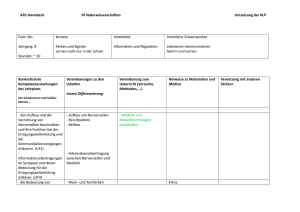
Pathogenetische Bedeutung von intrazellulären
Werbung

Neurologische Klinik und Poliklinik der Technischen Universität München Klinikum rechts der Isar (Direktor: Univ.-Prof. Dr. B. Conrad) Pathogenetische Bedeutung von intrazellulären Huntingtinaggregaten beim Morbus Huntington Immun- und enzymhistochemische Untersuchung menschlichen post-mortem-Gehirngewebes zu Vorkommen und Verteilung pathologischer Huntingtinaggregate beim Morbus Huntington Stefan Kümmerle Vollständiger Abdruck der von der Fakultät für Medizin der Technischen Universität München zur Erlangung des akademischen Grades eines Doktors der Medizin genehmigten Dissertation. Vorsitzender: Univ.-Prof. Dr. D. Neumeier Prüfer der Dissertation: 1. apl. Prof. Dr. A. Weindl, Ph.D. 2. Univ.-Prof Dr. B. Conrad Die Dissertation wurde am 22.02.2002 bei der Technischen Universität München eingereicht und durch die Fakultät für Medizin am 13.11.2002 angenommen. Diese Arbeit widme ich meinen Großeltern 1 ABKÜRZUNGEN: Abb. Abbildung AK Antikörper bzw. beziehungsweise ca. zirka DAB Diaminobenzidin DNA Desoxiribonukleinsäure et al. et alii etc. et cetera g Gramm GABA Gamma-Amino-Buttersäure h Stunde/Stunden Htt Huntingtin L Liter M Mol MH Morbus Huntington min Minuten mg Milligramm ml Milliliter µm Mikrometer NADPH Nicotinamid-Adenin-Dinukleotid-Hydrogen-Phosphat NADPH-d NADPH-Diaphorase N-terminal „Stickstoff-endig“ = am Stickstoffende gelegen resp. respektive SBMA Spinobulbäre Muskelatrophie SCA Spinozerebelläre Ataxie Tab. Tabelle usw. und so weiter z.B. zum Beispiel ZNS zentrales Nervensystem 2 INHALTSVERZEICHNIS Seite: 1. Einführung 6 1.1 Einleitung 6 1.2 Klinisches Bild 6 1.3 Epidemiologie 7 1.4 Neuropathologie 7 1.4.1 Striatum 8 1.4.1.1 Selektiver Nervenzellverlust im Striatum 10 1.4.1.2 Calbdindin-D28k-positive Nervenzellen sind besonders betroffen 11 1.4.1.3 NADPH-Diaphorase-haltige Neurone bleiben vermeintlich verschont 11 1.4.2 Zerebraler Kortex und subkortikale weiße Substanz 12 1.4.2.1 Laminare und zelluläre Pathologie der Großhirnrinde 12 1.4.2.2 Subkortikale weiße Substanz 13 1.4.3 Beteiligung weiterer Strukturen 13 1.5 Genetik 13 1.6 Genprodukt Huntingtin 14 1.6.1 Pathogenetische Überlegungen 15 1.6.2 Charakterisierung der Huntingtin-Expression 15 1.6.3 Interaktionen mit anderen Proteinen 16 1.6.4 Aggregationen von Huntingtin – Intrazelluläre Eiweißablagerungen 16 1.6.5 Pathogenetische Bedeutung der Huntingtinaggregate 18 1.6.6 Mechanismus der Proteinaggregation 18 1.7 Vergleich mit anderen CAG-Repeat-Krankheiten 19 1.8 Pathogenetische Rolle der Huntingtinaggregate ist weiterhin unklar 20 2. Problemstellung 21 3. Material und Methoden 22 3.1 Material 22 3.1.1 Gewebeproben 22 3.1.1.1 Untersuchtes Gehirngewebe 22 3.1.2 Verbrauchsmaterial 23 3.1.2.1 Antikörper 23 3.1.2.2 Lösungen für Immun- und Enzymhistochmie 24 3 3.2 Methoden 26 3.2.1 Gewebeaufbereitung 26 3.2.1.1 Immunhistochemie 26 3.2.1.2 Enzymhistochemie 27 3.2.1.3 Doppelfärbetechnik 28 3.2.1.4 Fertigstellung der Präparate 28 3.2.2 Auswertung 4. 28 Ergebnisse 30 4.1 EM48-Färbung 30 4.1.1 Darstellung und Charakterisierung von Huntingtinaggregaten 30 4.1.2 Topographische Verteilung der Huntingtinaggregate 31 4.1.3 Akkumulationen multipler kleiner Huntingtinaggregate 34 4.1.4 In Kontrollgehirnen sind keine Huntingtinaggregate nachweisbar 35 4.2 NADPH-Diaphorase-Neuronen und EM48-Färbung 4.2.1 Huntingtinaggregationen in NADPH-d-positiven Nervenzellen 36 4.2.2 NADPH-d- und EM48-Kombinationsfärbung in Kontrollen 36 4.3 Calbindin-Neuronen und EM48-Färbung 39 4.3.1 Calbindin-positive Nervenzellen enthalten nur selten Htt-Aggregate 39 4.3.2 Calbindin- und EM48-Kombinationsfärbung in Kontrollen 41 4.4 Kombinierte SMI32- und EM48-Färbung 5. 36 42 Diskussion 43 5.1 Kritische Beurteilung von Material und Methodik 43 5.1.1 Gewebeproben 43 5.1.2 Antikörper 43 5.1.3 Labortechniken 44 5.1.4 Auswahl der untersuchten Nervenzelltypen 45 5.2 Interpretation der Ergebnisse 45 5.2.1 Topographische Verteilung von Htt-Aggregaten 45 5.2.2 Mögliche pathogenetische Bedeutung von Neuropil-Htt-Aggregaten 47 5.2.3 Intrasomale Htt-Aggregate sind nicht auf vulnerable Nervenzellsubpopulationen beschränkt 47 5.2.3.1 Überraschend viele NADPH-d-Neurone enthalten Htt-Aggregate 48 5.2.3.2 Nur relativ wenige Calbindin-Neurone enthalten Htt-Aggregate 49 4 5.3 Zusammenfassung und Einordnung der Erkenntnisse in die vorhandene Literatur 49 5.3.1 Intrazelluläre Proteinaggregationen wurden lange als toxisch angesehen 50 5.3.2 Neuere in-vitro-Untersuchungen 50 5.3.3 Möglicherweise ist vor allem nicht-aggregiertes Huntingtin im Zellkern toxisch 51 5.3.4 Huntingtinaggregate per se sind wahrscheinlich nicht toxisch – vermutlich sind andere Faktoren für die Neurodegeneration verantwortlich 52 5.4 Andere wichtige potentielle Pathomechanismen 53 5.5 Ausblick 54 6. Zusammenfassung 56 7. Literaturverzeichnis 58 8. Danksagung 65 9. Lebenslauf 66 5 1. Einleitung 1.1 Einführung Unter den zahlreichen Krankheiten, die mit choreatischen Bewegungsstörungen einhergehen, ist der Morbus Huntington (Synonyma: Chorea Huntington, Chorea major, erbliche Chorea, erblicher Veitstanz) wahrscheinlich das bekannteste und wichtigste Leiden. Seinen Namen verdankt diese Krankheit George Huntington, einem damals auf Long Island/New York tätigen praktischen Arzt, der in einer weithin bekannt gewordenen Veröffentlichung 1872 durch eine äußerst sorgfältige und treffsichere, zugleich aber auch sehr anschauliche Beschreibung das Bild der erblichen Chorea charakterisiert hat (Huntington, 1872). Seine erste Begegnung mit „dieser Krankheit“ hatte Huntington jedoch schon im Alter von 8 Jahren, als er mit seinem ebenfalls als Arzt tätigen Vater auf Hausbesuch war. Seine Erinnerung daran beschrieb er später mit den folgenden Worten: „Over 50 years ago in riding with my father on his professional rounds, I saw my first cases of ‚that disorder‘. We suddenly came upon two women, mother and daughter, both bowing, twisting, grimacing. What could it mean? My father paused to speak with them and we passed on. From that point, my interest in the disease has never wholly ceased.“ (Huntington, 1910) Auch nach seinem Tod wurde dieser Krankheit viel Beachtung geschenkt, und sie rückte zunehmend in den Mittelpunkt zahlreicher, intensiver Forschungsbemühungen. Unter anderem diente der Morbus Huntington (MH) jahrelang als das Paradebeispiel für ein autsomal-dominantes Erbleiden schlechthin. Erstmals bei einer Erkrankung überhaupt, gelang es Forschern später durch Benutzung anonymer, genetischer Marker den für den MH verantwortlichen Gendefekt auf eine bestimmte Region innerhalb eines Chromosomes einzugrenzen (Gusella et al., 1983). Ein gutes Jahrzehnt danach konnte schließlich das eigentliche Gen, sowie das dazugehörige Protein identifiziert werden (Huntington’s Disease Collaborative Research Group, 1993). Doch trotz vieler, erstaunlicher Erkenntnisse und Fortschritte auf dem Gebiet des MH seither, kennt man bis heute nicht die eigentliche Ursache bzw. die pathophysiologischen Zusammenhänge, die dem stattfindenden Nervenzellverlust, und den damit einhergehenden, klinischen Symptomen zu Grunde liegen. 1.2 Klinisches Bild Der MH ist eine autosomal-dominante neurodegenerative Erkrankung, die durch choreatische Bewegungsstörungen, psychiatrische Auffälligkeiten, Abbau intellektueller Fähigkeiten und progrediente Demenz gekennzeichnet ist. Bei der weitaus häufigeren adulten Form manifestiert sich die Erkrankung für gewöhnlich um das 35.-50. Lebensjahr. Psychische bzw. kognitive Veränderungen 6 gehen dabei der neurologischen Symptomatik öfter voraus; doch entsteht der spezifische Verdacht auf MH zumeist erst dann, wenn choreatische Hyperkinesien hinzukommen. Die mentalen Störungen und neurologischen Beeinträchtigungen verschlechtern sich bei den Erkrankten gewöhnlich progredient und führen innerhalb von 10 bis 15 Jahren bis hin zu vegetativem Siechtum. Meist versterben die Betroffenen schließlich infolge von Schluckstörungen, Aspirationspneumonie und körperlicher Auszehrung. In 5-10% aller Fälle beobachtet man dagegen eine juvenile Manifestationsform mit einem Krankheitsbeginn vor dem 20. Lebensjahr. Im Gegensatz zur adulten Form ist bei diesem sogenannten juvenilen MH eine akinetisch-rigide Symptomatik, Dystonie mit dementivem Abbau und zerebralen Anfällen vorherrschend. Außerdem zeichnen sind diese Fälle durch eine raschere Progredienz aus (Weindl und Conrad, 1996; Adams et al., 1997). 1.3 Epidemiologie Epidemiologische Untersuchungen aus den USA und den meisten westeuropäischen Ländern ergaben eine Prävalenz der Erkrankung von ca. 4-8/100.000 Einwohner (Harper, 1992). Vergleichbare Prävalenzen finden sich auch in Deutschland, was unter anderem Untersuchungen aus dem südbayerischen Raum bestätigen (Knöbel, 1995). Damit zählt der MH in hiesigen Universitätskliniken zu den am häufigsten beobachteten hereditären Erkrankungen des zentralen Nervensystems. Relativ selten dagegen wird der MH in Finnland (0,1/100.000) und Japan (0,5/100.000) beobachtet, während die weltweit höchsten Prävalenzen vor allem in jenen Regionen zu finden sind, in denen betroffene Familien über Generationen hinweg ortsständig waren (ein bekanntes Beispiel hierfür ist die Gegend um den Maracaibosee in Venezuela (Penney et al., 1990)) Diese Abweichungen nach unten bzw. oben liegen somit wahrscheinlich hauptsächlich in der unterschiedlichen Abstammung und Vermischung der verschiedenen Bevölkerungsgruppen und Kulturen begründet. Davon jedoch abgesehen ist der MH eine weder durch Rasse, Geschlecht noch geographisch begrenzte Entität (Marshall und Shoulson, 1997). 1.4 Neuropathologie Der MH geht mit einer generalisierten Atrophie des Gehirnes einher, die jedoch in den verschiedenen Hirnregionen unterschiedlich stark ausgeprägt ist. Besonders stark von der Degeneration betroffen sind die Basalganglien (siehe Abb. 1.1). Das Gesamtgewicht eines MH-Gehirnes ist bei Autopsie um ca. 30% geringer als das eines Kontrollgehirns. Der neurodegenerative Prozeß scheint im Neostriatum zu beginnen und führt in fortgeschrittenen Fällen bis zu einer 50-65 prozentigen Verringerung dessen Querschnittsfläche. Doch auch beim Kortex beobachtet man eine Reduzierung um ca. 20-30%, ebenso wie bei der weißen Substanz (29-34%) (de la Monte et al., 1988). 7 C P 3cm Abb. 1.1 Makroskopischer Vergleich zweier fixierter Gehirnhälften in koronarer Schnittebene auf Höhe des rostralen Striatums: Rechts die Hemisphäre einer 49-jährigen Frau mit Morbus Huntington (pathologisches Stadium 3-4), links die Hemisphäre eines altersentsprechenden normalen Kontrollgehirns. Auffällig ist im MH-Gehirn die ausgeprägte Atrophie des Neostriatums (Nucleus caudatus, C und Putamen, P) mit konsekutiver Vergrößerung des Seitenventrikels, sowie eine deutliche Atrophie der Hirnrinde und ein markanter Verlust an weißer Substanz. 1.4.1 Striatum Das markanteste Charakteristikum des MH ist die auffallende, überdurchschnittlich stark ausgeprägte Atrophie des Neostriatums (Nucleus caudatus und Putamen). Während zu Beginn der Erkrankung in anderen Hirnregionen oft lange Zeit nur subtile, uncharakteristische neuropathologische Veränderungen zu beobachten sind, finden sich im Neostriatum meist schon deutliche Zeichen der Neurodegeneration. Die Degeneration schreitet dabei nach einer zeitlichen und topographischen Gesetzmäßigkeit voran. Besonders früh finden sich degenerative Veränderung in den caudo-dorsalen Anteilen des Nucleus caudatus, während der rostrale Caudatumkopf oft zunächst noch verschont zu bleiben scheint. Parallel dazu beobachtet man auch im Putamen die ersten Veränderungen in dessen caudalen Anteilen beginnend. Mit Fortschreiten der Krankheit breitet sich der neurodegenerative Prozeß zunehmend entlang einer Achse in dorso-ventraler, caudo-rostraler und medio-lateraler Richtung aus und involviert schließlich das gesamte Neostriatum. Lediglich der Nucleus accumbens bleibt bis in späte Stadien hinein relativ verschont (Vonsattel et al., 1985; Hersch und Ferrante, 1997). 8 Auf der Basis dieser charakteristischen neuropathologischen Veränderungen innerhalb des Striatums entwickelten Vonsattel et al. ein Schema bzw. ein Klassifikationssystem, nach welchem der Schweregrad der neuropathologischen Veränderungen in Stadien von 0-4 eingeteilt werden kann. Als Kriterien hierfür dienen sowohl makroskopische Aspekte des Striatums (siehe Abb. 1.2), als auch mikroskopische Einschätzungen des Nervenzellverlustes, sowie das Ausmaß der Gliose (Zunahme der Gliazelldichte) (Vonsattel et al., 1985). c1 c2 P A NA B C D Abb. 1.2 A-D Koronare Schnitte durch rostrale Striata verschiedener pathologischer Stadien des Morbus Huntington (Hämatoxylin/Eosin/Luxol-Fast-blue-Färbung). Abb. (A) zeigt ein normales Striatum eines Kontrollgehirns: Nucleus caudatus (c), dorso-medialer (c1) und ventro-lateraler Anteil (c2), Nucleus accumbens (NA) und Putamen (P). In den Stadien 0 und 1 sind keine makroskopischen Veränderungen zu erkennen (Abb. A). Ab Stadium 2 (Abb. B) ist eine beginnende striatale Atrophie zu beobachten, doch bleibt die Caudatumtaille medialwärts konvex ausgebuchtet. In Stadium 3 (Abb. C) ist die Atrophie schon soweit fortgeschritten, daß das Caudatum plan abgeflacht erscheint, und in Stadium 4 (Abb. D), dem Stadium schwerster Degeneration, nach medial hin eine konkave Oberfläche aufweist. Anmerkung: Neben makroskopischen Aspekten müssen für eine korrekte Stadieneinteilung ebenso auch mikroskopische Veränderungen berücksichtigt werden (siehe hierzu Text und Vonsattel et al., 1985). Zu Stadium 0 zählen die wenigen Fälle, in welchen trotz typischer, klinischer und familiärer Anamnese keine pathologischen Veränderungen bei der Autopsie entdeckt werden konnten. In Stadium 1 finden sich mikroskopische Veränderungen, mit bis zu 50 prozentigem Schwund striataler Neurone, ohne daß eine makroskopisch auffallende Atrophie zu beobachten ist. Durch zunehmenden Nervenzellverlust, einhergehend mit Astrogliose, wird jedoch ab Stadium 2 schon bei grober Betrachtung die atrophische Degeneration des Neostriatums erkennbar. Die Astrogliose entsteht dabei wahrscheinlich nicht durch eine primäre Astrozytose, sondern spiegelt lediglich ein relatives 9 Überleben der Astrozyten in einem ansonsten schrumpfenden Striatum wieder (Lange et al., 1976). Im Stadium 3 sind die oben beschriebenen pathologischen Veränderungen noch stärker ausgeprägt. Zunehmend ist auch das Pallidum von der Degeneration betroffen. Im schwersten Stadium 4 ist neben einer schon makroskopisch sehr eindrücklichen Atrophie des Striatums (siehe Abb. 1.2 D), ein Verlust striataler Nervenzellen von bis zu 90% mit fast ausschließlichem Übrigbleiben von Astrozyten zu beobachten (Vonsattel et al., 1985, 1998; Hersch und Ferrante, 1997). 1.4.1.1 Selektiver Nervenzellverlust im Striatum Morphologische, biochemische und immunhistologische Untersuchungen, speziell im Striatum, ergaben schon früh Hinweise dafür, daß auch auf zellulärer Ebene verschiedene Nervenzellsubpopulationen in unterschiedlichem Ausmaße von der Degeneration betroffen sind (Kowall et al., 1987). Striatale Nervenzellen können morphologisch in bedornte und unbedornte Neurone, sowie zusätzlich ihrer Größe nach in verschiedene Gruppen bzw. Nervenzellsubpopulationen unterteilt werden. Mit 7080% bilden mittelgroße bedornte Neurone den Hauptanteil aller striataler Nervenzellen, während 1020% zu den mittelgroßen bis großen unbedornten Neuronen zählen. Noch spezifischer lassen sich die verschiedenen Nervenzelltypen weiterhin durch diverse Neurotransmitter und Neuropeptide charakterisieren, welche in ihnen in unterschiedlichen Kombinationen lokalisiert zu finden sind (Übersicht, siehe Beal und Martin, 1986). Graveland et al. (1985) beobachteten mit Hilfe der Silberimprägnationstechnik nach Golgi, daß selektiv mittelgroße bedornte striatale Neurone („medium spiny neurons“) früh und in insgesamt stärkerem Maße pathologische Veränderungen aufweisen als mittelgroße bis große unbedornte Neurone („medium/large aspiny neurons“). Mittelgroße, bedornte Nervenzellen sind inhibitorische Projektionsneurone, die vom Neostriatum zu Pallidum und Substantia nigra projizieren und GABA (Gamma-Amino-Buttersäure) als primären Neurotransmitter benutzen. Neben GABA sind in ihnen zum Teil diverse Neuropeptide wie z.B. Substanz P, Enkephalin, Dynorphin und Calbindin D28k lokalisiert, durch die sich die Neuronen weiter subtypisieren lassen (Hersch und Ferrante, 1997). So fand man z.B. heraus, daß mittelgroße bedornte Neurone, welche Enkephalin enthalten, vornehmlich zum externen Segment des Globus pallidus projizieren, während Substanz P enthaltende Neurone vor allem zum internen Pallidumsegment hin ziehen. Das ist insbesondere interessant, weil sich darstellen ließ, daß in frühen Stadien des MH überproportional jene Nervenzellen degenerieren, die zum externen Pallidumsegment projizieren (Reiner et al., 1988). Man vermutet, daß es durch diesen differenzierten Verlust striataler inhibitorischer Projektionsneurone zu einer unausgeglichenen Aktivität zwischen den direkten und indirekten Bahnen des extrapyramidal-motorischen Systems kommt, was klinisch eine Chorea zur Folge haben kann (Albin et al., 1990; Wichmann und Delong, 1997). 10 1.4.1.2 Calbindin-D28k-positive Nervenzellen sind besonders betroffen Wie oben erwähnt, enthalten mittelgroße, bedornte Nervenzellen größtenteils auch Calbindin D28k (Gerfen et al., 1985; DiFiglia et al., 1989; Celio et al., 1990). Calbindin D28k ist ein Kalzium-bindendes Protein, von welchem angenommen wird, daß es eine Rolle in der neuronalen Kalziumpufferung spielt. Gerade diese Calbindin-D28K-immunreaktiven Neurone scheinen beim MH besonders vulnerabel zu sein und degenerieren unverhältnismäßig früh und insgesamt am stärksten (SetoOshima et al., 1988; Kiyama et al., 1990). Speziell in frühen pathologischen Stadien (Stadium 1-2) der Erkrankung fanden sich in dieser Nervenzellsubpopulation gelegentlich noch proliferative Veränderungen. So beobachtete man unter anderem ein anormales Dendritenwachstum mit teilweise sich rückwärts orientierenden distalen Dendritensegmenten („recurving“), sowie eine markante Zunahme der dendritischen Dornendichte. In weiter fortgeschrittenen Stadien (Stadium 3-4) hingegen waren neben einem massiven Zellverlust von bis zu 90% aller Calbindin-D28k-Neurone, vor allem auch degenerative Veränderungen, wie Verkürzung und Verplumpung der Dendriten, mit fokal irregulären Anschwellungen und deutlichem Dornenverlust zu finden. Über alle Stadien hinweg fiel den Autoren dabei jedoch das vermeintliche Verschontbleiben unbedornter Nervenzellen auf. (Ferrante et al., 1991). 