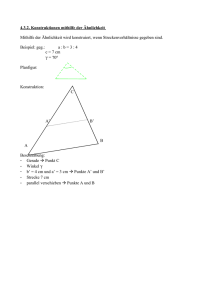
Lernen von Prototypen über Baumstrukturen Entwicklung und
Werbung

Lernen von Prototypen über
Baumstrukturen
Entwicklung und Evaluation
eines Verfahrens zum
Structural Incident Mining
Bachelorarbeit
im Studiengang Wirtschaftsinformatik der Fakultät
Wirtschaftsinformatik und Angewandte Informatik der
Otto-Friedrich-Universität Bamberg
Verfasser:
Gutachter:
Stefan Betzmeir
Prof. Dr. Ute Schmid
Zusammenfassung
Verschiedenste Data Mining Verfahren sind seit langem bewährte Werkzeuge, um aus einer Menge von Datensätzen Muster und damit neue
Erkenntnisse abzuleiten. Dabei sind die Quellen dieser Daten ebenso vielfältig wie deren Auswertungsmöglichkeiten. Standardverfahren
zur Datenaufbereitung und Analyse sind daher längst in nahezu allen
Bereichen von Forschung und Wirtschaft im Einsatz.
Typischerweise geht man dabei von Daten auf Basis eines festen
Datenschemas aus, deren Ausprägungen sich durch einen einfachen
Merkmalsvektor darstellen lassen, wie es z.B. bei einer Kundenadresskartei der Fall ist. Sobald jedoch Beziehungen zwischen verschiedenen
Entitäten abgebildet werden, entsteht dadurch strukturelle Dynamik.
Ein Kunde muss beispielsweise mit beliebig vielen Aufträgen und diese wiederum mit einer variablen Anzahl von Artikeln in Beziehung
gesetzt werden können. Um auf derartigen Daten Analysen mit den
eingangs genannten Verfahren durchführen zu können, ist es daher
gängige Praxis diese in eine flache und damit von Strukturinformationen bereinigte Darstellung zu bringen. Das ist nur dann möglich,
wenn die Semantik der Strukturellen Beziehungen bekannt, bzw. nicht
von Interesse bezüglich des Untersuchungsziels ist. In diesen Strukturinformationen verbirgt sich jedoch implizites Wissen über relationale
und kausale Zusammenhänge.
Deutlich wird dieser Aspekt am Beispiel des Customer-Supports einer Softwarefirma. Sie könnte ihre Software derart gestalten, dass beim
Auftreten eines Fehlers ein strukturiertes Abbild der Zustände ihrer
Komponenten gespeichert wird. Aufgabe des Service-Mitarbeiters ist
es dann, die Ursache eines Fehlers anhand dieser Daten zu lokalisieren.
In diesen Informationen, in Kombination mit dem Wissen über kausale
Zusammenhänge innerhalb der Software, kann der Service-Mitarbeiter
Muster erkennen, die ursächlich für den Fehler sind und einen Lösungsvorschlag anbieten. Derartige Geschäftsprozesse, die von Expertenwissen abhängig sind und manueller Datenverarbeitung bedürfen,
sind sehr Kostenintensiv und schlecht skalierbar, da sie üblicherweise
nicht automatisierbar sind. Mit Hilfe von Data Mining Verfahren könnte daher versucht werden Gemeinsamkeiten zwischen Fehlerprotokollen zu entdecken, denen der selbe Fehler zugrunde liegt. Da gängige
Verfahren, wie das Lernen von Entscheidungsbäumen oder das Trainieren neuronaler Netze, strukturelle Informationen ignorieren, wird
die Qualität der Analyse dadurch jedoch stark beeinträchtigt.
In der vorliegenden Arbeit soll daher ein bestehender Ansatz von
Schmid, Hofmann, Bader, Häberle und Schneider (2010), zum Erlernen
von Prototypen aus strukturierten Daten, erweitert werden. Ein solcher
Prototyp bildet strukturelle und merkmalsbezogene Eigenschaften einer Menge von Datensätzen ab, die vorher durch einen Experten als
ähnlich, im Bezug auf eine bestimmte Fragestellung, identifiziert wurden. Ein so gewonnener Prototyp kann anschließend verwendet werden, um neue Datensätze automatisch zu Klassifizieren. Dabei wird
ein Datensatz mit allen Prototypen verglichen und der Klasse zugeordnet, deren Prototyp den Datensatz am besten, hinsichtlich der Struktur
und den Merkmalen, beschreibt. Die Prototypentheorie ahmt dabei eine erfolgreiche kognitive Fähigkeit des Menschen nach, aus Objekten
mit übereinstimmenden Merkmalen, Konzepte abzuleiten.
I N H A LT S V E R Z E I C H N I S
Abbildungsverzeichnis
v
Algorithmenverzeichnis
vi
1
2
3
4
einführung
1.1 Bisheriger Ansatz . . . . . . . . . .
1.1.1 Case-Based Reasoning . . .
1.1.2 Lernen von Prototypen . .
1.1.3 Data Mining . . . . . . . . .
1.1.4 Structural Incident Mining
1.2 Aufbau der Arbeit . . . . . . . . .
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
1
1
1
1
2
2
2
analyse
2.1 Modellorientierter Ansatz . . . . . . . . . . . . .
2.1.1 Anti-Unifikation . . . . . . . . . . . . . .
2.1.2 Structure Dominance Tree Generalisation
2.1.3 Absolute Positionierung im Modell . . .
2.2 Instanzorientierter Ansatz . . . . . . . . . . . . .
2.2.1 Hierarchisch gewichtete Ähnlichkeit . . .
2.2.2 Ähnlichkeitsbasierte Alignierung . . . .
2.3 Multiple Sequence Alignment . . . . . . . . . . .
2.3.1 MSA in der Bioinformatik . . . . . . . . .
2.3.2 Progressive Alignierung . . . . . . . . . .
2.4 Paarweise Ähnlichkeit und Alignierung . . . . .
2.4.1 Needleman-Wunsch Algorithmus . . . .
2.4.2 Yangs Tree-Matching-Algorithmus . . . .
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
4
4
4
4
4
7
7
8
10
10
11
12
12
16
design
3.1 Definitionen . . . . . . . . . . . . . . . . . . . . . . . . .
3.1.1 Datenstrukturen . . . . . . . . . . . . . . . . . .
3.1.2 Funktionen . . . . . . . . . . . . . . . . . . . . .
3.2 Prototypextraktion . . . . . . . . . . . . . . . . . . . . .
3.2.1 Einstiegspunkt . . . . . . . . . . . . . . . . . . .
3.2.2 Rekursion . . . . . . . . . . . . . . . . . . . . . .
3.2.3 Prototypisierungsstrategie . . . . . . . . . . . . .
3.3 Progressive Alignierung . . . . . . . . . . . . . . . . . .
3.3.1 Hierarchisches Clustering . . . . . . . . . . . . .
3.3.2 Berechnung der Clusterdistanz . . . . . . . . . .
3.4 Alignierung . . . . . . . . . . . . . . . . . . . . . . . . .
3.4.1 Erzeugen der Vereinigungssequenz . . . . . . .
3.4.2 Alignierung assoziierter Sequenzelemente . . .
3.4.3 Übernahme nicht assoziierter Sequenzelemente
3.5 Tree-Matching . . . . . . . . . . . . . . . . . . . . . . . .
3.5.1 Parametrisiertes Tree-Matching . . . . . . . . . .
3.5.2 Backtracking . . . . . . . . . . . . . . . . . . . .
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
18
18
18
21
22
22
23
23
25
26
27
28
28
29
30
31
31
34
.
.
.
.
.
.
.
.
.
35
35
36
37
37
39
39
41
41
42
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
implementierung
4.1 Umgang mit generischen Datentypen . . . . . . . .
4.1.1 Datenschnittstelle . . . . . . . . . . . . . . . .
4.1.2 Prototypschnittstelle . . . . . . . . . . . . . .
4.1.3 Typspezifische Strategien . . . . . . . . . . .
4.2 Prototypextraktion . . . . . . . . . . . . . . . . . . .
4.2.1 Prototypextraktion aus strukturierten Daten
4.2.2 Prototypingstrategie . . . . . . . . . . . . . .
4.3 Alignierung . . . . . . . . . . . . . . . . . . . . . . .
4.4 Clustering . . . . . . . . . . . . . . . . . . . . . . . .
iii
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
4.4.1 Hierarchisches Clustering . . . .
4.4.2 Linkage . . . . . . . . . . . . . .
4.4.3 Cluster . . . . . . . . . . . . . . .
Sequence-Matching . . . . . . . . . . . .
4.5.1 Hierarchical Sequence-Matching
4.5.2 Caching von Ergebnissen . . . .
4.5.3 Hierarchical Tree-Matching . . .
4.5.4 Sequenz . . . . . . . . . . . . . .
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
42
43
44
46
46
47
47
47
resümee und ausblick
5.1 Evaluation . . . . . . . . . . . . . . . . .
5.2 Anknüpfungspunkte . . . . . . . . . . .
5.2.1 Empirische Ergebnisse . . . . . .
5.2.2 Iterative Alignierung . . . . . . .
5.2.3 Automatisches Clustering . . . .
5.2.4 Globale Alignierung . . . . . . .
5.2.5 Neuer modellorientierter Ansatz
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
49
49
49
49
50
50
50
50
4.5
5
Literaturverzeichnis
51
iv
ABBILDUNGSVERZEICHNIS
Abbildung 1
Abbildung 2
Abbildung 3
Abbildung 4
Abbildung 5
Abbildung 6
Abbildung 7
Abbildung 8
Abbildung 9
Abbildung 10
Abbildung 11
Abbildung 12
Abbildung 13
Abbildung 14
Abbildung 15
Abbildung 16
Abbildung 17
Abbildung 18
Abbildung 19
Abbildung 20
Abbildung 21
Abbildung 22
Abbildung 23
Abbildung 24
Abbildung 25
Abbildung 26
Abbildung 27
Modell als reguläre Grammatik . . . . . . . . . .
Modell in Baumdarstellung . . . . . . . . . . . . .
Instanzen eines Modells . . . . . . . . . . . . . . .
Ein SDTG Prototyp . . . . . . . . . . . . . . . . . .
Alignierung von Knoten . . . . . . . . . . . . . . .
Lokal optimale Alignierung von Knoten . . . . .
Initialzustand der Matrizen . . . . . . . . . . . . .
Alternative Pfade einer Alignierung . . . . . . . .
Matching zweier Sequenzelemente . . . . . . . . .
Fertig errechnete Matrizen . . . . . . . . . . . . .
Komponenten einer Sequenz . . . . . . . . . . . .
Unterschiedliche Datendarstellung . . . . . . . . .
Datenschnittstelle ITreeNode . . . . . . . . . . . . .
Prototypenschnittstelle IPrototype . . . . . . . . .
Typspezifische Strategien . . . . . . . . . . . . . .
Extraktionsschnittstelle IPrototypeExtraction . . .
Prototypextraktion aus Baumstrukturen . . . . .
Prototypingstrategien . . . . . . . . . . . . . . . .
Alignierungsschnittstelle IAlignment . . . . . . . .
Beziehung zwischen den Komponenten der Alignierung . . . . . . . . . . . . . . . . . . . . . . . .
Clusteringschnittstelle IClusterManager . . . . . .
ILinkage als Schnittstelle zur Berechnung der Clusterdistanz . . . . . . . . . . . . . . . . . . . . . . .
AbstractCluster als Cluster-Supertyp . . . . . . . .
LeafCluster, der einelementige Cluster . . . . . . .
LinkCluster als Verbindung zwischen zwei Clustern
Hierarchical Sequence-Matching . . . . . . . . . .
Die Sequenz als Komposition aus drei Datenstrukturen . . . . . . . . . . . . . . . . . . . . . . . . . .
v
5
5
6
6
9
9
14
14
15
15
20
36
36
37
38
39
40
40
41
42
43
43
45
45
45
46
48
ALGORITHMENVERZEICHNIS
1
2
Needleman-Wunsch Algorithmus . . . . . . . . . . . . .
Yangs Tree-Matching-Algorithmus . . . . . . . . . . . . .
12
17
3
4
5
6
7
8
9
10
11
12
13
14
Einstiegspunkt der Prototypenextraktion . . . .
Rekursive Prototypenextraktion . . . . . . . . . .
Prototypingstrategie “Anti-Unifikation” . . . . .
Prototypingstrategie “SDTG” . . . . . . . . . . .
Einstiegspunkt der progressiven Alignierung . .
Hierarchisches Clustering . . . . . . . . . . . . .
Berechnung der Distanz zweier Cluster . . . . .
Globale Alignierung der Sequenzen . . . . . . .
Assoziierte Sequenzelemente alignieren . . . . .
Nicht-Assoziierte Sequenzelemente hinzufügen
Hierarchical Tree-Matching-Algorithmus . . . .
Backtracking . . . . . . . . . . . . . . . . . . . . .
23
23
24
25
26
27
28
29
30
31
33
34
vi
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
1
EINFÜHRUNG
1.1
bisheriger ansatz
Die vorliegende Arbeit setzt auf den Ergebnissen einer Zusammenarbeit zwischen dem Lehrstuhl für Kognitive Systeme der Otto-FriedrichUniversität Bamberg und der Firma SAP auf, die im Rahmen der Masterarbeit von Bader (2009) stattgefunden hat. Dabei sollten Lösungen
erarbeitet werden, um Geschäftsprozesse im Kundenservice, bezüglich der Software ByDesign, durch maschinelle Lernverfahren zu automatisieren. Diese Software bietet dem Benutzer, beim Auftreten eines
Softwarefehlers, einen Dialog an, um eine Störungsmeldung an das
Service-Center zu schicken. Dort kann der Benutzer Informationen angeben, die für die Fehlerbehebung von Nutzen sein könnten. Zusätzlich wird der Zustand des Anwendungssystems, zum Zeitpunkt des
Fehlers, gespeichert. Mit Hilfe dieser Daten kann der Service-Mitarbeiter
feststellen, welche Ursachen den Fehler ausgelöst haben und dem Kunden entsprechend Hilfestellung leisten. Alle gelösten Störungsmeldungen werden zudem in einer Datenbank gespeichert, so dass bei ähnlichen Fällen in der Zukunft auf bereits ermittelte Lösungen zurückgegriffen werden kann. Das Bestreben war es nun, ein Verfahren zu
finden, mit dessen Hilfe man die Klassifikation neu eintreffender Störungsmeldungen, auf Basis bereits gefundener Lösungen, automatisieren kann, um so den personalintensiven Support zu entlasten und Kosten zu sparen.
1.1.1
Case-Based Reasoning
Als Grundlage diente hierfür der von Agnar und Plaza (1994) entwickelte Rahmen für das fallbasierte Schließen (case-based reasoning). Entsprechend dem case-based resoning Zyklus (Agnar & Plaza, 1994, S. 7 f.)
wird dabei zunächst für einen neu eintreffenden Fall in der Datenbank
nach einem bereits gelösten Fall gesucht, der dem neuen am ähnlichsten ist. Daraufhin wird die damit assoziierte Information wiederverwendet, die vorgeschlagene Lösung an den neuen Fall angepasst und
daraus resultierende Erkenntnisse wiederum für zukünftige Abfragen
gespeichert. Der Vorteil und zugleich der Nachteil eines case-based reasoning Verfahrens ist der Umfang der Datenbank, der mit jedem gelösten Fall zunimmt. Zwar nimmt so die Wahrscheinlichkeit zu, für
ein neues Problem eine bereits vorhandene Lösung zu finden, jedoch
dauert auch die Klassifikation eines neu eintreffenden Falles dadurch
immer länger.
1.1.2
Lernen von Prototypen
Da die ByDesign Störungsmeldungen von den SAP Service-Mitarbeitern
sogenannten “Master-Incidents” zugeordnet werden, also bereits gelösten Störungsmeldungen die von der Fehlerursache her ähnlich waren, wurde hierdurch bereits die Grundgesamtheit der Störungsmeldungen in Cluster ähnlicher Fälle unterteilt. Anstatt nun eine neu eintreffende Störungsmeldung mit allen Störungsmeldungen eines Clusters zu vergleichen, könnte sie stattdessen mit einem Clusterprototypen verglichen werden, der die gemeinsamen Merkmale der Clusterelemente repräsentiert, was die Laufzeit der Abfrage deutlich reduzieren würde.
1
1.1.3
Data Mining
Beispielsweise könnte für jedes Cluster von Daten, ein Entscheidungsbaum gelernt oder ein neuronales Netz trainiert werden. Eine neu eintreffende Störungsmeldung könnte dann automatisch der passendsten
Kategorie zugeordnet werden. Als Grundlage dieser und vieler anderer Data Mining Verfahren dienen hierzu Merkmalsvektoren. Prinzipiell könnte man die Störungsmeldungen, die im XML-Format vorliegen
und demnach eine Baumstruktur aufweisen, in eine “flache” Darstellung bringen. Dadurch würden jedoch implizite Strukturinformationen der Baumstruktur, wie Relationen und Abhängigkeiten, verlorengehen.
1.1.4
Structural Incident Mining
Um diese Strukturinformationen zu erhalten wurden verschiedene Verfahren zur Generalisierung gemeinsamer Strukturen, aus einer Menge
strukturierter Datensätze, evaluiert.
Anti-Unifikation
Mit der Anti-Unifikation wurde ein Verfahren ausgewertet, das ursprünglich zur Generalisierung von Termen entwickelt wurde. Die
Anti-Unifikation erzeugt aus einem Cluster strukturierter Datensätze deren Schnittmenge, d.h. nur die Strukturelemente, die in jedem
Datensatz an der selben Stelle vorkommen, werden Teil des so extrahierten Prototyps. Da dieses Vorgehen sehr empfindlich im Hinblick
auf verrauschte Daten ist, eignet es sich nur bedingt für die Prototypisierung von Störungsmeldungen, da die manuelle Klassifikation der
Daten durch Menschen fehlerbehaftet ist.
