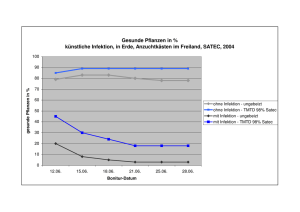
Entwicklung und Testung neuer DNA- und Protein
Werbung

Entwicklung und Testung neuer DNA- und Proteinbasierter Multikomponentenvakzinen sowie regulatorischer Adjuvanzien gegen eine Infektion mit B. anthracis in Auszucht-Mäusen und Ziegen Dissertation zur Erlangung des Doktorgrades der Naturwissenschaften (Dr. rer. nat.) Fakultät Naturwissenschaften Universität Hohenheim Institut für Nutztierwissenschaften (460) / PD Dr. med. vet. habil. W. Beyer und Institut für Zoologie (220) / Prof. Dr. rer. nat. U. Mackenstedt vorgelegt von Susanne Melanie Köhler aus Eisenach 2015 Dekan: Prof. Dr. rer. nat. Heinz Breer 1. berichtende Person: PD Dr. med. vet. habil. Wolfgang Beyer 2. berichtende Person: Prof. Dr. rer. nat. Ute Mackenstedt Eingereicht am: 11.03.2015 Mündliche Prüfung am: 10.07.2015 Die wurde vorliegende Arbeit am 12.05.2015 von der Fakultät Naturwissenschaften der Universität Hohenheim als „Dissertation zur Erlangung des Doktorgrades der Naturwissenschaften“ angenommen. Für meine Mutter Für Daniel Für mich „ALL WE HAVE TO DECIDE IS WHAT TO DO WITH THE TIME THAT IS GIVEN TO US.“ John Ronald R. Tolkien iii iv Inhalt 1 EINLEITUNG..........................................................................................................................1 1.1 ENTDECKUNG DER ÄTIOLOGIE VON MILZBRAND...........................................................................................1 1.2 TAXONOMIE UND MORPHOLOGIE................................................................................................................1 1.2.1 Aufbau der vegetativen Zelle...................................................................................................2 1.2.1.1 Die Kapsel......................................................................................................................................................... 3 1.2.1.2 Die Toxinkomponenten......................................................................................................................................4 1.2.2 Aufbau der Spore.....................................................................................................................5 1.3 VERBREITUNG UND LEBENSZYKLUS............................................................................................................6 1.4 PATHOGENESE (MENSCH UND TIER)...........................................................................................................8 1.5 IMPFSTOFFE..........................................................................................................................................10 1.5.1 Historische Entwicklung der Lebendvakzinen gegen B. anthracis........................................10 1.5.2 Entwicklung der lizenzierten Humanvakzinen.......................................................................11 1.5.3 Ansätze zur Entwicklung neuer Vakzinen gegen B. anthracis................................................13 1.5.3.1 Toxinkomponenten..........................................................................................................................................14 1.5.3.2 Antigene der vegetativen Zelle........................................................................................................................16 1.5.3.3 Antigene der Spore..........................................................................................................................................16 1.5.4 Genetische Immunisierung....................................................................................................17 1.5.5 Adjuvanzien...........................................................................................................................19 1.6 ZIEL DER ARBEIT..................................................................................................................................22 2 MATERIAL............................................................................................................................23 2.1 CHEMIKALIEN.......................................................................................................................................23 2.2 PUFFER, LÖSUNGEN UND MEDIEN............................................................................................................25 2.3 VERBRAUCHSMATERIALIEN......................................................................................................................26 2.4 VERSUCHSTIERE....................................................................................................................................26 2.5 GERÄTE...............................................................................................................................................27 2.6 ANTIKÖRPER.........................................................................................................................................27 2.7 UTENSILIEN..........................................................................................................................................28 2.8 ZELLLINIEN..........................................................................................................................................28 2.9 KITS...................................................................................................................................................28 2.10 GENETISCHES MATERIAL......................................................................................................................29 2.11 WIRKSTOFFE.......................................................................................................................................29 2.12 SOFTWARE.........................................................................................................................................29 3 METHODEN..........................................................................................................................30 3.1 STANDARDMETHODEN.............................................................................................................................30 3.2 HERSTELLUNG UND AUFBEREITUNG VON ANTIGENEN...................................................................................31 3.2.1 rPA83, rBclA und rLF............................................................................................................31 3.2.1.1 Anzucht von induzierten Bakterienkulturen.....................................................................................................31 3.2.1.2 Proteinaufreinigung mittels FPLC...................................................................................................................32 3.2.1.3 Dialyse............................................................................................................................................................. 33 3.2.2 Aufreinigung von Kapselmaterial aus einer Bakterienkultur.................................................34 3.2.3 Färbung mit Stains-All..........................................................................................................34 3.2.4 Herstellung eines vegetativen Vollantigen von B. anthracis..................................................35 3.2.5 Herstellung einer Sporensuspension von B. anthracis...........................................................35 3.2.6 Bestimmung der Sporenkonzentration...................................................................................36 3.2.7 Herstellung von Formalin inaktivierten Sporen (FIS)...........................................................36 i 3.3 AUF- UND VORBEREITUNG DER VAKZINEN.................................................................................................37 3.3.1 Endotoxinbestimmung...........................................................................................................37 3.3.2 Endotoxinaufreinigung..........................................................................................................38 3.3.3 DNA-Patronen-Herstellung...................................................................................................38 3.3.3.1 Präzipitation von DNA auf Goldstaub.............................................................................................................38 3.3.3.2 Schlauchpräparation und Beschichtung...........................................................................................................39 3.3.3.3 Qualitätskontrolle der DNA-Patronen.............................................................................................................40 3.4 VAKZINIERUNG, PROBENNAHME UND INFEKTION..........................................................................................40 3.4.1 Mausversuche........................................................................................................................42 3.4.1.1 Betäubung und Tötung....................................................................................................................................43 3.4.1.2 Impfung........................................................................................................................................................... 44 3.4.1.3 Probennahme und Diagnostik..........................................................................................................................44 3.4.1.4 Infektion.......................................................................................................................................................... 45 3.4.2 Ziegenversuch (Südafrika).....................................................................................................47 3.4.2.1 Impfung........................................................................................................................................................... 48 3.4.2.2 Probennahme...................................................................................................................................................49 3.4.2.3 Infektion.......................................................................................................................................................... 50 3.4.2.4 Diagnostik....................................................................................................................................................... 51 3.4.3 Wiederholung der Ziegenversuche (Türkei)...........................................................................52 3.5 SEROLOGISCHE ANALYSEVERFAHREN.........................................................................................................54 3.5.1 Blutaufbereitung....................................................................................................................54 3.5.2 Enzyme-linked immunosorbent Assay (ELISA)......................................................................54 3.5.3 Handhabung der Makrophagenzelllinie J774A.1..................................................................56 3.5.4 Toxin-Neutralisations-Assay (TNA).......................................................................................56 3.6 STATISTIK.............................................................................................................................................57 4 ERGEBNISSE UND DISKUSSION.....................................................................................59 4.1 TESTUNG VON VAKZINEKOMPONENTEN IM NMRI-MAUSMODELL..................................................................59 4.1.1 Einzelbetrachtungen der Impfung von Mäusen mit Vakzinen auf Proteinbasis.....................59 4.1.1.1 Mausgruppe LN-1000 (Lipopeptid).................................................................................................................60 4.1.1.2 Mausgruppe NC (Kapselkonjugat)..................................................................................................................60 4.1.1.3 Mausgruppe NCP (Kapselkonjugat; rPA83)....................................................................................................61 4.1.1.4 Mausgruppe LN-2000 (Lipopeptid).................................................................................................................62 4.1.1.5 Mausgruppe LB (Lipopeptid und rBclA).........................................................................................................62 4.1.1.6 Mausgruppe LP (Lipopeptid und rPA83).........................................................................................................64 4.1.1.7 Mausgruppe LBP (Lipopeptid, rBclA und rPA83)...........................................................................................65 4.1.1.8 Mausgruppe LBCP (Lipopeptid, rBclA, Kapselkonjugat und rPA83)..............................................................66 4.1.1.9 Mausgruppe LBCOP (Lipopeptid, rBclA, Kapselkonjugat, FIS und rPA83)...................................................67 4.1.2 Einzelbetrachtungen der Impfung von Mäusen mit Vakzinen auf DNA-Basis........................68 4.1.2.1 Mausgruppe TN (CIITA).................................................................................................................................68 4.1.2.2 Mausgruppe TM (CIITA; BclAD1D3-Uq)......................................................................................................69 4.1.2.3 Mausgruppe TK (CIITA; TPA-BclAD1D3-LAMP1).......................................................................................70 4.1.2.4 Mausgruppe TH (CIITA; TPA-rPA83-LAMP1)...............................................................................................71 4.1.2.5 Mausgruppe TA (CIITA; TPA-LFD1PAD4-LAMP1)......................................................................................72 4.1.2.6 Mausgruppe NS (TPA-LFD1PAD4-mIPS-1)...................................................................................................73 4.1.2.7 Mausgruppe TKS (CIITA; TPA-BclAD1D3-LAMP1, TPA-LFD1PAD4-mIPS-1)..........................................74 4.1.3 Überlebensdaten aller Mausversuche im Vergleich...............................................................76 4.1.4 Interner Vergleich der Titerdaten der Mausversuche............................................................79 4.2 DISKUSSION MAUSVERSUCHE..................................................................................................................83 4.2.1 Proteinvakzinen.....................................................................................................................83 Der Zusatz des Kapselkonjugats hatte keinen nachweisbaren Einfluss auf die Schutzwirkung.............................83 BclA und PA wirken einander unterstützend.........................................................................................................85 Zusatz von FIS bietet potentiell höheren Schutz, der nicht zwingend allein auf BclA beruht...............................87 4.2.2 DNA-Vakzinen........................................................................................................................89 Vektoren mit Toxinkomponenten zeigten eine geringere Immunogenität als vergleichbare Proteinvakzinen........89 Die internen Unterschiede der Toxinvektoren sind vermutlich dosisbedingt.........................................................90 ii DNA-Vektoren mit BclAD1D3 stimulierten eine sterile Immunität bei 50 % Schutzrate......................................91 Die kombinierte DNA-Vakzine TKS vermittelte nahezu vollständigen Schutz gegen eine voll virulente Infektion mit B. anthracis.......................................................................................................................................92 Zusammenfassung der Mausversuche...................................................................................................................94 4.3 TESTUNG VON VAKZINEKOMPONENTEN IN BURENZIEGEN..............................................................................96 4.3.1 Gesundheitszustand der Ziegen vor der Infektion..................................................................96 4.3.2 Überprüfung der Infektiösität der Sporen im BALB/c Mausmodell.......................................98 4.3.3 Kontrollgruppen....................................................................................................................98 4.3.4 Ermittlung der MLD..............................................................................................................99 4.3.5 Einzelbetrachtungen der Immunisierungen von Burenziegen mit der SSLV (Sterne Sporen Lebendvakzine)...................................................................................................................100 4.3.5.1 Ziegengruppe 1 (PBS)...................................................................................................................................101 4.3.5.2 Ziegengruppe 2 (SSLV).................................................................................................................................102 4.3.5.3 Ziegengruppe 3a/b (SSLV)............................................................................................................................104 Anti-rPA83 IgG-Titer und TNA-Titer..................................................................................................................104 Anti-rBclA und anti-FIS IgG-Titer......................................................................................................................108 Anti-vegetativ IgG-Titer......................................................................................................................................109 4.3.6 Zusammenfassung der SSLV-Gruppen.................................................................................111 4.3.7 Einzelbetrachtungen der Immunisierungen von Burenziegen mit NLV................................112 4.3.7.1 Ziegengruppe 4 (rPA83, rBclA und Lipopeptid)............................................................................................112 4.3.7.2 Ziegengruppe 5 (DNA-Vakzine)....................................................................................................................114 4.3.7.3 Ziegengruppe 6 (rPA83, rBclA, FIS und Lipopeptid)....................................................................................116 4.3.8 Versuchswiederholung der Gruppen 4 und 6 (Türkei).........................................................118 4.3.8.1 Negativkontrolle (Wiederholung)..................................................................................................................119 4.3.8.2 Ziegengruppe 4 (Wiederholung)....................................................................................................................120 4.3.8.3 Ziegengruppe 6 (Wiederholung)....................................................................................................................123 4.3.9 Zusammenfassung der Ergebnisse der NLV.........................................................................125 4.3.10 Interner Vergleich der Titerdaten der Ziegenversuche.......................................................128 4.3.11 Todesdatenübersicht der Ziegenversuche..........................................................................129 4.3.12 Diagnoseprozeduren..........................................................................................................132 4.3.12.1 Temperatur...................................................................................................................................................132 4.3.12.2 Zusammenhänge von Fieber, Antikörpertitern und Schutz..........................................................................137 4.3.12.3 Verhalten......................................................................................................................................................140 4.3.12.4 Detektion von Bakterien im Blut.................................................................................................................141 4.4 DISKUSSION ZIEGENVERSUCHE...............................................................................................................144 4.4.1 Versuchsdurchführung und Diagnoseprozeduren................................................................144 Das Auftreten von Fieber steht in Zusammenhang mit den Antikörpertitern gegen rPA83..................................144 Die Höhe der anti-rPA83 IgG-Titer vor der Infektion ist in Ziegen und Auszucht-Mäusen prädiktiv für das Überleben, aber möglicherweise abhängig von der Infektionsdosis......................................................146 Die MLD für das Feldisolat SD20 ist kleiner als 36 Sporen in Ziegen................................................................148 Beim Nachweis von Bakterien im Blut infizierter Ziegen ist mit einem Tod binnen 2 – 5 h zu rechnen.............148 Die hohen unspezifischen Hintergrundtiter bei Ziegenseren im ELISA lassen sich durch den Austausch von FKS mit Magermilchpulver stark reduzieren.................................................................................................149 Adverse Reaktionen von Ziegen auf die Immunisierung mit der SSLV beruhen möglicherweise auf parallel vorhandenen Infektionen.......................................................................................................................150 Verbesserung der Diagnoseprozeduren...............................................................................................................151 4.4.2 Impfversuche mit NLV.........................................................................................................153 Die DNA-Vakzine Kombination war in dieser Form weitestgehend wirkungslos...............................................153 FIS als Bestandteil einer Proteinvakzine erhöht die schützende Wirkung von rPA83.........................................154 4.4.3 Impfversuche mit der SSLV..................................................................................................155 Der Erfolg einer Erstimmunisierung mit der SSLV kann stark variieren.............................................................156 Die unspezifische phagozytotische Wirkung von Ziegenseren könnte die Wirkung der Immunisierung mit der SSLV beeinflussen................................................................................................................................157 Der Höchstpunkt der anti-rPA83 IgG-Titer ist 1 – 2 Wochen nach der Immunisierung zu erwarten...................158 Eine Vakzinierung mit der SSLV von Tieren mit erhöhten Antikörpertitern (gegen PA) ist wirkungslos............158 Eine gedoppelte Erstimmunisierung könnte die sporadischen Immunisierungsausfälle mit der SSLV beheben..159 Die Auffrischungsimpfung sollte 3 – 4 Monate nach der Erstimmunisierung erfolgen.......................................159 iii Eine jährliche Auffrischung der Impfung mit der SSLV könnte generell obsolet sein oder durch eine Änderung des Impfplans obsolet werden...............................................................................................................160 4.4.4 Generelle Anmerkungen zu den Ergebnissen.......................................................................161 BclA ist in Ziegen nicht bzw. wenig immunogen................................................................................................161 Die immunogene und möglicherweise auch Schutz vermittelnde Wirkung von FIS beruht nicht auf BclA........162 Eine überlebte Infektion erzeugt ein anderes Titerspektrum als eine Impfung mit der SSLV..............................164 BclA könnte im Ziegenmodell die gleiche Funktion für die Spore einnehmen wie die Kapsel für den vegetativen Zellkörper..............................................................................................................................................165 5 KONKLUSION UND AUSBLICK.....................................................................................166 6 ANHANG...................................................................................................................................I 6.1 SEQUENZEN............................................................................................................................................I 6.2 VERSUCHSDURCHFÜHRUNG.......................................................................................................................II iv Verzeichnis der Abbildungen Abbildung 1: Koloniemorphologie................................................................................................2 Abbildung 2: Kettenbildung..........................................................................................................2 Abbildung 3: Aufbau der vegetativen Zelle...................................................................................3 Abbildung 4: Kapselfärbung.........................................................................................................3 Abbildung 5: Sporenaufbau...........................................................................................................5 Abbildung 6: Zyklus der Anthrax Infektion..................................................................................7 Abbildung 7: Genetische Immunisierung....................................................................................18 Abbildung 8: synthetisches Lipopeptid (Bsp.)............................................................................20 Abbildung 9: Nomenklatur der Mausgruppen.............................................................................42 Abbildung 10: Rückstandbild von DNA-Patronen nach Verschuss............................................44 Abbildung 11: Burenziegen.........................................................................................................47 Abbildung 12: Außengehege für die Infektion............................................................................48 Abbildung 13: Blutentnahme für Serologie.................................................................................49 Abbildung 14: i. p. Injektion Maus..............................................................................................50 Abbildung 15: Färbung von Blutausstrichen mit Azur B............................................................52 Abbildung 16: ELISA-Schema....................................................................................................55 Abbildung 17: TNA-Belegungschema........................................................................................56 Abbildung 18: Mausgruppe NCP................................................................................................61 Abbildung 19: Mausgruppe LB...................................................................................................63 Abbildung 20: Mausgruppe LP...................................................................................................64 Abbildung 21: Mausgruppe LBP.................................................................................................65 Abbildung 22: Mausgruppe LBCP..............................................................................................66 Abbildung 23: Mausgruppe LBCOP...........................................................................................67 Abbildung 24: Mausgruppe TM..................................................................................................69 Abbildung 25: Mausgruppe TK...................................................................................................70 Abbildung 26: Mausgruppe TH...................................................................................................71 Abbildung 27: Mausgruppe TA...................................................................................................73 Abbildung 28: Mausgruppe NS...................................................................................................73 Abbildung 29: Mausgruppe TKS................................................................................................75 Abbildung 30:Überlebensdaten Mausgruppen (Proteinvakzinen, ~1000 Sporen)......................76 Abbildung 31: Überlebensdaten Mausgruppen (Proteinvakzinen, ~2000 Sporen).....................77 Abbildung 32: Überlebensdaten Mausgruppen (DNA-Vakzinen, ~1000 Sporen)......................78 Abbildung 33: Übersicht IgG- und TNA-Titer (Proteinvakzinen, Maus)...................................80 Abbildung 34: Übersicht IgG- und TNA-Titer (DNA-Vakzinen, Maus).....................................81 Abbildung 35: Ziegengruppe 2 (anti-rPA83 IgG-Titer).............................................................103 Abbildung 36: Ziegengruppe 2 (anti-FIS, -Vegetativ, -rBclA und TNA-Titer).........................103 Abbildung 37: Ziegengruppe 3a/b und 1 (anti-rPA83 IgG-Titer)..............................................105 Abbildung 38: Ziegengruppe 3a/b und 1 (TNA-Titer)..............................................................107 Abbildung 39: Ziegengruppe 3a/b und 1 (anti-rBclA und -FIS IgG-Titer)................................109 Abbildung 40: Ziegengruppe 3a/b (anti-vegetativ IgG-Titer)....................................................110 Abbildung 41: Ziegengruppe 4 (anti-rPA83 und -rBclA IgG-Titer)..........................................113 Abbildung 42: Ziegengruppe 5 (anti-rPA83 und -rLF IgG-Titer)..............................................115 Abbildung 43: Ziegengruppe 5 (anti-rBclA, -FIS, -vegetativ IgG- und TNA-Titer).................115 Abbildung 44: Ziegengruppe 6 (anti-rPA83 IgG-Titer).............................................................117 Abbildung 45: Ziegengruppe 6 (ELISA- und TNA-Titer).........................................................117 Abbildung 46: Ziegengruppe 4 (W) – anti-rPA83 und TNA-Titer............................................122 Abbildung 47: Ziegengruppe 4 (W) – anti-rBclA IgG-Titer......................................................122 Abbildung 48: Ziegengruppe 6 (W) – anti-rPA83 IgG- und TNA-Titer....................................124 v Abbildung 49: Ziegengruppe 6 (W) – anti-rBclA und -FIS IgG-Titer.......................................125 Abbildung 50: IgG- und TNA-Titer der NLV (Ziege)...............................................................126 Abbildung 51: Gesamtübersicht IgG- und TNA-Titer (Ziege)..................................................128 Abbildung 52: Überlebensdaten Ziegengruppen (Vergleich der Wiederholungen)...................130 Abbildung 53: Gesamtübersicht der Überlebensdaten der Ziegengruppen...............................131 Abbildung 54: Temperaturspektrum Ziegenversuch (Südafrika)..............................................133 Abbildung 55: Temperaturspektrum Ziegenversuch (Türkei)...................................................134 Abbildung 56: Temperaturverhältnisse......................................................................................136 Abbildung 57: BclA-Filament...................................................................................................163 Abbildung 58: GeneGun Patronenverschuss................................................................................II Verzeichnis der Tabellen Tabelle 1: Chemikalien...............................................................................................................23 Tabelle 2: Puffer, Lösungen und Medien....................................................................................25 Tabelle 3: Verbrauchsmaterialien................................................................................................26 Tabelle 4: Versuchstiere..............................................................................................................26 Tabelle 5: Geräte.........................................................................................................................27 Tabelle 6: Antikörper..................................................................................................................27 Tabelle 7: Utensilien...................................................................................................................28 Tabelle 8: Zelllinien....................................................................................................................28 Tabelle 9: Kits.............................................................................................................................28 Tabelle 10: Genetisches Material................................................................................................29 Tabelle 11: Wirkstoffe................................................................................................................29 Tabelle 12: Software...................................................................................................................29 Tabelle 13: Übersicht Vakzinekomponenten (Mausversuch).....................................................41 Tabelle 14: Impfgruppen Mausversuch......................................................................................43 Tabelle 15: Impfgruppen Ziegenversuch (Südafrika).................................................................48 Tabelle 16: Wiederholung der Ziegenversuche (Türkei) – Impfgruppen...................................53 Tabelle 17: Schwellenwerte........................................................................................................58 Tabelle 18: Mausgruppe LN-1000..............................................................................................60 Tabelle 19: Mausgruppe NC.......................................................................................................60 Tabelle 20: Mausgruppe NCP.....................................................................................................61 Tabelle 21: Mausgruppe