Lehrstuhl für Ökologische Chemie und Umweltanalytik
Werbung

Lehrstuhl für Ökologische Chemie und Umweltanalytik
der Technischen Universität München
Entwicklung von Online-Analyseverfahren auf der Basis von EinphotonenionisationsMassenspektrometrie
Fabian Mühlberger
Vollständiger Abdruck der von der Fakultät Wissenschaftszentrum Weihenstephan für
Ernährung, Landnutzung und Umwelt der Technischen Universität München zur Erlangung des akademischen Grades eines
Doktors der Naturwissenschaften (Dr. rer. nat.)
genehmigten Dissertation.
Vorsitzender:
Univ.-Prof. Dr.rer.nat., Dr.agr.habil. Harun Parlar
Prüfer der Dissertation:
1. Univ.-Prof. Dr.rer.nat., Dr.h.c.(RO) Antonius Kettrup
2. Univ.-Prof. Dr.rer.nat. R. Zimmermann (Universität
Augsburg)
3. Priv.-Doz. Dr. rer nat., Dr.rer.nat.habil. A. Ulrich
Die Dissertation wurde am 23.06.2003 bei der Technischen Universität München eingereicht und durch die Fakultät Wissenschaftszentrum Weihenstephan für Ernährung,
Landnutzung und Umwelt am 16.10.2003 angenommen.
Den Kindern im Irak
INHALTSANGABE
DANKSAGUNG ............................................................................................................6
1
EINLEITUNG UND AUFGABENSTELLUNG.................................................8
2
FLUGZEITMASSENSPEKTROMETRIE.......................................................14
3
4
2.1
LINEARES FLUGZEITMASSENSPEKTROMETER ..................................................17
2.2
REFLEKTRON-FLUGZEITMASSENSPEKTROMETER ............................................19
IONISATIONSMECHANISMEN .....................................................................21
3.1
RESONANZVERSTÄRKTE MEHRPHOTONENIONISATION (REMPI) ....................21
3.2
EINPHOTONENIONISATION (SPI) .....................................................................25
3.3
ELEKTRONENSTOßIONISATION (EI) .................................................................27
ERZEUGUNG UND DETEKTION VON VUV-LICHT.................................30
4.1
GRUNDLAGEN DER ERZEUGUNG VON LASERINDUZIERTEM VUV-LICHT .........32
4.2
ERZEUGUNG VON VUV-LICHT DURCH DEN STRAHLENDEN ZERFALL VON
EDELGASEXCIMEREN .....................................................................................40
4.2.1
Grundlagen der Excimerkinetik.............................................................41
4.2.2
Anregung durch Elektronenstrahl .........................................................47
4.3
5
DETEKTION VON VUV-LICHT .........................................................................50
4.3.1
Halbleiterdetektoren..............................................................................50
4.3.2
Ionisationsdetektoren.............................................................................51
4.3.3
Aufbau einer Zelle zum Nachweis von VUV-Strahlung.........................57
4.3.4
Anschluss und Ausleselektronik der Detektorzelle ................................58
4.3.5
Experimentelle Messungen von laserinduziertem VUV-Licht ...............60
MOBILES FLUGZEITMASSENSPEKTROMETER MIT DREI
VERSCHIEDENEN IONISATIONSMECHANISMEN ..................................67
5.1
AUFBAU DES MASSENSPEKTROMETERS ..........................................................69
5.2
LASER .............................................................................................................71
5.2.1
OPO-Laser.............................................................................................71
5.3
ZELLE ZUR ERZEUGUNG DER VUV-STAHLUNG ..............................................75
5.4
FLUGZEITMASSENSPEKTROMETER ..................................................................81
5.5
IONENQUELLE ZUR ELEKTRONENSTOßIONISATION ..........................................83
5.6
KOMBINATION DER IONISATIONSMETHODEN ..................................................87
5.7
MASSENFILTER ...............................................................................................93
5.8
KÜHLUNG .......................................................................................................95
5.9
GASSTANDARD ...............................................................................................96
5.10
EXPERIMENTE ZUR SPEZIFIZIERUNG DES MASSENSPEKTROMETERS ..............103
5.10.1
VUV-Erzeugung in der Edelgaszelle ...................................................103
5.10.2
Einphotonenionisation (SPI) ...............................................................107
5.10.3
Resonanzverstärkte Mehrphotonenionisation (REMPI)......................110
5.10.4
Elektronenstoßionisation (EI)..............................................................112
5.10.5
Massenfilter .........................................................................................116
5.11
PROBENAHME ...............................................................................................119
5.12
HOCHTEMPERATURSONDE ............................................................................121
5.13
GASANALYSEN MIT DEM REMPI-SPI-EI-TOFMS .......................................126
5.13.1
Online–Analyse von Zigarettenrauch..................................................126
5.13.2
Analyse von Proben der Benzinsynthese vor und nach der
Hydrierung...........................................................................................130
5.13.3
6
Online-Analyse von Rauchgas einer Müllverbrennungsanlage ..........135
MOBILES MASSENSPEKTROMETER MIT EXCIMERLICHTQUELLE ................................................................................................140
6.1
AUFBAU UND FUNKTION EINER EXCIMERLICHTQUELLE ................................140
6.2
KOMBINATION EINER EXCIMERLAMPE MIT EINEM TOFMS...........................145
6.3
MESSUNGEN ZUR SPEZIFIZIERUNG DER EXCIMER-TOFMS-KOMBINATION ..149
6.4
ERSTE GASANALYSEN MIT EINEM ELEKTRONENSTRAHL-GEPUMPTEN
EXCIMER-TOFMS .......................................................................................155
7
KONZEPTE ZUR STEIGERUNG DER EMPFINDLICHKEIT SOWIE
ZUR MODIFIKATION DES REMPI-SELEKTIVITÄTSPROFILS ...........159
7.1
ANREICHERUNG IN DER PROBENAHME .........................................................159
7.1.1
Modulator ............................................................................................160
7.1.2
Anreicherungskapillare .......................................................................163
7.1.3
Ultradicke Anreicherungskapillare .....................................................163
7.1.4
Online-Anreicherung ...........................................................................165
7.1.5
Offline-Anreicherung...........................................................................169
7.2
7.2.1
DERIVATISIERUNG ........................................................................................172
Vorversuche zur Derivatisierung.........................................................174
7.2.2
8
Derivatisieren in der Probenahme des TOFMS ..................................178
ZUSAMMENFASSUNG UND AUSBLICK ...................................................185
ABKÜRZUNGSVERZEICHNIS .............................................................................192
LITERATURVERZEICHNIS .................................................................................193
VERZEICHNIS EIGENER VERÖFFENTLICHUNGEN ...................................207
6
Danksagung
An dieser Stelle möchte ich mich bei allen bedanken, die zum Gelingen dieser Arbeit
beigetragen haben.
Herrn Prof. Dr. Antonius Kettrup danke ich für die freundliche Aufnahme an seinem
Institut.
Herrn Prof. Dr. Ralf Zimmermann danke ich herzlich für die Aufnahme in seine Arbeitsgruppe, die freundschaftliche Betreuung und die interessante Themenstellung.
Herrn Prof. Dr. Egmont Rohwer, vom Department of Chemistry der University of Pretoria, danke ich für die Betreuung während meines Aufenthalts in Südafrika. Insbesondere
danke ich ihm für die Vermittlung von Kenntnissen in Chromatographie.
Herrn Priv.-Doz. Dr. Andreas Ulrich vom Physik-Department der TU München danke
ich für die überaus erfolgreichen Versuche, die wir gemeinsam durchgeführt haben.
Herrn Prof. Dr. Ulrich Boesl-von Grafenstein danke ich für seine Diskussionsbeiträge.
Ich danke meinen ehemaligen Kollegen Ralph Dorfner, Dr. Martin Blumenstock und
Dr. Hans Jörg Heger für die Einarbeitung am Institut. Meinen Kollegen und Mitstreitern
Klaus Hafner, Thomas Ferge, Stephane Gallavardin, Thomas Adam, Thorsten Hauler,
Dr. Jürgen Mguhn, Dr. Maria Martinez, Katja Neuer-Etscheid und Dr. Thorsten Streibel
danke ich für ihre Beiträge, das gute Arbeitsklima und ihre Hilfe bei den Messkampagnen.
Frau Maria Fernandes-Whaley und Herrn Alexander Whaley von der University of Pretoria danke ich für die gute und freundschaftliche Zusammenarbeit während meines
Besuches in Pretoria und während ihres Aufenthaltes in München.
Ohne eine gute Werkstatt wäre der Aufbau des Gerätes nicht möglich gewesen. Daher
danke ich dem Werkstattleiter, Herrn Dietz, seinem Vorgänger, Herrn Seif, und ihrem
Team. Besonders loben möchte ich die Arbeiten von Herrn Lang, Herrn Huber, Herrn
Stocker, Herrn Taubert und Herrn Streitenberger. Herrn Frisch danke ich für seine Hilfe
bei all meinen Elektronik-Problemen.
Für die Hilfe bei den Messkampagnen und für ihre Diskussionsbeiträge möchte ich nicht
vergessen, den Kollegen vom Bayerischen Institut für Angewandte Umweltforschung
und –technik GmbH zu danken.
7
Ich danke dem BMBF für die Finanzierung des HGF-Strategiefonds-Projektes zur
Stickoxidminderung (99/11), in dessen Rahmen diese Arbeit entstehen konnte, und den
Kooperationspartnern vom Forschungszentrum Karlsruhe GmbH, vom CUTEC-Institut
GmbH, von der Fa. Martin GmbH und von der Fa. Lurgi Energie und Entsorgung
GmbH. Herrn Prof. Dr. Seifert danke ich für die Projektleitung und Herrn Dr. Warnecke
danke ich für die Zusammenarbeit bei Planung und Durchführung der Messkampagnen.
Für die Unterstützung bei den Messkampagnen danke ich außerdem den Mitarbeitern
des Müllheizkraftwerks in Coburg und den Angestellten des Gemeinschaftskraftwerks
Schweinfurt.
Großer Dank gebührt meinen Eltern, die mir das Studium ermöglicht und mich großzügig unterstützt haben, sowie meiner Freundin Michaela David und meinem Sohn Paul,
die mir liebevoll bei meiner Arbeit zur Seite gestanden haben.
8
Kapitel 1
1 Einleitung und Aufgabenstellung
Bisher werden in der Abfallwirtschaft, in der Petrochemie und in der Lebensmittelindustrie Spurenverbindungen in Prozessgasen mittels Offline-Analysenverfahren
nachgewiesen. Dazu werden Proben genommen, zum Teil angereichert, nasschemisch
vorbereitet
(extrahiert,
instrumentellen
vorgereinigt,
Analyse
eingeengt
unterzogen,
wie
etc.)
z.B.
und
mit
anschließend
einer
Gaschromatographie-
Massenspektrometrie (GC-MS) [1]. Die Gaschromatographie trennt die Inhaltsstoffe
einer Probe auf Grund von Flüchtigkeit oder Polarität zeitlich auf. Der Nachweis der
getrennten Verbindungen erfolgt mittels Elektronenstoß-Massenspektrometrie (EI-MS).
Die zeitlich aufeinanderfolgenden Massenspektren zeigen spezifische Fragmentmuster,
die anschließend chemischen Substanzen zugeordnet werden. Aufgrund der
aufwendigen und langwierigen Probenvorbereitung und der komplizierten Identifikation
der Verbindungen aus den Fragmentmustern ist dieses analytische Verfahren für eine
zeitnahe „Online“-Analytik nicht geeignet.
Durch die Online-Erfassung organischer und anorganischer Verbindungen in Prozessgasen im Spurenbereich könnten bei industriellen Prozessen Störsubstanzen detektiert,
Produkte überwacht und Prozesse gesteuert werden. In Müllverbrennungsanlagen wäre
es z.B. möglich, mit Hilfe von Online-Methoden Schadstoffe in Echtzeit nachzuweisen
und dadurch die Verbrennungsbedingungen entsprechend zu optimieren.
Da ein schnelles analytisches Verfahren ohne Probenvorbereitung und Anreicherung
auskommen sollte, sind die Anforderungen an die Selektivität und die Empfindlichkeit
sehr hoch. Daher eignen sich für Online-Messungen besonders Massenspektrometer
(MS) in Verbindung mit weichen Ionisationsverfahren.
Um Verbindungen aus einem komplexen Gasgemisch mit einem Massenspektrometer
zeitnah zu messen zu können, sollten die Zielverbindungen durch den Ionisationsschritt
möglichst wenig fragmentiert werden. Ionisationsverfahren, die bei der Ionisation nur
wenige Fragmente erzeugen, bezeichnet man als weiche Ionisationsverfahren. Nur mit
solchen Verfahren können Informationen für eine Vielzahl von Verbindungen direkt aus
den Massenspektren entnommen werden. Weiche Ionisationsverfahren sind z.B. die
chemische Ionisation (CI) [2], die Einphotonenionisation (engl.: snigle-photon
9
ionization, SPI) [3], die Feldionisation (FI) [4] und die resonanzverstärkte
Mehrphotonenionisation (engl.: resonance enhanced multiphoton ionization, REMPI)
[5]. Aufgrund der physikalischen Vorgänge bei der Ionisation unterscheiden sich diese
Ionisationsmechanismen in ihrer Selektivität. Um möglichst umfassende Informationen
über eine Probe zu erhalten, können mehrere dieser weichen Ionisationsverfahren
kombiniert werden.
Im Hinblick auf eine möglichst umfassende Gasanalyse wurde ein mobiles Massenspektrometer entwickelt, das den parallelen Einsatz mehrerer Ionisationsmethoden
ermöglicht. Das Konzept dieses Massenspektrometers sieht vor, sehr schnell aufeinanderfolgend
bestimmte
Bestandteile
des
Probengases
durch
unterschiedliche
Ionisationsmechanismen zu ionisieren und die so erzeugten Ionen mit einem Flugzeitmassenspektrometer (engl.: time-of-flight mass spectrometer, TOFMS) massenselektiv
nachzuweisen. Die in das Gerät integrierten Ionisationsmethoden sind die resonanzverstärkte Mehrphotonenionisation (REMPI), die Einphotonenionisation (SPI) und die
Elektronenstoßionisation (engl.: electon-impact ionization, EI).
1. Die laserbasierende resonanzverstärkte Mehrphotonenionisation (REMPI) ist als
selektive und sensitive analytische Methode bekannt [6-8]. Sie eignet sich für
Online-Messungen von Spurenbestandteilen in komplexen Gasgemischen. Bei
der REMPI-Technik werden Moleküle durch ein oder mehrere Photonen angeregt (UV-Spektroskopischer Schritt) und dann durch die weitere Absorption
eines oder mehrerer Photonen ionisiert (Ionisierungsschritt). Aufgrund der resonanten Anregung eines optischen Zustandes bei der REMPI-Ionisation findet
eine Ionisation nur unter Berücksichtigung der Lage und Art der Zwischenzustände statt. Je nach Temperatur des Analyten kann die REMPI-Technik
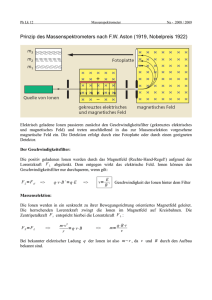
substanzklassenselektiv oder sogar isomerenselektiv sein (siehe Abbildung 1).
Die höchste Selektivität wird bei Verwendung der Überschalleinlasstechnik zur
adiabatischen Kühlung erreicht [9-12]. Durch Anpassung der Laserpulsleistungsdichte kann nahezu fragmentfrei ionisiert werden. Aufgrund der Lage der
Anregungszustände bei den aromatischen Verbindungen ist REMPI-TOFMS
zum Online-Nachweis dieser Verbindungen in Spurenkonzentrationen aus komplexen Gasgemischen prädestiniert [11, 13, 14]. In den vergangenen Jahren
wurde über den Einsatz von mobilen REMPI-TOFMS-Instrumenten für die Ana-
10
Kapitel 1
lyse von Müllverbrennungsrauchgasen [15-19], die Analyse von Kaffee-Röstgas
[14] sowie über die Analyse von Autoabgasen [20-22] berichtet.
2. Verglichen mit der REMPI-Methode ist die Einphotonenionisation (SPI) weniger selektiv (vgl. Abbildung 1). Bei dieser Technik wird vakuum-ultraviolettes
Licht (VUV-Licht) für die Einphotonenionisation verwendet [23]. Die Selektivität wird dabei durch die übertragene Ionisationsenergie bestimmt. Wird einem
Molekül durch die Absorption eines Photons mehr Energie als das spezifische
Ionisationspotential (IP) übertragen, so kommt es zur Ionisation. Eine typische,
mit einem Laser erzeugbare VUV-Photonenenergie ist 10,5 eV (118 nm). Mit
dieser Energie lassen sich viele organische und einige anorganische Moleküle
ionisieren. Laser-VUV-Licht kann durch Frequenzverdreifachung von intensiven
UV-Strahlen in einer Edelgaszelle erzeugt werden [24-26]. Eine stetig wachsende Zahl an Veröffentlichungen über die Erzeugung und Anwendung von
laserinduzierter VUV-Strahlung für die Massenspektrometrie zeigt das steigende
Interesse an dieser Methode [3, 23, 27-38].
3. Neben der REMPI- und SPI-Technik zur selektiven Ionisation von Spurenbestandteilen aus einem Gasstrom können die Bestandteile eines Gasgemisches
durch Elektronenstoß (EI) ionisiert werden. Das EI-Verfahren ist nicht geeignet,
Spurenbestandteile online zu detektieren, da es kaum selektiv ist und eine Vielzahl von Fragmentionen erzeugt. EI stellt aber eine wichtige Methode dar, um
die relativ stabilen Hauptbestandteile von Gasgemischen, wie z.B. N2, CO2, O2
und H2O, nachzuweisen.
Wie aus Abbildung 1 entnommen werden kann, sind REMPI, SPI und EI bezüglich
Selektivität sich ergänzende Techniken. Mit SPI sind je nach Intensität der Lichtquelle
Nachweisgrenzen bis in den ppb- (engl.: parts per billion, 1 Volumenbestandteil aus 109
Teilen) und mit REMPI sogar bis in den ppt- (engl.: parts per trillion, 1 Volumenbestandteil aus 1012 Teilen) Bereich erreichbar. Die Nachweisgrenzen bei EI liegen einige
Größenordungen höher. Bei einer Kombination dieser drei Ionisationsmethoden dient
die EI-Methode dazu, die typischen Hauptbestandteile von Prozessgasen, wie z.B. H2O,
N2 und CO2 nachzuweisen. Diese kleinen Moleküle haben hohe Ionisationspotentiale
und lassen sich mit EI gut ionisieren.
11
Die Aufgabenstellung dieser Promotionsarbeit ist die Entwicklung einer kompakten,
laserbasierenden Lichtquelle für VUV-Photonen. Die VUV-Lichtquelle soll so mit einem Flugzeitmassenspektrometer gekoppelt werden, dass das Analysegas in einem
Massenspektrometer alternierend und innerhalb kürzester Zeit mittels VUV-Photonen
durch den SPI-Prozess, mittels UV-Photonen durch den REMPI-Prozess und mittels
eines Elektronenstrahls durch den EI-Prozess analysiert werden kann. Dieses neue Massenspektrometer
soll
mobil
und
für
quantitative
Online-Analysen
von
Verbrennungsgasen aus der Flammenzone von Müllverbrennungsanlagen geeignet sein.
Des Weiteren soll in Hinblick auf die Entwicklung von industriell einsetzbaren Massenspektrometern
untersucht
werden,
ob
sich
elektronenstrahlgepumpte
Excimerlichtquellen anstelle von Lasern zum Bau kostengünstiger Flugzeitmassenspektrometer eignen.
Die Aufgabenstellung dieser Promotionsarbeit ist die Entwicklung einer kompakten,
laserbasierenden Lichtquelle für VUV-Photonen. Die VUV-Lichtquelle soll so mit einem Flugzeitmassenspektrometer gekoppelt werden, dass das Analysegas in einem Mas
Abbildung 1: Drei Ionisationsmethoden für die Online-Massenspektrometrie, die sich bei
gleichzeitigem oder kurz aufeinanderfolgendem Einsatz bezüglich Selektivität und Sensitivität gegenseitig ergänzen. Die Techniken sind nach steigender Selektivität der Ionisation
aufgelistet [39].
Die
vorliegende
Arbeit
beschreibt
die
Entwicklung
des
mobilen
Flugzeit-
Massenspektrometers, die Integration der Ionisationstechniken sowie die Erzeugung und
Detektion des VUV-Lichts. Es werden Versuche zur Spezifikation des Gerätes und die
daraus ermittelten Kenngrößen vorgestellt. Im Anschluss daran werden als Anwendungsbeispiele Messungen von Zigarettenrauch, von Mineralölfraktionen und vom
Abgas einer großtechnischen Müllverbrennungsanlage (MVA) dargestellt.
Für die Messung des Verbrennungsabgases einer MVA wurde das Instrument mehrere
Wochen im Kesselhaus einer Müllverbrennungsanlage betrieben. Um das bis zu
12
Kapitel 1
1300 °C heiße Abgas aus der Flammenzone der Anlage zu messen, musste für die Probenahme, im Rahmen dieser Arbeit, eine spezielle gekühlte Hochtemperatursonde
entwickelt werden. Die mit dem hier beschriebenem Spektrometer gewonnenen Ergebnisse der Messkampagne an einer MVA werden in [40] dargestellt und diskutiert.
Mit Hilfe von Standardgasen können TOFMS-Messungen quantifiziert werden (externe
Standardisierung). Für die Messungen an der MVA wurde ein Standardgas mit speziellen Verbindungen benötigt, das kommerziell nicht erhältlich war. Daher musste zur
Erzeugung eines Standardgasgemisches ein Gerät entwickelt werden. Der hier vorgestellte Standardgasgenerator basiert auf der Permeation von Substanzen durch eine
Membran oder Diffusion durch eine Kapillare und erzeugt einen konstanten Gasstrom,
in dem relevante Verbindungen in geeigneten, konstanten Konzentrationen enthalten
sind.
Um das Einsatzfeld des entwickelten Spektrometers zu erweitern und zur Verbesserung
der Nachweisgrenzen, wurden in Kooperation mit der Arbeitsgruppe von Prof. Rohwer
(University of Pretoria) Versuche zu Anreicherungseinheiten durchgeführt. Basierend
auf Kapillaranreicherungseinheiten mit absorptiven Innenbeschichtungen (stationäre
Phase) werden erste Versuche zur Steigerung der Nachweisempfindlichkeit eines
TOFMS-Systems vorgestellt und diskutiert. Weiterhin werden Konzepte zum Einsatz
von Kapillaranreicherungseinheiten zur externen Offline-Porbenahme mit anschließender thermischen Desorption und REMPI/SPI-TOFMS Analyse getestet. Es wird über
erste Tests der Kapillaranreicherungseinheiten, bei denen diese Einheiten zur In-situDerivatisierung von Aldehyden und Aminen verwendet werden, berichtet. Das In-SituDerivatisierungsverfahren ermöglicht es Amine und Aldehyde über entsprechende aromatische Derivate, die mit REMPI erfassbar sind, selektiv in einem komplexen
Gasgemisch nachzuweisen. Unveränderte Amine und Aldehyde sind hingegen mit
REMPI schwer nachzuweisen.
Im Hinblick auf die Entwicklung eines industrietauglichen Gerätes, das kontinuierlich
betrieben werden kann, einfach zu bedienen und wartungsarm ist, wurde während des
Aufbaus des beschriebenen Kombigerätes nach monochromatischen, intensiven VUVLichtquellen gesucht, die robuster und kostengünstiger sind als die laserinduzierte
VUV-Erzeugung. Eine mögliche Alternative stellt die elektronenstrahlgepumpte Excimer-Lampe dar. Ein Prototyp einer derartige Excimer-Lichtquelle wurde im Rahmen
dieser Arbeit erstmalig mit einem Massenspektrometer kombiniert. Die Experimente in
13
Kooperation mit dem Institut für Experimentalphysik (E12) der TU München verliefen
erfolgreich und lieferten vielversprechende Ergebnisse. Die VUV-Erzeugung durch den
Excimer-Prozess, der Aufbau einer Excimer-Lichtquelle und die ersten Experimente mit
der Kombination aus TOFMS und Excimer-Lampe sind ebenfalls Bestandteil dieser
Arbeit.
14
Kapitel 2
2 Flugzeitmassenspektrometrie
Die in dieser Arbeit beschriebenen Gasanalysegeräte, basieren auf der Flugzeitmassenspektrometrie. In diesem Abschnitt wird die Funktionsweise des linearen und des
Reflektron-Flugzeitmassenspektrometers beschrieben.
Werden z-fach geladene Ionen der Masse m im elektrostatischen Potenzialgradienten
erzeugt, nehmen sie aus dem Feld kinetische Energie auf und werden beschleunigt.
Ionen
unterschiedlicher
m/z-Verhältnisse
erreichen
im
E-Feld
verschiedene
Endgeschwindigkeiten. Eine sich anschließende feldfreie Flugstrecke legen Ionen mit
größerer Geschwindigkeit in einer kürzeren Zeit zurück als Ionen mit geringerer
Geschwindigkeit. Folglich werden Ionen mit unterschiedlichen m/z-Verhältnissen
getrennt.
Im Flugzeitmassenspektrometer (engl.: time-of-flight mass spectrometer, TOFMS) wird
das Detektorsignal zwischen der Erzeugung der Ionen und der Ankunft der voneinander
getrennten Spezies mit hoher Genauigkeit, z.B. durch Oszilloskope aufgezeichnet. Die
Ionensignale, über die Zeit aufgetragen, bilden die Flugzeitspektren, die durch Kalibrieren in Massenspektren umgewandelt werden können.
Gerade die Flugzeitmassenspektrometrie eignet sich besonders gut, wenn viele unterschiedliche Ionen in einem sehr kurzen Zeitintervall, und nicht kontinuierlich, erzeugt
werden und als Ionenverbände vom Entstehungsort extrahiert werden. Dieser Umstand
prädestiniert das TOFMS zur Kombination mit Ionenerzeugung durch gepulste Laser.
Bei der Laserionisation erzeugt ein ionisierender Laserstrahl im Zentrum der Ionenquelle in der Regel Ionen mit einfacher (z = 1) positiver Elementarladung (q). Alle Ionen
werden in etwa durch dieselbe Potenzialdifferenz (U) beschleunigt und erhalten damit
die gleiche kinetische Energie (E=1/2 mivi2).
qU =
1
mi vi2
2
[2.1]
Auf diese Weise erhält jedes Ion der Masse mi die spezifische Geschwindigkeit vi,
vi =
2qU
,
mi
[2.2]
15
mit der das Ion den Flug durch ein feldfreies Flugrohr antritt.
Aufgrund der unterschiedlichen Geschwindigkeiten trennen sich die Ionen auf ihrem
Flug entlang der feldfreien Strecke (d) und treffen schließlich nach ihrer Masse (m) getrennt auf dem Detektor auf. Durch Substitution von v durch d/t in der obigen Gleichung
wird deutlich, dass die Flugzeit (t) der Ionen proportional zur Wurzel ihrer Masse ist
( t ~ m ). Wenn die Massenabhängigkeit der Flugzeiten der zu untersuchenden Ionen
durch Ionen bekannter Masse kalibriert wird, lassen sich der gerätespezifische Parameter a und die Nachweisverzögerung b in Formel [2.3] durch lineare Regression
bestimmen.
t = a m +b
[2.3]
In die Steigung a fließen experimentelle Daten, wie z.B. Betriebsspannungen, die Geometrie der Abzugsfelder und die Länge der Driftstrecke ein, während b durch die
Ansprechzeiten des Detektors und der Elektronik verursacht wird.
Die Güte eines Massenspektrometers wird sowohl über sein oberes Massenlimit und
über die Transmission, als auch durch das Massenauflösungsvermögen R beschrieben.
Prinzipiell ist das obere Massenlimit nicht limitiert. Die Funktion des Detektors ist allerdings massenabhängig, kann aber experimentell bestimmt werden. Die Transmission
ist der Anteil detektierter Ionen von den erzeugten Ionen. Das Auflösungsvermögen R
beschreibt die Fähigkeit des Spektrometers, das Signal zweier Ionen mit kleinem Massenunterschied auch in Form voneinander getrennter Signalantworten zu detektieren.
In der Massenspektrometrie werden zwei Peaks als voneinander getrennt bezeichnet,
wenn das Signal zwischen den Peaks um 50% abfällt.
Wenn der feinste Massenunterschied zwischen zwei Peaks der Wert Dm ist, definiert
sich das Auflösungsvermögen zu
R=
m
Dm
[2.4]
16
Kapitel 2
In Flugzeitmassenspektrometern mit ausschließlich statischen elektrischen Feldern (für
welche t ~ m erfüllt ist) wird die Halbwertsbreite Dt (auch: full width at half maximum, FWHM) eines Flugzeitsignals zur Bestimmung des Auflösungsvermögens
herangezogen und es ergibt sich für die Massenauflösung R:
R=
m
t
=
Dm 2Dt
[2.5]
17
2.1 Lineares Flugzeitmassenspektrometer
In Abbildung 2A ist ein lineares TOFMS mit zweistufiger Ionenextraktion nach Wiley
und McLaren [41] skizziert. Bei einem linearen TOFMS [41, 42] gelangen die Ionen auf
direktem Weg durch das Flugrohr zum Ionendetektor (vgl. Abbildung 2A). Die Spannungen der Ionenquelle können so eingestellt werden, dass Ionen mit gleicher Masse
von verschiedenen Stellen des Ionisationsvolumens zur selben Zeit am Detektor ankommen. Dies ist gegeben, wenn der Detektor im 1. Ortsfokus liegt. Die Bedingungen
hierfür können nach Walter [43] berechnet werden.
Im Folgenden wird die Ionenfokussierung, die zu einer Erhöhung der Massenauflösung
führt, beschrieben (vgl. Abbildung 2). Je nach Ionisationsort, durchlaufen die erzeugten
Ionen eine größere oder kleinere Potenzialdifferenz. Demzufolge besitzen Ionen mit
gleichem m/z-Verhältnis beim Eintritt in den feldfreien Raum, je nach Ionisationsort,
eine größere oder kleinere kinetische Energie. Die Ionen mit gleichem m/z-Verhältnis,
die aufgrund ihres Entstehungsortes einen weiteren Weg im Feld zurücklegten, sind
schneller als die, die eine kürzere Strecke im Abzugsfeld zurückgelegt haben. Die
schnelleren Ionen müssen aber zum Detektor einen längeren Weg zurücklegen als die
langsameren Ionen. Genau im Ortsfokus werden die langsameren Ionen von den schnelleren Ionen eingeholt. Wenn der erste Ortsfokus so gewählt ist, dass er auf der
Oberfläche des Detektors liegt, erhält man scharfe Signale mit guter Massenauflösung.
Eine Besonderheit der von uns verwendeten TOFMS-Systeme ist, dass das Probengas
kurz vor der Ionisationszone aus einer heizbaren Metalleinlassnadel austritt (vgl.
Abbildung 2B). Damit das Abzugsfeld durch die geerdete Einlassnadel am Ionisationsort nicht gestört wird, muss der Ionisationsort, genau wie die Nadel, auf dem 0 VPotenzial liegen. Dies erreicht man, indem an die 1. und 2. Elektrode genau entgegengesetzte Potenziale anlegt (U1 = -U2).
18
Kapitel 2
Abbildung 2A: Schematischer Aufbau von linearen Flugzeit-Massenspektrometern nach Wiley und McLaren. Die Ionen werden zwischen der 1. und 2. Elektrode erzeugt und durch das
elektrische Feld E abgezogen. Zwischen der 2. und 3. Elektrode (s2) werden die Ionen nachbeschleunigt und gelangen anschließend in den feldfreien Raum. Nach dem Durchlaufen der
feldfreien Driftstrecke erreichen sie den Detektor und erzeugen das Detektorsignal. Die
schwarzen Punkte stellen Ionen mit gleichem m/z-Verhältnis dar, die trotz unterschiedlicher
Startpunkte den Detektor zur selben Zeit erreichen, da dieser im 1. Ortsfokus liegt. Wenn
man das Analysegas durch eine Einlassnadel bis kurz vor die Ionisationszone leitet, müssen,
wie in B dargestellt, die ersten beiden Elektroden auf entgegengesetzten Potenzialen liegen
(U1 = -U2).
19
2.2 Reflektron-Flugzeitmassenspektrometer
Die wahrscheinlich wichtigste Weiterentwicklung der Massenanalysatoren in der Flugzeitmassenspektrometrie geht auf die Arbeiten von Mamyrin [44] zurück. Zur
Kompensation der Energiefehler der Ionen wurde vorgeschlagen, Ionen mit Hilfe elektrostatischer Felder abzubremsen, deren Flugrichtung umzukehren und sie unter einem
bestimmten Winkel zum einfallenden Ionenstrahl auf einem Detektor zu fokussieren.
Ein solcher Aufbau wird als Reflektron-Flugzeitmassenspektrometer bezeichnet und ist
in der Abbildung 3 dargestellt.
Das Reflektron korrigiert die kinetischen Energieverbreiterungen von Ionen gleicher
m/z-Verhältnisse dadurch, dass höherenergetische Ionen tiefer in das Reflektronfeld
eintauchen und sich aus diesem Grund länger im Reflektron aufhalten als Ionen mit
geringerer kinetischer Energie. Gewöhnlich werden die Ionen im zweistufigen Reflektron zunächst durch ein Bremsfeld, das etwa zwei Drittel ihrer Energie kompensiert,
stark abgebremst, wobei sich positive Ionen durch einen positiven elektrischen Feldgradienten abbremsen lassen. An das durch die Bremselektroden aufgebaute Feld schließt
sich das Korrekturfeld an, welches von möglichst vielen scheibenförmigen Elektroden
aufgespannt wird. In Abbildung 3 wird der Einfachheit halber das Korrekturfeld nur von
2 Elektroden erzeugt.
In der Praxis werden relativ viele Elektroden benutzt, um das Korrekturfeld möglichst
homogen zu gestalten. Die Elektroden werden über eine Widerstandskette verschaltet,
so dass die Feldstärke zur Endplatte hin immer weiter zunimmt. Ist die kinetische Energie der Ionen in Vorwärtsrichtung kompensiert und die Geschwindigkeit folglich Null,
wirkt das Reflektronfeld in Rückwärtsrichtung beschleunigend. Nach Reflektion durch
den elektrostatischen Ionenspiegel treffen die Ionen auf den Detektor.
In einem Reflektron wird der 1. Ortsfokus, der hier näher an der Ionenquelle liegt als
beim linearen TOFMS, durch den Ionenspiegel auf den rückwärtigen Ionendetektor
abgebildet (Abbildung 3).
Mit einem Reflektron werden wesentlich höhere Massenauflösungen erreicht als mit
einem linearen TOFMS. Allerdings ist die Sensitivität beim Reflektron etwas geringer,
da durch die zusätzlichen implementierten Bauelemente Ionen verloren gehen.
20
Kapitel 2
Abbildung 3: Schematischer Aufbau eines gewinkelten Reflektrons mit Nadeleinlass. Die
schwarzen Punkte stellen Ionen mit gleichem m/z-Verhältnis dar, die trotz unterschiedlicher
Startpunkte den Detektor zur selben Zeit erreichen, da dieser im 2. Ortsfokus liegt.
Der interessierte Leser sei auf die Veröffentlichungen von Boesl et al. verwiesen [8, 45],
in denen die Entwicklung von Flugzeitmassenspektrometern detailliert beschrieben
wird.
21
3 Ionisationsmechanismen
In dieser Arbeit wird ein Flugzeitmassenspektrometer (TOFMS) vorgestellt, das in der
Lage ist, Gasmoleküle mittels drei verschieder Ionisationsmechanismen zu ionisieren
(siehe Kapitel 5). Die Ionisatismechanismen unterscheiden sich in den physikalischen
Vorgängen und sind bezüglich Sensitivität und Selektivität verschieden (Abbildung 4).
Nachfolgend werden die drei Ionisationsmechanismen beschrieben, durch die das im
Rahmen dieser Arbeit entwickelte TOFMS Gasproben analysieren kann.
3.1 Resonanzverstärkte Mehrphotonenionisation (REMPI)
Die
resonanzverstärkte
Mehrphotonenionisation
(engl.:
resonance
enhanced
multiphoton ionization, REMPI) ist eine Photoionisationsmethode, bei der die Atome
oder Moleküle durch Absorption mehrerer Lichtquanten ionisiert werden [7, 11, 15, 18,
45-56].
Dabei
ist
die
Energie
des
einzelnen
Photons
kleiner
als
das
Ionisierungspotenzial des Moleküls oder Atoms. Um eine Ionisation zu erreichen, muss
das Atom oder Molekül mehr als ein Photon absorbieren.
Bei resonanzverstärkter Multiphotonenionisation (REMPI) ist die Photonenenergie in
Resonanz mit einem angeregten Molekül- oder Atomzustand. Man klassifiziert daher
den REMPI-Prozess nach der Zahl der benötigten Photonen und der angeregten Zustände. Allgemein wird ein Prozess bei dem x-Photonen zu einem Zwischenzustand und yPhotonen zur Überschreitung des IP führen, als (x + y)-REMPI-Prozess bezeichnet.
Aromatische Verbindungen lassen sich z.B. mit dem (1 + 1)-REMPI-Prozess bei Wellenlängen zwischen 250 nm und 320 nm ionisieren. Aliphatische Verbindungen lassen
sich mit (1 + 1)-REMPI bei Wellenlängen deutlich unter 250 nm oder mit (2 + 1)REMPI ionisieren. Viele anorganischen Spezies können nur mit (2 + 1) oder mit noch
aufwendigeren REMPI-Prozessen erfasst werden. Zur Vertiefung sei auf die Veröffentlichungen [7, 55] hingewiesen.
22
Kapitel 3
Abbildung 4: Vergleich von REMPI, SPI und EI. Dargestellt sind von verschiedenen Molekülen, die mit A bis F bezeichnet sind, jeweils der Molekül-Grundzustand (X), ein
Anregungszustand (X*) und der Molekül-Ion-Grundzustand (X+). Rechts ist als Beispiel je
ein Massenspektrum dargestellt. Bei REMPI (oben) wird durch zwei Photonen (lUV) ionisiert. Das erste Photon bringt das Molekül in einen langlebigen angeregten Zustand. Durch
Absorption eines 2. Photons wird das angeregte Molekül ionisiert. Nach der SPI-Methode
(Mitte) wird ionisiert, wenn das IP eines Moleküls kleiner oder gleich der Photonenenergie
ist. Bei der Elektronenstoß-Ionisation (unten) ist der Energieübertrag der Elektronen (e-) so
groß, dass alle Moleküle ionisiert werden. Durch die hohe übertragene Energie kommt es
zusätzlich zur Bildung von Fragmenten.
Aufgrund des hohen Aufwandes, der bei Verwendung mehrere Laser erforderlich ist,
wurde mit dem in dieser Arbeit beschriebenen System nur nach der (1 + 1)-REMPIMethode ionisiert. Die Abbildung 4 (oben) zeigt für das Molekül A das Energie/Termschema eines resonanzverstärkten (1 + 1)-Ionisationsprozesses. Eine effiziente
23
und weiche (1 + 1)-Multiphotonenionisation kann wie im Fall A der Abbildung 4 (oben)
nur erfolgen, wenn die Laserwellenlänge in Resonanz mit der Anregungsenergie eines
UV-spektroskopischen Übergangs des Zielmolekühls ist, die Summe der Energien
zweier Photonen größer als das Ionisationspotenzial, d.h. eine Zweiphotonenionisation
energetisch möglich ist, und die Lebensdauer der angeregten Zustände nicht wesentlich
kürzer als die Laserpulsdauer (typischerweise 5 ns) ist.
Da Ionisationspotenziale und UV-Übergänge für verschiedene Moleküle unterschiedlich
sind, können bei einer bestimmten Laserwellenlänge jeweils nur einige bestimmte Verbindungen ionisiert werden. So absorbieren z.B. aromatische Verbindungen Photonen
mit Energien zwischen 5,0 eV (250 nm) und 3,9 eV (320 nm). Durch zwei Photonen
wird den Molekülen damit eine Energie zwischen 9,9 eV und 7,7 eV übertragen, welche
bei dieser Substanzklasse zum Ionisieren ausreicht. Ein großer und oft entscheidender
Vorteil dieser Ionisationsmethode gegenüber der Elektronenstoß-Ionisation (3.3) ist die
weitaus geringere Überschussenergie, die die Analyten bei der Ionisation erhalten, was
zu einem nahezu fragmentfreien Prozess führt [47].
Durch den selektiven Schritt über einen Anregungszustand ermöglicht das REMPIVerfahren den Nachweis von ausgewählten Spurenverbindungen aus komplex zusammengesetzten Proben. Aromatische, phenolische und heterozyklische Verbindungen
können bis in den ppt-Konzentrationsbereich hinein direkt aus einer Gasprobe nachgewiesen werden. Diese Methode eignet sich hervorragend für zeitaufgelöste OnlineMessungen, da kein vorhergehender Aufkonzentrierungsschritt nötig ist. Die mit einem
REMPI-TOFMS nachgewiesenen Ionen können durch zwei Kriterien identifiziert werden. Zum einen wird mit dem TOFMS die Masse des Ions bestimmt und zum anderen
ist die Ionisation nur möglich, wenn die verwendete Laserwellenlänge auf einen passenden Anregungszustand trifft. Damit wird Massen- und Wellenlängenselektivität erreicht
[46, 57, 58].
Die Wellenlängenselektivität lässt sich durch die Temperatur des Analyten beeinflussen.
Wenn z.B. das Probengas vor der Ionisation stark abgekühlt wird, befinden sich die
Moleküle statistisch verteilt öfter im rotatorischen Grundzustand und für den als
REMPI-Zwischenschritt benutzten 1. vibratorischen Anregungszustand gibt es weniger
rotatorische Entartungen. Dies macht die Absorptionslinien schmäler und bei gleicher
Wellenlänge werden bei gekühltem Probengas weniger Verbindungen ionisiert. Ist von
24
Kapitel 3
einem Molekül die genaue Energie einer UV-Absorptionslinie bekannt, kann dieses mit
Hilfe eines durchstimmbaren Lasers selektiv ionisiert werden.
Ein Probengas kann durch eine Überschall-Einlassdüse gekühlt werden. Zu dieser Technik, die im hier beschriebenen Gerät nicht vorhanden ist, verweise ich auf die
Literaturstellen [59-61].
Wie aus der Beschreibung des REMPI-Prozesses zu ersehen ist, ist für gezieltes Messen
spektroskopisches Wissen erforderlich. Um die UV-Wellenlänge zu ermitteln, bei der
mit REMPI ionisiert werden kann, wird im allgemeinen ein sehr schmalbandiger durchstimmbarer Laser verwendet. Mit diesem kann das REMPI-Ionisationsverhalten von
Substanzen in Abhängigkeit von der UV-Wellenlänge bestimmt werden (Abbildung 45
rechts). Für den analytischen Nachweis von Spurensubstanzen wird möglichst die Wellenlänge verwendet, bei der in der REMPI-Spektroskopie die meisten Ionen erzeugt
wurden. Damit steht die Photonenenergie fest, bei der die maximale Sensitivität für
diese Substanz erreicht wird. Gibt es eine andere Verbindung mit der selben Masse,
vergleicht man die spektroskopischen REMPI-Messungen dieser Substanzen und wählt
nach Möglichkeit eine Wellenlänge, bei der sich nur das für die Messung interessante
Molekül ionisieren lässt.
25
3.2 Einphotonenionisation (SPI)
Bei der Einphotonenionisationsmethode (engl.: single-photon ionization, SPI), werden
durch Absorption von nur einem Vakuum-Ultraviolet-Photon (VUV-Photon), Atome
oder Moleküle ionisiert [62, 63] (Abbildung 4, Mitte). Bedingung für die SPI z.B. von
einem Molekül ist, dass das Ionisationspotenzial (IP) des Moleküls unterhalb der Photonenenergie liegt (vgl. Abbildung 4).
In der analytischen Massenspektrometrie werden die 118 nm-VUV-Photonen für den
SPI-Prozess durch das Verdreifachen der dritten harmonischen Frequenz des Nd:YAGLasers (355nm) in einer Xenon Edelgaszelle durch einen nichtlinearen optischen Prozess erzeugt [24-26]. Diese Art der VUV-Erzeugung ist in Kapitel 4.1 beschrieben. Der
Verdreifachungsprozess in einer Edelgaszelle ist vergleichsweise ineffizient, aber da im
Vergleich zum REMPI-Prozess der Absorptionswirkungsquerschnitt für ein VUVPhoton sehr viel größer ist als der Absorptionswirkungsquerschnitt für zwei UVPhotonen bei REMPI, reicht eine relativ geringe Anzahl an VUV-Photonen, um ein
nachweisbares Ionensignal zu erzeugen.
Für die zeitauflösende Massenspektrometrie zur Untersuchung von chemischen Prozessen bietet sich die VUV-Einphotonenionisation an, da durch VUV-Ionisation
aliphatische und aromatische Kohlenwasserstoffe, als auch anorganische Verbindungen
wie Ammoniak und NO nachgewiesen werden können.
Die Selektivität der SPI-Methode ist geringer als bei REMPI und besteht darin, dass
Moleküle mit einem IP das höher liegt als die Photonenenergie nicht ionisiert werden.
Bei komplexen Prozessgasen kann es daher sinnvoll sein, dieses Verfahren mit der selektiveren REMPI-Methode zu kombinieren.
Die
meisten
Hintergrundgase,
wie
Stickstoff
(IP = 15.58 eV),
Sauerstoff
(IP = 12.06 eV), Kohlendioxid (IP = 13.77 eV), und Wasserdampf (IP = 12.62 eV), haben ein höheres Ionisationspotenzial als 10.49 eV der 118 nm-Photonen aus der
Nd:YAG-Laser–Xenonzellenkombination. Sie werden deshalb durch die 118 nm-VUVStrahlung nicht ionisiert. Dies ist ein Vorteil, da die in hohen Konzentrationen vorkommenden Hintergrundgase sonst zu Detektorsättigung führen würden, die auch die
anderen Massenkanäle beeinflusst. Außerdem würde es wegen der hohen Ionendichte in
der Ionenquelle und im Flugrohr des Massenspektrometers zu Raumladungseffekten
26
Kapitel 3
(Peakverbreiterung) kommen. Viele analytisch interessante Verbindungen hingegen, wie
z.B. NO (IP = 9.25 eV) oder die meisten organischen Moleküle, haben ein deutlich kleineres IP als 10.5 eV und lassen sich somit bis in den unteren ppb-Bereich nachweisen.
Aufgrund der Photonenenergie, die im Bereich der Molekül-IPs liegt, ist die SPIMethode mit laserinduziertem VUV-Licht eine sehr weiche Ionisationsmethode, die nur
sehr wenig zusätzliche Energie auf das Ion überträgt und somit zu geringer Fragmentation führt. Die große Anzahl der Veröffentlichungen, bei denen die SPI Methode
Anwendung findet, zeigt das Interesse der Analytiker an diesem Ionisationsverfahren [3,
23, 27-36, 38].
27
3.3 Elektronenstoßionisation (EI)
Die Elektronenstoßionisation (engl.: electron impact, EI) ist die Standardionisationstechnik für die organische Massenspektrometrie. Bei der EI werden Elektronen, die aus
einer Glühkathode emittiert werden, auf eine kinetische Energie von typischerweise
70 eV beschleunigt. Diese Elektronen stoßen auf die Moleküle und übertragen einen
Teil ihrer kinetischen Energie. Typischerweise werden – verglichen mit dem Ionisierungspotential (für organische Moleküle: IP ~ 6-11 eV) – relativ hohe Energien
übertragen. Das führt zu einer Fragmentierung vor allem fragilerer organischer Moleküle (vgl. Abbildung 4, unten). Diese Eigenschaft macht die EI ungeeignet zur OnlineBestimmung organischer Verbindungen aus komplexen Mischungen. Für den OnlineNachweis kleiner anorganischer Moleküle im ppm-Konzentrationsbereich, wie z.B. von
Schwefeldioxid (SO2), Blausäure (HCN) oder Schwefelwasserstoff (H2S), aber auch
von stabileren kleinen organischen Molekülen, wie z.B. Acetylen oder Methan, ist EI
jedoch sehr gut geeignet. Diese Spezies fragmentieren unter EI kaum und sind aufgrund
ihrer relativ hoch liegenden IPs nicht über die Photoionisationsmethoden SPI und
REMPI erfassbar.
In Abbildung 5 sind als Beispiel die Elektronenstoßspektren von HCN (27 amu) und
Benzol (78 amu) dargestellt. HCN fragmentiert kaum. Obwohl Benzol ein besonders
stabiles organisches Molekül ist, bilden sich bei EI mit 70 eV-Elektronen schon eine
Vielzahl an Fragmenten. Das 70 eV-EI-Spektrum in Abbildung 5 (rechts) zeigt für Ben-
Abbildung 5: Elektronenstoßspektren von HCN und Benzol. Die HCN- und das BenzolSpektrum wurden mit 70 eV-Elektronen erzeugt. Beide Spektren stammen vom National Institute of Standards and Technology (NIST) [64].
28
Kapitel 3
zol 23 Fragmentpeaks. Die Fragmentsignale erreichen dabei Höhen bis zu 20 % vom
Hauptsignal bei 78 m/z.
Da Verbrennungsabgase viele Verbindungen enthalten, die fragiler als Benzol sind, ist
offensichtlich, dass eine sehr große Anzahl von Fragmentpeaks die Identifikation von
Spurenbestandteilen erschwert oder sogar unmöglich macht.
Im Vergleich zur Photoionisation ist die Fragmentierung bei der Verwendung von Elektronen mit 70 eV kinetischer Energie sehr viel stärker (vgl. Abbildung 6). In der
Massenspektrometrie werden oft 70 eV-Elektronen verwendet, da sich mit dieser Energie effektiv ionisieren lässt und die Fragmente Aufschluss über die Struktur einer
Verbindung geben. Außerdem gibt es zu dieser Elektronenenergie, für eine Vielzahl von
Verbindungen, Vergleichsmassenspektren [64], die zur Identifizierung verwendet werden können. Verwendet man Elektronen mit niedrigerer kinetischer Energie, nimmt der
Fragmentierungsgrad bei Annäherung an das Ionisationspotential der zu untersuchenden
Verbindung ab (siehe Abbildung 6). Der Wirkungsquerschnitte von Molekühlen für
Elektronenstoßionisation ist bei Energien nahe des IPs aber sehr klein und steigt in diesem Bereich nur langsam an. Aus diesem Grund eignen sich relativ langsame
Elektronen schlecht für sensitive Messungen.
Die Sensitivität bei EI wird durch die Intensität des verwendeten Elektronenstroms bestimmt. Der Elektronenstrom korreliert mit der Heizspannung der Glühkatode und kann
daher in einem großen Bereich variiert werden.
Da die EI-Ionisierung im Prinzip eine kontinuierliche Ionenquelle darstellt und das
TOFMS ein gepulst arbeitender Massenanalysator ist, erwies sich die Implementierung
der EI-Methode in ein Lasermassenspektrometer als eine diffizile Optimierungsaufgabe
(siehe Kapitel 5.5).
