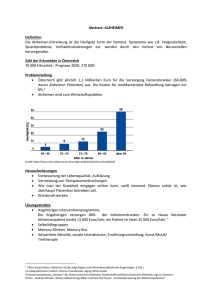
Morbus Alzheimer
Werbung

Diplomarbeit Morbus Alzheimer Aspekte der pharmakologischen Therapie eingereicht von Maximilian Fechner zur Erlangung des akademischen Grades Doktor der gesamten Heilkunde (Dr. med. univ.) an der Medizinischen Universität Graz ausgeführt am Institut für Experimentelle und Klinische Pharmakologie unter der Anleitung von Univ. Prof. i.R. Mag. pharm. Dr. phil. Eckhard Beubler und ao. Univ.-Prof. Dr. med. univ. Josef Donnerer Graz, 11. Januar 2017 Eidesstattliche Erklärung Ich erkläre ehrenwörtlich, dass ich die vorliegende Arbeit selbstständig und ohne fremde Hilfe verfasst habe, andere als die angegebenen Quellen nicht verwendet habe und die den benutzten Quellen wörtlich oder inhaltlich entnommenen Stellen als solche kenntlich gemacht habe. Graz, am 11. Januar 2017 Maximilian Fechner eh I Danksagung Zuerst und zuvorderst möchte ich meinen Eltern danken, die immer für mich da waren, die mich immer bedingungslos unterstützt haben, die mir wichtige Berater in jeder Lebenslage sind, ohne die mir dieses Studium gar nicht möglich gewesen wäre und denen gegenüber ich meinen Dank viel zu selten zum Ausdruck bringe. Univ. Prof. i.R. Mag. pharm. Dr. phil Eckhard Beubler möchte ich danken für die Ermöglichung, Betreuung und geduldige Korrektur dieser Arbeit. Auch ao. Univ.-Prof. Dr. med. univ. Josef Donnerer möchte ich meinen Dank für die Zweitbetreuung aussprechen. Schliesslich möchte ich hier noch meinen Dank an Lukas Lindinger für seine tatkräftige Unterstützung festhalten. II Zusammenfassung Der Morbus Alzheimer ist im Zuge steigender Lebenserwartung zur Volkskrankheit und sechsthäufigen Todesursache geworden. Diese häufigste aller Demenzen zeichnet sich durch pathognomonische Beta-Amyloid-Plaques und Tau-Fibrillen aus. Die ersten Medikamente wurden jedoch erst 90 Jahre nach Erstbeschreibung zugelassen und betrafen das bei der Erkankung pathologisch veränderte Neurotransmittersystem. Die Acetylcholinesterasehemmer Galantamin, Donepezil und Rivastigmin bewirken eine moderate symptomatische Besserung in leichten und mittleren Stadien der Alzheimer Demenz. Forschung im verwandten Bereich der cholinergen Agonisten konnte bislang keine wirksamen Medikamente hervorbringen. Der NMDA-Rezeptor-Antagonist Memantin zeigt ebenfalls nur geringe symptomatische Linderung in schweren Stadien der Erkrankung. In den letzten 10 Jahren konzentrierte sich die Forschung auf Hemmung und Abbau von BetaAmyloid, welches nach der Amyloid-Hypothese ursächlich für Entstehung und Verlauf der Krankheit ist. Die größten Hoffnungen ruhen auf β-Sekretase-Inhibitoren und passiver Immunisierung. Bisher konnte jedoch kein Durchbruch erzielt werden, so dass die Amyloid-Hypothese immer wieder in Frage gestellt wird. Fortschritte in der Diagnostik wie der Pittsburgh Compound B, sowie akkuratere Krankheitsmodelle in der frühen Pharmakoentwicklung lassen jedoch auf eine beschleunigte und verbesserte Entwicklung zukünfigter Wirkstoffe hoffen. III Abstract Alzheimer’s disease rose with increasing life expectancy to one of the most widespread illnesses and the sixth most important reason of death. This most common of all dementias counts beta-amyloid-plaques and tau-tangles among its hallmark features. The first drugs were only developed 90 years after first description of the disease and targeted the modified neurotransmitter-system. The Acetylcholinesterase-inhibitors Galantamine, Donepezil und Rivastigmine lead to a slight symptomatic improvement in light and moderate stages of the disease. Research in the related area of cholinergic agonists could not find effective medication either. The NMDA-receptor-antagonist Memantine shows only modest easing of symptoms in severe stages of this condition. The last 10 years saw a concentration on the inhibition and removal of beta-amyloid, which is the causing agent of the disease, according to the amyloid-hypothesis. The biggest hopes are placed on β-secretase-inhibitors and passive immunotherapy. So far, no breakthrough could be achieved, which led to the validity of the amyloid-hypothesis being questioned. Diagnostic advances like the Pittsburg Compound B, as well as more accurate models of the disease in the early stages of drug development give hope for an accelerated and improved development of future drugs. IV Inhaltsverzeichnis EIDESSTATTLICHE ERKLÄRUNG............................................................................................ I DANKSAGUNG ....................................................................................................................... II ZUSAMMENFASSUNG........................................................................................................... III ABSTRACT ............................................................................................................................. IV INHALTSVERZEICHNIS........................................................................................................... V ABKÜRZUNGSVERZEICHNIS ................................................................................................ VI ABBILDUNGSVERZEICHNIS ................................................................................................ VII EINLEITUNG............................................................................................................................ 1 MOTIVATION .......................................................................................................................... 1 GESCHICHTE ......................................................................................................................... 2 EPIDEMIOLOGIE ..................................................................................................................... 3 TERMINOLOGIE ...................................................................................................................... 6 SYMPTOMATIK........................................................................................................................ 8 PATHOPHYSIOLOGIE ............................................................................................................... 9 Amyloid ............................................................................................................................ 9 Tau ................................................................................................................................. 13 RISIKOFAKTOREN ................................................................................................................. 14 DIAGNOSTIK ........................................................................................................................ 16 Neuropsychologische Tests .......................................................................................... 17 Liquormarker ................................................................................................................. 18 Plasmamarker ................................................................................................................ 20 Elektroenzephalographie .............................................................................................. 20 Radiologie ..................................................................................................................... 21 Nuklearmedizin .............................................................................................................. 24 MATERIAL UND METHODEN ............................................................................................... 27 ERGEBNISSE ........................................................................................................................ 28 ÜBERBLICK DER PHARMAKOLOGISCHEN THERAPIE DER AD ..................................................... 28 CHOLINERGE PHARMAKA ...................................................................................................... 29 Cholinesterase Inhibitoren ............................................................................................. 30 Cholinerge Agonisten .................................................................................................... 32 GLUTAMATERGE PHARMAKA ................................................................................................. 35 NMDA Antagonisten ...................................................................................................... 35 ANTI-AMYLOIDE PHARMAKA .................................................................................................. 37 Alpha-Sekretase Aktivatoren ......................................................................................... 38 Beta-Sekretase Inhibitoren ............................................................................................ 40 Gamma-Sekretase Inhibitoren ....................................................................................... 41 Gamma-Sekretase Modulatoren ................................................................................... 43 Aktive Immunisierung .................................................................................................... 44 Passive Immunisierung ................................................................................................. 45 Apo E4 Inhibitoren ......................................................................................................... 47 Apo E2 Aktivatoren ........................................................................................................ 48 DISKUSSION ......................................................................................................................... 50 BEURTEILUNG DER CHOLINERGEN THERAPIE .......................................................................... 50 BEURTEILUNG DER GLUTAMATERGEN THERAPIE ...................................................................... 51 BEURTEILUNG DER AMYLOIDEN THERAPIE .............................................................................. 51 ZUSAMMENFASSENDE BEURTEILUNG ..................................................................................... 52 LITERATURVERZEICHNIS .................................................................................................... 54 V Abkürzungsverzeichnis 5-HT 5-Hydroxy-Tryptamin Ab Beta-Amyloid Abo Beta-Amyloid Oligomer AChE Acetyl-Cholin-Esterase AD Alzheimer Demenz ADAM A Disintegrin And Metalloproteinase (Alpha-Sekretase) ADAS-cog Alzheimer Disease Assessment Scale (cognitive part) ADDL Aβ-derived diffusible ligands AICD Aminoterminal Intra-Cellular Domain AMPA Aminohydroxy-Methylisoxazol-Propionic Acid APH1 Anterior Pharynx Defective (Teil der Gamma-Sekretase) APP Amyloid Precursor Protein BACE Beta-site APP Cleaving Enzyme (Beta-Sekretase) CT Computer Tomographie CTF-α Carboxy-Terminal Fragment CTF-β Carboxy-Terminal Fragment DGN Deutsche Gesellschaft für Neurologie FDG Fluordesoxyglucose LOAD Late Onset Alzheimer Demenz MCI Mild Cognitive Impairment MMSE Mini-Mental State Examination MRT Magnet Resonanz Tomographie NMDA N-Methyl-D-Aspartat NPI Neuro-Psychiatric Inventory PAM Positiver Allosterischer Modulator PEN Presenilin ENhancer enzyme (Teil der Gamma-Sekretase) PET Positronen Emissions Tomographie PS1 Präsenilin (Teil der Gamma-Sekretase) sAPP soluble APP, ein Teilfragment nach einem Sekretase Schnitt sAPP-α sAPP nach dem Alpha-Sekretase Schnitt sAPP-β sAPP nach dem Beta-Sekretase Schnitt SMD Standardized Mean Difference SPECT Single-photon Emission Computed Tomography α4β2 Nikotinischer Alpha-4-Beta-2-Rezeptor α7 Nikotinischer Alpha-7-Rezeptor VI Abbildungsverzeichnis FIGURE 1: ALTERSABHÄNGIGE INZIDENZ & PRÄVALENZ (LINKS) SOWIE REGIONALE PRÄVALENZVERTEILUNG (RECHTS) ....................................................................................... 3 FIGURE 2: HISTORIE & PROGNOSE ZUM ALTENANTEIL DER BEVÖLKERUNG (LINKS), SOWIE PROGNOSE DER FALLZAHLEN (RECHTS) ............................................................................... 4 FIGURE 3: WELTWEITE KOSTEN (LINKS), SOWIE KOSTENVERTEILUNG NACH SEKTOREN UND STAATSGRUPPEN (RECHTS) ................................................................................................. 4 FIGURE 4: KOSTEN- & PATIENTENVERTEILUNG (LINKS), SOWIE KOSTENUNTERSCHIEDE PRO PATIENT/IN (RECHTS) .......................................................................................................... 5 FIGURE 5: DSM-5 KLASSIFIKATION (AMERICAN PSYCHOLOGICAL ASSOCIATION, 2013) .................. 7 FIGURE 6: HÄUFIGKEIT VERSCHIEDENER DEMENTIELLER SYMPTOME (DEKOSKY, LOPEZ, JONES, FITZPATRICK, & BREITNER, 2002)........................................................................................ 8 FIGURE 7: ALPHA-, BETA- & GAMMA-SEKRETASE (LICHTENTHALER, 2012) .................................. 10 FIGURE 8: Α, Β UND g - SEKRETASEN (DE STROOPER, VASSAR, & GOLDE, 2010) .......................... 12 FIGURE 9: BRAAK STADIEN (ALZHEIMER-FORSCHUNG.DE, 2016) ................................................. 14 FIGURE 10: MINI-MENTAL STATUS (DEUTSCHE GESELLSCHAFT FÜR HÄMATOLOGIE UND ONKOLOGIE, 2016) .......................................................................................................... 17 FIGURE 11: ADAS-COG TEST (DEMENTIA COLLABORATIVE RESEARCH CENTERS, 2016) ............... 18 FIGURE 12: INTENSITÄTSVERTEILUNG ÜBER EEG-FREQUENZEN. LINKS KONTROLLGRUPPE, RECHTS ALZHEIMER-ERKRANKTEN (DAUWELS, ET AL., 2011) ........................................................... 21 FIGURE 13: VOXEL-BASIERTE (LINKS) VERSUS ATLAS-BASIERTE (RECHTS) MRT-ANALYSE. JE “WÄRMER” DIE FARBE, DESTO STÄRKER DIE KORTIKALE ATROPHIE UND DIE PLAQUEWAHRSCHEINLICHKEI (CUINGNET, ET AL., 2011) ................................................................ 22 FIGURE 14: FORM-ANALYSE DES ENTORHINALEN KORTEX, JE KÄLTER DIE FARBE, DESTO STÄRKER DIE EINZIEHUNG (DEVANAND, ET AL., 2012) ....................................................................... 22 FIGURE 15: DIFFUSIONSGEWICHTETES MRT, MAN SIEHT DIE DIFFUSIONSZUNAHME BEI DER ALZHEIMER-DEMENZ IM VERGLEICH ZUR KONTROLLGRUPPE (DOUAUD, ET AL., 2011) .......... 23 FIGURE 16: CT-ANALYSE, INTENSITÄTSMINDERUNGEN DER GRAUEN SUBSTANZ, ÄQUIVALENT ZU NERVENZELLVERLUSTEN, IN ORANGE (IMABAYASHI, ET AL., 2013) ....................................... 23 FIGURE 17: GLUKOSE STOFFWECHSEL, LINKS MCI PATIENT/IN, RECHTS ALZHEIMER PATIENT/IN (NORDBERG, RINNE, KADIR, & LAANGSTROM, 2010) .......................................................... 24 FIGURE 18: AMYLOID-AGGREGATION, LINKS ALZHEIMER-PATIENT/IN, RECHTS KONTROLLPERSON, VISUALISIERUNG MITTELS PITTSBURGH COMPOUND B (NORDBERG, RINNE, KADIR, & LAANGSTROM, 2010) ....................................................................................................... 25 FIGURE 19: TACRINE (CHEMBASE.CN) ........................................................................................ 30 FIGURE 20: GALANTAMINE (CHEMBASE.CN) ............................................................................... 30 FIGURE 21: DONEPEZIL (CHEMBASE.CN) .................................................................................... 30 FIGURE 22: RIVASTIGMINE (CHEMBASE.CN) ................................................................................ 31 FIGURE 23: PHENSERIN (CHEMNET.COM) ................................................................................... 31 FIGURE 24: HUPERZIN A (CHEMBASE.CN) .................................................................................. 32 FIGURE 25: XANOMELINE (WIKIPEDIA.ORG) ................................................................................ 33 FIGURE 26: BQCA (D KUDUK 2014) ........................................................................................ 34 FIGURE 27: ENCENICLIN (WIKIPEDIA.ORG) ................................................................................. 34 FIGURE 28: NELONICLIN (DRUGSPIDER.COM) ............................................................................. 34 FIGURE 29: VARENICLIN (CHEMBASE.CN) .................................................................................. 34 FIGURE 30: MEMANTIN (CHEMBASE.CN) .................................................................................... 36 FIGURE 31: KETAMIN (CHEMBASE.CN) ....................................................................................... 37 FIGURE 32: ETAZOLAT (WIKIPEDIA.ORG) .................................................................................... 39 FIGURE 33: EGCG (WIKIPEDIA.ORG) ......................................................................................... 39 FIGURE 34: BRYOSTATIN (WIKIPEDIA.ORG) ................................................................................. 39 FIGURE 35: LY-2811-376 (MEDCHEMEXPRESS.COM) ................................................................. 40 FIGURE 36: VERUBECESTAT (WIKIPEDIA.ORG) ............................................................................. 41 FIGURE 37: SEMAGACESTAT (WIKIPEDIA.ORG) ............................................................................ 42 FIGURE 38: BEGACESTAT (CHEMBASE.CN) ................................................................................ 42 FIGURE 39: AVAGACESTAT (SIGMAALDRICH.COM) ...................................................................... 42 FIGURE 40: E2012 (INTERNATIONAL JOURNAL OF ALZHEIMER’S DISEASE) ................................... 43 FIGURE 41: BEXAROTEN (WIKIPEDIA.ORG) .................................................................................. 