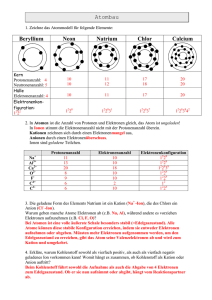
Praktische Einführung in die Chemie
Werbung

Praktische Einführung in die Chemie Versuchsanleitungen und Theoretische Grundlagen Vorwort Ein wesentlicher Bestandteil des Studiums der Chemie ist der Erwerb der Fähigkeit, mit chemischen Substanzen umzugehen, Stoffe zu synthetisieren oder zu analysieren und Messungen durchzuführen und auszuwerten. Diese praktischen Fähigkeiten werden, wie der Name sagt, in Praktika, die Sie während Ihres ganzen Chemiestudiums begleiten werden, gelehrt und eingeübt. Am Anfang dieser Reihe von Praktika steht das Integrierte Praktikum ”Praktische Einführung in die Chemie”, das deshalb als integriert bezeichnet wird, weil in ihm alle Bereiche der Chemie behandelt werden. In diesem Praktikum sollen Sie mit einfachen chemischen Operationen und Messungen vertraut werden. Die Versuche lehnen sich eng an die Inhalte der Vorlesung an. Gehörtes soll praktisch vertieft und selbst erlebt werden. Sie sollen Ihre Beobachtungsgabe schulen und lernen, chemische Apparaturen aufzubauen, Experimente und Messungen durchzuführen, sie zu protokollieren, zu interpretieren und auszuwerten. Die Versuche sind so ausgewählt, dass sie das Wesentliche hervortreten und Beziehungen allgemeiner Art erkennen lassen. Bei der Abfassung der Versuchsbeschreibungen wurde großer Wert darauf gelegt, die Durchführung des Versuchs und seine Auswertung detailliert zu beschreiben und die für diesen Versuch erforderlichen theoretischen Grundlagen darzustellen. Das heißt aber nicht, dass auf den Gebrauch von Lehrbüchern verzichtet werden kann. Das Praktikumsskript ersetzt kein Lehrbuch. Dieses Praktikumsskript ist die überarbeitete Fassung des Skripts aus dem Wintersemester 2012/13. Es enthält Versuchsbeschreibungen aus anorganisch-, organisch-, und physikalischchemischen Praktika, teilweise auch mit biochemischem Hintergrund, die nun auf die Erfordernisse der Studierenden im ersten Semester der Bachelor-Studiengänge Chemie, Materialwissenschaft, sowie Lebensmittelchemie und Chemie-Höheres Lehramt angepasst wurden. Auch mit sorgfältiger Überarbeitung ist es nicht möglich ein absolut fehlerfreies Skript zu erstellen. Informieren Sie uns also bitte, falls Sie Fehler entdecken und Verbesserungsvorschläge haben, Ihre Nachfolger werden es Ihnen danken. Die Fakultät Chemie wünscht Ihnen, dass Sie Ihr erstes chemisches Praktikum erfolgreich absolvieren, und hofft, dass Ihnen trotz der damit verbundenen Anstrengungen das Praktikum auch Spaß bereitet und Ihre Freude am chemischen Experimentieren wächst. Die Fakultät Chemie I Inhaltsverzeichnis Einleitung 1 1 Allgemeines 1 1.1 Termine und Anwesenheit . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1 1.2 Sicherheit im Labor . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1 1.3 Allgemeine Sicherheitshinweise . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 3 1.4 Verhalten bei Bränden . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 4 1.5 Umgang mit Chemikalien . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 4 1.6 Gefahrenbezeichungen . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 5 1.7 H- und P-Sätze . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 6 2 Ablauf eines Versuchstages 11 2.1 Vor dem Versuchstag . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 11 2.2 Versuchsbeginn und -durchführung . . . . . . . . . . . . . . . . . . . . . . . . . . . . 11 2.3 Ende eines Versuchstages . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 12 2.4 Protokoll . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 12 Versuch 1 (GMS) Gasgesetz, Molmassenbestimmung und Stöchiometrie 15 1 Theorie 15 1.1 Grundlagen der Stöchiometrie . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 15 1.2 Stoffmenge . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 16 1.3 Das ideale Gas . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 17 2 Aufgabenstellung und Versuchsanleitungen 18 2.1 Elektrolyse von Wasser/Synthese von Wasser in der Brennstoffzelle . . . . . . . . . 18 2.2 Bestimmung der molaren Masse von Calcium . . . . . . . . . . . . . . . . . . . . . . 19 2.3 Molmassenbestimmung nach Victor-Meyer . . . . . . . . . . . . . . . . . . . . . . . 19 3 Geräte und Chemikalien 21 4 Literatur 21 Versuch 2 (ABS) Optische Absorptionsspektroskopie 23 1 Theorie 23 1.1 Grundlagen der optischen Absorptionsspektroskopie . . . . . . . . . . . . . . . . . . 23 1.2 Teilchen im Kasten . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 23 2 Aufgabenstellung, Versuchsanleitung und -auswertung 26 2.1 Bestimmung der Länge eines eindimensionalen Potentialtopfs . . . . . . . . . . . . 26 2.2 Bestimmung der Dissoziationskonstante einer Säure . . . . . . . . . . . . . . . . . . 27 II 3 Geräte und Chemikalien 29 4 Literatur 29 Versuch 3 (BSP) Balmer-Serie und Spektroskopie 31 1 Theorie 31 1.1 Das Kern-Hülle-Modell . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 31 1.2 Wasserstoffspektrum und das Bohr’sche Atommodell . . . . . . . . . . . . . . . . . 33 1.3 Der Beginn der Quantenmechanik . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 36 1.4 Heisenberg’sche Unschärferelation und Schrödinger-Gleichung . . . . . . . . . 38 2 Aufgabenstellung, Versuchsanleitung und -auswertung 40 2.1 Balmer-Serie . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 40 2.2 Flammenfärbung und Emissionsspektren ausgewählter Elemente . . . . . . . . . . 43 3 Geräte und Chemikalien 44 4 Literatur 44 Versuch 4 (PSE) Periodisches System der Elemente 45 1 Theorie 45 1.1 Aufbau der Elektronenhülle – Aufbau des Periodensystems . . . . . . . . . . . . . . 45 1.2 Die Perioden . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 45 1.3 Die Gruppen . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 45 1.4 Die Orbitale . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 46 1.5 Das HSAB-Prinzip (auch: Pearson-Konzept) . . . . . . . . . . . . . . . . . . . . . 49 2 Aufgabenstellung, Versuchsanleitung und -auswertung 2.1 Beobachtung der unterschiedlichen Löslichkeit von Silberhalogeniden . . . . . . . . 2.2 Änderung der Eigenschaften beim Durchgang durch eine Periode – pH -Werte von ”Hydroxiden” der dritten Periode . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2.3 49 49 50 Vergleich der Farbigkeit von Übergangsmetallkomplexen/-verbindungen mit Hauptgruppenmetallkomplexen/-verbindungen . . . . . . . . . . . . . . . . . . . . . . . . . 50 3 Geräte und Chemikalien 51 4 Literatur 52 Versuch 5 (HGE) Hauptgruppenelemente 53 1 Theorie 53 1.1 Lewis-Fomeln (≡ Valenzstrichformeln) von Molekülen . . . . . . . . . . . . . . . . 53 1.2 Das VSEPR Konzept . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 53 1.3 Die Hauptgruppenelemente . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 54 III 1.4 Das Wasser . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2 Aufgabenstellung, Versuchsanleitung und -auswertung 62 63 2.1 Reaktion von Alkalimetallen in H2 O . . . . . . . . . . . . . . . . . . . . . . . . . . . 63 2.2 Verbrennung von Magnesium in CO2 . . . . . . . . . . . . . . . . . . . . . . . . . . . 63 2.3 Veresterung Anorganischer Säuren . . . . . . . . . . . . . . . . . . . . . . . . . . . . 64 2.4 Anionen des Kohlenstoffs . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 64 2.5 Hydrolyse von Nichtmetallverbindungen . . . . . . . . . . . . . . . . . . . . . . . . . 64 2.6 Oxidationsstufen von Nichtmetallen . . . . . . . . . . . . . . . . . . . . . . . . . . . 65 2.7 Oxidationswirkung von Halogenaten . . . . . . . . . . . . . . . . . . . . . . . . . . . 65 3 Geräte und Chemikalien 66 4 Literatur 66 Versuch 6 (NGE) Nebengruppenelemente 67 1 Theorie 67 1.1 Übersicht . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 67 1.2 Ausgewählte Elemente . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 67 2 Aufgabenstellung, Versuchsanleitung und -auswertung 71 2.1 Komplexes Redoxverhalten von Übergangsmetallen . . . . . . . . . . . . . . . . . . 71 2.2 Farbigkeit von Übergangsmetallen in Abhängigkeit vom Liganden . . . . . . . . . 71 2.3 Gemischtvalenz von Übergangsmetallverbindungen . . . . . . . . . . . . . . . . . . 72 3 Geräte und Chemikalien 73 4 Literatur 73 Versuch 7 (BPH) Bindungstheorie und Physikalische Eigenschaften 75 1 Theorie 75 1.1 Die Elektronegativität . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 75 1.2 Die Ionenbindung . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 75 1.3 Intermolekulare Kräfte . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 76 1.4 Die Elektronenpaarbindung, Atombindung, kovalente Bindung . . . . . . . . . . . 77 1.5 Die Molekülorbitaltheorie (MO-Theorie) . . . . . . . . . . . . . . . . . . . . . . . . . 77 1.6 Bändertheorie und Leitfähigkeit . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 79 1.7 Magnetismus . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 81 2 Aufgabenstellung, Versuchsanleitung und -auswertung 81 2.1 Schmelzen von Salzen und Salzmischungen . . . . . . . . . . . . . . . . . . . . . . . 81 2.2 Erhitzen, Abkühlen und Abschrecken von Schwefel . . . . . . . . . . . . . . . . . . 82 2.3 Bestimmung der Leitfähigkeit eines metallischen Leiters . . . . . . . . . . . . . . . 83 IV 2.4 Magnetismus . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 83 3 Geräte und Chemikalien 84 4 Literatur 84 Versuch 8 (MWG) Massenwirkungsgesetz 85 1 Theorie 85 1.1 Gleichgewichtsreaktionen . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 85 1.2 Die Ammoniaksynthese . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 86 2 Aufgabenstellung, Versuchsanleitung und -auswertung 2.1 90 Bestimmung der Gleichgewichtskonstanten Kp der Ammoniaksynthese in Abhängigkeit der Temperatur . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 90 3 Geräte und Chemikalien 93 4 Literatur 93 Versuch 9 (SBG) Säure-Base-Gleichgewichte 95 1 Theorie 95 1.1 Säure-Base-Theorien . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 95 1.2 Die Eigendissoziation des Wassers . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 95 1.3 Die Brønsted-Theorie . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 96 1.4 Die protochemische Spannungsreihe . . . . . . . . . . . . . . . . . . . . . . . . . . . . 97 1.5 Puffersysteme . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 99 1.6 Säure-Base Indikatoren . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 100 2 Aufgabenstellung, Versuchsanleitung und -auswertung 100 2.1 pH-Wert Abschätzung und Bestimmung . . . . . . . . . . . . . . . . . . . . . . . . . 100 2.2 Verwendung geeigneter Indikatoren . . . . . . . . . . . . . . . . . . . . . . . . . . . . 101 2.3 Titration von Essigsäure . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 102 2.4 Berechnung einer Puffermischung . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 102 3 Geräte und Chemikalien 103 4 Literatur 103 Versuch 10 (ROG) Redoxgleichgewichte 105 1 Theorie 105 1.1 Oxidation und Reduktion . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 105 1.2 Die elektrochemische Spannungsreihe . . . . . . . . . . . . . . . . . . . . . . . . . . . 105 1.3 Berechnung von Redoxpotentialen - Die Nernst’sche Gleichung . . . . . . . . . . . 106 V 1.4 Aufstellen von Redoxgleichungen . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 107 1.5 Kom- bzw. Synproportionierung und Disproportionierung . . . . . . . . . . . . . . 107 2 Aufgabenstellung, Versuchsanleitung und -auswertung 108 2.1 Elektrochemische Spannungsreihe . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 108 2.2 Abhängigkeit des Redoxpotentials vom pH-Wert . . . . . . . . . . . . . . . . . . . . 108 2.3 Redox-Amphoterie, Dis- und Synproportionierung . . . . . . . . . . . . . . . . . . . 109 2.4 Redox-Titration einer Cu2+ -Kationen-Lösung . . . . . . . . . . . . . . . . . . . . . . 110 3 Geräte und Chemikalien 111 4 Literatur 111 Versuch 11 (FLG) Fällungs- und Löslichkeitsgleichgewichte 113 1 Theorie 113 1.1 Löslichkeit und Löslichkeitsprodukt . . . . . . . . . . . . . . . . . . . . . . . . . . . . 113 1.2 Thermodynamik des Löslichkeitsprodukts . . . . . . . . . . . . . . . . . . . . . . . . 115 2 Aufgabenstellung, Versuchsanleitung und -auswertung 117 2.1 Einfluss verschiedener Zusätze auf das Löslichkeitsprodukt . . . . . . . . . . . . . . 117 2.2 Abhängigkeit des Löslichkeitsprodukts vom pH-Wert . . . . . . . . . . . . . . . . . 118 2.3 Fällungstitration einer Bromid-Lösung . . . . . . . . . . . . . . . . . . . . . . . . . . 118 3 Geräte und Chemikalien 119 4 Literatur 120 Versuch 12 (KOG) Komplexgleichgewichte 121 1 Theorie 121 1.1 Komplexreaktionen und Komplexstabilität . . . . . . . . . . . . . . . . . . . . . . . 121 1.2 Der räumliche Aufbau der Komplexe . . . . . . . . . . . . . . . . . . . . . . . . . . . 121 1.3 Die Ligandenfeldtheorie (Kristallfeldtheorie) . . . . . . . . . . . . . . . . . . . . . . 122 1.4 Die Valenzbindungstheorie . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 125 1.5 Eigenschaften von Komplexen . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 125 1.6 Die Nomenklatur von Komplexen . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 126 2 Aufgabenstellung, Versuchsanleitung und -auswertung 127 2.1 Beobachtung der Farben von Komplexen . . . . . . . . . . . . . . . . . . . . . . . . 127 2.2 Untersuchung der Stabilität von Komplexen . . . . . . . . . . . . . . . . . . . . . . . 127 2.3 Beobachtung der Hydratisomerie . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 128 2.4 Stabilität von Chelatkomplexen . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 128 3 Geräte und Chemikalien 129 VI 4 Literatur 129 Versuch 13 (LFG) Leitfähigkeit 131 1 Theorie 131 2 Aufgabenstellung, Versuchsanleitung und -auswertung 132 2.1 Aufgabenstellung . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 132 2.2 Versuchsdurchführung . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 132 2.3 Auswertung . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 133 3 Geräte und Chemikalien 133 4 Literatur 133 Versuch 14 (RKE) Reaktionskinetik - Enzyme: Aktivität und Stabilität 135 1 Theorie 135 1.1 Proteine, Struktur und Enzymaktivität . . . . . . . . . . . . . . . . . . . . . . . . . 135 1.2 Grundlagen der Reaktionskinetik . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 137 2 Aufgabenstellung, Versuchsanleitung und -auswertung 2.1 139 Bestimmung der Reaktionsgeschwindigkeit der Reduktion von Pyruvat zu Lactat in Abhängigkeit der Konzentration aktiver LDH . . . . . . . . . . . . . . . . . . . . 139 3 Geräte und Chemikalien 141 4 Literatur 141 Versuch 15 (DES) Destillationstechniken 143 1 Theorie 143 2 Aufgabenstellung, Versuchsaufbau und -auswertung 144 2.1 Trennung flüssiger Verbindungen durch Rektifikation bei Normaldruck . . . . . . 144 2.2 Trennung flüssiger Verbindungen durch Destillation unter vermindertem Druck am Rotationsverdampfer . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 145 3 Geräte, Chemikalien und Entsorgung 145 4 Literatur 146 Versuch 16 (SUB) Sublimation 147 1 Theorie 147 VII 2 Aufgabenstellung, Versuchsanleitung und -auswertung 2.1 147 Sublimation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 147 3 Geräte, Chemikalien und Entsorgung 148 4 Literatur 148 Versuch 17 (EXT) Extraktion 149 1 Theorie 149 2 Aufgabenstellung, Versuchsanleitung und -auswertung 149 2.1 Extraktion . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 149 3 Geräte, Chemikalien und Entsorgung 150 4 Literatur 150 Versuch 18 (CHR) Chromatographie 151 1 Theorie 151 2 Aufgabenstellung, Versuchsanleitung und -auswertung 152 2.1 DC-Chromatographie von Pflanzenfarbstoffen . . . . . . . . . . . . . . . . . . . . . . 152 2.2 DC- / Säulen-Chromatographie-Trennung von Benzoesäure und Benzophenon . . 153 3 Geräte, Chemikalien und Entsorgung 154 4 Literatur 154 Versuch 19 (UMK) Umkristallisation 155 1 Theorie 155 2 Aufgabenstellung, Versuchsanleitung und -auswertung 155 2.1 Umkristallisation von mit β-Naphtolorange gefärbter Benzoesäure aus Wasser. . . 155 3 Geräte, Chemikalien und Entsorgung 156 4 Literatur 156 Versuch 20 (ASP) Aspirin – Synthese von Acetylsalicylsäure 157 1 Theorie 157 2 Aufgabenstellung, Versuchsanleitung und -auswertung 157 2.1 Synthese von Acetylsalicylsäure . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 157 VIII 3 Geräte, Chemikalien und Entsorgung 159 4 Literatur 159 Versuch 21 (EAH) Vergleich der enzymatischen und alkalischen Hydrolyse von Triacylglyceriden 1 Theorie 161 161 1.1 Fette und Fettsäuren . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 161 1.2 Alkalische Fettverseifung . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 161 2 Aufgabenstellung, Versuchsanleitung und -auswertung 2.1 162 Alkalische und Enzymatische Hydrolyse von Tripalmitin . . . . . . . . . . . . . . . 162 3 Geräte und Chemikalien 164 4 Literatur 164 Einleitung 1 Einleitung 1 Allgemeines 1.1 Termine und Anwesenheit Das integrierte Praktikum findet an 21 Nachmittagen (à ca. 4h) im Zeitraum zwischen Ende Oktober und Mitte Februar im Wintersemester für Studierende der Fachrichtungen Chemie-Bachelor und Lebensmittelchemie statt; im Sommersemester dann zwischen Anfang April bis Mitte Juni für Studierende der Fachrichtungen Chemie- und NwT-Höheres Lehramt und Materialwissenschaft. Zum Praktikum gehören ferner 9 Nachmittage in denen ein ca. zweistündiges Seminar begleitend zum jeweiligen Block abgehalten wird. Die Teilnahme an jedem Praktikumsnachmittag ist Pflicht. Die Anwesenheit an den Seminarnachmittagen ist stark angeraten, jedoch keine Pflicht. Zum Bestehen des Praktikums müssen alle Versuche durchgeführt und bestanden werden. Für das Versäumen von Versuchstagen gelten folgende Regelungen: ● Entschuldigtes, attestiertes Fehlen (s. unten): 4 Nachmittage, ab dem 5. Nachmittag muss das gesamte Praktikum wiederholt werden. ● Unentschuldigtes Fehlen: 1 Nachmittag, ab dem 2. Nachmittag muss das gesamte Praktikum wiederholt werden. ● Nicht-Teilnahme aufgrund von Fehlverhalten: 3 Nachmittage, ab dem 4. Versäumnis muss das Praktikum wiederholt werden. Als Fehlverhalten wird folgendes bezeichnet: eine Verspätung von mehr als 5 Minuten, keine Betriebsanweisung für den Versuchsnachmittag vorbereitet, vermehrtes Nicht-Anlegen der persönlichen Schutzausrüstung (insbesondere der Schutzbrille, s. 1.2 Sicherheit im Labor), das Nicht-Bestehen des Vorkolloquiums (s. 1.2 Sicherheit im Labor) und das Nicht-Einhalten der Prokollabgabe / -korrekur-Prozedur (s. dort) Alle Fehlnachmittage müssen nach Ende der regulären Praktikumszeit nachgeholt werden. Bis zu 4 Nachmittage können entschuldigt bzw. ärztlich attestiert am regulären Termin versäumt werden, diese werden dann gegen Ende der Vorlesungszeit bzw. zu Beginn der vorlesungsfreien Zeit nachgeholt. Zum Entschuldigen müssen Sie sich am Praktikumstag bis spätestens 12:00 Uhr beim Praktikumsleiter Dr. Ingo Hartenbach telefonisch (0711/685-64254), per Fax (0711/68554254) oder E-mail ([email protected]) melden, Atteste können nachgereicht werden. Im Praktikum wird in Zweier- oder Dreiergruppen gearbeitet. Bei Fernbleiben eines Praktikumspartners führt der verbleibende Studierende den Versuch entweder alleine durch oder wird einer anderen Gruppe zugeteilt je nach Schwierigkeitsgrad bzw. Zeitaufwand des jeweiligen Versuchs. Die Entscheidung darüber liegt beim Assistenten. 1.2 Sicherheit im Labor Vor Beginn des Praktikums ist die Teilnahme an einer Allgemeinen Sicherheitsunterweisung, ebenso wie die Teilnahme an einer spezifischen Sicherheitsunterweisung Pflicht, die Termine dafür werden rechtzeitig per Aushang bzw. per Ankündigung in der Vorlesung und auch im Internet auf der Homepage des Praktikums angekündigt. Bei der spezifischen Sicherheitsunterweisung 2 Einleitung wird jeder Praktikant über die Sicherheitseinrichtungen im Labor unterrichtet. Die folgenden Broschüren stehen den Praktikanten jederzeit zum Nachschlagen zur Verfügung: Sicheres Arbeiten in Chemischen Laboratorien (Aufl. März 2006) (auch auf der DownloadWebseite des Integrierten Praktikums erhältlich) Richtlinien für Laboratorien (GUV R 120) Diese Broschüren informieren über die Richtlinien zur Unfallverhütung. Der Praktikant hat diese Richtlinien zur Kenntnis zu nehmen und einzuhalten. Dazu verpflichtet er sich mit der Unterschrift bei der Sicherheitsunterweisung. Ohne die Teilnahme an den beiden Sicherheitsunterweisungen ist eine Teilnahme am Praktikum nicht möglich. Das Betreten der Praktikumsräume ist nur mit persönlicher Schutzausstattung gestattet. Zu dieser gehören: ● eine Schutzbrille nach DIN 23, auch für Brillen- und Kontaktlinsenträger ● ein langärmliger baumwollener Arbeitsmantel ● geschlossenes Schuhwerk und lange Hosen/Röcke Einweg-Gummihandschuhe werden bei Versuchen, bei denen dies unabdingbar ist zur Verfügung gestellt. Wird ein Praktikant vermehrt (2-3 mal) ohne persönliche Schutzeinrichtung angetroffen, wird er/sie vom aktuellen Versuchstag ausgeschlossen und muss diesen wiederholen. Unbefugten oder Besuchern ist der Zutritt zum Labor strikt untersagt. Vor Beginn eines Versuchs hat der Praktikant sich anhand der Versuchsvorschrift im Skript über Versuchsaufbau, Durchführung und Reaktionsbedingungen zu informieren. Die Theorie entspricht hauptsächlich dem Stoff der Vorlesung ”Einführung in die Chemie” im Wintersemester. Da es nicht möglich ist, dass alle Studierenden die Versuche in der chronologischen Reihenfolge der Behandlung ihres Inhalts in der Vorlesung durchführen, muss der theoretische Inhalt der Versuche, ggf. auch mittels Literaturstudium, vorgearbeitet werden. Entsprechende Literaturhinweise finden sich am Ende der jeweiligen Versuchsbeschreibungen. Dazu gehört auch, sich über die Eigenschaften der verwendeten Chemikalien anhand der H- und P-Sätze und der ausliegenden Sicherheitsdatenblätter zu informieren und in der beiliegenden Betriebsanweisung (siehe Musterbetriebsanweisung) die entsprechenden Punkte (Umgang mit gefährlichen Stoffen, Schutzmaßnahmen, Erste Hilfe, Entsorgung) auszufüllen, und sich anhand von Literaturangaben über den Reaktionsmechanismus zu informieren. Über diese Kenntnisse werden von den Assistenten Kolloquien vor Beginn des Versuches durchgeführt. Wird das Vorkolloquium NICHT bestanden, muss der entsprechende Versuchstag nach Beendigung der regulären Praktikumszeit nachgeholt werden (s. 1.1 Termine und Anwesenheit). Essen, Trinken und Rauchen sind im Labor grundsätzlich untersagt. Bei Nichtbeachtung dieser Vorschrift droht ein Ausschluss vom aktuellen Versuchstag, welcher dann nach Beendigung der regulären Praktikumszeit wiederholt werden muss. Passiert dies ein zweites Mal, ist die Konsequenz der Praktikumsausschluss. Die gleichen Sanktionen gelten für die Einnahme von Drogen (auch Alkohol) bzw. für unter Drogen stehende oder betrunkene Praktikanten/Praktikantinnen. Straßenkleidung (jedoch keine Wertsachen!) muss in den bei Kursbeginn zugeteilten Garderobenschränken gelagert werden. Für im Labor beschädigte Kleidungsstücke kann keine Haftung übernommen werden. Es empfiehlt sich, im Garderobenschrank einen zweiten Satz älterer Klei- Einleitung 3 dungsstücke für den Notfall zu deponieren. Der Betrieb privater elektrischer Geräte (Mobiltelefone, Geräte der Unterhaltungselektronik wie Radios, CD-Player, MP3-Player usw.) ist im Praktikum aus Sicherheitsgründen nicht gestattet. Zuwiderhandlungen führen zum Laborverbot. 1.3 Allgemeine Sicherheitshinweise Was Sie vor Arbeitsbeginn wissen sollten: Wo befinden sich: Wissen Sie: Augenwaschflaschen ... wie die Medien (Gas, Wasser, Strom) abgeschaltet werden? Erste-Hilfe Schränke ... dass Feuerlöscher nach jeder Benutzung frisch gefüllt werden müssen? Feuerlöscher ... dass bei einem Großbrand kein Aufzug benutzt werden darf? Löschduschen ... dass Gasflaschen gegen Umfallen gesichert sein müssen? Fluchtwege ... was „Selbstschutz“ bedeutet? Arbeitsmedizinischer Dienst ... was bei schweren Unfällen zu tun ist? ... wer nach einem Unfall zu benachrichtigen ist? Darüber hinaus gilt: ● Jede Substanz ist in verschlossenen, eindeutig beschrifteten und nur für Chemikalien zugelassenen Gefäßen aufzubewahren (keine Trinkflaschen, etc.). ● Verschmutzte Gefäße, Gegenstände oder Laboratoriumseinrichtungen müssen sofort gesäubert werden. Substanzreste in Gefäßen nie stehen lassen. ● Beim Arbeiten mit giftigen, leicht entflammbaren, stark riechenden oder anderweitig belästigenden Substanzen muss immer ein Abzug benutzt werden. ● Entzündliche oder explosive Substanzen (z.B.: Ether, Alkohol) in der Nähe von offenen Flammen oder heißen Gegenständen bedeuten allerhöchste Gefahr!!! ● Glas bricht leicht! Glasgeräte immer kurz anfassen (kurzer Hebelarm). Herausragende Glasenden (T-Stücke, Krümmer an Kühlern usw.) nicht als Hebelarme beim Hantieren benutzen. Glasgefäße vorsichtig aufsetzen, nicht an Labortische anstoßen oder auf unebenen Stellen abstellen (punktförmig angreifende Kräfte, wie Sandkörner auf Tischen, sind für Glas gefährlich). ● In sich bewegende Anlagen niemals hineingreifen. Rührer oder Zentrifugen langsam Stufe um Stufe hinauf- oder herunterschalten. Bei ungewohnten Geräuschen sofort abschalten und nachsehen. ● Alle schweren Gegenstände vor dem Umfallen sichern. Gasflaschen mit Ketten oder schweren Riemen (nicht Draht oder Bindfaden) an der Wand oder am Labortisch befestigen bzw. auf einem speziell dafür vorgesehenen Flaschenwagen abstellen und transportieren. ● Achten Sie darauf, dass beim Verlassen des Laboratoriums die verwendeten Medien (z.B. Gas, Wasser, etc.) abgeschaltet sind, auch bei Ihren Nachbarn. 4 Einleitung ● Hauptursachen für Verbrennungen sind heiße Glasrohre, zurückschlagende Bunsenbrenner, eiserne Dreifüße, Tondreiecke u.ä., aber auch tiefgekühlte Apparate, Trockeneis oder flüssige Luft. Heiße Öfen können Brände auslösen, wenn in der Nähe mit organischen Lösungsmitteln gearbeitet wird. ● Alle Verbindungen, die zugleich oxidierende und reduzierende Bestandteile aufweisen (z.B.: Ammoniumnitrat), sind gefährlich. Auch das Zusammenbringen stark oxidierender Stoffe mit stark reduzierenden birgt große Gefahr. Instabile Verbindungen neigen zum Zerfall (z.B.: ● das aus KMnO4 und H2 SO4 (konz.) entstehende Mn2 O7 ). Elektrizität sieht man nicht, aber man fühlt sie, manchmal zu spät. 1.4 Verhalten bei Bränden In der Sicherheitsbelehrung werden die Standorte der Feuerlöscher und Notduschen gezeigt und deren Funktionsweise erklärt. Löschversuche mit nassen Tüchern und Wasser können in vielen Fällen (z.B. Ether) den Brand verstärken und sind daher zu unterlassen. Alle Lösungsmittel außer Wasser und halogenierten Kohlenwasserstoffen sind brennbar, die Dämpfe in bestimmten Konzentrationsbereichen explosiv. Beim Arbeiten mit brennbaren Lösungsmitteln dürfen keine Flammen in der Nähe sein. Lösungsmittelflaschen sind stets gut zu verschließen, es dürfen keine größeren Mengen an Lösungsmitteln am Arbeitsplatz vorrätig gelagert werden. Bei Ausbruch von Feuer sind alle leicht brennbaren Gegenstände rasch aus der Gefahrenzone zu entfernen. Haben die Kleider Feuer gefangen, wird die Notdusche benutzt. Lässt sich ein Brand mit den zur Verfügung stehenden Mitteln nicht mehr unter Kontrolle bringen, wird mittels des Brandmelders Feueralarm ausgelöst. Dann wird das Gebäude über die ausgeschilderten Fluchtwege (Treppenhaus) verlassen. 1.5 Umgang mit Chemikalien Bei allen Arbeiten sind Schutzbrille, Labormantel und ggf. Schutzhandschuhe zu tragen. Der Kontakt von Chemikalien mit Haut, Augen und Schleimhäuten ist auf jeden Fall zu vermeiden. Spritzer auf der Haut werden erst mit einem trockenen Lappen und anschließend ergiebig mit kaltem Wasser abgespült. Falls versehentlich Spritzer ins Auge gelangen, dieses sofort mit Wasser bzw. der Augendusche ausspülen und baldmöglichst einen Facharzt aufsuchen. Alle Arbeiten sollen, soweit möglich in einem gut ziehenden Abzug oder zumindest in gut belüfteten Räumen durchgeführt werden. Informieren Sie sich vor Beginn eines Versuches über gefährliche und gesundheitsschädliche Eigenschaften der eingesetzten Stoffe und tragen diese in die Betriebsanweisung ein. Flüssigkeiten nur mit dem Peleusball pipettieren. Fast alle Lösungsmittel, auch solche, die keine spezifischen Gifteigenschaften haben, wie z.B. Aceton oder Essigsäureethylester, wirken beim Einatmen berauschend, in größeren Konzentrationen narkotisierend. Die Giftigkeit von Stoffen, die über die Atemwege aufgenommen werden können, lässt sich anhand der MAK-Werte (Maximale Arbeitsplatzkonzentration) beurteilen. Diese Werte werden rechtsverbindlich in den Technischen Regeln für Gefahrenstoffe (TRGS 900) herausgegeben. Bei Präparaten, über die keine toxikologischen Daten vorliegen, gilt folgendes: Gefährliche Eigenschaften können nicht ausgeschlossen werden. Die Substanz ist mit der bei giftigen Chemikalien üblichen Vorsicht Einleitung 5 zu handhaben. Die ausgegebenen Chemikalien sind ausschließlich für Arbeiten im Labor vorgesehen. Es wird davon ausgegangen, dass die damit umgehenden Personen die notwendigen Vorsichtsmaßnahmen beim Umgang mit chemischen Substanzen kennen und anwenden. Da im Labor üblicherweise nur kleine Mengen verwendet werden, können Gefahren für die Gesundheit durch Beachtung der Empfehlungen weitgehend ausgeschaltet werden. Die Zuordnung krebserzeugender, erbgutveränderlicher oder die Fortpflanzungsfähigkeit gefährdenden Stoffe erfolgt in der Liste nach §4a Abs. 1 Gefahrstoffverordnung und der TRGS 905. Die TRGS 905 ist eine nationale Ergänzung der Liste nach §4a Gefahrstoffverordnung. Beide Listen sind zu beachten. Dabei werden krebserregende Stoffe mit K, erbgutverändernde Stoffe mit M und die Fortpflanzungsfähigkeit gefährdende Stoffe mit RF bzw. RE ausgewiesen. Die gefährlichen Eigenschaften werden in drei Kategorien unterteilt: Kategorie 1: Es ist bekannt, dass diese Stoffe beim Menschen zu den genannten Schäden bzw. Krankheiten führen. Kategorie 2: Anhand der Ergebnisse von Tierversuchen oder anderen Erkenntnissen ist davon auszugehen, dass diese Stoffe auch beim Menschen zu den genannten Schäden bzw. Krankheiten führen. Kategorie 3: Diese Stoffe können möglicherweise zu den genannten Schäden oder Krankheiten führen. Die Erkenntnislage ist jedoch noch unklar. Eine kurze Liste krebserregender Stoffe wurde Ihnen bei der Sicherheitsbelehrung vorgestellt. 1.6 Gefahrenbezeichungen Chemikalien mit deren Umgang besondere Gefahren verbunden sind werden zukünftig mit den Signalworten Achtung und Gefahr bezeichnet. Diese besonderen Gefahren werden durch die nachfolgend dargestellten Piktogramme genauer spezifiziert. Explosive Stoffe Entzündbare Stoffe Entzündend kende Stoffe GHS01 GHS02 GHS03 GHS05 Toxische Stoffe Gesundheitsgefährdende Stoffe GHS08 Umweltgefährdende Stoffe GHS09 Gase unter Druck GHS06 wir- Ätzende/Korrosive Stoffe GHS04 Achtung (Hinweis auf besondere Gefahren) GHS07 6 Einleitung 1.7 H- und P-Sätze Die H-Sätze (H = Hazard Statements) bezeichnen besondere Gefahren im Arbeiten mit Chemikalien, die P-Sätze (P = Precautionary Statements) beziehen sich auf Sicherheitsratschläge um diese Gefahren zu minimieren. Sollte trotz aller Vorsichtsmaßnahmen doch etwas passieren, ist sofort der Assistent zu benachrichtigen. Bezeichnung der besonderen Gefahren Physikalische Gefahren H200 Instabil, explosiv. H201 Explosiv, Gefahr der Massenexplosion. H202 Explosiv; große Gefahr durch Splitter, Sprengund Wurfstücke. H203 Explosiv; Gefahr durch Feuer, Luftdruck oder Splitter, Spreng- und Wurfstücke. H204 Gefahr durch Feuer oder Splitter, Spreng- und Wurfstücke. H205 Gefahr der Massenexplosion bei Feuer. H220 Extrem entzündbares Gas. H221 Entzündbares Gas. Bezeichnung der besonderen Gefahren Gesundheitsgefahren H300 Lebensgefahr bei Verschlucken. H301 Giftig bei Verschlucken. H302 Gesundheitsschädlich bei Verschlucken. H222 H223 H224 H225 H226 H228 H240 H241 Extrem entzündbares Aerosol. Entzündbares Aerosol. Flüssigkeit und Dampf extrem entzündbar. Flüssigkeit und Dampf leicht entzündbar. Flüssigkeit und Dampf entzündbar. Entzündbarer Feststoff. Erwärmung kann Explosion verursachen. Erwärmung kann Brand oder Explosion verursachen. Erwärmung kann Brand verursachen. Entzündet sich in Berührung mit Luft von selbst. H315 H317 H318 H319 H330 H331 H332 H334 Selbsterhitzungsfähig; kann in Brand geraten. In großen Mengen selbsterhitzungsfähig; kann in Brand geraten. In Berührung mit Wasser entstehen entzündbare Gase, die sich spontan entzünden können. In Berührung mit Wasser entstehen entzündbare Gase. Kann Brand verursachen oder verstärken; Oxidationsmittel. Kann Brand oder Explosion verursachen; starkes Oxidationsmittel. Kann Brand verstärken; Oxidationsmittel. Enthält Gas unter Druck; kann bei Erwärmung explodieren. Enthält tiefkaltes Gas; kann Kälteverbrennungen oder -Verletzungen verursachen. Kann gegenüber Metallen korrosiv sein. H340 H341 Umweltgefahren H373 H400 Sehr giftig für Wasserorganismen. 1 H410 Sehr giftig für Wasserorganismen mit langfristiger Wirkung. 2 H242 H250 H251 H252 H260 H261 H270 H271 H272 H280 H281 H290 H304 H310 H311 H312 H314 Kann bei Verschlucken und Eindringen in die Atemwege tödlich sein. Lebensgefahr bei Hautkontakt. H350 Giftig bei Hautkontakt. Gesundheitsschädlich bei Hautkontakt. Verursacht schwere Verätzungen der Haut und schwere Augenschäden. Verursacht Hautreizungen. Kann allergische Hautreaktionen verursachen. Verursacht schwere Augenschäden. Verursacht schwere Augenreizung. Lebensgefahr bei Einatmen. Giftig bei Einatmen. Gesundheitsschädlich bei Einatmen. Kann bei Einatmen Allergie, asthmaartige Symptome oder Atembeschwerden verursachen. Kann die Atemwege reizen. Kann Schläfrigkeit und Benommenheit verursachen. Kann genetische Defekte verursachen1 . Kann vermutlich genetische Defekte verursachen1 . Kann Krebs erzeugen1 . H351 Kann vermutlich Krebs erzeugen1 . H360 H362 H370 Kann die Fruchtbarkeit beeinträchtigen oder das Kind im Mutterleib schädigen1,2 . Kann vermutlich die Fruchtbarkeit beeinträchtigen oder das Kind im Mutterleib schädigen1,2 . Kann Säuglinge über die Muttermilch schädigen. Schädigt die Organe1,3 . H371 Kann die Organe schädigen1,3 . H372 Schädigt die Organe3 bei längerer oder wiederholter Exposition1 . Kann die Organe schädigen3 bei längerer oder wiederholter Exposition1 . Expositionsweg angeben, sofern schlüssig belegt ist, dass diese Gefahr bei keinem anderen Expositionsweg besteht. konkrete Wirkung angeben, sofern bekannt. H335 H336 H361 Einleitung H411 H412 H413 7 Giftig für Wasserorganismen, mit langfristiger Wirkung. Schädlich für Wasserorganismen, mit langfristiger Wirkung. Kann für Wasserorganismen schädlich sein, mit langfristiger Wirkung. Bezeichnung der besonderen Gefahren (nur EU) Physikalische Gefahren EUH001 In trockenem Zustand explosionsgefährlich. 3 oder alle betroffenen Organe nennen, sofern bekannt. EUH006 Mit und ohne Luft explosionsfähig. Bezeichnung der besonderen Gefahren (nur EU) Besondere Physikalische Gefahren EUH201 Enthält Blei. Nicht für den Anstrich von Gegenständen verwenden, die von Kindern gekaut oder gelutscht werden könnten. EUH210A Achtung! Enthält Blei. EUH014 Reagiert heftig mit Wasser. EUH202 EUH018 Kann bei Verwendung explosionsfähige/entzündbare Dampf/Luft-Gemische bilden. Kann explosionsfähige Peroxide bilden. EUH019 EUH044 Explosionsgefahr bei Erhitzen unter Einschluss. Gesundheitsgefahren EUH029 Entwickelt bei Berührung mit Wasser giftige Gase. EUH031 EUH070 EUH071 Entwickelt bei Berührung mit Säure giftige Gase. Entwickelt bei Berührung mit Säure sehr giftige Gase. Wiederholter Kontakt kann zu spröder oder rissiger Haut führen. Giftig bei Berührung mit den Augen. Wirkt ätzend auf die Atemwege. EUH059 Umweltgefahren Die Ozonschicht schädigend. EUH032 EUH066 Cyanacrylat. Gefahr. Klebt innerhalb von Sekunden Haut und Augenlider zusammen. Darf nicht in die Hände von Kindern gelangen. EUH203 Enthält Chrom (VI). Kann allergische Reaktionen hervorrufen. EUH204 Enthält Isocyanate. Kann allergische Reaktionen hervorrufen. EUH205 Enthält epoxidhaltige Verbindungen. Kann allergische Reaktionen hervorrufen. EUH206 Achtung! Nicht zusammen mit anderen Produkten verwenden, da gefährliche Gase (Chlor) freigesetzt werden können. EUH207 Achtung! Enthält Cadmium. Bei der Verwendung entstehen gefährliche Dämpfe. Hinweise des Herstellers beachten. Sicherheitsanweisungen einhalten. EUH208 Enthält Name des sensibilisierenden Stoffes. Kann allergische Reaktionen hervorrufen. EUH209 Kann bei Verwendung leicht entzündbar werden. EUH209A Kann bei Verwendung entzündbar werden. EUH210 EUH401 Sicherheitsdatenblatt auf Anfrage erhältlich. Zur Vermeidung von Risiken für Mensch und Umwelt die Gebrauchsanleitung einhalten. 8 Einleitung Vorsichtsmaßnahmen Allgemein P101 Ist ärztlicher Rat erforderlich, Verpackung oder Kennzeichnungsetikett bereithalten. P102 Darf nicht in die Hände von Kindern gelangen. P103 Vor Gebrauch Kennzeichnungsetikett lesen. Prävention P201 Vor Gebrauch besondere Anweisungen einholen. P202 Vor Gebrauch alle Sicherheitshinweise lesen und verstehen. P210 Von Hitze / Funken / offener Flamme / heißen Oberflächen fernhalten. Nicht rauchen. P211 Nicht gegen offene Flamme oder andere Zündquelle sprühen. P220 Von Kleidung / ... / brennbaren Materialien fernhalten/entfernt aufbewahren. P221 Mischen mit brennbaren Stoffen / ... unbedingt verhindern. P222 Kontakt mit Luft nicht zulassen. P223 Kontakt mit Wasser wegen heftiger Reaktion und möglichem Aufflammen unbedingt verhindern. P230 Feucht halten mit ... Vorsichtsmaßnahmen Reaktion P301 Bei Verschlucken: P231 Unter inertem Gas handhaben. P314 P232 Vor Feuchtigkeit schützen. P315 P233 Behälter dicht verschlossen halten. P320 P234 Nur im Originalbehälter aufbewahren. P321 P235 Kühl halten. P322 P240 P241 Behälter und zu befüllende Anlage erden. Explosionsgeschützte elektrische Betriebsmittel / Lüftungsanlagen / Beleuchtung / ... verwenden. Nur funkenfreies Werkzeug verwenden. Maßnahmen gegen elektrostatische Aufladungen treffen. Druckminderer frei von Fett und Öl halten. Nicht schleifen / stoßen / ... / reiben. Behälter steht unter Druck: Nicht durchstechen oder verbrennen, auch nicht nach der Verwendung. Staub / Rauch / Gas / Nebel / Dampf / Aerosol nicht einatmen. Einatmen von Staub / Rauch / Gas / Nebel / Dampf / Aerosol vermeiden. Nicht in die Augen, auf die Haut oder auf die Kleidung gelangen lassen. P335 P336 P263 Kontakt während der Schwangerschaft/und der Stillzeit vermeiden. P341 P264 P270 P271 Nach Gebrauch ... gründlich waschen. Bei Gebrauch nicht essen, trinken oder rauchen. Nur im Freien oder in gut belüfteten Räumen verwenden. P342 P350 P351 P242 P243 P244 P250 P251 P260 P261 P262 P302 P303 P304 P305 P306 Bei Bei Bei Bei Bei P307 Bei Exposition: P308 Bei Exposition oder falls betroffen P309 Bei Exposition oder Unwohlsein: P310 P330 P331 Sofort Giftinformationszentrum oder Arzt anrufen. Giftinformationszentrum oder Arzt anrufen. Bei Unwohlsein Giftinformationszentrum oder Arzt anrufen. Ärztlichen Rat einholen/ärztliche Hilfe hinzuziehen. Bei Unwohlsein ärztlichen Rat einholen/ärztliche Hilfe hinzuziehen. Sofort ärztlichen Rat einholen/ärztliche Hilfe hinzuziehen. Besondere Behandlung dringend erforderlich (siehe ... auf diesem Kennzeichnungsetikett). Besondere Behandlung (siehe ... auf diesem Kennzeichnungsetikett). Gezielte Maßnahmen (siehe ... auf diesem Kennzeichnungsetikett). Mund ausspülen. Kein Erbrechen herbeiführen. P332 P333 Bei Hautreizung: Bei Hautreizung oder -ausschlag: P334 In kaltes Wasser tauchen/nassen Verband anlegen. Lose Partikel von der Haut abbürsten. Vereiste Bereiche mit lauwarmem Wasser auftauen. Betroffenen Bereich nicht reiben. P311 P312 P313 Berührung mit der Haut: Berührung mit der Haut (oder dem Haar): Einatmen: Kontakt mit den Augen: kontaminierter Kleidung: P337 Bei anhaltender Augenreizung: P338 Eventuell vorhandene Kontaktlinsen nach Möglichkeit entfernen. Weiter ausspülen. Die betroffene Person an die frische Luft bringen und in einer Position ruhigstellen, die das Atmen erleichtert. Bei Atembeschwerden an die frische Luft bringen und in einer Position ruhigstellen, die das Atmen erleichtert. Bei Symptomen der Atemwege: Behutsam mit viel Wasser und Seife waschen. Einige Minuten lang behutsam mit Wasser ausspülen. P340 Einleitung P272 9 Kontaminierte Arbeitskleidung nicht außerhalb des Arbeitsplatzes tragen. Freisetzung in die Umwelt vermeiden. Schutzhandschuhe / Schutzkleidung / Augenschutz / Gesichtsschutz tragen. P352 Mit viel Wasser und Seife waschen. P353 P360 P361 P401 Vorgeschriebene persönliche Schutzausrüstung verwenden. Schutzhandschuhe / Gesichtsschild / Augenschutz mit Kälteisolierung tragen. Schwer entflammbare / flammhemmende Kleidung tragen. Atemschutz tragen. Bei unzureichender Belüftung Atemschutz tragen. Aufbewahrung ... aufbewahren. P402 An einem trockenen Ort aufbewahren. P374 P403 An einem gut belüfteten Ort aufbewahren. P375 P404 P405 In einem geschlossenen Behälter aufbewahren. Unter Verschluss aufbewahren. P376 P377 P406 In Korrosionsbeständigem / ... Behälter mit korrosionsbeständiger Auskleidung aufbewahren. Luftspalt zwischen Stapeln/Paletten lassen. Vor Sonnenbestrahlung schützen. P378 Haut mit Wasser abwaschen/duschen. Kontaminierte Kleidung und Haut sofort mit viel Wasser abwaschen und danach Kleidung ausziehen. Alle kontaminierten Kleidungsstücke sofort ausziehen. Kontaminierte Kleidung ausziehen und vor erneutem Tragen waschen. Kontaminierte Kleidung vor erneutem Tragen waschen. Bei Brand: Bei Großbrand und großen Mengen: Explosionsgefahr bei Brand. Keine Brandbekämpfung, wenn das Feuer explosive Stoffe/Gemische/Erzeugnisse erreicht. Brandbekämpfung mit üblichen Vorsichtsmaßnahmen aus angemessener Entfernung. Wegen Explosionsgefahr Brand aus der Entfernung bekämpfen. Undichtigkeit beseitigen, wenn gefahrlos möglich. Brand von ausströmendem Gas: Nicht löschen, bis Undichtigkeit gefahrlos beseitigt werden kann. ... zum Löschen verwenden. Bei Temperaturen von nicht mehr als ... °C / ... aufbewahren. Nicht Temperaturen von mehr als 50 °C aussetzen. Schüttgut in Mengen von mehr als ... kg bei Temperaturen von nicht mehr als ... °C aufbewahren. Von anderen Materialien entfernt aufbewahren. Inhalt in/unter ... aufbewahren. P390 P273 P280 P281 P282 P283 P284 P285 P407 P410 P411 P412 P413 P420 P422 Kombinationen von Vorsichtsmaßnahmen Kombinationen von P-Sätzen (Prävention) P231 + Unter inertem Gas handhaben. Vor FeuchtigP232 keit schützen. P235 + Kühl halten. Vor Sonnenbestrahlung schützen. P410 Kombinationen von P-Sätzen (Reaktion) P301 + P310 P301 + P312 P301 + P330 + P331 P302 + P334 BEI VERSCHLUCKEN: Sofort GIFTINFORMATIONSZENTRUM oder Arzt anrufen. BEI VERSCHLUCKEN: Bei Unwohlsein GIFTINFORMATIONSZENTRUM oder Arzt anrufen. BEI VERSCHLUCKEN: Mund ausspülen. KEIN Erbrechen herbeiführen. BEI KONTAKT MIT DER HAUT: In kaltes Wasser tauchen/nassen Verband anlegen. P362 P363 P370 P371 P372 P373 P380 P381 P391 P501 Umgebung räumen. Alle Zündquellen entfernen, wenn gefahrlos möglich. Verschüttete Mengen aufnehmen, um Materialschäden zu vermeiden. Verschüttete Mengen aufnehmen. Entsorgung Inhalt/Behälter ... zuführen. Kombinationen von Vorsichtsmaßnahmen P332 + Bei Hautreizung: Ärztlichen Rat einhoP313 len/ärztliche Hilfe hinzuziehen. P333 + Bei Hautreizung oder -ausschlag: Ärztlichen P313 Rat einholen/ärztliche Hilfe hinzuziehen. P335 + Lose Partikel von der Haut abbürsten. In kaltes P334 Wasser tauchen/ nassen Verband anlegen. P337 + Bei anhaltender Augenreizung: Ärztlichen Rat P313 einholen/ärztliche Hilfe hinzuziehen. P342 + Bei Symptomen der Atemwege: GIFTINFORP311 MATIONSZENTRUM oder Arzt anrufen. P370 + Bei Brand: Undichtigkeit beseitigen, wenn geP376 fahrlos möglich. P370 + P378 Bei Brand: ... zum Löschen verwenden. P370 + P380 Bei Brand: Umgebung räumen. 10 Einleitung P302 + P350 BEI KONTAKT MIT DER HAUT: Behutsam mit viel Wasser und Seife waschen. P302 + P352 BEI KONTAKT MIT DER HAUT: Mit viel Wasser und Seife waschen. P303 + P361 + P353 BEI KONTAKT MIT DER HAUT (oder dem Haar): Alle beschmutzten, getränkten Kleidungsstücke sofort ausziehen. Haut mit Wasser abwaschen/duschen. BEI EINATMEN: An die frische Luft bringen und in einer Position ruhigstellen, die das Atmen erleichtert. BEI EINATMEN: Bei Atembeschwerden an die frische Luft bringen und in einer Position ruhigstellen, die das Atmen erleichtert. BEI KONTAKT MIT DEN AUGEN: Einige Minuten lang behutsam mit Wasser spülen. Vorhandene Kontaktlinsen nach Möglichkeit entfernen. Weiter spülen. BEI KONTAKT MIT DER KLEIDUNG: Kontaminierte Kleidung und Haut sofort mit viel Wasser abwaschen und danach Kleidung ausziehen. BEI Exposition: GIFTINFORMATIONSZENTRUM oder Arzt anrufen. BEI Exposition oder falls betroffen: Ärztlichen Rat einholen/ärztliche Hilfe hinzuziehen. BEI Exposition oder Unwohlsein: GIFTINFORMATIONSZENTRUM oder Arzt anrufen. P304 + P340 P304 + P341 P305 + P351 + P338 P306 + P360 P307 + P311 P308 + P313 P309 + P311 P370 P380 P375 P371 P380 P375 + + + + Bei Brand: Umgebung räumen. Wegen Explosionsgefahr Brand aus der Entfernung bekämpfen. Bei Großbrand und großen Mengen: Umgebung räumen. Wegen Explosionsgefahr Brand aus der Entfernung bekämpfen. Kombinationen von P-Sätzen (Aufbewahrung) P402 + P404 In einem geschlossenen Behälter an einem trockenen Ort aufbewahren. P403 + P233 Behälter dicht verschlossen an einem gut belüfteten Ort aufbewahren. P403 + P235 Kühl an einem gut belüfteten Ort aufgewahren. P410 + P403 P410 + P412 P411 + P235 Vor Sonnenbestrahlung geschützt an einem gut belüfteten Ort aufbewahren. Vor Sonnenbestrahlung schützen und nicht Temperaturen von mehr als 50 °C aussetzen. Kühl und bei Temperaturen von nicht mehr als ... °C aufbewahren. Einleitung 11 2 Ablauf eines Versuchstages 2.1 Vor dem Versuchstag Vor Beginn des Versuchstages müssen Sie sich mit der Theorie und dem Ablauf des Versuches derart auseinandersetzen, dass Sie in der Lage sind das ca. 30 minütige Vorkolloquium zu bestehen. Dazu gehört auch, dass Sie sich mit den am Tag benötigten Chemikalien und deren sicherheitsrelevanten Eigenschaften (Giftigkeit, Brennbarkeit, Explosionsgefahr, etc.) vertraut machen. Darüber hinaus müssen die Reaktionsgleichungen erstellt werden und auch die sicherheitsrelevanten Eigenschaften der Produkte gewusst werden. Wie bereits zuvor erwähnt gilt: Wird das Vorkolloquium NICHT bestanden, gelten die Sanktionen, wie in 1.1 beschrieben. Darüber hinaus besteht bei Versuchen mit bekannten Chemikalien die Pflicht eine Betriebsanweisung (pro Gruppe) auszufüllen und an den Abzug des Arbeitsplatzes zu hängen. Ein Vordruck kann auf der Praktikumshomepage heruntergeladen werden. Ein Muster für eine Betriebsanweisung sieht folgendermaßen aus: Abb. 2.1: Muster einer Betriebsanwseisung 2.2 Versuchsbeginn und -durchführung Die Versuche werden gemäß der im Skript oder der im Platz ausliegenden Versuchsanleitung durchgeführt, wobei mit dem Assistenten abgesprochene Veränderungen stets beachtet werden. Während des Versuchsablaufs wird ein Laborjournal geführt, in dem der Ablauf eines Versuchs, die Beobachtungen und die erhaltenen Messwerte dokumentiert werden. Es soll kein perfekt formulierter Bericht, sondern ein Labortagebuch sein, jedoch sollte ein Außenstehender anhand 12 Einleitung des Laborjournals in der Lage sein den durchgeführten Versuch nachzuvollziehen. Damit hat man später eine exakte Beschreibung der Durchführung eines Versuchs. Auch aus einigen Fehlversuchen kann man Folgerungen und Schlüsse ziehen, aus denen sich anschließend ein erfolgreiches Experiment durchführen lässt. Im Laborjournal lassen sich rasch experimentelle und analytische Daten sowie Literaturhinweise zu jeder synthetisierten Verbindung finden. Dies ist auch für spätere Praktika, Diplom-, Doktorarbeiten, Berichte und Publikationen sehr wichtig. Für die Versuche des 3. Blocks (Organische Chemie und Arbeitstechniken) dient das Laborjournal als Protokoll. Wie führt man ein Laborjournal? Es wird leserlich und in einem leicht verständlichen Stil geschrieben, so dass jeder Fachkundige den Versuch anhand des Laborjournals reproduzieren kann. Das Laborjournal wird direkt während der Durchführung der Experimente am Arbeitsplatz geführt (keine lose Zettelwirtschaft) und darin steht der exakte beobachtete Verlauf eines Experiments. Während der Arbeiten im Praktikum hat der Praktikant zu seiner eigenen Sicherheit stets für Sauberkeit an seinem Arbeitsplatz zu sorgen sowie die zur Verfügung gestellten Geräte schonend zu behandeln. Zu Bruch gegangene Geräte müssen dem Assistenten unverzüglich gemeldet und am Ende des Praktikums gezahlt werden. 