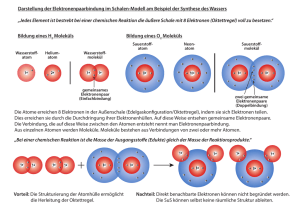
Stoffe – Valenzelektronen – Struktur
Werbung

Stoffe – Valenzelektronen – Struktur Chemiker teilen Materie, die ihnen in die Hände gerät, in verschiedene Kategorien ein: • Gemische o Heterogene Gemische (Mischung auf makroskopischer Ebene) o Homogene Gemische (Mischung auf molekularer Ebene, Beispiel: Lösung) • Reine Stoffe o Verbindungen o Elementare Verbindungen (z.B. Cl2) o Atomare Elemente (z.B. Edelgase) • Heterogene Gemische können Komponenten in verschiedenen Aggregatzuständen • Homogene Gemische und reine Substanzen können auch simultan in mehreren (flüssig, fest, gasförmig) enthalten. Aggregatzuständen vorliegen, man spricht dann von Phasen des jeweiligen Aggregatzustands (Beispiel: In Wasser schwimmendes Eis mit Wasserdampf darüber). Reine Stoffe lassen sich grob in 3 Kategorien einteilen: • Molekulare Substanzen o Eine relativ geringe Anzahl Atome ist fest miteinander über untereinander geteilte Valenzelektronen verbunden. Diese Bindung nennt man kovalent. Die Moleküle wiederum bilden mehr oder weniger feste Verbunde als weiche Festkörper, Flüssigkeiten oder Gase. Beispiele: Wasser, Ammoniak, Glucose, Octan, Kohlendioxid • Salze o Valenzelektronen sind praktisch vollständig von einer Atom- oder Molekülart auf den Bindungspartner übergegangen. Es herrschen grosse elektrische Potentiale zwischen den Komponenten, Kationen (Elektronenverlust, positiv geladen) und Anionen (Elektronengewinn, negativ geladen). Die starken 1 elektrostatischen Kräfte sorgen dafür, dass der Stoff meist fest oder höchstens flüssig ist. Beisiele: Natriumchlorid (Kochsalz), Titandioxid, Kaliumacetat • Metalle o Ein Teil oder auch alle Valenzelektronen sind von den Atomen gelöst und bewegen sich einigermassen frei zwischen den verbliebenen Kationen. Diese werden durch die bewegliche Ladungswolke ziemlich fest zusammengehalten. Solche Stoffe sind meist fest oder auch flüssig. Durch die beweglichen Elektronen leiten Metalle Wärme und elektrischen Strom. Beispiele: Eisen, Quecksilber, Natrium Selbstverständlich gibt es Grenzfälle und Mischformen. Salze, bei denen die Elektronen nicht so vollständig zwischen Kation und Anion ausgetauscht sind, haben einen gewissen kovalenten Anteil. Ebenso existieren Salze mit Metallcharakter, bei denen einige Elektronen nicht auf das Anion übergehen, sondern sich frei im Kristall bewegen. Ein Salz muss nicht notwendigerweise aus ionisierten Atomen bestehen, es kann sich auch aus geladenen molekularen Gruppierungen zusammensetzen, die in sich kovalent gebunden sind. Beispiel: Ammoniumnitrat. Schalenbau – Konsequenzen: Aufbauprinzip und Elektronegativität Metalle sind Elemente, deren Atome die Valenzelektronen eher schlecht binden. Sie sind im Periodensystem auf der linken Seite konzentriert. Der Grund dafür ist die gute Abschirmung der Kernladung durch die tiefer liegende Elektronenschalen bei Beginn einer neuen Schale. Mit steigender Kernladung in der Periode wird die Wirkung der Kernladung auf derselben Schale grösser, die Elektronen werden fester gebunden. Erst durch den Übergang zur nächsten Hauptquantenzahl vergrössert sich der mittlere Abstand der äusseren Elektronen zum Kern wieder so, dass die Abschirmung grösser wird. Eine Durchmischung der Schalen tritt allerdings ab der 3. Periode auf. Die 3d-Orbitalfunktionen sind besser abgeschirmt vom Kern als die 4s-Funktion, weil ihre grösste Elektronendichte relativ weit vom Kern liegt, während s-Funktionen immer eine hohe Dichte in Kernnähe aufweisen. Deshalb gehen 2 Valenzelektronen zuerst in den 4s-Zustand, bevor 3d-Zustände auftreten. Die Elemente mit Auffüllung der 3d-Zustände gehören deshalb zur 4. Periode, die mit 4d-Zuständen zur 5. 2 Periode etc. Für ein Atom mit nur einem Elektron (H, He+, Li2+ etc.) gilt allerdings das ideale Orbitalschema, weil es in einem solchen Atom keine Elektron-Elektron Abstossung gibt. Das lässt sich durch Anregung solcher Atome mit Licht (Spektroskopie) verifizieren, Die abnehmende Abschirmung nach rechts im Periodensystem hat auch zur Folge, dass die Elektronen von Bindungspartnern stärker gebunden werden. Damit wird die kovalente Bindung unsymmetrisch, ein statischer elektrischer Dipol entsteht: µ = qd , wobei q der Betrag der partiellen Ladung auf den Enden des Dipols ist und d der Abstand, hier gerichtet als Vektor. q+ q− d Diese Art Moleküle trägt immer ein kleines elektrisches Feld mit sich, das die gegenseitige Anziehung verstärkt. Stoffe dieser Art nennt man in der Chemie polar. Die Teilladungen q+ und q- sind stets kleiner als die Elementarladung, welche das kleinste freie Ladungsquant repräsentiert, und eine Eigenschaft der Elektronen und Protonen (Wasserstoff-Kerne) ist. Die Eigenschaft, Partnerelektronen stark zu binden, wird semiquantitativ durch die so genannte Elektronegativität beschrieben. Es gibt davon 5 Definitionen, gebräuchlich ist die des Amerikaners Linus Pauling, der ein fundamentales Werk der Chemie verfasst hat: The Nature of the Chemical Bond. Obwohl 1939 erschienen, ist es immer noch aktuell! Das Paulingsche Mass wird aus der Differenz von Bindungsenergien zweier Atomarten A und B bestimmt, wofür man die Dissoziationsenergien von A-B, A-A und B-B in die Atome misst. Als Referenzelement dient das Fluor, das am stärksten elektronenziehende Element, dem willkürlich der Wert 3.98 zugeordnet wird. In Molekülen, die mehr als zwei Atome enthalten, kann man das gesamte Dipolmoment durch Vektor-Addition der Bindungsdipolmomente der verbundenen atomaren Nachbarn darstellen. Das führt dazu, dass z.B. das gesamte Dipolmoment von BF3 gleich Null ist, weil die Fluoratome ein gleichseitiges Dreieck um das Bor bilden. Die Bindungsdipole sind stark, die EN von F ist 3.98, während Bor nur den Wert 2.04 aufweist. NH3 hingegen hat ein Netto-Dipolmoment, weil das N nicht in der Ebene der 3 H liegt. Der Grund für die andersartige Geometrie sind nicht gebundene Elektronen in der Valenzschale des N. 3 Chemischer Formalismus Die Elemente einer Verbindung werden in Formeln durch die im Periodensystem zugeordneten Abkürzungen repräsentiert. Die Anzahl der Atome einer Art wird durch einen nach und tief gestellten Index angegeben. Bei Salzen und metallischen Verbindungen gibt man die kleinste Einheit mit ganzzahligen Verhältnissen an, da sich diese Fragmente im Kristallgitter fortwährend wiederholen. Bei molekularen Verbindungen baut man etwas lokale Strukturinformation ein. Beispiel: Wasserstoffperoxid kann man stöchiometrisch als HO angeben, also eine Verbindung aus gleichviel Wasserstoff und Sauerstoff. In einem Wasserstoffperoxid-Molekül sind jedoch 2 Wasserstoffatome mit 2 Sauerstoffatomen verbunden, deshalb schreibt man H2O2. Man kann auch HOOH angeben, was die Lage der Bindungen zwischen den Atomen verdeutlicht. Die Brutto- oder Summenformeln sind nützlich, um aus den relativen Atommassen bequem die relative Molekülmasse zu berechnen, oder um einen Überblick über die Zusammensetzung zu bekommen. Strukturformeln Um die tatsächlichen Verhältnisse in einem Molekül oder Kristall genauer zu beschreiben, brauchen wir jedoch Strukturformeln. Dafür gibt es einfache Modelle, bei denen die Valenzelektronen durch Punkte und Striche symbolisiert werden, oder komplexe, die Orbtitaldiagramme verwenden. Wasser, brutto H2O, sieht dann so aus: H O H Die zwei Striche repräsentieren je ein Paar Elektronen die zwischen H und O geteilt werden. Jedes H steuert ein Elektron bei (mehr kann es nicht bieten), das O 2 Stück von den 6, die es besitzt (laut Ordnungszahl). Die Bindungselektronen gehören immer zu beiden Partneratomen. Also haben die beiden H nun 2 Elektronen, der Sauerstoff 8. Das ist die jeweilige maximale Elektronenzahl für die Hauptquantenzahl ihrer Valenzschalen. Beide Atomarten haben so die Elektronenkonfiguration ihres zugehörigen Edelgases, H die von He und O die von Ne. Dieser Zustand wird angestrebt, weil er die elektrostatische Energie für die Valenzschalen minimiert. Für Atome der zweiten Periode ist dies immer erfüllt, wenn sie 8 Elektronen in der Valenzschale tragen, man nennt dies auch Oktettregel. Für Wasserstoff mit seiner Mini- Schale ist es eine Duett-Regel. Ab der dritten Periode wird es kompliziert, weil die Schalen zu mischen beginnen. Es bleibt noch die kleine Frage, warum das H2O oben gewinkelt abgebildet wird und nicht etwa linear. Die Antwort liegt bei den 4 Elektronen des O, die nicht 4 für die Bindung verwendet wurden. Wohin damit? Elektronen stossen sich gegenseitig ab, so sehr sie sich auch vom Kern angezogen fühlen. Damit wandert wegen der Elektronendichte zwischen O und H, den Bindungen, die sich gegenseitig abstossen, Elektronenladungsdichte der Valenzschale des O möglichst weit davon weg. Elektronen bleiben dabei gepaart im nahezu gleichen Quantenzustand, sie unterscheiden sich nur durch ihre Spinquantenzahlen von + ½ und – ½. Diese Paare verhalten sich ähnlich wie Bindungen und stossen sich auch gegenseitig ab. Damit sich alle 4 Elektronenpaare möglichst weit voneinander entfernen und so die elektrostatische Abstossung minimieren, bildet sich ungefähr die Geometrie eines Tetraeders aus, wobei an 2 Ecken H sitzen und an den beiden andern ungebundene O : : Elektronenpaare. Man symbolisiert das einfach als H H wobei die Punkte die ungebundenen Elektronen darstellen, oder pseudo-dreidimensional H O Hier werden H die ungebundenen Elektronenpaare durch