LN-2000..............................................................................................62 Tabelle 22: Mausgruppe LB.......................................................................................................63 Tabelle 23: Mausgruppe LP........................................................................................................64 Tabelle 24: Mausgruppe LBP.....................................................................................................65 Tabelle 25: Mausgruppe LBCP...................................................................................................66 Tabelle 26: Mausgruppe LBCOP................................................................................................67 Tabelle 27: Mausgruppe TN.......................................................................................................68 Tabelle 28: Mausgruppe TM......................................................................................................69 Tabelle 29: Mausgruppe TK.......................................................................................................70 Tabelle 30: Mausgruppe TH.......................................................................................................71 Tabelle 31: Mausgruppe TA.......................................................................................................72 Tabelle 32: Mausgruppe NS.......................................................................................................74 Tabelle 33: Mausgruppe TKS.....................................................................................................75 Tabelle 34: Korrelation der Proteingruppen...............................................................................86 Tabelle 35: Gesundheitszustand nach Transport.........................................................................97 Tabelle 36: Kontrollgruppen (Ziege)..........................................................................................99 vi Tabelle 37: Bestimmung der MLD.............................................................................................99 Tabelle 38: Ziegengruppe 1......................................................................................................101 Tabelle 39: Ziegengruppe 2......................................................................................................102 Tabelle 40: Ziegengruppe 3a/b (anti-rPA83 IgG-Titer)............................................................105 Tabelle 41: Ziegengruppe 3a/b (TNA-Titer).............................................................................107 Tabelle 42: Ziegengruppe 3a/b (anti-rBclA und -FIS IgG-Titer)..............................................108 Tabelle 43: Ziegengruppe 3a/b (anti-vegetativ IgG-Titer)........................................................110 Tabelle 44: Ziegengruppe 4......................................................................................................113 Tabelle 45: Ziegengruppe 5......................................................................................................114 Tabelle 46: Ziegengruppe 6......................................................................................................116 Tabelle 47: Negativkontrolle (W) – Ziege (Türkei)..................................................................119 Tabelle 48: Ziegengruppe 4 (W) – ELISA- und TNA-Titer.....................................................121 Tabelle 49: Ziegengruppe 6 (W) – ELISA- und TNA-Titer.....................................................123 Tabelle 50: Zusammenhänge von Fieber, Antikörpertitern und Schutz (Ziegen).....................137 Tabelle 51: Zusammenhang zwischen Antikörpertiter und der Entwicklung von Fieber.........139 Tabelle 52: Detektion von Bakterien im Blut...........................................................................142 Tabelle 53: Probennahmepunkte Ziegenversuch (Südafrika).....................................................III Tabelle 54: Probennahmepunkte Ziegenversuch (Türkei - Wiederholungen)............................III Tabelle 55: exemplarische Temperaturerhebung in naiven Ziegen (Südafrika).........................IV Tabelle 56: Ziegenversuche (Südafrika), diagnostische Daten...................................................IV Tabelle 57: Ziegenversuche (Türkei), diagnostische Daten........................................................VI Tabelle 58: Zusammenhänge von Antikörpertitern und Schutz (Mäuse)..................................VII Tabelle 59: Test auf pTBC.......................................................................................................VIII vii Verzeichnis der Abkürzungen °C (Einheit) A ABTS APC APS AVA AVP B. anthracis B. cereus BCA BclA BHI bp (Einheit) BSA bzw. ca. CaCl2 cAMP CBA cfu (Einheit) CH CIITA CLR CO2 CSA D D1 D2 D3 D4 Dhc DMSO DNA DNas dpi DTT DVM E. coli EDTA Edtx EF EU (Einheit) F FIS FKS g (Einheit) GerD viii Grad Celsius Österreich 2,2'-Azino-di-(3-ethylbenzthiazolin-6-sulfonsäure) engl. antigen presenting cells Ammoniumperoxodisulfat eng. Anthrax vaccine adsorbed engl. Anthrax vaccine precipitated Bacillus anthracis Bacillus cereus sensu stricto engl. bicinchonin acid engl. Bacillus collagen-like protein of anthracis engl. brain heart infusion Basenpaar bovines Serumalbumin beziehungsweise cirka Calciumchlorid zyklisches Adenosinmonophosphat Columbia-Blutagar engl. colony forming units Schweiz engl. class II major histocompatibility complex transactivator engl. collagen-like region Kohlendioxid Casein-Soya-Agar Deutschland Domäne 1 Domäne 2 Domäne 3 Domäne 4 Dihydroxypropylcystein Dimethylsulfoxid engl. desoxyribonucleinacid engl. Desoxyribonuclease engl. days post infection 1,4-Dithiothreitol eng. doctor of veterinary medicine Escherichia coli engl. Ethylenediaminetetraacetic acid engl. Edema Toxin engl. Edema Factor engl. endotoxin unit Frankreich engl. formalin inacivated spores fötales Kälberserum Gramm engl. B. cereus spore germination protein h (Einheit) HCl HEPES hpi HRP i. c. i. d. i. m. IPS-1 IPTG kb (Einheit) KCl kDa (Einheit) KH2PO4 KLH l (Einheit) LAL LAMP1 Letx LF LPS M (Einheit) MAPKK mg (Einheit) MgCl2 MHC min (Einheit) ml (Einheit) MLD mm MTT MYA Na2HCO3 Na2HPO4 NaCl NaHCO3 NaHPO4 NaOH NLV nm (Einheit) Nr. OBP OD OMPC PA PAGE Pam PBS PCR Stunde engl. hydrogen chloride N-2-Hydroxyethylpiperazin-N'-2-ethansulfonsäure engl. hours post infection engl. horseradisch peroxidase intra cutan intra dermal intra muskulär engl. Interferon-ß-Promotor Stimulator 1 Isopropyl-β-D-thiogalactopyranosid Kilo-Basenpaar Kaliumchlorid Kilodalton Kaliumdihydrogenphosphat engl. keyhole limpet hemocyanin Liter Limulus Amöbuzyten Lysat engl. lysosomal-associated membrane protein 1 engl. Lethal Toxin engl. Lethal Factor Lipopolysaccharid Molar Mitogen-aktivierten Proteinkinasen Kinasen Milligramm Magnesiumchlorid engl. major histocompatibility complex Minute Milliliter Minimum letale Dosis Millimeter (3-(4,5-Dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium-bromide) engl. meat yeast agar Di-Natriumhydrogencarbonat Dinatriumhydrogenphosphat Natriumchlorid Natriumhydrogencarbonat Natriumhydrogenphosphat Natriumhydroxid Nicht-Lebendvakzine Nanometer Nummer engl. Onderstepoort Biological Products Optische Dichte engl. outer membrane protein complex engl. Protective Antigen Polyacrylamid-Gelelektrophorese Palmitinsäure engl. phosphat buffered saline engl. polymerase chain reaction ix PGA PLAP PMSF PRR pTBC PVP RLR RNA RNase rs s (Einheit) s. c. SASP SDS SSLV STI TAE TEMED TGB TLR TNF TPA TRIS TSA üN UpM (Einheit) Uq USA V (Einheit) x g (Einheit) z.T. ZA ZVH µg (Einheit) µl (Einheit) µm (Einheit) x Poly-γ-D-Glutaminsäure engl. placental alkaline phosphatase Phenylmethylsulfonylfluorid engl. pattern recognition receptor Pseudotuberkulose Polyvinylpyrrolidon engl. RIG-like receptor engl. Ribonucleinacid Ribonuklease Spearman Rangkorrelationskoeffizient Sekunde sub cutan engl. small acid-soluble proteins Natriumdodecylsulfat Sterne Sporen Lebendvakzine Sanitary-Technical Institute TRIS-Acetat-EDTA-Puffer N,N,N',N'-Tetramethylethylendiamin Trypton-Glukose-Bouillon engl. toll-like receptor engl. tumor necrosis factor engl. tissue plasminogen activator Tris-(hydroxymethyl)-aminomethan Trypton-Soja-Agar über Nacht Umdrehungen pro Minute Ubiquitin Vereinigte Staaten von Amerika Volt Vielfaches der Erdbeschleunigung zum Teil Süd Afrika Zentralen Einrichtung für Biologische und Biomedizinische Forschung mit Tierhaltung Mikrogramm Mikroliter Mikrometer Zusammenfassung Durch die Entdeckung und subsequenten Verwendung der Sterne Sporen Lebendvakzine (SSLV) in der Veterinärmedizin konnte die Zahl der globalen Milzbrandausbrüche seit 1930 stark reduziert werden. Dennoch sind auch heute noch Länder im mediterranen Raum, in Südund Zentralamerika, Afrika und Zentralasien, sowie einigen Gebieten der USA und Kanada endemisch mit Bacillus anthracis belastet. Unter anderem ist dies auf die noch immer ungeklärten Faktoren im Zyklus der wiederkehrenden Aus- und Verbreitung des Erregers und dessen Persistenz im Boden zurückzuführen. Zudem ist die Anwendbarkeit der SSLV durch die Notwendigkeit einer jährlichen Auffrischung, der Restvirulenz des Impfstammes bei einigen sensitiven Tierarten (Ziegen und Lamas) und der Unvereinbarkeit einer antibiotischen Behandlung mit einer gleichzeitigen Impfung eingeschränkt, sodass weiterhin nach alternativen Vakzinierungsmethoden geforscht wird. In dieser Arbeit wurden unter Verwendung von Vakzinen auf Protein- und DNA-Basis Möglichkeiten zur alternativen Immunisierung mit „nicht lebend Vakzinen“ (NLV) evaluiert. Zu testen waren in der Proteinstudie rPA83 als Toxinkomponente, Formalin inaktivierte Sporen (FIS) und rBclA als Sporenantigene, sowie ein Kapsel-Lipopeptid-Konjugat als Bestandteil der vegetativen Form von B. anthracis. Zur Unterstützung der Immunogenität wurde ein LipopeptidAdjuvans verwendet. Für die DNA-Vakzine kamen verschiedene Vektorrückgrate zum Einsatz, welche Signalsequenzen beinhalteten, die das integrierte Antigen (rPA83, PAD4LFD1 und BclAD1D3) über den MHCI, MHCII oder sekretorischen Weg dirigieren. Als Adjuvanzien wurden ein separater Vektor mit einem positiven MHCII-Regulator verwendet (CIITA) oder integriert in den Vektor des Antigens ein Interferon-ß Promotor Stimulator (mIPS1). Die aufgeführten Vakzinekomponenten wurden im Rahmen dieser Arbeit einzeln und in Kombination im NMRI-Mausmodell getestet, um eine geeignete Kombination für den Gebrauch in der Ziege auszuwählen. Die Applikation der Proteinkomponenten (25 µg pro Antigen, 108 FIS und 50 µg Lipopeptid) erfolgte s. c., die der DNA-Vakzine (3 µg pro Vektor) i. c. mittels GeneGun. Alle beteiligten Mausgruppen wurden 3 Mal im Abstand von 2 Wochen immunisiert und 3 Wochen nach der letzten Immunisierung mit ~1000 bzw. ~2000 Sporen eines voll virulenten Stammes (Ames) infiziert. Mit einigen Ausnahmen konnten für alle Gruppen, entsprechend der eingesetzten Antigene, Antikörpertiter nachgewiesen werden. In Bezug auf rPA83 waren hierbei die getesteten Proteinvakzinen effektiver als die DNA-Vakzinen, welche weitaus niedrigere Titer aufwiesen. Für rBclA waren äquivalent hohe Titer bei DNA- und Proteinvakzinen messbar. Den gemessenen anti-rBclA Titern entsprechende hohe IgG Titer gegen FIS konnten in alle Gruppen nachgewiesen werden, die sowohl nur rBclA als auch FIS oder beides in Kombination erhalten hatten. Das Kapselkonjugat war wenig immunogen, was retrospektiv vermutlich auf ein immunsupprimierendes Epitop zurückzuführen war. Die Überlebensraten der getesteten Gruppen bewegten sich zwischen 10 und 100 %, wobei alle Proteinvakzinen, die nur ein Antigen xi beinhalteten, mit 10 % Überlebenden nicht signifikant unterschiedlich zur eingesetzten Kontrolle waren. Durch Kombination von mindestens 2 verschiedenen Antigenen konnte eine Schutzrate von mindestens 60 % erreicht werden, wobei nur die Kombination aller Proteinvakzinen und FIS mit 100 % komplett schützte. Die DNA-Vakzinen zeigten einzeln Schutzraten von 30 – 50 %. Hervorstechend hierbei waren die getesteten Konstrukte mit rBclA, die ohne weitere Antigene mit einer Überlebensrate von 50 % die beste Schutzwirkung zeigten und zudem eine sterile Immunität aufwiesen. In Kombination mit dem besten Toxin-Vektor konnte eine Schutzrate von 90 % erreicht werden, womit DNA- und Proteinvakzine ein gleichwertiges Potential für weitere Versuche aufwiesen. Entsprechend wurden die erfolgversprechendsten Kombinationsvakzinen im Vergleich zur SSLV mit Hilfe von Kooperationspartnern in Südafrika und der Türkei in Ziegen getestet. Die schützende Wirkung der SSLV wurde in 3 Gruppen bestimmt, direkt nach der Erstimmunisierung, nach dem Verlauf von einem Jahr und direkt nach einer Auffrischungsimpfung. Im Bereich der Protein-basierten Vakzinen wurde auf die weitere Verwendung des Kapselkonjugats verzichtet, sodass eine Kombination aus Lipopeptid (500 µg), rPA83 und rBclA (je 25 µg bzw. 75 µg in den Wiederholungsversuchen), mit und ohne Zusatz von FIS (10 8 Sporen) im Ziegenmodell zum Einsatz kam. Für die DNA-Vakzine wurde eine Kombination aus je 1 mg NTC7382-TPA-LFD1PAD4-mIPS-1, pDNAVaccUltra1-TPA-BclAD1D3-LAMP1 und pDNAVacc-Ultra5-CIITA s. c. bzw. i. c. injiziert. Die Infektion der Vakzinegruppen wurde mit ~1000 Sporen eines voll virulenten Feldisolats der jeweiligen Region durchgeführt. Neben der Gegenüberstellung der Immunogenität und Schutzwirkung der Vakzinen im Ziegenmodell waren die Beobachtungen zum Verlauf der Immunreaktion von SSLV-immunisierten Ziegen im Zeitraum eines Jahres und die diagnostischen Daten zu Infektion von Ziegen (Verhalten, Temperatur, bakterielle Nachweise, Korrelationen und minimale Infektionsdosis) ebenfalls von Interesse. Im Vergleich zu den NLV erzeugten die SSLV-immunisierten Tiere ähnliche oder höhere Antikörpertiter gegen die gemessenen Antigene, wobei die höchsten Titer gegen rPA83 und FIS erreicht wurden. Die Protein-basierenden Vakzinen vermittelten äquivalente Schutzraten zur SSLV (60 – 100 %), wobei der Zusatz von FIS tendenziell eine höhere Schutzwirkung (80 %) erzielte als ohne (50 %). Die DNA-immunisierten Tiere wurden aufgrund der geringen Serokonversion keiner Belastungsinfektion ausgesetzt. Generell war die Immunreaktion auf das in Mäusen hoch immunogen BclA in Ziegen sehr niedrig, was in weiterführenden Studien untersucht werden sollte und eine Basis für eine Verbesserung der Vakzinekomposition der NLV und SSLV bietet. Die Immunisierungen mit der SSLV ergaben eine unterschiedliche Wirksamkeit der Erstimmunisierung, die sich ebenfalls im Antikörperspektrum deutlich von der Auffrischungsimpfung unterschied. Zusammen mit den erhobenen Daten zum Titerverlauf innerhalb eines Jahres konnte ein Vorschlag zur Optimierung des Impfplans der SSLV, auch in Kombination mit einer Protein-basierenden NLV erarbeitet werden. xii Summary The discovery of the Sterne spore live vaccine (SSLV) and subsequently its application in a veterinary context contributed to the global reduction of Anthrax related outbreaks since 1930. Nonetheless the causative agent Bacillus anthracis is still prevalent in some mediterranean countries, South and Central America, Africa and Central Asia, as well as the USA and Canada. Reasons for this are the persistence of the pathogen in the soil, as well as still undefined factors for an ongoing cycle of outbreak and spread of the disease and the limited applicability of the SSLV. This includes the necessity to revaccinate annually, the residual virulence in certain sensitive species (e. g. goats and llamas) and the incompatibility to treat and vaccinate simultaneously. To participate in the ongoing search for alternative vaccines this work was dedicated to evaluate protein- and DNA-based components as potential ingredients for a multi-component non-living vaccine formulation (NLV). For the protein-based NLV these included rPA83 as part of the Anthrax toxin, rBclA and Formalin inactivated spores (FIS) as spore specific antigens, a Capsule-Lipopeptide conjugate as part of the vegetative form of the pathogen and a Lipopeptide-adjuvant. The DNA-vaccines consisted of vector-backbones comprising signal sequences able to direct the integrated antigens (rPA83, PAD4LFD1 and BclAD1D3) to the MHCI, MHCII and the secretory pathway. A sperate vector encoding for a positive MHCIIregulator (CIITA) and a vector internal sequence for the Interferon-ß promotor stimulator (mIPS1) served as adjuvants for the DNA-vaccines. The specified components were tested in NMRI-mice alone and in combination to evaluate their potential for a vaccine combination to be used in goats. The protein components with 25 µg per antigen (108 spores in case of FIS) and 50 µg of Lipopeptide, were injected s. c., while the DNA-vaccines were applied i. c. via GeneGun. All mice were immunized 3 times in 2 week intervals and infected with ~1000 or ~2000 spores of a fully virulent strain (Ames) 3 weeks after the last immunisation. The majority of the groups showed detectable antibody titres against their respective antigens, with protein vaccines generally eliciting higher titres against rPA83 than the DNA-vaccines. Regarding rBclA equivalent high titres were measured for protein- and DNA-vaccines alike, which also corresponded to the anti-FIS titres for groups immunized with rBclA, FIS or both. The Capsule-Lipopeptide conjugate did not elicit high titres against the capsule, possibly due to an immune suppressing epitope. Survival rates ranged between 10 and 100 %, with all protein groups receiving only one antigen showing survival comparable to the process controls and the paramunized animals (< 10 %). Combinations of at least two protein components resulted in > 60 % survival, while full protection was only achieved if FIS was included. All DNA-vectors induced 30 – 50 % protectiveness when given alone. Notably DNA-vectors including BclAD1D3 elicited 50 % xiii survival and sterile immunity. A combination of the most promising vectors encoding for toxin and spore specific antigens achieved 90 % protectiveness in mice. According to the results from the mice trials, the auspicious protein- and DNA-vaccine combinations were tested in goats in comparison to the SSLV in cooperation with our project partners in South Africa and Turkey. The efficacy of the SSLV was assessed in 3 groups which were challenged shortly after the first immunisation, one year after the first immunisation or after the revaccination. The protein combination for the goat trials comprised of the Lipopeptide (500 µg), rPA83 and rBclA (25 µg respective 75 µg for the repetition) and was administered with or without the supplementation of FIS (108 spores). Due to the noted lack of immunogenicity the Capsule-Lipopeptide conjugate was not included. The vectors used for the DNA-vaccine combination included NTC7382-TPA-LFD1PAD4-mIPS-1, pDNAVaccUltra1-TPA-BclAD1D3-LAMP1 and pDNAVaccUltra5-CIITA (each 1 mg) which were injected s. c. respectively i. c. via syringe. The challenge was performed with ~1000 spores of fully virulent field isolates of the respective regions (Turkey and South Africa). Apart from the comparison of immunogenicity and protectiveness between SSLV and NLV in goats, assessment of data concerning the titre development of SSLV-immunized goats during the course of a year as well as detailed diagnostic data during the infection (behavior, temperature, bacterial loads, correlations and minimal infective dose) were integral part of this study. Compared to one another the SSLV-immunized animals showed equal or higher antibody titres against the measured antigens, with FIS and rPA83 being the most immunogen antigens. Utilizing a higher dose (75 µg) the protein-based NLV protected equivalently to the SSLV (60 – 100 %) yielding 50 % protectiveness without FIS and 80 % if FIS was included. The DNA-vaccines showed little to no immunogenicity in goats, thus no challenge was performed on these animals. The humoral reaction against BclA was generally poor in goats, which has not been noted before and could be a basis for further improvements concerning the SSLV and NLV alike. The different immunizations with the SSLV revealed a broad range for the efficacy of the first vaccination as well as a notable difference in the antibody spectrum between first vaccination and revaccination. Together with the recorded data of the antibody titre development throughout a year a more optimal protocol for immunisation with the SSLV, possibly in combination with an NLV was postulated. xiv 1. Einleitung 1 Einleitung 1.1 Entdeckung der Ätiologie von Milzbrand Die heute als Milzbrand, „Anthrax“, „charbon“ oder „woolsorter's disease“ bekannte Krankheit gehört zu einer der am frühsten dokumentierten Leiden der Menschheitsgeschichte. Beschreibungen von Milzbrand ähnelnden Krankheiten reichen bis weit ins Altertum zurück und finden sich in klassischen Werken, wie Homers „Ilias“ oder der „Ab Urbe Condita Libri“ von Livius, wieder [27][323]. Die Ergründung des ursächlichen Agens dieser Krankheit, welche als wiederkehrende Pest in Schafen und Rindern Weideland auf Dauer verseuchen konnte, gewann allerdings erst im Zuge der fortschreitenden Industrialisierung im 19. Jahrhundert an Brisanz. Durch den vermehrten Import von Wolle nach England kam es zwischen 1837 und 1880 zu vermehrten Ausbrüchen von Milzbrand unter Arbeitern der Wollindustrie, daher auch die Bezeichnung „woolsorter's disease“ [178]. Die Aufklärung der Ursache dieser grippeähnlichen Krankheit begann bereits 1823 mit Demonstrationen Éloy Berthelémys zur Infektiösität der Krankheit [223]. 1850 berichtete Pierre Rayer von fadenförmigen Gebilden im Blut verendeter Tiere. Auf diesen Beschreibungen aufbauend konnte Casimir Davaine 1863/64 die Übertragung der Krankheit durch infiziertes Blut nachweisen und Ernst Tiegel und Edwin Klebs (1864) die Infektiösität durch Filterung des infizierten Blutes negieren [323]. Diese Beobachtungen führten zu der Annahme, dass die Krankheit durch einen sich im Wirt vermehrenden, lebenden Organismus verursacht wird, der schlussendlich über eine Septikämie zum Tode führt. Die Bestätigung dieser Annahme, die Zuordnung der Krankheit Milzbrand zu einem spezifischen Bacillus anthracis (B. anthracis) und die Beschreibung des Lebenszyklus von B. anthracis gelang 1876 Robert Koch [171]. Durch R. Kochs Beobachtung der Germination und Sporulation konnte zudem erstmals, neben der Infektiösität von kontaminierten Blut, der Bezug der Krankheit zu kontaminierten Böden und leblosem Material hergestellt werden. Damit war Milzbrand die erste vollständig umschriebene Krankheit, die einem spezifischen Krankheitserreger zuordenbar war und untermauerte die damals noch umstrittenen Keimtheorie. 1.2 Taxonomie und Morphologie Die Gattung Bacillus ist eine sehr heterogene Gruppe von Endosporen bildenden Bakterien mit mehr als 200 Arten, die die Mehrheit der gefundenen Bakterien im Boden stellt [265]. Als pathogene Spezies dieser Gattung sind bisher nur Bacillus cereus sensu stricto (B. cereus), Bacillus thuringiensis, B. anthracis und Bacillus cytotoxicus bekannt [245][121]. Diese bilden zusammen mit den apathogenen Vertretern Bacillus mycoides, Bacillus pseudomycoides, Bacillus medusa und Bacillus weihenstephanensis, die genetisch sehr ähnliche B. cereus sensu lato 1 1. Einleitung Gruppe [245]. Durch phänotypische Klassifizierung, Nachweis der Virulenz und Genotypisierung lässt sich B. anthracis als eigenständige Spezies dieser Gruppe unterscheiden [136][324], wenngleich bereits mehrfach verschiedene B. cereus Stämme mit B. anthracis ähnlichen Eigenschaften beschrieben sind [140][165]. Für gewöhnlich zeichnet sich B. anthracis durch eine charakteristische Morphologie auf Blut- und Nähragar aus, auf denen flache, matte, weiße oder gräulich-weiße Kolonien gebildet werden, die eine klebrige Konsistenz mit meist fransigen Ausläufern aufweisen (Abbildung 1) [236]. Das Bakterium ist nicht hämolytisch, unbeweglich, sensitiv gegenüber Penizillin und dem diagnostischen „Gamma Phagen“ und bildet in Anwesenheit von Bicarbonat/CO2 eine charakteristische Kapsel [236]. Durch die Abklärung dieser Eigenschaften ist eine eindeutige Zuordnung in den meisten Fällen möglich und kann über eine diagnostiAbbildung 1: Koloniemorphologie B. anthracis Kolonien auf einer ColumbiaBlutagarplatte (CBA) nach über Nacht (üN) Inkubation bei 37 °C und 5 % CO2-Atmosphäre sche PCR, mittels Detektion der, die Pathogenitätsfaktoren kodierenden, Plasmide pX01 und pX02 und spezifischer chromosomaler DNA-Sequenzen, verifiziert werden [25] [238][289]. Mit 182 kb ist pX01 das größere der beiden charakteristischen „Milzbrandplasmide“ und kodiert für die Toxinkomponenten des Erregers [213]. Die Gene für den Auf- und Abbau der Kapsel sind auf einem zweiten, 95 kb großen Plasmid kodiert (pX02) [331][117]. Die Anwesenheit beider Plasmide sowie die Anzahl der vorhandenen Kopien haben direkten Einfluss auf die Virulenz von B. anthracis [86][59]. Neben diesen Virulenzfaktoren bestimmt die Fähigkeit des Bakteriums zur Überdauerung ungünstiger Umweltbedingungen mittels Sporulation die andauernde Verbreitung und Prävalenz des Erregers. 1.2.1 Aufbau der vegetativen Zelle B. anthracis ist ein grampositives, aerobes oder fakultativ anaerobes Bakterium, welches unter dem Mikroskop in vegetativer Form als 3 – 6 µm langes und 1 – 1,5 µm breites charakteristisch Stäbchen, mitunter in Anordnung von langen, bambusartigen Ketten zu erkennen ist [86] (Abbildung 2 und 4). Dem Aufbau der vegetativen Zelle von innen nach außen folgend (Abbildung 3), grenzt die zytoplasmatische Abbildung 2: Kettenbildung Vegetative Zellen von B. anthracis im Blutausstrich von infizierten Ziegen mittels Azur B angefärbt. Membran den Zellinhalt nach außen ab. Sie besteht aus Phospholipiden, Fettsäuren und min- 2 1. Einleitung destens 25 verschiedenen Membran- und 13 Lipoproteinen, welche zum Teil für die Faltung, Translokation und/oder Verankerung von extrazellulären Antigenen verantwortlich sind [342] [363]. Oberhalb der zytoplasmatischen Membran besitzt B. anthracis als Vertreter der grampositiven Bakterien eine dicke Zellwand. Sie ist aus Peptidoglycan und assoziierten Polymeren aufgebaut und dient vornehmlich der Stabilisierung der Zellform, sowie dem Schutz des Zellkörpers [96]. Außerhalb der Peptidoglycanschicht besitzt B. anthracis eine zweidimensionale, proteinhaltige, parakristalline Surface Layer (SLayer), welche die gesamte Zelle schützend umschließt [141][267][290][291]. Für B. anthracis sind gleich zwei, jeweils 94 kDa große S-Layer Proteine bekannt [212]: das Surface array protein (Sap) [87][81] und das Extractable Antigen 1 (EA1) [209]. Beide Proteine sind in geringen Mengen auch in der Humanvakzine AVP enthalten und fungieren sowohl Abbildung 3: Aufbau der vegetativen Zelle Elektronenmikroskopische Aufnahme eines Teilausschnitts einer bekapselten vegetativen Zelle von B. anthracis; Die markierten Oberflächenmerkmale sind die Kapsel (C – Capsule), die S-Layer (S – Surface-Layer), die Peptidoglycanschicht (P) und die zytoplasmatische Membran (m); Der Maßstab hat eine Länge von 250 nm [208] dort als auch in der Sterne Sporen Lebendvakzine (SSLV) oder bei natürlichen Infektionen als immunogene Antigene [9][84]. Eine partielle protektive Wirkung konnte allerdings bisher nur für EA1 in Mäusen festgestellt werden [332]. 1.2.1.1 Die Kapsel Im Gegensatz zu den meisten bakteriellen Kapseln ist die von B. anthracis gebildete äußerste Schicht nicht aus Polysacchariden [319], sondern aus γ-verketteten Proteinpolymeren der D-Glutaminsäure aufgebaut [143] [340]. Die Gene zur Bildung und Regulation dieser Poly-γ-D-Glutaminsäure (PGA) sind Abbildung 4: Kapselfärbung Färbung der Kapsel von B. anthracis indirekt mittels Tusche (links [208]) und direkt mit Azur B (rechts [231]) auf dem Plasmid pX02 kodiert [117][331], wobei die Kapselproduktion auf transkriptioneller Ebene von der Anwesenheit von Bicarbonat bzw. Indikatoren der Wirtsumgebung und des Mediums abhängig ist [288][97]. Entsprechend der Wachstumsbedingungen entsteht Kapselmaterial unterschiedlicher Länge (20 – 1500 kDa) [128][249], welches als schützende Schicht an der Zellwand oder der Zellmembran verankert ist [96]. Über Färbung mit Methylenblau [202] 3 1. Einleitung oder Azur B [231] oder indirekt mit Tusche [77] kann die Kapsel in mikroskopischen Aufnahmen sichtbar gemacht und zur Diagnostik hinzugezogen werden (Abbildung 4). Durch Degradierung werden Kapselfragmente in großer Zahl freigesetzt. Diese Fragmente dienen vermutlich als Täuschkörper zum Schutz vor dem Komplementsystem, zur Dämpfung der Reaktivität von Immunzellen und zur Behinderung der Ausreifung dendritischer Zellen [203][154]. Die Kapsel selbst wirkt nicht toxisch [292], erhöht aber die Virulenz von B. anthracis, da sie die Phagozytose der vegetativen Zelle durch Makrophagen beeinträchtigt [242] [163][275]. Die Opsonierung des Zellkörpers durch das Komplementsystem und spezifischer Antikörper wird ebenfalls behindert, da die Kapsel zum einen selbst kaum immunogen ist und zum anderen immunogene Epitope verdeckt [163][208]. Damit ist die Kapsel wesentlich an der Etablierung der Infektion beteiligt, da sie den Erreger vor dem Immunsystem abschirmt und dessen Vervielfältigung, die schlussendlich über Bakteriämie oder/und Toxämie zum Tode führt ermöglicht [242]. 1.2.1.2 Die Toxinkomponenten Neben der Kapsel stellen die sekretierten Toxinkomponenten von B. anthracis den zweiten Virulenzfaktor dar, der über pX01 ebenfalls plasmidkodiert ist [213]. Bereits 1954 fanden Harry Smith und James Keppie [293] eine „toxische, nicht zelluläre Komponente von Milzbrand“, die trotz Abtötung der Bakterien bei Tieren zum Tode führen konnte. Diese „Komponente“ besteht aus drei Teilen, die für sich genommen keine toxische Wirkung haben: dem schützenden Antigen (PA – engl. Protective Antigen), dem Letalfaktor (LF – engl. Lethal Factor) und dem Ödemfaktor (EF – engl. Edema Factor) [295][17]. Sie gehören zur Gruppe der binär oder ABToxine, die sich aus zwei funktionell ergänzenden Untereinheiten aufbauen, von der eine die enzymatische Aktivität beinhaltet (LF und EF) und die andere die Bindungsfähigkeit vermittelt (PA) [110]. Durch Kombination von PA mit LF wird das tödlich wirkende Letaltoxin (Letx) gebildet. Analog dazu bilden PA und EF das Ödemtoxin (Edtx), das nur bedingt tödlich ist aber großflächige Ödeme verursacht [295][17]. Dabei findet zunächst keine Bindung zwischen dem 83 kDa großen PA (PA83) [355] und EF oder LF statt. Zur Entfaltung der Wirksamkeit der Toxine muss PA83 zunächst proteolytisch in PA63 und PA20 gespalten werden [168][83]. Das entstandene PA63 heptamerisiert [216] und bildet eine ringförmige Präpore, die kompetitiv bis zu drei Moleküle LF und/oder EF binden kann [222]. Der entstehende Toxinkomplex gelangt durch rezeptorvermittelte Endozytose [80][37][274] [115] in die Wirtszelle, wo die sukzessive Freisetzung von EF und LF aus dem Endosom in das Zytosol [215][60] deren enzymatische Wirkung ermöglicht [370]. 4 1. Einleitung 1.2.2 Aufbau der Spore Bakterielle Sporen sind in nahezu allen Umweltproben vorhanden und können potentiell Millionen von Jahren in dieser resistenten Dauerform überleben [47][339]. Sie sind größtenteils metabolisch inaktiv, resistent gegenüber einer Vielzahl an natürlichen Stressfaktoren und somit ideal geeignet ungünstiger Bedingungen über längere Zeiträume zu überdauern [5][277][72][226]. Die ausgereifte Spore von B. anthracis ist im Durchschnitt 1,3 µm lang, 0,7 µm breit [51] und aus einer Reihe von Schichten mit spezifischer Funktion aufgebaut, die einem lichtundurchlässigen Kern umgeben (Abbildung 5). Der Kern enthält das dicht gepackte, genetische Material [278] und wird von einer inneren und äußere Membran umfasst. Der zwischen den Membranen liegende, lichtdurchlässige Kortex beinhaltet die Peptidoglykanschicht, die eine Schlüsselrolle in der Erhaltung des Ruhe- Abbildung 5: Sporenaufbau Elektronenmikroskopie einer Spore; sichtbar sind Kern (Cr – core), Kortex (Cx – cortex), Mantel (Ct – coat), Interner Zwischenraum (IS) und das Exosporium (Exo); Der Maßstab hat eine Länge von 700 nm [73] zustands der Spore und der Hitzebeständigkeit einnimmt [224]. Der mehrschichtige Sporenmantel grenzt den Kortex nach außen ab, besitzt eine selektive Durchlässigkeit und vermittelt der Spore durch die Bildung charakteristischer Rillen Flexibilität beim Ein- und Ausstrom von Wasser [346][5][51][72][74]. Die über 50 bisher für B. anthracis gefundenen, mit dem Mantel assoziierten Proteine [179][137] sind potentielle Kandidaten für zukünftige Vakzineformulationen [111]. Die äußerste Struktur der Spore ist ein ca. 10 – 20 nm vom Mantel entferntes, ballonartig aufgespanntes Exosporium, das aus einer parakristallinen Basalmembran mit aufgelagerten, haarähnlichen Strukturen aufgebaut ist [108][18][137]. Es ist elastisch, in der Größe variabel und nicht mit der Spore verbunden [72][137]. Ein Fehlen des Exosporiums wirkt sich nicht signifikant auf die Virulenz des Erregers aus [33][111], obgleich es für die Adhäsion an Makrophagen und anderen Oberflächen von Bedeutung ist [172][35]. Ebenso maskiert es Epitope der darunter liegenden Schichten, die von Makrophagen über Mustererkennungsrezeptoren erkannt werden können [16]. Generell sind alle Bestandteile des Exosporiums aufgrund ihrer direkten Assoziation mit der Umwelt als potentielle Vakzinekomponenten von Interesse. Besonders herauszustellen ist hierbei das „Bacillus collagen-like protein of anthracis“ (BclA) als primärer Bestandteil der peripheren Schicht aus feinen haarähnlichen Filamenten (Abbildung 5) [310][296]. Als immunodominantes Oberflächenprotein der Spore war es das erste identifizierte Protein des Exosporiums [310][296]. Es ist vermutlich bei der Adhäsion an das Wirtsgewebe involviert [254][34] und hat eventuell auch die Funktion andere immunstimulierende Antigene zu verdecken [279]. Die Virulenz beeinflusst es jedoch nicht direkt [33]. 