Structure Dominance Tree Generalisation
Darum wurde ein Ansatz auf Basis der Unifikation, also der Vereinigungsmenge der Strukturelemente aller Datensätze, entwickelt, in
dem sich dominante Strukturen ausprägen können, auch wenn diese
nicht in allen Instanzen vorhanden sind. Hierzu wird die Häufigkeit
des Auftretens eines Strukturelements relativ zu seinem Elternelement
gespeichert.
Modellorientierter Instanzvergleich
Der Vergleich zwischen den Datensätzen findet bei beiden Verfahren
mit Hilfe des Modells statt. Das Modell ist hierbei als ein Satz von Regeln zu verstehen, der die möglichen Merkmals- und Strukturausprägungen in den Instanzen bestimmt. Dabei muss das Modell als endlicher Baum darstellbar sein, da der Vergleich auf absoluten Positionen
innerhalb des Modells basiert. Diese Einschränkung bezüglich des Modells hat zur Folge, dass das bisherige Verfahren lediglich auf solche
strukturierten Daten angewendet werden kann, denen ein entsprechendes Modell zugrunde liegt, das diesen Anforderungen genügt.
1.2
aufbau der arbeit
Auf den bisherigen Ergebnissen aufbauend, wird in der vorliegenden
Arbeit ein modifizierter Ansatz zur Prototypextraktion aus strukturierten Daten entwickelt. Das Ziel ist es dabei die Restriktionen des modellorientierten Ansatzes aufzuheben, um die Prototypextraktion auf
jegliche Art baumartig strukturierter Daten anwenden zu können.
Dazu wird im Kapitel “Analyse” zunächst der bisherige, modellorientierte Ansatz analysiert, um die Gründe für dieser Restriktionen
2
Aufzudecken. Anschließend werden Verfahren aus der Bioinformatik,
zum Vergleich und zur Alignierung multipler Sequenzen, vorgestellt
und hinsichtlich der Eignung für die Prototypextraktion evaluiert. Um
das gewählte Verfahren für Baumstrukturen nutzbar zu machen, wird
zunächst dessen zentraler Algorithmus vorgestellt und mit einer für
Bäume geeigneten Variante verglichen.
Die Erkenntnisse aus der Analyse bilden das Fundament für den
Entwurf des neuen Verfahrens im Kapitel “Design”. Zunächst werden
die nötigen Datenstrukturen und Rahmenbedingungen definiert. Anschließend wird das gesamte Verfahren in Teilaufgaben zerlegt, in algorithmischer Form dargestellt und erläutert.
Im darauffolgenden Kapitel “Implementierung” wird die konkrete
Umsetzung des Verfahrens in der Programmiersprache Java erläutert.
Da der gesamte Ablauf bereits im Kapitel “Design” dargestellt wird,
liegt der Fokus bei der Implementierung auf den Designentscheidungen die getroffen wurden um die Software erweiterbar und wartbar
zu gestalten.
Abschließend folgt eine kritische Bewertung des entwickelten Ansatzes. Dabei werden Anknüpfungspunkte identifiziert die als Basis für
zukünftige Verbesserungen und Erweiterungen genutzt werden können.
3
2
A N A LY S E
Um das bisherigen Verfahre zum Lernen von Prototypen dahingehend
zu erweitern, um es potentiell auf jegliche Art baumartig strukturierter
Daten anwenden zu können, muss es zunächst analysiert und anschließend entsprechend angepasst werden.
2.1
modellorientierter ansatz
Die vorgestellten Lernverfahren, zur Erzeugung eines Prototypen aus
einem Cluster strukturierter Daten, setzen ein als Baum darstellbares
Modell voraus, das den Daten als Schema zugrunde liegt und damit
die Form seiner Instanzen vorgibt, wenn es heißt, “The incident model
– also called model tree – represents the general structure which is underlying each possible incident report and thereby restricts the form
of incident trees” (Schmid et al., 2010, S. 3). Die Funktion des Modells,
im Bezug auf die Prototyperstellung, zeigt sich, wenn man die Algorithmen au (Schmid et al., 2010, S. 4), auf Basis der Anti-Unifikation,
und sdtg (Schmid et al., 2010, S. 5), die Structure Dominance Tree Generalisation, näher betrachtet. Beide Algorithmen haben die Gemeinsamkeit, dass das Modell “top-down”, also bei der Wurzel beginnend,
traversiert und dabei jedes besuchte Modellelement M.p mit dem entsprechenden Element eines jeden Baumes I1 .p, ..., In .p im Cluster verglichen wird. Die Algorithmen unterscheiden sich jedoch in der Art
und Weise, wie sie aus den Korrespondierenden Elementen des Baumes das Prototypelement extrahieren.
2.1.1
Anti-Unifikation
Im Falle von au fließen die Merkmale eines Elements nur dann in den
Prototypen ein, wenn für die aktuell betrachtete Modellposition M.p
in jedem Baum des Clusters, an entsprechender Stelle, eine Übereinstimmung gefunden werden kann. Ist dies der Fall, wird der Algorithmus rekursiv mit den Kindknoten dieser Modellposition, sowie den
Kindknoten der entsprechenden Instanzelemente, fortgesetzt. Dieses
Verhalten führt jedoch zu einer hohen Anfälligkeit für verrauschte Daten, da für jede Position bereits eine Abweichung im Cluster zu einem
Ausschluss aus dem Prototypen führt (“Syntactic anti-unification is not
robust with respect to noise” (Schmid et al., 2010, S. 5)).
2.1.2
Structure Dominance Tree Generalisation
Im sdtg Algorithmus werden alle Elemente in den Prototypen aufgenommen. Dabei werden zusätzlich in jedem Prototypelement die Auftretenshäufigkeiten der repräsentierten Elemente, in Relation zu deren
Elternelementen, gespeichert. Der Algorithmus wird auch hier an jeder Position rekursiv mit den Kindknoten fortgesetzt. Somit erzeugt
der sdtg Algorithmus eigentlich eine Vereinigungsmenge der Instanzen eines Clusters, jedoch werden anhand der relativen Häufigkeiten
der Prototypelemente dominante Strukturen erkennbar.
2.1.3
Absolute Positionierung im Modell
Aus dem Vergleich der Algorithmen au und sdtg ist ersichtlich, dass
das Modell eine zentrale Rolle spielt, indem es anhand der Positio-
4
nen seiner Elemente festlegt, welche Elemente der Bäume miteinander
verglichen werden. Damit eine solche absolute Positionsbestimmung
möglich ist, wird in Definition 1 des model tree (Schmid et al., 2010, S.
3) gefordert, dass das Modell M als Baum fester Größe, mit Elementen
e des Typs τ, darstellbar sein muss. Dafür müssen im Modell folgende
Voraussetzungen erfüllt sein:
1. Für jeden Typ τ ist die Anzahl seiner Attribute, sowie deren Typ
fest
2. Ein Typ τ darf als Attribut nicht sich selbst enthalten
Zur Veranschaulichung dieser Gedanken soll ein einfaches Beispiel dienen.
Beispiel “Haustierbesitzer”
Es gilt das Datenmodell zur Speicherung von Haustierbesitzern zu
konzipieren. Von Interesse bezüglich der Person sind dabei der Name
sowie das Alter. Für das Haustier sind lediglich die Subklassen Hund
und Papagei zulässig. Die Klasse Hund erhält die Attribute Name und
Rasse. Ein Papagei soll die relevanten Attribute Name sowie Wörter – für
den Umfang des von ihm beherrschten Wortschatzes – enthalten. Als
Repräsentation des so definierten Modells bieten sich die Produktionen einer regulären Grammatik an.
σ → <BESITZER>
<BESITZER> → <NAME> <HAUSTIER> <ALTER>
<NAME> → [a-Z]+
<ALTER> → [0-9]+
<HAUSTIER> → <HUND>|<PAPAGEI>
<HUND> → <NAME> <RASSE>
<RASSE> → [a-Z]+
<PAPAGEI> → <NAME> <WÖRTER>
<WÖRTER> → [0-9]+
Abbildung 1: Modell als Produktionen einer regulären Grammatik
Diese Grammatik lässt sich als Baum fester Größe, d.h. als endlicher
Modellbaum darstellen (s. Abbildung 2). Zwei der daraus ableitbaren
Instanzen wären somit Instanz 1 und Instanz 2 (s. Abbildung 3).
Besitzer
Name
Haustier
Hund
Alter
Papagei
Name Rasse Name Wörter
Abbildung 2: Modell in Baumdarstellung
5
Besitzer
Besitzer
Name
Haustier
Alter
Name
Haustier
Alter
“Tim”
Hund
18
“John”
Papagei
40
Name
Rasse
“Struppi” “Terrier”
Instanz 1
Name
Wörter
“Captain Flint”
42
Instanz 2
Abbildung 3: Zwei Instanzen des Modells
Wendet man nun den sdtg Algorithmus auf den Cluster bestehend aus
Instanz 1 und Instanz 2 an, so ergibt sich der in Abbildung 4 dargestellte Prototyp.
Besitzer [2/2]
Name [2/2]
Tim [1/2]
John [1/2]
Haustier [2/2]
Hund [1/2]
Name [1/1]
Struppi [1/1]
Rasse [1/1]
Terrier [1/1]
Papagei [1/2]
Name [1/1]
Captain Flint [1/1]
Wörter [1/1]
42 [1/1]
Alter [2/2]
18 [1/2]
40 [1/2]
Abbildung 4: Der aus Instanz 1 und Instanz 2 abgeleitete Prototyp
Der bisherige sdtg Algorithmus ist somit in der Lage nicht nur relative Häufigkeiten in den Blättern zu zählen, was in einem Merkmalsvektor ohne Strukturinformationen ebenfalls möglich wäre, sondern
er bewahrt gleichzeitig strukturabhängige Informationen im Bezug
auf Merkmalsausprägungen die sich aus dem Auftreten verschiedener
Subtypen, wie im Beispiel Hund und Papagei als Subtypen von Haustier,
ergeben.
Einschränkungen des Modells
Die eingangs genannten Voraussetzungen bezüglich des Modells lassen sich nun leicht nachvollziehen, indem man neue Anforderungen
an unser Datenmodell stellt.
variable anzahl von attributen Beispielsweise könnte es interessant sein, nicht nur die Anzahl, sondern auch die konkreten Wörter zu speichern, die ein Papagei sprechen kann. Das könnte man beispielsweise durch folgende Produktionen ausdrücken:
6
<PAPAGEI> → <NAME> <WORT>*
<WORT> → [a-Z]+
Durch die Kleene’sche Hülle wären somit für jeden Papagei null bis beliebig viele Worte aufzählbar. Dadurch wäre das Modell jedoch nicht
mehr als Baum darstellbar, da dieser, um den Elementvergleich anleiten zu können, sozusagen für null bis beliebig viele Wortinstanzen
einen “Platz” vorsehen müsste. Der Baum würde sich an dieser Stelle
unendlich in die Breite ausdehnen und das Modell hiermit Voraussetzung 1 verletzen.
rekursive attribute Es könnte auch ein weiterer interessanter
Aspekt hinsichtlich der Datenanalyse sein, wie sich die Haustierhaltung innerhalb einer Familie über mehrere Generationen entwickelt
hat. Dazu könnte man jeder Person die Attribute Mutter und Vater verleihen, die wiederum vom Typ Person wären. Als Produktionen müsste
man das Modell, an der entsprechenden Stelle, wie folgt verändern:
<PERSON> → <NAME> <HAUSTIER> <ALTER>
<MUTTER> <VATER>
<MUTTER> → <PERSON>?
<VATER> → <PERSON>?
Es muss erlaubt sein, dass der Stammbaum ein absehbares Ende haben
darf, ausgedrückt durch den Quantoren ?, was der Kardinalität “null
bis eins” entspricht. Jedoch ergibt sich nun ein ähnliches Problem wie
schon im vorherigen Beispiel: Der Modellbaum müsste eine potentiell
unendliche Rekursion zurück durch die “Generationen” einer Person
vorsehen, wodurch er unendlich in die Tiefe wachsen würde. Hierdurch würde Voraussetzung 2 verletzt.
2.2
instanzorientierter ansatz
In der vorliegenden Arbeit soll nun versucht werden, auf Basis des
SDTG Ansatzes, eine neue Herangehensweise zu entwickeln die es erlaubt, ein breiteres Spektrum an strukturierten Daten zu verarbeiten.
Ziel soll es sein die bisherigen Einschränkungen im Bezug auf das Modell fallen zu lassen, wodurch es möglich sein wird das SDTG Verfahren auf alle Daten anzuwenden, dessen Modell als reguläre Grammatik (Typ 3 der Chomsky Hierarchie) darstellbar ist. Da hierdurch eine
Orientierung des Elementvergleichs auf Basis eines endlichen Modellbaumes nicht mehr möglich ist, muss eine andere Lösung gefunden
werden um strukturelle Entsprechungen zwischen 2 bis n Datensätzen
zu aufzudecken. Eine Möglichkeit ist der im folgenden betrachtete instanzorientierte Ansatz.
2.2.1
Hierarchisch gewichtete Ähnlichkeit
Setzt man für einen Cluster von Daten voraus, dass jedes seiner Elemente aus dem selben Datenmodell hervorgegangen ist, so können
diese auch ohne Kenntnis des Modells verglichen werden. Betrachtet
man die bisher genutzten Algorithmen, so fällt auf, dass zwar das
Modell bestimmt, an welcher Position der jeweils nächste Vergleich
stattfindet, jedoch dass an dieser Position letztendlich der Typ des Instanzelements das Aussehen des Prototypen und den Fortgang des
Verfahrens bestimmt. Es gilt also für jedes Element einer Instanz ein
entsprechendes Element in den anderen Instanzen zu finden, worauf
der jeweilige Algorithmus rekursiv, mit diesen Elementen als Wurzeln,
aufgerufen werden kann. Es ist naheliegend, dass man Elemente die
7
den gleichen Typ aufweisen als identisch betrachtet. Bei den denkbar
einfachsten Bäumen, also einelementigen Wurzeln, ist es trivial identische Elemente zu identifizieren:
A1
A2
B3
A1 hat den selben Typ wie A2 , B3 korrespondiert mit keinem anderen
Element. Fügt man den Wurzelknoten nun jeweils ein Kind hinzu, ist
die Frage nach der Identität nicht mehr intuitiv lösbar.
A1
A2
B3
C1
D2
C3
Nun korrespondiert zwar der Typ des Wurzelknotens A1 mit dem des
Wurzelknotens A2 , jedoch sind deren Kindknotentypen C1 und D2
nicht identisch. Auf der anderen Seite sind die Typen der Wurzelknoten A1 und B3 verschieden, jedoch gleichen sich deren Kindknotentypen C1 und C3 . Egal welche beiden Bäume man herausgreift, man
könnte nicht mehr behaupten sie seien identisch sondern man müsste
sie als ähnlich bezeichnen. Da man Identität auch als maximale Ähnlichkeit darstellen kann, wird, gerade im Hinblick auf die unterschiedlich Semantik des Begriffs “Identität”, im Folgenden bezüglich des Vergleichs von Elementen der Begriff “Ähnlichkeit” verwendet. Die Ähnlichkeit zweier Knoten, wird also nicht nur lokal durch die Ähnlichkeit
zweier Typen, sondern auch durch die Ähnlichkeit ihrer Kindknoten
beeinflusst. Die Ähnlichkeit zweier Knoten ist somit nichts anderes,
als der rekursive Vergleich zweier Bäume. Da Bäume jedoch eine Hierarchie beschreiben, würde man die lokale Ähnlichkeit der Wurzelknotentypen stets stärker gewichten als die durch Rekursion ermittelte
Ähnlichkeit ihrer Kinder, so dass deren Einfluss zu den Blättern hin
abnimmt. Durch die rekursive Bestimmung der Ähnlichkeit ist nun eine Beschränkung im Bezug auf das Modell aufgehoben und es darf
jetzt rekursive Produktionen wie diese enthalten:
<A> → <A>?
2.2.2
Ähnlichkeitsbasierte Alignierung
Die zweite Beschränkung des Modells lag darin, dass jeder Typ eine
feste Anzahl von Attributen aufweisen musste. Hebt man diese Einschränkung auf, so wäre beispielsweise folgende Produktion möglich:
<A> → <B>*
8
Jetzt ergibt sich ein neues Problem, denn nun muss zur Bestimmung
der Ähnlichkeit von A1 zu A2 zusätzlich geklärt werden, mit welchem
Knoten des zweiten Baumes B1.1 verglichen wird. Dazu müsste wiederum die Ähnlichkeit von B1.1 zu B2.1 und B2.2 bestimmt werden und
das Maximum daraus würde wiederum die Ähnlichkeit der Kinder
von A1 und A2 darstellen.
A1
A2
B1.1
B2.1
B2.2
Abbildung 5: Alignierung von Knoten
In Abbildung 5 ist die Lösung trivial, da die Knoten vom Typ B selber
keine Kinder haben und somit ist die Ähnlichkeit von B1.1 zu B2.1 und
zu B2.1 gleich. Erweitert man das Beispiel um weitere Knoten wird das
Problem deutlich.
A1
B1.1
A2
B1.2
B2.1
B2.2
C1
C2
D2
Abbildung 6: Lokal optimale Alignierung von Knoten
Nun ist im ersten Baum der Knoten B1.2 mit dem Kindknoten C1 hinzugekommen und im zweiten Baum haben die Knoten B2.1 und B2.2
jetzt Kinder. Beginnt man nun eine naive Alignierung mit dem Knoten
B1.1 so ergibt sich für die Ähnlichkeit mit den Knoten B2.1 und B2.2
jeweils der selbe Wert, da B1.1 keine Kinder hat die mit den Kindern
der anderen zwei Knoten verglichen werden könnten. Wird B1.1 mit
dem erstbesten Knoten B2.1 aligniert, so ergibt sich dadurch für B1.1
eine lokal optimale Alignierung. Für B1.2 bleibt nun jedoch nur noch
der Knoten B2.2 übrig, der sich auf Kindebene jedoch unterscheidet,
wohingegen B2.1 maximal Ähnlich zu B1.2 gewesen wäre. Eine global
optimale Alignierung kann also durch lokal optimale Entscheidungen
nicht garantiert werden.
