Marlies Michl Basics Hämatologie
Werbung

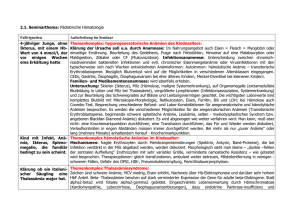
Marlies Michl Basics Hämatologie Leseprobe Basics Hämatologie von Marlies Michl Herausgeber: Elsevier Urban&Fischer Verlag http://www.narayana-verlag.de/b7609 Im Narayana Webshop finden Sie alle deutschen und englischen Bücher zu Homöopathie, Alternativmedizin und gesunder Lebensweise. Das Kopieren der Leseproben ist nicht gestattet. Narayana Verlag GmbH, Blumenplatz 2, D-79400 Kandern Tel. +49 7626 9749 700 Email [email protected] http://www.narayana-verlag.de Hämolytische Anämie II Angeborene hämolytische Anämien M e m b ra n d ef e kte Sphärozytose (hereditäre Kugelzellanämie) Sie ist die häufigste vererbte hämolytische Anämie in Mitteleuropa (Prävalenz in Deutschland 1:3000). Ursache Ursächlich ist ein Defekt oder Mangel der Zytoskelett-Strukturproteine Ankyrin (50 % der Fälle, autosomal-dominant), Bande 3 oder Spektrin (20 % der Fälle, autosomalrezessiv). Dadurch ist die Zellstabilität (Interaktion zwischen Zytoskelett und Zellmembran) reduziert und die bikonkave Form der Erythrozyten geht verloren. Die Erythrozyten nehmen eine energetisch günstigere Kugelform an (Sphärozyten). Die unflexiblen Sphärozyten behindern die MikroZirkulation und werden vorzeitig im MMS abgebaut. Klinik und Diagnostik Neben den Symptomen einer Anämie kommt es typischerweise zu Splenomegalie und rezidivierendem Ikterus. Hämolytische und schmerzhafte vasookklusive Krisen mit Organinfarkten sind häufige Komplikationen. Oft finden sich Pigmentgallensteine und damit verbunden Gallensteinkoliken. Neben einer positiven Familienanamnese und den typischen laborchemischen Hämolysezeichen ist der Nachweis einer verminderten osmotischen Resistenz der Kugelzellen beweisend für die Sphärozytose. Im Blutausstrich zeigen sich kleine Mikrosphärozyten, bei denen die zentrale Aufhellung fehlt. Therapie Bei symptomatischen Patienten ist die Splenektomie (nicht bei Kindern < 6 Jahren wegen Gefahr von Infektionen) angebracht. Hierdurch wird zwar weder die Zahl der Kugelzellen vermindert noch der Membrandefekt behoben, aber der Abbau der Erythrozyten deutlich verlangsamt und die hämolytische Anämie reduziert. Man hat herausgefunden, dass bei subtotaler Splenektomie (10-30 ml postoperatives Restvolumen) eine anhaltende Normalisierung des Hämoglobinwerts bei deutlich erniedrigter Hämolysestärke erreicht wird. Niemals vergessen, dass 2-3 Wochen vor Splenektomie eine Impfung gegen Pneumokokken und Haemophilus influenzae durchgeführt werden muss, um ein „OPSI" mit Pneumokokkensepsis und Verbrauchskoagulopathie zu verhindern. Elliptozytose Die Elliptozytose ähnelt klinisch der Sphärozytose mit mildem Verlauf. Sie ist Folge verschiedener genetischer Mutationen und wird autosomal-dominant vererbt. Es kommt zu einer defekten Zusammenlagerung von Spektrin-Heterodimeren zu -Heterotetrameren, was zur ellipsenförmigen Erythrozytenform führt (s. Bildanhang II, S. 150, l Abb. 13). Diagnose Sie wird morphologisch gestellt. Eine Splenektomie ist nur in 10 -15 % der Fälle nötig. Glukose-6-Phosphat-Dehydrogenase-Mangel (Favismus) Es handelt sich um eine der häufigsten Erbkrankheiten und die häufigste Enzymkrankheit weltweit. Das geographische Verteilungsmuster ähnelt dem der Sichelzellanämie (Afrika, Asien, Mittelmeerraum). Pathogenese Die Erkrankung, eine Punktmutation oder Deletion im Glukose-6-Phosphat-Dehydrogenase-Gen, wird X-chromosomal-rezessiv vererbt. Daher erkranken fast ausschließlich Männer, während Frauen Konduktorinnen sind, wobei deren Glukose-6-PhosphatDehydrogenase-Aktivität auf die Hälfte reduziert ist. Demnach variiert die klinische Symptomatik von absoluter Beschwerdefreiheit bis schwerer chronischer Hämolyse mit akuten lebensbedrohlichen hämolytischen Krisen. Wie bei der Sichelzellanämie besteht bei den Anlageträgern eine gewisse Resistenz gegenüber Malariaplasmodien (M. falciparum). Die Glukose-6-PhosphatDehydrogenase stellt ein wichtiges