5 1. Einleitung BclA ist aus 382 Aminosäuren [310] aufgebaut und lässt sich in drei Domänen unterteilen, von denen die mittlere Domäne eine kollagenähnliche Region (CLR) beinhaltet. Diese enthält eine stammspezifische Anzahl an GRX-Tripletts, die zu einer unterschiedlichen Länge der BclAFilamente führen [311]. Als glykosyliertes Trimer ist BclA über dessen aminoterminale Domäne mittels der Proteine ExsFA und ExsFB an der Basalmembran befestigt [310][309][297][32]. Die nach außen zeigende, immunodominante carboxyterminale Domäne weist eine runde Struktur mit Ähnlichkeit zum Komplement Faktor C1q auf und vermittelt die Adhäsionseigenschaften der Spore [32][254][296]. Weitere wichtige Proteine des Exosporiums umfassen neben BclB, das viele strukturelle Analogien zu BclA aufweist und eine Funktion in der Stabilisierung das Exosporiums hat [341][315], BxpA, BxpC, CotB-1, CotB-2, CotY und ExsK [111][251] [296][310]. Diese sind in ihrer Funktion und Relevanz als potentielle Bestandteile einer zukünftigen Vakzine allerdings noch nicht hinreichend charakterisiert. 1.3 Verbreitung und Lebenszyklus Durch strikte Regelungen im öffentlichen Gesundheitssystem und Routineimpfungen von Personal und Viehbeständen in Risikogebieten treten Neuerkrankungen in den Industriestaaten nur noch sehr sporadisch auf und sind meist mit historisch vorbelasteten Arealen, Einschleppung oder mutwilliger Freisetzung assoziiert [86][155]. In bestimmten Ländern Zentral- und Südamerikas, Zentralasiens und Afrikas, sowie im mediterranen Raum, einigen Gebieten Kanadas, der USA und Chinas ist der Erreger indes noch prävalent und verursacht regelmäßig große Epidemien mit immensen Verlusten an Mensch und Tier [357]. Dabei ist immer noch nicht uneingeschränkt geklärt, welche Ursachen und Faktoren in welchem Umfang zur Entstehung und Aufrechterhaltung einer Epidemie beitragen. Unter natürlichen Umständen befällt Milzbrand hauptsächlich wilde oder domestizierte Herbivoren wobei die Ansteckung von Menschen meist über den Umgang mit infizierten Tieren oder Tierprodukten stattfindet [357]. Durch Grasen auf kontaminierten Böden kommt es via Ingestion oder Inhalation zur Aufnahme von dormanten Sporen (Abbildung 6) [327]. Im infizierten Wirt entwickeln sich vegetative Zellen (Germination), die über ihre Vermehrung und Bildung der milzbrandspezifischen Toxine letztendlich zum Tod führen können [330]. Unter Laborbedingungen mittels Germinationsfaktoren [139] ist die Initiation der Germination bereits nach 2 – 10 min abgeschlossen [92][308] und stellt somit einen sehr schnellen Prozess dar. Der Germinationsprozess bei einer natürlich akquirierten Infektion, besonders Fragestellungen zum Ort der Germination und dessen zeitlichem Verlauf, ist noch immer Bestandteil der Forschung. Besonderer Fokus liegt dabei auf der Beteiligung von Makrophagen und dendritischen Zellen als Germinationsort und Transportvehikel [120][119][56][94]. 6 1. Einleitung Abbildung 6: Zyklus der Anthrax Infektion Vereinfachte Darstellung des Infektionszyklus mit Sporen als zentralem Element der Übertragung [357] Nach der Öffnung eines an Milzbrand verendeten Kadavers kommt es durch den Austritt der Bakterien in die Umwelt zu mangelinduziertem Stress, der die Bildung von Sporen (Sporulation) zur Folge hat [190]. Der Umfang und die Rate der Sporulation sind dabei abhängig von Umweltbedingungen wie Temperatur, Luftfeuchtigkeit, pH-Wert, Sonneneinstrahlung und dem Vorkommen von speziellen Kationen [65][217]. Verendete Tiere können bis zu 108 Bakterien pro ml Blut aufweisen und so das Erdreich mit hohen Sporenkonzentrationen verseuchen [8]. Die entstandenen Sporen verbleiben im Erdreich oder werden, durch Abschwemmung oder Vektoren, wie Insekten [321] und Aasfresser [190], weiter verschleppt. Durch orale, aerogene oder kutane Aufnahme der Sporen sind weitere Infektionen möglich wodurch Sporen das zentrale Element des natürlichen Zyklus des Organismus darstellen (Abbildung 6). Unklarheit besteht noch immer inwiefern eine Vervielfältigung der Sporen auch außerhalb des Wirts, im Boden [265], oder die Aufkonzentrierung von Sporen durch klimatische und geographische Bedingungen in sogenannten „concentrator areas“ [334] eine signifikante Rolle bei der Bildung von Infektionsherden und der Persistenz des Erregers spielen [67]. 7 1. Einleitung 1.4 Pathogenese (Mensch und Tier) Abhängig vom Eintrittsort der Sporen in den Körper unterscheidet man kutanen, pulmonalen, oral/gastrointestinalen und neuerdings injektionsbedingten Milzbrand [258][19][86]. Der Krankheitsverlauf ist dabei in Abhängigkeit von der Spezies, dem Infektionsweg, dem infizierenden Stamm, der Dosis und des gesamtheitlichen Gesundheitszustands subakut bis hyperakut oder chronisch. Der sogenannte Hautmilzbrand entsteht über Hautläsionen bei Kontakt zu infektiösem Material und stellt die häufigste Infektionsart beim Menschen dar [86][126]. Die Infektion bleibt dabei lokal, sodass nach 2 bis 7 Tagen an der ursprünglichen Läsion eine kleine Pustel entsteht. Diese kann sich unbehandelt weiter ausbreiten und bildet später eine Nekrose mit charakteristischem dunklen Schorf [126]. Erhebliche Gefahr geht hierbei von Ödemen aus, die potentiell die Luftwege abschnüren [86]. Die Sterblichkeitsrate bei unbehandelten Fällen beträgt 20 % [86] und lässt sich durch Behandlung mit gängigen Antibiotika auf 1 - 5 % reduzieren [104][170] [116]. Im Gegensatz dazu ist der pulmonal aufgenommene Lungenmilzbrand häufiger tödlich, da die Symptome bis kurz vor dem Tod einer milden Atemwegsinfektion ähneln und eine richtige Diagnose ohne Verdachtsmoment kaum möglich ist [86]. Akute Symptome treten plötzlich auf und umfassen Atemnot, Zyanose, hohe Temperatur und Kreislaufkollaps [126] und führen unbehandelt in > 80 % oder behandelt in 60 % der Fälle binnen 24 Stunden zum Tode [71][155] [116]. Es wird vermutet, dass inhalierte Sporen durch alveolare Makrophagen aufgenommen und in die Lymphknoten transportiert werden, wobei sie innerhalb der Makrophagen germinieren und nach Freisetzung in den Lymphknoten eine systemische Infektion auslösen [261][120]. Generell bedarf es eine um mehrere Größenordnungen höhere Dosis an Sporen als bei anderen Infektionswegen, um eine Infektion durch Aerosole herbeizuführen [14]. Eine Ansteckung auf diese Weise unter natürlichen Bedingungen wurde bei Tieren bisher nicht nachgewiesen und gilt als extrem unwahrscheinlich. Die Infektion über orale/gastrointestinale Aufnahme von Sporen ist vermutlich die häufigste natürliche Infektionsart. Im Menschen tritt diese Form nur beim Verzehr von infiziertem Fleisch auf und stellt daher vor allem in ärmeren Bevölkerungsschichten eine Problematik dar [126]. Ähnlich wie bei kutanem Milzbrand findet der Eintritt der Sporen in den Körper über eine Läsion in den Verdauungsorganen statt und verursacht eine Magen-Darm-Entzündung mit Symptomen wie Übelkeit, Erbrechen, Appetitlosigkeit, Fieber und teilweise blutigem Durchfall [116]. Unbehandelt liegt die Sterblichkeit bei 25 - 75 % [116]. Eine weitere sehr seltene Form bzw. symptomatische Ausprägung der genannten Infektionswege ist die Milzbrand-Meningitis mit typischen Symptomen wie Leukozytose und hämorrhagischer, eitriger Spinalflüssigkeit [126]. Sie kann als Zusatzkomplikation in allen genannten 8 1. Einleitung Milzbrandformen auftreten. Gerade die favorisierte Behandlungsmethode mit Penizillin schlägt bei Milzbrand Meningitis nicht an [175]. Weitere Antibiotika wie Erythromyzin, Chloramphenicol, Gentamicin, Ciprofloxacin und Tetracyclin können aber ebenfalls zur Behandlung verwendet werden [187]. Zusätzlich werden anti-Toxin Antikörper ebenfalls angewendet, da auch das Abtöten der Bakterien durch Antibiotika allein, ab einer gewissen Toxinkonzentration, ein Fortschreiten der Symptome durch die anhaltende Toxinwirkung nicht unterbindet [10] [283]. Bei Tieren, insbesondere Herbivoren verläuft die Krankheit häufig perakut oder sogar hyperakut, mit zunächst abwesenden oder unspezifischen Symptomen. Einem plötzlich auftretenden Schlaganfall (Apoplexie), der auf einer Septikämie beruht, folgt kurz nach dem Auftreten erster Symptome der Tod [126]. Bei einem akuten Krankheitsverlauf sind Krankheitssymptome bis zu 2 Tage vor dem Tod erkennbar und umfassen Herz-Kreislauf-Probleme, Fieber, Depression, hämorrhagische Schleimhäute, ödematöse Schwellung der Zunge und des Abdomens, sowie speziesbedingt Austritt von Blut aus den Körperöffnungen. Ein subakuter teils chronischer Verlauf ist ebenfalls möglich aber nicht die Regel [126]. Entsprechend der Sensitivität gegenüber Infektionen und der Schwere des Krankheitsverlaufs lassen sich am Beispiel von Mausinzuchtlinien mindestens zwei Gruppen unterscheiden [99][221][218][314]: Mauslinien mit Letx-sensitiven Makrophagen, die gegen eine Infektion resistenter sind als solche mit Letx-resistenten Makrophagen. Die Sensitivität der Makrophagen beruht dabei auf den entsprechenden Allelen für den Nalp1b Lokus [31], die bei sensitiven Makrophagen durch Letx zu einer Aktivierung des „Kaspase-1-abhängigen Inflammasoms [207]“ führen. Dies resultiert zum einen, über Ausschüttung inflammatorischer Zytokine, in einer potenten Immunreaktion des angeborenen Immunsystems gegen die Infektion [184][134][52] und zum anderen zum antimikrobiell induziertem Zelltod (Pyroptose) der Makrophagen [371]. Dabei besteht die Wirkung des Letx auf Makrophagen und anderen myeloiden Zellen im frühen Stadium der Infektion in der Behinderung der antibakteriellen Funktion der Phagozyten und somit der Unterstützung der Ausbreitung des Infekts. Die terminale Letalität von Letx und Edtx beruht allerdings auf deren systemischen Wirkung, die zur Schädigung von Zellen des HerzKreislauf-Systems und der Leber führt [197]. Neben den charakterisierten Inzuchtmodellen für Ratten und Mauslinien [99][354] sind als besonders sensitive Wildtiere Reh, Büffel, Gnu, Gazellen, Kudu und Rentier, sowie Alpakas, Lamas, Zebras und Ziegen [142][88][48][300][303] betroffen. 9 1. Einleitung 1.5 Impfstoffe Durch die Entdeckungen von R. Koch und John Bell waren die Voraussetzungen für eine Verbesserung der präventiven Maßnahmen zur Vermeidung von Milzbrandinfektionen [171][21] [178] gegeben. Sie bildeten die Grundlage der Folgearbeiten von Louis Pasteur und Max Sterne, die zur Entwicklung erster Veterinärimpfstoffe führten [237][300]. Diese und davon abgewandelte Impfstoffe sind seit dem Ende des 19. Jahrhunderts bis heute in der Tiermedizin im Einsatz [322] und ermöglichten eine rapide Verminderung der Milzbrandfälle in Tierbeständen und beim Menschen. Erst der mögliche Einsatz des Erregers als Biowaffe, der vermutlich erstmals im 1. Weltkrieg geplant wurde [356][252], rückte eine Verbesserung der vorhandenen Impfstoffe, vor allem in Hinblick auf die Anwendung im Menschen, wieder in den Vordergrund und führte letztendlich zur Entwicklung der heutigen gebräuchlichen humanen Milzbrandvakzinen. 1.5.1 Historische Entwicklung der Lebendvakzinen gegen B. anthracis Bereits 1880 wurde von William Smith Greenfield und später durch L. Pasteur gezeigt, dass eine verlängerte Inkubation von B. anthracis bei erhöhten Temperaturen (42 – 43 °C) zur Attenuierung und zum Verlust der Sporulationsfähigkeit führt [317]. Wie sich später herausstellte, basierte die festgestellte Attenuierung auf einem partiellen Verlust des hitzesensitiven Plasmids für die Toxinproduktion (pX01) [213]. Prinzipiell erhöhte sich durch die fortdauernde Passage bei hohen Temperaturen sukzessive der prozentuale Anteil an pX01-defizienten Bakterien, sodass die Restvirulenz der resultierenden Mischkultur in Abhängigkeit von der Inkubationszeit abnahm [213]. L. Pasteur nutzte dies aus und entwarf einen Impfplan, der eine Impfung mit zwei verschieden Inokula (Typ I und II) von unterschiedlichem Attenuierungsgrad im Abstand von 12 Tagen erforderte [237]. Die erfolgreiche Testung dieser als Duplex Lebendvakzine bekannten Impfstrategie fand 1881 in Pouilly-le-Fort an Schafen statt [237]. Diese erste Lebendvakzine wurde bis weit in die 1930er Jahre weltweit als Mittel der Wahl zur veterinären Milzbrandprophylaxe in Rindern und Schafen eingesetzt [126]. Deren Schutzwirkung beruhte unter anderem auch auf der minimalen Anwesenheit der Toxine, da Reinkulturen ohne Toxine nicht schützend sind [144]. Die Anwendbarkeit war aufgrund der umständlichen Präparation, der variablen Wirksamkeit, der Variationen im Attenuierungsgrad und der daraus resultierenden Krankheitsausbrüche durch die Immunisierung begrenzt [302][304][326]. Ebenso war eine Vakzinierung anfälliger Spezies aufgrund der Restvirulenz der Präparation nicht möglich [302]. Ein später von M. Sterne isolierter toxinogener Stamm (34F2) mit fehlendem Kapselplasmid (pX02) [302][300] bildete ab 1937 die Grundlage für eine neue Lebendvakzine (SSLV – Sterne Sporen Lebendvakzine), die für viele Spezies als unbedenklich galt und weitaus weniger Ne- 10 1. Einleitung benwirkungen aufwies [301][299][305][126]. Ursprünglich wurde der Stamm auf 50 % Nähragar für 24 Stunden unter 30 % CO2 angezogen und enthielt 105 – 106 Sporen pro ml in 0,5 % Saponin und 50 % Glyzerin [302][300][304]. Wenngleich die Sporenkonzentration heute etwas höher ausfällt (~ 107 Sporen pro ml), hat sich an der Zusammensetzung seither nicht viel verändert, sodass die SSLV als relativ sichere, effektive und kosteneffiziente Vakzine bis heute in Tieren verwendet wird [126]. Dennoch wird die Forschung nach Alternativen weiter vorangetrieben, da auch dieser Impfstoff mit einigen Nachteilen behaftet ist. So ist die Schutzdauer nur begrenzt und muss jährlich aufgefrischt werden, was vor allem beim Einsatz in Wildbeständen bedrohter Populationen problematisch ist [326]. Des Weiteren ist auch hier eine Restvirulenz in verschiedenen Spezies vorhanden [328][353][326], sodass es bei sensiblen Tieren (beispielsweise Ziegen) notwendig ist, 4 Wochen vor der eigentlichen Impfung, die Immunisierung durch eine Prä-Immunisierung mit einem Viertel der Dosis abzumildern [357]. Als wichtigstes Manko der SSLV gilt die Unvereinbarkeit mit einer antibiotischen Behandlung, da diese die Impfwirkung nahezu komplett aufhebt [348][298]. Dies ist besonders bei der Prävention einer Ausbreitung der Krankheit in einer befallenen Herde von Bedeutung. Daher hält die Suche nach alternativen Lebendvakzinen, Totimpfstoffen und azellulären Mehrkomponenten-Impfstoffen zur Gewährleistung eines sichereren, länger anhaltenden und universellen Veterinärimpfschutzes bis heute an. 1.5.2 Entwicklung der lizenzierten Humanvakzinen Aufgrund der möglichen Nebenwirkungen, wie Nekrosen an der Verabreichungsstelle und der gelegentlich vorkommenden Todesfälle in sensitiven Spezies, wurde von einer Verwendung der genannten Lebendvakzinen im Menschen abgesehen. Eine Ausnahme bildet die in der früheren Sowjetunion entwickelte und 1953 im Sanitary-Technical Institute (STI) lizenzierte Lebendvakzine für Menschen, die durch Auftragung eines SSLV-ähnlichen Stammes (STI-1) auf die zuvor aufgeraute Haut oder durch s. c. Applikation angewendet wird [282]. Die Vakzine wurde zwar umfangreich in Menschen und Tieren ohne Nebenwirkungen getestet, ist aber international nicht zur Verwendung im Menschen freigegeben, da entsprechende zugängliche, verifizierbare Veröffentlichungen fehlen [326]. Die Grundsteine zu den heute gängigen Humanvakzinen wurden bereits zu Beginn des 20. Jahrhunderts gelegt. 1904 wurde von Oskar Bail das schützendes Potential zellfreier Flüssigkeit eines Milzbrandödems entdeckt [7]. Diese Arbeiten wurden unter anderem von William Cromartie und Dennis Watson weitergeführt, welchen es bereits gelang Kaninchen, Meerschweinchen, Mäuse, Hamster und Schafe mit ähnlichen Formulationen zu schützen [63][347]. Allerdings war das verwendete, unprozessierte Ödemfluid immer noch toxisch und erzeugte 11 1. Einleitung Hautläsionen. Dieser toxische Effekt konnte später durch Adsorption mit Calciumphosphat entfernt werden, sodass nur noch das „schützende Antigen“ (Protective Antigen, PA) übrig blieb [63][316]. Derart filtrierte Kulturüberstände schützten in Abhängigkeit von der Größe des Inokulums und dem Alter der Kultur Kaninchen, Schafe, Affen und Meerschweinchen, nicht aber Mäuse [112][22]. Nachfolger waren die „Boor und Tresselt Vakzinen“, die für die Anzucht einen intermediär virulenten Stamm verwendeten und die zu impfenden Antigene durch Fällung und Trocknung aufkonzentrierten [29][30][320]. Dieser Ansatz funktionierte ebenfalls gut, war aber wie die Komposition von W. Cromartie und D. Watson, mit dem Makel behaftet, zu großen Teilen aus undefinierten Substanzen zu bestehen. In beiden Fällen war Serum essentieller Bestandteil des Inokulums und die resultierenden Vakzinen per se in ihrer Form als krude Kulturüberstände nicht vollständig ausdefiniert. Unter diesen Bedingungen war keine normierte Vakzine in großer Stückzahl und immer gleicher Qualität zu erzeugen. Dies änderte sich mit der Etablierung und Verfeinerung synthetischer, definierter Medien, die es erlaubten zwar niedrigere aber konstante Antigenkonzentrationen zu erzeugen [41][366][22]. Das von F. Belton und R. Strange in den 1950er Jahren entwickelte Hydrolysat Wachstumsmedium ermöglichte unter Zusatz von Aktivkohle, Kulturüberstände mit vergleichsweise hohem Gehalt an PA [22][326]. Dies bildete die Grundlage für die ab 1965 eingesetzte und 1979 lizenzierte Humanvakzine des Vereinigten Königreichs [326][325]. Diese ist auch unter dem Namen AVP – Anthrax vaccine precipitated – bekannt, da der gewonnene Antigenüberstand des verwendeten Kapsel-defizienten SSLV-Stamms (34F2) durch Präzipitation mit Kaliumaluminiumsulfat (Alaun) abgetrennt und aufkonzentriert wird [326] [116]. Die Vakzine enthält neben PA auch geringe Mengen an EF und LF, sowie zwei Proteine der S-Layer (Sap und EA1), wenngleich bisher unklar ist, welche Bedeutung diese Bestandteile (ausgenommen PA) für die Schutzwirkung haben [9]. Die in den USA in den 1960er Jahren entwickelte Humanvakzine AVA – Anthrax vaccine adsorbed – besteht aus Aluminiumhydroxid adsorbiertem zellfreiem Filtrat einer toxinogenen, unbekapselten, Protease-defizienten V770-NP1-R Kultur [244]. Im Gegensatz zu AVP enthält AVA nahezu kein EF und LF, da der verwendete Stamm hauptsächlich PA produziert und die Kulturbedingungen so ausgelegt sind, den Anteil an EF und LF weiter zu minimieren [244] [157]. Daher enthält AVA höhere Konzentrationen an PA und gilt gemeinhin als verträglicher als AVP [328]. Seit 1972 ist auch AVA für den Gebrauch im Menschen lizenziert [326][325]. Beide Vakzinen können milde Nebenwirkungen am Applikationsort zeigen und beinhalten einen umfangreichen Impfplan der eine jährliche Auffrischung vorsieht [36][244][116]. Dieser und die immer wieder auftretenden Lieferengpässe begründen die oft frühzeitig abgebrochenen oder nicht regelkonformen Impfverläufe [130][325]. Ebenso ist die tatsächliche Effizienz beider 12 1. Einleitung Vakzinen im Menschen bisher nicht hinreichend bewiesen, da die einzigen belastbaren Daten zur Schutzwirkung im Menschen mit einem Vorläufer der AVA Vakzine durchgeführt wurden [36][325]. Hinzu kommt, dass die Standardisierung sehr schwierig ist, die Vakzinen als nicht vollständig charakterisiert gelten und über die Entwicklung der Impfpläne keine lückenlose Dokumentation erhalten ist [325]. Hinweise wonach das Golfkriegssyndrom ebenfalls ursächlich mit der Impfung gegen Milzbrand verknüpft war [325], wie auch die Tatsache, dass die Humanvakzinen auf Grundlage von 30 Jahre alten Wissensstandards entwickelt wurden und aufgrund der Zulassung noch heute so produziert werden, sind der Grund für die andauernde Suche nach Alternativen. Vergleichende Tests in Tierversuchen zeigen indes deutlich, dass die Schutzwirkung der Vakzinen besonders bei Meerschweinchen abhängig vom eingesetzten Infektionsstamm variiert und bei Hamstern und Mäusen weit hinter den Erwartungen zurückbleibt [328] [192][146][89][90]. Dies unterstützend belegen zahlreiche Studien, dass rein auf PA bzw. den Toxinen basierende Impfstoffe nicht ausreichend für einen vollständigen Schutz sind [328] [329][326]. Überdies ist ein speziesübergreifender, universell verwendbarer Indikator zur Vorhersage einer Schutzwirkung nicht vorhanden. Die Bestimmung von Antikörpertitern gegen die Toxine und deren neutralisierenden Eigenschaften haben sich für einige Tiermodelle als gute prognostische Mittel erwiesen [147]. Berichte über die Korrelation der gemessenen Titer zum Überleben sind jedoch in Abhängigkeit vom Versuchsaufbau ambivalent [192][149][329][211], sodass zur endgültigen Evaluierung einer Vakzine ein direkter Test auf das Überleben einer letalen Infektion im avisierten Modellorganismus unabdinglich ist. 1.5.3 Ansätze zur Entwicklung neuer Vakzinen gegen B. anthracis Aus den vorherigen Kapiteln wird deutlich, dass die gängigen lizenzierten Vakzinen zwar einen wichtigen Anteil an der Eindämmung der ökologischen und ökonomischen Konsequenzen sowie der militärischen Bedrohung hatten und auch immer noch haben, ihre Verbesserung bzw. Ergänzung durch neue Vakzinen aber wünschenswert ist. Entsprechend der Entwicklung wissenschaftlicher Standards haben neue Impfstoffe für eine Zulassung in Mensch und Tier striktere Regularien zu erfüllen, als es noch für die SSLV, AVA und AVP der Fall war [325]. Idealerweise sollte ein neuer Impfstoff einfach zu applizieren sein und mit einem möglichst kurzen und unkomplizierten Impfplan eine langanhaltende Immunität vermitteln [329][326]. Zudem sollte er kaum Nebenwirkungen verursachen, vollständig avirulent und einfach und billig nach einem standardisierbaren Verfahren herzustellen sein [329][326][325]. Zur Annäherungen an diese Idealvorstellungen wird daher unentwegt nach neuen Wegen der Applikation, neuen immunogenen Antigenen, effektiveren Adjuvanzien, Methoden zur zielgerichteteren Freisetzung und der Wirkverbesserung vorhandener Antigene geforscht. 13 1. Einleitung 1.5.3.1 Toxinkomponenten Die Identifizierung der einzelnen Toxinkomponenten und die damit einhergehende Möglichkeit, diese aufzureinigen und einzeln zu betrachten, waren grundlegende Voraussetzungen, die eine Weiterentwicklung von azellulären Milzbrandvakzinen erst ermöglichten [295][294][183][259] [355][38]. Auf Basis dieser Erkenntnisse konnte bereits in den 70er Jahren gezeigt werden, dass PA in Abwesenheit von EF und LF eine schützende Wirkung hat, wohingegen für EF und LF allein nur eine marginale schützende Wirkung festgestellt werden konnte [295][294][147]. Daher gilt PA als essentieller Bestandteil neuer Vakzineentwicklungen [86]. Das monomere Polypeptid PA ist aus 735 Aminosäuren aufgebaut, enthält vier strukturelle Domänen [239] und fungiert als Bindungskomponente der enzymatisch aktiven Proteine LF und EF [220]. Wichtige Schlüsselpositionen auf PA zur Verbesserung von Behandlungsmethoden und Impfstrategien stellen sowohl die aminoterminale Domäne 1 (D1) als auch die rezeptorbindende, carboxyterminale Domäne 4 (D4) [287] dar. Wie unter 1.2.1.2 beschrieben wird PA83 innerhalb D1 proteolytisch in PA63 und PA20 gespalten, wodurch die Oligomerisierung von PA63 ermöglicht und die Bindungsstellen für EF/LF freigelegt werden. Eine Blockade der funktionellen Epitope von D1 und D4 kann die Aktivierung von PA83 (D1) bzw. die Bindung von PA83/PA63 an den Wirtszellrezeptor (D4) gänzlich verhindern und somit die Wirkungskaskade der Toxine unterbrechen [193][194]. Antikörpern, die gegen diese Epitope gerichtet sind, wird eine qualitativ höhere Wirkung zugesprochen, da sie neben der Markierung des Toxins als Fremdantigen auch neutralisierende Eigenschaften haben. Eine Reduktion der Toxinantigene auf die für die Blockade wesentlichen Epitope ist daher Inhalt diverser Studien [335][95][193] [194]. Aufgrund der Möglichkeit Antikörper gegen die Bindungsstellen für EF/LF zu generieren, die ebenfalls zu einer Blockade der Toxinwirkung führen können, ist auch die Verwendung von PA63 als Alternative zu PA83 in Vakzineformulationen von Interesse, wenngleich PA63 bisher in vergleichenden Studien eine geringere Immunogenität und Schutzwirkung als PA83 aufwies [124][95][1]. Die immunologische Relevanz des Spaltprodukts PA20 ist hierbei noch unklar. Es gibt Hinweise, wonach dieses Fragment eine apoptotische Wirkung auf Blutleukozyten [127] oder/und eine ablenkende Funktion als bevorzugtes Ziel bei der Bildung von Antikörpern gegen PA hat [248][247]. Weitere Strukturen, die für die Wirkverbesserung von PA von Bedeutung sein könnten, sind funktionale Epitope der Oligomerisation und der Translokation [227][285][286]. Ebenso bieten LF und EF als Bindungspartner von PA zusätzliche Ansatzpunkte zur Generierung neutralisierender Antikörper. LF ist eine hochspezialisierte zinkabhängige Metalloprotease [167], die diverse mitogenaktivierte Proteinkinase-Kinasen (MAPKKs) nahe ihrem aminoterminalen Ende spaltet und damit die Bindung und Phosphorylierung der Substrate der MAPKKs verhindert [54]. Die auf diese Art unterbrochenen regulative Signalwege in der Phosphorylierungskaskade eukaryotischer Zel- 14 1. Einleitung len [76][338][337] wirken sich auf die Reaktion gegen eine Reihe von stimulatorischen Signalen aus [220]. In Makrophagen, welche zunächst als hauptsächlicher Angriffspunkt der Milzbrandtoxine ausgemacht wurden [98][129], führt LF zur Unterbrechung der Signalwege der Antigenrezeptoren [232], sowie Caspase-1 induziert zur Zytokinausschüttung und zum Zelltod [235][184][134][52]. Darüber hinaus sind Kardiomyozyten und vaskuläre, glatte Muskelzellen als Zielzellen in der späten Phase der Infektion relevant, wenngleich auch bei anderen myeloiden Zellen, Hepatozyten und Endothelzellen eine Letx-Wirkung in vitro nachgewiesen werden konnte, deren Beeinträchtigung aber vermutlich nur eine untergeordnete Rolle spielt [198] [221]. Bei einer Größe von 90,2 kDa [38] beinhaltet LF vier unterscheidbare Domänen [233] von denen D1 und D4 für die Neutralisation relevante Angriffspunkte enthalten, da sie die funktionalen Epitope zur Bindung an PA (D1) beinhalten [6], bzw. die katalytische Aktivität vermitteln (D4) [193][233]. Zudem erzeugt LF im Menschen teilweise sogar eine stärkere immunogene Wirkung als PA [11], wobei die Mehrheit der neutralisierenden Antikörper die Epitope betreffen, die eine Bindung an PA vermitteln [193]. Eine marginal schützende Wirkung von LF allein ist ebenfalls bereits in Kaninchen und Mäusen nachgewiesen [243][138]. Zur Kombination der wirksamsten Eigenschaften von PA und LF wurde bereits eine atoxische Kombinationsvakzine bestehend aus D1 von LF und D4 von PA erfolgreich im Mausmodell getestet [11]. Von den drei Toxinkomponenten ist EF bisher am wenigsten erforscht. EF ist eine calmodulinabhängige Adenylatzyklase, die eine Erhöhung der Produktion des zyklischen Adenosinmonophosphats (cAMP) bewirkt [183][182][281] und damit auch die Konzentration von zellulär wirksamen ATP vermindert [197]. Die genauen Zusammenhänge der Edtx-induzierten Veränderungen im ATP-Haushalt der Zelle, die schlussendlich zur Schädigung von Geweben, Bildung von Ödemen und dem Zelltod führen können, sind noch nicht vollständig geklärt [93][198]. Eine koordinative Zusammenwirkung mit Letx wird ebenfalls diskutiert, da Edtx die Sensitivität der Wirtszelle gegenüber den Milzbrandtoxinen erhöht und deren raschen Abbau im Zytosol verhindert [204][269]. Neben myeloiden Zellen, die als Zielzellen der initialen Infektion relevant sind [219], wirkt die systemische Ausbreitung von Edtx im Verlauf der fortschreitenden Infektion vor allem schädigend auf Hepatozyten und Epithelzellen [198]. Mit 88,8 kDa besitzt EF eine ähnliche Größe wie LF, bildet aber nur drei Domänen [259]. Dabei hat die aminoterminale Domäne (D1) eine sehr hohe Ähnlichkeit zur aminoterminalen Domäne von LF und stellt ebenfalls die Bindestelle für PA dar [38]. Die enzymatische Aktivität ist auf den restlichen Domänen verankert [369][177][176]. Wenngleich auch EF Ansatzpunkte zur Entwicklung neuer, spezifischer Antigene bietet, wird ihm als Antigen für eine mögliche Vakzine eher wenig Bedeutung beigemessen, da es alleine nicht schützend ist [295] und bis auf wenige Ausnahmen nicht tödlich wirkt [93][197]. 15 1. Einleitung 1.5.3.2 Antigene der vegetativen Zelle Prinzipiell sind die Toxinkomponenten ebenfalls als vegetative Antigene anzusehen, da sie nur von vegetativen Zellen produziert und sezerniert werden. Zur Blockade einer weiteren Vermehrung der Bakterien und Detektion dieser sind aber Reaktionen gegen den Zellkörper selbst erforderlich, der durch die wenig immunogene Kapsel (PGA – Poly-γ-D-glumatic acid) nach außen abgegrenzt [292][86] ist. In ihre Rolle als Schutzhülle erhöht sie die Pathogenität durch ihre anti-phagozytotischen Eigenschaften [242][50][163][275], durch die Überdeckung immunogener Antigene [208][163], als Wirkungsverstärker in Assoziation mit Letx [85][152] und in freier Form als Täuschkörper [203][154]. Daher ist die Kapsel und die Erhöhung ihrer Immunogenität Bestandteil vieler Studien. Bisher konnte gezeigt werden, dass eine partielle Degradierung der PGA durch enzymatische Spaltung [275] oder eine entsprechende Optimierung der Länge der PGA [174][158] deren Immunogenität bereits verstärken können. Ebenso kann die Konjugation von PGA an andere immunogene Substanzen [50][49], wie z. B. PA [273][180] [105], die Kapsel als T-Zell unabhängiges Antigen zu einem Antigen mit hoher T-Zell-abhängiger Immunogenität konvertieren [345]. Neben der Kapsel rücken verstärkt auch darunter liegende Antigene immer weiter in den Fokus, da auch sie zur Schutzwirkung beitragen können [332]. 1.5.3.3 Antigene der Spore Die Bedeutung von neutralisierenden Antikörpern als bevorzugte Subpopulation ist in der jüngsten Zeit etwas abgemildert worden. So ist in mehreren Studien belegt, dass Antikörper gegen PA auch Sporen detektieren und opsonieren können [352], was die Germination von Sporen und die Abtötung der germinierten Zellen durch Makrophagen beeinflusst [351][62][298]. Andere vegetative Antigene wie EA1 sind ebenfalls auf der Spore nachweisbar und es scheint möglich [332], dass die Spore auch weitere vegetative Antigene beherbergen könnte, die vermutlich aus der Mutterzelle bei der Sporulation ihren Weg in bzw. auf die Spore gefunden haben. Dieser Umstand macht deutlich, dass die Bedeutung der anti-Toxin Antikörper vermutlich nicht alleinig auf ihrem toxinneutralisierendem Potential beruht, sondern dass sie auch an der Detektion und Beseitigung von Sporen beteiligt sind. Die Spore selbst und deren spezifische Antigene sind ebenfalls immunogen [42]. Antikörper, die direkt gegen die Spore gerichtet sind, beeinflussen ebenfalls deren Germination und Aufnahme durch Makrophagen, was die Etablierung einer Infektion verhindern kann und somit zu einer sterilen Immunität beiträgt [79][39][64]. Beide Arten von sporendetektierenden Antikörpern beschleunigen die Aufnahme durch Makrophagen. Ob dies im Einzelfall dem weiteren Infektionsverlauf durch den Transport und die Abschirmung der Sporen durch die Makrophagen zuträglich ist oder zu einer schnelleren Klärung der Infektion führt, muss noch geklärt werden. 16 1. Einleitung Die betreffenden, sporenspezifischen Antigene können Bestandteile des Exosporiums sein, welches wie unter 1.2.2 beschrieben die äußerste Schicht der Spore darstellt. Wenngleich einige der bisher identifizierten und charakterisierten Proteine des Exosporiums ebenfalls eine immunogene Wirkung haben, liegt der Hauptfokus innerhalb der Vakzineforschung bezüglich eines schützenden Sporenantigens auf BclA. Grund dafür ist die Dominanz der gegen BclA gerichteten Antikörper bei Immunisierungen mit Sporenpräparaten [296][297]. Zudem konnte bereits gezeigt werden, dass eine zusätzliche Gabe von BclA zu einer PA-Vakzine deren Schutzwirkung verbessert [125][39]. Eine weitere Möglichkeit, eine sporenspezifische Immunantwort zu erreichen, ist der Einsatz von Formalin inaktivierten Sporen (FIS), welche die Schutzwirkung in Kombination mit PA in bereits getesteten Tierarten erhöhen konnte [42]. Zwar stellen FIS-Präparationen bzw. andere inaktivierte Sporensuspensionen [79] ein undefiniertes Antigen dar, das zudem nicht synthetisch erzeugt wird und die Handhabung des Erregers erfordert. Bisherige publizierte Versuche zeigen aber, dass es möglicherweise einen wichtigen Zusatz als Antigen und Adjuvans in einer Kombinationsvakzine darstellt [106]. Als Bestandteil von intakten Sporen oder FIS-Präparationen überdeckt BclA andere immunogene Antigene und dämpft deren immunogene Wirkung [64] [61]. Entsprechende Präparationen ohne BclA wurden in den letzten Jahren auf Immunogenität und Schutzwirkung untersucht und zeigten vielversprechende Ergebnisse [336], sodass gegebenenfalls weitere Antigene des Exosporiums in den Fokus rücken. 1.5.4 Genetische Immunisierung Eine Alternative zu den Vakzineformulationen, die auf dem Einsatz des kompletten Erregers oder dessen einzelner Bestandteilen beruhen, stellt die genetische Vakzinierung dar. Im weitesten Sinne werden hierbei Gene, die für ein bestimmtes Protein kodieren, in Form von Vektoren (viral oder mittels Plasmid) injiziert, um eine immunologische Reaktion hervorzurufen. Im Idealfall werden professionelle Antigen präsentierende Zellen (APC) an der Injektionsstelle transfiziert. Dabei wird das eingebrachte Gen in den Zellkern eingeschleust, wie eigene DNA transkribiert und translatiert und die resultierenden Proteine über MHCI und auf MHCII [132] T-Zellen [234] präsentiert (Abbildung 7). Beim Vorhandensein von unmethylierten CpG-Motiven innerhalb der applizierten DNA kann die DNA selbst zusätzlich als Aktivator der angeborenen Immunabwehr dienen [173]. Eine funktionierende DNA-Vakzine bietet die Möglichkeit, sowohl zelluläre als auch humorale Immunantworten gleichermaßen zu aktivieren, hat eine hohe Antigenspezifität und bietet die Möglichkeit die Art der Immunantwort zu steuern. Darüber hinaus kann sie sehr viel schneller modifiziert werden und ist weitaus einfacher herzustellen und zu lagern als vergleichbare Prote- 17 1. Einleitung invakzinen [196][78]. Leider ist es momentan nur möglich, Protein-basierende Antigene auf genetische Vektoren zu übertragen, wodurch zuckerartige Antigene nicht in Frage kommen. Zudem kann die Effizienz genetischer Vakzinen geringer als die ihrer Protein-basierenden Gegenstücke sein, was auf Faktoren wie ineffiziente Zielgenauigkeit beim Einschleusung der Vektoren in die APCs, unzureichende Genexpression und schwache Antigenpräsentation zurückzuführen ist [102][262]. Daher werden DNA-Vakzine oft und erfolgreich innerhalb eines „Prime/Boost Ansatzes“ mit Proteinen oder Ganzzell-Vakzinen verwendet [45]. Damit genetische Vektoren auch eigenständig als äquivalente Alternative zu Protein-basierenden Vakzinen eingesetzt werden können, ist die Verbesserung der Applikationsart und die Optimierung der eingesetzten Vektoren und Antigensequenzen, sowie der Adjuvanzien entscheidend [103]. Abbildung 7: Genetische Immunisierung Mechanismus der genetischen Immunisierung: 1) Die Aufnahme von DNA kann frei erfolgen oder durch Integration in Vektoren bzw. die Applikationsart unterstützt werden. 