Strukturaufklärung der biotechnologisch interessanten Enzyme O
Werbung

Strukturaufklärung der
biotechnologisch interessanten Enzyme
O- Acetylserin-Sulfhydrylase
und
p-Aminobenzoat N-Oxygenase
INAUGURALDISSERTATION
zur Erlangung der Doktorwürde
Fakultät für Chemie, Pharmazie und Geowissenschaften
Albert-Ludwigs-Universität
Freiburg im Breisgau
vorgelegt von
Georg Ernst Zocher
Freiburg im Breisgau, Juni 2007
Tag der Bekanntgabe der Prüfungsergebnisse: 05.07.2007
Dekan: Prof. Dr. A. Bechthold
Referent: Prof. Dr. G. E. Schulz
Korreferent: Prof. Dr. Th. Friedrich
Science... never solves a problem
without creating ten more.
Wesentliche Teile dieser Arbeit wurden in folgenden Artikeln veröffentlicht:
Zocher, G., Wiesand, U. & Schulz, G. E. (2007). High resolution structure and catalysis of
O- acetylserine sulfhydrylase isozyme B from Escherichia coli, eingereicht.
Zocher, G., Winkler, R., Hertweck, C. & Schulz, G. E. (2007). Structure and action of the
N-oxygenase AurF from Streptomyces thioluteus, eingereicht.
Claus, M. T., Zocher, G. E., Maier, T. H. P. & Schulz, G. E. (2005). Structure of the O- acetyl­
serine sulfhydrylase isoenzyme CysM from Escherichia coli. Biochemistry 44, 8620-8626.
Alle relevanten Koordinaten und Strukturfaktoren wurden in der RCSB Protein Datenbank unter
den folgenden Eintragungen abgelegt:
AurF
2JCD
CysM
2V03
Inhaltsverzeichnis
1. Einleitung......................................................................................................................................... 1
1.1 O-Acetylserin-Sulfhydrylase aus Escherichia coli....................................................................1
1.1.1 Cystein-Biosynthese...........................................................................................................1
1.1.2 O-Acetylserin-Sulfhydrylasen............................................................................................3
1.1.3 Technische Aspekte der Sulfhydrylasen............................................................................ 6
1.2 p-Aminobenzoat N-Oxygenase aus Streptomyces thioluteus.................................................... 7
1.2.1 Streptomyceten................................................................................................................... 7
1.2.2 Biosynthese natürlicher Nitroverbindungen...................................................................... 8
1.2.3 Biosynthese von Aureothin................................................................................................ 9
1.2.4 Die N-Oxygenase AurF aus Streptomyces thioluteus...................................................... 10
1.3 Ausganspunkt und Ziel der Arbeit...........................................................................................12
2. Materialien......................................................................................................................................15
2.1 Geräte.......................................................................................................................................15
2.2 Chemikalien, Enzyme und Kits............................................................................................... 17
2.3 Bakterienstämme..................................................................................................................... 18
2.4 Plasmide...................................................................................................................................19
2.5 Medien, Puffer und Lösungen................................................................................................. 19
3. Methoden........................................................................................................................................ 22
3.1 Gentechnische Methoden.........................................................................................................22
3.1.1 Primer Design...................................................................................................................22
3.1.2 Mutagenese...................................................................................................................... 23
3.1.3 Plasmidpräparation...........................................................................................................25
3.1.4 Agarosegel-Elektrophorese.............................................................................................. 25
3.1.5 Transformation ................................................................................................................26
3.1.6 DNA-Sequenzierung........................................................................................................ 27
3.1.7 Bestimmung der DNA-Konzentration............................................................................. 28
3.2 Proteinpräparation....................................................................................................................28
3.2.1 Herstellung und Reinigung der Sulfhydrylase................................................................. 28
3.2.2 Herstellung und Reinigung von AurF.............................................................................. 32
3.3 Proteincharakterisierung.......................................................................................................... 33
3.3.1 Bestimmung der Proteinkonzentration.............................................................................33
3.3.2 Enzymassay von CysM.................................................................................................... 34
3.3.3 Diskontinuierliche SDS-Polyacryamidgel-Elektrophorese..............................................36
3.3.4 Dynamische Lichtstreuung...............................................................................................37
3.4 Proteinkristalle......................................................................................................................... 38
3.4.1 Kristallgitter und Symmetrie............................................................................................38
3.4.2 Kristallisation von Proteinen............................................................................................39
3.4.3 Lösungsmittelgehalt von Proteinkristallen.......................................................................42
3.4.4 Montage von Proteinkristallen......................................................................................... 43
3.5 Röntgenographische Methoden............................................................................................... 44
3.5.1 Röntgenstrahlung............................................................................................................. 44
3.5.2 Theorie der Röntgenstreuung...........................................................................................45
I
3.5.3 Reziprokes Gitter und Ewaldkonstruktion....................................................................... 48
3.5.4 Temperaturfaktoren..........................................................................................................50
3.5.5 Patterson-Funktion .......................................................................................................... 51
3.5.6 Kristallographische Datensammlung............................................................................... 52
3.5.7 Prozessierung und Datenreduktion.................................................................................. 54
3.5.8 Phasenbestimmung durch isomorphen Ersatz..................................................................55
3.5.9 Phasenbestimmung durch anomale Streuung...................................................................59
3.5.10 Bestimmung der Identität von Metallatomen durch anomale Streuung.........................63
3.5.11 Phasenbestimmung durch molekularen Ersatz...............................................................65
3.5.12 Nicht-kristallographische Symmetrie.............................................................................66
3.5.13 Dichtemodifikation........................................................................................................ 67
3.5.14 Modellbau und Elektronendichtekarten......................................................................... 70
3.5.15 Verfeinerung des Modells.............................................................................................. 72
3.6 Proteinstrukturen......................................................................................................................77
3.6.1 Qualität von Proteinstrukturen......................................................................................... 77
3.6.2 Darstellung von Proteinstrukturen................................................................................... 79
3.6.3 Proteinstrukturvergleiche................................................................................................. 79
4. Ergebnisse und Diskussion.............................................................................................................80
4.1 Ergebnisse zur Sulfhydrylase CysM........................................................................................80
4.1.1 Gentechnische Methoden zur Sulfhydrylase CysM......................................................... 80
4.1.2 Proteinpräparation und Kristallisation von CysM(K268A)............................................. 81
4.1.3 Enzymatische Aktivität von CysM ................................................................................. 85
4.1.4 Kristallographische Datensammlung von CysM(K268A)-Kristallen.............................. 85
4.1.5 Strukturlösung von CysM(K268A)..................................................................................87
4.1.6 Validierung der CysM(K268A)-Struktur.........................................................................89
4.1.7 Struktur von CysM(K268A)............................................................................................ 92
4.1.8 Das aktive Zentrum von CysM(K268A)..........................................................................97
4.2 Ergebnisse zur N-Oxygenase AurF........................................................................................102
4.2.1 Reinigung der N-Oxygenase.......................................................................................... 102
4.2.2 Kristallisation der N-Oxygenase.................................................................................... 104
4.2.3 Kristallographische Datensammlung von N-Oxygenase Kristallen...............................107
4.2.4 Strukturlösung der N-Oxygenase................................................................................... 109
4.2.5 Identität des Cofaktors der N-Oxygenase...................................................................... 113
4.2.6 Validierung des Proteinmodells der N-Oxygenase........................................................ 118
4.2.7 Strukturbeschreibung der N-Oxygenase ....................................................................... 123
4.2.8 Das aktive Zentrum der N-Oxygenase........................................................................... 130
5. Zusammenfassung und Ausblick..................................................................................................141
6. Literatur........................................................................................................................................ 143
7. Anhang......................................................................................................................................... 154
7.1 Eichgerade der Gelpermeation an S200 - 16/60.................................................................... 154
7.2 Selbstrotationsfunktion der N-Oxygenase Kristalle.............................................................. 154
8. Danksagung.................................................................................................................................. 155
II
Abkürzungen
AS
ASU
AurF
Ax
B-Factor
BFG
BSA
cisPt
CNP-Cystein
CysM
ddNTP
DLS
DMF
dNTP
GPC
IEC
IPTG
LBA
LMW
MAD
MIR
Mr
MWCO
NCS
OAS
OASS
OD600
PABA
PCR
PEG-X
PHABA
PNBA
Rcryst
Rcullis
Rfree
Riso
RKE
Rsym
RT
SAD
SeMet
SIR
TNB
v.e.H2O
v/v
w/v
WT
Aminosäure
Asymmetrische Untereinheit
p-Aminobenzoat N-Oxygenase aus Streptomyces thioluteus
Absorption bei der Wellenlänge x nm
kristallographischer Temperaturfaktor
Bakterienfeuchtgewicht
Rinderserumalbumin (bovine serum albumin)
cis-(NH3)2Cl2Pt(II)
S-(3-Carboxy-4-nitrophenyl)-L-Cystein
O-Acetylserin Sulfhydrylase aus E. coli
2´,3´-Didesoxynukleosid-5´-triphosphat
dynamische Lichtstreuung
Dimethylformamid
2´-Desoxynukleosid-5´-triphosphat
Gelpermeationschromatographie
Ionenaustausch-Chromatographie (ion exchange chromatography)
Isopropyl--Thiogalactosid
lysogeny broth Medium mit Ampicillin versetzt
Längenstandard für SDS-PAGE (low molecular weight standard)
multiple anomale Streuung (multi wavelength anomalous diffraction)
Multipler isomopher Ersatz (multiple isomorphous replacement)
relative Molekülmasse
Ausschlussgrenze für Molekularmassen (molecular weight cutoff)
Nicht-kristallographische Symmetrie (non-crstallographic symmetry)
O-Acetylserin
O-Acetylserin-Sulfhydrylase
optische Dichte bei 600 nm
p-Aminobenzoat
Polymerase-Kettenreaktion (polymerase chain reaction)
Polyethylenglycol mit einer mittleren Molekularmasse von X g/mol
p-Hydroxylaminobenzoat
p-Nitrobenzoat
kristallographischer R-Faktor
R-Faktor für gewichtete symmetrieverwandte Reflexintensitäten
freier kristallographischer R-Faktor
R-Faktor für isomorphe Differenzen
Dreifachmutante E57R Y148K R184E von CysM
R-Faktor für symmetrieverwandte Reflexintensitäten
Raumtemperatur
einfache anomale Streuung (single anomalous diffraction)
L-Selenomethionin
einfacher isomorpher Ersatz (single isomorphous replacement)
Thionitrobenzoesäure
voll entsalztes Wasser
Volumenanteile auf Gesamtvolumen (volume per volume)
Masse pro Volumen (weight per volume)
Wildtyp
Standardisierte Abkürzungen und Trivialnamen sind im European Journal of Biochemistry (mittlerweile
FEBS Journal) zu finden. (Information to authors (2000). Eur. J. Biochem. 267, 276-285.)
III
IV
Glossar
alignment
Anpassung von Proteinsequenzen zum Vergleich
batch
in Schüben oder größeren Bündeln
beamline
Messplatz am Synchrotron
bulk solvent
ungeordnetes Solvens im Kristall
constraints
Abweichungen vom Sollwert werden nicht zugelassen
cryo-loop
Nylonschleife zum Montieren von Kristallen im Stickstoffstrom auf dem
Goniometerkopf
cryo-protectant
Glasbildner, Zusatz zum Vermeiden von Eiskristallen beim Schockgefrieren
figure of merit
Gütekriterium berechneter Phasen
frame
Rotationsaufnahme über einen bestimmten Winkelbereich
grid
Gitter, Raster
hanging drop
Hängetropfen-Methode zum Kristallisieren
high oder low energy remote
bei MAD: Wellenlänge, die in Bezug auf die peak-Wellenlänge zu höherer oder
niedrigerer Energie verschoben ist
image plate
Bildplatte (Detektortyp für Röntgenstrahlung in der Proteinkristallographie)
inflection point
Wendepunkt einer Kurve
interface
Kontaktfläche
kit
Zusammenstellung von Lösungen und Reagenzien
linker
Verbindungsglied zwischen zwei Einheiten
loop
schleifenförmiger Verlauf der Polypeptidkette in Proteinstrukturen
maximum likelihood
statistische Methode der Wahrscheinlichkeitsrechnung
model bias
Modelleinfluss
omit
Deletion von Atomen bei der Elektronendichteberechnung
peak
Spitzenwerte von Verteilungen
pellet
sedimentierter Bodensatz
phasing power
Beitrag eines Schweratomderivats zur Phasierung
primer
real space
Oligonukleotid am Synthesebeginn eines komplementären
Stranges
Realraumvon
(imDNA
Gegensatz zum reziproken Raum)
rendering
Photorealistische Verbesserung von Graphiken durch Beleuchtung
restrained
eingeschränkt
rigid body
fester Körper in gleichnamiger Verfeinerungsmethode
scan
Reihenmessung, bei der ein Parameter kontinuierlich geändert wird
screen
Reihenexperiment, z. B. bei der Kristallisation
simmulated annealing
Simuliertes Tempern, Verfeinerungsmethode von Proteinstrukturen
sitting drop
Sitztropfen-Methode zum Kristallisieren
solvent flattening oder flipping
Methoden des Solvensglättens einer Elektronendichtekarte
superloop
Vorratsgefäß zum Auftragen von Proteinlösungen
template
Matrize
V
VI
Symbole und mathematische Zeichen
a ,
b ,c , , ,
*
*
*
*
Elementarzellparameter im realen Raum
*
a , b ,c , , ,
*
Elementarzellparameter im reziproken Raum
hkl
Phase des Strukturfaktors F(hkl)
Å
Ångström (10 m = 0.1 nm)
d
Netzebenenabstand
E
normierter komplexer Strukturfaktor
E
normierter Strukturfaktor
-10
f
Atomformfaktor
*
F ,F
komplexer Strukturfaktor (konjugiert komplexer Strukturfaktor)
Fcalc , Fobs
aus dem Modell berechnete bzw. gemessene Strukturfaktoramplituden
FP, FH, FPH (FP, FH, FPH)
Strukturfaktor (-amplitude) von Protein, Schweratomderivat bzw. Schweratom
g
Erdbeschleunigung
hkl hkl
Millersche Indices (Millersche Indices des Friedelpaars)
i
imaginäre Einheit (i = -1)
2
k
Richtungsvektor von Röntgenwellen
λ
Wellenlänge
m
figure of merit
P(u,v,w)
Pattersonfunktion
ρel
Elektronendichte
s
Ortsvektor im reziproken Raum
σ
Standardabweichung
σA
Gewichtungsfaktor für Strukturfaktoren
u,v,w
Koordinaten im Pattersonraum
VEZ
Volumen der Elementarzelle
VM
Matthews- oder Packungsparameter
x,y,z
Ortskoordinaten im realen Raum
VII
VIII
Einleitung
1. Einleitung
Im Fachgebiet Biochemie werden die molekularen Grundlagen des Lebens anhand der
codierenden Gene eines Organismus untersucht. Von zentraler Bedeutung ist dabei die Aufklärung
der im Organismus ablaufenden Stoffwechselwege und deren Mechanismen, die im Fall von Defek­
ten oft in schweren Krankheiten resultieren. Neben katalytisch aktiven RNA-Molekülen und RNAProtein-Komplexe wird der Stoffwechsel im Wesentlichen durch enzymatisch aktive Proteine
realisiert. Die Strukturaufklärung dieser molekularen Maschinen und insbesondere das Ableiten von
Struktur-Wirkungs-Beziehungen hat sich dabei als breites Aufgabengebiet etabliert. Atomare
Auflösungen dieser Strukturen werden größtenteils durch Röntgenstrukturanalyse an Protein­
kristallen erhalten. Die Strukturaufklärung von Zielproteinen pharmazeutischer Wirksubstanzen, die
die gezielte Generierung von Leitstrukturen im Rahmen der Entwicklung von Medikamenten
erlaubt, ist dabei ebenso von industriellem Interesse wie die Umsetzung einzelner oder kombinierter
Stoffwechselwege zur Synthese eines gewünschten Zielmoleküls. Dabei bietet die Anwendung
enzymatisch katalysierter Reaktionen im industriellem Maßstab eine elegante und kostengünstige
Altenative insbesondere zur Herstellung chiraler und enantiomerenreiner Produkte, die im Labor­
maßstab oft nur durch komplexe mehrstufige Synthesen zu realisieren sind. Diese Arbeit behandelt
die Strukturaufklärung der O-Acetylserin-Sulfhydrylase, die bereits zur biotechnologischen Produk­
tion von Cystein eingesetzt wird und die Strukturaufklärung einer N-Oxygenase, deren biotechno­
logisches Einsatzgebiet Teil intensiver Forschung ist.
1.1
1.1.1
O-Acetylserin-Sulfhydrylase aus Escherichia coli
Cystein-Biosynthese
Die Aminosäure Cystein nimmt durch Ihre Funktionsbreite innerhalb von Proteinstrukturen
eine wichtige Stellung ein. Neben der strukturstabilisierenden Wirkung von Cystein in
extrazellulären Proteinen durch Ausbildung kovalenter Disulfidbrücken wie beispielsweise im
Protein Keratin oder dem Hormon Insulin wird Cystein in vielen Proteinen als katalytischer oder
metallkoordinierender Rest beobachtet. Beispiele für die katalytische Funktion von Cysteinresten
sind die Cysteinprotease Papain und die Glutathion-Reduktase. Die metallkoordinierende Funktion
von Cystein kann am Beispiel der Eisen-Schwefel-cluster Proteine wie der UbiquinonOxidoreduktase aufgezeigt werden.
1
Einleitung
Die Ausgangsverbindung bei der Biosynthese von Cystein ist 3-Phosphoglycerat. Dieses
Zwischenprodukt der Glykolyse wird zunächst in einer dreistufigen Reaktion in Serin umgewandelt,
wobei das Edukt durch die 3-Phosphoglycerat-Dehydrogenase zu 3-Phosphohydroxypyruvat
oxidiert wird. Dieses wird anschließend von der PLP-abhängigen Phosphoserin-Amino-Transferase
zu Phosphoserin transaminiert und zur Bildung der Aminosäure von der Phosphoserin-Phosphatase
hydrolysiert (Abbildung 1).
Abbildung 1:
Biosynthese von Serin ausgehend vom Phosphoglycerat (Claus, 2004).
Der weitere Syntheseweg von Cystein unterscheidet sich bei Bakterien und Pflanzen von
demjenigen der in Säugetieren vorgefunden wird. Säugetiere stellen Cystein über einen komplexen
Stoffwechselweg her, während in Bakterien ein zweistufiger Prozess ausgehend vom Serin über
O-Acetylserin zum Cystein gefunden wurde. Die beiden Reaktionen werden dabei von der SerinAcetyl-Transferase (Genbezeichnung cysE) und der O-Acetylserin-Sulfhydrylase (cysK, cysM)
katalysiert (Abbildung 2).
Abbildung 2:
Zweistufiger Biosyntheseweg von Cystein. I) Im ersten Reaktionsschritt wird die Hydroxygruppe
von Serin acetyliert und im folgenden II) durch Hydrogensulfid substituiert (Claus, 2004).
Das für den zweiten Reaktionsschritt notwendige Hydrogensulfid wird von der Zelle durch die
Sulfat-Permease (cysA) in Form von Sulfat aufgenommen, von der ATP-Sulfurylase (cysD, cysN,
Heterodimer) unter Produktion von Adenosin-5'-phosphosulfat (APS) aktiviert und im folgenden
von der APS-Kinase (cysC) zu 3'-Phosphoadenosin-5'-phosphosulfat (PAPS) umgesetzt. Diese wird
in weiteren Reaktionsschritten über Sulfit zu Sulfid reduziert, woran die Enzyme PAPS-Reduktase
(cysH) und Sulfit-Reduktase (cysI, cysJ) beteiligt sind (Kredich, 1971; Kredich, 1996). Eine
alternative Schwefelquelle findet sich in Form von Thiosulfat, das ebenfalls von der SulfatPermease aufgenommen werden kann. Bemerkenswert ist die Tatsache, dass durch Aktivitäts­
2
O-Acetylserin-Sulfhydrylase aus Escherichia coli
studien nachgewiesen werden konnte, das CysM auch das Nukleophil Thiosulfat als Substrat für die
Produktion von S-Sulfocystein nutzen kann, während die analoge Reaktion für CysK nicht
beobachtet wird (Maier, 2003). Die Bildung von Cystein aus S-Sulfocystein erfolgt anschließend
durch Hydrolyse, wobei Sulfat entsteht.
1.1.2
O-Acetylserin-Sulfhydrylasen
Abbildung 3:
Katalytischer Zyklus der Sulfhydrylasen. I) Nukleophiler Angriff des Substrats O-Acetylserin, II)
geminale Diamin-Zwischenstufe, III) externes Imin, IV) α-Aminoacrylat-Zwischenstufe nach
Abspaltung von Acetat, V) externes Imin nach nukleophilem Angriff von Hydrogensulfid, VI)
geminales Diamin des Produkts (angepasst nach Burkhard et al., 1998 und Tai & Cook, 2001).
Bakterielle Sulfhydrylasen (O-Acetylserin(thiol)-lyase, EC 2.5.1.47) katalysieren den zweiten
Schritt der Biosynthese von Cystein aus O-Acetylserin und Hydrogensulfid (Abbildung 2). Dabei
existieren die zwei Isoformen CysK (O-Acetylserin-Sulfhydrylase A) und CysM (O-AcetylserinSulfhydrylase B), die zueinander eine Sequenzidentität von lediglich 43 % aufweisen. Als
charakteristisches Unterscheidungsmerkmal konnte ein loop in der Bindungstasche des Enzyms
ausgemacht werden, der ein Arg-Arg-Trp-Motiv aufweist (Claus et al., 2005). Die Isoformen A und
3
Einleitung
B der Sulfhydrylase scheinen unter unterschiedlichen Wachstumsbedingungen eine veränderte
Aktivität aufzuweisen. Bei aerobem Wachstum des Bakteriums wird der größte Anteil des Substrats
O-Acetylserin durch die Isoform A der Sulfhydrylasen umgesetzt. Unter diesen Bedingungen
konnte durch Aktivitätsstudien ein etwa zehnfacher Umsatz der Isoform A gegenüber der Isoform B
nachgewiesen werden. Demgegenüber erfolgt unter anaeroben Bedingungen und in Anwesenheit
höherer Thiosulfatkonzentrationen der Hauptumsatz über die Isoform B (Kredich, 1996). Diese
Funktion scheint das Resultat einer erforderlichen, schnellen Anpassung auf äußere Umwelt­
einflüsse zu sein. Beide Isoformen zeigen eine breite Substratspezifität, die verschiedene Schwefelund Stickstoffnukleophile umfasst und sogar einen Umsatz mit Cyanid als Kohlenstoff-Nukleophil
einschließt (Maier, 2003).
Der Reaktionsumsatz beider Isoformen erfolgt dabei über einem identischen, mehrstufigen
Reaktionszyklus (Abbildung 3), dessen Kinetik als Bi-Bi-Ping-Pong-Reaktion nachgewiesen wurde
(Burkhard et al., 1998). Die Bruttoreaktion kann dabei in zwei Halbreaktionen (I-III und IV-VI)
aufgeteilt werden. In der ersten Halbreaktion erfolgt die Bindung des Substrats OAS an das
kovalent am Protein fixierte Coenzym PLP unter Abspaltung von Acetat und Ausbildung eines
Aminoacrylats, während aus der zweiten das Endprodukt L-Cystein durch Addition von HS- an die
olefinische Doppelbindung hervorgeht. In der ersten Halbreaktion erfolgt dabei die Freisetzung des
Cofaktors zu einem externen Aldimin, der in der zweiten Halbreaktion wieder am Lys41 fixiert
wird.
Tabelle 1:
PDBa) /
Auflösung [Å]
Organismus
1Z7W / 2.20
Arabidopsis thaliana
41
A
- / Bonner et al., 2005
1Z7Y / 2.70
Arabidopsis thaliana
41
A
Externes Aldimin / Bonner et al., 2005
2ISQ / 2.80
Arabidopsis thaliana
41
A
2BHS / 2.80
E. coli
-
B
Peptid-Fragment der Acetyltransferase /
Francois et al., 2006
- / Claus et al., 2005
2BHT / 2.10
E. coli
-
B
- / Claus et al., 2005
1Y7L / 1.55
Haemophilus influenzae
36
A
1OAS / 2.20
Salmonella typhimurium
38
A
Peptid-Fragment der Acetyltransferase,
Externes Aldimin / Huang et al., 2005
- / Burkhard et al., 1998
1D6S / 2.30
Salmonella typhimurium
38
A
Externes Aldimin / Burkhard et al., 1999
1FCJ / 2.00
Salmonella typhimurium
38
A
Chlorid, Sulfat / Burkhard et al., 2000
2JC3 / 2.30
Salmonella typhimurium
94
B
- / Publikation in Vorbereitung
1O58 / 1.80
Thermotoga maritima
40
A
- / Heine et al., 2004
1VE1 / 1.45
Thermus thermophilus
42
A
- / Publikation in Vorbereitung
a)
b)
4
Bisher in der Protein-Datenbank veröffentlichte Sulfhydrylase-Strukturen
Sequenzidentitätb) Isoform Liganden / Literatur
[%]
Eintrag in der Protein-Datenbank
Sequenzidentität zur Sulfhydrylase CysM aus E. coli
O-Acetylserin-Sulfhydrylase aus Escherichia coli
Die O-Acetylserin-Sulfhydrylasen werden der β-Familie der PLP-abhängigen Enzyme
zugeordnet (Alexander et al., 1994). Es werden drei Familien unterschieden, die aufgrund der
Regioselektivität der katalysierten Reaktion α-, β- und γ-Familie genannt werden. Eine Suche inner­
halb der Proteindatenbank zeigt, dass diese Enzymklasse intensiv erforscht wurde. Im Kreis der
Sulfhydrylasen sind bisher zwölf Strukturen von Sulfhydrylasen aus sechs verschiedenen Organis­
men strukturell aufgeklärt. Tabelle 1 gibt einen Überblick über alle in der Protein Datenbank
aufgeführten Strukturen, die auch die in diesem Arbeitskreis aufgeklärten Strukturen der
Sulfhydrylase CysM umfasst (Claus et al., 2005). Interessanterweise findet sich darunter neben der
Sulfhydrylase CysM aus E.coli nur eine weitere Cystein-Synthase (PDB-Eintrag 2JC3), die eben­
falls zur Isoform B dieser Enzymklasse zählt. Daneben fallen die erreichten Auflösungen von bis zu
1.45 Å und die geringe Anzahl unterschiedlicher Substrat-Komplexe auf. Bisher konnten neben
dem Kondensationsprodukt aus Methionin und PLP (externes Aldimin) in den Strukturen mit den
PDB-Einträgen 1Z7Y, 1Y7L, und 1D6S keine Komplex-Struktur von Substrat- oder Substratähnlichen Molekülen beschrieben werden. Es konnten allerdings zwei Komplexe aus dem C-termi­
nalen Fragment einer Acetyltransferase und einer Sulfhydrylase strukturell bestimmt werden (2ISQ
und 1Y7L), die eine Wechselwirkung beider Enzyme beschreiben (Huang et al. 2005; Francois et
al., 2006). Zusätzlich konnte das Vorhandensein einer allosterischen Anionenbindungsstelle
nachgewiesen werden (Burkhard et al., 2000), die sich allerdings nicht in den Sulfhydrylasen der
Isoform B findet (Claus et al., 2005).
Die Kettenfaltung dieser Enzyme ist im wesentlichen identisch und im folgenden am Beispiel
von CysM aus E. coli beschrieben. Dieses Enzym besteht aus zwei nahezu gleich großen Domänen,
die analog zur β-Untereinheit der Tryptophan-Synthase mit N- und C-terminaler Domäne benannt
wurden. Die Tryptophan-Synthase ist das Enzym der β-Familie, dessen dreidimensionale Struktur
zuerst gelöst wurde (PDB-Code 1BKS; Hyde et al., 1988). Beide Domänen besitzen ein zentrales
β-Faltblatt, das auf beiden Seiten von α-Helices flankiert wird. Aufgebaut wird die N-terminale
Domäne von den Resten 1 bis 12 und 35 bis 144. Essentieller Bestandteil ist eine bewegliche
Subdomäne, die aus den Resten 85 bis 132 besteht. Das Motiv der etwas größeren C-terminalen
Domäne entspricht im Wesentlichen dem N-terminalen Teil, wobei das β-Faltblatt um zwei anti­
parallele β-Stränge erweitert ist, das aus den Resten 13 bis 34 besteht.
Zwischen den beiden Domänen befindet sich ein tiefer Spalt, an dessen Boden der PLPCofaktor kovalent in Form einer Schiff-Base an die ε-Aminogruppe von Lys41 gebunden ist. Im
Verlauf der Reaktion treten Konformationsänderungen auf, die in zwei kristallisierten Strukturen
nahegelegt werden konnten (PDB-Einträge: 2BHS, 2BHT). Die Konformationsänderung besteht
5
Einleitung
dabei in einer Bewegung der N-terminalen Subdomäne in Richtung der C-terminalen Domäne und
schließt damit das aktive Zentrum vom umgebenden Solvens soweit ab, dass nur noch ein schmaler
Tunnel zur Umgebung besteht. Diese Bewegung der Domänen gegeneinander beträgt dabei an der
äußersten Position 5 Å. Als Substrat-bindende Stelle konnte durch Strukturvergleiche mit den oben
beschriebenen Komplex-Strukturen (1Z7Y, 1Y7L und 1D6S) der loop mit den Aminosäureresten
68-72 ausgemacht werden. Ein weiterer loop im Bereich der Aminosäuren 208-213 wurde durch
Sequenzvergleiche als charakteristisches Merkmal der Isoform B der Sulfhydrylasen ausgemacht
und zudem für die relaxierte Substratspezifität von CysM verantwortlich gemacht.
1.1.3
Technische Aspekte der Sulfhydrylasen
Cystein wird als Zusatz verschiedener Produkten eingesetzt und erfüllt dabei unterschiedliche
technologische Aufgaben. Beispiele dafür sind der Zusatz zu Lebensmitteln (Backwaren und
Fleisch), zu Kosmetika (als Radikalfänger für Anti-Aging-Produkte) und auch als Rohstoff für
Arzeimittel wie etwa Acetylcystein, das als Hustenlöser fungiert. Die klassische Produktion von
Cystein beruht auf der Extraktion der Aminosäure aus Haaren und Federn mit Salzsäure, wobei das
das Oxidationsprodukt Cystin durch nachfolgende Elektrolyse zu Cystein gespalten wird. Ein neues
von der Firma Wacker Chemie AG erstmals eingesetztes Verfahren besteht in der Fermentation in
E. coli über den in Abschnitt 1.1.1 beschriebenen Stoffwechselweg. Bei diesem Prozess dienen
Glucose, Ammoniumsalze und Thiosulfat als Ausgangssubstanzen.
Neben der Herstellung von Cystein ist die Produktion von nicht-natürlichen Aminosäuren, die
als Bausteine in pharmazeutischen oder agrochemischen Produkten Verwendung finden, von
großen industriellen Interesse. Die Synthese nicht-natürlicher Aminosäuren und deren Derivate
verläuft bisher auf chemischem Weg und ist aufgrund der zu beachtenden Stereoselektivität der
einzusetzenden Reaktionen zeit- und kosteninstensiv. Durch Anpassung des Cystein-Stoffwechsels
von E. coli wurde ein neuartiger Biosyntheseweg zur Herstellung nichtnatürlicher Aminosäuren
etabliert. Dabei wird zunächst durch Fermentation O-Acetylserin produziert, das aufgrund einer
Mutation von den Zellen sekretiert wird. Nach Abtrennung der Zellen wird das konzentrierte
Medium zu einer Mischung eines Nukleophils und permeabilisierten Zellen gegeben, die über­
exprimiertes CysM enthalten. Nach wenigen Minuten ist die Konversion zur nicht-natürlichen
Aminosäure abgeschlossen. Möglich wird dieser Reaktionsablauf durch die relaxierte Substrat­
spezifität von CysM, das eine große Variabilität in Bezug auf das eingesetzbare Nukleophil zeigt.
Die Liste der erfolgreich getesteten Edukte enthält viele Schwefel- und Stickstoffverbindungen,
reicht aber auch zu den toxischen Reagenzien Cyanid und Azid. Diese Zellgifte sind der Grund für
6
O-Acetylserin-Sulfhydrylase aus Escherichia coli
die Trennung der Fermentation von der Synthese des Endprodukts (Maier, 2003). Ein Beispiel für
die erfolgreiche semisynthetische Produktion von nicht-natürlichen Aminosäuren ist S-Phenyl­
cystein, eine Ausgangsprodukt von Nelfinavirmesylat (Viracept) der spezifisch die HIV-1-Protease
inhibiert (Kaldor et al., 1997; Maier, 2003).
1.2
1.2.1
p-Aminobenzoat N-Oxygenase aus Streptomyces thioluteus
Streptomyceten
Streptomyceten gehören zu den Bakterien der Ordnung Actinomycetales und leben sowohl in
terrestrischen als auch in marinen Lebensräumen. Sie bilden bei guten Wachstumsbedingungen ein
fädiges Mycel aus, das an das Hyphengeflecht eukaryotischer Pilze erinnert. Streptomyceten
gehören zu den Gram-positiven Bakterien, einer taxonomischen Gruppe mit charakteristischem
Zellwandaufbau. Das Genom von Streptomyceten hat einen auffallend hohen GC-Gehalt von etwa
72% auf, wobei der Grund dafür bis heute unklar ist.
Streptomyceten sind für den Menschen in der Regel harmlos, bisher ist nur eine
humanpathogene Art bekannt. Der Nutzen dieser Organismen ist dagegen enorm. Die wohl
bedeutendste Eigenschaft der Streptomyceten ist deren Fähigkeit, eine Vielzahl komplexer
Naturstoffe herzustellen. Etwa 70% aller bekannten bioaktiven mikrobiellen Stoffwechselprodukte
werden von Streptomyceten gebildet (Gräfe, 1992; Kieser, 2000). Daher sind sie heute eine der
bedeutendsten Quellen für Substanzen oder Substanzvorläufer moderner Wirkstoffe mit anti­
bakterieller (beispielsweise Tetracyclin, Streptomycin), antifungaler (wie etwa Amphotericin B,
Nystatin), immunsuppressiver (beispielsweise Rapamycin) oder cytostatischer (beispielsweise
Bleomycin, Doxorubicin) Aktivität (Weber, 2003). Damit sind die Streptomyceten als Produzenten
pharmakologischer Wirkstoffe interessant. Der Grund für die Produktion dieser Substanzen, der
sogenannten Sekundärmetabolite, ist noch weitgehend unklar. Möglicherweise schützen sich die
Streptomyceten durch die Bildung von Antibiotika vor Nahrungskonkurrenten. Die Molekularbio­
logie dieser Organismen ist im Vergleich zu den gut charakterisierten Bakterien Escherichia coli
oder Bacillus subtilis wenig untersucht. Die Genome der Streptomyceten enthalten fast doppelt so
viele Gene (etwa 8000) wie die meisten anderen Bakterien, vermutlich infolge der zur Sekundär­
metabolit-Biosynthese benötigten Enzyme.
7
Einleitung
1.2.2
Biosynthese natürlicher Nitroverbindungen
Nitroaryleinheiten werden im Vergleich zur großen Anzahl beschriebener Naturstoffe zwar
selten aufgefunden, doch existiert eine beachtliche Anzahl dieser Verbindungen mit teils sehr
verschiedenen Funktionen. Als prominentester Vetreter dieser Substanzklasse kann das Antibio­
tikum Chloramphenicol (He et al., 2001) aufgeführt werden, das 1948 erstmals beschrieben wurde
und von dem Bakterium Streptomyces venezuelae sekretiert wird. Weitere Beispiele sind das anti­
fungale Pyrrolnitrin (Arima et al., 1964) und das cytostatische Aureothin (Hirata et al., 1961;
Cardillo et al., 1972), das sich aus dem p-Nitrobenzoat (im folgenden PABA genannt) ableitet.
Daneben findet sich die Aristolochiasäure, die der Auslöser der „chinesischen Kräuter-Nephro­
pathie“ ausgemacht wurde (Cosyns, 2003), das bakterielle Signalmolekül Hormaomycin (Rössner
et al., 1990), sowie das Pflanzenpathogen Thaxtomin aus Streptomyces turgidiscabies (Kers et al.,
2004). Abbildung 4 zeigt einige der Strukturen der aufgeführten natürlichen Nitroverbindungen.
Abbildung 4:
Beispiele für Naturstoffe, die Nitroaryleinheiten besitzen.
Über die Biosynthese dieser Nitroarylverbindungen ist wenig bekannt. In der Literatur werden
im wesentlichen zwei Stoffwechselwege beschrieben. Dies ist zum einen die direkte Nitrierung, die
die Nitrogruppe durch eine aromatische Substitution in das Substrat einführt und zum anderen die
als N-Oxidation beschriebene Reaktion, die eine vorhandene Aminogruppe durch Oxidation über
molekularen Sauerstoff in die entsprechende Nitrogruppe überführt.
Als Beispiel für die direkte Nitrierung kann Thaxtomin aufgeführt werden, bei dem die Betei­
ligung einer NO-Synthase an der direkten Nitrierung nachgewiesen wurde. Die Nitrierung der in
Thaxtomin enthaltenen Tryptophan-Einheit findet dabei an dem Komplex einer NO-Synthase mit
einer Trp-tRNA statt (Buddha et al., 2004). Wesentlich häufiger dürfte in der Natur der Biosynthe­
8
p-Aminobenzoat N-Oxygenase aus Streptomyces thioluteus
seweg die durch N-Oxygenasen vermittelte N-Oxidation beschritten werden, wie es auch im Fall der
Biosynthese von Chloramphenicol vermutet wird (He et al., 2001). Allerdings konnten bisher erst
zwei Enzyme zweifelsfrei als N-Oxygenasen classifiziert werden. Dies sind die N-Oxygenase PrnD
aus Pseudomonas fluorescens, die in der Biosynthese des oben genannten Pyrrolnitrin involviert ist
und auf Basis von Sequenzvergleichen der Rieske-Proteinfamilie zugeordnet wurde (Kirner et al.,
1998), sowie die N-Oxygenase AurF aus Streptomyces thioluteus, die die Startereinheit PABA zur
Biosynthese von Aureothin erzeugt und Gegenstand dieser Arbeit ist.
Abbildung 5:
1.2.3
A
Ar + NO2+
Ar-NO2
B
Ar -NH2 + O2
Ar-NO2
Einführung bzw. Modifiaktion der Nitrogruppe in Naturstoffe. A) Reaktion der direkten Nitrierung.
B) Reaktion, die über N-Oxygenasen vermittelt wird (Reaktion nicht ausgeglichen).
Biosynthese von Aureothin
Das Gencluster zur Bioynthese von Aureothin (Abbildung 4) aus Streptomyces thioluteus
konnte vollständig beschrieben werden (Abbildung 6A) und dem Gen aurF zweifelsfrei durch
Deletionsmutagenese die Funktion einer N-Oxygenase zugeordnet werden (He & Hertweck, 2003;
He & Hertweck 2004). Eine Deletion des aurF-Gens hatte die Bildung des Aureothin-Derivates
Aureonitrils zur Folge (Ziehl et al., 2005). Der vorgeschlagenene Biosyntheseweg umfaßt dabei im
ersten Schritt die von AurG katalysierte Substitution der Hydroxygruppe des Chorismats zur
entsprechenden Aminofunktion, wobei diese letztlich aus der Umsetzung von Glutamin zu Gluta­
mat resultiert. Nach Hydrolyse der Acetatgruppe schließt sich die von AurF katalysierte Oxidation
des zuvor gebildeten PABA zum p-Nitrobenzoat (PNBA) an (Abbildung 6B), das als Startereinheit
für die Polyketidsynthase (PKS) AurA fungiert und im folgenden durch die ebenfalls als Polyketid­
synthasen charakterisierten Genprodukte AurB und AurC zum Aureothin umgesetzt wird (Abbil­
dung 7).
9
Einleitung
Abbildung 6:
A) Organisation des Genclusters der Biosynthese von Aureothin im Bakterium Streptomyces
thioluteus (aus He & Hertweck, 2003). Die Pfeile indizieren die Transkriptionsrichtung der jeweiligen
Gene. In weiß sind die Polyketid Synthasen A bis C gezeigt, in schwarz die Gene die für die Produktion der Startereinheit PNBA verantwortlich sind. In grau sind Gene regulatorischer Protein
gezeigt. B) Vorgeschlagener Syntheseweg zur Bildung von PABA der durch die Proteine AurG und
AurF katalysiert wird (aus He & Hertweck, 2004).
Abbildung 7:
Modell der Biosynthese von Aureothin ausgehend von Chorismat, das durch die Genprodukte AurG
und AurF in die Startereinheit PNBA der Polyketid Synthasen transformiert wird (He & Hertweck,
2003). Die Genprodukte AurA bis AurC sind zudem auf der Basis von Sequenzhomolgie schematisch
in Domänen eingeteilt.
10
p-Aminobenzoat N-Oxygenase aus Streptomyces thioluteus
1.2.4
Die N-Oxygenase AurF aus Streptomyces thioluteus
Studien zum Mechanismus der von AurF katalysierten Reaktion (Abbildung 8) zeigen eine
sequentielle Oxidation der Aminogruppe, die über das detektierbare Zwischenprodukt Hydroxyl­
aminobenzoat (PHABA) verläuft und über postulierte Nitrosointermediate zum Produkt p-Nitro­
benzoat (PNBA) führt (Winkler & Hertweck, 2005).
Abbildung 8:
Reaktionsmechanismus der von AurF katalysierten N-Oxidation (Winkler & Hertweck, 2005).
Eine Analyse des Substratspektrums (Abbildung 9) zeigt die hohe Regioselektivität der Reak­
tion (Winkler et al., 2006). Diaminobenzoate werden von AurF hoch regioselektiv ausschließlich in
para-Position oxidiert (Abbildung 9A, B und C). Andere Substrate, die Substituenten in ortho- und
meta-Position tragen, werden allerdings akzeptiert (Abbildung 9J, K und L). Die Säurefunktion ist
essentiell (Abbildung 9M), kann aber auch durch eine Sulfonsäure (Abbildung 9E) substituiert sein
und eine zusätzliche Methylengruppe zwischen dem aromatischen Ring enthalten (Abbildung 9, D).
Insgesamt bietet AurF damit ein breites Substratspektrum an, das allerdings Raum für
Optimierungen bezüglich der Reaktionsgeschwindigkeiten der nicht-natürlichen Substrate lässt.
Einer Strukturaufklärung des Enzyms könnte daher eine gezielte Anpassung an ein erwünschtes
Substratspektrum durch gezielte Mutagenese der an der enzymatischen Reaktion beteiligten Amino­
säuren folgen.
Eine Klassifizierung der 336 Aminosäuren langen N-Oxygenase auf der Basis von Sequenz­
homologie konnte nicht erfolgen, da eine Datenbank-Suche lediglich in der Sequenzähnlichkeit zu
hypothetischen Proteinen resultierte und somit keine Einordnung in die möglichen Klassen der
Oxygenasen zuließ. Aufgrund der stufenweisen Oxidation konnte AurF allerdings den Mono­
oxygenasen zugeteilt werden. Insbesondere der vom Enzym verwendete Cofaktor war unklar,
obwohl erste Kolorimetrie- und Atomabsorption-Messungen auf eine Eisen- oder Mangan-abhäng­
ige Monooxygenase deuteten (persönliche Mitteilung Robert Winkler, 2006). Eine jüngst
11
Einleitung
erschienene Veröffentlichung teilte AurF den zweikernigen Eisen-abhängigen Monooxygenasen
(Simurdiak et al., 2006) zu, weil AurF das charakteristische Bindungsmotiv dieser Enzymklasse
enthält (Abschnitt 4.2.7).
Abbildung 9:
1.3
Substratspezifität der N-Oxygenase (Winkler et al., 2006). Die Reaktionsgeschwindigkeiten sind
auf die natürliche Reaktion von PABA zu PNBA normiert.
Ausgangspunkt und Ziel der Arbeit
In vorhergehenden Arbeiten konnte die Struktur der O-Acetylserin-Sulfhydrylase aus E. coli
gelöst werden (Claus et al., 2005). Die in diesen Arbeiten beschriebenen, bekannten Kristalli­
sationsbedingungen des Wildtyps von CysM (im folgenden CysM(WT) genannt) resultierten in
Kristallen mit großen Zellachsen und hohem Solvensgehalt und limitierten die Auflösung auf 2.7 Å
(Tabelle 2). Um die Kristallisation zu optimieren, wurden daher gezielt Oberflächenmutanten
12
Ausgangspunkt und Ziel der Arbeit
präpariert, um den vorherrschenden Kristallkontakt zwischen den Protomeren der genannten
Kristallform der Raumgruppe P6522 zu stören (Zocher, 2003). Durch die Dreifachmutante
E57R-Y148K-R184E (im folgenden CysM(RKE) genannt) konnte ein Teilerfolg hinsichtlich der
genannten Zielsetzung erreicht werden. Zudem konnte mit dieser Mutante die oben genannte
Struktur gelöst werden. Diese Kristallform mit Raumgruppe I41 zeigte ein verbessertes
Streuvermögen von 2.1 Å, beinhaltete allerdings weiterhin vier Protomere in der asymmetrischen
Einheit und zeigte insbesondere fast vollständige meroedrische Verzwilligung, was eine Struktur­
auswertung bzw. Verfeinerung deutlich erschwert und verschlechtert (Yeates, 1997). Auch diese zu
Beginn dieser Arbeit bekannte Kristallisationsbedingung sowie die Zellparameter sind in Tabelle 2
angegeben.
Tabelle 2:
Bekannte Kristallisationsbedingungen der O-Acetylserin-Sulfhydrylase, die zur Strukturlösung
führten (Claus et al., 2005). Zusätzlich sind die Zellparameter, sowie die Anzahl der Protomere und
die maximal erreichte Auflösung angegeben.
Kristallisationsbedingung
Raumgruppe
Zellparameter
Auflösung [Å]
Solvernsgehalt [%]
Protomere / ASU
a)
CysM(WT)a)
100 mM Na-Citrat pH 5.6
100 mM (NH4)2SO4
16-18 % (w/v) PEG 4000
P6522
a=b= 195.7 Å, c=235.8 Å
2.7
79
4
CysM(RKE)a)
100 mM Na-HEPES pH 7.6
150 mM CaCl2
28 % (v/v) PEG 400
I41
a=b= 150.0 Å, c=194.0 Å,
2.1
71
4
Die Sulfhydrylase in ihrer nativen Form wird im folgenden zu CysM(WT) abgekürzt, die Dreifachmutante
E57R-Y148K-R184E als CysM(RKE) bezeichnet.
Die Zielsetzung für die folgende Arbeit war die Produktion bzw. das Auffinden von Kristal­
lisationsbedingungen der Sulfhydrylase, die sich durch besonders gutes Streuvermögen und eine
geringere Anzahl von Protomeren in der asymmetrischen Einheit auszeichnete. Dazu sollten
ausgehend von den bekannten Strukturen gezielt Oberflächenmutanten präpariert und kristallisiert
werden. Diese Merkmale sind von besonderer Bedeutung für eine routinemäßige und einfache
Strukturlösung von Varianten, die für zukünftige Protein-engineering-Projekte im Rahmen der
gezielten Anpassung des Proteins in Bezug auf die Substratvariablität von Bedeutung sein sollte.
Zudem sollten Protein-Substrat-Komplexe kristallisiert werden, die einen zusätzlichen Nachweis für
den aufgestellten Enzym-Mechanismus ergeben.
Im zweiten Teil dieser Arbeit sollte in Zusammenarbeit mit dem Arbeitskreis von Professor
Hertweck (Hans-Knöll-Institut, Jena) versucht werden, die Kristallstruktur der N-Oxygenase AurF
zu bestimmen. Dabei war ein verlässliches Expressions- und Reinigungssystem für das Zielprotein
bereits etabliert, das für die Kristallisation ausreichende Mengen an Protein hoher Reinheit lieferte
13
Einleitung
(Winkler & Hertweck, 2005). Mit dem gereinigten Protein sollten Kristallisationsexperimente
durchgeführt werden, eventuell erhaltene Kristalle auf ihre Eignung für die röntgenkristallo­
graphische Strukturanalyse untersucht und gegebenenfalls die Qualität dieser Kristalle optimiert
werden. Zusätzlich sollten Substrat-Protein-Komplexe kristallisiert und deren Struktur bestimmt
werden, um Einblick in den Katalysemechanismus von AurF zu gewinnen. Aus diesen Ergebnissen
könnte in zukünftigen Studien durch gezielte Mutagenese eine Optimierung der enzymatischen
Aktivität erfolgen und gegebenenfalls die Substratspezifität verändert werden.
14
Materialien
2. Materialien
2.1
Geräte
Allgemeine Geräte
Pipetten
Reinwasseranlage
Zentrifugen
Waagen
Vortex
pH-Elektrode
pH-Meter
2 µl, 20 µl, 200 µL, 1000 µl, 5000 µL
Multipette® plus
Milli-Q-gradient
5415 C
5804 R
5417 R
Rotanta
AE 240
PE 3600
REAX 2000
IJ44/Meteo
761 Calimatic
Gilson
Eppendorf
Millipore
Eppendorf
Eppendorf
Eppendorf
Hettich
Mettler
Mettler
Heidolph
GAT
Knick
Gentechnik
Thermocycler
Zentrifugen
Touchdown
SpeedVac Concentrator
Hybaid
Savant
Agarosegel-Elektrophorese
Kammer
Netzgerät
Thermoblock
Wasserbad
Geldokumentation
GNA 100
ECPS 3000/150
50126101
R 10 Electronic
K 4 Electronic
Powershot A60
UV-Leuchtkasten
Pharmacia
Pharmacia
Liebisch
MGW Lauda
MGW Lauda
Canon
Intas
Bakterienkultivierung
Dampfsterilisator
Brutschrank
Bakterienschüttler
Photometer
Küvetten
Elektroporator
Elektroporationsküvetten
Varioklav 500 EV
Innova 4230
KS 125
KS 250
Novotron
PR 2210
K-Küvetten 407 1/1
Gene Pulser II mit
Pulse Controller II
Spaltbreite 0.2 cm
H & P Labortechnik
New Brunswick Sc. Co., Inc.
IKA Labortechnik
IKA Labortechnik
Infors AG
Eppendorf
Eppendorf
Biorad
Biorad
InVitrogen
15
Materialien
Proteinpräparation
Zentrifugen
Ultraschallbad
Ultraschallgenerator
Einmal-Filterhalter
Säulenmaterialien
Chromatographieanlagen
Pumpen
Schlauchpumpe
UV/Vis-Spektrophotometer
Quarzküvetten
Konzentratoren
Membranen
Dialyseschlauch
RC-2B
RC-5B
5415 D
Sonorex RK 106 Super
Modell 7100
0.20 µm und 0.45 µm, steril
Source-30Q
Source-15Q
Ni-NTA
Superdex-200 16/60 prep grade
ÄKTA Purifier 100
ÄKTA Explorer 100
ÄKTA Prime
Pump P-50
ISM321A
Lambda 40
104-QS
108-QS
Vivaspin 20 mL Concentrator
Amicon 250 mL
MWCO=30'000, ∅ = 70 mm
Servapor Ø 8, 16, 32 mm
Sorvall
Sorvall
Eppendorf
Bandelin
Measuring & Scientific Equipment
Renner GmbH
Amersham Biosciences
Amersham Biosciences
Amersham Biosciences
Amersham Biosciences
Amersham Biosciences
Amersham Biosciences
Amersham Biosciences
Amersham Biosciences
Ismatec
Perkin-Elmer
Hellma
Hellma
Vivascience
Amicon
Vivascience
Serva
Dynamische Lichtstreuung
DL-Apparatur
Software
Dyna Pro-801
Dyna Pro-801
Version 5.25.44, April 2000
Protein Solution
Protein Solution
SDS-PAGE
Elektrophorese-Apparatur
Spannungsquelle
Gel-Trockenrahmen
Mikrowellenofen
Mini-Protean II
Power Pac 3000
Eigenbau
FM B935
Biorad
Biorad
Institutswerkstatt
Moulinex
Kristallisation
Deckgläser
Agarose
Kristallisationsboxen
Pipetten
Mikroskop
Kristallaufnahmen
16
Durchmesser 21 mm
low melting
Linbro Zellkulturbox
Crystalclear Strips, 96 Wells
Research pro 100, 8 Kanal
Research pro 10, 1 Kanal
Laborlux 12 Pol S
Camedia C-3030 Zoom
Hecht-Assistent
Hampton Research
Flow Laboratories
Hampton Research
Eppendorf
Eppendorf
Leitz
Olympus
Geräte
Röntgenographische Methoden
Glaskapillaren
Hartwachs
Goniometerkopf
cryo loops
Röntgengenerator
Detektor
Tieftemperaturgenerator
2.2
∅ =0.3, 0.7 mm
Deiberit
Huber
0.1- 0.7 mm
RU200-B
RU-H2C
MAR Imaging Plate, ∅ =30 cm
X1000
600 series
W. Müller-Glas
Böhme, Bad Sachsa
Hampton Research
Hampton Research
Rigaku
Rigaku
Marresearch
Siemens
Oxford Cryosystems
Chemikalien, Enzyme und Kits
Allgemeine Chemikalien
Hersteller
β-Mercaptoethanol
Serva
Acrylamid, research grade
Fluka
Agar-Agar
Gibco
Agarose, research grade
Fluka
Ammoniumsulfat
Ampicillin, Natriumsalz
Merck
Sigma,Gerbu
Ammoniumperoxodisulfat
Merck
Caseine Pepton (Pepton 140)
Gibco
Coomassie Brilliant Blue R-250
Serva
DNA-Längenstandard (λ-Phagen-DNA, BstE verdaut)
Ethidiumbromid
New England Biolabs
Merck
Glycerin
Serva
Hefeextrakt
Gibco
HEPES
Gerbu
kb DNA-Längenstandard
LMW-Längenstandard
New England Biolabs
Amersham Biosciences
MES
Sigma
PEG (verschiedener mittlerer Molekularmasse)
Fluka
SDS
Fluka
Silikonisierlösung
Serva
TEMED
Tris
Merck
Fluka
Gängige Laborchemikalien sind nicht aufgeführt. Soweit nicht anders beschrieben, waren alle Chemikalien vom
Reinheitsgrad p.a.
