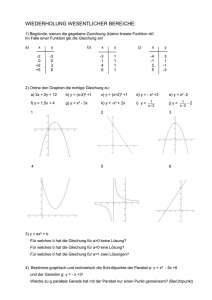
2. Bedienungsanleitungen mit praktischen Hinweisen
Werbung

Physikalisch-Chemisches Praktikum I Überarbeitete Version A. Kuhn / V. Epp (2011) Institut für Physikalische Chemie und Elektrochemie Leibniz-Universität Hannover Druck: 14.05.2016 Inhaltsverzeichnis 0. Einleitung .......................................................................................................... 3 1. Hinweise zum Ablauf des Praktikums .............................................................. 4 1.1. Praktikumsordnung ..................................................................................... 4 1.2. Inhalt der Leistungsnachweise .................................................................... 5 2. Bedienungsanleitungen mit praktischen Hinweisen ......................................... 7 2.1. Druck- und Vakuumtechnik ........................................................................ 7 2.2. Temperaturmessung und Temperaturregelung ........................................... 9 2.3. Stoffmengenbestimmung und Benutzung der Waagen ............................ 14 2.4. Volumenmessung ...................................................................................... 15 3. Versuche im Physikalisch-Chemischen Praktikum I mit Lageplan ............... 16 3.1. Wärmekapazität idealer Gase (Versuch Nr. 22) ....................................... 18 3.2. Joule-Thomson-Effekt (Versuch Nr. 9) .................................................... 25 3.3 Verbrennungsenergie einer aromatischen Verbindung (Versuch Nr. 2) ... 31 3.4 Verdampfungsenthalpie von Wasser (Versuch Nr. 1) ............................... 39 3.5 Verdampfungsenthalpie von Aceton (Versuch Nr. 6) ............................... 44 3.6. Gefrierpunktserniedrigung (Versuch Nr. 7) .............................................. 49 3.7. Schmelzdiagramm binärer Mischungen (Versuch Nr. 8) ......................... 59 3.8. Homogenes Dissoziationsgleichgewicht (Versuch Nr. 10) ...................... 65 3.9. Heterogenes Dissoziationsgleichgewicht (Versuch Nr. 11) ..................... 69 3.10. Adsorption (Versuch Nr. 5) .................................................................... 74 3.11. Oberflächenspannung (Versuch Nr. 23) ................................................. 79 3.12. Nernst’sche Gleichung (Versuch Nr. 24) ............................................... 85 3.13. Rohrzuckerinversion (Versuch Nr. 16) ................................................... 93 3.14. Moleküle in Bewegung (Versuch Nr. 25) ............................................... 99 3.15. Wasserstoffatom (Versuch 21) .............................................................. 107 3.16. Lichtabsorption von Farbstoffmolekülen (Versuch Nr. 26) ................. 115 4. Fehler- und Ausgleichsrechnung .................................................................. 123 [Text eingeben] 0. Einleitung Das Physikalisch-Chemische Praktikum I dient zusammen mit den beiden Vorlesungen in Physikalischer Chemie im Grundstudium dem Zweck, die Grundlagen der Physikalischen Chemie zu vermitteln. In der Auswahl der 10 Praktikumsversuche spiegelt sich die Schwerpunktsetzung im Bereich der Thermodynamik in der ersten Grundvorlesung wider und betont daher vor allem die Behandlung des chemischen Gleichgewichts. Trotzdem behandelt das Grundpraktikum nicht nur die Thermodynamik. Neben der Elektrochemie, der Bewegung von Ionen und Molekülen werden auch Versuche zur chemischen Kinetik und zum Aufbau der Materie durchgeführt. Da die theoretischen Grundlagen für diese Versuche erst in der zweiten Grundvorlesung behandelt werden, ist der Besuch der beiden Grundvorlesungen und die Nacharbeitung der dort behandelten Themen dringend notwendig für ein erfolgreiches Absolvieren des physikalisch-chemischen Grundpraktikums. Das Praktikumsskript setzt sich aus zwei Teilen zusammen. Der erste Teil enthält allgemeine Hinweise zum Ablauf des Praktikums und zu grundlegenden Experimentiertechniken, während der zweite Teil aus den Anleitungen für die verschiedenen Versuche mit einem Hinweis zur Fehler- und Ausgleichsrechnung besteht. In den Versuchsanleitungen wird nicht nur die praktische Durchführung der Versuche beschrieben, sondern es ist auch eine knappe Darstellung der relevanten theoretischen Grundlagen enthalten. Bei manchen Versuchen finden sich darüber hinaus noch theoretische Ergänzungen zum Bearbeiten der Praktikumsaufgaben. Das Praktikumsskript stellt jedoch keinen Ersatz zu einem Lehrbuch dar. Deshalb sind zu jedem Versuch die in der Vorbereitung zu erarbeitenden Lernziele stichpunktartig aufgeführt, so dass jeder Praktikumsteilnehmer die Möglichkeit hat, die benötigten theoretischen Grundlagen im Lehrbuch seiner Wahl aufzuarbeiten. Für die beiden am weitesten verbreiteten Lehrbücher, den „Atkins“ und den „Wedler“, sind zusätzlich explizit die entsprechenden Seitenzahlen als Literatur angegeben. Es wurde versucht, in den einzelnen Anleitungen eine Symbolik zu verwenden, die den Vorschlägen der IUPAC entspricht. Dadurch entsteht an manchen Stellen eine Diskrepanz zu der verwendeten Symbolik in den gängigsten Lehrbüchern. Es gibt sicher viele Dinge, die man am Praktikumsskript verbessern kann. Dies ist allerdings nur möglich, wenn entsprechende Verbesserungs- oder Änderungsvorschläge eingebracht werden. Deswegen soll dazu nochmals besonders ermutigt werden. Auch Schreib- und Tippfehler können nur beseitigt werden, wenn darauf aufmerksam gemacht wird. 1. Hinweise zum Ablauf des Praktikums 1.1. Praktikumsordnung Voraussetzungen zur Teilnahme am Praktikum sind: 1. Mathe-Schein I (Analysis) 2. PC1-Klausur (Thermodynamik-Schein) 3. Das AC-Praktikum soll soweit abgeschlossen sein, dass dort keine Experimente parallel zum PC-Grundkurs stattfinden müssen (Nachweis durch Laufzettel o. Prakt.-Schein) Die Anzahl der Praktikumsplätze pro Kurs ist begrenzt. Die Teilnehmer werden in der jeweiligen Vorbesprechung bestimmt. Das Praktikum umfasst 1. für die Studiengänge Bachelor, Chemie-Diplom und Biochemie-Diplom 10 Versuche. 2. für den Studiengang fächerübergreifender Bachelor, Höheres Lehramt 8 Versuche. Das Praktikum findet von 13.00-17.00 in den Räumen 040, 046 und 048 im Seitentrakt des Instituts für Physikalische Chemie und Elektrochemie statt. Es erstreckt sich über 4-5 Wochen, wobei von jedem Praktikanten 2-3 Versuche pro Woche zu absolvieren sind. Vor Beginn der praktischen Arbeiten müssen Sie sich über zu treffende Sicherheitsmaßnahmen bei der Durchführung des Versuches informieren. Spezielle Sicherheitsanweisungen zu den einzelnen Versuchen liegen am Praktikumsplatz aus. Sollten Sie mit den Sicherheitsmaßnahmen nicht vertraut sein, darf der Versuch nicht durchgeführt werden. Verstöße gegen Sicherheitsvorschriften (z. B. Schutzhandschuhe tragen, Schutzbrille aufsetzen) können zum Ausschluss vom Praktikum führen. Beim Umgang mit verflüssigten Gasen, anderen gefährlichen Substanzen und evakuierten oder unter Druck stehenden Glasapparaturen sind Schutzbrillen und Schutzhandschuhe zu tragen. Schutzhandschuhe und Schutzbrillen liegen im Praktikum aus. Besondere Sorgfalt ist beim Arbeiten mit Quecksilberthermometern und Probengefäßen, die mit Blei/Zinnmischungen gefüllt sind, zu üben. Wird ein entsprechendes Thermometer oder Probengefäß zerbrochen, so ist sofort die für das Praktikum zuständige chemisch-technische Assistentin zu benachrichtigen. Elektrische Schaltungen sind vor Benutzung dem Assistenten zur Prüfung vorzuzeigen. Stoppuhren können bei der Praktikumsassistentin ausgeliehen werden. Jeder Praktikant hat die Verantwortung für die pflegliche Behandlung der bereitgestellten Geräte. Die Waagen sind peinlich sauber zu halten. Für beschädigte Geräte wird im Falle grober Fahrlässigkeit Ersatz gefordert. Chemikalienreste, die nicht ins Abwasser gelangen dürfen, sind in die vorgesehenen Restebehälter zu füllen. Nach Abschluss des Versuches sind Geräte und Arbeitsplatz sorgfältig zu reinigen. Geräte und Substanzen sind an ihren ursprünglichen Platz zurückzustellen. Maßlösungen sind verschlossen zu halten. Vor Beginn des Praktikums muss durch Unterschrift die Kenntnisnahme der Praktikumsordnung sowie die Teilnahme an der Sicherheitsbelehrung bestätigt werden. Die weiblichen Kursteilnehmer werden zusätzlich über besondere Gefahren für werdende Mütter unterrichtet und bestätigen dies durch Unterschrift. 1.2. Inhalt der Leistungsnachweise 1.2.1 Antestate Zu Beginn eines jeden Versuchstages findet eine Vorbesprechung bzw. ein Antestat statt. Zur Durchführung des Versuches sollen Sie sich daher sowohl über den praktischen Teil als auch über die zum Versuch gehörenden theoretischen Grundlagen informieren. Im Antestat sollen Sie zeigen, dass Sie wissen, was das Ziel des Versuches ist, welche physikalisch-chemischen Größen gemessen werden und wie sie zusammenhängen, wie diese Größen gemessen werden und was man aus diesen Größen berechnen bzw. lernen kann. Sollten Sie nur unzureichend mit dem Versuchsstoff vertraut sein, dann muss das Antestat wiederholt werden. Falls im Antestat schwerwiegende Mängel vom Assistenten festgestellt werden, müssen Sie damit rechnen, dass die Praktikumsleitung Ihren Kenntnisstand überprüft. 1.2.2 Protokolle Für jeden durchgeführten Praktikumsversuch ist ein Protokoll anzufertigen. Die Protokolle werden von jeweils einem Gruppenmitglied geschrieben und müssen vor Beginn des nächsten Versuchs in die ausstehenden Protokollbehälter abgegeben werden. Die Auswertung kann ebenfalls mit Computer oder mit der Hand (Taschenrechner) geschehen. Zum Umfang und zum Anfertigen der Protokolle sollte folgende Gliederung beachtet werden: a) Protokollkopf: Gruppen-Nr.: Versuchs-Nr.: Protokollant: Teilnehmer: Datum: Assistent: Platz für Praktikumsstempel b) Versuchsbeschreibung: Kurze Beschreibung des Versuchs mit Zielsetzung in einem Satz. c) Theoretische Grundlagen: Dieser Abschnitt soll in kurzer und zusammenhängender Form die theoretischen Voraussetzungen für die Versuchsdurchführung wiedergeben (maximal 1 Seite). d) Versuchsaufbau und -durchführung: In diesem Abschnitt werden die durchgeführten praktischen Arbeiten beschrieben und anhand einer Versuchsskizze erläutert (maximal 1 Seite inklusive Skizze). e) Messprotokoll: Das vom Praktikumsassistenten oder technischen Assistenten abgezeichnete Messprotokoll (Messwerte in tabellarischer Form, DINA4) wird in das Protokoll eingeheftet. f) Auswertung: In diesem Abschnitt geht es darum, die eigenen Messergebnisse auszuwerten. Alle Zwischenergebnisse sollen mit den verwendeten Formeln übersichtlich dargestellt werden. Zum Schluss wird das Endergebnis angegeben. Für jeden Zwischenschritt muss eine entsprechende Fehlerrechnung durchgeführt werden, d. h. alle Größen sind mit einem Fehler anzugeben (siehe dazu auch Abschnitt über Fehler- und Ausgleichsrechnung auf den Seiten 123-129). Bei graphischen Darstellungen wird eine vollständige Achsenbeschriftung erwartet. Die Angaben müssen mit richtigen Einheiten versehen sein und die Messpunkte sind mit Fehlerbalken zu versehen. Außerdem soll die Abbildung zur Übersichtlichkeit beschriftet sein. Diese Punkte sind insbesondere beim Anfertigen der graphischen Darstellungen mit dem Computer zu beachten (maximal 5 Seiten inklusive graphischen Darstellungen). g) Fehlerbetrachtung: Hier werden die eigenen Messergebnisse mit Literaturangaben verglichen. Die verwendete Literaturquelle muss explizit angegeben werden (Titel, Verfasser, Verlag, Jahr, Seite). Im Praktikum gibt es dazu einen Ordner, der Literaturangaben zu jedem Versuch enthält. Zusätzlich findet man in dem Ordner weiterführende Literatur zum Versuch. Außerdem soll eine kritische Bewertung bzw. Diskussion der einzelnen Messergebnisse stattfinden. Dabei können noch nicht berücksichtigte Fehlerquellen angeführt werden oder es kann auf Vereinfachungen bzw. unzulässige Annahmen bei der Auswertung aufmerksam gemacht werden (maximal 1 Seite). 1.2.3. Abschlusskolloquium Das Abschlusskolloquium findet etwa 3-4 Wochen nach Beendigung des letzten Praktikumsversuches statt. Um zum Abschlusskolloquium zugelassen zu werden, muss der praktische Teil vollständig abgeschlossen werden, d. h. es müssen alle Protokolle von den jeweiligen Praktikumsassistenten abgezeichnet worden sein. Die Protokolle sind zum Abschlusskolloquium mitzubringen. Im Abschlusskolloquium wird von Ihnen erwartet, dass Sie mit den praktischen und theoretischen Grundlagen aller von Ihnen durchgeführten Versuche vertraut sind. 2. Bedienungsanleitungen mit praktischen Hinweisen 2.1. Druck- und Vakuumtechnik 2.1.1. Druckeinstellung am Reduzierventil a) Wenn kleine Drücke (bis Luftdruck) eingestellt werden sollen, ist folgendes zu beachten: 1. Hauptventil öffnen. 2. Auslassventil öffnen. 3. Apparatur anschließen (falls noch nicht geschehen). 4. Durch vorsichtiges Hereindrehen der Einstellschraube den gewünschten Druck einstellen, indem man den Druck am genauesten verfügbaren Manometer kontrolliert Abb. 1: Reduzierventil. b) Wenn größere Drücke eingestellt werden sollen und es nicht besonders auf die Genauigkeit ankommt, geht man folgendermaßen vor: 1. Hauptventil öffnen. 2. Durch Hereindrehen der Einstellschraube den gewünschten Druck einstellen, in dem man den Druck am Manometer des Reduzierventils kontrolliert. 3. Apparatur anschließen (falls noch nicht geschehen). 4. Auslassventil öffnen. c) Alle Reduzierventile müssen am Ende des Versuchstages „entlastet“ werden, d. h. das Gas muss herausgelassen werden, da sonst die Dichtungen im Ventil schnell verschleißen. Zum Entlasten eines Reduzierventils ist zu beachten: 1. Zunächst wird das Auslassventil geschlossen und der Gasweg nach außen geöffnet. 2. Hauptventil schließen. 3. Auslassventil wieder öffnen. 4. Sollte das Manometer nun immer noch einen Druck anzeigen, so muss die Einstellschraube etwas weiter herein gedreht werden, bis das Gas entweicht. 5. Auslassventil schließen. 6. Einstellschraube wieder bis locker vor den Anschlag herausdrehen. 2.1.2. Funktionsweise eines Dosenmanometers bzw. Federvakuummeters Das Innere eines kreisförmig gebogenen Rohres (sogenanntes Bourdonrohr) wird an den zu evakuierenden Behälter angeschlossen. Durch die Wirkung des äußeren Luftdruckes wird das Ende des Rohres beim Evakuieren mehr oder weniger gebogen. Dadurch wird das dort angreifende Zeigerwerk betätigt. Die Skala ist linear. Die Druckanzeige ist vom äußeren Luftdruck abhängig. Abb. 2: Aufbau eines Dosenmanometers mit Verbindung zum Rezipienten (1), Zeiger (2), Bourdon-Röhre (3) und Hebelsystem (4). 2.1.3. Betrieb und Funktionsweise einer Ölrotationspumpe Die Arbeitsweise von Ölrotations- bzw. Drehschieberpumpen ist in Abb. 3 dargestellt. In einem feststehenden Außenzylinder dreht sich exzentrisch ein Innenzylinder. Mit dem Innenzylinder sind zwei federnde Schieber verbunden, die den freien Raum zwischen Innen- und Außenzylinder in zwei Kammern I und II teilen. Abb. 3: Aufbau einer Ölrotationspumpe. Bei der eingezeichneten Drehrichtung wird in der Kammer I das Gas aus dem zu evakuierenden Gefäß angesaugt, bis der obere Schieber das Gaseinlassrohr passiert hat. Dann ist die eingesaugte Gasmenge zwischen beiden Schiebern eingeschlossen und wird über ein Auslassventil aus der Pumpe entfernt. Der Außenzylinder befindet sich in einem Ölbad, um eine sichere Abdichtung der beweglichen Teile (Durchführungen der Antriebswelle) zu erreichen. Mit solchen Ölrotationspumpen lässt sich ein Druck von etwa 10−2 mbar (1 Pa) erreichen. Bei den im Praktikum verwendeten Ölrotationspumpen sind folgende Punkte zu beachten: 1) Wenn sich eine Kühlfalle zwischen Versuchsaufbau und Pumpe befindet, muss diese mit flüssigem Stickstoff eingekühlt werden, bevor die Pumpe eingeschaltet wird. Nach dem Einkühlen sollte die Kühlfalle aber möglichst bald evakuiert werden, da sonst Luft in ihr kondensiert und den Gasweg zur Pumpe versperrt. 2) Vor jedem Ausschalten der Pumpe muss sie belüftet werden. Dazu ist an allen Apparaturen ein Dreiwegehahn zwischen Pumpe und Versuchsaufbau vorhanden. 3) Nach dem Belüften der Pumpe darf sie nicht in Betrieb bleiben, sondern muss ausgeschaltet werden, da sonst das Öl in der Pumpe aufschäumt und durch den Auspuff abgeblasen wird. 2.2. Temperaturmessung und Temperaturregelung 2.2.1. Flüssigkeitsthermometer Zur Temperaturmessung können im Prinzip alle Stoffeigenschaften herangezogen werden, die monoton von der Temperatur abhängen, wie z. B. die Volumenänderung einer Flüssigkeit. In diesen Flüssigkeitsthermometern verwendet man normalerweise Quecksilber als Thermometerfüllung. Eine spezielle Ausführung eines Quecksilberthermometers stellen die BeckmannThermometer dar, mit denen Temperaturdifferenzen sehr genau gemessen werden können. Dabei steigt das Quecksilber bei Erwärmung aus einem Vorratsgefäß durch eine sehr dünne Kapillare hoch. An der Kapillare ist ein Maßstab angebracht, dessen Teilstriche 1/100 °C voneinander entfernt sind, so dass Temperaturdifferenzen von 1/1000 °C noch geschätzt werden können. Diese Thermometer sind für verschiedene Messbereiche erhältlich (Kalorimeterthermometer). 2.2.2. Thermoelemente Bringt man zwei verschiedene Metalle I und II miteinander in Kontakt, so lädt sich eines der Metalle gegenüber dem anderen Metall elektrisch auf, weil die Austrittsarbeiten in den beiden Materialien verschieden sind. Die elektrische Spannung zwischen den beiden Metallen wird auch als Kontaktpotentialdifferenz bezeichnet. Man kann diese Potentialdifferenz selbst nicht direkt messen (siehe Abb. 1), weil beim Anschließen eines Messinstrumentes immer mindestens zwei Kontaktpotentialdifferenzen I/II und II/I auftreten, die sich jedoch gegenseitig kompensieren, so dass das Messinstrument die Spannung Null anzeigt. 1 II 2 I II Abb. 1: Geschlossener Stromkreis mit mehreren Kontaktpotentialen. Da die Kontaktpotentialdifferenzen jedoch empfindlich von der Temperatur abhängen, kann man sie zur Temperaturmessung benutzen. Hat nämlich die Verbindungs- oder Lötstelle 1 eine von der Lötstelle 2 verschiedene Temperatur, so zeigt das Messinstrument eine elektrische Spannung U an, die bei einigen Metallkombinationen in einem großen Temperaturbereich linear von der Temperaturdifferenz ΔT zwischen den Lötstellen abhängt, d. h. U = c·ΔT. Metall II Metall I Lötstelle 1 T1 T2 Lötstelle 2 Abb. 2: Messung der Temperatur mit einem Thermoelement. Ist die Temperatur T2 einer Lötstelle bekannt, so kann man aus der gemessenen Spannung Δϕ die Temperatur T1 der anderen Lötstelle bestimmen. Metallpaare, die zur Messung der Temperatur benutzt werden, bezeichnet man als Thermoelemente. Für die am häufigsten verwendete Metallkombination Chromel/Alumel (Chromel: 89% Ni, 10% Cr, 1% Fe; Alumel: 94% Ni, 2% Al, 1% Si, 3% Mn) ist die Spannung Δϕ in Abhängigkeit von T1 für den Fall T2 = 0 °C in Abb. 3 dargestellt. Der Kurve entnimmt man, dass die Proportionalitätskonstante c = 4.1∙10−2 mV/°C ist. Somit lassen sich problemlos Temperaturänderungen von einigen °C messen. Abb. 3: Thermospannung eines Chromel/Alumel-Thermoelements (T2 = 0 °C). Für eine größere Messgenauigkeit schaltet man mehrere Thermoelemente hintereinander. Mit einer solchen Thermosäule kann man die Genauigkeit ohne weiteres auf das 100-fache verbessern. 2.2.3. Heizregler Elektrische Heizregler dienen der Temperierung elektrischer Heizöfen oder Ölbäder. Sie werden zwischen das Heizgerät und die Spannungsquelle geschaltet, wobei ein Thermoelement die Temperatur im Heizgerät misst. Man stellt am Heizregler eine Solltemperatur ein. Die tatsächlich erreichte Temperatur misst man aber meistens mit einem genaueren Thermometer im Versuchsaufbau. Einstellen der Solltemperatur: Am Regler befindet sich ein Wahlschalter, der auf die Position "Soll"Temperaturanzeige-Regelung „Ein“-geschaltet gestellt werden muss. Dann stellt man mit dem Potentiometer die gewünschte Temperatur nach Skalenteilen am Anzeigeinstrument des Reglers ein. Der sogenannte Vollausschlag (100 Skalenteile) entspricht dabei der maximal möglichen Temperatur im jeweiligen Messbereich. Will man z. B. 120 °C im 300 °C Messbereich einstellen, so müsste man Solltemperatur Vollausschlag 120 °C 100 Skt. 40 Skt. Messbereich 300 °C einstellen. 2.2.4. Thermostate Es werden Thermostate benutzt, um die Messtemperatur konstant zu halten. Abb. 4: Schema eines Thermostaten. Ein Thermostat besteht aus einem Behälter, der mit einer Flüssigkeit, meist Wasser, gefüllt ist. In die Flüssigkeit tauchen ein Rührer, ein Tauchsieder, eine Kühlschlange und ein Temperaturregler. Mit Hilfe einer Kühlflüssigkeit (meist ebenfalls Wasser) deren Temperatur tiefer als die geforderte Temperatur liegen muss, wird die Thermostatenflüssigkeit gekühlt. Am Temperaturregler stellt man die gewünschte Temperatur ein. Sobald die Temperatur der Thermostatenflüssigkeit diesen Sollwert unterschreitet, wird vom Temperaturregler der Tauchsieder eingeschaltet, so dass die Temperatur der Thermostatenflüssigkeit wieder ansteigt. Beim Überschreiten der eingestellten Solltemperatur wird der Tauchsieder wieder abgeschaltet, und die Thermostatenflüssigkeit kühlt wieder ab. Da der Tauchsieder nicht unmittelbar nach dem Einschalten Wärme abgibt und nach dem Abschalten noch etwas weiterheizt, schwankt die Thermo- statentemperatur innerhalb enger Grenzen. Diese Grenzen sind normalerweise um so geringer, je besser die Kühlleistung der Kühlschlange und die Heizleistung des Tauchsieders aufeinander abgestimmt sind. Das Messobjekt, das thermostatisierent werden soll, kann direkt in die Thermostatenflüssigkeit eingetaucht werden. Es ist aber auch möglich, durch eine Pumpe dem Thermostaten Flüssigkeit zu entnehmen, sie mit dem Messobjekt in thermischen Kontakt zu bringen und anschließend in den Thermostaten zurückzuführen. Bei den im Praktikum verwendeten Thermostaten sind folgende Punkte zu beachten: 1) Wenn der Thermostat über eine zuschaltbare Dauerheizung verfügt, sollte sie in der Regel ausgeschaltet bleiben. Nur bei Solltemperaturen über 60 °C wird sie eingesetzt. 2) Die Wasserkühlung wird im Temperaturbereich unter Raumtemperatur und bis ca. 10 °C über Raumtemperatur mehr oder weniger stark aufgedreht. Bei noch höheren Solltemperaturen genügt die Wärmeabgabe über die Raumluft als Kühlung. 2.3. Stoffmengenbestimmung und Benutzung der Waagen Im Praktikum befinden sich insgesamt vier Waagen: Zwei elektronische Analysenwaagen mit einer Genauigkeit von 0.0001 g bzw. 0.00001 g und zwei elektronische Grobwaagen mit einer Genauigkeit von 0.1 g bzw. 0.01 g. 2.3.1. Elektronische Analysenwaage (Kern) Der Wägebereich beträgt maximal 150 g. 1) Waage einschalten (on/off) und warten bis 0.0000 g angezeigt werden. 2) Wägegut auflegen und nach ca. 5 Sekunden das Gewicht ablesen. 3) Durch Drücken der Tara-Taste (T) kann man die Anzeige bei Leergewichten von Probengläschen und Wägeschälchen wieder auf 0.0000 g bringen. Niemals die Mode-Taste betätigen! 2.3.2. Elektronische Analysenwaage (Sartorius) Sollte der Netzstecker dieser Waage nicht angeschlossen sein, benötigt die Waage 20 Min. Aufwärmzeit vor der ersten Benutzung, da die Genauigkeit sonst nicht garantiert werden kann. Um die Präzision zu erhalten ist die Wägeschale äußerst sauber zu halten. Die Genauigkeit beträgt bis 80 g Gesamtgewicht 0.01 mg und bis 220 g Gesamtgewicht 0.1 mg. 1) Waage einschalten (I/0) und abwarten bis 0.00000 g angezeigt wird. 2) Behälter für Wägegut auflegen und auf 0 tarieren (TARE). 3) Wägegut in Behälter legen und nach ca. 5 Sekunden das Gewicht ablesen. Auf keinen Fall dürfen mit den anderen Tasten die Grundeinstellungen der Waage verändert werden. 2.3.3. Elektronische Grobwaage (Kern) Bei einem Wägebereich bis 500 g beträgt die Genauigkeit 0.1 g, bei einem Wägebereich bis 1500 g ist die Genauigkeit nur noch 0.2 g. 1) Waage einschalten und warten bis 0.0 g angezeigt wird. 2) Durch Drücken der Tara-Taste kann man die Anzeige bei Leergewichten von Probengläschen und Bechergläsern wieder auf 0.0 g bringen. 3) Wägegut auflegen und nach ca. 5 Sekunden den Wert ablesen. 2.3.4. Elektronische Grobwaage (Denver MAXX) Bei einem Wägebereich bis 600 g beträgt die Ablesbarkeit 0.01 g und die Genauigkeit 0.03 g. 1) Waage einschalten und warten bis 0.00 g angezeigt wird. 2) Ggf. Waage auf 0 stellen (Taste „Zero“ drücken). Mit dieser Funktion kann auch das Gewicht von Wägeschälchen u. ä. nivelliert werden. 3) Wägegut auflegen und nach ca. 5 Sekunden den Wert ablesen. Auf keinen Fall dürfen mit den anderen Tasten die Grundeinstellungen der Waage verändert werden. 2.4. Volumenmessung 1) Die verwendeten Volumenmessgeräte, wie z. B. Pipetten, Messkolben und Pyknometer, gehören nicht in den Trockenschrank und dürfen auch nicht auf andere Weise erhitzt werden, da die Genauigkeit der Geräte darunter leidet. 2) Außerdem ist beim Einsatz von Peleusbällen darauf zu achten, dass sie nach Beendigung des Versuches wieder entlastet, d. h. nicht zusammengedrückt liegengelassen, werden. 3. Versuche im Physikalisch-Chemischen Praktikum I mit Lageplan Pflichtversuche: Thermochemie Versuch Nr. 2: Verbrennungsenergie Chemische Kinetik Versuch Nr. 16: Inversion von Rohrzucker Transport von Ionen und Diffusion von Molekülen Versuch Nr. 25: Moleküle in Bewegung Elektrochemisches Gleichgewicht Versuch Nr. 24: Nernst’sche Gleichung Wahlversuche: Eigenschaften von idealen und realen Gasen Versuch Nr. 22: Wärmekapazität von idealen Gasen oder Versuch Nr. 9: Joule-Thomson-Effekt Zustandsänderungen reiner Substanzen Versuch Nr. 1: Verdampfungsenthalpie von Wasser oder Versuch Nr. 6: Verdampfungsenthalpie von Aceton Zustandsänderungen einfacher Mischungen Versuch Nr. 7: Gefrierpunktserniedrigung oder Versuch Nr. 8: Schmelzdiagramm Chemisches Gleichgewicht Versuch Nr. 10: Homogenes Dissoziationsgleichgewicht oder Versuch Nr. 11: Heterogenes Dissoziationsgleichgewicht Chemisches Gleichgewicht unter Beteiligung von Grenzflächen Versuch Nr. 5: Adsorption oder Versuch Nr. 23: Oberflächenspannung von binären Lösungen Aufbau der Materie und Spektroskopie Versuch Nr. 26: Lichtabsorption von Farbstoffmolekülen oder Versuch Nr. 21: Wasserstoffatom Versuchs-Lageplan Praktikum I 3.1. Wärmekapazität idealer Gase (Versuch Nr. 22) Ziel des Versuches ist, die Wärmekapazitäten cV und cp von Luft zu bestimmen. Dabei wird angenommen, dass Luft sich unter den Versuchsbedingungen wie ein ideales Gasgemisch verhält. 3.1.1. Vorbereitung • • • • • Thermodynamik idealer / realer Gase Gibbs’sche Fundamentalgleichungen Hauptsätze der Thermodynamik Kalorimetrie Gaskinetische Erklärung der molaren Wärmekapazität bei 1-atomigen idealen Gasen • Bewegungsfreiheitsgrade von Molekülen / Gleichverteilungssatz 3.1.2. Literatur P. W. Atkins, Physikalische Chemie, 2. Auflage, VCH-Verlag, Weinheim, 1996, S. 23-46, 53-72, 153-161, 664-669. G. Wedler, Lehrbuch der Physikalischen Chemie, 4. Auflage, Wiley-VCHVerlag, Weinheim, 1997, S. 10-11, 13-22, 23-33, 69-75, 80-88, 272-283, 744753. 