die Bindungsstriche ohne Atom repräsentiert. Die Geometrie erklärt denn auch zusammen mit den Elektronegativitätsdifferenzen, warum H2O eine polare Substanz ist. Beispiele mit komplexeren Bindungsverhältnissen: Nitrit NO2−: N hat 5 Valenzelektronen (VE), jedes O hat 6. Dazu kommt ein extra Elektron wegen der negativen Ladung, total 18 VE. O O O O N N Die negative Ladung ist aufgrund der 2 gleichwertigen Formeln auf beide O gleichmässig verteilt. Die ungebundenen Valenzelektronen am N sorgen dafür, dass es gewinkelt ist. Ein Dipolmoment hat es hingegen nicht: Geladene Moleküle (= Ionen) haben kein Dipolmoment, sondern einfach eine Ladung. Mit einem weiteren O gelangt man zum Nitrat, das dann kein ungebundenes Elektronenpaar am N besitzt. O O O O + O + N N N O O O + O Die Repräsentation von Elektronen durch Punkte und von gepaarten Elektronen durch Striche ist die Notation nach Lewis. Die Ableitung der räumlichen Molekülstruktur aus der gegenseitigen Abstossung der Bindungselektronen und der ungebundenen Elektronen heisst 5 VSEPR-Modell (valence shell electron pair repulsion). Bisher haben wir Moleküle bzw. Ionen betrachtet, die Elemente der 1. und 2. Periode enthalten und die Oktettregel erfüllen. Beim Übergang zur 3. Periode können auch Bindungen aufgrund der Besetzung von d-Zuständen gebildet werden, z.B. im Sulfat: O O O S O Der Schwefel hat hier 10 Valenzelektronen, der Sauerstoff jedoch 8, wie es strikt für die 2. Periode gilt. Interessant ist, dass d-Zustände in einem Molekül schon in der 3. Periode besetzt werden können, in den freien Atomen aber nicht. Elemente können auch weniger als 8 Valenzelektronen tragen. Eine Variante ist der Verlust aller Valenzelektronen bei den Elementen niedriger Elektronegativität, wenn sie mit einem Element hoher Elektronegativität eine ionische Verbindung (Salz) eingehen. Die Valenzelektronen des schwach elektronegativen Elements werden gänzlich in die Valenzschale der stark elektronegativen Elements transferiert, da dann zu einem Anion mit entsprechender Edelgaskonfiguration wird. Das „Spenderatom“ wird zum Kation und trägt nun die Edelgaskonfiguration der vorgehenden Periode. Die Edelgaskonfiguration ist die energetisch am tiefsten liegende Besetzung einer Periode, deshalb streben molekül- oder ionenbildende Systeme dorthin. Beispiel: NaCl = [Ne]+ [Ar]–. Der andere Fall ist die Verbindung von Elementen ähnlicher Elektronegativität mit wenigen Valenzelektronen, z.B. BH3: H H B H Solche Verbindungen können ein Molekül mit einem ungebundenen Elektronenpaar durch dieses binden, indem sie es in ihre eigene Valenzschale einbauen. Lösungen Zur Durchführung einer Reaktion lieben die Chemiker nichts mehr als eine Lösung (die eines Problems sowieso). Lösungen sind homogene Mischungen und bieten deshalb einige Vorteile gegenüber Festkörpern oder Gasen als Reaktionsmedium, wobei der Nachteil der Festkörper schwerer wiegt. In Festkörpern sind Moleküle, Ionen oder Atome auf einer menschenkompatiblen Zeitskala ortsfest. Wenn man zwei Festkörperoberflächen 6 zusammenpresst, wird eine Reaktion dort höchstens sehr langsam ablaufen, und die Ansammlung der Produkte wird den Prozess zu Erliegen bringen. In Gasen ist die Vermischung von Reaktanden kein Problem, es kann sogar zu schnell gehen, so dass exotherme (wärmeproduzierende) Reaktionen explosionsartig verlaufen können. Eher Schwierigkeiten bieten die Behälter, die dicht sein müssen, weil die Reaktionsmischung sonst entweicht. Nachteilig ist auch, dass nur wenige Stoffe bei unsern Umweltbedingungen gasförmig sind, und dass das Verdampfen normalerweise flüssiger oder gar fester Stoffe viel Energie benötigt und dabei auch zur Zersetzung führen kann, bevor die erwünschte Reaktion eintritt. Lösungen lösen all diese Probleme: Die Moleküle sind mobil und können sich treffen, somit reagieren. In einer Lösung können Festkörper bzw. ihre Komponenten vorliegen, aber auch Gase, alle molekular verteilt. Im Falle heftiger exothermer Reaktion dient das Lösungsmittel als Moderator, es kann Energie absorbieren und durch Verdünnung die Reaktionsrate absenken. Nicht zuletzt ist auch die Sache mit dem Behälter viel einfacher als bei Gasen: Im einfachsten Fall genügt ein Becherglas, es braucht kein geschlossenes System mit Überdrucksicherung. Deshalb beginnen sehr viele chemische Arbeitsvorschriften mit: Man löst … In unserer nicht so perfekten Welt (eigentlich ist sie perfekt, wir sind nur zu ungeschickt) gibt es natürlich auch ein paar Haken bei der Nutzung von Lösungen. Es gibt z.B. Stoffe, die sich partout kaum in irgendeiner Flüssigkeit lösen. Es gibt Stoffe, die sind so reaktionsfreudig, dass sie mit allen bekannten Lösungsmitteln reagieren (oder