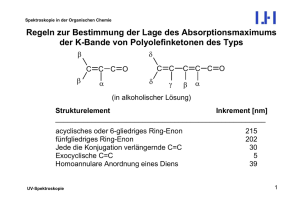
1 The principle of a 1D experiment The principle of the simplest 1D
Werbung

The principle of a 1D experiment The principle of the simplest 1D experiment is shown in Figure 1: The equilibrium zmagnetization (A) is rotated by 90° onto the y-axis (B) by the applied 90°x 1H pulse. During the acquisition time (C) or detection of the NMR signal, the magnetization precesses in the x,y-plane, but at different speeds (or frequencies ν i) for each proton i because of their different chemical environment. The receiver (on the y-axis) detects a signal for each proton, which looks like a cosine function. As, in addition, the magnetization returns to its equilibrium position along the zaxis in an exponential decay, each signal is modulated by an exponential function (inset D). Therefore, the resulting signal in the time domain for all protons is called Free Induction Decay (FID). The Fourier tansformation (FT) of this signal yields peaks with a Lorentzian lineshape at each frequency νi (inset E). (See H. Rattle, Chapter 2 for more information.) Figure 1: The simplest 1D experiment of FT-NMR. 1 Spektroskopie in der Organischen Chemie Die chemische Verschiebung Die Messfrequenz ν einer Kernsorte hängt bei einem isolierten Kern, z.B. 1H nur Magnetfeld (B0) und seinem magnetogyrischen Verhältnis γ ν(1H isoliert) = (γ(1H) /2π) · B0 Larmor-Beziehung Chemische Verschiebung δ (in ppm, unabhängig von Feldstärke): (νS - νref [in Hz]) / νref [in Hz] x 106 = δ (in ppm) νref z.B. Resonanzfreqenz von TMS Reale Moleküle -> Einfluss der umgebenden Elektronen (chemische Umgebung) auf ν(1H) -> Erzeugung lokaler Magnetfelder -> Veränderung von B0 am Ort des Kerns verändern (‚Abschirmung’): Beff = B0-σ B0 = B0(1 - σ) -> ν(1H) = (γ (1H)/2π) · B0 (1 - σ) = (γ (1H)/2π) · Beff Abschirmungskonstante σ -> klein (10-3 bis 10-6 Hz), aber ausreichend um Resonanzbereich mit separat auflösbaren1H-Signalen aufzuspannen. 1 H-NMR-Spektroskopie 1 Spektroskopie in der Organischen Chemie Bereich der 1H-chemischen Verschiebung (Abb. nach: Friebolin, mehr sieh Hesse, Meier, Zeeh) 1 H-NMR-Spektroskopie 2 Spektroskopie in der Organischen Chemie Die 1H-chemische Verschiebung – Strukturabhängigkeit Abgesehen von Lösungsmittel/Pufferbedingungen (Salzkonz., pH, T etc.), abhängig von Molekülstruktur über die Beeinflussung der Verteilung der Elektronendichte um einen bestimmten Kern durch seine Nachbarkerne. Der Resonanzbereich der Protonen in organischen Molekülen ist ca. 10 ppm breit. Nur saure Protonen (z.B. COOH oder SO3H) liegen darüber; Protonensignale von Formylgruppen (z.B. Aldehyde) liegen um δ = 9 bis 11. Auch die Amidprotonen des Indolrings (z.B. in Tryptophan) sind bei ∼10-11 ppm. Es gibt eine deutliche Abhängigkeit vom s-Charakter der wasserstofftragenden Kohlenstoffe, aber keine Korrelation! sp3 (Alkane): δ = 0.5 bis 4 sp2 (Alkene/Aromaten): δ = 4.5 bis 9 sp (Alkine): δ = ca. 2 bis 3 1 H-NMR-Spektroskopie 3 Spektroskopie in der Organischen Chemie Innerhalb des sp3-Bereiches werden Protonen durch benachbarte (geminale) elektronegative oder ungesättigte Substituenten entschirmt: δ(H-C-F) > δ(H-C-O) > δ(H-C-N) > δ(H-C-C=) > δ(H-C-C-) Aber auch der Substitutionsgrad spielt eine Rolle: δ(CHX) > δ(CH2X) > δ(CH3X) Cyclopropyl-1H-Signale haben ungewöhnlich kleine δ-Werte: -0.5 bis +0.5. (-> hohe Elektronendichte um Kern) Wie für die C-H Bindungen bewirkt auch bei anderen X-H Bindungen die Zunahme der Elektronegativität (EN) von X die Abnahme der Abschirmung (also eine Entschirmung) und entsprechend die Zunahme der chemischen Verschiebung von H, z.B. CH3-CH2-OH δ(HO) = 2.56 ppm > CH3-CH2-SH δ(HS) = 1.46 ppm da EN von O > EN von S 1 H-NMR-Spektroskopie 4 Spektroskopie in der Organischen Chemie Innerhalb des sp2-Bereiches sind zwei Einflüsse zu beachten: (a) Anisotropieeffekte z.B. in Aromaten -> δ(aromat.-H) > δ(olefin.