2.3 Ende eines Versuchstages Nach dem Arbeitsende sind die Arbeitstische und Schränke von den Praktikanten aufzuräumen und zu säubern. Abfälle sind nach entsprechender Behandlung in die dafür vorgesehenen Behälter einzufüllen. Darüber hinaus sind die gemeinschaftlich genutzten Einrichtungen (Waagen etc.) stets sauber und benutzbar zu halten. Störungen oder Defekte jeglicher Art sind unverzüglich den Assistenten zu melden. Geräte und Apparaturen müssen vor einer Reparatur gründlichst gereinigt werden, mit Chemikalien verunreinigte Geräte werden nicht repariert. Die Beendigung des Versuchstages kann nur erfolgen, wenn der Arbeitsplatz aufgeräumt und gereinigt ist und alle Geräte vollständig und voll funktionsfähig übergeben wurden. Verlässt ein Student den Arbeitsplatz bevor der Assistent diesen abgenommen hat, gilt der Versuchstag als nicht durchgeführt und muss nachgeholt werden. 2.4 Protokoll Zu jedem Kurstag ist pro Gruppe ein Protokoll anzufertigen. Das Protokoll kann sowohl handschriftlich als auch computergeschrieben dem Assistenten vorgelegt werden. Das Protokoll schließt die versuchsbezogene Betriebsanweisung mit ein. Im Protokoll sind die im Laborjournal gesammelten Daten und Beobachtungen auszuwerten, die erhaltenen Ergebnisse zu diskutieren und die weiterführenden Fragen bzw. Aufgaben zu beantworten. Das Protokoll muss am nächsten Versuchstag beim Assistenten vorliegen und spätestens am dritten Werktag nach Abgabe auch wieder abgeholt werden, außer der entsprechende Assistent nennt Ihnen einen anderen Termin. Ist eine Korrektur durchzuführen wird spätestens am nächsten Praktikumsnachmittag diese dem Assistenten übergeben, es sei denn es wird ein anderer Termin vereinbart. Ist das Protokoll dann noch nicht in Ordnung kann noch einmal nachkorrigiert werden, die Fristen werden vom Assisten- Einleitung 13 ten bestimmt. Wenn auch nach der zweiten Korrektur das Protokoll nicht in Ordnung ist, muss der Versuchstag wiederholt werden Die saubere, übersichtliche und verständliche Dokumentation der eigenen Arbeit ist ein sehr wichtiger und oft unterschätzter Teil wissenschaftlichen Arbeitens. Nachfolgend geben wir einige Hinweise, wie ein gutes Protokoll auszusehen hat bzw. welche häufigen Fehler unbedingt zu vermeiden sind: ● Kein Abschreiben vom Skript. Der Theorieteil darf 2 Seiten NICHT ÜBERSCHREITEN. Alles was darüber hinaus geht, wird vom Assistenten nicht korrigiert. ● Werden Passagen aus dem Internet wenig modifiziert oder gänzlich unverändert in das Protokoll übernommen, muss dies mit Quellenangabe erfolgen, ansonsten gilt dies als Betrugsversuch und wird gehandhabt wie unentschuldigtes NichtErscheinen zum Praktikumstag. ● Werden Passagen oder komplette Protokolle von Protokollen anderer Praktikanten oder aus sog. „Altmeistern“ direkt kopiert gilt dies ebenfalls als Betrugsversuch und wird gehandhabt wie unentschuldigtes Nicht-Erscheinen zum Praktikumstag. ● ● ● Ganze Sätze, jedoch so kurz wie möglich halten Kein Erlebnisaufsatz (Kein „ich“ oder „wir“; „man“ ist sehr sparsam zu benutzen) Keine Abkürzungen im Text (z. B. Lsg., Nd. etc.), außer sie werden zu Beginn des Protokolls definiert ● Zahlen im Textzusammenhang ausschreiben (drei Reagenzgläser), bei Angaben Ziffern verwenden (1,5 g, 100 °C) ● Für die meisten Versuche wird demineralisiertes (demin.) Wasser verwendet, kein destilliertes (dest.) Wasser ● ● ● NUR eigene Beobachtungen beschreiben und deuten! Reaktionsgleichungen aufstellen und ausgleichen - ggf. in der Literatur nachschlagen Genaue Mengenangaben (2 Tropfen, eine Spatelspitze, 3 g, ein Reagenzglas wird ca. 2 cm hoch mit ... gefüllt), unter dem Begriff „wenige“ werden hier 1-5 Tropfen aus einer Tropfpipette verstanden unter dem Begriff „einige“ 5-20 Tropfen. ● Einheitliche Zeit (alles im Präsens oder Präteritum) Bei computergeschriebenen Protokollen gilt zusätzlich: ● Wenn das Protokoll computergeschrieben wird, gilt dies für alle Bestandteile, d.h. keine Zeichungen oder Reaktionspfeile von Hand ● Abbildungen, Formeln, Tabellen, Diagramme sind auf dem Computer zu erstellen (sofern dies nicht während der Versuchsdurchführung geschehen ist) ● Blocksatz oder linksbündig, einheitliche, lesbare Schriftgröße und -art, Zeilenabstand 1 oder 1,5 Zeilen, Seitenzahlen, Einheitliche Formatierung (Überschriften hervorheben etc.) ● ● ● Reaktionspfeile Ð→ (kein Folgepfeil Ô⇒ o.ä.) bei Gleichgewichten Gleichgewichtspfeile ⇌ verwenden Hoch- und Tiefstellung der Zahlen beachten 14 Einleitung Musterprotokoll: Gruppennummer und Namen der Praktikanten Datum Versuch X: 1. Theorieteil: Zusammenfassung der wichtigsten theoretischen Grundlagen und Zusammenhänge, nicht mehr als zwei Seiten. 2. Aufgabenstellung, Versuch 1: Aufgabenstellung des jeweiligen Versuchs kurz darstellen. 3. Versuchsaufbau, Versuch 1: Skizze und Beschreibung der Versuchsanordnung, sofern dies notwendig ist. (Absprache mit dem Assistenten) 4. Versuchsdurchführung, Versuch 1: Beschreibung dessen was TATSÄCHLICH gemacht wurde, in eigenen Worten! Keine Abschrift aus dem Skript oder irgendwelchen Büchern! 5. Beobachtungen, Versuch 1: Auch hier wieder beschreiben was tatsächlich beobachtet wurde - Farbänderung, Gasentwicklung, Niederschlagsbildung etc. 6. Reaktionsgleichung, Versuch 1: ±0 ±0 +1 -2 O2 + 2 H2 Ð→ 2 H2 O wichtig hierbei: Die Reaktionsgleichung muss ausgeglichen sein und die Oxidationsstufen (wo sinnvoll) sind über die Edukte und Produkte zu schreiben. 7. Deutung / Auswertung, Versuch 1: Beschreibung des Resultats in eigenen Worten. Berechnungen angeben, ggf. Formeln, Abbildungen, Diagramme und Tabellen einfügen, diese gemäß Anweisungen im Skript beschriften. 8. Zusatzaufgaben, Versuch 1: Beantwortung der im Skript zusätzlich gestellten Aufgaben, bitte mit übertragener Fragestellung. 9. Aufgabenstellung, Versuch 2: ... (siehe Punkte 2-8) Gruppennummer Seite 1 Falls sich Messwerte im Laborjournal befinden, sind die entsprechenden Seiten in Kopie dem Protokoll beizufügen. Gasgesetz, Molmassenbestimmung und Stöchiometrie 15 VERSUCHSBLOCK 1: ATOMBAU UND PERIODENSYSTEM DER ELEMENTE Versuch 1 (GMS) Gasgesetz, Molmassenbestimmung und Stöchiometrie 1 Theorie 1.1 Grundlagen der Stöchiometrie Die wichtigste Grundlage chemischer Reaktionen ist, dass bei diesen die Gesamtmasse der an der Reaktion beteiligten Stoffe erhalten bleibt. Dieses Gesetz sagt jedoch noch nichts über die Massen- und Teilchenzahlenverhältnisse bei chemischen Reaktionen aus. Die sich damit befassenden Gesetze sind die stöchiometrischen Gesetze. 1.1.1 Gesetz der konstanten Proportionen (Joseph Louis Proust, 1799) „Das Massenverhältnis zweier sich zu einer Verbindung vereinigender Elemente ist konstant.“ Beim Zerlegen verschiedener Stoffe in deren Bestandteile werden daher immer gleiche Massenverhältnisse gefunden, z. B. die Zerlegung von Wasser in Wasserstoff und Sauerstoff ergibt ein Massenverhältnis 1:7,936 Wasserstoff : Sauerstoff (die Zerlegung von Ammoniak in Stickstoff und Wasserstoff ergibt ein Massenverhältnis von 1:4,632 Wasserstoff : Stickstoff) 1.1.2 Gesetz der multiplen Proportionen (John Dalton, 1803) „Die Massenverhältnisse zweier sich zu verschiedenen chemischen Verbindungen vereinigenden Elemente stehen im Verhältnis einfacher ganzer Zahlen zueinander.“ Elemente bilden häufig nicht nur eine, sondern mehrere Verbindungen miteinander. Ein Beispiel dafür ist die Reaktion von Stickstoff mit Sauerstoff aus der fünf verschiedene Verbindungen resultieren können. Deren Massenverhältnisse stellen jedoch nicht willkürliche, voneinander unabhängige Zahlenwerte dar, sondern stehen untereinander in einem einfachen Zusammenhang: Verbindung Verbindung Verbindung Verbindung Verbindung 1: 2: 3: 4: 5: Massenverhältnis Massenverhältnis Massenverhältnis Massenverhältnis Massenverhältnis Sauerstoff Sauerstoff Sauerstoff Sauerstoff Sauerstoff : : : : : Stickstoff Stickstoff Stickstoff Stickstoff Stickstoff = = = = = 0,571 1,142 1,713 2,284 2,855 : : : : : 1 1 1 1 1 = = = = = (1×0,571 (2×0,571 (3×0,571 (4×0,571 (5×0,571 : : : : : 1) 1) 1) 1) 1) Dieses Gesetz erweitert und beinhaltet daher das Gesetz der konstanten Proportionen. 16 Gasgesetz, Molmassenbestimmung und Stöchiometrie 1.1.3 Gesetz der äquivalenten Proportionen (Jeremias Benjamin Richter, 1791) „Elemente vereinigen sich stets im Verhältnis bestimmter Verbindungsmassen (Äquivalentmassen) oder ganzzahliger Vielfacher dieser Massen zu chemischen Verbindungen.“ Bei Verbindung 3 aus Kapitel 1.1.2 kann das Verhältnis 1,713 : 1 (= 3×0,571 : 1) auch durch 7,936 : 4,632 ausgedrückt werden, also genau diese Zahlen die auch die Massenverhältnisse der jeweiligen Elemente zu Wasserstoff darstellen (siehe Kapitel 1.1). Somit ergibt sich für die Stickstoff-Sauerstoff-Verbindungen: Verbindung Verbindung Verbindung Verbindung Verbindung 1: 2: 3: 4: 5: Massenverhältnis Massenverhältnis Massenverhältnis Massenverhältnis Massenverhältnis Sauerstoff Sauerstoff Sauerstoff Sauerstoff Sauerstoff : : : : : Stickstoff Stickstoff Stickstoff Stickstoff Stickstoff = = = = = 0,571 1,142 1,713 2,284 2,855 : : : : : 1 1 1 1 1 = = = = = (1×7,936) (2×7,936) (3×7,936) (4×7,936) (5×7,936) : : : : : (3×4,632) (3×4,632) (3×4,632) (3×4,632) (3×4,632) Hierbei stellen die Werte 7,936 und 4,632 die jeweiligen Äquivalentmassen von Sauerstoff und Stickstoff dar. Dieses Gesetz erweitert und beinhaltet daher die beiden zuvor erwähnten Gesetze. 1.1.4 Chemisches Volumengesetz (Joseph Louis Gay-Lussac, 1808) „Das Volumenverhältnis gasförmiger, an einer chemischen Reaktion beteiligter Stoffe lässt sich bei gegebener Temperatur und gegebenem Druck durch einfache ganze Zahlen wiedergeben.“ Man beobachtet, dass bei der Zerlegung von Wasser in seine Elemente diese zwar im Massenverhältnis 7,936 : 1 (Sauerstoff : Wasserstoff), jedoch in einem Volumenverhältnis von 1 : 2 (Sauerstoff : Wasserstoff) entstehen. Darüber hinaus zeigt das Experiment, dass aus zwei Volumenteilen elementaren Wasserstoffs und einem Volumenteil Sauerstoff zwei Volumenteile gasförmiges Wasser entstehen, jeder überschüssig angebotene Teil eines der beiden Edukte liegt nach der entsprechenden Reaktion unverändert vor. Daraus ergeben sich die folgenden Gleichungen: 2 Volumina Wasserstoff 3 Volumina Wasserstoff 1 Volumen Wasserstoff + + + 1 Volumen Sauerstoff 1 Volumen Stickstoff 1 Volumen Chlor Ð→ Ð→ Ð→ 2 Volumina Wasserdampf 2 Volumina Ammoniak 2 Volumina Chlorwasserstoff Aufgrund dieser Beobachtung postulierte Avogadro, dass es sich bei den Gasen Stickstoff, Sauerstoff, Wasserstoff und Chlor um zweiatomige Moleküle handelt und darüber hinaus, dass egal um welches Gas es sich handelt, dies bei festgelegten Bedingungen immer den gleichen Raum einnimmt. 1.2 Stoffmenge Zur Bestimmung des Begriffs Stoffmenge muss zuerst die Masse eines Atoms festgelegt werden. Dies geschah 1805 in Bezug auf ein Wasserstoffatom, dem John Dalton als relative Atommasse den willkürlichen Wert 1 gab. Dies erwies sich im Laufe der Zeit nicht als zweckmäßig und mit einem kurzen Umweg über Sauerstoff legte die IUPAC 1961 den Wert für die relative Atommasse des Kohlenstoffisotops 12 6C als 12 fest. Darauf basieren alle Atommassen im Periodensystem. Es Gasgesetz, Molmassenbestimmung und Stöchiometrie ergibt sich weiterhin, dass 12 g des Kohlenstoffisotops 17 12 6C die gleiche Teilchenanzahl besitzen, wie 18,998 g isotopenreines Fluor. Diese Teilchenzahl beträgt 6,02214×1023 und wird 1 Mol genannt (vergleichbar mit der Zahl 12, die 1 Dutzend genannt wird). Somit besitzen 6,02214×1023 12 6C-Kohlenstoffisotope die Masse 12 g, also ist deren molare Masse 12 g mol . Bei der Verwendung in Rechnungen kommt der Zahl 1 Mol die Bezeichnung Avogadro-Konstante zu (NA ). Sie besitzt die Einheit 1 mol . Ein Mol eines beliebigen Gases nimmt bei Standardbedingungen (1,013 bar Druck, 298 K Temperatur) immer ein Volumen von 24,46 Litern ein (siehe auch Versuch 8 MWG). 1.3 Das ideale Gas Der Zustand eines idealen Gases, d. h. eines Gases, bei welchem die Gasteilchen nicht untereinander wechselwirken und bei welchem das Eigenvolumen der Gasteilchen gegenüber dem Gesamtvolumen V vernachlässigbar ist, kann beschrieben werden durch Angabe des Volumens V , des Druckes p und der Temperatur T . Wie die allgemeine Erfahrung zeigt, nimmt bei konstanter Temperatur der Druck eines Gases zu, wenn das Volumen verringert wird: p∼ 1 V (T = konstant) (1) Wird das Volumen des Gases konstant gehalten und die Temperatur erhöht, so steigt der Druck linear mit der Temperatur T : p∼T (V = konstant) (2) Beide Beobachtungen können in einem Gesetz zusammengefasst werden, dem idealen Gasgesetz: p⋅V =n⋅R⋅T (3) R ist die allgemeine Gaskonstante und n die Stoffmenge bzw. Molzahl. Ein Mol eines Gases – so ergibt sich aus den Messungen – nimmt bei T = 273,15 K und p = 1 bar ein Volumen von 22,71⋅10−3 m3 ein. Setzt man die Größen in das ideale Gasgesetz ein, so ergibt sich für die allgemeine Gaskonstante der Wert: R = 8,314441 JK−1 mol−1 . Die Stoffmenge n ergibt sich aus der Masse m des Gases und seiner Molmasse M gemäß: n= m M (4) Einsetzen dieser Beziehung in die ideale Gasgleichung ergibt: p⋅V = m ⋅R⋅T M (5) bzw. aufgelöst nach der Molmasse M : M= m⋅R⋅T p⋅V (6) 18 Gasgesetz, Molmassenbestimmung und Stöchiometrie Wie aus dieser Gleichung folgt, kann man die Molmasse eines Gases bestimmen, wenn m, T, p und V bekannt sind. Diese Größen sind experimentell einfach zu ermitteln. Die meisten Gase und Dämpfe lassen sich bei Normalbedingungen in guter Näherung als ideale Gase beschreiben. Damit ist eine Molmassenbestimmung für leicht verdampfbare Flüssigkeiten mit Hilfe des idealen Gasgesetzes möglich. 2 Aufgabenstellung und Versuchsanleitungen 2.1 Elektrolyse von Wasser/Synthese von Wasser in der Brennstoffzelle 2.1.1 Aufgabenstellung Bei diesem Versuch soll durch Elektrolyse die Zusammensetzung von Wasser bestimmt werden. Weiterhin soll diese Zusammensetzung durch die Synthese mittels einer Brennstoffzelle verifiziert werden. 2.1.2 Versuchsanleitung Für die Durchführung werden die am Versuchsplatz ausliegenden Arbeitsanweisungen zur Elektrolyse befolgt. Es wird ca. 4-5 min. elektrolysiert und der Gasfüllstand in Abhängigkeit der Zeit abgelesen (alle 30 s). Nach Beendigung der Elektrolyse werden über eine Pneumatische Wanne zwei Reagenzgläser mit den entstandenen Gasen (Reagenzgläser nicht verwechseln) befüllt. Mit dem Gas, das am Plus-Pol entstanden ist, wird die Glimmspanprobe durchgeführt. Dazu wird ein Holzspan entzündet und die Flamme gelöscht, so dass der Span gerade noch glimmt. Der Span wird in das mit Gas gefüllte Reagenzglas gebracht. Mit dem Gas, das am Minus-Pol entstanden ist, wird die Knallgasprobe durchgeführt. Dazu wird das vollständig mit Gas gefüllte Reagenzglas mit dem Daumen verschlossen und das Gas an einer Flamme (Feuerzeug) entzündet. Für die Durchführung des zweiten Versuchsteils werden die am Versuchsplatz ausliegenden Arbeitsanweisungen zur Elektrolyse und zur Brennstoffzelle befolgt. Durch gezieltes Entweichen lassen der Gase, werden die Füllstände in beiden Gasauffangbehältern auf das gleiche Niveau gebracht (20 ml Gas). Die beiden Ablassschläuche werden nun an eine Brennstoffzelle mit Verbraucher angeschlossen, und am Ende der Reaktion werden beide Gasvolumina erneut abgelesen. Dieser Versuch wird mit folgenden Startvolumina an Gas erneut durchgeführt: 20 ml am Minus-Pol und 10 ml am Plus-Pol, 10 ml am Minus-Pol und 20 ml am Plus-Pol, dabei wird alle 5 Minuten der verbleibende Stand der Gasvolumina abgelesen. Welche Beobachtungen machen Sie, wie können Sie diese erklären? Gegebenenfalls müssen Sie zur Herstellung der Gase die Elektrolyse aus Versuchsteil 1 nochmals durchführen. (Dabei auf die maximalen Werte für Spannung und Stromstärke achten!) 2.1.3 Versuchsauswertung ● ● Welche Teilreaktionen finden während der Elektrolyse am Minus- bzw. Pluspol statt? Welche Teilreaktionen laufen bei Stromverbrauch am jeweiligen Pol ab? Gasgesetz, Molmassenbestimmung und Stöchiometrie ● 19 Ordnen Sie die Begriffe Kathode und Anode sowohl bei der Elektrolyse als auch beim Stromverbrauch den jeweiligen Polen (minus/plus) zu. ● Berechnen Sie die Anzahl an Molekülen der jeweiligen Gase, die nach 1, 2, 3 und 4 min. Elektrolyse entstanden sind. Welche Schlussfolgerungen können Sie daraus ableiten? 2.2 Bestimmung der molaren Masse von Calcium 2.2.1 Aufgabenstellung Aus der Reaktion von Calcium in Wasser soll dessen Molmasse bestimmt werden. 2.2.2 Versuchsanleitung Der Standzylinder wird vollständig mit Wasser gefüllt, mit einem Uhrglas verschlossen und umgedreht in die mit Wasser gefüllte Kristallisierschale eingebracht. Dort wird dieser mit einer Klemme am Stativ befestigt und nach Wegnahme des Uhrglases der anfängliche Wasserstand notiert. Achten Sie darauf, dass die Öffnung des Standzylinders möglichst nahe am Boden der Kristallisierschale ist, jedoch immer noch so weit entfernt, dass das vom Gas verdrängte Wasser aus dem Zylinder entweichen kann. Mit der Pinzette wird ein ca. 0,03 – 0,08 g schweres Calciumstück aus dem Vorratsgefäß entnommen und auf der Analysenwaage genau abgewogen. Das abgewogene Stück Calcium bringt man mit Hilfe der Pinzette zügig in den mit Wasser gefüllten Standzylinder. Es muss darauf geachtet werden, dass das Calciumstück auch während der Reaktion im Standzylinder verbleibt. Nach vollständiger Reaktion des Calciums wird das Gasvolumen abgelesen. Notieren Sie sich die Umgebungstemperatur und den momentanen Luftdruck. Außerdem werden noch wenige Tropfen Phenolphthalein ins Wasser gegeben. Der Versuch wird noch einmal wiederholt, um das zuerst erhaltene Ergebnis zu verifizieren. Benutzen Sie dazu frisches Wasser! 2.2.3 Versuchsauswertung ● ● Notieren Sie sich die Versuchsdurchführung. Beschreiben Sie Ihre Beobachtungen genau. Formulieren Sie eine Reaktionsgleichung für die Reaktion von Calcium in Wasser, um welche Art von Reaktion handelt es sich? ● ● ● ● Welches Gas entsteht bei der Reaktion von Calcium in Wasser? Wie ist das Stoffmengenverhältnis von Gas und Calcium? Wie wird die Stoffmenge eines Gases aus dem Volumen des entstandenen Gases berechnet? Berechnen Sie zuerst die Stoffmenge an gebildetem Gas (allgemeine Gasgleichung) und anschließend die molare Masse von Calcium. 2.3 Molmassenbestimmung nach Victor-Meyer 2.3.1 Aufgabenstellung Bei diesem Versuch wird die Molmasse einer unbekannten Flüssigkeit nach der Methode von Victor-Meyer bestimmt. 20 Gasgesetz, Molmassenbestimmung und Stöchiometrie 2.3.2 Versuchsaufbau Ein großes rohrartiges Gefäß ist mit einem Füllgas (z.B. Luft) höherer Temperatur T 2 gefüllt (siehe Abbildung 2.1), welches auch bei Zimmertemperatur T 1 der Gleichung für ideale Gase gehorcht. Wird am Boden eine kleine Menge von der zu untersuchenden Substanz eingebracht, die bei T 2 als Dampf das Volumen V 2 einnimmt, so entweicht oben eine gleiche Volumenmenge des Füllgases. Dieses wird auf die Zimmertemperatur T 1 abgekühlt und sein Volumen V 1 wird mit einer Gasbürette gemessen. Das Substanzgewicht m wird vor dem Einbringen bestimmt. V 2 und T 2 brauchen nicht gemessen zu werden, T 2 muss nur genügend hoch sein. V 1 ist dasjenige Volumen, welches die Substanz einnehmen würde, wenn sie auch bei Zimmertemperatur als Gas vorläge. Einsetzen von T 1 , V 1 , m und des herrschenden Druckes p in Gleichung 6 ergibt unmittelbar die Molmasse der verdampfbaren Substanz. Abb. 2.1: Schematische Darstellung einer Apparatur zur Bestimmung der Molmasse nach 2.3.3 Versuchsanleitung Victor-Meyer Ein leeres Probenkölbchen aus Glas wird mit Stopfen gewogen. Dann wird es mit Hilfe einer Spritze mit einer kleinen Menge der zu untersuchenden Flüssigkeit befüllt. Anschließend wird das Kölbchen mit Stopfen nochmals gewogen, die Einwaage soll 50 bis 80 mg betragen. Das Kölbchen K wird an der Stelle F (siehe Abbildung 2.1) in das Victor-Meyer-Rohr R eingebracht. Hierzu wird es mit dem am Stopfen befestigten Faden vorsichtig auf den Glasstab S gestellt. Dieser verhindert vorläufig das Hinabfallen. Als Heizung dient ein Wasserbad im Mantel M. Sobald sich ein Temperaturgleichgewicht eingestellt hat (Anzeige der Gasbürette B ändert sich nicht mehr), erzeugt man kurzzeitig durch Heben des Niveaugefäßes N einen Überdruck in R. Nach Wiederherstellung der Niveaugleichheit der beiden Flüssigkeitssäulen muss die Anzeige der Gasbürette gegenüber vorher unverändert sein, sonst ist das System undicht. Der Bürettenstand wird dann abgelesen und der Glasstab S soweit herausgezogen, dass das Kölbchen K nach unten auf die Glaswolle in R fällt. Wenn alle Substanz verdampft ist, wird wieder die Anzeige der Gasbürette bei Niveaugleichheit abgelesen. Die Volumendifferenz V 1 und Zimmertemperatur T werden in Gleichung 6 eingesetzt. Nach der Messung wird das Victor-Meyer-Rohr zum Entfernen von Dampfresten mit Luft gespült und eine kleine Menge Glaswolle auf das darin befindliche Kölbchen gegeben (mit Kunststoffstab nach unten schieben). Dieser Versuch wird noch zweimal wiederholt. Gasgesetz, Molmassenbestimmung und Stöchiometrie 21 2.3.4 Versuchsauswertung und weitere Aufgaben ● Mit dem bestimmten Volumen, der Einwaage, der aktuellen Zimmertemperatur und dem aktuellen Druck wird nach Gleichung 6 die Molmasse der unbekannten Substanz bestimmt. ● Welche Voraussetzungen müssen erfüllt sein, damit die Molmasse nach Victor-Meyer gemessen werden kann? 3 Geräte und Chemikalien Eine Geräteliste liegt am Platz aus Chemikalien Calcium Phenolphthalein H2 1) O2 1) Ca(OH)2 1) Gefahrenbezeichnung Gefahr (GHS02) Gefahr (GHS06) Gefahr (GHS02) Gefahr (GHS03) Gefahr (GHS08) H-Sätze 261 350, 341, 361f 220 270 315, 318, 335 Aceton2) Diisopropylether2) Gefahr (GHS03, 08) Gefahr (GHS02) Essigsäuremethylester2) n-Hexan2) Gefahr (GHS02, 08) Gefahr (GHS02, 08, 09) Methanol2) Gefahr (GHS02, 06) n-Pentan2) Gefahr (GHS02, 08, 09) 225, 319, 336, EUH066 225, 336, EUH019, EUH066 225, 319, 336, EUH066 225, 304, 315, 336, 361f, 373, 411 225, 301+311+331, 370 225, 304, 336, 411, EUH066 1) Produkte 2) Mögliche Substanzen zur Molmassenbestimmung nach Victor-Meyer P-Sätze 402+404 201, 281, 308+313 210, 377, 381, 403 244, 220, 370+376, 403 360, 302+352, 304+340, 305+351+338, 313 210, 233, 305+351+338 210, 233, 240, 403+235 210, 305+351+338, 403+233 210, 240, 273, 301+310, 302+352, 331, 403+235 210, 233, 280, 302+352, 304+340, 309+310, 403+225 273, 301+310, 331, 403+225 4 Literatur [1] [2] [3] G. Wedler: Lehrbuch der Physikalischen Chemie, 5. Aufl., Wiley-VCH, 2004. P. W. Atkins, J. de Paula: Physikalische Chemie, 4. Aufl., Wiley-VCH, 2008. A. F. Holleman, E. Wiberg: Lehrbuch der Anorganischen Chemie, 102. Aufl., Walter de Gruyter Verlag, 2007. [4] M. Binnewies, M. Jäckel, H. Willner, G. Rayner-Canham: Allgemeine und Anorganische Chemie, 2. Auflage, Spektrum Verlag, 2011. Optische Absorptionsspektroskopie 23 Versuch 2 (ABS) Optische Absorptionsspektroskopie 1 Theorie 1.1 Grundlagen der optischen Absorptionsspektroskopie Eine Lichtquelle liefert monochromatisches Licht der Wellenlänge λ mit der Intensität I0 (λ). Dieses Licht wird beim Durchstrahlen einer Küvette, die die Schichtdicke d hat, geschwächt. Nach dem Durchgang durch die Küvette hat der Lichtstrahl die Intensität I1 (λ). In der Küvette sei eine Substanz in einer Flüssigkeit gelöst. Die Konzentration der Substanz sei c 0 . Für die Absorption der Strahlung gilt dann das Lambert-Beer’sche Gesetz: I1 (λ) = I0 (λ) ⋅ e−(λ)⋅c0 ⋅d (1) Die Größe (λ) gibt das Absorptionsvermögen der gelösten Substanz an und wird als molarer Extinktionskoeffizient bezeichnet. Der molare Extinktionskoeffizient ist eine stoffspezifische Größe und kann zur Charakterisierung von Stoffen und zu ihrer Konzentrationsbestimmung benutzt werden. Sind der molare Extinktionskoeffizient eines Stoffes (λ) und die Schichtdicke d bekannt, so kann nach Gleichung (1) die Konzentration c0 bestimmt werden, wenn die Abb. 1.1: Prinzip der Absorptionsspek- Intensität des einfallenden Lichtes I0 (λ) und die Intensität des durchgelassenen Lichtes I1 (λ) bekannt troskopie sind. Beide Größen sind sehr einfach messbar. Die Absorption von Licht durch ein Molekül ist mit einem Energieübertrag verbunden. Das Molekül nimmt Energie auf. Je nach dem, wie groß die Energie des Lichtes ist, oder mit anderen Worten, welche Wellenlänge das Licht besitzt, können unterschiedliche Energieformen des Moleküls angeregt werden. So kann das Molekül z.B. schneller rotieren oder in Schwingung geraten. Es können aber auch wie bei den Atomen (siehe dazu Versuch 3 BSP) Elektronen angeregt werden. Im sichtbaren bis ultravioletten Bereich des Spektrums des Lichtes werden Elektronen angeregt, und wie bei den Atomen gibt es auch hier diskrete Energieniveaus. Die Kenntnis dieser Energieniveaus ist von großer Bedeutung, da sie Informationen über die elektronischen Zustände von Molekülen liefert, ist aber, wie leicht nachzuvollziehen ist, in den meisten Fällen sehr schwierig zu berechnen. Ein sehr einfacher Fall ist das Teilchen in einem Kasten. 1.2 Teilchen im Kasten Beim Teilchen im Kasten wird, wie aus dem Namen abzulesen ist, der Zustand, d.h. die Energie eines Elektrons, das in einem Kasten eingesperrt ist, berechnet. Der Kasten habe die Länge a 24 Optische Absorptionsspektroskopie und seine Wände seien unendlich hoch, damit das Elektron nicht entweichen kann. Mit diesem Modell lassen sich, wie in der Vorlesung behandelt wird, die Energiezustände des Elektrons durch Lösen der Schrödinger-Gleichung berechnen. Es ergeben sich für die Energien die Werte: En = n 2 ⋅ h2 8 ⋅ m ⋅ a2 (2) m ist die Masse des Elektrons, a, wie bereits erwähnt, die Länge des Kastens, h ist das Planck’sche Wirkungsquantum. Analog zum Bohr’schen Atommodell (siehe Versuch 3 BSP) ist n eine Quantenzahl, die folgende Werte annehmen kann: n = 1, 2, 3, ... (3) Befindet sich im Kasten nur ein Elektron im Grundzustand, so hat es die Quantenzahl: ng = 1. (4) Der nächstmögliche (angeregte) Energiezustand des Elektrons ist: na = 2. (5) Um das Elektron in diesen Zustand zu bringen ist die Energie: E = Ea − Eg (6) notwendig. Diese Energiedifferenz liegt im ultravioletten bis sichtbaren Bereich des elektromagnetischen Spektrums. Durch Absorption von Licht, dessen Energie mit der Energiedifferenz ∆E identisch ist, kann das Elektron von seinem Grundzustand ng = 1 in den angeregten Zustand na = 2 übergehen. Befinden sich im Kasten N -Elektronen, wobei die Einschränkung gilt, dass N geradzahlig sei, so muss beachtet werden, dass nach dem Pauliprinzip sich nur zwei Elektronen in jedem Energieniveau befinden dürfen. Die N -Elektronen besetzen also N /2-Energieniveaus im Kasten. Durch Absorption von Licht kann nun ein Elektron vom höchsten besetzten Zustand in den niedrigsten unbesetzten Zustand angehoben bzw. angeregt werden, also von: ng = N /2 (7) na = N /2 + 1 (8) auf Die Energiedifferenz ∆E ist dann nach Einsetzen von ng und na in Gleichung 2: ∆E = 2 h2 N N 2 {( + 1) − ( ) } 8 ⋅ m ⋅ a2 2 2 woraus sich ∆E = h2 (N + 1) 8 ⋅ m ⋅ a2 (9) (10) Optische Absorptionsspektroskopie 25 ergibt. Die Energie bzw. die Wellenlänge des Lichtes, das diesen Übergang erzeugt, ist ∆Eλ = h ⋅ c λga c = Lichtgeschwindigkeit (11) und entspricht der Energiedifferenz ∆E aus Gleichung 10. 1.2.1 Bestimmung der Länge eines π-Elektronensystems Organische Verbindungen mit alternierenden Einfach- und Doppelbindungen können im einfachsten Fall als ein eindimensionaler Kasten aufgefasst werden, in dem die π-Elektronen über die Bindungen delokalisiert sind. Die Länge dieses Kastens kann bestimmt werden, wenn die Zahl der Elektronen, also N , und die Energie Eλ bekannt ist, bei der die Substanz Licht absorbiert. Einsetzen dieser Größen in Gleichung 10 ergibt die gesuchte Größe a. Die Energie Eλ wird bestimmt, indem das Verhältnis von I1 (λ) zu I0 (λ) in Abhängigkeit von der Wellenlänge λ gemessen wird. Die Lage des Absorptionsmaximums in diesem Absorptionsspektrum liefert die Wellenlänge λga . 1.2.2 Bestimmung der Dissoziationskonstante einer Säure Wie bereits eingangs erwähnt, kann die optische Absorptionsspektroskopie auch zur Konzentrationsbestimmung benutzt werden, vorausgesetzt, dass die molaren Extinktionskoeffizienten der beteiligten Spezies bekannt sind. Auf diese Weise kann z.B. die Dissoziationskonstante einer Säure bestimmt werden. Eine Säure HA dissoziiert in Wasser gemäß: HA ⇌ A− + H + (12) Für die Dissoziationskonstante – die Gleichgewichtskonstante dieser Reaktion – gilt: KS = [H + ] ⋅ [A− ] [HA] (13) mit [H + ] = c(H + ), [A− ] = c(A− ) und [HA] = c(HA) wobei c die molare Konzentration der jeweiligen Stoffe bezeichnet. Aufgrund der Elektroneutralitätsbedingung sind die Konzentrationen von H + und A− gleich: [H + ] = [A− ] (14) Außerdem ist die Einwaage bzw. Ausgangskonzentration c0 (HA) bekannt. Die Summe aus nicht-dissoziierter und dissoziierter Säure muss gleich der Ausgangskonzentration sein. Es gilt also: [HA] + [A− ] = c0 (HA) (15) Gelingt es, die Konzentration A− zu bestimmen, so ist bei bekannter Ausgangskonzentration c0 (HA) die Konzentration von HA bestimmbar. Damit sind alle Größen in Gleichung 13 gegeben, um die Dissoziationskonstante einer Säure zu berechnen. (Siehe auch Versuch 9 SBG). 26 Optische Absorptionsspektroskopie 2 Aufgabenstellung, Versuchsanleitung und -auswertung 2.1 Bestimmung der Länge eines eindimensionalen Potentialtopfs 2.1.1 Aufgabenstellung Messung des Absorptionsspektrums einer β-Carotinlösung und Bestimmung der Länge des Potentialtopfs. 2.1.2 Versuchsanleitung Bedienung des Meßprogrammes UV-Winlab Einloggen: Nach dem Anschalten von Rechner, Drucker und Spektrometer loggen sich die Benutzer in der Domäne PRO unter dem Benutzernamen ”praktikum” ohne Kennwort ein und starten das Programm ”uvwinlab” über die Verknüpfung vom Desktop aus. Eingabe der Messparameter: Im Fenster ”methods” werden die Messparameter (z. B. ”pcanf.msc”) durch Doppelklick geladen. Die Messparameter auf der Seite ”Scan” dieses Fensters können gegebenenfalls angepasst werden. Am häufigsten anzupassen ist der Wellenlängenbereich in Nanometern, wobei ”Start wavelength” die maximale Wellenlänge, ”End” die minimale Wellenlänge und ”Data Interval” die Schrittweite/Auflösung (1 nm) sind. Weiterhin können Werte für die Anzahl der Messdurchgänge (”Number of cycles”, 1.) und den Ordinatenbereich des Anzeigefensters (”Ordinate min/max”, Intervall [0 ;1] ist guter Ausgangswert, der bei Bedarf später angepasst werden kann) angegeben werden. Die Seite ”Inst” des Fensters ”methods” enthält Angaben zum Aufnahmemodus (”A” für Absorptionsmessung), die Aufnahmegeschwindigkeit (”Scan speed”, 120 nm/min) und die verwendeten Strahlungsquellen. Diese Einstellungen dürfen nicht verändert werden. Die Seite ”Sample” enthält Angaben zu Anzahl der Proben und deren Zusammensetzung. Diese Eingaben dienen nur der Information des Benutzers und beeinflussen die Messung nicht. Gerätekalibrierung: Für jede Messreihe muss eine Gerätekalibrierung durchgeführt werden. Dazu werden die beiden Küvetten mit dem reinen Lösungsmittel etwa 2 cm hoch gefüllt und ins Spektrometer eingebracht (matte Seiten nach vorne und hinten, Beschriftung in eine gleichbleibende Richtung, z. B. links). Die Kalibrierung wird mit der Schaltfläche ”Autozero” gestartet und das Einbringen der Küvetten in einem Dialog bestätigt. Aufnahme der Messungen: Nach der Kalibrierung können alle Spektren dieser Messreihe aufgenommen werden. Dazu bleibt die hintere Küvette mit dem Lösungsmittel gefüllt und die vordere Küvette enthält die Probe im Lösungsmittel. Durch Drücken der Schaltfläche ”Start” wird die Messung durchgeführt. Bearbeitung der Diagramme: Das dadurch erzeugte Diagramm kann weiter bearbeitet werden, nachdem das entsprechende Fenster (z.B. ”Graph1”) mit der Maus markierte wurde. Ein Ausdruck ist über den Eintrag ”Print” im Menü ”File” möglich. Werden die Spektren weiterhin benötigt (z. B. für einen Vergleich mehrerer Spektren einer Messreihe), so Optische Absorptionsspektroskopie 27 können sie mit ”Save as...” abgespeichert werden. Die Schaltflächenleiste bietet verschiedene Möglichkeiten die Anzeige des Diagramms anzupassen und Maxima anzuzeigen. Die jeweilige Funktion wird beim Berühren der Schaltfläche mit der Maus in der Informationsleiste am unteren Fensterrand angezeigt. Über das Menü ”Data handling” können mit Hilfe des Eintrags ”List” die Werte in einem anzugebenden Intervall mit einer bestimmten Schrittweite (z. B. 10 nm) angezeigt und dann auch ausgedruckt werden. Der Eintrag ”Peak” dieses Menüs sucht automatisch nach Maxima. Im Menü ”View” können einzelne oder alle Kurven eines Diagramms gelöscht (”Remove ... curve(s)”) werden und mit ”Add Spectrum” gespeicherte Kurven hinzugeladen werden, um in einem gemeinsamen Diagramm anzuzeigen. Aufnahme des Absorptionsspektrums der β-Carotinlösung Führen Sie die Gerätekalibirierung mit reinem Cyclohexan durch und vermessen Sie die β-Carotinlösung im Wellenlängenbereich 500 − 330 nm nach obiger Anleitung. Drucken Sie das Diagramm aus und bestimmen Sie das Absorptionsmaximum. 2.1.3 Versuchsauswertung ● Ermitteln Sie die Lage des Maximums im Absorptionsspektrum des β-Carotins und berechnen Sie daraus die Energie des Lichts. Welchem elektronischen Übergang entspricht es vermutlich, wenn man das konjugierte π-System des β-Carotins als eindimensionalen Potentialtopf betrachtet (β-Carotin hat 22 π-Elektronen.)? ● Berechnen Sie mit diesen Angaben die Länge des Kastens. Die mittlere Bindungslänge in einem konjugierten π-System ist 139,8 pm. Welche Gesamtlänge ergäbe sich hieraus, wenn Sie berücksichtigen, dass der Bindungswinkel 120○ beträgt? Wie ist die Abweichung zu erklären? 2.2 Bestimmung der Dissoziationskonstante einer Säure 2.2.1 Aufgabenstellung Bestimmung der Dissoziationskonstante von 2,4-Dinitrophenol 2.2.2 Versuchsanleitung Achtung: 2,4 Dinitrophenol ist ein Hautgift! Bei Benetzen der Hände o. ä. sofort waschen! Führen Sie die Gerätekalibrierung mit demineralisiertem Wasser durch. Füllen Sie 50 ml der eingestellten Dinitrophenollösung (10−4 mol l ) in ein 100 ml Becherglas. Mit diesem Ansatz wird der gesamte Versuch durchgeführt. Nehmen Sie zunächst eine Messung dieser wässrigen Lösung im Wellenlängenbereich 500 – 300 nm auf und befüllen Sie dazu die Küvette. Nach der Messung wird die Probe wieder ins Becherglas zurückgegeben und auch bei allen folgenden Messungen wird ebenso verfahren. Dadurch bleibt die Gesamtstoffmenge des Dinitrophenols/Dinitrophenolats konstant. Die verschiedenen pH-Werte werden wie folgt gewählt, in einem Becherglas wird 28 ● ● ● Optische Absorptionsspektroskopie für die schwach saure Lösung 0.25 ml 0.1 molare HCl zugegeben. für die stark alkalische Lösung (pH > 10) zusätzlich 0.5 ml 0.1 molare NaOH zugegeben. für die stark saure Lösung zusätzlich (pH < 2) 0.4 ml 2 molare HCl zugegeben. Die sich durch diese Vorgehensweise ergebenden Verdünnungen sind in der Auswertung zu berücksichtigen. Nehmen Sie die Absorptionsspektren dieser vier Ansätze auf und speichern deren Diagramme in unterschiedliche Dateien. Drucken Sie jeweils die Extinktionswerte mit ”List” in der Schrittweite 10 nm aus. Laden Sie schließlich wie oben beschrieben die Kurven aller Messungen in ein Diagramm und drucken Sie dieses aus. 2.2.3 Versuchsauswertung 1. Messung der Absorptionsspektren von 2,4-Dinitrophenol-Lösungen (wässrig, stark alkalisch, stark sauer und schwach sauer). Anmerkung: Durch Änderung des pH-Wertes verschiebt sich das Gleichgewicht und es liegt mehr (bzw. weniger) 2,4-Dinitrophenol (HA) vor. Sie messen daher Absorptionsspektren von Lösungen mit unterschiedlichen Zusammensetzungen (fast nur HA, fast nur A− bzw. einer Mischung aus beiden). 2. Für die stark alkalische Lösung wird für fünf geeignet gewählte Messpunkte der Extinktionskoeffizient des Anions (λ)A− berechnet. 3. Für die stark saure Lösung wird ebenfalls für diese Messpunkte der Extinktionskoeffizient von 2,4-Dinitrophenol (λ)HA berechnet. 4. Für die wässrige Lösung wird ebenfalls für diese Messpunkte die Konzentration von A− und die Dissoziantionskonstante berechnet. Dissoziationskonstante des 2,4-Dinitrophenols In diesem Experiment wurde eine stark alkalische Lösung gemessen, in der das Dissoziationsgleichgewicht ganz auf die Seite des Anions A− verschoben ist, d.h. hier gilt: [A− ] = c0 (HA) (16) Da die Konzentration c0 - sie ist identisch mit der Einwaage - und die Schichtdicke bekannt sind, kann nach dem Lambert-Beer’schen Gesetz aus der Extinktion E(λ)A− der molare Extinktionskoeffizient (λ)A− berechnet werden: E(λ)A− = ln ( I0 (λ) ) = (λ)A− ⋅ c0 (HA) ⋅ d I1 (λ) (17) Die Extinktion wird an fünf geeignet gewählten Messpunkten aus dem Spektrum entnommen und daraus der molare Extinktionskoeffizient (λ)A− bei den jeweiligen Wellenlängen ermittelt. Auf gleiche Weise wird aus der dritten Messung, in der nur die undissoziierte Säure vorliegt, da die Lösung stark sauer ist, der molare Extinktionskoeffizient (λ)HA bestimmt. Es müssen die gleichen Messpunkte gewählt werden wie zuvor. Nachdem die molaren Extinktionskoeffizienten von HA und A− bekannt sind, wird die Extinktion – wieder an den identischen Messpunkten Optische Absorptionsspektroskopie 29 – aus dem Spektrum der wässrigen Lösung ermittelt. Die Extinktion der wässrigen Lösung ist gegeben durch: E(λ)Lsg = {(λ)HA ⋅ [HA] + (λ)A− ⋅ [A− ]} ⋅ d (18) Da außerdem Gleichung 15 gilt und c0 , d und die beiden molaren Extinktionskoeffizienten bekannt sind, sind für jeden Messpunkt die Konzentrationen von HA und A− und damit die Dissoziationskonstante berechenbar. Es werden für alle fünf Messpunkte die Dissoziationskonstanten berechnet. 3 Geräte und Chemikalien Eine Geräteliste liegt am Platz aus Chemikalien 2β-Carotin 2,4-Dinitrophenol Aceton Cyclohexan Gefahrenbezeichnung – Gefahr (GHS06, 09) Gefahr (GHS02, 08) Gefahr (GHS02, 08, 09) HCl aq Gefahr (GHS05) NaOH Gefahr (GHS05) H-Sätze P-Sätze EUH044 – 301, 311, 331, 373, 400 261, 273, 380, 301+310, 311 EUH066, 225, 319, 336 210, 233, 305+351+338 225, 304, 315, 336, 410 210, 240, 273, 301+310, 331, 403+235 314, 335, 290 301+330+331, 305+351+338, 280 314, 290 301+330+331, 305+351+338, 280 4 Literatur [1] [2] [3] G. Wedler: Lehrbuch der Physikalischen Chemie, 5. Aufl., Wiley-VCH, 2004. P. W. Atkins, J. de Paula: Physikalische Chemie, 4. Aufl., Wiley-VCH, 2008. C. Czeslik, H. Seemann, R. Winter: Basiswissen Physikalische Chemie, 2. Aufl., Teubner, 2007. [4] W. G. Moore, D. O. Hummel: Physikalische Chemie, 4. Aufl., Walter de Gruyter, 1986. Balmer-Serie und Spektroskopie 31 Versuch 3 (BSP) Balmer-Serie und Spektroskopie 1 Theorie Bei der optischen Wahrnehmung unserer Umwelt sind dem Menschen im makroskopischen wie im mikroskopischen Bereich Grenzen gesetzt. Will man Gegenstände, die kleiner als 0,1 mm sind, beobachten, benötigt man ein Lichtmikroskop. Ein Eindringen in die Mikrowelt ist mit Rastertunnelmikroskopen möglich, welche die Beobachtung von Atomen auf einer Metalloberfläche erlauben. Atomare Bausteine, also Elektronen, Protonen oder Neutronen, können jedoch auf keinen Fall direkt aufgelöst werden. Viele Erkenntnisse über Atome und ihre Bestandteile stammen aus Experimenten über das Verhalten von Materie unter bestimmten Versuchsbedingungen. Um die experimentellen Ergebnisse deuten zu können, entwickelt man bis heute Vorstellungen vom Aufbau der Atome. Es ist wichtig zu begreifen, dass ein solches Modell kein reales Abbild eines Atoms sein muss, sondern nur das Zustandekommen eines bestimmten Versuchsergebnisses zu erklären hat. Liegen also neue Erkenntnisse vor, die sich mit einem vorhandenen Modell nicht deuten lassen, muss dieses erweitert oder eventuell ein völlig neues Modell entwickelt werden. Auf diese Weise sind im Laufe der Zeit verschiedene Atommodelle entstanden, die teilweise nur noch historische Bedeutung besitzen. Der Begriff ”Atom” leitet sich von dem griechischen Wort ”atomos” für ”unteilbar” ab. Im theoretischen Teil wollen wir alte, überkommene Atommodelle überspringen und gleich mit dem ”Kern-Hülle-Modell” nach Ernest Rutherford (1871-1937) beginnen. 1.1 Das Kern-Hülle-Modell Der englische Physiker Rutherford bestrahlte eine dünne Goldfolie (etwa 500 nm ≡ 2000 Atomlagen dick) mit α-Strahlen (Helium-Kerne, Masse 4u (siehe 1.1.1 und Tab. 1), 2 positive Elementarladungen) und fand, dass nahezu alle α-Teilchen ungehindert die Metallfolie durchdringen. Nur ein sehr geringer Anteil wurde deutlich abgelenkt. Rutherford folgerte, dass α-Teilchen abgelenkt werden, wenn sie in die Nähe von positiv geladenen Teilchen gelangen, und schloss daher, dass die gesamte positive Ladung und fast die ganze Masse eines Gold-Atoms in einem winzigen Bereich im Zentrum des Atoms, dem Atomkern, vereinigt sein müssen. Die positive Ladung des Atomkerns wird durch die negativ geladenen Elektronen ausgeglichen, die sich in einem kugelförmigen Raum, der Elektronenhülle, die den Kern umgibt, bewegen. 1.1.1 Der Atomkern Der Kern ist im Vergleich zum gesamten Atom sehr klein, sein Durchmesser beträgt etwa ein Hunderttausendstel des Durchmessers des gesamten Atoms. Heutiger Wissensstand ist, dass der Kern eines Atoms aus positiv geladenen Protonen und ungeladenen Neutronen besteht. Die Kernbausteine Proton und Neutron bezeichnet man als Nukleonen (nucleus (lat.), Kern). Die Masse eines Protons und eines Neutrons beträgt jeweils ungefähr 1u (siehe Tab. 1). U ist die sog. 32 Balmer-Serie und Spektroskopie atomare Masseneinheit, die als ein Zwölftel der Masse des Kohlenstoff-Isotops (siehe Versuch 1 GMS): 1u = 1,6606⋅10 −27 12 C definiert ist kg. Atomkerne verschiedener Atomsorten (Elemente) unterscheiden sich in der Protonenzahl, die auch als Ordnungszahl bezeichnet wird. Betrachtet man die Elemente im Periodensystem, so nimmt die Protonenzahl von links nach rechts innerhalb der Perioden jeweils um einen Zähler zu. Der Kern des Wasserstoff-Atoms besteht nur aus einem Proton, der des Heliums aus 2 Protonen und 2 Neutronen. Isotope eines Elementes unterscheiden sich in der Neutronenzahl, was zu unterschiedlicher Nukleonenzahl und Masse des Atomkerns führt. Zur Kennzeichnung eines Atoms und seines Kerns wird die im Kasten unten erläuterte Schreibweise verwendet. Wegen der geringen Masse der Elektronen (siehe Tab. 1) ist die Masse des Atomkerns nahezu identisch mit der Masse des gesamten Atoms. Elementarteilchen Ladung (e) Masse (u) Proton +1 1,007276 Neutron 0 1,008665 Elektron -1 0,0005486 Tabelle 1.1: Ladung und Masse von Elementarteilchen (e = 1,6022 ⋅10−19 C) 1.1.2 Elektronenhülle und Atomspektren Im letzten Abschnitt standen der Atomkern und die ihn aufbauenden Komponenten im Vordergrund. Für den Chemiker ist jedoch in erster Linie die Elektronenhülle (und deren Aufbau) von Interesse, da ein Zusammenhang zwischen stofflichen Veränderungen bei chemischen Reaktionen und Veränderungen in der Elektronenhülle besteht. Da sich ein Elektron in der Hülle bewegt, besitzt es sowohl kinetische als auch potentielle Energie. Je weiter das Elektron im Mittel vom Kern entfernt ist, desto größer ist seine potentielle Energie. Erkenntnisse über den Aufbau der Elektronenhülle gehen auf Atomspektren zurück (siehe Kapitel 1.2.1), deren Besonderheiten uns noch beschäftigen werden. Zerlegt man weißes Sonnenlicht durch ein Prisma (ein Körper aus einem lichtbrechenden Stoff, der von zwei ebenen, nicht parallelen Flächen begrenzt wird), so erhält man ein kontinuierliches Spektrum. In einem Spektroskop zeigen sich solche Spektren als Farbbänder, bei denen die Farben kontinuierlich nacheinander in der Reihenfolge ihrer Wellenlänge λ erscheinen (z. B. Regenbogen). Neben der Wellenlänge λ, dies ist die Strecke zwischen Wellenberg und dem nächsten Wellenberg, wird eine Welle durch ihre Frequenz ν (Anzahl der Schwingungen pro Sekunde) charakterisiert. Es gilt der folgende Zusammenhang (c0 ist die Vakuumlichtgeschwindigkeit): ν= c0 λ (1) Kurzwelliges, ultraviolettes Licht ist besonders energiereich, rotes Licht ist energieärmer als blaues Licht. Die auf Max Planck zurückgehende Beziehung zwischen Energie E und Frequenz Balmer-Serie und Spektroskopie 33 ν bzw. Wellenlänge λ verdeutlicht das Gesagte (h ist das Planck’sche Wirkungsquantum): E =h⋅ν =h⋅ c0 λ mit h = 6, 6262 ⋅ 10−34 J ⋅ s (2) 1.2 Wasserstoffspektrum und das Bohr’sche Atommodell 1.2.1 Die Balmer-Serie Bereits 1859 zeigten Gustav Kirchhoff und Robert Bunsen mit Hilfe eines Prismenspektrographen, dass Atome nur Licht ganz bestimmter Wellenlängen λ absorbieren oder emittieren können. Diese diskontinuierlichen Absorptions- bzw. Emissionsspektren sind für jedes Atom charakteristisch und eindeutig. Johann Jakob Balmer gelang es 1885 mit Hilfe der Spektroskopie, auf rein empirischem Weg das Emissionsspektrum des Wasserstoffatoms durch eine einfache Formel zu beschreiben. Er stellte fest, dass das Spektrum aus einer Serie von Linien besteht, deren Frequenzen νn→m sich wie folgt quantitativ beschreiben lassen: νn→m = RH ⋅ c0 ⋅ ( 1 1 − 2) 2 m n (3) Für die Balmer-Serie gilt: m = 2 und n = 3, 4, 5, ... (Lyman und Paschen bestätigten die obige Formel, indem sie weitere Serien von Linien für m = 1 bzw. m = 3 fanden, die im UV(UltraViolett) bzw. im IR-(InfraRot) Bereich liegen). Die Konstante RH = 1, 0968 ⋅ 107 m−1 ist die sogenannte Rydberg-Konstante. Kennzeichnend für die Balmer-Serie ist, dass die Frequenzen für n = 3, 4, 5, 6 im sichtbaren Be- reich (zwischen λ = 400 − 800 nm) liegen und damit relativ einfach nachgewiesen werden können. Man bezeichnet diese Wellenlängen mit Hα , Hβ , Hγ und Hδ . Die nebenstehende Abbildung zeigt das kontinuierliche Spektrum weißen Lichts (a) sowie das Linienspektrum des atomaren Wasserstoffs im sichtbaren Bereich, die Balmer-Serie b). Zusätzlich sind die Elek- Abb. 1.1: Die Balmer-Serie tronenübergänge (Emissionen) der verschiedenen Serien des Wasserstoffs angegeben. 