zumindest mit denen, in denen sich der zweite gewählte Reaktand wohlfühlt). Aus diesen Gründen gibt es dann auch exotischere Arbeitsvorschriften, in denen Lösungen mit Festkörpern, Gase mit Festkörpern oder Gase mit Gasen zur Reaktion gebracht werden. Als besonders harte Methode bietet es sich auch an, zwei Festkörper zusammen zu schmelzen. Der Lösungsprozess Wenn man mit Lösungen arbeiten will, lohnt es sich, einmal den Lösungsprozess für Festkörper und Gase in unterschiedlichen Lösungsmitteln näher zu betrachten, ebenso den Zustand des gelösten Materials. Bei der Lösung von Gasen oder eines flüssigen Stoffs in Flüssigkeit handelt es sich um einen Transfer zwischen Phasen mit hoher Mobilität der Moleküle. Dieser geht ziemlich zwanglos per Diffusion vonstatten, wogegen das Aufbrechen eines Festkörpers ein komplexer Vorgang ist. Man muss hier zwischen dem Lösen polarer und apolarer Stoffe unterscheiden. Apolare Substanzen haben schwache nicht-kovalente 7 Bindungen zwischen den Molekülen in einem Kristall, diese Kristalle sind entsprechend weich. Moleküle treten relativ leicht aus der Oberfläche aus, viele solche Stoffe haben sogar einen deutlichen Dampfdruck (Iod, Naphthalin etc.). Kommt ein solches Material mit dem Lösungsmittel in Berührung, so treten Moleküle in die flüssige Phase über und verteilen sich darin. Gleichzeitig wird die „Lücke“ an der Kristalloberfläche mit Lösungsmittel „gestopft“, so dass eine Rückkehr sehr unwahrscheinlich ist. Der Vorgang setzt sich immer weiter fort, bis die Lösung gesättigt ist. Diese Bedingung umschreibt, dass die Dichte (Konzentration) des gelösten Stoffs so gross geworden ist, dass er wieder Aggregate bildet, d.h. kristallisiert. Dies ist das Lösungsgleichgewicht. Seine Lage hängt vom Verhältnis der Bindungskräfte zwischen zwei Stoffmolekülen zu denjenigen zwischen Stoffmolekülen und Lösungsmittelmolekülen ab. Hier sei kurz angemerkt, dass es dazu keine statischen Dipole braucht. Symmetrische Moleküle wie I2 lösen sich sehr gut in symmetrischen Lösungsmitteln wie CCl4. Die Kraft zwischen zwei solchen Partnern ist zwar schwach, doch immer anziehend, und wird Londonsche Dispersionskraft genannt. Dies ist nicht die van der Waals Kraft, sondern nur ein Teil davon! Sie kommt dadurch zustande, dass die Verteilung der Elektronen in einem Molekül (oder Atom) zeitlich nicht konstant ist. Es besteht ein zeitlich variables elektrisches Feld, das sich mit dem entsprechenden Feld des Nachbarmoleküls synchronisiert (grob vergleichbar mit dem makroskopischen Phänomen der elektrostatischen Influenz). Bei polaren Stoffen ist das Lösen mehr von den zwischenmolekularen Kräften abhängig, am extremsten wird das beim Lösen von Salzen oder starken Säuren unter elektrolytischer Dissoziation. Aus der Oberfläche eines Ionenkristalls treten praktisch keine Teilchen bei Raumtemperatur aus, zu gross sind die elektrostatischen Kräfte zwischen Kationen und Anionen. Ein Lösungsmittel, dessen Moleküle ein kräftiges statisches Dipolmoment besitzen, kann die Struktur jedoch aufbrechen. Man kann sich das wie folgt vorstellen: 8 Die Lösungsmitteldipole werden zunächst an der Oberfläche des Kristalls adsorbiert. Dabei richten sie sich antiparallel zu benachbarten Kationen und Anionen aus, was das lokale elektrische Feld eines solchen Paars schwächt. Die gesamte Bindungskraft an der Oberfläche wird vermindert. Durch die Schwingungen der Ionen im Kristallgitter können jetzt einzelne von ihnen austreten und werden sofort von Lösungsmitteldipolen eingehüllt. Verlässt ein Kation den Kristall oder umgekehrt ein Anion, muss wegen der Elektroneutralität gleich ein Gegenion austreten. Die Lücke wird mit einem Lösungsmitteldipol gefüllt, und der Abbauprozess geht weiter. Die bereits gelösten Ionen werden durch Dipole so eingehüllt, dass ihre wechselseitigen elektrostatischen Kräfte sehr gering werden: Wir haben eine Elektrolytlösung. Die Ionen sind zu einem grossen Grad gegeneinander beweglich, die Lösung leitet elektrischen Strom. Die Grenze der Löslichkeit wird auch hier erreicht, wenn die Konzentration der Ionen so gross wird, dass wieder Kristalle gebildet werden. Dies wiederum hängt davon ab, wie stark die Bindungskraft im Kristall im Vergleich zur Bindung der Ionen durch die Lösungsmitteldipole ist. Die Auflösung von polaren molekularen Stoffen in polaren Lösungsmitteln verläuft ähnlich, nur sind die Kräfte geringer. Die schlechte Löslichkeit von apolaren Gasen oder Flüssigkeiten in polaren Lösungsmitteln erklärt das auf den zwischenmolekularen Bindungskräften beruhende Modell ebenfalls: Im polaren Lösungsmittel herrschen stärkere Kräfte zwischen den Lösungsmittelmolekülen als die, die zwischen Gelöstem