Das Beispiel macht deutlich, wie sich die beiden Aufgaben der Ähnlichkeitsbestimmung sowie der Alignierung gegenseitig beeinflussen, denn
ohne eine optimale Alignierung der Kindebene mit anschließender
Ähnlichkeitsbestimmung kann auch die Ähnlichkeit der Elternknoten
nicht korrekt bestimmt werden, was wiederum die Voraussetzung für
deren Alignierung ist etc. Im Folgenden gilt es also einen effizienten
Ansatz zu finden, der Ähnlichkeitbestimmung und Alignierung in der
geforderten Weise verbindet.
9
2.3
multiple sequence alignment
Betrachtet man wissenschaftliche Arbeiten, die sich mit dem Vergleich
von Bäumen beschäftigen, so fällt auf, dass Forschung in diese Richtung betrieben wird, dass diese Ansätze jedoch meist den paarweisen
Vergleich beschreiben (Yang, 1991) oder domänenspezifische Merkmale der Daten ausnutzen (Zhai & Liu, 2005). Andererseits ist das Multiple Sequence Alignment ein Kernproblem der Bioinformatik und dort
sehr gut erforscht, da sich hierdurch Muster in der Evolution einer Familie von Proteinen aufdecken lassen (Notredame, 2002, S. 1). Weil sich
die optimale Alignierung der Knoten von Bäumen mit der Alignierung
einer Sequenz von Elementen vergleichen lässt, deren jeweilige Ähnlichkeit zueinander sich wiederum rekursiv durch Alignierung weiterer Sequenzen ergibt, nämlich die der Kindebenen, lohnt es sich, diese
wissenschaftliche Disziplin näher zu betrachten.
2.3.1
MSA in der Bioinformatik
Notredame (2002) Beschreibt die Schwierigkeit, im Bezug auf das Multiple Sequence Alignment (MSA), als Kombination von drei technischen
Schwierigkeiten (Notredame, 2002, S. 2):
1. Die Wahl der zu vergleichenden Sequenzen
2. Der Entwurf einer Vergleichsfunktion
3. Die Optimierung dieser Funktion
Der erste Punkt ist ist vom Anwendungsfall abhängig und daher im
Rahmen des Entwurfs eines generischen Ansatzes pauschal nicht lösbar. Bezüglich der Vergleichsfunktion wurde bereits festgestellt, dass
diese, im Falle von Bäumen, wiederum auf einer Alignierung beruht.
Abgesehen davon muss für den Vergleich auf Typebene lediglich definiert werden, wie ähnlich sich die Typen untereinander sind, was im
einfachsten Falle bedeuten könnte, dass gleiche Typen maximale und
ungleiche Typen keine Ähnlichkeit aufweisen. Die eigentliche Herausforderung stellt der dritte Punkt dar. Notredame (2002) stellt fest, dass
eine mathematisch optimale Alignierung, von mehr als drei Sequenzen, zu komplex ist und beruft sich dabei auf Wang und Jiang (1994).
Aus diesem Grund seien auch alle derzeitigen MSA Ansätze heuristisch und in drei Kategorien unterteilbar: exakte, progressive sowie iterative Ansätze.
Exakte Alignierung
Exakte Algorithmen finden also nicht, wie der Name vermuten lässt,
die optimale Alignierung von mehreren Sequenzen. Ihre Ergebnisse
sind jedoch laut Notredame (2002) “[...] usually close to optimality”, also von hoher Qualität. Der Umfang der Anzahl der zu vergleichenden
Sequenzen sei hier jedoch nicht genau abgesteckt, ist jedoch mit unter
20 Sequenzen eher gering. Da die Qualität eines Prototypen vor allem
von einer ausreichend großen Trainingsmenge abhängt, erscheint eine exakte Alignierung aufgrund der damit verbundenen Komplexität
unmöglich.
Progressive Alignierung
Die progressive Alignierung basiert auf der Berechnung der optimalen
Alignierung einer begrenzten Zahl von Sequenzen, woraufhin diese
nach und nach Zusammengefasst werden. Diese Herangehensweise
hat den Vorteil, dass für die Berechnung einer beschränkten Anzahl
10
von Sequenzen, sehr genaue Algorithmen basierend auf dynamic programming eingesetzt werden können. Der Nachteil ist jedoch, dass die
Zusammenfassung der Sequenzen wiederum auf Basis lokaler Maxima geschieht. Dadurch ist nicht gewährleistet, dass die daraus abgeleitete globale Alignierung optimal ist.
Iterative Alignierung
Die dritte von Notredame (2002) identifizierte Kategorie ist die der iterativen Alignierungsstrategien. Hierbei wird versucht eine gefundene
Alignierung in einer Serie von Iterationsschritten zu verfeinern. Diese
Modifikationen basieren auf stochastischen Methoden, wie simulated
annealing, genetischen Algorithmen oder deterministischen Ansätzen,
wobei versucht wird die Alignierung von Sequenzen, die bereits Teil
einer einer multiplen Alignierung sind, immer weiter zu optimieren,
bis keine Verbesserung mehr möglich ist. Dadurch können lokale Maxima, wie sie bei progressiven Verfahren auftreten können, vermieden
werden.
2.3.2
Progressive Alignierung
Für die vorliegende Arbeit wird von einer progressiven Alignierungsstrategie ausgegangen, da diese in der Bioinformatik die am weitesten
verbreitete Methode ist (“Progressive alignments are by far the most
widely used.” (Notredame, 2002, S. 3)) und sich durch eine hohe Geschwindigkeit und eine akzeptable Sensitivität bezüglich der Daten
auszeichnet (“This approach has the great advantage of speed and
simplicity, combined with reasonable sensitivity” (Notredame, 2002)).
Darüberhinaus ist es sinnvoll, zunächst die Qualität eines einfachen
progressiven Ansatzes im strukturellen Data Mining zu evaluieren. Sollte das Ergebnis unbefriedigend sein, kann in Zukunft darauf aufbauend ein iteratives Verfahren entwickelt werden, das die durch einen
progressiven Ansatz gefundene Alignierung verfeinert.
Der erste progressive Algorithmus wurde von Hogeweg und Hesper
(1984) beschrieben. Den größten Teil des dort beschriebenen Ansatzes
macht die Konstruktion des phylogentischen Baumes aus, also der aus
den Ähnlichkeiten der Sequenzen rekonstruierte evolutionäre “Stammbaum” . Es wird betont dass die Alignierung von Sequenzen und die
Konstruktion phylogenetischer Bäume aufeinander Aufbauen würden,
da ohne die Rechtfertigung dieser integrierten Betrachtungsweise das
Konzept einer “guten Alignierungn” keine Substanz habe (Hogeweg
& Hesper, 1984, S. 1). In der Beschreibung von CLUSTAL W, einer Erweiterung dieses Ansatzes, wird dieser phylogenetische Baum auch
als guide tree (Thompson, Higgins & Gibson, 1994, S. 4674) bezeichnet,
da dieser die globale Alignierung “leitet”.
Da es hier darum geht, einen generischen Ansatz zu entwerfen, in
dem die Entstehung von Instanzen strukturierter Daten unabhängig
voneinander allein durch ihr gemeinsames Modell gelenkt wird, das
darüber hinaus als bekannt vorausgesetzt wird, kann dieser Aspekt
vernachlässigt werden. Ignoriert man diese domänenspezifische Anforderung, ergibt sich laut Hogeweg und Hesper (1984) als auch bei
Thompson et al. (1994) folgender Ablauf:
1. Alle Sequenzen werden paarweise aligniert wobei gleichzeitig
deren Ähnlichkeit zueinander (match value) berechnet wird.
2. Das zueinander ähnlichste Paar wird zusammengefasst und im
weiteren Verlauf wie eine Sequenz behandelt.
3. Dieser Vorgang wird solange wiederholt, bis alle Sequenzen zueinander aligniert wurden.
11
2.4
paarweise ähnlichkeit und alignierung
Im Bezug auf die paarweise Ähnlichkeit und Alignierung verweisen
Hogeweg und Hesper (1984) als auch Notredame (2002) auf dynamic
programming Algorithmen, wie den von Needleman und Wunsch (1970),
da diese für eine kleine Anzahl von Sequenzen eine mathematisch optimale Alignierung gewährleisten (Thompson et al., 1994, S. 4673).
Definition 1 Für den Needleman-Wunsch Algorithmus werden die folgenden Annahmen getroffen:
• Eine Sequenz A der Länge n bestehe aus den Elementen A1 , A2 , ..., An
• M(i,j) sei ein zweidimensionales Feld, dessen Werte über die Koordinaten (i,j) angesprochen werden
• s( Ai , Bj ) sei eine Funktion welche die lokale Ähnlichkeit zwischen A
und B als Zahlenwert zurückliefert
• g sei die gap penalty, also der Wert der einer Lücke entspricht
• l(A) sei eine Funktion welche die Länge einer Sequenz A zurückliefert
• max(a,b) sei eine Funktion die die größte von zwei Zahlenwerten a und
b zurückliefert
2.4.1
Needleman-Wunsch Algorithmus
Der von Needleman und Wunsch (1970) entwickelte Algorithmus ist
dem von Levenshtein (1966) entwickelten Algorithmus, zur Berechnung der Edit-Distanz, sehr ähnlich. Er stellt eine “[...] general method
applicable to the search for similarities in the amino acid sequence
of two proteins” dar, also eine Methode um zwei Proteine, d.h. Sequenzen von Aminosäuren, zu alignieren. In Algorithmus 1 ist eine
mögliche Umsetzung abgebildet.
Algorithm 1 Needleman-Wunsch Algorithmus
1:
2:
3:
4:
5:
6:
7:
8:
9:
10:
11:
12:
13:
14:
15:
16:
17:
18:
19:
procedure sequence_matching(A, B, g)
m ← l ( A)
n ← l ( B)
for (i ← 0, i < m) do
M(i, 0) ← i · g
end for
for (j ← 0, j < n) do
M(0, j) ← j · g
end for
for (i ← 0, i < m) do
for (j ← 0, j < n) do
match ← M(i − 1, j − 1) + s( Ai , Bi )
delete ← M (i − 1, j) + g
insert ← M (i, j − 1) + g
M(i, j) ← max(max(match, delete), insert)
end for
end for
return M (i, j)
end procedure
Die Alignierungsmatrix
Dabei werden in einer Alignierungsmatrix sowohl jede Paarung von
Elementen der beiden Sequenzen, als auch Lücken (gaps) berücksich-
12
tigt. Lücken entstehen dann, wenn ein Element Ai nicht mit dem Element Bi aligniert werden kann und stattdessen versetzt, beispielsweise
auf Bi+1 , abgebildet wird. Geht man davon aus dass A i Elemente und
B j Elemente besitzt, so muss die Matrix M i + 1 Zeilen und j + 1 Spalten aufweisen, da eine Sequenz auch auf eine leere Sequenz abgebildet
werden kann, indem lauter Lücken eingefügt werden.
Somit stellt diese Matrix alle möglichen Alignierungen zwischen
zwei Sequenzen dar, welche sich durch das Einfügen von Lücken beschreiben lassen, wodurch die Reihenfolge der Elemente nicht verändert wird. Eine Alignierung ist dabei ein Pfad durch diese Matrix beginnend bei M (0, 0) und endend bei M (i, j) (Needleman & Wunsch,
1970, S. 443-446).
Das Scoring
Die Werte in den Zellen ergeben sich aus vorher festgelegten scores für
die Alignierung zweier Elemente, bzw. für das Einfügen einer Lücke.
Da in der Bioinformatik die ähnlichen Motive zweier Proteine aligniert
werden sollen, wird das Einfügen einer Lücke mit einem panalty factor, also einem negativen score belegt. Das führt dazu, dass Lücken
nur dann eingefügt werden, wenn die Elemente anders nicht aligniert
werden können. Der gap score, bzw. penalty factor, wird hier mit der
variablen g bezeichnet. Da die Funktion, welche die Ähnlichkeit zweier Elemente bestimmt, nicht pauschal definiert werden kann, wird die
Funktion s( Ai , Bj ) vorausgesetzt, welche die Ähnlichkeit zweier Elemente A und B liefert. In der Bioinformatik geschieht das beispielsweise Anhand einer amino acid weight matrix (Thompson et al., 1994, S.
4675), die festlegt, wie ähnlich sich zwei Aminosäuren sind.
Berechnung der Ähnlichkeit
Algorithmus 1 stellt den Ansatz von Needleman und Wunsch (1970)
dar. In Zeile 4 bis 9 wird die Ähnlichkeitsmatrix vorbereitet, d.h. die
Zellen für das Szenario, dass jede Sequenz sukzessive mit der leeren Sequenz aligniert wird. In der darauffolgenden doppelten Schleife,
werden die übrigen Zellen, also M(0, 0) bis M (i, j), befüllt. Dabei werden Werte für drei Szenarien berechnet:
• Übereinstimmung von Ai mit Bj (match)
• Lücke in A einfügen (insert)
• Lücke in B einfügen (delete)
Da eine Übereinstimmung bedeutet, dass im vorherigen Schritt des
Alignierungspfades keine Lücke eingefügt wurde, repräsentiert somit
die von der aktuellen Zelle aus gesehen nord-westliche Nachbarzelle
den vorherigen Schritt. Der Wert für eine Übereinstimmung ergibt sich
aus der Summe der nord-westlichen Zelle mit der Ähnlichkeit von A
und B (s. Zeile 12). Ähnlich verhält es sich in den anderen beiden Szenarien. Eine Lücke kann auf zwei Arten entstehen: Entweder hat man
in Sequenz A eine Lücke einfügt, dann war der vorherige Schritt die
westliche Nachbarzelle (s. Zeile 14), oder aber die Lücke wurde in Sequenz B eingefügt, dann war es die nördliche Nachbarzelle (s. Zeile
13). Der Wert für eine Lücke errechnet sich aus der Summe der westlichen bzw. nördlichen Zelle und dem gap score g. Die optimale Strategie
für die aktuelle Zelle ergibt sich somit durch das Maximum der drei
Werte match, insert und delete. In Zeile 15 wird dieser Wert der Zelle
zugewiesen. Wurden auf diese Weise die Werte aller Zellen berechnet,
so befindet sich am Ende in Zelle M (i, j) ein Wert, der die Ähnlichkeit
der Sequenzen A und B, bei optimaler Alignierung, ausdrückt (s. Zeile
18).
13
Berechnung der Alignierung
Eine optimale Alignierung lässt sich nun rekonstruieren, indem aus
der Zelle M (i, j) der Pfad zurückverfolgt wird, der zu den jeweiligen
Maxima geführt hat. Da dies aus der reinen Information der Zellenwerte nicht rekonstruierbar ist, muss hierzu bereits bei der Ermittlung
der Zellenwerte für jede Zelle gespeichert werden, ob das Maximum
aus einer Übereinstimmung von A und B und/oder einer Lücke in
A und/oder einer Lücke in B entstammt. Um den Algorithmus nicht
zu kompliziert zu gestalten, wurde dieser Schritt bewusst außen vor
gelassen und wird in einem späteren Kapitel behandelt.
Beispiel
Der Ablauf des Algorithmus lässt sich gut an einem kurzen Beispiel
erklären. Gegeben sei:
• Sequenz A = A,D,E
• Sequenz B = B,D,F
• s(A,B) liefert 1 für gleiche Zeichen, ansonsten -2
• Der gap score ist -1
Zunächst werden die Matrizen M, für das Scoring, und T für die Rekonstruktion einer optimalen Alignierung (Backtracking Matrix) vorbereitet, wie in Abbildung 7 zu sehen ist.
M
#
A
D
E
#
0
−1
−2
−3
B
−1
D
−2
F
−3
T
#
A
D
E
#
B
←
D
←
F
←
↑
↑
↑
Abbildung 7: Initialzustand der Matrizen
In Abbildung 8 wurden bereits einige Werte ermittelt. Im Falle von D
und B ergibt sich für die Ähnlichkeit s(’B’,’D’) = -2. In diesem Falle
sind die Werte für eine Lücke in A oder B sowie für eine Abbildung
von ’B’ auf ’D’ gleich:
• Übereinstimmung: -1 + (-2) = -3
• Lücke in Sequenz A: -2 + (-1) = -3
• Lücke in Sequenz B: -2 + (-1) = -3
Daher wird an der entsprechden Stelle in der Backtracking Matrix gespeichert dass ein optimaler Alignierungspfad an dieser Stelle nach
Norden, Nord-Westen oder Westen fortgesetzt werden kann.
M
#
A
D
E
#
0
−1
−2
B
−1
−2
−3
D
−2
−3
F
−3
−4
T
#
A
D
E
#
↑
↑
B
←
←-↑
←-↑
D
←
←-↑
Abbildung 8: Alternative Pfade einer Alignierung
14
F
←
←-↑
In der nächsten Zelle stimmen die Zeichen aus beiden Sequenzen überein. s(’D’,’D’) = 1 und somit ergeben sich folgende Werte:
• Übereinstimmung: -2 + 1 = -1
• Lücke in Sequenz A: -3 + (-1) = -4
• Lücke in Sequenz B: -3 + (-1) = -4
Das Maximum ist hier durch eine Alignierung des Zeichens ’D’ aus
A mit ’D’ aus B zu erreichen. Daher wird in der Backtracking Matrix
lediglich der Pfad nach Nord-Westen gespeichert.
M
#
A
D
E
#
0
−1
−2
B
−1
−2
−3
D
−2
−3
−1
F
−3
−4
T
#
A
D
E
#
↑
↑
B
←
←-↑
←-↑
D
←
←-↑
-
F
←
←-↑
Abbildung 9: Matching zweier Sequenzelemente
Wird der Algorithmus bis zum Ende ausgeführt erhält man die in Abbildung 10 dargestellten Matrizen. Beim Backtracking können sich alternative Pfade ergeben, deshalb sind die Zellen die in Frage kommen
grau hinterlegt.