1.2.2 Das Bohr’sche Atommodell Nach dem Bohr’schen Atommodell, das Niels Bohr 1913 ausgehend vom Rutherford’schen Kern-Hülle-Modell entwickelte, besteht das Wasserstoffatom aus einem positiv geladenen Proton als Kern, um den sich das negativ geladene Elektron auf einer Kepler-Bahn bewegt. Nach der klassischen Elektrodynamik müsste ein solches Gebilde jedoch Energie abstrahlen (sog. oszillieren- 34 Balmer-Serie und Spektroskopie der Hertz’scher Dipol), d. h. das Elektron würde ständig Energie verlieren und schließlich in den Kern stürzen. Atome könnten somit überhaupt nicht existieren. Zusätzlich könnte ein derartiges Gebilde gemäß der klassischen Elektrodynamik nur eine monochromatische Frequenz abstrahlen, nie jedoch mehrere unterschiedliche, diskrete Frequenzen. Auf der Basis der Experimente, die im letzten Kapitel angesprochen wurden, und der Arbeiten Plancks zur Quantelung der Energie stellte Bohr folgende Postulate auf, die mit der klassischen Elektrodynamik jedoch unvereinbar sind und die er auch nicht weiter begründete: 1. Für jedes Atom gibt es eine Anzahl stationärer Zustände En , in denen das Atom nicht strahlt. 2. Atome können Energie dann aufnehmen (Absorption) oder abgeben (Emission), wenn das betreffende Atom von einem Zustand in einen anderen übergeht. 3. Ist dieser Übergang mit Absorption oder Emission von Strahlung verbunden, dann gilt für die Frequenz der betreffenden Strahlung: νnm = 1 (Em − En ). h mit n, m = 1, 2, 3, ... (4) d. h. das Atom absorbiert oder emittiert beim Übergang vom Energiezustand Em nach En ein Lichtquant der entsprechenden Energie. 4. Der Bahndrehimpuls der stationären Elektronenbahnen ∣⃗l∣ darf nur ganze Vielfache von ̵ = h von annehmen (er ist somit gequantelt): h 2π ̵ = n ⋅ h. ∣⃗l∣ = n ⋅ h 2⋅π mit n = 1, 2, 3, ... (5) Aus den Bohr’schen Postulaten lassen sich mit Hilfe der klassischen Mechanik die Resonanzfrequenzen des Wasserstoffatoms berechnen. Das soll im Folgenden abgeleitet werden: Das Elektron der Ladung e und der Masse me bewegt sich mit der Winkelgeschwindigkeit ω auf einer Kreisbahn mit Radius r um den Atomkern; Zentrifugalkraft (sie ist der Zentripetalkraft entgegengesetzt, wirkt also nach außen) und Coulomb-Kraft sind im Gleichgewicht: e2 = me ⋅ r ⋅ ω 2 4 ⋅ π ⋅ 0 ⋅ r2 bzw. r3 = e2 4 ⋅ π ⋅ 0 ⋅ me ⋅ ω 2 (6) 0 ist die elektrische Feldkonstante (0 = 8, 8542 ⋅ 10−12 A ⋅ s ⋅ V −1 ⋅ m−1 ), me die Elektronenmasse (me = 9, 1095 ⋅ 10−31 kg) und ω die Winkelgeschwindigkeit. Weiter ergibt sich: ̵ = n⋅h ∣⃗l∣ = me ⋅ r2 ⋅ ω = n ⋅ h 2⋅π (7) Daraus ergibt sich, dass der Bahnradius des Elektrons nur diskrete Werte annehmen kann. Einsetzen von ω aus Gleichung 7 in Gleichung 6 liefert: rn = n2 ⋅ 0 ⋅ h2 π ⋅ me ⋅ e2 (8) Balmer-Serie und Spektroskopie 35 Die Zahl n ist die sog. Hauptquantenzahl, und es gilt n ≥ 1. Der kleinste Radius, r1 wird als Bohr-Radius bezeichnet. Die Energie des n-ten Zustands ergibt sich als Summe der potentiellen und kinetischen Energie: En = Epot + Ekin = − 1 me ⋅ e4 e2 + ⋅ me ⋅ rn2 ⋅ ω 2 = − 4 ⋅ π ⋅ 0 ⋅ rn 2 8 ⋅ 20 ⋅ h2 ⋅ n2 (9) Die Energie ist somit diskret, d. h. gequantelt, und in diesem Fall allein von der Hauptquantenzahl n bestimmt. Für die Frequenz eines abgestrahlten Lichtquants bei Änderung des Energiezustands gilt also: νnm = 1 me ⋅ e4 1 ⋅( 2 − 2) 2 3 m n 8 ⋅ 0 ⋅ h Das entspricht der empirischen Formel nach Balmer, der Vorfaktor (10) me ⋅e4 8⋅20 ⋅h3 stimmt jedoch nur ungefähr mit der Rydberg-Konstante RH überein. Exakte Übereinstimmung erhält man erst, wenn die Bewegung von Atomkern und Elektron um den gemeinsamen Schwerpunkt berücksichtigt wird. Der resultierende Korrekturfaktor 1 + me mp (mp ist die Masse des Protons) ergibt schließlich den richtigen Wert für RH . Nach dem Bohr’schen Atommodell entspricht die Balmer-Serie dem Übergang vom dritten, vierten, fünften, ... Energieniveau auf das zweite Energieniveau (siehe Abbildung 1.1). Zwar liefert das Bohr’sche Atommodell ein Ergebnis, das mit dem Experiment übereinstimmt, jedoch gelang dies nur unter Verletzung elementarer Gesetze der klassischen Physik. Außerdem funktioniert das Modell nach Bohr nur für das Wasserstoffatom, nicht jedoch bei Mehrelektronenatomen. Das Modell kann auch nicht erklären, warum Atome überhaupt stabil sind, denn ein sich auf einer Kreisbahn um den Kern bewegendes Elektron stellt eine beschleunigte Ladung dar, die nach den Gesetzen der Elektrodynamik Strahlung abgeben müsste und damit kontinuierlich Energie verlöre. Eine Abschätzung zeigt, dass ein Atom nur etwa 10−8 Sekunden existieren könnte. 1.2.3 Spektroskopie Elemente mit einer niedrigen 1. Ionisierungsenergie lassen sich durch die Flamme des Bunsenbrenners elektronisch anregen, d.h. ein Elektron der äußeren Schale wird durch Energiezufuhr in eine höhere Schale (= auf ein höheres energetisches Niveau) angehoben. Beim Zurückkehren aus diesem angeregten Zustand in den Grundzustand (= beim ”Zurückfallen” des Elektrons in seine ursprüngliche Schale) wird diese Energie in Form von Licht wieder abgegeben (= emittiert). Man beobachtet also eine charakteristische Flammenfärbung. In einem Spektroskop sind dabei die für dieses Element typischen Spektrallinien (= Energiestufen) zu sehen, das sog. charakteristische Spektrum. Hierbei handelt es sich um ein Emissionsspektrum (= Licht wird beim ”Zurückfallen” der Elektronen auf ihr Grundzustandsniveau ausgesendet) im Gegensatz zur Farbe von Komplexen (siehe Versuche 4 PSE und 12 KOG). Dort wird aus weißem Licht die Wellenlänge ”herausgefiltert”, die zur Anregung der Elektronen notwendig ist. Man sieht also die entsprechende Komplementärfarbe (siehe Versuch 2 ABS). Im analytischen Praktikum ist die Flammenfärbung v. a. wichtig um die Elemente Lithium, Natrium, Kalium, Calcium, Strontium und Barium in 36 Balmer-Serie und Spektroskopie Verbindungen nachzuweisen. Dabei wird in der Flamme des Bunsenbrenners das vorliegende Metallkation M n+ zuerst zum elementaren (und bei dieser Temperatur auch gasförmigen) Metall reduziert, welches dann das charakteristische Spektrum aussenden kann. 1.3 Der Beginn der Quantenmechanik Bis etwa 1900 glaubte man, dass die klassische Physik die Natur vollständig und in sich konsistent beschreiben könne. Es wurde zwischen Strahlung und Materie unterschieden. Materie bestand aus genau lokalisierbaren Teilchen, deren Bewegungen durch die Newton’sche Mechanik exakt beschrieben und vorhergesagt werden konnten, während Strahlung durch die elektromagnetische Theorie von Maxwell abgedeckt war, die Erkenntnisse von Coulomb, Ampère, Gauss, Volta und Faraday in sich vereinigte. Mit Hilfe dieser Theorie sagte Maxwell die Existenz elektromagnetischer Wellen voraus, die 1887 durch die Entdeckung der Radiowellen bestätigt wurde. Strahlung kann nach dem klassischen Bild der Physik nicht lokalisiert werden, sondern besitzt Wellencharakter und wird durch die Komponenten des elektrischen und magnetischen Feldes an jedem Ort des Raumes charakterisiert. Die Wellennatur des Lichts war durch die Fresnel’schen Interferenz-Experimente und die Young’schen Beugungsversuche bewiesen. Es gab zwar ungeklärte Phänomene (wie z. B. die Strahlung des Schwarzen Körpers), jedoch reichten diese nicht aus, das Formelgebäude der klassischen Physik und den sich daraus ableitbaren Determinismus in Frage zu stellen. Vielmehr meinte man, durch eine Verfeinerung der mathematischen Methoden die bestehenden Probleme lösen zu können. Das Zeitalter der Quantenmechanik beginnt im Jahre 1900. Max Planck stellt die fundamental neue Hypothese auf, dass Oszillatoren eines ”schwarzen Körpers” nur diskrete Energieniveaus annehmen und Licht nur in Energieeinheiten der Größe E = h ⋅ ν emittieren können (Strahlung des Schwarzen Körpers). ν ist die Oszillatorfrequenz, und h stellt das Planck’sche Wirkungsquantum, eine neue Fundamentalkonstante der Physik, dar. Damit war das Frequenzspektrum der Strahlung heißer Körper durch die Annahme einer sog. Energiequantelung erklärt. Einen weiteren Hinweis auf die Quantisierung der Energie ergab der photoelektrische Effekt, der 1905 von Albert Einsten erklärt wurde. Einstein fand, dass die maximale kinetische Energie eines Elektrons, das durch ein Photon der Energie h ⋅ ν aus einer Metalloberfläche herausgeschlagen wird, durch folgende Beziehung beschrieben wird: Tmax = h ⋅ ν − φ(M ) (11) Die Größe φ(M ) ist die Austrittsarbeit, die für das entsprechende Metall M charakteristisch ist und durch die Tiefe des Potentialtopfs U0 sowie die Fermi-Energie EF bestimmt ist: φ(M ) = U0 − EF (12) Ein direkter Beweis, dass sich ein Lichtquant (Photon) unter Umständen wie ein Teilchen verhält, wurde 1922 experimentell von Compton gefunden. Photonen haben, wie aus der Relativitätstheorie von Einstein abgeleitet werden kann, einen Impuls der Größe p = hν . Stößt ein Photon mit einem freien Elektron zusammen, so überträgt es einen Teil seiner Energie und damit seines Impulses auf das Elektron. Die Wellenlänge des Photons nach dem Stoß wird mit λ′ Balmer-Serie und Spektroskopie 37 bezeichnet. Mit Hilfe des Energie- und Impulserhaltungssatzes kann dann folgende Gleichung für den Compton-Effekt abgeleitet werden (θ ist der Streuwinkel, um den das Photon aus seiner ursprünglichen Bahn abgelenkt wird): λ′ − λ = h ⋅ (1 − cos θ) me ⋅ c0 (13) Der Nachweis des Compton-Effekts sowie des photoelektrischen Effekts zeigen, dass Licht aus Photonen besteht, die sich wie Teilchen mit genau definierter (gequantelter) Energie verhalten. Licht, also Strahlung, ist damit auch Materie und besitzt korpuskularen Charakter. Wesentlich für die moderne Physik ist aber die Wellennatur der Materie, die erst im Jahre 1927 experimentell von Davisson und Germer durch Elektronenbeugung an Einkristallen entdeckt wurde. Drei Jahre zuvor hatte de Broglie bereits postuliert, dass jedem bewegten Teilchen mit Impuls p eine Welle zugeordnet werden kann, welche die Wellenlänge λ = h p besitzt. Doppelspaltexperimente mit Elektronen zeigen das verblüffende Resultat eines klassischen Interferenzmusters mit Minima und Maxima der Elektronenverteilung auf dem Beobachtungsschirm. Die Wellenlänge eines Strahls von Elektronen mit dem Impuls p kann damit aus dem Interferenzmuster am Doppelspalt bestimmt werden. Die Entdeckung der Elektronen als Teilchen in Kathodenstrahlen gelang J. J. Thomson bereits 1897. Damit war der Welle-Teilchen-Dualismus der Materie formuliert, der schließlich zu den Postulaten der Quantenmechanik und der mathematischen Formulierung der Quantentheorie durch Heisenberg und Schrödinger führte. Der Welle-Teilchen-Dualismus führt bereits qualitativ zur Heisenberg’schen Unschärferelation. Man fragt sich, ob der Ort eines Teilchens, das Wellencharakter besitzt, exakt bestimmt werden kann. Ein lokalisiertes Wellenpaket (Einhüllende) kann durch Überlagerung sehr vieler Wellen mit verschiedenen Wellenlängen erhalten werden. Fast überall löschen sich die Wellen durch Interferenz aus, nur in einem bestimmten Bereich, der sich mit der Zeit bewegt, erfolgt Verstärkung. Wenn nun jeder Wellenlänge gemäß der de BroglieBeziehung ein Impuls p = h λ zugeordnet werden kann, bedeutet dies, dass eine Impulsverteilung vorliegt, die desto breiter ist, je genauer der Ort des Wellenpakets bestimmt werden soll. Allgemein konnte Werner Heisenberg zeigen, dass bestimmte Paare von Observablen (Messgrößen wie z. B. Ort und Impuls) nicht gleichzeitig exakt bestimmt werden können. Für den Impuls und den Ort eines Teilchens kann die Heisenberg’sche Unschärferelation also wie folgt formuliert werden: Es ist unmöglich, den Ort und den Impuls eines Teilchens gleichzeitig exakt zu bestimmen bzw. zu kennen. Diese Aussage wird durch die folgende Formel ausgedrückt: ∆x ⋅ ∆p ≥= ̵ h h = 2 4⋅π mit ∆x = Ortsunschärfe und ∆p = Impulsunschärfe (14) Es ist wichtig zu begreifen, dass die Unschärferelation nicht eine Unzulänglichkeit der Messung oder der Messapparatur widerspiegelt, sondern eine grundlegende Eigenschaft der Materie beschreibt, die aus den Postulaten der Quantenmechanik resultiert. Sie widerspricht dem klassischen Kausalitätsprinzip und bedeutet eine Zäsur im Weltbild der Physik. 38 Balmer-Serie und Spektroskopie 1.4 Heisenberg’sche Unschärferelation und Schrödinger-Gleichung Wir wollen noch einmal die Unschärferelation aufgreifen und eine Art Gedankenexperiment machen. Für ein ruhendes Teilchen gilt ∆p = 0, d. h. seine Impulsunschärfe ist null. Nun wollen wir mit Hilfe eines Mikroskops den Ort des Teilchens bestimmen, was bestenfalls mit einer Genauigkeit geschehen kann, die von der Größenordnung der Wellenlänge des benutzten Lichts ist. Für die Ortsunschärfe gilt also ∆x ≈ λ. Da aber ∆p = 0 ist, wäre das Produkt ∆x ⋅ ∆p gleich Null, und es läge eine Verletzung der Unschärferelation vor! Ist das tatsächlich der Fall, oder haben wir einen Fehler gemacht? Zur Ortsbestimmung müssen wir Licht benutzen, und die Quantenmechanik zeigt, dass Licht gequantelt ist und aus Photonen mit dem Impuls p = h λ besteht. Um das Teilchen nachweisen zu können, muss mindestens eines der Photonen aus dem konvergierenden Lichtstrahl hinter einer Sammellinse an dem Teilchen entweder gestreut oder von ihm absorbiert werden. Der Impulsübertrag an das Teilchen muss also mindestens Das Teilchen hat dann eine Impulsunschärfe ∆p ≥ h λ h λ betragen. zu dem Zeitpunkt, an dem sein Ort mit einer Unschärfe ∆x ≈ λ bestimmt worden ist. Die Multiplikation der beiden Unschärfen ergibt ∆x ⋅ ∆p ≥ λ ⋅ h =h λ Das stimmt mit der obigen Gleichung (bis auf einen Faktor (15) 1 4π ) überein. An diesem Beispiel sieht man, dass die Quantenmechanik in sich widerspruchsfrei ist. Wie verträgt sich nun das Bohr’sche Atommodell mit der Heisenberg’schen Unschärferelation? Wir rekapitulieren und erkennen, dass das Modell nach Bohr präzise, gleichzeitige Aussagen über Bahn bzw. Ort und Impuls (Geschwindigkeit) eines Elektrons erlaubt (mit anderen Worten: Die Bewegung des Elektrons ist durch eine klassische Bahnkurve beschrieben) und damit falsch sein muss, da es der Heisenberg’schen Unschärferelation widerspricht! Das Dilemma besteht also darin, dass das Bohr’sche Atommodell zwar das Wasserstoffspektrum richtig beschreibt, jedoch weder mit der klassischen Elektrodynamik noch der Quantenmechanik in Einklang zu bringen ist. Wir benötigen also ein widerspruchsfreies Atommodell! Die Tatsache, dass kleinste Teilchen immer auch Welleneigenschaften zeigen, legt die Möglichkeit nahe, ihr Verhalten mit Gleichungen zu beschreiben, die auch zur Darstellung anderer Arten von Wellen verwendet werden. Man geht von einer Wellenfunktion Φ aus, die von den Raumkoordinaten (x, y, z) und der Zeit t abhängt. Bei der Beschreibung stationärer Vorgänge (z. B. Elektronen im ungestörten Atom) ist der Zustand unabhängig von der Zeit, die Zeitabhängigkeit von Ψ braucht also nicht berücksichtigt werden. Bei einer Welle Ψ, die sich mit der Geschwindigkeit v dreidimensional im leeren Raum ausbreitet, lässt sich die Änderung von Ψ als Funktion von (x, y, z) und t durch eine Differentialgleichung zweiter Ordnung beschreiben. Die zeitunabhängige Amplitude der Welle wird durch die sog. Amplitudenfunktion ψ dargestellt: ( ∂2 ∂2 ∂2 4 ⋅ π2 ) + + ⋅ ψ = − ⋅ψ ∂x2 ∂y 2 ∂z 2 λ2 (16) Diese Gleichung gilt für beliebige dreidimensionale Wellen. Will man nun eine für kleinste Teilchen, wie z. B. Elektronen, gültige Wellengleichung erhalten, ersetzt man die Wellenlänge λ durch die de Broglie-Beziehung λ = h m⋅v : Balmer-Serie und Spektroskopie ( 39 ∂2 ∂2 ∂2 4 ⋅ π 2 ⋅ m2 ⋅ v 2 ) + + ⋅ ψ + ⋅ψ =0 ∂x2 ∂y 2 ∂z 2 h2 (17) Die kinetische Energie der Partikel Ekin ist gleich der Differenz zwischen Gesamtenergie E und der potentiellen Energie V : Ekin = 1 ⋅ m ⋅ v2 = E − V 2 (18) Einsetzen des Ausdruckes für die kinetische Energie in Gleichung 17 ergibt schließlich die von Schrödinger 1926 eingeführte Schrödinger-Gleichung: ( ∂2 ∂2 ∂2 8 ⋅ π2 ⋅ m ) + + ⋅ ψ + ⋅ (E − V ) ⋅ ψ = 0 ∂x2 ∂y 2 ∂z 2 h2 (19) Die Gleichung beschreibt das Verhalten von Mikropartikeln, insbesondere von Elektronen in Atomen und Molekülen vollständig. Die Schrödinger-Gleichung verbindet die Amplitudenfunktion ψ des Elektrons bzw. eines Teilchens mit seiner Energie und den Raumkoordinaten, die zur Beschreibung des Systems notwendig sind. ψ selbst besitzt keine anschauliche Bedeutung und ist nicht beobachtbar (keine Observable). Im Gegensatz dazu bildet der Ausdruck ∣ψ 2 ∣ dx dy dz ein Maß für die Wahrscheinlichkeit, das betreffende Elektron in einem Volumenelement dx dy dz anzutreffen (Bornsche Interpretation der Wellenfunktion Ψ). ∣ψ 2 ∣ gibt die Wahrscheinlichkeitsdichte an, d. h. den zeitlichen Durchschnitt der Ladungsverteilung, wie sie aus der Bewegung des Elektrons resultiert. Die Beschreibung des Verhaltens eines Teilchens mittels einer Wellengleichung darf nicht zu der falschen Vorstellung verleiten, das betreffende Teilchen ”sei eine Welle” oder bewege sich wellenförmig; vielmehr eröffnet ψ die Möglichkeit, die Aufenthaltswahrscheinlichkeit eines Teilchens berechnen zu können. Nach der Unschärferelation lassen sich nur Aussagen über den mehr oder weniger ”wahrscheinlichen” Ort eines Teilchens, jedoch nicht über die Art seiner Bewegung machen. Die Schrödinger-Gleichung lässt sich nicht streng deduktiv ableiten, sie ist nicht ”begründbar”, sondern eher die Folge der Anwendung der de Broglie-Beziehung auf ein sich bewegendes Teilchen. Die Richtigkeit der Gleichung wird quasi durch die Übereinstimmung theoretischer Ergebnisse mit experimentellen Beobachtungen untermauert. Im Prinzip sind unendlich viele Amplitudenfunktionen ψ möglich, die der besagten Schrödinger-Gleichung gehorchen. Da wir dem Quadrat von ψ aber eine bestimmte Bedeutung beimessen, sind nur diejenigen ψ-Funktionen physikalisch sinnvoll, die gewisse Bedingungen erfüllen. So muss ψ eine stetige Funktion sein und überall einen einzigen, endlichen Wert besitzen. Wäre ψ an irgendeinem Punkt ”unendlich”, so wäre die Wahrscheinlichkeit das Elektron dort anzutreffen unendlich groß, was der Unschärferelation widerspräche. Rechnungen zeigen, dass die Gesamtenergie E eines gebundenen Elektrons nur ganz bestimmte Werte annehmen kann, welche durch entsprechende ψ-Funktionen (sog. Eigenfunktionen) festgelegt sind. Die Quantelung der Energiezustände, also die Existenz bestimmter, ausgewählter Energiezustände, ergibt sich damit als mathematische Notwendigkeit und muss nicht - wie beim Bohrschen Atommodell - ad hoc eingeführt werden. In gleicher Weise erzwingen Randbedingungen eines schwingenden Systems Schwingungen mit 40 Balmer-Serie und Spektroskopie ganz bestimmten Frequenzen, man vergleiche mit der Länge einer Saite eines Musikinstruments. Anders ausgedrückt heißt das, dass zur Beschreibung stationärer Teilchen nur stehende Wellen erlaubt sind. Zuletzt bleibt zu erwähnen, dass die konsequente Anwendung der Schrödinger-Gleichung auf das Wasserstoffatom zu den gleichen, von der Hauptquantenzahl n abhängigen Energiewerten En führt, wie sie bereits aus der Bohr’schen Theorie bekannt sind. Die Schrödinger-Gleichung eröffnet damit den Zugang zu einem widerspruchsfreien Atommodell, das jedoch gegenüber dem Modell nach Bohr nur wenig anschaulich ist. 2 Aufgabenstellung, Versuchsanleitung und -auswertung 2.1 Balmer-Serie 2.1.1 Aufgabenstellung Bestimmung und Berechnung der Wellenlängen der Hα , Hβ und Hγ -Linien der Balmer-Serie 2.1.2 Versuchsanleitung Die Balmer-Lampe (siehe Abbildung 2.1) ist eine wechselstrombetriebene Gasentladungsröhre mit Wasserdampffüllung. Die abgeschmolzene Röhre wird durch einen an hygroskopischer Grundlage gebundenen Wasservorrat mit Wasserdampf versorgt. Die Wassermoleküle werden durch die elektrische Entladung in atomaren Wasserstoff und Hydroxylradikale aufgespalten. Eine hochtemperaturbeständige Kapillare im Innern der Lampe zwingt die Entladung auf einen engen Raum, so dass dort eine hohe Konzentration an atomarem Wasserstoff entsteht, der für die intensiven BalmerSpektrallinien verantwortlich ist; störende BanAbb. 2.1: Schematischer Aufbau der Balmer- den verursacht durch molekularen Wasserstoff Lampe H treten nicht auf. Eine oxidierende Substanz, 2 unterstützt von geeigneten Katalysatoren, sorgt dafür, dass während des Betriebs gebildeter Wasserstoff zu Wasser oxidiert wird, so dass im Innern der Röhre ein Wasserkreislauf stattfindet. Während des Kreislaufs kommt es zur Ablagerung rot-brauner Metalloxide, die sich jedoch nicht im kapillaren Teil der Röhre störend bemerkbar macht. Balmer-Serie und Spektroskopie 41 Sicherheitshinweise zur Balmer-Lampe: ● Manipulationen an der Balmer-Lampe ohne ausdrückliche Anweisung durch den Versuchsbetreuer sind verboten! ● Die heiße Balmer-Lampe darf unter keinen Umständen angefasst werden! Inbetriebnahme der Balmer-Lampe und Betriebshinweise ● Schalter am Hochspannungs-Netzgerät auf ”Ein” schalten. Die Balmer-Lampe sollte sofort zünden. ● Für ein stabiles Brennen der Balmer-Lampe ist eine bestimmte Betriebstemperatur (ca. 55°C, gemessen am Aluminiumsockel) erforderlich. Bei zu niedriger Betriebstemperatur können Entladungen außerhalb der Kapillare (siehe Abbildung 2.1) auftreten, was zu einem flackernden Betrieb der Lampe führt. Sorgen Sie für ein stabiles Brennen (stabile Entladungsbedingungen), indem Sie die Balmer-Lampe 10-15 Minuten vor dem Experiment einschalten! ● Kommt es infolge hoher Umgebungstemperaturen zu hohen Betriebstemperaturen (> 70°C), so kann die Balmer-Lampe (nach längerer Betriebsdauer) unter Umständen erlöschen. Schalten Sie die Lampe aus und warten Sie, bis sie sich abgekühlt hat. Danach sollte die ● Balmer-Lampe wieder normal weiterzubetreiben sein. Betreiben Sie die Balmer-Lampe nur in vertikaler Lage, mit dem Aluminium-Sockel nach unten. ● Beim Betrieb ist gelegentlich im Mittelteil der Kapillare die gelbe Natrium-Linie zu beobachten. Hier empfiehlt es sich, die oberen bzw. unteren Randzonen der Kapillare, in denen die störende gelbe Linie nicht auftritt, für das weitere Experimentieren zu verwenden. 2.1.3 Versuchsaufbau und Durchführung Bauen Sie die in Abbildung 2.2 dargestellte Anordnung zunächst ohne Rowland-Gitter (dispersives Element) auf. Dunkeln Sie den Raum zum Beobachten des Wasserstoffspektrums ab. Abb. 2.2: Versuchsaufbau zur Balmer-Serie 42 Balmer-Serie und Spektroskopie Bilden Sie die Balmer-Lampe durch Verschieben der Sammellinse f = 50 mm (f ist die Brennweite) scharf auf die Mitte des zunächst noch geschlossenen Spaltes ab. Öffnen Sie den Spalt und bilden Sie ihn mittels der Sammellinse f = 100 mm scharf auf den transparenten Schirm ab. In einem völlig verdunkelten Raum sollte die mattierte Seite des Schirms der Lichtquelle zugewandt sein. Bauen Sie dann das Gitter im Halter in die auf der Vorseite abgebildete Anordnung ein. Der Spalt sollte so weit geöffnet werden, dass ein Kompromiss zwischen Helligkeit und Auflösung zustande kommt. Betrachtet man nun die Balmer-Serie auf der der Strahlungsquelle abgewandten Seite des Schirms, so sollten symmetrisch um das Zentrum (sog. Weißlicht-Position, also das Beugungsmaximum 0. Ordnung) in abnehmendem Abstand zur zentralen Linie rote, türkisfarbene und blaue Hα , Hβ und Hγ -Linien zu beobachten sein. 2.1.4 Versuchsauswertung und weitere Aufgaben 1. Aufbau des Versuchs ”Balmer-Serie des Wasserstoffs”. 2. Bestimmung der Wellenlängen der Hα , Hβ und Hγ -Linien. 3. Berechnung der Wellenlängen der Hα , Hβ und Hγ -Linien mit Hilfe der BalmerSerienformel und Vergleich mit theoretisch berechneten Werten. Abb. 2.3: Liniengeometrie der BalmerSerie auf dem Schirm, mit der φ bestimmt werden kannt. Das Zentrum ist das Beugungsmaximum 0. Ordnung die Hα , Hβ und Hγ -Linien sind Beugungsmaxima 1. Ordnung. Abbildung 2.3 zeigt, welche geometrischen Größen für die Auswertung wichtig sind. L ist der (senkrechte) Abstand des Rowland-Gitters vom Schirm, x der jeweilige Abstand der Linien der Balmer-Serie zum Zentrum. Die Kopie des Rowland-Gitters wirkt wie sehr viele, äquidistante Spalte und bildet ein Beugungsgitter. Zylinderwellen, die aus den einzelnen Spalten hervorquellen, verschmelzen in einigem Abstand zu einer Wellenfront, die sich in der ursprünglichen Ausbreitungsrichtung weiterbewegt (Beugungsmaximum 0. Ordnung). Auch in anderen Richtungen laufen Wellenfronten, nämlich in solchen Richtungen, aus denen gesehen jeder Spalt um λ, 2λ, 3λ,... weiter entfernt ist als sein Nachbar. Für die Winkel φm der Beugungsmaxima m-ter Ordnung Balmer-Serie und Spektroskopie 43 (siehe Abbildung 2.3) gilt: sin φm = m⋅λ 1 mit λ = Wellenlänge und d = Gitterkonstante = mm d 600 (20) Abbildung 2.3 zeigt, wie der Beugungswinkel φ aus x und L ermittelt werden kann: tan φ = x x und damit φ = tan−1 ( ) L L (21) Damit können jetzt die Wellenlängen der Hα , Hβ und Hγ -Linien für m = 1 wie folgt berechnet werden: x λ = d ⋅ sin(tan−1 ( )) L (22) Darüber hinaus müssen folgende Aufgaben bearbeitet werden: ● ● ● Berechnen Sie der Ionisationsenergie des Wasserstoffatoms. Berechnen Sie den Bohr’schen Radius. Diskutieren Sie das Bohr’sche Atommodell aus der Sicht der modernen Physik bzw. der Quantenmechanik. Nach dem Bohr’schen Atommodell bewegt sich das Elektron der Ladung e auf einer Kreisbahn mit Radius r um den Atomkern, die Bewegung lässt sich also durch eine Bahnkurve beschreiben. Wie verträgt sich diese Aussage mit der Quantenmechanik? 2.2 Flammenfärbung und Emissionsspektren ausgewählter Elemente 2.2.1 Aufgabenstellung Bei diesem Versuch sollen die charakteristischen Flammenfärbungen und Emissionspektren ausgewählter Metallsalze beobachtet und dokumentiert werden. 2.2.2 Versuchsanleitung In acht Zerstäuberfläschchen befinden sich stark verdünnte wässrige Lösungen der folgenden Salze LiCl, NaCl, KCl, RbCl, CsCl, CaCl2 , SrCl2 und BaCl2 . In der neunten Zerstäuberflasche wird eine salzsaure Lösung von CuSO4 frisch aus einer Spatelspitze CuSO4 , wenig HClaq und demin. Wasser frisch zubereitet und nach Ende des Versuchs fachgerecht entsorgt. Diese Lösungen werden nacheinander in die Flamme des Bunsenbrenners gesprüht und die beobachteten Flammenfärbungen werden dokumentiert (hierbei wird in jedem Fall zu zweit gearbeitet: Eine(r) sprüht, die/der Andere beobachtet). In einer zweiten Runde schaut die/der Beobachtende durch das Spektroskop, prägt sich die beobachteten Linienspektren ein und dokumentiert diese so exakt wie möglich. Anhand des Spektrallinien-Posters im Spektroskopie-Raum kann mit Literaturangaben verglichen werden. Nach Beendigung dieser Versuche ist eine gründliche Reinigung der verwendeten Abzüge und Bunsenbrenner unabdingbar! Sollten Lösungen ausgehen, sind diese nach Absprache mit dem Assistenten wieder frisch aus den entsprechenden Metallsalzen und demineralisiertem Wasser zuzubereiten. 44 Balmer-Serie und Spektroskopie 2.2.3 Versuchsauswertung und weitere Aufgaben ● Die Flammenfarben und Emissionsspektren der zehn Metallsalzlösungen werden dokumentiert. 3 Geräte und Chemikalien Raum der Balmer-Serie: Balmer-Apparatur (keine Chemikalien) Spektroskopieraum: In zwei Abzügen je eine Metallgestell mit einem Bunsenbrenner und einem Handspektrokop, 2 × 9 Zerstäuberfläschchen mit den oben genannten Lösungen, 2 Spritzflaschen mit demineralisiertem Wasser Chemikalien LiCl NaCl KCl RbCl CsCl CaCl2 SrCl2 BaCl2 CuSO4 ⋅ 5 H2 O HClaq Gefahrenbezeichnung Achtung (GHS07) – – – – Achtung (GHS07) Achtung (GHS07) Gefahr (GHS06) Achtung (GHS07, 09) Gefahr (GHS05) H-Sätze 302, 315, 319 – – – – 319 302 332, 301 302, 315, 319, 410 314, 335, 290 P-Sätze 302+352, 305+351+338 – – – – 305+351+338 – 301+310 305+351+338, 302+352, 273 301+330+331, 305+351+338, 280 4 Literatur [1] [2] [3] G. Wedler: Lehrbuch der Physikalischen Chemie, 5. Aufl., Wiley-VCH, 2004. P. W. Atkins, J. de Paula: Physikalische Chemie, 4. Aufl., Wiley-VCH, 2008. A. F. Holleman, E. Wiberg: Lehrbuch der Anorganischen Chemie, 102. Aufl., Walter de Gruyter Verlag, 2007. [4] M. Binnewies, M. Jäckel, H. Willner, G. Rayner-Canham: Allgemeine und Anorganische Chemie, 1. Auflage, Spektrum Verlag, 2004. Periodisches System der Elemente 45 Versuch 4 (PSE) Periodisches System der Elemente 1 Theorie 1.1 Aufbau der Elektronenhülle – Aufbau des Periodensystems Das Periodensystem der Elemente wurde nach Vorarbeiten von Döbereiner erstmalig im Jahr 1869 von Lothar Meyer und Dimitrij Mendelejew vorgestellt. Die Elemente wurden seinerzeit nach steigenden Atommassen geordnet (an einigen Stellen stellte sich dies jedoch als falsch heraus). Aufgrund ähnlicher Eigenschaften, die ”periodisch” auftraten, wurden sie in Gruppen zusammengefasst. Es enthielt zwar noch einige Lücken, dadurch dass manche Elemente noch nicht entdeckt waren, allerdings war man sich darüber bewusst, dass an diesen Stellen noch Elemente fehlten. So bezeichnete man z. B. das Germanium bis zu seiner Entdeckung durch Clemens Winkler im Jahr 1886 als Eka-Silicium. 1.2 Die Perioden 1. Periode K-Schale 2 Elektronen Die Perioden sind die Reihen des Periodensys- 2. Periode L-Schale 8 Elektronen tems. Analog zu dem Fortschreiten der Reihen von 3. Periode M-Schale 8 Elektronen oben nach unten (1. Periode, 2. Periode, etc.) werden 4. Periode N-Schale 18 Elektronen die Elektronen der jeweiligen Elemente in Schalen ge- 5. Periode O-Schale 18 Elektronen füllt (K-Schale, L-Schale, etc.). Innerhalb der Periode 6. Periode P-Schale 32 Elektronen wächst die Zahl der Valenzelektronen der Atome von 7. Periode Q-Schale 32 Elektronen links nach rechts jeweils um eins, bis die betreffende Schale gefüllt ist (s. Tabelle 1.1). Das Volumen der Tabelle 1.1: Die Perioden Atome, die in einer Periode stehen, wird von links nach rechts kleiner, da der Bereich der Aufenthaltswahrscheinlichkeit der Elektronen räumlich derselbe bleibt, die Ladung des Kerns und damit sein Potential (bzw. die von ihm ausgehende Punktladungsdichte) jedoch größer wird. Damit ”zieht” der Kern die Valenzelektronen etwas mehr zu sich ”heran”, und das Volumen des Atoms wird dadurch kleiner. Beim Durchgang durch die Perioden von oben nach unten kommt immer wieder eine neue Schale hinzu und bringt damit eine sprunghafte Erhöhung des Atomvolumens mit sich. 1.3 Die Gruppen Elemente ähnlicher Eigenschaften werden in den Gruppen zusammengefasst. Es gibt 18 Gruppen, die die Spalten des Periodensystems darstellen. Sie werden nach IUPAC-Nomenklatur durchnumeriert, nach älterer, aber noch oft gebräuchlicher Nomenklatur in Haupt- und Nebengruppen unterteilt (s. Tabelle 1.2). Alle Elemente einer Gruppe besitzen dieselbe Zahl an Valenzelektronen und damit auch dieselbe maximale bzw. minimal mögliche Oxidationsstufe (auch wenn diese manchmal selten oder gar nicht realisiert wird, Sauerstoff tritt beispielsweise niemals in der Oxidationsstufe +6 auf). 46 Periodisches System der Elemente Oktett-Regel: Alle Elemente sind bestrebt durch Aufnahme, Abgabe oder das Teilen von Elektronen, die Elektronenkonfiguration des nächsten oder vorhergehenden Edelgases zu erhalten. IUPAC-Nomenklatur Haupt- und Nebengruppen Trivialnamen 1. Gruppe 2. Gruppe 3. Gruppe 4. Gruppe 5. Gruppe 6. Gruppe 7. Gruppe 8. Gruppe 9. Gruppe 10. Gruppe 11. Gruppe 12. Gruppe 13. Gruppe 14. Gruppe 15. Gruppe 16. Gruppe 17. Gruppe 18. Gruppe I. Hauptgruppe, Ia II. Hauptgruppe, IIa III. Nebengruppe, IIIb IV. Nebengruppe, IVb V. Nebengruppe, Vb VI. Nebengruppe, VIb VII. Nebengruppe, VIIb Alkalimetalle Erdalkalimetalle Selten-Erd-Metalle (incl. Ce-Lu) Titan-Gruppe Vanadium-Gruppe Chrom-Gruppe Mangan-Gruppe Eisen-Gruppe Cobalt-Gruppe Nickel-Gruppe, Platinmetalle Münzmetalle Zink-Gruppe Triele, Erdmetalle Tetrele, (Tattogene) Pentele, Pnik(t)ogene Chalkogene Halogene Edelgase VIII. Nebengruppe, VIIIb I. Nebengruppe, Ib II. Nebengruppe, IIb III. Hauptgruppe, IIIa IV. Hauptgruppe, IVa V. Hauptgruppe, Va VI. Hauptgruppe, VIa VII. Hauptgruppe, VIIa VIII. Hauptgruppe, VIIIa Tabelle 1.2: Die Gruppen 1.4 Die Orbitale Die Elektronenhülle eines Atoms wird grob unterteilt in die Schalen (K-Schale, L-Schale, etc.). Die Hauptquantenzahl n gibt darüber Auskunft in welcher Schale sich eine Elektron befindet,(n = 1 ⇒ K-Schale, n = 2 ⇒ L-Schale, etc.). Die Schalen werden aufgeteilt in die Orbitale. Ein Orbital ist ein bestimmter räumlicher (eigentlich rein rechnerischer Bereich), in dem sich die Elektronen eines Atoms am wahrscheinlichsten aufhalten. Vier verschiedene Sorten von Orbitalen, s-, p-, d- und f-Orbitale können besetzt werden. Die Nebenquantenzahl l (= Bahndrehimpulsquantenzahl) gibt darüber Auskunft in welcher Orbitalart sich ein Elektron befindet (l = 0 ⇒ s-Orbital, l = 1 ⇒ p-Orbital, l = 2 ⇒ d-Orbital und l = 3 ⇒ f-Orbital). Allerdings ist jede Orbitalsorte in verschiedene entartete (= energiegleiche) Orbitale aufgeteilt. Es existieren ein s-Orbital, drei p-Orbitale, fünf d-Orbitale und sieben f-Orbitale. In welchem der verschiedenen entarteten Orbitale einer Sorte sich ein Elektron befindet, wird durch die magnetische Quantenzahl ml angezeigt (mit −l ≤ ml ≤ +l). Diese gibt auch Informationen über die räumliche Anordnung (z. B. px , py oder pz ). Jedes Orbital kann letztlich maximal zwei Elektronen aufnehmen, deren Unterschied durch die magnetische Spinquantenzahl ms angegeben wird (ms = −1 /2 , +1 /2 ). Periodisches System der Elemente 47 Pauli-Prinzip: Zwei Elektronen eines Atoms können nie in allen 4 Quantenzahlen übereinstimmen. Die verschiedenen Orbitale unterscheiden sich in ihrer Energie sowohl durch die Schale, in der sie sich befinden als auch durch ihre Sorte. Hund’sche Regel: Die Orbitale werden aufsteigend von Orbitalen niedriger Energie zu denen höherer Energie mit Elektronen besetzt. Hierbei gilt, dass entartete Orbitale erst einfach und dann doppelt besetzt werden. In jeder Schale kommt eine neue Orbitalsorte hinzu, die theoretisch besetzt werden könnte. 1. 2. 3. 4. 5. 6. 7. Periode Periode Periode Periode Periode Periode Periode K-Schale L-Schale M-Schale N-Schale O-Schale P-Schale Q-Schale 1s-Orbital 2s-Orbital 3s-Orbital 4s-Orbital 5s-Orbital 6s-Orbital 7s-Orbital (1) (1), (1), (1), (1), (1), (1), 2p-Orbitale 3p-Orbitale 4p-Orbitale 5p-Orbitale 6p-Orbitale 7p-Orbitale (3) (3), (3), (3), (3), (3), 3d-Orbitale 4d-Orbitale 5d-Orbitale 6d-Orbitale 7d-Orbitale (5) (5), (5), (5), (5), 4f-Orbitale 5f-Orbitale 6f-Orbitale 7f-Orbitale (7) (7) (7) (7) Allerdings sind die pro Schale neu hinzugekommenen Orbitale oft energetisch so hochliegend, dass zuvor Orbitale aus einer höheren Schale besetzt werden. Die 3d-Orbitale werden beispielsweise erstmalig in der vierten Periode besetzt, die 4f-Orbitale erst in der sechsten Periode. Die Elemente, deren Valenzelektronen in d-Orbitalen liegen, werden als Übergangselemente (oder -metalle) bezeichnet, jene, deren Valenzelektronen in f-Orbitalen liegen, werden als Lanthanoide (4f) und Actinoide (5f) bezeichnet. 1. 2. 3. 4. 5. 6. 7. Periode Periode Periode Periode Periode Periode Periode K-Schale L-Schale M-Schale N-Schale O-Schale P-Schale Q-Schale 1s2 2s2 , 3s2 , 4s2 , 5s2 , 6s2 , 7s2 , 2p6 3p6 3d10 , 4p6 4d10 , 5p6 4f 14 , 5d10 , 6p6 5f 14 , 6d10 , 7p6 entspricht entspricht entspricht entspricht entspricht entspricht He Ne Ar Kr Xe Rn Energetisch günstige Elektronenkonfiguration sind entweder leere, halb- oder vollgefüllte entartete Orbitale einer Sorte (siehe auch Versuch 2 ABS). 1.4.1 s-Orbitale In jeder Periode existiert ein s-Orbital. Es ist kugelsymmetrisch, (siehe Abbildung 1.1), besitzt keine Knotenfläche und der einzige Orbitallappen weist ein positives Vorzeichen (aus der Lösung der Schrödinger-Gleichung, hat nichts mit Ladungen zu tun) auf. Abb. 1.1: s-Orbital 48 Periodisches System der Elemente 1.4.2 p-Orbitale Ab der Zweiten existieren in jeder Periode drei p-Orbitale. Diese werden häufig als hantelförmig bezeichnet (siehe Abbildung 1.2). Sie bestehen aus zwei Orbitallappen, die entlang einer Geraden ausgerichtet sind, wobei einer der Lappen ein positives und der andere Lappen Abb. 1.2: p-Orbitale ein negatives Vorzeichen aufweist (aus der Lösung der Schrödinger-Gleichung, hat nichts mit Ladungen zu tun). Zwischen diesen beiden Lappen existiert eine sogenannte Knotenfläche, an der ein Vorzeichenwechsel stattfindet. Dort ist die Aufenthaltswahrscheinlichkeit für Elektronen null. Die Knotenebene beinhaltet den Atomkern. Die drei gleichaussehenden Orbitale sind entlang der Achsen eines karthesischen Koordinatensystems angeordnet und werden dem entsprechent als px , py und pz bezeichnet. 1.4.3 d -Orbitale Ab der Dritten existieren in jeder Periode fünf d-Orbitale, die jedoch immer erst eine Periode später besetzt werden. Diese Orbitale haben im Unterschied zu den beiden vorgenannten Typen nicht alle die gleiche Form. Vier dieser Orbitale besitzen jeweils vier Lappen in Form eines vierblättrigen Kleeblatts (siehe Abbildung 1.3), die in drei Fällen zwischen den Achsen eines dreidimensionalen karthesischen Koordinatensystems ausgerichtet sind und als dxy , dxz und dyz bezeich- Abb. 1.3: d-Orbitale net werden. Die Lappen des vierten dieser kleeblattförmigen Orbitale sind entlang der x- und y-Achse ausgerichtet. Dieses Orbital wird als dx2 −y2 bezeichnet. Das fünfte d-Orbital besteht aus drei Lappen, von denen zwei hantelförmig entlang der z-Achse ausgerichtet sind und das gleiche Vorzeichen besitzen. Darüber hinaus besitzt dieses Orbital in der xy-Ebene einen ringförmigen (torusförmigen) Orbitallappen, der das zu den beiden hantelförmigen Lappen entsprechend entgegengesetzte Vorzeichen besitzt (siehe Abbildung 1.3). Dieses Orbital trägt die Bezeichnung dz 2 und besitzt wie die anderen d-Orbitale ebenfalls zwei Knotenflächen. Bei den d-Orbitalen weisen also immer gegenüberliegende Orbitallappen gleiche und benachbarte Orbitallappen unterschiedliche Vorzeichen auf (aus der Lösung der Schrödinger-Gleichung, hat nichts mit Ladungen zu tun). Somit werden bei allen d-Orbitalen zwei Knotenflächen beobachtet. Periodisches System der Elemente 49 1.5 Das HSAB-Prinzip (auch: Pearson-Konzept) Das HSAB-Prinzip ("Hard and Soft Acids and Bases") gibt einen Überblick darüber, welche Teilchen bei Lewis-Säure-Base-Reaktionen Koordinationsverbindungen miteinander eingehen. Entscheidend dabei ist die Größe der Elektronenhülle der jeweiligen Teilchen, die Ladung und die damit verbundene Polarisierbarkeit der entsprechenden Lewis-Säuren oder -Basen. Ist eine Teilchen leicht polarisierbar, spricht man von einem ”weichen” Teilchen, bei geringer Polarisierbarkeit wird es als ”hartes” Teilchen bezeichnet. Das Pearson-Konzept besagt dabei, dass ”harte” Säuren mit ”harten” Basen bevorzugt eine Verbindung eingehen und ”weiche” Säuren mit ”weichen” Basen. Verbindungen von ”harten” Säuren mit ”weichen” Basen und umgekehrt sind hingegen nicht so stabil. ”Harte” Säuren: besitzen eine hohe positive Ladung, eine kleine räumliche Ausdehnung, keine nicht-bindenden Valenzelektronenpaare und keine besetzten d-Orbitale. (z. B. Mg2+ , Al3+ , Ti4+ , etc.) ”Weiche” Säuren: besitzen eine kleine positive Ladung und nicht-bindende Valenzelektronenpaare oder gefüllte d-Orbitale (z. B. Zn2+ , Ag+ , Hg2+ , etc.) Die Donoratome der Basen sind im Allgemeinen umso härter, je kleiner (S2− härter als I− ), 2− je elektronegativer (Cl− härter als S2− ) bzw. je höher oxidiert (SO2− 3 härter als S ) sie sind. Bei Umsetzungen von verschiedenen Lewis-Säuren mit verschiedenen Lewis-Basen entstehen bevorzugt Produkte von ”harten” Säuren mit ”harten” Basen bzw. ”weichen” Säuren mit ”weichen” Basen. 2 Aufgabenstellung, Versuchsanleitung und -auswertung 2.1 Beobachtung der unterschiedlichen Löslichkeit von Silberhalogeniden 2.1.1 Aufgabenstellung Bei diesem Versuch soll das Löslichkeitsverhalten von Silberhalogeniden in Abhängigkeit des Halogenidanions untersucht werden (siehe auch Versuch 11 FLG). Hierbei sollen die Ähnlichkeiten im chemischen Verhalten von Elementen, die in derselben Gruppe stehen herausgearbeitet werden. 2.1.2 Versuchsanleitung In drei Reagenzgläsern werden Lösungen von KCl, KBr und KI hergestellt. Dazu wird eine Spatelspitze der jeweiligen Verbindung in ein Reagenzglas gegeben und mit demineralisierten Wasser bis zur Hälfte befüllt. Diese werden mit wenigen Tropfen AgNO3 -Lösung versetzt. Es wird zunächst versucht, die beobachteten Niederschläge in konzentrierter (NH4 )2 CO3 -Lösung (muss selbst hergestellt werden) wieder aufzulösen. Dieser Versuch wird wiederholt, jedoch wird versucht die beobachteten Niederschläge in konzentrierter Ammoniaklösung aufzulösen. 50 Periodisches System der Elemente 2.1.3 Versuchsauswertung ● ● Welche Farbe besitzen die Niederschläge von AgCl, AgBr und AgI? Formulieren Sie die Reaktionsgleichungen für die Fällung und das Lösen der entsprechenden Silberhalogenide und interpretieren Sie dieses Verhalten. ● ● Welche Löslichkeitsprodukte besitzen die drei Silberhalogenide (Literaturstudium). Welche weiteren periodischen Eigenschaften (neben der Bildung schwerlöslicher Silbersalze) besitzen die Halogene? Nennen Sie die, Ihrer Ansicht nach, drei wichtigsten. 2.2 Änderung der Eigenschaften beim Durchgang durch eine Periode – pH-Werte von ”Hydroxiden” der dritten Periode 2.2.1 Aufgabenstellung Bei diesem Versuch sollen die pH-Werte der Hydroxide bzw. der Oxid-Hydroxide der Elemente der dritten Periode bestimmt und damit der Übergang von alkalischem zu saurem Verhalten beim Übergang von metallischen zu nichtmetallischen Elementen in dieser Periode untersucht werden (siehe Versuch 9 SBG). 2.2.2 Versuchsanleitung In sieben Reagenzgläsern werden Lösungen von NaOH, Mg(OH)2 , Al(OH)3 , Si(OH)4 ∗ , PO(OH)3 , SO2 (OH)2 und ClO3 (OH) hergestellt. Bei festen Verbindungen wird eine Spatelspitze davon in ein Reagenzglas gegeben und dieses mit demineralisiertem Wasser bis zur Hälfte aufgefüllt. Bei flüssigen Verbindungen wird das Reagenzglas zur Hälfte mit demineralisiertem Wasser gefüllt und mit einer Tropfpipette von der entsprechenden Verbindung ca. 3–5 mm hoch zugegeben. Der pH-Wert der sieben Lösungen wird mit Universalindikatorpapier gemessen. ∗ Finden Sie Si(OH)4 auf Ihrem Chemikalienblech? Falls nein, warum nicht und was nehmen Sie stattdessen? 2.2.3 Versuchsauswertung ● Bestimmen Sie die pH-Werte der einzelnen Lösungen und erklären Sie diese Werte mit Hilfe von Dissoziationsgleichungen. ● ● Warum verhalten sich die Hydroxide so unterschiedlich? Es existieren keine ”reinen” Hydroxide der Elemente Phosphor, Schwefel und Chlor? Geben Sie anhand Ihres bisherigen Wissensstands eine plausible Erklärung. 2.3 Vergleich der Farbigkeit von Übergangsmetallkomplexen/-verbindungen mit Hauptgruppenmetallkomplexen/-verbindungen 2.3.1 Aufgabenstellung Dieser Versuch dient dem Vergleich von Verbindungen der Übergangsmetalle mit analogen Hauptgruppenmetallverbindungen bezüglich Farbigkeit und deren Entstehung. Periodisches System der Elemente 51 2.3.2 Versuchsanleitung a) In vier Reagenzgläsern wird je eine wässrige Lösung von KCl, CaCl2 , FeCl3 und CuSO4 hergestellt. Hierbei wird eine Spatelspitze der entsprechenden Verbindung in das Reagenzglas gegeben und dieses mit deminaraliserten Wasser bis zur Hälfte befüllt. Die beobachteten Farben werden dokumentiert. Die Lösungen werden mit Ammoniaklösung (ca. 2 cm hoch) versetzt und gut durchmischt. Lassen Sie eventuell entstandene Niederschläge absitzen und dokumentieren Sie dann die zu beobachtenden Farben. b) In vier Reagenzgläsern wird je eine wässrige Lösung von K2 SO4 , K2 CrO4 , KClO4 und KMnO4 hergestellt (siehe Versuchsteil a)). Die beobachteten Farben werden dokumentiert. 2.3.3 Versuchsauswertung ● ● Welche Moleküle bzw. Ionen sind für die Farben der Lösungen verantwortlich? Warum sind Übergangsmetallverbindungen häufig farbig, Hauptgruppenelementverbindungen jedoch praktisch nie? ● Wie werden die Farben dieser Verbindungen (und Komplexe) beeinflusst? (s. auch Versuche 2 ABS, 3 BSP und 12 KOG) 3 Geräte und Chemikalien Eine Geräteliste liegt im Platz aus Chemikalien KCl KBr KI (NH4 )2 CO3 AgNO3 NH3 aq NaOH Mg(OH)2 Al(OH)3 SiO2 ⋅x H2 O H3 PO4 H2 SO4 HClO4 CaCl2 FeCl3 ⋅ 6 H2 O CuSO4 ⋅ 5 H2 O K2 SO4 K2 CrO4 KClO4 KMnO4 Gefahrenbezeichnung – – – Achtung (GHS07) Gefahr (GHS03, 05, 09) Gefahr (GHS05, 09) Gefahr (GHS05) – – – Gefahr (GHS05) Gefahr (GHS05) Gefahr (GHS03, 05) Achtung (GHS07) Gefahr (GHS08) Achtung (GHS07, 09) – Gefahr (GHS06, 09) H-Sätze – – – 302 272, 314, 410 314, 335, 400 314, 290 – – – 314, 290 314, 290 271, 314 319 302, 315, 318 302, 315, 319, 410 – 350i, 340, 319, 335, 315, 317, 410 Gefahr (GHS03, 08) 271, 302 Gefahr (GHS03, 08, 09) 272, 302, 410 P-Sätze – – – – 301+330+331, 305+351+338, 273, 280 301+330+331, 305+351+338, 280, 273 301+330+331, 305+351+338, 280 – – 260 280, 301+330+331, 305+351+338, 309, 310 280, 301+330+331, 305+351+338, 309, 310 301+330+331, 305+351+338, 210, 280 305+351+338 280, 302+352, 305+351+338, 313 305+351+338, 302+352, 273 – 308+313, 305+351+338, 201, 280, 273, 302+352 210 210, 273 52 Periodisches System der Elemente 4 Literatur [1] A. F. Holleman, E. Wiberg: Lehrbuch der Anorganischen Chemie, 102. Aufl., Walter de Gruyter Verlag, 2007. [2] M. Binnewies, M. Jäckel, H. Willner, G. Rayner-Canham: Allgemeine und Anorganische Chemie, 2. Auflage, Spektrum Verlag, 2011. [3] G. Jander, E. Blasius: Lehrbuch der Analytischen und Präparativen Anorganischen Chemie, 16. Auflage, S. Hirzel Verlag, 2006. Hauptgruppenelemente 53 Versuch 5 (HGE) Hauptgruppenelemente 1 Theorie 1.1 Lewis-Fomeln (≡ Valenzstrichformeln) von Molekülen Gilbert Newton Lewis (1875-1946) entwickelte die Oktett-Regel und eine bis heute gültige Schreibweise für Moleküle mit Elektronenpaarbindungen und freien Elektronen. Jedes Elektron, das ein Atom umgibt, wird berücksichtigt. Zwei Elektronen (= ein Elektronenpaar) werden mit einem Strich, ein einzelnes Elektron mit einem Punkt angegeben. 