und Lösungsmittel möglich sind. Die Lösungsmittelmoleküle „kleben“ also zusammen und lassen das zu Lösende nicht herein. Die Löslichkeit ist also immer eine Frage der relativen Bindungskräfte zwischen und innerhalb von zwei Phasen. Lösung und Energie Lösungsvorgänge sind Zustandsänderungen und damit auch Energieumsatz begleitet. Die Energieänderung, gemessen in einem Kalorimeter mit Druckausgleich (p = const.) nennt man Enthalpie ∆H. Die Lösungsenthalpie ∆SH° für ein Salz setzt sich additiv zusammen aus der Gitterenergie ∆sublH°, die die Bindungsenergie des Kristalls darstellt, und der Solvatationsenthalpie ∆solvH°, die die Bindung der Lösungsmitteldipole an die Ionen repräsentiert. ∆sublH° steht bei Lösung eines Salze wie KCl(s) in H2O für KCl(s) → Der Vorgang für ∆solvH° ist Total ergibt sich K+(g) + Cl–(g) K+(g) + Cl–(g) → KCl(s) → K+(solv, l) + Cl–(solv, l) K+(solv, l) + Cl–(solv, l) ∆sublH° = 701.2 kJ/mol ∆solvH° = -684.1 kJ/mol ∆SH° = 17.1 kJ/mol 9 Hier sei noch angemerkt, dass ∆solvH° nicht nur die Energie beinhaltet, die beim Binden des Lösungsmittels an die Ionen frei wird, sondern auch den Energieverbrauch, um die an die Ionen zu bindenden Moleküle dem Lösungsmittelverband zu entziehen. Der zweite Anteil ist einiges kleiner als der erste, weil Dipol-Dipol Wechselwirkungen schwächer als Ion-Dipol Wechselwirkungen oder gar Ion-Ion Wechselwirkungen sind. Die Vorzeichen der ∆H-Werte sind systemegoistisch zu interpretieren: Negative Werte bedeuten, dass Wärme freigesetzt wird, positive, dass Wärme aufgenommen wird, d.h. das System Gelöstes - Lösungsmittel sich abkühlt. Obwohl der Lösungsvorgang für KCl externe Energie benötigt, läuft er spontan ab. Der Grund dafür ist der Gewinn an Beweglichkeit für die Ionen des Festkörpers, der durch die Entropieänderung ∆S ausgedrückt wird. Die Entropie ist eine Funktion, die die Freiheitsgrade der Bewegung in einem molekularen System repräsentiert bzw. im invertierten Sinn seinen Ordnungsgrad. Die Funktion, die beides, ∆H und ∆S zusammenfasst, ist ∆G = ∆H - T ∆S, genannt Gibbs-Energie. Damit ein Vorgang spontan abläuft, muss sein ∆G negativ sein. Im Gegensatz zur Lösung von Festkörpern oder Flüssigkeiten in Flüssigkeiten ist die Lösung von Gasen in Flüssigkeiten (oder Festkörpern) immer exotherm, d.h. ∆H ist negativ, weil der Übergang stets von schwach bis gar nicht gebundenem Zustand zu einem stärker gebundenen verläuft. Der gelöste Zustand Der flüssige Aggregatzustand ist der komplexeste, weil Ordnung und Chaos zugleich herrschen, und auch die strukturelle Komplexität verschiedener Flüssigkeiten ist sehr variabel. Apolare Flüssigkeiten gleichen einem sehr dichten Gas, gelöste Stoffe darin sind einfach dispergiert. Die Moleküle des Gelösten sind kaum stärker an die Lösungsmittelmoleküle gebunden als diese untereinander. Mit zunehmender Polarität erhöht sich die Ordnung im Lösungsmittel selbst, die Moleküle sind zwar immer noch sehr beweglich, aber es gibt bevorzugte gegenseitige Ausrichtungen. Im Extremfall treten so genannte „Wasserstoffbrücken“ auf. Diese Bedingung ist immer gegeben, wenn das Lösungsmittelmolekül ein sehr elektronegatives Element, an das H direkt gebunden ist, enthält, und das elektronegative Element mindestens ein ungebundenes Valenzelektronenpaar besitzt. Der Klassiker ist Wasser, H2O. Organische Abkömmlinge von H2O, die Alkohole, besitzen diese Eigenschaft ebenfalls, dazu kann man NH3(l) und seine organischen Derivate, die Amine, sowie HF und HCN(l) nennen. HF und HCN(l) gehören wie H2O zu den polarsten aller Lösungsmittel, allerdings sind die ersteren aus anderweitigen Gründen höchst unpopulär. 10 Die Wasserstoffbrücke ist eine Bindung zwischen einem der H-Atome eines Moleküls und dem ungebundenen Valenzelektronenpaar des elektronegativen Elements eines andern Moleküls. Diese Bindung ist stark gerichtet und praktisch linear. Sie sorgt dafür, dass Lösungsmittel, die sie bilden, stark strukturiert sind und hohe Siedepunkte haben. Beispiel H2O: O H O H O H H O H H O H H O H H O H H H H O H H Die Wasserstruktur ist natürlich in Wirklichkeit dreidimensional, mit den O-Atomen in Zentren von Tetraedern, die durch die H gebildet werden. Stoffe mit niedrigem Dipolmoment werden kaum eingelassen, weil das zu einer Erhöhung der Gesamtenergie führen würde, sie sind schlecht löslich in Wasser. Die Struktur wird gebrochen, wenn Ionen in sie eingebaut werden, weil die elektrostatische Kraft zwischen Ionen und Dipolen grösser ist als zwischen Dipolen allein. Sie wird auch gestört, wenn polare Substanzen mit ähnlichem Dipolmoment wie H2O eingefügt werden. Durch das Lösen von Ionen wird die Flüssigkeit lokal inhomogen. Die direkt am Ion liegenden Wassermoleküle