2) Betrifft die Aufnahme der DNA eine APC kann das Antigen direkt prozessiert und über MHC-Rezeptoren präsentiert werden. 3) Wird die DNA von anderen Zelle aufgenommen kann das produzierte Antigen beim Freiwerden durch Lyse oder Sekretion von anderen APC aufgenommen und ebenfalls präsentiert werden. 4) CpG-reiche Motive bakterieller DNA werden vom TLR9 erkannt und triggern die angeborene Immunität. [333] Da extrazelluläre DNA ineffizient aufgenommen und vornehmlich abgebaut wird, muss sie bestenfalls so appliziert werden, dass sie bereits intrazellulär vorliegt [313][263][57][333]. Durch Elektroporation kann, nach der Injektion von DNA-Molekülen, eine intrazelluläre Aufnahme durch elektrische Impulse und die damit einhergehende Permeabilisierung der Membranen forciert werden [200][230][201]. Neben der Elektroporation lassen sich DNA-Moleküle ebenfalls durch Partikel-Bombardement intrazellulär etablieren. Hierfür wird eine GeneGun verwendet, welche DNA-beschichtete metallische Mikropartikel (meist Gold) mit Hilfe hoher Beschleunigungen durch Zellmembranen und -wände befördert [101][349]. Die Form der Applikation beeinflusst hierbei auch die Art der resultierenden Immunantwort und kann unter Umständen ebenso Adjuvanscharakter haben [349]. So haben Feltquate et al. (1996) und später Weiss et al. (2002) gezeigt, dass die Injektion von DNA-Vakzinen eher eine T H1-Antwort mit erhöhten 18 1. Einleitung IgG2a-Titern bewirken kann, wohingegen die Applikation mittels GeneGun eine charakteristische TH2-Antwort mit IgG1-Gewichtung erzeugt [246][91][349]. Daher ist es sinnvoll, die Applikationsart der angestrebten Immunantwort anzupassen. Die erste erfolgreiche Vakzinierung durch ein Plasmid mit einem B. anthracis spezifischen Antigen wurde 1999 durchgeführt [118]. Hierbei wurde lediglich die Sequenz von PA63 flankiert von einem transkriptionalen Promotor und einem Polyadenylierungssignal i. m. eingesetzt und induzierte eine humorale und zelluläre Immunantwort, verbunden mit einer Schutzwirkung in BALB/c Mäusen gegen das Letx. Weitere Versuche mit Domänen von LF [364][243][102] und verschiedenen Vektoren, die ebenfalls i. m. appliziert wurden, waren gleichermaßen erfolgreich, wenngleich eine Auffrischungsimpfung mit Proteinkomponenten zur Verstärkung der Wirkung nötig war. Durch die Verwendung von Signalsequenzen, die das zu exprimierende Gen flankieren, kann die Effizient der eingesetzten Antigene weiter gesteigert werden, da sie eine gerichtete Translation des Antigens bewirken. Durch die Signalsequenz des gewebespezifischen Plasminogen Aktivators (TPA – engl. tissue-type plasminogen activator) ist es beispielsweise möglich, das integrierte Antigen nach der Translation zu sekretieren [186]. Ebenso sind Signalsequenzen zur Verankerung an der Zellmembran (PLAP – plazentarische alkaline Phosphatase [107], Einschleusung in den MHCII-Präsentationsweg über Verankerung in der endosomale Membran (LAMP1 – lysosomal-assoziiertes Membranprotein [368] und zur MHCI-spezifischen Präsentation über die Degradierung im Proteasom (Ubiquitin (Uq) [260]) anwendbar und bereits erfolgreich eingesetzt worden [124][125][123][211]. 1.5.5 Adjuvanzien Die immunogene Wirkung gereinigter, zellfreier Antigene ist gewöhnlich sehr gering und bedarf dem Zusatz von Adjuvanzien, die über lokale Gewebereizung oder Bindung der Antigene die Immunantwort gegenüber dem eingesetzten Antigen (meist durch Aktivierung von APC) unspezifisch steigern [151]. Darüber hinaus können Adjuvanzien und Pufferbedingungen die Zugänglichkeit gewisser Epitope verändern und steuern somit auch die Effizienz der Vakzine. Wenngleich auch einige Antigene selbst eine Adjuvanswirkung haben, so sind die wichtigsten Vertreter meist nicht infektiöse Bestandteile von Bakterien wie Zellwände, Polysaccharide, Hitzeschockproteine oder DNA. Durch Zusatz von unspezifischen, abgetöteten Corynebakterium ovis, Bordetella pertussis oder Mycobakterium tuberculosis (Freunds Adjuvans) zum Humanimpfstoff oder aufgereinigtem PA kann deren Schutzeffekt beispielsweise erheblich erhöht werden [329][330]. Allerdings verursachen Emulsionen oder Suspensionen aus Ganzzell-Präparationen meist eine sehr starke Entzündungsreaktion, sind sehr unspezifisch und beinhalten immer das Risiko der Verwendung 19 1. Einleitung eines vollständig abzutötenden Erregers [100]. Daher sind Bestrebungen zur Suche nach alternativen Adjuvanzien, welche auf die Form der Anwendung, die Art der Antigene und die angestrebte Immunantwort zugeschnitten sind, immer essentieller Bestandteil einer Impfstoffentwicklung. Zur Verfeinerung der Adjuvanswirkung gibt es Ansätze, die basierend auf den beschriebenen Ganzzell-Adjuvanzien nur einzelne bakterielle Bestandteile verwenden. Dazu gehören beispielsweise DeTox, eine Mischung aus detoxifiziertem Endotoxin und dem Zellwandskelett eines attenuierten Tuberkulosestammes, sowie verschiedene aufgereinigte Faktoren von Mycobakterium phlei oder monophosphoryl lipid A aus Salmonella minnesota, ein ebenfalls detoxifiziertes Lipopolysaccharid [3][257][326]. Diese und ähnliche mikrobiell erzeugte Wirkstoffe sind in Kombination mit PA erfolgreich erprobt [149][145], zeigen aber wie viele andere Ansätze nicht die gleiche Wirksamkeit in verschiedenen Tiermodellen. Zu den nicht mikrobiologisch hergestellten Adjuvanzien gehören die Aluminimsalze, wie Aluminiumhydroxid (Alhydrogel), Alaun (Alum) und Aluminiumphosphat, welche bisher als einzige für eine Anwendung im Menschen zugelassen sind. Das beispielsweise in AVA verwendete Alhydrogel erzeugt in Kombination mit PA eine starke humorale Immunantwort [122][95], ist aber in der Schutzwirkung stark speziesabhängig [90][149], sodass auch diese Adjuvanzien nicht universell einsetzbar sind. Weitere Adjuvanzien dieser Art sind verschiedene Lipide [4] [149], Saponine [162][161][145] oder andere Zellbestandteile, die in Kombination mit Antigenen in Form von Emulsionen, Liposomen, Microkapseln oder anderen Trägermaterialien verabreicht werden können [149][145]. Ein Beispiel hierfür sind Lipopeptide und Lipopeptid-Konjugate. Bakterielle Lipopeptide als Bestandteil der äußeren Membran von gramnegativen Bakterien sind charakteristisch aus der ungewöhnlichen Aminosäure Dihydroxypropylcystein (Dhc), acetyliert mit 2 – 3 Fettsäuren, aufgebaut [114][131]. Als Effektoren der angeborenen Immunität stellen sie hochaktive Immunadjuvanzien dar, die über Toll-like Rezeptoren (TLR) erkannt werden [133][2]. Die Art und Höhe der Adjuvanswirkung ist dabei stark von der Struktur des Lipopeptids und dessen Azetylierungsgrad abhängig [109]. Daher könAbbildung 8: synthetisches Lipopeptid (Bsp.) Synthetisches Lipopeptid Pam3Cys-SKKKK; dreifach acetyliert [358] nen durch gezielte Manipulation und Konjugation von Antigenen oder Epitopen am aminoterminalen Ende des Dhc spezifische, auf den jeweiligen Impfstoff zugeschnittene Adjuvanzien erstellt werden [359][271][358][109]. Im Speziellen sind hier Palmitinsäure (Pam) acetylierte Lipopeptide zu nennen (Abbildung 8), welche entsprechend des Acetylierungsgrads TLR-2 und TLR-6 (diazetyliert, [229]) bzw. TLR-1 und TLR-2 (triazetyliert, [312]) vernetzen und mit unterschiedlichen 20 1. Einleitung Modifikationen bereits erfolgreich als Adjuvans in antiviralen Vakzinestudien eingesetzt wurden [70][270][68][225]. Zur Unterstützung von DNA-Vakzinen können teilweise die bereits beschriebenen Adjuvanzien verwendet werden, wohingegen auch gänzlich andere Adjuvanzien möglich sind, welche sich zum Teil in Wirkung und Funktion vollkommen von den zuvor beschriebenen Adjuvanzien unterscheiden. Durch die charakteristische Spezifität der DNA-Vakzine selbst ergibt sich für die Adjuvanzien ein ähnlicher Ansatz, der weg von den unspezifischen Agitatoren der angeborenen Immunität und hin zu spezifischen, die Stoffwechselwege beeinflussenden Wirkmechanismen führt. Um die Einschleusung von Antigenen in den MHCI- und MHCII-Präsentationsweg weiter zu verstärken, kann eine künstlich erzeugte Hochregulierung dieser beiden Rezeptoren eine starke Adjuvanswirkung haben [164]. Der Klasse II Haupthistokompatibilitätskomplex Transaktivator (kurz CIITA) ist ein Hauptfaktor zur Kontrolle der Gene für die MHCII-Präsentation [318][253]. Er induziert die Expression von MHCI- und MHCII-Rezeptoren auf der Zelloberfläche [206][12] und resultierte bei Anwendung in Mäusen in verstärkter Antigenpräsentation auf diesen [164]. Die Sequenz für den Interferon-ß-Promotor Stimulator 1 (IPS-1) kann ebenfalls als Adjuvans eingesetzt werden [160][276]. Doppelsträngige und einzelsträngige RNAs werden von RIG-1like Rezeptoren (RLR) [159], einer Untergruppe von Mustererkennungsrezeptoren (PRR), zu denen auch die TLR gehören, erkannt [151] und stellen einen TLR-unabhängigen Weg der Erkennung viraler Infektionen dar [268]. PRR sind Teil der angeborenen Immunität, welche auch die adaptive Immunität stimulieren können. Detektieren diese RLR Fremd-DNA im Zytoplasma, interagieren sie mit IPS-1 und setzten so eine Signalkaskade in Gang, die ebenfalls Überschneidungspunkte mit anderen PRR Signalkaskaden aufweist [160]. Schlussendlich führt dies zur Sezernierung von TNF-alpha, IL-1, IL-6 und IL-12 durch Makrophagen und Monocyten, die wiederum die Th1-Zellentwicklung beeinflussen und die Aktivierung der adaptiven Immunität zur Folge haben [303]. Entsprechend führt die Überexpression von IPS-1 zur Verstärkung der nachgeschalteten Signalkaskaden, zur Aktivierung der Typ 1 Interferon Promotoren und Freisetzung inflammatorischer Zytokine [343][160]. Schließlich kann der verwendete DNA-Vektor ebenfalls auf eine Adjuvanswirkung ausgelegt werden. Eingebaute nicht methylierte CpG-Strukturen werden vom TLR9 als bakterielle DNA erkannt und wirken so stimulierend [26][135][173][13][169]. Daher kann eine Optimierung der Sequenzen unter Berücksichtigung von stimulierenden Mustern, sowie eine Vektorarchitektur, welche die Stabilität der generierten mRNA und des Plasmids selbst verbessert, die Effizienz der gesamten DNA-Vakzine erheblich steigern [361]. 21 1. Einleitung 1.6 Ziel der Arbeit Dem Aufruf der WHO folgend, Milzbrand als einer globalen „neglected zoonotic disease“ eine höhere Priorität in der wissenschaftlichen Forschung einzuräumen, sollten in dieser Arbeit neue Ansätze für einen Veterinärimpfstoff entwickelt und getestet werden. Die momentan verfügbare Veterinärvakzine ist in nahezu unveränderter Form seit ca. 1940 im Gebrauch und besteht aus einem attenuierten B. anthracis Stamm (34F2, SSLV, siehe Kapitel 1.5.1 ). Wenngleich der Lebendimpfstoff in vielen Tierspezies einen sehr guten Impfschutz bietet, so ist er jedoch mit einigen Nachteilen behaftet, was zum Teil seine Anwendbarkeit einschränkt. Zur Entwicklung einer alternativen Impfung wurden in dieser Arbeit, gefördert durch die DFG (BE 2157/3-1), verschiedene Komponenten für eine „nicht lebend Vakzine“ (NLV) getestet. Zielführend war dabei die Verwendung von Formulationen, deren Wirksamkeit auch bei gleichzeitiger antibiotischer Behandlung gewährleistet werden kann und deren Anwendbarkeit in sensiblen Tierarten unbedenklich ist. Diese beinhalteten neben neuen Proteinkomponenten sowohl neue Adjuvanzien als auch neue DNA-Vektoren. Immunogenität, Toxinneutralisationsfähigkeit und Schutzwirkung der Komponenten wurden einzeln und in Kombination unter Anwesenheit entsprechender Adjuvanzien im NMRI-Mausmodell bestimmt. Entsprechend den Ergebnissen der Mausversuche wurden geeignete Kombinationen vergleichend zur Veterinärvakzine (SSLV) im Ziegenmodell getestet und analysiert. Hierbei waren die Titerentwicklung über die Zeit und der Verlauf der Infektion von Interesse. Alle gesammelten Daten sollten auch im Hinblick auf prädiktive Korrelationen zur Schutzrate analysiert werden. Die Tierversuche wurden im Sinne der deutschen Tierschutzverordnung unter dem Aktenzeichen V 272/10 THy (Regierungspräsidium), der Abteilung für Tiergesundheit Südafrika unter Protokollnummer V41-10 (Verordnung 34 von 1984) und des ethischen Komitees für Tierversuche der Erciyes Universität Türkei (Protokollnummer 13/01 – 09.01.2013) autorisiert. 22 2. Material 2 Material 2.1 Chemikalien Tabelle 1: Chemikalien Name Herkunft 1-Butanol (> 99,5 %) Carl Roth GmbH & Co. KG (Karlsruhe, D) ABTS (2,2'-Azino-di-(3-ethylbenzthiazolin-6-sulfonsäure)) Roche Diagnostics GmbH (Mannheim, D) Acrylamid PAGE (40 %) GE Healthcare Europe GmbH (Freiburg, D) Agar-Agar Carl Roth GmbH & Co. KG (Karlsruhe, D) Agarose, Biozym LE Biozym Scientific GmbH (Hessisch Oldendorf, D) APS (Ammoniumperoxodisulfat, >98 %) Carl Roth GmbH & Co. KG (Karlsruhe, D) Azur B Sigma-Aldrich Chemie GmbH (Taufkirchen bei München, D) BCA (Bicinchoninsäure) Reagenz A Interchim SA – France (Montlucon, F) BCA (Bicinchoninsäure) Reagenz B Interchim SA – France (Montlucon, F) Bestatin Hydrochlorid (> 98 %) Interchim SA – France (Montlucon, F) BHI (Brain Heart Infusion) Agar Merck KGaA (Darmstadt, D) BHI (Brain Heart Infusion) Bouillon Merck KGaA (Darmstadt, D) Bisacrylamid (PlusOne N,N'-Methylene-bisacrylamide, 2 %) GE Healthcare Europe GmbH (Freiburg, D) Bromphenolblau Natriumsalz Carl Roth GmbH & Co. KG (Karlsruhe, D) BSA (Bovines Serumalbumin) Serva Electrophoresis GmbH (Heidelberg, D) Butanol Merck KGaA (Darmstadt, D) β-Mercaptoethanol CaCl 2 (Calciumchlorid) Merck KGaA (Darmstadt, D) CO2 (Kohlendioxid), gasförmig Westfalen Austria GmbH (Leobersdorf, A) Columbia-Agar Merck KGaA (Darmstadt, D) DMEM (mit L-Glutamin) Lonza Group AG (Basel, CH) DMSO (Dimethylsulfoxid) Merck KGaA (Darmstadt, D) DTT (1,4-Dithiothreitol, >99 %) Carl Roth GmbH & Co. KG (Karlsruhe, D) E-64 (Proteinase Inhibitor) Interchim SA – France (Montlucon, F) EDTA (Ethylendinitrilotetraessigsäure) Carl Roth GmbH & Co. KG (Karlsruhe, D) Essigsäure (100 %) Merck KGaA (Darmstadt, D) Ethanol (100 %) Thermo Fischer Scientific Inc. (Waltham, MA, USA) Ethanol (99,7 %, vergällt) Alkoholvertrieb Süd GmbH (Horb-Betra, D) Ethidiumbromid Carl Roth GmbH & Co. KG (Karlsruhe, D) FKS (fötales Kälberserum) Life Technologies GmbH (Darmstadt, D) Fleischextrakt BD BIOSCIENCES (Heidelberg, D) Formaldehydlösung (37 %) Merck KGaA (Darmstadt, D) Formamid Merck KGaA (Darmstadt, D) Gelatine Merck KGaA (Darmstadt, D) GeneRuler TM 1 kb DNA Ladder Fischer Scientific – Germany GmbH (Schwerte, D) GenLadder 250 bp – 10 kb Genaxxon Bioscience GmbH (Ulm, D) Glukose Carl Roth GmbH & Co. KG (Karlsruhe, D) Glycerin, 86 % Carl Roth GmbH & Co. KG (Karlsruhe, D) Goldstaub (1,0 µm) Bio-Rad Laboratories GmbH (München, D) Guanidin-HCl Alfa Aesar GmbH & Co. KG (Karlsruhe, D) Harnstoff VWR International GmbH (Darmstadt, D) HCl (Salzsäure) Merck KGaA (Darmstadt, D) Hefeextrakt Merck KGaA (Darmstadt, D) HEPES (N-2-Hydroxyethylpiperazin-N'-2-ethansulfonsäure, >99,5 %) Carl Roth GmbH & Co. KG (Karlsruhe, D) Histidin Carl Roth GmbH & Co. KG (Karlsruhe, D) Imidazol Carl Roth GmbH & Co. KG (Karlsruhe, D) Immersionsöl Carl Roth GmbH & Co. KG (Karlsruhe, D) IPTG (Isopropyl-β-D-thiogalactopyranosid) Carl Roth GmbH & Co. KG (Karlsruhe, D) Isopropanol VWR International GmbH (Darmstadt, D) KCl (Kaliumchlorid) KH2 PO4 (Kaliumdihydrogenphosphat) Merck KGaA (Darmstadt, D) LF (kommerziell) List Biological Laboratories Inc. (Campbell CA, USA) Merck KGaA (Darmstadt, D) Merck KGaA (Darmstadt, D) 23 2. Material Tabelle 1 Fortführung: Chemikalien 24 Name Herkunft Magermilchpuler AppliChem GmbH (Darmstadt, D) Methanol MgCl2 (Magnesiumchlorid) Sigma-Aldrich Chemie GmbH (Taufkirchen bei München, D) MnSO4 *H2 O (Mangan(II)-sulfat Monohydrat) Merck KGaA (Darmstadt, D) MTT (3-(4,5-Dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium-bromide) Na2 HCO3 (Di-Natriumhydrogencarbonat) Alfa Aesar GmbH & Co. KG (Karlsruhe, D) Na2 HPO4 (Dinatriumhydrogenphosphat) Merck KGaA (Darmstadt, D) NaCl (Natriumchlorid) NaHCO3 (Natriumhydrogencarbonat) Merck KGaA (Darmstadt, D) NaHPO4 (Natriumhydrogenphosphat) Merck KGaA (Darmstadt, D) NaN3 (Natriumazid) Sigma-Aldrich Chemie GmbH (Taufkirchen bei München, D) NaOH (Natriumhydroxid) Merck KGaA (Darmstadt, D) Nickel(II)-sulfat Hexahydrat Sigma-Aldrich Chemie GmbH (Taufkirchen bei München, D) PA (kommerziell) List Biological Laboratories Inc. (Campbell CA, USA) PBS (endotoxinfrei) Sigma-Aldrich Chemie GmbH (Taufkirchen bei München, D) Pepstatin A (> 90 %) Sigma-Aldrich Chemie GmbH (Taufkirchen bei München, D) Peroxyessigsäure (40 %) Merck KGaA (Darmstadt, D) PhastGel Blue R (Coomassie) Sigma-Aldrich Chemie GmbH (Taufkirchen bei München, D) Phosphoramidon Interchim SA – France (Montlucon, F) PMSF (Phenylmethylsulfonylfluorid) Carl Roth GmbH & Co. KG (Karlsruhe, D) Protein-Marker IV (10 – 170 kDa) PEQLAB Biotechnologie GmbH (Erlangen, D) PVP (Polyvinylpyrrolidon) Sigma-Aldrich Chemie GmbH (Taufkirchen bei München, D) RPMI-1640 Medium Sigma-Aldrich Chemie GmbH (Taufkirchen bei München, D) Saccharose Carl Roth GmbH & Co. KG (Karlsruhe, D) Schafsblut, defibriniert Thermo Fischer Scientific Inc. (Waltham, MA, USA) SDS (Natriumdodecylsulfat, >99,5 %) Carl Roth GmbH & Co. KG (Karlsruhe, D) Spermidin Sigma-Aldrich Chemie GmbH (Taufkirchen bei München, D) Stains-All (~ 95 %) Sigma-Aldrich Chemie GmbH (Taufkirchen bei München, D) Standard-Nähragar I Carl Roth GmbH & Co. KG (Karlsruhe, D) Standard-Nährmedium I Carl Roth GmbH & Co. KG (Karlsruhe, D) Stickstoff, flüssig Westfalen Austria GmbH (Leobersdorf, A) Stickstoff, gasförmig Westfalen Austria GmbH (Leobersdorf, A) TEMED (N,N,N',N'-Tetramethylethylendiamin) Carl Roth GmbH & Co. KG (Karlsruhe, D) Trichloressigsäure (>99 %) Carl Roth GmbH & Co. KG (Karlsruhe, D) TRIS (Tris-(hydroxymethyl)-aminomethan) Carl Roth GmbH & Co. KG (Karlsruhe, D) TRIS-HCl Carl Roth GmbH & Co. KG (Karlsruhe, D) Trypton (Bacto) BD BIOSCIENCES (Heidelberg, D) Trypton-Soja-Agar (TSA) Carl Roth GmbH & Co. KG (Karlsruhe, D) TWEEN 20 Sigma-Aldrich Chemie GmbH (Taufkirchen bei München, D) VENNO VET 1 super MENNO CHEMIE-VERTRIEB GmbH (Norderstedt, D) Wofasteril KESLA Hygiene AG (Bitterfeld-Wolfen, D) Xylol Merck KGaA (Darmstadt, D) Sigma-Aldrich Chemie GmbH (Taufkirchen bei München, D) Merck KGaA (Darmstadt, D) Merck KGaA (Darmstadt, D) 2. Material 2.2 Puffer, Lösungen und Medien Tabelle 2: Puffer, Lösungen und Medien Bezeichnung Zusammensetzung ABTS-Puffer 1 ABTS-Tablette in 50 ml sterilem Millipore Ampicillinlösung (1000fach) Beschichtungspuffer – ELISA (pH 9,5) 100 mg/ml Ampicillin-Natriumsalz in sterilem Millipore, Lagerung bei -20 °C 8,4 g/l NaHCO3 ; 3,56 g/l Na2 CO3 in sterilem Millipore Blocklösung – ELISA 10 % FKS (bzw. Magermilchpulver) in PBS Butanol (wassergesättigt) Butanol und steriles Millipore 1:1 vermengen und vor Gebrauch gut mischen CBA (Columbia-Blutagar) 5 % Schafsblut (Zugabe nach Sterilisation) in Columbia-Agar mit sterilem Millipore Chloramphenicollösung (1000fach) 25 mg/ml Chloramphenicol in Ethanol, Lagerung bei -20 °C Coomassie-Lösung 1 Färbetablette Phast Gel Blue R in 400 ml Entfärber durch Erhitzen auf 60 °C lösen und anschließend filtrieren CSA-Bicarbonat (Casein Soya Agar mit Bicarbonat) 0,7% NaHCO3 in CSA Dialysepuffer 1 (pH 7,5) 5 mM HEPES, 100 µl CaCl2 , 1 M Harnstoff, 10 % Glycerin in sterilem Millipore Dialysepuffer 2 (pH 7,5) 5 mM HEPES, 100 µl CaCl2 , 0,5 M Harnstoff, 10 % Glycerin in sterilem Millipore Dialysepuffer 3 (pH 7,5) 5 mM HEPES, 100 µl CaCl2 , 0,1 M Harnstoff, 10 % Glycerin in sterilem Millipore Dialysepuffer 4 / HEPES-Puffer (pH 7,5) 5 mM HEPES, 100 µl CaCl2 in sterilem Millipore Einfriermedium Elutionspuffer – FPLC (pH 4,5) 20 % DMSO in FKS 8 M Harnstoff, 0,1 M NaHPO4 , 0,01 M TRIS-HCl (pH 4,5) in sterilem Millipore Entfärber – SDS-PAGE 30 % (v/v) Methanol, 8% (v/v) Essigsäure in sterilem Millipore Ethidiumbromidlösung (20000fach) 10 mg/ml Ethidiumbromid in TAE-Puffer Fixierer – SDS-PAGE 40 % (v/v) Methanol, 10 % (v/v) Essigsäure in sterilem Millipore Gelpuffer B – SDS-PAGE (pH 8,8) 1,5 mol/l TRIS in sterilem Millipore Gelpuffer C – SDS-PAGE (pH 6,8) 1,0 mol/l TRIS in sterilem Millipore Histidin-Lösung (2fach) 40 g/l Histidin in sterilem Millipore IPTG-Lösung (1000fach) 1 M IPTG in sterilem Millipore, Lagerung bei -20 °C Kanamycinlösung (1000fach) 50 mg/ml Kanamycinsulfat in sterilem Millipore, Lagerung bei -20 °C 6 M Guanidin-HCl, 0,1 M NaHPO4 , 0,01 M TRIS-HCl (pH 8,0), 10 mM Imidazol in sterilem Millipore Lysepuffer – FPLC (pH 8,0; 4 °C) Lysepuffer – TNA MYA (Meat Yeast Agar, pH 7,0) NaCl-Gelatine 90 % (v/v) Isopropanol; 0,5 % (w/v) SDS; 25 mM HCl 10 g/l Fleischextrakt, 2 g/l Hefeextrakt, 0,04 g/l MnSO4 *H2 O, 15 g/l Agar in sterilem Millipore Nukleasepuffer (10fach) 0,9 % (w/v) NaCl, 0,1 % Gelatine in sterilem Millipore 0,1 M TRIS (pH 8,0); 10 mM MgCl 2 ; 0,1 M NaCl und 0,02% (w/v) NaN 3 in sterilem Millipore PBS (Phosphat buffered Saline, pH 7,4) 8 g/l NaCl, 0,2 g/l KCl, 0,12 g/l KH 2 PO4 , 0,91 g/l Na2 HPO4 in sterilem Millipore PBS-Gelatine 0,1 % Gelatine in PBS Probenpuffer (-DTT) SDS-PAGE (5fach, pH 6,8) 10 % SDS, 25 mg/l Bromphenolblau, 8 % (v/v) Gelpuffer C, 125 g/l Glycerin in sterilem Millipore Probenpuffer (+DTT) SDS-PAGE (5fach, pH 6,8) 15 g/l DTT, 2 % SDS, 25 mg/l Bromphenolblau, 8 % (v/v) Gelpuffer C, 125 g/l Glycerin in sterilem Millipore Probenpuffer Agarose-Gelelektrophorese (10fach) 0,25 % Bromphenolblau und 40 % Saccharose in sterilem Millipore Proteinasecocktail – FPLC 0,5 mg/ml Pepstatin A, 0,5 mg/ml Bestatin Hydrochlorid, 0,25 mg/ml E-64, 0,25 mg/ml Phosphoramidon, 1 mM PMSF in DMSO Puffer B – FPLC (pH 8,0) 8 M Harnstoff, 0,1 M NaHPO4 , 0,01 M TRIS-HCl (pH 8,0), 10 mM Imidazol in sterilem Millipore Puffer C – FPLC (pH 6,3) 8 M Harnstoff, 0,1 M NaHPO4 , 0,01 M TRIS-HCL (pH 6,3), 10 mM Imidazol in sterilem Millipore PVP-Lösung 0,1 mg/ml PVP in Ethanol (100 %) Sammelgel SDS-PAGE 900 µl Acrylamid PAGE (40 %), 900µl Bisacrylamid (2 %), 1,25 ml Gelpuffer C, 100 µl SDS (10 %), 75 µl APS (10 %), 7,5 µl TEMED, 6,8 ml steriles Millipore; ausreichend für 2-3 Gele SDS-PAGElektrophoresepuffer (10fach, pH 8,9) 75 g/l Glycerin in sterilem Millipore; Vor Gebrauch 1:10 verdünnen und mit SDS (Endkonzentration 0,1 %) versetzen Stains-All-Lösung (pH 8,8) 30 mM TRIS, 7,5 % (v/v) Formamid, 25 % (v/v) Isopropanol, 0,025 % (w/v) Stains-All in sterilem Millipore; pH-Wert 8,8 mit HCL einstellen TAE (TRIS-Acetat-EDTA) AgaroseGelelektrophorese-Puffer (50fach, pH 8,0) 342 g/l TRIS, 57,1 ml/l Essigsäure (100 %), 50 mM EDTA in sterilem Millipore TGB (Trypton-Glukose-Bouillon, pH 7,2) 2,5 g/l Hefeextrakt, 5 g/l Trypton, 1 g/l Glukose in sterilem Millipore Trenngel (15 %) SDS-PAGE 8,1 ml Acrylamid PAGE (40 %), 6 ml Bisacrylamid (2 %), 2,25 ml Glycerin (86 %), 5,62 ml Gelpuffer B, 225 µl SDS (10 %), 148,5 µl APS (10 %), 14,8 µl TEMED, 90 µl steriles Millipore; ausreichend für 2-3 Gele Trypsin-Lösung 0,5 g/l Trypsin, 0,2 g/l EDTA in sterilem Millipore Versuchsmedium – TNA 5 % FKS in DMEM Wachstumsmedium 1:1 Mischung aus DMEM und RPMI ergänzt mit 10% FKS Waschpuffer – ELISA 0,05 % TWEEN 20 in PBS 25 2. Material 2.3 Verbrauchsmaterialien Tabelle 3: Verbrauchsmaterialien 2.4 Name Herkunft Abdeckgläschen VWR International GmbH (Darmstadt, D) Armstulpen VWR International GmbH (Darmstadt, D) Atemschutzmaske (P3) Behr Labor-Technik (Düsseldorf, D) Baretthaube Behr Labor-Technik (Düsseldorf, D) Dialysemembran (Cellu-Sep T1, 3,5kDa) Membrane Filtration Products Inc. (Sequin, TX, USA) Dialysemembran (Cellu-Sep T2, 6 - 8kDa) Membrane Filtration Products Inc. (Sequin, TX, USA) Filterpapier Munktel & Filtrak GmbH (Bärenstein, D) Flaschenaufsatzfilter (500 ml, steril) Thermo Fischer Scientific Inc. (Waltham, MA, USA) Handschuhe, gepudert / ungepudert (M, L) megro GmbH & Co. KG (Wesel, D) Impföse (1, 10 µl) VWR International GmbH (Darmstadt, D) Injektionsnadel (blau, 23G x 1) TERUMO Medical Corporation (Summerset, NJ, USA) Injektionsnadel (braun, 26G x 1) TERUMO Medical Corporation (Summerset, NJ, USA) Injektionsnadel (gelb, 20G x 2) TERUMO Medical Corporation (Summerset, NJ, USA) Insulinspritze mit Nadel (1 ml) TERUMO Medical Corporation (Summerset, NJ, USA) Kryoröhrchen (2 ml) Thermo Fischer Scientific Inc. (Waltham, MA, USA) Küvette, Plastik (0,5 und 1 ml) ratiolab GmbH (Dreieich, D) Mikrotiterplatte, 96-Well (endotoxinfrei, steril, mit Deckel, round bottom, einzeln verpackt) Thermo Fischer Scientific Inc. (Waltham, MA, USA) Mikrotiterplatte, 96-Well (Maxisorp, flat bottom) Sigma-Aldrich Chemie GmbH (Taufkirchen bei München, D) Mikrotiterplatte, 96-Well (steril, mit Deckel, flat bottom, einzeln verpackt) BD BIOSCIENCES (Heidelberg, D) Objektträger Thermo Fischer Scientific Inc. (Waltham, MA, USA) Overall, Einweg ReWa GmbH (Bahlingen, D) Parafilm Sigma-Aldrich Chemie GmbH (Taufkirchen bei München, D) Petrischale, Plastik (92 x 16 mm, mit Entlüftungsnocken) nerbe plus GmbH (Winsen / Luhe, D) Pipettenspitzen (20, 200, 300, 1000 µl) Eppendorf AG (Hamburg, D) Pipettenspitzen, endotoxinfrei (200, 1000) Eppendorf AG (Hamburg, D) Plastikspatel (steril) VWR International GmbH (Darmstadt, D) Rasierer, Einweg Wilkinson Sword GmbH (Solingen, D) Reaktionsgefäß (1,5, 2 ml) Biozym Scientific GmbH (Hessisch Oldendorf, D) Röhrchen, Plastik (15, 50 ml) Greiner Bio-One International GmbH (Kremsmünster, A) Schürze ReWa GmbH (Bahlingen, D) Sepharosesäule (HiTrap Chelating HP, 5 ml) GE Healthcare Europe GmbH (Freiburg, D) Serumröhrchen BD Vacutainer (10 ml) und Zubehör BD BIOSCIENCES (Heidelberg, D) Spritze (1, 2, 5, 10, 20 ml) TERUMO Medical Corporation (Summerset, NJ, USA) Spritzenvorsatzfilter (0,2, 0,45 µM) Sartorius AG (Göttingen, D) Tefzel Schlauch Bio-Rad Laboratories GmbH (München, D) Überschuhe Behr Labor-Technik (Düsseldorf, D) Zellkulturflaschen (100, 200, 500 ml) BD BIOSCIENCES (Heidelberg, D) Versuchstiere Tabelle 4: Versuchstiere Spezies 26 Anzahl Zulieferer BALB/c Maus 6 SAVP (Sandrigham, ZA) Burenziege 37 diverse lokale Bauern (Pretoria, ZA) NMRI Maus 145 Charles River Laboratories (Köln, D) Ziegen (gemischte, türkische Rassen) 25 diverse lokale Bauern (Kars, TR) 2. Material 2.5 Geräte Tabelle 5: Geräte 2.6 Name Herkunft Agarose-Geldokumentation (Quantum) VILBER LOURMAT Deutschland GmbH (Eberhardzell, D) Agarose-Gelelektrophoresekammer (Agagel Mini) und Zubehör Biometra GmbH (Göttingen, D) Autoklav INTEGRA Biosciences GmbH (Fernwald, D) Bunsenbrenner CO2 -Inkubator Bochem Instrumente GmbH (Weilburg, D) CO2 -Inkubator INTEGRA Biosciences GmbH (Fernwald, D) GeneGun (Helios) und Zubehör Bio-Rad Laboratories GmbH (München, D) Heißluftsterilisator GE Healthcare Europe GmbH (Freiburg, D) Laminar-Flow Sicherheitswerkbank Klasse II BDK-Luft-und Reinraumtechnik GmbH (Sonnenbühl-Genkingen, D) Magnetrührer und Zubehör IKA-Werke GmbH & Co. KG (Staufen, D) Mauskäfig und Zubehör (Makrolon, 820 cm2 ) Zoo-Partner (Lunzenau, D) Messschreiber (REC-112) und Zubehör GE Healthcare Europe GmbH (Freiburg, D) Mikroskop, Hellfeld (Vilovert) Helmut Hund GmbH (Wetzlar, D) Mikrowelle Siemens-Electrogeräte GmbH (München, D) Millipore Milli-Q Biocel Wasser Aufreinigungssystem Merck KGaA (Darmstadt, D) Netzteil Biometra GmbH (Göttingen, D) pH-Meter WTW Wissenschaftlich-Technische Werkstätten GmbH (Weilheim, D) Photometer (Ultrospec 2100 pro) GE Healthcare Europe GmbH (Freiburg, D) Plattenlesegerät (Model 680) Bio-Rad Laboratories GmbH (München, D) Plattenleseinkubator, kinetisch (Endotoxin) Lonza Group AG (Basel, CH) Plattenwaschgerät (Model 1575) Bio-Rad Laboratories GmbH (München, D) Plattformschüttler, rotierend Heidolph Instruments GmbH & Co. KG (Schwabach, D) Polyacrylamid-Gelelektrophoresekammer (PAGE) und Zubehör Biometra GmbH (Göttingen, D) Protein-Aufreinigungssystem (ÄKTAprime plus) und Zubehör GE Healthcare Europe GmbH (Freiburg, D) Schlauchpumpe, peristaltisch (P-1) GE Healthcare Europe GmbH (Freiburg, D) Schlauchrotor (Tubing Prep Station) Bio-Rad Laboratories GmbH (München, D) Schlauchschnittmaschine (Tubing Cutter) Bio-Rad Laboratories GmbH (München, D) Schüttelinkubator Infors AG (Bottmingen, CH) Stickstoffkontainer (flüssig) Taylor-Wharton International LLC (Theodore, AL, USA) Thermoblock Biometra GmbH (Göttingen, D) Thermocycler Biometra GmbH (Göttingen, D) Tiefkühlschrank (HERAfreeze, -86 °C) Thermo Fischer Scientific Inc. (Waltham, MA, USA) Ultraschall-Homogenisator Hielscher Ultrasonics GmbH (Teltow, D) Vortexmischer Heidolph Instruments GmbH & Co. KG (Schwabach, D) Waage, fein Sartorius AG (Göttingen, D) Waage, grob OHAUS CORPORATION (Parsippany, NJ, USA) Wasserbad Köttermann GmbH &Co.KG (Uetze/Hänigsen, D) Wasserbad (Ultraschall) BANDELIN electronics GmbH & Co. KG (Berlin, D) Zentrifuge (Heraeus Labofuge 400) Thermo Fischer Scientific Inc. (Waltham, MA, USA) Zentrifuge (Heraeus Pico) Thermo Fischer Scientific Inc. (Waltham, MA, USA) Zentrifuge (Heraeus Sepatech Varifuge 3.2S) Thermo Fischer Scientific Inc. (Waltham, MA, USA) Zentrifuge (Heraeus Suprafuge 22) Thermo Fischer Scientific Inc. (Waltham, MA, USA) Zentrifuge (Spectrafuge Mini) Labnet International Inc (Edison, NJ, USA) Eppendorf AG (Hamburg, D) Antikörper Tabelle 6: Antikörper Name Hersteller eingesetzte Konzentration Kaninchen Anti-Ziege IgG HRP Thermo Fischer Scientific Inc. (Waltham, MA, USA) 1:6000 Ziege Anti-Maus IgG HRP Sigma-Aldrich Chemie GmbH (Taufkirchen bei München, D) 1:4000 Ziege Anti-Maus IgG1 HRP Acris Antibodies GmbH (Herford, D) 1:16000 Ziege Anti-Maus IgG2a HRP Acris Antibodies GmbH (Herford, D) 1:16000 27 2. Material 2.7 Utensilien Tabelle 7: Utensilien 2.8 Name Herkunft Becherglas (250, 500, 1000, 2000 ml) DURAN Group GmbH (Wertheim/Main, D) Erlenmeyerkolben (100, 250, 500, 1000 ml) DURAN Group GmbH (Wertheim/Main, D) Erlenmeyerkolben mit Schikanen (500, 1000 ml) DURAN Group GmbH (Wertheim/Main, D) Gesichtsschutzschirm Behr Labor-Technik (Düsseldorf, D) Glasflasche (50, 250, 500, 1000 ml) DURAN Group GmbH (Wertheim/Main, D) Glasperlen (Durchmesser 2 mm) Carl Roth GmbH & Co. KG (Karlsruhe, D) Glaspipette (1, 2, 5, 10, 20, 50 ml) Hilgenberg GmbH (Malsfeld, D) Impföse (Metall) häberle LABORTECHNIK GmbH & Co. KG (Lonsee-Ettlenschieß, D) Küvette, Quarz (0,5 und 1 ml) Hellma GmbH & Co. KG (Müllheim, D) Mehrkanalpipette, automatisiert, 8 Kanäle (15-300 µl) Eppendorf AG (Hamburg, D) Mehrkanalpipette, mechanisch, 8 Kanäle (20-200 µl) VWR International GmbH (Darmstadt, D) Messzylinder, glas (50, 100, 250, 500, 1000 ml) DURAN Group GmbH (Wertheim/Main, D) Neubauer Zählkammer und Zubehör LO – Laboroptik Ltd (Lancing, GB) Petrischale, Glas (120 x 20 mm) DURAN Group GmbH (Wertheim/Main, D) Pipette, mechanisch (20, 200, 1000 µl) Gilson Inc. (Middleton WI, USA) Pipettierhilfe (Easypet) Eppendorf AG (Hamburg, D) Reagenzglas und Zubehör (9, 16, 21, 32 ml) DURAN Group GmbH (Wertheim/Main, D) Roux-Flasche Dunn Labortechnick GmbH (Asbach, D) Saugpumpe DURAN Group GmbH (Wertheim/Main, D) Zelllinien Tabelle 8: Zelllinien 2.9 Bezeichnung Spezies Herkunft A1 (Tox+ ) (Kap- ) B. anthracis Universität Hohenheim (Stuttgart, D) A58 (Tox- ) (Kap- ) B. anthracis Universität Hohenheim (Stuttgart, D) A73 (Tox- ) (Kap+ ) B. anthracis Universität Hohenheim (Stuttgart, D) Ames (Tox+) (Kap+) B. anthracis Universität Hohenheim (Stuttgart, D) Bl21 pRep4 pQE30Lf E.coli Universität Hohenheim (Stuttgart, D) Bl21 pRep4 pQE30Pa83ec E.coli Universität Hohenheim (Stuttgart, D) Bl21 pRep4 Ril pQE30BclA1 E.coli Universität Hohenheim (Stuttgart, D) BL21-CodonPlus-RIL Competent Cells E.coli Agilent Technologies GmbH & Co.KG (Waldbronn, D) J774A.1 Maus Monozyten Makrophagen Zelllinie Sigma-Aldrich Chemie GmbH (Taufkirchen bei München, D) K-136 (Tox+) (Kap+) B. anthracis Kafkas Universität (Kars, TR) SD20 (Tox+) (Kap+) B. anthracis Universität Pretoria (Onderstepoort, ZA) SSLV 34F2 (Tox+) (Kap- ) B. anthracis OBP Onderstepoort Biological Products (Onderstepoort, ZA) Kits Tabelle 9: Kits 28 Name Herkunft Endotoxin-Reinigungs-Kit (EndoTrap blue) Hyglos GmbH (Bernried am Starnberger See, D) Gelextraktions-Kit (DNA) Genaxxon BioScience GmbH (Ulm, D) LAL (Limulus Amöbozyten Lysat) -Test-Kit, kinetisch, chromogen (Endochrome-K) Charles River Laboratories (Wilmington, MA, USA) Spritzen-Kit (GeneGun Patronen Präparation) Bio-Rad Laboratories GmbH (München, D) 2. Material 2.10 Genetisches Material Tabelle 10: Genetisches Material Name Herkunft BclaD1D3 Sequenz (Siehe Anhang) Thermo Fischer Scientific Inc. (Waltham, MA, USA) CIITA Sequenz (murin; Mn01492) GeneCopoeia (Rockville, MD, USA) LFD1PAD4 Sequenz zur Verfügung gestellt von L. Baillie NTC7382-TPA-LFD1PAD4-mIPS-1 Nature Technology Corporation (Lincoln, NE, USA) PA83 Sequenz (Siehe Anhang) zur Verfügung gestellt von W. Beyer pDNAVaccUltra1-TPA-BclAD1D3-LAMP1 Nature Technology Corporation (Lincoln, NE, USA) pDNAVaccUltra1-TPA-LFD1PAD4-LAMP1 Nature Technology Corporation (Lincoln, NE, USA) pDNAVaccUltra1-TPA-PA83-LAMP1 Nature Technology Corporation (Lincoln, NE, USA) pDNAVaccUltra4-BclA-Ubiquitin Nature Technology Corporation (Lincoln, NE, USA) pDNAVaccUltra5-CIITA Nature Technology Corporation (Lincoln, NE, USA) pQE-30 QIAGEN GmbH – Germany (Düsseldorf, D) pREP4 QIAGEN GmbH – Germany (Düsseldorf, D) Sequenzen von BclAD1D3 und PA83 im Anhang 2.11 Wirkstoffe Tabelle 11: Wirkstoffe Name Herkunft Albendazol Universitätsapotheke Universität Pretoria (Onderstepoort, ZA) Ampicillin-Natriumsalz Sigma-Aldrich Chemie GmbH (Taufkirchen bei München, D) Chloramphenicol Sigma-Aldrich Chemie GmbH (Taufkirchen bei München, D) Depomin Kafkas Universität (Kars, TR) Desoxyribonuklease I Sigma-Aldrich Chemie GmbH (Taufkirchen bei München, D) Diazepam-ratiopharm Injektionslösung (10 mg/2 ml) ratiopharm GmbH (Ulm, D) Isofluran Actavis Deutschland GmbH & Co. KG (München, D) Ivermectin Universitätsapotheke Universität Pretoria (Onderstepoort, ZA) Kanamycinsulfat Carl Roth GmbH & Co. KG (Karlsruhe, D) Kapsel-Konjugat (Pam3 Cys-AQEKEAKSELDYD-(isoE9)-NH 2) EMC microcollections GmbH (Tübingen, D) Kapselmaterial eigene Herstellung Ketamin (100 mg/ml) Lipopeptid (Mischung aus Pam3 Cys-SKKKK und Pam3 Cys-FISEAIIHVLHSRHPG) CP-Pharma Handelsgesellschaft mbH (Burgdorf, D) Niclosamid Universitätsapotheke Universität Pretoria (Onderstepoort, ZA) Penicillin/Streptomycin Kafkas Universität (Kars, TR) Pentobarbital Universitätsapotheke Universität Pretoria (Onderstepoort, ZA) rBclA eigene Herstellung Ribonuklease A Sigma-Aldrich Chemie GmbH (Taufkirchen bei München, D) rLF eigene Herstellung rPA83 eigene Herstellung T61 (5 mg/ml Tetracainhydrochlorid, 50 mg/ml Mebezoniumiodid und 200 mg/ml Embutramid) Intervet Deutschland GmbH (Unterschleißheim, D) Trypsin Sigma-Aldrich Chemie GmbH (Taufkirchen bei München, D) Xylavet (Xylazin, 20 mg/ml) CP-Pharma Handelsgesellschaft mbH (Burgdorf, D) EMC microcollections GmbH (Tübingen, D) 2.12 Software Tabelle 12: Software Name Hersteller Quantum.CAPT VILBER LOURMAT Deutschland GmbH (Eberhardzell, D) SigmaPlot Systat Software GmbH (Erkrath, D) WinKQCL Lonza Group AG (Basel, CH) 29 3. Methoden 3 Methoden Alle verwendeten Medien und Nährböden wurden, soweit nicht anders beschrieben, unter sterilen Bedingungen hergestellt und vor Benutzung für 15 min bei 121 °C autoklaviert oder steril filtriert. Die Entsorgung von regulärem, infektiösem und nicht infektiösem Material fand ebenfalls für 15 min bei 121 °C in getrennten Autoklaven für die Desinfektion statt. Die Reinigung von Oberflächen oder nicht autoklavierbaren Materialien erfolgte mit 1 %-iger VENNO VET Lösung (Einwirkzeit 0,5 – 2 h). Arbeiten mit voll virulentem B. anthracis wurden unter BSL-3 Bedingungen durchgeführt, wobei kontaminierte Oberflächen, Gerätschaften und Einwegartikel mit 1 %-iger frisch angesetzter Peroxyessigsäure oder 2,5 % Wofasteril mit einer Mindesteinwirkzeit von 30 min desinfiziert werden mussten [28]. Für Arbeiten mit B. anthracis wurden entsprechend Abfallbehälter vor Beginn der Arbeit frisch mit Desinfektionsmittel bereitgestellt. Im Anschluss wurden autoklavierbare Artikel zweifach bei 134 °C für 30 min autoklaviert und entsorgt bzw. für den weiteren Gebrauch gereinigt. Zur persönlichen Absicherung, insbesondere bei Gefahr auf Aerosolbildung und während der Tierversuche wurde mit P3-Atemschutzmaske, Gesichtsschutzschirm und Einwegschutzkleidung, bestehend aus doppelter Behandschuhung, Overall, Baretthaube, Überschuhen, Schürze und Armstulpen gearbeitet. 