-H) z.B. Benzol δ(H) 7.26 <-> Cyclohexan δ(H) 1.44 Dies ist eine Konsequenz der anisotropen Elektronenverteilung im Molekül und wird bei Aromaten nach Pople durch den Ringstrom-Effekt erklärt: Durch das äußere Magnetfeld B0 wird im Aromaten ein Ringstrom induziert, der seinerseits ein B0 entgegen gerichtetes Magnetfeld erzeugt. Dieses schwächt B0 oberhalb der Ringebene ab (Abschirmung; kleineres δ), während es B0 in der Ringebene verstärkt (Entschirmung = geringere Abschirmung; größeres δ). B0 1 H-NMR-Spektroskopie 5 Spektroskopie in der Organischen Chemie Beispiele für die Abschirmung oberhalb bzw. innerhalb der Ringebene: Cyclophan [18-Annulen] H δ = 9.28 in Ringebene δ = -4.03 C H H δ = -2.99 H ist oberhalb Ringebene Abschirmung, kleines δ über Ringebene Werte gemessen bei tiefer Temperatur; bei Raumtemperatur schneller Austausch mit Durchschnittssignal bei δ = 5.45. 1 H-NMR-Spektroskopie 6 Spektroskopie in der Organischen Chemie Auch andere funktionelle Gruppen üben Anisotropieeffekte aus, z.B. die Carbonylgruppe (C=O), Doppel-/Dreifachbindungen, Substituenten wie Nitrogruppe: (nach: Günther) Positives Vorzeichen -> Abschirmung (δ↓) & negatives -> Entschirmung (δ↑) Siehe z.B. Propan, Propen, Propin -> Hesse, Meier, Zeeh Anisotropieeffekt: allgemein aufgrund von magnetischer Anisotropie chemischer Bindungen -> entsprechend ist die magnetische Suszeptibilität X nicht in alle Raumrichtungen gleich (sogar C-C Einfachbindung zeigt Anisotropieeffekt). 1 H-NMR-Spektroskopie 7 Spektroskopie in der Organischen Chemie (b) Induktive und mesomere Effekte in ungesättigten Systemen, z.B. Phenol und Benzaldehyd: -> Einfluss auch abhängig von Ringposition relativ zu Substituent δ = 7.26 H OH δ = 6.70 OH OH H H δ = 7.14 CHO δ = 7.82 CHO H δ = 6.81 CHO H H δ = 7.48 1 H-NMR-Spektroskopie H δ = 7.55 8 Spektroskopie in der Organischen Chemie Einflüsse der Heteroatome in Heteroaromaten H H H N δ = 7.26 H δ = 8.59 N N δ = 7.38 δ = 7.75 und π-Elektronendichten in Aromaten + δ = 7.26 δ = 11.1 - + 2- δ = 5.5 δ = 7.7 δ = 5.7 Quasiaromatische Ionen: neben Anisotropie, Einfluss von Ladung auf e--Dichte 1 H-NMR-Spektroskopie 9 Spektroskopie in der Organischen Chemie In gewissen Grenzen ist es möglich, durch empirische Inkrementenregeln 1 H-chemische Verschiebungen mit befriedigender Präzision vorauszusagen. Diese Regeln gelten jeweils nur für ein ganz bestimmtes Molekülgerüst oder -fragment, dem ein Grundwert (δ-Wert für die unsubstituierte Verbindung) zugeordnet wird. Dazu werden dann – je nach vordefinierter Stellung – Inkrementenwerte von häufig auftretenden Substituenten hinzuaddiert. Beispiel: Disubstituierte Methane, R1–CH2–R2 (Shoolery-Regel) δ(CH2) = 1.25 + Σai Ri Alkyl Ph OH Cl Br I C≡N ai 0 1.3 1.7 2.0 1.9 1.4 1.2 Ph-CH2OH, berechnet: δ = 1.25 + 1.3 + 1.7 = 4.25; experimentell: δ = 4.4 Diese Regel kann sogar auf trisubstiuierte Methane ausgedehnt werden. 1 H-NMR-Spektroskopie 10 Spektroskopie in der Organischen Chemie Regel für olefinische Protonen; hier muss die relative Stellung der Substituenten beachtet werden, da es drei verschiedene Positionen gibt: Rcis H geminal H C C Ph R R trans δ = 5.25 + ΣS i R geminal cis trans -CO-R -Ph 1.10 1.38 1.12 0.36 0.87 -0.07 CO-CH3 H C C δ = 5.25 + 1.38 + 1.12 = 7.75 (exp.: ca. 7.5) δ = 5.25 + 1.10 + 0.36 = 6.71 (exp.: ca. 6.8) Eine eindeutige Zuordnung der beiden olefinischen Protonen ist also möglich. 