1.4.1.3 NADPH-Diaphorase-haltige Neurone bleiben vermeintlich verschont Abgesehen von den zahlenmäßig überwiegenden „medium spiny neurons“ (siehe 1.4.1.1) gibt es im Striatum sowie auch in der Hirnrinde zusätzlich eine Vielfalt verschiedener Interneurone, mit unterschiedlichen Charakteristika und entsprechend auch verschiedenen neuroaktiven Substanzen. Diese Interneurone sind mittelgroß bis groß, besitzen unbedornte bis nur sehr spärlich bedornte Dendriten („aspiny“ bis „sparsly spiny neurons“), und scheinen gegenüber der Neurodegeneration beim MH relativ resistent zu sein. Zu ihnen zählen vor allem große cholinerge Neurone, Substanz-Pexprimierende Neurone und solche, die Somatostatin und Neuropeptid Y enthalten und zusätzlich über eine NADPH-Diaphorase (NADPH-d) verfügen (Hersch und Ferrante, 1997). Durch diese NADPH-d lassen sich letztere Nervenzellen sehr leicht mittels einer enzymhistochemischen Reaktion darstellen. Diese NADPH-d-positiven Interneurone sind von mittlerer Größe, unbedornt, und machen ca. 3-4% aller striataler Nervenzellen aus. Speziell dieser Nervenzelltyp scheint beim MH sowohl im Striatum als auch in der Hirnrinde von der Degeneration relativ verschont zu bleiben (Ferrante et al., 1985; Albin et al., 1990; Cudkowicz und Kowall, 1990). Während sich die totale Anzahl striataler Neurone im Verlauf des pathologischen Prozesses stark verringert, nimmt die Dichte dieser NADPH-d-positiven Neuronen bei mikroskopischer Betrachtung um das 4- bis 5-fache zu. Dieses Phänomen ist auf einen kombinierten Effekt einer selektiven Resistenz dieser Nervenzellen, sowie auf das Schrumpfen des umliegenden Gewebes zurückzuführen (Ferrante et al., 1985, 1987). Entsprechende biochemische 11 und immunhistochemische Untersuchungen für Somatostatin und Neuropeptid Y bestätigen das Überleben gerade diesen Zelltyps (Dawbarn et al., 1985; Beal et al., 1988a). 1.4.2 Zerebraler Kortex und subkortikale weiße Substanz Der degenerative Prozeß ist keineswegs auf das Striatum begrenzt. Auch die Großhirnrinde atrophiert zunehmend im Krankheitsverlauf und ist klinisch wahrscheinlich maßgeblich an den kognitiven Defiziten und psychischen Störungen beteiligt. Doch sind die neuropathologischen Veränderungen der Hirnrinde bei weitem nicht so charakteristisch und einheitlich, wie das im Striatum der Fall ist. De la Monte et al. (1988) berichteten von einem durchschnittlich 21-29 prozentigen Gewebsverlust der Großhirnrinde, der von einer 29-34 prozentigen Reduktion an subkortikaler weißer Substanz begleitet wird. Das Ausmaß der kortikalen Atrophie korreliert dabei, ebenso wie die striatale Pathologie, mit dem klinischen Voranschreiten der Erkrankung (Vonsattel et al., 1985; de la Monte et al., 1988). Manche Autoren beschrieben, daß bestimmte kortikale Areale stärker degenerieren als andere, doch herrscht darüber im Detail noch Uneinigkeit (Hersch und Ferrante, 1997). 1.4.2.1 Laminare und zelluläre Pathologie der Großhirnrinde Die degenerativen Veränderungen der Großhirnrinde finden vor allem in Laminae III, V und VI statt. Eine Reduktion der Nervenzellen von 30-40% wurde für Lamina V resp. VI beschrieben. Der Zellverlust geht dabei proportional mit einem Volumenverlust einher, weshalb sich die Neuronendichte bei einfacher mikroskopischer Betrachtung nicht unbedingt ändern muß (Sotrel et al., 1991; Hedreen et al., 1991). Morphometrische Untersuchungen ergaben, daß die Durchschnittsgröße aller kortikaler Nervenzellen zusammengenommen beim MH abnimmt. Die Ursache hierfür mag zum einen in einem Schrumpfen der überlebenden Neuronen liegen. Andererseits konnte aber auch gezeigt werden, daß bevorzugt große pyramidale Nervenzellen, markierbar durch Antikörper für nicht-phosphorylisiertes Neurofilament (siehe 3.1.2.1), zu Grunde gehen (Sotrel et al., 1991; Cudkowicz und Kowall, 1990). Weiterhin konnte nachgewiesen werden, daß sich der kortikale Zellverlust nicht auf die Neurone beschränkt, die bevorzugt mit dem Striatum in Verbindung stehen. Diese Tatsache macht eine einfache retrograde Degeneration als Ursache für den kortikalen Zellverlust sehr unwahrscheinlich (Hersch und Ferrante, 1997). Ähnlich wie im Striatum scheinen auch im Kortex vor allem Interneurone, einschließlich der enthaltenen Substanzen, verschont zu bleiben (Beal et al., 1988b; Cudkowicz und Kowall, 1990). 12 1.4.2.2 Subkortikale weiße Substanz Auch die weiße Substanz bleibt nicht von der Atrophie verschont. Wie oben erläutert wird hier ein morphometrischer Gewebeverlust von ca. 30% beobachtet, der für einen beträchtlichen Anteil am gesamten Größen- bzw. Gewichtsverlust des Gehirnes verantwortlich ist. Doch erstaunlicherweise lassen sich trotz dieser markanten Gewebereduktion mikroskopisch, zumindest mit konventionellen Methoden, keine pathologischen Veränderungen erkennen (Vonsattel et al., 1998). 1.4.3 Beteiligung weiterer Strukturen Neben der Degeneration von Striatum und Hirnrinde wurden für den MH zahlreiche weitere pathologische Veränderungen anderer Hirnstrukturen beschrieben. Mitunter sehr stark ist oft auch das Pallidum betroffen. Ähnlich dem Neostriatum ist auch hier ist ein markanter Nervenzellverlust zu beobachten, der zu einer ausgeprägten Atrophie führen kann. Pathologische Veränderungen vergleichbaren Ausmaßes wurden unter anderem auch für die Substantia nigra, den Thalamus und Hypothalamus, und die Kleinhirnrinde beschrieben (Übersicht über die Neuropathologie beim MH, siehe Hersch und Ferrante, 1997). 1.5 Genetik Genetisch ist der MH ein autosomal-dominantes Erbleiden mit nahezu vollständiger Penetranz. Der dafür verantwortlich gemachte Gendefekt wurde auf Chromosom 4p16.3 lokalisiert und als Expansion einer meiotisch instabilen (CAG)n-Trinukleotid-Sequenz (Cytosin-Adenin-Guaninidin) identifiziert (Huntington’s Disease Collaborative Research Group, 1993). Während die entsprechenden Allele bei normalen Individuen zwischen 10 und 35 sich wiederholende CAG-Einheiten aufweisen, enthalten die Allele Betroffener von 36 bis über 100 dieser „CAG-Repeats“ (CAG-Wiederholungen). In einem Übergangsbereich von 36 bis 39 CAG-Repeats beobachtet man eine inkomplette Penetranz, mit Individuen, die dem klassischen Krankheitsbild erliegen und mit solchen, die bis ins Senium hinein symptomfrei bleiben. Doch ab einer Anzahl von 40 Basen-Tripletts und mehr manifestiert sich die Huntington-Krankheit zu 100 Prozent innerhalb einer normalen Lebensspanne (Rubinsztein et al., 1996; McNeil et al., 1997). Trotz einer teilweise erheblichen Streubreite, besteht zwischen der Länge der CAG-Repeats und dem Manifestations- bzw. Sterbealter eine eindeutige Korrelation. Je größer dabei die Anzahl der CAG-Tripletts ist, desto früher manifestiert sich die Erkrankung bzw. desto früher versterben die Betroffenen. Ab einer Anzahl von ca. 60 CAGEinheiten muß mit juvenilem Auftreten gerechnet werden (Gusella et al., 1997). 13 Aufgrund der meiotischen Instabilität kann sich die Länge der Trinukleotid-Sequenz bei der Ei- bzw. Samenzellbildung ändern; allerdings tut sie dies meist nur um eine kleine Anzahl (<6 CAG-Tripletts). Doch vor allem bei der Spermatogenese werden teilweise auch Expansionen erheblich größeren Ausmaßes beobachtet. Diese Erkenntnis stimmt mit der Tatsache überein, daß juvenile MH-Fälle praktisch immer von väterlich expandierten Allelen verursacht werden (paternale Transmission) (Zühlke et al., 1993). Für sporadisch auftretende Erkrankungen sind für gewöhnlich intermediäre Allele mit weniger als 36 CAG-Repeats verantwortlich, die selbst nicht krankheitsauslösend sind, aber aufgrund ihrer meiotischen Instabilität in den pathologischen Bereich hineinexpandieren und bei den Nachkommen zur MH-Manifestation führen können (Epplen et al., 1998). Eine erstaunliche Tatsache ist, daß Individuen mit homozygoter Anlage für den MH-Gendefekt klinisch nicht von typischen, heterozygoten Gendefekt-Trägern zu unterscheiden sind (Wexler et al., 1987; Myers et al., 1989). Das läßt zum einen vermuten, daß bei MH-Heterozygoten das verbleibende normale Allel den Krankheitsprozeß weder hinauszögert noch die Manifestierung wesentlich beeinflußt, und zum anderen, daß ein Defekt in beiden Allelen des MH-Gens nicht signifikant mehr Schaden anrichtet als eine heterozygote Anlage. 1.6 Genprodukt Huntingtin Beim MH befindet sich der mutierte Genabschnitt innerhalb einer kodierenden Sequenz für ein Protein, welches Huntingtin (Htt) genannt wird (Huntington’s Disease Collaborative Research Group, 1993). Auf Proteinebene wird die (CAG)n-Trinukleotid-Sequenz in eine entsprechend verlängerte (Glutaminsäure)n-Kette am N-terminalen, also Stickstoff-seitigen Ende des Huntingtins überschrieben. Doch welche pathogenetischen Auswirkungen dieser verlängerte sogenannte Polyglutamintrakt mit sich bringt, ist nicht bekannt. Bislang ist auch die physiologische Funktion von Htt weitestgehend ungeklärt. Eine Verwandtschaft mit anderen Proteinen wurde nicht entdeckt, doch findet man bei Homologen von z.B. Mäusen und Ratten, sowie bei anderen Arten, einen hohen Grad an Übereinstimmung (evolutionäre Konservierung) (Gusella et al., 1997). Htt wird weit verbreitet in den unterschiedlichsten Gewebetypen neuronalen wie nicht-neuronalen (z.B. in Nieren-, Leber-, Lymphoblasten-, Lungen-, Herzgewebe etc.) Ursprungs exprimiert. Auch innerhalb des Gehirns kann eine heterogene Genexpression beobachtet werden. So wird das Htt relativ stark in Nervenzellen exprimiert, während es in der Glia nicht oder nur in geringer Menge nachzuweisen ist (Li et al., 1993; Strong et al., 1993). 14 Auf ultrastruktureller Ebene ist Htt ein somatodendritisch lokalisiertes Protein und vor allem in Zellkörpern, Dendriten, Axonen und Nervenendigungen zu finden, normalerweise aber nicht innerhalb des Zellkerns. Eine Assoziation mit Mikrotubuli und synaptischen Vesikeln legt eine Rolle im Transport von Zellorganellen nahe (DiFiglia et al., 1995; Sapp et al., 1997; Velier et al., 1998). 1.6.1 Pathogenetische Überlegungen Beim Menschen konnte beobachtet werden, daß Individuen, die durch eine balancierte Translokation ein komplettes Htt-Allel verloren haben, und somit nur noch über 50% der normalen Htt-Produktion verfügen, keinen MH-Phänotypen entwickeln (Ambrose et al., 1994). Dementsprechend zeigen auch Mäuse, bei denen durch gezielte Mutagenese lediglich ein Huntingtin-Gen-Homologon inaktiviert wurde, ebenfalls keine Anomalien (Duyao et al., 1995). Diese Erkenntnisse sprechen dafür, daß nicht ein einfacher Funktionsverlust des Huntingtins für die Pathogenese verantwortlich sein kann. Dagegen führt eine komplette Inaktivierung beider Allele in Mäusen noch vor Entstehung des Nervensystems zu einem frühen embryonalen Tod (Duyao et al., 1995; Zeitlin et al., 1995). So scheint Htt, zumindest bei Mäusen, eine entscheidende Rolle in der ontogenetischen Entwicklung zu haben. Berücksichtigt man zusätzlich, daß es auch erwachsene Individuen mit homozygoter Anlage für die MH-Mutation gibt (siehe 1.5), darf angenommen werden, daß die Mutation an sich die normale Aktivität von Htt nicht entscheidend beeinträchtigt. Statt dessen wird wahrscheinlich dem Protein durch seinen verlängerten Polyglutamintrakt eine neue, toxische Eigenschaft zu Teil („toxic gain of function theory“) (Huntington’s Disease Collaborative Research Group, 1993; Ambrose et al., 1994; Ross, 1995; Sisodia et al., 1998) 1.6.2 Charakterisierung der Huntingtinexpression Es wurde gezeigt, daß Htt in verschiedenen Hirnregionen unterschiedlich stark exprimiert wird. Doch läßt sich kein eindeutiger Zusammenhang zwischen Intensität der Genexpression und jenen Regionen, die besonders von der Neurodegeneration betroffen sind (siehe 1.4), herstellen. So wurden gleichermaßen in Kontroll- wie auch in MH-Gehirnen z.B. innerhalb des Striatums lediglich mittlere HttNiveaus beobachtet (Li et al., 1993; Landwehrmeyer et al., 1995), während z.B. in der relativ verschont bleibenden Kleinhirnrinde das Htt auf vergleichsweise hohem Niveau exprimiert wird (Strong et al., 1993). Die unterschiedlich starke Degeneration diverser Hirnregionen läßt sich also nicht durch ein unterschiedlich hohes Htt-Vorkommen erklären. Auch auf zellulärer Ebene läßt eine unterschiedlich starke Genexpression keine Aussage über die zu beobachtende unterschiedliche Vulnerabilität der diversen Nervenzelltypen (siehe 1.4) zu. Die Ergebnisse und Interpretationen verschiedener Autorengruppen divergieren hierzu jedoch beachtlich. Während manche innerhalb des Striatums von überdurchschnittlich viel Htt in anfälligen „medium 15 spiny neurons“ und relativ wenig Htt in den vergleichsweise resistenten mittelgroßen und großen „aspiny neurons“ beobachteten (Ferrante et al., 1997), berichteten andere von einer gerade entgegengesetzten Relation (Gutekunst et al., 1995). Recht einhellig weisen die verschiedenen Autoren allerdings darauf hin, daß speziell in mittelgroßen striatalen Nervenzellen stets eine sehr große Variabilität an Htt-Immunreaktivität zu finden war. So färbten sich manche von ihnen intensiv an, während in anderen das Htt kaum nachzuweisen war (Ferrante et al., 1997; Sapp et al., 1997). Weitere Publikationen heben hervor, weder bei normalen noch bei MH-Gehirnen signifikante Unterschiede hinsichtlich des Htt-Gehalts bezüglich unterschiedlicher Nervenzelltypen gefunden zu haben (Landwehrmeyer et al., 1995). 1.6.3 Interaktion mit anderen Proteinen Auf molekularem Niveau interagiert Htt mit vielen verschiedenen Proteinen, deren Funktionen ebenfalls größtenteils unbekannt sind. Eines dieser Proteine ist z.B. das speziell im Gehirn angereicherte HAP1 (huntingtin-associated protein 1), welches interessanterweise je nach unterschiedlicher Länge des Polyglutamintrakts von Htt ein unterschiedlich starkes Bindungsverhalten aufweist. Obwohl bislang die genaue Funktion von HAP1 unbekannt ist, ließen sich durch solche Interaktionen mit spezifischen Proteinen trotz des ubiquitären Vorhandenseins von Htt z.B. eine selektive Hirnpathologie erklären (Li et al., 1995, 1996). Ähnliche Wechselwirkungen wurden auch für andere Proteine beschrieben. So z.B. für die GAPDH (Glycerinaldehyd-3-phosphat-Dehydrogenase), ein zentrales Enzym der Glykolyse (Burke et al., 1996). Doch konnte beim MH bislang keine konsekutiv anomale GAPDH-Aktivität belegt werden. Als anderer möglicher Kandidat zählt auch die Caspase 3, ein humanes Enzym, welches mit einem Apoptose-Gen (ced-3) von Nematoden verwandt ist. Es konnte gezeigt werden, daß Htt je nach Polyglutaminlänge spezifisch durch diese Caspase 3 gespalten wird. Als Folge davon könnte möglicherweise ein für bestimmte Zellen toxisch wirkendes Fragment von Htt entstehen (Goldberg et al., 1996). Ähnliche vielversprechende Beobachtungen wurden auch für zahlreiche andere Proteine beschrieben, doch welchen pathogenetischen Stellenwert Interaktionen mit diesen oder anderen Proteinen haben, ist noch nicht bekannt. 1.6.4 Aggregationen von Huntingtin – Intrazelluläre Eiweißablagerungen Mit der Kenntnis um die genetische Ursache, wurden diverse transgene Tiermodelle entwickelt. In einem Mausmodell (Stammlinie R6/2), bei welchem das humane Exon 1 von Htt inklusive einem expandierten (CAG)n-Abschnitt in das Mausgenom eingeschleust wurde, konnte ein dem MH ähnelnder progredienter neurologischer Phänotyp, mit charakteristischen Bewegungsstörungen, Gewichtsverlust und frühem Tod beobachtet werden (Mangiarini et al., 1996). Weitere Untersuchungen an dieser Linie ergaben, daß der klinischen Symptomatik die Formation neuronaler 16 intranukleärer Zelleinschlüsse („neuronal intranuclear inclusions“) vorausgeht. Diese Zellkerneinschlüsse wurden durch immunhistochemische Untersuchungen mit Antikörpern (AK) gegen das N-terminale Ende von Htt entdeckt (Davies et al., 1997). Auf Grundlage dieser Erkenntnis wurden mit entsprechenden, gegen N-terminales Htt gerichteten AK auch in menschlichem MH-Gehirngewebe ähnliche intranukleäre Einschlußkörperchen entdeckt (DiFilgia et al., 1997; Becher et al., 1998). Diese Zellkerneinschlüsse wurden vor allem in den tieferen Hirnrindenschichten (Laminae V und VI) beobachtet. In juvenilen MH-Fällen wurden sie in bis zu 52% aller kortikalen Neurone aufgefunden, in Gehirnen adulter MH-Patienten hingegen lediglich in 3-6% (DiFilgia et al., 1997). Weiterhin wurde berichtet, sie im Striatum bevorzugt in Zellkernen mittelgroßer Neurone angetroffen zu haben; nicht jedoch in Zellkernen der großen Nervenzellen, die typischerweise, wie oben schon erwähnt, vom pathologischen Prozeß relativ verschont bleiben (DiFilgia et al., 1997; Becher et al., 1998). Die Proteinaggregate waren signifikant größer als Nukleoli und befanden sich frei, ohne eine sie begrenzende Membran, innerhalb der Zellkerne. Auch extrazellulär wurden gelegentlich intensiv gefärbte Strukturen beobachtet, die an dystrophe Neuriten erinnerten (DiFilgia et al., 1997). Eine fehlende Immunreaktivität für AK gegen innere Epitope, sowie entsprechende Western-BlotUntersuchungen wiesen darauf hin, daß jene Proteinpräzipitationen lediglich N-terminale Bruchstücke von Htt enthalten, und sich kein normales Htt bzw. kein Htt voller Länge im Zellkern befindet. Dadurch ließe sich auch erklären, warum das Protein in den Zellkern gelangen kann. Denn Htt besitzt kein „nuclear localization signal“ (NLS), wäre aber in seiner vollen Länge zu groß, um frei in den Zellkern hinein zu diffundieren. Es wäre jedoch z.B. denkbar, daß das pathologische Htt durch ein Enzym, wie z.B. Caspase 3 (siehe 1.6.3), gespalten wird, und anschließend kleinere Bruchstücke davon passiv in den Zellkern wandern und dort akkumulieren könnten (Ross, 1997). In-vitro-Experimente zeigten, daß die Transfektion von Zellen mit N-terminalen Htt-Fragmenten inklusive einer verlängerten Polyglutaminkette sowohl im Zytoplasma als auch im Zellkern zu Aggregationen führen kann. Je kürzer dabei die Länge des Fragmentes war, desto häufiger bildeten sich Aggregationen im Zellkern aus. Weiterhin gab es Hinweise dafür, daß vor allem von kurzen HttFragmenten bzw. von den Zellkerneinschlüssen eine auf die Zellen toxische Wirkung ausgeht, die mit