M
#
A
D
E
#
0
−1
−2
−3
B
−1
−2
−3
−4
D
−2
−3
−1
−2
T
#
A
D
E
F
−3
−4
−2
−3
#
↑
↑
↑
B
←
←-↑
←-↑
←-↑
D
←
←-↑
↑
F
←
←-↑
←
←-↑
Abbildung 10: Fertig errechnete Matrizen
Das bedeutet das folgende Alignierungen gleichsam optimal sind:
-BD-F
A-DE-
-BDF
A-DE
BD-F
ADE-
BDF
ADE
Daran lässt sich gut erkennen, dass die Alignierung stark von der verwendeten Ähnlichkeitsfunktion s( A, B) und dem gewählten gap score
abhängt. Hätte man für die Alignierung nicht identischer Zeichen, eine solche Entscheidung mit einem hohen Abzug “bestraft”, so wären
die alternativen Pfade, welche die Alignierung von ’A’ und ’B’ bzw. ’E’
und ’F’ zulassen, nicht entstanden.
15
2.4.2
Yangs Tree-Matching-Algorithmus
Yang (1991) erweiterte den Algorithmus von Needleman und Wunsch
(1970) im Rahmen eines Ansatzes, um syntaktische Unterschiede zwischen zwei Versionen eines Programmes zu identifizieren. Die zwei
Versionen werden dabei als Parse-Tree dargestellt. Bei diesem Ansatz
soll zusätzlich Wissen über die Grammatik der Programmiersprache
zu besseren Ergebnissen verhelfen (“[...] we develop a comparison algorithm that exploits knowledge of the grammar” (Yang, 1991, S. 739)).
Ausgangspunkt hierbei ist ein dynamic programming Algorithmus von
Hirschberg (1977) der jedoch vom Prinzip her dem von Needleman
und Wunsch (1970) in der Darstellung von Algorithmus 1 entspricht.
Es fällt auf, dass es keine gap penalty gibt, da das Einfügen von
Lücken beim Vergleich zweier Programmversionen unkritisch, ja notwendig ist, da hier, im Gegensatz zur Evolution von Proteinsequenzen, von einer Programmversion auf die andere drastische Änderungen möglich sind.
Rekursion
Um Bäume vergleichen zu können, wird der Algorithmus 1 um einen
rekursiven Aufruf in Zeile 15 in Algorithmus 2 erweitert. Dadurch
wird die Ähnlichkeit der Elternknoten durch die Ähnlichkeit ihrer Kinder beeinflusst.
Vergleichbarkeit
Desweiteren führt Yang (1991) den interessanten Aspekt der Vergleichbarkeit (comparability) ein da er feststellt, dass der Vergleich zweier
Knoten nur dann stattfinden könne , wenn deren Elternknoten übereinstimmen (“[...] two nodes can match only if their parents contain
identical symbols” (Yang, 1991, S. 747)). Dabei könne es jedoch erwünscht sein die Kinder vergleichbarer Knoten ebenfalls aufeinander
abzubilden (“[...] we may want to match two nodes when their parents
contain ’comparable’ but not identical tokens” (Yang, 1991, S. 747).
Im Algorithmus 2 sieht man in Zeile 2 dass zwei Knoten nur dann
verglichen werden, wenn diese auch vergleichbar sind, jedoch tragen
diese nur zum score ihrer Kindknoten bei, wenn sie identisch sind (s.
Zeile 21 f.)
Definition 2 Für Yangs Tree-Matching Algorithmus müssen folgende Annahmen getroffen werden:
• Ein Knoten A habe die Kindknoten A0 , A1 , ..., An−1
• M(i,j) sei ein zweidimensionales Feld, dessen Werte über die Koordinaten (i,j) angesprochen werden
• cmp(A,B) sei eine Funktion, die 1 liefert, sofern A und B vergleichbar
sind, ansonsten aber 0 zurückliefert
• eq(A,B) sei eine Funktion, die 1 liefert, sofern A und B identisch (maximal ähnlich) sind, ansonsten aber 0 zurückliefert
• c(A,i) sei eine Funktion, die den i-ten Kindknoten des Knotens A liefert
• lc(A) sei eine Funktion, die die Anzahl der Kindknoten des Knotens A
liefert
16
Algorithm 2 Yangs Tree-Matching-Algorithmus
1:
2:
3:
4:
5:
6:
7:
8:
9:
10:
11:
12:
13:
14:
15:
16:
17:
18:
19:
20:
21:
22:
23:
24:
25:
26:
procedure tree_matching(A, B)
if cmp(A,B) = 0 then
return 0
end if
m ← lc( A)
n ← lc( B)
for (i ← 0, i < m) do
M(i, 0) ← 0
end for
for (j ← 0, j < n) do
M(0, j) ← 0
end for
for (i ← 0, i < m) do
for (j ← 0, j < n) do
match ← M(i − 1, j − 1) + tree_matching(c( A, i ), c( B, j))
delete ← M (i − 1, j)
insert ← M (i, j − 1)
M(i, j) ← max(max(match, delete), insert)
end for
end for
if eq(A,B)=1 then
return M (i, j) + 1
else
return M (i, j)
end if
end procedure
17
3
DESIGN
Mit den Erkenntnissen aus der Analyse lässt sich ein instanzbasierter Ansatz, zur Prototypenextraktion aus strukturierten Daten, formulieren. Dazu werden zunächst die verwendeten Datenstrukturen und
Funktionen definiert. Anschließend wird der gesamte Ablauf der Extraktion in Form von Algorithmen dargestellt.
3.1
definitionen
Um eine einheitliche Basis zur Beschreibung der zu entwickelnden
Verfahren zu schaffen, bedarf es einiger Definitionen.
3.1.1
Datenstrukturen
Zunächst müssen die verwendeten Datenstrukturen, sozusagen als Infrastruktur für die Prototypenextraktion, definiert werden. Grundsätzlich liegt hier der Betrachtung der Datenstrukturen die Metapher des
Objekts zugrunde. Ein Objekt stellt der Umwelt den Zugriff auf dessen
Attribute und Methoden zur Verfügung. Zur Darstellung der Attribute
und Methoden eines Objekts wird der Punkt-Operator “.” verwendet.
Auf das Attribut attr des Objekts o würde somit durch die Schreibweise o.attr verwiesen. Die Schreibweise einer Methode unterscheidet sich
von der eines Attributs durch die in Klammern gesetzte Argumentliste. Der Ausdruck o. f unc( arg1) stellt somit einen Aufruf der Funktion
func(), des Objekts o, mit dem Argument arg1 dar.
Element
Ein Element ist, im vorliegenden Kontext, ein Träger sachbezogener Informationen. Als solches ist es Teil einer Datenstruktur, die diese Elemente in Bezug zueinander setzt. In einer Datenstruktur sind somit die
reinen Strukturinformationen gespeichert. Generell würde man zwar
auch bei dem Element von einer Datenstruktur sprechen, jedoch ist die
Struktur seiner Informationen unveränderlich, so dass aus Sicht der
Prototypenextraktion diese Struktur keinen Informationsgehalt hat. Somit kann das Element als strukturell atomar angesehen werden.
Definition 3 Ein Element e sei ein Träger sachbezogener Informationen. Als
solches sei es eine Instanz des Typen τ.
Prototypelement
Ein aus strukturellen Daten gewonnener Prototyp ist nicht nur ein Repräsentant der dominierenden Elementausprägungen, sondern bildet
auch strukturelle Gemeinsamkeiten ab. Auch hier können wieder sachbezogene von strukturellen Informationen getrennt werden.
18
Definition 4 Ein Prototypelement pe bilde die gemeinsamen Merkmale einer
Menge von Elementen ab. Der Typ des Prototypelements sei PE.
• pe bildet die sachbezogenen Informationen eines Elements in seinem
Label pe.lbl ab
– pe.lbl ∈ D = {d/ d ist ein Zeichen des lateinischen Alphabets,
inkl. Sonderzeichen}
• Die relative Häufigkeit, des durch pe repräsentierten Elements und damit das Gewicht des dadurch beschriebenen Aspekts, ist die Ausprägung des Attributs pe.f
– pe. f ∈ Q
Liste
Die Liste ist eine wichtige Basisdatenstruktur, z.B. um 1 : n Relationen
abzubilden. Daher ist sie als Attribut oft Teil anderer Datenstrukturen.
Definition 5 Eine Liste l ← {} mit den Elementen e0 , ..., en schreibe sich
{e0 , ..., en }. Der Typ der Liste sei L.
• {} sei die leere Liste
– e : {} → L
• l.add(e) füge der Liste l das Element e hinzu
– L.add : τ → L
– {}.add(e) = {e}
• l.remove(e) entferne das Element e aus der Liste l
– L.remove : τ → L
– {e}.remove(e) = {}
• l.length sei die Länge der Liste l
– L.length ∈ N0
– {}.length = 0
– {e1 , ..., en }.length = n
• l.get(i ) liefere das i-te Element der Liste l
– L.get : N0 → τ
– {e1 , ..., en }.get(n) = en
• l.contains(e) überprüfe ob die Liste l das Element e enthält
– L.contains : τ → {>, ⊥}
– {e1 , ..., en }.contains(e1 ) = >
Baum
Es wird davon ausgegangen, dass sich strukturierte Daten in Form
eines Baumes darstellen lassen. Dabei kapselt jeweils ein Knoten ein
informationstragendes Element. Ein Baum lässt sich rekursiv beschreiben, deshalb genügt die Definition eines Baumknotens zur Beschreibung eines Baumes.
19
Definition 6 Ein Baum setze sich aus Baumknoten zusammen. Der Typ eins
Baumknotens und somit des ganzen Baumes sei T.
• Ein Baumknoten t kapsle ein Element in seinem Attribut t.e
– T.e ∈ τ
• Jeder Baumknoten habe einen Elternknoten t.t p , ein Wurzelknoten verweise hierbei auf das leere Element t.t p ← e
– T.t p ∈ T
• Jeder Knoten habe eine Liste von 0 bis n Kindknoten t.lt ← {t1 , ..., tn }
– T.lt ∈ L
Sequenz
Als Basis der Alignierungsverfahren dient die Sequenz. Eine Sequenz
hat 1 bis n Positionen, denen jeweils wiederum 1 bis n Sequenzelemente
zugeordnet sind. Dabei drückt die Zuordnung mehrerer Sequenzelemente zu einer Position deren Alignierung an dieser Position aus. Jedes Sequenzelement kapselt wiederum ein Element. Mit der in der gemeinsamen Position enthaltenen Information alleine könnte bereits eine paarweise Alignierung vorgenommen werden. Um jedoch aus den
paarweisen, also lokalen Alignierungen eine gültige globale Alignierung
der Sequenzen abzuleiten, werden zusätzlich die Informationen benötigt, aus welcher Sequenz ein Sequenzelement ursprünglich stammt
und mit welchen anderen Sequenzelementen es potentiell aligniert
werden kann.
Sequenz
A1
B1
A3
B2
Position
Sequenzelement
Abbildung 11: Komponenten einer Sequenz
20
Definition 7 Eine Sequenz setze sich aus Positionen zusammen, denen wiederum Sequenzelemente zugeordnet seien. Die Sequenz habe den Typen T, die
Position den Typen P und das Sequenzelement sei vom Typ SE.
• Eine Sequenz s hat 1 bis n Positionen s.l p ← { p1 , ..., pn }
– S.l p ∈ L
• Für eine Position p gilt:
– Ihr können 1 bis n Sequenzelemente
p.lse ← {se1 , ..., sen } zugeweisen werden
* P.lse ∈ L
– Sie gehört zu einer Sequenz p.s
* P.s ∈ S
• Für ein Sequenzelement se gilt:
– Es kapselt ein Element se.e
* SE.e ∈ τ
– Es ist mit 0 bis n weiteren Sequenzelementen
se.lse ← {se1 , ..., sen } assoziiert
* SE.lse ∈ L
– Es ist einer Position se.p zugeordnet
* SE.p ∈ P
– Es stammt ursprünglich aus einer Sequenz se.s
* SE.s ∈ S
3.1.2
Funktionen
Zum Verständnis der im folgenden präsentierten Algorithmen, werden
einige Funktionen vorausgesetzt, deren Funktionen lediglich deklarativ beschrieben werden, da es sich dabei nicht um Kernaufgaben des
zu entwickelnden Ansatzes handelt.
Instanziierung von Typen
Um die im vorhergehenden Abschnitt definierten Datentypen im Kontext eines Algorithmus verwenden zu können, muss es eine Möglichkeit geben diese zu instantiieren.
Definition 8 Die Funktion create(τ) erzeuge eine Instanz des Typen τ.
Sollte sich dieser Datentyp aus weiteren Datentypen zusammensetzen,
so gelten diese als ebenfalls instanziiert. Für Listen gelte die abgekürzte
Schreibweise {}
create : τ → τ
create( L) → {}
Lokale Ähnlichkeit
Wie schon in der Analyse erwähnt, kann keine Funktion pauschal bestimmt werden, die die lokale Ähnlichkeit zweier Instanzen eines generischen Typs τ zurückgibt. Daher wird die Funktion sim(τ, τ ) als
gegeben angenommen.
21
Definition 9 Die Funktion sim(τ, τ ) liefere die lokale Ähnlichkeit zweier
Instanzen des Typen τ als rationale Zahl zwischen 0.0 und 1.0 zurück.
• 1.0 sei die maximale Ähnlichkeit
• Bei Werten > 0.0 gelten die Instanzen als ähnlich
• 0.0 bedeute die Instanzen sind minimal ähnlich, also nicht vergleichbar
sim : τ × τ → Q
Sortieren
Da bei der globalen Alignierung die Ordnung der Knoten verloren
geht, ist es nötig diese, durch Sortierung der Knoten von Prototypenelementen einer Baumebene, wiederherzustellen. Wie bei der Ähnlichkeit ist die Ordnung der Elemente dabei wieder vom konkreten Anwendungsfall abhängig. Daher wird von einer Funktion sort(ltpe ←
{t pe1 , ..., t pen }) ausgegangen, die eine Liste von Knoten, deren Elemente Prototypenelemente sind, sortiert.
Definition 10 Die Funktion sort(ltpe ← {t pe1 , ..., t pen }) sortiere eine Liste
von Baumknoten, deren Elemente Prototypelemente sind.
sort : L → L
Labeling
Die Erzeugung eines Prototypen aus einer Menge alignierter Elemente, setzt eine Strategie voraus, in der definiert ist, wie die relevanten
Informationen aus Elementen vom Typ τ gewonnen werden können
und wie diese im Prototypen repräsentiert werden.
Definition 11 Die Funktion label_proto(p,lτ ) setze das Label p.lbl eines
Prototyps p entsprechend den Elementen einer Liste lτ .
label_proto : P × L → e
3.2
prototypextraktion
In der Analysephase wurde festgestellt, dass beide dort genannten Algorithmen au und sdtg auf eine Möglichkeit, zur Alignierung der Kindknoten zweier Knoten, angewiesen sind. Der Aspekt der Alignierung
lässt sich also getrennt von der Erzeugung der Prototypen betrachten.
3.2.1
Einstiegspunkt
Zunächst wird die Funktion extract_prototype() (s. Algorithmus 3)
als Einstiegspunkt definiert, in der lediglich die Rekursion vorbereitet
wird. extract_prototype() erwartet eine Liste von Baumknoten. Da der
rekursive Extraktionsvorgang im folgenden auf Listen von Listen von
Baumknoten, also auf den Kindknoten eines Knotens, arbeitet, wird
die initiale Liste von Wurzelknoten in den Zeilen 3 bis 5 in diese Form
gebracht. Anschließend beginnt der rekursive Abstieg in die Baumstrukturen durch Aufruf der Funktion process_children() mit der angepassten Liste. Da davon ausgegangen wird, dass die Bäume sich in
ihrer Wurzel nicht unterscheiden, wird bei der Prototypextraktion nur
ein Prototyp entstehen. Daher wird ab Zeile 6 gezielt der erste Prototyp zurückgegeben.
22
Algorithm 3 Einstiegspunkt der Prototypenextraktion
1:
2:
3:
4:
5:
6:
7:
8:
function extract_prototype(lt ← {t1 , ..., tn })
ltn ← {}
for all t ∈ lt do
ltr .add({t})
end for
t p ← process_children(ltn , lt .length, lt .length).get(1)
return t p
end function
3.2.2
Rekursion
Da im Zuge der rekursiven Prototypisierung globalen Informationen
über den gesamten Baum transportiert werden müssen, wird an process_children(),
zusätzlich zur Liste von Listen von Knoten llt , die Anzahl der Bäume
insgesamt tcount , sowie die Anzahl der Elternknoten t pcount der vorherigen Ebene übergeben.
Die Funktion process_children(), dargestellt in Algorithmus 4, aligniert zunächst in Zeile 2 die übergebenen Listen von Baumebenen
mit Hilfe der Funktion align_progressive(). Der Rückgabewert dieser
Funktion ist wiederum eine Liste von Listen, aber diesmal mit anderer Semantik: Die inneren Listen stellen hierbei alignierte Knoten dar.
Daher wird in Zeile 4 bis 7 mit proto_strategy() der Prototyp von jeder Liste alignierter Knoten erzeugt. Hierfür wird wiederum die Anzahl der Bäume, sowie die Anzahl der Elternknoten, weitergegeben.
Die Prototypen werden wiederum in einer Liste gesammelt, welche
schließlich den Rückgabewert der Funktion process_children() bildet.