1 H-NMR-Spektroskopie 11 Spektroskopie in der Organischen Chemie Regel für aromatische Protonen; auch hier muss die relative Stellung der Substituenten beachtet werden: H Rortho δ = 7.26 + ΣS i meta R R para Beispiel: o-Nitrobenzylalkohol 1 R ortho meta para -NO2 -CH2OH 0.95 -0.07 0.26 -0.07 0.38 -0.07 H-NMR-Spektroskopie (zufällig gleich) 12 Spektroskopie in der Organischen Chemie H CH2OH H δ = 7.26 + (-0.07) + 0.26 = 7.45 (exp.: ca. 7.5) δ = 7.26 + (-0.07) + 0.38 = 7.57 (exp.: ca. 7.8) NO2 H H δ = 7.26 + (-0.07) + 0.26 = 7.45 (exp.: ca. 7.5) δ = 7.26 + (-0.07) + 0.95 = 8.14 (exp.: ca. 8.1) Auch wenn die Präzision der Vorhersage nicht immer sehr überzeugend ist, gelingt es meist doch, zumindest die Reihenfolge der Signale nachzuvollziehen. Die Regel versagt aber leicht, wenn die Substituenten über den Benzolring sterisch oder elektronisch miteinander in Wechselwirkung treten (mangelnde Additivität der Inkremente). Merke: Inkrementenregeln sind häufig sehr nützlich, sollten aber immer nur als grobes Hilfsmittel angesehen werden. Sie können niemals experimentelle Befunde widerlegen. Mehr zu Inkrementregeln in Hesse, Meier, Zeeh. 1 H-NMR-Spektroskopie 13 Spektroskopie in der Organischen Chemie Die 13C-chemische Verschiebung Allgemein: Abschirmungskonstante hat verschiedene Komponenten σ = σdia + σpara + Σ σ´ (+ σMedium wenn intermol. WW) diamagnetischer Beitrag (σdia, Abschirmung): stammt von den sElektronen ist viel weniger strukturabhängig. Beruht allgemein auf der Erzeugung eines B0 entgegen gerichteten Feldes. Er ist der wichtigste Faktor für die 1H-chemische Verschiebung. paramagnetischer Abschirmungsbeitrags (σpara): eine Anregung von pElektronen durch B0 wirkt σdia entgegen diverse Nachbareffekte (σ´) – Beeinflussung von Feld am Kernort durch Nachbargruppen Im Vergleich zur 1H-chemischen Verschiebung ist der δ(13C)-Bereich sehr viel größer, weil er durch den paramagnetischen Beitrag zur Kernabschirmung (σpara, Entschirmung) bestimmt wird, der an die Existenz von p-Elektronen (ab 2. Hauptgruppe) gebunden ist. 13 C-NMR-Spektroskopie 1 Spektroskopie in der Organischen Chemie Wichtige Faktoren innerhalb des paramagnetischen Abschirmungsbeitrags sind die durchschnittliche (Elektronen-) Anregungsenergie ‹ΔE› und die Größe der p-Orbitale: σpara ~ 1 · (r-3)2p ΔE Wird ΔE kleiner, wird δ(13C) größer (Entschirmung, Tieffeldverschiebung). (Innerhalb ‹ΔE› dominiert die kleinste, die HOMO-LUMO-Anregungsenergie.) Der 13C-Resonanzbereich erstreckt sich über ca. 250 ppm. Die chemische Verschiebung von 13C Kernen hängt ganz allgemein stark von der Hybridsierung ab: sp3 (Alkane): δ = ≈10-80 ppm sp (Alkine): δ = ≈70-130 ppm sp2 (Alkene/Aromaten): δ = ≈100-160 ppm sp2 (Carbonylverbindungen): δ = ≈100-230 ppm 13 C-NMR-Spektroskopie 2 Spektroskopie in der Organischen Chemie Bereiche der 13C-chemischen Verschiebung (nach: Friebolin) 13 C-NMR-Spektroskopie 3 Spektroskopie in der Organischen Chemie Es sei erwähnt, dass es hier keine wesentlichen Unterschiede zwischen Alkenen und Aromaten gibt, weil der aus der 1H-NMR bekannte Anisotropieeffekt bei Aromaten (Ringstrom) hier aufgrund des viel größeren Resonanzbereichs nicht mehr identifizierbar ist. Die chemische Verschiebung der Alkan- und Alken-/Aromatenkohlenstoffe ist zudem von der Existenz benachbarter Heteroatome abhängig. Der Bereich für die Carbonylgruppen ist je nach Oxidationsstufe des CarbonylC´s sogar noch weiter unterteilt ist. Dadurch wird es sehr einfach, Kohlensäure- und Carbonsäurederivate (δ = 150-180) von Aldehyden (δ = 180-200) und Ketonen (δ = 190-220) zu unterscheiden. Noch größere δ-Werte haben nur Thioketone (δ = 250-270) und Carbeniumionen (δ = 200-400). Man beachte, dass alle genannten Bereiche nur als typische Bereiche anzusehen sind. Es gibt für alle Substanzklassen Molelülstrukturen, deren Resonanzen Ausreißer nach oben und unten sein können! 