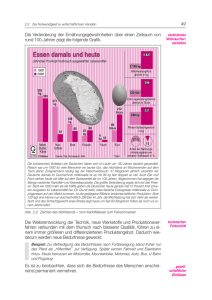
Adriano Aguzzi, Zįrich Heinz Bielka, Berlin Falko Herrmann
Werbung

Herausgeberbeirat Adriano Aguzzi, Zçrich Heinz Bielka, Berlin Falko Herrmann, Greifswald Florian Holsboer, Mçnchen Stefan H. E. Kaufmann, Berlin Peter C. Scriba, Mçnchen Gçnter Stock, Berlin Harald zur Hausen, Heidelberg Molekulare Medizin Aus dem Themenbereich der molekularen Medizin sind bereits folgende Titel der Herausgeber D. Ganten und K. Ruckpaul erschienen: Molekular- und Zellbiologische Grundlagen (1997) ISBN 3-540-61954-2 Tumorerkrankungen (1998) ISBN 3-540-62463-5 Herz-Kreislauf-Erkrankungen (1998) ISBN 3-540-62462-7 Immunsystem und Infektiologie (1999) ISBN 3-540-62464-3 Erkrankungen des Zentralnervensystems (1999) ISBN 3-540-64552-7 Monogen bedingte Erbkrankheiten 1 (2000) ISBN 3-540-65529-8 Monogen bedingte Erbkrankheiten 2 (2000) ISBN 3-540-65530-1 Molekularmedizinische Grundlagen von hereditåren Tumoren (2001) ISBN 3-540-67808-5 Molekularmedizinische Grundlagen von Endokrinopathien I (2001) ISBN 3-540-67788-7 Molekularmedizinische Grundlagen von nicht-hereditåren Tumoren (2002) ISBN 3-540-41577-7 Grundlagen der Molekularen Medizin, 2. çberarbeitete und erweiterte Aufl. (2003) ISBN 3-540-43207-8 Molekularmedizinische Grundlagen von håmatologischen Neoplasien (2003) ISBN 3-540-41640-4 Molekularmedizinische Grundlagen von rheumatischen Erkrankungen (2003) ISBN 3-540-43735-5 Molekularmedizinische Grundlagen von altersspezifischen Erkrankungen (2004) ISBN 3-540-00858-6 Detlev Ganten Klaus Ruckpaul gemeinsam mit Roland R. Wauer (Hrsg.) Molekularmedizinische Grundlagen von fetalen und neonatalen Erkrankungen Mit Beitrågen von Reinhard Berner, Lutz Bindl, Beate Brand-Saberi, Stephan Buderus, Jærg Bungert, Ingolf Cascorbi, Bodo Christ, Patrick Collombat, Christof Dame, Sæhnke Dammann, Klaus Diedrich, Jærg Dætsch, Stephan Ehl, Wilhelm Friedrich, Klaus Friese, Johann Gross, Annette Grçters, Sinuhe Hahn, Rudolf Happle, Michael Hofbeck, Wolfgang Holzgreve, Arne Kænig, Joachim Kreuder, Michael J. Lentze, Knud Linnemann, Volker von Loewenich, Michael Ludwig, Ahmed Mansouri, Peter Miny, Stefan Mundlos, Ioannis Mylonas, Andreas Plagemann, Christian Plank, Wolfgang Rascher, Anita Rauch, Christof Schaefer, Dietmar Schranz, Klaus Schwarz, Eberhard Schwinger, Sevgi Tercanli, Diana Tomi, Roland R. Wauer Mit 249 Abbildungen und 83 Tabellen 12 Professor Dr. med. Detlev Ganten Vorstandsvorsitzender Charit ± Universitåtsmedizin Berlin Schumannstr. 20/21 10117 Berlin Professor Dr. Klaus Ruckpaul Max-Delbrçck-Centrum fçr Molekulare Medizin (MDC) Robert-Ræssle-Straûe 10 13125 Berlin-Buch Professor Dr. Roland R. Wauer Universitåtsklinikum Charit Otto-Heubner-Centrum fçr Kinder- und Jugendmedizin Klinik fçr Neonatologie Schumannstraûe 20/21 10117 Berlin Legende zur Umschlagabbildung: 57 Tage alter Embryo mit sichtbar gemachtem Chondroskeleton. (Quelle: The Encyclopedia of Visual Medicine Series. An Atlas of the Human Embryo and Fetus, J. E. Jirasek (ed). The Parthenon Publishing Group, One Blue Hill Plaza, PO Box 1564, Pearl River, New York 10965, USA, 2001, 00-033639, ISBN 18570-659-X) ISBN 3-540-20138-6 Springer Berlin Heidelberg New York Bibliografische Information der Deutschen Bibliothek Die Deutsche Bibliothek verzeichnet diese Publikation in der Deutschen Nationalbibliografie; detaillierte bibliografische Daten sind im Internet çber <http://dnb.ddb.de> abrufbar. Dieses Werk ist urheberrechtlich geschçtzt. Die dadurch begrçndeten Rechte, insbesondere die der Ûbersetzung, des Nachdrucks, des Vortrags, der Entnahme von Abbildungen und Tabellen, der Funksendung, der Mikroverfilmung oder der Vervielfåltigung auf anderen Wegen und der Speicherung in Datenverarbeitungsanlagen, bleiben, auch bei nur auszugsweiser Verwertung, vorbehalten. Eine Vervielfåltigung dieses Werkes oder von Teilen dieses Werkes ist auch im Einzelfall nur in den Grenzen der gesetzlichen Bestimmungen des Urheberrechtsgesetzes der Bundesrepublik Deutschland vom 9. September 1965 in der jeweils geltenden Fassung zulåssig. Sie ist grundsåtzlich vergçtungspflichtig. Zuwiderhandlungen unterliegen den Strafbestimmungen des Urheberrechtsgesetzes. Springer ist ein Unternehmen von Springer Science+Business Media springer.de ° Springer-Verlag Berlin Heidelberg 2005 Printed in Germany Die Wiedergabe von Gebrauchsnamen, Handelsnamen, Warenbezeichnungen usw. in diesem Werk berechtigt auch ohne besondere Kennzeichnung nicht zu der Annahme, dass solche Namen im Sinne der Warenzeichenund Markenschutz-Gesetzgebung als frei zu betrachten wåren und daher von jedermann benutzt werden dçrften. Produkthaftung: Fçr Angaben çber Dosierungsanweisungen und Applikationsformen kann vom Verlag keine Gewåhr çbernommen werden. Derartige Angaben mçssen vom jeweiligen Anwender im Einzelfall anhand anderer Literaturstellen auf ihre Richtigkeit çberprçft werden. Herstellung: PRO EDIT GmbH, Elke Beul-Gæhringer, 69126 Heidelberg Umschlaggestaltung: design & production, 69121 Heidelberg Satz: K+V Fotosatz GmbH, 64743 Beerfelden-Airlenbach Gedruckt auf såurefreiem Papier 27/3150/gæh-5 4 3 2 1 0 Vorwort Die Neonatologie kann ebenso wie die Geriatrie als ein im Wesentlichen internistisches Fach definiert werden. Beide erhalten ihre fachspezifische Prågung durch die aus der besonderen Lebensphase sich ergebenden Besonderheiten in Øtiologie und Pathogenese von Erkrankungen. Dieser Logik folgend schlieût sich dem vorausgegangenen Band çber die molekularmedizinischen Grundlagen altersspezifischer Erkrankungen der vorliegende Band çber solche von fetalen und neonatalen Erkrankungen an. In wohl kaum eine andere medizinische Disziplin haben molekularmedizinische Methoden in hæherem Maûe Eingang gefunden als in die Prånatalmedizin und Neonatologie. Das betrifft die Reproduktionsmedizin ebenso wie gendiagnostische Verfahren zur Erkennung genetischer Erkrankungen und darauf basierende Pråventionen (z. B. Diåt bei der Phenylketonurie oder Substitutionen wie bei der Håmophilie) bzw. Behandlungsmethoden wie beispielsweise Gentherapie bei der Mukoviszidose. Neue Daten lassen die Annahme zu, dass zukçnftig die pluripotenten Stammzellen aus Nabelschnurblut oder aus adultem Knochenmark fçr den Zellersatz aller ausfallenden Organe verwendet werden kænnen. Sicher steht gegenwårtig bei der Stammzellforschung bei aller Zielgerichtetheit auf eine therapeutische Anwendung zunåchst die Grundlagenforschung an adulten Stammzellen im Vordergrund mit der Frage nach Gesetzmåûigkeiten der fetalen Entwicklung und deren Implikation in die Erkennung von molekularen und zellbiologischen Regulationsmechanismen. Die Geburt von Louise Brown im Jahre 1978 durch eine sterile Frau markiert den erfolgreichen Beginn der modernen Reproduktionsmedizin durch In-vitro-Befruchtung einer Eizelle durch eine våterliche Samenzelle (IVF). Dieses inzwischen als Methode der Wahl zur kçnstlichen Befruchtung entwickelte Standardverfahren wurde in den Folgejahren ergånzt durch die Etablierung einer weiteren Technik zur intrazytoplasmatischen Spermatozoeninjektion (ICSI) bei Våtern mit eingeschrånkter Spermienqualitåt. Beide Verfahren schlieûen jedoch bei familiårer Belastung durch monogenetische Erkrankungen pathologisch belastete Eizellen bzw. Spermatozoen nicht ein. Durch die Anwendung pråimplantationsdiagnostischer Verfahren ist heute der Ausschluss genetisch belasteter Embryonen mæglich geworden. Der erreichte Stand in der Reproduktionsmedizin ist bei allen Erfolgen sicher nur ein Anfang. Es bedarf noch groûer Anstrengungen, um einfachere, weniger belastende und risikoårmere Behandlungsmethoden zu entwickeln. In einigen Kapiteln des vorliegenden Bandes ist sowohl zu dieser Problematik ausfçhrlich Stellung genommen als auch zu damit im Zusammenhang stehenden bioethischen Fragen. Die beeindruckenden Entwicklungen der Pråimplantationsdiagnostik haben çber die engere Fachwelt hinaus groûe Resonanz in der breiten Úffentlichkeit gefunden und werden z. T. kontrovers diskutiert. Das gesamtgesellschaftliche Interesse an diesen komplexen Fragen reflektiert sich u. a. auch in der Bildung eines nationalen Ethikrates in Deutschland und in anderen Låndern, die sich mit diesen und anderen einschlågigen Fragen befassen. Dieses Interesse mag zum einen an den hierdurch mæglich gewordenen therapeutischen Aussichten und Hoffnungen liegen, VI Vorwort zum anderen jedoch an den hiermit aufgeworfenen Fragen nach einer mæglichen Verletzung ethischer Wertvorstellungen unseres Kulturkreises und mæglichem Missbrauch nicht zuletzt gerade in Deutschland unter dem Eindruck der Erfahrungen unserer jçngsten Geschichte. Die Anwendung der Pråimplantationsdiagnostik beispielsweise erfordert eine åuûerst verantwortungsvolle Indikationsstellung, deren prinzipielle Grenzen durch den Gesetzgeber u. a. im Embryonenschutzgesetz 1 festgelegt sind und vom nationalen Ethikrat2 sowie nationalen 3 und internationalen Organisationen 4 fachlich pråzisiert werden. Handlungsanweisungen per Gesetz oder Richtlinien aufzuzwingen ist aber keine dauerhafte Læsung, denn auf die brennenden Fragen, die durch die Anwendung der Gentechnologie am Menschen aufgeworfen werden, kann es keine endgçltigen Antworten geben 5. Unterschiedliche, sich wandelnde Wertmaûståbe und Bedçrfnisse fçhren zu unterschiedlichen moralischen Urteilen, die vor allem durch individuelle und æffentliche Auseinandersetzung mit den Chancen und Risiken der Biotechnologie und den Entwicklungen in der modernen Medizin erarbeitet werden kænnen 6. Jede Nation, jede Kultur und jede Religion hat andere Haltungen dazu 7. Nicht nur fçr Ørzte, sondern auch fçr Naturwissenschaftler ergibt sich daraus die Notwendigkeit des lebenslangen Lernens im Sinne der Erweiterung moralischer Kompetenz. Auch zu dieser Problematik wird in einigen Kapiteln des vorliegenden Bandes Stellung bezogen. In Deutschland starben vor ca. 100 Jahren von 1000 Geborenen ca. 200 vor ihrem ersten Geburtstag, vor 70 Jahren waren es ca. 70, vor 50 Jahren ca. 50, vor 30 Jahren 18±20. Gegenwårtig ist die Såuglingssterblichkeit auf unter 4,5½ gesunken. Rund 90% aller Todesursachen im Såuglingsalter sind auf perinatale Erkrankungen bzw. Komplikationen, Unreife und angeborene Fehlbildungen (> 70% der Todesfålle im 1. Lebensalter), den plætzlichen Kindstod (SIDS), auf Unfålle und