3.1.3. Theoretische Grundlagen 1) Prinzip der Messung 1a) CV-Bestimmung: Im Experiment werden die Stoffmenge n und die Zusammensetzung der Luft konstant gehalten. Daher hängt die Innere Energie U der Luft nur von zwei Variablen ab. Wählt man z. B. die Temperatur T und das Volumen V als unabhängige Variable, so ergibt sich U = U(T,V). Die Änderung der Inneren Energie U bei einer Zustandsänderung wird durch das totale Differential U U dU dT dV CV dT dV T V V T (1) beschrieben, wobei CV = (∂U/∂T)V die Wärmekapazität bei konstantem Volumen und π = (∂U/∂V)T der sog. innere Druck des Gases ist. Dieser lässt sich mit Hilfe des Schwarz’schen Satzes (Wedler, 4. Aufl. S. 29) auch ausdrücken als: U p T p V T T V (1a) Durch Einsetzen der idealen Gasgleichung ergibt sich, dass der innere Druck für das ideale Gas verschwindet. Somit hängt die Innere Energie U bei idealen Ga- sen nur von der Temperatur T ab und das totale Differential von U vereinfacht sich zu dU CV dT . (2) Nach dem 1. Hauptsatz der Thermodynamik lässt sich die Innere Energie eines Systems nur durch Wärme- oder Arbeitszufuhr bzw. -abfuhr verändern, d. h. dU Q W , (3) wobei das hier betrachtete Gas als geschlossenes System nur Volumenarbeit δW = −pdV mit der Umgebung austauschen kann. Aus (2) und (3) ergibt sich ein Ausdruck CV dT Q pdV , (4) der als Messvorschrift für CV genutzt werden kann. Der experimentelle Aufbau (siehe Abb. 1) zur Bestimmung der molaren Wärmekapazität cV = CV/n besteht aus einer 1-L-Glasflasche, die Luft enthält. In ihr befinden sich zwei elektrisch leitende Drähte, die als Heizung fungieren. Dabei wird elektrische Energie irreversibel in Wärmeenergie Q UIdt umgewandelt, wobei U die elektrische Spannung ist, die an den Drähten anliegt, und dt das Zeitintervall angibt, in dem die Stromstärke I durch die beiden Drähte fließt. Die aus der Erwärmung resultierende Druckerhöhung wird mit einem Feinmanometer gemessen. Da sich Druckmessungen jedoch experimentell nur schwierig ohne Volumenänderungen vornehmen lassen, muss der pdV-Term in Gleichung (4) berücksichtigt werden. Außerdem muss man für die Temperaturänderung dT in Gleichung (4) neben der Druck- auch die Volumenänderung mit einbeziehen, so dass man für das totale Differential der Temperatur T T dT dV dp p V p V (5) schreibt. In dem im Versuch verwendeten Feinmanometer kann die Volumenänderung aus der Druckänderung berechnet werden, weil dV = adp ist, wobei a eine von der Manometer-Flüssigkeit abhängige Proportionalitätskonstante ist. Bei dem im Versuch verwendeten Spezialöl ist a = 0.855 cm³/hPa. Aus Gleichung (4) erhält man damit für kleine Aufheizzeiten Δt die molare Wärmekapazität cV zu cV CV UI t app UI ap R R n ap V p ap V p t ap V , (6) wobei die entsprechenden Druckänderungen Δp am Feinmanometer abzulesen sind. 1b) Cp-Bestimmung: Am zweckmäßigsten stützt man sich auf das totale Differential der Enthalpie H = H(T, p) H H dH dT dp CP dT dp , T p p T (7) wobei Cp = (∂H/∂T)p die Wärmekapazität bei konstantem Druck und ε = (∂H/∂p)T den sogenannten isothermen Drosseleffekt bezeichnet. Dieser verschwindet wie der innere Druck π für ideale Gase: H V V T T V p T (7a) Somit hängt auch die Enthalpie bei idealen Gasen ausschließlich von der Temperatur ab und das totale Differential für H vereinfacht sich zu dH C p dT . (8) Mit Hilfe des 1. Hauptsatzes und der Definition der Enthalpie kann man die Änderung der Enthalpie dH auch schreiben als dH dU d ( pV ) Q pdV d ( pV ) dQ Vdp . (9) Somit erhält man C p dT Q Vdp . (10) Dabei ist dQ = UIdt wiederum die elektrisch erzeugte Wärme. Der Versuchsaufbau bei der Cp-Bestimmung besteht aus einem vertikal aufgebauten Kolbenprober, der über einen Dreiwegehahn mit der 1-L-Glasflasche verbunden ist (siehe Abb. 3). Die Temperaturänderung dT des Gases wird nun bei konstantem Druck durch die Bewegung des Kolbens im vertikalen Kolbenprober als Volumenänderung dV gemessen. Mit der Temperaturänderung dT p T dT dV dV nR V p (11) erhält man somit dQ UIdt C p dT C p p dV . nR (12) Für kleine Heizzeiten Δt ergibt sich damit die molare Wärmekapazität cp zu cp Cp n R UI , p V t (13) wobei ΔV die gemessene Volumenänderung und p = pA − pG der konstante, um den Druck pG = mg/AK verminderte atmosphärische Druck pA ist. Der zu berücksichtigende Druck pG kommt durch ein am Kolbenprober befestigtes Gewicht der Masse m zustande. Dabei bezeichnet g die Erdbeschleunigung und AK = 7.55.10−4 m2 die Kolbenquerschnittsfläche. 2) Differenz von cp und cV bei idealen Gasen Zunächst erhält man aus der Definition von Cp und H die Beziehung H U V Cp p . T p T p T p (14) Mit Hilfe der Kettenregel kann man umformulieren in U U T U V V CV . T p T V T p V T T p T p (15) Da für ideale Gase π = 0 ist, ergibt sich U U CV , T T p V (16) und mit Gleichung (14) erhält man V C p CV p CV nR bzw. c p cV R . T p (17) 3) Gleichverteilungssatz und angeregte Freiheitsgrade Der Gleichverteilungssatz besagt, dass die Energie eines Systems im Gleichgewicht gleichmäßig auf alle Freiheitsgrade verteilt ist. Daraus folgt, dass auch zur Wärmekapazität jeder Freiheitsgrad aus Translation, Rotation und Vibration den gleichen Anteil beiträgt. Jedem Term, der quadratisch in die Energie eingeht, kommt ein Betrag von R/2 für die Wärmekapazität cV zu. Daher muss jede Schwingung doppelt gezählt werden (Evib = mv2/2 + kx2/2). Tatsächlich gilt der Gleichverteilungssatz streng nur bei sehr hohen Temperaturen, wenn alle Freiheitsgrade angeregt sind. Dies ist eine Folge der Energiequantelung; bei kontinuierlicher Energie stimmt er exakt. Daher trägt die Translation (quasikontinuierliche Energie) schon bei sehr tiefen Temperaturen mit R/2 zur Wärmekapazität bei (vgl. Wedler, 4. Auflage, S. 84ff.). Die Verallgemeinerung des Gleichverteilungssatzes, dass jeder zugängliche Mikrozustand eines Systems im Gleichgewicht gleich wahrscheinlich ist, gilt exakt (vgl. statistische TD). Tabelle 1: Anzahl der Freiheitsgrade und Wärmekapazität Freiheitsgrade der Anzahl der Trans- RotaAtome lation tion 1 2 3, gestreckt 3, gewinkelt N, gewinkelt 3 3 3 3 3 — 2 2 3 3 Schwingung — 1 4 3 3N − 6 Gesamtzahl der quadratischen CV/R Beispiel Freiheitsgrade 3 1.5 He, Ar 7 3.5 N2, O2 13 6.5 CO2 12 6.0 H2O 6N − 6 3N − 3 CH4 3.1.4. Aufgaben 1) Man bestimme die molaren Wärmekapazitäten bei konstantem Volumen cV und bei konstantem Druck cp von Luft. 2) Man vergleiche die Differenz der experimentell bestimmten Werte von cV und cp mit dem für ideale Gase von der Thermodynamik geforderten Ergebnis. 3) Man diskutiere die experimentell gefundenen Einzelwerte von cV und cp im Zusammenhang mit Literaturwerten und berechneten Werten, die sich näherungsweise unter Verwendung des Gleichverteilungssatzes und dem Abzählen von angeregten Freiheitsgraden der Gasmoleküle ergeben. 3.1.5. Zubehör 1-L-Gasflasche mit eingebauter Heizung, Feinmanometer, Digitalzähler, Stromund Spannungsmessgerät, 1x 100-mL-Kolbenprober. 3.1.6. Durchführung a) Bestimmung von cV: Die 1-L-Glasflasche wird mit dem Feinmanometer mittels eines Dreiwegehahns verbunden. Abb. 1: Glasflasche mit Feinmanometer. Anschließend wird der Digitalzähler mit der Taste „Function“ in die Stellung „Timer“ gebracht. Mit der „Trigger“-Taste wählt man die unterste Einstellung. Um das Zeitintervall Δt zu messen, wird die „Reset“-Taste bedient und 1 Mal auf Start gedrückt. Es leuchtet nun eine grüne Lampe auf. Durch kurzes Antippen des Schalters (Δt < 1 s) fließt der Heizstrom und erwärmt die Luft. Es werden die Druckerhöhung Δp und die Zeitdauer Δt gemessen. Abb. 2: Schaltplan für elektrische Heizung. Die Messung wird insgesamt 20-mal mit verschiedenen Zeitintervallen, die alle jeweils kleiner als 1 Sekunde sein sollen, durchgeführt. Die Auftragung von Δp gegen Δt ergibt eine Gerade, aus deren Steigung man cV bestimmen kann. Dazu muss allerdings noch die Spannung U, die Stromstärke I und der Luftdruck p gemessen werden. b) Bestimmung von cp: Statt des Feinmanometers wird an die Glasflasche ein Kolbenprober über den Dreiwegehahn angeschlossen. Der Kolbenprober soll dabei vertikal ausgerichtet sein. Abb. 3: Glasflasche mit Kolbenprober. Zu Beginn des Versuchs wird der Kolbenprober bei offenem Dreiwegehahn soweit nach oben gedrückt, bis keine Luft mehr im Kolbenprober vorhanden ist. Dann wird der Dreiwegehahn so eingestellt, dass nur noch eine Verbindung zur Glasflasche besteht. Der Digitalcounter wird messbereit eingestellt. Der Kolben des Kolbenprobers wird nun in Drehung versetzt, so dass die Reibung möglichst gering ist. Ist der Versuch so weit vorbereitet, werden insgesamt 20 Messungen mit verschiedenen Heizzeiten Δt < 1 s durchgeführt. Dabei muss die Volumendifferenz ΔV vor und nach dem Betätigen des Digitalcounters gemessen werden. Aus der mittleren Steigung in der ΔV/Δt-Auftragung kann man cp bestimmen. Sollte der Kolben während einer Messung zur Ruhe kommen, so ist diese Messung ungültig, da die gemessene Volumenänderung ΔV zu gering ist. Für die Auswertung muss U, I, der äußere Luftdruck pA und die Masse m des Kolbens mit dem Gewichtsstückchen bestimmt werden. 3.1.6. Praktische Hinweise - Die Heizdrähte dürfen die Wand der Glasflasche auf keinen Fall berühren. - Der Stopfen zur Durchführung der Heizdrähte muss luftdicht abschließen. 3.2. Joule-Thomson-Effekt (Versuch Nr. 9) Das Ziel des Versuches ist, den Joule-Thomson-Koeffizienten JT für drei verschiedene Gase experimentell zu bestimmen. 3.2.1. Vorbereitung • • • • • • • Ursache des Joule-Thomson-Effektes Thermodynamik idealer / realer Gase Hauptsätze der Thermodynamik Gibbs’sche Fundamentalgleichungen Van der Waals / Virialgleichung Technische Bedeutung des Joule-Thomson-Effektes Bewegungsfreiheitsgrade von Molekülen / Gleichverteilungssatz 3.2.2. Literatur P. W. Atkins, Physikalische Chemie, 2. Auflage, VCH-Verlag, Weinheim, 1996, S. 23-46, 91-102, 153-161, 664-669. G. Wedler, Lehrbuch der Physikalischen Chemie, 4. Auflage, Wiley-VCHVerlag, Weinheim, 1997, S. 13-22, 23-33, 59-75, 80-88, 232-243, 248-252, 744-753. 3.2.3. Theoretische Grundlagen 1) Einleitung Der Joule-Thomson-Prozess ist eines der wichtigsten technischen Verfahren, um Gase abzukühlen. Im Linde-Verfahren wird er genutzt, um Luft zu verflüssigen. Bei dem Prozess wird ein komprimiertes Gas über eine Drossel adiabatisch ins Vakuum expandiert. Dabei werden attraktive (repulsive) Wechselwirkungen zwischen den Gasmolekülen aufgebrochen. Die hierfür nötige Energie muss aufgrund der adiabatischen Prozessführung aus dem Gas selbst stammen; es kühlt sich daher ab (wärmt sich auf). Da beim idealen Gas keine Wechselwirkungen zwischen den Gasmolekülen existieren, ist bei ihnen kein JouleThomson-Effekt zu erwarten. 2) Prinzip des Joule-Thomson-Prozesses Das Prinzip des Joule-Thomson-Prozesses ist in Abb. 1 dargestellt. Ein Gas befinde sich in einem thermisch isolierten Gefäß des Volumens V1 mit dem Druck p1 (vgl. Abb. 1-1). Durch eine poröse Trennwand strömt das Gas langsam in ein zweites Behältnis mit dem Innendruck p2. Die Kolben werden so bewegt, dass während des ganzen Prozesses p1 und p2 konstant bleiben. 1) p1, V1 2) p1 p2 3) p2, V2 Abb. 1: Schematische Darstellung des Joule-Thomson-Prozesses. Betrachtet man bei dieser Prozessführung die Änderung der inneren Energie dU = δQ + δW, so ändert sich diese nur aufgrund der mit den Kolben zugeführten/entnommenen Arbeit (adiabatisches Gefäß). Es gilt also: dU p1dV1 p1dV2 (1) Es sei an dieser Stelle noch erwähnt, dass diese einfache thermodynamische Diskussion nur bei extrem langsamer (quasistatischer) Prozessführung anwendbar ist; dies kann durch eine sehr schwer durchlässige Membran bewerkstelligt werden. Betrachtet man nun den Unterschied der inneren Energie von Zustand 1) und 3) in Abb. 1 so ergibt sich 0 V2 U p1dV1 p2dV2 p1V1 p2V2 . V1 (2) 0 Da die gesamte betrachtete Stoffmenge von Behältnis 1 zu Behältnis 2 übergegangen ist, kann geschrieben werden: U U 2 U1 p1V1 p2V2 (2a) H1 H 2 (2b) Das heißt, beim Joule-Thomson-Prozess bleibt die Enthalpie konstant, es handelt sich um einen isenthalpischen Effekt. Damit verschwindet die Änderung der Enthalpie dH. Für das totale Differential der Enthalpie gilt also: H H dH dT dp C p dT dp 0 p T p T (3) Hierbei sind Cp und ε die Wärmekapazität bei konstantem Druck bzw. der isotherme Drosseleffekt. Durch Umformung erhält man den Joule-ThomsonKoeffizient, der die Änderung der Temperatur mit dem Druck bei konstanter Enthalpie beschreibt: T Cp p H JT (4) Wie schon in Versuch 22 (S. 18) beschrieben, lässt sich der isotherme Drosseleffekt auch ausdrücken als: V V T T p (5) Betrachtet man in Zähler und Nenner von Gleichung (4) molare Größen und setzt Gleichung (5) ein, so erhält man für den Joule-Thomson-Koeffizienten: v T v T p JT . cp (6) Aus dieser Gleichung ist ersichtlich, dass der Joule-Thomson-Koeffizient für ideale Gase verschwindet. Bei realen Gasen kann JT je nach Temperatur und Druck verschiedene Vorzeichen besitzen. Das Vorzeichen von JT ist unmittelbar mit dem Vorzeichen von ε verknüpft. Ist ε negativ, so kühlt sich das Gas bei Expansion (p1 > p2) ab, für positive ε-Werte erwärmt es sich. Wie in der Einleitung beschrieben, hängt dies mit attraktiven bzw. repulsiven Wechselwirkungen der Teilchen zusammen. Um reale Gase zu beschreiben, kann man in (die exakt gültige) Gl. (6) Näherungsformeln für das molare Volumen v einsetzen, z.B. die van-der-WaalsGleichung oder einen Virial-Ansatz. Für den vereinfachten van-der-WaalsAnsatz gilt: a pv RT b RT p (7) Hierbei sind a und b die van-der-Waals-Parameter (a/v2…Binnendruck, b…Ausschlussvolumen). Damit ergibt sich für JT: 2a b RT JT cp (8) Aus dieser Formel ist direkt ersichtlich, dass der Joule-Thomson-Koeffizient einen Vorzeichenwechsel bei einer bestimmten Temperatur hat. Praktisch hat das zur Folge, dass man, will man Gase verflüssigen, unterhalb einer bestimm- ten Temperatur sein muss, da ansonsten Erwärmung auftritt. Bis zu dieser Temperatur muss also ggf. durch ein anderes Verfahren gekühlt werden. 2) Gleichverteilungssatz und angeregte Freiheitsgrade Siehe gleichnamiger Absatz in Versuch 22 (S. 21). Aus der Tabelle in diesem Absatz kann die Wärmekapazität bei konstantem Druck cpid = cVid + R für das ideale mehratomige Gas abgeschätzt werden, um den Nenner in Gleichung (8) näher zu definieren. 3) Berechnung von cp für reale Gase Natürlich interessieren uns hier reale Gase; um die Druckabhängigkeit der Wärmekapazität realer Gase zu erfassen (bei einem idealen Gas verschwindet sie selbstredend), geht man von Gleichung (3) aus. Da es sich bei dH um ein totales Differential handelt, ist der Satz von Schwarz erfüllt, d. h. es gilt: c p p T T p (9) Mit Hilfe der vereinfachten van-der-Waals-Gleichung (7) kann man nun die Temperaturabhängigkeit von ε berechnen. Einsetzen in (9) und anschließende Integration liefert c p cidp 2a p, RT 2 (10) wobei die Integrationsgrenzen so gewählt wurden, dass man für p 0 die Wärmekapazität des idealen Gases erhält. Man erhält also für den Joule-Thomson-Koeffizient: 2a b RT JT 2a cidp p RT 2 (11) 3.2.4. Aufgaben 1) Aus der Differenz zwischen Mess- und Nulllinie werden für die untersuchten Druckdifferenzen Δp = p1 − p2 die dazugehörigen Temperaturänderungen ΔT = T1 T2 abgelesen. Damit kann man den Joule-Thomson-Koeffizienten JT der drei untersuchten Gase für jede Druckdifferenz Δp gemäß T T JT . p H p H berechnen. Durch Mittelwertbildung bestimmt man anschließend den mittleren Joule-Thomson-Koeffizienten im untersuchten Druckbereich. 2) Man vergleiche die experimentell bestimmten Werte mit Literaturdaten. Außerdem diskutiere man die in den unterschiedlichen Werten für JT zum Ausdruck kommenden nicht-idealen Eigenschaften der Gase im Vergleich zueinander. 3) Man berechne JT für die drei untersuchten Gase mit Hilfe der vereinfachten van-der-Waals-Gleichung, indem man aus der Literatur Werte für a, b und cp verwendet. Anschließend diskutiere man die berechneten JT-Werte zusammen mit den Literaturwerten. 4) Für ideale Gase kann man mit Hilfe einer einfachen Faustregel, die vom Gleichverteilungssatz und dem Abzählen von angeregten Freiheitsgraden Gebrauch macht, die Werte von cV und cp abschätzen. Wie groß sind die entsprechenden Werte der molaren Wärmekapazitäten bei den drei untersuchten Gasen? Berechnen Sie mit Hilfe der vereinfachten van-der-Waals-Gleichung die Abweichung von dem idealen Verhalten für cp bei Stickstoff (N2). Diskutieren Sie diese Werte in Zusammenhang mit dem Literaturwert. Berechnen Sie ausgehend vom Literaturwert der Wärmekapazität cp von CO2 die Art und Anzahl der im Kohlendioxidmolekül bei Raumtemperatur angeregten Freiheitsgrade. 3.2.5. Zubehör Joule-Thomson-Zelle, Badthermostat mit Kühlaggregat, Thermostatisierspirale, Dosenmanometer, Schreiber, Reduzierventil mit Gasleitung. 3.2.6. Versuchsbeschreibung und Durchführung Das zu untersuchende Gas wird mit Hilfe eines in der Seitenwand fest installierten Reduzierventils V entnommen und strömt, bevor es die eigentliche JouleThomson-Zelle Z erreicht, zum Zwecke der Thermostatisierung durch eine aus langem Kupferrohr gebundene Spirale S, die sich in einem Wasserbad B bei Raumtemperatur befindet. Abb. 2: Versuchsaufbau zum Joule-Thomson-Prozess. Innerhalb der Zelle expandiert das Gas durch eine poröse Glasfritte G von seinem konstanten Anfangsdruck p1 auf den als konstant zu betrachtenden Druck p2, der gleich dem Druck der umgebenden Atmosphäre ist, mit der die obere Zelle über die Öffnung H in Verbindung steht. Der Druck p1, der nicht mehr als 800 mbar über dem Atmosphärendruck p2 liegen darf, wird mit Hilfe des Reduzierventils V vorgegeben und am Dosenmanometer M abgelesen (zur Funktionsweise eines Dosenmanometers siehe Seite 8). Direkt an der Unterseite der Glasfritte befindet sich das Thermoelement T1 zur Messung der Gastemperatur vor der Expansion abgelesen (zur Funktionsweise eines Thermoelementes siehe Seite 10). Das unmittelbar oberhalb der Trennwand angebrachte Thermoelement T2 misst die Temperatur des Gases nach der Expansion. Die Temperaturdifferenz ΔT zwischen den beiden Thermoelementen wird von einem Schreiber R aufgezeichnet. Nachdem man mit Hilfe des Reduzierventils einen Druckabfall von 800 mbar eingestellt hat, wird am Schreiber eine Messlinie aufgezeichnet. Wenn die Messlinie einen konstanten Verlauf zeigt, wird der Druck am Dosenmanometer abgelesen. Dann kann der Schreiber kurzgeschlossen werden, um die Nulllinie aufzuzeichnen. Danach wird der nächste Wert für den Druck eingestellt und die Messung wiederholt. Insgesamt werden pro Gas 7 Messwerte zwischen 200 und 800 mbar aufgenommen. Als Gase werden Wasserstoff (H2), Stickstoff (N2) und Kohlendioxid (CO2) verwendet. 3.2.7. Praktische Hinweise - Da die Skalenteile des Schreibers nicht mit der Graduierung des Papiers übereinstimmen, ist es am einfachsten, den Vollausschlag des Schreibers (0 100 Skalenteile) mit dem Lineal auszumessen und ebenso die gemessenen Spannungsdifferenzen. Man erhält den Differenzwert zwischen den beiden Thermospannungen dann gemäß folgender Beziehung Messwert (V) = Messbereich (V) × Messwert (cm) / Vollausschlag (cm). - Die Differenz der Thermospannungen kann dann mit Hilfe eines Proportionalitätsfaktors in die entsprechende Temperaturdifferenz ΔT umgerechnet werden. - Zur Druckeinstellung am Reduzierventil sind die Anweisungen in der Bedienungsanleitung auf Seite 7 für kleine einzustellende Drücke zu befolgen. Dabei ist beim Entlasten des Reduzierventils allerdings zu beachten, dass vorsichtshalber zuerst das Auslassventil geschlossen und der Schlauch zur Messapparatur abgezogen wird. Erst dann sollte man das Hauptventil zur Gasleitung schließen. 3.3 Verbrennungsenergie einer aromatischen Verbindung (Versuch Nr. 2) Das Ziel des Versuches ist, die Standardmesomerieenthalpie ΔMH° von Anthracen aus der kalorimetrisch gewonnenen Standardverbrennungsenthalpie ΔRH° abzuschätzen. 3.3.1. Vorbereitung • • • • • • Zustandsfunktionen und Zustandsänderungen Hauptsätze der Thermodynamik Thermochemie und Kalorimetrie Satz von Hess Zusammenhang zwischen ΔRH und ΔRU Druck- und Temperaturabhängigkeit von ΔRH, ΔRS, ΔRG. 3.3.2. Literatur P. W. Atkins, Physikalische Chemie, 2. Auflage, VCH-Verlag, Weinheim, 1996, S. 53-84. G. Wedler, Lehrbuch der Physikalischen Chemie, 4. Auflage, Wiley-VCHVerlag, Weinheim, 1997, S. 2-44. 3.3.3. Theoretische Grundlagen 1) Prinzip der Messung Bei diesem Versuch werden in einem Bombenkalorimeter verschiedene organische Verbindungen verbrannt. Der Autoklav (= Bombe) befindet sich in einem thermisch isolierten Wasserbad. Die Temperaturänderung wird gemessen; bei bekannter Wärmekapazität des ganzen Systems (Kalorimeter = Wasserbad + Bombe) kann man daraus die entstandene Reaktionswärme berechnen. Der prinzipielle Aufbau des Kalorimeters ist in Abb. 1 dargestellt. Q Kalorimeter K Bombe B Wasserbad W Abb. 1: Prinzipieller Aufbau eines Kalorimeters. In der Bombe wird eine Reaktion, hier eine Verbrennung, durchgeführt: SS O O2 CO CO2 H OH2O 2 2 2 (1) Dabei sind ν die stöchiometrischen Koeffizienten der Edukte (neg. Vorzeichen) und Produkte (pos. Vorzeichen). Bei der Reaktion werden Bindungen gebrochen und neue geknüpft. Die dabei frei werdende Energie ergibt sich aus den unterschiedlichen molaren Bildungsenergien ui der Edukte und Produkte. Für die entstehende Wärme δQ gilt daher: Q ui dni ui i d RUd i (2) i Hierbei ist ξ die Reaktionslaufzahl der Reaktion. Das Vorzeichen auf der rechten Seite kommt daher, dass Reaktionsenergien sich definitionsgemäß auf die reagierenden Stoffe beziehen. Sind die Produkte energieärmer, so wird Energie abgegeben (negatives Vorzeichen von ΔRU), die an die Umgebung abgegebene Wärme δQ ist allerdings positiv. Durch die entstehende Wärme wird das gesamte Kalorimeter K (Bombe + Wasserbad) erwärmt, da die Bombe mit der Umgebung (Wasserbad) Energie austauschen kann. Das gesamte Kalorimeter K bildet ein abgeschlossenes System, kann also mit der Umgebung weder Energie noch Stoffmenge austauschen. Daher ist die innere Energie des Kalorimeters K konstant: dU K 0 (3) Findet in der Bombe, die Teil des abgeschlossenen Systems ist, eine Reaktion statt, bei der sich die Energien der Edukte von denen der Produkte unterschieden, so kann diese Energieänderung innerhalb des abgeschlossenen Systems ausschließlich durch Temperaturänderung des Systems kompensiert werden: dU K RUd CV ,K dT 0 RUd CV ,K dT (4) Hierbei ist CV,K die Wärmekapazität des gesamten Kalorimeters bei konstantem Volumen. Vernachlässigt man die Temperaturabhängigkeiten von ΔRU und CV,K, so erhält man durch Integration aus Gleichung (3): RU CV ,K T (5) Vereinfachend wurde in der bisherigen Betrachtung angenommen, dass es sich um ideale Mischungen handelt. Ist dies nicht der Fall, müssen die molaren Enthalpien ui in ΔRU durch partielle molare Enthalpien ersetzt werden. 2) Zusammenhang zwischen ΔRU und ΔRH Wie aus Gleichung (1) leicht zu ersehen ist, gilt für ΔRU: U RU V ,T Entsprechend gilt für ΔRH: (5) U R H p ,T (6) Mit der Definition der Enthalpie H = U + pV ergibt sich bei konstantem Druck: H U ( pV ) U R H T , p T , p T , p T , p V p . T , p (6) Bei den hier untersuchten Reaktionen zwischen reinen Phasen (oder idealen Mischphasen) bezeichnet ΔRV = (V/ξ)T,p die bei der Reaktion stattfindende Volumenänderung pro Formelumsatz. Beachtet man, dass der Druck p vom Volumen V und der Reaktionslaufzahl ξ abhängt, dann kann man mit Hilfe der Kettenregel den Ausdruck für (U/ξ)T,p U U U V T , p p ( ,V ) T ,V T , p V T , T , p (7) umformen. Damit erhält man folgenden allgemeinen Zusammenhang U R H RU p RV . V T , (8) In der betrachteten Verbrennungsreaktion kann man in guter Näherung den Einfluss der Molvolumina der kondensierten Phasen auf ΔRV gegenüber den Molvolumina der beteiligten Gasphasen vernachlässigen. Nimmt man zusätzlich an, dass sich die beteiligten Gase unter den Verbrennungsbedingungen ideal verhalten, dann verschwindet die Volumenabhängigkeit der Inneren Energie bei beliebiger Temperatur und Zusammensetzung, d. h. (U/V)T,ξ = 0, und man erhält mit Hilfe des idealen Gasgesetzes: R H RU RT i (9) i 3) Druckabhängigkeit von ΔRH Die Verbrennung der Edukte zu den Produkten findet nicht unter Standardbedingungen, sondern typischerweise bei einem Druck von ca. 10 bar statt. Man muss also auf Standardbedingungen umrechnen, um aus der experimentell bestimmten Verbrennungsenergie ΔRU die Standardverbrennungsenthalpie ΔRH° berechnen zu können, d. h. man muss die Druckabhängigkeit von ΔRH kennen R H hi i . p p T T i (10) Die Druckabhängigkeit von ΔRH setzt sich hierbei additiv aus der Druckabhängigkeit der molaren Enthalpien der an der Reaktion beteiligten Komponenten zusammen. Da die Druckabhängigkeit von kondensierten Phasen vernachlässigt werden kann und vorausgesetzt wurde, dass sich die an der Reaktion beteiligten Gase ideal verhalten, verschwindet für die hier untersuchte Verbrennungsreaktion in guter Näherung die Druckabhängigkeit der Reaktionsenthalpie, d. h. R H R H RU RT i . (11) i 4) Inkrementsystem für (Standard-)Verbrennungsenthalpien Auf empirischem Wege hat man durch Messung der Bildungsenthalpien einer großen Anzahl von organischen Verbindungen gefunden, dass sich die Bildungsenthalpien vieler gasförmiger organischer Moleküle näherungsweise additiv aus Bindungsanteilen zusammensetzen lassen. Diese Bindungsanteile oder Bindungsinkremente sind charakteristisch für eine Bindung (z.B. C−H, C−C, C=C) und unabhängig davon, in welchem Molekül die Bindung vorliegt. Dadurch ergibt sich die Möglichkeit, die gesamte Verbrennungsenthalpie ΔRH von Anthracen aus den Verbrennungsenthalpieanteilen der einzelnen chemischen Bindungen des Anthracen-Moleküls zu berechnen (siehe Tabelle im Anhang). Bei Benzol und aromatischen Molekülen versagt allerdings dieses Vorgehen. Man nennt bei diesen Molekülen die Differenz zwischen der gemessenen und der durch Addition von Bindungsinkrementen berechneten Bildungsenthalpie Resonanz- oder Mesomerieenthalpie ΔMH. Die Mesomerieenthalpie ΔMH von gasförmigem Anthracen ist nach dem Satz von Hess identisch mit der Differenz der gemessenen und über das Inkrementsystem berechneten Verbrennungsenthalpien. Dabei ist allerdings zu berücksichtigen, dass die Inkremente nur für gasförmige Stoffe gelten, im Experiment aber festes Anthracen verbrannt wird, und man somit die Sublimationsenthalpie von Anthracen in der Berechnung von ΔMH berücksichtigen muss. 3.3.4. Aufgaben 1) Bestimmung der Kalorimeterkonstante CV,K durch Verbrennen von Benzoesäure. Den benötigten Wert für ΔRU kann man aus dem Literaturwert für die Standardverbrennungsenthalpie ΔRH° von fester Benzoesäure berechnen. 2) Experimentelle Bestimmung der Verbrennungsenergie ΔRU von festem Anthracen. 3) Berechnung der Standardverbrennungsenthalpie ΔRH° von festem Anthracen aus seiner experimentell bestimmten Verbrennungsenergie ΔRU und Vergleich mit Literaturdaten. 4) Ermittlung der Standardverbrennungsenthalpie ΔRH° von gasförmigem Anthracen mit Hilfe des Satzes von Hess unter Zuhilfenahme der Standardsublimationsenthalpie ΔsubH° von Anthracen. 5) Berechnung der Standardverbrennungsenthalpie ΔRH° von gasförmigem Anthracen aus dem Inkrementsystem für die (Standard)Verbrennungsenthalpien pro Bindung. 6) Berechnung der Standardmesomerieenthalpie ΔMH° von gasförmigem Anthracen aus der Differenz der beiden Werte für ΔRH° in Aufgabe 4 und 5. 