48 VII Einleitung Motivation Alzheimer ist eine Volkskrankheit. Und obwohl die Erstbeschreibung dieser Krankheit mittlerweile über 100 Jahre zurückliegt, ist noch immer keine effektive Therapie gefunden worden. Das könnte damit zusammenhängen, daß der Morbus Alzheimer über weite Teile des 20. Jahrhunderts als normaler Alterungsprozess abgetan wurde und sich Betroffene bestenfalls in Scham zurückgezogen haben. Dabei ist die Liste prominenter Erkrankter lang. Eine der ersten berühmten erkrankten Personen, die sich öffentlichkeitswirksam geäußert hat, war die ehemalige regierende Person der USA Ronald Reagan. Er teilte seine Gedanken in einem Brief mit der Öffentlichkeit. Dabei berührt er gleich mehrere wichtige Punkte. Er betont insbesondere die Bürde, die er seiner Frau und Familie damit auferlegt. Er spricht über das wichtige Thema der weiteren Lebensgestaltung nach Diagnosestellung. Aber auch das volle Bewusstsein der kommenden geistigen Regression findet eindrucksvollen Ausdruck: “At the moment, I feel just fine. I intend to live the remainder of the years God gives me on this earth doing the things I have always done. […] Unfortunately, as Alzheimer's disease progresses, the family often bears a heavy burden. I only wish there was some way I could spare [my wife] from this painful experience. […] I now begin the journey that will lead me into the sunset of my life.” - Ronald Reagan In der Tat ist diese Bürde so gross, dass sich die Mehrheit der Angehörigen im Verlauf der Erkrankung entscheidet, die Betreuung zu institutionalisieren, oft nach Jahren der erschöpfenden persönlichen Betreuung. Für die Erkrankten selbst sind die wachen Momente, in denen sie ihren eigenen Verfall bewusst wahrnehmen, vielleicht am schwersten zu verkraften. Und welche verheiratete Person würde nicht verzweifeln, wenn Jahrzehnte der gemeinsamen Erinnerung schon vor dem Leben selbst verrinnen. Und schliesslich, was bleibt vom Menschen, wenn seine Seele schon gegangen ist? Neben der Tragik der persönlichen Schicksale stellt die Alzheimer-Erkrankung aber auch eine große gesellschaftliche Herausforderung dar. Die Gesamtkosten belaufen sich auf bis zu einer halbe Million US-Dollar pro erkrankter Person (Alzheimer's Disease International, 2016) und im Angesicht der weiter fortschreitenden Alterung der westlichen Gesellschaften wird der Anteil der Erkrankten und somit die Belastung des Gesundheitssystems immer größer. 1 Geschichte Die Erkrankung ist nach der ärztlichen Person Alois Alzheimer benannt. Er bemerkte während seiner Tätigkeit an der städtischen “Irrenanstalt” in Frankfurt im Jahr 1901 an der 51-jährigen erkrankten Person Auguste Deter eine Symptomatik, die er keiner bekannten Erkrankung zuordnen konnte. Alzheimer wechselte kurze Zeit später zu Emil Kraeplin nach Heidelberg und nur wenig später gemeinsam mit diesem nach München. Den Fall der Auguste Deter verfolgte er aber aus der Entfernung weiter und ließ sich nach ihrem Tod ihr Gehirn schicken. In diesem fand er die für die Erkrankung typischen Plaques und beschrieb sie zum ersten Mal. In Abgrenzung zur “normalen” Degeneration des Alters und angesichts der relativen Jugend seiner Patientin benutzte er den Terminus “Präsenile Demenz”. Das war 1906. Schon wenige Jahre später fand Alzheimer die beschriebenen Plaques aber auch im Gehirn alter Menschen und hielt daraufhin die prä-senile und die senile Demenz für Varianten derselben Krankheit. Sein Chef Emil Kraeplin prägte ihm zu Ehren den Namen Morbus Alzheimer für die Erkrankung in seinem Lehrbuch “Psychatrie” und setzte seinen Schützling außerdem gegen andere kandidierende Personen auf einen Lehrstuhl in Breslau durch. Erst über 70 Jahre später, 1984 und 1986 wurden das Beta-Amyloid und das TauProtein als Schlüsselelemente der Alzheimer-Krankheit identifiziert. 1993, also 90 Jahre nach der Erstbeschreibung wurde das erste Medikament zugelassen, der Acetylcholinesterase-Hemmer Tacrin. Im selben Jahr fand man die APOe4 Mutation als genetischen Risikofaktor. Dann gelang 1995 die genetische Modifikation von Mäusen, die daraufhin Plaques wie bei Alzheimer-Erkrankten entwickelten. An diesen Mäusen ließen sich Medikamente nun leichter testen. 2002 wurde Memantin zugelassen, welches zusammen mit den CholinesteraseHemmern bis heute die Hauptmedikation bei Morbus Alzheimer darstellt. Die Diagnostik erfuhr 2004 einen wesentlichen Schub, als man den “Pittsburgh Compound B” fand, der sich an Beta-Amyloid anheftet und mittels PositronenEmisons-Tomographie sichtbar gemacht werden kann. 2010 rückte Alzheimer auf den 6. Platz der häufigsten Todesursachen in den Vereinigten Staaten (Alzheimer's Association, 2016). Trotz der intensiven Bemühung steht ein wirklicher Durchbruch in der Therapie noch aus. 2 Epidemiologie Der Morbus Alzheimer ist eine degenerative Erkrankung und betrifft vorwiegend den alten Menschen. Dementsprechend findet man in Inzidenzund Prävalenzuntersuchungen eine deutliche Zunahme mit steigendem Alter. Man geht momentan von einer Verdopplung der Erkrankungswahrscheinlichkeit ca. alle 6 Jahre nach dem 60. Lebensjahr (Alzheimer's Disease International, 2016) und somit einer exponentiellen Zunahme der Erkrankungswahrscheinlichkeit mit fortschreitendem Alter aus. Beobachtet man bei den 60-Jährigen noch eine Prävalenz von etwa 1%, so sind bei den 90-Jährigen bis zu 40% betroffen, je nach Region & Geschlecht sogar über 50%. Inzidenz&Prävalenznach AlterinEuropa Regionen 8% 7% 6% 5% 4% 3% 2% 1% 0% 50% 40% 30% 20% 10% 0% 60-64 65-69 70-74 75-79 80-84 85-89 90+ Inzidenz Regionen Prävalenz Figure 1: Altersabhängige Inzidenz & Prävalenz (links) sowie regionale Prävalenzverteilung (rechts) Die regionalen Unterschiede sind durchaus erheblich. Warum in Südamerika mehr als doppelt so viele Alzheimer-Erkrankte leben wie in Zentralafrika bleibt gegenwärtig Gegenstand der Spekulation. Unterschiede in der Diagnostik könnten genauso dafür verantwortlich zeichnen wie Differenzen im Lebensstil. Übergewicht und Bewegungsmangel werden als Risikofaktoren diskutiert. Auch eine Korrelation mit der Lebenserwartung ist denkbar. In Zentralfrika liegt sie mit rund 60 Jahren deutlich niedriger als in Europa (ca. 80 Jahre) oder Lateinamerika (ca. 70 Jahre) (WHO, 2016). Die durchschnittliche Lebenserwartung nach Diagnose beträgt 9 Jahre, ist aber individuell sehr unterschiedlich. Die Lebenserwartung im Einzelfall liegt zwischen 2 Jahren und 20 Jahren. (Alzheimer’s Foundation of America, 2016) 3 Anteilder65+&85+ander Gesamtbevölkerung PrognostizierteFallzahlen WeltweitinMillionen 30,0% 150 20,0% 100 10,0% 50 0,0% 0 2015 2020 2025 2030 2035 2040 2045 2050 1900 1920 1940 1960 1980 2000 2020 2040 65Jahreundälter AlzheimerFälle 85Jahreundälter Figure 2: Historie & Prognose zum Altenanteil der Bevölkerung (links), sowie Prognose der Fallzahlen (rechts) Der Anteil der Menschen an der Gesamtbevölkerung in den USA, die älter sind als 65 Jahre, hat sich von 4% im Jahr 1900 auf 13% im Jahr 2010 mehr als verdreifacht. Der Anteil der über 85-jährigen hat sich im gleichen Zeitraum von 0,2% auf 1,9% fast verzehnfacht. Bis zum Jahr 2050 wird mit einer weiteren Verdopplung der jeweiligen Anteile auf 20,2% bzw 4,3% gerechnet (Administration on Aging, 2016). Die gegenwärtigen weltweiten Fallzahlen liegen bei 46 Millionen Erkrankten und man rechnet mit einer Verdreifachung auf 131 Millionen bis 2050. (Alzheimer's Disease International, 2016) KosteninMillionenUSDollar Kostenverteilung 80% $1.500 60% $1.000 40% $500 20% $0 2010 2015 2018 0% Medizinisch Sozial ArmeLänder KosteninMillionenUS-Dollar Indirekt ReicheLänder Figure 3: weltweite Kosten (links), sowie Kostenverteilung nach Sektoren und Staatsgruppen (rechts) Die Kosten für die Betreuung von Alzheimer-Erkrankten sind enorm. Gegenwärtig liegen sie weltweit bei ca 800 Milliarden US-Dollar. Das liegt in der Größenordnung des Bruttoinlandsproduktes der Türkei (ca. 850 Milliarden US-Dollar) und deutlich 4 über dem Österreichs (ca. 400 Milliarden US-Dollar). Die symbolische Grenze von einer Billion US-Dollar wird nach extrapolierter gegenwärtiger Entwicklung bereits 2018 überschritten. Die Entwicklung eines Medikaments mit auch nur geringer progredienzhemmender Wirkung birgt also neben Lebensqualitätsgewinn großes volkswirtschaftliches Potenzial. Dabei muss man aber beachten, daß nur ca. die Hälfte dieser Kosten direkte Kosten für Medizin- & Sozialsystem darstellen. Die andere Hälfte geht als schwer kalkulierbare indirekte Kosten der Angehörigenbetreuung in die Rechnung ein. (Alzheimer's Disease International, 2016) RegionaleKosten- & Erkranktenverteilung Weltweite Kostenunterschiedepro erkrankterPerson 70% 60% $60.000 $50.000 $40.000 $30.000 $20.000 $10.000 $0 50% 40% 30% 20% 10% 0% G7 G20ohne G7 Kostenanteil Rest Erkranktenanteil Regionen Figure 4: Kosten- & Erkranktenverteilung (links), sowie Kostenunterschiede pro erkrankter Person (rechts) Die Kosten sind dabei weltweit sehr ungleich verteilt. Auf die G7-Staaten entfallen circa 20% der Erkrankten aber über 60% der Kosten. Das liegt einerseits am insgesamt deutlich höheren Lohnniveau der ersten Welt. Aber auch die Verteilung der Ausgaben unterscheidet sich. Die direkten Kosten liegen in der ersten Welt höher, in ärmeren Ländern wird dagegen ein höherer Anteil der Versorgung von Angehörigen geleistet, was als indirekte Kosten in die Gesamtrechnung eingeht. Wenn man sich die Kosten pro erkrankter Person anschaut, werden diese Unterschiede noch besser deutlich. Während eine an Alzheimer erkrankte Person in Zentralafrika 1000$ im Jahr kostet, in Osteuropa schon 8000$ veranschlagt werden, so belaufen sich die Kosten in Nordamerika auf bis zu 50.000$ pro Jahr (Alzheimer's Disease International, 2016). Das bedeutet bei einer gerundeten durchschnittlichen Lebenserwartung von 10 Jahren Gesamtkosten von bis zu einer halben Million USDollar pro erkrankter Person. 5 Terminologie Der Morbus Alzheimer ist eine chronisch progrediente kortikale Neurodegeneration mit pathognomonischen Beta-Amyloid-Plaques und Tau-Bündeln. Die AD (von English “Alzheimer’s Disease” oder auch “Alzheimer Demenz”) ist mit etwa 60% die mit Abstand häufigste Ursache für die Entwicklung einer Demenz (von lateinisch: “de-mens”, “abnehmender Verstand”). Die Begriffe “Alzheimer” und “Demenz” werden fälschlicherweise häufig synonym verwendet. Dabei beschreibt der Begriff AD zunächst einen neurodegenerativen Prozess, der in seiner Folge dementielle Symptome bewirkt. Eine Demenz hingegen ist ein Symptomen-Komplex, zu dem neben der typischen kognitiven Verschlechterung auch Persönlichkeitsveränderung, Agitiertheit, Wahnvorstellungen, Depressionen und mehr gehören. Anatomisch gehört die AD zu den kortikalen Demenzen mit Beginn und Fokus der Degeneration in Area Entorhinalis und Hippokampus. Die zweithäufigste kortikale Demenz ist der Morbus Pick (nach Arnold Pick, etwa 10% der Demenzen), bei dem man vor allem im Frontal-Lappen die typischen Pick-Körperchen findet. Davon abgrenzen kann man die subkortikalen Demenzen wie die Lewy-KörperDemenz (Frederik Lewy arbeitete im gleichen Labor wie Alois Alzheimer, 10% der Demenzen), die durch ihren Fokus auf die Basalganglien neben der dementiellen Symptomatik auch eine starke motorische Beeinträchtigung zeigt. Auch der Morbus Binswanger (nach Otto Binswanger, u.a. Therapeut Nietsches) als prominentestem Beispiel der vaskulären Demenzen gehört mit seiner arteriosklerotisch bedingten periventrikulären Demyelinisierung zu den subkortikalen Demenzen und stellt ebenfalls etwa 10% der Fälle. Von den oben genannten primären Demenzen, die zusammen für etwa 90% der Demenzen verantwortlich zeichnen (und interessanterweise alle im deutschsprachigen Raum erstbeschrieben wurden), kann man die sekundären Demenzen abgrenzen, die Folge einer anderen Grunderkrankung oder eines Drogen- oder Medikamentenmissbrauchs sind. Durch die grosse Sensibilität des Gehirns kann eine ganze Reihe von Erkankungen zumindest temporär dementielle Symptome hervorrufen, zuvorderst seien die grossen Volkskrankheiten Bluthochdruck, Diabetes mellitus und Hyperlipidämie genannt. Aber auch viele andere Stoffwechsel- und Mangelerkrankungen können sich als Demenz manifestieren. Der etwas diffuse, aber in der Literatur sehr häufig verwendete Begriff “Mild Cognitive Impairment” (MCI) bezeichnet ein prä-dementes Stadium, bei dem die kognitive 6 Leistung bereits unter dem Altersdurchschnitt liegt, jedoch noch keine Alltagseinschränkungen verursacht. Häufig entwickelt sich in Folge eine Demenz. Der Ausdruck “präsenile Demenz” schliesslich wurde zunächst zur Abgrenzung gegenüber der für normal befundenen “senilen Demenz” geprägt. Einige schreibende Personen halten die Trennung aufgrund möglicher Unterschiede in Ursache und Therapie für sinnvoll, andere streben eine Zusammenlegung der Begriffe an. Die ICD10 sieht eine Differenzierung vor (F00.0 & F00.1). Eine interessante Theorie besagt, dass nur bei der präsenilen AD die Plaques zum Gewebsuntergang führen, diese beim senilen Alzheimer nur Folge der ohnehin stattfindenen Degeneration seien. Die “American Psychiatric Association” (APA) hat in ihrer 2013 erneuerten DSM-5 Leitlinie den Begriff “Demenz” durch “Neurocognitive Disorder” ersetzt, der dann nach Ursache und Schweregrad subklassifiziert wird. Dafür werden zwei Gründe angeführt: Den Fokus auf “Störung” und damit Behandelbarkeit im Gegensatz zur als normal empfundenen Demenz zu legen, sowie die Stigmatisierung des Begriffes “Demenz” zu umgehen. Die für 2017 erwartete ICD-11 wird voraussichtlich eine ähnliche Begriffsänderung vornehmen. Figure 5: DSM-5 Klassifikation (American Psychological Association, 2013) 2011 haben das “Institute on Aging” (NIA) und die “Alzheimer's Association” (AA) nach 1984 die erste Anpassung ihrer Leitlinie vorgenommen, in der sie die AlzheimerDemenz in 3 Stadien unterteilen (National Institute of Aging, 2011) (McKhann, et al., 2011) 1. Vollausgeprägte Demenz 2. Prä-dementielles Stadium, mit milden Symptomen wie MCI 3. Prä-klinisches Stadium, ohne Symptome, aber diagnostizierbar Das präklinische Stadium trägt den Fortschritten der Diagnostik Rechnung und ist von entscheidender Bedeutung, da die Therapie in diesem Stadium wahrscheinlich am wirkungsvollsten ist. Abschliessend kann man sagen, dass die Terminologie der Bereiche Morbus Alzheimer und Demenz dank der erheblichen Fortschritte in Diagnostik und Therapie 7 seit einigen Jahren starken Änderungen unterliegt und wohl noch nicht abgeschlossen ist. Symptomatik Die Symptomatik des Morbus Alzheimer ähnelt in weiten Teilen der anderer Demenzen und die Abgrenzung zu diesen kann schwierig sein. Stärkere Vergesslichkeit, Verlegen von Gegenständen, Stimmungsschwankungen wie Aggressivität, Misstrauen und Depression, Affektverflachung, Wiederholen von Fragen, Wortfindungsstörungen, Orientierungsprobleme in vertrauter Umgebung und sozialer Rückzug sind die wichtigsten Auffälligkeiten, die Angehörige oder auch die Erkrankten selbst oft aufmerksam werden lassen (Alzheimer's Association, 2016) (Mayo Clinic, 2016). Häufigkeit Symptom 36% Apathie 32% Depression 30% Aggressivität 27% Schlafstörungen 21% Angst 20% Essstörungen 18% Wahnvorstellungen 13% Enthemmung 10% Halluzinationen 3% Euphorie Figure 6: Häufigkeit verschiedener dementieller Symptome (DeKosky, Lopez, Jones, Fitzpatrick, & Breitner, 2002) Dabei folgt das Auftreten der Symptome im allgemeinen einem bestimmten zeitlichen Verlauf. Schon in der Phase des MCI manifestiert sich dabei häufig eine Depression, wohl im Gleichschritt mit dem zunehmenden Bewusstsein der erkrankten Person über den eigenen kognitiven Verfall. Mit fortschreitender Demenz verschwinden die depressiven Symptome und werden durch Halluzination und Wahn abgelöst. Angst, Agitiertheit und Aggressivität finden sich vor allem im Endstadium der Demenz, wenn die erkrankte Person die einströmenden Wahrnehmungen nicht mehr verarbeiten und einordnen kann. 8 Pathophysiologie Die Pathophysiologie der Alzheimer Demenz ist noch nicht vollständig geklärt. Das grösste Interesse der Forschung richtet sich auf die beiden pathognomonischen Merkmale der AD, die Beta-Amyloid Plaques und die Tau-Protein-Fibrillen. Es ist strittig, welches dieser Proteine ursächlich für den neuronalen Zelltod ist. Amyloid Alois Alzheimer selbst entdeckte die perineuralen Amyloid-Ablagerungen bereits 1906 bei seiner prä-senil dementen Patientin, was ihn veranlasste, von einer neuen Krankheit zu sprechen. Amyloid-Ablagerungen waren schon vorher von anderen Stellen im Körper bekannt. Der Begriff geht auf Rudolf Virchow zurück, der die “Stärke-ähnliche” Anfärbbarkeit der copora amylacea 1854 beobachtete. Es sind verschiedene Krankheiten bekannt, deren Ursache in Amyloid-Ablagerungen im Gewebe liegt. Dafür wählte man den Oberbegriff “Amyloidosen”. Diese unterscheiden sich in den ihnen zugrunde liegenden Proteinen. Allen Amyloidosen gemein ist die Anfärbbarkeit mit Kongorot. Die pathogenen Wirkungen können dabei systemisch sein oder auch lokal wie beim Morbus Alzheimer. Ist zum Beispiel das Myokard betroffen, kann sich eine restrikive Kardiomyopathie entwickeln. Bei Lokalisation in den Nieren resultiert häufig ein nephrotisches Syndrom. Im Kontext der Alzheimer Demenz sind außerdem die peri-vaskulären Amyloid-Ablagerungen von Bedeutung, die zusätzlich zum direkten neuronalen Zelltod einen indirekten Zelltod über Mikroinfarkte verursachen können. Ursächlich für die ProteinAblagerungen der Amyloidosen ist in der Regel die Fehlfaltung eines Proteins, zum Beispiel aufgrund von Veränderungen in der Gen-Expression. Diese veränderten Proteine können dann nicht mehr abgebaut werden. Da die Neubildung in der Regel nicht gestört ist reichern sie sich in immer grösserer Menge im Gewebe an bis schliesslich die Primärstruktur des Gewebes zerstört wird. Die perineuralen Amyloid-Ablagerungen bei der Alzheimer Demenz nennt man “Plaques” oder “Drusen”. Sie bestehen zum größten Teil aus einer ganz bestimmten Form von Amyloid, dem Beta-Amyloid (abgekürzt “Aβ”). Aber auch Apolipoprotein E ist Teil der Plaques. Das Aβ entsteht aus dem Amyloid-Vorläufer-Protein (APP = Amyloid Precursor Protein), einem Protein der neuronalen Zellmembran. Zur physiologischen Funktion des APPs existieren bislang nur Spekulationen. Das APP kommt in sämtlichen 9 Wirbeltieren sowie vielen anderen Tierarten vor und gilt als phylogenetisch altes, stark konserviertes Protein (Tharp & Sarkar, 2013). Beim Menschen kennt man eine Reihe von Varianten des APP, die die Produktion von Aβ deutlich erhöhen können und ein starker Risikofaktor für die erbliche Form der Alzheimer Demenz sind. Andere APPMutationen hingegen können die Aβ-Produktion senken und somit Alzheimerpräventiv wirken (Jonsson, et al., 2012). APP kommt in den meisten Körperzellen vor, aber die AD-relevante Isoform APP-695 findet man nur in Neuronen. APP Schnitt Varianten Eine Reihe von Sekretasen kann das APP zerschneiden. Bislang identifiziert wurden die Alpha-, Beta- und Gamma-Sekretase. Diese drei Sekretasen kommen alle physiologisch in der neuronalen Zellmembran vor und schneiden das APP an unterschiedlichen Positionen. Figure 7: Alpha-, Beta- & Gamma-Sekretase (Lichtenthaler, 2012) Die Alpha- und Beta-Sekretase schneiden beide extra-zellulär und schliessen sich gegenseitig aus. D.h. wenn die Alpha-Sekretase geschnitten hat, wird die BetaSekretase nicht mehr schneiden und umgekehrt. Die extrazellulären Reste dieser Schnitte (sAPP-α und sAPP-β) haben bisher keine bekannte pathologische Wirkung. Die membranständigen Reste des Alpha- bzw Beta-Schnitts (CTF-α / CTF-83 und CTF-β / CTF-99, Carboxy-Terminales Fragment) werden anschliessend von der Gamma-Sekretase gespalten. Die Gamma-Sekretase arbeitet dabei innerhalb der Zellmembran und kann an zwei benachbarten Positionen schneiden. Die Kombination aus Alpha- und Gamma-Schnitt erzeugt das harmlose Fragment p3. Der für die Alzheimer Demenz relevante Schnitt, die Kombination aus Beta- und GammaSekretase, erzeugt das pathologische Beta-Amyloid (Aβ). 