3DModelling
Werbung

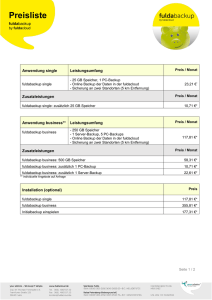
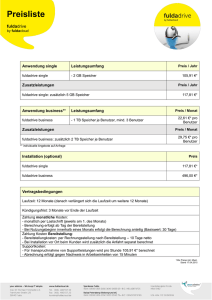
EDV-Anwendungen in der Organischen Chemie • 3D-Darstellung und Molekular Modeling Gliederung 3D-Darstellung und Molekular Modeling • Teil I: Übersicht • Teil II: Molekülmechanik • Teil III: Moleküldynamik • Teil IV: Quantenmechanik (ab initio, semiempirisch) Teil I: Übersicht 1. Was ist und wozu braucht man „Molekular Modeling"? 2. Theorien und Modelle des Molekular Modelings 3. Struktur-Energie-Beziehung A. Speicher EDV-OC: Molekular Modeling 1 1. Was ist und wozu braucht man „Molekular Modeling"? Ziel: Visualisierung/Beschreibung von Molekülen mit computerbasierten Methoden → insbesondere Moleküleigenschaften: • Struktur: die Molekülgeometrie Methoden: Kraftfelder bzw. welche Gestalt haben Moleküle ? Molekülmechanik (MM) Quantenmechanik (QM) • Konformationsraum von Molekülen welche Anordnungen der Atome sind sinnvoll ? • zeitliche Bewegung von Molekülen Energieminimierung Samplingmethoden Moleküldynamik (MD) wie finden Konformationsänderungen statt? • Berechnung von Interaktionsenergien Freie-Energie-Rechnungen wie stark bindet ein Ligand an ein Protein? A. Speicher EDV-OC: Molekular Modeling 2 2. Theorien und Modelle des Molekular Modelings • Molekülmechanik (MM) – Modell: Kerne werden durch "Elektronenfedern" zusammengehalten – Theorie: Hookesches Gesetz der "klassischen" Mechanik • Quantenmechanik (QM) – Modell: Elementarteilchen als Wellen (Wellenmechanik) – Theorie: Schrödinger Gleichung • Moleküldynamik (MD) – Modell: Bewegung der Atome im Raum in einem Zeitintervall – Theorie: Newtonsche Bewegungsgesetze • Quantitative Structure-Activity Relationships (QSAR) – Modell: neue Strukturen über einen "Baukasten" bekannter Systeme – Theorie: Beschreibung durch Deskriptoren und Statistiken A. Speicher EDV-OC: Molekular Modeling 3 Methoden der „Computational Chemistry“: ab initio Vollständig empirisch Quantenmechanik Molekülmechanik Neuronale Netze Kraftfelder semiempirische MO-Methoden Dichtefunktionaltheorie coupled cluster zunehmender Rechenaufwand machbare Größe des Molekülsystems Anzahl Atome 1.000.000 200 50 10 Zunehmende Spezialisierung auf bestimmte Eigenschaften A. Speicher EDV-OC: Molekular Modeling 4 3. Struktur-Energie-Beziehung • Energie als wichtigste Eigenschaft eines Moleküls. • Energie eines Systems (relativ zu einem anderen) bestimmt dessen Stabilität und Existenzfähigkeit. • Je niedriger die Energie, desto stabiler das System. • Zu jeder Struktur gehört genau ein Energiewert. Auftragung Energiewert gegen Strukturkoordinaten liefert: Energiehyperfläche A. Speicher EDV-OC: Molekular Modeling 5 Energiehyperfläche • Die Energiehyperfläche gibt den gesamten Konformationsraum eines Systems wieder. Energie • Minima sind stabile Strukturen. Sattelpunkt Maximum • Maxima sind instabile Strukturen. • Sattelpunkte sind Übergangszustände lokales Minimum A. Speicher EDV-OC: Molekular Modeling globales Minimum 6 Geometrieoptimierung Gradient berechnen Eingabestruktur "Wo geht's bergab?" Energie Atome entlang des Gradienten bewegen Energie berechnen Minimum? nein ja energieoptimierte Struktur A. Speicher global?! EDV-OC: Molekular Modeling 7 3D-Darstellung und Molekular Modeling • Teil I: Übersicht • Teil II: Molekülmechanik • Teil III: Moleküldynamik • Teil IV: Quantenmechanik (ab initio, semiempirisch) A. Speicher EDV-OC: Molekular Modeling 8 Molekülmechanik: Wichtige Strukturgrößen R Bindungslänge Bindungswinkel Torsionswinkel Für Berechnungen