1.2 Das VSEPR Konzept VSEPR bedeutet Valence Shell Electron Pair Repulsion (≡ Valenzelektronenpaarabstoßung). Diese Theorie zur Erklärung der Gestalt von Molekülen wurde von Gillespie und Nyholm gegen Ende der 50er Jahre des vorigen Jahrhunderts aufgestellt (daher auch: Gillespie-NyholmKonzept). Dieses Konzept stellt grundlegende Regeln zur Bestimmung der Molekülgestalt auf: 1. Alle ein Atom umgebenden Elektronenpaare (bindende und freie) ordnen sich aufgrund gegenseitiger Abstoßung in möglichst großem Abstand zueinander um ein Zentralatom an. Diese Anordnung geschieht derart, dass die Liganden sich ideal auf einer Kugeloberfläche um das Zentralatom verteilen. Dies wird durch folgende Geometrien realisiert: ∗ 2 Elektronenpaare linear 3 Elektronenpaare trigonal-planar 4 Elektronenpaare tetraedrisch 5 Elektronenpaare trigonal-bipyramidal oder quadratisch-pyramidal∗ 6 Elektronenpaare oktaedrisch energetisch liegen diese beiden Varianten nur wenige kJ mol auseinander. 2. Freie Elektronenpaare beanspruchen mehr Raum als bindende. Die Bindungswinkel nehmen daher ab (z. B.: CH4 besitzt einen Winkel H–C–H von 109, 5○ (= idealer Tetraederwinkel), der H–N–H-Winkel in NH3 beträgt 107○ , der H–O–H-Winkel in H2 O beträgt 104, 5○ ). Wenn freie Elektronenpaare zur Geometrie um ein Zentralatom beitragen, werden diese mit einem ψ bezeichnet, dieses wird mit der Anzahl der freien Elektronenpaare oben rechts versehen. Das ψ n wird dann dem Koordinationspolyeder voran gestellt (z. B. CH4 – Tetraeder, NH3 – ψ-Tetraeder oder ψ 1 -Tetraeder, H2 O – ψ 2 -Tetraeder). 3. Mehrfachbindungen nehmen mehr Raum ein als Einfachbindungen. Der Raumbedarf eines Valenzelektronenpaars sinkt mit steigender Elektronegativität des Liganden, dadurch nimmt der Bindungswinkel zwischen den anderen Liganden zu. 54 Hauptgruppenelemente 1.3 Die Hauptgruppenelemente 1.3.1 Die 1. Gruppe (= 1. Hauptgruppe): Wasserstoff und die Alkalimetalle Wasserstoff Das leichteste Element stellt auch gleichzeitig mit 90% Vorkommen das häufigste Element des Universums dar. Elementar kommt Wasserstoff als zweiatomiges Molekül vor, dessen Bindung relativ leicht durch einen Katalysator oder durch moderate Energiezufuhr gespalten werden kann, somit ist Wasserstoff höchst brennbar. Durch seine geringe Größe ist es dem Wasserstoff auch möglich, durch fast alle Arten von Stoffen mehr oder minder schnell durchzudiffundieren. Bei der Verbrennung von Wasserstoff an Luft entsteht Wasser, in dem Wasserstoff die Oxidationsstufe +1 aufweist. Alkalimetalle Als Alkalimetalle werden die Elemente der ersten Gruppe (bzw. ersten Hauptgruppe) bezeichnet: Lithium (Li), Natrium (Na), Kalium (K), Rubidium (Rb), Caesium (Cs) und das radioaktive Francium (Fr). Wasserstoff (H) steht zwar auch in der ersten Gruppe, teilt aber mit den erstgenannten nur wenig gemeinsame Eigenschaften und wird daher nicht zur Gruppe der Alkalimetalle gezählt. Lithium kommt zumeist in komplexanionischen Mineralien, wie Silicaten, Phosphaten oder Aluminaten vor. Elementares Lithium führt mit einem Standardpotential von −3, 045 V die Liste der besten Reduktionsmittel an und wird daher sehr oft in Batterien und Akkumulatoren verwendet. Darüber hinaus brennt Lithium extrem heiß und reagiert an Luft sowohl zu Li2 O, als auch zum Lithiumnitrid Li3 N. Das Lithium-Kation ist nach dem H+ das kleinste Kation. Ein wässriger Lithium-Ionen-Komplex besitzt daher eine Koordinationszahl von vier und nicht sechs, wie die anderen wässrigen Alkalimetall-Kationen-Komplexe. Lithium-Kationen 3– bilden nur mit OH– -, CO2– 3 - und PO4 -Anionen relativ schwerlösliche Salze, die zum Nachweis dienen könnten. Charakteristisch für Li+ ist die karminrote Flammenfärbung, die allerdings nur recht kurz bestehen bleibt. Natrium ist das sechsthäufigste Element in der Erdkruste. Die am weitesten verbreiteten Natriummineralien sind Feldspäte (Natronfeldspat Na[AlSi3 O8 ]), aber auch Steinsalz NaCl (Halit), Soda Na2 CO3 , Glaubersalz Na2 SO4 und Chilesalpeter NaNO3 . Elementares Natrium ist ein weiches, schnittfähiges Metall, das an feuchter Luft und in Wasser sofort oxidiert: Na + H2 O → NaOH + 1 /2 H2 ↑. Das Natrium-Kation liegt in wässriger Lösung als oktaedrischer Hexaquanatrium(I)-Komplex vor. Na+ -Salze sind fast alle leicht löslich (Ausnahme ist z. B. Natriummagnesiumuranylacetat NaMg(UO2 )3 (CH3 COO)9 ⋅ 9 H2 O). Zum Nachweis des Natriums eignet sich daher fast ausschließlich die Flammenfärbung. Kalium ist das siebthäufigste Element in der Erdkruste. Die am weitesten verbreiteten Kaliummineralien sind ebenfalls Feldspäte (Kalifeldspat K[AlSi3 O8 ]) und Kaliglimmer (Muskovit KAl2 (OH,F)2 [AlSi3 O10 ]), aber auch Sylvin KCl und Kaliumsulfat K2 SO4 . Elementares Kalium ist ebenfalls ein weiches, schnittfähiges Metall, das an feuchter Luft und im Wasser sofort analog zum Natrium, jedoch deutlich heftiger oxidiert. K+ liegt in wässriger Lösung als oktaedrischer Hexaquakalium(I)-Komplex vor. Kalium-Kationen bilden nur wenige schwerlösliche Salze, beispielsweise das Kaliumperchlorat KClO4 und das Kaliumnatriumhexanitrocobaltat(III) KNa2 [Co(NO2 )6 ], die beide auch zum Nachweis des K+ -Kations verwendet werden können. Im Vergleich zu Natrium und Kalium haben Hauptgruppenelemente 55 die Elemente Rubidium und Caesium einen geringen Anteil an der Erdkruste. Beide Metalle sind weich (Schmelzpunkte von 39○ C für Rubidium und 28, 5○ C für Caesium) und reagieren bereits an Luft extrem heftig, ebenso wie mit Wasser. An Luft reagieren die Alkalimetalle außer Lithium nicht zum Oxid gemäß A2 O (A = Alkalimetall). Im Fall des Natriums wird das Peroxid Na2 O2 erhalten, während Kalium, Rubidium und Caesium zum Hyperoxid AO2 reagieren. 1.3.2 Die 2. Gruppe (= 2. Hauptgruppe): Erdalkalimetalle Als Erdalkalimetalle werden die Elemente der zweiten Gruppe (bzw. zweiten Hauptgruppe) bezeichnet; diese sind Beryllium (Be), Magnesium (Mg), Calcium (Ca), Strontium (Sr), Barium (Ba) und das radioaktive Radium (Ra). Metallisches Beryllium ist ebenso wie seine Verbindungen hochgiftig und findet daher nur wenig Anwendung. Genutzt wird vor allem die Eigenschaft des Beryllium-Metalls, durchlässig für Röntgenstrahlung zu sein. Es wird als Material für Fenster in Röntgenröhren verwendet. Magnesium ist das achthäufigste Element in der Erdkruste. Als magnesiumhaltige Mineralien seien hier hauptsächlich die Carbonate (Bitterspat MgCO3 und Dolomit CaMg(CO3 )2 ) und die Silicate genannt (z. B.: Olivin (Mg,Fe)2 [SiO4 ]) wobei auch oxidische oder chloridische Doppelsalze nicht selten sind (z. B.: Spinell MgAl2 O4 ). Magnesium ist ein silberglänzendes Metall, das sich an Luft schnell mit einer Oxidschicht überzieht und dann eher weißsilbrig aussieht. Das Magnesium-Kation liegt in wässriger Lösung ebenfalls als oktaedrischer Hexaquamagnesium(II)-Komplex vor. Schwerlösliche Magnesiumverbindungen sind, in Analogie zum Lithium (sog. ”Schrägbeziehung” im PSE), das Magnesiumhydroxid, -carbonat und -phosphat. Aufgrund seiner geringen Dichte wird Magnesium sehr gerne in Leichtmetalllegierungen eingesetzt. Ebenso wie Lithium brennt Magnesium extrem heiß und ist in der Lage an Luft nicht nur zum Oxid MgO, sondern auch zum Nitrid Mg3 N2 zu reagieren. Calcium ist das fünfthäufigste Element in der Erdkruste. Die in der Natur vorherrschende Calciumverbindung ist das Carbonat in den Mineralien Kalkstein, Kreide, Marmor (alle CaCO3 ) und Dolomit CaMg(CO3 )2 . Darüber hinaus sind die Sulfate (Gips CaSO4 ⋅ 2 H2 O und Anhydrit CaSO4 ) wichtige calciumhaltige Mineralien, ebenso wie Phosphate Ca(OH,F)[PO4 ]3 (Apatit), Silicate (z.B.: Granate) und das Calciumfluorid CaF2 (Fluorit bzw. Flussspat). Ebenso wie Magnesium überzieht sich elementares Calcium an Luft schnell mit einer Oxidschicht und besitzt daher einen silbrig-weißen Glanz (siehe Versuch 1 GMS). Das Calcium-Kation liegt in wässriger Lösung als Hexaquacalcium(II)-Komplex vor. Strontium kommt in der Natur hauptsächlich in den Mineralien Coelestin SrSO4 und Strontianit SrCO3 vor. Sr2+ -Kationen bilden in Wasser, wie die übrigen Erdalkalimetalle auch einen oktaedrischen Hexaqua-Komplex. Wie das Strontium kommt auch das Barium in der Natur vorwiegend als Sulfat (Baryt oder Schwerspat BaSO4 ) oder Carbonat (Witherit BaCO3 ) vor. Barium bildet 2– 2– außer mit SO2– 4 und CO3 auch mit Chromat-Anionen CrO4 einen schwerlöslichen Niederschlag, der zum Nachweis und zur Abtrennung der Ba2+ - von den anderen Erdalkalimetall-Kationen verwendet werden kann. 1.3.3 Die 13. Gruppe (= 3. Hauptgruppe): Triele (Erdmetalle) Neben dem nichtmetallischen Bor (B) enthält die dreizehnte Gruppe (dritte Hauptgruppe) die Elemente Aluminium (Al), Gallium (Ga), Indium (In) und Thallium (Tl), die auch als Triele oder 56 Hauptgruppenelemente Erdmetalle bezeichnet werden. Die Mineralien des Elements Bor sind der Kernit Na2 [B4 O5 (OH)4 ] ⋅2 H2 O (= Na2 B4 O7 ⋅4 H2 O), Borax Na2 B4 O7 ⋅10 H2 O (Tinkal) und entsprechende Calciumbora- te. Bor besitzt ebenso wie das Silicium (sog. ”Schrägbeziehungen” im PSE) eine hohe Affinität zu Sauerstoff und kommt in der Natur ausschließlich an Sauerstoff gebunden vor. Das Boroxid B2 O3 , das Anhydrid der Borsäure, wird jedoch im Gegensatz zum SiO2 in der Natur nicht gefunden, da es sehr stark hygroskopisch ist. Mit seinen 3 Valenzelektronen neigt das Bor zu Elektronenmangelverbindungen, was sich in unter anderem der ortho-Borsäure H3 BO3 manifestiert. Diese ist KEINE Säure nach der Brønsted-Theorie, sondern eine Lewis-Säure (siehe Versuche 9 SBG und 12 KOG), die mit Wasser entsprechend H3 BO3 + H2 O → H+ + [B(OH)4 ]– reagiert. Die Reaktion als Lewis-Säure ist eine Möglichkeit für Bor seinem Elektronenmangel zu entgehen. Eine weitere Möglichkeit zur Bekämpfung des Elektronenmangels am Bor ist die Ausbildung von p − π − p − π-Rückbindungen durch Bindung zu elektronenreichen Partnern (F– , O2– ). Dies entspricht einer π-Bindung (siehe Versuch 7 BPH) eines freien Elektronenpaares des Liganden mit dem freien, senkrecht stehenden p-Orbital eines trigonal-planaren borzentrierten Moleküls (z. B.: BO3– 3 ). Die dritte Möglichkeit ist die Ausbildung von Zwei-Elektronen-Drei-Zentrenbindungen, die immer dann diskutiert werden, wenn das Bor zusammen mit Elementen, die NICHT als Lewis-Basen wirken (z.B.: H oder B selbst), eine Verbindung eingeht. Aluminium ist das dritthäufigste Element in der Erdrinde. Es kommt vorwiegend in der Oxidationsstufe +III vor. Die wichtigsten aluminiumhaltigen Mineralien sind Feldspäte und Glimmer (beides Alumosilicate), Korund Al2 O3 und Kryolith Na3 [AlF6 ]. Das technisch wichtige Bauxit ist ein Gestein, das aus Aluminiumoxiden und -hydroxiden besteht. Elementares Aluminium ist amphoter und bildet beim Lösen in Laugen den tetraedrischen Tetrahydroxoaluminat(III)-Komplex [Al(OH)4 ]– . In Wasser bildet Al3+ den oktaedrischen Hexaquaaluminium(III)-Komplex [Al(H2 O)6 ]3+ . Die Verwendung von Aluminium ist weit gefächert, in der Hauptsache ist Aluminium Bestandteil unzähliger Leichtmetalllegierungen. Die Bedeutung von Gallium liegt vor allem in der Dotierung von Halbleitermaterialen und LED’s. Dies gilt in geringerem Maße auch für Indium. Das giftige Thallium fand früher hauptsächlich als Rattengift Verwendung. Obwohl in der dritten Hauptgruppe die Oxidationsstufe +III vorherrschend ist, kommen die Elemente Indium und vor allem Thallium auch in der Oxidationsstufe +I mit gefülltem s-Orbital vor. Der Grund dafür liegt in dem von Periode zu Periode zunehmenden relativistischen Effekt, welcher es schwerer macht Elektronen aus den entsprechenden s-Valenzorbitalen zu entfernen. 1.3.4 Die 14. Gruppe (= 4. Hauptgruppe): Tetrele (Tattogene) Die 14. Gruppe des Periodensystems (vierte Hauptgruppe) enthält die Elemente Kohlenstoff (C), Silicium (Si), Germanium (Ge), Zinn (Sn, lat. Stannum) und Blei (Pb, lat. Plumbum). Diese Elemente werden auch als Tetrele (von tetra = vier) bezeichnet, eher selten auch als Tattogene (was soviel bedeutet wie Gerüstbildner). Obwohl Kohlenstoff das grundlegende Element der organischen Chemie darstellt ist es mit einem Anteil von 0, 02 % nur das 18. häufigste Element in der Erdkruste. Elementarer Kohlenstoff kommt natürlich in zwei Modifikationen vor, nämlich Diamant (aufgebaut aus einem Gerüst von Tetraedern aus Kohlenstoffatomen) und Graphit (aufgebaut aus Schichten von Sechsringen aus Kohlenstoffatomen). Aus letzterem können durch Hauptgruppenelemente 57 Spaltung Einzelschichten, das sog. Graphen erhalten werden. Weitere Modifikationen sind die Fullerene (kugelförmige Moleküle aus fünf- und sechsgliedrigen Kohlenstoffringen), die auch in beliebige weitere Formen gebracht werden können (z. B. Kohlenstoff-Nanoröhren). Neben einer Vielzahl organischer Moleküle werden einige kohlenstoffhaltige Verbindungen der anorganischen Chemie zugeschrieben. Zum einen sind das die Carbide, Stoffe, die Anionen aus elementarem Kohlenstoff enthalten. Die salzartigen Carbide werden in Methanide (C4– , ”Säureanion” des 4– Methans), Acetylenide (C2– 2 , ”Säureanion” des Acetylens) und Allenide (C3 ) unterschieden. Darüber hinaus sind kovalente Carbide bekannt (z. B.: SiC) sowie metallartige. Bei der Stahlherstellung wird Kohlenstoff in geringen Mengen in Eisen eingelagert um dieses zu härten. Weiterhin werden auch die Kohlenstoffoxide CO und CO2 zu den anorganischen Kohlenstoffverbindungen gezählt, ebenso wie die Kohlensäure und deren Salze die Carbonate. Das Carbonat-Anion ist das Säureanion der unbeständigen Kohlensäure H2 CO3 , die zu einem verschwindend geringen Prozentsatz beim Lösen ihres gasförmigen Anhydrids CO2 in H2 O entsteht. Bei der Kohlensäure handelt es sich um eine schwache Säure, welche mit einer starken Säure (Salzsäure, Schwefelsäure, etc.) aus ihren Salzen vertrieben werden kann, und dabei allerdings gleich in CO2 und H2 O zerfällt gemäß: H2 CO3 ⇌ CO2 + H2 O. Mit einer Elementhäufigkeit von 25, 8% ist das Silicium nach Sauerstoff (49, 4%) das zweithäufigste Element in der Erdrinde und kommt quasi immer in Verbindungen mit Sauerstoff vor. Es ist damit annähernd dreihundert Mal häufiger als sein kleineres Homologes Kohlenstoff. Das Siliciumdioxid SiO2 (Anhydrid der Kieselsäure H4 SiO4 , ortho-Kieselsäure, bzw. H2 SiO3 , meta-Kieselsäure) kommt in Form verschiedener Mineralien (Quarz, Bergkristall, Amethyst, Kieselstein, Seesand, etc.) vor, ebenso sind die entsprechenden Salze der Kieselsäure (Silicate) wie Magnesium-, Calcium-, Aluminium- und Eisensilicate von Bedeutung. Die Kieselsäure ist eine sehr schwache Säure und besitzt anders als Phosphor-, Schwefeloder Perchlorsäure eine hohe Neigung zur Kondensation durch Wasserabspaltung. Daher bildet sie nicht nur das reine ortho-Silicat, sondern auch aggregierter Einheiten über Silicattetraeder, die gemeinsame Ecken miteinander teilen (Eckenverknüpfung). Das Germanium besitzt große Ähnlichkeit mit dem Silicium und wird technisch auch in ähnlichen Bereichen verwendet. Das wichtigste Mineral des Elements Zinn ist der Zinnstein SnO2 , der sich nur mit Hilfe des Freiberger Aufschlusses als Thiosalz in Lösung bringen lässt. Die analoge Zinnsäure H2 SnO3 ist im freien Zustand nicht bekannt. Zinn ist ebenfalls ein wichtiger Bestandteil von Metalllegierungen. Ebenso, wie sein höheres Homologes Blei, kommt auch Zinn in der Oxidationsstufe +2 vor. Die wichtigste Oxidationsstufe des Elements Blei ist +2 (Erklärung siehe Indium und Thallium, 13. Gruppe), obgleich auch einige Pb(IV)-Verbindungen Anwendungsmöglichkeiten besitzen (PbO2 im Bleiakku, Mennige Pb3 O4 (≡ Pb2 [PbO4 ] Blei(II)-ortho-plumbat(IV)) als Rostschutzmittel, etc.). Das wichtigste Bleimineral ist der Bleiglanz (PbS, Galenit), der eine graue Farbe besitzt, während feinverteiltes gefälltes PbS eine schwarze Farbe aufweist. 1.3.5 Die 15. Gruppe (= 5. Hauptgruppe): Pentele (Pnik(t)ogene) Die 15. Gruppe des Periodensystems (fünfte Hauptgruppe) enthält die Elemente Stickstoff (N), Phosphor (P), Arsen (As), Antimon (Sb, von lat. Stibium) und Bismut (Bi), wobei N und P Nichtmetalle und As und Sb Halbmetalle darstellen. Bi besitzt hingegen metallischen Charakter. Diese 58 Hauptgruppenelemente Elemente werden als Pentele (von penta = fünf) bezeichnet, häufig auch als Pnik(t)ogene (was soviel bedeutet wie Erstickende). Elementarer Stickstoff N2 ist mit 78% der Hauptbestandteil der Erdatmosphäre, wobei diese auch die hauptsächliche Quelle für elementaren Stickstoff darstellt. Durch seine Dreifachbindung ist das N2 -Molekül sehr stabil. Bei einer Verbrennung an Luft reagieren nur sehr wenig Elemente mit Stickstoff (z. B. Magnesium und Lithium). Zwischen -3 und +5 kommt Stickstoff in jeder Oxidationsstufe vor, wobei die negativen überwiegend bei Ammoniakderivaten zu finden sind und die positiven hauptsächlich bei Stickoxiden und entsprechenden Sauerstoffsäuren auftreten. Die beiden wichtigsten Stickstoffverbindungen sind Ammoniak und Salpetersäure. Großtechnisch wird Ammoniak durch das Haber-Bosch-Verfahren dargestellt, dabei werden Stickstoff und Wasserstoff mit Hilfe eines Katalysators bei höherer Temperatur und hohem Druck miteinander zur Reaktion gebracht (siehe Versuch 8 MWG). Ammoniak (NH3 ) stellt eine gasförmige schwache Base dar, die sich aufgrund von Wasserstoffbrückenbindungen gut in Wasser löst. Durch katalytische Ammoniakverbrennung und weiterer Oxidation ist es möglich, Salpetersäure großtechnisch darzustellen. Salpetersäure (HNO3 ) ist eine starke, oxidierende Säure, die sich mit Wasser bis zu einem Gehalt von 65% mischt. Konzentriertere Mischungen (rauchende Salpetersäure) beinhalten zu einem beträchtlichen Teil gasförmige Stickoxide. Nitrate (die Salze der Salpetersäure) sind allesamt gut wasserlöslich, so dass natürlich vorkommende Nitrate recht selten und nur in extrem trockenen Gebieten zu finden sind (Chilesalpeter: NaNO3 ). Als schweres Homologes des Stickstoffs besitzt auch der Phosphor die Möglichkeit Oxidationsstufen zwischen -3 und +5 auszubilden, wobei +3 und +5 die bei weitem Häufigsten darstellen. Elementarer Phosphor kommt in vier farblich unterschiedlichen Modifikationen vor. Die gängigste Modifikation, roter Phosphor, ist amorph und findet Verwendung in Streichhölzern. Weißer Phosphor ist aus P4 -Tetraedern aufgebaut, leicht flüchtig und sehr giftig. Aufgrund seiner Brennbarkeit wird er unter Wasser aufbewahrt. Die thermodynamisch stabilste Form ist der schwarze Phosphor, der einen schichtartig-kristallinen Aufbau besitzt. Der violette Phosphor ist ebenso eine kristalline Modifikation, jedoch mit Raumnetzstruktur. P4 O10 (Phosphorpentoxid) ist das stabilste Oxid des Phosphors. Es ist molekular in einem adamantanartigen Gerüst aufgebaut und sehr hygroskopisch. Beim Lösen in Wasser entsteht die ortho-Phosphorsäure (H3 PO4 ), eine dreiprotonige Säure, die bei Wasserentzug eine Kondensationsreaktion zur meta-Phosphorsäure HPO3 (= H3 P3 O9 ) eingeht. H3 PO4 wirkt in ihrer ersten Protolysestufe als starke Säure (pKS ≈ 2), in der zweiten nur noch als schwache (pKS ≈ 7). Die dritte Protolysestufe ist extrem schwach (pKS ≈ 12) und daher auch nur im Alkalischen zu erreichen. Die bevorzugten Oxidationsstufen des Elements Arsen sind +5, +3 und etwas seltener -3. Eine der häufigsten Arsenverbindungen ist das hochgiftige Arsentrioxid As2 O3 (Arsenik), das beim Verbrennen von Arsen an Luft entsteht. As(V) ist relativ stabil, vor allem in Form der zur Phosphorsäure H3 PO4 analogen Arsensäure H3 AsO4 und ihrer Salze. Genau wie Arsen kommt Antimon hauptsächlich in den Oxidationsstufen +3, +5 und -3 vor, wobei die beiden letztgenannten eher selten sind. Das wichtigste Antimonerz ist der Grauspießglanz Sb2 S3 . Auch Antimon verbrennt an Luft wie Arsen nur zum Antimontrioxid Sb2 O3 . Im Gegensatz zur Phosphor- und Arsensäure ist die Antimon(V)säure der Formel H3 SbO4 nicht in freiem Zustand bekannt, sondern nur ihr ”Dihydrat”, der Summenformel H[Sb(OH)6 ] (≡ H3 SbO4 ⋅ 2 H2 O). In wässriger Lösung kann Sb3+ mit H2 S als orangefarbenes Antimonsulfid Hauptgruppenelemente 59 gefällt werden, welches sich mit der Zeit in die graue, thermodynamisch stabilere Modifikation von Sb2 S3 umwandelt (vgl. Ostwaldsche Stufenregel). Bismut ist das schwerste Element der 15. Gruppe und kommt eigentlich ausschließlich in der Oxidationsstufe +3 vor (+5 und -3 sind extrem selten). Ebenso, wie bei Thallium und Blei ist es mit zunehmender Periodennummer immer schwieriger das entsprechende s-Orbital zu ”entvölkern”. 1.3.6 Die 16. Gruppe (= 6. Hauptgruppe): Chalcogene Die 16. Gruppe des Periodensystems (sechste Hauptgruppe) enthält die Elemente Sauerstoff (O), Schwefel (S), Selen (Se), Tellur (Te) und das radioaktive Polonium (Po). Diese werden auch als Chalcogene bezeichnet, was soviel bedeutet wie Erzbildner. Der Sauerstoff ist mit ca. 50% Anteil an der Erdhülle das häufigste Element der Erdrinde. Er kommt zu ca. 20% als O2 in der Erdatmosphäre vor, sowie als Ozon O3 in einer dünnen Schicht der Stratosphäre, die einen großen Teil der solaren UV-Strahlung abblockt. Der größte Massenanteil des Sauerstoffs ist allerdings in den Gesteinen der Erdrinde als Oxid gebunden, in der Hauptsache an Silicium und Aluminium. Die bei weitem wichtigste Sauerstoffverbindung ist jedoch das Wasser (siehe Kapitel 1.4). In den meisten Verbindungen besitzt Sauerstoff die Oxidationsstufe –2, wobei in Peroxiden auch die Oxidationsstufe –1 mittels einer O–O-Bindung beobachtet wird, die aber leicht aufzuspalten ist. Aufgrund seiner Stellung in der 6. Hauptgruppe ist die niedrigstmögliche Oxidationszahl des Schwefels –2 im Sulfid-Anion S2– . Das Sulfid ist das Anion des Schwefelwasserstoffs, welcher in der ersten Protolysestufe eine schwache und in der zweiten Protolysestufe eine sehr schwache Säure darstellt. Sulfide bilden mit einigen (Schwer-)Metallen sehr schwer lösliche Sulfide. Als ”weiches” Anion (siehe HSAB-Prinzip, Versuch 6 NGE) besitzt S2− eine sehr große Affinität zu weichen Kationen, v. a. Übergangsmetallkationen, die nicht nur schwerlösliche Verbindungen, sondern auch recht stabile Komplexe mit Sulfid-Anionen bilden. Weitere stabile Oxidationsstufen sind +4 (z. B. in SO2 ) und +6 (z. B. in H2 SO4 ). In der Natur kommt Schwefel in Form von Metallsulfiden und Metallsulfaten aber auch gediegen (= elementar) vor. Fester elementarer Schwefel besitzt eine gelbe Farbe und kommt in zwei verschiedenen Modifikationen vor, deren zentrales Bauteil ein S8 -Ring in Kronenform ist. Schwefel schmilzt bei ≈ 120○ C sowohl unter Beibehaltung der S8 -Ringe (λ-Schwefel) als auch unter Bildung von Ringen unterschiedlicher Größe Sn (π-Schwefel, n = 6 − 26) und hochmolekularer Ketten, welche auch teilweise zu Ringen geschlossen sind Sx (µ-Schwefel, x = 103 − 106). Bei ≈ 445○ C verdampft Schwefel unter Bildung immer kleinerer molekularer (teils ringförmiger) Einheiten. An Luft verbrennt Schwefel zum SO2 , dem Anhydrid der schwefligen Säure H2 SO3 . Eine Lösung von SO2 in H2 O reagiert zu einem geringen Prozentsatz zur schwefligen Säure H2 SO3 , gemäß SO + H2 O ⇌ H2 SO3 . Die schweflige Säure ist zumindest in der ersten Protolysestufe noch als starke Säure zu bezeichnen (pKS1 = 1, 90), daher richtet von Industrieanlagen freigesetztes SO2 als saurer Regen auch große Schäden an. Die Weiterreaktion des SO2 zum SO3 , dem Anhydrid der Schwefelsäure H2 SO4 , funktioniert nur gut unter Verwendung eines Katalysators (vgl. Schwefelsäuredarstellung). Sulfat ist das Anion der Schwefelsäure H2 SO4 , die in konzentrierter Form eine sehr starke Säure darstellt, die ein nicht allzu hohes Oxidationspotential besitzt und sehr wasseranziehend (≡ hygroskopisch) ist. Aufgrund der geringen pKS -Werte beider Protolysestufen ist es nicht möglich, 60 Hauptgruppenelemente sie durch Zugabe einer starken Säure aus ihren Salzen auszutreiben, daher lassen sich die meisten schwerlöslichen Sulfate auch durch Zugabe konzentrierter Salzsäure nicht auflösen und können somit gut für Nachweisreaktionen eingesetzt werden. Die Elemente Selen und Tellur besitzen ebenso wie ihre kleineren Homologen nichtmetallischen (bei Te halbmetallischen) Charakter und weisen dieselben hauptsächlichen Oxidationsstufen wie der Schwefel auf. Die Säuren der höchsten Oxidationsstufe +6, H2 SeO4 und H6 TeO6 (eine Koordinationszahl von 4 um das vergleichsweise große Te6+ Kation ist zu niedrig, daher werden noch zwei Wassermoleküle zusätzlich angelagert, vgl. Antimon) werden jedoch seltener gebildet, wie jene der Oxidationsstufe +4. Diese weisen dann mit den Formeln H2 SeO3 und H2 TeO3 eine ψ 1 -tetraedrische Geometrie um das zentrale Se4+ bzw. Te4+ -Kation auf. 1.3.7 Die 17. Gruppe (= 7. Hauptgruppe): Halogene Die 17. Gruppe des Periodensystems (siebte Hauptgruppe) enthält die nichtmetallischen Elemente Fluor (F), Chlor (Cl), Brom (Br), Iod (I) und das radioaktive Astat (At). Diese werden auch als Halogene bezeichnet, was soviel bedeutet wie Salzbildner. Elementares Fluor, ein schwach gelblich-grünes Gas, ist das reaktionsfreudigste Nichtmetall, das stärkste Oxidationsmittel und besitzt die höchste Elektronegativität aller Elemente. Die hohe Reaktivität beruht nicht zuletzt auf der niedrigen Bindungsdissoziationsenergie des molekularen Fluors F2 . Die häufigsten fluoridhaltigen Mineralien sind der Flussspat (CaF2 , Fluorit), der Kryolith (Na3 AlF6 , wichtige Verbindung bei der Aluminiumherstellung) und der Fluorapatit Ca5 F[PO4 ]3 . Das Fluorid F– ist das Anion des Fluorwasserstoffs HF, dessen wässrige Lösung Flusssäure genannt wird. Wasserfreier Fluorwasserstoff siedet bei ca. 20○ C und besitzt aufgrund der Ausbildung von Wasserstoffbrückenbindungen eine unbegrenzte Mischbarkeit mit Wasser. Betrachtet man die Säurestärke von Flusssäure, so ist diese im Vergleich zu ihren ”größeren Brüdern” (wässrige Lösungen von) HCl, HBr und HI eine eher schwache Säure. Der Grund hierfür sind die zu den Wassermolekülen ausgebildeten Wasserstoffbrückenbindungen, welche bei einer Deprotonierung aufgebrochen werden müssen. Nichtsdestotrotz stellt die Flusssäure durch die hohe Affinität des F– -Anions zu Ca2+ -Kationen eine Bedrohung im Körper dar. Elementares Chlor ist ein gelbgrünes Gas, welches ebenfalls sehr reaktiv ist (gutes Oxidationsmittel). Chlor kann die Oxidationsstufen –1 (bevorzugt), +1, +3, +5, +7 annehmen (gerade Oxidationszahlen sind extrem selten), wobei es mineralisch immer als Chlorid Cl– vorkommt; Steinsalz (NaCl, Halit), Sylvin (KCl). Eine der wichtigsten Chlorverbindungen ist der gasförmige Chlorwasserstoff HCl, dessen wässrige Lösung Salzsäure genannt wird. Der Chlorwasserstoff ist im Gegensatz zur Salpetersäure und zur Schwefelsäure eine nicht-oxidierende Säure, sondern zeigt bestenfalls (im Fall hoch oxidierter Metallate) eine reduzierende Wirkung, worin das Cl– zum elementaren Chlor oxidiert wird. Das Chlor bildet in jeder seiner positiven Oxidationsstufen auch Sauerstoffsäuren, für +1 die hypochlorige Säure HClO (= HOCl), für +3 die chlorige Säure HClO2 (= HOClO), für +5 die Chlorsäure HClO3 (= HOClO2 ) und für +7 die Perchlorsäure HClO4 (= HOClO3 ). Die hypochlorige Säure entsteht beim Einleiten von Chlor in Wasser als Resultat einer Disproportionierungsreaktion gemäß: Cl2 + H2 O ⇌ HCl + HOCl. Die Perchlorsäure ist in wässriger Lösung die stärkste bekannte Säure (zusammen mit HI) und bildet die beständigsten Sauerstoffsalze Hauptgruppenelemente 61 des Chlors. Brom ist eines der beiden bei Raumtemperatur flüssigen Elemente (Quecksilber ist das andere). Es besitzt eine rote Farbe und einen recht hohen Dampfdruck. Daher ist der Gasraum in Gefäßen, die Brom beherbergen, auch immer mit Bromdampf gefüllt. Mit einer Dichte von 3, 14 g ⋅ cm−3 ist es rund 3× so schwer wie Wasser. Ebenso wie seine beiden leichteren Homologen ist auch das Brom ein recht gutes Oxidationsmittel, allerdings nicht ganz so stark wie Chlor oder Fluor. Natürlich vorkommendes Brom findet man stets in der Oxidationsstufe -1, (z. B. Bromargyrit AgBr). Die entsprechende Bromwasserstoffsäure HBr ist gasförmig und mit einem pKS -Wert von -8,9 um fast 2 pKS -Einheiten stärker als Chlorwasserstoff. Ebenso wie das Chlor kann auch Brom die positiven Oxidationsstufen +1, +3, +5 und +7 annehmen, wobei die Säuren der Oxidationsstufen +1 (hypobromige Säure) und +3 (bromige Säure) nicht besonders stabil sind. Die Bromsäure HBrO3 ist ein ebenso kräftiges Oxidationsmittel wie die Chlorsäure und zerfällt beim Eindampfen in Br2 und O2 . Die Perbromsäure lässt sich lediglich mit Hilfe starker Oxidationsmittel aus Bromaten gewinnen und explodiert beim Versuch, sie zu konzentrieren. Das Iod ist, anders als seine kleineren Homologen, bei Raumtemperatur ein (grauschwarzer, leicht violetter) Feststoff und besitzt einen geringen metallischen Charakter, ist aber nichtsdestotrotz als Nichtmetall anzusehen. Seine Reaktivität und Oxidationskraft sind auch wesentlich geringer als der leichteren Halogene, so kommt Iod in der Natur nicht nur als Iodid I– , sondern auch in Iodaten IO–3 vor. Elementares Iod bildet zusammen mit Iodid Addukte, die zu Ionen der Art (n× I2 ) + I– führt (z. B. I–3 , I–5 ) welche sich in die helicalen Amylosemoleküle der Stärke einlagern und dort eine tiefblaue Farbe hervorrufen. In Wasser und einigen organischen Lösungsmittel löst sich Iod mit brauner Farbe (in Wasser nur sehr geringe Löslichkeit), während eine Iodlösung mit aromatischen Kohlenwasserstoffen (z. B. Benzol) eine rote und mit chlorierten Kohlenwasserstoffen (z. B. CHCl3 ) eine violette Farbe besitzen. Auch Iod besitzt zu den anderen Halogenen analoge Sauerstoffsäuren, wobei hier die hypoiodige HIO und die iodige Säure HIO2 nicht besonders stabil sind. Die Iodsäure HIO3 ist wiederum ein kräftiges Oxidationsmittel, kann aber als einzige der Iod-Sauerstoffsäuren wasserfrei gewonnen werden. Die Periodsäure besitzt nur in ihrer meta-Form die Summenformel HIO4 . Da eine Koordination von 4 Oxid-Anionen um das große, hochgeladene Iod-Kation keine gute Abschirmung bietet, lagert die Periodsäure noch zwei Wassermoleküle um sich herum an und bildet die ortho-Periodsäure H5 IO6 mit oktaedrischer Sauerstoffkoordination um das I7+ -Kation (Zwischenstufe H3 IO5 ). Die ortho-Periodsäure zeigt die Tendenz Kondensationsreaktionen einzugehen, um die angelagerten Wassermoleküle wieder abspalten zu können. Hieraus resultieren die Di-, Tri- und Polyperiodsäuren, die alle die Molekülgestalt kantenverknüpfter Oktateder besitzen. 1.3.8 Die 18. Gruppe (= achte Hauptgruppe, auch 0. Gruppe): Edelgase Die 18. Gruppe des Periodensystems (achte Hauptgruppe) enthält die nichtmetallischen Elemente Helium (He), Neon (Ne), Argon (Ar), Krypton (Kr), Xenon (Xe) und das radioaktive Radon (Rn). Alle diese Elemente sind bei Zimmertemperatur gasförmig und besitzen sehr niedrige Kondensations- und Festpunkte. Aufgrund ihrer Reaktionsträgheit (Helium, Neon und Argon bilden keine Verbindungen) werden sie Edelgase genannt. Mit sehr starken Oxidationsmitteln können Fluoride (von Krypton und Xenon) und Oxide (von Xenon) erhalten werden. 62 Hauptgruppenelemente 1.4 Das Wasser Wasser ist nicht, wie man klassisch glaubte, ein Element, sondern eine Verbindung aus Wasserstoff und Sauerstoff mit der Formel H2 O. Wasser besitzt spezielle Eigenschaften, die Leben darin ermöglichen. Die erste Anomalie des Wassers ist bereits im Zustandsdiagramm (Abbildung 1.1) zu erkennen. Normalerweise besitzt eine Schmelzkurve eine positive Steigung, d.h. bei einer festgelegten Temperatur und steigendem Druck werden die meisten Stoffe fest. Die Schmelzkurve des Wassers besitzt eine negative Steigung, d.h. bei Abb. 1.1: Das Phasendiagramm des Wassers einer festgelegten Temperatur und steigendem Druck wird Wasser flüssig (Grundlage des Schlittschuh- und Skilaufens). Im festen Zustand (Eis) besitzt Wasser eine Gitterordnung, in der die Dipolmoleküle des Wassers in einer festen Anordnung analog zu Siliciumdioxid-Modifikationen vorliegen. Das bedeutet, dass das Sauerstoff-Zentralatom sowohl von zwei via kovalenter Bindung direkt gebundener Wasserstoffatome mit kurzem Abstand als auch von zwei Wasserstoffatomen anderer Wassermoleküle via Wasserstoffbrückenbindung mit langem Abstand umgeben ist. Das Sauerstoffatom besitzt somit eine Koordinationszahl von vier oder besser 2+2. Durch die beiden Wasserstoffatome mit langem Abstand weist die Kristallstruktur eine recht geringe Dichte (ρ ≈ 0, 92 g⋅cm−3 ) auf. Schmilzt das Eis nun, bricht die Kristallstruktur teilweise auf, die verhältnismäßig langen Wasserstoffbrückenbindungen verkürzen sich, die Dichte des Wassers nimmt zu bis zu einer Temperatur von 4 °C (ρ4○ C ≈ 1 g⋅cm−3 ). Steigt die Temperatur weiter, nimmt auch die Dichte wieder ab, allerdings nur bis zu einem Wert von ρ = 0, 96 g⋅cm−3 am Siedepunkt von 100 °C. D.h. Eis weist immer eine geringere Dichte und damit ein größeres Volumen auf, als flüssiges Wasser. Dies hat mehrere Bedeutungen für die Umwelt. Zum Einen besitzt flüssiges Wasser die Fähigkeit, Spalten und Ritzen von Gestein aufzufüllen. Beim Gefrieren dehnt sich das Wasser dann auf ca. 111% seines ursprünglichen Volumens aus und vergrößert die Risse oder sprengt gar das Gestein. Zum Anderen ist diese Dichteanomalie des Wassers lebensnotwendig für alle Wasserlebewesen, da das leichtere Eis sich immer an der Wasseroberfläche befindet, somit Flüsse, Seen, Teiche etc. meist nicht ganz bis zum Grund zufrieren und sich dort immer noch eine Temperatur von 4 °C hält. So ist auch das Zustandsdiagramm zu erklären, denn erhöht man den Druck auf das Eis (verdichtet es also), geht das Eis in seinen dichteren, nämlich flüssigen Zustand über. Hauptgruppenelemente 63 2 Aufgabenstellung, Versuchsanleitung und -auswertung 2.1 Reaktion von Alkalimetallen in H2 O 2.1.1 Aufgabenstellung Anhand dieser Reaktion soll die hohe Reaktivität der Alkalimetalle zusammen mit ihren recht niedrigen Schmelzpunkten beobachtet werden. 2.1.2 Versuchsanleitung Eine große Kunststoffschale wird zu ca. zwei Dritteln mit Wasser gefüllt und mit einigen Tropfen Phenolphthalein und etwas Spülmittel versetzt. Ein kleines Stück Lithium wird aus dem Paraffin genommen und in die Kunststoffschale gegeben (Geschlossener Abzug!!!). Das Verhalten des Metalls im Wasser wird dokumentiert. Dieses Experiment wird für Natrium wiederholt, wobei Natriumstücke eine Kantenlänge von 0,5 cm nicht überschreiten dürfen und ggf. von der Oxidhaut befreit werden müssen. (Vorsicht, sehr heftige, teilweise explosionsartige Reaktion zu erwarten; Geschlossener Abzug!!!). 2.1.3 Versuchsauswertung ● Stellen Sie die Gleichungen für die beiden Reaktionen auf und erklären Sie damit die beobachtete Farbänderung des mit Phenolphthalein versetzten Wassers. ● ● Warum ist es zu gefährlich diesen Versuch mit Kalium durchzuführen? Ermitteln Sie die Standardpotentiale der Alkali-Metalle Li, Na, K, Rb und Cs aus der Literatur; vergleichen Sie diese mit den beobachteten bzw. erwarteten Reaktionsgeschwindigkeiten/-intensitäten. ● Warum nimmt das Natrium nach wenigen Sekunden im Wasser Kugelform an? 2.2 Verbrennung von Magnesium in CO2 2.2.1 Aufgabenstellung Anhand dieser Reaktion soll die hohe Verbrennungstemperatur des Magnesiums erkannt werden. 2.2.2 Versuchsanleitung In einem zu etwa zwei Drittel mit Sand gefüllten Standzylinder wird etwas zerstoßenes Trockeneis gegeben (Trockeneis immer ausserhalb des Dewar-Transportbehälters zerkleinern!) und ca. zwei Minuten gewartet. Danach sollte die Atmosphäre im Standzylinder nur noch aus CO2 bestehen (Glimmspanprobe). Ein Stück Magnesiumband wird entzündet (vorher an der zu entzündenden Stelle anschmirgeln) und langsam mit der Tiegelzange in den Standzylinder abgelassen. 2.2.3 Versuchsauswertung ● ● Formulieren Sie die Reaktionsgleichung. Erklären Sie, warum elementares Magnesium so reagieren kann, andere Elemente oder brennbare Stoffe aber nicht. 64 Hauptgruppenelemente 2.3 Veresterung Anorganischer Säuren 2.3.1 Aufgabenstellung Bei diesem Versuch soll gezeigt werden, dass auch Ester anorganischer Säuren existieren. 2.3.2 Versuchsanleitung In eine Porzellanschale wird ein wenig feste Borsäure gegeben, die mit Methanol und einigen Tropfen konzentrierter Schwefelsäure vermischt wird. Diese Mischung wird angezündet. 2.3.3 Versuchsauswertung ● ● ● Stellen Sie die Reaktionsgleichung auf. Um welchen Reaktionstyp handelt es sich hierbei? Welchen Vorteil hat die Verwendung von H2 SO4 im Vergleich zu anderen Säuren? Handelt es isch bei der Schwefelsäure um einen ”reinen” Katalysator? 2.4 Anionen des Kohlenstoffs 2.4.1 Aufgabenstellung Bei diesem Versuch soll die starke Basenwirkung des Carbidanions gezeigt werden. 2.4.2 Versuchsanleitung In ein trockenes Reagenzglas werden ein bis zwei Spatelspitzen CaC2 gegeben. Nach der Zugabe von einigen Tropfen Wasser wird ein mit einer Pipette versehener durchbohrter Stopfen aufgesteckt und das austretende Gas entzündet wird. 2.4.3 Versuchsauswertung Formulieren Sie die Reaktionsgleichung der Gasentstehung und der Verbrennung und erklären Sie die Gasentstehung mit Hilfe der pKS -Werte des Säure-Base Paares C2 H2 /C2– 2 und des entsprechenden Reaktionspartners. 2.5 Hydrolyse von Nichtmetallverbindungen 2.5.1 Aufgabenstellung Bei diesem Versuch soll durch die Hydrolyse geklärt werden, in welchen Oxidationsstufen die Atome in der Ausgangsverbindung vorliegen. 2.5.2 Versuchsanleitung In einem Becherglas wird ein wenig Phosphorpentachlorid extrem vorsichtig mit Wasser versetzt. Nach Beendigung der Reaktion wird ein wenig der entstandenen Lösung in einem Reagenzglas mit Silbernitratlösung versetzt und in einem anderen Reagenzglas mit AmmoniumheptamolybdatLösung. Im ersten Reagenzglas sollte ein farbloser bzw. weißer und im zweiten Reagenzglas ein gelber Niederschlag zu beobachten sein (manchmal auch erst nach einigen Minuten). Hauptgruppenelemente 65 2.5.3 Versuchsauswertung ● ● Stellen Sie die Reaktionsgleichungen für die Hydrolyse und die Nachweisreaktionen auf. Erklären Sie die Struktur von Phosphorpentachlorid mit Hilfe des VSEPR-Konzepts. 2.6 Oxidationsstufen von Nichtmetallen 2.6.1 Aufgabenstellung Bei dieser Reaktion sollen die verschiedenen Oxidationsstufen des Schwefels beobachtet werden. 2.6.2 Versuchsanleitung Zwei Reagenzgläser werden jeweils zur Hälfte mit Natriumthiosulfatlösung gefüllt. In das erste Reagenzglas wird konzentrierte Salzsäure gegeben, in das zweite wenige Tropfen Silbernitratlösung. 2.6.3 Versuchsauswertung ● Stellen Sie die Reaktionsgleichungen für die Reaktionen auf, die in den beiden Reagenzgläsern stattfinden. ● Zeichnen Sie die Lewis-Formel des Thiosulfatmoleküls und beschriften Sie die einzelnen Atome mit deren Oxidationsstufen. 2.7 Oxidationswirkung von Halogenaten 2.7.1 Aufgabenstellung Bei dieser Reaktion soll die Oxidationswirkung von Bromat beobachtet werden. 2.7.2 Versuchsanleitung Es werden jeweils etwa 5 ml einer ca. 2 %igen Kaliumbromat-Lösung und einer ebenfalls 2 %igen Mangan(II)-Sulfat-Lösung hergestellt. Ein Reagenzglas wird ca. 1 cm hoch mit der Kaliumbromat-Lösung gefüllt und die gleiche Menge Mangan(II)-Sulfat-Lösung wird zugegeben. Danach wird tropfenweise (Pasteurpipette) konzentrierte Schwefelsäure zugegeben bis eine deutliche Farbänderung der Lösung sichtbar wird. Ein kleiner Teil dieser Lösung wird in ein zweites Reagenzglas dekantiert und schrittweise mit demineralisiertem Wasser unter regelmäßigem Schütteln verdünnt. Dokumentieren Sie Ihre Beobachtungen. 2.7.3 Versuchsauswertung ● Stellen Sie die Reaktionsgleichungen für die Reaktionen auf, die in den beiden Reagenzgläsern stattfinden und bestimmen die Oxidationsstufen der einzelnen Atome. ● Zeichnen Sie die Lewis-Formel des Bromatanions und beschriften Sie die einzelnen Atome mit deren Oxidationsstufen. 66 Hauptgruppenelemente 3 Geräte und Chemikalien Eine Geräteliste liegt im Platz aus Chemikalien Lithium Natrium Phenolphthalein H2 (Produkt) Magnesium CO2 (Trockeneis) H3 BO3 Methanol H2 SO4 CaC2 C2 H2 (Produkt) PCl5 Ag(NO3 ) (NH4 )6 Mo7 O24 ⋅ 4 H2 O Na2 S2 O3 HClaq KBrO3 MnSO4 ⋅ 4 H2 O Gefahrenbezeichnung Gefahr (GHS02, 05) H-Sätze EUH014, 260, 314 P-Sätze 301+330+331, 402+404, 305+351+338, 280 Gefahr (GHS02, 05) EUH014, 260, 314 301+330+331, 305+351+338, 280 Gefahr (GHS06) 350, 341, 361f 201, 281, 308+313 Gefahr (GHS02) 220 210, 377, 381, 403 Gefahr (GHS02) 228, 261, 252 210, 402+404 – – – Gefahr (GHS05) 360FD 201, 308+313 Gefahr (GHS02, 06) 225, 331, 311, 301, 370 210, 233, 280, 302+352 Gefahr (GHS05) 314, 290 280, 301+330+331, 309, 310, 305+351+338 Gefahr (GHS02) 260 223, 231+232, 370+378, 422 Gefahr (GHS02) EUH006, 220 210, 377, 381, 403 Gefahr (GHS06) EUH014, EUH029, 304+340, 301+330+331, 330, 302, 373, 314 305+351+338, 280 Gefahr (GHS03, 05, 09) 272, 314, 410 273, 280, 301+330+331, 305+351+338 – – – – – – Gefahr (GHS05) 314, 335, 290 301+330+331, 305+351+338, 280 Gefahr (GHS03, 06) 272, 301, 350 201, 309+310 Achtung (GHS07, 09) 373, 411 273, 314 4 Literatur [1] A. F. Holleman, E. Wiberg: Lehrbuch der Anorganischen Chemie, 102. Aufl., Walter de Gruyter Verlag, 2007. [2] M. Binnewies, M. Jäckel, H. Willner, G. Rayner-Canham: Allgemeine und Anorganische Chemie, 1. Auflage, Spektrum Verlag, 2004. [3] G. Jander, E. Blasius: Lehrbuch der Analytischen und Präparativen Anorganischen Chemie, 16. Auflage, S. Hirzel Verlag, 2006. Nebengruppenelemente 67 Versuch 6 (NGE) Nebengruppenelemente 1 Theorie 1.1 Übersicht Als Übergangsmetalle werden die Elemente der Gruppen 3-12 (Nebengruppen 3-10, 1 und 2) bezeichnet. Bei diesen werden nach dem Befüllen der s-Orbitale die d-Orbitale der vorigen Periode mit Elektronen bestückt. Daraus ergeben sich die folgenden Elektronenkonfigurationen der Elemente der Übergangsmetalle (siehe Versuch 4 PSE): 3. Nebengruppe Sc: 4s2 3d1 Y: 5s2 4d1 La: 6s2 5d1 8. Nebengruppe Fe: 4s2 3d6 Ru: 5s1 4d7 ∗ Os: 6s2 5d6 4. Nebengruppe Ti: 4s2 3d2 Zr: 5s2 4d2 Hf: 6s2 5d2 9. Nebengruppe Co: 4s2 3d7 Rh: 5s1 4d8 ∗ Ir: 6s2 5d7 5. Nebengruppe V: 4s2 3d3 Nb: 5s1 4d4 ∗ Ta: 6s2 5d3 10. Nebengruppe Ni: 4s2 3d8 Pd: 5s0 4d10 ∗ Pt: 6s1 5d9 ∗ 6. Nebengruppe Cr: 4s1 3d5 ∗ Mo: 5s1 4d5 ∗ W: 6s2 3d4 1. Nebengruppe Cu: 4s1 3d10 ∗ Ag: 5s1 4d10 ∗ Au: 6s1 5d10 ∗ 7. Nebengruppe Mn 4s2 3d5 Tc 5s1 4d6 ∗ Re 6s2 5d5 2. Nebengruppe Zn: 4s2 3d10 Cd: 5s2 4d10 Hg: 6s2 5d10 Unregelmäßigkeiten in den Elektronenkonfigurationen sind ein Resultat einer stabilen Konfiguration durch halb- oder vollgefüllte d-Orbitale (6. und 1. Nebengruppe) und weiterer, hier nicht besprochener Effekte. ∗ Die Lappen der d-Orbitale (d = diffuse) sind weit außen liegend und daher trotz des Umstandes, dass sie eigentlich eine Periode tiefer liegen in der Lage an Bindungen teilzunehmen. Diese räumlich näher am Kern befindliche Anordnung der d-Elektronen hat jedoch zur Folge, dass bei Ionisierung immer zuerst die Elektronen der s-Orbitale entfernt werden. Außer den Elementen der 3. Gruppe können die meisten Übergangsmetalle in verschiedenen Oxidationsstufen vorkommen, deren Stabilität nicht zuletzt von der Koordinationsumgebung des entsprechenden Metallkations abhängen. Z. B. besitzt Mn2+ die stabile Elektronenkonfiguration d5 (halbgefüllte d-Schale) und sollte daher stabiler sein als das Mn4+ -Kation mit d3 -Konfiguration, jedoch ist in oktaedrischer Koordinationsumgebung (siehe Versuch 12 KOG) die d3 -Konfiguration gegenüber der d5 -Konfiguration begünstigt. Die Chemie der Übergangsmetalle wird daher hauptsächlich durch das Verhalten der d-Orbitale bestimmt. 1.2 Ausgewählte Elemente 1.2.1 Vanadium Elementares Vanadium besitzt die Elektronenkonfiguration [Ar]3d3 4s2 und kann außer 0 noch die Oxidationsstufen +2, +3, +4 und +5 annehmen. Die wichtigsten vanadiumhaltigen Mineralien sind der Patronit VS4 und der Vanadinit Pb5 Cl[VO4 ]3 (isomorph zum Apatit Ca5 (OHF)[PO4 ]3 ). In jeder der vier Oxidationsstufen wird die oktaedrische Koordination bevorzugt, aber auch 68 Nebengruppenelemente tetraedrische und trigonal-bipyramidale Strukturen sind zu beobachten. Die Oxidationsstufe +5 wird beispielsweise in wässriger Lösung in Form des Vanadyl-Kations VO+ 2 ausgebildet, welches zusätzlich noch von vier Wasserliganden oktaedrisch koordiniert wird. Das [VO2 (H2 O)4 ]+ -Kation kann reduziert werden, wobei sich die Farbe je nach Oxidationsstufe des Vanadiums ändert: [VO2 (H2 O)4 ]2+ ⇌ VO(H2 O)5 ]2+ ⇌ [V(H2 O)6 ]3+ ⇌ [V(H2 O)6 ]2+ (+5, gelblich) (+4, blau) 2+ An Luft wird dann V (+3, grün) (+2, violett) wieder allmählich oxidiert, und die Farbspiele verlaufen rückwärts. 