17
Materialien
Enzyme
Hersteller
Benzonase (250 U/µL)
Merck
Kits
E.Z.N.A.®Plasmid Miniprep Kit II
Lösung-1, Lösung-2, Lösung-3, HB-Puffer, DNA-Waschpuffer, RNase
PeqLab
QuikChange Site-Directed Mutagenesis Kit
TurboPfu DNA-Polymerase, 10xReaktionspuffer,
Dpn I Restriktionsendonuklease, Kontroll-primer #1 und #2,
Kontroll-Plasmide, dNTP-Mix, EpicurianColiXL1-Blue
superkompetente Zellen
Stratagene
Crystal ScreenTM
50 verschiedene Pufferlösungen zur Proteinkristallisation
Hampton Research
Crystal Screen IITM
48 verschiedene Pufferlösungen zur Proteinkristallisation
Hampton Research
Crystal Screen CryoTM
48 verschiedene Pufferlösungen zur Proteinkristallisation
Hampton Research
Crystal Screen Cryo IITM
48 verschiedene Pufferlösungen zur Proteinkristallisation
Hampton Research
Wizard Screen I
48 verschiedene Pufferlösungen zur Proteinkristallisation
Emerald BioStructures
Wizard Screen II
48 verschiedene Pufferlösungen zur Proteinkristallisation
Emerald BioStructures
Wizard Screen Cryo I
48 verschiedene Pufferlösungen zur Proteinkristallisation
Emerald BioStructures
Wizard Screen Cryo II
48 verschiedene Pufferlösungen zur Proteinkristallisation
Emerald BioStructures
JBScreen Nr. 1 – 10
240 verschiedene Pufferlösungen zur Kristallisation
2.3
Bakterienstämme
Bakterienstamm
Genotyp
E. coli XL1-Blue
F', Tn10, proA+B+, lacIq, ∆(LacZ9M15/recA1, endA1, gyrA96(NaIr),
thi, hsdR17 (rK- mK-), supE44, relA1, lac
F', ompT, hsdSB (rB- mB-), gal, dcm (DE3), pLysS (CmR)
E. coli BL21(DE3)
18
Jena Bioscience
Hersteller
Stratagene
Novagen
Plasmide
2.4
Plasmide
pFL145stop
2.5
Klonierungsvektor, Expressionsvektor,
Ampicillinresistenz
Schwarz-Herion, 2002
Medien, Puffer und Lösungen
Agarosegel-Elektrophorese
50xTAE-Puffer
Tris
Essigsäure
EDTA
pH 8.1 (stellt sich ein)
2M
1M
100 mM
Ethidiumbromidlösung
1x TAE-Puffer mit Ethidiumbromid
1 mg/mL
Probenpuffer
Tris
EDTA
Glycerin
Bromphenolblau
pH 7.5 (RT, 6 M HCl)
kb-Standard
0.5, 1.0, 1.5, 2.0, 2.5, 3.0, 4.0,
5.0, 6.0, 8.0, 10.0 kb
10 mM
1 mM
50% (v/v)
0.05% (w/v)
New England Biolabs
Nährmedien
LB-Medium
Select Pepton 140
Hefeextrakt
NaCl
pH 7.5 (RT, 1 M NaOH)
10 g/L
5 g/L
170 mM
LB-Agar
LB-Medium mit Agar-Agar
LBA-Medium
LB-Medium mit Ampicillin (steril filtriert)
LBA-Agar
LBA-Medium mit Agar-Agar
15 g/L
2xYT-Medium
Select Pepton 140
Hefeextrakt
NaCl
pH 7.5 (RT, 1 M NaOH)
16 g/L
10 g/L
85 mM
2xYTA-Medium
2xYT-Medium mit Ampicillin (steril filtriert)
SOC-Medium
Hefeextrakt
Caseine-Pepton
NaCl
KCl
MgCl2
Glucose
pH 7.0 (RT, 1 M NaOH)
15 g/L
100 µg/mL
100 µg/mL
5 g/L
20 g/L
8.5 mM
2.5 mM
10 mM
20 mM
19
Materialien
Herstellung kompetenter Zellen
FSB-Puffer
Kaliumacetat
MnCl2
CaCl2
KCl
Hexammincobaltchlorid
Glycerin
DnD
Dithiothreitol
DMSO
KOAc
10 mM
45 mM
10 mM
100 mM
3 mM
10%
pH 6.4 (RT, 6 M HCl)
1M
90% (v/v)
pH 7.5 (RT, 0.01 M NaOH)
Proteinpräparation
IEC-Puffer-CysM-A
Tris
pH 8.0 (RT, 1 M HCl)
20 mM
IEC-Puffer-CysM-B
Tris
NaCl
pH 8.0 (RT, 1 M HCl)
20 mM
1000 mM
GPC-Puffer-CysM
HEPES
NaCl
pH 7.5 (RT, 1 M HCl)
50 mM
200 mM
GPC-Puffer-AurF
HEPES
NaCl
pH 7.5 (RT, 1 M HCl)
50 mM
150 mM
Phosphatpuffer
K2HPO4
pH 8.0 (RT, 1 M HCl)
100 mM
PLP-Lösung
Pyridoxal-5'-phosphat in v.e.H2O
Pyridoxin-Lösung
Vitamin-B6-Hydrochlorid in v.e.H2O
60 mg/mL
AHT-Lösung
AHT in v.e.H2O (gelagert bei -20 °C)
2 mg/mL
20 mM
SDS-PAGE
Acrylamid-Stammlösung
Acrylamid
Bisacrylamid
39% (w/v)
1% (w/v)
Elektrophoresepuffer
Tris
Glycin
SDS
pH 8.3 (stellt sich ein)
50 mM
380 mM
0.1% (w/v)
Sammelgelpuffer
Tris
SDS
pH 6.8 (RT, 6 M HCl)
0.5 M
0.4% (w/v)
20
Medien, Puffer und Lösungen
Sammelgelpuffer
Tris
SDS
pH 8.8 (RT, 6 M HCl)
1.5 M
0.4% (w/v)
Sammelgel 6% (w/v)
Acrylamid-Stammlösung
Sammelgelpuffer
v.e.H20
APS 10% (w/v)
TEMED
Tris
SDS
pH 6.8 (RT, 6 M HCl)
0.375 mL
0.625 mL
1.5 mL
25 µL
2.5 µL
1.5 M
0.4% (w/v)
Trenngel 12% (w/v)
Acrylamid-Stammlösung
Trenngelpuffer
v.e.H2O
APS 10% (w/v)
TEMED
1.5 mL
1.25 mL
2.25 mL
50 µL
5 µL
SDS-Probenpuffer
Sammelgelpuffer
SDS 16% (w/v)
Glycerin
Bromphenolblau 0.2% (w/v)
pH 6.8 (RT, 6 M HCl)
Färbelösung
Coomassie Brilliant Blue R-250
Ethanol
Essigsäure
Entfärbelösung
Ethanol
Essigsäure
LMW-Längenstandard
Phosphorylase B (94 kDa)
BSA(67kDa)
Ovalbumin (43 kDa)
Carboanhydrase (30 kDa)
Trypsin-Inhibitor (20 kDa)
α-Lactalbumin (14 kDa)
Sucrose
in SDS-Puffer
2 mL
2 mL
4 mL
1 mL
0.25% (w/v)
30% (v/v)
10% (v/v)
30% (v/v)
10% (v/v)
64 µg/mL
83 µg/mL
147 µg/mL
83 µg/mL
88 µg/mL
121 µg/mL
27 µg/mL
21
Methoden
3. Methoden
3.1
Gentechnische Methoden
3.1.1
Primer Design
Primer sind DNA-Oligonukleotide mit einer Länge von 15 bis 40 Basen. Diese werden für die
Polymerase-Kettenreaktion (PCR) benötigt, die die Grundlage der Mutagenese nach der Quik­
ChangeTM-Methode bildet. Zudem werden primer auch bei der DNA-Sequenzierung nach der
Sanger-Didesoxymethode (Sanger et al., 1977) eingesetzt.
Mutagenese-primer müssen bestimmte Voraussetzungen erfüllen. So sollten sie die nichtkomplementären Basen ungefähr in der Mitte enthalten und zwischen 25 und 45 Basen lang sein,
wobei die terminalen Basen jeweils mindestens ein Guanin oder Cytosin sein sollten. Der Guaninund Cytosingehalt sollte mindestens 40% betragen und der Schmelzpunkt (Tm) der Primer über
78 °C liegen (QuikChangeTM Site Directed Mutagenesis Kit, Firma Stratagene). Der Schmelzpunkt
wurde anhand folgender Gleichung berechnet:
Tm =85.10.41⋅GC−
N:
GC:
Fehler:
675
−Fehler
N
Gleichung 1
Anzahl der Basen
GC-Gehalt in %
Anzahl der mit dem Primer fehlgepaarten Basen in %
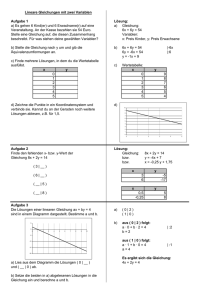
Für die ortsgerichtete Mutagenese nach der QuikChangeTM-Methode müssen unter Beachtung
der oben aufgeführten Bedingungen zwei exakt komplementäre Primer entworfen werden, einer für
den kodierenden und einer für den nicht kodierenden Strang der zu mutierenden DNA. Die
Synthese der benötigten primer wurde von der Firma Qiagen ausgeführt. Tabelle 3 gibt die primerSequenzen an.
Sequenzen der verwendeten primer.a) Die Positionen der Mutationen sind markiert.
Tabelle 3:
primer
Sequenz in 5'-3'-Richtunga)
K268A
GCA CTG CGG GTG GCA GCT GCT AAC CCT GAC GCG
80.5
69.7
R210A
GC AGC ATT CCC GGC ATT GCG CGC TGG CCT ACG
79.2
68.8
Q140E
GGA AAG CTG CTC GAT GAA TTC AAT AAT CCC GAT AAC
74.3
41.7
Q140A
GGA AAG CTG CTC GAT GCG TTC AAT AAT CCC GAT AAC
74.3
41.7
T68A
GTC TTA ATC GAA GCC GCG AGT GGT AAC ACC GGC
78.6
57.6
GTC TTA ATC GAA GCC AGC AGT GGT AAC ACC GGC
78.6
57.6
T68S
a)
22
TM [°C] GC-Gehalt [%]
Die Sequenzen der reverse-primer sind zu den hier aufgeführten komplementär.
Gentechnische Methoden
3.1.2
Mutagenese
Polymerase-Kettenreaktion
Ein DNA-Abschnitt, der von bekannten Sequenzen flankiert wird, lässt sich mit Hilfe der
Polymerase-Kettenreaktion (PCR) vervielfältigen (Mullis et al., 1986; Saiki et al., 1988). Dazu
werden zwei primer benötigt, die an den Enden des zu amplifizierenden doppelsträngigen DNAAbschnitts so hybridisieren, dass ihre 3´-Enden aufeinander zeigen. Die primer können auch
überhängende Enden oder fehlgepaarte Basen besitzen, so dass Restriktionsschnittstellen oder
Mutationen eingeführt werden können. Die PCR verläuft in drei sich wiederholenden Schritten. Im
ersten Schritt wird die template dsDNA zunächst bei Temperaturen von ca. 94-98 °C denaturiert,
um die beiden Stränge voneinander zu trennen. Anschließend erfolgt das annealing der primer bei
ca. 55 bis 70 °C gefolgt von der DNA-Synthese bei 68 °C (extension). Der Reaktionszyklus wird
bis zu 40 mal wiederholt, wodurch eine Amplifikation des von den primern begrenzten DNAAbschnitts erreicht wird.
Ortsgerichtete Mutagenese nach der QuikChangeTM-Methode
Die QuikChangeTM-Methode (Firma Stratagene) beruht auf dem Prinzip der PCR. Im
Anschluss
an
die
PCR
wird
hier
die
parentale,
nichtmutierte
DNA
durch
die
Restriktionsendonuclease DpnI verdaut, die nur die parentale methylierte DNA schneidet, während
die unmethylierte DNA, die durch die PCR generiert wurde, intakt bleibt. Danach wird die DNA in
E. coli XL1-Blue-Zellen transformiert und dort zu einem zirkulären Plasmid geschlossen.
Abbildung 10 zeigt schematisch den Ablauf.
Abbildung 10: Schematische Darstellung der ortsgerichteten Mutagenese nach der QuikChange-Methode (nach Pro
duktinformation, Firma Stratagene).
23
Methoden
Tabelle 4
Temperaturprogramm der QuikChange™-Mutagenese
Vorgang
Temperatur [°C]
Zeit [min]
Zyklen
Denaturierung
95
1
1
Denaturierung
95
1
16
55 - 70
1
16
Extension (1.5 min/kb)
68
7
16
Vervollständigung der Extension
68
15
1
Annealing
Die drei Schritte der PCR annealing, Denaturierung und extension wurden in einem
Thermocycler durch ein Temperaturprogramm realisiert, das Tabelle 4 entnommen werden kann.
Nach Vorschrift des Herstellers wurden die in Tabelle 5 aufgeführten Lösungen und Enzyme in
einem 200 µL-PCR-Reaktionsgefäß zusammengegeben. Um das Schmelzen der DNA zu
erleichtern, wurde DMSO bis zu einer Konzentration von 2% (v/v) zugegeben. Die Menge an
dsDNA-template wurde zwischen 5 ng und 50 ng pro Ansatz variiert. Nach Abschluss der PCR
wurde die methylierte Templat-DNA mit DpnI verdaut, indem 1 µL (10 U) dieses Enzyms
zugesetzt und für 1-3 h bei 37 °C inkubiert wurde. Nach PCR und Verdau wurde die DNA nach
dem Transformationsprotokoll der Firma Stratagene in superkompetente E. coli XL1-Blue-Zellen
transformiert. Selektion auf Zellen, die das Plasmid tragen, erfolgte durch Kultivierung auf LBAAgarplatten. Hierzu wurde der gesamte Transformationsansatz mit einem Drigalski-Spatel
gleichmäßig auf einer LBA-Agarplatte ausgestrichen und ü. N. bei 37 °C inkubiert. Je Ansatz
wurden zwei der so erhaltenen Kolonien in Flüssigkulturen übergeimpft und deren Plasmid-DNA
wie in Abschnitt 3.1.3 beschrieben isoliert. Die Sequenzierung der erhaltenen Plasmide zur Über­
prüfung des Mutagenese-Ergebnisses wurde von der Firma Seqlab durchgeführt (Abschnitt 3.1.6).
Tabelle 5
Zusammensetzung eines Quikchange™-Mutagenese-Ansatzes
Inhalt
10xReaktionspuffer
5
Plasmidlösung (5 ng/µL – 50 ng/µL)
1
+ -Primer (100 ng/µL)
1,25
- -Primer (100 ng/µL)
1,25
dNTP-Mix
v.e.H2O
Pfu DNA-Polymerase (2.5 U/µL)
24
Volumen [µL]
1
41
1
Gentechnische Methoden
3.1.3
Plasmidpräparation
Die Plasmidpräparation wurde mit dem E.Z.N.A.® Plasmid MiniprepKit II der Firma PeqLab
durchgeführt. Prinzip dieser Methode ist es, zunächst im alkalischen Milieu die Zellen zu lysieren
sowie die chromosomale DNA zu denaturieren (Birnboim et al., 1979). Bei der anschließenden
Neutralisation wird die DNA präzipitiert und kann so zusammen mit der bei hoher Ionenstärke
unlöslichen hochmolekularen RNA durch Zentrifugation abgetrennt werden. Niedermolekulare
RNA wird durch Zugabe von RNase A abgebaut. Die zurückbleibende Plasmid-DNA kann durch
Bindung an geeignete Säulenmaterialien chromatographisch gereinigt werden. Die Durchführung
erfolgte nach Anleitung des Herstellers. Eluiert wurde mit 2x20 µL sterilem v.e.H2O, das zuvor auf
90 °C erhitzt wurde. Die Konzentration der so erhaltenen Plasmidlösung wurde bestimmt, die unter
Abschnitt 3.1.6 beschriebene Menge Plasmid zum Sequenzieren gegeben und der Rest der Lösung
bei -20 °C gelagert.
3.1.4
Agarosegel-Elektrophorese
Durch die Agarosegel-Elektrophorese ist es möglich, DNA-Fragmente gemäß ihrer Länge
elektrophoretisch aufzutrennen. Agarose besteht aus einem linearen Polymer aus alternierenden
D-Galaktose- und 3,6-Anhydro-L-Galaktoseeinheiten. Löst sich Agarose unter Erhitzen in einem
geeigneten Puffer, so erstarrt sie beim Abkühlen unter Ausbildung einer Polymermatrix, deren
Porengröße eine Funktion der Agarosekonzentration ist. Die elektrophoretische Mobilität linearer
DNA-Fragmente im Gel ist dabei bis zu einem gewissen Grad umgekehrt proportional zum
Logarithmus der Anzahl der Basenpaare. Außerdem ist die Wanderungsgeschwindigkeit der DNA
im Agarosegel von deren Größe und Konformation abhängig. Dies ermöglicht neben einer
Trennung aufgrund unterschiedlicher Länge auch eine Trennung von zirkulär-superhelicaler,
superhelicaler und linearer Plasmid-DNA. Durch Vergleich mit einem DNA-Standard ist eine
Abschätzung der Länge der DNA-Fragmente möglich. Die Detektion der DNA im Gel erfolgt durch
Anfärben mit Ethidiumbromid, einem Fluoreszenzfarbstoff, der zwischen den DNA-Basen
interkaliert. Bei Anregung mit UV-Licht der Wellenlänge 254 nm zeigt der interkalierte im Gegen­
satz zum freien Farbstoff eine starke rot-orange Fluoreszenz (Sharp et al., 1973).
Es wurden 1%-ige Gele verwendet. Die Agarose wurde hierzu in 1xTAE-Puffer suspendiert
und solange erhitzt, bis eine klare Lösung entstanden war. Nach Ausgleich des durch Verdunstung
verlorenen Wassers und Abkühlung auf 50 °C wurde die Lösung mit Ethidiumbromid bis zu einer
Endkonzentration von 0.5 µg/mL versetzt und in eine Gelkammer gegossen. Zur Ausbildung der
Probentaschen wurde ein Kamm eingesetzt. Nach Erstarren des Gels wurde die Gelkammer in die
25
Methoden
mit 1xTAE-Puffer gefüllte Elektrophoreseapparatur eingesetzt und der Kamm entfernt. Die Proben
(8 µL) wurden mit Probenpuffer (5 µL) versetzt und in die Geltaschen pipettiert. Als Längen­
standard wurde 1-kB-Standard verwendet. Die Elektrophorese erfolgte horizontal im elektrischen
Feld bei 100 V und 120 mA. Zur Auswertung wurden die Gele unter UV-Licht bei 254 nm
photographiert.
3.1.5
Transformation
Als Transformation wird das Einschleusen von exogener DNA in Bakterienzellen in Form
eines Plasmids bezeichnet. Die Fähigkeit zu diesem Prozess, die sogenannte Kompetenz, ist
abhängig vom Bakterienstamm, dem Wachstumsstadium, den Wachstumsbedingungen und der
Transformationsmethode.
Herstellung kompetenter Zellen
Für die Herstellung kompetenter E. coli Zellen des Stamms BL21(DE3) wurde nach der
Calciumsalz-Methode vorgegangen (Hanahan, 1983). Dazu wurden je 10 mL LB-Medium mit
20 mM MgCl2 mit einer Glycerinkultur von E. coli Bakterien angeimpft und bei 37 °C und 400 rpm
inkubiert. Beim Erreichen einer optischen Dichte bei 600 nm von 0.4 wurden die Bakterien
abzentrifugiert (3000*g, 4 °C, 10 min) und der Überstand verworfen. Alle folgenden Schritte
wurden auf Eis durchgeführt. Die Zellen wurden 10 min auf Eis inkubiert, dann wurde 4 mL
eisgekühlter FSB-Puffer zugegeben und die Zellen durch vorsichtiges Pipettieren resuspendiert.
Erneut wurde 10 min auf Eis inkubiert, die Suspension 10 min bei 3000*g zentrifugiert und der
Überstand verworfen. Die erhaltenen Bakterienzellen wurden in 0.8 mL FSB-Puffer resuspendiert
und 28 µL DnD-Lösung zugegeben. Es wurde 15 min auf Eis inkubiert, nochmals 28 µL DnD
zugegeben und vorsichtig umgeschwenkt. Die auf Eis gehaltenen Zellen behielten so ca. 16 h ihre
Kompetenz. Sollten kompetente Zellen eingefroren werden, wurde auf die selbe Weise
vorgegangen, aber anstatt dem FSB-Puffer wurde der FSB-Puffer und statt DnD-Lösung reines
Dimethylsulfoxid (Sambrook et al., 1989) verwendet. Das Einfrieren unter Erhaltung der
Kompetenz wurde durch Schockfrieren von aliquotierten Zellen in flüssigem Stickstoff und
Lagerung bei –80 °C erreicht. Die in hohem Maße kompetenten („superkompetente“), kommerziell
erhältlichen, tiefgefrorenen Zellen des Stamms XL1-Blue und die kompetenten Zellen des Stammes
BL21(DE3) wurden auf die selbe Art und Weise zur Transformation verwendet.
26
Gentechnische Methoden
Transformation durch Hitzeschock
Die bei –80 °C gelagerten Zellen wurden langsam auf Eis aufgetaut. Zu 40 µL der Zell­
suspension wurde 1 µL (etwa 100 ng) der zu transformierenden DNA-Lösung gegeben und 20 min
auf Eis inkubiert. Anschließend wurde der Transformationsansatz 45 s bei 42 °C im Wasserbad
einem Hitzeschock ausgesetzt und danach nochmals für 2 min auf Eis inkubiert. Abschließend
wurden 400 µL SOC-Medium zum Transformationsansatz gegeben und 1 h bei 37 °C zur
Expression der plasmidkodierten Ampicillinresistenz inkubiert. Zur Selektion wurden 200 µL des
Ansatzes mit Hilfe eines Drigalski-Spatels auf eine LBA-Agarplatte ausgestrichen und ü. N. bei
37 °C inkubiert.
Glycerinstammkulturen
Zur Herstellung von Glycerinstammkulturen für die Proteinpräparation wurde von der LBAAgar-Platte eine einzelne Kolonie ausgewählt und mit dieser 15 mL 2YTA-Medium angeimpft.
Nachdem die Kultur eine OD600 von ungefähr 0.4 erreicht hatte, wurden 500 µL 1:1 mit 90%-igem
Glycerin vermischt und anschließend bei -80 °C eingelagert.
3.1.6
DNA-Sequenzierung
Die DNA-Sequenzierungen wurden von den Firmen Seqlab (Göttingen) und GATC Biotech
(Konstanz) durchgeführt. Die Firmen benutzen zur Sequenzierung eine Variante der Didesoxy­
methode nach Sanger (1977). Ein primer, der komplementär zu einem Bereich in 3'-Richtung der zu
sequenzierenden DNA ist, wird mit Hilfe von DNA-Polymerase, dNTPs und mit verschiedenen
Fluoreszenzfarbstoffen markierte ddNTPs zu einem DNA-Tochterstrang verlängert. Die Synthese
dieses Tochterstranges wird abgebrochen, sobald ein ddNTP eingebaut wird, dem die 3´-OHGruppe und somit der Anknüpfungspunkt für ein neues dNTP fehlt. Zur Amplifizierung werden,
ähnlich wie bei einer PCR, mehrere Zyklen durchlaufen. Die so generierten unterschiedlich langen
Stücke werden elektrophoretisch getrennt und durch die Detektion der vier unterschiedlichen
Fluoreszenzmarker ausgelesen.
Es wurden jeweils 3 µg der zu untersuchenden Plasmid-DNA in 15 µL v.e. H2O und 10 µL
primer-Lösung der Konzentration 10 pmol/µL abgegeben. Der primer war so gewählt, dass er etwa
100 bp vor der zu sequenzierenden Stelle anlagern konnte. Für das CysM-Gen aus E. coli wurde die
Sequenzierungsmethode für GC-reiche Sequenzen gewählt, da dadurch die Qualität der Sequen­
zierungsergebnisse verbessert wurde.
27
Methoden
3.1.7
Bestimmung der DNA-Konzentration
Die Bestimmung der DNA-Konzentration erfolgte durch Messung der Absorption bei einer
Wellenlänge von 260 nm. Eine exakte Bestimmung ist nur bei Kenntnis der Zusammensetzung und
der Extinktionskoeffizienten der Nukleotide möglich. Der Extinktionskoeffizient eines Oligo­
nukleotids berechnet sich nach Gleichung 2 zu:
= n AMP⋅ AMP nCMP⋅ CMP n GMP⋅ GMPnTMP⋅TMP
Gleichung 2
nXMP = Anzahl des jeweiligen Nukleotids im Oligonukleotid
εAMP = 15'400 M-1cm-1
εCMP = 7400 M-1cm-1
-1
-1
εGMP = 11'700 M cm
εTMP = 8800 M-1cm-1
Die Berechnung der DNA-Konzentration kann durch folgende Näherungen vereinfacht
werden: die mittlere Molmasse der Nukleotide beträgt 340 g/mol, alle Nukleotide kommen gleich
häufig vor, und der Absorptionswert von 1.0 entspricht einer Konzentration von 50 μg/mL dsDNA
(bei ssDNA 35 μg/mL) (Sambrook et al., 1989). Damit ergeben sich folgende Gleichungen:
c 1 [ μg / mL] = A260⋅X⋅F
c 1 [ μg / mL] =
c1 [ μg /mL]⋅1000
N⋅340 g /mol
Gleichung 3
Gleichung 4
X = Konzentration bei A260 = 1.0 (dsDNA: 50 µg/mL, ssDNA: 35 µg/mL)
F = Verdünnungsfaktor
N = Gesamtzahl der Nukleotide
3.2
3.2.1
Proteinpräparation
Herstellung und Reinigung der Sulfhydrylase
Das Reinigungsprotokoll wurde aus einer vorhergehenden Arbeit adaptiert (Zocher, 2003). Es
ist dort detailliert beschrieben. Durch gezielte Optimierung der Parameter der einzelnen
Reinigungsschritte und durch Verwendung geringerer Proteinmengen war es möglich, die Zeit für
die Proteinpräparation durch Verzicht auf die erste Anionenaustausch-Chromatographie an der
Source 30Q bei annähernd gleichbleibender Proteinqualität erheblich zu verkürzen. Eine kurze
Zusammenfassung der CysM-Reinigungen findet sich in Abbildung 11.
28
Proteinpräparation
Animpfen von 300 mL LBA-Medium
mit ü.N.-Kultur (E. coli BL21(DE3) mit pFL145stop)
Animpfen von 300 mL LBA-Medium
mit ü.N.-Kultur (E. coli BL21(DE3) mit pFL145stop)
Bakterienkultivierung, Induktion mit AHT und Zugabe Bakterienkultivierung, Induktion mit AHT und Zugabe
von Vitamin B6-Hydrochlorid, weitere Inkubation ü. N. von Vitamin B6-Hydrochlorid, weitere Inkubation ü. N.
Zellernte und Aufschluss mittels Ultraschall
Zellernte und Aufschluss mittels Ultraschall
Ionenaustausch-Chromatographie
an Source 30Q
Ionenaustausch-Chromatographie
an Source 15Q
Konzentration bei einer Ausschlussmasse von 10 kDa
Dialyse gegen Phosphat-Puffer und
anschließende Rekonstitution ü. N.
Dialyse gegen Phosphat-Puffer und
anschließende Rekonstitution ü. N.
Dialyse gegen IEC-Puffer A
Ionenaustausch-Chromatographie
an Source 15Q
Konzentration bei einer Ausschlussmasse von 10 kDa
Konzentration bei einer Ausschlussmasse von 30 kDa
Gelpermeationschromatographie an
Superdex-75 16/60 prep grade
Gelpermeationschromatographie an
Superdex-200 16/60 prep grade
Konzentration bei einer Ausschlussmasse
von 10 kDa und Dialyse gegen v.e.H2O
Konzentration bei einer Ausschlussmasse
von 30 kDa und Dialyse gegen Dialyse-Puffer
Kristallisation und Proteincharakterisierung
Kristallisation und Proteincharakterisierung
Abbildung 11:
Schematische Darstellung der Produktion und Reinigung von CysM aus E. coli; links nach Zocher
(2003), rechts das für diese Arbeit verwendete Reinigungsverfahren.
Bakterienkultivierung
Zur Expression wurde der Stamm BL21 (DE3) und das CysM-Gen auf dem Vektor pFL145stop (Schwarz-Herion, 2002) verwendet. Die Proteinproduktion ist im verwendeten Vektor durch
Anhydrotetracyclin (AHT) induzierbar. Zur Bakterienkultivierung in Luftschüttlern wurden je
300 mL LBA-Medium in zwei 1-L-Erlenmeyerkolben mit Schikane mit 3 mL Übernachtkultur des
plasmidtragenden Expressionsstammes angeimpft. Es wurde bei 37 °C unter Schütteln mit 190 rpm
inkubiert. Bei Erreichen einer OD600 von 0.6 - 0.8 wurde die Temperatur auf 25 °C gesenkt und mit
30 µL AHT-Lösung induziert. Die Bakterien wurden über Nacht kultiviert und im Anschluss
zentrifugiert (20 min, 13'700*g, 4 °C). Das überstehende klare Medium wurde verworfen. Das
Bakterienpellet wurde gewogen und bis zur weiteren Verarbeitung bei -20 °C eingefroren.
29
Methoden
Tiefgefrorene Zellen wurden bei 4 °C aufgetaut und mit dem dreifachen Volumen (v/w) des
Bakterienfeuchtgewichtes IEC-Puffer-CysM-A resuspendiert. Zum Abbau von DNA wurde pro
15 mL Suspension 1 µL Benzonase zugegeben. Der Aufschluss erfolgte durch Ultraschall unter Eis­
kühlung. Insgesamt wurde 5 min beschallt, wobei jeder Beschallung von 1 s eine Pause von 2 s
folgte, um eine zu starke Erwärmung der Suspension zu vermeiden. Der Aufschluss wurde
abschließend zentrifugiert (38'000*g, 20 min, 4 °C) und vor Auftrag auf die Ionenaustauschchro­
matographie durch eine Membran (0.45 µm) filtriert.
Ionenauschtauschchromatographie
Bei der Ionenaustauschchromatographie (IEC) werden geladene Stoffe reversibel an eine
unlösliche, chemisch inerte und geladene Matrix gebunden. Zu diesen Stoffen zählen auch Proteine,
die eine vom pH-Wert abhängige Nettoladung besitzen. Die Trennung basiert darauf, dass die zu
trennenden Proteine mit verschiedenen Affinitäten an die Säulenmatrix binden und anschließend
durch Anlegen eines linear ansteigenden Salzgradienten (die Salzionen konkurrieren mit den
Proteinen um die Matrix) sukzessiv wieder vom Säulenmaterial gelöst und dadurch zu
unterschiedlichen Zeitpunkten eluiert werden.
Im ersten Reinigungsschritt wurde der Rohextrakt der IEC an Source 15Q zugeführt. Dieses
zu den starken Anionentauschern zählende Material trägt über einen Linker quartäre AminoethylSubstituenten und erreicht aufgrund der kleinen Partikelgröße von 15 µm eine hohe Trennleistung
bei dennoch hoher Kapazität.
Tabelle 6
30
Parameter der Reinigung von CysM mittels IEC an Source 15Q
Säule
Source 15Q
Material
monodisperse Polystyrolkügelchen (15 µm) mit quartären
Aminen als funktionelle Gruppen
Abmessungen
∅: 15 mm, H: 57 mm, V: 6 mL
Äquilibierung
3 Vt mit IEC-Puffer-CysM-A
Auftragsfluss
1.0 mL/min
Waschen
10 Vt mit IEC-Puffer-CysM-A
Salzgradient
0 - 100 % IEC-Puffer-CysM-B, Gradient über 80 Vt
Flussrate
2 mL/min
Fraktionsvolumen
2.5 mL
Detektion
280 / 412 nm
Temperatur
4 °C
Proteinpräparation
Nach Äquilibrierung der Säule mit 3 Vt (Vt=30 mL) IEC-PufferCysM-A wurde der Rohextrakt
bei einer Flussrate von 1.0 mL/min aufgetragen. Anschließend wurde über 10 Vt bei einem Fluss
von 2 mL/min ungebundene Proteine ausgewaschen, bevor die Sulfhydrylase mit einem NaClGradienten von 29 % IEC-Puffer-CysM-B Puffer (entsprechend 290 mM IEC-Puffer-CysM-B)
eluierte. Tabelle 6 fasst die Parameter der verwendeten Säule, sowie den eingestellten Gradienten
zur Elution zusammen. Leitfähigkeit und Absorption bei 280 nm und zusätzlich bei 412 nm zur
Detektion des Cofaktors wurden kontinuierlich verfolgt und aufgezeichnet. Die Fraktionen wurden
mittels SDS-PAGE untersucht. Die reinen CysM-Fraktionen wurden vereinigt und mittels Konzen­
trationszellen (MWCO 30 kDa) auf etwa 2 mL eingeengt.
Gelpermeationschromatographie
Bei der Gelpermeationschromatographie (GPC) werden unter der Voraussetzung einer
globulären Form Proteine aufgrund ihrer Größe bzw. Molekularmasse voneinander getrennt. Daher
eignet sich dieses Trennverfahren auch dazu, die Molekularmasse von Proteinen in ihrer nativen
Form abzuschätzen. Die Gelmatrix besteht aus Polydextrankugeln mit Poren definierter Größe, in
die Moleküle diffundieren können. Dadurch werden kleine Moleküle stark retardiert, während
große Moleküle nahezu ungehindert durch die Matrix wandern. Zur Vermeidung unspezifischer
Protein-Protein-Wechselwirkungen sowie von Interaktionen zwischen Proteinen und Säulenmaterial
wurden dem GPC-Puffer-CysM 200 mM NaCl zugesetzt. Um Schwebeteilchen zu entfernen, wurde
die Proteinprobe vor dem Auftrag zentrifugiert (15'000*g, 4 °C, 10 min) und durch eine 0.2 µm
Membran filtriert.
Tabelle 7
Parameter der Reinigung von CysM mittels GPC an Superdex S200
Säule
Superdex-200 16/60
Säulenmaterial
Hochvernetztes Agarosegel mit kovalent gebundenem Dextran
Trennbereich
10 kDa bis 600 kDa
Abmessungen
∅: 15 mm, H: 68 mm, V: 120 mL
Kapazität
bis zu 30 mg Protein
Äquilibierung
2 Vt, GPC-Puffer-CysM
Flussrate
0.5 - 0.8 mL/min
Fraktionsgröße
1.5 mL
Temperatur
4 °C
Zur GPC wurde eine Säule mit Superdex-200-Material verwendet. Die Parameter der verwen­
deten Säule sind in Tabelle 7 aufgeführt. Zuvor wurde die CysM-haltige Lösung auf etwa 2% des
Säulenvolumens konzentriert. Die GPC wurde mit einem Äkta Prime FPLC-System durchgeführt,
31
Methoden
die Elution der Proteinlösung bei 280 nm mit der PrimeView Software verfolgt und Fraktionen zu
je 1.5 mL gesammelt. Diese wurden mittels SDS-PAGE untersucht und CysM-haltige Fraktionen
vereinigt. Die resultierende CysM-Lösung wurde daraufhin gegen Dialyse-Puffer-CysM dialysiert
und nach Einstellen der Proteinkonzentration entweder zur Kristallisation eingesetzt, dem
Aktivitätstest zugeführt oder bis zur Verwendung bei -20 °C eingelagert.
3.2.2
Herstellung und Reinigung von AurF
Die Proteinproduktion und Reinigung von AurF wurde zu wesentlichen Teilen von Robert
Winkler
(Hans-Knöll
Institut,
Jena)
durchgeführt.
Eine
kurze
Zusammenfassung
des
Reingungsprotokolls wird an dieser Stelle gegeben und kann detailliert in der Literatur nachge­
schlagen werden (Winkler et al., 2006).
AurF wurde in das Plasmid pMAL-c2x kloniert und als Fusionsprotein bestehend aus einem
N-terminalen maltosebindenden Protein und dem C-terminal gelegenen AurF in E.coli BL21(DE3)Zellen produziert. Nach Aufschluss des Bakterienpellets erfolgte die Reinigung mittels
Affinitätschromatographie an Amylose-Material und Ionenaustauschchromatographie an einer
HiTrapQ-Säule. Nach proteolytischem Verdau mit Faktor-Xa Protease erfolgte die Isolierung des
nativen AurF durch erneute IEC an einer HiTrap Q-Säule, der zur Entfernung der Faktor-Xa
Protease eine Benzamidin FastFlow-Säule nachgeschaltet war. Das derart isolierte AurF wurde zur
Strukturbestimmung übergeben.
Gelpermeationschromatographie
Zur Entfernung von Aggregaten und restlichem Maltose-bindenden Protein erfolgte im finalen
Reinigungschritt eine GPC an Superdex S200-Material. Das native AurF wurde mittels einer Zentri­
fugalkonzentrationzelle der Firma VivaScience (MWCO 30 kDa) auf etwa 1-3% des
Säulenvolumens konzentriert und durch eine 0.22 µm Membran steril filtriert. Die GPC wurde mit
einer Äkta Prime FPLC Einheit durchgeführt und die UV-Absorption bei 280 nm verfolgt. Die
Säulendimensionen und Charakteristika der Chromatographie entsprechen der in Tabelle 7
durchgeführten Chromatographie, wobei GPC-Puffer-AurF verwendet wurde. Nach SDS-PAGE
wurden die AurF-haltigen Fraktionen vereinigt, konzentriert und kristallisiert. Überschüssiges
Protein wurde nach Zugabe von Glycerin (Endkonzentration 20% v/v) bei -80 °C bis zur weiteren
Verwendung eingelagert.
32
Proteincharakterisierung
3.3
Proteincharakterisierung
3.3.1
Bestimmung der Proteinkonzentration
Ist der molare dekadische Extinktionskoeffizient εx eines Proteins bekannt, so kann die
Konzentration von gereinigten Proteinlösungen durch Messung bei einer Wellenlänge von 280 nm
bestimmt werden. Da ε280 der einzelnen Aminosäuren bekannt sind, läßt sich bei Kenntnis der
Zusammensetzung eines Proteins ein theoretischer Wert für dessen Extinktionskoeffizienten
berechnen. Bei einer Wellenlänge von 280 nm absorbieren die Aminosäuren Tryptophan, Tyrosin
und Cystein in Disulfidbrücken (Gill & von Hippel, 1989), weshalb sich die in angegebene Formel
ergibt:
280 = n Trp⋅5690n Tyr⋅1280n Cystin⋅120⋅M−1 cm −1
nx
ε280
Gleichung 5
Anzahl der entsprechenden Aminosäuren im Protein
molarer dekadischer Extinktionskoeffizient des Proteins bei 280 nm
Die Anzahl der absorptionsaktiven Aminosäurereste sind für das jeweilige Protein im
folgenden beschrieben. Aus diesen errechnen sich unter Anwendung des Lambert-Beer´sche
Gesetzes nach Gleichung 6 die Umrechnungsfaktoren zur Proteinbestimmung.
c [mg /mL] =
A280:
Mr:
d:
Fprot:
A280⋅M r [ Da]
ε M −1 cm−1 ⋅d [cm]
=F Prot⋅A280
Gleichung 6
Absorption bei λ = 280 nm
Molekularmasse
Schichtdicke der Küvette
Umrechnungsfaktor für das jeweilige Protein (siehe unten)
CysM
CysM enthält drei Tryptophan-, sechs Tyrosin- und zwei Cystein-Reste, die jedoch keine
Disulfidbrücken ausbilden. Damit ergibt sich ein Extinktionskoeffizient bei 280 nm von
24'750 M-1cm-1. Aufgrund des im CysM kovalent gebundenem Kofaktors Pyridoxalphosphat (PLP)
muß dieser neben der Absorption der Aminosäuren gesondert berücksichtigt werden. Das in Form
einer Schiffschen Base an das Protein gebundene Pyridoxal-5'-phosphat zeigt ein Absorptions­
maximum sowohl bei 280 nm als auch bei 412 nm. Der Extinktionskoeffizient beider Maxima des
PLP wurde zu ε280=ε412=7600 M-1cm-1 bestimmt (Hidalgo et al., 1994). Damit ergibt sich der
Extinktionskoeffizient bei 280 nm von CysM durch Summation der Extinktionskoeffizienten von
33
Methoden
Protein und Cofaktor. Ebenso kann das Verhältnis von Holo- zu Apoenzym aus dem
Absorptionsverhältnis A280:A412 abgeschätzt werden. Für den CysM-Wildtyp beträgt dieses 4.26 bei
100% reinem Holoenzym. Der Faktor zur direkten Umrechnung der Proteinkonzentration aus der
gemessenen Absoprtion ergibt sich damit zu FProt(CysM) = 1.01.
AurF
AurF besitzt acht Tryptophane, sechs Tyrosine und keine Disulfidbrücke, wenngleich zwei
Cysteine-Reste in der Aminosäuresequenz zu finden sind. Damit ergibt sich der Umrechnungsfaktor
zur Konzentrationsbestimmung aus der Extinktion zu FProt(AurF) = 0.72
3.3.2
Enzymassay von CysM
Michaelis-Menten Kinetik
Enzymatische Aktivitäten werden durch die Michaelis-Menten-Gleichung beschrieben, wobei
dieser Kinetik die unter Gleichung 7 beschriebene Reaktion zu Grunde liegt. Das Enzym bildet mit
dem Substrat einen Enzym-Substrat-Komplex, der im Weiteren zum Produkt umgesetzt wird.
E+S
E:
S:
P:
ES:
kn:
k1
k-1
ES
k2
P +E
Gleichung 7
Enzym
Substrat
Produkt
Enzym-Substrat-Komplex
Geschwindigkeitskonstante
Unter den Annahmen eines präexistierenden Gleichgewichtes zwischen Enzym und Substrat
(k-1 >> k2) und eines Fließgleichgewichtes ([ES]=const.) sowie weiterer Vereinfachungen, lässt sich
aus obigem Reaktionsablauf die Michaelis-Menten-Gleichung (Gleichung 8 ) ableiten:
v=
v:
Vmax:
kcat:
[E]t:
[S]:
KM:
34
V max⋅[S ]
K M [S ]
Reaktionsgeschwindigkeit
maximale Reaktionsgeschwindigkeit; Vmax=kcat*[E]t
katalytische Konstante, Wechselzahl
Gesamtkonzentration des Enzyms
Substratkonzentration
Michaelis -Menten-Konstante; KM=(k-1+k2/k1)
Gleichung 8
Proteincharakterisierung
Die kinetischen Parameter werden normalerweise nicht mit einem v gegen [S] Diagramm
ausgewertet, da Vmax experimentell oft nicht erreicht werden kann und daher die aus der Messung
resultierende Hyperbel im Bereich der Substratsättigung derart flach verläuft, dass in Folge dessen
der Fehler von Vmax relativ groß wird und sich dann auf KM überträgt. Die Michaelis-MentenGleichung kann jedoch auf verschiedene Arten in eine lineare Form überführt werden. Die
gebräuchlichste Linearisierung ist der doppelt reziproke Auftrag nach Lineweaver-Burk
(Gleichung 9). Zur Auswertung wird 1/v gegen 1/[S] aufgetragen. Aus Steigung und
Achsenabschnitt der erhaltenen Gerade können KM und Vmax errechnet werden.
1 KM 1
1
=
⋅
v V max [ S ] V max
Gleichung 9
Werden die kinetischen Parameter eines Enzyms über die Michaelis-Menten-Gleichung und
deren Linearisierung ermittelt, muss die angewendete Messmethode bestimmten Bedingungen
genügen. So soll zum Beispiel Substratsättigung vorliegen. Als Richtwert sollte für die
Enzymkonzentration [E] < 0.001[S]0 ([S]0: Anfangskonzentration des Substrats) gelten (Lottspeich
& Zorbas, 1998). Auch sollten stets initiale Geschwindigkeiten gemessen werden, d. h. der
gemessene Umsatz sollte max. 10% betragen. Dies verhindert einerseits einen Fehler der aus einer
Produktinhibition resultieren könnte, andererseits werden die Grundannahmen, die zur Ableitung
der Michaelis-Menten-Gleichung getroffen wurden, kaum verletzt. Im Rahmen der Messungen der
kinetischen Parameter der OASS wurden diese Bedingungen soweit als möglich beachtet.
Die Wechselzahl kkat gibt an, wieviele Moleküle Substrat von einem aktiven Zentrum des
Enzyms je Zeiteinheit umgesetzt werden. Damit bestimmt die Wechselzahl zusammen mit der
Michaliskonstanten die Effizienz eines Enzyms. Die Wechselzahl kkat wird durch Bestimmung der
Aktivität dividiert durch die Enzymkonzentration (Gleichung 10) erhalten.
k kat =
V max
[ E ]T
Gleichung 10
Aktivität der Sulfhydrylase
Um die enzymatischen Aktivität der OASS photometrisch bestimmen zu können, wurde ein
Aktivitätstest verwendet, der an einer Sulhydrylase aus Salmonella typhimurium etabliert wurde
(Tai et al., 1993). Im Gegensatz zur der von CysM katalysierten Reaktion wird in diesem Test das
chromophore S-Nukleophil 5-Mercapto-2-nitrobenzoat (TNB) anstatt des Hydrogensulfids als
35
Methoden
Substrat umgesetzt, so dass sich die Eduktabnahme photometrisch bei λ = 412 nm und 500 nm
verfolgen lässt (εTNB,412 = 13`600 M-1 cm-1). Die Produkte S-(3-Carboxy-4-nitrophenyl)-L-cystein und
Acetat resultieren dabei aus der Umsetztung von TNB mit OAS (Abbildung 12).
S-
O
H3C
O
COO-
COO +
NH3
OASS
+
COO-
O2N
S
+
NH3
+
O
H3C
O-
-OOC
NO 2
O-Acetyl-L-Serin
Abbildung 12:
TNB
S-(3-Carboxy-4-nitrophenyl)-L-cystein
Acetat
Reaktion der OASS mit TNB als nukleophiles Substrat, im Gegensatz zur natürlichen Reaktion
wird eine chromophore Gruppe als Edukt eingesetzt, so dass sich durch Detektion der Absorptionsabnahme der Reaktionsverlauf verfolgen läßt (aus Wiesand, 2007).
Für die Aktivitätsmessung wurden 950 µL Puffer (100 mM HEPES pH 7.0, 10 mM
O-Acetlyserin und 10-1000 µM TNB) bei 37 °C und bei 25 °C für 3 min inkubiert und die Messung
nach Zugabe von 50 µL der zu untersuchenden Proteinlösung gestartet. Die zugegebene
Proteinmenge wurde im Bereich von 0.5 µg bis 80 µg variiert. Die eingesetzte Proteinmenge wurde
frisch präpariert und die Zeit der benötigten Inkubation bei 37 °C und 25 °C minimiert. Das
verwendete TNB wurde durch Zugabe von 2 mM DTT zu einer Lösung von 0.5 mM DTNB in
HEPES-Puffer (50 mM, pH 7.0) frisch hergestellt. Ein Einfluss des Schwefelnukleophils DTT auf
die Reaktion konnte ausgeschlossen werden. Die Absorption wurde bei 412 nm verfolgt und zur
Auswertung der Extinktionskoeffizient von TNB (ε412=13600 M-1cm-1) verwendet (Tai et al., 1993).
Um eine Substratsättigung zu erreichen, wurde die Absorption zusätzlich bei 500 nm verfolgt. Der
Extinktionkoeffizient folgte dabei aus der Messung von zehn verschiedenen TNB-Stammlösungen
im zu messenden Substratbereich zu 970 M-1cm-1. Das entstehende Produkt Cysteinnitrobenzoat
(CNP) zeigte bei 312 nm ein Absorptionsmaximum, hatte allerdings bei 412 nm keine nennens­
werte Absorption, so dass ein störender Einfluss nicht vorhanden war.
3.3.3
Diskontinuierliche SDS-Polyacryamidgel-Elektrophorese
Die diskontinuierliche SDS-Polyacryamidgel-Elektrophorese (SDS-PAGE) wurde zur Ab­
schätzung der Reinheit und des Proteingehalts einer Lösung verwendet (Laemmli, 1970). Die Gele
werden durch Copolymerisation von Acrylamid mit N,N‘-Methylenbisacrylamid hergestellt. Durch
Variation der Konzentrationen und des Verhältnisses der beiden Monomeren ist es möglich, die
Porengröße und den Vernetzungsgrad des Gels zu beeinflussen (Westermeyer, 1990).
36
Proteincharakterisierung
Die Trennung der Proteine erfolgte in 12%igen Gelen bei pH 8.8. Die Fokussierung der
Proteinbanden kann erheblich verbessert werden, wenn die Proben vor dem kleinporigen Trenngel
durch ein großporiges 6%iges Sammelgel bei pH 6.8 laufen (Righetti et al., 1990).
Das anionische Detergenz SDS (Natriumdodecylsulfat, sodium dodecylsulfate) bindet Proteine
im festen Massenverhältnis 1.4:1 in Mizellen und maskiert so deren Eigenladung. Dadurch erfolgt
eine Trennung der Proteine ausschließlich aufgrund der Masse des Polypeptids. Durch Vergleich
mit einem bei der SDS-PAGE mitlaufenden Proteinstandard kann die Molmasse der zu
analysierenden Proteine bestimmt werden. Die SDS-PAGE wurde mit 6%-igen Sammelgelen und
12%igen Trenngelen durchgeführt. Die Proben wurden mit SDS-Probenpuffer verdünnt, kurz auf
95°C erhitzt und 15 µL der Mischung je Geltasche aufgetragen. Die Elektrophorese erfolgte bei
konstanter Stromstärke von 45 mA. Die Gele wurden in heißer Coomassie-Lösung 5 min gefärbt
und anschließend mit Entfärberlösung behandelt. Zur Archivierung wurden die Gele photographiert
und zur Konservierung zwischen zwei Cellophanfolien getrocknet.
3.3.4
Dynamische Lichtstreuung
Die dynamische Lichtstreuung (DLS) wird zur Bestimmung von Molekularmasse, Reinheit
und Homogenität einer Proteinprobe eingesetzt. Bei der DLS werden Fluktuationen der Intensität
des Streulichts eines monochromatischen Lichtstrahls in einer Probe unter einem bestimmten
Winkel (90°) gemessen. Verursacht werden die Fluktuationen im Streuverhalten durch statistische
Bewegungen der Moleküle, wodurch die Mobilität und der Diffusionskoeffizient D meßbar werden.
Unter
Berücksichtigung
der
Brownschen
Molekularbewegung
und
mit
Kenntnis
des
Diffusionskoeffizienten D läßt sich der hydrodynamische Radius RH über die Stokes-EinsteinBeziehung nach Gleichung 11 berechnen (Burchard, 1985). Aus RH kann unter Annahme einer
mittleren Dichte ρ für Protein von 1.377 g·cm-3 (Herstellerangabe Firma Protein Solutions) und der
Annahme eines globulären Teilchens die Masse eines Proteinmoleküls, wie in Gleichung 12
beschrieben, berechnet werden.
RH=
RH
k
T
η
D
ρ
k⋅T
6 ⋅D⋅
Gleichung 11
hydrodynamischer Radius [m]
Boltzmann Konstante [J·K-1]
absolute Temperatur [K]
Viskosität des Lösungsmittels [kg·m-1·s-1]
Diffusionskoeffizient [m2·s-1]
Dichte des Partikels [kg·cm-3]
37
Methoden
4
M r = ⋅⋅R H3⋅
3
Gleichung 12
Die DLS gibt Aufschluss über die Kristallisierbarkeit eines Proteins (D'Arcy, 1994; FerréD'Amaré & Burley, 1997). Falls die Proteinprobe monomodal vorliegt, ist die Wahrscheinlichkeit,
dass das Protein kristallisiert, wesentlich höher als bei polydispersen Proben.
Die Messung der DLS erfolgte mit Proteinkonzentrationen von etwa 1 mg/mL. Dazu wurde
das Protein (10 mg/mL) 1:10 mit dem entsprechenden DLS-Puffer (pH 5.0, 6.0 und 7.0) verdünnt
und anschließend zentrifugiert (16'000*g, 30 min, 4 °C). Insgesamt wurden 13 µL Lösung in eine
Quarzküvette gegeben. Es wurden etwa 20 Messungen durchgeführt und mit der Auswertesoftware
DynaPro-801 (Firma Protein Solutions) analysiert.