erforderlich: • Atomtypen • Kraftfelder mit Kraftfeldparametrisierung: empirisch • „Sterische Energie“ Geometrieoptimierung A. Speicher EDV-OC: Molekular Modeling 9 Modell der Molekülmechanik Modell: starre "Atomkugeln", über "Elektronenfedern" verbunden Theorie: Kugel-Federn-Ensemble folgt Gesetzen der klassischen Mechanik Hooke-Gesetz: O F x x0 x C=O H F D x C sp2 x C sp3 H H H Epot F dx x Epot x0 x x A. Speicher 1 D x 2 2 D = Federkonstante Parameter k für jede Bindung im Molekül EDV-OC: Molekular Modeling 10 Estr Ebend Ecross Eesp Etors Eoop Evdw Kraftfeld: Satz an geeigneten „Parametern“ für möglichst viele Effekte Kraftfeldenergie (engl. force field energy): EFF Estr Ebend Etors Eoop Ecross Eesp Evdw A. Speicher EDV-OC: Molekular Modeling 11 Kraftfelder Parameter: Kraftfeldparametrisierung: • Ein Satz an geeigneten „Parametern“ wird in einem „Kraftfeld“ zusammengefasst. • Sogenannte „empirische“ Kraftfelder sind mit Hilfe von experimentellen Daten parametrisiert. k1ABC k 2AB k 2ABC Z A ZB R k iABC weitere Atomtyp bestimmt, welche Parameter des Kraftfeldes verwendet werden sollen, z. B. Das Kraftfeld „kennt“ einen Satz optimaler Bindungslängen, Bindungswinkel und Torsionswinkel sowie zusätzliche Potentiale sowie dazugehörige Energiewerte. A. Speicher EDV-OC: Molekular Modeling 12 Zusätzliche Potentiale: Kreuzterme zwischen • Winkel – Bindungslängen, • Bindung – Torsion, • Winkel – Torsion, • Winkel – Winkel,... Nichtbindende Wechselwirkungen • Elektrostatische Kräfte (Coulomb-Potential): bis 250 kcal/mol („Salzbrücken“) Atome 4 oder mehr Bindungen entfernt (Konzept der partiellen Atomladungen) • Elektrische Multiplole • Koordinative Bindung von Metallen (Komplexe): ≈ 200 kcal/mol • Wasserstoffbrücken: 1-8 kcal/mol • van der Waals Kräfte: ≈ 0.5 kcal/mol (pro Atompaar) A. Speicher EDV-OC: Molekular Modeling 13 Kraftfelder – ein (unvollständiger) Stammbaum "In theory, there is no difference between theory and practice; in practice, there is.“ (C. Reid) MM Allinger 1973 MM2 Allinger 1977 Karplus, Gelin AMBER Weiner, Kollman 1981 Assisted Model Building with Energy Refinement CHARMM Brooks, Karplus 1982 GROMOS van Gunsteren, Berendsen 1987 MM3 Allinger 1989 OPLS Jorgensen 1989 Dreiding Olafson, Goddard 1990 MMFF MM4 Allinger 1996 GAFF Caldwell, Case 2004 UFF Rappé, Goddard 1992 Chem3D Ultra: MM2, MMFF94 (Merck Molecular Force Field…) A. Speicher EDV-OC: Molekular Modeling 14 Minimierungsalgorithmen zur Geometrie-/Energieoptimierung vorgegebene Molekülgeometrie (Konformer) spezifische Kraftfeldenergie energetisch günstigere Geometrie ? Kraftfeldenergie EFF berechnen (single point) Gradient F berechnen Ableitung der Energie nach der Geometrie = Atomkoordinaten. Gradient (= "Steigung") berechnen: EFF F EFF r Minimum? E F FF 0 r Atome entlang des Gradienten bewegen 2 EFF 0 EFF 2 r Minimum? ja energieoptimierte Struktur Kraftfeldenergie nein Konfomationsparameter r A. Speicher EDV-OC: Molekular Modeling 15 Lokale Minima und Globales Minimum: Generierung und Optimierung zufälliger Veränderungen der Atomkoordinaten energetisch günstigere Koordinaten als neue Geometrie akzeptieren ähnliche Vorgehensweise: Monte Carlo Simulationen Moleküldynamik - Rechnungen Hinweise: Kraftfeldenergie: • KEINE direkte physikalische Bedeutung • Je nach verwendetem Kraftfeld erhält man unterschiedliche Energien, Energievergleiche nur INNERHALB eines Kraftfeldes möglich. • nur isomere Strukturen vergleichbar (mit gleicher Anzahl und Art der Bindungen, z. B. Stereoisomere und