1.2.2 Chrom Die wichtigsten Chrommineralien sind Chromeisenstein FeCr2 O4 (Chromit, im Spinell-Typ) und Rotbleierz Pb[CrO4 ] (Krokoit). Die Elektronenkonfiguration von elementarem Chrom ist [Ar]3d5 4s1 , trotzdem ist die Oxidationsstufe +1 sehr selten. Die hauptsächlichen Oxidationsstufen des Chroms sind +3 und +6. In Oxidationsstufe +6 wirkt Chrom als sehr gutes Oxidationsmittel. Hierbei liegt das gelbe Tetraoxochromat(VI)-Anion vor (Chromat [CrO4 ]2– ) welches bei Säurezugabe eine Kondensationsreaktion eingeht und zum orangefarbenen Dichromat-Anion [Cr2 O7 ]2– wird: 2 [CrO4 ]2– + 2 H+ ⇌ [Cr2 O7 ]2– + H2 O. Die hohe Stabilität der Oxidationsstufe +3 ist hauptsächlich darauf zurückzuführen, dass die 3d3 -Elektronenkonfiguration im oktaedrischen Ligandenfeld sehr gut stabilisiert wird (siehe Versuch 12 KOG). Der im Wasser vorhandene violette Hexaquachrom-Komplex [Cr(H2 O)6 ]3+ ist fast nie zu beobachten, da schon der Austausch eines Wasserliganden gegen einen anderen Liganden (z. B. OH– , Cl– , SO2– 4 , etc.) eine Grünfärbung der Komplexe hervorruft. Der dabei ablaufende Vorgang wird Hydratisomerie genannt. In einer Lösung von CrCl3 ⋅ 3 H2 O beispielsweise finden folgende Ligandenaustauschprozesse statt: [CrCl3 (H2 O)3 ] ⋅ 3H2 O ⇌ [CrCl2 (H2 O)4 ]+ ⋅ 2H2 O + Cl– ⇌ [CrCl(H2 O)5 ]2+ ⋅ H2 O + 2Cl– ⇌ Cr(H2 O)6 ]3+ + 3Cl– dunkelgrün hellgrün hellgrün violett 1.2.3 Mangan Elementares Mangan besitzt die Elektronenkonfiguration [Ar]3d5 4s2 und kann in folgenden Oxidationsstufen vorkommen: +2 +3 [Ar]3d5 4s0 : [Ar]3d4 4s0 : +4 +5 +6 +7 [Ar]3d3 4s0 : [Ar]3d2 4s0 : [Ar]3d1 4s0 : [Ar]3d0 4s0 : 2+ in wässriger Lösung [Mn(H2 O)6 ] Hexaquamangan(II), blaßrosa 3+ in wässriger Lösung [Mn(H2 O)6 ] Hexaquamangan(III), granatrot, sehr oxidationsempfindlich in Wasser schwerlöslich (Braunstein MnO2 bzw. MnO(OH)2 ), braunschwarz 3– als Anion [MnO4 ] (Tetraoxo)Manganat(V), hellblau, disproportioniert sehr leicht 2– als Anion [MnO4 ] (Tetraoxo)Manganat(VI), dunkelgrün, disproportioniert leicht – als Anion [MnO4 ] (Tetraoxo)Manganat(VII) oder Permanganat, tiefviolett, stabil in wässriger Lösung, aber gutes Oxidationsmittel Das Permanganat-Anion lässt sich leicht reduzieren in alkalischer Lösung zu Braunstein (0 = 0,59 V) und in saurer Lösung zu Mangan(II) (0 = 1,51 V). Dieser Umstand (und natürlich die tiefviolette Farbe) lassen sich zum Nachweisen vorhandener [MnO4 ]– -Anionen gut nutzen. Außerdem findet das Permanganat als Maßlösung für Redoxtitrationen Verwendung. Mn4+ ist vor allem als Braunstein mit einer Zusammensetzung von MnO2 bis MnO(OH)2 (unterschiedlicher Nebengruppenelemente 69 Wassergehalt) bekannt. Mn4+ besitzt in Komplexen ausschließlich oktaedrische Koordinationsumgebung die bei einer d3 -Elektronenkonfiguration besonders stabil ist. Mn2+ geht mit fast allen Liganden (Ausnahme sind nur die ganz starken) high-spin-Komplexe ein. Neben oktedrischen sind bei Mn2+ auch tetraedrische Komplexe bekannt. Hierbei weisen die oktaedrischen Komplexe eine blassrosa Farbe, die tetraedrischen eine grüne Farbe auf. 1.2.4 Eisen Eisen ist das vierthäufigste Element in der Erdkruste und nach Aluminium das zweithäufigste Metall. Elementares Eisen besitzt die Elektronenkonfiguration [Ar]3d6 4s2 und kommt bevorzugt in den Oxidationsstufen +2 ([Ar]3d6 4s0 ) und +3 ([Ar]3d5 4s0 ) vor. Die wichtigsten Eisenmineralien sind Magneteisenstein (Magnetit) Fe3 O4 , Rot- und Brauneisenstein (Hämatit Fe2 O3 (rot) und Fe2 O3 ⋅ 1,5 H2 O (braun)) und Eisenkies (Pyrit FeS2 ). In wässriger Lösung (schwaches Ligandenfeld, high-spin-Komplexe) bietet Fe3+ die energetisch günstigste Konfiguration, beim Übergang zu starken Liganden mit hoher Feldaufspaltung (low-spin-Komplexe) ist Fe2+ deutlich stabiler. Physiologisch wichtig ist Eisen in der Oxidationsstufe +2, das im Blut (im HämoglobinMolekül) den elementaren Sauerstoff komplexiert und ihn an den Stellen, an denen er benötigt wird, wieder abgibt. Gelangt ein starker Komplexligand in die Blutbahn (CO, CN– ), bildet dieser einen sehr stabilen Eisen(II)-Komplex, blockiert die Hämoglobinmoleküle und der Sauerstoff wird nicht mehr transportiert (tödlich). Eine wässrige Lösung von Fe2+ besitzt eine grüne Farbe ([Fe(H2 O)6 ]2+ -Komplex). Eine wässrige Lösung von Fe3+ weist aufgrund der Bildung des Pentaquahydroxoeisen(III)-Komplexes [Fe(H2 O)5 (OH)]2+ (und höher polymerisierter Aggregate, sog. Isopolyoxo-Kationen) eine gelbe Farbe auf. Der Hexaquaeisen(III)-Komplex [Fe(H2 O)6 ]3+ ist mit einem pKS -Wert von 2,46 noch als starke Säure zu bezeichnen und existiert nur bei einem pH-Wert < 0 mit blassvioletter Farbe. Das Redoxsystem Fe2+ ⇌ Fe3+ + e– ist ein in der Analytik sehr beliebtes System, welches auch in alkalischer Lösung ein recht hohes Normalpotential besitzt. In saurer Lösung ist es möglich, mit Hilfe von Luftsauerstoff Fe2+ in Fe3+ zu überführen. 1.2.5 Cobalt Elementares Cobalt besitzt die Elektronenkonfiguration [Ar]3d7 4s2 und kommt bevorzugt in den Oxidationsstufen +2 ([Ar]3d7 4s0 ) und +3 ([Ar]3d6 4s0 ) vor. Cobalt ist für den Menschen als zentraler Bestandteil des Vitamins B12 lebenswichtig. Co2+ bildet sowohl oktaedrische (JahnTeller-Effekt im low-spin-Fall) als auch tetraedrische, sowie quadratisch-pyramidale Komplexe. Als Beispiel für letztere sind v.a. die Pentacyanocobaltat(II)-Komplexe [Co(CN)5 ]3– von Bedeutung, die zur Wahrung einer oktaedrischen Geometrie am Zentralatom eine Dimerisierung zum zweikernigen Cluster Decacyanodicobaltat(II) [Co2 (CN)10 ]6– durchführen können. Diese Co–CoBindung ist nicht besonders stark, und so können einige Lewis-Basen dazwischen eingelagert 3+ werden, z.B. das Peroxid-Anion O2– bevorzugt hingegen 2 oder selbst elementarer Sauerstoff. Co mit fast allen Liganden den low-spin Fall mit oktaedrischer Koordination. Als Test auf Cobalt eignet sich die sog. Boraxperle sehr gut, aufgrund der charakteristischen tiefen Blaufärbung des entsprechenden Cobaltborats. 70 Nebengruppenelemente 1.2.6 Nickel Elementares Nickel besitzt die Elektronenkonfiguration [Ar]3d8 4s2 und kommt bevorzugt in der Oxidationsstufe +2 ([Ar]3d8 4s0 ) vor. Wichtige Nickelerze sind Gelbnickelkies (Nickelblende, Millerit) NiS, Rotnickelkies (Nickelit) NiAs und Weißnickelkies (Chloanthit) NiAs2-3 . Ni2+ bildet im high-spin Fall oktaedrische Komplexe, mit starken Liganden (low-spin) jedoch quadratischplanare. Sowohl mit H2 O, als auch mit NH3 bildet Ni2+ oktaedrische Komplexe, den grünen Hexaquanickel(II)- [Ni(H2 O)6 ]2+ und den blauen Hexamminnickel(II)-Komplex [Ni(NH3 )6 ]2+ . Mit sterisch anspruchsvollen Liganden ist selbst bei recht ungünstiger Ligandenfeldaufspaltung eine tetraedrische Umgebung möglich. 1.2.7 Kupfer Elementares Kupfer besitzt eine Elektronenkonfiguration von [Ar]4s1 3d10 und kommt hauptsächlich in den Oxidationsstufen +1 (stabilste Oxidationsstufe im Festkörper) und +2 (stabilste Oxidationsstufe in wässriger Lösung) vor. Die wichtigsten Erze sind sulfidhaltige Verbindungen (”Kiese”, ”Glanze’) und das Rotkupfererz (Cu2 O, Cuprit). Elementares Kupfer überzieht sich an Luft sogleich mit einer dünnen Schicht an Cu2 O und erscheint dadurch ”kupferrot”, wobei das eigentliche Metall eine hellrote Farbe besitzt. Kupfermetall ist ein sehr guter elektrischer und Wärmeleiter. Cu(I) in wässriger Lösung ist nur mit starken oder sterisch sehr anspruchsvollen (= großen) Liganden stabil, ansonsten disproportioniert es zu elementarem Kupfer und Cu(II), da dieses eine erheblich größere Hydratationsenthalpie besitzt. Cu(I) bildet stabilisierbar, farblose Komplexe mit entweder tetraedrischer oder linearer (vgl. Ag(I)) Koordination (CN = 2 oder 4). Cu(II) bildet in wässriger Lösung eigentlich oktaedrische Komplexe, die meistens als quadratisch-planar bezeichnet werden, weil durch die 3d9 -Elektronenkonfiguration kein ideales, sondern ein Jahn-Teller-verzerrtes, elongiertes Oktaeder (eher als quadratische Bipyramide zu bezeichnen) vorliegt, in dem die beiden axialen Liganden recht weit vom zentralen Cu2+ -Kation entfernt sind (vgl. Versuch 12 KOG). Somit liegt hier sozusagen eine Koordinationszahl von 4+2 vor und der [Cu(NH3 )4 ]2+ -Komplex ist in Wirklichkeit ein Tetrammindiaquakupfer(II)-Komplex [Cu(NH3 )4 (H2 O)2 ]2+ . 1.2.8 Quecksilber Elementares Quecksilber besitzt die Elektronenkonfiguration [Xe]6s2 5d10 und kommt außer in der Oxidationsstufe +2 auch in der Oxidationsstufe +1 vor, dann allerdings als zweiatomige ○ molekulare Spezies Hg2+ 2 . Es ist das einzige bei Raumtemperatur flüssige Metall (Smp.: −38, 8 C), ist giftig, weist mit 13,6 g⋅cm−3 eine recht hohe Dichte auf und besitzt einen ausreichend hohen Dampfdruck, um beim Einatmen Vergiftungen hervorrufen zu können. Viele Metalle sind in elementarem Quecksilber löslich und bilden dabei Legierungen, die sog. Amalgame. Die meisten Element-Quecksilberverbindungen sind nicht etwa salzartig zu betrachten, sondern besitzen einen recht hohen kovalenten Anteil, bedingt auch durch die vergleichsweise hohe Elektronegativität des Quecksilbers von ca. 2. Das bekannteste Quecksilbererz ist das Zinnober HgS (rot). Das mit H2 S gefällte schwarze Quecksilbersulfid (Metacinnabarit) stellt die thermodynamisch weniger stabile Modifikation dar und wandelt sich in die rote Form um (vgl. Ostwald-Volmer Stufenregel). Nebengruppenelemente 71 1.2.9 Wolfram Elementares Wolfram besitzt, im Unterschied zu seinem kleineren Homologen Chrom die Elektronenkonfiguration [Xe]6s2 5d4 . In der Natur kommt Wolfram fast ausschließlich in der Oxidationsstufe +6 in Wolframaten [WO4 ]2– vor (Scheelit: CaWO4 , Wolframit: (MnFeII )WO4 und Stolzit: PbWO4 ). Glühdrähte für Glühlampen, Halogenlampen und Röntgenröhren bestehen aus elementarem Wolfram, da der hohe Schmelzpunkt von 3410○ C eine lange Lebensdauer verspricht. Eine teilweise Reduktion von Wolfram von Oxidationsstufe +6 nach +5/+4 führt zu gemischtvalenten Verbindungen und in diesen zu einer intensiven Blaufärbung (”Wolframblau”). 2 Aufgabenstellung, Versuchsanleitung und -auswertung 2.1 Komplexes Redoxverhalten von Übergangsmetallen 2.1.1 Aufgabenstellung Bei dieser Reaktion soll das Redoxverhalten von Mangan in wässriger Lösung beobachtet und ausgewertet werden. 2.1.2 Versuchsanleitung 500 mg Kaliumpermanganat werden in 150 ml Wasser gelöst. 5 ml dieser Lösung werden in einen Standzylinder eingebracht und mit Wasser auf 250 ml aufgefüllt. In einem Becherglas werden 10 ml 50%ige Natronlauge mit 10 ml 1%iger Natriumformiatlösung vermischt. Diese Mischung wird in einem Schwung in den Standzylinder gegeben, und nicht umgerührt. Die alkalische NaHCOO-Lösung sinkt nach unten. Nach kurzer Zeit wird die violette KMnO4 -Lösung, von unten beginnend, blau. Die blaue Farbe schlägt allerdings nach einiger Zeit, wieder von unten nach oben fortschreitend, in eine grüne Farbe um. Sobald die untere Hälfte der Lösung grün gefärbt ist, wird diese mit 25 ml halbkonz. Schwefelsäure mit Hilfe einer 25 ml Vollpipette unterschichtet. Der untere Teil der grünen Farbschicht färbt sich rotbraun. Anschließend wird mit 25 ml einer einprozentiger Natriumsulfitlösung unterschichtet. Der untere Teil der rotbraunen Schicht wird entfärbt. 2.1.3 Versuchsauswertung ● ● Stellen Sie die Reaktionsgleichungen für alle ablaufenden Prozesse auf. Ermitteln Sie die Standardpotentiale für die stattfindenden Redoxprozesse (Literaturstudium). 2.2 Farbigkeit von Übergangsmetallen in Abhängigkeit vom Liganden 2.2.1 Aufgabenstellung Bei diesem Versuch soll die Farbigkeit von Übergangsmetallkomplexen in Abhängigkeit vom Liganden ermittelt werden. 72 Nebengruppenelemente 2.2.2 Versuchsanleitung 0,5 g wasserhaltiges NiCl2 wird in einem Becherglas in 25 ml demin. Wasser gelöst. Dann werden 5 vorbereitete Reagenzgläser zu je 1 /4 mit dieser NiCl2 -Lösung gefüllt. Reagenzglas 1 zeigt die grüne Farbe des Ausgangssalzes. Zu der Lösung in Reagenzglas 2 werden wenige Tropfen NH3 -Lösung zugegeben. In Reagenzglas 3 wird mehr Ammoniaklösung zugegeben. In Reagenzglas 4 wird NH3 -Lösung im Überschuss zugegeben. In das 5. Reagenzglas wird solange festes KCN zugegeben bis eine Farbänderung der Lösung auftritt. 2.2.3 Versuchsauswertung ● ● Formulieren Sie die Reaktionsgleichungen. Erklären Sie die verschiedenen Farben mit Hilfe der spektrochemischen Reihe und der Ligandenfeldtheorie (siehe Versuch 12 KOG). ● Berechnen Sie die Ligandenfeldaufspaltungsenergien ∆ für den Hexaaquanickel(II)- und den Hexaamminnickel(II)-Komplex. 2.3 Gemischtvalenz von Übergangsmetallverbindungen 2.3.1 Aufgabenstellung Bei diesem Versuch soll der Einfluss von Gemischtvalenz auf die Farbigkeit von Übergangsmetallverbindungen studiert werden. 2.3.2 Versuchsanleitung a) Zu einer wässrigen Lösung von FeCl3 werden im Reagenzglas wenige Tropfen einer verdünnten Lösung des gelben Blutlaugensalzes K4 [Fe(CN)6 ] gegeben. b) Zu einer schwach salzsauren Lösung von Na2 WO4 wird im Reagenzglas eine Zinkgranalie gegeben und kurze Zeit gewartet. 2.3.3 Versuchsauswertung ● ● ● Stellen Sie die Reaktionsgleichungen für die beiden Reaktionen auf. Geben Sie eine Erklärung für die Farbigkeit der gemischtvalenten Verbindungen. Nennen Sie für die verschiedenen charge-transfer Prozesse je ein Beispiel, welches nicht in diesen Versuchen vorkommt. Nebengruppenelemente 73 3 Geräte und Chemikalien Eine Geräteliste liegt im Platz aus Chemikalien KMnO4 NaOH Gefahrenbezeichnung Gefahr (GHS03, 08, 09) Gefahr (GHS05) NaHCOO H2 SO4 – Gefahr (GHS05) Na2 SO3 NiCl2 – Gefahr (GHS06, 09) NH3 aq Gefahr (GHS05, 09) KCN FeCl3 K4 [Fe(CN)6 ] Na2 WO4 Zink HClaq Gefahr (GHS06, 09) Achtung (GHS07) – Achtung (GHS07) – Gefahr (GHS05) H-Sätze 272, 302, 410 314, 290 P-Sätze 210, 273 301+330+331, 305+351+338, 280 – – 314, 290 280, 301+330+331, 309, 310, 305+351+338 – – 350i, 360D, 341, 372, 315, 273, 281, 302+352, 304+340 301+331, 334, 317, 410 314, 335, 400 301+330+331, 305+351+338, 280, 273 EUH032, 300+310+330, 410 273, 280, 302+352, 304+340 302, 315, 318, 317 280, 302+352, 305+351+338 412 273 302 – – – 314, 335, 290 301+330+331, 305+351+338, 280 4 Literatur [1] A. F. Holleman, E. Wiberg: Lehrbuch der Anorganischen Chemie, 102. Aufl., Walter de Gruyter Verlag, 2007. [2] M. Binnewies, M. Jäckel, H. Willner, G. Rayner-Canham: Allgemeine und Anorganische Chemie, 1. Auflage, Spektrum Verlag, 2004. [3] G. Jander, E. Blasius: Lehrbuch der Analytischen und Präparativen Anorganischen Chemie, 16. Auflage, S. Hirzel Verlag, 2006. Bindungstheorie und Physikalische Eigenschaften 75 Versuch 7 (BPH) Bindungstheorie und Physikalische Eigenschaften 1 Theorie 1.1 Die Elektronegativität Die Elektronegativität (EN) ist ein Maß für die Tendenz der Atome eines Elements Elektronen zu sich zu ”ziehen”. Sie ist eine berechnete Größe, die innerhalb einer Periode von links nach rechts zunimmt und innerhalb einer Gruppe von oben nach unten abnimmt. Das elektronegativste Element ist daher Fluor, das am wenigsten elektronegative (nicht-radioaktive) Element ist Caesium. Wichtig für die Art der Bindung zwischen zwei und mehreren Elementen ist daher die Elektronegativitätsdifferenz zwischen den verschiedenen Bindungspartnern. Faustregel: EN > 2 ⇒ Ionenbindung; EN < 2 ⇒ kovalente Bindung. 1.2 Die Ionenbindung Ist die Elektronegativitätsdifferenz zwischen den Reaktionspartnern größer als ca. 2, so gibt der weniger elektronegative Partner ein oder mehrere Valenzelektronen an den elektronegativeren Partner ab (in den allermeisten Fällen zur Erfüllung der Oktett-Regel), so dass positive geladene Ionen (= Kationen) des weniger elektronegativen Partners und negativ geladene Ionen (= Anionen) des elektronegativeren Partners entstehen. Zwischen diesen gegensätzlich geladenen Ionen herrschen Coulomb’sche Anziehungskräfte (siehe Kapitel 1.3.3). Dadurch ordnen sich Kationen und Anionen regelmäßig zueinander an; ein fester Stoff (sog. Salz) entsteht. Es werden zwei verschiedene Ordnungstypen unterschieden: 1. Die Nahordnung beschreibt die direkte sog. Koordinationsumgebung eines Kations mit Anionen bzw. eines Anions mit Kationen (erste Koordinationssphäre), manchmal aber auch noch die zweite oder dritte Koordinationssphäre. 2. Die Fernordnung beschreibt die regelmäßige Anordnung dieser Koordinationsumgebungen in die drei Raumrichtungen. Dies führt zu zwei Arten von Festkörpern: 1. Ein kristalliner Festkörper besitzt sowohl Nah- als auch Fernordnung 2. Ein amorpher Festkörper besitzt eine Nah-, aber keine Fernordnung Salzartige Verbindungen besitzen folgende Eigenschaften: hoher bis sehr hoher Schmelzpunkt, farbig oder farblos durchsichtig, Löslichkeit in H2 O, wenn auch manchmal nur sehr schlecht, im festen Zustand: praktisch nicht leitend, im geschmolzenen Zustand und in Lösung: leitend, aber mit Zersetzungserscheinungen (Elektrolyse). Beispiele für salzartige Stoffe: Steinsalz (NaCl), Kaliumnitrat (KNO3 ), Soda (Na2 CO3 ), Marmor (CaCO3 ), Natriumhydroxid (NaOH), Kalk (CaO). 76 Bindungstheorie und Physikalische Eigenschaften 1.3 Intermolekulare Kräfte 1.3.1 Van der Waals-Kräfte Dies ist die schwächste Art intermolekularer Wechselwirkungen. Hierbei handelt es sich um Kräfte zwischen unpolaren oder annähernd unpolaren Molekülen oder Atomen, die darauf zurückzuführen sind, dass innerhalb einer eigentlich gleichmäßigen Verteilung von Elektronen in der Hülle um das Atom oder Molekül Fluktuationen entstehen, so dass es für kurze Zeit zu einer Trennung der beiden Ladungsschwerpunkte kommt (fluktuierender Dipol). Dieser kann nun bei einem anderen Molekül oder Atom, das sich in der Nähe aufhält (die Kräfte nehmen mit wachsendem Abstand r zwischen den Teilchen sehr schnell ab, da die Wechselwirkungsenergie proportional zu r−6 ist) ein Dipolmoment induzieren (induzierter Dipol). Dies ist der sog. Dispersionseffekt. So entsteht elektrostatische Anziehung, die allerdings im Vergleich zu Wechselwirkungen zwischen Ionen sehr schwach ist. Daher besitzen Verbindungen, deren Moleküle nur über van der Waals-Kräfte zusammengehalten werden meist niedrige Schmelz- und Siedepunkte und eine hohe Flüchtigkeit. Je größer diese Moleküle sind, desto einfacher ist es, die Elektronen innerhalb der dann auch großen Hülle zu verschieben. Große Moleküle weisen somit eine größere Polarisierbarkeit auf. Van der Waals-Kräfte können auch entstehen, wenn ein Molekül mit permanentem Dipolmoment bei einem unpolaren Molekül oder Atom ein Dipolmoment induziert und dadurch dann die zuvor beschriebenen Anziehungskräfte zustande kommen (sog. Induktionseffekt). 1.3.2 Dipol-Dipol-Wechselwirkungen Die Wechselwirkungen zwischen zwei permanenten Dipolen (sog. Richteffekt) gehören auch in die Kategorie der van der Waals-Kräfte, werden hier aber gesondert behandelt. Im Vergleich zu einem fluktuierenden oder induzierten Dipolmoment ist das permanente Dipolmoment eines Moleküls abhängig von der Molekülgeometrie und weist damit eine bestimmte Vorzugsrichtung auf. Außerdem sind die Kräfte zwischen permanenten Dipolen deutlich stärker als zwischen fluktuierenden oder induzierten. Im Vergleich zum Induktionseffekt ist der Richteffekt abhängig von der Temperatur, was bedeutet, dass bei hoher Temperatur die zunehmende Bewegung der Moleküle ihrer Ausrichtung entgegen wirkt. Eine besonders starke Form von Dipol-DipolWechselwirkungen sind Wasserstoffbrückenbindungen. Wenn ein oder mehrere Wasserstoffatome an ein sehr elektronegatives Element gebunden (N, O, oder F) sind, entsteht aufgrund der hohen Elektronegativitätsdifferenz ein hohes Dipolmoment. Somit entstehen sehr starke Dipol-DipolWechselwirkungen zwischen gleichartigen Molekülen (z. B. zwischen H2 O- oder NH3 -Molekülen, aber auch zwischen H2 O- und NH3 -Molekülen). 1.3.3 Ionenbindung Eine Ionenbindung entsteht durch Coulomb’sche Anziehungskräfte zwischen (positiv geladenen) Kationen und (negativ geladenen) Anionen. Im Unterschied zu den Dipol-Dipol-Wechselwirkungen ist die Ionenbindung nicht gerichtet, sondern radialsymmetrisch. Sie stellt die stärkste intermolekulare Bindung dar. Die Kraft, die zwischen den Ionen wirkt, berechnet sich über das Bindungstheorie und Physikalische Eigenschaften Coulomb’sche Gesetz: F= zA ⋅ e ⋅ zK ⋅ e 1 ⋅ 4 ⋅ π ⋅ 0 r2 77 (1) 0 = Dielektrizitätskonstante des Vakuums; e = Elementarladung; r = Abstand zw. den Ionen; zA = Ladung des Anions; zK = Ladung des Kations Das bedeutet, die Kraft zwischen den Ionen nimmt bei Vergrößerung des Abstands nicht so stark ab wie die van der Waals-Kraft. 1.4 Die Elektronenpaarbindung, Atombindung, kovalente Bindung Ist die Differenz der Elektronegativitäten der einzelnen Bindungspartner < 2, versuchen die Atome ein Elektronenoktett zu erreichen, indem sie sich gemeinsam ein oder mehrere Elektronenpaar(e) teilen. Hierzu stellen beide Partner mindestens ein Elektron für eine Bindung zur Verfügung. Diese werden zu Elektronenpaaren zwischen den beiden Bindungspartnern (z. B. H● + ●Cl ⇌ H–Cl, ●O● + ●O● ⇌ O=O, etc.). Hierbei werden wieder zwei Fälle unterschieden: 1. Bei einer Bindung zwischen zwei Partnern derselben Atomsorte ist die Differenz der Elektronegativitäten 0, daher wird im Fall einer Bindungsspaltung homolytisch gespalten, d. h. das(die) bindende(n) Elektronenpaar(e) wird(werden) so gespalten, dass jedem Partner (jeweils) ein Elektron aus jeder Bindung verbleibt. 2. Bei einer Bindung zwischen zwei Partnern unterschiedlicher Atomsorte ist die Differenz der Elektronegativitäten ≠ 0, daher wird im Fall einer Bindungsspaltung heterolytisch gespalten, d. h. das (die) bindende(n) Elektronenpaar(e) wird (werden) dem elektronegativerem Partner zugeteilt, der weniger elektronegative geht leer aus. Dies bedeutet auch, dass selbst im Fall der Bindung das Elektronenpaar sich nicht gleichberechtigt (sozusagen ”in der Mitte”) zwischen den beiden Bindungspartnern befindet, sondern zum elektronegativeren Partner hin verschoben ist. Um überhaupt eine Bindung zwischen zwei Atomen zu erreichen müssen je ein Orbital oder Orbitallappen eines Atoms mit demjenigen eines zweiten Atoms überlappen. Damit entstehen aus den ursprünglichen Atomorbitalen (siehe Versuch 4 PSE) durch Linearkombination die gleiche Anzahl von Molekülorbitalen. 1.5 Die Molekülorbitaltheorie (MO-Theorie) Zur Beschreibung der Bindungsverhältnisse in einem Molekül kann die Molekülorbitaltheorie herangezogen werden. Die Grundlage ist das Entstehen von Molekülorbitalen aus den bereits existierenden Atomorbitalen. Dies geschieht durch eine Linearkombination aus den Atomorbitalen gemäß des sog. LCAO-Konzepts (LCAO = Linear Combination of Atomic Orbitals). Es gibt mehrere Möglichkeiten zur Orbitalüberlappung, die zwei wichtigsten werden nachfolgend beschrieben. 78 Bindungstheorie und Physikalische Eigenschaften 1.5.1 Die σ-Bindung Zwei Orbitale überlappen mit jeweils einem Orbitallappen gleichen Vorzeichens, so dass entlang der Bindungsachse keine Knotenfläche vorliegt. Eine σ-Bindung kann dadurch erreicht werden, dass zwei Atome mit ihren s-Orbitalen oder mit jeweils einem Lappen eines p- oder d-Orbitals sich entlang einer Achse (der Bindungsachse) nähern, so dass diese Orbitale dann (mathematisch gesehen) addiert werden können. Dieser Vorgang ist nur dann konstruktiv, wenn beide Lappen das gleiche Vorzeichen besitzen. Somit entsteht zwischen den beiden Bindungspartnern ein großer Orbitallappen mit gleichen Vorzeichen in dem sich die Bindungselektronen aufhalten; dieses Orbital wird als bindendes σ-Orbital bezeichnet. Um mathematisch korrekt linear zu kombinieren muss jedoch ein zweites Orbital entstehen, bei dem die beiden Lappen gleichen Vorzeichens Abb. 1.1: Die σ-Bindung voneinander subtrahiert werden. Somit entsteht ein zweites Orbital bei dem sich senkrecht zur Bindungsachse eine Knotenebene befindet. Dieses Orbital begünstigt die Bindung nicht, eine Besetzung des Orbitals wirkt einer Bindung entgegen. Somit wird es als antibindendes σ-Orbital oder als σ ∗ -Orbital bezeichnet. 1.5.2 Die π-Bindung Zwei Orbitale überlappen mit jeweils zwei Orbitallappen gleichen Vorzeichens, so dass entlang der Bindungsachse eine Knotenfläche vorliegt. Dies wird dadurch erreicht, dass zwei Atome mit ihren p-Orbitalen oder zwei d-Orbitallappen, die senkrecht zu einer bestehenden σBindung liegen sich einander so nähern, dass eine Linearkombination möglich ist. Auch hier ist wieder eine positive Linearkom- Abb. 1.2: Die π-Bindung bination möglich; es entsteht ein bindendes π-Orbital. Bei negativer Linearkombination entsteht analog zur Betrachtung der σ-Orbitale auch hier ein antibindendes π-Orbital oder π ∗ -Orbital mit einer Knotenfläche zwischen den beiden Bindungspartnern. Im Unterschied zu den σ-Orbitalen existiert bei den π-Orbitalen eine Knotenfläche entlang der Bindungsachse, d. h. die Aufenthaltswahrscheinlichkeit von Elektronen auf der Bindungsachse ist null. Bindungstheorie und Physikalische Eigenschaften 79 1.5.3 Das Molekülorbitalschema Die zuvor angesprochenen Linearkombinationen können in einem Energieschema aufgetragen werden. Bindende Molekülorbitale sind immer energetisch günstiger als die aus den gleichen Atomorbitalen gebildeten, antibindenden Molekülorbitale. Die Bilanzenergie aus den gebildeten Molekülorbitalen muss dabei der Bilanzenergie aus den dafür verwendeten Atomorbitalen entsprechen. Die Molekülorbitale werden dann im Mole- Abb. 1.3: Molekülschemata von O2 (links) und HF (rechts) külorbitalschema (MO-Schema) mit Elektronen gemäß der Hund’schen Regel (siehe Versuch 4 PSE) befüllt, bis alle ursprünglich vorhandenen Elektronen, die zur Bindung beitragen können, verbraucht sind. Sind mehr bindende als anti-bindende Molekülorbitale gefüllt findet eine Bindung statt, sind bindende und antibindende Molekülorbitale zu gleichen Anteilen gefüllt findet keine Bindung statt. Die Schemata in Abbildung 1.3 zeigen links die Bindungsverhältnisse im O2 - und rechts im HF-Molekül. Bei O2 werden aus den jeweils drei 2p-Orbitalen der Sauerstoffatome ein σ-bindendes und zwei π-bindende Orbitale generiert sowie die gleiche Anzahl an antibindenden Orbitalen. Die drei voll gefüllten, bindenden Orbitale suggerieren eine Dreifachbindung, jedoch müssen auch noch zwei antibindende Orbitale mit jeweils einem Elektron gefüllt werden, so dass sich die Bindungsordnung um 1 verringert und somit zwischen den beiden Sauerstoffatomen im O2 -Molekül eine Doppelbindung herrscht. Bei HF entsteht aus dem 1s-Orbital des Wasserstoffs und einem der drei 2p-Orbitale des Fluors ein bindendes und ein antibindendes σ-Molekülorbital. Die beiden anderen gefüllten p-Orbitale des Fluors können nichts zur Bindung beitragen und werden somit als nichtbindende Orbitale bezeichnet. Da nun nur das bindende σ-Molekülorbital mit Elektronen gefüllt wird, das antibindende leer bleibt, existiert im HF-Molekül eine Einfachbindung, deren Energie jedoch nur wenig unter derjenigen der p-Orbitale des Fluors liegt. Somit ist diese Bindung als nicht sonderlich stabil zu bezeichnen und die Energie, die dafür notwendig ist, diese zu spalten, ist auch nicht sehr hoch. 1.6 Bändertheorie und Leitfähigkeit Die bislang gezeigten Beispiele zur Molekülorbitaltheorie beschränkten sich auf zweiatomige Moleküle. Bereits bei diesen ist zu beobachten, dass die bindenden und antibindenden Orbitale relativ dicht beieinander liegen und dazwischen sich ein energetischer Bereich befindet in dem keine Orbitale angesiedelt sind. Beim Übergang zu mehratomigen Molekülen oder gar zu einem Festkörper aus sehr vielen Atomen ist die Bindungssituation nicht mehr so differenziert zu betrachten. Hier ist 80 Bindungstheorie und Physikalische Eigenschaften es nicht mehr möglich in den Energiebereichen der bindenden und antibindenden Orbitale einzelne Energieniveaus zu unterscheiden und so wird die Vereinfachung getroffen diese Bereiche als Bänder zu bezeichnen. Das Band im bindenden Energiebereich ist üblicherweise ganz mit Elektronen gefüllt und wird daher als Valenzband bezeichnet. Jenes im antibindenden Energiebereich ist zumeist leer oder nur mit wenigen Elektronen gefüllt, die sich dann innerhalb dieses Bandes auch frei bewegen können, daher wird es auch als Leitungsband bezeichnet. Der Energiebereich dazwischen, in dem sich keine Orbitale befinden ist die sogenannte verbotene Zone (auch Bandlücke). Die Größe der Bandlücke ist stoffspezifisch und anhand dieser werden drei verschiedene Stoffklassen unterschieden: Abb. 1.4: Übersicht zur Bändertheorie Isolatoren: Stoffe deren Valenz- und Leitungsband mehr als 4 eV auseinander liegen und keine Elektronen durch Energiezufuhr vom Valenz- in das Leitungsband angehoben werden können. Sie leiten somit den elektrischen Strom nicht (siehe Abbildung 1.4 c). Halbleiter: Stoffe deren Valenz- und Leitungsband weniger als 4 eV auseinander liegen. Bei diesen ist es möglich durch Energiezufuhr Elektronen vom Valenz- in das Leitungsband anzuheben (siehe Abbildung 1.4 b). Leiter: Dies sind Stoffe, deren Valenz- und Leitungsband miteinander überlappen und somit bereits ohne Energiezufuhr elektrische Leitfähigkeit aufweisen. Dies ist der Fall in Metallen (siehe Abbildung 1.4 a). Ungeachtet dessen, dass in einem metallischen Leiter durch die Überlappung von Valenz- und Leitungsband die Grundvoraussetzung für die Bewegung von Elektronen gegeben ist (die Valenzelektronen sind hierbei nicht mehr an ein bestimmtes Atom gebunden, sondern frei durch das ganze Metall beweglich, sog. Elektronengas; dies wird auch als Metallbindung bezeichnet), müssen sich die Elektronen immer noch vorbei an den in einem regelmäßigen Gitter angeordneten Metallatomrümpfen bewegen. Da diese der Elektronenbewegung ”im Weg” sind, geht diese nicht ganz reibungsfrei vonstatten und dies macht sich als elektrischer Widerstand bemerkbar. Je höher nun die Temperatur, desto mehr schwingen die Atomrümpfe auf ihren Gitterplätzen und erschweren den Elektronen das Durchkommen. Die Folge davon ist ein Anstieg des elektrischen Widerstands. Bei Leitern sinkt also die elektrische Leitfähigkeit mit der Temperatur. Im Gegensatz dazu steigt die elektrische Leitfähigkeit bei Halbleitern mit der Temperatur, da diese ja eine gewisse Mindestenergie benötigen um die Elektronen vom Valenz- in das Leitungsband anzuheben. Innerhalb gewisser Grenzen gilt hierbei auch, dass bei höherer Temperatur immer mehr Elektronen angehoben werden können und sich daher die Leitfähigkeit verbessert, der Widerstand verringert. Bindungstheorie und Physikalische Eigenschaften 81 1.7 Magnetismus Eine weitere Eigenschaft, die durch die Valenzelektronen eines Stoffes bestimmt wird, ist der Magnetismus. Da gemäß des Pauli-Verbots (siehe Versuch 4 PSE) nicht alle vier Quantenzahlen eines Elektrons mit denen eines anderen Elektrons übereinstimmen können, müssen sich zwei Elektronen im gleichen Orbital durch die Spinrichtung unterschieden. Diese wird durch die magnetische Spinquantenzahl, die die Werte ms = + 1 /2 und − 1 /2 annehmen kann, angezeigt. Während sich der Spin eines Elektrons mit ms = + 1 /2 mit demjenigen eines Elektrons mit ms = − 1 /2 zu null kompensiert, addieren sich die Spins von Elektronen, die gemäß der Hund’schen Regel in entarteten Orbitalen einfach besetzt werden, da diese die gleiche magnetische Spinquantenzahl besitzen. Entsprechend der Besetzung der Elektronen in den Valenzorbitalen werden verschiedene magnetische Phänomene unterschieden: Diamagnetismus: Besitzt ein Stoff ausschließlich gepaarte Elektronen zeigt dieser keinen (bzw. einen sehr schwach abstoßenden) Effekt bezüglich einem äußeren Magnetfeld. Paramagnetismus: Besitzt ein Stoff ein oder mehrere ungepaarte Elektronen zeigt er schwache Anziehung in ein von außen angelegtes Feld durch Ausrichtung der Spinvektoren der Elektronen. Bei Wegfall des äußeren Feldes ordnen sich diese wieder rein zufällig zueinander an. Eine besondere Form des Paramagnetismus ist der Ferromagnetismus: Stoffe die Ferromagnetismus zeigen besitzen ungepaarte Elektronen, die sich in einem Magnetfeld ausrichten und bewirken, dass der Stoff in das Feld hineingezogen wird. Bei Wegnahme des Feldes verbleiben die Spinvektoren der Elektronen jedoch in den sogenannten Weiss’schen Bezirken ausgerichtet. Der Stoff bleibt magnetisch. Das ist das Prinzip von Permanentmagneten. Bei Einwirkung von Energie (mechanisch oder thermisch) geht diese Ausrichtung allerdings wieder verloren, der permanente Magnetismus verschwindet. 2 Aufgabenstellung, Versuchsanleitung und -auswertung 2.1 Schmelzen von Salzen und Salzmischungen 2.1.1 Aufgabenstellung Bei diesem Versuch soll dokumentiert werden, wie viel Energie nötig ist, um Salze und Salzmischungen zum Schmelzen zu bringen. Da eine direkte Temperaturmessung in der Flamme des Bunsenbrenners schlecht möglich ist, wird die Zeit ermittelt, welche die einzelnen Salze/Salzmischungen zum Erreichen des Schmelzpunkts bei konstanter Leistung des Bunsenbrenners benötigen. 2.1.2 Versuchsanleitung Eine geringe Menge festes wasserfreies Natriumsulfat Na2 SO4 wird auf eine Magnesiarinne gegeben. Diese wird mit der Tiegelzange in die wärmste Zone der Bunsenbrennerflamme (Spitze des Innenkegels) gehalten und die Zeit bis zum Schmelzen wird gemessen. (Falls es überhaupt 82 Bindungstheorie und Physikalische Eigenschaften schmilzt, falls nicht wird der Versuch nach ca. 5 Minuten abgebrochen). Die Magnesiarinne wird mit demineralisiertem Wasser gewaschen und somit von den Resten des Natriumsulfats befreit. Dieser Versuch wird für festes wasserfreies Kaliumnitrat KNO3 wiederholt. Dieser Versuch wird zum dritten Mal für eine Mischung (Mischungsverhältnis ca. 1:1) von Na2 SO4 und KNO3 wiederholt, die gegebenenfalls zuvor im Mörser vermengt wurde. 2.1.3 Versuchsauswertung ● Dokumentieren Sie die Zeit, welche die Salze / Salzmischungen bei den jeweiligen Versuchen zum Schmelzen benötigen. Ziehen Sie Rückschlüsse auf die Schmelztemperaturen. ● Erklären Sie den Begriff Eutektikum bzw. eutektisches Gemisch (Literaturstudium). 2.2 Erhitzen, Abkühlen und Abschrecken von Schwefel 2.2.1 Aufgabenstellung Bei diesem Versuch sollen die einzelnen Modifikationen des elementaren Schwefels beim Durchlaufen der Temperaturen von Raumtemperatur bis zum Siedepunkt und zurück beobachtet, dokumentiert und den literaturbekannten Modifikationen zugewiesen werden. Außerdem wird elementarer Schwefel in seiner flüssig-plastischen Modifikation abgeschreckt, um zu zeigen, dass es möglich ist eine feste Modifikation zu erhalten, die normalerweise nicht bei Standardbedingungen existiert. 2.2.2 Versuchsanleitung Ca. 3 g Schwefel werden in ein großes Reagenzglas gegeben. (Dabei ist darauf zu achten, dass kein Schwefel innen oder außen an den Reagenzglaswänden anhaftet). Zu Beginn wird das Reagenzglas vorsichtig erhitzt (Brennerflamme ohne Sauerstoffzufuhr). Nach kurzem Halten des Reagenzglases in die Brennerflamme wird das Reagenzglas immer wieder leicht geschwenkt um eine gleichmäßige Wärmeausbreitung zu gewährleisten. Wenn der Schwefel zu schmelzen beginnt, wird ein Teil des nun dünnflüssigen Schwefels in ein Becherglas mit Eiswasser gegossen. Danach wird das Reagenzglas mit der heißen Brennerflamme (mit Sauerstoffzufuhr) so vollständig wie möglich erhitzt, um die weiteren Modifikationen des Schwefels zu erhalten. 2.2.3 Versuchsauswertung ● Dokumentieren Sie die einzelnen erkennbaren Modifikationen des festen und flüssigen Schwefels und weisen Sie diesen die in der Literatur beschriebenen Modifikationen zu. ● Zeichnen Sie die kleinste Baueinheit des festen α-Schwefels. Welche Kräfte herrschen zwischen den Schwefelatomen in dieser kleinsten Baueinheit? ● ● Welche Kräfte herrschen zwischen den kleinsten Baueinheiten des Schwefels im Festkörper? Welche Aggregatzustände können für Stoffe beobachtet werden? Wie nennt man die Übergänge zwischen diesen? Handelt es sich bei diesen Übergängen um chemische Vorgänge (Begründung)? Bindungstheorie und Physikalische Eigenschaften 83 2.3 Bestimmung der Leitfähigkeit eines metallischen Leiters 2.3.1 Aufgabenstellung Bei diesem Versuch soll der Widerstand eines metallischen Leiters (Kupferdraht) sowohl bei Raumtemperatur als auch bei erhöhter Temperatur gemessen und die Ergebnisse interpretiert werden. 2.3.2 Versuchsanleitung Der Versuch wird gemäß der am Platz ausliegenden Versuchsbeschreibung aufgebaut und durchgeführt. 2.3.3 Versuchsauswertung ● ● Erstellen Sie eine Widerstandskurve für den Kupferdraht. Ist die Widerstandsänderung mit der Temperatur ein linearer Vorgang? 2.4 Magnetismus 2.4.1 Aufgabenstellung Bei diesem Versuch soll das magnetische Verhalten eines Ferromagnets in Abhängigkeit von der Temperatur gemessen werden. 2.4.2 Versuchsanleitung An einem Ende eines (mit Plastikfolie umhüllten) Gadoliniumstabs wird bei Raumtemperatur ein Permanentmagnet gehalten. Indem das andere Ende des Gadoliniumstabs über das Gläschen mit Eisenpulver geführt wird, werden die magnetischen Eigenschaften des Gadoliniumstabs geprüft. Der Gadoliniumstab wird in einem Reagenzglas in ein Becherglas mit Eiswasser gestellt. Nach dem Abkühlen wird an einem Ende des (mit Plastikfolie umhüllten) Gadoliniumstabs ein Permanentmagnet gehalten. Indem das andere Ende des Gadoliniumstabs über das Gläschen mit Eisenpulver geführt wird, werden die magnetischen Eigenschaften des Gadoliniumstabs erneut geprüft. Der Gadoliniumstab wird in der Hand wieder aufgewärmt. An einem Ende des handwarmen Gadoliniumstabs wird ein Permanentmagnet angebracht. Indem das andere Ende des Gadoliniumstabs über das Gläschen mit Eisenpulver geführt wird, werden die magnetischen Eigenschaften des Gadoliniumstabs erneut geprüft. 2.4.3 Versuchsauswertung ● Wie wird die Temperatur bezeichnet bei der ein ferromagnetischer Stoff diese Eigenschaft verliert? Wie hoch ist diese für Gadolinium? 84 Bindungstheorie und Physikalische Eigenschaften 3 Geräte und Chemikalien Eine Geräteliste liegt im Platz aus Benötigte Chemikalien Na2 SO4 (wasserfrei) KNO3 (wasserfrei) Schwefel Gefahrensymbole – Achtung (GHS03) Achtung (GHS07) H-Sätze – 272, 210 315 P-Sätze – – 302+352 4 Literatur [1] A. F. Holleman, E. Wiberg: Lehrbuch der Anorganischen Chemie, 102. Aufl., Walter de Gruyter Verlag, 2007. [2] [3] [4] G. Wedler: Lehrbuch der Physikalischen Chemie, 5. Aufl., Wiley-VCH, 2004. P. W. Atkins, J. de Paula: Physikalische Chemie, 4. Aufl., Wiley-VCH, 2008. Einschlägige Lehrbücher zur Experimentalphysik. Massenwirkungsgesetz 85 VERSUCHSBLOCK 2: DAS CHEMISCHE GLEICHGEWICHT, ELEKTROCHEMIE UND REAKTIONSKINETIK Versuch 8 (MWG) Massenwirkungsgesetz 1 Theorie 1.1 Gleichgewichtsreaktionen Jede chemische Reaktion ist im Grunde eine Gleichgewichtsreaktion. Bei Erreichen des Gleichgewichts wird der Anschein erweckt, dass dies den völligen Stillstand der Reaktion bedeuten würde. Das stimmt zwar für den Nettoumsatz (bei konstanten äußeren Bedingungen), jedoch nicht für die Betrachtung der molekularen Ebene, hier findet ständig fortschreitender Umsatz in beide Richtungen, also eine ständige Umwandlung statt. Die Lage des Gleichgewichts wird durch die Gleichgewichtskonstante angegeben. 1.1.1 Herleitung der Gleichgewichtskonstanten Betrachtung einer Beispielreaktion: 2 B + 3 I2 ⇌ 2 BI3 . Die Reaktionsgeschwindigkeit v ist proportional zum Produkt der Konzentrationen der Reaktionspartner, k ist der entsprechende Proportionalitätsfaktor. Dieser beschreibt den Zusammenhang zwischen der Entstehung des Produkts bzw. der Vernichtung der Edukte und der messbaren Geschwindigkeit. Die Geschwindigkeitsgesetze lauten für die Hinreaktion v1 = k1 ⋅ c2B ⋅ c3I2 (1) v2 = k2 ⋅ c2BI3 . (2) und für die Rückreaktion Im Gleichgewicht gilt v1 = v2 und daraus folgt: 2 cBI k1 = 2 33 k2 cB ⋅ cI2 (3) Dies ist das Massenwirkungsgesetz, welches 1867 von Guldberg und Waage formuliert, 1873 von Bodenstein bewiesen und 1883 von van’t Hoff theoretisch abgeleitet wurde. 86 Massenwirkungsgesetz 1.1.2 Definition der Gleichgewichtskonstanten Die Gleichgewichtskonstante ist das Verhältnis aus den Proportionalitätsfaktoren für die Hinund Rückreaktion. Dies ist definiert als: K= Produkt der Konzentrationen der Produkte Produkt der Konzentrationen der Edukte (4) Die stöchiometrischen Koeffizienten gehen hierbei als Exponenten ein. Wird die Reaktionsrichtung vertauscht (Produkte ↔ Edukte), wandelt sich die Gleichgewichtskonstante in ihren Kehrwert um. Bei Gasreaktionen wird anstelle der Konzentration des Stoffes sein Partialdruck verwendet. (Partialdruck = Druckanteil eines bestimmten Stoffes am Gesamtdruck der Gasmischung). Der Partialdruck hängt mit der Konzentration über das ideale Gasgesetz (p ⋅ V = n ⋅ R ⋅ T ) zusammen. Mit c = n V gilt hierfür: c = p R⋅T , das heißt für eine konstante Temperatur ist c ∼ p. 1.2 Die Ammoniaksynthese 1.2.1 Die Gleichgewichtskonstante Kp Leitet man gasförmigen Stickstoff N2 und gasförmigen Wasserstoff H2 über einen Katalysator, reagieren beide nach folgender Reaktionsgleichung zu gasförmigem Ammoniak NH3 : 1 3 N2 + H2 ⇌ NH3 2 2 (5) Die Lage des Gleichgewichtes dieser Reaktion kann mit Hilfe des Massenwirkungsgesetzes für Gase durch Gleichung 6 beschrieben werden. Kp = [ [ pNH3 ] p0 pN2 12 ] p0 ⋅[ pH2 32 ] p0 (6) mit Kp = Gleichgewichtskonstante, p0 = Standarddruck und pNH3 , pN2 und pH2 = Gleichgewichtspartialdrücke der entsprechenden Komponenten. Diese Gleichung gilt näherungsweise allerdings nur bei kleinen Partialdrücken. Meist wird der Standarddruck p0 = 1 bar gesetzt und die Konstante folgendermaßen formuliert: Kp = [pNH3 ] [pN2 ] 2 ⋅ [pH2 ] 2 1 3 (7) Dabei darf allerdings nicht vergessen werden, dass die Gleichgewichtskonstante Kp nach wie vor dimensionslos ist. Die Freie Reaktionsenthalpie ∆r G im Gleichgewichtszustand lässt sich wie folgt ausdrücken: ∆r G = ∆r G0 + R ⋅ T ⋅ ln K (8) wobei ∆r G0 die Standardreaktionsenthalpie ist, die sich ergibt, wenn alle Gase im Standardzustand vorliegen, R ist die allgemeine Gaskonstante und T die absolute Temperatur in Kelvin. Wenn für eine Reaktion ∆r G positiv ist, muss Nutzarbeit zugeführt werden, damit die Reaktion überhaupt abläuft. Ist jedoch ∆r G negativ, so kann die Reaktion spontan, unter Abgabe von Nutzarbeit, Massenwirkungsgesetz 87 ablaufen. Befindet sich die Reaktion dagegen im Gleichgewicht, so gilt: ∆r G = 0 (9) das heißt, je stärker negativ ∆r G ist, umso weiter ist die Reaktion vom Gleichgewichtszustand entfernt und umso mehr Nutzarbeit wird abgegeben, bis der Gleichgewichtszustand erreicht ist. Daher wird ∆r G auch als die ”Triebkraft” der Reaktion bezeichnet. Aus den Gleichungen 8 und 9 erhält man die wichtige Beziehung zwischen ∆r G und K: ∆r G0 = −R ⋅ T ⋅ ln K bzw. ln K = − ∆r G0 R⋅T (10) 1.2.2 Die Temperaturabhängigkeit der Gleichgewichtskonstanten K Um die Temperaturabhängigkeit von ln K zu erhalten, muss Gleichung 10 nach der Temperatur abgeleitet und mit Hilfe der Gibbs-Helmholtz-Gleichung umgeformt werden. Daraus ergibt sich dann die van’t Hoff’sche Gleichung: 1 dK d ln K ∆r H 0 ⋅ = = K dT dT R ⋅ T2 (11) An der obigen Form der sogenannten van’t Hoff’schen Reaktionsisobaren erkennt man, dass bei exothermen Reaktionen (∆r H 0 < 0) die Steigung ( dK dT ), d.h. die Änderung der Gleichge- wichtskonstanten bei zunehmender Temperatur negativ ist, was wiederum bedeutet, dass das Gleichgewicht, analog zum Prinzip von Le Chatelier, sich bei höheren Temperaturen auf die Seite der Edukte verschiebt. Analoges gilt im umgekehrten Fall, dem einer endothermen Reaktion (∆r H 0 > 0), wobei sich das Gleichgewicht entsprechend auf die Seite der Produkte verschiebt. Das heißt anschaulich, die Reaktion versucht, dem äußeren Zwang auszuweichen. Bei Temperaturerhöhung wird die Reaktion begünstigt, welche Wärme verbraucht. Die unbestimmte Integration der Gleichung 11, wobei ∆r H 0 als temperaturunabhängig betrachtet wird, führt zu folgendem Ausdruck: ln K = − ∆r H 0 1 ⋅ + const. R T (12) Die van’t Hoff’sche Gleichung wird zum einen bei der Umrechnung einer Gleichgewichtskonstanten von einer Temperatur auf eine andere und zum anderen bei der Ermittlung der Reaktionsenthalpie aus Gleichgewichtsmessungen bei verschiedenen Temperaturen angewendet. 1.2.3 Ermittlung von Kp nach einer dynamischen Methode Wie aus dem Versuchsaufbau ersichtlich wird hier das Ammoniak-Gleichgewicht nach einer Strömungsmethode bestimmt. Zur schnellen Einstellung des Gleichgewichtes befindet sich ein industrieller Eisenkatalysator im Strömungsrohr, der die Lage des Gleichgewichtes nicht beeinflusst. Das Gasgemisch, das den Strömungsreaktor verlässt, wird durch verdünnte Schwefelsäure geleitet. Die Neutralisation der Schwefelsäure durch das entstandene Ammoniak wird durch einen Farbumschlag des Indikators angezeigt. Die Zeit, die bis zum Farbumschlag vergeht, dient als 88 Massenwirkungsgesetz Umsatzparameter. Weitere Parameter sind die Strömungsgeschwindigkeiten von Stickstoff und Wasserstoff, die Temperatur des Reaktors und der Druck. Wie schon in Kapitel 1.2.1 erwähnt, lautet für das hier zu untersuchende Gleichgewicht 1 3 N2 + H2 ⇌ NH3 2 2 (5) und die Gleichgewichtskonstante Kp : Kp = [pNH3 ] [pN2 ] 2 ⋅ [pH2 ] 2 1 3 (7) Wie kann nun Kp über die zuvor genannten Parameter bestimmt werden? Es sei nNH3 die Stoffmenge in mol Ammoniak, die in einer gegebenen Zeit den Reaktor verlassen. Dann ist die Stoffmenge von Stickstoff nN2 und von Wasserstoff nH2 , die in der gleichen Zeit den Reaktor verlassen, gleich den Differenzen zwischen den Stoffmengen n0,N2 und n0,H2 , die während der Zeit t in den Reaktor einströmen und der Stoffmengen, die verbraucht werden, um Ammoniak zu bilden. Hieraus ergibt sich: 3 nH2 = n0,H2 − nNH3 2 1 nN2 = n0,N2 − nNH3 2 (13) (14) Die Gesamtstoffmenge nges , die den Reaktor während der Zeit t verlässt, ist also: nges = nN2 + nH2 + nNH3 = n0,N2 + n0,H2 − nNH3 (15) Unter den in diesem Versuch angewandten Bedingungen ist nNH3 mindestens 100 mal kleiner als n0,N2 und n0,H2 , sodass die Strömungsgeschwindigkeiten von N2 und H2 durch die Reaktion praktisch nicht geändert werden. Daher gilt für n in guter Näherung: nges ≅ n0,N2 + n0,H2 (16) Nun können die Stoffmengen nach dem idealen Gasgesetz p⋅V =n⋅R⋅T (17) und mit der Strömungsgeschwindigkeit V̇ = V t (18) durch die Temperatur T (in K), der Zeit t (in s), dem Druck p (in bar) und den Strömungsgeschwindigkeiten V̇ (in l⋅s−1 )ersetzt werden. Die Strömungsgeschwindigkeit ist die erste Ableitung des Volumens V nach der Zeit t (vgl. Geschwindigkeit). Sie gibt an, welches Volumen (eines Gases oder einer Flüssigkeit) in einer definierten Zeit ein Rohr etc. durchströmt. Will man nun verschiedene Gasströme miteinander vergleichen, muss man beachten, dass die Dichte von Gasen sehr stark druck- und temperaturabhängig ist. Daher ist es unumgänglich normierte bzw. standardisierte Größen einzuführen. So wird die Strömungsgeschwindigkeit V̇ meist in Normliter Massenwirkungsgesetz 89 pro Sekunde (Nl⋅s−1 ) angegeben, diese gilt nur bei einer definierten Temperatur T0 (= 293,2 K) und einem definierten Druck p0 (= 1 bar). Werden nun n0,H2 = p0 ⋅ V̇H2 ⋅ t R ⋅ T0 und (19) p0 ⋅ V̇N2 ⋅ t R ⋅ T0 (20) p0 ⋅ (V̇H2 + V̇N2 ) ⋅ t R ⋅ T0 (21) n0,N2 = in Gleichung 16 eingesetzt, ergibt sich nges = Nun sind die Partialdrücke von N2 , H2 und NH3 , die man zur Berechnung von Kp benötigt, nach dem Dalton’schen Gesetz immer ein bestimmter Bruchteil des Gesamtdrucks p (am Reaktorausgang). In diesem Versuch entspricht der Gesamtdruck dem aktuellen Luftdruck. pN2 = ( pH2 = ( V̇N2 nN2 )⋅p=( )⋅p nges V̇N2 + V̇H2 V̇H2 nH2 )⋅p=( )⋅p nges V̇N2 + V̇H2 pNH3 = ( (22) und (23) nNH3 )⋅p nges (24) Setzt man nun Gleichung 21 in Gleichung 24 ein, ergibt sich: R ⋅ T0 )⋅p p0 ⋅ (V̇N2 + V̇H2 ) ⋅ t pNH3 = nNH3 ⋅ ( (25) mit R = 0, 08319 l⋅bar⋅mol−1 ⋅K−1 , p0 = 1 bar und T0 = 293,2 K. Somit wird Gleichung 25 zu: pNH3 = l 24, 391 mol ⋅ nNH3 ⋅ p (26) (V̇N2 + V̇H2 ) ⋅ t Zum Schluss setzt man die Gleichungen 22, 23 und 26 in Gleichung 6 ein und man erhält die Konstante Kp in den von uns gewünschten Parametern: Kp = p 3 [ NH ] p0 [ pN2 21 ] p0 ⋅[ pH2 32 ] p0 oder Kp = = l 24,391 mol ⋅nNH3 ⋅p (V̇N2 +V̇H2 )⋅t [( V̇ 1 V̇N2 2 ) ⋅ p] N2 +V̇H2 ⋅ [( V̇ 3 V̇H2 2 ) ⋅ p] N2 +V̇H2 l 24, 391 mol ⋅ nNH3 ⋅ (V̇N2 + V̇H2 ) 1 3 V̇N22 ⋅ V̇H22 ⋅ t ⋅ p ⋅ p0 ⋅ p0 (27) (28) mit V̇H2 , V̇N2 = eingestellte Strömungsgeschwindigkeiten von H2 und N2 in (Nl⋅s−1 ); p = gemessener Luftdruck, t = gemessene Zeit, nNH3 = Stoffmenge an NH3 (= 2, 5 ⋅ 10−5 mol) und p0 = Standarddruck. 90 Massenwirkungsgesetz 2 Aufgabenstellung, Versuchsanleitung und -auswertung 2.1 Bestimmung der Gleichgewichtskonstanten Kp der Ammoniaksynthese in Abhängigkeit der Temperatur 2.1.1 Aufgabenstellung In diesem Versuch sollen Sie Zeitmessungen bei vier verschiedenen Temperaturen in einem Temperaturbereich von 500 bis 700°C und unterschiedlichen Gasgeschwindigkeiten durchführen. 2.1.2 Versuchsdurchführung Versuchsaufbau Abb. 2.1: Schematischer Aufbau der Apparatur zur Ammoniaksynthese Erster Teil (Vorbereitung) 1. Dreiwegehahn so drehen, dass die Gase nur in den Abzug geleitet werden. 2. Hauptventile weit aufdrehen und an den Druckminderventilen 1 bar einstellen. 3. Am N2 -Rotameter den Stickstoffstrom auf ca. 10 Skalenteile (SkT) einstellen. Massenwirkungsgesetz 4. 