3.1 Standardmethoden Sofern nicht anders beschrieben, wurden mikrobiologische und gentechnische Standardmethoden entsprechend der publizierten, gängigen Protokolle durchgeführt [15][153]. Diese umfassten: • Methoden zur Anzucht, Überimpfung und Lagerung von Bakterien- und Zellkulturen • Absorptionsmessungen zur Bestimmung der Zelldichte oder DNA-Konzentration mittels Photometer 30 • Messung und Einstellung des pH-Werts • SDS-PAGE und Agarosegelelektrophorese • Färbung von Gelen mittels Coomassie-Lösung • Bestimmung der Proteinkonzentration mittels BCA-Methode 3. Methoden 3.2 Herstellung und Aufbereitung von Antigenen 3.2.1 rPA83, rBclA und rLF Zur Herstellung großer Mengen von rPA83, rBclA und rLF für die Diagnostik im ELISA oder zur Anwendung als Impfstoff wurden bereits vorhandene E. coli Stämme vom Typ BL21CodonPlus verwendet. Diese waren zuvor durch Mitarbeiter der Universität Hohenheim mit entsprechenden Plasmiden zur Synthese der genannten rekombinanten Antigene transformiert worden. In Kürze zusammengefasst, wobei die einzelnen Arbeitsschritte im Folgenden detailliert beschrieben werden, sind entsprechend Bakterienkulturen in großen Volumina angesetzt und zur Produktion ihrer spezifischen Proteine mit IPTG induziert worden. Nach dem Ernten der Bakterien kam es zu Lyse und weiteren Aufreinigung der Proteine mittels „fast protein liquid chromatography“ (FPLC) über das integrierte His-Tag. Zur weiteren Verwendung der aufgereinigten Proteine fand ein Medienwechsel über Dialyse und eine qualitative Überprüfung über SDS-PAGE statt. Die Konzentration wurde im Anschluss quantitativ mittels Bicinchoninsäure (BCA) bestimmt. Kamen die Präparationen zur Verwendung als Impfstoff in Frage, schlossen sich weitere, reinigende Maßnahmen an (Kapitel 3.3). 3.2.1.1 Anzucht von induzierten Bakterienkulturen Aus einer Reinkultur wurde mittels Überimpfung einer Einzelkolonie eine 50 ml Vorkultur (Standard I Medium mit entsprechendem Zusatz von Antibiotika) angelegt. Die Inkubation erfolgte schüttelnd (120 UpM) üN bei 37 °C. Am Folgetag war eine analog angesetzte Hauptkultur (500 ml) durch Zufügen von 5 ml der Vorkultur zu beimpfen. Diese war dann bei 37 °C unter Schütteln in einem Erlenmeyerkolben mit Schikanen bis zu einer OD 600 nm von 0,6 zu inkubieren. Nach dem Erreichen der gewünschten Zelldichte wurde die Kultur mit IPTG (Endkonzentration 1 mM) induziert und weitere 3 h bei 37 °C schüttelnd inkubiert. Bei den Bakterienstämmen für rBclA und rLF wurden teilweise bessere Ergebnisse erzielt, wenn das Wachstum nach der Induktion üN bei 28 °C geschah. Nach Beendigung der Induktion war die Bakteriensuspension bei 4000 x g für 15 min zu zentrifugieren. Das resultierende Pellet wurde bis zur weiteren Verwendung bei -80 °C eingefroren. Zur Verfolgung der Proteinproduktion wurden vor dem Beginn der Induktion und an deren Ende je eine 1 ml Probe entnommen. 31 3. Methoden 3.2.1.2 Proteinaufreinigung mittels FPLC Die über IPTG induzierten Bakterienkulturen produzierten je nach Plasmid rPA83, rBclA und rLF im Überschuss. Zur Trennung der Proteine von den restlichen Bestandteilen der produzierenden Bakterien waren die Proteine mit einem His-Tag versehen. Diese nicht natürlich vorkommende Aneinanderreihung von sechs Histidinen besitzt im basischen Bereich eine extrem negative Ladung und kann daher eine starke Bindung mit Ni 2+-Ionen oder anderen stark positiven Ionen eingehen. Durch Auftragung der lysierten Bakterienkultur auf eine Säule mit gebundenen Ni2+-Ionen lassen sich die gebildeten Proteine aufreinigen, da andere Bestandteile ohne das His-Tag nicht anhaften und somit heruntergewaschen werden. Durch anschließendes graduelles Senken des pH-Werts ändert das His-Tag seine Ladung über neutral zu positiv und kann so inklusive des angehängten Proteins von der Säule in nahezu reiner Form eluiert werden. Mittels FPLC konnte diese Aufreinigung automatisiert durchgeführt werden. Hierbei wurde eine zuvor präparierte Säule in einem vorgefertigtem Programm mit der Bakteriensuspension beladen, durch Puffer gespült und in Fraktionen eluiert. Während des gesamten Prozesses wurde kontinuierlich die Konzentration der die Säule verlassenden Proteine erfasst, sodass schlussendlich die Fraktionen mit der höchsten Konzentration weiterverwendet werden konnten. Da die verwendeten Säulen durch Gaseinschlüsse oder Schmutzpartikel leicht verstopfen, mussten alle Flüssigkeiten und zuführenden Schläuche gas- und partikelfrei eingesetzt werden und wurden daher vor Verwendung steril filtriert und entgast. Im Detail war die Aufreinigung mittels FPLC folgendermaßen durchzuführen: 1. Messung und Einstellung des pH-Werts aller einzusetzenden Puffer (Lysepuffer, Puffer B, Puffer C, Elutionspuffer), sowie Filtration und Entgasung dieser. 2. Einrichten des FPLC-Geräts: a) Spülung und Entgasung aller angeschlossenen Schläuche. b) Einlauf der Puffer in die zugehörigen Schläuche. c) Beladung der HiTrap Sepharose Säule mit 0,1 M NiSO4 mittels Spritze wie vom Hersteller angegeben. d) Säule in das Gerät einsetzen und Dokumentationsschreiber in Betrieb nehmen. e) Für die fraktionierte Elution 60 1,5 ml Eppendorfgefäße in das Auffangrad einsetzen und mit je 100 µl TRIS (pH 8,5) zur Neutralisation befüllen. 3. Zugabe von 1 ml Proteinasecocktail zu 100 ml eisgekühltem Lysepuffer und Resuspension eines induzierten Bakterienpellets mit diesem. 4. Lyse der Bakteriensuspension unter Rühren für 1 h im Eisbad. 5. Zentrifugation des Lysats bei 12000 x g und 4 °C für 20 min. 6. Überstand des Lysats abnehmen und in einen frischen, hohen, gekühlten Zylinder (100 ml) überführen, Pellet verwerfen. 32 3. Methoden 7. Lysat und Lysepuffer an die vorgesehenen Schläuche anschließen und Programm starten. 8. Programm (soweit nicht anders vermerkt, liefen alle Flüssigkeiten mit 3 ml/min über die Säule): a) Einlauf von 50 ml Lysepuffer zur Spülung und Äquilibrierung der Säule. b) Einlauf des kompletten Lysats (~100 ml) mit 1 – 3 ml/min. c) Waschgang B mit Einlauf von 50 ml Puffer B d) Waschgang C mit Einlauf von 50 ml Puffer C e) Elution in 1 ml Fraktionen durch Einlauf von 50 ml Elutionspuffer mit 1 – 3 ml/min. f) Spülung des Geräts und der Säule mit 100 ml sterilem Millipore. 9. Markieren, Schließen, Schwenken und Einfrieren von Fraktionen mit adäquaten Proteinkonzentrationen. 10. Nachbereitung des Geräts durch Spülen aller Schläuche mit sterilem Millipore. 11. Zur Lagerung der Säule die NiSO 4-Ionen, mittels Spülung (Lysepuffer versetzt mit 0,05 M EDTA) entsprechend den Anweisungen des Herstellers entfernen. 3.2.1.3 Dialyse Die aus der FPLC resultierenden Fraktionen wurden entsprechend ihrer Konzentration vereinigt und über Dialyse einem Pufferwechsel mit graduell sinkender Harnstoffkonzentrationen [1 M, 0,5 M, 0,1 M] hin zu einer HEPES-Puffer Formulation ohne Harnstoff (Dialysepuffer 4) unterzogen. Dafür wurden Dialyseschläuche mit Porengrößen zwischen 3,5 und 8 kDa verwendet, welche nach dem Befüllen dicht verschlossen und vollständig bedeckt sukzessive in Dialysepuffer 1 bis 4 inkubiert wurden. Alle Dialyseschritte fanden in einem hohen Becherglas (2 l) gekühlt und unter sachtem Rühren statt. Die Inkubationszeiten betrugen 3 h für Dialysepuffer 1, 1,5 h für Dialysepuffer 2, 1 h für Dialysepuffer 3 und je dreimal 30 min für Dialysepuffer 4. Zur späteren Analyse und Dokumentation von Verlusten wurden auch hier vor und nach der Dialyse Proben genommen. Bei einer Verwendung der Proteine für den ELISA waren die Reinigungsschritte mit der Dialyse beendet, sodass diese nach Bestimmung der Konzentration und Verifikation der Größe und Reinheit mittels SDS-PAGE eingesetzt werden konnten. 33 3. Methoden 3.2.2 Aufreinigung von Kapselmaterial aus einer Bakterienkultur Um Kapselmaterial als Antigen für die Verwendung im ELISA herzustellen, wurde der B. anthracis Stamm A73 verwendet, welcher aufgrund des Fehlens des Plasmids pX01 keine Toxinkomponenten produzieren kann und damit attenuiert ist. Durch die fehlenden Toxinproduktion ist ebenfalls eine Verunreinigung der Kapselpräparation mit den Toxinen von vornherein ausgeschlossen. Das Protokoll folgte mit kleineren Abwandlungen den von Joyce et al. (2006) [158] publizierten Aufreinigungsschritten. Pro Präparation war der A73 auf 6 – 10 Platten CSA-Bicarbonat aus einer frischen Kultur auszuplattieren und bei 37 °C mit 20 % CO2 für 24 h zu bebrüten. Danach waren die Platten unter einer Sterilbank mit möglichst geringem Volumen an PBS abzuschwemmen. Die vereinigte Bakterienkultur wurde durch autoklavieren für 30 min bei 121 °C abgetötet und anschließend für 10 min bei 10000 x g abzentrifugiert. Der resultierende Überstand war abzunehmen, in ein frisches Gefäß zu überführen, auf Sterilität zu überprüfen und bei 4 °C üN in Nukleasepuffer, versetzt mit 140 U DNase und 10 U RNase zu inkubieren. Zur Fällung der löslichen Proteine wurde am Folgetag Trichloressigsäure mit einer Endkonzentration von 5 % zugefügt, auf Eis für 30 min inkubiert und anschließend bei 10000 x g für 30 min zentrifugiert. Zur Neutralisation erfolgte eine Zugabe von NaOH zum überführten Überstand bis zu einem pH-Wert von 7. Die Quantität und Reinheit der Präparation wurde durch die Bestimmung der Proteinkonzentration und die Analyse in der SDS-PAGE bestimmt. Hierbei mussten im Vergleich zwei identisch beladene Gele jeweils mit Coomassie und Stains-All gefärbt werden, da die Kapsel nur mit Stains-All sichtbar gemacht werden kann [113]. Die Abwesenheit der entsprechenden Bande für die Kapsel bei der Coomassiefärbung galt als zusätzlicher, absichernder Nachweis der Identität des Präparats. 3.2.3 Färbung mit Stains-All Die Färbung von SDS-Gelen mit Stains-All wurde ausschließlich für die Verifikation der Kapselpräparation durchgeführt und hielt sich mit kleineren Abweichungen an das von Goldberg et al. (1997) [113] publizierte Protokoll. Bei Färbungen zur Analyse von Größe und Reinheit anderer Präparate wurde standardmäßig Coomassie [153] verwendet. Zur Entfernung von SDS mussten die Gele einem dreifachen Waschzyklus unterzogen werden, welcher vorsah, die Gele jeweils dreimal mit einer 25 %igen Isopropanol-Lösung zu spülen und anschließend in selbiger für 10 min schwenkend zu inkubieren. Die Isopropanol-Lösung wurde anschließend durch die Stains-All-Lösung ersetzt und schwenkend unter Lichtausschluss für 2 h inkubiert. Die Entfärbung erfolgte graduell durch Lichteinfluss, wobei die Färbung der Banden pH-abhängig und die Kapselpräparation entsprechend als blaue Bande erkennbar war. 34 3. Methoden 3.2.4 Herstellung eines vegetativen Vollantigen von B. anthracis Zum Nachweis von spezifischen Antikörpern, die gegen den vegetativen Zellkörper und dessen nicht-toxische Produkte gerichtet sind, wurde ein vegetatives Vollantigen produziert, dessen Zusammensetzung nicht genau definiert ist. Zur Vermeidung von Verunreinigung mit Kapseloder Toxinantigenen wurde hierfür der toxin- und kapseldefiziente B. anthracis Stamm A58 verwendet. Zunächst war der A58 in BHI Bouillon versetzt mit 0,7 % Bicarbonat anzuziehen und üN bei 37 °C zu inkubieren. Anschließend wurden die Zellen zweimal mit PBS gewaschen und bei 4000 x g für 30 min bei 4 °C pelletiert. Das resultierende Pellet musste für eine kombinierte mechanische und chemische Lyse kurz in flüssigem Stickstoff schockgefroren und direkt im Anschluss mit 20 ml PBS versetzt mit SDS (Endkonzentration 1 %) resuspendiert werden. Durch zehnmaliges Beschallen mit Ultraschall für 5 – 10 s wurde die Lyse weiter verstärkt. Anschließend fand eine weitere Zentrifugation zur Pelletierung der unlöslichen Bestandteile bei 4000 x g für 15 min bei 4 °C statt. Der Überstand wurde mit einem Spritzenfilter steril filtriert und danach auf Sterilität überprüft. Nach einer Bestimmung der Konzentration mittels BCA konnte die Präparation im ELISA als vegetatives Antigen verwendet werden. 3.2.5 Herstellung einer Sporensuspension von B. anthracis Für die Infektion im Tierversuch und die Präparation von FIS war zunächst die Erstellung einer Sporensuspension nach einem Standardprotokoll (prEN 13697:2000 [357]) mit leichten Abwandlungen nötig. Eine Vorkultur des gewünschten Stammes wurde in TGB mit 106 Sporen beimpft und bei 30 °C bis zum Eintreten in die exponentielle Wachstumsphase bei einer Konzentration von 107 cfu/ml inkubiert. Zur Inokulation von Roux-Flaschen mit MYA waren jeweils 2 – 3 ml der Vorkultur überführt und durch Schwenken mit Hilfe von 5 – 10 sterilen Glasperlen verteilt worden. Überstehende Flüssigkeit wurde am Ende wieder entfernt. Die Inkubation erfolgte für 14 Tage bei 30 °C, wobei die Sporulation bereits nach 3 Tagen und nochmals nach dem Ablauf der vollen Inkubationszeit durch Phasenkontrastmikroskopie überprüft wurde. Die Abschwemmung der Sporen erfolgte mit NaCl-Gelatine ebenfalls über Schwenken mit Hilfe steriler Glasperlen. Zur vollständigen Abtötung vorhandener, vegetativer oder nicht vollständig sporulierter Zellen war die gewonnene Suspension im Wasserbad für 30 min bei 65 °C zu erhitzen (Hitzeinaktivierung). Im Anschluss wurden die Sporen fünfmal mit NaCl-Gelatine gewaschen, final in einem gewünschten Volumen NaCl-Gelatine aufgenommen und bis zur Verwendung bei 4 °C gelagert. Zur Feststellung der Konzentration wurde abschließend eine Sporenauszählung vorgenommen und gegebenenfalls direkt vor der Verwendung, nebst erneuter Hitzeinaktivierung wiederholt. 35 3. Methoden 3.2.6 Bestimmung der Sporenkonzentration Zur Bestimmung der Sporenkonzentration musste die Sporensuspension zunächst gründlich unter Vermeidung einer Aerosolbildung vermischt werden, wobei dies auch für jede weitere anzulegende Verdünnung zutraf. Die anschließende Reihenverdünnung wurde in 15 ml Röhrchen durchgeführt in die je 9,0 ml NaCl-Gelatine vorgelegt waren. Entsprechend wurden aus der durchmischten Ausgangslösung mittels 1 ml Glaspipette 1 ml auf die erste Verdünnungsstufe übertragen, mit einer frischen 1 ml Glaspipette durchmischt und 1 ml auf die nächste Stufe übertragen. Abhängig von der erwarteten Konzentration schlossen sich wiederholend weitere Verdünnungsschritte an. Bei niedrigen Ausgangsvolumina konnte der erste Verdünnungsschritt auch durch Übertragung von 0,1 ml Sporensuspension auf 9,9 ml NaCl-Gelatine erfolgen. Üblicherweise wurden entsprechend der erwartenden Konzentration je 100 µl der Verdünnungsstufen zwischen 10-4 und 10-7 im Doppelansatz auf CBA ausplattiert und üN bei 37 °C inkubiert. Auszählbare Konzentrationen lagen im Bereich von 10 – 300 cfu pro Platte und ergaben entsprechend der Verdünnungsstufen einen Durchschnittswert für die Sporenkonzentration der Ausgangssuspension. Für die Infektionsversuche waren zudem zusätzlich abgefüllte Infektionsdosen zur Verifikation der tatsächlich verabreichten Sporenzahl auszuzählen. Hierfür wurden die überzähligen Dosen in gleicher Art wie die zur Infektion verwendeten Dosen in eine Spritze aufgezogen und über die Spritze in ein entsprechendes Volumen an NaCl-Gelatine zur Reihenverdünnung gegeben oder direkt ausplattiert, wenn die erwartete Sporenzahl ohne weitere Verdünnung auszählbar war. Auf diese Art sollten etwaige Rückstände in der Spritze oder dem Portionierungsgefäß berücksichtigt werden. Die angegebenen Konzentrationen der Infektionsdosen der Tierversuche beziehen sich, soweit nicht anders vermerkt, auf diese Auszählung. 3.2.7 Herstellung von Formalin inaktivierten Sporen (FIS) Die FIS-Präparation wurde entsprechend der von Guidi-Rontani et al. (1999) [120] vorgestellten Methodik mit leichten Abwandlungen durchgeführt. Eine vorher hergestellte, ausgezählte und gut durchmischte Sporensuspension des kapseldefizienten B. anthracis A1 Stammes (SSLV) war in möglichst hoher Konzentration zu je 5 ml vorsichtig auf eine Formalinlösung (Endkonzentration 4 %) aufzuschichten. Die Vermischung erfolgte durch vorsichtiges Schwenken, um eine Bildung von Lufteinschlüssen zu vermeiden, da diese für gewöhnlich eine vollständige Inaktivierung verhinderten. Die Inkubation erfolgte üN bei 37 °C. Am Folgetag wurde die Suspension bei 4000 rpm für 15 min bei RT abzentrifugiert und viermal mit PBSGelatine im Maximalvolumen gewaschen. Nach dem letzten Waschgang war das Pellet im 36 3. Methoden ursprünglich eingesetzten Volumen der Sporensuspension an NaCl-Gelatine wieder aufzunehmen und vorerst bei 4 °C in einem Glasgefäß aufzubewahren. Zur Kontrolle der Sterilität wurde ein Aliquot von 50 µl abgenommen, mit 50 µl einer Histidin-Lösung (Endkonzentration: 20 g/l) versetzt und 30 min bei RT inkubiert, um die Wirkung von potentiell vorhandenem Formalin zu neutralisieren. Das gesamte Volumen war auf eine CBA-Platte auszustreichen und üN bei 37 °C zu bebrüten. Die Präparation wurde verworfen, wenn keine Sterilität vorhanden war. Etwaige Parallelansätze waren bis zu diesem Schritt vollkommen getrennt behandelt und betrachtet worden und wurden erst nach Gewährleistung der Sterilität vereinigt. Zur permanenten Lagerung einer einheitlichen FIS-Präparation musste diese unter ständigem Rühren in 1,5 ml Eppendorfgefäße aliquotiert und anschließend bei -80 °C weggefroren werden. Bei der Verwendung im ELISA oder als Antigen innerhalb einer Vakzine wurden stets Aliquots einer einheitlichen Präparation verwendet, die zudem maximal zweimal aufgetaut wurden. Die vermerkte Konzentration der FIS-Präparate bezog sich immer auf die ursprünglich eingesetzte Anzahl an lebensfähigen Sporen. 3.3 Auf- und Vorbereitung der Vakzinen 3.3.1 Endotoxinbestimmung Zur Verwendung der aufgereinigten Proteine (rBclA und rPA) im Tierversuch musste sichergestellt werden, dass die zu applizierende Dosis insgesamt einen Endotoxingehalt von 5 EU/kg Körpergewicht mit 10 ng Endotoxin pro EU [205] nicht überschritt. Entsprechend mussten Antigenpräparationen vor der Verwendung als Vakzine via LAL-Test-Kit auf Endotoxingehalt getestet und gegebenenfalls weiter gereinigt werden. Alle Arbeitsschritte waren möglichst in Glaswaren auszuführen, welche zuvor bei 180 °C für 6 h trocken-sterilisiert werden mussten, um Endotoxinfreiheit zu gewährleisten. Verwendete Plastikgefäße wurden üN in 1 M NaOH inkubiert, gründlich mit Endotoxin-freiem Wasser gespült, getrocknet und bei 121 °C sterilisiert oder entsprechend endotoxinfrei erstanden. Zur Evaluierung der Endotoxinkonzentration musste für jeden Test eine Standardreihe mit einer Endotoxinlösung von bekannter Konzentration erstellt werden. Diese war Bestandteil des Test-Kits und hatte eine Konzentration von 50 EU/ml. Die daraus zu erstellende Standardreihe beinhaltete Konzentrationen von 50 bis 0,005 EU/ml, welche in Glasröhrchen mit endotoxinfreiem Wasser angesetzt wurden. Die zu testenden Proben wurden ebenfalls in verschiedenen Verdünnungen getestet. Das gesamte Protokoll sah für jeden Mischschritt ein einminütiges Vortexen zur vollständigen Durchmischung vor, welches vor der Belegung der verwendeten sterilen, endotoxinfreien 96-Well-Platte mit den präparierten Proben und der Standardreihe wiederholt wurde. Die Blindprobe (endotoxinfreies Wasser) und die Standardreihe waren zu je 100 µl 37 3. Methoden im Doppelansatz in die Kavitäten der Platte aufzutragen. Eine vierfache Auftragung war für die Proben nötig, da jeweils zwei Ansätze mit Endotoxin (5 EU/ml) versetzt wurden, um eine hemmende oder amplifizierende Wirkung der Bestandteile sichtbar zu machen. Die fertig belegte Platte, mit einem Deckel abgedeckt, inkubierte im Anschluss für 10 min bei 37 °C. Danach erfolgte die Zugabe von je 100 µl frisch angesetztem LAL zu jeder Kavität und die kinetische Messung des Farbumschlags bei OD405 nm und 37 °C. Die Errechnung der Konzentration geschah über das verwendete Programm (WinKQCL) automatisch. 3.3.2 Endotoxinaufreinigung Zur weiteren Aufreinigung mittels EndoTrap blue Endotoxin-Reinigungs-Kit wurden nur Präparationen verwendet, die eine hohe Proteinkonzentration in Kombination mit niedrigem Endotoxingehalt aufwiesen. Die verwendete Reinigungssäule hatte ein Volumen von 1 ml und wurde manuell entsprechend der Vorgaben des Herstellers zur Säuberung von maximal 15 ml Proteinlösung verwendet. Zur Vorbereitung der Säule war diese je zweimal mit 3 ml Regenerationspuffer und 3 ml Equilibrierungspuffer zu spülen. Die Auftragung der Proteinlösung erfolgte sukzessive im Anschluss, wobei das Eluat nach dem Ausfluss des Säulenvolumens wieder aufgefangen wurde. Durch Nachspülen mit 1 ml Equilibrierungspuffer konnte die aufgetragene Probe komplett heruntergespült werden. Die Elution wurde in endotoxinfreien Gefäßen aufgefangen, auf Protein- und Endotoxingehalt geprüft und gegebenenfalls erneut gereinigt. Durch Regeneration der Säule mit zweimal 3 ml Equilibrierungspuffer konnte die Säule bis zu 10 Mal wiederverwendet werden. Im Schnitt war eine 10 – 100fache Reduzierung des Endotoxingehalts pro Aufreinigung erreichbar. 3.3.3 DNA-Patronen-Herstellung Zur nadelfreien Applikation der DNA-Vakzine via GeneGun waren goldbeschichtete Patronen herzustellen. Dabei wurde DNA mit dem Ziel, sie intrazellulär einzuschleusen, an Goldpartikel gebunden und über eine Druckluftpistole in die Haut geschossen. 3.3.3.1 Präzipitation von DNA auf Goldstaub Hierfür wurden zunächst 30 mg Goldstaub mit 250 µl Spermidin (0,05 M) versetzt und durch mehrmaliges Vortexen und Beschallen im Ultraschallbad für je 3 – 5 s so vermischt, dass eine homogene Dispersion frei von Verklumpungen vorlag. Danach erfolgte die Zugabe der DNAVektoren zu je 150 bis 200 µl bei einer Konzentration von 1 µg/µl. Bei der Erstellung von Mischpatronen, also Patronen mit zwei oder mehr unterschiedlichen Vektoren musste die Ver- 38 3. Methoden mischung vor der Zugabe erfolgen, um eine gleichförmige Beschichtung zu gewährleisten. Anschließend wurde die Dispersion kurz gevortext (5 s) und schließlich durch tropfenweise Zugabe von 250 µl Calciumchlorid (1 M) und unter sachtem Vortexen gefällt. Zum vollständigem Setzen der gefällten Goldpartikel wurde die Dispersion 10 min bei RT inkubiert und anschließend für 15 – 60 s bei 3000 – 4000 x g abzentrifugiert. Das verbliebene Pellet wurde nach Abgießen des Überstands durch mehrmaliges Anschnipsen resuspendiert und fünfmal mit 1 ml frischen 100 %igem Ethanol gewaschen. Nach Beendigung der Waschschritte war das überstandsfreie Pellet in insgesamt 3 ml PVP-Lösung zu resuspendieren. Diese erfolgte schrittweise mit je 0,5 ml PVP-Lösung, wobei eine leichte Ultraschallbehandlung zur besseren Resuspension verwendet werden konnte. Die so hergestellte Gold-DNA-Dispersion konnte bis zur Verwendung luftdicht verschlossen bei -20 °C bis zu 2 Monate gelagert werden. 3.3.3.2 Schlauchpräparation und Beschichtung Zur Beschichtung eines Tefzel Schlauchs mit der präparierten DNA-Dispersion musste dieser zunächst auf 76 cm zurechtgeschnitten und kurz mit Ethanol (100 %) gespült werden. Die anschließende Trocknung fand im Schlauchrotor für mindestens 5 min mittels Stickstoffbegasung (1 bar) ohne Rotation statt. Nach dem Entfernen des getrockneten Schlauchs aus der Apparatur wurde die zuvor präparierte Gold-DNA-Dispersion kurz gevortext und beschallt und anschließend schnell unter Vermeidung von Luftblasen in den Tefzel Schlauch aufgezogen. Der Schlauch musste danach vorsichtig in die Apparatur eingespannt und zur gleichmäßigen Ablagerung der Partikel im Inneren des Schlauchs sofort in einem streng einzuhaltenden Rhythmus rotiert werden: 1. Vollständige, ununterbrochene Rotation für 1 min. 2. ¼ Umdrehungen alle 15 s für 2 min. 3. ¼ Umdrehungen alle 30 s für 4 min. 4. ¼ Umdrehungen alle 60 s für 4 min. Nach der letzten Bewegung des Schlauches und der darauf folgenden finalen Ablagerung für 1 min wurde die Flüssigkeit durch den Anschluss einer Schlauchpumpe mit 6 ml/min vollständig abgezogen. Die Trocknung erfolgte unter Rotation mit 1 bar Stickstoff für mindestens 5 min bzw. bis der Schlauch vollständig durchgetrocknet war. Die Endtrocknung geschah für 2 – 5 min bei 3 – 4 bar. Der fertige Schlauch wurde durch eine Schlauchschnittmaschine in gleichförmige Patronen geschnitten, die bis zur Verwendung luftdicht und trocken bei 4 °C bis zu 8 Monate gelagert werden konnten. 39 3. Methoden 3.3.3.3 Qualitätskontrolle der DNA-Patronen Zur Kontrolle der präparierten DNA-Patronen wurde das Vorhandensein und die Konzentration von DNA pro Patrone, sowie der Qualität des Verschusses getestet. Zur Feststellung des Inhalts der Patronen erfolgte eine Spülung von insgesamt 5 Patronen einer Präparation mit 200 µl Millipore. Die Beschichtung wurde dabei durch kurzes Vortexen und Beschallen von der Patrone gelöst. Je 1, 5 und 10 µl der resultierenden Dispersion 1 : 1 versetzt mit Probenpuffer wurden anschließend im Vergleich zum 1 kb DNA-Marker und der ursprünglich eingesetzten DNALösung in einer Agarosegelelektrophorese in einem 1 %igem Agarosegel auf Anwesenheit der Plasmide überprüft. Die Auswertung der Geldokumentation erfolgte mit dem Programm Quantum.CAPT. Das Restvolumen der 200 µl Probe wurde kurz anzentrifugiert, um das Absetzen etwaiger Goldpartikel zu beschleunigen. Danach waren 100 µl abzunehmen, mit Millipore auf 1000 µl aufzufüllen und in einem Photometer zur Bestimmung der DNA-Konzentration zu messen. Anhand der gemessenen OD konnte über die Formel OD 260 nm = 1 ≙ 50 µg/ml, der eingerechneten Verdünnung und verwendeten Anzahl an ausgespülten Patronen auf die durchschnittliche Menge an beschichteter DNA pro Patrone geschlossen werden. Zur finalen Kontrolle der Qualität wurden mindestens 2 Patronen einer Präparation probeweise mit einer GeneGun auf Filterpapier verschossen. Dabei waren die Intensität der Färbung des Einschusses, die Verteilung der Einfärbung und die Rückstände in der Patrone zu bewerten und galten ggf. als Ausschlusskriterium für eine Verwendung. 3.4 Vakzinierung, Probennahme und Infektion Die Protein-basierenden Antigene umfassten rPA83, rBclA, ein Kapselkonjugat und FIS, sowie ein kommerzielles Lipopeptid als Adjuvans. Das in dieser Arbeit verwendete Lipopeptid stellt eine Mixtur aus Pam3Cys-SKKKK, einem TLR2/1 Aktivator [46] und Pam3Cys-FISEAIIHVLHSRHPG einem promiskuitiven T-Helferzellepitop des Pottwal Myoglobins (AA 106-121) dar [271]. Beim Kapselkonjugat handelt es sich ebenfalls um ein künstliches Konstrukt. Basierend auf den vielversprechenden Ergebnissen zur Erhöhung der Immunogenität der Kapsel durch Konjugation (siehe auch Kapitel 1.5.3.2 ) wurde in der Hoffnung Adjuvans und Antigen in einer Substanz zu vereinen ein Kapsel-Lipopeptid-Konjugat auf Grundlage des Lipohexadekapeptids des B. cereus Sporen Germinationsprotein (GerD) konstruiert. Das resultierende Nonamer der PGA Pam3Cys-AQEKEAKSELDYD-(isoE9)-NH2 wie auch das Lipopeptid Adjuvans wurde extern durch EMC Microcollections hergestellt. Die Produktion und Aufreinigung von rPA83 und rBclA aus Escherichia coli (E. coli) und FIS aus Sporen der SSLV war Bestandteil dieser Arbeit. 40 3. Methoden Für die DNA-basierenden Vakzinen wurden bis auf eine Ausnahme Vektorrückgrate der pDNAVaccUltra Familie verwendet, welche zur Verwendung als Vakzinevektoren optimiert sind [361][360]. Je nach integriertem Antigen beinhalteten die Vektoren verschiedene flankierende Signalsequenzen zur Sekretion (TPA [372], endosomalen Lokalisation (LAMP1 [53] [185][362] oder Einschleusung in das Proteasom (Uq [69]). Das pDNAVaccUltra Vektorrückgrat ohne Signalsequenzen wurde als separater Vektor auch für das Adjuvans CIITA verwendet und entsprechend in Mischung mit den jeweiligen Vakzinevektoren appliziert. Für mIPS-1, als weiteres zu testendes Adjuvans, wurde ein anderes Vektorrückgrat verwendet, in das ebenfalls das zu testende Antigen eingebaut war. Die zu testenden Antigene wurden durch die vertreibende Firma NATX in die entsprechenden Vektoren kloniert und in den benötigten Mengen hergestellt. Die Antigene umfassten LFD1PAD4, PA83 und BclAD1D3. Das Fusionsprotein LFD1PAD4, bestehend aus der PA-bindenden Domäne von LF und der rezeptorbindenen Domäne von PA wurde vergleichend zu PA83 getestet. Im Gegensatz zum Proteingegenstück, wurde im Falle von BclA für die DNA-Versuche eine verkürzte Version von BclA verwendet, um die Effizienz des Konstruktes zu steigern. Die mittlere, kollagenähnliche Domäne von BclA fehlte daher vollständig, da sie kaum Anteil an den gebildete sporenspezifischen Antikörpern hat [195]. Die resultierenden Vektoren NTC7382-TPA-LFD1PAD4-mIPS-1, pDNAVaccUltra1TPA-LFD1PAD4-LAMP1, pDNAVaccUltra1-TPA-PA83-LAMP1, pDNAVaccUltra1-TPA- BclAD1D3-LAMP1, pDNAVaccUltra4-BclAD1D3-Uq und pDNAVaccUltra5-CIITA wurde innerhalb der Mausversuche via GeneGun appliziert und entsprechend i. m. im Ziegenversuch. Tabelle 13: Übersicht Vakzinekomponenten (Mausversuch) Proteinbasierende Vakzinekomponenten Bezeichnung Abkürzung Beschreibung FIS O Formalin inaktivierte Sporen der SSLV Kapselkonjugat C Nonamer der Kapsel konjugiert an ein Lipopeptid: Pam3 Cys-AQEKEAKSELDYD-(isoE9)-NH 2 als Vakzine mit Adjuvanscharakter Lipopeptid L Mixtur aus Pam3 Cys-SKKKK und Pam3 Cys-SFISEAIIHVLHSRHPG als Adjuvans rBclA B rekombinantes BclA in voller Länge als Bestandteil des Exosporiums rPA83 P rekombinantes PA in voller Länge als Bestandteil des Letaltoxins DNA-basierende Vakzinekomponenten Bezeichnung Abkürzung Beschreibung NTC7382-TPA-LFD1PAD4-mIPS-1 S Fusionsprotein LFD1PAD4 als Bestandteil des Letaltoxins innerhalb eines sekretierenden Vektors inklusive des vektorinternen Adjuvans mIPS-1 pDNAVaccUltra1-TPA-BclAD1D3-LAMP1 K BclA ohne Collagen-ähnliche Domäne als Bestandteil des Exosporiums innerhalb eines sekretierenden und endosomal einschleusenden Vektors pDNAVaccUltra1-TPA-LFD1PAD4-LAMP1 A Fusionsprotein LFD1PAD4 als Bestandteil des Letaltoxins innerhalb eines sekretierenden und endosomal einschleusenden Vektors pDNAVaccUltra1-TPA-PA83-LAMP1 H PA in voller Länge als Bestandteil des Letaltoxins innerhalb eines sekretierenden und endosomal einschleusenden Vektors pDNAVaccUltra4-BclAD1D3-Uq M BclA ohne Collagen-ähnliche Domäne als Bestandteil des Exosporiums innerhalb eines Proteasom einschleusenden Vektors pDNAVaccUltra5-CIITA T murines CIITA als vektorexternes Adjuvans Die Abkürzungen beziehen sich auf die Gruppenkürzel für den Mausversuch. 41 3. Methoden Die beschriebenen Protein- und DNA-Vakzine sollten im Tierversuch auf Immunogenität und Schutzwirkung getestet werden. Hierfür war zunächst eine Testung aller Komponenten einzeln und in Kombination im NMRI-Mausmodell vorzunehmen, um die erfolgversprechendsten Kompositionen für die Anwendung im Ziegenmodell zu ermitteln. Protein und DNA Vakzine wurden hierbei als eigenständige, parallel verlaufende und separate Versuchszweige angesehen, deren Kombination zunächst nicht vorgesehen war. Eine Übersicht der verwendeten Vakzinekomponenten ist in Tabelle 13 zu sehen. 