Enzym im Pentosephosphatweg, einem Nebenweg der Glykolyse, dar und dient im Erythrozyten der Produktion von NADPH. Dieses benötigt der Erythrozyt zur Reduktion von Glutathion, das den Erythrozyten vor oxidativem Stress schützt. Ist die Glukose-6-Phosphat-Dehydrogenase defekt, fehlt der Oxidationsschutz des Erythrozyten und er wird frühzeitig abgebaut. Klinik Es kommt zu akuten hämolytischen Krisen als Reaktion auf oxidativen Stress bei Infek- tionen, durch den Genuss von Saubohnen (Favabohnen) oder durch die Einnahme von Medikamenten (Malariamedikamente, Sulfonamide, Chloramphenicol, hohe Dosen ASS, Vitamin-K-Analoga). Die Patienten leiden unter starken Schmerzkrisen (z.B. Bauch-, Rückenschmerzen), Schüttelfrost und Fieber. Diagnostik Wegweisend sind die beschriebenen laborchemischen Anämie- und Hämolyseparameter sowie der Nachweis von Heinz-Innenkörperchen, die oxygeniertes denaturiertes Hämoglobin darstellen (s. Bildanhang II, S. 150, l Abb. 16), fragmentierte Zellen und Poikilozyten (abnorm geformte Erythrozyten, z. B. keulen-, sichel-, hantel-, birnenförmig) im Blutausstrich. Im Intervall zwischen zwei hämolytischen Krisen normalisiert sich das Blutbild wieder. Diagnostisch beweisend ist der Nachweis einer verminderten Glukose-6Phosphat-Dehydrogenase-Aktivität im Erythrozyten. Therapie Eine kurative Therapie gibt es nicht. Betroffene sollten die auslösenden Noxen meiden, da eine Hämolyse im Allgemeinen nur bei Exposition oben genannter Auslöser erfolgt. Bei schweren Anämien können Bluttrans fusionen verabreicht werden. Bei Säuglingen können eine Phototherapie und Austauschtransfusionen indiziert sein. Pyruvatkinasemangel Der Pyruvatkinasemangel ist ein relativ seltener Glykolysedefekt und wird autosomalrezessiv vererbt. Gestört ist der letzte Schritt der aeroben Glykolyse, dessen Endprodukte Pyruvat und ATP sind. Durch die verringerte ATP-Synthese kommt es - nur bei homozygoten Merkmalsträgern - zur erhöhten Zellsteifigkeit der Erythrozyten mit hämolytischer Anämie und Splenomegalie. Diagnostik Der Blutausstrich zeigt Akanthozyten (geschrumpfte Erythrozyten mit typischer „Stechapfelform") und eine Retikulozytose. Beweisend ist der Nachweis einer verminderten Pyruvatkinase-Aktivität im Erythrozyten. Therapie Sie besteht aus Bluttransfusionen v. a. während Infektionen und Schwangerschaft. Eine Splenektomie kann die Transfusionshäufigkeit verringern. Eine kurative Therapie gibt es nicht. Leseprobe von Marlies Michl „BASICS Hämatologie“ Herausgeber: Elsevier Urban & Fischer Leseprobe erstellt vom Narayana Verlag, 79400 Kandern, Tel: 0049 (0) 7626 974 970-0 Anämien Störungen der Hämoglobinsynthese Zu Thalassämie und Sichelzellanämie siehe Seite 58/59 und 60/61. Erworbene hämolytische Anämien Immunologische Genese Autoimmunhämolytische Anämie (AIHA) Autoimmunhämolytische Anämien entstehen durch vorzeitige Erythrozytolyse infolge Autoantikörperbildung. Die Art der Hämolyse ist abhängig von der Klasse der Autoantikörper (IgM bzw. IgG). Die AIHA kann in jedem Alter unabhängig vom Geschlecht auftreten. 70 % der Patienten haben Wärmeautoantikörper, 15 % Kälteautoantikörper und 15% beide. Ätiologie In etwa der Hälfte der Fälle tritt die AIHA primär bzw. idiopathisch auf. Sekundäre AIHA entstehen infolge von Infektionen, durch die Einnahme von Medikamenten oder im Zusammenhang mit malignen lymphoproliferativen oder rheumatischen Erkrankungen (l Tab. 5). Diagnostik Im Labor finden sich Zeichen einer intra-und extravasalen hämolytischen Anämie. Alle AIHA sind durch einen positiven CoombsTest (s. S. 8/9), dem wichtigsten Test bei Verdacht auf AIHA, gekennzeichnet. Er weist IgG (Wärmeautoantikörper) oder Komplement (Kälteautoantikörper) auf der Erythrozytenoberfläche nach. Einteilung Sie erfolgt nach der Aktivität der Autoantikörper in Abhängigkeit vom Temperaturoptimum: 54 l 55 AIHA durch Wärmeantikörper: Sie gehören zur Klasse der IgG, ihr Temperaturoptimum liegt bei 37 °C. Die