29
Abbildung 6: Gemessene Elektronenstoß- und Photoionisations-Massenspektren von Diethylether [65]. Die Elektronenstoß-Massenspektren wurden mittels Elektronen mit einer
kinetischen Energie von 70, 12 oder 10 eV erzeugt.
30
Kapitel 4
4 Erzeugung und Detektion von VUV-Licht
Es ist in der Flugzeitmassenspektrometrie nicht möglich, Schadstoffe in ppmKonzentrationen oder weit darunter gleichzeitig mit N2, O2 und CO2, die in Konzentrationen von einigen Prozent vorhanden sind, zu detektieren. Für die Umweltanalytik
wäre es daher gut, ein teilselektives Verfahren zu haben, das die Hauptbestandteile der
Luft nicht ionisiert.
Die Photoionisation mit Vakuum-UV-Licht (VUV-Licht) stellt solch eine teilselektive
Methode dar. Verwendet man z.B. Licht mit einer Wellenlänge von 103 nm und somit
mit einer Photonenenergie von 12 eV, würden aufgrund des Ionisationspotenzial (IP)
von N2 (IP = 15,58), O2 (IP = 12,06), H2O (IP = 12,62) und CO2 (IP = 13,77) [64] diese
Gasbestandteile nicht ionisiert werden. Andere Moleküle mit einem IP von höchstens
12 eV wären nachweisbar.
In technischer Hinsicht ist zu beachten, dass unterhalb von 120 nm auch spezielle Optiken wie z.B. MgF2- und LiF-Kristalle eine relativ geringe Transmission zeigen
(Abbildung 7). Unter 110 nm ist MgF2 praktisch nicht mehr durchlässig und LiF weist
schon hohe Transmissionsverluste auf. Es ist zwar möglich, einen Photonenstrahl durch
Reflexion zu formen und zu leiten, doch ist es oft notwendig, den Erzeugungsort der
VUV-Strahlung durch eine Trennschicht vom Vakuum der Ionenquelle zu trennen. Dafür wird ein VUV-Licht-transmittierendes Material benötigt.
Aufgrund der IPs der Luftbestandteile, der verfügbaren optischen Materialien und der
Fortschritte in der Festkörperlasertechnik bietet sich in Verbindung mit einem TOFMS
die laserindizierte 118,2 nm-Strahlung, die man durch das Summenfrequenzmischen der
3. harmonischen Wellenlänge (355 nm) eines Nd:Yag-Lasers erhält, an. Die 118,2 nmStrahlung mit einer Photonenergie von 10,49 eV nützt die Transmissionsgrenzen moderner Materialien optimal aus und besitzt, wie im Weiteren gezeigt wird, genügend
Energie, um eine Vielzahl interessanter Moleküle zu ionisieren und damit der Flugzeitmassenspektrometrie zugänglich zu machen.
Die ersten SPI-Massenspektrometer wurden in den 60er Jahren gebaut. Zur Massenbestimmung wurden damals Sektorfeldmassenanalysatoren verwendet [66, 67]. Bevor es
möglich war laserinduzierteres VUV-Licht zu erzeugen, wurden Gasentladungslampen
in unterschiedlichen Ausführungen und mit verschiedenen Gasen verwendet, die bis ins
31
tiefe VUV und darunter emittieren. Zum trennen der Lichtquelle vom Vakuum der Ionenquelle wurden schon damals die oben genannten Kristalle verwendet. Bei den
Spektroskopieversuchen mit Wellenlängen unter 120 nm wurden dünnen Kapillaren
(ähnlich Lichtleiter) in Verbindung mit differenziellen Druckstufen verwendet [68-70].
Die Wellenlängenselektion aus dem breitbandigen Emissionsspektren der Entladungslampen erfolgte über einen Monochromator [66, 67, 71]. Aufgrund der vergleichsweise
geringen Photonendichten, die mit den Lampen am Ionisationsort erzeugt werden konnten, wurden nur Nachweisgrenzen bis 10 ppm erreicht [72].
Um Spurenbestanteile eines Gasstroms in der Ionenquelle eines TOFMS mittels SPI zu
ionisieren, benötigt man intensive VUV-Strahlung. Lässt sich VUV-Strahlung fokussieren, so sind ausreichende Photonendichten am Ionisationort erzeugbar. Nicht nur mit
Laser induzierter VUV-Strahlung könnten die erforderlichen Photonendichten erzeugt
werden, sondern auch mit brillanten Excimer-Punktlichtquellen. Deren intensiver
Leuchtpunkt lässt sich durch ein Linsensystem in die Ionisationszone abbilden.
Im Folgenden werden die theoretischen Grundlagen der VUV-Lichtquellen beschrieben,
die sich für den Einsatz in mobilen Flugzeitmassenspektrometern eignen. Anschließend
wird die Detektion der VUV-Strahlung behandelt.
Abbildung 7: Relative Transmission durch 2-3 mm MgF2 (links) und LiF (rechts) in Abhängigkeit von der Wellenlänge. [73]
32
Kapitel 4
4.1 Grundlagen der Erzeugung von laserinduziertem VUV-Licht
v
Wird von außen an ein Gas ein hochfrequentes elektromagnetisches Feld ( E ) angelegt,
werden die quasifreinen Elektronen in Schwingung versetzt. Wird die Stärke des elektromagnetischen Feldes erhöht, kommt irgendwann der Punkt, bei dem die Elektronen
v
dem Feld nicht mehr folgen können. Damit ist eine Stärke des E -Feldes erreicht, bei
der die Elektronen nicht mehr harmonisch, sondern unharmosch schwingen. Die
v
Reaktion eines Mediums auf ein angelegtes Feld wird Polarisation ( P ) genannt.
Da das Verschieben der Elektronen gegenüber dem Atommittelpunkt selber wieder ein
v
elektrisches Feld erzeugt, das dem von außen angelegten Feld ( E ) entgegengerichtet ist
und damit das gesamt Feld im Medium beeinflusst, ist
v
v v
P = e 0 * c (E) * E .
[4.1]
v
Dabei ist e 0 die elektrische Feldkonstante und c (E ) die elektrische Suszeptibilität.
v
Dadurch, dass die Elektronen dem E -Feld bei geringer Stärke harmonisch folgen könv
nen, ist c (E ) in diesem Bereich eine Konstante. Erst wenn das im Medium erzeugte
Gegenfeld nicht mehr proportional mit dem angelegten Feld steigt, ist c eine Funktion
v
von E .
Abbildung 8: Schematische Darstellung des Aufbaus einer Xenonzelle mit zwei Linsen zur
Erzeugung von 118 nm VUV-Strahlung aus einem 355 nm Laserstrahl. Die VUV-Strahlung
entsteht durch Frequenzverdreifachung im Fokus des Pumplaserstrahls.
Photonen sind elektromagnetische Wellen. Fokussiert man einen Laserstrahl in ein Gas
(siehe Abbildung 8), so kann man im Fokalbereich so große Feldstärken erzeugen, dass
die Elektronen dort unharmonisch schwingen. Für diesen Fall kann die Funktion [4.1]
vereinfacht als Potenzreihe geschrieben werden.
33
v
v
v
v
P = e 0 * c 1 * E + c 2 * E 2 + c 3 * E 3 + ...
(
)
[4.2]
Aufgrund der relativ kleinen Werte von c2 , c3,... liefern die nichtlinearen Terme
v
v
v
c 2 * E 2 + c 3 * E 3 + ... erst bei sehr großen Feldstärken ( E ) einen relevanten Beitrag.
Bei Festkörpern ergibt sich für die Größenordnung der Konstanten c1 » 1, c2 »
10−10 cm/V und c3 » 10-17 cm2/V2 [74]. Die höheren Ordnungen der Suszeptibilität
werden sehr schnell kleiner und sehr große Feldstärken wären nötig, damit diese Terme
zur Polarisation beitragen.
Durch den Einfluß der Nachbaratome auf die Bewegung der quasifreien Elektronen sind
die Suszeptibilitäten c1, c2 , c3, ... eigentlich Tensoren. In isotropen Medien wie Gläsern
und Gasen, in denen alle physikalischen Vorgänge in alle Richtungen gleich ablaufen,
ist c eine skalare Größe.
Wird das elektromagnetische Feld im Medium durch Photonen erzeugt, deren Strahlung
sich wie eine harmonische Welle ausbreitet, ist
v
v
E = E 0 cos(wt).
[4.3]
Die resultierende elektrische Polarisation ist damit
v
v
v
P = e 0 c 1 E 0 cos(w t ) + e 0 c 2 E 02 cos 2 (w t ) +
[4.4]
v
+ e 0 c 3 E03 cos3 (w t ) + ... .
Dies kann geschrieben werden als
v
v
v
P = e 0 c 1 E 0 sin(w t ) + 1 / 2e 0 c 2 E 02 [1 + cos(2w t )] +
[4.5]
v
1 / 4e 0 c 3 E 03 [3 cos(w t ) + cos(3w t )] + ... .
Hätte man nur den linearen ersten Term von [4.5], würde die elektrische Polarisationswelle einem Wechselstrom entsprechen, der dem einfallenden Licht folgt. Die
elektromagnetische Strahlung bzw. das Licht, welches vom Medium abgestrahlt werden
würde, wäre die gebrochene Welle, die sich im allgemeinen mit geringerer Geschwindigkeit v fortpflanzt als die erzeugende Welle und dieselbe Frequenz wie das einfallende
Licht hat. Im Gegensatz dazu liefern die Terme höherer Ordnung (n) in Gleichung [4.5]
Polarisationswellen, die mit der Frequenz nw schwingen.
34
Kapitel 4
Aus der Maxwellgleichung für elektromagnetische Wellen
[4.6]
v
v
¶2E
DE - ee 0 m 0 2 = 0
¶t
und
[4.7]
r r
r
r
r v
D = P + e 0 E bzw. ee 0 E = e 0 E + P
ergibt sich
v
v
v
2
2
¶ E
¶ E ¶ P
m
(
e
+ 2 )=0
0
0
¶t 2
¶t
¶z 2
[4.8]
2
und dann in mehreren Schritten aus [4.5]
[4.9]
v
v
¶ E
¶2E
- e 0 m0 2 =
¶z 2
¶t
2
= m0
v
v
v
¶ 2 (e 0 c 1 E 0 cos(wt ) + 1 / 2e 0 c 2 E 02 [1 + cos(2wt )] + 1 / 4e 0 c 3 E 03 [3 cos(wt ) + cos(3wt )] + ...)
¶t 2
v
v
v
v
¶ 2 PNL
¶2E
¶2E
¶2E
und
- e 0 m 0 2 = c 1e 0 m 0 2 + m 0
¶z 2
¶t
¶t
¶t 2
[4.10]
die Wellengleichung
v
v
v
¶ 2 PNL
¶2E
¶2E
- ( c 1 + 1)e 0 m 0 2 = m 0
¶z 2
¶t
¶t 2
mit
v
v
v
¶ 2 (1 / 2e 0 c 2 E 02 [1 + cos(2wt )] + 1 / 4e 0 c 3 E 03 [3 cos(wt ) + cos(3wt )] + ...)
¶ 2 PNL
m0
= m0
¶t 2
¶t 2
[4.11]
35
bei der die nichtlineraren (NL) Glieder der Polarisation auf der rechten Seite zusammengefasst sind.
Durchdringt eine elektromagnetische Welle ein isotropes Medium, wie in Abbildung 9
dargestellt, so kann aufgrund der Tatsache, dass alle materialspezifischen Vorgänge hier
in allen Richtungen gleich ablaufen, eine Freuquenzverdopplung der Pumpstrahlung
nicht stattfinden. Wie Abbildung 9A zu entnehmen ist, kann auf eine Welle mit doppelter Frequenz von der eingestrahlten Welle kein positiver Energiebetrag übertragen
werden. In anisotropen Kristallen hingegen ist dies möglich. Abbildung 9B zeigt, dass
auf eine Welle mit 3-facher Frequenz in einem isotropen Medium ein positiver Energiebetrag übertragen werden kann. Daher ist es möglich in isotropen Medien die
3. harmonische Wellenlänge zu erzeugen (Third Harmonic Generation, THG). Die
geradzahligen Potenzen von [4.5], die eine geradzahlige Vervielfachung der
Eingangswelle beschreiben, führen zu keinem Energieübertrag und sind somit zu
ignorieren.
Abbildung 9: Modell zur Erzeugung der 3. harmonischen Welle in einem isotropen Gas
durch nichtlineare Polarisation der Atome. A: Frequenzverdopplung kann nicht stattfinden da
bei einer Welle mit doppelter Frequenz in einem isotropen Medium kein positiver Energiebetrag in die verdoppelte Welle übergeht. Die Energie die kurzzeitig übertragen wird, wird
während der nächsten Halbwelle wieder entzogen (ç+Eç = ç-Eç). B: Bei Frequenzverdreifachung hingegen ist der übertragene Energiebetrag (durch +/-E gekennzeichnet) positiv. Die
3. harmonische Wellenlänge kann also konstruktiv erzeugt werden.
36
Kapitel 4
Wird dies berücksichtigt, wird bei den relevanten Termen von [4.5] coswt durch
½ exp(iwt) ersetzt und die Ableitungen gleich dem Term für die nichtlinieare
Polarisation aus Gleichung [4.11] gesetzt, ergibt sich
r
¶ 2 PNL 1
= e 0 c 3w 2 E 03 [9 exp (i3wt ) + 3 exp (iwt )].
2
8
¶t
[4.12]
Dies in [4.11] eingesetzt ergibt die neue Funktion
r
r
¶2E
¶2E
- ( c 1 + 1)e 0 m 0 2 =
¶z 2
¶t
[4.13]
1
e 0 c 3w 2 E 03 [9 exp(i3w t ) + 3 exp(iwt )]
8
und mit der Definition der Brechzahl n = c + 1 für die eingestrahlte Wellenlänge die
Funktion
n12e 0 m 0w 2 exp(iw t - ikz ) =
[4.14]
1
e 0 c 3w 2 E02 [9 exp(i3w t - i3kz ) + 3 exp(iw t - ikz )] .
8
In diesem vereinfachten Modell erfährt die eingestrahlte Welle keine Dämpfung im Medium und daher liefert die zweifache Ortsableitung aus [4.11] hier keinen Beitrag. Der
erste Term auf der rechten Seite von [4.14] oszilliert mit 3w und beschreibt damit die
Erzeugung der 3. harmonischen Wellenlänge aus der Grundwellenlänge w. Stark vereinfacht betrachtet wird in Abhängigkeit von der vom Medium abhängigen Konstante c3,
die Energie von drei Photonen mit der Frequenz w verwendet, um ein Photon der 3.
harmonischen Wellenlänge mit der Frequenz 3w zu bilden. Die ausführliche Behandlung dieser VUV-Erzeugung ist in den Fachbücher [74-77] und Veröffentlichungen [25,
26, 78] zu finden.
Der Brechungsindex (n) einer elektromagnetischen Welle in einem Medium hängt von
deren Frequenz ab. Nach
c
= nw
vv
[4.15]
ist auch die Ausbreitungsgeschwindigkeit (vw) einer elektromagnetischen Welle von
ihrer Frequenz abhängig. Nach der normalen Dispersion breitet sich die von außen ein-
37
v
v
gestrahlte elektromagnetische Welle ( E w) schneller aus als die erzeugte E 3w-Welle mit
dreifacher Frequenz. Dies hat zur Folge, dass nach einem bestimmten Weg die erzeugte
v
v
E 3w-Welle nicht mehr in Phase ist mit der einige Millimeter zuvor erzeugten E 3wv
Welle. Das Resultat ist, dass sich die E 3w-Wellen auf diese Weise selber schwächen
oder sogar aufheben. Wenn man Laserstrahlung sehr stark fokussiert, muss dieser Effekt
nicht berücksichtigt werden, da die 3. harmonische Wellenlänge quasi in einem Punkt
erzeugt wird. Bei sanfterer Fokussierung, wie sie bei besonders starker Laserleistung
v
üblich ist, kann die E 3w-Welle in der Strahltaille über einige Millimeter erzeugt werden
und daher muss dort dieser Effekt beachtet werden.
Nimmt man vereinfacht an, dass das kurzwelligere Licht der 3. harmonischen Wellenlänge konstant über eine Länge (l) erzeugt wird, so ist die Intensität (I3) der
3. harmonischen Wellenlänge gegeben durch die Funktion
é
l ù
sin2 ê(2p (nw - n3w ) ú
l0 û
ë
I3 (l ) µ
.
2
(nw - n3w )
[4.16]
Sind die Brechungsindizes nw und n3w für die erzeugende und die 3. harmonische Wellenlänge bekannt, kann man mit Gleichung [4.16] und der magnetischen Feldkonstante
l0 die Länge (l) ausrechnen, bei der die Intensität der 3. harmonischen Wellenlänge
maximal ist. Diese Länge (lc) wird Kohärenzlänge genannt.
1 l0
lc =
4 n1 - n3
[4.17]
Da ein fokussierter Laserstrahl, auf Grund von Beugung, im Fokus nicht spitz zusammen und danach wieder spitz auseinander läuft, sondern wie bei einer Taille in einer
Kurve zusammen und auseinander geht, ist es schwer, mit obiger vereinfachter Methode
die optimalen Fokusbedingungen einzustellen. Bei energiereichen Laserstrahlen fokussiert man eher weniger stark, um im Fokus durch die dann zu hohen Feldstärken keine
Gasdurchbrüche (Gas geht durch starkes elektrisches Feld über in den Plasmazustand)
zu erzeugen. Wenn man die 3. harmonische Wellenlänge auf einer Strecke erzeugt, die
länger als die Kohärenzlänge ist, reduziert sich die Effektivität der VUV-Erzeugung.
38
Kapitel 4
Abbildung 10: Schematische Darstellung des Brechungsindex in Abhängigkeit von der Frequenz: Die Sprünge (schraffierte Bereiche) in diesen Dispersionskurven werden von
optischen Übergänge hervorgerufen. Argon zeigt zwischen den Frequenzen w und 3w ein
positives Dispersionsverhalten. Da bei Xenon zwischen w und 3w ein Bereich mit negativer
Dispersion (schraffierter Bereich) liegt, ist hier nw > n3w.. Mischt man Xenon und Argon im
richtigen Verhältnis, kann nw = n3w sein.
Dieses Problem lässt sich durch Anpassung der Ausbreitungsgeschwindigkeiten der
Grundwellenlänge vw und der 3. harmonischen v3w und damit auch die Brechungsindizes
nw und n3w in dem Medium lösen.
Zwischen zwei optischen Übergängen zeigt ein Gas normale Dispersion. Das bedeutet,
dass in diesem Bereich nw < n3w und damit wegen Gleichung [4.15] vw > v3w ist (vgl.
Abbildung 10). Liegt zwischen w und 3w ein optischer Übergang mit negativer Dispersion, so kann nw > n3w sein. Xenon, das man in der Praxis zur Erzeugung von 118 nmPhotonen aus 355 nm-Laserstahlung verwendet, hat eine relativ hohe Suszeptibilität c3
und einen wie in Abbildung 10 (unten) dargestellten Bereich negativer Dispersion zwischen 355 nm und 118 nm. Argon zeigt in diesem Bereich nur einen positiven
Dispersionsverlauf. Für eine Mischung von Xenon und Argon, im Verhältnis 1 zu 10,5
39
[78, 79], ergibt die Dispersionsbeziehung nw = n3w. Das heißt: gleiche Ausbreitungsgeschwindigkeiten (vw = v3w) für die Strahlen, und damit kann die 3. harmonische
Wellenlänge entlang eines beliebig langen Weges mit konstruktiver Überlagerung erzeugt werden.
40
Kapitel 4
4.2 Erzeugung von VUV-Licht durch den strahlenden Zerfall von
Edelgasexcimeren
Neben der zuvor beschriebenen laserindizierten VUV-Erzeugung, sind prinzipiell weitere VUV-Lichtquellen als Photonenquelle für den SPI-Prozess denkbar. Es gibt z.B.
Deuterium-Gasentladungslampen, deren VUV-Spektrum aus einem schwachen breitbandigen Kontinuum, den Werner- und den Lyman-Molekühlbanden etwa zwischen
130 nm und 160 nm und der sehr schmalbandigen Lyman-a-Emission bei 121,567 nm
besteht. Das breite Emissionsspektrum und die geringe Lichtausbeute schränken die
Anwendung der Deuteriumlampe für die analytische Massenspektrometrie ein.
Bei dem Zerfall von Edelgas-Excimeren (gebundene Edelgasatome) entsteht je nach
Edelgas Strahlung im VUV-Bereich. Mit diesem weiter unten ausführlicher beschriebenen Prozess wurden, basierend auf den Arbeiten von Tanaka [80], verschiedene
Excimer-Lichtquellen entwickelt.
Eine schon länger bekannte Excimer-VUV-Lampe ist die Barriere-Entladungslampe
[81]. Bei dieser werden Edelgasatome durch eine Gasentladung hoch angeregt. Dies
führt zu einer effizienten, aber flächigen Excimer-Emission, die schlecht fokussierbar
ist. Daher ist dieser Typ als VUV-Lichtquelle für die SPI-MS nicht geeignet.
Die Entladung in Mikrohohlkathoden [82, 83], bei der unter hohem Druck in einem,
durch die Hohlkathoden begrenzten, Bereich VUV-Licht emittierende Excimere erzeugt
werden, ist wegen der Qualität und der geringen Stabilität des Spektrums ebenfalls nicht
geeignet. Die Qualität der Emission wird hier beeinträchtigt, da in der ExcimerLeuchtzone das Excimer-Gas durch Elektrodenabbrand permanent verunreinigt wird.
Ausgehend von den Arbeiten zur Ionenstrahlanregung von Edelgasen [80, 84-88] wurde
eine mit einem Elektronenstrahl betriebene brillante VUV-Lichtquelle entwickelt (siehe
Abbildung 11) [84, 89]. Solch eine Excimer-Lichtquelle wurde im Rahmen dieser Arbeit erstmalig erfolgreich zum Ionisieren eines Gases in der Ionenquelle eines TOFMS
eingesetzt und wird daher nachfolgend genauer beschrieben.
41
Abbildung 11: Skizze einer elektronenstrahlgepumpten Excimerlichtquelle. Der Elektronenstrahl tritt durch eine spezielle Folie in ein dichtes Gas. Im Gas verlieren die Elektronen ihre
kinetische Energie und erzeugen so Excimermolekühle, bei deren Zerfall die Excimerstahlung frei wird.
4.2.1 Grundlagen der Excimerkinetik
Zum Bau einer effizienten Excimer-Lichtquelle müssen eine Vielzahl von Faktoren
beachtet werden, die aus der Theorie der Excimer-Strahlung abgeleitet werden können.
Deshalb folgt hier eine kurze Einführung in die Excimer-Vorgänge, wie sie bei allen
reinen Edelgasen stattfinden.
Angeregte Edelgasatome ( X * ), können mit nicht angeregten Edelgasatomen (X) eine
Bindung eingehen und ein Edelgasmolekül bilden, das als Excimer bezeichnet wird.
Solche diatomaren Moleküle sind nur in einem angeregten Zustand stabil und zerfallen
aus dem gebundenen Molekülzustand in einen repulsiven Grundzustand (vgl.
Abbildung 12). Bei dem Zerfall der Excimere wird die Anregungsenergie in Form von
Photonen frei.
Das Emissionsspektrum der reinen Edelgas-Excimere liegt im VUV-Spektralbereich
und ist bestimmt durch zwei verschiedene Arten von Strahlungsübergängen. Die erste
Art (1. Kontinuum) ist bestimmt durch den Zerfall von vibratorisch angeregten Zuständen und führt zu schmalen hochenergetischen Kontinua. Dieser Prozess findet
überwiegend in Gasen mit geringer Dichte statt. Bei dichten Gasen kann sich das angeregte Excimer durch Stöße schnell abregen und es finden überwiegend Excimerzerfälle
aus dem vibratorischen Grundzustand statt. Man spricht vom 2. Kontinuum [80]. Die
Kinetik dieses Prozesses soll nun für reine Edelgase beschrieben werden.
Das 2. Kontinuum der Excimer-Emission wird beim Übergang eines Edelgasmoleküls
(X2*) aus den niedrigsten angeregten Zuständen 1 S u+ und 3 S u+ in den repulsiven Grund-
42
Kapitel 4
zustand 1 S +g emittiert (vgl. Abbildung 12). Dabei steht S für den elektronischen Bahndrehimpuls des Molekülzustandes und das Pluszeichen für die Spiegelsymmetrie an der
Ebene, die die Molekülachse beinhaltet. Das Vorzeichenverhalten der Wellenfunktion
bei einer Inversion am Molekülzentrum wird durch u bzw. g beschrieben. Dabei steht u
für einen Vorzeichenwechsel und g für Inversionssymmetrie. Die hochgestellte Zahl vor
dem Bahndrehimpuls gibt die Multiplizität des Zustandes an.
Abbildung 12: Potenzialkurven der elektronischen Zustände des Xenonexcimers in Abhängigkeit vom intermolekularen Abstand nach Brodmann [90]. Eingezeichnet sind das
1. Kontinuum bei 147 nm, das 2. Kontinuum bei 172 nm und die dazugehörigen atomaren
Energieniveaus mit dem ersten Grundzustandsübergang bei 146,96 nm.
Abbildung 12 zeigt links bei kleinen Abständen zwischen zwei Atomen das Energieniveau-Schema des Xenon-Excimers und rechts bei größeren Abständen die
dazugehörigen Atomniveaus. Der strahlende Übergang eines Excimermoleküls vom
1
S u+ - und 3 S u+ -Zustand in den repulsiven 1 S +g -Zustand führt zu breitbandiger Emission
bei 172 nm. Dieser Zerfall aus dem 2. Kontinuum ist bei hohen Drücken (>100mbar)
dominant. Bei niedrigeren Drücken tritt schmalbandige 147 nm -Emission in den Vordergrund, die dem Übergang aus dem höheren Vibrationsniveau des
1,3
S u+ -Zustands in
43
den
repulsiven
1
S +g -Grundzustand
des
Excimers
zugeordnet
werden
kann
(1. Kontinuum).
Wie aus Abbildung 12 ersichtlich ist, liegt das Emissionsmaximum des 2. Kontinuums
bei einer niedrigeren Energie als das der niedrigsten Resonanzlinie des Atoms. Im Fall
von Xe kann die Emission des 2. Kontinuums, bei 172 nm nicht durch die niedrigste
¾® 5 p 5 6 s ( 3 P1 , 3 P2 ) } bei 146,96 nm absorbiert werden.
Xenon-Resonanz { 5 p 6 (1 S 0 ) ¾
Verluste durch Selbstabsorption können somit nicht auftreten.
Bei der Erzeugung von Excimerlicht durch Anregung eines Edelgases mit einem Elektronenstrahl geben die Elektronen ihre kinetische Energie an die Atome ab und erzeugen
so ionisierte Edelgasatome (X+) und angeregte Atome (X* und X**).
ì X + + 2e - ü
ï
ï
X + e- ¾
¾® í X * + e - ý
ï X ** + e - ï
î
þ
[4.18]
Die dabei entstehenden Ionen bilden mittels eines Dreiteilchenstoßes mit zwei Edelgasatomen im Grundzustand Molekülionen ( X 2+ ). Eines der Edelgasatome trägt dabei die
überschüssige Energie fort (siehe Abbildung 13).
X + + 2X ¾
¾® X 2+ + X
[4.19]
Diese Molekülionen ( X 2+ ) zerfallen durch den Einfang von freien Elektronen zu hoch
angeregten Atomen ( X ** ).
X 2+ + e - ¾
¾® X ** + X
[4.20]
Diese hoch angeregten Atome, die aus Edelgasionen hervorgingen und die direkt durch
die eingestrahlten Elektronen erzeugten hochangeregten Atome gelangen durch Stöße
mit Atomen im Grundzustand, wie in Abbildung 13 dargestellt, in den atomaren
X * ( 3 P1 ) - oder den X * ( 3 P2 ) -Zustand.
X ** + 2 X ¾
¾® X 2** + X
[4.21]
44
X 2** + X ¾
¾® X ( 3 P1, 2 ) + 2 X
Kapitel 4
[4.22]
Aus den Atomen in den X * ( 3 P1 ) - bzw. X * ( 3 P2 ) -Zuständen entstehen wiederum
durch Dreierstoß Excimer-Moleküle, durch deren strahlende Abregung in den repulsiven X 2 (1 S) -Zustand, wie oben diskutiert, die Strahlung (hn) des 2. Kontinuums
entsteht. Der ungebundene X 2 (1 S) -Zustand zerfällt in zwei Atome.
X * ( 3 P1, 2 ) + 2 X ¾
¾® X 2* (1,3 S u+ ) + X
[4.23]
X 2* (1,3 S u+ ) ¾
¾® X 2 (1 S ) + hu
X 2 (1 S ) ¾
¾® X + X
Der steile Potenzialverlauf des repulsiven X 2 (1 S) -Zustandes unterhalb des Ausgangspunktes für die Excimer-Emission ist verantwortlich für die Breite der Excimerspektren
von ca. 10 nm.
Abbildung 13: Vereinfachtes Schema der Entstehung des 2. Kontinuums. Gültig für alle reinen Edelgasatome (X) außer Helium.
45
Tabelle 1: Edelgase und Gasgemische mit ihren bei Anregung emittierenden Spezies, die dazugehörigen Wellenlängen/Photonenenergien des Emissionsmaximums, der Bandbreite der
Emission und die Energieumwandlungseffizienz bei Elektronenstoßanregung.
Gas/Gasgemisch
Emittierende
Spezies
Emissionsprozess
He
He2*
Ne
Ne2*
Ar
Ar2*
Kr
Kr2*
Xe
Xe2*
Ne/H2
H*
Ar/Xe
Kr/Xe
Xe*
Ar/O2
O*
Ne/Ar/Kr
Kr*
He/F2
Ne/F2
F2*
Ar/F2
ArF*
strahlende
Excimerabregung
strahlende
Excimerabregung
strahlende
Excimerabregung
strahlende
Excimerabregung
strahlende
Excimerabregung
atomare
Emission
(nach Energietransver)
atomare
Emission
(nach Energietransver)
atomare
Emission
(nach Energietransver)
atomare
Emission
(nach Energietransver)
strahlende
Excimerabregung
strahlende
Excimerabregung
a
Zentrale
Wellenlänge
[nm]
60 nm /80 nm
Effizienza Bandbreite
[%]
[nm]
Photonenenergie
des Emissionsmaximums [eV]
20,7 eV / 15,5 eV
~40 %
20 nm
14,9 eV
~40 %
4,5 nm
126 nm
9,8 eV
~40 %
9 nm
147 nm
8,4 eV
~40 %
11 nm
172 nm
7,2 eV
~40 %
14 nm
121,57 nm
10,20 eV
~10 %
<0,1 nm
147 nm
8,4 eV
~10 %
<0,1 nm
130 nm
9,5 eV
~10 %
<0,1 nm
124 nm
10,0 eV
~10 %
<0,1 nm
157 nm
7,9 eV
~10 %
2 nm
193 nm
6,4 eV
~10 %
2 nm
83 nm
Prozentualer Anteil der kinetischen Elektronenenergie, die in VUV-Strahlung umgewandelt wird
Die Bildung von hoch angeregten Edelgasatomen aus Ionen [4.20] erfolgt relativ langsam und trägt bei hohen Gasdichten wenig zur gesamten Excimer-Strahlung bei. Die
angeregten X * ( 3 P1 ) - bzw. X * ( 3 P2 ) -Atomzustände werden nicht nur direkt durch
Dreistoß über den X 2** -Molekülzustand aus hoch angeregten X ** -Atome, sondern auch
durch strahlende Zerfälle der X ** -Atome besetzt. Dieser strahlende Übergang ist im
Übrigen auch für das Leuchten im sichtbaren Bereich einer Excimer-Lichtquelle ver-
46
Kapitel 4
antwortlich. Durch den Zerfall, der sich aus einem Edelgasatom im Grundzustand und
einem angeregten Atome ( X * ) bildenden Singlett-Excimere X 2* (1 S) und TriplettExcimere X 2* ( 3 S) , entsteht die Excimer-Strahlung. Die Geschwindigkeit, mit der sich
die VUV emittierenden Excimere bilden, hängt wegen des Dreierstoßes stark vom Edelgasdruck ab. Die Zerfallszeiten dieser Zustände sind aus der Literatur bekannt [91]. Der
Zerfall des Tripletts in den Grundzustand ist aufgrund der Quantenzahlen ein verbotener
Zerfall und dauert daher länger. Der Xenon (1 S) -Zustand hat z.B. eine mittlere Zerfallszeit von 5,5 ns und der ( 3 S) -Zustand eine Zerfallszeit von ca. 100 ns [91]. Details
zur Lebensdauer der beteiligten Spezies finden sich in der Literatur [91, 92]. In letzterer
sind auch die Potenzialkurven des Argonexcimers zu finden.
Anhand der Reaktionen durch die die zerfallenden Excimermoleküle entstehen, lässt
sich erkennen, welchen Einfluss die Dichte des Edelgases auf die Strahlung des
2. Kontinuums hat. Bei hohen Drücken ist die Wahrscheinlichkeit für die Dreierstöße
größer und die X 2 (1,3 S u+ ) -Zustände werden schneller besetzt. Dadurch reduziert sich
die Strahlung des 1. Kontinuums und mehr der durch die Elektronen eingebrachten Energie führt zur Excimerstrahlung des 2. Kontinuums [84].
Excimer-Lichtquellen beschränken sind nicht nur auf das breitbandige 2. Kontinuum
von Edelgasexcimeren. So ist z.B. die Emission der 2p-1s-Lyman-a-Linie (121,5 nm)
von atomarem Wasserstoff bei einer Neon-Wasserstoff-Mischung möglich [87]. Dabei
führt die Excimerbildung des Neons letztlich zur Anregung des Wasserstoffmoleküls
und damit zur Emission der 1. Resonanzlinie des Wasserstoffatoms. Das Spektrum einer
Lyman-a-Lampe zeigt eine um Größenordnungen schmalbandigere Emission als die
Edelgas-Eximere (2. Kontinuum). Wie aus Tabelle 1 ersichtlich ist, lassen sich über
einen derartigen Energietransfer und Beimischungen anderer Gase weitere schmalbandige Lichtquellen realisieren. Tabelle 1 zeigt für die bekannten Edelgase und
Gasgemische durch welchen Prozess bei welcher Wellenlänge das Emissionsmaximum
liegt und gibt Auskunft über die Effizienz bei Elektronenstoßanregung und die Bandbreite der Emission. Die Tabelle entspricht dem augenblicklichen Forschungsstand.
Weitere Emissionswellenlängen durch Energietransfer sind denkbar. Die ExcimerEmissionsspektren der einzelnen reinen Edelgase sind in Abbildung 14 zu sehen.
47
Abbildung 14: Excimer-Emissionsspektren der reinen Edelgase [93].
4.2.2 Anregung durch Elektronenstrahl
Für die hier beschriebene intensive und brillante Excimerlichtquelle wird ein Elektronenstrahl zur Anregung benötigt (vgl. Abbildung 11). Dieser Elektronenstrahl soll in
einem möglichst kleinen Volumen des Edelgases möglichst viel Energie in Form von
Atomanregungen und Ionisationen deponieren. Dieser Energieverlust, den die Elektronen in einem Gas erleiden, kann durch die Bethe-Bloch-Formel beschrieben werden.
Stellt man damit den Verlauf der Energiedeposition eines Elektronenstrahls mit einigen
keV kinetischer Energie in einem Gas dar, so ergibt sich ein wie in Abbildung 15 dargestellter Verlauf ähnlich einer Gaußschen Kurve [94]. Zu sehen ist, dass der
Energieverlust schnellerer Elektronen örtlich unschärfer ist als der von langsameren
Elektronen.
48
Kapitel 4
Abbildung 15: Schematische Darstellung des Energieverlustes zweier Elektronenstrahlen mit
unterschiedlicher Ausgangsenergie (E1 < E2) in Abhängigkeit von der Eindringtiefe in ein
Gas.
Durch die größere räumliche Verteilung der Energiedeponierung eines höherenergetischen Strahls im Excimergas (vgl. Abbildung 15) werden aus einem größeren Volumen
Photonen emittiert. Da für die TOFMS-Anwendung der Leuchtpunkt mit Hilfe einer
Linse in das Zentrum der Ionenquelle abgebildet werden muss und dort eine möglichst
hohe Photonendichte benötigt wird, ist ein kleiner intensiver Leuchtpunkt vorteilhaft.
Weiterhin ist diesbezüglich zu beachten, dass der Elektronenstrahl im Vakuum erzeugt
werden muss und durch ein Eintrittsfenster in das Excimergas gelangt. Da dieses Fenster nur eine bestimmte thermische Last aufnehmen kann und langsamere Elektronen
mehr Energie an das Fenster abgeben, bedarf es einer Optimierung, um möglichst viele
relativ langsamere Elektronen in das Excimergas zu bringen, ohne das Eintrittsfenster
thermisch zu zerstören.
Der Abstand des Maximums (Xmax) der Energiedeposition vom Eintrittsort (X0 = 0)
wird als Energiedepositionstiefe bezeichnet und errechnet sich, abgeleitet von der
Bethe-Formel [85], aus
Re =
Vm 6,1 × 1017 1, 75
E .
N AZ
[4.24]
49
Vm ist hier das molare Normvolumen in cm3, Na die Avogadro-Konstante, Z die Ordnungszahl des Targetgases und E die kinetische Elektonenenergie in keV. Für 1 bar
Argon ergibt sich daraus z.B. bei 10 keV Elektronenenergie eine mittlere Energiedepositionstiefe von 0,7 mm [95].
Die Anregungselektronen in unserer Anwendung mit einer kinetischen Energie von ca.
10 keV verlieren unmittelbar nach Eintritt in das Gas ihre Energie hauptsächlich durch
unelastische Stöße und durch die Erzeugung von schnellen Sekundärelektronen mittels
Ionisation. Dies führt zu einer deutlichen Aufweitung des Strahls und somit auch zu
einem größeren Emissionsgebiet. Die Aufweitung hängt mit der Reichweite der Elektronen im Gas zusammen und vergrößert sich mit der Eindringtiefe der Elektronen [94].
Das Volumen (V), in dem die Leistung (P) des Pump-Elektronenstrahls deponiert wird,
kann durch die starke Streuung angenähert als Kugel betrachtet werden, deren Radius
von der Energiedepositionstiefe abhängt. Aus der Formel für die Energiedepositionstiefe
ergibt sich, dass die deponierte Leistung eines Elektronenstrahls pro Volumen des Excimergases proportional zu P/V ~ r3/E4,25 (mit r: Gasdichte; E: Elektronenenergie) ist
[85].
50
Kapitel 4
4.3 Detektion von VUV-Licht
Sowohl bei laserinduzierten als auch bei Excimer-VUV-Licht ist für den Anwender
wichtig zu wissen, ob die zur VUV-Lichterzeugung notwendigen Betriebsparameter
optimal gewählt sind. Die Effizienz, z.B. einer VUV-Edelgaszelle, hängt unter anderem
von der Reinheit des Edelgases oder Gasgemisches, von den Fokusbedingungen und den
Pumpstrahlparametern ab. Die emittierte Lichtintensität einer Excimerlampe hängt
hauptsächlich von den Elektronenstrahlparametern und dem Füllgas ab. Zur Optimierung all dieser Parameter bei Prototypen, wie auch im täglichen Betrieb, ist ein VUVPhotonendetektor erforderlich. Mit ihm können alle Parameter auf maximale Photonenemission optimiert werden.
4.3.1 Halbleiterdetektoren
Zur Detektion von VUV-Licht kann man Halbleiterdetektoren verwenden. Bei diesen
wird im Halbleiter durch ein auftreffendes Photon ein Elektron/Loch-Paar erzeugt. Geschieht dies im Bereich des Überganges p-dotierter und n-dotierter Halbleiter, so werden
diese Ladungsträger getrennt. Über einen externen Stromkreis fließt ein Strom, der proportional zu den erzeugen Elektron/Loch-Paaren ist (Abbildung 16A). Photonen ab
einer Energie von 1,12 eV können in Silizium (Si) ein Elektron/Loch-Paar erzeugen.
Photonen mit höherer Energie können entsprechend mehr Paare erzeugen. An der Eintrittsschicht zum Halbleiter wird je nach Wellenlänge ein bestimmter Anteil der
Photonen reflektiert. Die Anzahl der Ladungen, die in einem Halbleiter pro auftreffendem Photon mit der Energie E erzeugt werden, nennt man Quanteneffizienz. Wie in
Abbildung 16B zu sehen ist, liegt diese bei einem Siliziumdetektor im Bereich
100 nm – 200 nm in der Größenordnung um 1. Solch ein Detektor (Typ AXUV-100,
IRD Inc., Torrance, USA) wurde bei den Messungen mit der Excimerlampe verwendet.
Problematisch bei den Halbleiterphotodetektoren ist, dass Strahlung bis 1100 nm Ladungen erzeugt und somit z.B. auch Sonnenlicht und Licht von durch Elektronenstoß
angeregten Atomen zum Photostrom beitragen. Es gibt zwar für Halbleiterphotodetektoren
filtrierende
Beschichtungen,
die
nur
Strahlung
aus
einem
bestimmten
Wellenlängenfenster zum Halbleiter gelangen lassen. Es ist aber zu bedenken, dass die
51
erzeugte VUV-Strahlung oft sehr viel schwächer als die übrigen vorhandenen Strahlungen ist.
Im Fall der laserinduzierten VUV-Strahlung ist die 355 nm-Pumpstrahlung um bis zu
5 Größenordnungen intensiver. Diese intensive Strahlung kann durch dünne
Beschichtungen nicht vollständig separiert werden. Ein weiterer Nachteil dieser
Detektoren ist, dass ihre aktive Fläche sehr klein ist und somit bei einem Punktstrahler
nur ein kleiner Teil des Raumwinkels erfassbar ist. Außerdem muss bei einem parallelen
VUV-Strahl dieser genau auf den Detektor justiert werden, was bei der schwachen,
nicht sichtbaren VUV-Strahlung schwierig ist.
Abbildung 16A: Schaltbild eines Halbleiterphotodetektors. B: Quanteneffizienzverlauf eines
Silizium-Photodetektors [64].
4.3.2 Ionisationsdetektoren
Besser geeignet zur Detektion von schwachem VUV-Licht sind gasgefüllte Ionisationszellen. Bei dieser Art von Detektoren wird ein spezielles Gas, das sich zwischen zwei
Elektroden befindet, durch die VUV-Strahlung ionisiert. An den Elektroden liegt eine
Spannung an, wodurch die freigesetzten Elektronen zur positiven Elektrode und die
positiven Ionen zur negativen Elektrode hin beschleunigen. Die Ladungen, die auf den
Elektroden auftreffen, können nachgewiesen werden und sind ein Maß für die eingestrahlte Lichtintensität.
Die Wellenlängenselektivität wird bei dem Gasdetektor durch das Ionisationspotenzial
(IP) des Füllgases bestimmt. Durch das gegebene Ionisationspotenzial trägt sämtliche
52
Kapitel 4
Strahlung von Photonen mit einer Energie unterhalb des IP nicht zur Ionenerzeugung
bei. Dies gilt solang, wie bei Energiedichten gemessen wird, bei denen keine Mehrphotonenionisationen auftritt (vgl. REMPI Abschnitt 3.2). Liegt die Photonenenergie über
dem IP und ist aufgrund des Absorptionswirkungsquerschnittes der Gasatome oder Moleküle bekannt, dass die gesamte eingestrahlte VUV-Strahlung auf dem Weg durch die
Detektorzelle absorbiert wird, kann davon ausgegangen werden, dass jedes der eingestrahlten Photonen mit der Effektivität des spezifischen Ionisationsausbeutefaktors (Y)
Elektron/Ion-Paare erzeugt. Der spezifische Ionisationsausbeutefaktor, der sich aus der
Anzahl der erzeugten Ionen geteilt durch die Anzahl der absorbierten Photonen ergibt,
wird bei Photonenenergien über dem IP größer als Null und nähert sich dann dem Wert
1. Der Anstieg rührt daher, dass bei Energien nahe dem IP noch Anregungen zu hochenergetischen Rydbergzuständen möglich sind [70, 96].
Als Füllgas für Detektorzellen ist NO besonders geeignet. NO hat ein IP von 9,26 eV
[64], was einer Photonenwellenlänge von 134 nm entspricht. Es ist damit gut geeignet
sowohl für laserinduziertes VUV-Licht mit 118 nm als auch für die Excimerstrahlung
von Edelgasen und einigen Gasgemischen (vgl. Tabelle 1). Tabelle 2 stellt einige Ionisationsausbeutefaktoren und Absorptionswirkungsquerschnitte von NO [97] bei VUVWellenlängen dar, die mittels Nd:YAG-Laser oder Excimerlampe erzeugt werden können. Für die Werte in Tabelle 2 wurden die von Watanabe [97] gemessenen
Absorptionskoeffizienten verwendet und umgerechnet. In seinen Versuchen stellte Watanabe
fest,
dass
bei
NO-Drücken
über
4 mm
Hg
(ca.
5mbar)
der
Ionisationsausbeutefaktor (Y) wahrscheinlich aufgrund von Elektronenrekombinationen
druckabhängig ist.
Die Absorptionswirkungsquerschnitte von NO sind ab einer Wellenlänge von 58,43 nm
bekannt [97]. Mit der Extinktionsformel [4.3.1] bei der I0 für die Anzahl der eingestrahlten Photonen, s für den Absorptionswirkungsquerschnitt, n für die Moleküldichte und l
für die Eindringtiefe steht, lässt sich z.B. errechnen, nach welcher Eindringtiefe l(99%)
99% der eingestrahlten Photonen (I0) in Abhängigkeit von der Dichte absorbiert sind
(Formel [4.3.3]).
I = I 0 e -snl
[4.3.1]
I = I 0 0,01 = I 0 e -snl ( 99%)
[4.3.2]
53
l (99 %) =
ln 0,01
- sn
[4.3.3]
Die Moleküldichte n ergibt sich aus dem Quotienten aus der Anzahl der Gasmoleküle
pro Mol und dem Molvolumen. Unter Annahme eines idealen Gases ist dies unter
Normalbedingungen
1
mol = 2,69 × 1019 1
n=
cm 3
cm 3 .
2,24 × 10 4
mol
6,02 × 10 23
[4.3.4]
Für geringere Drücke (P) und andere Temperaturen (T) lässt sich mit der Zustandsgleichung von Clapeyron und der molaren Gaskonstante (R) das Molvolumen (V) im
Nenner von Gleichung [4.3.4] errechnen.
V =
RT
P
[4.3.5]
Mit Hilfe dieser Formeln und den Wirkungsquerschnitten von NO aus Tabelle 2 lässt
sich somit die Zellenlänge berechnen, die notwendig ist, damit bei gegebenem Gasdruck
innerhalb der Zelle 99% der Strahlung absorbiert wird und diese Strahlung aufgrund des
Ionisationsausbeutefaktors (Y) die Ladungen (Q) erzeugt.
Q = YI 0
[4.3.6]
Somit lässt sich aus den Gleichungen [4.3.6] und [4.3.2] durch die gemessenen Ladungen Q in einer idealer Weise mindestens l = l(99%) langen Zelle die Anzahl an
eingestrahlten Photonen (I0) bestimmen.
Der Gasdruck in der Detektorzelle wird entsprechend Bedingung [4.3.3] so eingestellt,
dass eine günstige Detektorlänge gewählt werden kann. Weiterhin muss beachtet werden, dass nicht alle Ionen direkt am Eingangsfenster zum Detektor erzeugt werden, da
hier Ladungen verloren gehen können. Für einen geringen Arbeitsdruck in der Zelle
spricht auch noch, dass die Driftzeit der Ladungen zu den Elektroden aufgrund von weniger Stoßpartnern kürzer ist. Wenn bei niedrigen Drücken die Elektroden nicht lang
genug gemacht werden können, um den Großteil der Strahlung zwischen den Platten zu
54
Kapitel 4
absorbieren, kann dies über Gleichung [4.3.1] berücksichtigt werden.
Wird ein einfacher und billiger Detektor benötigt, um z.B. die VUV-Erzeugung zu optimieren, so kann die Detektorzelle, die ohne Vakuumeinrichtung auskommt, verwendet
werden. Um verschiedene Eindringtiefen zu erhalten, kann das Detektorgas mit einem
im benötigten VUV-Bereich transparenten und inerten Gas (z.B. N2 oder He) verdünnt
werden. Abbildung 17 stellt den nach [4.3.1] errechneten Intensitätsverlauf von 118 nmVUV-Strahlung in einer Detektorzelle bei verschiedenen NO-Konzentrationen unter
Atmosphärendruck dar. Anhand der Abbildung 17 lässt sich erkennen, dass eine Konzentration von 2% NO einen günstigen Intensitätsverlauf ergibt, bei dem die Detektorlänge von ca. 4 cm gut ausgenützt ist. Ferner werden nicht alle Ionen, wie es bei 20%
NO der Fall ist, direkt am Eintrittsfenster gebildet.
Tabelle 2: Absorptionskoeffizienten und Ionisationsausbeutefaktoren von NO für VUVWellenlängen von verschiedenen Lichtquellen [97].
Lichtquelle
Zentrale Emissions- Absorptionswellenlänge
wirkungs-querschnitt
(s)
Laser VUV
118nm
Ionisationsausbeutefaktor
(Y)
2,41×10-18 cm2 @
88% @
118,05 nm
118,05 nm
Excimerlampe mit 121,57 nm
2,45×10-18 cm2 @
81% @
Ne/H2
121,57 nm
121,57 nm
Excimerlampe mit 124 nm
1,91×10-18 cm2 @
86% @
N2/Ar/Kr
123,96 nm
122,35 nm
Excimerlampe mit 126 nm
2,14×10-18 cm2 @
86% @
Ar2
125,99 nm
122,35 nm
Excimerlampe mit 130 nm
2,15×10-18 cm2 @
22% @
Ar/O2
130,18 nm
130,74 nm
55
Abbildung 17: Mit Gleichung [4.3.1] errechneter theoretischer Intensitätsverlauf von
118 nm-VUV-Strahlung in einer Detektorzelle mit verschiedenen Konzentrationen von NO
in N2 bei Atmosphärendruck.
Die durch das absorbierte Licht erzeugten Ladungen werden durch die angelegte Spannung (U) zu den Elektroden hin beschleunigt. Der resultierende Strom, der sich wie in
Abbildung 18 dargestellt verhält, wächst zunächst mit der angelegten Spannung bis die
Sättigungsstromstärke erreicht ist. In diesem 1. Bereich werden die Elektronen durch
das elektrische Feld so schwach getrennt, dass sie wieder mit dem Molekülion rekombinieren können. Im Sättigungsbereich gelangen alle erzeugten Ladungen zu den
Elektroden und der resultierende Strom ist gleich den erzeugten Ladungen. Bei weiterer
Steigerung der Spannung nimmt der Strom dann wieder proportional zur Spannung zu.