3.4.1 Mausversuche Für die Testung der Vakzinekomponenten im Mausmodell wurden 8 – 12 Wochen alte, weibliche NMRI-Mäusen in verschiedenen Versuchsgruppengrößen benutzt. Vakzinegruppen beinhalteten je 10 Tiere, negative Kontrollgruppen nur fünf. Die Benennung der Gruppen setzte sich aus dem Kürzel für das Adjuvans als ersten Buchstaben und mindestens einem weiteren Buchstaben für eine Vakzinekomponente zusammen (Abbildung 9). In Tabelle 14 sind alle Impfgruppen der Mausversuche zur Übersicht aufgeführt. Entsprechend der durchgeführten Versuche waren insgesamt drei Negativkontrollen vorhanden, die Abbildung 9: Nomenklatur der Mausgruppen als Vergleichsmaßstab für die ihnen zugeordneten Gruppen Benennung der Mausgruppen entsprechend der verabreichten Adjuvanzien (1. Buchstabe; rot) und Vakzinen (2. und folgende Buchstaben; blau) mit Angabe von mindestens je einem Buchstaben für Adjuvans und Vakzine galten. Alle Negativkontrollen wurden analog der Vakzinegruppen geimpft, erhielten aber nur das jeweilige gruppen- spezifische Adjuvans (Lipopeptid oder CIITA). Entsprechend sind jeder Gruppe nach einer Eingewöhnungszeit von 2 Wochen drei Impfungen im Abstand von 2 Wochen verabreicht worden. In diesem Zeitraum oblag die Pflege den Mitarbeitern der „Zentralen Einrichtung für Biologische und Biomedizinische Forschung mit Tierhaltung“ (ZVH) der Universität Hohenheim, welche auch die Markierung über Ohrlochung und die Entnahme von Blut zur Analyse durchführten. Die Infektion wurde 3 Wochen nach der letzten Impfung im Institut für Umwelt und Tierhygiene in einem behördlich zugelassenen Milzbrandlabor durchgeführt. Für den Zeitraum der Infektion wurde daher die Pflege, Beobachtung und Nachbereitung durch die Verfasserin der Arbeit und die Mitarbeiter des Instituts gewährleistet. Für den gesamten Versuchszeitraum waren die Mäuse in Käfigen mit solidem Böden auf Einstreu zu je 5 Tieren pro Käfig, mit entsprechendem Enrichment untergebracht. Nahrung und Wasser standen ad libitum zur Verfü- 42 3. Methoden gung. Umgebungstemperatur, Luftfeuchtigkeit und Hell/Dunkelzyklus (12 h) wurden entsprechend den Vorschriften eingerichtet und kontrolliert. Tabelle 14: Impfgruppen Mausversuch Gruppe Tierzahl Impfung Infektionsdosis LN-1000 5 50 µg Lipopeptid (Negativkontrolle der Proteinvakzinen für Infektion mit ~1000 Sporen) 840 LN-2000 5 50 µg Lipopeptid (Negativkontrolle der Proteinvakzinen für Infektion mit ~2000 Sporen) 1990 TN 5 ~3 µg pDNAVaccUltra5-CIITA (Negativkontrolle für DNA-Vakzine) 894 Proteinvakzinen mit ~1000 Sporen infiziert: NC 10 25 µg Kapselkonjugat 840 NCP 10 je 25 µg Kapselkonjugat und rPA83 901 Proteinvakzinen mit ~2000 Sporen infiziert: LB 10 50 µg Lipopeptid und 25 µg rBclA 2020 LP 10 50 µg Lipopeptid und 25 µg rPA 2020 LBP 10 50 µg Lipopeptid und je 25 µg rBclA und rPA 2020 LBCP 10 50 µg Lipopeptid und je 25 µg rBclA, rPA und Kapselkonjugat 2020 LBCOP 10 50 µg Lipopeptid, je 25 µg rBclA, rPA und Kapselkonjugat und 108 FIS 2020 NS 10 ~3 µg NTC7382-TPA-LFD1PAD4-mIPS-1 894 TA 10 insgesamt ~3 µg DNA zu gleichen Teilen bestehend aus pDNAVaccUltra1-TPA-LFD1PAD4-LAMP1 und pDNAVaccUltra5-CIITA 894 TH 10 insgesamt ~3 µg DNA zu gleichen Teilen bestehend aus pDNAVaccUltra1-TPA-PA83-LAMP1 und pDNAVaccUltra5-CIITA 894 TM 10 insgesamt ~3 µg DNA zu gleichen Teilen bestehend aus pDNAVaccUltra4-BclAD1D3-Uq und pDNAVaccUltra5-CIITA 901 TK 10 insgesamt ~3 µg DNA zu gleichen Teilen bestehend aus pDNAVaccUltra1-TPA-BclAD1D3-LAMP1 und pDNAVaccUltra5-CIITA 901 TKS 10 ~3 µg NTC7382-TPA-LFD1PAD4-mIPS-1 und je ~1,5 µg pDNAVaccUltra5-CIITA und pDNAVaccUltra1-TPABclAD1D3-LAMP1 864 DNA-Vakzinen: Die Infektionsdosis bezieht sich auf die Anzahl tatsächlich applizierter Sporen, die über die Auszählung von mindestens zwei überzähligen Infektionsdosen ermittelt wurde (siehe auch Kapitel 3.2.6) 3.4.1.1 Betäubung und Tötung Zur Vorbereitung der Mäuse für den DNA-Beschuss, Applikation der Impfstoffe und die Infektion war eine kurzzeitige Betäubung der Tiere notwendig. Hierfür ist eine belüftete Box verwendet worden, in der die Tiere einzeln bis zur Bewusstlosigkeit mit Isofluran begast wurden. Die Gasmenge war so gewählt, dass eine schnelle Wirkung ohne Exitation erzielt werden konnte. Bis zur Wiedererlangung des Bewusstseins standen die Versuchstiere unter Beobachtung und wurden, wenn nötig durch Herzmassage wiederbelebt. Zur Tötung der überlebenden Versuchstiere nach dem Versuchsende oder zur Euthanasie moribunder Tiere wurde in gleicher Art CO 2 verwendet. Hierbei verblieben die zu tötenden Tiere im Begasungsgefäß bis zum Stillstand der Atmung und der Absetzung von Harn. Die Sicherstellung des eingetretenen Todes hatte oberste Priorität vor der weiteren Präparation des Leichnams. 43 3. Methoden 3.4.1.2 Impfung Alle Mausgruppen wurden dreimal im Abstand von 2 Wochen immunisiert. Hierfür waren für die Proteinvakzinen entsprechend der eingesetzten Antigene je 50 µg Lipopeptid und 108 Sporen FIS, sowie je 25 µg rPA83, rBclA und Kapselkonjugat anzusetzen. Die Impfdosen wurden für jede Impfung und Gruppe gemeinschaftlich in Mischung angesetzt, um die Gleichförmigkeit der Impfdosen zu gewährleisten. Das angesetzte Volumen wurde entsprechend so mit sterilem endotoxinfreiem PBS aufgefüllt, dass sich eine 200 µl Dosis ergab, die s. c. in den Nacken zu verabreichen war. Für die Applikation der DNA-Vakzinen war eine Rasur des Abdomens notwendig. Hierfür wurden alle Versuchstiere initial im entsprechenden Bereich geschoren und rasiert und direkt vor der Applikation der Dosis nochmals rasiert und mit Ethanol (70 %) desinfiziert. Jedes Versuchstier erhielt zwei DNA-Patronen pro Dosis i. c. appliziert mit 400 bar via GeneGun. Die Zusammensetzung beider Patronen war gleich und entsprach zusammengenommen einer Mindestmenge von 3 µg. Alle Versuchstiere einer Gruppe erhielten die gleiche Kombination an DNA-Patronen. Bei Versuchsgruppen mit 10 Tieren war eine Patronenpräparation nicht ausreichend. Um eine Schwankung der Impfdosis zu vermeiden, bestanden die Impfungen aus je einer Patrone einer Präparation, sodass zu jeder Immunisierung mit der gleichen Menge pro Tier gearbeitet werden konnte. Die Patronen Abbildung 10: Rückstandbild von DNA-Patronen nach Verschuss wurden vor der Benutzung jeweils probeweise auf Filter- Skala von 1 – 5 der verwendeten DNA-Patronen nach Verschuss; Einfärbung durch GoldRückstände papier verschossen und auf Rückstände begutachtet (siehe Anhang, Abbildung 58). Wie in Abbildung 10 zu sehen, konnten die Patronen rückstandslos (1) bis unbenutzt aussehend (5) verschossen werden, wenngleich bei allen Rückstandsbildern eine deutliche Einfärbung des Filterpapiers bei Probeschüssen zu sehen war. 3.4.1.3 Probennahme und Diagnostik Zur serologischen Analyse wurde vor dem Beginn der Impfung und direkt vor der Infektion Blut über die Vena facialis oder retrobulbär durch die Mitarbeiter der ZVH entnommen. Außerdem erfolgte eine Blutentnahme über Herzpunktion nach der Beendigung des Infektionsversuchs. Hierfür wurden überlebende Tiere getötet, identifiziert und anschließend durch Öffnung des Brustraums und Herzpunktion entblutet. Verstorbenen und getöteten Tieren waren überdies Milz und Leber für einen Erregernachweis zu entnehmen. Die entnommenen Organe wurden gequetscht, auf CBA ausplattiert und üN 44 3. Methoden bei 37 °C und 5 % CO2 inkubiert. Die Detektion des Erregers in Leber und Milz diente der Verifikation der Todesursache bei den der an der Infektion verstorbenen Tieren. Die entsprechende Abwesenheit des Erregers in Überlebenden galt als aussagekräftiger Beweis für die Klärung der Infektion in diesen Tieren. 3.4.1.4 Infektion Die Infektion erfolgte 3 Wochen nach der letzten Impfung unter Betäubung mit einer s. c. Injektion von ~25LD50 (~1000 Sporen [24]) bzw. ~50LD50 (~2000 Sporen [24]) einer Sporensuspension eines voll virulenten Ames Stammes in den Nacken. Aufgrund von Kapazitätsbeschränkungen bezüglich der Versuchstierhaltung im BSL-3 wurden die Versuche teilweise gestaffelt durchgeführt. Die Sporensuspension war dabei für alle Versuche gleich und in entsprechendem Umfang zuvor angesetzt worden. Vor jedem Infektionszyklus wurde sie erneut hitzebehandelt (siehe Kapitel 3.2.5) und ausgezählt, wobei sich die Konzentration nur marginal veränderte. Um die tatsächlich applizierte Dosis abzuschätzen, wurden zusätzlich nach der Infektion überzählige Dosen auf CBA ausplattiert und ausgezählt (siehe Kapitel 3.2.6). Die entsprechenden Mittelwerte dieser Auszählungen sind bei den Gruppen vermerkt und unterlagen leichten Schwankungen. Angestrebt wurden aber Dosen von ~1000 und ~2000 Sporen in einem Volumen von 150 – 200 µl. Die Infektion der Mausgruppen geschah mehrheitlich mit ~1000 Sporen (~25LD 50). Eine erhöhte Dosis von ~2000 Sporen (~50LD50) wurde nur für die Proteingruppe LN-2000, LP, LBP, LBCP und LBCOP angewendet. Hintergrund für die unterschiedliche Infektionsdosis einzelner Gruppen, waren die Ergebnisse hier nicht beschriebener Vorversuche in denen dieselben Gruppen mit ~25LD50 infiziert wurden. Eine Unterscheidung der Schutzwirkung war mit dieser Dosis für die genannten Gruppen nicht möglich, da alle Vakzinekompositionen 80 – 100 % Schutzraten erzeugten. Außerdem war bei Gruppe LN-1000 eine verlängerte Überlebenszeit beobachtet worden. Diese Restschutzwirkung des Lipopeptids war ebenfalls bereits in anderen Studien aufgefallen [24]. Für eine erkennbare Unterscheidung der betreffenden Gruppen und eine stärkere Absetzung von der Negativkontrolle wurde daher eine höhere Infektionsdosis für diese Proteingruppen angewendet. Eine äquivalente Erhöhung der Infektionsdosis für die DNAbasierenden Vakzinen schien nicht sinnvoll, da die dort eingesetzten Adjuvanzien keine Restschutzwirkung aufwiesen und eine ausreichende Unterscheidung der Gruppen gegeben war. Gleiches galt für die Vorversuche der Protein-basierenden Vakzinen mit dem Kapselkonjugat (Gruppe LN-1000, NC und NCP), deren Wiederholung mit einer höheren Infektionsdosis in Anbetracht der Ergebnisse überdies nicht sinnvoll schien. 45 3. Methoden Das Überwachen der Sterberate vollzog sich bis 3 – 4 Wochen nach der Infektion, wobei todgeweihte Tiere und Tiere, die den Versuchszeitraum überlebten mit CO 2, wie unter 3.4.1.1 beschrieben, euthanasiert wurden. Die Beobachtungsintervalle betrugen 3 Stunden, wobei dies auch nachts und am Wochenende gewährleistet werden musste. Bei Auffälligkeiten wie rauem, gesträubtem Fell, Zittern, leichter Abgeschlagenheit und hoher Atemfrequenz war die Beobachtungsfrequenz auf 1 Stunde zu erhöhen. Waren Versuchstiere bewegungslos, apathisch, lethargisch, krampfend oder moribund, musste eine Euthanasie durchgeführt werden. Da Mäuse oft symptomlos an einer B. anthracis Infektion versterben, konnte für viele Tiere der genaue Todeszeitpunkt nicht ermittelt werden. Die Überlebenszeit umfasste daher den Zeitraum von der Applikation der Infektion bis zur Feststellung des Todes, respektive der Durchführung der Euthanasie. Die Identifizierung und eindeutige Zuordnung von Individuen zur Überlebenszeit erfolgte über Ohrmarkierungen. 46 3. Methoden 3.4.2 Ziegenversuch (Südafrika) Abbildung 11: Burenziegen Burenziegen mit Ohrmarkierungen innerhalb des Versuchsaufbaus Die Ziegenversuche in Südafrika wurden in Kooperation mit SANParks und der Universität von Pretoria, Abteilung für veterinäre, tropische Krankheiten, in Zusammenarbeit mit Okechukwu C. Ndumnego, Masterstudent im DFG-Projekt und Tierarzt aus Nigeria, und Dr. Henriette van Heerden durchgeführt. Dabei oblag Herrn Ndumnego die Vorauswahl, Testung, Impfung und Blutung der Ziegen, sowie die Vorbereitung der Sporensuspension zur Infektion. Die Applikation und Überwachung der Infektion wurde gemeinschaftlich in Südafrika durchgeführt. Die serologische Auswertung erfolgt durch die Verfasserin der Arbeit an der Universität Hohenheim. Die Herstellung der Antigene zur Vakzinierung und serologischen Analyse vollzog sich analog zum Mausversuch. Für den Erwerb der Versuchstiere wurden Burenziegen ohne Krankheits- oder Impfhistorie bezüglich B. anthracis aus lokalen Beständen auf anti-rPA83 IgG-Titer überprüft und, wenn keine die Titer niedrig genug oder nicht nachweisbar waren, erstanden. Die Versuchsgruppen bestanden daher aus einer zufällig ausgesuchten, heterogenen Mischung von 2 – 5 weiblichen und kastrierten männlicher Burenziegen mit ähnlichem Alter. Für eine eindeutige Identifizierung waren alle Tiere mit Ohrplaketten und entsprechenden Nummern (Nr.) markiert (Abbildung 11). Vor dem Start der Versuchsreihe war der Gesundheitszustand aller Versuchstiere überprüft und eine Wurmkur mit Albendazol, Niclosamid und Ivermectin durchgeführt worden. Die Haltung und Versorgung der Tiere während der Akklimatisierung, Vakzinierung und Blutung oblag den Mitarbeitern der Universität Pretoria. Die Infektion fand in überdachten Außengehegen im Krüger Nationalpark statt (Abbildung 12), da dort aufgrund der Prävalenz von B. anthracis eine Kontamination durch den Versuch selbst unerheblich war. Der Transport zur Versuchsanlage fand 5 bis 14 Tage vor der Infektion statt. Die Pflege der Tiere wurde gemein- 47 3. Methoden schaftlich unter Mithilfe der Mitarbeiter des Nationalparks (SANParks) übernommen. Die Versorgung mit Wasser erfolgte während des gesamten Versuchs ad libitum. Heu und Pellets wurden mengenkontrolliert zu geregelten Zeiten verfüttert. In Tabelle 15 sind alle in Südafrika durchgeführten Gruppen aufgelistet. Es wurden zwei ProteinKombinationen, eine DNA-Kombination, drei Kontrollgruppen und drei verschiedene Impfgruppen mit der kommerziellen SSLV vergleichend in Burenziegen getestet. Ebenfalls Teil des Ziegenversuchs waren zwei BALB/c Mausgruppen, bestehend aus je drei naiven, 8 – 12 Wochen alten, weiblichen Abbildung 12: Außengehege für die Infektion Mäusen, die zur Verifizierung der Infektiösität des Infektionsgehege im Krüger Nationalpark eingesetzten Infektionsstammes verwendet wurden. Eine detaillierte Übersicht zu den Probennahmezeitpunkten, Immunisierungen, Transporten und Infektionen findet sich im Anhang (Tabelle 53). Tabelle 15: Impfgruppen Ziegenversuch (Südafrika) Gruppe Tierzahl Impfung Infektionsdosisa Infektionszeitpunkt b 1 3 1 ml PBS (1 Mal) 36 62 2 5 SSLV, kommerziell (1 Mal) 850 6 3a 5 SSLV, kommerziell (1 Mal) 850 62 3b 5 SSLV, kommerziell (2 Mal im Abstand von 58 Wochen) 850 4 4 5 Protein-Kombination mit 500 µg Lipopeptid und je 25 µg rPA83 und rBclA (3 Mal im Abstand von 3 Wochen) 844 4 5 5 DNA-Kombination mit je 1 mg NTC7382-TPA-LFD1PAD4-mIPS-1, pDNAVaccUltra5-CIITA und pDNAVaccUltra1-TPA-BclAD1D3-LAMP1 (3 Mal im Abstand von 3 Wochen) und 8 Wochen nach der letzten Impfung ein Protein-Boost entsprechend der Dosis von Gruppe 6 nicht infiziert - 6 5 Protein-Kombination mit 500 µg Lipopeptid, 108 Sporen FIS und je 25 µg rPA83 und rBclA (3 Mal im Abstand von 3 Wochen) 844 4 LK 2 500 µg Lipopeptid (1 Mal) 844 11 NK 2 nicht geimpft 172 0 a Anzahl tatsächlich applizierter Sporen, durch Auszählung der Infektionsdosis ermittelt b in Wochen nach der letzten Immunisierung Die Infektionsdosis bezieht sich auf die Anzahl tatsächlich applizierter Sporen, die über die Auszählung von mindestens zwei überzähligen Infektionsdosen ermittelt wurde (siehe auch Kapitel 3.2.6) 3.4.2.1 Impfung Die Durchführung der Impfung oblag Herrn DVM Ndumnego. Für die jeweiligen Injektionen wurde das Tier durch Pflegepersonal liegend am Boden fixiert, sodass der Oberschenkel des Hinterlaufs frei zugänglich war. Nach der Desinfektion mit Ethanol (70 %) erfolgte die Injektion s. c. in eine Hautfalte im Inguinalbereich. Die Konzentration der zu applizierenden Komponenten für die Proteinvakzinen betrug entsprechend 500 µg Lipopeptid, je 25 µg rPA83 und 48 3. Methoden rBclA, sowie 108 Sporen FIS, wenn diese ein Bestandteil der Vakzine waren. Die Verdünnung der Vakzinekomponenten erfolgte in sterilem endotoxinfreiem PBS, wobei die Impfdosen einer Gruppe zusammengefasst angesetzt wurde. Die Planung sah vor, alle Bestandteile der Impfung zu vermischen und in einem Endvolumen von 1 ml zu applizieren, um eine Zusammenwirkung aller Bestandteile sicherzustellen. Dies wurde durch die Mitarbeiter vor Ort missverstanden, sodass alle Vakzinekomponenten der Proteinapplikationen einzeln an die gleiche Stelle, jeweils in einem Volumen von 1 ml appliziert wurden. Dieser Fehler wurde erst nach Beendigung des Versuchs und dessen Auswertung realisiert und zog eine Versuchswiederholung (Kapitel 3.4.3) nach sich. Die Impfdosen der DNA-Vakzinen umfassten je 1 mg DNA pro Vektor und waren in einer einzigen 1 ml Dosis i. c. mittels Spritze appliziert worden. Der Protein-Boost, fand analog zu den Proteinvakzinen statt und war entsprechend ebenfalls suboptimal durchgeführt. Zur Testung der kapseldefizienten SSLV wurde ein kommerzielles Präparat verwendet, welches entsprechend der Gebrauchsanweisung des Herstellers zu je 1 ml s. c. injiziert wurde. Gruppe 1 diente dabei als Vergleichsgruppe, da dieser nur der Verdünnungspuffer (PBS) der Vakzine injiziert wurde. Einer weiteren Kontrollgruppe waren einmalig 500 µg Lipopeptid verabreicht worden, um adverse Effekte und die unspezifische Schutzwirkung des Adjuvans nachzuprüfen. 3.4.2.2 Probennahme Zur serologischen und diagnostischen Blutabnahme wurde den Tieren im Versuchszeitraum Blut aus der Halsvene (Vena jugularis externa) entnommen (Abbildung 13). Abgesehen von Gruppe 3a/b und 1, die über den Verlauf von einem Jahr zusätzlich auch monatlich beprobt wurden, sind alle Ziegen vor dem Beginn der Immunisierung, vor jeder weiteren Immunisierung, sowie vor und nach der Infektion geblutet worden (Details siehe Anhang, Tabelle 53). Diese Probennahmen umfassten ca. 10 ml Blut, die mittels Serumröhrchen aufgefangen wurden und der serologischen Analyse auf Antikörper Titer gegen, rPA83, rBclA, rLF, FIS und vegetativem Antigen, sowie TNA dienten. Weitere Blutentnahmen folgten Abbildung 13: Blutentnahme für Serologie Blutentnahme vor der Infektion über die Vena jugularis externa während der Überwachung der Infektion. Bei Verdachtsmomenten auf eine durchschlagende 49 3. Methoden Infektion mit B. anthracis waren durch Insulinspritzen zur Diagnostik kleinste Mengen (~100 µl) an Blut aus der Halsvene zu entnehmen und mittels Blutausstrich (siehe Kapitel 3.4.2.4) auf Bakterien zu überprüfen. Die Verwendung von Insulinspritzen für diese Entnahme war ohne weiteres möglich und ermöglichte die Durchführung wiederholter Blutentnahmen. 3.4.2.3 Infektion Die Infektion wurde in zwei Etappen in einer Region des Krüger Nationalparks durchgeführt, welche für B. anthracis endemisch ist. Der eingesetzte Stamm war ein Feldisolat (SD20), welches 2001 aus dem Ohr eines Schafes isoliert wurde, das im Gebiet von Standerton, Provinz Mpumalanga, Südafrika an Milzbrand verendet war. Der Stamm enthielt nachweislich die Plasmide pX01 und pX02 und wurde weiter über mikrobiologische und biochemische Methoden, sowie Sequenzanalyse charakterisiert und als B. anthracis verifiziert [181]. Die endgültige Verifikation der Letalität des Stammes konnte erst im Versuchsgehege des Krüger Nationalparks durchgeführt werden. Hierfür waren Abbildung 14: i. p. Injektion Maus zwei Gruppen von je drei naiven BALB/c Mäusen Infektion von BALB/c-Mäusen via i. p. Injektion mit 500 (200 µl) und 1000 (400 µl) Sporen, der zur Infektion verwendeten Suspension, i. p. infiziert worden (Abbildung 14). Die Freigabe der Ziegen zur Infektion geschah erst nach der einwandfreien Feststellung der Letalität des Stammes. Der Zeitraum zwischen Transport und Infektion betrug 5 Tage für die Negativkontrolle (NK), 9 Tage für Gruppe 4 und 6 und 14 Tage für alle anderen Versuchsgruppen (siehe Anhang, Tabelle 53). Die Infektion der Ziegen erfolgte wie zuvor beschrieben mit verschiedenen Sporenkonzentrationen im Bereich von 36 – 850 Sporen. Analog zum Vorgehen in den Mausversuchen wurden auch hier überzählige Infektionsdosen ausgezählt, um die tatsächliche Menge an applizierten Sporen abschätzen zu können (siehe Kapitel 3.2.6). Der Beobachtungszeitraum nach der Infektion umfasste bis zu 14 Tage, wobei todgeweihte Tiere und Tiere die den Versuchszeitraum überlebten mit Pentobarbital euthanasiert wurden. Aufgrund der Haltungsbedingungen in einem offenen Gehege konnten die Tiere tagsüber ohne Unterbrechung überwacht werden. Die Beobachtungsintervalle nachts betrugen 3 Stunden, wobei kranke und sterbende Tiere gewöhnlich auf sich aufmerksam machten. Daher war für die 50 3. Methoden meisten Tiere der Todeszeitpunkt eindeutig feststellbar. Eine Euthanasie war vorzunehmen, wenn Symptome wie abnormale Atem- und Herzfrequenz, Krampfanfälle, Lethargie, Hämorrhagie und Apathie auftraten und ein Nachweis des Erregers im Blut erbracht wurde (siehe Kapitel 3.4.2.4) bzw. der bevorstehende Tod eindeutig war. Davon ausgeschlossen waren die Tiere der Kontrollgruppen, da für diese das eindeutige Eintreten des Todes durch den Erreger, sowie die Feststellung des Todeszeitpunkts durch diesen für die Auswertung essentiell notwendig waren. Die notierte Überlebenszeit umfasste den Zeitraum von der Applikation der Infektion bis zur Feststellung des Todes, respektive der Durchführung der Euthanasie. Verstorbene Tiere inklusive des sie umgebenden Bodens wurden nach der Identifikation des Tiers und weiteren diagnostischen Maßnahmen mit Formalin (10 %) besprüht. Die in Leichensäcke verpackten Kadaver wurden noch vor Ort kremiert. 3.4.2.4 Diagnostik Zur Evaluation des Fortschreitens der Infektion wurde das Fress- und Sozialverhalten, die Temperatur und der physiologische Zustand jedes Tieres regelmäßig dokumentiert. Hierfür war täglich morgens bei jedem Tier rektal die Temperatur zu bestimmen. Anschließend und nochmals nachmittags wurde die Fütterung unter Beobachtung und Dokumentation des Verhaltens vorgenommen. Die Beobachtung der Tiere war tagsüber uneingeschränkt möglich und wurde entsprechend fortdauernd protokolliert. Bei auffälligen Tieren mit erhöhter Temperatur (> 40 °C) oder gestörtem Sozial- und Fressverhalten wurde die Frequenz der Temperaturmessung erhöht und jeweils im Zuge der Messung eine kleine Menge Blut (~100 µl) zur Analyse entnommen. Ein Tropfen des frischen Blutes wurde auf einen Objektträger aufgetragen und mittels eines zweiten Objektträgers verschmiert, um einen Blutausstrich anzufertigen (Abbildung 15). Diese möglichst dünne Blutschicht musste zunächst luftgetrocknet und anschließend in purem Methanol für 1 – 2 min fixiert werden. Danach erfolgte die Inkubation mit Azur B [231] für 1 – 5 min. Nach dem Abspülen der Färbelösung und Trocknung des Objektträgers konnte dieser unter dem Mikroskop auf angefärbte, bekapselte Bakterien untersucht werden. Über diese Methode war eine Detektion von Bakterien im Blut in weniger als einer Stunde möglich, wobei in Einzelfällen zu Verifikation ebenfalls ein Ausstrich auf CBA durchgeführt wurde. Im Falle der einwandfreien Detektion von Bakterien im Blut war eine Euthanasie des jeweiligen Tieres umgehend vorzunehmen. Davon ausgeschlossen waren die Kontrollgruppen, da für diese der Todeszeitpunkt unverfälscht ermittelt werden musste. Zur Verifikation der Todesursache wurde bei verstorbenen Tieren über das Anritzen des Ohres ebenfalls ein Blutausstrich angefertigt, um den Nachweis einer systemischen Infektion mit B. anthracis zu erbringen. Die entsprechende 51 3. Methoden Abwesenheit des Erregers in Überlebenden galt als aussagekräftiger Beweis für deren Klärung der Infektion und Klassifizierung als Überlebende. Abbildung 15: Färbung von Blutausstrichen mit Azur B Abgebildet sind die verschiedenen Schritte einer Färbung mit Azur B, beginnend mit dem Trocknen und Fixieren eines dünnen Blutausstriches (A), der Färbung mit Azur B und anschließender Spülung (B) und abschließend mit der Mikroskopie (C). Entsprechend der Färbeintensität war der Zellkörper und die umgebende Kapsel als ballonartige, rosafarbene Struktur angefärbt. 3.4.3 Wiederholung der Ziegenversuche (Türkei) Bei der Nachbetrachtung der Ziegenversuche in Südafrika war aufgefallen, dass die Immunisierung der Gruppen 4 und 6 nicht wie geplant durchgeführt worden war, das heißt die Impfkomponenten einzeln und nicht wie erforderlich in Mischung appliziert worden waren. Da sich die Möglichkeit ergab, in Kooperation mit der Kafkas Universität (Kars, Türkei) unter der Leitung von Prof. Dr. Mitat Şahin einen weiteren Infektionsversuch vorzunehmen, wurden die Gruppen 4 und 6 zu je 10 Tieren zusammen mit einer Negativkontrolle (NK (W)) bestehend aus 5 Tieren in einem verbesserten Ansatz wiederholt (Tabelle 16). Der generelle Versuchsaufbau im Sinne der Vorbereitung, Durchführung, Probennahme, Diagnostik, Nachbereitung und Auswertung erfolgte analog der zuvor beschriebenen Ziegenversuche mit geringfügigen Optimierungen. Die Vorauswahl, Testung, Impfung und Blutung der Ziegen, sowie die Vorbereitung der Sporensuspension zur Infektion oblag den Mitarbeitern der Kafkas Universität, angeleitet durch die Tierärzte Dr. Fatih Büyük und Dr. Özgür Ҫelebi. Die Applikation und Überwachung der Infektion wurde zusammen mit Herrn O.C. Ndumnego in den Versuchsanlagen der Kafkas Universität durchgeführt. Die Herstellung der Antigene und die serologische Auswertung der Versuche 52 3. Methoden erfolgte durch die Verfasserin der Arbeit an der Universität Hohenheim, analog zum Maus- und Ziegenversuch in Südafrika. Tabelle 16: Wiederholung der Ziegenversuche (Türkei) – Impfgruppen Gruppe 4 (W) Tierzahla 10 (8) Impfung Protein-Kombination mit 500 µg Lipopeptid und je 75 µg rPA83 und rBclA (3 Mal im Abstand von 3 Wochen) Infektionsdosisb Infektionszeitpunkt d 918 5 8 6 (W) NK (W) 10 (10) 5 (4) Protein-Kombination mit 500 µg Lipopeptid, 10 Sporen FIS und je 75 µg rPA83 und rBclA (3 Mal im Abstand von 3 Wochen) nicht geimpft a initial immunisierte Tierzahl (infizierte Tierzahl) b Anzahl tatsächlich applizierter Sporen, durch Auszählung der Infektionsdosis ermittelt c wurde in 2 Etappen zu je 2 Tieren infiziert d in Wochen nach der letzten Immunisierung 918 1027 (918) 5 c 0 Die Infektionsdosis bezieht sich auf die Anzahl tatsächlich applizierter Sporen, welche über die Auszählung von mindestens zwei überzähligen Infektionsdosen ermittelt wurde (siehe auch Kapitel 3.2.6); (W) – dient zur Unterscheidung der wiederholten Gruppen Die zur Immunisierung verwendeten Tiere stellten eine heterogene Gruppe aus männlichen und weiblichen Ziegen unterschiedlichen Alters (ca. 1 – 3 Jahre) und unterschiedlicher Rassen dar. Im Gegensatz zu den Bedingungen des Versuchs in Südafrika wurden die Ziegen während der gesamten Versuchsdurchführung in einer abgeschlossenen Stallanlage gehalten, bei der für den Zeitraum der Immunisierung ebenfalls ein großzügiges Außengehege frei zugänglich war. Damit entfiel die Notwendigkeit für einen Transport innerhalb des Versuchs. Die Impfung der zufällig zusammengestellten Gruppen erfolgte dreimal im Abstand von 3 Wochen nach einer Eingewöhnungszeit von einem Monat, während der eine Wurmkur mit Ivermectin und Depomin vorgenommen wurde. Die Dosis der Antigene rPA83 und rBclA wurde im Vergleich zum Vorversuch von 25 µg auf 75 µg pro Dosis angehoben, um eine verbesserte Wirksamkeit zu erreichen. Damit betrug die Konzentration der zu applizierenden Komponenten entsprechend 500 µg Lipopeptid, je 75 µg rPA83 und rBclA für Gruppe 4 (W), sowie zusätzlich 108 FIS für Gruppe 6 (W). Die NK (W) erhielt keinerlei Immunisierung. Zur Vermeidung erneuter Applikationsfehler wurden die Impfdosen an der Universität Hohenheim angefertigt und die Applikation der ersten Immunisierung durch die Verfasserin der Arbeit angeleitet. Probennahmen zur Serumanalyse wurden vor jeder Impfung, vor der Infektion und nach dem Überleben der Infektion vorgenommen. Eine detaillierte Übersicht zum Ablauf befindet sich im Anhang (Tabelle 54). Im Zeitraum der Immunisierungen fielen insgesamt 3 Ziegen durch den gewährten Freigang im Außengehege einheimischen Prädatoren zum Opfer. Zwei Tiere aus Gruppe 4 (W) verstarben im Zeitraum zwischen zweiter und dritter Immunisierung und ein Tier der NK (W) verstarb zwischen der dritten Immunisierung und der Infektion. Die serologischen Daten dieser Ziegen wurden bis zu Ihrem Ausscheiden aus dem Versuch in alle Betrachtungen mit einbezogen und sind entsprechend gekennzeichnet. 53 3. Methoden Zur Infektion standen daher nur 8 Tiere für Gruppe 4 (W), 10 Tiere für Gruppe 6 (W) und 4 Tiere für die NK (W) zur Verfügung. Die Infektion erfolgte mit Stamm K-136, einem Feldisolat der Region. Dieses war durch die Mitarbeiter der Kafkas Universität aus der Milz eines an Milzbrand verstorbenen Rinds isoliert und mittels Nachweis der virulenzvermittelnden Plasmide pX01 und pX02 identifiziert worden. Der Nachweis der Virulenz erfolgte in Mäusen durch Prof. Dr. Mehmet Doğanay an der Erciyes Universität (Kayseri, Türkei). In Anlehnung an die verwendete Infektionsdosis der Ziegenversuche in Südafrika wurde auch hier eine Dosis von ~1000 Sporen angestrebt. Die Infektion aller Tiere wurde mittels einer Sporensuspension durchgeführt, die eine Konzentration von 1255 Sporen/ml aufwies. Entsprechend der vorhergehenden Versuche waren auch hier überzählige Infektionsdosen bei jeder Infektion ausgezählt worden, wodurch sich die in Tabelle 16 angegebenen leicht veränderten Konzentrationen ergaben. Da der Infektionsstamm K-136 bisher nur in Mäusen erprobt worden war, wurde die Infektion zunächst exemplarisch an zwei zufällig ausgewählten Ziegen der NK (W) erprobt. Hierfür wurden die Ziegen von der Herde abgetrennt und in einem abgeschlossenen Bereich der Stallungen gehalten. Nach der Verifikation der Virulenz waren die verbleibenden Ziegen der NK (W) sowie die Ziegen der Gruppe 4 (W) und 6 (W) in gleicher Art infiziert worden. Aufgrund dieser Staffelung ergaben sich für die Angaben zu diesem Probenzeitpunkt und die Konzentration der ausgezählten Infektionsdosen Schwankungen. 3.5 Serologische Analyseverfahren 3.5.1 Blutaufbereitung Blutentnahmen für die Serologie wurden entweder bereits mit Serumröhrchen durchgeführt oder nach der Entnahme zur Gerinnung und Absetzung für 1 – 2 h bei 4 °C inkubiert. Die anschließende Zentrifugation bei 2000 x g und 4 °C für 10 min diente zur Sedimentation des Blutkuchens, wonach das aufliegende Serum in ein frisches Gefäß überführt wurde. Die Lagerung der gewonnenen Seren erfolgte bis zur Verwendung bei -80 °C, wobei größere Mengen zuvor gemischt und aliquotiert wurden. 3.5.2 Enzyme-linked immunosorbent Assay (ELISA) Zur Evaluation der Wirksamkeit der eingesetzten Impfstoffe wurden Seren der immunisierten Tiere auf detektierbare Antikörper gegen verschiedene Antigene im ELISA überprüft. Zunächst mussten hierfür 96-Well-Platten mit 0,5 µg Antigen in 100 µl Beschichtungspuffer pro Kavität befüllt und üN abgedeckt bei 4 °C inkubiert werden. Beschichtungen mit FIS wurden mit 10 8 Zellen pro Kavität bei 37 °C durchgeführt. Am Folgetag wurden die Platten zweimal mit 54 3. Methoden Waschpuffer durch ein Plattenwaschgerät gewaschen und danach mit 100 µl Blocklösung pro Kavität für 1 h bei RT inkubiert. Nach zwei weiteren Waschzyklen konnten die Platten mit den vorbereiteten Seren und Kontrollen belegt werden. Die Seren wurden auf den Platten, gewöhnlich beginnend mit einer Konzentration von 1 : 100, im Doppelansatz über 8 bzw. 12 log2 Stufen in Blocklösung reihenverdünnt (siehe Abbildung 16). Abweichungen von der Startkonzentration waren durch das Volumen des vorhandenen Serums oder den zu erwartenden Titerwerten bedingt. Ein Positivserum mit bekanntem Titer war zu Vergleichszwecken im Einfachansatz austitriert auf jeder Platte mitzuführen, ebenfalls ein entsprechendes Negativserum als Einzelpunktmessung im Doppelansatz. Jede Platte enthielt zudem mindestens sechs Parallelansätze für Blindproben, denen anstatt einer Serumprobe nur die Blocklösung zugefügt wurde. Die Belegung der Platten erfolgte so, dass das Endvolumen in allen Kavitäten 100 µl betrug. Nach einer Inkubation von 2 h bei RT wurden die Platten fünfmal gewaschen. Im nächsten Schritt war der sekundäre Antikörper in Blocklösung zu je 100 µl pro Kavität aufzutragen, welcher entsprechend der zu testenden Tierart und nachzuweisenden Antikörperklasse variabel war (Details siehe Material, S. 27). Alle sekundären Antikörper waren HRP konjugiert und wurden in unterschiedlichen Konzentrationen entsprechend der individuellen Wirksamkeit eingesetzt. Diese war zuvor durch einen Schachbretttest für jeden Abbildung 16: ELISA-Schema sekundären Antikörper ermittelt wor- Titration über 8 (links) und 12 (rechts) Stufen; orange/gelb – Reihenverdünnung der Testseren; rot/rosa – positives Kontrollserum; grün – Negativkontrolle; grau – Blindprobe den. Hierbei wurde ein bekanntes po- sitives Serum in 10 Parallelansätzen (entsprechend Abbildung 16, links) austitriert und der zu testende sekundäre Antikörper um 90° versetzt ebenso (ähnlich Abbildung 16, rechts). Die Konzentration des sekundären Antikörpers, die den Titer des Vergleichsserums korrekt wiedergab, bzw. am effektivsten funktionierte, ergab die einzusetzende Konzentration, wenn diese auch bei einem Negativserum unreaktiv blieb. Nach einer weiteren Inkubation für 1 h bei RT wurden die Platten sechsmal gewaschen. Anschließend erfolgte die Zugabe von je 100 µl ABTS-Lösung zu jeder Kavität, welche durch die Anwesenheit des sekundären Antikörpers einen Farbumschlag zu dunkelgrün zeigt. Da diese Reaktion ebenfalls durch Licht katalysiert werden kann, fand die Inkubation für 30 min im Dunkeln statt. Abschließend wurde die OD bei 414 nm in einem Plattenlesegerät gemessen. Für die Auswertung war der Mittelwert der Blindproben von allen Werten abzuziehen. Die so normierten OD-Werte der Seren wurden gemittelt und anschließend in SigmaPlot gegen die Verdünnungsstufen aufgetragen. Der ELISA-Titer war als reziproker Wert der Verdünnung 55 3. Methoden deklariert, welcher eine OD414 nm von 0,1 zeigte, da dies der maschinelle Schwellenwert für die Nachweisgrenze ist. Das eingesetzte positive Kontrollserum musste auf jeder Platte den korrekten Titerwert mit einem Spielraum von einer Titerstufe anzeigen, damit die Ergebnisse der Platte gewertet wurden. Entsprechend musste das Negativserum unter der Nachweisgrenze bleiben bzw. dem etabliertem unspezifischen Hintergrundwert entsprechen. 3.5.3 Handhabung der Makrophagenzelllinie J774A.1 Alle Arbeiten mit der murinen Makrophagenzelllinie J774A.1 wurden unter einer Sterilbank durchgeführt. Die Zellkultur betreffende Gerätschaften, sowie die Arbeitsfläche und Handschuhe sind vor Gebrauch mit Ethanol (70 %) desinfiziert worden. Vor der Verwendung in der Zellkultur waren die einzusetzenden Medien im Wasserbad auf ca. 37 °C zu erhitzen. Die Anzucht, Erhaltung und Vermehrung der Zelllinie erfolgte entsprechend des Bedarfs nach standardisierten Methoden [15] in Zellkulturflaschen mit 100, 200 und 500 ml Volumen bei 37 % und 5 % CO2. 