Infektionen zurçckzufçhren. Obwohl diese Erfolge die Såuglingssterblichkeit als wesentliche gesundheitspolitische Zielgræûe zugunsten der Zielstellung ¹Reduktion der Frçh- und Langzeitmorbiditåtª immer stårker in den Hintergrund treten låsst, zeigen die aktuellen Ursachen der Såuglingssterblichkeit immer noch wesentliche Schwerpunkte der Prånatalmedizin und Neonatologie. Aus der jçngeren Vergangenheit und der Gegenwart sind zahlreiche Beispiele fçr die erfolgreiche Anwendung molekularmedizinischer Erkenntnisse und deren gezielte Umsetzung in die tågliche Routineanwendung bekannt. In der Prånatalmedizin haben vor allem Tripel- und AFP-Test sowie Ultraschallscreening zur frçhzeitigen Identifikation von fetalen Erkrankungen und Fehlbildungen (z. B. Aneuploidie) sowie zur Einleitung geeigneter Maûnahmen gefçhrt, die u. a. wesentlich zur Senkung der Såuglingssterblichkeit infolge Fehlbildungen beigetragen haben. Hier sei auf die Kapitel zu den håufigsten Organfehlbildungen von ZNS, Herz, Niere und Skelett, aber auch auf die nichthereditåren Fehlbildungen und fetalen Schådigungen verwiesen. Ein anderes hervorragendes Beispiel ist das heute weitgehend molekularmedizinisch begrçndete Management der multikausalen Hyperbilirubinåmie, bei der die 1 Gesetz zum Schutz von Embryonen (Embryonenschutzgesetz ± ESchG) 13. Dezember 1990 ± BGBl. I, S. 2747 Nationaler Ethikrat: Stellungnahme ¹Genetische Diagnostik vor und wåhrend der Schwangerschaftª www.ethikrat.org 3 Wissenschaftlicher Beirat der Bundesårztekammer http://www.aerztetag.de/30/Richtlinien/Empfidx/Schwanger.html 4 ESHRE Recommendations: http://humrep.oupjournals.org/cgi/content/full/16/4/790 5 J. Reich (2001) In Mattel J-F (co-ordinator) The human genome. Council of Europe Publishing 6 z. B. das Bioethik-Projekt www.1000fragen.de, G. Fabry: Bildung einer årztlichen Identitåt. Dt Ørztebl (2002) 99:2690±2693 7 Umgang mit dem vorgeburtlichen Leben in anderen Kulturen. Jahrestagung 2003, Nationaler Ethikrat, www.ethikrat.org 2 a Vorwort in der Vergangenheit mit dem Kernikterus bzw. mit dem Hydrops fetalis infolge fetaler Erythroblastose verbundene schlechte Prognose heute ihren Schrecken verloren hat. Die zahlreichen Facetten der Diagnose, Pråvention, der symptomatischen bzw. kausalen Therapie betreffen sowohl die Rh-Blutgruppen-Inkompatibilitåt (beherrscht durch prånatales Screening, Rh-Immunprophylaxe und postnatale Phototherapie), die metabolische Hyperbilirubinåmie infolge passagerem hepatischen Glukuronyltransferasemangel (behandelt mit Phototherapie), abnorme Håmolyse infolge Pathomorphologie (Sichelzellen) oder Enzymdefekt (G-6-PDH-Mangel) der Erythrozyten als auch die sehr seltenen hepatozellulåren Transportproteindefekte und posthepatischen Gallengangsatresien. Detaillierter wird auf die molekularmedizinisch begrçndete Diagnostik bzw. Therapie im Abschnitt ¹Molekulare Ursachen von Wachstums- und Entwicklungsstærungenª eingegangen. Der Nachweis einer Mutation ist nicht gleichbedeutend mit der klinischen Diagnose einer Krankheit. So entwickelten nur etwa 1% der homozygoten Merkmalstråger ¹Håmochromatoseª auch tatsåchlich die Erkrankung 8. Ursache einer anderen, wesentlich håufigeren monogenen Erkrankung, der Mukoviszidose, sind Mutationen des cftr-Gens, von dem çber 1000 verschiedene Mutationen beschrieben werden. Das Gen kodiert das CFTR-Protein, das den Cl±-Transport sekretorischer Epithelzellen reguliert. Fehlt das CFTR oder ist es defekt, werden Sekretion und Absorption von Cl± beeintråchtigt. Diese Kenntnisse werfen eine Reihe von Fragen im Hinblick auf die zukçnftige Gentherapie auf, aber auch im Hinblick darauf, vorsichtig mit der prådiktiven Diagnostik bei Kindern und Jugendlichen umzugehen. 9 In der Neonatologie hat der Kenntniszuwachs çber die reifeabhångigen molekularen Mechanismen zu einer Senkung der Sterblichkeit gefçhrt. Als Beispiel sei das Atemnotsyndrom der Frçhgeborenen genannt, dessen kausale Behandlung erst durch den kombinierten Einsatz 1.) der prånatalen Lungenreifeinduktion mit Glukokortikoiden und 2.) von Surfactant, also durch ein molekularmedizinisches Therapiekonzept, seine Schrecken verlor. Mit den Erkenntnissen aus der grundlagenorientierten und angewandten Surfactantforschung konnten spezielle Entitåten des Atemnotsyndroms wie die Alveolarproteinosen identifiziert, weitere neonatologische, pådiatrische und adulte Krankheiten mit Atemnot pathogenetisch orientiert behandelt (Mekoniumaspirationssyndrom, ARDS, Pneumonie, RSV-Bronchiolitis) und neue synthetische Surfactantpråparate entwickelt werden. Der Kenntniszuwachs çber die reifeabhångigen molekularen Mechanismen der spezifischen und unspezifischen Immunabwehr hat sicher auch zur erheblichen Senkung der Såuglingssterblichkeit beigetragen. Die Pråventions- und Therapiestrategien blieben aber bisher rein symptomatisch und bestehen in der Regel aus allgemeinen Hygienemaûnahmen, in einer kalkulierten bzw. erregerspezifischen Antibiotikaapplikation und einer evtl. erforderlichen Kreislauftherapie. Spezielle Behandlungsmethoden der Neugeborenensepsis, beispielsweise mit Antizytokinen oder in Form von Vakzinen gegen Staphylokokken bzw. von speziellen monoklonalen Antikærpern, existieren noch nicht. Neben dem engen Zusammenhang zwischen neurologischen Entwicklungsstærungen extrem unreifer Frçhgeborener und systemischen Infektionen besteht der zusåtzliche Problemkreis Pråvention bzw. Therapie von hypoxischen Hirnschåden. Obwohl inzwischen viele Mechanismen der Pathogenese dieser perinatalen Hirnschådigung geklårt sind, mçssen die auf dieser Basis entwickelten neuen Strategien zur Pråvention und Therapie erst ihre Effektivitåt in der Praxis nachweisen. Der vorliegende Band der Buchreihe ¹Molekulare Medizinª geht auf einige dieser Probleme detailliert ein. Die Themen spannen einen groûen Bogen von ethischen Problemen, die sich aus der Anwendung der prånatalen Diagnostik und der Pråim8 9 Beutler (2002) Lancet 359:211±218 http://www.bundesaerztekammer.de/30/Richtlinien/Richtidx/Praediktiv/ VII VIII Vorwort plantationsdiagnostik ergeben, bis hin zu Besonderheiten von molekularen Ursachen von Hauterkrankungen. In 6 Abschnitten mit insgesamt 23 Kapiteln wird der aktuelle Stand der molekularmedizinischen Erkenntnisse in der Prånatalmedizin und Pådiatrie dargelegt. Einfçhrend werden allgemeine Aspekte wie Mechanismen der Steuerung der Embryonalentwicklung, der Morbiditåt und Mortalitåt aus epidemiologischer Sicht und ethische Fragen in der prånatalen Diagnostik und Pråimplantationsdiagnostik bei der Anwendung heute mæglicher molekulardiagnostischer Techniken aufgezeigt. Ausfçhrlich werden die Ursachen von Organfehlbildungen hereditårer und nichthereditårer Ursachen, wie beispielsweise durch Umweltschadstoffe verursachte Schådigungen, sowie die molekularen Ursachen von Wachstumsund Entwicklungsstærungen erærtert. Breiter Raum wird der neonatalen Infektabwehr sowie der Erærterung der Ursachen von Herz-, Kreislauf- und Erkrankungen des Atmungssystems gewidmet. Schlieûlich werden in einem abschlieûenden Kapitel pharmakologische Interventionen unter Berçcksichtigung entwicklungsphysiologischer Besonderheiten dargelegt. Ein gesonderter Abschnitt, der als Zeittafel ausgewiesen ist, gibt einen Ûberblick çber wichtige Etappen in der Entwicklung der Pådiatrie. Dieser Band sei allen medizinisch interessierten Lesern empfohlen, welche die stçrmische Entwicklung der pådiatrischen Grundlagenforschung bis hin zur Entwicklung neuer gendiagnostischer Verfahren verfolgen mæchten. Die Herausgeber bedanken sich bei den Autoren, bei der Redaktion Biomedizin des Springer-Verlages und bei dem Hersteller fçr eine stets fruchtbare Zusammenarbeit auch bei diesem Band in der Buchreihe ¹Molekulare Medizinª. Berlin, im Herbst 2004 Die Herausgeber Inhaltsverzeichnis 1 Allgemeine Aspekte 1.1 Mechanismen der Steuerung der Embryonalentwicklung . . . . . . . . . . . . . . . 3 1.2 Morbiditåt und Mortalitåt in der Neugeborenen- und Såuglingsperiode ± Grundlagen fçr die molekulare Epidemiologie in der Neonatologie . . . . . . . . 49 1.3 Prånatale Diagnostik . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 81 1.4 Pråimplantationsdiagnostik . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 97 Bodo Christ, Beate Brand-Saberi Roland R. Wauer Wolfgang Holzgreve, Sevgi Tercanli, Sinuhe Hahn, Peter Miny Eberhard Schwinger, Michael Ludwig, Diana Tomi, Klaus Diedrich 1.5 Ethische Probleme bei der Pråimplantationsdiagnostik (PID) aus pådiatrischer Sicht . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 121 Volker von Loewenich 1.6 Ethische Probleme in der Neonatologie . . . . . . . . . . . . . . . . . . . . . . . . . . . . 129 Volker von Loewenich 2 Organfehlbildungen 2.1 Herzfehlbildungen . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 141 Anita Rauch, Michael Hofbeck 2.2 Molekulare Ursachen von Anomalien der Nieren und Harnwege bei Neugeborenen . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 183 Jærg Dætsch, Christian Plank, Wolfgang Rascher 2.3 Molekulare Genetik von Fehlbildungen und Wachstumsstærungen des Skeletts bei Neugeborenen . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 199 Stefan Mundlos X Inhaltsverzeichnis 3 Nichthereditåre Fehlbildungen und Schådigungen 3.1 Umweltbedingte vorgeburtliche Entwicklungsschåden . . . . . . . . . . . . . . . . . 231 3.2 Infektionsbedingte fetale Schådigungen . . . . . . . . . . . . . . . . . . . . . . . . . . . 265 3.3 Fetale Programmierung und funktionelle Teratologie . . . . . . . . . . . . . . . . . 325 Christof Schaefer Ioannis Mylonas, Klaus Friese Andreas Plagemann 4 Molekulare Grundlagen von Wachstumsund Entwicklungsstærungen 4.1 Molekulare Mechanismen von Fehlbildungen, Wachstums-, Differenzierungs- und Entwicklungsstærungen des Zentralnervensystems . . . 347 4.2 Molekulare Ursachen von Entwicklungsstærungen des Endokriniums . . . . . . . 365 4.3 Molekulare Ursachen der Stærungen der Håmoglobinsynthese beim Embryo, Fetus und Neugeborenen . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 393 4.4 Molekulare Ursachen von Stærungen des hepatogastroenterologischen Systems bei Neugeborenen . . . . . . . . . . . 409 Patrick Collombat, Ahmed Mansouri Annette Grçters Christof Dame, Jærg Bungert Lutz Bindl, Stephan Buderus, Sæhnke Dammann, Michael J. Lentze 5 Molekulare Grundlagen der neonatalen Infektabwehr 5.1 Stærung der Reifung und Differenzierung des Immunsystems als Krankheitsursache . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 455 5.2 Molekulare Mechanismen der neonatalen Abwehr von bakteriellen Infektionen . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 477 5.3 Molekulare Mechanismen der Abwehr konnataler und perinataler Virusinfektionen . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 507 Wilhelm Friedrich, Klaus Schwarz Reinhard Berner Stephan Ehl a Inhaltsverzeichnis 6 Neonatale Erkrankungen des Herz-Kreislaufund des Atmungssystems sowie der Haut 6.1 Molekulare Ursachen myokardialer Insuffizienz beim Neugeborenen . . . . . . . 533 Joachim Kreuder, Dietmar Schranz 6.2 Molekulare Grundlagen von Hypoxie und Asphyxie . . . . . . . . . . . . . . . . . . . 573 Johann Gross 6.3 Molekulare Ursachen von Hauterkrankungen bei Neugeborenen . . . . . . . . . . 607 Arne Kænig, Rudolf Happle 7 Neonatale Pharmakologie . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 643 Ingolf Cascorbi, Knud Linnemann Zeittafel . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 661 Sachverzeichnis . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 667 XI Autorenverzeichnis Priv.-Doz. Dr. med. Reinhard Berner Universitåt Freiburg Zentrum fçr Kinderheilkunde und Jugendmedizin Mathildenstraûe 1, 79106 Freiburg E-Mail: [email protected] Patrick Collombat Max-Planck-Institut fçr Biophysikalische Chemie Abteilung Molekulare Zellbiologie AG Molekulare Zelldifferenzierung Am Fassberg 11, 37077 Gættingen Dr. Lutz Bindl Klinikum der RWTH Aachen Pauwelsstraûe 30, 52074 Aachen Prof. Dr. med. Christof Dame Molekulare Neonatologie Klinik fçr Neonatologie, Campus Virchow-Klinikum Charit ± Universitåtsmedizin Berlin Augustenburger Platz 1, 13353 Berlin E-Mail: [email protected] Dr. Beate Brand-Saberi Albert-Ludwigs-Universitåt Freiburg Institut fçr Anatomie und Zellbiologie Lehrstuhl II Albertstraûe 17, 79104 Freiburg E-mail: [email protected] Dr. Stephan Buderus Kinderklinik St. Marien-Hospital Robert-Koch-Str. 1, 53115 Bonn Prof. Dr. rer. nat. Jærg Bungert Department of Biochemistry and Molecular Biology College of Medicine, University of Florida 1600 SW Archer Road, Gainesville, FL 32610, USA E-mail: [email protected] Prof. Dr. Dr. Ingolf Cascorbi Universitåtsklinikum Schleswig-Holstein Campus Kiel Institut fçr Pharmakologie Hospitalstraûe 4, 24105 Kiel E-Mail: [email protected] Prof. Dr. Bodo Christ Albert-Ludwigs-Universitåt Freiburg Institut fçr Anatomie und Zellbiologie Lehrstuhl II Albertstraûe 17, 79104 Freiburg E-Mail: [email protected] Dr. Sæhnke Dammann Abt. fçr Kinder- und Jugendmedizin Oberschwaben-Klinik Nikolausstraûe 10, 88212 Ravensburg Prof. Dr. Klaus Diedrich Universitåtsklinikum Schleswig-Holstein Campus Lçbeck Klinik fçr Frauenheilkunde und Geburtshilfe Ratzeburger Allee 160, 23538 Lçbeck E-Mail: [email protected] Priv.-Doz. Dr. Jærg Dætsch Friedrich-Alexander-Universitåt Universitåtsklinik fçr Kinder und Jugendliche Loschgestraûe 15, 91054 Erlangen E-Mail: [email protected] Priv.-Doz. Dr. Stephan Ehl Universitåt Freiburg Zentrum fçr Kinderheilkunde und Jugendmedizin Mathildenstraûe 1, 79106 Freiburg E-Mail: [email protected] XIV Autorenverzeichnis Prof. Dr. Wilhelm Friedrich Universitåtsklinik fçr Kinder- und Jugendmedizin Prittwitzstraûe 43, 89075 Ulm E-Mail: [email protected] Prof. Dr. med. Dr. h.c. mult. Wolfgang Holzgreve Universitåtsfrauenklinik Spitalstraûe 21, 4031 Basel, Schweiz E-Mail: [email protected] Prof. Dr. Klaus Friese Ludwig-Maximilians-Universitåt Klinik und Poliklinik fçr Frauenheilkunde und Geburtshilfe Maistraûe 11, 80337 Mçnchen Priv.-Doz. Dr. med. Arne Kænig Philipps-Universitåt Klinik fçr Dermatologie und Allergologie Deutschhausstraûe 9, 35033 Marburg E-Mail: [email protected] Prof. Dr. Johann Gross Humboldt-Universitåt zu Berlin Charit ± Universitåtsmedizin Berlin Hals-Nasen-Ohrenklinik Molekularbiologisches Forschungslabor Spandauer Damm 130, Haus 31 14050 Berlin E-Mail: [email protected] Prof. Dr. Joachim Kreuder Justus-Liebig-Universitåt Abteilung Kinderkardiologie Zentrum fçr Kinderheilkunde und Jugendmedizin Feulgenstraûe 12, 35392 Gieûen E-Mail: [email protected] Prof. Dr. Annette Grçters Charit ± Universitåtsmedizin Berlin Campus Virchow-Klinikum Humboldt-Universitåt zu Berlin Pådiatrische Endokrinologie, Allgemeine Poliklinik Otto-Heubner-Centrum fçr Kinder- und Jugendmedizin Augustenburger Platz 1, 13353 Berlin E-Mail: [email protected] Priv.-Doz. Dr. Sinuhe Hahn Universitåtsfrauenklinik Kantonsspital Basel Schanzenstraûe 46, 4031 Basel, Schweiz E-Mail: [email protected] Prof. Dr. Rudolf Happle Philipps-Universitåt Klinik fçr Dermatologie und Allergologie Deutschhausstraûe 9, 35033 Marburg E-Mail: [email protected] Prof. Dr. Michael Hofbeck Eberhard-Karls-Universitåt Tçbingen Abteilung Kinderheilkunde II Klinik fçr Kinderheilkunde und Jugendmedizin Hoppe-Seyler-Straûe 3, 72076 Tçbingen E-Mail: [email protected] Prof. Dr. Michael J. Lentze Zentrum fçr Kinderheilkunde des Universitåtsklinikums Bonn Allgemeine Kinderheilkunde und Poliklinik Adenauerallee 119, 53113 Bonn E-Mail: [email protected] Dr. Knud Linnemann Ernst-Moritz-Arndt-Universitåt Institut fçr Pharmakologie Abteilung Neonatologie der Klinik fçr Kinder- und Jugendmedizin Friedrich-Læffler-Straûe 23 d, 17487 Greifswald E-Mail: [email protected] Prof. Dr. med. Volker von Loewenich Kinderarzt, Neonatologe Bruno Stçrmer Straûe 27, 60529 Frankfurt E-Mail: [email protected] Priv.-Doz. Dr. med. Michael Ludwig Zentrum fçr Hormon- und Stoffwechselerkrankungen, Gynåkologie, Endokrinologie und Reproduktionsmedizin Lornsenstr. 6, 22767 Hamburg E-Mail: [email protected] Dr. Ahmed Mansouri Max-Planck-Institut fçr Biophysikalische Chemie Abteilung Molekulare Zellbiologie AG Molekulare Zelldifferenzierung Am Fassberg 11, 37077 Gættingen E-Mail: [email protected] a Prof. Dr. Peter Miny Universitåtskinderspital beider Basel (UKBB) Abteilung Medizinische Genetik Departement Klinisch-Biologische Wissenschaften Postfach, 4005 Basel, Schweiz E-Mail: [email protected] Prof. Dr. Stefan Mundlos Humboldt-Universitåt zu Berlin Charit ± Universitåtsmedizin Berlin Campus Virchow-Klinikum Institut fçr Medizinische Genetik Augustenburger Platz 1, 13353 Berlin E-Mail: [email protected] Dr. Ioannis Mylonas Ludwig-Maximilians-Universitåt Klinik und Poliklinik fçr Frauenheilkunde und Geburtshilfe Maistraûe 11, 80337 Mçnchen med.uni-muenchen.de E-Mail: [email protected] Prof. Dr. med Andreas Plagemann Charit ± Universitåtsmedizin Berlin Campus Virchow-Klinikum Klinik fçr Geburtsmedizin Leiter ¹Experimentelle Geburtsmedizinª Augustenburger Platz 1 13353 Berlin E-mail: [email protected] Dr. Christian Plank Friedrich-Alexander-Universitåt Klinik fçr Kinder und Jugendliche Loschgestraûe 15, 91054 Erlangen E-Mail: [email protected] Autorenverzeichnis Dr. med. Christof Schaefer Pharmakovigilanz- und Beratungszentrum fçr Embryonaltoxikologie Berliner Betrieb fçr Zentrale Gesundheitliche Aufgaben (BBGes) Spandauer Damm 130, Haus 10 14050 Berlin E-Mail: [email protected] Prof. Dr. Dietmar Schranz Justus-Liebig-Universitåt Abteilung Kinderkardiologie Zentrum fçr Kinderheilkunde und Jugendmedizin Feulgenstraûe 12, 35392 Gieûen E-Mail: [email protected] Dr. Klaus Schwarz Universitåt Ulm Institut fçr klinische Transfusionsmedizin und Immungenetik und Abteilung Transfusionsmedizin Helmholtzstraûe 10, 89081 Ulm E-Mail: [email protected] Prof. Dr. Eberhard Schwinger Universitåtsklinikum Schleswig-Holstein Campus Lçbeck Institut fçr Humangenetik Ratzeburger Allee 160 23538 Lçbeck E-Mail: [email protected] Priv.-Doz. Dr. Sevgi Tercanli Universitåtsfrauenklinik Spitalstraûe 21 4031 Basel, Schweiz E-Mail: [email protected] Prof. Dr. Wolfgang Rascher Friedrich-Alexander-Universitåt Klinik fçr Kinder und Jugendliche Loschgestraûe 15, 91054 Erlangen E-Mail: [email protected] Dr. Diana Tomi Universitåtsklinikum Schleswig-Holstein Campus Lçbeck Institut fçr Humangenetik Ratzeburger Allee 160 23538 Lçbeck Priv.-Doz. Dr. Anita Rauch Friedrich-Alexander-Universitåt Erlangen-Nçrnberg Institut fçr Humangenetik Schwabachanlage 10, 91054 Erlangen E-Mail: [email protected] Prof. Dr. Roland R. Wauer Universitåtsklinikum Charit Otto-Heubner-Centrum fçr Kinder- und Jugendmedizin Klinik fçr Neonatologie Schumannstraûe 20/21, 10117 Berlin E-Mail: [email protected] XV Abkçrzungen und Erlåuterungen Aa a-AT a-1,3-Integrin a-TIF Abort ACEInhibitoren actA ADAR Adipositas ADO Aminosåurezahl Alpha-Antitrypsin Adhåsionsmolekçl Alpha trans-inducing factor: wichtiges Tegumentprotein, das bei der HSV-Replikation eine bedeutende Rolle spielt (andere Bezeichnungen: VP16, VMW65, UL48) BRD Personenstandsgesetz v. 24.03.1994: Leibesfrucht, die keine der o. g. Lebenszeichen (s. Lebendgeborenes) und ein Geburtsgewicht von < 500 g aufweist. Falls das Geburtsgewicht nicht vorliegt, gelten 22 vollendete SW bzw. eine Kærperlånge von < 25 cm als gleichwertig wie 500 g Geburtsgewicht. BRD Personenstandsgesetz 157: Leibesfrucht mit einem Gewicht von unter 1000 g bei Fehlen aller fçr eine Lebendgeburt maûgeblichen Lebenszeichen. DDR: Anordnung çber die årztliche Leichenschau vom 4. Dez. 1978 (GBl I 1979 Nr. 1 S. 4 ; § 4 [2]). Keine menschliche Leiche ist eine Leibesfrucht mit einem Gewicht unter 1000 g, bei der nach vollståndigem Verlassen des Mutterleibes Herzund Atemtåtigkeit nicht oder nur eines der beiden Lebenszeichen vorhanden waren Inhibitoren des Angiotensinkonversionsenzyms (Bluthochdruckmedikamente) Verschlçsselt das ActA-Protein der Listerien, das fçr die actinabhångige intra- und interzellulare Motilitåt der Bakterien verantwortlich ist RNA-spezifische Adenosindeaminase Fettsucht Allel-Drop-out ADPKD Autosomal-dominante polyzystische Nierenerkrankung: durch Mutation im Polyzystin 1 oder 2 hervorgerufen; fçhrt in aller Regel im Erwachsenenalter zur chronischen Niereninsuffizienz ADS Attention deficit syndrome AER Apikale ektodermale Randleiste AFP Alpha-Fetoprotein: Protein, das normalerweise von der Leber und dem Dottersack des Fetus produziert wird. Wåhrend der Schwangerschaft ist der AFP-Wert erhæht. Sein Spiegel vermindert sich bald nach der Geburt. Im Kærper des Erwachsenen ist es nur noch in kleinsten Restmengen vorhanden und hat wahrscheinlich keine Funktion