersten beiden Banden können den C-H-Schwingungen des Trioctylphosphinoxids zugeordnet werden. Tabelle 3: Übersicht über die Reaktionen zum Austausch von TOPO gegen Thionaphtylthiol an CdSeNanopartikeln. Lösungsmittel Temperatur Zeit Thiol A Pentan RT 4d 20 mg Nd, Aufarbeitung B Pentan + 0.1 mL RT 1d 20 mg Nd, Aufarbeitung Pyridin C Pyridin RT 4d 16 mg Lösung, Aufarbeitung D Pyridin 70 °C 15 h - Lösung, Aufarbeitung Pyridin 70 °C 4d 30 mg Lösung, Aufarbeitung D* (nicht in Pyridin 60 °C 15 h 20 mg Lösung, Aufarbeitung Abb. 14) Pyridin 60 °C 15 h 20 mg Lösung, Aufarbeitung E Pyridin 60 °C 1d 18 mg Lösung, Aufarbeitung Dies weist zusammen mit dem Fehlen einer Bande über 3000 cm-1, die bei einer Koordination von Naphtylthiol zu erwarten wäre, darauf hin, dass kein Austausch gegen das Thiol stattgefunden hat. Die dritte Bande (1093 cm-1) stammt von der P-O-Streckschwingung des koordinierten TOPOs. Sie ist sowohl im Vergleich zu den dazugehörigen C-H-Schwingungen als auch im Vergleich zu unbehandelten CdSe-TOPO-Nanopartikeln schwach ausgeprägt, d. h. nur ein Teil der Alkylgruppen stammt von den Octylresten des TOPOs. Becerra et al. haben durch 31P NMR Untersuchungen zeigen können, dass nach der Synthese von CdSeNanopartikeln unter Verwendung von TOPO und TOP (Trioctylphosphin) die Oberfläche 36 / CdSe sowohl mit TOPO als auch mit TOP bedeckt ist, wobei TOPO an Cd2+ koordiniert und TOP als TOP-Se vorliegt [59]. 1000 3000 -1 W ellenlänge [cm ] Abb. 14: Ausschnitte aus den IR-Spektren der oberflächenmodifizierten CdSe-Nanopartikel. Die Reaktionen A bis E sind von oben nach unten dargestellt. Die markierten Wellenlängen entsprechen von links nach rechts 3048, 2960, 2927, 1093 und 1071 cm-1. Für keine der Proben wurden S-H-Schwingungen bei ~ 2600 cm-1 beobachtet. In der von uns verwendeten Synthesevorschrift wird anstelle von Trioctylphosphin Tributylphosphin (TBP) verwendet, das durch Reaktion mit elementarem Selen TBPSe bildet. Diese Spezies kann analog zu TOPSe an der Oberfläche der Partikel vorliegen. Offensichtlich ist also durch die Reaktionsbedingungen ein Teil des TOPOs entfernt worden, ohne dass Thiol diesen Platz eingenommen hat, und die beobachteten C-H-Schwingungen stammen sowohl von TOPO als auch von TBP. Aufgrund der bekannten Säureempfindlichkeit von CdSe-Nanopartikeln[60], die möglicherweise zu dem beobachteten Verlust von TOPO geführt hat, wurde die obige Reaktion zur Aufnahme von frei werdenden H+-Ionen in Gegenwart von 0.1 mL Pyridin durchgeführt (Reaktion B). Wie in der oben beschriebenen Reaktion wird ein Niederschlag gebildet. Die IR-Banden der Partikel unterscheiden sich jedoch leicht: es sind deutliche Banden für die CH-Schwingungen des TOPOs bei 2960 und 2927 cm-1 zu erkennen, zusätzlich ist aber die PO-Streckschwingung bei 1093 cm-1 im Vergleich zu Reaktion A wesentlich ausgeprägter. Demnach wird die Oberfläche auch nach der Reaktion noch von Trioctylphosphinoxid koordiniert. Dies wird durch das Verhalten der Nanopartikel bestätigt: Die gewaschenen Partikel sind im Gegensatz zu denen aus Reaktion A teilweise in Toluol löslich und das UV-Spektrum dieser Lösung weist eine um etwa 10 nm zu höheren Wellenlängen verschobene Absorption auf, d. h. die Partikel sind leicht gewachsen. Demnach ist zwar ein Verlust an koordiniertem CdSe / 37 TOPO eingetreten (und damit ein Verlust an sterischer Stabilisierung, was Niederschlagsbildung und geringfügige Agglomeration zur Folge hat), ein Rest an Löslichkeit kann jedoch erhalten werden. Darüber hinaus deutet sich eine sehr schwache Bande bei 3048 cm-1 an, d. h. es könnte eine geringe Menge Naphtylthiol an die Oberfläche koordiniert haben. Insgesamt lässt sich also die Reaktion in Bezug auf die Stabilität der Partikel durch Zugabe von Pyridin zwar verbessern, aber das Ziel, ein kompletter Austausch, kann auf diese Weise nicht verwirklicht werden. Die Stabilisierung lässt sich auf der einen Seite durch die basischen Eigenschaften des Pyridins erklären, aber es ist zusätzlich auch denkbar, dass Pyridin über das freie Elektronenpaar an die Oberfläche koordiniert: Alivisatos et al.[28] konnten zeigen, dass TOPOgecappte CdSe-Nanopartikel, die aus Pyridin auf einer Oberfläche abgeschieden und im Vakuum getrocknet wurden, in ihren XPS-Spektren keinen Stickstoffpeak und nur noch einen sehr kleinen Anteil an Phosphor aufweisen. Das heißt, ein Großteil des Phosphors kann durch Auflösen in Pyridin entfernt werden und nur ein kleiner Anteil bleibt auf der Oberfläche zurück (die Abwesenheit des Stickstoffpeaks kam durch Entfernen des Pyridins im Vakuum zustande). Darüber hinaus ist aus 1H NMR-Untersuchungen an CdS-Nanopartikeln, die mit Thiophenol und Pyridin gecappt sind, bekannt, dass Pyridin höchstwahrscheinlich mit der Nanokristall-Oberfläche assoziiert ist und rasch ad- und desorbiert (die ortho-, meta-, und para-Resonanzen entsprechen denen des reinen Pyridins).[61] Da Pyridin die Partikel offensichtlich stabilisiert, wurde die Base in Reaktion C als einziges Lösungsmittel verwendet. Während des mehrtägigen Rührens bei Raumtemperatur wird nun im Gegensatz zu Reaktionen A und B im Reaktionsverlauf kein Niederschlag mehr gebildet. Die Partikel können ausgefällt, gewaschen und anschließend in Pyridin oder Chloroform gelöst werden. Das IR des Reaktionsprodukts zeigt sowohl die C-H-Schwingungen des TOPOs bei 2960 und 2927 cm-1 als auch die des Naphtylthiols bei 3048 cm-1. Auf diese Weise kann also erstmals ein zumindest partieller Austausch des TOPOs gegen Naphtylthiol erreicht werden. In Übereinstimmung dazu findet sich im niederfrequenten Bereich des IRSpektrums eine deutliche Bande des Naphtylthiols bei 1071 cm-1. Das Vorliegen von freiem Naphtylthiol durch ungenügendes Waschen kann ausgeschlossen werden, da weder in den Reaktionen A und B, in denen die Aufarbeitung auf analogem Weg erfolgte, C-HSchwingungen des Thiols beobachtet noch S-H-Schwingungen im Bereich ~ 2600 cm-1 detektiert werden konnten. Um das Ausmaß des Austauschs weiter zu steigern, wurden verschiedene Strategien ausgewählt (Reaktionen D, D* und E). Zunächst wurde die Reaktionstemperatur von Raumtemperatur auf 60-70 °C erhöht (Reaktion E). Der Reaktionsverlauf ist analog zu Reaktion C, d. h. es wird kein Niederschlag gebildet. Die im IR-Spektrum sichtbaren CH-Schwingungen des TOPOs (2960 und 2927 cm-1) und des Naphtylthiols (3048 cm-1) belegen, dass auch hier ein Austausch des TOPOs stattgefunden hat. Allerdings ist hier erstmals der Anteil von Naphtylthiol größer als der des TOPOs. Das Verhältnis der Banden bei 1093 cm-1 (TOPO) und 1071 cm-1 (Thionaphtol) bestätigt diesen Befund. Offensichtlich kann also der Anteil an Thio38 / CdSe naphtol auf der Oberfläche der CdSe-Nanopartikel durch eine höhere Temperatur erfolgreich vergrößert werden, wobei die Wärme vermutlich die Dissoziation des koordinativ gebundenen TOPOs von der Oberfläche erleichtert. Unter der Annahme, dass der Austauschprozess möglicherweise durch das nach der Dissoziation in Lösung befindliche TOPO zu einem Gleichgewicht führt, wurde der Austausch sukzessive durchgeführt (Reaktionen D und D*). Dafür wurden die Partikel in einem Zwischenschritt aufgereinigt und ein weiteres Mal in die Austauschreaktion eingesetzt, wobei in Reaktion D das Ziel war, zunächst TOPO gegen Pyridin und dann gegen das Thiol ausgetauscht werden sollte und in Reaktion D* in beiden Reaktionen Thiol anwesend war. Die IR-Spektren der aus diesen Reaktionen erhaltenen CdSe-Nanopartikel sind praktisch identisch (Abb. 14 zeigt daher nur das IR-Spektrum von Reaktion D). In beiden Reaktionen hat ein Austausch stattgefunden, der Anteil an Thionaphtol konnte jedoch im Vergleich zu Reaktion E nicht gesteigert werden. Die Partikel aus der im Sinne eines kompletten Austauschs erfolgreichsten Reaktion E wurden neben der IR-Spektroskopie weiteren analytischen Methoden unterzogen. Vergleicht man das UV/VIS-Spektrum der Edukt-Partikel (CdSe-TOPO) mit den nach dem Austausch erhaltenen Partikeln (CdSe-Naphtylthiol/TOPO) (Abb. 15), so erkennt man, dass das Absorptionsmaximum zu etwas höheren Wellenlängen verschoben und die Bande verbreitert worden ist. D. h., die Partikel sind während der Reaktion minimal gewachsen und haben eine breitere Größenverteilung bekommen. Die dazu gehörigen elektronenmikroskopischen Aufnahmen zeigen zunächst, dass sowohl vor als auch nach der Reaktion Nanopartikel mit einem Durchmesser von etwa 3 nm vorliegen. Sie bestätigen außerdem den Befund der UV/VIS-Spektren, verdeutlichen aber durch ihre Ähnlichkeit auch, dass dieser Effekt sehr gering ist. 4 Absorption [a. u.] 3 2 CdSe-TOPO CdSe-Naphtylthiol/TOPO 1 0 500 600 700 CdSe-TOPO CdSe-SNaph W ellenlänge [nm] Abb. 15: UV/VIS-Spektren von CdSe-TOPO Nanopartikeln vor (oberer Graph bzw. linkes TEM-Bild, bar = 30 nm) und nach (unterer Graph bzw. rechtes TEM-Bild, bar = 27 nm) der Oberflächen-Modifizierung unter den Reaktionsbedingungen E. CdSe / 39 3.5 Zusammenfassung Nach der Etablierung der Synthese von CdSe-Nanopartikeln durch die Thermolyse von Me2Cd und TBPSe konnte gezeigt werden, dass Me2Cd als die am häufigsten in der CdSeSynthese verwendete Cd-Quelle erfolgreich durch andere organometallische Precursor ausgetauscht werden kann. Die unter Verwendung von Dineopentylcadmium, Bis(3-diethylaminopropyl)cadmium und (2,2’-Bipyridin)dimethylcadmium hergestellten TOPO-bedeckten CdSe-Nanopartikel zeigen keinerlei Qualitätsverlust im Vergleich zur Standardreaktion. Weder die Größe der Partikel noch ihre Größenverteilung, Oberfläche oder Kristallinität hängen von der Cd-Quelle ab. Dies ermöglicht das Arbeiten mit weniger giftigen, stabileren und kristallinen Cd-Precursoren. Insbesondere (2,2’-Bipyridin)dimethylcadmium ist einfach zugänglich und am besten für die Substitution von Dimethylcadmium geeignet. Kurz nach Abschluss dieser Arbeiten erschien eine Arbeit von Peng et al., in denen die Bildung von qualitativ hochwertigen CdSe-Nanopartikeln unter Verwendung des maximal einfachen Precursors CdO beschrieben wird. Die in-situ Reaktion des Cadmiumoxids mit Hexylphosphonsäure (HPA) oder Tetradecylphosphonsäure (TDPA) führt zur intermediären Bildung eines HPA/TDPA-Cadmium-Komplexes, der mit dem durch Injektion zugeführten Selenprecursor zu nahezu monodispersen CdSe-Partikeln und –Stäbchen reagiert. Die Reaktion ist durch eine langsame Nukleation nicht nur gut reproduzierbar, sondern auch bei niedrigeren Temperaturen durchführbar und hängt darüber hinaus nicht von der Injektionsdauer ab. Diese Methode erfüllt somit alle Kriterien für eine einfache Synthese von hochwertigen CdSe-Nanopartikeln und wird von unseren Ergebnissen natürlich nicht übertroffen. Dennoch stellen die hier durchgeführten Arbeiten einen wichtigen Beitrag zur Variation der organometallischen Cd-Quelle dar und sind darüber hinaus für die Substitution von Me2Cd in CVD-Prozessen (insbesondere für die Gasphasensynthese von CdSe-Partikeln) von Bedeutung, in denen CdO wegen seiner geringen Flüchtigkeit nicht als Precursor verwendet werden kann. Die Versuche zum Austausch der nach der Reaktion an die Oberfläche der Nanokristallite koordinierten TOPO-Moleküle haben gezeigt, dass ein kompletter Austausch der Oberfläche weder durch erhöhte Temperaturen noch durch wiederholten, sukzessiven Austausch noch über die Zwischenstufe des schwach koordinierenden Pyridins erreicht werden kann. Von ähnlichen Ergebnissen ist in der Literatur bereits berichtet worden. ES-MS Untersuchungen von Bowmaker[62] zum Ligandaustausch an dem definierten CdS Nanocluster [S4Cd17(SPh)28]2- haben gezeigt, dass in fast allen Fällen nur ein partieller Austausch der Thiophenolatliganden gegen andere Thiole, Dithiole oder Thioalkohole stattfindet. Darüber hinaus entstehen keine Cluster mit einer festen Anzahl n an ausgetauschten Liganden, sondern immer eine Verteilung [S4Cd17(SPh)28-n(SR)n]2- mit einem Wertebereich für n. Bawendi et al.[63] berichten, dass sie die Oberfläche von CdSe-TOPO-Nanopartikeln nicht komplett gegen 4-Picolin, 4-(trifluoromethyl)thiophenol oder Tris(2-Ethylhexyl)phosphat austauschen konnten: die Festkörper NMR-Spektren aller Proben zeigten restliche Alkylprotonen des 40 / CdSe TOPOs. Der Anteil der ursprünglich koordinierten Liganden wurde aufgrund der NMRIntensitäten mit 10-15 % angegeben. In Kapitel 3.4.2 wurde außerdem bereits auf den Restgehalt an Phosphor in SAMs von CdSe-Nanopartikeln verwiesen. Die Gruppe um Brus konnte dieses Ergebnis bestätigen und zudem den Anteil an TOPO nach Ligandaustausch mit Pyridin auf 11 % angeben.[64] Der komplette Austausch der Oberfläche ist aufgrund von Resten an sehr fest gebundenem TOPO und möglicherweise aufgrund von weiteren Oberflächenspezies, die neben Cd←O=PR3 vorliegen, nicht möglich. Eine plausible Spezies ist das aus der Literatur bekannte Selenid Se=PR3. Dennoch konnten wir zeigen, dass man erstens eine Base benötigt, um bei der Koordination von Thiol freiwerdende Protonen zu binden und zweitens höhere Temperaturen, um eine Dissoziation des TOPOs von der Oberfläche zu erreichen. Aufgrund dieser Ergebnisse wurde in Erwägung gezogen, vergleichende Austausch-Experimente durchzuführen, um eine Aussage darüber machen zu können, mit welchen Liganden wieviel der Oberfläche modifiziert werden kann und welchen Einfluss die Größe der Partikel auf den Austausch hat. Die Voraussetzung für solche Experimente ist allerdings, dass die EduktPartikel immer die gleiche Größe und Größenverteilung aufweisen müssen. Eine exakte Reproduktion dieser Eigenschaften ist jedoch nicht möglich (siehe Ergebnisse aus Kapitel 3.2.3) und daher wurde dieser Ansatz nicht weiter verfolgt. CdSe / 41 4 Zinkoxid-Nanopartikel 4.1 Stand der Forschung und Aufgabenstellung Wenn Zink-Aquaionen durch Hydroxidionen in wässriger Lösung gefällt werden, wird Zinkhydroxid gebildet, das im kolloidalen Zustand nicht dehydratisiert werden kann. Die freie Bildungsenthalpie lässt sich unter Verwendung der thermodynamischen Daten der beteiligten Substanzen gemäß Zn(OH)2 (s) → ZnO (s) + H2O (l) zu ∆Gf0=-0.39 kJ/mol berechnen. D. h. die Dehydratisierung von Zn(OH)2 in Wasser ist praktisch thermoneutral bei Raumtemperatur, weswegen die Synthese von ZnO-Nanopartikeln in nicht-wässrigen Medien durchgeführt werden muss. 1985 gelang Henglein und Mitarbeitern[65] erstmals die Synthese von ZnOKolloiden durch die Fällung von Zn(ClO4)2 mit NaOH in Methanol bzw. i-PrOH unter Ausnutzung der dehydratisierenden Wirkung von Alkoholen. Es entstanden sehr kleine Partikel (Ø< 2.5 nm), die durch kleine Mengen an Polyphosphat stabilisiert wurden. Zwei Jahre später berichteten Bahnemann et al.[66] über die Synthese von ZnO-Kolloiden aus Zinkacetat in 2Propanol, Wasser und Acetonitril nach Zugabe von NaOH. Durch die Verwendung von Zinkacetat wird das Acetatanion als Ligand für die ZnO-Oberfläche direkt in die Synthese eingeführt. Verwendet man andere Salze wie Zinkperchlorat, Zinkchlorid oder Zinknitrat wird ZnO in Form von Aggregaten gebildet, da keine geeigneten Stabilisatoren für die Oberfläche anwesend sind. Dies ist seitdem eine weit verbreitete Methode, die mit leichten Modifizierungen[67-70] standardmäßig für die Herstellung von ZnO-Kolloiden zu physikalischen Untersuchungen verwendet wird. Neben Acetatgruppen sind Stabilisatoren wie Hexametaphosphat[65], Propionsäure[69], Thiole[71] oder Octadecylphosphonsäure[71] verwendet worden. ZnO ist ein Standardmaterial in der Halbleiter-Elektrochemie und Photochemie, aber auch in der Katalyse. Außerdem wurden ZnO-Dispersionen benutzt, um chemische Reaktionen mit Licht zu katalysieren (Photokatalyse). Dabei wird von der Bandlücke des ZnO Gebrauch gemacht, deren Größe wie oben beschrieben im UV-Bereich liegt. Die aufgenommene Energie kann dann in einem zweiten Schritt an weitere Moleküle abgegeben werden. Auf diese Weise kann z. B. Cu(II) in Lösung zu metallischem Kupfer reduziert werden[72] oder metallisches Silber aus einer AgNO3-Lösung abgeschieden werden[73] oder H2O2 aus einer mit Sauerstoff gesättigten wässrigen Dispersion von ZnO synthetisiert werden[74]. Die physikalischen Eigenschaften von ZnO-Kolloiden sind in der Literatur eingehend untersucht worden.[65-69, 75-81] Wie CdSe ist auch ZnO ein Halbleiter, dessen Ladungsträger bei abnehmender Partikelgröße eingeengt werden (quantum confinement). Die Folge sind energiereichere Excitonen und eine größere Bandlücke im Vergleich zum Festkörper: Die Absorption von FK-ZnO beginnt bei 372 nm bzw. 3.3 eV[67], kleine ZnO-Partikel absorbieren hingegen bei etwa 310 nm. Die Fluoreszenz von ZnO sollte demnach im UV-Bereich liegen, 42 / ZnO insbesondere angesichts der Anwendungsfelder für Zinkoxid, doch die ersten Kolloide[65] zeigten blau-grüne Fluoreszenz bei λ = 460 nm, die sich durch Alterungsprozesse noch zu grün und schließlich gelb-grün (540 nm) verschiebt und nur eine schwache Emission im Bereich der Bandkante (λ=370 nm). Die beiden Emissionen haben unterschiedliche Lebenszeiten von τ = 10 ns für die sichtbare Emission und τ < 1 ns für die UV-Emission und die geringe Quantenausbeute lässt außerdem darauf schließen, dass die Emission durch strahlungslose Rekombination der Ladungsträger begleitet wird.[66] Da die Intensität der sichtbaren Emission durch Zugabe von Zn2+-Ionen erhöht wird[65] bzw. Anionen wie Iodid, Thiocyanat oder Sulfid diese quenchen[76] geht man davon aus, dass die sichtbare Emission durch Anionen-Vakanzen verursacht wird. Man nimmt an, dass Anionen-Vakanzen V02+ Elektronen aus dem Valenzaband aufnehmen können, wobei V0+ und ein Loch h+ entstehen. V0+-Niveaux fungieren als deep traps für die Leitungsbandelektronen und erhöhen damit die Wellenlänge des emittierten Lichts vom UV in den sichtbaren Bereich. Die Dominanz der sichtbaren Emission gegenüber der wünschenswerten UV-Emission wird für nahezu alle ZnO-Kolloide beschrieben, insbesondere solche, die durch Fällung hergestellt wurden und mit Acetatgruppen stabilisiert werden. Dies gilt ausdrücklich nur für Kolloide, denn von auf Saphir gewachsenen nanowires aus ZnO wurde kürzlich über die Anwendung als UV-Nanolaser berichtet.[82] Zhou et al. konnten zeigen, dass mit Acetatgruppen gecappte ZnO-Partikel aus einem ZnO Kern und einer Zn(OH)2 Hülle bestehen.[83, 84] Sie vermuten, dass die schwache Bandgapemission mit der Anwesenheit von Zinkhydroxid zusammenhängt, da Annealing (T > 125 °C), das die Zersetzung von Zn(OH)2 auf der Oberfläche zu ZnO bewirkt, zu einer verstärkten UV-Emission führt. Dieser Vermutung wird dadurch verstärkt, dass ZnO-Kolloide mit einer verringerten OH-Bedeckung, die durch die oben beschriebene Fällungsmethode unter Verwendung von Polyvinylpyrrolidon als Oberflächenligand hergestellt wurden, ebenfalls eine wesentlich verstärkte UV-Lumineszenz zeigen.[81, 85, 86] Insgesamt konzentrieren sich trotz zahlreicher Veröffentlichungen über Zinkoxid-Kolloide die meisten auf eine einzige Methode, und zwar die Fällung von Zink(II) Salzen mit Alkalihydroxiden. Nur wenige Gruppen berichten über alternative Methoden, die intrinsisch die Bildung von Zinkhydroxid auf der Oberfläche bzw. Sauerstoffdefizienz zu vermeiden suchen, wie zum Beispiel die elektrochemische Synthese in Acetonitril/THF mit Tetraoctylammoniumbromid (TOAB) sowohl als Elektrolyt als auch als Ligand.[87] Obwohl bekannt ist, dass sich organometallische Precursor hervorragend für die Synthese von sowohl metallischen[50, 51, 88, 89] als auch nicht-metallischen[28, 59] Nanopartikeln in nicht-wässrigen Medien eignen, gibt es nur drei Veröffentlichungen, die davon Gebrauch machen: (1) die Hydrolyse von Zinkbuthanolat in reversen Mizellen[90] (2) die Thermolyse von Diethylzink in TOPO[91] und (3) die Oxidation von Zinknanopartikeln, die durch Pyrolyse von [Zn(C6H11)2] hergestellt wurden[92]. Methode (1) leidet unter Schwierigkeiten bei der Aufarbeitung der Additive. Bei Methode (2) wird Sauerstoff durch Einleiten von O2 in das Lösungsmittel für Diethylzink ZnO / 43 zugeführt, wobei die entstehenden Partikel vermutlich aufgrund von Sauerstoffdefizienz hauptsächlich sichtbar emittieren. Methode (3) liefert qualitativ hochwertige ZnO-Nanokristallite, wenn auch methodisch etwas unbefriedigend in zwei Schritten. Über einen definitiv nicht-hydrolytischen Weg zu ZnO-Kolloiden unter Verwendung eines maßgeschneiderten Einkomponentenprecusors (single-source precursor, SSP) ist bisher nicht berichtet worden. Ziel dieser Arbeit war es daher, unter bekannten organometallischen Zn-OVerbindungen, die unter anderem im Arbeitskreis von Prof. M. Drieß hergestellt werden, einen geeigneten SSP auszuwählen und ihn unter optimierten Bedingungen zu ZnO-Kolloiden umzusetzen. 4.2 Synthese von ZnO-Nanopartikeln durch Thermolyse von organometallischen Precursoren 4.2.1 Auswahl der Precursor In der Arbeitsgruppe Drieß der Ruhr-Universität Bochum wurden drei verschiedene Zinksiloxide 1-3 hergestellt (siehe Schema 6). Me3Si Me O Me Zn Zn O SiMe3 Me Zn O O Me 1 SiMe3 Me3Si SiMe3 O I O Zn Zn Zn O O SiMe3 SiMe3 I O I Zn O SiMe3 Zn O n Zn Me3Si SiMe3 2 O Me3Si SiMe3 Zn 3 I Schema 6: Zinksiloxide als mögliche Precursoren für die Synthese von ZnO-Kolloiden Das Konzept für diese Art von Precursoren ist die Verwendung von Me3Si- bzw. Me3SiOals leicht flüchtige, chemisch inerte Abgangsgruppen. Sie stabilisieren einerseits die oligomere Struktur der Verbindungen und erhöhen auf der anderen Seite sowohl die Löslichkeit in organischen Solventien als auch die Flüchtigkeit der entsprechenden Verbindung. 1 und 2 sind über eine Brønsted Säure/Base Reaktion zwischen Dimethylzink und HOSiMe3 zugänglich, 3 kann durch die Reaktion von 1 mit Iod erhalten werden.[93] Aus der Festkörper-Pyrolyse von 1[94] ist bekannt, dass es zwei verschiedene, klar getrennte und erstaunlich saubere Reaktionskanäle gibt: bei niedrigen Temperaturen (160 °C) entsteht wie erwartet ZnO (Weg A in Schema 7), erhöht man die Temperatur jedoch auf etwa 300 °C, ist die Bildung von elementarem Zink bevorzugt (Weg B in Schema 7). Obwohl nicht klar ist, ob der Zerfall sowohl nach A als auch nach B uni- oder höher molekular verläuft, ist jedoch in jedem Fall eine formale 1,2-Eliminierung von SiMe4 gegenüber einer 1,1-Eliminierung von Me3SiOMe kinetisch bevorzugt. Das stimmt gut mit der Hypothese überein, dass der Abbau des Cubangerüsts, wie er im Falle einer 1,1 Eliminierung (Weg A) stattfinden muss, sehr 44 / ZnO wahrscheinlich eine höhere Aktivierungsenergie benötigt. Außerdem müssen drei Zn-O-Bindungen gebrochen werden, im Gegensatz zu nur Zn-C und Si-O auf Weg B. Me3Si Me Zn A ZnO Me O Me - Me4Si Zn O Zn O O SiMe3 B SiMe3 - Me3SiOMe Zn Zn Me3Si Me 1 Schema 7: Zerfallswege für [MeZnOSiMe3]4 1. Die polymere Verbindung [Zn(OSiMe3)2]n (2) verhält sich ähnlich: bei langsamem Erwärmen auf moderate Temperaturen (80 - 230 °C innerhalb von 7 h) wird ZnO gebildet, während schnelles Erhitzen zur Bildung von elementarem Zink führt. Durch Verfolgen des Zerfallsprozesses mit MS weiß man, dass [MeZnOSiMe3]4 und (Me2SiO)n entstehen, und es wird folglich angenommen, dass der Zerfallsprozess von 2 über das tetramere Zinksiloxid 1 verläuft (siehe Schema 8). Aufgrund der intermediären Bildung von 1 bietet Verbindung 2 daher keinen Vorteil gegenüber 1, zumal die Verbindung eine sehr schlechte Löslichkeit in organischen Solventien aufweist und sich daher schlecht für die Thermolyse in heißen, koordinierenden Lösungsmitteln eignet. SiMe3 SiMe3 O O ∆ Zn Zn O - (Me2SiO)n O [(MeZnOSiMe3)4] SiMe3 - Me3SiOMe B n SiMe3 Zn A 1 - Me4Si ZnO 2 Schema 8: Zerfallsweg von [Zn(OSiMe3)2]n 2. Eine thermogravimetrische Analyse des Iod-substituierten Kubans 3 schließlich zeigte, dass sich die Verbindung in einem zweistufigen Prozess zu ZnO zersetzt, wobei der größte Massenverlust bei Temperaturen zwischen 320 und 400 °C auftritt. Für eine Thermolyse in einem Lösungsmittel ist diese Zerfallstemperatur eindeutig zu hoch, weswegen sich Verbindung 3 als Precursor nicht eignet. Aus den Untersuchungen zur Festkörper-Pyrolyse der Verbindungen 1-3 wird klar, dass sich nur eine der drei möglichen Verbindungen, nämlich Kuban 1, für eine nass-chemische Thermolyse zu ZnO-Kolloiden eignet. Darüber hinaus haben wir einen weiteren Zinkoxid-SSP untersucht, der sich aus der Systematik der in den Kapiteln zu Kupfer, Nickel und Palladium beschriebenen Precursor ergeben hat, nämlich [Zn(OCH(Me)CH2NMe2)2] 4 (Schema 9). Obwohl diese Art von Precursor ursprünglich entworfen wurde, um das zentrale Metallatom durch die Koordination von ZnO / 45 Reduktionsmitteln, nämlich HO-CH(Me)CH2NMe2, von der Oxidationsstufe +II in den elementaren Zustand zu reduzieren, ist es interessant zu wissen, ob Metalle, die in der Spannungsreihe unter den unedlen Metallen zu finden sind, ebenso reagieren oder ob nicht vielmehr die Affinität für Sauerstoff den Zerfallsprozess dominiert. In den folgenden Unterkapiteln werden die Thermolysen der Verbindungen 1 und 4 in heißen koordinierenden Lösungsmitteln sowie ihre Analytik beschrieben. Me3Si Me O Me Zn Zn O SiMe3 Me Zn O O Zn Me3Si Me2 N O Zn SiMe3 O N Me2 Me 1 4 Schema 9: Untersuchte SSP aus den Reihen [MeZnOSiMe3]n und M(OR)2. 4.2.2 Thermolyse von [MeZnOSiMe3]4 bei niedrigen Temperaturen [MeZnOSiMe3]4 muss sublimiert oder umkristallisiert verwendet und unbedingt mit MS und H NMR auf Reinheit überprüft werden, da die Ergebnisse der Pyrolysen sonst nicht reproduzierbar sind. Die Synthese der ZnO-Kolloide erfolgte durch Lösen des Precursors in n-Octylamin und Injektion dieser Lösung in 160 °C heißes Hexadecylamin. Nach einer Wachstumszeit von 30 Minuten ließ man die Reaktionsmischung auf Raumtemperatur abkühlen, arbeitete die Partikel durch Waschen mit Methanol auf und löste sie schließlich in Toluol. Die klare, farblose Lösung ist über Wochen stabil, d. h. es ist keine Bildung eines Niederschlags zu beobachten. Das UV/VIS-Spektrum der in Toluol gelösten Partikel (Abb. 16) zeigt ein lokales Maximum bei einer Wellenlänge von λ = 310 nm (4.0 eV). Der onset ergibt sich durch Extrapolieren des steilen Teils des Anstiegs zu 340 nm (3.65 eV) und liegt damit eindeutig bei niedrigeren Wellenlängen als der von makrokristallinem ZnO (372 nm bzw. 3.3 eV). Die Blauverschiebung von 32 nm spiegelt einen Anstieg der Bandlücke wieder und kann dem Auftreten von quantum confinement Effekten zugeschrieben werden. Da diese Effekte bei ZnO nur für Partikel mit einem Durchmesser ≤ 6 nm beobachtet werden[65], kann aus dem UV/VIS Spektrum bereits auf das Vorhandensein von nanoskaligem Material geschlossen werden. Da die Größe der Partikel auch die Größe der Bandlücke und damit die Lage der Absorptionsbande bestimmt, kann man umgekehrt aus dem onset der Kurve die Partikelgröße berechnen. Im Fall von ZnO ergibt sich unter Verwendung von Gleichung 7 eine Partikelgröße von 2.4 nm, was deutlich unter dem oben genannten Wert für den Bohrschen Radius liegt. Obwohl sowohl die hier verwendete Methode für die Bestimmung der Bandlücke[Trindade, 1997 #287] als auch die Verwendung von Gleichung 7[Winkler, 1999 #74][Éfros, 1982 #285][Weller, 1993 #286] publiziert worden sind, hat einer der Referees unserer Veröffentlichung[Hambrock, 2002 #56] ihre Gültigkeit angezweifelt. Die Existenz 1 46 / ZnO eines quantum confinements lässt sich jedoch auch unabhängig davon nachweisen: erstens ist das Absorptionsmaximum unserer kolloidalen Lösung (λ = 310 nm) im Vergleich zur Absorption von makrokristallinem ZnO (λ § QP GHXWOLFK EODXYHUVFKREHQ XQG ]ZHLWHQV wird für alle bereits publizierten ZnO-Nanopartikel im Bereich des quantum confinements (≤ 6nm) ein Absorptionsmaximum im Bereich von 300-325 nm beobachtet. 1,5 4x10 5 382 nm (3.18 eV) 3x10 1,0 0,5 2x10 1x10 5 5 5 Intensität [cps] Absorption [a. u.] Fluoreszenz Absorption 300 350 400 450 500 550 0 600 Wellenlänge [nm] Abb. 16: Links: UV/VIS Spektrum (links) und Photolumineszenz (rechts, Anregung bei 250 nm) von gewaschenen ZnO-Kolloiden in Toluol, hergestellt durch Pyrolyse von 1 in HDA. Rechts: Zum Vergleich dieselben Spektren von ZnO-Kolloiden, die durch Fällung von Zn(OAc)2 mit NaOH ohne bzw. mit PVP (Polyvinylpyrrolidon) erhalten wurden.[85] Wie in Kapitel 4.1 beschrieben, besitzt nanokristallines ZnO ein charakteristisches Fluoreszenzspektrum mit einem Maxima im sichtbaren und einem zweiten im UV-Bereich. Das Emissionsspektrum der kolloidalen ZnO-Lösung in Abb. 16 zeigt eine breite Emission mit einem Maximum bei 382 nm (3.18 eV), das der Bandkantenemission zugeordnet werden kann. Im Vergleich zur Absorption ist die Emission zu höheren Wellenlängen verschoben (Stokes Shift) und shiftet darüber hinaus zu höheren Wellenlängen mit steigender Anregungswellenlänge. Diese Charakteristika können einer breiten Partikelverteilung (siehe Diskussion TEM) bzw. Defektniveaus des ZnO (siehe SAED, XRD und Kapitel 4.1) zugeordnet werden, da angenommen wird (siehe z. B. [68] bzw. Kapitel 2.3.2), dass Lichtabsorption ElektronenLoch-Paare erzeugt, die sehr schnell in shallow und deep traps eingefangen werden. Diese Ladungsträger können zueinander tunneln und dabei entweder strahlungslos unter Kopplung zu Gitterphononen oder strahlend unter Emission eines Lichtquants rekombinieren. Elektronen-Loch Paare in shallow traps emittieren dabei Photonen mit einer höheren Wellenlänge als solche in deep traps. Je breiter nun die Größenverteilung der Partikel ist, desto breiter ist auch die Verteilung der traps und damit auch der Emission. Zum Vergleich ist in Abb. 16 rechts ein Spektrum aus der Literatur abgebildet. Man kann deutlich erkennen, dass die mit Acetat bedeckten ZnO-Partikel eine breite Emissionsbande im sichtbaren Bereich zeigen, während die Anwesenheit von PVP eine bessere Oberflächenpassivierung und damit eine erhöhte Emission im UV-Bereich ermöglicht. Letzteres dürfte der state of the art für ZnO-Kolloide sein. Die Emissionsbande der von uns hergestellten Kolloide liegt damit zwar ZnO / 47 im Bereich der PVP-modifizierten Kolloide von Yang et al., weist aber eine wesentlich größere Halbwertsbreite auf. Die TEM-Aufnahmen, die in Abb. 17a gezeigt sind, lassen separierte, sphärische Partikel mit einem Durchmesser von 2-3 nm erkennen. Die Größe stimmt sehr gut mit dem Durchmesser überein, der aus den Daten des Absorptionsspektrums erhalten wurde. Allerdings ist der Kontrast sehr gering und die entsprechende SAED zeigt nur schwache und diffuse Reflexe. Es ist daher anzunehmen, dass die Kristallinität der ZnO-Kolloide niedrig ist, was aufgrund des Emissionsverhaltens bereits vermutet wurde. Abb. 17: TEM-Aufnahmen von ZnO-Nanopartikeln in Toluol, erhalten durch die Pyrolyse von 1 in HDA. (a) nach Aufarbeitung mit Methanol, Balken = 66 nm; (b) nach Trocknen bei 80 °C, Balken = 117 nm. 4 2,0x10 4 1,5x10 4 1,0x10 4 5,0x10 3 (103) (110) (102) (200, 112, 201) (100) (002) Intensity [a. u.] (101) 2,5x10 0,0 20 30 40 50 60 70 2 Theta [°] Abb. 18: Pulver XRD von ZnO Nanopartikeln nach Trocknen der gewaschenen Partikel bei 80 °C. Die Partikel entstanden durch Pyrolyse von 1 in HDA. Proben für die Röntgenbeugung wurden durch Trocknen der Partikel bei 80 °C im Trockenschrank unter Normaldruck erhalten. Das Beugungsmuster (siehe Abb. 18) des so erhaltenen Pulvers entspricht hexagonalen ZnO-Partikeln mit einer Größe von etwa 8 nm, die sich aus der Halbwertsbreite der Reflexe über die Scherrer-Gleichung abschätzen lässt. Das Anwachsen der Partikelgröße ist unerwartet, da sowohl Spanhel[68] als auch Eychmüller[95] 48 / ZnO berichtete, dass Änderungen von Oberflächenkomposition und Teilchendurchmesser erst bei einer Temperatur über 300 °C beobachtet wurden. Wir nehmen an, dass unsere davon abweichende Beobachtung durch die Verwendung eines anderen Liganden stammt. Im Gegensatz zu den aufgrund von ionischen Wechselwirkungen fest bindenden Acetatgruppen wie sie in Eychmüllers Arbeiten verwendet wurden, bindet HDA nur schwach durch koordinative Wechselwirkung an die ZnO-Oberfläche unserer Partikel und ermöglicht dadurch wesentlich leichter Änderungen in Größe und Kristallinität der Partikel. Wie später ausgeführt wird (Seite 49) ist anzunehmen, dass die Partikeloberfläche auch nach dem Trocknungsprozess mit HDA oder anderen hydrophoben Resten bedeckt ist, da sich das Pulver komplett in Toluol auflösen lässt. Die TEM-Aufnahmen dieser Lösung (Abb. 17b) zeigen Partikel mit einer Größe von 7-8 nm, was gut mit der Abschätzung aus der Röntgenbeugung übereinstimmt. Im dazugehörigen SAED sind klare Beugungsringe zu erkennen, die (von unten nach oben) den Gitterebenen (100), (002/101), (102), (110), (103) und (112) von Zinkoxid entsprechen. Der erhöhte Kontrast im Realbild lässt sich einer verbesserten Kristallinität zuordnen bzw. bestätigt, dass die qualitativ schlechteren Aufnahmen der ungetrockneten Partikel wahrscheinlich durch einen niedrigeren Kristallinitätsgrad bedingt sind. Die Veränderung der Partikelmorphologie wirkt sich auch auf die optischen Eigenschaften der Lösung aus. Die Maxima der UV/VIS Absorption bzw. Photolumineszenz verschieben sich zu 340 bzw. 420 nm. Der onset der Absorption liegt nun bei 378 nm (3.3 eV) und entspricht damit dem von makrokristallinem Zinkoxid, d. h. es sind keine quantum confinement Effekte mehr zu beobachten. Dies zeigt eine Partikelgröße an, die über dem Bohrschen Radius, d. h. > 6 nm liegt, und entspricht damit den Ergebnissen aus XRD und SAED. ZnO-Pulver 100 ZnO-Nanopartikel (nach Aufarbeitung) Absorption [a. u.] -1 3408 cm 50 -1 -1 3328 cm 3165 cm -1 3249 cm 0 4000 ZnO-Nanopartikel (nach Trocknen) -1 2919 cm 2852 cm 3000 -1 2000 1000 -1 W ellenzahl [cm ] Abb. 19: IR-Spektren von ZnO-Pulvern. Oben: durch Fällung hergestelltes ZnO-Pulver, Mitte: ZnO-Kolloide durch Thermolyse von 1, Unten: dieselben ZnO-Kolloide nach Behandlung im Trockenschrank. ZnO / 49 Um zu untersuchen, ob die von uns verwendete Syntheseroute tatsächlich das Auftreten von OH-Gruppen an der Oberfläche vermeidet, wurden drei verschiedene Proben mittels IRSpektroskopie untersucht: (1) ZnO-Pulver, das durch alkalische Fällung hergestellt wurde, (2) die von uns synthetisierten ZnO-Kolloide und (3) dieselben Kolloide nach Trocknen bei erhöhten Temperaturen. Das IR-Spektrum (Abb. 19) zeigt für das durch Fällung hergestellte ZnO-Pulver, das stellvertretend für OH-bedeckte Nanopartikel steht, eine deutliche Bande bei 3408 cm-1, die der Schwingung von OH-Gruppen zuzuordnen ist. Aus der Literatur[83, 84] ist eine ähnliche Schwingungsfrequenz (ν = 3400 cm-1) für die OH-Gruppen auf ZnO-Nanopartikeln bekannt, so dass das verwendete Pulver als Vergleich geeignet ist. Das IR-Spektrum der von uns über die Thermolyse von 1 in HDA synthetisierten Kolloide ist in der Mitte der Abbildung zu sehen. Es zeigt drei Schwingungsbanden oberhalb und zwei unterhalb von 3000 cm-1, die alle von koordiniertem Hexadecylamin stammen. Es liegen eindeutig keine Schwingungsbanden bei 3400 cm-1 vor, so dass davon ausgegangen werden kann, dass die Kolloide in der Tat keine (d. h. keine nachweisbaren) OH-Gruppen an der Oberfläche aufweisen. Durch das Erhitzen während des Trocknungsvorgangs scheint jedoch die Aminfunktion des koordinierten HDAs beeinflusst zu werden wie das Fehlen von Banden im Bereich von 3100 bis 3300 cm-1 im darunter abgebildeten Spektrum belegt. Dies beruht offensichtlich nicht auf einer Desorption von intaktem HDA, denn die C-H-Schwingungen der Alkylkette (< 3000 cm-1) sind nach wie vor zu sehen. Eine mögliche Erklärung für diesen Sachverhalt kann das Auftreten eines C-N-Bindungsbruchs sein, wobei die Alkylketten auf der Oberfläche verbleiben und damit wahrscheinlich für die Löslichkeit in Toluol verantwortlich sind (und nicht HDA wie zunächst angenommen). Über den Verbleib der Aminofunktion kann bislang keine Aussage gemacht werden. Denkbar wären jedoch ein Entweichen als Ammoniak oder aber ein Einbau von Stickstoff in die Oberfläche der ZnO-Nanopartikel. 