10 Je nach genauer Gamma-Schnittposition am CTF sind die zwei Formen Aβ-40 und Aβ-42 bekannt. Die Aβ-42-Variante ist dabei stärker hydrophob, schwerer löslich, neigt stärker zu Plaque-Bildung und gilt als stärker pathologisch als Aβ-40. Die durch die Gamma-Sekretase entstehenden Fragmente p3, bzw Aβ werden in den Extrazellularraum abgegeben, die andere Hälfte des Schnittes nennt man AICD (Aminoterminale Intra-Cellulare Domäne) und diese wird intrazellulär abgebaut. Die Kombination aus Alpha- und Gamma-Sekretase erzeugt also die 3 Fragmente sAPP-α, p3 und AICD. Die Kombination aus Beta- und Gamma-Sekretase erzeugt die 3 Fragmente sAPP-β, Aβ und AICD. APP Sekretasen Die Alpha-Sekretasen gehören zur Familie der ADAM (A Disintegrin And Metalloproteinase). 1998 wurde bei einem Tumor-Nekrose-Faktor-ConvertingEnzyme (TACE) die Fähigkeit zum Alpha-Schnitt festgestellt und ADAM17 genannt. 1999 wurden ADAM9 und ADAM10 als Alpha-Sekretasen identifiziert. Nachdem lange Zeit unbekannt war, welche der Sekretasen in Bezug auf den APP-Schnitt die grösste Spezifität hat, erachtet man mittlerweile die ADAM10 als wesentlich (Kuhn, et al., 2010). Neben der verringerten Bildung von Beta-Amyloid scheint ein Spaltprodukt der Alpha-Sekretasen, das sAPP-α, zusätzlich neuroprotektive und synaptogene Wirkung zu haben (Thornton, Vink, Blumbergs, & Heuvel., 2006). Man kennt zwei Subtypen der Beta-Sekretase. BACE1 erzeugt Beta-Amyloid, BACE2 nicht. BACE2 kommt im Gehirn kaum vor. Die beiden Enzyme haben jedoch eine Übereinstimmung in etwa 64% ihrer Aminosäuren-Sequenz. Das erschwert die Entwicklung eines BACE1-spezifischen Effektors. 1999 konnte die BACE1 zum ersten Mal synthetisiert und die beta-amyloidogene Wirkung in vitro demonstriert werden (MacLeod, Hillert, Cameron, & Baillie, 2015). Nur membranständige BACE1 kann APP schneiden, lösliche BACE1 hat diese Fähigkeit nicht. Mäuse mit deaktivierter BACE1 produzieren kein Beta-Amyloid (Luo, et al., 2001). Das bedeutet im Umkehrschluss, dass nur die BACE1 Beta-Amyloid erzeugt. Die Beta-Sekretase ist aber nicht nur bei AD-Erkrankten aktiv, sondern auch bei gesunden Personen. Möglicherweise ist eine Kombination aus erhöhter BetaSekretase-Aktivität, verminderter Alpha-Sekretase-Aktivität und vermehrter APPExpression für die Zunahme von Aβ verantwortlich. 11 Da komplett inaktivierte BACE1 zu Problemen wie Myelinisierungsstörungen führt, gibt es Versuche der Teilinhibition der BACE1 bei heterozygoten Mäusen. Dies führt erstaunlichweise nur bei weiblichen Mäusen zu einer Plaque-Reduktion (Sadleir, Eimer, Cole, & Vassar, 2015). Figure 8: α, β und g - Sekretasen (De Strooper, Vassar, & Golde, 2010) Die BACE-Expression scheint durch Beta-Amyloid gefördert zu werden (Mamada, et al., 2015). Das wäre eine (sich selbst verstärkende) positive Feedback-Schleife, die man normalerweise in Organismen sehr selten findet. Die Gamma-Sekretase besteht aus Presenilin 1 & 2 (PS1, PS2) und dem Presenilin Enhancer Enzyme 2 (PEN2), sowie Nicastrin und der Anterior Pharynx-Defective 1 (APH1) (Yonemura, et al., 2011). Sie wurde als letztes der drei APP-Schnitt-Enzyme entdeckt und scheint einen hohen Grad an Heterogenität aufzuweisen. Ausserdem ist die Gamma-Sekretase deutlich länger und komplexer als die beiden anderen Sekretasen. PS1 scheint etwa 24 mal mehr Beta-Amyloid zu produzieren als PS2 (Yonemura, et al., 2011) 12 Beta-Amyloid Pathomechanismus Der genaue Pathomechanismus des Beta-Amyloids selbst ist noch ungeklärt. Ungewöhnlich für ein Protein ist der schwache strukturelle Zusammenhalt. So konnte man zeigen, dass Beta-Amyloid keine feste dreidimensionale Struktur aufweist, sondern sporadisch zwischen einer Reihe von Faltungen wechselt. Das schwerer lösliche und pathologischere Aβ-42 Monomer ist dabei konformationell stabiler als das Aβ-40 (Yang & Teplow, 2008). Möglicherweise sind aber gar nicht die Plaques selber pathologisch, sondern schon deren Vorstufe, die ADDLs (A-beta-Derived Diffusible Ligands, lösliche BetaAmyloid-Oligomere) (Catalano, et al., 2006). Nach einer Theorie ist das lösliche Aβ direkt verantwortlich für die neuronale Degeneration, indem es mit Insulin um Rezeptorstellen konkurriert und so den Glukosemetabolismus einschränkt (Xie, et al., 2002). Manche Arbeitsgruppen gehen sogar soweit, die Einschränkung des Glukosemetabolismus als Diabetes Typ 3 zu bezeichnen (Steen, et al., 2005). Aβ-Plaques finden sich dabei nicht nur peri-neuronal sondern auch peri-vaskulär. Die vaskulären Plaques enthalten einen größeren Anteil and Aβ-40, während die neuronalen Plaques hauptsächlich aus Aβ-42 bestehen. Ebenfalls ungeklärt ist, ob Amyloid oder Tau für den kognitiven Verfall ursächlich sind. Seit Aufstellung der Amyloid-Hypothese Mitte der 80er Jahre konzentriert sich die Pharmakoentwicklung stark auf diesen Aspekt der Erkrankung. Tau Tau-Protein hilft normalerweise durch Anlagerung an Mikrotubuli bei der räumlichen Stabilisierung des Zytoskeletts. Bei zu starker Phosphorylierung (Hyperphosphorylierung) verliert es seine physiologische Funktion, reichert sich zunächst im Zytosol an und bildet schliesslich funktionslose Knäuel, so genannte “neurofibrillary tangles”. Die Menge dieser Knäuel korreliert direkt mit dem Krankheitsfortschritt und wird seit 1991 nach Braak in 6 Stadien eingeteilt. Im Endstadium findet man die Tau-Knäuel auch ausserhalb der Zellen und spricht dann von “Ghost-Tangles”. Es konnte gezeigt werden, dass das Tau-Protein bei Alzheimer-Erkrankten in konformationell veränderter Form vorliegt. Ob dies die Ursache für die verstärkte Phosphorylierung ist, oder ein Missverhältnis von Kinasen und Phosphatasen (Bindung & Entfernung von Phosphor) konnte noch nicht geklärt werden. Neben dem 13 Tau-Protein findet man bei Alzheimer-Erkankten auch hyperphosphorylierte Tubuline und Neurofilamente. Während das “geknäulte” (“tangled”) Tau wohl inert ist und keine weiteren pathologischen Veränderungen verursacht, führt das zytosolische hyperphosophorylierte Tau in vitro zur Lösung selbst des normalen Tau-Proteins von den Mikrotubuli. Figure 9: Braak Stadien (alzheimer-forschung.de, 2016) In vitro konnte durch Dephosphorylierung des abnormen Taus ein funktionsfähiges Microtubli-bindendes Protein wiederhergestellt werden. Der Verlust an funktionsfähigem Tau und in Konsequenz eines stabilen Zytoskeletts ist die wahrscheinliche Ursache für einen schliesslichen Zelluntergang. Neben dem Morbus Alzheimer findet sich hyperphosphoryliertes Tau-Protein auch in einigen weiteren „Tauopathien“, wie der frontotemporalen Demenz, dem Morbus Pick, der progressiven nuklearen Blickparese oder der kortikobasalen Degeneration. Risikofaktoren Der mit Abstand größte Risikofaktor ist zunehmendes Lebensalter. Daneben gibt es aber eine Reihe weiterer Faktoren, die die Ausbildung der AD begünstigen können. Die drei wichtigsten genetischen Faktoren sollen hier kurz beschrieben werden. Darüberhinaus haben Lebenstilfaktoren wie sportliche Aktivitäten, soziale Interaktion, Bildung oder Tabakkonsum wahrscheinlich ebenfalls Einfluss auf die Krankheitsentstehung. 14 1992 entdeckte man, dass Mutationen des Amyloid Precurser Protein (APP) ein erhöhtes Erkrankungsrisiko für die AD darstellen. Die ersten entdeckten Mutationen nannte man “London” und “Dutch” Mutationen. Sie verändern das APP insbesondere in der Nähe des Gamma-Sekretase-Schnitts und führen so zu erhöhter Produktion von Beta-Amyloid-42 (Citron, et al., 1992). Weitere bekannte Mutationen sind die “Schweden”-Mutation, die die Beta-Sekretase-Bindung erleichert und die “Flamen”Mutation, die die Alpha-Sekretase-Bindung erschwert (MacLeod, Hillert, Cameron, & Baillie, 2015). Das APP-Gen ist auf Chromosom 21 lokalisiert und so erklärt sich auch die erhöhte Erkankungswahrscheinlichkeit von Trisomie 21 Erkankten. Das exprimierte Protein selbst ist Teil von Zellmembranen, sowohl von Neuronen als auch anderen Körperzellen. Kürzlich wurde eine weitere Mutation im Bereich des BetaSekretase-Schnitts entdeckt (Typ “Island”), die die Beta-Amyloid-Produktion um 40% verringert und so Alzheimer-protektiv wirkt (Jonsson, et al., 2012). Diese “Island”Mutation ist auch ein neuer starker Indikator für die Richtigkeit der AmyloidHypothese. 1993 fanden Strittmatter et al., dass Apolipoprotein E an Beta-Amyloid bindet, und dass die E4 Variante des ApoE bei Alzheimer-Erkankten deutlich häufiger zu finden ist (Strittmatter, et al., 1993). Tatsächlich liegt ApoE auf Chromosom 19 und ist physiologischerweise am Fettstoffwechsel beteiligt. An Neuronen bindet es mittels eines Rezeptors der LDL-Familie. Die drei beim Menschen bekannten Allele des ApoE bezeichnet man als E2, E3 & E4. Diese 3 Varianten unterscheiden sich jeweils nur in einer Aminosäure voneinander, sind in ihrer Wirkung bezogen auf die AD aber stark verschieden. So ist das Riskio, als ApoE4-Träger an AD zu erkranken bei Heterozygoten auf das 2-5fache und bei Homozygoten auf das 5-10fache erhöht. 50% der Alzheimer Erkankten sind mindestens heterozygot E4. Dabei ist das E3-Allel mit 77% Verbreitung mit Abstand am häufigsten. Das AD-Risiko von ApoE2-Trägern ist hingegen verringert. Hier könnte ein interessanter pharmakologischer Ansatzpunkt liegen (Castellano, et al., 2011). 1995 fand man die ersten Mutationen im Präsenilin 1 & 2 Gen (PS1, PS2), die unausweichlich zur AD führen (Duff, et al., 1996). Mäuse ohne PS1 stellten sich als nicht lebensfähig heraus. 1999 erkannte man dann, dass PS1 & PS2 Teile der bis dahin unbekannten Gamma-Sekretase sind (Wolfe, et al., 1999). Die drei oben genannten Mechanismen beziehen sich alle auf Beta-Amyloid. Das stellt eine starke Unterstützung für die Amyloid-Hypothese dar. Dabei führen APP und PS1/2 Mutationen zu erhöhter Amyloid-Produktion, ApoE4 hingegen zu verstärkter Aggregation und zu verringertem Abbau des Amyloids. 15 Diagnostik Demenzen sind so weit verbreitet, dass die kognitive Degeneration häufig mit dem normalen Alterungsprozess verwechselt und in der Folge keine medizinische Hilfe gesucht wird. Dabei behält immerhin die Hälfte der Bevölkerung ihre geistigen Fähigkeiten bis zuletzt. Für die primären Demenzen wie die Alzheimer-Erkrankung ist zwar bislang keine kausale Therapie verfügbar, doch lassen sich der Verlauf verzögern und die Symptome mildern. Von besonderer Bedeutung ist die differentialdiagnostische Abgrenzung zu den sekundären Demenzen, da diese in besonders starkem Maße von einer Therapie profitieren. In Hinblick auf gewonnene Lebensqualität sowie persönliche und auch finanzielle Belastung gilt es frühestmöglich eine Diagnose zu stellen, da die gegenwärtig verfügbare Medikation in den Anfangsstadien der Krankheit die besten Resultate erzielt. Für die verwandten Krankheitsbilder MCI und AD erfolgt traditionell zunächst die Diagnose einer Demenz mittels sorgfältiger Anamnese und Neuropsychologischer Tests (s.u.). Die Symptome müssen dabei seit mindestens 6 Monaten vorliegen, um kurzfristige Ursachen ausschliessen zu können. Falls eine Demenz vorliegt, folgt als zweites Kriterum der Ausschluss anderer Ursachen wie zum Beispiel Enzephalitiden, Parkinson, Multiple Sklerose, Tumoren, Stoffwechselerkrankungen, Mangelernährung, Alkoholmissbrauch, etc. Dafür erfolgen in der Regel LiquorProben, Blutbild und morphologische Bildgebung. Liegt keine andere Ursache für eine Demenz vor, so kann man die Diagnose Alzheimer stellen. Definitiv läßt sich diese Diagnose zwar erst histologisch post mortem stellen, die Zuverlässigkeit der klinischen Diagnose liegt mittlerweile jedoch schon bei etwa 90%. (Alzheimer’s Foundation of America, 2016) 2013 hat die NIH/AA nach 27 Jahren ihre Leitlinien angepasst und das präklinische Stadium der Alzheimer Demenz aufgenommen (McKhann, et al., 2011). Dies ist ein symptomfreier Zustand mit nuklearmedizinisch meßbaren beginnenden Plaques. Ermöglicht wurde die Beschreibung dieses Zustandes erst durch die Entdeckung des “Pittsburgh Compound B”. Das hat zunächst nur theoretische Relevanz, da symptomfreie Personen keine behandelnde Person aufsuchen. Diese Änderung könnte aber große Wirkung haben, da nun Screening-Programme möglich werden, die Risikogruppen wie alte Menschen, Boxer oder Träger bestimmter Gene konsequent und regelmäßig auf das Vorliegen von Plaques und beginnender neuronaler Degeneration zu testen und frühzeitig einer Therapie zuzuführen. 16 Neuropsychologische Tests Erstaunlicherweise wurde der erste systematische und standardisierte Test zur Erfassung demenzieller Symptome, der sogenannte Mini-Mental-Status-Test, erst 1975 erstellt, etwa 70 Jahre nach der Erstbeschreibung des Morbus Alzheimer. Dieser Test lässt sich relativ rasch in etwa 15 Minuten durchführen und hat sich so zur vermutlich meistverwendeten Ersteinschätzung etabliert. Gerade in frühen Demenzstadien ist er aber nicht sehr sensitiv. Sehr weit verbreitet in der Diagnostik und Therapieevaluation ist auch der kognitive Teil des Alzheimer Disease Assessment Scale, kurz ADAS-cog. Die Durchführung dauert mit circa 30 Minuten etwa doppelt so lang, dafür liegen sowohl Sensitivität als auch Spezifität nahe bei 100%. Gerade in den frühen Stadien einer möglichen Demenz ist dieser Test also dem Mini-Mental Status vorzuziehen. Figure 10: Mini-Mental Status (Deutsche Gesellschaft für Hämatologie und Onkologie, 2016) 17 Figure 11: ADAS-cog Test (Dementia Collaborative Research Centers, 2016) Neben diesen beiden häufigen Tests existiert eine große Zahl weiterer Tests, ohne dass bisher ein klarer Standard auszumachen wäre. Um eine Vergleichbarkeit in Studien und Diagnostik zu gewährleisten, wäre ein solcher Standard aber sinnvoll. Seit 2012 versucht das amerikanische “National Institute of Health” mit seinem wissenschaftlichen Gewicht, einen Standard zu etablieren, die “NIH Toolbox”. (National Institute of Health, 2012) Es bleibt abzuwarten, ob dieser Versuch erfolgreich verläuft. Die verschiedenen neuropsychologischen Tests eignen sich dabei vorwiegend, die Verdachtsdiagnose einer Demenz zu erhärten, und weniger, um zwischen verschiedenen Demenzformen zu differenzieren. Liquormarker Eine Liquor Entnahme ist einfach durchzuführen, risikoarm und kostengünstig. Es ist also naheliegend nach Biomarkern im Liquor zu suchen, die die Alzheimer-Diagnose verifizieren oder falsifizieren, bzw den Schweregrad angeben können. Traditionell wurde die Liquor-Diagnostik im Kontext der AD nur zum Ausschluss anderer 18 Ursachen wie zum Beispiel Multipler Sklerose verwendet. Das ändert sich aber gerade, da einige neue Marker eine sehr hohe Aussagekraft aufweisen. Schliesslich sind Liquor-Marker nicht nur geeignet zur Diagnostik, sondern auch zur Verlaufskontrolle einer pharmakologischen Therapie. Der Liquor/Serum – Albumin Quotient ist ein genereller Indikator für die Intaktheit der Blut-Hirn-Schranke. Er ist zum Beispiel erhöht bei Infektionen, Entzündungen, Tumoren und vaskulärer Demenz, nicht aber bei Morbus Alzheimer. Daher eignet sich dieser Parameter zum Ausschluss verschiedener Differential-Diagnosen, die ebenfalls dementielle Symptome hervorrufen können. In gleicher Weise sind die intrathekalen Immunglobuline zum Beispiel bei Multipler Sklerose oder Neuroborreliose erhöht, bei Alzheimer jedoch nicht. Aus der Korrelation mit PET-Scans vermutet man eine verringerte Menge an BetaAmyloid-42 im Liquor bei zunehmender Aggregation desselben im Gehirn. Man nimmt an, dass in Folge der Ablagerung im Gehirn weniger verstoffwechseltes Amyloid im Liquor ankommt. Studien zeigen eine Abnahme des Liquor-BetaAmyloids-42 auf etwa 50% der Altersnorm bei Alzheimer-Erkankten (Blennow, 2004). Die Gesamtmenge an Tau-Protein im Liquor korrelliert direkt mit neuronaler Degeneration. So findet man erhöhte Werte dieses Proteins unter anderem bei Erkankten nach Schlaganfällen, Schädel-Hirn-Traumen und Creutzfeld-Jakob. Bei Alzheimer ist das Liquor-Tau ebenfalls erhöht und es gibt sogar einen quantitativen Zusammenhang: je höher die Tau-Werte, desto rapider verläuft die kognitive Degeneration. Durchschnittlich liegen die Tau-Werte etwa 3-fach über der Altersnorm. In Zukunft wird die Betrachtung des phosphorylierten Tau-Proteins die Aussagekraft möglicherweise weiter erhöhen. Erste Studien deuten darauf hin, dass dieses nur bei Alzheimer-Erkankten erhöht ist, nicht aber bei anderen neuronalen Erkrankungen. (Blennow, Hampel, Weiner, & Zetterberg, 2010) Gegenwärtig experimentiert man erfolgreich mit der Kombination von LiquorMarkern. Insbesondere die Tau- & Amyloid-Marker sind hier vielversprechend. Eine Studie zeigt zum Beispiel eine Zunahme der Sensitivität von 80% auf 86% und der Spezifität von 84% auf 97% im Vergleich zur Verwendung nur eines Markers. (Maddalena, et al., 2003). Ein solcher Test verursacht auch lediglich Kosten von circa 200$, MRT und PET sind wesentlich teurer. (Blennow, Hampel, Weiner, & Zetterberg, 2010) Nach neuen Markern wird weiterhin gesucht. Kandidaten sind unter anderem verschiedene Beta-Amyloid Vorläufer, Isoformen und Antikörper. Bislang sind die Ergebnisse diesbezüglich aber noch inkonsistent. 