Länge der Polyglutaminsequenz zunimmt (Cooper et al., 1998; Hackam et al., 1998; Martindale et al., 1998). Bei post-mortem-Gewebe von MH-Patienten und transgenen Tiermodellen, sowie auch in Zellen aus in-vitro-Experimenten, ist den Proteinaggregationen Ubiquitin angelagert (Davies et al., 1997; DiFiglia et al., 1997; Cooper et al., 1998). Ubiquitin selbst ist ein kleines stabiles Protein, mit der Aufgabe andere nicht mehr benötigte Proteine zu markieren und der Degradation bzw. dem proteolytischen Abbau zuzuführen (Jentsch und Schlenker, 1995). Dies legte die Vermutung nahe, daß das anomale Htt bzw. dessen Bruchstücke zwar als pathologisch erkannt, jedoch in Form dieser Aggregate nicht oder nur inkomplett dem proteolytischen Abbau zugänglich sind. 17 1.6.5 Pathogenetische Bedeutung der Huntingtinaggregate Trotz intensiver Forschungsbemühungen ist es bis heute nicht gelungen, die beim MH verantwortlichen pathogenetischen Mechanismen gänzlich aufzuklären und eine schlüssige kausale Erklärung für den selektiven Nervenzellverlust zu finden. Mit Entdeckung der neuronalen Zellkerneinschlüsse, sowie den anfangs vermeintlich eindeutigen Hinweisen auf deren Assoziation mit Nervenzelldegeneration und deren neurotoxische Potenz, glaubte man nun endlich das pathogenetische Korrelat für den Zellverlust gefunden zu haben (DiFiglia et al., 1997; Becher et al., 1998; Cooper et al., 1998). So wurde spekuliert, die intranukleären Proteineinschlüsse könnten zu Störungen intranukleärer Transportvorgänge oder auch zu subtilen Auswirkungen auf die Transkription führen (Davies et al., 1997; DiFiglia et al., 1997; Cha, 2000). Das könnte wiederum zur Folge haben, daß z.B. die Expression spezieller, für Nervenzellen zentraler Elemente, wie z.B. Rezeptoren, mitochondriale Proteine oder andere essentielle Moleküle entscheidend beeinflußt werden (Ross, 1997). Obwohl diese Meinung, daß Zellkerneinschlüsse maßgeblich für den Nervenzelluntergang verantwortlich sind, zunächst weithin Akzeptanz fand, wies unter anderem Ordway et al. (1997) auf die Möglichkeit hin, daß diese Eiweißaggregate eventuell lediglich ein Beiprodukt bzw. ein unschuldiges Kennzeichen anderer toxischer Vorgänge darstellen, und selbst nicht für den Zelltod verantwortlich sein könnten. Neuere Experimente weisen ebenfalls in diese Richtung. So berichten Saudou et al. (1998), daß in vitro intranukleäre Einschlüsse weder den Zelltod auslösen noch ihn voraussagen. Nach Transfektion von in-vitro-Zellen mit mutantem Htt, entwickelten striatale gleichermaßen wie hippokampale Zellen intranukleäre Proteinablagerungen. Doch während sich die Degenerationsrate bei den striatalen Zellen verdoppelte, blieb sie bei hippokampalen Zellen unverändert. Genauere Betrachtung ergab zudem, daß speziell Enkephalin-haltige mittelgroße Striatumzellen, die beim MH ganz besonders stark betroffen sind (siehe 1.4.1.1), bevorzugt degenerieren. Weiterhin war es durch Blockierung Ubiquitinkonjugierender Enzyme möglich, die Bildung von Proteinaggregaten größtenteils zu unterbinden, gleichzeitig aber die Expression von mutantem Htt unbeeinflußt zu lassen. Und obwohl nun als Folge davon weitaus weniger intranukleäre Einschlußkörperchen ausgebildet wurden, erhöhte sich die Rate der Zelldegeneration erheblich (Saudou et al., 1998). 1.6.6 Mechanismus der Proteinaggregation Über die Ursache der eigentlichen Aggregation von mutantem Htt wurden verschiedene Hypothesen vorgeschlagen. So beobachteten z.B. Perutz et al. (1994), daß lange Polyglutaminstränge selbst oder im Verbund mit anderen Polyglutamin-haltigen Molekülen durch starke Wasserstoffbrückenbildung eine β-Faltblattstruktur ausbilden und unter Umständen präzipitieren können. Das ist insbesondere deshalb interessant, da bekannt ist, daß auch 18 zahlreiche Transkriptionsfaktoren lange Polyglutaminsequenzen enthalten. So wäre es z.B. denkbar, daß essentielle Transkriptionsfaktoren durch die Einbindung in Aggregationen möglicherweise aus dem Verkehr gezogen werden und somit ihrer Funktion nicht mehr nachgehen könnten. Scherzinger et al. (1997) bestätigten in einem in-vitro-Modell, daß N-terminale Htt-Fragmente für sich genommen ab einer bestimmten Schwellenlänge des Polyglutamintrakts unlösliche Aggregate ausbilden. Diese Aggregate setzen sich aus fibrillären, β-Amyloid-artigen Strukturen zusammen. Ein anderer möglicher Aggregationsmechanismus leitet sich aus der Tatsache ab, daß Proteine mit zunehmender Polyglutaminlänge bessere Substrate für Transaminasen darstellen, und dadurch unter Umständen zahlreiche kovalente Verbindungen mit anderen essentiellen Peptiden eingehen könnten, bis die immer größer werdenden Moleküle schließlich ihre Löslichkeit verlieren, ausfällen und so nicht mehr ihre eigentliche Aufgabe erfüllen können (Kahlem et al., 1996). Ob diese oder andere Mechanismen für die Präzipitation bzw. Ausbildung von Aggregaten wirksam sind, ist noch nicht abschließend geklärt. 1.7 Vergleich mit anderen CAG-Repeat-Krankheiten Außer für den MH sind für mindestens 7 weitere neurodegenerative Erkrankungen ähnliche Expansionen einer instabilen CAG-Trinukleotid-Sequenz verantwortlich. Im einzelnen konnte dies bis heute unter anderem für die Spinobulbäre Muskelatrophie (SBMA oder auch Kennedy-Erkrankung), die Dentatorubropallidoluysiane Atrophie (DRPLA), Spinozerebelläre Ataxie 3 (SCA3, MachadoJoseph-Krankheit), sowie für die Spinozerebellären Ataxien 1, 2, 6 und 7 (SCA1, 2, 6 bzw. 7) nachgewiesen werden (Young, 1998; Schöls et al., 2001). Diese Erkrankungen werden auch unter dem Begriff der CAG-Repeat- bzw. Polyglutaminkrankheiten zusammengefaßt. Die jeweils davon betroffenen, strukturell sehr verschiedenen Gene sind untereinander nicht verwandt, und kodieren für ganz verschiedenartige Proteine, deren Funktionen bislang größtenteils unbekannt sind. Eine Ausnahme davon bilden lediglich die SBMA und die SCA6, bei welchen die Gene für einen AndrogenRezeptor bzw. einen spannungsabhängigen Kalziumkanal kodieren. Obwohl die verschiedenen neurodegenerativen CAG-Repeat- bzw. Polyglutaminkrankheiten jeweils für sich, hinsichtlich Neurodegeneration und klinischer Ausprägung, recht spezifische Charakteristika aufweisen, findet man doch auch viele Gemeinsamkeiten unter ihnen: z.B. sind sie alle (mit Ausnahme von SBMA) autosomal-dominant, haben einen bilateralen symmetrischen Nervenzellverlust zur Folge, manifestieren sich für gewöhnlich erst im mittleren Lebensalter und entwickeln einen fatalen progredienten neurologischen Phänotyp (Young, 1998). Doch noch eine weitere frappierende Gemeinsamkeit verbinden diese verschiedenen Polyglutaminkrankheiten miteinander: Wie oben für den MH beschrieben, konnten inzwischen für fast alle Polyglutaminkrankheiten (ausgenommen für SCA2) pathologische Aggregationen der mutanten Proteine innerhalb von Nervenzellen gefunden werden (Sieradzan und Mann, 2001). Das führte schon 19 bald zu der naheliegenden Hypothese, daß, so unterschiedlich die Krankheiten und ihre jeweils mutierten Proteine auch sein mögen, diese intranukleären Einschlußkörperchen in Nervenzellen ein gemeinsames, zentrales Element der Pathogenese darstellen (Ross, 1997; Davies et al., 1998; Becher et al., 1998) Inwieweit die Eigenschaften der jeweils betroffenen Proteine hinsichtlich der Pathogenese von Bedeutung sind, ist noch unklar. Einen Hinweis dazu gibt jedoch ein Experiment, in welchem Mäusen eine lange CAG-Sequenz in ein ektopes, vermeintlich unbeteiligtes Gen eingeschleust wurde. Dabei handelte es sich um das Gen für die Hypoxanthin-Guanin-Phosphoribosyltransferase. Erstaunlicherweise entwickelten auch diese Mäuse einen progredienten neurologischen Phänotyp mit Ataxien, Krampfanfällen, und frühem Tod. Außerdem wurden ebenfalls intranukleäre Einschlußkörperchen beobachtet (Ordway et al., 1997). Diese Tatsache legt deshalb die Vermutung nahe, daß die Polyglutaminsequenzen an sich zum einen für die Zellkerneinschlüsse und zum anderen für die Neurotoxizität verantwortlich sind, und das betroffene Protein eventuell lediglich die Verteilung der Eiweißaggregate und das selektive Muster der Neurodegeneration bestimmen. 1.8 Pathogenetische Rolle der Huntingtinaggregate ist weiterhin unklar Wie oben angedeutet sprechen viele Argumente für die Vermutung, daß sowohl beim MH, als möglicherweise auch bei diversen anderen neurodegenerativen Erkrankungen, die pathologischen Eiweißaggregationen zumindest mitverantwortlich für den Zelluntergang sind. Trotz dieser verlockenden und vermeintlich naheliegenden These, konnten dafür bislang keine eindeutigen Beweise erbracht werden. In der vorliegenden Untersuchung soll nun mit Hilfe von immun- und enzymhistochemischen Methoden versucht werden, auch an menschlichem post-mortem-Gehirngewebe von MH-Patienten die oft postulierte pathogenetische Bedeutung von Htt-Aggregationen für den Nervenzelluntergang zu überprüfen. 20 2. Problemstellung Es ist allgemeinhin anerkannt, daß beim MH ein selektiver Nervenzellverlust stattfindet. Bislang ist jedoch unklar, welche pathogenetischen Mechanismen für die differenzierte Degeneration verantwortlich sind. Seit Entdeckung von pathologischen Htt-Aggregationen innerhalb der Zellkerne bzw. dem Zytoplasma von Nervenzellen, wurde weithin vermutet, daß diese Zelleinschlüsse die eigentliche Ursache für den Nervenzelluntergang sind. Die hier vorliegende Disserations-Arbeit soll eine mögliche Beziehung zwischen der Ausbildung von Htt-Aggregaten und einer konsekutiven Degeneration bzw. dem Nervenzelluntergang untersuchen. Dazu sollen folgende Überlegungen angestellt werden: • Sollten Htt-Aggregationen tatsächlich direkt für den Nervenzelluntergang verantwortlich sein, dann sollte erwartet werden können, die Htt-Aggregate im Verlauf der Erkrankung bevorzugt in den Hirnregionen und in den Nervenzellsubpopulationen anzutreffen, die besonders früh bzw. stark von der Degeneration betroffen sind. • Hirnareale bzw. Nervenzellsubpopulationen die von der Degeneration verschont bleiben, sollten dagegen entsprechend keine bzw. weniger pathologische Htt-Aggregationen enthalten. Um dies zu überprüfen soll in dieser Studie Gehirngewebe von MH-Patienten immun- und enzymhistochemisch aufbereitet und mikroskopisch ausgewertet werden. Im Speziellen soll hierfür das Vorkommen und die Verteilung von Htt-Aggregationen in Striatum und Hirnrinde bzw. in Calbindin-D28k-positiven Nervenzellen und NADPH-d-haltigen Neuronen untersucht und miteinander verglichen werden. 21 3. Material und Methoden 3.1 Material 3.1.1 Gewebeproben Die für diese Untersuchungen verwendeten menschlichen Gewebeproben stammen aus dem Bedford VA Medical Center Brain Tissue Archive (Bedford/USA), der Harvard Brain Bank (Cambridge/USA), und von der Emory University (Atlanta/USA). 3.1.1.1 Untersuchtes Gehirngewebe Für die Untersuchung werden striatale und kortikale Gewebeproben von 12 klinisch bzw. genetisch diagnostizierten MH-Patienten verwendet. Die Diagnose MH wurde jeweils post mortem durch die neuropathologische Untersuchung bestätigt und in ihre pathologischen Stadien nach Vonsattel et al. (1985) eingeteilt (siehe Tab. 3.1). Als Kontrollen dienen entsprechende Gewebeproben von 4 Patienten vergleichbaren Alters ohne bekannte neurologische Leiden, sowie Hirnschnitte von 6 Patienten mit bekannten neurologischen Erkrankungen (siehe Tab 3.1). Nach einem mittleren post-mortem-Intervall von 9,2h (siehe Tab. 3.1) wurden alle Gehirne frisch seziert und in 4°C kalter 2%iger Paraformaldehyd-Lysin-Periodat-Lösung über 24-36h fixiert. Anschließend wurden die Gewebeblöcke mit 0,1M „phosphate buffered saline“ (= PBS-Puffer) gespült und über 36h in Frostschutzmittel mit einer ansteigenden Konzentration von 10% Glycerol / 2% Dimethylsufoxid (DMSO)-Lösung bzw. 20% Glycerol / 2%DMSO-Lösung eingelegt. Von den Striata wurden mit einem Gefriermikrotom (Zeiss) Serienschnitte in koronarer Schnittebene mit einer Dicke von 50µm angefertigt. Vom Neostriatum werden aufgrund der differenzierten Pathologie vorwiegend Schnitte in Höhe des Nucleus accumbens septi (Abb. 1.2) untersucht. Entsprechend wurden kortikale Schnitte gleicher Dicke vertikal zur Hirnoberfläche angefertigt. Die in dieser Studie untersuchte Hirnrinde stammt dafür aus insulären sowie temporalen Kortexarealen. Für die nachfolgende histochemische Bearbeitung wurden die Gewebeschnitte in 0,1M PBS-Puffer / 0,08% Natriumazid bei 4°C aufbewahrt. 22 Verwendetes Gehirngewebe: (Gehirne der Einfachheit halber durchnumeriert) Gehirn MH1 MH2 MH3 MH4 MH5 MH6 MH7 MH8 MH9 MH10 MH11 MH12 Mittelwert CT1 CT2 CT3 CT4 Mittelwert NK1 NK2 NK3 NK4 NK5 NK6 Mittelwert Alter (Jahre) 29 40 53 66 62 71 74 68 n.b. 73 17 22 52,3 68 31 60 64 55,8 67 78 71 77 69 53 69,2 Geschlecht w w m w m m w w m m m m w m m m m m m m m m - PMI (Stunden) 6 17,4 12 20 12,5 2 10 13 7 3,5 5 3 9,2 3 26 20 11 15 16 14 5 10 14 7 11 CAGrepeats Pathologie 48 MH Stadium 1 42 MH Stadium 1 44 MH Stadium 1 44 MH Stadium 2 n.b. MH Stadium 2 46 MH Stadium 3 45 MH Stadium 3-4 47 MH Stadium 3-4 45 MH Stadium 4 47 MH Stadium 4 (89) JMH Stadium 4 n.b. JMH Stadium 4 45,3 n.b. Kontrolle n.b. Kontrolle n.b. Kontrolle n.b. Kontrolle n.b. M. Alzheimer n.b. M. Alzheimer n.b. M. Parkinson n.b. Multiple Sklerose n.b. Multiinfarktdemenz n.b. Schizophrenie - Tab. 3.1 Erklärungen: PMI = post-mortem-Intervall; w = weiblich, m = männlich; n.b. = nicht bekannt; MH = Morbus Huntington; JMH = juveniler Morbus Huntington, CT = normales Kontrollgehirn, NK = neurologisches Kontrollgehirn. 3.1.2 Verbrauchsmaterial 3.1.2.1 Antikörper • EM48 – Antikörper gegen N-terminales Huntingtin Erhalten in Kooperation von Dr. Xiao-Jiang Li, Emory University School of Medicine, Atlanta/USA; verwendet in einer Verdünnung in PBS von 1:500. EM48 ist ein polyklonales Kaninchen-Antiserum, welches spezifisch gegen das N-terminale Ende von Htt gerichtet ist. Eine Kreuzreaktivität gegen andere Proteine ist nicht bekannt. EM48 markiert vorwiegend aggregierte N-terminale Htt-Fragmente, wohingegen es mit nicht aggregiertem bzw. normalem Htt nicht reagiert. Die Affinität von EM48 ist von der Länge des Polyglutamintrakts unabhängig. Ausgewählte Literaturstellen: Li und Li (1998); Gutekunst et al. (1999). 23 • Calbindin D28k – Antikörper gegen Kalzium-bindendes Protein Bezogen von Swiss Antibodies, Bellinzona/Schweiz; verwendet in einer Verdünnung in PBS von 1:3000. Calbindin D28k erkennt als monoklonaler Maus-Antikörper ein neuronales Kalzium-bindendes Protein, welches in einer Untergruppe von mittelgroßen bedornten Neuronen im Striatum vorkommt. Dabei konnte gezeigt werden, daß diese Calbindin-D28k-positive Nervenzellen zu einer beim MH bevorzugt degenerierenden Nervenzellsubpopulation gehören. Ausgewählte Literaturstellen: Gerfen et al. (1985); DiFiglia et al. (1989); Celio et al. (1990); Ferrante et al. (1991). • SMI32 – Antikörper gegen nicht-phosphoryliertes Neurofilament H Bezogen von Sternberger Monoclonals Inc, Lutherville/USA; verwendet in einer Verdünnung in PBS von 1:1000. SMI32 ist ein monoklonaler Maus-Antikörper gegen nicht-phosphoryliertes Neurofilament H der meisten Säugetiere, inklusive das des Menschen. SMI32 markiert neuronale Zellkörper, Dendriten und (vornehmlich dicke) Axone im zentralen wie peripheren Nervensystem. Andere Gewebe- und Zelltypen dagegen sind nicht reaktiv. Besondere Affinität weist SMI32 gegenüber einer Untergruppe kortikaler Neuronen auf, die beim MH vorzugsweise degenerieren. Ausgewählte Literaturstellen: Sternberger und Sternberger, 1983; Morrison et al., 1987; Cudkowicz und Kowall, 1990. 3.1.2.2 Lösungen für Immun- und Enzymhistochemie • 2%ige Paraformaldehyd-Lysin-Periodat-Lösung 1L Aqua dest. auf 60°C erhitzen und darin lösen: 5ml einer 10M NaOH = Natronlauge 60g Paraformaldehyd 33g dibasisches Na2PO4 = Dinatriumhydrogenphosphat auf Raumtemperatur abkühlen lassen und darin lösen: 9g monobasiches NaHPO4 = Natriumhydrogenphosphat 6,45g Na-m-Periodat 41,1g Lsyin-Salzsäure-Salz filtrieren, mit Aqua dest. auf 3L auffüllen, und mit 10M Natronlauge auf pH7,4 einstellen • 10% (bzw. 20%) Glycerol / 2% DMSO-Lösung (DMSO = Dimethylsulfoxid) zu 2,7L (bzw. 2,4L) 0,1M Phosphat-Puffer (pH7,4) hinzufügen 300ml (bzw. 600ml) Glycerol 60ml DMSO-Lösung 24 • PBS-Puffer 0,1Mol, pH7,35 (PBS = phosphate buffered saline) in 1L Aqua dest. lösen: 11g dibasisches Na2PO4 = Dinatriumhydrogenphosphat 3g monobasiches NaHPO4 = Natriumhydrogenphosphat 8,5g NaCl = Kochsalz mit Salzsäure auf pH7,35 einstellen • Methanol / 0,3% Wasserstoffperoxid- Lösung 100ml reines Methanol 1ml 33%iges H2O2 = Wasserstoffperoxid • Tris-HCl-Puffer 50mM (bzw. 0,1M), pH7,4 in 1L Aqua dest. lösen: 6,1g (bzw. 12,2g) Tris-(Trishydroxymethylaminomethan)-Base 3ml konzentrierte HCl = Salzsäure (37%ig) mit Salzsäure auf pH7,4 einstellen • Primär-Antikörper-Lösung (für 100ml) 90ml PBS-Puffer (0,1M, pH7,35) 10ml normales Ziegen-Serum (GIBCO, Grand Island, NY/USA) 0,3ml Triton X-100 (Sigma Chemicals, St. Louis, MO/USA) 1,25ml 8%iges Natriumazid (zur Haltbarmachung) dazu in entsprechender Verdünnungen (siehe 3.1.2.1) die Primär-AK-Seren • Sekundär-Antikörper-Lösung (für 100ml mit 1:300 Verdünnung) 90ml PBS-Puffer (0,1M, pH7,35) 10ml normales Ziegen-Serum (GIBCO, Grand Island, NY/USA) 0,33ml Peroxidase-konjugierte Ziegen-anti-Kaninchen- bzw. Ziegen-anti-Maus-IgG (Boehringer Mannheim, Indianapolis, IN/USA) • NADPH-Diaphorase-Färbe-Lösung 10ml Tris-HCl-Puffer (0,1M, pH7,4) 4mg NADPH (Sigma Chemicals, St. Louis, MO/USA) 10mg Nitroblau Tetrazoliumsalz (Sigma Chemicals, St. Louis, MO/USA) 0,08ml Triton X-100 • DAB-Färbe-Lösung 110ml Tris-HCl-Puffer (50mM, pH7,6) 70mg Natrium-Imidazol (Sigma Chemicals, St. Louis, MO/USA) 1mg DAB = 3,3‘-Diaminobenzidin (Sigma Chemicals, St. Louis, MO/USA) 4 Mikrotropfen H2O2= Wasserstoffperoxid (abgemessen per Insulinspritze) 25 3.2 Methoden 3.2.1 Gewebeaufbereitung 3.2.1.1 Immunhistochemie Für die immunhistochemische Färbung wird die indirekte Methode (siehe Abb. 3.1) mit Peroxidasekonjugiertem zweitem Antikörper (AK) verwendet (Polak et al., 1987). Alle folgenden Färbeschritte werden bei Raumtemperatur durchgeführt, sofern nicht explizit anderweitig erwähnt. Dazu werden die Hirnschnitte zunächst zur Unterdrückung gewebeständiger (=endogener) Peroxidase für 30 min in eine Methanol / 0,3% Wasserstoffperoxid-Lösung gegeben. Überschüssiges Wasserstoffperoxid wird durch dreimaliges zehnminütiges Waschen in PBS-Puffer eliminiert. Um unspezifische Bindungsstellen zu blockieren, erfolgt eine einstündige Vorinkubation in 10%igem normalem Ziegen-Serum. Anschließend werden die Gewebsschnitte freischwimmend mit Primär-AK-Lösung (Primär-Antikörper = EM48, Calbindin D28k, SMI32) bei Raumtemperatur für 12-18h inkubiert. Dabei enthalten alle Primär-AK-Lösungen 0,3% Triton X-100 (zur besseren Gewebepenetration durch Zerstören von Zellmembranlipiden), sowie 10%iges normales Ziegen-Serum (um eine unspezifische Hintergrundfärbung zu vermindern). Daran schließt sich wiederum dreimaliges zehnminütiges Waschen in PBS-Puffer an. Nachfolgend werden die Präparate für 3-5h mit Peroxidase-konjugierten Sekundär-AK (Ziegen-antiKaninchen-AK im Falle von EM48 bzw. Ziegen-anti-Maus-AK für Calbindin D28k und SMI32) inkubiert. Die Sekundär-AK werden dazu in einer Verdünnung von 1:300 verwendet. Bei diesem Schritt fungieren die Fc-Fragmente der Primär-AK als Antigen für die Enzym-konjugierten Sekundär-AK (siehe Abb. 3.1). 