3.3.5. Zubehör Kalorimeter mit Berthelot-Malerscher Bombe mit Wasserbad und Rührer, Pastillenpresse, Zünddraht, Zündtrafo, Thermoelement, Schreiber, Vielfachmessgerät, Benzoesäure und Anthracen. 3.3.6. Durchführung Die Kalorimeterkonstante CV,K wird durch Verbrennung von Benzoesäure mit bekannter Verbrennungswärme ΔRU bestimmt. Dazu werden ca. 0,25 g Benzoesäure zu einer Pastille (A) gepresst, wobei ein ca. 10 cm langer Draht (B), der zur Zündung (Aufhebung der Reaktionshemmung) dient, mit eingepresst wird. Abb. 2: Pastille (A) mit eingepresstem Zünddraht (B). Der Zünddraht muss vorher gewogen werden, um die Substanzmasse aus der Gewichtsdifferenz zwischen Draht und fertiger Pastille zu ermittelten (siehe Bedienungsanleitung zum Benutzen der Waagen auf S. 14). Daraus kann man die umgesetzte Stoffmenge Δξ an Benzoesäure berechnen. Die Pastille wird in die kalorimetrische Bombe eingehängt, indem die Enden des Zünddrahtes mit den Elektroden verbunden werden. Anschließend wird die Bombe mit ca. 10 bar O2 gefüllt und in das mit Wasser gefüllte Kalorimeter gestellt. Zur Druckeinstellung am Reduzierventil sind die Anweisungen in der Bedienungsanleitung auf Seite 7 zum Einstellen größerer Drücke zu befolgen. Abb. 3: Berthelot-Mahler’sche Verbrennungsbombe mit gepresster Pastille (A) und Zünddraht (B). Analog wird mit Anthracen als zu untersuchender Substanz verfahren. Beide Substanzen werden sowohl mit dem älteren Versuchsaufbau als auch mit dem neuen Gerät untersucht. Bei dem älteren Kalorimeter wird der Temperaturverlauf (Vorperiode - Zündung - Nachperiode) mit einem Thermoelement gemessen, das an einen Schreiber angeschlossen ist (zur Funktionsweise eines Thermoelementes siehe Seite 10). Das zur Auswertung benötigte ΔT erhält man aus dem Temperaturverlauf durch Extrapolation von Vor- und Nachperiode bei Gleichheit der beiden schraffierten Flächen, wie es in Abb. 4 gezeigt ist. Abb. 4: Temperaturverlauf im Kalorimeter. Bei dem neueren Kalorimeter (Typ C4000) ist zu beachten, dass das Gerät mindestens 1 Stunde vor Versuchsbeginn durch den Assistenten eingeschaltet wird, damit sich konstante thermische Verhältnisse einstellen. Nach dem Einbringen der Kalorimeterbombe und dem Schließen des Deckels erfolgt eine automatische Überwachung des Temperaturverlaufs. Sobald sich die Temperaturdrift innerhalb eines erlaubten Spielraumes bewegt, wird der Bediener durch ein akustisches Signal zum Ablesen der Temperaturanzeige und zum Betätigen der Zündtaste aufgefordert. Nachdem die Taste „Zündung“ betätigt wurde, erfolgt die automatische Überwachung des Temperaturverlaufes. Wenn die Temperaturdrift wieder innerhalb der erlaubten Toleranzgrenzen ist, erfolgt erneut ein akustisches Signal. Die Temperatur muss dann innerhalb von 3 Minuten abgelesen werden. Durch Öffnen des Deckels wird der Versuch beendet. 3.3.7. Praktische Hinweise - Verrußte Quarztöpfchen können mit Aceton gereinigt werden. - Der Draht darf in der Pastille nicht verdreht sein. - Beim Pastillenpressen ist darauf zu achten, dass der Draht in der vorhandenen Nute fixiert wird, damit er nicht flachgepresst wird. - Die Pastillenpresse nicht zu fest anziehen, da sonst der Draht in der Pastille reißt. - Vor dem Verschließen der Kalorimeter-Bombe überprüfen, ob sich die beiden Zuleitungskontakte nirgends berühren und so den Zündstromkreis kurzschließen. - Vor dem Verschließen der Bombe und nach dem Gaseinleiten den Zündstromkreis in der Bombe mit dem Ohm-Meter auf Durchgang prüfen. - Das verwendete Vielfachmessgerät nicht zu lange in Ohm-Stellung lassen, da sich bei zufälliger Berührung der beiden Kontakte die Batterie des Gerätes entlädt. - Vor dem Einleiten des Sauerstoffs wird die Kalorimeterbombe mit Sauerstoff vorgespült. Das Auslassventil der Bombe muss zunächst geschlossen sein. Es wird erst beim Einleiten des Gases vorsichtig für einige Sekunden geöffnet. - Die Kalorimeterbombe sollte bis 3 mm über dem flachen Teil des Deckels im Wasser stehen, wenn sie sich im Kalorimeter befindet. Bei der älteren Apparatur - Darauf achten, dass einer der Zündstecker in den isolierten Kontakt im Deckel der Kalorimeterbombe gesteckt wird. - Wenn bei der Zündung ein Strom von ca. 8 A angezeigt wird, herrscht ein Kurzschluss in der Bombe. - Die Messung darf auf keinen Fall zu früh abgebrochen werden, da man sonst nicht vernünftig extrapolieren kann. (Faustregel: Maximum im ersten Viertel) Beim neuen Kalorimeter - Der isolierte Kontakt muss sich unbedingt vorn befinden, wenn die Bombe in das Kalorimeter eingesetzt wird - Beim Schließen des Deckels nochmals kontrollieren, ob die Zündkontakte richtig auf dem Bombendeckel aufsetzen - Am Ende des Versuchstages das Reduzierventil entlasten. 3.3.8. Anhang Tabelle 1: Verbrennungsenthalpien pro Bindung und Korrekturen für spezielle Strukturelemente nach F. Klages, Chem. Ber. 82, 358 (1949). Bindungstyp/ Strukturelemente C−H C−C C=C C=C C=C C=C C=C C=C Korrektur 5-Ring Korrektur 6-Ring Korrektur für CC C C Konfigurationen alle alle H2C=CH2 RHC=CH2 cis-RHC=CHR trans-RHC=CHR R2C=CHR R2C=CR2 alle alle alle ΔRH (kJ/mol) 226.0 206.4 509.0 498.6 491.4 487.3 484.3 483.1 25.1 4.2 −7.1 3.4 Verdampfungsenthalpie von Wasser (Versuch Nr. 1) Das Ziel des Versuches ist die elektrokalorimetrische Bestimmung der Verdampfungsenthalpie von Wasser. 3.4.1. Vorbereitung • • • • • • • Zustandsänderungen / Fundamentalgleichungen Hauptsätze der Thermodynamik Kalorimetrie und Thermochemie Kirchhoff’scher Satz Phasengleichgewichte / Phasengrenzlinie Phasendiagramme (insbesondere bei Wasser) Kritischer Punkt / Prinzip der übereinstimmenden Zustände (Pictet-Trouton’sche Regel) 3.4.2. Literatur P. W. Atkins, Physikalische Chemie, 2. Auflage, VCH-Verlag, Weinheim, 1996, S. 40-47, 55-72, 139-145, 153-164, 173-188. G. Wedler, Lehrbuch der Physikalischen Chemie, 4. Auflage, Wiley-VCHVerlag, Weinheim, 1997, S. 23-41, 232-248, 271-288, 299-309. 3.4.3. Theoretische Grundlagen 1) Prinzip der Messung Bei diesem Versuch wird Wasser in einem Dewar-Gefäß (adiabatisch) mit Hilfe eines Tauchsieders zum Kochen gebracht. Die eigentliche Messung findet im quasistationären Zustand statt, wenn das Wasser siedet, und die Temperatur konstant bleibt. Es wird weiterhin mit dem Tauchsieder Energie zugeführt. Dies führt nun allerdings nicht mehr zur Erwärmung, sondern ausschließlich zu Verdampfung, d.h. die Stoffmenge flüssigen Wassers wird geringer, die gasförmigen Wassers steigt. In dem Versuchsaufbau wird der entstandene Dampf aufgefangen, kondensiert und gewogen. Die mit dem Tauchsieder zugeführte Energie wird ebenfalls gemessen. Aus diesen Informationen kann man die Verdampfungsenthalpie bestimmen. 2) Theorie Die durch den Tauchsieder zugeführte Wärme kann man aus Strom und Spannung berechnen, Q UIdt (1) wobei U und I Spannung und Strom des Tauchsieders sind. Beim Sieden führt diese Wärme ausschließlich zu Verdampfen von Wasser, d. h. die Stoffmenge des flüssigen Wassers nimmt ab, die des gasförmigen nimmt zu. Da der Druck des Systems bei der Messung im Gleichgewicht mit der Umgebung (Atmosphärendruck) steht, ist es sinnvoll, energetische Änderungen mit Hilfe der Enthalpien H zu beschreiben. Für die Änderung der Enthalpie durch das Verdampfen am Siedepunkt gilt: H H dH l dnl g dng hl dnl hg dng n n (2) wobei hl und hg die molaren Enthalpien der Flüssigkeit und des Dampfes sind und dnl und dng die Stoffmengenänderungen in den beiden Phasen bezeichnen. Da die Stoffmengenänderungen dnl und dng im vorliegenden Experiment nicht unabhängig voneinander sind, kann man Gleichung (2) durch die Änderung der Umsatzvariable dξ= dng = −dnl in dH hld hg d vap H d , (3) umformulieren, wobei ΔvapH =hg − hl die molare Verdampfungsenthalpie ist. Da sich die Enthalpie des Systems nur aufgrund der elektrisch zugeführten Wärme δQ ändert, kann man Gleichung (1) und Gleichung (3) gleichsetzen: UIdt vap H d , (4) Durch Integration erhält man: UI t vap H , (4a) Im Experiment geht ein gewisser Anteil an Wärme verloren (nicht perfekter Dewar, Dampf nicht perfekt im Gleichgewicht mit dem Wasser, sondern etwas wärmer …). Dazu führt man im einfachsten Fall einen konstanten Verlustfaktor C ein und erhält somit UI vap H C. t (5) Die Messung des Umsatzes Δξ pro Zeiteinheit Δt wird im Experiment durch Auskondensieren und anschließendes Auswiegen des gebildeten Wasserdampfes bestimmt. 3) Temperaturabhängigkeit der Verdampfungsenthalpie Die Temperaturabhängigkeit der Verdampfungsenthalpie kann mit Hilfe des Kirchhoff’schen Satzes beschrieben werden: T2 vap H (T2 ) vap H (T1 ) (c gp c lp ) dT (6) T1 Für nicht zu große Temperaturintervalle kann man die Temperaturabhängigkeit der Wärmekapazitäten c gp und c lp in guter Näherung vernachlässigen, so dass man für die Temperaturabhängigkeit der Verdampfungsenthalpie vap H (T2 ) vap H (T1 ) (c gp c lp )(T2 T1 ) (7) erhält. 4) Prinzip der übereinstimmenden Zustände (Pictet-Trouton’sche Regel) Mit Hilfe der kritischen Größen eines realen Gases kann man sogenannte reduzierte Variablen für Druck, Temperatur und Molvolumen einführen und die stoffspezifischen Parameter a und b in der van-der-Waals-Gleichung ersetzen. Als Ergebnis erhält man eine Zustandsgleichung, die frei von stoffspezifischen Parametern ist und eine Art „universelle Zustandsgleichung“ darstellt. Man spricht auch vom Prinzip der übereinstimmenden Zustände und meint damit, dass bestimmte physikalisch-chemische Eigenschaften, wie z. B. der Kompressionsfaktor am kritischen Punkt oder die Verdampfungsentropie, universellen Gesetzmäßigkeiten folgen. Dazu stellt man sich die Verdampfung als einen Übergang von der flüssigen Phase mit dem Molvolumen vl in die gasförmige Phase mit dem Molvolumen vg bei konstanter Temperatur T vor. Betrachtet man also für diesen Übergang die molare Innere Energie u in Abhängigkeit von T und v, so ergibt sich u du dv . v T (8) Bei einem Stoff, der durch die van der Waals-Gleichung beschrieben wird, ist die Volumenabhängigkeit der Inneren Energie durch a u 2 v T v (9) gegeben. Somit erhält man durch Integration entlang der Isothermen vapU u g u l a a , vl vg (10) wobei ΔvapU die Innere Verdampfungsenergie ist. Beachtet man, dass das Molvolumen der Flüssigkeit klein gegenüber dem Molvolumen des Dampfes ist, dann ergibt sich ΔvapU ≈ a/vl. Da sich das Molvolumen der Flüssigkeit näherungsweise durch das Kovolumen b beschreiben lässt, erhält man im Rahmen der van-derWaals’schen Näherung für das Verhältnis der inneren Verdampfungsenergie und der kritischen Temperatur Tk = 8a/(27bR) vapU Tk 27 R, 8 (11) d. h. einen konstanten Wert. Da außerdem die Verdampfungstemperatur Tvap bei p° zur kritischen Temperatur Tk ≈ (5/3)Tvap proportional ist, ergibt sich somit für die Verdampfungsentropie ΔvapS vap S vap H Tvap vapU Tvap 135 R 1 R 7 R 24 (12) ein Wert, der unabhängig von stoffspezifischen Parametern ist. Für alle die Stoffe, für die der van-der-Waals’sche Ansatz eine gute Näherung darstellt, wird dieses universelle Verhalten für die Verdampfungsentropie auch beobachtet. Die experimentellen Werte liegen allerdings in der Regel eher bei ΔvapS ≈ 10R (PictetTrouton’sche Regel). 3.4.4. Aufgaben 1) Aus der Auftragung der zugeführten elektrischen Leistung P = UI gegen die pro Zeiteinheit verdampfte Stoffmenge an Wasser Δξ/Δt bestimmt man den Verlustfaktor C und die Verdampfungsenthalpie ΔvapH. 2) Aus dem gemessenen Luftdruck, der gleich dem Dampfdruck des siedenden Wassers ist, und der experimentell bestimmten Verdampfungsenthalpie ΔvapH kann man mit Hilfe der Clausius-Clapeyron’schen-Gleichung abschätzen, bei welcher Temperatur das Wasser im Experiment verdampft (vgl. Theorie „Verdampfungsenthalpie von Aceton“). Aus der Temperaturabhängigkeit der Verdampfungsenthalpie berechne man dann ΔvapH für eine Verdampfungstemperatur, bei der der Dampfdruck p gleich dem Normaldruck ist, und vergleiche den so erhaltenen ΔvapH-Wert mit Literaturwerten. 3) Man berechne den Wert für ΔvapH mit Hilfe der Pictet-Trouton’schen Regel. Was lässt sich im Vergleich zu den experimentellen Werten über die Tragfähigkeit des Prinzips der übereinstimmenden Zustände bei Wasser aussagen? 3.4.5. Zubehör Dewar-Gefäß, Tauchsieder, Kühler mit Dampfableitungsrohr, Netzgerät für Heizung, Becherglas. 3.4.6. Durchführung Um das Kühlwasser zu kühlen, wird in einem Behälter ein Eis-Wasser-Gemisch bereitet und die Kupfer-Kühlschlange hineingelegt. Dann wird das DewarGefäß bis 6 cm unter dem Rand mit destilliertem Wasser gefüllt. Das Wasser wird mit dem Tauchsieder zum Kochen gebracht (220 V Netzsteckdose). Danach wird der Tauchsieder an das Gleichspannungsnetzgerät angeschlossen und dem siedenden Wasser wird eine zeitlich konstante elektrische Leistung P = UI zugeführt, wobei U und I zu notieren sind. Abb. 1: Versuchsaufbau zur elektrokalorimetrischen Bestimmung der Verdampfungsenthalpie von Wasser. Der entstehende Wasserdampf wird im Kühler vollständig auskondensiert. Die Wassertropfen fängt man in einem Becherglas auf und ermittelt die gebildete Wassermasse Δm durch Wägung (siehe Bedienungsanleitungen zum Benutzen der Waagen auf Seite 14). Pro Leistungseinstellung wartet man 15 Minuten auf die Gleichgewichtseinstellung (gleichmäßig tropfender Kühler) und nimmt dann jeweils 5 Minuten lang 2 Messwerte auf. Dies geschieht bei sechs verschiedenen Leistungseinstellungen zwischen 40 und 80 Watt. Am Ende der Messung darf man nicht vergessen, den herrschenden Luftdruck mit dem Barometer zu messen. 3.5 Verdampfungsenthalpie von Aceton (Versuch Nr. 6) Ziel des Versuches ist, den Dampfdruck von Aceton für verschiedene Temperaturen zu messen und daraus dessen Verdampfungsenthalpie zu bestimmen. 3.5.1. Vorbereitung • • • • • Gibbs’sche Fundamentalgleichungen Allgemeine Gleichgewichtsbedingungen Phasengleichgewichte / Phasengrenzlinie Kirchhoff’scher Satz Zustandsänderungen und Zustandsdiagramme idealer und realer Gase: (p,Vm)-, (pVm,p)-, (p,T)- und (Vm,T)-Auftragung • Kritischer Punkt / Theorem der übereinstimmenden Zustände (Pictet-Trouton’sche Regel) 3.5.2. Literatur P. W. Atkins, Physikalische Chemie, 2. Auflage, VCH-Verlag, Weinheim, 1996, S. 40-47, 139-145, 153-164, 173-188. G. Wedler, Lehrbuch der Physikalischen Chemie, 4. Auflage, Wiley-VCHVerlag, Weinheim, 1997, S. 33-41, 232-248, 271-288, 299-309. 3.5.3. Theoretische Grundlagen 1) Prinzip der Messung Man betrachtet flüssiges Aceton im Gleichgewicht mit gasförmigem Aceton, das sich in einem geschlossenen Behälter bei konstantem Druck p befindet. Der Behälter selbst sei mit einem Wärmebad der Temperatur T in thermischem Kontakt. Die freie Verdampfungsenthalpie ist bei den beiden sich im Gleichgewicht befindenden Phasen gleich null: vapG g l 0 g l (1) Die chemischen Potentiale sich im Gleichgewicht befindender Phasen sind also identisch. Für währendes Gleichgewicht gilt außerdem: dg dl (2) Mit der charakteristischen Gleichung des chemischen Potentials ergibt sich: s g dT v g dp s l dT v l dp (3) Hierbei sind si und vi die molare Entropie bzw. das molare Volumen der Phase i = g,l. Umstellen ergibt die Clapeyron’sche Gleichung: dp s g s l vap S dT v g v l vapV (4) Durch Vernachlässigen des molaren Volumens der flüssigen Phase gegenüber der Gasphase und durch Einsetzen der idealen Gasgleichung vg = RT/p erhält man d ln p vap S dT RT (5) Mit der Gibbs-Helmholtz-Gleichung und der Gleichgewichtsbedingung (Gl. 1) gilt ΔvapH = T∙ΔvapS. Es ergibt sich die Clausius-Clapeyron’sche Gleichung: d ln p vap H dT RT 2 (6) Vernachlässigt man die Temperaturabhängigkeit der Verdampfungsenthalpie, so kann man die Clausius-Clapeyron’sche Gleichung integrieren und es ergibt sich der gesuchte Zusammenhang zwischen dem Dampfdruck und der Verdampfungsenthalpie ln p vap H RT c, (7) wobei c eine Integrationskonstante darstellt. 2) Temperaturabhängigkeit der Verdampfungsenthalpie Die Temperaturabhängigkeit der Verdampfungsenthalpie kann mit Hilfe des Kirchhoff’schen Satzes beschrieben werden: T2 vap H (T2 ) vap H (T1 ) (c gp c lp ) dT (8) T1 Für nicht zu große Temperaturintervalle kann man die Temperaturabhängigkeit der Wärmekapazitäten c gp und c lp in guter Näherung vernachlässigen, so dass man für die Temperaturabhängigkeit der Verdampfungsenthalpie vap H (T2 ) vap H (T1 ) (c gp c lp )(T2 T1 ) (9) erhält. 3) Prinzip der übereinstimmenden Zustände (Pictet-Trouton‘sche Regel) Mit Hilfe der kritischen Größen eines realen Gases kann man sogenannte reduzierte Variablen für Druck, Temperatur und Molvolumen einführen und die stoffspezifischen Parameter a und b in der van-der-Waals-Gleichung ersetzen. Als Ergebnis erhält man eine Zustandsgleichung, die frei von stoffspezifischen Parametern ist und eine Art „universelle Zustandsgleichung“ darstellt. Man spricht auch vom Prinzip der übereinstimmenden Zustände und meint damit, dass bestimmte physikalisch-chemische Eigenschaften, wie z. B. der Kompressionsfaktor am kri- tischen Punkt oder die Verdampfungsentropie, universellen Gesetzmäßigkeiten folgen. Dazu stellt man sich die Verdampfung als einen Übergang von der flüssigen Phase mit dem Molvolumen vl in die gasförmige Phase mit dem Molvolmen vg bei konstanter Temperatur T vor. Betrachtet man also für diesen Übergang die molare Innere Energie u in Abhängigkeit von T und v, so ergibt sich u du dv . v T (10) Bei einem Stoff, der durch die van der Waals-Gleichung beschrieben wird, ist die Volumenabhängigkeit der Inneren Energie durch a u 2 v T v (11) gegeben. Somit erhält man durch Integration entlang der Isothermen vapU u g u l a a , vl vg (12) wobei ΔvapU die Innere Verdampfungsenergie ist. Beachtet man, dass das Molvolumen der Flüssigkeit klein gegenüber dem Molvolumen des Dampfes ist, dann ergibt sich ΔvapU ≈ a/vl. Da sich das Molvolumen der Flüssigkeit näherungsweise durch das Kovolumen b beschreiben lässt, erhält man im Rahmen der van-derWaals’schen Näherung für das Verhältnis der inneren Verdampfungsenergie und der kritischen Temperatur Tk = 8a/(27bR) vapU Tk 27 R, 8 (13) d. h. einen konstanten Wert. Da außerdem die Verdampfungstemperatur Tvap bei p° zur kritischen Temperatur Tk ≈ (5/3)Tvap proportional ist, ergibt sich somit für die Verdampfungsentropie ΔvapS vap S vap H Tvap vapU Tvap 135 R 1 R 7 R 24 (14) ein Wert, der unabhängig von stoffspezifischen Parametern ist. Für alle die Stoffe, für die der van-der-Waals’sche Ansatz eine gute Näherung darstellt, wird dieses universelle Verhalten für die Verdampfungsentropie auch beobachtet. Die experimentellen Werte liegen allerdings in der Regel eher bei ΔvapS ≈ 10R (PictetTrouton’sche Regel). 3.5.4. Aufgaben 1) Aus den experimentell bestimmten Dampfdrücken bei den verschiedenen Temperaturen bestimme man die Verdampfungsenthalpie ΔvapH für Aceton. 2) Man berechne die Verdampfungsenthalpie ΔvapH am Siedepunkt Tvap von Aceton bei p = p° und vergleiche das Ergebnis mit Literaturwerten. 3) Man berechne den Wert für ΔvapH mit Hilfe der Pictet-Trouton’sche Regel. Was lässt sich im Vergleich zu den experimentellen Werten über die Tragfähigkeit des Prinzips der übereinstimmenden Zustände bei Aceton aussagen? 3.5.5. Zubehör Glasapparatur mit Manometer, Ölrotationspumpe, 2 Dewargefäße, Aceton als Kühlbadflüssigkeit, Kühlautomat, Heizung (220 V), 2 Thermometer. 3.5.6. Durchführung Zum Schutz der Ölrotationspumpe (siehe Betrieb und Funktionsweise einer Ölrotationspumpe auf Seite 8) wird die Kühlfalle zwischen dieser und der Apparatur vor Versuchsbeginn mit flüssigem Stickstoff eingekühlt. Die Apparatur wird bis zum Hahn H1 mit der Ölrotationspumpe evakuiert. Dann wird das Messkölbchen soweit mit Aceton gefüllt, dass das Schliffthermometer gut eintaucht, außerdem sollten noch zwei Siedesteinchen hinzugefügt werden. Das Kölbchen wird mit den Spannfedern an der Apparatur befestigt. Die Schliffe müssen gut gefettet sein. Nun wird das Messkölbchen vorsichtig evakuiert. Nach Schließen des Hahnes H1 stellt sich am Manometer ein konstanter Zeigerausschlag (d. h. konstanter Druck) ein, sofern die Apparatur dicht ist und die Temperatur konstant bleibt. Um Luftreste zu entfernen, wird das Messkölbchen noch zweimal kurzzeitig evakuiert. Anschließend wird das Aceton für das Kühlbad in ein Dewargefäß gefüllt und mit dem Kühlautomaten auf −23 °C abgekühlt (Thermometer im Acetonbad beachten). Dann wird der Kühlfinger aus dem Bad entfernt und der Kühlautomat abgestellt. Das Dewargefäß wird unter das Messkölbchen gestellt und mit der Hebebühne auf die richtige Höhe gebracht. Der Messkolben sollte bis ca. 0,5 cm über dem inneren Flüssigkeitsspiegel in das Kühlbad eintauchen. Abb. 1: Messapparatur zur Dampfdruckbestimmung. Wenn sich das Aceton im Messkolben auf die Temperatur des Kühlbades abgekühlt hat, muss es noch einmal evakuiert werden. Nachdem sich der Gleichgewichtsdruck eingestellt hat, kann man den ersten Druckwert am Manometer ablesen. Da das Manometer nur die Differenz zum Luftdruck anzeigt, muss man für die Angabe des absoluten Dampfdruckes den Luftdruck mit dem Barometer im Praktikum messen. Die Temperatur wird nun mit Hilfe der Heizung zunächst um ca. 10 °C, dann (ab −10 °C) um jeweils ca. 5 °C erhöht und der Dampfdruck ermittelt. Im letzten Durchgang wird der Dampfdruck bei Raumtemperatur gemessen. Höhere Temperaturen als Raumtemperatur sind zu vermeiden, da ansonsten Aceton an den kälteren Stellen der Apparatur einkondensiert. Nach Beendigung der Messung wird die Apparatur vorsichtig belüftet. Anschließend wird das Aceton aus dem Messkolben zur Kühlbadflüssigkeit dazugegeben und in die Vorratsflasche zurückgefüllt. Praktische Hinweise - Das Acetonbad muss hin und wieder vorsichtig umgerührt werden, da sich sonst ein Temperaturgefälle darin einstellt. - Temperaturkonstanz im Messkolben ist zu erwarten, wenn die Temperatur des Kühlbades annähernd mit der Temperatur der Messsubstanz übereinstimmt. - Die Heizung darf nicht heiß in das kalte Acetonbad gestellt werden. Sie darf also erst nach dem Eintauchen in das Acetonbad eingeschaltet werden und muss vor dem Herausnehmen wieder ausgeschaltet werden. - Die Rotationspumpe muss vor dem Ausschalten am Hahn H2 belüftet werden, da sonst Öl zurückströmen kann. 3.6. Gefrierpunktserniedrigung (Versuch Nr. 7) Im Versuch wird das Schmelzverhalten von verdünnten wässrigen Lösungen untersucht, um die Molmasse der gelösten Substanzen zu bestimmen. 3.6.1. Vorbereitung • • • • • • Allgemeine Gleichgewichtsbedingungen Phasengleichgewichte bei reinen Stoffen und binären Mischungen Kolligative Eigenschaften Gibbs’sche Phasenregel Schmelzdiagramme von binären Mischungen Thermische Analyse von Schmelzdiagrammen 3.6.2. Literatur P. W. Atkins, Physikalische Chemie, 2. Auflage, VCH-Verlag, Weinheim, 1996, S. 139-145, 153-165, 184-188, 225-230, 239-241. G. Wedler, Lehrbuch der Physikalischen Chemie, 4. Auflage, Wiley-VCHVerlag, Weinheim, 1997, S. 271-290, 299-345, 362-368. 3.6.3. Theoretische Grundlagen 1) Kolligative Eigenschaften und ihre mikroskopische Ursache Kolligative Eigenschaften werden solche Eigenschaften von gelösten Stoffen genannt, die (in erster Näherung) nur von der Anzahl der gelösten Teilchen, nicht aber von deren chemischer oder physikalischer Natur (Masse, intermolekulare Wechselwirkungen) abhängen. Diese Eigenschaft kann genutzt werden, um molare Massen zu bestimmen. Zu den kolligativen Eigenschaften gehören neben der Gefrierpunktserniedrigung, die Gegenstand dieses Versuchs ist, auch die Siedepunktserhöhung und die Osmose (s. Abb. 1). Osmose tritt an einer semipermeablen Membran, bei der an der einen Seite reines Lösungsmittel, auf der anderen Seite Lösungsmittel mit gelöster Substanz vorliegt. Die Membran sei so beschaffen, dass sie das Wasser durchlässt, nicht aber die gelöste Substanz (z. B. NaCl). Pro Zeiteinheit treffen daher auf der Seite mit reinem Wasser mehr Wassermoleküle auf die Membran und passieren sie, als auf der anderen Seite, wo auch Fremdteilchen vorhanden sind, die einen Teil der an die Membran stoßenden Moleküle ausmachen, aber reflektiert werden. Folge ist der osmotische Druck. Ähnlich ist bei der Siedepunktserhöhung die Reduktion des Dampfdrucks die Ursache. Dieser ist dadurch geringer, dass ein Teil der Oberfläche von nichtflüchtigen Fremdteilchen belegt ist. Pro Zeiteinheit treffen also weniger Wasserteilchen mit ausreichender Energie, um in die Gasphase überzugehen, auf die Oberfläche und verlassen die kondensierte Phase. Die Kondensation ist dagegen nicht beeinträchtigt ist. Folge ist ein niedrigerer Dampfdruck, der erst bei höherer Temperatur als im reinen Lösungsmittel den Atmosphärendruck erreicht. Ganz ähnlich ist es bei der Gefrierpunktserniedrigung. In der linken Abbildung ist Wasser bei 0 °C gezeigt. Pro Zeiteinheit verlassen gleich viele Moleküle den Keim, wie angelagert werden. Daneben ist dieselbe Situation mit Fremdteilchen zu sehen. Diese behindern die Anlagerung von Wassermolekülen am Keim, nicht aber das Verlassen von Wassermolekülen aus dem Keim. Folge ist, dass bei dieser Temperatur der Keim schrumpft, das Eis schmilzt also bei 0 °C, der Gefrierpunkt ist erniedrigt. Abb. 1: Kolligative Eigenschaften. 2) Gefrierpunktserniedrigung in ideal verdünnten Lösungen Um die im vorhergehenden Absatz entwickelten (zugegebenermaßen einfachen) Überlegungen mathematisch zu erfassen, müssen zunächst die Modellvorstellungen klarer entwickelt werden. Am einfachsten ist dies im Falle des Phasengleichgewichts Gas/Flüssigkeit. Für das Gas können wir ohne großen Fehler das ideale Gasgesetz zur Beschreibung verwenden und können aus der Gleichgewichtsbedingung Rückschlüsse auf thermodynamische Größen in der Flüssigkeit ziehen. Diese können wir dann auch für das Phasengleichgewicht fest/flüssig benutzen, das eigentlich Gegenstand dieses Versuchs ist. Man betrachte nun also das Phasengleichgewicht Gas/Flüssigkeit einer flüchtigen Komponente bei konstantem Gesamtdruck p. In die flüssige Komponente werden nun nicht-flüchtige Teilchen eingeführt, deren intermolekulare Wechselwirkungen mit den Lösungsmittelmolekülen in der flüssigen Phase sich aber nicht von denen der Lösungsmittelmoleküle untereinander unterscheiden. Der Dampfdruck des Lösungsmittels L kann dann mit dem Raoult’schen Gesetz beschrieben werden: pL xL pL (1) Hierbei sind pL der Dampfdruck der Lösung, xL der Molenbruch des Lösungsmittels und pL* der Dampfdruck des reinen Lösungsmittels. Da Gleichgewicht zwischen flüssiger und fester Phase herrscht, sind die chemischen Potentiale sowie (bei währendem Gleichgewicht) deren totalen Differentiale identisch: Lg Ll (2) d Lg d Ll Die charakteristische Gleichung für das chemische Potential lautet: d sdT vdp (3) Wir wollen nun die Druckabhängigkeit des chemischen Potentials in der Gasphase bei konstanter Temperatur (dT = 0)betrachten. Es ergibt sich mit dem idealen Gasgesetz v = RT/p: d RT dp RTd ln p p (4) Als nächstes wird nun die Änderung des chemischen Potentials in der Gasphase betrachtet, die damit einhergeht, dass sich der Dampfdruck der Lösung im Vergleich zu dem des reinen Lösungsmittels pL ändert. Dies kann einfach durch Integration von Gleichung (4) geschehen: Lg pL Lg pL d pL g g RTd ln p L pL L pL RT ln pL pL pL (5) Da die chemischen Potentiale der Gasphase und der flüssigen Phase im GGW identisch sind (Gl.