Agent Orange Trichlorphenoxyessigsåure: vor allem im Vietnamkrieg von den USA eingesetztes ¹Entlaubungsmittelª AHR Arylhydrocarbonrezeptoren AIDS Acquired immune deficiency syndrome: Erkrankung, hervorgerufen durch HIV, die mit einer Zerstærung der lymphatischen Organe einhergeht aire-Gen Autoimmunregulatorgen Allel Gleiche oder unterschiedliche Zustandsform eines Gens. In diploiden Organismen sind fçr jedes autosomale Gen ein maternales und ein paternales Allel vorhanden AMH Anti-Mçller-Hormon Amnion Extraembryonale Membran bei Vægeln, Reptilien und Såugern, die einen mit Flçssigkeit (Fruchtwasser) gefçllten Sack bildet, der den Embryo umschlieût Amniozentese Fruchtwasserentnahme ab der 13. bis 14. SSW. Mit einer Hohlnadel (Durchmesser < 1 mm) wird unter Ultraschallsicht durch die Bauch- XVIII Abkçrzungen und Erlåuterungen decke (transabdominal) die Fruchthæhle punktiert und Fruchtwasser aspiriert Aneuploidie Sprich An-eu-ploÒdie: ploÒd = -fach; di-ploÒd ist zweifach (wie der normale Chromosomensatz einer Kærperzelle); eu-ploÒd ist ¹gut-fachª, d. h. die Zahl der Chromosomen ist regelrecht; an-eu-ploÒd bedeutet das Gegenteil Angiogenese Ausreifung bereits pråformierter primitiver Gefåûstrukturen Anlage Kondensation aus Chondrozyten, die in Form und relativer Græûe dem zukçnftigen Knochen entspricht und wåhrend der Organogenese entsteht Anorexigen Die Nahrungsaufnahme hemmend ANR Anterior neural ridge (anteriorer neuraler Kamm) ANS Atemnotsyndrom, Surfactantmangelsyndrom, ¹respiratory distress syndromeª ANT Adeninnukleotid-Translokasen: sind an der inneren Membran der Mitochondrien lokalisiert und kontrollieren die Freisetzung von Zytochrom C wåhrend der Apoptose Anti-HBc Antikærper gegen HBcAg des HBV Anti-HBs Antikærper gegen HBsAg des HBV AP-1 Aktivatorprotein 1: wie NF-jB ein nukleårer Transkriptionsfaktor APAF-1 Apoptotic protease activating factor 1 APC Antigen pråsentierende Zelle: in der Regel Makrophagen oder dendritische Zellen, aber auch B-Zellen APECED Autoimmune polyendocrinopathy candidiasis ectodermal dystrophy Apoptose Genetisch programmierter Zelltod, der in der Ontogenese weit verbreitet ist. Ein Indikator fçr den ¹Selbstmordª der Zelle ist die Fragmentierung der DNA ARC AIDS-related complex: Spåtfolgen einer HIV-Infektion ARDS Adultes RDS, sekundåres ANS ARLI Autosomal-rezessive lamellåre Ichthyose ARND Alcohol-related neurodevelopment disorder ARNT Aryl hydrocarbon receptor nuclear translocator: identisch mit HIF-1b ARPKD Autosomal-rezessive polyzystische Nierenerkrankung: meist durch Mu- ART AS Asymmetrische Zellteilungen ATII AT-II-Antagonisten ATF A-terminal ATR-16Syndrom ATR-X Syndrom AUC Autosome AVE Axone AZT BAG tation im Gen pkhd1 (polycystic and hepatic disease 1) hervorgerufen, fçhrt håufig im Kindesalter bereits zur chronischen Niereninsuffizienz Assisted reproductive technologies Aminosåuren Zellteilungen, bei denen aufgrund unterschiedlicher Verteilung von Zellbestandteilen unterschiedliche Tochterzellen entstehen Alveolarepithelzellen vom Typ II Angiotensin-II-Antagonisten (Bluthochdruckmedikamente) Activating transcription factor Aminoterminales Ende eines Proteins, gekennzeichnet durch die freie NH2-Gruppe der endståndigen Aminosåure Symptomkombination aus a-Thalassåmie, mentaler Retardierung und assoziierten Entwicklungsstærungen, der ein Rearrangement auf dem Chromosom 16p13.3 zugrunde liegt, bei welchem neben den a-Globin-Genen die kodierende Sequenz fçr den Transkriptionsfaktor sox8 deletiert ist a-Thalassåmie, schwere mentale Retardierung, Genitalfehlbildungen sowie Fehlbildungen oder Funktionsstærungen anderer Organsysteme (Skelett, Niere, Herz, Gastrointestinaltrakt) bei einer Mutation des xh2- (atrx-)Gens auf dem X-Chromosom (Xq13.3) Area under the curve: Flåche unter der Kurve im Konzentration-ZeitDiagramm eines Medikamentes, entspricht mathematisch dem Integral und dient zur Bestimmung der durchschnittlichen Konzentration Alle Chromosomen auûer den Geschlechtschromosomen X (weiblich) und Y (månnlich) Anteriores viszerales Endoderm Aus den Nervenzellen auswachsende lange Fortsåtze, die mit anderen Nervenzellen, Muskel-, Haut- oder Drçsenzellen in Kontakt treten Azidothymidin: Medikament zur Therapie und Prophylaxe einer HIVInfektion Bradyzoitantigen: Oberflåchenantigen von Toxoplasma gondii a BAT-Werte Biologische Arbeitsstofftoleranzwerte BCG Bacille Calmette-Gurin BCL2 Familie von Proteinen, die an der Regulation des Zelltodes beteiligt sind. Erstmals in B-Zell-Lymphomen als Ergebnis einer chromosomalen Translokation nachgewiesen BDGF Brain-derived growth factor: spezifischer Wachstumsfaktor bdr Borrelia direct repeat: Gene von Borrelien, welche wahrscheinlich eine Rolle in der Pathogenese der Borreliose spielen BF-1 Brain factor-1 BfR Bundesinstitut fçr Risikoabschåtzung bhlh Basische Helix-Loop-Helix-Gene: Transkriptionsfaktoren, die sehr frçh in der Neurogenese und in der Myogenese eine Rolle spielen. Dazu gehæren Neurogenin 1, 2 u. 3 (Nervensystem u. endokrines Pankreas), myf5 u. myod (Skelettmuskel) bHLH-PAS Basic helix-loop-helix, Helix-Schleife-Helix-Struktur von DNA: PAS leitet sich von Period, ARNT und SIM (¹single-mindedª) ab. Diese Strukturen sind charakteristisch fçr Gene, deren Produkte u. a. an der Differenzierung und ontogenetischen Entwicklung beteiligt sind BHS Blut-Hirn-Schranke: durch nicht fenestrierte Endothelien gebildete Grenzschicht zwischen meningealen Blutgefåûen und Liquorraum BID BH3-interacting domain death agonist Biokyberneto- Entwicklung und Vorprogrammiegenese rung kybernetischer Regelsysteme des Organismus; çberwiegend pråund frçhpostnatal Biopsie Entnahme einer lebenden Gewebeprobe zu diagnostischen Zwecken BLAST Basic local alignment search tool: spezieller Algorithmus, um Gene und Ûbereinstimmung (Genvergleich) in unterschiedlichen Datenbanken zu finden Blastomere Durch Teilung der Zygote (s. dort) und weiterer Zellteilungen entstehender kugeliger Zellhaufen (daher frçher auch Morula = Maulbeere) Abkçrzungen und Erlåuterungen Blastozyste Blasenfærmiges Stadium des frçhen Embryos, frçher auch als Gastrula bezeichnet (wærtlich: Båuchlein). In dieser Blastozyste entwickelt sich die sog. innere Zellmasse, in der dann die Embryonalplatte (s. dort, auch Primitivstreifen genannt) ausgebildet wird Blutgruppen- Globosid: Rezeptor fçr den ParvoP-Antigen virus B19, besteht aus den P1-, Pund Pj-Antigenen. Es existieren vier Phånotypen: die P1- und P2-Phånotypen sind infektionsgefåhrdet; die P1j- und P-Phånotypen, die nicht das P-Antigen auf den Erythrozyten besitzen, sind dagegen gegen eine Infektion resistent Blutstamm- Bei der BSZT werden håmatozelltransplan- poetische Vorlåuferzellen eines getation sunden Spenders çbertragen, deren Ansiedlung im Empfånger zu einer Rekonstitution des Blut bildenden Systems sowie des Immunsystems fçhrt. Das Verfahren ist fçr die Behandlung angeborener und erworbener Stærungen dieser Systeme geeignet B-Lympho- B-Zellen synthetisieren und pråsenzyten tieren Antikærper auf der Zelloberflåche. Nach Antigenerkennung differenzieren B-Zellen zu Plasmazellen, welche diese Antikærper sezernieren BMP Bone morphogenetic protein: wåhrend der Embryonalentwicklung aktive Signalmolekçle (Proteine) der TGF-b-Superfamilie bp Basenpaare BPD Bronchopulmonale Dysplasie, chronische Atemstærungen BPI Bactericidal/permeability-increasing protein: kationisches, etwa 50 kD groûes Protein, das zu den antimikrobiellen Defensinen gehært BrachyVerkçrzung der Phalangen und/oder daktylie der Metacarpalia/Metatarsalia Bradyzoiten Asexuelle, langsam wachsende Vermehrungsform von Toxoplasma (T.) gondii BRIC Benigne rekurrierende intrahepatische Cholestase bsnd Untereinheit des Chloridkanals: kodiert fçr Barttin, Mutation ver- XIX XX Abkçrzungen und Erlåuterungen BSR BSZT C Cadherine CAR CAT1, CAT2 CATCH22Syndrom cbfa1 CBMNC CBP cccDNA CCR/CXCR CD14 CDGSyndrome cdk5 ursacht das Bartter-Syndrom mit Taubheit (Typ 4) Bradyzoite-specific recombinant: Oberflåchenantigen von Toxoplasma gondii Blutstammzelltransplantation (s. dort) Komplementfaktoren: Das Komplementsystem besteht aus mehr als 20 Plasmaproteinen, den Komplementfaktoren, die in einer komplexen Reaktionskaskade interagieren. Die Aktivierung des Komplementsystems ist nicht zuletzt fçr die rasche Elimination insbesondere von bekapselten bakteriellen Erregern von Bedeutung Kalziumabhångige Zelladhåsionsmolekçle (z. B. N-Cadherin, B-Cadherin, E-Cadherin) Konstitutiver Androstanrezeptor: zytosolischer Phenobarbitalrezeptor, der die Induktion von z. B. CYP3A4 oder UGT1A1 moduliert Transmembranåre Kationentransporter 1 u. 2 DiGeorge-Syndrom cdk5r1 Gen fçr core binding factor alpha1: Transkriptionsfaktor Cord blood mononuclear cells: mononukleåre Zellen aus Nabelschnurblut CREB binding protein Covalently closed-circular DNA: kreisfærmiges DNA-Molekçl, das in der Replikation des HBV-Virus vorkommt Chemokinrezeptoren: CC-Chemokine besitzen 2 benachbarte Zystinreste, bei den CXC-Chemokinen sind die beiden Zystine durch eine Aminosåure getrennt Glykoprotein, das von Monozyten/ Makrophagen exprimiert wird und als Rezeptor fçr Endotoxin/LPS fungiert Gruppe von hereditåren Erkrankungen, die durch Stærungen der Glykosilierung hervorgerufen werden (¹carbohydrate deficient glycoproteinsª) Gen der cyclin-dependent kinase 5 CHILD CDPX2 CDR c-fos, c-jun CFTR CFU-GM CGH Chaperon Chemotaxis Chondrozyten Chorda dorsalis Chorionbiopsie Chorionzotten Chromatin Gen der cyclin-dependent kinase 5, regulatory subunit 1 X-chromosomal-dominante Chondrodysplasia punctata (ConradiHçnermann-Happle-Syndrom) Complementarity determining regions: hypervariable Regionen am Aminoterminus der Immunglobulinketten, welche die spezifische Antigenerkennungsstelle im Wesentlichen bestimmen Transkriptionsfaktoren Cystic fibrosis transmembrane conductance regulator (MRP7): cAMPabhångiger Chloridkanal. Hereditåre Defekte fçhren zum Krankheitsbild der zystischen Fibrose Colony-forming units-granulocyte/ monocyte: pluripotente myeloische Progenitorzellen, die sich zu den verschiedenen håmatologischen Zelllinien differenzieren kænnen Comparative genome hybridization Proteinkomplexe, die zunåchst ungefaltete, spåter teilweise gefaltete Polypeptidketten enthalten Gerichtete Wanderung von Leukozyten zum Ort der Infektion Congenital hemidysplasia