sind in seinem elektrostatischen Feld ausgerichtet: H2O H2O OH2 H2O OH2 H2O eigentlich 3-D 1. Solvathülle 1. + 2. Solvathülle Für Anionen liegen die Wasser-Dipole natürlich umgekehrt. Wie dick die Lösungsmittelschicht, die durch die Ionen ausgerichtet wird, tatsächlich ist, hängt von der Art 11 der Ionen selbst ab. Je kleiner der Radius und je grösser die Ladung, desto grösser die Ladungsdichte an der „Oberfläche“ eines Ions, und desto stärker die Solvatationsbindung. Dies führt zum nur scheinbar paradoxen Effekt, dass Ionen, die im Kristallgitter wenig Raum einnehmen, in Messungen an Lösungen grösser erscheinen als Ionen, die im Kristall viel Platz brauchen. Eine solche Messung betrifft z.B. die elektrische Leitfähigkeit, bei der die Ionen durch ein externes elektrisches Feld zur Wanderung gezwungen werden. Anionen haben bei gleicher Ladung meist eine kleinere Ladungsdichte als Kationen, weil der negative Ladungsüberschuss auf Grund der gegenseitigen Elektronen-Abstossung die Elektronenhülle aufbläht, während in Kationen der positive Ladungsüberschuss die Elektronenhülle kontrahiert. Die Folge davon ist, dass die partielle Solvatationsenthalpie des Kations die Löslichkeit von Salzen stark beeinflusst. Konzentrationsbegriff Um die Präsenz eines gelösten Stoffs im Lösungsmittel zu charakterisieren, genügt eine einfache Mengenangabe, auch als sehr praktischer Wert in Mol, nicht mehr. Etwas geschickter ist es, die Zahl der Mole auf eine bestimmte Masse des Lösungsmittels anzugeben. Die Zahl der Mole pro kg Lösungsmittel heisst Molalität. Das hat den Vorteil, dass die Zahl Lösungsmittelmoleküle pro Mol Gelöstes temperaturunabhängig ist, im Gegensatz zum Mass der Molarität, das als Mole pro Liter Lösungsmittel definiert ist. Die Zahl der Lösungsmittelmoleküle pro Volumen ist temperaturabhängig, weil die Massendichte der meisten Stoffe sich stark mit der Temperatur ändert. Eine sehr clevere Konzentrationsdefinition ist der Molenbruch: Er ist bestimmt als die Molzahl der anzugebenden Komponente im Verhältnis zur Summe der Mole aller Komponenten eines Lösungs- oder Gasgemischs. Für Arbeiten in wässriger Lösung hat sich die Molarität eingebürgert, trotz der Schwäche mit der Temperaturabhängigkeit. Man behilft sich, indem man thermochemische oder kinetische Werte auf so genannte Standardbedingungen, in diesem Fall T = 298.15 K (25°C) und p = 101.3 kPa (1 atm), bezieht. Bei physikalisch-chemischen Bestimmungen wird eher die Molalität bzw. der Molenbruch verwendet. Allen Konzentrationsmassen ist gemeinsam, dass sie beschreiben, wie wahrscheinlich man eine bestimmte Molekülart in einer normierten Teilmenge eines Gemischs antrifft. Diese Eigenschaft ordnet die Konzentrationsmasse allgemeiner als Masse einer Dichte im physikalischen Sinn ein. In Gasen kann man als Konzentrationsmass den Partialdruck verwenden: Der Gesamtdruck eines Gasgemischs setzt sich additiv aus den Mol-Anteilen der 12 Komponenten zusammen, was direkt aus Avogadros Befunden hervorgeht. Der Teildruck entspricht also genau dem Molanteil, gleicht somit als Mass dem Molenbruch. In den folgenden Betrachtungen werden wir als Konzentrationsmasse die Molarität M der Dimension [M] = mol l-1 und den Partialdruck p (Pa) verwenden. Da die Definition der Konzentration c= n für die Molarität ist, ist die Menge leicht als n=cV zu errechnen. Molare V Konzentrationen während einer Reaktion A+B → C werden als [A], [B] und [C] symbolisiert. Die totalen analytischen Anfangskonzentrationen werden mit cA und cB bezeichnet. cC ist gleich Null. Verläuft die Reaktion genau wie notiert, so gilt die ganze Zeit [A] + [C] = cA und [B] + [C] = cB wegen der Massenerhaltung. Konzentrationsabhängige Phänomene in der Chemie: Reaktionsraten Anstelle des Begriffs Reaktionsrate ist im deutschsprachigen Raum auch „Reaktionsgeschwindigkeit“ üblich. „Geschwindigkeit“ ist aber intuitiv so sehr an mechanische Ortsveränderungen gebunden, dass Reaktionsrate eher der Beobachtung gerecht wird, wenn ein Stoffumsatz gerade abläuft. Im Englischen heisst es denn auch „reaction rate“. Definieren kann man das auf zwei Arten: Entweder ist es der Mengenumsatz pro Zeiteinheit, oder ein Konzentrationsumsatz pro Zeiteinheit. Es stellte sich empirisch früh heraus, dass sich die mathematische Beschreibung des Mengenumsatzes schlecht zur Verallgemeinerung und zur Übertragung auf Messgrössen eignet. Im Gegensatz dazu haben sich konzentrationsbasierte Beschreibungen sehr bewährt. Die Pioniere der chemischen Kinetik sind heute ziemlich vergessen: Der Erste, der den zeitlichen Verlauf einer chemischen Reaktion beschrieb, war Ludwig Wilhelmy. Er leitete schon 1850 auch die korrekte mathematische Beschreibung