4.2.3 Thermolyse von [MeZnOSiMe3]4 bei hohen Temperaturen Die Durchführung der Thermolyse bei höheren Temperaturen führte wie die Ergebnisse der Festkörperpyrolyse von 1 bereits erwarten ließen zu einem unterschiedlichen Reaktionsverhalten. Bei der Injektion der Precursors bei 300 °C bildete sich ein grauer Niederschlag. Nach dem Waschen mit Methanol kann das Material zwar in Toluol fein dispergiert werden, aber es entsteht keine klare Lösung. Die TEM-Aufnahmen des Materials erklären dieses Verhalten: es sind Stäbchen mit einer Länge von ungefähr 1µm bzw. 50 nm Breite entstanden, die sich aufgrund ihrer Dimensionen selbstverständlich nicht mehr lösen lassen. Die Stäbchen bestehen aufgrund der Röntgenbeugung aus elementarem Zink: Abb. 20 zeigt die (002), (100), (101) und (102) von hexagonalem Zink. Die Bildung des Metalls stimmt hervorragend mit der Bildung von Zink in der Festkörperpyrolyse von 1 überein. Das recht ähnliche Verhalten von 1 in der Festphase und „in Lösung“ legt nahe, dass es sich bei dem Zerfall von 1 sehr wohl um einen unimolekularen Prozess handeln kann. Die Kolloidsynthese wird in hoch verdünnter Lösung durchge50 / ZnO führt, unter Bedingungen also, unter denen ein Methyltransfer zwischen zwei Kubanen sehr unwahrscheinlich ist. 1000 (002) 800 (101) 400 200 (102) (100) Intensity [a. u.] 600 0 30 35 40 45 50 55 60 2 Theta [°] Abb. 20: TEM Aufnahme (links, Balken = 300 nm ) und XRD von Zinkstäbchen aus der Pyrolyse von 1 in HDA bei 300 °C. Die Injektion von [MeZnOSiMe3]4 in HDA bei hohen Temperaturen könnte auch, insbesondere in Anwesenheit eines Lewis-basischen, Zink koordinierenden Lösungsmittels wie HDA, zu einer Deoligomerisierung von 1 in Dimere oder sogar Monomere führen. Dieser Mechanismus würde konsequenterweise eine geminale 1,1 reduktive Eliminierung von Me3SiOMe bevorzugen und elementares Zink als Produkt liefern. 4.2.4 Thermolyse von [Zn(OCH(Me)CH2NMe2)2] Das Zinkalkoxid 4 ist ein zähflüssiges, farbloses Öl, das analog zur Literatur synthetisiert wurde.[96] Da die thermischen Eigenschaften inklusive ihrer Zersetzungstemperatur nicht bekannt waren, wurde eine thermogravimetrische Analyse durchgeführt, um eine Reaktionstemperatur für eine Thermolyse in HDA zu ermitteln. 100 TG [%] 80 60 40 20 0 100 200 300 400 500 Temperatur [°C] Abb. 21: TGA von 5.5 mg [Zn(OCH(Me)CH2NMe2)2] von RT bis 600 °C in einer Argonatmosphäre. Die Heizrate betrug 10 K/Min. ZnO / 51 Aus Abb. 21 ist zu erkennen, dass zunächst ein geringer, aber kontinuierlicher Gewichtsverlust stattfindet, der dann von einem zweistufigen Gewichtsverlust zwischen 150 und 275 °C gefolgt wird. Die Restmasse liegt bei 26.5 % der Einwaage und somit etwas unter dem theoretisch berechneten Rückstand von Zinkoxid (30 %). Dies wird vermutlich durch einen Verdampfungsverlust des Precursors im Temperaturbereich bis 150 °C verursacht. Aus der TG-Kurve wurde die Temperatur für die Thermolyse auf 300 °C festgesetzt. Der Precursor wurde in n-Octylamin gelöst und bei 300 °C in die heiße Lösung aus Hexadecylamin injiziert. Nach einer Wachstumszeit von 30 Minuten bei etwas verringerter Temperatur wurde die Reaktionsmischung abkühlen gelassen. Durch Zugabe von Methanol konnte ein weißer Feststoff ausgefällt werden, der durch Zentrifugation abgetrennt, mit Methanol gewaschen wurde und schließlich in Toluol gelöst wurde. 1,5 4x10 5 382 nm (3.18 eV) 3x10 1,0 0,5 2x10 1x10 5 5 5 Intensität [cps] Absorption [a. u.] Fluoreszenz Absorption 300 350 400 450 500 550 0 600 Wellenlänge [nm] Abb. 22: Links: UV/VIS-Spektrum und Photolumineszenz von ZnO-Kolloiden in Toluol nach Thermolyse von 4 in heißem HDA; Rechts: Zum Vergleich dieselben Spektren aus der Thermolyse von 1 in heißem HDA. Das UV/VIS Spektrum dieser toluolischen Lösung ist in Abb. 22 gezeigt. Man erkennt deutlich eine Schulter, die bei λ = 310 (4.0 eV) nm liegt. Der onset ergibt sich durch Extrapolieren des steilen Teils des Anstiegs zu 342 nm (3.64 eV), d. h. im Rahmen der Messgenauigkeit sind die Absorptionseigenschaften identisch mit denen der Kolloide aus der Thermolyse von 1 und können folglich analog interpretiert werden: es liegt nanoskaliges Material mit einem nach Gleichung 7 zu 2.4 nm berechneten Durchmesser vor. Auch das Emissionsspektrum unterscheidet sich nicht von dem aus der Thermolyse von 1 erhaltenen: wie dort wird ein Stokes Shift beobachtet sowie ein Shiften des Maximums mit der Anregungswellenlänge, d. h. auch hier muss von einer breiten Partikelverteilung wie auch Defektleveln ausgegangen werden. Wie nicht anders zu erwarten spiegelt sich die Ähnlichkeit in den optischen Eigenschaften auch in den elektronenmikroskopischen Untersuchungen wider. Die in Abb. 23 gezeigten Partikel sind annähernd sphärisch, deutlich voneinander separiert und haben einen Durchmesser von etwa 3 nm. Die SAED zeigt hingegen etwas intensivere Reflexe, d. h. die untersuchten Partikel weisen einen höheren Grad an Kristallinität auf und lassen damit eine eindeutige Zuordnung zu ZnO zu. Die erhöhte Kristallinität lässt sich möglicherweise auf eine 52 / ZnO höhere Temperatur während Injektion und Wachstum zurückführen und erklärt auch den etwas höheren Kontrast des Realbildes im Vergleich zu den Aufnahmen aus der Thermolyse von 1. Abb. 23: TEM Aufnahme (links, Balken = 66 nm) und Elektronenbeugung (rechts) von ZnO-Kolloiden. 4.3 Zusammenfassung Es wurden drei verschiedene Zinksiloxide als SSP für ZnO untersucht. Für eine Synthese von kleinen Zinkoxid-Kolloiden bei niedrigen Temperaturen (aber auch für die Herstellung von ZnO Pulver mit extrem hoher Oberfläche, siehe Arbeiten des AK Drieß[94]) ist das tetramere Zinksiloxid [MeZnOSiMe3]4 am besten geeignet. Der Zerfall von 1 kann selektiv durch die Temperatur gesteuert werden: bei hohen Temperaturen wird metallisches Zink in Form von mikrometer großen Stächen gebildet, während bei niedrigen Temperaturen kolloidales, mit HDA stabilisiertes Zinkoxid entsteht. Ganz offensichtlich ist die Konkurrenz von Silicium und Zink um den Sauerstoff der Grund für das ambivalente Verhalten des Zinksiloxids. Im Gegensatz zu 1 kann das Zinkalkoxid 4 auch bei hohen Temperaturen zu ZnOKolloiden thermolysiert werden, wobei sich die Eigenschaften der aus beiden Precursoren erhaltenen Kolloide im UV/VIS, PL, TEM oder SAED nicht voneinander unterscheiden. Die höhere Zersetzungstemperatur von 4 ist allerdings für die Synthese von Cu/ZnO-Systemen (siehe Kapitel 8) unter Verwendung von M(OR)2 mit M=Cu, Zn von Vorteil, da sich Cu(OR)2 erst ab 200 °C zersetzt und schmale Größenverteilungen noch höhere Temperaturen benötigen. Bei diesen Temperaturen würde das Zinksiloxid sich jedoch nicht zu ZnO, sondern zu Zink zersetzen. Bei der Verwendung des Alkoxids 4 können die Precursor jedoch gleichzeitig bei derselben Temperatur zu ZnO und Cu zersetzt werden. Interessant ist, dass es in der Reihe der M(OR)2-Precursoren einen Bruch gibt, wenn von edlen Metallen wie Cu, Ni oder Pd zu unedlen Metallen wie Zink übergegangen wird. Während erstere sauber zu metallischen Partikeln thermolysiert werden, ist im Falle von Zink das Reduktionspotential von HO-CH(Me)CH2NMe2 nicht ausreichend bzw. die Bildung des Oxids offensichtlich energetisch wesentlich günstiger. ZnO / 53 Der „Beitrag der (Organometall-) Chemie zur Synthese von Nanopartikeln“ wie er in der Einleitung dieser Arbeit als Anspruch formuliert wurde, ist hier in mehreren Aspekten gezeigt worden. Im Vergleich zu den Standardmethoden für die Präparation von Zinkoxid-Kolloiden wurden in dieser Arbeit bei beiden Precursoren die konstituierenden Elemente über einen Einkomponenten-Precursor, kombiniert mit einem nicht-hydrolytischen Ansatz eingeführt. Dadurch wird auf der einen Seite die Bildung von hydroxylierten Oberflächen von vornherein vermieden wie durch IR-Spektroskopie gezeigt werden konnte und auf der anderen Seite die Wahrscheinlichkeit für das Auftreten einer Sauerstoffdefizienz minimiert. Dass dieser Zugang über die Organometallchemie erfolgversprechend ist, konnte an den Emissionsspektren der ZnO-Kolloide in Lösung gezeigt werden: im Vergleich zu Kolloiden, die aus Zn(OAc)2 in Ethanol hergestellt wurden, liegt die Emission hier nahe des UV-Bereichs. Aufgrund der großen Halbwertsbreite der Emissionsbande ist jedoch anzunehmen, dass die Kristallinität der Partikel (noch) sehr gering ist und weitere Experimente erforderlich sind, um die Reaktionsbedingungen so zu optimieren, dass die Partikel weniger Baufehler aufweisen. Ein weiterer Vorteil der von uns hergestellten Partikel ist der, dass sich der Durchmesser der als Pulver isolierten Partikel schon bei Erhitzen auf etwa 80 °C vergrößert. Bei der Herstellung von Schichten aus Acetat-gecappten Partikeln (für optische Bauteile) werden Temperaturen von 300 °C benötigt, um dies zu erreichen, was die Auswahl an möglichen Substraten auf diejenigen beschränkt, die unter solch hohen Temperaturen stabil bleiben. Diese Bedingung könnte durch Verwendung der von uns hergestellten Kolloide, die durch wesentlich schwächer und koordinativ bindende Amindonoren stabilisiert sind, umgangen werden. 54 / ZnO 5 Kupfer-Nanopartikel 5.1 Metallische Nanopartikel: ein Überblick Die Forschung an metallischen Partikeln im Größenbereich von Nanometern verfolgt im Wesentlichen zwei Interessen: (1) mögliche Anwendungen in der Nanotechnologie und (2) das fundamentale Interesse an den metallischen Eigenschaften während des Übergangs vom Molekül zum Festkörper (von G. Schmid treffend mit dem Schlagwort „embryonale Metalle“ belegt worden). Die Einleitung dieses Kapitels umfasst zunächst die Gesamtheit der metallischen Nanopartikel, stellvertretend für alle in den Kapiteln 5 bis 7 behandelten Metalle (Kupfer, Palladium und Nickel). Im nächsten Abschnitt wird dann auf die speziellen Synthesemethoden für Kupfer-Nanopartikel eingegangen. Eine sehr charakteristische Eigenschaft von Nanopartikeln, darunter auch einigen metallischen, ist ihre spezifische, intensive Farbe. Seit der Antike werden deswegen z. B. Goldpartikel für die Färbung von Gläsern verwendet. Das aus Kirchenfenstern bekannte rubinrote Glas (ruby glass) enthält fein verteilte Goldkolloide, die während der Glasherstellung erzeugt werden. Bis heute gibt es keine großen Änderungen in der Herstellung von roten Gläsern, nur die Farbpalette ist seitdem wesentlich erweitert worden. Aus Gründen, die in Kapitel 2 ausführlich behandelt worden sind, ist vorauszusehen, dass metallische quantum dots für die Nanoelektronik eine große Rolle spielen werden: sie können einzelne Elektronen speichern. Liganden-stabilisierte Nanopartikel können mit einem Elektron aufgeladen werden, das solange auf dem Partikel bleibt, bis die entsprechende Spannung an der Gegenelektrode angelegt wird, um es wieder zu entfernen. Das heißt, diese Partikel verhalten sich wie miniaturisierte Transistoren. Abb. 24: Rechts: SEM-Bild eines 17 nm großen, mit H2N-C6H4-SO3Na stabilisierten Palladiumpartikels zwischen zwei Platinspitzen. Links: Strom-Spannungs-Kurve.[97] In Abb. 24 ist dies für ein Palladiumpartikel mit 17 nm Durchmesser gezeigt. Bei Raumtemperatur verhält sich dieses Partikel zwar noch wie der entsprechende Festkörper, d. h. es zeigt das lineare Ohmsche Verhalten, bei 4.2 K jedoch kann eine deutliche CoulombKupfer / 55 Blockade von ~ 55 mV beobachtet werden. Für noch kleinere Partikel, wie z. B. Phosphinstabilisierte Au55-Cluster mit einem Durchmesser von 1.4 nm, kann dieses Verhalten sogar bei Raumtemperatur beobachtet werden.[98] Bis zur Verwendung von solchen Einelektronentransistoren in elektronischen Bauteilen ist jedoch noch ein weiter Weg zurückzulegen, da bisher weder die Problematik der Addressierbarkeit der einzelnen Partikel noch die der Verknüpfung der Minitransistoren zu geordneten Strukturen gelöst worden ist. Die gewaltige Leistungssteigerung, die zu erwarten ist, rechtfertig allerdings die intensiven Forschungen auf diesem Gebiet. Metallische Partikel eignen sich aufgrund ihrer Farbigkeit und ihres hohen Kontrasts im Elektronenmikroskop außerdem hervorragend zur Markierung von z. B. DNA-Molekülen. Modifiziert man Gold-Nanopartikel mit zwei verschiedenen, nicht-komplementären DNASträngen, und vereinigt sie miteinander, so liegen sie getrennt in Lösung vor und die Lösung ist rot gefärbt (siehe Abb. 25, links). Gibt man nun ein DNA-Stück dazu, das zwei sticky ends hat, die jeweils komplementär zu den zwei verschiedenen DNA-Strängen an den Goldpartikeln sind, so findet eine Basenpaarung statt und die Lösung zeigt einen Farbumschlag von rot nach blau, wobei die Goldpartikel agglomerieren und ausfallen. Dies bietet nicht nur eine Möglichkeit, metallische Partikel mit Hilfe von DNA zu ordnen, sondern ist außerdem ein empfindlicher Nachweis für den entsprechenden DNA-Strang und kann damit für die Analytik von Erbmaterial verwendet werden (siehe Abb. 25, rechts und ganz rechts).[99-101] Abb. 25: Links: Strategie der Anordnung von Gold-Nanopartikeln mittels DNA.[99] Rechts: Weiterentwicklung dieser Strategie zum Nachweis von Anthrax oder einzelnen Basen. Das wohl eindeutig vielversprechendste Anwendungsfeld für nanopartikuläres metallisches Material ist die Katalyse, sowohl heterogen als auch homogen. Je kleiner nämlich die Partikel werden, desto größer wird der Anteil der Oberfläche im Verhältnis zum Volumen, d. h. je kleiner die Partikel, desto mehr Atome sind dem katalytischen Prozess zugänglich, was zur Folge hat, dass die katalytische Effizienz pro Atom mit abnehmender Größe der Partikel ebenfalls zunimmt. Unter den Oberflächenatomen sind diejenigen, die an Kanten und Ecken platziert sind, aktiver als die auf Flächen. Der Prozentsatz von solchen Ecken- und Kanten56 / Kupfer atomen steigt mit zunehmender Krümmung der Oberfläche und deshalb sind sehr kleine Metallpartikel bevorzugte Katalysatoren. Die Wahrscheinlichkeit für eine Aggregation der Partikel steigt allerdings ebenfalls mit abnehmender Größe. Das Ausmaß kann aber effektiv reduziert werden, indem man die Oberflächenenergie durch Adsorption von Liganden verringert oder aber die metallischen Partikel auf einem Träger verankert. Die katalytische Aktivität selber beruht auf der Übertragung und anschließenden Speicherung von Elektronen auf den gelösten Partikeln. Die gespeicherten Elektronen können durch MultielektronTransferprozesse Substanzen in Lösung reduzieren. Grundsätzlich hängt die Aktivität von der Partikelgröße, der Partikelmorphologie und von der Art der verwendeten Stabilisatoren ab. Deshalb ist eine wesentliche Anforderung an die Synthese von Metall-, aber auch allen anderen Nanopartikeln, diese Eigenschaften gezielt zu steuern. Nanopartikel, deren Oberfläche nicht durch Liganden stabilisiert wird, sind zwar durch Trägerung der heterogenen Katalyse zugänglich, für die homogene Katalyse sind sie jedoch aufgrund von Agglomeration ungeeignet. Stabilisierte Partikel hingegen sind (wie in Kapitel 2 ausgeführt worden ist) in organischen oder anorganischen Solventien löslich und können daher für die homogene Katalyse genutzt werden. Eine Ligandenhülle kann allerdings auch ein Nachteil sein, da die Liganden katalytisch aktive Zentren besetzen. Auf der anderen Seite können stabilisierte Partikel aber auch dazu benutzt werden, den Katalysator in Bezug auf seine Selektivität zu optimieren. Da Ligandenmoleküle nie die gesamte Oberfläche eines Partikels bedecken, gibt es genug Kanäle, die es Reaktanden und Produktmolekülen erlaubt, die Metalloberfläche zu erreichen bzw. von ihr zu desorbieren. Beispielsweise sind Palladium-Nanopartikel für die katalytische Hydrierung von 1-Butenyltrimethylammoniumbromid[102] Allylalkohol[103], Styrol[104] (hier ist die katalytische Aktivität der mit TOAB gecappten Pd-Nanopartikel 35 Mal höher als 10%Pd/C), Isopropylacrylamid[105] und in der Suzuki-Reaktion[106] als Katalysatoren verwendet worden. Weitere Aufmerksamkeit erhalten Metallpartikel schließlich (analog zu ihren halbleitenden Kollegen), wenn es um die Erforschung der „vernachlässigten Dimension“, den Nanometerund Subnanometer-Bereich, geht. Auch im metallischen Bereich interessiert die Fragestellung nach dem Übergang von Molekül- zu Festkörpereigenschaften: 1981 konnte Henglein zum Beispiel zeigen, dass eine der metallischen Eigenschaften, nämlich die Oberflächenplasmonenresonanz, schon bei Clustern mit nur 15 Atomen auftritt. Kleine Metallpartikel sind grundsätzlich wesentlich schwieriger zu handhaben als Halbleiter-Nanopartikel, da sie (mit Ausnahme der Edelmetalle) im Allgemeinen extrem empfindlich gegenüber Sauerstoff sind. Die meisten Effekte, die von den Halbleitern bekannt sind, gelten aber auch für Metallpartikel. So werden z. B. auch Metalle durch Anwachsen der Bandlücke schließlich zu Halbleitern, allerdings erst bei sehr kleinen Durchmessern von wenigen Nanometern. Kupfer / 57 5.2 Stand der Forschung und Aufgabenstellung Fast alle Synthesemethoden (mit Ausnahme der physikalischen Techniken, die in der Regel nicht in kondensierter Phase ablaufen) beruhen auf einem Reduktionsprozess von Metallkationen durch verschiedenste Reduktionsmittel. Die verwendeten Lösungsmittel reichen von Wasser bis hin zu unpolaren Medien, je nach Polarität des verwendeten Metallsalzes bzw. Metallkomplexes. Die Art des verwendeten Edukts bestimmt auch das dazu gehörige chemische Reduktionsmittel. Wasserstoff, anorganische Hydrid-Überträger, organische Reduktionsmittel wie Alkohole, aber auch Photoreduktion, Ultraschall und radiolytische Methoden sind in den Synthesen von Metallpartikeln verwendet worden. Analog zu Halbleiterpartikeln müssen auch bei Metallpartikeln Liganden vorhanden sein, die das Wachstum bei einer Größe im Nanometerbereich terminieren. Solche Reduktionen in Gegenwart von stabilisierenden Liganden werden unter dem Begriff controlled chemical reduction[107, 108] oder auch kinetic trapping verstanden. Im Folgenden werden einige nasschemische und physikalische Methoden, sowie Templatmethoden für die Synthese von Kupfer-Nanopartikeln vorgestellt. Nasschemische Methoden. Eine der typischen Reduktionsmittel für die Synthese von metallischen Nanopartikeln sind Borhydride. Bönnemann hat auf diese Weise eine Vielzahl von Metallpartikeln hergestellt, im Falle von Kupfer geht man dabei von [N(octyl)4]2[CuCl2Br2] in Toluol aus und reduziert mit Li[BEt3H].[109] Die entstehenden, etwa 5-10 nm großen Partikel werden durch das als Gegenion der Kupferquelle eingeführte NR4+ stabilisiert. Durch Hexanthiol stabilisierte Kupfer-Nanopartikel können durch eine ähnliche Reduktion von wässrigem Kupfernitrat mit LiEt3BH in Gegenwart eines Gemischs aus Tetraoctylammoniumbromid (TOABr) und Hexanthiol synthetisiert werden.[110] Die Partikel sind anfangs sphärisch mit einem Durchmesser von 1-2 nm, können aber durch thermisches Annealing auf >10 nm wachsen, wobei sich u. a. dreieckige Formen und rods von Cu2O herausbilden. Der Nachteil, mit zwei möglichen Stabilisatoren zu arbeiten (TOABr und Hexanthiol), lässt sich sehr elegant vermeiden, wenn das Thiol selber als Reduktionsmittel verwendet wird. Dies ist für die Reduktion von Kupfernitrat in superkritischem Wasser gezeigt worden.[111] In Abwesenheit des Thiols entstehen oxidische Partikel, während in seiner Anwesenheit die Oberfläche wirksam bei einer Größe von 7 nm stabilisiert wird. Oft werden auch Polymere, insbesondere Polyvinylpyrrolidon (PVP) zur Stabilisierung von Metall-Nanopartikeln herangezogen. So kann Kupferacetat in Gegenwart von PVP durch Hydrazin zu 7 – 23 nm großen Kupfer-Nanopartikeln reduziert werden. Die Menge des PVPs bestimmt dabei sowohl die Größe als auch die Größenverteilung der Metallpartikel.[112] Die Reduktion von Kupferacetat durch Ethanol in Gegenwart von PVP führt hingegen zur Bildung von Kupferoxiden. Metallische Partikel entstehen nur, wenn Magnesium als Katalysator anwesend ist, sind stark agglomeriert und weisen eine breite Größenverteilung auf (20100 nm).[113] Neben PVP eignet sich auch Polyvinylalkohol als Stabilisator, während Polyethylenglycol keine stabilisierenden Eigenschaften aufweist und in Gegenwart von Polyacryl58 / Kupfer amid keine Reduktion der Kupferkationen möglich ist.[114] Von der Verwendung von Polyvinylsulfat, Dextrin, Amylopektin, Methylamylopektin, Methylcellulose, Ethylcellulose und 2-Hydroxyethylcellulose wird ebenfalls berichtet.[115] Kupfer-Nanopartikel können durch thermische Reduktion (80 °C) von Cu(N2H3-COO)2 2H2O hergestellt werden. Diese Methode macht Gebrauch von dem autokatalytischen, exothermen Zerfall von Metall-Hydrazin-Carboxylaten, liefert aber große, agglomerierte Partikel mit einer sehr breiten Größenverteilung (200-300 nm). Die Reduktion der Kupfer-Hydrazin-Carboxylate kann ebenso durch Behandlung mit Ultraschall erfolgen, wobei man annimmt, dass in situ gebildete Wasserstoffradikale als Reduktionsmittel fungieren. In diesem Fall entstehen kleinere Partikel (50 nm), die aber ebenfalls eine Größenverteilung aufweisen, agglomeriert sind und darüber hinaus Cu2O enthalten.[116] Setzt man der Reaktion ein zwitterionisches Tensid bei, nämlich Cetyltrimethylammonium p-Toluolsulfonat, findet ein Übergang von sphärischen zu elongierten Partikeln statt, da die Tenside während der Reaktion fadenartige Mizellen bilden, die als Formvorlage für die Partikel dienen (s. Templatmethoden).[117] Wesentlich reinere Partikel entstehen, wenn an Stelle des Ultraschalls Gammastrahlen als Reduktionsmittel verwendet werden. Der Vorteil dieser Methode liegt darin, dass das Reduktionsmittel ein solvatisiertes Elektron mit einem sehr niedrigen Redoxpotential ist und keine oxidischen Verunreinigungen entstehen können. Die Partikel, die mit dieser Methode aus Kupferacetat oder -chlorid hergestellt wurden, sind stabile, mit PVP, PVA, Cetyltrimethylammoniumbromid, Natriumdodecylsulfat oder Polyether gecappte, kristalline Cu-Nanopartikel mit einem Durchmesser von 17 bis 80 nm.[118] Physikalische Methoden. Auch physikalische Methoden sind verwendet worden, um Kupferpartikel herzustellen, wie z. B. die gepulste Laserdeposition, die sich gut für die Herstellung von Cu-Al2O3-Filmen eignet[119] oder die Inertgas-Kondensation mit in-situ Wärmeverdichtung[120]. Bei letzterer Technik werden zunächst Nanopartikel durch die Verdampfung von Kupfer bei hohen Temperaturen und 400 Pa Argondruck hergestellt (Inertgas-Kondensation) und dann bei 200 °C unter einem Druck von 1.0 GPa verdichtet (insitu Wärmeverdichtung). Templat-Methoden. Ebenso eignen sich Templat-Methoden für die Synthese von nanostrukturiertem Material. Es sind Arbeiten bekannt, in denen Dendrimere, DiblockCopolymere, reverse Mizellen oder Kohlenstoff Nanotubes als „Nano-” oder „MikroReaktoren“ dienen. Als Dendrimer wurde PAMAM (Poly(amidoamin)) verwendet. Die Methode beruht darauf, Metallkationen in das Innere der Dendrimere einzubringen und dort mit NaBH4 oder Hydrazin zu reduzieren. Dabei kann die Größe der entstehenden Metallpartikel durch die chemische Struktur und Größe der Dendrimere gesteuert werden.[121, 122] Nach einem ähnlich Prinzip verläuft die Synthese von Cu-Partikeln in DiblockCopolymeren: Das Polymer, bestehend aus polymerisiertem Methyltetracyclododecen und Norbornendicarbonsäure) wird mit Cu-Kationen (Cu(OAc)2) beladen, wobei ein Ionenaustausch mit den Carboxylatgruppen der Netzwerkstruktur stattfindet. Nach Reduktion mit Kupfer / 59 H2 findet man Kupfercluster mit einer breiten Größenverteilung von 4 bis 12 nm Durchmesser eingebettet in den Polymerfilm.[123] In reversen Mizellen können Kupfer-Nanopartikel mit einer schmalen Größenverteilung durch die Reduktion von Cu(AOT)2 synthetisiert werden.[124] Hierbei lassen sich Größe (2 bis 10 nm) und Form der Partikel (sphärisch oder zylindrisch) durch die Wahl des Reduktionsmittels (Hydrazin oder NaBH4), den WasserGehalt oder die Konzentration an Cu(AOT)2 steuern. Ist der Wassergehalt zu hoch, werden jedoch Kupferoxid-Partikel gebildet. M. Pileni et al. konnten in einer weiteren Arbeit zeigen, dass sich die Form der Partikel auch durch die Anwesenheit von verschiedenen Salzen steuern lässt.[125] Höhere Wachstumsraten lassen sich erzielen, wenn man mit Mizellen in komprimierten Gasen oder überkritischen Flüssigkeiten arbeitet.[126] Bei der Verwendung von Kohlenstoffröhrchen als Templat führt die Reduktion von Kupfersalzen durch Wasserstoff augrund des Durchmessers der Röhrchen zu einer Größenkontrolle der sphärischen Cu-Nanopartikel im Bereich von wenigen bis 35 nm. Die Partikel können im Gegensatz zu anderen Templatsynthesen sehr einfach durch Ultraschall von den Kohlenstoff Nanotubes abgelöst werden. Erhöht man den Anteil des Kupfersalzes, werden die Nanoröhrchen mit einer dicken Kupferschicht überzogen und es lassen sich gezielt metallische Fäden herstellen.[127] Trotz der vielen Arbeiten über die Synthese von Kupfer-Nanoapartikeln, gibt es einen großen Bedarf an nicht-wässrigen und chemisch einfachen Systemen, die wenige Reagenzien benötigen, insbesondere im Hinblick auf Zusätze zur Reduktion und zur Stabilisierung, die keine zusätzliche technische Ausrüstung erfordern und die vor allem keine Salze oder andere, schwer zu entfernende Nebenprodukte liefern wie sie bei der Verwendung von Mizellen oder Hydriden als Reduktionsmittel anfallen. Ziel unserer Arbeiten auf dem Gebiet der nanopartikulären Kupferpartikel war es daher, mit Hilfe des anorganisch-chemischen Wissens einen Precursor zu finden, der mit möglichst wenigen (oder gar keinen) Zusätzen zu Nanokristalliten führt, die nicht wie in vielen der oben beschriebenen Arbeiten agglomerieren, eine breite Größenverteilung aufweisen oder mit Kupferoxiden verunreinigt sind. Analog zu den Arbeiten mit CdSe-Nanopartikeln sollte dann versucht werden, die chemischen Eigenschaften der Oberfläche durch eine Oberflächenmodifizierung der Partikel zu steuern. 5.3 Synthese von Kupfer-Nanopartikeln durch Thermolyse des organometallischen Precursors [Cu(OCH(Me)CH2NMe2)2] 5.3.1 Vorstellung des Precursors Das Know-how für die Precursorchemie lässt sich in einem scheinbar entfernt verwandten Bereich finden: der chemischen Dampfabscheidung (CVD). Dort werden die Eigenschaften von Precursoren für die Abscheidung von dünnen Metallfilmen maßgeschneidert, z. B. ihre Flüchtigkeit, kinetische und thermische Stabilität und Zersetzung zu Metallatom bzw. –ion 60 / Kupfer und einfachen Abgangsgruppen. Da Kolloide auf der anderen Seite oft als „Oberfläche in Lösung“ bezeichnet werden, ist es vernünftig, die Konzepte der Precursor-Entwicklung in der CVD auf die Synthese von Metallpartikeln in Lösung zu übertragen. So sollte es möglich sein, das Reduktionsmittel zur Bildung von nullwertigem Metall aus Metallkationen bereits in den Precursor einzubauen und es zudem so zu wählen, dass einfache, möglichst gasförmige Produkte entstehen. Die Verbindung sollte ferner kristallin, einfach zugänglich sein und bei erhöhten Temperaturen das Metall freisetzen. Der Vorteil bei der Verwendung solcher metallorganischer Verbindungen ist, dass sie sich prinzipiell in ein bereits bekanntes und gut kontrollierbares System einsetzen lassen, das gleichzeitig Lösungsmittel und Stabilisator ist: hochsiedende, langkettige Alkane mit einer zur Koordination befähigten Kopfgruppe (Amin oder Phosphinoxid) wie ursprünglich für die Synthese von II-VI Halbleitern entwickelt (siehe Kapitel 3). Damit ist eine Kontrolle über beide Parameter, die die Eigenschaften der entstehenden Nanopartikel bestimmen, möglich: das Ausgangsmaterial (Precursor und verwendetes Lösungsmittel) und das Reaktionssystem (Temperatur, Zeit, Konzentration). Unseres Wissens nach sind in der Literatur bislang keine metallorganischen Precursor für die Synthese von Kupfer-Nanopartikeln verwendet worden. Als Precursor für die Synthese von metallischen Kupfer-Nanopartikeln wurde die organometallische Kupferverbindung [Cu(OCH(Me)CH2NMe2)2] ausgewählt. Sie ist bereits als CVD-Precursor bekannt[128, 129], einfach zugänglich durch eine Alkohol-Austausch-Reaktion von Kupfermethanolat mit dem entsprechenden Aminoalkohol[130] und lässt sich durch Sublimation bei 55 °C im Vakuum (10-2 mbar) als violetter Feststoff in hoher Reinheit erhalten. Die kristalline Substanz ist einfach handhabbar und nur wenig licht- und luftempfindlich und entspricht somit den oben genannten Kriterien. Es ist bereits gezeigt worden, dass die Pyrolyse von [Cu(OCH(Me)CH2NMe2)2] in einer Stickstoffatmosphäre bei Normaldruck laut XRD metallisches Kupfer als einziges Produkt ergibt.[130] Young et al. haben die Thermolyse des Kupfer-Precursors mit Hilfe von IR-Spektroskopie untersucht und fanden, dass die Bildung des Kupfers von der Entwicklung von 1-Dimethylaminopropanol und 1-Dimethylaminopropan-2-on im Verhältnis 1:1 begleitet wird.[131] Auch bei der Verwendung dieses Precursors zur chemischen Dampfabscheidung konnten die oben genannten Verbindungen mittels 1H NMR als einzige flüchtige Produkte identifiziert werden.[129] Der erhebliche Vorteil dieser Verbindung liegt also in den koordinierten Liganden: der Alkohol kann ganz offensichtlich zum Keton oxidiert werden und damit als Reduktionsmittel für Cu(II) dienen. Das hat für die Synthese von metallischen Nanopartikeln durch Reduktion der entsprechenden Metallkationen einen großen Vorteil: der Synthese müssen keine zusätzlichen Reduktionsmittel zugefügt werden, was das System beträchtlich vereinfacht: • Jedem Kation steht ein Reduktionsmittel in unmittelbarer Umgebung zur Verfügung, d. h. die Probleme der exakten Zugabe und anschließenden homogenen Verteilung entfallen, Kupfer / 61 • im Gegensatz zu der Reduktion von Metallsalzen mit beispielsweise Hydridüberträgern ist die Reduktion hier unimolekular, d. h. die Nukleation ist unabhängig von der Konzentration der Edukte, alle Kationen werden bei Erreichen der kritischen Temperatur gleichzeitig reduziert, d. h. weder die Dauer einer Injektion noch Inhomogeneitäten in der Konzentration beeinflussen die Nukleation und das Wachstum, und das Oxidationsprodukt 1-Dimetyhlaminopropan-2-on wird durch seine Flüchtigkeit und die hohen Temperaturen schon während der Reaktion entfernt, d. h. aufwändige Reinigungsschritte wie bei der Verwendung von Mizellen oder bei Salzfracht entfallen. • • 5.3.2 Thermolyse Als Reaktionsmedium wurde Hexadecylamin (HDA) ausgewählt, eine bei 45-48 °C schmelzende, wachsartige Substanz, die sich bis zu Temperaturen > 300 °C ohne Zersetzung erhitzen lässt. HDA dient gleichzeitig als Lösungsmittel und Stabilisator, sowohl elektronisch durch die Koordination des freien Elektronenpaars am Stickstoff an Metallatome der Oberfläche als auch sterisch durch die dann nach außen weisenden Alkylketten. Die Pyrolyse wurde unter Schutzgas bei Temperaturen zwischen 200 und 300 °C durch Injektion einer Lösung von [Cu(OCH(Me)CH2NMe2)2] in n-Octylamin durchgeführt. Für die Auswahl der Reaktionstemperatur während der Thermolyse in HDA wurden Daten aus TG/DTA herangezogen (siehe Abb. 26, Messung Ralf Becker[129]). Der Gewichtsverlust erfolgt in einem einzigen Schritt, wobei der Massenrückstand von 18 % aufgrund von teilweiser Verdampfung des Precursors etwas unter dem theoretisch berechneten Anteil an Kupfer liegt. Das exotherme Signal der DTA entspricht der Zersetzung von [Cu(OCH(Me)CH2NMe2)2], die damit bei einem Wert von 185 °C liegt und die Mindesttemperatur für die Thermolyse festlegt. Die idealen Eigenschaften dieses Precursors, d. h. seine scharf definierte Zersetzung, eröffnen darüber hinaus lohnenswerte Perspektiven für weiteres Liganden-Egineering mit dem Ziel, die Zersetzungstemperatur weiter zu senken. 100 TG Masse [%] 80 60 DTA 40 20 50 100 150 200 250 300 Temperatur [°C] Abb. 26: TG/DTA von ~ 20 mg [Cu(OCH(Me)CH2NMe2)2] in einer Argonatmosphäre. Die Heizrate betrug 5 K/min. in einem Temperaturbereich von RT bis 350 °C.[129] 62 / Kupfer Bei Temperaturen < 200 °C liegt der Cu(II)-Komplex entgegen der mittels TG/DTA ermittelten Zersetzungstemperatur unzersetzt in HDA vor, eine Erhöhung der Temperatur bzw. eine Injektion bei Temperaturen > 200 °C führt hingegen zur sofortigen Gasentwicklung und tiefroten Verfärbung der Reaktionsmischung innerhalb von einer Minute. Die Gasentwicklung wird durch die bei diesen Temperaturen flüchtigen Nebenprodukte (Alkohol und Keton) verursacht, während die rote Farbe auf die Entstehung von Kupferpartikeln bzw. deren Oberflächenplasmonenresonanz (siehe UV/VIS) zurückzuführen ist. Nach einem Wachstumsprozess von 30 Minuten wurde die Reaktion beendet, indem sie auf Raumtemperatur abkühlen gelassen wurde. Die Zugabe von Toluol zum festen Rückstand ergibt tiefrote Lösungen, die extrem empfindlich gegen Sauerstoff sind (Verfärbung von rot nach blau innerhalb weniger Minuten). Das Entfernen von überschüssigem HDA gelingt durch Zugabe von Methanol, Zentrifugation und Waschen der ausfallenden Nanopartikel. Die gereinigten Partikel können wieder in Toluol gelöst werden und sind über Monate bei Raumtemperatur und Abwesenheit von Luft stabil. 