26 konjugierte Peroxidase Sekundärantikörper DAB-Färbereaktion Primärantikörper Durch Fixierung demaskierte Epitope Abb. 3.1 Indirekte immunhistochemische Färbemethode mit Peroxidase-konjugiertem Sekundärantikörper Zur Sichtbarmachung der gebundenen, Peroxidase-konjugierten Sekundär-AK werden die Gewebsschnitte in eine Lösung von DAB (3,3‘-Diaminobenzidin 1mg/ml) in Tris-HCl-Puffer (50mM, pH7,6) mit 0,005% Wasserstoffperoxid gegeben. Dabei reduziert die Peroxidase Wasserstoffperoxid zu freiem Sauerstoff, welcher wiederum das zunächst farblose DAB oxidiert und als braunes Polymer präzipitieren läßt. Durch vorheriges hinzufügen von 0,01M Imidazol kann die Peroxidase-Reaktion bei Bedarf intensiviert werden (Straus, 1982). Die gewünschte Färbeintensität wird nach 2-5min erreicht. Um die unspezifische Hintergrundfärbung, sowie die Spezifität der AK bzw. Antiseren zu beurteilen, werden einige Kontrollschnitte unter Weglassen der Primär-AK bzw. nach vorheriger Absorption jener mit bekanntem Antigen / Protein dem gleichen Färbeprozeß unterzogen. Dabei wird lediglich eine homogene, unspezifische und diffuse Hintergrundfärbung des gesamten Gewebsschnittes beobachtet. 3.2.1.2 Enzymhistochemie Um in Schnitten das gewebeständige Enzym NADPH-Diaphorase (NADPH-d) nachweisen zu können, wird bei ausgewählten Hirnschnitten auf eine vorige Inkubation mit Methanol / Wasserstoffperoxid verzichtet, um eine Enzym-Denaturierung zu verhindern. Zur Darstellung NADPH-d-haltiger Neuronen wird eine an den direkten NADPH-d-Enzm-Nachweis (Vincent et al., 1983) angelehnte Methode (Ferrante et al., 1987) verwendet. Dazu werden die Präparate frei schwimmend bei 37°C je nach gewünschter Färbeintensität zwischen 0,5 und 3 Stunden lang in einer Lsg. von 4mg NADPH, 10mg Nitroblau-Tetrazolium, 0,8% Triton X-100 in 10ml 0,1M Tris-HCl-Puffer (pH7,4) gefärbt. Bei dieser Methode wird das farblose Nitroblau- Tetrazoliumchlorid NADPH-abhängig reduziert und dadurch in das unlösliche blauviolette Formazan umgewandelt, welches dann präzipitiert. 27 Um die Spezifität der NADPH-d-Reaktion nachzuweisen, werden Kontrollschnitte nach Enzyminaktivierung (Denaturierung durch Überhitzung des Gewebes 60°C für 2h) gefärbt, sowie unversehrte Präparate lediglich alleine mit Nitroblau-Tetrazoliumsalz inkubiert. In beiden Fällen wurde keine Färbung beobachtet. 3.2.1.3 Doppelfärbetechnik Für die Doppelfärbung eines Präparates mit EM48-AK und Calbindin-D28k- bzw. SMI32-AK werden die gewünschten Hirnschnitte einer doppelten immunhistochemischen Färbung unterzogen. Dazu werden die Gewebeproben zuerst komplett für EM48 gefärbt (wie unter 3.2.1. beschrieben), dann dreimalig (x10min) in PBS-Puffer gewaschen, und anschließend einem kompletten zweiten immunhistochemischen Färbeprozeß unterzogen. Da EM48 nur eine geringe Hintergrundfärbung aufweist (siehe unten), gibt es hinsichtlich einem unspezifischen „Nachdunkeln“ des Präparates keine Probleme. Um eine Kombination von Immun- und Enzymhistochemie durchführen zu können, wird eine gewisse Hintergrundfärbung in Kauf genommen und an ausgewählten Gewebeschnitten zur Verhinderung einer Enzymdenaturierung auf eine Vorinkubation in Methanol / Wasserstoffperoxid verzichtet. Zuerst werden die Präparate nach der enzymhistochemischen Methode für den NADPH-d-Nachweis gefärbt (siehe 3.2.2), anschließend dreimalig (x10min) in PBS-Puffer gewaschen, und dann dem zweiten Färbeschritt, der Immunhistochemie für EM48, unterzogen. 3.2.1.4 Fertigstellung der Präparate Die Schnitte werden nach Abschluß der Färbeprozesse nochmals in PBS-Puffer gewaschen und anschließend in nassem Zustand auf Gelatine-beschichtete Objektträger aufgetragen. Nach Trocknen an freier Luft über Nacht werden den Präparaten mit ParaMount® als Medium Deckgläser aufgelegt und dadurch versiegelt. 3.2.2 Auswertung Die mikroskopische Auswertung der Hirnschnitte erfolgt mit einem Leitz-Lichtmikroskop. Benutzt werden Objektive mit Vergrößerungsfaktor 4x, 10x, 20x und 40x. Für Interferenzkontrastuntersuchungen nach Nomarski wird das Videomikroskop (MicroVideo Instruments, Avon, MA/USA) eingesetzt. Fotografische Aufnahmen werden mit einem Nikon Labophot-2 Mikroskop angefertigt. 28 Für die quantitative Auswertung werden pro MH-Fall, wann immer möglich, jeweils 10-15 Serienschnitte eingescannt (MicroVideo Instruments, Avon, MA/USA), und mit einer speziellen Histologiebildbearbeitungssoftware (NIH Image, W. Rasband, NIH, Bethesda, MD) ausgewertet. Von dem extrem seltenen MH-Gehirngewebe der frühen pathologischen Stadien 1 und 2 stehen teilweise lediglich 6-10 Serienschnitte zur Verfügung. Um die Anzahl an Htt-Aggregaten annähernd festzustellen, werden per Zufallsraster 2500µm² große Ausschnitte aus dem Zielgebiet (dorsales / ventrales Neostriatum bzw. Kortex Laminae V-VI) ausgewählt und anschließend alle EM48-immunreaktiven Aggregate ausgezählt. Zur Quantifikation und Beurteilung perikaryaler bzw. intranukleärer Htt-Aggregate wird das Vorhandensein von EM48-immunreaktiven Aggregaten innerhalb von immunhistochemisch bzw. enzymhistochemisch differenziert angefärbten Nervenzellsubpopulationen computerunterstützt geprüft und in Bezug zur absoluten Anzahl der jeweils untersuchten Nervenzellen ausgewertet. 29 4. Ergebnisse 4.1 EM48-Färbung 4.1.1 Darstellung und Charakterisierung von Huntingtinaggregaten Nach alleiniger immunhistochemischer Färbung der Gehirnschnitte mit EM48 beobachtet man makroskopisch, sowohl bei Kontrollen als auch bei MH-Präparaten, lediglich eine unspezifische, schwache Hintergrundfärbung. Doch schon unter geringer lichtmikroskopischer Vergrößerung werden in den MH-Präparaten zahlreiche intensiv markierte punkt- bis ovalförmige Strukturen unterschiedlicher Größe und Lokalisation sichtbar (Abb. 4.1). Es handelt sich hierbei um EM48immunreaktive Htt-Aggregate, welche zumindest teilweise aus pathologischen Htt-Fragmenten zusammengesetzt sind. Mit Hilfe der Nomarski-Optik läßt sich ein reliefartiger Bildkontrast herstellen, durch welchen die Konturen der Nervenzellkörper verdeutlicht werden können (Abb. 4.1 A). Dadurch läßt sich erkennen, daß sich der bei weitem größte Anteil an Htt-Aggregaten (>98%) außerhalb der Zellkörper befindet, und gleichzeitig auch nur sehr wenige Nervenzellen pathologische HttAggregationen enthalten. Noch deutlicher zeigt sich diese Beobachtung nach einer Kombinationsfärbung von EM48 mit SMI32 (Abb. 4.1 B). SMI32 markiert ein neuronales Filament, wodurch die einzelnen Strukturen wie Zellkörper, Zellkern und größere Dendriten bzw. Axone der Nervenzellen differenzierbar werden. Und auch hier bestätigt sich, daß die meisten Htt-Aggregate außerhalb der Nervenzellkörper lokalisiert sind, sowie daß nur sehr wenige Neuronen Htt-Aggregationen enthalten. Aggregate, die weder im Zellkern, noch im somalen Zytoplasma lokalisiert werden können, werden im Weiteren als Neuropilaggregate definiert. 30 A B 75µm 100µm Abb. 4.1 A,B A zeigt einen Ausschnitt aus dem EM48-gefärbten Insel-Kortex eines MH-Gehirnes (pathologisches Stadium 4). Man erkennt deutlich die intensiv dunkelbraun bis schwarz gefärbten punkt- bis ellipsoidförmigen Htt-Aggregate mit einer Größe von durchschnittlich 250µm. Gelegentlich werden auch tubuläre Formen beobachtet, die teilweise bis über mehrere 100µm Länge aufweisen. Durch Nomarski-Optik stellen sich die Zellkörper in A reliefartig dar. Dadurch kann man erkennen, daß sich die meisten Aggregate außerhalb der Zellkörper befinden, und außerdem nur wenige der Nervenzellen Htt-Aggregate in ihrem Soma enthalten (Pfeil). Diese Beobachtung wird durch eine kombinierte Färbung mit EM48 und SMI32 in B verdeutlicht. Auch hier zeigt sich, daß die meisten Aggregate außerhalb der Nervenzellkörper lokalisiert sind. 4.1.2 Topographische Verteilung der Huntingtinaggregate EM48-immunreaktive Htt-Aggregate sind heterogen über das gesamte Gehirn verteilt zu beobachten. Dabei befinden sich weitaus mehr dieser pathologischen Proteinkomplexe innerhalb der grauen Substanz als z.B. in Marklager und anderen nervenzellarmen Strukturen. Doch sind die Htt-Aggregate auch innerhalb einer definierten Hirnregion ungleich verteilt. So kann man selbst innerhalb eines einzigen Präparates, z.B. der Hirnrinde, direkt neben Bereichen mit scheinbar lokal angehäuften Aggregaten, Bereiche finden, die vermeintlich frei von Aggregaten sind. 31 In der Hirnrinde läßt sich keine eindeutige Relation zwischen verschiedenen Kortexarealen (frontal, temporal, parietal usw.) und unterschiedlicher Aggregatdichte feststellen. Einheitlich finden sich jedoch stets mehr Htt-Aggregate innerhalb der in der Tiefe liegenden Kortexschichten gegenüber mehr oberflächlich gelegenen Schichten. So sieht man auch in Abb. 4.2 deutlich mehr Htt-Aggregate in Laminae V und VI, gegenüber sehr viel weniger und kleineren Aggregaten in Lamina III. A 300µm B 125µm C 125µm Abb. 4.2 A-C Die Abbildungen zeigen ein EM48-immunhistochemisch gefärbtes Präparat vom Insel-Kortex eines fortgeschrittenen Huntington-Falles (pathologisches Stadium 3). In A erkennt man bei 4-facher lichtmikroskopischer Vergrößerung zahlreiche EM48-immunreaktive HttAggregationen, die sich vor allem in den tieferen kortikalen Schichten befinden. Bei 20facher Vergrößerung erkennt man nur wenige, meist vereinzelt vorkommende EM48immunreaktive Strukturen in Lamina III (B) gegenüber deutlich mehr pathologischen Proteinaggregaten in Lamina V-VI (C). 32 Im Neostriatum (Abb. 4.3) markiert EM48 signifikant (p≤0,001) weniger immunreaktive Htt-Aggregate als im Kortex. Auch hier beobachtet man eine ungleichmäßige Verteilung der Htt-Ablagerungen. So finden sich signifikant (p≤0,01) mehr Htt-Aggregate in ventralen als in dorsalen Bereichen des Nucleus caudatus. Dieser Verteilungsgradient bleibt über die verschiedenen pathologischen Stadien hinweg erhalten (Tab. 4.1). A B C D 125µm Abb. 4.3 A-D Die EM48-Immunhistochemie im dorsalen (A) und ventralen (C) Nucleus caudatus eines MH-Patienten im frühen Stadium (Stadium 1), sowie im dorsalen (B) und ventralen (D) Nucleus caudatus eines fortgeschrittenen Falles (Stadium 4) macht deutlich, daß sich unabhängig vom Stadium zum einen bedeutend weniger Htt-Aggregate im Neostriatum befinden als z.B. in tiefen Hirnrindenschichten (siehe zum Vergleich hierzu Abb. 4.1), und zum anderen innerhalb des Neostriatums ein Gradient mit mehr Aggregaten in ventralen und weniger Aggregaten in dorsalen Aspekten zu beobachten ist. In frühen Stadien sind HttAggregate im Striatum zahlreicher und deren durchschnittliche Größe geringer als in späten Stadien (siehe dazu Tab. 4.1). (Maßstab in A-D gleich) 33 Insgesamt sind im Neostriatum in frühen Stadien mehr Htt-Aggregate nachweisbar als in fortgeschritteneren Fällen, während dagegen im Kortex mit zunehmender Pathologie auch die Anzahl der Aggregationen anzusteigen scheint (Tab. 4.1). Unabhängig von der Hirnregion nimmt die Durchschnittsgröße der einzelnen immunreaktiven Proteinkomplexe mit Fortschreiten der Pathologie zu. So beträgt die Größe der Aggregate im Insellappenkortex im Stadium 1 durchschnittlich noch 10,8µm, im Stadium 4 hingegen durchschnittlich ca. 24µm. Kortex (insulär/temporal) Nucleus caudatus dorsal ventral MH Stadium 1 & 2 172 ± 10,9 12,6 ± 3,2 65 ± 6,2 MH Stadium 3 & 4 203 ± 12,1 3,1 ± 0.8 29,4 ± 5,6 Tab. 4.1 Die angegebenen Zahlen verstehen sich als die Anzahl immunreaktiver HttAggregate pro 2500µm² ± mittlere Standardabweichung. Dazu wurden jeweils nach einem Zufallsraster 2500µm²-große Felder ausgesucht, und die immunreaktiven HttAggregate innerhalb der Laminae V-VI von insulärem (n=76) bzw. temporalem Kortex (n=65), sowie in dorsalen (n=130) und ventralen (n=127) Bereichen des Nucleus caudatus auf Höhe des Nucleus accumbens ausgezählt. 4.1.3 Akkumulationen multipler kleiner Huntingtinaggregate Unter stärkerer lichtmikroskospischer Vergrößerung fällt das Augenmerk unter anderem auf Akkumulationen zahlreicher, kleiner punktförmiger EM48-immunreaktiver Aggregate (Abb. 4.4 A, B). Mit Hilfe von Nomarski-Optik kann man erkennen, daß sich diese Aggregatanhäufungen innerhalb von Zellkörpern befinden, und dabei vermeintlich den Zellkern aussparen. Das Zytoplasma einer einzigen Zelle kann so von einigen wenigen bis zu über fünfzig, durchschnittlich 0,1-1µm große Htt-Aggregate enthalten. Man beobachtet sie sowohl in striatalen als auch in kortikalen Bereichen. Deutlich mehr dieser intrazellulären Htt-Aggregatanhäufungen finden sich in frühen MH-Stadien (Stadium 1 bzw. 2), verglichen mit pathologisch fortgeschritteneren Fällen (Stadium 3 bzw. 4). 34 4.1.4 In Kontrollgehirnen sind keine Huntingtinaggregate nachweisbar In keinem der untersuchten Kontrollgehirne (siehe Tab. 3.1) werden oben beschriebene EM48immunreaktive Aggregate bzw. Strukturen beobachtet. Lediglich eine sehr schwache, diffuse Färbung neuronalen Zytoplasmas ist zu sehen. Interessanterweise werden bei neurologischen Kontrollpräparaten von Alzheimer-Gehirnen auch einige der sogenannten Alzheimer-Plaques durch die EM48-Immunhistochemie angefärbt. Die Anfärbung ist allerdings sehr unregelmäßig und bei weitem nicht so intensiv wie die der Htt-Aggregate. 35 4.2 NADPH-Diaphorase-Neuronen und EM48-Färbung 4.2.1 Huntingtinaggregationen in NADPH-d-positiven Nervenzellen Wie in anderen Studien schon berichtet wurde (siehe 1.4.1.3), beobachtet man in MH-Präparaten eine Zunahme der Dichte vermeintlich verschont bleibender NADPH-d-positiver Nervenzellen. Kombinierte NADPH-d-Enzymhistochemie und EM48-Färbung offenbart, daß die bei alleiniger EM48-Färbung gefundenen punktförmigen Htt-Aggregatanhäufungen (siehe 4.1.3) ausnahmslos in eben diesen NADPH-d-positiven Nervenzellen anzutreffen sind (Abb. 4.4 C-F). Dabei enthalten in frühen Stadien (pathologisches Stadium 1 und 2) annähernd alle kortikalen (>96%) und knapp die Hälfte (49,2%) der striatalen NADPH-d-Neuronen einzelne bis multiple perikaryal gelegene Htt-Aggregationen. Relativ selten hingegen (<2%) finden sich in frühen Stadien in den NADPH-d-positiven Zellen intranukleäre Htt-Einschlußkörperchen. Mit fortschreitender Pathologie nimmt die Häufigkeit der multiplen perikaryal gelegenen HttAggregationen deutlich ab, die Häufigkeit intranukleärer Aggregationen hingegen deutlich zu. So finden sich in MH-Präparaten der pathologischen Stadien 3 und 4 nur noch in knapp 60% aller kortikalen und 20% aller striataler Nervenzellen einzelne bis multiple perikaryal gelegenen HttAggregationen. Dagegen enthalten in diesen fortgeschrittenen Fällen durchschnittlich 6,3% aller NADPH-d-positiven Nervenzellen im Striatum und in 8% aller kortikalen NADPH-d-Zellen intranukleäre Einschlußkörperchen (Abb. 4.5). Beide untersuchten juvenilen MH-Fälle entsprechen diesbezüglich Gewebeproben vom pathologischen Stadium 4 (siehe Tab. 3.1). Hier werden bis auf gelegentliche einzelne, größere zytoplasmatische Htt-Aggregate keine dieser perikaryalen Htt-Aggregatanhäufungen in NADPH-dZellen beobachtet. Dagegen enthalten ca. 10-12% aller NADPH-d-Nervenzellen, sowohl im Striatum als auch im Kortex, intranukleäre Htt-Aggregate. 4.2.2 NADPH-d- und EM48-Kombinationsfärbung in Kontrollen In Kontroll-Präparaten mit kombinierter Färbung für NADPH-d und EM48 werden keine vermeintlichen immunreaktiven Aggregationen sichtbar. Zu differenzieren sind gelegentliche intensiv-dunkelblaue Formazankristalle als Artefakte der Enzymhistochemie, die sich jedoch eindeutig vom braunen DABFärbeendprodukt der Htt-Aggregate unterscheiden lassen. 36 A B C D E F 25µm Abb. 4.4 A-F In allein für EM48 immunhistochemisch gefärbten MH-Präparaten können sowohl im Kortex (A), als auch im Striatum (B) intrazelluläre Akkumulationen bzw. Anhäufungen zahlreicher, punktförmiger Htt-Aggregate beobachtet werden (Pfeile). Kombinierte EM48und NADPH-d-Enzym-Färbung zeigt, daß sich diese Aggregatakkumulationen ausnahmslos innerhalb NADPH-d-positiver Nervenzellen befinden. In frühen pathologischen Stadien enthalten fast alle kortikalen (C), sowie annähernd 50% aller striatalen (D) NADPH-d-Neurone einzelne bis multiple punktförmige, perikaryal gelegene Htt-Aggregate (Maßstab in A-D gleich). E und F zeigen Ausschnittsvergößerungen aus C resp. D. 37 A 50µm B 50µm C 25µm D 25µm Abb. 4.5 A-D Kombinierte EM48-Immunhistochemie und NADPH-d-Enzymhistochemie des Nucleus caudatus eines MH-Patienten im pathologischem Stadium 1 (A und C) und eines MHPatienten im pathologischem Stadium 4 (B und D). Beide NADPH-d-positive Neurone in A enthalten Anhäufungen zahlreicher punktförmiger Htt-Aggregate innerhalb des Perikaryon. Außerdem sieht man kleine und große Htt-Aggregate innerhalb des Neuropils (Pfeilspitzen). In frühen Stadien scheinen die Zellkerne jedoch meist frei von Htt-Aggregationen zu bleiben (C). In MH-Hirnschnitten der Stadien 3 und 4 beobachtet man signifikant weniger perikaryale Htt-Aggregationen, dagegen eine Tendenz zu mehr intranukleären EM48-immunreaktiven Aggregaten (B und D) (Pfeile). 