Konformere) Beachte: • MM-Rechnungen enthalten keine Elektronen! • keine delokalisierte π-Elektronensysteme! (jenseits der Aromaten) • Die Atomtypen müssen korrekt bestimmt sein! • Alle Systeme befinden sich im Grundzustand! A. Speicher EDV-OC: Molekular Modeling 16 Zusammenfassung: Molekülmechanik • Modell: Atome als starre Kugeln, Bindungen als Federn • Anwendung der "klassischen Mechanik" • Jedes Atom muss einem korrekten Atomtyp zugeordnet werden. • Geometrieoptimierung durch Energieminimierung • optimierte Struktur entspricht z.B. der Gleichgewichtslage • Sterische Energien haben keine direkte physikalische Bedeutung • MM-Rechnungen sind relativ wenig rechenintensiv, es können ganze Enzyme „gemodellt“ werden. • allgemein liefern MM-Rechnungen erste, gute Molekülgeometrien A. Speicher EDV-OC: Molekular Modeling 17 Chem3D Ultra 15.1 Die wichtigsten Funktionen sind: Menü File: Model Settings: Darstellung von Molekülen Preferences: Allgemeine Einstellungenusw. Menü View: sehr umfangreich, u.a. ChemDraw Panel Menü Structure: Daten zur Geometrie anzeigen Menü Calculations: MM und MD-Berechnungen zur Geometrie-Optimierung Menü Surfaces: „Moleküloberflächen“ anzeigen Tool-Bars: Standard: Öffnen, speichern, drucken, … Building: Markierung (Aufheben durch Klick auf freie Fläche) Verschiebung Rotation (häufig benötigt) Zoom-Funktion (vergrößern, verkleinern) Verschieben von Teilen Zeichenstift: Bindungen Texteingabe von Atomen Alternative: ChemDraw Panel Radiergummi A. Speicher Model Display: Bindungen und Atome („Rendering“): Wire Frame (Linien) Sticks (Röhren) Balls and Sticks (Atome als Kugeln, Bindungen als Röhren, Cylinders Space Filling: „echte“ Atomradien, Hintergrund Stereodarstellung Koordinaten Atomsymbole und –Nummerierung Calculations: Rechenverfahren, siehe Menü Demo: Automatische Drehbewegungen, .. Surfaces: siehe Menü Calculations: Rechenverfahren: Molecular mechanics: Kraftfeld MM2, MMFF94 MM2 - Compute Properties: Berechnet die Energie des aktuellen Molekülzustandes („Single Point“) - Minimize Energy: Optimiert die Struktur unter den anzugebenden Algorithmen zur Findung des lokalen Minimums sowie den Bedingungen der Terminierung. - Molecular Dynamics: hilft bei Auffinden des globalen Minimums („Simulated Annealing“) EDV-OC: Molekular Modeling 18 Einfache Übungen: A. Speicher EDV-OC: Molekular Modeling 19 Allgemeine ÜBUNG • Jede Gruppe: • Laden Sie das „Übungsblatt_MM“ herunter und drucken es aus. • Bearbeiten Sie die Übung mit ChemDraw 3D und tragen Sie die Werte ein ! Protokoll ! Spezielle ÜBUNG Entwerfen / versuchen Sie Modeling-Studien an „Ihrem“ Molekül Protokoll !! A. Speicher EDV-OC: Molekular Modeling 20 3D-Darstellung und Molekular Modeling • Teil I: Übersicht • Teil II: Molekülmechanik • Teil III: Moleküldynamik • Teil IV: Quantenmechanik (ab initio, semiempirisch) Teil III: Moleküldynamik Newtonsche Bewegungsgleichungen, Molekülmechanik, Trajektorie MD-Algorithmus If we knew the initial position and velocity of every atom in the universe, and all of the forces acting on them, we could determine the future of the universe! Isaac Newton A. Speicher EDV-OC: Molekular Modeling 23 Moleküldynamik (MD) Was ist MD ? • MD berechnet das zeitliche Verhalten eines molekularen Systems. • MD-Simulationen enthalten detaillierte Informationen über Fluktuationen und Konformationsänderungen. • Für große Moleküle (Proteine, Nukleinsäuren) sind diese Rechnungen besonders aufwendig, die Resultate aber entsprechend aussagekräftig. MD-Simulationen haben mehrere