Algorithm 4 Rekursive Prototypenextraktion
1:
2:
3:
4:
5:
6:
7:
8:
9:
function process_children(llt ← {lt1 , ..., ltn }, tcount , t pcount )
llta ← align_progressive(llt )
ltpe ← {}
for all lta ∈ llta do
t pe ← proto_strategy(lta , tcount , t pcount )
ltpe .add(t pe )
end for
return sort(ltpe )
end function
3.2.3
Prototypisierungsstrategie
Da sich die Anti-Unifikation und das SDTG Verfahren lediglich in der
Art und Weise unterscheiden, wie der Prototyp aus einer Menge alignierter Knoten extrahiert wird, kann auch diese Logik in einer separaten Funktion untergebracht werden. Diese Trennung ist von Vorteil, um in Zukunft verschiedene Ansätze objektiv miteinander vergleichen zu können. Die folgenden Funktionen proto_strategy_au() sowie
proto_strategy_sdtg() stellen daher zwei Prototypisierungsstrategien
dar, die in Algorithmus 4 in Zeile 5 verwendet werden können.
Anti-Unifikation
In der Prototypisierungsstrategie proto_strategy_au() (s. Algorithmus
5) wird gleich in Zeile 2 getestet, ob die Anzahl der alignierten Knoten mit der Anzahl tcount der Bäume übereinstimmt. Ist dies nicht der
Fall, so wird das leere Element e ausgegeben, da nur Elemente, die in
allen anderen Bäumen eine Entsprechung haben, Teil des Prototypen
werden.
23
In den Zeilen 5 bis 8 wird ein neues Prototypelement erzeugt. Dabei
wird in Zeile 6 die Funktion label_proto () aufgerufen um das Label des
Prototypen, anhand der Merkmale der alignierten Knoten aus Liste lt ,
zu setzen. Anschließend wird ein neuer Baumknoten erzeugt und das
Prototypelemen als dessen Element gesetzt.
In den Zeilen 9 bis 12 wird zunächst eine Liste erzeugt, der anschließend alle Listen mit Kindknoten der alignierten Knoten hinzugefügt
werden. Danach werden die Prototypelementknoten der Kinder, durch
rekursiven Aufruf der Funktion process_children(), extrahiert und dem
soeben erzeugten Prototypknoten als Kindknoten angehängt (s. Zeile 12). Dabei wird wiederum die Gesamtanzahl der Bäume weitergereicht. Die Anzahl der Elternknoten, für die Ebene ihrer Kinder, ist
jetzt lt .length, also die Anzahl der alignierten Knoten.
Algorithm 5 Prototypingstrategie “Anti-Unifikation”
1:
2:
3:
4:
5:
6:
7:
8:
9:
10:
11:
12:
13:
14:
15:
function proto_strategy_au(lt ← {t1 , ..., tn }, tcount , tpcount )
if lt .length < tcount then
return e
end if
pe ← create( PE)
label_proto( pe, lt )
t p ← create( T )
t p .e ← pe
ltc ← {}
for all t ∈ lt do
ltc .add(t.lt )
end for
if ltc .length > 0 then
t p .lt ← process_children(ltc , tcount , lt .length)
end if
return t p
17: end function
16:
Structure Dominance Tree Generalisation
Die Structure Dominance Tree Generalisation (SDTG) unterscheidet sich
nur an wenigen Punkten von der Anti-Unifikation. Gleich zu Beginn
fällt auf, dass es hier keine Abbruchbedingung wie in Algorithmus
5 gibt, da ein SDTG-Prototyp keine Schnittmenge, sondern vielmehr
eine gewichtete Vereinigungsmenge der Bäume darstellt.
Die Erzeugung des Prototypelementknotens in den Zeilen 2 bis 6
ist identisch mit der der Anti-Unifikation, bis auf die Tatsache, dass
in Zeile 4, die relative Häufigkeit des Auftretens eines Elements, zur
Anzahl der Elternknoten, im Prototypelement gespeichert wird. Der
weitere Verlauf entspricht wieder dem der Anti-Unifikation.
24
Algorithm 6 Prototypingstrategie “SDTG”
1:
2:
3:
4:
5:
6:
7:
8:
9:
10:
11:
12:
13:
14:
15:
3.3
function proto_strategy_sdtg(lt ← {t1 , ..., tn }, tcount , tpcount )
pe ← create( PE)
label_proto( pe, lt )
l .length
pe. f ← ttpcount
t p ← create( T )
t p .e ← pe
ltc ← {}
for all t ∈ lt do
ltc .add(t.lt )
end for
if ltc .length > 0 then
t p .lt ← process_children(ltc , tcount , lt .length)
end if
return t p
end function
progressive alignierung
Wie sich herausstellt, verbirgt sich die meiste Logik in der progressiven
Alignierung. Bereits in der Analyse wurde dieses Verfahren vorgestellt,
das aus einem hierarchischem Clusteringverfahren und einem dynamic
programming Algorithmus, zur Berechnung der paarweisen Ähnlichkeit und Alignierung zweier Sequenzen, besteht. Das konkrete Vorgehen wird nun in Algorithmus 7 dargestellt.
Zunächst werden in den Zeilen 2 bis 16 die Listen von Baumknoten
in Form Sequenzen überführt. Dazu wird in Zeile 4 für jede Liste eine neue Sequenz erzeugt. Die Einzelnen Elemente der Liste, also die
Baumknoten, werden dabei von jeweils einem in Zeile 6 neu erzeugtem Sequenzelement gekapselt (s. Zeile 7). Dem Sequenzelement wird
in Zeile 8 die Sequenz s als Ursprungssequenz se.s zugewiesen. Daraufhin wird in Zeile 9 eine neue Position erzeugt, diese wird mit der
neu erzeugten Sequenz verknüpft (s. Zeile 10 und 11). Schließlich wird
auch die Position mit dem Sequenzelement in beide Richtungen verknüpft (s. Zeilen 12 bis 13). Jede so erzeugte Sequenz wird der Liste ls
hinzugefügt.
Im Zuge des hierarchischen Clusterings der Sequenzen in der Funktion clusterh() wird bei der Berechnung der paarweisen Ähnlichkeiten
gleichzeitig die Assoziation zwischen den Sequenzelementen hergestellt. Diese Assoziationen werden anschließend in der Funktion merge() genutz, um die Sequenzen zu vereinigen und somit eine globale
Alignierung herzustellen.
In den Zeilen 19 bis 26 wird die global alignierte Sequenz wieder in
eine Listendarstellung gebracht. Dazu wird für jede Position eine Liste
erstellt (s. Zeile 21), die alle Baumknoten aufnimmt (s. Zeile 23). Diese
Liste von Listen von alignierten Baumknoten wird schließlich in Zeile
27 zurückgegeben.
25
Algorithm 7 Einstiegspunkt der progressiven Alignierung
1:
2:
3:
4:
5:
6:
7:
8:
9:
10:
11:
12:
13:
14:
15:
16:
function align_progressive(llt ← {lt1 , ..., ltn })
ls ← {}
. Liste von Baumknoten in Sequenz umwandeln
for all lt ∈ llt do
s ← create(S)
for all t ∈ lt do
se ← create(SE)
se.e ← t
se.s ← s
p ← create( P)
s.l p ← { p}
p.s ← s
p.lse ← {se}
se.p ← p
end for
ls .add(s)
end for
llsa ← clusterh(ls ) . Clustering und paarweise Assoziierung
s a ← merge(llsa )
. Globale Alignierung
17:
18:
19:
20:
21:
22:
23:
24:
25:
26:
27:
28:
llt ← {} . Sequenz alignierter Positionen in Liste umformen
for all p ∈ s a .l p do
lt ← {}
for all se ∈ p.lse do
lt .add(se.e)
end for
llt .add(lt )
end for
return llt
end function
3.3.1
Hierarchisches Clustering
Das von Hogeweg und Hesper (1984) beschriebene progressive Verfahren, lässt sich direkt als hierarchisches Clustering von Sequenzen
darstellen, wobei als Distanzmaß die Ähnlichkeit der Sequenzen herangezogen wird.
Die Funktion clusterh() implementiert ein hierarchisches Clusteringverfahren und erwartet eine Liste von Sequenzen. Zunächst wird eine
Liste lclu erzeugt, welche die einzelnen Cluster enthalten wird. Da zu
Beginn jede Sequenz einen eigenen Cluster darstellt, werden diese jeweils einer Liste hinzugefügt, die wiederum der Liste lclu angehängt
werden (s. Zeile 2 bis 5).
Da das Clustering erst dann fertig ist, sobald alle Cluster zu einem
verschmolzen wurden, terminiert die Schleife in Zeile 6 erst, sobald die
Liste der Cluster lclu nur noch ein Element enthält. In jedem Schleifendurchlauf wird sodann die Distanz jeder Paarung von Clustern mittels
avg_linkage() berechnet. Das Cluster-Paar mit der geringsten Distanz,
also der größten Ähnlichkeit, wird schließlich in der Zeile 22 in einem
neuen Cluster vereint und die vorher eigenständigen Cluster werden
entfernt. Den Rückgabewert der Funktion clusterh() bildet somit der
vereinigte Cluster.
26
Algorithm 8 Hierarchisches Clustering
function clusterh(ls ← {s1 , ..., sn })
lclu ← {}
3:
for all s ∈ ls do
4:
lclu .add({s})
5:
end for
1:
2:
while lclu .length > 1 do
max_score ← −1
lbest a ← e
lbestb ← e
6:
7:
8:
9:
for i ← 1, lclu .length do
for j ← i + 1, lclu .length do
10:
11:
la ← lclu .get(i )
lb ← lclu .get( j)
curr_score ← avg_linkage(la , lb )
if curr_score > max_score then
max_score ← curr_score
lbest a ← la
lbestb ← lb
end if
12:
13:
14:
15:
16:
17:
18:
19:
end for
end for
20:
21:
22:
23:
24:
25:
26:
27:
lclu .add({lbest a , lbestb })
lclu .remove(lbest a )
lclu .remove(lbestb )
end while
return lclu .get(1)
end function
3.3.2
Berechnung der Clusterdistanz
In Zeile 14 des Algorithmus 8 wird die Distanz zwischen zwei Clustern mit Hilfe der Funktion avg_linkage (s. Algorithmus 9) berechnet.
Gleich zu Beginn werden die beiden übergebenen Cluster in eine flache Darstellung gebracht. In der Schleife von Zeile 5 bis 9 werden die
einzelnen Sequenzen der beiden Cluster miteinander über die Funktion htm() verglichen (s. Zeile 7). Die Ähnlichkeiten werden alle in der
Variablen score aufsummiert. Der Rückgabewert ist der auf die Anzahl
der Vergleichsoperationen normierte score.
27
Algorithm 9 Berechnung der Distanz zweier Cluster
1:
2:
3:
4:
5:
6:
7:
8:
9:
10:
function avg_linkage(la , lb )
la f ← flatten_cluster(la , {})
lb f ← flatten_cluster(lb , {})
score ← 0
for all s a ∈ la f do
for all sb ∈ lb f do
score ← score + htm(s a , sb )
end for
end for
score
return l .length
·l .length
af
bf
11:
end function
12:
function flatten_cluster(lc , l f )
if lc .length = 1 then
l f .add(lc .get(1))
return l f
end if
flatten_cluster(lc .get(1), l f )
flatten_cluster(lc .get(2), l f )
return l f
end function
13:
14:
15:
16:
17:
18:
19:
20:
3.4
alignierung
Der nächste Schritt in Algorithmus 7 nach dem hierarchischen Clustering der Sequenzen in Zeile 17 ist die Ableitung der globalen Alignierung der Sequenzen zueinander durch die Funktion merge().
3.4.1
Erzeugen der Vereinigungssequenz
Diese Funktion lässt sich gut rekursiv gestalten, da das Ergebnis der
Vereinigung zweier Cluster stets eine Liste mit zwei Elementen ist (s.
Zeile 22 Algorithmus 8). Nur die ursprünglichen Cluster, also die Blätter des Dendrogramms, bestehen lediglich aus einem Element. Diese
Tatsache bildet zugleich die Voraussetzung für die Abbruchbedingung
der Rekursion in den Zeilen 1 bis 4 des Algorithmus 10: Ist die Rekursion in den Blättern angekommen, so wird der Inhalt dieses Clusters,
also das Element, ausgegeben und die Funktion arbeitet sich von den
Blättern zur Wurzel (bottom up) durch den gesamten Cluster.
Zunächst wird in Zeile 7 eine neue Sequenz sm erstellt, die am Ende
die Vereinigungssequenz von s a und sb enthalten soll. In der äußeren
Schleife von Zeile 8 bis 17 wird für jede Position p a der Sequenz s a
eine neue Position pm erzeugt und mit der neuen Sequenz assoziiert
(s. Zeile 9 bis 12). In der inneren Schleife von Zeile 13 bis 16 werden
schließlich die Sequenzelemente von s a an die neue Position in sm gesetzt. Bildlich gesprochen “hängen” die Sequenzelemente der Sequenz
s a nun nicht mehr an den Positionen von s a , sondern an den Positionen
von sm . Die einfachen Referenzen von den Positionen der Originalsequenz s a zurück auf die Sequenzen bleiben jedoch erhalten.
Anschließend werden die Sequenzelemente aus s a mit denen aus
sb anhand ihrer Assoziationen aligniert. Dazu wird auf die Funktion
align_assoc() verwiesen. Sollten dabei Sequenzelemente in sb keine Assoziation zu s a haben, so werden diese letzendlich mit Hilfe der Funktion add_orphans() an die gemeinsame Sequenz angehängt.
28
Algorithm 10 Globale Alignierung der Sequenzen
1:
2:
3:
4:
5:
6:
7:
8:
9:
10:
11:
12:
13:
14:
15:
16:
17:
18:
19:
20:
21:
function merge(llc ← {lc1 , ..., lcn })
if llc .length = 1 then
return llc .get(1)
end if
s a ← merge(llc .get(1))
sb ← merge(llc .get(2))
sm ← create(S)
. neue Sequenz erzeugen
for all p a ∈ s a .l p do
. s a Positionen kopieren
pm ← create( P)
sm .l p .add( pm )
pm .s ← sm
pm .lse ← {}
for all sea ∈ p a .lse do . Elemente an neue Position setzen
sea .p ← pm
pm .lse .add(sea )
end for
end for
align_assoc(sm , s a , sb )
add_orphans(sm , sb )
return sm
end function
3.4.2
Alignierung assoziierter Sequenzelemente
Die Umschließende Schleife in Zeile 2 bis 27 der Prozedur align_assoc()
(Algorithmus 11) iteriert nun über alle Sequenzelemente von s a . Die aktuelle Position (in der neuen gemeinsamen Sequenz sm ) eines Sequenzelements sea wird in Zeile 4 zunächst einer Liste p pool hinzugefügt,
die als Pool möglicher Positionen für die mit sea direkt und indirekt
assoziierten Sequenzelemente dient.
Die folgende Schleife iteriert nun über alle direkt mit sea assoziierten Sequenzelemente. Sind diese nicht bereits einer Position in der
neuen Sequenz zugewiesen worden (s. Zeile 7), so könnten sie (se peerd )
und die Sequenzelemente, die sich jeweils eine Position mit ihnen teilen (se peerd .p.lse ), potentiell in der neuen Sequenz mit Sequenzelement
sea aligniert werden. Anschließend wird im Pool möglicher Positionen
l pool , für jedes der direkt und indirekt assoziierten Sequenzelemente,
nach einer Position in sm gesucht, an die das jeweilige Sequenzelement
gesetzt werden kann, ohne die Integrität der globalen Alignierung zu
zerstören. Dafür muss in Zeile 12, für jedes dieser Sequenzelemente,
mit der Funktion valid_pos() getestet werden, ob sich nicht bereits an
der Zielposition ein Sequenzelement aus der selben Ursprungssequenz
befindet. Falls dabei keine Position des Pools in Frage kommt, so wird
in Zeile 18 bis 20 eine neue Position erzeugt und der gemeinsamen
Sequenz hinzugefügt. Auf diese Position kann das assoziierte Sequenzelement nun gesetzt werden (s. Zeile 21 und 22). Anschließend wird
in Zeile 23 die neue Position Teil des Pools.
29
Algorithm 11 Assoziierte Sequenzelemente alignieren
1:
2:
3:
4:
5:
6:
7:
8:
9:
10:
11:
12:
13:
14:
15:
16:
17:
18:
19:
20:
21:
22:
23:
24:
25:
26:
27:
28:
29:
30:
31:
32:
33:
34:
35:
36:
procedure align_assoc(sm , s a , sb )
for all p a ∈ s a .l p do . Assoziierte Elemente aus sb alignieren
for all sea ∈ p a .lse do
l pool ← {sea .p}
NEXT_PEER :
for all se peerd ∈ sea .lse do
if se peerd .p.s = sm then
goto NEXT_PEER
end if
for all se peer ∈ se peerd .p.lse do
for all p align ∈ l pool do
if valid_pos(se peer , p align ) then
se peer .p ← p align
p align .lse .add(se peer )
goto NEXT_PEER
end if
end for
pnew ← create( P)
sm .l p .add( pnew )
pnew .s ← sm
pnew .lse ← {se peer }
se peer .p ← pnew
l pool .add( pnew )
end for
end for
end for
end for
end procedure
function valid_pos(se, p)
for all se pos ∈ p.lse do
if se pos .s = se.s then
return ⊥
end if
end for
return >
end function
3.4.3
Übernahme nicht assoziierter Sequenzelemente
Der letzte Schritt in der Funktion merge() (s. Algorithmus 10, Zeile 19)
ist das Hinzufügen nicht mit s a assoziierter Sequenzelemente aus sb
zur gemeinsamen Sequenz sm . Dies wird mittels der Prozedur add_orphans() erreicht. Hier wird ausgehend von der Sequenz sb , über all
deren Positionen iteriert, wobei in den Zeilen 3 bis 5 jeweils eine neue
Position erzeugt, jedoch noch nicht in die gemeinsame Sequenz sm
eingehängt wird. Für alle Sequenzelemente jeder Position in sb wird
in Zeile 7 geprüft, ob diese noch mit einer Position in sb verknüpft
sind. Wenn dies der Fall ist, so wird dieses Sequenzelement in die
Liste der Positionselemente der neu erzeugten Position aufgenommen
(Zeile 8) und umgekehrt wird das Sequenzelement mit der Position
verknüpft (Zeile 9). Nach dem Durchlauf der Sequenzelemente einer
Position in sb wird in Zeile 12 getestet ob der neu erzeugten Position
Sequenzelemente hinzugefügt wurden. Wenn ja, wird sie in an die
neue gemeinsame Sequenz angehängt.