13 C-NMR-Spektroskopie 4 Spektroskopie in der Organischen Chemie In aromatischen und heteroaromatischen Systemen spielt die π-Ladungsdichte eine signifikante Rolle; sie korreliert gut mit der 13C-chemischen Verschiebung: 13 C-NMR-Spektroskopie 5 Spektroskopie in der Organischen Chemie Wegen des großen Resonanzbereichs ist die Signalzuordnung ein wesentliches Problem, ohne dessen Lösung Strukturbestimmungen oft fragwürdig bleiben. Es ist daher sehr hilfreich, empirisch ermittelte Strukturabhängigkeiten und Inkrementenregeln zu kennen, die oft spezifisch für bestimmte Substanzklassen sind. Im folgenden werden einige dieser Regeln vorgestellt. Man beachte, dass in den letzten Jahrzehnten ein- und zweidimensionale Multipuls-NMR-Experimente entwickelt wurden, die empirische Zuordnungen teilweise oder sogar ganz entbehrlich machen. Sie beruhen auf Spin-Spin-Korrelationen durch die zwischen den Kernen liegenden Bindungen (skalare Kopplung) oder durch den Raum (dipolare Kopplung, NOE) und erlauben dadurch, ein dreidimensionales Netzwerk der 1H- und 13C-Kerne aufzustellen. Zu den wichtigsten dieser Methoden gehören COSY, HETCOR, HMQC, HMBC, NOESY und einige andere (mehr dazu in IMIII). Obwohl die Messung dieser 2D-NMR-Spektren heutzutage zur Routine gehört, ist es dennoch sehr nützlich, die Strukturabhängigkeit der 13C-chemischen Verschiebung zu kennen. Häufig ist die Anwendung einfacher Inkrementenregeln der Anforderung weiterer Messzeit für 2D-NMR-Messungen vorzuziehen. 13 C-NMR-Spektroskopie 6 Spektroskopie in der Organischen Chemie GRANT-PAUL-Regel zur Berechnung der 13C-chemischen Verschiebung von Alkanen: δ(Ci) = -2.3 + ∑ Ak + ∑ Siα k Zu dem Grundwert (-2.3) addiert man Inkremente Ak für alle benachbarten CAtome, die sich je nach Entfernung unterscheiden; k = α, β, γ, δ, ε ... Aα = +9.1 , Aβ = +9.4 , Aγ = -2.5 , Aδ = +0.3 , Aε = +0.2 Dazu müssen aber noch sog. sterische Korrekturfaktoren Siα berücksichtigt werden, die die Nichtadditivität der Ak-Werte darstellen. Man sucht den Siα-Wert des höchstsubstituierten α-Atoms heraus. 13 Ci α-CH3 α-CH2 α-CH α-C CH3 CH2 CH C 0 0 0 -1.5 0 0 -3.7 -8.4 -1.1 -2.5 -9.5 (-15) -3.4 -7.5 (-15) (-25) C-NMR-Spektroskopie 7 Spektroskopie in der Organischen Chemie Die in Klammern gesetzten Siα-Faktoren, die nur bei sehr stark verzweigten und sterisch besonders anspruchsvollen Alkanen benötigt werden, sind nur relativ unpräzise Schätzwerte. Beispiel: δ(C) von Isooctan H3C CH H3C CH2 CH3 CH3 C CH3 δ(ber.) = -2.3 + 4 · 9.1 + 1 · 9.4 + 2 · (-2.5) - 8.4 = 30.1 [δ(exptl.) = 31.1] Abweichungen von ± 1 zwischen den berechneten und experimentellen 13Cchemischen Verschiebungen sind normal; genauer können diese empirischen Regeln nicht sein. Es gibt eine verbesserte empirische Regel, die LINDEMAN-ADAMS-Regel, die aber nur bei stark verzweigten Alkanen überlegen ist. 13 C-NMR-Spektroskopie 8 Spektroskopie in der Organischen Chemie Die GRANT-PAUL-Regel wurde erweitert durch Einführung von Inkrementen für andere Kohlenwasserstoffreste und Heterosubstituenten, durch die aliphatische Kohlenstoffatome in Alkoholen, Alkylhalogeniden, Aminen, Nitroalkanen, Thiolen, Alkenen, Alkinen, Ketonen, Aldehyden, Carbonsäuren, Nitrilen etc. berechnet werden können ( Hesse, Meier, Zeeh, es gibt auch PC-basierte Rechenprogramme (Pretsch)). Es ist zu beachten, dass diese Regeln bei cyclischen und insbesondere bei oligocyclischen Verbindungen oft nur unpräzise Voraussagen erlauben oder sogar ganz versagen. Man erkennt, dass bei den α-Effekten die Elektronegativität des Heteroatoms bzw. die Gruppenelektronegativität die bestimmende Rolle spielt, während dies für die β-Effekte nicht so eindeutig ist. Besonders interessant sind die γ-Effekte, die eine deutliche sterische Abhängigkeit zeigen und somit sehr gut für die Unterscheidung von Stereoisomeren geeignet ist: Wenn an einem C-Atom (α) ein Substituent X hängt, erzeugt dieser auf ein C-Atom in γ-Position ein sog. γ-Substituenteneffekt Δδγ = δγ(X) - δγ(X=H). 