5.3.3 Analytische Charakterisierung Die analytische Charakterisierung wurde an Partikeln durchgeführt, die unter den folgenden Bedingungen synthetisiert wurden: 322 mg (1.2 mmol) Precursor in 4 mL Octylamin (c = 0.3 mol/L), injiziert bei T = 300 °C in 7 g HDA und bei T = 225 °C für t = 30 Min. wachsen gelassen. Dies ist wichtig für den Vergleich mit Pyrolysen aus Kapitel 5.3.4. Das UV/VIS Spektrum der roten Lösungen in Toluol (c § µmol/mL, Molalität § 0.01 µmol/mg) zeigen eine ausgeprägte Absorptionsbande bei 558 nm (Abb. 27). Diese Resonanz wurde von anderen Gruppen ebenfalls für die Absorption von Kupferpartikeln gefunden (siehe z. B. [109, 116, 132, 133]) und kann der Anregung von „plasmon polaritons“ (siehe Kapitel 2.2.1) zugeordnet werden, d. h. Oszillationen des Elektronengases durch Wechselwirkung mit dem elektromagnetischen Feld; allgemein werden sie als Oberflächen-Plasmonen-Resonanz (OPR) bezeichnet. Absorption [a. u.] 2 mit Aufarbeitung 558 nm ohne Aufarbeitung 1 566 nm 0 400 500 600 700 Wellenlänge [nm] Abb. 27: UV/VIS-Spektrum von aufgereinigten (oben) und nicht aufgereinigten, d. h. mit Überschuss HDA (unten) Cu-Nanopartikeln in Toluol. Kupfer / 63 Im Vergleich zur Absorption von Festkörper-Kupfer (600 nm) ist die Absorption um 42 nm blauverschoben und liefert damit einen ersten Hinweis darauf, dass nanoskaliges Material entstanden ist: OPR kann an Festkörpern aufgrund des geringen Anteils der Oberfläche vernachlässigt werden, bei Nanopartikeln ist das Verhältnis von Oberfläche zu Volumen jedoch so groß, dass sie zum charakteristischen Merkmal wird. Genau diese Eigenschaft hat zur schon Eingangs erwähnten Verwendung von Goldpartikeln zur Färbung von Gläsern geführt. Eine Verschiebung der energetischen Lage der OPR lässt sich in zwei Fällen beobachten: (1) bei der Aufarbeitung und (2) bei Kontakt mit Luftsauerstoff (siehe Kapitel 5.4.2). Wenn die Partikel ohne weitere Aufarbeitung direkt nach der Reaktion, d. h. mit einem Überschuss an Hexadecylamin, in Toluol gelöst und UV-spektroskopisch vermessen werden ist die Lage des Absorptionsmaximums im Vergleich zu aufgearbeiteten Partikeln um 8 nm auf 566 nm rotverschoben (siehe Abb. 27). Eine Ursache dafür könnte das Vorliegen von großen Partikeln, d. h. größer als 25 nm, sein, da ab dieser Größe eine Verschiebung der Absorption zu erwarten ist. Wird jedoch das Reaktionsprodukt ohne Aufarbeitung in Toluol gelöst und auf ein TEM-Grid aufgebracht, lassen sich im Elektronenmikroskop erstens keine Partikel dieses oder größeren Durchmessers finden und zweitens lässt sich kein Unterschied zu den aufgearbeiteten Partikeln feststellen, d. h. die Verschiebung im UV/VIS Absorptionsspektrum lässt sich nicht auf Veränderungen der Eigenschaften der Partikel durch den Prozess der Aufarbeitung zurückführen. Während der Aufarbeitung wird jedoch durch Waschen mit Methanol der Überschuss an HDA entfernt. Da die Absorption sowohl durch die Dielektrizitätskonstante der Partikel als auch durch die des sie umgebenden Mediums bestimmt wird, ist anzunehmen, dass die Dielektrizitätskonstante des HDAs für die Verschiebung der OPR in Proben mit einem Überschuss an HDA verantwortlich ist. Das Vorliegen von nanoskaligem Material wird durch die elektronenmikroskopischen Aufnahmen der Probe bestätigt (siehe Abb. 28). Auf dem gesamten Netz sind wohldefinierte, sphärische Partikel von etwa 7.5 nm Durchmesser zu sehen. Abb. 28: TEM-Aufnahme von Cu-Nanopartikeln, bar = 110nm. Die Vergrößerung zeigt die Tendenz der Partikel, sich in hexagonalen Netzwerken selbst zweidimensional anzuordnen. Sowohl Größe als auch Form der Partikel sind sehr einheitlich und die einzelnen Partikel sind von ihren Nachbarn aufgrund der Alkylketten ihrer Liganden etwa 2 nm entfernt. Die Teilver64 / Kupfer größerung in Abb. 28 zeigt, dass die schmale Größenverteilung der Partikel offensichtlich auch zur Bildung eines regelmäßigen hexagonalen 2-D Netzwerks aus freistehenden Kolloiden führt. Dennoch scheinen die Partikel bei höherer Vergrößerung (> 300 k) instabil zu sein, denn es gelingt nicht, Gitterauflösung zu erreichen. Die zugehörige SAED ist in Abb. 29 links gezeigt. Man erkennt klar getrennte Beugungsringe, die den Gitterebenen (111), (200), (220) und (311) des Kupfers in der kubisch dichtesten Packung zugeordnet werden können. Demnach liegen nicht einfach aggregierte nullwertige Kupfer-Atome vor, sondern eine geordnete Kugelpackung wie sie in metallischen Festkörperstrukturen ausgebildet wird. Neben den scharfen Beugungsringen von metallischem Kupfer, zeigt die Probe weitere, diffuse Reflexe. Diese können aufgrund ihrer Lage den Oxiden des Kupfers zugeordnet werden, d. h. den (111) und (220) Netzebenen des CuO bzw. den (-111) und (-113) Ebenen von Cu2O. Da die Probe vor der Untersuchung im Elektronenmikroskop in Kontakt mit Luftsauerstoff war, ist nicht eindeutig klar, ob die Oxide schon während der Synthese der Partikel oder erst durch Luftoxidation gebildet werden. Abb. 29: Elektronenbeugung (SAED) der Kupfer-Nanopartikel aus Abb. 28. Links nach Luftkontakt, rechts unter Luftausschluss, realisiert durch die Präparation in einer Glovebox und Benutzung eines VakuumtransferDoppelkipphalters. Um dies zu klären, wurde die Probe unter Argonatmosphäre präpariert und mit Hilfe eines Vakuumtransfer-Doppelkipphalters unter Schutzgas transportiert. Das resultierende SAEDMuster zeigt nun keine Beugungsringe mehr für oxidisches Kupfer. Wird diese Probe für etwa 10 Minuten Luftkontakt ausgesetzt, beobachtet man dieselben Oxidringe wie oben beschrieben. Daraus kann geschlossen werden, dass die synthetisierten Partikel aus metallischem Kupfer bestehen und dass ihre Oberfläche durch Luftkontakt leicht oxidiert wird, wodurch diffuse Beugungsringe neben den intensiven und scharfen Reflexen des intakten Cu(0)-Kerns entstehen. Die Existenz der oxidischen Beugungsringe weist darauf hin, dass die Oxidschicht nicht amorph ist, sondern bereits Festkörperstrukturen ausbildet. Dass es sich um eine Oxidation der Oberfläche handelt, wird außerdem durch die SAED Aufnahmen von Partikeln verschiedener Größe bestätigt: je größer die Partikel sind, desto geringer ist die Intensität der oxidischen Reflexe relativ zu der der metallischen Reflexe. Dies korreliert ausgezeichnet mit der Tatsache, dass das Verhältnis von Oberfläche zu Volumen mit steigendem Partikelradius kleiner wird. Kupfer / 65 Unabhängig von der Elektronenbeugung bestätigt das EDX-Spektrum der Nanopartikel die Zusammensetzung der Probe. Neben dem dominierenden Signal für Cu sind Signale für C, N und Au sichtbar, die vom koordinierenden Lösungsmittel bzw. dem verwendeten Kohlenstoffbeschichteten Goldgrid stammen. Die Lösung der Partikel in Toluol wurde zudem mittels Dynamischer Lichtstreuung (DLS) vermessen (Abb. 30). Im Gegensatz zur Elektronenmikroskopie, die immer nur einen Ausschnitt aus einer Probe analysiert, wird in der DLS die Streueigenschaft der gesamten Probe vermessen. Sie liefert den gemittelten hydrodynamischen Radius durch Fitten der experimentell erhaltenen Korrelationsfunktion an eine theoretische Funktion, die die Diffusionskonstante D und über die Stokes-Einstein-Gleichung den Radius r enthält. Die Verteilungsfunktion zeigt einen einziges Signal bei r = 5 nm, wobei die Beiträge von größeren Partikeln aufgrund ihres geringen Streubeitrags (Gewichtung 10-9 bis 10-14) vernachlässigt werden können. Das heißt, der Durchmesser der Partikel ist im Vergleich zu dem aus den TEM-Aufnahmen ermittelten um 2.5 nm größer. Da in der DLS hydrodynamische Radien bestimmt werden, kann diese Differenz den Liganden (HDA) zugeordnet werden, die den metallischen Kupferkern umgeben. Die Differenz stimmt sehr gut mit den im Elektronenmikroskop beobachteten Abständen zwischen zwei benachbarten Partikeln überein. 7 Intensität [a. u.] 1x10 2.11 nm 0 3 4 5 6 7 8 9 10 Radius [nm] Abb. 30: Lichtstreuung von Kupfer-Nanopartikeln in Toluol. Das Modell des Hexadecylamins wurde mit Molecular Mechanics 2 berechnet (Farbkennzeichnung: Kohlenstoff dunkelgrau, Wasserstoff hellgrau, Stickstoff blau, freies Elektronenpaar rot). Berechnet man mit Hilfe eines einfachen Modelling-Programms die Struktur des langkettigen Amins, kann seine Länge zu 2.11 nm abgeschätzt werden. Daraus ergibt sich der maximal mögliche Abstand zwischen zwei Partikeln zu 4.2 nm. Da der tatsächlich beobachtete Abstand (2.5 nm) weit unter dem maximal möglichen liegt, kann davon ausgegangen werden, dass (1) die Moleküle nicht in der oben abgebildeten Konformation, sondern zumindest teilweise in cis-Konformation vorliegen und/oder (2) zwischen den Ketten Verzahnungen vorliegen, die durch vdW-Wechselwirkungen stabilisiert werden. 66 / Kupfer 5.3.4 Variation der Reaktionsparameter Da Größe und Form von Nanopartikeln einen Einfluss auf ihre physikalischen und chemischen Eigenschaften haben (siehe Kapitel 2.2), ist es ein erklärtes Ziel von synthetischen Zugängen, diese Charakteristika zu steuern. In dem von uns gewählten System ist dies durch Variation von Precursor und Reaktionsparametern möglich. [Cu(OCH(Me)CH2NMe2)2] ist durch seine Kristallinität, Reinheit, reduzierenden Liganden und Zerfallstemperatur und –mechanismus bereits hervorragend für die Synthese von Kupfer-Nanopartikeln geeignet. Daher wurde das Augenmerk auf die Variation der Reaktionsparameter, nämlich die Wahl von Lösungsmittel, Temperatur, Zeit und Konzentration, gelegt. Variation des Lösungsmittels Führt man die Reaktion in TOPO an Stelle von Hexadecylamin unter ansonsten identischen Bedingungen durch, beobachtet man die Bildung eines dunklen Niederschlags, der nicht in Toluol löslich ist. Die zugehörigen TEM-Aufnahmen (Abb. 31 links) zeigen größere, meist sphärische Partikel mit einer breiten Größenverteilung von 50 nm bis 3.7 µm. Die Elektronenbeugung (nicht abgebildet) bestätigt die Bildung von metallischem Kupfer. Das heißt, die Verwendung von TOPO führt zu einem Anwachsen der Partikel um mindestens den Faktor 10 im Vergleich zur Reaktion in HDA. Dies erklärt auch die beobachtete Unlöslichkeit in Toluol. Abb. 31: TEM-Aufnahmen von unlöslichen Kupfer-Partikeln. Links: durch Pyrolyse von [Cu(OCH(Me)CH2NMe2)2] in 6 g TOPO (Bedingungen bis auf die Verwendung von TOPO wie in Kapitel 5.3.3). Bar = 200 nm. Rechts: durch Pyrolyse von 4 mL 0.3-M [Cu(OCH(Me)CH2NMe2)2] (in TBP) in 5 g TOPO (JH-88a). Bar = 950 nm. Es spielt jedoch nicht nur die Wahl des koordinierenden Lösungsmittel eine Rolle, sondern auch die des Lösungsmittels für den Precursor. Wird die obige Reaktion unter Verwendung von Tributylphosphin (TBP) an Stelle von Octylamin durchgeführt, sinkt der Radius der Partikel und verschiedenste Formen können in den TEM-Aufnahmen (Abb. 31 rechts) beobachtet werden: sphärische, ovale und überraschenderweise auch Stäbchen und Zylinder. Die Größe reicht von 8 bis 60 nm für die sphärischen und von 15-40 nm (kurze Achse) und 50-100 nm (lange Achse) für die länglichen Partikel. Auch hier bestätigt die Elektronenbeugung die Bildung von metallischen Kupfer-Partikeln. Kupfer / 67 Variation der Zeit Um den Einfluss der Wachstumszeit auf Größe und Form der gebildeten Partikel zu prüfen, wurde die Reaktion aus Kapitel 5.3.3 bereits nach 10 statt nach 30 Minuten durch Entfernen des Heizpilzes beendet. Die Lösung der aufgearbeiteten Partikel in Toluol absorbiert im Bereich des sichtbaren Lichts mit einem Absorptionsmaximum von λmax = 558 nm (Spektrum nicht abgebildet) und ist damit identisch mit der Reaktion, die 30 Minuten wachsen gelassen wurde. Dies war zu erwarten, da die Lage der Absorption für Partikel kleiner 20 nm nicht von der Größe abhängt. Die TEM-Aufnahme der Partikel (Abb. 32) zeigt jedoch, dass die Länge der Wachstumszeit durchaus einen Einfluss hat: die Größenverteilung ist wesentlich breiter bzw. bimodal mit kleinen Partikeln von 3-4 nm und etwa doppelt so großen Partikeln von 67 nm im Durchmesser. Die Partikel bestehen aus kristallinem Kupfer mit den bereits oben beschriebenen zusätzlichen Reflexen durch CuxO der an Luft oxidierten Oberfläche (SAED nicht abgebildet). Das heißt, die Partikel, die nach einer Reaktionszeit von 10 Minuten in der Lösung vorhanden sind, unterscheiden sich nicht in ihrer Zusammensetzung, Kristallinität oder Form, sondern lediglich in ihrer Größe von den nach 30 Minuten erhaltenen, sehr einheitlichen Partikeln. Abb. 32: Links: TEM-Aufnahme von Cu-Nanopartikeln, erhalten durch Pyrolyse von 4 mL 0.3-M [Cu(OCH(Me)CH2NMe2)2] (in Octylamin) in 7 g HDA (JH-92b) nach einer Wachstumszeit von t = 10 Minuten. Bar = 45 nm. Rechts zum Vergleich dieselbe Reaktion mit einer Wachstumszeit von 30 Minuten. Bar = 65 nm. Sie stellen vielmehr einen Ausschnitt dieser Reaktion kurz nach der Injektion der Precursor dar und es ist daher anzunehmen, dass hier ein früher Zeitpunkt des Wachstums abgebildet ist. Die großen Partikel in Abb. 32 haben bereits nahezu die Größe der fertigen Partikel (7-8 nm), während die kleineren Partikel am Ende der Reaktion nicht mehr vorhanden sind. Da die Monomerkonzentration zu diesem Zeitpunkt sehr niedrig ist, ist aufgrund der Wachstumskinetik von Halbleiterpartikeln, die am System CdSe intensiv erforscht worden ist und in Kapitel 2.1 vorgestellt wurde, anzunehmen, dass der kritische Radius für ein Auflösen der Partikel zwischen dem der großen und dem der kleinen Partikel liegt, d. h. 3 nm < rc < 7 nm. In Folge dieser Konstellation tritt ostwald ripening ein, d. h. die kleineren Partikel lösen sich auf und 68 / Kupfer das Material lagert sich an die vorhandenen größeren Partikel an. Dieser Effekt der Instabilität von kleinen Partikeln, d. h. die Abhängigkeit der Energie des Metallatoms in einem Nanopartikel von der Größe des Partikels, ist sogar noch weitaus stärker als bisher angenommen wie Campbell et al.[134] durch kalorimetrische Messungen der Adsorptionswärme in dem System Pb/MgO (110) zeigen konnten. Bis zu einer Größe von etwa 2.5 nm ist beispielsweise die Stabilität von Bleipartikeln auf MgO (110) vergleichbar der des Festkörpers (die Adsorptionswärme nimmt nur um etwa 7 kJ/Mol zu 188 kJ/Mol ab). Verringert sich die Größe jedoch weiter, nimmt die Adsorptionswärme und damit die Stabilität der Partikel drastisch ab. Für ein Bleipartikel mit einem Durchmesser von 1 nm beträgt sie nur noch etwa 130 kJ/Mol und liegt damit um ein Drittel niedriger als die des Festkörpers. Weitere Untersuchungen dieses Wachstumsprozesses werden in Kapitel 5.5 vorgestellt. Variation der Temperatur Der Einfluss der Temperatur wurde durch Ausführung der Reaktion aus Kapitel 5.3.3 bei Injektionstemperaturen von 200 °C und 250 °C beleuchtet. Die Reaktionen verlaufen analog zu der oben beschriebenen, wobei jedoch eine Injektion der 25 °C kalten Kupfer-Lösung bei 200 °C zu einer Absenkung der Temperatur unter 200 °C führt und der Precursor so lange unzersetzt in Lösung vorliegt bis die Temperatur wieder 200 °C erreicht hat. In allen Fällen entstanden rote, in Toluol lösliche Partikel mit einer UV/VIS Absorption von λ = 558 nm. Die dazugehörigen TEM-Bilder (nicht abgebildet) zeigen alle sphärische separierte Partikel, deren Elektronenbeugung metallisches Kupfer mit einer oxidierten Oberfläche zeigt. Der Unterschied jedoch liegt in der Größenverteilung der entstandenen Partikel. Während die Injektion bei hohen Temperaturen, d. h. bei 250 - 300 °C, zu hoch monodispersen, etwa 8 nm großen Partikeln führt, entstehen bei niedrigen Temperaturen Partikel in einem Größenbereich von etwa 3 bis 8 nm. Die Beobachtungen aus der Wachstumskinetik bei unterschiedlichen Wachstumszeiten haben bereits darauf hinweisen, dass ostwald ripening in den Wachstumsprozess hin zu der erwünschten schmalen Größenverteilung involviert ist und dass es eine Funktion der Wachstumszeit ist. Ganz offensichtlich bestimmt also auch ein anderer Reaktionsparameter, nämlich die Temperatur, in welchem Maß die Größenverteilung fokussiert wird. Es sind Temperaturen zwischen 250 bis 300°C erforderlich, um den Materialtransfer von kleinen zu großen Partikeln zu aktivieren. Es ist allerdings auch denkbar, dass ostwald ripening bei niedrigeren Temperaturen nur mit einer geringeren Geschwindigkeit abläuft. Um dies zu überprüfen, wurde die Injektion bei 200 °C durchgeführt und die Reaktionsmischung für etwa 4 Stunden bei dieser Temperatur gehalten. Das Ergebnis sind Kupfer-Nanopartikel mit einer wesentlich schmaleren Größenverteilung, die der der bei 250 oder 300 °C gewachsenen Partikel entspricht (nicht abgebildet). Das heißt, ostwald ripening ist im Falle des Wachstums von Kupfer-Nanopartikeln in HDA sowohl eine Funktion der Temperatur als auch der Zeit. Kupfer / 69 Variation der Konzentration Führt man die Reaktion aus Abb. 31, rechts, mit einem Zehntel der Menge an Precursor unter ansonsten konstanten Bedingungen durch, wird ebenfalls ein dunkler Feststoff gebildet, der in Toluol unlöslich ist. Die elektronenmikroskopischen Aufnahmen (Abb. 33) zeigen im Gegensatz zu der Reaktion mit hoher Konzentration an Precursor keine elongierten Partikel mehr. Die Partikel haben vielmehr eine einheitliche, leicht ovale Form in einem Größenbereich von 30-60 nm. Die Bildung von elongierten Partikeln bei erhöhten Konzentrationen an Monomer ist ein bei II-VI-Halbleiterpartikeln wohl bekanntes Phänomen (siehe Kapitel 3.2.1). Es wird davon ausgegangen, dass bei einer hohen Konzentration an Monomer die Reaktivität der verschiedenen Oberflächen zum Tragen kommt, so dass das Wachstum anisotrop verläuft. Abb. 33: Links: TEM-Aufnahmen von Kupfer-Nanopartikeln, hergestellt durch Pyrolyse von 0.4 mL 0.3-M [Cu(OCH(Me)CH2NMe2)2] (in TBP) in 6 g TOPO. Bar = 130 nm. Rechts zum Vergleich eine TEM-Aufnahme der Reaktion aus Abb. 31 rechts. Bar = 950 nm. Dieses Konzept lässt sich offensichtlich auf die Synthese von Kupfer-Nanopartikeln übertragen, denn eine Erniedrigung der Konzentration führt offensichtlich zur Ausbildung von Partikeln mit kleineren Seitenverhältnissen (aspect ratios). Aufgrund der Tatsache, dass sich die Partikel bei hoher Vergrößerung als instabil erweisen, ist es jedoch nicht möglich gewesen, die bevorzugte Wachstumsrichtung bzw. reaktive Oberfläche mittels TEM zu bestimmen. 5.4 Oberflächenmodifizierung von HDA-gecappten Cu-Nanopartikeln 5.4.1 Einleitung Nach der Synthese liegen die Kupfer-Partikel umgeben von einer Hülle aus koordiniertem Hexadecylamin vor. Wie in Kapitel 3.4.1 beschrieben, eignen sich koordinierende Liganden für den Austausch mit Liganden, die ihrerseits eine kovalente Bindung zu den Oberflächenatomen der Partikel ausbilden können. In der Literatur sind zwar Kupferpartikel mit den verschiedensten Oberflächenbedeckungen synthetisiert worden, jedoch ist bislang nichts über die 70 / Kupfer postsynthetische Oberflächenmodifizierung von Kupfer-Nanopartikeln berichtet worden. Es sei hinzugefügt, dass sich die meisten der bekannten Systeme für solche Untersuchungen auch nicht eignen, da die Nanokristallite z. B. in Polymeren oder Mizellen vorliegen (siehe Kapitel 5.1). Das Ziel dieser Arbeit war es daher, die Oberflächenreaktivität der Kupfer-Kolloide zu untersuchen und möglicherweise für eine Oberflächenmodifizierung auszunutzen. Mit Hilfe von geeigneten Modifizierungen wäre es dann möglich, die chemischen Eigenschaften (wie z. B. die Löslichkeit) gezielt zu steuern. Viele Gruppen berichten über eine starke Oxidationsempfindlichkeit der Kupferpartikel wie wir sie in der SAED beobachtet haben.[110, 112, 115, 122] Aufgrund der extremen Sauerstoff-Empfindlichkeit der Partikel war ein weiteres Ziel dieser Austauschexperimente der bessere Schutz der Kupferkristallite vor Oxidation. Wir haben daher zunächst die Reaktivität gegenüber Wasser und Sauerstoff näher untersucht und dann aufgrund der starken Wechselwirkung von Thiolatgruppen mit Übergangsmetallen, die bis zur Ausbildung einer kovalenten M-SBindung führt, Thiole für Untersuchungen zum Austausch des koordinativ gebundenen Hexadecylamins ausgewählt. 5.4.2 Verhalten gegenüber Wasser und Sauerstoff Setzt man eine Lösung von Kupfer-Nanopartikeln in Toluol dem Kontakt mit Luft aus und verfolgt die Veränderungen mittels UV/VIS-Sepktroskopie, beobachtet man eine kontinuierliche Rotverschiebung von 558 nm auf 599 nm innerhalb von mehreren Stunden (Abb. 34). 2 45 Min. (585 nm) Absorption [a. u.] 0 Min. (559 nm) 4 h (599 nm) 2d (ca. 613 nm) 1 0 500 550 600 650 700 Wellenlänge [nm] Abb. 34: Verlauf des UV/VIS-Spektrums von Kupfer-Nanopartikeln nach Kontakt mit Luftsauerstoff. Dabei lassen sich drei Phasen unterscheiden: in den ersten 45 Minuten nimmt die Absorption bei gleichzeitiger Verschiebung des Absorptionsmaximums um 24 nm auf 585 nm zunächst zu und fällt dann im Laufe der nächsten Stunden wieder ab, wobei das Absorptionsmaximum nur noch um 14 nm verschoben wird und mit einer sichtbaren Verbreiterung der Bande verbunden ist. Nach zwei Tagen schließlich ist keine Absorption mehr detektierbar. Der gesamte Vorgang wird begleitet von einer Farbänderung von rot nach Kupfer / 71 blau. Aufgrund der Elektronenbeugung (SAED), die an luftoxidierten Partikeln durchgeführt worden ist (siehe Kapitel 5.3.3), ist bekannt, dass (1) die Kupferoberfläche leicht durch Luftsauerstoff oxidiert wird, (2) dieser Prozess sehr schnell abläuft (wenige Minuten) und (3) die entstehende Oxidschicht als kristalline Festkörperstruktur vorliegt. Es ist anzunehmen, dass die erste Phase des Luftkontakts der Lösung ebenfalls mit einer Oxidation der Kupferoberfläche einhergeht. Die dabei beobachtete Rotverschiebung wird daher auf die Bildung von CuxO zurückgeführt. Da das Oxid als kristalline Schicht vorliegt, tragen seine Absorptionseigenschaften signifikant zum UV/VIS Spektrum bei: CuO ist ein Halbleiter mit einer Bandlücke von 1.85 eV (670 nm, Festkörper), die aufgrund des quantum confinements mit abnehmendem Radius anwächst. Für 6 nm-Partikel wurde eine Bandlücke von 2.18 eV (568 nm) gemessen.[135] Die Absorption der nanopartikulären Lösung von 8 nm großen Kupfer-Nanopartikeln in Toluol verschiebt sich zu Anfang sehr rasch zu höheren Wellenlängen, nähert sich dann jedoch einem Maximum bei 599 nm an. Diese Absorption kann CuO-Partikeln bzw. Kupferpartikeln mit einer CuO-Schicht zugeordnet werden, denn die literaturbekannte Absorption von 570 nm ist für 6 nm gemessen worden und müsste für 8 nm rotverschoben sein. Nach vier Tagen ist abgesehen von einer sehr breiten Bande zwischen 550 und 700 nm keine Absorption mehr zu erkennen. Dies lässt darauf schließen, dass die Partikel nicht über einen längeren Zeitraum in Lösung stabil sind, und lässt sich durch eine TEM-Untersuchung bestätigen. Allerdings gilt diese Aussage nur für verdünnte Lösungen, denn konzentriertere Lösungen von Cu-Nanopartikeln, die an Luft oxidiert worden sind, bestehen nach wie vor aus Partikeln wie elektronenmikroskopische Aufnahmen bewiesen haben. Dabei ist auffällig, dass weder die Größe der Partikel sich während der Oxidation verändert hat noch die Intensität der oxidischen Reflexe von denen der in Abb. 29 gezeigten abweicht. Dies ist ein weiterer Hinweis darauf, dass die Absorption bei λ = 599 nm Kupferpartikeln mit einer Kupferoxidschicht entspricht und dass die Absorption aufgrund der Anregung von Elektronen über die Bandlücke des Halbleiters zustande kommt. Die Absorption kann nicht von metallischem Kupfer stammen, da sich die Größe der Partikel nicht geändert hat und sie damit nach wie vor ein Absorptionsmaximum von λ = 558 nm aufweisen müssten, wenn man davon ausgeht, dass die Oberfläche der Partikel mit der elektromagnetischen Strahlung wechselwirkt, um die entsprechenden Oberflächenplasmonen anzuregen. Versetzt man eine toluolische Lösung von aufgearbeiteten Kupfer-Nanopartikeln hingegen mit dem gleichen Volumen an entgastem Wasser, so ist keinerlei Veränderung zu verzeichnen. Selbst nach Wochen liegen eine klare wässrige Phase und eine rote organische Phase nebeneinander vor. Die die Partikel umgebende, hydrophobe Schicht aus Alkylketten verhindert also wirkungsvoll den Kontakt von Wassermolekülen mit der ansonsten extrem reaktiven Kupfer-Oberfläche. 72 / Kupfer 5.4.3 Verhalten gegenüber Thiolen Die Untersuchungen in dieser Reihe wurde mit Hilfe einer Stammlösung von Kupfer-Nanopartikeln in Toluol durchgeführt, um die Menge an benötigtem Thiol möglichst genau abschätzen zu können (siehe Experimentalteil). Die Kolloidlösung wurde mit einem leichten Überschuss des Thiols versetzt und bei Raumtemperatur unter Schutzgas rühren gelassen. Als Thiole wurden tert-Butylthiol, Naphtylthiol und Octadecanthiol verwendet. Bei der Verwendung von tert-Butylthiol beobachtet man eine Farbveränderung von rot nach blau innerhalb von wenigen Minuten. Die Lösung ist in dieser Form über Wochen stabil, d. h. es wird kein Niederschlag gebildet. Die optischen Eigenschaften der Lösung unterscheiden sich deutlich von denen des Edukts: die Oberflächenplasmonenresonanz des Kupfers bei λ = 558 nm ist nicht mehr detektierbar und es ist keine neue Absorptionsbande entstanden. Das bedeutet, dass zumindest an der Oberfläche der Partikel keine Kupfer-Atome mehr vorliegen und dass kein Material mit einer definierten Absorption entstanden ist wie zum Beispiel Nanopartikel von Cu2S, die bei einer vergleichbaren Größe von 7 nm Licht der Wellenlänge 620 nm absorbieren sollten.[136] Nicht auszuschließen ist zunächst jedoch die Bildung von nanopartikulärem CuS, das eine charakteristische Absorption bei λ = 900 nm zeigt[136] und damit außerhalb des von uns messbaren Bereichs liegt. Aus der elektronenmikroskopischen Untersuchung geht allerdings hervor, dass die blaue Lösung keine Nanopartikel mehr enthält, d. h. auch die Bildung von CuS kann ausgeschlossen werden. Man kann also (1) aufgrund der fehlenden OPR von Kupfer davon ausgehen, dass tert-Butylthiol zur Oberfläche der Partikel vordringen und dort mit Kupfer reagieren kann. Es ist darüber hinaus anzunehmen, dass dies (2) aufgrund der Abwesenheit von Partikeln im TEM zu einem Auflösen der CuPartikel führt und dass am Ende dieses „Ätz-Prozesses“ mit hoher Wahrscheinlichkeit molekulare Kupferverbindungen vorliegen. Bei der Verwendung von Naphtylhiol beobachtet man an Stelle eines Farbumschlags die Bildung eines Niederschlags nach etwa 30 Minuten. Dieser wurde durch mehrmaliges Waschen gereinigt und elektronenmikroskopisch untersucht. Die TEM-Aufnahmen (Abb. 35 rechts) zeigen agglomerierte Partikel, mit einem Durchmesser von 5 bis 13 nm. Der Vergleich mit der Aufnahme vor der Oberflächenmodifizierung (Abb. 35 links) zeigt, dass die Größenverteilung der Partikel durch die Reaktion breiter geworden ist. Das Beugungsbild (SAED) der modifizierten Partikel (nicht abgebildet) bestätigt, dass es sich um kristalline Kupferpartikel handelt, d. h. die Zusammensetzung der kristallinen Phase hat sich durch die Reaktion mit Naphtylthiol nicht verändert. Darüber hinaus ist festzustellen, dass keine zusätzlichen Reflexe gefunden wurden. Das bedeutet, dass (1) keine kristallinen Phasen von CuxS entstanden sind und (2) die Oberfläche im Gegensatz zu den nicht-modifizierten Partikeln nicht oxidiert worden ist. Nach der Behandlung mit Naphtylthiol liegen also nach wie vor Kupfer-Partikel vor, aber die Oberfläche muss sich definitiv verändert haben. Da im Beugungsbild keine Oxide zu sehen sind, heißt das, dass die Oberfläche besser gegen Oxidation geschützt ist. Aus der Tatsache, dass die Partikel trotz geschützter Oberfläche einen Kupfer / 73 Niederschlag ausbilden, lässt sich aber schließen, dass kein Naphtylthiolat die Oberfläche terminiert. Abb. 35: Links: TEM-Aufnahme der Kupfer-Nanopartikel vor der Oberflächenmodifizierung (stabilisiert durch HDA), bar = 125 nm. Rechts: TEM-Aufnahmen des Niederschlags aus der Reaktion dieser Nanopartikel mit Naphtylthiol. Bar = 68 nm. Ganz ähnliche Beobachtungen wurden bei der Behandlung von HDA-bedeckten Kupferpartikeln mit Octadecanthiol gemacht. Auch hier bildet sich innerhalb von etwa 30 Minuten ein dunkler Niederschlag, der im Elektronenmikroskop aggregierte Partikel mit einer im Vergleich zu den Eduktpartikeln verbreiterten Größenverteilung (6-19 nm) zeigt. Ebenso wie bei der Behandlung mit Naphtylthiol liegen nach Behandlung mit Octadecanthiol kristalline Kupferpartikel vor, deren SAED weder oxidische noch andere zusätzliche Reflexe aufweist. Wie oben ist also die Oberfläche verändert worden, jedoch nicht durch Terminierung mit Octadecanthiolat, da die Partikel nach außen weiterhin mit einer Alkylkette terminiert und damit in Toluol löslich sein müssten. Abb. 36: Links: TEM-Aufnahme der Kupfer-Nanopartikel vor der Oberflächenmodifizierung (stabilisiert durch HDA). Bar = 77 nm. Rechts: TEM-Aufnahmen des Niederschlags aus der Reaktion dieser Nanopartikel mit Octadecanthiol. Bar = 80 nm. Aus den Arbeiten mit Alkanthiolen auf definierten (100)-, (110)- und (111)- Kupferoberflächen, die von S. Vollmer aus der Gruppe um Ch. Wöll durchgeführt wurden[137, 138] ist be74 / Kupfer kannt, dass die Bedeckung und Zusammensetzungen von Alkanthiolat-Monolagen auf Cu (110) empfindlich von der Temperatur abhängt. Alkanthiole HS-(CH2)n-1-CH3 der Kettenlängen n = 1-10 bilden bei niedrigen Temperaturen (< 200 K) eine (2 x 2) Thiolatschicht auf Kupfer (100) aus. Die Sättigung der Oberfläche bei Raumtemperatur führt hingegen zu einer dichter gepackten c(6 x 2) Thiolatmonolage. Dieser Film ist metastabil und lagert sich bei Raumtemperatur durch partielle Desorption von Alkylketten in einen stabilen Thiolat-SulfidFilm um, der ebenfalls c(6 x 2)-strukturiert ist. Die Bildung von Sulfid beginnt bei Temperaturen um 250 K und endet bei etwa 400 K, wenn die Alkylketten komplett desorbiert sind. Oberhalb von 500 K findet eine Schwefel-induzierte Rekonstruktion der Oberfläche statt. Der Grund für diesen Prozess liegt in der starken Cu-S-Wechselwirkung, die für identische Strukturen der Monolagen unabhängig von der Kettenlänge des Thiols verantwortlich ist, während die Wechselwirkung von benachbarten Ketten eine untergeordnete Rolle spielt. Aus früheren Arbeiten mit Alkanthiol-Filmen auf polykristallinem Kupfer[139] ist außerdem bekannt, dass längeres Einwirken des Alkanthiols auf die Kupferoberfläche zu einer Reaktion des Thiols mit der Oberfläche und zur Bildung von Kupfersulfid führt. Aufgrund dieser Arbeiten liegt die Vermutung nahe, dass bei der Behandlung von Cum(HDA)n mit Thiolen zwar zunächst ein Austausch stattfindet (der wahrscheinlich auch für die Verbreiterung der Größenverteilung verantwortlich ist), dann jedoch der Kohlenwasserstoff-Rest des Thiols desorbiert und es zur Ausbildung von CuS kommt. Gegen diese Vermutung spricht, dass weder EDX-Spektren noch SAED einen direkten Nachweis für die Anwesenheit von CuS erbrachten. Auf der anderen Seite ist diese Tatsache nicht erstaunlich, da (1) nicht zu erwarten ist, dass eine kristalline Sulfid-Phase ausgebildet wird und (2) die Dicke der Schicht aufgrund der geringen Menge an Thiol (pro OF-Atom Cu wurde ein Molekül Thiol zugegeben) wesentlich schmaler sein wird als z. B. eine Oxidschicht. Darüber hinaus kann (3) die Abwesenheit von oxidischen Reflexen als indirekter Nachweis gedeutet werden, denn auch eine dünne Schicht an CuS kann die Partikeloberfläche vor der Oxidation durch Sauerstoff schützen. 5.4.4 Verhalten gegenüber Carbonsäuren Da die Behandlung von HDA-geschützten Kupferpartikeln mit Thiolen aufgrund der starken Cu-S-Bindung bzw. der im Vergleich dazu schwachen S-C-Bindung wahrscheinlich zur Ausbildung von Oberflächen-Kupfersulfid führt, wurde getestet, ob sich Carbonsäuren für eine Oberflächenmodifizierung eignen. Als Vertreter dieser Verbindungsklasse wurde Ölsäure ausgewählt, da sie in der Literatur häufig zur Stabilisierung von metallischen Nanopartikeln verwendet wird.[50] Im Gegensatz zu den obigen Reaktionen wurde kein Niederschlag ausgebildet, sondern eine langsame Entfärbung der Lösung beobachtet. Die elektronenmikroskopischen Untersuchungen bestätigen das, was die fehlende OPR von Kupfer bzw. jeglicher Farbigkeit bereits impliziert: es sind keine Partikel mehr vorhanden. Neben dem kurzkettigen Thiol HS-C(CH3)3 führt also auch die Einwirkung von Ölsäure zu einer Auflösung der Kupfer / 75 Kupferpartikel und zur Ausbildung von molekularen Strukturen. Allerdings besteht ein farblicher Unterschied zwischen den mit t-Butylthiol und mit Ölsäure behandelten Proben. Erstere bildet blaue Lösungen aus, letztere farblose. Es ist jedoch bekannt, dass Cu(II) je nach Ligand blaue oder grüne und Cu(I) farblose Lösungen ergibt. Dies legt die Vermutung nahe, dass die metallischen Cu-Kolloide je nach zugegebenem Liganden in unterschiedlichem Maße oxidiert werden. Bei der Zugabe von Thiol demnach zu Cu(II) und bei Zugabe von Ölsäure zu Cu(I). 5.4.5 Verhalten gegenüber Kohlenmonoxid Aus den bisherigen Untersuchungen ist zu schließen, dass kleine Moleküle wie O2 und tertButylthiol leicht bis zur Oberfläche der Partikel vordringen und mit ihr entsprechend ihrer Reaktivität reagieren können. Dies führt damit zwar auch zu einer Oberflächenmodifizierung, aber nicht im Sinne eines Austauschs, sondern einer Reaktion. Unter Verwendung eines zwar kleinen, aber weniger reaktiven Moleküls wie z. B. Kohlenmonoxid könnte es aber dennoch möglich sein, einen Austausch oder zumindest eine Adsorption auf der Oberfläche zu erreichen. Abb. 37: Aufbau zur IR-Spektroskopie von Cu-Kolloiden in Toluol (Apparate der Technischen Chemie der Ruhr-Universität Bochum, Wilma Busser und Lamy Khodeir). 1 = Gaszufuhr (Argon oder CO), 2 = Reaktor für kolloidale Cu-Lösung, 3 = Pumpe, 4 = ATR-Messzelle (Circle Cell der Firma Sensir, Modell: Dura Sampl IR II, die in einen FTIR-Spektrometer der Firma Nicolet eingebaut wurde). Dafür werden etwa 25 mL einer toluolischen Lösung mit einer vergleichsweise hohen Konzentration von 24 mmol/L an HDA-gecappten Kupfer-Nanopartikeln hergestellt und unter Luftausschuss in den geschlossenen Kreislauf eines ATR-Messgeräts integriert (siehe Abb. 37). Die kolloidale Lösung befindet sich in einem Glasgefäß, wird von dort mit Hilfe einer Pumpe über PVC-Schläuche zur Messzelle gesaugt und fließt anschließend wieder zurück in das Vorratsbehältnis. Die unveränderte weinrote Farbe der kolloidalen Lösung zeigt an, dass keinerlei Luftkontakt stattgefunden hat. Die Lösung wurde etwa eine Stunde unter Argon 76 / Kupfer zirkulieren gelassen, um den Diamantkristall in der ATR-Zelle vollständig zu benetzen. Nach einer Stunde wird das System unter einen CO-Druck von circa 5 bar gesetzt, wobei die Lösung der Kupferpartikel weiterhin zirkuliert, und mittels IR-Spektroskopie die Veränderungen des Systems verfolgt. Die Vermischung der Lösung mit Kohlenmonoxid wird durch das verwendete Mikropumpsystem gewährleistet, dessen Pumpenkopf aus zwei ineinander greifenden Zahnrädern aufgebaut ist. 102 100,0 nach 1 min 99,8 Transmission [%] nach 30 min Transmission [%] 101 nach 60 min 100 nach 90 min 99,6 99,4 2160 99,2 2045 1967 99,0 nach 120 min 2200 2150 2100 2050 2000 1950 1900 -1 Wellenzahl [cm ] 99 2600 2500 2400 2300 2200 2100 2000 1900 -1 Wellenzahl [cm ] Abb. 38: Infrarotspektren von Kupfer-Kolloiden in Toluol (c § PPRO / EHL 5DXPWHPSHUDWXU XQWHU HLQHP Druck von 5 bar Kohlenmonoxid (Background ist die kolloidale Lösung vor der Zudosierung von CO). Die Messungen erfolgten in einem Messbereich von 6000 – 400 cm-1 und einer Auflösung von 4 cm-1. Die Apertur betrug 92 %. Weiterhin wurde eine Verstärkung von 4 eingestellt. Die Erfassung der Analytik erfolgte über ein OMNICE.S.P. 5.2-Programm der Firma Nicolet. Die Messungen wurden in Zusammenarbeit mit Lamy Khodeir (Technische Chemie, Prof. Muhler) durchgeführt. In Abb. 38 unten ist das Schwingungsspektrum der kolloidalen Lösung gezeigt. Nach etwa einer Stunde erscheinen im Bereich der CO-Schwingungen drei Banden bei 2160, 1953 bzw. 2044 cm-1. Die Carbonyle von Cu2+-Kationen sind instabil und absorbieren bei tiefen Temperaturen oder unter hohem CO-Druck zwischen 2236 und 2160 cm-1. Cu+-Kationen bilden wesentlich stabilere Carbonyle, deren CO-Banden zwischen 2160 und 2130 cm-1 liegen.