19 Plasmamarker Da Blutplasma noch leichter zu gewinnen ist als Liquor, wird natürlich auch in dieser Richtung nach möglichen Markern für die Alzheimer-Krankheit gesucht. Zwischen Plasma-Amyloid-Spiegel und Krankheits-Stadium konnte aber bisher kein Zusammenhang hergestellt werden. Eine Kombination aus 18 Plasma-Proteinen führte jedoch zu einer 90% diagnostischen Genauigkeit. Die untersuchten Proteine stehen in Zusammenhang mit Hämatopoese, Apoptose und Immunregulation. (Ray, et al., 2007) Eine Vielzahl von Messgrößen automatisiert mit dem Auftreten einer Krankheit zu korrelieren, könnte ein vielversprechender Weg für die Zukunft sein. Elektroenzephalographie Die EEG-Forschung zum Thema Alzheimer konzentriert sich auf die Bereiche • Intensitätsverschiebungen • Reduzierte Komplexität • Reduzierte Synchronität Zunächst ist in jedem Falle eine Vorverarbeitung notwendig, um Artefakte wie Muskelbewegungen herauszufiltern. Die Intensitätsverschiebungen sind im folgenden Schaubild sehr schön dargestellt. Man sieht deutlich eine Verstärkung der Intensität in den Frequenzen unterhalb von 8Hz (Delta & Theta – Bänder) und eine Verringerung der Intensität in den Frequenzen oberhalb von 8Hz (Alpha & Beta – Bänder). (Dauwels, Vialatte, & Cichocki, 2010) Die reduzierte Komplexität der EEG-Bänder kann man mit verschiedenen EntropieMaßen erfassen. Beispielhaft sei hier das Lempel-Ziv-Welch-Verfahren genannt. Dieses findet in Daten Abschnitte, die sich gleichen. Je mehr gleiche Abschnitte gefunden wurden desto geringer ist die Komplexität und umgekehrt. Bei AlzheimerErkankten wiederholen sich EEG-Abschnitte häufiger als bei gesunden Versuchspersonen, was wohl auf die verringerte neuronale Interaktion durch generelle Neurodegeneration bedingt ist. Dieser Algorithmus wird interessanterweise auch in der Informatik verwendet, zum Beispiel in der bekannten GIF-Kompression für Bilder. In diesem Falle wird die Bilddatei umso stärker komprimiert, je mehr gleiche Muster sich in den Ausgangsdaten finden (Zhao, et al., 2007). Auch die Synchronität der verschiedenen EEG-Ableitungen ist bei AlzheimerErkankten im Allgemeinen reduziert. Man führt dies weniger auf einen Verlust an 20 Neuronen, als vielmehr auf Diskonnektionen der verschiedenen kortikalen Zentren zurück. Figure 12: Intensitätsverteilung über EEG-Frequenzen. Links Kontrollgruppe, rechts Alzheimer-Erkrankten (Dauwels, et al., 2011) Die existierenden Studien zur Alzheimer-Diagnostik mittels EEG sind in gegenwärtiger Form jedoch noch nicht ausreichend sensitiv und spezifisch, um einen klinischen Einsatz empfehlen zu können. (Dauwels, Vialatte, & Cichocki, 2010) Möglicherweise kann eine Kombination der verschiedenen Marker einen Zugewinn an Reliabilität bringen, analog zum Vorgehen bei Liquor- und Plasma-Markern. Dazu konnten jedoch keine Studien gefunden werden. Radiologie Makroskopisch lässt sich sowohl bei der Autopsie post-mortem, als auch mittels morphologischer Bildgebung wie der Magnetresonanztomographie und der Computertomographie eine Atrophie des gesamten Kortex bei Alzheimer-Erkrankten beschreiben, mit sichtbarer Vergröberung der Gyri über der Altersnorm. Das Problem in der Befundung liegt bei der morphologischen Bildgebung also zum einen in der Beurteilung des Atrophiegrades im Vergleich zur Altersnorm als auch in der Objektivierung des Befundes. Da sich die Befunde verschiedener behandelnder Personen selbst bei gleichen Rohdaten unterscheiden, sucht man nach automatisierten Möglichkeiten der Beurteilung eines Schnittbildes. In der gegenwärtigen Befundung des Morbus Alzheimer werden MRT & CT daher hauptsächlich zum Ausschluss anderer Erkrankungen wie Tumoren oder Blutungen verwendet, sowie zur groben Beurteilung der Hirnatrophie. 21 Die Herangehensweisen zur automatisierten Analyse lassen sich dabei zunächst nach betrachteter Region gruppieren. Man kann das gesamte Hirnvolumen, nur die Rinde oder nur bestimmte häufig betroffene Bereiche wie den Hippocampus betrachten. Figure 13: Voxel-basierte (links) versus Atlas-basierte (rechts) MRT-Analyse. Je “wärmer” die Farbe, desto stärker die kortikale Atrophie und die Plaque-Wahrscheinlichkei (Cuingnet, et al., 2011) Hat man sich für ein Gebiet entschieden, kann man dieses dann Voxel für Voxel (Volumen Pixel) betrachten oder nach Funktionsbereichen (Atlas-basiert) gruppieren. Dabei kann man die untersuchte Region dann zum Beispiel nach der veränderten Dicke des Kortex oder nach der Veränderung der Konstellation aus Neuronen, Axonen und Liquor klassifizieren. (Cuingnet, et al., 2011) Figure 14: Form-Analyse des Entorhinalen Kortex, je kälter die Farbe, desto stärker die Einziehung (Devanand, et al., 2012) Alternativ kann man auch das Volumen bestimmter Bereiche messen (in der Regel Hippokampus) oder die genaue Form dieser Bereiche analysieren, was die Sensitivität erhöht, da so selbst Einziehungen in kleinsten Regionen auffällig werden (Devanand, et al., 2012). 22 Mit Hilfe der diffusionsgewichteten Magnetresonanztomographie (DT-MRT oder auch nur DTI) kann man die Diffusionsrichtung von Wasser visualisieren. Da dieses leichter entlang der axonalen Bahnen verläuft als quer zu diesen, kann man mit diesem Verfahren sehr schön die großen axonalen Bahnen sichtbar machen, auch Traktographie genannt. Da bei der AD Neurone inklusive ihrer Axone absterben, führt das zu einer messbar erhöhten Quer-Diffusion in der Traktographie. Figure 15: Diffusionsgewichtetes MRT, man sieht die Diffusionszunahme bei der Alzheimer-Demenz im Vergleich zur Kontrollgruppe (Douaud, et al., 2011) Die meisten Studien sind zwar MRT-basiert, aber manche Arbeitsgruppen meinen, in CT-Messungen das überlegene Instrument zu haben, unter anderem, da sich durch die erhöhte Messgeschwindigkeit weniger Artefakte ergeben. (Imabayashi, et al., 2013) Zum Ausschluss anderer Demenz-Ursachen wird die CT wegen ihrer größeren Verfügbarkeit und schnelleren Durchführung sogar häufiger verwendet als die MRT. Figure 16: CT-Analyse, Intensitätsminderungen der grauen Substanz, äquivalent zu Nervenzellverlusten, in Orange (Imabayashi, et al., 2013) 23 Nuklearmedizin Die Szintigraphie bietet sei 40 Jahren die Möglichkeit, Stoffwechselprozesse im Körper sichtbar zu machen. Die beiden verwendeten Verfahren PET (=PositronenEmissions-Tomographie) und SPECT (=single photon emission computed tomography) sind sich sehr ähnlich. Beide verwenden Radionukleotide, die sich in stoffwechselaktivem Gewebe vermehrt anreichern und messen die emittierte Strahlung. PET verwendet dazu Beta- und SPECT Gamma-Strahlung. PET bietet eine genauere Auflösung ist aber auch etwas teurer in der Durchführung. Für beide Methoden stehen rotierende Aufnahmeköpfe und somit 3D-Bilder zur Verfügung. Weiterhin gibt es die Möglichkeit der räumlichen Verknüpfung mittels CT/MRT, da die szintigraphischen Methoden selbst nur die Positionen ihrer Marker erfassen, nicht aber das restliche Gewebe. Im Rahmen der Alzheimer-Diagnostik ist bereits eine Vielzahl von Radiopharmaka verfügbar, die verschiedenste Teilaspekte der Krankheit visualisieren können. So lässt sich die Aktivität verschiedener Neurotransmittersysteme darstellen, der Glukoseverbrauch und die Amyloid- & Tau-Aggregation. Auch aktivierte Mikroglia und Lipidstoffwechsel sind Gegenstand der Forschung. Schliesslich wird wohl in Zunkunft auch der Therapieerfolg mittels funktioneller Bildgebung kontrolliert werden können. Figure 17: Glukose Stoffwechsel, links an MCI erkrankte Person, rechts an Alzheimer erkrankte Person (Nordberg, Rinne, Kadir, & Laangstrom, 2010) 24 Zur Messung des Glukosestoffwechsels verwendet man die FluorDesoxyGlukose (FDG). Im Gleichschritt mit dem Untergang neuronalen Gewebes nimmt der Zuckerstoffwechsel ab, FDG ist also auch ein Marker für die Progredienz der Erkrankung. Weiterhin besteht eine hohe Sensitivität (ca. 90%), da eine Veränderung im Zuckerstoffwechsel schon lange vor klinischen Symptomen sichtbar wird. (Nordberg, Rinne, Kadir, & Laangstrom, 2010) Auch die Lokalisation der hauptsächlich betroffenen Areale kann so sichtbar gemacht werden, das sind im wesentlichen der frontale und temporale Kortex sowie der posteriore Gyrus Cinguli. Besonders interessant sind neuere Studien zur getrennten Betrachtung der verschiedenen Neurotransmittersysteme. Bei der Betrachtung des cholinergen Neurotransmittersystems kann man sowohl das Enzym Acetylcholinesterase mittels des Markers C-PMP betrachten als auch die verschiedenen Rezeptorsubtypen selbst, namentlich den nikotinischen (F-fluoro-A85380) und den muskarinischen (C-NMPB). Die AChE-Marker korellieren dabei in bisherigen Studien sehr gut mit der Erkrankung und können möglicherweise bald als Marker eingesetzt werden. Beim Versuch, die Rezeptoren statt der Transmitter zu markieren, konnte beim muskarinischen Rezeptor bislang kein Zusammenhang entdeckt werden. Die Ergebnisse zum nikotinischen Rezeptor sind zumindest vielversprechend. (Nordberg, Rinne, Kadir, & Laangstrom, 2010) Figure 18: Amyloid-Aggregation, links an Alzheimer erkrankte Person, rechts Kontrollperson, Visualisierung mittels Pittsburgh Compound B (Nordberg, Rinne, Kadir, & Laangstrom, 2010) 25 Das dopaminerge System zeigt zwar Veränderungen bei der Autopsie, es konnte bislang aber keine Visualisierung in vivo erfolgen. Allerdings schlägt der SPECTMarker I-FP-CIT bei Lewy-Body-Demenz an und wird aus diesem Grund zur Differentialdiagnose zum Morbus Alzheimer eingesetzt. Forschung zu Radiopharmaka der Rezeptorsubtypen D1-D5 läuft noch und ist bislang widersprüchlich. Im serotonergen System konnte in einzelnen Studien eine Reduktion der 5-HT-1 und 5-HT-2 Subtypen gezeigt werden. Seit 2004 der Amyloid-Marker Pittsburgh Compound B (PIB) gefunden wurde, erfuhr die Diagnostik in diesem Bereich einen wesentlichen Schub. Der PiB ist ein radioaktives Analogon des Thioflavin T, das an Beta-Amyloid bindet und dieses so im PET sichtbar macht. Der Zusammenhang zwischen erhöhter Amyloid-Aggregation und der Alzheimer-Erkankung konnte in zahlreichen Studien gut belegt werden. Die Empfindlichkeit mittels PET-PIB entspricht der Liquor-Diagnostik des BetaAmyloids, ist aber etwa 8 Mal so teuer. Das stellt die künftige breite Anwendbarkeit durchaus in Frage. 26 Material und Methoden Zur Bewertung verschiedener Aspekte der pharmakologischen Therapie des Morbus Alzheimer wurde systematisch vorhandene Literatur gesichtet. Dazu wurden im wesentlichen die Suchmaschinen PubMed und Google Scholar herangezogen. Eine zeitliche Eingrenzung wurde per se nicht vorgenommen, neuste Ergebnisse wurden jedoch in besonderem Maße in Betracht gezogen. Ausserdem wurde gängige Fachliteratur gesichtet, sowie Leitlinien neurologischer Gesellschaften eingearbeitet. Es wurden insbesondere neuste Studien zu cholinerger, glutamaterger und amyloider Therapie recherchiert. Die betrachteten Studien wurden ausserdem auf Widersprüchlichkeiten und Gemeinsamkeiten untereinander bewertet. 27 Ergebnisse Überblick der pharmakologischen Therapie der AD Mitte der 1970er Jahre stellte man zum ersten mal eine Verbindung her zwischen dem cholinergen Neurotransmittersystem und der Gedächtnisleistung auf der einen Seite, sowie dem Untergang des zerebralen cholinergen Systems beim Morbus Alzheimer. Die AD wurde als cholinerge Erkrankung betrachtet, so wie der Morbus Parkinson als dopaminerge Erkrankung verstanden wurde. Die pharmakologische Forschung konzentrierte sich in der Folge auf die Verbesserung der cholinergen Funktion. Bis zur Zulassung des ersten Cholinesterasehemmers Tacrin 1993 verstrichen dennoch beinahe 20 Jahre. Aufgrund dessen Hepatotoxizität wurde diesem zugunsten der im folgenden genauer beschriebenen Cholinergika die Zulassung wieder entzogen. Neben dem cholinergen System ist bei der AD auch das glutamaterge Neurotransmittersystem betroffen. Dort findet man eine chronische Überaktivierung. Diese versucht man pharmakologisch zu mindern. Die Suche nach einem wirksamen Glutamat-Rezeptor-Antagonisten führte 2002 zur Zulassung von Memantin für moderate und schwere Formen der AD. (Danysz & Parsons, 2012) Die Aufstellung der Amyloid-These 1991 führte zu einer weitgehenden Verlagerung der pharamakologischen Forschung. Diese zielte nun auf die Hemmung von Synthese, Aggregation, sowie Lyse der Beta-Amyloid-Plaques. (Schneider, et al., 2014) Bislang sind die Möglichkeiten der pharmakologischen Therapie sehr begrenzt. Zugelassen zur Behandlung der AD sind aktuell nur drei Cholinesterasehemmer (Donepezil, Galantamin, Rivastigmin) sowie der NMDA-Rezeptor-Antagonist Memantin (Beubler, 2011). Da die kognitiven Unterschiede zwischen Placebo und getestetem Pharmakon sehr gering sein können, werden die Testzeiträume verlängert, um Effekte besser sichtbar werden zu lassen. Wurden die Cholinesterasehemmer und Memantin noch in 3–6 monatigen Studien erprobt, erstrecken Versuche mit neuen Pharmaka sich heute meist über 12-24 Monate. Aus einem ähnlichen Grund nehmen auch die TestKollektive stetig an Größe zu, denn so können auch geringe kognitive Verbesserungen signifikant beschrieben werden. Nach grossen diagnostischen Fortschritten wie dem Pittsburgh Compound B, aber auch der Messung von intrathekalem Beta-Amyloid (siehe Kapitel Diagnostik) verlagern sich Studien zudem zunehmend darauf, Ihren Erfolg an Biomarkern anstatt 28 klinischen neuropsychologischen Tests zu messen. Auf diese Weise lassen sich ebenfalls bereits subtilere Verbesserungen darstellen. Ausserdem werden die Ergebnisse so reproduzierbarer. Die verbesserte Diagnostik wird aber nicht nur zur Bewertung des Studienerfolgs verwendet, sondern bereits vor Studienbeginn zur Auswahl der Versuchspersonen. So kann man die Studien mittlerweile auch auf vorklinische Stadien der AD ausweiten, die mit neuropsychologischen Tests gar nicht erfasst werden können. Cholinerge Pharmaka Bereits früh bemerkte man Defekte im cholinergen System bei Alzheimer-Erkrankten und erklärte sich damit auch die dementiellen kognitiven Defizite. Der Neurotransmitter Acetylcholin wird physiologischerweise durch eine Esterase wieder aus dem synaptischen Spalt entfernt. Wenn die Ausschüttung des Transmitters abnimmt, kann man wahlweise die Produktion des Transmitters verstärken, den Transmitter selbst nachahmen oder seinen Abbau hemmen. Zunächst wurden Acetylcholin-Vorstufen wie Lecithin erprobt, jedoch ohne Erfolg. Muskarinische Rezeptor-Agonisten wie Xanomelin zeigten im Labor eine gewisse Wirksamkeit, werden wegen starker Nebenwirkungen aber bisher nicht verwendet. Die beste Wirkung am synaptischen Spalt konnte man mit Acetylcholin-Esterase-Hemmern erzielen. Entsprechende Medikamente waren schon auf dem Markt um zum Beispiel die Myastenia gravis oder Glaukome zu behandeln. Für die Behandlung des Morbus Alzheimer sind momentan die drei neueren AChE-Hemmer Donepezil, Galantamin und Rivastigmin zur Behandlung der leichten und moderaten AD zugelassen. Physiostigmin und Tacrin werden nicht mehr verwendet. Da Cholinesterasehemmer auf das Vorhandensein endogenen Acetylcholins angewiesen sind, nimmt ihre Wirksamkeit mit zunehmendem Untergang der cholinergen Neurone ab. Weiterhin wirkt diese Substanzgruppe rein symptomatisch, die Krankheit wird so weder am Fortschreiten gehindert, noch geheilt. Die Acetylcholinesterase liegt im Gehirn von Säugetieren in zwei Formen vor, der häufigeren, tetrameren G4-Form und der selteneren, monomeren G1-Form. Pharmaka können eine unterschiedlich starke Affinität zu diesen beiden Formen aufweisen. In ihren parasympathischen Nebenwirkungen sind sich die Cholinesterasehemmer sehr ähnlich. Im wesentlichen kommt es zu Übelkeit, Erbrechen, Durchfall, Muskelkrämpfen und Schlaflosigkeit. 29 Cholinesterase Inhibitoren Tacrin war unter dem Handelsnamen Cognex 1995 das erste zugelassene Medikament zur Behandlung der AD. Bereits bei der Zulassung war bekannt, dass bei mehr als 40 Prozent der Erkrankten reversible Transaminasenerhöhungen auftreten. Wegen der Hepatotoxizität wurde Tacrin in Deutschland bald wieder vom Markt genommen. In den USA war es bis noch 2013 erhältlich. Figure 19: Tacrine (chembase.cn) Der reversible AChE-Hemmer Galantamin wurde 1950 in Russland entwickelt und wird seitdem in Osteuropa zur Therapie der Myasthenie verwendet. Im Westen ist es seit 2001 zur Behandlung der AD und vaskulären Demenz zugelassen. Es wurde zunächst aus dem kaukasischen Schneeglöckchen gewonnen, wird heute aber synthetisch hergestellt. Es bindet sowohl direkt an nikotinische Acetylcholinrezeptoren als auch hemmend an die AChE. Seit 2014 gibt es Generika. Figure 20: Galantamine (chembase.cn) Da es hepatisch durch die CYP3A4 und CYP2D6 metabolisiert wird ist die Gabe bei Leber- aber auch bei Niereninsuffizienz kontraindiziert. Wechselwirkungen sind bekannt mit Parasympathomimetika, Parasympatholytika, Digoxin, Betablockern und Muskelrelaxantien. Die Halbwertszeit beträgt 7 Stunden, die Proteinbindung 18%, die Bioverfügbarkeit etwa 90%. Der älteste noch verwendete AChE-Hemmer Donepezil wurde 1996 zugelassen, wie Galantamin bindet es reversibel. Es ist das meistverkaufte AlzheimerMedikament und seit 2010 auch als Generikum erhältlich. Als einziges der AChE-Hemmer ist es auch zur Behandlung der schweren AD zugelassen, bisher aber nur in den USA. Figure 21: Donepezil (chembase.cn) Es wird wie Galantamin durch CYP3A4 und CYP2D6 metabolisiert und weist vergleichbare Wechselwirkungen und Kontraindikationen auf. Weitere häufige 30 Interaktionen sind mit Levodopa, Anticholinergika und Suxamethoniumchlorid beschrieben. Die Halbwertszeit beträgt 70 Stunden, die Proteinbindung 96%, die Bioverfügbarkeit 100%. Rivastigmin unterscheidet sich deutlich von den beiden oben genannten Substanzen. Durch seine lange Bindung von etwa 10 Stunden an Acetylcholinesterase bezeichnet man Rivastigmin Figure 22: Rivastigmine (chembase.cn) auch als pseudo-irreversiblen AChE-Hemmer. Ausserdem bindet Rivastigmin bevorzugt an die G1-Isoform der AChE und wirkt so stärker im Cortex als peripher, wodurch sich etwas geringere Nebenwirkungen ergeben. Zusätzlich zur AChE hemmt Rivastigmin ausserdem die Butyrylcholinesterase, ein verwandtes Enzym. Schliesslich wird Rivastigmin direkt im synaptischen Spalt metabolisiert und kann auch bei nieren- und leberinsuffizienten Erkrankten verwendet werden und es bestehen keine Wechselwirkungen mit anderen Medikamenten. Es wurde im Jahr 2000 zur Behandlung der leichten und mittleren AD zugelassen. (Inglis, 2002) (Jann, 2000). Seit 2014 gibt es Generika. Es ist als Kapsel, Lösung und seit 2008 auch als Pflaster erhältlich. Die Bioverfügbarkeit bei oraler Einnahme beträgt circa 40%. Nach 2 - 3 Stunden wird der maximale Wirkspiegel im Liquor erreicht. Die Halbwertszeit liegt bei 1,5 Stunden, die Proteinbindung bei 40%, die maximale Tagesdosis bei 12 mg. Die Applikation des Pflasters erfolgt einmal täglich bei einer maximalen Dosis von 9,5 mg. Die Dosis sollte über mehrere Wochen langsam gesteigert werden, um Unverträglichkeiten früh zu erkennen. Phenserin ist ein Derivat des Cholinesterasehemmers Physiostigmin. Es gibt zwei Enantiomere “-“ und “+”. Nur das “-“ Enantiomer wirkt auf die Cholinesterase. Beide Enantiomere haben jedoch die Eigenschaft, die Amyloid-Precursor-Protein-Synthese zu modulieren und so effektiv auch Amyloid-Beta zu reduzieren. Das “+” Enantiomer kann man wegen seiner fehlenden Cholinesterasewirkung deutlich höher 31 Figure 23: Phenserin (chemnet.com) dosieren und so einen ausgeprägteren Effekt auf die Plaquebildung erzielen. “-“ Phenserin zeigte in vitro zwar gute Wirksamkeit, konnte in klinischen Phase 3 Studien jedoch keine signifikante Verbesserung zeigen und wurde 2009 vom Hersteller eingestellt. Die Verbindung bleibt aber Ausgangspunkt für Modifikationen, die möglicherweise bessere Ergebnisse bringen (Shinada, et al., 2012). Das “+” Enantiomer von Phenserin wird unter dem Namen Posiphen klinisch erprobt. Erste Studien sahen vielversprechend aus. So konnte dieses cholinerg inaktive Enantiomer im Mausversuch die APP und Amyloid-Last deutlich reduzieren (Lahiri, et al., 2007) (Klein, 2007). 