with ichthyosiform nevus and limb defects Zellen des Knorpels Primitivstruktur der embryonalen Wirbelsåule Entnahme von Chorionzotten der Plazenta (Mutterkuchen) ab der 10. SSW. Mit einer Hohlnadel (Durchmesser < 1 mm) wird unter Ultraschallsicht heute meist durch die Bauchdecke (transabdominal) die Plazenta punktiert und etwa 20 mg Gewebe aspiriert. Manchmal wird das Verfahren als CVS (¹chorionic villus samplingª) bezeichnet Ausstçlpungen der åuûeren Eihaut, aus denen spåter die Plazenta wird und die bereits deren Funktion wahrnehmen Besteht im Wesentlichen aus dem DNA-Strang, einer Gruppe basischer Proteine (Histone) und einer groûen Zahl von Nicht-Histon-ChromatinProteinen. Die Struktur des Chromatins wird in erster Linie durch a Wechselwirkungen zwischen DNA und Histonen bestimmt CIPO Chronische intestinale Pseudoobstruktion Cis-aktiv Regulator-DNA-Sequenz, die Gene beeinflusst, die sich auf demselben Chromosom befinden. So ist z. B. die HRE-Sequenz cis-aktiv CLCNKA Chloridkanal vom Typ A CLCNKB Chloridkanal vom Typ B: Mutation verursacht das Bartter-Syndrom Typ 3 c-met Protoonkogen: Tyrosinkinaserezeptor, Rezeptor fçr ¹scatter factorª (SF) cMOAT Canalicular multisystemic organic anion transporter: Transportprotein, dessen Defekt zum Dubin-JohnsonSyndrom fçhrt CLP C-type lectin-like proteins: ATPasen, welche in die Regulation und Degradierung von Peptiden involviert sind CMV Zytomegalievirus col1a1 Gen fçr Kollagen Typ 1 a1-Kette col2a2 Gen fçr Kollagen Typ 2 a2-Kette Connexine Membranproteine, aggregieren zu hexamerischen Hemikanålen (Connexone); diese bilden mit Connexonen der Nachbarzellen gap junctions, welche den Durchtritt von Ionen und kleinen Molekçlen erlauben Cordozentese Entnahme von fetalem Blut aus der Nabelschnur ab der 18. bis 20. SSW. Mit einer Hohlnadel (Durchmesser < 1 mm) wird unter Ultraschallsicht durch die Bauchdecke (transabdominal) die Nabelschnur punktiert und 12 ml fetales Blut aspiriert COX-1 Zyklooxygenase 1, katalysiert die Bildung von Prostaglandinen, Prostazyklinen und Thromboxan aus Arachidonsåuren COX-2 Zyklooxygenase 2, bei Knock-out im Mausmodell entwickelt sich eine Nierenhypoplasie CPAP Continuous positive airway pressure, d. h. es wird kontinuierlich ein positiver Druck in den Atemwegen mit speziellen Nasen- oder Gesichtsmasken aufgebaut CpG-Inseln Genomabschnitte mit Anreicherung der Dinukleotidsequenz mit den Ba- Abkçrzungen und Erlåuterungen sen Guanin/Cytosin, vornehmlich in Promotorbereichen gelegen CR3 Komplementrezeptor 3: entsprechend dem a2-Integrin CD11b/CD18 oder MAC-1, auf Makrophagen, dendritischen Zellen, NK- und T-Zellen lokalisiert CR4 Komplementrezeptor 4: entsprechend dem Molekçl CD11c/CD18 oder p150,95 CRABP Zellulåres Retinsåure bindendes Protein CRBP Zellulåres Retinol bindendes Protein Cre Causes recombination CRE cAMP response elements: Gene und einzelne Abschnitte des HSV-Genoms, welche durch cAMP-phosphorylierte Proteine aktiviert werden CREB CRE-binding protein CRH Corticotropin releasing hormone CriglerSchwere Form der HyperbilirubinåNajjarmie auf der Basis einer ugt1a1Syndrom Defizienz Crossing-over s. unter Reifeteilung CRS Congenital rubella syndrome (Rætelnembryopathie): fetale Schådigung, wie z. B. Herzfehlbildungen, Augenanomalien und Innenohrschwerhærigkeit, die durch das Rætelnvirus verursacht werden CST1 Zellwandglykoprotein von Toxoplasma-gondii-Zysten CT Computertomographie CTL CD8+-zytotoxische T-Zelle CumarinPrånatale Entwicklungsstærung an Syndrom Nase, Epiphysen der Ræhrenknochen, Augen und Ohren mit IUGR und ggf. mentaler Entwicklungsretardierung CVS Kongenitales Varizellensyndrom: fetale Schådigungen, wie Hauterscheinungen, ZNS-Schåden und Augenanomalien, die durch das VZV verursacht werden CYP Zytochrom-P450-Enzyme: wichtige Phase-I-Enzym-Familie der Leber und weiterer Organe, die endogene Stoffe wie Steroide, Lipide, aber auch Arzneimittel und Fremdstoffe metabolisiert CYP1A2 Zytochrom P450 1A2 CYP2C9 Zytochrom P450 2C9 (Tolbutamid-Hydroxylase) XXI XXII Abkçrzungen und Erlåuterungen CYP2C19 Zytochrom P450 2C19 (Mephenytoin-Hydroxylase) CYP2D6 Zytochrom P450 2D6 (Debrisoquin-Hydroxylase) CYP3A4 Zytochrom P450 3A4: wichtigstes Arzneimittel metabolisierendes P450-Enzym. Es unterliegt vielen Interaktionen CYP3A7 Enzym, das ausschlieûlich von Feten exprimiert wird D1R, D2R Dopaminrezeptoren: D1R wirkt aktivierend, D2R inhibitorisch auf die Synthese von cAMP dab1 Disabled homolog 1 (Drosophila) dax11 DSS-AHC critical region on the X-chromosome 1, gene 1 DC Dendritische Zelle DC-SIGN Dendritic-cell-specific ICAM3-grabbing non-integrin: vermittelt Rollen und transendotheliale Migration von DC DCX Doublecortin Defensine Kationische Peptide von etwa 30±33 Aminosåuren, die eine antibiotikaåhnliche Wirkung aufgrund ihrer Poren bildenden Eigenschaften ausçben Deepithelia- Ausgliederung von Zellen aus einem lisierung epithelialen Verband verbunden mit dem Verlust funktioneller Zell-ZellKontakte und der Polarisierung Deformation Durch mechanische Kraft auf den Embryo verursachte Verånderung Deformitåten Lagebedingte Form- oder Strukturanomalien nach normaler Embryogenese Delta Transmembranprotein, das an Notch bindet. Delta-Notch-Interaktionen laufen an benachbarten Zellen ab und sind an zahlreichen Grenzziehungen wåhrend der Embryonalentwicklung beteiligt Dermis Bindegewebiger Bestandteil der Haut DermoDorsales Somitenkompartiment, myotom das Skelettmuskulatur, Dermis und Endothelzellen bildet DesignerUtopie eines Kindes mit nach Baby Wunsch zusammengestelltem Erbgut Desmale Knochenbildung und -wachstum Ossifikation durch direkte Differenzierung von Vorlåuferzellen in Osteoblasten Deszendenz, maternal Determination Nachkommenschaft auf der mçtterlichen Familienseite Festlegung der Zellen auf ein bestimmtes Entwicklungsschicksal hin dfna7 Das Gen wurde mit einem autosomal-dominant vererbbaren, progressiven Hærverlust assoziiert. Eventuelle Rolle in der Pathogenese des CMV (Hærverlust) Diabetes, Diabetes mellitus mit absolutem Typ 1 Insulinmangel infolge immunologischer oder idiopathischer Zerstærung der pankreatischen B-Zellen Diabetes, Diabetes mellitus mit relativem Typ 2 Insulinmangel infolge eines sekretorischen Defizits und/oder einer Insulinresistenz DifferenAusprågung zelltypspezifischer zierung Merkmale diHETE Dihydroxyeicosatriensåure: durch Lipoxygenasen oxidierte Derivate der Arachidonsåure Disposition Veranlagung im Sinne von Krankheitsveranlagung, Empfånglichkeit fçr bestimmte Erkrankungen Disruptionen Sekundår nach normaler Embryogenese aufgrund Durchblutungsstærungen oder mechanischer Barrieren verkçmmerte Organbereiche DMS Diffuse mesangiale sklerosetypische histologische Verånderungen beim kongenitalen nephrotischen Syndrom des nichtfinnischen Typs. Øhnliche histologische Verånderungen finden sich in der Niere beim Denys-Drash-Syndrom 1 DNA Desoxyribonukleinsåure (englisch und international, deutsch auch DNS): sehr groûes Molekçl, das als Tråger der Erbinformation dient DNAKovalente Methylierung von Cytosin Methylierung zu 5-Methylcytosin, çberwiegend in der Basensequenz CpG DNMT DNA-Methyltransferase dNTP Desoxynucleosidtriphosphat: Einzelbaustein der DNA Domåne Teilstçck einer DNA, der man bestimmte Funktionen zuordnen kann DominantEin Allel ist mutiert, das andere negativ normal. Ûberwiegt die Wirkung des mutierten Allels, ist die Funktion a des normalen Gens negativ beeinflusst DominantGenmutation, die zur Produktion negative eines aberranten Genproduktes Mutation fçhrt, welches die Funktion des Wildtyp-Genproduktes stært DOP Degenerate oligonucleotide primed dra Down regulated in adenoma: menschliches Gen, das den zur Familie der ¹solute carrierª gehærenden Chloridtransporter kodiert dsDNA Doppelstrångige DNA dtdst Gen des diastrophen Dysplasiesulfattransporters DuchenneFortschreitende Degeneration der MuskelMuskulatur, immer tædlich durch dystrophie spåt im Verlauf auftretenden Ausfall der Atemmuskulatur. Geschlechtsgebundene Vererbung wie bei Håmophilie (s. dort) Dyslipidåmie Gestærte Blutfettwerte Dysostose Auf ein bestimmtes Skelettelement begrenzte Verånderung Dysplasie Alle Zellen eines bestimmten Gewebes betreffend E Early, (auch b-)Genexpression von Herpesviren Embryonaler Tag (z. B. E8,5) oder Exon (z. B. E1) E1, E2 Die E1- und E2-Strukturproteine des Rætelnvirus sind fçr die Induktion der humoralen håmagglutinierenden, håmolysierenden und neutralisierenden Aktivitåt verantwortlich ebp Emopamil binding protein: Gen, welches eine D(8)-D(7)-Sterolisomerase kodiert und bei CDPX2 mutiert ist EBS Epidermolysis bullosa simplex EbsteinFehlanlage der TrikuspidalklappenAnomalie segel: håufig kombiniert mit Vorhofseptumdefekt oder offenem Foramen ovale EBV Epstein-Barr-Virus: Virus der Herpesgruppe ECM Extrazellulåre Matrix: Material im Zwischenzellraum (Intrazellularraum), das die ungeformte Grundsubstanz (Proteoglykane, Glykoproteine) und Fasern (Kollagen) umfasst Ecogenetik Kombination und Interaktion exogener und endogener Faktoren Abkçrzungen und Erlåuterungen E. coli EDNRB EET Escherichia coli Rezeptor fçr Endothelin B Epoxyeicosatriensåure: durch Lipoxygenasen oxidierte Derivate der Arachidonsåure EGF Epidermaler Wachstumsfaktor: Nullmutationen bei der Maus haben eine zystische Sammelrohrformation zur Folge EGFR Rezeptor des epidermalen Wachstumsfaktors EHK Epidermolytische Hyperkeratose Typ Brocq EIA Enzymimmunoassays EKLF Erythroid Krçppel-like factor (s. dort) Ektoderm Øuûeres Keimblatt, das den epithelialen Teil der Haut (Epidermis) sowie das zentrale und periphere Nervensystem bildet Ektrodaktylie Spalthand/-fuû ELBW Extremely low birth weight, extrem untergewichtiges Lebendgeborenes, Geburtsgewicht < 1000 g ELISA Enzyme-linked immunosorbent assay Embryo Frçhestes Stadium eines Organismus, Stadium der Anlage der Organe Embryonal- Streifenfærmiges Gebilde im Zentplatte rum der inneren Zellmasse der Blastozyste (s. dort), frçheste Ausbildung des von seinen åuûeren Hilfsgeweben abgrenzbaren Menschen Embryonale Diese Zellen werden aus der inneStammzellen ren Zellmasse eines Såugerembryos gewonnen und kænnen unbegrenzt in Kultur gehalten werden EMSA Electrophoretic mobility shift assay: Nachweisverfahren fçr Protein-DNAWechselwirkungen. Protein-DNAKomplexe laufen bei elektrophoretischer Auftrennung langsamer als freie DNA