30
Algorithm 12 Nicht-Assoziierte Sequenzelemente hinzufügen
1:
2:
3:
4:
5:
6:
7:
8:
9:
10:
11:
12:
13:
14:
15:
16:
procedure add_orphans(sm , sb )
for all pb ∈ sb .l p do
pnew ← create( P)
pnew .s ← sm
pnew .lse ← {}
for all seb ∈ pb .lse do
if seb .p.s = sb then
pnew .lse .add(seb )
seb .p ← pnew
end if
end for
if pnew .lse .length > 0 then
sm .l p .add( pnew )
end if
end for
end procedure
3.5
tree-matching
Im letzen Abschnitt wurde die Funktion der Sequenz im Alignierungsprozess dargestellt. Für die globale Alignierung müssen zunächst die
Assoziationen zwischen den Sequenzelementen hergestellt werden. In
Abschnitt 2.4 wurden bereits dynamic programming Algorithmen besprochen, mit denen sich die Ähnlichkeit zweier Sequenzen berechnen lässt. Da dies die Bestimmung einer mathematisch optimalen Alignierung voraussetzt, lassen sich an diesem Punkt auch die Assoziationen zwischen den Sequenzelementen vornehmen. Hierzu ist das
Backtracking als zusätzlicher Schritt nötig. Diese Kombination, aus
paarweiser Ähnlichkeitsberechnung und Assoziation der alignierten
Sequenzelemente, wird im Folgenden als “Matching” bezeichnet.
3.5.1
Parametrisiertes Tree-Matching
Mit der progressiven Alignierungsstrategie von Hogeweg und Hesper
(1984) als Rahmen und dem von Yang (1991) auf Bäume angepassten
dynamic programming Algorithmus ließe sich theoretisch ein Vergleich
von 2 bis n Bäumen durchführen. Jedoch muss bedacht werden, dass
die von Hogeweg und Hesper (1984) entwickelte Strategie zwar gewährleistet, dass zu jedem Zeitpunkt maximal zwei Sequenzen verglichen werden, und der von Yang (1991) beschriebene Algorithmus den
Vergleich von Baumknoten wie den Vergleich von Sequenzen behandelt, jedoch bedeutet das auch, dass für den Vergleich zweier Knoten
der gesamte Algorithmus rekursiv für jede mögliche Alignierung ihrer
Kinder augerufen wird. Die Komplexität erhöht sich also mit der Tiefe
der Bäume, bzw. mit der Anzahl der Kinder pro Knoten.
Komplexität
Die Komplexität eines solchen Ansatzes lässt sich leicht überschlagen,
wenn man davon ausgeht, dass der paarweise Vergleich von Sequenzen, auf Basis eines dynamic programming Ansatzes, einen Aufwand
von O(n2 ) hat, da hierbei zwei Sequenzen eine Matrix aufspannen,
deren Zellen im Zuge der Berechnung gefüllt werden. Dazu wird im
folgenden von einem Baum, mit festem branching factor b ausgegangen.
Der branching factor ist dabei die Anzahl der Kinder, die jeder Knoten
hat. Die Tiefe, also die Anzahl der Ebenen eines Baumes, wird mit d
beschrieben. Vergleicht man nun zwei Knoten miteinander (Ebene 0),
so hängt dieser Vergleich vom paarweisen Alignment ihrer Kindkno-
31
ten ab (Ebene 1) ab, also von einem Alignment zweier Sequenzen. Der
Aufwand, zwei Knoten zu alignieren, ist also O(b2 ), da der konstante
Aufwand von 1 für die Wurzelebene entfallen kann. Dieser Aufwand
fällt rekursiv nun wieder für jeden Kindknoten an, wodurch auf Ebene
1 bereits ein Aufwand von O(b2 · b2 ) entsteht. Somit gelangt man zu
einer Komplexität von O(b2·(d−1) ). Hierdurch wird deutlich dass der
branching factor sowie die Tiefe der Bäume sehr starken Einfluss auf die
Komplexität des Verfahrens haben.
Lookahead
Wie bereits in den ersten Beispielen deutlich wurde, erhöht sich die
Qualität des Alignments mit jeder zusätzlich betrachteten Ebene von
Kindknoten. Es bietet sich daher an, die Zahl der betrachteten Ebenen, im Zuge der Alignierung zweier Knoten, zu beschränken, was im
Folgenden als Lookahead mit der Variablen l bezeichnet wird.
Parent weight
Im Zuge der Einführung einer Tiefenbegrenzung durch den Lookahead,
erscheint es konsequent, bei der Berechnung der Ähnlichkeit zweier
Knoten, die Ähnlichkeit der Kindknoten sukkzessive mit weniger Gewicht einfließen zu lassen. Dazu wird das Konzept des parent weight,
ausgedrückt durch die Variable p, eingeführt. Es legt fest, in welchem
Verhältnis die Ähnlichkeit der Kindknoten zu der lokalen Ähnlichkeit
ihrer Elternknoten steht.
Identität, Vergleichbarkeit und Ähnlichkeit
Yang (1991) führte in seinem Ansatz den Aspekt der Vergleichbarkeit
ein, um auch Knoten Alignieren zu können, die zwar nicht den identischen Typ aufweisen, jedoch von der Semantik her ähnlich sind. Da
Yang (1991), wie auch Needleman und Wunsch (1970), nicht normierte
Werte verwenden um die Ähnlichkeit auszudrücken, sind für die Konzepte Identität, Vergleichbarkeit sowie Ähnlichkeit unterschiedliche
Funktionen nötig, welche die Klassifikation übernehmen. Normiert
man jedoch die Ähnlichkeit auf einen Bereich zwischen einschließlich
0.0 und 1.0, so lassen sich die drei genannten Konzepte durch das der
Ähnlichkeit ausdrücken:
• Die Elemente A und B sind identisch, wenn sie die Ähnlichkeit
1.0 aufweisen
• Die Elemente A und B sind vergleichbar sobald sie eine Ähnlichkeit > 0.0 aufweisen
• Die Elemente A und B sind nicht vergleichbar und damit auch
nicht identisch wenn sie eine Ähnlichkeit von 0.0 aufweisen
Diese Vergleichsoperation wird, wie bereits in der Definition erwähnt,
von der Funktion sim() bereitgestellt.
Hierarchical Tree-Matching
Die neuen Anforderungen lassen sich auf Basis von Yangs Tree-Matching
Algorithmus (Algorithmus 2) leicht umsetzen. Beim Aufruf der Funktion muss nun der Lookahead l sowie das Parent Weight p mit übergeben
werden (s. Zeile 1). Die Verarbeitung kann bereits in Zeile 4 abgebrochen werden, falls A und B nicht vergleichbar sind, deren lokale Ähnlichkeit also 0.0 beträgt. Bei der Berechnung der Matrix wird nun beim
Rekursionsschritt zur Ermittlung der Ähnlichkeit der Kindknoten (s.
Zeile 17) der Lookahead l um eins verringert. Da jetzt Ähnlichkeiten
32
errechnet werden sollen, deren Gewicht zu den Blattknoten hin abnehmen soll, wird in Zeile 35 die Ähnlichkeit der Elternknoten A und
B im Verhältnis p zu 1 − p zu deren Kindknoten gewichtet. Die Normierung der Ähnlichkeit ergibt sich, indem die Ähnlichkeit der Kindknoten durch das Maximum der Anzahl der Kinder geteilt wird. Falls
sowohl A als auch B keine Kinder haben oder der Lookahead kleiner
oder gleich 0 ist, so zählt nur deren Ähnlichkeit (s. Zeile 33). Da hier
zur Bestimmung der Ähnlichkeit zweier Knoten eine Hierarchie von
Kindknoten betrachtet werden muss, wir dieses Verfahren als Hierarchical Tree-Matching bezeichnet.
Algorithm 13 Hierarchical Tree-Matching-Algorithmus
1:
2:
3:
4:
5:
6:
7:
8:
9:
10:
11:
12:
13:
14:
15:
16:
17:
18:
19:
20:
21:
22:
23:
24:
25:
26:
27:
28:
29:
30:
31:
32:
33:
34:
35:
36:
37:
38:
39:
40:
41:
42:
43:
44:
function htm(s a , sb , l, p)
m ← s a .lt .length
n ← sb .lt .length
if sim(s a , sb ) = 0 then
return 0
end if
for i ← 0, m do
am (i, 0) ← 0
end for
for j ← 0, n do
am (0, j) ← 0
end for
for i ← 0, m do
for j ← 0, n do
tca ← s a .tt .get(i )
tcb ← sb .tt .get( j)
match ← am (i − 1, j − 1) + htm(tca , tcb , l − 1, p)
delete ← am (i − 1, j)
insert ← am (i, j − 1)
max_score ← max(max(match, delete), insert)
am (i, j) ← max_score
if insert = max_score then
at (i, j) ← ’i’
else if delete = max_score then
at (i, j) ← ’d’
else
at (i, j) ← ’m’
end if
end for
end for
trackback(at , s a , sb )
if (m = n = 0) ∨ (l <= 0) then
return sim(s a , sb )
else
am (i,j)
return p · sim(s a , sb ) + (1 − p) · max
(m,n)
end if
end function
function max(a,b)
if a > b then
return a
else
return b
end if
end function
33
3.5.2
Backtracking
Zusätzlich wird in Algorithmus 13 das Backtracking implementiert
um nicht nur die Ähnlichkeit zu berechnen, sondern auch eine mathematisch optimale Alignierung zu erzeugen. Hierzu werden in Zeile 22 bis 29 die Werte der Backtracking-Matrix at gesetzt. Prinzipiell
kann es mehr als eine optimale Alignierung geben. Es ergeben sich immer dann verschiedene Pfade durch die Matrix sobald mehrere mögliche Operationen (Einfügung, Löschung und Abbildung) dem maximal
möglichen Wert der jeweiligen Zelle entsprechen. Diese alternativen
Pfade könnte man theoretisch für spätere Verbesserungen der globalen Alignierung nutzen. Im vorliegenden Ansatz wird darauf verzichtet und eine optimale Alignierung anhand der Vereinbarung gewählt,
dass Einfügeoperationen vor Löschungen und diese wiederum vor Abbildungen präferiert werden. Hierdurch werden nur Varianten zugelassen, in denen keine unvergleichbaren Sequenzelemente aufeinander
abgebildet werden. Diese Problematik zeigte sich in dem Beispiel zum
Abschnitt 2.4.1.
Algorithm 14 Rückverfolgung der Alignierung und Assoziation der
Sequenzelemente
1:
2:
3:
4:
5:
6:
7:
8:
9:
10:
11:
12:
13:
14:
15:
16:
17:
18:
19:
20:
procedure trackback(at , s a , sb )
i ← sa + 1
j ← sb + 1
while i > 1 ∧ j > 1 do
if at = ’i’ then
j ← j−1
else if at = ’d’ then
i ← i−1
else
sea ← s a .l p .get(i − 1)
seb ← sb .l p .get( j − 1)
if ¬sea .lse .contains(seb ) then
sea .lse .add(seb )
end if
if ¬seb .lse .contains(sea ) then
seb .lse .add(sea )
end if
end if
end while
end procedure
Das Backtracking ist separat im Algorithmus 14 dargestellt. Die Funktion trackback() erwartet die Backtracking-Matrix at sowie die zu alignierenden Sequenzen s a und sb . In der Schleife wird die Matrix rückwärts durchlaufen, beginnend in der äußersten Zelle. Der Pfad ergibt
sich durch die in den Zellen gespeicherten Werte: Hat eine Einfügebzw. eine Löschoperation in die aktuell betrachtete Zelle geführt, so
wird zwischen den Sequenzen keine Assoziation hergestellt. Bei einer
Einfügeoperation führt der Pfad in westlicher Richtung weiter (s. Zeile 6), bei einer Löschoperation in nördlicher (s. Zeile 14). Bei einer
Abbildung eines Sequenzelements auf ein anderes, wird das der Zelle
entsprechende Sequenzelement aus seb in die Liste der Assoziierten Sequenzelemente von sea aufgenommen (s. Zeile 13). Das selbe geschieht
analog mit Sequenzelement sea in Zeile 16. Diese Assoziationen werden schließlich, wie weiter oben beschrieben, bei der Vereinigung der
Sequenzen zur Herstellung einer globalen Alignierung verwendet (s.
Algorithmus 10).
34
4
IMPLEMENTIERUNG
Im letzten Kapitel wurden die in der Analyse identifizierten Anforderungen algorithmisch ausgearbeitet. Dabei haben sich bereits Komponenten mit unterschiedlichem Fokus ergeben. Das Ziel ist es nun das
gesamte Verfahren konkret in Java, also einer objektorientierten Programmiersprache umzusetzen. Um möglichst unabhängig von konkreten Datenstrukturen zu sein muss sich dieser Aspekt auch im Entwurf
der Softwarearchitektur widerspiegeln.
Laut Sommerville (2006) hängen Stil und Struktur der eingesetzten
Softwarearchitektur von den nicht-funktionalen Systemanforderungen
ab (s. (Sommerville, 2006, S. 242)). Als solche werden Performance, Security, Safety, Availability und Maintainability genannt.
Der Fokus dieser Arbeit liegt auf der Implementierung einer Softwarebibliothek, zur Prototypenextraktion aus strukturierten Daten. Da
die mit diesem Aufgabenkomplex verbundenen Teilaufgaben noch nicht
klar voneinander abgegrenzt wurden und er sich durch Weiterentwicklungen in der Zukunft noch verändern wird, ist die wichtigste nichtfunktionale Systemanforderung, dass das System klar strukturiert und
wartbar ist, was der Maintainability entspricht.
Sommerville (2006) beschreibt, dass eine darauf ausgerichtete Architektur aus kleinen, in sich abgeschlossenen Komponenten bestehen
muss, die sich einfach auswechseln lassen, sowie dass Produzenten
und Konsumenten von Daten getrennt werden müssen, wodurch eine
Abhängigkeit von einer gemeinsamen Datenstruktur vermieden wird
(Sommerville, 2006, S. 243).
Zunächst wird daher eine Schnittstelle zu strukturierten Daten definiert, ohne eine konkreten Typen vorauszusetzen. Analog zum Design
werden anschließend die für das Verfahren nötigen Datenstrukturen
vorgestellt. Schließlich wird die bisher algorithmisch definierte Logik
passenden Komponenten zugewiesen und die Interaktionen zwischen
ihnen erläutert.
4.1
umgang mit generischen datentypen
Schmid et al. (2010) beschreibt ein Element folgendermaßen: “[...] we
write an element e as τ (name) where name is a variable which can
be instantiated by a constant name-string [...] ” (Schmid et al., 2010,
S. 3). In dieser Definition stellt ein Element bereits einen Typen τ mit
dem Merkmal name dar. Diese Sichtweise rührt von der Orientierung
an der Domäne der SAP Business ByDesign Incidents, welche den Ausgangspunkt der Entwicklung des SDTG Ansatzes darstellt. Bei den Incidents handelt es sich um Fehlerprotokolle, die als strukturierte Daten
in Form von XML-Dateien persistiert werden. Eine Besonderheit der
Darstellung ist es, dass jedes Element lediglich über ein Attribut name
verfügt, worauf man beschloss die Ausprägung dieses Attributs mit
der Ausprägung des Elements e gleichzusetzen und dies im Typen τ
zusammenzufassen (Siehe auch (Bader, 2009, S. 39)). Der in Abbildung
12 dargestellte Baum entspricht dabei dieser Sichtweise.