3.4
3.4.1
Proteinkristalle
Kristallgitter und Symmetrie
Kristalle sind periodische Gefüge von identischen, sich wiederholenden Einheiten. Diese
Einheiten werden Elementarzellen genannt und sind per Definition das kleinste Volumen innerhalb
eines Kristalls, das durch eine reine Translations-Operation in seinen benachbartes Äquivalent
überführt werden kann. Die Beschreibung der Elementarzelle erfolgt durch die drei Raumvektoren
a , b ,c . Der ganze Kristall kann aus dabei aus den auftretenden Symmetrieelementen in
Kombination mit den Gitter-Translationen beschrieben werden. Es gibt 32 Möglichkeiten, die in
Kristallen erlaubten Symmetrien zu kombinieren, woraus 230 mögliche Raumgruppen resultieren
(International Tables for Crystallography, 1996). Jede Raumgruppe wird dabei durch einen
bestimmten Satz von Symmetrie-Operationen definiert. Werden die Symmetrie-Operationen auf
einen bestimmten Teil der Zelle, die so genannte asymmetrische Einheit, angewendet, entsteht
daraus die gesamte Elementarzelle. Die asymmetrische Einheit entspricht dabei dem kleinsten
Volumen innerhalb der Elemantarzelle, der nicht weiter durch Symmetrie-Operationen reduziert
werden kann. Ziel der Röntgenstrukturanalyse ist daher die Bestimmung der atomaren Anordnung
innerhalb der asymmetrischen Einheit.
Unter Berücksichtigung der 32 möglichen Punktgruppen lassen sich sieben Kristallsysteme
unterscheiden: triklin, monoklin, orthorhombisch, tetragonal, trigonal, hexagonal und kubisch.
Durch Kombination dieser Kristallsysteme mit den möglichen Kristallpackungen (primitv,
38
Proteinkristalle
innenzentriert, flächenzentriert) ergeben sich die 14 Bravais-Gitter. Für die Kristallographie von
Proteinen sind nicht alle möglichen Raumgruppen von Bedeutung. Da biologische Makromoleküle
Chiralität aufweisen, reduzieren sich die möglichen Symmetrieelemene auf Dreh- und Schrauben­
achsen. Damit reduziert sich die Zahl der möglichen Punktgruppen auf elf und die daraus
resultierenden Raumgruppen auf 65.
3.4.2
Kristallisation von Proteinen
Für die Röntgenstrukturanalyse werden hochgeordnete Einkristalle ausreichender Größe
benötigt. Bis heute ist es allerdings nicht möglich, geeignete Bedingungen für die Kristallisation
eines Proteins vorherzusagen. Die Herstellung geeigneter Proteinkristalle ist daher ein
zeitintensiver, empirischer Prozess, der große Mengen hochreines Protein benötigt, um
systematische Kristallisationsexperimente durchzuführen. Die heute üblichen Kristallisations­
methoden beruhen sämtlich auf dem Prinzip, dem Protein durch die sukzessive Erhöhung eines
geeigneten Fällungsmittels Solvens zu entziehen und so die Ausbildung von Protein-ProteinWechselwirkungen zu favorisieren. Wird die Sättigungsgrenze des Proteins erreicht, entsteht ein
metastabiler Zustand, in dem Kristallwachstum, aber keine Keimbildung möglich ist. Erst durch
weitere Erhöhung der Fällungsmittelkonzentration wird ein labiler Zustand erreicht, in dem sich
spontan Kristallisationskeime ausbilden können (McPherson, 1990). Ideale Bedingungen zur
Kristallisation eines Proteins sind dann gegeben, wenn in der labilen Phase lediglich wenige Keime
entstehen und das System anschließend durch Kristallwachstum sowie die dadurch abnehmende
Proteinkonzentration in die metastabile Phase übergeht. Übliche Fällungsmittel zur Protein­
kristallisation sind anorganische Salze, wasserlösliche Polymere sowie organische Lösungsmittel.
Die Bildung von Kristallen wird durch eine Vielzahl von Parametern wie der Protein-,
Fällungsmittel- und Salzkonzentration, den verwendeten Puffersubstanzen, dem pH-Wert und der
Temperatur beeinflusst (Gilliland & Ladner, 1996). Eine systematische Variation aller Parameter ist
aufgrund des dazu notwendigen Zeit- und Materialaufwandes kaum möglich. Daher werden bei der
Suche nach initialen Kristallisationsbedingungen Reihenexperimente (screenings) durchgeführt, um
den vieldimensionale Parameterraum punktuell abzutasten (Carter & Carter, 1979, Cudney et al.,
1994). Häuig wird dabei ein von Jancarik & Kim (1991) entwickeltes screening verwendet, das auf
der statistischen Analyse erfolgreicher Kristallisationsbedingungen basiert. Heute sind von
zahlreichen Herstellern eine ganze Reihe solcher screenings kommerziell erhältlich. Protein­
39
Methoden
kristalle, die daraus initial gewachsen sind, können anschließend durch Variationen der Bedingun­
gen optimiert werden. Eine Möglichkeit, das Kristallisationsverhalten eines Proteins zu verändern,
besteht darin, die Oberfläche des Moleküls durch gezielte Mutationen zu modifizieren.
Die für die Kristallisation von Proteinen am häufigsten verwendeten Methoden sind die
Gasphasendiffusion, die Dialyse und das Batchverfahren (Ducruix & Giegé, 1992). In dieser Arbeit
wurde für initiale screening-Experimente die Sitztropfen-Methode (sitting drop) eingesetzt,
während die Hängetropfen-Methode (hanging drop) bei der Verfeinerung initial erhaltener
Bedingungen Verwendung fand (Abbildung 13). Beide Methoden sind Varianten der Gasphasen­
diffusion (McPherson, 1990). Ein Tropfen einer Proteinlösung wird an einem Deckplättchen über
einer Reservoirlösung aufgehängt oder auf eine dafür vorgesehene Stelle gesetzt und das Gefäß
luftdicht verschlossen. Die Konzentration des Fällungsmittels ist in der Proteinlösung geringer als
in der Reservoirlösung. Daher wird dem Proteintropfen solange durch Diffusion über die Gasphase
Wasser entzogen, bis ein Konzentrationsausgleich zwischen den Lösungen erreicht ist. Dadurch
steigen die Konzentrationen des Fällungsmittels und des Proteins langsam an und die Lösung
erreicht den übersättigten Bereich. Die Geschwindigkeit dieses Prozesses wird durch den
Konzentrationsunterschied zwischen Protein- und Reservoirlösung gesteuert. Da das Volumen des
Reservoirs um ein Vielfaches größer ist als das des Tropfens, entspricht die Endkonzentration des
Fällungsmittels ungefähr der Konzentration im Reservoir.
A
B
Abbildung 13: Schematische Darstellung der Kristallisation nach der Sitztropfen-Methode A) und der HängetropfenMethode B).
Kristallisationsexperimente nach der Sitztropfen-Methode wurde in den kommerziell
erhältlichen Crystal Clear Boxen (Hampton Research) durchgeführt, wobei 70 µL Kristallisations­
puffer in das Reservoir vorgelegt wurden. Anschließend wurde je 1 µL Proteinlösung mit 1 μL
Reservoirpuffer auf der dafür vorgesehenen Auftragsstelle vermischt. Eine Austrocknung der
Proteintropfen wurde durch Abkleben der Box mit Crystal Clear Klebeband verhindert. Für die
Kristallisationsexperimente
40
nach
der
Hängetropfen-Methode
wurden
Kristallisationsboxen
Proteinkristalle
verwendet, die ursprünglich zur Kultivierung von Zellen entwickelt worden waren. Der über­
stehende Rand der Vertiefungen wurde mit Sandpapier geglättet und mit Silikonöl bestrichen. Die
Vertiefungen wurden mit 500 µl Reservoirpuffer gefüllt und ein silikonisiertes Deckplättchen mit
anhängendem Tropfen aufgesetzt. Durch das Silikonisieren der Glasoberfläche kann das Verlaufen
des Tropfens auf dem Deckplättchen verhindert werden. Dazu wurden diese kurz in eine
ethanolische Silikonlösung getaucht, an der Luft getrocknet und anschließend für 1 h auf 180 °C
erhitzt. Die Tropfen wurden durch Vermischen von 2 µL Reservoirlösung mit 2 µL Proteinlösung
mit einer Proteinkonzentration von 5 - 15 mg/mL hergestellt. Die Kristallisationsansätze wurden bei
20 °C inkubiert.
Agarosegelkristallisation
Konnten initiale Kristalle, die aus Screening Experienten erhalten wurden, durch
konventionelle Verfeinerung der Kristallisationsbedingungen wie Fällungsmittelkonzentration,
Salzkonzentration oder pH-Wert nicht optimiert werden, so wurde versucht, die Kristalle durch
Einsatz von Agarosegelen zu verbessern. Gelartige Medien erhöhen dabei den Diffusionsko­
effizienten, wodurch sowohl Konvektion als auch das Sedimentieren von Kristallen verhindert wird
(Biertüpfel et al., 2002). Zudem ist das Kristallwachstum verlangsamt aufgrund der höheren
Viskosität innerhalb des Gel, so dass die resultierenden Kristalle häufig von höherer Qualität sind.
Die Agarosegelkristallisation wurde wie die Hängetropfen-Methode durchgeführt. Allerdings
wurde das Protein zuvor mit einer speziellen, bei tiefen Temperaturen schmelzenden AgaroseLösung (2% (w/v), die zuvor auf 35 °C erhitzt wurde, im Verhältnis 1:4 mit Protein-Lösung
vermengt und 2 µL davon zügig auf die Deckplättchen pipettiert, bevor das Gel erstarrte. Nach
kurzer Wartezeit wurde zu der gelartigen Masse 2 µL des Reservoirpuffers hinzupippetiert, ohne
den Tropfen zu durchmischen. Das Deckplättchen wurde über dem Reservoir positioniert und bei
20 °C gelagert.
Tränkexperimente
Proteinkristalle weisen im allgemeinen einen hohen Anteil an Lösungsmittel auf und sind von
vernetzten Lösungsmittelkanälen durchzogen. Daher ist es Molekülen mit geringer Molekularmasse
möglich, an spezifische Bindestellen im Protein zu diffundieren. Somit können Schweratomderivate
oder Protein-Substrat-Komplexe durch Tränken von Kristallen in geeigneten Lösungen erhalten
werden (Petsko, 1985). Befinden sich Bindestellen im Bereich von Kristallkontakten oder werden
Konformationsänderungen des Proteins induziert, so wird häufig die Ordnung im Kristall gestört,
41
Methoden
was zur Verminderung des Streuvermögens bis hin zum Bruch des Kristalls führen kann. Schwer­
atomderivate von Kristallen wurden durch Zugabe von Schweratomlösungen in Reservoir- oder
Aufbewahrungspuffer sowie durch Zugabe von Schweratomverbindungen in ungelöster Form zu
Kristallen in Mutterlauge oder nach Umsetzen dieser in einen Aufbewahrungspuffer hergestellt. Die
Endkonzentration der Schweratomverbindung im Tropfen betrug dabei 1-10 mM, sofern nicht mit
Verbindungen in fester Form gearbeitet wurde und keine Quantifizierung des Schwermetallgehalts
der Tränklösungen erfolgen konnte. Die Tränkzeit wurde zwischen wenigen Minuten und mehreren
Stunden variiert und die Integrität der Kristalle durch visuelle, regelmäßige Überprüfung im
Lichtmikroskop bestätigt.
Cokristallisation
Der Zugang zu Bindestellen im Protein kann durch Kristallkontakte blockiert sein. Zudem
können bestimmte Konformationsänderungen von nöten sein, ohne die ein entsprechendes Substrat
keinen Zugang zur Bindestelle findet. Beide Problemstellungen lassen ein Einbringen eines
gewünschten Moleküls in das Protein durch Tränkexperimente unwahrscheinlich erscheinen. Als
Alternative zu den Tränkexperimenten bietet sich in diesen Fällen die Cokristallisation an. Dabei
wird das Protein gegebenenfalls nach Präinkubation mit dem Substrat, Inhibitor, Cofaktor oder
ähnlichem im Gemisch zu Kristallisationsexperimenten eingesetzt.
Die Cokristallisation wurde an der N-Oxygenase zum Einbringen verschiedener Substrate,
Zwischenprodukte und auch Substrat-Analoga eingesetzt. Dabei wurde zur gereinigten AurFLösung die zwei- bis fünfache molare Menge des gelöstem Zielmoleküls zugegeben. Der Ansatz
wurde für 20 min bei 45 °C inkubiert, um eine Besetzung der aktiven Zentren zu gewährleisten.
Daraufhin wurde sofort auf 4 °C abgekühlt, die Lösung über eine Membran filtriert (100 µm) und
direkt zur Kristallisation eingesetzt. Je nach erwünschter Proteinkonzentration wurde der Ansatz
durch Zugeben der entsprechenden Kristallisationslösung verdünnt.
3.4.3
Lösungsmittelgehalt von Proteinkristallen
Proteinkristalle bestehen neben den Polypeptidketten in hohem Maße auch aus Lösungsmittel­
molekülen, die zum überwiegenden Teil ungeordnet vorliegen. Die statistische Analyse des
Solvensgehalts von zahlreichen Kristallstrukturen führte zur Definition des sogenannten Matthewsoder Packungsparameters VM (Gleichung 13, Matthews, 1968).
42
Proteinkristalle
V M=
VM :
VEZ:
Mr :
z:
n:
V EZ
Mr ⋅ z ⋅ n
Gleichung 13
Packungsparameter [ų/Da]
Volumen der Elementarzelle [ ų]
Molekularmasse des Proteins [Da]
Anzahl der Moleküle pro asymmetrischer Einheit
Anzahl der asymmetrischen Einheiten pro Elementarzelle
Der Packungsparameter VM gibt das Volumen in ų an, das im Kristall jeder Molekular­
masseneinheit in Da durchschnittlich zur Verfügung steht. Bei löslichen Proteinen beträgt der VMWert 1.7 - 3.5 ų/Da mit einem Maximum der Verteilung bei etwa 2.2 ų/Da. Bei einer nicht zu
großen Anzahl von Molekülen in der asymmetrischen Einheit kann diese daher durch Einsetzen
verschiedener Werte für z ermittelt werden. Unter Annahme einer mittleren Proteindichte ρProt von
1.35 g/cm³ lässt sich nach Gleichung 4 aus dem Packungsparameter der Solvensgehalt χ eines
Proteinkristalls berechnen.
Der Solvensgehalt χ von Proteinkristallen beträgt nach dieser statistischen Auswertung zwi­
schen 30 % und 70 %, im Mittel etwa 50 %. Zudem zeigten diese Untersuchung, dass der Solvens­
gehalt eines Kristalls im Zusammenhang mit der Molekularmasse des Proteins steht (Kantardjieff &
Rupp, 2003).
z⋅n⋅M r
V
Prot⋅N
1.23
=1− Prot =1−
=1−
V EZ
z⋅n⋅V M⋅M r
VM
χ:
VProt :
N:
3.4.4
Gleichung 14
Solvensgehalt des Kristalls
Volumen des Proteinanteils im Kristall
Avogadro-Zahl [6.022 · 1023 mol-1]
Montage von Proteinkristallen
Aufgrund ihres hohen Solvensgehaltes müssen Proteinkristalle während röntgenographischer
Messungen vor dem Dehydrieren geschützt werden. Für Messungen bei Raumtemperatur geschieht
dies gewöhnlich durch Montieren des Kristalls in einer Kapillare, bei Messungen um 100 K durch
Montage in einem cryo loop und anschließendem Schockgefrieren im kalten Stickstoffstrom oder
flüssigem Stickstoff. Durch die Messung bei tiefen Temperaturen werden primäre und sekundäre
Strahlenschäden im Kristall drastisch vermindert (Garman, 1999), womit die Datensammlung an
Synchrotrons überhaupt erst möglich wird. Zur Vermeidung von Eiskristallen werden der
Mutterlauge Glasbildner wie Glycerin oder niedermolekulare PEGs in ausreichender Konzentration
43
Methoden
hinzugefügt, oder alternativ die Mutterlauge bzw. der Aufbewahrungspuffer gegen Öle wie
Paratone-N oder Paraffinöl ausgetauscht. Beide Methoden können die Kristallqualität stark
vermindern, vor allem die Mosaizität der Kristalle kann beträchtlich zunehmen (Mitchell &
Garman, 1994). Durch geschickte Wahl des cryo protectant, Optimierung seiner Konzentration
sowie eine langsame Erhöhung der Konzentration des Glasbildners können Schäden am Kristall
meist vermieden werden.
Kristallmontage in Glaskapillaren
Zur röntgenographischen Untersuchung von Proteinkristallen bei Raumtemperatur wurden
diese mit etwas Mutterlauge in eine silikonisierte Kapillare aus Quarzglas (Durchmesser
0.5 – 0.7 mm) überführt. Danach wurde die Mutterlauge um den Kristall mit Filterpapierstreifen
weitestgehend entfernt. Flüssigkeit, die keinen direkten Kontakt zum Kristall hatte, wurde in der
Kapillare belassen, um ein Austrocknen des Kristalls zu verhindern. An den Enden wurde die
Kapillare mit Hartwachs luftdicht verschlossen.
Kristallmontage in cryo loops
Um einen Proteinkristall cryokristallographisch zu untersuchen, wurde dieser schrittweise mit
steigenden Konzentrationen des Glasbildners bis zur gewünschten Endkonzentration versetzt und
anschließend mit einem cryo loop aus dem Cryopuffer entnommen. Dieser loop bestand aus einem
Nylonfaden, der in Form einer Schleife mit 0.2 bis 1.0 mm Durchmesser auf einer Halterung
montiert war. Um ein Austrocknen des Kristalls zu verhindern, wurde der loop unmittelbar nach der
Entnahme des Kristalls in den kalten Stickstoffstrom verbracht und so bei 100 K schockgefroren.
3.5
3.5.1
Röntgenographische Methoden
Röntgenstrahlung
Für die Proteinkristallographie wird Röntgenstrahlung mit einer Wellenlänge von etwa 0.1 nm
verwendet. Diese kann auf unterschiedliche Weise erzeugt werden. Zum einen stehen
Röntgengeneratoren zur Verfügung, die mittels Drehanoden die gewünschte Strahlung erzeugen.
Dazu werden Elektronen aus einem Glühdraht (Filament) emittiert und in einem elektrischen Feld
von 45 kV beschleunigt, bevor sie auf eine Kupferanode treffen. Durch die hohe Energie der
beschleunigten Elektronen werden Cu-Elektronen aus Schalen mit niedrigem Energieniveau
herausgeschlagen. Diese werden durch Elektronen aus höheren Schalen unter Emission von
Röntgenstrahlung ersetzt. Da dabei der größte Teil der Energie in Wärme umgesetzt wird, wird der
44
Röntgenographische Methoden
Fokus der Elektronen durch eine Rotation der Anode dauernd versetzt, wobei die Anode intensiv
gekühlt wird. Die emittierte Röntgenstrahlung wird mit Hilfe von Nickelfiltern- oder Graphitmono­
chromatoren auf die Wellenlänge der Cu Kα-Strahlung von 1.5418 Å eingestellt und über einen
Kollimator zu einem Strahl mit 0.3 mm Durchmesser gebündelt. Es wurden die Röntgengeneratoren
RU200 B und RU H2C der Firma Rigaku bei einer Spannung von 44 kV und einer Stromstärke von
118 mA betrieben.
Diese Röntgenstrahlung ist jedoch für die Proteinkristallographie nur eingeschränkt zu
verwenden, da die Intensität und die Brillanz des erzeugten Strahl oft nicht ausreichen, um
Datensätze mit guter Qualität zu messen. Daher wird Synchrotronstrahlung eingesetzt, die z. B. an
der Swiss Light Source in Villigen (Schweiz) oder BESSY (Berlin) zur Verfügung steht. Diese wird
erzeugt, indem im Hochvakuum Elektronen oder Positronen auf einer Kreisbahn auf relativistische
Geschwindigkeiten beschleunigt werden. Die Teilchen werden dazu mit starken Magneten
abgelenkt, wobei bei jeder Richtungsänderung Strahlung in einem breiten Wellenlängenbereich
(600 nm bis 0.01 nm) emittiert wird. Durch Reflektion an Einkristallen kann die gewünschte
Wellenlänge sehr genau eingestellt werden. Synchrotronstrahlung bietet im Vergleich zu Röntgen­
generatoren mit Drehanode eine wesentlich höhere Intensität, eine deutlich fokussierteren Strahl
sowie die Möglichkeit zur freien Wahl der Wellenlängen im Bereich von 0.7 bis 2.0 Å, was
insbesondere für die Phasenbestimmung nach der MAD-Methode von entscheidender Bedeutung
ist. Die Synchrotron-Messungen wurden an den beamlines X06SA des SLS in Villigen (Schweiz)
und den beamlines BL14.1 und 14.2 am BESSY (Berlin) durchgeführt.
3.5.2
Theorie der Röntgenstreuung
Tritt Röntgenstrahlung in Wechselwirkung mit Materie, so werden die Elektronen im
Wechselfeld der elektromagnetischen Strahlung zu Schwingungen angeregt. Als oszillierende
Dipole emittieren sie ihrerseits eine kohärente Streustrahlung, d. h. ihre Wellenlänge ist mit der
Wellenlänge der Primärstrahlung identisch und ihre Phase steht in eindeutiger Beziehung zur Phase
der Primärstrahlung. Diese Art von Streuung wird elastische oder Thomson-Streuung genannt.
Durch inelastische Streuung, auch Compton-Streuung genannt, hervorgerufene Effekte werden bei
der Röntgenstrukturanalyse vernachlässigt. Da die Wellenlänge der verwendeten Primärstrahlung in
der Größenordnung wie die Bindungsabstände der Atome im Streuer besitzt, kommt es zu
Interferenzerscheinungen. Ist die Materie ein Kristall, so wird die Symmetrie des entstandenen
Interferenzbildes (Reflexmusters) durch dessen Gittersymmetrie bestimmt, während die Intensitäten
der Reflexe durch die Anordnung der Streuzentren, also durch die Elektronendichteverteilung in der
45
Methoden
asymmetrischen Einheit, bedingt wird. Wird die einfallende Röntgenstrahlung als ebene Welle
betrachtet, so kann der Streuvorgang an Materie anhand des in Abbildung 14 dargestellten Modells
veranschaulicht werden. Dabei wird die Röntgenstrahlung als ebene Welle betrachtet. Besitzt die
einfallende Strahlung den Richtungsvektor k0 und die gestreute Welle den Richtungsvektor k , so
lässt sich die Streuamplitude E am Ort x durch Integration über alle streuenden Volumenelemente
d r mit der Elektronendichte el r nach Gleichung 15 berechnen werden.
te
De
r
kto
Streuer
einfallende
Welle
Abbildung 14:
Schematische Darstellung der Röntgenstreuung an Materie. Die einfallende Strahlung wird als ebene
Welle betrachtet, die zum Detektor hin gestreut wird.
0
E=konst⋅e i ⋅t−k R ∫ el r ⋅e i k− k r d r
Vol
ω = 2πc/λ:
t:
k0 :
k :
el r :
Gleichung 15
Kreisfrequenz ω und Wellenlänge λ der Röntgenstrahlung
Zeit
Wellenvektor des einfallenden Strahls ∣k0∣=∣k ∣=2 /
Wellenvektor des gestreuten Strahls
Elektronendichte am Ort d r
Im Term vor dem Integral sind physikalische Informationen über die Ausbreitung der ebenen
Welle im Raum enthalten. Das Integral selbst ist eine Funktion der Elektronendichteverteilung im
streuenden Volumenelement und wird daher als Strukturfaktor bezeichnet. Dieser kann durch
Einführung des Streuvektors s mit 2 s=k − k0 vereinfacht werden (Gleichung 16)
F s = ∫ el r ⋅e 2 s r d r
Vol
Gleichung 16
In dieser Gleichung wird die ortsabhängige Elektronendichte el r aus dem realen Raum in
den reziproken Raum überführt, in dem die komplexen Strukturfaktoren F s von s abhängig
sind. Aus mathematischer Sicht ist dieses Integral eine Fourier-Analyse. Bei der Röntgenstreuung
an Kristallen kommt es nur dann zu positiven Interferenzen, wenn die Phasenverschiebung zwi­
schen gestreuten Sekundärwellen ein Vielfaches von 2π ist. Daraus resultieren die LaueBedingungen:
46
Röntgenographische Methoden
a⋅s=h
b⋅s =k
c⋅s =l
Gleichung 17
h, k, l ganze Zahlen
Die ganzen Zahlen h, k und l werden als Millersche Indizes bezeichnet und beschreiben die
Periodizitäten des Proteinkristalls im realen Raum. Wird der Ortsvektor r des realen Raums in
fraktionellen Koordinaten x,y,z dargestellt, ergibt sich für den Strukturfaktor Gleichung 18.
1
F hkl =V EZ
1
1
∫ ∫ ∫ el x , y , z ⋅e 2 i hxkylz dxdydz
Gleichung 18
x=0 x=0 x=0
Durch Auflösen von Gleichung 18 nach der Elektronendichte am Ort x,y,z und FourierRücktransformation (Fourier-Synthese) der Strukturfatoren Fhkl lässt sich die Elektronendichte aus
den komplexen Strukturfaktoren berechnen. Da Diffraktion nur in diskreten Richtungen (LaueBedingungen), geht die Integration in eine Summation über.
el x , y , z =
1
∑ ∑ ∑ F h , k , l ⋅e−2 i hx kylz
V EZ h k l
Gleichung 19
Der Strukturfaktor F wird als komplexe Zahl durch seine Amplitude F(hkl) und Phase α(hkl)
beschrieben (Gleichung 20). Die Strukturfaktoramplituden sind dabei experimentell durch Messung
der Intensitäten Iobs(hkl) zugänglich (Gleichung 21), die Phasen hingegen können nicht direkt aus
dem Röntgenexperiment ermittelt werden. Dieses Problem wird in der Kristallographie als Phasen­
problem der Röntgenstrukturanalyse beschrieben, zu dessen Lösung indirekte Methoden entwickelt
wurden (vgl. Abschnitte 3.5.8, 3.5.9, 3.5.11).
F h , k , l =F hkl exp[i hkl ]
Gleichung 20
I obs hkl∝∣F h , k ,l 2∣= F h , k , l⋅F ∗ h , k , l
Gleichung 21
Zur vereinfachten Beschreibung des Streuprozesses an geordneten kristallinen Systemen kann
das Braggsche Gesetz (Gleichung 22) herangezogen werden (Bragg & Bragg, 1913). Der Streu­
prozess wird dabei Reflexion der Röntgenstrahlung an einer Schar paralleler Netzebenen (hkl)
beschrieben. Die Netzebenen verlaufen dabei durch Gitterpunkte des Kristallgitters und werden
durch die Millerschen Indizes h, k und l bezeichnet, die jeweils eine Schar paralleler Netzebenen
beschreiben. Der Abstand zwischen zwei parallelen Netzebenen beträgt dhkl. Einfallende mono­
chromatische Röntgenstrahlung trifft unter dem Glanzwinkel Θhkl auf eine Netzebenenschar und
47
Methoden
wird an den einzelnen Netzebenen “reflektiert“. Positive Interferenz, die als Reflex gemessen
werden kann, tritt allerdings nur dann auf, wenn der Weg, den die an verschiedenen Netzebenen
gestreuten Photonen zurücklegen, ein ganzzahliges Vielfaches der Wellenlänge λ ist. In
Abbildung 15 sind die geometrischen Verhältnisse zum Braggschen Gesetz dargestellt.
n⋅=2 d hkl⋅sin hkl
n:
λ:
dhkl:
Gleichung 22
natürliche Zahl
Wellenlänge [Å]
Netzebenenabstand [Å]
einfallender
Strahl
Θ
gestreuter
Strahl
hkl
.
dhkl . sin Θ
Abbildung 15:
3.5.3
hkl
Θ
hkl
dhkl
.
dhkl. sin Θ
hkl
Schematische Darstellung des Braggschen Gesetzes. Eine konstruktive Interferenz zwischen zwei
gestreuten Strahlen tritt nur dann auf, wenn der Gangunterschied zwischen ihnen einem Vielfachen
der Wellenlänge des einfallenden Strahls entspricht.
Reziprokes Gitter und Ewaldkonstruktion
Die Millerschen Indizes h, k, l beschreiben Scharen von parallelen Netzebenen im realen
Raum. Die Streuung von Röntgenstrahlen im realen Raum erzeugt den Streuvektor s im
reziproken Raum, welcher als Betrag definitionsgemäß den Kehrwert des Netzebenenabstandes
besitzt und senkrecht zu den zugehörigen Netzebenen steht. Durch die so genannte Laue-Bedingung
kann eine Beziehung zwischen s und dem reziproken Raum aufgestellt werden (Gleichung 41)
Die für s erlaubten diskreten Werte definieren dabei das reziproke Gitter mit den Basisvektoren
∗
a ∗ ,
b und c ∗ :
s = h
a k
b l c
∗
∗
∗
Gleichung 23
Unter Berücksichtigung der Laue-Bedingungen (Gleichung 17), ergibt sich nach Gleichung 24
eine definierte räumliche Beziehung zwischen realem und reziprokem Raum. Dabei sind die
Dimensionen des reziproken Raums umgekehrt proportional zu den Dimensionen des realen
48
Röntgenographische Methoden
∗
Raums. Die Basisvektoren des reziproken Gitters a ∗ ,
b und c ∗ leiten sich nach Gleichung 24
aus den Basisvektoren des realen Raums a
,
b und c ab, wobei im Falle rechtwinkliger Elemen­
tarzellen die Vektoren beider Systeme kolinear sind.
a=
∗
∗
∗
∗
∗
∗
b ×
c
c ×
a
a × b
,
b=
, c =
V EZ
V EZ
V EZ
Abbildung 16:
Gleichung 24
Darstellung der Ewald-Konstruktion. Diese zeigt die Beziehung zwischen dem Realraum und dem
reziproken Raum eines Streuexperiments bei Verwendung monochromatischer Strahlung. Der
Kristall ist in der Mitte der Ewald-Kugel mit einem Radius von 1/λ montiert. Der Ursprung des
reziproken Raumes wird durch den Schnittpunkt des Primärstrahls mit der Ewaldkugel definiert.
Ein Reflex wird beobachtet, wenn der Endpunkt des Diffraktionsvektors (reziproker Gitterpunkt)
auf der Oberfläche der Ewaldkugel zu liegen kommt.
Der Zusammenhang zwischen reziprokem und realem Gitter sowie das Auftreten von
Reflexen nach dem Braggschen Gesetz (Gleichung 22) wird durch die Ewald-Konstruktion
veranschaulicht (Abbildung 16). In der Ewald-Konstruktion wird der streuende Kristall in den
Mittelpunkt der Ewald-Kugel mit dem Radius 1/λ gelegt. Der Ursprung des reziproken Gitters liegt
im Austrittspunkt des Primärstrahls aus der Ewald-Kugel. Ein Reflex wird nur dann beobachtet,
wenn der Streuvektor s des reziproken Gitterpunktes P(hkl) auf die Oberfläche der Ewald-Kugel
fällt und F(hkl) ≠ 0 ist. Durch Drehung des Kristalls wird auch das reziproke Gitter gedreht,
weshalb nach und nach alle reziproken Gitterpunkte P(hkl) auf die Oberfläche der Ewald-Kugel
49
Methoden
gelangen. Unter Vernachlässigung der anomalen Röntgenstreuung besitzt das reziproke Gitter in
Bezug auf die Reflexintensitäten im Gegensatz zum realen Kristallgitter ein zusätzliches Inversions­
zentrum. Diese Eigenschaft wird durch das Friedelsche Gesetz beschrieben (Gleichung 25).
∣F hkl ∣2=∣F ∗ hkl∣2
Gleichung 25
Damit ist die Intensität eines gemessenen Reflexes hkl identisch mit der Intensität des
invertierten Reflexes hkl . Sogenannte Friedel-Paare weisen also die gleiche Intensität auf, so dass
bei der Messung der Reflexintensitäten nur die Hälfte des reziproken Raums erfasst werden muß.
Zusätzlich wird durch Symmetrien des Kristallgitters das zu erfassende Volumen des reziproken
Raums verkleinert.
3.5.4
Temperaturfaktoren
Der Strukturfaktor F(hkl) (Gleichung 18) kann auch durch die Summe der Beiträge aller
Atome in der Elementarzelle beschrieben werden. Aufgrund thermischer Bewegung der Atome und
statistischer Fehlordnungen im Kristall resultiert eine auflösungsabhängige Verringerung der
Intensitäten der Streustrahlung (Debye, 1914), die durch Korrekturterme berücksichtigt wird. Der
Korrekturterm exp(1/4 Bs2) beschreibt dabei die isotrope Versetzung der Atome, wobei B zum
mittleren Verschiebungsquadrat <u2> der Atome entlang einer Raumachse proportional ist
(Gleichung 26). Ein B-Faktor von 30 Å2 entspricht somit einem mittleren Verschiebungsquadrat
von 0.62 Å. Kann während der Messung eine ausreichende Zahl an Reflexen gesammelt werden, so
können auch anisotrope Versetzungen bestimmt werden, so dass die in drei Raumrichtungen unter­
schiedlichen Schwingungsamplituden gesondert betrachtet werden können. Dies ist im allgemeinen
nur für hochaufgelöste Strukturen möglich, da nur diese ein ausreichendes Observablen-zuParameter Verhältnis liefern.
natom
1 2
− Bs
4
F hkl =∑ f j s ⋅e
⋅e
2 i⋅s⋅
rj
Gleichung 26
j
fj(s)
B
Streufaktor für ein punktförmiges Atom j im Ruhezustand
isotroper Temperaturfaktor (B-Faktor)
B=82 ⟨u 2 ⟩
Gleichung 27
Der absolute Skalierungsfaktor C der Intensitäten kann ebenso wie der mittlere Temperatur­
faktor aus den gemessenen Intensitäten abgeschätzt werden (Wilson, 1949), wenn Auflösungen
besser als 3 Å erreicht werden. Dabei kann der Skalierungsfaktor sowie der mittlere B-Faktor durch
50
Röntgenographische Methoden
lineare Regression ermittelt werden, wenn der natürliche Logarithmus des Quotienten aus den
gemessenen mittleren Intensitäten und den berechneten Intensitäten gegen (sinθ/λ)2 aufgetragen
wird (Gleichung 28).
ln
⟨ I s ⟩
sin 2
=ln
C−2
B
2
∑ f 2i s
Gleichung 28
i
3.5.5
Patterson-Funktion
Die Patterson-Funktion (Gleichung 29) ist eine Fourier-Summation der gemessenen Reflex­
intensitäten, wobei Phasenwinkel von Null angenommen werden (Patterson, 1934). Damit kann die
Patterson-Funktion direkt aus den gemessenen Daten berechnet werden, ohne Kenntnis über die
Phase zu besitzen. Da die resultierende Funktion einen Satz von Vektoren repräsentiert, wird die
u beschrieben, um eine
Patterson-Funktion als Funktion der Komponenten u,v,w des Vektors
Verwechslung mit den relativen Koordinaten x,y,z zu verhindern. Die Patterson-Funktion kann
zudem durch Umformen in Gleichung 30 geschrieben werden. Ansschaulich gesehen handelt es
sich hierbei um eine Summation aller Vektoren zwischen allen Atomen der Einheitszelle, da das
Produkt der Elektronendichte am Ort
r1 und r1u
nur dann von Null verschieden ist, wenn
dieses dem Differenzvektor zweier Atome entspricht. Damit ergeben sich für eine PattersonFunktion mit N Atomen N(N+1) Maxima, die nicht auf dem Ursprung liegen. Da zu jedem
Atompaar zwei Vektoren mit gegensätzlicher Richtung existieren, ist die Patterson-Funktion
zentrosymmetrisch. Die Schraubenachsen des Kristalls werden zu einfachen Rotationsachsen im
Pattersonraum transformiert. Die Höhe der Patterson Maxima ist vom Produkt der Anzahl der
Atome der jeweiligen Atompaare abhängig.
P u , v , w=
1
∣F h , k , l∣2⋅cos[ 2hukvlw]
V EZ ∑
h
P
u =∫ r1⋅−[ u1 −r1 ] d r1
r1
Gleichung 29
Gleichung 30
Die Atompositionen können für kleine Moleküle, die nur wenige Atome in der Elementarzelle
besitzen, häufig ab initio allein die Differenzvektoren der Patterson-Funktion bestimmt werden.
Dieses Verfahren scheitert in der Protein-Kristallographie hingegen aufgrund der großen Anzahl der
51
Methoden
Atome in der Zelle. Außerdem ist die erreichte Auflösung ist meist zu gering. Allerdings kann die
Patterson-Funktion zur Bestimmung der Atompositionen der Schweratom-Substruktur von derivati­
sierten Kristallen (Abschnitt 3.5.8), bei der Phasierung durch molekularen Ersatz und zu
Bestimmung des Rotations- und Translationsoperatoren beim Auftreten nicht-kristallographischer
Symmetrie genutzt werden.
3.5.6
Kristallographische Datensammlung
Die Qualität der gemessenen Daten und damit die Vollständigkeit und Genauigkeit der
gemessenen Reflexe samt ihrer Intensitäten ist für die Röntgenstrukturanalyse von entscheidender
Bedeutung. Die Qualität der gemessenen Daten ist ausschlaggebend für den Gang der Struktur­
lösung und die erreichbare Genauigkeit des Strukturmodells. In dieser Arbeit wurden die Reflexe
und ihre Intensitäten mittels der Rotationsmethode aufgenommen (Arndt et al., 1973). Zur Daten­
sammlung wird der Kristall hierbei in kleinen Intervallen um eine definierte Achse rotiert, die
gestreuten Röntgenquanten durch einen Detektor aufgenommen und zu einem image oder frame
zusammengesetzt, wobei die ermittelten Intensitätswerte sowie die Daten zur Aufnahmegeometrie
gespeichert werden. Als Röntgenquellen kamen bei den Messungen wassergekühlte KupferDrehanoden (RU-200B und RU-H2C, Firma Rigaku) zum Einsatz, die mit einer Stromstärke von
118 mA und einer Spannung von 44 kV betrieben wurden. Die intensive Kupfer Kα-Linie (1.54 Å)
wurde dabei durch einen Graphit-Monochromator selektiert und auf einen Strahl von 300 µm
Durchmesser fokussiert. Zur Detektion kristallographischer Daten wurden zweidimensional
ortsempfindliche Detektoren wie Drahtflächenzähler, Bildplatten (image plates) und CCDDetektoren (charge coupled device) verwendet.
Messungen wurden neben den Experimenten am Synchrotron im Rahmen dieser Arbeit an den
beiden vorhandenen Hausanlagen durchgeführt. Hausanlage-1, die zum Testen der Streukraft
hergestellter Kristalle verwendet wurde, bestand aus einer Kupfer-Drehanode (RU-200B, Firma
Rigaku) und einem Drahtflächenzähler (X-1000, Firma Siemens) mit Dreikreis-Goniometersystem
(Abbildung 17). Vor jeder Messung musste der Detektor bezüglich inhomogener Empfindlichkeit
sowie geometrischer Verzerrungen kalibriert werden. Dazu wurde ein Plättchen amorphen Eisens in
den Röntgenstrahl eingebracht und die isotrope Röntgenfluoreszenzstrahlung detektiert. Aus den
dabei gemessenen Intensitätswerten wurde eine Korrekturtabelle für die ortsabhängige
Empfindlichkeit (flood-field correction) des Detektors erstellt. Anschließend wurde eine Lochgitter­
platte aus Messing (brass-plate) vor dem Detektor montiert und erneut die Röntgenfluoreszenz­
strahlung des Eisenplättchens detektiert. Das dabei entstandene Punktmuster diente der Korrektur
geometrischer Verzerrungen des Detektors. Zur Datensammlung wurde die Orientierung des
52
Röntgenographische Methoden
Kristalls relativ zum einfallenden Strahl über die Winkel φ, χ, und ω festgelegt, wobei χ beim
Dreikreis-Goniometersystem bei 45° fixiert ist. Der zu detektierende Auflösungsbereich wurde
durch Ausschwenken in der 2θ-Ebene gewählt. Der Kristall-Detektor-Abstand wurde so eingestellt,
dass aufgenommene Reflexe durch den Detektor noch klar getrennt abgebildet werden konnten.
Dabei kann der richtige Abstand (in cm) bei der Verwendung von Cu-Kα-Strahlung aus den
Zellachsen abgeschätzt werden, indem die Detektordistanz auf etwa 1/8 bis 1/10 der längsten
Elementarzellachse (in Å) eingestellt wird (Howard et al., 1987). Der Kristall wurde während der
Bestrahlung um den Winkel ω von 0.2° pro frame gedreht. Die Belichtungszeit wurde abhängig
von der Streukraft des montierten Kristalls gewählt und betrug 2 bis 10 min.
Abbildung 17:
Datensammlung an der Hausanlage-1 am Drahtflächenzähler mit Dreikreis-Goniometersystem
(χ=45°). Der Kristall kann während der Messung um die Winkel ω und φ gedreht werde. Der
Auflösungbereich wird durch Ausschwenken um den Winkel 2Θ eingestellt..
Die Meßstation der Hausanlage-2 ist schematisch in Abbildung 18 dargestellt. Als Röntgenquelle
diente eine Kupfer-Drehanode (RU-H2C, Firma Rigaku). Als Detektor kam eine image plate mit
einem Durchmesser von 30 cm zum Einsatz. Die Belichtungszeit wurde abhängig von der Streu­
kraft der Kristalle gewählt und betrug 10 bis 20 min pro gemessenem frame, wobei der Kristall um
den Winkel φ von 0.25° bis 1° gedreht wurde.
Messungen mit Synchrotronstrahlung wurden an den beamlines PX und PX-II der Swiss Light
Source (SLS, Villigen, Schweiz), sowie an den beamlines BL14.1 und BL14.2 am Berliner
Elektronenspeicherring (BESSY, Berlin) durchgeführt. Die Wellenlänge wurde mit einem Si(111)Monochromator selektiert und die gestreuten Röntgenquanten mit CCD-Detektoren (Firma Marre­
search) detektiert. Aufgrund der hohen Intensität des Röntgenstrahls betrug die Belichtungszeit pro
aufgenommenen frame zwischen 1 s (SLS) und 5 s (BESSY), wobei der Rotationswinkel φ
zwischen 0.3° und 1° eingestellt wurde.
53
Methoden
Abbildung 18:
3.5.7
Datensammlung an der Hausanlage-2 mit installierter image plate. Der Kristall wird während der
Messung um den Winkel φ gedreht.
Prozessierung und Datenreduktion
Die Rohdaten wurden mit dem die Programmpaket XDS (Kabsch, 1988; 1993) prozessiert.
Dabei werden in einem ersten Schritt starke Reflexe auf wenigen frames bzw. images identifiziert
und die Orientierungsmatrix des Kristalls, die die Lage und Dimension eines primitiven
Kristallgitters im Verhältnis zu den Laborkoordinaten beschreibt, anhand von Differenzvektoren
bestimmt. Das reziproke Gitter wird anschließend anhand der stärksten Reflexe indiziert. Zur
Integration der Daten werden in einem ersten Schritt dreidimensionale Profile starker vollständig
erfasster Reflexe für unterschiedliche Detektorregionen sowie unterschiedliche Sätze von frames
(batches) erstellt. Im drauf folgenden Schritt werden dann alle Reflexe unter Verwendung des
bestmöglichen Profils (profile fitting) integriert. Anschließend werden die Zellparameter nochmals
anhand von Reflexen optimiert, deren Schwerpunkte ausreichend nahe an ihren vorhergesagten
Positionen liegen (post refinement). Die erhaltenen Rohintensitäten und deren Standardabweichung
werden abschließend in Bezug auf Lorentz-Polarisation und Absorption korrigiert und durch eine
einfache Skalierung aneinander angepasst.
Die experimentellen Bedingungen können sich im Verlauf der Messung ändern (schwankende
Intensität des Röntgenstrahls, Nachlassen der Streukraft durch Strahlenschäden oder Anisotropie
des Kristalls). Daher wird eine genaue Skalierung der gemessenen Intensitäten nötig, so dass
mehrfache Messungen eines Reflexes eine möglichst geringe Abweichung von dessen mittlerer
Intensität aufweisen (Evans, 2006). Über eine zero-dose Extrapolation lassen sich darüber hinaus
die durch Strahlenschäden verursachten Effekte korrigieren. Abschließend werden die skalierten
Daten auf die asymmetrische Einheit des reziproken Raumes reduziert und in Intensitäten
umgewandelt. Hierzu wurden die Programme XSCALE und XDSCONV verwendet, die ebenfalls
54
Röntgenographische Methoden
Teils des Programmpakets XDS sind. Im Laufe dieser Arbeit wurde neben der manuellen
Auswertung insbesondere bei bekannten Zellparametern und Raumgruppen aufgenommener
Datensätze auf das Programm APRV zurückgegriffen (Kroemer et al., 2004), das die oben
genannten Schritte automatisiert durchführt und zusätzlich die Parameter optimiert.
Als Maß für die Qualität der Daten werden üblicherweise eine Reihe statistischer Kriterien in
Auflösungsschalen angegeben. So ist der für symmetrieverwandte Reflexintensitäten I(hkl)
definierte Rsym-I ein Maß für die Übereinstimmung mehrfach gemessener, identischer Reflexe
(Gleichung 31). Allerdings ist er stark von der Anzahl der gemessenen Reflexe (Multiplizität)
abhängig. Ein robusteres Kriterium findet sich im Rmeas (Gleichung 32; Diederichs & Karplus,
1997), der jeden Reflex mit einer Funktion seiner Multiplizität wichtet. Weitere Kriterien für die
Qualität der gemessenen Daten liefern das Signal-zu-Rausch-Verhältnis I/σ(I), sowie die Vollstän­
digkeit und Multiplizität der Daten.
R sym−I =
∑hkl ∑i ∣I hkl i −⟨ I hkl ⟩∣
∑hkl ∑i I hkli
Gleichung 31
I(hkl)
Intensität des i-ten Reflexes
<I(hkl)> mittlere Intensität aller Reflexe bei gegebenen hkl
∑
Rmeas =
hkl
n hkl n
∑∣I hkl i−⟨ I hkl⟩∣
n hkl −1 i=1
hkl
n hkl
Gleichung 32
∑ ∑ ∣I hkl i∣
hkl i=1
nhkl
3.5.8
Anzahl gemessener Reflexe (hkl)
Phasenbestimmung durch isomorphen Ersatz
Diese Methode beruht auf der Eigenschaft, dass elektronenreiche Schweratome an definierte
Positionen eines Proteins binden können. Es ist allerdings notwendig, dass keine dauerhafte
Änderung der Proteinstruktur oder der Kristallpackung auftritt. Die zusätzlichen Elektronen des
gebundenen Schwermetallatoms bewirken eine Änderung der Reflexintensitäten relativ zum nativen
Kristall, was eine Bestimmung initialer Phasen ermöglicht (Green et al., 1954). Die Herstellung
eines ausreichend isomorphen Schwermetallatomderivats lässt sich nicht durch eine standardisierte
Vorgehensweise erreichen, sondern ist ein empirisches Verfahren, das häufig nur durch Testen einer
Vielzahl von verschiedenen Schwermetallverbindungen und Anwendung unterschiedlicher
Protokolle zu erreichen ist. Durch Skalieren der Daten eines Derivats auf den korrespondierenden
55
Methoden
nativen Datensatz kann abgeschätzt werden, ob eine isomorphe Bindung des Schwermetallatoms
stattgefunden hat. Der hierzu benötigte Skalierungsfaktor k findet sich in Gleichung 33. Zum
Skalieren wurde das Programm SCLAEIT (CCP4, 1994) verwendet, das unter anderem den
R-Faktor Riso (Gleichung 34) in den Auflösungsschale angibt. Riso-Werte zwischen 10% und 30%
deuten dabei auf ein brauchbares Derivat hin. Demgegenüber muss bei höheren Werten von NichtIsomorphie ausgegangen werden, während bei kleineren Werten keine definierte Bindung des
Schwermetallatoms vorliegt.
k=
∑ F P⋅F PH
1
hkl
2
∑F
hkl
2
PH
{[
∑ F 2P⋅∑ F 2PH
3
hkl
1− 1− ⋅ hkl
4
∑ F P⋅F PH
hkl
2
]}
1
2
∑∣F PH − F P∣
Riso = hkl
∑ FP
Gleichung 33
Gleichung 34
hkl
FPH Strukturfaktoramplitude des Derivats
FP Strukturfaktoramplitude des nativen Kristalls
Der Verlauf der isomorphen Differenzen in Abhängigkeit von der Auflösung gibt einen
Hinweis auf die Qualität des Derivats. Die isomorphen Differenzen nehmen mit zunehmender
Auflösung aufgrund der Proportionalität zu FP ab, wohingegen der nicht-isomorphe Anteil mit
steigender Auflösung gleich bleibt oder zunimmt (McRee, 1992).
Bestimmung der Schweratompositionen
Für eine Phasierung durch isomorphen Ersatz ist die Kenntnis der Schweratom-Substruktur
notwendig. Eine initiale Bestimmung der Positionen ist über die Differenz-Patterson-Funktion
möglich (Abschnitt 3.5.5), wobei zur Berechnung der Patterson-Funktion (Gleichung 29) die
Quadrate der isomorphen Differenzen ΔFiso2= (FPH – FP)2 eingesetzt werden. Bei dieser
Differenzbildung bleiben im idealen Fall ausschließlich die Anteile FH übrig, wobei die derart
berechnete Patterson-Funktion somit im Wesentlichen aus den Vektoren zwischen den gebundenen
Schweratomen besteht. Durch Vektoren symmetriegekoppelter Schweratome entstehen Maxima der
Funktion, die sich in bestimmten Ebenen häufen. Diese Ebenen werden Harker-Ebenen genannt.
Aus diesen Maxima und deren Lage lassen sich die Positionen der Schwermetallatome ermitteln
und in Koordinaten umsetzten. Die Differenz-Patterson-Funktion (Gleichung 35) wurde unter
Verwendung des Programms FFT (CCP4, 1994) berechnet und mit Hilfe der Programme
XCONTOUR und XPATPRED, die Teil des Programmpaketes XTALVIEW (McRee, 1992) sind,
56
Röntgenographische Methoden
analysiert. Aus diesen initialen Phasen können weitere Schweratompositionen, auch von anderen
Derivaten durch die Analyse der Differenz-Fourier-Funktion ermittelt werden, die ebenso eingesetzt
werden kann, um strukturelle Änderungen isomorpher Protein-Ligand-Komplexe zu untersuchen.
Die Differenz-Fourier-Funktion wurde unter Verwendung des Programmes FFT (CCP4, 1994)
berechnet. Bei komplexen Patterson-Funktionen wurde eine automatische Interpretation der Daten
mit dem Programm SHELXD (Schneider & Sheldrick, 2002) durchgeführt.
x , y , z =∑ F PH −F P exp −i exp [ −2 i hxky lz ]
hkl
Gleichung 35
Phasierung und Verfeinerung der Schweratomparameter
Wenn die Schwermetallatompositionen eines Derivats i bekannt sind, können die Phasen der
Strukturfaktoren FP des nativen Proteins unter Anwendung der Harker-Konstruktion (Harker, 1956)
aus den Strukturfaktoren FHi eines Derivats berechnet werden (Abbildung 19). Hierbei wird um den
Ursprung des Vektor-Diagramms ein Kreis mit dem Radius FP gezogen. Die aus dem vorläufigen
Schweratommodell berechneten Strukturfaktoren FHi von Derivat i werden negativ vom Ursprung
aus abgetragen und um ihre Endpunkte Kreise mit dem Radius FPHi gezeichnet. Die Phase des
Strukturfaktors FP ergibt sich aus dem Schnittpunkt aller Kreise. Folglich ist bei nur einem
Schwermetallderivat (single isomorphous replacement, SIR) die Phase des Strukturfaktors FP nicht
eindeutig bestimmt. Eine eindeutige Lösung kann allerdings durch zusätzliche Phaseninformation
aus anomaler
Streuung (SIRAS,
Abschnitt 3.5.9),
Dichtemodifikation (Abschnitt 3.5.13),
molekularem Ersatz (Abschnitt 3.5.11), direkten Methoden oder weitere Schweratomderivate
(multiple isomorphous relacement, MIR) erhalten werden.
Die Fehler der Phasenbestimmung durch isomorphen Ersatz resultieren im allgemeinen durch
schlechte Isomorphie der Derivate zueinander und wurden als lack of closure (ε) beschrieben (Blow
& Crick, 1959). Dabei wird zur Vereinfachung die Fehlerfreiheit der Strukturfaktoren FP und FHi
angenommen und der Fehler εi vollständig zu FPHi addiert (Abbildung 20).
57
Methoden
Abbildung 19:
Argand-Diagramm der Harker-Konstruktion. A) Im SIR-Fall resultieren zwei gleich wahrscheinliche Lösungen für αP, die durch die Schnittpunkt G und H festgelegt sind. B) Die MIRPhasierung erlaubt hingegen die eindeutige Bestimmung von FP.
Abbildung 20:
Fehlerbehandlung bei Phasierung durch isomorphen Ersatz. A) Im ideal isomorphen Fall
addieren sich die Strukturfaktoren FP und FHi exakt zu FPHi und erfüllen damit die Gleichung des
isomorphen Ersatzes FP + FHi = FPHi. B) Im realen Fall treten Fehler durch Messung, Nicht-Isomorphie und Schweratomparameter auf, so dass das Vektordreieck nicht geschlossen ist. FHi und FP
werden als fehlerfrei angenommen. Der resultierende Fehler wird als lack of closure bezeichnet.