EMT Epitheliomesenchymale Transition: das Entstehen eines mesenchymalen Zellverbandes aus einem Epithel emx2 Empty spiracles homolog 2 (Drosophila): Gen auf Locus 10q26.1 en1 Engrailed homolog 1: Gen auf Locus 2q13q21 Enchondrale Knochenwachstum durch UmwandOssifikation lung von Knorpel in Knochen XXIII XXIV Abkçrzungen und Erlåuterungen Endothelin Endothelial und endokardial sezerniertes Peptid, das neben seiner direkten vasopressorischen Wirkung an der Morphogenese des Herzens und der groûen herznahen Gefåûe beteiligt ist, vermittelt durch divergente ETA- und ETB-Rezeptoren ENI und ENII Enhancer elements I and II: regulieren die vier Promotoren (¹core proteinª, ¹large surfaceª S1, ¹major surfaceª S2 und X-Gen-Promoter) des HBV ENTIS European Network of Teratology Information Services Entoderm Inneres Keimblatt, das den MagenDarm-Trakt auskleidet und die Drçsen bildet, die sich vom Darm aus entwickeln EntwickGen, das die Aktivitåt anderer Gene lungskonwåhrend der Entwicklung kontroltrollgen liert, auch Meistergen genannt env Gene auf dem HIV-Genom, welche Proteine fçr die HIV-Hçlle (¹envelopeª) kodieren EPAS1 Endothelial PAS domain protein 1: identisch mit HIF-2a Epiblast Øuûere Zellschicht der zweiblåttrigen Keimscheibe, aus der spåter die drei definitiven Keimblåtter (Ektoderm, Entoderm und Mesoderm) hervorgehen Epigenese Schrittweise erfolgende Entwicklung, durch die eine zunehmende Komplexitåt des Organismus erreicht wird. Sie vollzieht sich durch Signalaustausch zwischen verschiedenen Strukturelementen. Gegensatz: Pråformation Epigenetisch In umweltabhångiger Weise zusåtzlich zu genetischen Faktoren, aber nicht unabhångig von diesen; gebråuchlich im Sinne erworbener Disposition Epikanthus Sichelartige Falte am inneren Rand des oberen Lides Ephrine Sie stellen mit ihren Rezeptoren, den Eph, ein wichtiges Signalsystem dar, das Informationen auf benachbarte Zellen çbertrågt EPO Erythropoetin ER Endoplasmatisches Retikulum Estrogen-Rezeptoren ERK Extrazellulår regulierte Kinase Erythroid Der Transkriptionsfaktor EKLF wird Krçppel-like ausschlieûlich in erythroiden Zellen factor der ¹primitivenª und ¹definitivenª Håmatopoese exprimiert und bindet an das DNA-Motiv CACC Erythropoese Blutbildungsprozess von einer pluripotenten Stammzelle bis zum Erythrozyten. In diesem Prozess sind spezielle Aspekte der zellulåren Proliferation und Differenzierung relevant ErythroEin neuer Parvovirus, der sich virus V9 > 11% vom B19 unterscheidet, aber åhnliche Krankheitsbilder wie dieser verursacht ES Embryonale Stammzellen (s. dort) ESHRE European Society of Human Reproduction and Embryology ESchG Embryonenschutzgesetz EST Expressed sequence tags: rapide und effiziente Methode, die auf dem Zusammenfçhren von Daten und der Erstellung einer Datenbank basiert. Ermæglicht die Charakterisierung von Genen, welche von Geweben oder Mikroorganismen exprimiert werden EU Europåische Union EUROCAT European Registry of Congenital Anomalies and Twins Exon DNA-Abschnitt eines Gens, der einen Teil der Proteinstruktur kodiert Expansion Verlångerung eines Repeats Expressivitåt Art der phånotypischen Auswirkung eines genetischen Merkmals FAA Fumarylacetoacetat FanconiFamiliårer Schwund des Blut bildenAnåmie den Knochenmarkes (Blutungsneigung, Blutarmut, Abwehrschwåche), kombiniert mit weiteren Fehlbildungen FAS Fetales Alkoholsyndrom FAS-L FAS-Ligand: Zytokin, das an der Zellapoptose beteiligt ist. Ligand fçr den FAS-Rezeptor FcRn IgG-Transporter-Protein, das in der Plazenta exprimiert wird Fehlgeburt s. Abort Fetus Organismus vor der Geburt nach Abschluss der Organanlage, im Stadium der Ausdifferenzierung der Organe Fcc-Rezeptor Rezeptor fçr Immunglobulin G FGF Fibroblasten growth factor: Gruppe spezifischer Wachstumsfaktoren. Sie a stellt eine wichtige Familie von Signalproteinen dar und ist an zahlreichen Entwicklungsprozessen beteiligt fgf8 Gen des Fibroblastenwachstumsfaktors 8 FGFR Rezeptoren fçr Fibroblastenwachstumsfaktoren fgfr1 Gen des Fibroblastenwachstumsfaktor-Rezeptors 1 Fibronektin Ein Glykoprotein der extrazellulåren Matrix, das fçr Zell-Matrix-Interaktionen sowie fçr die Migration von Zellen wichtig ist FIDD Frequency of Inherited Disorders Database FISH Fluoreszenz-in-situ-Hybridisierung: Mit einem Fluoreszenzfarbstoff markierte DNA-Sonden werden unter geeigneten Bedingungen auf Chromosomenpråparate oder Interphasezellen aufgebracht. Diese lagern sich ausschlieûlich an ihre durch die Sondensequenz definierte Referenzregion an (Hybridisierung). Methode zum Nachweis spezifischer Chromosomenregionen FISH-Analyse Fluoreszenz-in-situ-Hybridisierungsanalyse: In der Regel ist damit die Hybridisierung einer fluoreszenzmarkierten DNA-Sonde auf Chromosomen gemeint, mit der z. B. zytogenetisch nicht sichtbare Aneusomien nachgewiesen werden kænnen FloppyLethargie, Trinkschwåche, TachyInfantpnoe, Tachykardie, Zyanose, TemSyndrom peraturregulationsstærung, Muskelhypertonie FOG Friend of GATA (FOG-1 und FOG-2) sind komplexe Zinkfingerproteine, die mit der aminoterminalen Kette der GATA-Transkriptionsfaktoren physikalisch interagieren und deren Funktion steuern. Diese Kofaktoren sind sowohl fçr die erythrozytåre als auch fçr die megakaryozytåre Entwicklung essenziell. Zudem sind sie bedeutsam fçr die Valvulogenese des Herzens und Entwicklung des herzeigenen Gefåûsystems foxp3 Forkhead-Box-P3-Gen foxn1 Forkhead-Box-N1-Gen Abkçrzungen und Erlåuterungen Frameshift Mutation, die zu einer Ønderung des Leserasters der DNA und zu einer verånderten Aminosåuresequenz des Proteins fçhrt. Der Startpunkt der Translation wird um eine oder zwei Basen nach links oder rechts verschoben Frçhgebore- Wucherung heranwachsender Netznenretinohautgefåûe sehr unreifer Frçhgebopathie rener. Kann zu Erblindung fçhren FrçhgeNeugeborenes mit einem Gestatiborenes onsalter < 37 vollendete Wochen (< 259 Tage) FSH Follikel stimulierendes Hormon FTA-ABS Fluoreszenz-Treponema-pallidumAntikærper-Absorptionstest FTA-ABSFluoreszenz-Treponema-pallidumIgM Antikærper-Absorptionstest mit isolierter IgM-Fraktion F-Zellen Erythroide Zellen, die a2c2 Håmoglobin-Tetramere enthalten, welche durch einen Ûberschuss an a-Globinen bei verminderter b-Globin-Synthese entstehen G6PDH Glukose-6-Phosphat-Dehydrogenase gag Gene auf dem HIV-Genom, die gruppenspezifische Antigene kodieren GAG Glykosaminoglykane, die an Proteine gebunden sind und mit diesen Proteoglykane bilden. Sie stellen einen Bestandteil der extrazellulåren Matrix dar GAL Galanin: die Nahrungsaufnahme stimulierendes Peptidhormon (29 Aminosåuren) GALE UDP-Galaktose 4'-Epimerase GALK Galaktokinase GALT Galaktose-1-Phosphat-Uridyltransferase Gameten Keimzellen: Sammelbegriff sowohl fçr Eizellen als auch fçr Spermien (¹Samenfådenª) A c, GcDiese beiden c-Globine (Ac, Gc) Globin-Gene unterscheiden sich in ihrer Primårstruktur durch die Aminosåure in Position 136, bei der es sich entweder um Alanin (Ac) oder Glyzin (Gc) handelt. Das quantitative Verhåltnis von Gc zu Ac ist wåhrend der Gestation 75 : 25 und kehrt sich wåhrend der ersten zehn Monate postpartal in ein Verhåltnis von 40 : 60 um XXV XXVI Abkçrzungen und Erlåuterungen GAP GATA gata4 GBE GBS-F gC1-qR gcm2 G-CSF GDF-5 GDNF Geburtsgewicht Guanosintriphosphatase aktivierende Proteine Zinkfinger-Transkriptionsfaktor: Gruppe von 6 Zinkfingerproteinen, die an die (T/A)GATAG/A Konsensussequenz binden und eine bedeutende Rolle bei der Differenzierung und Proliferation von Zellen spielen. Basierend auf Sequenzhomologien und den Expressionsmustern wurden die GATA-Transkriptionsfaktoren ursprçnglich in zwei Untergruppen eingeteilt. GATA-1, -2 und -3 wurden dabei als eine Gruppe von Faktoren zusammengefasst, die primår fçr die Regulation der Håmatopoese relevant ist. Måuse mit einer Knock-out-Mutation im gataGen zeigen eine starke Anåmie, da die Differenzierung der roten Blutzellen gestært ist Gen der gata-Familie, das wesentlich an der frçhen kardialen Morphogenese und der Myokardhypertrophie beteiligt ist Glycogen debranching enzyme Læslicher Faktor von Gruppe-BStreptokokken mit einer mutmaûlich sehr ausgeprågten immunstimulatorischen Potenz Stellt das gC1q vom C1q der ersten Komponente der Komplementkaskade dar. Beteiligt an der Bindung mit InIB von L. monocytogenes Glial cells missing homolog 2: Gen Granulozytenkolonie stimulierender Faktor: håmatopoetischer Wachstumsfaktor. Polypeptid, das die Proliferation und Differenzierung håmatopoetischer Stammzellen in Richtung neutrophiler Granulozyten stimuliert Growth and differentiation factor 5 Glia cell line-derived neurotrophic factor: Fçr die von oral nach aboral erfolgende embryonale Einwanderung des Plexus myentericus in den Darmtrakt erforderlicher Ligand der RTK (s. RTK) Das nach der Geburt eines Feten oder Neugeborenen zuerst festgestellte Gewicht. Dieses sollte mæglichst innerhalb der ersten Stunde nach der Geburt ermittelt Gemeinschaftseffekt Genetik Genlocus Genom Genomik Genotypus Gestationsalter Gestationsdiabetes GG GilbertSyndrom gjb2, gjb3 gli gli3 GLUT1 GLUT5 GM-CSF GnRH GP G-Proteine Gi-Protein Gs-Protein GPCR werden, bevor ein postnataler Gewichtsverlust eingetreten ist Community effect: Erscheinung, dass die Differenzierung der Zellen von einer ausreichenden Anzahl der Zellen abhångt Analyse und Beschreibung eines Einzelgens und dessen Funktion Ort eines Genes auf der DNS (Desoxyribonukleinsåure) Summe des Erbgutes Analyse und Beschreibung von Funktion und Wechselwirkung aller Gene im Genom untereinander und mit der Umwelt Gesamtheit aller Erbanlagen eines Organismus Schwangerschaftsdauer, gerechnet vom ersten Tag der letzten normalen Menstruation, ausgedrçckt in vollendeten Wochen und Tagen Wåhrend der Schwangerschaft manifestierter und/oder erstmalig diagnostizierter Diabetes mellitus Grundgesetz (deutsche Verfassung) Erblich bedingte milde Form der Hyperbilirubinåmie basierend auf Polymorphismen der UGT1A1 (s. dort) Gap junction protein beta 2, beta 3: alternative Bezeichnung fçr Connexingene Das Gen wurde aus einem Glioblastom isoliert Amplified in Glioma assoziiertes Gen Glukosetransporter 1 u. a. der Plazenta Glukosetransporter 5 Granulocyte monocyte colony stimulating factor: håmatopoetischer Wachstumsfaktor, der die Proliferation und Differenzierung von håmatopoetischen Stammzellen in Neutrophile und Monozyten stimuliert