35
A(“Anton”)
A
name
B
C
“Anton”
name
name
B(“Berta”) C(“Caesar”)
“Berta” “Caesar”
Abbildung 12: Unterschiedliche Datendarstellung
Da die zu entwickelnde Software prinzipiell auf jegliche Art strukturierter Daten angewendet werden können soll, wird darauf verzichtet
Annahmen bezüglich deren Eigenschaften zu treffen. Es wird stattdessen von Strukturen ausgegangen, die über baumartige Zugriffsmechanismen verfügen wie sie bereits in der Definition des Baumknotens in
Abschnitt 3.1.1 vorausgesetzt wurden. Um vom konkreten Datentypen
zu abstrahieren, wird daher lediglich eine Schnittstelle definiert, die
eine baumtypische Navigation innerhalb der zu verarbeitenden Datenstruktur zulässt. Dieser Ansatz bringt einige Vorteile mit sich:
• Die geforderte Trennung zwischen Produzent und Konsument
ist gewährleistet
• Einmal definiert kann der Zugriff auf ein bestimmtes Quellformat ohne Vorverarbeitung durchgeführt werden
• Verschiedene Ansätze können objektiv miteinander verglichen
werden, da alle die selbe Schnittstelle zu den Daten nutzen
4.1.1
Datenschnittstelle
Die Schnittstelle zu den Eingabedaten definiert sich analog zur Definition des Baumknotens aus Abschnitt 3.1.1. Die in Abbildung 13 dargestellte generische Schnittstelle ITreeNode entspricht daher der abstrakten Definition des Baumknotens. Demnach muss eine Klasse die diese
Schnittstelle realisiert den generischen Typen E binden. Er entspricht
dabei dem Element, also dem Träger der sachbezogenen Informationen.
data.tree
E: Object
«interface»
ITreeNode
getParent(): ITreeNode
getChildren(): List< ITreeNode<E> >
getElement(): E
addChildNode(elem: E): ITreeNode<E>
setElement(elem: E)
sort(cmp: Comparator<E>)
Abbildung 13: Datenschnittstelle ITreeNode
Wie bereits für die Definition des abstrakten Baumknotens gefordert,
so ist auch hier das Element nicht gleichzeitig der Baumknoten, son-
36
dern es wird weiterhin sachbezogene von struktureller Information getrennt. ITreeNode definiert dabei folgende Methoden:
• getParent() liefert den Elternknoten
• getChildren() liefert eine Liste der Kindknoten
• getElement() liefert das gekapselte Element des generischen Typs
E zurück
• addChildNode() fügt dem Knoten einen Kindknoten hinzu, in
dem das übergebene Element gekapselt wird
• setElement() legt das Element fest das durch den Knoten gekapselt werden soll
• sort() sortiert den Baum mit Hilfe einer Strategie, die die Priorität festlegt. Diese muss die Schnittstelle Comparator implementieren
4.1.2
Prototypschnittstelle
Auch der Prototyp ist wieder eine Baumstruktur und setzt sich daher auch aus Strukturinformationen und sachbezogenen Informationen zusammen. Der Zugriff auf die Struktur kann dabei wiederum
über die Schnittstelle ITreeNode erfolgen. Das Prototypelement muss
entsprechend der abstrakten Definition Zugriff auf sein Label und die
Häufigkeit zulassen. Dessen Schnittstelle IPrototype ist in Abbildung
14 dargestellt.
data.prototype
«interface»
IPrototype
getLabel(): String
getFrequency(): double
setLabel(label: String)
setFrequency(freq: double)
Abbildung 14: Prototypenschnittstelle IPrototype
Folgende Methoden werden von IPrototype definiert:
• getLabel() liefert das Label des Prototypen als Zeichenkette zurück
• getFrequency() gibt die relative Auftrittshäufigkeit des Elements
als rationale Zahl aus, das durch das Prototypelement repräsentiert wird
• setLabel() setzt das Label
• setFrequency() setzt die relative Auftrittshäufigkeit
4.1.3
Typspezifische Strategien
Jegliche Logik die sich auf die Struktur der Daten bezieht hat nun die
nötigen Zugriffsmöglichkeiten auf die Eingabedaten. Element- und damit typspezifische Operationen können nicht pauschal festgelegt werden. Da diese der Nutzer der Software bereitstellen muss, werden auch
37
hierfür Schnittstellen definiert. In Abschnitt 3.1.2 wurden in diesem
Zusammenhang folgende Aspekte genannt:
• Der Vergleich von Elementen (Ähnlichkeit)
• Die Ordnung/Sortierung von Elementen (Priorität)
• Die relevanten Merkmale eines Elements und wie diese im Prototypen abgebildet werden (Labeling)
Diese drei Strategien werden durch die Schnittstellen ISimilarity, Comparable und ILabeling definiert, welche sich im gemeinsamen Paket logic.comparison befinden. Die Paketstruktur und die Schnittstellen sind
in Abbildung 15 dargestellt.
logic.comparison
similarity
E: Object
«interface»
ISimilarity
+getSimilarity(E o1, E o2): double
priority
E: Object
«interface»
Comparator
+compare(E o1, E o2): int
priority
E: Object
«interface»
ILabeling
+labelPrototype(proto: IPrototype, elements List<E>)
Abbildung 15: Typspezifische Strategien im Paket logic.comparison
Ähnlichkeit (ISimilarity)
Um die lokale Ähnlichkeit zwischen zwei Elementen des generischen
Typs E zu ermitteln, greift die Programmlogik auf eine, durch den
Benutzer bereitgestellte, Strategie in Form einer Klasse zu, die ISimilarity für den verwendeten Elementtypen E implementiert. Das geschieht über die Methode getSimilarity(), die der Funktion sim() der
Designphase entspricht. Sie gibt für zwei Elemente des gleichen Typs
deren Ähnlichkeit als rationale Zahl zurück. Als Konvention wird davon ausgegangen dass das Ergebnis auf den Bereich zwischen 0.0 und
1.0 normiert ist (s. auch 3.1.2)
Priorität (Comparator)
Eine Implementierung der Schnittstelle Comparator gibt beim Aufruf
der Methode compare() für zwei Instanzen, o1 und o2, des gleichen Typs
38
einen ganzzahligen Wert zurück, der die Priorität von o1 gegenüber o2
widerspiegelt:
• Ein Wert < 0.0 bedeutet o1 hat eine niedrigere Priorität als o2
• 0.0 bedeutet o1 und o2 sind gleichwertig
• Ein Wert > 0.0 bedeutet o1 hat eine höhere Priorität als o2
Wie bereits erwähnt ist diese Funktionalität nötig, um die durch den
Alignierungsprozess verlorengegangene Ordnung der Elemente wieder herzustellen. Die Methode compare() erfüllt damit die selbe Aufgabe wie die Funktion sort().
Labeling (ILabeling)
Die Schnittstelle ILabeling definiert die für die Erzeugung des Prototypen notwendige Methode labelPrototype(), die wie die Funktion label_proto() die relevanten gemeinsamen Merkmale einer Liste alignierter Elemente auf das Label eines Prototypelements überträgt.
4.2
prototypextraktion
Die Funktionalität die extract_prototype() zur Verfügung stellt, soll in
der Implementierung in Java zunächst als Schnittstelle definiert werden. Dadurch soll ein Rahmen für zukünftige Erweiterungen geschaffen werden. Die Abbildung 16 zeigt die Schnittstelle IPrototypeExtraction.
logic.extraction
I: Object, O: Object
«interface»
IPrototypeExtraction
+extractPrototype(clusterMembers: List<I>): O
Abbildung 16: Extraktionsschnittstelle IPrototypeExtraction
Wie extract_prototype() erwartet ihre einzige Methode extractPrototype()
eine Liste von Elementen. Jetzt ist jedoch der Datentyp der Elemente nicht mehr auf den des Baumknotens festgelegt. Stattdessen weist
IPrototypeExtraction die generischen Typen I und O auf. Damit ist das
allgemeine Konzept der Prototypextraktion beschrieben: Anhand einer
Menge von Elementen eines beliebigen Typs wird ein Objekt erzeugt,
das diese Menge repräsentiert.
4.2.1
Prototypextraktion aus strukturierten Daten
Der entwickelte Ansatz zur Prototypextraktion aus strukturierten Daten füllt diesen Rahmen nun für Daten mit einer bestimmten Eigenschaft aus. Die abstrakte Klasse TreePrototypeExtraction konkretisiert
die Schnittstelle entsprechend für baumartig Strukturierte Daten und
bindet damit den generischen Typen E an ITreeNode<E>. In Abbildung
17 wird dieser Zusammenhang deutlich.
TreePrototypeExtraction implementiert die Funktionalität von extractPrototype() jetzt entsprechend extract_prototype(). Die Methode processChildNodes() entspricht der Funktion process_children und greift demnach
zum erzeugen des Prototypelements auf die Methode applyPrototypeStrategy() zurück. Diese abstrakte Methode muss jedoch erst von einer
39
logic.extraction
I: Object, O: Object
«interface»
IPrototypeExtraction
+extractPrototype(clusterMembers: List<I>): O
«bind»
I -> ITreeNode<E>
O -> ITreeNode< IPrototype >
TreePrototypeExtraction
- alignmentStrategy: IAlignment<E>
- sortingStrategy: Comparator<E>
- labelingStrategy: ILabeling<E>
+ extractPrototype(clusterMembers:List< ITreeNode<E> >)
: ITreeNode< IPrototype >
- processChildNodes(childNodeList:List< List< ITreeNode<E> > >)
: List< ITreeNode<IPrototype> >
# applyPrototypingStrategy(clusterMembers: List< ITreeNode<E> >,
treeCount: int, parentCount: int) : ITreeNode<IPrototype>
ILabeling
Comparator IAlignment
Abbildung 17: TreePrototypeExtraction konkretisiert IPrototypeExtraction
für baumartig strukturierte Daten
konkreten Klasse, die TreePrototypeExtraction erweitert, implementiert
werden. Die weiter oben beschriebenen typspezifischen Strategien ILabeling und Comparator tauchen hier als Abhängigkeiten wieder auf, da
diese für die Prototypenextraktion für den jeweiligen Eingabedatentyp
extern bereitgestellt werden müssen. Zusätzlich wird in der Methode processChildNodes() eine Alignierungsstrategie zur Alignierung der
in den Knoten gekapselten Elemente benötigt. Diese wird durch die
Schnittstelle IAlignment definiert.
TreePrototypeExtraction
- alignmentStrategy: IAlignment<E>
- sortingStrategy: Comparator<E>
- labelingStrategy: ILabeling<E>
+ extractPrototype(clusterMembers:List< ITreeNode<E> >)
: ITreeNode< IPrototype >
- processChildNodes(childNodeList:List< List< ITreeNode<E> > >)
: List< ITreeNode<IPrototype> >
# applyPrototypingStrategy(clusterMembers: List< ITreeNode<E> >,
treeCount: int, parentCount: int) : ITreeNode<IPrototype>
AuPrototypeExtraction
SdtgIoPrototypeExtraction
# applyPrototypingStrategy(...
# applyPrototypingStrategy(...
Abbildung 18: Die Prototypingstrategien Anti-Unifikation und SDTG
als Erweiterung der Klasse TreePrototypeExtraction
40
4.2.2
Prototypingstrategie
Die durch die Funktion proto_strategy() realisierte Prototypisierungsstrategie, wobei die Funktionen proto_strategy_au() und proto_strategy_sdtg()
verschiedene Varianten darstellen, wird durch eine Erweiterung der
abstrakten Klasse TreePrototypeExtraction bereitgestellt. Wie schon in
der Designphase, so soll als Alternative zur SDTG-Strategie ebenfalls
die Anti-Unifikation umgesetzt werden.
In der Abbildung 18 sieht man deutlich dass AuPrototypeExtraction
sowie SdtgIoPrototypeExtraction lediglich die in TreePrototypeExtraction
abstrakt definierte Methode applyPrototypeStrategy() implementieren.
4.3
alignierung
Da die Alignierungsfunktion eine eigenständige Komponente darstellt
soll auch dieser Aspekt durch eine Schnittstelle zugänglich gemacht
werden. Im letzten Kapitel wurde die Funktion align_progressive() definiert, welche die einzelnen Komponenten der progressiven Alignierung steuert.
logic.alignment
E: Object
«interface»
IAlignment
+ align(sequences: List< List<E> >)
: List< List<E> >
Abbildung 19: Alignierungsschnittstelle IAlignment
In Abbildung 19 ist die Schnittstelle IAlignment im Kontext des Pakets
logic.alignment abgebildet. Wie align_progressive() erwartet die Methode align() eine Liste von Listen mit Elementen des generischen Typs E
und gibt auch eine solche zurück, jedoch mit anderer Semantik. Wie
bereits bei IPrototypeExtraction soll diese generelle Definition einer Alignierungsstrategie für strukturierte Daten konkretisiert werden.
Progressive Alignierung
In der Klasse TreeAlignmentProgressive ist das progressive Alignierungsverfahren für Baumknoten, entsprechend der Funktion align_progressive(),
realisiert. Dabei werden auch hier die Listen von Baumknoten in Form
von Sequenzen verwaltet. Die allgemeine Beziehung zwischen den genannten Komponenten wird in Abbildung 20 dargestellt.
Das nötige Clusterverfahren wird wiederum als komplett eigenständige Komponente ausgelagert. Die Funktionalität der Prozedur merge()
wird dabei als Methode eines Clusters realisiert, die dafür wiederum
auf eine typspezifische Strategie zurückgreift.
41
nodeSeq: Sequence
logic.alignment
«instanceOf»
sequence
E: Object
Sequence
«creates»
E: Object
«interface»
IAlignment
+ align(sequences: List< List<E> >)
: List< List<E> >
«bind»
E -> ITreeNode<E>
2..
TreeAlignmentProgressive
ISimilarity<E>
Abbildung 20: Beziehung zwischen den Komponenten TreeAlignmentProgressive und Sequence
4.4
clustering
Die Schnittstelle für das Clustering orientiert sich an der Funktion clusterh(). Die Methode cluster() erwartet ebenfalls eine Liste von Elementen. Wieder ist der Typ der Elemente nicht festgelegt. Da typabhängige
Operationen des Clusterings nur in der Funktion avg_linkage() genutzt
werden, kann man von Linkage als Komponente sprechen. Aus diesem Grund erwartet die Methode cluster(), zusätzlich zur Liste der
Elemente, eine Strategie zur Berechnung des Abstands zwischen zwei
Clustern, in Form einer Implementierung der Schnittstelle ILinkage.
4.4.1
Hierarchisches Clustering
Wie in Abbildung 21 dargestellt ist das hierarchische Clusteringverfahren, wie es für die progressive Alignierung benötigt wird, eine Implementierung der Schnittstelle IClusterManager. Die Methode cluster()
von ClusterManagerHierarchical entspricht somit der Funktion clusterh(),
mit dem Unterschied, dass hiermit das Clustering unabhängig vom
Elementtyp möglich ist, sofern eine passende Linkage-Strategie bereitgestellt wird.
42
logic.clustering
ILinkage
«interface»
IClusterManager
+ cluster(elements: List<I>, distanceFunction: ILinkage<I>)
: AbstractCluster<I>
ClusterManagerHierarchical
+ cluster(elements: List<I>, distanceFunction: ILinkage<I>)
: AbstractCluster<I>
Abbildung 21: Clusteringschnittstelle IClusterManager
4.4.2
Linkage
Im Design des Verfahrens ist in avg_linkage() ein Verfahren dargestellt,
dass die Distanz zwischen zwei Clustern dadurch berechnet, indem
die paarweisen Ähnlichkeiten der Sequenzen in den Clustern zueinander berechnet wurden und das Ergebnis auf die Anzahl der Vergleichsoperationen normiert wurde. Prinzipiell sind hier jedoch verschiedene
Verfahren denkbar und daher sollen Freiheitsgrade an dieser Stelle zugelassen werden.
logic.clustering.linkage
I: Object
«interface»
ILinkage
+ getDistance(c1: AbstractCluster<I>,
c2: AbstractCluster<I>): double
I: Object
AverageLinkage
- scoringStrategy: ISimilarity<I>
+ getDistance(c1: AbstractCluster<I>,
c2: AbstractCluster<I>): double
ISimilarity
Abbildung 22: ILinkage als Schnittstelle zur Berechnung der Clusterdistanz
Deshalb definiert die Schnittstelle ILinkage lediglich die Methode getDistance(), die für zwei Cluster vom Typ AbstractCluster deren Distanz
berechnet und diese als rationale Zahl zurückgibt. Per Konvention ist
diese auf den Wertebereich zwischen 0.0 bis 1.0 normiert. ILinkage ist
wieder über den generischen Typ I parametrierbar, wodurch der Typ
der Clusterlemente nicht eingeschränkt wird.
43
Die Methode getDistance() der Klasse AverageLinkage entspricht der
Funktion avg_linkage(), im Gegensatz zu ihr ist die Logik, zur Bestimmung der Ähnlichkeit zwischen den Elementen, nicht auf das Hierarchical Tree Matching festgelegt. Wie in Abbildung 22 zu sehen, benötigt AverageLinkage hierfür wiederum eine externe Strategie vom Typ
ISimilarity, dessen Referenz ihr über den Konstruktor mitgegeben werden muss.
4.4.3
Cluster
In der algorithmischen Definition des Extraktionsverfahrens ist man
davon ausgegangen, dass man Cluster durch verschachtelte Listen darstellen kann, wie dies bei funktionalen Programmiersprachen der Fall
ist. Bei einer objektorientierten Programmiersprache wie Java ist dies
jedoch nicht möglich. Zwar ist der Elementtyp einer parametrierbaren
Listenklasse variabel, die Schachtelung dieser Parameter muss dabei
jedoch eindeutig sein. Daher wird ein Cluster als konkrete Datenstruktur eingeführt.
Prinzipiell gibt es zwei Arten von Clustern: Verbindungscluster und
Blattcluster. Blattcluster sind einelementige Cluster, sie kapseln also lediglich ein Element. Ein Verbindungscluster ist ein Cluster der zwei
andere Cluster miteinander verbindet. Damit ein Verbindungscluster
also z.B. einen Blattcluster mit einem anderen Verbindungscluster vereinigen kann, müssen beide Arten von Clustern einen gemeinsamen
Supertyp haben.
AbstractCluster
AbstractCluster ist eine abstrakte Klasse und stellt diesen Supertyp dar.
Sie implementiert die Methode add() mit der einem Cluster ein anderer
hinzugefügt werden kann. Diese Methode ist damit automatisch auch
in seinen Subklassen LeafCluster und LinkCluster verfügbar. Die Methode erwartet ein Objekt vom Typ AbstractCluster, erstellt intern einen
LinkCluster, der den Cluster auf dem die Methode aufgerufen wurde und den zu verbindenden Cluster verknüpft, und gibt diesen als
neuen Cluster zurück. Dadurch ist wieder eine beliebige Schachtelung
der Cluster möglich, so wie dies im Design vorgesehen war. Weiterhin werden folgende abstrakte Methoden definiert, die von konkreten
Unterklassen implementiert werden müssen:
• getMergedElements() gibt den Inhalt des Clusters vereinigt aus
– Wird dabei eine Vereinigungsstrategie vom Typ IMerge übergeben, so bestimmt diese, wie die Elemente vereint werden,
und von welchem Typ dieses vereinigte Objekt ist
– Ansonsten werden die Elemente des Clusters als Menge
vom Typ Set zurückgegeben
• getSize() gibt die Anzahl, der im Cluster enthaltenen Elemente,
zurück
Die Methode getMergedElements() arbeitet sich dabei ebenfalls rekursiv von den Blattclustern nach oben, wie es in dem Algorithmus zur
Funktion merge() beschreiben ist. Da jedoch auch hier die eigentliche
Merging-Strategie, also wie zwei Elemente miteinander vereint werden, nicht pauschal festgelegt werden kann, erwartet die Methode diese Strategie in Form einer Implementierung der Schnittstelle IMerge.