38 4.3 Calbindin-Neuronen und EM48-Färbung 4.3.1 Calbindin-positive Nervenzellen enthalten nur selten Htt-Aggregate Durch Calbindin-D28k-Immunhistochemie wird eine repräsentativ vulnerable Nervenzellsubpopulation sichtbar gemacht. Wie zuvor berichtet, beobachtet man über die verschiedenen pathologischen Stadien hinweg einen progredienten Verlust Calbindin-positiver Neuronen, mit deutlich ausgeprägterer Degeneration im dorsalen als im ventralen Striatum. In den MH-Hirnschnitten werden nach alleiniger Calbindin-D28k-Färbung keine Htt-Aggregate bzw. aggregatähnlichen Strukturen beobachtet. Nach Kombinationsfärbung für Calbindin D28k und EM48 werden jedoch die Htt-Aggregate wie zuvor deutlich sichtbar (Abb. 4.6 und 4.7). Obwohl gleichermaßen für Calbindin D28k als auch für EM48 die immunhistochemische Sichtbarmachung durch die DAB-Färbereaktion erfolgt, lassen sich die EM48immunreaktiven Htt-Aggregate eindeutig durch ihre Morphologie und weitaus intensivere Immunreaktivität, und somit intensiveres Färbeverhalten, von der Calbindin-Immunreaktivität unterscheiden. Durch Manipulation der Lichtquelle, sowie durch unterschiedliches Fokussieren zum Erlangen eines räumlichen Eindruckes, lassen sich die Htt-Aggregate außerdem leicht dem Perikaryon bzw. dem Zellkern zuordnen. Auch unter Calbindin- und EM48-Doppelfärbung erkennt man, daß sich das Gros der Htt-Aggregate im Neuropil befindet; unabhängig davon ob im Striatum oder Kortex, sowie über alle pathologischen Stadien hinweg. In frühen Stadien des MH (pathologisches Stadium 1 und 2) enthalten weniger als 4% aller Calbindinpositiver Nervenzellen im Striatum intranukleäre oder perikaryale Htt-Aggregationen (Abb. 4.6 A+C). Bei genauer Betrachtung zeigt sich, daß jeweils ungefähr die Hälfte dieser Htt-Aggregationen im Zellkern bzw. im Perykaron lokalisiert sind. Somit enthalten also insgesamt nur weniger als 2% aller Calbindin-Neuronen intranukleäre Htt-Aggregate. Akkumulationen kleiner punktförmiger Htt-Aggregate wie sie in NADPH-d-Zellen gefunden wurden (siehe 4.2.1), werden in Calbindin-Neuronen nicht beobachtet. Um ein mögliches Übersehen kleinster Aggregationen in Calbindin-positiven Zellen auszuschließen, werden probeweise einige Präparate einer Doppelfärbung mit EM48/Hämatoxylin resp. EM48/Nissl-Färbung unterzogen. Durch die unterschiedlichen Farbqualitäten dieser Methoden, lassen sich die EM48-immunreaktiven HttAggregate noch leichter von zellulären Strukturen unterscheiden (hierzu keine Abbildung gezeigt). Anhand morphologischer Kriterien (vor allem Kernmembrancharakteristika) lassen sich ebenfalls, wenn auch mit bedeutend mehr Aufwand, bedornte von unbedornten Neuronen unterscheiden. Und auch in diesen Kontrolluntersuchungen bestätigt sich, daß in frühen Stadien weniger als 4% aller mittelgroßen bedornten Neurone, die bekanntermaßen zum großen Teil Calbindin-positiven Nervenzellen entsprechen, Htt-Aggregate enthalten, und weniger als die Hälfte davon sich in den Zellkernen befinden. In fortgeschritteneren Fällen (pathologisches Stadium 39 3 und 4) ist erwartungsgemäß der massive Verlust Calbindin-positiver Nervenzellen zu beobachten. Vor allem in dorsalen Bereichen des Nucleus caudatus finden sich nur noch äußerst spärlich übriggebliebene Calbindin-Neurone (Abb. 4.6 B+D). In diesen späteren Stadien werden nur noch äußerst selten intranukleäre bzw. perikaryale Htt-Aggregationen beobachtet. Eine weitere bemerkenswerte Beobachtung ist, daß in allen Präparaten zahlreiche Calbindin-haltige Nervenzellen mit eindeutig degenerativen Veränderungen zu finden sind, die weder perikaryale noch intranukleäre Htt-Aggregationen aufweisen. A B C D 50µm Abb. 4.6 A-D Kombinierte Färbung für EM48 und Calbindin D28k in dorsalem (A) und ventralem (C) Nucleus caudatus eines Stadium 1 MH-Gehirnes, und in dorsalem (B) und ventralem (D) Nucleus caudatus eines MH-Gehirnes im pathologischem Stadium 4. Sowohl in frühen als auch in späten Stadien der Degeneration finden sich in weniger als 4% der bedornten, Calbindin-positiven Striatumneurone EM48-immunreaktive Aggregate in deren Zellkörpern (Pfeile in C und D). Auch hier sieht man den Großteil der Htt-Aggregate im Neuropil lokalisiert (Pfeilspitzen in A-D). (Maßstab in A-D gleich) EM48-markierte Htt-Aggregate können leicht durch ihre Morphologie und ihr intensiveres DAB-Färbeverhalten von der Calbindin-Färbung unterschieden werden. 40 Innerhalb der Insellappen- bzw. Temporallappenhirnrinde enthalten nur sehr wenige Calbindin-positive Nervenzellen Htt-Aggregate (Abb. 4.7 A). Die meisten EM48-immunreatkiven Htt-Aggregate befinden sich auch hier, in frühen wie in späten Stadien im Neuropil. Gelegentlich werden sie dabei in einer linearen bzw. perlschnurartigen Anordnung (Abb. 4.7 B) angetroffen, die die Vermutung nahelegt, daß diese Aggregationen mit Nervenfasern bzw. Dendriten assoziiert sind. A 40µm B 40µm Abb. 4.7 A,B A und B zeigen eine doppelimmunhistochische Färbung für EM48 und Calbindin D28k innerhalb der Inselhirnrinde eines fortgeschrittenen (Stadium 4) MH-Gehirnes. Die HttAggregate sind von der Calbindin-Immunreaktivität morphologisch sowie durch ihr intensiveres DAB-Färbeprodukt leicht differenzierbar. Intranukleäre (Pfeil in A) sowie perikaryale Htt-Aggregate sind in Calbindin-positiven kortikalen Neuronen nur sehr selten zu beobachten. Aggregatakkumulationen wie in NADPH-d-Zellen (siehe Abb. 4.4) sind in ihnen nicht zu finden. Gelegentlich kann man EM48-immunoreaktive Htt-Aggregate linear aneinandergereiht beobachten (B), was die Vermutung nahelegt, daß diese Htt-Aggregate mit Dendritenstämmen bzw. Nervenfasern assoziiert sein könnten (siehe 4.4). 4.3.2 Calbindin- und EM48-Kombinationsfärbung in Kontrollen Weder in den neurologischen noch den normalen Kontrollgehirnen wurden nach kombinierter Färbung für Calbindin D28k und EM48 vermeintliche immunreaktive Aggregationen von Htt beobachtet. Das Calbindin-Färbeverhalten der mittelgroßen bedornten Neurone scheint jedoch im Vergleich zu MHPräparaten etwas homogener zu sein, was wahrscheinlich auf die bessere Integrität dieser hier nicht degenerativ veränderten Zellen zurückzuführen ist. 41 4.4 Kombinierte SMI32- und EM48-Färbung Um letztere Vermutung zu überprüfen, werden einige Präparate kombiniert für EM48 und SMI32 gefärbt. SMI32 markiert nicht-phosphoryliertes Neurofilament und färbt dadurch Zellkörper, Axone und Dendriten von Nervenzellen an, die beim MH bevorzugt degenerieren (siehe 3.1.2.1). In Abb. 4.8 sieht man linear angeordnete EM48-immunreaktive Htt-Aggregate die scheinbar in enger Beziehung zu Dendritenstämmen bzw. dendritischen Strukturen stehen. Obwohl lichtmikroskopisch kein eindeutiger Aufschluß über deren Lokalisation auf feinstruktureller Ebene möglich ist, vermittelt es den Eindruck, als ob sie sich innerhalb geschwollener Dendritendornen oder innerhalb von Axonendknöpfen befinden. A 40µm B 40µm Abb. 4.8 A,B A und B stammen aus dem Insellappenkortex eines MH-Patienten (Stadium 4). Die Präparate sind kombiniert immunhistochemisch für EM48 und SMI32 gefärbt. SMI32Antikörper erkennen dabei das Neurofilament von Nervenzellen, die beim MH besonders vulnerabel sind. Wie in beiden Abb. zu sehen ist, stehen die Htt-Aggregate, zumindest teilweise, in enger Beziehung zu dendritischen Strukturen. Möglicherweise sind sie innerhalb von Axonendknöpfen oder angeschwollenen Dendriten bzw. Dornen lokalisiert. 42 5. Diskussion 5.1 Kritische Beurteilung von Material und Methodik 5.1.1 Gewebeproben Für eine Studie dieser Art ist offensichtlich das Vorhandensein von ausreichend, gut erhaltenem postmortem-Gehirngewebe eine unabdingbare Voraussetzung. Dazu wurde Gehirngewebe aus drei verschiedenen neuropathologischen Zentren rekrutiert. Während Gewebeproben von fortgeschrittenen MH-Fällen (pathologische Stadien 3 bzw. 4) in uneingeschränkt ausreichender Menge vorhanden waren, konnten von MH-Fällen in frühen Stadien (pathologische Stadien 1 bzw. 2) teilweise nur weniger Gehirnschnitte untersucht werden, als wünschenswert gewesen wäre. Der Grund hierfür ist, daß besonders Gewebeproben von frühen pathologischen MH-Stadien äußerst rar sind. Das liegt daran, daß der MH zum einen generell eine relativ seltene Erkrankung ist; zum anderen erliegen Betroffene in präsymptomatischen Stadien bzw. Individuen mit Gehirnen mit noch nicht weit fortgeschrittener Neurodegeneration nicht direkt der Erkrankung, sondern versterben nur dann, wenn zusätzliche Begebenheiten, wie z.B. Unfall, Suizid, Begleiterkrankungen etc. hinzukommen. Weiterhin spielen post mortem oft logistische Probleme eine Rolle, die dazu führen, daß die Verstorbenen nicht adäquat einer Obduktion zugeführt werden. Das trifft insbesondere dann zu, wenn die MH-Patienten bzw. -Betroffenen außerhalb des Krankenhauses versterben. Aus mangelnder Aufklärung und Unwissen liegt zum Zeitpunkt des Todes oft auch keine Einwilligung des Patienten bzw. der Angehörigen für eine wissenschaftliche Untersuchung des Gehirnes vor. Stimmen die Angehörigen dann doch zu, so geht zumindest oft wertvolle Zeit bis zur Entnahme des Gehirnes verloren, durch die die Qualität der Gewebeproben stark beeinträchtigt wird. Nicht selten wird die Spende des Gehirns für die Forschung gänzlich abgelehnt. Um diesen Engpaß an essentiellem Untersuchungsmaterial zu beseitigen, bedarf es einer intensiven Aufklärung Betroffener und Angehöriger, sowie die Sicherstellung einer schnellen und für die Forschung adäquaten Entnahme der Gehirne vor allem auch präsymptomatischer bzw. ambulanter MH-Patienten, die außerhalb des Krankenhauses versterben. Denn gerade in frühen pathologischen Stadien, solange die Degeneration noch nicht allzu weit fortgeschritten ist, ist es wahrscheinlich noch am ehesten möglich, Erkenntnisse über die pathogenetischen Vorgänge herausfinden zu können. 5.1.2 Antikörper Diese Arbeit basiert auf dem Nachweis der pathologischen Htt-Aggregate in den Gewebeproben von MH-Patienten. Für die immunhistochemische Darstellung wurde hierfür das Antiserum EM48 43 verwendet, welches für uns in Kooperation mit Wissenschaftlern der Emory University, Atlanta/USA hergestellt wurde (siehe 3.1.2.1). Dies stellt jedoch vor allem hinsichtlich der Vergleichbarkeit mit anderen Studien ein Problem dar, da die unterschiedlichen Forschergruppen jeweils mit verschiedenen Antiseren bzw. Antikörpern arbeiten, die teilweise sehr unterschiedliche Charakteristika aufweisen. Diese Tatsache liegt darin begründet, daß für die Herstellung der Antikörper unterschiedliche DNA-Sequenzen bzw. Fusionsproteine verwendet wurden, die verschiedene Epitope des Huntingtins repräsentieren. So scheint das EM48-Antiserum im Vergleich zu den zuvor benutzten Antikörpern (Davies et al., 1997; DiFiglia et al., 1997; Becher et al., 1998) sensitiver Htt-Aggregationen beim MH zu erkennen und dadurch weitaus mehr und auch kleinere Htt-Aggregate zu markieren (Li und Li, 1998; Gutekunst et al., 1999). Außerdem werden auch die weitaus häufigeren im Neuropil lokalisierten Htt-Aggregate sichtbar, die bislang nur sehr wenig Beachtung gefunden haben. Wie unter 4.1.4 erwähnt, wurden mittels EM48-Immunhistochemie auch einige Alzheimer-Plaques angefärbt. Da die Färbung allerdings sehr unregelmäßig, relativ schwach und wenig einheitlich war, legt das die Vermutung nahe, daß es sich hierbei lediglich um ein unspezifisches Artefakt handelt. So könnten sich mutante Htt-Moleküle oder eventuell die EM48-Antikörper selbst in den fibrillären Strukturen der Alzheimer-Plaques verfangen, und somit für eine unspezifische Färbung verantwortlich sein. Trotzdem sollte man nicht ausschließen, daß Htt auch beim Morbus Alzheimer eine Rolle spielen könnte, und möglicherweise an der Ausbildung von Alzheimer-Plaques bzw. Fibrillen beteiligt ist. Weitere Experimente und die Herstellung neuer AK gegen verschiedene Epitope des Huntingtins werden es sicherlich ermöglichen, viele neue Erkenntnisse über die Verteilung, Funktion und potentielle pathogenetische, aber natürlich auch physiologische Bedeutung von aggregiertem bzw. nicht-aggregiertem Htt zu erlangen. Mit zunehmendem Wissen wird es dann auch möglich sein noch sensitivere bzw. spezifischere AK für den jeweils gewünschten Nachweis herzustellen. 5.1.3 Labortechniken Hinsichtlich Fixierungstechnik und immun- bzw. enzymhistochemischen Färbemethoden wurde versucht, nach den weithin gängigsten Laborprotokollen vorzugehen. Doch aufgrund mangelnder Standardisierung findet man bei näherer Betrachtung der Literatur je nach Autorengruppe teilweise deutliche Variationen innerhalb der Versuchsanordnung bzw. Laborprotokolle. So werden teilweise z.B. unterschiedliche Fixierungstechniken (Formalin oder Azeton in verschiedenen Konzentrationen), andere Puffersysteme (TBS, PBS, o.a.), abweichende physikalische Parameter (Temperatur, Osmolarität, pH-Wert etc.) sowie verschiedene Antikörper bzw. Antiseren in uneinheitlichen Konzentrationen eingesetzt. Um diese Variabilität einzuschränken und dadurch eine bessere Vergleichbarkeit von Ergebnissen verschiedener Forschergruppen zu erreichen, wünschenswert, wann immer möglich, eine Standardisierung der Methodik voranzutreiben. 44 wäre es 5.1.4 Auswahl der untersuchten Nervenzelltypen Es sind zahlreiche verschiedene neuroaktive Substanzen und entsprechende Nachweismöglichkeiten bekannt, durch die unterschiedliche Nervenzelltypen differenziert werden können. Wir entschieden uns in dieser Studie dafür, Calbindin-D28k-positive Neuronen repräsentativ für eine vulnerable Nervenzellsubpopulation zu untersuchen, und jene mit den als resistent geltenden NADPH-dNeuronen zu vergleichen. Die Entscheidung fiel auf diese zwei Nervenzelltypen, da sie beide zum einen beim MH sehr gut untersucht und beschrieben wurden (siehe 1.4.1.2-3), und zum anderen weil es sich dabei um jeweils relativ einfache, gängige Nachweisverfahren handelt, die sich gut mit der EM48-Immunhistochmie kombinieren ließen. Trotzdem gäbe es noch zahlreiche andere Subtypen mit unterschiedlicher bzw. gegensätzlicher Vulnerabilität (wie z.B. Enkephalin-haltige versus cholinerge Neuronen), die in weiteren Studien zu untersuchen sicherlich sehr interessant wären. 5.2 Interpretation der Ergebnisse 5.2.1 Topographische Verteilung von Htt-Aggregaten Die neurodegenerativen Veränderungen beim MH schreiten nach einem charakteristischen topographischen Muster voran. Ein besonders früher und der insgesamt stärkste Gewebs- und Nervenzellverlust ist im Striatum zu beobachten. Erste degenerative Veränderungen finden sich vor allem in dorso-medialen Bereichen des Striatums, um sich dann im Verlauf der Erkrankung immer mehr in ventro-lateraler Richtung auszuweiten (Vonsattel et al., 1998). Parallel dazu findet sich auch im Neokortex ein signifikanter Zellverlust, der jedoch weniger stark ausgeprägt ist und insgesamt variabler zu sein scheint (Cudkowicz und Kowall, 1990; Hersch und Ferrante, 1997). Diese Untersuchungen zeigen nun, daß sich in Gebieten bevorzugter Degeneration keineswegs mehr Htt-Aggregate befinden als in Gebieten relativer Aussparung. Stattdessen sind im Striatum nur außerordentlich wenige Htt-Aggregate zu beobachten, während deutlich mehr Htt-Aggregate im vergleichsweise weniger stark atrophierenden Neokortex zu finden sind. Und auch im Striatum selbst werden im überproportional stark degenerierenden dorsalen Anteil des Nucleus caudatus signifikant weniger Htt-Aggregate beobachtet, als in weniger stark betroffenen ventralen Bereichen (siehe Tab. 4.1). Diese Diskrepanz zwischen Regionen mit nur spärlichem Htt-Aggregataufkommen und starker Degeneration, sowie gleichzeitig das vermeintliche Verschontbleiben von Hirnarealen trotz hoher Aggregatdichte, sprechen gegen eine ausschlaggebende direkt wirkende Toxizität dieser HttAggregationen bzw. gegen einen direkten Zusammenhang zwischen dem Auftreten der Htt-Aggregate und dem Zelluntergang. 45 Für diese Überlegung muß jedoch auch der stattfindende Nervenzellverlust berücksichtigt werden. So kann es offensichtlich keine intraneuronalen Htt-Aggregate geben, wenn gar keine Nervenzellen mehr vorhanden sind. Ein einzelnes Präparat stellt gewissermaßen lediglich eine Momentaufnahme in einem langdauernden pathologischen Prozeß dar. So wäre es denkbar, daß im untersuchten Gewebe zu einem früheren Zeitpunkt einmal mehr intrazelluläre Htt-Aggregate vorhanden waren, aber durch eine konsekutive Nervenzelldegeneration die Nervenzelldichte abgenommen hat, und parallel dazu auch die Htt-Aggregate wieder verschwunden sind bzw. sich aufgelöst haben. So sind in weit fortgeschrittenen MH-Fällen gerade im dorsalen Striatum nur sehr wenige Htt-Aggregate zu beobachten; gleichzeitig ist in dieser Region aber auch der Nervenzellverlust so stark ausgeprägt, daß eine quantitative Korrelation zwischen Htt-Aggregaten und Neuronen nicht mehr sinnvoll erscheint. Dennoch läßt sich die Dissoziation zwischen regionalem Htt-Aggregataufkommen und Ausmaß der Nervenzelldegeneration nicht allein durch einen Nervenzellverlust erklären. Von entscheidender Bedeutung hierfür ist die Berücksichtigung des zeitlichen Verlaufs der Degeneration. Bei der Untersuchung von Gewebeproben präsymptomatischer Patienten bzw. von MH-Gehirnen in frühen pathologischen Stadien (Stadium 1-2) finden sich ausreichend, noch gut erhaltene Gewebsabschnitte mit annähernd normaler Nervenzelldichte. Je nach der topographischen Lage darf, unter der Annahme die Erkrankung ginge ihren normalen Weg, davon ausgegangen werden, daß diese Gewebsabschnitte im weiteren Verlauf der Erkrankung zunehmend degeneriert wären. Und gerade auch in diesen frühen Stadien, zu Beginn der Neurodegeneration zeigt sich, daß nur sehr wenige HttAggregate in den prädestiniert degenerierenden Regionen, wie z.B. im Striatum, anzutreffen sind. Zur gleichen Zeit sind in scheinbar verschont bleibenden Regionen teilweise außerordentlich viele HttAggregate zu finden. Ein weiterer Hinweis dafür, daß sich Htt-Aggregate im Verlauf des MH mit zunehmendem Nervenzellverlust nicht einfach auflösen bzw. verschwinden, ist die Beobachtung, daß mit Fortschreiten der Krankheit und der damit einhergehenden Degeneration, die Anzahl der HttAggregationen zumindest in der Hirnrinde ansteigt und gleichzeitig deren Durchschnittsgröße insgesamt zunimmt. So hat es also den Anschein, daß, wenn sich Htt-Aggregate einmal ausgebildet haben, diese dann im weiteren Verlauf der Degeneration größtenteils erhalten bleiben und zunehmend größer werden. Die topographische Verteilung der Htt-Aggregate läßt also nicht direkt auf den zu beobachtenden Nervenzellverlust schließen. So sind diese pathologischen Proteinaggregationen möglicherweise nur Merkmale oder unschuldige Nebenerscheinungen, die zumindest nicht auf direkte Weise ursächlich mit dem Nervenzelluntergang verknüpft bzw. für ihn verantwortlich sind. 46 5.2.2 Mögliche pathogenetische Bedeutung von Neuropil-Htt-Aggregaten Es hat sich bestätigt, daß der bei weitem größte Anteil an Htt-Aggregaten (>98%) außerhalb der Nervenzellkörper im Neuropil lokalisiert ist. Durch ultradünne Gewebsschnitte und elektronenmikroskopische Untersuchungen hat unsere Arbeitsgruppe schon an anderer Stelle zeigen können, daß sich diese Neuropil-Htt-Aggregate hauptsächlich intrazellulär innerhalb von dendritischen Strukturen befinden (Gutekunst et al., 1999). Durch Markierung von Neurofilamenten (siehe Abb. 4.8) erkennt man die enge Assoziation vieler dieser Htt-Aggregate mit Dendriten. Lichtmikroskopisch läßt sich nicht eindeutig feststellen, wo genau auf ultrastruktureller Ebene diese Htt-Aggregate lokalisiert sind. Doch macht es den Eindruck, daß sie sich innerhalb geschwollener Dendritendornen bzw. innerhalb von Axonendknöpfen befinden. Interessanterweise wurden für den MH schon zuvor markante degenerative wie proliferative Veränderungen der Dendriten im Kortex und Striatum beschrieben (Graveland et al., 1985; Ferrante et al., 1991). Im einzelnen handelte es sich dabei um fokale Dendritenanschwellungen, scheinbar abgeknickt bzw. rückwärts wachsende Dendritenstämme, sowie Veränderungen von Anzahl und Größe der Dornen. Das Aufspüren zahlreicher im Neuropil gelegener Htt-Aggregate legt nun die Vermutung nahe, daß diese Proteinaggregate für die Ausbildung der pathologischen Veränderungen der Dendriten zumindest mitverantwortlich sind. Wie wir durch Untersuchungen präsymptomatischer MH-Fälle zeigen konnten, bilden sich diese Neuropilaggregate schon sehr früh im Verlauf des MH aus. Viele der beobachteten Htt-Aggregate sind dabei so groß, daß mechanische Störungen im intrazellulären bzw. intradendritischen Stofftransport sehr wahrscheinlich sind, und es vermutlich noch vor dem eigentlichen Nervenzelluntergang zu funktionellen Störungen der betroffenen Nervenzellen kommt. Wie oben erläutert, sind im Striatum selbst nur wenige Htt-Aggregate lokalisiert, im Kortex dagegen relativ viele. Mit dem Wissen, daß das Striatum jedoch Zielgebiet zahlreicher, meist exzitatorischer, kortikostriataler Projektionen ist, wäre es denkbar, daß Htt-Aggregate im Kortex indirekt für eine striatale Pathologie verantwortlich sind. So könnten z.B. Neuropilaggregate im Kortex zu einem veränderten Erregungsmuster bzw. einer anomalen Aktivität kortikaler Nervenzellen und ihrer Efferenzen führen, und somit konsekutiv den striatalen Input verändern. Ein alterierter Input, wie z.B. eine erhöhte exzitatorische Aktivität, könnte dann im weiteren Verlauf über andere Pathomechanismen (siehe 5.4) zu einer striatalen Neurodegeneration führen. So gibt es unter anderem zahlreiche Hinweise und Experimente dafür, daß exzitotoxische Phänomene bei der Pathogenese des MH eine zentrale Rolle spielen könnten (siehe 5.4). 