Gonadotropin-Releasing-Hormon Glykoproteine, die von Viren produziert werden und fçr deren Vermehrungszyklus verantwortlich sind Guanine nucleotide binding proteins inhibitorisches G-Protein stimulatorisches G-Protein Receptor-like G-coupled receptors, G-Protein-gekoppelte Rezeptoren a gpr54 GR G-protein-coupled receptor 54: Gen Zytoplasmatischer Glukokortikoidrezeptor GRE Glucocorticoid response element GvHD Graft-versus-Host-Krankheit: klinische Komplikation, die besonders im Rahmen der BSZT beobachtet wird und Folge einer durch T-Zellen des Spenders ausgelæsten Reaktion gegen Zellen des Transplantatempfångers ist (s. BSZT) HAH Håmagglutinationshemmtest hairy Segmentierungsgen mit oszillierender Expression Hamartom Geschwulst aus atypisch differenziertem Keimgewebe Håmoglobin Das menschliche Håmoglobin besteht aus zweimal zwei Globinketten (Tetramer) mit vier Håmgruppen. Das Håm besteht aus einem Protoporphyringerçst mit einem zentralen zweiwertigen Eisenion. Der çberwiegende Teil des mit dem Blut transportierten Sauerstoffs wird chemisch an Håm gebunden. Verschiedene Håmoglobinvarianten zeichnen sich durch eine unterschiedliche Sauerstoffaffinitåt aus HåmoGenetisch bedingte Verånderungen globinoder Aminosåurezusammensetzung pathien der Globinketten, die mit einer Funktionsstærung des Håmoglobins einhergehen Håmophilie Fehlen des Blutgerinnungsfaktors VIII (A) oder seltener IX (B). An das X-Chromosom gebundene, rezessive Vererbung. Da die entsprechende Anlage auf dem Y-Chromosom fehlt, erkranken nur Månner, da sie der rezessiven Krankheitsanlage auf dem X-Chromosom nichts entgegenzusetzen haben. Theoretisch mægliche Bluterinnen kommen nicht vor, da sie vorgeburtlich absterben Håmorrhagi- Untergegangenes Gewebe, in das es sche Infarkte sekundår eingeblutet hat HaploHaploinsuffizienz liegt vor, wenn an insuffizienz einem Genort das Vorhandensein einer intakten Genkopie nicht ausreicht, um eine normale Funktion zu gewåhrleisten HAS HIF-1 ancillary sequence Abkçrzungen und Erlåuterungen HbA HbA2 Hb-Bart'sSyndrom HBeAg HbF Hb Gower 1 Hb Gower 2 HbHSyndrom Hb Portland I Hb Portland II HBsAg HCC HCF hCG HED-ID Adultes Håmoglobin, das die Hauptkomponente der Håmoglobine beim Erwachsenen darstellt und aus zwei a- und zwei b-Ketten zusammengesetzt ist (a2b2) Adultes Håmoglobin, das aus zwei a- und zwei d-Ketten zusammengesetzt ist (a2d2) Homozygote a-Thalassåmie, bei der alle vier a-Globin-Gene deletiert sind; aus dem Ûberschuss von c-Globinen resultiert die Bildung von c-Homotetrameren (c4, Hb Bart's). Bei erhaltenem f-Globin-Locus kænnen auûerdem Hb Portland 1 (f2c2) mit einem Anteil von 10 bis 20% am Gesamthåmoglobin sowie eine geringe Menge Hb Portland 2 (b2c2) gebildet werden HBV-e-Antigen Fetales Håmoglobin, das aus zwei aund zwei c-Globin-Ketten gebildet wird (a2c2) Embryonales Håmoglobin, das aus zwei f- und zwei e-Globin-Ketten zusammengesetzt ist (f2e2) Embryonales Håmoglobin, das aus zwei a- und zwei e-Globin-Ketten zusammengesetzt ist (a2e2) Struktureller oder funktioneller Verlust von drei der vier a-GlobinGene. Aufgrund der çberschçssigen b-Globin-Molekçle kommt es zur Bildung von b4-Homotetrameren (HbH) Embryonales bzw. fetales Håmoglobin, das aus zwei f- und zwei c-Globinen zusammengesetzt ist (f2c2) Hb Portland II (f2b2) ist eine Form des embryonalen bzw. fetalen Håmoglobins, das nur bei der homozygoten a-Thalassåmie gebildet wird HBV-Hçllenantigen Hepatozellulåres Karzinom Host cell factor: formt zusammen mit a-TIF und OCT1 einen Proteinkomplex, der die Promotoren fçr die a-immediate genes des HSV aktiviert Humanes Choriongonadotropin Hypohidrotische ektodermale Dysplasie mit Immundefekt XXVII XXVIII Abkçrzungen und Erlåuterungen Hemizygotie Vorhandensein von nur einer Genkopie bei diploiden Zellen HensenPrimitivknoten: Struktur am vorKnoten deren Ende des Primitivstreifens, die das Zellmaterial fçr die Chorda dorsalis liefert. Der Knoten ist dem Spemann-Organisator der Amphibien homolog Hereditår Erblich, im Sinne intergenerativer Weitergabe durch das Erbmaterial Heterozygot Vorliegen verschiedener genetischer Merkmale (Allele) am selben Genlocus HFI Hereditåre Fruktoseintoleranz HID Hystrix-like ichthyosis with deafness HIF Hypoxia inducible factor: Transkriptionsfaktor HIG-Test Håmolysis-in-Gel-Test Histon Histone sind eine Gruppe von fçnf kleinen Proteinen, die durch einen hohen Anteil basischer Aminosåuren (Arginin und Lysin) gekennzeichnet sind. Ihre Struktur weist eine zentrale, annåhernd globulåre Domåne und flexible C- und N-terminale Arme auf. Die Seitenketten kænnen durch Acetylierung, Phosphorylierung oder Methylierung modifiziert werden HIV Humanes Immundefizienzvirus HLA Humanes Leukozytenantigen HIgR Nectin1-alpha: Immunglobulin, ein Rezeptor fçr HSV-1 und HSV-2 hIy Gen, welches das Listeriolysin (LLO) der Listerien kodiert HMG Humanes Menopausengonadotropin HNF-3b Hepatocyte nuclear factor 3b: Transkriptionsfaktor, der in der frçhen Embryonalentwicklung exprimiert wird HO Håmoxygenase Homologe Gene, die aufgrund eines evolutioGene åren Zusammenhangs weitreichende Sequenzåhnlichkeiten aufweisen Homæobox DNA-Region in homæotischen Genen, die eine Homæodomåne kodiert. Sie kommt in sehr vielen Entwicklungskontrollgenen wie hoxund pax-Genen vor und wurde in der Evolution wenig veråndert HomæoKonservierte, funktionelle Domåne domåne der Homæoboxproteine, die essen- zielle Funktionen bei der Embryonalentwicklung ausçbt Homæostase Konstanz des inneren Milieus des Kærpers Homozygot Vorliegen identischer genetischer Merkmale (Allele) am selben Genlocus HOX Transkriptionsfaktoren, die durch eine Homæoboxdomåne, die an DNA bindet, gekennzeichnet sind hox-Kode Muster entlang der Kærperachse exprimierter hox-Gene hox-Gene Homæotische Gene, welche die Identitåt von Kærperabschnitten kontrollieren, z. B. hoxa3 (homeodomain-gene a3) HPE Holoprosenzephalie HPFH Hereditary persistence of fetal hemoglobin: hereditåre persistierende çberschieûende c-Globin-Expression bei Ûberschuss an a-Globinen infolge verminderter b-Globin-Synthese hpt Kodiert einen Phosphattransporter, der fçr die Replikation von L. monocytogenes im Zytosol von infizierten Zellen verantwortlich gemacht wird HRE Hypoxia response element: DNA-Abschnitt von Genen, an die HIF-1-Protein bindet (s. HIF) HS Hypersensitive sites: Hypersensitive Stellen sind 200 bis 400 Basenpaare lange DNA-Elemente, die sehr gehåuft Bindungsstellen fçr spezifisch erythroid/håmatopoetisch oder ubiquitår mit der DNA interagierende Transkriptionsfaktoren tragen. Diese Sequenzen haben eine extreme Sensitivitåt gegençber Nukleasen HSV Herpes-simplex-Virus HveA Herpes virus entry mediator A: Mitglied der TNF-Rezeptor-Familie, kommt nur in wenigen Zellen vor (z. B. T-Lymphozyten) und dient als Rezeptor von HSV Hydrops Bildung von Údemen und Wasserfetalis einlagerungen im Gewebe (Aszites, Hydrothorax, Hydroperikard) Hypertonie Bluthochdruck Hypertropher Differenzierungsstufe der ChondroKnorpel zyten, die morphologisch durch eine massive Græûenzunahme des Chondrozyten gekennzeichnet ist IAP Inhibitor of apoptosis protein a ICAM-1 ICBDMS ICD ICP ICSI ID id2 IDO IE IEG IFN IFN-a IFT IgA IGF IGFBP IgG IgM IGT ihh IjB IKK IL Abkçrzungen und Erlåuterungen Interzellulåres Zelladhåsionsmolekçl 1: Adhåsionsmolekçl, u. a. auf Endothelzellen, das als Rezeptor fçr das Molekçl LFA-1 auf Leukozyten fungiert (s. LFA-1) International Clearinghouse for Birth Defects Monitoring Systems Internationale Klassifikation der Diagnosen Infected cell polypeptide: Bezeichnung von HSV-1- und HSV-2-Genen Intrazytoplasmatische Spermieninjektion: Labortechnik, um mit einem Spermium eine Eizelle zu befruchten. Wird bei der PID der einfachen IVF vorgezogen, da ein Mehr an Zellen die molekulargenetische Diagnostik stært Inhibitory domain Gehært zu einer Familie von Genen, die als Inhibitoren der Differenzierung genannt werden, daher auch die Abkçrzung ¹idª. Sie sind in der Lage, die Aktivitåt der bHLH-Faktoren zu inhibieren Indoleamin-2,3-dioxygenase Internationale Einheit Immediate early gene Interferon Alpha-Interferon, Zytokin Immunfluoreszenztest Immunglobulin A Insulin-like growth factor: insulinåhnlicher Wachstumsfaktor, dessen Aktivitåt çber Bindungsproteine reguliert wird IGF bindendes Protein Immunglobulin G Immunglobulin M Impaired glucose tolerance (gestærte Glukosetoleranz) Indian hedgehog homolog. Signalmolekçl mit Homologie zum Drosophila-Gen Hedgehog Inhibitorisches Molekçl: hemmt NFjB durch Sequestrierung IjB-Kinase: ein Multiproteinkinasekomplex, bestehend aus den Untereinheiten IKKa, IKKb, IKKc, letztere ist identisch mit NEMO (s. dort); der Komplex phosphoryliert IjB (s. dort) und aktiviert dadurch NF-jB Interleukin: immunregulatorische Mediatoren, die von Leukozyten se- zerniert werden und auûerordentlich vielfåltige Funktionen in der Regulation des Immunsystems haben IL-1b Interleukin 1b: produziert von aktivierten Makrophagen, Endothelzellen, B-Zellen und Fibroblasten; Induktion der Inflammation mit Údem. Færdert die Produktion von Prostaglandinen, IL-2 und das Leukozytenwachstum IL-6 Interleukin 6: 26-kD-Protein, Zytokin mit vielfåltiger biologischer Wirkung, wird von T-Zellen, Monozyten und Fibroblasten produziert IL-8 Interleukin 8: basisches proinflammatorisches Zytokin, produziert von Monozyten, Gefåûendothelien u. a. Zelltypen, Chemoattraktant und Aktivator von Neutrophilen; Modulator der endothelialen Adhåsion und Transmigration ILS Isolierte Lissenzephalie Implantation ¹Einpflanzungª, hier eines Embryos in den Uterus (Gebårmutter) Imprinting Ein Gen wird als ¹imprintedª bezeichnet, wenn seine Aktivierung oder Inaktivierung im Embryo von der Herkunft des Allels (vom Vater oder von der Mutter) abhångt Induktion Vorgang, bei dem eine Zellgruppe einer anderen ihr Entwicklungsschicksal durch Signalçbermittlung diktiert InIA Internalin Locus A, das mit der Phagozytose von Listeria monocytogenes assoziiert ist InIB Internalin B, das ein Invasin darstellt und nicht kovalent an der Zelloberflåche gebunden ist InIC-H Internaline von L. monocytogenes, welche wahrscheinlich an der Pathogenese beteiligt sind iNOS Induzierbare Stickstoffmonoxidsynthase In-situDarstellung der råumlichen VerteiHybridilung spezieller mRNA im Gewebe sierung oder Darstellung eines Genortes am Chromosom durch komplementåre