Wärmeantikörper binden an die Erythrozytenoberfläche. Die opsonierten Erythrozyten werden von den Makrophagen extravasal im MMS von Milz und Leber zerstört. Diese AIHA manifestiert sich als (chronische) hämolytische Anämie mit variablem Schweregrad. Meist kommt es zu unspezifischen Anämiesymptomen (Abgeschlagenheit, Müdigkeit, Kurzatmigkeit, Herzrasen). Bei der körperlichen Untersuchung zeigen sich Zeichen einer Anämie (Tachypnoe, Tachykardie, Schleimhautblässe) und Splenomegalie. Es kommt infolge verschiedenster Auslöser plötzlich zu schweren hämolytischen Krisen mit Fieber, Ikterus und „bierbraunem" Urin. Im Blutausstrich fällt bei vorliegenden Wärmeautoantikörpern eine Sphärozytose auf. An erster Stelle stehen therapeutisch die Behandlung der Grunderkrankung und die Immunsuppression (hochdosiert Kortikoide z. B. Prednisolon). Ist der Hauptabbauort der Erythrozyten die Milz (szintigraphischer Nachweis), ist eine Splenektomie zu diskutieren. Bei vital bedrohlicher Hämolyse sind Erythrozytenkonzentrate zu transfundieren. AIHA durch Kälteantikörper: Sie gehö-ren zur Klasse der IgM, ihr Temperaturoptimum liegt bei 4 °C. Die Kälteautoantikörper (IgM) fixieren Komplement auf der Erythrozytenoberfläche, wodurch die Erythrozyten v. a. intravasal über den klassischen Komplementweg zerstört werden. Die Klinik gleicht der der AIHA durch Wärmeautoantikörper. Die Anämie verläuft meist milde und verschlechtert sich nach Kälteexposition. Hochtitrige Kälteagglutinine können bei Kälteexposition zu einer Agglutination von Erythrozyten in den Kapillaren führen und somit eine Akrozyanose (violette Hautfärbung an Fingern, Zehen, Nase und Ohren, Abb. l) oder sogar akrale Ulzerationen verursachen. Die schmerzhafte Akrozyanose bei Kälte im Vergleich zur Beschwerdefreiheit in warmer Umgebungstemperatur ist ein wegweisendes Symptom. Im Blutaus- I Abb. 1: Akrozyanose der Finger bei AIHA durch Kälteautoantikörper. [16b] strich fällt bei vorliegenden Kälteautoantikörpern eine starke Erythrozyten-Agglutination auf (s. Bildanhang II, S. 150, Abb. 14). Therapie: Da der Krankheitsverlauf meist milde ist, reicht es häufig aus, wenn die Patienten die Kälte meiden. Steroide und Splenektomie sind hier nicht wirksam! Zytostatika wie Chlorambucil oder Cyclophosphamid sind schweren Fällen vorbehalten. Abgegrenzt wird die paroxysmale Kältehämoglobinurie vom Typ Donath-Landsteiner (IgG), die nur bei Kindern und nach Virusinfektionen auftritt. Alloimmunhämolytische Anämie Bei der alloimmunhämolytischen Anämie reagieren Antikörper eines Organismus mit den antigenen Strukturen auf Erythrozyten eines anderen Organismus der gleichen Spezies. Die Erythrozyten werden also durch fremde, nicht körpereigene Antikörper (AlloVIsoantikörper) zerstört. Diese Anämieform kommt vor bei Rhesus-Inkompatibilität des Neugeborenen und ABO-Inkompatibilität (Ak der Mutter gegen Erythrozyten des Kindes, s. S. 56/57). Weitere Auslöser für alloimmunogene Reaktionen sind Transfusionszwischenfälle und allogene Knochenmarktransplantationsreaktionen (s. S. 112/113 und 120/121). Wärme-AIHA Primär: idiopathisch (IgG) Sekundär: lymphoproliferative Erkrankungen (maligne Lymphome) und andere Neoplasien, rheumatische Erkrankungen (z. B. rheumatoide Arthritis oder Kollagenosen wie systemischer Lupus erythematodes), Medikamente (Methyldopa, NSAR, Penicillin, Chinidin), Infektionen (Viren). Kälte-AIHA Primär: idiopathisch (IgM) Sekundär: lymphoproliferative Erkrankungen {maligne Lymphome), Infektionen (Mykoplasmen, Mononukleose), monoklonale Gammopathie (z.B. M. Waldenström). Tab. 5: Ätiologie der AIHA; häufige Ursachen fett ausgezeichnet. Leseprobe von Marlies Michl „BASICS Hämatologie“ Herausgeber: Elsevier Urban & Fischer Leseprobe erstellt vom Narayana Verlag, 79400 Kandern, Tel: 0049 (0) 7626 974 970-0 Marlies Michl Basics Hämatologie 176 Seiten, kart. erschienen 2010 Mehr Bücher zu Homöopathie, Alternativmedizin und gesunder Lebensweise www.narayana-verlag.de