2012 gab es erste Phase I Studien mit gesunden Versuchspersonen, die eine gute Verträglichkeit zeigten. (Maccecchini, et al., 2012). Die weitere Entwicklung bleibt spannend. Huperzin wird aus einem chinesischen Moosfarn gewonnen und in der traditionellen chinesischen Medizin schon lange verwendet, unter anderem zur Behandlung der Myasthena gravis. Auch die keltischen Druiden verwendeten diesen Wirkstoff, der auch im Tannenbärlapp enthalten ist. Figure 24: Huperzin A (chembase.cn) In China wird die AD bereits mit Huperzin behandelt. Besonders interessant ist, dass Huperzin sowohl die Cholinesterase hemmt, als auch den NMDA-Rezeptor antagonisiert. Es wirkt also gleichzeitig auf beide bei der AD betroffenen Neurotransmittersysteme. Es wurden bereits Phase 2 Studien durchgeführt, die aber bisher keine signifikanten Wirkungen nachweisen konnten. Möglicherweise wurde die Wirkdosis zu gering angesetzt. Weitere Studien laufen noch. Eine Metaanalyse von 20 klinischen Studien mit insgesamt 1823 Versuchspersonen bescheinigt Huperzin signifikante kognitive Effekte, hält aber weitere Studien aufgrund methodischer Mängel für notwendig (Yang, Wang, Tian, & Liu, 2013). Cholinerge Agonisten Neben den Cholinesterasehemmern wurden auch Versuche unternommen, cholinerge Rezeptoren direkt zu stimulieren. Problematisch ist bei deren Entwicklung insbesondere, eine spezifische Wirkung in geschädigten Cortexarealen zu erreichen ohne dabei starke periphere Nebenwirkungen auszulösen. Als Ziele in Frage kommen dabei vor allem die muskarinischen M1 und M4 Rezeptoren, die 32 hauptsächlich zerebral verteilt sind, sowie die nikotinischen Alpha-7 (α7) und Alpha4-Beta-2 (α4β2) – Rezeptoren (Toyohara & Hashimoto, 2010). Da sich die verschiedenen cholinergen Rezeptoren-Subtypen nicht sehr stark voneinander unterscheiden, stellt sich die Entwicklung spezifisch wirkender Pharmaka bislang schwierig dar. Neue Ansätze, wie die Positiven Allosterischen Modulatoren (PAM), lösen dieses Problem möglicherweise. Die PAM binden nicht an der physiologischen Bindungsstelle des Agonisten, sondern an anderer Position. Dadurch ergeben sich für die Entwicklung spezifischer M1/M4 Pharmaka viele mögliche neue Kopplungspunkte am Rezeptor (Nickols & Conn, 2014). Beta-Amyloid blockiert möglicherweise den α7 -Rezeptor bei AD-Erkrankten (Kihara & Shimohama, 2004). Diskutiert wird auch, ob Beta-Amyloid über den α7 - Rezeptor intrazellulär aufgenommen wird, und dort die Tau-Phosphorylierung induziert (Nagele, D’andrea, Anderson, & Wang., 2002). Die α4β2 Ausprägung scheint bei Alzheimer-Erkrankten deutlich verringert zu sein. Dies lässt sich mittlerweile sogar in vivo mittels SPECT beobachten (O’Brien, et al., 2007). Das würde eine pharmakologischen Korrektur nahe legen. Ausserdem scheinen sowohl α4β2 als auch α7 relevant am Gedächtnis und Lernfähigkeit beteiligt zu sein (Levin, 2013). Problematisch bei der Entwicklung eines langfristig wirksamen nikotinischen Agonisten ist die schnelle Gewöhnung und somit die ständige Notwendigkeit der Dosiserhöhung (Levin, 2013). Muskarinrezeptor Agonisten Xanomelin wirkt an muskarinischen M1 und M4 Rezeptoren und ist als einziger cholinerger Agonist bislang in Phase 3 Studien erprobt worden. Dabei zeigte der Wirkstoff nur eine geringe kognitive Verbesserung und starke gastro-intestinale Nebenwirkungen. Auffällig waren Figure 25: Xanomeline (wikipedia.org) die starken positiven Effekte auf Halluzinationen, Verwirrung und Sprachstörungen. Daraufhin began man, Xanomelin und verwandte Muskarin-Rezeptor-Agonisten auch bei Schizophrenie-Erkrankten zu testen. Der Wirkstoff selbst wird wegen der starken Nebenwirkungen nicht eingesetzt und auch nicht mehr weiterentwickelt (Foster, Choi, Conn, & Rook, 2014). 33 BQCA (Benzyl Quinolone Carboxylic Acid) (auch SML0497) war einer der ersten positiven allosterischen Modulatoren (PAM) mit Affinität für M1 – Rezeptoren (Kuduk & Beshore, 2014). Als PAM aktiviert er den Rezeptor nicht direkt, sondern erleichtert die Bindung von Acetylcholin (Nickols & Conn, 2014). Der verwandte PAM PQCA zeigte im Mausmodell eine Kognitionsverbesserung vergleichbar der von Donepezil (Puri, Wang, Vardigan, Kuduk, & Uslaner, 2015). Menschliche Studien für PAMs liegen noch nicht vor. Figure 26: BQCA (D Kuduk 2014) Nikotinrezeptor Agonisten Enceniclin (auch EVP-6124) von Envivo Pharmaceuticals ist ein partieller Alpha-7-Agonist. In Phase 1 Studien wurden zunächst keine Nebenwirkungen berichtet, Phase 2 Studien bescheinigten kognitive Verbesserungen (Deardorff, Shobassy, & Grossberg, 2015). In Phase 3 traten starke gastrointestinale Nebenwirkungen auf. Drei Phase 3 Studien wurden ohne Veröffentlichung von Ergebnissen abgebrochen. Figure 27: Enceniclin (wikipedia.org) Neloniclin (auch ABT-126) von Abbott ist ein allosterischer Alpha-7-Agonist. Nachdem eine Phase 2 Studie zunächst kognitive Verbesserungen angegeben hatte, konnte in einer 2b Studie mit 400 Erkrankten kein Nachweis mehr erbracht werden, so dass die Entwicklung eingestellt wurde (Gault, et al., 2014). Figure 28: Neloniclin (drugspider.com) Vareniclin ist ein Partialagonist am Alpha-4-Beta-2 – Rezeptor. Es wirkt ausserdem in geringerem Masse an Alpha-7 und 5-HT3 – Rezeptoren. Es ist bereits zugelassen zur Nikotinentwöhnung von rauchenden Personen. Es wurde 2010 in einer Phase 2 Studie an Alzheimer-Erkrankten erprobt, konnte aber keine kognitive Verbesserung erweisen (Kim, et al., 2013). 34 Figure 29: Vareniclin (chembase.cn) PHA-543613 ist ein weiterer Agonist am nikotinischen alpha-7-Rezeptor und ein Beispiel für einen neuen Wirkstoff der bisher nur im Tiermodell gestestet wurde. Diese Versuche zeigen bisher eine deutliche kognitive Verbesserung. Die Wirksamkeit scheint stärker als bei Galantamin zu sein (Sadigh-Eteghad, Mahmoudi, Babri, & Talebi, 2015). Glutamaterge Pharmaka Neben dem cholinergen ist insbesondere auch das glutamaterge System bei Alzheimer-Erkrankten betroffen. Der Neurotransmitter Glutamat bindet an eine Reihe von Rezeptoren, die zunächst nach Typ in die Ionotropen und die metabotropen Rezeptoren unterteilt werden. Die Ionotropen Rezeptoren sind nach demjenigen Molekül benannt, welches spezifisch an diese bindet. So kennt man den NMDARezeptor (N-Methyl-D-Aspartat), den AMPA-Rezeptor (AminohydroxyMethylisoxazol-Propionic Acid) und die Kainat-Rezeptoren. Die zweite Gruppe der metabotropen Rezeptoren wird einfach in mGlu1 bis mGlu8 unterteilt. Bei Alzheimer-Erkrankten unterliegt das glutamaterge System einer chronischen Überaktivierung durch die NMDA-Rezeptoren. Das führt zum Zelltod durch CalciumÜberladung. Sowohl Unter- als auch Überaktivierung führen zum Verlust neuronaler Plastizität und konsekutiv schweren Einschränkungen beim Lernen. Man diskutiert ob lösliches Beta-Amyloid für die Überaktivierung der NMDA-Rezeptoren direkt verantwortlich ist. (Parsons, Stöffler, & Danysz, 2007) Ein natürlicher Antagonist des NMDA-Rezeptors ist Magnesium. Dessen Konzentration im Blutplasma ist bei der AD signifikant verringert (ionisiertes Magnesium: 0,50 mmol/L bei AD-Erkrankten gegenüber 0,53 mmol/L bei Gesunden) (Barbagallo, Belvedere, Di Bella, & Dominguez, 2011). Der Magnesium-Mangel lässt sich sogar direkt korrelieren mit dem kognitiven Verfall (genauer, dem MMSE-Score) von Alzheimer-Erkrankten (Barbagallo, Belvedere, Di Bella, & Dominguez, 2011). Die chronische NMDA-Überaktivierung könnte also sowohl durch eine direkte Aktivierung als auch durch eine verringerte Hemmung oder durch eine Kombination beider Faktoren verursacht sein. NMDA Antagonisten Die Magnesiumkonzentration medikamentös zu erhöhen stellte sich bislang problematisch dar. Orale Magnesiumgabe führt zumindest bei gesunden Individuen nicht zu erhöhten Magnesium-Spiegeln in Plasma oder Blutzellen (Wary, et al., 1999). 35 Hohe parenterale Dosen würden ausserdem zu Hypermagnesiämie mit Muskelschwäche, Bradykardie, Vasodilatation, Verwirrtheit, Atemdepression, etc, führen (Parsons, Stöffler, & Danysz, 2007). 2010 wurde eine neue Magnesium-Verbindung getestet, Magnesium-L-Threonat (MgT). Diese führte im Rattenmodel zu Verbesserungen im Lernen, sowie im Kurzund Langzeitgedächtnis (Slutsky, et al., 2010). Weiterführende Studien an transgenen Alzheimer Mäusen scheinen zu einem Rückgang an Amyloid-Beta, weniger Neuronenuntergängen und verbesserter Kognition zu führen (Li, et al., 2014). Hier könnte ein vielversprechender Weg für neue pharmakologischen Entwicklungen liegen. Magnesium fördert aber beispielsweise auch die Aggregation von Tau-Fibrillen (Yang & Ksiezak-Reding, 1999), was einer wirksamen Therapie im Weg stehen könnte. Memantin wurde 1968 zunächst als DiabetesMedikament entwickelt, stellte sich diesbezüglich aber als ineffektiv heraus. Erst in den 1980er Jahren erkannte man die Wirkung von Memantin auf NMDA-Rezeptoren. Die Zulassung folgte bald darauf in Deutschland (1989) aber erst 10 Jahre später in den USA (2002). Seit 2014 sind auch Generika erhältlich. Bisher ist Memantin nur für die Behandlung der mittleren und schweren AD zugelassen. Figure 30: Memantin (chembase.cn) Memantin ist ein unkompetitiver Antagonist am NMDA-Rezeptor. Es konkurriert also nicht direkt am NMDA-Rezeptor um einen Platz, sondern bildet mit dem EnzymSubstrat-Komplex eine neue Verbindung mit veränderter Reaktionsgeschwindigkeit. (Chen & Lipton, 1997). Im Unterschied zur nicht-kompetitiven Hemmung kann Memantin erst dann an den Rezeptor binden, nachdem ein Substrat angedockt hat. Auf diese Weise wirkt Memantin umso besser, je stärker der Rezeptor aktiviert wird. Eine völlige Blockade, die kognitiv nachteilig wäre, wird so verhindert. Memantin hemmt den NMDA-Rezeptor aber nicht nur direkt, sondern auch indirekt über eine Verminderung der Glutamat-Ausschüttung durch Blockierung von spannungsabhängigen Calcium-Kanälen (Lu, Lin, & Wang, 2010). 36 Ein verwandtes interessanterweise Pharmakon ist Ketamin. Der Wirkmechanismus ist sehr ähnlich zu Memantin; beide wirken nur am offenen NMDA-Ionen-Kanal blockierend und beide sind spannungsabhängig in ihrer Wirkung. Die klinische Wirkung ist jedoch sehr unterschiedlich. Ketamin wird als Anästhetikum Figure 31: Ketamin (chembase.cn) verwendet und bewirkt Schlaf und Schmerzfreiheit und führt zu dissoziativen Rauschzuständen. Memantin wird im Gegensatz gut vertragen mit milden positiven Effekten auf Kognition und Gedächtnis. (Johnson, Glasgow, & Povysheva, 2015) Ketamin wird neben der Schmerztherapie neuerdings auch zur Behandlung der Depression eingesetzt, bei der es erstmals Ergebnisse innerhalb Stunden anstatt bisher Wochen produzieren kann (Krystal, Sanacora, & Duman, 2013) Hier wirkt Memantin gar nicht. Es liegt also nahe, dass diese beiden verwandten Pharmaka an verschiedenen NMDA-Rezeptor-Subtypen wirken. Auch die Wirksamkeit von Memantin an weiteren neuronalen Rezeptoren könnte zum speziellen Wirkungsprofil beitragen. So werden neben NMDA-Rezeptoren ausserdem 5-HT-3-Rezeptoren antagonisiert, nikotinische Alpha-7-AcetylcholinRezeptoren antagonisiert und Dopamin D2-Rezeptoren aktiviert. Interessanterweise wirkt es also über den nikotinischen Alpha -7-Acetylcholin-Rezeptor der cholinergen Funktion und somit den AChE-Hemmern entgegen. Dabei führt die Alpha-7-Blockade zu einer initialen Verschlechterung der Kognition und konsekutiv einer verstärkten Expression dieser Rezeptoren und daraufhin kognitiven Besserung (Aracava, Pereira, Maelicke, & Albuquerque, 2005). Möglicherweise wirkt Memantin ausserdem hemmend auf die Amyloid- und Tau-Bildung (Wu & Chen, 2009). Die Bioverfügbarkeit beträgt 100%, die Halbwertszeit 60-100 Stunden. Memantin ist relativ gut verträglich, insbesondere treten die Nebenwirkungen der Acetylcholininhibitoren nicht auf. Es sind aber Nebenwirkungen wie Kopfschmerzen und Schwindel beschrieben. Anti-Amyloide Pharmaka Die Beta-Amyloid-Hypothese als wesentlicher pathogenetischer Mechanismus des Morbus Alzheimer ist mittlerweile über 25 Jahre alt. Es gibt viele kritische Personen, aber ein grosser Teil der Forschung setzt noch immer hier an. 37 Dabei werden verschiedene Ansätze verfolgt, um die Menge an Beta-Amyloid im Gehirn zu reduzieren. Die APP-Sekretasen (Alpha, Beta und Gamma) sind ein guter Ansatzpunkt, um die Produktion einzuschränken. Die Sekretasen können dabei in jedem Fall nur die Entstehung neuen Beta-Amyloids einschränken, nicht jedoch bestehendes entfernen. Ihr Einsatz sollte also frühest möglich, idealerweise noch vor den ersten Symptomen begonnen werden. Mit Hilfe von Antikörpern versucht man, die Phagozytose des Beta-Amyloids anzuregen. Dabei unterscheidet man die aktive Immunisierung mit Antigenen die Teile des Beta-Amyloids nachbilden und eine körpereigene Immunantwort provozieren sollen von der passiven Immunisierung mit künstlich erzeugten Antikörpern, die dann regelmässig appliziert werden müssen. Mutationen des Apolipoprotein E haben starken Einfluss auf Enstehung und Verlauf der AD. So ist bekannt, dass die E4 Variante des ApoE die AD begünstigt und die E2 Variante hemmend wirkt. Alpha-Sekretase Aktivatoren Die Familie der Alpha-Sekretasen schneidet das Amyloid-Precursor-Protein auf eine Weise, dass kein kein Beta-Amyloid mehr entstehen kann (Es entstehen stattdessen die unproblematischen Fragmente sAPP-α, CTF-α und p3, siehe Kapitel Pathophysiologie). Zur Verringerung der Plaque-Bildung liegt es also nahe, die Aktivität der Alpha-Sekretasen anzuregen und auf diese Weise dem Beta-Schnitt das Substrat zu entziehen. Im Mausmodell führte die Überexpression der Alpha-Sekretase ADAM10 zu einer Zunahme von sAPP-α, einer Reduktion von Beta-Amyloid-Plaques und kognitiver Verbesserung. Ausserdem scheint die ADAM10 – Aktivität bei Alzheimer-Erkrankten verringert zu sein. Diese Erkenntnisse lieferten die Grundlage für weitere pharmakologische Forschung. Gegenwärtig werden Strategien untersucht, die Alpha-Sekretasen direkt zu aktivieren oder indirekt über Vitamin A, Tyrosinkinase oder die Proteinkinase C. (De Strooper, Vassar, & Golde, 2010). Aber auch die Hemmung der vermehrten Endozytose der Alpha-Sekretase bei Alzheimer-Erkrankten wird untersucht. 38 Etazolat (auch EHT-0202) von Exonhit Therapeutics ist ein GABA-Rezeptor-Modulator und war der erste in Bezug auf gesteigerte Alpha-Sekretase-Aktivität untersuchte Wirkstoff. Dieser zeigte in einer Phase 2 Studie mit 159 Erkrankten 2011 zunächst gute Verträglichkeit (Vellas, et al., 2011) (Yiannopoulou & Papageorgiou, 2013). Aus nicht genannten Gründen wurde die Entwicklung 2012 jedoch eingestellt. Figure 32: Etazolat (wikipedia.org) EpiGalloCatechinGallat (NCT00951834) ist in grossen Mengen in grünem Tee enthalten. Im Mausmodell konnte gezeigt werden, dass diese Verbindung die AlphaSekretase stimuliert (vermutlich mittels der Proteinkinase C) und so die Beta-Amyloid-Produktion reduziert und sogar Plaques abbaut. Ausserdem hemmt EGCG wahrscheinlich direkt die Aggregation von Beta-AmyloidOligomeren zu Plaques. Gegenwärtig laufen noch klinische Phase 2 Studien (Mähler, et al., 2013). Figure 33: EGCG (wikipedia.org) Bryostatin wurde bereits in den 60er Jahren erstmals aus Bryozoen, auch Moostierchen genannt, extrahiert. Dieses Molekül hat eine starke Wirkung auf die Proteinkinase C und so mittelbar auch auf die Regulation der AlphaSekretase. Zunächst konnte in Ratten eine kognitive Verbesserung gezeigt werden. Auch die positiven Effekte auf depressive Verstimmungen können für AlzheimerErkrankte von Relevanz sein. (Sun & Alkon, 2005). Phase 1 Studien mit Menschen verliefen nebenwirkungsarm. Ergebnisse aus bis 2017 laufenden Phase 2 Studien stehen noch aus (clinicaltrials.gov, 2016). 39 Figure 34: Bryostatin (wikipedia.org) Beta-Sekretase Inhibitoren Die Beta-Sekretase (BACE) schneidet das Amyloid-Precursor-Protein. Nach einem weiteren Schnitt durch die Gamma-Sekretase ensteht Aβ. In der Pharmakonentwicklung sucht man nach spezifisch auf die zerebale BACE1 wirkenden Substanzen. Da die BACE neben APP noch weitere Proteine schneidet, könnte man auch mit einer Teilinhibition arbeiten, um starke Nebenwirkungen zu vermeiden (siehe Kapitel Pathophysiologie) Da die katalytische Aminosäuresequenz der Beta-Sekretase sehr lang ist, waren die ersten Moleküle, die die Bindungsstelle der Beta-Sekretase an das APP in vitro wirksam blockieren konnten, zu gross, um die Blut-Hirn-Schranke zu passieren. Daraufhin konzentrierte sich die Forschung auf die Entwicklung kleinerer Moleküle. Auch bei dieser zweiten Generation der Beta-Sekretase-Inhibitoren war das Hauptproblem das Erreichen suffizienter Wirkspiegel im Gehirn. Seit kurzem wird die dritte Generation in ersten klinischen Studien erprobt (Riqiang Yan & Robert Vassar, 2014). In diesen ersten Studien konnte bereits die grundsätzliche Wirksamkeit der BetaSekretase-Inhibitoren auf Beta-Amyloid im menschlichen Gehirn gezeigt werden. Aufgrund von Nebenwirkungen und der erst in jüngerer Zeit begonnenen klinischen Studien, konnte aber noch kein Medikament zur Marktreife entwickelt werden. Anstatt bestehende BACE1 zu blockieren, könnte ein weiterer interessanter Ansatz darin bestehen, ihre Expression zu hemmen, denn diese ist bei AD-Erkrankten erhöht. Die Entwicklung bleibt spannend. Einige Substanzen dieser Gruppe sollen im folgenden exemplarisch kurz vorgestellt werden. LY-2811-376 von Eli Lilly war der erste BetaSekretase-Inhibitor der gute Bioverfügbarkeit mit einer tatsächlichen Beta-Amyloid-Reduktion im Menschen demonstrierte (May, et al., 2011). Aufgrund starker Nebenwirkungen wurden die Versuche nach einer Phase 1 Studie eingestellt und mit dem modifizierten Nachfolger LY-2886-721 wieder aufgenommen. Phase 1 verlief nebenwirkungsfrei bei 47 Versuchspersonen und konnte eine etwa 70% Reduktion von Beta-Amyloid im Liquor demonstrieren. In Phase 2 traten LeberSchädigungen auf, so dass man die Studie abbrach (May, et al., 2015). 