Dieser proportionale Zustand beginnt, wenn die erzeugten Ladungsträger im elektrischen Feld eine so hohe Geschwindigkeit zwischen den Stößen mit den Gasatomen oder
Molekühlen erreichen, dass sie neutrale Gasmoleküle durch Stöße ionisieren. Auch diese sekundär erzeugten Ionen und Elektronen, deren Anzahl um so größer ist, je größer
die Feldstärke ist, beteiligen sich am Ladungstransport. In diesem Zustand ist der Strom
an den Platten proportional zur Spannung verstärkt. Bei noch größeren Feldern in der
Kammer kommt es dann zur Gasentladung. Dieser Bereich wird Geiger-Müller-Bereich
genannt.
56
Kapitel 4
Abbildung 18: Stromcharakteristik einer Ionisationskammer in Abhängigkeit von der Abzugsspannung bei konstanter Anzahl primär erzeugter Ionen.
Der Proportional-Bereiche beginnt etwa bei einer Feldstärke von 104 V/cm [98] und ist
Druck- und Gasabhängig. Folglich muss bei einer Detektorzelle mit zwei parallelen
Elektroden im Abstand von 1 cm eine Spannung größer 10 kV angelegt werden, um in
den verstärkenden Bereich zu gelangen. Beim Anlegen sehr hoher Spannungen an einen
Detektor mit 1 cm getrennten Elektroden, ist zu beachten, dass ab einer Spannung ab
10 kV es zu einem spontanen Ladungsübersprung (aufgrund der hohen Feldstärke geht
das Gas in den Plasmazustand über und wird leitend) kommen kann. Bei Plattengeometrie und Atmosphärendruck wird demzufolge der Detektor im Rekombinations- oder
Sättigungs-Bereich betrieben. Anders verhält es sich, wenn die Detektorzelle im Bereich
unter 100 Pa betrieben wird. Bei 100 Pa (1 mbar) ist die mittlere freie Weglänge der
Elektronen zwischen den Stößen so groß, dass schon bei einigen 100 V der Detektor im
Poroportional-Bereich arbeitet.
Erwähnt seien an dieser Stelle zylindrisch aufgebaute Detektoren [98]. Bei diesen ist
eine innere Elektrode, aus einem sehr dünnen Draht, umschlossen von einem Metallzylinder mit einem Durchmesser in der Größenordnung von einigen Zentimetern. Bei
dieser Detektorgeometrie werden schon beim Anlegen einiger 100 V in der Nähe der
inneren Elektrode Feldstärken erzeugt, bei denen eine Gasverstärkung bei hohen Drücken statt findet [98].
57
4.3.3 Aufbau einer Zelle zum Nachweis von VUV-Strahlung
Für die Versuche zum Nachweis von VUV-Licht wurde eine Zelle, wie in Abbildung 19
dargestellt, verwendet. Diese Zelle besteht aus einem Kunstoffzylinder, in dem zwei
parallele Edelstahlplatten als Elektroden mit den Abmessungen 40 mm x 40 mm im
Abstand von 1 cm angebracht sind. Das VUV-Licht gelangt durch ein geklebtes MgF2Fenster in die Zelle. Durch das Kleben erspart man sich den Fensterandrückring, der das
E-Feld in der Zelle stört und zu einem Verlust an Ladungsträgern führen könnte. Außerdem können die Elektroden ohne diesen Ring näher am Fenster angebracht werden. Die
Zelle kann durch zwei Gasanschlüsse mit je einem Nadelventil befüllt und gespült werden. Die Flanschplatte, an der das Eintrittsfenster angebracht wurde, ist aus Aluminium
und wird an die jeweilige Lichtquelle angeflanscht. Damit liegt dieser Flansch, wie in
Abbildung 19A und B zu sehen ist, auf dem 0 V-Potenzial. Liegt die Potenzialdifferenz
von 0 V zu +500 V, wie in Abbildung 19A dargestellt, an den beiden Platten an, gelangen einige positive Kationen nicht zur Elektrode. Wenn hingegen zwei zum
Nullpotenzial symmetrische Spannungen angelegt sind (Abbildung 19B), werden positive wie auch negative Ladungsträger vom Entstehungsort vor dem Fenster weg zu den
Elektroden gezogen.
Abbildung 19: Schematische Darstellung der Ionisationszelle mit Poterntialverlauf und Ionenflugbahn bei A mit den Elektrodenpotenzialen von 0 V und +500 V und bei B mit den
Elektrodenpotenzialen +250 V und -250 V
58
Kapitel 4
4.3.4 Anschluss und Ausleselektronik der Detektorzelle
Zum Auslesen des Gasdetektors kann, wie in Abbildung 20A dargestellt, ein Oszilloskop verwendet werden. Mit diesem können auf zwei unterschiedliche Arten die
erzeugten Ladungen nachgewiesen werden.
1. Bei der einfachen Methode wird aus der Elektrodenfläche (A), dem Plattenabstand (s) und der elektrischen Feldkonstante (e) die Kapazität (C) der
Detektorzelle berechnet.
C=
eA
s
[4.3.7]
Wird der Widerstand R (vgl. Abbildung 20A) sehr groß gewählt, so ergibt sich
mit der durch das Oszilloskop gemessenen Spannungsänderung DV mit Formel
[4.3.8] die Anzahl (N) der Elementarladungen (q), die auf die Elektrode auftreffen.
Nq =
DV
C
[4.3.8]
Diese Anzahl (N) dividiert durch den Ionisationsausbeutefaktor (Y) ergibt bei
vollständiger Lichtabsorption und Sättigungsspannung ein Maß für den Photonstrom. Da der Widerstand allerdings sofort mit dem Nachladen der Kapazität (C)
beginnt, ist diese Methode nur für sehr kurze Ionenpulse geeignet.
Bei einer mittleren freien Weglänge (l) der Ladungen in der Detektorzelle von
ca. 500 nm, wie sie in etwa bei 1 bar auftritt, und einem E-Feld von 500 V/cm
ergibt sich mit [4.3.9] und der jeweiligen Teilchenmasse (m) eine mittlere Geschwindigkeit (v) der Elektronen von 4,7·104 m/s und eine mittlere
Geschwindigkeit (v) der NO+ Moleküle von 2,0·102 m/s.
v=
qE
2
l
m
2
[4.3.9]
59
Abbildung 20A: Schaltbild Photonendetektor mit Oszilloskop. B: Elektronensignal aus Detektorzelle mit 100 V Abzugsspannung und einem NO zu N2 Mischungsverhältnis von 9 zu 1
bei 1 bar Zellendruck.
Für die 5 mm zu den Elektroden benötigen die Elektronen somit im Mittel
100 ns und die Kationen 25 µs. Bei den Elektronen ist daher auch die zeitliche
Verteilung des Auftreffens geringer. Bei dieser ersten Methode sollten daher
die Elektronen gemessen werden. Dazu musste wie in Abbildung 19A dargestellt, eine positive Hochspannung (HV) angelegt werden. Wenn hingegen bei
negativer Hochspannung gemessen wird, ist nicht nur die schlechtere zeitlich
Verteilung der Ionen, sondern auch noch einen Verlust an Ionen zu berücksichtigen (vgl. Abbildung 19A).
2. Bei der zweiten Methode wird wieder über einen Entkoppelkondensator die
Spannung am Widerstand (R) gemessen. Wird bei bekanntem Widerstand (R)
über den Spannungspeak (vgl. Abbildung 20B) integriert, so ergibt sich mit
[4.3.10] die Anzahl der Ladungen (Nq), die durch den Widerstand (R) nachgeladen werden und auch ein Maß für die Photonen sind.
V (t ) dt
ò
Nq =
[4.3.10]
R
Der Vorteil dieser zweiten Methode besteht darin, dass die Kapazität (C) und
somit die druck- und gasabhängige elektrische Feldkonstante (e) nicht bekannt
60
Kapitel 4
sein müssen. Ein länger andauernder Ladungsstrom ist hier unproblematisch. Allerdings muss bei dieser Methode ein elektronischer Integrator nachgeschaltet
werden, damit ein Spannungswert erzeugt wird, der mit einer spezifischen Eichkonstante in direkte Relation mit der Photonenzahl gesetzt werden kann.
Zum Messen von kontinuierlichen Lichtquellen muss der Koppelkondensator,
der auch zum Schutz des Messgerätes dient, entfernt werden. Der Photonenfluss
(IPh) ist dann gegeben durch
I Ph =
U
YeR
[4.3.11]
4.3.5 Experimentelle Messungen von laserinduziertem VUV-Licht
Die nachfolgend beschriebenen Experimente sind beschränkt auf Versuche bei Umgebungsdruck in der Zelle, da das Ziel die Entwicklung einer billigen und einfachen
Detektorzelle ist, die ohne Vakuumvorrichtung auskommt. Für die Experimente stand
ein 20 %tiges NO-Gasgemisch (vorgemischte Druckflasche) zur Verfügung. Mittels
zweier Flußregler konnte das NO-Gemisch mit reinem Stickstoff weiter verdünnt werden. Mit dem Gasgemisch aus NO und N2 wurde die Zelle bei sehr kleinem Durchfluss
während der Messung ständig gespült.
Abbildung 21: Relatives Ionensignal bei verschiedenen NO Konzentrationen in 1 bar Stickstoff.
61
Abbildung 21 zeigt den relativen Verlauf der Signalhöhen (vgl. Abbildung 20B) bei
einer Ausleseelektronik wie in Abbildung 20A dargestellt und -100 V Abzugsspannung.
Als einfache Näherung an die Fläche des Ionensignalpeaks und damit der Ionenzahl
wurde hier die Peakhöhe verwendet. Ein Maximum des Ionensignals ergab sich bei einer NO-Konzentration von etwas unter 10 %. Dieses Maximum befindet sich im selben
Bereich, wie durch Gleichung [4.3.2] für eine Zelle mit 4 cm langen Elektroden vorhergesagt. Theoretisch wurde das Maximum bei etwa 2 % NO erwartet.
Messungen mit einer Zelle mit einem nachgeschalteten Elektrodenpaar, wie in
Abbildung 22 dargestellt, ergaben bei 20 % NO schon gegenüber den vorderen Elektroden am zweiten Elektrodenpaar ein Ionensignal in Höhe von 3 %. Bei 6,2 % NO lag das
Ionensignal schon bei 9 %. Damit liefert die obige Theorie für die Lichtabsorption in
einem dichten Gasgemisch einen guten Anhaltspunkt und Ausgangswert für eine experimentelle Optimierung.
Abbildung 22: Skizze eines Ionendetektors mit zwei Elektroden-Paaren für Messungen zur
Eindringtiefe von VUV-Strahlung in ein Gas.
Zielvorgabe für ein späteres Messinstrument ist ein Spannungswert, der über einen bekannten Zusammenhang mit den eingestrahlten VUV-Photonen korreliert. Bei der
Auslesemethode, bei der die Fläche unter dem Spannungspeak ein Maß für die erzeugten Ionen ist, wird eine Integratorschaltung verwendet, die in der Lage ist, das Signal
über die Dauer des Pulses zu integrieren. Diese Schaltung gibt dann eine Spannung aus,
die proportional zur Peakfläche ist. Mit einer weiteren einfachen Schaltung, die auf einem Display jeweils den maximalen Spannungswert des Integrators anzeigt, ergibt sich
62
Kapitel 4
eine mit der VUV-Photonenzahl korrelierte Spannung. Abbildung 23A zeigt solch eine
Integratorschaltung mit einem Operationsverstärker (LF 355) als zentralem Bauteil und
einem Oszilloskop als Auslesegerät. Zum Test dieser Schaltung wurde ein Rechtecksignal
aus
einem
Funktionsgenerator
am
Eingang
des
Integrators
angelegt
(Abbildung 23B). Der Integratorausgang zeigt einen linearen Anstieg bis zum Ende des
Signals. Damit ist die am Integratorausgang anliegende Maximalspannung ein Maß für
Abbildung 23A: Schaltbild der Detektorzelle mit Integrator. B: Oben ist das Rechtecksignal
eines Funktionsgenerators dargestellt und darunter der daraus resultierende Spannungsverlauf des Integrators. C: Oben ist das Ionensignal dargestellt, welches der Integrator erhält und
darunter der Integratorausgang. Zu beachten ist dabei, dass das RC-Glied der Integratorsschaltung dem Zeitverhalten des Signals angepasst sein muss. Für das Beispiel in C ist das
RC-Glied nicht optimal gewählt. Das Integrationssignal fällt zu früh ab (vgl. gestrichelte Line), was zu einem Integrationsfehler führt.
63
die Rechteckfläche. Abbildung 23C zeigt unten das resultierende Integratorsignal eines
typischen Ionensignals. Der Ionenzahl wird hier beispielsweise der Integratorspannungswert –0,24 V zugeordnet. Der benötigte Integrator-Kallibrationsfaktor für eine
absolute
Photonenmessung
kann
bei
festen
Integratoreinstellungen,
wie
in
Abbildung 23B dargestellt, leicht errechnet werden.
Ein weiterer Parameter einer Detektorzelle ist die Abzugsspannung. Das Ionensignal in
Abhängigkeit von der Abzugsspannung sollte ein Verhalten, wie in Abbildung 18 dargestellt, zeigen. Wie in Absatz 4.3.2 erwähnt, ist der Proportional-Bereich bei
Atmosphärendruck mit der Plattengeometrie nicht zu erreichen. Abbildung 24 zeigt den
Verlauf des integrierten Ionensignals bei einer Gasfüllung mit 10 % NO in Abhängigkeit
von der Abzugsspannung. Wird Abbildung 18 mit Abbildung 24, verglichen, so beginnt
unter den gegebenen Umständen der Sättigungs-Bereich am Ende der Abzugsspannungsskala von Abbildung 24. Höheren Spannungen wurden aus technischen Gründen
nicht verwendet.
Abbildung 24: Verschieden Abzugsspannungen und die daraus resultierenden Ionensignale.
64
Kapitel 4
Geht man anhand von Abbildung 24 davon aus, dass bei etwa 1000 V der Sättigungsbereich beginnt (alle erzeugten Ladungen erreichen die Elektroden), so lässt sich mit
Abbildung 24 abschätzen, dass bei 500 V Abzugsspannung etwa 80 % der Ladungsträger zu den Elektroden gelangen.
In Abbildung 25 ist eine Messung von verschiedenen Strahlungsintensitäten bei 118 nm
dargestellt. Für diese Messung wurde die Intensität der 355 nm-Pumpstrahlung, aus der
in einer Xenon-Edelgaszelle ein kleiner Anteil in 118 nm-Strahlung umgewandelt wird,
mittels mehrerer Quarzglasplättchen reduziert. Die Energie der 5 ns langen 355 nmPulse wurde mit einem herkömmlichen Energiemessegerät bestimmt. Die erzeugte
VUV-Strahlung wurde durch eine MgF2-Linse und ein MgF2-Fenster in die Detektorzelle mit 6,4 % NO bei 500 V Abzugsspannung nachgewiesen. Anhand der angepassten
Funktion, dargestellt durch die durchgezogene Linie in Abbildung 25, zeigt sich die
kubische Abhängigkeit zwischen Pumpstrahlung und erzeugtem VUV-Licht bei der
Konversion in einer Edelgaszelle. Die Konstanten A, B, und C dieser Funktion
Y=(A + (B + X)^3) C, bei der X für die Intensität bei 355 nm und Y für das Ionensignal
steht, sind begründet durch die Energieschwelle (B), ab der in einer Edelgaszelle VUVLicht erzeugt wird sowie durch Absorption oder Reflexion z.B. an den Fenstern, den
Vorgängen in der Zelle und der Elektronik (A und c). Anhand dieser Funktion lässt sich
abschätzen, wie sich die VUV-Erzeugung in einem kleinen Bereich oberhalb von 6,3 mJ
355 nm-Strahlung verhält. Zu der verwendeten Methode zur Energiereduzierung der
355 nm-Strahlung mit Glasplättchen, die in den Strahlengang gelegt werden, sei noch
vermerkt, dass neben der Energie des Lichtpulses auch noch das Strahlprofil etwas verändert wird, indem die energiereicheren Bereiche des Strahls an den Glasplättchen
prozentual stärker geschwächt werden. Da die Strahlbedingungen einen Einfluss auf die
Umwandlungseffizienz haben, wird hier auch in einem kleinem Maß die Umwandlungseffizienz durch das veränderte Stralprofil beeinflusst und nicht nur durch die
Energie.
In der Literatur sind für die Verdreifachung von 355 nm-Strahlungenergie zu 118 nm
Stahlungsenergie in einer Edelgaszelle Effizienzen von 10-5 zu finden [27, 99]. Bei
6,3 mJ 355 nm-Strahlung sollte daher in etwa 6,3·10-8 Joule 118 nm-Strahlung entstehen und somit ca. 3,7·1010 Photonen pro Laserschuss. Die Integratorspannung aus
Abbildung 25 von 680 mV bei 6,3 mJ 355 nm-Strahlenergie steht für eine Signalfläche
von 6,7·10-6 Vs und somit bei einem Widerstand (R) von 470 kW für 9·107 an der Anode gesammelter Elektronen (vgl. [4.3.10]). Berücksichtigt man dann das unvollständige
65
Einsammeln der Elektronen durch die geringe Abzugsspannung von +500 V mit dem
Faktor 0,8, die Ionisationsausbeute mit dem Faktor 0,88 und die Verluste an der Linse
und dem Fenster mit je Faktor 0,4, so ergibt sich aus dem Ionensignal eine VUVAusbeute von 7,9·108 Photonen. Damit beträgt das Detektorsignal nur etwa 1/50 vom
theoretisch erwarteten Wert. Aufgrund dieser Diskrepanz zwischen erwarteten und
nachgewiesenen Photonen, die in der Druckabhängigkeit des Ionisationsausbeutefaktors
Y=Y(P) begründet ist, ist für eine Absolutmessung von VUV-Photonen eine Kalibrierung einer solchen Zelle notwendig. Dies könnte an einem Synchrotron oder mit einer
geeichten Excimer-Lampe vorgenommen werden.
Bei den Experimenten hat sich gezeigt, dass die Ergebnisse gut reproduzierbar sind. Die
Detektorzelle eignet sich somit sehr gut für die tägliche Justierung der Laser- oder Excimer-Lampen-Parameter und zum relativen Photonenflussvergleich.
Für die einfache Optimierung von Lichtquellen bietet sich an, ein kleines offenes
Fläschchen mit z.B. Toluol (IP = 8,8 eV [64]) in die Zelle zu stellen und die Zelle dann
gleichmäßig bei kleinem Fluss (einige ml/min) mit Stickstoff zu spülen. Nachdem sich
Abbildung 25: 118 nm Photoionensignal bei verschiedenen 355 nm Pumpenergien und eine
dazugehörige Kurvenanpassung um das Photoionisationssignal außerhalb der Messpunkte
vorherzusagen.
66
Kapitel 4
dann auf diese Weise eine N2-Toluol-Gleichverteilung in der Zelle eingestellt hat, kann
VUV-Strahlung unterhalb von 140 nm Wellenlänge reproduzierbar nachgewiesen werden. Mit Anilin (IP = 7,72 eV [64]), welches bei Raumtemperatur ausreichend flüchtig
ist, sind dann sogar Photonen mit Wellenlängen kleiner als 160 nm nachzuweisen und
damit auch ein Großteil der bis jetzt bekannten Excimer-Wellenlängen. In der NIST
Datenbank [64] findet man auch noch eine Vielzahl von Molekülen mit einem IP von
weniger als 7,0 eV, die somit geeignet sind für Licht mit einer Wellenlänge über
177 nm.
Mit der hier beschriebenen Ionisationszelle, die mit bis zu einem bar mit dem Ionisationsgas
gefüllt ist, lässt sich keine Aussage über die zeitliche Verteilung der eintreffenden Photoenen
machen. Eine Messung der Strahlintensität ist daher nicht möglich.
67
5 Mobiles Flugzeitmassenspektrometer mit drei verschiedenen Ionisationsmechanismen
Kernpunkt der vorliegenden Doktorarbeit ist die Entwicklung eines mobilen Flugzeitmassenspektrometers zur umfassenden Online-Analyse von Gasen. Das Gerät soll in der
Lage sein, eine Vielzahl von Verbindungen unterschiedlicher Spezies online und quantitativ nachzuweisen. Das Massenspektrometer soll sich für Feldmessungen in
industrieller Umgebung eignen. Mit dem Gerät sollte unter widrigen Bedingungen, wie
sie an Müllverbrennungsanlagen vorherrschen, über einen längeren Zeitraum gemessen
werden können. Zu den widrigen Bedingungen gehören Vibrationen, feinster Staub,
hohe Umgebungstemperatur und geringer Platz an den Messstellen.
Schon bei der Konzeptionierung des Laser-Massenspektrometers mussten einige grundlegende Dinge berücksichtigt werden.
1. Um aromatische, aliphatische und anorganische Verbindungen gleichzeitig und
online zu messen, müssen die Ionisationsmethoden REMPI, SPI und EI so in das
neue Gerät integriert werden, dass eine Gasprobe sehr schnell durch die verschiedenen Ionisationsmethoden mit ihren unterschiedlichen Selektivitäten
analysiert werden kann. Dazu wird eine spezielle Ionenquelle benötigt.
2. Damit aromatische Verbindungen selektiv nachgewiesen werden können, muss
für den REMPI-Prozess durchstimmbares UV-Licht zwischen 200 und 350 nm
zur Verfügung stehen.
3. Für eine hohe Empfindlichkeit des Gerätes musste eine intensive VUVLichtquelle entwickelt und eingebaut werden.
4. Für die Quantifizierung muss ein geeignetes Standardgas zu Verfügung stehen.
5. Die Probenahme muss dafür ausgelegt sein, heiße und reaktive Gase zur Ionenquelle des TOFMS zu leiten.
6. Die äußeren Abmessungen müssen es erlauben, das vollständige Gerät durch eine Standardtür zu fahren.
7. Die einzelnen Komponenten sollten so angebracht sein, dass diese auch bei erhöhten Umgebungstemperaturen nicht ausfallen. Außerdem darf staubige
68
Kapitel 5
Umgebungsluft nicht zu sensiblen Komponenten gelangen, vor allem nicht zu
den optischen Komponenten.
8. Das Gerät muss über einen längeren Zeitraum zuverlässig die Verbindungen
nachweisen können, zu deren Nachweis es gebaut wurde.
Unter Beachtung dieser Punkte wurde das in Abbildung 26 abgebildete Gerät entwickelt
und gebaut. Es wird nachfolgend detailliert beschrieben und spezifiziert. Messungen
von unterschiedlichen Proben am Ende dieses Kapitels zeigen, welche Ergebnisse mit
dem Gerät gewonnen werden können und geben ein Bild von den vielfältigen Einsatzmöglichkeiten des Instrumentes.
Abbildung 26: Mobiles TOFMS mit Beschriftung der Hauptkomponenten.
69
5.1 Aufbau des Massenspektrometers
Abbildung 26 zeigt das fertige Massenspektrometer kurz vor einem 5-wöchigen Einsatz
an einer Müllverbrennungsanlage. Anhand von Abbildung 26 werden kurz die Hauptkomponenten des Gerätes vorgestellt, aus denen es aufgebaut ist. Neu entwickelte
Komponenten werden dann im Anschluss detailliert beschrieben.
Ganz oben in der Abbildung sind der 2 m lange Heizschlauch der Probenahme-Einheit
(vgl. Kapitel 7) und eine metallener Kasten zu erkennen. In dem Kasten befindet sich
der Nd:YAG-Laser und das OPO-System (optischer parametrischer Oszillator) der Firma GWU Lasertechnik (Erftstadt, Deutschland). Diese beiden Laserkomponenten
werden anschließend genauer beschrieben.
Hinter der schwarzen Verkleidung befindet sich die Ionenquelle und das Flugrohr des
TOFMS, die THG-Zelle mit Xenon-Druckflasche, ein Wellenlängen-Kalibriersystem
(Typ LRL 005, Fa. Athos, Pfungstadt, Deutschland), ein steuerbarer Signalvorverstärker
und die optischen Systeme zum Einkoppeln der 355 nm-Strahlung in die THG-Zelle und
der UV-Strahlung für den REMPI-Prozess in die Ionenquelle. Darunter ist auf der linken
Seite hinter einer Plexiglasscheibe ein 19“-Einschub mit der Vakuumkontrolleinheit und
den Temperaturreglern für den Probeneinlass. Der schmale Einschub darunter versorgt
die Elektronenkanone mit den nötigen Spannungen. Es folgt ein leerer Einschub (Reserve) und dann zwei 19“-Computer. Der erste steuert den OPO-Laser und die
Wellenlängenkalibriereinheit. Der zweite Rechner übernimmt die gesamte Datenaufnahme.
Im mittleren Teil des Gerätes befindet sich oben das Hochspannungsnetzteil für die
Spannungen der Ionenquelle und des Flugrohrs, darunter der Einschub mit Druckmesser
und Getterpumpennetzteil, ein Netzteil für den Signalvorverstärker und ein Notschalter.
Weiter unten befindet sich ein 4-Kanal-Pulsgenerator, der den zeitlichen Ablauf bei
Messungen mit alternierenden Ionisationsmethoden regelt. Darunter ist noch ein Steuergerät für die Laserstrahlsteuerung eingebaut und der Freiraum ganz unten ist für den
Gasstandard.
Unter dem Gerät ist die Vakuumvorpumpe federnd aufgehängt. Sie ist an dieser Stelle
angebracht, damit ihre Abwärme nicht in das Innere des Gerätes gelangt. Außerdem
befindet sich dort ein Pressluft-Entfeuchter, ein Ölabscheider und ein Feinpartikelfilter.
70
Kapitel 5
Nicht sichtbar ist die Bedieneinheit für die Computer, bestehend aus einem Flachbildschirm und einer 19“-Tastatur, mit welcher beide Rechner des Gerätes bedient werden.
Diese kann an jeder Seite des Gerätes angebracht werden. Dies ist nötig, um auch bei
rückwärtigen Einstellarbeiten freien Blick auf den Bildschirm zu haben. Bei Feldmessungen herrscht oftmals Platzmangel, so dass die Kontrolle des Gerätes nicht immer von
der Frontseite des Gerätes aus möglich ist.
71
5.2 Laser
Sowohl für die Erzeugung von 118 nm VUV-Strahlung als auch für die Erzeugung von
durchstimmbarer UV-Strahlung mit einem OPO-System wird 355 nm-Strahlung benötigt. Diese Ausgangsstrahlung wird bereitgestellt von einem Surelite-III-Laser der Firma
Continuum (Santa Clara, USA). Der Nd:YAG-Laser liefert mit 10 Hz 3 – 5 ns lange
355 nm-Lichtpulse mit 225 mJ.
Die 355 nm-Strahlung wird durch einen Strahlteiler (vgl. Abbildung 32) in zwei Strahlen mit 11 % und 89 % der Ausgangsenergie aufgeteilt. 11 % werden zur VUVErzeugung verwendet, die restlichen 89 % pumpen den OPO-Laser.
5.2.1 OPO-Laser
OPO ist die Abkürzung für optischer parametrischer Oszillator. Der dieser Methode zur
Erzeugung von Laserlicht zugrundeliegende Prozess wird im folgenden stark vereinfacht dargestellt.
Mit Hilfe von speziellen Kristallen ist es möglich, durchstimmbares Laserlicht in einem
großen Wellenlängenbereich zu erzeugen [100]. Diese speziellen Kristalle sind doppelbrechend. Dies bedeutet, dass aufgrund der Kristallanisotropie die Brechungsindizes
von Licht mit unterschiedlichen Polarisationsrichtungen, verschieden sind. Strahlt man
in diese Kristalle intensives Laserlicht ein, so kommt es zu nichtlinearen Effekten. Für
ein genaueres Studium sei auf die Veröffentlichung von Harris [101] verwiesen. Durch
diese nichtlinearen Effekte im Kristall wird ein Teil des eingestrahlten Laserlichts in
Licht mit zwei anderen Wellenlängen umgewandelt. Bei dieser Umwandlung, bei der
die Energieerhaltung gilt, entstehen aus einem eingestrahlten Photon mit der Frequenz
wp je ein Photon mit wS und ein Photon mit wI.
wP = ws +wI
[5.1]
Diese resultierende Strahlung nennt man Signal- und Idlerstrahlung. Die beiden Strahlungen sind linear polarisiert. Die Polarisationsebenen dieser Strahlungen stehen
senkrecht zueinander.
72
Kapitel 5
Neben der Energieerhaltung gilt für die Photonen auch der Impulserhaltungssatz
r
r r
kP = ks + kI ,
[5.2]
der sich bei kollinearen Strahlen mit den jeweiligen Brechungsindizes nS, nI und nP
schreiben lässt als
w P n p = w s nS + w I nI .
[5.3]
Abbildung 27: Abhängigkeit der Laserenergie von der Wellenlänge des GWU-Lasersystems.
Oben ist die Energie der verdoppelten Signal-OPO-Strahlung dargestellt, unten der Verlauf
am Ausgang der SFM-Einheit. Da in der Mitte der Verläufe der Kristall gewechselt werden
muss, ergibt sich ein oben erkennbarer Sprung.
73
Die Bedingungen [4.15] und [5.3] können in einem OPO-Kristall bei bestimmter Kristallgitterausrichtung nur von einer Signal- bzw. Idlerwellenlänge erfüllt werden. Dreht
man den Kristall gegenüber dem einfallendem Laserstrahl um eine bestimmte Achse, so
verändern sich aufgrund der Anisotropie der Kristallstruktur die Brechungsindizes und
wegen der Bedingungen [5.3] und [4.15] damit auch die Frequenzen von Signal- und
Idlerstrahlung.
Mit einem Beta-Bariumborat- (β-BaB2O4, BBO) Kristall können bei einer Pumpwellenlänge von 355 nm Wellenlängen von 412 nm bis 2250 nm erzeugt werden [102].
Wenn nach dem OPO-Kristall ein Verdopplerkristall angebracht ist, kann prinzipiell
z.B. aus der Signalstrahlung (412 nm – 710 nm) durch Verdopplung UV-Licht von
206 nm bis 355 nm erzeugt werden. Unterhalb 220 nm nimmt die Effizienz das verwendeten OPO-Kristalls rapide ab, so dass mit diesem Prozess im blauen Spektralbereich
nur bis 220 nm gemessen werden konnte. Die Linienbreite im UV lag bei 0,07 nm. Das
OPO-System der Firma GWU erzeugt aus 190 mJ 355 nm-Strahlung 2 – 4 mJ UV-Licht
(Abbildung 27).
Zur Erweiterung des Wellenlängenbereichs in den blauen Bereich besitzt das GWULasersystem eine SFM-Einheit (Summen-Frequenz-Mischung). In dieser Einheit wird
die 266 nm-Strahlung, die durch Verdoppeln der 532 nm-Strahlung erhalten wird, welche die verdoppelte 1064 nm-Nd:YAG-Strahlung (Nd:YAG-Grundwellenlänge) ist, mit
der Idlerstrahlung des OPOs gemischt. Im Photonenbild wird aus einem 266 nm-Photon
und einem Idler-Photon ein SFM-Photon.
w SFM = w 266 + w I
[5.4]
bzw.
1
lSFM
=
1
1
+
l266 lI
[5.5]
Mit Hilfe des SFM-Prozesses kann der Laser UV-Licht von 230 – 198,5 nm erzeugen
(Abbildung 27). Bei Annäherung an 198,5 nm fällt die Laserenergie allerdings auf
0,3 mJ ab. Beim SFM-Prozess ist die Polarisation der erzeugenden Strahlungen zu beachten. Durch den OPO-Prozess ist die Polarisationsebene der Idlerstrahlung gedreht
74
Kapitel 5
und muss für den SFM-Prozess mit einem Polarisator der 266 nm-Strahlung angepasst
werden.
Als problematisch erwiesen sich bei den Wellenlängen unter 220 nm die Optiken. Die
Absorptionsverluste bei Optiken mit schlechter UV-Qualität stiegen sehr schnell an im
Wellenlängenbereich unter 220 nm. Was die OPO-Strahlung betrifft, sollte nachfolgend
mit so wenig optischen Bauteilen wie möglich gearbeitet werden, da es für den gesamten
Wellenlängenbereich
(189 nm - IR)
eines
OPO-Lasers
keine
geeigneten
Antireflexbeschichtungen gibt und somit mit Verlusten von 10 % pro durchdrungener
Oberfläche gerechnet werden muss. Nicht ganz optimal ist das längliche Strahlprofil des
GWU-Lasersystems. Zur Ionisation im Massenspektrometer kann dieses Strahlprofil mit
Hilfe eines Zylinderlinsenteleskop verbessern werden. Es ist aber auch möglich dieses
Strahlprofil auszunützen, indem auf eine Weise in die Ionenquelle eingestrahlt wird,
dass das Probengas entlang seiner Einlassrichtung bestrahlt wird.
75
5.3 Zelle zur Erzeugung der VUV-Stahlung
Durch die vorgegebenen Außenabmessungen des mobilen TOFMS-Systems, in das die
Zelle zur VUV-Erzeugung aus Laserstrahlung integriert werden musste, war die maximale Größe der Zelle vorgegeben. Damit das fertige Massenspektrometer durch eine
Standardtür von 80 cm Breite passt, stand für die Zelle mit Verbindungsflansch und
Optik eine Länge von etwas weniger als 55 cm zur Verfügung (vgl. Abbildung 30).
Abbildung 28: Bildschirmanzeige des unter LabViewTM erstellten Programms zur Berechnung des Strahlengangs durch eine THG-Zelle mit anschließender Fokussierung. Von links
kommt ein paralleler 355 nm-Strahl, der bei x = 20 mm von einer nicht dargestellten Linse
mit f = 100 mm fokussiert wird. Nach dem Fokus gelangt der Strahl bei x = 200 mm zur
MgF2-Linse. Durch die Verschiebung der Linse um 4 mm und die zwei unterschiedlichen
Brechungsindizes wird der 118 nm-Strahl in den Punkt x = 340 mm, y = -11 mm fokussiert.
Die 355 nm-Strahlung läuft divergent am Fokalpunkt vorbei. Durch die verschiedenen frei
wählbaren Parameter konnte der Strahlengang der THG-Zelle veranschaulicht und optimiert
werden.
76
Kapitel 5
Außerdem musste es möglich sein, die THG-Zelle so an der Ionenquelle anzubringen,
dass auch ein UV-Strahl für die REMPI-Ionisation durch das Zentrum der Ionenquelle
geführt werden kann. In Abbildung 30 ist dieser Strahl als OPO-Strahl gekennzeichnet.
Um den Strahlverlauf durch die THG-Zelle mit ihren beiden Linsen darstellen und vorab simulieren zu können, wurde ein LabVIEWTM-Programm geschrieben. Das
Programm arbeitet mit der Abhängigkeit sin a / sin b = n2 / n1, wobei a der Einfallswinkel ins Medium mit n1 zum Lot der optischen Fläche und b der Ausfallswinkel zum
Lot auf der anderen Seite im Medium mit n2 ist. Mit dieser Formel wird der Strahlengang durch jede der optischen Flächen errechnet. Mit der Steigung, der Position des
Durchtritts durch die Grenzfläche und dem Abstand der Grenzflächen, kann das Programm nach mehreren Durchläufen einer Routine den Strahlenverlauf darstellen.
Das LabVIEW-Programm ist außerdem in der Lage, den Entstehungsort und die Richtung der erzeugten 118 nm-Strahlung zu simulieren und durch Beachtung der
verschiedenen Brechzahlen bei verschiedenen Wellenlängen den Strahlverlauf durch die
Ionenquelle darzustellen. Sämtliche Abstände, Krümmungsradien, Brechzahlen und
Positionen können mit dem Programm kontinuierlich getestet werden. Mit dieser Software, deren Oberfläche in Abbildung 28 wiedergegeben ist, konnte simuliert werden,
wie die Optiken gewählt werden müssen, um unter den gegebenen geometrischen Randbedingungen den VUV-Strahl zu erzeugen, ihn in die Ionenquelle zu leiten und ihn von
der 355 nm-Strahlung zu trennen.
Getestet wurde das Programm mittels einiger ausgewählter Optiken. Die errechneten
Strahlwege wurden mit denen des Programms Beams 2.4 (Simulation Gauß'sche Optik)
[103] erfolgreich verglichen.
Die VUV-Ionisation ist eine sehr weiche Ionisationsmethode, bei der es zu relativ geringer Fragmentierung der Ionen kommt. Problematisch bei dieser Art der VUVErzeugung ist, dass von der eingestrahlten 355 nm-Primärstrahlung nur ca. 0.001 % [27]
in VUV-Strahlung umgewandelt wird. Dies bedeutet, dass ein Strahl in die Ionenquelle
geschickt wird, der zu 99.999 % aus der 355 nm-Primärstrahlung und nur zu 0.001 %
aus der VUV-Strahlung besteht. Einerseits ist der Wirkungsquerschnitt für die VUVIonisation sehr viel größer als bei der Mehrphotonenionisation, andererseits ist die Intensität der 355 nm-Primärstrahlung, die zu einer Mehrphotonenionisation führen kann,
um Größenordnungen größer. Dies kann sogar zur Ionisation von Substanzen wie Stickstoff, Sauerstoff und Wasser führen, deren IP über 10.5 eV liegt [62]. Die 355 nm-
77
Primärstrahlung ist darüber hinaus in der Lage, einem durch SPI erzeugten Ion, dessen
Anregungsniveaus niedriger als beim neutralen Molekül liegen, Energie resonant zu
übertragen. Dies kann eine Fragmentierung des Ions bewirken.
All diese durch die Fundamentale hervorgerufenen Effekte wirken sich negativ auf die
Selektivität des Spektrums aus. In der Literatur findet man unterschiedliche Methoden,
um den negativen Auswirkungen der 355 nm-Strahlung entgegenzuwirken. Die meisten
nutzen den Unterschied im Brechungsindex der 118 nm- und 355 nm-Strahlung für
MgF2 oder LiF aus, um die Strahlen zu trennen. In einigen Arbeiten [30, 31, 37, 62, 99,
104-107] wird dabei wie in Abbildung 29 auf der linken Seite dargestellt verfahren.
Dabei wird die Krümmung der Linse zur Fokussierung des VUV-Lichts so wählen, dass
der VUV-Strahl fokussiert wird, der 355 nm-Strahl aber divergent bleibt. Damit ist die
Energiedichte am Ionisationsort der Fundamentalen relativ gering und nur wenig Ionen
können durch Mehrphotonenabsorption gebildet werden. In anderen Arbeiten [23, 27,
33, 38, 108] hingegen wurde die Apparatur so aufgebaut, dass der gemischte Strahl aus
118 nm und 355 nm die fokussierende Linse, wie auf der rechten Seite von
Abbildung 29 dargestellt, außermittig trifft. Dieser Aufbau hat den Vorteil, dass beide
Strahle am Ionisationsort getrennt verlaufen und die Ionen nicht mit der Fundamentalen
in Berührung kommen. Bei diesem Verfahren besteht darüber hinaus die Möglichkeit,
die Fundamentale vor der Ionisationszone abzuschatten.
Neben einer Linse zur Strahltrennung kann auch ein Prisma aus LiF oder MgF2 zur
Strahltrennung verwendet werden [78, 109]. Da die VUV-Strahlung in einem Strahlfokus entsteht und danach divergent verläuft, ist allerdings auch hier eine fokussierende
Linse notwendig. Indes ist zu Beachten, dass für 2-3 mm dicke Kristalle aus LiF die
Transmission von 118 nm-Strahlung nur ca. 55 % und durch MgF2 nur ca. 40% beträgt
[73]. Bei Strahltrennung mit Hilfe eines Prismas sind daher große Transmissionsverluste
zu erwarten.
Es konnten keine effektiven Beschichtungen gefunden werden, die 118 nm-Strahlung
reflektieren und 355 nm-Strahlung durchlassen. Bei Beschichtungen, die dagegen die
UV-Strahlung reflektieren und die VUV-Strahlung durchlassen, besteht wieder das
Problem, dass ein Glas oder Kristall mit hoher VUV-Absorption passiert werden muss.
Der hier beschriebene Aufbau verfolgt das Konzept der Strahltrennung durch eine außermittig bestrahlte Linse wie in Abbildung 29 (links) dargestellt. Das Besondere des
verwirklichten Aufbaus besteht darin, dass durch eine vakuumdichte Verschiebeeinrich-
78
Kapitel 5
tung die VUV-Linse, die das VUV-Licht nach der Entstehung fokussiert, auch zentral
angestrahlt werden kann. Damit sind Versuche zur Auswirkung der UV-Strahlung ermöglicht.
Die oben beschriebene Software wurde so programmiert, dass das Verschieben der
VUV-Linse im rechten Winkel zu den einfallenden Strahlen simuliert werden kann.
Abbildung 29: Darstellung der mögliche Strahlführungen zur Separation der 355 nmStrahlung von der 118 nm-Strahlung. Links trifft der gemischte 355 nm- und 118 nm-Strahl
mittig auf die MgF2-Linse. Durch die Linse wird der 118 nm-Strahl in das Zentrum der Ionenquelle fokussiert. Der divergente 355 nm-Strahl passiert hier auch die Ionisationszone.
Rechts durchdringt der gemischte Strahl die Linse dezentral. Die 118 nm-Strahlung wird
wiederum in das Zentrum der Ionenquelle fokussiert. Der 355 nm-Strahl läuft aus dem
118 nm-Strahl heraus und kann mit einer Blende vor der Ionenquelle abgeblockt werden.
79
Abbildung 30: Schematische Darstellung der VUV-Zelle in Verbindung mit der Ionenquelle
eines TOFMS. Senkrecht von oben wird der 355 nm-Strahl in die Zelle fokussiert, wo im
Zentrum (THG-Zone) 118 nm-Photonen erzeugt werden. Von der THG-Zone aus gelangen
die 355 nm- und die 118 nm-Strahlung zur MgF2-Linse, die auch gleichzeitig das Austrittsfenster der Zelle ist. Aufgrund der unterschiedlichen Brechungsindizes wird die 118 nmStrahlung in das Zentrum der Ionenquelle fokussiert, wohingegen die 355 nm-Strahlung divergent am Zentrum vorbeistrahlt. Die THG-Zelle ist so konstruiert und angebracht, dass ein
weiterer Strahl (OPO-Strahl) für den REMPI-Prozess in das Zentrum der Ionenquelle gelenkt werden kann.
80
Kapitel 5
Die Zelle, wie sie letztendlich nach allen Überlegungen und Simulationen gebaut wurde,
ist in Abbildung 30 dargestellt. Von oben kommt 355 nm-Strahlung und wird durch eine
bewegliche Linse mit f = 100 bei 355 nm durch ein Fenster in das Zentrum der Zelle
fokussiert, wo die VUV-Strahlung entsteht. Nach dem Fokus (THG-Zone) gelangt die
divergente VUV- und UV-Strahlung zu einer MgF2-Linse, die den VUV-Strahl in das
Zentrum der Ionenquelle fokussiert. Bei der Einstellung, wie sie in Abbildung 30 dargestellt ist, gelangt das UV-Licht nicht in die Ionisationszone sondern wird durch eine
vorgeschaltete Blende abgefangen.
Die Zelle wurde so schlank gebaut, dass durch denselben Flansch, an dem die THGZelle angeschlossen ist, ein UV-Strahl (OPO-Strahlung) eingestrahlt werden kann, um
in der Ionisationszone alternativ Probenmolekühle auch durch den REMPI-Prozess zu
ionisieren.
In Abbildung 30 sind noch die weiter unten beschriebene Getterpumpe zum Reinigen
des Edelgases sowie die benötigten Anschlüsse der THG-Zelle dargestellt. Durch die
Einlassnadel im unteren Teil der Abbildung gelang das Probengas in die Ionisationszone, wo es durch die UV- oder die VUV-Strahlung ionisiert wird. Die Ionen werden von
dort durch ein elektrisches Feld in des Flugrohr beschleunigt, dort massenspezifisch
getrennt und dann nachgewiesen.
81
5.4 Flugzeitmassenspektrometer
Das Flugzeitmassenspektrometer mit Ionenquelle wurde nach vorgegebenem Konzepten
gebaut und geliefert (Fa. Stefan Kaesdorf, München, Deutschland). Das verwendete
Flugzeitmassenspektrometer besitzt eine zweistufige Ionenquelle nach Wiley und McLaren [41]. Im Anschluss an die Ionenquelle folgt eine elektrostatische Ionenlinse, um
den Ionenstrahl zu kollimieren und eine ebenfalls elektrostatische X-Y-Ablenkeinheit,
um den Ionenstrahl auf den MCP–Detektor (multi-channel plates) hin auszurichten.
Nach diesen Ionenoptiken und einem durchlaufenen Potenzialgefälle von maximal 3 kV
gelangt der Ionenstrahl in den feldfreien Raum des Flugrohres.
Das TOFMS kann als lineares TOFMS und als Reflektron betrieben werden. Wird das
TOFMS in linearer Betriebsweise [41, 42] betrieben, so gelangen die Ionen auf direktem
Weg durch das Flugrohr auf den Ionendetektor am anderen Ende des Massenspektrometers. Im Reflektron-Betrieb ist der Ionenspiegel, der sich vor dem Detektor, der bei
linearem Betrieb die Ionen detektiert, aktiv und lenkt die Ionen zurück auf einen 2. Detektor am vorderen Ende des Flugrohrs (vgl. Abbildung 3). Die Spannungsversorgung
der Abzugselektroden ist so ausgelegt, dass wie in Abbildung 2B die Ionisationszone
auf 0 V-Potenzial liegt. Damit kann mit Hilfe eines Nadeleinlasses am Ionisationsort
eine hohe Gasdichte erzeugt werden, was die Sensitivität das Aufbaus positiv beeinflusst.
Tabelle 3: Technische Daten des verwendeten Flugzeitmassenspektrometers.
freie Driftstrecke des Reflektrons
801 mm
Länge des Ionenreflektors
95 mm
Winkel zwischen ein- und auslaufendem Ionenstrahl im 6,1°
Ionenreflektor
Durchmesser der Eintrittsblende
6 mm
maximales Feld in der Ionisationszone
2000 V / 14 mm
Maximale Beschleunigungsspannung
3000 V (2 stufig)
Ionendetektoren (multi channel plates)
CHEVRON,
S304-10-D
Typ
82
Kapitel 5
Die Ionenquelle des verwendeten Flugzeitmassenspektrometers ist mit einer Elektronenkanone ausgestattet. Diese Elektronenkanone ist so angeordnet, dass in die
Ionisationszone sowohl Laserlicht als auch Elektronen aus der Elektronenkanone eingestrahlt werden können. Dadurch kann das Probengas in der Ionisationszone sowohl mit
Hilfe von Photonen als auch mit Elektronen ionisiert werden.
Technische Kenndaten des Flugzeitmassenspektrometers mit integrierter Elektronenstoßionenquelle sind in Tabelle 3 zusammengefasst.
83
5.5 Ionenquelle zur Elektronenstoßionisation
Bei den TOFMS-Messungen mit effusivem Einlasssystem wurden Ionen bis zu einer
Masse von maximal 350 m/z detektiert. Diese schweren Ionen benötigen bei den üblichen Beschleunigungsspannungen in etwa 20 ms vom Ionisationsort bis zum Detektor.
Bei Verwendung eines 10 Hz-Lasersystems bedeutet dies, dass nach Auftreffen der
schwersten Ionen auf dem Detektor im TOFMS für die nächsten 99980 ms nichts geschieht. Selbst mit modernen Nd:YAG-Lasern sind nur Pulsrate bis 50 Hz möglich.
Daher wurde von der Fa. Käsdorf eine Elektronenkanone in der Ionenquelle so angebracht, dass die Ionisationszone neben den VUV- und UV-Strahlen auch von einem
variierbaren Elektronenstrahl getroffen werden kann.
Der Elektronenstrahl dieser Kanone kann mit bis zu 20 kHz gepulst werden. Da die
Elektronen, mit denen das Probengas in der Ionisationszone beschossen wird, geladene
Teilchen sind, muss das Abzugsfeld, das zwischen Repeller und 1. Extraktionsplatte bei
Laserionisation anliegt, während der Elektroneneinstrahlung abgeschaltet sein. Bei anliegendem Feld würden die leichten Elektronen von der Elektronenkanone direkt zum
positiven Repeller gezogen werden. Mit Hilfe zweier Hochspannungspulser (Fa. Stefan
Kaesdorf, München, Deutschland) über die Repeller und 1. Extraktionsblende mit der
Hochspannungsversorgung verbunden sind, kann das Abzugsfeld zwischen Repeller
und 1. Extraktionsblende innerhalb einiger ns an- und ausgeschaltet werden.
Abbildung 31 zeigt das Trigger-Signal für die Elektronenextraktion der Elektronenkanone und das Trigger-Signal, das die Hochspannungspulser steuert. Gibt ein
Funktionsgenerator (z.B. Typ DG 535, Fa. Stanford Research Instruments Inc., Sunnyvale, USA oder Typ PDG 204, Fa. Scientific Instruments, Gilching, Deutschland)
Trigger-Signale vor, wie in Abbildung 31 dargestellt, so wird das Probengas in der Ionisationszone für 1 ms mit Elektronen beschossen und anschließend werden die erzeugten
Ionen nach einer Pause von 50 ns durch das angeschaltete Abzugsfeld beschleunigt.
Geht man davon aus, dass die Gasatome bei effusivem Gaseinlass langsamer als die
Schallgeschwindigkeit (320 m/s) sind [110], so legen die Gasatome in 1 ms weniger als
0,3 mm zurück. Bezogen auf das Abzugsvolumen der Ionenquelle von einigen Kubikmillimetern gehen damit durch die schnelle zeitliche Abfolge von Ionenerzeugung und
Extraktion kaum Ionen verloren.
84
Kapitel 5
Abbildung 31: Trigger-Signale zur Steuerung des Elektronenstrahls (oben) und der Abzugsspannungen (unten) bei EI-Betrieb.
Durch den Heizstom der Elektronenkanone lässt sich die Intensität des Elektronenstrahls
und damit auch die Anzahl der erzeugten Ionen regulieren. Die Elektronenbeschleunigungsspannung lässt sich durch das Netzteil stufenlos einstellen. Somit können
Elektronen mit verschiedenen kinetischen Energien zur Ionisation verwendet werden.
Dies ist besonders interessant, weil damit auch 70 eV-Elektronenstoßspektren aufgenommen werden können, deren Fagmentmuster mit den Spektren der NIST-Datenbank
[64] vergleichbar sind. Außerdem können z.B. Spektren mit 20 eV-Elektronen erzeugt
werden, für die Stickstoff einen kleinen Wirkungsquerschnitt zeigt als andere Verbindungen. Damit lässt sich die Anzahl der Stickstoffionen relativ zu den Ionenzahlen
anderer Verbindungen etwas reduzieren.
Aufgrund der hohen möglichen Repetitionsrate und wegen der vergleichbar geringeren
Empfindlichkeit wird bei der Elektronenstoßionisation eine andere Signalerfassung als
bei der Laserionisation verwendet. Zur Aufnahme der Spektren wird eine Zählkarte eingesetzt, die den Zeitbereich eines Spektrums, in dem Ionensignale vom Detektor
erwartet werden, in lauter gleiche Zeitfenster (Zeitkanäle) aufteilt. Bei der verwendeten
Zählkarte (Typ P7886, Fa. Fast ComTec, München, Deutschland) beträgt die Kanalbreite mindestens 500 ps. Die 20 ms, in denen Ionen am Detektor pro Schuss erwartet
werden, sind somit in 40.000 Kanäle unterteilt. Jeder Kanal zählt nur den Wert 0 oder 1.