5.2.3 Intrasomale Htt-Aggregate sind nicht auf vulnerable Nervenzellsubpopulationen beschränkt Über alle pathologischen Stadien hinweg enthalten insgesamt nur relativ wenige Nervenzellen intranukleäre bzw. perikaryale Htt-Aggregationen. Um jedoch die pathogenetische Bedeutung dieser intrasomalen Htt-Aggregate weiter zu beurteilen, sollen verschiedene Nervenzellsubpopulationen, die beim MH unterschiedlich stark degenerieren, differenziert betrachtet werden. 47 Folgende Überlegung sollte in dieser Untersuchung im Speziellen überprüft werden: Sollten, wie in der gegenwärtigen Literatur häufig postuliert, die pathologischen intranukleären bzw. perikaryalen HttAggregate direkt toxisch wirken und für den Zelltod verantwortlich sein, dann sollte man erwarten können, diese Htt-Aggregate vor allem in jenen Nervenzellsubpopulationen anzutreffen, die dem Untergang geweiht sind; repräsentativ für einen vulnerablen Nervenzelltypen werden hierfür Calbindinpositive Neurone untersucht. Nervenzellen die beim MH jedoch der Degeneration trotzen können und vom Untergang relativ verschont bleiben, sollten entsprechend keine bzw. weniger Htt-Aggregate aufweisen; stellvertretend für einen solchen resistenten Nervenzelltypen werden NADPH-d-Neurone näher betrachtet. 5.2.3.1 Überraschend viele NADPH-d-Neurone enthalten Htt-Aggregate Es ist inzwischen gut belegt, daß NADPH-d-Neurone zu einer Nervenzellgruppe gehören, die beim MH vom degenerativen Prozeß relativ verschont bleibt. Entgegen der Erwartung finden sich aber scheinbar gerade in diesem Nervenzelltyp außerordentlich häufig intrasomale Htt-Aggregate. Vor allem in frühen pathologischen Stadien werden in fast allen kortikalen und in annähernd 50% aller striatalen NADPH-d-Neuronen Aggregationen pathologischen Huntingtins gefunden. Meist handelt es sich hierbei um multiple, kleinste, perikaryal gelegene Htt-Aggregate (Aggregatanhäufungen) unter Aussparung des Zellkerns. Relativ selten (<2%) sind zu diesem Zeitpunkt dagegen eindeutig intranukleäre Htt-Aggregate zu beobachten. Mit Fortschreiten der Erkrankung und zunehmender Pathologie nimmt die Anzahl perikaryaler Htt-Aggregationen deutlich ab, die Zahl intranukleärer Aggregate jedoch leicht zu. Mit dem Wissen, daß die absolute Anzahl an NADPH-d-Zellen im Verlauf des MH annähernd unverändert bleibt, spricht diese Beobachtung dafür, daß ein Großteil der Zellen mit zytoplasmatischen Htt-Aggregatanhäufungen überlebt und anscheinend einen Mechanismus zur Entfernung zumindest der zytoplasmatischen Aggregationen besitzt. Vor allem in späteren pathologischen Stadien werden zunehmend auch Htt-Aggregate, mit teils beträchtlicher Größe, in den Zellkernen beobachtet, ohne daß jedoch die betroffenen NADPH-d-Zellen besondere Zeichen der Degeneration aufweisen. Das zeigt, daß auch bei diesem Nervenzelltypen Htt-Fragmente in den Zellkern gelangen und sich intranukleäre Htt-Aggregate ausbilden können. Diese Beobachtungen sprechen somit dafür, daß zumindest bei diesem Nervenzelltyp von den intrasomalen Htt-Aggregationen per se keine maßgebliche Toxizität ausgeht. Vielmehr scheint es sogar so, daß möglicherweise gerade die Ausbildung von Htt-Aggregaten diese Nervenzellen vor der Degeneration bewahren kann. 48 5.2.3.2 Nur relativ wenige Calbindin-Neurone enthalten Htt-Aggregate Mittelgroße Calbindin-haltige Nervenzellen im Striatum zählen zu den Neuronen, die beim MH besonders früh Zeichen der Degeneration aufweisen und im Verlauf des pathologischen Prozesses insgesamt am stärksten vom Zelluntergang betroffen sind. In diesen Calbindin-positiven Nervenzellen werden allerdings weder in frühen noch in fortgeschrittenen Stadien auffallend viele intranukleäre HttAggregationen beobachtet. Vielmehr befinden sich in frühen pathologischen Stadien, prozentual gesehen, annähernd gleich häufig intranukleäre Htt-Aggregationen in vulnerablen CalbindinNervenzellen wie in resistenten NADPH-d-Neuronen. So werden zu Beginn des degenerativen Prozesses auch in Calbindin-positiven Striatumneuronen in nur weniger als 2% von ihnen intranukleäre Htt-Aggregate gefunden. Mit anderen Worten enthält diese prädestiniert degenerierende Nervenzellsubpopulation nicht mehr intranukleäre Htt-Aggregate als resistente NADPH-d-Neuronen. In fortgeschrittenen Stadien beobachtet man in Calbindin-Zellen prozentual gesehen noch seltener Zellkerneinschlußkörperchen als zu Beginn der Degeneration, wobei der massive Zellverlust dieses Nervenzelltyps eine quantitative Korrelation problematisch macht. Über alle pathologischen Stadien hinweg findet man in diesen Calbindin-Neuronen nur sehr selten perikaryale bzw. zytoplasmatische Htt-Aggregate, worin sie sich von NADPH-d-Neuronen unterscheiden. So sind auch die zuvor beschriebenen Anhäufungen von multiplen Htt-Aggregaten in ihnen überhaupt nicht anzutreffen. Diese Erkenntnisse legen die Vermutung nahe, daß für den Zelluntergang dieser speziell vulnerablen Nervenzellsubpopulation das Auftreten von perikaryalen bzw. intranukleären Htt-Aggregate nicht von entscheidender Bedeutung sein kann. Bestärkt wird diese Überlegung auch durch die Beobachtung, daß zahlreiche eindeutig degenerativ veränderte Nervenzellen scheinbar frei von intrasomalen HttAggregaten sind. 5.3 Zusammenfassung und Einordnung der Erkenntnisse in die vorhandene Literatur Aus den Ergebnissen dieser Untersuchungen an menschlichem post-mortem-Gehirngewebe lassen sich nochmals zusammenfassend für den MH folgende zentrale Aussagen ableiten: Das Auftreten von Htt-Aggregaten in einer bestimmten Hirnregion bedeutet nicht, daß diese Hirnregion im weiteren Verlauf des MH degenerieren wird. Gleichzeitig bleiben die Hirnareale, in denen keine bzw. nur wenige Htt-Aggregate zu beobachten sind, nicht von der Neurodegeneration verschont. Die topographische Verteilung von Htt-Aggregaten läßt also keinen Rückschluß über das Ausmaß der regional unterschiedlich starken Degeneration zu. Entsprechend geht, auf zellulärer Ebene betrachtet, beim MH scheinbar ein Großteil der Nervenzellen zu Grunde, ohne daß sich je in ihnen perikaryale bzw. intranukleäre Htt-Aggregationen ausgebildet hätten. Insbesondere die am stärksten degenerierende Nervenzellsubpopulation enthält nur sehr wenige Htt-Aggregationen. Zudem hat sich gezeigt, daß Nervenzellen, die intrasomale Htt-Aggregationen enthalten, nicht automatisch dem Untergang geweiht sind. So wurden gerade in resistenten Nervenzelltypen auffallend häufig Htt- 49 Aggregationen beobachtet. Das legt die Vermutung nahe, daß von den Htt-Aggregationen möglicherweise sogar eine zytoprotektive Wirkung ausgeht. 5.3.1 Intrazelluläre Proteinaggregationen wurden lange als toxisch angesehen Beim MH wurden erstmals 1997 intranukleäre Proteinaggregationen in Nervenzellen entdeckt und beschrieben (siehe 1.6.4). In den darauffolgenden Jahren fand man auch bei den meisten anderen Polyglutaminkrankheiten ähnliche, neuropathologisch charakteristische Aggregationen der mutanten Proteine innerhalb von Nervenzellen bzw. deren Zellkernen (Sieradzan und Mann, 2001). Die pathogenetische Bedeutung dieser Proteinaggregationen war und ist bislang jedoch noch weitgehend ungeklärt. Zunächst wurde mit deren Entdeckung allgemein vermutet, daß sie für den Nervenzelluntergang verantwortlich sind; so auch beim MH. Es wurde spekuliert, daß die Ausbildung von intranukleären Proteinablagerungen bzw. Aggregationen z.B. zu mechanischen Störungen im Stoffwechsel führen könnte, oder daß funktionell wichtige Proteine, wie z.B. Transkriptionsfaktoren sequestriert werden, und dadurch möglicherweise die Gentranskription entscheidend beeinflußt wird (Davies et al., 1997; DiFiglia et al., 1997; Ross, 1997). Erste in-vitro-Experimente diesbezüglich ergaben, daß sich mit abnehmender Gesamtgröße der Htt-Fragmente, sowie mit zunehmender Länge des Polyglutamintraktes, die Tendenz zur Ausbildung von Htt-Aggregaten erhöht und gleichzeitig damit die Überlebensfähigkeit der Zellen sinkt. Weiterhin konnte gezeigt werden, daß mit mutantem Htt transfizierte Zellen, die intranukleäre Htt-Aggregate enthalten, weniger überlebensfähig sind und empfindlicher auf Apoptose-induzierende Reize („apoptotic stress“) reagierten, als Zellen ohne Aggregationen (Martindale et al., 1998; Cooper et al., 1998; Hackam et al., 1998). Trotz dieser zunächst vermeintlich eindeutigen Erkenntnisse wurde schon früh der ursächliche Zusammenhang zwischen Htt-Aggregation und Nervenzelltod in Frage gestellt. So stellte Ordway et al. (1997) die provokative Überlegung an, daß die Htt-Aggregate für den Zelltod nicht verantwortlich sind, sondern unter Umständen einen Mechanismus der Zelle darstellen mutantes Htt zu sequestrieren, damit dessen toxische Wirkung zu blockieren, und somit sogar eventuell zytoprotektiv wirken. Neuere Untersuchungen sowohl zum MH als auch zu anderen Polyglutaminkrankheiten scheinen diese These zu bekräftigen. 5.3.2 Neuere in-vitro-Untersuchungen Bemerkenswerte Erkenntnisse dazu erlangten Saudou et al. (1998) anhand von in-vitroUntersuchungen. Verschiedene Nervenzelltypen wurden mit unterschiedlichen Htt-Genkonstrukten transfiziert. Die Expression eines Htt-Fragmentes mit verlängerter Polyglutaminsequenz resultierte erwartungsgemäß in der Ausbildung von intranukleären sowie zytoplasmatischen Htt-Aggregaten. Doch in dieser Versuchsanordnung war das Vorhandensein von Htt-Aggregaten für die Zellen nicht 50 gleichbedeutend mit Neurodegeneration bzw. Zelltod. Bestimmte Nervenzelltypen blieben von den HttAggregationen scheinbar unbeeinflußt, während andere, insbesondere striatale Projektionsneurone, verstärkt zu Grunde gingen. Weiterhin stellte sich heraus, daß durch Ausschleusen des mutanten Huntingtins aus dem Zellkern (mit Hilfe eines „nuclear export signal“), dessen neurotoxische Potenz vollkommen verloren geht. Durch Transfektion mit verschieden langen N-terminalen Htt-Fragmenten konnte außerdem gezeigt werden, daß mit abnehmender Fragmentlänge die Anzahl an HttAggregaten in den Nervenzellen zwar ansteigt, die Absterberate der Nervenzellen davon jedoch unbeeinflußt bleibt. Mit dem Wissen darum, daß Ubiquitin ein Bestandteil der Htt-Aggregate ist (siehe 1.6.4) und möglicherweise zentral an der Formation von Aggregaten beteiligt ist, untersuchten Saudou et al. (1998) zusätzlich was geschieht, wenn das Ubiquitin-konjugierende Enzym blockiert wird. Tatsächlich konnte dadurch die Ausbildung von Htt-Aggregaten massiv reduziert werden; doch überraschenderweise stieg gleichzeitig die Absterberate in diesen Zellkulturen deutlich an. Diese in-vitro-Beobachtungen weisen somit recht eindeutig darauf hin, daß mutantes Htt vor allem im Zellkern toxisch wirkt und dort vermutlich apoptotische Prozesse in Gang setzt. Der Zelltod scheint jedoch von der Ausbildung von Htt-Aggregaten unabhängig zu sein; vielmehr hat es den Anschein, daß das Unterbinden von Aggregationen mit einer erhöhten Toxizität assoziiert ist. Ungeklärt bleibt die Tatsache, warum bestimmte Nervenzelltypen anfälliger zu sein scheinen als andere. 5.3.3 Möglicherweise ist vor allem nicht-aggregiertes Huntingtin im Zellkern toxisch Schon zuvor wurde vermutet, daß das mutante Polyglutamin-haltige Htt erst nach Eintritt in den Zellkern toxisch wirkt (Davies et al., 1997; Ross, 1997; DiFiglia et al., 1997; Ordway et al., 1997). Wie oben ausgeführt, zytoplasmatische, wurden perikaryal in NADPH-d-Neuronen gelegene in frühen Htt-Aggregationen Stadien angetroffen, zunächst und erst vor allem spät im neurodegenerativen Prozeß vereinzelt auch intranukleäre Einschlußkörperchen. Ein Grund dafür, daß viele NADPH-d-Neuronen vor allem zytoplasmatische Aggregate ausbilden, könnte z.B. darin liegen, daß dieser Nervenzelltyp über andere bzw. weniger aktive Mechanismen der proteolytischen Spaltung von Htt verfügt. Durch Aggregation der deshalb hypothetisch relativ gering anfallenden Menge von HttFragmenten im Zytoplasma, könnten toxisch wirkende Moleküle unter Umständen ausreichend neutralisiert bzw. aus dem Verkehr gezogen werden, noch ehe sie in den Zellkern gelangen und dort ihr Unheil anrichten könnten. Mit solch einer Überlegung ließe sich z.B. auch erklären, warum gerade dieser Nervenzelltyp trotz bzw. gerade wegen der zahlreichen zytoplasmatischen Htt-Aggregationen von der Neurodegeneration verschont bleibt. So spekulierte auch Sisodia (1998), daß die unterschiedliche Vulnerabilität der einzelnen Nervenzelltypen möglicherweise nicht von den HttAggregationen abhängt, sondern von der Menge des nicht-aggregierten Huntingtins innerhalb der Zellkerne. 51 Viele Hinweise sprechen dafür, daß mutantes Htt erst nach einer proteolytischen Spaltung in den Zellkern gelangen, und damit erst toxisch werden kann. Es konnte gezeigt werden, daß intranukleäre Htt-Aggregate hauptsächlich gespaltene N-terminale Htt-Fragmente enthalten, während zytoplasmatische Aggregate sowohl aus Htt-Fragmenten als auch aus intakten Htt-Molekülen bestehen (Kim et al., 1999). Es wurde vermutet, daß für die enzymatische Spaltung mutanten Huntingtins, und somit folglich für die Ausbildung von Htt-Aggregaten, Caspasen verantwortlich sind (siehe 1.6.3; Goldberg et al., 1996; Wellington et al., 1998). Caspasen sind Proteasen, die vor allem in Zusammenhang mit apoptotischen Prozessen gebracht werden konnten. Durch Blockieren von Caspasen mit verschiedenen Inhibitoren konnte in vitro an geklonten striatalen Nervenzellen eindeutig eine differenzierte Dissoziation zwischen der Ausbildung von Aggregaten und dem neuronalem Zelltod demonstriert werden (Kim et al., 1999). Außerdem gibt es auch Anzeichen dafür, daß intrazelluläre Einschlußkörperchen bzw. Proteinablagerungen möglicherweise lediglich das Resultat einer Überlastung der normalen Proteindegradation darstellen (Johnston et al., 1998). Die Aggregate sind demnach vielleicht nur eine unspezifische Nebenerscheinung, die mit den pathogenetisch bedeutsamen Vorgängen nichts zu tun hat. 5.3.4 Huntingtinaggregate per se sind wahrscheinlich nicht toxisch – vermutlich sind andere Faktoren für die Neurodegeneration verantwortlich Wie oben (siehe 1.7) kurz erläutert, weisen die diversen CAG-Repeat- bzw. Polyglutaminkrankheiten zahlreiche Parallelen auf. Für die meisten unter ihnen konnten, entsprechend dem MH, pathologische intrazelluläre Proteinaggregationen gefunden werden; so auch bei fast allen bekannten Spinozerebellären Ataxien (SCA). Eine Ausnahme hiervon stellt dabei lediglich die SCA vom Typ 2 dar. Hier geht die Neurodegeneration vonstatten, ohne daß pathologische Proteinaggregationen gefunden werden konnten (Huynh et al., 1999). Unter der Annahme, daß die verschiedenen Polyglutaminkrankheiten eine gemeinsame pathogenetische Ursache haben, legt diese Beobachtung jedoch die Vermutung nahe, daß die Proteinaggregationen an sich nicht der eigentliche Auslöser für den Nervenzelluntergang sein können, sondern möglicherweise lediglich ein sekundäres Phänomen darstellen. Wie aus der Einleitung und der Diskussion ersichtlich wird, gehen die wissenschaftlichen Meinungen und Hypothesen über die pathogenetische Bedeutung von zytoplasmatischen bzw. intranukleären Proteinaggregationen beim MH, wie auch bei zahlreichen anderen Polyglutaminkrankheiten, deutlich auseinander. Trotzdem scheint sich zunehmend die Vermutung zu erhärten, daß intrazelluläre Einschlußkörperchen nicht ausschlaggebend für den Nervenzelluntergang verantwortlich sind, sondern ganz im Gegenteil von ihnen möglicherweise eine zytoprotektive Wirkung ausgeht. Die meisten Erkenntnisse hierzu stammen aus neueren in-vitro-Untersuchungen und aus transgenen Tiermodellen. Die vorliegende Arbeit steht mit diesen neueren Erkenntnissen im Einklang und ergänzt diese Hypothese durch Untersuchungen von post-mortem-Gehirngewebe von MH-Patienten. 