( 2)), kann man mit Gleichung (1) für das chemische Potential der flüssigen Phase schreiben: Ll Ll RT ln xL (6) Dies ist das gesuchte Ergebnis für die Änderung des chemischen Potentials der flüssigen Phase durch Zugabe von Fremdteilchen. Ll*, das chemische Potential der reinen Phase ist hierbei der Referenzzustand. Bei diesem äußerst wichtigen Ergebnis sollte erwähnt werden, dass es ohne weiteres so abgewandelt werden kann, dass die Vereinfachungen, die wir in den bisherigen Überlegungen gemacht haben, beseitigt werden können, indem der Molenbruch durch die Aktivität ersetzt wird. Durch Kenntnis der Abhängigkeit des chemischen Potentials einer flüssigen Phase bei Zugabe von Fremdteilchen, können wir uns nun dem eigentlichen Problem, dem Phasengleichgewicht fest/flüssig zuwenden. Wir entnehmen Gleichung (6), dass das chemische Potential der flüssigen Phase sich durch Zugabe von Fremdteilchen verringert. Da die feste Phase nur das Lösungsmittel enthält, und keine Fremdatome, bleibt das chemische Potential der festen Phase durch Zugabe von Fremdatomen in die flüssige Phase unberührt. Wir können also schreiben: Ls Ll Ls Ll RT ln xL (7) d Ls d Ll RT d ln xL [Gleichung (7.2) gilt ohne Einschränkungen auch für das Gas/FlüssigkeitsPhasengleichgleichgewicht, wenn man Ls* durch Lg* ersetzt.] Am besten lässt sich die Bedeutung der Gleichungen (3), (6) und (7b) in einem (T)-Diagramm (konstanter Druck, dp = 0) verstehen. Die Temperaturabhängigkeit des chemischen Potentials ist gerade die negative partielle molare Entropie –s (vgl. Gl. (3)). Es gilt für das für die molaren Entropien der Aggregatszustände sg* > sl* > ss*. Ist die flüssige Phase eine Mischphase, so kommt ein zusätzlicher Entropiebetrag hinzu, die molare Mischungsentropie. Durch (neg.) Ableitung von Gl. (6) nach der Temperatur erhält man die molare partielle Entropie der Lösung als sl = sl* − R lnx mit dem zusätzlichen Betrag der Mischungsentropie − R lnx (> 0). Mit diesen Informationen kann ein (T)-Diagramm skizziert werden. Vernachlässigt man die Temperaturabhängigkeiten der molaren partiellen Entropien sg*, sl*, sl und ss*, so können die jeweiligen chemischen Potentiale als Geraden der Steigung –s eingezeichnet werden. Abb. 2: Chemisches Potential und Gefrierpunktserniedrigung bzw. Siedepunktserhöhung. Im nächsten Abschnitt soll der Betrag der Gefrierpunktserniedrigung thermodynamisch abgeleitet werden. Dafür muss die Temperaturabhängigkeit des Phasengleichgewichts von Gl. (7c) bei konstantem Druck (dp = 0) mit Hilfe von Gleichung (3) betrachtet werden: sLsdT sLldT RT d ln xL Mit der molaren Schmelzentropie ΔfusSL* = sLl*−sLs* ergibt sich: fus SL dT RT d ln xL (8) (9) Mit der Gibbs-Helmholtz-Beziehung im Gleichgewicht (ΔfusGL* = 0) ergibt sich: fus H L dT RT d ln xL T (10) fus H L dT d ln xL RT 2 Integration unter Annahme unveränderter Schmelzenthalpie (mit der Randbedingung: Integration von Reinphase zu Phase mit xL): Tfus Tfus L fus H L dT d ln xL RT 2 1 x fus H L RTfus fus H L RTfus (11) ln xL Bezieht man Gl. (11) auf den Stoffmengenanteil der zugegebenen Substanz B, xB, so ergibt sich mit Taylor-Reihenentwicklung erster Ordnung ln(1 − xB) ≈ −xB: fus H L fus H L fus H L xB Tfus 2 RTfus RTfus RTfus (12) Die zweite Vereinfachung ergibt sich aus der Annahme, dass die Gefrierpunkts 0 ist abnahme relativ zum absoluten Gefrierpunkt gering ist. Tfus Tfus Tfus die Differenz der Gefrierpunkte des reinen Lösungsmittels und der Lösung. Zur Bestimmung von molaren Massen aus der Gefrierpunktserniedrigung lässt sich Gl. (12) mit Hilfe der Definition des Molenbruchs zweckmäßig umformen. Der Molenbruch kann für große Verdünnung vereinfacht werden: xB nB n m M B B L nL nB nL M BmL (13) Aus den Gleichungen (12) und (13) ergibt sich: mB M L fus H L Tfus 2 M B mL RTfus (13a) 2 RTfus M L mB MB fus H L Tfus mL 3) Abkühlverhalten von Schmelzen Zur Ermittlung des Schmelzdiagramms verfolgt man das Abkühlverhalten, d. h. die Abkühlgeschwindigkeiten, von Schmelzen verschiedener Zusammensetzung. Betrachtet man zunächst die reine Schmelze A bei einer Temperatur T, die größer ist als die Temperatur der Umgebung TU, dann wird sich die Schmelze durch Abgabe von Wärme an die Umgebung abkühlen. Dabei ist die abgegebene Wärmemenge und damit die Änderung der Temperatur der Temperaturdifferenz T TU zwischen Probe und Umgebung proportional: Cp dT (T TU ) , dt (14) wobei α ein Proportionalitätsfaktor ist. Mit der Anfangstemperatur T0 der Schmelze erhält man durch Integration von Gl. (1) das sogenannte Newton’sche Abkühlungsgesetz T TU (T0 TU )e t C p , (15) das ein exponentielles Abkühlverhalten für die reine Schmelze vorhersagt. Sobald die Probe soweit abgekühlt ist, dass der Gefrierpunkt Tfus,A der reinen Schmelze erreicht wird, bleibt die Temperatur T der Schmelze konstant, weil beim Phasenübergang erster Ordnung die Schmelzenthalpie freigesetzt wird. Erst wenn die gesamte Schmelze erstarrt ist, sinkt die Temperatur T der Probe wieder rasch ab (Abkühlkurve A). Kühlt man eine Lösung mit einem großen Überschuss an Substanz A ab, dann beobachtet man zunächst das gleiche Verhalten wie bei der reinen Schmelze (Abkühlkurve AB). Wenn die Temperatur Tfus,A erreicht wird, beginnt die Komponente A auszukristallisieren, wobei die Temperatur Tfus,A gegenüber der Schmelztemperatur der reinen Substanz erniedrigt ist. Durch das Auskristallisieren von A wird die Lösung in Bezug auf die Komponente B konzentrierter, und der Gefrierpunkt sinkt weiter ab, allerdings langsamer als zuvor. Die Lösung verarmt mit sinkender Temperatur nun solange an der auskristallisierenden Komponente A, bis eine in beiden Komponenten gesättigte Lösung erreicht wird. Dann scheidet sich neben dem Lösungsmittel A auch der gelöste Stoff B ab, und die Temperatur TE der Schmelze bleibt konstant bis sämtliche Substanz in den festen Zustand übergegangen ist. An dieser Stelle befindet man sich im sogenannten eutektischen Punkt E (altgr. εὔτηκτος ‚leicht schmelzend‘), bei dem das binäre System seinen niedrigsten isobaren Schmelzpunkt aufweist und die Schmelze mit zwei verschiedenen Feststoffen im Gleichgewicht steht. Sowohl die eutektische Temperatur als auch die eutektische Zusammensetzung sind druckabhängig. Die Gibbs’sche Phasenregel F = C – P + 2 wird unter isobaren Bedingungen (konstanter Druck) F = C – P + 1, so dass dann der eutektische Punkt ein invarianter Punkt (F = 0) wird, an dem für ein zweikomponentiges System drei Phasen miteinander im Gleichgewicht stehen. Schmelzdiagramm A-B T Abkühlungskurven A, AB, E TT AB A E Tfus,A* AB Schmelze Tfus,A E A(s) + Schmelze B(s) + Schmelze TE A(s)+B(s) B xA xB A Zeit Abb. 3: Isobares Schmelzdiagramm mit dazugehörigen Abkühlungskurven. Geht man umgekehrt von einer Schmelze aus, in der die Komponente B in großem Überschuss vorliegt, so kristallisiert zunächst ausschließlich B aus, bis der eutektische Punkt E erreicht ist und dann neben B auch A ausgeschieden wird. Eine Schmelze, die von vornherein die Zusammensetzung des eutektischen Gemisches besitzt, verhält sich im Abkühlverhalten wie eine chemisch reine Substanz (Abkühlkurve E). Eine solche Mischung ist von einem reinen Stoff durch das Schmelzverhalten nicht zu unterscheiden, erst die Untersuchung der ausgeschiedenen Kristalle zeigt, dass es sich um ein binäres System handelt. Bei der Aufnahme der Erstarrungskurven lässt es sich trotz guter Rührung oft nicht vermeiden, dass die Schmelze unterkühlt wird und die Temperatur deshalb nach Bildung der ersten Kristalle wieder ansteigt. Die Erstarrungstemperatur Tfus lässt sich in diesem Fall bestimmen, wenn man den geradlinigen Teil der Abkühlungskurve bis zum Schnittpunkt mit dem steil abfallenden Ast nach links verlängert. T reiner Stoff / Eutektikum T Lösung Tfus Tfus Zeit Zeit Abb. 4: Auswertung der Erstarrungskurven bei Unterkühlung. Trägt man die Temperatur Tfus, bei der eine der beiden Komponenten gerade auskristallisiert, z. B. in Abhängigkeit vom Molenbruch xA der Komponente A auf, so erhält man das Schmelzdiagramm, d. h. eine Kurve, die für das betrachtete binäre System charakteristisch ist. 3.6.4. Aufgaben 1) Ermitteln Sie aus den gemessenen Abkühlkurven graphisch die Gefrierpunktserniedrigung ΔTfus,A bei Wasser als Lösungsmittel für die untersuchten Stoffe mit den verschiedenen Zusammensetzungen. 2) Durch Auftragen der gemessenen Gefrierpunktserniedrigung ΔTfus,A gegen das Verhältnis aus mB/mA und unter Verwendung des in der Literatur bekannten Wertes für ΔfusHA* bestimme man die Molmassen Mm,B der unbekannten Verbindungen. 3) Vergleichen Sie die experimentell bestimmten Molmassen mit den tatsächlichen Werten. 3.6.5. Zubehör Weites, dickwandiges Reagenzglas mit Stopfen und Rührer, Motor, Thermoelement, Kühlthermostat mit Temperiergefäß, Aceton, Versuchssubstanzen. 3.6.6. Durchführung Das Aceton wird im Temperiergefäß des Kühlaggregates auf ungefähr −15 °C gekühlt. Dann werden 20 mL destilliertes H2O in das Versuchsgefäß eingefüllt und die Versuchsanordnung wie in Abb. 5 aufgebaut. Der Rührer muss sich leicht bewegen lassen, da er sonst leicht verbiegt. Der Schreiber wird so eingestellt, dass der Nullpunkt ungefähr bei 30 Skalenteilen liegt. Zum Messen wird der Messbereich 0,5 mV (cal.) eingestellt. Sollte die Thermospannung (zur Funktionsweise eines Thermoelementes siehe Seite 10) anfangs noch zu groß für diesen Bereich sein, den Messbereich entsprechend höher einstellen, bis sich das Wasser im Versuchsgefäß weit genug abgekühlt hat. Die Papiergeschwindigkeit sollte 2 cm/min betragen. Der Papiervorlauf sollte aber immer erst dann eingeschaltet werden, wenn man den empfindlichsten Messbereich einstellen kann. Abb. 5: Apparatur zur Gefrierpunktserniedrigung. Zunächst wird die Abkühlkurve von reinem Wasser der Masse mA zweimal aufgenommen. Dann wird die empfohlene Einwaagemenge mB (siehe Aufschrift des Probenglases) an Substanz zugegeben und wieder eine Abkühlkurve aufgenommen. Danach wird die Messung noch einmal mit der Hälfte der empfohlenen Einwaagemenge wiederholt. 3.6.7. Praktische Hinweise - Damit sich das Wasser im Versuchsgefäß zwischen den Messungen immer wieder erwärmen kann, wird das Temperiergefäß mit dem Kühlbad abgesenkt. - Bei der Substanzzugabe muss darauf geachtet werden, dass nichts an den Gefäßwandungen haften bleibt. - Die Substanz muss sich restlos aufgelöst haben, bevor mit der Messung begonnen wird. - Beim Auftreten einer Unterkühlung während des Abkühlvorgangs muss die Messung lange genug weitergeführt werden, um den exakten Wert des Gefrierpunktes ermitteln zu können. 3.7. Schmelzdiagramm binärer Mischungen (Versuch Nr. 8) Ziel des Versuches ist die thermische Untersuchung des Schmelzverhaltens von Pb/Sn-Mischungen unterschiedlicher Zusammensetzung, um daraus das Schmelzdiagramm des binären Systems zu konstruieren. 3.7.1. Vorbereitung • • • • • • Allgemeine Gleichgewichtsbedingungen Phasengleichgewichte bei reinen Stoffen und binären Mischungen Kolligative Eigenschaften Gibbs’sche Phasenregel Schmelzdiagramme von binären Mischungen Thermische Analyse von Schmelzdiagrammen 3.7.2. Literatur P. W. Atkins, Physikalische Chemie, 2. Auflage, VCH-Verlag, Weinheim, 1996, S. 139-145, 153-165, 184-188, 225-230, 239-241. G. Wedler, Lehrbuch der Physikalischen Chemie, 4. Auflage, Wiley-VCHVerlag, Weinheim, 1997, S. 271-290, 299-345, 362-368. 3.7.3. Theoretische Grundlagen 1) Abkühlverhalten von Schmelzen Zur Ermittlung des Schmelzdiagramms verfolgt man das Abkühlverhalten, d. h. die Abkühlgeschwindigkeiten, von Schmelzen verschiedener Zusammensetzung. Betrachtet man zunächst die reine Schmelze A bei einer Temperatur T, die größer ist als die Temperatur der Umgebung TU, dann wird sich die Schmelze durch Abgabe von Wärme an die Umgebung abkühlen. Dabei ist die abgegebene Wärmemenge und damit die Änderung der Temperatur der Temperaturdifferenz T TU zwischen Probe und Umgebung proportional: Cp dT (T TU ) , dt (1) wobei α ein Proportionalitätsfaktor ist. Mit der Anfangstemperatur T0 der Schmelze erhält man durch Integration von Gleichung (1) das sogenannte Newton’sche-Abkühlungsgesetz T TU (T0 TU )e t C p , (2) das ein exponentielles Abkühlverhalten für die reine Schmelze vorhersagt. Sobald die Probe soweit abgekühlt ist, dass der Gefrierpunkt Tfus,A der reinen Schmelze erreicht wird, bleibt die Temperatur T der Schmelze konstant, weil beim Phasenübergang erster Ordnung die Schmelzenthalpie freigesetzt wird. Erst wenn die gesamte Schmelze erstarrt ist, sinkt die Temperatur T der Probe wieder rasch ab (Abkühlkurve A). Kühlt man eine Lösung mit einem großen Überschuss an Substanz A ab, dann beobachtet man zunächst das gleiche Verhalten wie bei der reinen Schmelze (Abkühlkurve AB). Wenn die Temperatur Tfus,A erreicht wird, beginnt die Komponente A auszukristallisieren, wobei die Temperatur Tfus,A gegenüber der Schmelztemperatur der reinen Substanz erniedrigt ist. Durch das Auskristallisieren von A wird die Lösung in Bezug auf die Komponente B konzentrierter, und der Gefrierpunkt sinkt weiter ab, allerdings langsamer als zuvor. Die Lösung verarmt mit sinkender Temperatur nun solange an der auskristallisierenden Komponente A, bis eine in beiden Komponenten gesättigte Lösung erreicht wird. Dann scheidet sich neben dem Lösungsmittel A auch der gelöste Stoff B ab, und die Temperatur TE der Schmelze bleibt konstant bis sämtliche Substanz in den festen Zustand übergegangen ist. An dieser Stelle befindet man sich im sogenannten eutektischen Punkt E (griech.: gut schmelzend), bei dem das binäre System seinen niedrigsten isobaren Schmelzpunkt aufweist und die Schmelze mit zwei verschiedenen Feststoffen im Gleichgewicht steht. Sowohl die eutektische Temperatur als auch die eutektische Zusammensetzung sind druckabhängig. Die Gibbs’sche Phasenregel F = C – P + 2 wird unter isobaren Bedingungen (konstanter Druck) F = C – P + 1, so dass dann der eutektische Punkt ein invarianter Punkt (F = 0) wird, an dem für ein zweikomponentiges System drei Phasen miteinander im Gleichgewicht stehen. Schmelzdiagramm A-B T Abkühlungskurven A, AB, E TT AB A E Tfus,A* AB Schmelze Tfus,A E A(s) + Schmelze B(s) + Schmelze TE A(s)+B(s) B xA xB A Zeit Abb. 1: Isobares Schmelzdiagramm mit dazugehörigen Abkühlungskurven. Geht man umgekehrt von einer Schmelze aus, in der die Komponente B in großem Überschuss vorliegt, so kristallisiert zunächst ausschließlich B aus, bis der eutektische Punkt E erreicht ist und dann neben B auch A ausgeschieden wird. Eine Schmelze, die von vornherein die Zusammensetzung des eutektischen Gemisches besitzt, verhält sich im Abkühlverhalten wie eine chemisch reine Substanz (Abkühlkurve E). Eine solche Mischung ist von einem reinen Stoff durch das Schmelzverhalten nicht zu unterscheiden, erst die Untersuchung der ausgeschiedenen Kristalle zeigt, dass es sich um ein binäres System handelt. Bei der Aufnahme der Erstarrungskurven lässt es sich trotz guter Rührung oft nicht vermeiden, dass die Schmelze unterkühlt wird und die Temperatur deshalb nach Bildung der ersten Kristalle wieder ansteigt. Die Erstarrungstemperatur Tfus lässt sich in diesem Fall bestimmen, wenn man den geradlinigen Teil der Abkühlungskurve bis zum Schnittpunkt mit dem steil abfallenden Ast nach links verlängert. T reiner Stoff / Eutektikum T Lösung Tfus Tfus Zeit Zeit Abb. 2: Auswertung der Erstarrungskurven bei Unterkühlung. Trägt man die Temperatur Tfus, bei der eine der beiden Komponenten gerade auskristallisiert, z. B. in Abhängigkeit vom Molenbruch xA der Komponente A auf, so erhält man das Schmelzdiagramm, d. h. eine Kurve, die für das betrachtete binäre System charakteristisch ist. 2) Gefrierpunktserniedrigung in ideal verdünnten Lösungen Am Erstarrungspunkt liegen feste und flüssige Phase im Gleichgewicht vor: Ls Ll d d s L l L (3) Wie in Absatz 1) und 2) des Versuchs „Gefrierpunktserniedrigung“ hergeleitet wird, kann das chemische Potential einer flüssigen Phase A, die mit gelösten Fremdteilchen versetzt ist, beschrieben werden durch: Al Al RT ln aA (4) Al* ist das chemische Potential der reinen Phase und hierbei der Referenzzustand. Gleichung (4) ist zu entnehmen, dass das chemische Potential bei Zugabe von Fremdteilchen sinkt. Im weiteren wird die Aktivität durch den Molenbruch xA ersetzt, was für ideal verdünnte Lösungen gerechtfertigt ist. Mit Gl. (3) und (4) ergibt sich aus der Tatsache, dass nur der reine Feststoff A auskristallisiert: As Al RT ln xA s A l A d d RT d ln xA (5) Durch Zugabe der Fremdteilchen ist eine Gefrierpunktserniedrigung zu erwarten, da das chemische Potential der flüssigen Phase abgesenkt ist (vgl. auch Versuch „Gefrierpunktserniedrigung“). Im nächsten Abschnitt soll der Betrag der Gefrierpunktserniedrigung thermodynamisch abgeleitet werden. Dafür muss die Temperaturabhängigkeit des Phasengleichgewichts von Gl. (5) bei konstantem Druck (dp = 0) mit Hilfe der Fundamentalgleichung des chemischen Potentials d sdT vdp betrachtet werden: sAsdT sAldT RT d ln xA (6) Mit der molaren Schmelzentropie ΔfusSA* = sAl* − sAs* ergibt sich: (7) fus SA dT RT d ln xA * Mit der Gibbs-Helmholtz-Beziehung im Gleichgewicht (ΔfusGA = 0) ergibt sich: fus H A dT RT d ln xA T (8) fus H A dT d ln xA RT 2 Integration unter Annahme unveränderter Schmelzenthalpie mit Randbedingung der Reinphase zur Phase mit xA: Tfus Tfus fus H A dT RT 2 fus H A RTfus xA d ln xA 1 fus H A RTfus (9) ln xA Bezieht man Gleichung (11) auf den Stoffmengenanteil der zugegebenen Substanz B, xB, so ergibt sich: fus H A 1 1 ln 1 xB R Tfus Tfus (10) Eine entsprechende Gleichung kann man aufstellen, um den Einfluss von gelöstem A auf den Gefrierpunkt von B zu erfassen. 3.7.4. Aufgaben 1) Durch Aufnehmen von 2 Abkühlkurven des reinen Zinn wird das verwendete Thermoelement unter der Annahme, dass die Thermospannung im Bereich zwischen 100 °C und der Schmelztemperatur von reinem Zinn linear mit der Tem- peratur zunimmt, kalibriert. Anschließend werden die Abkühlkurven von verschiedenen Sn/Pb-Mischungen gemessen, um daraus das entsprechende Schmelzdiagramm zu konstruieren. 2) Man bestimme die Zusammensetzung des eutektischen Gemisches aus dem Schmelzdiagramm. 3) Das experimentell bestimmte Schmelzdiagramm und die Werte des eutektischen Gemisches werden zusammen mit Literaturdaten diskutiert. 4) Das Schmelzdiagramm für die Pb/Sn-Gemische wird ausgehend von reinem Pb und Sn nach Gleichung (8) aus der Gefrierpunktserniedrigung berechnet. Was lässt sich bei metallischen Schmelzen (flüssigen Legierungen) im Vergleich mit den experimentellen Daten über die Leistungsfähigkeit und Gültigkeit des verwendeten Modells zur Berechnung des Schmelzdiagramms aussagen? 3.7.5. Zubehör Probengefäße mit verschiedenen Pb/Sn-Mischungen, Probenschmelzofen mit Rührmotor, Chromel-Alumel-Thermoelement mit Ofen für 100 °C-Vergleichsmessstelle, Computer mit Drucker. 3.7.6. Durchführung Die beiden Öfen werden 1 Stunde vor Beginn des Versuches durch den Assistenten eingeschaltet. Abb. 3: Apparatur zur Schmelzdiagrammbestimmung. Nachdem die Vergleichsstelle die Temperatur von siedendem Wasser erreicht hat, wird eine Probe im Ofen geschmolzen. Dabei wird der Temperaturanstieg mit dem Thermoelement verfolgt und die Messwerterfassung am Computer gestartet und abgelesen (zur Funktionsweise eines Thermoelementes siehe Seite 10). Allerdings sollen die Daten der Aufheizkurven nicht abgespeichert werden. Nachdem die Probe vollständig geschmolzen ist, wird sie aus dem Ofen genommen und die Abkühlkurve aufgenommen, indem man die Messwerterfassung erneut startet. Die dabei aufgenommenen Werte werden auf Diskette gespeichert, wobei automatisch der Temperaturverlauf auf dem Drucker ausgedruckt wird. 3.7.7. Praktische Hinweise - Die Proben sollten nicht zu stark und nicht zu schwach aufgeheizt werden. Beim Schmelzen kann man sich dabei an der jeweils vorausgehenden Probe orientieren. Der Abstand der Schmelzpunkte liegt bei ca. 1 mV Thermospannung (zwischen Probe 4 und 5 allerdings bei ca. 3 mV). - Das Reagenzglas mit dem Wasser darf niemals aus dem Ofen genommen werden. Wasser braucht erst nachgefüllt zu werden, wenn der Wasserspiegel bis unter den Ofenrand gesunken ist. Zum Nachfüllen von Wasser den Stopfen herausnehmen und das Reagenzglas mit der Spritzflasche bis zur Markierung auffüllen. - Unterkühlungen kann man unter Umständen vermeiden, wenn man dem Probenröhrchen leichte Erschütterungen (mit Hilfe des Rührmotors) zufügt. - Die Bleiprobe sollte zum Schluss gemessen und zur schonenden Abkühlung im wärmeisolierten Becherglas stehen gelassen werden. 3.8. Homogenes Dissoziationsgleichgewicht (Versuch Nr. 10) Ziel des Versuches ist, die Gleichgewichtskonstante für das NO2/N2O4Dissoziationsgleichgewicht bei verschiedenen Temperaturen zu messen, um daraus die Standardreaktionsenthalpie zu bestimmen. 3.8.1. Vorbereitung • Gibbs’sche Fundamentalgleichungen • Chemisches Gleichgewicht • Abhängigkeit des chemischen Potentials von Druck, Temperatur und Zusammensetzung • Zusammenhang zwischen den verschiedenen Gleichgewichtskonstanten • Massenwirkungsgesetz • Druck- und Temperaturabhängigkeit der Gleichgewichtskonstanten (Le Chatelier-Braun’sches Prinzip) • Ideales Gasgesetz / Gesetz von Dalton / Dissoziationsgrad 3.8.2. Literatur P. W. Atkins, Physikalische Chemie, 2. Auflage, VCH-Verlag, Weinheim, 1996, S. 153-165, 195-201, 216-219, 225-230, 255-272. G. Wedler, Lehrbuch der Physikalischen Chemie, 4. Auflage, Wiley-VCHVerlag, Weinheim, 1997, S. 271-290, 299-303, 329-336, 372-391, 406-408. 3.8.3. Theoretische Grundlagen Man betrachte die allgemeine homogene Dissoziationsreaktion A(g) 2B(g) . (1) Die Freie Enthalpie G des Gesamtsystems hängt neben der Temperatur T und dem Druck p auch von den beiden Stoffmengen nA und nB ab. Bei konstantem p und T ergibt sich somit für die Änderung der Freien Enthalpie dG A dnA BdnB . (2) Mit Hilfe der Umsatzvariable (Reaktionslaufzahl) ξ kann man Gleichung (2) umformulieren und erhält dG (2B A ) d . (3) Im chemischen Gleichgewicht ist die Freie Enthalpie G minimal, d. h. ΔRG = (∂G/∂ξ)T,p = 0, so dass sich das chemische Gleichgewicht für die Dissoziationsreaktion auch mit Hilfe der chemischen Potentiale A und B als A 2 B (4) schreiben lässt. Nimmt man an, dass sich die beiden gasförmigen Komponenten A und B in der Mischung ideal verhalten, dann erhält man pA pB 2 RT ln B p . p A RT ln (5) Man kann Gleichung (5) mit der Freien Standardreaktionsenthalpie ΔRG° = 2°B − °A in pB2 (2 B A ) R G ln ln K RT RT p p A (6) umformen, wobei K die dimensionslose sogenannte thermodynamische Gleichgewichtskonstante ist. Nimmt man an, dass zu Beginn der Reaktion eine bestimmte Stoffmenge n0 von A und kein B vorhanden war, dann kann man die Stoffmengen von A und B im Gleichgewicht nA = (1 − α)n0 und nB = 2αn0 durch den Dissoziationsgrad α ausdrücken. Über die Molenbrüche von A und B xA pA 1 p 1 und xB pB 2 p 1 (7) lassen sich die Partialdrücke pA und pB und somit auch die Gleichgewichtskonstante K durch den Dissoziationsgrad α und den Gesamtdruck p pB2 4 2 p K pA p 1 2 p (8) beschreiben. Eine Messung des Dissoziationsgrades α für verschiedene Temperaturen T kommt somit einer temperaturabhängigen Bestimmung von K bzw. von ΔRG° gleich. Aus der Temperaturabhängigkeit der Freien Standardreaktionsenthalpie ΔRG° erhält man die Standardreaktionsentropie −ΔRS° = (∂ΔRG°/∂T)ξ und über die Gibbs-Helmholtz-Gleichung auch die Standardreaktionsenthalpie ΔRH° R H ( R G / T ) . T T2 (9) Mit Hilfe der Gleichgewichtskonstanten K aus Gleichung (6) ergibt sich die van’t Hoff’sche Reaktionsisobare zu R H ln K , RT 2 T (10) aus der man durch Integration unter der Annahme, dass ΔRH° nicht von der Temperatur abhängt, zu R H (11) const. RT gelangt. Dabei ist allerdings zu beachten, dass sich im hier beschriebenen Experiment die Zusammensetzung ξ des Gasgemisches mit der Temperatur T veränln K dert. Somit darf man Gleichung (10) im Allgemeinen nur näherungsweise zur Bestimmung von ΔRH° anwenden, falls chemisches Gleichgewicht vorliegt. Da jedoch im untersuchten Fall davon ausgegangen wird, dass sich die beiden Komponenten im Gasgemisch ideal verhalten, hängt ΔRH° nicht von der Zusammensetzung ξ ab und Gleichung (10) kann ohne Einschränkung zur Bestimmung von ΔRH° verwendet werden. Die Messung des Dissoziationsgrades α erfolgt unter Ausnutzung des idealen Gasgesetzes pV = nRT, wobei n = nA + nB = n0(1 + α) die gesamte Stoffmenge ist. Da das Volumen V des gesamten Gasgemisches bei verschiedenen Temperaturen im Experiment annähernd konstant bleibt, ergibt sich für das Verhältnis des Gesamtdruckes p zu einem Referenzgesamtdruck p∞ bei zwei verschiedenen Temperaturen T und T∞ p (1 )T . p (1 )T (12) Ist die Temperatur T∞ groß genug, dann liegt das chemische Gleichgewicht bei endothermen Reaktionen, wie z. B. der N2O4-Dissoziation, praktisch vollständig auf der Seite von B, d. h. α∞ = 1. Somit kann man den Dissoziationsgrad α 2 pT 1 pT (13) und gemäß Gleichung (8) auch die Gleichgewichtskonstante K für beliebige Temperaturen T < T∞ bestimmen. 3.8.4. Aufgaben 1) Man berechne Kp und Kx für das NO2/N2O4-Dissoziationsgleichgewicht und zeige, wie Kp und Kx mit der thermodynamischen Gleichgewichtskonstante K zusammenhängen. 2) Man trage den Gesamtdruck p gegen die Temperatur T auf. Man zeichne in die Abbildung den hypothetischen Verlauf der Drücke von reinem NO 2 und N2O4 mit ein. Wie hängt die Differenz zwischen dem Gesamtdruck p und dem hypothetischen Druck von reinem N2O4 bzw. NO2 mit dem Dissoziationsgrad α zusammen? Außerdem zeichne man bei einer Temperatur in der Graphik ein, wie groß die Partialdrücke von NO2 und N2O4 sind. 3) Man nehme an, dass bei der größten Temperatur im Experiment die Dissoziation in NO2 vollständig ist und berechne den Dissoziationsgrad α und die thermodynamische Gleichgewichtskonstante K für alle Temperaturen T < T∞. 4) Man trage ln(K) gegen 1/T auf und berechne aus der Steigung die Standardreaktionsenthalpie ΔRH° für die Dissoziation von N2O4 in NO2 und vergleiche das Ergebnis mit Literaturdaten. Hat die Temperaturabhängigkeit von ΔRH° einen großen Einfluss auf Ihr Ergebnis? 3.8.5. Zubehör Vakuumapparatur mit Quarzspiralmanometer, Ölrotationspumpe, Rührer, Heizung, Heizregler, Transformator, Dosenmanometer, Thermometer. 3.8.6. Durchführung Die Hauptmenge des N2O4 befindet sich in der Doppelspirale D, die durch ein Glasrohr mit dem Quarzspiralmanometer MQ verbunden ist. Durch Druckunterschiede zwischen dem Inneren der Quarzspirale und dem umgebenden Raum weitet sich die Spirale auf bzw. zieht sich zusammen. Durch vorsichtiges Evakuieren oder Belüften der Spirale mit dem Dreiwegehahn werden die Zeiger des Quarzspiralmanometers genau übereinander gestellt. Dann ist der Druck innen und außen gleich und kann am Differenzdruckmanometer M abgelesen werden. Das Thermometer hängt frei im Zentrum der Doppelspirale. Die gesamte Versuchsanordnung befindet sich in einem Silikonölbad, das mit dem Rührer R durchmischt und über die elektrische Heizung E erwärmt werden kann. Abb. 1: Dissoziationsgleichgewicht von N2O4 und NO2. Der Gesamtdruck p wird zuerst bei Raumtemperatur bestimmt, dann werden am Heizregler ca. 8 Temperaturen zwischen 40 und 180 °C eingestellt. Wenn die Temperatur konstant ist, wird das Quarzspiralmanometer MQ abgeglichen. Nach 10 bis 15 Min. kann der Gleichgewichtsdruck p abgelesen werden, wenn der Zeiger des Manometers MQ keinen Ausschlag mehr anzeigt. 