[140] Die von uns beobachtete, hochfrequente Bande bei 2160 cm-1 weist damit auf die Anwesenheit von kationischen Kupferatomen hin. Dies bedeutet entweder, dass die Nanokristallite nicht vollständig reduziert vorliegen oder aber, dass trotz vorsichtiger Überführung der kolloidalen Lösung in das ATR-System ein Kontakt mit Luftsauerstoff stattgefunden hat, der zur Oxidation der Kupferatome zu Cu1+/2+ geführt hat. Aufgrund der SAED-Messungen an Kupferpartikeln ohne Kontakt mit Sauerstoff (siehe Kapitel 5.3.3) bzw. aufgrund der Tatsache, dass eine Oxidation der Oberfläche sehr schnell abläuft (siehe Kapitel 5.4.2), ist letztere Möglichkeit jedoch wahrscheinlicher. Die Schwingungsfrequenz von auf metallischem Kupfer adsorbiertem CO kann Werte bis hinunter zu 2070 cm-1 annehmen[141], Kupfer / 77 während die Schwingungsfrequenzen von terminal gebundenem CO in der metallorganischen Chemie einen Bereich von 2120 bis 1850 cm-1 abdecken[142]. Demnach sollten die von uns beobachteten Banden terminal gebundenem CO entsprechen, wobei allerdings die Frage entsteht, warum zwei Banden in diesem Bereich liegen. Knözinger et al. berichten ebenfalls von zwei Banden (bei 2045 und 2018 cm-1), die sie nach Absorption von CO auf einem reduzierten Cu/SiO2-Katalysator beobachten und einem geminalem Cu0(CO)2-Komplex (2045 cm1 ) bzw. einem linearen Cu0-CO (2018 cm-1) zuordnen.[140] Unter der Annahme, dass die von uns hergestellten Partikel einen größeren Durchmesser aufweisen als die in der dort verwendeten Cu/SiO2-Probe, könnte man annehmen, dass die CO-Schwingungsfrequenz aufgrund einer stärkeren Elektronendonierung (größere Anzahl von nullwertigen Metallatomen) zu niedrigeren Wellenzahlen verschoben wird. Allerdings beobachten die Autoren nach Reduktion ihrer Probe bei höheren bzw. niedrigeren Temperaturen (wobei höhere Temperaturen größere und niedrigere Temperaturen kleinere Partikel erzeugen) eine Verschiebung von nur + bzw. -10 cm-1. Da die von uns beobachteten Banden jedoch 100 cm-1 auseinander liegen und zudem gleichzeitig und nicht nacheinander auftauchen, liegt es näher, zwei Spezies mit unterschiedlicher Verbrückung anzunehmen und die höherfrequente Bande einem terminal gebundenem CO bzw. die niederfrequente Bande einem verbrückenden CO zuzuordnen. Normalerweise liegt die Schwingungsfrequenz für verbrücktes CO auf Kupfer wesentlich niedriger (1835 bis 1814 cm-1)[143], aber es wurde auch schon eine Absorption von 2003 cm-1 für verbrücktes CO in einem Cu/Al2O3-Katalysator berichtet[141]. Darüber hinaus sind verbrückte Carbonyle weder typisch für Kupfer noch exisitieren Berichte über CO-Adsorption auf Kupferkolloiden in der Literatur, so dass ein Vergleich mit Ergebnissen anderer Gruppen schwierig ist. Es ist jedoch über die CO-Absorption auf wohl charakterisierten NickelKolloiden berichtet worden: die Autoren berichten unabhängig von der Größe der Partikel (!) über Schwingungsbanden bei 2140 cm-1 (im verwendeten LM gelöstes CO), 2030 cm-1 (terminales CO) und 1915 cm-1 (verbrückendes CO). Diese Ergebnisse sind sehr konsistent mit den von uns beobachteten Schwingungsbanden. 5.5 Untersuchungen zum Wachstumsmechanismus: Mie-Ellipsometrie Die ersten Schritte in der Synthese von Metall-Kolloid-Partikeln durch Reduktion von Metallkationen mit Bildung von metallischen Nukleationskeimen und anschließender Agglomeration, die zu Metallclustern mit wachsender Nuklearität führen, sind immer noch Spekulation. Mit Hilfe von in-situ X-Ray Absorptions-Untersuchungen (XANES) bei der Bildung von Cu-Kolloiden mit Tetraalkylammoniumsalzen als Stabilisator wurde erstmals Licht auf diese ersten Schritte geworfen. Demnach wird in einem ersten Schritt vierfach koordiniertes, quadratisch-planares Cu(II) zu einem linearen, zweifach koordinierten Cu(I)Komplex reduziert. Diesem schnellen Schritt folgt ein langsamerer Prozess der Agglomeration durch weitere Reduktion unter Ausbildung von Cu-Cu-Bindungen. Nach der 78 / Kupfer Reaktion ist der metallische Charakter vorherrschend und nur für die Oberflächenatome wird noch ein Oxidationszustand ungleich Null vermutet.[109] Eine weitere Untersuchung beschäftigt sich mit der Reduktion von Cu2+ durch COO-. Dazu wurden wässrige Lösungen von Cu(ClO4)2 und Natriumformiat einer Pulsradiolyse unterzogen und Änderungen in der optischen Absorption und Leitung aufgezeichnet. Drei verschiedene Abläufe können voneinander unterschieden werden: (1) Bildung und Auflösen von ersten Intermediaten (CuCO2 und Cu2+) innerhalb von etwa 100 ms. (2) Bildung von Nukleationskeimen innerhalb von Millisekunden bis Sekunden und (3) plötzliche Bildung von größeren Aggregaten im Sekundenbereich; zu Beginn ist nur eine Absorption im UV-Bereich zu sehen, wahrscheinlich durch nichtmetallische Oligomere, später zeigen die größeren Partikel Absorption im sichtbaren Bereich.[144] Innerhalb der physikalischen Fakultät der Ruhr-Universität Bochum gibt es eine weitere Messmethode, die sich sehr gut eignet, Wachstumsprozesse zu untersuchen: Partikel werden mit Laserlicht einer definierten Polarisationsrichtung bestrahlt und das von den Partikeln gestreute Licht unter einem festen Winkel gemessen. Die Veränderung des Polarisationszustandes des gestreuten Lichts wird unter Anwendung der Mie-Ellipsometrie analysiert. Die Mie-Ellipsometrie wurde erstmals 1994 von Hayashi und Tachibana[145] zur insitu Partikeldiagnostik in Plasmen vorgeschlagen Unter Verwendung dieses Ansatzes wurde von Gebauer[146] dieses Verfahren an der Ruhr-Universität Bochum für die in-situ Diagnostik der Partikelgrößenverteilung weiterentwickelt. Der ursprünglich für eine Plasmakammer entwickelte Aufbau wurde für die Untersuchung des Wachstums von Kupfer-Nanopartikeln in Lösung leicht modifiziert. Das von einem Argon-Ionen-Laser erzeugte kohärente Licht mit einer Wellenlänge von 488 nm wird zunächst durch einen Polarisator in einen definierten Polarisationszustand gebracht und anschließend in ein rechteckiges Glasgefäß mit den Maßen 4 cm x 4 cm x 10 cm eingestrahlt, das als Reaktionsraum diente. Das Glasgefäß war mit zwei NS14.5-Schliffen und einem Stickstoffhahn ausgestattet, so dass es wie in der Standardreaktion (Kapitel 5.3) mit Rückflusskühler und Temperaturfühler versehen werden konnte. Statt eines Heizpilzes, der sich für rechteckige Gefäße nicht eignet, wurde ein Heizdraht verwendet, mit dem das Glasgefäß umwickelt wurde. Dabei wurden Ein- und Austrittsöffnungen für das eingestrahlte Laserlicht bzw. das gestreute Licht freigelassen. Die Synthese der Kupfer-Nanopartikel erfolgte durch Injektion von [CuOCH(Me)CH2NMe2)2] (gelöst in n-Octylamin) in HDA bei T=200 °C. Es wurden die ellipsometrischen Winkel Ψ und ∆ während des Entstehens und Anwachsens der Nanopartikel gemessen. Die Messwerte sind in Abb. 39 als Funktion der Zeit aufgetragen. Man erkennt, dass die Messwerte bis zur Injektion des Precursors stabil sind. Nach der Injektion des Precursors ändert sich der Messwert von Ψ innerhalb von 40 Sekunden von 23° auf einen Minimalwert von 7°. Der Messwert steigt dann auf einen Wert von 9° an (1 Minute) und anschließend innerhalb von 20 Minuten linear bis auf einen Wert von 11.5°, wobei ein lokales Minimum Kupfer / 79 durchlaufen wird. Der Winkel ∆ ändert sich nach Injektion des Precursors ebenfalls sprunghaft von 355° auf 325° (45 Sekunden) und läuft dann nach einer kurzen Phase mit sehr geringer Steigung (1 Minute) asymptotisch auf einen Wert von ca. 260° zu, der nach etwa 32 Minuten konstant bleibt. Nach der Messung wurde die erkaltete Reaktionsmischung in der Glovebox aufgearbeitet und im TEM vermessen. Wie in den Messungen ohne Lasereinstrahlung sind kristalline, sphärische und wohl separierte Kupfer-Nanopartikel entstanden. 400 22 380 20 360 Psi [grad] 18 340 16 320 14 12 300 Psi 10 Delta [grad] 24 280 Delta 8 260 6 0 20 40 Zeit [min] Abb. 39: Gemessener zeitlicher Verlauf von Ψ und ∆ in HDA. Der Pfeil bezeichnet den Zeitpunkt der Injektion von [CuOCH(Me)CH2NMe2)2]. Diese Messungen wurden in Zusammenarbeit mit Thomas Galka und Gerd Gebauer (Experimentalphysik II, Prof. Winter) an der Ruhr-Universität Bochum durchgeführt. Die Entwicklung einer Theorie zur quantitativen Beschreibung der Messwerte und ihrer physikalischen Ursachen ist Gegenstand der aktuellen Forschung unserer Koorperationspartner in der Experimentalphysik II der Ruhr-Universität Bochum (Gerd Gebauer, Prof. Winter). Eine Aussage lässt sich aber auch schon zum jetzigen Zeitpunkt machen: nach der Injektion laufen zwei bis drei verschiedene Prozesse von unterschiedlicher Zeitdauer ab, zunächst ein oder zwei sehr schnelle von etwa 40 Sekunden (bis Punkt 1), bzw. 60 Sekunden (bis Punkt 2) und dann ein langsamer Prozess, der nach etwa 30 Minuten abgeschlossen ist (Punkt 3). Nach 30 Minuten sind sowohl für Ψ als auch für ∆ konstante Werte erreicht, d. h. das System ändert seine optischen Eigenschaften nicht mehr. Ein separat durchgeführtes Experiment, bei dem sofort nach der Injektion des Precursors eine kleine Probe der Reaktionsmischung entnommen wurde, liefert einen Hinweis darauf, welchen Charakter der erste Schritt haben könnte: die entnommene Probe löst sich vollständig in Methanol auf und die elektronenmikroskopische Aufnahme zeigt separierte Partikel mit einer Größe von 4-8 nm. Aufgrund der Löslichkeit in Methanol gehen wir davon aus, dass die Oberfläche noch nicht mit HDA bedeckt ist (die Alkylkette des HDAs bewirkt in Methanol eine Niederschlagsbildung der Kupferkolloide). Ein Möglichkeit, die Löslichkeit in Methanol zu erklären ist es anzunehmen, dass die Partikel mit noch in dem System anwesenden Liganden des Precursors, HO-CH(Me)CH2NMe2, bedeckt sind. Dies wird durch die 80 / Kupfer Beobachtung unterstützt, dass die Partikel keine Wechselwirkung miteinander eingehen: im Gegensatz zu dem sonst beobachteten Abstand von etwa 2 nm zwischen zwei benachbarten HDA-bedeckten Kupferpartikeln sind die Partikel mindestens 5 nm voneinander entfernt. Eine nach 2 Minuten entnommene Probe bildet bei Zugabe von Methanol sofort einen Niederschlag. Die TEM-Aufnahmen zeigen separierte Partikel mit einem Abstand von 2 nm. Dies weist auf eine Koordination mit einem Liganden hin, der Wechselwirkungen mit dem Liganden der ihm benachbarten Partikel eingehen kann, also auf HDA, dessen Alkylketten über vdW-Kräfte miteinander wechselwirken. Die Verteilung der Partikelgrößen ist etwas breiter als bei der direkt nach der Injektion entnommenen Probe. Eine nach 5 Minuten entnommene Probe schließlich zeigt die bereits auf Seite 68 beschriebene Charakteristik: die Partikel liegen nach wie vor mit einem minimalen Abstand von etwa 2 nm vor, weisen jedoch eine bimodale Größenverteilung mit 4 bzw. ≥ 8 nm auf. Diese Charakteristik ändert sich während der nächsten Stunde nicht wesentlich, erst nach mehreren Stunden wird eine allmähliche Fokussierung der Größenverteilung beobachtet. Somit scheinen drei Prozesse eine Rolle zu spielen: zunächst die Nukleation von Kupfer, die zur drastischen Veränderung des Messsignals bis Punkt 1 führt. Daran anschließend die Bedeckung der Partikel mit dem koordinierenden Lösungsmittel HDA, die möglicherweise den leichten Anstieg zu Punkt 2 verursacht, und schließlich der länger andauernde Prozess des Ostwald-Ripenings, der zu der bimodalen Verteilung führt (Punkt 3). Die Erhöhung der Reaktionszeit (besser noch der Temperatur) führt schließlich zu einer einheitlichen Partikelverteilung wie sie in Kapitel 5.3.3 gezeigt worden ist. Es sei angemerkt, dass die externe Messung bei sehr viel höheren Konzentrationen durchgeführt worden ist, da die entnommene Probenmenge ansonsten nicht für eine TEM-Untersuchung ausgereicht hätte. Die Konzentrationen entsprechen dabei denen des Kapitels 5.3.3, während die in der MieEllipsometrie etwa um den Faktor 100 kleiner sind. Die extern beobachteten Zeiten für die unterschiedlichen Prozesse liegen demnach wesentlich niedriger als die mit Hilfe der MieEllipsometrie bestimmten. 5.6 Zusammenfassung Unter der Ausnutzung des anorganisch-chemischen Wissens über maßgeschneiderte Precursor ist die organometallische Kupferverbindung [Cu(OCH(Me)CH2NMe2)2] als Edukt für die Synthese von metallischen Kupfer-Nanopartikeln eingeführt worden. Durch Thermolyse dieses Precursors in Hexadecylamin konnten nicht-wässrige Kolloide (non-aqueous colloids, NACs) mit nahezu monodisperser Größenverteilung hergestellt werden. Die sphärischen Partikel sind gut voneinander separiert und neigen zur Anordnung in zweidimensionalen, hexagonalen Mustern aus freistehenden Kolloiden. Eine solche Selbstordnung ist unseres Wissens nach in der Literatur bislang nicht für Kupferpartikel beschrieben worden. Der Vorteil dieses Precursors liegt darin, dass keine weiteren Zusätze, insbesondere von Kupfer / 81 Reduktionsmitteln, benötigt werden, da die koordinierenden Liganden das Zentralatom während des Zerfallsprozesses reduzieren. Darüber hinaus ist die Verbindung leicht und kostengünstig zugänglich. Aufgrund der Verwendung dieses organometallischen Precursors sind als Reaktionsmedium hochsiedende organischen Lösungsmittel (HDA, TOPO etc.) zugänglich, die gleichzeitig als Stabilisator fungieren. Dies reduziert nicht nur das System um einen weiteren Zusatz, sondern ermöglicht auch das Arbeiten unter Schutzgas in einem nichtwässrigen System. Die auf diese Weise hergestellten Nanopartikel weisen daher keinerlei oxidische Verunreinigungen auf. Es konnte gezeigt werden, dass die Reaktion sehr stark vom verwendeten Lösungsmittel abhängt. TOPO ist zwar ein ähnlich hochsiedendes Lösungsmittel wie HDA und wie dieses zur Koordination über freie Elektronenpaare befähigt, in allen Experimenten wurde jedoch Material mit Abmessungen erhalten, die weit über den in HDA beobachteten Werten liegen. Trotz des sauerstoff-haltigen Lösungsmittels wurde jedoch metallisches Kupfer gebildet und nicht eines seiner Oxide, wie aufgrund der leichten Oxidierbarkeit des Kupfers durchaus denkbar wäre. Das heißt, die Bildung von metallischen Agglomeraten und ihr Wachstum verlaufen in TOPO wesentlich schneller als in HDA, was darauf zurückzuführen ist, dass TOPO metallisches Kupfer wesentlich schlechter stabilisiert. Falls TOPO für Kupfer ein schlecht koordinierendes Lösungsmittel ist, muss die Konzentration an Metall-Nanopartikeln geringer sein, um Stabilisierung zu erreichen. Die Injektion von weniger Stock-Solution (0.4 mL statt 4 mL) des Precursors (als Lösung in Tributylphosphin) führt zu einer einheitlicheren Formverteilung der resultierenden Kupfer-Partikel, aber nach wie vor zu Agglomeration. Die entstandenen Partikel haben jedoch wesentlich kleinere Abmessungen, so dass die Annahme, dass die Stabilisierung durch TOPO schwächer ist, bestätigt werden konnte. Somit eignet sich TOPO zwar für die Synthese von Kupferpartikeln, erfüllt aber nicht die Qualitätsanforderungen in Bezug auf Monodispersität, Größensteuerung und Schutz der Oberfläche. Da bei der Verwendung von HDA jedoch keine Partikel hergestellt werden können, die größer als etwa 15 nm sind, eröffnet die Verwendung von TOPO die Möglichkeit, größere metallische Partikel herzustellen. Im Laufe der Arbeiten zur Oberflächenmodifizierung hat sich ein klares Bild der Reaktivität von ligandbedeckten Kupfernanopartikeln ergeben. Kleine Moleküle wie CO und O2 können durch die Ligandenhülle hindurch bis zur Oberfläche der Partikel durchdringen und dort entsprechend ihrer Reaktivität mit der Oberfläche reagieren. Sauerstoff bewirkt eine Oxidation der Oberflächenatome und führt zur Ausbildung von core-shell Cu/CuxO-Partikeln, die in höher konzentrierten Lösungen als HDA-stabilisierte Kolloide vorliegen. Die ausgebildete Oxidschicht schützt die Partikel dabei vor einer weiteren Oxidation. Da der Oxidationsprozess bereits kurz nach Luftkontakt beginnt, weisen die von uns hergestellten metallischen Partikel eine extrem reaktive Oberfläche auf. Kohlenmonoxid reagiert als reduktiv wirkendes Molekül natürlich nicht mit metallischem Kupfer, sondern wird auf der Oberfläche der Partikel adsorbiert. Die entsprechenden Schwingungsbanden konnten in-situ 82 / Kupfer durch ATR-Spektroskopie beobachtet werden und wurden terminal und verbrückend gebundenem CO zugeordnet. Die Beobachtung der CO-Adsorption auf HDA-bedeckten Kupferpartikeln hat erhebliche Bedeutung für katalytische Prozesse, z. B. für die Methanolsynthese mit einem Cu/ZnO/Al2O3-Katalysator. Es ist oft argumentiert worden, dass die für die Stabilisierung nötigen Liganden die Nutzung der Oberfläche eines Nanopartikels für einen katalytischen Prozess verhindern und dass deshalb Kolloide keine guten Modelle für einen Katalysator darstellen. Hiermit konnte gezeigt werden, dass diese Argumentation nicht richtig ist, da CO (und auch O2) durch die Ligandenhülle hindurch an die Oberfläche gelangen können. Kleine, aber sehr polare Moleküle wie H2O können hingegen nicht zur Oberfläche vordringen, da sie von den hydrophoben Alkylketten, die die Partikel umgeben, abgestoßen werden. Bei der Zugabe von Ölsäure bzw. tert-Butylthiol wird eine dritte Reaktionsmöglichkeit beobachtet: die Partikel werden in einer Art Ätzprozess aufgelöst und liegen in einer (molekularen) Form vor, die mit Hilfe des Elektronenmikroskops nicht mehr aufzulösen sind. Die Zugabe von Naphtylthiol bzw. Octadecanthiol führen schließlich zur Bildung eines Niederschlags, wobei wir annehmen, dass analog zu einkristallinen Kupferoberflächen zunächst eine Thiolschicht auf der Oberfläche gebildet wird, dann aber unter den Reaktionsbedingungen eine C-S-Bindungsspaltung erfolgt und die Oberfläche durch eine dünne Schicht Kupfersulfid bedeckt wird. Da die Beugungsbilder dieses Materials keine CuxO-Reflexe mehr zeigen, ist es auf diese Weise gelungen, die Partikel vor Oxidation durch Luftsauerstoff zu schützen. Für die Bewertung dieser Ergebnisse lohnt sich auch ein Vergleich mit Goldoberflächen: Es gibt eine Reihe von Untersuchungen zur Ausbildung von SAMs (Self Assembled Monolayers) auf Goldoberflächen und es problemlos möglich, GoldNanopartikel mit Thiolen zu modifizieren. Alkanthiole auf Goldoberflächen haben sich sogar zu dem Standardsystem für selbstordnende Monolagen entwickelt. Der Grund dafür liegt in der relativ inerten Reaktivität des Edelmetalls, wodurch Präparation und Analytik von SAMs ohne Schutzgasatmosphäre möglich ist. Die Wechselwirkung zwischen Gold und Schwefel wird aufgrund der Bindungsenergie Eb(S-Au(111)) ≈ 1.6 eV[147] als schwach bezeichnet und die Ausbildung von SAMs auf Gold daher der vdW-Wechselwirkungen von benachbarten Alkylketten zugeschrieben. Warum also lassen sich diese Ergebnisse nicht auch bei Verwendung mit Kupfer erhalten? Im Vergleich zu dem Edelmetall Gold sind andere Übergangsmetalle wie z. B. Kupfer wesentlich reaktiver. Die Folge ist, dass die meisten organischen Substanzen dissoziativ absorbiert werden und verschiedene absorbierte Spezies bilden.[148] Daher kann man nicht davon ausgehen, dass Experimente mit Gold repräsentativ für die Ausbildung von SAMs sind und auf andere Systeme wie Kupfer übertragen werden können. Unsere Ergebnisse zeigen, dass dies auch für die Oberflächenmodifizierung von Nanopartikeln gilt. Kupfer / 83 M e2 N O Cu N M e2 O N uk leatio n OH Cu M e 2N K oo rd inatio n vo n H D A Cu H 2N 13 O stw ald-R ip en ing O stw ald-R ip en ing Obwohl es wegen der zahlreichen Reaktionsparameter sehr schwer ist, einen allgemeingültigen Reaktionsmechanismus für die Entstehung der Kupferkolloide aufzustellen, liefern die zur Abhängigkeit von der Zeit und der Konzentration durchgeführten Experimente sowie die Mie-ellipsometrischen Messungen Anhaltspunkte für einen möglichen Ablauf, der in Schema 10 dargestellt ist. Wir nehmen an, dass die Reduktion des Cu(+II) zu nullwertigem Kupfer sehr rasch verläuft und zur sofortigen Bildung von kleinen Nukleationskeimen führt. Diese sind – wie die Löslichkeit in Methanol vermuten lässt – von noch nicht entwichenen Liganden umgeben. Nach dem Austausch dieses Liganden gegen das in hohem Überschuss vorliegende und nicht flüchtige HDA beginnt ein Reifungsprozess (Ostwald Ripening), bei dem Material von größeren zu kleineren Partikeln übertragen wird. Er führt zur Ausbildung einer bimodalen Verteilung mit großen und sehr kleinen Partikeln. Bei sehr langen Reaktionszeiten oder höheren Temperaturen führt dieser Prozess schließlich zu einer homogenen Partikelgrößenverteilung. Schema 10: Postulierter Wachstumsmechanismus für die Bildung von Kupferkolloiden durch Injektion von [Cu(OCH(Me)CH2NMe2)2] in heißes Hexadecylamin. 84 / Kupfer 6 Palladium-Nanopartikel 6.1 Stand der Forschung und Aufgabenstellung Die Forschungsinteressen an Palladium-Kolloiden sind wie in Kapitel 5 bereits beschrieben auf der einen Seite das Interesse an fundamentalen Eigenschaften beim Übergang Molekül – Cluster – Festkörper und auf der anderen Seite die möglichen technischen Anwendungen, vor allem in der Katalyse. Aufgrund des weitaus edleren Charakters des Palladiums eignen sich Palladium-Nanopartikel insbesondere für die Realisierung von miniaturisierten Bauteilen. In der Einleitung zu metallischen Nanopartikeln in Kapitel 5.1 ist bereits ein solcher Einelektronentransistor beschrieben worden. Kleine Edelmetall-Nanopartikel werden oft mit der Bezeichnung giant clusters belegt. Dies beruht auf massenspektroskopischen Untersuchungen, die gezeigt haben, dass die Bindungsenergie von Metallnanopartikeln mit weniger als 1000 Atomen aufgrund des Größenquantisierungseffekts periodisch variieren. Im Fall der Edelmetalle sind diejenigen mit sogenannten magic numbers stabil, d. h. dichtest hexagonal oder kubisch gepackte Cluster mit n Schalen um ein Zentralatom und damit Cluster mit einer abgeschlossenen äußeren Form. In Abb. 40 ist ein typischer Cluster mit n=5 Schalen gezeigt. Abb. 40: Idealisiertes Modell des giant clusters § phen§ OAc§ .[149] Tabelle 4: Übersicht über giant cluster mit n = 1 bis 8 Schalen. Cluster mit n = 1 bis 6 Schalen. Anzahl Schalen Anzahl Atome 1 1 + 12 = 13 2 13 + 42 = 55 3 55 + 92 = 147 4 147 + 162 = 309 Beispiel Größe [nm] Au55(PPh3)12Cl6 [150] 1.4 Pt309phen*36O30+10 [151] 1.8 Pd561phen36O200 [152] 5 309 + 252 = 561 2.44 6 561 + 362 = 923 7 923 + 492 = 1415 Pd1415phen60O~1100 [152] 3.15 8 1415 + 642 = 2057 Pd2057phen84O~1600 [152] 3.6 Palladium / 85 Die Zahl der Atome pro Schale berechnet sich nach der Summe über 10n2 + 2. In Tabelle 4 ist die Gesamtzahl der Atome im Metallkern für eine bis acht Schalen berechnet. Daraus ergeben sich (angenäherte) Summenformeln für die entsprechenden Partikel, was dazu geführt hat, dass sie eher den Clustern zugeordnet worden sind als Nanopartikeln, die im Allgemeinen eine Größenverteilung aufweisen. Die Herstellung dieser Materialien kann analog zum Kupfer-System auf verschiedenen Wegen erfolgen, nämlich durch physikalische Techniken, durch Reduktion des Metallsalzes mit den verschiedensten Reduktionsmitteln oder durch Verwendung von Templaten. Zum Schutz vor Agglomeration werden die Partikel in Gegenwart von Stabilisatoren hergestellt. Hierzu zählen Polymere, Thiole und Alkylammoniumsalze. Einige Beispiele seien kurz vorgestellt. Physikalische Techniken. Reetz und Mitarbeiter[153-155] stellten Palladium-Cluster auf elektrochemischem Wege her. Dabei werden anodisch aus einem Palladiumblech Metallkationen erzeugt, die zur Kathode wandern (Platin) und dort zu elementarem Palladium reduziert werden. Die Elektrolytlösung enthält Tetraoctylammoniumbromid (TOAB) oder andere quartäre Ammoniumsalze, die die entstehenden Cluster stabilisieren. Da der kritische Radius von Clustern umgekehrt proportional zur Überspannung ist, lässt sich durch Variation der Stromdichte die Größe der Partikel in einem Bereich von 1.4 bis 3.1 nm einstellen. Ebenfalls sehr kleine Partikel im Bereich von 1 bis 3 nm lassen sich durch Elektronenstrahlverdampfung und anschließender Kondensation zusammen mit organischen Lösungsmitteln als Stabilisatoren herstellen.[156] Reduktion der Metallsalze. Die Verwendung von Wasserstoff als Reduktionsmittel für Palladiumacetat wurde 1993 von Schmid publiziert.[152] Als Stabilisator wird Phenanthrolin verwendet und es entstehen Palladiumcluster mit 5, 7 und 8 Schalen, d. h. idealisierte Zusammensetzungen mit 561, 1415 bzw. 2057 Palladiumatomen, die einen Durchmesser von 2.5-4 nm aufweisen. Typisch chemische Reduktionsmittel sind hingegen Borhydride. Reduziert man allerdings Metallsalze mit Trialkylborhydriden in organischen Medien, ohne dass stabilisierende Liganden anwesend sind, entstehen amorphe Metallpulver, z. B. für Palladium mit Partikeln von 12-28 nm Größe.[157] Erst die Reduktion in Gegenwart von kationischen lipophilen Tensiden wie Tetralkylammoniumsalzen R4NX führt zu MetallKolloiden.[158] Dabei können Reduktionsmittel und Tensid entweder separat oder als kombiniertes Ammoniumborat NR4(BR3H) zugegeben werden oder aber Metallsalz und NR4X zu NR4MXn gekoppelt werden. Dieses Prinzip lässt sich auch auf wasserlösliche Liganden übertragen wie z. B. Betaine.[159] Thiol-stabilisierte Palladium-Partikel sind von mehreren Gruppen synthetisiert worden. Ulman et al.[160] reduzierten Palladiumacetat in einem einphasigen System (THF) in der Gegenwart von Octadecanthiol mit LiEt3BH4 und erhielten 2.25 nm große kristalline Pd-Partikel. Chen et al.[161] hingegen synthetisierten Palladiumpartikel mit einer Größe zwischen 1 und 5 nm in einem zweiphasigen Wasser-Toluol System. Dazu wird zunächst PdCl2 aus der salzsauren wässrigen Phase mittels Tetraoctylammoniumbromid in die 86 / Palladium organische Phase transferiert, dort mit Octanthiol versetzt und schließlich mit NaBH4 reduziert. Die Autoren beobachten in den ersten 15 Minuten die schnelle Bildung von großen Partikeln, die dann im Laufe der nächsten 60 Minuten zu kleineren und stabileren Partikeln zerfallen. Nach 90 Minuten Reaktionszeit sind im Elektronenmikroskop, wahrscheinlich aufgrund von vollständigem Zerfall, keine Partikel mehr zu sehen. Die sehr milde Thermolyse von Pd(NO3)2 durch Tetraalkylammoniumcarboxylate, die gleichzeitig als Reduktionsmittel und Liganden dienen, macht die größenselektive Synthese von Palladiumpartikeln (2.5 bis 6.8 nm) abhängig von der elektronischen Natur des Carboxylats möglich. Elektronenziehende Substituenten erniedrigen beispielsweise das Reduktionsvermögen der Carboxylate, was eine langsame Nukleation und daher große Partikel zur Folge hat.[162] Bei der Reduktion von PdCl2 durch Alkohole[163] in Gegenwart des zur Stabilisierung weit verbreiteten Polymers PVP kann die Größe der Partikel (1.7 bis 3 nm) durch die Menge des Polymers und die Art bzw. Konzentration des verwendeten Alkohols gesteuert werden. Beim Wechsel von Methanol über Ethanol zu 1-Propanol wächst die Größe der Partikel, wenn auch der Bereich sehr klein ist (diese Abhängigkeit trifft ebenso für Platin zu, doch für Rhodium wurde genau die umgekehrte Beobachtung gemacht). Größere (>3 nm) Partikel können durch schrittweises Wachstum erhalten werden. PdCl2 kann auch durch Erzeugung von Radikalen in einer mit Ultraschall behandelten wässrigen Lösung des Salzes zum Metall reduziert werden.[164, 165] In Gegenwart eines Stabilisators, z. B. SDS oder einem wässrigen Polymer, werden stabile Palladiumpartikel gebildet, deren Radius mit Hilfe des Stabilisators und der Konzentration an Pd(II)-Ionen gesteuert werden kann. In der Literatur wird dazu folgender Mechanismus angenommen: Durch die Einwirkung der Schallwellen entstehen in wässriger Lösung kleine Bläschen, die wachsen und schließlich kollabieren. Das Innere dieser Bläschen ist durch extrem hohe Temperaturen (mehrere tausend °C) und Drücke (mehrere hundert atm) charakterisiert, so dass Wasser sonolytisch in H- und OH-Radikale gespalten wird. Diese reagieren mit den Stabilisatoren zu sekundären, reduzierenden Radikale, die schließlich die Reduktion der Metallkationen bewirken. Ebenfalls durch Ultraschall, aber unter Verwendung von Pd(OAc)2 als Pd(II)-Quelle und einem quartären Ammoniumsalz als Stabilisator/Reduktionsmittel haben Gedanken et al.[166] Pd-Nanopartikel mit einem Durchmesser von 10-20 nm synthetisiert. Daneben ist es auch möglich, zunächst PdO-Nanopartikeln aus Pd(NO3)2 herzustellen und sie in einem nachfolgenden Schritt vollständig durch Ethanol zu reduzieren.[167] Obwohl eine Vielzahl von organometallischen Palladium-Precursoren inklusive (!) ihrer Anwendung zur Abscheidung von metallischen Filmen in CVD-Prozessen bekannt ist, gibt es kaum Berichte über ihre Verwendung als Precursoren für Nanopartikel. Giorgio und seine Mitarbeiter berichteten zwar über die Verwendung von Palladiumacetylacetonat, jedoch nur im Zusammenhang mit der Zersetzung auf MgO Mikrokristalliten.[168] Darüber hinaus wurde von der Verwendung von Allyl(cyclopentadienyl)palladium, CpPd(Allyl), berichtet, das mit Hilfe von Wasserstoff zu Palladiumclustern reduziert wurde.[169] Doch auch hier ist die Synthese mit Hilfe eines Templats, dem Zeolith NaY, durchgeführt worden und es sind Partikel Palladium / 87 mit einer sehr breiten Größenverteilung entstanden. Unseres Wissens existiert in der Literatur kein Bericht über die Verwendung von organometallischen Precursoren zur Herstellung von Liganden-stabilisierten, freistehenden Pd-NACs. Das Ziel war daher, einen geeigneten und einfach zugänglichen Precursor zu synthetisieren und ihn mittels Thermolyse in einem dafür besonders geeigneten System, nämlich einem heißen koordinierenden Lösungsmittel, zu Nanopartikeln umzusetzen. 6.2 Synthese von Palladium-Nanopartikeln unter Verwendung des organometallischen Precursors [(Cp)(Allyl)Pd] 6.2.1 Precursor Für die Synthese der metallischen Nanopartikel wurde zunächst eine organometallische, bereits als CVD-Precursor bekannte Verbindung, nämlich Cyclopentadienyl(allyl)palladium, verwendet. Die Synthese dieser Verbindung erfolgte in zwei Schritten über (1) die Umsetzung von Tetrachloropalladat mit Allylchorid zum Allylpalladiumchlorid-Dimer und (2) die Umsetzung des Dimers mit NaCp. Cp(Allyl)Pd ist eine leicht flüchtige Verbindung mit einem Dampfdruck von 25 torr bei 100 °C[170], die bei Raumtemperatur in Form von dunkelroten Kristallen vorliegt. Ihre Kristallinität und auch ihre relativ hohe Stabilität gegenüber Feuchtigkeit und Luft erleichtern die Handhabung des Precursors und führten erstmals 1988 zur Anwendung als CVD-Precursor für Palladiumschichten.[171] Die Zersetzung von Cp(Allyl)Pd bei 250°C und 10-4 Torr in Abwesenheit eines reaktiven Gases wie Wasserstoff ergab amorphe Palladiumschichten, die mit etwa 5% Kohlenstoff verunreinigt waren. War bereits elementares Palladium in Form von Nukleationskeimen vorhanden, so fand der Precursorzerfall bei niedrigeren Temperaturen statt, was dafür spricht, dass es sich bei der Zersetzung um einen autokatalytischen Prozess handelt. Als Nebenprodukte entstanden neben geringen Mengen Hexadien in erster Linie Propen und Cyclopentadien, die auf einen radikalischen Zerfallsmechanismus hinweisen.[171] Bei Zusatz von Wasserstoff lässt sich die Abscheidungstemperatur auf 30-60°C (bei 60 torr) erniedrigen und des weiteren der Reinheitsgrad der Schicht deutlich verbessern (< 1% Verunreinigungen), da in diesem Fall die Liganden in einem wahrscheinlich konzertiert ablaufenden Reaktionsmechanismus zu den entsprechenden gesättigten Kohlenwasserstoffen hydriert werden und vollständig von der Oberfläche desorbieren.[172] Pd 250 °C 10-4 Torr Pd (0) + + + 43 % 38 % -4 Schema 11: Zerfall von (Cyclopentadienyl)(Allyl)Pd bei 250 °C und 10 Torr. 88 / Palladium < 1% [171] 6.2.2 Thermolyse von (η η3-Allyl)Pd η5-Cyclopentadienyl)(η Thermolyse in Hexadecylamin Für eine Thermolyse unter Bedingungen analog zu denen der erfolgreichen Präparation von Kupfer-Nanopartikeln wurde eine 0.33-M Stammlösung des Precursors in Octylamin hergestellt. Bei der Injektion von 4 mL dieser Stammlösung in trockenes, O2-freies Hexadecylamin bei einer Temperatur von 100 °C wurde ein sofortiger Farbumschlag nach braun/schwarz beobachtet. Die Reaktionsmischung wurde für weitere 30 Minuten bei dieser Temperatur gehalten, bevor sie abkühlen gelassen wurde und die Partikel durch Waschen mit Methanol aufgearbeitet wurden. Im Gegensatz zu den Kupfer-Nanopartikeln war das aus (Cp)(Allyl)Pd hergestellte Material nicht in Toluol löslich. Dies deutet bereits darauf hin, dass in diesem Fall keine mit organischen Molekülen, d. h. mit HDA, geschützten Partikel entstanden sind, denn die langen Alkylketten des Amins würden eine Löslichkeit der Partikel in organischen Solventien gewährleisten. Die TEM-Aufnahmen dieses Materials bestätigen den ersten Befund: der Großteil des Materials liegt in großen, etwa 0.5 µm großen, annähernd sphärischen Agglomeraten vor (siehe Abb. 41 links). Bei einer höheren Vergrößerung zeigt sich jedoch, dass diese Agglomerate sich aus einzelnen Partikeln zusammen setzen, deren Durchmesser im Bereich von Nanometern liegt (etwa 3-4 nm mit einer relativ schmalen Größenverteilung). Die einzelnen Partikel sind kaum, und wenn, dann nur in Bereichen von geringer Konzentration, z. B. an den Rändern solcher Agglomerate, voneinander getrennt, d. h. eine Koordination von HDA an die Oberfläche der Partikel kann aufgrund einer fehlenden sterischen Abschirmung ausgeschlossen werden. Durch Röntgenbeugung (nicht abgebildet) konnte nachgewiesen werden, dass die Partikel aus kristallinem Palladium bestehen. Agglomerat Kolloidales Material Abb. 41: TEM-Aufnahmen von Palladiumpartikeln durch die Pyrolyse von (Cp)(Allyl)Pd in HDA. Länge der Bars von links nach rechts: 460 nm, 78 nm, 36 nm, 23 nm. Aus der Breite der Reflexe konnte ihre Größe mit Hilfe der Scherrer-Formel zu etwa 3 nm bestimmt werden, was sehr gut mit dem im Elektronenmikroskop gefundenen Wert übereinPalladium / 89 stimmt. Ein sehr geringer Teil des Materials wurde abseits der Agglomerate in Form von teilweise separierten und teilweise (scheinbar) agglomerierten Partikeln gefunden (Abb. 41 rechts). Die agglomerierten Partikel erweisen sich jedoch bei höherer Vergrößerung ebenfalls als einzelne Partikel, wenn auch mit einer von der sphärischen Form abweichenden Morphologie, die sich am ehesten als elongiert oder wurmartig beschreiben lässt. Die Größenverteilung dieser Partikel reicht von 3 bis 7 nm. Der für die Präparation von Pd-Partikeln in HDA gefundene hohe Grad an Agglomeration lässt sich durch eine schwache Wechselwirkung zwischen Amin und nullwertigem Edelmetall erklären. Aus Arbeiten, die sich mit der Bildung von SAMs von Dodecylamin auf Goldoberflächen beschäftigen, weiß man, dass sich Aminfilme nur unter ganz bestimmten Bedingungen ausbilden und auch dann nur für wenige Stunden unter Stickstoff stabil sind.