Die Verfeinerung der Schweratomparameter erfolgte in dieser Arbeit durch das Programm
SHARP/ AUTOSHARP (Bricogne et al., 2003). Dieses Programm arbeitet mit maximum likelihood
Zielfunktionen und verfeinert dabei sowohl die Koordinaten, die Besetzungszahlen (basierend auf
isomorpher und/oder anomaler Differenz), die Temperaturfaktoren (isotrop oder anisotrop) der
Schweratome, als auch die Skalierungsfaktoren und Nicht-Isomorphie Parameter der analysierten
Datensätze. Sowohl der Verlauf der Verfeinerung als auch die Qualität wurden anhand des Cullis RFaktor Rcullis, des figure of merit (FOM), sowie der phasing power bewertet. Der ursprünglich nur
58
Röntgenographische Methoden
für zentrische Reflexe definierte Rcullis (Cullis et al., 1962) liegt für brauchbare Derivate unter einem
Wert 0.8, für sehr gute unter 0.6. Der FOM liegt für gute Derivate über 0.5, die phasing power
sollte größer als 1 sein. Neben diesen Qualitätsindikatoren wurden die Parameter der SchweratomSubstruktur durch Analyse der log-likelihood gradient residual maps überprüft, die von SHARP
berechnet wurden. Diese dreidimensionale Darstellung des Gradienten des log-likelihood zeigt
Fehler der Schweratompositionen auf, die als negative oder positive Maxima erscheinen.
Schließlich wurden Projektionen von (Fobs)-Elektronendichtekarten auf eine Solvensabgrenzung hin
untersucht.
3.5.9
Phasenbestimmung durch anomale Streuung
Der bisherigen Betrachtung des Streuprozesses lag eine elastische Streuung an freien Elektronen zu
Grunde. Diese Sichtweise verliert ihre Gültigkeit, sobald die Energie der eingesetzten Röntgen­
strahlung der Bindungsenergie von Elektronen im Streuer ähnelt. In diesem Fall treten Resonanz­
erscheinungen auf. Diese sind für Elektronen der K- und L-Schale schwerer Elemente besonders
ausgeprägt. Als Folge der inelastischen Wechselwirkungen verändern sich sowohl Intensität als
auch Phasenbeziehung der Streustrahlung gegenüber der Primärstrahlung. Dieser Effekt wird als
anomale Streuung bezeichnet und kann durch zusätzliche wellenlängenabhängige Terme des
Streufaktors berücksichtigt werden (Gleichung 36). Des weiteren unterscheiden sich die Friedel­
paare FPA(+) und FPA(-) der symmetrieverwandten Reflexe in einem Kristall des Proteins P, der
anomale Streuer enthält, aufgrund der anomalen Beiträge von H sowohl in Amplitude als auch
Phase (Gleichung 37). Das Friedelsche Gesetz verliert damit seine Gültigkeit (Abbildung 21).
f = f 0 f ' i f ' '
f0
f '(λ), f ''(λ)
Gleichung 36
wellenlängenunabhängiger Streufaktor
reeller bzw. imaginärer Anteil der anomalen Streuung
F PA + =F P F A +=F P F ' ' AF ' ' A
Gleichung 37
F PA - =F P F A -=F P F ' ' A−F ' ' A
FH
normaler Anteil der Strukturfaktoren der anomalen Streuung
F'A = (f '(λ)/f0) FA reeller anomaler Anteil
F''A = (f ''(λ)/f0) FA imaginärer anomaler Anteil
Die Absorptionskanten der leichten Elemente (C, N, O, P und S), die den wesentlichen Teil
der Atome in Proteinen ausmachen, liegen bis auf Schwefel und Phosphor nicht im Bereich der
Energie der zur Röntgenstrukturanalyse verwendeten Strahlung. Die Energien der K-Kanten von
59
Methoden
Schwefel und Phosphor können jedoch bei guter Auflösung, Datenqualität und Multiplizität zur
Phasierung genutzt werden (Hendrickson & Teeter, 1981; Dauter & Adamiak, 2001). Die Energien,
die üblicherweise bei der Röntgenstrukturanalyse Verwendung finden, entsprechen ab den
Elementen der vierten Periode des Periodensystems den Übergangsenergien der K- und L-Schalen
und die anomalen Beiträge zur Streuung werden so groß, dass sie auch bei mittleren Auflösungen
messbar sind. Da der anomale Anteil an der Streuung üblicherweise nur etwa 3% bis 5% ausmacht,
ist eine präzise Datensammlung und eine hohe Multiplizität der Daten essentiell. Sind keine
anomalen Streuer im Protein enthalten, müssen diese inkorporiert werden. Dabei hat sich als
günstige Methode die direkte Inkorporation von Selen als Selenomethionin (SeMet) ins Zielprotein
erwiesen (Hendrickson et al., 1990). Neben dem sicheren Einbau der anomalen Streuer und der im
Verlauf der Strukturlösung möglichen Idenifikation von Sequenzbereichen ist ein weiterer Vorteile
der SeMet-Inkorporation die meist identische Kristallisationseigenschaft. Zudem können Tränkex­
perimente vermieden werden, die oft einen Verlust der Kristallqualität mit sich bringen.
Abbildung 21: Darstellung der Zusammensetzung der Strukturfaktoren der Friedelpaare FPA(+) und FPA(-) bei
anomaler Streuung. Die Bezeichnung der Strukturfaktoren erfolgte wie in den Gleichungen. Zur
Verdeutlichung ist der Anteil der anomale Streuung stark übersteigert dargestellt.
Die Differenzen der Friedel-Paare können als zusätzliche (single/multiple isomorphous
replacement with anomalous scattering, SIRAS/MIRAS) oder alleinige (single-wavelength
anomalous diffraction) SAD, oder multiwavelength anomalous diffraction, MAD) Quelle von
Phaseninformation genutzt werden. Im Unterschied zu SIR werden bei der SIRAS-Methode die
anomalen Differenzen des Derivat-Datensatzes berücksichtigt, falls diese gemessen wurden. Die
zusätzliche Phaseninformation kann zur Unterscheidung der Händigkeit herangezogen werden.
Abbildung 22 (Seite 63) zeigt eine schematische Darstellung der Harker-Konstruktion für die
genannten Methoden.
60
Röntgenographische Methoden
Bei einem MAD-Experiment werden Datensätze bei verschiedenen Wellenlängen aufgenom­
men, die sich durch unterschiedliche Werte für f ' und f '' des anomalen Streuers auszeichnen (vgl.
Abbildung 23, Seite 64). Vor allem im Bereich der Absorptionskante des anomalen Streuers sind
der reele und imaginäre Anteil an der Streuung stark von der Wellenlänge der eingesetzten
Strahlung abhängig. Im Vergleich zur SAD-Methode (Messung bei nur einer Wellenlänge) wird für
jede Wellenlänge ein Satz anomaler Differenzen erhalten. Zudem kann die Abhängigkeit des reelen
Teils der anomalen Streuung von der Wellenlänge zur Phasenbestimmung verwendet werden (dis­
persive Differenzen). Um die unterschiedlichen Anteile von f ' und f '' optimal auszunutzen, werden
üblicherweise drei Messungen ausgeführt: zum einen bei einer Wellenlänge, die dem Maximum der
f ''-Kurve entspricht (peak), einer zweiten im Minimum der f '-Kurve, also dem Wendepunkt der
f ''-Kurve (inflection point), und einer dritten von der Absorptionskante weit entfernten (high oder
low energy remote). Die genaue Lage der Absorptionskante ist von der Umgebung des anomalen
Streuers im Kristall abhängig und wird vor der Messung experimentell durch einen Fluores­
zenzscan ermittelt. Alle Datensätze müssen bei dieser Methode von einem Kristall aufgenommen
werden, da die Unterschiede, zwischen F(+) und F(-), die durch anomale Streuung verursacht
werden, nur etwa 5% des Gesamtsignals ausmachen und daher Fehler, die durch Nicht-Isomorphie
bei Verwendung mehrerer Kristalle auftreten könnten, zu vermeiden sind.
Bestimmung der Schweratompositionen
Die Differenzen der Strukturfaktoren FPA(+) und FPA(-) der Friedelpaare (Gleichung 38), die
auch anomale Differenzen oder Bijvoet-Differenzen genannt werden, resultieren aus den unter­
schiedlichen Vorzeichen des imaginären anomalen Anteils der Strukturfaktoren der anomalen
Streuer (Gleichung 37).
F ano= F PA + −F PA -
1
1
Gleichung 38
mit F PA + und F PA - Strukturfaktoramplituden der Friedelpaare bei λ1
1
1
Findet die Messung bei mehreren Wellenlängen statt, so können diese durch den realen Anteil
der anomalen Streuung zusätzlich zur Phaseninformation beitragen. Dabei kann eine starke
Abhängigkeit der dispersiven Differenzen (Gleichung 39) in analoger Weise wie bereits bei der
Methode des isomorphen Ersatzes ausgenutzt werden. Die Bestimmung der Substruktur der
anomalen Streuer erfolgt wie im Fall des isomorphen Ersatzes beschrieben unter Anwendung der
Patterson-Funktion. Als Koeffizienten können sowohl anomale als auch dispersive Differenzen
verwendet werden.
61
Methoden
F disp=< F >−< F >
1
< F >
< F >
1
2
Gleichung 39
2
Mittel von F PA + und F PA - bei λ1
Mittel von F PA + und F PA - bei λ2
1
1
2
2
Die Skalierung und Analyse von SAD- oder MAD-Daten zur Schweratomsuche erfolgte durch
Verwendung des Programms XPREP (Firma Bruker AXS GmbH, Karlsruhe, Deutschland, 2003).
Als Maß für die Güte der Daten wurde dabei das Verhältnis von anomalem Signal und Rauschen
(für SAD größer 1.3 σ, für MAD größer 1 σ) sowie die Korrelationskoeffizienten des anomalen
Signals bei verschiedenen Wellenlängen (größer 30%) in den Auflösungsschalen bestimmt.
Weiterhin wurde XPREP benutzt, um die ΔF und FA Werte für die Substrukturbestimmung mit
SHELXD (Schneider & Sheldrick, 2002) zu berechnen.
Phasierung und Verfeinerung der Schweratomparameter
Aus den initial erhaltenen Positionen der anomalen Streuer können die Strukturfaktoren
FPA(+) und FPA(-) der anomalen Streuer für jede Wellenlänge berechnet werden. Die Phasierung
erfolgt im folgenden analog zur Phasierung durch SIR/MIR, dabei können die Strukturfaktoren FP,
also die Strukturfaktoren der “reinen“ Proteinstruktur, durch eine Harker-Konstruktion ermittelt
werden (Abbildung 22). Da für diese das Friedelsche Gesetz gilt, ergeben sich die Phasen der
“reinen“ Proteinstruktur wieder aus den Schnittpunkten der Kreise. Eine eindeutige Lösung für die
Phasen kann bei SAD durch Verwendung von SIRAS oder durch das Einsatz von weiteren
Wellenlängen ähnlich wie im MIR-Fall erhalten werden. Eine alternative Möglichkeit die Protein­
phasen direkt zu bestimmen, ist durch so genannte MAD observational equations (Hendrickson et
al., 1985) gegeben (Gleichung 40). Für drei Wellenlängen können entsprechend sechs Gleichungen
geschrieben werden. Da die Strukturfaktoramplituden FPA und FA von der Wellenlänge unabhängig
sind, ist dieses Gleichungssystem für die drei unbekannten Parameter FPA, FA, und (αPA -α A)
überbestimmt. Wenn die anomale Substruktur bekannt ist, kann daher der gewünschte Protein­
phasenwinkel direkt bestimmt werden.
Die Verfeinerung der Parameter der anomalen Streuer erfolgt analog zu der unter
Abschnitt 3.5.8 beschriebenen Art und Weise, wobei jedoch gegen anomale und dispersive
Differenzen verfeinert wird. Im Falle von SIRAS/MIRAS kann zusätzlich noch gegen isomorphe
Differenzen verfeinert werden, was zu einer hohen Qualität der Phasen führt.
62
Röntgenographische Methoden
Abbildung 22:
2
Harker-Konstruktion für die SAD/SIRAS- und die MAD/MIRAS-Methode. A) In Gegenwart eines
anomalen Streuers A. B) Im SAD-Fall mit einem anomalen Streuer A gibt es zwei Möglichkeiten G
und H als Endpunkt von FPA (nicht gezeigt). Kann ein nativer Datensatz hinzugefügt werden
(SIRAS), so wird eine eindeutige Lösung für die Phase erhalten. B) Phasierung durch MAD oder
MIRAS: Durch Messung bei einer zweiten Wellenlänge (gestrichelt) kann FP eindeutig bestimmt
werden. Ein zusätzlicher nativer Datensatz (gepunktet) kann die Phasenbestimmung erleichtern.
2
2
1
F + =F
1
F - 2 =F 2PAa 1 F 2 Ab 1 F PA F A cos PA− A −c 1 F PA F A sin PA− A
2
F +2=F 2 PAa 2 F 2 Ab 2 F PA F A cos PA−A c 2 F PA F A sin PA− A
2
F - =F
2
mit
PA
2
a 1 F Ab 1 F PA F A cos PA− A c 1 F PA F A sin PA− A
Gleichungen 40
2
PA
a 2 F Ab 2 F PA F A cos PA− A −c 2 F PA F A sin PA− A
2
2
a n = f ' n f ' ' n / 2
b n= 2 f ' n / f 0
c n =2 f ' ' n / f 0
3.5.10
Bestimmung der Identität von Metallatomen durch anomale Streuung
Neben der Möglichkeit der Phasenbestimmung durch Messung anomaler Daten kann ein
MAD-Experiment auch zur Identifikation eines Metallatoms eingesetzt werden. Ein derartig
Nachweis ist auch über andere Verfahren wie EXAFS (extended X-ray absorption fine structure)
oder EPR (electron paramagnetic resonance) zu erhalten, allerdings hat die hier eingesetzte
Methode den Vorteil, neben der Identifikation des zu untersuchenden Metalls auch dessen genaue
Lokalisation innerhalb der Proteinstruktur zu bestimmen. Die hier beschriebene Methode wurde in
etwas abgewandelter Form bereits erfolgreich eingesetzt, um am Protein gebundene Calciumionen
von Kaliumionen zu unterscheiden (Than et al., 2005). Es wurden hierzu die anomalen Daten eines
63
Methoden
zu untersuchenden Metalls sowohl an dessen Maximum der f ''-Kurve (peak) als auch low energy
remote davon aufgenommen. Wie schon in Abschnitt 3.5.9 beschrieben, muss diese Messung an
einem einzelnen Kristall bei zwei Wellenlängen für jedes zu bestimmende Metall durchgeführt
werden, um zum einen den bei der Messung auftretenden Fehler zu minimieren, aber auch, um für
eine Abschätzung des Gehalts der einzelnen Metalle die anomalen Daten exakt aufeinander
skalieren zu können. Abbildung 23 zeigt schematisch das Absorptionsverhalten der zur Struktur­
lösung der N-Oxygenase untersuchten Metallionen. Die genaue Lage der Absorptionskanten muss
allerdings wie bereits beschrieben vor der Messung experimentell durch einen Fluoreszenzscan
bestimmt werden.
W ellenlänge [Å]
4
2.0
2.1
1.9
1.8
1.6
1.5
1.4
1.3
f ''
2
f', f'' (E lektronen)
1.7
1.2
4
2
0
0
Fe,pk
M n,rm
M n,pk Fe,rm
-2
-2
f'
-4
M angan
Eisen
-4
-6
-6
-8
-8
6000
Abbildung 23:
7000
8000
Energie (eV)
9000
10000
Theoretischer Verlauf der anomalen Streuung für die Metalle Eisen und Mangan. Die vier Wellenlängen an denen die Messung zur Identifikation dieser Metalle durchgeführt wurde sind durch
Pfeile markiert. Es wurde sowohl an den Kanten (peak, mit pk abgekürzt) der jeweiligen Metalle als
auch jeweils im Energiebereich vor diesen Kanten (low energy remote, mit rm abgekürzt) gemessen.
Diese Strategie erlaubte die Berechnung doppelter anomaler Differenzen, die nur für die entsprechenden Metalle charakteristische Maxima aufweist.
Für die Auswertung werden so genannte doppelt anomale Differenz-Werte (double anomalous
difference, DDANO) berechnet. Diese beschreiben die Änderung der Differenzen der Struktur­
faktoren ΔFANO nach Gleichung 38 bei einer Änderung der Wellenlänge von λpeak zu λlrem. Die
resultierenden Strukturfaktoren ΔΔFANO = ΔFANO(λpeak) – ΔFANO(λlrem) werden dabei zur Berechnung
64
Röntgenographische Methoden
der metallspezifischen DDANO-Elektronendichte nach Gleichung 41 verwendet. Ein Maximum an
einer definierten Position dieser Elektronendichte ist somit nur zu finden, wenn das zu bestimmende
Metall M an der entsprechenden Position im Protein gebunden vorliegt.
xyz =
1
V
∑ [F hkl+,M , pk − F hkl- ,M , pk −F hkl+,M , rm −F hkl- , M ,rm ]×exp [−2 i hx ky lz i hkl − 2 ]
Gleichung 41
hkl
Zur Berechnung einer derartigen DDANO-map wurden die Daten zunächst wie im Abschnitt
3.5.7 beschrieben prozessiert. Die Phasen resultierten aus einer rigid body Verfeinerung mit
REFMAC (Murshudov et al., 1997) und wurden zusammen mit den Datensätzen aller vier Wellen­
längen durch das Programm CAD (CCP4, 1994) zur Skalierung mit SCALEIT vorbereitet. Zur
Berechnung der finalen DDANO-Elektronendichte wurden die Programme SFTOOLS (CCP4,
1994) und FFT (CCP4, 1994) verwendet.
3.5.11
Phasenbestimmung durch molekularen Ersatz
Neben den bisher beschriebenen Methoden zur Strukturaufklärung findet die Methode des
molekularen Ersatzes (molecular replacement, MR) breite Anwendung. Diese kann genutzt werden,
wenn die Struktur des Zielproteins oder die eines homologen Proteins bekannt ist. Das Haupt­
einsatzgebiet von MR ist zum einen bei der Phasierung eines Proteins, das in unterschiedlichen
Kristallformen kristallisiert und dessen Struktur nur in einer Kristallform gelöst wurde, zu finden.
Zum anderen kann ausgenutzt werden, dass Proteine mit einer Sequenzidentität von mehr als 30%
meist eine vergleichbare Faltung besitzen (Sander & Schneider, 1991). Die Strukturen verwandter
Proteine dienen bei der Strukturlösung durch MR als Suchmodelle des unbekannten Proteins.
Aufgrund statistischer Überlegungen ist davon auszugehen, dass bei löslichen Proteinen die Zahl
der möglichen Kettenfaltungsmuster auf 500 bis 1000 begrenzt ist (Schulz, 1981; Benner et al.,
1997). Da die Zahl der bekannten Proteinstrukturen in den letzten Jahren stark angestiegen ist, wird
es immer wahrscheinlicher, durch Sequenzvergleiche und Sekundärstrukturvorhersagen ein
geeignetes Suchmodell zu finden.
Die Methode des molekularen Ersatzes plaziert dabei ein Suchmodell in die Einheitszelle einer
unbekannten Kristallstruktur und kann als sechsdimensionales Problem betrachtet werden, das in
eine jeweils dreidimensionale Rotations- und Translationssuche separiert werden kann (Rossmann
& Blow, 1962). Der Vergleich der Patterson-Funktionen von Suchmodell und Röntgendaten der
unbekannten Kristallstruktur stellt dabei das grundlegende Prinzip des molekularen Ersatzes dar
65
Methoden
und dient als Kriterium für die Molekülplatzierung. Für eine erfolgreiche Suche sind neben dem
Grad der strukturellen Ähnlichkeit zwischen Suchmodell und Zielmolekül auch die Anzahl an
Monomeren in der Zelle sowie die Komplexizität der Raumgruppe von großer Bedeutung. Stehen
initiale Phasen zur Verfügung, so können diese durch einen phasierten isomorphen Ersatz
berücksichtigt werden. In der vorliegenden Arbeit wurde für die Methode des molekularen Ersatzes
das mit maximum likelihood Zielfunktionen arbeitende Programm PHASER (McCoy et al., 2005)
verwendet. Korrekte Lösungen der Translationsfunktion zeichnen sich bei der Verwendung von
PHASER durch eine Höhe von mindestens sechs Standardabweichungen über dem Mittelwert aller
Maxima (Z-score) aus und führen zu einem signifikanten Anstieg des log-likelihood.
Demgegenüber heben sich Lösungen der Rotationsfunktion meist deutlich weniger voneinander ab.
3.5.12
Nicht-kristallographische Symmetrie
Enthält die asymmetrische Einheit mehrere identische Moleküle, so können diese durch lokale
Symmetrieoperationen ineinander überführt werden. Die sich daraus ergebende lokale Symmetrie
wird als nicht-kristallographische Symmetrie (NCS) bezeichnet. Im Fall von oligomeren Proteinen
liegt häufig eine geschlossene Punktgruppensymmetrie vor, so dass die über NCS verwandten
Protomere durch die Anwendung einer n-zähligen Rotation ineinander überführt werden können.
Die NCS kann allerdings auch aus einer Kombination von Rotations- und Translationsanteilen
bestehen.
R=∫ P u P urot d u
Gleichung 42
U
U
P
u
P
urot
Volumen der Patterson-Funktion mit dem Radius |u|
Patterson-Funktion am Ort u
rotierte Patterson-Funktion am Ort urot
Die Rotationskomponente R der NCS lässt sich aus den Maxima der Selbstrotationsfunktion
bestimmen (Rossmann & Blow, 1962). Dazu wird die Patterson-Funktion eines Datensatzes mit
sich selbst überlagert, wobei eine der Funktionen in vorgegebenen Schritten um den Ursprung
rotiert wird (Gleichung 42). Die Integrationsgrenzen werden so gewählt, dass lediglich Selbstvektoren, also Vektoren zwischen Atomen innerhalb eines Protomers, genutzt werden und störende
Kreuzvektoren zwischen Atomen unterschiedlicher Protomere sowie das Maximum im Ursprung
der Patterson-Funktion unberücksichtigt bleiben.
66
Röntgenographische Methoden
Die Translationskomponente T der NCS kann nur in Spezialfällen ohne vorherige Phasen­
information bestimmt werden. Üblicherweise wird die Translationskomponente jedoch mit Hilfe
bekannter Schweratompositionen und/oder initialer Phasen bestimmt. Sind die Bindestellen der
Schweratompositionen symmetrisch über die NCS-verwandten Protomere der kristallographisch
asymmetrischen Einheit verteilt, kann daraus in Näherung das Zentrum der NCS-Kopie und damit
die Position der Rotationsachse bestimmt werden. Alternativ kann auch versucht werden, die
Protomere der asymmetrischen Einheit durch geeignete Masken voneinander abzugrenzen und
anschließend die Symmetrieoperatoren zu bestimmen. In der vorliegenden Arbeit wurden sowohl
Rotations- als auch Translationskomponenten vorhandener NCS mit dem Programm RESOLVE aus
bekannten Schweratompositionen ermittelt (Terwilliger, 2002) und während der Dichtemodifikation
mit RESOLVE (Abschnitt 3.5.13) verfeinert (Terwilliger, 2000).
3.5.13
Dichtemodifikation
Die initial erhaltenen Phasen sind meist noch zu ungenau, um eine (vollständig) in Form eines
atomaren Modells interpretierbare Elektronendichtekarte zu erhalten. Zur Verbesserung der Phasen
kann das Wissen um allgemeine Charakteristika der Elektronendichtekarten von Makromolekülen
eingesetzt werden. Die sich daraus ergebenden Einschränkungen im realen Raum – etwa die geringe
Variation der Elektronendichte in Solvensregionen – führen zu Einschränkungen der möglichen
Phasen der Strukturfaktoren. Durch die Wahl von Phasen, die mit den experimentell ermittelten
Daten vereinbar sind und gleichzeitig zu einer plausibleren Elektronendichtekarte (im Sinne der
gemachten Einschränkungen) führen, kann eine erhebliche Verbesserung der initialen Phasen
herbeigeführt werden. Zusätzlich können durch ein iteratives Verfahren Phasen auch de novo über
ihre ursprüngliche Auflösungsgrenze hinaus bestimmt werden (phase extension).
Zur Zeit existieren zwei prinzipiell unterschiedliche Ansätze zur Dichtemodifikation. Bei der
konventionellen Vorgehensweise wird eine Modifikation der Elektronendichtekarte im Realraum
vorgenommen und die durch Rücktransformation der Karte erhaltenen modifizierten Phasen mit
den experimentellen kombiniert. Problematisch dabei sind die angemessene relative Wichtung der
modifizierten und experimentellen Phasen sowie das Fehlen robuster Stoppkriterien für den iterativ
durchgeführten Prozess. Die von Terwilliger (1999) eingeführte Methode der statistischen
Dichtemodifikation eliminiert diese Schwächen durch eine konsequente statistische Behandlung
aller Quellen an Phaseninformation. Dabei wird eine Wahrscheinlichkeitsfunktion für jeden Satz
möglicher Strukturfaktoren {Fh} formuliert, die sich aus den folgenden zwei Anteilen zusammen­
67
Methoden
setzt: einer Wahrscheinlichkeitsverteilung für die observierten Strukturfaktoren (Unsicherheiten der
Amplituden und Wahrscheinlichkeitsverteilung für die experimentell ermittelten Phasen) sowie
einer Wahrscheinlichkeitsverteilung der Werte der aus diesen resultierenden Elektronendichte an
jedem Punkt der Elektronendichtekarte. Letztere misst, wie wahrscheinlich der momentane Wert
unter Berücksichtigung bekannter Charakteristika (etwa der Glätte des Solvens) von Elektronen­
dichtekarten ist. Der Prozess der Dichtemodifikation, also das Auffinden der Strukturfaktoren, die
sowohl mit den experimentellen Daten als auch den Erwartungen an die Eigenschaften der Elektro­
nendichtekarte konsistent sind, entspricht somit dem Auffinden des Maximums der zusammen­
gesetzten Wahrscheinlichkeitsfunktion. Die im Folgenden aufgeführten Methoden der Dichtemodi­
fikation werden üblicherweise eingesetzt. In der vorliegenden Arbeit wurden dazu die Programme
DM (Cowtan, 1994), SOLOMON (Abrahams & Leslie, 1996) und RESOLVE (Terwilliger, 2000)
verwendet.
Solvensglätten (solvent flattening und solvent flipping)
Physikalische Grundlage der Methoden des solvent flattening oder solvent flipping ist die
Tatsache, dass die interferierend streuende Elektronendichte in Lösungsmittelbereichen wenig
Variation aufweist. Der Grund dafür ist die hohe thermische Beweglichkeit der Solvensmoleküle
und ihre Unordnung im Kristall. So entstehen definierte Solvensbereiche mit geringer Elektronen­
dichte, die sich von Bereichen des Proteins abgrenzen lassen. Dies wiederum führt zu einer starken
Einschränkung der Phasen der Strukturfaktoren. Die Abgrenzung von Solvens und Protein erfolgt
mit Hilfe einer Maske, die nach unterschiedlichen Algorithmen erstellt werden kann (Wang, 1985;
Leslie 1987; Abrahams & Leslie, 1996, Terwilliger, 1999). Mit Hilfe der Maske wird daraufhin bei
der konventionellen Dichtemodifikation die Elektronendichtekarte dadurch verändert, dass
Lösungsmittelbereiche auf einen konstanten Wert gesetzt werden (solvent flattening), oder der
jeweilige aktuelle Wert mit -1 multipliziert wird (solvent flipping). Bei der statistischen Dichte­
modifikation
wird
die
Wahrscheinlichkeit
der
momentanen
Elektronendichtewerte
im
Solvensbereich durch den Grad der Abweichung vom Mittelwert abgeschätzt. Eeffektiver ist es
allerdings, mit einer Verteilungsfunktion der möglichen Elektronendichtewerte statt mit dem
Mittelwert zu arbeiten (siehe Histogramm-Anpassung). Die Methode des Solvensglättens funktio­
niert um so effektiver, je höher der Lösungsmittelgehalt des Kristalls ist.
68
Röntgenographische Methoden
Histogramm-Anpassung (histogram matching)
Die Methode der Histogramm-Anpassung wird meistens in Kombination mit der Methode des
Solvensglättens angewendet, um die Elektronendichte innerhalb der Proteinmaske direkt zu
verbessern. Dabei wird ausgenutzt, dass die Häufigkeit beobachteter Elektronendichtewerte von den
in der Elementarzelle vorhandenen Atomtypen und den auftretenden interatomaren Abständen
abhängt. Da diese für Proteine in der Regel ähnlich sind, kann die Verteilung der
Elektronendichtewerte einer unbekannten Proteinstruktur an ein ideales Dichtehistogramm
angepasst
(konventionell;
Zhang
&
Wahrscheinlichkeitsverteilungsfunktion
Main,
der
1990)
oder
für
Elektronendichtewerte
die
Bestimmung
herangezogen
einer
werden
(statistisch; Terwilliger, 2000).
Nicht-kristallographische Symmetrie
Liegen mehrere identische oder zumindest weitgehend ähnliche Protomere in der
asymmetrischen Einheit vor, so ergibt sich aus den daraus resultierenden Einschränkungen für die
Elektronendichte und die sich daraus ergebenden Einschränkungen möglicher Phasen eine
wirkungsvolle Möglichkeit zur Dichtemodifikation. Bei der konventionellen Dichtemodifikation
werden Dichtewerte NCS-verwandter Positionen gemittelt, wobei gegebenenfalls noch eine
Wichtung durch die erwartete Ähnlichkeit der NCS-Kopien erfolgt (Abrahams & Leslie, 1996). Bei
der statistischen Dichtemodifikation wird die Information aus NCS mit zur Bestimmung der
Wahrscheinlichkeitsverteilungsfunktion der Elektronendichtewerte herangezogen (Terwilliger,
2002). Da dabei für jeden Punkt der Elektronendichtekarte eine eigene Wahrscheinlichkeitsver­
teilung der Elektronendichtewerte ermittelt wird, werden mögliche lokale Abweichungen von der
NCS automatisch berücksichtigt.
Lokale Mustererkennung (local pattern matching)
Das Auftreten charakteristischer lokaler Muster in der Elektronendichte von Proteinen kann
zur statistischen Dichtemodifikation herangezogen werden (Terwilliger, 2003c). Dabei wird die
Übereinstimmung von Mustern innerhalb einer Kugel mit 2 Å Radius für jeden Punkt der Elektro­
nendichtekarte mit Vorlagen aus einer Bibliothek verglichen, um einen neuen, idealen Schätzwert
für die Elektronendichte im Zentrum der Kugel zu erhalten (der ursprüngliche Elektronendichtewert
im Zentrum der Kugel bleibt dabei unbeachtet). Als Ergebnis dieser Analyse werden idealisierte
Elektronendichtekarte erhalten, ein so genanntes image, deren Fehler nahezu unkorreliert mit denen
69
Methoden
der ursprünglichen Karte sind. Durch eine der statistischen Dichtemodifikation verwandten
Verfahrensweise können aus dieser idealisierten Karte neue Schätzwerte für Phasen und deren
Wahrscheinlichkeitsverteilung ermittelt werden. Diese Phaseninformation wird anschließend mit
den ursprünglichen Phasen kombiniert.
3.5.14
Modellbau und Elektronendichtekarten
Modellbau
Resultiert aus den initial bestimmten oder dichtemodifizierten Phasen eine zumindest teilweise
interpretierbare Elektronendichtekarte, so kann mit dem Bau eines Proteinmodells begonnen
werden. Abhängig von Qualität und Auflösungsgrenze der Phasen kann dies auch automatisch
erfolgen, wozu in der vorliegenden Arbeit die Programme ARP/wARP (Perrakis et al., 1999) und
RESOLVE (Terwilliger, 2003a, b) verwendet wurden. Die beiden Programme verwenden dabei
deutlich unterschiedliche Verfahrensweisen. ARP/wARP baut und verfeinert zunächst ein Modell
aus freien Atomen mit einem Abstand von 1.1 Å in Bereichen hoher Elektronendichte (free atoms
model). Anschließend werden die Atome unter Berücksichtigung bekannter Geometrien des
Peptidrückgrats auf mögliche Cα-Positionen hin untersucht und auf einen Satz der wahrschein­
lichsten, nicht verzweigten, nicht überlappenden Kettenverläufe reduziert. Das hybride Modell aus
Cα-Ketten und freien Atomen wird verfeinert und wenn möglich die Proteinsequenz anhand der
überbleibenden, nicht dem Rückgrat zugehörigen, freien Atome zugeordnet. Zuletzt werden noch
Seitenketten gebaut und deren Konformation verfeinert.
RESOLVE hingegen nutzt eine FFT-basierte Suche mit einer Bibliothek an Fragmenten
unterschiedlicher Länge, um die Hauptkette aufzubauen. Die aufgefundenen Positionen werden
anschließend durch eine Maximierung der Korrelation von Fragmentdichte und beobachteter Dichte
verfeinert. Die Zuordnung der Sequenz und das Bauen der Seitenketten erfolgt auf ähnliche Art und
Weise, wobei eine Bibliothek an Standard-Rotameren aller möglichen Seitenketten zum Einsatz
kommt. Beide Programme nutzen den Modellbau zusätzlich, um die initiale Phaseninformation zu
korrigieren. Es reicht deshalb häufig stark fehlerbehaftete Phaseninformation aus, um einen
Großteil des Proteinmodells automatisch zu bauen oder zumindest die Qualität der Phasen so weit
zu verbessern, dass manuell modelliert werden kann. In der vorliegenden Arbeit wurde zum
manuellen Modellbau das interaktive Grafikprogramm COOT (Emsley & Cowtan, 2004) verwen­
det. Zur Unterstützung des manuellen Modellbaus, vor allem in loop-Bereichen, wurde zudem das
Programm RAPPER (de Bakker et al., 2003) verwendet, das einen automatischen Modellbau auf
70
Röntgenographische Methoden
Basis aminosäurespezifischer Bibliotheken bevorzugter Kombinationen der Hauptkettentorsions­
winkel φ und ψ ermöglicht. Der manuelle Modellbau erfolgte im Wechsel mit der Verfeinerung des
Modells. Dabei wurden die im Folgenden aufgeführten Elektronendichtekarten verwendet.
(Fobs * FOM)-Elektronendichtekarte
Fourierkarten mit den Koeffizienten mF obs expia (m = figure of merit) werden genutzt, falls
kein Modell vorliegt. Die Phasen α können entweder direkt aus der Schweratomverfeinerung oder
der Dichtemodifikation erhalten werden.
(Fobs – Fcalc)-Elektronendichtekarte
Differenz-Fourierkarten mit den Koeffizienten F obs− F calc expi calc zeigen Unterschiede
zwischen Modell und tatsächlicher Struktur auf. Fehlende Bereiche des Modells erscheinen mit
positiver Differenzdichte, falsch positionierte mit negativer. Die Differenzen erscheinen mit halber
tatsächlicher Höhe und sind ab einem Konturniveau von drei Standardabweichungen (3 σ) der
durchschnittlichen Elektronendichte signifikant. (Fobs-Fcalc)-Dichtekarten dienen als Basis für die
Korrektur und Ergänzung des Proteinmodells sowie für den Einbau von Wassermolekülen oder
Liganden.
(2Fobs – Fcalc)-Elektronendichtekarte
Diese mit den Koeffizienten 2 F obs −F calc exp i calc berechnete Dichtekarten zeigen die
Elektronendichte des Modells und die Differenzdichte in voller Höhe und werden auf einem
Konturniveau von etwa 1 σ dargestellt. Aufgrund des starken Modelleinflusses (model bias) werden
sie üblicherweise nicht genutzt und stattdessen gewichtete Elektronendichtekarten eingesetzt.
Gewichtete (mFobs – DFcalc)- und (2mFobs – Dfcalc)-Elektronendichtekarten
Stellt das Modell nur einen Teil der Gesamtstruktur dar oder ist es stark fehlerbehaftet, so
empfiehlt es sich, die berechneten Strukturfaktoren entsprechend zu gewichten. Zur Wichtung
werden nach Read (1986; 1990) so genannte σA-Terme herangezogen, die die gemessenen Struktur­
faktoramplituden mit den berechneten in Beziehung setzen (Abbildung 24). In dieser Arbeit wurden
σA-gewichtete Elektronendichtekarten mit Modulen aus dem Programmpaket CNS (Brünger et al.,
1998) sowie den CCP4-Programmen SIGMAA, REFMAC und FFT (CCP4, 1994) berechnet.
prime-and-switch Elektronendichtekarten
Eine weitere Methode, den Einfluss eines (fehlerbehafteten) Modells auf die Berechnung einer
(2Fobs-Fcalc)-Elektronendichte zu minimieren, stellt neben σA-gewichteten Karten die so genannten
prime-and-switch Elektronendichtekarten dar (Terwilliger, 2001). Dabei wird zunächst ausgehend
71
Methoden
von den besten vorhandenen Strukturfaktoramplituden, Phasen und Wichtungsfaktoren eine initiale
Elektronendichte berechnet. Anschließend werden durch eine der statistische Dichtemodifikation
ähnelnde
Verfahrensweise
(Abschnitt 3.5.13)
neue
Schätzwerte
für
Phasen
und
deren
Wahrscheinlichkeitsverteilung bestimmt, die abschließend zur Berechnung der prime-and-switch
Elektronendichtekarte herangezogen werden. Die resultierenden Phasen sind weniger stark vom
Modell beeinflusst als die zugrunde liegenden, da sie stärker durch die Eigenschaften der
Elektronendichtekarte beeinflusst sind als durch das Modell selbst.
Abbildung 24:
3.5.15
Koeffizienten zur Berechnung von gewichteten Elektronendichtekarten. Fehler im Modell sind auf
Fehler von Atompositionen und Streubeiträgen zurückzuführen und haben Gaußsche Fehler in Phase
sowie Amplitude der daraus berechneten Strukturfaktoren zur Folge. Der Erwartungswert der sich
daraus ergebenden Verteilungsfunktion für einen aus dem Modell berechneten Strukturfaktor ist
durch eine zweidimensionale Gaußverteilung um DFcalc (0≤D≥1) gegeben. Der Faktor D schließt
neben Fehlern von Atompositionen und Streubeiträgen auch Effekte durch ein unvollständiges Modell
sowie Unterschiede der absoluten Skalierung und der B-Faktoren mit ein.
Verfeinerung des Modells
Ein initial erhaltenes Proteinmodell stellt in der Regel noch nicht die best mögliche
Interpretation der gemessenen Daten dar. Die aufgrund der Modellparameter berechneten
Strukturfaktoramplituden Fcalc zeigen eine schlechte Übereinstimmung mit den Observablen Fobs.
Ziel der Verfeinerung des Modells ist es daher, durch eine Variation der Modellparameter diese
Übereinstimmung zu maximieren. Ein Maß der Übereinstimmung ist der kristallographische
R-Faktor Rcryst (Gleichung 43). R-Faktoren um 50% sind für initiale, unverfeinerte Proteinstrukturen
keine Seltenheit. Zufallsstrukturen besitzen einen R-Faktor von 59% (Wilson, 1949), gut verfeinerte
Röntgenstrukturen hingegen weisen R-Faktoren zwischen 10 und 25% auf.
72
Röntgenographische Methoden
∑ ∣F obs hkl−k⋅F calc hkl ∣
Rcryst = hkl
∑∣F obs hkl ∣
Gleichung 43
hkl
k
auflösungsabhängiger Skalierungsfaktor
Prinzipiell lassen sich Modelle nur dann verfeinern, wenn die Zahl der Observablen
(Röntgenreflexe) die der zu verfeinernden Parameter übersteigt. Da die Zahl der Observablen durch
Auflösungsgrenze, Vollständigkeit und Raumgruppe des Kristalls beschränkt ist, müssen in der
Proteinkristallographie meist noch Nebenbedingungen für eine erfolgreiche Verfeinerung eingeführt
werden. Diese Einschränkung der Werte (restraints), die freie Parameter annehmen dürfen, wirken
dabei wie zusätzliche Observablen. Bei der Verfeinerung von Proteinstrukturen werden abhängig
von der Auflösungsgrenze der experimentellen Daten unterschiedlich gewichtete stereochemische
Einschränkungen für die geometrischen Parameter des Modells eingeführt (Engh & Huber, 1991)
und die Temperaturfaktoren (siehe unten) benachbarter Atome aneinander gekoppelt, um
übermäßige Schwankungen innerhalb kurzer Distanzen zu vermeiden. Zudem kann die Ähnlichkeit
durch nicht-kristallographische Symmetrie (NCS) verknüpfter Moleküle berücksichtigt werden.
Die Zahl der freien Parameter sollte zu Beginn der Verfeinerung möglichst niedrig sein und
erst allmählich erhöht werden (Kleywegt & Jones, 1995). Ein gutes Modell weist am Ende der
Verfeinerung chemisch sinnvolle Stereochemie und Temperaturfaktoren auf, erklärt die kristallo­
graphischen und biochemischen Daten und benutzt dazu eine minimale Anzahl freier Parameter.
Werden zu viele Parameter gleichzeitig freigegeben, besteht die Gefahr, experimentelles Rauschen
zu modellieren (overfitting). Um dies zu vermeiden, wird ein kleiner, zufällig ausgewählter Teil der
Reflexe, der so genannter Testdatensatz (etwa 5-10 % aller Reflexe, mindestens jedoch 500), nicht
mit in die Verfeinerung einbezogen. Mit diesen Reflexen wird in Analogie zum kristallogra­
phischen R-Faktor (Gleichung 43) der freie R-Faktor Rfree berechnet (Gleichung 44; Brünger, 1992).
Der freie R-Faktor korreliert mit dem mittleren Fehler der Modellphasen und dient daher als Maß
für den Fortschritt der Verfeinerung (Kleywegt & Brünger, 1996).
∑ ∣F obs hkl−k⋅F calc hkl ∣
R free =
∑ ∣F obs hkl∣
hkl ∈T
Gleichung 44
hkl ∈T
73
Methoden
Parametrisierung des Modells: Koordinaten
Generell wird das Modell als eine Einheit aus einzelnen Atomen interpretiert, die durch die
drei Ortskoordinaten x,y,z einen Temperaturfaktor B sowie eine Besetzungszahl pro Atom para­
metrisiert werden. Kann davon ausgegangen werden, dass die Orientierung und Positionierung des
Modells oder von Teilen des Modells noch sehr ungenau ist, so lohnt sich eine Parametrisierung als
rigide Gruppe. Dabei werden Atomgruppen als starre Körper (rigid bodies) definiert und deren
sechs Rotations- und Translationsfreiheitsgrade unabhängig voneinander verfeinert. Da lediglich
eine geringe Anzahl an Parametern benötigt wird, kann die Verfeinerung bei niedriger Auflösung
durchgeführt werden, wodurch sich der Konvergenzradius erhöht. Eine ähnlich drastische
Reduktion der Modellparameter kann erreicht werden, wenn mehrere NCS-verwandte Moleküle
vorliegen und diese eng aneinander gekoppelt werden (NCS-constraints). Alternativ kann auch eine
mehr oder weniger starke Variation der Parameter innerhalb der NCS-Gruppe zugelassen werden,
um Unterschieden zwischen den Molekülen gerecht zu werden (NCS-restraints). Da die Verteilung
der Bindungswinkel und -längen eine sehr geringe Streuung aufweist (Engh & Huber, 1991),
können diese vor allem bei Strukturen niedriger und mittlerer Auflösung als Konstanten behandelt
werden. Die Struktur lässt sich nach einer Idealisierung der Bindungswinkel und -längen allein
durch Torsionswinkel zwischen Atomgruppen parametrisieren, was ebenfalls zu einer beträcht­
lichen Verringerung der Anzahl an Parametern führt. Diese Art der Parametrisierung wird vor allem
in Verbindung mit einer simulated annealing Verfeinerung in CNS (Brünger et al., 1998) genutzt,
wobei eine Molekulardynamik-Simulation bei erhöhter Temperatur durchgeführt wird. Durch die
geringere Zahl an Freiheitsgraden können dabei deutlich höhere Simulationstemperaturen
verwendet werden, was den Konvergenzradius der Methode erhöht.
Parametrisierung des Modells: Temperaturfaktoren
Fehlordnungen und thermische Bewegungen der Atome im Proteinkristall werden durch den
Temperaturfaktor B beschrieben (Abschnitt 3.5.4). Abhängig von der Auflösung des Kristalls
werden unterschiedlich viele Parameter verfeinert: Bei geringer Auflösung kann aufgrund des
schlechten Verhältnisses von Observablen zu Parametern lediglich ein mittlerer isotroper B-Faktor
für das ganze Protein angegeben werden. Im mittleren Auflösungsbereich werden isotrope
B-Faktoren gruppenweise, unterhalb von ca. 2.5 Å individuell für jedes Atom bestimmt. Erst bei
hoch aufgelösten Kristallstrukturen kann die Schwingung der Atome in alle drei Raumrichtungen
durch einen anisotropen Temperaturfaktor beschrieben werden.
74
Röntgenographische Methoden
Eine annähernde Parametrisierung der anisotropen Verschiebungen bei mittleren Auflösungen
lässt sich durch die TLS-Methode (Schomaker & Trueblood, 1968; Abbildung 25) erreichen. Dabei
werden Parameter für eine Gruppe von Atomen (Domänen, Sekundärstrukturelemente oder Seiten­
ketten) verfeinert, wodurch sich die Anzahl benötigter Parameter gegenüber einer Verfeinerung
anisotroper B-Faktoren jedes einzelnen Atoms stark reduziert. Die TLS-Gruppen werden als
pseudo-starre Körper behandelt. Die mittlere quadratische Verschiebung U eines Atoms wird im
TLS-Formalismus durch die drei Tensoren T (translation), L (libration) und S (screw rotation)
beschrieben und in Abhängigkeit von seiner Entfernung zum Schwerpunkt der TLS-Gruppe
angegeben. Bei einer maximalen Auflösung von 2.0 Å können ein bis fünf TLS-Gruppen definiert
werden, bei 1.5 Å bis zu 50. Die Zuordnung einzelner Atome zu Gruppen sollte sich an der
Domänenstruktur bzw. an Sekundärstrukturelementen des Proteins orientieren. Die Entscheidung,
ob eine Parametrisierung durch TLS zulässig ist, sollte sich immer am Einfluss auf die R-Faktoren
Rcryst und Rfree sowie die Qualität der erhaltenen Elektronendichte orientieren. In der vorliegenden
Arbeit wurden TLS-Parameter in einem späten Stadium der Strukturverfeinerungen mit dem
Programm REFMAC (Murshudov et al., 1997; Winn et al., 2001) bestimmt und nachfolgend mit
Hilfe des Programms TLSANL (Howlin et al., 1993) analysiert. Die vorherige Einteilung der TLSGruppen wurde automatisiert durch das Programm TLSMD (Painter & Merritt, 2006) vorgenom­
men, das die Proteinstruktur auf Flexibilität hin untersucht.
Abbildung 25:
Schematische Darstellung der TLS-Verfeinerung. Jede TLS-Gruppe schwingt dabei um ihren
Schwerpunkt (Winn et al., 2003)
maximum likelihood Verfeinerung
Moderne Verfeinerungsprogramme, wie die in der vorliegenden Arbeit verwendeten Program­
me REFMAC (Murshudov et al., 1997) und CNS (Brunger et al., 1998) arbeiten nach dem
maximum likelihood Prinzip. Dabei wird das Maximum einer komplexen Wahrscheinlichkeits­
75
Methoden
funktion gesucht, welche die Wahrscheinlichkeit der Modellparameter zum einen daran misst, wie
gut sie mit chemisch/physikalischen Erwartungen übereinstimmen, und zum anderen, wie gut die
aus dem Modell berechneten Erwartungswerte der Strukturfaktorenamplituden (oder Intensitäten)
mit den observierten übereinstimmen. Die Zielfunktion berücksichtigt zudem Unsicherheiten der
Modellparameter, der Phasen und gemessenen Strukturfaktoramplituden.
Solvenskorrektur
Kristalle von Makromolekülen enthalten einen hohen Anteil an ungeordnetem Solvens (bulk
solvent), das lediglich zur Röntgenstreuung bei niedriger Auflösung (ab etwa 6 Å) beiträgt. Da
Strukturmodelle üblicherweise keine Beschreibung des ungeordneten Solvens enthalten, können
Reflexe in einem Auflösungsbereich größer 6 Å nur ungenügend erklärt werden. Für eine stabile
Verfeinerung unter Einbeziehung aller gemessener Daten wird deshalb üblicherweise eine Solvens­
korrektur vorgenommen. Dazu können zum einen zusätzliche Strukturfaktoren für das ungeordnete
Solvens eingeführt werden, oder die anhand des Modells berechneten Strukturfaktoren durch
zusätzliche Skalierungsfaktoren an die observierten Strukturfaktoren angepasst werden.
Einbau von Wasser und Liganden
Wassermoleküle wurden mit dem Programm ARP/wARP (Perrakis et al., 1999) sowie mit
COOT (Emsley & Cowtan, 2004) eingebaut. Grundlage für die automatische Suche mit
ARP/wARP sind (mFobs - DFcalc)- und (2mFobs - DFcalc)-Elektronendichtekarten auf Konturniveaus
von 3.2 bzw. 0.9 σ sowie eine Analyse der unmittelbaren Umgebung der hinzugefügten Wasser­
moleküle. Die aufgefundenen Positionen wurden manuell überprüft und gegebenenfalls korrigiert
oder ergänzt. Beim Einbau von Wassermolekülen mit COOT wurden zunächst automatisch Maxima
in (mFobs - DFcalc)-Elektronendichtekarten bei 3.2 σ gesucht, die eine annähernde Kugelgestalt auf­
wiesen, und aufgefundene Positionen manuell überprüft. Zudem wurde in der jeweiligen folgenden
Verfeinerungsrunde automatisch eine Analyse der unmittelbaren Umgebung aller Wassermoleküle
durchgeführt und überprüft, ob die jeweilige Position (mFobs - Dfcalc)-Elektronendichte von mindes­
tens 0.9 σ aufwies.
Die vom Protein gebundenen Liganden wurden vom PRODRG2 Server (Schuettelkopf &
van Aalten, 2004) modelliert und energieminimiert oder bereits optimierte Koordinaten der CCP4Standardbibliothek entnommen. Ebenso wurden die benötigten restraints zur Verfeinerung aus der
CCP4-Standardbibliothek oder vom PRODRG2 Server eingesetzt. Der Einbau der Liganden in das
Proteinmodell erfolgte mit dem Programm COOT (Emsley & Cowtan, 2004).
76
Proteinstrukturen
3.6
3.6.1
Proteinstrukturen
Qualität von Proteinstrukturen
Das finale Strukturmodell enthält auch nach der Verfeinerung noch Fehler, die sich aber meist
auf eng begrenzte Bereiche beschränken. Zur Beurteilung der Qualität eines Strukturmodells gibt es
zahlreiche Kriterien, die sich entweder auf die Gesamtstruktur oder aber auf lokale Bereiche wie
etwa einzelne Aminosäuren beziehen (Dodson et al., 1998). Zur Validierung von Proteinstrukturen
sollten nur solche Kriterien herangezogen werden, die nicht in den Zielfunktionen der zur
Verfeinerung verwendeten Programme enthalten sind. Validierungsprogramme wie PROCHECK
(Laskowski et al., 1993), WHATCHECK (Hooft et al., 1996) oder MOLPROBITY (Lovell et al.,
2000) verwenden möglichst viele unabhängige Kriterien sowie statistische Analysen, um Fehler im
Strukturmodell sicher aufzufinden.
Kristallographischer R-Faktor
Der kristallographische R-Faktor Rcryst (Gleichung 43) beschreibt die Übereinstimmung
zwischen den gemessenen Strukturfaktoramplituden Fobs mit den aus dem Strukturmodell
errechneten Strukturfaktoramplituden Fcalc. Der kristallographische R-Faktor ist als Maß für die
Modellqualität alleine betrachtet nur beschränkt aussagekräftig, da er stark vom Verhältnis der
Anzahl der Observablen zur Anzahl der verfeinerten Parameter abhängig ist (Kleywegt & Jones,
1995). In Verbindung mit dem freien R-Faktor Rfree (Gleichung 44) hingegen kann er Rückschlüsse
auf den Verlauf der Verfeinerung zulassen.
Elektronendichte-Korrelation im Realraum
Die Bestimmung der Korrelation zwischen beobachteter Elektronendichte ρobs und der aus dem
Modell berechneten Elektronendichte ρcalc erlaubt auf atomarer Ebene eine Beurteilung des
vorhandenen Modells im Realraum. Durch Analyse der Elektronendichte-Korrelation können
unzureichend verfeinerte oder schlecht definierte Segmente innerhalb der modellierten
Proteinstruktur aufgefunden werden. Allerdings muss auf eine generelle schlechtere Korrelation in
Strukturbereichen, die sich durch hohe Temperatur-Faktoren und damit hohe Beweglichkeit
auszeichnen, geachtet werden. Gleichung 45 zeigt die Formel zur Berechnung des Korrelations­
koeffizienten CC mit dem Programm OVERLAPMAP (CCP4, 1994).
CC =
⟨ obs⋅ calc ⟩−⟨ obs ⟩⋅⟨calc ⟩
⟨
2
obs
⟩−⟨2obs ⟩− ⟨2calc−2calc ⟩
Gleichung 45
77
Methoden
Stereochemie
Der von Engh & Huber (1991) abgeleitete Satz idealer Bindungslängen und -winkel für
Proteine beruht auf einer Analyse hoch aufgelöster Kristallstrukturen niedermolekularer
Verbindungen. Die mittlere quadratische Abweichung (rmsd) von diesen Idealwerten wurde in der
vorliegenden Arbeit mit den Programmen PROCHECK (Laskowski et al., 1993) und
WHATCHECK (Hooft et al., 1996) analysiert. Da diese stereochemischen Parameter in den Ziel­
funktionen aller üblichen Verfeinerungsprogramme enthalten sind, ist ihre Aussagekraft als
Qualitätskriterium eher beschränkt, es können jedoch Hinweise auf Verlauf und Qualität der Ver­
feinerung erhalten werden.