3.5.4 Toxin-Neutralisations-Assay (TNA) Der hier beschriebene Test richtet sich nach den publizierten Angaben des USAMRIID mit entsprechenden Labor-bedingten Anpassungen. Während im ELISA die anti-Toxin Antikörper quantitativ bestimmt werden können, wird im TNA eine Aussage über deren Qualität möglich. Behindern gebildete Antikörper, neben ihrer Eigenschaft die Antigene zu markieren, die Funktion der Milzbrandtoxine, spricht man von neutralisierenden Antikörpern. Im TNA kann daher die Fähigkeit von Seren oder spezifischen Antikörpern ermittelt werden, Makrophagen konzentrationsabhängig vor der Giftwirkung des Letaltoxins zu schützen. Zunächst wurde eine Konzentration von 5 * 105 Zellen/ml in Wachstumsmedium hergestellt und zu 200 µl pro Kavität in einer sterilen 96-Well-Platte ausgesät, die danach üN inkubierte. Dabei waren auf jeder Platte drei Kavitäten für die Blindproben zellfrei zu belassen (Abbildung 17). Zur Vorbereitung Abbildung 17: TNA-Belegungschema orange/gelb – Reihenverdünnung der Testseren; rot/rosa – positives Kontrollserum; grün – Mediumkontrolle; blau – Toxinkontrolle; grau – Blindprobe der zu testenden Seren waren diese vor der Verwendung im TNA im Heizblock für 30 min bei 56 °C zu inaktivieren. Am Folgetag wurde auf einer separaten, sterilen 96-Well-Platte eine Reihenverdünnung über 8 log2 Stufen der zu testenden Seren im Versuchsmedium, versetzt mit 500 ng/ml PA und 100 ng/ml LF, angefertigt (Startkonzentration 1 : 50 bzw. 1 : 100). Abweichungen von der Startkonzentration waren durch das Volumen 56 3. Methoden des vorhandenen Serums oder den zu erwartenden Titerwerten bedingt. Zusätzlich zu den zu testenden Seren im Doppelansatz enthielt jede Platte (Abbildung 17) ein positives Kontrollserum im Einfachansatz, sowie nicht titrierte Einzelmessungen für die Toxinkontrolle (Versuchsmedium mit PA und LF, Zellen, kein Serum), die Mediumkontrolle (Versuchsmedium ohne PA und LF, Zellen, kein Serum) und die Blindproben zum Abgleich des unspezifischen Hintergrunds (Versuchsmedium ohne PA und LF, keine Zellen, kein Serum). Die so vorgelegte Platte wurde für eine Stunde bei 37 °C und 5 % CO2 inkubiert, um die Antikörper-Toxin-Wirkung vor dem Kontakt mit den Makrophagen zu gewährleisten. Im Anschluss wurden beginnend mit der niedrigsten Serumkonzentration je 100 µl dieser vor-inkubierten Toxinplatte 1 : 1 auf die Platte mit den angewachsenen Makrophagen übertragen, nachdem das Wachstumsmedium dort vollständig von den Zellen abgenommen war. Nach einer weiteren Inkubation für 3 h bei 37 °C und 5 % CO2, erfolgte die Zugabe von 25 µl MTT (5 mg/ml in PBS) zu jeder Kavität. Dieser Farbstoff wird von lebenden Zellen in dunkelviolette Farbkristalle umgewandelt und ermöglicht so den Nachweis der schützenden Wirkung der Antikörper, so sie vorhanden sind. Für die Umsetzung des Farbstoffs wurden die Zellen weitere 2 h bei 37 °C und 5 % CO2 inkubiert. Vor der photometrischen Messung des Farbumschlags (gelb/braun) erfolgte eine Lyse der Zellen durch Zugabe von 100 µl Lysepuffer zu jeder Kavität. Nach Bereinigung der durch die Mischung entstandenen Bläschen wurde die OD über ein Plattenlesegerät bei 540 nm gemessen. Zur Ermittlung des Neutralisationstiters aus den gemessenen Werten war der Durchschnittswert der Blindproben von allen Werten abzuziehen. Die gemittelte Mediumkontrolle stellte den 100 %-Wert für das Überleben dar, analog die Toxinkontrolle den 0 %-Wert. Alle gemessenen Werte der Reihenverdünnung für die unbekannten Seren und die Positivkontrolle wurden mit der Formel: (ODProbe - ODToxinkontrolle) / (ODMediumkontrolle - ODToxinkontrolle) * 100 normalisiert wodurch sich für jede eingesetzte Konzentration ein prozentualer Wert für das Überleben der Zellen ergab. Dabei wird der Neutralisationstiter als reziproker Wert der Serumverdünnung mit 50 % Schutzwirkung definiert. Diese Verdünnungsstufe wurde mit Hilfe des SigmaPlot Regression Wizard über eine sigmoide Regressionskurve (mit 4 Parametern) bestimmt. 3.6 Statistik Für die Betrachtung des Ausgangs der Infektion wurden die Sterbedaten zwischen verschiedenen Gruppen im Log-rank-Test miteinander verglichen. Als ausschlaggebende Zeitintervalle galten volle überlebte Tage. Die Überlebenszeit ergab sich aus dem Zeitintervall vom Beginn der Infektion bis zu dem Zeitpunkt an dem das Tier tot aufgefunden bzw. euthanasiert oder behandelt wurde. Zum Vergleich der serologischen Daten der Gruppen untereinander wurden im Falle der Ziegenversuche die zu verschiedenen Zeitpunkten gemessenen Titer zunächst gegen die eigenen 57 3. Methoden unspezifischen Hintergrundtiter der Woche 0 normalisiert und im ungepaarten, zweiseitigen t-Test auf signifikante Unterschiede geprüft. Bei den Mausversuchen wurde auf eine Normalisierung verzichtet, da nahezu keine unspezifischen Hintergrundtiter detektierbar waren. Bei gruppeninternen Vergleichen wurden die in den Tabellen ange- Tabelle 17: Schwellenwerte Anzahl der Individuen rs - Schwelle 4 1,000 5 0,900 6 0,829 7 0,714 8 0,643 9 0,600 10 0,564 rs-Schwellen für p ≤ 0,05 ; rs-Werte unterhalb der angegebenen Werte für die entsprechende Tierzahl sind nicht signifikant gebenen, nicht normalisierten Werte verwendet, damit ein statistische Abgleich zu den unspezifischen Hintergrundtitern erfolgen konnte. Hierfür war ein gepaarter, zweiseitige t-Test bzw. der Spearman Rangkorrelationskoeffizient (rs) zu verwenden. Der rs diente zur Abschätzung von Korrelationen zwischen gemessenen Titern untereinander, sowie der Überlebenszeit. Dabei wird jedem gemessenen Wert einer Datenreihe entspre- chend der Höhe ein Rang zugewiesen und paarweise mit anderen, gleichgroßen Datenreihen verglichen. Eine Korrelation ist um so stärker, je näher der r s-Wert an ±1 liegt. Die Signifikanz dieser Korrelation ist schwellenabhängig bezüglich der Anzahl der Werte der Datenreihe, im vorliegenden Fall also der Individuenzahl. Allen statistischen Berechnungen wurde eine Signifikanzschwelle von p ≤ 0,05 zugrunde gelegt. Die entsprechenden rs-Werte sind Tabelle 17 zu entnehmen. Die Berechnungen erfolgten mittels SigmaPlot. Angegebenen Mittelwerte sind arithmetische Mittel, welche wenn nötig in den Abbildungen durch einen positiv gerichtete Standardabweichung ergänzt sind. 58 4. Ergebnisse und Diskussion 4 Ergebnisse und Diskussion 4.1 Testung von Vakzinekomponenten im NMRI-Mausmodell Zur serologischen Evaluation der Vakzine wurden Antikörpertiter gegen rPA83, rBclA, rLF, die Kapsel und FIS mit den Subklassen IgG, IgG1 und IgG2a entsprechend der eingesetzten Vakzinekomponenten im Serum jeder Maus mittels ELISA gemessen. Zur qualitativen Bewertung der Toxin-Antikörpertiter ist außerdem der Neutralisationstiter über einen TNA bestimmt worden. Die Messungen wurden, soweit nicht anders beschrieben, für die Seren vor der Impfung (Negativseren) und direkt vor der Infektion durchgeführt. Detaillierte Titer-Werte der Negativseren sind allerdings nur in Einzelfällen angegeben. Soweit nicht anders vermerkt, konnten keine unspezifischen Hintergrundtiter für die Negativseren nachgewiesen werden. Generell wurden für jedes Tier Einzelwerte gegen diejenigen Antigene bestimmt, mit denen die jeweilige Gruppe immunisiert wurde. Eine Mischprobe der vereinigten Seren einer Gruppe kam zum Einsatz als Abgleich zum Negativserum oder zusätzlicher Antigene, die nicht Bestandteil der jeweiligen Vakzinekomposition waren. Diese sind zusammen mit den Einzelmessungen als Gruppengesamttiter in den Wertetabellen der jeweiligen Gruppe vermerkt. Die Darstellung der Messwerte innerhalb der Gruppenbeschreibung erfolgte nur für die entsprechend der jeweiligen Immunisierung relevanten Titer jeder Gruppe. Zunächst sind die serologischen Ergebnisse aller Mausgruppen (unterteilt in Protein- und DNA-Vakzinen) einzeln aufgeführt und werden in den nachfolgenden Kapiteln gemäß der Schutzraten und Immunogenität untereinander verglichen. 4.1.1 Einzelbetrachtungen der Impfung von Mäusen mit Vakzinen auf Proteinbasis Für diesen Teil der Studie wurden Protein-basierende Vakzine zunächst einzeln und später in Kombination im NMRI-Mausmodell geprüft. Die eingesetzten Adjuvanzien umfassten das Lipopeptid und, als Kombination von Vakzine und Adjuvans, das Kapselkonjugat. Das Lipopeptid wurde mit 50 µg und FIS mit 108 Sporen pro Dosis eingesetzt, so sie Bestandteil der Impfung waren. Alle weiteren Komponenten waren in einer Menge von 25 µg pro Dosis beigefügt. Die Applikation der Dosen mit einem Gesamtvolumen von 200 µl erfolgte unter Betäubung s. c. in den Nacken. Die Infektion fand analog mit ~1000 bzw. ~2000 Sporen eines voll virulenten Stammes (Ames) statt. Für Vergleichszwecke wurden zwei Kontrollgruppen mit entsprechend unterschiedlichen Infektionsdosen mitgeführt. 59 4. Ergebnisse und Diskussion 4.1.1.1 Mausgruppe LN-1000 (Lipopeptid) Eine Gruppe von 5 Mäusen wurde wie zuvor beschrie- Tabelle 18: Mausgruppe LN-1000 ben dreimal mit dem Lipopeptid paramunisiert und mit Maus Nr. 840 Sporen (~25LD50) eines voll virulenten Ames Stam- rBclA IgG rPA83 IgG TNA Kapsel IgG FIS IgG 501 Tod in dpi 4 mes infiziert. Diese Gruppe diente als negative Ver- 493a 3 gleichsgruppe für alle Proteingruppen, die ebenfalls mit 489 ~1000 Sporen infiziert wurden. Für diese Gruppe wurde M - - - - - 3,8 S - - - - - 1,0 nur einmal direkt vor der Infektion Blut abgenommen a 494b <200 <100 <100 360 <100 4 3 490 5 verlor zwischen 2. und 3. Impfung ein Auge erlitt bei der Infektion einen Herzstillstand und konnte wiederbelebt werden b und in einer Mischprobe der Gruppe analysiert. ELISA- und TNA-Titer der Mausgruppe LN-1000 vor der Infektion als vereinigte Mischprobe der Gruppe gemessen, sowie Eintritt des Todes in Tagen nach der Infektion (dpi); Titer-Werte ergeben sich als reziproker Wert der letzten Verdünnungsstufe mit einer OD404nm ≥ 0,1; Verstorbene sind entsprechend orange hinterlegt; M – arithmetischer Mittelwert; S – Standardabweichung Wie in Tabelle 18 ersichtlich, waren die gemessenen Titer mit < 100/200 bis auf den anti-Kapsel IgG-Titer, der mit 360 ebenfalls nur gering ausfiel, unter der Nachweisgrenze der jeweiligen Tests. Da bereits beim Test auf IgG keine bzw. nur geringe Titer gemessen wurden, ist von einem Test auf IgG1- oder IgG2a-Antikörper abgesehen worden. Alle eingesetzten Mäuse dieser Gruppe verstarben 3 – 5 Tage nach der Infektion (dpi). Da nur mit dem Adjuvans paramunisiert wurde, waren keinerlei Titer oder Überlebende zu erwarten. 4.1.1.2 Mausgruppe NC (Kapselkonjugat) Eine Gruppe von 10 Mäusen wurde wie zuvor beschrieben dreimal mit einem Kapselkonju- Tabelle 19: Mausgruppe NC Maus Nr. rBclA rPA83 IgG IgG TNA Kapsel FIS Tod in IgG dpi IgG IgG1 IgG2a 481a <200 <200 <200 - 482 <400 <400 <400 4 483 <100 <100 <100 3 Die gemessenen Titer vor der Infektion sind in 484 100 <100 <100 <100 <100 <100 Tabelle 19 einzusehen. Durch die Immunisie- 486 552 <100 <100 487 <100 <100 <100 3 rung konnten die anti-Kapsel IgG-Titer in eini- 488 100 <200 <200 3 499 257 <100 <100 4 gen Individuen bestenfalls leicht erhöht wer- 502 142 <100 <100 gat geimpft und mit 840 Sporen (~25LD50) eines voll virulenten Stammes (Ames) infiziert. den, wenngleich sie nur in einem Bereich knapp oberhalb der Nachweisgrenze lagen. Bei allen anderen getesteten Antigenen ergab sich durch die Immunisierung keine Veränderung. Ebenso gab es keine nachweisbaren Sub- 485 <400 <100 <100 2 <100 3 3 - M - - - 115 - - - 3,1 S - - - 56 - - - 0,6 erhielt zur 2. Impfung versehentlich 2 Dosen und starb bei der Applikation der Infektion am Narkotikum; serologische Daten dieser Maus flossen in die Betrachtungen mit ein, Daten zum Überleben entsprechend nicht a ELISA- und TNA-Titer der Mausgruppe NC vor der Infektion, sowie Eintritt des Todes in dpi. Titer-Werte ergeben sich als reziproker Wert der letzten Verdünnungsstufe mit einer OD404nm ≥ 0,1. Die Überwachung des Überlebens erfolgte bis 21 dpi. Überlebende sind entsprechend grün hinterlegt, Verstorbene orange; M – arithmetischer Mittelwert; S – Standardabweichung klassentiter gegen die Kapsel. Einige Seren konnten aufgrund ihres geringen Volumens nicht mit optimalen Ausgangskonzentrationen getestet werden. Von neun infizierten Mäusen verstarben acht nach 2 – 4 dpi. Maus Nr. 502 überlebte die Infektion komplett. Auf eine grafische Darstellung der Werte wurde auf60 4. Ergebnisse und Diskussion grund der niedrigen Titer verzichtet. Serologisch waren keine Unterschiede zwischen überlebenden und verstorbenen Tieren zu erkennen. 4.1.1.3 Mausgruppe NCP (Kapselkonjugat; rPA83) Eine Gruppe von 10 Mäusen wurde wie zuvor beschrieben drei Mal mit einer Mischung aus dem Kapselkonjugat und rPA83 geimpft und mit 901 Sporen (~25LD50) eines voll virulenten Stammes (Ames) infiziert. Durch die Immunisierung konnten bei allen Mäusen im Vergleich zum Negativserum signifikant höhere und relativ homogene anti-rPA83 IgG-, IgG1-Titer, sowie Neutralisationstiter erzeugt werden (Tabelle 20; Abbildung 18). Die IgGTiter gegen rPA83 waren dabei auch signifikant höher als die IgG1- und IgG2a-Titer, wel- Abbildung 18: Mausgruppe NCP Halblogarithmische Auftragung der Antikörpertiter; Titerwerten unterhalb der Nachweisgrenze wurde ein Betrag von 1 zugewiesen; * markiert signifikant erhöhte Titer im Vergleich zum Negativserum che sich untereinander ebenfalls signifikant unterschieden (p ≤ 0,004). Hierbei schien der Anteil der IgG1- bzw. IgG2a-Titer nur einen kleinen Teil der Gesamt-IgG auszumachen. Bezüglich rPA83 und in Tendenz auch für das Kapselkonjugat war allerdings eine deutliche Gewichtung zur IgG1 Subklasse sichtbar. Eine Korrelation zwischen TNA-Titern und anti-rPA83 IgG- und IgG1-Titern gab es nicht (rs ≤ +0,361). Tabelle 20: Mausgruppe NCP Maus Nr. rBclA rPA83 TNA IgG IgG1 IgG2a 57 350000 165161 1858 58 256000 57806 59 122181 60 197818 61 IgG Kapsel FIS Tod in IgG dpi IgG IgG1 IgG2a 16119 648 3200 <400 - 400 19353 <400 1973 <400 - 71019 <400 2281 <400 <200 <400 - 38812 <400 3909 1241 6400 <400 13 453818 51200 658 5046 <400 <200 <400 256000 112309 903 4218 <400 200 <400 63 512000 62761 <400 3909 <400 <200 <400 64 350000 87535 <400 7628 <400 200 <400 - 65 256000 14864 <400 4189 <400 <200 <400 2 62a 347 66 <200 6 512000 67716 <400 5507 <400 <200 <400 M - 326582 72918 382 7216 189 1197 - - 7 S - 133063 41689 615 5761 422 2130 - - 6 a - Negativserum wies anti-rPA83 IgG-Titer von 217 auf ELISA- und TNA-Titer der Mausgruppe NCP vor der Infektion, sowie Eintritt des Todes in dpi. Titer-Werte ergeben sich als reziproker Wert der letzten Verdünnungsstufe mit einer OD 404nm ≥ 0,1. Die Überwachung des Überlebens erfolgte bis 21 dpi. Überlebende sind entsprechend grün hinterlegt, Verstorbene orange; M – arithmetischer Mittelwert; S – Standardabweichung 61 4. Ergebnisse und Diskussion In Abbildung 18 sind die für diese Gruppe wichtigen Titer noch einmal graphisch dargestellt. Die gemessenen anti-Kapsel Titer, wie auch die anti-rPA83 IgG2a-Titer waren zwar erhöht, allerdings nicht bei allen Individuen und auch in keinem Fall signifikant (p ≥ 0,098). Die Infektion überlebten 7 von 10 Mäusen, wobei ebenfalls die Anzahl der überlebten Tage für die verstorbenen Tiere durch die Impfung teilweise erhöht werden konnte. Eine signifikante Korrelation zwischen der Überlebenszeit und den gemessenen Titern bestand nur für die anti-rPA83 IgG1-Titer, welche schwach korrelierten (rs = +0,697). 4.1.1.4 Mausgruppe LN-2000 (Lipopeptid) Eine Gruppe von 5 Mäusen wurde wie zuvor beschrieben dreimal mit dem Lipopeptid paramunisert und mit 1990 Sporen (~50LD50) eines voll virulenten Stammes (Ames) infiziert. Diese Gruppe diente zum Negativabgleich aller Proteingruppen bei denen ebenfalls eine Infektion mit ~2000 Sporen erfolgte. Wie in Tabelle 21 ersichtlich, waren die gesamtheitlich als vereinigte Mischprobe der Gruppe gemessenen Titer mit < 100/200 bis auf den anti-Kapsel IgG-Titer unter der Nachweisgrenze der jeweiligen Tests. Da bereits beim Test auf IgG keine bzw. nur geringe Titer gemessen wurden, ist von einem Test auf IgG1- oder IgG2a-Antikörper abgesehen worden. Im Gegensatz zur Gruppe LN-1000 wurde hier ebenso das Serum vor der Tabelle 21: Mausgruppe LN-2000 Maus Nr. rBclA IgG rPA83 IgG TNA Paramunisierung als vereinigte Mischprobe der Gruppe Kapsel IgG FIS IgG 1 3 2 3 Tod in dpi 3 <200 <100 <100 340 <200 3 4 2 5 10 M - - - - - 4,2 S - - - - - 3,3 ELISA- und TNA-Titer der Mausgruppe LN-2000 vor der Infektion als vereinigte Mischprobe der Gruppe gemessen, sowie Eintritt des Todes in dpi; Titer-Werte ergeben sich als reziproker Wert der letzten Verdünnungsstufe mit einer OD404nm ≥ 0,1; Verstorbene sind entsprechend orange hinterlegt; M – arithmetischer Mittelwert; S – Standardabweichung untersucht und ergab keine relevanten IgG-Titer gegen die jeweiligen Antigene. Hierbei ist hervorzuheben, dass dies auch für den anti-Kapsel IgG-Titer (< 100) galt, der nach der Paramunisierung immerhin oberhalb der Nachweisgrenze messbar war. Von den eingesetzten Mäusen sind vier bis zum 3. Tag nach der Infektion verstorben, Maus Nr. 5 zeigte zwar ab dem 3. Tag großflächige Ödeme und andere leichte Krankheitssymptome, verstarb aber erst am Tag 10. 4.1.1.5 Mausgruppe LB (Lipopeptid und rBclA) Eine Gruppe von 10 Mäusen wurde wie zuvor beschrieben dreimal mit einer Mischung aus dem Lipopeptid und rBclA geimpft und mit 2020 Sporen (~50LD50) eines voll virulenten Stammes (Ames) infiziert. Durch die Immunisierung mit rBclA konnte in allen Individuen ein signifikant (p ≤ 0,001) erhöhter IgG- bzw. IgG1-Titer gegen rBclA erzeugt werden (siehe Tabelle 22). Die entsprechenden IgG2a-Titer waren ebenfalls erhöht, aber stark gestreut und nicht signifikant 62 4. Ergebnisse und Diskussion (p = 0,08). Interessanterweise konnte ebenso ein hoher anti-FIS IgG-Titer über die Detektion von BclA-Filamenten im FIS erzeugt werden. Wie auch schon in anderen Gruppen beschrieben, war zudem ein leichter Titer gegen die Kapsel messbar, der bereits vor der Immunisierung im Negativserum als unspezifischer Hintergrundtiter nachweisbar war. Tabelle 22: Mausgruppe LB Maus Nr. rBclA rLF rPA83 IgG IgG TNA Kapsel FIS Tod in IgG IgG dpi IgG IgG1 IgG2a 6 341333 483555 21000 <500 3 7 192000 341333 3500 <500 3 8 366933 568888 <250 <500 3 9 192000 265481 1111 <500 4 10 61866 68740 2777 324 11 247466 251259 833 12 512000 777481 375 <500 3 13 93866 106666 1777 <500 5 14 426666 426666 4500 <500 3 15 136533 184888 2875 <500 M 257066 347496 3875 - - - - - 3,4 S 149351 220316 6187 - - - - - 0,7 <500 <1600 <500 390 84676 3 3 ELISA- und TNA-Titer der Mausgruppe LB vor der Infektion, sowie Eintritt des Todes in dpi. Titer-Werte ergeben sich als reziproker Wert der letzten Verdünnungsstufe mit einer OD404nm ≥ 0,1; Die Überwachung des Überlebens erfolgte bis 25 dpi; Überlebende sind entsprechend grün hinterlegt, Verstorbene orange; M – arithmetischer Mittelwert; S – Standardabweichung Obwohl diese Versuchsgruppe keine Toxinkomponenten erhalten hatte, wurde ein TNA mit allen Einzelseren durchgeführt, da der TNA für das vereinigte Serum der Gruppe einen leichten Anstieg bei der Startverdünnung von 1 : 100 zeigte. Nach einem exemplarischen Test mit allen Seren bei einer Startkonzentration von 1 : 500 war nur das Serum von Maus Nr. 10 auffällig und wurde in höherer Konzentration nochmal getestet, woraus sich dann der in Tabelle 22 aufgeführte Titer ergab. Zum Abgleich wurde das Negativserum von vor der Immunisierung ebenfalls als Mischprobe der Gruppe getestet und zeigte bei einer Startkonzentration von 1 : 100 einen ähnlichen Anstieg wie das Serum vor der Infektion. Demnach wies Maus Nr. 10 möglicherweise bereits vor der Immunisierung einen niedrigen unspezifischen TNA-Titer auf, der den Ausgang der Infektion beeinflusst haben könnte. In Abbildung 19 sind alle relevanten Titer noch einmal grafisch dargestellt, von 10 Versuchstieren waren neun innerhalb von 5 dpi verstorben. Die überlebende Maus Nr. 10 hatte die niedrigsten IgG und IgG1 Titer dieser Versuchsgruppe. Entsprechend gab es keine signifikante Korrelation zwischen den anti-rBclA Titern und der Überlebenszeit, wobei die errechnete Tendenz sogar eher antagonistischer Abbildung 19: Mausgruppe LB Halblogarithmische Auftragung der Antikörpertiter; Titerwerten unterhalb der Nachweisgrenze wurde ein Betrag von 1 zugewiesen; * markiert signifikant erhöhte Titer im Vergleich zum Negativserum 63 4. Ergebnisse und Diskussion Natur war (rs ≤ -0,43). Die Titer für anti-rBclA IgG und IgG1 hatten eine ähnliche Höhe und Verteilung und zeigten eine hohe Korrelation rs = +0,924), womit eine entsprechend starke IgG1-Gewichtung vorlag. 4.1.1.6 Mausgruppe LP (Lipopeptid und rPA83) Eine Gruppe von 10 Mäusen wurde wie Tabelle 23: Mausgruppe LP zuvor beschrieben dreimal mit einer Maus Nr. Mischung aus dem Lipopeptid und rPA83 geimpft und mit 2020 Sporen (~50LD50) eines voll virulenten Stammes (Ames) infiziert. Durch die Immunisierung konnte in allen Individuen ein signifikant erhöhter (p ≤ 0,02) IgG-, IgG1- bzw. IgG2a-Titer gegen rPA83, sowie TNA-Titer erzeugt werden (Tabelle 23). Wie auch schon in anderen rBclA rPA83 TNA Kapsel FIS Tod in IgG IgG dpi IgG IgG1 IgG2a 36 660945 448000 22303 13398 7 37 442181 304000 37818 24064 4 38 432872 368000 5575 10421 - 39 660945 240000 147393 20095 4 623709 448000 44606 16871 414254 304000 1909 13150 42 623709 304000 23757 10669 3 43 512000 256000 67878 12406 3 44 512000 256000 30545 14390 3 45 735418 448000 2181 10173 40 41 IgG <200 400 <100 3 3 2 M - 561803 337600 38397 14564 - - 3,7 S - 113102 84142 43517 4544 - - 1,4 ELISA- und TNA-Titer der Mausgruppe LP vor der Infektion, sowie Eintritt des Todes in dpi. Titer-Werte ergeben sich als reziproker Wert der letzten Verdünnungsstufe mit einer OD404nm ≥ 0,1. Die Überwachung des Überlebens erfolgte bis 25 dpi. Überlebende sind entsprechend grün hinterlegt, Verstorbene orange; M – arithmetischer Mittelwert; S – Standardabweichung Gruppen beschrieben, war zudem ein unspezifischer Hintergrundtiter gegen die Kapsel messbar, der auch bereits vor der Immunisierung nachweisbar war. Die anti-rPA83 IgG-, IgG1- und IgG2a-Titer unterschieden sich untereinander signifikant, obwohl IgG und IgG1 (Abbildung 20) ähnlich hoch und homogen verteilt waren (p ≤ 0,001). Die anti-rPA83 IgG2a-Titer streuten sehr stark, sodass einige Individuen einen fast nicht nachzuweisenden Titer zeigten, während andere nahezu äquivalente IgG1 und IgG2a-Titer aufwiesen. Daher war die Tendenz zu einer IgG1 gerichteten Immunantwort noch immer vorhanden, wobei IgG1 und IgG2a nur einen Teil der gemessenen IgG auszumachen schienen. In dieser Versuchsgruppe verstarben 9 von 10 Versuchstieren bis zum 7. Tag nach der InfekAbbildung 20: Mausgruppe LP Halblogarithmische Auftragung der Antikörpertiter; Titerwerten unterhalb der Nachweisgrenze wurde ein Betrag von 1 zugewiesen; * markiert signifikant erhöhte Titer im Vergleich zum Negativserum 64 tion. Keine der gemessenen Werte korrelierten untereinander oder zur überlebten Zeit in Stunden (rs ≤ ±0,62). 4. Ergebnisse und Diskussion 4.1.1.7 Mausgruppe LBP (Lipopeptid, rBclA und rPA83) Eine Gruppe von 10 Mäusen wurde wie zuvor beschrieben dreimal mit einer Mischung aus dem Lipopeptid, rBclA und rPA83 geimpft und mit 2020 Sporen (~50LD50) eines voll virulenten Stammes (Ames) infiziert. Wie in Tabelle 24 zu sehen, konnten durch die Impfung in allen Individuen erhöhte anti-rBclA, -rPA83 und TNA-Titer erzeugt werden (p ≤ 0,048). Die TNA-Titer korrelierten signifikant mit den IgG-Titern gegen rPA83 (rs = +0,85). Außerdem waren IgGTiter gegen FIS und unspezifische Hintergrundtiter gegen die Kapsel nachweisbar, wobei letztere bereits vor der Immunisierung nachweisbar waren. Tabelle 24: Mausgruppe LBP Maus Nr. rBclA rPA83 TNA Kapsel FIS Tod in IgG IgG dpi IgG IgG1 IgG2a IgG IgG1 IgG2a 16 102787 160000 <250 496484 436148 <250 12406 3 17 114424 160000 6666 589575 398222 85333 13398 9 18 128000 201142 <250 310303 175407 397 10669 - 19 186181 320000 <250 364606 256000 1000 9181 - 20 77575 91428 1500 473212 251259 136533 27041 21 151272 246857 <250 837818 625777 3647 35722 22 135757 160000 1444 310303 265481 16000 12158 580 46276 3 - 23 61090 70857 <250 143515 184888 1833 7693 - 24 91151 114285 1611 213333 208592 6133 5212 - 25 186181 246857 3333 411151 237037 76800 22328 M 123442 177143 1455 415030 303881 32768 15581 - - 5,2 - S 42637 77589 2142 199480 141720 48757 9186 - - 3,2 ELISA- und TNA-Titer der Mausgruppe LBP vor der Infektion, sowie Eintritt des Todes in dpi. Titer-Werte ergeben sich als reziproker Wert der letzten Verdünnungsstufe mit einer OD 404nm ≥ 0,1. Die Überwachung des Überlebens erfolgte bis 25 dpi. Überlebende sind entsprechend grün hinterlegt, Verstorbene orange; M – arithmetischer Mittelwert; S – Standardabweichung In Abbildung 21 sind alle wichtigen Einzelmessungen dieser Gruppe noch einmal grafisch dargestellt. Es ist zu erkennen, dass alle IgG-, IgG1- und TNA-Titer homogen verteilt sind, während die IgG2a-Titer stark verstreut liegen und nicht für alle Individuen messbar waren. Daher ergab sich eine starke IgG1-Gewichtung, sowohl für rPA83 als auch für rBclA, die beide eine starke Korrelation zwischen IgG und IgG1 Titern aufwiesen (rs ≥ +0,81). In dieser Versuchsgruppe verstarben 3 von 10 Versuchstieren, wobei bei einem Individuum eine verlängerte Überlebenszeit von 9 Tagen zu beobachten war. Keiner der gemessenen Titer war ausschlaggebend für eine ver- Abbildung 21: Mausgruppe LBP Halblogarithmische Auftragung der Antikörpertiter; Titerwerten unterhalb der Nachweisgrenze wurde ein Betrag von 1 zugewiesen; * markiert signifikant erhöhte Titer im Vergleich zum Negativserum längerte Überlebenszeit (rs ≤ ±0,53). 65 4. Ergebnisse und Diskussion 4.1.1.8 Mausgruppe LBCP (Lipopeptid, rBclA, Kapselkonjugat und rPA83) Eine Gruppe von 10 Mäusen wurde wie zuvor beschrieben dreimal mit einer Mischung aus dem Lipopeptid, rBclA, dem Kapselkonjugat und rPA83 geimpft und mit 2020 Sporen (~50LD50) eines voll virulenten Stammes (Ames) infiziert. Als Auffälligkeit bei dieser Gruppe ist zu vermerken, dass nach den Impfungen bei einem Teil der Versuchstiere kleine Verkapselungen im Injektionsbereich zu spüren waren, welche bei den anderen Versuchsgruppen, mit Ausnahme von LBCOP nicht aufgetreten sind. Tabelle 25: Mausgruppe LBCP Maus Nr. rBclA rPA83 TNA Kapsel FIS Tod in IgG IgG dpi IgG IgG1 IgG2a IgG IgG1 IgG2a 26 143360 197818 1382 407272 337454 1230 11914 1344 27 40320 55272 1000 180363 113454 9692 11914 <400 - 28 110080 232727 4680 471272 241454 5538 10425 4561 - - 29 48640 145454 24170 180363 74181 11692 4472 2859 - 30 143360 197818 20765 226909 64000 14615 8937 1225 - 31 110080 113454 11063 349090 241454 3692 10425 6808 32 194560 232727 6382 308363 162909 30153 5960 3131 - 33 99840 113454 2595 442181 215282 31384 23324 987 4 34 48640 113454 <1000 663272 241454 5076 11417 1106 3 35 12960 29818 1000 768000 215272 43692 39199 2655 24 M 95184 143200 7304 399709 190691 15676 13799 2468 - 9,6 S 56849 70901 8677 196943 86179 14382 10239 2022 - 9,8 18707 7 ELISA- und TNA-Titer der Mausgruppe LBCP vor der Infektion, sowie Eintritt des Todes in dpi. Titer-Werte ergeben sich als reziproker Wert der letzten Verdünnungsstufe mit einer OD 404nm ≥ 0,1. Die Überwachung des Überlebens erfolgte bis 25 dpi. Überlebende sind entsprechend grün hinterlegt, Verstorbene orange; M – arithmetischer Mittelwert; S – Standardabweichung Das Negativserum dieser Gruppe wurde als Mischprobe der Gruppe auf IgG-Titer gegen rPA83, rBclA, FIS und die Kapsel getestet und zeigte nur bei der Kapsel einen unspezifischen Hintergrundtiter. Ein TNA-Titer war ebenfalls nicht nachweisbar. Durch die Immunisierung konnten signifikante IgG-, IgG1- bzw. IgG2a-Titer gegen rPA83 und rBclA, sowie signifikante anti-Kapsel IgG- und TNA-Titer erzeugt werden (p ≤ 0,016, Tabelle 25). Ein erhöhter anti-FIS IgG-Titer war ebenfalls messbar. Wie in Abbildung 22 zu sehen, waren die gemessenen Titer bezüglich rBclA und die anti-rPA83 IgG2a-Titer stark gestreut. Für beide Antigene ergab sich eine ähnlich hohe IgG1-Gewichtung, wobei sich für rBclA eine signifikante starke (rs = +0,85) und für rPA83 eine schwache (rs = +0,65) Korrelation zwischen IgG- und IgG1-Titern ergab. TNA- und rPA83-Titer korrelierten nicht signifikant miteinander (rs ≤ +0,59), ließen aber eine Tendenz erkennen. 66 Abbildung 22: Mausgruppe LBCP Halblogarithmische Auftragung der Antikörpertiter; Titerwerten unterhalb der Nachweisgrenze wurde ein Betrag von 1 zugewiesen; * markiert signifikant erhöhte Titer im Vergleich zum Negativserum 4. Ergebnisse und Diskussion Die Infektion überlebten 6 von 10 Versuchstieren, wobei ein Tier erst kurz vor Beendigung des Versuchs am 24. Tag nach der Infektion verstarb. Da auch hier der Erreger aus der Milz dieses Tieres nachgewiesen werden konnte, ist als Todesursache die Infektion mit dem Erreger anzusehen. Keiner der gemessenen Titer korrelierte signifikant zum Überleben (rs ≤ ±0,61). 4.1.1.9 Mausgruppe LBCOP (Lipopeptid, rBclA, Kapselkonjugat, FIS und rPA83) Eine Gruppe von 10 Mäusen wurde wie zuvor beschrieben dreimal mit einer Mischung aus dem Lipopeptid, rBclA, dem Kapselkonjugat, rPA83 und FIS geimpft und mit 2020 Sporen (~50LD50) eines voll virulenten Stammes (Ames) infiziert. Diese Gruppe stellte die Kombination aller vorhandenen Impfkomponenten auf Proteinbasis dar. Die schon beschriebenen Verkapselungen Immunisierungen waren durch hier die ebenfalls bemerkbar. Wie in Tabelle 26 aufgeführt, konnten Abbildung 23: Mausgruppe LBCOP Halblogarithmische Auftragung der Antikörpertiter; Titerwerten unterhalb der Nachweisgrenze wurde ein Betrag von 1 zugewiesen; * markiert signifikant erhöhte Titer im Vergleich zum Negativserum durch die Immunisierung gegen jedes eingesetzte Antigen signifikante Antikörpertiter für alle gemessenen Subklassen, sowie TNA-Titer erzeugt werden (p ≤ 0,044), obgleich die Titer für einige Messreihen stark streuten (Abbildung 23). Die Gewichtung der Subklassen für rPA83 und rBclA lag hierbei auf IgG1, welcher zudem stark mit den jeweiligen gesamt IgG korrelierte (rs ≥ +0,82). Tabelle 26: Mausgruppe LBCOP Kapsel FIS Tod in IgG IgG dpi 28781 2991 51200 - 15489 17867 660 8000 - 229209 13106 14890 973 24400 - 381387 163720 110297 33742 5147 92800 - 11272 433632 351255 61957 16378 1356 43200 - 175627 7636 381387 273860 18042 12906 3756 40800 - 68000 145860 <1000 512000 988279 1489 23820 3200 25600 - 53 42000 61023 2363 412734 470325 73531 70453 895 5688 - 54 124000 363162 7636 172408 273860 3617 8441 852 65422 - 56 58000 175627 14545 214204 247069 32000 5960 400 16711 - M 103866 171534 14427 440947 445618 37106 23324 2023 37382 - S 82158 121105 20707 285533 302547 35188 18680 1625 27277 - Maus Nr. rBclA rPA83 TNA IgG IgG1 IgG2a IgG IgG1 IgG2a 46 170666 256000 24000 1191183 988279 41531 47 19333 52837 2000 454530 470325 48 35333 61023 5000 256000 49 272000 363162 69818 50 62666 61023 51 186666 52 ELISA- und TNA-Titer der Mausgruppe LBCOP vor der Infektion, sowie Eintritt des Todes in dpi. TiterWerte ergeben sich als reziproker Wert der letzten Verdünnungsstufe mit einer OD 404nm ≥ 0,1. Die Überwachung des Überlebens erfolgte bis 25 dpi. Überlebende sind entsprechend grün hinterlegt, Verstorbene orange; M – arithmetischer Mittelwert; S – Standardabweichung 67 4. Ergebnisse und Diskussion Die TNA-Titer korrelierten in dieser Gruppe schwach mit den anti-rPA83 IgG-Titern (rs = +0,65). Eine starke Korrelation konnte für die Titer gegen FIS und rBclA gezeigt werden und betraf alle Subklassen (rs ≥ +0,65). In dieser Versuchsgruppe konnten alle 10 Versuchstiere gegen eine Infektion mit B. anthracis geschützt werden. 4.1.2 Einzelbetrachtungen der Impfung von Mäusen mit Vakzinen auf DNA-Basis Für diesen Teil der Studie wurden die zur Impfung eingesetzten Vektoren vorerst einzeln und (resultierend aus den Ergebnissen der Einzelapplikationen) in Kombination getestet. Als Adjuvans, sofern es nicht integraler Bestandteil der Vakzinevektoren selbst war, wurde neben den antigenkodierenden Vektoren ein zusätzlicher, für CIITA-kodierender Vektor mit Adjuvanswirkung verabreicht. War mehr als ein Vektor Bestandteil der Impfung, sind Mischpatronen erstellt worden, um eine simultane Wirkung zu gewährleisten. Pro Dosis wurden mindestens 3 µg DNA mittels 2 Patronen appliziert. Schwankungen ergaben sich durch die unterschiedlichen Präparationen, wobei innerhalb der Gruppen stets die gleiche Kombination an Patronen pro Individuum verwendet wurde. Die Infektion erfolgte mit ~1000 Sporen unter Betäubung s. c. in den Nacken. 4.1.2.1 Mausgruppe TN (CIITA) Eine Gruppe von 5 Mäusen wurde wie zuvor beschrieben Tabelle 27: Mausgruppe TN dreimal mit je zwei DNA-Patronen paramunisiert, welche Maus Nr. mit pDNAVaccUltra5-CIITA 31 beschichtet waren und 3 Wochen nach der letzten Paramu- 33 nisierung mit 894 Sporen (~25LD50) eines voll virulenten 36 dem Adjuvans-Vektor Stammes (Ames) infiziert. Diese Gruppe diente als negative Vergleichsgruppe für alle DNA-Gruppen. Für die Paramunisierung wurden nur Patronen einer Präparation benutzt, deren DNA-Menge 2,4 µg pro Patrone betrug. Die verwendeten Patronen verschossen sich bei Probeschüssen rBclA rLF rPA83 IgG IgG IgG TNA Tod in dpi 2 32 3 174 <200 <200 <100 35 4 3 3 M - - - - 3,0 S - - - - 0,7 ELISA- und TNA-Titer der Mausgruppe TN vor der Infektion, sowie Eintritt des Todes in dpi; Titer-Werte ergeben sich als reziproker Wert der letzten Verdünnungsstufe mit einer OD 404nm ≥ 0,1; Die Überwachung des Überlebens erfolgte bis 25 dpi; Überlebende sind entsprechend grün hinterlegt, Verstorbene orange; M -arithmetischer Mittelwert; S – Standardabweichung auf Filterpapier gleichmäßig mit intensiver Färbung und nahezu ohne Rückstände (Rückstandbild 2 – 3, siehe Abbildung 10 und 58). Das Blut vor Beginn der Paramunisierung und vor der Infektion (aufgeführt in Tabelle 27) wurde als vereinigte Mischprobe der Gruppe auf Antikörper gegen rBclA, rPA83 und rLF, sowie TNA-Titer untersucht. Es konnten zu beiden Zeitpunkten keine relevanten Titer gegen die getesteten Antigene gefunden werden. Einzig für rBclA waren nach der Paramunisierung 68 4. Ergebnisse und Diskussion unspezifische Hintergrundtiter messbar. Erwartungsgemäß verstarben alle Individuen dieser Gruppe bis zum 4. Tag nach der Infektion. 