3.8.7. Praktische Hinweise - Nachdem alle Drücke für die zu untersuchenden Solltemperaturen gemessen wurden, wird der Temperatur-Regler auf einen Wert < 15 °C gestellt und ausgeschaltet. - Bevor man die Ölrotationspumpe abschaltet, muss sie unbedingt belüftet werden. 3.9. Heterogenes Dissoziationsgleichgewicht (Versuch Nr. 11) Ziel des Versuches ist, die Gleichgewichtskonstante für ein heterogenes Dissoziationsgleichgewicht bei verschiedenen Temperaturen zu messen, um daraus die Standardreaktionsenthalpie zu bestimmen. 3.9.1. Vorbereitung • Gibbs’sche Fundamentalgleichungen • Chemisches Gleichgewicht • Abhängigkeit des chemischen Potentials von Druck, Temperatur und Zusammensetzung • Zusammenhang zwischen den verschiedenen Gleichgewichtskonstanten • Massenwirkungsgesetz • Druck- und Temperaturabhängigkeit der Gleichgewichtskonstanten • Le Chatelier-Braun’sches Prinzip • Gibbs’sche Phasenregel mit und ohne chemische Gleichgewichte 3.9.2. Literatur P. W. Atkins, Physikalische Chemie, 2. Auflage, VCH-Verlag, Weinheim, 1996, S. 153-165, 195-201, 216-219, 225-230, 255-272. G. Wedler, Lehrbuch der Physikalischen Chemie, 4. Auflage, Wiley-VCHVerlag, Weinheim, 1997, S. 271-290, 299-303, 329-336, 372-391, 406-408. 3.9.3. Theoretische Grundlagen Man betrachte die Dissoziation von festem [Ni(NH 3)6]Br2 in Ammoniak und festes [Ni(NH3)2]Br2, die man durch die Reaktionsgleichung A(s) B(s) 4C(g) (1) beschreiben kann. Dabei handelt es sich um eine heterogene Reaktion, an der zwei feste Phasen und eine gasförmige Phase beteiligt sind. Nimmt man nun an, dass sich die 3 Phasen im thermischen und mechanischen Gleichgewicht befinden, dann hängt die Freie Enthalpie G des Gesamtsystems von der Temperatur T, dem Druck p und den Stoffmengen nA, nB und nC ab. Bei konstantem p und T ergibt sich somit die Änderung der Freien Enthalpie zu dG A dnA BdnB C dnC . (2) Mit Hilfe der Umsatzvariable (Reaktionslaufzahl) ξ kann man Gleichung (2) umformulieren und erhält dG ( B 4C A ) d . (3) Im chemischen Gleichgewicht ist die Freie Enthalpie G minimal, d. h. es gilt (∂G/∂ξ)T,p = 0, so dass sich das chemische Gleichgewicht für die heterogene Dissoziationsreaktion auch mit Hilfe der chemischen Potentiale A, B und C als A B 4 C (4) schreiben lässt. Vernachlässigt man nun den Dampfdruck der beiden festen Phasen A und B, dann handelt es sich bei der Gasphase um eine reine Phase. Nimmt man zusätzlich an, dass sich die gasförmige Komponente C ideal verhält, dann ergibt sich für das chemische Potential C der reinen gasförmigen Phase pC , p C C RT ln (5) wobei pC der Partialdruck von C ist. Die chemischen Potentiale A und B der beiden reinen, festen Phasen A und B ergeben sich zu A A und B B . (6) Hierbei wurde die Druckabhängigkeit der chemischen Potentiale der beiden Feststoffe vernachlässigt. Somit kann man die Gleichgewichtsbedingung (4) in 4 p G ( B 4 C A ) R ln C ln K RT RT p (7) umformen, wobei ΔRG° die Freie Standardreaktionsenthalpie und K die dimensionslose thermodynamische Gleichgewichtskonstante ist. Da der Partialdruck pC der Komponente C in der reinen Gasphase gleich dem Gesamtdruck p ist, kann man durch Messen des Gesamtdruckes p die Gleichgewichtskonstante bestimmen. Im hier betrachteten Fall ist p eindeutig durch die Temperatur T festgelegt. Man spricht daher auch von einem univarianten System, d. h. das System hat im chemischen Gleichgewicht nur noch einen Freiheitsgrad. Eine Messung des Partialdruckes pC bei verschiedenen Temperaturen T kommt einer temperaturabhängigen Bestimmung von K bzw. von ΔRG° gleich. Aus der Temperaturabhängigkeit der Freien Standardreaktionsenthalpie ΔRG° erhält man die Standardreaktionsentropie −ΔRS° = (∂ΔRG°/∂T)ξ und über die GibbsHelmholtz-Gleichung auch die Standardreaktionsenthalpie ΔRH° R H ( R G / T ) . T T2 (8) Mit Hilfe der Gleichgewichtskonstanten K aus Gleichung (7) ergibt sich die van’t Hoff’sche Reaktionsisobare zu R H ln K d ln( p C p) , 4 2 dT RT T (9) aus der man durch Integration unter der Annahme, dass ΔRH° nicht von der Temperatur abhängt, zu p H ln C R const. p 4 RT (10) gelangt. Da es sich bei dem hier untersuchten Fall, um eine reine gasförmige Phase handelt, deren Dampfdruck eindeutig durch die Temperatur festgelegt ist, konnte im zweiten Schritt in Gleichung (9) das partielle Differential durch ein totales Differential ersetzt werden. 3.9.4. Aufgaben 1) Man berechne Kp und Kx für die Dissoziation des Ammoniaks und zeige, wie Kp und Kx mit der thermodynamischen Gleichgewichtskonstante K zusammenhängen. 2) Man bestimme den Partialdruck von Ammoniak in Abhängigkeit der Temperatur T bei der Dissoziation von festem [Ni(NH3)6]Br2. 3) Man trage ln(K) gegen 1/T auf und berechne aus der Steigung die Standardreaktionsenthalpie ΔRH° für die Dissoziation von festem [Ni(NH3)6]Br2 in NH3 und festem [Ni(NH3)2]Br2. 4) Vergleichen Sie Ihr Ergebnis mit Literaturdaten. 3.9.5. Zubehör Vakuumapparatur mit Quarzspiralmanometer, Ölrotationspumpe, Rührer, Heizung, Heizregler, Transformator, Dosenmanometer, Thermometer. 3.9.6. Durchführung Die Glasapparatur ist zu Versuchsbeginn evakuiert. Abb. 1: Glasapparatur zur Untersuchung des heterogenen Dissoziationsgleichgewichtes. Das Kölbchen mit der Substanz befindet sich in einem Ofen mit Temperaturregler, dessen Bedienung vom Assistenten erklärt wird (siehe Bedienungsanleitung zum Betrieb von Heizreglern auf Seite 11). Zur Druckmessung dient ein Quarzspiralmanometer. Durch Druckunterschiede zwischen dem Inneren der Quarzspirale und dem umgebenden Raum weitet sich die Spirale auf bzw. zieht sich zusammen. Durch vorsichtiges Evakuieren (siehe Betrieb und Funktionsweise von Ölrotationspumpen auf Seite 8) oder Belüften der Spirale mit dem Dreiwegehahn C werden die Zeiger des Quarzspiralmanometers genau übereinander gestellt. Dann ist der Druck innen und außen gleich und kann am Differenzdruckmanometer D abgelesen werden (zur Funktionsweise eines Dosenmanometers siehe Seite 8). Da das Gleichgewicht der Reaktion bei Raumtemperatur auf der Seite des Hexamin-Nickel-(II)-bromids liegt, notiert man sich den vor dem Aufheizen am Manometer D angezeigten Druck, um später alle gemessenen Druckwerte darauf zu beziehen. Dann wird am Heizregler eine Temperatur von ca. 90 °C eingestellt. Nachdem das Thermometer am Glaskolben einen konstanten Wert anzeigt, muss noch der Gleichgewichtsdruck bestimmt werden. Dazu werden entstehende Zeigerausschläge am Quarzspiralmanometer immer wieder abgeglichen bis sich der Zeiger nicht mehr bewegt. Dann kann der Gleichgewichtsdruck am Dosenmanometer D abgelesen werden. Die Gleichgewichtseinstellung dauert 10-15 min. Abb. 2: Probenofen. Es werden für die Dissoziation ca. 8 Messwerte bei Temperaturen zwischen 90 und 195 °C aufgenommen. Dabei sollten die Temperaturerhöhungen umso kleiner sein, je höher die Temperatur ist, z. B.: 90, 120, 150, 160, 170, 180, 185, 190, 192, 195 °C. Die letzten Temperaturen sind nur dann messbar, wenn der Luftdruck sehr hoch und damit der Messbereich des Manometers D entsprechend groß ist. Zur Beendigung des Versuches wird der Ofen ausgeschaltet, damit die Reaktion in umgekehrter Richtung ablaufen kann. 3.9.7. Praktische Hinweise - Vor dem Abschalten muss die Ölrotationspumpe belüftet werden (Hahn links an der Apparatur). - Die Hähne A und B dürfen nicht betätigt werden, sondern müssen immer geschlossen bleiben. - Der Heizregler mit dem Thermoelement dient nur zur Temperierung der Probe, gemessen wird mit dem Thermometer. - Der Schalter am Heizregler muss unbedingt auf „Soll - Reg. Ein“ eingestellt sein, da sonst entweder keine Temperaturregelung stattfindet oder man den Sollwert nicht einstellen kann. - Die Temperatur der Substanz darf auf keinen Fall so hoch eingestellt werden, dass in der Apparatur ein höherer Druck als Luftdruck entsteht bzw. das Ende der Messskala am Dosenmanometer überschritten wird, da die Apparatur sonst undicht werden und im schlimmsten Fall zerbrechen kann. 3.10. Adsorption (Versuch Nr. 5) Ziel des Versuches ist, den Verlauf der Adsorptionsisotherme von in Wasser gelöstem Methylenblau an Aktivkohle zu bestimmen. 3.10.1. Vorbereitung • • • • • • • Adsorption Chemisches Gleichgewicht / Grenzflächengleichgewichte Chemisches Potential von verdünnten Lösungen Mischungsvorgänge (Mischungsentropie) Verlauf der Adsorptionsisothermen nach Henry, Langmuir und Freundlich Kinetische Beschreibung von chemischen Gleichgewichten Wesen der Adsorptionskräfte (Physi- oder Chemisorption) 3.10.2. Literatur P. W. Atkins, Physikalische Chemie, 2. Auflage, VCH-Verlag, Weinheim, 1996, S. 195-203, 216-219, 255-266, 929-937. G. Wedler, Lehrbuch der Physikalischen Chemie, 4. Auflage; Wiley-VCHVerlag, Weinheim, 1997, S. 288-292, 329-336, 372-386, 428-435. 3.10.3. Theoretische Grundlagen An einer Grenzfläche zwischen zwei Phasen wird oft eine gewisse Stoffmenge an Gas oder gelöster Substanz angereichert. Diesen Effekt der Anreicherung von Molekülen eines Stoffes an einer Oberfläche bezeichnet man als Adsorption. Bei der Adsorption handelt es sich um einen chemischen Gleichgewichtsvorgang, an dem als ein Reaktionspartner eine feste oder flüssige Phasengrenzfläche beteiligt ist. Dabei interessiert man sich dafür, wie viele Adsorptionsplätze auf der Oberfläche von einem Stoff in Abhängigkeit seines Partialdruckes in der Gasphase oder seiner Konzentration in der Lösung belegt werden. Bei einer gegebenen Temperatur spricht man dabei von der sogenannten Adsorptionsisotherme. Betrachtet man z. B. die Adsorption einer in Wasser gelösten Substanz B(aq) an einer festen Oberfläche, kann man dies als Reaktion einer Oberflächenposition S(vac) mit der gelösten Substanz S(vac) zur adsorbierten Substanz BS(ads) formulieren: B(aq) S(vac) BS(ads) (1) Im Gleichgewicht gilt ΔRG = 0, also: ads Baq Svac BS . (2) Betrachtet man eine ideal verdünnte Lösung, dann erhält man für Baq Baq Baq RT ln xB . (3) Dabei gibt der Molenbruch xB = nB/(nB + nA) das Stoffmengenverhältnis von gelöster Substanz B zur Gesamtstoffmenge aus gelöster Substanz B und Lösungsmittel A an und Baq ∞ bezeichnet das chemische Potential der reinen gelösten Substanz in einem hypothetischen Standardzustand, bei dem die gelöste Substanz als Reinstoff vorliegt, aber die Eigenschaften wie bei unendlicher Verdünnung hat (vgl. Wedler, 4. Auflage, S. 333). In einer thermodynamischen Beschreibung der Adsorption kann man sich die mit Stoffmenge belegte Grenzfläche ebenfalls als eine Art „Lösung“ vorstellen. Dabei übernimmt die reine, d. h. unbelegte, Grenzfläche die Rolle des „Lösungsmittels“ und die adsorbierte Stoffmenge entspricht in dieser Beschreibung der „gelösten Substanz“. Ist die Grenzfläche nun ebenfalls ideal verdünnt, d. h. ist das Verhältnis von adsorbierter Stoffmenge nads zur Stoffmenge nmax, die maximal adsorbiert werden kann, gering, kann man das chemisches Potential von B auf der Oberfläche in Analogie zu Gleichung (3) als Bads Bads RT ln (4) ansetzen, wobei θ = nads/nmax der Molenbruch für die adsorbierte Substanz, der sogenannte Bedeckungsgrad, ist. Oftmals spricht man anstatt von adsorbierter Stoffmenge nads bzw. maximal adsorbierter Stoffmenge nmax von den besetzten Adsorptionsplätzen Nads und der maximalen Anzahl an möglichen Adsorptionsplätzen Nmax. Für das chemische Potential der vakanten Position kann man in Anlehnung an Gleichung (4) schreiben (dieser Ausdruck kann ebenso wie Gleichung (4) aus statistischen Überlegungen hergeleitet werden): Svac Svac* RT ln 1 (5) Referenzzustand ist hier die vollkommen unbesetzte Oberfläche. Aus Gleichung (5), (4) und (3) ergibt sich aus Gleichung (2): cB vac* ads S RT ln 1 B RT ln , c Baq RT ln (6) wobei der Molenbruch durch Konzentrationen ersetzt wurde. Mit Hilfe der Freien Standardadsorptionsenthalpie ΔadsG° lässt sich Gleichung (6) umformen, adsG ( ads Baq Svac* ) BS ln ln K , RT RT 1 c c B (7) wobei K die thermodynamische Gleichgewichtskonstante ist. Damit ergibt sich für die Gleichgewichtskonstante K: K 1 cB c (8) Umgeformt ergibt sich die sogenannte Langmuir’sche Adsorptionsisotherme cB c . c 1 K B c K (9) Für den Grenzfall sehr kleiner Konzentrationen (K(cB/c°) << 1) geht sie in das Henry’sche Adsorptionsgesetz K cB c0 (10) über, während man bei sehr großen Konzentrationen (K(cB/c°) >> 1) Sättigung beobachtet. Dies hängt an der Voraussetzung, dass die maximale Anzahl an Adsorptionsplätzen Nmax als konstant angenommen wurde, d. h. maximal nur eine Monolage an gelöster Substanz adsorbiert werden kann. Die Langmuir’sche Adsorptionsisotherme vermag das Adsorptionsverhalten an Grenzflächen mit einheitlichen Adsorptionsplätzen gut zu beschreiben. Bei vielen festen Grenzflächen unterscheiden sich jedoch die Adsorptionsplätze hinsichtlich ihrer Adsorptionsenthalpie, d. h. die Annahme, ΔadsH als unabhängig von θ zu betrachten, ist nicht mehr gerechtfertigt. Um die Adsorption an festen Grenzflächen besser zu beschreiben, muss man daher über mehrere Langmuir’sche Adsorptionsisothermen mit unterschiedlichen K-Werten mitteln. Die historisch zunächst empirisch von Freundlich gefundene Adsorptionsisotherme c a B c b (14) mit zwei adsorptionsspezifischen Konstanten a und b entspricht einer solchen Mittelung. Im Versuch wird die Adsorption von Methylenblau an Aktivkohle untersucht. Dazu wird die adsorbierte Stoffmenge an Methylenblau nB pro eingewogener Menge an Aktivkohle mK aus der Differenz der Konzentration an Methylenblau vor und nach Zugabe von Aktivkohle bestimmt. Die Konzentrationsmessung von Methylenblau erfolgt mit einem Spektralphotometer. Abb. 1: Schematische Darstellung des Spektralphotometers. Dabei wird ausgenutzt, dass eine bestimmte Farbstoffkonzentration cB eine wellenlängenabhängige Abschwächung der eingestrahlten Lichtintensität I bewirkt. Die Abnahme dI der Intensität, die eintritt, wenn die Lichtstrahlung die Strecke dz in einer Probe zurücklegt, ist proportional zur Länge des zurückgelegten Weges, der Konzentration des Farbstoffs und der Intensität des Lichtstrahls, d.h. dI ~ −cFIdz. Mit Hilfe der wellenlängenabhängigen Proportionalitätskonstante κ ergibt sich dI/I = −κcFdz. Durch Integration mit geeigneten Anfangsbedingungen erhält man daraus das sogenannte Lambert-Beer’sche Gesetz ln I cF zK , I0 (15) wobei I0 die anfängliche Lichtintensität und zK die gesamte Dicke der Probe ist. Das Verhältnis aus I/I0 wird als Transmission bezeichnet. Oftmals verwendet man allerdings anstatt des natürlichen den dekadischen Logarithmus und führt die sogenannte dekadische Absorbanz bzw. Extinktion A ein I0 (16) cF zK . I Dabei ist ε = κ/ln10 der (dekadische) Extinktionskoeffizient. Die gemessene Extinktion ist also direkt zur Konzentration an Methylenblau proportional. A lg 3.10.4. Aufgaben 1) Wie hängt die pro Gramm Aktivkohle adsorbierte Substanzmenge an Methylenblau nB/mK mit dem Bedeckungsgrad θ zusammen? 2) Man bestimme die Adsorptionsisotherme von in Wasser gelöstem Methylenblau an Aktivkohle und überprüfe, ob sich der Verlauf der Messwerte besser durch die Freundlich- oder die Langmuir-Adsorptionsisotherme erklären lässt. Dazu muss man bei der Langmuir-Isotherme cB/(nB/mK) gegen cB auftragen und bei der Freundlich-Isotherme ln(nB/mK) gegen ln(cB/c°). Durch die Methode der kleinsten Fehlerquadrate muss herausgefunden werden, welche der beiden Isothermen die experimentellen Daten besser beschreibt. 3) Man ermittle die Konstanten für die Freundlich-Isotherme und vergleiche die Werte mit Literaturangaben. 3.10.5. Zubehör 2 Magnetrührer, Spektralphotometer mit Farbfilter, Bechergläser, Messkolben, Pipetten, Filterpapier, Aktivkohle, Methylenblau. 3.10.6. Durchführung Zuerst wird das Spektralphotometer eingeschaltet (Drehschalter 13 und Schiebeschalter 6, links hinten). In der Aufwärmphase des Photometers (10-20 Min.) darf sich keine Küvette im Gerät befinden. Das Filterrad 14 wird auf „gelb“ gestellt. Mit Drehknopf 9 werden 585 nm eigeregelt. Mit der Folientaste 8 wird „Transmission/Absorption“ auf dem Display 3 eingestellt. Mit dem Drehregler 13 wird die Transmission auf 0% geregelt (ohne Küvette). Dann wird eine Rundküvette mit destilliertem Wasser in den Küvettenschacht 2 eingesetzt und mit dem Drehregler 12 die Anzeige auf 100% Transmission eingestellt. Das Gerät ist jetzt betriebsbereit. Bei längeren Messpausen sollte sich keine Küvette im Küvettenschacht befinden. Aus der bereitgestellten wässrigen Methylenblau-Stammlösung von ungefähr 5·10−3 mol/L wird eine Verdünnungsreihe von 6 Lösungen mit Ausgangskonzentration zwischen cB0 = 8·10−6 und 3·10−4 mol/L hergestellt. Mit den vier am schwächsten konzentrierten Lösungen wird die Extinktion mit Hilfe des Photometers gemessen. Daraus erhält man eine Eichkurve, die einem erlaubt aus der Messung der Extinktion die Farbstoffkonzentration zu bestimmen. Dazu wird zunächst nochmals die Einstellung von 0% und 100% Transmission überprüft (siehe oben). Dann wird die Küvette mit der gefärbten Lösung in den Strahlengang gebracht und die entsprechende Extinktion (am Gerät: Absorption) abgelesen. Für jede Lösung ist der Vorgang zweimal zu wiederholen. Nach der Messung sollte die Lösung aus der Küvette zur Weiterverwertung wieder in das jeweilige Messkölbchen zurückgefüllt werden. Um die Adsorption von Methylenblau an Aktivkohle zu untersuchen, werden jeweils 50 mL der zu untersuchen Farbstofflösung einer bestimmten Anfangskonzentration cB0 mit 1 g Aktivkohle versetzt und 15 Minuten auf dem Magnetrührer gerührt. Anschließend filtriert man die Aktivkohle ab und bestimmt die Rest- bzw. Gleichgewichtskonzentration cB an Methylenblau durch zwei Extinktionsmessungen. Die pro Gramm Aktivkohle adsorbierte Stoffmenge an Methylenblau ergibt sich aus nB/mK = VL(cB0 − cB)/mK, wobei VL das zur Adsorption verwendete Volumen der Farbstofflösung und mK die Einwaage an Aktivkohle darstellt. Beim Filtrieren ist allerdings zu beachten, dass das Filterpapier ebenfalls Methylenblau adsorbiert. Deshalb muss man zunächst das Filterpapier sorgfältig mit der verbleibenden Farbstofflösung anfeuchten und die ersten Tropfen des Filtrats verwerfen. Erst danach wird der Rest der Lösung in ein sauberes und trockenes Gefäß filtriert. 3.11. Oberflächenspannung (Versuch Nr. 23) Ziel des Versuches ist, den Platzbedarf eines Ethanol-Moleküls in der Grenzfläche zwischen Dampfphase und Lösung aus der Konzentrationsabhängigkeit der Oberflächenspannung bei wässrigen Ethanol-Lösungen zu ermitteln. 3.11.1. Vorbereitung • • • • • Gibbs’sche Fundamentalgleichungen Grenzflächenthermodynamik und Grenzflächeneigenschaften Oberflächenüberschuss, Gibbs’sche Gleichung der Oberflächenspannung Oberflächenaktive Substanzen Bedeckungsgrad und Langmuir’sche Adsorptionsisotherme (Theorie der Adsorption) • Verdünnte Lösungen 3.11.2. Literatur P. W. Atkins, Physikalische Chemie, 2. Auflage, VCH-Verlag, Weinheim, 1996, S. 216-219, 907-916, 929-933. G. Wedler, Lehrbuch der Physikalischen Chemie, 4. Auflage; Wiley-VCHVerlag, Weinheim, 1997, S. 329-336, 411-425. 3.11.3. Theoretische Grundlagen Man betrachte eine flüssige Mischphase α mit den beiden Komponenten A und B, die mit ihrer Dampfphase β im chemischen Gleichgewicht ist. Die beiden Phasen α und β seien durch eine Grenzfläche Aαβ voneinander separiert. Man kann sich nun vorstellen, dass die beiden Phasen α und β den Raum vollständig mit Stoffmenge ausfüllen, so dass das Volumen V des Gesamtsystems sich aus den Volumina Vα bzw. Vβ der beiden Phasen α und β gemäß V = Vα + Vβ zusammensetzt. Wenn die Konzentrationen der beiden Komponenten in der Phase α und β bis an die Grenzfläche konstant wären, dann würde sich auch die Freie Enthalpie G des Gesamtsystems additiv aus den Freien Enthalpien Gα und Gβ der beiden Phasen zusammensetzen. Allerdings unterscheiden sich die intermolekularen Wechselwirkungen in der Grenzfläche sowohl zu denen in der flüssigen Phase als auch zu denen in der Gasphase. Dies kann in einem mehrkomponentigen System auch dazu führen, dass sich eine Komponente in der Grenzfläche anreichert. Die Freie Enthalpie G des Gesamtsystems setzt sich also nicht nur aus den Beiträgen der Volumenphasen Gα und Gβ zusammen, sondern es kommt die freie Grenzflächenenthalpie Gαβ hinzu. G G α G β G αβ (1) Bei den nachfolgenden Überlegungen wird vorausgesetzt, dass zwischen den beteiligten Volumenphasen α bzw. β sowie der Grenzflächenphase αβ thermisches und mechanisches Gleichgewicht herrscht, d. h. Tα = Tβ = Tαβ = T und pα = pβ = pαβ = p. Darüber hinaus sollen die beiden Komponenten A und B sich auch im chemischen Gleichgewicht befinden, d. h. α = β = αβ = und Bα = Bβ = Bαβ = B. Die Freie Enthalpie G des Gesamtsystems hängt nun neben dem Druck p, der Temperatur T und den Stoffmengen nA und nB auch von der Grenzfläche Aαβ ab, so dass sich für die Änderung der Freien Enthalpie dG SdT Vdp A dnA BdnB dAαβ (2) ergibt, wobei σ die sogenannte Oberflächenspannung der Lösung ist. Die Oberflächenspannung gibt somit an, um wie viel sich die Freie Enthalpie G des Gesamtsystems verändert, wenn bei konstantem Druck, konstanter Temperatur und konstanter Zusammensetzung die Oberfläche verändert wird. Für die beiden Volumenphasen ergibt sich die Änderung der Freien Enthalpie zu dG α S α dT V α dp A dnAα BdnBα dG S dT V dp β β β β A dnA β B dnB . (3) Dabei bezeichnen die dnAα und dnBα bzw. dnAβ und dnBβ die Änderungen der Stoffmenge von A und B in den beiden ausgedehnten Phasen α und β. Somit lässt sich die Änderung der Freien Grenzflächenenthalpie dGαβ durch dG αβ S αβ dT V αβ dp A dnAαβ BdnBαβ dAαβ (4) beschreiben, wobei Sαβ die Grenzflächenentropie ist und nAαβ bzw. nBαβ die Stoffmengen an A und B in der Grenzflächenphase αβ sind. Bei konstanter Temperatur T und konstantem Druck reduziert sich Gl. (4) zu: dG αβ A dnAαβ BdnBαβ dAαβ (5) Durch Integration von unendlich kleiner Grenzfläche ausgehend bis zur Fläche Aαβ, ergibt sich: G αβ A nAαβ BnBαβ Aαβ . (6) Bildung des totalen Differentials ergibt: dG αβ A dnAαβ nAαβ d A BdnBαβ nBαβ d B dAαβ Aαβ d . (7) Vergleich von Gl. (5) und (7) ergibt eine Gibbs-Duhem-Relation nAαβ d A nBαβ d B Aαβ d 0 , (8) die die Änderung der chemischen Potentiale von A und B mit der Änderung der Oberflächenspannung verknüpft. Unter Einführung der Oberflächenkonzentration der beiden Komponenten ΓA = nAαβ/Aαβ und ΓB = nBαβ/Aαβ bezeichnet man Gleichung (8) auch als die Gibbs’sche Adsorptionsisotherme oder auch als die Gibbs’sche Gleichung der Oberflächenspannung d ( A d A B d B ) . (9) Wir wollen nun den speziellen Fall betrachten, dass sich eine der beiden Komponenten (B) sehr stark bevorzugt in der Grenzfläche anreichert. Dann kann man in Gleichung (9) die Oberflächenkonzentration der anderen Komponente (A) vernachlässigen und erhält dσ = −ΓBdB. Wenn man nun weiß, wie sowohl das chemische Potential B von B in der Lösung als auch der Oberflächenkonzentration ΓB von der Konzentration cB abhängen, kann man die Konzentrationsabhängigkeit von σ berechnen. Dazu muss man beachten, dass viele oberflächenaktive Substanzen (das sind die Substanzen, die sich bevorzugt in der Grenzfläche anreichern) meistens nur eine Monolage ausbilden. Daher bietet es sich an, den sogenannten Bedeckungsgrad einzuführen, der angibt, welcher Bruchteil der Monolage bereits mit oberflächenaktiver Substanz besetzt ist. Bei vorgegebener Grenzfläche Aαβ ist der Bedeckungsgrad θ gemäß B Bmax (10) mit der Oberflächenkonzentration ΓB verknüpft, wobei ΓBmax die Oberflächenkonzentration für die vollständig belegte Monolage bezeichnet. Diese Größe wiederum ist mit der Fläche aB aB 1 N A Bmax (11) verbunden, die ein einzelnes Molekül B in der gefüllten Monoschicht einnimmt. Dabei ist NA die Avogadro-Konstante. Für das einfachste Adsorptionsmodell an einer völlig einheitlichen Grenzfläche nach Langmuir (vgl. Versuch „Adsorption) hängt der Bedeckungsgrad über cB c KaB c 1 K B 1 KaB c K (12) von der Konzentration cB des gelösten Stoffes ab. Hierbei ist K die thermodynamische Gleichgewichtskonstante für den Anreicherungs- bzw. Adsorptionsvorgang und aB = cB/c° die Aktivität von B in der Flüssigkeit. Hat man es mit einer ideal verdünnten Lösung von B in A zu tun, dann gilt für das chemische Potential von B und für sein Differential: B B RT ln aB d B RTd ln aB (13) beschreiben. Somit kennt man alle Abhängigkeiten um die Gibbs’scheAdsorptionsisotherme dσ = −ΓBdB zu integrieren: aB aB d RT aB 0 aB aB max B aB RT Bmax KaB d ln aB 1 Ka B 0 aB aB (14) K 1 KaB daB a 0 B Es ergibt sich: aB aB aB 0 RT Bmax ln 1 KaB bzw. * RT Bmax ln 1 KaB (15) wobei σ* die Oberflächenspannung des reinen Lösungsmittels, d. h. der reinen Komponente A, bezeichnet. Durch experimentelle Untersuchung des Verlaufs von σ = σ(cB) ist man in der Lage neben der thermodynamischen Gleichgewichtskonstanten auch ΓBmax zu bestimmen und erhält daraus den Flächenbedarf aB eines Moleküls B in der vollständig gefüllten Monolage. Die Oberflächenspannung σ der verschiedenen Lösungen wird mit der Ringmethode nach du Noüy untersucht. Dazu wird mit einer Feder(-Waage) die Kraft gemessen, die notwendig ist, um eine Flüssigkeitslamelle, die sich beim Herausziehen des Ringes aus der Lösung bildet, abzureißen. Die Änderung der Freien Enthalpie dG, die benötigt wird, um den Ring mit dem Radius r bei konstanter Temperatur, Druck und Zusammensetzung um ein kleines Stückchen dz nach oben zu ziehen, ist gegeben durch Aαβ dG dAαβ dz F dz . z T , p ,nA ,nB (16) Bis zum Abreißen ist der Kraft Fσ, die auf die Oberfläche wirkt, immer eine entsprechende Federkraft Ff = −Fσ entgegen gerichtet. Da beim Bügelring in unserem Experiment die Änderung der Oberfläche beim Herausziehen konstant ist Aαβ 2 2 r , z T , p ,nA ,nB (17) erhält man die Oberflächenspannung direkt aus der gemessenen maximalen Federkraft Ff,max beim Abreißen Ff,max 2 2 r . (18) Dabei ist berücksichtigt worden, dass der Ring eine Innen- und Außenseite besitzt. 3.11.4. Aufgaben 1) Man bestimme die maximale Abreißkraft Ff,max bei den verschieden EthanolWasser-Lösungen und berechne daraus die Oberflächenspannung σ. Man vergleiche die σ-Werte von reinem Wasser und reinem Ethanol mit Literaturdaten. 2) Man trage die gemessene Oberflächenspannung σ gegen die Konzentration von Ethanol cB auf. 3) Man bestimme aus der Konzentrationsabhängigkeit von σ den Flächenbedarf aB eines einzelnen Ethanol-Moleküls in der vollständig gefüllten Monolage. Dazu muss man mit Hilfe eines nicht-linearen Kurvenregressionsprogramms die Messdaten mit Hilfe von Gl. (15) anpassen. 4) Man schätze den Wert von aB im Rahmen eines einfachen geometrischen Modells ab, das von einer dichtesten Packung an Ethanol-Molekülen in der vollständig gefüllten Monolage ausgeht und vergleiche diesen Wert mit dem aus der Konzentrationsabhängigkeit von σ experimentell bestimmten Wert. 3.11.5. Zubehör 100-mL-Becherglas, höhenverstellbares Stativ, Tensiometer, Bügelring (mittlerer Umfang = 0,061 m), Pipetten, Pipettierball, Ethanol, destilliertes Wasser, 100-mL-Fläschchen mit Ethanol-Wasser-Lösungen. 