Torsionswinkel
Aufgrund der sterischen Hinderung von Peptidbindungen existieren in der Hauptkette nur zwei
Rotationsfreiheitsgrade pro Rest. Die zwei Hauptkettenwinkel φ und ψ sind auf bestimmte Werte
durch die Geometrie der Peptidgruppe festgelegt. Für alle Reste außer Glycin führt das Vorhanden­
sein einer voluminösen Seitenkette am Cα zu Überschneidungen mit dem Wasserstoff am Haupt­
kettenstickstoff oder mit dem Carbonylsauerstoff. Im Ramachandran Diagramm φ wird gegen ψ für
alle Reste des Modells aufgetragen. Die Reste sollten in erlaubten Regionen zu liegen kommen, die
ursprünglich durch Modellierung (Ramachandran & Sasisekharan, 1968) und später durch Biblio­
theken von in hoch aufgelösten Strukturen beobachteten Hauptkettentorsionswinkeln (Lovell et al.,
2003) definiert wurden.
Eine ähnliche Situation liegt auch für die Seitenkettentorsionswinkel vor. Durch Analyse von
hochaufgelösten Strukturen wurde auch hier eine Bibliothek von bevorzugten Rotameren erstellt
(Lovell et al., 2000), gegen die das Modell verglichen werden kann. In Proteinstrukturen taucht nur
eine beschränkte Anzahl von Seitenkettenrotameren auf, so dass ein Satz von Torsionswinkeln einer
Seitenkette, der einer häufig gefundenen Konformation entspricht, mit hoher Wahrscheinlichkeit
korrekt ist. Da es für die Seitenkettenkonformationen während der Verfeinerung mehrere äquiva­
lente Energieminima gibt, können sie nur schwach oder gar nicht beschränkt werden. Dadurch sind
sie ein sehr guter unabhängiger Qualitätsindikator in der Strukturvalidierung. In dieser Arbeit
wurden Hauptkettentorsionswinkel-(Ramachandran)-Analysen mit PROCHECK (Laskowski et al.,
1993) und RAMPAGE (Lovell et al., 2003) und Seitenkettentorsionswinkel (Rotamer) Analysen
mit COOT (Emsley & Cowtan, 2004) und MOLPROBITY (Lovell et al., 2000) durchgeführt.
78
Proteinstrukturen
3.6.2
Darstellung von Proteinstrukturen
Eine atomare Darstellung von Proteinstrukturen ist aufgrund der Molekülgröße nur in kleinen
Ausschnitten sinnvoll. Die Darstellung größerer Bereiche erfordert aus Gründen der Übersicht­
lichkeit einen gewissen Abstraktionsgrad, wozu Polypeptidketten häufig als Bändermodelle
dargestellt werden. Zudem sinnvoll ist die Darstellung der Oberfläche des Proteins, da sie einen
besseren Eindruck von der tatsächlichen Gestalt eines Makromoleküls vermittelt. Die Zuordnung
der Aminosäuren zu Sekundärstrukturelementen wurde in der vorliegenden Arbeit automatisch mit
den Programmen DSSP (Kabsch & Sander, 1983) und STRIDE (Frishman & Argos, 1995)
vorgenommen. Die Programme analysieren dabei die Umgebung der polaren Atome in der Protein­
hauptkette im Hinblick auf charakteristische Muster im Netzwerk der Wasserstoffbrücken. Zudem
werden die Diederwinkel der Hauptkettenatome für die Zuordnung zu den Sekundärstruktur­
elementen mit in die Analyse einbezogen. Zur Berechnung von molekularen Oberflächen wurden
das Programm MSMS (Sanner et al., 1996) verwendet. Sämtliche Darstellungen wurden mit dem
Programm POVSCRIPT+ (Fenn et al., 2003) erstellt und mit POVRAY (http://www.povray.org)
photorealistisch aufbereitet. Kristall- und Multimerkontakte sowie Wechselwirkungen zwischen
Protein und Liganden wurden mit dem Programm PISA (Krissinel & Henrick, 2005) analysiert.
3.6.3
Proteinstrukturvergleiche
Die meisten Proteine besitzen keine einzigartige Faltung, sondern lassen sich Strukturfamilien
zuordnen. Häufig werden ähnliche Strukturen auch dann beobachtet, wenn die betreffenden Pro­
teine keinerlei Ähnlichkeit in Sequenz oder Funktion aufweisen. Der strukturelle Vergleich erfor­
dert eine genaue Überlagerung der zu vergleichenden Moleküle. Als Kriterien für die Ähnlichkeit
zweier Moleküle werden die mittleren quadratischen Abweichungen (rmsd) der Positionen sich
entsprechender Atome (Maiorow & Crippen, 1994) oder Distanzmatrizes (Holm & Sander, 1993)
verwendet. Entscheidend für eine optimale strukturelle Überlagerung ist dabei die Zuordnung sich
entsprechender Aminosäurereste. In dieser Arbeit wurde eine nicht redundante Strukturdatenbank
mit dem Programm DALI (Holm & Sander, 1993) nach strukturell ähnlichen Proteinen durchsucht
und diese anschließend mit dem Programm LSQMAN (Kleywegt, 1996) mit der Suchstruktur
überlagert. Alternativ wurde mit dem Programm SSM (Krissinel & Henrick, 2004) gearbeitet,
wobei zunächst eine vorläufige Überlagerung durch den Vergleich von Sekundärstrukturelementen
erfolgt und erst dann eine iterative, dreidimensionale Überlagerung von Cα-Positionen. Für eine
Suche nach bestimmten Charakteristika einer Proteinstruktur wurde das Programm PINTS (Stark et
al., 2003) verwendet, das eine Überlagerung definierter Teile einer Proteinstruktur gegen die
Protein-Datenbank erlaubt.
79
Ergebnisse und Diskussion
4. Ergebnisse und Diskussion
4.1
4.1.1
Ergebnisse zur Sulfhydrylase CysM
Gentechnische Methoden zur Sulfhydrylase CysM
In vorangegangen Arbeiten war ein zuverlässiges Protokoll zu Einführung von Punktmuta­
tionen in den Wildtyp über die QuikChange Methode etabliert worden (Zocher, 2003). Für eine
ausführliche Beschreibung wird daher auf diese Literatur verwiesen. Eine kurze Zusammenfassung
der experimentellen Vorgehensweise findet sich in Abschnitt 3.1.2.
Zur Verbesserung der Kristallisationsfähigkeit der Sulfhydrylase wurden Oberflächenmu­
tanten hergestellt. Da eine gezielte Mutagenese anhand vorherrschender Kristallkontakte vorhan­
dener Strukturen (Abschnitt 1.3) nicht zum gewünschten Erfolg führte, wurde eine andere Strategie
bezüglich der weiteren Mutantenplanung verfolgt. Durch Austausch langer, geladener Seitenketten,
die an der Proteinoberfläche gelegen sind, gegen kurze unpolare Aminosäuren sollte ein
entropischer Vorteil für die Kristallisation des Proteins geschaffen werden (Derewenda, 2004). Auf
dieser Grundlage wurden verschiedene an der Protein-Oberfläche gelegene Lysin- und GlutamatReste gegen Alanin ausgetauscht. Unter den fünf hergestellten Mutanten (K59A, K115A, R165A,
K268A und E201A) zeigte die Oberflächenmutante K268A (im folgenden CysM(K268A) genannt),
ein besonders gutes Kristallisationsvermögen und hohe Streufähigkeit.
Um den Einfluss einzelner Aminosäuren auf die Aktivität der Sulfhydrylase zu untersuchen,
wurden Einzelmutanten innerhalb des aktiven Zentrum hergestellt und präpariert. Insbesondere der
Einfluss der Aminosäuren, welche das Substrat während des katalytischen Mechanismuses fixieren
oder stabilisieren, sollte überprüft werden. Für die Planung der Mutanten wurde auf die Aktivitäts­
studien von Sulfhydrylasen aus anderen Organismen zurückgegriffen (Bonner et al., 2005;
Burkhard et al., 1998). Als Modell für die Mutantenplanung diente dabei die zum Zeitpunkt der
Planung bereits vorhandene Struktur CysM(K268A) (Abschnitt 4.1.7). Da bekannt war, dass die
Ausbildung stabilisierender Wasserstoffbrücken zum Substrat zum großen Teil auf der Wechsel­
wirkung mit dem Peptidrückgrat beruhte, begrenzte sich die Zahl der möglichen Mutationen auf die
folgenden Positionen: T68, S69, Q140 und R210. Die Aminosäure T68 bildet dabei eine Wasser­
stoffbrücke zur Carboxylat-Gruppe des Citrats aus (siehe Tabelle 15, Seite 98), woraus die gleiche
Eigenschaft für das Substrat O-Acetylserin abgeleitet wurde und daher die Mutanten T68S und
T68A geplant wurden. Die gleiche Funktion erfüllen auch die Reste Q140 und S69, wobei Serin
80
Ergebnisse zur Sulfhydrylase CysM
zusätzlich eine stabilisierenden Einfluss auf einen Zwischenzustand des Katalysezyklus haben
könnte (Abschnitt 4.1.8). Daher wurden die Mutanten Q140E, Q140A und S69A geplant. Der Rest
Arg210 wurde in vorhergehenden Arbeiten für die breite Substratspezifität von CysM verantwort­
lich gemacht (Claus et al., 2004), so dass zudem die Mutante R210A hergestellt werden sollte. In
Abbildung 39 (Seite 100) sind die Positionen der erfolgreich mutierten Aminosäuren in der
Bindetasche von CysM(K268A) dargestellt.
Alle Mutanten bis auf Ser69A konnten unter Einsatz des etablierten Protokolls erfolgreich
eingeführt und präpariert werden. Der Erfolg der QuikChange wurde durch Sequenzierung bestätigt
(Abschnitt 3.1.6). Die Mutation S69A konnte trotz mehrmaliger Versuche nicht eingeführt werden.
Es zeigte sich, das die PCR-Reaktion regelmäßig den Einbau zusätzlicher Basen verursachte, wobei
eine Erklärung dieses Phänomens nicht gefunden werden konnte. Für Aktivitätsmessungen mußte
daher auf diese Mutante verzichtet werden.
4.1.2
Proteinpräparation und Kristallisation von CysM(K268A)
Die Reinigung der Sulfhydrylase wurde wie in Abschnitt 3.2.1 beschrieben durchgeführt.
Dabei diente als Basis der Reinigung das in der Literatur beschriebene Protokoll (Zocher, 2003;
Claus et al., 2005). Auf eine ausführliche Beschreibung der einzelnen Reinigungsschritte wird hier
verzichtet und nur die finale Gelpermeation an Superdex S200 gezeigt, die ein Abschätzen des
Reinheitsgerades erlaubt. Abbildung 26 zeigt exemplarisch das Elutionsprofil der GPC der K268AMutante an Superdex S200-16/60. Dieses zeigt ein einzelnes Absorptionsmaximum bei einem
Elutionsvolumen von 83.8 mL und kann der dimeren Sulfhydrylase zugeordnet werden. Die mittels
SDS-PAGE untersuchten Fraktionen (Abbildung 27) zeigen die Reinheit des isolierten Enzyms. Die
CysM-haltigen Fraktionen wurden vereinigt, mittels Zentrifugalkonzentrationszelle bei einer Aus­
schlussmasse von 30 kDa eingeengt und nach Dialyse gegen 5 mM HEPES-Puffer (pH 7.5) spek­
troskopisch untersucht. Hierzu wurde die Absorption der CysM-haltigen Lösung in einem
Wellenlängenbereich von 250 nm bis 500 nm gemessenen. Ein derartiges Spektrum für alle in
dieser Arbeit hergestellten Mutanten ist in Abbildung 28 gezeigt und erlaubt aus dem Verhältnis der
peak-Höhen bei 280 nm und 412 nm das Abschätzen des Holo- zu Apoenzym-Verhältnisses
(Abschnitt 3.3.1). Die Absorptionsverhältnisse A280:A412 sind für alle präparierten Mutanten in
Tabelle 8 aufgeführt. Die Ausbeuten betrugen für die gereinigten Mutanten zwischen 30-60 mg pro
Liter eingesetztes Kulturmedium.
81
Ergebnisse und Diskussion
Absorptionsverlauf der K268A M utante
vereinigte Fraktionen
300
59 kDa
Absorption [m AU]
250
200
150
100
50
0
0
10
20
30
40
50
60
70
80
90
Elutionsvolumen [mL]
Abbildung 26:
Exemplarisches Elutionsprofil von CysM(K268A) an Superdex 200-16/60. Die hergestellten Aktivitätsmutanten eluierten im selben Volumenbereich, der den dimeren Oligomerisierungsgrad der
Sulfhydrylase belegt. Im Unterschied zur K268A-Mutante zeigten die präparierten Aktivitätsmutanten allerdings teilweise eine deutlich geringere Stabilität. Die resultierende Aggregation war
durch Absorptionsmaxima im Ausschlussvolumen der GPC nachweisbar (Daten nicht gezeigt).
Bahn M:
Bahn 1:
Bahn 2:
Bahn 3:
Bahn 4:
Bahn 5:
Bahn 6:
Bahn 7:
Bahn 8:
Bahn 9:
Bahn 10:
Bahn 11:
Bahn 12:
Bahn 13:
Bahn 14:
Abbildung 27:
82
SDS-Gel ausgewählter Fraktionen der GPC an S200-16/60.
LMW-Standard
Fraktion 76.5-78.0 mL
Fraktion 78.0-79.5 mL
Fraktion 79.5-81.0 mL
Fraktion 81.0-82.5 mL
Fraktion 82.5-84.0 mL
Fraktion 84.0-85.5 mL
Fraktion 85.5-87.0 mL
Fraktion 87.0-88.5 mL
Fraktion 88.5-90.0 mL
Fraktion 90.0-91.5 mL
Fraktion 91.5-93.0 mL
Fraktion 93.0-94.5 mL
Fraktion 94.5-95.0 mL
Fraktion 95.0-96.5 mL
Ergebnisse zur Sulfhydrylase CysM
K268A
WT
R210A
Q140E
Q140A
T68A
T68S
1.0
Absorption [mAU]
0.8
0.6
0.4
0.2
0.0
300
Abbildung 28:
350
W ellenlänge[nm]
400
450
Absorptionsverlauf der präparierten Mutanten. Das auftretenden Maximum bei 280 nm ist das
Resultat der Absorption der aromatischen Aminosäurereste und des Kofaktors. Die Absorption
bei 412 nm ist hingegen allein auf den Cofaktor PLP zurückzuführen (Abschnitt 3.3.1).
Die zur Aktivitätsmessung hergestellten Mutanten R210A, Q140E, Q140A, T68A und T68S
zeigten bis auf R210A alle eine deutlich reduzierte Enzym-Stabilität, was sich in der Aggregation
des Proteins bereits nach 1-2 Tagen äußerte. Demgegenüber konnte der Wildtyp, wie auch die
Mutanten K268A und R210A problemlos auch über einen längeren Zeitraum von ein bis zwei
Wochen bei 4 °C gelagert werden, ohne dass nennenswerte Proteinaggregation zu erkennen war.
Für die Präparation der Mutanten war eine zügige Arbeitsweise nötig und stets auf eine gute
Kühlung zu achten. Alle Proteinlösungen wurden zur Kristallisation auf 8 mg/mL eingestellt und
diese Lösung der Kristallisation unterworfen oder als Stammlösung zur Bestimmung der enzyma­
tischen Aktivität eingesetzt. Die Mutante Q140E zeigte zudem ein deutlich schlechteres Absorp­
tionsverhältnis A280:A412 (Tabelle 8), das auch nach erneuter Rekonstitution nicht verbessert werden
konnte.
83
Ergebnisse und Diskussion
Tabelle 8:
Absorptionsverhältnisse A280:A412 der präparierten Mutanten von CysM
Absorptionsverhältnis
A280:A412
Holo-zu-ApoenzymVerhältnis
Wildtyp
4.19
98 %
T68S
4.48
95 %
R210A
4.37
97 %
Q140A
4.50
94 %
T68A
4.30
99 %
Q140E
5.04
82 %
Für die K268A-Mutante konnten zahlreiche Kristallisationsbedingungen gefunden werden,
allerdings zeigten lediglich die Kristalle, die aus dem Wizard Screen I-6 erhalten wurden, nach
Verfeinerung der Kristallisationsparameter ein außerordentlich gutes Streuvermögen. Die verfei­
nerte Kristallisationsbedingungen sind in Tabelle 9 aufgeführt und resultierten in Kristallen hexago­
naler Form mit maximalen Abmessungen von 1000 x 400 x 400 µm3. Der Kristall der zur Daten­
sammlung am SLS (Villigen, Schweiz) verwendet wurde, ist in Abbildung 29 gezeigt.
Tabelle 9
Parameter der Kristallisationverfeinerung von Kristallform A
a)
b)
Abbildung 29:
84
screen
Fällungsmittel
Puffer [0.1 M]
WS I-6
17-19% (v/v) PEG 8000a)
Na-Citrat pH 5.2-5.7b)
Variation der Konzentration in 1.0%-Schritten
Variation des pH-Wertes in 0.1 pH-Einheiten
Kristall der Oberflächenmutante K268A der Sulfhydrylase. Nach Verfeinerung der Kristallisationsparameter konnten Kristalle mit Abmessungen von bis zu 1000 x 400 x 400 µm3 erhalten werden. Charakteristisch für diese Kristallisatinsbedingung war der hexagonale Querschnitt.
Ergebnisse zur Sulfhydrylase CysM
4.1.3
Enzymatische Aktivität von CysM
Neben der strukturellen Charakterisierung der Sulfhydrylase wurden Aktivitätsmutanten prä­
pariert, deren Einfluss auf die Aktivität getestet werden sollte. Im Rahmen einer Diplomarbeit wur­
de hierzu ein einfacher assay entwickelt (Ulrich Wiesand, 2007). Eine genaue Beschreibung der
Durchführung findet sich in der angegebenen Literatur und ist in Abschnitt 3.3.2 zusammengefasst.
Die präparierten Mutanten zeigten, wie bereits beschrieben, eine wesentlich geringere
Stabilität sowohl gegenüber dem CysM(WT) als auch gegenüber der Oberflächenmutante
CysM(K268A). Noch deutlicher war der Einfluss dieser Mutationen auf die beobachtete Enzym­
aktivität. Nur für die Mutante T68S konnte noch ein merklicher Umsatz festgestellt werden. Die
weiteren hergestellten Mutanten zeigten Restaktivitäten von 2% und weniger. Dieses Resultat war
nicht weiter verwunderlich, da die Muatanten explizit so gewählt wurden, dass ein möglichst großer
Effekt auf die Enzymaktivität erzielt wurde (Abschnitt 4.1.1). Eine detailierter Diskussion dieser
Mutagenese-Studien wird in Abschnitt 4.1.8 bei der Betrachtung des aktiven Zentrums der
Sulfhydrylase gegeben.
Tabelle 10:
Enzymatische Aktivität der CysM-Mutanten bei 37 °C unter Verwendung von TNB als Substrata)
Enzym
kcat [s-1]
Wildtyp
24
0.7
T68Sb)
11 b)
0.6 b)
55
R210A
-
-
2
Q140A
-
-
0.4
T68A
-
-
0.1
Q140E
-
-
< 0.1
a)
b)
4.1.4
KM [mM]
kcat / KM [%]
100
-1 -1
Der kcat / KM -Wert wurde für den Wildtyp zu 34000 M s bestimmt.
Zur Stabilisierung von CysM(T68S) wurden diese Daten unter Verwendung von 300 mM HEPES
pH 7.0 bestimmt.
Kristallographische Datensammlung von CysM(K268A)-Kristallen
Die Messung am Synchrotron erfolgte bei 100 K. Daher wurden die Kristalle zur Vermeidung
von Eisbildung zuvor mit einem Frostschutz versehen, der durch Tränken des Kristalls in
Kristallisationspuffer mit 28% (v/v) Glycerin erreicht wurde. Die Konzentration des Glasbildners
wurde dabei in vier Stufen um jeweils 7% (v/v) bis zur Endkonzentration erhöht, bevor der Kristall
im Gasstrom bei 100 K schockgefroren wurde.
85
Ergebnisse und Diskussion
Tabelle 11:
Parameter und Statistik der gemessenen Datensätze der O-Acetylserin-Sulfhydrylase CysM(K268A)
Wellenlänge λ [Å]
0.90500
Detektor
marCCD
Detektor Abstand [mm]
Drei Messungen: 148, 217, 297
Auflösung
63-1.33 (1.37-1.33)
Raumgruppe
P6522
Zellparameter[Å]
A = 76.6, b = 76.6, c = 209.8
Anzahl der Reflexe
gemessen
1325909 (49092)
unabhängig
83156 (6220)
Rsym-I [%]
5.9 (35.0)
Vollständigkeit [%]
98.6 (87.9)
Multiplizität
7.4 (7.9)
Mosaizität [°]
0.150
<I>/<σ(I)>
21.4 (3.5)
3
Packungsparameter [Å /Da]
2.72
Solvensgehalt [%]
54.8
Protomer / ASU
1
Die Werte in den Klammern beziehen sich auf die letzte Auflösungsschale.
Die Datensammlung erfolgte an der beamline PXII der Swiss Light Source (Villigen,
Schweiz). Vor der Aufnahme eines kompletten Datensatzes wurden Testbilder in verschiedenen
Regionen des Proteinkristalls und unter verschiedenen Winkelbereichen aufgenommen, um die
optimalen Parameter für die Messung zu ermitteln. Anhand dieser Bilder wurde die Existenz von
mindestens einer langen Zellachse ersichtlich, die aufgrund des hervorragenden Streuvermögens
des Kristalls eine sorgfältige Aufnahmestrategie erforderte. Um die maximale Auflösung des
Kristalls nicht zu beschneiden und dennoch eine gute Separation der Reflexe zu erhalten, wurden
drei Datensätze mit unterschiedlichen Abständen zum Detektor aufgenommen. Für den
hochaufgelösten Datensatz wurde der Detektorabstand auf 148 mm eingestellt, während für die
low-resolution-Reflexe der Abstand des Detektors auf 217 mm und abschließend auf 297 mm mit
entprechenden off-set entlang der y-Achse eingestellt wurde. Während der Messung streute der
Kristall isotrop und zeigte erst während der Datensammlung der letzten Bilder eine rapide Abnahme
der Streukraft, obwohl nach jedem Wechsel des Detektorabstandes der Kristall im beam translatiert
wurde.
Die Prozessierung erfolgte mit XDS/XSCALE (Kabsch, 1998) wie in APRV (Kroemer et al.,
2004) implementiert (Abschnitt 3.5.7). Die Parameter und Statistiken der Datensammlung sind in
Tabelle 11 zusammengefasst. Der Kristall mit Raumgruppe P6522 aus Abbildung 29 zeigte eine
86
Ergebnisse zur Sulfhydrylase CysM
Auflösungsgrenze von 1.33 Å mit einem einzelnen Protomer in der asymmetrischen Untereinheit.
Der Packungsparameter liegt mit 2.72 Å3/Da nahe am statistischen Mittel von 2.69 Å3/Da
(Kantardjieff & Rupp, 2003). Diese Kristallform erfüllt somit hinsichtlich der geforderten Ziel­
setzung die Ansprüche sowohl in Bezug auf die Streukraft als auch in Bezug auf die Anzahl der
Protomere, die die asymmetrische Untereinheit belegen.
4.1.5
Strukturlösung von CysM(K268A)
Phasierung durch molekularen Ersatz
Zur Phasierung wurde die Methode des molekularen Ersatzes herangezogen. Als Suchmodell
diente das Protomer der Kette A der Struktur CysM(WT), das aus dem Modell mit dem PDBEintrag 2BHS erhalten wurde. Die Rotations- und Translationssuche wurde mit dem Programm
PHASER (McCoy et al., 2005) durchgeführt. Die initial erhaltene Lösung besaß einen Z-Score von
16.2 und einen log-likelihood von 3300. Diese Werte zeigen eine eindeutige Lösung.
Modellbau und Verfeinerung
Obwohl die Methode des molekularen Ersatzes bei Vorhandensein homologer Strukturen eine
schnelle und einfache Methodik zur Phasierung von Datensätzen darstellt, beinhaltet sie jedoch den
Nachteil, dass die resultierenden Phasen lediglich Annäherungen an die realen Phasen darstellen
und durch das verwendete Suchmodell fehlerbehaftet sind. Dieses Phänomen wird in der Kristallo­
graphie als model bias bezeichnet. Um diese Fehlerbehaftung bei weiterer Verfeinerung auszu­
schließen, wurde das Proteinmodell mit dem Programm ARP/wARP (Perrakis et al., 1999) durch
automatisierte Neupositionierung der Proteinatome neu aufgebaut. Die im aktiven Zentrum
vorhandene, klar definierte positive Differenzelektronendichte wurde zweifelsfrei einem Citrat­
molekül zugeordnet, das aus dem Kristallisationspuffer aufgenommen wurde. Das Citrat wurde von
Hand in die Elektronendichte eingepasst und anschließend eine simulated annealing Verfeinerung
mit CNS (Brünger et al., 1998) durchgeführt. Die hierfür erforderlichen Topologie-Parameter
wurden mit dem Programm PRODRG (Schuettelkopf & van Aalten, 2004) generiert. Eine zweite
Konformation des Citratmoleküls innerhalb des aktiven Zentrums wurde ersichtlich, die ebenfalls
manuell gesetzt wurde. Die beiden Konformationen wurden im folgenden mit unterschiedlicher
Besetzung von 60:40 verfeinert. Der weitere Verlauf der Verfeinerung bestand aus mehreren
Zyklen, in denen das Modell von Hand an die jeweils erzeugte Elektronendichte angepasst und
mittels REFMAC (Murshudov et al., 1997) erneut verfeinert wurde. Wassermoleküle wurde mit
87
Ergebnisse und Diskussion
ARP_WATERS (Lamzin & Wilson, 1993) eingefügt und manuell mit COOT (Emsley & Cowtan,
2004) überprüft. Dies resultierte in einem Modell, das bei isotroper Verfeinerung der B-Faktoren
einen R-Faktor von 17.0% und einen freien R-Faktor von 18.6% aufwies.
Tabelle 12:
Statisktik und Daten des verfeinerten Modells der CysM(K268A)
Auflösung [Å]
25-1.33
R-Faktor
0.158
Rfree (test set of 1.5%)
0.172
Anzahl der Nicht-H-Atome
2647
davon
2290
Protein
Glycerin
12
Citrat
16
Wassermoleküle
329
2
mittlerer Temperatur-Factor [Å ]
Protein-Hauptkette
16.5
Protein-Seitenkette
19.4
Glycerin
23.0
Citrat
16.8
Wassermoleküle
32.9
rms-Abweichungen
Bindungslängen [Å]
0.016
Bindungswinkel [°]
1.677
Ramachandran-Winkel
in bevorzugten Bereichen [%]
98.0
in erlaubten Bereichen [%]
2.0
Aufgrund der erreichten hohen Auflösung wurde versucht, die B-Faktoren des Proteinmodells
abschließend anisotrop zu verfeinern. Eine derartige Verfeinerung gegen den gemessenen Datensatz
ist nur für eine hohe Anzahl observierter Reflexe möglich, da bei Anwendung anisotroper B-FaktorVerfeinerung die Zahl der pro Atom zu verfeinernden Parameter um sechs zunimmt. Als Richtwert
kann ein Observablen-zu-Parameter-Verhältnis von 2.5 als Mindestanforderung an anisotrope
Verfeinerung genannt werden. Obwohl diese Anforderung für die erhaltene CysM-Struktur bei
weitem erfüllt war, zeigte sich eine derartige Verfeinerung weder mit SHELXL (Sheldrick, 1997)
noch mit REFMAC stabil. Daher wurde statt der vollständigen anisotropen Beschreibung des
Proteinmodells ein TLS-refinement durchgeführt, wobei die Parametrisierung der 12 TLS-Gruppen
mit dem Programm TLSMD (Painter & Merritt, 2006) durchgeführt wurde, das eine Analyse der
Kristallstruktur auf flexible Bereiche hin unternimmt. Die derartige Verfeinerung verbesserte die
R-Faktoren von 17.0% respektive 18.6 % für den freien R-Faktor auf 15.8% respektive 17.2 % für
den freien R-Faktor.
88
Ergebnisse zur Sulfhydrylase CysM
4.1.6
Validierung der CysM(K268A)-Struktur
Das finale Modell konnte bis zu einem R-Faktor von 15.8% verfeinert werden. Der freie
R- Faktor betrug 17.2%. Das Protomer, das mit den Lösungsmittelmolekülen den Inhalt der
asymmetrischen Einheit ausmacht, bestand aus den Aminosäureresten 1-294. Die C-terminalen
Reste 295-303 der Proteinkette waren ungeordnet und daher in der Elektronendichte nicht zu
erkennen. Das Proteinmodell zeigte zudem klar definierte Elektronendichte für den Cofaktor PLP,
das über den Rest Lys41 kovalent an das Enzym gebunden war und für ein Citrat-Molekül, das in
zwei Konformationen im aktiven Zentrum des Enzyms eingelagert wurde.
Abbildung 30: Ramachandran-Diagramme der Struktur von CysM(K268A). Die Positionen aller Aminosäurereste
sind durch Kreuze (Glycinreste) und Quadrate (alle anderen Aminosäuren) gekennzeichnet. Es sind
separate Diagramme für generell bevorzugte Winkelkombinationen A), für Glycin typische Kombinationen B) sowie für charakteristische Winkelkombinationen von Aminosäuren, die direkt vor Prolinen
gelegenen sind C) und für Proline D) dargestellt. Die Farbgebung des Hintergrundes markiert die
energetisch unterschiedlich klassifizierten Bereiche, dunkle Regionen sind am meisten bevorzugt
(Lovell et al., 2003).
89
Ergebnisse und Diskussion
Die Qualität des Proteinmodells wurde während der Verfeinerung stetig anhand der Güte der
R-Faktoren sowie der Abweichung von geometrischen Parametern (Abschnitt 3.5.15) überprüft. In
Tabelle 12 sind die Verfeinerungsstatistiken der finalen Struktur aufgeführt, die die gute Qualität
des Proteinmodells belegen. Der Vergleich mit den Standardbindungslängen und -winkeln (Engh
und Huber, 1991) zeigte keine nenneswerten Abweichungen. Die Geometrie des Proteinmodells
wurde mit den Programmen RAMPAGE (Lovell et al., 2003) und WHATCHECK (Hooft et al.,
1996) durchgeführt, wobei alle Ergebnisse dieser Analysen innerhalb gängiger Toleranzgrenzen
liegen. Fast alle dihedralen Hauptkettentorsionswinkel (98%) befinden sich in energetisch günstigen
Bereichen des Ramachandran-Diagramms, was in Abbildung 30 veranschaulicht ist. Neben den
Hauptkettentorsionswinkeln wurden auch die Konformationen und die daraus resultierenden
Torsionswinkel als Qualitätskriterium herangezogen und überprüft. Diese Vorgehensweise ist hilf­
reich bei der Validierung der Proteinstruktur, da die Verfeinerung der Seitenkettenwinkel nicht in
den Algorithmen der Verfeinerungsprogramme enthalten ist (Abschnitt 3.6.1). Verglichen mit Bib­
liotheken von Standard-Rotameren aller Aminosäuren (Laskowski et al., 1993) liegen auch hier alle
beobachteten Winkelkombinationen innerhalb gängiger Toleranzgrenzen.
Zur Analyse der Temperatur-Faktoren wurden die residual-B-Faktoren, die aus der TLS-Ver­
feinerung mit dem Programm TLSANL (Howlin et al., 1993) erhalten wurden, zu isotropen
B-Faktoren zusammengeführt und anschließend mit BAVERAGE (CCP4, 1994) gemittelt. Abbil­
dung 31 gibt den Verlauf der Temperatur-Faktoren entlang der Proteinkette wieder. Hohe Beweg­
lichkeit innerhalb der Proteinkette findet sich in der Subdomäne (Aminosäuren 86-133), die die
Reste 112-133 umfasst, was bereits anhand der beiden anderen bekannten Kristallformen dokumen­
tiert wurde (Claus et al., 2005). Allerdings fällt der Unterschied bei einem mittleren B-Faktor von
16.5 Å2 gegenüber den maximal 37 Å2 in diesem Bereich nicht so deutlich aus wie für die anderen
Kristallformen beschrieben. Die höchsten Temperatur-Faktoren finden sich allerdings im Bereich
der Aminosäuren 210-220. Dieser loop fungiert wie im folgenden gezeigt als Deckel über der
Bindetasche des Enzyms und wurde für die Substratspezifität von CysM verantwortlich gemacht,
insbesondere das darin enthaltene Arg-Arg-Trp-Motiv. Die B-Faktoren dieses Segments steigen
dabei auf bis zu 51 Å2 an, was die strukturelle Flexibilität des loops belegt.
Die Dichtekorrelation aufgetragen gegen die Aminosäurereste der Proteinkette zeigt die Güte
der Übereinstimmung der Elektronendichte mit dem Proteinmodell auf. Diese wurde mit dem
Programm OVERLAPMAP (CCP4, 1994) berechnet und ist ebenfalls in Abbildung 31 gezeigt.
Diese Dichtekorrelation ist ein weiterer Hinweis auf die insgesamt gute Qualität des Proteinmodell.
Leichte Abweichungen finden sich nur in den Bereichen, die auch durch hohe Temperatur-Faktoren
auffallen.
90
Ergebnisse zur Sulfhydrylase CysM
0
20
40
60
80
100
120
140
160
180
200
220
240
260
280
70
0.95
50
B-Faktor
D ichtekorrelation
0.90
40
0.85
Dichtekorrelation
2
B-Faktor [Å ]
60
1.00
30
0.80
20
10
0.75
0
20
40
60
80
100
120
140
160
180
200
220
240
260
280
Am inosäurerest
Abbildung 31: Temperatur-Faktoren (unten) und Dichtekorrelation (oben) der Proteinkette von CysM(K268A).
V ergleich C ysM (RK E) K ette A m it C ysM (K 286A )
V ergleich C ysM (W T) K ette A m it C ysM (K 268A )
3.0
∆ Cα
[Å]
2.5
2.0
1.5
1.0
0.5
0.0
0
20
40
60
80
100 120 140 160 180 200 220 240 260 280
Aminosäurerest
Abbildung 32: Abstände zwischen den Cα-Atomen von CysM(K268A) mit CysM(WT) und CysM(RKE).
91
Ergebnisse und Diskussion
Unter Verwendung von LSQMAN (Kleywegt, 1996) wurden die Protomere der Kristallform
CysM(WT) (Kette A, offene Konformation) und der Kristallform CysM(RKE) (Kette A,
geschlossene Konformation) überlagert und die Abweichungen zur Struktur CysM(K268A)
analysiert. Die Überlagerung lieferte im Mittel geringe Abweichungen von 0.51 Å für den
Vergleich mit CysM(WT), respektive 0.88 Å für CysM(RKE). Daher kann festgestellt werden, dass
das Modell für CysM(K268A) eher der geöffneten Konformation der Sulfhydrylase ähnelt. Ein
entsprechender Vergleich der Cα-Abweichungen findet sich in Abbildung 32. Dennoch finden sich
auch hier größerer Abweichungen im Bereich der Subdomäne an den Aminosäuren 86-100, 108121 und 127-132. Dagegen entspricht der für die Substratspezifität verantwortliche loop 208-213
weitestgehend der Konformation, die auch in der Struktur CysM(WT) gefunden wurde. Zudem
kann noch eine signifikante Änderung im Substrat-bindenden loop beobachtet werden, der die Reste
68 bis 72 umfasst. Dieser nimmt einen Konformation ein, die eine Interpretation als Zwischen­
zustand der Strukturen CysM(WT) und CysM(K268A) nahe legt.
4.1.7
Struktur von CysM(K268A)
Sekundär- und Tertiärstruktur
Die Sekundärstruktur von CysM(K268A) unterscheidet sich kaum von der Anordnung der
Sekundärstrukturelemente wie sie für CysM(WT) beschrieben wurde (Claus, 2004). Ein Vergleich
mit den bekannten Strukturen CysM(WT) und CysM(RKE) ist in Abbildung 33 gegeben. Kleine
Unterschiede finden sich an den Helices α1, α5, α6, α7 und 3104. Diese Unterschiede sind aber
vernachlässigbar und wohl am ehesten auf die höhere Auflösung des Datensatzes und der daraus
resultierenden höheren Genauigkeit der Struktur zurückzuführen.
Die Kettenfaltung von CysM(K268A) entspricht ebenso den bereits bekannten Strukturen
CysM(WT) und CysM(RKE) und kann detailliert in der angegebenen Literatur nachgelesen werden
(Claus et al., 2005). Abbildung 34 zeigt die Gesamtfaltung für die Struktur CysM(K268A). Zum
besseren Vergleich wurde für diese Abbildung die identische Orientierung mit Blickrichtung ent­
lang der zweizähligen Achse des Dimers gewählt, wie sie in oben genannter Literatur beschrieben
wird. Im aktiven Zentrum des Enzyms findet sich zusätzlich ein Citratmolekül, das die Bindetasche
der Sulfhydrylase nahezu vollständig ausfüllt und aus dem Kristallisationspuffer aufgenommen
wurde. Wie bereits beschrieben, besteht die Sulfhydrylase aus einer C-terminalen und einer Nterminalen Domäne, die wiederum durch eine bewegliche Subdomäne aufgeteilt wird (Burkhard et
92
Ergebnisse zur Sulfhydrylase CysM
al., 1998; 1999; 2000). Diese Domäne zeigt während der Katalyse der Sulfhydrylase hohe
Beweglichkeit (siehe Abbildung 31), wobei die Bewegung gegen die C-terminale Domäne das
Öffnen und Schließen des aktiven Zentrums im Verlauf der enzymatischen Reaktion bewirkt.
.
.
.
.
50
.
MSTLEQTIGNTPLVKLQRMGPDNGSEVWLKLEGNNPAGSVKDRAALSMIVEAEKRGRIKPGDVLIEATS
ss
3101
β1
β2 3102
α1
β3
Sequenz
CysM(K268A)
CysM(WT)
CysM(RKE)
.
.
.
100
.
.
.
.
GNTGIALAMIAALKGYRMKLLMPDNMSQERRAAMRAYGAELILVTKEQGMEGARDLALEMANRGEGKLLDQFNNP
s s
-------- ---
α2
β4
α3
β5
α4
β6
Sequenz
CysM(K268A)
CysM(WT)
CysM(RKE)
150
.
.
.
.
200
.
DNPKAHYTTTGPEIWQQTGGRITHFVSSMGTTGTITGVSEFMREQSKPVTIVGLQPEEGSSIPGIRRWPTEYLPG
ssss
α5
α6
β7
α7
β8
Sequenz
CysM(K268A)
CysM(WT)
CysM(RKE)
.
.
.
250
.
.
.
.
IFNASLVDEVLDIHQRDAENTMRELAVREGIFCGVSSGGAVAGALRVAKANPDAVVVAIICDRGDRYLSTGVFGE
a
3103
β9
α8
α9
β10
3104
Sequenz
CysM(K268A)
CysM(WT)
CysM(RKE)
Abbildung 33: Sekundärstruktur der Sulfhydrylase-Strukturen CysM(WT), CysM(RKE) und CysM(K268A) im Vergleich. Die zweite Zeile gibt die Sequenz der Sulfhydrylase wieder. In der dritten Zeile sind die SekundärstukturElemente von CysM(K268A), in der vierten Zeile von CysM(WT) und schließlich in der fünften Zeile von CysM(RKE)
gezeigt. Helices sind als Zylinder und β-Stränge als Pfeile symbo- lisiert. Weiterhin sind die Bezeichnungen der Sekun­
därstrukturelemente angegeben (sechste Zeile). Die Einfärbung erfolgte nach Aufteilung der Domänen. Die C-terminale
Domäne ist schwarz, die N-terminale Domäne grau dargestellt, wobei die bewegliche Subdomäne hellgrau gezeigt ist.
Abbildung 34: Stereodarstellung des CysM(K268A)-Dimers. Die zweizählige Achse ist in schwarz gezeigt. Die beiden Protomere sind rot und blau eingefärbt und die N-terminalen Subdomänen sind in grün bzw.
orange dargestellt. Dabei sind die zentralen β-Faltblattstränge dunkler dargestellt, während die
flankierenden Helices hell hervorgehoben wurden. Der Cofaktor PLP und das Citratmolekül, die die
aktive Tasche des Enzyms belegen, sind in der ball-and-stick-Darstellung gezeichnet. Die Oberflächenmutation K268A, die zur besseren Kristallisation eingeführt wurde, ist durch eine gelbe Kugel
markiert.
93
Ergebnisse und Diskussion
Abbildung 35: Überlagerung der Protomere von CysM(K268A) (rot), CysM(WT) (blau) und CysM(RKE) (grün)
sowie der einzigen in der PDB-Datenbank aufgeführten Sulfhydrylase der Isoform B (schwarz) (PDBEintrag 2JC3). Die größten Unterschiede finden sich im Substrat-bindenden loop und der beweglichen Subdomäne.
Eine Überlagerung der drei Strukturen CysM(K268A), CysM(WT) und CysM(RKE) ist in
Abbildung 35 dargestellt und zeigt den Unterschied dieser Strukturen bezogen auf die Kettenfaltung
nochmals sehr deutlich. Dabei kann die C-terminale Domäne der Sulfhydrylasen sowohl mit
CysM(WT) als auch CysM(RKE) sehr gut überlagert werden, wobei der loop im Segment 208-213
innerhalb der Struktur von CysM(K268A) die Konformation der Wildtyp-Kristallform einnimmt.
Diese Konformation wurde als wesentlicher Unterschied der C-teminalen Domänen der Strukturen
CysM(WT) und CysM(K268A) mit CysM(RKE) identifiziert. Demgegenüber zeigen sich erheb­
liche Abweichungen im Vergleich der N-terminalen Domänen untereinander. Die neue Kristallform
zeigt hier vor allem im Bereich der beweglichen Subdomäne größere Abweichungen. Diese Subdo­
mäne nimmt in dieser Struktur eine Konformation ein, die einem Zustand zwischen der offenen und
geschlossenen Konformation des Enzyms gleichkommt. Die größte Distanz, die bei der Bewegung
der N-terminalen Domäne hin zur C-terminalen Domäne gemessen werden konnte, befindet sich am
Rest Glu116 mit einem Abstand von 5 Å zwischen offenem und geschlossenem aktiven Zentrum.
Demgegenüber nimmt der Rest 116 der CysM(K268A)-Struktur mit einem Abstand von 2.5 Å zu
beiden Extremen nahezu eine perfekte Mittelstellung ein. Aus diesem Bild lässt sich klar ein Über­
gangszustand des Enzyms zwischen offener und geschlossener Konformation folgern. In Anbe­
tracht der Tatsache, dass sich zudem ein Substrat-ähnliches Molekül in der Bindetasche des Enzyms
findet, kann diese Kristallform zur Beschreibung der strukturellen Änderungen während eines
Katalysezykluses dienen. Insbesondere die Änderungen innerhalb des aktiven Zentrums sind hierbei
von Interesse und werden in Abschnitt 4.1.8 diskutiert.
94
Ergebnisse zur Sulfhydrylase CysM
Quartärstruktur und Kristallpackung
Obwohl die asymmetrische Einheit der CysM(K268A)-Struktur nur auf das Protomer
beschränkt ist, findet sich auch in dieser Kristallform die Dimer-Kontaktfläche. Anders als bei den
bisherigen Kristallformen von CysM(WT) und CysM(RKE) liegt die C2-Achse des funktionalen
Dimers in der Struktur CysM(K268A) exakt auf einer kristallographischen, zweizähligen Achse.
Die Analyse der Kristallkontakte erfolgte mit PISA (Krissinel & Henrick, 2005). Das Dimer­
interface (Kontaktfläche I) weist mit einer Kontaktfläche von 1517 Å2 nahezu die gleiche Größe
auf, wie sie in den anderen Strukturen beschrieben wurde. Es treten 20 Wasserstoff-, zwei
Salzbrücken und 34 hydrophobe Wechselwirkungen auf, die die Monomere zusammenhalten. Eine
Liste der beteiligten Aminosäuren findet sich in Tabelle 13 und unterscheidet sich nur marginal von
der bisher beschriebenen Kontaktfläche. Der größte Unterschied besteht in einem etwas effizienter
in das andere Protomer verzahnten N-Terminus. Die hexagonale Raumgruppe P6522 enthält zwölf
asymmetrische Einheiten in der Elementarzelle und dementsprechend zwölf Protomere. Alle
auftretenden Symmetrien sind ausschließlich kristallographischer Natur. Abbildung 36 gibt eine
schematische Darstellung der Symmetrieelemente, die sich in der genannten Raumgruppe finden
lassen. Die Abbildung 37 zeigt den Inhalt einer Elementarzelle, der durch Anwendung der
Symmetrie-Operationen auf CysM(K268A) entsteht. Die Packung des Kristalls kann dabei als
Doppel-Helix zweier um eine zweizählige Achse versetzter Protomere beschrieben werden, wobei
die Kontaktfäche III die Protomere entlang der z-Achse verknüpft und Kontaktfläche II die
versetzten Protomere innerhalb der Doppelhelix zusammenhält. Das Dimerinterface wird über eine
weitere zweizählige Achse erzeugt. Daneben sind nur noch unwesentliche Kristallkontakte geringer
Fläche vorhanden. Alle Kristallkontakte, die das Protomer innerhalb der Kristallpackung aufbaut,
sind in Tabelle 13 aufgelistet.
Abbildung 36: Die Raumgruppe P6522 in der schematischen Darstellung mit Blickrichtung entlang der z-Achse
(International Tables of Crystallography 1996).
95
Ergebnisse und Diskussion
Tabelle 13:
Kristallkontakte von CysM(K268A).
Name Sym-Op;
Oberfläche Interagierende Aminosäurereste
Translation
[ Å2]
I
-y+1,-x+1,-z+1/6 a)
1517
1-7, 12-14, 17,18, 31, 33-38, 60, 78,
81-84, 98, 101, 102, 104-107, 161,
242, 245-251
b)
II
x-y,-y,-z;
-1,-1,0
624
53-57, 59, 131, 136, 144, 145, 147,
148, 151, 184, 188, 214, 215, 217, 222
b)
III
-x+1,y-1/2,-z+1/2;
-1,-1,-2
608
86, 88, 97, 100, 101, 104, 109-112,
114, 117, 125, 129, 132, 134
IV
-x+1,-x+y,-z+1/3;
0,-1,0
121
165, 167, 191, 272, 274
VI
-y,-x,-z+1/6;
-1,-1,0
27
214, 215
a)
b)
Kette 1
Kette 2
21, 23, 167, 168, 187, 194,
199, 201, 214, 223, 226-231,
265, 267, 268-273
290, 293, 294
b)
Dieser Kontakt entspricht dem Interface des funktionalen Dimers.
Aufgrund der zweizähligen Symmetrieachse sind jeweils die gleichen AS am Kontakt beteiligt.
Abbildung 37: Kristallpackung von CysM(K268A). Gezeigt ist der Inhalt einer Elementarzelle. Die Kolorierung erfolgte nach angewandter Symmetrieoperation. A) Blickrichtung entlang der z-Achse, B) um 90°
gedreht. In schwarz sind exemplarisch zwei zweizählige Achsen eingezeichnet.
96
Ergebnisse zur Sulfhydrylase CysM
Die verbesserten Kristallisationseigenschaften von CysM(K268A) sind das Resultat der Ein­
führung der Oberflächenmutation und der Einlagerung des Citrats in das aktive Zentrum. Der Rest
K268A trägt mit 58 Å2 zu fast 10% der Gesamtfläche dieses Kontaktes bei. Zusätzlich scheint auch
das Substrat-ähnliche Citrat Einfluss auf die Streukraft der resultierenden Kristalle zu haben. Es
konnten auch für den Wildtyp Kristalle erhalten werden, die ein verbessertes Streuvermögen
aufwiesen. Allerdings zeigte sich die Reproduktion dieser Kristalle als wesentlich aufwendiger.
4.1.8
Das aktive Zentrum von CysM(K268A)
Der Cofaktor PLP zeigt in der Struktur CysM(K268A) die gleiche Konformation und die
selben Wechselwirkungen, wie sie bereits von den Strukturen CysM(WT) und CysM(RKE) bekannt
sind. In Bezug auf die oben genannten Strukturen treten lediglich marginale Änderungen der
Abstände zwischen den koordinierenden Aminosäuren und PLP auf, die aufgrund der im
allgemeinen etwas kürzeren Bindungsabstände auf eine etwas festere Bindung des Cofaktors
schließen lassen. Dieser Umstand kann als weiteres Indiz für das Vorliegen einer Enzymkonfor­
mation angesehen werden, die während der enzymatischen Reaktion eingenommen wird. Da im
Verlauf der von CysM katalysierten Reaktion die kovalente Bindung des Coenzyms PLP
aufgegeben wird und ein Aminoacrylat gebildet wird, scheint eine festere Anbindung des Cofaktors
über die verbleibenden Wasserstoffbrücken schlüssig, um den Verlust des externen Aldimins aus
der Bindetasche des Enzyms zu verhindern. Tabelle 14 fasst die Abstände und Wechselwirkungen
des Cofaktors zur Proteinkette zusammen und vergleicht diese mit den veröffentlichten Bindungs­
längen (Claus, 2004). Für eine detaillierte Beschreibung der Bindung des Kofaktors wird auf die
genannte Literatur verwiesen.
Tabelle 14:
Wasserstoffbrücken des PLP in CysM(K268A)
Abstand [Å]
CysM(WT) Abstand [Å]a)
PLP
Protein
O3
Asn71-Nδ2
2.7
3.0
N1
Ser255-Oγ
3.1
2.7
OP1
Thr175-N
2.8
2.8
OP1
Thr175-Oγ1
2.4
2.6
O2P
Gly174-N
2.8
3.1
O2P
Thr176-N
2.9
3.0
O3P
Thr178-N
2.8
3.1
O3P
Thr178-Oγ1
2.7
2.6
Zum Vergleich sind die Abstände der CysM(WT)-Struktur aufgeführt (Claus, 2004).
a)
97
Ergebnisse und Diskussion
Abbildung 38:
Nummerierung der Atome im Citratmolekül.
Wie bereits beschrieben, fand sich im aktiven Zentrum ein Citratmolekül, das aus dem Kristal­
lisationspuffer im aktiven Zentrum des Enzyms gebunden wurde. Dieses konnte zweifelsfrei durch
die resultierende positive Differenzelektronendichte zugeordnet werden. Das Citrat bedeckt eine
Proteinoberfläche von 315 Å2 und wird über elf Wasserstoffbrücken fixiert (Tabelle 15). Dabei liegt
das Citrat im Reaktionszentrum der Sulfhydrylase in zwei unterschiedlichen Konformationen vor,
die sich durch die Konfiguration der Carboxygruppe am C4-Atom (Abbildung 38)unterscheidet.
Tabelle 15:
Wasserstoffbrücken des Citrats im CysM(K268) für die 40% besetzte Konformation
Citrat
Protein
Abstand [ Å]
O2'
LLP41-Nζ a)
2.9
O1'
Asn70-N
3.0
O1'
Asn71-N
3.0
O1'
Thr72-N
2.9
O1''
Thr68-Oγ1
2.7
O1''
Ser69-N
3.0
O1''
Gln140-Nε2
2.9
O4''
Arg210-Nε
2.3
O4'
Arg210-Nφ2
2.3
O6'
Arg210-Nφ2
3.4
O6'
Ser69-Oγ
2.6
Als LLP wird das kovalente Addukt aus PLP und Lysin bezeichnet, das über das Nζ-Atom des
Lys41 gebunden ist.
a)
In der Konformation, die mit einer Besetzung von 0.4 verfeinert wurde, wird die Carboxyfunktion über Wasserstoffbrücken zu beiden Stickstoffatomen der Seitenkette des Arg210 stabilisiert, während in der Konformation, die mit einer Besetzung von 0.6 verfeinert wurde, lediglich die
Ausbildung einer sehr schwachen Wechselwirkung zum Peptid-Stickstoff des Ile209 resultiert.
Zudem zeigt sich in dieser Konformation ein außergewöhnlich kurzer Abstand von 2.2 Å zum Car­
bonyl-Sauerstoff des Restes Gly208, der aufgrund des abstoßenden Charakters zweier negativer
98
Ergebnisse zur Sulfhydrylase CysM
Partialladungen deutlich größer ausfallen sollte. Allerdings könnte das Peptidrückgrat in diesem
Segment auch in einer weiteren Konformation verfeinert werden, die einen Abstand von 2.4 Å
ermöglicht.
Die erste Carboxyfunktion wird über die Reste 68-72 fixiert. Auffällig dabei ist die Anzahl der
Wechselwirkungen zwischen dem substratbindenden loop (Reste 68 bis 72) und dieser Carboxy­
funktion, die auf eine sehr gute Fixierung des Substrats schließen lässt. In der Tat zeigt die Elek­
tronendichte in diesem Bereich exakt definierte Grenzen. Eine Wasserstoffbrücke, die in diesem
Segment über eine Seitenkette (Thr68) vermittelt wird, zeigt bei Mutation zu Alanin einen
vollständigen Verlust der Aktivität, während ein Ersatz dieser Aminosäure durch Serin die Aktivität
lediglich halbiert (Tabelle 10). Es wurde bereits anhand der Kristallstruktur anderer Sulfhydrylasen
im Komplex mit Substrat-ähnlichen Molekülen gezeigt, dass dieser loop für die Fixierung der
Carboxyfunktion im O-Acetylserin verantwortlich ist (Burkhard et al., 2000). Dieser Substratbindende loop zeigt auch zu beiden bekannten Strukturen CysM(WT) und CysM(RKE) die größten
Abweichungen (Abbildung 39). Ebenso wie bei der Betrachtung der Kettenfaltung zeigt die
CysM(K268A)-Struktur auch in diesem Bereich eine Konformation, die einen Zwischenzustand der
beiden anderen Strukturen entspricht.