Aufgaben: • Generierung von Konformationen, Suche im Konformationsraum • Dynamische Simulation von Systemen im Gleichgewicht • Nicht-Gleichgewichts-Simulationen Beispiel: Dynamische Prozesse in kleinen Molekülen • Auffinden von Minimumstrukturen • lokale Minima und globales Minimum A. Speicher EDV-OC: Molekular Modeling 24 MD Algorithmus zufällig Richtung und Geschwindigkeit auf alle Atome (sodass im Mittel die „Simulationstemperatur“ vorliegt) Eingabestruktur und Simulationsdauer eingeben Atome für ein festes Zeitintervall bewegen Speichern neue Geometrie (Kraft auf ein Atom durch umliegende Atome berechnen) Trajektorie ja Ende der Simulation A. Speicher neue Richtung und Geschwindigkeit bestimmen Simulationsdauer erreicht? EDV-OC: Molekular Modeling nein 25 Zusammenfassung: Moleküldynamik • MD dient der Simulation eines Systems in einem Zeitfenster • MD baut auf der Molekülmechanik auf • Modell: Atome als starre Kugeln, Bindungen als Federn • zu Beginn der Simulation werden die Geschwindigkeiten zufällig auf die Atome verteilt, so dass eine gegebene mittlere Temperatur vorliegt • Atome bewegen sich in einem (MM-)Kraftfeld • Die Trajektorie liefert einen "Film" über den Bewegungsverlauf des Systems. A. Speicher EDV-OC: Molekular Modeling 26 3D-Darstellung und Molekular Modeling • Teil I: Übersicht • Teil II: Molekülmechanik • Teil III: Moleküldynamik • Teil IV: Quantenmechanik (ab initio, semiempirisch) Teil IV: Quantenmechanik • • • • • • Schrödinger Gleichung Hamilton-Operator Born-Oppenheimer Näherung Wellenfunktionen Basissätze (ab initio Verfahren) Semiempirische Verfahren A. Speicher PC3: Quantenchemie und Spektroskopie "Gar manches rechnet Erwin schon Mit seiner Wellenfunktion Nur wissen möcht' man gerne wohl, Was man sich dabei vorstell'n soll." EDV-OC: Molekular Modeling Erich Hückel (1896-1980) 29 Was kann man mit QM berechnen? • Molekulare Geometrie • Bindungsenthalpien • Dipolmoment • Elektronenverteilung • Elektrostatisches Potential • IR-Spektren • HOMO/LUMO Energien und UV/Vis-Spektren • chemische Verschiebungen in der NMR-Spektroskopie • Reaktionspfade • Struktur und Energie von Zwischenstufen/ Übergangszuständen A. Speicher EDV-OC: Molekular Modeling 30 Semiempirische Methoden Theorie: (elektronische) Schrödinger Gleichung, aber: • vereinfachende Annahmen (Wegfallen der Rumpfelektronen) und • Verwendung empirisch gewonnener Parameter • wesentlich schneller, u. U. aber nur für parametrisierte Systeme gut Beachte: • Energien semiempirischer Rechnungen haben keine direkte physikalische Bedeutung! (sind nur innerhalb des Modells relevant) • Energiedifferenzen semiempirischer Rechnungen entsprechen jedoch wieder realen/physikalischen Energieunterschieden! Gängige semiempirische Methoden: • AM1 (Austin Model 1) • PM3 (Parameterization Method 3) • Dichtefunktionaltheorie (DFT) A. Speicher EDV-OC: Molekular Modeling 31 Zusammenfassung Molekular Modeling • Molekülmechanik (MM) – – – – sehr schnell: CPU-Zeit Anzahl der Atome erste, gute Strukturen auch für große Moleküle (z. B. Enzyme) geeignet nur Strukturinformationen, keine elektronischen Informationen • Quantenmechanik (QM, ab initio) – sehr langsam: CPU-Zeit (Anzahl der Basisfunktionen)4 – sehr große Bandbreite an berechenbaren Eigenschaften • Quantenmechanik (QM, semiempirisch) – Mittelweg zwischen Kraftfeld- und ab-initio-Methoden – parametrisierte Methode muss zur Problemstellung passen • Moleküldynamik (MD) – erlaubt Simulationen in einem Zeitfenster A. Speicher EDV-OC: Molekular Modeling 32