her. Diese Arbeit wurde bestätigt durch die gemeinsamen Studien von Augustus Harcourt und William Esson. Für eine Reaktion wie oben beschrieben, wurde gefunden, dass A+B → C ∆[C] = k[A][B] , wenn man [A], [B] und [C] in regelmässigen, im ∆t Vergleich zur gesamten Reaktionszeit kurzen Zeitintervallen bestimmte. Das funktionierte natürlich nur mit recht langsamen Reaktionen, war aber dennoch eine sehr wichtige Erkenntnis. Aus der Abhängigkeit vom Produkt der Konzentrationen der Reaktanden wurde durch Max Trautz die Kollisionstheorie der chemischen Reaktionen entwickelt, welche auch 13 auf der statistischen Mechanik von Ludwig Boltzmann basiert. Die Konzentration beschreibt, wie wahrscheinlich es ist, eine bestimmte Molekülart in einem normierten Volumen anzutreffen. Das Produkt zweier Konzentrationen beschreibt deshalb die Anzahl aller möglichen Kontakte zweier Molekülarten im Einheitsvolumen. Man kann das aus der Kombinatorik ableiten: Haben wir ein geschlossenes Gefäss (das Einheitsvolumen) und darin beispielsweise 6 rote und 3 blaue Kugeln, so berechnet sich die Anzahl aller möglichen Berührungen zwischen roten und blauen Kugeln, falls wir das Gefäss bewegen, zu 6 x 3. Diese Zahlen repräsentieren aber gerade auch die „Konzentration“ roter und blauer Kugeln im Einheitsvolumen. Der Faktor k ist die so genannte Geschwindigkeitskonstante. Wie schon Wilhelmy fand, hängt sie von der Temperatur (und auch vom Druck) ab. Gemäss Kollisionstheorie ist k eine Erfolgswahrscheinlichkeit. Um eine Kollision von A mit B erfolgreich zum Produkt C zu bringen, müssen noch mehr Bedingungen als die schlichte Tatsache der Begegnung von A und B erfüllt sein. Die kinetische Bewegungsenergie der Moleküle muss einen Mindestwert besitzen, und nicht alle räumlich verschiedenen Begegnungen führen zum Erfolg, weil Moleküle stark strukturiert sein können. Wilhelmy hat sein Gesetz auch richtigerweise in eine Differentialgleichung überführt: d [C] d [A] d [B] =− =− = k[A][B] dt dt dt Dies ist die noch heute übliche Art, Gesetze für Reaktionsraten zu notieren. Die Differentialquotienten stehen für die Rate, der Term k[A][B] für die Konzentrationsabhängigkeit der Rate. Die Zeitabhängigkeit lässt sich durch Integration erhalten, dazu jedoch später. Das chemische Gleichgewicht Es gibt Reaktionen, die nie im Sinne von A+B → C A und B gänzlich zu C umwandeln. Die Reaktion scheint „abzubrechen“ doch das kann nicht sein, weil es keine physikalischen Gründe für einen Stopp gibt (Magie wäre noch möglich). Die einzige rationale Erklärung besteht darin, eine Gegenreaktion C → A+B in Betracht zu ziehen. Nach einem gewissen Umsatz von A und B zu C steigt die Konzentration von C so sehr, dass sie die Rate der Gegenreaktion die Bildungsrate von C kompensiert: Wir haben Gleichgewicht, genau genommen dynamisches Gleichgewicht, die einzige Form in der Chemie, denn statische Gleichgewichte wurden bis heute nicht gefunden. 14 Man könnte so ein dynamisches Gleichgewicht auch mit „rasendem Stillstand“ umschreiben, weil die Stoffumsätze in beiden Richtungen sehr hoch sein können, ohne dass äusserlich etwas geschieht. Wir schreiben jetzt d [C] = 0 = k f [A][B] − kr [C] dt um auszudrücken, dass sich [C] nicht mehr ändert. –kr stammt vom Gesetz für die Rückreaktion, d [C] = − kr [C] , das den Zerfall von C beschreibt. kf ist das k aus der dt ursprünglichen Formulierung, da wir jetzt zwei Reaktionen betrachten, müssen wir ihre kFaktoren unterscheiden. f steht für forward und r für reverse. 0 = k f [A][B] − kr [C] kann man umformen zu k [C] = f = K eq [A][B] kr Das ist das Massenwirkungsgesetz (MWG, Law of Mass Action), wie es zuerst 1864 von Guldberg und Waage formuliert wurde. Es besagt, in welchem Verhältnis [A], [B] und [C] für konstante Umgebungsbedingungen (Temperatur, Druck) zueinander stehen, sobald Gleichgewicht erreicht ist. Die Reaktionsgleichung wird jetzt auch entsprechend ⇀ C A+B ↽ geschrieben. Die Gleichgewichtskonstante Keq ist keine echte Konstante, sondern eigentlich eine Funktion der Temperatur und des Drucks, genauso wie die Geschwindigkeitskonstanten, aus denen sie errechnet wird. Das Massenwirkungsgesetz ist auch die exakte Form, mit der sich die Konzentrationsabhängigkeit des Prinzips von Le Châtelier begründen lässt. Dieses besagt, dass ein chemisches Gleichgewichtssystem immer auf einen ausgeübten Zwang hin ausweicht. Erhöhung der Temperatur verschiebt ein Gleichgewicht in Richtung der endothermen Teilreaktion, Abkühlen in Richtung der exothermen. Bei Gasreaktionen treibt Erhöhung des Drucks das Gleichgewicht auf die Seite mit der kleineren Anzahl Moleküle, weil dann das Volumen abnimmt. Bei gleicher Zahl Moleküle geschieht nichts. Die Wirkung von Konzentrationsänderungen lässt sich mit dem Massenwirkungsgesetz illustrieren: Für [C] = K eq erhöht die Zugabe von A die Konzentration [C], und [B] nimmt ab. Gibt [A][B] man C zu, steigen [A] und [B]. 