Die Mutante Q140E und Q140A zeigen einen T68A entsprechenden Effekt. Die Amidfunktion
des Gln140 bildet eine Wasserstoffbrücke zur Carboxygruppe des Citrats aus. Dieser stabilisierende
Einfluss geht durch Mutation zur negativ geladenen Carboxyfuktion eines Glutamats bzw. zur
hydrophoben Methylgruppe eines Alanins verloren, da diese entweder in einer Abstoßung gleicher
Ladungen resultiert (Q140E) oder in einer Aminosäure, die nicht in der Lage ist, eine Wasserstoff­
brücke auszubilden (Q140A). Dies spiegelt sich in einer kaum messbaren Aktivität der Mutanten
Q140A und Q140E von 0.4%, respektive 0.1% wider.
Die dritte Carboxyfunktion des Citrats ist aus dem aktiven Zentrum heraus gerichtet und geht
Wechselwirkungen zur Hydroxyfunktion des Ser69, sowie zur Guanidiniumgruppe des Arg210 ein.
Die Hydroxygruppe des Citrats zeigt Wechselwirkungen zur Hydroxy-Funktion des Pyridoxal­
phosphats und zum Nζ-Atom des kovalent am PLP verknüpften Lysins und kommt bei einem
Abstand von 2.92 Å exakt in der Position zu liegen, die für einen Angriff des Substrats
O- Acetylserin erwartet wird. Abbildung 39 verdeutlicht diese Ergebnisse.
Zusammenfassend kann das Citrat damit als Substrat-ähnliches Molekül deklariert werden, das
anscheinend einen Zwischenzustand im Katalyse-Mechanismus stabilisiert. Unter Berücksichtigung
des enzymatischen Zyklus kommt für diesen Zwischenzustand am ehesten die Additionsreaktion
vom Hydrogensulfid an das Aminoacrylat zum iminischen Zwischenprodukt (Abbildung 3,
Reaktion IV zu V) in Frage, da die zwei zusätzlichen negativen Ladungen des Citrats, die im
99
Ergebnisse und Diskussion
natürlichen Substrat keine Entsprechung finden, als mögliche Kandidaten eines nukleophilen
Angriffs auf die Doppelbindung des kondensierten Zwischenproduktes dienen könnten. Im
folgenden wurde aus den Gegebenheiten der bekannten Strukturen sowie den Aktivitätsstudien ein
Modell für diese Teilreaktion aufgestellt.
Abbildung 39:
Stereodarstellung des aktiven Zentrums von CysM(K268A). Alle Reste, die die Bindetasche des Enzyms auskleiden, sind dargestellt (braun). Der kovalent an das Lys41 gebundene Cofaktor PLP
(orange) ist wie in den Strukturen CysM(WT und CysM(RKE) orientiert. Die mutierten Reste, deren
Aktivität bestimmt wurde, sind cyan hervorgehoben. Das Citrat (orange), das in zwei Konformationen vorliegt, füllt das aktive Zentrum aus und kommt so über dem Cofaktor zu liegen, wie es für
ein potentielles Substrat erwartet würde. Die Wasserstoffbrücken des Citratmoleküls sind durch rote
Linien gekennzeichnet. Der bewegliche loop der Reste 68-72 ist für die Strukturen CysM(WT) und
CysM(RKE) in grau gezeigt. In grün ist die (FO-FC)-Differenzelektronendichte bei einem Konturniveau von 3 σ gezeigt.
Ausgehend von CysM(K268A) und dem darin gebundenen Citrat wurde das Zwischenprodukt
Aminoacrylat modelliert. Dabei wurden im wesentlichen zwei Fixpunkte gewählt, an denen dieses
Molekül im Enzym justiert ist. Dies war zum einen die Phosphatgruppe des PLP, das mit dem loop
der Reste 174 bis 176 wechselwirkt, und zum anderen die Carboxygruppe des Citrats, die über den
substratbindenden loop koordiniert ist. Das Aminoacrylat wurde über den PRODRUG Server
erzeugt und anschließend so in das aktive Zentrum modelliert, dass Zusammenstöße mit Proteinsei­
tenketten vermieden wurden und Bindungsabstände zwischen potentiellen Wasserstoffbrückenbin­
dungen optimiert wurden. Dabei waren die möglichen Konformationen des Aminoacrylats aufgrund
der weitestgehend planaren Struktur beschränkt. Für das finale Modell wurden die Seitenketten von
Thr175, Asn271, sowie Arg210 leicht verdreht, wobei darauf geachtet wurde, das die erhaltenen
Konformationen nicht in energetisch ungünstigen Bereichen zu liegen kamen.
Das finale Modell ist in Abbildung 40 gezeigt. Das Aminoacrylat erfährt hierbei nahezu die
selben Wechselwirkungen an der Phosphatgruppe sowie an der Carboxygruppe, wie es für PLP (an
den Resten 174-176) und Citrat (an den Resten 68-72) der Fall war. Das Nukleophil konnte dabei so
100
Ergebnisse zur Sulfhydrylase CysM
positioniert werden, dass ein Angriff auf die Doppelbindung fast senkrecht aus dem Halbraum
erfolgen kann. Das Hydrogensulfid erfährt in diesem Modell eine stabilisierende Wasserstoff­
brücken zum Arg210-Nφ2 (2.6 Å), die das Nukleophil in diesem Zustand positioniert. Dieses Modell
liefert damit eine Erklärung für den nahezu vollständigen Verlust der Aktivität durch Einführung
der Mutante R210A. Die Protonierung der Doppelbindung erfolgt dementsprechend aus dem gegen­
überliegenden Halbraum durch das im Verlauf der Reaktion protonierten Lys41, so dass diese
Reaktion wie erwartet als stereochemisch reine Anti-Addition an die olefinische Doppelbindung des
Aminoacrylats angesehen werden kann. Der Substrat-bindende loop und insbesondere der Rest
Ser69 scheint dabei im Reaktionszyklus durch seine Bewegung sterischen Druck auf das
angreifende Nukleophil auszuüben, indem das Nukleophil in den Halbraum über der Doppelbin­
dung des Aminoacrylats gedrückt wird.
Abbildung 40:
Stereodarstellung der Geometrie des postulierten Modells der Reaktion vom Aminoacrylat mit dem
Nukleophil Hydrogensulfid aus dem gleichen Blickwinkel wie in Abbildung 39. Reste, die die Bindetasche des Enzyms umschließen, sind braun dargestellt. Das Aminoacrylat (grün) wurde in das
aktive Zentrum modelliert. Der nukleophile Angriff des Schwefels (orange Kugel) wird durch die
Aminosäure Arg210 stabilisiert. Zudem ist ein mögliches Modell für den nukleophilen Angriff des
im Enzymassay verwendeten TNB-Moleküls (ebenfalls grün) gezeigt.
Neben dem Schwefel-Nukleophil konnte ein Modell für den Angriff des im Enzymassay
verwendeten TNB-Moleküs aufgestellt werden. Dazu wurde das TNB manuell in die aktive Tasche
von CysM gedockt, so dass die Sulfidgruppe ebenfalls über dem anzugreifenden Kohlenstoff zu
liegen kam. Das TNB konnte dabei kollisionsfrei in das aktive Zentrum gebaut werden.
Wesentliche Wechselwirkung tritt dabei zum Arg210 auf, das sowohl Wasserstoffbrücken zur
Sulfidgruppe ausbildet, als auch die Carboxy-Funktion des TNB-Moleküls fixiert. Dieses Modell
könnte als Ausgangspunkt für weitere Anpassungen des Enzyms an große Substrate durch
Mutationen dienen, deren Einsatz die Herstellung weiterer nicht-natürlicher Aminosäuren ermög­
licht.
101
Ergebnisse und Diskussion
4.2
4.2.1
Ergebnisse zur N-Oxygenase AurF
Reinigung der N-Oxygenase
Wie bereits in Abschnitt 3.2.2 beschrieben, erfolgte die Produktion und weitestgehend auch
die Reinigung von AurF durch Robert Winkler am Hans-Knöll-Institut in Jena. Eine detaillierte
Beschreibung der Enzymisolierung ist in der Literatur gegeben (Winkler et al., 2006). Die
Proteinlösung wurde in Chargen zu etwa 15-20 mg Protein aus Jena geliefert. Das Protein enthielt
zusätzlich zur Aminosäuresequenz der N-Oxygenase einen N-terminalen linker von neun Amino­
säuren, der aus dem proteolytischen Verdau des Fusionsprotein durch Faktor-Xa resultierte. Die
Präsens dieses linkers wurde sowohl durch ESI-MS, als auch durch Sequenzierung der gereinigten
N-Oxygenase nachgewiesen (persönliche Mitteilung Dr. Schiltz, 2006).
100
61 kDa
Absorption bei 280 nm
Absorption [m AU]
90
80
70
60
50
M BP
40
30
0
Abbildung 41:
10
20
30
40
50
60
70
Elutionsvolum en [m L]
80
90
100
Exemplarisches GPC-Elutionsprofil der N-Oxygenase an Superdex 200-16/60. Die schwarze Linie
zeigt den Absorptionsverlauf bei 280 nm. Vereinigte AurF-haltige Fraktionen sind durch einen
Strich unterhalb des peaks mit einem Elutionsvolumen von 84.4 mL gekennzeichnet, was einer
Molekularmasse von etwa 60.8 kDa entspricht. Das in geringen Mengen vorhandene Maltosebindende Protein eluiert in Übereinstimmung des Molekularmasse von etwa 26.7 kDa bei einem Volumen von 93.7 mL und konnte Basislinien-getrennt vom Zielprotein separiert werden. Die Identifikation des MBP erfolgte anhand des Vergleichs der SDS-PAGE-Gele des vorhergehenden proteolytischen Verdaus.
Die gelieferte proteinhaltige Lösung enthielt geringe Mengen verunreinigendes MaltoseBinde-Protein (MBP) und zudem eine zweite AurF-Spezies (AurF2 in Abbildung 42), die sowohl
durch N-terminale als auch C-terminale Sequenzierung zweifelsfrei dem durch proteolytischen
102
Ergebnisse zur N-Oxygenase AurF
Verdau erzeugten Zielprotein zugeordnet werden konnte (persönliche Mitteilung Robert Winkler,
2006). Die N-Oxygenase (AurF in Abbildung 42), die als ausgeprägte Bande im SDS-Gel erscheint,
zeigt auf Basis der Sequenz des N- und C-Terminus keinen Unterschied zur oben beschriebenen
Spezies AurF2, so dass das differierende Laufverhalten der SDS-PAGE zwischen AurF und AurF2
unerklärt blieb. Es zeigte sich jedoch, dass die verunreinigende AurF-Spezies (AurF2) eine
wesentlich geringere Stabilität aufwies und insbesondere bei der Konzentration der Proteinlösung
präzipitierte, so dass dieser Spezies keine weitere Beachtung geschenkt wurde.
Die aus Jena erhaltene Proteinlösung wurde mittels Zentrifugal-Konzentrationszelle (MWCO
30 kDa) auf ein Volumen von etwa 2 mL eingestellt und nach Filtration über eine Membran
(200 µm) der Gelpermeation an Superdex 200 zugeführt. Das exemplarische Elutionsprofil (siehe
Abbildung 41) zeigt zwei Absorptionsmaxima. Der peak bei einem Elutionsvolumen von
VE= 84.4 mL ist auf die N-Oxygenase zurückzuführen und bescheinigt dieser Spezies eine moleku­
lare Masse von etwa 61 kDa, während der peak bei VE= 93.7 mL (26.7 kDa) dem verunreinigenden
MBP zugeordnet werden kann. Der Oligomerenstatus von AurF, der basierend auf der Eichung der
GPC (Anhang 7.1) als Dimer abgeschätzt wurde, konnte durch DLS bestätigt werden, die in der
gereinigten Proteinlösung eine monomodale Spezies mit einer Molekularmasse von 60 kDa
detektierte. Der dimere Status der N-Oxygenase wurde im folgenden anhand der Kristallstruktur
manifestiert (Abschnitt 4.2.7).
Abbildung 42:
Exemplarische SDS-PAGE ausgewählter Fraktionen der GPC der N-Oxygenase an Superdex 200-16/60. Jede Bahn wurde mit 10 µL Protein beladen. Die AurF-haltigen Fraktionen mit
einem Elutionsvolumen von 78.7 mL bis 89.2 mL wurden vereinigt und zur Kristallisation eingesetzt.
Bahn 1:
Bahn 3:
Bahn 5:
Bahn 7:
Bahn 9:
Fraktion
Fraktion
Fraktion
Fraktion
Fraktion
78.7-80.2 mL
81.7-83.2 mL
84.7-86.2 mL
87.7-89.2 mL
90.7-92.2 mL
Bahn 2:
Bahn 4:
Bahn 6:
Bahn 8:
Bahn M:
Fraktion 80.2-81.7 mL
Fraktion 83.2-84.7 mL
Fraktion 86.2-87.7 mL
Fraktion 89.2-90.7 mL
LMW-Standard
103
Ergebnisse und Diskussion
Das zugehörige SDS-Gel der GPC an Superdex 200-16/60 ist exemplarisch in Abbildung 42
wiedergegeben. Die Fraktionen mit einem Elutionsvolumen von V= 78.7 mL bis V= 89.2 mL wur­
den vereinigt und mittels Zentrifugal-Konzentrationszelle (MCWO 30 kDa) konzentriert. Diese
Proteinlösung wurde entweder direkt zur Kristallisation eingesetzt oder nach Zugabe von Glycerin
(Endkonzentration 10% (v/v)) bei -80 °C bis zur weiteren Verwendung eingelagert.
4.2.2
Kristallisation der N-Oxygenase
Die frisch präparierte, zur Homogenität gereinigte N-Oxygenase wurde durch photometrische
Bestimmung auf eine Konzentration von 8 mg/mL eingestellt. Wurde eingefrorenes Protein
verwendet, erfolgte die Konzentrationsbestimmung nach Dialyse ü. N. gegen GPC-Puffer. Das
Einfrieren des Proteins hatte auf das Kristallisationsverhalten des Proteins keinen erkennbaren
Einfluss, was durch Vergleich der Kristallisationseigenschaften von frisch präparierten AurF mit
eingefrorenen Protein anhand des Crystal Screen I überprüft wurde.
Zum Auffinden initialer Kristallisationsbedingungen wurden die Kristallisationsscreens
Crystal Screen I+II (CS I+II), Crystal Screen Cryo (CSC), Wizard Screen I+II (WS I+II), Wizard
Cryo Screen I+II (WSC I+II) und die Jena Bioscience Screens I-V (JB I-VIII) in der SitztropfenMethode verwendet. Hierfür wurden 1.5 µL Tropfen der Proteinlösung und 1.5 µL der Reservoir­
lösung vermischt. Das Reservoirvolumen betrug 60 µL. Nach vier Tagen konnten in 15 Bedingun­
gen initiale Kristalle gefunden werden. Die Ergebnisse dieser Reihen-Experimente sind in
Tabelle 16 und der Abbildung 43 zusammengefaßt.
Aus dem Vergleich der initialen Bedingungen wurde ersichtlich, dass AurF in einem weiten
pH-Bereich von pH 6.5 bis pH 10.5 in Gegenwart von Polyethylenglykolen verschiedener mittlerer
Molekularmassen eine Kristallbildung aufwies (CS I-41, WS I-21, WS I-28, WS I-41, WS II-16,
WS II-39). Daher erfolgte eine Verfeinerung dieser Bedingungen nach pH-Wert (ΔpH 0.1) und
Konzentration von PEG 8000 (Δ(w/v) = 2%). Die anderen initialen Kristallisationsbedingungen
wurden in gleicher Weise bezüglich der Fällungsmittelkonzentration, dem pH-Wert und der
Salzkonzentration verfeinert und auf Röntgentauglichkeit getestet. Lediglich zwei der Kristal­
lisationsbedingungen zeigten nach Optimierung der Kristallisationsparameter ein hinreichendes
Streuvermögen. Dies waren zum einen die Bedingung CSC II-22 und zum anderen die Bedingung
WS I-21. Die optimalen Bedingungen zur Herstellung dieser Kristalle zeigt Tabelle 17.
104
Ergebnisse zur N-Oxygenase AurF
Tabelle 16:
Kristallisationbedigungen der intitialen Kristalle der N-Oxygenase. Die Kristallformen sind exemplarisch sind in Abbildung 43 gezeigt.
screen
Kristallisationsbedingung
Kristallform [µm]
Crystal Screen I-41
100 mM HEPES pH 7.5
10% (v/v) 2-Propanol, 20% (w/v) PEG 8000
Kristallform G
50 x 30x 30
Crystal Screen I-43
30% PEG 8000
Kristallform F
40 x 40x 40
Crystal Screen II-25
10 mM Cobalt(II)-chlorid-Hexahydrat
100 mM MES pH 6.5
1.8 M Ammoniumsulfat
Kristallform E
400 x 200x 200
Crystal Screen II-32
100 mM Natriumchlorid
100 mM HEPES pH 7.5
1.6 M Ammoniumsulfat
Kristallform C
1000 x 10x 10
Crystal Screen Cryo -22
0.17 M Natriumacetat-Trihydrat
0.085 M Tris pH 8.5
25.5% (w/v) PEG 4000, 15% (v/v)
Kristallform B
80 x 80x 30
Wizard Screen I-21
100 mM HEPES pH 7.5
20% (w/v) PEG 8000
Kristallform A
600 x 30x 30
Wizard Screen I-28
200 mM Natriumchlorid
100 mM HEPES pH 7.5
20% (w/v) PEG 3000
Kristallform A
60 x 30x 30
Wizard Screen I-41
100 mM CHES pH 9.5
30% (w/v) PEG 3000
Kristallform D
10 x 10x 10
Wizard Screen II-16
100 mM CHES pH 9.5
30% (v/v) PEG 400
Kristallform E
300 x 160x 160
Wizard Screen II-37
200 mM Lithiumsulfat
100 mM Tris pH 7.0
1.0 M Na/K-Tartrat
Kristallform H
900 x 20x 20
Wizard Screen II-39
100 mM CAPS pH 10.5
20% (w/v) PEG 8000
Kristallform I
100 x 100x 100
Jena Bioscience Screen I-C1
100 mM Natriumchlorid
100 mM Bicine 9.0
30% (w/v) PEG 550 MME
Kristallform D
10 x 10x 10
Jena Bioscience Screen I-D5
200 mM Natriumacetat
100 mM HEPES pH 7.5
20% (w/v) PEG 3000
Kristallform A
40 x 20x 20
Jena Bioscience Screen V-D2
100 mM HEPES pH 7.5
20% w/v PEG 10000
Kristallform A
60 x 30x 30
105
Ergebnisse und Diskussion
Abbildung 43:
Kristallformen der intitialen Kristallisationsbedingungen der N-Oxygenase (Tabelle 16). Die Kristalle
wurden nach etwa zwei bis vier Tagen erhalten.
Tabelle 17
Optimierte Kristallisationsbedingungen der N-Oxygenase. Die Kristallisation erfolgte in Agarosegelen mit einer Endkonzentration von 0.5% (w/v).
screen
Fällungsmittel
Puffer
Salz
CSC I-22
22-23% (w/v) PEG 4000
0.085 M Tris pH 8.3
0.17 M Natriumacetat
WS I-21
17-18% (w/v) PEG 8000
0.1 M HEPES pH 7.7
0.05 M NaBr
Das Reflexmuster dieser Kristalle zeigte in beiden Fällen eine Überlagerung von mindestens
einem weiteren Gitter, welches auf ein ungeordnetes Kristallwachstum schließen ließ. Dies wird
zudem anhand des Kristallbildes in Abbildung 44A ersichtlich. Um die Kristallqualität zu
verbessern wurden Agarosegel-Kristallisationsexperimente wie in Abschnitt 3.4.2 beschrieben
durchgeführt. Diese Versuche erfolgten im Hängetropfen-Verfahren. Die Leistungsfähigkeit dieser
Methode ist in Abbildung 44 dargestellt. Ohne die Verwendung von Agarose erreichten die
Kristalle der Bedingung WS I-21 trotz Optimierung der Parameter eine maximale Größe von
120 x 100 x 100 µm3 und hatten einen schichtförmigen Aufbau. Durch Anwendung der Gelkristallisation konnte die Wachstumszeit der Kristalle von drei auf etwa zehn Tage verlangsamt werden,
106
Ergebnisse zur N-Oxygenase AurF
was zu einer erheblich verbesserten Qualität und Größe der Kristalle führte. Diese zeigten an der
Hausanlage-2 ein Streuvermögen von bis zu 3.5 Å mit einem klar definierten Streumuster. Ein
weiterer Vorteil der Kristallisation im Gel wurde aus den Tränkexperimenten mit Schwer­
metallatom-Derivaten ersichtlich. Während Kristalle, die aus Tropfen ohne Verwendung von
Agarose wuchsen, sofort zerbrachen, waren Kristalle in Gel-Konditionen zur Handhabung aus­
reichend stabil. Ein Grund für dieses Verhalten dürfte in der deutlich reduzierten Diffusionsge­
schwindigkeit der zugegeben Lösungen zu finden sein.
Abbildung 44:
4.2.3
Agarosegel-Kristallisation der Kristallisationsbedingung WS I-21. Trotz Optimierung der Parameter konnte eine zufriedenstellende Kristallgröße und -qualität nur durch Verwendung der Agarosegel-Kristallisation erreicht werden. Abbildung 44A zeigt den Kristall, der zur Messung des nativen
Datensatzes verwendet wurde.
Kristallographische Datensammlung von N-Oxygenase Kristallen
Da alle Messungen am Synchrotron bei 100 K erfolgten, mußten die Kristalle zur Vermeidung
von Eisbildung zuvor mit einem Frostschutz versehen werden. Hierzu wurden die nativen Kristalle
mit 30 % (v/v) Ethylenglykol im Reservoirpuffer getränkt. Die Konzentration des Glasbildner wur­
de dabei in fünf Stufen sequentiell erhöht, um eine Erhöhung der Kristallmosaizität zu vermeiden.
Da eine Phasierung durch molekularen Ersatz aufgrund des Fehlens eines geeigneten
Suchmodells nicht durchgeführt werden konnte und ein Selenomethionin-Derivat zum Zeitpunkt
der Messung noch nicht vorhanden war, wurde nach Schwermetallderivaten gesucht, um eine
Phasierung nach der Methode der multiwavelength anomalous diffraction (MAD) zu erreichen.
Hierzu wurden verschiedene gesättigte Schwermetalllösungen hergestellt und diese zu den in Gel­
konditionen kristallisierten Kristallen pipettiert. Das Platin-Derivat, das zur Phasenbestimmung
herangezogen wurde, konnte durch Tränken der nativen Kristalle erzeugt werden, indem zu den
5 µL großen Agarosegeltropfen 0.5 µL einer gesättigten cisPlatin-Lösung zugegeben wurden. Der
Kristall wurde nach einer Inkubationszeit von 10 min in Frostschutz-Puffer überführt, in einem
cryo-loop befestigt und bis zur Messung an der beamline bei 100 K eingefroren. Als Glasbildner
wurde für das Platin-Derivat Glycerin (30% (v/v)) statt Ethlenglykol verwendet. Das cryo-Protokoll
entsprach dem der nativen Kristalle.
107
Ergebnisse und Diskussion
Die Datensammlung wurde an der beamline BL14.1 von BESSY (Berlin) durchgeführt.
Hierzu wurden Datensätze von zwei Kristallen aufgenommen, ein hoch-aufgelöster Datensatz des
nativen Proteinkristalls und ein MAD-Datensatz an der peak-, inflection point- und high energy
remote-Wellenlänge der Platin L-Kante.
Tabelle 18:
Parameter und Statistik der gemessenen Datensätzea),b) der N-Oxygenase
cisPlatin getränkter AurF-Kristall
Nativer AurF-Kristall
peak
infl
hrem
Wellenlänge λ [Å]
0.90814
1.07225
1.07258
0.91841
Detektor
marCCD 165
Detektor Abstand [mm]
150
250
250
250
Auflösung
62-2.11 (2.18-2.11)
62-3.77
(3.88-3.77)
66-3.81
(3.93-3.81)
86-3.77
Raumgruppe
P212121
Zellparameter [Å]
a = 62.2, b = 115.4, c = 125.8
a = 62.7 b = 114.4, c = 131.8
gemessen
392134 (36328)
70164 (6323)
69979 (6339)
71289 (5796)
unabhängig
52924 (4863)
17939 (1628)
17908 (1633)
18397 (1493)
Rsym-I [%]
6.9 (28.3)
11.5 (32.2)
9.4 (27.7)
12.5 (37.3)
Vollständigkeit [%]
100.0 (100.0)
100.0 (100.0)
99.9 (99.9)
99.9 (99.3)
Multiplizität
7.4 (7.5)
3.9 (3.9)
3.9 (3.9)
3.9 (3.9)
Mosaizität [°]
0.101
0.134
0.139
0.150
<I>/<σ(I)>
18.7 (5.3)
12.5 (4.4)
15.0 (5.2)
12.3 (4.0)
marCCD165
P212121
Anzahl der Reflexe
Protomer / ASU
2
3
2
Packungsparameter [Å /Da] 2.96
3.10
Solvensgehalt [%]
60
a)
b)
59
high energy remote und inflection wurde zu hrem bzw. infl abgekürzt.
Die Angaben in den Klammern beziehen sich auf die letzte Auflösungschale.
Die Wellenlängen, bei denen die MAD-Messungen (Abschnitt 3.5.9) durchgeführt wurden,
waren vorher durch eine Fluoreszenz-Messung des Kristalls bestimmt worden, ebenso wie die
Streufaktoren f ' und f ''. Für die MAD-Messung des Platin-Derivats wurde die Wellenlänge des
peak-Datensatzes zu λ =1.07225 Å (Maximum von f ′′) und des inflection point-Datensatzes zu
λ = 1.07258 Å (Minimum von f ′) bestimmt. Für den high energy remote-Datensatz gibt es keine
Vorgaben, außer dass der Unterschied zwischen den Streufaktoren möglichst gering sein sollte. Hier
wurde ein high energy remote-Datensatz bei λ = 0.91841 Å aufgenommen.
Alle Datensätze wurden mit der Software APRV prozessiert und skaliert, die die Programme
XDS, XSCALE implementiert. Die Parameter der Datensammlung und die Statistik der Auswer­
tung der gemessenen Datensätze ist in Tabelle 18 zusammengefasst. Die Datensätze konnten
eindeutig indiziert werden und gehörten alle zur Raumgruppe P212121 mit zwei Protomeren in der
108
Ergebnisse zur N-Oxygenase AurF
ASU und einem Solvensgehalt von etwa 60%. Die Zellparameter sind in Tabelle 18 wiedergegeben
und zeigten Unterschiede zwischen den gemessenen Kristallen. Insbesondere die Zellachsen des
nativen Datensatzes waren im Vergleich zum Platin-Derivat derart verschieden, dass die daraus
resultierende Nicht-Isomorphie die Strukturlösung erschwerte.
4.2.4
Strukturlösung der N-Oxygenase
Analyse der Daten
Alle für AurF aufgenommenen Datensätze wurden mit dem Programm SHARP aufeinander
skaliert, wobei zunächst statistische Ausreißer eliminiert wurden. Das Ergebnis der Analyse ist in
Tabelle 19 zusammengefasst. Die MAD-Datensätze des cisPt-Derivats und der Mangan-Eisen-Mes­
sung zeigten intern gute Riso-Werte (durch Schattierung hervorgehoben), wobei die Riso-Werte der
cisPt-MAD-Datensätze mit über 7% höher ausfielen als für perfekt isomorphe Kristalle erwartet.
Eine mögliche Ursache könnten lokale Änderungen in der Proteinstruktur sein, die durch Strahlen­
schaden verursacht wurden. Des weiteren trat zwischen den cisPt-MAD- und dem nativen Daten­
satz Nicht-Isomorphie mit Riso-Werten von über 52% auf, so dass dieser nicht zur Phasierung bei­
tragen konnte. Dies war anhand der Abweichung der Zellparameter, insbesondere der c-Achse mit
6 Å, zu erwarten.
Tabelle 19:
Riso-Werte b) der gemessenen Datensätzea) der N-Oxygenase
cisPlatin Derivat von AurF
natives AurF zur Bestimmung der Metallatome
Nativ
Pt-hrem
Pt-infl
Pt-peak
Mn-lrem
Mn-peak
Fe-lrem
Fe-peak
---
0.526
0.525
0.523
0.319
0.326
0.318
0.329
Pt-hrem
0.526
---
0.076
0.071
0.479
0.471
0.469
0.477
Pt-infl
0.525
0.076
---
0.077
0.477
0.470
0.468
0.475
Pt-peak
0.523
0.071
0.077
---
0.474
0.467
0.465
0.472
Mn-lrem
0.319
0.479
0.477
0.474
---
0.036
0.035
0.037
Mn-peak
0.326
0.471
0.470
0.467
0.036
---
0.040
0.030
Fe-lrem
0.318
0.469
0.468
0.465
0.035
0.040
---
0.052
Fe-peak
0.329
0.477
0.475
0.472
0.037
0.030
---
---
Nativ
a)
b)
high energy remote, low energy remote und inflection wurde zu hrem, bzw. lrem, bzw. infl abgekürzt.
siehe Gleichung 34 in Abschnitt 3.5.8, der Riso wurde im Auflösungsbereich von 20-4.0 Å bestimmt.
Die Datensätze des Platin-Derivates wurden nach Prozessierung und Skalierung mit dem
Programm XPREP analysiert, das eine auflösungsabhängig Abschätzung der anomalen Differenzen
der Daten erlaubt. Die hierzu ermittelten anomalen Korrelationskoeffizienten sind ebenso wie das
anomale Signal-zu-Rausch-Verhältnis in Tabelle 20 aufgelistet. Die Datensätze zeigen bis zu einem
Auflösungsbereich von 4.4 Å eine ausreichend hohe anomale Korrelation von über 30%, weshalb
zur Schweratomsuche und Phasierung alle drei Datensätze genutzt wurden.
109
Ergebnisse und Diskussion
Tabelle 20:
Auflösung [Å]
Anomales Signal-zu-Rausch-Verhältnis und anomale Korrelationskoeffizienten der gemessenen MADDatensätzea)
Anomales Signal-zu-Rausch-Verhältnis
Korrelationskoeffizient [%]
infl
peak
hrem
Infl / peak
Infl / hrem
peak / hrem
62 - 8.0
5.04
5.88
5.58
98.6
95.9
97.0
8.0 - 6.0
2.91
3.39
2.88
88.7
84.5
86.5
6.0 - 5.8
2.06
2.31
1.93
71.7
62.3
58.6
5.8 - 5.6
2.13
2.36
2.02
72.3
58.7
66.9
5.6 - 5.4
1.92
2.03
1.75
66.4
55.5
62.0
5.4 - 5.2
1.80
1.96
1.58
61.4
46.1
56.3
5.2 - 5.0
1.66
1.82
1.65
55.5
57.8
51.5
5.0 - 4.8
1.57
1.64
1.51
54.9
36.8
46.7
4.8 - 4.6
1.44
1.60
1.45
39.2
39.2
44.4
4.6 - 4.4
1.36
1.47
1.37
43.4
35.7
34.6
17.4
21.9
4.4 - 4.2
1.25
1.39
1.20
29.4
a)
high energy remote und inflection wurde zu hrem bzw. infl abgekürzt.
Schweratomsuche und Phasierung
Die Schweratomsuche wurde initial mit dem Programm SHELXD durchgeführt (Abschnitt
3.5.9) und die Ergebnisse zusätzlich über anomale Differenz-Patterson-Dichtekarten (Gleichung 38)
überprüft. Dabei konnten drei Platin-Positionen bestimmt werden. Diese wurden in das Programm
SHARP/AUTOSHARP transferiert und nach zusätzlichen Platinpositionen gesucht. Hier konnten
weitere sechs Platin-Positionen eindeutig bestimmt werden. Die Parameter der Schwermetallatome
wurden anschließend gegen die MAD-Daten verfeinert, wobei der Datensatz, der am inflection
point gemessen wurde, als Referenzdatensatz genutzt wurde. In Tabelle 21 sind Statistiken der
Verfeinerung und Phasierung zusammengefasst.
Die neun Platin-Positionen erlaubten die Bestimmung des NCS-Operators mit dem Programm
RESOLVE, der beide Protomere über diese Symmetrie-Operation in Beziehung setzt. Diese
Operation bestand in einer Rotation von 180°, die auf ein C2-Dimer hinwies. Die Existenz dieser
zweizähligen Achse war bereits aus der Berechnung der Selbstrotationsfunktionen ersichtlich (siehe
Anhang). Obwohl die Daten jenseits von 4.2 Å nicht zur Phaseninformation beitrugen, wurden die
Proteinphasen bis zur vollen Auflösung von 3.8 Å berechnet. Die resultierende Elektronendichte
war unter Berücksichtigung dieser Auflösungsgrenze von guter Qualität und ließ bereits kurze
helikale Segmente erkennen.
110
Ergebnisse zur N-Oxygenase AurF
Tabelle 21: Schweratomparameter und Statistiken des cisPt-Derivats
Auflösungsbereich [Å]
phasing power
isomorph – azentrisch
isomorph - zentrisch
anomal – azentrisch
Rcullis
isomorph – azentrisch
isomorph - zentrisch
anomal – azentrisch
FOM
zentrisch
azentrisch
peak
86 - 3.77
0.697
0.556
1.600
0.745
0.787
0.639
cisPt-MAD
infla)
1.411
1.261
1.076
1.325
0.687
0.722
0.682
0.714
0.414
0.583
Pt-Positionen
x
y
z
1
0.255
0.162
0.327
2
0.006
0.021
0.794
3
0.300
0.345
0.349
4
1.000
0.150
0.278
5
0.380
0.275
0.637
6
0.321
0.494
0.019
7
0.005
0.379
0.596
8
0.096
0.193
0.747
9
0.345
0.048
0.176
a)
high energy remote und inflection wurde zu hrem bzw. infl abgekürzt.
Abbildung 45:
hrema)
occupancy
2.33
1.57
1.24
1.19
1.22
1.21
0.76
1.10
0.64
B-Faktor
155
171
67
195
109
189
128
164
173
Projektion der Elektronendichte vor und im Verlauf der Dichtemodifikationen der Strukturlösung
durch cisPt-MAD. Die Elektronendichte ist jeweils bis zu einer maximalen Auflösung von 3.8 Å
und einem Konturniveau von minimal 1.5 σ entlang der x-Achse der Elementarzelle gezeigt, wobei
über einen Bereich von 20 % der c-Achse projiziert wurde. A) zeigt die initiale Elektronendichte
nach der Phasierung mit SHARP, B) die mit DM / SOLOMON geglättete Elektronendichte und C)
mit RESOLVE und nicht-kristallographischer Symmetrie erhaltene modifizierte Elektronendichte.
111
Ergebnisse und Diskussion
Abbildung 46:
Stereodarstellung eines Ausschnittes der Elektronendichte nach der Dichtemodifikation mit
RESOLVE zusammen mit einem Ausschnitt des finalen Modells von AurF im Bereich der
Aminosäuren 228 bis 239. Die Elektronendichte ist jeweils bis zu einer maximalen Auflösung von
3.8 Å bei einem Konturniveau von 1.5 σ dargestellt. Durch die Dichtemodifikation wurde die
Phaseninformation aus der MAD-Phasierung deutlich verbessert.
Dichtemodifikation, Modellbau und Strukturverfeinerung
Die initialen SHARP-Phasen des Pt-MAD wurden durch Dichtemodifikation (Abschnitt 3.5.9)
mit den Programmen DM/SOLOMON, wie sie in AUTOSHARP implementiert sind, weiter verbes­
sert, so dass eine klare Solvensabgrenzung zu erkennen war (Abbildung 45). Da aber lediglich bis
etwa 3.8 Å eine ausreichende Qualität der Phasen vorlag, konnte nicht automatisiert mit
Programmen wie ARP/wARP oder RESOLVE gebaut werden. Stattdessen wurden die Sekundär­
strukturelemente von AurF manuell mit Sekundärstruktur-Fragmenten aus einer Fragmentbibliothek
aufgebaut. Das resultierende Modell von etwa 160 nicht zusammenhängenden poly-Ala-Resten in
der ASU war als Suchmodell allerdings nicht ausreichend, um unter Verwendung des Programms
PHASER eine eindeutige Lösung zu erhalten. Die Elektronendichte konnte aber nochmals
wesentlich durch Anwendung der zweizähligen NCS mittels RESOLVE verbessert werden. Der
automatisierte Modellbau wurde anschließend durch RESOLVE_build initiert und und lieferte
durch manuelles Vervollständigen ein Modell von etwa 340 Aminosäureresten (Abbildung 47) aus
mehreren Fragmenten, wobei die Sequenz der Seitenketten partiell in die Elektronendichte gedockt
werden konnte. Dieses Modell konnte mit dem Programm PHASER auf den nativen Datensatz
übertragen werden. Zur Verringerung von model bias wurde das Modell mit ARP/wARP automa­
tisiert neu aufgebaut. Zusätzlich wurde eine prime-and-switch Elektronendichtekarte (Abschnitt
3.5.14) berechnet und das Modell manuell mit COOT korrigiert. Im weiteren Verlauf der
Verfeinerung folgten mehrere Zyklen, in denen das Modell von Hand an die jeweils erzeugte
Elektronendichte angepasst und mittels REFMAC erneut verfeinert wurde. Dabei wurde die
zunächst strenge NCS sukzessive gelockert, bis sie am Ende der Verfeinerung vollständig freigege­
112
Ergebnisse zur N-Oxygenase AurF
ben wurde. Die Termini beider Ketten sind ungeordnet und in der Elektronendichte nicht sichtbar.
Die in der Elektronendichte sehr schlecht definierte loop-Region der Aminosäuren 48-54 wurde in
beiden Proteinen mit dem Programm RAPPER (de Bakker et al., 2003), das besondere Rücksicht
auf die Geometrie der Aminosäurereste legt, neu gebaut. Wassermoleküle wurde mit dem Pro­
gramm ARP_WATERS (Lamzin & Wilson, 1993) eingefügt und manuell mit COOT überprüft.
Dies resultierte im finalen Modell mit Rcryst und Rfree von 19.6% bzw. 24.3%.
Abbildung 47:
4.2.5
Initiales, aus Sekundärstrukturfragmenten aufgebautes Modell der N-Oxygenase (rot) im Vergleich
mit dem finalen Modell (grau). Das initiale Modell umfasst etwa 55 % aller erwarteten Reste und war
ausreichend, um auf den nativen Datensatz übertragen zu werden.
Identität des Cofaktors der N-Oxygenase
Wie bereits beschrieben ist die N-Oxygenase AurF ein Metalloenzym, genauer eine Monooxy­
genase mit einem zweikernigen metallischem Cofaktor. Aus vorhergehenden Arbeiten war bekannt,
dass AurF sowohl Eisen, als auch Mangan inkorporierte (Winkler et al., 2006). Eine eisenabhängige
Arbeitsweise des Enzyms war vor Veröffentlichung dieser Arbeit postuliert worden (Simurdiak et
al., 2006) und stützte sich im Wesentlichen auf das konservierte Bindungsmotiv (Abschnitt 4.2.8)
von AurF und ESR-Daten. Diese Arbeiten konnten die Identiät des als Cofaktor fungierenden
Metalls jedoch nicht eindeutig belegen, so dass kam dieser Aspekt der Strukturaufklärung der
N-Oxygenase besondere Bedeutung zukam. Dieser Abschnitt befasst sich mit den Ergebnissen der
in dieser Arbeit erhaltenen ESR-Messungen und durchgeführten Röntgenexperimenten hinsichtlich
der Identifikation des Cofaktors des zur Kristallisation eingesetzten Enzyms. Eine detailierte
Diskussion, die Rücksicht auf bisherige Erkenntnisse nimmt und einen Vergleich mit
strukturverwandten Enzymen zieht, ist in Abschnitt 4.2.7 gegeben.
113
Ergebnisse und Diskussion
ESR-Untersuchungen
ESR-Messungen des Fusionsproteins der N-Oxygenase wurden in Kooperation mit Prof.
Th. Friedrich (Institut für Organische Chemie und Biochemie, Freiburg) durchgeführt. Hierzu
wurden drei Proben des Enzyms untersucht, zum einen das ungeschnittene Fusionsprotein MBPAurF, das Fusionsprotein in Gegenwart von Wasserstoffperoxid und schließlich das Fusionsprotein
in Gegenwart von H2O2 und natürlichen Substrat p-Aminobenzoesäure (PABA). Die ESR-Spektren
dieser drei Messungen sind in den Abbildungen 48A bis 48C wiedergegeben.
A
750
3+
Fe
500
250
2+
Mn
Intensität
0
-250
-500
-750
-1000
AurF
-1250
0
B
1000
2000
3000
Magnetfeld [G]
3+
Fe
600
4000
5000
Radikalbildung
400
200
Mn
?+
Intensität
0
-200
-400
-600
-800
Probe Aurf + H2O2
-1000
0
1000
2000
3000
4000
5000
6000
4000
5000
6000
Magnetfeld [G]
C
1250
1000
Fe
750
3+
500
Intensität
250
0
Mn
2+
-250
-500
-750
-1000
-1250
Aurf + H 2 O 2 + Substrat
-1500
0
Abbildung 48:
114
1000
2000
3000
M agnetfeld [G ]
ESR-Spektren des Fusionsprotein MBP-AurF. A) Das Fusionsprotein ohne Zusätze B) MBP-AurF
mit Wasserstoffperoxid und c) MBP-AurF mit Wasserstoffperoxid und natürlichem Substrat PABA.
Ergebnisse zur N-Oxygenase AurF
Das Spektrum des Fusionsproteins zeigt ein intensives Signal zwischen 3076.5 G und
3540.9 G, das zu einem Sextett mit durchschnittlichem Abstand der Maxima von 93 G aufgespalten
ist. Das Zentrum des Signals liegt bei 2.05 g. Dieses Signal ist charakteristisch für ein
mononukleares Mn2+-Zentrum mit einem Spin-Zustand von 5/2 (Chang et al., 2004), kann aber
auch für binukleare Mn-Zentren beobachtet werden (Samples et al., 2005). Des weiteren kann aus
diesem Spektrum auf die Anwesenheit von Eisen geschlossen werden. Dieses zeigt ein schwaches
Signal bei 1567.1 G, respektive 4.31 g (Chang et al., 2004; Keyer et al., 1996). Die Aufspaltung des
Mangansignals geht in Anwesenheit von H2O2 teilweise verloren. Stattdessen ist das Spektrum
durch einen ausgeprägten peak bei 3360.3 G (1.98 g) charakerisiert, welcher als Radikalspezies am
Manganzentrum interpretiert werden kann. Dass das ESR-Spektrum in Gegenwart von H2O2 und
Substrat wieder das ursprüngliche Absorptionsmaximum des Fusionsproteins MBP-AurF bei 2.05 g
einschließlich der Sextett-Aufspaltung eines Mn2+ aufweist, gibt Anlass zu der Annahme, dass die
enzymatische Aktivität der N-Oxygenase auf der Einlagerung von Mangan in das aktive Zentrum
des Enzyms basiert. Es ist hierbei anzumerken, das dem Fusionsprotein nach Behandlung mit Was­
serstoffperoxid enzymatische Aktivität nachgewiesen werden konnte, die zuvor nach Zellaufschluss
der kultivierten E. coli-Zellen verlorengegangen war. Ein derartiges als Peroxide-Shunt bezeichne­
tes Verfahren wurde auch für AurF beschrieben (Winkler et al., 2006).
MAD-Experiment an der Mangan und Eisen K-Kante
Die ESR-Messungen gaben berechtigten Zweifel an einer eisenabhängigen Arbeitsweise von
AurF. Um zu klären, welche Metallatome welche Positionen innerhalb der Proteinstruktur
einnehmen, wurde ein spezielles MAD-Experiment durchgeführt (Abschnitt 3.5.10). Die
Bestimmung der Identität der Metallatome beruht auf der Änderung der anomalen Differenzen
zwischen zwei Wellenlängen, so dass zur Unterscheidung der Metalle zusätzlich zu den
Datensätzen an den peak-Wellenlängen von Eisen und Mangan jeweils ein Datensatz im
Energiebereich vor der Absorptionskante (low energy remote) des jeweiligen Metalls aufgenommen
werden mußte. Insgesamt wurden daher vier Datensätze an einem einzelnen Kristall von nativem
AurF gesammelt: an den K-Kanten der zu untersuchenden Metalle Mangan und Eisen sowie jeweils
an einer low energy remote-Wellenlänge der jeweiligen Metalle. Die Strategie dieser Messung ist in
Abbildung 23 wiedergegeben und wurde bereits zur Unterscheidung von Ca2+ und K+ erfolgreich
angewandt (Than et al., 2005).
115
Ergebnisse und Diskussion
2600
M n- peak ,
1.89276 Å ,
6550 eV
2400
2200
Fluoreszenz [m AU]
2000
1800
1600
Fe- peak ,
1.73673 Å ,
7139 eV
1400
1200
1000
800
M n- lrem ,
1.89850 Å,
6540 eV
600
Fe- lrem ,
1.74504 Å ,
7105 eV
400
200
0
6530
6540
6550
7110
7120
7130
7140
Energie (eV)
Abbildung 49: Fluoreszenzscans an den Absorptionskanten der zu untersuchenden Metalle Eisen und Mangan. Die
Wellenlängen an denen die Datensätze zur Identifikation der Metalle aufgenommen wurden, sind
durch Pfeile markiert.
Tabelle 22:
Statistik und Parametera) der Datensammlung an einem nativen N-Oxygenase Kristall zur Bestimmung
der Identität und Positionen der Metalle Eisen und Mangan innerhalb der Proteinkette.
Fe-peak
Fe-lremb)
Mn-peak
Mn-lremb)
Wellenlänge λ [Å]
1.73673
1.74504
1.89276
1.89580
Auflösung
64-2.57
(2.68-2.57)
63-2.58
(2.69-2.58)
63-2.80
(2.92-2.80)
57.6-2.80
(2.92-2.80)
gemessen
203922 (6643)
111774 (6885)
165288 (11334)
158416 (10570)
unabhängig
50944 (3798)
48343 (3776)
39495 (3411)
40080 (3484)
Rsym-I [%]
8.2 (34.6)
5.4 (20.4)
6.2 (19.4)
6.0 (18.6)
Vollständigkeit [%]
90.7 (57.4)
87.4 (58.0)
91.1 (66.9)
92.5 (68.1)
Multiplizität
4.0 (1.7)
2.3 (1.8)
3.8 (3.3)
4.0 (3.0)
Mosaizität [°]
0.400
0.639
0.590
0.518
<I>/<σ(I)>
16.8 (3.3)
15.8 (5.46)
19.1 (5.3)
19.1 (5.6)
Anzahl der Reflexe
a)
b)
116
Die Daten wurden an der beamline BL14.2 (BESSY, Berlin) bei einem Detektorabstand von 110 mm gesam
melt. Die Datensätze wurden der Raumgruppe P212121 mit Zellachsen von a = 62.3 Å, b = 115.5 Å und
c = 126.7 Å zugeordnet. Es finden sich zwei Protomere in der ASU. Dies entspricht einem Packungsparameter
von 2.99 und einem Solvensgehalt von 59%.
low energy remote wurde zu lrem abgekürzt.
Ergebnisse zur N-Oxygenase AurF
Die Absorptionskanten wurden mittels Fluoresenzscans bestimmt und peak-Datensätze an
den Wellenlängen λ(Mn-peak) = 1.89276 Å und λ(Fe-peak) = 1.73673 Å gesammelt. Abbildung 49
zeigt den experimentell bestimmten Fluoreszenzverlauf an den K-Kanten von Mangan und Eisen.
Da die Datensammlung an einem einzelnen Kristall erfolgen mußte und der durch Röntgenstrahlung
verursachte Strahlenschaden mit steigender Wellenlänge zunimmt, wurde die Dosis der einge­
setzten Strahlung durch Einsatz von Filtern und kurzen Belichtungszeiten reduziert. Dies begrenzte
die Auflösung auf etwa 2.8 Å für die Mangan-Datensätze bzw. 2.6 Å für die Datensammlung an der
Eisen-Kante und Eisen low energy remote. Die erzielten Auflösungen waren allerdings zur Berech­
nung der anomaler Differenzen vollkommend zufrieden stellend. Die Statistik dieser Datensätze ist
in Tabelle 22 zusammengefasst.
Abbildung 50: DDANO-Elektronendichtekarten im Bereich des aktiven Zentrums mit den Seitenketten der koordinierenden Aminosäuren und dem zweikernigen Metallcluster in der ball-and-stick-Darstellung. Die
DDANO-Karte von Mangan ist bei einem Konturniveau von 10 σ dargestellt (violett), demgegenüber
zeigt die in grün gezeichnete DDANO-Karte die Eisenpositionen bei einem Konturniveau von 4 σ.
Die Datensätze wurden mit XDS/XSCALE prozessiert und skaliert, wie sie in APRV imple­
mentiert sind. Die Phasen resultierten aus einer rigid-body-Verfeinerung mit dem Programm
REFMAC, wobei das finale AurF-Modell als Startpunkt verwendet und gegen den Mn-lremDatensatz verfeinert wurde. Anschließend wurden die Strukturfaktoramplituden F+ und F- aller vier
Wellenlängen unter Verwendung des Programms CAD (CCP4, 1994) zusammengefasst und im
folgenden auf den Mn-lrem-Datensatz skaliert. Dieser Datensatz wurde als Referenz ausgewählt da
er wenig anomale Streuung bezüglich der zu untersuchenden Metallatome Mangan und Eisen
aufweist (Abschnitt 3.5.10). Die double-difference-anomalous-Elektronendichtekarte (DDANOmap) resultierte im folgenden aus der Berechnung der Differenzen der Strukturfaktoren nach
Gleichung 41 und Kalkulation der Elektronendichtekarte unter Verwendung des Programmes
117
Ergebnisse und Diskussion
FFTOOLS (CCP4, 1994). Der Auflösungsbereich dieser Dichtekarten wurde zur Berechnung auf
4.5 Å beschränkt, um das Signal-zu-Rausch-Verhältnis zu optimieren, da die anomalen Daten nur
bis zu dieser Auflösung zu verwenden waren. Abbildung 50 zeigt die resultierende DDANO-map
für beide erwarteten Metalle.
Aus den DDANO-Elektronendichtekarten geht zweifelsfrei die Anwesenheit beider Metalle
hervor, weitere Metall-Positionen innerhalb der Proteinstruktur konnten nicht detektiert werden.
Der Metall-Gehalt wurde unter Verwendung des Programms MAPMAN (CCP4, 1994)berechnet, in
dem die DDANO-Elektronendichte für das jeweilige Metall über das Volumen der beide
Metallzentren integriert wurde. Der verwendete Integrationsradius betrug mit 1.6 Å in etwa die
Hälfte des Metall-Metall-Abstandes. Zudem wurde das Mangan-zu-Eisen-Verhältnis durch
manuelle Integration der peak-Flächen abgeschätzt. Hierzu wurden die peak-Maxima der Elektro­
nendichten bestimmt und anschließend die Volumina der Elektronendichte bei 2/3 der maximalen
peak-Höhe durch Ausmessen der Dimensionen der resultierenden ellipsoiden Dichtekarte ermittelt.
Beide Methoden sind nicht frei von Fehlern. Während die Verwendung des Programms MAPMAN
durch Integration über den halben Metall-Metall-Abstand Teile der vorhandenen Elektronendichte
nicht berücksichtigt, resultiert der Fehler der manuellen Methode zum einen aus der Abschätzung
des Volumina des Ellipsoids und zum anderen aus einen systematischen Fehler an sich, der aus dem
vorhanden Rauschen in den DDANO-maps hervorgeht. Unter Berücksichtigung der Fehlerquellen
beider Methoden kann das Mangan-zu-Eisen-Verhältnis zu etwa 8:1 angegeben werden, bei einem
Fehler von etwa ±5% unabhängig von der eingesetzten Methode zur Berechnung dieser
Metallverhältnisse. Die Schlußfolgerungen dieser Messungen werden in Kapitel 4.2.8 diskutiert.
4.2.6
Validierung des Proteinmodells der N-Oxygenase
Geometrie des Modells
Das verfeinerte Modell der AurF besteht aus dem funktionalen Dimer, das den Inhalt der ASU
bildet. Bei beiden Protomeren fehlen die ersten 6 Aminosäurereste, sowie der zusätzliche N-term­
inale linker (Abschitt 4.2.1). Ebenso sind die C-terminalen Reste 322-336 der Kette A sowie die
Reste 320-336 der Kette B nicht in der Struktur enthalten.
Die Validierung des Strukturmodells von AurF erfolgte während der Verfeinerung anhand der
Güte der R-Faktoren sowie der Abweichung von geometrischen Parametern (Abschnitt 3.6.1).