Kommt vom Detektor ein Signal, während ein bestimmter Kanal aktiv ist, und überschreitet dieses Signal eine vorgegebene Diskriminatorschwelle, so wird für diesen
Kanal im Speicher der Wert 1 addiert. Wenn das Signal unter der Diskriminatorschwelle
85
liegt, wird dem Speicher nichts hinzugefügt. Die Diskriminatorschwelle wird so gewählt, dass das Signal eines einzigen Ions diese gerade übersteigt. Nach einer
vorgegebenen Anzahl von Durchläufen bzw. Schüssen der Elektronenkanone werden
die Werte aus den Kanälen als Spektrum abgespeichert und die Kanalspeicher für eine
neue Messung gelöscht. Wird die Elektronenkanone mit 20 kHz betrieben und nimmt
man 20.000 Durchgänge auf, so erhält man 1 Spektrum pro Sekunde.
Dieses Verfahren funktioniert sehr gut und ist Stand der Technik bei Ionenquellen, in
denen sich das Analysengas am Ionisationsort unter Drücken bis 10-5 mbar befindet.
Beim verwendeten Aufbau gelangt das Probengas durch die Einlassnadel direkt zum
Ionisationsort, wo es ins Vakuum entlassen wird. In der Ionisationszone herrschen damit
sehr viel höhere Drücke. Damit kann es leicht zur gleichzeitigen Erzeugung mehrerer
gleich schwerer Ionen kommen. Diese Ionen, die den Detektor zur selben Zeit erreichen,
erzeugen ein ihrer Anzahl proportionales Signal. Der Kanal dieser Ionen addiert aber
nur 1. Durch diese Nichtbeachtung des Mehrionensignals können die Gesamtspektren in
diesem Fall nicht für quantitative Aussagen gebraucht werden.
Für quantitative Messungen ist daher z.B. der Elektronenstrom über die Heizspannung
der Elektronenkanone so weit zu reduzieren, dass mit großer statistischer Wahrscheinlichkeit höchstens ein Ion pro Sorte und Schuss erzeugt wird. Bei einer dynamischen
Online-Messung muss dies auch noch für die höchste zu erwartende Konzentration einer
Substanz erfüllt sein. Ist man an Substanzen im ppm-Bereich interessiert und stellt den
Elektronenstrom auf diese ein, so werden Substanzen in Prozentkonzentrationen zwar
auch detektiert, ihr Signalverlauf ist aber nicht mit ihrem Konzentrationsverlauf korreliert.
Zur Einstellung des optimalen Elektronenstroms bei ausreichend kleiner Koinzidenz
mehrerer Ionen in einem Zeitkanal, wird wie folgt verfahren. Zum Erreichen der maximalen Nachweisgrenze ist ein Probenstandard nötig, der die zu analysierende Substanz
in
der
maximal
vorkommenden
Konzentration
enthält
und
das
NIST-EI-
Referenzspektrum [64] bei gleicher Elektronenenergie. Zeigt sich beim Vergleich des
Referenzspektrums mit dem aufgenommenen Spektrum, dass die relativen Signalhöhen
der Fragmente übereinstimmen, ist der Elektronenstrom noch nicht zu groß.
Das Signal eines Ion beträgt je nach Detektorspannung etwa 5 - 10 mV. Gleich nach der
Vakuumdurchführung des Signalkabels wird das Detektorsignal durch einen Vorverstärker (Fa. Stefan Kaesdorf, München, Deutschland) um den Faktor 15 verstärkt. Die
86
Kapitel 5
Diskriminatorschwelle kann damit bei ca. 50 mV liegen. Damit verursachen Störungen,
die nach dem Vorverstärker eingefangen werden und viel kleiner als 50 mV sind, kein
Rauschen auf der Grundlinie des EI-Spektrums.
87
5.6 Kombination der Ionisationsmethoden
Ziel dieser Arbeit ist die Entwicklung eines Flugzeitmassenspektrometers, das nahezu
gleichzeitig ein Probengas mit drei unterschiedlichen Ionisationsmechanismen ionisieren kann und es dadurch möglich ist ein Gas umfassend und mit hoher Zeitauflösung zu
analysieren. Das hier vorgestellten TOFMS-Systems kann ein Probengas, gewissermaßen gleichzeitig mit dem REMPI-, SPI- und EI-Verfahren analysieren.
Dadurch, dass der OPO-Laser und die VUV-Zelle vom selben Nd:YAG-Laser mit
355 nm-Strahlung versorgt werden, stehen 118 nm-VUV-Licht für den SPI-Prozess und
UV-Licht für den REMPI-Prozess zur selben Zeit zur Verfügung. Setzt man in den
355 nm-Strahlengang zur VUV-Zelle und in den UV-Strahlweg vom OPO-Laser zur
Ionenquelle je einen Hubmagneten mit Teflonplättchen zur Strahlabblockung (vgl.
Abbildung 32), so kann je nach freigegebenem Strahl mit dem SPI- oder REMPIProzess ionisiert werden. Werden diese Magneten mit den vom Laser vorgegebenen
10 Hz gesteuert, so kann abwechselnd je ein SPI- und ein REMPI-Spektrum aufgenommen werden. Da der verwendete Rechner (400 MHz, 128 MByte RAM, NT4) nicht
schnell genug war, um mit 10 Hz einzelne Spektren zu speichern, wurden in der Regel
5 Spektren zu einem gemittelt und dieses gespeichert. Bei der parallelen Aufnahme von
REMPI- und SPI-Spektren geben die Hubmagneten (Fa. Bürklin, München, Deutschland) mit 10 mm Hub z.B. alle 0,5 s einen anderen Strahl frei. Damit die erfassten Daten
der jeweiligen Ionisationsart exakt zugeordnet werden, wurde das Datenerfassungsprogramm dahingehend erweitert, dass die Magneten über die serielle Schnittstelle
(RS 232) gesteuert werden. So ist die Software in der Lage während einer Messung z.B.
ein aus 5 Einzelspektren gemitteltes SPI-Spektrum in ein Verzeichnis zu speichern und
das gemittelte REMPI-Spektrum in ein anderes. Auf diese Weise können pro Sekunde je
ein REMPI- und ein SPI-Spektrum abgespeichert werden.
Aufgrund der verschiedenen Parameter des VUV- und UV-Strahls und der verschiedenen Strahlenwege durch die Ionenquelle (siehe Abbildung 30) existiert für jede
Ionisationsmethode ein optimaler Satz an Hochspannungen für das Massenspektrometer. Da dieser Satz Spannungen für REMPI und SPI nicht gleich ist und in dieser
Ausführung nicht mit der erforderlichen Frequenz umgeschaltet werden konnte, sind
unter Verwendung eines Spannungssatzes bei Messungen mit alternierender Ionisationsmethode Verluste bei einer der Ionisationsmethoden in Kauf zu nehmen. In der
88
Kapitel 5
Regel wurde der Spannungssatz derjenigen Ionisationsmethode verwendet, von der die
interessantesten Ergebnisse zu erwarten waren.
Bei Laser-Massenspektrometern, bei denen der Pumplaser nicht genügend Energie liefert, die durch einen Strahlteiler auf VUV-Zelle und OPO-Laser verteilt werden kann,
kann mittels eines schnellen, präzisen Klapp- oder Drehspiegels der ganze 355 nmStrahl umgeleitet werden. Damit erübrigen sich die alternierenden Hubmagnete.
Begnügt man sich damit, den REMPI-Prozess nur mit 266 nm durchzuführen, um sich
den teuren OPO-Laser zu sparen, so kann durch einen steuerbaren Spiegel abwechselnd
355 nm für die VUV-Erzeugung und 266 nm für den REMPI-Prozess bereitgestellt werden. Dieses Verfahren wird in [20] beschrieben und wurde patentiert [111]. Es
funktioniert, weil die 266 nm-Strahlung für REMPI durch zweimaliges Verdoppeln der
1064 nm-Strahlung des Nd:YAG-Lasers in je einem Kristall erzeugt wird. Die 355 nmStrahlung entsteht hingegen, wie in Abbildung 33 dargestellt, durch die einmalige Ver-
Abbildung 32: Skizze der Laserstrahlen zwischen Nd:YAG-Laser und VUV-Zelle sowie zwischen OPO-Laser und Ionenquelle. In beiden Strahlwegen befindet sich je ein Hubmagnet
mit Strahlblocker zum alternierenden Unterbrechen der Strahlen. Dadurch ist es möglich,
zwischen der SPI- und der REMPI-Methode zu wechseln. Gesteuert werden die Elektromagnete durch den Messrechner.
89
dopplung der Fundamentalen (532 nm) und Mischung der 532 nm-Strahlung mit der
Fundamentalen zur 355 nm-Strahlung. Montiert man, wie in Abbildung 33 zu sehen ist,
einen steuerbaren Spiegel in den 1064 nm- und 532 nm-Strahlengang und lenkt damit
die Strahlen über einen 532 nm reflektierenden dicroiden Spiegel zu einem weiteren
Verdopplerkristall, so kann 266 nm-Strahlung für den REMPI-Prozess erzeugt werden.
Lässt man die 1064 nm- und die 532 nm-Strahlung ungehindert zu einem Kristall, der
aus einem 1064 nm-Photon und einem 532 nm-Photon ein 355 nm-Photon generiert, so
erhält man die zur VUV-Erzeugung notwendige 355 nm-Strahlung.
Abbildung 33: Laseraufbau mit Mischkristall, Verdopplungskristallen, dicroiden Spiegeln
und Klappspiegel zur abwechselnden Erzeugung von 266 nm-Strahlung für REMPI und
355 nm-Strahlung für die Vakuum-UV-Erzeugung.
90
Kapitel 5
Wie oben erwähnt, liegt es nahe, das Gerät so zu betreiben, dass zwischen den 10 HzLaserschüssen für REMPI und SPI mit 20 kHz EI-Transienten erzeugt werden.
Will man EI, SPI und REMPI quasi gleichzeitig in einem TOFMS betreiben, so sind
einige zeitliche Abläufe aufeinander abzustimmen und das Signal ist je nach Ionisationsart der entsprechenden Datenerfassung zuzuführen. Um das Signal zwischen
Transientenrekorder-Karte und Vielkanalanlysator hin und her zu schalten, besitzt der
Vorverstärker eine Signalweiche, die mittels eines einfachen Trigger-Signals angesteuert werden kann. Bei „high“ gelangt das Signal zum Vielkanalanalysator, sonst zur
Transientenrekorderkarte.
Da ein Nd:YAG-Laser schlecht zu triggern ist, wird das Trigger-Signal des Lasers als
Mastertrigger verwendet und alle anderen Vorgänge werden darauf abgestimmt. Der
Lasertrigger wird verwendet zum Starten der Transientenrekorderkarte (Datenaufnahme
bei Laserionisation) und zum Triggern eines Funktionsgenerators (siehe Abbildung 34).
Für diese Art des Betriebes benötigt man einen Funktionsgenerator (Typ PDG 204, Fa.
Scientific Instruments, Gilching, Deutschland) mit mindestens 3 unabhängigen Kanälen,
Signalzählfunktion und Signalunterdrückungsfunktion. Der Funktionsgenerator muss so
programmiert werden, dass er nach dem Startsignal des Lasers (in Abbildung 34 die
negative Flanke von A) z.B. mit 10 kHz oder 20 kHz beginnt, eine vorgegebene Anzahl
von Sequenzen abzuarbeiten.
Nach dem Lasertrigger findet je nach Laserstrahlfreigabe durch den Magnetschalter in
der Ionenquelle SPI oder REMPI statt. Die Elektronenkanone darf jetzt keine Elektronen freisetzen. Es wird aber ein Abzugsfeld benötigt. Daher geht das Trigger-Signal B
in Abbildung 34 in der ersten Sequenz nicht auf „an“ (high), wohl aber der Trigger C für
das Abzugsfeld. Das Signal D für die Signalweiche bleibt auf „aus“ (low) für die Tansientenrekorder-Karte. Bei der Laserionisation ist es günstiger, wenn das Abzugsfeld
schon während der kurzen Ionenerzeugung eingeschaltet ist. Die Hochspannungspulser
so anzusteuern, dass sowohl für Laserionisation (LI) als auch für EI ein optimales Feld
anliegt, wäre sehr aufwendig. Daher werden hier bei der LI kleine Verluste in Kauf genommen.
Der verwendete Funktionsgenerator PDG 204 bietet die Möglichkeit, am Anfang oder
Ende einer vorgegebenen Anzahl von Sequenzen (bursts) 2 oder mehr Sequenzen eines
bestimmten Kanals zu unterdrücken (burst-suppress-mode). Mit dieser Möglichkeit
werden Kanal B und D nach dem Lasertrigger erst einmal für 2 Sequenzen in den Ruhe-
91
stand versetzt. Nach der 1. Sequenz ist das LI-Massenspektrum aufgenommen und da
mindestens 2 Sequenzen unterdrückt werden, geschieht in der 2. Sequenz nichts.
Von der 3. bis zur 1000. Sequenz wird durch das Trigger-Signal B der Elektronenstrahl
und durch C das benötigte gepulste Abzugsfeld erzeugt. Das Signal D für die Signalweiche steht während dieser Zeit permanent auf „an“.
Mit der in Abbildung 34 dargestellten Zeitsteuerung kann das TOFMS-System ein Probengas 10 mal in der Sekunde mit LI und 9920 mal mit EI analysieren. Da bei vielen
Messungen auch noch der weiter unten beschriebene Massenfilter aktiv war, der nur bis
maximal 10 kHz betrieben werden kann, arbeitet der Funktionsgenerator im Beispielschema der Abbildung 34 nur mit 10 kHz anstelle der möglichen 20 kHz.
Bei gleichzeitiger EI, SPI und REMPI war die Einstellung der TOF-Spannungen noch
etwas kritischer. Auf die Signalhöhe bei EI brauchte nicht geachtet zu werden. Sie konnte durch die Heizspannung immer getrennt von den anderen Ionisationsformen
eingestellt werden. Wegen der unterschiedlichen Ausbreitung von Elektronenstrahl und
Abbildung 34: Triggerpulse die bei quasi gleichzeitiger Elektronenstoß- und Laserionisation
benötigt werden. A ist die Spur das Mastertriggers vom Nd:YAG-Laser, auf den alle anderen
Trigger synchronisiert sind. B ist der Trigger für die Elektronenextraktion, C der Trigger für
die Hochspannungspulser der Ionenextrakionsplatten und D sind die Pulse für den Vorverstärker mit Signalweiche.
92
Kapitel 5
Laserstrahlen am Ionisationsort ist es viel schwieriger, bei EI in Kombination mit LI in
die Nähe der Massenauflösung (siehe Seite 109) zu kommen als bei EI allein. Auch hier
ist zu empfehlen, den Satz TOF-Spannungen zu verwenden, der den interessantesten
Prozess begünstigt. Außerdem gibt es die Möglichkeit, von außen einen Permanentmagneten zu verschieben, der die Elektronenflugbahn beeinflusst. Damit kann der
Elektronenstrahl etwas auf einen Laserspannungssatz hin optimiert werden.
93
5.7 Massenfilter
Die im TOFMS verwendeten MCP-Detektoren haben nur einen begrenzten dynamischen Bereich. Ist z.B. in einem Analysengas eine Substanz in einer Konzentration von
10 ppm enthalten und eine etwas schwerere Substanz mit 0,1 ppm und beide Substanzen
haben für das verwendete Laserlicht in etwa denselben Wirkungsquerschnitt, so unterscheiden sich die Signalspannungen am Detektor um den Faktor 100. Verursacht die
schwere Substanz 10 mV, so sind es bei der anderen schon 1 V.
Da ein MCP-Detektor nach Überschreiten einer bestimmten Signalhöhe etwas Zeit benötigt, um sich nachzuladen, fällt seine normalerweise konstante Verstärkung für einige
Zeit etwas ab. Bei den verwendeten Detektoren (Typ Chevron S304-10-D, Fa. Burle,
Lancaster, USA) hat sich herausgestellt, dass nach einem Signalpeak, der höher als 1 V
ist, die Signalpeaks der schwereren Ionen durch den Verstärkungsabfall kleiner bzw.
unterdrückt werden.
Aufgrund dieser technisch bedingten Signalbeeinflussung ist es in manchen Fällen erforderlich, Ionen einer Substanz, die in verhältnismäßig hoher Konzentration vorhanden
ist, vom Detektor fernzuhalten. Dies kann man dadurch erreichen, dass auf der Flugbahn
der Ionen zum Detektor an einer Stelle, an der die verschieden schweren Ionen bereits
örtlich getrennt fliegen, kurzzeitig ein in der Ausdehnung begrenztes Ablenkfeld erzeugt
wird.
Bei einem Reflektron-TOFMS eignet sich als Ort der Bereich des 1. Ortsfokus besonders, weil hier die gleich schweren Ionen zeitlich fokussiert fliegen. Ein räumlich
begrenztes Ablenkfeld kann durch ein enges Gitter erzeugen werden, bei dem die
benachbarten Gitterdrähte immer genau auf dem entgegengesetzten Potenzial liegen.
Zwischen den Gitterdrähten herrscht damit ein sehr starkes Feld. Etwas davor und dahinter kompensieren sich aber diese Poteniale. Wie man in der Simulation von
Abbildung 35 sieht [112], werden Ionen daher nur zwischen den Gitterdrähten abgelenkt. Davor oder dahinter bleibt ihre Flugbahn unbeeinflusst. Schaltet man mit
schnellen Hochspannungsschaltern die Spannungen der sonst neutralen Gitterdrähte
kurz an, während sich die Ionen der störenden Masse zwischen den Drähten befindet,
und danach gleich wieder aus, so bleibt der Flug der leichteren Ionen davor und der
schwereren dahinter unbeeinflusst. Die abgelenkten Ionen gelangen nicht zum Detektor.
94
Kapitel 5
Auf den Massenspektren an der Stelle des störenden Massenpeaks ist nach Ablenken der
Störionen lediglich eine etwas unruhigere Grundlinie zu sehen.
Durch die Abzugsspannungen der Ionen lässt sich die Position des 1. Ortsfokus bestimmen. Optimiert man damit die Positon des 1. Ortsfokus auf den Massenfilter, so können
mit dem Massenfilter des TOFMS (Fa. Stefan Kaesdorf, München, Deutschland) einzelne Massen eliminiert werden, ohne die Nachbarmassen zu beeinflussen.
Abbildung 35: Potenziallinienverlauf um ein dünnes, abwechselnd positiv und negativ geladenes Massenfiltergitter. Von links nach rechts sind Flugbahnen von Ionen zu sehen, die
durch das Nahfeld des Massenfilters bei angelegten Potenzialen vom Weg zum Detektor abgelenkt werden.
95
5.8 Kühlung
Für die optischen und einige andere Komponenten des TOFMS stellt der feine Staub im
Kesselhaus von Müllverbrennungsanlagen, ein Problem dar. Zum Schutz gegen den
Staub, müsste das gesamte TOFMS idealerweise luftdicht gekapselt sein. Da sich aber
im Gerät viele Komponenten befinden, die bis zu 2 kW Abwärme produzieren, wie die
Turbomolekularpumpen, die Rechner mit der 60°C heißen Transientenrekorderkarte,
das Lasernetzteil und einige weitere Netzteile, würde eine Kapselung zu einem Hitzestau und damit zum Ausfall der Komponenten führen.
Bei der Konstruktion des Gerätes wurden mehrere Methoden eingesetzt, um diese Probleme zu lösen. Der Bereich des Nd:YAG-Laserkopfes und der gesamte OPO-Laser, die
am empfindlichsten auf Staub reagieren, wurden komplett gekapselt und werden durch
einen Wärmetauscher, der am Kühlwasserkreislauf des Lasers angeschlossen ist, temperiert. Die sehr viel Wärme produzierende, aber staubunempfindliche Vakuumvorpumpe
wurde von allen anderen Komponenten getrennt außerhalb des Gerätes angebracht (siehe Abbildung 26).
Das restliche Gerät wird mit gereinigter Pressluft gekühlt. Dazu wird Pressluft, die an
Industrieverbrennungsanlagen reichlich zur Verfügung steht, erst mit einem Membrantrockner (Drypoint, Fa. BEKO, Neuss, Deutschland) entfeuchtet und mit zwei
Filterelementen von Öl und Feinpartikeln befreit. Die so gereinigte Pressluft gelangt
über zwei Schalldämpfer in den rechten und linken Teil des Gerätes. Durch Luftauslässe
mit Filtermatten gelangt die erwärmte Luft wieder nach außen. Bei dieser Anordnung
fließt permanent gereinigte Luft vom Inneren des Gerätes nach außen. Staubpartikel
können dadurch nicht in das Gerät gelangen. Auch bei Wartungsarbeiten am offenen
Gerät wirkt sich dieser Luftfluss positiv aus. Im Rahmen einer fünf Wochen andauernden Messkampagne im Kesselhaus einer Müllverbrennungsanlage war das Innere des
Gerätes nahezu staubfrei.
Die Pressluftmenge, die notwendig ist, um bei Laserbetrieb die Laserwärme abzuführen,
ist beträchtlich, es sind mehr als 10 l / s notwendig.
96
Kapitel 5
5.9 Gasstandard
Quantifizieren und Kalibrieren sind wichtige Vorgänge bei der analytischen Lasermassenspektrometrie. Die Anzahl der nachgewiesenen Ionen einer Sorte bei gleicher
Konzentration im Analysengas hängt von einer Vielzahl von Geräteparametern ab. Da
sich die Geräteparameter des hoch komplexen Messsystems im Laufe einer Messkampagne oder sogar innerhalb eines Tages ändern können, sollte vor jeder Messsequenz ein
Standardgas mit relevanten Substanzen in bekannten Konzentrationen analysiert werden. Ähnlich wie schon in anderen Arbeiten [15, 18] beschrieben, wurde Standardgas
mit relevanten Substanzen im unteren ppm- bis ppb-Bereich zur externen Massenkalibration
und
Quantifikation
der
Photoionisationsmassenspektren
verwendet.
Gasstandards in Druckflaschen können als Sondergase kommerziell gekauft werden.
Aufgrund von Wandeffekten (z.B. Adsorption) können diese Gase allerdings altern und
sind nur in Konzentrationen im ppm-Bereich und höher erhältlich. Ein weiteres Problem
dieser statischen Gasstandards ist, dass nicht beliebig viele verschiedene Substanzen
gemischt werden können und sich auch nicht alle Substanzen miteinander kombinieren
lassen. Umfangreichere Standardgemische müssen daher selbst hergestellt werden.
Hierzu wurde ein kompakter Standardgasgenerator in 19“-Bauweise entwickelt
(Abbildung 37).
Abbildung 36: Schematische Darstellung des Standardgenerators. Eine Membranpumpe
saugt durch einen Flussregler Luft und bläst diese durch einen Glasbehälter in dem sich die
speziell eingeschlossenen Standardsubstanzen befinden. Der Behälter mit den Standardsubstanzen wird durch Peltiertechnik temperiert.
97
Abbildung 37: 19“-Gasstandardgenerator
In diesem Standardgenerator mischen sich Substanzen aus definiert ausgasenden Behältern mit einem definierten Gasstrom zum Standardgas. Je nach Flüchtigkeit der
Substanz werden aus ihnen Permeationsstandards oder Diffusionsstandards hergestellt
[15, 113-116]. Solche Diffussions- und Permeationsstandardröhrchen wurden mit relevanten Substanzen gefüllt. Lagert man die Standardröhrchen bei konstanter Temperatur
und unter konstantem Luftfluss (f), so kann man durch den gewogenen Gewichtsverlust
(Dm) der Substanz mit dem Molgewicht (M), bekannter Zeitspanne (Dt) zwischen den
Wägungen und dem molaren Normvolumen (Vmn = 22,4 l/mol) die Konzentration (c)
der einzelnen Substanzen im austretenden Gas errechnen.
c=(
Dm / D t
Vmn ) / f
M
[5.6]
Um konstante Umgebungsbedingungen für die Standards herzustellen, wurde in einem
19“-Gehäuse eine kleine elektrische Membranpumpe an der Saugseite mit einem Durchflusskontroller verbunden (vgl. Abbildung 36). Es erwies sich im Hinblick auf die
98
Kapitel 5
Lebensdauer der Pumpe als sinnvoll, die Luftflussrestriktion und Kontrolle auf dieser
Seite der Pumpe vorzunehmen. Der konstante Luftstrom gelangt dann in einen Aluminiumquader (150x60x60mm), an dessen vier langen Seiten je ein Peltierelement
(40x40mm) zur Temperierung sitzt und nach außen hin mit einem Kühlkörper verbunden ist. Die Luft wird in dem Quader einige Male hin und her geführt, um dessen
Temperatur anzunehmen. Der Aluminiumkörper besitzt eine große Bohrung (Æ 51mm),
in die ein Glasbehälter mit einem Glasschliff am einen Ende und einer kleinen Öffnung
am anderen Ende passend eingelassen ist. Die temperierte Luft gelangt durch die kleine
Öffnung in das Innere des Glasrohres, in dem sich die Standardröhrchen befinden. Am
Ausgang des Glasrohres erhält man so einen konstant temperierten und gleichmäßigen
Luftstrom mit konstanten Anteilen der Standardsubstanzen im Konzentrationsbereich
von 10 ppb aufwärts. Im Aluminiumkörper steckt ein Temperatursensor, durch den das
Peltierregelgerät (Typ PRG 400, Fa. Peltron, Furth, Deutschland) die Temperatur überwacht und durch Umpolung der Versorgungsspannung für die Peltierelemente den
Körper sowohl kühlen als auch heizen kann. Die Konzentrationen der Standards kann
durch Erhöhung der Temperatur etwas gesteigert werden.
Abbildung 38: Aufbau eines Permeations- und eines Diffusionsstandard
99
Abbildung 39: Gewichtsabnahme verschiedener Permeations- und Diffusionsstandards (vgl.
Tabelle 4) in einem Standardgenerator bei 25 °C (A) und 30 °C (B).
100
Kapitel 5
Die verwendeten Permeationsstandards bestehen aus kleinen Glasröhrchen, die mit einer
festen oder flüssigen reinen Substanz gefüllt und mit einer Teflonmembran verschlossen
sind. Durch den Konzentrationsunterschied der eingeschlossenen Substanz zu seiner
Umgebung erhält man einen ständigen Permeationsfluss aus dem Standard nach außen.
Substanzen mit einem niedrigeren Dampfdruck wurden in kleine Glasröhrchen mit einer
kleinen Öffnung eingeschmolzen. Diese kleine Öffnung kann ein sehr kleines Loch
(Æ < 0,5 mm) oder eine eingeklebte Kapillare sein (1 – 2 cm lang, ID 0,2 mm). Diese
verschiedenen Standardbauweisen (Abbildung 38) werden benötigt, um die Konzentration der einzelnen Komponenten im Standardgas der Nachweisempfindlichkeit des
Gerätes anzupassen.
In
Abbildung 39
ist
der
Gewichtsverlust
von
einigen
Permeationsstandards
(Abbildung 38) dargestellt. Diese Standardsubstanzen befanden sich in kleinen Glasröhrchen mit einem Innendurchmesser von etwa 2 mm. An einem Ende war das
Röhrchen zugeschmolzen, am anderen Ende wurde es mit einem Teflonschrumpfschlauch (Wanddicke ca. 0,15 mm) und einem weiteren kurzen Stück zugeschmolzenen
Glasrohrs verschlossen. Für flüssige Sandardsubstanzen wurde das Glasröhrchen an
einer Stelle mit einem feinen Bunsenbrenner erwärmt und durch blasen erweitert (Flaschenbauch). Die befüllten Röhrchen lagerten über 2 Jahre bei 25 °C in einem Luftstrom
von 10 ml/min. Aus Gleichung [5.6] ergibt sich z.B. für Benzol aus den Werten von
Abbildung 39 eine Konzentration von 1,1 ppm und für Naphthalin eine Konzentration
von 150 ppb. Phenanthren, Butanal, Phenol, Pyren, Dibenzofuran und Biphenyl verlieren als Permeationsstandards durch die Teflonmembran zu wenig Gewicht, als dass ein
gleichmäßiger Gewichtsverlust gemessen werden konnte. Außerdem sind von diesen
Substanzen Konzentrationen im 100 ppb-Bereich erwünscht. Von diesen Substanzen
sollten daher in Zukunft Diffusionsstandards hergestellt werden.
Von einigen Substanzen, die bei einer Neubestückung des Standardgenerator verwendet
wurden, sind daher Diffusionsstandards (Abbildung 38) erstellt worden. Diese Standards besaßen anstatt eines verschlossenen Teflonschlauchs eine ca. 1 cm lange
Kapillare (ID 0,2 mm), die mit UHU Endfest eingeklebt war. Ein Vorversuch hatte
gezeigt, das UHU Endfest nach dem Aushärten nicht weiter ausgast und daher im späteren Verlauf kein Gewicht verliert. Das Glasröhrchen mit Anilin wurde sogar
unverschlossen mit den anderen in den Generator bei 30 °C und 20 ml/min Luftfluss
gestellt.
101
In Tabelle 4 sind die mittleren Konzentrationen der Substanzen im Standardgas in Abhängigkeit
von
den
Umgebungsbedingungen
aufgetragen,
um
künftig einen
Anhaltspunkt zu haben, welche Konzentration in diesem Standardgenerator unter den
selben Bedingungen zu erwarten sind. Die Stoffe, die trotz Diffusionsstandard nicht
merklich an Gewicht verloren, könnten wie Anilin in einem offenen Röhrchen bei erhöhter Temperatur gelagert werden. Dazu muss allerdings untersucht werden ob sich
diese Stoffe dann hygroskopisch verhalten.
Bei dieser Art eines dynamischen Standards müssen sich die Standards über mehrere
Wochen hinweg im temperierten Glasrohr befinden, damit die Konzentrationen der
Substanzen in der austretenden Luft verlässlich bestimmt werden können. Die Behälter
mit den Standardsubstanzen sollten zum Wiegen nur kurzzeitig aus dem temperierten
Lagerbehälter genommen werden. Es empfiehlt sich, ein leeres Standardröhrchen mit im
Generator zu lagern, an dessen unverändertem Gewicht sich die Waage (min. 0,1 mg
Genauigkeit) und das Wiegen überprüfen lässt.
102
Kapitel 5
Tabelle 4: Konzentrationen von Permeations- und Diffusion-Standards bei verschiedenen
Umgebungsbedingungen.
Leck
Temp
Fluss
Permeation
Permeation
Permeation
Diffussion
Permeation
Permeation
Permeation
Permeation
Diffusion
Permeation
Permeation
Diffusion
Permeation
Permeation
Diffusion
Permeation
Diffusion
Diffusion
Teflon
Teflon
Teflon
Kapillare
Teflon
Teflon
Teflon
Teflon
Kapillare
Teflon
Teflon
Loch
Teflon
Teflon
Kapillare
Teflon
Kapillare
Kapillare
30 °C
25 °C
25 °C
30 °C
25 °C
30 °C
25 °C
25 °C
30 °C
30 °C
25 °C
30 °C
25 °C
30 °C
30 °C
25 °C
30 °C
30 °C
20 ml/min
10 ml/min
10 ml/min
20 ml/min
10 ml/min
20 ml/min
10 ml/min
10 ml/min
20 ml/min
20 ml/min
10 ml/min
20 ml/min
10 ml/min
20 ml/min
20 ml/min
10 ml/min
20 ml/min
20 ml/min
Mittlere
Konzentration
5 ppm
4 ppm
0,8 ppm
2 ppm
nicht messbar
1 ppm
1 ppm
1 ppm
0,5 ppm
0,3 ppm
0,5 ppm
0,8 ppm
0,02 ppm
0,1 ppm
4 ppm
0,3 ppm
0,03 ppm
nicht messbar
Diffusion
Kapillare
30 °C
20 ml/min
nicht messbar
Permeation
Permeation
Diffusion
Permeation
Permeation
Permeation
Permeation
Teflon
Teflon
Kapillare
Teflon
Teflon
Teflon
Teflon
30 °C
25 °C
30 °C
25 °C
25 °C
25 °C
25 °C
20 ml/min 0,02 ppm
10 ml/min 0,15 ppm
20 ml/min 0,04 ppm
10 ml/min nicht messbar
10 ml/min nicht messbar
10 ml/min nicht messbar
10 ml/min nicht messbar
Substanz
Masse Standard
[amu] Typ
Ammoniak
17
Acetaldehyd
Pyrrol
Butanal
Benzol
44
67
72
Benzol
Pyridin
78
79
Toluol
92
Anilin
Phenol
Benzonitril
93
94
103
MCB
112
78
6-Caprolactam 113
Indol
117
Pyrimidin
123
2-Amino 4,6
Naphthalin
128
Chinolin
Biphenyl
Dibenzofuran
Phenanthren
Pyren
129
154
168
178
202
103
5.10 Experimente zur Spezifizierung des Massenspektrometers
Nachfolgend beschriebene Experimente wurden durchgeführt, um das Massenspektrometer und seine neu entwickelten Komponenten zu spezifizieren. Es werden die
Experimente beschrieben und die ermittelten Spezifikationsdaten aufgeführt.
5.10.1 VUV-Erzeugung in der Edelgaszelle
Entscheidend für die Umwandlung der 355 nm-Pumpstrahlung in 118 nm-VUVStrahlung in der Edelgaszelle ist der Xenon-Druck in der Zelle. Nur in einem kleinen
Xe-Druckbereich lässt sich VUV-Strahlung effektiv erzeugen. Neben dem Xe-Druck
spielt die Reinheit des Xenons eine bedeutende Rolle. Verunreinigungen können schon
bei relativ niedrigen Energiedichten im 355 nm-Fokus Gasdurchbrüche hervorrufen. Ein
weiterer Grund für die Verwendung besonders reinen Xenons ist die Absorption von
VUV-Licht durch Verunreinigungen, wie z.B. Sauerstoff.
Trotz Adsorptionsgetter (siehe Kapitel 6.1), gereinigtem Edelstahl, Metalldichtungen
und wenig ausgasenden Dichtungen aus Perfluorelastomer (Kalrezâ) für die Fenster,
muss die Zelle alle paar Monate neu mit Xenon der Reinheit 5.0 oder höher befüllt werden. Das verwendete Xenon (Fa. Messer Griesheim, Krefeld, Deutschland) wurde in
einer 10 bar-Druckdose mit 1 l Inhalt geliefert. Bei einem Zellenvolumen von ca. 1 l und
einem Xe-Arbeitsdruck von einigen mbar reicht diese Menge für ca. 100 Befüllungen.
Um nicht schon beim Befüllen die Zelle zu kontaminieren, wurde ein spezielles Ventilund Rohrleitungssystem aufgebaut, bei dem alle Volumina evakuiert werden können,
bevor sie mit Xe gefüllt werden (siehe Abbildung 40). Dieses System besteht aus 4 Ventilen (A, C, D und E), wobei das Ventil A einen großen Rohrleitungsquerschnitt hat, um
die THG-Zelle möglichst gut durch das Vakuum der Ionenquelle abpumpen zu können.
Ventil B ist ein Absperr- und Reduzierventil, mit dem dosiert werden kann.
Zum Abpumpen der Zelle öffnet man die Ventile A und B und senkt langsam den Druck
in der Zelle. Bei einem Druck von unter 10-1 mbar kann das große Absperrventil A geöffnet werden, ohne eine große Druckwelle in der Ionenquelle zu erzeugen. Noch
104
Kapitel 5
Abbildung 40: Rohrleitungs- und Ventil-Plan zum Befüllen und Evakuieren der THG-Zelle.
günstigere Bedingungen für das Befüllen mit Xenon werden dadurch geschaffen, dass
während Ventil A offen ist, die THG-Zelle mit einem Heizband erwärmt wird.
Zum Befüllen vergewissert man sich, ob alle Ventile geschlossen sind. Dann werden
Ventil A und E geöffnet, um das Leitungssystem zu evakuieren. Danach werden beide
Ventile wieder geschlossen. Öffnet man anschließend Ventil D, gelangt eine kleine
Menge Xenon in die Leitung zwischen Ventil D und E. Dann wird D wieder geschlossen und durch Ventil E und B das Xenon in die Zelle gelassen. Diese kleine Menge an
Xenon reicht aus, um die Zelle zwischen 50 mbar und 100 mbar zu füllen. Alle Ventile
werden anschließend wieder geschlossen.
Während der Versuche zeigte es sich, dass es am günstigsten ist, den optimalen XeDruck in der Zelle nicht beim Befüllen sondern durch Abpumpen einzustellen. Wenn
Ventil C und das Regulierventil B etwas geöffnet werden, kann das Xenon langsam und
kontrolliert entweichen. Ist der ideale Druck erreicht, werden alle Ventile geschlossen.
Der optimale Xe-Druck für die THG-Zelle wurde durch Messungen an einem Standardgemisches aus 10 ppm Benzol, Toluol und p-Xylol (BTX) ermittelt. Das Standardgas
wurde permanent eingelassen und mittels VUV-SPI analysiert. Da die Ionenzahl proportional zur Fläche unter den Signalpeaks des SPI-Spektrums ist, wurde die Peakfläche
und damit die Ionenzahl über dem Xe-Druck aufgetragen. Der Druck wurde mit einer
gasartunabhängigen Druckmessröhre (Typ CMR 261, Fa. Pfeiffer, Asslar, Deutschland)
105
Abbildung 41: SPI-Ionenausbeute in Abhängigkeit vom Xe-Druck in der THG-Zelle. Bei den
zwei Messungen wurden jeweils 10 ppm Benzol, Toluol und p-Xylol in die Ionenquelle eingelassen. Um die Anzahl der erzeugten Ionen zu ermitteln, wurde über die Fläche unter den
Signalpeaks integriert.
aufgenommen. Die Ergebnisse sind in Abbildung 41 dargestellt. Aufgrund der Ergebnisse dieser Messungen sollte die THG-Zelle mit ca. 8 mbar Xenon betrieben werden.
Es ist zu erwähnen, dass bei einer ähnlichen Edelgaszelle mit einem Laser mit weniger
Energie und einem anderen Strahlprofil die maximale VUV-Erzeugung bei einem XeDruck von 12 – 14 mbar erreicht wurde. Daraus ist zu schließen, dass aufgrund der unterschiedlichen Fokusbedingungen der Xe-Druck an jeden Pumpstrahl angepasst werden
muss. Außerdem sei noch vermerkt, dass durch Heizen der Getterpumpe zum Zwecke
der Adsorption die Temperatur in der Zelle langsam ansteigt und sich damit auch der
Xe-Druck erhöht. Während des Laserbetriebes sollte daher die Getterpumpe nicht beheizt werden.
Neben dem Xe-Druck spielt die 355 nm-Pumpenergie eine entscheidende Rolle. In
Abbildung 42 ist die relative Zahl der mit SPI erzeugten 10 ppm-BTX-Ionen gegenüber
der eingestrahlten 355 nm-Energie dargestellt. Dem Verlauf der Ionenzahl von Toluol
und Xylol ist eine links oben in der Abbildung dargestellte Funktion mit einer speziellen
106
Kapitel 5
Software angepasst worden (Originâ, Fa. Microcalä, Northampton, USA). Das diese
Funktion eine kubische Abhängigkeit der Ionenzahl und damit auch der VUVPhotonenzahl beschreibt, wird nach Gleichung 4,12 erwartet. Mit dieser Formel lässt
sich in etwa vorhersagen, wie sich die Ionenausbeute bei höherer 355 nm-Energie verhält. Zu bedenken ist jedoch, dass die 355 nm-Energie nicht beliebig erhöht werden
kann. Ab einer bestimmten Energiedichte finden auch bei reinem Xenon im Fokalbereich Gasdurchbrüche statt.
Abbildung 42: SPI-Ionensignal von 10 ppm Benzol, Toluol und p-Xylol in Abhängigkeit von
der Intensität der 355 nm-Pumpstrahlung. Die gepunktete Linie ist mit Hilfe einer Fitfunktion
erstellt worden und zeigt die kubische Abhängigkeit der VUV-Erzeugung von der Pumpenergie.
Bei der Messung in Abbildung 42 ist zu beachten, dass die Energieänderung durch
Glasplättchen, wie sie für Mikroskope verwendet werden, erzeugt wurde. Für die Messung stand ein 14 mJ 355 nm-Laserstrahl zu Verfügung, der durch Einlegen der
Glasplättchen geschwächt wurde. Durch Reflexion beim Eindringen in jedes Plättchen
verliert der Strahl schrittweise an Energie. Da das Reflexionsverhalten auch etwas energieabhängig ist, verändert man durch diese Methode das Strahlprofil eines Strahls mit
gaussförmiger Energieverteilung. Durch ein verändertes Strahlprofil ändern sich auch
die Fokusbedingungen in der Zelle und damit auch die VUV-Erzeugung.
Wieviel VUV-Photonen in der Zelle bei den verschiedenen 355 nm-Energien erzeugt
wurden, kann nicht genau angegeben werden. In der Literatur wird von EnergieumUm-
107
wandlungseffizienzen von 10-4 bis 10-5 gesprochen [27, 99]. Bei 20 mJ/Puls 355 nmStrahlung -soviel stand für die VUV-Erzeugung bei dem hier beschriebenen Gerät maximal zur Verfügung- ergibt dies 200 nJ/Puls bis 2 mJ/Puls und damit 1011 bis 1012
118 nm-Photonen pro Puls.
Der Versuch, die VUV-Strahlung mit einem Halbleiterdetektor (siehe Kapitel 4.3.1)
hinter der Ionisationszone zu messen, scheiterte an der um Größenordnungen höheren
Intensität an 355 nm-Strahlung in der Nähe des VUV-Strahls. Trotz Trennung der
355 nm- und 118 nm-Strahlen war es aufgrund von diffuser 355 nm-Strahlung nicht
möglich, die VUV-Strahlung zu quantifizieren. Mit einer geeichten Ionisationszelle
(Kapitel 4.3.5) sollte dieses in Zukunft jedoch möglich sein.
5.10.2 Einphotonenionisation (SPI)
Zur Spezifikation eines neuen TOFMS-Systems gehören z.B. die erreichbaren Nachweisgrenzen und die Massenauflösung. In dieser Arbeit wird die Nachweisgrenze (d)
nach der Formel [5.7] von Williams et al. [50] berechnet. Es wird auch davon ausgegangen, dass eine Substanz nachgewiesen werden kann, wenn ihr Signalpeak 3 mal
größer als das Rauschen auf der Grundlinie ist (S/N > 3). Das Rauschen wird dabei über
die Standardabweichung (s) in einen Bereich neben dem Massenpeak ermittelt, in dem
sich kein Massenpeak befindet (siehe Abbildung 43). Der Mittelwert (m) des Signals in
diesem Bereich liefert das Grundlinienniveau, das von der Signalpeakhöhe (p) subtrahiert werden muss, um die absolute Höhe des Signals zu erhalten.
3s
d=
c.
p-m
[5.7]
Für die Messungen zur Bestimmung der Nachweisgrenzen stand zum einen ein Standardgas aus der Flasche mit 10 ppm-Benzol, Toluol und p-Xylol (BTX), später auch
noch mit 10 ppm Dekan, zur Verfügung und zum anderen ein in einem Standardgasgenerator (siehe Kapitel 5.9) erzeugtes Gasgemisch mit mehreren Komponenten in
niedrigerer Konzentration (vgl. Tabelle 5).
Abbildung 43 zeigt ein 118 nm-SPI-Massenspektrum des 10 ppm-BTX-Standards. Die
Grundlinie liegt hier bei –0,67 mV und die Streuung bzw. das Rauschen bei 1,2 mV.
Aus den Signalhöhen und der Formel [5.7] ergeben sich die in Abbildung 43 und in
108
Kapitel 5
Abbildung 43: SPI-Massenspektrum eines 10 ppm-BTX-Standardgases. Angegeben ist die
Signalhöhe und die aus der Standardabweichung und Grundlinienhöhe errechnete Nachweisgrenze (NWG) bei einer Mindestsignalhöhe von 3 mal dem Grundlinienrauschen.
Tabelle 5 dargestellten Nachweisgrenzen (NWG). Für das Spektrum in Abbildung 43
wurden 5 Einzelspektren zusammengefasst. Da die Einzelspektren mit 10 Hz erzeugt
werden, benötigt man für diese Nachweisgrenzen eine Aufnahmezeit von 0,5 s.
In Abbildung 44 ist das Massenspektrum abgebildet, mit dem der überwiegende Rest
der NWGs von Tabelle 5 ermittelt wurde. Etwas schwierig erwies sich die Ermittlung
der NWG von Pyridin. Benzol mit einem 13C-Atom (6,6 % aller Benzol-Moleküle), wie
auch Pyridin haben die Masse 79 m/z. Aus dem Spektrum in Abbildung 44 kann der
Pyridin-Anteil des 79 m/z-Peaks abgeschätzt werden, indem man das Peakverhältnis
(78 m/z / 79 m/z) von Benzol aus Abbildung 43 nimmt und danach den Pyridinanteil
des 79 m/z-Peaks in Abbildung 44 abschätzt.
Abbildung 44: SPI-Massenspektrum einer Standardgasmischung zur Bestimmung einzelner
Nachweisgrenzen.
109
Tabelle 5: SPI-Nachweisgrenzen (NWG) einiger Substanzen, die in bekannter Konzentration zur Verfügung standen. Aufgelistet ist auch die Konzentration der Substanzen im
Standardgas bei der Messung und die Anzahl der Spektren, die zusammengefasst wurden,
um die NWG zu ermitteln.
Substanz
Masse
IP
Konzentration im
gemittelte
Nachweisgrenze
Standard
Spektren
bei S/N > 3
Ammoniak
17 amu
10,2 eV 2,2 ppm
100
100 ppb
Pyrrol
67 amu
8,20 eV 1,0 ppm
100
4 ppb
Benzol
78 amu
9,24 eV 10,0 ppm
5
45 ppb
1,4 ppm
100
2 ppb
Pyridin
79 amu
9,26 eV 0,5 ppm
100
~ 3* ppb
Toluol
92 amu
8,5 eV
10,0 ppm
5
60 ppb
0,4 ppm
100
3 ppb
7,72 eV 0,9 ppm
100
3 ppb
Anilin
93 amu
Benzonitril
103 amu
9,73 eV 0,02 ppm
100
1 ppb
p-Xylol
106 amu
8,4 eV
5
60 ppb
MCB
112 amu
9,07 eV 4,0 ppm
100
10 ppb
Indol
117 amu
7,76 eV < 0,02 ppm
100
< 5 ppb
Naphthalin
128 amu
8,12 eV 0,03 ppm
100
5 ppb
Chinolin
129 amu
8,63 eV 0,03 ppm
100
4 ppb
Dekan
142 amu
9,65 eV 10,0 ppm
5
40 ppb
*
10,0 ppm
Die Nachweisgrenze von Pyridin wurde nach Substraktion des 13C-Anteils von Benzol ermittelt.
Das Spektrum der Abbildung 44 stellt den Mittelwert von 100 Einzelspektren dar. Um
die damit erhaltenen NWGs zu erzielen, muss also mindestens 10 s lang gemessen werden. Geht man davon aus, dass das Rauschen auf der Grundlinie statistischen Ursprungs
ist, so verbessert sich die NWG mit der Wurzel der Anzahl von gemittelten Spektren.
Verwendet man 100 Spektren anstelle von 5 Spektren, verbessert sich damit die Nachweisgrenze um den Faktor 4,5.
Würde man in Abbildung 43 die Ionensignale nicht über der Massenachse sondern über
der Flugzeitachse darstellen, so würde der p-Xylol Peak bei t = 16,23 ms liegen und auf
halber Höhe eine Breite von DtFWHM = 4,5 ns haben. Errechnet man damit die Massenauflösung (R50%) mit [5.8], ergibt sich eine R50% von ca. 1800 m/z.
R50% =
t
2Dt FWHM
[5.8]
110
Kapitel 5
Ermittelt man die Massenauflösung mit Hilfe eines schwächeren Ionensignals, wie z.B.
mit dem MBC-Peak in Abbildung 44, so erhält man das vergleichbare Ergebnis
R50% = 1900.
5.10.3 Resonanzverstärkte Mehrphotonenionisation (REMPI)
Um die REMPI-Nachweisgrenzen der verschiedenen Substanzen in einem Standardgas
zu bestimmen, bedarf es mehrerer Messungen. Schließlich hat jede Substanz ihren maximalen
REMPI-Wirkungsquerschnitt
bei
einer
anderen
Wellenlänge
(vgl.
Abbildung 45 rechts). Bezüglich des REMPI-Wirkungsquerschnitts ist aber noch zu
beachten, dass der Laser bei verschiedenen Wellenlängen verschiedene Energien liefert.
Da beim REMPI-Prozess zwei Photonen benötigt werden, hängt die Ionisationsausbeute
quadratisch von der Energiedichte am Ionisationsort ab. Die an der GSF mit einem besonders schmalbandigen durchstimmbaren Laser aufgenommenen Wellenlängenspektren in Abbildung 45 (rechts) [40] wurden daher auf eine Laserenergie normiert.
Will man die Nachweisgrenze (NWG) [5.8] des Geräts für eine bestimmte Substanz
ermitteln, kann es sinnvoll sein, nicht mit der Wellenlänge zu ionisieren, für die das
Molekül den größten REMPI-Wirkungsquerschnitt hat, sondern bei einer Wellenlänge
mit einem Optimum an Energie und Wirkungsquerschnitt.
In Abbildung 45 (links) sind zwei Massenspektren desselben Standardgases bei zwei
verschiedenen Wellenlängen des Ionisationslasers dargestellt. In den Wellenlängenspektren von Abbildung 45 (rechts) sind die zwei verschiedenen Wellenlängen, bei
denen das Standardgas gemessen wurde, mit gepunkteten Linien eingezeichnet.
Die scharfen Massenpeaks in Abbildung 45 (links) unterhalb von 70 m/z sind Fragmente von Anilin und haben eine maximale Signalhöhe von 1,2 % des Hauptsignals bei
93 m/z. Zum Vergleich: Bei 70 eV-EI hätte der Peak bei 66 m/z eine relative Höhe gegenüber dem Hauptpeak von 32 % [64]. Bei Verringerung der Energiedichte am
Ionisationsort, würden auch die Signale bei 65 m/z und 66 m/z verschwinden. Kann dies
durch Aufweiten des Strahls und damit größerem Strahldurchmesser in der Ionisationszone und demzufolge größerem Ionisationsvolumen erreicht werden, so ist kein Verlust
an Signalhöhe beim Hauptpeak zu erwarten.
111
Abbildung 45: Zwei REMPI-Spektren des Gasstandards bei zwei verschiedenen Wellenlängen (links). Je nach Wellenlänge erhält man eine andere Sensitivität für das im Standard
enthaltenen Benzol (78 m/z), Toluol (92 m/z), Anilin (93 m/z), Indol (117 m/z) und Naphthalin (128 m/z). Rechts sind die Wellenlängenspektren von einigen der Substanzen dargestellt,
die die relative REMPI-Ionisierbarkeit in Abhängigkeit von der Wellenlänge zeigen.