91 Das Temperatur-Kontrollgerät einschalten und die gewünschte Temperatur (in °C) einstellen. (Fangen Sie mit der niedrigsten Temperatur an, da der Ofen nur sehr langsam abkühlt) 5. Am H2 -Rotameter den Wasserstoffstrom ebenfalls auf ca. 10 Skalenteile (SkT) einstellen. 6. Lesen Sie den Luftdruck am Barometer ab und notieren Sie ihn. Es dauert ca. 45 min bis sich das Temperaturgleichgewicht eingestellt hat. In dieser Zeit können Sie den zweiten Teil des Versuches bearbeiten. Zweiter Teil 7. Geben Sie 10 ml einer 0,05 mol l (0,05 M bzw. früher 0,1 N) H2 SO4 -Lösung in einen 2l- Messkolben und füllen Sie diesen mit demin. Wasser bis zur Markierung auf. (10 ml-Pipette verwenden). 8. Geben Sie 50 ml der unter 7. hergestellten H2 SO4 -Lösung und 5 Tropfen Methylrot-Lösung in einen 100 ml-Erlenmeyerkolben. 9. Die H2 SO4 -Lösung von 8. wird nun mit einer 0,01 mol l (0,01 M bzw. früher 0,01 N) NaOH- Lösung titriert. Dazu füllen Sie die Mikrobürette mit der NaOH-Lösung bis zur Markierung (0,00 ml) auf. Lassen Sie langsam, unter leichtem Schwenken des Erlenmeyerkolbens, die NaOH-Lösung zur H2 SO4 -Lösung tropfen (Verbrauch ca. 2,5 ml). Am Umschlagpunkt von Rot nach Zitronengelb stoppen Sie. Diese Probe bewahren Sie auf, um später (im dritten Teil) den Umschlagspunkt besser erkennen zu können. In der Zwischenzeit sollte die erste Temperatur erreicht sein und Sie können den dritten Teil des Versuches durchführen. Dritter Teil 10. Geben Sie jeweils 50 ml der H2 SO4 -Lösung von 7., je 5 Tropfen Methylrot-Lösung und je einen Rührfisch in vier 100 ml-Erlenmeyerkolben. 11. Stellen Sie am H2 -Rotameter den Wasserstoffstrom zwischen 9 und 60 Nl⋅h−1 ein und notieren Sie den Wert. (Die zur Umrechnung von Nl⋅h−1 in SkT notwendigen Tabellen liegen beim Versuch aus. Bitte beachten Sie, dass für H2 und N2 unterschiedliche Tabellen gelten.) 12. Stellen Sie am N2 -Rotameter den Strom ein, allerdings zwischen 3 und 23 Nl⋅h−1 . Der Wert wird notiert. 13. Spannen Sie einen Erlenmeyerkolben von 9. mit einer Klammer über den Magnetrührer, setzen den Glaseinsatz ein und schalten Sie den Magnetrüher ein (nicht zu schnell rühren). 14. Jetzt drehen Sie schnell den Dreiwegehahn so, dass das Gasgemisch nur durch die H2 SO4 Lösung geleitet wird und starten gleichzeitig die Zeitmessung. (Es sind deutlich Gasblasen am Einleitungsrohr des Schliffkolbens zu sehen.) 15. Verfolgen Sie die Farbänderung der Lösung und stoppen die Zeitmessung wenn der Farbumschlag erfolgt (vergleichen Sie mit der in 9. beiseite gestellten Lösung). Notieren Sie die Zeit in Sekunden. 16. Drehen Sie den Dreiwegehahn zügig in die ursprüngliche Position (siehe 1.) 92 Massenwirkungsgesetz 17. Spülen Sie das Gaseinleitungsrohr mit demin. Wasser. 18. Wiederholen Sie die Schritte 10. bis 17. dreimal, allerdings bei unterschiedlichen Gasgeschwindigkeiten (aber bei gleicher Temperatur). 19. Jetzt können Sie am Temperaturkontrollgerät die nächste Temperatur einstellen. Um nicht so viel Gas zu verschwenden, stellen Sie bitte beide Gasströme auf ca. 10 SkT ein. Es dauert nun ca. 15 - 20 min bis sich das Temperaturgleichgewicht eingestellt hat; die neue Temperatur wird notiert. 20. Führen Sie bei dieser Temperatur ebenfalls die Schritte 10. bis 17. durch. Den ganzen dritten Teil sollten Sie solange wiederholen, bis Sie bei 4 verschiedenen Temperaturen jeweils 4 Messungen bei verschiedenen Gasgeschwindigkeiten durchgeführt haben. (insgesamt 16 Messungen) Ausschalten der Apparatur Nach Ihrer letzten Messung gehen Sie wie folgt vor: 21. Drehen Sie den Dreiwegehahn so, dass das Gasgemisch nur in den Abzug geleitet wird. 22. Schalten Sie das Temperaturkontrollgerät aus. 23. Schalten Sie den Magnetrührer aus. 24. Drehen Sie den H2 -Haupthahn zu. 25. Stellen Sie am N2 -Rotameter 0,0 SkT ein. 26. Spülen Sie bitte alle verwendeten Glasgeräte, falls dies noch nicht erfolgt ist. 2.1.3 Versuchsauswertung ● Berechnen Sie die Gleichgewichtskonstanten Kp nach Gleichung 28 für die jeweiligen Messungen und stellen Sie Ihre Messergebnisse entsprechend des nachfolgend stehenden Beispiels dar. ● Bilden Sie bei jeder Temperatur einen Mittelwert von Kp und aus diesem ln Kp . Tragen Sie in einem Schaubild ln Kp gegen 0 1 T auf. Aus der Steigung der entstandenen Geraden können Sie ∆r H bestimmen (siehe Gleichung 12). ● Vergleichen Sie Ihren ermittelten Wert mit dem Literaturwert und diskutieren Sie eventuelle Abweichungen. Beispiel einer Messung: T = 873,2 K (= 600°C), Luftdruck p = 0,981 bar, nNH3 = 2, 5 ⋅ 10−5 mol V̇H2 V̇N2 (l⋅h−1 ) (l⋅s−1 ) 23, 029 6, 40 ⋅ 10 28, 005 39, 316 28, 005 −3 7, 78 ⋅ 10 −3 1, 09 ⋅ 10 −2 7, 78 ⋅ 10 −3 t (l⋅h−1 ) (l⋅s−1 ) 3, 707 1, 03 ⋅ 10 8, 329 10, 325 12, 290 (s) −3 2, 31 ⋅ 10 −3 2, 87 ⋅ 10 −3 3, 41 ⋅ 10 −3 172 118 86 108 Kp ln Kp (−) 1, 63 ⋅ 10 −3 1, 61 ⋅ 10 −3 1, 63 ⋅ 10 −3 1, 61 ⋅ 10 −3 (−) −6, 416 −6, 431 −6, 419 −6, 433 Massenwirkungsgesetz 93 3 Geräte und Chemikalien Eine Geräteliste liegt am Platz aus Chemikalien H2 N2 NH3 (Produkt, g) Gefahrenbezeichnung Gefahr (GHS02, 04) Achtung (GHS04) Gefahr (GHS05, 09) H-Sätze 220, 280 280 EUH071, 331, 221, 314, 280, 400 H2 SO4 Gefahr (GHS05) 314, 290 NaOH Methylrotlösung Gefahr (GHS05) – 314, 290 – P-Sätze 210, 377, 381, 403 403 210, 260, 273, 280, 304+340+315, 303+361+353+315, 377, 381, 403, 305+351+338+315, 405 280, 301+330+331, 305+351+338, 309, 310 301+330+331, 305+351+338, 280 – 4 Literatur [1] [2] [3] G. Wedler: Lehrbuch der Physikalischen Chemie, 5. Aufl., Wiley-VCH, 2004. P. W. Atkins, J. de Paula: Physikalische Chemie, 4. Aufl., Wiley-VCH, 2008. G. Huybrechts, G. Petre: Journal of Chemical Education, Vol. 53, Nr. 7, 1976. Säure-Base-Gleichgewichte 95 Versuch 9 (SBG) Säure-Base-Gleichgewichte 1 Theorie 1.1 Säure-Base-Theorien Arrhenius-Theorie: Säuren sind H+ -Donatoren (oder Donoren) und Basen sind OH– -Donatoren (oder Donoren). Diese Theorie ist auf die wässrige Lösung beschränkt. Brønsted-Theorie: Säuren sind H+ -Donatoren (oder Donoren) und Basen sind H+ -Akzeptoren. Diese Theorie gilt auch in der Gasphase, z. B.: HCl + NH3 ⇌ NH4 Cl. Lewis-Theorie: Säuren sind Elektronenpaarakzeptoren und Basen sind Elektronenpaardonatoren (oder -donoren) 1.2 Die Eigendissoziation des Wassers H2 O ist ein schwach amphoterer Elektrolyt (Ampholyt) und kann daher sowohl als Säure, als auch als Base fungieren. Die Dissoziation erfolgt in hydratisierte H+ und OH– -Ionen gemäß H2 O ⇌ H+ + OH– , dieser Vorgang wird auch als Autoprotolyse bezeichnet. Für reinstes Wasser gilt bei 298 K (25°C): K= [H+ ] ⋅ [OH− ] [H2 O] (siehe Versuch 8 MWG, Schreibweise: [H2 O] = c(H2 O) = (1) n V, Einheit: mol l ) Das Gleichgewicht liegt hierbei so weit auf der Seite des Edukts Wasser, dass die Konzentration von H2 O durch die Autoprotolyse nicht beeinflusst wird. Ein Liter Wasser besitzt ungefähr die Masse 1000 g (1 kg), 1 mol Wasser besitzt die Masse 18 g, daraus ergibt sich eine Konzentration von Wasser in Wasser von 1000 g⋅mol 18 g⋅l = 55, 52 mol l . Diese Konzentration ist näherungsweise als konstant zu betrachten und damit ergibt sich aus Gleichung 1: K ⋅ [H2 O] = KW = [H+ ] ⋅ [OH− ] mit KW = 10−14 mol2 l2 (2) H+ und OH– entstehen hierbei immer im gleichen Verhältnis, somit gilt [H+ ] = [OH− ]. Daraus folgt aus Gleichung 2 KW = [H+ ]2 = [OH− ]2 (3) und für die Konzentrationen von H+ und OH– ergibt sich: [H+ ] = [OH− ] = √ mol KW = 10−7 l Um die Schreibweise zu vereinfachen gilt folgendes: (4) 96 Säure-Base-Gleichgewichte 1. Für die Angabe der Konstanten werden die 10er Logarithmen verwendet, für KW = 10−14 ergibt sich somit log KW = −14 2. In wässriger Lösung sind die Konzentrationen meist kleiner als 1 mol l , ebenso sind die Dissoziationskonstanten überwiegend < 1, daher ist log K < 0, somit wird auch das (−)-Zeichen mit in die Schreibweise einbezogen. Generelle Schreibweise: − log KX = pKX So gilt für reines Wasser (unabhängig von der Menge extern zugegebener H+ - und OH– -Ionen) bei der Bildung des negativen dekadischen Logarithmus von Gleichung 2: − log KW = − log([H+ ] ⋅ [OH− ]) = − log[H+ ] − log[OH− ] bzw. pKW = pH + pOH (5) (6) Analog ergibt sich aus den Gleichungen 3 und 5: − log KW = − log[H+ ]2 = − log[OH− ]2 (7) und aus den Gleichungen 3 und 6: pKW = 2 ⋅ pH = 2 ⋅ pOH (8) Die Autoprotolyse ist ein endothermer Prozess, d.h. die Gleichgewichtskonstante ist temperaturabhängig. Bei steigender Temperatur wird KW größer. T (in ○ C) 0, 13 ⋅ 10−14 pKW pH = pOH 14, 89 7, 45 −14 14, 07 7, 04 13, 26 6, 63 12, 26 6, 13 KW 0○ C 0, 86 ⋅ 10 ○ 20 C 5, 47 ⋅ 10 ○ −14 50 C 5, 45 ⋅ 10 ○ −13 100 C Beispiele für andere Substanzen, die ebenfalls Autoprotolyse zeigen: Schwefelsäure: – 2 H2 SO4 ⇌ H3 SO+ 4 + HSO4 Phosphorsäure: 2 Flusssäure: 3 Ammoniak: 2 H3 PO4 ⇌ H4 PO+ 4 + + HF ⇌ H2 F + HF–2 – NH3 ⇌ NH+ 4 + NH2 H2 PO–4 pKH2 SO4 = 3, 6 pKH3 PO4 = 2, 0 pKHF = 11, 1 pKNH3 = 33, 0 1.3 Die Brønsted-Theorie In wässriger Lösung wird diejenige Substanz als Säure bezeichnet, die die Konzentration an H+ -Ionen erhöht (und da [H+ ] ⋅ [OH– ] = 10−14 , automatisch die an OH– -Ionen verringert). Für die Base gilt das Umgekehrte. Das konstitutionelle Merkmal von Brønsted-Säuren bzw. -Basen ist, dass das Säuremolekül mindestens 1 Proton enthalten muss (H-Atom, das als H+ -Proton abgespalten werden kann) und ein Basenmolekül mindestens ein freies Elektronenpaar benötigt. Säure-Base-Gleichgewichte In saurer Lösung gilt: In alkalischer Lösung gilt: 97 [H+ ] > 10−7 [H+ ] < 10−7 mol l (d.h. der pH-Wert ist kleiner als 7 und mol l (d.h. der pH-Wert ist größer als 7 und [H+ ] > [OH– ]) [H+ ] < [OH]) Nach Hydrolyse einer Säure entsteht die deprotonierte Form, die potentiell selbst wieder ein Proton aufnehmen kann, also eine Base ist; man bezeichnet diese als die zur Säure HA konjugierte (oder korrespondierende) Base A− . Umgekehrt nennt man die aus einer Base B hervorgehende Säure HB + , die zur Base B konjugierte (oder korrespondierende) Säure. Es gilt: Je stärker die Säure, desto schwächer ist die konjugierte Base, je schwächer die Säure, desto stärker die konjugierte Base und umgekehrt. 1.4 Die protochemische Spannungsreihe Je nach Lage des Gleichgewichts werden Säuren bzw. Basen in Kategorien unterteilt, jedoch nicht scharf voneinander abgegrenzt. Die Säurekonstanten (pKS -Werte) der Säuren sind tabellarisch ihrer Stärke entsprechend zusammengefasst. Ebenso wie bei der elektrochemischen Spannungsreihe (siehe Versuch 10 ROG) gilt, dass stärker protolysierende Substanzen es vermögen, schwächere zu protonieren. 1.4.1 Sonderfall Wasser In einigen Lehrbüchern wird für die Reaktion H3 O+ ⇌ H2 O + H+ in der protochemischen Spannungsreihe der pKS -Wert 0 angegeben, in anderen der Wert −1, 76. Die Erklärung hierfür ist die Möglichkeit, die ablaufende Reaktion ebenso mit der Gleichung H3 O+ + H2 O ⇌ H2 O + H3 O+ zu beschreiben. Das dazu entsprechende Massenwirkungsgesetz würde folgendermaßen lauten: KS = [H3 O+ ] ⋅ [H2 O] [H2 O] ⋅ [H3 O+ ] (9) Da bei dieser Gleichung alle Konzentrationen gekürzt werden können, ergibt dies KS = 1 ⇒ pKS = 0. Wendet man allerdings die bisher immer getroffene Vereinfachung, dass die Konzentration des Wassers als Edukt sich dabei nicht ändert an und bezieht diese in KS mit ein, führt das zu folgendem Massenwirkungsgesetz: KS = [H3 O+ ] ⋅ [H2 O] [H3 O+ ] (10) Nur die Konzentrationen an H3 O+ können hier gekürzt werden, und der Ausdruck KS = [H2 O] verbleibt. Daraus ergibt sich KS ≈ 55, 52 mol l und pKS hätte den Wert von −1, 76. 1.4.2 Sehr starke Säuren und Basen Säuren, für die gilt: pKS ≤ 2, werden für gewöhnlich als starke Säuren (z. B. HCl, HBr, HI, HNO3 , H2 SO4 (beide Protolysestufen), HClO4 , HMnO4 , H2 CrO4 ) bezeichnet; das Gleiche gilt für Basen mit pKS ≥ 12 (z. B. NaOH, KOH). Bei diesen Säuren/Basen findet ein vollständiger 98 Säure-Base-Gleichgewichte Übertrag des Protons (vollständige Protolyse) statt. Allgemein gilt: [H+ ] = 10−7 + c0 (HA) bzw. [OH– ] = 10−7 mol l mol l + c0 (B) (mit c0 (X) = Ausgangskonzentration des Stoffes X). Wenn für die Konzentration einer sehr starken Säure bzw. Base die Bedingung c0 (HA) bzw. c0 (B) >> 10−7 mol l erfüllt ist, kann die folgende Vereinfachung getroffen werden: [H ] = c0 (HA) bzw. [OH ] = c0 (B). Daraus ergibt sich: + – pH = − log[H+ ] = − log c0 (HA) für starke Säuren bzw. pH = − log[H+ ] = 14 + log[OH− ] = 14 + log c0 (B) für starke Basen. 1.4.3 Mäßig starke / schwache Säuren bzw. Basen Der pKS -Bereich zwischen 2 und ungefähr 3, 5 wird meistens als Bereich mittelstarker Säuren bezeichnet, pKS > 4 dann als schwach. Bei Basen ist der Bereich von 7 bis 10 der schwache Bereich, 10, 5 bis 12 der mittelstarke. Bei diesen Säuren/Basen findet immer unvollständige Protolyse statt. Für eine Säure mit dem Protolysegleichgewicht HA + H2 O ⇌ H3 O+ + A− gilt das folgende Massenwirkungsgesetz: K= [H3 O+ ] ⋅ [A− ] [HA] (11) Nach der Gleichgewichtseinstellung liegt folgendes vor: [H3 O+ ] = [A− ] = α ⋅ c0 (HA) und (12) [HA] = c0 (HA) − α ⋅ c0 (HA) (13) wobei α der Protolysegrad ist, also der Bruchteil der Säure, der protolysiert ist (mit 0 ≤ α ≤ 1). Daraus ergibt sich für das MWG: KS = α2 ⋅ c20 (HA) (1 − α) ⋅ c0 (HA) woraus folgt: (14) entspricht (15) (1 − α) ⋅ KS = α2 ⋅ c0 (HA) α2 ⋅ c0 (HA) + α ⋅ KS − KS = 0 (16) Für α ergibt sich somit folgende Beziehung: α= −KS ± √ KS2 + 4 ⋅ KS ⋅ c0 (HA) (17) 2 ⋅ c0 (HA) Diese exakte Ableitung benötigt man nur für Säuren mit KS c0 (HA) des mittelstarken Bereichs (z.B. HClO, H2 SO3 , H3 PO4 ) Für > 10−2 , also vor allem diejenigen KS c0 (HA) < 10−2 vereinfacht sich die Rechnung, da die Protolyse nur in sehr geringem Umfang stattfindet. Dabei gilt: [HA] ≈ c0 (HA) und außerdem [H3 O+ ] = [A− ]. Gleichung 11 vereinfacht sich dadurch zu: K= [H3 O+ ] ⋅ [A− ] [H3 O+ ]2 ≈ [HA] c0 (HA) (18) Säure-Base-Gleichgewichte 99 Für [H3 O+ ] gilt somit: [H3 O+ ] = √ KS ⋅ c0 (HA) (19) Somit ergibt sich für den pH-Wert: 1 pH = (pKS − log c0 (HA)) 2 Für eine schwache Base mit KB c0 (B) (20) < 10−2 und damit [B] ≈ c0 (B) gilt analog: 1 pOH = (pKB − log c0 (B)) 2 (21) Für den pH-Wert ergibt sich mit Hilfe von Gleichung 6: 1 pH = 14 − ( (pKB − log c0 (B))) 2 (22) 1.5 Puffersysteme Ein Puffer ist eine Mischung aus einer Säure und ihrer konjugierten Base (bzw. einer Base und ihrer konjugierten Säure). Puffergemische haben große praktische Bedeutung, da sie den pH-Wert bei Säure- bzw. Basenzugabe nur wenig ändern. Der pH-Wert von Puffersystemen wird durch die Henderson-Hasselbalch-Gleichung beschrieben (entspricht dem Massenwirkungsgesetz für die Protolyse der Säure HA in Gegenwart ihrer konjugierten Base A− : HA + H2 O ⇌ H3 O+ + A− wie in Gleichung 11 beschrieben. Daraus ergibt sich: pKS = pH − log [A− ] [HA] (23) Für den pH-Wert einer Pufferlösung ergibt sich somit folgende Beziehung: pH = pKS + log [A− ] [HA] (24) Im Zähler des Arguments des Logarithmus steht dabei immer die Konzentration der Base und im Nenner immer die Konzentration der Säure. Für äquimolare Mengen an [A− ] und [HA] gilt [A− ] = [HA]. Daraus folgt pH = pKS . Werden also äquimolare Mengen einer Säure und ihrer konjugierten Base (bzw. umgekehrt) gemischt, so entspricht der pH-Wert der Lösung dem pKS -Wert der Säure. Die Änderung des pH-Werts bei Zugabe von starker Säure/Base zu einem Puffersystem verläuft folgendermaßen: Zugabe von starker Säure: A − ⇒ + HX ⇌ HA + X [A− ] [HA] − [A− ] und damit log [HA] wird kleiner ⇒ pH-Wert sinkt Zugabe von starker Base: B + HA ⇌ HB + + A− ⇒ [A− ] [HA] [A− ] und damit log [HA] wird größer ⇒ pH-Wert steigt Für jede 10-fache Veränderung des Verhältnisses [A− ] [HA] ändert sich der pH-Wert nur um eine Einheit. Ein Puffer aus HA und der konjugierten Base A− kann daher externe, stärkere Säuren 100 Säure-Base-Gleichgewichte HX bzw. Basen B durch die Reaktionen A− + HX ⇌ HA + X − bzw. B + HA ⇌ HB + + A− abfangen. Dabei gelten nach Henderson-Hasselbalch folgende Grenzen: [A− ] [HA] [A− ] [HA] von 10 ∶ 1 bis 1 ∶ 10: von 100 ∶ 1 bis 1 ∶ 100: 1 bzw. + log 10 pH = pKS + log 10 1 pH = pKS + log 1 100 bzw. + log pH = pKS ± 1 pH = pKS ± 2 100 1 Ein Maß für die Fähigkeit eines Puffers externe Säuren und Basen gleichermaßen abzufangen ist die Pufferkapazität. Sie entspricht der Stoffmenge einer starken Säure bzw. Base, die man zu einem Puffer zugeben kann, ohne dass sich der pH-Wert um mehr als eine Einheit ändert. Die Pufferkapazität ist bei [A− ] = [HA] am höchsten. Dann gilt mit der Henderson-HasselbalchGleichung 23 pH = pKS . Aus diesem Grund sollte man sich bei der Wahl eines Puffers für Substanzen entscheiden, deren pKS -Wert möglichst nahe am erforderlichen pH-Wert liegt. Die wichtigsten Puffer sind: H3 PO4 / H2 PO−4 H2 PO−4 HPO2− 4 / / HPO2− 4 PO3− 4 pKS = 2, 15 pKS = 7, 20 pKS = 12, 15 pKS = 3, 74 HCOOH / HCOO− − H3 C–COOH / H3 C–COO NH3 / NH+4 pKS = 4, 76 pKS = 9, 24 Puffermischungen besitzen eine große Bedeutung in der Analytik. Sehr häufig kann man sich die Schwerlöslichkeit von Verbindungen zum Abtrennen und späteren Identifizieren zunutze machen (siehe Versuch 11 FLG). Oft hängt die Konzentration der zur Fällung erforderlichen Gegenionen 2− vom pH-Wert ab (z. B.: CrO2− 4 , S ). 1.6 Säure-Base Indikatoren Indikatoren sind meist organische Farbstoffe, die selbst mittelstarke oder schwache Säuren bzw. Basen sind und bei denen sich entweder die protonierte und die deprotonierte Form farblich voneinander unterscheiden (zweifarbige Indikatoren) oder nur eine Form farbig ist (einfarbiger Indikator). Der pH-Wert, bei dem der Farbumschlag erfolgt, hängt dabei vom pKS Wert der Indikatorsäure bzw. vom pKB -Wert der Indikatorbase ab. Bei zweifarbigen Indikatoren erfolgt der Umschlag innerhalb eines Konzentrationsintervalls, bei einfarbigen Indikatoren ist der Farbumschlag durch den Wegfall oder das Hinzukommen der Farbe meist schärfer. 2 Aufgabenstellung, Versuchsanleitung und -auswertung 2.1 pH-Wert Abschätzung und Bestimmung 2.1.1 Aufgabenstellung Bei diesem Versuch sollen die pH-Werte verschiedener Lösungen abgeschätzt und danach bestimmt werden, um festzustellen welche Verbindungen in Wasser sauer, alkalisch oder neutral reagieren. Säure-Base-Gleichgewichte 101 2.1.2 Versuchsanleitung In 10 Reagenzgläser werden folgende Lösungen gegeben: 0,1 molare Salzsäure, 0,01 molare Salzsäure, Leitungswasser, demineralisiertes Wasser, Natriumcarbonatlösung, Natriumhydrogencarbonatlösung, Natriumacetatlösung, Kaliumnitratlösung, Ammoniumchloridlösung, Hydroxylammoniumchloridlösung (Alle Lösungen müssen aus den entsprechenden Chemikalien und demineralisiertem Wasser hergestellt werden. Die zur Verfügung stehende Salzsäure ist 37%ig (Massenprozent) und ist damit ungefähr 10 molar). Die pH-Werte dieser Lösungen werden zunächst mit Ihrem Vorwissen abgeschätzt und dann anschließend sowohl mit Indikatorpapier (mit Glasstab auftüpfeln, sparsam verwenden) als auch mit dem pH-Meter bestimmt (Glaselektrode des pH-Meters vor jeder Verwendeung sehr gut mit demineralisiertem Wasser spülen). 2.1.3 Versuchsauswertung ● Formulieren Sie die Reaktionsgleichungen zur Reaktion aller oben genannten Substanzen in Wasser. ● ● ● Welche pKS -Werte besitzen die oben genannten Substanzen? (Literaturstudium) Erklären Sie den leicht sauren pH-Wert des demineralisierten Wassers. Berechnen Sie die pH-Werte von 0,1 und 0,01 molarer Salzsäure. 2.2 Verwendung geeigneter Indikatoren 2.2.1 Aufgabenstellung Bei diesem Versuch soll ermittelt werden, welche Indikatoren für welchen pH-Bereich geeignet sind. 2.2.2 Versuchsanleitung In jeweils fünf Reagenzgläser wird 0,1 molare Salzsäure, 0,1 molare Essigsäure, 0,1 molare Ammoniaklösung und 0,1 molare Natronlauge gegeben und die pH-Werte dieser Lösungen werden mit dem pH-Meter bestimmt. Jede dieser Lösungen wird nun (in unterschiedlichen Reagenzgläsern) mit 5 verschiedenen Indikatorlösungen versetzt, nämlich Methylorangelösung, Methylrotlösung, Bromthymolblaulösung, Phenolphthaleinlösung und Universalindikatorlösung. (Die verwendeten Lösungen werden selbst aus den vorhandenen Lösungen bzw. Feststoffen hergestellt. Die zur Verfügung stehenden Lösungen sind 37% Salzsäure, 25% Ammoniaklösung und 96% Essigsäure (alle Angaben in Massenprozent). 2.2.3 Versuchsauswertung ● ● Bestimmen Sie den Umschlagsbereich der einzelnen Indikatoren. Wie funktioniert der Universalindikator? 102 Säure-Base-Gleichgewichte 2.3 Titration von Essigsäure 2.3.1 Aufgabenstellung Bei diesem Versuch soll die Stoffmenge einer Essigsäurelösung unbekannter Konzentration mit Hilfe von Säure-Base-Titration mit 0,1 molarer Natronlauge bestimmt werden. 2.3.2 Versuchsanleitung Der vom Assistent ausgegebene Analysenkolben wird (vorsichtig) bis zur 100 ml Marke mit demineralisiertem Wasser aufgefüllt und durch Umschütteln durchmischt. Mit Hilfe einer 25 ml Vollpipette werden 25 ml dieser Lösung in einen Erlenmeyerkolben gegeben und auf ca. 100 ml mit demineralisiertem Wasser aufgefüllt, mit wenigen Tropfen Phenolphthaleinlösung versetzt und wieder durch vorsichtiges Schütteln gemischt. Die Bürette wird bis zur Nullmarke mit NaOH-Maßlösung befüllt (etwas überbefüllen und in ein Becherglas ablassen bis die Nullmarke erreicht wird). Es wird bis zum Farbumschlag der Lösung von farblos nach rotviolett titriert (erfahrungsgemäß können die ersten 5 ml der Natronlauge etwas schneller abgelassen werden, ab dann vorsichtig titrieren und den Erlenmeyer-kolben kontinuierlich schwenken). Diese Titration wird ein zweites Mal durchgeführt um das zuvor erhaltene Ergebnis zu verifizieren, wobei die Bürette nicht wieder neu befüllt wird, sondern der Inhalt bis zur nächsten vollen ml Marke abgelassen und von dort aus weitertitriert wird. 2.3.3 Versuchsauswertung ● ● Wie hoch ist die Stoffmenge der unbekannten Essigsäurelösung? Bei welchem pH-Wert findet die Neutralisation statt (Erklärung)? 2.4 Berechnung einer Puffermischung Bei diesem Versuch soll mit Hilfe der Henderson-Hasselbalch-Gleichung eine Pufferlösung berechnet werden, die bei einem vorgegebenen pH-Wert ihre maximale Pufferwirkung entwickelt. Aus Natriumacetat-Trihydrat-Lösung und einer 0,1 molaren Essigsäure soll eine Puffermischung hergestellt werden, die bei einem pH-Wert von 5 ihre maximale Pufferwirkung entfaltet. Berechnen Sie die Konzentration der dafür benötigten Natriumacetat-Trihydrat-Lösung in 10 ml einer 0,1 molaren Essigsäure. Welche Einwaage von Natriumacetat-Trihydrat muss in 10 ml 0,1 molarer Essigsäure gelöst werden, um die geforderte Pufferlösung herzustellen? Säure-Base-Gleichgewichte 103 3 Geräte und Chemikalien Eine Geräteliste liegt im Platz aus Chemikalien HClaq Gefahrenbezeichnung Gefahr (GHS05) Na2 CO3 NaHCO3 NaCH3 COO ⋅ 3 H2 O KNO3 NH4 Cl (NH3 OH)Cl Achtung – – Achtung Achtung Achtung Essigsäure Gefahr (GHS05) NH3 Gefahr (GHS05, 09) aq (GHS07) (GHS03) (GHS07) (GHS07, 09) NaOH Gefahr (GHS05) Methylorange Methylrot Bromthymolblau Phenolphthalein Universalindikator Gefahr (GHS06) – – Gefahr (GHS06) – H-Sätze 314, 335, 290 P-Sätze 301+330+331, 305+351+338, 280 319 260, 305+351+338 – – – – 272, 210 – 302, 319 305+351+338 351, 315, 319, 317, 373, 273, 281, 302+352, 308+313, 400, 290, 302+312 305+351+338 226, 314 301+330+331, 305+351+338, 280 314, 335, 400 301+330+331, 305+351+338, 280, 273 314, 290 301+330+331, 305+351+338, 280 301 – – – – – 350, 341, 361f 201, 281, 308+313 – – 4 Literatur [1] A. F. Holleman, E. Wiberg: Lehrbuch der Anorganischen Chemie, 102. Aufl., Walter de Gruyter Verlag, 2007. [2] M. Binnewies, M. Jäckel, H. Willner, G. Rayner-Canham: Allgemeine und Anorganische Chemie, 1. Auflage, Spektrum Verlag, 2004. [3] G. Jander, E. Blasius: Einführung in das Anorganisch-Chemische Praktikum, 15. Auflage, S. Hirzel Verlag, 2005; Lehrbuch der Analytischen und Präparativen Anorganischen Chemie, 16. Auflage, S. Hirzel Verlag, 2006. [4] E. Fluck, M. Becke-Göhring: Einführung in die Theorie der quantitativen Analyse, 7. Auflage, Steinkopff Verlag, 1990. Redoxgleichgewichte 105 Versuch 10 (ROG) Redoxgleichgewichte 1 Theorie 1.1 Oxidation und Reduktion In früheren Zeiten wurden die Begriffe Oxidation und Reduktion mit der Aufnahme und der Abgabe von Sauerstoff in Verbindung gebracht. Ebenso wie die Arrhenius’sche Säure-Base Theorie s. Versuch 9, SBG) ist dies zwar nicht falsch, aber umfasst auch nicht alle Reaktionen, die zur Klasse der Redoxreaktionen zu zählen sind. Die erweiterten Definitionen sind daher: Oxidation: Abgabe von Elektronen eines Atoms oder Ions, die Oxidationsstufe wird größer (die Oxidationszahl wird erhöht) Reduktion: Aufnahme von Elektronen eines Atoms oder Ions, die Oxidationsstufe wird kleiner (die Oxidationszahl wird verringert). Oxidationsmittel: Ein Stoff, der in der Lage ist, andere Stoffe zu oxidieren (wird dabei selbst reduziert). Reduktionsmittel: Ein Stoff, der in der Lage ist, andere Stoffe zu reduzieren (wird dabei selbst oxidiert). In einer Reaktionsgleichung findet niemals nur eine Oxidation oder nur eine Reduktion statt. Immer wenn einer der Stoffe oxidiert wird, muss ein anderer somit reduziert werden. Analog zur Säure-Base Theorie mit konjugierten Säure-Base Paaren existieren auch bei Redox-Reaktionen entsprechende Redox-Paare, in denen ein Atom in einer oxidierten Form (also mit einer höheren Oxidationsstufe) und einer reduzierten Form (also mit einer niedrigeren Oxidationsstufe) vorliegt. Ebenfalls analog zur Säure-Base Reaktion (nach der Brønsted-Theorie) bei der Protonen vom einen auf den anderen Stoff übergehen, ist die Redoxreaktion als eine Reaktion zu betrachten, in der Elektronen vom einen auf den anderen Stoff übertragen werden. 1.2 Die elektrochemische Spannungsreihe Ebenso wie die Säuren und Basen können die Oxidations- und Reduktionsmittel entsprechend ihrer Oxidations- bzw. Reduktionskraft aufgelistet werden. Das Maß hierfür ist das sog. Normalpotential eines Redoxpaares. Dies kann nur relativ zu demjenigen eines anderen Redoxpaares bestimmt werden. Wird beispielsweise ein Zinkstab in eine Kupferlösung getaucht, findet eine Reaktion gemäß Cu2+ + Zn ⇌ Cu + Zn2+ statt, d. h. es wird sich elementares Kupfer auf dem Zinkstab abscheiden. Wird umgekehrt ein Kupferstab in eine Zinklösung getaucht, findet keine Reaktion statt. Es ist dem elementaren Zink also offensichtlich möglich, Kupfer von seiner oxidierten Form Cu2+ in seine elementare Form Cu0 zu reduzieren. Dabei wird Zink selbst vom elementaren Zustand zu Zn2+ oxidiert. Dem Kupfer ist dies im umgekehrten Fall nicht möglich. Daraus 106 Redoxgleichgewichte folgt, dass Zink ein stärkeres Reduktionsmittel als Kupfer ist, wobei mit dieser Anordnung jedoch keine Quantifizierung möglich ist. Die Quantifizierung, wie viel stärker das Zink als Reduktionsmittel im Vergleich zum Kupfer ist, kann mit Hilfe einer gelvanischen Zelle ermittelt werden. Die Potentialdifferenz wird als Spannung gemessen und als elektromotorische Kraft EM K bezeichnet. Um eine reproduzierbare Reihe aufstellen zu können, muss ein Redoxpaar als Nullpunkt gewählt werden und alle anderen Potentiale als Differenz dazu gemessen werden. Daraus ergeben sich dann die Normalpotentiale. Da die meisten der wichtigen Redoxreaktionen in wässriger Lösung ablaufen, wird das Redoxpaar H2 ⇌ 2 H+ + 2 e− als Standard zu Null gesetzt. Üblicherweise wird die reduzierte Form eines Redoxpaares mit negativem Normalpotential als Reduktionsmittel bezeichnet, die oxidierte Form eines Redoxpaares mit positivem Potential als Oxidationsmittel, wobei dies jedoch vom betrachteten System abhängig ist. Die reduzierte Form eines Redoxpaares mit positivem Potential ist durchaus in der Lage gegenüber der oxidierten Form eines Redoxpaares mit höher positivem Potential als Reduktionsmittel zu wirken. Alle Metalle deren M etall / M etallx+ -Redoxpaare ein negatives Normalpotential aufweisen, lösen sich daher in Säuren unter Bildung von H2 . Dies zeigt, dass es dem stärkeren Reduktionsmittel möglich ist, das schwächere aus seiner oxidierten Form in die reduzierte zu bringen, wobei das Reduktionsmittel selbst oxidiert wird (gilt analog für starke Oxidationsmittel). 1.3 Berechnung von Redoxpotentialen - Die Nernst’sche Gleichung Die Abhängigkeit des Potentials eines Redoxsystems (= einer Halbzelle eines galvanischen Elements) von Konzentration und Temperatur wird durch die Nernst’sche Gleichung beschrieben: E = E0 + R⋅T c(oxidierte Form) ⋅ ln z⋅F c(reduzierte Form) (1) mit R = Gaskonstante (8,314 J⋅K−1 ⋅mol−1 ), T = Temperatur (bei Normalbedingungen 298 K), F = Faraday-Konstante (berechnet sich aus e ⋅ NA = 1, 602 ⋅ 10−19 C ⋅ 6, 022 ⋅ 1023 mol−1 = 96485 C⋅mol−1 ), z = Zahl der bei dem betrachtetem Redoxsystem übergehenden Elektronen. Bei Normalbedingungen und Verwendung des dekadischen Logarithmus anstelle des natürlichen vereinfacht sich die Gleichung zu: E = E0 + c(oxidierte Form) 0, 059 V ⋅ log z c(reduzierte Form) (2) Beispiel: Berechnung des Potentials des Daniell-Elements (Galvanisches Element aus einer Cu/Cu2+ - und einer Zn/Zn2+ -Halbzelle): Redox-Paar Redoxpotential bei 25°C Normalpotential Zn ⇌ Zn V 0 EZn = EZn + 0,059 ⋅ log[Zn2+ ] 2 V 0 ECu = ECu + 0,059 ⋅ log[Cu2+ ] 2 0 EZn = −0, 76 V 2+ + 2 e− Cu ⇌ Cu2+ + 2 e− 0 ECu = +0, 34 V Redoxgleichgewichte 107 Die EM K erhält man aus der Differenz der beiden Halbelemente: 0 0 ∆E = ECu − EZn = ECu − EZn + 0, 059 V [Cu2+ ] ⋅ log 2 [Zn2+ ] (3) 0 0 Für [Cu2+ ] = [Zn2+ ] gilt: ∆E = ECu − EZn = ECu − EZn = 1, 10 V. Da während des Betriebs des Daniell-Elements die [Zn2+ ] Konzentration wächst und die [Cu2+ ] Konzentration abnimmt, muss nach obiger Gleichung auch die Spannung des Elements abnehmen. 1.4 Aufstellen von Redoxgleichungen Reaktionsgleichungen von Redoxreaktionen sind wichtig für das Reaktionsverständnis in der Chemie. Auch hierbei muss natürlich beachtet werden, dass zu jeder Oxidation immer eine Reduktion stattfindet und die Elektronenbilanz ausgeglichen sein muss. Hierfür stellt man am besten Teilgleichungen auf. Nachfolgend ein Beispiel, die Oxidation von Natrium in Wasser: Oxidation: Na Ð→ Na+ + e− Reduktion: 2 H2 O + 2 e− Ð→ 2 OH– + H2 Um eine ausgeglichene Elektronenbilanz zu erreichen muss die Gleichung der Oxidation verdoppelt werden: Bilanz: 2 Na + 2 H2 O Ð→ 2 Na+ + 2 OH– + H2 Befinden sich die entstehenden Ionen nach der Reaktion in Lösung, muss dem Rechnung getragen werden, was bedeutet, dass die Formulierung 2 NaOH anstelle von 2 Na+ + 2 OH– in obiger Gleichung als falsch zu erachten wäre. Werden zum richtigen Beschreiben des Produkts Oxid3+ Anionen benötigt (z. B. das Entstehen von CrO2– 4 aus der Oxidation von Cr ), werden bei Reaktion in alkalischer Lösung so viele OH– -Ionen wie nötig zugeführt, die verbleibenden Protonen werden ebenfalls mit OH– -Anionen abgefangen: z. B. 2 Cr3+ + 3 H2 O2 + 10 OH– Ð→ 2 CrO2– 4 + 8 H2 O. Der gleiche Vorgang in saurer Lösung benötigt dann so viele H2 O-Moleküle wie Oxid-Anionen benötigt werden, die H+ -Kationen werden frei: – 2– + z. B. 2 Mn2+ + 5 S2 O2– 8 + 8 H2 O Ð→ 2 MnO4 + 10 SO4 + 16 H . Müssen bei einer Redoxreaktion freiwerdende Oxid-Anionen abgefangen werden, werden im Sauren soviele H+ -Kationen eingesetzt, dass alle Oxid-Anionen zu Wasser reagieren: z. B. 2 MnO–4 + 10 I– + 16 H+ Ð→ 2 Mn2+ + 5 I2 + 8 H2 O. Der gleiche Vorgang im Alkalischen benötigt so viele Wasser-Moleküle als Edukt, dass freiwerdende O2– -Anionen als OH– auf der Produktseite abgefangen werden können: 2– – z. B. 2 MnO–4 + 3 SO2– 3 + H2 O Ð→ 2 MnO2 + 3 SO4 + 2 OH . In saurer Lösung entstehen niemals OH– -Anionen und können auch nicht als Edukt eingesetzt werden. Das Gleiche gilt für H+ -Kationen in alkalischer Lösung. 1.5 Kom- bzw. Synproportionierung und Disproportionierung Liegen in einer Reaktionsmischung Atome eines Elements sowohl in einer hohen als auch in einer niedrigen Oxidationsstufe vor und zeigen nach der Redoxreaktion alle Atome dieses Elements eine gemeinsame ”mittlere” Oxidationsstufe, dann nennt man diesen Vorgang Kom- bzw. Synproportionierung (z. B. 2 MnO–4 + 3 Mn2+ + 4 OH– Ð→ 5 MnO2 + 2 H2 O). Befindet sich ein 108 Redoxgleichgewichte Atom in einer ”mittleren” Oxidationsstufe, so ist es möglich, dass es eine Redoxreaktion mit sich selbst eingeht, so dass danach die Atome dieses Elements zum Einen in einer höheren und zum Anderen in einer niedrigeren Oxidationsstufe als zuvor vorliegen. Diesen Vorgang nennt man Disproportionierung (z. B. Zerfall von Wasserstoffperoxid H2 O2 Ð→ H2 O + 1 2 O2 ). 2 Aufgabenstellung, Versuchsanleitung und -auswertung 2.1 Elektrochemische Spannungsreihe 2.1.1 Aufgabenstellung Eine Einordnung der untersuchten Redoxpaare in die elektrochemische Spannungsreihe soll untersucht werden. 2.1.2 Versuchsanleitung a) Vier Reagenzgläser werden jeweils zu ca. 2 cm hoch mit verdünnter Salzsäure (1 Teil Wasser : 1 Teil konz. Salzsäure) gefüllt. Danach werden ein Stück Magnesium-Band, eine Zink-Granalie oder -Raspel, etwas Nickelpulver und einige blanke Kupferspäne zugegeben (ggf. diese vorher etwas anschleifen) b) In drei Reagenzgläsern werden Zinkstücke vorsichtig mit wenigen Tropfen konzentrierter Salzsäure, verdünnter (1 Teil Wasser : 1 Teil konz. Salpetersäure) und konzentrierter Salpetersäure versetzt. Dieser Versuch wird mit blanken Kupferspänen wiederholt. 2.1.3 Versuchsauswertung Formulieren Sie die Reaktionsgleichungen zu den einzelnen Versuchsteilen mit Teilgleichungen unter Angabe aller Oxidationsstufen und dokumentieren Sie die verschiedenen Reaktionsgeschwindigkeiten. 2.2 Abhängigkeit des Redoxpotentials vom pH-Wert 2.2.1 Aufgabenstellung Bei diesem Versuch soll erkannt werden, dass das Redoxpotential recht häufig auch vom pH-Wert der entsprechenden Lösung abhängig ist. 2.2.2 Versuchsanleitung a) Eine KNO3 -Lösung wird 3 cm hoch in ein Reagenzglas gegeben und mit wenig Schwefelsäure angesäuert. Zu dieser Lösung wird ca. 1 cm hoch (NH4 )2 Fe[SO4 ]2 -Lösung zugegeben und mit der KNO3 -Lösung durchmischt. Diese Mischung wird nun mit konzentrierter Schwefelsäure vorsichtig unterschichtet, indem man das Reagenzglas schräg hält und die konzentrierte Schwefelsäure an der inneren Wand herunterfließen lässt. An der Berührungszone entsteht ein brauner bis violetter Ring. Redoxgleichgewichte b) 109 In einem Becherglas wird etwas Fe[SO4 ] in wenig Wasser gelöst und eine Spatelspitze KNO3 sowie 2-3 NaOH-Plätzchen hinzugegeben. Das Becherglas wird mit einem Uhrglas, an dessen Unterseite ein Stück feuchtes Indikatorpapier geklebt ist, bedeckt. Das Indikatorpapier wird mit der Zeit blau, falls nicht kann das Becherglas vorsichtig mit dem Bunsenbrenner erhitzt werden (Sieden der Reaktionsmischung ist zu verweiden, da sonst Spritzer der alkalischen Reaktionsmischung das Indikatorpapier blau färben würden). 2.2.3 Versuchsauswertung ● Formulieren Sie die Reaktionsgleichungen zu den einzelnen Versuchsteilen mit Teilgleichungen unter Angabe aller Oxidationsstufen. ● Welche Normalpotentiale besitzt das Redoxpaar Fe2+ /Fe3+ in saurer und alkalischer Lösung (Literaturstudium)? 2.3 Redox-Amphoterie, Dis- und Synproportionierung 2.3.1 Aufgabenstellung Bei diesem Versuch sollen die Begriffe Redoxamphoterie, Disproportionierung und Kom- bzw. Synproportionierung anhand eines Experiments geklärt werden. 2.3.2 Versuchsanleitung a) Wenige ml einer KMnO4 -Lösung (ca. 2 cm hoch) werden in einem Reagenzglas mit wenigen Tropfen einer alkalischen MnSO4 -Lösung (eine Spatelspitze MnSO4 und eine NaOH-Perle werden vollständig in Wasser gelöst) versetzt. b) Zu wenigen ml einer 30 % H2 O2 -Lösung (nicht höher als 1 cm im Reagenzglas) wird in einem Reagenzglas eine Spatelspitze MnO2 hinzugegeben. In das entstehende Gas wird ein glimmender Span gehalten. c) Einige ml einer stark verdünnten 30 % H2 O2 -Lösung (ca. 1 cm hoch) werden in einem Reagenzglas mit NaOH stark alkalisch gemacht. Dazu werden einige Tropfen einer MnSO4 Lösung gegeben. d) Einige ml KMnO4 -Lösung (ca. 1 cm hoch) werden mit wenigen Tropfen Schwefelsäure angesäuert und tropfenweise mit 30% H2 O2 -Lösung versetzt. 2.3.3 Versuchsauswertung ● Formulieren Sie die Reaktionsgleichungen zu den einzelnen Versuchsteilen mit Teilgleichungen unter Angabe aller Oxidationsstufen. ● Wie werden diese Reaktionen aus den Versuchsteilen a) und b) bezeichnet und wozu dient der glimmende Span beim zweiten Reaktionsteil? ● Wie wirkt H2 O2 in den Versuchsteilen c) und d)? 110 Redoxgleichgewichte 2.4 Redox-Titration einer Cu2+ -Kationen-Lösung 2.4.1 Aufgabenstellung Bei diesem Versuch, soll die Stoffmenge einer unbekannten Lösung von Cu2+ mit Hilfe einer iodometrischen Redoxtitration bestimmt werden. 2.4.2 Versuchsanleitung Die Analysenlösung wird im Meßkolben bis zur 100 ml Marke mit demineralisiertem Wasser aufgefüllt und gut umgeschüttelt. Anschließend werden 25 ml der Analysenlösung in einen Erlenmeyerkolben abpipettiert (mit einer 25 ml Vollpipette), mit demineralisiertem Wasser auf etwa 150 ml aufgefüllt, mit ca. 5 ml Essigsäure versetzt und mit 2 g in 10 ml demineralisiertem Wasser gelöstem Kaliumiodid versetzt. Es entsteht ein Niederschlag (um welche Verbindung handelt es sich hierbei?). Die Lösung wird mit ca. 3 ml einer gesättigten Stärkelösung (2 Spatelspitzen mit Stärkepulver werden dafür in einem mit Wasser zu 1 3 gefüllten Reagenzglas in der Hitze gelöst), die als Indikator dient, versetzt und färbt sich dabei blau. Die Bürette wird bis zur Nullmarke mit Na2 S2 O3 -Maßlösung befüllt (etwas überbefüllen und in ein Becherglas ablassen bis die Nullmarke erreicht wird) und die Analysenlösung wird titriert bis zum Umschlag der Farbe von blau nach farblos (die Blaufärbung sollte mindestens eine Minute verschwunden bleiben). Diese Titration wird ein zweites Mal durchgeführt, um das zuvor erhaltene Ergebnis zu verifizieren, wobei die Bürette nicht wieder neu befüllt wird, sondern der Inhalt bis zur nächsten vollen ml Marke abgelassen und von dort aus weitertitriert wird. 2.4.3 Versuchsauswertung ● Berechnen Sie noch am Versuchstag die Stoffmenge an Cu2+ , die in der Analysenlösung enthalten ist. ● Formulieren Sie die Reaktionsgleichungen zu den einzelnen Versuchsteilen mit Teilgleichungen unter Angabe aller Oxidationsstufen. Zeichnen Sie die Strukturformel des Thiosulfatund des Tetrathionat-Anions mit Angabe der Oxidationsstufen der einzelnen Atome. ● Formulieren Sie unter Angabe der Normalpotentiale (Literatur) das Redoxpotential der bei2– den Redoxpaare Cu2+ /Cu+ und I– /ceI2, sowie I– /I2 und S2 O2– 3 /S4 O6 (Nernst-Gleichung). Redoxgleichgewichte 111 3 Geräte und Chemikalien Eine Geräteliste liegt im Platz aus Chemikalien HClaq Magnesium Zink Nickel Kupfer H2 (Produkt) HNO3 Gefahrenbezeichnung Gefahr (GHS05) Gefahr (GHS02) – Gefahr (GHS06) – Gefahr (GHS02) Gefahr (GHS05) H-Sätze 314, 335, 228, 261, – 251, 372, – 220 272, 314, P-Sätze 290 301+330+331, 305+351+338, 280 252 210, 402+404 – 317, 412 281, 273, 308+313, 302+352 – 210, 377, 381, 403 301+330+331, 305+351+338, 260, 290 280 KNO3 Achtung (GHS03) 272, 210 – H2 SO4 Gefahr (GHS05) 314, 290 301+330+331, 305+351+338, 280, 309, 310 (NH4 )2 Fe[SO4 ]2 – – – FeSO4 Achtung (GHS07, 09) 302, 319, 315 305+351+338, 302+352 NH3 (Produkt, g) Gefahr (GHS05, 09) EUH071, 331, 221, 210, 260, 273, 280, 304+340+315, 314, 280, 400 303+361+353+315, 377, 381, 403, 305+351+338+315, 405 NaOH Gefahr (GHS05) 314, 290 301+330+331, 305+351+338, 280 MnSO4 Achtung (GHS07, 09) 373, 411 273, 314 MnO2 Gefahr (GHS08) 272, 302+332 – KMnO4 Gefahr (GHS03, 08, 272, 302, 410 210, 273 09) H2 O2 -Lösung (30 %) Gefahr (GHS03, 05) 272, 302,318 210, 305+351+338, 313 O2 (Produkt) Gefahr (GHS03) 270 244, 220, 370+376, 403 CuSO4 ⋅ 5 H2 O Achtung (GHS07, 09) 302, 315, 319, 410 305+351+338, 302+352, 273 Essigsäure Gefahr (GHS05) 226, 314 301+330+331, 305+351+338, 280 KI – – – Stärke – – – Na2 S2 O3 – – – 4 Literatur [1] A. F. Holleman, E. Wiberg: Lehrbuch der Anorganischen Chemie, 102. Aufl., Walter de Gruyter Verlag, 2007. [2] M. Binnewies, M. Jäckel, H. Willner, G. Rayner-Canham: Allgemeine und Anorganische Chemie, 1. Auflage, Spektrum Verlag, 2004. [3] G. Jander, E. Blasius: Einführung in das Anorganisch-Chemische Praktikum, 15. Auflage, S. Hirzel Verlag, 2005; Lehrbuch der Analytischen und Präparativen Anorganischen Chemie, 16. Auflage, S. Hirzel Verlag, 2006. [4] E. Fluck, M. Becke-Göhring: Einführung in die Theorie der quantitativen Analyse, 7. Auflage, Steinkopff Verlag, 1990. Fällungs- und Löslichkeitsgleichgewichte 113 Versuch 11 (FLG) Fällungs- und Löslichkeitsgleichgewichte 1 Theorie 1.1 Löslichkeit und Löslichkeitsprodukt Die maximale Menge eines Stoffes, die sich in einem Lösemittel lösen kann, ist eine charakteristische Eigenschaft dieses Stoffes. Sie wird als Löslichkeit L bezeichnet und entspricht der Sättigungskonzentration dieses Stoffes in dem Lösemittel. Die Löslichkeit einer Verbindung ist von der Temperatur und sehr stark von der Art des Lösemittels abhängig. Bei den folgenden Betrachtungen soll Wasser das Lösemittel sein und die Temperatur soll den Standardbedingungen (298,15 K bzw. 25°C) entsprechen. Als Beispiel in Abbildung 1.1 ist hier das Gleichgewicht einer gesättigten wässrigen Lösung von AgCl gezeigt, die einen Bodensatz an festem AgCl enthält (nur dann ist die Lösung auch wirklich gesättigt). Im Gleichgewicht werden pro Zeiteinheit ebenso viele Ionenpaare Ag+ + Cl– aus dem Feststoff in Abb. 1.1: Fällungs- / Löslichkeitsgleichgewicht am Beispiel von AgCl Lösung gehen, wie aus der Lösung in das Kristallgitter des festen AgCl eingebaut werden (dynamisches Gleichgewicht). Dieser Prozeß wird durch die Reaktionsgleichung AgClfest ⇌ Ag+ aq + Cl– aq beschrieben. Das Massenwirkungsgesetz mit der stöchiometrischen Gleichgewichtskonstante K für diesen Lösevorgang lautet: [(Ag+ )aq ] ⋅ [(Cl− )aq ] K= [AgClfest ] (1) Der feste Bodenkörper an AgCl ist zwar am Löse-/Kristallisationsvorgang beteiligt, er gehört aber nicht zum homogenen Gleichgewicht der Lösung selber, sondern zu einer zweiten (festen) Phase. Seine Konzentration wird unabhängig davon, wie viel Bodenkörper vorhanden ist, als konstant angesehen und in die Gleichgewichtskonstante mit einbezogen. Daraus resultiert das MWG in der Form: K ⋅ [AgClfest ] = KL = [(Ag+ )aq ] ⋅ [(Cl− )aq ] (2) wobei KL als Löslichkeitsprodukt bezeichnet wird. Das Löslichkeitsprodukt KL ist also das Produkt aus den Konzentrationen an Ag+ und Cl– in der gesättigten Lösung. Der Zahlenwert von KL für AgCl in Wasser bei 25°C beträgt 1, 77 ⋅ 10−10 mol2 . l2 Wird eine gesättigte AgCl-Lösung mit Wasser verdünnt, so wird das Produkt [Ag ] ⋅ [Cl ] in der Lösung kleiner als KL und der Bodenkörper + − beginnt sich so lange aufzulösen, bis [Ag+ ] ⋅ [Cl– ] wieder den Wert des Löslichkeitsprodukts erreicht hat. Falls eine Lösung Ag+ - und Cl– -Ionen in so geringen Konzentrationen enthält, dass das Löslichkeitsprodukt unterschritten ist, so ist diese Lösung nicht gesättigt. Werden nun zusätzlich Ag+ - oder Cl– -Ionen oder beide hinzugegeben, so wird ab dem Moment, ab dem das Löslichkeitsprodukt erreicht wird, die Lösung gesättigt und AgCl beginnt auszufallen. Es kann vorkommen, dass eine Lösung übersättigt ist, d. h. es fällt kein Niederschlag aus, obwohl 114 Fällungs- und Löslichkeitsgleichgewichte KL überschritten ist. Dies hat kinetische Gründe, d. h. das Gleichgewicht (nachdem sich ein Niederschlag bilden sollte) stellt sich nicht oder nur sehr langsam ein. Ist das Löslichkeitsprodukt für eine Verbindung bekannt, kann aus ihm die Löslichkeit der Verbindung berechnet werden. Für die Löslichkeit von AgCl gilt: L(AgCl) = [AgCl]gel. = [Ag+ ] = [Cl− ] (3) KL = [Ag+ ] ⋅ [Cl− ] (4) [Ag+ ] und [Cl− ] in Gleichung 4 durch L(AgCl) aus Gleichung 3 ersetzt ergibt: KL = L2 (AgCl) (5) woraus sich folgende Beziehung für die Löslichkeit L(AgCl) ergibt: L(AgCl) = √ KL Die Löslichkeit von AgCl (in Wasser bei 25°C) beträgt also (6) √ 1, 77 ⋅ 10−10 = 1, 33 ⋅ 10−5 mol l . Gleichung 3 zufolge sind die Konzentrationen an Ag+ und an Cl– der gesättigten Lösung ebenfalls 1, 33 ⋅ 10−5 mol l . Aus der Berechnung der Löslichkeit aus dem Löslichkeitsprodukt ergibt sich zwangsläufig die Stoffmengenkonzentration als Einheit für die Löslichkeit. Die Angaben Löslichkeit und Löslichkeitsprodukt einer Verbindung beinhalten im Grunde die gleiche Information, nämlich ”wie gut (oder schlecht) löst sich eine Verbindung”. Aus dem Löslichkeitsprodukt kann die Löslichkeit berechnet werden und umgekehrt aus der Löslichkeit das Löslichkeitsprodukt. Eine grobe Einteilung der Löslichkeit von Stoffen erfolgt so, dass Verbindungen die eine Löslichkeit L > 1 mol l besitzen als leicht löslich bezeichnet werden und solche die eine Löslichkeit L < 0, 1 mol l besitzen als schwer löslich bezeichnet werden. Üblicherweise wird aber für leicht lösliche Verbindungen die Löslichkeit angegeben (und zwar wieviel Gramm der Verbindung sich in 100 ml Wasser lösen) und für schwerlösliche Verbindungen das Löslichkeitsprodukt KL . Die Zahlenwerte der Löslichkeiten L oder Löslichkeitsprodukte KL sind für sehr viele Verbindungen bekannt und in Lehrbüchern oder Tabellenwerken der Analytischen Chemie zu finden. Im folgenden ist eine Ableitung des Zusammenhangs zwischen Löslichkeitsprodukt und Löslichkeit für den allgemeinen Fall einer Verbindung Ax By , die sich gemäß Ax By ⇌ x Ay+ + y B x− löst beschrieben. Das Löslichkeitsprodukt KL ergibt sich zu: KL = [Ay+ ]x ⋅ [B x− ]y (7) Für die Löslichkeit L(Ax By ) ergibt sich: L(Ax By ) = [Ax By ]gel. = 1 y+ 1 [A ] = [B x− ] x y (8) [B x− ] = y ⋅ L(Ax By ) (9) Damit gilt für [Ay+ ] und für [B x− ]: [Ay+ ] = x ⋅ L(Ax By ) und Fällungs- und Löslichkeitsgleichgewichte 115 Werden die Beziehungen aus 9 in Gleichung 8 eingesetzt ergibt sich: KL = (x ⋅ L(Ax By ))x ⋅ (y ⋅ L(Ax By ))y (10) Somit ergibt sich für das Löslichkeitsprodukt: KL = xx ⋅ y y ⋅ L(Ax By )(x+y) und für die Löslichkeit: L(Ax By ) = √ (x+y) (11) KL ⋅ yy (12) xx Mit Hilfe von Gleichung 11 kann leicht das Löslichkeitsprodukt aus der Löslichkeit und von Gleichung 12 die Löslichkeit aus dem Löslichkeitsprodukt errechnet werden. 1.2 Thermodynamik des Löslichkeitsprodukts 1.2.1 Beeinflussung durch Temperaturänderung Der Wert von KL hängt thermodynamisch gesehen einerseits von der relativen Größe der Gitterenergie (diese beschreibt energetisch den ”Zusammenhalt” der Ionen im Festkörper) und der Summe der Solvatationsenergien (in wässriger Lösung: Hydratationsenergie) ab. Wie jede Gleichgewichtskonstante ist auch KL entsprechend der Gibbs-Helmholtz-Gleichung ∆G = ∆H − T ∆S von der Temperatur abhängig. Die Stärke der Temperaturabhängigkeit wird von der relativen Größe für ∆H und ∆S bestimmt. Es gilt, je größer ∆S; desto größer ist die Temperaturabhängigkeit. 