[173] Dass sich trotz der schwachen Gold-N-Bindung SAMs ausbilden, wird den zusätzlichen vdWWechselwirkungen der Alkylketten untereinander zugeschrieben. Diese Stabilisierung ist jedoch auf einer gekrümmten Oberfläche wie sie Nanopartikel aufweisen wesentlich schwächer ausgeprägt und führt zu thermodynamischer und/oder kinetischer Instabilität von beispielsweise Gold-[174] oder Platin-[175] Nanopartikeln. Die Bildung von wurmartigen Formen ist in der Literatur der metallischen Nanopartikel nicht unbekannt. Chaudret[176] berichtet über derartig geformte Ruthenium-Partikel, sobald die Hydrierung des Precursors [(COD)(COT)Ru] in Gegenwart von unverzweigten Aminen mit einer Kettenlänge von 12 und mehr Kohlenstoffatomen durchgeführt wird, und erklärt diese Beobachtung mit einem schnellen Aminaustausch auf der Oberfläche der Partikel, in Übereinstimmung mit der oben beschriebenen schwachen Wechselwirkung. Wenn jedoch HDA nur schwach an die Partikeloberfläche koordiniert, sollte es möglich sein, das Amin als intermediären Liganden zu verwenden und ihn gegen andere Liganden auszutauschen, die stärkere Wechselwirkungen mit Palladium eingehen können wie z. B. Thiole. Die direkte Verwendung von Thiolen, beispielsweise als Zusatz zu HDA, ist aufgrund der Reaktivität des Precursors nicht möglich: (Cp)(Allyl)Pd reagiert mit Thiolen zu (Allyl)(Thiolato)Palladium-Dimeren[177], d. h. die Zugabe von Thiol kann erst nach der Zersetzung des Precursors erfolgen. Dafür wurde der Palladium-Precursor zunächst wie oben beschrieben in HDA zersetzt, dann jedoch wurde kurz vor Beginn des Abkühlens die Reaktionsmischung mit Dodecanthiol versetzt. Im Gegensatz zu dem ohne Thiolzusatz synthetisierten Material ist nun ein Großteil des gewaschenen Materials in Toluol löslich. Die TEM-Aufnahmen in Abb. 42 zeigen, dass das Metall in diesem Fall in deutlich voneinander separierten Partikeln mit einer relativ breiten Größenverteilung von 2 bis 5 nm vorliegt. Im Gegensatz zu HDA ist Dodecanthiol aufgrund einer stärkeren Pd-S-Wechselwirkung zur Stabilisierung der edelmetallischen Partikel offensichtlich wesentlich besser geeignet und führt durch sterische Abschirmung gegenüber benachbarten Partikeln zu freistehenden Kolloiden. Auffällig ist, dass die Größenverteilung ähnlich breit ist wie die der wenigen freistehenden Partikeln aus der Präparation mit HDA ohne Zusätze. Da dies jedoch in beiden Fällen nur Partikel betrifft, 90 / Palladium die durch Liganden stabilisiert werden (HDA oder Dodecanthiol), scheint die Koordination der Oberfläche eine Voraussetzung für das Auftreten eines ripenings bzw. einer Defokussierung der Größe zu sein. HS N N Abb. 42: TEM-Aufnahmen von Palladium-Nanopartikeln, die in Gegenwart von Dodecanthiol (links, bar = 75 nm) und Phenanthrolin (rechts, bar = 83 nm und ganz rechts, bar = 26 nm) unter Verwendung von HDA als intermediärem Liganden synthetisiert wurden. Partikel, die nicht koordiniert werden, agglomerieren beim Abkühlen und weisen einen kleineren Durchmesser mit einer schmaleren Größenverteilung auf. Da der Reaktionsverlauf bis kurz vor Beginn des Abkühlens identisch ist, heißt das aber auch, dass die Partikel, bevor es zu einer Koordination kommt, in dieser Form vorliegen müssen und erst durch Zusatz von Thiol einer Defokussierung unterliegen. Die ersten Palladium-Cluster, die isoliert wurden (giant cluster, siehe Einleitung) wurden durch Phenanthrolin, einen Stickstoff-Donor, stabilisiert. In der Annahme, dass die stabilisierenden Fähigkeiten auf noch größere „Cluster“, nämlich Palladium-Nanopartikel übertragen werden kann, wurde die für Dodecanthiol beschriebene Syntheseroute unter Verwendung von Phenanthrolin durchgeführt. Ähnlich jedoch wie bei primären Aminen als Stabilisator reicht die Donor-Kapazität bzw. die Stärke der Wechselwirkung zwischen Stickstoff und Palladium nicht aus, um freistehende Kolloide zu erzeugen (siehe Abb. 42 rechts). Die TEM-Aufnahmen zeigen, dass ähnlich wie dort 3-4 nm große Partikel entstehen, die hochgradig agglomeriert vorliegen, auch wenn die Agglomerate etwas weniger dicht gepackt sind und mit einem Durchmesser von 30-50 nm etwas kleinere sphärische Strukturen ausbilden als dort. Thermolyse in Toluol In der Regel liegen die Zersetzungstemperaturen von organometallischen Verbindungen in einem Bereich, der weit über den Siedetemperaturen von typischen organischen Lösungsmitteln liegt. Z. B. zersetzt sich [Cu(OCH(Me)CH2NMe2)2] ab einer Temperatur von 200 °C und kann daher nur zusammen mit hochsiedenden Lösungsmitteln, die bei RT fest sind und erst weit über 300 °C sieden, verwendet werden (HDA, TOPO etc.). Dies führt zwar zu qualitativ hochwertigen Partikeln mit einer perfekten Schutzhülle gegenüber Agglomeration, erfordert jedoch eine etwas aufwändigere Aufreinigung, nämlich das Entfernen des Feststoffs Palladium / 91 durch Abzentrifugation unter Schutzgasatmosphäre, als z. B. das einfache Abdestillieren eines niedrig siedenden Lösungsmittels. (Cp)(Allyl)Palladium hat jedoch eine wesentlich niedrigere Zersetzungstemperatur (100 °C in Hexadecylamin, in der Gegenwart von SAMs aus Thiolen sogar schon bei 60 °C oder RT). Daher sollte es möglich sein, die Synthese von PalladiumNanopartikeln in organischen Lösungsmitteln durchzuführen. Dazu wurden 0.5 Äquivalente eines Stabilisators (siehe Abbildungstexte) in Toluol gelöst und auf 50 °C erhitzt. In diese Mischung wurde eine toluolische Lösung von (Cp)(Allyl)Pd injiziert und bei derselben Temperatur rühren gelassen. Im Gegensatz zur Injektion in HDA tritt kein Farbumschlag ein, der eine Zersetzung des roten Precursors anzeigen würde. Auch nach mehreren Stunden ist noch keine Farbveränderung zu beobachten und erst nach 24 h bildet sich ein schwarzer Film an der Glaswand des Reaktionsgefäßes. Entfernt man das Lösungsmittel und analysiert den Rückstand mittels 1H NMR, erhält man ein sehr sauberes Spektrum des unzersetzten Precursors neben den Signalen des Stabilisators. Die Bildung eines metallischen Films deutet jedoch darauf hin, dass zumindest ein Teil des Precusors zersetzt worden sein muss. Um zu überprüfen, ob es zur Bildung von Nanopartikeln gekommen ist, wurden Lösung und Niederschlag getrennt voneinander im Elektronenmikroskop untersucht. Der Niederschlag besteht eindeutig aus Agglomeraten, die sich ihrerseits aus 2-3 nm großen Partikeln zusammensetzen. Die SAED dieser Partikel zeigt in Übereinstimmung mit der Größe verbreiterte Ringe, die den (111), (200), (220) und (311) Netzebenen von Palladium entsprechen (nicht abgebildet). Die überstehende Lösung scheint auf den ersten Blick zunächst keine Partikel zu enthalten. Erst bei hoher Vergrößerung (120 bis 400 k) lassen sich Partikel erkennen, die einen sehr kleinen Durchmesser von etwa 1-3 nm haben. Agglomerat Kolloidales Material Abb. 43: TEM-Aufnahmen von Palladium-Nanopartikeln aus der Thermolyse von Cp(Allyl)Pd in Toluol in Gegenwart von TOPO als Stabilisator. Links: Niederschlag (bar = 65 bzw. 40 nm), rechts: Lösung (bar = 78 nm). Demnach ist also eine Temperatur von 50 °C nicht ausreichend, um eine komplette Zersetzung des Precursors zu erreichen. Wird die Reaktion jedoch in siedendem Toluol durchgeführt, finden sich dieselben Charakteristika, sowohl im Reaktionsverlauf als auch in der Analyse des Materials: Die Reaktion läuft zwar schneller ab, angezeigt durch die Abscheidung eines metallischen Films nach wenigen Stunden, doch der Precursor liegt laut 1H NMR nach wie vor unzersetzt in der Lösung vor. Die elektronenmikroskopischen Aufnahmen be92 / Palladium stätigen, dass der Niederschlag aus metallischen Pd-Nanopartikeln besteht, die größere Agglomerate ausbilden, und auch bei dieser Reaktion liegen in der Lösung sehr kleine Partikel mit einem Durchmesser ≤ 3 nm vor. Neben der Auswahl des Stabilisators ist die Temperatur des Lösungsmittels ein entscheidender Parameter, der die Form und Größe der entstehenden Partikel beeinflusst. Beispielsweise führt die Injektion bei hoher Temperatur zu einer sofortigen Zersetzung des gesamten Materials und ermöglicht damit die Bildung von Agglomeraten, sofern kein geeigneter Stabilisator anwesend ist, um dies zu verhindern (vgl. Thermolyse in HDA). Da hier, bei der Verwendung von Toluol als LM, jedoch nach Beendigung der Thermolyse unzersetztes (Cp)(Allyl)Pd nachgewiesen werden konnte, kann jedoch davon ausgegangen werden, dass die Reaktion sehr langsam abgelaufen ist, d. h. dass nur nach und nach Palladium freigesetzt und eine Agglomeration durch zu hohe Konzentrationen vermieden worden sein muss. Durch diese optimalen Bedingungen war es möglich, kolloidale Nanopartikel mit einem sehr kleinen Durchmesser herzustellen. Dass dennoch Agglomerate gebildet worden sind, weist darauf hin, dass nicht (nur) die Temperatur bzw. die Reaktionsgeschwindigkeit der entscheidende Faktor für die Agglomeration ist, sondern die Auswahl des Stabilisators. Dass die Größe der kolloidalen Partikel im unteren Nanometerbereich liegt, weist darauf hin, dass auf diese Weise eher giant cluster mit etwa 5 ± 2 Schalen hergestellt werden. Diese in der Literatur veröffentlichten Cluster dieser Art konnten durch Koordination eines eher schwachen Stabilisators (phen), der in unseren Experimenten, d. h. bei Zusatz zu HDA, keine Stabilisierung bewirkte, erhalten werden. Es scheint also, als ob schwache, koordinierende Stabilisatoren nur dann wirkungsvoll stabilisieren können, wenn (1) die Partikel klein sind und (2) die Bildung von Palladiumatomen sehr langsam erfolgt. Thermolyse in höhersiedenden organischen Lösungsmitteln (HDA < Sdp. > Toluol) Um die Hypothese zu überprüfen, dass niedrige Temperaturen bzw. eine langsame Zersetzung erforderlich sind, um eine Stabilisierung zu erreichen, wurde dieselbe Reaktion statt in Toluol in höher siedenden Lösungsmitteln, nämlich o-Dichlorbenzol (Sdp. 181 °C) und Ethylenglycol (Sdp. 196-198 °C) durchgeführt. In beiden Fällen trat sofort nach der Injektion ein Farbumschlag nach braun/schwarz ein und das nach der Reaktion isolierte Material war nicht in Toluol löslich. Im Falle der in Ethylenglycol synthetisierten Partikel war allerdings eine kurzfristige Löslichkeit in Methanol zu beobachten. Die elektronenmikroskopischen Aufnahmen zeigen durchweg agglomerierte Partikel, wobei sich die Form der Agglomerate nach der Art des verwendeten Lösungsmittels richtet. Jedes der Agglomerate ist aus kleinen Partikeln mit einem Durchmesser von 3 ± 1 nm aufgebaut und keine der Proben weist kolloidale, also freistehende Partikel auf. Das heißt zunächst einmal, dass bei höheren Temperaturen in der Tat keine Stabilisierung der Partikel erfolgt. Durch Vergleich mit den analogen Thermolysen bei niedrigeren Temperaturen kann vermutet werden, dass die Ursache hierfür eine zu hohe Konzentration an Palladiumatomen durch Zersetzung des Precursors direkt nach der Injektion ist. Das heißt aber auch, dass weder der StaPalladium / 93 bilisator noch das Lösungsmittel einen Einfluss auf die Größe/Morphologie dieser Partikel haben. Ethylenglycol / TOPO Dichlorbenzol / HDA Ethylenglycol / HDA Dichlorbenzol / TOPO Dichlorbenzol / Ölsäure Abb. 44: TEM-Aufnahmen von Palladium-Nanopartikeln. Angabe des Lösungsmittels / des Stabilisators in den jeweiligen Aufnahmen. Um diese Überlegung zu erhärten, wurde (Cyclopentadienyl)(Allyl)Palladium in Substanz erhitzt und der Rückstand elektronenmikroskopisch untersucht. Die Realbilder des Pyrolyserückstands zeigen Partikel mit genau derselben Charakteristik wie die Partikel, die aus der Thermolyse von Cp(Allyl)Pd in HDA erhalten wurden: es sind circa 3 nm große Partikel entstanden, die sich zu Mikrometer großen Agglomeraten zusammenlagert haben. Ähnliche Beobachtungen wurde auch für ein anderes Edelmetall, nämlich Platin gemacht. Pyrolysiert man [(cod)PtMe(N3)] durch Erhitzen in einer inerten Atmosphäre (TGA-Experiment)[178] und analysiert den Rückstand im TEM, so findet man Agglomerate, die sich aus kleinen, etwa 1.5 bis 4 nm großen metallischen Partikeln zusammensetzen. Es ist interessant, dass beide organometallischen Verbindungen dieser Edelmetalle auch ohne die Anwesenheit eines Stabilisators zu nanopartikulärem Material führen. Auch wenn die durchgeführten Experimente keinerlei statistisches Gewicht haben, ist es möglicherweise ein allgemein gültiges Phänomen, dass über (wahrscheinlich radikalische) Zerfallsmechanismen nanopartikuläres Material entsteht. Die Ursache für dieses Verhalten ist in der Stabilität von metallischen Partikeln zu sehen: Partikel mit einer bestimmten Anzahl an Atomen (oder auch einer bestimmten Ladung) weisen eine erhöhte Stabilität auf und die Addition eines weiteren Atoms führt zu einem Anstieg der potentielle Energie, die diesen Prozess thermodynamisch ungünstig macht. 94 / Palladium Thermolyse in Gegenwart von kationischen Stabilisatoren Unter der Annahme, dass ein Zerfallsmechanismus, der von unpolaren Medien unbeeinflusst verläuft, möglicherweise ionische bzw. polare Zwischenstufen bildet, wurde die Reaktion in o-Dichlorbenzol, dem 1 Äquivalent Tetraoctylammoniumbromid zugesetzt wurden, durchgeführt. Die elektronenmikroskopischen Aufnahmen dieses Materials sind in Abb. 45 gezeigt. Die Größe der Partikel beträgt zwischen 20 und 40 nm, wobei die Formen der einzelnen Partikel sehr unregelmäßig sind. Dennoch lässt sich festhalten, dass hier erstmalig Partikel gebildet worden sind, die einen Durchmesser von deutlich über 3 nm aufweisen. (111 ) (2 0 0 ) (2 2 0 ) (3 11 ) Abb. 45: TEM-Aufnahme (bar = 87 nm) und SAED von Palladium-Nanopartikeln durch Pyrolyse von (Cp)(Allyl)Pd in HDA in Gegenwart von R4NBr. Rechts zum Vergleich eine Thermolyse in HDA in Abwesenheit des Ammoniumsalzes (bar = 43 nm). Da der Unterschied der Synthesen lediglich in der zusätzlichen Anwesenheit von R4NBr besteht, muss das Ammoniumkation auch die Ursache für diese Beobachtung sein, d. h. in diesem Fall ist eine Wechselwirkung zwischen dem Stabilisator und den Partikeln möglich. Eine denkbare Erklärung wäre die, dass die gebildeten Palladiumpartikel auf ihrer Oberfläche eine Schicht von positiv geladenem Palladium ausbilden bzw. eine positive Ladung über das gesamte Partikel verteilt ist. Auch in Anbetracht eines fehlenden Reduktionsmittels ist es sehr gut denkbar, dass nicht alle Palladiumionen zum Metall reduziert werden. (Über die Abscheidung von Pd2+ bei der Bedampfung von SAMs aus Dithiolen mit Cp(Allyl)Pd ist bereits berichtet worden[177]). Besteht das Reaktionsmedium lediglich aus HDA bzw. anderen, nur koordinativ bindenden Molekülen, ist die Wechselwirkung zwischen Partikeloberfläche und Stabilisator relativ gering. Darüber hinaus werden sich annähernde Palladiumionen von der gleichnamigen Ladung abgestoßen. Die Folge ist, dass die Partikel nur eine geringe Größe erreichen, die von einer maximalen Ladung pro Partikel bestimmt wird und nicht von den Reaktionsbedingungen. Anders in der Anwesenheit von R4NBr: Hier ist die Ausbildung einer elektrischen Doppelschicht gemäß Pdδ+ - Br¯ (- R4N+) möglich. Sich annähernde Ionen werden durch die Anwesenheit von negativ geladenen Bromidionen nicht mehr abgestoßen und die Partikel können bis zu den beobachteten Größen wachsen, die etwa um den Faktor 10 über denen liegen, die ohne ein quartäres Ammoniumsalz synthetisiert worden sind. Palladium / 95 6.2.3 Oberflächenmodifizierung von Palladium-Nanopartikeln Aufgrund der oben beschriebenen Thermolyse-Experimente mit und ohne Zugabe eines ionischen Stabilisators gehen wir davon aus, dass der Zerfall von Cp(Allyl)Pd zu geladenen Partikeln führt. Falls diese Annahme richtig sein sollte, müsste es möglich sein, Partikel, die zunächst ohne einen ionischen Stabilisator synthetisiert worden sind, postsynthetisch so zu modifizieren, dass sie in einem Solvens löslich sind, d. h. kolloidal vorliegen. DMAP (Dimethylaminopyridin) ist eine Verbindung, die eine tertiäre, stark basische Aminogruppe in Konjugation zu einer elektronenschiebenden (schwach basischen) Gruppe enthält, weswegen folgende mesomere Grenzstrukturen denkbar sind: N N N N Aufgrund dieser Eigenschaften gelang es Caruso et al., TOAB-bedeckte Palladium-Partikel mit Hilfe von DMAP aus der organischen in die wässrige Phase zu transferieren.[56] Die aus unseren Thermolysen in HDA o. ä. erhaltenen Partikel liegen zwar nicht stabilisiert vor, doch wenn ihre Oberfläche eine positive Ladung trägt, kann das partiell negative Stickstoffatom des Pyridinrings durch elektrostatische Wechselwirkung an die Oberfläche koordinieren. Die positive (Teil)Ladung der Dimethylaminogruppe sollte dann eine Löslichkeit in hydrophilen Solventien wie Wasser ermöglichen. Abb. 46: TEM-Aufnahmen der wässrigen Phase (0.1-M Lösung von DMAP) nach Überschichten mit einer toluolischen Dispersion aus Palladium-Nanopartikeln. Bar = 52 nm. Überschichtet man eine 0.1-molare Lösung aus DMAP in Wasser mit einer toluolischen Dispersion aus Palladium-Nanopartikeln, so beobachtet man bereits nach etwa 15 Minuten eine Entfärbung der organischen Phase und eine intensive Verfärbung der wässrigen Phase. Die elektronenmikroskopischen Aufnahmen der wässrigen Lösung zeigen kleine, zwischen 2 96 / Palladium und 4 nm große Partikel, die separiert voneinander vorliegen (siehe Abb. 46). Die EDX-Analyse dieser Partikel bestätigt, dass es sich dabei um Palladium handelt (neben Signalen für Gold und Kohlenstoff, die von dem als Träger verwendeten C-beschichteten Goldgrid stammen). Auf diese Weise ist es nicht nur gelungen, agglomerierte Palladium-Nanopartikel kolloidal in Lösung zu bringen. Es konnten außerdem die Eingangs beschriebenen Schlussfolgerungen aus den Thermolysen bestätigt werden. Wenn nun die Ursache für das Vorliegen von geladenen Partikeln tatsächlich eine unvollständige Reduktion der Palladiumkationen während der Zersetzung von (Allyl)(Cp)Pd ist, sollte es möglich sein, mit Hilfe der organometallischen Chemie einen Precursor maßzuschneidern, der dieses Problem umgeht. Dieser Ansatz wird im nächsten Kapitel verfolgt. 6.3 Synthese von Palladium-Nanopartikeln unter Verwendung des organometallischen Precursors [Pd(OCH(Me)CH2NMe2)2] 6.3.1 Vorstellung des Precursors Als zweiter Precursor wurde das Alkoxid [Pd(OCH(Me)CH2NMe2)2] verwendet. Der Aufbau dieser Verbindung aus einem 2-wertigen Metallkation und zwei oxidierbaren, N,O-chelatisierten Liganden hat sich bereits für die Herstellung metallischer Kupfer-Nanopartikel (siehe Kapitel 5) bewährt. Aufgrund der Neigung des Palladiums, quadratisch-planare Komplexverbindungen mit der Oxidationsstufe II zu bilden, sollte die zum Kupfer analoge, jedoch in der Literatur unbekannte Verbindung [Pd(OCH(Me)CH2NMe2)2] ebenfalls leicht herzustellen sein und ähnliche Vorteile für die Herstellung von metallischen Nanopartikeln aufweisen. Me2 N O Pd(OAc)2 + K2CO3 2 HO * Pd Toluol, RT NMe2 * N Me2 O Schema 12: Synthese des Palladiumprecursors [Pd(OCH(Me)CH2NMe2)2] 2. Die Synthese gelingt in einem Syntheseschritt und guten Ausbeuten durch Umsetzen von Palladiumacetat mit 2 Äquivalenten des entsprechenden Aminoalkohols in Gegenwart einer Base. Die gelben Kristalle lassen sich aufgrund des edlen Charakters des Metallkations nicht mehr unzersetzt sublimieren wie die analoge Kupfer-Verbindung, da das Standardpotential von Palladium mit E = 0.99 V wesentlich positiver ist als das von Kupfer oder Nickel (ECu = 0.34 V und ENi = -0.22 V), d. h. die Reduktion zum Metall ist aufgrund des Zusammenhangs ∆G = -nFE thermodynamisch begünstigt. Sie werden stattdessen mit Pentan gewaschen und Palladium / 97 aus Toluol kristallisiert. [Pd(OCH(Me)CH2NMe2)2] wurde durch Elementaranalyse, 1H und 13 C NMR, FAB und TGA charakterisiert und seine Struktur mittels Röntgenbeugung bestimmt. Aus der Röntgenstrukturanalyse geht hervor, dass das Metallzentrum durch zwei N,O-chelatisierende Aminoalkoholat-Liganden umgeben ist und eine nahezu quadratisch-planare Koordination mit O-Pd-N-Winkeln von etwa 90° annimmt. Die Anordnung der 5-gliedrigen Chelatringe ist derart, dass sich die anionischen Sauerstoffatome, und demzufolge auch die beiden Stickstoff-Donoratome trans zueinander befinden. Solche trans-Anordnungen werden häufig in Palladiumkomplexen mit anionischen O-Donoren und zwei monodentaten Aminliganden gefunden wie z. B. in [Pd(OAc)2(Et2NH)2].[179] Die C-O-Bindungslänge (1.420 Å) ist bedeutend länger als die C-O-Bindungslängen in Alkoxid-Komplexen von Platin und Iridium (1.33-1.37 Å)[180, 181]. Wiberg hat gezeigt, dass bei Alkoxiden von späten Übergangsmetallen eine Wechselwirkung zwischen dem freien Elektronenpaar am Sauerstoff des Alkoxidliganden und der C-H-Bindung einer benachbarten CH3- oder CH2R-Gruppe zur Verkürzung des C-O-Abstandes führt.[182] Die Methylgruppe eines Methoxids kann sich beispielsweise so anordnen, dass es zu einer maximalen Wechselwirkung der beteiligten Orbitale kommt, die im Extremfall zu O-C-Bindungslängen im Bereich einer Doppelbindung führen können wie in [Pt(Me)(OMe)(dppe)] mit d(O-C) = 1.258 Å.[183] Ist der Sauerstoff jedoch Teil eines Rings wie in [Pd(OCH(Me)CH2NMe2)2], wird diese Wechselwirkung wesentlich kleiner sein, da sich die C-H-Bindungen eclipsed zu den freien Elektronenpaaren anordnen. Abb. 47: Röntgenstruktur des quadratisch-planaren [Pd(OCH(Me)CH2NMe2)2]. Tabelle 5: Ausgewählte interatomare Distanzen [Å] und Winkel [°] für [Pd(OCH(Me)CH2NMe2)2]. Bindungslängen Bindungswinkel Pd-O1 1.9849(16) O1-Pd1-N2 94.54(7) Pd-N1 2.0505(18) O1-Pd1-N1 85.46(7) O1-C1 1.420(4) N1-Pd1-N2 179.2(6) 98 / Palladium Die koordinierenden Aminoalkohole haben ein stereogenes Zentrum, das in Schema 12 mit * gekennzeichnet ist, weswegen 2 in drei verschiedenen stereoisomeren Formen vorliegt, nämlich (R,S)-2, das aufgrund der C2-Symmetrie identisch mit dem (S,R)-Isomeren ist, und dem dazu diastereomeren Enantiomerenpaar (R,R)-2 und (S,S)-2. Da der Aminoalkohol als Racemat eingesetzt wurde, sollte 2 bei statistischer Verteilung, d. h. im Fall, dass die Stereoisomere sich sowohl thermodynamisch als auch kinetisch wenig voneinander unterscheiden, im Verhältnis 2:1:1 = (R,S):(R,R):(S,S) vorliegen. NMR-spektroskopisch unterscheidbar sind prinzipiell nur Diastereomere, d. h. (R,R) bzw. (S,S) ist von (R,S) bzw. (S,R) unterscheidbar, sofern die magnetische Umgebung der jeweiligen Kerne von der Stereochemie beeinflusst ist. Betrachtet man das NMR-Spektrum in Abb. 48, sind zwei Signale, nämlich die des Protons an C-2 und die der Methylgruppe C-1 nur einfach vorhanden, d. h. in diesem Fall sind die Verschiebungen für beide Diastereomere identisch. Die übrigen Signale sind jedoch von der räumlichen Orientierung der C-1 Methylgruppe beeinflusst und scheinen daher doppelt vorzuliegen. Für die diastereotopen Methylgruppen am Stickstoff sind insgesamt vier Signale zu finden. Jeweils zwei der peaks gehören zu einem Diastereomer, wobei die chemische Verschiebung davon abhängt, ob sie cis oder trans zur 1-Methylgruppe steht. Aufgrund der Signalverhältnisse lassen sich die innen liegenden Singuletts zu einem der Diastereoisomere und die außen liegenden dem anderen zuordnen. Die in Abb. 48 vorgenommene absolute Zuordnung zu A oder B wurde willkürlich gemacht. Eine Integration von A-5 und B-5 ergibt ein Verhältnis der Diastereomere in Lösung von 1.0:1.1. A/B-1 A-4 A-5 1 B-4 B-5 Me 2 N O * 2 Pd 3 N 4 5 A: (R,R) oder (S,S) A/B-3a A/B-3b A/B-2 O 1 Me 2 N O * 2 Pd 3 } } X 4 3 N Y 2 1 Shift [ppm] 4 O 5 B: (R,S) oder (S,R) Abb. 48: 1H NMR Spektrum von [Pd(OCH(Me)CH2NMe2)2] in C6D6. Für die diasterotopen Protonen an C-3 sind ebenfalls vier Signalgruppen zu finden. Man könnte annehmen, dass ähnlich zur Situation der NMe2-Gruppe zwei mal zwei Signale vorliegen, doch eine Betrachtung der Kopplungskonstanten schließt diese Interpretation aus. Die mit X bezeichnete Signalgruppe besteht aus einem dd mit Kopplungskonstanten von 4 und 10 Palladium / 99 Hz, während die Signalgruppe Y (in dieser Auflösung) aus einem td mit 2 und 11 Hz besteht. Die größere der beiden Kopplungskonstanten kann der geminalen Kopplung zum zweiten Proton an C-3 zugeordnet werden, d. h. es liegen nicht 2x2 Signale, sondern 2x1 Signal vor. Für die Zuordnung der kleineren Kopplungskonstanten ist es hilfreich, das Kopplungsmuster des C-2 Multipletts zu betrachten. Es handelt sich um ein ddquar, das aber aufgrund der identischen Kopplungskonstante eines Dubletts mit dem Quartett ein pseudo-dquin ist. Die Kopplungskonstanten betragen 2 Hz (d) bzw. 5 Hz (d und q). Das C-2 Proton weist also zwei verschiedene Kopplungskonstanten zu den benachbarten diastereotopen Protonen an C-3 auf, wobei die größere Kopplungskonstante dem trans-angeordneten Proton und die kleinere dem cis-Proton zugeordnet werden kann. Demnach besteht die X-Gruppe aus den Signalen der jeweils zum C-2 Proton trans stehenden Protonen beider Diastereoisomere, während die YGruppe aus denen der cis stehenden Protonen ebenfalls beider Isomere besteht. Die Feinaufspaltung dieses Protons kommt vermutlich durch 1,4-Kopplung der zu diesem Proton trans stehenden C-1-Methylgruppe zustande. Die thermischen Eigenschaften von [Pd(OCH(Me)CH2NMe2)2] wurden mit Hilfe von TG/DTA (siehe Abb. 49) näher untersucht. Die TG-Kurve zeigt zunächst einen schwachen Gewichtsverlust bei etwa 75 °C an, begleitet von einem endothermen Signal der DTA-Kurve, und fällt in einem Temperaturbereich zwischen 120 und 140 °C steil ab. Die Restmasse von 34 % stimmt sehr gut mit dem theoretischen Gewichtsanteil des Palladiums von 34.4 % überein. Daraus ergibt sich, dass für die Thermolyse eine Reaktionstemperatur 150 bis 200 °C gewählt werden sollte. 12 100 10 8 6 60 4 DTA [µV] TG [%] 80 2 40 0 20 0 50 100 150 200 250 300 350 Temperatur [°C] Abb. 49: TG/DTA von [Pd(OCH(Me)CH2NMe2)2] unter Argonatmosphäre von RT bis 400 °C mit einer Heizrate von 10 K/Minute. Die Massenspektroskopie bestätigt die Bildung von Palladium durch eine schrittweise Abspaltung von einem der chelatisierenden Liganden und Palladium. Ausgehend vom Molpeak (m/z = 311) ist ein Massenverlust von ∆m = 103 zu beobachten, der zu einem Palladiumfragment mit nur einem Liganden führt (m/z = 208). Die Massendifferenz zu einem der intensiven peaks des Spektrums (m/z = 102) entspricht mit ∆m = 106 einer Abspaltung 100 / Palladium von Palladium, wobei davon auszugehen ist, dass das Metall neutral ist, da der peak bei m/z = 106 in der Größenordnung des Rauschens liegt. Die Abspaltung von NMe2 schließlich führt zum Basispeak des Spektrums mit m/z = 58. Me2 N O Me2 N Pd N Me2 Me2N Pd C3H6O m/z = 58 O OH m/z = 311 O m/z =208 O Pd ∆m = 106 m/z = 102 NMe2 ∆m = 44 NMe2 ∆m = 103 Schema 13: Postulierter Zerfallsmechanismus für [Pd(OCH(Me)CH2NMe2)2] nach Elektronen-Stoß-Ionisation bei 70 eV im Massenspektrometer. 6.3.2 Thermolyse von [Pd(OCH(Me)CH2NMe2)2] Die Thermolyse des Alkoxidprecursors wurde unter Bedingungen durchgeführt, die analog zu der entsprechenden Synthese von Kupfer-Nanopartikeln gewählt wurden. Dazu wurde eine 0.33-M Stammlösung des Precursors in Octylamin hergestellt und bei T = 200 °C in HDA injiziert. Nach der Injektion war eine sofortige Schwarzfärbung zu beobachten, gefolgt von einer deutlichen Gasentwicklung, was auf eine Zersetzung des Precursors bzw. die Bildung von metallischem Material hinweist. (111 ) (2 0 0 ) (2 2 0 ) (3 11 ) Abb. 50: TEM- und SAED-Aufnahmen des Thermolyseprodukts von Pd(OR)2 in HDA (bar = 98 bzw. 110 nm). Zum Vergleich rechts dieselbe Thermolyse unter der Verwendung von (Cp)(Allyl)Pd (bar = 43 nm). Abb. 50 zeigt die elektronenmikroskopischen Real- und Beugungsbilder des aufgearbeiteten Materials. Sowohl SAED als auch EDX (nicht abgebildet) weisen darauf hin, dass nachweislich metallisches Palladium gebildet worden ist, angezeigt durch (1) die Beugungsreflexe des SAEDs, die den (111), (200), (220) und (311)-Netzebenen von Palladium entsprechen und (2) einem Signal für Palladium in der EDX-Analyse, die von Signalen des Grids (Kupfer) und des Probenhalters (Aluminium) begleitet werden. Die Partikel, die im Realbild zu erkennen sind, haben eine Größe von etwa 8 nm. Sie sind eindeutig voneinander separiert, wobei die Palladium / 101 Distanz zwischen zwei benachbarten Partikeln durch die Hülle der organischen Liganden (HDA) bedingt ist. Ein Teil der Partikel liegt jedoch auch in Form von größeren Partikeln bzw. Agglomeraten vor (siehe mittleres Bild). Vergleicht man diese Ergebnisse mit der Thermolyse des (Cyclopentadienyl)(Allyl)Palladiums, die in Abb. 50 rechts abgebildet sind, so wird der Unterschied sehr deutlich: Hier stabilisierte, separierte, ca. 8 nm große Partikel und dort Agglomerate aus 3 nm großen, „nackten“ Partikeln. Wir nehmen an, dass die Ursache für diese Beobachtung die bereits erwähnte unvollständige Readuktion der Palladiumionen des Cp(Allyl)Pd während der Thermolyse ist, während die Palladiumkationen des Alkoxids durch das Ligandendesign eine vollständige Reduktion erfahren. Dies führt zum Wachstum von neutralen Partikeln (im Gegensatz zu einer Aufladung im Falle von [Cp(Allyl)Pd] ), die analog den Kupfer-Nanopartikeln durch HDA stabilisiert werden. 6.4 Zusammenfassung Es sind – unseres Wissens nach erstmals – Palladium-Nanopartikel und kolloidales Palladium (NACs) unter Verwendung von organometallischen Precursoren hergestellt worden. Beide in dieser Arbeit vorgestellten Precursor, sowohl [(η3-Allyl)(η5-Cyclopentadienyl)Palladium] als auch [Pd(OCH(Me)CH2NMe2)2], sind leicht zugänglich, kristallin und zerfallen bei moderaten Temperaturen zu metallischem Palladium, wobei flüchtige Nebenprodukte entstehen, die leicht zu entfernen sind. Setzt man die Verbindungen durch Injektion thermolytischen Bedingungen aus, d. h. erhöhten Temperaturen und hochsiedenden, koordinierenden Lösungsmitteln, kommt es zur Abscheidung von Palladium in Form von nanopartikulärem Material. Die Entstehung von Kolloiden bzw. nicht-stabilisierten Nanopartikeln hängt ganz entscheidend von dem verwendeten Precursor und den gewählten Reaktionsbedingungen ab: • In unpolaren Reaktionsmedien und bei Pyrolyse zerfällt Cp(Allyl)Pd völlig unabhängig von Reaktionsmedium und Stabilisator zu kleinen, 3-4 nm großen Partikeln, die sich in Form von Agglomeraten organisieren. Die Wahl des Lösungsmittels beeinflusst die Form dieser Agglomerate. • [Pd(OCH(Me)CH2NMe2)2] hingegen bildet bei Thermolyse in HDA 8 nm große Partikel, die durch das Amin stabilisiert sind. • Sehr kleine kolloidale Partikel lassen sich bei Verwendung von Cp(Allyl)Pd nur durch Thermolyse bei niedrigen Temperaturen in Gegenwart eines schwach koordinierenden Stabilisators herstellen. • Größere kolloidale Partikel entstehen bei Zusatz eines kovalent bindenden Ligandens (Thiol) nach der Wachstumsphase bzw. vor dem Abkühlvorgang der Thermolyse von Cp(Allyl)Pd Größere kolloidale Partikel entstehen außerdem, wenn der Thermolyse von Cp(Allyl)Pd ionische Liganden (quartäres Ammoniumsalz) zugesetzt werden. • 102 / Palladium • Der Zusatz von ionischen Liganden bewirkt hingegen keine Veränderungen in der Thermolyse von [Pd(OCH(Me)CH2NMe2)2] Aus dem unterschiedlichen Verhalten der Precursor wird auf folgenden Mechanismus geschlossen: Direkt nach der Injektion kommt es (in Abhängigkeit von der Temperatur) mehr oder weniger schnell zur Zersetzung des organometallischen Precursors und der Reduktion des Palladium-Kations. Im Falle des Palladiumalkoxids ist für jedes Palladium-Kation ein Äquivalent Reduktionsmittel vorhanden, so dass in einer Redoxreaktion aus jedem Pd2+ metallisches Pd0 entsteht. Im Falle von Cp(Allyl)Pd ist jedoch kein Reduktionsmittel vorhanden und es ist anzunehmen, dass unter den Reaktionsbedingungen ein Teil der Palladiumionen nicht zum Metall reduziert wird. In den nun folgenden Schritten der Nukleation und des Wachstums lagern sich die Metallatome bzw. –ionen zusammen. Sind nur Pd(0)-Atome vorhanden, verlaufen diese Prozesse bis zum Erreichen des kritischen Radius bzw. bis zur Terminierung des Wachstums durch Koordination von (neutralen) Liganden. Werden jedoch Pd2+Ionen in diese Prozesse mit einbezogen, wächst mit zunehmender Größe die positive Partialladung der Partikel. Dies schützt die Partikel vor weiterem Wachstum und führt zu Partikeln, die aufgrund ihrer gleichnamigen Ladung voneinander separiert sind. Setzt man dieser Reaktion R4NBr zu, kann die Ladung ausgeglichen werden und das Wachstum wird erst zu einem wesentlich späteren Zeitpunkt terminiert. Hier konnte die organometallische Chemie einen wesentlichen Beitrag zur Synthese von Nanopartikeln leisten. Durch den maßgeschneiderten Precursor [Pd(OCH(Me)CH2NMe2)2] ist es möglich, ohne den Zusatz von Reduktionsmitteln (im Falle von CH-Liganden typischerweise Wasserstoff) zu arbeiten, und dennoch eine vollständige Reduktion des zentralen Metallatoms zu gewährleisten. Durch den gesamten Reaktionsaufbau wird den Reduktionsmethoden von Pd-Salzen in wässrigen Lösungen ein einfacher Zugang zu nicht-wässrigen Palladium-Kolloiden an die Seite gestellt. Palladium / 103 7 Nickel-Nanopartikel 7.1 Stand der Forschung und Aufgabenstellung Die Gründe für das große Interesse an metallischen Nanopartikeln sind bereits in Kapitel 5.1 ausführlich vorgestellt worden. An dieser Stelle sei deshalb nur auf die Synthesemethoden speziell für Nickel-Nanopartikel eingegangen. Wie bei allen Synthesen von Nanopartikeln kann man auch hier zwischen nasschemischen, physikalischen und Templatmethoden unterscheiden. Templatsynthesen. Die meisten Methoden konzentrieren sich auf die Herstellung von metallischen Drähten im Nanometerbereich und beruhen auf der direkten Imprägnierung von mesoporösen Materialien mit Precursormolekülen oder –ionen und anschließender thermischer oder chemischer Reduktion, sind aber bislang nur für edelmetallische nanowires verwendet worden. Dai et al. haben aufbauend auf dieser Methodik eine neue Strategie entwickelt, indem sie zunächst Palladium in den Poren von SBA-15 abgeschieden haben, das in Form von Nanopartikeln in den Kanälen vorliegt. Diese Partikel dienen anschließend als Katalysatoren für die stromlose Abscheidung von Nickel aus Ni(NO3)2 in Form von sehr schmalen (wenige Nanometer) Nanodrähten.[184] Wesentlich größere Poren entstehen bei der Anodisierung von Aluminiumoxid (d > 30 nm). Nickelstäbe mit einem Durchmesser von 50 nm sind durch elektrochemische Abscheidung in den Poren von anodischem Aluminiumoxid (AAO) synthetisiert worden.[185] Modifiziert man die Wände der Poren vor der Abscheidung des Nickels mit einem organischen Amin, z. B. Methyl-γ-diethylentriaminopropyldimethoxysilan, führt die Affinität von Nickel zur Aminogruppe zur bevorzugten Abscheidung des Metalls an den Porenwänden, so dass keine soliden Drähte, sondern Röhrchen entstehen.[186] Abhängig von den Reaktionsbedingungen beträgt der Durchmesser solcher Nanoröhren z. B. 160 nm, ihre Länge 20 µm und ihre Wanddicke 30 nm. Darüber hinaus sei die Abscheidung von Nanodrähten mit nur 4 nm Durchmesser in dem Hohlraum von Kohlenstoffnanoröhrchen erwähnt.