13 C-NMR-Spektroskopie 9 Spektroskopie in der Organischen Chemie H X X Cα α C antiperiplanar C C γ gauche γ Sind Cα und Cγ antiperiplanar, ist Δδγ zwischen -2 und +2, meist um 0, während Δδγ bei gauche- oder synperiplanarer Stellung zwischen -4 und -7 liegt. Dies bezeichnet man als den „diamagnetischen γ-gauche-Effekt“, durch den Stereoisomere unterschieden werden können: H3C δ = 16.8 CH3 13 C-NMR-Spektroskopie H3C CH3 δ = 11.4 Unterschied: -5.4 ppm 10 Spektroskopie in der Organischen Chemie Ferner abhängig von Art des Substituenten: OH CH3 Δδ = -6.2 Δδ = -6.6 C C Δδ = -6.1 C Br Dies lässt sich zur Strukturbestimmung ausnutzen. So kann man zum Beispiel α- und β-anomere Monosaccharide voneinander unterscheiden: HO HO HO δ = 72.6 HO O C H 13 C-NMR-Spektroskopie α HO OH HO HO δ = 75.6 (gauche) O C H HO OH β (antiperiplanar) 11 Spektroskopie in der Organischen Chemie Auch für Alkene und Benzole gibt es leistungsfähige Inkrementenregeln ( Hesse, Meier, Zeeh). Die Werte für die diversen Substituenten sind hier wie bei den entsprechenden 1H-Regeln von der relativen Position abhängig und reflektieren induktive und mesomere Effekte. Die Regel für Alkene (Grundwert: δ = 123.3) hat pro Substituenten zwei Inkremente zu beachten, direkt gebunden und geminal: I1–CH(α)=C(β)H–I2 Einige Beispiele für Substitutenteneffekte bei Benzolderivaten (Grundwert: δ = 128.5); vier Inkremente pro Substituent, ipso, ortho, meta und para: CH3 OH C≡N 13 C-NMR-Spektroskopie ipso ortho meta para +9.3 +26.9 -16.0 +0.6 -12.6 +3.5 0.0 +1.6 +0.7 -3.1 -7.6 +4.3 12 Spektroskopie in der Organischen Chemie Man erkennt den Einfluss der Elektronegativität (I-Effekte) an den direkt gebundenen Kohlenstoffatomen (ipso). Andererseits sind die M-Effekte von OHund Nitrilgruppe durch die Signalverschiebungen der ortho- und para-Kohlenstoffatome ablesbar; meta-Effekte hingegen sind meist vernachlässigbar klein, was anhand der mesomeren Grenzstrukturen veranschaulichbar ist: O H +O H +O H CN - CN CN - + + Die Substituenteneffekte sind weitgehend additiv und können zur Berechnung von di- oder trisubstituierten Benzolen genutzt werden. 13 C-NMR-Spektroskopie 13 Spektroskopie in der Organischen Chemie Die 13C-chemische Verschiebungen der para-Kohlenstoffatome monosubstituierter Benzole korrelieren gut mit der HAMMETT-Konstanten σ+: δ(13C) σ+ [Die δ-Werte sind hier auf den von Benzol (δ = 128.5) bezogen.] 13 C-NMR-Spektroskopie 14 Spektroskopie in der Organischen Chemie Carbonyl-Kohlenstoffatome – Struktureinflüsse Konjugation einer Carbonylgruppe mit einer C,C-Doppelbindung wirkt sich stark aus: O O δ = 208.5 δ = 199.5 α δ = 129.5 β δ = 150.6 δ = 127.4 Das Carbonyl-Kohlenstoffatom wird durch die Konjugation um ca. 10 ppm abgeschirmt (diamagnetische Signalverschiebung, ppm-Wert↓), eine konjugationstypische Beobachtung, die auch für Aldehyde, Carbonsäuren und deren Derivate gilt. Das α-Olefinkohlenstoffatom wird durch die Konjugation kaum beeinflusst, wogegen das β-Atom stark entschirmt wird (paramagnetische Signalverschiebung, ppm-Wert↑) und gelegentlich sogar bis in der Resonanzbereich der Carbonylgruppe (δ > 160) verschoben werden kann. 13 C-NMR-Spektroskopie 15 Spektroskopie in der Organischen Chemie Auch diese Signalverschiebungen können qualitativ sehr einfach durch Mesomerie-Grenzstrukturen erklärt werden: O O - + Für die Carbonylgruppe wird der Doppelbindungscharakter geschwächt, was mit einer Abschirmung korrespondiert. Das β-C wird positiviert (geringere Elektronendichte), was grundsätzlich mit einer Entschirmung einhergeht. Vergleicht man dagegen die beiden folgenden Acetophenone, CH3 O C δ = 195.7 CH3 13 C-NMR-Spektroskopie CH3 O C δ = 205.5 CH3 16 Spektroskopie in der Organischen Chemie findet man, dass in dem 2,6-Dimethylderivat offenbar die Konjugation erschwert ist. Dies ist in der Tat der Fall; durch sterische Wechselwirkung dreht sich die Acetylgruppe aus der Ebene heraus. Wasserstoffbrücken und Dipol-Dipol-Wechselwirkungen haben ebenfalls einen signifikanten Einfluss: H C O δ = 191.5 H C O H δδ+ O H δδδ = 196.9 C + N δδ- δδ+ O - δ = 188.6 O O Sie verändern ein wenig die Polarisation der Doppelbindung (δδ+ bzw. δδ-), was sich sofort in der Änderung der 13C-chemischen Verschiebung niederschlägt. 