40 Figure 35: LY-2811-376 (medchemexpress.com) Verubecestat von Merck (auch MK-8931) ist der bislang erfolgreichste Beta-Sekretase-Inhibitor und wird als bisher einziger Wirkstoff dieser Klasse bereits in Phase 3 Studien erprobt. In Phase 1 & 2 Studien wurde gute Verträglichkeit und eine BetaAmyloid-Reduktion um etwa 80% demonstriert (Forman, et al., 2012). 2018 werden die Ergebnisse der gegenwärtigen klinischen Phase 3 Studie erwartet. Figure 36: Verubecestat (wikipedia.org) E-2609 von Eisai konnte in acht Phase 1 Studien eine Beta-Amyloid-Reduktion von bis zu 90% in der höchsten Dosis von 400mg ohne schwerere Nebenwirkungen demonstrieren (Bernier, et al., 2013). Eine Phase 2 Studie mit 700 Versuchspersonen, die nach Amyloid-PET-Scans ausgewählt wurden endet Anfang 2017. Die Rekrutierung zu einer Phase 3 Studie hat im November 2016 begonnen. Gamma-Sekretase Inhibitoren Die Gamma-Sekretase ist die längste und komplexeste der APP-Sekretasen und wurde als letzte identifiziert (siehe Kapitel Pathophysiologie). Die Entwicklung von hirngängigen Pharmaka scheint für die Gamma-Sekretase leichter zu gelingen als beispielsweise für die Beta-Sekretase-Inhibitoren. Das größte Problem stellen die starken Nebenwirkungen dar. Da bei Veränderungen der Gamma-Sekretase auch der Notch-Signalweg stark betroffen ist, entwickeln Mäuse Hirn-, Skelett-, Gastrointestinal-Deformitäten. Um diesen Nebenwirkungen zu begegnen, entwickelt man so genannte “Notch-Sparing Inhibitors”, die eine höhere Affinität zur Verhinderung des APP-Schnitts haben als zu Notch. Auch mit der Modifikation von Teilen der Gamma-Sekretase wird experimentiert. Im Mausversuch wurde durch die genetische Elimination der APH-Subtypen APH-1b und APH-1c eine verringerte Beta-Amyloid-Produktion ohne größere Nebenwirkungen erreicht (De Strooper, Vassar, & Golde, 2010). Ausserdem scheint PS1 für einen größeren Teil der Beta-Amyloid-Produktion verantwortlich zu sein, als PS2. (De Strooper, et al., 1998). Dementsprechend gibt es Überlegungen spezifische Inhibitoren für die PS1, beziehungsweise APH1 zu entwickeln und so möglicherweise Nebenwirkungen reduzieren zu können. Bei Nagetieren unterscheidet sich dieses Enzym jedoch stark von der menschlichen Variante. So besitzen Menschen nur APH-1a und APH-1b, Mäuse zusätzlich die APH41 1c (De Strooper, Vassar, & Golde, 2010). Dadurch werden die Versuche mit neuen Pharmaka in ihrer Aussagekraft eingeschränkt. Semagacestat (auch LY-450-139) ist ein unspezifischer Gamma-Sekretase-Inhibitor und war das erste Medikament dieser Art, das bis in klinische Phase 3 Studien vorgedrungen ist. Die Beta-Amyloid-Level konnten dabei in der höchsten Figure 37: Semagacestat (wikipedia.org) Dosis um bis zu 84% reduziert werden (Bateman, et al., 2009). Erstaunlicherweise besserte sich die Kognition der Erkrankten dabei jedoch nicht, sondern verschlechterte sich sogar. Darüberhinaus entwickelten etwa 5% der Versuchspersonen Basal-Zell-Karzinome, möglicherweise in Folge der Beeinträchtigung des Notch Signalwegs (Doody, et al., 2013). Nach der dritten Phase 3 Studie wurde die Entwicklung dieses Pharmakons 2011 eingestellt. Begacestat (auch GSI-953) ist einer der ersten so genannten Notch Sparing GSI. Der wichtige Notch Signalweg soll bei Applikation also möglichst unbeinträchtigt bleiben. Begacestat hat eine 16fach höhere Affinität zur Verhinderung des APP-Schnitts als zum Notch-Schnitt (Hopkins, 2012). Die Entwicklung wurde nach Phase 1 im Jahr 2010 jedoch eingestellt. Figure 38: Begacestat (chembase.cn) Avagacestat (auch BMS-708-163) von BristolMyers Squibb wurde ein 137fach erhöhte Selektivität an APP gegenüber Notch bescheinigt. Es bindet direkt an PS1 und konkurriert dort mit anderen GSI um Aktivität. Mit GSM bestehen keine Wechselwirkungen (Crump, SV, & Wang, 2012). In Ratten und Hunden konnte eine Amyloid-Reduktion ohne Notch-Nebenwirkungen demonstriert werden. 2009 wurden Phase 2 Studien mit verschiedenen Figure 39: Avagacestat Dosierungen begonnen, aus denen die meisten (sigmaaldrich.com) Erkrankten wegen starker gastrointestinaler Nebenwirkungen ausschieden. Kognitive Verbesserungen wurden ebenfalls nicht erreicht. Die Entwicklung wurde daraufhin im Jahr 2013 beendet. 42 Gamma-Sekretase Modulatoren Neben Gamma-Sekretase-Inhibitoren wird auch mit Gamma-Sekretase-Modulatoren (GSM) experimentiert. Der dahinterstehende Gedanke ist, die Beta-AmyloidProduktion nicht zu verhindern, sondern sie zu verschieben und zwar entgegen der Richtung, die man bei Alzheimer-Erkrankten beobachtet, das heißt mehr Aβ40 auf Kosten von Aβ42. Dabei wirken die GSM nicht direkt an der katalytisch aktiven Stelle des Enzyms, sondern sind allosterische Modulatoren. Auf diese Weise bleibt auch der wichtige und therapeutisch nebenwirkungssensible Notch Signalweg unbeeinflusst. Die ersten GSMs waren Nicht-Steroidale-Anti-Rheumatika. Wegen geringer Hirngängigkeit, wurde auf lipophilere Substanzen ausgewichen. Diese befinden sich gerade in den ersten Stadien klinischer Untersuchungen. Tarenflurbil (ein Enantiomer des NSAR Flurbiprofen, wirkungslos auf COX 1&2) war der erste getestete Gamma-Sekretase-Modulator. Die klinische Phase 3 Studie zeigte zwar keine Nebenwirkungen, aber auch keine signifikanten Verbesserungen im ADAS-cog, so dass die Entwicklung eingestellt wurde (Green, et al., 2009). Da eine schlechte Penetration der Blut-Hirn— Figure 18: Tarenflurbil (wikipedia.org) Schranke als ursächlich für die schwache Wirksamkeit angenommen wird, gibt es Versuche Tarenflurbil in Polyactide einzubetten, um die Hirngängigkeit zu verbessern. Erste in vitro Studien verliefen vielversprechend (Meister, et al., 2013). E-2012 von Eisei ist ein GSM der zweiten Generation. Diese binden wahrscheinlich an den PEN2-Teil der Gamma-Sekretase und ausserdem an APP (Borgegard, et al., 2012). E-2012 demonstrierte eine Halbierung des Beta-Amyloids im Plasma. Nachdem Figure 40: E2012 (International Journal of Alzheimer’s Disease) bei einigen Versuchspersonen ein Katarakt aufgetreten war, wurde die Entwicklung eingestellt (Nakano-Ito, et al., 2013). Der modifizierte Nachfolger E-2212 zeigt ebenfalls eine Halbierung der Beta-Amyloid Level und bisher keine Nebenwirkungen in einer klinischen Phase 1 Studie. EVP-0962 ist ein weiteres NSAR, das im Mausmodell Wirksamkeit demonstrieren konnte, in Phase 1 Studien keine schweren Nebenwirkungen zeigte und gegenwärtig in einer Phase 2 Studie mit 52 Versuchspersonen erprobt wird. 43 Aktive Immunisierung Die aktive Immunisierung mit Antigenen hat den Vorteil, dass der Körper bei Erfolg kontinuierlich eigene Antikörper produziert. Das größte Risiko besteht momentan in einer überschiessenden Immunantwort, die sich zum Beispiel in einer Enzephalitis äußern kann. Eine weitere Schwierigkeit stellt die Passage der Blut-Hirn-Schranke dar. Die aktive Immuno-Therapie für den Morbus Alzheimer wurde 1999 von Dale Schenk et al begonnen. Im Mausmodell führten Beta-Amyloid-Antigene zu einer deutlichen Plaque-Reduktion. Die ersten klinischen Studien wurden mit AN-1792 im Jahr 2000 durchgeführt. Phase 1 Studien zeigten gute Ergebnisse ohne Nebenwirkungen. 2001 mussten die Phase 2 Studien jedoch wegen gehäuften Auftretens von Meningoenzephalitis bei ca. 6% der Versuchspersonen wieder abgebrochen werden. Die Beta-Amyloid-Plaques wurden zwar reduziert, aber ohne signifikanten Effekt auf die Kognition (Lambracht-Washington & Rosenberg, 2013). In einer Subgruppe der abgebrochenen AN-1792-Studie fand man eine deutliche Reduktion des Beta-Amyloids sowie eine verbesserte Kognition (Fettelschoss, Zabel, & Bachmann, 2014). Diese Phase 2 Studien brachten trotz Nebenwirkungen den Nachweis, dass sich Beta-Amyloid im menschlichen Gehirn mittels Immunisierung reduzieren liess. Aufgrund der überschiessenden Entzündungsreaktion wurden in der Folge Antigene entwickelt, die keine T-Zell-Aktivierung nach sich zogen. Dazu konzentrierte man sich auf Peptide von weniger als 8 Aminosäuren Länge, da diese keine T-Zell-Reaktion auslösen. ACC-001 koppelt die letzten sieben Aminosäuren des Beta-Amyloids-42 an das CRM197 Trägermolekül (ein modifiziertes Diphterietoxin). Im Mausmodell zeigte diese Substanz bereits Plaque-reduzierende und Kognitions-verbessernde Wirkung ohne T-Zell-iniziierte Entzündungsreaktionen. (Fettelschoss, Zabel, & Bachmann, 2014) CAD-106 von Novartis exprimiert die ersten sechs Aminosäuren des Beta-Amyloids auf einem Virus-like-Particle (VLP) names Qβ. Auch dieser Impfstoff ist ähnlich vielversprechend und bisher nebenwirkungsfrei wie ACC-001 (Fettelschoss, Zabel, & Bachmann, 2014). Eine Phase 2 Studie wurde 2015 begonnen, die Resultate stehen noch aus. 44 Affitope (AD01, AD02) von AFFiRiS besteht aus 6 Aminosäuren die das N-terminale Ende des Beta-Amyloids nachbilden. Diese werden an das SchlitzschneckenHämocyanin (KLH, Keyhole Limpet Hemocyanin) als Trägermolekül gekoppelt. In Mausmodellen wurde eine Beta-Amyloid-Reduktion demonstriert, sowie kognitive Verbesserungen. Wegen der Kürze des Antigens von nur 7 Aminosäuren, sind bisher keine T-Zell assoziierten Nebenwirkungen bekannt (Mandler, et al., 2014). Phase 2 Studien konnten bisher keinen Wirknachweis erbringen. Weitere Studien sind aber geplant. Die DNS-Immunisierung ist eine Variante der aktiven Immunisierung. Dabei wird das gewünschte Antigen nicht direkt appliziert, sondern die DNS-Sequenz des Antigens subkutan oder intramuskulär verabreicht. Erste Versuche zeigten bei einer DNS-BetaAmyloid-42-Immunisierung eine Reduzierung der Beta-Amyloid-Mengen im Mausmodel um über 60% (Qu, Boyer, Johnston, Hynan, & Rosenberg, 2006). 2013 demonstrierte eine Arbeitsgruppe bei prophylaktischer DNA-Immunisierung eine Beta-Amyloid-Reduzierung um 42% im Maus-Vergleichs-Experiment (Rosenberg & Lambracht-Washington, 2013). Dieselbe Gruppe erzielte mit der Kombination aus gewöhnlicher Antigen-Immunisierung und zusätzlicher DNA-Immunisierung (PrimeBoost-Protocol) deutlich höhere Antikörper-Titer als üblich, 250 μg/mL. Passive Immunisierung Die relativ kleinen Antikörper passieren meist problemlos die Blut-Hirn-Schranke und führen in bisherigen Versuchen zu deutlich weniger Nebenwirkungen als aktive Immunkomplexe. Der Wirkspiegel dieser monoklonalen Antikörper (-mab) muss jedoch regelmässig aufgefrischt werden, was sowohl teuer als auch aufwendig ist, sowie die Compliance senkt. Häufige Nebenwirkungen sind Ödeme und Mikroblutungen. Antikörper erschweren einerseits direkt die Amyloid-Aggregation und führen andererseits zu Phagozytose durch Mikroglia. Ein weiterer Effekt ist die Verteilung der Antikörper-Antigen-Komplexe auf den ganzen Blutkreislauf entlang eines Konzentrationsgradienten weg vom Gehirn (Lambracht-Washington & Rosenberg, 2013). Ausserdem scheinen sie auch die Wirkung des löslichen Beta-Amyloids auf die Neurotransmittersysteme zu unterbinden. (Gouras, 2009). Auch scheinen Antikörper, welche an das extra-zelluläre APP-Fragment binden, intrazellulär aufgenommen zu werden und dort auf Beta-Amyloid-Aggregationen zu wirken. (Gouras, 2009) 45 Die Nebenwirkungen sind umso geringer, je weniger Beta-Amyloid im Erkrankten vorhanden ist und scheinen also wesentlich aus der Interaktion mit Beta-Amyloid zu resultieren. Dementsprechend wäre ein frühestmöglicher Therapiebeginn sinnvoll, durchaus noch vor Eintreten erster Symptome. Diese wahrscheinlich Erkrankenden lassen sich zum Beispiel mittels PET-Screenings von Risikogruppen erfassen. Solanezumab (auch LY2062430) von Ely Lilly ist ein humanisierter Maus-Antikörper und bindet an die Beta-Amyloid-Teilsequenz 13-28. Er bindet stärker an BetaAmyloid-Oligomere als an Plaques. Phase 1 & 2 Studien zeigten kaum Nebenwirkungen. Zwei grosse Phase 3 Studien konnten keine signifikanten kognitiven Verbesserungen zeigen. Nachträgliche Auswertungen dieser Studien, die nur milde Alzheimer-Fälle betrachteten, fanden jedoch eine 34 prozentige Verbesserung der Kognition im Placebo-Vergleich in dieser Subgruppe. Daraufhin wurden neue Phase 3 Studien angesetzt, die sich nur auf die Behandlung der milden Form der AD beziehen (Selkoe & Hardy, 2016) (Lambracht-Washington & Rosenberg, 2013). Im November 2016 wurden jedoch auch diese Studien wegen Wirkungslosigkeit abgebrochen. Angesichts eines erwarteten Umsatzes von etwa 10 Milliarden Dollar, fiel der Aktienkurs des Entwicklers Eli Lilly daraufhin um 10% (NZZ, 2016). Crenezumab stammt ebenfalls von Mäusen und bindet an eine ähnliche Sequenz, die Aminosäuren 12-23 des Beta-Amyloids. In Phase 1 Studien wurden vergleichsweise geringe Nebenwirkungen bescheinigt (Lambracht-Washington & Rosenberg, 2013). Vier Phase 1, 2 & 3 Studien laufen momentan. Gantenerumab ist ein rein menschlich produzierter Antikörper, der an die Epitope 311 sowie 19-28 binden kann. Bei AD-Erkrankten konnte in der PET-Kontrolle mit Pittsburgh Compound B eine 30%ige Reduktion von Beta-Amyloid festgestellt werden (Lambracht-Washington & Rosenberg, 2013). Drei Phase 3 Studien laufen noch. Aducanumab wird aus B-Zell-Klonen gesunder Personen gewonnen. Man dachte, dass die B-Zellen der gesunden Personen die Beta-Amyloid-Plaques erfolgreich verhindert hätten und das gleiche auch bei erkrankten Personen leisten könnten. Es bindet an Plaques und Oligomere, aber nicht an monomeres Beta-Amyloid. In einer erfolgreichen Phase 1 Studie wurden die Versuchspersonen erstmals mittels PETScan selektiert. 20% der Versuchspersonen entwickelten kleinere Ödeme. Die Testgruppe mit der höchsten Dosis erlitt keinen kognitiven Verfall. Seit 2015 befindet dieses Pharmakon in einer Phase 3 Studie mit 1300 Versuchspersonen mit MCI und milder AD, die mittels PET-Scan ausgewählt werden. (Selkoe & Hardy, 2016) 46 Intravenöse Immunglobuline (IVIG) werden aus gepoolten Plasmaspenden gewonnen. Man extrahiert nur die IgG – Antikörper. Diese verleihen der empfangenden Person eine breite humorale Immunität gegen alle Antigene, gegen die der Pool der Spender immun war. Die Idee dabei ist, dass man die natürlich vorkommenden Anti-Amyloid-Antikörper auffrischen möchte. Diese sind bei Alzheimer-Erkrankten verringert. Die IVIGs scheinen ausserdem speziell auf fehlgefaltetes Beta-Amyloid zu wirken und zusätzlich Mikroglia zu aktivieren. Schliesslich sind die Nebenwirkungen sehr gering im Vergleich zu anderen Immunisierungsstrategien (Relkin, 2014). Octagam und Gammagard sind IVIGs die zunächst vielversprechend aussahen, aber in Phase 2 und Phase 3 Studien keine signifikante kognitive Verbesserung zeigen konnten (Loeffler, 2013). Momentan wird Flebogamma in einer Phase 3 Studie getestet. Zur Verbesserung dieser Methode könnte man unter anderem gezielt die Konzentration an Alzheimer-relevanten Antikörpern erhöhen. APP-Antikörper sind Antikörper, die nicht an Beta-Amyloid-Sequenzen binden, sondern an die Aminosäure-Sequenz 663 bis 671 des APP. Die Beta-Sekretase, die an dieser Position schneidet wird dadurch blockiert und die Produkton von BetaAmyloid unterbunden. So braucht man die Beta-Sekretase selbst gar nicht zu verändern oder zu inaktivieren und sie kann ihrer weiteren physiologischen Funktion noch nachkommen. In vitro konnte mit dem Antikörper BBS1 (Blocking Beta Site 1) eine Beta-Amyloid-Reduktion um 22% nach nur 9 Stunden erreicht werden. Im Gegensatz zu anderen Antikörpern, wirkt BBS1 nur dem Beta-Schnitt entgegen, wird rasch metabolisiert und führt zu keiner Immunantwort und zu keinen damit verbundenen Nebenwirkungen (Arbel, Yacoby, & Solomon, 2005) (RabinovichNikitin, Rakover, Becker, & Solomon, 2012). Dieser Ansatz scheint vielversprechend, menschliche Studien liegen jedoch bislang nicht vor. Apo E4 Inhibitoren Wie Im Kapitel Risikofaktoren beschrieben, spielt das E4 Allel des ApoE eine grosse Rolle bei der Entstehung von Beta-Amyloid-Plaques. 50% der Alzheimer-Erkrankten sind homo- oder heterozygot für E4, im Kontrast zu 15% der Gesamtbevölkerung. Es stellt den grössten Risikofaktor für die Late-Onset-Alzheimer-Demenz (LOAD) dar. ApoE fördert wahrscheinlich die Aggregation und hemmt den Abbau von BetaAmyloid. Der genau Wirkmechanismus ist jedoch noch unbekannt (Kline, 2012). Mäuse ohne ApoE bilden kaum Amyloid-Plaques. Es liegt also nahe, Pharmaka zu entwicklen, die ApoE4 direkt verringern oder die Interaktion zwischen ApoE und Beta47 Amyloid blockieren. Die Forschung steht in diesem Bereich noch in einem sehr frühen Stadium, aber einige interessante Ansätze sollen hier erwähnt werden: Beta-Amyloid 12-28 Peptid: Die Aminosäuren 12-28 des Beta-Amyloids sind die Region der Interaktion mit ApoE. Dieses Molekül ist klein genug, um die Blut-HirnSchranke zu überwinden. Es bindet an ApoE und verhindert so wirksam dessen Interaktion mit Beta-Amyloid. Im Mausmodell konnte eine Plaquereduktion sowie positive Wirkung auf Kognition und Gedächtnis demonstriert werden (Kuszczyk, et al., 2013). LDL-Rezeptor Überexpression: Der LDL-Rezeptor ist wesentlich an der Regulation von ApoE beteiligt. Überexpression dieses Rezeptors an Astrozyten führt im Mausmodell zu deutlich verringerten ApoE-Mengen und konsekutiv auch zu reduzierten Plaques. Ausserdem scheint Beta-Amyloid auch direkt an den LDLRezeptor zu binden und von Astrozyten endozytiert zu werden (Basak, Verghese, Yoon, Kim, & Holtzman, 2012). ApoE – Antikörper: Ein Beispiel eines monoklonalen Antikörpers der an ApoE4 binden und dieses so der Phagozytose zuführen kann ist HJ-63. Dieser MAB zeigte eine 60-80 prozentige Plaque-Reduktion im Mausmodell (Liao, et al., 2014) (Kim, et al., 2012) Klinische Studien wurden noch nicht durchgeführt. Apo E2 Aktivatoren Es ist bekannt, dass Menschen, die das ApoE2 Allel tragen, deutlich seltener an Alzheimer erkranken (siehe Kapitel Risikofaktoren). Der genaue Wirkmechanismus ist unbekannt. Möglicherweise ist einfach die Affinität zu Beta-Amyloid geringer, so dass sich weniger Plaques ausbilden. Insofern führte eine erhöhte ApoE2 Produktion nur zur einer kompetitiven Verdrängung des E4. Bei einer unspezifischen ApoE Verstärkung gilt es die Häufigkeit des E4-Allels in der Alzheimer-Population zu bedenken. Ein solcher unspezifischer ApoE-Verstärker würde in der E4-Population negativ wirken und könnte also nur bei AlzheimerErkrankten ohne E4 eingesetzt werden. Alternativ wäre die Entwicklung eines spezifischen E2-Verstärkers denkbar. Bexaroten von Eisei ist ein Agonist am Retinoid X Rezeptor (RXR). Es wird seit 1999 zur Behandlung des kutanen T-Zell-Lymphoms verwendet. Über den Retinoid Rezeptor bewirkt das Medikament eine verstärkte Expression der ApoE RNA und Proteinbildung. (Cramer, 48 Figure 41: Bexaroten (wikipedia.org) et al., 2012) berichten über eine 50%ige Beta-Amyloid-Reduktion innerhalb von 72 Stunden im Mausmodell. In einer Phase 2 Studie aus dem Jahr 2014 konnte ebenfalls eine Plaque-Reduktion und MMSE-Score Verbesserung gefunden werden (clinicaltrials.gov, 2016). Viele Arbeitsgruppen zweifeln jedoch an diesen Ergebnissen (O'Hare, et al., 2016). Bisher ist sind keine weiteren Studien geplant. 