52 Doch bleibt somit weiterhin die Frage offen, was die eigentliche Ursache für die Neurodegeneration beim MH ist. Wie oben diskutiert, mehrt sich der Verdacht, daß möglicherweise gerade das nichtaggregierte Htt von größerer pathogenetischer Bedeutung ist, als die Aggregationen an sich. So werden zukünftige Studien sicherlich vermehrt auch das Augenmerk auf die Untersuchung von freien, nicht-aggregierten Htt-Molekülen bzw. Htt-Fragmenten richten. 5.4 Andere wichtige potentielle Pathomechanismen Der MH steht seit geraumer Zeit im Mittelpunkt zahlreicher intensiver Forschungsbemühungen. Lange noch vor Identifikation des Htt-Gens sind aus unterschiedlichsten Experimenten und Beobachtungen Hinweise und Vermutungen für verschiedene potentielle Pathomechanismen hervorgegangen. Ohne direkte Implikation mit dieser Studie, soll hier der Vollständigkeit halber eine der vermeintlich bedeutendsten Hypothesen, nämlich die mögliche Rolle der Exzitotoxizität kurz angesprochen werden. Das Striatum erhält zahlreiche exzitatorische Afferenzen aus fast dem gesamten zerebralen Kortex. Es konnte gezeigt werden, daß exzitatorisch wirkende Substanzen bzw. Aminosäuren (wie z.B. Glutamat), sowie bestimmte Analoga davon auf Neuronen toxisch wirken und deren Zelltod initiieren können. Dieses Phänomen wird „Exzitotoxizität“ genannt. Durch intrastriatale Injektion des Glutamatagonisten Kaininsäure erzeugten McGeer und McGeer (1976), sowie Coyle und Schwarcz (1976) erstmals in einem Tiermodell (Ratten) Läsionen im Gehirn, die an den MH erinnern ließen. In den darauffolgenden Jahren wurden zahlreiche andere exzitatorisch wirkende Substanzen untersucht. Es stellte sich zunehmend heraus, daß durch Stimulation eines bestimmten Glutamatrezeptorsubtypen, nämlich des N-methyl-D-Aspartat- (NMDA)-Rezeptors, in Ratten wie auch in Affen eine für den MH noch ähnlichere charakteristische Neuropathologie mit vergleichbarem, selektivem Nervenzellverlust induziert werden kann (Beal et al., 1986; 1989). Auch post-mortemUntersuchungen an Gehirnen von MH-Patienten deuteten darauf hin, daß an der Pathogenese auch exzitotoxische Prozesse eine Rolle spielen könnten. So wurde z.B. festgestellt, daß bevorzugt gerade jene Nervenzellen zu Grunde gehen, die normalerweise NMDA-Rezeptoren besitzen (Young et al., 1988; Arzberger et al., 1997). Doch wie es im MH-Gehirn zu einer NMDA-Rezeptor-vermittelten Toxizität kommen könnte, ist unklar. Denkbar wäre z.B., daß durch einen fehlerhaften Abtransport oder eine ineffektive Inaktivierung exzitatorischer Aminosäuren, die Rezeptoren einer verlängerten Stimulation ausgesetzt sind; eine andere Möglichkeit wäre, daß die Rezeptoren selbst überempfindlich auf äußere Reize reagieren, oder daß sie eventuell auch in ihrer Anzahl verändert sind. Eine daraus folgende Überstimulation der Rezeptoren könnte einen veränderten Ioneneinstrom nach sich ziehen, der im weiteren durch eine gestörte Kalziumhomöostase bzw. einen gestörten Energiehaushalt im Nervenzelluntergang enden könnte (Whetsell, 1996). Eine interessante mögliche Verbindung zwischen dem mutantem Htt und einer NMDA-Rezeptor-vermittelten Toxizität stellten kürzlich Sun et al. (2001) her. Sie fanden Hinweise dafür, daß normales Htt indirekt die Aktivität von NMDA53 Rezeptoren beeinflussen kann, indem es mit einem die Rezeptoren modulierenden Protein (PSD-95) eine Bindung eingeht. Durch eine verlängerte Polyglutaminsequenz im Htt verändert sich das Bindungsverhalten mit jenem PSD-95 derart, daß konsekutiv NMDA-Rezeptoren empfindlicher auf exzitatorische Reize reagieren, und somit die Glutamat-vermittelte Exzitotoxizität erhöht wird. Verstärkte Expression von normalem Htt hingegen vermindert deutlich die NMDA-Rezeptor-abhängige Exzitotoxizität. So könnte demnach das normale Htt beim Gesunden eine Rolle bei der Regulation bzw. Modulation von bestimmten Nervenzellrezeptoren spielen und eine Überstimulation bestimmter Neuronen verhindern. Durch eine verlängerte Polyglutaminsequenz innerhalb des Proteins, könnte das mutante Htt dieser Aufgabe möglicherweise nicht mehr gerecht werden. Es gibt auch Hinweise dafür, daß primär Störungen im Energiehaushalt einer langsamen, exzitotoxischen Nervenzelldegeneration zu Grunde liegen könnten (Albin und Greenamyre, 1992). So wurde gezeigt, daß durch eine chemisch induzierte Störung in der Energiebereitstellung bzw. durch Inhibition der Adenosintriphosphat- (ATP)-Produktion Nervenzellen sensibler auf exzitotoxische Reize reagieren bzw. anfälliger sind und eher absterben. Diese erhöhte exzitotoxische Vulnerabilität ließ sich durch Applikation von NMDA-Antagonisten blockieren (Zeevalk und Nicklas, 1990; Beal et al., 1993). Interessanterweise konnten auch beim Menschen in vivo, vor allem durch radiologische und nuklearmedizinische Methoden (Magnetresonanzspektroskopie, Positronenemissionstomographie) Anzeichen für einen frühen striatalen Hypometabolismus bzw. Störungen im Energiehaushalt gefunden werden (Kuwert et al., 1990; Jenkins et al., 1993). (Für eine Übersicht über die diversen Hypothesen einer exzitotoxischen und metabolischen Pathogenese siehe Massieu und Garcia, 1998). 5.5 Ausblick Trotz gewaltiger Fortschritte in der MH-Forschung, ist es bis heute nicht gelungen die pathophysiologischen Zusammenhänge, die für die Neurodegeneration verantwortlich sind, endgültig zu klären. Ein entscheidender Durchbruch war sicherlich die Identifikation des genetischen Defektes 1993 und die daraus folgende Entdeckung des Htt-Proteins. Durch diese Errungenschaft war es nunmehr nicht nur möglich einen direkten Gentest zu entwickeln, sondern auch grundlegend neuartige, experimentelle Forschungsansätze zu entwerfen. Doch trotz zahlreicher neuer histologischer, histochemischer Untersuchungsmöglichkeiten, Herstellung verschiedener transgener Tiermodelle und in-vitro-Versuchsanordnungen seither, sind wir noch immer weit davon entfernt, die Pathogenese des MH im einzelnen zu verstehen. Für die Klinik bedeutet das, daß man heutzutage durch einen direkten Gentest die Diagnose MH bzw. die Anlage dafür eindeutig und ohne großen, methodischen Aufwand feststellen kann. Doch gibt es bislang keine wirklich wirksame, kausale Therapie, die in der Lage ist, dem Nervenzelluntergang Einhalt zu gebieten oder ihn entscheidend zu verzögern. So steht auch heute noch, zumindest 54 außerhalb von experimentellen Studien, die symptomatische und supportive Behandlung im Vordergrund (Übersicht, siehe Marshall und Shoulson, 1997). Nicht zuletzt auf diesem Hintergrund ist die Durchführung eines solchen Gentests, speziell auch bei genetisch vorbelasteten aber noch gesunden Individuen, aus ethischen und psychologischen Gründen äußerst kritisch zu bewerten und anzuwenden (Epplen und Przuntek, 1998). Mit zunehmender Aufklärung und dem Verstehen der fatalen, pathogenetischen Vorgänge beim MH, eröffnen sich immer neue Ansätze und potentielle Möglichkeiten die Krankheit eines Tages wirksam therapieren zu können. Vor allem auch im Hinblick auf die Gentherapie eröffnen sich ganz neue Perspektiven. So wird es vielleicht einmal möglich sein, das defekte Gen zu reparieren, auszutauschen bzw. zu neutralisieren, und dadurch die Krankheit komplett aufzuhalten oder gar zu heilen. Doch bis zu diesem Schritt ist es sicherlich noch ein weiter Weg. Eine interessante Studie im Hinblick auf eine generelle Therapierbarkeit soll hier noch kurz erwähnt werden. Yamamoto et al. (2000) erzeugten ein transgenes Mausmodell, in welchem das Exon1 des Htt-Gens mit einer verlängerten Polyglutaminsequenz in das Mausgenom eingeschleust wurde. Zusätzlich wurde auch ein Regulatorgen miteingeschleust, durch welches es nun möglich war, durch Zufütterung einer bestimmten Substanz zu einem beliebigen Zeitpunkt das Gen gewissermaßen auszuschalten, und somit die Htt-Genexpression zu unterbinden. Entsprechend anderen Tiermodellen (siehe 1.6.4), entwickelte auch diese transgene Mauslinie einen dem MH ähnelnden progredienten neurologischen Phänotyp mit Ausbildung von pathologischen intrazellulären Proteinaggregaten. Wurde nun zu einem bestimmten Zeitpunkt, zu dem sich bei den Mäusen schon charakteristische Symptome, neurologische Merkmale, sowie auch intrazelluläre Aggregationen ausgebildet hatten, das Gen ausgeschaltet, konnte der Krankheitsprozeß gestoppt und zu einem großen Teil sogar rückgängig gemacht werden. So bildeten sich die motorischen Störungen fast vollständig zurück. Gleichzeitig verschwanden auch die intranukleären und zytoplasmatischen Proteinaggregationen wieder. Dieses Tiermodell legt somit die Vermutung nahe, daß Nervenzellen beim MH nicht automatisch dem Untergang geweiht sind, sondern erst durch den anhaltenden Einfluß des mutanten Proteins zu Grunde gehen. Außerdem scheint es, daß zumindest die frühen Symptome wahrscheinlich nicht auf einen Nervenzelltod, sondern ersteinmal auf funktionelle Störungen zurückzuführen sind, und somit auch potentiell reversibel sind. Sollte es nun therapeutisch gelingen auch beim Menschen die Expression des mutanten Proteins zu unterbinden bzw. zu reduzieren, oder entscheidend in dessen pathogenetische Vorgänge einzugreifen, dann ließe sich die Krankheit eventuell nicht nur verzögern, sondern in den frühen Stadien möglicherweise sogar heilen (Bates, 2000). 55 6. Zusammenfassung Die pathogenetische Bedeutung von intranukleären bzw. zytoplasmatischen Htt-Aggregationen beim MH ist bislang weitgehend unklar. Erste Berichte und Interpretationen hierzu stammten vor allem von Beobachtungen aus in-vitro-Experimenten und von transgenen Tiermodellen, ohne daß jedoch eindeutig ein direkter Zusammenhang zwischen den pathologischen Proteinaggregationen und der Neurodegeneration hergestellt werden konnte. Vor allem jüngere in-vitro-Untersuchungen deuten darauf hin, daß die Aggregationen an sich wahrscheinlich nicht ausschlaggebend für den Nervenzelluntergang verantwortlich sind. Die hier vorliegende Arbeit versucht nun, ergänzend zu den Erkenntnissen aus in-vitro-Experimenten und von transgenen Tiermodellen anderer Forschergruppen, die pathogenetische Bedeutung von HttAggregationen beim MH auch in menschlichem post-mortem-Gewebe zu untersuchen. Hierfür wurden Gewebeproben von 12 unterschiedlichen MH-Gehirnen, von 4 neuropathologisch unauffälligen, sowie von 6 neuropathologisch vorbelasteten Kontrollgehirnen untersucht. Im einzelnen sollte das Vorkommen und die Verteilung von Htt-Aggregaten in verschiedenen Hirnregionen, sowie innerhalb verschiedener Nervenzellsubpopulationen beurteilt und dokumentiert werden, um damit eine mögliche Korrelation zwischen Htt-Aggregatausbildung und Degeneration bzw. Zelltod zu überprüfen. Dazu wurden von den Gewebeproben Gefrierschnitte angefertigt, die anschließend mit Hilfe kombinierter immun- und enzymhistochemischer Färbemethoden aufbereitet und mikroskopisch ausgewertet wurden. Zur Darstellung der Htt-Aggregationen wurde hierzu EM48, ein polyklonales Antiserum gegen N-terminales Htt verwendet. Durch dieses Antiserum wurden deutlich mehr HttAggregationen sichtbar, als in anderen Publikationen zuvor beschrieben wurde. Das Gros dieser Aggregationen ist dabei außerhalb der Nervenzellkörper im Neuropil lokalisiert anzutreffen. Außerdem konnte eine deutliche Dissoziation zwischen bevorzugt degenerierenden Hirnregionen und Regionen hoher Aggregatdichte aufgezeigt werden. Durch kombinierte Doppelfärbung wurde weiterhin festgestellt, daß Htt-Aggregationen auch auf zellulärer Ebene nur relativ selten in den bevorzugt degenerierenden Nervenzellsubpopulationen anzutreffen sind, dagegen in den als resistent geltenden Nervenzelltypen überdurchschnittlich häufig gefunden werden können. Schlußfolgernd weisen die Ergebnisse dieser histologischen Untersuchung menschlichen postmortem-Gewebes darauf hin, daß die pathologischen intranukleären bzw. perikaryalen Proteinaggregationen nicht direkt für die Neurodegeneration verantwortlich zu machen sind. Vielmehr hat es sogar den Anschein, daß die Ausbildung von Aggregaten die betroffenen Nervenzellen vor dem Zelltod schützen können. In der Diskussion wird erläutert, daß diese Schlußfolgerung durchaus im Einklang mit diversen aktuellen Forschungsergebnissen und Publikationen zu sehen ist. Doch kritisch betrachtet ist festzustellen, daß die tatsächliche pathogenetische Bedeutung der intrazellulären Proteinaggregationen noch immer weitgehend ungeklärt ist. Weiterführende Untersuchungen 56 menschlicher Gewebeproben, die Entwicklung neuer transgener Tiermodelle, sowie in-vitroVersuchsanordnungen werden jedoch in Zukunft sicherlich noch mehr Aufschluß darüber geben. 57 7. Literaturverzeichnis 1. Adams RD, Victor M, Ropper AH (Eds.). Degenerative Diseases of the Nervous System, th Huntington Chorea. In: „Principles of Neurology“, McGraw-Hill, New York, 1997, 6 edition: 75,1060-1064 2. Albin RL, Reiner A, Anderson KD, Penney JB, Young AB. Striatal and nigral neuron subpopulations in rigid Huntington's disease: implications for the functional anatomy of chorea and rigidity-akinesia. Ann Neurol. 1990; 27:357-65. 3. Albin RL, Greenamyre JT. Alternative excitotoxic hypotheses. Neurology. 1992; 42:733-8. 4. Ambrose CM, Duyao MP, Barnes G, Bates GP, Lin CS, Srinidhi J, Baxendale S, Hummerich H, Lehrach H, Altherr M, Wasmuth J, Buckler A, Church D, Housman D, Berks M, Micklem G, Durbin R, Dodge A, Read A, Gusella J, MacDonald ME. Structure and expression of the Huntington's disease gene: evidence against simple inactivation due to an expanded CAG repeat. Somat Cell Mol Genet. 1994; 20:27-38. 5. Arzberger T, Krampfl K, Leimgruber S, Weindl A. Changes of NMDA receptor subunit (NR1, NR2B) and glutamate transporter (GLT1) mRNA expression in Huntington's disease--an in situ hybridization study. J Neuropathol Exp Neurol. 1997; 56:440-54. 6. Bates G. Huntington's disease. In reverse gear. Nature. 2000; 404(6781):944-5. 7. Beal MF, Martin JB. Neuropeptides in neurological disease. Ann Neurol. 1986; 20:547-65. 8. Beal MF, Kowall NW, Ellison DW, Mazurek MF, Swartz KJ, Martin JB. Replication of the neurochemical characteristics of Huntington's disease by quinolinic acid. Nature. 1986; 321(6066):168-71. 9. Beal MF, Mazurek MF, Ellison DW, Swartz KJ, McGarvey U, Bird ED, Martin JB. Somatostatin and neuropeptide Y concentrations in pathologically graded cases of Huntington's disease. Ann Neurol. 1988a; 23:562-9. 10. Beal MF, Swartz KJ, Finn SF, Bird ED, Martin JB. Amino acid and neuropeptide neurotransmitters in Huntington's disease cerebellum. Brain Res. 1988b; 454:393-6. 11. Beal MF, Kowall NW, Swartz KJ, Ferrante RJ, Martin JB. Differential sparing of somatostatinneuropeptide Y and cholinergic neurons following striatal excitotoxin lesions. Synapse. 1989; 3:3847. 12. Beal MF, Brouillet E, Jenkins BG, Ferrante RJ, Kowall NW, Miller JM, Storey E, Srivastava R, Rosen BR, Hyman BT. Neurochemical and histologic characterization of striatal excitotoxic lesions produced by the mitochondrial toxin 3-nitropropionic acid. J Neurosci. 1993; 13:4181-92. 13. Becher MW, Kotzuk JA, Sharp AH, Davies SW, Bates GP, Price DL, Ross CA. Intranuclear neuronal inclusions in Huntington's disease and dentatorubral and pallidoluysian atrophy: correlation between the density of inclusions and IT15 CAG triplet repeat length. Neurobiol Dis. 1998; 4:387-97. 14. Burke JR, Enghild JJ, Martin ME, Jou YS, Myers RM, Roses AD, Vance JM, Strittmatter WJ. Huntingtin and DRPLA proteins selectively interact with the enzyme GAPDH. Nat Med. 1996; 2:347-50. 15. Celio MR. Calbindin D-28k and parvalbumin in the rat nervous system. Neuroscience. 1990; 35:375-475. 58 16. Cha JH. Transcriptional dysregulation in Huntington's disease. Trends Neurosci. 2000; 23:387-92. 17. Cooper JK, Schilling G, Peters MF, Herring WJ, Sharp AH, Kaminsky Z, Masone J, Khan FA, Delanoy M, Borchelt DR, Dawson VL, Dawson TM, Ross CA. Truncated N-terminal fragments of huntingtin with expanded glutamine repeats form nuclear and cytoplasmic aggregates in cell culture. Hum Mol Genet. 1998; 7:783-90. 18. Coyle JT, Schwarcz R. Lesion of striatal neurones with kainic acid provides a model for Huntington's chorea. Nature. 1976; 263(5574):244-6. 19. Cudkowicz M, Kowall NW. Degeneration of pyramidal projection neurons in Huntington's disease cortex. Ann Neurol. 1990; 27:200-4. 20. Davies SW, Turmaine M, Cozens BA, DiFiglia M, Sharp AH, Ross CA, Scherzinger E, Wanker EE, Mangiarini L, Bates GP. Formation of neuronal intranuclear inclusions underlies the neurological dysfunction in mice transgenic for the HD mutation. Cell. 1997; 90:537-48. 21. Davies SW, Beardsall K, Turmaine M, DiFiglia M, Aronin N, Bates GP. Are neuronal intranuclear inclusions the common neuropathology of triplet-repeat disorders with polyglutamine-repeat expansions? Lancet. 1998; 351(9096):131-3. 22. Dawbarn D, De Quidt ME, Emson PC. Survival of basal ganglia neuropeptide Y-somatostatin neurones in Huntington's disease. Brain Res. 1985; 340:251-60. 23. de la Monte SM, Vonsattel JP, Richardson EP Jr. Morphometric demonstration of atrophic changes in the cerebral cortex, white matter, and neostriatum in Huntington's disease. J Neuropathol Exp Neurol. 1988; 47:516-25. 24. DiFiglia M, Christakos S, Aronin N. Ultrastructural localization of immunoreactive calbindin-D28k in the rat and monkey basal ganglia, including subcellular distribution with colloidal gold labeling. J Comp Neurol. 1989; 279:653-65. 25. DiFiglia M, Sapp E, Chase K, Schwarz C, Meloni A, Young C, Martin E, Vonsattel JP, Carraway R, Reeves SA, Boyce FM. Huntingtin is a cytoplasmic protein associated with vesicles in human and rat brain neurons. Neuron. 1995; 14:1075-81 26. DiFiglia M, Sapp E, Chase KO, Davies SW, Bates GP, Vonsattel JP, Aronin N. Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science. 1997; 277(5334):1990-3 27. Duyao MP, Auerbach AB, Ryan A, Persichetti F, Barnes GT, McNeil SM, Ge P, Vonsattel JP, Gusella JF, Joyner AL, MacDonald ME. Inactivation of the mouse Huntington's disease gene homolog Hdh. Science. 1995; 269(5222):407-10. 28. Epplen JT, Przuntek H. Morbus Huntington: Im Spannungsfeld zwischen Klinik, Gendiagnostik und ausstehender Gentherapie. Deutsches Ärzteblatt. 1998; 95:A-32 29. Ferrante RJ, Kowall NW, Beal MF, Richardson EP Jr, Bird ED, Martin JB. Selective sparing of a class of striatal neurons in Huntington's disease. Science. 1985; 230(4725):561-3. 