13 C-NMR-Spektroskopie 17 Spektroskopie in der Organischen Chemie 13 C,1H-Kopplungskonstanten NMR-Signalaufspaltungen aufgrund von skalaren Kopplungen treten grundsätzlich bei beiden Kopplungspartnern auf. Entsprechend kann man 13C,1HKopplungen sowohl an 13C- als auch an 1H-Signalen ( IM1) ablesen. Dabei ist aber ein wesentlicher Unterschied zu beachten: Da die natürliche Häufigkeit des 13C-Kerns nur ca. 1.1%, die des 1H-Kern aber praktisch 100% ist, befinden sich fast 99% aller Protonen nicht an 13C-, sondern an NMR-inaktiven 12C-Isotopen. Das 1H-NMR-Signal solcher Protonen wird also gar nicht durch eine 13C-Kopplung beeinflusst (Hauptsignal). Nur die ca. 1% der an 13C befindlichen Protonen erzeugen ein 1H-Signal, das durch die große Kopplung mit 13C über eine Bindung (120-250 Hz; ) in ein Dublett aufgespalten wird. Diese kleinen Signale links und rechts des Hauptsignals werden als 13C-Satelliten bezeichnet. Sie sind meist wegen ihrer geringen Intensität im spektralen Rauschen gar nicht erkennbar. Umgekehrt ist praktisch jedes 13C-Atom an einen 1H-Kern gebunden. Das bedeutet, dass alle 13C-Signale durch die 1H-Kopplungen aufgespalten sind und 13 C,1H-Kopplungen hier leichter erkannt werden können. 13 C-NMR-Spektroskopie 1 Spektroskopie in der Organischen Chemie 1 H-NMR-Signal von trans-1,2-Dichlorethen: Cl H 12 C Cl 13 C-NMR-Spektroskopie 12 C 13 C H Cl H Cl 12 C H 2 Spektroskopie in der Organischen Chemie Hauptsignal: von dem Isotopomer mit zwei 12C-Kernen. Es ist ein Singulett, weil es zu einem A2-Spinsystem gehört ( Zwei-Spinsysteme); keines der beiden Kohlenstoffeatome ist NMR-aktiv. 13 C-Satelliten-Signale: Dubletts sind, aufgespalten durch die vicinale Kopplung der beiden, in diesem einfach 13C-markierten Alkan unterschiedlichen Wasserstoffe. Die beiden Protonen sind magnetisch nicht äquivalent; es ist ein AA´X-Spinsystem (streng genommen wegen des Isotopeneffekts ein ABX-System)! Nebensignal von grün markierten Proton: ist ebenfalls ein Dublett, das jedoch wegen der nur sehr kleinen 13C,1H-Kopplung über zwei Bindungen unter dem Hauptsignal versteckt ist. Auch 13C-NMR-Spektren weisen natürlich 13C-Satelliten auf, die zu zweifach 13Cmarkierten Molekülen gehören [ J(13C,13C)]. Wiederum ist deren Intensität um etwa zwei Größenordnungen kleiner als die der Hauptsignale. Weil das Problem des Signal-Rausch-Verhältnisses in der 13C-NMR-Spektroskopie viel gravierender ist als in der 1H-NMR, ist es hier noch schwieriger, die 13C-Satelliten zu beobachten. Gewisse Multipuls-NMR-Techniken (Doppelquanten-Kohärenz-Experimente, INADEQUATE IM3) erlauben ihre Messung unter Ausblendung der Hauptsignale; allerdings ohne nennenswerten Intensitätsgewinn. 13 C-NMR-Spektroskopie 3 Spektroskopie in der Organischen Chemie 13 C,1H-Kopplungen über eine Bindung, 1J(13C,1H) Diese Kopplung wird häufig – physikalisch nicht ganz korrekt – „direkte Kopplung“ genannt, weil die Kopplungspartner direkt gebunden sind. Sie ist praktisch immer positiv und zeigt sehr große Werte, die eine gute Korrelation zum s-Charakter des Kohlenstoffatoms zeigen. Sie betragen: sp3: 120 bis 150 Hz ; sp2: 150 bis 180 Hz ; sp: 200 bis 250 Hz Man kann sogar die Hybridisierung aus der 1J(13C,1H)-Kopplungskonstanten errechnen: 1 λ2 1 13 1 J( C, H) = 500 · s [Hz] mit s = 1 + λ2 aus dem Hybrid sp ; s ist der s-Anteil in den Hybriden: s = 0.25 bei sp3, 0.33 bei sp2, 0.5 bei sp. Bei Kohlenwasserstoffen ergeben sich so 120-130 Hz, 160-170 Hz bzw. 250 Hz. Bei gespannten Ringsystemen wie Cyclopropanen oder -butanen finden sich Zwischenwerte: 160 Hz (sp 2.13) 13 C-NMR-Spektroskopie 134 Hz (sp 2.73) 4 Spektroskopie in der Organischen Chemie Elektronegative Substituenten am CH-Fragment, dessen Kopplung gemessen wird, vergrößeren 1J(13C,1H) deutlich. So sind wegen der Sauerstoffatome in den Sacchariden die Kopplungen i.a. 140-145 Hz. Geminale 13C,1H-Kopplungen, 2J(13C,1H) Diese Kopplungskonstanten über 2 Bindungen können positiv und negativ sein; ihre Absolutwerte sind bei Alkanen i.a. nur wenige Hz groß, bei