Die leichteste Substanz im Standard war Benzol in einer Konzentration von 1,4 ppm
(vgl. Tabelle 6). Wie man am Benzol-Wellenlängenspektrum in Abbildung 45 (rechts)
sieht, kann Benzol besonders gut bei 259 nm mit REMPI ionisiert werden. Aus diesem
Grund ist Benzol auch nur im Massenspektrum mit 259 nm-Strahlung sichtbar. Toluol
lässt sich auch mit 259 nm-Strahlung gut ionisieren. Im 290 nm-Spektrum resultiert das
Signal bei 92 m/z (~1 % von 93 m/z) aus der Wasserstoffabspaltung des Anilins
(93 m/z). Wie aus dem Wellenlängenspektrum ersichtlich ist, wird Anilin bei 290 nm
gut ionisiert. Bei einer Messung mit 283 nm-Licht wäre die NWG von Anilin, wie in
112
Kapitel 5
Tabelle 6: REMPI-Nachweisgrenzen (NWG) einiger Substanzen, die in bekannter Konzentration zur Verfügung standen. In der Tabelle sind außerdem die Konzentration der
Substanz im Standardgas, die Wellenlänge, bei der die NWGs ermittelt wurden, und die
Anzahl der Spektren aufgelistet, die zur Ermittlung der NWG zusammengefasst wurden.
Substanz
Masse
gemessen bei Konzentration im gemittelte
Nachweisgrenze
Benzol
78 amu
Wellenlänge
Standard
Spektren
bei S/N > 3
259 nm
1,4 ppm
100
3 ppb
Toluol
92 amu
259 nm
0,3 ppm
100
4 ppb
Anilin
93 amu
290 nm
0,8 ppm
100
350 ppt
p-Xylol
106 amu
266 nm
10 ppm
20
25 ppb
Indol
117 amu
280 nm
< 0,03 ppm
100
< 600 ppt
Naphthalin
128 amu
290 nm
0,02 ppm
100
2 ppb
Tabelle 6 angegeben, noch besser. Bei Indol (117 m/z) ist das Signal erwartungsgemäß
bei 259 nm größer. Naphthalin (128 m/z) verhält sich auch entsprechend seines Wellenlängenspektrums und ist im 290 nm-Spektrum nicht zu erkennen.
Sämtliche REMPI-NWGs, die mit dem hier beschriebenen Gerät ermittelt werden konnten, sind in Tabelle 6 aufgeführt. Diese NWGs können bei anderen Wellenlängen, bei
denen das Verhältnis zwischen Laserenergie und Wirkungsquerschnitt günstiger ist,
durchaus noch niedriger liegen.
Es sei angemerkt, dass diese NWGs unter günstigen Umständen im Labor ermittelt
wurden. Bei weniger günstigen Umgebungsbedingungen, wie bei den Messungen an
einer Müllverbrennungsanlage, waren die erzielten NWGs bis zu einer Größenordnung
schlechter. Dies betraf besonders den REMPI-Prozess, da hier die Laserenergie quadratisch in die Ionenausbeute eingeht und der Laser unter solchen Feldbedingungen schwer
zu justieren ist.
5.10.4 Elektronenstoßionisation (EI)
Für die Elektronenstoßionisation befindet sich an der Ionenquelle des Massenspektrometers eine Elektronenkanone in der durch Glühemission Elektronen frei gesetzt
werden. Die Elektronen werden in einem elektrostatischen Feld auf eine bestimmte kinetische Energie beschleunigt und glangen anschließend zur Ionisationszone. Durch
113
Stöße mit den Gasmolekülen wird kinetische Energie der Elektronen auf die Moleküle
übertragen, wodurch es zur Ionisatation kommen kann (vgl. Kapitel 3.3).
Das Einstellen der Elektronenstoßionisation gestaltet sich bei diesem Kombi-Gerät besonders schwierig. Grund dafür ist, wie zuvor beschrieben, der hohe Gasdruck am
Ionisationsort in Kombination mit einem Vielkanal-Analysator. Der hohe Gasdruck
wird von der Einlassnadel verursacht, die das Probengas kurz vor der Ionisationszone
ins Vakuum entlässt. Der Vielkanal-Analysator ist notwendig zur Aufnahme der Spektren, die mit bis zu 20 kHz erzeugt werden.
Bei kurzen Messzyklen, wie man sie zur Bestimmung von dynamischen Prozessen benötigt, wird der Elektronenfluss aus der Elektronenkanone mit dem Heizstrom so
eingestellt, dass z.B. eine Substanz in einer Konzentration von 100 ppb relativ häufig
ein Einionensignal, aber keine Mehrionensignale erzeugt. Eine Substanz in einer Konzentration von 10 % und einem ähnlichen EI-Wirkungsquerschnitt verursacht bei
denselben Einstellungen ein 106-fach so intensives Signal, das aber im entsprechenden
Kanal des Vielkanalanalysators genauso als ein Ereignis gezählt wird wie ein einzelnes
Ion. Der Wert, den der Vielkanalanalysator für eine konzentrierte Substanz ausgibt, ist
daher ohne quantitativen Wert. Diese hohen Signale können durch ihre steilen Flanken
zu elektrischen Schwingungen in den Signalkabeln führen, deren Amplituden als schwerere Moleküle von der Datenerfassung erkannt und gewertet werden. In Abbildung 47 B
verursacht der Stickstoff bei 28 m/z ein so starkes Signal, dass es bis 31 m/z noch zu
elektronischen Störungen kommt.
Tabelle 7: Hauptbestandteile der Luft mit deren Konzentrationen
Gas
Molare Masse
Volumenanteil
Stickstoff (N2)
28 amu
78,1 %
Sauerstoff (O2)
32 amu
21,0 %
Argon (Ar)
40 amu
0,9 %
Kohlendioxid (CO2)
44 amu
340 ppm
Neon (Ne)
20 amu
18 ppm
Zum Einstellen der Heizspannung der Elektronenkanone und damit des Elektronenstroms eignet sich Luft sehr gut. Wie in Tabelle 7 dargestellt, enthält Luft eine Reihe
von Substanzen in unterschiedlichen Konzentrationen. Zur Messung einer Substanz im
100 ppm-Konzentrationsbereich, ist es z.B. sinnvoll, die Parameter des Gerätes so zu
setzen, dass CO2 mit einer Konzentration von 340 ppm in Luft gut detektiert wird. In
114
Kapitel 5
den EI-Spektren der Abbildung 46 A und Abbildung 47 A, die jeweils Summenspektren
von 100.000 Einzelspektren sind, ist Argon (40 m/z) und CO2 (44 m/z) gut zu erkennen,
in Abbildung 47 A sogar schwach das Neon (20 m/z).
Ob die vom Vielkanalanalysator ausgegebenen Werte auf Ein- oder MehrionenEreignissen beruhen, ist nicht ohne weiteres zu erkennen. Zur Überprüfung kann die
Anzahl von Ereignissen, aus denen sich ein Massenpeak zusammensetzt, bestimmt werden. Setzt sich ein Massenpeak aus einer Anzahl von Ereignissen zusammen, die größer
ist als 70 % der Anzahl der für das Spektrum verwendeten Einzelspektren, so kann mit
einer gewissen statistischen Wahrscheinlichkeit davon ausgegangen werden, dass zumindest in einigen Fällen mehr als ein Ion einer Sorte den Ionendetektor während der
Aufnahme für ein Einzelspektrum erreicht hat. Aus diesem Grund ist dann ein quantitativer Vergleich zwischen zwei Messungen nicht möglich.
Abbildung 46: 70 eV-EI-Massenspektrum von Benzol in Luft (A). Zum Vergleich dazu das
70 eV-EI-Massenspektrum von NIST (B). Wenn die Fragmentmuster identisch sind, sind der
Elektronenstrom und die Vielkanalanalysator-Karte richtig eingestellt und die Massenpeaks
von (A) werden durch Aufaddieren von Einzelionen erzeugt. Bei der Messung des Spektrums
(A) wurde N2 durch den Massenfilter unterdrückt. Das starke Rauschen in (A) von 0 bis
10 m/z resultiert aus den Hochspannungspulsern des Massenfilters.
Neben der Kontrolle der Höhe der Massenpeaks kann man das Fragmentmuster einer
Substanz auch dazu verwenden, die Einstellungen der Elektronenkanone zu kontrollieren. Wenn das Fragmentmuster einer Substanz z.B. mit dem Fragmentmuster der
Substanz vom National Institute of Standards and Technology (NIST) [64] bei gleicher
Elektronenenergie vergleichbar ist, sind die Einstellungen der Ionenquelle günstig. In
Abbildung 46 ist das Ergebnis einer 70 eV-EI-Benzolmessung dargestellt (links) und
mit dem 70 eV-Fragmentmuster von NIST (rechts) verglichen. Da bei dieser Messung
Benzol in Luft gemessen wurde, können auch die Bestandteile von Luft, wie H2O, O2,
115
Abbildung 47: 23 eV-EI-Massenspektren von Luft (A) und 1000 ppm HCN in N2 (B). Die
Signale im Massenbereich von 28 bis 31 m/z werden durch den hohen N2-Anteil hervorgerufen.
Ar und CO2 erkannt werden. Stickstoff wurde bei dieser Messung mit Hilfe eines Massenfilters unterdrückt.
Die Elektronenstoßionisation wurde für die Messungen an einer industriellen Müllverbrennungsanlage sehr wichtig, weil mit ihr Cyanwasserstoff (HCN) bis in den
unteren ppm-Bereich nachgewiesen werden konnte. Mit SPI konnte HCN wegen des
hohen Ionisationspotenzials von 13,60 eV nicht ionisiert werden. Auch wurde keine
UV-Wellenlänge gefunden, um HCN mit Einfarben-REMPI zu ionisieren.
Aus der in Abbildung 47 B dargestellten Messung von 1000 ppm HCN in N2 ergab sich
aus 100.000 Einzelspektren eine NWG von 5 ppm. Die Elektronen hatten bei dieser
Messung eine kinetische Energie von 23 eV. Für die Analyse von HCN ist diese niedrige Elektronenenergie vorteilhaft, da der EI-Wirkungsquerschnitt von N2 gegenüber z.B.
HCN unterhalb von 30 eV überproportional abfällt.
Bei allen vorangegangenen EI-Messungen wurde die Elektronenkanone mit relativ geringer Heizspannung betrieben, um nicht zu viele Ionen pro Schuss der
Elektronenkanone (pro Transient) zu erzeugen. Bei Reduktion der Repetitionsrate der
Elektronenkanone auf etwa 100 Hz und gleichzeitig maximalem Elektronenstrom können die Ionensignale auch mit einem Transientenrekorder oder Oszilloskop
aufgenommen werden.
Für die in Abbildung 48 dargestellten Transientenrekorder-Messungen wurde wieder ein
Massenfilter verwendet, um Stickstoff bei 28 m/z zu unterdrücken. Trotz des Massenfil-
116
Kapitel 5
ters gelangt noch eine große Anzahl von N2-Ionen zum Detektor. Dennoch war es möglich, HCN auch mit dieser Methode zu messen (siehe Abbildung 48 A). Die NWG für
HCN lag bei 50 ppm und wurde bei einer Messdauer von 1 s erreicht. Die Spektren in
Abbildung 48 sind das Resultat von 100 Einzelspektren. Abbildung 48 B stellt zum
Vergleich die Messung von reiner Luft unter denselben Bedingungen dar.
Abbildung 48: 20 eV-EI-Massenspektren von Stickstoff mit 1000 ppm HCN (A) und ohne
HCN (B). Die Spektren wurden nicht mit dem Vielkanalanalysator, sondern mit der Transientenrekorder-Karte aufgenommen. Die Anzahl der Stickstoffionen (28 m/z) ist durch den
Massenfilter reduziert.
5.10.5 Massenfilter
Typischerweise werden für den zeitaufgelösten Nachweis von Ionen im TOFMS-System
MCPs (engl.: multi-channel plates) verwendet. Diese Detektoren verstärken intern
durch einen Sekundäreffekt die Ionensignale. Der Verstärkungsfaktor eines MCPDetektors ist abhängig von der angelegten Betriebsspannung. Beim Nachweis von Ionen
fließt kurzzeitig ein Strom im MCP-Detektor. Wenn eine sehr große Anzahl von Ionen
gleichzeitig auf den Detektor trifft, wie dies für die Benzol-Ionen (78 m/z) bei der Messung für Abbildung 49 A1 der Fall war, sinkt durch den Stromfluss im Detektor die
anliegende Spannung kurzzeitig ab. Dies bewirkt, dass für nachfolgende schwerere Ionen der Detektor mit einem unbekannten, geringerem Verstärkungsfaktor arbeitet. Wenn
man Abbildung 49 A1 mit A2 vergleicht, sieht man, dass durch das große BenzolIonensignal in A1 höhere Massen kaum mehr nachgewiesen werden können. In
Abbildung 49 A2 wurde durch den Massenfilter verhindert, dass die Benzol-Ionen zum
117
Detektor gelangen, was zu einem besseren Nachweis der schwereren Substanzen dieser
Öldestillationsprobe führte.
Beim Vergleich der Messung in Abbildung 49 B1, in der das Benzol nur ein Signal von
1 V verursachte, mit der Messung in B2, bei der Benzol nicht zum Detektor gelangte,
wird deutlich, dass die schwereren Ionen in beiden Fällen gleich effektiv nachgewiesen
werden.
Abbildung 49: Messung eines Rohöl-Destillationsproduktes mit sehr hohem Benzolanteil.
Bei Messung A1 verursacht das Benzol ein Ionensignal von 3,5 V. Wird die Probe unter denselben Bedingungen gemessen, aber mit Unterdrückung der Benzol-Ionen durch den
Ionenfilter, erkennt man in A2 den negativen Einfluss des hohen Signals auf die Detektion
der schwereren Ionen. Wenn das maximale Ionensignal nur eine Höhe von 1 V (B1) erreicht,
werden die nachfolgenden Massen nicht beeinflusst (B2).
Bei den von der Firma Kaesdorf in das TOFMS eingebauten MCP-Detektoren (Typ
CHEVRON S3040-10-D, Fa. Burle, Lancaster, USA) musste dafür gesorgt werden, dass
einzelne Ionensignale nicht über 1 V anstiegen, um nachfolgende Massen nicht undefiniert zu beeinflussen. Mit Hilfe des Massenfilters kann somit der dynamische Bereich
des TOFMS erhöht werden. Dies ist besonders dann vorteilhaft, wenn trotz geeignetem
Ionisationsmechanismus andere, ebenfalls vorkommende Substanzen um Größenord-
118
Kapitel 5
nungen höhere Signale verursachen als die Substanzen, die im Zentrum des Interesses
liegen.
Theoretisch wäre es auch denkbar, mit dem Massenfilter nur einen konstanten Anteil
von Ionen einer Sorte vom Detektor fern zu halten und damit das Signal von Ionen selektiv und definiert zu schwächen. Versuche zu dieser Art der Anwendung eines
Massenfilters, speziell zum linearen Intensitätsverhalten, stehen noch aus.
Abbildung 50: EI-Spektren von Luft mit Wasserdampf. Durch die Elektronenenergie von
70 eV fragmentieren N2 und O2 in der Ionisationszone zu N und O. Durch den Massenfilter
kann man die atomaren Sauerstoffionen in Messung B vom Detektor fernhalten, ohne die
Nachbarmassen zu beeinflussen. Messung A wurde zum Vergleich ohne Massenfilter durchgeführt. Bei Messung C wurden die Wasserionen vom Detektor ferngehalten.
Wie in Abschnitt 5.7 beschrieben, lassen sich mit dem Massenfilter einzelne Ionen einer
Masse besonders scharf ausblenden, wenn der 1. Ortsfokus (siehe 5.7) der Ionen im
Massenfilter liegt. Bei der EI-Ionisation fragmentieren N2 (28 amu) und O2 (32 amu) in
der Ionisationszone zu 2N (14 amu) und 2O (16 amu). Außerdem lässt sich H2OWasserdampf (18 amu) mit EI nachweisen.
In Abbildung 50 A sind die Massenpeaks von N, O, und H2O bei 14, 16 und 18 m/z zu
erkennen. Mit dem Massenfilter konnte nach Optimierung der Abzugsspannungen in der
Ionenquelle das O-Signal bei 16 m/z unterdrückt werden, ohne die beiden Nachbarsignale bei 14 und 18 m/z zu beeinflussen (siehe Abbildung 50 B). In Abbildung 50 C
wurde H2O eliminiert, ohne Einfluss auf die anderen Signale zu nehmen.
119
5.11 Probenahme
Zur Probenahme gehören alle Teile, die benötigt werden, um das Analysengas vom Entnahmeort zum Massenspektrometer zu transportieren. In der Regel werden eine
Probenahmesonde, eine Partikelfiltereinheit und eine Verbindungsleitung benötigt. Es
ist sicherzustellen, dass das Messgas möglichst schnell und ohne chemische Veränderungen zum Analysator gelangt.
Zur Messung komplexer und korrosiver Gase stark unterschiedlicher Flüchtigkeit, wie
sie z.B. im Feuerraum einer Müllverbrennungsanlage vorkommen, unter Vermeidung
von Memory-Effekten, sollte die Probenahmestrecke vollständig aus inertem Quarzglas
aufgebaut und mindestens auf 250 °C beheizbar sein.
Es hat sich gezeigt, dass bei Temperaturen unterhalb von 250 °C schwerflüchtige teerartige Substanzen im Probenahmesystem kondensieren. Dieses Kondensat würde über
einen längeren Zeitraum Substanzen freisetzen, so dass es zu Interferenzen kommen
könnte.
Zur kontinuierlichen Entnahme einer Gasprobe aus einem Feuerraum oder Reaktor wird
eine Probenahmesonde verwendet. Diese Sonde, die nachfolgend detailliert beschrieben
wird, übergibt das Gas an eine Partikelfiltereinheit. Das Abtrennen der Partikel ist notwendig, da das Gas anschließend durch eine biegsame dünne Quarzglaskapillare zum
Massenspektrometer strömt, die sonst verstopfen würde. Der Ort und das Temperaturniveau des Filters muss geschickt gewählt werden, damit sich nach dem Filter keine neuen
Partikel bilden und bei der relativ niedrigen Temperatur der Probenahmestrecke Zielsubstanzen nicht von Partikeln adsorbiert werden. Der Partikelfilter besteht aus
gepresster Quarzglas-Wolle und ist zur Oberflächenvergrößerung zylindrisch geformt.
Das Probengas wird mit Hilfe einer Saugpumpe mit einem Volumenstrom von
2 − 5 l/min durch den Filter gezogen. Dieser relativ hohe Fluss ist notwendig, um das
Gasvolumen des Filtergehäuses und der Glasteile der Sonde möglichst schnell auszutauschen.
Der
geringe
Teilstrom
durch
die
unbelegte
Quarzkapillare
zum
Massenspektrometer reicht dazu nicht aus. Der Fluss durch die Kapillare wird bedingt
durch das Vakuum des Massenspektrometers am anderen Ende der Probnahme. Mit dem
Durchmesser der Kapillare wird der Fluss zum TOFMS festgelegt. Der Fluss in das
Massenspektrometer betrug bei einer 2 m langen Kapillare mit Innendurchmesser (ID)
120
Kapitel 5
0,32 mm bei 250 °C 6,7 ml/min. Er verhält sich aufgrund der Brownschen Bewegung
der Gasmolekühle in Abhängigkeit von der Temperatur wie in Abbildung 51 dargestellt
und beeinflusst damit auch die Güte des Vakuums in der Ionenquelle. Geschützt und
temperiert wird die Transferkapillare durch einen Heizschlauch (Fa. Horst, Lorsch,
Deutschland).
Zwischen Filter und Massenspektrometer kann zur Steigerung der Empfindlichkeit je
nach Bedarf noch ein Thermodesorber eingefügt werden. Ein solch InlineThermodesorber zum Anreichern und schnellen Desorbieren ist nachfolgend beschrieben (Kapitel 7.1).
Abbildung 51: Fluss des Probengases durch eine 2 m lange Kapillare (ID 0,32 mm) bei verschiedenen Temperaturen und die sich dadurch ergebenden Drücke in der Ionenquelle
121
5.12 Hochtemperatursonde
Ein Einsatzgebiet des beschriebenen Laser-TOFMS sind Messungen von Müllverbrennungsrauchgasen
(siehe
Kapitel
5.13.3).
Für
die
Entnahme
von
heißen
Verbrennungsgasen aus dem Feuerraum einer Müllverbrennungsanlage (MVA) wurde
eine spezielle Probenahmesonde entwickelt und erfolgreich getestet (Abbildung 52).
Eine derartige Sonde wird benötigt, um die Probe bei Temperaturen von über 1300 °C
in ausreichender Entfernung von der Kesselwand zu entnehmen. Im Bereich nahe der
Kesselwand unterscheidet sich die Abgaszusammensetztung von der in der Mitte des
Kessels. Gas, das an der Wand entlang aufsteigt, erreicht nicht dieselben Temperaturen
wie das Gas direkt über dem Feuer, was zu unterschiedlichen Reaktionsverläufen führt.
Außerdem kommt das „Randgas“ mit der Wand in Kontakt und kann dort katalytisch
verändert werden. Des weiteren verteilt sich der Müll auf dem Brennbett so, dass im
mittleren Bereich mehr brennender Müll liegt als direkt an den Wänden. Von unten im
Randbereich des Brennbetts eingeblasene Luft steigt daher mit einem relativ hohen Sauerstoffgehalt an den Wänden auf. Um nicht Randeffekte zu untersuchen, muss das
Probengas in einigem Abstand von der Wand entnommen werden.
Da das heiße Probengas auf dem Weg zum Analysator nur mit inerten Oberflächen in
Berührung kommen darf, hat die Sonde die weitere Aufgabe, ein dünnes Quarzglasrohr
zu stützen und zu schützen. Des Weiteren muss die Sonde so aufgebaut sein, dass das
Gas auf dem Weg durch die Kesselwand nach außen nicht so stark abkühlt, dass es zu
Kondensationen kommt. Weiterhin sollte das Probengas möglichst schnell auf eine
Temperatur zwischen 250 °C und 300 °C gekühlt werden, um schnellen Reaktionen im
Gas und an der Propenahmeoberfläche nur ein kleines Zeitfenster zu geben.
Das entscheidende Problem beim Bau von Hochtemperatursonden stellen die Materialien dar. Bei normalen Edelstählen beginnt bereits zwischen 300 °C und 400 °C die
Verzunderung. Das heißt, zwischen der Stahloberfläche und der Gasatmosphäre kommt
es zu Reaktionen, bei denen sich chemische Verbindungen bilden, die man als Zunderschicht bezeichnet. Die Zunderschicht wächst durch die Diffusion von Eisen aus dem
Stahl an die Oberfläche, wodurch sich das Metall zersetzt. Werden dem Stahl Elemente
zulegiert, die leichter als Eisen oxidieren und außerdem eine diffusionshemmende Oxidschicht bilden, wie das bei Chrom und Silizium der Fall ist, kann die
122
Kapitel 5
Zunderbeständigkeit wesentlich erhöht werden. Der austenitische Chromnickelstahl
1.4841 (nach DIN 17007) hat eine Zundergrenztemperatur von 1150 °C in Luft und
behält seine Zugfestigkeit bis zu hohen Temperaturen. Dieser Stahl ist daher trotz seiner
hohen Kosten das Material der Wahl für die tragenden Rohre der in Abbildung 52 I dargestellten Sonde.
Die in Abbildung 52 dargestellte Sonde zeigt die 3. Generation von Sonden, die für
MVAs entwickelt und getestet wurden. Damit sich die bis zu einem Meter frei und
waagrecht in das Feuer ragende Sonde aus hitzebeständigem Stahl, der bei 1450 °C
schmilzt, bei 1200 °C nicht verbiegt, wurde eine Sonde mit Luftkühlung entwickelt. Bei
dieser Sonde gelangt kalte Pressluft durch einen Anschluss (A) in die Lanze. Gleich
nach dem Eintritt in die Sonde wird die kalte Luft auf 8 Röhrchen (B) mit einem
Durchmesser von 5 mm verteilt und gelangt durch diese an die Spitze der Sonde. Dort
strömt die kalte Luft aus den Röhrchen in den Hohlraum der Doppelrohrsonde. Dass die
kalte Luft an der Spitze der Lanze den Ring anbläst, durch den die zwei Hauptrohre
dicht miteinander verschweißt sind, ist besonders günstig, da diese Schweißnähte eine
Schwachstelle sein können. Beim Schweißen kann es zu Legierungsentmischungen und
Gefügeveränderungen kommen und somit zu Veränderungen der Materialeigenschaften.
Von der Spitze der Sonde strömt die Luft zwischen den Röhrchen zurück und kühlt die
hitzebeständigen Hauptrohre (D und C). Die Lanze ist so konstruiert, dass die heiße Luft
ohne sich mit der kalten zu vermischen zum hinteren Ende der Lanze gelangt, wo sie
aus der Sonde austritt (A). Diese Konstruktion ist vorteilhaft, weil durch die Luftführung die Wärme, die hauptsächlich am vorderen Teil der Sonde aufgenommen wird, den
hinteren Teil der Lanze temperiert. An Vorgängertypen dieser Lanze hat sich gezeigt,
dass die heiße Luft hinter dem Zutritt der kalten Luft ganz am Ende der Lanze austreten
muss, damit ihr hinterstes Ende nicht zu kalt wird.
Das innere (D) und das äußere Hauptrohr (C) sind am hinteren Ende, das in das Kesselhaus ragt, nicht miteinander verschweißt, sondern mit einem konischen Graphitring (E)
und der Anzugsmutter (F) zentrisch und dicht verbunden. Dies ist notwendig, weil vor
allem beim Einführen der Lanze in den Feuerraum die Temperaturdifferenz zwischen
innerem und äußerem Rohr beträchtlich ist und damit auch die Differenz der Längendehnung. Eine starre Schweißverbindung an beiden Enden würden die Schweißnähte
nicht aushalten. Die gezeigte Konstruktion erlaubt es dem inneren Rohr, durch den Graphitring zu gleiten und seine Länge unabhängig vom äußeren Rohr zu verändern. Hierzu
sei noch erwähnt, dass sich beim schnellen Einbringen der Lanze ohne Wärmeisolier-
123
Abbildung 52 I: Foto der Hochtemperatursonde mit eingezeichneter Kühlluftführung vor
dem Verschweißen. II, Verschraubtes Ende der Sonde mit Wärmeisolierung.
124
Kapitel 5
schicht das äußere Rohr um 6,8 mm mehr als das innere Rohr ausdehnte. Bei einem
Wärmeausdehnungskoeffizienten von 10-6 1/K [117] für Nickelstahl ergibt dies bei einer
Länge von 1 m einen mittleren Temperaturunterschied von 680 °C zwischen dem inneren und dem äußeren Rohr.
Es zeigte sich, dass sich wegen der geringen Wärmekapazität von Luft nicht genügend
Wärme abführen lässt, um die Lanze kühl genug zu halten. Aus diesem Grund wurde
die Sonde, wie in Abbildung 52 II dargestellt, mit einer Wärmeisolierung (G) aus
3,2 mm dickem Keramik-Papier der Firma KAGER GmbH (Dietzenbach, Deutschland)
versehen. Das einlagige, bis 1260 °C verwendbare Isoliermaterial wurde mit einem
Edelstahlrohr (H) fixiert und geschützt. Aufgrund der langen Lieferzeiten für
Hochtemperaturstähle konnte für den fünfwöchigen Dauertest der Lanze kein
Spezialstahl verwendet werden. Nach der Testzeit war dieser Schutzmantel so
korrodiert, dass er seine Aufgaben gerade noch erfüllen konnte. Ansonsten war die
Lanze, die zur Kühlung an eine 8 bar-Druckluftversorgung angeschlossen war,
unversehrt. Die Druckluft für die Lanze, die bei bis zu 1300 °C 90 cm frei von der
Kesselwand ins Feuer ragte (vgl. Abbildung 53), musste mit einem Absperrhahn
reduziert werden, damit sich das Probengas in dem 50 cm langen Stück der Lanze, das
sich in der Kesselwand befand, nicht unter 250 °C abkühlte.
In der Metallsonde befindet sich ein Quarzglasrohr (Schmelztemperatur 1710 °C) mit
6 mm Innendurchmesser, durch das das Probengas aus dem Kessel zum Filter gelangt.
Das Quarzrohr ragt vorne einige Zentimeter aus der Sonde heraus, um keine Falschluft
aus dem Innenrohr der Lanze zu ziehen. Weiterhin wird, um das Ansaugen von Falschluft zu verhindern, am hinteren Ende der Sonde der Raum zwischen Quarzrohr und
Stahlrohr möglichst dicht mit Glaswolle verpresst, damit durch den Unterdruck im Kessel nicht Kesselhausluft (Falschluft) durch die Lanze zur Spitze gelangt, dort in das
Quarzrohr eingesaugt wird und das Messergebnis verfälscht.
Vorne kann das Quarzrohr nur ein kurzes Stück ins Feuer ragen, da Quarz (b-Quarz) bei
diesen Temperaturen seine Kristallstruktur langsam erst zu a-Quarz (ab 573 °C) und
dann zu b-Tridymit (ab 870 °C) ändert und dabei durch das unterschiedliche Ausdehnungsverhalten der Gefüge milchig und sehr porös wird.
Das Innenrohr, in dem das Quarzrohr frei liegt, zeigte bei einer Messung im Feuerraum
einer MVA das in Abbildung 53A dargestellte Temperaturverhalten, das mit einem 2 m
langen NiCr/Ni-Thermoelement ermittelt wurde. Die Temperatur des Metallrohrs fiel
125
Abbildung 53: Darstellung der Hochtemperatursonde bei der Probenahme im Brennraum einer MVA. A: Temperatur des hitzebeständigen inneren Stahlrohrs in Abhängigkeit von der
Position in der Lanze. B: Temperaturverlauf im inneren Quarzrohr bei der Probenahme.
hier sogar knapp unter 200 °C. Das Quarzglas, durch das die heißen Gase gesaugt werden und das keinen guten Kontakt zum Stahlrohr hat, kühlt beim Ansaugen des Gases
nur auf 250 °C ab. Dieses in Abbildung 53B dargestellte Temperaturverhalten wurde im
Quarzrohr bei einer Saugvolumenstrom von 5 l/min ermittelt.
Bei diesem Volumenstrom und einem Quarzrohr-Innendurchmesser von 6 mm wird das
Probengas innerhalb von 0,4 Sekunden auf eine Temperatur von unter 300 °C abgekühlt, bei der das Gas weitgehend nicht mehr reaktiv ist.
Die Lanze passt mit einem Außendurchmesser von 50 mm gut durch die an MVAs üblichen 2,5’’-Stutzen. Die bis zu 250 °C heiße Kühlabluft kann entweder durch einen
weiteren freien Stutzen an anderer Stelle zurück in den Feuerraum geblasen, oder durch
einen Schalldämpfer in das Kesselhaus entlassen werden.
126
Kapitel 5
5.13 Gasanalysen mit dem REMPI-SPI-EI-TOFMS
Im folgenden Kapitel werden die Ergebnisse der ersten Messungen mit dem hier beschriebenen REMPI-SPI-EI-TOFMS-Instrument vorgestellt. Bis Ende 2002 wurden
Messungen an Fischer-Tropsch-Destillationsproben einer Ölraffinerie und an Zigarettenrauch durchgeführt.
Finanziert wurde der Aufbau des Kombigeräts durch ein Forschungsprojekt mit dem
Ziel, an Müllverbrennungsanlagen im Brennraum online stickstoffhaltige Substanzen
messen zu können. Das TOFMS-Gerät wurde in einem 5-wöchigen Dauereinsatz an
einer großen deutschen Hausmüllverbrennungsanlage getestet. Hierzu werden am Ende
dieses Kapitels einige exemplarische Messung vorgestellt. Weitere Messungen und eine
detaillierte Analyse der Ergebnisse sind an anderer Stelle zu finden [40].
5.13.1 Online–Analyse von Zigarettenrauch
Etwa 21 Millionen Menschen in Deutschland rauchen. Nach Angaben der WHO verringert jahrelanges Rauchen die Lebenserwartung. Das Krebsrisiko wird durch Rauchen
erhöht und eine Reihe weiterer Krankheiten können begünstigt oder hervorgerufen werden. Die Wahrscheinlichkeit, durch Zigarettenrauchen zu erkranken, ist um ein
Vielfaches höher, als durch Luftschadstoffe in einem Ballungsgebiet.
Zigarettenrauch enthält unter anderem Teer, Nikotin, Kohlenmonoxid, Blausäure, Phenole, Schwefelwasserstoff, Ammoniak, Stickoxide, Kohlenwasserstoffe und Aldehyde.
Durch moderne Analysetechniken konnte nachgewiesen werden, dass im Zigarettenrauch einige tausend Verbindungen enthalten sind [118-121]. Viele der Verbindungen
sind krebserregend oder stehen im Verdacht kanzerogen zu sein. Viele weitere Stoffe im
Zigarettenrauch sind als giftig oder reizend einzustufen.
Durch Variation der Tabaksorten, Tabakmischungen, des Zigarettenpapiers und durch
den Einsatz von Filtern kann auf die Konzentration einiger Substanzen Einfluss genommen werden. Verbesserungen in der Rauchzusammensetzung stellen daher ein
weites Forschungsgebiet dar.
127
Ein TOFMS-System, wie es in dieser Arbeit entwickelt wurde, kann für die OnlineAnalyse von Zigarettenrauch eingesetzt werden. Es lässt sich damit vor allem die Veränderung des Rauches von vor dem Filter bis in den Mundraum und bis zum Ausatmen
darstellen. Die Online-Analyse des Rauches ist vor allem deshalb interessant, weil sich
von Zug zu Zug eine Reihe von Parametern bei der Zigarette ändert [53]. Je nachdem
wie der Raucher an seiner Zigarette zieht, ändert sich z.B. die Glühtemperatur, je näher
die Glutzone zum Filter rückt, desto mehr vom Tabak zuvor absorbierter Substanzen
werden wieder desorbiert und auch der Filter ändert sein Verhalten. Nachfolgend ist
eine exemplarische Messung von Zigarettenrauch dargestellt.
In der Glutzone der Zigarette findet bei sehr hoher Temperatur von etwa 900 °C die
Verbrennung und Pyrolyse von Tabak, Zusatzstoffen und Papier statt [122]. Zieht der
Raucher an der Zigarette, wird der sog. Hauptstromrauch gebildet. Zwischen den Zügen,
wenn die Zigarette im Aschenbecher liegt, entsteht Nebenstromrauch. Dieser Nebenstrom wurde in den nachfolgend beschriebenen Messungen analysiert.
Zwei Nebenstrom-Massenspektren, aufgenommen mit SPI (118 nm) und REMPI
(266 nm), zeigt Abbildung 54. Eine mögliche Zuordnung der Peaks ist in Tabelle 8 zu
finden. Die Verbindungen sind dort nach aufsteigender Masse aufgelistet. Die Art der
Ionisation ist in der letzten Spalte aufgeführt. Die Zuordnung der einzelnen Massen zu
bestimmten Verbindungen ist nur selten eindeutig. So sitzen z.B. bei den Kohlenwasserstoffen auf den Massen der Alkine auch die Diene und durch Kettenverzweigungen sind
Massen mehrfach besetzt.
Die Spektren in Abbildung 54 zeigen die gemessenen Pyrolyse- und VerbrennungsProdukte der Zigarette. Bei der Ionisation mit VUV-Photonen (Abbildung 54 oben) sind
die Signale von Furan (68 m/z) und Phenol (94 m/z) dominant. Phenol ist ein typisches
Pyrolyseprodukt. Weiterhin werden Kohlenwasserstoffketten ab 3 Kohlenstoffatomen,
Cyclohexan und Benzolderivate nachgewiesen. Von den kurzen Kohlenwasserstoffketten können einige wegen ihres IPs von mehr als 10,5 eV nicht nachgewiesen werden.
Das REMPI-Spektrum (Abbildung 54 unten) zeigt mono- und polyzyklische aromatische Kohlenwasserstoffe. Dies sind typische Verbrennungsprodukte. Die Hauptpeaks
werden hier Toluol (92 m/z), Phenol (94 m/z), Xylol (106 m/z) Indol (117 m/z), Indan
(118 m/z), den Alkyl-Indenen, Alkyl-Indolen und den Alkyl-Styrolen zugeordnet. Insgesamt ergibt sich in den Massenspektren ein Ensemble aus Verbindungen, die
überwiegend zu einem der oben genannten Grundgerüste gehören und einer Progression
128
Kapitel 5
mit dem Intervall 14 m/z folgen. Das Vorkommen von Indol (117 m/z), das das stärkste
Signal verursacht, ist typisch für Rauchgas von Pflanzenmaterial mit einem hohen Gehalt an Alkaloiden.
Die dargestellte Messung zeigt, dass sich ein Massenspektrometer mit REMPI und SPI
für die Studie von Verbrennungs- und Pyrolyse-Prozessen eignet. Da bis zu 10 komplette Massenspektren in der Sekunde aufgenommen werden können, ist diese Technik
besonders geeignet für das Untersuchen von dynamischen Prozessen, wie das Rauchen
einer Zigarette.
Abbildung 54: SPI und REMPI-(266 nm) Massenspektrum von Nebenstromrauch einer Zigarette. Beide Spektren wurden mit demselben TOFMS innerhalb von 2 Sekunden
aufgenommen.
129
Tabelle 8: Mögliche Verbindungen für die in Abbildung 54 dargestellten Zigarettenrauchpeaks. Aufgrund der Vielzahl der Verbindungen ist eine genaue Zuordnung nicht möglich.
Es ist der Name der Verbindung, die atomare Masse und das Ionisationpotenzial aufgelistet.
In der letzten Spalte ist angegeben, ob die Masse mit SPI (118 nm) oder mit REMPI
(266 nm) nachgewiesen wurde.
Bezeichnung
Propen
Ethylamin
Acetaldehyd
1,3-Butadien
Buten
Acrolein
Aceton
Propanal
Furan
Penten
Crotonaldehyd
Methylvinylketon
Benzol
Cyclohexan
Cyclopentanon
Toluol
Phenol
Heptadien
Styrol
m-Xylol
Benzaldehyd
Cresol (o,m,p)
Indol
Methyl-Benzonitril
Indan
1,3,5-Alkylbenzol
Methylindol
Alkylstyrol
Methylnaphthalin
Alkyl-Inden
Alkyl-Indol
Alkyl-Styrol
4-Vinylguaiacol
Alkyl-Naphthalin
Alkyl-Inden
Alkyl-Indol
Alkyl-Styrol
Nikotin
Anabasin
Dibenzofuran
Alkyl-Naphthalin
Alkyl-Inden
Phenanthren
Anthranzen
Alkyl-Biphenyl
Alkyl-Inden
Formel
C3H6
C2H7N
C2H4O
C4H6
C4H8
C3H4O
C3H6O
C3H6O
C4H4O
C5H10
C4H6O
C4H6O
C6H6
C6H12
C5H8O
C7H8
C6H6O
C7H12
C8H8
C8H10
C8H10
C7H8O
C8H7N
C8H7N
C9H10
C9H12
C8H7N
C10H12
C11H10
C11H12
C10H11N
C11H14
C9H10O2
C12H12
C12H14
C11H13N
C12H16
C10H14N2
C10H14N2
C12H8O
C13H14
C13H16
C14H10
C14H10
C14H14
C14H18
Masse (m/z)
42
43
44
54
56
56
58
58
68
70
70
70
78
84
84
92
94
96
104
106
106
108
117
117
118
120
131
132
142
144
145
146
150
156
158
159
160
162
162
168
170
172
178
178
182
186
IP (eV)
9,7
8,9
10,2
9,0
9,6
10,1
9,7
10,0
9,6
9,5
9,7
9,65
9,2
9,9
9,26
8,5
8,5
9,5
8,47
8,55
9,5
8,3
7,76
9,32-9,58
8,6
8,40
7,5–7,7
8,0-8,1
7,91-7,96
8,77
7,89
7,44
Ionisationsverfahren
SPI
SPI
SPI
SPI
SPI
SPI
SPI
SPI
SPI
SPI
SPI
SPI
SPI / REMPI
SPI
SPI
SPI / REMPI
SPI / REMPI
SPI
REMPI
SPI / REMPI
SPI / REMPI
REMPI
REMPI
REMPI
REMPI
REMPI
REMPI
REMPI
REMPI
REMPI
REMPI
REMPI
REMPI
REMPI
REMPI
REMPI
REMPI
SPI
SPI
REMPI
REMPI
REMPI
REMPI
REMPI
REMPI
REMPI
130
Kapitel 5
5.13.2 Analyse von Proben der Benzinsynthese vor und nach der Hydrierung
In Südafrika wird ein großer Teil des benötigten Öls und Benzins aus Kohle gewonnen.
Gründe dafür sind unter anderem die billige Förderung von Kohle im Tagebau, eine
größere Unabhängigkeit von Ölimporten und das Einsparen von Devisen.
Bei diesem großindustriellen Prozess wird Kohle unter hohem Druck und bei hoher
Temperatur mit Sauerstoff und Wasserdampf in ein Rohgas umgewandelt, das Pyrolyseprodukte der Kohle, Wasserdampf, Methan, Kohlendioxid, Kohlenmonoxid,
Wasserstoff und einige Stickstoff- und Schwefelverbindungen enthält.
Dieses Rohgases wird anschließend gereinigt, wobei unter anderem Pech anfällt, das
weiter zu Koks verarbeitet wird. In Reaktoren reagieren das Kohlenmonoxid und der
Wasserstoff des gereinigten Gases unter hohem Druck nach dem Fischer-Tropsch Verfahren katalytisch zu Kohlenwasserstoffen mit bis zu 20 Kohlenstoffatomen (C 20).
Dieses Verfahren wurde zwischen 1923 und 1925 von Fischer und Tropsch entwickelt,
um großtechnisch Kohlenwasserstoffe für die Benzin-, Paraffin- und Motorenölherstellung zu erzeugen. Damals sollte es helfen, Deutschland von Ölimporten unabhängig zu
machen.
Das gereinigte Rohgas mit den Pyrolyse-Kohlenwasserstoffen und dem erhöhten Kohlenwasserstoffanteil aus der Fischer-Tropsch-Synthese wird stufenweise abgekühlt.
Durch die unterschiedlichen Dampfdrücke der Kohlenwasserstoffe erhält man je nach
Abkühltemperatur verschiedene Fraktionen, die unterschiedliche Anteile an verschieden
schweren Kohlenwasserstoffen haben. In einem bestimmten Temperaturbereich kondensiert die Fraktion, die reich an Kohlenwasserstoffen mit 6 bis 12 Kohlenstoffatomen ist.
Diese Fraktion ist Ausgangsprodukt für die Benzinherstellung. Die nächstschwere Fraktion wird zur Herstellung von Kerosin und die noch schwerere zur Herstellung von Ölen
und Dieselkraftstoffen verwendet.
Damit die Benzin-Fraktion die Ansprüche der Autoindustrie erfüllt, muss der Gehalt an
ungesättigten aliphatischen Kohlenwasserstoffen mit 2 Doppelbindungen (Diene) durch
Hydrieren reduziert werden. Die katalytische Hydrierung ist ein wichtiger Prozess und
beeinflusst die Oktanzahl des Kraftstoffs. Hydriert man zu stark, so werden auch die
aromatischen Verbindungen hydriert, was zu einer Verringerung der Oktanzahl führt mit
131
der Folge, dass dem Destillat wieder teure Zusatzstoffe beigemischt werden müssen, um
die geforderte Oktanzahl zu erreichen.
Die Oktanzahl ist eine charakteristische Kerngröße für die Selbstzündeigenschaft
(Klopffestigkeit) eines Kraftstoffes. Ein Kraftstoff mit einer Oktanzahl von z.B. 87 hat
dieselben Selbstzündeigenschaften wie ein Gemisch aus 87 % Oktan und 13 % Heptan.
Zur Steuerung der chemischen Prozesse sind Online-Messverfahren notwendig, die innerhalb von Sekunden Aufschluss über die Gaszusammensetzung geben. Auf diese
Weise kann es ermöglicht werden, die Prozesse optimal zu steuern und den Wirkungsgrad möglichst hoch zu halten. Wie aus der kurzen Prozessbeschreibung zu entnehmen
ist, spielen sich die wichtigsten Reaktionen in der Gasphase ab. Darüber hinaus können
viele der interessanten organischen Verbindungen mit REMPI oder SPI selektiv nachgewiesen werden. Aus diesen Gründen würde sich das in dieser Arbeit vorgestellte
Massenspektrometer hervorragend zur Steuerung der beschriebenen Prozesse eignen.
Einige Vorversuche, für die Kondensatproben von einer Raffinerie zur Verfügung gestellt wurden, sind im folgenden Abschnitt dargestellt.
Kondensatproben, die einem laufenden Syntheseprozess entnommen wurden, werden in
diesem Abschnitt mit REMPI bei 248 nm und SPI analysiert. Im Gegensatz zur Analyse
von Schadstoffen in Luft befinden sich bei den Messungen der unterschiedlich flüchtigen Kondensate in ihnen Anteile von Substanzen in Prozent-Konzentrationen, die mit
REMPI oder SPI ionisiert werden. Benzol, Toluol und Xylol sind in der Regel in Konzentrationen von einigen Prozenten enthalten und liefern, auch wenn man eine für diese
Substanzen ungünstige REMPI-Wellenlänge wählt, ein sehr intensives Signal. Deshalb
ist es schwierig, Substanzen in geringen Konzentrationen daneben nachzuweisen. Um
bei den Messungen nicht den Detektor und den Signalvorverstärker zu schädigen, wurden einige der nachfolgenden Messungen mit verringerter Detektorspannung und damit
geringerer Detektorverstärkung durchgeführt. Dies hat allerdings zur Folge, dass sich
die Nachweisempfindlichkeit für alle Substanzen dieser Messungen verringert.
Da nicht direkt an den Anlagen in der Gasphase gemessen werden konnte, sondern mit
flüssigen Kondensatproben gearbeitet wurde, musste berücksichtigt werden, dass zuerst
überwiegend die flüchtigeren Bestandteile aus dem Kondensat austreten und später relativ stärker die schwerer flüchtigen Anteile. Um dem Rechnung zu tragen und um
Spektren zu erhalten, die die Konzentrationsverteilung im Kondensat wiedergeben,
wurde jeweils 1 ml des Kondensats in einem Fläschchen verdampft und so lange gemes-
132
Kapitel 5
Abbildung 55: SPI-TOFMS-Headspace-Messung eines Kondensats vor der Hydrierung. Das
untere Spektrum ist eine Vergrößerung (andere Ordinate) vom unteren Bereichs des oberen
Spektrums.
Abbildung 56: SPI-TOFMS-Headspace-Messung eines Kondensats nach der Hydrierung.
Das untere Spektrum ist eine Vergrößerung (andere Ordinate) vom unteren Bereichs des oberen Spektrums.
133
sen, bis es vollständig verdampft war. Dann wurde aus den Einzelspektren ein Summenspektrum errechnet, das die Konzentrationsverhältnisse der Kondensatprobe wiedergibt.
Messungen der flüchtigen Bestanteile über dem flüssigen Kondensat werden hier als
Headspace-Messungen bezeichnet.
Abbildung 55 zeigt die Messung der Probe, die vor dem Hydrierungsreaktor gezogen
wurde. Deutlich sind im oberen Teil der Abbildung die Benzolderivate Benzol (78 m/z),
Toluol (92 m/z) und Xylol (106 m/z) (BTX) zu erkennen. In der darunterliegenden, vergrößerten Darstellung findet man aus der Reihe der Benzolderivate noch Alkyl-Benzol.
In der Vergrößerung sind außerdem noch die Reihen der Alkyl-Styrole (C8+nH8+2n), Alkane (CnH2n+2),
Alkene (CnH2n),
Alkine (CnH2n-2)
und
Phenole (C6+nH6+2nO)
zu
erkennen. Die Massenpeaks bei (54 + n 14) m/z werden durch Alkine und Diene hervorgerufen. Beide Substanzgruppen können mit 10.5 eV-VUV-Photonen ionisiert
werden.
Beim Vergleich der in Abbildung 55 dargestellten Messung mit der in Abbildung 56,
bei der die Probe nach der Hydrierung entnommen wurde, erkennt man bei genauerer
Betrachtung, dass die Phenole bei der 2. Messung nicht mehr detektiert werden und die
Alkine bzw. Diene sehr stark abgenommen haben. Die Abnahme der Diene und Alkine
entspricht genau dem, was durch den Prozess der Hydrierung erreicht werden soll.
Um zu erfahren, zu welchen Teilen das SPI-Signal auf der Masse 54 m/z durch Butin
bzw. Butadien hervorgerufen wird, könnte in Zukunft mit Hilfe der Excimerlampe mit
den 130 nm-Photonen des Ar/O2-Gemisches ionisiert werden. 1-Butin und 2-Butin haben ein IP von 10,20 eV und 9,58 eV, 1,3-Butadien und 1,2-Butadien hingegen ein IP
von 9,07 eV und 9,03 eV. Mit der 130 nm-Strahlung mit 9,5 eV kann daher nur das
Dien ionisieren werden.
N2 und O2 können nicht durch den SPI-Prozess ionisiert werden. Diese beiden Moleküle
werden durch Elektronen ionisiert, die durch Streulicht, das die Einlassnadel trifft, aus
dem Metall austreten und im Abzugsfeld so viel kinetische Energie erhalten, dass sie
ineffektiv ionisieren können.
Für die hier beschriebenen Untersuchungen an Proben mit hohem BTX-Anteil ist die
Verwendung des Massenfilters besonders hilfreich. Abbildung 57 oben zeigt die Messung eines Kondensats der Kohlepyrolyse vor der Hydrierung ohne Massenfilter. Um
nicht zu hohe Signale zu erhalten, musste dabei der Ionendetektor in seiner Verstärkung
134
Kapitel 5
stark reduziert werden. Abbildung 57 unten zeigt eine Messung derselben Probe, aber
mit Massenfilter. Der Massenfilter war so eingestellt, dass Moleküle bis 106 m/z den
Ionendetektor nicht erreichten. Da damit die Hauptbestandteile eliminiert wurden, konnte mit einer sehr viel höheren Detektorverstärkung gearbeitet werden. Die
Nachweisgrenzen der Moleküle mit Massen über 106 m/z wurden dadurch deutlich erhöht.
Abbildung 57: SPI-TOFMS-Headspace-Messung eines Kondensats vor der Hydrierung mit
und ohne Massenfilter. Durch die Verwendung des Massenfilters konnte der Ionendetektor
mit einer höheren Verstärkungsspannung betrieben werden.
Abbildung 58: REMPI-TOFMS-Headspace-Messung eines Kondensats vor der Hydrierung
mit 248 nm-UV-Licht. Im Gegensatz zu den oberen SPI-Messungen sind hier nur die aromatischen Verbindungen zu sehen.
135
Untersucht man die Probe des Pyrolysegases vor der Hydrierung mit REMPI z.B. bei
248 nm, so lassen sich die aromatischen Verbindungen sehr gut nachweisen (siehe
Abbildung 58).
Die Messungen zu diesem Kapitel zeigen, dass sich SPI-TOFMS und REMPI-TOFMS
mit Ionenmassenfilter eignen, um komplexe Mischungen petrochemischen Ursprungs zu
analysieren. Es müsste daher mit diesem Verfahren möglich sein, die Zusammensetzung
des Endprodukts konstant zu halten und somit die Produktion kostengünstiger zu gestalten.