1.2.2 Weitere Faktoren, die die Löslichkeit beeinflussen a) Fremdioniger Zusatz - Einführung der Aktivität Wird die Löslichkeit einer Verbindung nicht in reinem Wasser betrachtet, sondern in einer wässrigen Lösung, die größere Mengen anderer (fremder) Ionen als die der betrachteten Verbindung enthält, wird ihre Löslichkeit erhöht. Dies liegt daran, dass die Ionen der betrachteten Verbindung nicht mehr ideal hydratisiert und unabhängig voneinander vorliegen, sondern sich zu Aggregaten zusammenschließen, was eine scheinbar geringere Konzentration zur Folge hat. Diese scheinbaren Konzentrationen werden als Aktivitäten bezeichnet und sind als effektive oder als wirksame Konzentrationen zu verstehen. Nach der Debye-Hückel-Theorie über die elektrostatischen Wechselwirkungen von Ionen in Elektrolyten gilt: a(X) = f (X) ⋅ c(X) (Einheit mol l ) mit f (X) als dimensionslosem Aktivitätskoeffizient für den gilt (0 ≤ f ≤ 1) und a(X) als Aktivität eines Ions X. Für ideal verdünnte Lösungen wird der Aktivitätskoeffizient zu 1 und die Aktivität entspricht genau der Konzentration. Abweichungen vom idealen Verhalten werden durch einen kleiner werdenden Aktivitätskoeffizienten berücksichtigt. Das Massenwirkungsgesetz gilt streng genommen auch nur dann, wenn Aktivitäten betrachtet werden. Für eine Reaktion, wie das + − Auflösen eines Salzes in Wasser wie beispielsweise KBrf est ⇌ Kaq + Braq gilt dann: Ka = a(K + ) ⋅ a(Br− ) = f (K + ) ⋅ c(K + ) ⋅ f (Br− ) ⋅ c(Br− ). 116 Fällungs- und Löslichkeitsgleichgewichte b) Schwerlösliche Verbindungen, die Komplexe bilden Die Löslichkeit schwerlöslicher Verbindungen, die lösliche Komplexe bilden können, wird sehr stark durch die Anwesenheit von Komplexbildnern beeinflusst. Dies soll am Beispiel von Silberchlorid, welches sich in konzentrierter Ammoniaklösung auflöst beschrieben werden. Ag+ -Ionen können aufgrund der Schwerlöslichkeit von AgCl gut mit Cl– -Ionen gefällt werden. Gibt man allerdings Ammoniak hinzu, löst sich der Niederschlag sukzessive wieder auf. Die zugrunde liegenden Gleichgewichte lauten: AgCl ⇌ Ag+ + Cl− (13) Ag+ + 2 NH3 ⇌ [Ag(NH3 )2 ]+ (14) AgCl + 2 NH3 ⇌ [Ag(NH3 )2 ]+ + Cl− (15) Gleichung 13 beschreibt den Lösungsvorgang von AgCl, Gleichung 14 beschreibt die Komplexbildung von Ag+ -Kationen mit Ammoniak zum Silberdiammin-Komplex und Gleichung 15 die Summe aus 13 und 14. Durch den Komplexierungsprozess 14 werden laufend Ag+ -Ionen aus dem Gleichgewicht entfernt. Dies führt dazu, dass sich das Gleichgewicht 13 nach rechts verschiebt und die Löslichkeit von AgCl steigt. Ob sich eine schwerlösliche Silberverbindung aber tatsächlich in Ammoniak völlig auflöst, hängt vom Löslichkeitsprodukt, von der Komplexbildungskonstante, von der Konzentration des Ammoniaks und natürlich auch von der Menge der Silberverbindung ab, welche sich auflösen soll.Dieselben Zusammenhänge gelten auch für die Komplexbildung von Ag+ mit Cl– -Ionen zum Dichloroargentat-Komplex [AgCl2 ]– . In Abbildung 1.2 ist die maximal mögliche Ag+ - Ionenkonzentration einer Lösung in Abhängigkeit der Cl– -Konzentration wiedergegeben. Wird eine 10−4 molare Ag+ -Lösung hergestellt (z. B. durch Auflösen von AgNO3 ) so enthält die klare Lösung eben gerade 10−4 mol l Ag+ . Werden nun Cl– -Ionen zugegeben (z. B. in Form von HCl), so beginnt ab einer Cl– -Konzentration von etwa 10−6 mol l fes- tes AgCl auszufallen (Punkt 1 in Abbildung 1.2). Bei weiterer Zugabe von Cl− fällt immer mehr AgCl aus, bis bei einer Cl− -Konzentration von etwa 10−3 mol l Abb. 1.2: Komplexierungsverhalten von AgCl bei Zugabe von Salzsäure so viel AgCl ausgefallen ist, dass die Ag+ - Konzentration der Lösung nur noch 10−6 mol l beträgt (Punkt 2 in Abb. 1.2). Wird nun weiter Cl− zugegeben löst sich der Niederschlag unter Bildung des Dichloroargentat-Komplexes [AgCl2 ]− sukzessive wieder auf, bis bei einer Cl− -Konzentration von etwa 0, 5 mol l der Niederschlag wieder völlig aufgelöst ist und eine klare Lösung entstanden ist (Punkt 3 in Abb. 1.2). c) Schwerlösliche Verbindungen, die protolysieren können Die Löslichkeit von Verbindungen, die protolysieren können sind sehr stark vom pH-Wert abhängig. Dies soll am Beispiel von CaCO3 beschrieben werden. CaCO3 ist zwar eine schwerlösliche Verbindung, das beim Lösen entstandene Carbonat-Anion kann jedoch zum Hydrogencarbonat- Fällungs- und Löslichkeitsgleichgewichte 117 Anion und weiter zur gelösten Kohlensäure protolysieren. Die relevanten Gleichungen sind: CaCO3 ⇌ Ca2+ + CO2− 3 (16) + − CO2− 3 + H3 O ⇌ HCO3 + H2 O (17) HCO−3 + H3 O+ ⇌ H2 CO3 + H2 O (18) H2 CO3 ⇌ CO2 + H2 O (19) CaCO3 + 2 H3 O+ ⇌ Ca2+ + CO2 + 3 H2 O (20) – Ca2+ bildet nur mit dem CO2– 3 -Ion eine schwerlösliche Verbindung, mit dem HCO3 -Ion oder der gelösten Kohlensäure nicht. Wird durch Zugabe einer Säure der pH-Wert und damit die H3 O+ Konzentration erhöht, wird das Carbonat-Ion sukzessive protoniert und damit dem Gleichgewicht 16 entzogen. Das Gleichgewicht 16 verschiebt sich nach rechts und CaCO3 löst sich. Ob und wie weit sich eine schwerlösliche Verbindung, deren Anion protolysieren kann, auflöst, hängt von seinem Löslichkeitsprodukt, vom pH-Wert der Lösung und von den Säurekonstanten der zum Anion korrespondierenden Säure ab. Das für CaCO3 beschriebene gilt für alle schwerlöslichen Carbonate, aber z. B. auch für schwerlösliche Sulfide, Oxalate, Hydroxide und Sulfate. Das unterschiedliche Löseverhalten von Verbindungen kann man sich bei Trennungen von Ionen zunutze machen. Beispielsweise ist HgS so schwer löslich, dass Hg2+ -Ionen durch Zugabe von Sulfidionen auch im stark Sauren als HgS ausfallen. Mn2+ -Ionen können unter diesen Bedingungen nicht gefällt werden, erst wenn die Lösung alkalisch gemacht wird fällt auch Mn2+ als MnS aus. 2 Aufgabenstellung, Versuchsanleitung und -auswertung 2.1 Einfluss verschiedener Zusätze auf das Löslichkeitsprodukt 2.1.1 Aufgabenstellung Bei diesem Versuch soll der Einfluss verschiedener Zusätze auf die Löslichkeit erkannt und mit Hilfe des Löslichkeitsgleichgewichts erklärt werden. 2.1.2 Versuchsanleitung a) Einige ml gesättigter NaCl-Lösung werden in jeweils vier Reagenzgläser (ca. 2 cm hoch) gegeben. Es wird dann je Reagenzglas die gleiche Menge an konzentrierter Salzsäure, konzentrierter Natronlauge, konzentrierter Ammoniaklösung und konzentrierter KaliumnitratLösung hinzugefügt. b) Ein bis zwei Tropfen (nicht mehr) Silbernitratlösung werden in einem Reagenzglas (ca. 1 cm hoch) erst mit wenig und danach mit so viel konzentrierter Salzsäure versetzt, dass sich der zuerst beobachtete Niederschlag auflöst. 118 Fällungs- und Löslichkeitsgleichgewichte c) Eine Magnesiumchloridlösung wird im Reagenzglas (ca. 2 cm hoch) mit konzentrierter Natronlauge versetzt. Es fällt ein weißer Niederschlag von Mg(OH)2 aus. Bei einem Überschuss an OH– -Ionen stellt dies praktisch eine quantitative Fällung dar. Die überstehende Lösung wird vom Niederschlag abdekantiert und verworfen. Der Niederschlag wird dann mit einigen ml konzentrierter NH4 Cl-Lösung versetzt. In einem zweiten Reagenzglas wird die analoge Menge Magnesiumchloridlösung mit konzentriertem Ammoniak versetzt. Auch hier entsteht ein Niederschlag, die Fällung ist jedoch auch bei einem Überschuss an Ammoniak nicht quantitativ. 2.1.3 Versuchsauswertung ● Formulieren Sie die Reaktionsgleichungen der Versuchsteile a) und b) und erklären Sie anhand dieser Gleichungen die Vorgänge bei Zugabe der verschiedenen Lösungen. ● Formulieren Sie die Löslichkeitsprodukte für die entstandenen schwerlöslichen Verbindungen. ● Formulieren Sie die Reaktionsgleichungen aus Versuchsteil c) und nennen Sie Gründe für die nicht-quantitative Menge an Niederschlag. 2.2 Abhängigkeit des Löslichkeitsprodukts vom pH-Wert 2.2.1 Aufgabenstellung Bei diesem Versuch soll gezeigt werden, dass das Löslichkeitsprodukt auch von anderen Gleichgewichten, wie z. B. Säure-Base-Gleichgewichten abhängig sein kann. 2.2.2 Versuchsanleitung Zu je einigen ml Kupfer- und Nickelsulfatlösung (ca. 2 cm hoch im jeweiligen Reagenzglas) werden einige Tropfen konzentrierte Salzsäure gegeben (pH ca. 0). Beide Lösungen werden daraufhin mit H2 S-Wasser versetzt. Zu der mit Schwefelwasserstoff versetzten Nickelsulfat-Lösung werden dann einige ml Ammoniaklösung gegeben. 2.2.3 Versuchsauswertung ● Ermitteln Sie die Reaktionsgleichung und die Löslichkeitsprodukte von Nickel- und Kupfersulfid (Literaturstudium). ● Geben Sie eine Erklärung, warum Kupfersulfid im Sauren ausfällt, Nickelsulfid hingegen erst im Alkalischen. 2.3 Fällungstitration einer Bromid-Lösung 2.3.1 Aufgabenstellung Bei diesem Versuch, soll die Stoffmenge Br– -Lösung unbekannter Konzentration mit Hilfe einer Fällungstitration mit einer AgNO3 -Maßlösung bestimmt werden. Fällungs- und Löslichkeitsgleichgewichte 119 2.3.2 Versuchsanleitung Der Bromid-Anionen enthaltende Analysenkolben wird mit demineralisiertem Wasser auf genau 100 ml aufgefüllt. Daraus werden 25 ml der Analysenlösung entnommen (mit Hilfe einer 25 ml Vollpipette) und mit demineralisiertem Wasser in einem Erlenmeyerkolben auf 100 ml verdünnt. Zu dieser Lösung werden ca. 1 ml 0,1 molare Essigsäure und ca. 5 Tropfen Eosin-Lösung zugegeben. Dann wird mit 0,1 molarer AgNO3 -Maßlösung nicht zu schnell und unter ständigem Umschütteln von orange über milchig rosa bis zum Farbumschlag nach pink titriert. Ein langsames Herantasten an den Äquivalenzpunkt ist ratsam, da der Umschlag von orange-rosa nach pink schlagartig erfolgt. (Ein Tropfen kann unter Umständen schon zu viel sein). Wird übertitriert, dann ist ein Ausflocken in der Lösung zu beobachten, es bildet sich ein farbiger Niederschlag, die überstehende Lösung wird transparent. 2.3.3 Versuchsauswertung ● ● Formulieren Sie die Reaktionsgleichungen der Fällungsreaktion. Berechnen Sie noch am Versuchstag die Bromid-Ionen-Stoffmenge der Analysenlösung. 3 Geräte und Chemikalien Eine Geräteliste liegt im Platz aus Chemikalien NaCl HClaq NaOH NH3 aq KNO3 AgNO3 MgCl2 NH4 Cl CuSO4 ⋅ 5 H2 O NiSO4 H2 S Essigsäure Eosin Gefahrenbezeichnung – Gefahr (GHS05) H-Sätze – 314, 335, 290 P-Sätze – 301+330+331, 305+351+338, 280 Gefahr (GHS05) 314, 290 301+330+331, 305+351+338, 280 Gefahr (GHS05, 09) 314, 335, 400 301+330+331, 305+351+338, 280, 273 Achtung (GHS03) 272, 210 – Gefahr (GHS03, 05, 09) 272, 314, 410 301+330+331, 305+351+338, 273, 280 – – – Achtung (GHS07) 302, 319 305+351+338 Achtung (GHS07, 09) 302, 315, 319, 410 305+351+338, 302+352, 273 Gefahr (GHS05, 08) 350i, 341, 360D, 372, 315, 201, 280, 273, 308+313, 334, 317, 410, 302+332 342+311, 302+352 Gefahr (GHS02, 06, 09) 330, 220, 400 260, 210, 273, 304+340+315, 377, 381, 405, 403 Gefahr (GHS05) 226, 314 301+330+331, 305+351+338, 280 Achtung (GHS07) 319 260, 305+351+338 120 Fällungs- und Löslichkeitsgleichgewichte 4 Literatur [1] A. F. Holleman, E. Wiberg: Lehrbuch der Anorganischen Chemie, 102. Aufl., Walter de Gruyter Verlag, 2007. [2] M. Binnewies, M. Jäckel, H. Willner, G. Rayner-Canham: Allgemeine und Anorganische Chemie, 1. Auflage, Spektrum Verlag, 2004. [3] G. Jander, E. Blasius: Einführung in das Anorganisch-Chemische Praktikum, 15. Auflage, S. Hirzel Verlag, 2005; Lehrbuch der Analytischen und Präparativen Anorganischen Chemie, 16. Auflage, S. Hirzel Verlag, 2006. [4] E. Fluck, M. Becke-Göhring: Einführung in die Theorie der quantitativen Analyse, 7. Auflage, Steinkopff Verlag, 1990. Komplexgleichgewichte 121 Versuch 12 (KOG) Komplexgleichgewichte 1 Theorie Als Komplexverbindungen werden für gewöhnlich Koordinationsverbindungen bezeichnet, bei denen ein oder mehrere Liganden an ein kationisches (oder neutrales) Metallzentrum koordinieren. Allerdings ist es auch möglich, komplexe Säuren bzw. deren Anionen (z. B.: H2 SO4 = SO2 (OH)2 , SO2– 4 ) als Komplexe zu bezeichnen. Der gesamte Komplex kann hierbei entweder positiv, negativ oder auch ungeladen sein. 1.1 Komplexreaktionen und Komplexstabilität Die Reaktion zur Bildung eines Komplexes ist eine klassische Säure-Base-Reaktion nach der LewisTheorie. Hierbei stellt das Zentralkation (oder -atom) (meistens ein Metallkation, am häufigsten Übergangsmetalle) die Lewis-Säure dar; die Lewis-Base ist der Komplexligand (ein Teilchen, welches mindestens ein freies Elektronenpaar zur Bindungsbildung zur Verfügung stellen kann). Auch diese Reaktion ist eine Gleichgewichtsreaktion, die jedoch in einzelne Reaktionsschritte unterteilt werden muss (siehe Versuch 8 MWG). Das Produkt der Gleichgewichtskonstanten aus den Einzelreaktionen (sog. Elementarreaktionen) ergibt dann die Gleichgewichtskonstante für die Gesamtreaktion, z. B.: Ni2+ + CN− ⇌ [NiCN]+ K1 = [Ni(CN)]+ + CN− ⇌ [Ni(CN)2 ] K2 = [Ni(CN)2 ] + CN− ⇌ [Ni(CN)3 ]− K3 = [Ni(CN)−3 ] + CN− ⇌ [Ni(CN)4 ]2− K4 = K1 ⋅ K2 ⋅ K3 ⋅ K4 = KA [[Ni(CN)]+ ] [Ni2+ ]⋅[(CN)− ] [[Ni(CN)2 ]] [[Ni(CN)]+ ]⋅[(CN)− ] [[Ni(CN)3 ]− ] [[Ni(CN)2 ]]⋅[(CN)− ] [[Ni(CN)4 ]2− ] [[Ni(CN)3 ]− ]⋅[(CN)− ] Diese resultierende Konstante nennt man Komplexbildungskonstante. Sie ist von großer Bedeutung, denn diese Konstante gibt auch an, wie stabil der Komplex ist bzw. ob er zur Dissoziation neigt. Daher wird die Komplexbildungskonstante auch Komplexstabilitätskonstante oder Komplexassoziationskonstante KA genannt und ihr reziproker Wert wird als Komplexdissoziations- −1 konstante KD bezeichnet, somit gilt KA = KD . 1.2 Der räumliche Aufbau der Komplexe Ein Zentralatom oder -kation hat das Bestreben, seine verhältnismäßig hohe Punktladung durch seine Liganden so gut wie möglich nach außen abzuschirmen. Dies ist, ähnlich wie beim VSEPR-Konzept (siehe Versuch 5 HGE) v. a. dann möglich, wenn sich die Liganden auf einer Kugeloberfläche möglichst gleichmäßig um das Zentralatom anordnen. Die beliebtesten Geometrien sind das Oktaeder, das Tetrader, die trigonale Bipyramide, aber auch quadratisch- und trigonal-planare sowie lineare Geometrien werden realisiert. Andererseits muss bei der ”besten” 122 Komplexgleichgewichte Abschirmung auch berücksichtigt werden, dass sich um ein Kation einer gewissen Größe nicht beliebig viele Liganden anordnen können. Zuletzt müssen dabei auch die möglichen Bindungsverhältnisse, die durch freie und besetzte Orbitale gegeben sind, beachtet werden. Um eine Erklärung zu finden, wie die Liganden sich in einem konkreten Fall um ein Zentralatom anordnen und wie viele es sind, bedient man sich verschiedener Theorien. 1.3 Die Ligandenfeldtheorie (Kristallfeldtheorie) Die Ligandenfeldtheorie (Kristallfeldtheorie) zur Erklärung der Komplexgestalt kommt dann zum tragen, wenn das Zentralatom eine d-Elektronenkonfiguration von minimal d1 und maximal d9 besitzt. Die Basis dieser Theorie ist, dass die Entartung der fünf d-Orbitale bei der Annäherung von Liganden durch das Feld ihrer Elektronenpaare aufgehoben wird. Wichtig dabei ist, dass das Zentralatom Elektronen besitzt, die nicht zur Bindung beitragen und die irgendwo ”untergebracht” werden müssen. Dies geschieht dann in den Orbitalen, deren Lappen nicht in Richtung des Liganden zeigen und somit vom Feld der Ligandenelektronen also auch nicht beeinträchtigt werden. 1.3.1 Oktaedrische Komplexe Es wird davon ausgegangen, dass sich sechs Liganden dem Zentralatom auf den Achsen eines dreidimensionalen Koordinatensystems nähern. Von den fünf d-Orbitalen des Zentralatoms besitzen das dz 2 - und das dx2 −y2 -Orbital Orbitallappen, die auf den Koordinatenachsen liegen (siehe Versuch 4 PSE), die Orbitallappen der anderen drei Orbitale dxy , dxz und dyz liegen zwischen den Koordinatenachsen. Die Entartung wird dann derart aufgehoben, dass die drei Orbitale (dxy , dxz und dyz ), die dem Abb. 1.1: Ligandenfeldaufspaltung bei oktaedrischer Koordination Liganden nicht ”im Weg” (= sterisch günstig) sind, energetisch abgesenkt werden und daher gut für die ”Restelektronen” des Zentralatoms verwendet werden können. Die anderen Orbitale, die im Weg des Liganden liegen, werden energetisch angehoben und können gut für die Bindung zum Liganden verwendet werden (s. Kapitel 1.4). Energetisch ergibt sich dann das in Abbildung 1.1 gezeigte Schema. Mit ∆o wird hierbei die Aufspaltung der Orbitalenergie (sog. Ligandenfeldaufspaltungsenergie) im Oktaederfeld beschrieben. Um dem Energieerhaltungssatz genüge zu tun, beträgt die Absenkung der Energie der Orbitale dxy , dxz und dyz 2 5 ∆o und die Energieerhöhung des dz 2 - und des dx2 −y2 -Orbitals 35 ∆o . In den ”unteren” Orbitalen können jetzt die ”Restelektronen” des Zentralatoms untergebracht werden (energetisch günstigste Elektronenkonfigurationen d3 und d6 ). Komplexgleichgewichte 123 1.3.2 Tetraedrische Komplexe Es wird davon ausgegangen, dass sich vier Liganden dem Zentralatom innerhalb eines dreidimensionalen Koordinatensystems entsprechend Abbildung 1.2 (oben) annähern. Die vier weißen (oder die vier schwarzen) Kugeln nähern sich dem Zentralkation aus den in der Abbildung angegebenen Richtungen. Dabei sind die”weißen” Lappen der dxy , dxz und dyz Orbitale sterisch ungünstiger als die ”grauen” der dz 2 - und dx2 −y2 -Orbitale. Daher werden jetzt die letztgenannten energetisch abgesenkt und bevorzugt für die ”Restelektronen” des Zentralatoms verwendet, während die erstgenannten drei energetisch angehoben werden und dann für die Ausbildung von Bindungen zur Verfügung stehen (s. Kapitel 1.4). Beim tetraedrischen Ligandenfeld sieht die Energieaufspaltung der d-Orbitale daher gemäß Abbildung 1.2 (unten) aus. Die Ligandenfeldaufspaltungsenergie wird hier mit ∆t bezeichnet und ist verglichen mit einem analogen okta- Abb. 1.2: Annäherung der Liganden zum tetredrischen Feld (oben) und Ligandenfeldaufspaltung bei tetraedrischer Koordination (unten) edrischen Ligandenfeld (gleiches Zentralatom, gleiche Liganden) nur 4 9 so groß wie diejenige des Oktaederfelds. Außerdem ist das Aufspaltungverhältnis genau umgedreht im Vergleich zur Oktaederfeldaufspaltung. 1.3.3 Stärke von Komplexliganden - Die spektrochemische Reihe Die Größe der Ligandenfeldaufspaltungsenergie (z. B. ∆o , ∆t ) ist von mehreren Dingen abhängig. Zum ersten ist dies die Geometrie, in der die das Zentralatom umgebenden Liganden angeordnet sind (s. Kap. 1.3.1 und 1.3.2). Dann natürlich vom Zentralatom (-kation) bzw. dessen Elektronenkonfiguration. Allgemein gilt, dass bei gleichem Element die Aufspaltung bei einer höheren Oxidationsstufe größer wird im Vergleich zu derjenigen bei kleiner Oxidationsstufe. Innerhalb einer Periode nimmt die Aufspaltungsenergie von links nach rechts leicht zu, beim Durchgang durch eine Gruppe nimmt die Aufspaltungsenergie von oben nach unten stärker zu. Der sichtbarste Einfluß auf die Aufspaltungsenergie kommt jedoch dem Liganden zu. Die Liganden sind ihrer Stärke entsprechend in der spektrochemischen Reihe aufgelistet. Die Größe der Aufspaltung der d-Orbitale (= Größe der Ligandenfeldaufspaltungsenergie = Ligandenfeldstabilisierungsenergie) hat direkte Auswirkungen auf die Besetzung der d-Orbitale mit den Restelektronen des Zentralkations. Laut der Hund’schen Regel (siehe Versuch 4 PSE) müssen entartete Orbitale zuerst einzeln besetzt werden. Bei einer oktaedrischen Ligandenfeldaufspaltung beispielsweise werden 124 Komplexgleichgewichte aus den fünf ursprünglichen entarteten d-Orbitalen drei entartete, die energetisch tiefer, und zwei entartete, die energetisch höher liegen. Fängt man an mit drei Elektronen zu besetzten, werden zuerst die 3 energetisch tiefliegendsten d-Orbitale einzeln besetzt. Nach der Hund’schen Regel müsste das vierte Elektron mit umgekehrtem Spin eines der drei tiefliegenden Orbitale doppelt besetzen. Für das ”Paaren” der Elektronen in einem Orbital ist allerdings auch Energie nötig, die sog. Spinpaarungsenergie. Diese tritt mit der Ligandenfeldaufspaltungsenergie folgendermaßen in Konkurrenz: Spinpaarungsenergie < Ligandenfeldaufspaltungsenergie: In diesem Fall werden zuerst die energetisch tiefer liegenden Orbitale einfach, dann doppelt und dann erst die energetisch höher Orbitale liegenden einfach, dann doppelt besetzt. Hier ist dann die Ligandenfeldaufspaltungsenergie so groß, dass trotz Spinpaarung ein Energiegewinn erzielt wird. Somit werden Komplexe mit minimaler Anzahl an ungepaarten Elektronen (= ungepaarten Spins) erhalten, die sogenannten low-spin Komplexe. Spinpaarungsenergie > Ligandenfeldaufspaltungsenergie: In diesem Fall werden zuerst die energetisch tiefer liegenden Orbitale einfach, dann die energietisch höher Orbitale liegenden einfach besetzt, danach erst die energetisch tiefer liegenden doppelt, dann die höher liegenden Orbitale doppelt besetzt. Da der Energiegewinn durch die Ligandenfeldaufpaltungsenergie den Verlust durch die Spinpaarungsenergie nicht kompensieren kann, werden Komplexe mit maximaler Anzahl an ungepaarten Elektronen (= ungepaarten Spins) erhalten, die sogenannten high-spin Komplexe. 1.3.4 Low-spin- und high-spin-Komplexe Um überhaupt low-spin- und high-spin-Komplexe zu unterscheiden, muss die Ligandenfeldaufspaltungsenergie ein gewisses Maß aufweisen. Da die Aufspaltung des Tetraederfelds grundsätzlich kleiner ist als die des Oktaederfelds, sind tetraedrische low-spin-Komplexe quasi nie zu beobachten. So genügt es, Aufspaltungen des Oktaederfelds zu betrachten und da erst zu entscheiden, bei welchen Elektronenkonfigurationen eine solche Entscheidungsfindung überhaupt nötig ist. Die Elektronenkonfigurationen d1 -d3 besetzen in jedem Fall die energetisch tiefliegenden Orbitale einfach, und bei den Elektronenkonfigurationen d8(∗) und d9 sind die tiefliegenden in jedem Falle doppelt besetzt. Also gibt es nur für die Elektronenkonfigurationen d4 -d7 eine Entscheidungsmöglichkeit ob ein low-spin- oder ein high-spin-Komplex vorliegt. (*) Auch für die Elektronenkonfiguration d8 kann der low-spin- und der high-spin-Fall unterschieden werden (s. nächstes Kapitel). 1.3.5 Das quadratisch-planare Ligandenfeld Im Gegensatz zu Verbindungen, die dem VSEPR-Konzept gehorchen, werden bei Komplexverbindungen auch Geometrien erhalten, die keine ideale kugelförmige Abschirmung um das Zentralkation gewährleisten. Die häufigste hierbei ist die quadratisch-planare. Wird ein Zentralkation der Elektronenkonfiguration d8 mit einem starken Liganden ”gepaart”, bietet das oktaedrische Feld keinen ausreichenden Energiegewinn. Besser ist die energetische Trennung der beiden höher liegenden Orbitale in ein sehr energiereiches (hier dx2 −y2 ), das bei einer d8 - Komplexgleichgewichte 125 Konfiguration unbesetzt verbleiben kann und ein energetisch abgesenktes (hier dz 2 ), das mit zwei Elektronen voll besetzt werden kann. Dies hat zur Folge, dass nur vier Liganden sich dem Zentralatom nähern können und zwar auf der x- und der y-Achse. Das dx2 −y2 -Orbital ist hierbei das sterisch ungünstigste und wird energetisch stark erhöht. Die zweitgrößte energetische ”Belastung” erfährt das dxy -Orbital, liegt aber energetisch schon in der Nähe des Energieniveaus der fünf entarteten d-Orbitale des Kations im kugelsymmetrischen Feld. Tiefer liegen dann noch das dxz , das dyz und das dz 2 -Orbital. 1.3.6 Der Jahn-Teller Effekt Sind die energetisch höchstliegenden Orbitale aufgrund der Elektronenkonfiguration des Zentralkations unterschiedlich besetzt (z. B. bei d7 low-spin im Oktaeder ist von den beiden energetisch hochliegenden Orbitale (dz 2 und dx2 −y2 ) eines einfach und das andere gar nicht mit Elektronen besetzt, bei d9 im Oktaeder ist eines einfach und das andere doppelt mit Elektronen besetzt), wird versucht, durch eine Aufhebung der Entartung dieser Orbitale (energetische Absenkung des höher besetzten und energetische Anhebung des niedriger besetzten Orbitals) einen Energiegewinn zu erzielen. Dies hat sichtbare Auswirkungen auf die Komplexgeometrie, nämlich dahingehend, dass die Liganden, die in Richtung des energetisch abgesenkten Orbitals liegen, weiter vom Zentralatom entfernt werden. Ist bei einem oktaedrischen Komplex das dz 2 -Orbital höher besetzt als das dx2 −y2 )-Orbital, entfernen sich die Liganden in z-Richtung weiter vom Zentralatom und das Oktaeder sieht gestreckt aus; sind die Besetzungsverhältnisse umgekehrt, entfernen sich die Liganden in x- und y-Richtung weiter, und man erhält ein gestauchtes Oktaeder (wobei letzterer Fall bislang noch nicht gesichert beobachtet wurde). Dieser Effekt wird als Jahn-Teller Effekt bezeichnet (auch: Jahn-Teller Verzerrung). 1.4 Die Valenzbindungstheorie Die Bindungsverhältnisse der Komplexe können auch mit der Valenzbindungstheorie beschrieben werden. Es wird hierbei, zur Vereinfachung, davon ausgegangen, dass die Entartung der fünf dOrbitale beibehalten wird, aber trotzdem entsprechend der Ligandenstärke low-spin und high-spin bei der Befüllung der d-Orbitale unterschieden wird. Nun wird für die Bindung zum Liganden von einer entsprechenden Hybridisierung des Zentralatoms ausgegangen unter Berücksichtigung der noch freien d-Orbitale. 1.5 Eigenschaften von Komplexen 1.5.1 Magnetismus in Komplexen Besitzt ein Komplex keine ungepaarten Elektronen (kann nur im low-spin Zustand der Fall sein), besitzt er annähernd kein magnetisches Moment, er ist diamagnetisch. Besitzt ein Komplex ein oder mehrere ungepaarte Elektronen (kann im low-spin Zustand der Fall sein, ist immer der Fall im high-spin Zustand) können sich diese in einem Magnetfeld ausrichten, er ist paramagnetisch. Je mehr ungepaarte Elektronen ein Komplex besitzt, desto stärker ist sein magnetisches Moment. (siehe Versuch 7 BPH) 126 Komplexgleichgewichte 1.5.2 Farben von Komplexen Farbigkeit von Komplexen kann verschiedene Ursachen haben. Allen gemein ist jedoch, dass es sich bei der Farberscheinung in Komplexen um das sogenannte Absorptionsspektrum handelt, d.h. Elektronen werden durch eine bestimmte Wellenlänge des sichtbaren Lichts auf ein höheres Niveau angeregt. Dabei wird diese Wellenlänge aus dem weißen Licht herausgefiltert: die Komplementärfarbe bleibt sichtbar. (siehe Versuche 2 ABS, 3 BSP und 6 NGE) 1.5.3 Chelatkomplexe Besitzt ein Ligand mehrere Koordinationsstellen, die auch gleichzeitig für die Koordination am gleichen Metallzentrum genutzt werden können, spricht man von einem Chelatliganden (z. B. Oxalat, Tartrat, EDTA ≡ Ethylendiamintetraacetat). Diese Komplexe besitzen sowohl thermodynamisch als auch kinetisch eine höhere Stabilität. Die hohe thermodynamische Stabilität beruht auf der Erhöhung der Entropie des Systems, da zur Bildung eines beispielsweise oktaedrischen Komplexes mit einem zweizähnigen Liganden (= Ligand mit zwei Koordinationsstellen) in wässriger Lösung folgende Reaktion abläuft: [M (H2 O)6 ] + 3 X ⇌ [M X3 ] + 6 H2 O, bei der aus vier freien Teilchen (auf der linken Seite) sieben freie Teilchen (auf der rechten Seite) werden (= Erhöhung der Unordnung = Erhöhung der Entropie). Die kinetische Stabilität beruht darauf, dass zur Bildung eines Chelatkomplexes im Vergleich zu einem Komplex mit einem nicht-Chelatliganden Komplexes sich (nach der kinetischen Gastheorie) weniger Teilchen treffen müssen und bei der Dissoziation alle Bindungen eines Liganden zum Zentralatom gleichzeitig geöffnet werden müssen. 1.6 Die Nomenklatur von Komplexen 1. Die Namen aller anionischen Liganden enden auf ”o”. Enden die Namen der den Liganden zugrundeliegenden Anionen bzw. Molekülen auf ”-id”, ”-it” oder ”-at”, werden sie in Komplexen mit ”-ido”, ”-ito’ oder ”-ato” bezeichnet. Leider gibt es hier einige traditionell bedingte Ausnahmen: Wichtige neutrale Liganden: H2 O – aqua, NH3 – ammin, NO – nitrosyl, CO – carbonyl Wichtige anionische Liganden: F– – fluorido (fluoro), Cl– – chlorido (chloro), Br– – bromido (bromo), I– – iodido (iodo), O2– – oxido (oxo), OH– – hydroxido (hydroxo), NO–2 – nitrito-N (nitro) bei Koordination über N bzw. nitrito-O (nitrito) bei Koordination über O, S2– – sulfido (thio), CN– – cyanido-C (cyano) bei Koordination über c bzw. cyanido-N (isocyano) bei Koordination über N, SCN– – thiocyanato bei Koordination über S bzw. isothiocyanato – bei Koordination über N, C2 O2– 4 – oxalato, H3 CCOO – acetato. 2. Kommt ein Ligand mehrfach in einem Komplex vor, wird seine Häufigkeit mit folgenden (griechischen) Vorsilben vor dem entsprechenden Liganden bezeichnet: 2 (di), 3 (tri), 4 (tetra), 5 (penta), 6 (hexa), 7 (hepta), 8 (octa), 9 (ennea) 3. In einem Komplex werden die Liganden alphabetisch geordnet, ohne die Häufigkeitsvorsilbe zu berücksichtigen. Komplexgleichgewichte 4. 127 Bei neutralen oder kationischen Komplexen endet der Name mit der Nennung des Metalls, bei anionischen Komplexen endet der Name auf die Silbe ”-at”. Diese folgt auf den Namen, den das entsprechende Metall in seiner Oxosäure besitzt (meist Latein): Al (-aluminat), As (-arsenat), Pb (-plumbat), Cd (-cadmat), Fe (-ferrat), Au (-aurat), Cu (-cuprat), Hg(-mercurat), Ag (-argentat), V (-vanadat), Sn (stannat). 2 Aufgabenstellung, Versuchsanleitung und -auswertung 2.1 Beobachtung der Farben von Komplexen 2.1.1 Aufgabenstellung Bei diesem Versuch soll die Farbigkeit von Komplexen in Abhängigkeit von deren Liganden ermittelt werden. 2.1.2 Versuchsanleitung a) In zwei Reagenzgläsern wird je eine Spatelspitze CuSO4 zuerst in wenig Wasser gelöst und anschließend ca. 2 cm hoch mit Ammoniaklösung bzw. konzentrierter Natronlauge versetzt. Alle entstehenden Farben werden dokumentiert. b) Eine Spatelspitze wasserfreies CoCl2 wird in einem Reagenzglas mit wenig konz. Salzsäure versetzt, und die entstehende blaue Lösung wird dann mit H2 O bis zum Farbwechsel nach rosa verdünnt. 2.1.3 Versuchsauswertung ● ● ● Formulieren Sie die Reaktionsgleichungen und benennen Sie die Komplexe systematisch. Ordnen Sie die verwendeten Liganden nach ihrer Stärke in die spektrochemische Reihe ein. Wie können Sie die beobachteten Farben erklären? 2.2 Untersuchung der Stabilität von Komplexen 2.2.1 Aufgabenstellung Bei diesem Versuch soll mit Hilfe unterschiedlicher Komplexstabilität zwei verschiedene Ionen in wässriger Lösung voneinander getrennt werden. 2.2.2 Versuchsanleitung Jeweils eine Spatelspitze CuSO4 und CdSO4 werden zusammen in ein Reagenzglas gegeben und in wenig demineralisiertem Wasser gelöst (ca. 1-2 cm hoch mit Wasser versetzten und so lange schwenken bis sich alles gelöst hat). In zwei weiteren Reagenzgläsern werden wässrige Lösungen von CuSO4 und CdSO4 bereitgestellt. Anschließend wird in alle drei Reagenzgläser so viel konz. Ammoniaklösung zugegeben bis der pH-Wert größer als 10 ist. Nun wird in das Reagenzglas mit der Mischung aus CuSO4 und CdSO4 festes KCN zugegeben, so lange bis sich der anfangs gebildete Niederschlag gerade wieder auflöst. Zu allen drei Lösungen wird nun H2 S-Wasser gegeben (ca. 1 cm hoch). 128 Komplexgleichgewichte 2.2.3 Versuchsauswertung ● Formulieren Sie die Reaktionsgleichungen VOR der Durchführung. Mit welchen Gefahren ist zu rechnen und wie werden diese beseitigt? ● ● Benennen Sie die entstandenen Komplexe systematisch. Vergleichen Sie die Fällung der Mischung aus CuSO4 und CdSO4 mit den jeweiligen Einzelfällungen. 2.3 Beobachtung der Hydratisomerie 2.3.1 Aufgabenstellung Bei diesem Versuch soll ermittelt werden, dass auch ein teilweiser Ligandenaustausch die Farbe von Komplexen ändern kann. 2.3.2 Versuchsanleitung Eine kleine Menge feingepulverten Chromalauns (KCr[SO4 ]2 ⋅ 12 H2 O) wird in der Kälte (Raumtemperatur ist ausreichend) in einem Reagenzglas in einigen ml Wasser aufgelöst. Die Lösung ist blau-violett gefärbt. Beim Erhitzen der Lösung tritt eine Farbänderung nach tiefgrün ein. 2.3.3 Versuchsauswertung ● Welche Vorgänge spielen sich in der Lösung ab? Ermitteln Sie die Reaktionsgleichung und benennen Sie die Komplexe systematisch. ● ● Geben Sie eine Definition für ”Hydratisomerie”. Welche zwei verschiedenen Isomere sind denkbar für die oktaedrischen Komplexe [CrCl2 (H2 O)4 ]+ und [CrCl3 (H2 O)3 ]? Zeichnen und benennen Sie diese Isomere. 2.4 Stabilität von Chelatkomplexen 2.4.1 Aufgabenstellung Bei diesem Versuch soll die Anwendung von Chelatkomplexen in der qualitativen (Versuchsteil a) und der quantitativen Analytik (Versuchsteil b) ermittelt werden. 2.4.2 Versuchsanleitung a) Eine Spatelspitze NiCl2 wird in einem Reagenzlas in wenigen Tropfen konz. Ammoniaklösung gelöst. 2-4 Tropfen dieser Lösung werden auf eine Tüpfelplatte gegeben und mit 1-2 Tropfen Dimethylglyoximlösung (= Diacetyldioximlösung) versetzt. b) Komplexometrische Titration einer Lösung unbekannter Stoffmenge an Ca2+ . Der Kolben mit der Analysenlösung wird bis zur Markierung auf 100 ml mit demineralisiertem Wasser aufgefüllt und homogenisiert. Anschließend werden 25 ml der so vorbereiteten Analysenlösung in einen Erlenmeyerkolben pipettiert (25 ml Vollpipette) und mit demineralisiertem Wasser auf ca. 100 ml aufgefüllt. Danach wird eine Indikatorpuffertablette und nach deren Auflösung eine Tropfpipette voll Ammoniaklösung hinzugegeben. Es wird mit 0,1 molarer EDTA-Maßlösung bis zum scharfen Umschlag von braun-orange nach grün titriert. Komplexgleichgewichte 129 2.4.3 Versuchsauswertung ● ● ● ● Formulieren Sie die Reaktionsgleichungen und benennen Sie die Komplexe systematisch. Zeichnen Sie den roten Ni2+ -Komplex aus Versuchsteil a) geometrisch korrekt. Wie hoch ist die Stoffmenge an Ca2+ -Kationen in der Analysenlösung? Zeichnen Sie den Ca-EDTA-Komplex. Was bedeutet EDTA? 3 Geräte und Chemikalien Eine Geräteliste liegt im Platz aus Chemikalien CuSO4 NH3 aq Gefahrenbezeichnung Achtung (GHS07, 09) Gefahr (GHS05, 09) H-Sätze 302, 315, 319, 410 314, 335, 400 NaOH Gefahr (GHS05) 314, 290 CoCl2 Gefahr (GHS06, 09) HCl Gefahr (GHS05) 350i, 341, 360F, 302, 334, 317, 410 314, 335, 290 aq CdSO4 Gefahr (GHS06, 09) KCN Gefahr (GHS06, 09) H2 S Gefahr (GHS02, 06, 09) KCr[SO4 ]2 ⋅ 12 H2 O NiCl2 Achtung (GHS07) Gefahr (GHS06, 09) Dimethylglyoxim EDTA-Lösung Indikatorpuffertabletten Achtung (GHS02) – Achtung (GHS07) 350, 340, 360FD, 330, 301, 372, 410 EUH032, 300+310+330, 410 330, 220, 400 P-Sätze 273, 305+351+338, 302+352 273, 280, 301+330+331, 305+351+338 280, 301+330+331, 305+351+338 201, 273, 308+313, 304+341, 281 280, 301+330+331, 305+351+338 201, 273, 304+340 273, 280, 302+352, 304+340 260, 210, 273, 304+340+315, 377, 381, 405, 403 302+352, 305+351+338 273, 281, 302+352, 304+340 315, 319 350i, 360D, 341, 372, 315, 301+331, 334, 317, 410 228 – – – 302, 317, 319 280, 305+351+338 4 Literatur [1] A. F. Holleman, E. Wiberg: Lehrbuch der Anorganischen Chemie, 102. Aufl., Walter de Gruyter Verlag, 2007. [2] M. Binnewies, M. Jäckel, H. Willner, G. Rayner-Canham: Allgemeine und Anorganische Chemie, 1. Auflage, Spektrum Verlag, 2004. [3] G. Jander, E. Blasius: Einführung in das Anorganisch-Chemische Praktikum, 15. Auflage, S. Hirzel Verlag, 2005; Lehrbuch der Analytischen und Präparativen Anorganischen Chemie, 16. Auflage, S. Hirzel Verlag, 2006. [4] E. Fluck, M. Becke-Göhring: Einführung in die Theorie der quantitativen Analyse, 7. Auflage, Steinkopff Verlag, 1990. Leitfähigkeit 131 Versuch 13 (LFG) Leitfähigkeit 1 Theorie Löst man einen Elektrolyten, wie z.B. ein Salz, in Wasser, so entsteht eine Lösung in der sich positiv geladene Kationen, sowie negativ geladene Anionen befinden. Wird nun ein elektrisches Feld angelegt, so können sich diese Ionen in der Lösung aufgrund ihrer Ladung zu den Elektroden hinbewegen. Durch diese Bewegung der Ionen wird die Lösung leitfähig. Durch Leitfähigkeitsmessungen kann die Wanderung der Ionen untersucht werden, hierbei wird der elektrische Widerstand R einer Lösung mit bekannter Konzentration gemessen. Dazu werden gut leitende Platinelektroden in einem definierten Abstand verwendet und ein Wechselfeld angelegt, um einen Aufbau von Ladungen an den Elektroden und somit das Elektrolysieren der Lösung zu vermeiden. Für den elektrischen Widerstand gilt nach dem Ohmschen Gesetz: U =R⋅I (1) Wird ein homogener Leiter der Länge l und der Querschnittsfläche A verwendet, so ergibt sich für den spezifischen Widerstand ρ: ρ=R⋅ A l (2) ρ hat die SI-Einheit Ω ⋅ m. Die spezifische Leitfähigkeit κ ist der Kehrwert des spezifischen Widerstandes ρ mit der SI-Einheit Ω−1 ⋅ m−1 bzw. S ⋅ m−1 (S = Siemens). Mit dem Leitfähigkeitsmessgerät kann nun κ in S ⋅ cm−1 abgelesen werden. Durch Multiplikation mit der Messzellenkonstante, die auf der Messzelle angegeben ist, ergibt sich daraus die spezifische Leitfähigkeit κ in S ⋅ cm−1 . Die Äquivalentleitfähigkeit Λc bei der Äquivalentkonzentration des Elektrolyten c (in val ⋅ l−1 , auch als Normalität n bezeichnet; val ist die Einheit der Äquivalentmenge) berechnet sich daraus nach: Λc = κ c (3) (Λc wird in S⋅cm2 ⋅val−1 angegeben) Bei starken Elektrolyten, wie z.B. Salzen, kann man mit einem nahezu vollständigen Zerfall in Ionen rechnen, deren Beweglichkeit jedoch (besonders bei hohen Konzentrationen) durch Wechselwirkungen (= elektrostatische Kräfte) behindert ist. Obwohl die spezifische Leitfähigkeit bereits auf die Konzentration normiert wurde (= durch die Konzentration geteilt), zeigt sich hier trotzdem noch eine Konzentrationsabhängigkeit. Diese Abhängigkeit der Leitfähigkeit von der Konzentration wird durch Wechselwirkungskräfte hervorgerufen. Für diese starken Elektrolyte fand Kohlrausch empirisch das nach ihm benannte Quadratwurzelgesetz: Λc = Λ0 − a ⋅ √ c (4) Hierbei sind Λc die Äquivalentleitfähigkeit bei der Konzentration c, Λ0 die Äquivalentleitfähigkeit 132 Leitfähigkeit bei unendlicher Verdünnung (Konzentration 0) und a eine Konstante. Trägt man nun Λc gegen √ c auf, so erhält man im Falle eines starken Elektrolyten eine Gerade. Bei schwachen Elektrolyten, die nur teilweise dissoziieren, wie z.B. Essigsäure, kommt es zwischen den Ionen infolge ihrer kleinen Konzentration zu keinen nennenswerten Wechselwirkungen. Es gilt das Massenwirkungsgesetz für das Gleichgewicht zwischen Ionen und undissoziierten Molekülen. Es wird hier auch als Ostwald’sches Verdünnungsgesetz bezeichnet und besitzt die Form: K′ = Λ2c ⋅ c Λ0 ⋅ (Λ0 − Λc ) (5) Neben den Äquivalentleitfähigkeiten Λc und Λ0 und der Konzentration c wird hier die Gleichgewichtskonstante K ′ der Dissoziation des Elektrolyten verwendet. Durch geeignetes Umformen erhält man aus Gleichung 5 das Ostwald’sche Verdünnungsgesetz in der folgenden Form: 1 1 c ⋅ Λc = ′ 2+ Λc K ⋅ Λ0 Λ0 Trägt man nun 1 Λc (6) gegen c ⋅ Λc auf, so erhält man im Falle eines schwachen Elektrolyten eine Gerade. 2 Aufgabenstellung, Versuchsanleitung und -auswertung 2.1 Aufgabenstellung 1. Es sind die Leitfähigkeiten von zwei unbekannten Elektrolyten in Abhängigkeit von der Konzentration zu messen und daraus die Äquivalentleitfähigkeit zu bestimmen. Es soll festgestellt werden, welcher der beiden Elektrolyte der starke und welcher der schwache ist. Außerdem sind die Äquivalentleitfähigkeiten bei unendlicher Verdünnung sowie die Konstanten des Ostwald’schen Verdünnungsgesetzes bzw. des Quadratwurzelgesetzes von Kohlrausch zu bestimmen. 2. Es ist eine Leitfähigkeitstitration eines Gemisches einer starken und einer schwachen Säure durchzuführen. Die ausgegebene Menge von starker und schwacher Säure ist zu bestimmen. 2.2 Versuchsdurchführung 1. Für jeden der beiden unbekannten Elektrolyten werden zwei Konzentrationsreihen angesetzt. Für die erste Konzentrationsreihe werden 250 mL bidestilliertes H2 O und für die zweite Reihe 50 mL bidestilliertes H2 O vorgelegt. Dazu wird jeweils mit einer Messpipette die 0,1 N Elektrolytlösung zugegeben und zwar in Portionen zu 0,5 mL, 0,5 mL, 1 mL, 3 mL, 5 mL und 10 mL, um die Elektrolytkonzentration in der Lösung schrittweise zu erhöhen. Nach jeder Zugabe wird umgerührt und die Leitfähigkeit gemessen. Die Leitfähigkeit des bidestillierten Wassers ist von den gemessenen Werten zu subtrahieren. Leitfähigkeit 2. 133 Der vom Assistent ausgegebene Analysenkolben wird (vorsichtig) bis zur 100 ml Marke mit bidestilliertem Wasser aufgefüllt und durch Umschütteln durchmischt. Mit Hilfe einer 25 ml Vollpipette werden 25 ml dieser Lösung in ein Becherglas gegeben. Die Bürette wird bis zur Nullmarke mit NaOH-Maßlösung befüllt (etwas überbefüllen und in ein Becherglas ablassen bis die Nullmarke erreicht wird). Die Maßlösung wird in 1mL-Schritten zugegeben und nach jeder Zugabe wird umgerührt und die Leitfähigkeit gemessen (WICHTIG: Zur Leitfähigkeitsmessung muss der Magnetrührer unbedingt abgestellt werden, da sonst der Rührfisch gegen die Elektrode schlägt. Schäden, die durch Nicht-Beachten dieser Anweisung entstehen, sind von den Studierenden zu bezahlen!). Diese Titration wird ein zweites Mal durchgeführt um das zuvor erhaltene Ergebnis zu verifizieren. 2.3 Auswertung 1. Für beide Elektrolyte ist in einem Diagramm die Äquivalentleitfähigkeit Λc gegen die Konzentration c aufzutragen. In zwei weiteren Diagrammen sind Λc gegen und 1 Λc gegen c ⋅ Λc aufzutragen. Aus diesen Diagrammen lässt sich entscheiden, welches der schwache und welches der starke Elektrolyt ist. Durch Extrapolation c → 0 lässt sich die Äquivalentleitfähigkeit bei unendlicher Verdünnung bestimmen. Aus den Steigungen der Geraden lassen sich die Dissoziationskonstante K ′ und die Konstante a des Gesetzes von Kohlrausch entnehmen. 2. Die Leitfähigkeit ist gegen die Zugabe von NaOH in einem Diagramm aufzutragen. Aus diesem sind die beiden Äquivalenzpunkte zu bestimmen. 3 Geräte und Chemikalien Eine Geräteliste liegt am Platz aus Chemikalien NaOH Essigsäure HClaq H2 SO4 Bernsteinsäure Phenolphthalein Gefahrenbezeichnung Gefahr (GHS05) Gefahr (GHS05) Gefahr (GHS05) Gefahr (GHS05) Achtung (GHS07) Gefahr (GHS06) H-Sätze 314, 290 226, 314 314, 335, 290 314, 290 319 350, 341, 361f P-Sätze 301+330+331, 305+351+338, 280 301+330+331, 305+351+338, 280 301+330+331, 305+351+338, 280 280, 301+330+331, 305+351+338, 309, 310 305+351+338 201, 281, 308+313 4 Literatur [1] [2] [3] G. Wedler: Lehrbuch der Physikalischen Chemie, 5. Aufl., Wiley-VCH, 2004. P. W. Atkins, J. de Paula: Physikalische Chemie, 4. Aufl., Wiley-VCH, 2008. C. Czeslik, H. Seemann, R. Winter: Basiswissen Physikalische Chemie, 2. Aufl., Vieweg + Teubner, 2007. [4] W. G. Moore, D. O. Hummel: Physikalische Chemie, 4. Aufl., Walter-de-Gruyter, 1986. Reaktionskinetik - Enzyme: Aktivität und Stabilität 135 Versuch 14 (RKE) Reaktionskinetik - Enzyme: Aktivität und Stabilität 1 Theorie 1.1 Proteine, Struktur und Enzymaktivität 1.1.1 Proteine Proteine sind die wichtigsten Funktionseinheiten der lebenden Zelle. Sie sind aus Aminosäuren aufgebaut, die über Peptidbindungen zu Ketten verknüpft sind. Die Peptidketten haben üblicherweise Längen von mindestens 50 bis über mehrere hundert Aminosäuren. Für den Aufbau von Proteinen werden in der Zelle 20 verschiedene Aminosäuren verwendet. Diese besitzen Seitenketten unterschiedlicher Eigenschaft (sauer, basisch, polar, hydrophob, aromatisch). Die Abfolge der Aminosäuren in einem Protein (sog. Primärstruktur) ist über die genetische Information genau definiert. Im wässrigen Medium falten sich die Peptidketten im Verlauf ihrer Synthese Abb. 1.1: Nativstruktur eines Proteins in definierte Strukturen. Kürzere Abschnitte nehmen dabei entweder schraubenartige Strukturen (α-Helices) oder gestreckte, übereinander geschichtete Strukturen (β-Faltblätter) ein (sog. Sekundärstruktur). Diese ordnen sich (zur Erreichung eines Energieminimums) in eine genau definierte dreidimensionale Struktur (sog. Tertiärstruktur). Hydrophobe Bereiche werden dazu im Kern des Proteins verborgen, polare und geladene Gruppen sind in Richtung Lösungsmittel exponiert und vermitteln Bindungen zu den Wassermolekülen. Ionische Bindungen, van-der-WaalsKräfte und Wasserstoffbrückenbindungen – in besonderen Fällen auch kovalente Bindungen (siehe Versuch 7 BPH) – innerhalb der Peptidkette stabilisieren zusätzlich die native Struktur des Proteins. Korrekt gefaltete Proteinketten können sich zu Komplexen aus mehreren (auch unterschiedlichen) Untereinheiten zusammenlagern (sog. Quartärstruktur). Proteine erfüllen nur in der korrekten Nativstruktur (siehe Abbildung 1.1) ihre biologischen Aufgaben/Aktivität. Proteine sind relativ labil. Unter bestimmten Bedingungen (z.B. extreme pH-Werte, erhöhte Temperaturen, Detergenzien, hydrophobe Lösungsmittel, mechanische Kräfte) kann die native Struktur zerstört werden. Die Peptidketten entfalten dann zu Zufallsstrukturen; die biologische Funktion/Aktivität geht verloren. Zumeist verlieren die entfalteten Moleküle zusätzlich ihre Löslichkeit, bilden Bindungen zu anderen Peptidmolekülen und damit Aggregate, die aus der wässrigen Phase ausfallen. Organismen die in relativ lebensfeindlicher Umgebung existieren (zum Beispiel thermophile Bakterien) sind aber durchaus in der Lage Proteine zu synthetisieren, die auch unter solch extremen Bedingungen ihre Struktur und damit biologische Aktivität bewahren. 136 Reaktionskinetik - Enzyme: Aktivität und Stabilität 1.1.2 Enzyme Viele Proteine, sog. Enzyme, wirken in der Zelle als hocheffiziente Biokatalysatoren. Diese beschleunigen (bio-)chemische Reaktionen und ermöglichen so die Umsetzung von Stoffen unter den milden Temperaturbedingungen des Lebens. Enzyme enthalten zusätzlich zur Pepidkette oft sog. Cofaktoren. Diese sind für den Katalysemechanismus wichtig. Cofaktoren sind oft komplexe organische Moleküle oder Metallionen (z.B. Mg2+ , Ca2+ , Mn2+ , Fe2+ , Zn2+ , Co2+ ). Sind Cofaktoren fest (hier: dauerhaft) an das Enzym gebunden spricht man von prostethischen Gruppen, transient bindende (für kurze Zeit gebundene) Cofaktoren nennt man Coenzyme. Enzyme sind wichtig für den chemischen Stoffwechsel der Zelle (Metabolismus), haben aber auch eine zentrale Bedeutung in der Regulation zellulärer Prozesse. Im vorliegenden Versuch sollen Enzymreaktionen am Beispiel der Lactatdehydrogenase (LDH) untersucht und die Denaturierung dieses Enzyms bei erhöhten Temperaturen verfolgt werden. 1.1.3 Physiologische Bedeutung der Lactatdehydrogenase Lactatdehydrogenase (LDH) ist ein wichtiges Enzym des Energiestoffwechsels der Zelle. Unter anaeroben Bedingungen (Fehlen von O2 ) gewinnt die Zelle Energie (gespeichert in Form von ATP-Molekülen) durch Spaltung von Glucose (Traubenzucker) zu Pyruvat (Anion der Brenztraubensäure), der sogenannten Glykolyse (siehe Abbildung 1.2). In einem Zwischenschritt dieses metabolischen Weges wird Gycerinaldehyd-3-Phosphat oxidiert. Dazu werden Reduktionsäquivalente (in Form von negativ geladenem Wasserstoff H– (1 Proton + 2 Elektronen) auf das Coenzym NAD+ übertragen und dieses zu NADH reduziert. Um ein Zusammenbrechen der Reaktion durch vollständigen Verbrauch der in der Zelle nur begrenzt vorhandenen NAD+ -Moleküle zu ver- Abb. 1.2: Glykolyse-Zyklus hindern wird NAD+ durch Oxidation regeneriert; die Reduktionsäquivalente werden dazu auf Pyruvat übertragen und dieses zu Lactat (Anion der Milchsäure) reduziert. Dieser Fall tritt beim Menschen bei kurzzeitiger Überlastung der Skelettmuskeln zum Beispiel beim Sport ein. Bei Sauerstoffmangel gewinnen die Muskelzellen weiter ATP durch Glykolyse und produzieren über den oben beschriebenen Mechanismus als Endprodukt Lactat. Der Milchsäurespiegel steigt an (der Athlet übersäuert). Die Bildung von Milchsäure in Reaktion auf Muskelbeanspruchung dient deshalb in der Sportmedizin als Maß der Belastung bzw. Belastbarkeit. In der Regenerationsphase wird Lactat mit Hilfe der LDH in der Leber zu Pyruvat oxidiert, welches dann in den aeroben Energiestoffwechsel eingeschleust und ”verbrannt” werden Reaktionskinetik - Enzyme: Aktivität und Stabilität 137 kann. Bestimmte (fakultativ anaerobe) Mikroorganismen, z.B. Milchsäurebakterien setzen die LDH-Reaktion zur ständigen Energiegewinnung unter Sauerstoffmangel ein; diesen Prozess nennt man Milchsäuregärung. Hefen haben eine alternativen Weg entwickelt: Sie reduzieren Pyruvat unter Abspaltung von CO2 zu Ethanol (alkoholische Gärung). Beide Mechanismen sind von zentraler Bedeutung in bestimmten ”biotechnologischen” Prozessen der Nahrungsmittelbereitung: Milchsäuregärung: Joghurt, Sauermilch, Käse, Sauerkraut, Alkoholische Gärung: Wein, Bier, Spirituosen, Backen. 