30. Ferrante RJ, Kowall NW, Beal MF, Martin JB, Bird ED, Richardson EP Jr. Morphologic and histochemical characteristics of a spared subset of striatal neurons in Huntington's disease. J Neuropathol Exp Neurol. 1987; 46:12-27. 31. Ferrante RJ, Kowall NW, Richardson EP Jr. Proliferative and degenerative changes in striatal spiny neurons in Huntington's disease: a combined study using the section-Golgi method and calbindin D28k immunocytochemistry. J Neurosci. 1991; 11:3877-87. 32. Ferrante RJ, Gutekunst CA, Persichetti F, McNeil SM, Kowall NW, Gusella JF, MacDonald ME, Beal MF, Hersch SM. Heterogeneous topographic and cellular distribution of huntingtin expression in the normal human neostriatum. J Neurosci. 1997; 17:3052-63. 59 33. Gerfen CR, Baimbridge KG, Miller JJ. The neostriatal mosaic: compartmental distribution of calcium-binding protein and parvalbumin in the basal ganglia of the rat and monkey. Proc Natl Acad Sci U S A. 1985; 82:8780-4. 34. Goldberg YP, Nicholson DW, Rasper DM, Kalchman MA, Koide HB, Graham RK, Bromm M, Kazemi-Esfarjani P, Thornberry NA, Vaillancourt JP, Hayden MR. Cleavage of huntingtin by apopain, a proapoptotic cysteine protease, is modulated by the polyglutamine tract. Nat Genet. 1996; 13:442-9. 35. Graveland GA, Williams RS, DiFiglia M. Evidence for degenerative and regenerative changes in neostriatal spiny neurons in Huntington's disease. Science. 1985; 227(4688):770-3. 36. Gusella JF, Wexler NS, Conneally PM, Naylor SL, Anderson MA, Tanzi RE, Watkins PC, Ottina K, Wallace MR, Sakaguchi AY, Young AB, Shoulson I, Bonilla E, Martin JB. A polymorphic DNA marker genetically linked to Huntington's disease. Nature. 1983; 306(5940):234-8. 37. Gusella JF, MacDonald ME. Genetics and Molecular Biology of Huntington’s Disease In: „Movement Disorders: Neurologic Principles and Practice“, Watts RL, Koller WC (Eds.), McGraw-Hill, New York, 1997: 477-490 38. Gutekunst CA, Levey AI, Heilman CJ, Whaley WL, Yi H, Nash NR, Rees HD, Madden JJ, Hersch SM. Identification and localization of huntingtin in brain and human lymphoblastoid cell lines with anti-fusion protein antibodies. Proc Natl Acad Sci USA. 1995; 92:8710-4. 39. Gutekunst CA, Li SH, Yi H, Mulroy JS, Kuemmerle S, Jones R, Rye D, Ferrante RJ, Hersch SM, Li XJ. Nuclear and neuropil aggregates in Huntington's disease: relationship to neuropathology. J Neurosci. 1999; 19:2522-34. 40. Hackam AS, Singaraja R, Wellington CL, Metzler M, McCutcheon K, Zhang T, Kalchman M, Hayden MR. The influence of huntingtin protein size on nuclear localization and cellular toxicity. J Cell Biol. 1998; 141:1097-105. 41. Harper PS. The epidemiology of Huntington's disease. Hum Genet. 1992; 89:365-76. 42. Hedreen JC, Peyser CE, Folstein SE, Ross CA. Neuronal loss in layers V and VI of cerebral cortex in Huntington's disease. Neurosci Lett. 1991; 133:257-61. 43. Hersch SM, Ferrante RJ. Neuropathology and Pathophysiology of Huntington’s Disease In: „Movement Disorders: Neurologic Principles and Practice“, Watts RL, Koller WC (Eds.), McGraw-Hill, New York, 1997: 503-526. 44. Huntington's Disease Collaborative Research Group. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. Cell. 1993; 72:97183. 45. Huntington G. On chorea. Med Surg Reporter Philadelphia. 1872; 26:317-321. 46. Huntington G. Recollections of Huntington’s chorea as I saw it at East Hampton, Long Island, during my boyhood. J Nerv Ment Dis. 1910; 37:255. 47. Huynh DP, Del Bigio MR, Ho DH, Pulst SM. Expression of ataxin-2 in brains from normal individuals and patients with Alzheimer's disease and spinocerebellar ataxia 2. Ann Neurol. 1999; 45:232-41. 48. Jenkins BG, Koroshetz WJ, Beal MF, Rosen BR. Evidence for impairment of energy metabolism in vivo in Huntington's disease using localized 1H NMR spectroscopy. Neurology. 1993; 43:268995. 49. Jentsch S, Schlenker S. Selective protein degradation: a journey's end within the proteasome. Cell. 1995; 82:881-4. 60 50. Johnston JA, Ward CL, Kopito RR. Aggresomes: a cellular response to misfolded proteins. J Cell Biol. 1998; 143:1883-98. 51. Kahlem P, Terre C, Green H, Djian P. Peptides containing glutamine repeats as substrates for transglutaminase-catalyzed cross-linking: relevance to diseases of the nervous system. Proc Natl Acad Sci U S A. 1996; 93:14580-5. 52. Kiyama H, Seto-Ohshima A, Emson PC. Calbindin D28K as a marker for the degeneration of the striatonigral pathway in Huntington's disease. Brain Res. 1990; 525:209-14. 53. Kim M, Lee HS, LaForet G, McIntyre C, Martin EJ, Chang P, Kim TW, Williams M, Reddy PH, Tagle D, Boyce FM, Won L, Heller A, Aronin N, DiFiglia M. Mutant huntingtin expression in clonal striatal cells: dissociation of inclusion formation and neuronal survival by caspase inhibition. J Neurosci. 1999; 19:964-73. 54. Knöbel KKJ. Die Huntington’sche Krankheit – Epidemiologische Studie in Südbayern und somatosensibel evozierte Potentiale in der Diagnostik. Dissertation, Technische Universität München, 1995. 55. Kowall NW, Ferrante RJ, Martin JB. Patterns of cell loss in Huntington’s disease. Trends Neurosci. 1987; 10:24-29. 56. Kuemmerle S, Gutekunst CA, Klein AM, Li XJ, Li SH, Beal MF, Hersch SM, Ferrante RJ. Huntingtin aggregates may not predict neuronal death in Huntington's disease. Ann Neurol. 1999; 46:842-9. 57. Kuwert T, Lange HW, Langen KJ, Herzog H, Aulich A, Feinendegen LE. Cortical and subcortical glucose consumption measured by PET in patients with Huntington's disease. Brain. 1990; 113:1405-23. 58. Landwehrmeyer GB, McNeil SM, Dure LS 4th, Ge P, Aizawa H, Huang Q, Ambrose CM, Duyao MP, Bird ED, Bonilla E, de Young M, Avila-Gonzalez AJ, Wexler NS, DiFiglia M, Gusella JF, MacDonald ME, Penney JB, Young AB, Vonsattel JP. Huntington's disease gene: regional and cellular expression in brain of normal and affected individuals. Ann Neurol. 1995; 37:218-30. 59. Lange H, Thorner G, Hopf A, Schroder KF. Morphometric studies of the neuropathological changes in choreatic diseases. J Neurol Sci. 1976; 28:401-25. 60. Li SH, Schilling G, Young WS 3rd, Li XJ, Margolis RL, Stine OC, Wagster MV, Abbott MH, Franz ML, Ranen NG, Folstein S, Hedreen J, Ross C. Huntington's disease gene (IT15) is widely expressed in human and rat tissues. Neuron. 1993; 11:985-93. 61. Li XJ, Li SH, Sharp AH, Nucifora FC Jr, Schilling G, Lanahan A, Worley P, Snyder SH, Ross CA. A huntingtin-associated protein enriched in brain with implications for pathology. Nature. 1995; 378(6555):398-402. 62. Li XJ, Sharp AH, Li SH, Dawson TM, Snyder SH, Ross CA. Huntingtin-associated protein (HAP1): discrete neuronal localizations in the brain resemble those of neuronal nitric oxide synthase. Proc Natl Acad Sci U S A. 1996; 93:4839-44. 63. Li SH, Li XJ. Aggregation of N-terminal huntingtin is dependent on the length of its glutamine repeats. Hum Mol Genet. 1998; 7:777-82. 64. Mangiarini L, Sathasivam K, Seller M, Cozens B, Harper A, Hetherington C, Lawton M, Trottier Y, Lehrach H, Davies SW, Bates GP. Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell. 1996; 87:493506. 65. Marshall FJ, Shoulson I. Clinical Features and Treatment of Huntington’s Disease. In: „Movement Disorders: Neurologic Principles and Practice“, Watts RL, Koller WC (Eds.), McGraw-Hill, New York, 1997: 493-498 61 66. Martindale D, Hackam A, Wieczorek A, Ellerby L, Wellington C, McCutcheon K, Singaraja R, Kazemi-Esfarjani P, Devon R, Kim SU, Bredesen DE, Tufaro F, Hayden MR. Length of huntingtin and its polyglutamine tract influences localization and frequency of intracellular aggregates. Nat Genet. 1998; 18:150-4. 67. Massieu L, Garcia O. The role of excitotoxicity and metabolic failure in the pathogenesis of neurological disorders. Neurobiology (Bp). 1998; 6:99-108. 68. McGeer EG, McGeer PL. Duplication of biochemical changes of Huntington's chorea by intrastriatal injections of glutamic and kainic acids. Nature. 1976; 263(5577):517-9. 69. McNeil SM, Novelletto A, Srinidhi J, Barnes G, Kornbluth I, Altherr MR, Wasmuth JJ, Gusella JF, MacDonald ME, Myers RH. Reduced penetrance of the Huntington's disease mutation.Hum Mol Genet. 1997; 6:775-9. 70. Morrison JH, Lewis DA, Campbell MJ, Huntley GW, Benson DL, Bouras C. A monoclonal antibody to non-phosphorylated neurofilament protein marks the vulnerable cortical neurons in Alzheimer's disease. Brain Res. 1987; 416:331-6. 71. Myers RH, Leavitt J, Farrer LA, Jagadeesh J, McFarlane H, Mastromauro CA, Mark RJ, Gusella JF. Homozygote for Huntington disease. Am J Hum Genet. 1989; 45:615-8. 72. Ordway JM, Tallaksen-Greene S, Gutekunst CA, Bernstein EM, Cearley JA, Wiener HW, Dure LS 4th, Lindsey R, Hersch SM, Jope RS, Albin RL, Detloff PJ. Ectopically expressed CAG repeats cause intranuclear inclusions and a progressive late onset neurological phenotype in the mouse. Cell. 1997; 91:753-63. 73. Penney JB Jr, Young AB, Shoulson I, Starosta-Rubenstein S, Snodgrass SR, Sanchez-Ramos J, Ramos-Arroyo M, Gomez F, Penchaszadeh G, Alvir J, Esteves J, DeQuiroz I, Marsol N, Moreno H, Conneallyy PM, Bonilla E, Wexler NS. Huntington's disease in Venezuela: 7 years of follow-up on symptomatic and asymptomatic individuals. Mov Disord 1990; 5:93-9 74. Perutz MF, Johnson T, Suzuki M, Finch JT. Glutamine repeats as polar zippers: their possible role in inherited neurodegenerative diseases. Proc Natl Acad Sci U S A. 1994; 91:5355-8. 75. Polak JM, Van Noorden S. An Introduction to Immunocytochemistry: Current Techniques and Problems. In: „Microscopy Handbooks #11“, Royal Microscopy Society Publications, Oxford University Press, Oxford (Ed.), 1987, revised edition, 5.1. 76. Reiner A, Albin RL, Anderson KD, D'Amato CJ, Penney JB, Young AB. Differential loss of striatal projection neurons in Huntington disease. Proc Natl Acad Sci U S A. 1988; 85:5733-7. 77. Ross CA. When more is less: pathogenesis of glutamine repeat neurodegenerative diseases. Neuron. 1995; 15:493-6. 78. Ross CA. Intranuclear neuronal inclusions: a common pathogenic mechanism for glutaminerepeat neurodegenerative diseases? Neuron. 1997; 19:1147-50. 79. Rubinsztein DC, Leggo J, Coles R, Almqvist E, Biancalana V, Cassiman JJ, Chotai K, Connarty M, Crauford D, Curtis A, Curtis D, Davidson MJ, Differ AM, Dode C, Dodge A, Frontali M, Ranen NG, Stine OC, Sherr M, Abbott MH, Franz ML, Graham CA, Harper PS, Hedreen JC, Hayden MR. Phenotypic characterization of individuals with 30-40 CAG repeats in the Huntington disease (HD) gene reveals HD cases with 36 repeats and apparently normal elderly individuals with 36-39 repeats. Am J Hum Genet. 1996; 59:16-22. 80. Sapp E, Schwarz C, Chase K, Bhide PG, Young AB, Penney J, Vonsattel JP, Aronin N, DiFiglia M. Huntingtin localization in brains of normal and Huntington's disease patients. Ann Neurol. 1997; 42:604-12. 81. Saudou F, Finkbeiner S, Devys D, Greenberg ME. Huntingtin acts in the nucleus to induce apoptosis but death does not correlate with the formation of intranuclear inclusions. Cell. 1998; 95:55-66. 62 82. Scherzinger E, Lurz R, Turmaine M, Mangiarini L, Hollenbach B, Hasenbank R, Bates GP, Davies SW, Lehrach H, Wanker EE. Huntingtin-encoded polyglutamine expansions form amyloidlike protein aggregates in vitro and in vivo. Cell. 1997; 90:549-58. 83. Schöls L, Riess O, Schmidt T. Autosomal dominant vererbte spinozerebellare Ataxien: Klinik, Genetik und Pathogenese. Deutsches Ärzteblatt. 2001; 98:A-1546 84. Seto-Ohshima A, Emson PC, Lawson E, Mountjoy CQ, Carrasco LH. Loss of matrix calciumbinding protein-containing neurons in Huntington's disease. Lancet. 1988; 1(8597):1252-5. 85. Sieradzan KA, Mann DM. The selective vulnerability of nerve cells in Huntington's disease. Neuropathol Appl Neurobiol. 2001; 27:1-21. 86. Sisodia SS. Nuclear inclusions in glutamine repeat disorders: are they pernicious, coincidental, or beneficial? Cell. 1998; 95:1-4. 87. Sotrel A, Paskevich PA, Kiely DK, Bird ED, Williams RS, Myers RH. Morphometric analysis of the prefrontal cortex in Huntington's disease. Neurology. 1991; 41:1117-23. 88. Sternberger LA, Sternberger NH. Monoclonal antibodies distinguish phosphorylated and nonphosphorylated forms of neurofilaments in situ. Proc Natl Acad Sci U S A. 1983; 80:6126-30. 89. Straus W. Imidazole increases the sensitivity of the cytochemical reaction for peroxidase with diaminobenzidine at a neutral pH. J Histochem Cytochem. 1982; 30:491-3. 90. Strong TV, Tagle DA, Valdes JM, Elmer LW, Boehm K, Swaroop M, Kaatz KW, Collins FS, Albin RL. Widespread expression of the human and rat Huntington's disease gene in brain and nonneural tissues. Nat Genet. 1993; 5:259-65. 91. Sun Y, Savanenin A, Reddy PH, Liu YF. Polyglutamine-expanded huntingtin promotes sensitization of N-methyl-D-aspartate receptors via post-synaptic density 95. J Biol Chem. 2001; 276:24713-8. 92. Velier J, Kim M, Schwarz C, Kim TW, Sapp E, Chase K, Aronin N, DiFiglia M. Wild-type and mutant huntingtins function in vesicle trafficking in the secretory and endocytic pathways. Exp Neurol. 1998; 152:34-40. 93. Vincent SR, Staines WA, Fibiger HC. Histochemical demonstration of separate populations of somatostatin and cholinergic neurons in the rat striatum. Neurosci Lett. 1983; 35:111-4. 94. Vonsattel JP, Myers RH, Stevens TJ, Ferrante RJ, Bird ED, Richardson EP Jr. Neuropathological classification of Huntington's disease. J Neuropathol Exp Neurol. 1985; 44:559-77. 95. Vonsattel JP, DiFiglia M. Huntington disease. J Neuropathol Exp Neurol. 1998; 57:369-84. 96. Weindl A, Conrad B. Chorea und choreatische Bewegungsstörungen. In „Bewegungsstörungen in der Neurologie – Richtig erkennen und behandeln“, Conrad B, Ceballos-Baumann AO (Hrsg.), Thieme Verlag, Stuttgart, 1996: 155-172 97. Wellington CL, Ellerby LM, Hackam AS, Margolis RL, Trifiro MA, Singaraja R, McCutcheon K, Salvesen GS, Propp SS, Bromm M, Rowland KJ, Zhang T, Rasper D, Roy S, Thornberry N, Pinsky L, Kakizuka A, Ross CA, Nicholson DW, Bredesen DE, Hayden MR. Caspase cleavage of gene products associated with triplet expansion disorders generates truncated fragments containing the polyglutamine tract. J Biol Chem. 1998; 273:9158-67. 98. Wexler NS, Young AB, Tanzi RE, Travers H, Starosta-Rubinstein S, Penney JB, Snodgrass SR, Shoulson I, Gomez F, Ramos Arroyo MA, Penchaszadeh GK, Moreno H, Gibbons K, Faryniarz A, Hobbs W, Anderson MA, Bonilla E, Conneally PM, Gusella JF. Homozygotes for Huntington's disease. Nature. 1987; 326(6109):194-7. 99. Whetsell WO Jr. Current concepts of excitotoxicity. J Neuropathol Exp Neurol. 1996; 55:1-13. 63 100. Wichmann T, Delong MR. Physiology of the Basal Ganglia and Pathophysiology of Movement Disorders of Basal Ganglia Origin. In: „Movement Disorders: Neurologic Principles and Practice“, Watts RL, Koller WC (Eds.), McGraw-Hill, New York, 1997: 87-94 101. Yamamoto A, Lucas JJ, Hen R. Reversal of neuropathology and motor dysfunction in a conditional model of Huntington's disease. Cell. 2000; 101:57-66. 102. Young AB, Greenamyre JT, Hollingsworth Z, Albin R, D'Amato C, Shoulson I, Penney JB. NMDA receptor losses in putamen from patients with Huntington's disease. Science. 1988; 241(4868):981-3. 103. Young AB. Huntington's Disease and Other Trinucleotide Repeat Disorders. In: "Molecular Neurology", Martin JB (Ed.), Scientific American Medicine, New York, 1998: 35-54 104. Zeevalk GD, Nicklas WJ. Chemically induced hypoglycemia and anoxia: relationship to glutamate receptor-mediated toxicity in retina. J Pharmacol Exp Ther. 1990; 253:1285-92. 105. Zeitlin S, Liu JP, Chapman DL, Papaioannou VE, Efstratiadis A. Increased apoptosis and early embryonic lethality in mice nullizygous for the Huntington's disease gene homologue. Nat Genet. 1995; 11:155-63. 106. Zühlke C, Riess O, Bockel B, Lange H, Thies U. Mitotic stability and meiotic variability of the (CAG)n repeat in the Huntington disease gene. Hum Mol Genet. 1993; 2:2063-7. 64 8. Danksagung Bedanken möchte ich mich besonders bei Herrn Prof. Dr. Adolf Weindl, Ph.D., der sich meiner als Doktorvater an der Technischen Universität angenommen hat, und mir mit Rat und Tat beim Schreiben und Fertigstellen der Dissertationsschrift zur Seite stand. Für die Überlassung des Themas für meine Doktorarbeit, die zur Verfügungstellung des Labors und der Gewebeproben, und die Einarbeitung und Betreuung bei der Durchführung der Untersuchungen bin ich Herrn Robert J. Ferrante, Ph.D. zu großem Dank verpflichtet. Als Leiter des neuropathologischen Labors in Bedford/Mass. und mein Betreuer vor Ort, konnte ich mir seiner Hilfe und tatkräftigen Unterstützung stets sicher sein. Mein besonderer Dank gilt Herrn Prof. Dr. Godehard Oepen, M.D., Ph.D., dessen persönliche Verbindungen und Engagement es mir ermöglicht hatte, ein Jahr als Research Fellow an der Boston University zu verbringen und dort den experimentellen Teil meiner Doktorarbeit zu realisieren. Sein Enthusiasmus für die Sache und seine großzügige Hilfsbereitschaft - auch in persönlichen Belangen half mir dabei über so manch schwierige Phase hinweg. Außerdem möchte ich all den anderen Personen, Freunden und meiner Familie danken, die es mir ermöglicht haben, dieses Projekt zu verwirklichen. 65 9. Lebenslauf Name: Stefan Kümmerle Geburtsdatum/-ort: 10. März 1973 in Freiburg im Breisgau Schule: Sept. 1983- Juni 1992 Gymnasium Schule Birklehof, Abitur im Juni 1992 Studium: April 1994 - März 1996 Medizinstudium an der Universität Mainz (Vorklinik) März 1996 Physikum / Ärztliche Vorprüfung (Gesamtnote: „gut“) Mai 1996 - Mai 2001 Medizinstudium an der TU München (Klinik), Universitätsklinikum Rechts der Isar Mai 2001 Praktisches Jahr: April 2000 - März 2001 3. Staatsexamen / Ärztlichen Prüfung (Gesamtnote: „gut“) am Universitätsklinikum „Evaristo Garcìa“ „Universidad del Valle“, Cali / Kolumbien, jeweils 16 Wochen: Chirurgie, Innere Medizin und Gynäkologie & Geburtshilfe (Wahlfach) Arzt im Praktikum: Dez. 2001 – Feb. 2002 Parkklinik Weißensee, Berlin in der Abteilung für Innere Medizin seit Juni 2002 Vivantes Klinikum am Urban, Berlin, Abteilung für Psychiatrie und Psychotherapie Research Fellowship / Experimenteller Teil der Dissertation: März 1998 - März 1999 1 Jahr Arbeit als Research Fellow in der Abteilung für Experimentelle Neurologie und Neuropathologie, Boston University Medical School, Boston / USA Fremdsprachenkenntnisse: Englisch (fließend in Wort und Schrift) Spanisch (fließend in Wort und Schrift) Französisch (Grundkenntnisse) 66