3.11.6. Durchführung Aus dem entsprechenden Vorrats-Fläschchen füllt man ca. 50 mL EthanolWasser-Lösung in das 100-mL-Becherglas (siehe Hinweise zur Volumenmessung auf Seite 15). Dann wird das Glas in das verstellbare Stativ eingespannt. Der Messring wird an die linke Seitenstange des Tensiometers gehängt und das Becherglas so positioniert, dass der Ring mindestens 0,5 cm in die Lösung eintaucht. Mit der Stellschraube hinten am Tensiometer wird die Seitenstange so eingestellt, dass sie sich zwischen den Markierungen befinden. Das Tensiometer muss dabei auf 0 mN eingestellt sein. Dann wird das Becherglas mit der Stellschraube des Stativs vorsichtig abgesenkt und das Tensiometer gleichzeitig so verstellt, das die Seitenstange beständig zwischen den Markierungen bleibt, bis der Kontakt des Rings mit der Flüssigkeitsoberfläche reißt. Die jetzt zu sehende Einstellung am Tensiometer entspricht der maximalen Federkraft Ff,max beim Abreißen für die entsprechende Lösung. Allerdings ist der so erhaltene Wert noch etwas ungenau. 0 mN 9 3 6 Tensiometer Bügelring Becherglas mit Lösung Abb. 1: Apparatur zur Bestimmung der Oberflächenspannung nach du Noüy. Um die genaue Abreißkraft zu ermitteln, sollte man das Tensiometer noch einmal um 0,05-0,10 mN zurückstellen und die Seitenstange vorsichtig soweit herunterdrücken, dass der Ring wieder in die Flüssigkeit taucht. Dann stellt man am Tensiometer ganz langsam höhere Werte ein, bis der Kontakt zwischen Ring und Oberfläche erneut reißt, ohne das Becherglas weiter abzusenken. Da die Flüssigkeitsoberfläche ruhiger ist, wenn man das Becherglas nicht bewegen muss, ist der so ermittelte Wert der genauere. Diese Messung wird für alle bereitstehenden Lösungen 5 Mal bei Raumtemperatur durchgeführt. Die Messlösungen sollen wiederverwendet werden, d. h. sie müssen nach Gebrauch in das richtige Fläschchen zurückgefüllt werden. 3.12. Nernst’sche Gleichung (Versuch Nr. 24) Ziel des Versuches ist, das Standardpotential E° einer elektrochemischen Reaktion bei verschiedenen Temperaturen zu messen und daraus die Standardreaktionsenthalpie ΔRH° zu bestimmen. Vorbereitung: • • • • • • Elektrochemisches Gleichgewicht Elektrochemisches Potential EMK, Temperaturabhängigkeit der EMK Aktivität, Aktivitätskoeffizienten Standardzustände Konzentrationsabhängigkeit von Aktivitätskoeffizienten (Debye-Hückel-Theorie) Literatur: P. W. Atkins, Physikalische Chemie; 2. Auflage, Wiley-VCH, Weinheim, 1996, S. 216-219, 300-329. G. Wedler, Lehrbuch der Physikalischen Chemie, 4. Auflage; Wiley-VCH, Weinheim, 1997, S. 187-196, 329-344, 448-485. Theoretische Grundlagen: 1) Prinzip der Messung: Im Versuch geht es um die Reaktion Ag (s) Cl (aq ) Fe(CN ) 36 (aq ) Fe(CN ) 64 (aq ) AgCl (s) . (1) Da es sich um eine Redox-Reaktion handelt, kann man versuchen, die Reaktion durch eine elektrochemische Zelle zu realisieren Ag | AgCl(s) | Cl (aq) ¦ Fe(CN) 64 (aq) , Fe(CN)36 (aq) | Pt . (2) Dabei wird eine Ag/AgCl-Elektrode über eine Spannungsquelle an eine Fe(II)/Fe(III)-Redoxelektrode angeschlossen. Befindet sich die elektrochemische Zelle zusammen mit der äußeren Spannungsquelle im Gleichgewicht, d. h. fließt kein Strom durch die elektrochemische Zelle, dann kann man die elektrische Spannung E, die an der äußeren Spannungsquelle anliegt, mit der Freien Reaktionsenthalpie ΔRG der chemischen Reaktion R G FE (3) in Verbindung bringen. Dabei wird E auch als die elektromotorische Kraft (EMK) der elektrochemischen Zelle bezeichnet und ν gibt an, wie viele Elektronen pro Formelumsatz fließen. Für die obige Reaktion ist ν=1. Durch eine temperaturabhängige Messung der EMK bei konstantem Druck p und konstanter Zusammensetzung ξ kann man die Reaktionsentropie ΔRS=−(∂G/∂T)p,ξ=νF(∂E/∂T)p,ξ und über die Gibbs-Helmholtz-Beziehung R H T2 (E T) F T p, (4) auch die Reaktionsenthalpie ΔRH gewinnen. Häufig interessiert man sich aber für den Wert von ΔRH, wenn die Produkte und Edukte in ihren Standardzuständen vorliegen. Bei der hier zu untersuchenden Reaktion ist der Standardzustand bei allen Stoffen durch ein Druck von p=p°=105 Pa festgelegt, wobei die Stoffe in der wässrigen Lösung zusätzlich als ideal verdünnt bei einer Konzentration von c=c°=1 mol/L betrachtet werden. Man erhält nun die Standardreaktionsenthalpie ΔRH° aus der Temperaturabhängigkeit der Standard-EMK E°, wobei die Standard-EMK E° bei obiger Reaktion über die Nernst’sche Gleichung mit E verknüpft ist E E RT a Fe ( II ) a AgCl ln F a Fe ( III ) a Ag a Cl . (5) Liegen alle Stoffe in ihrem jeweiligen Standardzustand a=1 vor, dann ist E=E°. Normalerweise wird die Spannung E zwischen den beiden Halbzellen jedoch bei Konzentrationen gemessen, wo a≠1 ist und man muss die Standard-EMK E° aus konzentrationsabhängigen Messungen bestimmen. Dazu führt man das Experiment zweckmäßigerweise mit einer Standard-Ag/AgCl-Elektrode durch, so dass die Abweichungen der gemessenen Spannung E vom Standardwert E° allein von den Aktivitäten der Fe(II)- bzw. Fe(III)-Ionen herrühren. Die Aktivitäten aJ=J(cJ/c°) der Fe(II)- bzw. Fe(III)-Ionen hängen von den Konzentrationen cJ der Ionen in der Lösung ab, wobei J der Aktivitätskoeffizient des entsprechenden Ions ist. Damit ergibt sich die EMK der elektrochemischen Zelle zu E E RT Fe ( II ) c Fe ( II ) RT c Fe ( II ) RT Fe ( II ) . ln E ln ln F Fe ( III ) c Fe ( III ) F c Fe ( III ) F Fe ( III ) (6) In ideal verdünnten Lösungen sind die Aktivitätskoeffizienten gleich Eins und Gleichung (6) geht in die vereinfachte Form der Nernst’schen Gleichung über, in der nur noch Konzentrationen auftreten. Bei konzentrierteren Lösungen muss man jedoch die von Eins abweichenden konzentrationsabhängigen Aktivitätskoeffizienten berücksichtigen, weshalb die EMK in komplizierter Weise von der Elektrolytkonzentration abhängt. In stark verdünnten Elektrolytlösungen lässt sich die Konzentrationsabhängigkeit der Aktivitätskoeffizienten durch das Debye-Hückel-Grenzgesetz beschreiben ln J A z 2J (I / c)1 2 . (7) Dabei ist A eine vom Lösungsmittel abhängige Proportionalitätskonstante, z J die Ladung des betrachteten Ions und I 12 z 2J c J (8) J bezeichnet die gesamte Ionenstärke der Elektrolytlösung. Daher kann man erwarten, dass im Gültigkeitsbereich des Debye-Hückel-Grenzgesetzes auch eine entsprechende Abhängigkeit für die EMK beobachtet wird, so dass man E E RT c Fe ( II ) RT ln 7A (I / c)1 2 . F c Fe ( III ) F (9) erhält. Einerseits kann man nun das Konzentrationsverhältnis c Fe(II)/cFe(III) bei konstanter Ionenstärke I variieren, um die Konzentrationsabhängigkeit der EMK gemäß der Nernst’schen Gleichung in ihrer vereinfachten Form zu überprüfen. Andererseits ist man gemäß Gleichung (9) in der Lage, durch Messen der EMK von äquimolaren Fe(II)/Fe(III)-Lösungen mit verschiedenen Konzentrationen c=cFe(II)=cFe(III), d. h. verschiedenen Ionenstärken I, den Standard-Wert der EMK E° über eine geeignete Extrapolation zu gewinnen. 2) Elektrochemisches Gleichgewicht: Die gesamte Redoxreaktion (1) kann in zwei Teilreaktionen zerlegt werden L : AgCl(s) e Ag(s) Cl (aq) R : Fe(CN) 36 (aq) e Fe(CN) 64 (aq) , (10) die in der linken (L) bzw. rechten (R) Halbzelle der elektrochemischen Zelle ablaufen sollen. Die Änderung der Freien Enthalpie dG L in der linken Halbzelle bei konstantem Druck p und Temperatur T ist gegeben durch ~ ~ ~ ~ dG L AgCl dn AgCl e dn e Ag dn Ag Cl dn Cl , (11) ~ i die elektrochemischen Potentiale der einzelnen Komponenten darwobei die ~ i=i+ziFϕi setzen sich aus dem chestellen. Die elektrochemischen Potentiale mischen Potential i der entsprechenden Komponente und dem elektrischen Potential ϕi der jeweiligen Phase zusammen, wobei zi die Ladung der Komponente angibt. Die Stoffmengenänderungen dni der verschiedenen Komponenten können mit der Änderung einer Umsatzvariable dξ=dni/νi in Verbindung gebracht werden, so dass sich die Änderung der Freien Enthalpie in der linken Halbzelle als ~ ~ ~ ~ dG L ( Ag AgCl e )d Cl (12) schreiben lässt. Die linke Halbzelle befindet sich im elektrochemischen Gleichgewicht, wenn die Freie Enthalpie GL minimal ist, d. h. ~ ~ ~ ~ Ag AgCl e 0 . Cl (13) Setzt man für die elektrochemischen Potentiale die entsprechenden chemischen und elektrischen Potentiale ein, dann ergibt sich eine Potentialdifferenz ΔϕL zwischen der Ag-Elektrode und der wässrigen Lösung L ( Ag Cl AgCl e ) / F R G L / F , (14) wobei ΔRGL die Freie Reaktionsenthalpie für die Reaktion in der linken Halbzelle ist. Eine entsprechende Potentialdifferenz ΔϕR entsteht auch in der rechten Halbzelle zwischen der verwendeten Pt-Elektrode und der wässrigen Lösung R ( FeII FeIII e ) / F R G R / F . (15) Dabei ist ΔRGR die Freie Reaktionsenthalpie für die entsprechende Reaktion in der rechten Halbzelle. Somit baut sich eine elektrische Spannung Δϕ=ΔϕRΔϕL zwischen den beiden Elektroden auf R G R G R R G L F ( R L ) F , (16) die durch die Freie Reaktionsenthalpie ΔRG der gesamten elektrochemischen Reaktion bestimmt ist. Ist ΔRG für die Reaktion negativ, dann ist die elektrische Spannung Δϕ der galvanischen Zelle positiv und die Reaktion läuft in der angegebenen Richtung freiwillig ab. Um zu verhindern, dass die Redoxreaktion freiwillig abläuft und sich somit das chemische Gleichgewicht in der elektro- chemischen Zelle einstellt, muss man die Spannung der galvanischen Zelle durch eine äußere Spannungsquelle kompensieren. Mit Hilfe der äußeren Spannungsquelle kann die Einstellung des chemischen Gleichgewichts in der elektrochemischen Zelle gehemmt werden und es fließt kein Strom durch die elektrochemische Zelle. Dann wird die elektrische Spannung Δϕ der galvanischen Zelle mit der elektromotorischen Kraft E identisch. Somit beschreibt Gleichung (16) den gesuchten Zusammenhang zwischen der elektromotorischen Kraft E und der Freien Reaktionsenthalpie ΔRG. Berücksichtigt man, wie ΔRG von der Zusammensetzung abhängt, dann ergibt sich die Nernst’sche Gleichung (5) für die zu untersuchende Redoxreaktion. Im Versuchsaufbau ist die äußere Spannungsquelle durch ein hochohmiges Voltmeter ersetzt worden. Dabei wird angenommen, dass der Strom, der durch die Zelle fließt, so gering ist, dass die mit dem Voltmeter gemessene Spannung Δϕ gleich der EMK der galvanischen Zelle ist. 3) Konzentrationsabhängigkeit des Aktivitätskoeffizienten J: Um zu verstehen, wie der Aktivitätskoeffizient J von der Konzentration der Elektrolytlösung abhängt, betrachtet man das chemische Potential J eines Ions in der realen Elektrolytlösung J J RT ln J , (17) wobei J° sich auf das chemische Potential eines hypothetisch ungeladenen idealen Lösungszustandes bezieht. Die Differenz (J−J°) ist somit mit der (molaren) elektrischen Arbeit gleichzusetzen, die man braucht um die ungeladene ideale Lösung aufzuladen. Um die molare elektrische Aufladearbeit auszurechnen, benötigt man das elektrische Potential des betrachteten Ions J mit der Ladung zJe. Dazu stellt man sich vor, dass das Zentral-Ion von einer kugelsymmetrischen Ionenwolke mit der Ladung −zJe umgeben ist, die sich in einem mittleren Abstand rD vom Zentral-Ion befindet. Das Coulomb-Potential dieser Ionenwolke im Lösungsmittel mit der Dielektrizitätskonstanten ε ist durch J zJe 1 , 4 rD (18) gegeben, so dass sich die molare Aufladearbeit (J−J°) zu NA J J 4 rD zJe N A (z J e) 2 z J e d(z J e) 2 4 rD , 0 (19) berechnet. Damit ergibt sich der Aktivitätskoeffizient des betrachteten Ions zu N A ( z J e) 2 , ln J 2 4 rD RT (20) d. h. um so kleiner der Abstand rD zwischen dem Zentral-Ion und der ihn umgebenden Ionenwolke ist, desto kleiner wird auch der Aktivitätskoeffizient J. Man kann erwarten, dass der mittlere Abstand rD mit zunehmender Konzentration der Elektrolytlösung c, d. h. zunehmender Ionenstärke I, abnimmt und somit auch der Aktivitätskoeffizient J kleiner wird. Eine genauere Rechnung für sehr verdünnte Lösungen zeigt, dass rD umgekehrt proportional zur Quadratwurzel der Ionenstärke I ansteigt rD 1 (I / c)1 2 . (21) Damit ergibt sich das Debye-Hückel-Grenzgesetz (7) für den Aktivitätskoeffizienten ln J z 2J (I / c)1 2 . (22) Aufgaben: 1) Man überprüfe die Nernst’sche-Gleichung, indem man die EMK E bei konstanter Temperatur T für verschiedene Zusammensetzungen der RedoxHalbzelle misst und E gegen ln(cFe(II)/cFe(III)) aufträgt. Man bestimme aus der Geradensteigung die Faraday-Konstante F und vergleiche mit Literaturdaten. 2) Inwieweit bleibt die Ionenstärke der Elektrolytlösung bei den verschiedenen Konzentrationsverhältnissen cFe(II)/cFe(III) wirklich konstant? 3) Man messe für unterschiedliche äquimolare Fe(II)/Fe(III)-Konzentrationen die EMK. Man berechne die Ionenstärke I für die verschiedenen Konzentrationen c=cFe(II)=cFe(III) und bestimme aus der Auftragung von E gegen (I/c°) 1/2 den Standard-Wert der EMK bei den 3 untersuchten Temperaturen. 4) Man ermittle aus der Temperaturabhängigkeit von E° den Wert für ΔRS° und ΔRH° und berechne damit den Wert für ΔRG° bei 298 K. Man vergleiche das Ergebnis mit Literaturdaten für E°, ΔRG°, ΔRS° und ΔRH°. Zubehör: Thermostat, Pt-Elektrode, Ag/AgCl-Elektrode, Stativ mit Elektrodenhalter, Magnetrührer, div. Bechergläser, Messkolben mit Stammlösungen, Fläschchen mit Fe(II)/Fe(III)-Lösungen, Küchentücher, Schutzhandschuhe. Durchführung: a) Es wird eine Messreihe mit unterschiedlichen Konzentrationsverhältnissen cFe(II)/cFe(III) von Fe(II)- und Fe(III)-Lösungen bei Raumtemperatur durchgeführt. Dazu werden die angesetzten Lösungen verwendet, die aus 0.001 M Fe(II)- und Fe(III)-Ausgangslösungen hergestellt wurden. b) Anschließend wird für verschiedene äquimolare Konzentrationen zwischen c=0.01 und 0.0005 mol/L jeweils eine Messreihe bei Raumtemperatur, bei 30 °C und bei 40 °C aufgenommen. Dazu sollte man die Lösungen im Thermostaten vortemperieren, damit sich die gewünschte Temperatur im Messgefäß nachher schneller einstellen kann (siehe Bedienungsanleitung zum Betrieb von Thermostaten auf Seite 12). Für eine EMK-Messung wird ein 100 mL Becherglas mit ca. 50 mL Lösung gefüllt und auf den Magnetrührer gestellt. Die Elektroden werden in die Halteklammer eingesetzt und soweit abgesenkt, dass sie möglichst tief eintauchen ohne das Magnetrührstäbchen zu berühren (siehe Abb. 1). Dann wird mit dem Voltmeter die Spannung zwischen den Elektroden gemessen. Bevor sich eine konstante Spannung einstellt, driftet der Wert einige Zeit. Diese Zeit muss vor der Messung unbedingt abgewartet werden. Nach Aufnahme des Messwertes wird die Lösung in das Fläschchen zurückgefüllt. Dieses Fläschchen kann dann zur Vortemperierung für weitere Messungen in den Thermostaten gestellt werden. Im Thermostaten haben 5-6 Fläschchen Platz. Bevor man mit der Messung der nächsten Lösung beginnen kann, sind die Elektroden unbedingt mit einem sauberen Küchentuch abzutrocknen. Auch im Glasschutz der Pt-Elektrode darf keine Lösung mehr vorhanden sein. Da die Eisenverbindungen als reizend eingestuft sind, müssen dabei entsprechende Schutzhandschuhe getragen werden. Bei der 30 °C und 40 °C-Messung werden die Lösungen in das temperierte Messgefäß gefüllt und die Temperatur wird mit dem Thermometer kontrolliert. Nach jeder Messung wird das Messgefäß genauso abgetrocknet wie die Elektroden. Ansonsten verläuft diese Messreihe genau wie die vorhergehende. Abb. 1: Aufbau der elektrochemischen Messzelle. Praktische Hinweise: - Die benutzten Küchentücher werden wie verunreinigtes Filterpapier entsorgt und müssen daher gesammelt werden (Plastikbeutel im Abzug). - Spülwasser, in dem sich Reste der Lösungen befindet, wird gesammelt und zum Schluss in den bereitstehenden Restebehälter gefüllt. - Die Ag/AgCl-Elektrode muss am Versuchsende wieder in das Reagenzglas mit KCl-Lösung zurückgestellt werden. 3.13. Rohrzuckerinversion (Versuch Nr. 16) Ziel des Versuches ist, aus der Untersuchung des Polarisationswinkels in Abhängigkeit der Zeit die Geschwindigkeitskonstanten für die Rohrzuckerinversion bei 2 verschiedenen Temperaturen zu messen und daraus die Aktivierungsenergie zu bestimmen. 3.13.1. Vorbereitung • • • • • • • • • Chemische Reaktionskinetik Reaktionsgeschwindigkeit und Geschwindigkeitskonstante Geschwindigkeitsgesetze Elementarreaktionen Stationäre Zustände und vorgelagerte Gleichgewichte Kinetische Formulierung des Massenwirkungsgesetzes Temperaturabhängigkeit der Geschwindigkeitskonstanten (Arrhenius-Gesetz) Theorie des aktivierten Komplexes Polarimetrie 3.13.2. Literatur P. W. Atkins, Physikalische Chemie, 2. Auflage, VCH-Verlag, Weinheim, 1996, S. 813-842, 886-896. G. Wedler, Lehrbuch der Physikalischen Chemie, 4. Auflage; Wiley-VCHVerlag, Weinheim, 1997, S. 166-171, 179-184, 868-873, 876-878, 907-925. 3.13.3. Theoretische Grundlagen Man betrachte die Dissoziation von Saccharose (S) zu Glucose (G) und Fructose (F) in wässriger Lösung C12H 22O11 (aq) H 2O C6H12O6 (aq) C6H12O6 (aq). Saccharose Glucose Fructose (1) Die Freie Standardreaktionsenthalpie ΔRG° für die Dissoziation ist stark negativ und man erwartet daher, dass die Reaktion ausgehend von den Edukten freiwillig abläuft und im chemischen Gleichgewicht fast ausschließlich Glucose und Fructose vorliegen. Trotzdem beobachtet man, dass eine wässrige Lösung von Saccharose stabil ist. Dies hängt damit zusammen, dass die Aktivierungsenergie für die Reaktion hoch und dadurch die Einstellung des chemischen Gleichgewichtes gehemmt ist. Erst wenn man einen geeigneten Katalysator zusetzt, der die Reaktionshemmung vermindert, kann sich der chemische Gleichgewichtszustand in beobachtbarer Zeit einstellen. Bei der Rohrzuckerspaltung wird das durch die Zugabe einer Brönstedt-Säure bewirkt. Die Zugabe von z. B. verdünnter Salzsäure zur wässrigen Saccharose-Lösung führt demnach dazu, dass die Aktivierungsenergie erniedrigt und somit die Reaktionsgeschwindigkeit für die Hydrolyse von Saccharose erhöht wird. Aus der Untersuchung der Reakti- onsgeschwindigkeit kann man daher etwas über die Größe der Aktivierungsenergie lernen. Um die zeitliche Veränderung der Saccharose-Konzentration mit der Geschwindigkeitskonstanten k in Verbindung zu bringen, benötigt man eine Vorstellung über den Mechanismus der Hydrolyse-Reaktion. Für den Ablauf der Reaktion kann man sich nach der Zugabe von Säure vorstellen, dass sich zunächst ein schnelles vorgelagertes Gleichgewicht ausbildet S H+ k1 k1 SH+ , (2) bei dem die Saccharose protoniert wird. Anschließend kann im zweiten Schritt die protonierte Saccharose mit Wasser zu Glucose und Fructose reagieren k2 SH + H 2O G F H+ , (3) wobei das chemische Gleichgewicht fast vollständig auf der Produktseite liegt. Wir können also für die Bildung der protonierten Saccharose SH + nach dem allgemeinen Formalismus der Kinetik folgende Gleichung aufstellen: d [SH+ ] k1[S][H+ ] k2[H2O] k1 [SH + ] dt (4) Für die Produktbildung kann entsprechend formuliert werden: d [G] k2[SH + ][H 2O] dt (5) Für die Geschwindigkeitskonstanten gilt hier k2[H2O] << k−1 (Deprotonierung schneller als Produktbildung) und k1<< k−1 (vorgelagertes Gleichgewicht liegt auf der Eduktseite). Also kann in der Summe (k2[H2O] + k-1) von Gl. (4) der Summand k2[H2O] vernachlässigt werden. Unter diesen Bedingungen ist über einen weiten Zeitraum die Konzentration des Zwischenprodukts nahezu konstant, d. h. d[SH+]/dt ≈ 0 (vorgelagertes Gleichgewicht). Damit vereinfacht sich Gleichung (4) zu: 0 k1[S][H + ] k1[SH + ] [SH + ] k1 [S][H + ] k1 (6) Eingesetzt in Gleichung (5) ergibt sich: d [G] k k2 1 [S][H + ][H 2O] dt k1 (7) Da Wasser als Lösungsmittel im großen Überschuss vorliegt, verändert sich die Wasserkonzentration nur unmerklich, wir können sie als konstant annehmen. Da Protonen hier einerseits im Überschuss vorliegen, andererseits als Katalysator zurückgebildet werden, können wir auch die Protonenkonzentration als konstant annehmen und alle Konstanten zusammenfassen: d [G] k[S] dt (8) Aufgrund der Quasistationarität des Zwischenproduktes gilt für den Eduktverbrauch entsprechend den stöchiometrischen Koeffizienten νs = −νG: d [S] k[S] dt (9) Diese einfache Differentialgleichung für die Reaktion (Pseudo-)1. Ordnung lässt sich durch Variablentrennung integrieren: [S]( t t ) d [S] [S] k dt [S]( t 0) 0 t (10) Es ergibt sich für die Reaktion 1. Ordnung eine exponentielle Abnahme der Eduktkonzentration: [S](t ) [S](0) e kt (11) Mit Hilfe von Gl. (11) kann durch zeitabhängige Konzentrationsmessung die Geschwindigkeitskonstante k bestimmt werden. Diese ist stark temperaturabhängig. Die Temperaturabhängigkeit wird in vielen Fällen und nicht zu großen Temperaturintervallen befriedigend durch das empirische Gesetz von Arrhenius beschrieben: k (T ) A e EA kBT (12) Hierbei sind EA die Aktivierungsenergie für die Reaktion und kB die BoltzmannKonstante. Der Exponentialterm gibt nach Boltzmann den Anteil der Moleküle an, die genügend thermische Energie haben, um die Aktivierungsschwelle zu überwinden, während der prä-exponentielle Faktor A formal die Geschwindigkeitskonstante bei unendlich hoher Temperatur (alle Moleküle haben genügend thermische Energie) angibt. Dieser wird auch als Stoßfaktor bezeichnet, der die Stoßfrequenz (bei für die Reaktion geeigneter geometrischer Ausrichtung) enthält. Temperaturabhängige Messung der Geschwindigkeitskonstanten k erlaubt gemäß Gleichung (10) die Bestimmung der Aktivierungsenergie der Reaktion. Neben dem empirischen Gesetz von Arrhenius gibt es auch theoretisch fundierte Gesetze; hier sei die Theorie des aktivierten Komplexes von Eyring erwähnt. Mit einigen Vereinfachungen und Annahmen kommt man für einfache Reaktionen in der Gasphase zu folgendem Ausdruck: k (T ) kBT e h G kBT kBT e h H T S kBT A T e H kBT (13) Hierbei ist h die Planck’sche Konstante, G ist die freie Aktivierungsenthalpie. Im letzten Schritt wurde angenommen, dass die Aktivierungsentropie über den betrachteten Temperaturbereich konstant bleibt. Die Ähnlichkeit zu Gleichung (12) ist offensichtlich. Die lineare Temperaturabhängigkeit des Vorfaktors, der den Hauptunterschied markiert, fällt gegenüber der Temperaturabhängigkeit des Exponentialterms in kleinen Temperaturintervallen nicht ins Gewicht. 3.13.4. Aufgaben 1) Man untersuche mit dem Polarimeter den Drehwinkel α in Abhängigkeit der Zeit t bei 25 °C und 35 °C und überprüfe durch eine geeignete Auftragung, ob das vorgeschlagene Geschwindigkeitsgesetz pseudo-erster Ordnung richtig ist. 2) Man bestimme die beiden Geschwindigkeitskonstanten k für 25 °C und 35 °C und berechne daraus die Aktivierungsenergie EA und den Stoßfaktor A. Anschließend vergleiche man die erhaltenen Werte mit Literaturdaten. 3.13.5. Zubehör Polarimeter mit Na-Dampf-Lampe, Umwälzthermostat, Bechergläser, Pipetten, Rohrzucker, Salzsäure. 3.13.6. Durchführung Die Konzentration des Rohrzuckers cS wird mit einem Halbschattenpolarimeter gemessen (Abb. 1). Das Charakteristische dieses Apparates ist der Polarisator, der aus zwei Polarisationsprismen besteht, deren Größe so bemessen ist, dass sie jeweils die Hälfte des Gesichtsfeldes einnehmen. Entscheidend für die Anordnung ist, dass der Hauptschnitt H' des Halbprismas P' um einen kleinen Winkel ε gegenüber dem Hauptschnitt H des Halbprismas P gedreht ist (Abb. 2a). Kreuzt man den Analysator A mit P', so ist die rechte Seite des Gesichtsfeldes dunkel und die linke mäßig hell (Abb. 2c). Dreht man den Analysator A, so dass er senkrecht zur Winkelhalbierenden zwischen den beiden Hauptschnitten H und H' steht, erscheinen beide Gesichtsfelder gleich hell (Abb. 2d), was das Auge mit großer Genauigkeit feststellt. Die Analysatorstellung lässt sich mit Hilfe von Nonien auf etwa 0,1 Grad ermitteln. Dabei berücksichtigt man, dass bei nicht zu konzentrierten Lösungen der Drehwinkel α der Ebene des linear polarisierten Lichtes proportional zur Konzentration an gelöster (optisch aktiver) Substanz ist. Zu Beginn der Reaktion ergibt sich ein Drehwinkel von α0 = βcS0, während man am Ende der Reaktion α∞ = β'cS0 erhält. Dabei ist β der spezifische Drehwinkel von reiner Saccharose und β' der von Invertzucker. Für beliebige Reaktionszeiten t ergibt sich der Drehwinkel zu α = βcS +β'cS0−cS). Damit ist man in der Lage das Konzentrationsverhältnis von cS/c S0 durch cS , cS0 0 (14) auszudrücken. Es wird daher eine Messung bestehend aus der Bestimmung des Ausgangsdrehwinkels α0, dem zeitlichen Verlauf der Drehung des Winkels α und der Bestimmung des Enddrehwinkels α∞ bei Raumtemperatur und 35 °C ausgeführt. (2 Nicol-Prismen) (2 Nicol-Prismen) Okular Abb. 1: Halbschattenpolarimeter. Abb. 2: Okularbilder beim Abgleich (d → Abgleichsposition). Dazu wird zunächst aus 22.5 g Rohrzucker und 127.5 g dest. H2O eine 15%-ige Lösung hergestellt und diese in einem der beiden Temperiergefäße des Thermostatkreislaufs vortemperiert (siehe Bedienungsanleitung zum Betrieb von Thermostaten auf Seite 12). Dann wird der Drehwinkel α0' gemessen. Zum Starten der Reaktion bei t = 0 werden 20 mL der Zuckerlösung mit 20 mL ca. 2N HCl, die ebenfalls in einem Temperiergefäß vorgewärmt wurde, verdünnt und schnell intensiv vermischt (siehe Hinweise zur Volumenmessung auf Seite 15). Daher ist der Ausgangsdrehwinkel α0 = α0'/2. Die Küvette des Polarimeters (Inhalt ca. 20 ml) wird zunächst mit dem Reaktionsgemisch vorgespült und anschließend damit gefüllt. Dann wird der Drehwinkel α in folgenden Zeitintervallen gemessen: 5 × 3 Min., 4 × 5 Min., 1 × 10 Min. Weitere 40 mL Reaktionsmischung werden während dieser Zeit in einem Wasserbad kurz (5-10 Min.) auf ca. 70 °C erwärmt (nicht überhitzen), um damit später die Enddrehung β (bei der gleichen Temperatur wie die vorhergehende Messung) bestimmen zu können. 3.13.7. Zusatzinformation Warum drehen optisch aktive Medien die Polarisationsebene linear polarisierten Lichts? Auch wenn die exakte Wechselwirkung zwischen den Elektronen eines Licht brechenden Mediums und den Photonen des Lichts kompliziert ist und klassisch nicht beschrieben werden kann, kann die Drehung der Polarisationsebene dennoch leicht prinzipiell verstanden werden. In einem Licht brechenden Medium ist die Ausbreitungsgeschwindigkeit des Lichts gemäß Gl. (14) herabgesetzt: cMedium cVakuum nMedium (15) Hierbei ist nMedium der Brechungsindex des Mediums. Er hängt von der Photonen-Elektronen-Wechselwirkung ab. Diese Wechselwirkung ist in einem chiralen Medium ebenfalls chiral, da die Elektronenverteilung chiral ist. Man kann sich linear polarisiertes Licht als Überlagerung von linksdrehend zirkular polarisiertem und rechtsdrehend zirkular polarisiertem Licht vorstellen. Diese beiden chiralen Komponenten wechselwirken in einem chiralen Medium unterschiedlich mit den Elektronen; der Brechungsindex unterscheidet sich also für linksdrehend und rechtsdrehend zirkular polarisiertes Licht. Die Ausbreitungsgeschwindigkeit der beiden Komponenten ist also unterschiedlich; es entsteht eine Phasenverschiebung. Vektoraddition der beiden phasenverschobenen Lichtkomponenten ergibt eine Drehung der Polarisationsebene. 3.14. Moleküle in Bewegung (Versuch Nr. 25) Dieser Versuch besteht aus zwei Teilen. Das Phänomen der Diffusion soll über einerseits makroskopisch über die Drift von farbigen Permanganat-Ionen im elektrischen Feld, andererseits mikroskopisch über Betrachtung der Brown’schen Bewegung von Latex-Partikeln unter dem Mikroskop erfahrbar gemacht werden. 