5.13.3 Online-Analyse von Rauchgas einer Müllverbrennungsanlage
Die Entsorgung von Siedlungs- und Industrieabfällen wird weltweit zum wachsenden
Problem. Während die Müllproduktion auch in Europa kontinuierlich steigt, werden
zunehmend restriktive Gesetze über die zur Deponierung zugelassene Abfallmenge verabschiedet. Gleichzeitig wurden in den letzten Jahren viele Müllverbrennungsanlagen
(MVAs) aufgrund neuer, strengerer Bestimmungen über deren Emissionsbegrenzung
geschlossen.
Zur Einhaltung der hohen Umweltanforderungen ist bei Abfallverbrennungsanlagen eine
aufwändige und kostenintensive Rauchgasreinigung notwendig. Komplexe und Rauchgasreinigungssysteme sind erforderlich, um aus einer Vielzahl an Verbindungen, die bei
der Verbrennung von Müll freigesetzt werden, Schadstoffe wie z.B. Dioxine, Quecksilber, aber auch Stickoxide (NOx) zurückzuhalten.
Zur primärseitigen Reduktion der NOx-Emission industrieller Müllverbrennungsanlagen
soll von Abfallverbrennungsanlagen ein reaktionskinetisches Modell erstellt werden.
Zur Entwicklung und Validierung dieses Modells ist neben der Kenntnis der NO2- und
NO-Konzentrationen auch die der Konzentrationen der möglichen Vorläuferverbindungen wie z.B. NH3, HCN und organischer Stickstoffverbindungen in Abhängigkeit von
ihrer Verteilung im Brennraum unter realen und wechselnden Verbrennungsbedingungen notwendig.
Auf der Suche nach einer Zielsubstanz oder Leitgröße für eine geplante Prozesssteuerung ist es erforderlich, relevante chemische Substanzen online zu messen.
Veränderungen von Betriebsbedingungen und deren Einfluss auf die chemische Signatur
136
Kapitel 5
des Rauchgases spielen dabei eine wichtige Rolle und müssen mit hoher Zeitauflösung
erfasst werden.
Verbrennungsrauchgas enthält mehrere hundert Substanzen im Spurenbereich. Die Messung einer Substanz oder Substanzklasse in einem derart komplexen Abgasgemisch
erfordert eine hochselektive Messtechnik.
Während der Messungen, die in diesem Abschnitt beschrieben sind, befand sich das
TOFMS-System
(Abbildung 26)
im
Kesselhaus
einer
industriellen
Müllverbrennungsanlage unterhalb der Sekundärlufteindüsung. Um das ca. 800 kg
schwere Gerät dort aufzustellen, mussten im Kesselhaus einige Gitterböden entfernt
werden, damit es mit einer Winde auf die gewünschte Eben gebracht werden konnte.
Das Gerät wurde in ca. 1,5 m Entfernung vom Messstutzen, in dem sich die in Kapitel
[5.12] beschriebene Hochtemperatursonde befand, aufgebaut. Zum Betrieb musste es
nur mit einphasigem Netzstrom und mit Pressluft zur Kühlung versorgt werden.
Bei der in Abbildung 59 und Abbildung 60 dargestellten Messung wurde das Rauchgas,
welches aus dem Bereich der Flammenspitzen entnommen wurde, im 10 s-Rhythmus
abwechselnd mit SPI und REMPI (224 nm) ionisiert. Zwischen den Laserpulsen wurde
das Rauchgas mit einem 10 kHz- 23 eV-Elektronenstrahl ionisiert. Jedes REMPI- und
SPI-Spektrum ist der Mittelwert aus 100 Einzelspektren. Das EI-Spektrum entstand aus
100.000 EI-Transienten. Damit benötigte das Gerät für jedes gespeicherte Spektrum
10 s.
Wie man anhand des SPI-Spektrums der Abbildung 59 (A) erkennt, konnten mit der
VUV-Ionisation Alkane, Alkene und Alkine analysiert werden. Benzolderivate, Cyclohexan (84 m/z), Phenol (94 m/z) und Naphthalin (128 m/z) werden ebenfalls sehr
empfindlich nachgewiesen. Besonders wichtig für das oben erwähnte Projekt zur Stickoxidminderung ist der Nachweis von Ammoniak (17 m/z) und Benzonitril (103 m/z).
Abbildung 59 (B) zeigt, dass sich mit REMPI bei 224 nm vor allem die polyzyklischen
Aromaten wie Alkyl-Naphthaline, Acenaphthylen, Alkyl-Biphenyle, Phenanthren und
Pyren gut detektieren lassen. Sehr gut konnte bei dieser Wellenlänge der Konzentrationsverlauf von Naphthalincarbonitril verfolgt werden.
137
Abbildung 59: Online-SPI (118 nm) (A), REMPI (224 nm) (B) und EI-(23 eV)-Messung (C)
von Rauchgas einer MVA aus der Flammzone. Das Massenspektrometer befand sich für diese Messung im Kesselhaus. Alle drei Spektren wurden innerhalb von 10 s aufgenommen.
Dargestellt sind die unterschiedlichen Substanzen und Substanzklassen, die mit den unterschiedlichen Ionisationsverfahren gemessen wurden.
Abbildung 59 (C) zeigt das 23 eV-Elektronenstoß-Spektrum. Hier ist HCN (27 m/z) gut
zu erkennen. Das bei dieser Darstellung abgeschnittene Signal bei 36 m/z stammt von
Salzsäure (HCl). Das Signal bei 64 m/z wird durch Schwefeldioxid und das bei 26 m/z
von Acetylen hervorgerufen. C2H2, HCN, HCl und SO2 können aufgrund ihrer IPs von
11,4; 13,6; 12,7 und 12,3 mit 118 nm-SPI nicht nachgewiesen werden. Wie auch bei den
70 eV-EI-Referenzspektren des NIST [64] ist für SO2 ein Fragmentsignal bei 48 m/z
und für C2H2 bei 25 m/z zu finden. Durch das Chlor-37-Isotop (24,2 %) erscheint für
HCl auch ein Signal bei 38 m/z. Benzol bei 78 m/z wird ebenfalls nachgewiesen. Die
138
Kapitel 5
starken Signale bei 28, 32, 40 und 44 m/z stammen von Stickstoff, Sauerstoff, Argon
und Kohlendioxid.
Während der Messung, aus der die Spektren von Abbildung 59 9 min nach Messbeginn
erhalten wurden, wurde 20 min lang mehrt als 1200 °C heißes Rauchgas analysiert. In
Abbildung 60 ist der Konzentrationsverlauf einiger ausgewählter Substanzen dargestellt.
Die Verläufe sind nach der Masse der Verbindungen geordnet und nicht nach der Art
ihrer Ionisierung. Bei den Substanzen, die bei der Messung an der MVA als quantifizierter Gasstandard zu Verfügung standen, ist der Konzentrationsverlauf in ppm angegeben.
Bei den anderen Substanzen ist der relative Konzentrationsverlauf dargestellt. Man erkennt, dass nicht alle Verbindungen einen gleich großen Konzentrationsanstieg bei
5 min zeigen.
Das SPI-Benzolsignal bei 5 min überschreitet den zu dieser Zeit eingestellten Messbereich der Transientenrekorder-Karte von 50 mV. Um dennoch den Benzolverlauf
komplett quantifizieren zu können, wurde die Benzolkonzentration über die Masse
79 m/z ermittelt. Bei 6 Kohlenstoffatomen beträgt das Verhältnis vom 78 m/z- zum
79 m/z-Signal 14,6 : 1. Dies resultiert aus der Isotopenverteilung von 98,9 %
12
C zu
1,1 % 13C. Das gegenüber 78 m/z 14,6 mal kleinere 79 m/z-Signal überschritt nicht den
Messbereich der Messkarte. Da auch bei der Messung des 10 ppm-BTX-Gasstandards
der 79 m/z-Peak deutlich zu erkennen war, konnte durch den linearen Vergleich der
Peakflächen Benzol direkt quantifiziert werden.
Die hier präsentierten Messergebnisse und die Art ihrer Darstellung, für die eine spezielle Software entwickelt wurde, sind insbesondere geeignet, Reaktionsmechanismen zu
erkennen und zu validieren.
Mit dem TOFMS-System in Verbindung mit der hier beschriebenen Hochtemperatursonde konnten viele neue Erkenntnisse über die Zusammensetzung des Rauchgases
einer MVA in der Flammzone gewonnen werden [40].
139
Abbildung 60: Konzentrationsverläufe einer 20 min-Messung. Die ausgewählten Verbindungen wurden mit REMPI (224 nm), SPI (118 nm) und EI (23 eV) gemessen. Die Substanzen,
für die ein Probenstandard zur Verfügung stand, wurden quantifiziert und hier in ppmKonzentrationen dargestellt. Die durch die gestrichelte Linie bei 9 min gekennzeichnete Zeit
stellt die Zeit dar, bei der die in Abbildung 59 dargestellten Spektren aufgenommen wurden.
140
Kapitel 6
6 Mobiles Massenspektrometer mit Excimer-Lichtquelle
Die Entwicklung von Flugzeitmassenspektrometern soll dahin führen, dass TOFMSSysteme zu einem kostengünstigen Preis hergestellt werden können. Solch ein kommerzielles Gerät sollte bezüglich der Selektivität einstellbar sein. Es müsste von einem
Laien zu bedienen und wartungsarm sein.
Ein großer Schritt in diese Richtung gelang mit der anschließend beschriebenen und
getesteten Kombination aus TOFMS und Excimer-Lichtquelle. Nachfolgend ist der
Aufbau einer Excimer-Lichtquelle und die Adaption mit einem TOFMS beschrieben. Im
Anschluss an die Gerätebeschreibung werden erste Online-Messungen gezeigt.
6.1 Aufbau und Funktion einer Excimerlichtquelle
Bei der verwendeten Excimerlichtquelle werden durch einen kollminierten Elektronenstrahl in einem Punkt eines Gasraums Excimeratome erzeugt. Beim Zerfall der
Excimeratome wird von diesem Punkt aus VUV-Strahlung emittiert.
Der Schlüssel zum Bau einer intensiven und brillanten Lichtquelle liegt im Eintrittsfenster, durch das ein Elektronenstrahl in das Volumen mit dichtem Edelgas oder
Gasgemischen gelangt. In dem Gasvolumen verlieren die Elektronen durch Stöße ihre
kinetische Energie und regen damit Edelgasatome an. Angeregte Edelgasatome können
Excimere bilden, bei deren Zerfall die Excimerstrahlung frei wird (vgl. 4.2). Es musste
daher ein Fenster entwickelt werden, welches Elektronen mit einigen keV an kinetischer
Energie möglichst ungehindert passieren können. Außerdem muss dieses Fenster den
Vakuumbereich der Elektronenerzeugung von der Excimerkammer, in der sich Gase mit
bis zu einigen bar Druck befinden, gasdicht trennen.
Es ist gelungen ein Fenster aus Siliziumnitrid herzustellen [84], das den obigen Ansprüchen genügt. Technologien aus der Halbleiterfertigung ermöglichten es, 300 nm dünne
Folien aus diesem Material zu fertigen. Hierzu wird ein Siliziumwaver auf einer Seite
mit einer Nitridschicht versehen und die Rückseite photolithographisch strukturiert.
Anschließend wird dann das Silizium von der Rückseite her bis zur Nitridschicht durch
ein Ätzverfahren abgetragen. Zurück bleibt eine durch die Photolithographie definierte
141
Nitridfolie auf einem Siliziumträger. Ein 10 keV-Elektronenstrahl verliert in solch einer
Folie etwa 5-10 % seiner Energie. Die bisher verwendeten SiN-Folien mit einem
Schmelzpunkt bei 1900 °C und einer Dimension von 1 mm x 1 mm halten thermisch bis
zu 0,1 W/mm2 bei 4 bar Druckdifferenz aus. Demnach kann durch dieses Fenster ein bis
zu 1 W intensiver 10 keV-Elektronenstrahl in ein Excimergas einkoppeln. Bei höheren
Elektronenstrahlleistungen führen thermische Spannungen zum Reißen der Folie. Damit
ist die Folie das limitierende Teil für den Elektronenstrom und den Gasdruck solch einer
Excimerlichtquelle.
Solange diese Technologie der Folienherstellung noch unbekannt war, mussten wegen
der Gasdichtigkeit 10 mal dickere Metallfolien verwendet werden und damit auch
schnellere Elektronen, was ab 15 keV zu Isolations- und Röntgenstrahlproblemen führt.
Mit den neuen Folien können jetzt kompakte und brillante VUV-Lichtquellen, wie in
Abbildung 61 dargestellt, gebaut werden. Von einer elektronenstrahl-gepumpten Excimerlampe, bei der das Excimergas nicht durch erodierende Prozesse, wie z.B. bei der
Mikrohohlkathodenlampe, verunreinigt wird, erhält man saubere Emissionsspektren
über einen sehr langen Zeitraum (siehe Abbildung 64 links).
Eine Excimerlampe, die wie in Abbildung 61A mit einem Flugzeitmassenspektrometer
verbunden ist, besteht aus einer Elektronenkanone, die unter Vakuum einen Elektronenstrahl erzeugt. Dieser Strahl muss an der Stelle der Spezialfolie eine dem Fenster
entsprechende Ausdehnung haben. Dies wird durch eine elektromagnetische Ablenkund Kollimations-Einheit erreicht. Diese besteht aus einer Fokussierelektrode und vier
Elektromagneten zwischen der Elektronenkanone und der Folie (Abbildung 63B). Wie
in Abbildung 61B ist der Siliziumträger mit der Durchtrittsfolie über ein Loch im
Trennflansch zwischen Elektronenerzeugungsvakuum und Excimergas geklebt. In Zukunft wird gelötet statt geklebt. Auf der Excimergasseite wird nun durch den
Elektronenstrahl direkt hinter der Folie ein etwa 1 mm3 großes Volumen zur Emission
von VUV-Photonen angeregt. Um den Leuchtpunkt ist ein Parabolspiegel mit einer speziellen AlMgF2 -Beschichtung zur Reflexion des Lichts angebracht. Die Position und
Krümmung des Parabolspiegels wurde so gewählt, dass der Leuchtpunkt im Ortsfokus
des Parabolspiegels liegt und so das auf den Spiegel auftreffende Licht parallelisiert
wird. Die Excimergaszelle kann durch zwei Ventile befüllt und gespült werden (siehe
auch Abbildung 63B). Als Austrittsfenster für das Licht aus der Excimerzelle dient eine
plankonvexe MgF2-Linse mit einer Brennweite von 79 mm bei 121 nm Wellenlänge.
Diese Linse ist so ausgelegt, dass sie das parallele Licht vom Parabolspiegel auf das
142
Kapitel 6
Zentrum der Ionenquelle eines TOFMS fokussiert. Die Linse trennt gasdicht das Volumen des Excimergases vom Vakuum der Ionenquelle. Auf diese Weise wird kein
weiteres VUV -durchlässiges Fenster benötigt. Dies ist vorteilhaft, da im VUV-Bereich
auch der Lichtdurchgang durch spezielle Materialien mit hohen Verlusten verbunden ist.
Die Effizienz, mit der aus der Elektronenenergie Lichtenergie erzeugt wird, beträgt in
etwa 40 % (vgl. Tabelle 1) und kann theoretisch bis zu 60 % erreichen [123]. Die Lichtquelle kann kontinuierlich und mit einer geeigneten Elektronenkanone auch gepulst
betrieben werden [88]. Bei gleicher mittlerer thermischer Last für die Eintrittsfolie können damit kurzzeitig, entsprechend der Pulslänge, auch höhere Elektronenströme
eingestrahlt und damit auch die Lichtintensität erhöht werden. Die minimale Pulsdauer
des Lichtes ist, wie in Kapitel 4.2 beschrieben, druckabhängig und reicht bei den reinen
Edelgasen von einigen hundert Nanosekunden bei 1 bar Edelgasdruck bis unter hundert
Nanosekunden bei 8 bar. Um für den kurzen Ionisationsprozess ein Maximum an VUVPhotonen zur Verfügung zu haben, wurde in den nachfolgend beschriebenen Experimenten die Lichtquelle ausschließlich gepulst betrieben. Dazu wurde die Extraktion
einer Elektronenkanone, wie sie früher bei Fernsehgeräten benutzt wurde, gepulst.
Durch eine für ein übliches Flugzeitmassenspektrometer üblicherweise günstige Lichtpulsrepetitionsrate
von
20 kHz
(maximale
Ionenflugzeit
50 ms)
und
eine
Elektronenstrahldauer von 100 ns könnte der Elektronenstrom und die Lichtausbeute
theoretisch gegenüber dem kontinuierlichen Betrieb um das 500-fache erhöht werden.
Da die Folie kurzzeitig aber nicht so viel Wärme abführen kann, werden weitere Experimente zeigen müssen, wo die Grenzen des Fensters liegen.
Während der Experimente, für die nur eine Folie zur Verfügung stand, wurde die Lampe
sehr vorsichtig mit einem 1 mA 10 keV-Elektronenstrahl gepulst betrieben. Die Repetitionsrate des Elektronenstrahls lag bei 120 Hz. Bei Strömen von 1 mA bei 10 keV
Elektronen wurde somit das Gas mit einem 10 W-Elektronenstrahl gepumpt, was bei
den reinen Edelgasen zu einer VUV-Lichtemission von 4 W führte. Relative Lichtintensitäten und Lichtpulslängen konnten mittels eines VUV-sensitiven Halbleiterdetektors
gemessen werden, der nahe der Lichtquelle angebracht war.
Das Edelgas, das zur Erzeugung von VUV-Licht verwendet wird, muss wesentlich reiner als 99,999 % (Qualität 5.0) sein und ist nicht in Flaschen erhältlich. Während der
Versuche zirkulierte daher das Edelgas durch einen externen 800 °C heißen TitanGasreiniger. Diese Aufgabe des Gasreinigens, für eine lange Standzeit einer Gasfüllung,
143
Abbildung 61: (A) Schematische Zeichnung von einer elektronenstrahl-gepumpten Excimerlichtquelle kombiniert mit einem Flugzeitmassenspektrometer. (B) Vergrößerung des
Bereichs der Lichtquelle, in dem das VUV-Licht vor einer 300 nm dicken SiN-Folie durch
den Elektronenstrahl erzeugt wird.
144
Kapitel 6
wird in Zukunft eine kleine Getterpumpe übernehmen (siehe Abbildung 61A und
Abbildung 63B).
Ein Getter ist eine gasbindende Vakuumpumpe [124]. Das Gettermaterial reinigt das
Edelgas von Verunreinigungen. Es gibt zwei Mechanismen, die für das Festhalten der
Moleküle verantwortlich sind: die Chemiesorption, bei der die angelagerten Moleküle
eine chemische Bindung mit den Getteratomen der Oberfläche eingehen und die Physisorption, bei der die Gasmoleküle durch physikalische Wechselwirkung an das
Gettermaterial schwach gebunden werden. Bei der Chemisorption ist durch Zuführung
von Energie ein Wandern der Moleküle vom Ort des Einfangens zu einem anderen Ort
auf der Oberfläche oder ins Innere des Getters möglich.
Hat sich auf der Getteroberfläche, beispielsweise bei 20 °C, durch chemische Adsorption eine Monolage aus aktiven Gasen gebildet, ist die Oberfläche gegen weitere
Gasanlagerung geschützt. Sie kann aber durch Reaktivierung wieder adsorptionsfähig
gemacht werden. Bei der Reaktivierung durch „Tempern“ gelangen die oxidierenden
Verbindungen in tiefere Atomlagen des Getters. Reduzierende Verbindungen, wie zum
Beispiel Wasserstoffverbindungen, neigen dazu, bei hohen Temperaturen in die Gasphase überzugehen. Nach jeder Reaktivierung steigt die Konzentration der
verunreinigenden Atome im Inneren des Getters. Durch die höhere Konzentration der
Verunreinigungen im Getter wird deren Konzentrationsgradient geringer und verringert
damit die Aufnahmefähigkeit des Getters. Die verwendeten Getterpumpen (Fa. SAES
getters, Mailand, Italien) sind im Neuzustand mit einer Oxidschutzschicht belegt. Diese
ermöglicht den Einbau des Getters unter Atmosphäre. Zur Aktivierung des Getters muss
dieser dann kurz bis 900 °C erhitzt werden. Dadurch gelangt die Schutzschicht in tiefere
Atomlagen und der Getter ist in der Lage, schon bei Zimmertemperatur H2 sowie H2O,
CO, CO2, N2 und O2 gut zu adsorbieren.
Außer zum Reinigen des Edelgases sorgt eine weitere Getterpumpe dafür, dass die Güte
des Vakuums im Bereich der Elektronenkanone erhalten bleibt (vgl. Abbildung 61A und
Abbildung 63B).
145
6.2 Kombination einer Excimerlampe mit einem TOFMS
Für die TOFMS-Experimente mit elektronenstrahl-gepumpter Excimerlampe wurde ein
TOFMS-System verwendet, das ursprünglich für die Ionisation mit ca. 5 ns langen Laserpulsen konzipiert wurde. Mittels eines Spezielflansches wurde die oben beschriebene
Excimerlampe, wie in Abbildung 61 dargestellt, mit dem TOFMS verbunden. Die
MgF2-Linse der Excimergaszelle wurde so gewählt, dass der Ortsfokus der ExcimerVUV-Strahlung im Zentrum der Ionenquelle liegt. Die Strahlung der Lampe kann aufgrund der Excimerkinetik bei weniger als 1 bar Edelgasdruck nicht so kurz wie ein
Laser gepulst werden. Bei den Versuchen hatten die Excimerlichtpulse eine Länge von
ca. 8 ms (vgl. Abbildung 62A). Bei Laserionisation definiert der kurze Laserstrahl, während dessen Dauer die Ionen erzeugt werden, die Startzeit für den Flug der Ionen durch
das Flugzeitmassenspektrometer. Um bei den langen Excimerlichtpulsen akkurate Startbedingungen zu erhalten, ist es notwendig, die Hochspannung (HV) an den Abzugsbzw. Beschleunigungsplatten zu pulsen (Abbildung 61A). Die Abzugsplatten wurden
mittels schneller Hochspannungsschalter („push-pull“-Schalter Typ HTS 31-GSM mit
einer Schaltzeit von 15 ns / 3 kV, Fa. Behlke Elektronik, Germany), die wegen der von
ihnen ausgehenden elektromagnetischen Strahlung mit möglichst kurzen, geschirmten
Kabeln mit den Vakuumdurchführungen verbunden wurden, mit HV versorgt. Trotz
solcher Vorkehrungen konnte nicht verhindert werden, dass der Multi-Channel-Plate
(MCP) -Detektor, wie in Abbildung 62C sichtbar, einen Teil dieser Strahlung einfing.
Allerdings verursacht die Störstrahlung ein starkes Rauschen am Detektor zu einer Zeit,
in der kein Ionensignal erwartet wird. Sogar Wasserstoff würde aufgrund seiner kurzen
Flugzeit erst nach dem Rauschen am Detektor ein Signal verursachen. Es hat sich als
günstig erwiesen, das Beschleunigungsfeld, wie in Abbildung 62B dargestellt, erst nach
dem Abklingen der Lichtemission anzulegen. Dadurch werden in der Ionenqelle während des Lichtstrahls Ionen erzeugt und dann bei eingeschaltetem Abzugsfeld
gesammelt auf die Flugstrecke geschickt. Das gesamte Zusammenspiel von Zeit und
Dauer des Lichtpulses, des Abzugpulses und des resultierenden Detektor-Signals, hier
am Beispiel von Piperidin und Toluol, ist in Abbildung 62A-C dargestellt. Durch die
unterschiedliche Masse von Piperidin und Toluol erreicht Piperidin (amu 85) den Detektor nach 13 ms und Toluol (amu 92) nach 14 ms.
146
Kapitel 6
Abbildung 62: (A) VUV-Excimerlichtpuls aufgezeichnet mit einem Photodetektor. (B) Triggerpuls für die schnellen „push-pull“-Hochspannungsschalter, die den Repeller und den
Extraktor des TOFMS mit Spannung versorgen. (C) Excimerlampen-TOFMS Signal von Piperidin und Toluol, wobei die elektromagnetische Strahlung der Hochspannungspulse durch
den Detektor eingefangen wurde.
Die Spannungsversorgung der Elektronenkanone, die HV-Pulser, wie auch die Datenerfassungskarte (250 MHz, 1 Gs/s Transientenrekorderkarte, Typ DP 110, Fa. acqiris,
Genf, Schweiz), mit der das Detektorsignal aufgezeichnet wird, werden durch zwei
Pulsgeneratoren (Typ DG535, Stanford Research Instruments Inc., Sunnyvale, USA)
zeitlich gesteuert.
Aus dem Kathodenfilament, das kontinuierlich mit 6 V und 20 mA versorgt wurde, wurden mit 120 Hz 12 ms lang Elektronen extrahiert. Nach der Extraktion der Elektronen
und damit der Lichterzeugung wurde für 350 ns das Abzugsfeld in der Ionenquelle angeschaltet. Die Datenerfassungskarte bekommt zur selben Zeit einen Puls und zeichnet
von dieser Zeit an das Detektorsignal auf. Durch die Pulsgeneratoren konnten alle zeitlichen Abläufe optimiert werden. Es wurden auch unterschiedliche Zeiten für die positive
HV des Repellers und die negative HV der Extraktorplatte getestet. Es erwies sich dann
147
aber als optimal, beide für das Abzugsfeld verantwortlichen Platten gleichzeitig zu pulsen.
Das verwendete TOFMS ist ein kompaktes Reflektron-TOFMS mit einer feldfreien
Flugstrecke von 634 mm. Die Ionenquelle des TOFMS wird mit einer 210 l/s und das
Flugrohr mit einer 60 l/s Turbomolekularpumpe unter Vakuum gehalten. Der Einfachheit halber wird dieses TOFMS in Abbildung 61A als lineares TOFMS dargestellt.
Die Betriebsspannungen des Flugzeitmassenspektrometers mit Wiley-McLarenIonenquelle [41] waren für den Repeller +400 V (gepulst), für den Extraktor -400 V
(gepulst) und für die Linerspannung des Reflektrons –2500 V (statisch). Der zweistufige
Ionenspiegel besteht aus 13 Ringelektroden über eine Länge von 117 mm. Darin werden
die Ionen vom Linerpotenzial (-2500 V) zum Potenzial der 1. Elektrode (-500 V) in
einer ersten Stufe abgebremst und dann zwischen den 13 Elektroden, in denen das Potenzial von -500 V auf +450 V ansteigt, reflektiert.
Der Repeller und die Extraktorplatte liegen auf dem gleichen Potenzial, nur mit
unterschiedlichem Vorzeichen (vgl. Abbildung 61A). Wie schon früher beschrieben,
[18] befindet sich dadurch die Ionisationszone am Ende der auf 0 Potenzial liegenden
Einlassnadel, auch auf 0-Potenzial. Auf diese Weise wird das Abzugsfeld in der
Umgebung der Einlassnadel am wenigsten gestört und die Ionen werden von dort aus
gerade zum Flugrohr beschleunigt. Nach einer zweiten Extraktionsblende fliegen die
Ionen durch eine in Abbildung 61A nicht dargestellten elektrostatische Einzellinse und
eine y-x-Ablenkeinheit. Der Detektor des Reflektrons ist ringförmig mit einem 1,57“Innenloch, durch das die Ionen aus der Ionenquelle in das Flugrohr gelangen, bevor sie
nach Spiegelung im Ionenspiegel auf selbigen Ringdetektor treffen (nicht in
Abbildung 61A dargestellt).
Das verwendete Nadeleinlassystem ist in Veröffentlichungen bereits detailliert beschrieben [18, 59] worden. Es besteht aus einer Edelstahlnadel, in der eine
Quarzglaskapillare verläuft. Die Kapillare endet bündig mit der Nadel, wo das Probengas aus der Kapillare etwa 2 mm vor dem Zentrum der Ionenquelle effusiv in das
Vakuum strömt. Im Zentrum der Ionenquelle findet dann die Ionisation durch das VUVLicht statt. Für den Weg vom Probenahmeort zur Einlassnadel sei auf Kapitel 5.11 verwiesen. Allerdings konnte bei den hier präsentierten ersten Versuchen weder die
Probenahmestrecke noch die Einlassnadel geheizt werden.
148
Kapitel 6
In Abbildung 63A ist das gesamte, etwa 1,5 m hohe kompakte TOFMS mit Excimerlampe
zu
sehen.
Die
zu
diesem
Zeitpunkt
noch
nicht
integrierten
Spannungsversorgungen für die Elektronenkanone können ohne weiteres in den freien
19“-Einschubplatz integriert werden. Mit den Getterpumpen benötigt das Gerät auch für
längere Messungen nur einen normalen 220 V-Stromanschluss mit 16 AmperAbsicherung. Abbildung 63B zeigt ein Foto der für diese ersten Versuche verwendeten
Excimerlampe. Vor Abgabe dieser Arbeit konnte aufgrund eines im Rahmen der Bayerischen Forschungsstiftung geförderten Projekts die Größe der Lampe bereits halbiert
werden.
Abbildung 63: (A) Foto des mobilen 19“ TOFMS mit elektronenstrahl-gepumpter Excimerlichtquelle. (B) Vergrößerter Ausschnitt der Excimerlampe.
149
6.3 Messungen zur Spezifizierung der Excimer-TOFMS-Kombination
Während der nachfolgend beschriebenen Messungen zur Spezifizierung der ExcimerTOFMS-Kombination und bei der Messung von Motorradabgas wurde mit der in
Abbildung 63 dargestellten Excimerlampe bei 120 Hz Repetitionsrate 4 W VUV-Licht
erzeugt. Um das neue, noch nicht getestete Fenster, durch das der Elektronenstrahl zur
Excimererzeugung in das Edelgas gelangt, nicht zu zerstören, wurde die Excimerkammer nur bis etwa 500 mbar mit Edelgas gefüllt. Das Fenster kann theoretisch Drücken
bis über 1 bar standhalten. Mangels Ersatz wurde hier aber sehr vorsichtig vorgegangen.
Bei einem Druck von 1 bar wäre die Lichtausbeute viel höher gewesen. Auch die Brillanz und damit die Strahleigenschaften sind bei höherem Druck besser.
Bei den nachfolgend beschriebenen Messungen wurden mit der Excimerlampe bei
120 Hz VUV-Lichtimpulse erzeugt. Da die Datenerfassungskarte aber nicht mit 120 Hz
ganze Massenspektren auf die Festplatte des Messrechners schreiben kann, wurden zur
Reduzierung der Datenmenge immer eine größere Anzahl von Einzelspektren gemittelt
und das daraus resultierende gemittelte Spektrum gespeichert.
Ein großer Vorteil der elektronenstrahl-gepumpten Excimerlichtquelle gegenüber einem
VUV-Laser ist, dass die VUV-Wellenlänge durch Austausch des Excimergases verändert werden kann. Dadurch kann, wie in Abbildung 64 dargestellt, die Selektivität
verändert werden. Für den in Abbildung 64 dargestellten Versuch wurde eine Excimerlampe einmal mit Argon (Ar) und einmal mit Krypton (Kr) befüllt und mit dem
erzeugten Excimerlicht ein Gasgemisch mit Benzol und Toluol untersucht. Durch das
unterschiedliche Emissionsspektrum von Ar bzw. Kr mit einem Emissionsmaximum bei
lcent = 126 nm bzw. lcent = 147 nm und dementsprechend mit einem Maximum in der
Photonenenergie bei 9,8 eV bzw. 8,4 eV (vgl. Tabelle 1), konnte aus dem Probengas mit
Benzol und Toluol, deren IP bei 9,2 eV bzw. 8,5 eV liegt, Benzol selektiv ausgeblendet
werden. Auf diese Art und Weise ist es bedingt möglich, sich bei den Massenspektren
auf interessante Verbindungen zu konzentrieren und das Massenspektrum von uninteressanten Verbindungen frei zu halten. Durch Variation des Excimerlichts können auf
Grund der verschiedenen Ionisationspotenziale der Verbindungen Massensignale identifiziert werden. Die bei der Analyse von Spurenbestandteilen störenden Bestandteile von
Luft wie, z.B. N2, O2 und H2O, werden bei keiner der VUV-Wellenlängen aus Tabelle 1
ionisiert und damit nicht nachgewiesen.
150
Kapitel 6
Abbildung 64: (A) Excimer Emissionsspektrum von Argon (Ar2*, oben) und und Krypton
(Kr2*, unten). Zusätzlich eingezeichnet sind die Ionisationspotenziale (IP) von Benzol und
Toluol. (B) TOF-Massenspektrum von Benzol und Toluol ionisiert mit Argon- (Ar2*, lcent =
126 nm, oben) und Krypton- (Kr2*, lcent = 147 nm, unten) Excimerlicht. Mit Kryptonexcimerlicht ist in diesem Fall eine selektive Ionisation von Toluol möglich (rechts, unten).
Für die in Abbildung 64B dargestellten Massenspektren von Benzol und Toluol wurden
je 500 Einzelspektren zu einem gemittelt. Solch ein gemitteltes Spektrum ist hier dargestellt.
Zum Test, wie weich man mit VUV-Licht ionisiert, wurde Luft mit Piperidin direkt in
die Ionenquelle des TOFMS eingelassen und dort mittels VUV-Licht ionisiert. Das SPIMassenspektrum von Piperidin ist in Abbildung 65A dargestellt. Die Ionisation erfolgte
hier mittels 126 nm-Licht des Argon-Excimers. Abbildung 65 zeigt einen Piperidin- und
einen Toluol-Peak. Bei diesem Spektrum wird der Peak bei der Masse 92 m/z durch
eine Verunreinigung der Probenamestrecke mit Toluol hervorgerufen, welches zuvor in
hohen Konzentrationen eingelassen wurde. Zum Vergleich sind in Tabelle 9 die Massen
aufgeführt, bei denen bei einer Elektronenstoßionisation mit 70 eV-Elektronen Fragmente auftreten. Dabei zeigt sich, dass bei der Elektronenstoßionisation zum einen der
größte Peak nicht bei der ursprünglichen Molekülmasse von 85 m/z, sondern bei der
151
Fragmentmasse 84 m/z auftritt und zum anderen, dass 6 Fragmente vorkommen, die
gegenüber dem dominierenden Peak bei 84 m/z eine relative Signalstärke von mehr als
20 % haben. Bei der SPI von Piperidin mit Excimerlicht ist kein Fragment zu erkennen.
Der einzige Peak ist bei der Piperidinmasse 85 m/z.
Die Nachweisempfindlichkeit der VUV-Lampen-TOFMS-Kombination wurde mittels
eines Gasstandards ermittelt. Das Standardgas aus der Flasche (10 l, 200 bar, Fa. Messer
Griesheim, Krefeld, Deutschland) enthielt 10 ppm Benzol, Toluol und p-Xylol (BTX) in
Stickstoff. Dieses Gas wurde auch mit 126 nm-Argon-Excimerlicht ionisiert.
500 Einzelspektren wurden für das Spektrum in Abbildung 65B gemittelt, welches zur
Bestimmung der Nachweisgrenzen in Tabelle 10 diente. Um das Spektrum vom
hochfrequenten Rauschen zu befreien, wurde es zusätzlich noch gleitend über 0,25 m/z
geglättet.
Die Nachweisgrenzen werden hier nach der von Williams et al [50] vorgeschlagenen
Methode ermittelt. Danach ist die Nachweisgrenze das Verhältnis von Signal (S) zum
Rauschen (R). Es gibt verschiedene Meinungen darüber, in welchem Verhältnis das
Signal zum Rauschen (S/N) mindestens stehen muss, damit ein echter Massenpeak erkannt wird. In dieser Arbeit soll S/N mindestens 3 sein. Die Nachweisgrenze (d) kann
mittels [6.1] berechnet werden. Dabei ist s die Standardabweichung des Signals zwischen zwei echten Peaks, c die Konzentration der Substanz im Analysegas, m das
Grundlinienniveau des Spektrums und p die Signalhöhe.
3s
d=
c.
p-m
[6.1]
Da dieses Spektrum eine Mittelung von 500 Einzelspektren ist und die Nachweisgrenze
mit der Wurzel aus der Anzahl der gemittelten Spektren korreliert, ist die Nachweisgrenze für ein einzelnes Massenspektrum in diesem Fall 22 mal schlechter. Zu bedenken
ist allerdings, dass bei 120 Hz solch ein gemitteltes Spektrum in 4,1 s erzeugt wird.
Der Grund für die schlechte Massenauflösung von R50% = 88 (bei 85 m/z) ist die mangelnde Erfahrung mit einer Excimerlampen-TOFMS-Kombination. Zudem war die
Ionenquelle den Excimerstrahlgegebenheiten nicht angepasst. Vor allem das Pulsen der
Abzugsplatten bereitete Schwierigkeiten.
Um in Zukunft bessere Massenauflösungen zu erzielen, sollten spezielle Ionenquellen
eingesetzt werden. Z.B. könnte die Lampe in Verbindung mit einem Orthogonal-
152
Kapitel 6
TOFMS [125-128] betrieben werden. In Verbindung mit einem Orthogonale-TOFMS
kann die Excimerlampe sogar kontinuierlich betrieben werden. Daneben gibt es noch
die Möglichkeit, die erzeugten Ionen kurzzeitig in der Ionenquelle zu speichern und erst
danach zu beschleunigen. Solche Konzepte mit Ionenspeicher, in einer statischen Potenzialmulde [129, 130], oder in einem elektromagnetischen Hochfrequenzfeld [131]
wurden für ähnliche Problematik bereits verwendet. Zukünftige Arbeiten werden hier
ansetzen. Es ist auch geplant, die Excimerlampe mit einem FT-ICR MS (engl.: Fourier
Transform - Ion Cyclotron Resonance Mass Spectrometer), einem IMS (engl.: Ion Mobility Spectrometer) und einem Quadrupolmassenspektrometer zu koppeln. Das Konzept
mit einem Quadrupolmassenspektrometer bietet sich an, da hier die Lampe nicht gepulst
werden muss.
Für ein TOFMS-System mit Ionenquellen ohne Ionenanreicherungsschritt ist eine geringe zeitliche Lichtpulsbreite wichtig, um eine hohe Massenauflösung zu erreichen.
Abbildung 65: (A) Excimerlampen (Ar2*, lcent = 126 nm) TOFMS-Massenspektrum einer
Piperidinprobe mit Toluolverunreinigung. (B) Massenspektrum eines 10 ppm-Benzol-, Toluol- und p-Xylol (BTX)-Standards.
153
Entscheidend für die Lichtpulsbreite einer Excimerlampe, ist der Gasdruck in der Edelgaszelle [92]. Wie sich schon jetzt abzeichnet, wird der ungünstige zeitliche Verlauf der
Excimerlichtemission (Abbildung 62A) durch neue Folien, die bis 8 bar Druckdifferenz
aushalten, sehr verbessert werden können. Es wird erwartet, dass sich mit solchen Folien Lichtpulse von unter 100 ns erzeugen lassen.
Durch Messungen mit einem Halbleiterphotodetektor für VUV-Photonen (Typ AXUV20. IRD Inc., Torrance, USA) am Ionisationsort hat sich gezeigt, dass von den 2,6·1018Photonen/s, die bei 4 W VUV-Lichtleistung entstehen, nur 2,6·1014 Photonen/s am Detektor, mit 20 mm2 aktiver Fläche, nachgewiesen werden. Daraus ergab sich ein
enttäuschender Lichtsammelfaktor von 8,8·10-5. Verglichen mit der Photonenzahl, die
man bei selbem Abstand in der Ionenquelle ohne optische Komponenten, also durch die
4 P Strahlung, erhalten würde, ist diese Anzahl nur um den Faktor 10 kleiner als die
nachgewiesene Anzahl. Da es theoretisch möglich sein sollte, einen Lichtsammelfaktor
von 10-2 zu erreichen, könnte mit Verbesserungen in diesem Bereich noch viel Nachweisempfindlichkeit gewonnen werden. Insbesondere müsste dann die Beschichtung des
Parabolspiegels verbessert werden. Es ist aber auch eine Methode denkbar, bei der eine
Linse sehr nahe vor den Leuchtpunkt gesetzt wird, um damit einen möglichst großen
Anteil der 4 P-Strahlung zu sammeln und den Leuchtpunkt in das Zentrum der Ionenquelle abzubilden.
Tabelle 9: Fragmentliste von Piperidin ionisiert mit VUV-Licht der Excimerlampe (SPI, Ar2*,
λcent. = 126 nm) und durch 70 eV-Elektronen [64]. Verglichen werden die Massenpeaks
(> 20 % rel. Grösse) relativ zum größten vorkommenden Peak. Bei SPI tritt keine Fragmentation auf.
Massenpeaks von Piperidin
[m/z]
42
43
44
56
57
84
85
(M+)
Relatives SPI-Signal
(Ar2*, λcent. = 126 nm) [%]
-
-
-
-
-
-
100
Relatives EI-Signal (70 eV)
[%]
25
22
33
44
44
100
53
154
Kapitel 6
Tabelle 10: Mit VUV-Licht einer Excimerlampe (Ar2*,
λcent. = 126 nm) erreichte Nachweisempfindlichkeiten für
Benzol, Toluol und Xylol (BTX).
Substanz
S/N für 10
ppm Standard
Nachweisgrenze (S/N > 3)
Benzol
10
3 ppm
Toluol
9
3 ppm
1,4-Xylen
7
4 ppm
155
6.4 Erste Gasanalysen mit einem elektronenstrahl-gepumpten Excimer-TOFMS
Der Einsatzbereich des beschriebenen TOFMS mit Excimerlampe wird zweifelsohne im
Bereich der Online-Messungen und Online-Überwachung von industriellen Prozessen
liegen, wie sie z.B. in Raffinerien stattfinden. Weiterhin sind Anwendungen in der Umweltanalytik, in der medizinischen Diagnostik und der Chemiewaffenaufspürung
denkbar. Um die Möglichkeiten dieses einfach zu bedienenden, stabil laufenden und
wartungsfreundlichen Systems aufzuzeigen, wurden aus Platzgründen Messungen an
einem Motorrad (Yamaha, Typ 43F) im Labor durchgeführt. Dabei wurde das Abgas am
Ende des Auspuffs entnommen (vgl. Kapitel 5.11).
Abbildung 66A zeigt ein Motorradabgasspektrum, das kurz nach dem Motorstart aufgenommen wurde. Zum Ionisieren wurde wieder Argon-Excimerlicht (126 nm) verwendet.
Um Änderungen in der Zusammensetzung besser verfolgen zu können, wurden nur
60 Einzelspektren zu einem Spektrum gemittelt. Dazu wurden zwei Spektren in der
Sekunde gespeichert. Trotz der schlechten Massenauflösung ist die Reihe der ethylierten
Benzole von Benzol (78 m/z) über Toluol (92 m/z) und Xylol/Ethylbenzol (106 m/z) bis
Trimethylbenzol (120 m/z) gut zu erkennen. Weiterhin wurden bei 70 m/z und 84 m/z
Massenpeaks sichtbar, die wahrscheinlich aufgrund der Verbrennungsbedingungen im
Motor, Krotonaldehyd [28] und Pentenal zugeschrieben werden können.
In Abbildung 66B (unten) ist eine kontinuierliche Motorradabgasmessung über 60 s in
3D-Ansicht dargestellt. Die Messung begann vor dem Start des Motors und dauerte bis
nach dem Abstellen des Motors. Dazwischen wurde die Drehzahl des Motors ohne Last
zweimal kurzzeitig erhöht. Diese Messung ist Teil einer längeren Messung, bei der drei
Startzyklen aufgenommen wurden. Diese drei Zyklen sind in Abbildung 66B (oben) in
Konturdarstellung zu sehen. Dabei entspricht die Grauskalierung zu helleren Tönen
einem zunehmenden Signal. Man erhält diese Art von Konturdarstellung, wenn man
senkrecht von oben auf eine 3D-Darstellung, wie in Abbildung 66B (unten) dargestellt,
fotografiert. Trotz der nicht geheizten Probenahmestrecke sind die zwei Drehzahlerhöhungen in beiden Darstellungen gut zu erkennen.
156
Kapitel 6
Abbildung 66: (A) Excimerlampen (Ar2*, lcent = 126 nm) SPI-Massenspektrum von Motorradabgas kurz nach dem Start des Motors. (B) 3D Darstellung einer Excimerlampen-(Ar2*,
lcent = 126 nm) SPI-TOFMS-Motorradabgasmessung. Bei ca. 60 s wird das Motorrad gestartet, dann wird der Motor im Stand zweimal auf Touren gebracht und bei etwa 90 s wieder
abgestellt. Man erkennt das Emissionsverhalten bezüglich Benzol, Toluol und Xylol (BTX).
Rechts darüber sind in der Konturdarstellung drei Motorstartzyklen dargestellt.
157
Erstellt wurden diese Spektren durch das Aneinanderreihen der gespeicherten Spektren.
Bei dieser Messung wurden 2 Spektren pro Sekunde gespeichert. Ein großer Teil dieser
auf LabView basierenden Daten Erfassungs- und Auswertesoftware wurden schon früher an der GSF entwickelt [18] und dieser Anwendung angepasst.
Abbildung 67: Kalibrierte Konzentrations-Zeitprofile von Benzol, Toluol, Xylol und Trimethylbenzol einer Excimerlampen-TOFMS (Ar2*, lcent = 126 nm) Motorradabgasmessung.
Die Kalibration wurde mit Hilfe eines externen Gasstandards vorgenommen.
In Abbildung 67 werden von derselben Messung, wie in Abbildung 66B (unten) dargestellt, 4 Konzentrationsverläufe gezeigt. Diese Konzentrationsverläufe wurden durch
Integration der einzelnen Massenpeakts aller gespeicherten Spektren gewonnen. Stellt
man die Werte der Flächen (Integral der Massenpeaks) im zeitlichen Verlauf dar, so
ergeben sich wie in Abbildung 67 dargestellte Konzentrationsverläufe. Misst man davor
oder danach den Probenstandard, der diese Substanzen in bekannter Konzentration enthält, unter denselben Gerätebedingungen, so kann durch den Vergleich der Flächen
unter den Peaks die Konzentration der Substanz im Motorradabgas bestimmt werden
(vgl. Tabelle 10). Auf diese Weise konnten die Verläufe von Benzol und Toluol quantifiziert werden. Die Xylol- bzw. Ethylbenzol-Spur wurde näherungsweise durch das
p−Xylol im Gasstandard quantifiziert, da die Photoabsorptionswirkungsquerschnitte der
158
Kapitel 6
Xylol-Isomeren und des Ethylbenzols als vergleichbar angenommen werden können. Da
Trimethylbenzol nicht im Standard enthalten war, zeigt dieser Verlauf nur relative Änderungen.
159
7 Konzepte zur Steigerung der Empfindlichkeit sowie zur
Modifikation des REMPI-Selektivitätsprofils
Zur Steigerung der Empfindlichkeit eines TOFMS-Systems wird in diesem Abschnitt
untersucht, ob mit einem Modulator, ähnlich wie er zur GC/GC-Kopplung verwendet
wird, sich Bestandteile eines Probengases kurzzeitig anreichern, diese konzentriert frei
setzen lassen und sich damit die Empfindlichkeit eines TOFMS erhöhen lässt.
Weiterhin wird untersucht, ob sich mit einem Modulator in situ gesundheitsrelevante
Luftschadstoffe wie Aldehyde und Amine mit einem geeigneten aromatischen Reagenz
derivatisieren lassen, um sie so der selektiven laserinduzierten resonanzverstärkten Multiphotonenionisations-Massenspektrometrie zugänglich zu machen.
7.1 Anreicherung in der Probenahme
Um die Nachweisempfindlichkeit eines REMPI-TOFMS-System für Gasphasenmessungen zu erhöhen, ist es möglich, die Probe vor der massenspektrometrischen
Untersuchung über einen kurzen Zeitraum anzureichern und dann thermisch zu desorbieren. Dazu ist ein Verfahren gefragt, bei welchem das Aufkonzentrieren der Probe
möglichst schnell geht, um die hohe Zeitauflösung eines Massenspektrometers weiterhin
nutzen zu können.
Silikongummi z.B. ist geeignet zum Anreichern von Proben. Durch die Struktur von
Silikon gelangen Substanzen wie bei einer Flüssigkeit in das Material und können dort
stoffspezifisch gespeichert werden. Ein geeignetes Verfahren, die Substanzen schnell
und konzentriert aus dem Silikon freizusetzen, ist die Thermodesorption (TD). Um die
Möglichkeiten zu untersuchen, mit dieser Methode die Empfindlichkeit eines TOFMSSystems zu steigern, wurde eine kleine Thermodesorptionseinheit in die Probenahme
des beschriebenen Laser-TOFMS eingefügt. Die Thermodesorptionseinheit besteht aus
einem Modulator und einer Kapillare mit stationärer Silikonphase.
160
Kapitel 7
7.1.1 Modulator
Bei dem verwendeten Modulator handelte es sich um den von Burger et al. beschriebenen Typ [132]. Der Modulator (Abbildung 68) besteht aus einem dünnen
Edelstahlröhrchen (1), das durch seinen elektrischen Widerstand mit Strom geheizt
wird. Das Heizen erfolgt segmentweise. Die Temperatur und die Heizdauer jedes Segmentes kann durch das von einem Mikroprozessor gesteuerte Netzgerät eingestellt
werden. Der Heizstrom wird dem ca. 10 cm langen Röhrchen (1) durch 11 Stege (2)
zugeführt. Die Stege bestehen aus dem selben Material wie das Hauptröhrchen (1). Dadurch heizen sich die Stege selber genau so auf wie das Hauptröhrchen und es entstehen
keine kalten Stellen an den Stromzuführungen. Durch das Modulatorröhrchen (1) verläuft eng anliegend eine GC-Kapillare mit stationärer Phase.
Die stationäre Phase, die in der Regel aus einer Silikonschicht auf der Innenseite der
Kapillare besteht, absorbiert im kalten Zustand Bestandteile des Probengases. Durch
Heizen der Kapillare werden die aufgenommenen Bestandteile wieder frei gesetzt. Da
mit dem Modulator ein Segment nach dem anderen geheizt werden kann, schiebt man
beim Heizen die desorbierten Substanzen durch den Modulator und bekommt nach dem
Abbildung 68: Schematische Darstellung des Modulators mit Anreicherungskapillare. Der
Modulator besteht aus 10 Segmenten, die nacheinander geheizt werden können. Das Heizen
der Segmente geschieht nach dem Prinzip des elektrischen Widerstands. Die Ströme zum
Heizen liefert ein durch einen Mikroprozessor gesteuertes Netzgerät. Das Edelstahlröhrchen
(1) hat 11 Stege (2), über die der Strom durch die Leitungen (3) zugeführt wird. In dem Modulator steckt eine Kapillare (4) mit örtlich begrenzter stationärer Phase (5).
161
Heizen des letzten Segmentes einen konzentrierten Puls der angereicherten Substanzen.
Der Puls mit den angereicherten Substanzen wird vom Modulator zur Ionenquelle des
TOFMS gezogen und bewirkt dort kurzzeitig einen Signalanstieg und damit auch eine
Verbesserung der Nachweisgrenze für Substanzen, die vom Silikon absorbiert wurden.
In gewissen Grenzen kann durch längere Anreicherung im Modulator die Nachweisempfindlichkeit erhöht werden. Andererseits verliert man an Zeitauflösung bei langen
Absorptionsphasen.