49 Diskussion Beurteilung der cholinergen Therapie Seit dem Erscheinen der cholinergen Medikamente wird Ihre Wirksamkeit und Effektivität immer wieder kontrovers diskutiert. Hansen et al. betrachten über 300 klinische Studien und beschreiben eine durchschnittliche Verbesserung um 3 Punkte auf dem Alzheimer Disease Assessment Scale (ADAS, 0-70 Punkte möglich) im Vergleich zu Placebo-Gruppen. Wenn man bedenkt, dass die Erkrankten durchschnittlich auf 20-30 Punkte kommen, entspricht das einer Verbesserung um etwa 10%. Hansen et al. berichten von differierenden Aussagen zu Wirksamkeitsunterschieden. Manche Studien berichteten über bessere Effektivität von Donepezil, andere von Galantamin, andere fänden keine Unterschiede (Hansen, et al., 2008). Die AChE-Hemmer zeigen insgesamt eine schwache, aber dennoch signifikante Wirksamkeit in allen Stadien der AD, sind aber bisher nur zugelassen für leichte bis moderate AD. Die Gabe in schweren Stadien wird jedoch von der DGN mittlerweile auch empfohlen (Deutsche Gesellschaft für Neurologie, 2016). Eine weitere umfangreiche Meta-Studie von Di Santo et al. betrachtet insbesondere die Wirksamkeit in den verschiedenen Stadien der AD, unterteilt mittels der MiniMental State Examination in leichte, mittlere und schwere AD. Donepezil wirkt demnach besser bei schwerer AD und Galantamin, Rivastigmin etwas besser bei leichter AD (Di Santo, Prinelli, Adorni, Caltagirone, & Musicco, 2013). Die Kosten-Nutzen-Debatte zum Einsatz von Cholinesterase-Hemmern bei Alzheimer-Erkrankten wurde eine Zeitlang intensiv geführt. Eine Meta-Analyse aus dem Jahr 2006 (Loveman, et al., 2006) sieht die Kosten von Donepezil bei 80.000 Pfund pro “quality adjusted life year” (QALY) bei einer durchschnittlichen Reduzierung des Vollzeitpflegebedarfs um nur etwa 1,5 Monate. Die Mittelwerte für Rivastigmin (57.000 Pfund; 1,43-1,63 Monate Pflegereduktion) und Galantamin (68.000 Pfund; 1,42-1,73 Monate Pflegereduktion) sehen ähnlich aus. Die Arbeitsgruppe hält diese Kosten im Angesicht der moderaten Effekte nicht für angemessen. Die Situation hat sich spätestens seit 2014 jedoch grundlegend geändert, da nun alle momentan zugelassenen Cholinesterasehemmer als Generika frei verfügbar und somit deutlich billiger sind. Der Einsatz dieser gering symptomatisch wirksamen Medikamente dürfte nun fraglos zu empfehlen sein. 50 Beurteilung der glutamatergen Therapie Genau wie die cholinergen Pharmaka wird der Effekt von Memantin unterschiedlich beurteilt. Eine Auswertung von 9 randomisierten klinischen Studien der MemantinMonotherapie mit insgesamt 2433 Erkrankten (Matsunaga, Kishi, & Iwata, 2015) fand eine standardisierte Mittelwertsdifferenz (SMD) von 0,09 – 0,27, also einen geringen aber doch signifikanten positiven Effekt auf die Kognition. Die SMD ist definiert als (Pharmakon-Effekt - Placebo-Effekt) / Standardabweichung. Werte von 0,2 bezeichnet man als geringen Effekt, 0,5 als mittleren Effekt und 0.8 als starken Effekt. Dies ist ein sehr gutes Mass, um therapeutische Effekte unabhängig von der Menge an Versuchspersonen vergleichen zu können. Leider wird die SMD noch nicht überall verwendet. Eine weitere Metastudie (Molino, Colucci, Fasanaro, Traini, & Amenta, 2013) fand 13 verschiedenene Studien zu Memantin und Donepezil aufgrund methodischer Unterschiede nur schwer direkt quantitativ vergleichbar, bewertet aber die Summe der Resultate als eindeutig kognitiv relevant für beide Pharmaka. Die Kombinationstherapie ergebe keine Verbesserung gegenüber einer jeweiligen Monotherapie. Eine andere Auswertung über 7 klinische Studien mit insgesamt 2182 Versuchspersonen zur Kombinationstherapie von Memantin und Cholinesterasehemmern (Matsunaga, Kishi, & Iwata, 2014) fand eine standardisierte Mittelwertsdifferenz von 0,13 für die kognitive Verbesserung gegenüber einer Cholinesterasehemmer Monotherapie. Die Ergebnisse sind also widersprüchlich. In den aktuellen Leitlinien wird die Gabe von Memantin bei mittlerer und schwerer AD aber empfohlen. Beurteilung der amyloiden Therapie Neben der rein symptomatischen Behandlung durch cholinerge und glutamaterge Pharmaka, verspricht die Amyloid-Therapie eine kausale Behandlung und somit nachhaltige Besserung. In 20 Jahren der Forschung in diesem Bereich ist bisher noch kein Medikament zur Marktreife gelangt. Nicht einmal entscheidende Linderung der Symptome konnte in Studien erreicht werden. Das führte dazu, dass die AmyloidHypothese selbst immer wieder angegriffen und in Frage gestellt wurde. Verschiedene Experimente (s.u.), die deren Richtigkeit scheinbar bestätigen, und ein 51 Mangel an echten Alternativen bedingen jedoch eine anhaltende Fokussierung auf diesen Bereich. Amyloid-Studien beruhen meistens auf Versuchen mit doppelt transgenen Mäusen, die sowohl ein modifiziertes Amyloid-Precursor-Protein besitzen als auch ein mutiertes Präsenilin 1 Gen. Mäuse dieser Art beginnen nach etwa einem halben Jahr Beta-Amyloid-Plaques zu bilden, jedoch keine Tau-Fibrillen. Diese Mäuse eigneten sich nur sehr eingeschränkt für Amyloid-Studien. Cohen et al. entwickelten 2013 ein Modell mit modifiziertem APP und PS1 an Ratten statt Mäusen. Dieses scheint sich viel näher am Menschen zu bewegen. Beispielsweise besitzt die Ratte die gleichen sechs Tau-Protein-Isoformen wie der Mensch, die Maus jedoch nur drei. Die Beta-Amyloid-Level steigen bei diesen transgenen Ratten deutlich stärker an und in der Folge entwickelt sich die komplette vom Menschen bekannte Alzheimer-Kaskade mit Tau-Fibrillen, Amyloid-Plaques, Gliose, Neuronenverlust und kognitiven Störungen (Cohen, et al., 2013). Choi et al. gelang 2014 die Anlage einer dreidimensionalen neuronalen Zellkultur, mit der erstmals in vitro die komplette Alzheimer-Kaskade inklusive induzierter TauFibrillen nachgebildet werden konnte (Choi, et al., 2014). Eine solcher Versuchsaufbau wäre für initiale Studien noch deutlich nützlicher als das verbesserte Tiermodell. In diesen neuen Modellen kann man zum einen eine weitere Bestätigung der Amyloid-Hypothese sehen, zum anderen lässt sich die zukünftige Pharmakonentwicklung möglicherweise verbesseren und beschleunigen. Viele bisherige Medikamenten-Studien verwendeten klinische Kriterien zur Auswahl von Versuchspersonen. Im Nachhinein wurde mit PET-Scans festgestellt, dass bis zu 30% der Versuchspersonen gar keine erhöhten Beta-Amyloid-Spiegel aufwiesen. Das verfälscht das Gesamtergebnis von Studien zu Beta-Amyloid reduzierenden Medikamenten natürlich deutlich. Mittlerweile geht man dazu über die Versuchspersonen schon im Vorfeld mittels PET-Scan zu selektieren (Selkoe & Hardy, 2016). Das könnte den Erfolg zukünftiger Studien deutlich verbessern. Zusammenfassende Beurteilung Die pharmakologische Therapie des Morbus Alzheimer steht über 100 Jahre nach der ersten Beschreibung der Krankheit noch immer in den Anfängen. Die einzigen zugelassenen Medikamente verschaffen bestenfalls eine leichte Linderung der Symptome, beziehungsweise einen kurzen Aufschub des unweigerlich fortschreitenden kognitiven Verfalls. Eine kausale Therapie ist bisher nicht möglich. 52 Im Rahmen dieser Arbeit wurden drei pharmakologische Ansatzstellen exemplarisch betrachtet. Das cholinerge und glutamaterge Neurotransmittersystem stellen die Basis für die existierende Medikation. Es gibt anhaltende Forschung in diesem Bereich, die aufgrund der zu erwartenden rein symptomatischen Resultate jedoch eher beschränkt ist. Das Hauptaugenmerk der Forschung richtet sich trotz vieler Rückschläge nach wie vor auf die Amyloid-Therapie. In jüngster Zeit gab es einige wichtige Fortschritte. Dazu gehören verbesserte Tiermodelle und akkuratere Zellkulturen sowie diagnostische Fortschritte, die bessere Einschlusskriterien für klinische Studien ermöglichen. Die Effekte dieser Fortschritte kommen gerade erst in der pharmakologischen Forschung an und ermöglichen zukünftig eventuell eine deutliche Beschleunigung und qualitative Verbesserung neuer Medikamentenstudien. Die grössten Hoffnungen ruhen momentan auf Beta-Sekretase-Hemmern und passiver Immunotherapie. Weitere Ansätze sind beispielsweise Tau-Therapie, Antihypertensive Therapie oder Insulintherapie. Da Alzheimer eine Volkskrankheit ist und mittlerweile die sechsthäufigste Todesursache darstellt, fliessen noch immer grosse Mittel in die pharmakologische Entwicklung. Eine entscheidende Verbesserung der Therapie in den nächsten Jahren ist zwar momentan nicht absehbar aber durchaus möglich. 53 Literaturverzeichnis Administration on Aging. (2016). Older Population as a Percentage of the Total Population. Retrieved 12 30, 2016, from http://www.aoa.acl.gov/aging_statistics/future_growth/docs/By_Age_Total_Po pulation.xls alzheimer-forschung.de. (2016). Die Alzheimer-Krankheit und ihre Stadien. Retrieved 12 30, 2016, from https://www.alzheimer-forschung.de/alzheimerkrankheit/aktuelles.htm?showid=3197 Alzheimer’s Foundation of America. (2016). Alzheimer diagnosis. Retrieved 12 30, 2016, from http://www.alzfdn.org/AboutAlzheimers/diagnosis.html Alzheimer’s Foundation of America. (2016). Alzheimer Life Expectancy. Retrieved 12 30, 2016, from http://www.alzfdn.org/AboutAlzheimers/lifeexpectancy.html Alzheimer's Association. (2016). Alzheimer Symptoms. Retrieved 12 30, 2016, from http://www.alz.org/national/documents/checklist_10signs.pdf Alzheimer's Association. (2016). Major Milestones. Retrieved 12 30, 2016, from http://www.alz.org/research/science/major_milestones_in_alzheimers.asp Alzheimer's Disease International. (2016). World Alzheimer Report 2015. Retrieved 12 30, 2016, from http://www.worldalzreport2015.org American Psychological Association. (2013). DSM 5 Coding Update. Retrieved 12 30, 2016, from https://psychiatryonline.org/pb/assets/raw/dsm/pdf/DSM5%20Coding%20Update_Final.pdf Aracava, Y., Pereira, E. F., Maelicke, A., & Albuquerque, E. X. (2005). Memantine blocks α7 nicotinic acetylcholine receptors more potently than N-methyl-daspartate receptors in rat hippocampal neurons. Journal of Pharmacology and Experimental Therapeutics, 312(3), 1195-1205. Arbel, M., Yacoby, I., & Solomon, B. (2005). Inhibition of amyloid precursor protein processing by beta-secretase through site-directed antibodies. Proceedings of the National Academy of Sciences of the United States of America, 102(21), 7718-7723. Barbagallo, M., Belvedere, M., Di Bella, G., & Dominguez, L. J. (2011). Altered ionized magnesium levels in mild-to-moderate Alzheimer's disease. Magnesium Research, 24(3), 115-121. Basak, J. M., Verghese, P. B., Yoon, H., Kim, J., & Holtzman, D. M. (2012). Lowdensity lipoprotein receptor represents an apolipoprotein E-independent pathway of Aβ uptake and degradation by astrocytes. Journal of Biological Chemistry, 287(17), 13959-13971. Bateman, R. J., Siemers, E. R., Mawuenyega, K. G., Wen, G., Browning, K. R., & Sigurdson, W. C. (2009). A gamma-secretase inhibitor decreases amyloidbeta production in the central nervous system. Annals of Neurology, 66(1), 48-54. Bernier, F., Sato, Y., Matijevic, M., Desmond, H., McGrath, S., Burns, L., . . . Albala, B. (2013). Clinical study of E2609, a novel BACE1 inhibitor, demonstrates target engagement and inhibition of BACE1 activity in CSF. Alzheimer's & Dementia: The Journal of the Alzheimer's Association, 9(4), 886. Beubler, E. (2011). Kompendium der Pharmakologie: gebräuchliche Arzneimittel in der Praxis (3. ed.). Springer, Wien. Blennow, K. (2004). Cerebrospinal fluid protein biomarkers for Alzheimer's disease. NeuroRX, 1(2), 213-225. 54 Blennow, K., Hampel, H., Weiner, M., & Zetterberg, H. (2010). Cerebrospinal fluid and plasma biomarkers in Alzheimer disease. Nature Reviews Neurology, 6(3), 131-144. Borgegard, T., Juréus, A., Olsson, F., Rosqvist, S., Sabirsh, A., Rotticci, D., & Paulsen, K. (2012). First and second generation γ-secretase modulators (GSMs) modulate amyloid-β (Aβ) peptide production through different mechanisms. Journal of Biological Chemistry, 287(15), 11810-11819. Castellano, J. M., Kim, J., Stewart, F. R., Jiang, H., DeMattos, R. B., Patterson, B. W., & Fagan, A. M. (2011). Human apoE isoforms differentially regulate brain amyloid-β peptide clearance. Science translational medicine, 3(89), 89ra57. Catalano, S. M., Dodson, E. C., Henze, D. A., Joyce, J. G., Krafft, G. A., & Kinney., G. G. (2006). The role of amyloid-beta derived diffusible ligands (ADDLs) in Alzheimer's disease. Current topics in medicinal chemistry, 6(6), 597-608. Chen, H., & Lipton, S. A. (1997). Mechanism of memantine block of NMDAactivated channels in rat retinal ganglion cells: uncompetitive antagonism. The journal of physiology, 449(1), 27. Choi, S. H., Kim, Y. H., Hebisch, M., Sliwinski, C., Lee, S., D’Avanzo, C., & Chen, H. (2014). A three-dimensional human neural cell culture model of Alzheimer/'s disease. Nature, 515(7526), 274-278. Citron, M., Oltersdorf, T., Haass, C., McConlogue, L., Hung, A. Y., Seubert, P., . . . Selkoe, D. J. (1992). Mutation of the β-amyloid precursor protein in familial Alzheimer's disease increases β-protein production. Nature, 360(6405), 672674. clinicaltrials.gov. (2016). Bryostatin Phase 2. Retrieved 12 30, 2016, from https://clinicaltrials.gov/ct2/show/NCT02431468 clinicaltrials.gov. (2016). NCT01782742. Retrieved 12 30, 2016, from https://clinicaltrials.gov/ct2/show/NCT01782742 Cohen, R. M., Rezai-Zadeh, K., Weitz, T. M., Rentsendorj, A., Gate, D., Spivak, I., . . . Breunig, J. J. (2013). A transgenic Alzheimer rat with plaques, tau pathology, behavioral impairment, oligomeric Aβ, and frank neuronal loss. The Journal of neuroscience, 33(15), 6245-6256. Cramer, P. E., Cirrito, J. R., Wesson, D. W., Lee, C. D., Karlo, J. C., Zinn, A. E., & Casali, B. T. (2012). ApoE-directed therapeutics rapidly clear β-amyloid and reverse deficits in AD mouse models. Science, 335(6075), 1503-1506. Crump, C., SV, C., & Wang, F. (2012). BMS-708,163 Targets Presenilin and Lacks Notch-Sparing Activity. Biochemistry, 51(37), 7209-7211. Cuingnet, R., Gerardin, E., Tessieras, J., Auzias, G., Lehericy, S., Habert, M.-O., . . . Colliot, O. (2011). Automatic classification of patients with Alzheimer's disease from structural MRI: a comparison of ten methods using the ADNI database. Neuroimage, 56(2), 766-781. Danysz, W., & Parsons, C. G. (2012). Alzheimer's disease, beta-amyloid, glutamate, NMDA receptors and memantine--searching for the connections. British journal of pharmacology, 167(2), 324-352. Dauwels, J., Srinivasan, K., Ramasubba Reddy, M., Musha, T., Vialatte, F.-B., Latchoumane, C., . . . Cichocki, A. (2011). Slowing and loss of complexity in Alzheimer's EEG: two sides of the same coin? International journal of Alzheimer’s disease, 2011(p1), 539621. Dauwels, J., Vialatte, F., & Cichocki, A. (2010). Diagnosis of Alzheimer's disease from EEG signals: where are we standing? Current Alzheimer Research, 7(6), 487-505. 55 De Strooper, B., Saftig, P., Craessaerts, K., Vanderstichele, H., Guhde, G., & Annaert, W. (1998). Deficiency of presenilin-1 inhibits the normal cleavage of amyloid precursor protein. Nature, 391(6665), 387-390. De Strooper, B., Vassar, R., & Golde, T. (2010). The secretases: enzymes with therapeutic potential in Alzheimer disease. Nature Reviews Neurology, 6(2), 99-107. Deardorff, W. J., Shobassy, A., & Grossberg, G. T. (2015). Safety and clinical effects of EVP-6124 in subjects with Alzheimer's disease currently or previously receiving an acetylcholinesterase inhibitor medication. Expert review of neurotherapeutics, 15(1), 7-17. DeKosky, L. C., Lopez, O., Jones, B., Fitzpatrick, A., & Breitner, J. (2002). Prevalence of neuropsychiatric symptoms in dementia and mild cognitive impairment: Results from the cardiovascular health study. JAMA, 288(12), 1475-1483. Dementia Collaborative Research Centers. (2016). ADAS cog scale. Retrieved 12 30, 2016, from http://www.dementia-assessment.com.au Deutsche Gesellschaft für Hämatologie und Onkologie. (2016). Mini Mental Status Test. Retrieved 12 30, 2016, from https://www.dgho.de/informationen/dokumente-derarbeitskreise/geriatrische-onkologie/MMSE%20Folstein.pdf Deutsche Gesellschaft für Neurologie. (2016). S3 Leitlinie Demenzen. Devanand, D., Bansal, R., Liu, J., Hao, X., Pradhaban, G., & Peterson, B. S. (2012). MRI hippocampal and entorhinal cortex mapping in predicting conversion to Alzheimer's disease. Neuroimage, 60(3), 1622-1629. Di Santo, S. G., Prinelli, F., Adorni, F., Caltagirone, C., & Musicco, M. (2013). A meta-analysis of the efficacy of donepezil, rivastigmine, galantamine, and memantine in relation to severity of Alzheimer's disease. Journal of Alzheimer's Disease, 35(2), 349-361. Doody, R. S., Raman, R., Farlow, M., Iwatsubo, T., Vellas, B., Joffe, S., & Aisen, P. S. (2013). A phase 3 trial of semagacestat for treatment of Alzheimer's disease. New England Journal of Medicine, 369(4), 341-350. Douaud, G., Jbabdi, S., Behrens, T. E., Menke, R. A., Gass, A., Monsch, A. U., . . . Matthews, P. M. (2011). DTI measures in crossing-fibre areas: increased diffusion anisotropy reveals early white matter alteration in MCI and mild Alzheimer's disease. Neuroimage, 55(3), 880-890. Duff, K., Eckman, C., Zehr, C., Yu, X., Prada, C.