3.14.1. Vorbereitung • • • • • • • Fick’sche Gesetze und Diffusion Wahrscheinlichkeitsdichte Brown’sche Molekularbewegung Langevin-Gleichung Stokes-Einstein-Gleichung Nernst-Einstein-Gleichung Einstein-Smoluchowski-Gleichung 3.14.2. Literatur P. W. Atkins, Physikalische Chemie, 2. Auflage, VCH-Verlag, Weinheim, 1996, S. 771-783, 787-807. G. Wedler, Lehrbuch der Physikalischen Chemie, 4. Auflage; Wiley-VCHVerlag, Weinheim, 1997, S. 187-215, 814-821. 3.14.3. Theoretische Grundlagen a) Die Fick’schen Gesetze Im Jahre 1855 veröffentlichte der Physiologe A. Fick seine bekannten Gesetze zur Diffusion. Das 1. Fick’sche Gesetz besagt, dass der Teilchenfluss über eine Fläche A proportional zum Konzentrationsgradient ist. jn dn dc D Adt dx (1) Die temperaturabhängige Proportionalitätskonstante D wird Diffusionskoeffizient genannt und ist ein Maß für die Beweglichkeit der diffundierenden Teilchen. (Es sei erwähnt, dass auch konzentrationsabhängig sein kann, und im allgemeinen nicht-isotropen 3d-Fall kein Skalar, sondern ein Tensor 2. Ordnung ist.) Das 1. Fick’sche Gesetz beschreibt also das aus dem Alltag bekannte Phänomen, dass sich Konzentrationen angleichen bzw. Stoffe sich vermischen. Aus dem 1. Fick’schen Gesetz folgt bei nicht-stationären Bedingungen das 2. Fick’sche Gesetz, das die zeitliche Entwicklung von Konzentrationsprofilen angibt (hier für den eindimensionalen Fall): c 2c D 2 t x (2) Diese allgemeine Diffusionsgleichung kann man kann man für verschiedene Randbedingungen lösen, um die zeitliche Fortentwicklung von Konzentrationsprofilen c(x, t) zu berechnen. Um die Charakteristik der Diffusion zu erfassen, wählt man als Randbedingung, dass zum Zeitpunkt t = 0 die gesamte Stoffmenge am Ort x = 0 ist (mathematisch wird dies durch eine sogenannte δ-Funktion ausgedrückt). Damit ergibt sich für einen in x-Richtung unendlich ausgedehnten Quader der Fläche A Raum folgende Lösung: c( x,0) c( x, t ) n ( x) A n A 4 Dt x2 4 e Dt (3) (Es sei hier noch erwähnt, dass das Integral der Konzentration über den gesamten Raum (hier die Dimension x) unabhängig von t immer die Stoffmenge n ergibt.) Die Charakteristische Lösung der Diffusionsgleichung sind also GaußFunktionen. In Abb. 1 sind die Konzentrationsprofile c(x, t) für den oben formulierten Fall dargestellt: Abb. 1: Charakteristische Konzentrationsprofile für 1dDiffusion, Startbedingung: Dirac’sche δ-Funktion. Die Tatsache, dass Gauß-Funktionen, die auch stochastische (zufällige) Prozesse beschreiben, charakteristische Lösung der Differentialgleichung sind, ist schon ein erster Hinweis auf die stochastische Ursache der Diffusion. Tatsächlich ist wird Diffusion heute als der Translationsanteil der Wärmebewegung verstanden. Sie findet also permanent statt, unabhängig davon, ob ein Konzentrationsgradient vorliegt, oder nicht. Falls ein Konzentrationsgradient vorliegt, führt diese stochastische, ungerichtete Bewegung zu einem NettoTeilchenfluss (vgl. 1. Fick’sches Gesetz), der das Phänomen einfach messbar und erfahrbar macht. Für diese mikroskopische Betrachtungsweise der Diffusion muss das 2. Fick’sche Gesetz lediglich uminterpretiert werden. An die Stelle der Konzentra- tion (Teilchendichte) c(x, t) tritt die Wahrscheinlichkeitsdichte p(Δx, t), die angibt, wie wahrscheinlich es ist, dass ein Teilchen sich bis zum Zeitpunkt t = t′ um die Strecke Δx von dem Ort entfernt hat, an dem es sich zum Zeitpunkt t = 0 aufgehalten hat. p t D 2 p (4) x 2 Es ergibt sich die charakteristische Lösung p (x, t ) 1 e Dt x 2 4 Dt . (5) (Um von der Wahrscheinlichkeitsdichte zur Wahrscheinlichkeit zu gelangen, muss man die Wahrscheinlichkeitsdichte über ein bestimmtes Segment, das man betrachtet, integrieren. Man erhält dann die Wahrscheinlichkeit, das Teilchen in diesem Segment anzutreffen.) Eine charakteristische Größe für die Breite Verteilung zu einem bestimmten Zeitpunkt ist das 2. Moment oder mittlere Verschiebungsquadrat (Δx)²: x 2 (t ) 2 Dt (6) Zusammenfassend lässt sich also festhalten: i) Diffusion ein stochastischer, ungerichteter Prozess. ii) Diffusion findet als Translationsanteil der Wärmebewegung immer statt. iii) Diffusion führt infolge von i) und ii) zu Vermischung und Konzentrationsausgleich. iv) Alle bisherigen Überlegungen beziehen sich auf ideale Mischungen. Für nicht-ideale Mischungen sollten chemische Potentiale anstelle von Konzentrationen betrachtet werden. Spätestens dann sollte auf die Theorie der irreversiblen Prozesse zurückgegriffen werden. Im Rahmen dieses Versuchs werden zwei Experimente durchgeführt, um die bisherigen Überlegungen zu überprüfen: a) Diffusion eines Farbstoffes in einer Petrischale zur Betrachtung der Diffusion bei Vorliegen eines Konzentrationsgradienten. Hierbei kann mit einer für die Versuchsbedingungen angepassten Lösung von Gleichung (2) gearbeitet werden, um D zu bestimmen (wird zum jetzigen Zeitpunkt noch nicht angeboten). b) Brown’sche Bewegung von suspendierten Partikeln zur Betrachtung der Diffusion ohne Konzentrationsgradient. Hierbei wird erfahrbar, dass Moleküle und auch sehr kleine suspendierte Partikel sich tatsächlich im Gleichgewicht weiterhin bewegen. Es wird mit Gleichung (6) gearbeitet, um den Diffusionskoeffizienten D zu bestimmen. b) Die Brown’sche Bewegung suspendierter Partikel Im nächsten Teil wollen wir versuchen, die Diffusionskoeffizienten D mit anderen physikalischen Eigenschaften zu korrelieren. Dazu betrachten wir zunächst die Brown’sche Bewegung suspendierter Partikel: Betrachtet man kleine suspendierte Partikel unter dem Mikroskop, so sieht man, dass diese zitternde Zick-Zack-Bewegungen durchführen. Diese Bewegung fällt umso stärker aus, je kleiner die Partikel sind. Man führt die Zitterbewegung darauf zurück, dass die Stöße der diffundierenden Lösungsmittelmoleküle (hier Wasser), die von allen Seiten auf das Teilchen wirken, sich nicht zu jedem Zeitpunkt vollständig kompensieren. Daher wirkt eine zeitlich veränderliche Kraft auf das suspendierte Partikel. Mathematisch lässt sich das über eine Newton’sche Gleichung ausdrücken: dp mdv F (t ) dt dt (7) p bezeichnet hier den Impuls, nicht den Druck(!). Die Impulsänderung ist also gleich der zeitlich veränderlichen Kraft F(t). Zunächst einmal können wir nicht viel über die Kraft wissen. Wir können allerdings überlegen, dass ein Teilchen, das sich mit einer bestimmten Geschwindigkeit fortbewegt, von „vorne“ mehr Stöße erfahren wird als von „hinten“. Wir können die zeitabhängige Kraft F(t) also in zwei Terme aufspalten; einen abbremsenden Term, der von der Geschwindigkeit des Partikels abhängt, und einen, der die rein stochastischen Stöße beschreibt, die ein (gedachtes) ruhendes Teilchen erfahren würde. dp Fstoch (t ) v dt (8) Für den abbremsenden Term wurde eine Taylor-Reihen-Entwicklung 1. Ordnung angesetzt. In erster Näherung kann man für den Koeffizienten α das Stokes’sche Reibungsgesetz annehmen, das für makroskopische Teilchen bei laminarer Strömung gilt: dp Fstoch (t ) 6 rv dt (9) Hierbei sind die Viskosität des Lösungsmittels und r der Radius des Partikels. Als nächstes versuchen wir, Aussagen über die stochastische Kraft Fstoch(t) zu treffen. Aus Symmetriegründen sollte sie im zeitlichen Mittel null ergeben; ansonsten wäre damit ein Impuls des gesamten Lösungsmittels verbunden. Ebenfalls aus Symmetriegründen sollte sie im Mittel ortsunabhängig sein. Bildet man das Mittel über viele Teilchen, gilt für dieses ergodische System: Fstoch (t ) 0 x Fstoch (t ) 0 (10) Die Gleichungen (7) bis (9) beschreiben jeweils die Impulsänderung durch eine Kraft. Würde man die Kraft explizit kennen, könnte man über Integration den Ort des Partikels zu jedem beliebigen Zeitpunkt bestimmen. Dies entzieht sich unseren Möglichkeiten, da wir nur eine mittlere Kraft kennen. Allerdings können wir versuchen, die Differentialgleichung (9), die auch Langevin’sche Gleichung genannt wird, im Mittel zu lösen, also z. B. ein mittleres Verschiebungsquadrat abzuleiten. Dafür formulieren wir Gleichung (9) um: m d 2x dx F ( t ) 6 r stoch dt dt 2 (11) Nach Multiplikation mit x und Anwendung der Kettenregel ergibt sich: 2 m d 2 x2 dx 2 dx m x Fstoch (t ) 3 r 2 dt 2 dt dt (12) Diese nicht ganz triviale Umformung ist „rückwärts“ leichter nachzuvollziehen. Nun mitteln wir über sehr viele Teilchen und erhalten mit Hilfe des Gleichverteilungssatzes mv 2 2 kBT 2 und Gl. (10.2) x Fstoch (t ) 0 : 1 2 md x 2 dt 2 2 kBT 3 r d x2 dt (13) Diese Differentialgleichung hat folgende allgemeine Lösung: d x2 dt k T B Ce 3 r 6 r t m (14) Der zweite Exponentialterm mit der unbestimmten Konstante C klingt sehr schnell ab. Für nicht zu kurze Zeiten ergibt sich durch weitere Integration: x 2 (t ) kBT t 3 r (15) Vergleich mit Gleichung (6) ergibt die Stokes-Einstein-Beziehung: D kBT 6 r (16) Nehmen wir an, dass das Stokes’sche Reibungsgesetz auch bei noch kleineren Dimensionen eine sinnvolle erste Abschätzung ist, so bietet Gleichung (16) die Möglichkeit, Molekülgrößen beispielsweise farbiger Moleküle aus Diffusionsexperimenten abzuleiten. 1 Man überzeuge sich, dass für stetige und differenzierbare Funktionen das Mittel ihrer Ableitungen gleich der Ableitung der gemittelten Funktion ist, indem man eine Taylor-Entwicklung annimmt und über die Koeffizienten mittelt. c) Die Bewegung von gelösten Ionen im elektrischen Feld Eine andere Möglichkeit, Diffusionskoeffizienten zu bestimmen, ist Messung der Driftgeschwindigkeit im elektrischen Feld. Um die Bewegung von gelösten Ionen im elektrischen Feld zu beschreiben, muss man die Langevin-Gleichung (9) um den Term der elektrischen Kraft erweitern: dp Fstoch (t ) 6 rv qE dt (17) Mittelung über sehr viele Teilchen ergibt: dp Fstoch (t ) 6 rv qE dt dv m 6 r v qE dt (18) Die mittlere Geschwindigkeit der Ionen v (= Driftgeschwindigkeit) steigt an, bis sich die beiden Summanden auf der rechten Seite kompensieren. Dann bleibt die Driftgeschwindigkeit konstant: v qE uE 6 r (19) Hierbei werden die stoff- und lösungsmittelspezifischen Konstanten zur Driftbeweglichkeit u zusammengefasst. Vergleich von Gleichung (16) und (19) ergibt die Nernst-Einstein-Beziehung: D ukT q (20) 3.14.4. Aufgaben 1) Man verfolge die zweidimensionale Zickzackbewegung von zwei suspendierten Partikeln, die einen unterschiedlichen Radius r besitzen. 2) Man bestimme die mittleren Verschiebungsquadrate x2 und y2 in der xund y-Richtung und berechne daraus für die beiden Teilchen den Diffusionskoeffizienten D. Anschließend kann man mit Hilfe der Stokes-Einstein Gleichung die Avogadro-Konstante bestimmen. Dabei nimmt man an, dass für die Viskosität der Lösung der entsprechende Wert des Lösungsmittels eingesetzt werden darf. 3) Man bestimme die Driftgeschwindigkeit v von MnO4−-Anionen in Wasser und berechne daraus die Ionenbeweglichkeit u, die molare Ionenleitfähigkeit λ = zFu und den effektiven Radius r von MnO4−. Man vergleiche die Werte mit Literaturdaten. 4) Man bestimme den Diffusionskoeffizient von Permanganat-Ionen in Wasser aus der Messung der Driftgeschwindigkeit. 5) Man bestimme den Diffusionskoeffizient von Permanganat-Ionen aus dem Diffusionsversuch in der Petrischale und vergleiche sie mit dem aus 4) erhaltenen Diffusionskoeffizienten. 6) Man trage alle Diffusionskoeffizienten aus 1) bis 5) gegen den Radius auf und extrapoliere auf den abgeschätzten Radius von Wasser. Den damit erhaltenen Selbstdiffusionskoeffizienten von Wasser vergleiche man mit einem Literaturwert. 3.14.5. Zubehör Zu a): Flachkammer, Gleichspannungsnetzgerät, Thermometer, KMnO4, KNO3. Zu b): Lichtmikroskop mit Kamera und Netzgerät, Fernseh-Monitor, Zählkammer, Objektträger, Deckplättchen, Latex-Suspension. 3.14.6. Durchführung Zu a): Es werden zwei Elektrolytlösungen benötigt: Elektrolyt I (farblos): 0,01 molare Kaliumnitrat-Lösung Elektrolyt II (farbig): 0,06 molare Kaliumpermanganat-Lösung. Eine Deckplatte auf die Laufrinne legen. Die Vertiefung an der Anodenseite her tropfenweise mit dem Elektrolyt I füllen, bis sich durch Kapillarwirkung auch die Laufrinne füllt. Dieser Vorgang kann ggf. durch leichtes Hin- und Herbewegen der Deckplatte in Längsrichtung unterstützt werden. Die Laufrinne muss bis kurz vor der kathodenseitigen Vertiefung blasenfrei gefüllt sein, keinesfalls darf der farblose Elektrolyt in die Vertiefung gelangen. In die kathodenseitige Vertiefung mit einer zweiten Pipette tropfenweise den farbigen Elektrolyt II füllen, bis die Lösung gerade die Übergangsstelle zwischen Vertiefung und Laufrinne erreicht. Damit der Kontakt der beiden Lösungen möglichst genau an der Übergangsstelle erreicht wird, nun vorsichtig nochmals 1 bis 2 Tropfen farblosen Elektrolyt I in die anodenseitige Vertiefung einfüllen. Die beiden Kammern sollten vor dem Einschalten der Spannung ungefähr gleich hoch gefüllt sein und die Schichtgrenze nicht wandern (eventuell ca. 10 min. warten). Nach Anlegen der Spannung (30 V) wandern die violetten MnO4−-Ionen in Richtung auf die Anode. Es ist eine breite Farbsäule mit etwas diffuser Frontlinie zu sehen. Fließt nach Anlegen der Spannung kein Strom, so kann der Flüssigkeitsstand in den Vertiefungen zu niedrig und somit der Stromkreis unterbrochen sein. Nach jedem Versuch ist die Flachkammer mit einer Spülmittellösung zu reinigen und anschließend mit destilliertem Wasser abzuspülen. Mit organischen Lösungsmitteln darf die Kammer nicht behandelt werden, da diese das Kammermaterial (Plexiglas) angreifen. Abb. 1: Flachkammer mit Abdeckplättchen. Zu b): In einem optischen Mikroskop wird die Brown’sche Molekularbewegung von suspendierten Latex-Kügelchen untersucht. Dazu wird das Bild des Mikroskops über eine Videokamera auf einen Fernseh-Monitor übertragen. Dort werden auf Klarsichtfolien die Trajektorien von zwei Teilchen unterschiedlicher Größe nacheinander für 15 Minuten verfolgt. Dazu wird nach jeweils 30 Sekunden das untersuchte Teilchen durch einen Punkt auf der Folie markiert. Die Kalibrierung des zurückgelegten Weges erfolgt mit einem in die Zählkammer eingeritzten Maßstab, der ebenfalls auf der Folie eingezeichnet werden muss. Damit kann dann auch der Durchmesser 2aB der untersuchten Teilchen ausgemessen werden. Die Temperatur der Suspension muss abgeschätzt werden. 3.15. Wasserstoffatom (Versuch 21) Ziel des Versuches ist, die Ionisierungsenergie und das Termschema des Wasserstoffatoms spektroskopisch zu bestimmen. 3.15.1. Vorbereitung • • • • • Bohr’sches Atommodell Ionisierungsenergie und Rydbergkonstante von Wasserstoff De-Broglie-Beziehung Quantenmechanische Beschreibung des H-Atoms Beugung am Spalt bzw. Strichgitter 3.15.2. Literatur P. W. Atkins, Physikalische Chemie, 2. Auflage, VCH-Verlag, Weinheim, 1996, S. 343-353, 395-410, 418-427. G. Wedler, Lehrbuch der Physikalischen Chemie, 4. Auflage; Wiley-VCHVerlag, Weinheim, 1997, S. 108-111, 119-140, 514-547, 556-560. 3.15.3. Theoretische Grundlagen 1) Emissionsspektrum von atomarem Wasserstoff Das Emissionsspektrum von Wasserstoff, dessen Anteil im sichtbaren Bereich (Balmer-Serie) in diesem Versuch aufgenommen wird, sieht folgendermaßen aus: Abb. 1: Emissionsspektrum von atomarem Wassertsoff (Bild entnommen aus http://www.pci.uzh.ch/teaching/Download/grundlagenchemieseeger.html). Empirisch wurde schon im 19. Jahrhundert von J. Balmer festgestellt, dass die Wellenlängen folgendem Zusammenhang gehorchen: 1 1 RH 2 2 n n 1 n n n (1) Hierbei ist λ die Wellenlänge der emittierten Strahlung, RH ist die sog. Rydbergkonstante. Der Physikalische Hintergrund war damals noch vollkommen unklar. Allein die Tatsache, dass ein diskretes Linienspektrum und nicht ein kontinuierliches Spektrum auftritt, war unverstanden, geschweige denn, dass für Formel (1) eine theoretische Begründung geliefert werden konnte. 2) Bohr’sches Atommodell Mit dem Ende des 19. und im ersten Jahrzehnt des 20. Jahrhunderts hinzugekommenen Erkenntnissen (Existenz von Proton und Elektron, sowie Quantelung der Lichtenergie gemäß EPhoton = hν) versuchte Niels Bohr, eine theoretische Erklärung für die Balmer’sche Formel zu finden. In seinem Modell zum H-Atom nimmt Bohr an, dass das Elektron um das Proton kreist – ähnlich wie die Erde um die Sonne. An die Stelle der Gravitationskraft tritt hier die Coulombkraft, die mit der Zentrifugalkraft im Gleichgewicht steht: me v(r ) 2 e2 FZ =FC r 4 0 r 2 mv(r ) 2 e2 (2) 4 0 r Hierbei sind me die Masse des Elektrons, v(r) die bahnabhängige Geschwindigkeit des kreisenden Elektrons, r der Radius der Bahn, e die Ladung des Elektrons und ε0 die Dielektrizitätskonstante des Vakuums. Betrachtet man die Gesamtenergie E(r), die einem Elektron auf einer Bahn mit Radius r zukommt, so setzt sich diese aus der kinetischen und aus der potentiellen Energie des Elektrons T(r) und V(r) zusammen: E (r ) T (r ) V (r ) m v(r ) 2 e FC dr 2 r me v(r ) 2 e2 dr 2 2 4 r 0 r (3) me v(r ) 2 e2 2 4 0 r Die Integration in der zweiten Zeile beschreibt, wie sich die Potentielle Energie V(r) ändert, wenn man ein Elektron von unendlicher Entfernung bis zum Radius r an ein Proton annähert. Durch Vergleich mit der Bahnbedingung Gleichung (2) kann man den ersten Summanden ersetzen. Es ergibt sich für die Gesamtenergie des Elektrons: E (r ) e2 8 0 r (4) Das Vorzeichen bedeutet, dass das Atom stabil ist – es handelt sich um einen energieärmeren Zustand als die Dissoziation. Allerdings erklärt Gl. (4) noch nicht das Auftreten eines Linienspektrums, da es keine Einschränkung gegenüber der Wahl von r gibt, und damit auch eine kontinuierliche Energie. Ein noch gravierenderes Problem des Modells ist, dass gemäß den Maxwell’schen Gleichungen (Elektrodynamik) eine rotierende Ladung Strahlung aussendet. Demnach müsste das System Energie verlieren und kollabieren, was im Widerspruch zur Existenz der Atome steht. N. Bohr stellte daher folgende Postulate auf, um seine Modellvorstellungen mit der Realität in Einklang zu bringen: 1. Nur bestimmte Bahnradien, deren Drehimpuls ein Vielfaches von ħ = h/2π beträgt, sind erlaubt. (Diese Bedingung ist nicht willkürlich gewählt, sondern ergibt sich aus dem Bohr’schen Korrespondenzprinzip.) 2. Die Bahnen sind strahlungslos. 3. Wenn ein Elektron von einer höheren Bahn in eine niedrigere springt, wird die Energiedifferenz zwischen den Bahnenergien E(r) − E(r′) als Photon ausgestrahlt. Durch Absorption eines Photons kann das Elektron entsprechend in eine höhere Bahn springen. Mit der Quantenbedingung des ersten Postulats mvr = nħ (n = 1, 2, 3, …) und Gl. (3) und (4) ergibt sich für die Gesamtenergie der erlaubten Bahnen: En e4me 1 8 02h2 n2 (5) Aus dem 3. Postulat folgt nun für die Energie des abgestrahlten Photons: EPhoton e4me 1 1 En En 2 2 2 2 8 0 h n n (6) Durch Vergleich mit der empirischen Formel von Balmer (Gl. 1) ergibt unter Benutzung von c = λν erhält man für die Rydbergkonstante: e4me RH 8 0 2h3c (7) 3) Erfolge und Probleme des Bohr’schen Modells Erfolge: Unter Einführung eines einfachen Modells und einer einfachen Quantenbedingung kann die empirische Formel wiedergegeben werden. Rückführung der Rydbergkonstanten auf Naturkonstanten Entwicklung des Konzepts gequantelter Atomenergien und Absorption/ Emission als Quantensprünge. Probleme: Widerspruch zu Elektrodynamik letztlich nicht befriedigend gelöst. Quantenbedingung nicht ausreichend theoretisch begründet. Modell versagt bei Mehrelektronensystemen. Aufspaltung von Linien in Magnetfeld nicht erklärbar. 4) Der Weg zu Schrödingergleichung und Quantenmechanik Das Bohr’sche Atommodell hatte eines klar gezeigt: Die Energieniveaus in Atomen sind gequantelt. Diskrete Energieniveaus kannte man schon aus einem ganz anderen Gebiet der Physik: aus der Wellenmechanik. Bei einer schwingenden Saite beispielsweise werden können sich nur bestimmte Schwingungen ausbilden und andere nicht. Die Schwingungsfrequenz ist also gequantelt. Stehende Wellen der Art Ψ(x, t) = ψ(x) ∙ cos(ωt + φ) werden mathematisch allgemein durch folgende Differentialgleichung ausgedrückt: d 2 ( x) 4 2 2 ( x) dx 2 (8) Die Idee war nun, diese Differentialgleichung, die bei der Einführung von Randbedingungen nur bestimmte Lösungen hat, zu nutzen, um Atome zu beschreiben. 1922 stellte L. de Broglie in seiner Dissertation die These auf, dass der Welle-Teilchen-Dualismus sich nicht nur auf Photonen bezieht, sondern auch auf Teilchen mit Ruhemasse, z. B. Elektronen (dies konnte erst 1929 experimentell bestätigt werden). Er stellte folgende Beziehung zwischen der Welleneigenschaft λ (Wellenlänge) und der Teilcheneigenschaft p (Impuls) auf: h p (9) Setzt man die de-Broglie-Beziehung in Gl. (8) ein, so ergibt sich die eindimensionale Schrödingergleichung. Dazu betrachtet man zunächst den Faktor 1/λ2: p2 2m 2m T E V 2 h2 h2 h2 1 (10) Eingesetzt in Gleichung 8 ergibt sich: d 2 ( x) 8 2m E V 2 ( x) dx 2 h (11) Nach einigen Umstellungen ist dies die eindimensionale stationäre Schrödingergleichung: d 2 ( x) V ( x) ( x) E ( x) 2m dx 2 2 (12) 5) Ausblick: Quantenmechanische Beschreibung des H-Atoms Um das H-Atom mit Hilfe von Gl. (12) beschreiben zu können, muss man einerseits berücksichtigen, dass es sich um ein 3-dimensionales Problem handelt, und andererseits die bekannte potentielle Energie des Elektrons (Gleichung (3)) einfügen: e2 E 2m 4 0r 2 (13) Hierbei steht Δ für den Laplace-Operator, dessen Projektion in kartesische Koordinaten ergibt: kart d2 d2 d2 2 2 2 dy dz dx Für die Lösung dieser Differentialgleichung sei auf andere Lehrveranstaltungen oder auf Lehrbücher verwiesen. Als Lösung ergeben sich in vollkommener Übereinstimmung mit dem Bohr’schen Modell folgende Energieeigenwerte: e4me 1 En 2 2 2 8 0 h n (5) Die zugehörigen Wellenfunktionen (= Eigenfunktionen), die sich in einen Radialanteil Rn,l(r) (Laguerre-Polynome) und einen winkelabhängigen Teil Yl,m(θφ) (Kugelflächenfunktionen) aufspalten lassen, lauten: (r , , ) Rn,l (r ) Yl ,m ( , ) (14) Wie bei für eine 3d-Welle eines gebundenen Teilchens zu erwarten, ergeben sich 3 Quantenzahlen. Dies sind 1. die Hauptquantenzahl n, die möglichen Zustände der Radialkomponente der Wellenfunktion beschreibt und der Quantenzahl des Bohr’schen Modells entspricht; 2. die Nebenquantenzahl l, die die Symmetrieeigenschaften der Wellenfunktion (s, p, d, ...) beschreibt; 3. die Magnetquantenzahl m, die die Ausrichtung der Wellenfunktion beschreibt (px, py, pz). Außerdem gibt es noch eine zusätzliche Quantenzahl, die auf den Spin des Elektrons zurückzuführen ist, was hier nicht betrachtet wurde. Das Betragsquadrat der (normierten) Wellenfunktion hat die physikalische Bedeutung einer Wahrscheinlichkeitsdichte. Sie ist also ein Maß für die Aufenthaltswahrscheinlichkeit des Elektrons. Gleichzeitig kann sie verwendet werden, um Erwartungswerte aller physikalischen Größen zu berechnen. 3.15.4. Aufgaben 1) Mit Hilfe der gelben Na-D-Linie kalibriert man das Goniometer und bestimmt anschließend die Wellenlängen aller sichtbaren Linien im Wasserstoffspektrum. 2) Im nächsten Schritt muss man den gemessenen Wasserstofflinien die Quantenzahlen n und n' zuordnen. Dazu betrachte man das Frequenzverhältnis νn+1/νn+2 von zwei benachbarten Emissionsübergängen mit n' = n + 1 n und n' = n + 2 n einer Spektralserie n1n n 2 n 1 1 n 2 (n 1) 2 (2n 1)(n 2) 2 . 2 1 1 4( n 1)( n 1) n 2 (n 2)2 (10) Man nehme an, dass zwei der benachbarten Linien im beobachteten Spektrum zu Übergängen von diesem Typ gehören und finde durch Ausprobieren, d. h. durch Einsetzen von verschiedenen Werte für n, heraus, zu welcher Spektralserie die beobachteten Linien gehören. 3) Man vergleiche die den Quantenzahlen n und n' zugeordneten Wellenlängen bzw. Frequenzen der beobachteten Spektrallinien mit Literaturdaten. 4) Durch Auftragen der gemessenen Frequenzen ν der einzelnen Spektrallinien gegen 1/n'2 bestimme man die Rydbergkonstante RH und die Ionisierungsenergie EIP des H-Atoms und vergleiche die experimentell bestimmten Werte mit Literaturwerten. Außerdem vergleiche man die Werte mit theoretisch berechneten Werten ohne und mit Berücksichtigung der endlichen Masse des Atomkerns. 5) Man konstruiere das Termschema (Grotrian-Diagramm) des H-Atoms und diskutiere die Unterschiede zum Termschema der Alkalimetalle. 6) Man vergleiche das Bohr’sche Atommodell mit der quantenmechanischen Beschreibung des H-Atoms durch die stationäre Schrödingergleichung. Welche Vorstellungen des Bohr’schen Modells müssen korrigiert werden? 3.15.5. Zubehör Natriumdampflampe, Wasserstofflampe, Gitter, Spektralapparat mit Goniometer. 3.15.6. Durchführung Das Strichgitter wird zunächst im Gitterhalter fest eingeklemmt und dann auf dem arretierten Prismentisch so positioniert, dass es senkrecht zur Spaltrohrachse steht. Man muss nun die Spektrallampe so einrichten, dass man die verschiedenen Beugungsordnungen im Fernrohr beobachten kann. Der Drehwinkel αz, den man am Winkelmesser (Goniometer) einstellen muss, um eine Spektrallinie im Fadenkreuz zu beobachten, ist über folgende Beziehung mit der Wellenlänge λ der Spektrallinie verknüpft G (9) sin( z ) , z wobei z die Beugungsordnung und G die Gitterkonstante, d. h. der mittlere Abstand zwischen zwei Spalten im Strichgitter, ist. Um den gemessenen Drehwinkel einer Wellenlänge zuordnen zu können, muss man die Gitterkonstante des Strichgitters bestimmen. Dazu beobachtet man bei der Na-Spektrallampe die hell-leuchtende gelbe Na-D-Linie in 1. Beugungsordnung. Mit der bekannten Wellenlänge von λD = 589.29 nm kann man G bestimmen. Dabei geht man allerdings am besten immer so vor, dass man in positiver und negativer Richtung den Beugungswinkel misst und davon den arithmetischen Mittelwert bestimmt. Strichgitter Spaltrohr Fernrohr Spektrallampe Goniometer Beobachter Abb. 2: Spektralapparatur mit Goniometer. Die experimentelle Aufgabe besteht nun darin, die Wellenlängen aller Spektrallinien bei der Wasserstofflampe, die man im sichtbaren Spektralbereich beobachten kann, zu bestimmen, indem man die entsprechenden Drehwinkel misst. 3.15.7. Anhang Abb. 3: Emissionsspektrum von Natrium. Abb. 4: Balmer-Spektralserie von atomarem Wasserstoff. 3.16. Lichtabsorption von Farbstoffmolekülen (Versuch Nr. 26) Ziel des Versuches ist, die Absorptionsspektren von organischen Farbstoffen zu untersuchen und im Rahmen eines einfachen quantenchemischen Modells zu diskutieren. 3.16.1. Vorbereitung • • • • • • • Teilchen im Kasten De-Broglie-Beziehung und stehende Elektronenwellen Energieniveauschema und Wellenfunktionen für Teilchen im Kasten Pauliprinzip Quantenmechanische Beschreibung des Teilchens im Kasten Optische Auswahlregeln Absorptionsspektroskopie 3.16.2. Literatur P. W. Atkins, Physikalische Chemie, 2. Auflage, VCH-Verlag, Weinheim, 1996, S. 343-353, 365-371, 513-519, 557-563. G. Wedler, Lehrbuch der Physikalischen Chemie, 4. Auflage; Wiley-VCHVerlag, Weinheim, 1997, S. 108-111, 119-126, 134-147, 598-611, 628-634. 