1.2 Grundlagen der Reaktionskinetik Bei diesem Versuch soll der zeitliche Verlauf einer chemischen Reaktion verfolgt werden. Als Maß für den zeitlichen Ablauf einer chemischen Reaktion dient allgemein die Reaktionsgeschwindigkeit, die als Konzentrationsänderung pro Zeiteinheit definiert ist (Grundbegriffe der Reaktionskinetik). Betrachtet man die Reaktion: a A + b B Ð→ c C + d D (1) so werden die Konzentrationen der beiden Stoffe A und B im Verlaufe der Reaktion immer kleiner, während die Konzentrationen von C und D zunehmen. Die Zunahme der Produkte entspricht nach Gleichung 1 der Abnahme der Ausgangsstoffe. Es ist also im Prinzip gleichgültig, ob man die Abnahme von Stoff A oder B oder die Zunahme von C oder D betrachtet. Allgemein wird man immer den Stoff wählen, dessen Konzentration am einfachsten bestimmt werden kann. Da sich die Konzentration kontinuierlich ändert, wird die Reaktionsgeschwindigkeit oder die Reaktionsrate r einer chemischen Reaktion als die Änderung der Konzentration mit der Zeit definiert. Mathematisch entspricht diese Änderung der ersten Ableitung der Konzentration eines Reaktionspartners nach der Zeit. Für die obige Reaktion gilt: 1 dcB 1 dcC 1 dcD 1 dcA − ⋅ =− ⋅ =+ ⋅ =+ ⋅ a dt b dt c dt d dt (2) Gleichung 2 bringt zum Ausdruck, dass z.B. die Abnahme der Konzentration des Stoffes A (−dcA ) in dem Zeitintervall dt gleich der Zunahme des Stoffes C (+dcC ) ist. Im Praktikumsversuch soll dies indirekt am Beispiel der LDH-katalysierten Reduktion von Pyruvat zu Lactat gemäß nachfolgender Reaktionsgleichung ermittelt werden: Pyruvat + NADH + H+ ⇌ Lactat + NAD+ (3) Der Reaktionsverlauf kann mit Hilfe eines Photometers anhand der Reaktion von NADH zu NAD+ (bei einer Wellenlänge von λ = 366 nm absorbiert nur NADH) gemäß Abbildung 1.3 verfolgt werden. Die Geschwindigkeit der Reaktion – d. h. die zeitliche Änderung der Konzentration eines Reaktionsteilnehmers (Substrat) im Verlauf der Reaktion – kann anhand der Änderung der Extinktion gemessen werden. Die Extinktion (optische Auslöschung eines Lichtsignals durch Absorption von Lichtquanten an Molekülen) ist proportional zur Konzentration der absorbierenden Moleküle (siehe Versuch 2 ABS). E ist also ein Maß für die Konzentration c des verfolgten Stoffes 138 Reaktionskinetik - Enzyme: Aktivität und Stabilität Abb. 1.3: Redoxreaktion NAD+ ⇌ NADH (Lambert-Beer’sches Gesetz, siehe Versuch 2 ABS): E = − log I1 =⋅d⋅c I0 (4) mit I0 = Intensität des eingestrahlten Lichts, I1 = Intensität des austretenden Lichts, = Extinktionskoeffizient, d = Schichtdicke der Lösung, c = Konzentration des absorbierenden Stoffes. Aus der Steigung der erhaltenen Messkurve kann man die Geschwindigkeit v der Abnahme der NADH Konzentration errechnen. Diese ist ein direktes Maß für die vorhandene Enzymmenge und wird in der Enzymologie deshalb auch als Volumenaktivität AV bezeichnet. Durch Auflösen von Gleichung 4 nach c erhält man: c= E ⋅d Somit folgt für die Reaktionsgeschwindigkeit (v = v = AV = (5) dc dt ): ∆E ∆c = ∆t ∆t ⋅ ⋅ d (6) mit v = Reaktionsgeschwindigkeit, AV = Enzymaktivität in der Küvette (1 µmol Substratumsatz / (min⋅ml) = U(unit) /ml), ∆E = Extinktionsänderung (2 cm = 0,1 bei 100 mV bzw. 2 cm = 0,01 bei 10 mV), ∆t = Zeitänderung (in min), d = Lichtweg durch Küvette (1 cm), = Extinktionskoeffizient (3,3 ml/(µmol⋅cm)). Allerdings ist die Enzymaktivität in der Probe (und nicht in der Messküvette) von Interesse, somit muss die Verdünnung mit eingerechnet werden. Daraus folgt: AV = ∆E VT ⋅ ∆t ⋅ ⋅ d VP (7) mit VT = Testvolumen (1000 µl), VP = Probenvolumen (20 µl), der Quotient entspricht dem Verdünnungfaktor. Reaktionskinetik - Enzyme: Aktivität und Stabilität 139 2 Aufgabenstellung, Versuchsanleitung und -auswertung 2.1 Bestimmung der Reaktionsgeschwindigkeit der Reduktion von Pyruvat zu Lactat in Abhängigkeit der Konzentration aktiver LDH 2.1.1 Aufgabenstellung Es soll die thermische Zersetzung der LDH mittels ihrer bei thermischer Behandlung geringer werdenden katalytischen Aktivität auf die Reduktion von Pyruvat zu Lactat untersucht werden. 2.1.2 Versuchsanleitung Durchführung des LDH-Aktivitätstests Das Spektrometer wird im abs-Modus (absorbance (engl.) = Extinktion, nicht zu verwechseln mit absorption (engl.) = Absorption) betrieben. Vor Beginn der Messung wird das Spektrometer und der angeschlossene Schreiber mit dem Nullwert geeicht (set reference am Photometer + Schreiber auf Nulllinie; Eingangsspannung 10 mV: Papierbreite entspricht einer Extinktionseinheit). Der Maximalausschlag des Schreibers (Startposition zu Beginn der Reaktion) wird mit dem Maximalwert festgelegt. Zur Messung wird folgender Testansatz verwendet: 67 mM Phosphatpuffer pH 7.2 940 µl Pyruvatlösung (2,75 mg/ml) 20 µl NADH-Lösung (11 mg/ml) 20 µl Probenlösung (LDH) 20 µl Zur Herstellung der Pyruvatlösung ca. 1 mg einwiegen und mit der zum Erreichen der geforderten Konzentration notwendigen Menge Puffer auffüllen. Zur Herstellung der NADH-Lösung ca. 4 mg einwiegen und mit der zum Erreichen der geforderten Konzentration notwendigen Menge Puffer auffüllen. Um Pipettierfehler möglichst gering zu halten kann für den Testansatz folgende Lösung (Mix) vorbereitet werden (bei RT aufbewahren!): 67 mM Phosphatpuffer pH 7.2 9,4 ml Pyruvatlösung (2,75 mg/ml) 200 µl NADH-Lösung (11 mg/ml) 200 µl Die Reaktion wird mit der Zugabe der LDH-Probenlösung gestartet. Dafür werden in die Küvette 980 µl Testlösung vorgelegt und 20 µl LDH-Probenlösung dazu pipettiert. Die Küvette wird mit Parafilm abgedichtet, gemischt (zum Entfernen von Luftbläschen einige Male auf den Tisch klopfen), ins Photometer gestellt und dann wird sofort mit Hilfe des Schreibers die Abnahme der Extinktion verfolgt. Thermische Denaturierung der LDH Wird LDH erhitzt entfalten schon bei relativ milden Temperaturen die Peptidketten, das Enzym verliert seine natürliche Struktur (Konformation) und wird inaktiv. Die Denaturierungsreaktion Eaktiv → Einaktiv ist eine Reaktion 1. Ordnung; die Reaktionsgeschwindigkeit ist proportional 140 Reaktionskinetik - Enzyme: Aktivität und Stabilität Abb. 2.1: Aktivitäts-Zeit-Diagramme zur Konzentration des Edukts: vIA = dcE = k ⋅ cE dt (8) mit vIA = Geschwindigkeit der Inaktivierungsreaktion, k = Reaktionskonstante, cE = Enzymkonzentration. Durch Integration erhält man: cE = cE0 ⋅ e−kt siehe linke Kurve (A) in Abbildung 2.1 (9) mit cE0 = Enzymkonzentration zum Zeitpunkt 0. Daraus folgt: ln cE = ln cE0 − k ⋅ t oder ln cE = −k ⋅ t + ln cE0 (10) was einer Geradengleichung mit der negativen Steigung k und dem Achsenabschnitt ln cE0 entspricht (siehe rechte Kurve (B) in Abbildung 2.1). Die enzymkatalysierte Reaktion, also die Reduktion von Pyruvat zu Lactat, gehorcht einer anderen Regel, der sogenannten MichaelisMenten Kinetik. Bei hohen Substratkonzentrationen sind alle Enzymmoleküle mit Substrat belegt, d.h. die Reaktion ist gesättigt. Die Reaktionsgeschwindigkeit ist unabhängig von der Substratkonzentration und bleibt über einen größeren Bereich konstant. Das erklärt den geraden Verlauf der ∆E-Messkurven. 2.1.3 Versuchsdurchführung Die LDH aus Schweinherzmuskeln wird so verdünnt, dass die Ausgangslösung eine Volumenaktivität von ca. 2 – 4 U/ml (∆E/min = 0, 13 − 0, 26) aufweist. Es werden ca. 15 Tests in den Küvetten angesetzt. Damit werden mehrere Tests zur Messung der Volumenaktivität in der Ausgangslösung durchgeführt. Daraus wird ∆E/min ermittelt und die Volumenaktivität errechnet. Die Vorgehensweise beim Test sollte danach verinnerlicht sein und die Abweichungen der Messwerte sollte unter 10 % sein. 200 µl der LDH-Lösung werden in einem Eppendorf-Reaktionsgefäß bei 55°C in einen Thermoblock gestellt. Nach definierten Zeitabschnitten 0 – 30 min wird die Restaktivität zu acht Zeitpunkten (2, 5, 8, 12, 16, 20, 25, 30 min) bestimmt. Reaktionskinetik - Enzyme: Aktivität und Stabilität 141 2.1.4 Versuchsauswertung ● Theoretischer Teil: Wie sind Proteine aufgebaut? Was ist ein Enzym? Welche Bedingungen sind von Bedeutung für die Stabilität eines Proteins? Wie funktioniert eine Reaktion erster Ordnung? Definieren Sie die Begriffe Aktivierungsenergie (Achtung: kommt NICHT im Theorieteil des Skripts vor) und Katalysator. ● Die Messergebnisse werden in ein Diagramm der Auftragung Aktivität gegen Zeit und ein Diagramm der Auftragung log(Aktivität) gegen Zeit eingetragen. Hieraus wird die Halbwertszeit der Denaturierung und die Geschwindigkeitskonstante k bestimmt. ● Zeichen Sie die Strukturen von Brenztraubensäure und Milchsäure und bestimmen Sie unter Angabe der jeweiligen Oxidationszahlen welches Teilchen reduziert wird. 3 Geräte und Chemikalien Eine Geräteliste liegt am Platz aus Chemikalien Pyruvat-Lösung NADH-Lösung LDH-Lösung Phosphat-Pufferlösung Lactat-Lösung (Produkt) NAD+ -Lösung (Produkt) Gefahrenbezeichnung – – – – – – H-Sätze – – – – – – P-Sätze – – – – – – 4 Literatur [1] [2] [3] G. Wedler: Lehrbuch der Physikalischen Chemie, 5. Aufl., Wiley-VCH, 2004. P. W. Atkins, J. de Paula: Physikalische Chemie, 4. Aufl., Wiley-VCH, 2008. J. M. Berg, L. Stryer, J. L. Tymoczko: Biochemie, 5. Aufl. Spektrum, 2007. Destillationstechniken 143 VERSUCHSBLOCK 3: ORGANISCHE CHEMIE UND ARBEITSTECHNIKEN Versuch 15 (DES) Destillationstechniken 1 Theorie Ein Gemisch zweier flüssiger Substanzen a, b mit unterschiedlichen Siedepunkten Ts (a) < Ts (b) lässt sich durch Destillation trennen. Die Zusammensetzung der Flüssigkeit wird durch den Molenbruch xa = ya = n∗a ∗ na +n∗b na na +nb beschrieben (na = Stoffmenge der leichter flüchtigen Substanz a in mol). bezeichnet die Zusammensetzung der Dampfphase. Die Diagramme in Abbildung 1.1 zeigen den Verlauf des Siedepunkts und des Taupunkts in Abhängigkeit von xa bzw. ya für die Gemische 2-Propanol/1-Butanol (A) und Cyclohexan/Benzylalkohol (B). Abb. 1.1: Siedediagramme Trägt man die Zusammensetzung des Dampfs ya über der siedenden Mischung xa auf, so erhält man die sogenannten Gleichgewichtskurven: Verdampft man eine Mischung B mit dem Gehalt xa = 0, 2 an leichter siedendem Cyclohexan, so enthält der Dampf bereits über 90% der Substanz. Eine Solche Mischung lässt sich durch einfache Destillation trennen. (siehe Kapitel 2.2). Bei Gemisch A erhält man dagegen eine Reinheit von unter 50%. Lässt man jedoch den Dampf kondensieren und verdampft erneut, so erhält man 80% und in einer dritten Stufe über 90% Reinheit (siehe Pfeile im Diagramm in Abbildung 1.2). Dieses Verfahren wird Rektifikation genannt und mit einer sogenannten Kolonne durchgeführt. (siehe Kapitel 2.1) 144 Destillationstechniken Abb. 1.2: Gleichgewichtsdiagramme 2 Aufgabenstellung, Versuchsaufbau und -auswertung 2.1 Trennung flüssiger Verbindungen durch Rektifikation bei Normaldruck 2.1.1 Versuchsanleitung In einer NS 14-Destillationsapparatur (50 ml Destillationskolben, Vigreux Kolonne (30 cm), Schliffthermometer, gebogener Vorstoß, graduierter Tropftrichter und 25 ml Vorlagekolben) werden 20 ml der Mischung von 1-Butanol und 2-Propanol (1/1 V /V ) zusammen mit einem Magnetrührstab vorgelegt. Man heizt das Ölbad auf die Siedetemperatur des Gemisches (siehe Diagramm in Abbildung 1.1). Dann wird die Temperatur langsam gesteigert, bis erstes Destillat übergeht. Während der Destillation wird die Ölbadtemperatur so geregelt, dass das Destillat mit möglichst konstanter Geschwindigkeit übergeht. (ca. 1 Tropfen pro Sekunde). Der Verlauf der Destillation wird in regelmäßigen Abständen zusammen mit der Destillatmenge, der Siedetemperatur und der Ölbadtemperatur protokolliert. Zum Schneiden einer Fraktion wird Abb. 2.1: Versuchsauf- das gesammelte Destillat aus dem Tropftrichter in einen gewogenen Vorlagekolben abgelassen. bau 2.1.2 Versuchsauswertung ● Erstellen Sie eine Stoffbilanz, messen Sie die Brechungsindices der Reinsubstanzen 20 20 n20 D (a), nD (b) und der einzelnen Fraktionen nD . Berechnen Sie die Zusammensetzung der Fraktionen nach der Formel xa = ● 20 n20 D −nD (b) 20 (b) n20 (a)−n D D Zeichnen Sie die Siedekurve der Destillation (Siedetemperatur in Abhängigkeit vom Destillatvolumen). Zeichnen Sie in dieses Diagramm die Fraktionsschnitte und den Verlauf der Ölbadtemperatur ein. Diskutieren Sie die Effektivität der Destillation. Destillationstechniken 145 2.2 Trennung flüssiger Verbindungen durch Destillation unter vermindertem Druck am Rotationsverdampfer 2.2.1 Versuchsanleitung Ein 250 ml NS 29-Rundkolben wird leer gewogen. Anschließend werden 100 ml einer Mischung von Benzylalkohol in Cyclohexan (1/1 V /V , mit Messzylinder abmessen!) mit der Hilfe eines Trichters (keine Substanzreste auf Schlifffläche!) in den Kolben gefüllt. Der Vorlagekolben wird ebenfalls leer gewogen. Der Destillationskolben wird am Dampfrohr des Rotationsverdampfers befestigt (Schliff nicht fetten, Sicherungsbügel nur leicht festziehen). Der Dampfdruck von Cyclohexan B Abb. 2.2: Ver- lässt sich nach folgender Formel berechnen: ln(p/mbar) = A − T (in °C)+C mit A suchsaufbau = 16,02; B = 2753, C = 221,94. Die Destillation wird bei einer Badtemperatur von 40°C durchgeführt. Der Motor wird eingeschaltet und am Vakuumcontroller der gewünschte Solldruck eingestellt. Danach wird der Controller gestartet und der Destillationskolben in das auf ca. 60°C geheizte Wasserbad abgesenkt. Das Ende der Destillation ist erreicht wenn kein Destillat mehr übergeht. Zum Abschalten wird zuerst der Destillationskolben aus dem Heizbad gehoben dann der Vakuumcontroller gestoppt. Anschließend wird die Apparatur belüftet. Nach vollständigem Druckausgleich (Druckanzeige beachten!) wird der Motor abgeschaltet und Destillationskolben und Vorlage abgenommen. 2.2.2 Versuchsauswertung Destillationskolben und Vorlagekolben werden zurückgewogen und die erhaltene Masse berechnet. Der Brechungsindex von Destillat und Rückstand wird bestimmt und die Reinheit abgeschätzt. 3 Geräte, Chemikalien und Entsorgung Eine Geräteliste liegt am Platz aus. Die Destillate und Rückstände werden in die entsprechenden Vorratsflaschen zurückgegeben. Chemikalien 2-Propanol 1-Butanol Cyclohexan Benzylalkohol Gefahrenbezeichnung Gefahr (GHS02, 08) Gefahr (GHS08) Gefahr (GHS02, 08, 09) Achtung (GHS07) H-Sätze 225, 319, 336 226, 302, 318, 315, 335, 336 225, 304, 315, 336, 410 332, 302 P-Sätze Sdp. (○ C) 305+351+338, 210, 82 233 280, 313, 302+352, 117 305+351+338 210, 240, 273, 331, 81 301+310, 403+235 271 205 n20 D ρ (g/cm3 ) 0,78 1,3792 0,81 1,3993 0,78 1,4263 1,04 1,5396 146 Destillationstechniken 4 Literatur S. Hünig, P. Kreitmeier, G. Märkl, J. Sauer: Arbeitsmethoden in der Organischen Chemie, 1. Aufl., Lehmanns, 2006. Sublimation 147 Versuch 16 (SUB) Sublimation 1 Theorie Die meisten Feststoffe gehen beim Erwärmen unter Normaldruck zuerst in den flüssigen (Schmelzpunkt), dann in den gasförmigen Zustand über (Siedepunkt). Wasser beispielsweise schmilzt bekanntermaßen bei 0°C und siedet bei 100°C. Hingegen geht festes Kohlendioxid (Trockeneis) bei −78°C ohne zu schmelzen direkt in den gasförmigen Zustand über. Diesen Vorgang nennt man Sublimation. Ermittelt man Schmelz- und Siedepunkt eines Stoffes bei unterschiedlichem Druck, so erhält man ein sogenanntes Abb. 1.1: Beispiel eines Zustandsdiagramms Phasendiagramm (s. Abb. 1.1). Hier zeigt sich, dass an einem besonderen Punkt, dem sogenannten Tripelpunkt, Schmelz- und Siedepunkt zusammenfallen. Bei geringerem Druck tritt keine flüssige Phase mehr auf, der Stoff sublimiert. Wasser beispielsweise hat einen Tripelpunkt von 0,01°C bei 6 mbar. Campher hat bei Normaldruck einen Schmelzpunkt von 179°C und siedet bei 209°C. Der Tripelpunkt liegt bei 181,6°C und 538 mbar. Die Sublimationskurve kann nach folgender Formel berechnet werden: ln(p/mbar) = A − B T (in °C)+C mit A = 19,72; B = 6106, C = 273,15. 2 Aufgabenstellung, Versuchsanleitung und -auswertung 2.1 Sublimation 2.1.1 Aufgabenstellung Trennung eines Gemisches von Campher und Aktivkohle durch Sublimation 2.1.2 Versuchsanleitung Es ist zunächst der Schmelzpunkt der verunreinigten Probe zu bestimmen. Anhand der Sublimationskurve (siehe Abbildung 1.1) kann der geeignete Druck bestimmt werden, bei dem unterhalb des Tripelpunkts sublimiert werden kann. Zur präparativen Reinigung der Substanz werden etwa 2–5 g der verunreinigten Substanz in das Sublimationsgefäß eingewogen, und der Kühlfinger so aufgesetzt, dass er sich etwa 2 bis 3 cm oberhalb der Substanzoberfläche befindet. Nun wird der ermittelte Unterdruck angelegt (Vakuummessgerät im Nebenschluss). Das Sublimationsgefäß im Heizbad wird langsam erwärmt (das Sublimationsgut darf Abb. 2.1: Versuchsaufbau auf keinen Fall schmelzen!). Die Apparatur sollte möglichst weit in das Heizbad eintauchen, gegebenenfalls muss die Apparatur mit Aluminiumfolie isoliert werden, um das Abscheiden des Sublimats an der Außenwand zu verhindern. 148 Sublimation Nach vollständiger Sublimation wird das Ölbad entfernt, die Sublimationsapparatur belüftet und der Kühlfinger vorsichtig herausgenommen. Das Sublimat wird mit einem Spatel auf ein Uhrglas überführt, gewogen und der Schmelzpunkt bestimmt. Der Rückstand wird ebenfalls gewogen. 2.1.3 Versuchsauswertung ● Protokollieren Sie die durchgeführten Vorproben, den Verlauf der Sublimation (Druck, Badtemperatur, Dauer, Ausbeute, Schmelzpunkte und Aussehen von Roh- und Reinprodukt). ● Vergleichen Sie den Schmelzpunkt der Camphermischung vor der Sublimation mit dem der gereinigten Probe. 3 Geräte, Chemikalien und Entsorgung Eine Geräteliste liegt am Platz aus Chemikalien Campher Aktivkohle Gefahrenbezeichnung Gefahr (GHS02, 08) – H-Sätze 228, 315, 319, 335 – P-Sätze 210, 302+352, 304+340, 305+351+338 – Die gereinigten Substanzen werden wieder in die aufgestellten Vorratsflaschen zurückgegeben. Achten Sie darauf, dass keine Verwechslungen erfolgen! Die festen Rückstände im Sublimationsgefäß werden mechanisch (z. B. mit dem Spatel) entfernt und in den festen organischen Sondermüll gegeben. Substanzreste werden mit etwas Aceton in den halogenfreien organischen Sonderabfall gespült. 4 Literatur S. Hünig, P. Kreitmeier, G. Märkl, J. Sauer: Arbeitsmethoden in der Organischen Chemie, 1. Aufl., Lehmanns, 2006. Extraktion 149 Versuch 17 (EXT) Extraktion 1 Theorie Viele Stoffe sind sowohl in Wasser als auch in organischen Lösungsmitteln löslich. Bringt man eine wässrige Lösung eines solchen Stoffes mit einem organischen Lösungsmittel in Kontakt, das mit Wasser nur wenig mischbar ist (hydrophob), so wird der Stoff aus der wässrigen Phase extrahiert, das heißt herausgelöst. Da der Stoffübergang an der Phasengrenze stattfindet, werden wässrige und organische Phase intensiv durchmischt, um die Phasengrenze zu vergrößern. Im Labor geschieht dies in einem sogenannten Scheidetrichter (siehe Abbildung 2.1) durch Schütteln. Daher wird dieses Verfahren auch Ausschütteln genannt. Der gelöste Stoff verteilt sich zwischen den beiden Phasen bis zu einer Gleichgewichtseinstellung. Für ein gegebenes organisches Lösungsmittel bei einer bestimmten Temperatur ist das Verhältnis der Konzentration in der organischen Phase co und der wässrigen Phase cw konstant: K = co cw (Nernst’sches Verteilungsgesetz). Ist K > 100 so kann mit einem Extraktionsschritt ein vollständiger Stoffübergang erreicht werden. Ist K < 100, so muss die Extraktion nach Abtrennen der organischen Phase mit frischem Lösungsmittel mehrmals wiederholt werden um den Stoff vollständig aus der Wasserphase zu entfernen. Die Löslichkeit von Carbonsäuren und Aminen lässt sich durch Wahl des pH-Wertes steuern. Carboxylate (basisch) und Ammoniumsalze (sauer) sind gut wasserlöslich, die Carbonsäuren und Amine dagegen gut löslich in organischen Lösungsmitteln. 2 Aufgabenstellung, Versuchsanleitung und -auswertung 2.1 Extraktion 2.1.1 Aufgabenstellung Trennung einer Stoffmischung in ihre Einzelbestandteile durch Extraktion. Die dabei benötigten verdünnten Säuren und Laugen müssen zunächst frisch hergestellt werden. 2.1.2 Versuchsanleitung 1. Extraktion von 4’-Aminoacetophenon 3 g eines Gemisches aus gleichen Gewichtsteilen Benzoesäure, 4’-Aminoacetophenon und Naphthalin werden in 50 ml Diethylether gelöst und in einem Scheidetrichter zweimal mit jeweils 25 ml 5 % wässriger Salzsäure kräftig geschüttelt, wobei der Scheidetrichter zum Druckausgleich öfters belüftet werden muss. Die wässrige Phase wird von der Etherphase abgetrennt und unter Kühlung mit einem Eisbad mit soviel 5 M Natronlauge versetzt (etwa 20 ml), bis die Lösung alkalisch reagiert (Kontrolle mit pH-Papier). Der Niederschlag von 4’-Aminoacetophenon wird abfiltriert, mit Wasser gewaschen und getrocknet. Ausbeute und Schmelzpunkt werden bestimmt. Abb. 2.1: Versuchsaufbau 150 Extraktion 2. Extraktion von Benzoesäure Die Etherphase wird im Scheidetrichter zweimal mit jeweils 25 ml 5 % wässriger Natronlauge ausgeschüttelt (Scheidetrichter dabei öfters belüften). Die wässrige Phase wird von der Etherphase abgetrennt und mit soviel halbkonzentrierter Salzsäure versetzt (etwa 20 ml), bis die Lösung sauer reagiert (Kontrolle mit pH-Papier). Der farblose Niederschlag von Benzoesäure wird abfiltriert, mit Wasser gewaschen und getrocknet. Ausbeute und Schmelzpunkt werden bestimmt. 3. Isolierung von Naphthalin Die Etherphase wird im Scheidetrichter einmal mit etwa 50 ml gesättigter wässriger Natriumchloridlösung geschüttelt, die wässrige Phase wird abgetrennt und verworfen. Nach dem Trocknen mit Na2 SO4 und Abfiltrieren des Trockenmittels wird der Diethylether am Rotationsverdampfer abdestilliert. Der gelbliche feste Rückstand wird anhand des Schmelzpunkts als Naphthalin identifiziert. 2.1.3 Versuchsauswertung Ermitteln Sie anhand Ihrer ausgewogenen Substanzen die Ausbeuten für die Extraktion und vergleichen Sie die Schelzpunkte mit den Literaturwerten. 3 Geräte, Chemikalien und Entsorgung Eine Geräteliste liegt am Platz aus Chemikalien Benzoesäure 4’-Aminoacetophenon Naphthalin Diethylether Gefahrenbezeichnung Achtung (GHS07) Achtung (GHS07) Achtung (GHS07, 09) Gefahr (GHS02, 08) H-Sätze P-Sätze 302, 319 302 HClaq Gefahr (GHS05) 302, 350, 410 EUH019, EUH066, 224, 302, 336 314, 335, 290 NaOH Gefahr (GHS05) 314, 290 Na2 SO4 – – 305+351+338 – Smp. (°C) 122 106 M (g/mol) 122,12 135,17 273, 281, 308+313 210, 240,403+235 80 k. A. 128,17 74,14 280, 301+330+331, 305+351+338 280, 301+330+331, 305+351+338 – k. A. 36,45 k. A. 40,00 k. A. 142,04 Die isolierten Substanzen werden in die entsprechenden Vorratsbehälter zurückgegeben. Der am Rotationsverdampfer abdestillierte Ether wird in den Abfallbehälter für halogenfreie Lösungsmittel gegeben. 4 Literatur S. Hünig, P. Kreitmeier, G. Märkl, J. Sauer: Arbeitsmethoden in der Organischen Chemie, 1. Aufl., Lehmanns, 2006. Chromatographie 151 Versuch 18 (CHR) Chromatographie 1 Theorie Flüssigkeitschromatographie ist ein Verfahren zur Stofftrennung, bei der Substanzen in einer mobilen Phase (Laufmittel) gelöst an einer stationären Phase (hier: Kieselgel) vorbeigeführt werden. Durch Adsorption an der stationären Phase werden die verschiedenen Substanzen unterschiedlich stark zurückgehalten und wandern deshalb mit verschiedenen Geschwindigkeiten mit der mobilen Phase. Dies führt zu einer räumlichen Auftrennung der Verbindungen. Die Verzögerung des Stoff- Abb. 1.1: Verlauf einer Chromatographie transports wird Retention genannt. Der Retentionsfaktor Rf = d d0 ist bei gegebener stationärer und mobiler Phase für jede Substanz eine Konstante. Bei der Dünnschichtchromatographie ist die stationäre Phase als dünne Schicht auf einer Trägerfolie fixiert. Das Laufmittel wird durch Kapillarkräfte in die Dünnschicht hineingesaugt. Bei farbigen Substanzen können die Laufstrecken der Substanzen direkt gemessen werden, bei farblosen Substanzen ermöglicht ein Fluoreszenzindikator in der Schicht die Detektion unter UV-Licht. Bei der Säulenchromatographie ist die stationäre Phase in einem Rohr gepackt. Das Laufmittel wird durch den Druck der Flüssigkeitssäule oder mittels Überdruck durch die Packung gepresst. Die getrennten Substanzen werden am Auslauf der Säule in Fraktionen aufgefangen. 152 Chromatographie 2 Aufgabenstellung, Versuchsanleitung und -auswertung 2.1 DC-Chromatographie von Pflanzenfarbstoffen 2.1.1 Aufgabenstellung DC-Chromatographie von Pflanzenfarbstoffen 2.1.2 Versuchsanleitung Herstellung des Pflanzen-Extrakts 2-3 frische Blätter (Gras o.ä.) werden mit etwas gereinigtem Seesand und einer Spatelspitze CaCO3 (zur Neutralisation der Pflanzensäuren!) in einer Reibschale zerrieben. Man gibt 15 ml Aceton zu, verreibt und lässt im Dunkeln (der Extrakt ist lichtempfindlich!) 5 min in einem Eisbad stehen. Es wird hierauf nochmals gründlich verrieben. Dünnschichtchromatographie Der dunkelgrüne Extrakt wird auf die Startlinie einer DC-Kieselgel-Alufolie konzentrierend (d. h. mehrfach nacheinander) aufgetragen. Das Chromatogramm wird mit einem Gemisch aus Ligroin, 2-Propanol und Wasser (V /V = 400 ∶ 40 ∶ 1) in einem dunklen Raum entwickelt. Die DC-Folie kann aus der Chromatographiekammer genommen werden, wenn die Laufmittelfront etwa 1 cm unterhalb der oberen Kante der DC-Platte liegt. Die Solvensfront und die farbigen Flecken werden sofort mit einem Bleistift markiert und die Rf -Werte bestimmt. Reihenfolge der Farbstoffe mit abnehmendem Rf -Wert: Gelbrotes β-Carotin > Phäophytin a > blaugrünes Abb. 2.1: Dünnschichtchromatogramm Chlorophyll a > gelbgrünes Chlorophyll b > gelbes Xanthophyll. Es ist zügiges Arbeiten erforderlich, da die Farbstoffe im Licht schnell verblassen. 2.1.3 Auswertung Im Laborjournal werden alle Dünnschichtchromatogramme abgezeichnet, die Rf -Werte berechnet und die Durchführung protokolliert. Chromatographie 153 2.2 DC- / Säulen-Chromatographie-Trennung von Benzoesäure und Benzophenon 2.2.1 Aufgabenstellung Trennung von Benzoesäure und Benzophenon mittels DC- und Säulen-Chromatographie 2.2.2 Versuchsanleitung Dünnschichtchromatographietest Je eine Spatelspitze Benzoesäure und Benzophenon werden in ca. 1 ml Essigsäureethylester/Cyclohexan (1/2 V /V ) gelöst. Mit einer Glaskapillare wird je eine Probe auf einer KieselgelDC-Alufolie aufgetragen. Auf einem dritten Punkt wird ein Gemisch beider Substanzen aufgetragen (je eine Spatelspitze in 1 ml Laufmittel gelöst). Unter UV-Licht (λ = 254 nm), werden die Flecken mit Bleistift markiert und die Rf -Werte berechnet (Laborjournal). Säulen-Chromatographie Präparation der Säule Eine Pasteur-Pipette wird so an einer Stativstange befestigt, dass man noch bequem einen Reagenzglasständer darunter stellen kann. In die Verjüngung der Pipette wird ein kleiner Wattebausch eingebracht, der das Auslaufen der Säulenfüllung verhindert. Anschließend wird die Pasteur-Pipette mit Kieselgel gefüllt (ca. 4/5 der Höhe) und auf die Oberfläche der Säulenpackung wird ca. 5 mm Seesand gegeben. Mit Hilfe einer Handpumpe wird die Säule mit 10 ml der Laufmittelmischung (Cyclohexan/Essigsäureethylester 2:1) gewaschen. Dabei darf die Säule nie trocken laufen! Um Risse und Luftblasen in der Säule zu verhindern, wird während des Absetzens der Säulenfüllung an die Säule geklopft. Chromatographie Wenn das Laufmittel in der Säule soweit abgelaufen ist, dass die Oberkante des Flüssigkeitsspiegels bündig mit der Oberkante der Seesandschicht ist, werden 3 bis 5 Tropfen der Mischung von Benzoesäure und Benzophenon vorsichtig entlang der Innenwand der Säule aufgetragen. Nachdem der Flüssigkeitsspiegel gerade wieder die Seesandoberkante erreicht hat, wird zur Entwicklung des Chromatogramms Laufmittel portionsweise nachgefüllt, wobei auf einen konstanten Auslauf zu achten ist. Dabei darf die Säule nie trocken laufen! Das Eluat wird in kleinen Reagenzgläsern etwa 1 cm hoch aufgefangen. Je eine Probe der erhaltenen Fraktion wird auf eine DC-Platte getüpfelt. Unter der UV-Lampe wird kontrolliert, welche Fraktionen Substanz enthalten. Nachdem 10-15 Fraktionen gesammelt wurden (abhängig von der Trennwirkung der Säule) kann die Säule zusätzlich mit 5 ml reinem Essigsäureethylester eluiert werden. Danach wird die Entwicklung der Chromatographie abgebrochen. Reinheitskontrolle Die DC-Platte mit Proben aller Fraktionen und einer Probe der Substanzmischung (Referenz) wird in einer Chromatographiekammer mit Cyclohexan/ Essigsäureethylester (2:1) entwickelt. Die Substanzflecken sind unter der UV-Lampe (λ = 254 nm) sichtbar und werden mit einem weichen Bleistift markiert. 154 Chromatographie 2.2.3 Auswertung Im Laborjournal werden alle Dünnschichtchromatogramme abgezeichnet, die Rf -Werte berechnet und die Durchführung protokolliert. Als Fazit soll die Trennleistung der Säule beurteilt und ggf. Verbesserungsvorschläge gemacht werden. 3 Geräte, Chemikalien und Entsorgung Eine Geräteliste liegt am Platz aus Chemikalien Aceton Gefahrenbezeichnung Gefahr (GHS02, 08) Ligroin (Petroleumben- Gefahr (GHS02, 08, zin) 09) 2-Propanol Gefahr (GHS02, 08 Benzoesäure Achtung (GHS07 Benzophenon Achtung (GHS09) Gefahr (GHS02, 08) Essigsäureethylester Cyclohexan Gefahr (GHS02, 08, 09) H-Sätze EUH066, 225, 336 EUH066, 225, 336, 411 225, 319, 336 302, 319 410 EUH066, 225, 336 225, 304, 315, 410 P-Sätze Sdp. (○ C) 77 319, 210, 233, 305+351+338 304, 210, 273, 243, 301+310, 331 305+351+338, 210, 233 305+351+338 273 210, 240, 305+351+338 100-140 210, 240, 273, 301+310, 403+235, 331 81 319, 336, 82 k. A. 49 k. A. Die verbliebenen Lösungsmittel (Essigsäureethylester und die Mischung Cyclohexan/Ethylacetat) werden in den Abfallbehälter für halogenfreie Lösungsmittel gegeben. Die Chromatographiesäule kann in den aufgestellten Sammelbehälter für verunreinigte Betriebsmittel gegeben werden. 4 Literatur S. Hünig, P. Kreitmeier, G. Märkl, J. Sauer: Arbeitsmethoden in der Organischen Chemie, 1. Aufl., Lehmanns, 2006. Umkristallisation 155 Versuch 19 (UMK) Umkristallisation 1 Theorie Zur Reinigung von Feststoffen wird im Syntheselabor die Umkristallisation eingesetzt. Dabei wird in einem geeigneten Lösungsmittel eine heiß gesättigte Lösung der verunreinigten Substanz hergestellt. Unlösliche Verunreinigungen werden durch heiße Filtration entfernt. Beim Abkühlen kristallisiert die Substanz in reiner Form während die Verunreinigungen in Lösung bleiben. Die Löslichkeit eines Stoffes hängt von den molekularen Eigenschaften von Lösungsmittel und gelöstem Stoff ab (siehe Versuch 11 FLG). Sie nimmt meist mit der Temperatur zu. Die Sättigungskonzentration steigt beim Siedepunkt des Lösungsmittels in der Regel stark an, deshalb wird meist aus siedendem Lösungsmittel umkristallisiert. Haben die Verunreinigungen eine ähnliche Struktur wie der zu reinigende Stoff, so ist ihre Löslichkeit ähnlich. Da die Konzentration der Verunreinigungen in der Lösung gering ist, wird beim Abkühlen ihre Sättigungskonzentration nicht überschritten, sodass nur der reine Stoff auskristallisiert. 2 Aufgabenstellung, Versuchsanleitung und -auswertung 2.1 Umkristallisation von mit β-Naphtolorange gefärbter Benzoesäure aus Wasser. 2.1.1 Aufgabenstellung Mit Methylrot gefärbte Benzoesäure soll durch Umkristallisation aus Wasser gereinigt werden. 2.1.2 Versuchsanleitung Vorbereitung Ein 250 ml Erlenmeyerkolben wird zusammen mit einem Glastrichter und einem Faltenfilter im Trockenschrank bei ca. 60°C erwärmt. Lösen der Substanz In einem zweiten 250 ml Erlenmeyerkolben werden 6 g der verunreinigten Benzoesäure in 80 ml Wasser suspendiert, ein Magnetrührstäbchen zugegeben und mit einem Uhrglas abgedeckt. Es wird unter Rühren auf dem Heizrührer erwärmt. Wenn die Lösung siedet, wird portionsweise Wasser zugegeben, bis eine klare Lösung entstanden ist. Die genaue Lösungsmittelmenge wird protokolliert. Der Erlenmeyerkolben wird Abb. 2.1: vom Heizrührer genommen, damit die Lösung ein wenig abkühlt. Anschließend werden Versuchsca. 0,3 g gekörnte Aktivkohle zugegeben und nochmals unter Rühren zum Sieden aufbau erhitzt. 156 Umkristallisation Heiße Filtration Die heiße gesättigte Lösung wird durch den im Trockenschrank erwärmten Trichter filtriert. Wenn die Benzoesäure im Filter auskristallisiert, wird der Erlenmeyerkolben auf dem Heizrührer erhitzt, so dass der im Trichter kondensierende Wasserdampf die ausgefallene Säure wieder auflöst. Kristallisation und Filtration Man lässt die gesättigte, heiße Lösung langsam auf Raumtemperatur abkühlen, dabei kristallisiert die Benzoesäure aus. Der erhaltene Kristallbrei wird hierauf auf einem Büchnertrichter abgesaugt, zweimal mit je 10 ml eiskaltem Wasser gewaschen, der Filterkuchen mit einem Glasstopfen abgepresst und trocken gesaugt. Die Kristalle werden quantitativ in eine Porzellanschale überführt und im Exsikkator über Phosphorpentoxid bis zur Gewichtskonstanz getrocknet. Abb. 2.2: Versuchsaufbau 2.1.3 Versuchsauswertung ● ● Die Umkristallisation wird protokolliert Der Schmelzpunkt der verunreinigten Benzoesäure und des umkristallisierten Produkts werden bestimmt. Anhand der Auswage wird die Ausbeute des Verfahrens ermittelt. 3 Geräte, Chemikalien und Entsorgung Eine Geräteliste liegt am Platz aus Chemikalien Benzoesäure β-Naphtolorange P4 O10 Gefahrenbezeichung Achtung (GHS07) Achtung (GHS07) Gefahr (GHS05) H-Sätze 302, 319 315, 319, 335 314 Smp. (○ C) 305+351+338 122 261, 305+351+338 167 260, 280, 301+330+331, k. A. 305+351+338 P-Sätze M (g/mol) 122,12 350,32 283,92 Strukturformeln ausgewählter Moleküle: Benzoesäure β-Naphtolorange 4 Literatur S. Hünig, P. Kreitmeier, G. Märkl, J. Sauer: Arbeitsmethoden in der Organischen Chemie, 1. Aufl., Lehmanns, 2006. Aspirin – Synthese von Acetylsalicylsäure 157 Versuch 20 (ASP) Aspirin – Synthese von Acetylsalicylsäure 1 Theorie Abb. 1.1: Reaktionsgleichung der Aspirinsynthese Durch Umsetzung von Alkoholen mit Anhydriden können Ester gewonnen werden. Die Reaktion folgt einem Additions-Eliminierungs-Mechanismus. Abb. 1.2: Reaktionsmechanismus der Aspirinsynthese 2 Aufgabenstellung, Versuchsanleitung und -auswertung 2.1 Synthese von Acetylsalicylsäure 2.1.1 Aufgabenstellung Acetylisierung von Salicylsäure durch Umsetzung mit Essigsäureanhydrid. 2.1.2 Versuchsanleitung Durchführung der Reaktion In einen trockenen 50 ml Zweihalskolben, ausgestattet mit Rührfisch, Rückflusskühler (ohne Wasserkühlung), Hebebühne, Magnetrührer, Heizbad und Temperaturfühler, werden 2,08 g (15 mmol) Salicylsäure eingewogen (Feststofftrichter benutzen!). Mit einer Messpipette werden 1,75 ml (18,5 mmol) Essigsäureanhydrid aus einer Vorratsflasche entnommen und in den Reaktionskolben gegeben. Mit einer Pasteurpipette werden unter Rühren 2 Tropfen (nicht mehr!) konz. Schwefelsäure zugefügt. Die Mischung wird 15 Minuten (nicht länger!) bei 50°C gerührt (die 158 Aspirin – Synthese von Acetylsalicylsäure Temperatur darf 60°C nicht überschreiten, ansonsten finden Nebenreaktionen statt). Während der Reaktion geht die Salicylsäure zuerst in Lösung und die gebildete Acetylsalicylsäure kristallisiert als farbloser Feststoff aus. Nach dem Abkühlen wird der feste Kuchen mit einem Spatel zerkleinert und mit 20 ml einer Eis-Wasser-Mischung aufgeschlämmt. Isolierung und Reinigung Das Rohprodukt wird über einem Büchnertrichter abgesaugt und dreimal mit je 20 ml kaltem demineralisiertem Wasser gewaschen. Das Filtrat wird Verworfen. Der Filterkuchen wird mit einem breiten Spatel fest auf die Filterplatte gepresst und durch längeres Durchsaugen von Luft weitgehend getrocknet. Zum Umkristallisieren (siehe auch Versuch 19 UMK) wird das feuchte Rohprodukt in einen 50 ml NS 29 Einhalsrundkolben überführt, mit 8 ml Ethanol/Wasser Mischung (1:2) suspendiert und unter Rückfluss erhitzt (Badtemperatur: 85°C), bis eine klare Lösung entsteht. Nun wird das Heizbad entfernt und die Lösung (animpfen) langsam (ca. 10 min) abgekühlt. Der Feststoff wird wieder über einen Büchnertrichter abgesaugt, portionsweise mit insgesamt 20 ml kalter Mischung aus Ethanol/Wasser (1:2) gewaschen und scharf abgepresst. Nach dem Trocknen im Vakuumexsikkator über Sicapent werden Ausbeute und Schmelzpunkt bestimmt. 2.1.3 Versuchsauswertung ● Laborjournal: Zeichnen Sie die Reaktionsgleichung mit Summenformeln und Molmassen. Geben Sie kurz den Apparaturaufbau an (mit Kolbengrößen) und alle verwendeten Chemikalien mit genauen Mengenangaben. Beschreiben Sie die Durchführung des Versuchs mit allen Arbeitschritten, Zeit- und Temperaturangaben sowie allen Beobachtungen. Protokollieren Sie die erhaltene Menge an Rohprodukt zusammen mit dem Schmelzpunkt. Geben Sie auch die Art und Weise der Trocknung (Trockenmittel, Dauer) an. ● Protokollieren Sie die Reinigung des Rohprodukts: Menge eingesetzter Substanz, benötigtes Lösungsmittel, Isolierung und Trocknung des Kristallisats. Erstellen Sie eine Massenbilanz. Geben Sie abschließend die Ausbeute an Reinprodukt in g, mol und % der Theorie und allen physikalischen Daten des Produkts (Smp., Aussehen) an. ● Vergleichen Sie die analytischen Daten Ihrer Substanz mit den Literaturdaten. Aspirin – Synthese von Acetylsalicylsäure 159 3 Geräte, Chemikalien und Entsorgung Eine Geräteliste liegt am Platz aus Chemikalien Gefahrenbezeichnung H-Sätze P-Sätze Smp. (°C) Sdp. (°C) ρ (g/cm3 ) Salicylsäure Gefahr (GHS08) Gefahr (GHS05) Gefahr (GHS05) 302, 318 305+351+338, 313 157-160 k. A. k. A. 226, 332, 302, 314, 335 314, 290 −73 140 1,08 k. A. 335 1,84 225 280, 301+330+331, 305+351+338 301+330+331, 305+351+338, 280, 309, 310 210 k. A. 78 k. A. 302 – 136 k. A. k. A. Essigsäureanhydrid1) H2 SO4 Ethanol2) Acetylsalicylsäure (Produkt) Gefahr (GHS02) Gefahr (GHS08) 1) Dampfdruck bei 20○ C: 4hPa 2) Dampfdruck bei 20○ C: 59hPa 4 Literatur Div. Autoren: Organikum, 22. Aufl., Wiley-VCH, 2004. Vergleich der enzymatischen und alkalischen Hydrolyse von Triacylglyceriden 161 Versuch 21 (EAH) Vergleich der enzymatischen und alkalischen Hydrolyse von Triacylglyceriden 1 Theorie 1.1 Fette und Fettsäuren Fett ist eine Sammelbezeichnung für feste, halbfeste oder flüssige Produkte des Pflanzen- oder Tierkörpers, die chemisch im Wesentlichen aus gemischten Triacylglyceriden von höheren Fettsäuren mit gerader Anzahl von Kohlenstoff-Atomen bestehen. Triacylglyceride sind die Fettsäureester mit dem Dreifachalkohol Glycerin (siehe Abbildung 1.1). Je nach Herkunftsorganismus variiert die durchschnittliche Kettenlänge und Anzahl der Unsättigungen der Fettsäuren zum Teil erheblich. Abb. 1.1: Ein Triacylglycerid 1.2 Alkalische Fettverseifung Die Herstellung von Seife gehört zu den ältesten chemischen Verfahren. Schon vor 4500 Jahren haben die Sumerer und Ägypter Pflanzenöle mit Pottasche zur Herstellung von Seife verkocht. Auch die Germanen und Gallier haben einfache Seifen hergestellt, während den Römern das Seifensieden unbekannt war. Die traditionelle Herstellung, bei der Pottasche zunächst mit gebranntem Kalk in Kalilauge überführt wurde, diese dann in erster Linie mit Rindertalg zu Seife verkocht und die entstandene Seife anschließend mit Kochsalz ausgesalzt wurde, hatte über fast zwei Jahrtausende Bestand. Gewöhnliche Seife ist ein halbfestes oder festes Gemisch von Natrium- oder Kaliumsalzen langkettiger Fettsäuren. Die Zusammensetzung des Gemisches hängt von den verwendeten Fetten ab. Natriumsalze der Fettsäuren ergeben harte Seifen (Kernseifen), Kaliumsalze weiche Seifen (Schmierseifen). In einem Triacylglycerid zeigen die einzelnen Esterbindungen unterschiedliche Reaktivität (siehe Abbildung 1.2. Die Esterbindung an Position 2 kann im Vergleich zu Position 1 relativ leicht gespalten werden. Sie wird daher bei Verseifungsreaktionen (= Esterhydrolyse) bevorzugt angegriffen. Die anderen Bindungen haben eine geringere Reaktivität. Bei der Hydrolyse von Triacylglyceriden entsteht zunächst ein Diacylglycerid. Hierbei kann entweder die Fettsäure an Position 1, 2 oder 3 abgespalten werden, es entstehen die drei Regioisomere 1,2-Diacylglycerid, 2,3-Diacylglycerid oder 1,3-Diacylglycerid. Handelt es sich um ein Triacylglycerid bei dem an allen Positionen die gleiche Fettsäure verestert ist, so erhält man nach der Hydrolyse auch zwei Stereoisomere; 1,2- und 2,3-Diacylglycerin. 162 Vergleich der enzymatischen und alkalischen Hydrolyse von Triacylglyceriden Abb. 1.2: Schema der Alkalischen Triglycerid-Hydrolyse R = C15 H31 , Tripalmitin (Fettsäure: Palmitinsäure) 1.2.1 Enzymatische Fettverseifung Lipasen (EC 3.1.1.3), eine Untergruppe der Hydrolasen, sind eine Enzymklasse, die Triacylglyceride spalten. Sie kommen im Verdauungstrakt fast aller Tierarten (Pankreas, Darmschleimhaut), in Milch, Leber von Wirbeltieren, in ölhaltigen Samen (z.B. Soja, Erdnuß), sowie in zahlreichen Mikroorganismen vor. Aufgrund ihres definierten räumlichen Aufbaus (Schlüssel-Schloss-Prinzip) wird bei der Hydrolyse die terminale bzw. die subterminale Position bevorzugt angegriffen. Je nach Enzym kann also bevorzugt das 1,3- bzw. das 2,3-Diacylglycerid entstehen. Die im Versuch verwendete Lipase aus dem Pilz Rhizomucor miehei weist eine 1,3-Selektivität auf und bildet somit in der ersten Stufe ein 2,3-Diacylglycerid. Bei längeren Reaktionszeiten wird auch die zweite terminale Fettsäure hydrolysiert und es entsteht das 2-Monoacylglycerid. In der organischen Synthese können Lipasen auch zur Umsetzung prochiraler und racemischer Verbindungen verwendet werden. Lipasen zeigen eine hohe Aktivität in organischen Lösungsmitteln bei gleichzeitiger Stereoselektivität. Vorteile von enzymatisch katalysierten Reaktionen sind: die Umsetzungen können in der Regel bei niedriger Temperatur und zum Teil in wässrigen Lösungen durchgeführt werden, was Energie und organische Lösungsmittel einspart. Hinzu kommt, dass Enzyme aufgrund ihrer räumlichen Struktur meist eine hohe Regio- und Stereoselektivität zeigen. Nachteile von Enzymen sind, dass nicht für jede Reaktion ein Enzym existiert, ihre oft teure Herstellung, geringe Stabilität oder die Abhängigkeit von Cofaktoren. Einige dieser Probleme kann man jedoch durch genetische Veränderung, also der Herstellung von maßgeschneiderten Enzymen, lösen. 2 Aufgabenstellung, Versuchsanleitung und -auswertung 2.1 Alkalische und Enzymatische Hydrolyse von Tripalmitin 2.1.1 Aufgabenstellung Es soll Tripalmitin durch Umsetzung mit Natronlauge (Teil 1) und mit einer Lipase (Teil 2) hydrolysiert werden. Vergleich der enzymatischen und alkalischen Hydrolyse von Triacylglyceriden 163 2.1.2 Versuchsanleitung Herstellung der Tripalmitinsuspension Die Reaktionslösung besteht aus 5 Gew.-% Tripalmitin suspendiert in Wasser. Die Suspension wird mit 2 Gew.-% Gummi arabicum stabilisiert. Alkalische Fettverseifung In einem 10 ml Rundkolben wird 2 ml der Tripalmitinsuspension mit 100 µl 3 M Natronlauge versetzt und für 90 min bei 45°C gerührt. Nach 5, 45 und 90 min werden Proben von 400 µl entnommen. Die Probe wird mit 200 µl Salzsäure (1 M) angesäuert. Danach wird je zwei Mal mit 400 µl eines Gemisches aus tert-Butylmethylether und n-Hexan (1:1) extrahiert. Zur besseren Phasentrennung wird das Gemisch kurz zentrifugiert und die organische Phase in ein neues Gefäß überführt. Enzymatische Fettverseifung In einem 10 ml Rundkolben werden 100 µl Lipase aus Rhizomucor miehei, 4,5 ml Tripalmitinsuspension und 500 µl TRIS (c = 1 mol l , pH 7, TRIS: Tris(hydroxymethyl)-aminomethan, Puffer) gemischt und 90 min bei 30°C gerührt. Alle 30 min wird eine Probe von 400 µl entnommen. Diese wird zwei Mal mit 400 µl eines Gemisches aus tert-Butylmethylether und n-Hexan (1:1) extrahiert (nicht ansäuern! Warum?). Zur besseren Phasentrennung wird das Gemisch kurz zentrifugiert und die organische Phase in ein neues Gefäß überführt. Reaktionsverfolgung mit Dünnschichtchromatographie Der Reaktionsverlauf wird mit Hilfe der Dünnschichtchromatographie (DC) verfolgt. Dabei wird die Probe auf eine mit Kieselgel beschichtete Aluminiumfolie, ca. 1 cm vom unteren Rand (Bleistiftlinie), aufgetragen. Die Folie wird dann in die zum Teil mit Lösungsmittel (Hexan, Diethylether, Essigsäure, 55:45:1) gefüllte DC-Kammer gestellt. Das Fließmittel wird dabei durch die Kapillarwirkung langsam durch die Kieselgelschicht gesogen (siehe Versuch 18 CHR). Die Trennwirkung kommt dadurch zustande, dass je nach Stärke der Wechselwirkung der stationären Phase (Kieselgel) mit dem Analyt (Palmitat, Tri-, Di- und Monoacylglycerid) dieser mehr oder weniger weit von der mobilen Phase mitgenommen wird. Wenn die Fließmittelfront etwa 1 cm von der Oberkante der DC-Folie entfernt ist, wird diese aus der Kammer genommen, die Laufmittelfront markiert und getrocknet. Die farblosen Analyten werden mit einem Tauchreagenz (Kupfer(II)-sulfat-Lösung) und anschließendem Erhitzen mit dem Heißluftföhn sichtbar gemacht. Die Rf -Werte (Verhältnis Substanzlaufstrecke zur Gesamtlaufstrecke) werden bestimmt. 2.1.3 Versuchsauswertung ● Berechnen Sie die Rf -Werte und identifizieren Sie die Substanzen anhand von Literaturwerten. Beschreiben Sie die Ergebnisse der DC und benennen Sie Unterschiede zwischen den beiden Reaktionsansätzen. ● Interpretieren Sie die Ergebnisse und stellen Sie Unterschiede heraus. 164 Vergleich der enzymatischen und alkalischen Hydrolyse von Triacylglyceriden 3 Geräte und Chemikalien Eine Geräteliste liegt im Platz aus Chemikalien Tripalmitin Gummi Arabicum Lipase NaOH HClaq t-Butylmethylether n-Hexan Diethylether Essigsäure CuSO4 ⋅ 5 H2 O Gefahrenbezeichnung H-Sätze – – – – – – Gefahr (GHS05) 314, 290 Gefahr (GHS05) 314, 335, Gefahr (GHS02, 08) 225, 315 Gefahr (GHS02, 08, 09) 225, 304, 315, 336, Gefahr (GHS02, 08) EUH019, 224, 302, Gefahr (GHS05) 226, 314 Achtung (GHS07, 09) 302, 315, P-Sätze – – – 301+330+331, 305+351+338, 280 290 301+330+331, 305+351+338, 280 210, 302+352 361, 373, 210, 240, 273, 301+310, 302+352, 411 403+235, 331 EUH066, 210, 240, 403+235 336 301+330+331, 305+351+338, 280 319, 410 305+351+338, 302+352, 273 4 Literatur [1] [2] Div. Autoren: Organikum, 22. Aufl., Wiley-VCH, 2004. R. D. Schmid, R. Verger: ”Lipasen: Grenzflächen-Enzyme mit attraktiven Anwendungen”, Angew. Chem. 110, 1998, S. 1694-1720.




![ÜBERGANGSMETALLKOMPLEXE Na2[Fe(CN)5NO] ⋅ 2H2O](http://s1.studylibde.com/store/data/002682926_1-4994fab9e6ff9a7ce3deb92323ee5953-300x300.png)