Der Modulator wurde nach dem Konzept von Burger aufgebaut [132] und ist geeignet
Kapillaranreicherungseinheiten bis zu einem Außendurchmesser von 0,53 mm zu desorbieren. Besonders schwierig bei der Herstellung des neuen Modulators war die
leitende Befestigung der Stege (2) an das Hauptröhrchen (1). Weichlot konnte aufgrund
seiner niedrigen Schmelztemperatur nicht verwendet werden. Daher wurde Hartlot aus
Silber eingesetzt. Das Hartlöten von Röhrchen mit sehr dünnen Wandstärke mit einer
Mikrobunsenbrennerflamme erfordert Fingerspitzengefühl, da die Röhrchen schnell
verglühen. Außerdem darf nur sehr wenig Silberlot verwendet werden, um nicht durch
eine dicke Silberschicht an den Verbindungsstellen der Röhrchen den elektrischen Widerstand zu erniedrigen. Ein niedrigere Widerstand an der Lötstelle hätte zu Folge, dass
sich bei der Erwärmung durch elektrischen Strom, diese Stelle weniger erwärmt als der
Rest der Röhrchen.
Mit dem maximalen Strom von 10 A bei 5 V ließ sich mit dem neuen Modulator innerhalb von 2-3 Sekunden eine Segmenttemperatur von 200 °C erreichen.
Die Temperatur in der Anreicherungskapillare im Modulator konnte mit einem selbstgebauten NiCrNi-Thermoelement gemessen werden. Das Thermoelement wurde aus
einem Ni- und einem CrNi-Draht mit je 0,025 mm Durchmesser gefertigt. Verbindet
man die beiden Drähte durch eine Punktschweißung, so erhält man ein NiCrNiThermoelement und damit temperaturabhängige Spannungen. Die Drähte wurden in
eine Kapillare mit ca. 0,3 mm Außendurchmesser gefädelt. Die Kapillare mit dem
Thermoelement konnte in die Kapillare mit der stationären Phase gesteckt werden. So
konnte die Temperatur an verschiedenen Stellen im Modulator gemessen werden.
Bei Kopplung des Modulators mit einem TOFMS-System kann die Anreicherungskapillare durch permanentes Heizen über die gesamte Länge deaktiviert werden. Bei heißer
stationärer Phase werden Gasbestandteile vom Silikongummi absorbiert und treten
gleich wieder zurück in den Gasstrom. Dadurch ist die Wirkung des Modulators aufge-
162
Kapitel 7
hoben. Es ist also möglich, wie in Abbildung 69 dargestellt, den Modulator und seine
Wirkung ein und aus zu schalten. Beim Heizen des Modulators über die ganze Länge
misst das TOFMS die Verbindungen mit sehr hoher Zeitauflösung, wie sie von der Probenahme aufgenommen werden (Abbildung 69A). Ist der Modulator aktiv, indem er
z.B. permanent segmentweise von vorne nach hinten geheizt wird, so werden im Modulator Substanzen angereichert und je nach Zykluslänge des Modulators konzentrierte
Pulse in Richtung der Ionenquelle geschickt (Abbildung 69B). Die Zeitauflösung solch
einer Messung hängt in diesem Fall von der Zykluslänge des Modulators ab.
Hinsichtlich des Anschlusses des Modulators an die Probenahme des TOFMS ist es sehr
wichtig, dass die permanente Heizung der Probenahme bis ganz zum Modulator reicht.
Eine kalte Stelle zwischen Anreicherung und Analysator bewirkt eine zeitliche Verschmierung der Konzentrierung.
Abbildung 69: Konzept zur Kopplung eines Modulators mit einem REMPI-TOFMS zur Steigerung der Empfindlichkeit. Wird der Modulator während der Probenahme permanent über
die gesamte Länge geheizt, wird ohne Anreicherung der Probe mit hoher Zeitauflösung gemessen (A). Wird der Modulator sequenziell beheizt, so findet eine Anreicherung in den
kalten Segmenten statt und nach jedem Heizdurchgang gelangen die angereicherten Substanzen gepulst zum TOFMS.
163
In dem verwendeten Modulator wurden Kapillaren mit verschiedenen Schichtstärken
der stationären Phase eingesetzt. Die Kapillaren basierten auf herkömmlichen GCKapillaren und wurden von uns für den Einsatz als Anreicherungseinheit modifiziert.
7.1.2 Anreicherungskapillare
Anreicherungskapillaren für den Modulator können aus belegten GC-Kapillaren gefertigt werden. Im Modulator kamen Anreicherungskapillare mit einem ID von ca. 0,5 mm
und einer Dicke der inneren Silikonbeschichtung von 0,35 mm oder 2,65 mm (z.B. SE 30
mit 100 % Polydimethylsiloxan-Phase) zum Einsatz (siehe Abbildung 70).
Zur Vorbereitung der Kapillaren für den Modulatoreinsatz wird die stationäre Phase an
den Enden der Kapillare entfernt. Die im Modulator eingebrachte Kapillare darf nur
innerhalb des Modulators eine Phase haben, damit außerhalb an den nicht modulierten
Stellen keine Substanzen absorbiert werden.
Taucht man die etwa 20 cm lange Kapillare für den Modulator in eine Mischung aus
30 % Azeton und 70 % Hexan, so steigt die Flüssigkeit durch den Kapillareffekt in der
Kapillare bis zu einer bestimmten Höhe über die Flüssigkeit. Lässt man die Flüssigkeit
bis zu der Stelle steigen, bis zu der man die Phase auswaschen möchte, so bricht das
Azeton bis dort hin die Silikonstruktur auf. Lässt man die Kapillare etwa 10 min in der
Mischung, kann man anschließend die stätionäre Phase durch mehrmaliges Aufsteigenlassen von reinem Hexan entfernen. Dies macht man auf beiden Seiten der Kapillare.
Hält man die Kapillare gegen das Licht und betrachtet die Stelle bis zu der die Phase
entfernt wurde, so erkennt man bei vollständiger Auswaschung der Phase den Übergang.
Bevor die Kapillaren eingesetzt werden können, müssen sie jetzt nur noch gereinigt
werden. Dies geschieht, indem man unter Edelgasfluss die Anreicherungskapillaren bei
ca. 250 °C einige Stunden lang ausheizt.
7.1.3 Ultradicke Anreicherungskapillare
Zur Herstellung von Anreicherungskapillaren mit ultradicker stationärer Phase verwendet man unbelegte Kapillaren, in die man einen Silikongummischlauch einfädelt. Das
Herstellungsverfahren dieser Anreicherungskapillaren stammt von Burger [133].
164
Kapitel 7
Der Silikongummischlauch hat im Normalzustand einem Außendurchmesser (AD) von
0,65 mm und einem ID von 0,3 mm und wird für medizinische Zwecke von der Firma
Technical Products Inc. (Georgia, USA) vertrieben. Um den weichen Silikongummischlauch in die Kapillare mit einem ID von 0,53 mm zu bekommen, muss der Schlauch
etwa bis zu seiner doppelten Länge gedehnt und dann in flüssigem Stickstoff steif gefroren werden. Die gefrorenen Enden des Schlauches müssen unter flüssigem Stickstoff
entfernt werden, da sie durch die Pinzetten, mit denen der Schlauch gehalten wurde,
verformt sind. Der steife und durch das Dehnen jetzt etwas dünnere Schlauch lässt sich
untergetaucht in den flüssigen Stickstoff in die Kapillare schieben. Mit einer weiteren
dünneren Kapillare, lässt sich das hintere Ende des gefrorenen Schlauches in die Kapillare schieben. Nimmt man die Kapillare mit dem Schlauch aus dem flüssigen Stickstoff
heraus, so wird das Silikon wieder weich und der Schlauch zieht sich zusammen. Das
Resultat ist eine Kapillare, in der ein Silikongummischlauch überall eng anliegt. Die
Dicke der stationären Phase beträgt hier etwas mehr als 100 mm. Da die Länge des Silikonschlauchs nicht genau der Länge vor dem Dehnen entspricht, sind einige Versuche
nötig um die Länge des Silikongummischlauchs in der Kapillare zu erhalten, die der
Länge des Modulators entspricht.
Der Transfer der angereicherten Probe in das Flugzeitmassenspektrometer wird über
eine unbelegte Kapillare mit ID 0,32 mm realisiert. Zu beachten ist, dass diese Anreicherungskapillare mit ultradicker stationärer Phase einen freien ID von unter 0,3 mm hat
(vgl. Abbildung 70B). Damit stellt diese Anreicherungskapillare eine kleine Flussre-
Abbildung 70: Querschnitt durch ein Quarzkapillare mit 2,65 mm dicker stationärer Phase
(A). Im Vergleich dazu der Querschnitt durch eine Kapillare mit 100 mm dicker Silikongummiphase (B). Beide Querschnitte sind etwa um den Faktor 100 vergrößert dargestellt.
165
striktion dar. Ein geringerer Gaseinlass in das TOFMS hat eine negative Auswirkung
auf die Nachweisgrenzen. Auch diese Anreicherungskapillare muß wie oben beschrieben gereinigt werden.
7.1.4 Online-Anreicherung
Eine wichtige Voraussetzung für den Einsatz des Modulators mit Anreicherungskapillare ist, dass das Modulatorsystem sich gegenüber unterschiedlichen Konzentrationen von
Substanzen im Analysengas linear verhält. Die Konzentration einer im Modulator desorbierten Substanz soll in einem möglichst weiten Konzentrationsbereich linear von
der eingebrachten Konzentration abhängen. Dieses Verhalten ist erforderlich, um über
eine Probenstandard quantifizieren zu können.
Abbildung 71: Darstellung der linearen Beziehung zwischen der auf die Anreicherungseinheit injizierte Menge Formaldehyd-Oxim (HCHO-Oxim) und dem anschließend
nachgewiesenen Gesamtionensignal.
Ab einem bestimmten durchstömten Volumen kann eine Substanz je nach Modulatorlänge und stationärer Phase nicht mehr vollständig von der kalten Phase gehalten
werden. Es kommt dann zum so genannten Durchbruch der Substanz durch die Anreicherungseinheit. Das Durchbruchsvolumen, nach dem eine Substanz auch ohne heizen
166
Kapitel 7
aus der Anreicherungseinheit gelangt, ist für jede Verbindung unterschiedlich und hängt
von ihrer Flüchtigkeit und Polarität ab. Für quantitative Messungen mit dem Modulator
ist es erforderlich, dieses Volumen zu kennen, da sich die Absorption des Modulators
bei Volumen über dem Durchbruchsvolumen nicht mehr linear verhält.
Um die Linearität der Trap unterhalb des Durchbruchsvolumen nachzuweisen, wurden
verschiedene Mengen in unterschiedlichen Konzentrationen des für spätere Messungen
interessanten O−Benzyloximformaldehyde (HCHO-Oxim) mit einer Spritze in den Modulator mit Anreicherungskapillare injiziert. Nach Einbringen der Substanz wurde der
Modulator gestartet und die desorbierte Verbindung mit REMPI bei 266 nm nachgewiesen. Der Modulator machte solange einen Desorptionszyklus nach dem anderen, bis die
Anreicherungskapillare vollkommen frei vom HCHO-Oxim war. Nach dem ersten Modulatorzyklus wird das meiste HCHO-Oxim detektiert. Danach nimmt das Ionensignal
von Zyklus zu Zyklus stark ab.
Für die Untersuchung zum linearen Verhalten der Modulatorsystems muss das gesamte
HCHO-Signal verwendet werden. Dazu wurde das Ionensignal vom ersten bis zum letzten Modulatorzyklus zusammenaddiert und mit den Gesamt-Ionensignalen der anderen
injizierten Mengen HCHO-Oxim verglichen (Abbildung 71).
Das Gesamt-REMPI-Signal in Abhängigkeit der Injizierung von 3,3 ng bis 67 ng
HCHO-Oxim in den 10 cm langen Modulator mit 10 Segmenten und einer präparierten
SE 30 Kapillare (2,65 nm stationäre Phase) ist in Abbildung 71 zu sehen. Es zeigt sich
ein lineares Desorptionsverhalten der Anreicherungskapillare gegenüber der injizierten
Mengen HCHO-Oxim. Auch bei Injizierung verschiedener Verdünnungen des HCHOOxims mit Hexan zeigte sich das lineare Verhalten gegenüber der injizierten HCHOOxim Menge.
Bei Online-Messungen wird das Analysengas permanent durch den Modulator zur Ionenquelle des TOFMS gesaugt. Solange die Segmente des Modulators nicht geheizt
werden, werden Substanzen in der stationären Phase des Segments angereichert. Verschiedenen Substanzen lassen sich aufgrund ihrer unterschiedlichen Flüchtigkeit und
Polarität unterschiedlich gut anreichern. Um die Online-Messeigenschaften des Modulators mit verschiedenen Substanzen zu untersuchen, wurde das TOFMS mit dem
Modulator und einen Probenstandard verbunden. Der Probestandard enthielt unter anderem 0,8 ppm Anilin (93 amu), einige ppb Indol (117 amu) und 0,03 ppm Naphthalin
(128 amu).
167
Abbildung 72 zeigt eine Messung bei der 5 Minuten lang bei einem Fluss von
10 ml/min das Standardgas durch den kalten Modulator gezogen wurde. Anschließend
wurde der Modulator gestartet. Zum Zeitpunkt t = 0 s begann der Modulator seinen ersten von 15 Heizzyklen. Während der ganzen Messung war der Modulator auf der
Eingangsseite mit dem Standardgas verbunden. Der Modulator wurde somit permanent
mit Standardgas versorgt. Die Segmente wurden jeweils 2,5 s mit 10 A bis 200 °C aufgeheizt.
Abbildung 72: Zeitlicher SPI-Signalverlauf einer Messung von Anilin, Indol und Naphthalin
nach Anreicherung der Substanzen im Modulatur über 5 Minuten. Die einzelnen Peaks
stammen von je einem Desorptionsdurchlauf des Modulators. Während der ganzen Messung
gelangen die Substanzen kontinuierlich aus einem Standardgasgemisch zum Modulator. Aus
diesem Grund bleiben die Signalerhöhungen im hinteren Verlauf der Messung konstant.
Aufgrund der langen Probenahmezeit vor dem Modulationsbeginn sind die ersten
Signalpeaks von Abbildung 72 am höchsten. Die Überhöhung des ersten Anilin-Peaks
ist sehr gering. Anilin ist sehr flüchtig und bricht selbst bei einer Konzentration von
0,8 ppm innerhalb von Sekunden durch die Anreicherungskapillare mit einer 2,65 mmSilikonphase. Erkennbar ist dies auch daran, dass das Signal zwischen den Modulatorzyklen nicht bis zur Grundlinie zurück geht. Aufgrund dieser schlechten Retention des
Anilins lässt sich die Nachweisgrenze mit der verwendeten Anreicherungskapillare
nicht sehr erhöhen. Durch die schwache Wechselwirkung zwischen Anilin und der stati-
168
Kapitel 7
onären Phase lässt sich am Konzentrationsverhalten in Abbildung 72 (oben) bei genauer
Betrachtung der Temperaturanstieg in den einzelnen Segmenten erkennen. Beim Heizen
jedes Segments kann etwas Anilin den Modulator verlassen. Daher stammen die scharfen Spikes auf den Signalanstiegen.
Die Konzentration von Indol im Standardgas war bei weitem am geringsten. Aus diesem
Grund und aufgrund seiner Flüchtigkeit ist die Signalüberhöhung des ersten Peaks am
größten. Bei Anreicherung von Substanzen unterhalb des spezifischen Durchbruchvolumens erwartet man bei einer Probenahmezeit von 5 Min gegenüber einer
Probenahmezeit von 30 s eine Signalvergrößerung um den Faktor 10. Bei der in
Abbildung 72 dargestellten Messung hatten die Modulatorzyklen eine Länge von 30 s.
Das erste Signal sollte daher etwa 10 mal so groß sein wie die weiteren Signalpeaks im
Abstand von 30 s. Dass der erste Signalpeak nicht die 10-fache Fläche der folgenden
Peaks hat, liegt wahrscheinlich daran, dass das Durchbruchsvolumen für die Substanzen
überschritten wurde. Denkbar ist auch, dass die Substanzen nich vollsändig desorbiert
werden mit einem Heizzyklus. Hierzu sind künftig weitere Versuche notwendig.
Mit Naphthalin verhält es sich ähnlich wie mit Indol. Hier ist jedoch auffällig, dass die
Peaks etwas schärfer sind und ca. 4 s früher auftreten als die von Indol. Grund dafür
kann entweder die etwa 2 m lange, unbelegte 250 °C heiße Transferkapillare zwischen
Modulator und TOFMS sein, die trotz fehlender Beschichtung für verschiede Substanzen verschiedene Retentionszeiten zeigt, oder eine kalte Stelle an der Verbindung
zwischen Modulator und Transferkapillare. Aufgrund von Adsorptions- und Kondensationseffekten verursacht eine kalte Stelle in der Probenahme zwischen Modulator und
Ionenquelle in Abhängigkeit von der Flüchtigkeit der Substanzen eine Verzögerung und
Verbreiterung der Pulse.
Zusammenfassend lässt sich sagen, dass sich durch das 5-minütige Vorkonzentrieren die
Nachweisempfindlichkeit gegenüber einer Messung ohne Modulator für Anilin um den
Faktor 1,6 verbessern lies. Für Indol und Naphthalin war es hingegen schon das 23bzw. 22-fache. Bei einer Modulator-Zykluszeit von 30 s verbesserte sich die Nachweisempfindlichkeit für Anilin um das 1,5-fache. Für Indol und Naphthalin um das 3,5- und
7,1-fache. Mit der Anreicherungskapillare mit 0,35 mm dicker stationärer Phase wurden
schlechtere Ergebnisse erzielt. Die Anreicherungskapillare mit ultradicker Phase brachte
keine Verbesserung, da durch sie der Gasfluss geringer ist und damit die Gasdichte am
Ionisationsort niedriger ist, was die Empfindlichkeit vermindert. In Zukunft sollten da-
169
her dickere Anreicherungseinheiten mit mehreren parallelen Silikonschläuchen getestet
werden [134, 135]. Diese würden den Fluß nur gering beeinflussen, das Gas hätte eine
relativ geringe Geschwindigkeit in der Anreicherungseinheit und könnte damit gut mit
der dicken Silikongummiphase wechselwirken.
7.1.5 Offline-Anreicherung
Mit einem TOFMS-Systems dauert die Analyse einer Gasprobe nur wenige Sekunden.
Aus diesem Grund sind die TOFMS-Systeme zum Online-Messen von dynamischen
Prozessgasen gut geeignet.
Manchmal ist es allerdings, z.B. aus Platzgründen, nicht möglich, das Massenspektrometer nahe genug am Messpunkt aufzubauen. In diesem Fall ist es hilfreich, wenn das
Massenspektrometer auch für die Analyse von offline gesammelten Proben geeignet ist.
Um dies mit Hilfe des Modulators zu testen, wurden Anreicherungskapillaren auf Basis
einer SE 30-Kapillare extern mit Autoabgas beladen. Mit Hilfe einer kleinen Saugpumpe wurden bei einem Fluss von 1 l / min Abgas vom Kaltstart eines Ottomotor 1 Minute
lang durch die Anreicherungskapillare gesaugt. Anschließend wurden die Anreicherungskapillaren in den Modulator gesteckt und die Desorptionssequenz gestartet.
Abbildung 73 zeigt das REMPI-Messergebnis bei 266 nm von zwei desorbierten Anreicherungskapillaren. Das untere Spektrum ist ein wenig schwächer als das obere. Dies
liegt daran, daß der Aromatenausstoß eines Benzinmotors beim Erwärmen stark zurück
geht und zuerst die Anreicherungskapillare der oberen Messung mit Abgas beladen
wurde und erst danach die Kapillare für die untere Messung.
Abbildung 74 zeigt eine Online-REMPI-Messung vom Abgas eines kalten Benzinmotors. Für diese Messung wurde mit einem REMPI-TOFMS Abgas eines Autos direkt
vom Auspuff entnommen und online analysiert [20]. Zu beachten ist hier, dass die Online-Messung von Abbildung 74 und die Probenentnahme für die Offline-Messung nicht
am selben Tag und am selben Auto stattfanden. Beim Vergleich von Abbildung 73 mit
Abbildung 74 erkennt man aber, dass sich die sehr flüchtigen Bestandteile wie Benzol
und Toluol nur schlecht auf den Anreicherungskapillaren festhalten ließen. Diese beiden
Substanzen sind bei der Offline-Messung kaum zu sehen. Schwere flüchtige Substanzen
wie die methylierten Naphthaline, lassen sich hingegen gut auf der Anreicherungskapil-
170
Kapitel 7
lare speichern und erzeugen bei der Offline-Messung ein im Vergleich zu den anderen
Substanzen stärkeres Signal als bei der Online-Messung.
Abbildung 73: Offline-REMPI-Messungen (@266 nm) von kaltem Benzinmotorenabgas eines Autos. Für die Messungen wurde Abgas durch zwei Anreicherungskapillaren gesaugt.
Danach wurden die Kapillaren in den Modulator gesteckt und so lange sequenziell beheizt,
bis die Anreicherungskapillaren wieder frei von den Bestandteilen des Autoabgases waren.
Das dargestellte REMPI-Signal entspricht dem Summensignal über die ganze Desorptionszeit. Die zwei Messungen zeigen die Reproduzierbarkeit dieser Methode.
Abbildung 74: Online-REMPI-Messungen (@266 nm) von kaltem Benzinmotorenabgas eines Autos.
171
Vor einer routinemäßigen Verwendung der TD-REMPI-TOFMS-Methode sind noch
weitere Versuche notwendig. Man erkennt, dass durch das unterschiedliche Absorptionsverhalten der Anreicherungskapillare gegenüber unterschiedlichen Substanzen die
Quantifizierung auch nur über eine Vergleichsmessung mit Modulator und identischer
Anreicherungskapillare vorgenommen werden kann. Zu beachten ist dabei, dass die
Quantifizierung nur in dem Bereich stattfinden kann, in dem das spezifische Durchbruchsvolumen der Anreicherungskapillare für die zu quantifizierende Substanz nicht
überschritten wird.
172
Kapitel 7
7.2 Derivatisierung
Die laserinduzierte resonanzverstärkte Multiphotonenionisations-Massenspektrometrie
(REMPI-MS) eignet sich besonders für den selektiven Nachweis aromatischer Kohlenwasserstoffe. Durch die hohe Selektivität und Empfindlichkeit können relevante
aromatische Spurensubstanzen aus Prozeßgasen oder Umweltproben online und in
Echtzeit nachgewiesen werden.
Um die selektive REMPI-MS-Methode auch für einen Nachweis anderer gesundheitsrelevanter
Luftschadstoffe
anwendbar
zu
machen,
sind
spezielle
Derivatisierungsreaktionen erforderlich, die Tracermoleküle, welche gut mittels REMPI
ionisiert werden können, an die Analytmoleküle koppeln. Diese Reaktionen sollen ohne
Wechsel in die flüssige Phase realisiert werden (dies ist der Fall bei den klassischen
Derivatisierungsreaktionen für die organische Spurenanalytik), um langwierige Aufkonzentrierungs-Prozeduren, sowie Kontaminationen und Probenverluste zu vermeiden.
Sinnvoll ist daher die Integration einer Anreicherungs- und Derivatisierungs-Einheit in
die Probenahmestrecke.
Die im Arbeitskreis von Prof. Burger (Universität von Stellenbosch, RSA) und Prof.
Rohwer (Universität von Pretoria, RSA) entwickelten Anreicherungs- und Derivatisierungstechniken auf der Basis der sog. Silikon-Channeltraps sind ideal für diese
Aufgabenstellung.
Im Rahmen eines Projektes vom BMBF (Bundesministerium für Bildung und Forschung, Deutschland) und der NRF (National Research Foundation, Südafrika) zur
wissenschaftlichen und technischen Zusammenarbeit (WTZ) wurde in Kooperation mit
der Gruppe von Prof. Rohwer die Anreicherungs- und Derivatisierungstechnik weiterentwickelt und dem schnellen Spurennachweis von Aldehyden und Aminen mit der
REMPI-MS-Methode angepasst.
Um aliphatische Verbindungen nach dem REMPI-Prozess zu ionisieren, wird entweder
Laserstrahlung unter 200 nm benötigt, oder die Verbindungen müssen gleichzeitig von
Laserstrahlen mit unterschiedlichen Wellenlängen bestrahlt werden (z.B. 2 + 1-REMPI).
Aliphatische Verbindungen können mit REMPI bei Verwendung nur einer Wellenlänge
nicht ionisiert werden.
173
Derivatisierungsreaktionen können angewandt werden, um die Flüchtigkeit von Molekühlen, die thermische Stabilität von instabilen Verbindungen und die Sensitivität von
Nachweisverfahren zu erhöhen. Außerdem kann durch Derivatisierung die Komplexität
einer Probe reduziert werden. Im Prinzip können spezifischen Substanzklassen durch
Derivatisierungsreaktionen ein mit (1 + 1)-REMPI ionisierbares Chromophor angehängt
werden [111, 136]. Mit einer geeigneten Derivatisierungsreaktion lässt sich z.B. Aldehyden und Aminen ein aromatischer Ring anhängen. Das mit einem „REMPI-Label“
(aromatischer Ring) versehene Derivat kann mit (1 + 1)-REMPI bei geeigneter Wellenlänge selektiv ionisiert werden.
Eine Derivatisierungsverbindung lässt sich in zwei Teile unterteilen. Dies sind der organische Hauptbestandteil, z.B. ein aromatischer Ring, und der reaktive Substituent. Der
organische Hauptteil verringert die Flüchtigkeit und ist verantwortlich für die chemische
und thermische Stabilität des Produkts. Zu beachten ist, dass auch dieser Hauptbestandteil die Derivatisierungsreaktion beeinflusst. Der reaktive Substituent bestimmt die
Selektivität der Derivatisierungreaktion. Dies geschieht, indem funktionelle Gruppen
nur mit bestimmten anderen Gruppen reagieren [137].
Wir wählten Benzylhydroxylamin (BHA), um Aldehyde zu derivatisieren und Benzaldehyd (BD) für die Reaktion mit den Aminen. Beide Verbindungen verleihen dem
Derivat thermische Stabilität und einen aromatischen Ring.
Abbildung 75: Derivatisierungsreaktion von Aldehyden mit Benzylhydroxylamin (A) und
von Aminen mit Benzaldehyd (B). Der mit R gekennzeichnete Rest ist eine CxH2x+1-Gruppe
(x D 0):
174
Kapitel 7
Die Reaktion der Derivatisierung eines Aldehyds mit BHA ist in Abbildung 75 (oben)
zu sehen. Der von BD mit einem Amin im unteren Teil der Abbildung 75. Der mit R
gekennzeichnete Rest kann ein Wasserstoffatom oder eine CxH2x+1-Gruppe sein.
Für die TOFMS-Anwendung soll die Derivatisierung in der Probenahme in der stationären Phase einer Kapillare stattfinden. Aus mehreren Gründen eignet sich eine Phase aus
Silikongummi (Polydimethylsiloxan) für das Anreichern und Derivatisieren von Umweltgiften. Silikongummi ist inert, beeinflusst nicht die Derivatisierungsreaktion und
freigesetzte Bestandteile des Silikons werden durch den REMPI-Prozess nicht nachgewiesen. Der oben vorgestellte Modulator mit den Anreicherungskapillaren sollte daher
auch für die Derivatisierung von Aminen und Aldehyden geeignet sein. Diesbezüglich
wurden im Rahmen der Kooperation mit Südafrika mit Hilfe eines Gaschromatographen
(GC) nachfolgend beschriebene Vorversuche unternommen und anschließend mit dem
hier beschriebenen TOFMS und Modulator erste Online-Derivatisierungsversuche
durchgeführt.
7.2.1 Vorversuche zur Derivatisierung
Die ersten Derivatisierungsversuche wurden mit Anreicherungskapillaren mit ultradicker Phase (wie oben beschrieben) in Verbindung mit einem GC-Massenspektrometer
(GC-MS) unternommen. Für diese Versuche wurde ein spezielles gewinkeltes Glasrohr
mit einem Durchmesser von ca. 2 cm angefertigt (vgl. Abbildung 76). Durch dieses
wurde eine Anreicherungskapillare gesteckt. Die Anreicherungskapillare enthielt einen
27 cm langen Silikonschlauch als stationäre Phase. In dem Glasrohr konnte die Anreicherungskapillare zum Desorbieren durch heiße Luft erwärmt werden. Mit einem kalten
Luftstrom wurde die Anreicherungskapillare nach dem Desorbieren wieder abgekühlt.
Die Anreicherungskapillare mit ultradicker Phase in dem Glasreaktor bestand, wie oben
beschrieben, aus einer unbelegten Kapillare (ID 0,53 mm) mit einem Silikonschlauch im
Inneren (vgl. Kapitel 7.1.3).
175
Abbildung 76: Versuchsaufbau zur Derivatisierung mit Anreicherungs- und Derivatisierungseinheit (Anreicherungskapillare), 6-Wege-Ventil, Saugpumpe und Gebläse für kalte und
heiße Luft. Mit diesem Aufbau und einem GC-MS wurden die ersten Versuche zur Derivatisierung von Aminen und Aldehyden in der stationären Phase der oben beschriebenen
Anreicherungskapillaren durchgeführt.
In der in Abbildung 76 dargestellten Position des 6-Wege-Ventils befindet sich der Versuchsaufbau in dem Mode, in dem die Anreicherungskapillare kalt ist und beladen
werden kann. Dabei saugt die Saugpumpe mit 5 ml / min die Substanzen durch die Anreicherungskapillare. Gleichzeitig wird das GC-MS mit Helium gespült, um die
Kapillare des GC-MS zu reinigen und die Elektronenquelle zu schützen.
Um die Substanzen in der Anreicherungskapillare zu desorbieren, dreht man das
6−Wege-Ventil um 60° in die andere Position und lässt heiße Luft durch den Reaktor
strömen. Dadurch kann das Helium die von der Anreicherungskapillare thermisch
desorbierten Substanzen in das GC drücken.
Der Temperaturverlauf in der Anreicherungskapillare beim Desorbieren wurde zuvor
mittels eines oben erwähnten sehr dünnen Thermoelementes ermittelt. Es zeigte sich,
dass bei diesem Aufbau die Anreicherungskapillare innerhalb von ca. 5 s eine Temperatur von 100 °C erreicht und sich dann innerhalb der nächsten 300 s auf 160 °C erwärmt.
176
Kapitel 7
Abbildung 77A: Chromatogramm der Desorption einer Anreicherungseinheit, die zuvor mit
Benzylhydroxylamin (BHA) und dann mit Formaldehyd beladen wurde. Das Signal bei
1,1 min stammt vom flüchtigen Formaldehyd. Das Signal bei 3,9 min vom FormaldehydOxim und das Signal bei 5,2 min vom BHA. B: 70 eV-EI-Massenspektrum aufgenommen bei
3,9 min (gepunktete Linie). Durch das EI-Massenspektrum lässt sich das Signal des Chromatogramms bei 3,9 min dem Derivat Formaldehyd-Oxim zuordnen.
Mehrere verschiedene Verbindungen wurden mit diesem Aufbau getestet. Zum Derivatisieren der Aldehyde verwendeten wir BHA, welches wir als erstes in die
Anreicherungskapillare gebracht haben. Aus einem Fläschchen wurde verdampftes
BHA eine bestimmte Zeit lang durch die Anreicherungskapillare gesaugt. Danach wurde
z.B. Formaldehyd, welches von Paraformaldehyd freigesetzt wurde, durch die Anreicherungskapillare gesaugt. Anschließend wurde das 6-Wege-Ventil auf Desorption gestellt
und Heißluft durch den Reaktor geblasen. Die Substanzen in der Anreicherungskapillare
wurden dadurch desorbiert und gelangten durch geheizte Transverkapillaren zum GC,
177
wo die Substanzen auf einer 30 m langen Kapillare mit einer dünnen Innenbeschichtung
von 95 % Dimethylsiloxan und 5 % Phenylmethylsiloxan und einem GC-OfenTemperaturprogramm getrennt wurden. Das so gewonnene Chromatogramm ist in
Abbildung 77A zu sehen.
Das Chromatogramm (Abbildung 77A) zeigt, dass das Gas von der Desorption überwiegend aus 3 Verbindungen bestand. Durch das MS, welches dem GC nachgeschaltet
war, konnten diese drei Hauptbestandteile identifiziert werden. Durch den Vergleich der
70 eV-EI-Spektren mit Vergleichsspektren zeigte sich, dass das Formaldehyd die erste
der drei Verbindungen ist. Auf Grund seiner Flüchtigkeit kommt es schon nach 1,1 min
aus der GC-Kapillare. BHA verlässt das GC 5,2 min nach Beginn der Desorption. Durch
die EI-Messung konnte die dritte Verbindung bei 3,9 min dem Formaldehyd-Oxim zugeordnet werden.
Abbildung 77B zeigt das EI-Massenspektrum des Gases, welches nach 3,9 min das GC
verließ. Aufgrund des Fragmentmusters lässt sich im Vergleich mit dem FormaldehydOxim-Referenzspektrum von NIST [64] erkennen, dass das Signal des Chromatogramms bei 3,9 min vom Formaldehyd-Oxim stammt (gepunktete Linie in
Abbildung 77A). Dies zeigt, dass in der Anreicherungskapillare Formaldehyd mit BHA
reagierte und zu einem beträchtlichen Teil, als Produkt (vgl. Abbildung 75 oben) das
Derivat Formaldehyd-Oxim entstand.
Zur Derivatisierung der Amine wurde Benzaldehyd (BD) verwendet. Bei den in
Abbildung 75 gezeigten Reaktionen werden die verschieden großen Amine und Aldehyde durch das R für Rest dargestellt. Dieser Rest kann auch eine OH-Gruppe enthalten,
wie z.B. bei Ethanolamin. Ethanolamin derivatisierten wir mit BD in der Anreicherungskapillare, indem wir zuerst BD und dann das Ethanolamine in die
Anreicherungskapillare gebracht haben. Anschließend wurden die bei 220 °C desorbierten Substanzen wieder mit dem GC-MS analysiert. Das Chromatogramm dieser
Messung ist in Abbildung 78A zu sehn. Die Messung wies Ethanolamin bei 0,8 min,
BD bei 1,5 min und das Derivat Benzylpropan-Oxim nach 4,4 min nach. Das EIMassenspektrum, welches das Derivat bei 4,4 min identifiziert, ist in Abbildung 78B zu
sehen.
178
Kapitel 7
Abbildung 78 A: Chromatogramm der Desorption einer Anreicherungseinheit, die zuvor mit
Benzaldehyd (BD) und dann mit Propylamin beladen wurde. Das Signal bei 0,8 min stammt
vom flüchtigen Propylamin, das bei 1,5 min vom BD und das bei 4,4 min vom BenzylpropanOxim. B: 70 eV-EI-Massenspektrum aufgenommen bei 4,4 min (gepunktete Linie). Durch
das EI-Massenspektrum lässt sich das Signal des Chromatogramms bei 4,4 min dem Derivat
Benzylpropan-Oxim zuordnen.
Mit diesen ersten Derivatisierungstests, die mit relativ hohen Konzentrationen der Edukte durchgeführt wurden, konnte gezeigt werden, dass in der stationären Phase einer
Kapillare die Derivatisierungsreaktion stattfindet. Es zeigte sich, dass die Derivatisierung mit den gewählten Derivatisierungsreagenzien BHA und BD effizient in der
Silikonphase abläuft und dass die erwarteten Derivate entstehen. Anhand dieser Ergebnisse ist zu erwarten, dass mit einer Kombination aus TOFMS-System und Modulator
mit Anreicherungskapillare, Amine und Aldehyde effektiv zu derivatisieren sind und
diese damit selektiv mit dem REMPI-Prozess in einem komplexen Probengas nachgewiesen werden können.
7.2.2 Derivatisieren in der Probenahme des TOFMS
Aliphatische Aldehyde und Amine werden mit REMPI unter Verwendung nur einer
Wellenlänge schlecht ionisiert. Mit SPI bei 118 nm können sie hingegen ionisiert werden. In komplexen Gasen gibt es allerdings oft mehrere Verbindungen mit der Masse
179
der Amine und Aldehyde, die mit 118 nm VUV-Licht ionisiert werden, was den Nachweis mit diesem Verfahren oft verhindert. Durch geeignete Derivatisierungsreaktion
können Aldehyde und Amine mit (1 + 1)-REMPI nachgewiesen werden. Dadurch, dass
diese Verbindungen über ihre Derivate nachgewiesen werden können, kann die Derivatisierungsreaktion so gewählt werden, dass das Derivatisierungsprodukt eine Masse
besitzt, bei der es nicht zu Überlagerungen mit anderen Verbindungen kommt. Der SPIMS-Nachweis dieser Verbindungen ist dann möglich.
Ein Gasanalysegerät sollte in der Lage sein, Amine und Aldehyde in einem komplexen
Gas mit großen Konzentrationsunterschieden nachzuweisen. Massenspektrometer mit
Elektonenstoßionisation sind in diesem Fall ungeeignet, weil die Fragmentierung der
Molekühle bei der Ionisierung es sehr schwer macht, die einzelnen Massenpeaks den
Molekülen zuzuordnen. Gelingt es den Aminen oder Aldehyden einen aromatischen
Ring innerhalb der Probenahme anzuheften, ist es möglich, diese Substanzen über die
Derivate selektiv und online mit einem REMPI-TOFMS zu messen. Benutzt man Benzylhydroxylamin (BHA, 123 amu) zum derivatisieren von Formaldehyd (30 amu) bzw.
Acethaldehyd (44 amu), so erhält man ein aromatisches Derivat mit der Masse 135 amu
bzw. 149 amu. Das Derivat lässt sich mit REMPI durch die Wellenlänge, bei der es
ionisiert werden kann, und durch seine Masse identifizieren. Steht dem Aldehyd in ausreichendem Maß BHA zur Verfügung, so ist die nachgewiesene Menge des Derivates
direkt proportional zur aufgenommenen Menge des Aldehyds.
Für die nachfolgenden Versuche wurde das TOFMS über eine 2 m lange geheizte Transferkapillare mit dem Modulator verbunden. Die Verbindung zwischen Transferkapillare
und Anreicherungskapillare erfolgte mit einem Pressfit-Kapillarverbinder. Wie in
Abbildung 79 zu sehen ist, befand sich auf der Eingangsseite der Anreicherungskapillare ein T-Pressfit. Über diesen konnte gleichzeitig Derivatisierungssubstanz und
Analysengas durch den Modulator in Richtung des Massenspektrometers gesaugt werden. Die Verwendung von Anreicherungskapillaren, basierend auf SE 30 Kapillaren mit
2,65 mm stationärer Phase, brachte die besten Ergebnisse. Die Derivatisierungssubstanz
befand sich in einem Fläschchen, welches mit einem Septum verschlossen war. In das
Fläschchen wurden zwei Kapillaren gesteckt. Die eine wurde mit einem Ende des TPressfits verbunden, durch die zweite wurde mit ca. 5 ml / min Stickstoff geblasen. So
gelangte kontinuierlich eine konstante Menge an Derivatisierungssubstanz zum Modulator.
180
Kapitel 7
Abbildung 79: Probenahmeaufbau zur Online-, In-situ-Derivatisierung. Von links kann
gleichzeitig eine Gasprobe und die Derivatisierungssubstanz (BHA oer BD) auf die Anreicherungskapillare im Modulator geladen werden. Die Derivatisierung findet in der
stationären Phase der Anreicherungskapillare im Modulator statt. Bei der Desorption der Anreicherungskapillare durch den Modulator gelangt das in der stationären Phase gebildete
Derivatisierungsprodukt zum TOFMS. Die Transferwege zu dem Modulator und vom Modulator zur Ionenquelle werden permanent geheizt.
Für die in Abbildung 80 dargestellten Messungen wurde der zweite Arm des T-Pressfits
mit einem Probenstandard verbunden, der 1,3 ppm Propylamin und 0,3 ppm Butylamin
in Stickstoff lieferte. Somit gelangten Amine und BD gleichzeitig auf die Anreicherungskapillare und konnten dort reagieren. Nach Desorption durch den Modulator
gelangten die restlichen nicht derivatisierten Amine, das BD und die Produkte der
Derivatisation etwas zeitlich versetzt zum TOFMS (vgl. Abbildung 81).
Für die in Abbildung 80 und Abbildung 81 dargestellte Messung wurde für 10 s BD
gleichzeitig mit Propylamin und Butylamin aufgenommen. Anschließend wurde der
Modulator gestartet. Mit dem Start des Modulators begann auch die Analyse des Gases
vom Modulator mit dem TOFMS im SPI-Mode. 56 s nach Start des Modulators erhielt
man das in Abbildung 80A dargestellte Massenspektrum. Hier erkennt man bei 147 m/z
das derivatisierte Propylamin. Die Peaks bei 118, 119 und 146 m/z sind Fragmente des
Derivates (vgl. 70 eV-Reverenzspektrum von NIST [64]). Da man mit SPI (@ 118 nm)
auch Propylamin und Butylamin ionisieren kann, sind diese auch in Abbildung 80A zu
sehen. Die maximal Konzentration der Butylamin-Derivates (161 amu) erreicht das
TOFMS 7 s später, zu sehen in Abbildung 80B.
181
Der zeitliche Signalverlauf der Amine, des BD und der Derivate ist in Abbildung 81
dargestellt. Die Messung beginnt mit Beginn der Desorption im 1. Modulatorsegment.
In Abbildung 81 A ist der Konzentrationsverlauf von Propyl- und Butylamin dargestellt.
Die Verläufe beginnen mit hohen Signalen, die gleich zu Beginn abfallen. Das wird von
den Aminen, die vor der Modulation beim Ziehen der Probe durch die Anreicherungskapillare durchbrechen, verursacht. Da mit Beginn der Modulation der Probenstandard
und das BD (106 amu) abgehängt werden, fällt dieses Signal sofort ab. Während der
Modulation wird permanent eine große Menge des zuvor hoch konzentriert eingebrachten BD freigesetzt. Da dabei auch Fragmente der Masse 59 amu entstehen, steigt die
Konzentration von Propylamin (59 amu) parallel zu BD, dessen Verlauf in
Abbildung 81 B dargestellt ist, an. Der grau/schwarz dargestellte Abschnitt des Propylaminverlaufes wird daher vom BD hervorgerufen. Die höchste Konzentration der nicht
derivatisierten Amine erreichten das TOFMS nach 54 s.
Abbildung 80: SPI-Massenspektren des Desorptionsgases einer Anreicherungskapillare, die
zuvor mit Propyl-, Butylamine und Benzaldehyd (BD) beladen wurde. Das Massenspektrum
von A zeigt das Produkt der Derivatisierung von Propylamin und BD bei 147 m/z. Da das
Derivat von Butylamin und BD 7 s später das TOFMS erreicht, wird dieses Derivat
(161 m/z) etwas später nachgewiesen (Spektrum B). Das SPI-Signal/Zeit-Verhalten der beteiligten Substanzen ist in Abbildung 81 zu sehen.
182
Kapitel 7
Abbildung 81: SPI-Signal/Zeit-Verläufe bei der Desorptionsmessung einer Anreicherungskapillare auf die Propyl-, Butylamine und Benzaldehyd aufgebracht wurden. Zum Zeitpunkt
t = 0 beginnt der Modulator mit der Desorption. Der Signalverlauf der desorbierten, nicht
derivatisierten Amine ist in Teil A zu sehen. B zeigt das Verhalten von BD. Das Verhalten
der Derivate ist in Teil C zu sehen. Zum Zeitpung t = 56 s und t = 63 s (gepunktete Linie)
wurden die in Abbildung 80 dargestellten Spektren gewonnen. Aus den Ionensignalen 80
solcher Einzelspektren wurden diese Verläufe ermittelt.
Wie schon erwähnt, gelangt ein Teil des BD beim Heizen jedes Modulatorsegments aus
der Anreicherungskapillare. Daher rührt die lang andauernde sehr hohe BDKonzentration. Während des Nachweises der hohen BD-Konzentration von 5 bis 45 s
wird durch das hohe Signal die Grundlinie der Massenspektren, aus deren Ionensignalen
die Verläufe erstellt wurden, auf der Seite der schwereren Massen beeinflusst. Darin
liegt der Abfall der Verläufe der Derivate (Abbildung 81 C) im Bereich 5 bis 45 s begründet. Die höchsten Konzentrationen der Derivate werden bei 56 und 63 s
nachgewiesen. Die Massenspektren zu diesen Zeitpunkten sind in Abbildung 80 dargestellt.
183
Aus den Massenspektren bei 56 und 63 s, bei denen die Signale der jeweiligen Derivate
am höchsten waren, wurde die Nachweisgrenze für Propyl- und Butylamin über deren
Derivate errechnet. Bei einen Verhältnis Signal zu Rauschen von 1 zu 3 ergibt sich aus
den eingebrachten Konzentrationen der Amine für Propylamin eine Nachweisempfindlichkeit von 60 ppb und für Butylamin von 80 ppb.
Die In-situ-Derivatisierung von Aminen mit Benzaldehyd war erfolgreich. Die verschiedenen Parameter der Derivatisierung müssen noch optimiert werden. Außerdem muss
dieses Verfahren noch mit kleineren Aminen getestet werden. Derivatisierte Aldehyde
konnten bis zum Zeitpunkt der Fertigstellung dieser Arbeit noch nicht in situ und online
nachgewiesen werden. Hier stehen in Zukunft noch Tests mit anderen Derivatisierungsverbindungen wie Anilin an. Eine aktive Kühlung des Modulators während der
Absorption der Verbindungen zur Verbesserung der Aufnahmefähigkeit der Anreicherungskapillare sollte untersucht werden. Es sollte getestet werden, ob für diese
Anwendung Anreicherungskapillaren mit mehreren parallelen Silikonschläuchen [134,
135] von Vorteil sind, da mit diesen dem Reaktionspartner mehr Volumen der stationären Phase zur Verfügung steht.
Abbildung 82: Einfarben-REMPI-Spektrum von Formaldehyd-Oxim. Demnach lässt sich
Formaldehyd-Oxim, das bei der Derivatisierungsreaktion aus Formaldehyd und Benzylhydroxylamin entsteht, am besten mit REMPI bei 266 nm ionisieren. Die scharfen Spikes bei
259 nm und 272 nm stammen von Verunreinigungen in der Probe (z.B. Benzol)
Um gleiche oder bessere Empfindlichkeiten mit REMPI zu erhalten, wie sie hier mit SPI
gezeigt wurden, ist es notwendig, den REMPI-Wirkungsquerschnitt der Derivate in Abhängigkeit von der Wellenlänge zu kennen. Mit Hilfe von Wellenlängenspektren wird
dann eine günstige UV-Wellenlänge gewählt, bei der das Derivat effektiv ionisiert wird
[40].
184
Kapitel 7
In Abbildung 82 ist das REMPI-Wellenlängen-Spektrum des Derivates von Formaldehyd und Benzylhydroxylamin (BHA) dargestellt. Demnach würde man versuchen, das
Derivat Formaldehyd-Oxim mit REMPI bei 266,3 nm nachzuweisen. Für die restlichen
Derivate der Amine und Aldehyde mit BHA oder BD liegen die REMPIWellenlängenspektren noch nicht vor. Aus diesem Grunde können noch keine REMPI
Messungen präsentiert werden, wie es ursprünglich geplant war.
Die hier dargestellten Versuche zur Anreicherung und Derivatisierung gehören zu einer
noch nicht abgeschlossenen Versuchsreihe, die in Zusammenarbeit mit Herrn Prof.
Rohwer von der Universität von Pretoria in Südafrika durchgeführt wird. Gefördert wird
dieses Vorhaben durch das Bundesministerium für Bildung und Forschung in Deutschland und der National Resurch Foundation in Südafrika. Schon zum jetzigen Zeitpunkt
lässt sich an den ersten Ergebnissen erkennen, dass die Anreicherungs- und Derivatisierungs-Technik mit einem Modulator bei bestimmten Anforderungen eine sinnvolle
Ergänzung für ein TOFMS ist und damit seine Einsatzmöglichkeiten erweitert.
185
8 Zusammenfassung und Ausblick
In der vorliegenden Arbeit wurde das Konzept eines Flugzeitmassenspektrometers vorgestellt, mit dem durch den Einsatz verschiedener Ionisationsmethoden eine Gasprobe
umfassend analysiert werden kann. Es wurde ein mobiles TOFMS-System, mit dem das
zu untersuchende Gas (quasi) gleichzeitig durch die Ionisationsmethoden REMPI, SPI
und EI analysiert werden kann, aufgebaut und an einer Müllverbrennungsanlage (MVA)
erfolgreich getestet. Messungen verschiedener Proben aus unterschiedlichen Prozessen
(Tabakrauch, Benzinherstellung, Müllverbrennung) zeigen die vielfältigen Einsatzmöglichkeiten des neuen Systems. In Zusammenhang mit den Messungen an einer MVA
werden ein neu entwickelter Standardgasgenerator und eine Hochtemperatursonde vorgestellt. Zum Nachweis der VUV-Photonen, die für das SPI-Verfahren benötigt werden,
wurde ein simpler VUV-Detektor, basierend auf einer NO-Gaszelle, aufgebaut. In der
Arbeit wird weiterhin gezeigt, dass mit einer thermischen Modulatoreinheit, wie sie bei
der GC/GC-Kopplung Verwendung findet, die Nachweisempfindlichkeit des TOFMS
gesteigert werden kann. Außerdem können mit einem thermischen Modulator Verbindungen derivatisiert und so der selektiven REMPI-Technik zugänglich gemacht werden.
Im Hinblick auf die Entwicklung eines kommerziellen Prozessgasanalysegerätes wird
das erste TOFMS-Gerät mit elektronenstrahlgepumpter Excimer-Lampe vorgestellt. Es
werden die Kopplung der Excimer-Lichtquelle mit einem TOFMS beschrieben und erste
Online-Messungen gezeigt.
Im Folgenden werden die in dieser Arbeit beschriebenen Konzepte und Entwicklungen
im Einzelnen zusammengefasst dargestellt.
Flugzeitmassenspektrometer mit drei verschiedenen Ionisationsmechanismen
Es wurde ein Flugzeitmassenspektrometersystem entwickelt, mit dem die Zusammensetzung eines Probengases quasi gleichzeitig mittels VUV-Photonen durch die
Einphotonenionisation (SPI), mittels UV-Photonen durch die resonanzverstärkte Mehrphotonenionisation
(REMPI)
und
mittels
eines
Elektronenstrahls
durch
die
Elektronenstoßionisation (EI-Ionisation) analysiert werden kann. Mit dem Instrument ist
es möglich, je Sekunde 1 vollständiges REMPI-Massenspektrum (Summenspektrum aus
5 Einzelspektren), 1 SPI-Massenspektrum (Summenspektrum aus 5 Einzelspektren) und
1 EI-Massenspektrum (Summenspektrum aus 9900 Einzelspektren) eines Gases aufzu-
186
Kapitel 8
nehmen. Die UV-Wellenlänge für den REMPI-Prozess kann dabei zwischen 200 nm
und 340 nm betragen. Für den SPI-Prozess steht VUV-Licht mit einer Wellenlänge von
118 nm (10.5 eV) zur Verfügung. SPI und REMPI können mit einer Repetitionsrate von
10 Hz durchgeführt werden. Die Elektronenstrahlpulse stammen von einer Elektronenkanone und können mit bis zu 20 kHz aufeinander folgen. Die kinetische Energie der
Elektronen kann in einem großen Bereich von 6 bis 90 eV variiert werden.