markierbare Nukleinsåuresonden Insulin Den Blutzucker senkendes Hormon der Bauchspeicheldrçse (51 Aminosåuren) XXIX XXX Abkçrzungen und Erlåuterungen Integrine Transmembranproteine, die zwischen der extrazellulåren Matrix und dem Zytoskelett vermitteln. Sie bestehen aus einer a- und b-Untereinheit und binden auûen z. B. Laminin und Fibronektin, innen Talin Intermediåres Schmaler Mesodermstreifen Mesoderm zwischen dem paraxialen Mesoderm und dem Seitenplattenmesoderm, aus dem sich die Harn- und Geschlechtsorgane bilden Intron DNA-Abschnitt eines Gens zwischen zwei Exons: Introns werden durch Spleiûen aus der Primår-mRNA entfernt Inversion Herausbrechen eines chromosomalen Abschnittes mit Wiedereinbau in umgekehrter Orientierung In vitro ¹im Glaseª IP Incontinentia pigmenti IPAS Inhibitory PAS domain protein IPEXX-chromosomaler kongenitaler Syndrom Immundefekt mit Autoimmunenteropathie und Polyendokrinopathie infolge von Mutationen im foxp3-Gen, welches das immunregulatorische Protein Scurfin kodiert IRD Infantile Refsum-Erkrankung IRES Internal ribosomal entry site: eine Region des 5' -UTR-Endes des HCV, welches die Translation des HCV beeinflussen kann IRF Interferonregulatorische Faktoren Iriskolobom Lçcke in der Iris irx Iroquois, nach der in Drosophila melanogaster bekannten Mutation, die ein irokesenåhnlichen Phånotyp aufweist IUGR Intrauterine growth retardation IVF In-vitro-Fertilisation: Zusammenfçhren von Eizelle und Spermien auûerhalb des weiblichen Kærpers, nåmlich im Glas/im Labor, zwecks spåterer Implantation der befruchteten Eizelle in den Uterus IVH Intraventrikulåre Hirnblutung IVIG intravenæses Immunglobulin, Hyperimmunglobulinpråparat IZ Intermediårzone jag1 Menschliches Gen auf 20p11±12, analog dem murinen Jagged1-Gen, dessen Mutation eine Ursache des Alagille-Syndroms darstellt K1, K10 Keratin 1, Keratin 10: ersetzen die basalen Keratine K5 und K14 bei zunehmender Differenzierung der Keratinozyten in den suprabasalen Zellschichten K5, K14 Keratin 5, Keratin 14: intrazellulåre Strukturproteine, bilden Heterodimere (Intermediårfilamente) der Basalzellschicht, bauen das Zytoskelett auf und stehen mit den Desmosomen in Verbindung kal1 Kallmann syndrome gene 1 kb Kilobasen KBR Komplementbindungsreaktion Keimscheibe Frçhe Embryonalanlage, die zwischen Amnionhæhle und Dottersack gelegen ist und zunåchst aus Epiblast und Hypoblast besteht KID Keratitis-Ichthyosis-Taubheit-Syndrom Klonale Die Abkæmmlinge einer Zellgruppe Restriktion verbleiben innerhalb eines Bereichs und vermischen sich nicht mit Zellen benachbarter Kompartimente Knock-out Gezielte Inaktivierung eines Gens nach gerichteter Mutagenese Kollagen Aus einer Triplehelix bestehendes Strukturprotein KompartiAbgegrenzter Bereich im Embryo, ment in dem sich såmtliche Nachkommen einer kleinen Gruppe von Progenitorzellen befinden Kompetenz Fåhigkeit von Zellen oder Geweben, sich auf bestimmte Signale hin unterschiedlich zu differenzieren Kondensation Erste morphologisch erkennbare Ansammlung von Zellen, welche die Anlage einzelner Knochen bilden Konsensus- Sequenzen der DNA: kommen in sequenzen mehreren Genen vor und sind fçr bestimmte Eigenschaften verantwortlich KP Kortikale Platte L Late (auch c-)Genexpression von Herpesviren Lamina Subepitheliale Bindegewebspropria schicht Laminin Glykoprotein, das insbesondere in der Basallamina vorkommt. Es ist fçr Zell-Matrix-Interaktionen von Bedeutung LAMP Limbic system-associated membrane protein a LAS LAT Lateralplattenmesoderm LBP LBW LCR LCR-Holokomplex LDH LDL Lebendgeborenes Abkçrzungen und Erlåuterungen Lymphadenopathie, durch HIV verursacht Transmembranåres Transportprotein fçr groûe Aminosåuren (large amino acids) Bereich an der Flanke des Embryo, aus dem die Zellen stammen, welche die Extremitåten bilden LPS bindendes Protein: Plasmaprotein, das freies LPS im Serum abfångt, bindet und zu den LPS-Rezeptoren an der Phagozytenzelloberflåche wie z. B. CD14 transportiert Low birth weight: untergewichtiges Lebendgeborenes, Geburtsgewicht < 2500 g Locuskontrollregion: prominentestes regulierendes DNA-Element im b-Globin-Genlocus, das aus mindestens fçnf hypersensitiven Stellen (s. HS) besteht. Sie erstreckt sich çber eine Distanz von mehr als 15 kb stromaufwårts vom embryonalen e-Globin-Gen. Sie reguliert die Chromatinstruktur und vermittelt die Aktivierung der Globinexpression im b-Globin-Genlocus in allen Stadien der Entwicklung erythroider Zellen Råumliche Annåherung der HS der LCR durch Bindung von Proteinkomplexen und Protein-Protein-Interaktionen Lactatdehydrogenase: Oberflåchenantigene von Toxoplasma gondii Low-Density-Lipoprotein: wesentliches Lipoprotein fçr den Transport von Cholesterin zu den Zellen Eine aus der Empfångnis stammende Frucht, die unabhångig von der Schwangerschaftsdauer vollståndig aus dem Mutterleib ausgestoûen oder extrahiert ist, nach Verlassen des Mutterleibes atmet oder irgendein anderes Lebenszeichen erkennen låsst, wie Herzschlag, Pulsation der Nabelschnur oder deutliche Bewegung der willkçrlichen Muskulatur, gleichgçltig, ob die Nabelschnur durchtrennt oder die Plazenta ausgestoûen wurde oder nicht. Jedes unter diesen Voraussetzungen neugeborene Kind ist als lebend geboren zu betrachten. LEF1 lekti Leptin Letalitåt Leukomalazie, periventrikulåre Leviticus LFA-1 DDR: Anordnung çber die årztliche Leichenschau vom 4. Dez. 1978 (GBl I 1979 Nr. 1 S. 4; § 4): Lebend geboren ist, wenn nach vollståndigem Verlassen des Mutterleibes, unabhångig vom Durchtrennen der Nabelschnur oder von der Ausstoûung der Plazenta die bei den Lebenszeichen Herztåtigkeit und Lungenatmung vorhanden waren. Das normale durchschnittliche Geburtsgewicht von reifgeborenen deutschen Neugeborenen wird fçr 1992 von Voigt et al. (1996) fçr Jungen mit 3600 g und fçr Mådchen mit 3450 g angegeben Lymphoid enhancer factor 1: ist in den WNT-Signalweg involviert Lympho-epithelial Kazal type-related inhibitor (s. spink5) Die Nahrungsaufnahme hemmendes Hormon des Fettgewebes (167 Aminosåuren) Anteil der durch die Krankheit verursachten Todesfålle an der Anzahl der Erkrankten. Die Letalitåt bezeichnet im Gegensatz zur Sterblichkeit (Mortalitåt) die Wahrscheinlichkeit, dass ein an einer bestimmten Krankheit Leidender an dieser Krankheit stirbt Leukomalazie bedeutet Erweichung (Malazie) weiûer (leukos) Hirnsubstanz, die bei Frçhgeborenen vorzugsweise um die seitlichen Hirnkammern (Seitenventrikel) herum (periventrikulår) auftritt, da diese Gegend noch weniger mit Blutgefåûen als andere Gehirnregionen versorgt und damit besonders empfindlich gegen Durchblutungsstærungen ist. Periventrikulår verlaufen die langen motorischen Bahnen. Resultat ihrer Beschådigung sind spastische Låhmungen 3. Buch Mose: Dieses und das 5. Buch Mose (Deuteronomion) enthalten die alttestamentlichen Gesetze Leukocyte function-associated antigen-1: Molekçl auf Leukozyten, das im Rahmen der Auswanderung von Leukozyten aus der Blutbahn in infiziertes Gewebe das Anheften an Endothelzellen ermæglicht XXXI XXXII Abkçrzungen und Erlåuterungen LH LHA lhx3, lhx4 LIF Luteinisierendes Hormon Area lateralis hypothalami LIM-Homæobox-Gene 3, 4 Leukemia inhibitory factor: pleiotropes Zytokin LIM Transkriptionsfaktoren, die nach ihrem ersten Vorkommen in den Genen Lin11 und Mec3 des Fadenwurms Caenorhabditis elegans bezeichnet werden. Sie kænnen verschiedene konservierte Proteindomånen enthalten, wie Homæobox oder Zinkfinger. Einige von ihnen werden auch mit lhx benannt lis1 Gen fçr Lissenzephalie 1 LLO Listeriolysin: notwendig fçr den bakteriellen Schutz der Listeria monocytogenes vor einer Phagozytose LOH Loss of heterozygosity: Verlust der Heterozygotie Lox Locus of X-ing-over LPH Laktase-Phlorizin-Hydrolase (Laktase spaltende Disaccharidase) LPS Lipopolysaccharidstruktur (sog. Endotoxin): læst in einem infizierten Wirt nach Phagozytose der Makrophagen eine akute Entzçndungsreaktion aus lrp8 Gen fçr den Apolipoprotein-E-Rezeptor LTA Lipoteichonsåure: Bestandteil der Zellwand grampositiver Bakterien lunatic fringe Segmentierungsgen mit oszillierender Expression Lymphatische Man unterscheidet primåre und Organe sekundåre lymphatische Organe. Die primåren lymphatischen Organe sind Knochenmark und Thymus, hier werden Lymphozyten gebildet. Milz, Lymphknoten, Tonsillen und lymphatisches Gewebe der Schleimhåute sind sekundåre lymphatische Organe, hier findet eine Einwanderung reifer Lymphozyten zur weiteren funktionellen Differenzierung statt MAA Maleylacetoacetat MAG Matrixantigen von Toxoplasma gondii MAK-Werte Maximale Arbeitsplatzkonzentration MalformaFehlbildungen tionen MALT Mukosaassoziiertes Lymphgewebe MAP Mitogenaktiviertes Protein MAPK MAS mash1 Mitogenaktivierte Proteinkinase Mekoniumaspirationssyndrom Mammalian achaete-scute homolog 1: wurde als ein homologes Gen zum Drosophila-melanogasterachaete-scute-Gen isoliert MBL Mannose bindendes Lektin: Plasmaprotein, welches das bakterielle Zuckermolekçl Mannose erkennt und zur Opsonisierung und Komplementaktivierung fçhrt MCP-1 Monocyte chemoattractant protein-1: Chemokin, das in der Chemotaxis eine Rolle spielt M-CSF Makrophagenkolonie stimulierender Faktor MDCR Miller-Dieker-Syndrom der chromosomalen Region MDR1 Multidrug resistance gene 1: P-Glykoprotein (alias abcb1) MDS Miller-Dieker-Syndrom MEF Myocyte enhancing factor: wirkt bei der myogenen Determination mit sowie verstårkt und stabilisiert die Expression der myogenen Determinationsfaktoren MeHg Methylquecksilber Meistergen Master gene: Gen, das die Aktivitåt anderer Gene reguliert. Auch Selektor- oder Entwicklungskontrollgen genannt Mesenchym Lockeres embryonales Bindegewebe, dessen Zellen wandern kænnen Metabolisches Syndrom X: Symptomenkomplex Syndrom çberwiegend diabetogener und atherogener Stærungen vor dem Hintergrund von Ûbergewicht und Insulinresistenz Metabolizer, Individuum mit hoher Aktivitåt extensive eines bestimmten metabolisierenden Enzyms Metabolizer, Individuum mit hereditårem Defekt poor eines bestimmten metabolisierenden Enzyms. Die Aktivitåt ist entweder gering oder komplett defizient METHepatocyte growth factor receptor: Rezeptor InlB von L. monocytogenes bindet an diesen Rezeptor und fçhrt zu einer PI-3-Kinase-Aktivierung und zur Invasion MG Molekulargewicht MHC Haupthistokompatibilitåtskomplex: Seine Gene kodieren die Proteine,