Falls diese nicht bereitgestellt wird, so werden die Elemente des Clusters als Menge ausgegeben.
44
logic.clustering.cluster
E: Object
AbstractCluster
+ add(leaf: AbstractCluster<E>): AbstractCluster<E>
+ getMergedElements(): Set<E>
+ <O> getMergedElements(IMerge<E,O> mergeStrategy): O
+ getSize(): int
E:Object
E:Object
LinkCluster
LeafCluster
...
...
Abbildung 23: AbstractCluster als Cluster-Supertyp
LeafCluster
In Abbildung 24 sieht man wie AbstractCluster als Blattcluster in der
Klasse LeafCluster erweitert wurde. Im Attribut leaf wird hierbei das
Element gespeichert, daher wird beim Aufruf von getMergedElements()
lediglich dieses zurückgegeben. Dieses Verhalten entspricht der Abbruchbedingung der Rekursion in Zeile 3 der Funktion merge() in Algorithmus 10.
E:Object
LeafCluster
- leaf: E
+ getMergedElements(): Set<E>
+ <O> getMergedElements(IMerge<E,O> mergeStrategy): O
+ getSize(): int
Abbildung 24: LeafCluster, der einelementige Cluster
LinkCluster
Ein Verbindungscluster kombiniert zwei Cluster zu einem. Bei einem
Aufruf von getMergedElements() auf einem LinkCluster wird die selbe Methode auf den beiden verbundenen Clustern aufgerufen. Die
zwei entstandenen Objekte werden schließlich mit Hilfe der MergingStrategie verschmolzen. Das entspricht den Zeilen 5 und 6 der Funktion merge() in der sie sich selbst rekursiv aufruft.
E:Object
LinkCluster
- leftLink: AbstractCluster<E>
- rightLink: AbstractCluster<E>
+ getMergedElements(): Set<E>
+ <O> getMergedElements(IMerge<E,O> mergeStrategy): O
+ getSize(): int
Abbildung 25: LinkCluster als Verbindung zwischen zwei Clustern
45
4.5
sequence-matching
Wie die Funktion avg_linkage(), so bestimmt auch die Methode getDistance() der Klasse AverageLinkage die Distanz zwischen zwei Elementen nicht selbstständig, sondern legt lediglich fest, wie die einzelnen
Distanzen zwischen den Elementen zu einem Wert kombiniert werden. Die Funktion avg_linkage() ruft zur Bestimmung der Distanz die
Funktion htm() auf, die auch gleichzeitig die paarweisen Assoziationen
zwischen den Sequenzelementen vornimmt. Anstatt nun ein weiteres
Konzept wie die Distanz einzuführen, kann man stattdessen auch hier
das der Ähnlichkeit einsetzen.
4.5.1
Hierarchical Sequence-Matching
Das in der Funktion htm() realisierte Hierarchical Tree-Matching ist, wenn
man es genau nimmt, lediglich ein rekursiver Sequenzvergleich, bei
dem sich die Ähnlichkeit zwischen zwei Sequenzelementen durch deren “Kindsequenzen” ergibt. Deshalb wird hier, etwas allgemeiner, ein
Hierarchical Sequence-Matching implementiert, dass erst bei der Anwendung auf Baumknoten zu einem Hierarchical Tree-Matching wird.
logic.comparison.similarity
E: Object
«interface»
ISimilarity
+getSimilarity(E o1, E o2): double
«bind»
Sequence<E>
E: Object
AbstractSimilarityCaching
- cache: Map< Pair<I>,Double >
+ getSimilarity(obj1: I, obj2: I): double
# calculateSimilarity(obj1: I, obj2: I): double
«bind»
E -> Sequence<E>
hierachical
E: Object
HierarchicalSequenceMatching
- similarityStrategy: ISimilarity<E>
- translationStrategy: ITranslator<E, Sequence<E> >
- lookahead: int
- parentWeight: double
# calculateSimilarity(obj1: E, obj2: E): double
+ matchSequence(s1: Sequence<E>,
s2: Sequence<E>, la: int): double
- trackBack(seq1 List< SequenceElement<E> >,
seq2 List< SequenceElement<E> >, tbm int[][])
nodeSeq: Sequence
ISimilarity ITranslator
«modifies»
Abbildung 26: Hierarchical Sequence-Matching
46
4.5.2
Caching von Ergebnissen
In Abbildung 26 sieht man, dass die Klasse HierarchicalSequenceMatching nicht direkt die Schnittstelle ISimilarity implementiert. Vielmehr
ist sie eine Erweiterung der abstrakten Klasse AbstractSimilarityCaching.
Der Hintergrund hierbei ist, dass die Ähnlichkeit eines Sequenzpaares, im Zuge des hierarchischen Clusterings, oft wiederholt nachgefragt wird. Da diese Berechnung jedoch, wie bereits in Abschnitt 3.5
erwähnt, sehr teuer ist, ist es sinnvoll sie für ein Sequenzpaar nur einmal durchzuführen, das Ergebnis zu speichern und im Falle einer wiederholten Anfrage das gespeicherte Ergebnis zurückzugeben.
Diese Caching-Funktionalität wird von AbstractSimilarityCaching bereitgestellt. Als Cache verwendet die Klasse eine Instanz von Map, einem assoziativen Speicher, mit Hilfe dessen einem Paar von Elementen
eine rationale Zahl zugeordnet werden kann. Wird die Methode calculateSimilarity() zur Berechnung der Ähnlichkeit zwischen zwei Elementen aufgerufen, so wird zunächst im Cache geprüft ob die Ähnlichkeit
dieses Elementpaars bereits berechnet wurde. Wenn ja, wird die Zahl
direkt zurückgegeben, wenn nicht, so wird die Ähnlichkeit mit Hilfe
der Methode calculateSimilarity() der Subklasse berechnet und anschließend im Cache gespeichert.
4.5.3
Hierarchical Tree-Matching
Um das Zusammenspiel der einzelnen Komponenten des Hierarchical
Sequence-Matchings zu erläutern, bietet es sich an, dies am Beispiel des
Hierarchical Tree-Matchings zu tun.
Lokale Ähnlichkeit der Elemente
Den Kern stellt die Klasse HierarchicalSequenceMatching dar. Ihre Methode matchSequence() weist im Grunde die selbe Logik auf wie die
Funktion htm() (s. Algorithmus 13). Die Aufgabe der Funktion sim()
übernimmt hierbei eine typspezifische Strategie zur Ähnlichkeitsbestimmung der Klasse ISimilarity. Gefragt ist nun ein Matching von Sequenzen, dessen Elemente Baumknoten (Typ ITreeNode) sind. Hierfür
wurde die Ähnlichkeitsstrategie SimilarityTreeNode, vom Typ ISimilarity), implementiert. Da ein Baumknoten jedoch wiederum ein Element
eines beliebigen Typen kapseln kann, muss SimilarityTreeNode im Konstruktor wiederum eine Ähnlichkeitsstrategie passend zum Elementtyp mitgegeben werden. Diese Schachtelung von Strategien macht den
Ansatz sehr flexibel, ohne dabei die einzelnen Komponenten unnötig
stark an einen bestimmten Typen zu binden.
Translationsstrategie
Da der in matchSequence() eingesetzte Algorithmus, für die Bestimmung des Matchings zweier Sequenzelemente, rekursiv das selbe auch
für deren Kindsequenzen tun muss, wird hier wieder eine externe Strategie benötigt. Die Schnittstelle dafür bildet ITranslator. Sie definiert die
Methode translate(), die der Übersetzung von einem Objekt in ein anderes dient. Für das Tree-Matching wird daher mit TranslatorTreeNode
eine Übersetzungsstrategie implementiert, die einen Baumknoten in
eine Sequenz seiner Kindknoten “übersetzt”.
4.5.4
Sequenz
Die Sequenz als Basisdatenstruktur kann entsprechend der Definition im Kapitel Design direkt als Komposition der Komponenten Se-
47
quenz, Position und Sequenzelement dargestellt werden. Dieser Zusammenhang ist in Abbildung 27 dargestellt.
logic.alignment.sequence
E: Object
Sequence
- positions: List< SequencePosition<E> >
+ merge(Sequence<E>): Sequence<E>
+ getAlignedObjects(): List< List<E> >
...
E: Object
SequencePosition
- sequence: Sequence<E>
# members: List<SequenceElement>
...
baseSequence
E: Object
SequenceElement
- baseSequence: Sequence<E>
- position: SequencePosition<E>
- peers: List< SequenceElement<E> >
- element: E
...
1
0..
peers
Abbildung 27: Die Sequenz als Komposition aus drei Datenstrukturen
Sequence
Die Klasse Sequence verwaltet eine Liste mit den Sequenzpositionen
vom Typ SequencePosition. Auffällig ist, dass die Funktion merge() (s.
Algorithmus 10) jetzt Teil der Klasse Sequence geworden ist, dabei hat
sich an der Funktionsweise jedoch nichts geändert.
SequencePosition
Eine SequencePosition referenziert die ihr zugeordneten Sequenzelemente ebenfalls über eine Liste. Die Sequence Instanz der die Position zugeordnet ist, wird über die Variable sequence referenziert.
SequenceElement
Ein Sequenzelement vom Typ SequenceElement ist mit keinen bis beliebig vielen anderen Sequenzelementen assoziiert. Diese Referenzen
werden in der Liste peers gespeichert. Ein Sequenzelement stammt aus
einer bestimmten Ursprungssequenz und hält eine Referenz auf diese in der Variablen baseSequence. Die Position der ein Sequenzelement
zugeordnet ist, kann sich im Zuge der Vereinigung von Sequenzen
ändern. Die Referenz auf die aktuelle Position wird in der Variablen
position gespeichert.
48
5
RESÜMEE UND AUSBLICK
Der modellorientierte Ansatz zur Prototypextraktion setzt ein Modell
voraus, dass als Baum darstellbar ist um es als Richtschnur für den
Vergleich der Instanzen zu verwenden. In Abschnitt 2.1 wurde gezeigt, dass diese Voraussetzung die Ausdruckskraft, im Hinblick auf
die möglichen Instanzen des Modells, sehr stark einschränkt. Im Vergleich zu den Produktionen einer regulären Grammatik (Typ 3) erlaubt
das eingeschränkte Modell keine Kleene’sche Hülle sowie keine rekursiven Produktionen. Dadurch ergeben sich strukturelle Unterschiede
zwischen den Instanzen lediglich durch Wahlmöglichkeiten bezüglich
der Produktionen, bildlich gesprochen also indem verschiedene “Pfade” im Modellbaum eingeschlagen werden.
5.1
evaluation
Der im Rahmen dieser Arbeit entwickelte instanzorientierte Ansatz
hingegen, setzt das gemeinsame Modell als gegeben voraus, nutzt dieses jedoch nicht um den Vergleich der Instanzen zu führen. Da das
Matching, d.h. der Vergleich, in Verbindung mit der Alignierung der
Baumstrukturen, nun ausschließlich über die Ähnlichkeit der Instanzen geschieht, musste ein Verfahren zum effizienten Matching multipler Bäume entwickelt werden. Mit einem modifizierten Verfahren
aus der Bioinformatik, dass die Hierarchie der Bäume nutzt, um die
Komplexität des Vergleichs einzuschränken, ist dies schließlich möglich geworden. Das Modell muss nun nicht mehr als endlicher Baum
dargestellt werden können, wodurch das Ziel, Prototypen aus Instanzen einer regulären Grammatik zu extrahieren, erreicht ist.
Bei der Umsetzung des Ansatzes als Java Softwarebibliothek wurde
ein besonderes Augenmerk auf die Erweiterbarkeit gelegt. Nahezu alle
Komponenten der Software lassen sich austauschen und unabhängig
voneinander nutzen, wodurch eine Plattform für zukünftige Weiterentwicklungen geschaffen wurde. Auch der objektive Vergleich verschiedener Ansätze und Teilaspekte lässt sich so einfach durchführen. Um
diese Flexibilität zu erreichen wurde darauf geachtet, jegliche typspezifische Logik, in Form definierter Strategien, auszulagern, so dass die
Softwarebibliothek leicht in konkrete Anwendungen eingebunden werden kann. Dies ist wichtig, um neben der theoretischen Auswertung
auch empirische Daten über den Nutzen dieser neuen Verfahren sammeln zu können. Auf diese Weise ist ein schneller Erfahrungsaustausch
zwischen Forschung und Anwendung möglich.
5.2
anknüpfungspunkte
Das instanzorientierte Verfahren zu Prototypextraktion bietet viel Potential in Hinblick auf dessen Anwendung, Auswertung und Optimierung.
5.2.1
Empirische Ergebnisse
Es gilt nun vielfältige Anwendungsmöglichkeiten zu finden, in denen
sich das Verfahren bewähren kann. Interessant wäre beispielsweise zu
testen, inwiefern sich die unterschiedlichen Parameter des Verfahrens,
wie der Lookahead, auf die Qualität des Prototyps und die Laufzeit
auswirken.
49
Auch die Datendarstellung hat erheblichen Einfluss auf die Komplexität der Analyse. Tiefe Hierarchien, die sich erst unterhalb des
durch den Lookahead abgedeckten Bereich unterscheiden, können zu
schlechten Ergebnissen führen. Hier wären beispielsweise automatische verlustfreie Transformationen in einem Vorverarbeitungsschritt
denkbar.
5.2.2
Iterative Alignierung
Auf Basis des entwickelten progressiven Verfahrens zur Alignierung,
könnte ein iteratives Verfahren realisiert werden, das lokale Maxima
durch ständige Verfeinerung der Alignierung vermeidet und so zu einer besseren globalen Alignierung führt. Hierbei ließen z.B. sich die
alternativen Pfade, die bei der paarweisen Alignierung entstehen, nutzen.
5.2.3
Automatisches Clustering
Weiterhin stellt das automatische Clustering auf Basis des hierarchischen Tree-Matchings eine interessante Möglichkeit dar, Teilaspekte
der Arbeit weiterzuentwickeln. Damit wäre es möglich, eine Menge
von Daten, auf Basis struktureller Ähnlichkeiten zu partitionieren. Dadurch ließen sich bisher unbekannte Zusammenhänge und Kategorien
in Daten aufdecken. Im Rahmen der Softwarebibliothek müsste hierzu
lediglich das implementierte Clusteringverfahren dahingehend erweitert werden, dass es z.B. ab einer bestimmten Clustergröße stoppt und
die so erzeugten Cluster ausgibt.
5.2.4
Globale Alignierung
Hinsichtlich der Ableitung der globalen aus den paarweisen Alignierungen wurde im vorliegenden Ansatz eine eher rechenintensive, dafür aber anschauliche Lösung vorgestellt, die zu vielen Vergleichsoperationen führt. Hier lassen sich mit Sicherheit effizientere Abläufe finden.
5.2.5
Neuer modellorientierter Ansatz
Der Nachteil des instanzorientierten Ansatzes ist, dass die Informationen, die das Modell bietet, nicht genutzt werden. Der modellorientierte
Ansatz hingegen nutzt diese Informationen, schränkt dabei jedoch die
Ausdruckskraft des Modells ein. Anstatt also die Idee eines modellorientierten Ansatzes an sich fallen zu lassen, sollte dieser vielmehr unter
anderen Prämissen neu angegangen werden. Beispielsweise wäre es
vorstellbar, die zur Erzeugung der Instanzen eines Clusters angewandten Regeln, statt die daraus resultierenden Strukturen zu generalisieren.
50
L I T E R AT U R V E R Z E I C H N I S
Agnar, A. & Plaza, E. (1994). Case-based reasoning: Foundational
issues, methodological variations, and system approaches. AI
Communications, 7 (1), 39–59.
Bader, F. (2009). Modellbasierte Klassifikation von Störungsmeldungen in
betriebswirtschaftlichen Softwaresystemen - Ein Ansatz zum PrototypLernen durch strukturbasierte Generalisierung.
Hirschberg, D. S. (1977). Algorithms for the longest common subsequence problem. Journal of the ACM (JACM), 24 (4), 664–675.
Hogeweg, P. & Hesper, B. (1984). The alignment of sets of sequences
and the construction of phyletic trees: An integrated method.
Journal of Molecular Evolution, 20 (2), 175–186.
Levenshtein, V. I. (1966, Februar). Binary Codes Capable of Correcting
Deletions, Insertions and Reversals. Soviet Physics Doklady, 10,
707.
Needleman, S. B. & Wunsch, C. D. (1970, März). A general method applicable to the search for similarities in the amino acid sequence
of two proteins. Journal of Molecular Biology, 48 (3), 443–453.
Notredame, C. (2002). Recent progress in multiple sequence alignment:
a survey. Pharmacogenomics, 3 (1), 131–144.
Schmid, U., Hofmann, M., Bader, F., Häberle, T. & Schneider, T. (2010).
Incident mining using structural prototypes (Bericht). University of
Bamberg.
Sommerville, I. (2006). Software engineering (8. Aufl.). Addison Wesley.
Thompson, J. D., Higgins, D. G. & Gibson, T. J. (1994, November). CLUSTAL w: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific
gap penalties and weight matrix choice. Nucleic Acids Research,
22 (22), 4673 –4680.
Wang, L. & Jiang, T. (1994). On the complexity of multiple sequence
alignment. Journal of Computational Biology, 1 (4), 337–348.
Yang, W. (1991, July). Identifying syntactic differences between two
programs. Software - Practice and Experience, 21 (7), 739-755.
Zhai, Y. & Liu, B. (2005). Web data extraction based on partial tree
alignment. In Proceedings of the 14th international conference on
world wide web (S. 76–85).
51
Ich erkläre hiermit gemäß § 17 Abs. 2 APO, dass ich die vorstehende
Bachelorarbeit selbstständig verfasst und keine anderen als die
angegebenen Quellen und Hilfsmittel benutzt habe.
21. Oktober 2010
Datum
Unterschrift
52