Mit Hilfe neu entwickelter Elektronik und Software werden REMPI-, SPI- und EIMassenspektren getrennt gespeichert. Die Auswertungssoftware erlaubt es, Konzentrationen von Verbindungen, die mit unterschiedlichen Ionisationsprozessen nachgewiesen
wurden, zu vergleichen.
Das Massenspektrometer ist mobil und sehr robust, was während einer 5-wöchigen
Messkampagne an einer MVA unter Beweis gestellt worden ist. Mit der speziell entwickelten Edelgaszelle zur Erzeugung von Laser-VUV-Licht konnte über die gesamte Zeit
der Messkampagne mit nur einer Xe-Gasfüllung effektiv VUV erzeugt werden.
Die mit Hilfe dieses Gerätes erzielten Messergebnisse zeigen, dass mit SPI Spurengasbestandteile effektiv und weich ionisiert werden können. Mit SPI wurden aliphatische,
aromatische und anorganische Verbindungen, wie Ammoniak und NO, nachgewiesen.
Somit ließ sich mit SPI das spezifische massenspektrometrische 118 nm-SPI-Muster
einer Probe aufnehmen. Die erzielten Nachweisgrenzen reichen bis in den unteren ppbBereich.
Mit REMPI konnten bei beliebiger UV-Wellenlänge gezielt aromatische Verbindungen
nachgewiesen werden. Je nach REMPI-Wellenlänge konnten von einem komplexen
Analysengas spezifische REMPI-Spektren aufgenommen werden. Im REMPI-Mode des
Massenspektrometers wurden Nachweisgrenzen bis in den oberen ppt-Bereich erzielt.
Vor allem Verbindungen mit mehr als einem aromatischen Ring ließen sich im Vergleich zu SPI mit REMPI sensitiver nachweisen.
Mit EI lassen sich die zur Charakterisierung eines Prozesses wichtigen Hauptbestandteile eines Gases messen. Bei Verbrennungsprozessen können dies H2O, CO, O2 und CO2
sein. In dieser Arbeit wurde EI aber hauptsächlich dazu verwendet, Verbindungen zu
detektieren, die mit REMPI oder SPI nicht erfasst werden konnten. Im Abgas einer
MVA konnten mit EI Acetylen, HCN, HCl und SO2 nachgewiesen werden. Die Nach-
187
weisgrenze für HCN konnte mittels eines Gasstandards bestimmt werden und betrug
5 ppm (S/N = 3).
Aufgrund der TOFMS-Einstellungen im Kombi-Betrieb reduziert sich die Sensitivität
bei gleichzeitiger Untersuchung eines Gases mit REMPI, SPI und EI um einige Prozent.
Dasselbe gilt für die maximale Massenauflösung von 1800 m/z (gemessen im SPIMode).
Im Rahmen dieser Arbeit wurden zum ersten Mal quasi gleichzeitige (innerhalb einer
Sekunde) Online-Analysen eines Gases mit REMPI, SPI und EI durchgeführt. Die präsentierten Messungen eines Standardgases und die Anwendungsbeispiele Tabakrauch,
Kohlevergasungsdestillat (Fischer-Tropsch-Verfahren) und Müllverbrennungsabgas
zeigen eine Vielzahl an Verbindungen, die sich mit dem TOFMS-System nachweisen
ließen. Bei der Messung des Müllverbrennungsabgases wurde gezeigt, dass mit dem
TOFMS-System mehrere Verbindungen gleichzeitig online mit einer Zeitauflösung im
Sekundenbereich und quantitativ gemessen werden können, obwohl mit unterschiedlichen Verfahren ionisiert wird.
Denkbar ist auch der Einsatz das entwickelten Messgerätes in der Lebensmittelindustrie
zur Überwachung und Steuerung des Kaffeeröstprozesses. Bei der Motoren- und Katalysatorenentwicklung könnte mit dem Massenspektrometer eine Vielzahl der
Abgasbestandteile mit hoher Zeitauflösung gleichzeitig gemessen werden; sogar die
Integration in ein Fahrzeug zur Messung unter Realbedingungen scheint möglich. In der
Halbleiterherstellung könnten mit Hilfe des Gerätes Prozessräume auf kleinste Verunreinigungen hin überwacht werden. Auch die Überwachung von Lüftungsanlagen
bezüglich gesundheitsschädlicher Verbindungen wurde angedacht. In der chemischen
Industrie ist eine Vielzahl von Prozessen zu finden, die durch die schnelle Messung mit
dem TOFMS-System optimiert und gesteuert werden könnten. Als Beispiele sind petrochemische Prozesse und spezifische Synthesen z.B. in der Arzneimittelherstellung zu
nennen. Im Bereich des Umweltschutzes können mit dem Gerät Belastungen ermittelt
und durch die umfassende Analyse auch Hinweise auf den Verursacher gewonnen werden.
Gasstandard
Das zur Quantifizierung der MVA-Messungen verwendete Standardgas wurde mit einem eigens entwickelten Standardgasgenerator erzeugt. In dem Generator wird einem
188
Kapitel 8
konstanten Luftstrom eine konstante Menge an Substanzen zugemischt. Die Substanzen
befinden sich in Glasröhrchen, die mit einer Teflonmembran oder einer dünnen Kapillare verschlossen sind. Durch Permeation oder Diffusion gelangen geringe Mengen der
Verbindungen in den Luftstrom.
Damit über einen langen Zeitraum die Konzentrationen der Verbindungen konstant
bleiben, muss neben dem Luftstrom auch die Temperatur der Röhrchen konstant gehalten werden. Der entwickelte Standardgasgenerator besteht daher im Wesentlichen aus
einer Membranpumpe, einem Gasflussregler und einer sehr genau temperierbaren
Kammer mit den Standardsubstanzen.
Mit dem beschriebenen Gasstandard konnte über 1 Jahr lang ein Luftstrom (20 ml/min)
erzeugt werden, in dem bis zu 20 für die Messungen an der MVA relevante Verbindungen in konstanten Konzentrationen von 20 ppb – 5 ppm enthalten waren.
Hochtemperatursonde
Bei Abgasmessungen in der Flammenzone einer MVA kann das Probengas nicht mit
einem einfachen Rohr entnommen werden.
·
Das Probengas muss auch bei 1300 °C mindestens 50 cm entfernt von der Kesselwand entnommen werden.
·
Da das Probengas zur Vermeidung von Kondensationen in der Probenahme nicht
unter 250 °C abkühlen darf, muss die Probenahme in der bis zu 60 cm dicken Kesselwand aktiv geheizt werden.
·
Das reaktive Probengas soll möglichst nur mit inerten Oberflächen in Berührung
kommen und möglichst schnell auf eine Temperatur abgekühlt werden, bei der Reaktionen im Analysengas stark verlangsamt werden.
Die im Rahmen dieser Arbeit entwickelte Hochtemperatursonde kann bei 1300 °C mehrere Wochen lang bis zu 1 m waagrecht ins Feuer ragen. In ihr verläuft ein inertes
Quarzglasrohr, in dem das Probengas schnell auf ca. 250 °C abgekühlt wird. Die Lanze
ist durch Druckluft gekühlt. Die Wärme, die die Kühlluft an der Spitze der Sonde aufnimmt, wird dazu verwendet, die Lanze in der Kesselwand auf Temperatur zu halten.
Durch Verändern des Kühlluftstroms kann die Lanze an verschiedene Kesseltemperaturen und Wanddicken angepasst werden. Da das Konzept der neu entwickelten Lanze
189
gegenüber herkömmlichen wassergekühlten Lanzen viele Vorteile bietet, wird es zum
Patent angemeldet.
Während der Messkampagne an einer großen Müllverbrennungsanlage konnte mit Hilfe
dieser Lanze für mehrere verschiedene Gasanalysengeräte Probengas in ausreichender
Entfernung von der Kesselwand entnommen werden.
VUV-Detektor
Bei der VUV-Erzeugung aus einem intensiven 355 nm-Laserstrahl wird nur ein geringer
Anteil der Pumpstrahlung in 118 nm-VUV-Strahlung umgewandelt. Der Strahl, der die
frequenzverdreifachende Edelgaszelle verlässt, besteht deshalb nur zu einem kleinen
Teil aus VUV-Photonen. Der überwiegende Anteil besteht aus 355 nm-Photonen. Bei
der Excimer-Erzeugung entsteht neben den VUV-Photonen durch Atomabregungen
Licht im sichtbaren Bereich. Das intensive UV-Licht, das bei der VUV-Erzeugung
durch Laserstrahlung beteiligt ist, und das sichtbare Licht, das der Excimer-Prozess emittiert, machen für den selektiven Nachweis von VUV-Photonen einen Detektor
erforderlich, der gegenüber sichtbarem und UV-Licht unempfindlich ist.
Halbleiterdetektoren, auch mit speziellen Beschichtungen zur Wellenlängenfilterung,
erwiesen sich wegen der relativ geringen VUV-Photonenzahl in einem intensiven UVStrahl als ungeeignet.
Die Versuche mit einer NO gefüllten Ionisationszelle haben gezeigt, dass sich VUVStrahlung mit einer Wellenlänge kürzer als 134 nm aufgrund des Ionisationspotenzials
(IP) von NO (9,26 eV) unbeeinflusst von sichtbarem und von UV-Licht nachweisen
lässt. Bei Verwendung von Gasen mit einem niedrigeren IP lässt sich diese Technik
auch für längerwelliges VUV-Licht verwenden. Die Ionisationszelle erwies sich als ein
wichtiges Hilfsmittel zur Optimierung der VUV-Erzeugung. Mit ihrer Hilfe ist es möglich, die vielen Parameter der VUV-Erzeugung mit direktem Feedback auf eine
maximale VUV-Photonenausbeute einzustellen. Die beschriebene Ionisationszelle ist
einfach und billig herzustellen.
Da die mit der NO-Zelle erzielten Messergebnisse reproduzierbar waren, ist eine Kalibrierung der Zelle möglich. Quantitativ konnte VUV-Strahlung mit der Ionisationszelle
noch nicht gemessen werden, da hierzu erst Messungen an einer Referenzlichtquelle
erfolgen müssen.
190
Kapitel 8
Anreicherung und Derivatisierung
In Kooperation mit der Arbeitsgruppe von Prof. Rohwer in Pretoria wurden Versuche
zur Anreicherung von Gasbestandteilen in der Probenahmestrecke des TOFMS vorgenommen. Mit Hilfe von kurzen Anreicherungseinheiten, basierend auf GC-Kapillaren
mit dicker stationärer Phase, und einem sogenannten Modulator zur thermischen Desorption
dieser
Traps
konnten
je nach Flüchtigkeit einer Substanz
deren
Nachweisgrenze um bis zu einer Größenordnung gesteigert werden.
Anhand von Abgasproben eines Otto-Motors zeigte sich, dass die Traps geeignet sind,
um extern (offline) Proben zu nehmen, die dann in der Probenahme des TOFMS desorbiert und mit dem TOFMS analysiert werden.
Es wurde gezeigt, dass Amine und Aldehyde in den Traps in situ derivatisiert werden
können. Zur Derivatisierung der Amine wurde Benzaldehyd und für die Aldehyde Benzylhydroxylamin verwendet. Durch die Derivatisierung der Amine und Aldehyde mit
den genannten Substanzen werden diese mit einem aromatischen Ring verknüpft, wodurch sie sich prinzipiell mittels REMPI selektiv in einem komplexen Gasgemisch
nachweisen lassen.
Die effektive in-situ-Derivatisierung von Aldehyden mit Benzylhydroxylamin in den
Traps konnte mit Hilfe von SPI-Messungen gezeigt werden. Die Derivatisierung von
Aminen mit Benzaldehyd in den Traps wurde mit GC-MS nachgewiesen.
Excimer-TOFMS
Das präsentierte TOFMS mit Excimerlampe und die damit vorgenommenen Messungen
zeigen, dass das neu entwickelte und zum Patent angemeldete System, bestehend aus
elektronenstrahlgepumpter Excimerlampe und TOFMS ein vielseitig einsetzbares und
sehr kompaktes Gerät zur Analyse von komplexen Gasgemischen ist. Selbst mit dem
hier beschriebenen Prototyp konnten nach kürzester Zeit aromatische Verbindungen
quantitativ im unteren ppm-Bereich nachgewiesen werden. Die Ionisation mit VUVLicht ist sehr weich und damit fragmentarm. Durch den Wechsel des Excimergases ist
es möglich, verschiedene Lichtwellenlängen bzw. Photonenenergien zu erhalten und
damit verschiedene SPI-Selektivitäten einzustellen.
Die niedrigen Kosten und die geringe Wartungsanfälligkeit der Excimerlampe verhelfen
diesem System gegenüber TOFMS-Systemen, die mit laserinduziertem VUV-Licht ar-
191
beiten, zu einem großen Vorteil. Auch gegenüber Systemen mit chemischer Ionisierung
(CI) dürfte das Excimer-Konzept aufgrund der geringen Kosten und der hohen
Selektivität Vorteile haben.
Mit den präsentierten Versuchen konnte erstmalig das Potenzial dieser Lichtquelle für
die Massenspektrometrie gezeigt werden. Für die Ecximer-TOFMS-Anwendung haben
die Versuche gezeigt, wie die Nachweisgrenzen und die Massenauflösung dieses Konzepts verbessert werden können.
Die Entwicklung von Excimer-TOFMS-Systemen wird in einem von der Bayerischen
Forschungsstiftung finanzierten Projekt fortgesetzt.
192
Abkürzungsverzeichnis
AD
Außenduchmesser
amu
atomare Masseneinheit
arb. Units
willkürliche Einheiten
BD
Benzaldehyd
BHA
Benzylhydroxylamin
BTX
Benzol, Toluol und p-Xylol
EI
Elektronenstoß-Ionisation
GC
Gaschromathograph
GC-MS
Gaschromatographie-Massenspektrometrie
HCHO-Oxim
Formaldehyd-Oxim
HGF
Helmholz Forschungsgemeinschaft
ID
Innenduchmesser
IP
Ionisationspotenzial
LI
Laser-Ionisation
MCB
Monochlorbenzol
MCP
multi-channel-plate
MS
Massenspektrometrie
MVA
Müllverbrennungsanlage
m/z
Masse pro Ladung
NWG
Nachweisgrenze
OPO
Optischer Parametrischer Oszillator
ppb
1 Volumenbestandteil aus 10E9 Teilen
ppm
1 Volumenbestandteil aus 10E6 Teilen
ppt
1 Volumenbestandteil aus 10E12 Teilen
REMPI
Resonanzverstärkte Mehrphotonen-Ionisation
SFM
Summen Frequenz-Mischung
SPI
Einphotonen-Ionisation
TD
Thermodesorption
TOFMS
Flugzeitmassenspektrometer
UV
ultravioletes Licht
VUV
Vakuum ultravioletes Licht (100 nm < VUV < 200 nm)
WHO
Welt Gesundheitsorganisation
193
Literaturverzeichnis
1.
Bjørseht, A., ed. Handbook of Polycyclic Aromatic Hydrocarbons. 1983, Marcel
Dekker, Inc.: New York and Basel.
2.
Munson, B., Chemical Ionization Mass Spectrometry: Ten Years Later. Analytical Chemistry, 1977. 49(9): 772A-778A.
3.
Miller, J.C. und R.N. Compton, Vacuum ultraviolet spectroscopy of molecules
using third-harmonic generation in rare gases. Journal of Chemical Physics,
1982. 76(8): 3967-3973.
4.
Mueller, E.W., Zeitschrift für Physik, 1951. 131: 136-142.
5.
Hager, J.W. und S.C. Wallace, Two-Laser Photoionization Supersonic Jet Mass
Spectrometry of Aromatic Molecules. Analytical Chemistry, 1988. 60: 5-10.
6.
Hurst, G.S., M.G. Payne, S.D. Kramer und J.P. Young, Resonance ionization
spectroscopy and one-atom detection. Reviews of Modern Physics, 1979. 51(4):
767-819.
7.
Lubman, D.M., ed. Lasers and Mass Spectrometry. 1990, Oxford University
Press: New York.
8.
Boesl, U., R. Weinkauf, C. Weickhardt und E.W. Schlag, Laser ion sources for
time-of-flight mass spectrometry. International Journal of Mass Spectrometry
and Ion Processes, 1994. 131: 87-124.
9.
Cool, T.A. und B.A. Williams, Ultrasensitive Detection of Chlorinated Hydrocarbons by Resonance Ionization. Combustion Science and Technology, 1992.
82: 67-85.
10.
Zimmermann, R., C. Lermer, D. Lenoir und U. Boesl. Isomer selective ionization of chlorinated PAH´s: Detection of impurities in technical 9monochloranathracene. 7th International Symposium on Resonance Ionization
Spectroscopy. 1994. Bernkatel-Kues: American Institute of Physics, AIP-Press,
New York, 527-530.
194
11.
Oser, H., R. Thanner und H.-H. Grotheer, Jet-REMPI for the detection of trace
gas compounds in complex gas mixtures, a tool for kinetic research and incinerator process control. Combustion Science and Technology, 1996. 116-117:
567-582.
12.
Tembreull, R. und D.M. Lubman, Use of Two-Photon Ionization with Supersonic Beam Mass Spectrometry in the Discrimination of Cresol Isomers.
Analytical Chemistry, 1984. 56: 1962-1967.
13.
Gittins, C.M., M.J. Castaldi, S.M. Senkan und E.A. Rohlfing, Real-Time Quantitative Analysis of Combustion-Generated Polycyclic Aromatic Hydrocarbons by
Resonance-Enhanced Multiphoton Ionization Time-of-Flight Mass Spectrometry.
Analytical Clemistry, 1997. 69(3): 286-293.
14.
Zimmermann, R., H.J. Heger, C. Yeretzian, H. Nagel und U. Boesl, Application
of Laser Ionization Mass Spectrometry for On-line Monitoring of Volatiles in the
Headspace of Food Products: Roasting and Brewing of Coffee. Rapid Communications in Mass Spectrometry, 1996. 10: 1975-1979.
15.
Zimmermann, R., H.J. Heger, A. Kettrup und U. Boesl, A Mobile Resonanceenhanced Multiphoton Ionization Time-of-Flight Mass Spectrometry Device for
On-line Analysis of Aromatic Pollutants in Waste Incinerator Flue Gases: First
Results. Rapid Communications in Mass Spectrometry, 1997. 11: 1095-1102.
16.
Zimmermann, R., H.J. Heger, R. Dorfner, U. Boesl, M. Blumenstock, D. Lenoir
und A. Kettrup, A Mobile Laser Mass Spectrometer (REMPI-TOFMS) for Continuous Monitoring of Toxic Combustion Byproducts: Real-Time On-Line
Analysis of PAH in Waste Incineration Flue Gases. Combustion Science and
Technology, 1998. 134(1-6): 87.
17.
Zimmermann, R., H.J. Heger, A. Kettrup und U. Nikolai, Direct observation of
the formation of aromatic pollutants in waste incineration flue gases by on-line
REMPI-TOFMS laser mass spectrometry. Fresenius Journal of Analytical Chemistry, 2000. 366: 368-374.
18.
Heger, H.J., R. Zimmermann, R. Dorfner, M. Beckmann, H. Griebel, A. Kettrup
und U. Boesl, On-line Emission Analysis of Polycyclic Aromatic Hydrocarbons
down to pptv Concentration Levels in the Flue Gas of an Incineration Pilot
195
Plant with a Mobile Resonance-Enhanced Multiphoton Ionization Time-ofFlight Mass Spectrometer. Analytical Chemistry, 1999. 71: 46-57.
19.
Thanner, R., H. Oser und H.-H. Grotheer, Time-resolved monitoring of aromatic
compounds in an experimental incinerator using an improved jet-resonanceenhanced multi-photon ionization system Jet-REMPI. European Mass Spectrometry, 1998. 4: 215-222.
20.
Mühlberger, F., R. Zimmermann und A. Kettrup, A Mobile Mass Spectrometer
for Comprehensive On-Line Analysis of Trace and Bulk Components of Complex
Gas Mixtures: Parallel Application of the Laser-Based Ionization Methods VUV
Single-Photon Ionization, Resonant Multiphoton Ionization, and Laser-Induced
Electron Impact Ionization. Analytical Chemistry, 2001. 73(15): 3590-3604.
21.
Franzen, J., R. Frey, A. Holle, H. Betzold, W. Ulke und U. Boesl, Fast Multicomponent Exhaust Gas Analyzer for Motor Development Workplaces. SAE
Technical Papers, 1993. 930082: 55.
22.
Weickhardt, C., U. Boesl und E.W. Schlag, Laser Mass Spectrometry for TimeResolved Multicomponent Analysis of Exhaust Gas. Analytical Chemistry, 1994.
66: 1062-1069.
23.
Pallix, J.B., U. Schühle, C.H. Becker und D.L. Huestis, Advantages of SinglePhoton Ionisation over Multiphoton Ionisation for Mass Spectometric Surface
Analysis of Bulk Organic Polymers. Analytical Chemistry, 1989. 61: 805-811.
24.
Vidal, C.R., Four-Wave Frequency Mixing in Gases, in Tunable Lasers, L.F.
Mollenauer and J.C. White, Editors. 1987, Springer Verlag: Berlin: 56-113.
25.
Maker, P.D. und R.W. Terhune, Study of Optical Effects Due to an Induced Polarization Third Order in the Elektric Field Strength. Physical Review, 1965.
137(3A): 801-818.
26.
Bjorklund, G.C., Effects of Focusing on Third-Order Nonlinear Processes in
Isotropic Media. IEEE Journal of Quantum Electronics, 1975. QE-11, No. 6:
287-296.
27.
Becker, C.H., On the use of single-photon ionization for inorganic surface
analysis. Fresenius Journal of Analytical Chemistry, 1991. 341: 3-6.
196
28.
Butcher, D.J., D.E. Goeringer und G.B. Hurst, Real-Time Determination of Aromatics in Automobile Exhaust by Single-Photon Ionization Ion Trap Mass
Spectrometry. Analytical Chemistry, 1999. 71(2): 489-496.
29.
Kornienko, O., E.T. Ada, J. Tinka, M.B.J. Wijesundara und L. Hanley, Organic
Surface Analysis by Two-Laser Ion Trap Mass Spectrometry. 2. Improved Desorption/Photoionisation Configuration. Analytical Chemistry, 1998. 70: 12081213.
30.
Materer, N., R.S. Goodman und S.R. Leone, Laser single-photon ionization
mass spectrometry measurements of SiCl and SiCl2 during thermal etching of
Si(100). Journal of Vacuum Science and Technolgy, 1997. 15(A4): 2134-2142.
31.
McEnally, C.S., L.D. Pfefferle, R.K. Mohammed, M.D. Smooke und M.B.
Colket, Mapping of Trace Hydrocarbon Concentrations in Two-Dimensional
Flames Using Single-Photon Photoionization Mass Spectrometry. Analytical
Chemistry, 1999. 71: 364-372.
32.
Shi, Y.J., X.K. Hu, D.M. Mao, S.S. Dimov und R.H. Lipson, Analysis of Xanthate
Derivatives
by
Vacuum
Ultraviolet
Laser-Time-of-Flight
Mass
Spectrometry. Analytical Chemistry, 1998. 70: 4534-4539.
33.
Steenvoorden, R.J.J.M., P.G. Kistemaker, A.E. Vries, L. Michalak und N.M.M.
Nibbering, Laser single photon ionization mass spectrometry of linear, branched
and cyclic hexanes. International Journal of Mass Spectrometry and Ion Processes, 1991. 107: 475-489.
34.
Trevor, J.L., L. Hanly und K.R. Lykke, Laser Desorption/Vacuum Ultraviolet
Photoionization of Alkanethiolate Self-assembled Monolayers. Rapid Communications in Mass Spectrometry, 1997. 11: 587-589.
35.
Vries, M.S. und H.E. Hunziker, Vacuum UV photoionization mass spectrometry
of small polymers using jet cooling. Journal of Photochemistry and Photobiology
A - Chemistry, 1997. 106: 31-36.
36.
Werner, J.H. und T.A. Cool, Flame sampling photoionization mass spectrometry
of dichloroethenol. Chemical Physics Letters, 1998. 290: 81-87.
197
37.
Werner, J.H. und T.A. Cool. The Combustion of Trichloroethylene Studied with
Vacuum Ultraviolet Photoionisation Mass Spectrometry. 27th Symposium (International) on Combustion. 1998: The Combustion Institiute, 413-423.
38.
Zoller, D.L., S.T. Sum und M.V. Johnston, Determination of Polymer Type and
Comonomer Content in Polyethylenes by Pyrolysis-Photoionization Mass Spectrometry. Analytical Chemistry, 1999. 71: 866-872.
39.
Heger, H.J., U. Boesl, R. Zimmermann, R. Dorfner und A. Kettrup, On-line
REMPI-TOFMS Laser Mass Spectrometry for Combined Multi-ComponentPattern Analysis and Target-Compound Monitoring: Non-Chlorinated Aromatics and Chlorobenzene in Flue Gases of Combustion Processes. European Mass
Spectrometry, 1999. 5: 51-57.
40.
Hafner, K., Untersuchungen zur Bildung brennstoffabhängiger Stickoxide bei
der Abfallverbrennung mittels on-line analytischer Messmethoden, bisher
unveröffentliche Doktorarbeit am Wissenschaftszentrum Weihenstephan, TU
München.
41.
Wiley, W.C. und I.H. McLaren, Time-of-Flight Mass Spectrometer with Improved Resolution. Review of Scientific Instruments, 1955. 26(12): 1150-1157.
42.
Katzenstein, H.S. und S.S. Friedland, New Time-of-Flight Mass Spectrometer.
Review of Scientific Instruments, 1955. 26: 324-327.
43.
Walter, K., Anwendung von Flugzeit-Massenspektrometrie und LaserSpektroskopie zur Untersuchung von großen Biomolekülen und zur Spektroskopie polyatomarer organischer Radikal-Kationen, Dissertation, 1989, Fakultät für
Chemie, Biologie, Geowissenschaften, TU München.
44.
Mamyrin, B.A., V.I. Karataev, D.V. Shmikk und V.A. Zagulin, The massreflectron, a new nonmagnetic time-of-flight mass spectrometer with high resolution. Journal of Experimental and Theoretical Physics, 1973. 37: 45-48.
45.
Boesl, U., R. Weinkauf und E.W. Schlag, Reflectron time-of-flight mass spectrometry and laser excitation for the analysis of neutrals, ionized molecules and
secondary fragments. International Journal of Mass Spectrometry and Ion Processes, 1992. 112: 121-166.
198
46.
Boesl, U., H.J. Neusser und E.W. Schlag, Two-Photon Ionization of Polyatomic
Molecules in a Mass Spectrometer. Zeitschrift für Naturforschung, 1978. 33 a:
1546-1548.
47.
Boesl, U., H.J. Neusser und E.W. Schlag, Multi-Photon Ionization in the Mass
Spectrometry of Polyatomic Molecules: Cross Sections. Chemical Physics, 1981.
55: 193-204.
48.
Zimmermann, R., C. Weickhardt, U. Boesl und E.W. Schlag, Influence of Chlorine Substituent Positions on the Torsional Potentials of Dichlorinated
Biphenyls: R2PI-Spectra of the First Singulet Transition. Journal of Molecular
Structure, 1994. 327: 81-97.
49.
Zimmermann, R., H.J. Heger, E.R. Rohwer, E.W. Schlag, A. Kettrup und U.
Boesl. Coupling of Gas Chromatography with Jet-REMPI Spectroscopy and
Mass Spectrometry. 8th International Symposium on Resonance Ionization Spectroscopy. 1996. Penn State University, USA: AIP Conference Series 388,
American Institute of Physics, 119-122.
50.
Williams, B.A., T.N. Tanada und T.A. Cool, Resonance Ionization Detection
Limits for Hazardous Emissions, in 24th Symposium (International) on Combustion. 1992, The Combustion Institute: Pittsburgh: 1587-1596.
51.
Marshall, A., A. Clark, R. Jennings, K.W.D. Ledingham, J. Sander und R.P.
Singhal, Laser-induced dissociation, ionization and fragmentation processes in
nitroaromatic molecules. International Journal of Mass Spectrometry and Ion
Processes, 1992. 116: 143-156.
52.
Weickhardt, C., R. Zimmermann, U. Boesl und E.W. Schlag, Laser Mass Spectrometry
of
Dibenzodioxin,
Dibenzofuran
and
Two
Isomers
of
Dichlorodibenzodioxins: Selective Ionization. Rapid Communications in Mass
Spectrometry, 1993. 7: 183-185.
53.
Zimmermann, R., H.J. Heger und A. Kettrup, On-Line monitoring of traces of
aromatic-, phenolic- and chlorinated components in flue gases of industrial
scale incinerators and cigarette smoke by direct-inlet laser ionization-mass
spectrometry (REMPI-TOFMS). Fresenius Journal of Analytical Chemistry,
1999. 363: 720-730.
199
54.
Rohlfing, E.A., Resonantly Enhanced Multiphoton Ionization for the Trace Detection of Chlorinated Aromatics, in 22nd Symposium (International) on
Combustion. 1988, The Combustion Institute: Pittsburgh: 1843-1850.
55.
Letokhov, S., Laser Photoionization Spectroscopy. 1987, Orlando: Academic
Press.
56.
Tanada, T.N., J. Velazquez, N. Hemmi und T.A. Cool, Surrogate Detection for
Continuous Emission Monitoring by Resonance Ionization. Combustion Science
and Technology, 1994. 101: 333-348.
57.
Herrmann, A., S. Leutwyler, E. Schumacher und E. Wöste, Multiphoton ionization: Mass selctive laser-spectroscopy of Na2 and K2 in molecular beams.
Chemical Physics Letters, 1977. 52: 418.
58.
Antonov, V.S., I.N. Knyazev, V.S. Letokhov, V.M. Matuik, V.G. Movshev und
V.K. Potapov, Stepwise laser photoionization of molecules in a mass spectrometer: A new method for probing and detection of polyatomic molecules. Optics
Letters, 1978. 3(2): 37.
59.
Hafner, K., R. Zimmermann, E.R. Rohwer, R. Dorfner und A. Kettrup, A Capillary-Based Supersonic Jet Inlet System for Resonance-Enhanced Laser
Ionization Mass Spectrometry: Principle and First On-line Process Analytical
Applications. Analytical Chemistry, 2001. 73(17): 4171-4180.
60.
Rohwer, E.R., R. Zimmermann, H.J. Heger, R. Dorfner, U. Boesl und A.
Kettrup, Verfahren und Vorrichtung zur Erzeugung eines gerichteten Gasstrahls,
20.05.1998, Deutschland, Patent Nr. 198 22 672.1, Deutsches Patentamt.
61.
Rohwer, E.R., R. Zimmermann, R. Dorfner, U. Boesl und A. Kettrup, Gaseinlaß
zur Erzeugung eines gerichteten und gekühlten Gasstrahls, Deutschland, zum
Patent angemeldet.
62.
Butcher, D.J., Vacuum Ultraviolet Radiation for Single-Photonionization Mass
Spectrometry: A Review. Microchemical Journal, 1999. 62: 354-362.
63.
Becker, U. und D.A. Shirley, eds. VUV and Soft X-Ray Photoionization. Physics
of atoms and molecules. 1996, Plenum Press: New York.
200
64.
Mallard, W.G. und P.J. Linstrom, NIST Chemistry WebBook, NIST Standard
Reference Database (http://webbook.nist.gov). 2000, National Institute of Standards and Technology (NIST): Gaithersburg MD, 20899.
65.
Roboz, J., Introduction to MASS SPECTROMETRY : Instrumentation and Techniques. ASMS Publications Classic Works in Mass Spectrometry. Vol. 3. 1986,
New York: John Wiley & Sons, Inc.
66.
Poschenrieder, W.P. und P. Warneck, Gas Analysis by Photo-Ionization Mass
Spectrometry. Jornal of Applied Physics, 1966. 37(7): 2812-2820.
67.
Diebler, V.H. und R.M. Reese, Mass Spectrometric Study of the Photoionization
of Acethylene and Acetylene-d2*. Jornal of Chemical Physics, 1964. 40: 20342035.
68.
Brion, C.E., A Windowless Photoionization Source for High Resolution Analytical Mass Spectrometry. Analytical Chemistry, 1965. 37(13): 1706-1709.
69.
Brion, C.E., An Improved Mass Spectrometer Photoionization Sourze. Analytical
Chemistry, 1966. 38(13): 1941-1942.
70.
Watanabe, K., Photoionization and Total Absorption Cross Section of Gases. II.
O2 and N2 in the Region 850-1500Å. Journal of Chemical Physics, 1956. 25(5):
965-971.
71.
Schönheit Von, E., Massenspektrometrische Untersuchung der Photoionisation
von Wasserstoff. Zeitschrift für Naturforschung, 1960. 15a: 841-842.
72.
Poschenrieder, W.P. und P. Warneck, Mass Spectrometric Gas Analysis Utilizing Selective Photoionization. Analytical Chemistry, 1968. 40(2): 385-390.
73.
Acton Research Corporation, Optics and Coatings. 1997: Datenblatt.
74.
Gobrecht, H., Bergmann-Schaefer Lehrbuch der Experimentalphysik. 1974.
75.
Körner, H.J. und W. Zinth, Physik III, Optik, Quantenphänomene und Aufbau
der Atome. Vol. 3. 1994, München: R. Oldenburg Verlag.
76.
Hecht, E., Optik. 1989, Bonn: Addison Verlag.
77.
Vidal, C.R., Four-Wave Frequency Mixing in Gases. Tunable Lasers, ed. L.F.
Mollenauer and J.C. White. Vol. 59. 1987, Berlin: Springer Verlag.
201
78.
Kung, A.H., J.F. Young und S.E. Harris, Generation of 1182-A radiation in
phase-matched mixtures of inert gases. Applied Physics Letters, 1973. 22: 301303.
79.
Kung, A.H., J.F. Young und S.E. Harris, Erratum: Generation of 1182-A radiation in phase-matched mixtures of inert gases. Applied Physics Letters, 1976.
28: 239.
80.
Tanaka, Y., A.S. Jursa und F.J. Blanc, Continuous Emission Spectra of Rare
Gases in the Vacuum Ultraviolet Region II. Neon and Helium. Journal of the Optical Society of America B - Optical Physics, 1958. 48: 304.
81.
Gellert, B.B. und U. Kogelschatz, Generation of Excimer Emission in Dielectric
Barrier Discharges. Applied Physics B, 1991. 52: 14.
82.
El-Habachi, A. und K.H. Schoenbach, Emission of excimer radiation from direct
current, high-pressure hollow cathode discharges. Applied Physics Letters,
1998. 72(1): 22-24.
83.
El-Habachi, A. und K.H. Schoenbach, Generation of intense excimer radiation
from high-pressure hollow cathode discharges. Applied Physics Letters, 1998.
73(7): 885 - 887.
84.
Wieser, J., D.E. Murnick, A. Ulrich, H.A. Huggins, A. Liddle und W.L. Brown,
Vacuum ultraviolet rare gas excimer light source. Review of Scientific Instruments, 1997. 68(3): 1360-1364.
85.
Ulrich, A., J. Wieser, M. Salvermoser und D. Murnick, Anregung dichter Gase
mit niederenergetischen Elektronenstrahlen - Neue Wege zu brillanten Lichtquellen und Excimer-Lasern. Physikalische Blätter, 2000. 56(6): 49-52.
86.
Ulrich, A. und J. Wieser, eds. Low-Energy Electon-Beam-Induced XUV Excimeremission. Jahresbericht des Beschleunigerlabaratoriums der Universität und
der Technischen Universität München. 2000.
87.
Wieser, J., M. Salvermoser, L.H. Shaw, A. Ulrich, D.E. Murick und H. Dahi,
Lyman alpha emission via resonant energy transfer. Journal of Physics B, 1998.
31: 4589-4597.
202
88.
Ulrich, A., C. Nießl, J. Wieser, H. Tomizawa, D.E. Murnick und M. Salvermoser, Lasers in dense gases pumped by low-energy electron beams. Journal of
Applied Physics, 1999. 86: 3525.
89.
Ulrich, A., J. Wieser und W. Krötz, VUV-Lampe, 19.9.1996, Deutschland, Patent
Nr. 44 38 407, Deutsches Patentamt.
90.
Brodmann, R. und G. Zimmerer, Vacuum-ultraviolet fluorescence under monochromatic exitation and collision processes in gaseous Kr and Xe. Physica,
1977. 82C: 3395.
91.
Lorents, D.C., The Physics of Elektron Beam Excited Rare Gases at High Densities. Physica B+C, 1976. 82: 19-26.
92.
Ribitzki, G., A. Ulrich, B. Busch, W. Krötz, J. Wieser und D.E. Murnick, Electron densities and temperatures in a xenon afterglow with heavy-ion excitation.
Physical Review E, 1994. 50: 3973.
93.
Ulrich, A., Zur Verfügung gestellt. 2003, Institut für Experimentalphysik (E12),
Technische Universität München: München.
94.
Valkealahti, S., J. Schou und R.M. Nieminen, Energy deposition of keV electrons in light elements. Journal of Applied Physics, 1988. 65(6): 2258 - 2271.
95.
Ulrich, A., J. Wieser, M. Salvermoser und D. Murnick, Anregung dichter Gase
mit niederenergetischen Elektronenstrahlen. Physikalische Blätter, 2000. 56(6):
49-52.
96.
Watanabe, K., Photoionization and Total Absorption Cross Section of Gases. I.
Ionization Potentials of Several Molecules. Cross Section of NH3 and NO. Journal of Chemical Physics, 1954. 22(9): 1564-1570.
97.
Watanabe, K., F.M. Matsunaga und H. Sakai, Absorption Coefficient and
Photoionization Yield of NO in the Region 580-1350 Å. Applied Optics, 1967.
6(3): 391-396.
98.
Kleinknecht, K., Detektoren für Teilchenstrahlung. Teubert Studienbücher: Physik. 1992, Stuttgart: B. G. Teubert.
203
99.
Boyle, J.G., L.D. Pfefferle, E.E. Gulcicek und S.D. Colson, Laser driven electron ionization for a VUV photoionization time-of-flight mass spectrometer.
Combustion Science and Technology, 1990. 70: 187-203.
100.
Giordmaine, J.A. und R.C. Miller, Tunable coherent parametric oscillation in
LiNbO3 at optical frequencies. Physical Review, 1965. 14: 973.
101.
Harris, S., Tunable Optical Parametric Oszillators. Proceedings of the IEEE,
1969. 57(12): 2096-2113.
102.
Fan, Y.X., R.C. Eckardt, R.L. Beyer, J. Nolting und R. Wallenstein, Visible
BaB2O4 optical parametric oscillator pumped at 355 nm by a single-axial-mode
pulsed source. Applied Physics Letters, 1988. 53: 2014-2016.
103.
Wolfrum, K., Beams., TU München.
104.
Suto, K., Y. Sato, C.L. Reed, V. Skorokhodov, Y. Matsumi und M. Kawasaki,
Ion Fragment free Imaging of the Ion-Pair Photodissociation of CH§Cl, CH3Br,
C2H5Cl and C2H5Br at 118 nm. Journal of Physical Chemistry, 1997. 101:
1222-1226.
105.
Bäßmann, C., Entwicklung einer effizienten Ionenquelle unter Einsatz verschiedener Ionisationsprozesse für die Analytik und für Experimente an
massenselektierten molekularen Ionen, Diplomarbeit, 1991, Technische Universität München.
106.
Van Bramer, S.E. und M.V. Johnston, 10.5-eV Photoionization Mass Spectrometry of Aliphatic Compounds. Journal of the American Society for Mass
Spectrometry, 1990. 1: 419-426.
107.
Werner, J.H. und T.A. Cool, Flame sampling photoionization mass spectrometry
fo CH3PO2 and CH3OPO2. Chemical Physics Letters, 1997. 275: 278-282.
108.
Schühle, U., J. Pallix und C.H. Becker, Sensitive Mass Spectrometry of Molecular Adsorbates by Stimulated Desorption and Single-Photon Ionization. Journal
of the American Chemical Society, 1988. 110: 2323-2324.
109.
Feldmann, D., J. Laukemper und K.H. Welge, IR-MPE/D dynamics studied by
pulsed VUV laser photoionization. Journal of Chemical Physics, 1983. 79(1):
278-282.
204
110.
Levy, D.H., The Spectroscopy of Very Cold Gases. Science, 1981. 214: 263.
111.
Zimmermann, R., H.J. Heger, A. Kettrup, F. Mühlberger, K. Hafner und U.
Boesl, Verfahren und Vorrichtung zum Nachweis von Verbindungen in einem
Gasstrom,05.04.2000 Eingang, Deutschland, Patent Nr. DE 100 14 874.6, Deutsches Patentamt.
112.
Dahl, D.A. und J.E. Delmore, SIMION. 1988, Idaho National Engineering Laboratory: Idaho Falls.
113.
O´Keeffe, A.E. und G.C. Ortman, Primary Standards for Trace Gas Analysis.
Analytical Chemistry, 1966. 38(6): 760-763.
114.
Namiesnik, J., Permeation Devices for the Preparation of Standard Gaseous
Mixtures. Chromatographia, 1983. 17: 47-48.
115.
Namiesnik, J., Generation of Standard Gaseous Mixtures. Journal of Chromatography, 1984. 300: 79-108.
116.
Scarangelli, F.P., A.E. O'Keefe, E. Rosenberg und J.P. Bell, Preparation of
known concentrations of gases and vapours with permeation devices calibrated
gravimetrically. Analytical Chemistry, 1970. 42: 871-876.
117.
Kuchling, H., Taschenbuch der Physik. 1991, Thun und Frankfurt/Main: Harri
Deutsch Verlag.
118.
Klus, H. und H. Kuhn, Verteilung verschiedener Tabakrauchbestandteile auf
Haupt- und Nebenstromrauch (Eine Übersicht). Beiträge zur Tabakforschung International, 1982. 11(5): 229-265.
119.
Sakuma, H., M. Kusama, K. Yamaguchi, T. Matsuki und S. Sugawara, The Distribution of Cigarette Smoke Components between Mainstream and Sidestream
Smoke. Beiträge zur Tabakforschung International, 1984. 12(4): 199-209.
120.
Dallüge, J., L.L.P. Van Stee, J. Williams, J. Beens, R.J.J. Vreuls und U.A.T.
Brinkman, Unravelling the composition of very complex samples by comprehensive gas chromatography coupled to time-of-flight mass spectrometry: Cigarette
smoke. Journal of Chromatography A, 2002. 974: 169-184.
205
121.
Rodgman, A., Studies of Polycyclic Aromatic Hydrocarbons in Cigarette Mainstream Smoke: Identification, Tobacco Precursors, Control of Levels: A Review.
Beiträge zur Tabakforschung International, 2001. 19(7): 361-379.
122.
Zimmermann, R., R. Dorfner und A. Kettrup, Direct analysis of products form
plant material pyrolysis. Journal of Analytical and Applied Pyrolysis, 1999.
49(Elsevier): 257-266.
123.
Boichenko, A.M., S.I. Yakovlenko und V.F. Trasenko, Electron beam-excited Xe
excilamp's optimal characteristicse. Laser and Particle Beams, 2000. 18(04):
655-660.
124.
Kerspe, Vakuumtechnik in der industriellen Praxis. Sonderdruck aus Themenband Kerspe u.a. 1987, Ehningen: Expert Verlag.
125.
Laiko, V.V. und A.F. Dodonov, Resolution and Spectral-line Shapes in the Reflecting Time-of-flight Mass Spectrometer with Orthogonal Injected Ions. Rapid
Communications in Mass Spectrometry, 1994. 8: 720-726.
126.
Dawson, J.H.J. und M. Guilhaus, Orthogonal-acceleration Time-of-flight Mass
Spectrometer. Rapid Communications in Mass Spectrometry, 1989. 3(5): 155159.
127.
Coles, J. und M. Guilhaus. Ortogonal Acceleration TOF-MS - Making TOF
Work with Continuous Ion Sources. The Ion Trap Storage/Reflectron Time-ofFlight Mass Spectrometer. 1992. New York: Le Croy Corporation.
128.
Olthoff, J.K., I.A. Lys und R.J. Cotter, A Pulsed Time-of-flight Mass Spectrometer for Liquid Secondary Ion Mass Spectrometry. Rapid Communications in
Mass Spectrometry, 1988. 2(9): 171-175.
129.
Grix, R., U. Grüner, G. Li, H. Stroh und H. Wollnik, An Electron impact Storage
Ion Source for Time-of-flight Mass Spectrometers. International Journal of Mass
Spectrometry and Ion Processes, 1989. 93: 323-330.
130.
Grix, R., R. Kutscher, G. Li, U. Grüner und H. Wollnik, A Time-of-flight Mass
Analyzer with High Resolving Power. Rapid Communications in Mass Spectrometry, 1988. 2(5): 83-85.
206
131.
Paul, W., Electromagnetic traps for charged and neutral particles. Reviews of
Modern Physics, 1990. 62: 531-540.
132.
Burger, B.V., T. Snyman, W.J.G. Burger und W.F. Van Rooyen, Thermal modulator array for analyte modulation and comprehensive two-dimensional gas
chromatography. Journal of Separation Science, 2003. 26: 123-129.
133.
Burger, B.V., M. Le Roux und W.J.G. Burger, Headspace Analysis: A Novel
Method for the Production of Capillary Traps with Ultra-Thick Stationary Phase
Layers. Journal of High Resolution Chromatography, 1990. 13: 777-779.
134.
Ortner, E.K. und E.R. Rohwer, Trace Analysis of Semi-Volatile Organic Air
Pollutants Using Thick Film Silicone Rubber Traps with Capillary Gas Chromatography. Journal of High Resolution Chromatography, 1996. 19: 339-344.
135.
Ortner, E.K. und E.R. Rohwer, Trapping efficiency of aqueous pollutants using
multichannel thick film silicone rubber traps with capillary gas chromatography. Journal of Chromatography, 1999. 863 A: 57-68.
136.
Zimmermann, R., E.R. Rohwer und H.J. Heger, In-Line Catalytic Derivatization
Method for Selective Detection of Chlorinated Aromatics with a Hyphenated
Gas Chromatography Laser Mass Spectrometry Technique: A Concept for Comprehensive Detection of Isomeric Ensembles. Analytical Chemistry, 1999. 71:
4148-4153.
137.
Poole, C.F. und S.K. Poole, Chromatography Today. 1991, Amsterdam: Elsevier.
207
Verzeichnis eigener Veröffentlichungen
1.
Heger, H. J., R. Zimmermann, R. Dorfner, F. Mühlberger, K. Hafner, A. Kettrup,
U. Boesl, Multikomponenten- und Zielsubstanzen-Monitoring mit Lasermassenspektrometrie, Frühjahrstagung der Deutschen Physikalischen Gesellschaft
(MS), 1999 Heidelberg, Deutschland.
2.
Zimmermann, R., H. J. Heger, R. Dorfner, M. Blumenstock, Th. Hauler, U.
Boesl, F. Mühlberger, K. Hafner, T. Ferge, A. Kettrup, Entwicklung und Anwendung von MS-Verfahren mit selektiver Ionisation zur direkten Analyse komplexer
Proben, Frühjahrstagung der Deutschen Physikalischen Gesellschaft (MS), 2000
Bonn, Deutschland.
3.
Zimmermann, R., H. J. Heger, A. Kettrup, F. Mühlberger, K. Hafner, U. Boesl:
Verfahren und Vorrichtung zum Nachweis von Verbindungen in einem Gasstrom; Deutsche Patent Nr. DE 100 14 874.6, 2000.
4.
Mühlberger, F., Entwicklung einer Vakuum-UV-Ionenquelle für die Massenpektrometrie an Umweltproben, Diplomarbeit, 2000, Technische Universität
München, Deutschland.
5.
Ulrich, A., F. Mühlberger, R. Zimmermann und A. Kettrup, Ionenquelle bei der
UV-VUV-Licht zur Ionisation verwendet wird, eingereicht 20.09.2000, Deutschland, Patent Nr. 100 44 655.8, Deutsches Patentamt.
6.
Mühlberger, F., R. Zimmermann und A. Kettrup, A Mobile Mass Spectrometer
for Comprehensive On-Line Analysis of Trace and Bulk Components of Complex
Gas Mixtures: Parallel Application of the Laser-Based Ionization Methods VUV
Single-Photon Ionization, Resonant Multiphoton Ionization, and Laser-Induced
Electron Impact Ionization. Analytical Chemistry, 2001. 73(15): 3590-3604.
7.
Mühlberger, F., J. Wieser, R. Zimmermann, A. Ulrich, Time of Flight Mass
Spectrometry (TOFMS) using Single Photon Ionisation (SPI) via Particle Beam
Induced VUV Excimer Light, Jahresbericht 2001, Maier-Leibnitz-Laboratorium
der LMU und TU München, Deutschland.
8.
Zimmermann, R., F. Mühlberger, A. Ulrich, Anwendung verschiedener VUVLichtquellen zur weichen und selektiven Ionisation für die analytische Massenspektrometrie: Elektronenstrahlgepumpte Edelgas-Excimer Lampe und
208
Frequenzverdreifachung von UV-Laserpulsen, 34. Tagung der deutschen Gesellschaft für Massenspektrometrie, 2000 München, Deutschland.
9.
Mühlberger, F., J. Wieser, A. Ulrich und R. Zimmermann, Coupling of a novel
Electron Beam Pumped Rare Gas-Excimer VUV-Light Source for Single Photon
Ionization (SPI) to a Mobile Time-of-Flight Mass Spectrometer: A New Concept
for a Robust and Compact On-Line Real-Time Industrial Process Gas Analyzer.
Analytical Chemistry, 2002. 74(15): 3790-3801.
10.
Ulrich, A., A. Görtler, G. Kornfeld, R. Krücken, A. Morozov, F. Mühlberger, A.
Peters, R. Steinhübl, J. Wieser, R. Zimmermann, Plasma parameters and radiative properties of low-energy electron beam excited gases, International
Conference on Phenomenia in ionized Gases (ICPIG), 2003 Greifswald,
Deutschland
11.
Zimmermann, R., F. Mühlberger, K. Hafner, Ein TOF-Massenspektrometer mit
paralleler VUV-Einphotonenionisation, 36. Tagung der deutschen Gesellschaft
für Massenspektrometrie, 2003 Münster, Deutschland
12.
F. Mühlberger, A. Ulrich, P. Wallenta, Moleküle im Anflug: Anwendung inkohärenter
Vakuum-Ultraviolett-Lichtquellen
zur
Fotoionisation
in
der
Massenspektrometrie. Laser + Photonik, 2003. 3: 12-13.
13.
L. Cao, F. Mühlberger, T. Adam, T. Streibel, H. Z. Wang, A. Kettrup, R.
Zimmermann, Application of on-line soft laser ionisation in time of flight-mass
spectrometry based on resonance enhanced multi-photon und VUV single photon ionisation (REMPI/SPI-TOFMS): Investigation of the pyrolysis of typical
organic contaminants in the steel recycling process. Analytical Chemistry, in
press.