[187] Analog zur oben beschriebenen Pd-katalysierten Abscheidung wird auch hier die poröse Struktur von AAO ausgenutzt, und zwar durch thermischen Zerfall von Propen, der zur einheitlichen Bedeckung der Poren mit Kohlenstoff führt. Die sich anschließende Abscheidung von Nickel wurde durch MOCVD von Nickelocen realisiert. Nach Auflösen des AAO mit Hilfe von NaOH erhält man Kohlenstoffröhrchen mit einem Durchmesser von etwa 30 nm, die einen einzelnen metallischen Nanodraht von 500 nm Länge und 4 nm Durchmesser enthalten. Physikalische Methoden. Nickelpartikel sind bei tiefen Temperaturen durch die Kodeposition von Nickeldampf und verschiedenen organischen Lösungsmitteln synthetisiert worden.[188] Die meisten der so hergestellten Kolloide sind aufgrund der nur schwach koordinie- 104 / Nickel renden Lösungsmittel wie Aceton oder Ethanol sehr instabil. Stabile Kolloide mit einem Durchmesser von 19 nm entstehen nur bei der Verwendung von 2-Methoxyethanol. Reduktive Methoden. Zwischen den physikalischen und den reduktiven Methoden sind zunächst zwei elektrochemische Methoden zu nennen.[153, 154, 189] Die erste Methode verläuft analog zu der von Reetz publizierten Synthese von Palladiumpartikeln. Aus einem Nickelblech werden anodisch Metallkationen erzeugt, die zur Kathode wandern (Platin) und dort zu elementarem Nickel reduziert werden Auch hier werden die enstehenden Cluster durch TOABr stabilisiert, das gleichzeitig als Elektrolyt dient. Bei der zweiten Methode, der „H2 coevolution method“ wird Nickel unter gleichzeitiger Bildung von Wasserstoff auf einer Graphitoberfläche abgeschieden. Die Bildung von Wasserstoff und Nickel läuft bei jedem Potential < -1.2 V parallel ab, da die abgeschiedenen Nickelpartikel sofort als Katalysatoren für die Bildung von Wasserstoff dienen. Die entstehenden Partikel weisen eine schmale Größenverteilung im Bereich von 50 bis 100 nm auf und ihre Morphologie kann durch die Abscheidungsspannung gesteuert werden. Rein chemische reduktive Methoden sind bei der Synthese von Nickelpartikeln durch die Reduktion von NiCl2 mit Hydrazin in der Gegenwart von Cetyltrimethylammouniumbromid[190] oder bei der Reduktion von NiCl2 mit NaBH4 in superkritischem Ethanol[191] angewendet worden. Triphenylphosphin-stabilisierte Nickel-Kolloide sind durch die Reduktion von Nickelacetylacetonat mit Et2AlH in der Gegenwart von PPh3 hergestellt worden.[192] Auch wenn die reduktiven Methoden zweifelsohne das größte synthetische Potential haben, da sie weder auf die Verfügbarkeit von geeigneten Templaten angewiesen sind noch den Einschränkungen von rein physikalischen Methoden unterliegen, leiden sie doch unter dem Nachteil, dass die Zugabe von Reduktionsmitteln nötig ist und damit Nebenprodukte gebildet werden, die die Aufarbeitung stören oder die Oberfläche der entstehenden Partikel kontaminieren können. Dies kann durch die Verwendung von nullwertigen organometallischen Precursoren mit flüchtigen Abgangsgruppen wie z. B. Biscyclooctadiennickel, [(COD)2Ni], umgangen werden. De Caro und Bradley[193] haben durch Zersetzung dieser Verbindung in Methylenchlorid in der Gegenwart von PVP polymergeschützte Nickel-Kolloide hergestellt, während Chaudret[194] die Zersetzung in THF in der Gegenwart von Wasserstoff durchgeführt hat. Vor kurzem wurden von derselben Gruppe auf diese Weise nanoskalige Nickel-Stäbchen hergestellt.[49] In der vorliegenden Arbeit wurde ein Ansatz verfolgt, der die reduktiven Methoden mit der Verwendung von organometallischen Precursoren verbindet. Bei der Synthese von Kupferund Palladiumpartikeln ist bereits ein Strukturelement verwendet worden, dass sich außerordentlich gut eignet, zweiwertige Kationen durch eine koordinierte Alkoholfunktion zum entsprechenden Metall zu reduzieren: [M(OCH(Me)CH2NMe2)2] mit M = Cu, Pd. Insbesondere für das Kupfer-System können durch Verwendung dieser Verbindung hochwertige, nahezu monodisperse Nanopartikel erhalten werden, ohne dass weitere Zusätze erforderlich sind. Nickel / 105 7.2 Synthese von Nickel-Nanopartikeln unter Verwendung des organometallischen Precursors [Ni(OCH(Me)CH2NMe2)2] 7.2.1 Vorstellung des Precursors Der Precursor [Ni(OCH(Me)CH2NMe2)2] 1 ist bereits literaturbekannt und wurde von HubertPfalzgraf et al. durch die Reaktion von Nickelchlorid mit dem entsprechenden Natriumsalz des Aminoalkohols hergestellt.[195, 196] Diese Synthese wurde von uns leicht modifiziert (siehe Schema 14). Der zeitaufwändige erste Schritt lässt sich wesentlich verkürzen, wenn statt elementarem Natrium Natriumhydrid verwendet wird. Darüber hinaus kann die Aufreinigung statt durch Kristallisation bei –15 °C über Nacht durch Sublimation im Vakuum bei 65 °C innerhalb von wenigen Stunden erfolgen. Die IR-Schwingungen[195] und die Kristallstrukturdaten[196] dieser Verbindung sind von der Gruppe um Hubert-Pfalzgraf bereits veröffentlicht worden. Im experimentellen Teil der vorliegenden Arbeit sind ergänzend dazu die 1H NMR und MS Daten von 1 angegeben. N + NaH - H2 Me2 N O ONa OH N + 0.5 eq NiCl2 Ni - NaCl N Me2 O Schema 14: Synthese des verwendeten Nickel-Precursors [Ni(OCH(Me)CH2NMe2)2] 1. Um den Zerfall von 1 und damit seine Eignung für die Freisetzung von nullwertigem Nickel zu analysieren, wurden Daten der Massenspektroskopie und TG/DTA verwendet. Der Molpeak des Massenspektrums liegt bei m/z = 262. Berechnet man jedoch das Isotopenmuster von [Ni(C5H12ON)2] (siehe Tabelle 6) und vergleicht die Werte mit den beobachteten, stellt man fest, dass sie nicht übereinstimmen. Insbesondere die Intensität bei m/z = 263 liegt mit 88 % relativer Intensität wesentlich höher als der berechnete (12 %). Geht man jedoch davon aus, dass neben M auch M+H gebildet wird, wird die Intensität bei m/z = 263 erhöht. Tabelle 6: Berechnete und beobachtete Isotopenmuster. Der erste Wert gibt die Masse (in g/mol) an, der zweite die relative Intensität (in %). NiC10H24N2O2 NiC10H24N2O2 1 eq NiC10H24N2O2 (M) Beobachtete Werte (M) (M+H) + 0.8 eq NiC10H24N2O2 (M+H) Massenspektrum 262: 100.0 262: 100 262: 100.0 263: 11.9 263: 100.0 263: 92 263: 88 264: 39.2 264: 11.9 264: 49 264: 53 265: 6.2 265: 39.2 265: 38 265: 36 266: 5.7 266: 6.2 266: 11 266: 13 106 / Nickel Bei einer Gewichtung von 1 : 0.8 = M : M+H werden die beobachteten Werte gut widergegeben. Ausgehend von diesen beiden Signalen wird der folgende Zerfall sehr kompliziert: Das nächste intensive Signal bei m/z = 161 setzt sich aus mindestens 3 Verbindungen zusammen (NiC5H11ON, NiC5H12ON, NiC5H13ON), die durch einen Massenverlust von ∆M = 101-104 zustande kommen können. Eine Gewichtung dieser Komponenten von 0.7 : 0.4 : 0.7 reproduziert die beobachteten Intensitätswerten recht gut (siehe Tabelle 7). Eine Zuordnung dieser Werte zu Strukturfragmenten oder die Postulation eines Zerfallsschemas lässt sich auf dieser Grundlage jedoch nicht aufbauen. Tabelle 7: Beobachtete und berechnete Isotopenmuster für den ersten Massenverlust beim Zerfall von [Ni(OCH(Me)CH2NMe2)2]. Der erste Wert gibt die Masse (in g/mol) an, der zweite die relative Intensität (in %). NiC5H11ON NiC5H12ON NiC5H13ON Berechneter Wert (A) (B) (C) mit A : B : C = 0.7 : 0.4 : 0.7 159: 100.0 Beobachete Werte 159: 70 159 73 160: 44 160 39 160: 5.9 160: 100.0 161: 38.5 161: 6.0 161: 100.0 161: 99 161 100 162: 3.8 162: 38.5 162: 6.0 162: 22 162 18 163: 5.2 163: 3.8 163: 38.5 163: 32 163 31 Die thermogravimetrische Analyse des Precursors zeigt im Temperaturbereich von 120 bis 190 °C einen relativ steilen Gewichtsverlust in einem einzigen Schritt. Der Masserest von 16 % entspricht einem kompletten Umsatz von 1 in metallisches Nickel und flüchtige Nebenprodukte (berechneter Rückstand: 22 %). Der etwas kleinere Wert im Vergleich zu dem berechneten könnte auf eine teilweise Verdampfung des Precursors zurückzuführen sein. Ein starkes und zwei schwache endotherme Signale der DTA begleiten die Zersetzung von 1, wobei das scharfe Signal bei 147 °C der Zersetzungstemperatur der Verbindung zugeordnet wird. 6 100 4 2 80 -2 -4 40 DTA [µV] TG [%] 0 60 -6 20 -8 0 0 100 200 300 400 -10 500 Temperatur [°C] Abb. 51: TG/DTA von 10.4 mg [Ni(OCH(Me)CH2NMe2)2] in einer Argonatmosphäre. Die Heizrate betrug 5 K/min. in einem Temperaturbereich von RT bis 500 °C. Nickel / 107 Des weiteren wurde 1 bei erhöhten Temperaturen im Vakuum einer geschlossenen Apparatur zersetzt und die nach Abkühlen flüssigen Rückstände der Zersetzung von 1 in CDCl3 gelöst und mittels 1H (siehe Abb. 52)und 13C NMR (nicht abgebildet) untersucht. Der Vergleich mit den Resonanzen von 1-Dimethylamino-propan-2-ol und 1-Dimethylaminopropan-2-on[197] zeigt, dass diese Verbindungen entstanden sind und der Zerfall von 1 auf reduktivem Weg erfolgt. 5/8 3 OH O N 1 2 4 N 5 6 7 8 1 3 6 7 4 2 4,0 3,5 3,0 2,5 2,0 1,5 1,0 Shift [ppm] 1 Abb. 52: H NMR der Rückstände aus der thermischen Zersetzung von 1. Vergleicht man jedoch die Integrale der Signale für die Positionen Nr. 1 (Alkohol) und 7 (Keton) stellt man fest, dass die Verbindungen nicht wie erwartet im Verhältnis Alkohol:Keton = 1:1, sondern 2:1 enstanden sind. Der Unterschuss des Ketons kann entweder durch eine Zersetzung bei den hohen Temperaturen erklärt werden oder weist auf eine unvollständige Reduktion der Nickelkationen zu Nickel hin. Da die Nanopartikel, die wie später gezeigt wird aus dieser Verbindung durch Thermolyse erhalten werden können, jedoch metallisch sind und kein NiO enthalten, ist anzunehmen, dass Ersteres der Grund für den beobachteten Unterschuss ist. Dies Annahme wird durch die braune Verfärbung der Flüssigkeit, die bereits auf eine Zersetzung hindeutet, unterstützt. Darüber hinaus berichten Davi et al.[131] von der Bildung eines weiteren Produkts bei der Zersetzung von [Cu(OCHCH2NMe2)2] bei hohen Temperaturen, das sie zwar nicht identifizieren konnten, jedoch auf thermische Zersetzung der primären Nebenprodukte (Alkohol und Keton) zurückführen. Unterstützende GC/MS-Untersuchungen, die auf die Entstehung von Bruchstücken hinweisen könnten, wurden nicht durchgeführt. Aufgrund dieser Daten wird angenommen, dass 1 gemäß Schema 15 zerfällt. Das heißt, dass auch hier keine zusätzlichen Reduktionsmittel erforderlich sind und das Reaktionssystem auf die gleiche Weise wie in Kapitel 5.3.1 bereits beschrieben vereinfacht wird. Es sei hinzugefügt, dass Schema 15 keine Aussage über den Reaktionsmechanismus impliziert. 108 / Nickel Me2 N O ∆T Ni N Me2 OH Ni (0) + O N + N O Schema 15: Zerfall von 1 zu elementarem Nickel, 1-Dimethylaminopropanol und 1-Dimethylaminopropanon. 7.2.2 Pyrolyse Für die Pyrolyse von 1 in heißen koordinierenden Lösungsmitteln wurde HDA als Lösungsmittel verwendet. Während starke π-Donor-Liganden ein Zusammenbrechen der Magnetisierung zur Folge haben, ist für σ-Donor-Liganden wie Amine zunächst angenommen[198], später aber auch bewiesen worden[49], dass sie die Oberflächenmagnetisierung nicht reduzieren. Die qualitativ besten Nickelpartikel (bezogen auf die Monodispersität der Partikel) lassen sich durch Verwendung von 7 g HDA, in das bei 180 °C Ölbadtemperatur eine 0.3-M Lösung von 1 in Octylamin injiziert wird, herstellen. Nach der Aufarbeitung erhält man eine gold-braune Lösung in Toluol, die unter Schutzgas über Wochen hinweg stabil ist und sogar an Luft einige Tage ohne Bildung eines Niederschlags aufbewahrt werden kann. Die elektronenmikroskopischen Aufnahmen der kolloidalen Lösung sind in Abb. 53 gezeigt. Man erkennt sphärische Partikel mit einem Durchmesser von 6.5 nm. Die Größe und die Form der Partikel sind sehr einheitlich und die einzelnen Partikel sind deutlich von den benachbarten Teilchen getrennt. Der Abstand wurde zu 2.5 nm bestimmt und kann der Hülle aus Liganden zugeordnet werden, die den metallischen Kern umgeben. (111 ) (2 0 0 ) (2 2 0 ) (3 11 ) 1 .4 8 A Abb. 53: Links: TEM-Aufnahme von Nickel-Nanopartikeln. Die Probe wurde durch Auftropfen der toluolischen Lösung auf ein mit einem Kohlenstofffilm bedecktes Goldnetz präpariert (Bar = 84 nm). Rechts: SAED der links abgebildeten Partikel. Die Vergrößerung zeigt, dass die schmale Größenverteilung ganz offensichtlich zu einer Selbstanordnung von freistehenden Kolloiden in zweidimensionale, hexagonale Gitter führt. Die dazugehörige SAE Beugung ist in Abb. 53 rechts abgebildet. Die hellen, klaren Reflexe können den (111), (200), (220) und (311) Netzebenen von kubischem Nickel zugeordnet Nickel / 109 werden. Zusätzlich lässt sich bei manchen Beugungsbildern ein zusätzlicher sehr schwacher Reflex bei einem Netzebenenabstand von 1.48 Å beobachten, der von Nickeloxid verursacht wird. Da dieser Reflex (1) nur für kleine Partikel auftritt und (2) bei Präparation und anschließendem Transport unter Schutzgas (Vakuum-Transfer-Doppelkipphalter) nicht beobachten lässt, wird angenommen, dass Nickeloxid wie bei Kupfernanopartikeln durch oberflächliche Oxidation nach Kontakt mit Luftsauerstoff entstehen. Die einzelnen Beugungsringe der Nickelkolloide weisen außerdem Punkte mit Intensitätsmodulation auf. Punkte entstehen, wenn eine begrenzte Anzahl an Partikeln vorliegen und sind bei einer statistischen Anordnung der Partikel normalerweise gleichmäßig verteilt. Die Intesitätsmodulation weist daher auf ein bestimmtes Maß an Ordnung hin und stimmt damit mit der im Realbild beobachteten Selbstordnung überein. Im Gegensatz zu der deutlich sichtbaren OPR von Kupferkolloiden sind für Nickelkolloide keine definierten Absorptionsbanden zu erkennen, d. h. Nickel zeigt keine OPR im sichtbaren Bereich der elektromagnetischen Strahlung (Spektrum nicht abgebildet). In der Literatur wird deswegen auch selten über die Absorptionseigenschaften dieses Nanomaterials berichtet, ähnlich für die schwereren d10-Elemente Palladium und Platin. Ausnahme bildet eine theoretische Berechnung aus dem Jahre 1981[199], die eine Absorption von 235 nm für sphärische Partikel von 10 nm Durchmesser in wässrigem Medium vorhersagt. Die Lösung in Toluol zeigt jedoch keinerlei Absorption in diesem Bereich. Führt man die Reaktion bei höheren Temperaturen durch, wird im Gegensatz zu der Thermolyse von [Cu(OCH(Me)CH2NMe2)2], bei der bei höheren Temperaturen eine Fokussierung der Größenverteilung eintritt, beobachtet, dass die Bildung von Stäbchen und Quadern begünstigt ist. Bei einer Injektionstemperatur von 250 °C ist zwar die Mehrheit der Partikel sphärisch, aber die Größenverteilung ist verbreitert (8-15 nm) und es entstehen Stäbchen mit 7-10 nm Breite und 15-25 nm Länge. Die Anwesenheit von Partikeln anderer Morphologie stört darüber hinaus auch die Selbstordnung in zweidimensionale Reihen. Injektion bei 300 °C erhöht den Anteil von Stäbchen und viereckigen Nickelpartikeln. Abb. 54: : TEM Aufnahmen von Nickel-Nanopartikeln, erhalten durch die Thermolyse von 1 bei (links) 250 °C (Balken = 85 nm) und (rechts) 300 °C (Balken = 76 nm). 110 / Nickel 7.3 Zusammenfassung Der organometallische Precursor [Ni(OCH(Me)CH2NMe2)2] wurde über eine vereinfachte Syntheseroute hergestellt, mittels MS und NMR charakterisiert und thermogravimetrisch untersucht. Durch die Thermolyse dieser Verbindung in HDA konnten qualitativ hochwertige metallische Nickel-Nanopartikel synthetisiert werden. Wie im Falle von Kupfer und Palladium gewährleisten die Liganden, die an das Zentralatom des organometallischen Precursors koordiniert sind, eine Reduktion des zweiwertigen Metallions, während das Lösungsmittel koordinierende Funktion übernimmt und die entstandenen Partikel vor Agglomeration schützt. Die kolloidalen Partikel tendieren aufgrund ihrer sehr schmalen Größenverteilung zu einer Selbstordnung in ein hexagonales Muster. Auf diese Weise konnte mit Hilfe der organometallischen Chemie ein kristalliner, einfach herstellbarer Precursor für die Synthese von Nanopartikeln entwickelt, der im Gegensatz zu den meisten literaturbekannten Verfahren keines Zusatzes an Reduktionsmittel bedarf. Die Qualität der hierdurch zugänglichen Partikel kann sich mit den besten aus der Literatur messen. [Cu(OCH(Me)CH2NMe2)2] ist eine Verbindung, die ursprünglich für die Abscheidung von Kupfer in einem CVD-Prozess verwendet worden ist. Durch unsere Arbeiten ist gezeigt worden, dass dieser, aber auch andere organometallische CVD-Precursoren für Metalle unter den geeigneten Bedingungen ebenso als Precursoren für Nanopartikel geeignet sind. Umgekehrt sind nun Untersuchungen gestartet worden, um zu zeigen, dass die analoge Ni-Verbindung [Ni(OCH(Me)CH2NMe2)2] als Precursor für metallische Nickelpartikel geeignet ist und folgerichtig ebenso für die Abscheidung von dünnen Nickelfilmen in der chemischen Dampfabscheidung verwendet werden kann. Die ersten Versuche zeigten bereits eine Abscheidung von amorphem Nickel (als Schicht bzw. als Kugeln), es sind jedoch weitere Untersuchungen nötig, um die Versuchsbedingungen zu optimieren und die Ergebnisse zu validieren. Nickel / 111 8 Modellsysteme für den Cu/ZnO Katalysator in der Methanolsynthese 8.1 Einleitung und Aufgabenstellung Kohlenmonoxid und Wasserstoff können, je nach Katalysator, zu Methan, Kohlenwasserstoffen, Methanol, höheren Alkoholen, Aldehyden, Säuren oder auch zu Kohlenstoff und Wasser reagieren. Die Synthese von Methanol ist insbesondere im Hinblick auf den zukünftig vielleicht möglichen Ersatz von Verbrennungsmotoren durch Elektromotoren interessant. Heute wird der meiste Teil noch als Lösungsmittel oder gasoline extender (Streckmittel für Benzin) verwendet und ist Ausgangschemikalie für viele Chemikalien wie z. B. Formaldehyd, Methyltertbutylether, Essigsäure, Methylmetacrylat und Dimethyltherphtalat. Weltweit wurde Methanol 1995 in einer Menge von 27 Mio. Jahrestonnen produziert, Tendenz steigend, und liegt damit unter den Top-10 Chemikalien. Die Synthese wurde bereits 1923 von der BASF[200] kommerzialisiert, damals noch unter hohen Temperaturen und Drücken aus Synthesegas unter Verwendung eines ZnO-Cr2O3 Katalysators. Obwohl schon kurze Zeit später bekannt war, dass Kupfer ein extrem reaktives Metall für die katalytische Produktion von Methanol ist, insbesondere in Kombination mit ZnO und Al2O3, dauerte es bis 1966, bevor ICI[201-203] eine Katalyse einführte, die bei niedrigen Temperaturen und Drücken unter Verwendung eines heterogenen Cu-ZnO Katalysators arbeitet. Dabei liegt metallisches Kupfer feinverteilt auf einem gemischten Trägermaterial aus Al2O3 und ZnO vor, eine Kombination, die seitdem nahezu ausschließlich verwendet wird. Einige mechanistische Details des Katalyseprozesses konnten bereits geklärt werden: Durch Experimente, in denen eine kleine Menge CO2 einem stationären Gasstrom an CO und H2 zugegeben wurde, ist bekannt, dass Methanol aus CO2 wesentlich schneller gebildet wird als aus CO. Außerdem konnte gezeigt werden, dass die Oberfläche des Metalls während der Synthese nicht mit atomarem Sauerstoff bedeckt ist. Eine ganze Reihe von Fragen werden jedoch immer noch kontrovers diskutiert, wie z. B. die Natur der aktiven Spezies oder die Rolle des Zinkoxids und anderer Bestandteile des Katalysators. Angesichts dieser Uneinigkeit in der Literatur über solch zentrale Aspekte besteht nach wie vor ein Bedarf an Untersuchungen, die diese Fragen klären. Da sich Realkatalysatoren aufgrund ihrer Komplexität nicht zur Untersuchung und Charakterisierung von Metall-Substrat-Wechselwirkungen eignen, spielen Modellsysteme für das Cu/ZnO/Al2O3-System eine wichtige Rolle. Genau dies, nämlich die Entwicklung von Modellsystemen mit dem Ziel, die Wechselwirkungen, die in diesem System auftreten, zu verstehen, ist Thema des Sonderforschungsbereichs 558 „Metall-Substrat-Wechselwirkungen in der heterogenen Katalyse“. Im Verbund der unterschiedlichen Teilprojekte dieses SFBs ist es Aufgabe der anorganischen Chemie, metallorganische Precursor-Routen für die Katalysator-Präparation zu entwickeln. Insbesondere sollten 112 / Messing kolloid-, photo- und sonochemische Ansätze für nanopartikuläre Metall/Substrat-Komposite in den SFB eingebracht werden. Das von uns bearbeitete und in diesem Kapitel vorgestellte Projekt über nanopartikuläre Metall/Substrat-Komposite zielt darauf ab, die offenbar entscheidende Cu/ZnO-Grenzfläche gewissermaßen in einem freistehenden (oberflächenstabilisierten) Kompositpartikel "heraus zu präparieren" und in Lösung "handhabbar zu machen". Um dieses Ziel zu realisieren, wurden basierend auf precursor- und kolloidchemischen Methoden drei verschiedene Wege für die Präparation von Cu@ZnO-Nanokompositen untersucht. Diese werden in den folgenden drei Kapiteln vorgestellt. 8.2 Cu und ZnO in räumlicher Trennung: Imprägnierung von ZnO mit Kupfer-Nanopartikeln Nachdem es gelungen ist, qualitativ hochwertige kolloidale Kupfer-Partikel zu synthetisieren, ist es denkbar, eine Cu/Zn(O)-Grenzfläche zu realisieren, indem eine einkristalline ZinkoxidOberfläche mit stabilisierten Kupferpartikeln imprägniert wird. Die Imprägnierung wurde durch einfaches Versetzen einer Suspension von Zinkoxid-Pulver (Alpha) in trockenem Toluol mit einer Lösung von 8 nm großen kolloidalen HDA-stabilisierten Kupfer-Nanopartikeln in Toluol realisiert. Die entstehende Suspension wurde auf ein mit Kohlenstoff bedecktes Goldgrid getropft und elektronenmikroskopisch untersucht. Die beiden Bilder in Abb. 55 zeigen zwei mögliche Situationen für dieses System: die Kupferpartikel können sich entweder auf den im Vergleich wesentlich größeren ZnO Partikeln absetzen oder räumlich von ihnen getrennt vorliegen. 8 nm ZnO ZnO 380 nm 95 nm Cu Abb. 55: TEM-Aufnahme von Zinkoxidpulver nach Impregnation mit kolloidalen Kupferpartikeln. Die links abgebildete Konstellation ließ sich nur durch Einwirken von Ultraschall auf die Cu/ZnO-Suspension erreichen, wobei der Anteil von Cu@ZnO immer noch sehr gering war. Man kann klar Partikel von etwa 8 nm Durchmesser erkennen, die sich auf einem 380 x 95 nm2 großen Stäbchen befinden. Die kleinen Partikel bestehen aus Kupfer, ihre Größe hat sich durch die Impregnation nicht geändert. Die großen rechteckigen Partikel sind oxidisches Material wie aus separaten Messungen des verwendeten ZnO-Pulvers bekannt ist. Die überwiegende Mehrheit der Partikel befindet sich jedoch in der rechts abgebildeten Messing / 113 Situation, also ohne Kontakt zwischen Kupfer und Zinkoxid. Auch hier lassen sich aufgrund von Größe und Morphologie die Zuordnungen zu Kupfer bzw. Zinkoxid leicht treffen. Ganz offensichtlich ist die Wechselwirkung zwischen der Oberfläche des Zinkoxids und den Alkylketten, die die Kupfer-Nanopartikel umgeben, so gering, dass die Impregnationsmethode nicht zu dem gewünschten Cu@ZnO-System führt. Die HDA-Stabilisierung wird nur durch sonochemischen Einfluss und nur sehr unvollständig aufgebrochen. Möglicherwiese ist das Anhaften der Kupferpartikel auf ZnO im sonochemischen Experiment durch teilweisen Verlust der HDA Schutzhülle zurückzuführen. 8.3 Cu und ZnO in räumlicher Nähe: Mischungen von Cu- und ZnOKolloiden Nachdem offenbar die Stabilisierung des Kupfers mit organischen Liganden die Impregnation von Mikrometer großen ZnO-Partikeln mit HDA-stabilisiertem nano-Kupfer verhindert, wurde in einem zweiten Ansatz versucht, die Cu/ZnO-Grenzfläche in core-shell Cu/ZnOPartikeln zu erzeugen. So sollte es möglich sein, zunächst ZnO-Partikel herzustellen (diese Arbeiten wurden bereits in Kapitel 4 vorgestellt) und diese dann als Keime für das Aufwachsen von Kupfer zu verwenden. Praktisch wurde dies durch eine 2-Stufen Synthese realisiert, indem zunächst ZnO-Partikel durch Injektion von [EtZn(OiPr)][204] in 170 °C heißes HDA hergestellt wurden. In dieser Reaktionsmischung wurde anschließend bei etwas höheren Temperaturen (200 °C) eine äquimolare Menge [Cu(OCH(Me)CH2NMe2)2] thermolysiert. Die TEM-Aufnahmen dieser Reaktion sind zusammen mit der Elektronenbeugung in Abb. 56 gezeigt. (11 2 ) (1 0 3 ) (11 0 ) (1 0 1 )/ (0 0 2 ) (1 0 0 ) (111 ) (3 11 ) (2 2 0 ) (3 11 ) Z nO K u p fe r Abb. 56: TEM Aufnahme (links, bar = 78 nm) und SAED (rechts) von Kupfer- und Zinkoxid-Nanopartikeln durch Pyrolyse von [EtZn(OiPr)3] und [Cu(OCH(Me)CH2NMe2)2] in HDA. Das Realbild lässt klar eine bimodale Partikelverteilung mit kleineren (Ø = 4 nm) und größeren (Ø = 12 nm) Partikeln erkennen. Die einzelnen Partikel haben sphärische Formen und sind mit einem Abstand von etwa 4 nm voneinander separiert. Das Beugungsmuster dieser Probe weist eine hohe Anzahl von Reflexen auf, die sich den in Abb. 56 rechts aufge114 / Messing listeten Gitterebenen von Kupfer und ZnO zuordnen lassen, d. h. es sind separate metallische und oxidische Phasen entstanden. Demnach lässt sich vermuten, dass die oben beschriebene Synthesemethode nach der Zersetzung von [Cu(OCH(Me)CH2NMe2)2] zu Kupferatomen entweder zu einem Wachstum von Kupfer auf bereits vorhandenen Zinkoxidpartikeln oder zu einer separaten Nukleation geführt hat. Die Tatsache, dass eine bimodale Verteilung in der Partikelgröße vorliegt, lässt allerdings Letzteres wahrscheinlicher sein. Um diese Annahme zu überprüfen, wurden Pyrolyseexperimente mit den einzelnen Precursoren durchgeführt. Während bei der Pyrolyse von [EtZn(OiPr)] ZnO-Partikel mit einem Durchmesser von 3-4 nm entstehen, bildet [Cu(OCH(Me)CH2NMe2)2] Cu-Kristallite mit einem Durchmesser ≥ 8 nm. Aufgrund dieser Ergebnisse wurden die kleinen Partikel in Abb. 56 Zinkoxid zugeordnet bzw. die größeren Partikel Kupfer. Insgesamt ist damit ein System realisiert worden, in dem Kupfer und Zink bzw. Zinkoxid mit einem minimalen Abstand von 4 nm zueinander vorliegen, jedoch keine Cu/Zn-Grenzfläche ausbilden. Dieses Verhalten weicht deutlich von der eigentlichen Vorstellung ab, dass die primär vorliegenden ZnO-Partikel als Nukleationskeime für Kupfer dienen. Damit aber eine Nukleation von Kupfer auf ZnO möglich ist, müsste das an die Oberfläche der ZnO-Partikel koordinierte HDA desorbieren, damit eine Annäherung des Kupfer-Precursors gewährleistet ist. Dies scheint nun definitiv nicht der Fall zu sein. Offenbar sind die ZnO-Partikel stark durch HDA geschützt. Diese Interpretation wird durch die Tatsache gestützt, dass die HDA-ZnO-Partikel vergleichsweise klein sind. Im Größenbereich von 2-3 nm sind ca. 30-40% aller Atome Oberflächenatome. Es liegt daher nahe, eine starke Adsorption des HDAs an den Lewisaciden Zn2+-Zentren der Oberfläche bzw. auch Wasserstoff-Brücken zu den O2--Zentren zu vermuten. 8.4 Inniger Kontakt zwischen Kupfer und Zink: Coating von KupferNanopartikeln mit Zink Da die zweistufige Thermolyse von erst [EtZn(OiPr)]n und dann [Cu(OCH(Me)CH2NMe2)2] zu separaten Kupfer- bzw. ZnO-Partikeln führt, muss für die Realisierung eines direkten Kontakts zwischen Cu und Zn ein anderer Weg eingeschlagen werden. Aus der Beobachtung, dass primär erzeugte HDA-stabilisierte ZnO-Partikel nicht als Keime für die Nukleation von Kupfer-Partikeln dienen bzw. so keine Cu@ZnO core-shell Kompositpartikel herstellbar sind, ist abzuleiten, dass die Bildung von Kompositartikeln durch weniger unterschiedliche Adsorptionsenergien des Stabilisators auf verschiedenen Partikeloberflächen begünstigt werden sollte. So entstand die Idee, eine entsprechende Synthese von Cu/Zn-Legierungspartikeln zu versuchen, da HDA auf Kupfer bzw. Zink als Metalloberflächen wahrscheinlich weniger unterschiedlich stark adsorbiert als auf ZnO- versus Kupfer-Oberflächen. Wir vermuteten außerdem, dass der Energiegewinn durch eine Legierungsbildung zwischen Kupfer und Zink eine zusätzliche Triebkraft für die Bildung von Cu/Zn-Nanokompositen sein kann. Messing / 115 8.4.1 Precursor und Synthese Als Quelle für elementares Zink wurde Et2Zn ausgewählt. Um seine Eignung, Zink freizusetzen, zu testen, wurde es in Octylamin gelöst und in heißes HDA injiziert. Es bildet sich ein grauer Niederschlag, der sich genau wie das Material aus der Thermolyse von [MeZnOSiMe3]4 nicht in Toluol lösen lässt. XRD-Messungen bestätigten die Bildung von metallischem Zink (die Abbildungen und weitere Analytik sind in den Kapiteln 8.4.2 bis 8.4.4 zu finden) und somit die Tauglichkeit von Et2Zn als Zinkquelle. Für die Untersuchung von gemischten Cu/Zn Systemen wurden fünf verschiedene molare Verhältnisse von Kupfer zu Zink in den Vorratslösungen ausgewählt, nämlich Cu/Zn = 100/0, 90/10, 50/50, 30/70 und 0/100. Die Synthese von Cu/Zn 100/0 und 0/100 erfolgte wie in Kapitel 5 bzw. oben beschrieben. Für das System mit dem geringsten Zinkanteil wurden zunächst Kupferpartikel synthetisiert und ohne Aufarbeitung auf 180 °C erhitzt. Die entsprechende Menge Et2Zn wurde tropfenweise zugegeben und die Reaktionsmischung nach einer Reaktionszeit von weiteren 10 Minuten abgekühlt und standardmäßig aufgearbeitet. Für die äquimolare Mischung wurden die Precursor vor der Zugabe in Octylamin gelöst, miteinander vermischt, und dann in heißes HDA injiziert. Die Reaktionsmischung wurde analog zur Versuchsvorschrift für Kupfernanopartikel 30 Min. bei dieser Temperatur gehalten, dann abkühlen gelassen und aufgearbeitet. Das Zink-reiche System wurde auf dieselbe Art und Weise präpariert. Es kann auch analog zum 90/10 System hergestellt werden, wobei weder in der Partikelmorphologie noch in der Beugung eine Veränderung zur erstgenannten Methode zu bemerken ist, so dass die beiden Methoden wahlweise verwendet werden können. Alle Systeme (mit Ausnahme von Cu/Zn 0/100) bilden nach Aufarbeitung mit Methanol und Zentrifugation rot gefärbte Lösungen in Toluol. 116 / Messing 558 Absorption [a. u.] 2 1 0 400 500 600 700 Wavelength [nm] Abb. 57: (a) UV/VIS (b) TEM (bar = 110 nm) und (c) SAED der Lösung des Cu/Zn = 100/0 Systems in Toluol. 4 557 Absorption [a. u.] 3 2 1 0 400 500 600 700 Wavelength [nm] Absorption [a. u.] 564 Abb. 58: (a) UV/VIS (b) TEM (bar = 130 nm) und (c) SAED der Lösung des Cu/Zn = 90/10 Systems in Toluol. 0,5 400 500 600 700 Wavelength [nm] Absorption [a. u.] Abb. 59: (a) UV/VIS (b) TEM (bar = 116 nm) und (c) SAED der Lösung des Cu/Zn = 50/50 Systems in Toluol. 1 400 500 600 700 Wavelength [nm] Abb. 60: (a) UV/VIS (b) TEM (bar = 175 nm) und (c) SAED der Lösung des Cu/Zn = 70/30 Systems in Toluol. 1000 800 (002) (101) 400 200 (102) (100) Intensity [a. u.] 600 0 30 35 40 45 50 55 60 2 Theta [°] Abb. 61: (b) TEM (bar = 500 nm) und (c) XRD des Systems Cu/Zn = 0/100. Messing / 117 8.4.2 Analytische Charakterisierung Zusammensetzung (EDX) Wie nicht anders zu erwarten, zeigt die EDX-Analyse der 100/0 und 0/100-Systeme neben den vom Grid stammenden Signalen (Gold, Kohlenstoff) nur Signale für Kupfer bzw. Zink. Bei den gemischten Systemen sind jedoch durchweg Signale für beide Metalle, Kupfer und Zink, zu sehen. Da die Proben vor der Messung aufgereinigt worden sind und deshalb jegliches mikrokristallines Material, das (wie später gezeigt wird) durch eine separate Nukleation von Zink entstehen würde, abgetrennt worden ist, kann man davon ausgehen, dass in allen Fällen gemischte Cu/Zn-Systeme gebildet worden sind. Das Verhältnis von Kupfer zu Zink, das in den Proben mit Cu/Zn = 90/10, Cu/Zn = 50/50 bzw. Cu/Zn = 30/70 gemessen wurde, betrug Cu/Zn = 95/5 (±10%), Cu/Zn = 70/30 (±10%) bzw. Cu/Zn = 35/65 (±10%). Die in die Synthese eingebrachten Verhältnisse liegen damit (bis auf Cu/Zn = 50/50) innerhalb der Messgenauigkeit einer EDX-Analyse, sind aber tendentiell etwas niedriger als die in den fertigen Komposit-Partikeln gefundenen. In der folgenden Beschreibung wird jedoch die alte Systematik beibehalten, um keine Verwirrung zu erzeugen. UV/VIS Absorption Reine, metallische Kupfer-Nanopartikel zeigen eine definierte Oberflächenplasmonenresonanz (OPR), die auch die intensive rote Farbe ihrer Lösungen erzeugt. Sie liegt bei λ = 558 nm und ist bis zu einer Partikelgröße von etwa 25 nm größenunabhängig und damit charakteristisch für nanoskaliges Kupfer (siehe Abb. 57a). Das UV/VIS Spektrum des 90/10-Systems ist in Abb. 58a gezeigt. Das (lokale) Absorptionsmaximum liegt bei λ = 558 nm, d. h. bei demselben Wert wie das reine Kupfersystem. Das heißt, dass ein geringer Anteil an Zink die Oberflächeneigenschaften von Kupferpartikeln nicht beeinflusst. Natürlich kann dieses Verhalten auch durch völlige Abwesenheit von Zink entstehen, doch die oben beschriebenen Ergebnisse der EDX-Messungen widerlegen diese Möglichkeit. Wenn das Verhältnis von Kupfer zu Zink auf 1.0 erhöht wird, zeigt das UV/VIS-Spektrum der entsprechenden toluolischen Lösung eine Absorptionsbande bei λ = 564 nm (Abb. 59a). Im Vergleich zu reinen Kupferpartikeln ist der Wert damit klar zu höheren Wellenlängen verschoben. Die Rotverschiebung von 6 nm zeigt, dass die OPR nicht länger ausschließlich von Kupfer verursacht wird, sondern dass zusätzliche Effekte eine Rolle spielen. Da die Liganden aufgrund der synthetischen Bedingungen dieselben sind wie im 100/0 System und aus EDXMessungen bereits bekannt ist, dass die Partikel zu 30% aus Zink bestehen, kann dieser Effekt der Anwesenheit von Zinkatomen zugeschrieben werden. Bei einem Anteil von 70 mol-% Zink schließlich wird keine OPR mehr im UV/VISSpektrum beobachtet (Abb. 60a). Das heißt, es ist offenbar ein neues System mit veränderten 118 / Messing charakteristischen Eigenschaften entstanden, das keine Oberflächenelektronen mehr für die Wechselwirkung mit sichtbarem oder UV-Licht zur Verfügung stellt. Es ist festzuhalten, dass die Injektion von Et2Zn in heißes HDA Material ergibt, das nicht in Toluol löslich ist. Das heißt, wenn die Thermolyse von Et2Zn zu Zinkpartikeln führen würde, ohne dass mit den ebenfalls vorhandenen Kupferpartikeln oder Kupferatomen eine Reaktion eingegangen würde, würde das Material durch die Aufarbeitung abgetrennt werden und die UV/VIS Absorption nicht beeinflussen. Es ist also anzunehmen, dass die beobachteten optischen Eigenschaften aus den gemischten Cu/Zn-Systemen stammen und nicht von separaten Kupfer- und ZinkPartikeln. 