15 Heterogene Gleichgewichte Dieser Fall liegt vor, wenn bei einer Reaktion Moleküle, Atome oder Ionen zwischen Phasen bzw. Aggregatzuständen übertreten. Typische Fälle sind Bildung oder Zersetzung von Festkörpern in Lösung, Austausch zwischen Gas und Festkörper oder Gas und Lösung. In der Lösung können wir unser normales Konzentrationsmass verwenden, in der Gasphase den Partialdruck. Ein sehr praktischer Zusammenhang besteht auch darin, dass die gelöste Konzentration eines Gases in einer Flüssigkeit oder einem Festkörper in erster Näherung proportional zu seinem Partialdruck ist. Die schwierigere Frage ist die nach der Konzentration eines Festkörpers. Dazu kann man feststellen, dass, weil ein Festkörper nur Austauschreaktionen an der Grenzfläche zu einer Lösung oder einem Gas eingehen kann, sich die Konzentration innerhalb der festen Phase nicht ändert, bis der Festkörper aufgebraucht ist. Das ist anders für Gas in Kontakt mit Lösung oder Kontakt zwischen zwei nicht mischbaren Lösungen. Dort führt die molekulare Bewegung zu einem ständigen Konzentrationsausgleich in den Phasen, wenn etwas über die Grenzfläche ein- oder auswandert. Eine Lösung oder ein Gas über einem Festkörper „sieht“ hingegen immer eine konstante Konzentration des Feststoffs. Das schlägt sich dann in einer einfacheren Schreibweise des MWG nieder. Beispiel: Die Auflösung von CaCO3 in CO2-haltigem Wasser. Das ist das Phänomen, das zur Wasserhärte führt und zur Bildung von Karst-Erscheinungen. Das Gleichgewicht besteht zwischen 3 Phasen. ⇀ Ca2+ + 2 HCO3– CaCO3 + H2O + CO2 ↽ [Ca 2+ ][HCO3− ]2 K= [CO 2 ][H 2O] Man sieht hier, dass CaCO3 gar nicht im MWG auftaucht. Da seine „Konzentration“ nicht variiert, solange festes Material da ist, wurde es gleich in die Konstante einbezogen. Manche Lehrbücher schreiben, die Konzentration eines Festkörpers sei immer gleich 1, was aber so nicht stimmt (Mortimer hat’s richtig). Weil Chemiker gern vereinfachen, kann man an diesem MWG noch weiter basteln. Falls H2O das Lösungsmittel ist, ist seine Konzentration im Gegensatz zum Gelösten so gross, dass sie selbst bei einem gewissen Umsatz nahezu konstant bleibt. Also weg damit und rein in die Konstante [Ca 2+ ][HCO3− ]2 K'= [CO 2 ] Wie schon erwähnt, ist die Konzentration eines gelösten Gases etwa proportional zum Partialdruck, und CO2 ist ein Gas, darum endet man bei 16 [Ca 2+ ][HCO3− ]2 pCO 2 K"= Ein Beispiel für ein Festkörper-Gasphase Gleichgewicht ist das Exponieren von NH4HS in einer Vakuumkammer. Dabei treten NH3 und H2S aus dem Festkörper in die Gasphase über: ⇀ NH3(g) + H2S(g) NH4HS(s) ↽ MWG: K p = pNH3 pH 2 S Gibt man NH3 und H2S gleichmässig zu (entspricht Kompression der Gasphase), so bildet sich einfach mehr NH4HS, bis die Partialdrucke wieder gleich sind wie vorher. Gibt man NH3 allein zu, so wird so lange NH4HS gebildet, bis die Partialdrucke wieder Kp erfüllen, dabei ist dann pNH 3 > pH 2 S . Zugabe von nicht reagierenden Gasen oder NH4HS ändert nichts an den Partialdrucken. Wegnahme von NH3 oder H2S bewirkt Verdampfen von NH4HS, bis das Produkt der Partialdrucke wieder Kp erfüllt. Temperaturerhöhung verstärkt die Verdampfung, Kp und damit die Partialdrucke werden grösser, weil Kp bezüglich Temperatur eben keine Konstante ist! Die Konstante des Lösungsgleichgewichts von in Wasser schwerlöslichen Salzen besitzt eine spezielle Bezeichnung: Löslichkeitsprodukt. Der Name rührt daher, dass der Nenner des MWG eigentlich nur die „Festkörperkonzentration“ enthält, die aber in der Konstante verschwindet und somit immer ein Produkt verbleibt. Beispiele: ⇀ Ba2+ + SO42− BaSO4 ↽ ⇀ 2 Ag+ + CrO42− Ag2CrO4 ↽ L(BaSO4 )=[Ba 2+ ][SO 2-4 ] L(Ag 2CrO 4 )=[Ag + ]2 [CrO 2-4 ] Das Löslichkeitsgleichgewicht spielt nur, solange Festkörper vorhanden ist! Ist die Verdünnung so gross, dass kein Salz ausfällt, ist das Löslichkeitsprodukt bedeutungslos. Gekoppelte Gleichgewichte Falls für eine Molekül- oder Ionenart in einer Lösung 2 (oder gar mehr) unterschiedliche Reaktionen möglich sind, so sind die dazugehörigen Gleichgewichte gekoppelt. Beispiel: ⇀ NH3 + B(OH)3 ↽ NH3 + H+ ⇀ ↽ H + HN H O H - B O O NH4+ H (H3NB(OH)3) K1 = [H 3 NB(OH)3 ] [NH 3 ][B(OH)3 ] K2 = [NH +4 ] [NH 3 ][H + ] H 17 Beide MWGs enthalten die Konzentration [NH3] und können somit unter Elimination dieser Variablen ineinander eingesetzt werden: K1 [H 3 NB(OH)3 ][H + ] = = K3 [NH +4 ][B(OH)3 ] K2 Dieses Gleichgewicht ist zentral für die Bestimmung von Amin-Stickstoff in organischem Material nach Kjeldahl. 18