3.16.3. Theoretische Grundlagen Verantwortlich für die Lichtabsorption eines Farbstoffes im sichtbaren Bereich und im UV-Bereich sind elektronische Übergänge vom elektronischen Grundzustand in den ersten angeregten Zustand. Bei organischen Molekülen sind diese Übergänge im sichtbaren Bereich meist π-π*-Übergänge oder n-π*-Übergänge in ausgedehnten delokalisierten π-Bindungssystemen. σ-Elektronen sind fester gebunden. Daher bedürfen σ-σ*-Übergänge viel größerer Energiebeträge. Bei den hier zu untersuchenden Cyanin-Farbstoffen sind sämtliche Atome des mesomeren Systems über trigonal planare Bindungen miteinander verknüpft, wie es in Abb. 1 gezeigt ist. Das Molekül verfügt also über ein ausgedehntes delokalisiertes π-Bindungssystem. Der energieärmste Übergang ist ein π-π*Übergang. N + N k Abb. 1: Cyanin-Farbstoff-Kationen mit k = 0, 1, 2, 3, ... . In Abb. 2 ist der Vorgang der Lichtabsorption in diesem Falle prinzipiell gezeigt. Abb. 2: Vorgang der Absorption im einfachen quantenmechanischen Bild. Im Grundzustand ist das HOMO (highest occupied molecular orbital) mit zwei Elektronen unterschiedlichen Spins besetzt. Es handelt sich um den sog. Singulett-Grundzustand S0, in dem keine ungepaarten Elektronen auftreten. Trifft ein Photon geeigneter Energie (Resonanzbedingung) auf das Elektronensystem, so kann ein Elektron ohne Spinumkehr in das LUMO (lowest unoccupied molecular orbital) überführt werden. Es entsteht der angeregte SingulettZustand S1. Der Übergang mit Spinumkehr in den angeregten Triplett-Zustand T1 ist verboten. In dem in Abb. 2 gezeigten Bild stecken schon sehr viele quantenmechanische Ergebnisse. Wir wollen in diesem Versuch versuchen, mit einem einfachen quantenmechanischen Modell die Energiedifferenz ΔE für den π-π*-Übergang abzuleiten. Zunächst einmal muss man allerdings etwas tiefer einsteigen. Aus den Absorptionsspektren und noch deutlicher aus den Emissionsspektren von Molekülen und Atomen kann man schließen, dass in Atomen und Molekülen keine kontinuierlichen Energien angenommen werden können, die Energie ist „gequantelt“, d. h. es gibt diskrete Energieniveaus. Diese zunächst verblüffende Tatsache, die man aus der Welt makroskopischer Körper nicht kennt, wird so erklärt, dass mikroskopische Teilchen auch Wellencharakter haben (Welle-TeilchenDualismus). Aus der Wellenmechanik ist bekannt, dass sich in schwingenden Systemen mit gewissen Randbedingungen (befestigte Saite, Fell einer Trommel, Wasser im Schwimmbecken, usw.) nur bestimmte Schwingungszustände ausbilden können; das Auftreten von diskreten Zuständen ist also zumindest aus der Welt der Wellen ein bekanntes Phänomen. Wellen werden durch die allgemeine Wellengleichung, eine partielle Differenzialgleichung 2. Ordnung beschrieben. Beim Einführen von Randbedingungen, hat diese Wellengleichung nur noch diskrete Lösungen. Das Auftreten von diskreten Energieniveaus in Atomen und Molekülen ist also ein Hinweis auf den Wellencharakter der elektronischen Zustände. Die Wellengleichung, die diese elektronischen Zustände beschreibt ist die (zeitunabhängige) Schrödingergleichung. Für ein Teilchen im eindimensionalen Raum mit der ortsabhängigen Potentiellen Energie V(x) lautet sie: h 2 d 2 n ( x) 2 V ( x) n ( x) En n ( x) 8 m dx 2 (1) Hierbei sind h das Planck’sche Wirkungsquantum, m die Masse des Teilchens, Ψn die Wellenfunktion des Zustandes n und En die zugehörige Energie. Das Betragsquadrat der Wellenfunktion hat die physikalische Bedeutung einer Wahrscheinlichkeitsdichte, ein Elektron anzutreffen. Natürlich besteht das Cyanin-Molekül, das wir hier betrachten aus sehr vielen Teilchen (Atomkerne, Rumpfelektronen, Bindungselektronen, …). Alle diese Teilchen und ihre kinetischen Energien und Wechselwirkungen müsste man bei einer exakten Behandlung berücksichtigen. Wir werden uns auf die Betrachtung eines einzelnen π-Elektrons im Potentialfeld der Atomrümpfe des Moleküls beschränken und annehmen, dass die Elektron-Elektron-Wechselwirkung im πSystem keine prinzipiellen Änderungen nach sich ziehen. Um die Schrödingergleichung (1) für ein Elektron in einem Potentialfeld lösen zu können, benötigt man dessen potentielle Energie mit V(x) in ebendiesem Potentialfeld. Schematisch in ein realistisches V(x) in Abb. 3 dargestellt. Da die Atomrümpfe positiv geladen sind, kann man für das π-Elektron annehmen, dass die potentielle Energie im Bereich des Moleküls geringer ist als außerhalb des Moleküls. Definitionsgemäß hat das Elektron weit außerhalb des Moleküls eine Potentielle Energie von null (Dissoziation als Referenzzustand). Abb. 3: Schematische Darstellung der Potentiellen Energie eines π-Elektrons. Die Rechnung wird erheblich vereinfacht, wenn man die Wände dieses Potentialtopfs unendlich hoch macht, d. h. Dissoziation unterbindet. Die Fehler, die man damit macht, sind für niedrig angeregte Zustände (hier betrachten wir den Grundzustand und den ersten angeregten Zustand) gering. Wir nähern das tatsächliche Potential also durch ein Kastenpotential mit unendlich hohen Wänden an. Da wir nur an Energiedifferenzen interessiert sind, können wir außerdem der Potentiellen Energie im Kasten den Wert null zuordnen. Abb. 1: Schematische Darstellung der Potentiellen Energie eines π-Elektrons. Damit vereinfacht sich Gleichung (1) innerhalb des Kastens (V = 0) zu: h 2 d 2 n ( x) 2 En n ( x) 8 me dx 2 (2) Der Tatsache, dass das Potential außerhalb des Kastens (x < −L/2 und x > L/2) unendlich ist, werden wir als Randbedingung Rechnung tragen. Wenn die Potentielle Energie unendlich ist, kann sich das Elektron dort nicht aufhalten, d. h. seine Aufenthaltswahrscheinlichkeit ist dort null. Die Wellenfunktion Ψn, deren Quadrat die Wahrscheinlichkeitsdichte für den Aufenthaltsort ist, muss dann dort ebenfalls null sein. Es muss also gelten: n ( L / 2) 0 n ( L / 2) 0 (3) Betrachten wir nun die Differentialgleichung (2). Es kommen die Funktion und ihre negative 2. Ableitung vor. Als allgemeine Lösung könnte man also ansetzen: n ( x) An sin kn x n (4) Die erste Randbedingung lautet: k L An sin n n 0 2 (5) Man wird ihr gerecht durch: kn L kL n 0 n n 2 2 Für die angesetzte Funktion ergibt sich also: (6) k L An sin kn x n 0 2 (7) Die zweite Randbedingung ergibt: n k L k L mit n = 1, 2, 3, … (8) An sin n n 0 kn L n kn 2 L 2 Als endgültigen Ansatz nehmen wir also: n x n n ( x) An sin L L (9) Einsetzen in Gleichung (2) ergibt: n x d 2 An sin h L n x 2 En An sin 2 8 me dx L 2 (10) h 2 n 2 2 En 8 2 me L2 Wir erhalten also folgende Energie-Eigenwerte, die ein Elektron im Potentialkasten mit unendlich hohen Wänden annehmen kann, in Abhängigkeit von der „Quantenzahl“ n: n2h2 En 8me L2 (11) Nun müssen wir nachträglich die anderen Elektronen, die in dem π-System vorkommen, wieder einführen und besetzten die Energieniveaus beginnend bei n = 1 mit jeweils zwei Elektronen unterschiedlichen Spins. Man kann die Anzahl der π-Elektronen N mit der Anzahl an Kettengliedern k des CyaninFarbstoffes (vgl. Abb. 1) in Verbindung bringen. Dazu berücksichtigt man, dass das π-System auch die Stickstoffatome in umfasst, d. h. N = 2(k + 3). Damit ergibt sich für die Quantenzahlen nHOMO=k + 3 bzw. nLUMO=k + 4, so dass man die Anregungsenergie ΔE h2 E ELUMO EHOMO 2(k 3) 1 8me L2 (12) in Abhängigkeit von k darstellen kann. Schwieriger ist es, die Kastenlänge L durch die Anzahl k auszudrücken, weil man zunächst nicht genau weiß, über wie viele Bindungen sich das π-System erstreckt. Daher setzt man die Kastenlänge L als L (2 k a) d (13) an, wobei d der mittlere C−C-Bindungsabstand im konjugierten π-System ist und a ein Parameter darstellt, der die effektive Ausdehnung des π-Systems beschreibt. Damit ergibt sich die Wellenlänge λmax des langwelligsten Übergangs mit Hilfe von Gleichung (12) zu hc 8mec (2k a) d . E h 2(k 3) 1 2 max (14) Um die k-Abhängigkeit im Experiment zu bestätigen, formt man Gleichung (14) in 2(k 3) 1 max 12 12 8m c e (2k a) d h (15) um und erwartet bei Auftragen der linken Seite von (13) gegen k eine Gerade, aus der man den mittleren C-C-Bindungsabstand d und den Parameter a, der die effektive Ausdehnung des π-Systems beschreibt, gewinnen kann. Um das Absorptionsmaximum λmax zu bestimmen, wird ein Spektral-Photometer benutzt. Dabei bewirkt eine vorgegebene Farbstoffkonzentration cF eine wellenlängenabhängige Abschwächung der eingestrahlten Lichtintensität I. Die Abnahme dI der Intensität, die eintritt, wenn die Lichtstrahlung die Strecke dz in einer Probe zurücklegt, ist proportional zur Länge des zurückgelegten Weges, der Konzentration des Farbstoffs und der Intensität des Lichtstrahls, d. h. dI ~ −cFIdz. Mit Hilfe der wellenlängenabhängigen Proportionalitätskonstante κ ergibt sich dI/I = −κcFdz. Durch Integration mit geeigneten Anfangsbedingungen erhält man daraus das sogenannte Lambert-Beer’sche Gesetz ln I cF zK , I0 (16) wobei I0 die anfängliche Lichtintensität und zK die gesamte Dicke der Probe ist. Das Verhältnis aus I/I0 wird als Transmission bezeichnet. Oftmals verwendet man allerdings anstatt des natürlichen den dekadischen Logarithmus und führt die sogenannte dekadische Absorbanz bzw. Extinktion A ein I0 (17) cF zK . I Dabei ist ε = κ/ln10 der (dekadische) Extinktionskoeffizient. Die Untersuchung der Extinktion A in Abhängigkeit der Wellenlänge λ erlaubt somit eine Bestimmung von ε = ε(λ) und des Absorptionsmaximums λmax für die verschiedenen Farbstoffe. A lg 3.16.4. Aufgaben 1) Man bestimme die Absorptionsspektren der 3 vorhandenen CyaninFarbstoffe mit k = 1, 2 und 3 mit dem Spektrophotometer. 2) Für den Cyanin-Farbstoff mit k = 3 überprüfe man am Absorptionsmaximum das Lambert-Beer’sche Gesetz. Man bestimme den entsprechenden Extinktionskoeffizienten ε = ε(λmax) und vergleiche mit Literaturdaten. 3) Aus den Absorptionsmaxima λmax der 3 untersuchten Farbstoffe berechne man die linke Seite von Gleichung (10) und trage sie gegen k auf. Wird die the- oretisch vorhergesagte Gesetzmäßigkeit erfüllt? Man bestimme aus der Auftragung a und d. Hierbei ist zu berücksichtigen, dass wir mit einem eindimensionalen Modell gearbeitet haben. Damit ist d die Projektion der tatsächlichen Bindungslänge auf die x-Achse ist. Da das Molekül 120°-Winkel aufweist, ist die tatsächliche Bindungslänge um den Faktor 1/cos(30°) größer. Was lässt sich über die Ausdehnung des π-Systems aussagen? Wie stimmt der ermittelte C-CBindungsabstand d mit Literaturdaten überein? Diskutieren Sie in diesem Zusammenhang die Anwendbarkeit des Modells und die Vereinfachungen, die mit dem Modell gemacht werden. 4) Man berechne die Energien der verschiedenen Zustände mit n ≤ nLUMO relativ zur potentiellen Energie und konstruiere das Energieniveaudiagramm. 5) Wie sehen die Wellenfunktionen des Elektrons im eindimensionalen Kasten der Länge L aus? (Kann man aus der Symmetrie der Elektronenwellenfunktionen auf die optischen Auswahlregeln schließen?) 3.16.5. Zubehör Photometer, Küvetten, Cyanin-Farbstoffe, Ethanol, Messkolben, Vollpipetten. 3.16.6. Durchführung Die Extinktion wird mit einem sogenannten Spektralphotometer gemessen. Abb. 4: Schematische Darstellung des Spektralphotometers. Dazu wird am Photometer zunächst bei einer Küvette, die nur Lösungsmittel enthält, die Transmission auf 100% abgeglichen. Anschließend wird die Küvette mit der zu untersuchenden Lösung in den Strahlengang gebracht und die dazugehörige Transmission I/I0 der Lösung abgelesen. Für jede Lösung und Wellenlänge muss dieses Vorgehen wiederholt werden. Die gewünschte Wellenlänge kann dazu mit einem Handrad am Photometer eingestellt werden. Im Versuch werden die Absorptionsspektren von 3 Cyanin-Farbstoffen mit k = 1, 2 und 3 gemessen. Dazu werden die bereitgestellten ca. 10 −5 M Farbstofflösungen benutzt. Nach jeder Extinktionsmessung wird die ethanolische Farbstofflösung aus der Küvette in das jeweilige Messkölbchen zurückgefüllt. Bei dem Farbstoff mit k = 1 wird der Wellenlängenbereich zwischen 350 und 550 nm untersucht, bei dem Chromophor mit k = 2 der Bereich zwischen 400 und 700 nm und bei dem Cyanin-Farbstoff mit k = 3 wird zwischen 500 und 800 nm gemessen. Wichtig ist dabei vor allem die Absorptionsmaxima λmax sehr genau zu bestimmen. Anschließend wird bei dem Farbstoff mit k = 3 eine Verdün- nungsreihe mit 5 Konzentrationen zwischen 1∙10 −5 und 5∙10−7 mol/L untersucht (siehe Bedienungsanleitungen zum Benutzen der Waagen auf Seite 14 und die Hinweise zur Volumenmessung auf Seite 15). Für alle Konzentrationen wird die Transmission I/I0 am Absorptionsmaximum λmax bestimmt. Außerdem muss die Dicke zK der verwendeten Küvette mit einer Schieblehre ausgemessen werden. 4. Fehler- und Ausgleichsrechnung 1) Zufällige und systematische Fehler Messungen sind gewöhnlich mit Fehlern behaftet. Diese können durch falsches Ablesen der Messwerte, Unvollkommenheiten der Messgeräte, Schwankungen der Messbedingungen und vieles andere verursacht werden. Je nach ihrer Auswirkung kann man Messfehler allgemein in zwei Gruppen unterteilen: • Systematische Fehler, die dadurch charakterisiert sind, dass sie auch bei mehrmaligem Messen in erster Näherung immer gleich bleiben. • Zufällige Fehler, die von Messung zu Messung verschieden groß sind und zu einer Streuung der Messwerte führen. Während zufällige Fehler an der Streuung der Messergebnisse beim mehrmaligen Messen derselben Größe erkannt werden, treten systematische Fehler nicht so offensichtlich zutage, sondern werden gewöhnlich erst durch eine gründliche und kritische Untersuchung des Messvorgangs bemerkt. Da es sich bei den zufälligen Fehlern im mathematischen Sinne um zufällige Ereignisse handelt, gehorchen sie den Gesetzen der Wahrscheinlichkeitsrechnung. Man kann daher ihren Einfluss auf das Messergebnis untersuchen, indem man die einzelnen Messresultate nach statistischen Methoden auswertet. 2) Mittelwert und mittlerer Fehler der Einzelmessungen Man betrachtet den Fall, dass irgendeine physikalisch-chemische Größe insgesamt n mal gemessen wurde. Den wahren Wert, den diese Größe besitzt, bezeichnet man mit xw, die n Messwerte mit x1, x2, ... , xn. Bei der einzelnen Messung möge jeweils eine große Zahl von zufälligen Einflüssen wirksam sein, die zur Folge haben, dass die xi von xw abweichen. Eine gewisse Anzahl von Faktoren wirkt auf eine Vergrößerung des Messwertes hin, eine Reihe von anderen Faktoren auf eine Verkleinerung. Es erhebt sich nun die Frage, wie man aus den einzelnen Messungen xi denjenigen Wert bestimmt, der mit größter Wahrscheinlichkeit dem wahren Wert xw entspricht. Diesen Wert bezeichnet man mit x . Die Bedingung zur Bestimmung des gesuchten wahrscheinlichsten Wertes x von xw ist durch das Minimum der Summe der quadratischen Abweichungen von xw gegeben, d. h. n ( xw xi )2 Minimum i 1 für xw x . (1) Das Minimum liegt nun an der Stelle, an der die Ableitung der Summe in Gleichung (1) nach xw gleich Null wird. Somit erhält man zur Bestimmung von x die Gleichung n d n 2 ( x x ) 2 w i ( xw xi ) 0 für xw x , dxw i1 i 1 (2) aus der sich durch Umformen 1 n x xi , n i 1 (3) ergibt. Man sieht also, dass derjenige Wert x , der mit größter Wahrscheinlichkeit dem wahren Wert xw entspricht, durch das arithmetische Mittel der einzelnen Messwerte gegeben ist. Die Abweichungen der einzelnen Messwerte xi vom Mittelwert nennt man die Fehler der einzelnen Messungen und bezeichnet sie mit δi = xi − x . Ein geeignetes Maß für die Größe dieser Fehler stellt die Streuung dar, die man erhält, indem man die Summe der Abweichungsquadrate durch n teilt und anschließend aus dem Resultat die Wurzel zieht. Man nennt diese Größe den mittleren Fehler m' der Einzelmessungen bezüglich des Mittelwertes m 1 n 2 1 n i ( xi x )2 . n i 1 n i 1 (4) Den mittleren Fehler m der Einzelmessungen bezüglich des wahren Wertes xw erhält man zu m 1 n 2 1 n 1 n 2 2 2 ( x x ) xi nx . i i n 1 i 1 n 1 i 1 n 1 i 1 (5) 3) Fortpflanzung des mittleren Fehlers und des maximalen Fehlers einer Einzelmessung Gewöhnlich führt man eine Vielzahl von Messungen der Größen x und y durch und fragt danach, wie sich die mittleren Fehler in x und y auf den mittleren Fehler in z = f(x,y) auswirken. Es sollen nun r Messungen der Größe x und s Messungen der Größe y durchgeführt werden. Die erhaltenen Werte werden mit x1, x2, ... xr bzw. y1, y2, ... ys, die Mittelwerte mit x bzw. y und die mittleren Fehler mit mx bzw. my bezeichnet. Den Mittelwert von z kann man dann gemäß Gleichung (3) 1 r s z zik (r s) i1 k 1 (6) berechnen. Für relativ kleine Fehler geht diese Formel in z f (x, y) (7) über, d. h. man kann den Mittelwert von z = f(x,y) berechnen, indem man in f(x,y) die Mittelwerte von x und y einsetzt. Den mittleren Fehler mz von z erhält man in Analogie zu Gleichung (5) aus r s 1 mz ( zik z )2 (r s 1) i 1 k 1 (8) Auch hier lässt sich für relativ kleine Fehler der mittlere Fehler mz näherungsweise aus den mittleren Fehlern von x und y berechnen mz f x2 ( x , y )mx2 f y2 ( x , y )m 2y , (9) wobei fx( x , y ) die partielle Ableitung der Funktion f(x,y) nach x an der Stelle x = x und y = y ist und fy( x , y ) die entsprechende partielle Ableitung nach y ist. Dies ist das sogenannte Fehlerfortpflanzungsgesetz. Die Gleichung lässt sich auf eine beliebige Anzahl von Messgrößen verallgemeinern. Im Praktikum wird in den meisten Fällen aus Zeitgründen eine physikalischchemische Größe nur einmal gemessen. Das bedeutet, dass man versuchen muss, den maximalen Fehler Δx der Messgröße x abzuschätzen bzw. ihn aus Informationen über das Messinstrument herauszulesen. Der maximale Fehler Δx wird dabei so gewählt, dass das Messergebnis mit Sicherheit in dessen Grenzen liegt. Betrachtet man nun den Fall, dass zwei verschiedene Größen x und y gemessen werden und die neue Größe z=f(x,y) eine Funktion von x und y ist, dann stellt sich die Frage, wie groß der maximale Fehler in z ist, wenn die Fehler in x und y bei einer einmaligen Messung Δx und Δy betragen? Dazu kann man eine zum Fehlerfortpflanzungsgesetz ähnliche Formel verwenden z | f x ( x , y ) | x | f y ( x , y ) | y , (10) wobei an die einzelnen Faktoren Betragsstriche gesetzt wurden, um zu vermeiden, dass sich die Fehler in x und y teilweise kompensieren. Gewöhnlich versucht man diese Gleichung so umzuformen, dass rechts und links die sogenannten relativen Fehler Δx/x, Δy/y bzw. Δz/z stehen. 4) Mittlerer Fehler des Mittelwertes Als Maß für die mögliche Abweichung des berechneten Mittelwertes x vom wahren Wert xw führt man den mittleren Fehler von x ein, der mit m bezeichnet wird. Der mittlere Fehler des Mittelwertes m ist keineswegs mit dem mittleren Fehler m der Einzelmessungen identisch, da mit wachsendem n die Wahrscheinlichkeit dafür, dass sich die Fehler in den xi kompensieren, immer größer und somit der Fehler von x immer kleiner wird. Um m zu berechnen, geht man von Gleichung (3) aus. Dieser Formel gemäß ist x eine Funktion f(x1, x2, ... , xn) von n Veränderlichen x1, x2, ..., xn. Jede dieser Größen ist mit einem mittleren Fehler m behaftet. Der mittlere Fehler in x lässt sich somit mit Hilfe des Feh- lerfortpflanzungsgesetzes (9) aus den Fehlern der xi berechnen. Man erhält so den mittleren Fehler des Mittelwertes zu m m , n (11) indem man den mittleren Fehler m der n Einzelmessungen durch n1/2 dividiert. Das Ergebnis der Messreihe wird dann in der Form x m dargestellt. 5) Ausgleichsrechnung (lineare Regression) Oftmals interessiert im Praktikum der Fall, dass zwei Messgrößen x und y linear voneinander abhängen, d. h. y = a + bx. Die einzelnen gemessenen Wertepaare werden mit x1y1, x2y2, ... , xnyn bezeichnet. Wenn man die erhaltenen Wertepaare in ein Koordinatensystem einträgt, erhält man z. B. die in Abb. 1 angegebenen Punkte. Diese Punkte liegen wegen der zufälligen Fehler, die bei den Messungen auftreten können, nicht auf einer Geraden. Man möchte aber die wahre Gerade finden, um so die Konstanten a und b zu bestimmen. Auf welche Weise kann man nun diejenige Gerade finden, die mit größter Wahrscheinlichkeit mit der tatsächlich vorliegenden Geraden übereinstimmt? Es lässt sich zeigen, dass man diese Gerade so wählen muss, d. h. die Konstanten in der Weise bestimmen muss, dass die Summe der quadratischen Abweichungen der Punkte von der Gerade ein Minimum wird. Abb. 1: Messwerte yi in Abhängigkeit der xi und nach (12) und (14) berechnete Ausgleichsgerade. Die Abweichungen des Punktes xiyi von der Geraden in y-Richtung ist durch yi – a − bxi gegeben, d. h. die Summe der quadratischen Abweichungen n ( yi a bxi )2 (12) i 1 muss minimal sein. Dies ist gleichbedeutend damit, dass die beiden partiellen Ableitungen der Summe in (12) nach a und b verschwinden müssen, d. h. n n 2 ( y a bx ) 0 und ( yi a bxi )2 0 . i i a i1 b i1 (13) Ausführen der partiellen Differentiation und umformen ergibt die gesuchten Bestimmungsgleichungen für a und b yi xi2 xi xi yi i i i a i 2 n xi ( xi ) 2 i und b n xi yi xi yi i n i i xi2 i i ( xi ) 2 . (14) i Mit Hilfe von a und b lässt sich nun die Fehlerquadratsumme (12) berechnen und somit die Streuung my, d. h. der mittlere Fehler der einzelnen y-Werte, angeben my 1 n ( yi a bxi )2 . n 2 i 1 (15) Die mittleren Fehler, mit denen a und b behaftet sind, ergeben sich zu ma m y xi2 i 2 n xi ( xi ) 2 i und mb m y n , n xi2 ( xi ) 2 i i (16) i so dass der y-Achsenabschnitt bzw. die Steigung der Ausgleichsgeraden durch a ± ma bzw. b ± mb gegeben sind. Als Qualitätsmaß für die Ausgleichsgerade wird oftmals der sogenannte Korrelationskoeffizient r2 2 n xi yi xi yi i i i r2 2 2 2 2 n xi ( xi ) n yi ( yi ) i i i i (17) verwendet. Je näher r2 bei Eins liegt, um so besser beschreibt die Ausgleichsgerade die Messpunkte. 6) Fehlerrechnung und Fehlerbetrachtung in den Protokollen Für jeden Versuch muss eine Fehlerrechnung durchgeführt werden. Dazu werden entweder die Mittelwerte, die mittleren Fehler der Einzelmessungen, die mittleren Fehler der Mittelwerte und die sich fortpflanzenden mittleren Fehler berechnet, falls die verschiedenen physikalisch-chemischen Größen mehrmals gemessen wurden, oder es werden maximale Fehler der Messgrößen abgeschätzt und daraus die maximalen Fehler der interessierenden Größen berechnet. Anschließend wird eine Abbildung erstellt, in der die Mittelwerte zusammen mit Fehlerbalken dargestellt werden. Die Auftragung in der Abbildung ist so zu wählen, dass eine lineare Regression durchgeführt werden kann, so dass Achsenabschnitt und Steigung mit den entsprechenden Fehlern bestimmt werden können. Daraus lassen sich dann die interessierenden physikalischchemischen Größen gewinnen. Danach werden die Ergebnisse in Zusammenhang mit Literaturdaten diskutiert. Daran schließt sich eine sogenannte Fehlerbetrachtung, d. h. eine kritische Untersuchung des Messvorgangs und des Vorgehens bei der Auswertung, an. Das bedeutet, man muss sich z. B. Gedanken über mögliche systematische Fehler machen oder auf Modellannahmen bzw. Näherungen in der Auswertung hinweisen, die die möglichen Abweichungen vom Literaturwert erklären können. 7) Beispiel Der elektrische Widerstand R = R(T) eines Halbleiters hängt von der Temperatur ab. Bei einer bestimmten Temperatur T wurden die folgenden Messwerte für die elektrische Spannung U und die Stromstärke I unabhängig voneinander erhalten: U = 5.0, 4.7, 5.2, 5.1, 5.3 und 5.1 V; I = 0.27, 0.29, 0.30, 0.25 und 0.28 mA. Man berechne den Mittelwert für den elektrischen Widerstand und den mittleren Fehler des Mittelwertes mit Hilfe des Ohm’schen-Gesetzes. A) Der Widerstand wird aus dem Ohmschen Gesetz R = U/I berechnet. Aus den 6 unabhängigen Messwerten für die Spannung U und den 5 Messwerten für die Stromstärke I erhält man als Zwischenergebnis insgesamt 30 Messwerte für den Widerstand R = 18.52, 17.24, 16.67, 20.00, 17.86, 17.41, 16.21, 15.67, 18.80, 16.79, 19.26, 17.93, 17.33, 20.80, 18.57, 18.89, 17.59, 17.00, 20.40, 18.21, 19.63, 18.28, 17.67, 21.20, 18.93, 18.89, 17.59, 17.00, 20.40 und 18.21 k. Nun kann man gemäß Gleichung (6) den Mittelwert zu R =18.3 k berechnen. Den mittleren Fehler der Einzelmessung erhält man gemäß (7) zu mR = 1.4 k und der mittlere Fehler des Mittelwertes ergibt sich aus (11) zu m R = 0.3 k. Damit erhält man das Endergebnis R = (18.3 ± 0.3) k. B) Unter der Annahme, dass die Fehler in U und I relativ klein sind, kann man auch alternativ vorgehen. Dazu berechnet man zunächst die Mittelwerte, die mittleren Fehler und die Fehler des Mittelwertes für U und I gemäß den Beziehungen (3), (5) und (11). Damit erhält man U = (5.1 ± 0.1) V bei einem mittleren Fehler für die Einzelmessungen von mU = 0.2 V. Für die Stromstärke ergibt sich I = (0.28 ± 0.01) mA bei einem mittleren Fehler für die Einzelmessung von mI = 0.02 mA. Nun berechnet man den Mittelwert des Widerstandes R U I = 18.2 k gemäß Gleichung (7) und den mittleren Fehler mR = 1.5 k mit Hilfe des Fehlerfortpflanzungsgesetzes (9). Der mittlere Fehler des Mittelwertes ergibt sich dann aus (11) zu m R = 0.3 k, d. h. R = (18.2 ± 0.3) k. Die Unterschiede zwischen den Werten in den beiden Vorgehensweisen beruhen darauf, dass im Fall A mit den exakten Beziehungen (6) und (8) gerechnet wurde, während im Fall B nur die genäherten Beziehungen (7) und (9) verwen- det wurden. Solange die Fehler klein genug sind, stimmen die beiden Ergebnisse im Rahmen der Fehlergrenzen überein. Das Vorgehen im Fall B hat dann den Vorteil, dass es mit deutlich weniger Rechenaufwand verbunden ist. Das Ergebnis wurde dabei auf die kleinste Zahl der Nachkommastellen gerundet, die in den Ausgangswerten vorkommt. In den Rechnungen wurden die erhaltenen Zwischenergebnisse allerdings auf eine signifikante Stelle mehr gerundet (siehe z. B. P. W. Atkins, Physikalische Chemie, 2. Auflage, VCHVerlag, Weinheim, 1996, S. 9-10). Wenn man die Spannung U = 5.0 V und die Stromstärke I = 0.27 mA jeweils nur einmal gemessen hat, dann muss man versuchen den maximalen Fehler der beiden Größen abzuschätzen, um somit den maximalen Fehler des Widerstandes zu berechnen. Da der maximale Fehler so gewählt sein soll, dass der Messwert mit Sicherheit in seinen Grenzen liegt, würde man die maximalen Fehler mit ΔU = 0.3 V bzw. ΔI =0.03 mA ansetzen. Damit ergibt sich der Mittelwert des Widerstandes zu 18.5 k und der maximale Fehler des Widerstandes wird gemäß (11) zu ΔR = 3.2 k berechnet. Somit erhält man R = (18.5 ± 3.2) k, d. h. der relative maximale Fehler beträgt ΔR/R = ΔU/U + ΔI/I = 17.3 %. Im Vergleich zur Vorgehensweise unter A und B ist der resultierende Fehler im Widerstand deutlich größer, weil durch ein mehrmaliges Messen von Größen der Fehler des Mittelwertes deutlich herabgesetzt werden kann. Wenn man nun die Spannung U und Stromstärke I bei verschiedenen Temperaturen T gemessen hat, dann trägt man den jeweiligen mittleren Widerstand R als Punkt in das entsprechende Diagramm R-T-Diagramm ein und fügt die maximalen Fehler ΔR und ΔT als Fehlerbalken dazu. Falls man die Werte für R und T aus einer echten Mittelwertbildung gewonnen hat, dann gibt man die mittleren Fehler der Einzelmessungen mR bzw. mT als Fehlerbalken an. Zweckmäßigerweise wird dazu eine Auftragung gewählt, in der man eine lineare Abhängigkeit zwischen Ordinate und Abszisse erwartet. So hängt in vielen Fällen der elektrische Widerstand eines Halbleiters exponentiell von der Temperatur ab, d. h. R = R0exp(−a/T). Um zu überprüfen, ob die gemessenen Widerstands-Werte in Abhängigkeit der Temperatur T diese Gesetzmäßigkeit befolgen, wird ln(R/) gegen 1/T aufgetragen. Falls in der Abbildung eine lineare Abhängigkeit sichtbar wird, lassen sich a, R0 und die entsprechenden Fehler von a und R0 mit Hilfe der linearen Regression bestimmen. Die Fehlerbalken Δln(R/) = ΔR/R und Δ(1/T) = ΔT/T2 in der Abbildung lassen sich wieder aus den maximalen Fehlern ΔR und ΔT gemäß Gleichung (11) berechnen.