8.4.3 Elektronenmikroskopie (TEM) Wie aus Kapitel 5 bekannt ist, bildet Kupfer nach der Thermolyse von [Cu(OCH(Me)CH2NMe2)2] in HDA sphärische, gut voneinander separierte und hoch monodisperse Partikel von etwa 8 nm aus, die in Abb. 57b zum Vergleich gezeigt sind. Am anderen Ende des Systems bildet das 100% Zinksystem Material mit einer ganz anderen Morphologie. Et2Zn entwickelt nach der Thermolyse in HDA unregelmäßige Stäbchen mit einer Länge von etwa 1 µm und einer Breite von 50 nm aus (Abb. 61b). Da keine der TEM-Aufnahmen der gemischten Systeme (Abb. 58b bis Abb. 60b) die Anwesenheit von Stäbchen zeigt, kann davon ausgegangen werden, dass Zink, wenn es im System vorhanden ist, wahrscheinlich in Form einer Legierung mit Kupfer vorliegt. Abb. 58b zeigt die elektronenmikroskopischen Aufnahmen des Cu/Zn 90/10 Systems. Auch hier liegen klar voneinander separierte Partikel vor, wenn auch mit einer wesentlich breiteren Größenverteilung zwischen 5 und 10 nm. Die höhere Dispersität kommt durch die Verwendung von bereits breiter verteilten Kupferpartikeln als Augangsmaterial zustande und hängt somit nicht mit der Zugabe des Zinks zusammen. Die EDX Analyse dieser Probe zeigte ein Spektrum mit Signalen für Kupfer und Zink, was beweist, dass in der Tat beide Elemente vorhanden sind. Allerdings ergab die quantitative Analyse der Kα Linien von Kupfer bzw. Zink, dass das Verhältnis größer ist als angenommen, nämlich Cu/Zn = 95/5. Für das System mit äquimolaren Mengen Cu und Zn werden ebenfalls sphärische Partikel mit einem Durchmesser von etwa 10 nm beobachtet, die jedoch facettiert sind, eine breite Größenverteilugn aufweisen und zum Teil dreieckige Formen annehmen wie Abb. 59b zeigt. Auch hier ist der Anteil an Zink niedriger als die zugegebene Menge, d. h. Cu/Zn = 70/30. Bei einem Anteil an Zink von 70 % werden wieder sphärische Partikel ausgebildet (Abb. 60b), wobei keine dreieckigen Formen zu beobachten sind. Die Partikel haben eine Größe von 10 nm und sind wie aufgrund ihrer Löslichkeit in Toluol zu erwarten, voneinander separiert. Die Zusammensetzung wurde mittels EDX zu 35/65 = Cu/Zn bestimmt. Messing / 119 8.4.4 Elektronenbeugung (SAED und XRD) Unter Schutzgas präparierte und mit Hilfe eines Vakuumtransferhalters transportierte Kupferpartikel zeigen ein Beugungsmuster, dessen Reflexe eindeutig den (111), (200), (220) und (331) Reflexen von kubischem Kupfer zugeordnet werden können. Aus Beugungsbildern, die nach Kontakt dieser Proben mit Luft aufgenommen wurden, ist bekannt, dass die Oberfläche des Kupfers sehr leicht oxidiert wird, was zur Folge hat, dass zusätzliche Reflexe auftreten, die den Gitterebenen von Cu(I)- und Cu(II)oxid zugeordnet werden können. Es konnte bereits gezeigt werden (Kapitel 5.4.2), dass nur die Oberfläche oxidiert wird und nicht die gesamten Partikel, da (1) die intensiven Reflexe von metallischem Kupfer bestehen bleiben und die zusätzlichen Reflexe schwach und diffus erscheinen und (2) die oxidischen Reflexe auch für größere Partikel beobachtet werden, dort jedoch aufgrund des abnehmenden Oberflächen-Volumen Verhältnisses wesentlich schwächer in ihrer Intensität sind. Die Anwesenheit von 10 % Zink im System hat das in Abb. 58c gezeigte Beugungsbild zur Folge. Die den Reflexen entsprechenden Gitterabstände sind in Tabelle 8 angegeben und lassen sich eindeutig den (111), (200), (220) und (331) Ebenen von metallischem Kupfer zuordnen. Obwohl die Probe nach der Präparation in der Glovebox Kontakt zu Luftsauerstoff hatte, zeigt sie keinerlei Reflexe für oxidisches Kupfer. Da von reinen Kupferkolloiden bekannt ist, dass die Oberfläche oxidiert wird, kann hier davon ausgegangen werden, dass die Kupferoberfläche eines Systems durch die Legierung mit Zink effektiv vor der Oxidation geschützt ist. Das Vorliegen von einzig kristallinem Kupfer stimmt gut mit der Beobachtung überein, dass die OPR dieser Lösung im Vergleich zum reinen Kupfersystem nicht verschoben ist. Auf den ersten Blick sieht auch das Beugungsbild des 50/50 Systems (Abb. 59) nach metallischem Kupfer ohne CuxO aus. Eine genauere Untersuchung der Reflexe deckt jedoch fünf zusätzliche Beugungsringe mit geringer Intensität auf, nämlich bei Gitterabständen von 2.35, 1.61, 1.37, 1.21 und 1.16 Å (jeweils ± 0.02 Å). Durch Vergleich mit Literaturwerten kann die Existenz von kristallinem Zink bzw. Zinkoxid, was eine durchaus wahrscheinliche Form für anwesendes Zink wäre, ausgeschlossen werden, da mindestens zwei der dafür charakteristischen Reflexe fehlen (siehe Tabelle 8). Anstatt separate Phasen auf den Kupferpartikeln zu bilden, kann Zink jedoch auch – wie ursprünglich beabsichtigt – mit Kupfer legieren. Die Gitterabstände für verschiedene Cu/Zn Legierungen sind in Tabelle 8 angegeben, aber einzig die Reflexe von CuZn2, Danbait, passen zu den Daten aus der SAED. Dies führt zu der Vermutung, dass der größte Teil der Nanokristallite aus elementarem Kupfer bestehen, mit einer zusätzlichen Phase aus CuZn2. Dies würde also einem heterogen zusammengesetzten Nanokomposit entsprechen. Ein Vorliegen von Kupfer neben CuZn2 kann hingegen ausgeschlossen werden, da separat vorliegendes Kupfer (wie aus dem System Cu/Zn = 100/0 bekannt ist) oxidische Reflexe im SAED-Muster zeigen müsste. Da (1) die Oberflächenplasmonenresonanz dieser Partikel zu höheren Wellenlängen verschoben ist und 120 / Messing (2) keine Reflexe für CuxO detektiert wurden, kann darüber hinaus vermutet werden, dass CuZn2 zumindest teilweise an der Oberfläche lokalisiert ist. Das Beugungsbild des Kupfersystems mit 30/70 Cu/Zn zeigt ein völlig neues Beugungsmuster. Während reine Kupfersysteme und solche mit einem geringen Anteil an Zink vier charakteristische Reflexe für Kupfer zeigen, weist das SAED in Abb. 60 zwei Reflexe mit sehr hoher Intensität (2.09 und 1.21 Å), einen mit mittlerer, aber dennoch scharfer Intensität (1.48 Å) und mehrere diffuse und schwache Signale auf. Tabelle 8: Gemessene und theoretische Gitterabstände (Intensitäten in Klammern) für unterschiedliche Cu/Zn Systeme (in Å bzw. %). 100/0a 2.09 90/10 2.09 50/50 30/70 2.35 diffuse Reflexe (b) 2.10 2.09 (b) Kupfer Zink 2.47 (53) 2.31 (40) 2.09 (100) Zinkoxid 2.81 (57) 2.60 (44) 2.48 (100) 2.09 (100) Cu5Zn8 2.09 (100) 1.91 (23) 1.81 1.80 1.80 1.82 1.61 1.63 1.48 1.38 1.81 (46) Messing 2.12 (100) 2.06 (17) 1.83 (35) CuZnb CuZn2c 2.35 (50) 2.16 (40) 2.09 (100) 2.08 (100) 1.69 (28) 1.37 1.62 (32) 1.48 (29) 1.38 (23) 1.59 (30) 1.47 (15) 1.37 (40) 1.34 (25) 1.33 (21) 1.28 1.28 1.27 1.21 1.16 1.10 1.28 1.21 1.28 (20) 1.21 (10) 1.20 (29) 1.23 (30) 1.16 (30) 1.10 1.09 (17) 1.09 1.09 (b) = breit Fett gedruckte Zahlen entsprechen Reflexen mit hoher Intensität; Für die Vergleichsmaterialien sind nur Intensitäten > 5% (für CuZn2 > 10%) angegeben; a kein Kontakt mit Luftsauerstoff; bZhangengite, cDanbaite Aufgrund fehlender Reflexe kann die Existenz von Zink und Zinkoxid ausgeschlossen werden und das Vorliegen einer Cu-Zn-Legierung angenommen werden. Cu5Zn8 passt sehr gut zu den gefundenen Gitterabständen, da es lediglich zwei Reflexe mit hinreichender Intensität besitzt (2.09 und 1.21 Å). Allerdings besitzt der Reflex bei 1.21 Å nur eine Intensität von 10 % im Vergleich zu 100 % für den Reflex bei 2.09 Å. Die tatsächlich gefundene Intensität bei 1.21 Å ist jedoch wesentlich größer, d. h. es muss einen weiteren Beitrag an dieser Stelle geben. Die Verbindung, die dafür in Frage kommt, ist CuZn, das mit einer relativen Intensität von 29 % zu dem Reflex beiträgt und darüber hinaus den Reflex bei 1.48 Å erklärt. Messing selber (CuZn) kann hingegen ausgeschlossen werden, weil es einen Reflex bei 1.83 Å mit einer relativen Intensität von 35 % aufweisen sollte. Es gibt zwar einen Reflex bei 1.82 Å, aber im Gegensatz zu dem bei 2.09 Å, der aufgrund seiner Breite die theoretischen Reflexe bei 2.12 und 2.06 Å einschließt, ist er kaum ausgeprägt und diffus. Die schwachen Reflexe bei 1.82 und 1.28 Å können einem kleinen Anteil an kristallinem Kupfer zugeordnet werden. Messing / 121 8.5 Zusammenfassung Unter Ausnutzung der synthetischen Kenntnisse zur Herstellung von stabilisierten, freistehenden Kolloiden ist es uns gelungen, mehrere Systeme aufzubauen, in denen Cu und Zn bzw. ZnO in unterschiedlichen Konstellationen vorliegen. Dabei führen sowohl die Impregnation von ZnO-Pulver mit kolloidalem Kupfer als auch die sequentielle Thermolyse von Kupferund ZnO-Precursoren zu separat vorliegenden Cu- und ZnO-Systemen. Ein inniger Kontakt zwischen Kupfer und Zink kann auf diese Weise nicht realisiert werden. Die Herstellung von Systemen, die Kupfer und Zink enthalten, gelingt jedoch, wenn man die Entstehung von ZnO-Partikeln, die sehr effektiv durch organische Liganden stabilisiert werden können, vermeidet und statt dessen einen Zink-Precursor verwendet. Auf diese Weise konnte nicht nur der Stabilisierungseffekt umgangen werden, sondern dem System ein zusätzlicher Weg zur Stabilisierung, nämlich der der Legierungsbildung, eröffnet werden. Es konnte mittels EDX gezeigt werden, dass die entstandenen Partikel sowohl Zink als auch Kupfer enthalten. Bei einem geringen Anteil an Zink wird weder die OPR noch die Kristallinität der Partikel beeinflusst, aber es konnte beobachtet werden, dass die Partikel wesentlich besser gegen Oxidation durch Luftsauerstoff geschützt sind als reine Kupferpartikel. Dies weist auf eine Legierungsbildung zwischen Kuper und Zink hin, da aus Studien des Oxidationsmechanismus von Messing bekannt ist, dass bei einer Oxidation zunächst Zink oxidiert wird und dann erst Kupfer[205] und analog bei einer Reduktion einer oxidierten Cu-Zn-Legierung zuerst CuO reduziert wird und dann erst ZnO[206]. Bei einem höheren Anteil an Zink entstehen (neben kristallinem, metallischen Kupfer) in den Beugungsmustern der Partikel erkennbare Cu-Zn-Phasen, nämlich CuZn2 (Danbait), Cu5Zn8 und CuZn (Zhangengite). Diese Daten lassen schließen, dass Kupfer in innigem Kontakt mit Zink vorliegt und dass es somit gelungen ist, eine Cu-Zn-Grenzfläche zu erzeugen. Ob und wie die so hergestellten Cu/Zn-Systeme für die Präparation von heterogenen Katalysatoren nützlich sind, ist noch offen. Katalytische Testreaktionen sind (noch) nicht durchgeführt worden, sind aber Bestandteil der aktuellen Forschung im Sonderforschungsbereich (SFB) 558 „Metall-Substrat Wechselwirkungen in der heterogenen Katalyse“ der Ruhr-Universität Bochum, insbesondere in Zusammenarbeit mit den Arbeitskreisen von Prof. Muhler und Prof. Schüth. Es ist für Kupfer zwar bereits gezeigt worden, dass trotz der Schutzhülle Kohlenmonoxid an die Oberfläche vordringen kann, aber es besteht natürlich die Frage, ob und wie die organischen Stabilisatoren die katalytische Aktivität beeinflussen. Jedoch erscheint die Hinwendung zu kolloidchemischen Ansätzen für Metall/SubstratSysteme nicht nur aus grundlegender Sicht, sondern auch aus verfahrenstechnischer Perspektive interessant. So bietet die dreiphasige Methanolsynthese (kommerzieller Prozess LPMEOH), wobei der feste Katalysator als feines Pulver in einem Lösungsmittel dispergiert ist ("Slurry-Reaktor"), gegenüber dem zweiphasigen gas-fest Prozess einen zusätzlichen Freiheitsgrad (z.B. Wahl des Lösungsmittels usw.).[207, 208] 122 / Messing Abgesehen von der Realisierung eines Modellkatalysators, in dem Kupfer und Zink direkt nebeneinander vorliegen, ist auf diesem Wege auch die Synthese von bimetallischen Cu/Zn Partikeln gelungen. Bimetallische Partikel stehen im Fokus des wissenschaftlichen Forschungsinteresses, sowohl im akademischen Bereich als auch in der Industrie. Akademische Forscher sind im wesentlichen an der Abhängigkeit der optischen, physikalischen und strukturellen Eigenschaften von der Zusammensetzung interessiert, während sich die Industrie auf potentielle Anwendungen konzentriert, insbesondere die Katalyse (nicht nur die MethanolSynthese durch Cu-ZnO) und zukünftige Datenspeichersysteme (magnetische Nanolegierungen). Die meisten Veröffentlichungen beziehen sich jedoch auf mehr oder weniger edle Metalllegierungen bzw. metallische core-shell Strukturen wie z. B. Pd/Ni[209], Au/Pd[210, 211], Au/Pt[212], Co/Pt[88, 213, 214], Pd/Cu[215], Pt/Ru[216], Fe/Pt[217, 218], Ag/Au[219], and Ag/Pd[211] (diese Aufzählung berücksichtig nur Kolloide, d. h. lösliche Nanopartikel). Aufgrund ihrer Stellung in der Spannungsreihe sind diese Metalle wesentlich stabiler gegenüber der Oxidation mit Luftsauerstoff, was ihre Synthese und Analyse wesentlich vereinfacht. Unseres Wissens nach gibt es praktisch keinen Bericht über kolloidale Legierungen, die aus mindestens einem reaktiven Metall bestehen. Die hier vorgestellten Cu/Zn-Kolloide stellen somit den ersten Zugang zu kolloidalen Messing-Phasen dar und es ist gut vorstellbar, dass es in der Fortsetzung dieser Arbeit (Marie-Kathrin Schröter) möglich ist, durch gezielte Oxidation auch Cu/ZnO-Kolloide herzustellen. Messing / 123 9 Zusammenfassung Die vorliegende Arbeit ist in enger Zusammenarbeit mit dem Sonderforschungsbereich 558 „Metall-Substrat-Wechselwirkungen in der heterogenen Katalyse“ entstanden. Der SFB 558 beschäftigt sich mit dem Cu/ZnO/Al2O3-Katalysator, der für die großtechnische Synthese von Methanol verwendet wird. Die wesentliche Fragestellung, die hinter den zahlreichen Forschungsarbeiten steht, ist, wie Aktivität, Selektivität und Stabilität von Metall/MetalloxidKatalysatoren von der Art der Metall-Substratwechselwirkung abhängen. Bei der Katalysatorpräparation muss ein inniger, chemischer Kontakt zwischen Metall und Substrat und eine optimale Dispersion des Metalls erreicht werden. In diesem Sinne handelt es sich also um spezielle Nanokomposite. Genau diesen Zugang zu nanopartikulärem Material für den SFB 558 zu erschließen, war Aufgabe der anorganischen Chemie und im Besonderen Aufgabe dieser Arbeit. Das Ziel, das dadurch erreicht werden sollte, war die Präparation von nanokompositären Cu/Zn(O)-Systemen, die einen innigen Kontakt der beteiligten Metalle, Kufer und Zink, aufweisen sollten. Dabei sollte insbesondere das Wissen der (organometallischen) Chemie über das Design von molekularen Precusoren ausgenutzt und die Synthese von nicht-wässrigen Kolloiden, sog. NACs (non-aqueous colloids), ermöglicht werden. Um diesen Zugang zu Nanopartikeln und Kolloiden, also in einem Lösungsmittel dispergierten Nanopartikeln, zu eröffnen, wurde mit einem System begonnen, das in der Literatur wohlbekannt und im Hinblick auf Stabilität und Oxidationsempfindlichkeit vergleichsweise leicht zu handhaben ist: dem II-VI-Halbleiter Cadmiumselenid. Me Cd Me + Se P 3 Me Cd Me TOPO ∆T CdSe O P Cd T O P O /T B P S e T O P O /T B P S e T O P O /T B P S e E t2N Cd N E t2 7 T O P O /T B P S e Me Me Cd N N Abb. 62: Synthese von CdSe-Nanopartikeln unter Verwendung von verschiedenen organometallischen Precursoren. Bar der TEM-Aufnahmen = 56 nm. 124 / Zusammenfassung Neben der Einarbeitung in die Synthese von Nanopartikeln war es Ziel dieser Arbeiten, den weit verbreiteten, jedoch extrem toxischen und darüber hinaus sicherheitsgefährdenden Cadmiumprecursor Me2Cd durch weniger flüchtige, möglichst kristalline organometallische Verbindungen zu ersetzen. Um dies zu erreichen, wurden drei verschiedene Konzepte der anorganischen Chemie angewendet: die Verwendung von sterisch anspruchsvollen Liganden Dineopentylcadmium, das Ausnutzen von intramolekularer Stabilisierung Bis(3-diethylaminopropyl)cadmium und die Bildung von Addukten (2,2’-Bipyridin)dimethylcadmium. In allen Fällen entstehen weitaus stabilere und damit ungefährlichere Verbindungen, im Falle des Bipyridin-Addukts sogar in kristalliner Form. Die Verwendung der neuen Precursor führt zu Nanopartikeln, die sich qualitativ nicht von denen aus der Standardreaktion mit Me2Cd als Precursor unterscheiden. Mit der Erfahrung, die damit über den Halbleiter CdSe vorlag, haben wir uns dem ebenfalls halbleitenden Zinkoxid zugewandt, das einer der Bestandteile des relevanten Cu/ZnO/Al2O3Systems ist. Zinkoxid ist (im Gegensatz zu CdSe) in seiner nanopartikulären Form in der Regel durch toxikologisch eher unbedenkliche Precursoren zugänglich. Allerdings führt die weit verbreitete Synthese durch Dehydratisierung von kolloidalem Zinkhydroxid zu Partikeln, die aufgrund von Sauerstoffdefizienz unbefriedigende optische Eigenschaften aufweisen (Lichtemission im sichtbaren statt im ultravioletten Bereich). Erstaunlicherweise wurden bislang jedoch nicht über die Verwendung von Einkomponentenprecursoren berichtet, mit denen dieser Nachteil vermieden werden kann. Hier greift wieder die Precursorchemie, die unter anderem auf ein ausgedehntes Wissen darüber zurückgreifen kann, wie Metall und Sauerstoff in einer molekularen Verbindung miteinander verknüpft werden können. Me3Si Me O Me Zn Zn O SiMe3 Me Zn O O HDA SiMe3 Me3Si ZnO ∆T Zn H 2N 15 Me 1 5 2,5x10 4 (101) 5,0x10 4 3 (200, 112, 201) 0 1,0x10 4 (103) A bsorption 5 1,5x10 (102) 1x10 5 4 (100) 2x10 2,0x10 Intensity [a. u.] 1 (002) 5 Intensität [cps] Absorption [a. u.] 3x10 (110) 4x10 Fluoreszenz 0 300 350 400 450 500 550 W ellenlänge [nm] 600 650 0,0 20 30 40 50 60 70 2 Theta [°] Abb. 63: Synthese von Zinkoxid-Kolloiden unter der Verwendung des Zinksiloxids 1. Bar (TEM) = 48 nm. Im Rahmen dieser Arbeit konnte gezeigt werden, dass sich aus dem von der Arbeitsgruppe Prof. Drieß zur Verfügung gestellte Zinksiloxid [ZnOSiMe3]4, aber auch aus dem Zinkalkoxid Zusammenfassung / 125 [Zn(OCH(Me)CH2NMe2)2] durch Thermolyse in HDA Zinkoxid-Kolloide herstellen lassen, deren Emission nahe der Bandkante liegt. Metallische Nanopartikel werden in der Literatur hauptsächlich durch die Reduktion von Metallsalzen synthetisiert, wobei das Reduktionsmittel dem Reaktionssystem zusätzlich zugeführt werden muss und nach der Synthese als Salzfracht vorliegt. Auch hier bietet die Organometallchemie einen neuen Lösungsansatz: Metallkationen lassen sich mit Liganden so stabilisieren, dass das Reduktionsmittel bereits im Precursor vorhanden ist. Aminoalkohle sind solche Liganden und die entsprechenden Komplexe M(OR)2 bewiesen sich in dieser Arbeit als hervorragend geeignete Precursoren für die Synthese von metallischen Nanopartikeln. Me2 N O HDA ∆T M O Metall H2N 15 N Me2 M = Cu, Pd, Ni, Zn Kupfer Nickel Palladium Zinkoxid Abb. 64: Synthese von metallischen Nanopartikeln unter der Verwendung von organometallischen Precursoren der Form [M(OCH(Me)CH2NMe2)2]. Bar (Kupfer) = 110 nm. Bar (Palladium) = 98 nm. Bar (Nickel) = 138 nm. Bar (Zinkoxid) = 57 nm. Sie liefern durch Thermolyse in hochsiedenden organischen Lösungsmitteln nicht nur qualitativ hochwertige Kupfer-Nanopartikel, die den zweiten Bestandteil des im Fokus stehenden Cu/ZnO-Systems ausmachen. Das Prinzip lässt sich darüber hinaus auch auf andere Metalle wie Nickel, Palladium und Zink übertragen. Dabei werden die Alkoxide der edleren 126 / Zusammenfassung Metalle zum nullwertigen Metall reduziert und bilden, insbesondere im Fall von Kupfer und Nickel, extrem hochwertige, wohl definierte und nahezu monodisperse Nanopartikel. Das Alkoxid des wesentlich unedleren Zinks wird hingegen zu Nanopartikeln des entsprechenden Oxids thermolysiert, deren Eigenschaften denen der aus [ZnOSiMe3]4 synthetisierten Partikel entsprechen. Nachdem nun fundierte Kenntnisse über die einzelnen Komponenten des Cu-/ZnO-Katalysators vorlagen, wurde begonnen, ein gemischtes System zu präparieren. Es stellte sich jedoch heraus, dass dies nicht wie ursprünglich geplant durch einfaches Zusammensetzen der beiden Komponenten möglich ist, sondern dazu führt, dass Kupfer und Zinkoxid bevorzugt getrennt voneinander in Form von Kolloiden vorliegen. Das heißt, sie bilden keine Grenzfläche aus, in der Kupfer und Zink in innigem Kontakt miteinander existieren. Deshalb wurde die Neigung der beteiligten Metalle, Legierungen zu bilden, ausgenutzt und die Synthese so gestaltet, dass ein Precursor für metallisches Zink anstelle eines Zinkoxid-Precursors verwendet werden konnte. In diesem Fall scheint die Bildung einer Legierung energetisch vorteilhafter zu sein als die Ausbildung von getrennten Zink- und Kupferpartikeln, die sich durch Koordination von organischen Liganden stabilisieren. Es konnte nachgewiesen werden, dass die entstandenen, kolloidal gelösten Partikel sowohl aus Zink als auch aus Kupfer bestehen und ihr Beugungsbild auf die Existenz von verschiedenen CuxZny-Verbindungen hinweist. Damit ist ein Schritt in Richtung auf die Erzeugung definierter Cu/ZnO-Grenzflächen in freistehenden, kolloidal-stabilisierten Nanopartikeln gelungen. Im Hinblick auf die katalytischen Eigenschaften solcher Partikel konnte darüber hinaus bewiesen werden, dass die nach der Synthese auf der Oberfläche der Partikel koordinativ verankerten Liganden die Oberfläche für den Zutritt kleiner Moleküle nicht vollständig abschirmt: Kohlenmonoxid, eine der Schlüsselverbindungen in der Methanolsynthese, kann durch die Liganden zur Oberfläche gelangen und dort physisorbiert werden. Dies ist nicht nur erstmals für Kupferkolloide gezeigt worden, sondern bietet darüber hinaus die Möglichkeit, die CO-Wechselwirkung getrennt vom Substratmaterial zu beobachten, was mit geträgerten Materialien prinzipiell nicht möglich ist. Des Weiteren sind die Grundlagen gelegt worden, diese Ergebnisse mit denen der anderen Komponenten zu vergleichen: es sind ZnO-Kolloide mit derselben Oberflächenbedeckung (HDA) synthetisiert worden, es ist ein System realisiert worden, in dem sowohl Cu- als auch ZnO-Kolloide vorliegen und es wurden Kolloide hergestellt, in denen Cu und Zn in innigem Kontakt vorliegen, also in einer Situation, die gemeinhin für den Cu/ZnO-Katalysator angenommen wird. Somit ist es nun prinzipiell möglich, die Beiträge der Komponenten am Beispiel der CO-Adsorption voneinander zu separieren. Im Rahmen der synthetischen Arbeiten konnten außerdem Kenntnisse bezüglich des Wachstumsmechanismus (am Beispiel von Kupfer und Palladium) und der Oberflächenreaktivität von Kolloiden (am Beispiel von CdSe und Kupfer) gewonnen werden. Dem geneigten Leser seien daher zusätzlich die Zusammenfassungen der einzelnen Kapitel auf den Seiten 40f, 53f, 81ff, 102f, 111 und 122f nahegelegt. Zusammenfassung / 127 10 Experimenteller Teil 10.1 Allgemeine Arbeitsvorschriften Alle Synthesen der Nanopartikel durch Pyrolyse von organometallischen Precursoren (Überschreitung der Flammpunkte!) sowie die meisten präparativen Arbeiten wurden unter Inertgasatmosphäre in ausgeheizten Glasgeräten durchgeführt (Schlenck-Rohr-Techniken, GloveBox, 4.9 Argon (Reinheit > 99.998 Vol-%) mit Kupferkatalysator und Molsieb). Die für die Synthesen verwendeten Lösungsmittel wurden nach gängigen Methoden getrocknet, mit Argon gesättigt und über Molekularsieb aufbewahrt (Restwassergehalt < 3ppm, Karl-Fischer). Lösungsmittel, die für die Reinigung der Nanopartikel verwendet wurden, wurden ohne Molekularsieb aufbewahrt und im Falle von Kupfer-Nanopartikeln zusätzlich durch EinfrierAuftau-Zyklen entgast. Die Aufarbeitung erfolgte in der Regel durch Ausfällen der Nanopartikel mit Methanol und Zentrifugation (Hettich, EBA 8). Dieser Waschvorgang wurde drei Mal durchgeführt, wobei die agglomerierten Partikel vor der nächsten Zentrifugation mit Hilfe von Ultraschall (Branson, 2210) in frischem Methanol aufgeschlämmt wurden. Anschließend wurden die Partikel je nach Analytik oder folgenden Syntheseschritten entweder in Toluol gelöst oder im Vakuum getrocknet. 10.2 Charakterisierung 1 H und 13 C NMR-Spektren wurden an einem Bruker DPX 200 gemessen. Die Bezeichnungen werden wie folgt abgekürzt: s, Singulett, d, Dublett, t, Triplett, q, Quartett, quin, Quintett, m, Multiplett, b, breit. IR-Spektroskopie. IR-Spektren wurden als KBr-Presslinge mit einem Perkin Elmer FT-IR 1720 X Spektrometer aufgenommen. Proben von Nanopartikeln wurden entweder durch Verreiben der gewaschenen und getrockneten Partikel in KBr oder durch Imprägnierung von KBr mit einer toluolischen Lösung der Partikel und anschließendem Trocknen im Vakuum präpariert. Die Lage der Banden wird in cm-1 angegeben. Die Intensitätsangaben werden wie folgt abgekürzt: w, schwach, m, mittel, s, stark, vs, sehr stark; sh, scharf, b, breit. UV-VIS-Spektroskopie. Für die optische Spektroskopie wurde ein Perkin Elmer Lambda 9 UV/VIS/NIR Spektrophotometer verwendet. Nanopartikuläre Proben wurden in Toluol gelöst und in Küvetten aus optischem Spezialglas mit 1 cm Pfadlänge vermessen. Die Spektren wurden, wenn nicht anders vermerkt, gegen Luft gemessen, da die Absorption des Lösungsmittels nicht im Bereich der Absorption der Proben lag. Photolumineszenz. PL-Spektren wurden mit Hilfe eines Fluoro Max-2 Instruments mit einer kontinuierlichen Ozon-freien Xenon-Lampe (150 Watt) und einem R928P Photomultiplier aufgenommen. Die Breite der slits betrug 2.5 nm und die Integrationszeit 1 s. Die Proben 128 / Experimenteller Teil wurden als toluolische Lösungen in Küvetten aus optischem Spezialglas und 1 cm Pfadlänge vermessen. Die Anregungswellenlänge variierte zwischen 250 und 350 nm und zu den einzelnen Spektren angegeben. Transmissionselektronenmikroskopie (TEM). TEM-Untersuchungen wurden an einem Hitachi H-8100 Raster- und Transmissionselektronenmikroskop mit LaB6 Filament durchgeführt, das mit einer Beschleunigungsspannung von 200 kV betrieben wurde. EDX (energydispersive X-ray) Spektren wurden mit einem Oxford Link System mit einem Si(Li) Kristall und ultradünnem Fenster ATW 2 aufgenommen. Für die Probenpräparation wurden die Nanopartikel in Toluol entweder gelöst oder suspendiert und auf ein mit einem Kohlenstofffilm überzogenes Gold- oder Kupfergrid getropft. Die Proben wurden einen Tag vor der entsprechenden Messung präpariert, um eine vollständige Trocknung des Grids in der Glove-Box oder an Luft zu gewährleisten (dies reduziert einerseits die Kontamination der Probe während der Messung und ermöglicht andererseits eine Selbstordnung der Partikel während des langsamen Trocknungsvorgangs). Massenspektrometrie. Die massenspektroskopischen Untersuchungen wurden an einem VG Instruments AutoSpec durchgeführt. Als Matrix für die Pulverproben wurde m-Nitrobenzylalkohol verwendet. Dynamische Lichtstreuung. Die Lösungen der Partikel (in der Regel in Toluol) wurden mit Hilfe eines He-Ne Lasers (Uniphase, 22 mW) in einem ALV PhotonenkorrelationsSpektrometer (ALV/DLS/SLS/5000E Goniometer-System der ALV Laser Vertriebsgesellschaft mbH Lagen, Deutschland) vermessen und über einen ALV-5000E Korrelator ausgewertet. Röntgenpulverdiffraktometrie. Die Diffraktogramme wurden mit CuK.-Strahlung ( = 1.541Å) auf einem Bruker-AXS D8-Diffraktometer aufgenommen. Einkristallröntgenstrukturanalyse. Die Einkristall-Röntgenstrukturanalysen wurde von Dr. Klaus Merz an einem Bruker-axs-SMART 100 Diffraktometer der Firma Bruker AXS unter Verwendung von Mo-K.-Strahlung ( = 71.07 pm, Graphit-Monochromator) durchgeführt. Thermogravimetrische Analysen. Thermogravimetrische Messungen wurden von Silvia Grum, Urmila Patil und Harish Parala auf einem TG/DTA6200/SII EXSTAR6000-Gerätes der Firma Seiko Instruments durchgeführt. Experimenteller Teil / 129 10.3 Chemikalien Chemikalie Hersteller Aufbewahrung/Reinigung (CH3)3CCH2MgBr Standard-Synthese Schutzgasatmosphäre, TK-Truhe 2,2’-Bipyridin unbek. Sublimiert, Glovebox 2-Thionaphtol 99 % Aldrich Für OF-Modifizierung 30 Min. bei RT im Vakuum 3-Chlor-1-brompropan 99 % Acros - Allylchlorid 98 % Aldrich - Cadmiumchlorid, wasserfrei Sigma Glovebox Diethylamin (für die Synthese) Merck - Diethylzink Strem Glovebox Kaliumcarbonat (p. A.) Riedel-de Haen - Kupferchlorid, wasserfrei Glovebox Lithium-Pulver unbek. Glovebox Lithium-Stäbe unbek. Schutzgasatmosphäre MeMgI 3-M in Et2O Fluka Kühlschrank N,N-Dimethylamino-2-propanol Aldrich Getrocknet über Molsieb, destilliert, Argon NaCp U. Weckenmann Schutzgasatmosphäre Natriumhydrid Fluka - n-Hexadecylamin 90 % Acros Getrocknet und entgast bei 100 °C im Vakuum Nickelchlorid Aldrich Schutzgasatmosphäre N-Tributylphosphin 96 % Acros Destilliert, Schutzgasatmosphäre, TK-Truhe Octylamin 99+ % Acros Getrocknet 99+ % über Molsieb, destilliert, entgast, Schutzgasatmosphäre, TK-Truhe Ölsäure 99+ % Aldrich Entgast im Vakuum Palladiumacetat unbek. - Palladiumchlorid unbek. - Pyridin (z. A.) Merck Destilliert (NaOH), Schutzgasatmosphäre Selen Pulver Merck Glovebox tert-Butylthiol Aldrich Destilliert, Argon, TK-Truhe Trioctylphosphinoxid 90 % Fluka Getrocknet und entgast bei 100 °C im Vakuum ZnO 99.9995 % Nanotek Entgast im Vakuum 130 / Experimenteller Teil 10.4 Synthesen 10.4.1 Vorschriften zum Kapitel Cadmiumselenid Dimethylcadmium (modifiziert nach [220]) In eine kräftig gerührte Suspension von 12.0 g CdCl2 in 60 mL Diethylether wurden 50 mL Methylmagnesiumiodid (~ 3M in Et2O) getropft, so dass ein stetiger Rückfluss eintrat. Nach Beenden des Zutropfens wurde eine weitere Stunde zum Rückfluss erhitzt. Nach Abkühlen der Reaktionsmischung wurde die Lösung von den entstandenen Salzen abkanüliert und destilliert. Dabei war darauf zu achten, dass zunächst bei Normaldruck auf 120 °C erhitzt wurde, um das in etherischer Lösung gebildete Addukt CdCl2 2 Et2O zu spalten. Dadurch ließ sich der Anteil an Diethylether im Produkt auf <4 % reduzieren. Anschließend wurde das Produkt bei etwa 50 °C unter leichtem Vakuum als farblose, klare und luftempfindliche Flüssigkeit destilliert und bei 0 °C im Dunklen aufbewahrt. Ausbeute: 7.83 g (84 %). H NMR (C6D6): δ = -0.43 (s) und Signale für Et2O C NMR (C6D6): δ = -2.5 (s) Alternativ lässt sich Dimethylcadmium durch die Pyrolyse von (2,2’-Bipyridin)-dimethylcadmium herstellen (s. dort). 1 13 (2,2’-Bipyridin)-dimethylcadmium[220] 6.1 g 2,2’-Bipyridin in 20 mL Et2O wurden zum Sieden erhitzt und langsam mit Me2Cd 2 Et2O (hergestellt aus 7.2 g CdCl2) versetzt. Es entstand eine klare, gelbe Lösung, die langsam auf –80 °C gekühlt wurde. Durch Kühlen für 1.5 h bei dieser Temperatur könnte das Produkt komplett kristallisiert werden. Die überstehende, farblose Lösung wurde abkanüliert und die gelben Kristalle mit –80 °C kaltem Ether gewaschen. Nach Trocknen im Vakuum wurden das Produkt bei 0 °C im Dunklen aufbewahrt. Ausbeute: 8.15 g (70 %). 1 H NMR (d8-Toluol): δ = -0.54 (s, 6H, CH3), 6.75 (m, 2H, Bipy), 7.22 (m, 2H, Bipy), 8.54 (m, 2H, Bipy), 8.67 (m, 2H, Bipy) Freisetzen von Dimethylcadmium: 1 g Me2Cd Bipy wurden im Vakuum auf 60 °C erhitzt und das entstandene Dimethylcadmium als klare, farblose Flüssigkeit aufgefangen. Eventuelle gelbe Verunreinigungen durch Sublimation des Addukts lassen sich durch wiederholte flaskto-flask Destillation entfernen. Das Produkt ist im Gegensatz zur Synthese mit anschließender Destillation (etwa 4 % Et2O) völlig lösemittelfrei. Ausbeute: 0.5 g (84 %). 1 H NMR (C6D6): δ = -0.44 (s), keine Signale für Et2O Dineopentylcadmium (modifiziert nach [221]) Zu einer Suspension von 1.68 g (9.16 mmol) CdCl2 in 20 mL Et2O wurden bei Raumtemperatur tropfenweise 21 mL (CH3)3CCH2MgBr (1.08-M in Et2O) gegeben. Die Reaktionsmischung wurde 14 h rühren gelassen. Danach wurde die Lösung vom Niederschlag abkanüExperimenteller Teil / 131 liert und im Vakuum eingeengt. Aus dem zurückbleibenden Feststoff wurde das Produkt durch Sublimation als kristalliner, weißer, luftempfindlicher Feststoff erhalten. Ausbeute: 1.5 g (64 %). 1 H NMR (C6D6): δ = 0.94 (s, 9H), 1.21 (s, 2H). Masse: 256 (M+1), 241 (M-CH3), 185 (M-Neopentyl), 114 (M-2xNeopentyl). 3-(Diethylamino)propyllithium Zu einer Lösung von 250 g 3-Chlor-1-brompropan in 100 mL Diethylether wurden unter leichter Kühlung (Wasserbad) 312 mL Diethylamin getropft und bei Raumtemperatur über Nacht rühren gelassen. Es bildete sich ein weißer Niederschlag, der abfiltriert und mit Ether gewaschen wurde. Die organische Phase wurde im Wasserstrahl-Vakuum eingeengt und anschließend mit 400 mL 2-n HCl ausgeschüttelt. Die entstandene wässrige Phase wurde abgetrennt und vorsichtig mit 100 g KOH versetzt, um 3-(Diethylamino)-1-chlorpropan als farblose, klare Flüssigkeit freizusetzen (130 g). Zur Lithiierung wurden während 1 Stunde 50 g 3(Diethylamino)-1-chlorpropan in 100 mL Et2O in eine Suspension von 5.0 g Lithium Pulver in 200 mL trockenem Diethylether getropft (Rückflusskühlung, Schutzgasatmosphäre). Die Reaktion dauerte noch ca. 1 Stunde nach Ende der Zugabe an und wurde anschließend 3 h bei Raumtemperatur rühren gelassen. Die Lösung wurde von entstandenem LiCl abkanüliert und im Vakuum eingeengt. Der weiße Feststoff wurde in ca. 200 mL trockenem Hexan gelöst, nochmals von LiCl abkanüliert und die Lösung auf die Hälfte ihres Volumens eingeengt. Durch Kühlen auf –20 °C kristallisierte das Produkt als weißer Feststoff aus. Ausbeute: 21.5 g (53 %). 1 H NMR (C6D6): δ = -0.61 (t, 2H, 1-CH2), 0.92 (t, 6H, N-CH2-CH3), 2.34 (m, 2H, 2-CH2), 2.56 (t, 2H, 3-CH2), 2.57 (q, N-CH2-CH3) Bis(3-diethylaminopropyl)cadmium (modifiziert nach [222]) Zu einer Suspension von 0.54 g CdCl2 (2.95 mmol) in Et2O (25 mL) wurde bei Raumtemperatur tropfenweise eine Lösung von 0.71 g 3-(Diethylamino)propyllithium (5.86 mmol) in 25 mL Et2O gegeben. Die Reaktionsmischung wurde 5 h zum Rückfluss erhitzt und bei Raumtemperatur über Nacht rühren gelassen. Danach wurde die Lösung vom Niederschlag abkanüliert und im Vakuum eingeengt. Destillation (Turbomolekularpumpe) ergab 0.76 g (76%) eines farblosen Öls. 1 H NMR (C6D6): δ = 0.52 (t, 2H, CdCH2), 0.89 (t, 6H, N-CH2-CH3), 2.03 (m, 2H, CdCH2CH2), 2.25 (m, 2H, CH2NEt2), 2.37 (q, 4H, N-CH2-CH3). 13 C NMR (C6D6): δ = 11.1 (s, NCH2CH3), 12.5 (s, CdCH2), 27.3 (s, CdCH2CH2), 46.6 (s, N-CH2-CH3), 57.9 (s, CH2NEt2). Herstellung der Vorratslösungen für die Synthese von CdSe-Nanopartikeln 0.4 g elementares Selen (5 mmol) und 7 mmol Me2Cd (Menge abhängig vom Ether-Gehalt) wurden in jeweils 10 g n-Tributylphosphin gelöst. Im Gegensatz zur Literatur wurden die 132 / Experimenteller Teil Lösungen nicht vereinigt, sondern getrennt in dunklen Gläsern bei 0° C in der Glove-Box aufbewahrt. Dadurch konnte ein Verlust an Precursor durch Bildung von Niederschlag vermieden werden. Synthese von CdSe-Nanopartikeln[28, 29] 4 g Trioctylphosphinoxid (TOPO, 90 %) wurden im Vakuum bei etwa 200 °C getrocknet und entgast. Nach Abkühlen wurde der 100 mL 2-Hals-Schlenckkolben mit einem mit Überdruckventil verschlossenen Rückflusskühler und einem Septum versehen. Der Rückflusskühler diente dem Rückfluss von n-Tributylphosphin und zum Zurückhalten von TOPO im Argonstrom. Über das Septum wurde ein Temperaturfühler (GTF 601 Pt100 1/10-DIN) in den Kolben eingeführt. TOPO wurde unter Normaldruck und Argonatmosphäre erhitzt und die Temperatur mit Hilfe eines über einen taktenden Temperatur-Regler (GIR 1002 / Pt 100 T) gesteuerten Heizmantels auf 350.0 °C eingestellt. Jeweils 0.2 mL der Se- und Me2Cd-Vorratslösungen wurden vereinigt und in die heiße Lösung injiziert. Dabei war darauf zu achten, dass die Injektion möglichst schnell erfolgte (< 1s) und die Lösung heftig gerührt wurde. Durch die Injektion sank die Temperatur ab und wurde für 10 Min. auf 315 °C gehalten. Die Reaktion wurde durch Entfernen des Heizpilzes beendet und unter Argonstrom auf Raumtemperatur abkühlen gelassen. Durch Zugabe eines Überschusses an trockenem Methanol (ca. 20 mL) wurden die Nanokristallite ausgefällt und durch Zentrifugation abgetrennt. Überschüssiges TOPO wurde durch mehrmaligem Waschen und Zentrifugieren mit Methanol entfernt, wobei die Suspension vor der Zentrifugation mit Hilfe von Ultraschall aufgeschlämmt wurde. Nach der Reinigung wurden die Nanopartikel in Toluol gelöst. Anmerkung: Bei der Verwendung von reinem TOPO (• %) wurden pro 6 g TOPO 0.2 mL Vorratslösung verwendet (jeweils 0.1 mL Cd- und Se-Lösung). Oberflächenmodifizierung von CdSe-TOPO-Nanopartikeln Für die Austauschexperimente an CdSe-TOPO-Nanopartikeln wurden grundsätzlich mit Methanol gewaschene Partikel verwendet. Austausch von TOPO gegen Naphtylthiol in Pyridin: Die gewaschenen Partikel wurden in Toluol gelöst, filtriert (200 µm Filter) und die Hälfte der Lösung für die Analyse des Edukts abgetrennt. Die andere Hälfte wurde getrocknet, gewogen (40 mg) und in trockenem Pyridin gelöst. Nach Zugabe von 15 mg Naphthylthiol wurde die Lösung 1 Tag lang auf 70 °C (Ölbadtemperatur) erwärmt. Die Aufarbeitung erfolgte durch Ausfällen der Partikel mit Pentan, Zentrifugation und mehrmaligem Waschen mit Pentan. Die aufgearbeitetn Partikel waren nicht mehr in Toluol löslich. Für den Austausch von TOPO gegen sukzessive Pyridin und Naphtylthiol wurden die gewaschenen Partikel in absolutem Pyridin aufgenommen und über Nacht auf 70 °C (Ölbadtemperatur) erhitzt. Nach Ausfällen und wiederholtem Waschen mit Pentan wurden die Nanokristallite erneut in trockenem Pyridin aufgnommen, mit 15 mg Naphtylthiol versetzt und über Experimenteller Teil / 133 einen Zeitraum von 4 Tagen auf 70 °C erhitzt. Die Aufarbeitung erfolgte durch Ausfällen und Waschen mit Pentan. 