Tabelle 23 fasst die Verfeinerungsstatistiken des fertigen Modells zusammen. Das Strukturmodell
konnte zu guten R-Faktoren verfeinert werden. Im Vergleich zu Standardbindungslängen und
118
Ergebnisse zur N-Oxygenase AurF
-winkeln (Engh und Huber, 1991) ergeben sich jeweils nur geringe Abweichungen. Zudem befinden
sich alle Ergebnisse der mit den Programmen RAMPAGE und WHATCHECK durchgeführten
Analysen innerhalb gängiger Toleranzgrenzen. In der N-Oxygenase-Struktur liegen 97.8% aller
dihedralen Hauptkettentorsionswinkel in energetisch günstigen Bereichen des Ramachandran-Dia­
gramms, was in Abbildung 51 veranschaulicht ist. Neben den Hauptkettentorsionswinkeln können
auch Seitenkettentorsionswinkel als Qualitätskriterium herangezogen werden, da sie nicht in den
Zielfunktionen der Verfeinerungsprogramme formuliert sind (Abschnitt 3.6.1). Verglichen mit Bib­
liotheken an Standard-Rotameren aller Aminosäuren (Laskowski et al., 1993) liegen auch hier alle
auftretenden Winkelkombinationen innerhalb gängiger Toleranzgrenzen wie in Abbildung 52
anhand der Reste Isoleucin, Leucin und Phenylalanin gezeigt. Nur die Seitenkettenwinkel, von
Aminosäuren in loop-Regionen, die eine außerordentlich schlechte Elektronendichte aufweisen,
zeigen Abweichungen von den Vorzugskonformationen.
Tabelle 23:
Statistik und Daten des verfeinerten Modells der N-Oxygenase
Auflösung [Å]
35 - 2.11
Raumgruppe
P212121
Rcryst
0.196
Rfree (test set of 5%)
0.243
Anzahl der nicht-H-Atome
Protein
5316
Mangan
4
Ehtlenglykol
Wassermoleküle im aktiven Zentrum
28
a)
Weitere Wassermoleküle
Durchschnittlicher isotroper B-Faktor [Å2]
Hauptkette A / Hauptkette B
5
434
34.4 / 48.2
Seitenketten A / Seitenketten B
36.8 / 49.5
Mangan
24.5 / 46.5
Ethylenglykol
46.1
Wassermoleküle
48.6
bond lengths [Å]
0.015
bond angles [°]
1.417
rmsd
Ramachandran-Statistik der Aminosäuren [%]
favorisierte Geometrie
erlaubte Geometrie
a)
97.8
2.2
Zwei Wassermoleküle waren 50% besetzt
119
Ergebnisse und Diskussion
Abbildung 51:
Ramachandran-Diagramme für das Modell der N-Oxygenase. Die Positionen aller Aminosäurereste
sind durch Kreuze (Glycinreste) und Quadrate (alle anderen Aminosäuren) gekennzeichnet. Es sind
separate Diagramme für A) generell bevorzugte Winkelkombinationen, B) für Glycin typische Kom
binationen sowie für C) charakteristische Winkelkombinationen von Aminosäuren, die direkt vor
Prolinen gelegenen sind und für D) Proline dargestellt. Die Farbgebung des Hintergrundes markiert
die energetisch unterschiedlich klassifizierten Bereiche, dunkle Regionen sind am meisten bevorzugt
(Lovell et al., 2003).
Abbildung 52:
Verteilung der Seitenkettenwinkel χ1 und χ2 im AurF-Modell am Beispiel der Aminosäuren Isoleucin,
Leucin und Phenalalanin.Bevorzugte Bereiche sind zunehmend dunkel dargestellt. Die einzelnen Seitenketten sind durch Quadrate symbolisiert, wobei hellere bevorzugten Konformationen entsprechen.
120
Ergebnisse zur N-Oxygenase AurF
Analyse der Temperaturfaktoren und der Dichtekorrelation
Die Übereinstimmung des Proteinmodells mit der Elektronendichte wird über den real-space
Dichtekorrelationskoeffizienten beschrieben und mit dem Programm OVERLAPMAP (CCP4,
1994) berechnet. In Abbildung 53 sind die Korrelationkoeffizienten der Hauptkette für die beiden
Protomere der asymmetrischen Untereinheit aufgetragen. Diese zeigen in allen Bereichen ein gute
Übereinstimmung mit der Elektronendichte. Die größten Abweichungen befinden sich in loopRegionen der Proteinstruktur, deren Elektronendichte aufgrund der konformationellen Freiheits­
grade derartiger Bereiche schlechter definiert ist.
120
0
20
40
60
80
100 120 140 160 180 200 220 240 260 280 300 320
1.0
110
100
0.8
0.6
2
B-Faktor [Å ]
80
70
60
0.4
D ichtekorrelation
K ette A
K ette B
90
50
40
0.2
30
20
0.0
0
20
40
60
80
100 120 140 160 180 200 220 240 260 280 300 320
A m inosäurerest
Abbildung 53: B-Faktoren und Dichtekorrelation der N-Oxygenase für beide Proteinketten.
Abbildung 53 zeigt zudem die Auftragung der B-Faktoren der Hauptkette für die jeweilige
Aminosäure. Die durchschnittlichen B-Faktor unterscheiden sich für die beiden Proteinketten A und
B und wurden zu 34.4 Å2 respektive 48.2 Å2 errechnet, wobei der Verlauf dieser Temperaturfak­
torkurven für beide Ketten identisch ist. Die durchgängig höheren B-Faktoren in Kette B sind das
Resultat der Kristallpackung, da die Kette B im Vergleich zum Protomer der Kette A aufgrund der
geringeren Kontaktfläche zu benachbarten Molekülen verstärkt dem Solvens ausgesetzt ist. Weitere
121
Ergebnisse und Diskussion
Auffälligkeiten im Bezug auf die B-Faktoren finden sich innerhalb der Proteinstruktur nicht. Hohe
B-Faktoren zeigen sich ausschließlich in den Bereichen der Struktur, die solvensexponiert sind oder
sich in loop-Regionen befinden. Insbesondere die Aminosäuren, die das Metallzentrum definieren
und die aktive Tasche auskleiden, zeigen keine auffallenden Temperaturfaktoren. Allerdings
könnten die hohen B-Faktoren im Bereich der Aminosäuren 287-303 auf ein variables Segment
hindeuten, was die Lage des postulierten Substratkanals (Abschnitt 4.2.8) stützen würde.
Eine Übereinstimmung der beiden Protomere in der asymmetrischen Untereinheit wird über
die rms-Abweichung der Cα-Positionen beschrieben (Abbildung 54). Bei einem Mittelwert von
0.54 Å zeigen vor allem Segmente in loop-Regionen, die durch schlecht definierte Elektronendichte
auffallen, größere Abweichungen zwischen den Protomeren. Auch hier zeigt das bereits beschriebe­
ne Segment im Bereich der Aminosäuren 287 bis 303 eine höhere Abweichung der C α-Positionen
zum Mittelwert. Ebenso zeigen sich große Abweichungen an den N- und C-Termini, die auf die
geringe Fixierung dieser Bereiche innerhalb der Proteinstrukutur zurückzuführen sind.
2.2
2.0
1.8
1.6
∆ Cα
[Å]
1.4
1.2
1.0
0.8
0.6
0.4
0.2
0.0
0
20
40
60
80
100
120
140
160
180
200
220
240
260
280 300
320
Aminosäurerest
Abbildung 54:
122
Abweichungen der Cα-Positionen der beiden Protomere zueinander in Abhängigkeit des Kettenverlaufs.
Ergebnisse zur N-Oxygenase AurF
4.2.7
Strukturbeschreibung der N-Oxygenase
Sekundärstruktur
Die Analyse der Sekundärstrukturelemente der Struktur von AurF erfolgte anfänglich mit
Hilfe der Programme DSSP und STRIDE (Abschnitt 3.6.2), die endgültige Zuordnung dann durch
Vergleich der jeweiligen Zuordnung in den beiden Protomeren der asymmetrischen Untereinheit.
Diese ist in Abbildung 55 dargestellt. Das aus dieser Einteilung resultierende Topologie-Diagramm
(Abbildung 56) zeigt schematisch die Faltung der N-Oxygenase.
•
•
• α1
•
•
•
• α2
llllllllllllllllllllasssssssssssdlllllllllllllllllllllllllllllllllasssdll
mreeqPHLATTWAARGWVEEEGIGSATLGRLVRAWPRRAAVVNKADILDEWADYDTLVPDYPLEIVPFAEHPLFLAAE
88
•
•
α3
•
•
•
•
• α4
•
•
assssssssssssssssssssssssssssssssssdllllllllassssssssssssssssssssssssssssssdlllllllllll
PHQRQRVLTGMWIGYNERVIATEQLIAEPAFDLVMHGVFPGSDDPLIRKSVQQAIVDESFHTYMHMLAIDRTRELRKISERPPQPEL 165
•
α5
•
• α6
•
•
•
• α7
•
•
assssssssssdlllassssssssssssssssssssssssssdlllllasssssssssssssssssssssssssssssdllasssss
VTYRRLRRVLADMPEQWERDIAVLVWGAVAETCINALLALLARDATIQPMHSLITTLHLRDETAHGSIVVEVVRELYARMNEQQRRA 252
α8 •
•
α9
•
α10 •
•
α11 •
•
•
ssssssssssssdlllasssssssssdllllassssssssdllllllllllasssssssssdllllll
LVRCLPIALEAFAEQDLSALLLELNAAGIRGAEEIVGDLRSTAGGTRLVRDFSGARKMVEQLGLDDAVdfdfperpdwsphtpr
336
Abbildung 55:
Sekundärstrukturelemente der N-Oxygenase. Die Aminosäuresequenz befindet sich in der dritten
Zeile. Die verwendete Sekundärstrukturzuordnung, die aufgrund der Programme STRIDE und
DSSP getroffen wurden, ist in der 2. Zeile graphisch dargestellt. Die 1. Zeile enthält die Nummerierung der Sekundärstrukturelemente sowie einen Punkt an jeder 10. Aminosäure. Grau hinterlegt
sind die metallkoordinierenden Aminosäuren.
Abbildung 56:
Topologie-Diagramm der N-Oxygenase aus Streptomyces thioluteus. Die Kreise symbolisieren α-Helices in der Aufsicht auf das Protein. Die erste und letzte Aminosäure der jeweiligen Helix ist angegeben. Ungeordnete Teile der Struktur sind gestrichelt gezeichnet. Das charakteristische vier-HelixBündel, das das Metallzentrum beinhaltet, ist grau hinterlegt. Der schwarze Punkt symbolisiert das
Metallzentrum innerhalb des vier-Helix-Bündels.
123
Ergebnisse und Diskussion
Anhand der Sekundärstruktur wird deutlich, dass AurF ein fast ausschließlich α-helikales
Protein ist; 208 Aminosäurereste je Protomer nehmen innerhalb von elf Helices eine α-helikale
Konformation ein. Dies entspricht einem helikalen Anteil von 62%. Demgegenüber existiert nur ein
sehr kurzes β-Faltblatt am Dimerinterface des Proteins, das aus zwei Strängen der drei
Aminosäuren 14-16 des N-terminalen Arms eines Protomers und den drei Aminosäuren 160-162
des anderen Protomers gebildet wird. Aufgrund der sehr geringen Extension dieses β-Faltblattes
wurde dieses in den Abbildungen 55 und 56 nicht weiter berücksichtigt.
Tertiärstruktur
Auf der Basis der Strukturhomologie, die aus der Verwendung der Programme DALI, SSM
und PINTS hervorging, kann die Faltung der N-Oxygenase denen der Di-Eisen-Oxo-Proteine
zugeordnet werden, auch wenn die Sequenzidentität zu diesen O2-aktivierenden Enzymen nicht
signifikant ist. Ein Vergleich mit den ermittelten Strukturverwandten wird weiter unten gegeben.
Abbildung 57 zeigt die Tertiärstruktur der N-Oxygenase. Das Protomer besteht aus einer
einzelnen Domäne mit einem aus den Helices α3, α4, α6 und α7 aufgebauten zentralen HelixBündel. Im Zentrum dieses Bündels ist das zweikernige Metallcluster koordiniert. Um dieses
charakteristische Merkmal dieser Enzym-Klasse sind sieben weitere Helices angeordnet, die das
aktive Zentrum vom Lösungsmittel abschirmen und so schädigende Oxidationen innerhalb der Zelle
in Form von Nebenreaktionen mit nicht-natürlichem Substrat verhindern.
Quartärstruktur
Sowohl die GPC- als auch die DLS-Experimente wiesen die N-Oxygenase als Dimer aus.
Dieser Oligomerisierungsgrad konnte durch Analyse der Kristallstruktur bestätigt werden. AurF
kristallisiert in seiner funktionalen Einheit als Dimer mit C2-Symmetrie. Das Dimerinterface wird
dabei von den N-Termini beider Protomere gebildet, die verzahnend in das andere Protomer
reichen. Neben dem N-Terminus sind vor allem Reste der Helices α1, α3 und α4 an dem DimerKontakt beteiligt. Der Kontakt verdeckt eine solvenszugängliche Fläche von 2336 Å2, was 15% der
Oberfläche eines isolierten Protomers entspricht und wird durch 25 Wasserstoffbrücken und
38 nonpolare Wechselwirkungen vermittelt. Eine Salzbrücke kann im Dimer-Kontakt nicht
gefunden werden. Eine statistische Auswertung der Kontaktflächen von physiologischen Dimeren
in der Protein-Datenbank (Jones & Thornton, 1996) weist für homodimere Proteine im Mittel eine
Kontaktfläche von 1685 Å2 aus, was gut mit einem AurF Dimer übereinstimmt. Eine detaillierte
Auflistung aller am Dimer-Kontakt beteiligten Aminosäuren findet sich im der Tabelle 24 (Kon­
takt I), die zusätzlich alle weiteren Kontaktflächen, die im Kristall beobachtet wurden, zusammen­
fasst.
124
Ergebnisse zur N-Oxygenase AurF
Abbildung 57:
Stereodarstellung eines Untereinheit der N-Oxygenase. Die Sekundärstrukturelemente sind analog
zur Abbildung 55 bezeichnet. Das zentrale Helix-Bündel ist farblich hervorgehoben (gelb). Die
flankierenden Strukturelemente sind in grün dargestellt. Die Aminosäuren, die die Kavität des
aktiven Zentrum auskleiden sind in cyan gezeigt, das modellierte Substrat in rot. Die nichtkristallographische zweizählige Achse ist in schwarz gezeigt. Der Zugang zum aktiven Zentrum des
Enzyms wird durch einen Stab (violett) dargestellt.
Abbildung 58:
Funktionales Dimer der N-Oxygenase. Die gezeigten Merkmale sowie die Farbgebung entsprechen
denen aus Abbildung 57. Zudem ist das zweite Protomer dargestellt.
125
Ergebnisse und Diskussion
Der Abstand beider Metallzentren beträgt 22 Å. Da keine Aminosäuren innerhalb des DimerKontaktes identifiziert werden konnten, die eine Kommunikation der beiden aktiven Zentren
untereinander vermitteln könnten, kann davon ausgegangen werden, dass diese unabhängig
voneinander arbeiten. Abbildung 58 zeigt die Organisation der N-Oxygenase zum physiologisch
relevanten Dimer.
Kristallpackung
AurF kristallisiert in der Raumgruppe P212121, die vier asymmetrische Untereinheiten pro Ele­
mentarzelle besitzt. In jeder ASU befinden sich zwei Protomere, die nicht-kristallographisch mitein­
ander verwandt sind. Über die nicht-kristallographische Symmetrieoperation ergibt sich aus den
Protomeren das funktionale Dimer. Die Lage der Symmetrieachsen ist schematisch in Abbildung 59
gezeigt.
Abbildung 59:
126
Schematische Darstellung der Raumgruppe P212121 aus International Tables of Crystallography,
(1996), mit Blickrichtung entlang der Achsen x, y, z. Rechts unten ist die Lage der asymmetrischen
Einheiten innerhalb der Elementarzelle gezeigt.
Ergebnisse zur N-Oxygenase AurF
Neben dem Dimerkontakt existieren zwei weitere vorherrschende Kristallkontakte (II und III)
die aus der Anwendung der Symmetrieoperationen resultieren und damit die wesentlichen Ver­
netzungen der Untereinheiten untereinander darstellen (Tabelle 24). Die Analyse der Kristall­
kontakte mit dem Programm PISA (Krissinell & Henrick, 2005) zeigt zudem die größere
Gesamtfläche, die das Protomer A in Form von Kontaktflächen zu Verfügung stellt. Während
Protomer B mit einer summierten Kristallkontaktfläche 725 Å2 beiträgt, stellt das Protomer A mit
einer Gesamtoberfläche von 1319 Å2 eine deutlich bessere Vernetzung zu seinen umgebenden
Molekülen her. Dieser rigidere Halt der Untereinheit A innerhalb des Kristalls ist somit für die
niedrigeren Temperaturfaktoren und die besser definierte Elektronendichte im Vergleich zum
Protomer B verantwortlich. Abbildung 60 verdeutlicht die Vernetzung der Untereinheit durch
Darstellung des Inhalts einer Elementarzelle.
Tabelle 24:
Kristallkontakte der N-Oxygenase
Name Kontakt Sym-Op;
Translation
Oberfläch Interagierende Aminosäurereste
e
[ Å2]
Kette 1
Kette 2
2336
7-20, 33, 35-37, 41, 42, 45, 46,
48,49, 91, 95, 98, 99, 102, 103,
107, 110, 113, 121, 123, 126,
127, 130, 131, 133, 134, 137,
138, 140, 141, 144, 145, 147,
148, 151, 156-165, 168, 172,
175
7-20, 33, 35-37, 41, 42, 45, 46,
48, 49, 91, 95, 98, 99, 102, 103,
107, 110, 113, 121, 123, 126,
127, 130, 131, 133, 134, 137,
138, 140, 141, 144, 145, 147,
148, 151, 156-165, 168, 172
x-1/2,-y+1/2,-z ;
-1,0,0
594
37, 43 ,44, 47, 50, 53, 208, 214,
215, 218, 221, 222, 224, 225,
228
20-26, 30, 33, 123, 236, 239,
240, 243-247, 250
A-B
-x+1,y-1/2,z+1/2;
1,-1,0
576
273, 277, 280-286, 289, 290,
292-295, 298
169, 170, 173, 174, 177, 259,
262, 263, 266, 267, 269, 270,
289, 292, 293, 298, 299,
IV
A-A
x-1,y,z;
-1,0,0
167
52-57, 155
180, 246-248, 251
V
A-B
x-1/2,-y+1/2,-z;
-1,0,0
140
64, 208, 220, 211
51, 151-153, 155,
VI
B-A
a)
B-A
NCS
II
A-A
III
I
x-1/2,-y+1/2,-z ;
9 44, 46
-1,0,0
a)
Dieser Kontakt entspricht dem Interface des funktionalen Dimers.
250, 318
127
Ergebnisse und Diskussion
Abbildung 60: Kristallpackung der N-Oxygenase in der Raumgruppe P212121. Gezeigt ist der Inhalt einer Elementarzelle. Die Kolorierung erfolgte nach angewandter Symmetrieoperation. A) Blickrichtung entlang der
x-Achse, B) entlang der y-Achse.
Vergleich der N-Oxygenase mit strukturverwandten Proteinen
Eine globale Sequenzhomologie zu strukturell bekannten Enzymen war nicht gegeben. Eine
Sequenzhomologie-Suche mit dem Programm BLAST (Altschul et al., 1997) resultierte lediglich in
hypothetischen Proteinen unbekannter Funktion aus verschiedenen Bakterien mit einer Sequenz­
identität von 32-20%. Demgegenüber ergab eine Suche nach strukturverwandten Proteinen mit den
Programmen DALI, SSM und PINTS die Ribonukleotid Reduktase Komponente R2 aus E. coli und
anderen Organismen als strukturell ähnlichste Proteinfamilie (Nordlund & Eklund, 1993; Eriksson
et al. 1998; Voegtli et al., 2001; Högböm et al., 2004; Strand et al., 2004). Als weitere strukturell
homologe Enzyme wurden zwei Methan-Hydroxylasen (Elango et al., 1997, Rosenzweig et al.,
1997), eine Toluol-Hydroxylase (Sazinsky et al., 2004), einer Phenol-Hydroxylase (Sazinsky et al.,
2006) und zwei Acyl-Desaturasen (Lindqvist & Huang, 1996; Dyer et al., 2005) gefunden. Eine
Auflistung dieser Strukturverwandten mitsamt den Z-Score-Werten und der Anzahl der über­
lagerten Reste, die aus der DALI-Suche resultierten, findet sich in Tabelle 25.
128
Ergebnisse zur N-Oxygenase AurF
Tabelle 25:
PDB-Eintraga)
Auszug der Ergebnisse aus der Struktur-Homologie-Suche mit dem Programm DALI
Z-Score
Anzahl überlagerter Aminosäurereste
RMSD [Å]
Sequence Identity [%]
1W69 (A)
15.5
232
3.5
13
1RIB (A)
14.1
239
3.9
11
1MHY (D)
14.1
231
3.8
10
1MTY (C)
13.2
240
4.3
8
2NIP (A)
12.1
222
3.5
10
1ZAO (A)
11.6
189
3.6
14
1TOS (A)
10.9
221
3.8
12
1AFR (A)
10.7
215
4.2
12
a)
Es wurden die PDB-Einträge in der Proteinstrukturdatenbank verwendet. Die Überlagerung erfolgte auf die
Polypeptidkette A der N-Oxygenase mit den in den Klammern aufgeführten Ketten des jeweiligen Strukturverwandten. Diese sind mit folgenden Strukturen gleichzusetzen:
1W69:
1RIB:
1MHY:
1MTY:
2NIP:
1ZAO:
1T0S:
1AFR:
Ribonukleotid Reduktase Komponente R2 aus Maus
Ribonukleotid Reduktase Komponente R2 aus E.coli
Methan Monooxygenase-Hydroxylase aus Methylosinus trichosporium OB3b
Methan Monooxygenase-Hydroxylase aus Methylococcus Capsulatus
Phenol-Hydroxylase-Regulator-Proteinkomplex aus Pseudomonas spezies OX1
Acyl-ACP Desaturase DesA2 aus Mycobacterium tuberculosis H37Rv
Toluol/o-Xylol-Monooxygenase Hydroxylase aus Pseudomonas stutzeri OX1
Acyl-ACP Desaturase aus Rizinussamen
Das gemeinsame Merkmal aller gefundenen Strukturverwandten reduziert sich allerdings auf
das Vorhandensein des zentralen Helix-Bündels, welches das zweikernige Metallzentrum ein­
schließt. Abbildung 61 zeigt ein strukturbasiertes Sequenzalignment im Bereich des charakteris­
tischen vier Helix-Bündels. Aus diesem geht nochmals überzeugend die geringe Sequenzidentität
der Strukturverwandten hervor, obwohl diese dennoch ein nahezu identisches Metall-BindungsMotiv aufweisen. Die koordinierenden Aminosäuren sind nicht nur innerhalb der eigenen Enzym­
familie streng konserviert (Resultat einer Homologie-Suche mit BLAST), sondern zeigen auch
untereinander keine Abweichungen. Alle strukturverwandten Enzyme dieser Klasse weisen als
konserviertes Bindemotiv des Metallclusters einen (D/E)-x2-H-xn-(D/E)-x2-H fingerprint auf, wobei
beide (D/E)-x2-H-Motive in einem Abstand von bis zu 100 Aminosäureresten zu finden sind. Für
AurF findet sich ein Abstand der beiden E-x2-H-Motive mit n = 87. Einzige Ausnahme im koordi­
nierenden Metall-Bindungs-Motiv ist der Rest His223, der keine vergleichbare Aminosäure an
entsprechender Position der überlagerten Strukturen findet. Eine detaillierte Betrachtung in Bezug
auf His223 wird in Abschnitt 4.2.8 anhand der Diskussion des aktiven Zentrum und insbesondere in
Bezug auf die Metallidentiät des Cofaktors der N-Oxygenase gegeben.
129
Ergebnisse und Diskussion
AurF
1W69
1T0S
1MTY
1AFR
•
•
α3
•
•
•
•
α4 •
•
lassssssssssssssssssssssssssssssssssdllllllllassssssssssssssssssssssssssssssdl
EPHQRQRVLTGMWIGYNERVIATEQLIAEPAFDLVMHGVFPGSDDPLIRKSVQQAIVDESFHTYMHMLAIDRTRELRK
ERHFISHVLAFFAASDGIVNENLVERFSQE 153
TEARCFYGFQIAMENIHSEMYSLLIDTYIK
WVSTMQLHFGAIALEEYA-ASTAEARMAR 117
PGNRNMATFGMMDENRHGQIQLYFPYANVK
WNETMKVVSNFLEVGEYN-AIAATGMLWDS 127
AEQKNGYLAQVLDEIRHTHQCAYVNYYFAKNQ
DDYFVVLVGDMITEE
88
YSWAIWTRAWTAEENRHGDLLNKYLYLSG
AurF
1W69
1T0S
1MTY
1AFR
•
•
α7 •
•
•
α7 •
•
lassssssssssss ssss s sssssssssdlllllasssssssssssssssssssssssssssssdll
EQWERDIAVLVWGA-VAET-C-INALLALLARDATIQPMHSLITTLHLRDETAHGSIVVEVVRELYARMNEQ
RVVAFA-AVEGIFFSGSFASIFW 246 PGLTFSNELISRDEGLHCDFACLMFKHL
VSIMLTFAFETGF-TNMQFLGLAA 210 HTFASLISSIQTDESRHAQQGGPSLKILVEN
VECSLNLQLVGEA-CFTNPLIVAV
219 EITPTVFLSIETDELRHMANGYQTVVSI-AN
YLGFIYTS-FQER-A-TFISHGNTAR 209 IKLAQICGTIAADEKRHETAYTKIVEKLFEI
155
186
150
162
158
249
281
248
259
246
Abbildung 61: Strukturbasiertes Sequenzalignment der strukturell verwandten Proteine (siehe Tabelle 25) auf das charakteristische Helix-Bündel. Ein Alignment abseits dieser Helices konnte nicht vorgenommen werden.
Die erste Zeile markiert jede 10. Aminosäure durch einen Punkt, zudem sind die Helices wie in
Abbildung 56 nach ihrer Sekundärstruktur bezeichnet. Die zweite Zeile stellt die Grenzen der Sekundärstrukturelemente dar. Zeile drei zeigt die Sequenz von AurF. Aminosäuren, die mit AurF innerhalb eines Cα-Abstandes von 3 Å überlagert werden konnten sind grau markiert bzw. bei mehr als 3 Å
nicht hinterlegt. Die metallkoordinierenden Aminosäuren sind schwarz gezeigt.
Das Struktur-basierte Sequenzalignment ging aus dem Programm LSQMAN hervor, das ein
optimiertes Überlagern zweier Strukturen aufeinander erlaubt. Zudem erfolgte eine visuelle Kon­
trolle des automatisiert erhaltenen Resultats unter Verwendung des Programmes COOT. Eine Über­
lagerung in anderen Bereichen jenseits des Helix-Bündels konnte nicht in angemessener Weise
erstellt werden. Die verbleibenden Strukturelemente unterscheiden sich ebenso wie der Oligo­
merisierungszustand deutlich von AurF. Allerdings lieferte die Überlagerung einen zusätzlichen
Hinweis auf den Zugang zum aktiven Zentrum des Enzyms. Alle aufgeführten strukturell ver­
glichenen Enzyme sind zu einem Halbraum des Metallzentrum geöffnet. Insbesondere konnte dieser
Hinweis anhand einer Substratstruktur der Toluol-Hydroxylase (Sazinsky et al., 2004) verifiziert
werden.
4.2.8
Das aktive Zentrum der N-Oxygenase
Das Metallzentrum
Wie bereits beschrieben wird das aktive Zentrum durch ein zweikerniges Metallcluster
definiert. Die Elektronendichte innerhalb des aktiven Zentrum ist klar definiert und erlaubt eine
exakte Positionierung der koordinierenden Aminosäuren E101, E136, H139, E196, H223, E227 und
H230 (Abbildung 62). Ein Halbraum des clusters ist zu einer kleinen Kavität hin geöffnet, die
ausreichend Platz für wenige Wassermoleküle bietet. Wie bereits diskutiert, zeigen die beiden
Untereinheiten keine Abweichungen in Bezug auf ihre Polypeptidstruktur. Auch zeigen beide
Metall-zentren keinen signifikanten Unterschied zueinander (Abbildung 62). Ein bemerkenswerter
Unterschied findet sich allerdings in der Lösungsmittelstruktur innerhalb der beiden aktiven
130
Ergebnisse zur N-Oxygenase AurF
Zentren. In Untereinheit B können zwei Wassermoleküle zweifelsfrei in die Elektronendichte
positioniert werden, während in Untereinheit A eine ellipsoide Elektronendichte resultiert, die das
Setzen eines einzelnen Wassermoleküls nicht erlaubt. Daher wurden hier zwei Wassermoleküle mit
jeweils halber Besetzung verfeinert. Zudem wurde die Struktur testweise mit einem am aktiven
Zentrum der Untereinheit fixierten Sauerstoffmolekül verfeinert. Dies resultierte zwar in einer
vergleichsweise geringeren Differenz-Elektronendichte, doch kann die Identifikation eines O2Moleküls bei der erhaltenen Auflösung von 2.1 Å nicht gesichert beschrieben werden. Das aktive
Zentrum der Kette A wurde daher mit zwei Wassermolekülen halber Besetzung belegt, lässt aber
dennoch Interpretationsspielraum für die Anbindung des Substrates Sauerstoff und die Geometrie
dieser Koordination.
Abbildung 62:
Das aktive Zentrum in der ball-and-stick-Darstellung. Gezeigt ist die (2Fo-Fc)-Elektronendichte
(grün) bei einem Konturniveau mit 2.5 σ und die (Fo-Fc)- Elektronendichte (braun) bei einem Konturniveau mit 3.0 σ dargestellt, die aus der Verfeinerung der Proteinstruktur in Abwesenheit der
Wassermoleküle im aktiven Zentrum resultierte.
Abbildung 63 zeigt die Koordinationsgeometrie der Metalle sowie die Abstände zu den Ko­
ordinationspartnern in der Untereinheiten A. Außerdem sind diese für die Untereinheit A nach
Verfeinerung mit einem möglicherweise gebundenen O2-Molekül gezeigt. Aus diesen Abbildungen
geht hervor, dass beide Metallatome eine unterschiedliche erste Koordinationsphäre aufweisen und
somit asymmetrisch koordiniert sind. Während das mit Nummer eins bezeichnete Metall pseudooktaedrisch koordiniert ist, kann am zweiten Metall eine pseudo-trigonal-bipyramidale Umgebung
gefunden werden. Alle Sauerstoff-Mangan- und Stickstoff-Mangan-Distanzen der Proteinliganden
zeigen übliche Werte von etwa 2.0 bis 2.2 Å. Innerhalb der Lösungsmittelstruktur befindet sich das
an beide Metalle koordinierte Wasser der ersten Koordinationssphäre in einem Abstand von etwa
2.6 Å zu den Metallen (respektive 2.6 Å und 2.0 Å für die beiden halbbesetzten H2O-Moleküle der
Untereinheit A). Das zweite Wasser ist demgegenüber nochmals weitere 2.5 Å entfernt positioniert
und wird lediglich über eine Wasserstoffbrücke zur Säurefunktion des Glu196 im aktiven Zentrum
131
Ergebnisse und Diskussion
fixiert. Der Temperaturfaktor aus dem sich die Beweglichkeit des Moleküls ableiten lässt, zeigt
demzufolge einen deutlich höheren Wert von 50 Å2 für dieses Wasser, was mit dessen loser
Bindung korrespondiert.
Abbildung 63:
Geometrie der Metallkoordination. Es sind lediglich die koordinierenden Atome der Aminosäuren
am Metallzentrum mitsamt den Bindungsabständen gezeigt mit Ausnahme der beiden verbrückenden Glutamate E136 und E227. A) zeigt die Koordination des Metallzentrum in der Untereinheit A,
die sich durch zwei halbbesetzte Wassermoleküle innerhalb der ersten Koordinationssphäre von
Untereinheit B unterscheidet, in der sich an entsprechender Position ein einzelnes, vollbesetztes
Wassermolekül findet. Der Metall-Metall-Abstand beträgt 3.56 Å in Untereinheit A bzw. 3.65 Å in
Untereinheit B. B) zeigt das entsprechende Schaubild nach Verfeinerung gegen ein putativ gebundenes Sauerstoffmolekül. Zudem sind die resultierenden Differenz-Elektronendichtekarten bei einem
Konturniveau von 3 σ mit zwei halbbesetzten Wassermolekülen (blau) und mit einem Sauerstoffmolekül (grün) gezeigt.
Unterschiede zu den strukturverwandten Proteinen
Wie bereits beschrieben, beschränken sich die Gemeinsamkeiten der strukturellen Homologen
der N-Oxygenase auf das zentrale Helix-Bündel, das in guter Übereinstimmung für die verwandten
Enzyme mit AurF überlagert werden konnte. Der zentrale Unterschied gegenüber den strukturverwandten Proteinen ist in der Identität des koordinierten Metalls begründet. Alle Enzyme, die zu
AurF strukturelle Homologie zeigen, wurden als Eisen-abhängige Enzyme postuliert oder explizit
132
Ergebnisse zur N-Oxygenase AurF
nachgewiesen. Für AurF konnte zweifelsfrei sowohl durch ESR-Spektroskopie als auch durch
Röntgenkristallographie gezeigt werden, dass der überwiegende Anteil gebundenen Metalls mit
etwa 85% Mangan zuzuordnen ist (siehe Abschnitt 4.2.5). Allerdings kann nicht exakt geklärt
werden, ob die Einlagerung an Mangan ein Artefakt der heterologen Proteinproduktion in E.coli ist,
oder ob es sich im Fall der N-Oxygenase um eine neue Unterklasse der O2-aktivierenden Enzyme
der RNR-Familie Klasse II handelt, die sich durch eine Mangan-abhängige Arbeitsweise
auszeichnet. Es ist also zu klären, welche der möglichen Metallspezies die katalytische Aktivität
verursacht.
Obwohl die Röntgenstrukturanalyse eindeutig die Metallidentität, die Positionen der jeweili­
gen Metallspezies und auch deren stöchiometrisches Verhältnis im zur Kristallisation eingesetzten
AurF bestimmen konnte, kann die Frage nach der aktiven Form des Enzyms durch diese Methode
nicht zweifelsfrei geklärt werden. Der naheliegende Nachweis, der durch eine vergleichende Aktivi­
tätsmessung zweier Proteine zu führen wäre, die vollständig mit einer einzigen Metallspezies be­
setzt sind (also AurF-Mn2, bzw. AurF-Fe2), konnte nicht erbracht werden, da es nicht gelang, AurF
im Bezug auf den Metallgehalt rein zu produzieren. Auch führte der Versuch den Metallgehalt
durch gezielte Inkorporation mit einer Metallsorte zu beeinflussen, nicht zum Erfolg. Alle Versuche
das Protein von seinen Metallen zu befreien und durch andere gezielt zu ersetzen, resultierten in der
Aggregation der N-Oxygenase. Auf Basis des Bindungsmotivs (D/E)-x2-H-xn-(D/E)-x2-H sowie
Aktivitäts- und ESR-Messungen wurde die N-Oxygenase als Eisen-abhängiges Enzym klassifiziert
(Simurdiak et al., 2006). Diese Arbeiten, in denen AurF als N-terminales His6-Fusionsprotein in
sehr geringen Mengen produziert werden konnte, stehen allerdings zu großen Teilen im Wider­
spruch zu den Ergebnissen, die im Laufe dieser Arbeit erhalten wurden. Aus den unternommenen
Untersuchungen kann zusammenfassend festgestellt werden:
1. Die N-Oxygenase inkorporiert sowohl im hier verwendeten Expressionssystem als auch im
Expressionssystem nach Simurdiak et al. (2006) mit hoher Präferenz Mangan. Das für diese
Arbeite benutzte System zur Proteinproduktion erbrachte dabei ein Mangan-zu-Eisen-Verhältnis
von etwa 8:1. Unter Berücksichtigung des dreifachen Überschusses an Eisen im Kultivierungs­
medium resultiert eine etwa 20-fache Präferenz der Mangan-Inkorporation gegenüber Eisen.
2. Die produzierte N-Oxygenase ist aktiv. Dies konnte sowohl in vivo als auch in vitro durch
Messung der Enzymaktivität nachgewiesen werden (Winkler et al., 2006). Für das zur
Kristallisation eingesetzte Enzym erbrachte der in vitro-Assay eine spezifische Aktivität von
0.0032 U/mg bei 23 °C, in vivo betrug die spezifische Aktivität bei 30 °C 0.036 U/mg
(persönliche Mitteilung Robert Winkler, 2007). Die sehr geringen Aktivitäten decken sich mit
den gemessenen Aktivitäten der zweiten bisher charakterisierten N-Oxygenase PrnD (Lee &
133
Ergebnisse und Diskussion
Zhao, 2006), deren spezifische Aktivität AurF vergleichbar ist. Der Unterschied in der in vivound in vitro-Aktivität kann sowohl durch die deutlich reduzierte Reaktionstemperatur als auch
durch die speziellen Reaktionbedingungen erklärt werden, die von den natürlichen Konditionen
abweichen. Als Substrat fand beispielsweise H2O2 Verwendung, während die natürliche vom
Enzym katalysierte Reaktion Sauerstoff umsetzt. Ein Vergleich der Enzymaktivität mit der von
Simurdiak et al. (2006) produzierten N- Oxygenase, die mit einem drastischen Überschuss an
Eisen kultiviert wurde und ebenfalls Aktivität zeigte konnte nicht erfolgen, da hier keine
quantitativen Aussagen zur Enzymaktivität gemacht wurden.
Tabelle 26: Kultivierungsserie von AurF unter Verwendung verschiedener Mangan-zu-Eisen-Verhältnisse a)
Eisen : Mangan
molares Verhältnis
Relative Aktivität (%)
nach Kultivierung über 5 h
Relative Aktivität (%)
nach Kultivierung über 24 h
48
:
1
170±50
90±50
12
:
1
120±50
105±50
3
:
1
100
100
3
:
4
120±50
180±50
3
:
16
160±50
170±50
b)
a)
In Anbetracht er großen Fehler des in vivo-Assays kann zusammenfassend nur eine in etwa gleichbleibende Aktivität des Enzyms auch unter der Zufuhr großer Eisenmengen festgestellt werden.
b)
Diese Metallverhältnisse führten zum Protein, das zur Kristallisation eingesetzt wurde und einen
Mangan-zu-Eisen-Gehalt von etwa 8:1 aufweist.
3. Der Versuch, durch unterschiedliche Mangan-zu-Eisen-Verhältnisse im Kulturmedium einen
Einfluss auf die Enzymaktivität nachzuweisen, schlug fehl. Tabelle 26 zeigt die erhaltenen
Ergebnisse für Kultivierungsserien bei molaren Mangan-zu-Eisen-Verhältnissen von 16:3 bis zu
1:48 (persönliche Mitteilung Robert Winkler, 2007). Trotz der drastisch veränderten Metallver­
hältnisse konnte kein Einfluss auf die Enzymaktivität festgestellt werden. Erfolgt die Protein­
produktion durch eine fünfstündige Kultivierung mit anschließender Bestimmung der Enzym­
aktivität, so kann sowohl ein Anstieg der Aktivität mit erhöhte Eisen- als auch mit erhöhtem
Mangan-Gehalt festgestellt werden. Eine 24-stündige Kultivierung resultiert demgegenüber in
einer ansteigenden Aktvität mit zunehmenden Mangan-Gehalt. Unter Berücksichtigung der
großen relativen Unsicherheit des in-vivo-assays, wurde der Fehler der angegeben Daten zu
50% in Bezug auf die Standard-bedingungen abgeschätzt, was obige Aussagen relativiert.
Allerdings bleibt festzuhalten, dass eine erhöhte Eisenzufuhr in das Kulturmedium mit einem
bis zu 50-fachen Überschuss an Eisen nicht in einer erhöhten Aktivität resultierte. Dies ist um so
erstaunlicher, als dass ein derartiger Einfluss beschrieben wurde (Simurdiak et al., 2006). Ein in
134
Ergebnisse zur N-Oxygenase AurF
LB-Medium produziertes Protein zeigte demzufolge keine messbare Aktivität, die allerdings
nach Produktion des Proteins in Gegenwart von 100 mM Fe3+ nachgewiesen werden konnte. Da
genaue Angaben in Bezug auf die erhaltene Aktvität nicht gemacht wurden, können hieraus
keine weiteren Rückschlüsse gezogen werden.
4. Die ESR-Messungen geben Aufschluss über den katalytischen Zyklus der enzymatischen in
vitro-Aktivität (Abschnitt 4.2.5). Während in Abwesenheit von H2O2 und Substrat die
röntgenographischen Resultate bezüglich der Metallgehalte bestätigt werden, zeigt sich in
Anwesenheit von Wasserstoffperoxid die Ausbildung eines Radikals am Mangan-Zentrum, das
wiederum in Gegenwart des Substrates von diesem abstrahiert wird. Dies steht im Einklang mit
einem möglichen enzymatischen Katalyse-Zyklus, der zunächst durch Koordi-nation und
Spaltung des H2O2-Moleküls die Bildung einer radikalischen Spezies begünstigt, die im
folgenden auf das Substrat übertragen wird. Da die beobachtete Reaktion allerdings nicht den
nativen Katalyse-Bedingungen entspricht und zudem in Anwesenheit von H2O2 auch ein
ausgeprägteres Eisensignal zu verzeichnen ist, sind diese Ergebnisse allein nicht hineichend, um
eine Mangan-abhängige Funktionsweise zu beweisen.
Zusammenfassend geben diese Resultate begründeten Zweifel an einem postulierten
zweikernigen Eisen-Kofaktor und lassen stattdessen eine Mangan-abhängige Enzymreaktion
möglich erscheinen. Jedoch wurden alle strukturverwandten Proteine als Di-Eisen-Enzyme
postuliert. Da AurF und seine Strukturverwandten allerdings nahezu ein identisches Bindungsmotiv
am Metallzentrum aufweisen, muss die These einer mangan-abhängigen Reaktion auch eine
Erklärung für die bevorzugte Inkorporation des Mangans und dessen katalytische Funktion im
Vergleich zu bisherigen Strukturen liefern können. Die Frage nach dem Unterschied im Bindungs­
motiv zwischen den eisenabhängigen Strukturverwandten und der N-Oxygenase kann offensichtlich
in der ersten Koordinationsphäre gefunden werden. Demzufolge besitzt die N- Oxygenase einen
zusätzlichen Proteinliganden in Form von His223, während keines der strukturell homologen
Proteine einen entsprechenden Rest aufweisen kann (Abbildung 64). Die freie Koordinationstelle
wird in diesen Enzymen meist von einem Wassermolekül besetzt. Die zu His223 äquivalente
Position wird innerhalb aller strukturell verwandter Proteine von einen hydrophoben Rest in Form
eines Ile, Val oder in seltenen Fällen eines Ala besetzt. All diese Reste sind aufgrund ihrer
hydrophoben Funktion nicht in der Lage, an einer Metall-Koordination mitzuwirken.
Eine Datenbanksuche mit dem Programm BLAST zeigte für jedes zu AurF untersuchten,
strukturell verwandten Proteins (Tabelle 25), dass innerhalb der 15 nächsten homologen Strukturen
aus verschiedenen Organismen die zu His223 korrespondierende Aminosäure streng bzw. stark
135
Ergebnisse und Diskussion
konserviert ist und ausschließlich von hydrophoben Aminosäureresten belegt wird. Zudem zeigte
diese Datenbanksuche, dass auch der Rest His223 in allen gelisteten hypothetischen Proteinen, die
Sequenz-Homologie zu AurF besitzen, streng konserviert ist, was wiederum die Schlussfolgerung
erlaubt, dass dieser Rest His223 ein charakteristisches Merkmal der N-Oxygenase darstellt.
Abbildung 64:
Exemplarische Überlagerung der strukturverwandten Proteine mit AurF. In blau ist die Struktur der
N-Oxygenase dargestellt, in orange die R2-Komponente der Ribonukleotid Reduktase aus Maus. Zur
vereinfachten Darstellung wurde auf eine vollständige Überlagerung aller koordinierenden
Aminosäurereste von allen strukturell homologen Enzymen verzichtet und zusätzlich nur die zu
His223 korrespondierende Aminosäure der Methan-Monooxygenase-Hydroxylase aus Methylococcus capsulatus (grün), der Toluol/o-Xylol-Monooxygenase-Hydroxylase aus Pseudomonas stutzeri
OX1 (cyan) und der ACP-Desaturase aus Rizinussamen (grau) dargestellt. Aus dieser Abbildung geht
die besondere Bedeutung dieses koordinierenden Restes His223 hervor.
Dass große Effekte auf die Metallspezifität von Metalloproteinen schon bei geringen
Änderungen der Koordinationssphäre des Metalls zu erwarten sind, konnte sehr anschaulich am
Beispiel der Superoxid-Dismutasen gezeigt werden. Anzumerken ist, dass diese Proteinfamilie
weder Sequenzverwandtschaft noch strukturelle Verwandtschaft zur N-Oxygenase aufweist. Die
Superoxid-Dismutasen fungieren als biologische Radikalfänger, die die Disproportionierung von
Superoxiden katalysieren. Innerhalb dieser Enzymfamilie werden vier Klassen unterschieden, die
sich durch die Art des zur Katalyse verwendeten Metalls und ihrer Faltung differenzieren. Zwei
Klassen weisen allerdings hohe strukturelle Homologie auf und werden lediglich anhand des
einkernigen Metallkofaktors differenziert. Dies sind die Mangan-abhängigen Superoxid Dismutasen
(Mn-SOD) bzw. die Eisen-abhängigen Superoxid Dismutasen (Fe-SOD), die nur Aktivität auf­
weisen, wenn das entsprechende Metall im Enzym gebunden wurde.
Ein bemerkenswertes Beipiel für den großen Effekt auf die Spezifität der Metall-Inkorporation
in Bezug auf die Koordinationssphäre des untersuchten Enzyms findet sich in der Literatur. Es
konnte gezeigt werden, dass durch Mutation eines einzelnen Restes die Spezifität der Metall-
136
Ergebnisse zur N-Oxygenase AurF
Einlagerung nahezu vollständig umgekehrt werden konnte (Edward et al., 1998; Whittaker &
Whittaker, 1998). Im vorliegenden Fall der Mn-SOD wurde eine hohe Spezifität für Mangan bei
Kultivierung in LB-Medium gefunden, die durch Austausch eines Glutamats (Glu170), das ein
metall-koordinierendes Histidin (His171) in der Struktur fixiert, zu hoher Eisen-Spezifität
umgekehrt werden konnte. Abbildung 65 zeigt einen Ausschnitt der Struktur. Die Mutation E170A
hat dabei unmittelbaren Einfluss auf die Koordinationssphäre des Metallzentrums, und zeigt daher
eindrucksvoll, dass die Metallspezifität eines Enzyms schon durch kleine Strukturänderungen stark
beeinflusst werden kann.
Abbildung 65: Darstellung eines Ausschnitts der dimeren Mangan-abhängigen Superoxid Dismutase aus E. coli (Bild
in Anlehnung an Whittaker & Whittaker (1998)). Die beiden Untereinheiten des Dimers sind in grün,
respektive cyan gezeigt. Der Austausch des His171-fixierenden Glu170 zu Alanin bewirkt die Destabiliserung der Konformation des His171 im aktiven Zentrum der anderen Untereinheit und hat eine
nahezu vollständige Umkehr der Spezifität der Metall-Inkorporation unter sonst identischen Kultivierungsbedingungen zur Folge.
Modell der Substratbindung
Neben der Struktur eines Proteins ist dessen Komplexstruktur mit dem korrespondierenden
Substrat bzw. Substraten ein wichtiges Ziel der Strukturaufklärung, da nur die geometrische
Anordnung der Edukte und/oder Produkte Rückschlüsse auf die genauen geometrischen Verhält­
nisse und stabilisierenden Effekte innerhalb des Katalysezyklus eines Enzyms zulassen. Es wurden
mannigfaltige Versuche unternommen, derartige Komplexstrukturen zu erzeugen. Unter anderem
wurden hierzu neun verschiedene Substrate bzw. substratähnliche Verbindungen getestet
(Abbildung 66), die zusammen mit der N-Oxygenase unter Verwendung verschiedener Kristallisa­
tionstechniken in Form von Tränkexperimenten, Kokristallisation und Cokristallisation mit vor­
heriger Inkubation der AurF bei hohen Temperaturen (40 °C) zu Substratkristallen führen sollten.
Es konnten verschiedene Kristallisationbedingungen erhalten und verfeinert werden. Auch war es
zum Teil möglich, diese Kristalle röntgenographisch zu vermessen, allerdings zeigte die Auswer­
tung der neun aufgenommenen Datensätze für keinen eine Elektronendichte innerhalb der Protein­
struktur, die auf eines der eingesetzten Substrate zurückzuführen gewesen wäre.
137
Ergebnisse und Diskussion
O
O
O
OH
H2N
SO3H
O
NH2
PNBA
OH
H2N
H2N
O 2N
PABA
OH
OH
p-Aminosulfonsäure
O
2,3-Diaminobenzoesäure
SO3H
CH3
O
OH
H3C
OH
H2N
p-Aminophenylessigsäure
Abbildung 66:
Benzoeäure
Sulfonsäure
3-Methylbutansäure
Verwendete Substrate bzw. Substrat-ähnliche Verbindungen zur Herstellung von Protein-SubstratKomplexen durch Tränkexperimente bzw. Kokristallisationsversuche.
Da keine Kristalle des Proteins im Komplex seinem Substrat oder einer analogen Verbindung
erhalten werden konnten, wurde das Substrat im folgenden modelliert (Abbildung 67). Das resul­
tierende Modell wurde anhand von Mutagenese-Studien in Kombination mit Aktivitäts-messungen
verifiziert. Das zweikernige Metallzentrum zeigt eine kleine Kavität. Ausgehend von dieser Kavität,
konnte ein mit Lösungsmittel gefüllter Kanal ausgemacht werden, der von der Oberfläche des
Proteins bis zum Metallzentrum reicht. In beiden Protomeren wird dieser Kanal durch ein Arginin
blockiert, so dass das aktive Zentrum nicht direkt zugänglich ist. Diese Blockade scheint unter
Berücksichtigung der während der Reaktion auftretenden, reaktiven, oxidierenden Spezies eine
Schutzfunktion darzustellen, die eine Übertragung derartiger Radikale nur auf das Substrat
innerhalb des aktiven Zentrums erlaubt. Zudem findet sich im Rest Arg96 eine positiv geladene
Kopfgruppe, die eine Stabilisierung der unter physiologischen Bedingungen negativen Ladung der
Carboxylatgruppe des natürlichen Substrats Benzoesäure ermöglicht. Im postulierten Modell der
Substratbindung nimmt dieser Rest eine zentrale Rolle ein, indem das Arginin das Benzoat in der
Kavität oberhalb des zweikernigen Metallclusters fixiert. Um das Substrat in diese Kavität einzu­
passen, waren lediglich geringe Änderungen an den Seitenkettenkonformationen der Reste Arg96,
Thr100, Leu202 und Phe264 notwendig. Alle Änderungen resultierten entweder in einer weiteren
Vorzugskonformation der Seitenkette oder zumindest in energetisch erlaubten Geometrien dieser
Aminosäurereste. Die kleinsten Abstände zum Substrat betrugen im finalen Modell 3.2 Å, was für
van-der-Waals-Kontakte angemessen ist. Auf der Basis dieses Modells wurden die in Tabelle 27
aufgeführten Mutanten geplant und deren Aktivität bestimmt. Die Präparation der Mutanten und
auch die Aktivitätsmessung wurde von Robert Winkler (Hans-Knöll Institut, Jena) durchgeführt.
138
Ergebnisse zur N-Oxygenase AurF
Abbildung 67:
Tabelle 27:
Modell der Substratbindung in der Stereoansicht im aktiven Zentrum der Untereinheit A. Das modellierte Substrat ist in grün gezeigt. Die in grau gezeigte Oberfläche wurde für die Mutante R96A
berechnet, um den Kanalgang zu verdeutlichen, der sonst aufgrund der Arginin-Seitenkette verschlossen wäre. Die violette Linie zeigt den Zugang von der Proteinoberfläche in den KanalWechselwirkungen der Liganden mit dem Metall sind in Form von roten Strichen dargestellt.
Hydrophobe Kontakte zum Substrat sind als grün gepunktete Linien und Wasserstoffbrücken als
schwarz gepunktete Linien gezeigt. Die mutierten Aminosäuren sind in orange gezeigt, die
Konformation von Leu300 der Untereinheit B ist transparent und die Konformationen der Aminosäuren, die eine Platzierung des Substrats in der Kavität ermöglichen, in grün dargestellt.
Relative Enzymaktivität der Einzelmutanten der N-Oxygenase aus in vivo Messungena)
Aurf-Mutante
Wildtyp
R96A
Relative Enzymaktivität [%]
100
0
T100A
314
T100L
14
L202F
357
F264A
105
L300W
80
a)
Die spezifische Aktivität des Wildtyps von AurF wurde zu 0.036 U/mg bestimmt.