Alkenen und Aromaten können Werte bis 10 Hz erreicht werden. Bei Alkinen sind noch größere Werte möglich, und auch Aldehyde gehen weit über 10 Hz hinaus, aber dies sind eher Ausnahmen: H H H C C H Cl3 C H3 C O O +49.6 Hz +26.7 Hz +46.3 Hz 13 C-NMR-Spektroskopie 5 Spektroskopie in der Organischen Chemie Vicinale 13C,1H-Kopplungen, 3J(13C,1H) Für diese Kopplungen über 3 Bindungen existiert eine KARPLUS-analoge Abhängigkeit () vom Torsionswinkel τ des entsprechenden Strukturelements. Für 3J(1H,1H) (siehe auch IM1): 13 C-NMR-Spektroskopie 6 Spektroskopie in der Organischen Chemie Aus: Skript über 3J(1H,1H) von J.M. Reich, U. Wisconsin Bothner-By Gleichung: empirische Karplusgleichung, die keine unterschiedlichen J0 Werter für 0-90° und 90-180° erfordert. Für 3J(1H,13C): Aus: R.Aydin, J.-P. Loux, H. Günther, Angew. Chem. Int. Ed. Engl. 21(6):449, 1982 13 C-NMR-Spektroskopie 7 Spektroskopie in der Organischen Chemie Koplanare Anordnungen (τ = 00 oder 1800): Werte bis zu 10-15 Hz Nichtkoplanarität: kleine bis nicht detektierbare Werte stereochemische Differenzierung basierend auf 3J-Kopplungen möglich (Beispiele für 3J(1H,1H) siehe Reich Skript & Protein Rückgrat Φ-Winkel) Basis für die zweidimensionale, weitreichende 13C,1H-KorrelationsSpektroskopie ( COLOC, HMBC, IM3). 13 C,13C-Kopplungen über eine Bindung, 1J(13C,13C) Wie bereits erwähnt, können 13C,13C-Kopplungen wegen der geringen natürlichen Häufigkeit dieses Isotops nur über die 13C-Satelliten von 13C-Signalen mit ihren sehr geringen Intensitäten gemessen werden, was aber mit Hilfe der heutigen Messtechniken und der Anwendung von Pulsfolgen, die DoppelquantenÜbergänge selektieren, durchaus machbar ist. 13 C-NMR-Spektroskopie 8 Spektroskopie in der Organischen Chemie Natürlich ist es auch möglich, durch Isotopenmarkierung die Intensität der 13CSatelliten zu erhöhen, sodass sie die der 13C-Hauptsignale erreichen können. Allerdings ist hier eine gezielte Synthese mit kostspieligen 13C-markierten Reagenzien erforderlich. Die Kopplungskonstante 1J(13C,13C) ist wie die 1J(13C,1H) eine Funktion der Hybridisierung, die hier wegen der Existenz zweier C-Atome sogar eine quadratische Abhängigkeit: 1 13 J( C,13C) ~ s1 · s2 (bei Symmetrie: ~ s2) Einige Werte einfacher Kohlenwasserstoffe: sp3 - sp3 H H H C C H H H +34.6 Hz H3C H C H C H +41.9 Hz sp2 - sp2 sp3 - sp sp3 - sp2 H3C C C H H C H +67.5 Hz H C H +67.6 Hz sp - sp H C C H +171.5 Hz C C +56.0 Hz 13 C-NMR-Spektroskopie 9 Spektroskopie in der Organischen Chemie NMR-Lösungsmittel (s. auch IM1) In der NMR-Spektroskopie werden i.a. deuterierte Lösungsmittel verwendet. Die Substitution der leichten durch die schweren Wasserstoffatome hat zwei Vorteile: - Deuterium als Spin-1-Kern hat auch magnetisches Moment, aber seine Resonanzfrequenz ist sehr weit von der der Protonen entfernt. Es kann deshalb dazu benutzt werden, das Verhältnis von Magnetfeld und Radiofrequenz konstant zu halten (Lock), ein Routineverfahren zur elektronischen Stabilisierung der Messmethode. - Das Lösungsmittel hat bei 100%iger Substitution kein eigenes Signal in der 1 H-NMR. Bei der Verwendung der normalen, nichtdeuterierten Lösungsmittel wäre bei den üblichen Substratkonzentrationen das Lösungsmittelsignal das bei weitem größte, was zu unerwünschten Signalüberlagerungen, aber auch zu Problemen bei der Darstellung kleinerer Signale nach der FourierTransformierung des FIDs führen kann. Schreibweisen am Beispiel des Aceton: (CD3)2CO, Aceton-d6 oder [D6]-Aceton. NMR-Spektroskopie 1 Spektroskopie in der Organischen Chemie Dennoch haben die üblicherweise verwendeten deuterierten Lösungsmittel ein – wenn auch kleines – 1H-Signal, weil der Deuterierungsgrad der üblichen kommerziellen Lösungsmittel nicht 100%, sondern nur 99% bis 99,9% ist. 1 H-chemische Verschiebungen häufig benutzter Lösungsmittel (bei RT): CHCl3 (Chloroform) ! = 7.24 Methanol-d3 (CHD2OD) ! = 3.35 