-M., Perez-Tur, J., & Hutton, M. (1996). Increased amyloid-β42 (43) in brains of mice expressing mutant presenilin 1. Nature, 383(6602), 710-713. Fettelschoss, A., Zabel, F., & Bachmann, M. F. (2014). Vaccination against Alzheimer disease: an update on future strategies. Human vaccines & immunotherapeutics, 10(4), 847-851. Forman, M., Palcza, J., Tseng, J., Leempoels, J., Ramael, S., Han, D., & Jhee, S. (2012). The novel BACE inhibitor MK-8931 dramatically lowers cerebrospinal fluid Aβ peptides in healthy subjects following single-and multiple-dose administration. Alzheimer's & Dementia, 8(4), 704. Foster, D., Choi, D., Conn, P., & Rook, J. (2014). Activation of M1 and M4 muscarinic receptors as potential treatments for Alzheimer’s disease and schizophrenia. Neuropsychiatric Disease and Treatment, 2014(10), 183-191. Gault, L. M., Meier, A., Florian, H., Gauthier, S., Lin, Y., Tang, Q., & Othman, A. (2014). A Phase 2 trial of the efficacy and safety of the alpha7 agonist ABT56 126 as an add-on treatment in mild-to-moderate Alzheimer's dementia. Alzheimer's & Dementia: The Journal of the Alzheimer's Association, 10(4), 917-918. Gouras, G. K. (2009). Immunotherapy for Alzheimer disease. mAbs, 1(2), 112-114. Green, R. C., Schneider, L. S., Amato, D. A., Beelen, A. P., Wilcock, G., Swabb, E. A., & Zavitz, K. H. (2009). Effect of Tarenflurbil on Cognitive Decline and Activities of Daily Living in Patients With Mild Alzheimer Disease. Jama, 302(23), 2557-2564. Hansen, R. A., Gartlehner, G., Webb, A. P., Morgan, L. C., Moore, C. G., & Jonas, D. E. (2008). Efficacy and safety of donepezil, galantamine, and rivastigmine for the treatment of Alzheimer’s disease: a systematic review and metaanalysis. Clinical interventions in aging, 3(2), 211. Hopkins, C. R. (2012). ACS Chemical Neuroscience Molecule Spotlight on Begacestat (GSI- 953). ACS chemical neuroscience, 3(1), 3-4. Imabayashi, E., Matsuda, H., Tabira, T., Arima, K., Araki, N., Ishii, K., . . . Iwatsubo, T. (2013). Comparison between brain CT and MRI for voxel-based morphometry of Alzheimer's disease. Brain and behavior, 3(4), 487-493. Inglis, F. (2002). The tolerability and safety of cholinesterase inhibitors in the treatment of dementia. International journal of clinical practice, Supplement(127), 45-63. Jann, M. W. (2000). Rivastigmine, a new-generation cholinesterase inhibitor for the treatment of Alzheimer's disease. Pharmacotherapy: The Journal of Human Pharmacology and Drug Therapy, 20(1), 1-12. Johnson, J. W., Glasgow, N. G., & Povysheva, N. V. (2015). Recent insights into the mode of action of memantine and ketamine. Current opinion in pharmacology, 20, 54-63. Jonsson, T., Atwal, J. K., Steinberg, S., Snaedal, J., Jonsson, P. V., Bjornsson, S., . . . Maloney, J. (2012). A mutation in APP protects against Alzheimer's disease and age-related cognitive decline. Nature, 488(7409), 96-99. Kihara, T., & Shimohama, S. (2004). Alzheimer's disease and acetylcholine receptors. Acta neurobiologiae experimentalis, 64(1), 99-106. Kim, J., Eltorai, A. E., Jiang, H., Liao, F., Verghese, P. B., Kim, J., . . . Holtzman, D. M. (2012). Anti-apoE immunotherapy inhibits amyloid accumulation in a transgenic mouse model of Aβ amyloidosis. The Journal of experimental medicine, 209(12), 2149-2156. Kim, S. Y., Choi, S. H., Rollema, H., Schwam, E. M., McRae, T., Dubrava, S., & Jacobsen, J. (2013). Phase II crossover trial of varenicline in mild-tomoderate Alzheimer's disease. Dementia and geriatric cognitive disorders, 37(3), 232-245. Klein, J. (2007). Phenserine. Expert opinion on investigational drugs, 16(7), 10871097. Kline, A. (2012). Apolipoprotein E, amyloid-ß clearance and therapeutic opportunities in Alzheimer's disease. Alzheimer's research & therapy, 4(4), 1. Krystal, J. H., Sanacora, G., & Duman, R. S. (2013). Rapid-acting glutamatergic antidepressants: the path to ketamine and beyond. Biological psychiatry, 73(12), 1133-1141. Kuduk, S., & Beshore, D. C. (2014). SAR Studies on Carboxylic Acid Series M1 Selective Positive Allosteric Modulators (PAMs). Current topics in medicinal chemistry, 14(15), 1738-1754. 57 Kuhn, P.‐H., Wang, H., Dislich, B., Colombo, A., Zeitschel, U., Ellwart, J. W., . . . Lichtenthaler, S. F. (2010). ADAM10 is the physiologically relevant, constitutive α‐secretase of the amyloid precursor protein in primary neurons. The EMBO journal, 29(17), 3020-3032. Kuszczyk, M. A., Sanchez, S., Pankiewicz, J., Kim, J., Duszczyk, M., Guridi, M., . . . Sadowski, M. J. (2013). Blocking the interaction between apolipoprotein E and Aβ reduces intraneuronal accumulation of Aβ and inhibits synaptic degeneration. The American journal of pathology, 182(5), 1750-1768. Lahiri, D. K., Chen, D., Maloney, B., Holloway, H. W., Yu, Q.-s., Utsuki, T., . . . Greig, N. H. (2007). The experimental Alzheimer's disease drug posiphen [(+)phenserine] lowers amyloid-beta peptide levels in cell culture and mice. Journal of Pharmacology and experimental therapeutics, 320(1), 386-396. Lambracht-Washington, D., & Rosenberg, R. N. (2013). Anti-amyloid beta to taubased immunization: developments in immunotherapy for Alzheimer disease. ImmunoTargets and therapy, 2013(2), 105. Levin, E. D. (2013). Complex relationships of nicotinic receptor actions and cognitive functions. Biochemical pharmacology, 86(8), 1145-1152. Li, W., Yu, J., Liu, Y., Huang, X., Abumaria, N., Zhu, Y., . . . Liu, X.-G. (2014). Elevation of brain magnesium prevents synaptic loss and reverses cognitive deficits in Alzheimer’s disease mouse model. Molecular brain, 7(1), 1. Liao, F., Hori, Y., Hudry, E., Bauer, A. Q., Jiang, H., Mahan, T. E., & Lefton, K. B. (2014). Anti-ApoE antibody given after plaque onset decreases Aβ accumulation and improves brain function in a mouse model of Aβ amyloidosis. The Journal of Neuroscience, 34(21), 7281-7292. Lichtenthaler, S. F. (2012). Alpha-secretase cleavage of the amyloid precursor protein: proteolysis regulated by signaling pathways and protein trafficking. Current Alzheimer research, 9(2), 165-177. Retrieved from http://www.biochemie.abi.med.unimuenchen.de/pictures/publications/car_20110523_002.jpg Loeffler, D. A. (2013). Intravenous immunoglobulin and Alzheimer’s disease: what now? Journal of neuroinflammation, 10(1), 1. Loveman, E., Green, C., Kirby, J., Takeda, A., Picot, J., Payne, E., & Clegg, A. (2006). The clinical and cost-effectiveness of donepezil, rivastigmine, galantamine and memantine for Alzheimer's disease. NIHR Journals Library, 10(1), 1-160. Lu, C.-W., Lin, T.-Y., & Wang, S.-J. (2010). Memantine depresses glutamate release through inhibition of voltage-dependent Ca 2+ entry and protein kinase C in rat cerebral cortex nerve terminals: an NMDA receptor-independent mechanism. Neurochemistry international, 57(2), 168-176. Luo, Y., Bolon, B., Kahn, S., Bennett, B. D., Babu-Khan, S., Denis, P., & Fan, W. (2001). Mice deficient in BACE1, the Alzheimer's β-secretase, have normal phenotype and abolished β-amyloid generation. Nature neuroscience, 4(3), 231-232. Mähler, A., Mandel, S., Lorenz, M., Ruegg, U., Wanker, E. E., Boschmann, M., & Paul, F. (2013). Epigallocatechin-3-gallate: a useful, effective and safe clinical approach for targeted prevention and individualised treatment of neurological diseases? EPMA Journal, 4(1), 1. Maccecchini, M. L., Chang, M. Y., Pan, C., John, V., Zetterberg, H., & Greig, N. H. (2012). Posiphen as a candidate drug to lower CSF amyloid precursor protein, amyloid-beta peptide and tau levels: target engagement, tolerability 58 and pharmacokinetics in humans. Journal of Neurology, Neurosurgery & Psychiatry, 83(9), 894-902. MacLeod, R., Hillert, E.-K., Cameron, R. T., & Baillie, G. S. (2015). The role and therapeutic targeting of α-, β-and γ-secretase in Alzheimer's disease. Future Science, 1(3). Maddalena, A., Papassotiropoulos, A., Müller-Tillmanns, B., Jung, H. H., Hegi, T., Nitsch, R. M., & Hock, C. (2003). Biochemical diagnosis of Alzheimer disease by measuring the cerebrospinal fluid ratio of phosphorylated tau protein to βamyloid peptide42. Archives of neurology, 60(9), 1202-1206. Mamada, N., Tanokashira, D., Hosaka, A., Kametani, F., Tamaoka, A., & Araki, W. (2015). Amyloid β-protein oligomers upregulate the β-secretase, BACE1, through a post-translational mechanism involving its altered subcellular distribution in neurons. Molecular brain, 8(1), 1. Mandler, M., Santic, R., Gruber, P., Cinar, Y., Pichler, D., Funke, S. A., . . . Mattner, F. (2014). Tailoring the antibody response to aggregated Aß using novel Alzheimer-vaccines. PLoS one, 10(1), e0115237. Matsunaga, S., Kishi, T., & Iwata, N. (2014). Combination therapy with cholinesterase inhibitors and memantine for Alzheimer’s disease: a systematic review and meta-analysis. International Journal of Neuropsychopharmacology, 18(5), pyu115. Matsunaga, S., Kishi, T., & Iwata, N. (2015). Memantine monotherapy for Alzheimer’s disease: a systematic review and meta-analysis. PloS one, 10(4), e0123289. May, P. C., Dean, R. A., Lowe, S. L., Martenyi, F., Sheehan, S. M., Boggs, L. N., & Monk, S. A. (2011). Robust central reduction of amyloid-β in humans with an orally available, non-peptidic β-secretase inhibitor. The Journal of neuroscience, 31(46), 16507-16516. May, P. C., Willis, B. A., Lowe, S. L., Dean, R. A., Monk, S. A., Cocke, P. J., & Audia, J. E. (2015). The potent BACE1 inhibitor LY2886721 elicits robust central Aβ pharmacodynamic responses in mice, dogs, and humans. The Journal of Neuroscience, 35(3), 1199-1210. Mayo Clinic. (2016). Alzheimer Symptoms. Retrieved 12 30, 2016, from http://www.mayoclinic.org/diseases-conditions/alzheimersdisease/symptoms-causes/dxc-20167103 McKhann, G. M., Knopman, D. S., Chertkow, H., Hyman, B. T., Jack, C. R., Kawas, C. H., . . . Mayeux, R. (2011). The diagnosis of dementia due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimer's & Dementia, 7(3), 263-269. Meister, S., Zlatev, I., Stab, J., Docter, D., Baches, S., Stauber, R. H., & Deutsch, M. (2013). Nanoparticulate flurbiprofen reduces amyloid-β 42 generation in an in vitro blood–brain barrier model. Alzheimer's research & therapy, 5(6), 1. Molino, I., Colucci, L., Fasanaro, A. M., Traini, E., & Amenta, F. (2013). Efficacy of memantine, donepezil, or their association in moderate-severe Alzheimer’s disease: A review of clinical trials. The Scientific World Journal, 2013(1). Nagele, R. G., D’andrea, M. R., Anderson, W. J., & Wang., H.-Y. (2002). Intracellular accumulation of β-amyloid 1–42 in neurons is facilitated by the α7 nicotinic acetylcholine receptor in Alzheimer’s disease. Neuroscience, 110(1), 199211. 59 Nakano-Ito, K., Fujikawa, Y., Hihara, T., Shinjo, H., Kotani, S., Suganuma, A., . . . Tsukidate, K. (2013). E2012-induced cataract and its predictive biomarkers. Toxicological sciences, 137(1), 249-258. National Institute of Aging. (2011). Alzheimer Diagnostic Guidelines. Retrieved 12 30, 2016, from https://www.nia.nih.gov/newsroom/2011/04/alzheimersdiagnostic-guidelines-updated-first-time-decades National Institute of Health. (2012). NIH Toolbox. Retrieved 12 30, 2016, from http://www.nihtoolbox.org/ Nickols, H. H., & Conn, P. J. (2014). Development of allosteric modulators of GPCRs for treatment of CNS disorders. Neurobiology of disease, 61, 55-71. Nordberg, A., Rinne, J. O., Kadir, A., & Laangstrom, B. (2010). The use of PET in Alzheimer disease. Nature Reviews Neurology, 6(2), 78-87. NZZ. (2016). Alzheimer-Medikament ohne Wirkung. Retrieved 12 30, 2016, from http://www.nzz.ch/wirtschaft/fehlschlag-in-der-pharmabranche-alzheimermedikament-ohne-wirkung-ld.130293 O'Hare, E., Jeggo, R., Kim, E.-M., Barbour, B., Walczak, J.-S., Palmer, P., & Lyons, T. (2016). Lack of support for bexarotene as a treatment for Alzheimer's disease. Neuropharmacology, 100, 124-130. O’Brien, J., Colloby, S., Pakrasi, S., Perry, E., Pimlott, S., Wyper, D., . . . Williams, E. (2007). alpha-4-beta-2 nicotinic receptor status in Alzheimer’s disease using 123I-5IA-85380 single-photon-emission computed tomography. Journal of Neurology, Neurosurgery & Psychiatry, 78(4), 356-362. Parsons, C. G., Stöffler, A., & Danysz, W. (2007). Memantine: a NMDA receptor antagonist that improves memory by restoration of homeostasis in the glutamatergic system-too little activation is bad, too much is even worse. Neuropharmacology, 53(3), 699-723. Puri, V., Wang, X., Vardigan, J. D., Kuduk, S. D., & Uslaner, J. M. (2015). The selective positive allosteric M1 muscarinic receptor modulator PQCA attenuates learning and memory deficits in the Tg2576 Alzheimer's disease mouse model. Behavioural brain research, 287, 96-99. Qu, B., Boyer, P. J., Johnston, S. A., Hynan, L. S., & Rosenberg, R. N. (2006). Aβ 42 gene vaccination reduces brain amyloid plaque burden in transgenic mice. Journal of the neurological sciences, 244(1), 151-158. Rabinovich-Nikitin, I., Rakover, I. S., Becker, M., & Solomon, B. (2012). Beneficial effect of antibodies against β-secretase cleavage site of APP on Alzheimer’slike pathology in triple-transgenic mice. PloS one, 7(10), e46650. Ray, S., Britschgi, M., Herbert, C., Takeda-Uchimura, Y., Boxer, A., Blennow, K., . . . Karydas, A. (2007). Classification and prediction of clinical Alzheimer's diagnosis based on plasma signaling proteins. Nature medicine, 13(11), 1359-1362. Relkin, N. (2014). Intravenous immunoglobulin for Alzheimer's disease. Clinical & Experimental Immunology, 178(S1), 27-29. Riqiang Yan, P., & Robert Vassar, P. (2014). Targeting the β secretase BACE1 for Alzheimer’s disease therapy. Lancet Neurology, 13(3), 319-329. Rosenberg, R. N., & Lambracht-Washington, D. (2013). DNA Aβ42 vaccination as possible alternative immunotherapy for Alzheimer disease. JAMA neurology, 70(6), 772-773. Sadigh-Eteghad, S., Mahmoudi, J., Babri, S., & Talebi, M. (2015). Effect of alpha-7 nicotinic acetylcholine receptor activation on beta-amyloid induced 60 recognition memory impairment. Possible role of neurovascular function. Acta Cirurgica Brasileira, 30(11), 736-742. Sadleir, K. R., Eimer, W. A., Cole, S. L., & Vassar, R. (2015). Aβ reduction in BACE1 heterozygous null 5XFAD mice is associated with transgenic APP level. Molecular neurodegeneration, 10(1), 1. Schneider, L. S., Mangialasche, F., Andreasen, N., Feldman, H., Giacobini, E., Jones, R., . . . Winblad, B. (2014). Clinical trials and late-stage drug development for Alzheimer's disease: an appraisal from 1984 to 2014. Journal of internal medicine, 275(3), 251-283. Selkoe, D. J., & Hardy, J. (2016). The amyloid hypothesis of Alzheimer's disease at 25 years. EMBO molecular medicine, 8(6), 595-608. Shinada, M., Narumi, F., Osada, Y., Matsumoto, K., Yoshida, T., Higuchi, K., . . . Satoh, M. (2012). Synthesis of phenserine analogues and evaluation of their cholinesterase inhibitory activities. Bioorganic & medicinal chemistry, 20(16), 4901-4914. Slutsky, I., Abumaria, N., Wu, L.-J., Huang, C., Zhang, L., Li, B., . . . Zhuo, M. (2010). Enhancement of learning and memory by elevating brain magnesium. Neuron, 65(2), 165-177. Steen, E., Terry, B. M., J Rivera, E., Cannon, J. L., Neely, T. R., Tavares, R., . . . de la Monte, S. M. (2005). Impaired insulin and insulin-like growth factor expression and signaling mechanisms in Alzheimer's disease--is this type 3 diabetes? Journal of Alzheimer's Disease, 7(1), 63-80. Strittmatter, W. J., Saunders, A. M., Schmechel, D., Pericak-Vance, M., Enghild, J., Salvesen, G. S., & Roses, A. D. (1993). Apolipoprotein E: high-avidity binding to beta-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proceedings of the National Academy of Sciences, 90(5), 1977-1981. Sun, M.-K., & Alkon, D. L. (2005). Dual effects of bryostatin-1 on spatial memory and depression. European journal of pharmacology, 512(1), 43-51. Tharp, W. G., & Sarkar, I. N. (2013). Origins of amyloid-beta. BMC genomics, 14(1), 1. Thornton, E., Vink, R., Blumbergs, P. C., & Heuvel., C. V. (2006). Soluble amyloid precursor protein α reduces neuronal injury and improves functional outcome following diffuse traumatic brain injury in rats. Brain research, 1094(1), 38-46. Toyohara, J., & Hashimoto, K. (2010). α7 nicotinic receptor agonists: potential therapeutic drugs for treatment of cognitive impairments in schizophrenia and Alzheimer’s disease. The open medicinal chemistry journal, 4(1). Vellas, B., Sol, O., Snyder, P. J., Ousset, P.-J., Haddad, R., Maurin, M., . . . Pando, M. P. (2011). EHT0202 in Alzheimer's disease: a 3-month, randomized, placebo-controlled, double-blind study. Current Alzheimer Research, 8(2), 203-212. Wary, C., Brillault-Salvat, C., Bloch, G., Leroy-Willig, A., Roumenov, D., Grognet, J., . . . Carlier, P. (1999). Effect of chronic magnesium supplementation on magnesium distribution in healthy volunteers evaluated by 31-P-NMRS and ion selective electrodes. British journal of clinical pharmacology, 48(5), 655662. WHO. (2016). Life expectancy Data by WHO region. Retrieved 12 30, 2016, from http://apps.who.int/gho/data/view.main.SDG2016LEXREGv?lang=en Wolfe, M. S., Xia, W., Ostaszewski, B. L., Diehl, T. S., Kimberly, W. T., & Selkoe, D. J. (1999). Two transmembrane aspartates in presenilin-1 required for 61 presenilin endoproteolysis and γ-secretase activity. Nature, 398(6727), 513517. Wu, T.-Y., & Chen, C.-P. (2009). Dual action of memantine in Alzheimer disease: a hypothesis. Taiwanese Journal of Obstetrics and Gynecology, 48(3), 273277. Xie, L., Helmerhorst, E., Taddei, K., Plewright, B., Van Bronswijk, W., & Martins, R. (2002). Alzheimer’s ß-amyloid peptides compete for insulin binding to the insulin receptor. The Journal of Neuroscience, 22(10). Yang, G., Wang, Y., Tian, J., & Liu, J.-P. (2013). Huperzine A for Alzheimer’s disease: a systematic review and meta-analysis of randomized clinical trials. PloS one, 8(9), e74916. Yang, L.-s., & Ksiezak-Reding, H. (1999). Ca2+ and Mg2+ selectively induce aggregates of PHF-tau but not normal human tau. Journal of neuroscience research, 55(1), 36-43. Yang, M., & Teplow, D. B. (2008). Amyloid-beta-protein monomer folding: freeenergy surfaces reveal alloform-specific differences. Journal of molecular biology, 384(2), 450-464. Yiannopoulou, K. G., & Papageorgiou, S. G. (2013). Current and future treatments for Alzheimer’s disease. Therapeutic advances in neurological disorders, 6(1), 19-33. Yonemura, Y., Futai, E., Yagishita, S., Suo, S., Tomita, T., Iwatsubo, T., & Ishiura, S. (2011). Comparison of presenilin 1 and presenilin 2 γ-secretase activities using a yeast reconstitution system. Journal of Biological Chemistry, 286(52), 44569-44575. Zhao, P., Van-Eetvelt, P., Goh, C., Hudson, N., Wimalaratna, S., & Ifeachor, E. (2007). Characterization of EEGs in Alzheimer's disease using information theoretic methods. 29th Annual International Conference of the IEEE Engineering in Medicine and Biology Society, 2007, pp. 5127-5131. 62