Die durchgeführten Mutagenesestudien bestätigen das aufgestellte Modell der Edukt-Bindung,
in dem das Substrat PABA zwischen die Reste Arg96, Thr100, L202 und Phe264 eingezwängt
wird. Insbesondere zeigt die Mutation R96A die essentielle Bedeutung dieser Aminosäure. Ohne
die stabilisierende Wirkung der positiv geladenen Guanidiniumgruppe konnte keine Aktivität
139
Ergebnisse und Diskussion
beobachtet werden. Auch zeigen die Mutationen am Thr100 mit T100A und T100L die erwarteten
Effekte auf die Enzymaktivität. Eine Verkleinerung der Seitenkette an entsprechender Position
resultiert in einer deutlich erhöhten Katalysegeschwindigkeit um den Faktor drei, demgegenüber
resultiert eine Vergrößerung dieser Seitenkette in einer relativen Aktivität von 14%. Die 3.5-fach
erhöhte Enzymaktivität, die durch die Mutation L202F verursacht wird, kann durch die geringere
sterische Belastung der durch die Aromatizität verursachten planaren C-Atome des Phenylalanin
erklärt werden, die im Vergleich zu den Cδ-Atomen der Leu202 dem Substrat mehr Freiraum für die
Substratbindung gewähren. Die fast gleichbleibende Aktivität der Mutante F264A war auf den
ersten Blick unerwartet, resultiert jedoch daraus, dass der raumfordernde aromatische Ring des
Phenylalanins parallel zum Substrat ausgerichtet ist und das Cβ-Atom beider Reste (Alanin und
Phenylalanin) ein vergleichbares Volumen in Richtung zum Substrat aufweist. Die Mutante L300W
wurde geplant, um den Eintrittskanal von der Proteinoberfläche hin zum aktiven Zentrum zu
verengen. Jedoch war die beobachtete Reduktion der Aktivität marginal, vermutlich aufgrund der
Tatsache, dass die Kette um den Rest L300 hohe Mobilität aufweist, so dass der Indolring des
Tryptophans leicht zur Seite bewegt werden kann und den Zugang zur Kavität freigibt.
Das Modell erklärt zudem die hohe Regioselektivität der Reaktion. Die Oxidation des Stick­
stoffs findet ausschließlich in para-Position statt, ortho- und meta-Aminogruppen werden nicht
oxidiert. Zudem ist durch dieses Modell der geringe bzw. nicht vorhandene Umsatz von
Aminobenzoaten erklärt, die an den meta- und/oder ortho-Positionen zusätzliche Substituenten
tragen. Aufgrund der kleinen Kavität können substituierte p-Aminobenzoesäure-Derivate wegen der
resultierenden sterischen Behinderung nur wesentlich langsamer umgesetzt werden als das
natürliche Substrat. Für eine detaillierte Beschreibung des Substratspektrums der N-Oxygenase wird
auf Winkler et al. (2006) verwiesen. Eine zusammengefasste Form dieser Ergenisse ist in Abschnitt
1.2.4 aufgeführt.
140
Zusammenfassung und Ausblick
5. Zusammenfassung und Ausblick
Die Arbeit befasste sich mit der Strukturaufklärung zweier Enzyme, die bereits in der groß­
industriellen, biotechnologischen Produktion eine Rolle spielen (Cystein-Produktion mit CysM)
bzw. dessen Einsatz für derartige Verfahren geprüft wird (N-Oxidationen mit AurF). Dabei konnten
folgende Ergebnisse erhalten werden:
Zunächst wurde durch Protein-engineering an der O-Acetylserin-Sulfhydrylase CysM aus
Escherichia coli eine neue Kristallform hergestellt. Diese konnte durch Einführen der Oberflächen­
mutante K268A erhalten werden. Diese Kristallform gehört zur Raumgruppe P6522 und enthält ein
einzelnes Protomer in der asymmetrischen Untereinheit. Damit bietet es sich für zukünftige Struk­
turuntersuchungen an. Mit der erzielten Auflösung von 1.33 Å ist diese Struktur die zur Zeit bestaufgelöste Struktur aller Sulfhydrylasen in der Proteindatenbank. In dieser Struktur konnte eine
Konformation gefunden werden, die zweifelsfrei einen Zwischenzustand des Proteins aus den be­
kannten Strukturen der offenen und geschlossenen Form gleichkommt. Da im aktiven Zentrum des
Proteins zusätzlich ein Substrat-ähnliches Citratmolekül gefunden werden konnte, wurde auf der
Basis dieser Bindung ein Modell für den Teilschritt der enzymkatalysierten Reaktion vom Amino­
acrylat zum Produkt aufgestellt. Zusätzlich konnte ein Modell für den nukleophilen Angriff des im
Aktivitätstest verwendeten 5-Mercapto-2-nitrobenzoat aufgestellt werden. Der in einer Diplom­
arbeit etablierte Enzymassay (Wiesand, 2007) bestätigte dieses Modell durch Messung der Aktivität
gezielt hergestellter Mutanten. Dabei konnte die besondere Bedeutung der bereits in vorhergehen­
den Arbeiten (Claus et al., 2005) beschriebenen Aminosäure Arg210 bestätigt werden, die für die
relaxierte Substratspezifität verantwortlich gemacht wurde. Basierend auf den Ergebnissen dieser
Arbeit sind die Grundlagen für eine gezielte Anpassung des Enzyms an weitere unnatürliche
Substrate geschaffen worden. Diese Anpassung sowie die Optimierung der aktiven Bindestelle an
die einsetzbaren Substrate sollten Gegenstand weiterer Untersuchungen sein.
Weiterhin wurde eine neuartige N-Oxygenase aus Streptomyces thioluteus untersucht. Dieses
zu den Monooxygenasen zählende Enzym, das keine Sequenzhomologie zu Proteinen bekannter
Funktion zeigt, katalysiert die hoch regioselektive Oxidation von p-Aminobenzoat zu p- Nitro­
benzoat. Jedoch werden auch Derivate der p-Aminobenzoesäure als Substrate akzeptiert und
machen das Enzym für die biotechnologische Produktion dieser Stoffklasse interessant. Das Protein
konnte kristallisiert und die Struktur durch MAD-Phasierung mit einem cisPlatin-Derivat bis zu
einer Auflösung von 2.1 Å gelöst werden. Die dimere N-Oxygenase besteht aus einer einzelnen
Domäne mit elf α-Helices. Auf der Basis von Strukturvergleichen konnte die Faltung der N-Oxyge­
141
Zusammenfassung und Ausblick
nase den anderer sauerstoffaktivierender, zweikerniger Eisen-Enzyme und letzlich der Ribo­
nukleotid-Reduktase-Familie zugeordnet werden. Die charakteristische Eigenschaft dieser Enzyme
ist ein zentrales vier-Helix-Bündel, das den als Cofaktor fungierenden zweikernigen Metallcluster
beinhaltet. Die Identität des Metalls wurde anhand eines speziellen MAD-Experiments bestimmt
und durch ESR bestätigt. Dabei konnte zweifelsfrei ein Mangan-zu-Eisen-Verhältnis von 8:1 nach­
gewiesen werden. Die hohe Präferenz der N-Oxygenase zu Mangan steht damit im Widerspruch zur
postulierten Eisen-Abhängigkeit des Enzyms. Aufgrund dieser Untersuchungen wurde AurF als
Mangan-abhängiges Enzym postuliert. Als charakteristisches Merkmal dieser neuen Enzymklasse
wurde ein koordinieriendes Histidin identifiziert, das in den zu AurF homologen Proteinen hoch
konserviert ist und in keinem der strukturverwandten Proteine gefunden wurde. Ein Modell der
Substratbindung des Enzyms konnte erstellt und durch Aktivitätsstudien ausgewählter Mutanten
bestätigt werden. Dabei zeigten die mutierten Aminosäuren einen großen Einfluss auf die
Geschwindigkeit der Reaktion und könnten als Ansatzpunkt der gezielten Anpassung des Enzyms
für den biotechnologischen Einsatz dienen. Zudem bieten sich auf der Basis dieser Strukturdaten
weitere Mutationsstudien in Kombination mit Aktivitätmessungen für eine gezielte Optimierung
von AurF an nicht-natürliche Substrate an. Desweiteren bleibt die Mangan-Abhängigkeit der
N- Oxygenase zu bestätigen. Ein wichtiger Ansatzpunkt ist hierbei die Mutation des für AurF
charakteristischen His223, das im Zuge weiterer Untersuchungen zu hydrophoben Resten Ile, Val
und Ala mutiert werden könnte. Diese Reste finden sich an entsprechender Position bei den
strukturverwandten Eisen-abhängigen Enzymen.
142
Literatur
6. Literatur
Abrahams, J. P. & Leslie, A. G. (1996). Methods used in the structure determination of bovine mitochondrial F1
ATPase. Acta Crystallogr. Sect. D 52, 30-42.
Alexander, F. W., Sandmeier, E., Mehta, P. K. & Christen, P. (1994). Evolutionary relationships among pyridoxal5'-phosphate-dependent enzymes. Regio-specific α, β and γ families. Eur. J. Biochem. 219, 953-960.
Altschul, S. F., Madden, T. L., Schaffer, A. A., Zhang, J., Zhang, Z., Miller, W. & Lipman, D. J. (1997). Gapped
BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids. Res. 25, 33893402.
Arima, K., Imanaka, H., Kousaka, M., Fukuda, A. & Tamura, G. (1965). Studies on pyrrolnitrin, a new antibiotic. J.
Antibiot. (Tokyo) 18, 201-204.
Arima, K., Imanaka, H., Kousaka, M., Fukuda, A. & Tamura, G. (1964). Pyrrolnitrin, a new antibiotic substance,
produced by Pseudomonas. Agric. Biol. Chem. 28, 575–576.
Arndt, U. W., Champess, J. N., Phizackerly, R. P. & Wonacott, A. J. (1973). A single-crystal oscillation camera for
large unit cells. J. Appl. Cryst. 6, 457-463.
Barik, S. (1993). Methods in Molecular Biology, Vol. 15: PCR-Protocols: Current Methods Applic., pp. 277-286.
Humana Press Inc., Totowa, NJ.
Benner, S. E., Chothia, C. & Hubbard, T. J. (1997). Population statistics of protein structures: Lessons from structural
classifications. Curr. Opin. Struct. Biol. 7, 369-376.
Berman, H. M., Westbrook, Z., Feng, G., Gilliland, T. N., Bhat, H., Weissig, I. N., Shindyalov, P. E. & Bourne, P. E.
(2000). The Protein Data Bank. Nucleic Acids Res. 28, 235-242.
Biertümpfel, C., Basquin, J., Suck, D. & Sauter, C. (2002). Crystallization of biological macromolecules using agarose
gel. Acta Crystallogr. Sect. D 58, 1657-1659.
Birnboim, H. C. & Doly, J. (1979). A rapid alkaline lysis procedure for screening recombinant plasmid DNA. Nucleic
Acids Res. 7, 1531-1522.
Blow, D. M. & Crick, F. H. C. (1959). The treatment of errors in the isomorphous replacement method. Acta
Crystallogr. Sect. D 12, 794-802.
Bonner, E. R., Cahoon, R. E., Knapke, S. M. & Jez, J. M. (2005). Structural and functional analysis of O-acetyserine
sulfhydrylase from Arabidopsis thaliana. J. Biol. Chem. 280, 38803-38813.
Bragg, W. H. & Bragg, W. L. (1913). The reflection of X-rays by crystals: I. Proc. Roy. Soc. (London) A 88, 428-438.
Bricogne, G., Vonrhein, C., Flensburg, C., Schiltz, M. & Paciorek, W. (2003). Generation, representation and flow of
phase information in structure determination: recent developments in and around SHARP 2.0. Acta
Crystallogr. Sect. D 59, 2023-2030.
Brünger, A. T. (1992). Free R value: A novel statistical quantity for assessing the accuracy of crystal structures. Nature
355, 472-475.
Brünger, A. T. (1993). Assessment of phase accuracy by cross validation: the free R value. Methods and applications.
Acta Crystallogr. Sect. D 49, 24-36.
Brünger, A. T., Adams, P. D., Clore, G. M., DeLano, W. L., Gros, P., Grosse-Kunstleve, R. W., Jiang, J. S.,
Kuszewski, J., Nilges, M., Pannu, N. S., Read, R. J., Rice, L. M., Simonson, T. & Warren, G. L. (1998).
Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta
Crystallogr. Sect. D 54, 905-921.
143
Literatur
Buddha M. R., Tao T., Parry R. J. & Crane B. R. (2004). Regioselective nitration of tryptophan by a complex between
bacterial nitric-oxide synthase and tryptophanyl-tRNA synthetase. J. Biol. Chem. 279, 49567–49570.
Burchard, W. (1985). New aspects of polymer characterization by dynamic light scattering. Chimia 39, 10-18.
Burkhard, P., Rao, G. S. J., Hohenester, E., Schnackerz, K. D., Cook, P. F. & Jansonius, J. N. (1998). Threedimensional structure of O-acetylserine sulfhydrylase from Salmonella typhimurium. J. Mol. Biol. 283, 121133.
Burkhard, P., Tai, C.-H., Ristoph, C. M., Cook, P. F. & Jansonius, J. N. (1999). Ligand binding induces a large
conformational change in O-acetylserine sulfhydrylase from Salmonella typhimurium. J. Mol. Biol. 291, 941953.
Burkhard, P., Tai, C.-H., Jansonius, J. N. & Cook, P. F. (2000). Identification of an allosteric anion-binding site on
O-acetylserine sulfhydrylase: Structure of the enzyme with chloride bound. J. Mol. Biol. 303, 279-286.
Cardillo, R., Fuganti, C., Ghiringhelli, D., Giangrasso, D. & Grasselli, P. (1972). On the biological origin of the
nitroaromatic unit of the antibiotic aureotine. Tetrahedron Lett. 13, 4875–4878.
Carter, C. W., Jr. & Carter, C. W. (1979). Protein crystallization using incomplete factorial experiments. J. Biol. Chem.
254, 12219-12223.
CCP4 (Collaborative Computational Project 4) (1994). The CCP4 suite: programs for protein crystallography. Acta
Crystallogr. Sect. D 50, 760-763.
Chang, C. H., Svedruzic. D., Ozarowski, A., Walker, L., Yeagle, G., Britt, R. D., Angerhofer A. & Richards, N. G. J.
(2004). EPR spectroscopic characterization of the manganese center and a free radical in the oxalate
decarboxylase reaction: Identification of a tyrosyl radical during turnover. J. Biol. Chem. 279, 52840–52849.
Chothia, C. (1992). One thousand families for the molecular biologist. Nature 357, 543-544.
Claus, M. T. (2004). Kristallstrukturen der O-Acetylserin-Sulfhydrylase CysM aus Escherichia coli,der humanen
cytosolischen Thymidin-Kinase sowie eines Komplexes einer viralen Thymidin-Kinase. Dissertation,
Universität Freiburg im Breisgau.
Claus, M. T., Zocher, G. E., Maier, T. H. P. & Schulz, G. E. (2005). Structure of the O-acetylserine sulfhydrylase
isoenzyme CysM from Escherichia coli. Biochemistry 44, 8620-8626.
Cosyns, J.P. (2003). Aristolochic acid and ‘chinese herbs nephropathy’: A review of the evidence to date. Drug Saf. 26,
33–48.
Cowtan, K. (1994). 'dm': An automated procedure for phase improvement by density modification. Joint CCP4 and
ESF-EACBM Newsletter on Protein Crystallography 31, 34-38.
Cowtan, K. (1998). Modified phased translation functions and their application to molecular-fragment location. Acta
Crystallogr. Sect. D 54, 750-756.
Cudney, R., Patel, S., Weisgräber, K., Newhouse, Y. & McPherson, A. (1994). Screening and optimization strategies
for macromolecular crystal growth. Acta Crystallogr. Sect. D 50, 414-423.
Cullis, A. F., Muirhead, H., Perutz, M. F., Rossmann, M. G. & North, A. T. C. (1962). A threedimensional Fourier
synthesis at 5.5 Å resolution. Determination of the phase angles. Proc. Roy. Soc. London Ser. A 265, 15-38.
D’Arcy, A. (1994). Crystallizing proteins – a rational approach? Acta Crystallogr. Sect. D 50, 469-471.
Das, S., Incarvito, C. D., Crabtree, R. H. & Brudvig, G. W. (2006). Molecular recognition in the selective oxygenation
of saturated C-H bonds by a dimanganese catalyst. Science, 312, 1941-1943.
Dauter, Z. & Adamiak, D. A. (2001). Anomalous signal of phosphorus used for phasing DNA oligomer: importance of
data redundancy. Acta Crystallogr. Sect. D 57, 990-995.
144
Literatur
de Bakker, P. I. W., DePristo, M. A., Burke, D. F. & Blundell, T. L. (2003). Ab initio construction of polypeptide
fragments: Accuracy of loop decoy discrimination by an all-atom statistical potential and the AMBER force
field with the Generalized Born solvation model. Proteins 51, 21-40.
de la Fortelle, E. & Bricogne, G. (1997). Maximum-likelihood heavy-atom parameter refinement for multiple isomorphous replacement and multiwavelength anomalous diffraction methods. Methods Enzymol. 276, 472-494.
Debye, P. (1914). Interferenz von Röntgenstrahlen und Wärmebewegung. Annal. Phys. 43, 49-95.
Derewenda, Z. S. (2004). Rational protein crystallisation by mutational surface engineering. Structure 12, 529-535.
Diederichs, K. (2006). Some aspects of quantitative analysis and correction of radiation damage. Acta Crystallogr.
Sect. D 62, 96-101.
Diederichs, K. & Karplus, P. A. (1997). Improved R-factors for diffraction data analysis in macromolecular
crystallography. Nat. Struct. Biol. 4, 269-275.
Drenth, J. (1994). "Principles of protein X-ray crystallography.," Springer-Verlag, New York.
Ducruix, A. & Giegé, R. (1999) Methods of crystallization. In: Crystallization of nucleic acids and proteins. A
practical approach. 2nd ed. (Ducruix, A. & Giegé, R., eds.), pp. 121-147, Oxford University Press, Oxford,
UK.
Dyer, D. H., Lyle, K. S., Rayment, I. & Fox, B. G. (2005). X-ray structure of putative acyl-ACP desaturase DesA2
from Mycobacterium tuberculosis H37Rv. Protein Sci. 14, 1508-1517.
Edward, R. A., Whittaker, M. M., Whittaker, J. W., Jameson, G. B. & Baker, E. N. (1998). Distinct metal environment
in Fe-substituted manganese superoxide dismutase provides a structural basis of metal specificity. J. Am.
Chem. Soc. 120, 9684-9685.
Elango, N., Radhakrishnan, R., Froland, W. A., Wallar, B. J., Earhart, C. A., Lipscomb, J. D. & Ohlendorf, D. H.
(1997). Crystal structure of the hydroxylase component of methane monooxygenase from Methylosinus
trichosporium OB3b. Protein Sci. 6, 556-568.
Emsley, P. & Cowtan, K. (2004). Coot: model-building tools for molecular graphics. Acta Crystallogr. Sect. D 60,
2126-2132.
Engh, R. & Huber, R. (1991). Accurate bond and angle parameters for X-ray protein structure refinement. Acta
Crystallogr. Sect. A 47, 392-400.
Eriksson, M., Jordan, A. & Eklund, H. (1998). Structure of Salmonella typhimurium nrdF ribonucleotide reductase in
its oxidized and reduced forms. Biochemistry 37, 13359-13369.
Evans, P. (2006). Scaling and assessment of data quality. Acta Crystallogr. Sect. D 62, 72-82.
Fenn, T. D., Ringe, D. & Petsko, G. A. (2003). POVScript+: a program for model and data visualization using
persistence of vision ray-tracing. J. Appl. Crystallog. 36, 944-947.
Ferré-D’Amaré, A. R. & Burley, S. K. (1997). Dynamic light scattering in evaluating crystallizability of
macromolecules. Methods Enzymol. 276, 157-166.
Fox, B. G., Shanklin, J., Ai, J., Loehr, T. M. & Sanders-Loehr, J. (1994). Resonance Raman evidence for an Fe-O-Fe
center in stearoyl-ACP desaturase. Primary sequence identity with other diiron-oxo proteins. Biochemistry 33,
12776-12786.
Francois, J. A., Kumaran, S. & Jez, J. M. (2006). Structural basis for interaction of O-acetylserine sulfhydrylase and
serine acetyltransferase in the Arabidopsis cysteine synthase complex. Plant Cell 18, 3647-3655.
Frishman, D. & Argos, P. (1995). Knowledge-based protein secondary structure assignment. Proteins 23, 566-579.
145
Literatur
Gallagher, D. T., Gilliland, G. L., Xiao, G., Zondlo, J., Fisher, K. E., Chinchilla, D. & Eisenstein, E. (1998). Structure
and control of pyridoxal phosphate dependent allosteric threonine deaminase. Structure 6, 465-475.
Garman, E. (1999). Cool data: quantity AND quality. Acta Crystallogr. Sect. D 55, 1641-53.
Gill, S. C. & von Hippel, P. H. (1989). Calculation of protein extinction coefficients from amino acid sequence data.
Anal. Biochem. 182, 319-326.
Gilliland, G. L. & Ladner, J. E. (1996). Crystallization of biological macromolecules for X-ray diffraction studies.
Curr. Opin. Struct. Biol. 6, 595-603.
Glykos, N. M. & Kokkinidis, M. (2000). On the distribution of the bulk-solvent correction parameters. Acta
Crystallogr. Sect. D 56, 1070-1072.
Gräfe, U. (1992). Biochemie der Antibiotika. Spektrum Akademischer Verlag, Heidelberg.
Green, D. W., Ingram, V. M. & Perutz, M. F. (1954). The structure of haemoglobin IV. Sign determination by the
isomorphous replacement method. Proc. Roy. Soc. A 225, 287-307.
Hanahan, D. (1983). Studies on transformations of Escherichia coli with plasmids. J. Mol. Biol. 166, 557-580.
Harker, D. (1956). The determination of phases of the structure factors of noncentrosymmetric crystals by the method
of double isomorphous replacement. Acta Crystallogr. 9, 1-9.
He, J. & Hertweck, C. (2003). Iteration as programmed event during polyketide assembly; molecular analysis of the
aureothin biosynthesis gene cluster. Chem. Biol. 10, 1225-1232.
He, J. & Hertweck, C. (2004). Biosynthetic origin of the rare nitroaryl moiety of the polyketide antibiotic aureothin:
involvement of an unprecedented N-oxygenase. J. Am. Chem. Soc. 126, 3694-3695.
He, J., Magarvey, N., Piraee, M. & Vining, L. C. (2001). The gene cluster for chloramphenicol biosynthesis in
Streptomyces venezuelae ISP5230 includes novel shikimate pathway homologues and a monomodular nonribosomal peptide synthetase gene. Microbiology 147, 2817-2829.
Heine, A., Canaves, J. M., von Delft, F., Brinen, L. S., Dai, X., Deacon A. M., Elsliger, M. A., Eshaghi, S., Floyd, R.,
Godzik, A., Grittini, C., Grzechnik, S. K., Guda, C., Jaroszewski, L., Karlak, C., Klock, H. E., Koesema, E.,
Kovarik, J. S., Kreusch, A., Kuhn, P., Lesley, S. L., McMullen, D., McPhillips, T. M., Miller, M. A., Miller,
M. D., Morse, A., Moy, K., Ouyang, J., Page, R., Robb, A., Rodriguez, K., Schwarzenbacher, R., Selby, T. L.,
Spraggon, G., Stevens, R. C., van den Bedem, H., Velasquez, J., Vincent, J., Wang, X., West, B., Wolf, G.,
Hodgson, K. O., Wooley, J. & Wilson, I. A. (2004). Crystal structure of O-acetylserine sulfhydrylase
(TM0665) from Thermotoga maritima at 1.8 Å resolution. Proteins 56, 387-391.
Hendrickson, W. A. & Teeter, M. M. (1981). Structure of the hydrophobic protein crambin determined directly from
the anomalous scattering of sulphur. Nature 290, 107-113.
Hendrickson, W. A., Horton, J. R. & LeMaster, D. M. (1990). Selenomethionyl proteins produced for analysis by
multiwavelength anomalous diffraction (MAD): a vehicle for direct determination of three-dimensional
structure. EMBO J. 9, 1665-1672.
Hendrickson, W. A., Smith, J. L. & Sheriff, S. (1985). Direct phase determination based on anomalous scattering.
Methods Enzymol. 115, 41-55.
Hidalgo, C., Sevilla, J. M., Pineda, T. & Blazquez, M. (1994). Enolimine and geminaldiamine forms in the reaction of
pyridoxal phosphate with ethylenediamine. An electrochemical and spectroscopic contribution. J. Phys. Org.
Chem. 7, 227-233.
Hirata, Y., Nakata, H., Yamada, K., Okuhara, K. & Naito, T. (1961). The structure of aureothin, a nitro compound
obtained from Streptomyces thioluteus. Tetrahedron 14, 252-274.
146
Literatur
Högbom, M., Huque, Y., Sjöberg, B.-M. & Nordlund, P. (2002). Crystal structure of the di-iron/radical protein of
ribonucleotide reductase from Corynebacterium ammoniagenes. Biochemistry 41, 1381-1389.
Högbom, M., Stenmark, P., Voevodskaya, N., McClarty, G., Gräslund, A. & Nordlund, P. (2004). The radical site in
chlamydial ribonucleotide reductase defines a new R2 subclass. Science 305, 245-247.
Holm, L. & Sander, C. (1993). Protein structure comparison by alignment of distance matrices. J. Mol. Biol. 233, 123138.
Holton, T., Ioerger, T. R., Christopher, J. A. & Sacchettini, J. C. (2000). Determining protein structure from electrondensity maps using pattern matching. Acta Crystallogr. Sect. D 56, 722-734.
Hooft, R. W., Vriend, G., Sander, C. & Abola, E. E. (1996). Errors in protein structures. Nature 381, 272-272.
Horn, U., Strittmatter, W., Krebber, A., Knüpfer, U., Kujau, M., Wenderoth, R., Müller, K., Matzku, S., Plückthun, A.
& Riesenberg, D. (1996). High volumetric yields of functional dimeric miniantibodies in Escherichia coli,
using an optimized expression vector and high-cell density fermentation under non-limited growth conditions.
Appl. Microbiol. Biotechnol. 46, 524-532.
Howard, A. J., Gilliland, G. L., Finzel, B. C., Poulos, T. L., Ohlendorf, D. H. & Salemme, F. R. (1987). The use of an
imaging proportional counter in macromolecular crystallography. J. Appl. Crystallog. 20, 383-387.
Howlin, B., Butler, S. A., Moss, D. S., Harris, G. W. & Driessen, H. P. C. (1993). TLSANL: TLS parameteranalysis
program for segmented anisotropic refinement of macromolecular structures. J. Appl. Crystallog. 26, 622-624.
Huang, B., Vetting, M. W. & Roderick, S. L. (2005). The active site of O-acetylserine sulfhydrylase is the anchor
point for bienzyme complex formation with serine acetyltransferase. J. Bacteriol. 187, 3201-3205.
Hyde, C. C., Ahmed, S. A., Padlan, E. A., Miles, E. W. & Davies, D. R. (1988). Threedimensional structure of the
tryptophan synthase α2β2 multienzyme complex from Salmonella typhimurium. J. Biol. Chem. 263, 1785717871.
International Tables for Crystallography (1996). Vol. A, Space-Group Symmetry. Kluwer Academic Publisher,
Dordrecht.
Ioerger, T. R., Holton, T., Christopher, J. A. & Sacchettini, J. C. (1999). TEXTAL: a pattern recognition system for
interpreting electron density maps. Proc. Int. Conf. Intell. Syst. Mol. Biol., 130-137.
Jancarik, J. & Kim, S. H. (1991). Sparse matrix sampling: a screening method for crystallization of proteins. J. Appl.
Cryst. 24, 409-411.
Jancarik, J., Scott, W. G., Milligan, D. L., Koshland, D. E., Jr. & Kim, S. H. (1991). Crystallization and preliminary
X-ray diffraction study of the ligand-binding domain of the bacterial chemotaxis-mediating aspartate receptor
of Salmonella typhimurium. J. Mol. Biol. 221, 31-34.
Jones, T. A., Zou, J.-Y., Cowan, S. W. & Kjeldgaard, M. (1991). Improved methods for building protein models in
electron density maps and the location of errors in these models. Acta Crystallog. Sect. A 47, 110-119.
Kabsch, W. (1976). A solution for the best rotation to relate two sets of vectors. Acta Crystallog. Sect. A 32, 922-923.
Kabsch, W. (1978). A discussion of the solution for the best rotation to relate two sets of vectors. Acta Crystallog. Sect.
A 34, 827-828.
Kabsch, W. (1988). Evaluation of single-crystal X-ray diffraction from a position-sensitive detector. J. Appl.
Crystallog. 21, 916-924.
Kabsch, W. (1993). Automatic processing of rotation diffraction data from crystals of initially unknown symmetry and
cell constants. J. Appl. Crystallog. 26, 795-800.
147
Literatur
Kabsch, W. & Sander, C. (1983). Dictionary of protein secondary structures: pattern recognition of hydrogen-bonded
and geometrical features. Biopolymers 22, 2577-2637.
Kaldor, S. W., Kalish, V. J., Davies, J. F., Shetty, B. V., Fritz, J. E., Appelt, K., Burgess, J. A., Campanale, K. M.,
Chirgadze, N. Y., Clawson, D. K., Dressman, B. A., Hatch, S. D., Khalil, D. A., Kosa, M. B., Lubbehusen,
P. P., Muesing, M. A., Patick, A. K., Reich, S. H., Su, K. S. & Tatlock, J. H. (1997). Viracept (nelfinavir
mesylate, AG1343): A potent, orally bioavailable inhibitor of HIV-1 Protease. J. Med. Chem. 40, 3979-3985.
Kantardjieff, K. A. & Rupp, B. (2003). Matthews coefficient probabilities: Improved estimates for unit cell contents
of proteins, DNA, and protein-nucleic acid complex crystals. Protein Sci. 12, 1865-1871.
Kers, J. A., Wach, M. J., Krasnoff, S. B., Widom, J., Cameron, K. D., Bukhalid, R. A., Gibson, D. M., Crane, B. R. &
Loria, R. (2004). Nitration of a peptide phytotoxin by bacterial nitric oxide synthase. Nature 429, 79–82.
Keyer K. & Imlay J.A.(1996). Superoxide accelerates DNA damage by elevating free-iron levels. Proc. Natl. Acad.
Sci. USA 93, 13635–13640.
Kieser, T., Bibb, M., Buttner, M. J., Chater, K. F. & Hopwood, D. A. (2000). Practical Streptomyces Genetics. The
John Innes Foundation, Norwich, U.K.
Kirner, S., Hammer, P. E., Hill, D. S., Altmann, A., Fischer, I., Weislo, L. J., Lanahan, M., van Pée, K.-H. & Ligon, J.
M. (1998). Functions encoded by pyrrolnitrin biosynthetic genes from Pseudomonas fluorescens. J. Bacteriol.
180, 1939-1943.
Kleywegt, G. J. (1996). Use of non-crystallographic symmetry in protein structure refinement. Acta Crystallogr. Sect.
D 52, 842-857.
Kleywegt, G. J. & Jones, T. A. (1995). Where freedom is given, liberties are taken. Structure 3, 535-540.
Kleywegt, G. J. & Brünger, A. T. (1996). Checking your imagination: applications of the free R value. Structure 4,
897-904.
Kleywegt, G. J., Henrick, K., Dodson, E. J. & van Aalten, D. M. (2003). Pound-wise but penny-foolish: How well do
micromolecules fare in macromolecular refinement? Structure 11, 1051-1059.
Klinman, J. P. (2001). Life as aerobes: are there simple rules for activation of dioxygen by enzymes? J. Biol. Inorg.
Chem. 6, 1-13.
Kraut, J., Sieker, L. C., High, D. F. & Freer, S. T. (1962). Chymotrypsinogen: a three-dimensional Fourier synthesis
at 5 angstrom resolution. Proc Natl. Acad. Sci. USA 48, 1417-1424.
Kredich, N. M. (1971). Regulation of L-cystein biosynthesis in Salmonella typhimurium. J. Biol. Chem. 246, 34743484.
Kredich, N. M. (1996). Biosynthesis of cysteine. In: Escherichia coli and Salmonella typhimurium. Cellular and
molecular biology. (Neidhard, F. C., Curtiss, R., Ingraham, J. L., Lin, E. C. C., Low, K. B., Magasanik, B.,
Reznikoff, W. S., Riley, M., Schaechter, M. & Umberger E., eds.), pp. 514-527, Washington DC ASM Press,
USA.
Krissinel, E. & Henrick, K. (2004). Secondary-structure matching (SSM), a new tool for fast protein structure
alignment in three dimensions. Acta Crystallogr. Sect. D 60, 2256-2268.
Krissinel, E. & Henrick, K. (2005). Detection of protein asemblies in crystals. In "CompLife 2005." (M. S. Berthold,
ed.), pp. 163-174. Springer-Verlag, Berlin.
Kroemer, M., Dreyer, M. K. & Wendt, K. U. (2004). APRV - a program for automated data processing, refinement
and visualization. Acta Crystallogr. Sect. D 60, 1679-1682.
Laemmli, U. K. (1970). Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature
227, 680-685.
148
Literatur
Lamzin, V. S. & Wilson, K. S. (1993). Automated refinement of protein models. Acta Crystallog. Sect. D 49, 129-149.
Laskoswki, R. A., MacArthur, M. W., Moss, D. S. & Thornton, J. M. (1993). PROCHECK - a programm to check the
stereochemical quality of protein structures. J. Appl. Crystallog. 26, 283-291.
Lee, B. & Richards, F. M. (1971). The interpretation of protein structures: estimation of static accessibility. J. Mol.
Biol. 55, 379-400.
Lee, J. & Zhao, H. (2006). Mechanistic studies on the conversion of arylamines into arylnitro compounds by
aminopyrrolnitrin oxygenase: identification of intermediates and kinetic studies. Angew. Chem. Int. Ed. 45,
622-625.
Lee, J.-K., Ang, E.-L. & Zhao, H. (2006). the substrate specificity of aminopyrrolnitrin oxygenase (PrnD) by
mutational analysis. J. Bacteriol. 188, 6179-6183.
Leslie, A. G. W. (1987). A reciprocal space method for calculation of a molecular envelope using the algorithm of B.
C. Wang. Acta Crystallogr. Sect. A 43, 134-136.
Leslie, A. G. W. (1992). Recent changes to the MOSFLM package for processing film and image plate data. Joint
CCP4 & ESF-EAMCB Newsletter on Protein Crystallography, No. 26.
Lindqvist, Y. & Huang, W. (1996). Crystal structure of D9 stearoyl-acyl carrier protein desaturase from castor seed and
its relationship to other di-iron proteins. EMBO J. 15, 4081-4092.
Liu, Y. D., Gu, Y. X., Zheng, C. D., Hao, Q. & Fan, H. F. (1999). Combining direct methods with isomorphous
replacement or anomalous scattering data. VIII. Phasing experimental SIR data with the replacing atoms in a
centrosymmetric arrangement. Acta Crystallogr. Sect. D 55, 846-848.
Lottspeich, F. & Zorbas, H. (1998). Bioanalytik , Spektrum Akademischer Verlag, Heidelberg, Berlin.
Lovell, S. C., Davis, I. W., Arendall, W. B. III, de Bakker, P. I., Word, J. M., Prisant, M. G., Richardson, J. S. &
Richardson, D. C. (2003). Structure validation by Cα geometry: φ, ψ and Cβ deviation. Proteins 50, 437-450.
Lovell, S. C., Word, J. M., Richardson, J. S. & Richardson, D. C. (2000). The penultimate rotamer library. Proteins
40, 389-408.
Maier, T. H. P. (2003). Semisynthetic production of unnatural L-α-amino acids by metabolic engineering of the cystein
biosynthetic pathway. Nat. Biotechnol. 21, 422-427.
Maiorov, V. N. & Crippen, G. M. (1994). Significance of root-mean square deviation in comparing threedimensional
structures of globular proteins. J. Mol. Biol. 235, 625-634.
Matthews, B. W. (1968). Solvent content of protein crystals. J. Mol. Biol. 33, 491-497.
Matthews, B. W. (1977). X-ray structure of proteins. In "The Proteins" (H. Neurath and R. L. Hill, eds.), pp. 404-590.
Academic Press, New York, USA.
McCoy, A. J., Grosse-Kunstleve, R. W., Storoni, L. C. & Read, R. J. (2005). Likelihood-enhanced fast translation
functions. Acta Crystallogr. Sect. D 61, 458-464.
McPherson, A. (1990). Current approaches to macromolecular crystallization. Eur. J. Biochem. 189, 1-23.
McRee, D. E. (1992). A visual protein crystallographic software system for X11/XView. J. Mol. Graph. 10, 44-46.
McRee, D. E. (1993). Practical Protein Crystallography. Academic Press, San Diego, USA.
McRee, D. E. (1999). XtalView/Xfit--A versatile program for manipulating atomic coordinates and electron density. J.
Struct. Biol. 125, 156-165.
149
Literatur
Mitchell, E. P. & Garman, E. F. (1994). Flash freezing of protein crystals: investigation of mosaic spread and
diffraction limit with variation of cryoprotectant concentration. J. Appl. Crystallog. 27, 1070-1074.
Mullis, K., Faloona, F., Scharf, S., Saiki, R., Horn, G. & Erlich, H. (1986). Specific enzymatic amplification of DNA in
vitro, the polymerase chain reaction. Cold Spring Harbor Symp. Quant. Biol. 51, 263-273.
Murshudov, G. N., Vagin, A. A. & Dodson, E. J. (1997). Refinement of macromolecular structures by the maximumlikelihood method. Acta Crystallogr. Sect. D 53, 240-255.
Nordlund, P. & Eklund, H. (1993). Structure and function of the Escherichia coli ribonucleotide reductase protein R2.
J. Mol. Biol. 232, 123-164.
Painter, J. & Merritt, E. A. (2006). Optimal description of a protein structure in terms of multiple groups undergoing
TLS motion. Acta Crystallogr. Sect. D 62, 439-450.
Patterson, A. L. (1934). A Fourier series method for the determination of the components of interatomic distances in
crystals. Phys. Rev. 46, 372-376.
Perrakis, A., Morris, R. & Lamzin, V. S. (1999). Automated protein model building combined with iterative structure
refinement. Nat. Struct. Biol. 6, 458-463.
Petsko, G. A. (1985). Preparation of isomorphous heavy-atom derivatives. Methods Enzymol. 114, 147-156.
Rabeh, W. M. & Cook, P. F. (2004). Structure and mechanism of O-actylserine sulfhydrylase. J. Biol. Chem. 279,
26803-26806.
Ramachandran, G. N. & Sasisekharan, V. (1968). Conformation of polypeptides and proteins. Adv. Protein Chem. 23,
283-438.
Read, R. J. (1986). Improved Fourier coefficients for maps using phases from partial structures with errors. Acta
Crystallogr. Sect. A 36, 878-884.
Read, R. J. (1990). Structure factor probabilities for related structures. Acta Crystallogr. Sect. A 46, 900-912.
Reynolds, J. A. & Tanford, C. (1970). The gross conformation of protein-sodium dodecyl sulfate complexes. J. Biol.
Chem. 245, 5161-5165.
Rice, L. M. & Brünger, A. T. (1994). Torsion angle dynamics: reduced variable conformational sampling enhances
crystallographic structure refinement. Proteins 19, 277-290.
Richardson, J. S. (1985). Describing patterns of protein tertiary structure. Methods Enzymol. 115, 341-358.
Richardson, J. S. (1985). Schematic drawings of protein structures. Methods Enzymol. 115, 359-380.
Righetti, P. G., Gianazza, E., Gelfi, C. & Chiari, M. (1990). In Gel electrophoresis of proteins: a practical approach
(Hames, B. D. & Rickwood, D., eds.) 2nd ed., pp. 149-214, Oxford University Press, Oxford.
Rosenbaum, G., Holmes, K. G. & Witz, J. (1971). Synchrotron radiation as a source for X-ray diffarction. Nature 230,
434-437.
Rosenzweig, A. C., Brandstetter, H., Whittington, D. A., Nordlund, P., Lippard, S. J. & Frederick, C. A. (1997). Crystal
structures o f the methane monooxygenase hydroxylase from Methylococcus capsulatus (Bath): implications
for substrate gating and component interactions. Proteins 29, 141-152.
Rossmann, M. G. & Blow, D. M. (1962). The detection of sub-units within the crystallographic asymmetric unit.
Acta Crystallogr. Sect. D 15, 24-31.
Rössner E., Zeeck A. & König W.A. (1990). Elucidation of the structure of hormaomycin. Angew. Chem. Int. Ed. Engl.
29, 64–65.
150
Literatur
Saiki, R. K., Gelfandt, D. H., Stoffel, S., Scharf, S. J., Higuchi, R., Horn, G. T., Mullis, K. B. & Ehrlich, H. A. (1988).
Primer-directed enzymatic amplification of DNA with a thermostable DNA polymerase. Science 239, 487491.
Sambrook, J., Fritsch, E. F. & Maniatis, T. (1989). Molecular cloning: A laboratory manual. 2nd ed., Cold Spring
Harbor Laboratory Press, Cold Spring Harbor, USA.
Samples C. R., Howard T., Raushel F. M. & DeRose V. J. (2005). Protonation of the binuclear metal center within the
active site of phosphotriesterase. Biochemistry 44, 11005–11013.
Sander, C. & Schneider, R. (1991). Database of homology-derived protein structures and the structural meaning of
sequence alignment. Proteins 9, 56-68.
Sanger, F., Nicklen, S. & Coulson, A. R. (1977). DNA sequencing with chain-terminating inhibitors. Proc. Natl.
Acad. Sci. USA 74, 5463-5467.
Sanner, M. F., Olson, A. J. & Spehner, J. C. (1996). Reduced surface: an efficient way to compute molecular surfaces.
Biopolymers 38, 305-320.
Sazinsky, M. H., Bard, J., Di Donato, A. & Lippard, S. J. (2004). Crystal structure of the toluene/o-xylene monooxygenase hydroxylase from Pseudomonas stutzeri OX1. J. Biol. Chem. 279, 30600-30610.
Sazinsky, M. H., Dunten, P. W., McCormick, M. S., Di Donato, A. & Lippard, S. J. (2006). X-ray structure of a
hydroxylase – regulatory protein complex from a hydrocarbon-oxidizing multicomponent monooxygenase,
Pseudomonas sp. OX1 phenol hydroxylase. Biochemistry 45, 15392-15404.
Schnackerz, K. D. & Cook, P. F. (1995). Resolution of pyridoxal 5'-phosphate from O-acetylserine sulfhydrylase from
Salmonella typhimurium and reconstitution of apoenzyme with cofactor and cofactor analogues as a probe of
the cofactor binding site. Arch. Biochem. Biophys. 324, 71-77.
Schneider, T. R. (2002). A genetic algorithm for the identification of conformationally invariant regions in protein
molecules. Acta Crystallogr. Sect. D 58, 195-208.
Schneider, T. R. (2004). Domain identification by iterative analysis of error-scaled difference distance matrices. Acta
Crystallogr. Sect. D 60, 2269-2275.
Schneider, T. R. & Sheldrick, G. M. (2002). Substructure solution with SHELXD. Acta Crystallogr. Sect. D 58, 17721779.
Schomaker, V. & Trueblood, K. N. (1968). On the rigid-body motion of molecules in crystals. Acta Crystallogr. Sect.
B 24, 63-76.
Schulz, G. E. (1981). Protein-Differenzierung: Entwicklung neuartiger Proteine im Laufe der Evolution. Angew. Chem.
93, 143-151.
Schüttelkopf, A. W. & van Aalten, D. M. (2004). PRODRG: a tool for high-throughput crystallography of proteinligand complexes. Acta Crystallogr. Sect. D 60, 1355-1363.
Schwarz-Herion, K. (2002). Klonierung, Expression, Reinigung, Kristallisation und röntgenographische
Charakterisierung der O-Acetyl-Serin-Sulfhydrylase CysM aus Escherichia coli. Diplomarbeit, Universität
Freiburg im Breisgau.
Sharp, P. A., Sudgen, B. & Sambrook, J. (1973). Detection of two restriction endonuclease activities in Haemophilus
parainfluenzae using analytical agarose ethidium bromide electrophoresis. Biochemistry 12, 3055-3063.
Sheldrick, G. (1997) SHELXL-97 Program for crystal structure refinement, Institut für Anorganische Chemie der
Universität, Tammanstrasse 4, D-3400 Göttingen, Germany.
151
Literatur
Simurdiak, M., Lee, J. & Zhao, H. (2006). A new class of arylamine oxygenases: evidence that p-aminobenzoate
N-oxygenase (AurF) is a di-iron enzyme and further mechanistic studies. ChemBioChem 7, 1169-1172.
Southern, E. M. (1979). Gel electrophoresis of restriction fragments. Methods Enzymol. 68, 152-176.
Spain, J. C. (1995). Biodegradation of nitroaromatic compounds. Annu. Rev. Microbiol. 49, 523-555.
Stark, A., Sunyaev, S. & Russell, R.B. (2003). A model for statistical significance of local similarities in structure J.
Mol.Biol. 326, 1307-1316.
Sternbach, H. (1991). Chromatographische Methoden in der Biochemie. Thieme, Stuttgart.
Strand, K. R., Karlsen, S., Kolberg, M., Rohr, A. K., Görbitz, C. H. & Andersson, K. K. (2004). Crystal structural
studies of changes in the native dinuclear iron center of ribonucleotide reductase protein R2 from Mouse. J.
Biol. Chem. 279, 46794-46801.
Tai, C. H. & Cook, P. F. (2001). Pyridoxal 5'-phosphate-dependent alpha, beta-elimination reactions: mechanism of
O-acetylserine sulfhydrylase. Acc. Chem. Res. 34, 49–59.
Tai, C. H., Nalabolu, S. R., Jacobson, T. M., Minter, D. E. & Cook, P. F. (1993). Kinetic mechanisms of A and B
isozymes of O-acetylserine sulfhydrylase from Salmonella typhimurium LT-2 the natural and alternative
reactants. Biochemistry 32, 6433-6442.
Terwilliger, T. C. (1999). Reciprocal-space solvent flattening. Acta Crystallogr. Sect. D 55, 1863-1871.
Terwilliger, T. C. (2000). Maximum-likelihood density modification. Acta Crystallogr. Sect. D 56, 965-972.
Terwilliger, T. C. (2001). Maximum-likelihood density modification with pattern recognition of structural motifs. Acta
Crystallogr. Sect. D 57, 1755-1762.
Terwilliger, T. C. (2002). Rapid automatic NCS identification using heavy-atom substructures. Acta Crystallogr. Sect.
D 58, 2213-2215.
Terwilliger, T. C. (2003a). Automated main-chain model building by template matching and iterative fragment
extension. Acta Crystallogr. Sect. D 59, 38-44.
Terwilliger, T. C. (2003b). Automated side-chain model building and sequence assignment by template matching. Acta
Crystallogr. Sect. D 59, 45-49.
Terwilliger, T. C. (2003c). Statistical density modification using local pattern matching. Acta Crystallogr. Sect. D 59,
1688-1701.
Than, M. E., Henrich, S., Bourenkov, G. P., Bartunik, H. D., Huber, R. & Bode, W. (2005). The endoproteinase furin
contains two essential Ca2+ ion stabilizing its Nterminus and the unique S1 specificity pocket. Acta
Crystallogr. Sect. D 61, 505-512.
Thompson, J. D., Higgins, D. G. & Gibson, T. J. (1994). CLUSTALW: improving the sensitivity of progressive
multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix
choice. Nucleic Acids Res. 22, 4673-4680.
Tronrud, D. E. (2004). Introduction to macromolecular refinement. Acta Crystallogr. Sect. D 60, 2156-2168.
Vagin, A. & Teplyakov, A. (1997). MOLREP: an automated program for molecular replacement. J. Appl. Crystallog.
30, 1022-1025.
Vetting, M. W., Wackett, L. P., Que, Jr., L., Lipscomb, J. D. & Ohlendorf, D. H. (2004). Crystallographic comparison
of manganese- and iron-dependent homoprotocatechuate 2,3-dioxygenases. J. Bacteriol. 186, 1945-1958.
Voegtli, W. C., Ge, J., Perlstein, D. L., Stubbe, J. & Rosenzweig, A. C. (2001). Structure of the yeast ribonucleotide
reductase Y2Y4 heterodimer. Proc. Natl. Acad. Sci. USA 98, 10073-10078.
152
Literatur
Wallar, B. J. & Lipscomb, J. D. (1996). Dioxygen activation by enzymes containing binuclear non-heme iron clusters.
Chem. Rev. 96, 2625-2657.
Wang, B. C. (1985). Resolution of phase ambiguity in macromolecular crystallography. Methods Enzymol. 115, 90112.
Wang, J. W., Chen, J. R., Gu, Y. X., Zheng, C. D., Jiang, F., Fan, H. F., Terwilliger, T. C. & Hao, Q. (2004). SAD
phasing by combination of direct methods with the SOLVE/RESOLVE procedure. Acta Crystallogr. Sect. D
60, 1244-1253.
Weber, T., Welzel, K., Pelzer, S., Vente, A. & Wohlleben, W. (2003). Exploiting the genetic potential of polyketide
producing streptomycetes. J. Biotechnol. 106, 221 - 232.
Westermeyer, R. (1990). "Electrophorese-Praktikum," VCH, Weinheim.
Whittaker, M. M. & Whittaker, J. W. (1998). A glutamate bridge is essential for dimer stability and metal selectivity in
manganese superoxide dismutase. J. Biol. Chem. 273, 22188-22193.
Wiesand (2007). Kinetische und kristallographische Untersuchungen an Mutanten der O-Acetylserin-Sulfhydrylase
Isoform CysM aus Escherichia coli sowie Expressions- und Reinigungsversuche der Landomycin Glykosyltransferase 2 aus Streptomyces cyanogenus. Diplomarbeit, Universität Freiburg im Breisgau.
Willing, A., Follmann, H. & Auling, G. (1988). Ribonucleotide reductase of Brevibacterium ammoniagenes is a
manganese enzyme. Eur. J. Biochem. 170, 603-611.
Wilson, A. J. C. (1949). The probability distributions of X-ray intensities. Acta Crystallogr. 2, 318-321.
Winkler, R. & Hertweck, C. (2005). Sequential enzymatic oxidation of aminoarenes to nitroarenes via hydroxylamines.
Angew. Chem. Int. Ed. 44, 4083-4087.
Winkler, R., Richter, M. E. A., Knüpfer, U., Merten, D. & Hertweck, C. (2006). Regio- and chemoselective enzymatic
N-oxygenation in vivo, in vitro, and in flow. Angew. Chem. Int. Ed. 45, 8016-8018.
Winn, M. D., Isupov, M. N. & Murshudov, G. N. (2001). Use of TLS parameters to model anisotropic displacements
in macromolecular refinement. Acta Crystallogr. Sect. D 57, 122-133.
Winn, M. D., Murshudov, G. N. & Papiz, M. Z. (2003). Macromolecular TLS refinement in REFMAC at moderate
resolutions. Methods Enzymol. 374, 300-321.
Yeates, T. O. (1997). Detecting and overcoming crystal twinning. Methods Enzymol. 276, 344-358.
Zhang, K. Y. J. & Main, P. (1990). Histogram matching as a new density modification technique for phase refinement
and extension of protein molecules. Acta Crystallogr. Sect. A 46, 41-46.
Ziehl M., He J., Dahse, H. M. & Hertweck C. (2005). Mutasynthesis of aureonitrile: An aureothin derivative with
significantly improved cytostatic effect. Angew. Chem. Int. Ed. Engl. 44, 1202–1205.
Zocher, G. E. (2003). Mutagenese, Präparation, Kristallisation und röntgenographische Untersuchungen der O-Acetylserin-Sulfhydrylase CysM aus Escherichia coli. Diplomarbeit, Universität Freiburg im Breisgau.
153
Anhang
7. Anhang
7.1
Eichgerade der Gelpermeation an S200 - 16/60
3,0
2,8
Ferritin
440 kDa
Catalase
232 kDa
2,6
2,4
log MR
2,2
BSA
67 kDa
2,0
Ovalbumin
43 kDa
1,8
1,6
Chymotrypsionogen
25 kDa
1,4
Cytochrom C
12 kDa
1,2
1,0
Regressionsgerade y= -0.039x + 5.0
0,0
60
65
70
75
80
85
90
95
Elutionsvolumen [mL]
7.2
154
Selbstrotationsfunktion der N-Oxygenase Kristalle
100
105
110
Danksagung
8. Danksagung
Die vorliegende Arbeit wurde im Arbeitskreis von Prof. Dr. Georg E. Schulz am Institut für
Organische Chemie und Biochemie der Albert-Ludwigs-Universität Freiburg im Breisgau ange­
fertigt. Prof. Dr. Georg E. Schulz danke ich für die Vergabe des interessanten Themas, sein
Interesse am Fortgang der Arbeit und seine stete Diskussionsbereitschaft.
Besonderen Dank schulde ich auch Michael Claus. Er hat wesentlich zum Gelingen dieser Arbeit
beigetragen.
Den Diplomanden Ulrich Wiesand und Johannes Bauer danke ich für Ihr Engagement und Ihre
Einsatzbereitsschaft während ihrer Arbeiten an der Sulfhydrylase bzw. der Cyclase.
In diesem Zusammenhang gilt auch meinen motivierten Mitarbeiterpraktikanten Petra Waiz, Peter
Schmider und Claudia Blattner bei den Arbeiten an der Sulfhydrylase mein Dank.
Ulrike Weiser, Antonio Espin und Bärbel Dirr haben durch ihre engagierte Arbeit stets für ein
einsatzbereites Labor gesorgt, wofür ich ihnen herzlich danken möchte.
Sehr herzlich möchte ich Linda Böhm für ihre Hilfsbereitschaft, Freundlichkeit und das große
Engagement für den Arbeitskreis danken.
Christian Schleberger danke ich für gute Teamarbeit bei der Wartung und Pflege des ComputerNetzwerks.
Einen ganz besonderen Dank schulde ich Stefan Büdenbender, Saskia Kissel Michael Mittler,
Franziska Zähringer, Friederike Schmidt und Nora Treiber. Sie haben erheblich dazu beigetragen,
dass mir die Jahre im Arbeitskreis in guter Erinnerung bleiben werden.
Zuguterletzt gilt großer Dank meiner Familie: Jacqueline, Michaela und Andrea für ihre
Unterstützung und ihr Verständnis über meine gesamte Studienzeit.
155
Danksagung
156