Aceton-d5 ! = 2.04 Benzol-d5 ! = 7.27 CHDCl2 (Dichlormethan) ! = 5.32 CHD2CN (Acetonitril) ! = 1.93 HDO ! = 4.65 Merke: Die Lösungsmittelmoleküle, deren 1H-Signale man beobachtet, besitzen einen 1H-Kern und eine um 1 verminderte Zahl von Deuteriumatome, also z.B. Aceton-d5. NMR-Spektroskopie 2 Spektroskopie in der Organischen Chemie Die 1H-Signale der deuterierten Lösungsmittel sind jedoch keine Singuletts, wenn mehr als 1 Wasserstoffatome im Molekül, also auch noch Deuteriumatome (2H) vorhanden sind, da die Kerne dann skalar koppeln. Beispiel CHDCl2: Deuterium hat die Spinquantenzahl 1. Das bedeutet, dass es für einen Deuteriumkern drei praktisch gleich populierte Energieniveaus gibt: -1, 0 und +1. Das Signal eines 1H-Kern, der mit einem 2H koppelt, besteht also aus drei äquidistanten, praktisch gleich intensiven Linien (1:1:1-Triplett): 2J (2H,1H) !(1H) 2 H,1H-Kopplungskonstanten sind ungefähr um den Faktor 1/6.5 kleiner als Jx(1H,1H), weil "(1H)/ "(2H) # 6.5. 3 NMR-Spektroskopie Spektroskopie in der Organischen Chemie Bei mehr als einem Deuterium-Kopplungspartner wird das 1H-Signal mit jedem neuen Deuterium-Kopplungspartner in ein 1:1:1-Triplett aufgespalten. Beispiel: Methanol-d, CHD2-OD !(1H) Es entsteht ein 1:2:3:2:1-Quintett. NMR-Spektroskopie 4 Spektroskopie in der Organischen Chemie Die Multiplizitäten und die relativen Peakintensitäten können bei Kopplung mit Spin-1-Kernen aus einer dem PASCALschen Dreieck ähnlichen Darstellung abgeleitet werden: n=0 n=1 n=2 n=3 n=4 n=5 n=6 1 1 1 4 1 5 15 6 21 50 1 3 10 30 90 1 2 6 16 45 126 1 1 3 7 19 51 141 1 2 6 16 45 126 1 3 10 30 90 1 4 1 15 5 1 50 21 6 1 Jede Zahl ist immer die Summe der unmittelbar über ihr sowie der beiden links und rechts davon stehenden Zahlen; keine Zahl (außerhalb des Dreiecks) bedeutet 0. 5 NMR-Spektroskopie Spektroskopie in der Organischen Chemie Schwieriger ist es bei Lösungsmitteln, die chemisch unterschiedliche Deuteriumpositionen enthalten. Beispiel: Deuterobenzol, C6HD5 H D D D D D NMR-Spektroskopie Das Proton (oben) koppelt mit zwei ortho-, zwei meta- und einem para-ständigen Deuteriumkern. Da die ortho- und die meta-2H-Kerne paarweise auch noch magnetisch nichtäquivalent (!) sind, resultiert ein außerordentlich komplexes 1H-Signal das aus vielen Einzellinien besteht (siehe links simuliertes Signal). Die D,H-Kopplungskonstanten sind aber recht klein, nämlich nur ca. 15% (1/6.5) der entsprechenden J(1H,1H)-Werte. Deshalb beobachtet man i. a. ein deutlich verbreitertes Singulett (die Einhüllende). 6 Spektroskopie in der Organischen Chemie Lösungsmittelsignale in der 13C-NMR Für die Aufspaltungen der 13C-NMR-Signale von deuterierten Lösungsmitteln gilt Analoges wie bei Protonen, weil beide Kerne einen Kernspin von 1/2 haben. Allerdings bekommt man für nicht oder nur teilweise deuterierte Spezies separate Signale (Isotopenverschiebung) mit der jeweiligen Deuterium-Aufspaltung. Angesichts der üblicherweise sehr hohen Deuterierungsgrade kommerzieller Lösungsmittel spielt dies aber kaum eine Rolle. Das 13C-Signal des gebräuchlichsten Lösungsmittels CDCl3 (Deuterochloroform) ist ein Triplett bei ! = 77.0: 1J (13C,2H) !(13C) Auch hier gilt: 1 J(13C,1H) # 6.5 · 1J(13C,2H) 7 NMR-Spektroskopie Spektroskopie in der Organischen Chemie 13 C-chemische Verschiebungen, Multiplizitäten und 1J(13C,2H)-Werte (in Hz) einiger deuterierter Lösungsmittel: CDCl3 (Chloroform) CD2Cl2 (Dichlormethan) CD3OD (Methanol) CD3-CO-CD3 (Aceton) C6D6 (Benzol) CD3-SO-CD3 (DMSO) CD3CN (Acetonitril) Pyridin-d5 NMR-Spektroskopie 77.0 53.5 49.3 29.3 206.3 128.0 39.7 1.3 117.7 123.5 135.5 149.5 tert quint sept sept mult tert sept sept mult tert tert tert 32 27 21 20 24 21 21 25 24 27 8 Spektroskopie in der Organischen Chemie Beispiele für Lösungsmittelsignale (Aceton) mit unterschiedlichem Deuterierungsgrad: *: C1HD2-CO-CD3 o: C1H2D-CO-CD3 NMR-Spektroskopie *: CD3-CO-CD3 o: C1HD2-CO-CD3 9