10.4.2 Vorschriften zum Kapitel ZnO Synthese von ZnO-Nanopartikeln 7 g Hexadecylamin wurden bei 100 °C im Vakuum während 30 Min. getrocknet und entgast. Der Aufbau erfolgte analog zur Synthese von CdSe-Nanopartikeln. 224 mg [ZnOSiMe3]4[93] wurden in 4 mL Octylamin gelöst und in 180 °C (Ölbad) heißes HDA injiziert. Nach 30 Minuten bei dieser Temperatur wurde die Reaktion abkühlen gelassen und das Thermolyseprodukt mit Methanol ausgefällt. Die Partikel wurden zentrifugiert, mit Methanol gewaschen und in Toluol aufgenommen. Für die Synthese von Zinkstäbchen wurde die Reaktion bei T=300 °C unter ansonsten analogen Bedingungen durchgeführt. Bei der Verwendung von [Zn(OCH(Me)CH2NMe2)2] wurden 324 mg des Precursors in 4 mL Octylamin gelöst und bei 300 °C in HDA injiziert. 10.4.3 Vorschriften zum Kapitel Kupfer [Cu(OCH(Me)CH2NMe2)2] (modifiziert nach [130] 0.48 g Lithium wurden bei Raumtemperatur in 30 mL Methanol gelöst und die entstandene Lösung mit einer Lösung von 4.84 g CuCl2 in 30 mL Methanol versetzt. Der Niederschlag wurde abfiltriert, drei Mal mit Methanol gewaschen und im Vakuum getrocknet. (Bei unzureichender Reinigung wurde die nachfolgende Reaktion durch Chloridionen gestört). Das getrocknete Cu(OMe)2 wurde in 30 mL Toluol suspendiert und mit 11 mL N,N-Dimethylamino2-propanol versetzt. Dabei entstand eine tief violett gefärbte Lösung, die etwa 0.5 h bei Raumtemperatur rühren gelassen und dann im Vakuum eingeengt wurde. Die Sublimation des trockenen Rückstandes bei 65 °C Ölbadtemperatur ergab das Produkt als dunkle, violette Kristalle. Synthese von Cu-Nanopartikeln 7 g n-Hexadecylamin (HDA) wurden bei etwa 100 °C Ölbadtemperatur während 0.5 h getrocknet und entgast. Analog zur Synthese der CdSe-Nanopartikel wurde der Schlenck-Kolben mit Rückflusskühler und Septum versehen. 322 mg des Precursors [Cu(OCH(Me)CH2NMe2)2] wurden in 4 mL n-Octylamin gelöst und bei 300 °C unter kräftigem Rühren in HDA injiziert. Es trat eine heftige Gasentwicklung ein und die farblose Aminlösung verfärbte sich dunkelrot. Nach 30-minütigem Rühren bei 225 °C wurde die Reaktion durch Entfernen des Heizpilzes gestoppt und im leichten Argonstrom auf Raumtemperatur abkühlen gelassen. Die Reinigung der Partikel erfolgte analog zu CdSe. Wegen der Oxidations134 / Experimenteller Teil empfindlichkeit der Kupfer-Partikel wurde sie jedoch in der Glove-Box und mit entgasten Lösungsmitteln durchgeführt. Die Reaktionsbedingungen für die anderen Synthesen sind in den einzelnen Kapiteln angegeben. Oberflächenmodifizierung von Cu-Nanopartikeln Das Reaktionsprodukt einer Pyrolyse von 322 mg [Cu(OCH(Me)CH2NMe2)2] (siehe obige Vorschrift), wurde komplett durch Waschen mit Methanol gereinigt und in 50.0 mL Toluol gelöst. Unter der Annahme, dass ein vollständiger Umsatz des Precursors zu Kupfer-Nanopartikeln stattgefunden hat, ist diese Lösung 0,024-M, 1 mL enthält 1.5 mg Kupfer. Bei einer durchschnittlichen Größe von 8 nm im Durchmesser haben die einzelnen Partikel eine Oberfläche von 201 nm2, was bei einer Oberfläche von 0.15 nm2 pro Cu-Atom 1333 Kupferatomen entspricht. Mit Hilfe der Dichte des Festkörpers lässt sich daraus die Menge an OberflächenCu-Atomen zu 1.4 µmol pro mL berechnen. Dies entspricht der maximalen Menge an OF-CuAtomen, da erstens aufgrund von während der Reaktion gebildeten Kupferfilmen die Pyrolyse offensichtlich nicht vollständig zu Nanopartikeln führt und zweitens aus sterischen Gründen nicht jedes Cu-Atom mit HDA koordiniert und damit in einen Austausch involviert sein wird Für die Reaktionen zur Oberflächenmodifizierung wurden 1.0 mL der Stammlösung mit 1.4 µmol, d. h. einem leichten Überschuss, des neuen Liganden versetzt und unter Schutzgas bei Raumtemperatur rühren gelassen. Die nachfolgende Analytik orientierte sich am Verlauf der Reaktion und ist aus Kapitel 5.4.3 ersichtlich. 10.4.4 Vorschriften zum Kapitel Palladium [Pd(OCH(Me)CH2NMe2)2] 0.5 g (2.2 mmol) Palladiumacetat in 50 mL Toluol wurde mit 0.44 g Dimethylaminopropanol (4.4 mmol) sowie dem 10-fachen Überschuss an Kaliumcarbonat versetzt und bei Raumtemperatur einen Tag rühren gelassen. Dabei trat ein Farbumschlag von orange nach gelb ein. Die gelbe Suspension wurde filtriert und das Filtrat im Vakuum getrocknet. Der zurückbleibende Feststoff wurde 3 Mal mit Pentan gewaschen (je 10 mL) und im Vakuum getrocknet. Das Produkt entstand als gelber Feststoff mit einer Ausbeute von 81 %. 1 H NMR (C6D6): δ = 1.37 (d, 3H, 1-CH3), 1.82 (td, 1H, 3-CH2), 2.40 (dd, 1H, 3-CH2), 2.45, 2.49, 2.59, 2.60 (s, 6H, N-CH3), 4.33 (m, 1H, 2-CH). Zuordnung zu den Isomeren siehe Seite 97. 13 C NMR (C6D6): δ = 47.6 (s, 1-CH3), 51.6, 52.0 (s, N-CH3), 71.3 (s, 2-CH), 76.6 (s, 3-CH2) Masse: Siehe Seite 97. Cp(Allyl)Pd Die Synthese wurde nach Literaturvorschrift durchgeführt.[223, 224] Experimenteller Teil / 135 Synthese von Palladium-Nanopartikeln Siehe Synthese von Kupfer-Nanopartikeln. Für die Thermolyse wurden 7 g Hexadecylamin verwendet. 250 mg des Precursors wurden in 2 mL Octylamin suspendiert und bei T = 200 °C in HDA injziert. Die Reaktionszeit betrug 30 Minuten. 10.4.5 Vorschriften zum Kapitel Nickel [Ni(OCH(Me)CH2NMe2)2] (modifiziert nach [195, 196] 10 mL trockenes N,N-Dimetylamino-2-propanol wurden in 50 mL Toluol gelöst und vorsichtig mit 2.2 g Natriumhydrid versetzt. Nach Ende der Zugabe wurde für 30 Minuten bei Raumtemperatur rühren gelassen und anschließend das Lösungsmittel im Vakuum entfernt. Der Feststoff wurde mit 50 mL THF und 6.2 g Nickelchlorid (wasserfrei) versetzt und für 2 Stunden zum Rückfluss erhitzt. Nach Entfernen des Lösungsmittels erhielt man einen grünen Feststoff, der durch Sublimation im Vakuum (65 °C) gereinigt wurde. Ausbeute: 30 %. H NMR (C6D6): δ = 1.02 (d, 3H, 1-CH3), 1.32 (td, 1H, 3-CH2), 1.85 (m, 1H, 3-CH2), 1.93, 1.96, 2.53 mit Schulter (s, 6H, N-CH3), 3.60 (m, 1H, 2-CH). 13 C NMR (C6D6): δ = 46.2 (s, 1-CH3), 49.6, 50.1 (s, N-CH3), 67.9 (s, 2-CH), 75.5 (s, 3-CH2) Masse: siehe Kapitel 7.2.1. 1 Synthese von Nickel-Nanopartikeln Siehe Synthese von Kupfer-Nanopartikeln. Für die Thermolyse wurden 7 g Hexadecylamin verwendet. 320 mg des Precursors wurden in 4 mL Octylamin suspendiert und bei T = 180 °C in HDA injziert. Die Reaktionszeit betrug 30 Minuten. 10.4.6 Vorschriften zum Kapitel Cu/ZnO-Systeme Imprägnierung von ZnO mit Kupfer-Nanopartikeln Etwas eine Spatelspitze kommerzielles ZnO (Nanotek) wird unter Schutzgasatmosphäre mit 3 mL einer Lösung von Cu-HDA-NACs in Toluol versetzt. Zur besseren Verteilung des unlöslichen ZnO wird die Mischung 15 Minuten im Ultraschallbad behandelt und anschließend bei Raumtemperatur rühren gelassen. Nach wenigen Stunden ist ein Farbumschlag von rot nach grau eingetreten. Für die Präparation der TEM-Probe wurde die Suspension gut durchmischt und ein Tropfen auf ein Kohlenstoff-beschichtetes Kupfergrid gegeben. Pyrolyse von Et2Zn 7 g Hexadecylamin wurden im Vakuum bei 100 °C Ölbadtemperatur getrocknet und entgast und mit Hilfe eines Heizpilzes auf 325 °C geheizt. 150 mg Et2Zn wurden in 4 mL Octylamin gelöst und rasch in die heiße Lösung injiziert. Dabei fiel die Temperatur auf knapp unter 300 °C und wurde für 30 Minuten bei diesem Wert gehalten. Direkt nach der Injektion wurde ein Farbumschlag von farblos nach gelb beobachtet. Die Reaktionsmischung wurde im Laufe 136 / Experimenteller Teil der Reaktion dunkler und nahm schließlich eine graue Farbe an. Die Reaktion wurde durch Entfernen des Heizpilzes beendet und unter leichtem Argonstrom auf Raumtemperatur abkühlen gelassen. Das Reaktionsprodukt wurde durch Zugabe von Methanol, Zentrifugation und mehrmaligem Waschen mit Methanol an Luft aufgereinigt. Der graue Rückstand löste sich nicht in Toluol. Synthese von Cu/Zn-Material (Methode A) 7 g im Vakuum bei 100 °C getrocknetes und entgastes HDA wurden unter Schutzgasatmosphäre auf 250 °C erhitzt. In diese Lösung wurden 320 mg [Cu(OCH(Me)CH2NMe2)2] in 4 mL Octylamin injiziert. Nach 10 Minuten wurden über einen Tropftrichter langsam 4 mL einer Lösung von 1.3 g Et2Zn in 10 mL Toluol zugetropft (Cu/Zn = 30/70). Alternative: Nach obiger Vorschrift aus 320 mg Precursor synthetisierte Cu-HDA-NACs wurden (inklusive des nach der Synthese vorhandenen überschüssigen HDAs) auf 200 °C erhitzt und über eine Spritze langsam (8 Minuten) mit 14 mg Et2Zn in 0.5 mL Octylamin (Cu/Zn = 90/10) versetzt. Nach weiteren 10 Minuten wurde die Reaktion durch Entfernen des Heizpilzes beendet und unter leichtem Argonstrom auf Raumtemperatur abkühlen gelassen. Die Aufarbeitung erfolgte in der Glovebox durch Zugabe von Methanol, Zentrifugation des Rückstandes und mehrmaliges Waschen mit Methanol. Die gereinigten Partikel wurden in Toluol gelöst. Synthese von Cu/Zn-Material (Methode B) 7 g HDA wurden bei 100 °C im Vakuum getrocknet und entgast und unter Schutzgasatmosphäre auf 250 °C erhitzt. Zu einer Lösung von 320 mg [Cu(OCH(Me)CH2NMe2)2] in 3.0 mL Octylamin wurden 147 mg Et2Zn in 1 mL Octylamin gegeben. Bei Zugabe entfärbte sich die dunkle Lösung des Cu-Precursors und es bildete sich ein gelblich-weißer Niederschlag. Dieser wurde in das heiße Hexadecylamin injiziert. Kurz nach der Injektion konnte ein Farbumschlag von farblos nach dunkelrot beobachtet werden und die Temperatur fiel auf 230 °C. Nach weiteren 30 Minuten dieser Temperatur wurde die Reaktion durch Entfernen der Heizquelle beendet und unter leichtem Argonstrom auf Raumtemperatur abkühlen gelassen. Die Aufarbeitung erfolgte analog zu Methode A in der Glovebox. Synthese von Cu/Zn-Material (Methode C) 7 g HDA wurden durch Erhitzen im Vakuum getrocknet und entgast. Nach Erhitzen unter Normaldruck (Argon) auf 250 °C erhitzt, wurden 320 mg [Cu(OCH(Me)CH2NMe2)2] in 4 mL Octylamin injiziert. Nach 10 Minuten wurden bei T = 230 °C 1.12 mg (2,2’-Bipyridin)Diethylzink in 3 mL Octylamin über einen Zeitraum von 10 Minuten zugetropft. Nach weiteren 10 Minuten wurde die Reaktion durch Entfernen des Heizpilzes beendet und unter leichtem Argonstrom auf Raumtemperatur abkühlen gelassen. Die Aufarbeitung erfolgte analog zu Methode A in der Glovebox. Experimenteller Teil / 137 11 Literaturverzeichnis [1] [2] [3] [4] [5] [6] [7] [8] [9] [10] [11] [12] [13] [14] [15] [16] [17] [18] [19] [20] [21] [22] [23] [24] [25] [26] [27] [28] [29] [30] R. Feynman, Annual Meeting of the American Physical Society 29.12.1959. M. Faraday, Philos. Trans. R. Soc. London 1857, 147, 154. J. Turkevich, P. C. Stevenson, J. Hillier, Discuss. Faraday Soc. 1951, 55. J. Turkevich, G. Kim, Science 1970, 169, 873. J. Turkevich, Gold Bull. 1985, 18, 86. A. Henglein, Top. Curr. Chem. 1988, 143, 113. M. Michaelis, A. Henglein, J. Phys. Chem. 1992, 96, 4719. A. Henglein, D. Meisel, Langmuir 1998, 14, 7392. A. Henglein, M. Giersig, J. Phys. Chem. B 1999, 103, 9533. M. Maase, Doktorarbeit an der Universität Aachen 1999. E. R. W. U. Gasser, A. Schofield, P. N. Pusey, D. A. Weitz, Science 2001, 292, 258. X. Peng, J. Wickham, A. P. Alivisatos, J. Am. Chem. Soc. 1998, 120, 5343. D. V. Talapin, A. L. Rogach, A. Kornowski, M. Haase, H. Weller, Nano Lett. 2001, 1(4), 207. S. Link, M. A. El-Sayed, J. Phys. Chem. B 1999, 103, 8410. A. N. Goldstein, C. M. Echer, A. P. Alivisatos, Science 1992, 256, 1425. T. Castro, R. Reifenberger, E. Choi, R. P. Andres, Phys. Rev. B 1990, 13, 8548. V. I. Klimov, D. W. McBranch, C. A. Leatherdale, M. G. Bawendi, Phys. Rev. B 1999, 60 (19), 13740. D. B. Williams, C. B. Carter, in Transmission electron microscopy, Plenum Press. M. Bruchez, M. Moronne, P. Gin, S. Weiss, A. P. Alivisatos, Science 1998, 281, 2013. W. C. W. Chan, S. M. Nie, Science 2998, 281, 2016. D. L. Klein, R. Roth, A. K. L. Lim, A. P. Alivisatos, P. L. McEuen, Nature 1997, 389, 1749. H. Weller, Angew. Chem. 1998, 37 (12), 1658. Y. Zhang, G. Hodes, I. Rubinstein, E. Grünbaum, R. R. Nayak, J. L. Hutchison, Adv. Mater. 1999, 11 (17), 1437. N. L. Pickett, F. G. Riddell, D. F. Foster, D. J. Cole-Hamilton, J. R. Fryer, J. Mater. Chem. 1997, 7 (9), 1855. J. R. Babcock, R. W. Zehner, L. R. Sita, Chem. Mater. 1998, 10 (8), 2027. Y. Wada, H. Kuramoto, J. Anand, T. Kitamura, T. Sakata, H. Mori, S. Yanagida, J. Mater. Chem. 2001, 11, 1936. T. Torimoto, H. Kontani, Y. Shibutani, S. Kuwabata, T. Sakata, H. Mori, H. Yoneyama, J. Phys. Chem. B 2001, 105 (29), 6838. J. E. Bowen Katari, V. L. Colvin, A. P. Alivisatos, J. Phys. Chem. 1994, 98, 4109. C. B. Murray, D. J. Norris, M. G. Bawendi, J. Am. Chem. Soc. 1993, 115, 8706. Z. A. Peng, X. Peng, J. Am. Chem. Soc. 2001, 123, 183. 138 / Literatur [31] [32] [33] [34] [35] [36] [37] [38] [39] [40] [41] [42] [43] [44] [45] [46] [47] [48] [49] [50] [51] [52] J. Hambrock, A. Birkner, R. A. Fischer, J. Mater. Chem. 2001, 11, 3197. P. S. Nair, N. Revaprasadu, T. Radhakrishnan, G. A. Kolawole, J. Mater. Chem. 2001, 11, 1555. B. Ludolph, M. A. Malik, P. O´Brien, N. Revaprasadu, Chem. Commun. 1998, 1849. T. Trindade, P. O´Brien, J. Mater. Chem. 1996, 6 (3), 343. S.-H. Yu, Y.-S. Wu, J. Yang, Z.-H. Han, Y. Xie, Y.-T. Qian, X.-M. Liu, Chem. Mater. 1998, 10, 2309. U. K. Gautam, M. Rajamathi, F. Meldrum, P. Morgan, R. Seshadri, Chem. Commun. 2001, 629. W. Wang, Y. Geng, P. Yan, F. Liu, Y. Xie, Y. Qian, J. Am. Chem. Soc. 1999, 121, 4062. M. S. Steigerwald, A. P. Alivisatos, J. M. Gibson, T. D. Harris, R. Kortan, A. J. Muller, A. M. Thayer, T. M. Duncan, D. C. Douglass, L. E. Brus, J. Am. Chem. Soc. 1998, 110, 3046. V. Ptatschek, B. Schreder, K. Herz, U. Hilbert, W. Ossau, G. Schottner, O. Rahäuser, T. Bischof, G. Lermann, A. Materny, W. Kiefer, G. Bacher, A. Forchel, D. Su, M. Giersig, G. Müller, L. Spanhel, J. Phys. Chem. B 1997, 101, 8898. S. W. Haggata, D. J. Cole-Hamilton, J. R. Fryer, J. Mater. Chem. 1997, 7 (10), 1969. Z. Zhang, S. Dai, X. Fan, D. A. Blom, S. J. Pennycook, Y. Wei, J. Phys. Chem. B 2001, 106 (40), 10338. H. Parala, H. Winkler, M. Kolbe, A. Wohlfart, R. A. Fischer, R. Schmechel, H. von Seggern, Adv. Mater. 2000, 12 (14), 1050. J. D. Klein, R. D. Herrick, D. Palmer, M. J. Sailor, C. J. Brumlik, C. R. Martin, Chem. Mater. 1993, 5 (7), 902. E. Lifshitz, I. Dag, I. Litvin, G. Hodes, S. Gorer, R. Reisfeld, M. Zelner, H. Minti, Chem. Phys. Lett. 1998, 288, 188. X. Peng, L. Manna, W. Yang, J. Wickham, E. Scher, A. Kadavanich, A. P. Alivisatos, Nature 2000, 404, 59. Z. A. Peng, X. Peng, J. Am. Chem. Soc. 2001, 123 (7), 1389. L. Manna, E. C. Scher, A. P. Alivisatos, J. Am. Chem. Soc. 2000, 122, 12700. S.-M. Lee, Y.-W. Jun, S.-N. Cho, J. Cheon, J. Am. Chem. Soc. 2002. N. Cordente, M. Respaud, F. Senocq, M.-J. Casanove, C. Amiens, B. Chaudret, Nano Lett. 2001, 1 (10), 565. V. F. Puntes, K. M. Krishnan, A. P. Alivisatos, Science 2001, 291, 2115. K. Soulantica, A. Maisonnat, F. Senocq, M.-C. Fromen, M.-J. Casanove, B. Chaudret, Angew. Chem. 2001, 113, 3072. W. U. Huynh, J. J. Dittmer, A. P. Alivisatos, Science 2002, 295, 2425. Literatur / 139 [53] [54] [55] [56] [57] [58] [59] [60] [61] [62] [63] [64] [65] [66] [67] [68] [69] [70] [71] [72] [73] [74] [75] [76] [77] [78] [79] H. Winkler, A. Birkner, V. Hagen, I. Wolf, R. Schmechel, H. von Seggern, R. A. Fischer, Adv. Mater. 1999, 11 (17), 1444. R. P. Andres, J. D. Bielefeld, J. I. Henderson, D. B. Janes, V. R. Kolagunta, C. P. Kubiak, W. J. Mahoney, R. G. Osifchin, Science 1996, 273, 1690. X. Peng, T. E. Wilson, A. P. Alivisatos, P. G. Schultz, Angew. Chem. Int. Ed. Engl. 1997, 36 (1-2), 145. D. I. Gittins, F. Caruso, Angew. Chem. 2001, 113 (16), 3089. B. O. Dabbousi, J. Rodriguez-Viejo, F. V. Mikulec, J. R. Heine, H. Mattousi, R. Ober, K. F. Jensen, M. B. Bawendi, J. Phys. Chem. B 1997, 101, 9463. M. A. Hines, P. Guyot-Sionnest, J. Phys. Chem. 1996, 100, 468. L. Becerra, C. B. Murray, R. G. Griffin, M. G. Bawendi, J. Chem. Phys. 1994, 100, 3297. J. G. C. Veinot, J. Galloro, L. Pugliese, V. Bell, R. Pestrin, W. J. Pietro, Can. J. Chem. 1998, 1998. J. R. Sachleben, E. W. Wooten, L. Emsley, A. Pines, V. L. Colvin, A. P. Alivisatos, Chem. Phys. Lett. 1992, 198 (5), 431. T. Lover, W. Henderson, G. A. Bowmaker, J. M. Seakins, R. P. Cooney, Chem. Mater. 1997, 9, 1878. M. Kuno, J. K. Lee, B. O. Dabbousi, F. V. Mikulec, M. G. Bawendi, J. Chem. Phys. 1997, 103 (23), 9869. B. S. Kim, L. Avila, L. E. Brus, I. P. Herman, Appl. Phys. Lett. 2000, 76 (25), 3715. U. Koch, A. Fojtik, H. Weller, A. Henglein, Chem. Phys. Lett. 1985, 122, 507. D. W. Bahnemann, C. Kormann, M. R. Hoffmann, J. Phys. Chem. 1987, 91, 3789. M. Haase, H. Weller, A. Henglein, J. Phys. Chem. 1988, 92, 482. L. Spanhel, M. A. Andersen, J. Am. Chem. Soc. 1991, 113, 2826. K. Borgohain, S. Mahamuni, Semicond. Sci. Technol. 1998, 13, 1154. E. A. Meulenkamp, J. Phys. Chem. B 1998, 102, 5566. E. M. Wong, P. G. Hoertz, C. J. Liang, B.-M. Shi, G. J. Meyer, P. C. Searson, Langmuir 2001, 17, 8362. L. Lu, unpublished results 2002. E. Baur, A. Perret, Helv. Chim. Acta 1924, 7, 910. J. R. Harbour, M. L. Hair, J. Phys. Chem. 1979, 83, 652. J. Rabani, D. Behar, J. Phys. Chem. 1989, 93, 2559. P. V. Kamat, B. Patrick, J. Phys. Chem. 1992, 1992, 6829. S. Sakohara, M. Ishida, M. A. Anderson, J. Phys. Chem. B 1998, 102, 10169. S. Monticone, R. Tufeu, A. V. Kanaev, J. Phys. Chem. B 1998, 102, 2854. A. van Dijken, E. A. Meulenkamp, D. Vanmaekelbergh, A. Meijerink, J. Phys. Chem. B 2000, 104, 1715. 140 / Literatur [80] A. van Dijken, E. A. Meulenkamp, D. Vanmaekelbergh, A. Meijerink, J. Phys. Chem. B 2000, 104, 4355. [81] C. L. Yang, J. N. Wang, W. K. Ge, L. Guo, S. H. Yang, D. Z. Shen, J. Appl. Phys. 2001, 90, 4489. [82] M. H. Huang, S. Mao, H. Feick, H. Yan, Y. Wu, H. Kind, E. Weber, R. Russo, P. Yang, Science 2001, 292, 1897. H. Zhou, H. Alves, D. M. Hofmann, W. Kriegseis, B. K. Meyer, G. Kaczmarczyk, A. Hoffmann, Appl. Phys. Lett. 2002, 80, 210. H. Zhou, H. Alves, D. M. Hofmann, B. K. Meyer, G. Kaczmarczyk, A. Hoffmann, C. Thomsen, Phys. Stat. Sol. B 2002, 229, 825. L. Guo, S. Yang, C. Yang, P. Yu, J. Wang, W. Ge, W. K. L. Wong, Appl. Phys. Lett. 2000, 76 (20), 2901. L. Guo, S. Yang, Chem. Mater. 2000, 12, 2268. S. Mahamuni, K. Borgohain, B. S. Bendre, V. J. Leppert, S. H. Risbud, J. Appl. Phys. 1999, 85, 2861. J.-I. Park, J. Cheon, J. Am. Chem. Soc. 2001, 123, 5743. J. Hambrock, R. Becker, A. Birkner, J. Weiß, R. A. Fischer, Chem. Commun. 2002, [83] [84] [85] [86] [87] [88] [89] 68. [90] D. Kaneko, H. Shouji, T. Kawai, K. Kon-No, Langmuir 2000, 16, 4086. [91] M. Shim, P. Guyot-Sionnest, J. Am. Chem. Soc. 2001, 123, 11651. [92] F. Rataboul, C. Nayral, M.-J. Casanove, A. Maisonnat, B. Chaudret, J. Organomet. Chem. 2002, 643-644, 307. [93] M. Driess, K. Merz, S. Rell, Eur. J. Inorg. Chem. 2000, 2517. [94] J. Hambrock, S. Rabe, K. Merz, A. Birkner, A. Wohlfart, R. A. Fischer, M. Driess, J. Mater. Chem. 2002, submitted for publication. [95] V. Noack, A. Eychmüller, Chem. Mater. 2002, 14, 1411. [96] S. C. Goel, M. Y. Chiang, W. E. Buhro, Inorg. Chem. 1990, 29, 4646. [97] A. Bezryadin, C. Dekker, G. Schmid, Appl. Phys. Lett. 1997, 71, 1273. [98] L. F. Chi, M. Hartig, T. Drechsler, T. Schwaack, C. Seidel, H. Fuchs, G. Schmid, Appl. Phys. A 1998, A66, 187. [99] C. A. Mirkin, R. L. Letsinger, R. C. Mucic, J. L. Storhoff, Nature 1996, 382, 607. [100] R. Elghanian, J. J. Storhoff, R. C. Mucic, R. L. Letsinger, C. A. Mirkin, Science 1997, 277, 1078. [101] T. A. Taton, C. A. Mirkin, R. L. Letsinger, Science 2000, 289, 1757. [102] J. Liu, J. Alvarez, W. Ong, E. Román, A. E. Kaifer, Langmuir 2001, 17, 6762. [103] Y. Niu, L. K. Yeung, R. M. Crooks, J. Am. Chem. Soc. 2001, 123 (28), 6840. [104] C.-B. Hwang, Y.-S. Fu, Y.-L. Lu, S.-W. Jang, P.-T. Chou, C. R. C. Wang, S. J. Yu, J. Catal. 2000, 195, 336. Literatur / 141 [105] [106] [107] [108] [109] [110] [111] [112] [113] [114] [115] [116] [117] [118] [119] [120] [121] [122] [123] [124] [125] [126] [127] [128] [129] [130] [131] [132] [133] M. Zhao, R. N. Crooks, Angew. Chem. 1999, 111 (3), 375. Y. Li, M. Q. El-Sayed, J. Phys. Chem. B 2001, 105 (37), 8938. M. P. Pileni, Langmuir 1997, 13, 3266. M. P. Pileni, Langmuir 1997, 13, 172. J. Rothe, J. Hormes, H. Bönnemann, W. Brijoux, K. Siepen, J. Am. Chem. Soc. 1998, 120, 6019. S. Chen, J. M. Sommers, J. Phys. Chem. B 2001, 105, 8816. K. J. Ziegler, R. C. Doty, K. P. Johnston, B. A. Korgel, J. Am. Chem. Soc. 2001, 123, 7797. H. H. Huang, F. Q. Yan, Y. M. Kek, C. H. Chew, G. Q. Xu, W. Ji, P. S. Oh, S. H. Tang, Langmuir 1997, 13, 172. S. Ayyappan, R. S. Gopalan, G. N. Subbanna, C. N. R. Rao, J. Mater. Res. 1997, 12 (2), 398. E. R. Savinova, A. L. Chuvilin, V. N. Parmon, J. Mol. Catalysis 1988, 48, 217. H. Hirai, H. Wakabayashi, M. Komiyama, Bull. Chem. Soc. Jpn. 1996, 59, 367. N. A. Dhas, C. P. Raj, A. Gedanken, Chem. Mater. 1998, 10, 1446. R. A. Salkar, P. Jeevanandam, G. Kataby, S. T. Aruna, Y. Koltypin, O. Palchik, A. Gedanken, J. Phys. Chem. B 2000, 104, 893. S. S. Joshi, S. F. Patil, V. Iyer, S. Mahumuni, Nanostrcutured Mater. 1998, 10 (7), 1135. R. S. J. Gonzalo, C. N. Afonso, J. Bosbach, T. Wenzel, F. Stietz, F. Träger, D. Babonneau, D. E. Hole, J. Appl. Phys. 2001, 89 (10), 5734. X. J. Wu, L. G. Du, H. F. Zhang, J. F. Liu, Y. S. Zhou, Z. Q. Li, L. Y. Xiong, Y. L. Bai, Nanostructured Mater. 1999, 12, 221. M. Zhao, L. Sun, R. M. Crooks, J. Am. Chem. Soc. 1998, 120, 4877. L. Balogh, D. A. Tomalia, J. Am. Chem. Soc. 1998, 120, 7355. R. T. Clay, R. E. Cohen, New. J. Chem. 1998, 745. M. P. Pileni, J. Am. Chem. Soc. 1993, 115, 3887. A. Filankembo, M. P. Pileni, Appl. Surf. Sci. 2000, 260. J. P. Cason, C. B. Roberts, J. Phys. Chem. B 2000, 104 (6), 1217. P. Chen, X. Wu, J. Lin, K. L. Tan, J. Phys. Chem. B 1999, 22, 4559. R. Becker, J. Weiß, A. Devi, R. A. Fischer, J. Phys. IV France 2001, 11, 569. R. Becker, A. Devi, J. Weiß, U. Weckenmann, M. Winter, C. Kiener, H.-W. Becker, R. A. Fischer, Chem. Vap. Dep. 2002, submitted for publication. S. C. Goel, K. S. Kramer, M. Y. Chiang, W. E. Buhro, Polyhedron 1990, 9, 611. V. L. Young, D. F. Cox, M. E. Davis, Chem. Mater. 1993, 5, 1701. T. Sosebee, M. Giersig, A. Holzwarth, P. Mulvaney, Ber. Bunsenges. Phys. Chem. 1995, 99 (1), 40. I. Lisiecki, F. Billoudet, M. P. Pileni, J. Phys. Chem. 1996, 100, 4160. 142 / Literatur [134] [135] [136] [137] [138] [139] C. T. Campbell, S. C. Parker, D. E. Starr, Science 2002, 298, 811. R. V. Kumar, Y. Diamant, A. Gedanken, Chem. Mater. 2000, 12, 2301. R. H. Kore, J. S. Kulkarni, , S. K. Haram, Chem. Mater. 2001, 13 (5), 1789. S. Vollmer, Dissertation an der Ruhr-Universität Bochum 2001. S. Vollmer, G. Witte, C. Wöll, Langmuir 2001, 17 (24), 7560. P. E. Laibinis, G. M. Whitesides, D. L. Allara, Y.-T. Tao, A. N. Parikh, R. G. Nuzzo, J. Am. Chem. Soc. 1991, 113, 7152. K. Hadjiivanov, T. Venkov, H. Knözinger, Catalysis Letters 2001, 75 (1-2), 55. A. Dandekar, M. A. Vannice, J. Catal. 1998, 178, 621. C. Elschenbroich, A. Salzer, Organometallchemie, Teubner Studienbücher, 1993. B. Hayden, K. Kretzschmar, A. Bradshaw, Surf. Sci. 1985, 155, 553. B. G. Ershov, E. Janata, A. Henglein, J. Phys. Chem. 1991, 95, 8996. Y. Hayashi, K. Tachibana, Jpn. J. Appl. Phys. 1994, 33, L476. G. Gebauer, Dissertation an der Ruhr-Universität Bochum 2001. A. J. Leavitt, J. T. P. Beebe, Surf. Sci. 1994, 314, 23. B. C. Wiegand, C. M. Friend, Chem. Rev. 1992, 92, 491. [140] [141] [142] [143] [144] [145] [146] [147] [148] [149] M. N. Vargaftik, V. P. Zargorodnikov, I. P. Stolarov, I. I. Moiseev, D. I. Kochubey, V. A. Likholobov, A. L. Chuvilin, K. I. Zarnaraev, J. Mol. Catalysis 1989, 53, 315. [150] G. Schmid, R. Pfeil, R. Boese, F. Bandermann, S. Meyer, G. H. M. Calis, J. A. W. van der Velden, Chem. Ber. 1981, 114, 3634. [151] G. Schmid, B. Morun, J.-O. Malm, Angew. Chem. 1989, 101 (6), 772. [152] G. Schmid, M. Harms, J.-O. Malm, J.-O. Bovin, J. van Ruitenbeck, H. W. Zandbergen, W. T. Fu, J. Am. Chem. Soc. 1993, 115, 2046. [153] M. T. Reetz, W. Helbig, J. Am. Chem. Soc. 1994, 116, 7401. [154] M. T. Reetz, M. Winter, R. Breinbauer, T. Thurn-Albrecht, W. Vogel, Chem. Eur. J. 2001, 7 (5), 1084. [155] M. T. Reetz, W. Helbig, S. A. Quaiser, U. Stimming, N. Breuer, R. Vogel, Science 1995, 267, 367. [156] R. W. Devenish, T. Goulding, B. T. Heaton, R. Whyman, J. Chem. Soc., Dalton Trans. 1996, 673. [157] H. Bönnemann, W. Brijoux, R. Brinkmann, E. Dinjus, Angew. Chem. 1993, 103, 1344. [158] J. Kiwi, M. Grätzel, J. Am. Chem. Soc. 1979, 101, 7214. [159] H. Bönnemann, G. Braun, W. Brijoux, R. Brinkmann, A. Schulze Tilling, K. Seevogel, K. Siepen, J., Organomet. Chem. 1996, 520, 143. [160] C. K. Yee, R. Jordan, A. Ulman, H. White, A. King, M. Rafailovich, J. Sokolov, Langmuir 1999, 15, 3486. [161] S. Chen, K. Huang, J. A. Stearns, Chem. Mater. 2000, 12, 540. [162] M. T. Reetz, M. Maase, Adv. Mater. 1999, 11, 773. Literatur / 143 [163] T. Teranishi, M. Miyake, Chem. Mater. 1998, 10, 594. [164] K. H. B. Okitsu, Y. Maeda, Chem. Mater. 1996, 8, 315. [165] K. O. Mizukoshi, Y. Maeda, T. A. Yamamoto, R. Oshima, Y. Nagata, J. Phys. Chem. B 1997, 101 (36), 7033. [166] N. A. Dhas, A. Gedanken, J. Mater. Chem. 1998, 8 (2), 445. [167] C. Hérard, P. Bowen, J. Kemaître, J. Dutta, Nanostruct. Mater. 1995, 6, 313. [168] S. Giorgio, C. Chapon, C. R. Henry, Langmuir 1997, 13, 2279. [169] L. Sordelli, G. Martra, R. Psaro, C. Dossi, S., Coluccia, J. Chem. Soc., Dalton Trans. 1996, 765. [170] J.-C. Hierso, P. Serp, R. Feurer, P. Kalck, Appl. Organomet. Chem. 1998, 12, 161. [171] J. E. Gozum, D. M. Pollina, J. A. Jensen, G. S. Girolami, J. Am. Chem. Soc. 1988, 110, 2688. J.-C. Hierso, C. Satto, R. Feurer, P. Kalck, Chem. Mater. 1996, 8, 2481. C. Xu, L. Sun, L. J. Kepley, M. R. Crooks, Anal. Chem. 1993, 65, 2102. D. V. Leff, L. Brandt, J. R. Heath, Langmuir 1996, 12, 4723. H. Perez, J.-P. Pradeau, P.-A. Albouy, J. Perez-Omil, Chem. Mater. 1999, 11, 3460. [172] [173] [174] [175] [176] C. Pan, K. Pelzer, K. Philippot, B. Chaudret, F. Dassenoy, P. Lecante, M.-J. Casanove, J. Am. Chem. Soc. 2001, 123 (31), 7584. [177] U. Weckenmann, Dissertation an der Ruhr-Universität Bochum 2002. [178] N. Oberbeckmann, unveröffentlichte Ergebnisse 2001. [179] T. A. Stephenson, S. M. Morehouse, A. R. Powell, J. P. Heffer, G. Wilkinson, J. Chem. Soc. 1965, 3632. [180] R. C. Mehrotra, S. K. Agarwal, Y. P. Singh, Coord. Chem. Rev. 1985, 68, 101. [181] H. E. Bryndza, W. Tam, Chem. Rev. 1988, 88, 1163. [182] K. B. Wiberg, J. Am. Chem. Soc. 1990, 112, 3379 und 8765. [183] H. E. Bryndza, J. C. Calabrese, M. Marsi, D. C. Roe, W. Tam, J. E. Bercaw, J. Am. Chem. Soc. 1986, 108, 4805. [184] Z. Zhang, S. Dai, D. A. Blom, J. Shen, Chem. Mater. 2002, 14, 965. [185] J. L. J. T. Yao, D. Y. Wu, D. M. Sun, K. H. Xue, B. Ren, B. W. Mao, Z. Q. Tian, Surf. Sci. 2002, 514 (1-3), 108. [186] J. Bao, C. Tie, Z. Xu, Q. Zhou, D. Shen, Q. Ma, Adv. Mater. 2001, 13 (21), 1631. [187] B. K. Pradhan, T. Kyotani, A. Tomita, Chem. Commun. 1999, 1317. [188] G. Cárdenas, J. Acuna, Colloid Polym. Sci. 2001, 279, 442. [189] M. P. Zach, R. M. Penner, Adv. Mater. 2000, 12 (12), 878. [190] D.-H. Chen, S.-H. Wu, Chem. Mater. 2000, 12, 1354. [191] Y.-P. Sun, H. W. Rollins, R. Guduru, Chem. Mater. 1999, 11, 7. [192] A. Duteil, G. Schmid, W. Meyer-Zaika, J. Chem. Soc., Chem. Commun. 1995, 31. [193] D. de Caro, J. S. Bradley, Langmuir 1997, 13 (12), 3067. [194] T. Ould Ely, C. Amiens, B. Chaudret, Chem. Mater. 1999, 11, 526. 144 / Literatur [195] L. G. Hubert-Pfalzgraf, V. G. Kessler, J. Vaissermann, Polyhedron 1997, 16 (24), 4197. [196] P. Werndrup, S. Gohil, V. G. Kessler, M. Kritikos, L. G. Hubert-Pfalzgraf, Polyhedron 2001, 20, 2163. [197] R. Rittner, J. A. Vanin, B. Wladislaw, Magn. Reson. Chem. 1988, 26, 51. [198] S. Pick, Dreyssé, H., Surf. Sci. 2000, 460, 153. [199] J. A. Creighton, D. G. Eadon, J. Chem. Soc., Faraday Trans. 1981, 87, 3881. [200] BASF, German Patents 1923, 415 686; 441 433; 462 837. [201] D. Cornthwaite, UK Patent,, 1 296 212 (ICI). [202] P. Davies, F. F. Snowdon, G. W. Bridger, D. O. Huges, P. W. Young, UK Patent,, 1 010 871 (ICI). [203] J. T. Gallagher, J. M. Kidd, UK Patent,, 1 159 035 (ICI). [204] C. G. Kim, unpublished results 2001. [205] B. S. Kim, T. Piao, S. N. Hoier, S. M. Park, Corr. Sci. 1995, 37 (4), 557. [206] S. Chaturvedi, J. A. Rodriguez, J. Hrbek, Surf. Sci. 1997, 384 (1-2), 260. [207] A. Cybulski, Catal. Rev.-Sci. Eng. 1994, 36, 557. [208] G. P. van der Laan, A. A. C. M. Beenackers, B. Ding, J. C. Strikwerda, Catal. Today 1999, 48, 93. [209] T. Teranishi, M. Miyake, Chem. Mater. 1999, 11, 3414. [210] M. L. Wu, D.-H. Chen, T.-C. Huang, Langmuir 2001, 17, 3877. [211] Y.-H. Chen, Y.-H. Tseng, C.-S. Yeh, J. Mater. Chem. 2002, 12, 1419. [212] A. Henglein, J. Phys. Chem. B 2000, 104, 2201. [213] T. Ould Ely, C. Pan, C. Amiens, B. Chaudret, F. Dessanoy, P. Lecante, M.-J. Casanove, A. Mosset, M. Respaud, J.-M. Broto, J. Phys. Chem. B 2000, 104, 695. [214] E. E. Carpenter, C. T. Seip, C. J. O´Connor, J. Appl. Phys. 1999, 85, 5184. [215] J. S. Bradley, E. W. Hill, B. Chaudret, A. Duteil, Langmuir 1995, 11 (3), 693. [216] C. Pan, F. Dassenoy, M.-J. Casanove, K. Philippot, C. Amiens, P. Lecante, A. Mosset, B. Chaudret, J. Phys. Chem. B 1999, 103, 10098. [217] S. Sun, C. B. Murray, D. Weller, L. Folks, A. Moser, Science 2000, 287, 1989. [218] Z. R. Dai, S. Sun, Z. L. Wang, Nano Lett. 2001, 1 (8), 443. [219] S. Link, Z. L. Wang, M. A. El-Sayed, J. Phys. Chem. B 1999, 103, 3529. [220] D. F. Foster, D. J. Cole-Hamilton, Inorg. Synth. 1997, 29. [221] P. O´Brien, J. R. Walsh, Polyhedron 1990, 9 (12), 1483. [222] H. W. W. Schumann, T. Seuss, O. Just, L. Pohl, World Pat., WO 9322472 1993, Merck GmbH. [223] Y. Zhang, Z. Yuan, R. J. Puddephatt, Chem. Mater. 1998, 10 (8), 2293. [224] Y. Tatsuno, T. Yoshida, Seiotsuka, Inorg. Synth. 1979, 19, 220. Literatur / 145 12 Anhang 12.1 Publikationen, Vorträge, Posterbeiträge Publikationen [1] J. Hambrock, A. Birkner, R. A. Fischer „Synthesis of CdSe nanoparticles using various organometallic cadmium precursors” J. Mater. Chem. 2001, 11, 3197. [2] J. Hambrock, R. Becker, A. Birkner, J. Weiss, R. A. Fischer „A non-aqueous organometallic route to highly monodispersed copper nanoparticles using [Cu(OCH(Me)CH2NMe2)2]” Chem. Commun. 2002, 69. [3] R. Pietschnig, T. Langkau, R. Neuser, F. Hipler, J. Hambrock, S. Schäfer "Synthesis, Characterization and AC Impedance Properties of a Novel Ion Conducting Polymer" Macromol. Rapid Commun., 2003, 24, 303. [4] J. Hambrock, S. Rabe, K. Merz, A. Birkner, A. Wohlfart, R. A. Fischer, M. Drieß „Low-temperature approach to high surface ZnO nanopowders and a non-hydrolytic synthesis of ZnO colloids using the single-source precursor [MeZnOSiMe3]4 and related zinc siloxides” Zur Publikation angenommen von J. Mater. Chem. [5] J. Hambrock, M. K. Schröter, A. Birkner, Ch. Wöll, R. A. Fischer „Nano-Brass: Bimetallic Cu/Zn colloids by a non aqueous organometallic route” Eingereicht an Chem. Mater. [6] A. Wohlfart, J. Khanderi, A. Devi, J. Hambrock, A. Birkner, R. A. Fischer „Controlled growth and characterization of ordered and disordered GaN nanostructures using Bisazido(dimethylaminopropyl)gallium as single molecule precursor“ Eingereicht an J. Cryst. Growth [7] M. K. Schröter, J. Hambrock, L. Khodeir, W. Busser, E. Löffler, M. Muhler, R. A. Fischer „Study of CO adsorption on copper, zinkoxide and mixed Cu/Zn colloids in solution“ In Vorbereitung [8] J. Hambrock, M. K. Schröter, A. Birkner, R. A. Fischer „Synthesis of non-aquoeus palladium colloids using the organometallic precursors [Cp(Allyl)Pd] and [Pd(OCH(Me)CH2NMe2)2] In Vorbereitung Anhang / V Vorträge [1] „Synthese und Oberflächenmodifizierung von Nanopartikeln“ Zukunftsperspektiven in der Anorganischen Chemie, Symposium der Universitäten Dortmund und Bochum, Ruhr-Universität Bochum, 31.5.-1.6.2001. [2] Vortragsreihe über Nanotechnologie, Nanopartikel, CVD und Selbstordnung Februar bis April 2002 King Mongkuts Institute of Technology, Faculty of Applied Science, Bangkok, Thailand. Konferenzen und Poster [1] MRS Fall Meeting Boston, USA, 26.-30.11.1999 [2] 38th Tutzing Symposion, „Chemical Nanotechnology – From Visions to Products Tutzing, 6.-9.2.2000 [3] 3. Materialwissenschaftlicher Tag an der Ruhr-Universität Bochum Posterbeitrag: H. Parala, J. Hambrock, A. Birkner, M. Kolbe, R. A. Fischer, „CdSe: Mesoskopische Komposite und oberflächenmodifizierte Kolloide“ Bochum, 28.4.2000 [4] MRS Fall Meeting Boston, USA, 27.11.-1.12.2000 [5] Dechema Nanotechnology Talks II Posterbeitrag: J. Hambrock, H. Parala, A. Birkner, R. A. Fischer, „Synthesis of Metal and Semiconductor Nanoparticles via a non-aqueous organometallic route“ Frankfurt, 8.-9.10.2001 VI / Anhang 12.2 Lebenslauf Persönliche Daten Name Geburtsdatum Geburtsort Nationalität Julia Hambrock 21.01.1974 Hannover Deutsch Ausbildung 1980/81 1981-83 1983/84 1984-93 15.6.1993 Grund- und Hauptschule Elze, Niedersachsen Österfeldschule, Stuttgart, Baden-Württemberg Martinusschule, Mainz, Rheinland-Pfalz Rabanus-Maurus-Gymnasium, Mainz, Rheinland-Pfalz Abitur 1993-95 25.8.1995 1995-98 30.9.1998 Chemiestudium an der Johannes-Gutenberg-Universität, Mainz Vordiplom Chemiestudium an der Ruprecht-Kars-Universität, Heidelberg Diplom. Diplomarbeit bei Prof. Dr. G. Helmchen, „Palladiumkatalysierte allylische Aminierung und ihre Anwendung in der Synthese von chiralen Carbonukleosiden Seit 1999 Promotion bei Prof. Dr. R. A. Fischer an der Ruhr-Universität Bochum Stipendien 9/1998 5/1999 – 6/2001 Seit 4/2002 Stipendium für hervorragende Studienleistungen aus den Mitteln des Dr.-Sophie-Bernthsen-Fonds Stipendium im Rahmen des Graduierten Kollegs „Dynamische Prozesse an Festkörperoberflächen“ Stipendiatin von e-fellows.net Anhang / VII Ich erkläre hiermit, dass ich die voliegende Dissertation selbst verfasst und mich dabei keiner anderen als den von von mir ausdrücklich bezeichneten Quellen und Hilfen bedient habe. Ich erkläre hiermit, dass ich an keiner anderen Stelle ein Prüfungsverfahren beantragt bzw. die Dissertation in dieser oder anderer Form bereits anderweitig als Prüfungsarbeit verwendet oder einer anderen Fakultät als Dissertation vorgelegt habe. Julia Hambrock. VIII / Anhang