4.1.2.2 Mausgruppe TM (CIITA; BclAD1D3-Uq) Eine Gruppe von 10 Mäusen wurde wie Tabelle 28: Mausgruppe TM zuvor beschrieben dreimal mit je zwei Maus Nr. DNA-Patronen, welche mit den Vektoren pDNAVaccUltra5-CIITA und pDNAVaccUltra4-BclAD1D3-Uq beschichtet waren, geimpft und 3 Wochen nach der letzten Immunisierung mit 901 Sporen (~25LD50) eines voll virulenten Stammes (Ames) infiziert. Eine Impfdosis bestand aus je einer Patrone mit 1,51 und 1,99 µg DNA. Die Patronen konnten bei allen Impfungen nahezu rückstandsfrei mit intensiver rBclA rLF rPA83 IgG IgG TNA FIS Tod in IgG dpi IgG IgG1 IgG2a 47 75636 143238 <250 6 48 279272 512000 3380 - 49 96000 195047 <250 - 50 384000 768000 1861 3 51 67716 28160 <250 52 331636 707047 <250 53 279272 633904 5714 - 54 477090 902095 6095 3 55 46545 85333 <250 6 56 174545 274285 3619 M 221171 424911 2067 - - - - 4,5 S 150564 316907 2473 - - - - 1,7 <500 <100 <100 204800 4 - - ELISA- und TNA-Titer der Mausgruppe TM vor der Infektion, sowie Eintritt des Todes in dpi. Titer-Werte ergeben sich als reziproker Wert der letzten Verdünnungsstufe mit einer OD 404nm ≥ 0,1. Die Überwachung des Überlebens erfolgte bis 21 dpi. Überlebende sind entsprechend grün hinterlegt, Verstorbene orange; M – arithmetischer Mittelwert; S – Standardabweichung Färbung verschossen werden (Rückstandbild 1 – 2, siehe Abbildung 10 und 58). Wie in Tabelle 28 aufgeführt, konnten durch die Impfungen signifikante IgG-, IgG1- und IgG2a-Antikörpertiter gegen rBclA erzeugt werden (p ≤ 0,017). Hervorzuheben ist, dass ebenfalls ein sehr hoher, den anti-rBclA IgG-Titern entsprechender IgG-Titer gegen FIS vorhanden war. Damit konnte gezeigt werden, dass die spezifisch gegen BclAD1D3 gebildeten Antikörper auch natürliches BclA auf Sporen detektieren können. Wie in Abbildung 24 zu sehen, schwankten die erzeugten Titer zwischen den Individuen stark. Allerdings ähnelte sich die Verteilung der IgG- und IgG1-Titer gegen rBclA sehr. Entsprechend stark war die Korrelation beider Werte (rs = +0,98). IgG2a-Titer gegen rBclA waren im Vergleich sehr viel niedriger und für die Hälfte der getesteten Seren nicht nachweisbar. Daraus ergab sich für die gesamte Gruppe Abbildung 24: Mausgruppe TM Halblogarithmische Auftragung der Antikörpertiter; Titerwerten unterhalb der Nachweisgrenze wurde ein Betrag von 1 zugewiesen; * markiert signifikant erhöhte Titer im Vergleich zum Negativserum eine deutliche Gewichtung zur IgG1-Subklasse. Belegbare Korrelationen zwischen der Überlebenszeit und den gemessenen antirBclA Titern gab es nicht (rs ≤ ±0,07). 69 4. Ergebnisse und Diskussion In dieser Gruppe überlebten 5 der 10 Tiere, wobei zwei verstorbene Tiere eine verlängerte Überlebenszeit von 6 Tagen aufwiesen. Daher wurde in dieser Gruppe auch das Serum der überlebenden Mäuse nach der Infektion untersucht. Von Interesse war hierbei, die Entwicklung von anti-rPA83 Titern durch die Infektion. Diese würden eine vollständig verlaufene Infektion mit Anwesenheit von vegetativen Zellen und Ausschüttung der Milzbrandtoxine belegen, gegen die über eine reine BclA Immunisierung keine Schutzwirkung bestehen sollte. Für alle Individuen konnte nach der überlebten Infektion kein IgG-Titer gegen rPA83 gemessen werden (< 100/200). 4.1.2.3 Mausgruppe TK (CIITA; TPA-BclAD1D3-LAMP1) Eine Gruppe von 10 Mäusen wurde wie Tabelle 29: Mausgruppe TK zuvor beschrieben dreimal mit je zwei Maus Nr. DNA-Patronen, welche mit den Vektoren rBclA rLF rPA83 IgG IgG TNA FIS Tod in IgG dpi IgG IgG1 IgG2a 37 200727 363162 35200 4 38 91454 160744 500 3 39 218181 303627 301 - 40 418909 446511 39200 - 41 168727 238139 60800 4 42 45090 49860 1046 43 142545 226232 5176 7 44 66909 110139 1441 - 45 119272 208372 1735 - rung mit 901 Sporen (~25LD50) eines 46 192000 267906 6235 3 M 166381 237469 15163 - - - - 4,2 voll virulenten Stammes (Ames) infi- S 106094 116938 21718 - - - - 1,6 pDNAVaccUltra5-CIITA und pDNAVaccUltra1-TPA-BclAD1D3LAMP1 beschichtet waren, geimpft und 3 Wochen nach der letzten Immunisie- ziert. Jede Dosis umfasste je eine Patrone mit 1,51 und 1,3 µg DNA. Die <500 <100 <100 68923 - ELISA- und TNA-Titer der Mausgruppe TK vor der Infektion, sowie Eintritt des Todes in dpi. Titer-Werte ergeben sich als reziproker Wert der letzten Verdünnungsstufe mit einer OD404nm ≥ 0,1. Die Überwachung des Überlebens erfolgte bis 21 dpi. Überlebende sind entsprechend grün hinterlegt, Verstorbene orange; M – arithmetischer Mittelwert; S – Standardabweichung Patronen konnten bei allen Impfungen nahezu rückstandsfrei mit intensiver Färbung verschossen werden (Rückstandbild 1 – 2, siehe Abbildung 10 und 58). Es konnten durch die Impfungen signifikante IgG-, IgG1- und IgG2a-Antikörpertiter gegen rBclA erzeugt werden (p ≤ 0,04; Tabelle 29). Ebenso waren hohen IgG-Titer gegen FIS gemessen worden, während IgG-Titer gegen rPA83 und rLF, sowie TNA-Titer nicht nachweisbar waren. Wie zuvor bei Gruppe TM bemerkt, belegten die gemessenen Antikörpertiter gegen FIS die Abbildung 25: Mausgruppe TK Funktionalität der gebildeten Antikörper gegen Halblogarithmische Auftragung der Antikörpertiter; Titerwerten unterhalb der Nachweisgrenze wurde ein Betrag von 1 zugewiesen; * markiert signifikant erhöhte Titer im Vergleich zum Negativserum BclAD1D3 auch bei natürlichem BclA. Wie in 70 Abbildung 25 zu sehen, ist die Verteilung der 4. Ergebnisse und Diskussion IgG- und IgG1-Titer gegen rBclA nahezu identisch und korrelierte entsprechend stark (rs = +0,99). Die IgG2a-Titer gegen rBclA waren im Vergleich zu den IgG1-Titern sehr viel niedriger, aber im Gegensatz zur Gruppe TM für alle Individuen nachweisbar. In dieser Gruppe überlebten ebenfalls 5 von 10 Mäusen, wobei eine Korrelation der gemessenen Titer zur überlebten Zeit in Stunden nicht gegeben war (rs ≤ ±0,16). Analog zur Mausgruppe TM wurde auch hier das Blutserum der Überlebenden nach der Infektion auf rPA83-Antikörper getestet und zeigten keinerlei nachweisbare Titer (< 100). 4.1.2.4 Mausgruppe TH (CIITA; TPA-rPA83-LAMP1) Eine Gruppe von 10 Mäusen wurde wie zuvor Tabelle 30: Mausgruppe TH beschrieben dreimal mit je zwei DNA-Patronen, Maus Nr. die mit den Vektoren pDNAVaccUltra5-CIITA und rBclA rLF IgG IgG rPA83 TNA Tod in IgG IgG1 IgG2a 21 51200 18773 <800 652 5 22 37485 12800 <800 <250 4 23 12800 5120 <800 <250 - 24 19657 10453 <800 <250 3 21028 4693 <800 <250 5 56685 15360 <800 <250 - 27a 800 <800 <800 <250 4 28 28342 8320 <800 125 3 29 84114 25600 1300 1614 - infiziert. Jede Dosis enthielt je eine Patrone mit 30 4342 1440 <800 <250 3 M - - 31645 10256 - 239 4,0 2,4 und 1,5 µg DNA. Die Patronen mit 2,4 µg S - - 26068 8059 - 524 1,0 pDNAVaccUltra1-TPA-rPA83-LAMP1 beschichtet waren, geimpft und 3 Wochen nach der letzten Immunisierung mit 894 Sporen (~25LD50) eines voll virulenten Ames Stammes DNA verschossen sich sehr gleichmäßig mit einer intensiven Färbung, wiesen aber bei allen Patronen Rückstände auf (Rückstandbild 3, siehe Abbildung 10 und 58). Die andere Präparati- 25 26 a 131 <100 dpi Negativserum zeigte anti-rPA83 IgG-Titer von 564 ELISA- und TNA-Titer der Mausgruppe TH vor der Infektion, sowie Eintritt des Todes in dpi. Titer-Werte ergeben sich als reziproker Wert der letzten Verdünnungsstufe mit einer OD404nm ≥ 0,1. Die Überwachung des Überlebens erfolgte bis 28 dpi. Überlebende sind entsprechend grün hinterlegt, Verstorbene orange; M – arithmetischer Mittelwert; S – Standardabweichung on hatte weniger Rückstände (1 – 2), aber eine weniger intensive, diffuse Färbung. Beim Test der Negativseren war nur ein Serum auffällig (Maus Nr. 27). Es zeigte einen unspezifischen Hintergrundtiter von 546 gegen rPA83. In Tabelle 30 sind alle gemessenen Titer vor der Infektion für diese Gruppe aufgeführt. Für rBclA war nach der Immunisierung ein detekt ierbarer, unspezifischer Hintergrundtiter messbar, der vor der Immunisierung nicht vorhanden war. Die IgG- und IgG1-Titer gegen Abbildung 26: Mausgruppe TH rPA83 waren nach der Immunisierung signifi- Halblogarithmische Auftragung der Antikörpertiter; Titerwerten unterhalb der Nachweisgrenze wurde ein Betrag von 1 zugewiesen; * markiert signifikant erhöhte Titer im Vergleich zum Negativserum kant erhöht (p ≤ 0,001), wobei die IgG1-Titer zwar niedriger als die IgG-Titer waren, aber 71 4. Ergebnisse und Diskussion beide stark miteinander korrelierten (rs = +0,927). Dies und die starke Streuung aller Titerwerte ist in der grafischen Darstellung der Werte in Abbildung 26 ebenfalls deutlich erkennbar. Der niedrige Anteil der IgG1-Antikörper an den gesamt IgG-Antikörpern lässt aber Raum für Spekulationen zu anderen involvierten Subklassen. Nachweisbare IgG2a-Titer konnten in nur einem Individuum erzeugt werden und auch dort nur marginal. Entsprechend lies sich die Gewichtung nicht beziffern, lag aber eindeutig auf einer IgG1-Antwort. Des Weiteren wiesen die Seren dieser Gruppe nur geringe oder nicht detektierbare TNA-Titer auf. Trotzdem war eine schwache Korrelation der TNA-Titer mit den antirPA83 Titern vorhanden (rs ≥ +0,67). Es überlebten 3 von 10 Versuchstieren, wobei keine verlängerte Überlebenszeit ersichtlich war. Signifikante Korrelationen zwischen der überlebten Zeit und den gemessenen Titern waren nicht vorhanden (rs ≤ +0,61). 4.1.2.5 Mausgruppe TA (CIITA; TPA-LFD1PAD4-LAMP1) Eine Gruppe von 10 Mäusen wurde wie zuvor beschrieben dreimal mit je zwei DNA-Patronen, welche mit den Vektoren pDNAVaccUltra5-CIITA und pDNAVaccUltra1-TPA-LFD1PAD4LAMP1 beschichtet waren, geimpft und 3 Wochen nach der letzten Impfung mit 894 Sporen (~25LD50) eines voll virulenten Stammes (Ames) infiziert. Jede Dosis enthielt je eine Patrone mit 2,1 und 1,8 µg DNA. Beide Präparationen ließen sich sehr gut verschießen und hatten kaum Rückstände (Rückstandbild 2 – 3, siehe Abbildung 10 und 58), wobei die höher konzentrierte Patrone intensivere Verfärbungen erzeugte. Die Analyse der Negativseren offenbarte nur für ein Serum (Maus Nr. 19) einen unspezifischen Hintergrundtiter gegen rPA83 von 400. In Tabelle 31 sind die gemessenen Titer für die Seren vor der Infektion zu sehen. Tabelle 31: Mausgruppe TA Maus Nr. rBclA rLF rPA83 TNA Tod in IgG IgG1 IgG2a IgG IgG1 IgG2a 11 4303 4266 <800 <800 <800 <800 <250 5 12 11696 9813 <800 <800 <800 <800 <250 3 13 12137 32426 <800 <800 <800 <800 <250 4 14 6289 6400 <800 <800 <800 <800 <250 4 15 4303 5760 <800 <800 <800 <800 <250 4 22058 25600 <800 1600 <800 <800 <250 - 17 32662 32426 1306 12800 3200 <800 <250 - 18 18096 17920 1013 2096 <800 <800 <250 - 19a 25158 25600 <800 4855 800 <800 <250 - 20 7724 11520 <800 <800 <800 <800 <250 4 16 IgG <200 dpi M - 14443 17173 232 2135 400 - - 4,2 S - 9708 11090 494 4066 1015 - - 0,7 a hatte bereits vor der Immunisierung einen anti-rPA83 IgG-Titer von 400 ELISA- und TNA-Titer der Mausgruppe TA vor der Infektion, sowie Eintritt des Todes in dpi. Titer-Werte ergeben sich als reziproker Wert der letzten Verdünnungsstufe mit einer OD404nm ≥ 0,1. Die Überwachung des Überlebens erfolgte bis 28 dpi. Überlebende sind entsprechend grün hinterlegt, Verstorbene orange; M – arithmetischer Mittelwert; S – Standardabweichung 72 4. Ergebnisse und Diskussion Die erzeugten Antikörpertiter gegen rPA83 waren im Gruppenmittel nicht signifikant erhöht (p ≥ 0,11). Einzig die Überlebenden hatten einen sehr niedrigen, nachweisbaren IgG-Titer, der entsprechend stark mit dem Überleben korrelierte (rs = +0,88). Gegen rLF konnten dagegen in allen Individuen signifikante IgG- und IgG1-Titer erzeugt werden (p ≤ 0,001). Die IgG2a-Titer waren für beide Antigene in den meisten Tieren nicht nachweisbar. Die Gewichtung der IgG Antwort bezüglich LF lag bei IgG1. Dies spiegelte sich auch in einer starken Korrelation (rs = +0,89) und der nahezu identischen Verteilung der Titerwerte (Abbildung 27) wieder. Beim Vergleich der IgG-Titer gegen rPA83 und rLF ergab sich, trotz der teilweise nicht nachweisbaren Titer gegen rPA83, ebenfalls eine starke Korrelation (rs = +0,88). Das heißt, Tiere mit einem detektierbaren anti-rPA83 IgG-Titer wiesen auch die höchsten anti-rLF IgG-Titer auf. Ein detektierbarer TNA-Titer war für keines der getesteten Seren vorhanden. In dieser Gruppe verstarben 6 von 10 Individuen innerhalb der ersten 5 Tage nach der Infektion. Eine Korrelation zur überlebten Zeit in Stunden ergab sich neben den anti-rPA83 IgG-Titern auch etwas schwächer zu den anti-rLF IgGTitern (rs = +0,685). Abbildung 27: Mausgruppe TA Halblogarithmische Auftragung der Antikörpertiter; Titerwerten unterhalb der Nachweisgrenze wurde ein Betrag von 1 zugewiesen; * markiert signifikant erhöhte Titer im Vergleich zum Negativserum 4.1.2.6 Mausgruppe NS (TPA-LFD1PAD4-mIPS-1) Eine Gruppe von 10 Mäusen wurde wie zuvor beschrieben dreimal mit je zwei DNA-Patronen, die mit dem Vektor NTC7382-TPALFD1PAD4-mIPS-1 beschichtet waren, geimpft und 3 Wochen nach der letzten Immunisierung mit 894 Sporen (~25LD50) eines voll virulenten Stammes (Ames) infiziert. In dieser Gruppe war kein zusätzlicher Vektor als Adjuvans beigefügt, da der Vakzinevektor selbst regulatorische Sequenzen mit Adjuvanscharakter (mIPS-1) aufwies. Jede Dosis setzte sich aus je einer Patrone mit 2,1 und 1,3 µg Abbildung 28: Mausgruppe NS Halblogarithmische Auftragung der Antikörpertiter; Titerwerten unterhalb der Nachweisgrenze wurde ein Betrag von 1 zugewiesen; * markiert signifikant erhöhte Titer im Vergleich zum Negativserum DNA zusammen. Damit war die Konzentration des antigentragenden Vektors doppelt so hoch 73 4. Ergebnisse und Diskussion wie in den anderen Versuchsgruppen, die zusätzlich den CIITA-Vektor enthielten. Die serologische Untersuchung der Seren vor der Infektion ergab die in Tabelle 32 aufgelisteten Titer, die teilweise in Abbildung 28 dargestellt sind. Tabelle 32: Mausgruppe NS Maus Nr. rBclA rLF rPA83 TNA Tod in IgG IgG1 IgG2a IgG IgG1 IgG2a 1 66560 88746 <800 21557 25600 <800 1296 6 2 36693 40106 <800 6400 3986 <800 397 5 3 293546 266240 4114 94315 5440 4457 2630 - 4 204800 119466 <800 16168 22186 <800 844 3 41813 24320 <800 2610 9386 <800 498 4 41813 37546 <800 45810 102400 <800 447 - 7 21333 12373 <800 21557 22186 <800 400 3 8 21333 8106 <800 800 14080 <800 149 4 9 8960 8106 <800 4884 63146 <800 50 - 10 69973 42666 <800 10778 39253 <800 199 3 5 6 IgG 200 dpi M - 80682 64768 - 22488 30766 - 691 4,1 S - 93200 79441 - 28511 30768 - 772 1,2 ELISA- und TNA-Titer der Mausgruppe NS vor der Infektion, sowie Eintritt des Todes in dpi. Titer-Werte ergeben sich als reziproker Wert der letzten Verdünnungsstufe mit einer OD404nm ≥ 0,1. Die Überwachung des Überlebens erfolgte bis 28 dpi. Überlebende sind entsprechend grün hinterlegt, Verstorbene orange; M – arithmetischer Mittelwert; S – Standardabweichung Für rLF und rPA83 waren signifikante aber generell stark gestreute IgG- und IgG1-, sowie TNA-Titer messbar (p ≤ 0,023), wohingegen die jeweiligen IgG2a-Titer nur in einem Individuum nachweisbar waren. Dabei korrelierten die IgG1-Titer gegen rPA83 nicht mit den entsprechenden IgG-Titern (rs = +0,19), für rLF indes stark (rs = +0,94). Ebenfalls ergab sich eine signifikante Korrelation der TNA-Titer zu den anti-rPA83 und anti-rLF IgG-Titern (r s ≥ +0,66). Da keine aussagekräftigen IgG2a-Titer messbar waren, ist eine IgG1-Gewichtung sowohl für rLF, als auch für rPA83 anzunehmen (Abbildung 28). Eine signifikante Korrelation der rPA83 und rLF Titer untereinander war nicht vorhanden (rs = +0,55). In dieser Gruppe überlebten 3 von 10 Tieren die Infektion, wobei zwischen den gemessenen Titern und der überlebten Zeit in Stunden ebenfalls keine erkennbare Korrelation bestand (rs ≤ ±0,30). 4.1.2.7 Mausgruppe TKS (CIITA; TPA-BclAD1D3-LAMP1, TPA-LFD1PAD4-mIPS-1) Eine Gruppe von 10 Mäusen wurde wie zuvor beschrieben dreimal mit je zwei DNA-Patronen, die mit den Vektoren pDNAVaccUltra5-CIITA, pDNAVaccUltra1-TPA-BclAD1D3-LAMP1 und NTC7382-TPA-LFD1PAD4-mIPS-1 beschichtet waren, geimpft. Jede Dosis setzte sich aus je einer Patrone mit 3,65 und 3,51 µg DNA zusammen. Das Mischverhältnis der Vektoren entsprach einer 1 : 1 : 2 Mischung, sodass die applizierte Menge jedes Vektors der Menge in den Einzelapplikationen entsprach. Daher betrug die Gesamtmenge an DNA pro Dosis mindestens 6 µg für diese Gruppe. Die Auswahl der Vektoren stützte sich auf die gewonnenen Daten der Vorversuche und ist unter 4.1.4 erläutert. Die Infektion fand 3 Wochen nach der letzten Immu- 74 4. Ergebnisse und Diskussion nisierung mit 864 Sporen (~25LD50) eines voll virulenten Stammes (Ames) statt. Durch die Immunisierung konnten signifikante IgG- und IgG1-Titer gegen rBclA, rLF und rPA83 erzeugt werden (p ≤ 0,004; Tabelle 33), wobei die IgG2a-Titer nicht für alle Individuen messbar waren. Ein TNA-Titer war für nahezu alle Individuen nachweisbar, ist aber aufgrund der hohen Standardabweichung und der generell sehr niedrigen Titer nicht signifikant erhöht (p = 0,06). Ein leicht erhöhter Titer gegen FIS, sowie ein unspezifischer Abbildung 29: Mausgruppe TKS Halblogarithmische Auftragung der Antikörpertiter; Titerwerten unterhalb der Nachweisgrenze wurde ein Betrag von 1 zugewiesen; * markiert signifikant erhöhte Titer im Vergleich zum Negativserum Hintergrundtiter gegen die Kapsel wurde ebenfalls gemessen, beide waren im Negativserum nicht nachweisbar. Eine signifikante Korrelation der TNA-Titer ergab sich für anti-rLF IgG und IgG1 (rs ≥ +0,85), sowie anti-rPA83 IgG1 (rs = +0,66). Wie in Abbildung 29 ersichtlich, ähneln sich die Titerspektren für IgG und IgG1 bei allen Antigenen, was sich für rPA83 und rLF auch in entsprechenden, signifikanten Korrelationen widerspiegelt (rs ≥ +0,77). IgG2a-Titer gegen die verschiedenen Antigene waren nicht in allen Tieren nachweisbar und deutlich niedriger als die IgG- und IgG1-Titer. Daher ist von einer starken IgG1-Gewichtung aller relevanten Antigene auszugehen. Eine Korrelation der Antigene untereinander war nicht gegeben (rs ≤ +0,56). Tabelle 33: Mausgruppe TKS Maus Nr. rBclA rLF rPA83 TNA Kapsel FIS Tod in IgG IgG dpi IgG IgG1 IgG2a IgG IgG1 IgG2a IgG IgG1 IgG2a 274 15680 22531 653 31360 55040 <400 15872 19918 <400 <100 275 31680 31346 1224 44800 87040 <400 81920 82286 4114 150 - 276 41600 27755 7706 24320 42880 400 29696 34612 5812 <100 - 277 37760 31346 514 26240 55040 1000 27136 32000 <400 150 - 281 60160 35918 <400 55040 92160 491 82286 73143 3004 199 9 286 31040 42449 <400 128000 363520 <400 76800 77061 400 646 287 13440 21551 <400 48000 92160 582 32768 23837 759 348 - 288 25920 30041 400 46080 104960 <400 81920 55510 498 199 - 289 31680 35918 <400 50560 138240 <400 113633 159347 <400 2291 - 292 56320 58122 612 122880 281600 873 58122 88816 <400 646 M 34528 33698 1111 57728 131264 335 60015 64653 1459 463 - - - S 15256 10620 2351 37181 106317 392 32158 41909 2093 682 - - - - 229 2204 - - ELISA- und TNA-Titer der Mausgruppe TKS vor der Infektion, sowie Eintritt des Todes in dpi. Titer-Werte ergeben sich als reziproker Wert der letzten Verdünnungsstufe mit einer OD 404nm ≥ 0,1. Die Überwachung des Überlebens erfolgte bis 18 dpi. Überlebende sind entsprechend grün hinterlegt, Verstorbene orange; M – arithmetischer Mittelwert; S – Standardabweichung 75 4. Ergebnisse und Diskussion 4.1.3 Überlebensdaten aller Mausversuche im Vergleich Aufgrund der unterschiedlichen Infektionsdosis und der Unterscheidung zwischen Protein- und DNA-Vakzinen wurden die Überlebensdaten in drei Gruppen zusammengefasst und analysiert. Auffällige Vergleiche zwischen diesen Gruppen werden aber ebenfalls beschrieben. Bei keinem Individuum, das die Infektion überlebte und am Ende euthanasiert wurde, konnte der Erreger in Leber oder/und Milz nachgewiesen werden. Alle in diesem Zeitraum selbstständig verstorbenen Tiere wiesen B. anthracis in Leber oder/und Milz auf. Folglich ist davon auszugehen, dass durch die Immunisierung eine komplette Klärung der Infektion bei allen Überlebenden stattgefunden hat und entsprechend die Todesursache bei allen verstorbenen Tieren eine fortschreitende Infektion mit B. anthracis war. In Abbildung 30 sind die Überlebenskurven für die Proteingruppen aufgeführt, die mit ~25LD50 eines voll virulenten Stamm (Ames) infiziert wurden. NC unterschied sich nicht signifikant von der Kontrollgruppe und LN-1000, während die Versuchstiere der Gruppe NCP mit 70 % Überlebenden signifikant besser geschützt waren als beide anderen Gruppen. Abbildung 30:Überlebensdaten Mausgruppen (Proteinvakzinen, ~1000 Sporen) Prozentuale Auftragung der Überlebenden über die Zeit in dpi; Angegebene p-Werte über den jeweiligen Kurven beziehen sich auf den Vergleich mit der Kontrollgruppe (LN-1000); Kap – Kapselkonjugat 76 4. Ergebnisse und Diskussion In Abbildung 31 sind die restlichen Versuchsgruppen, welche mit Protein-Komponenten immunisiert wurden, aufgeführt. Diese waren mit ~50LD50 eines voll virulenten Stamm (Ames) infiziert worden. Die Gruppen LB und LP mit je 10 % Überlebenden unterschieden sich nicht signifikant von der negativen Vergleichsgruppe LN-2000 und voneinander. Da für LN-2000 eine leicht verlängerte Überlebenszeit aufgetreten ist, muss angemerkt werden, dass es für LB und LP ebenfalls keine signifikanten Unterschiede zu den anderen Kontrollgruppen (LN-1000 und TN) gab. Mit 70 % und 60 % Überlebenden unterschieden sich LBP und LBCP nicht signifikant voneinander (p = 0,696). Bei einer Überlebensrate von 100 % unterschied sich LBCOP mit Ausnahme von LBCP (und NCP) von allen anderen Proteingruppen signifikant (p ≤ 0,029) und stellte somit die beste Proteinkombination dar. Abbildung 31: Überlebensdaten Mausgruppen (Proteinvakzinen, ~2000 Sporen) Prozentuale Auftragung der Überlebenden über die Zeit in dpi; Angegebene p-Werte über den jeweiligen Kurven beziehen sich auf den Vergleich mit der Kontrollgruppe (LN-2000); Kap – Kapselkonjugat In Abbildung 32 sind die Überlebenskurven nach einer Infektion mit ~25LD 50 eines voll virulenten Stammes (Ames) der Gruppen zur Testung der DNA-Vakzine aufgeführt. Alle DNAGruppen hatten eine signifikant höhere Überlebensrate als die Kontrollgruppe, die nur den Adjuvansvektor CIITA erhalten hatte (TN). Innerhalb der DNA-Gruppen, die nur mit Toxinkomponenten immunisiert wurden, gab es mit 30 – 40 % ähnliche Überlebensraten (p ≥ 0,573). Analog verhielt es sich für die Gruppen TK und TM, welche jeweils mit einem Vektor immunisiert wurden, der BclAD1D3 als Antigensequenz enthielt (p = 0,983) und 50 % Schutzwirkung 77 4. Ergebnisse und Diskussion erzielte. Die Kombination der erfolgversprechendsten Toxin- und Sporenvektoren resultierte in einer im Vergleich zu allen anderen DNA-Vakzinen signifikant besseren Überlebensrate von 90 % und verlängerten Überlebenszeit für das verstorbene Tier. Gesamtheitlich betrachtet, waren die Kontrollgruppen LN-1000, LN-2000 und TN trotz unterschiedlicher Infektionsdosis nicht voneinander zu unterscheiden (p ≥ 0,137). Innerhalb der Proteinvakzinen konnte keines der getesteten Antigene alleine eine Schutzwirkung erzielen, die sich von den Kontrollgruppen unterschied. Erst eine Kombination von rPA83 mit mindestens einem weiteren Antigen, erzeugte einen partiellen Schutz, der nur bei zusätzlicher Anwesenheit von FIS komplettiert wurde. Abbildung 32: Überlebensdaten Mausgruppen (DNA-Vakzinen, ~1000 Sporen) Prozentuale Auftragung der Überlebenden über die Zeit in dpi; Angegebene p-Werte über den jeweiligen Kurven beziehen sich auf den Vergleich mit der Kontrollgruppe (TN); LFD1PAD4, PA83 und BclAD1D3 ohne weitere Anhänge beziehen sich auch die Vektorrückgrate mit TPA und LAMP1 Der potentielle Beitrag des Kapselkonjugats zur Schutzwirkung ist indes fraglich. Im Gegensatz dazu konnten die DNA-Vektoren mit BclA (TM und TK) ohne zusätzliche Antigene eine signifikant höhere Überlebensrate erzeugen (p ≤ 0,023) als die Kontrollgruppe und die vergleichbare Proteinvakzine (LB) und erreichten äquivalente Ergebnisse wie die Kombinationsvakzinen LBP und LBCP (p ≥ 0,375). Die Mehrheit der Toxinvektoren erzeugten ohne weitere Antigene ähnliche Überlebensraten wie die Proteingruppen LP, NCP, LBP und LBCP. Es gilt allerdings zu beachten, dass eine geringere Infektionsdosis bei den DNA-Vakzinen verwendet wurde und die Vergleiche zwischen Gruppen mit unterschiedlicher Infektionsdosis kritisch zu betrachten sind. Dennoch konnten auf beiden Versuchszweigen Kombinationsvakzinen erstellt werden, deren Schutzwirkung nahezu komplett war und sich nicht voneinander unterschied (p = 0,317). 78 4. Ergebnisse und Diskussion 4.1.4 Interner Vergleich der Titerdaten der Mausversuche Zum Vergleich der gemessenen Titerdaten der Mausversuche untereinander wurden die gemittelten IgG Antikörpertiter in Reihenfolge der Komplexität der eingesetzten Impfkomponenten, unterteilt in Protein- und DNA-Vakzinen, einander gegenübergestellt. Insgesamt waren bei einigen Individuen oder Messpunkten sporadisch niedrige unspezifische Hintergrundtiter für einige Antigene messbar, obwohl die Messmethoden so eingestellt waren, dass entsprechend naive Seren keinen Titer ergaben. Dies betraf vornehmlich das Kapselpräparat, aber auch rPA83 und rBclA. Die rPA83-assoziierten unspezifischen Hintergrundtiter traten ausnahmslos vor der Immunisierung auf und betrafen nur vereinzelte Individuen. Im Falle von rBclA verhielt es sich entgegengesetzt, da erst nach einer Vakzinierung ohne BclA oder FIS ein unspezifischer Hintergrundtiter messbar war. Eine Verunreinigung ist ausgeschlossen. In beiden Fällen waren die Messwerte unerheblich (<600) und haben in Anbetracht der Höhe der durch die Vakzinierung generierten Antikörpertiter gegen rPA83 und rBclA keine statistische Relevanz. Die gemessenen unspezifischen Hintergrundtiter gegen die Kapsel waren indes sowohl vor der Immunisierung, als auch nach der Immunisierung bei den Gruppen erhöht, die nicht mit dem Kapselkonjugat immunisiert wurden. Wie in Abbildung 33 ersichtlich, waren unspezifische Titer gegen die Kapsel in ähnlicher Höhe in allen Gruppen, die mit dem Lipopeptid immunisiert wurden, detektierbar. Da dies entsprechend auch die Kontrollgruppen betraf, ist davon auszugehen, dass es sich bei den gemessenen Titern nicht um spezifische gegen die Kapsel gerichtete Antikörpertiter handelt. Davon ausgenommen sind nur die Gruppen LBCP und LBCOP, welche identische und weitaus höhere Titer gegen die Kapsel aufwiesen (p = 0,594). Die anti-rPA83 IgG-Titer aller Protein-Gruppen, die entsprechend mit rPA83 immunisiert wurden, waren signifikant erhöht und lagen alle in einem ähnlich hohen Bereich (~ 5 * 105). Damit waren sie bis auf Gruppe LP, die einen noch höheren Titer aufwies, nicht signifikant unterschiedlich (p ≥ 0,259). Ein ähnliches Verhältnis ergab sich auch im Bezug auf IgG1, wobei diese in Abhängigkeit von der applizierten Vakzinekomposition nur einen Teil der gesamt IgG ausmachten und entsprechend auch Unterschiede in der Korrelation zeigten. Etwaige Effekte durch zusätzliche Vakzinekomponenten oder Adjuvanzien könnten demnach nicht in der Höhe der IgG, sondern über Veränderungen der Subklassengewichtung detektierbar sein. Zur Abschätzung der Qualität der anti-rPA83 Antikörper wurden die Seren auf ihre neutralisierende Wirkung im TNA getestet. Für die Proteingruppen, denen unter anderem rPA83 verabreicht wurde, ergaben sich mit Ausnahme von Gruppe NCP, welche niedrigere Titer aufwies, einheitlich hohe, signifikante TNA-Titer (p ≥ 0,167). Korrelationen zu den anti-rPA83 Titern waren uneinheitlich und nur für LBP und LBCOP signifikant, die beide auch die höchsten TNA-Titer zeigten. 79 4. Ergebnisse und Diskussion Abbildung 33: Übersicht IgG- und TNA-Titer (Proteinvakzinen, Maus) Logarithmische Auftragung der ELISA-Titer (IgG) und TNA-Titer der Seren aller Proteingruppen vor der Infektion; Die Standardabweichung ist über den Balken als positiver Wert angegeben (bei Messwerten aus vereinigten Mischproben entsprechend nicht); Werten unterhalb der Nachweisgrenze wurde für eine bessere Darstellung ein Wert von 100 zugewiesen; Kap – Kapselkonjugat Ähnlich den gemessenen IgG-Titern gegen rPA83 verhielten sich die anti-rBclA IgG-Titer, die für alle entsprechend immunisierten Proteingruppen äquivalente Werte zeigten (~10 5; p ≥ 0,225). Ausgenommen hierbei war Gruppe LB, die weitaus höhere Titer als die restlichen Gruppen aufwies (p ≤ 0,014). Die generell starke IgG1 Gewichtung war bei den anti-rBclA Titern noch deutlicher sichtbar und wurde in einer entsprechend starken Korrelation der IgGund IgG1-Titer reflektiert. Dies war mit Ausnahme der Gruppe TKS für alle getesteten Gruppen, inklusive der DNA-Vakzinen signifikant. Die DNA-Vakzinegruppen TK und TM waren über die gemessenen rBclA-Titer nicht zu unterscheiden (p ≥ 0,074, Abbildung 34), wenngleich TM einen messbaren IgG2a-Titer für alle Individuen aufwies und TK nicht. Für eine DNA-Vakzine erstaunlich, hatten TM und TK äquivalente oder sogar höhere Titer als die vergleichbare Proteinvakzinen. Da auch die Überlebensraten beider Gruppen gleich waren, musste die Auswahl einer der Vektoren für eine Kombinationsvakzine auf anderer Grundlage erfolgen. Schlussendlich wurde der Vektor pDNAVaccUltra1-TPA-BclAD1D3-LAMP1, aufgrund der möglichen antagonistischen Wirkung des Ubiquitins in Kombination mit anderen DNA-Vektoren [211] ausgewählt. Trotzdem erzeugte die 80 4. Ergebnisse und Diskussion Kombinationsvakzine TKS niedrigere anti-rBclA IgG- und IgG1-Titer als die Gruppen TK und TM, sowie alle Proteinvakzinen mit rBclA als Bestandteil (p ≤ 0,017) und zeigte somit schlechtere Immunogenität als die korrespondierende Einzelapplikation des BclA-Vektors (TK). Allerdings waren die IgG Titer dieser Gruppe bezogen auf alle eingesetzten Antigene uniformer als in den Einzelapplikationen. Abbildung 34: Übersicht IgG- und TNA-Titer (DNA-Vakzinen, Maus) Logarithmische Auftragung der ELISA-Titer (IgG) und TNA-Titer der Seren aller DNA-Gruppen vor der Infektion; Die Standardabweichung ist über den Balken als positiver Wert angegeben (bei Messwerten aus vereinigten Mischproben entsprechend nicht); Werten unterhalb der Nachweisgrenze wurde für eine bessere Darstellung ein Wert von 100 zugewiesen; LFD1PAD4, PA83 und BclAD1D3 ohne weitere Anhänge beziehen sich auch die Vektorrückgrate mit TPA und LAMP1; n.v. – nicht vorhanden (Wert wurde nicht gemessen) Alle Gruppen, die mit rBclA immunisiert wurden, wiesen neben dem anti-rBclA Titer auch einen anti-FIS IgG-Titer auf. Dieser war in teilweise ähnlicher Höhe wie der anti-rBclA IgGTiter vorhanden und somit nicht allein der Gruppe vorbehalten, die zusätzlich auch FIS erhielt. Da der anti-FIS Titer für alle Gruppen außer LBCOP nur als Mischprobe der jeweiligen Gruppe exemplarisch gemessen wurde, können nur Tendenzen angegeben und analysiert werden. Augenscheinlich erhöht die gleichzeitige Gabe von FIS und rBclA den anti-FIS Titer aber nicht weiter, da kein merklicher Unterschied zwischen den Gruppen LBP, LBCP und LBCOP vorliegt. Die Kombination der Toxin- und Sporenvektoren in der Gruppe TKS schien außerdem einen negativen Einfluss auf die anti-FIS Titer gehabt zu haben. 81 4. Ergebnisse und Diskussion Die IgG-Antikörpertiter gegen rPA83 der DNA-Vakzinen mit Toxinkomponenten hatten im Vergleich zu den analogen Proteingruppen einen ca. zehnfach niedrigeren Wert (p ≤ 0,001). Für IgG1 und IgG2a waren bis auf wenige Ausnahmen ebenfalls signifikant höhere Titer bei den Proteingruppen vorhanden. Innerhalb der DNA-Gruppen waren die IgG- und IgG1-Titer gegen rPA83 für NS und TH gleich (p ≥ 0,056), wobei beide signifikant höhere Titer als TA zeigten (p ≤ 0,038), da in dieser Gruppe nur sehr niedrige, nicht bei allen Individuen nachweisbare Titer gegen rPA83 erzeugt werden konnten. Für eine Kombinationsvakzine wurde daher dieser Toxinvektor nicht in Erwägung gezogen. Da beide verbliebenen Vektoren ähnlich gute antirPA83 Titer erzeugten und der Vektor NTC7382-TPA-LFD1PAD4-mIPS-1 der Gruppe NS neben PA auch eine LF-Komponente besaß, lag die Entscheidung für dessen Einsatz in der Kombinationsvakzine nahe. Entsprechend konnte die Kombinationsvakzine TKS sogar signifikant höhere anti-rPA83 IgG-Titer als alle Einzelapplikationen der DNA-Vakzine (p ≤ 0,038) erzeugen. Antikörpertiter gegen rLF, das ebenfalls zu den Toxinkomponenten zählt, wurden nur bei den DNA-Vakzinen gemessen. Die IgG-Titer von Gruppe NS waren dabei signifikant höher als die von Gruppe TA (p = 0,038), wobei in beiden Fällen die IgG-Titer gegen rLF signifikant höher waren als die Titer gegen rPA83 (p ≤ 0,035). Dies war für die Kombination TKS nicht der Fall (p = 0,861). Der anti-LF IgG-Titer von Gruppe TKS hatte zudem die gleiche Höhe, wie die Einzelapplikation desselben Vektors in Gruppe NS (p = 0,479). Generell lag für die anti-rLF Titer eine starke Gewichtung zu IgG1 vor. Bezüglich der Neutralisierungsfähigkeit der ToxinAntikörper waren in den DNA-Gruppen nur sehr niedrige TNA-Titer detektierbar. Einzig die Gruppen NS und TKS hatten signifikant erhöhte Titer, die signifikant niedriger als die der Proteingruppen waren (p ≤ 0,001). 82 4. Ergebnisse und Diskussion 4.2 Diskussion Mausversuche Mit der Zielführung für DNA- und Proteinvakzinen geeignete Kombinationen zu finden, die eine Erprobung im Ziegenmodell rechtfertigen, sollen im Folgenden die Ergebnisse der Mausversuche anhand der Fachliteratur diskutiert werden. 4.2.1 Proteinvakzinen Wie unter 4.1.4 bereits angerissen, konnte keines der erprobten Antigene für sich genommen eine signifikante Schutzwirkung erzielen, gleichwohl, mit Ausnahme der Kapsel, hohe Antikörpertiter gegen die eingesetzten Antigene erzeugt wurden. Erst eine Kombination von rPA83 mit mindestens einem weiteren Antigen erzeugte einen partiellen Schutz, der nur bei zusätzlicher Anwesenheit von FIS komplettiert wurde. Während Einzelimmunisierungen mit PA umfangreich erprobt sind und in Abhängigkeit vom eingesetzten Adjuvans, dem Tiermodel, dem Infektionsstamm und dessen eingesetzter Dosis stark variieren [149][145], sind Studien zur Schutzwirkung von BclA, FIS und der Kapsel oder deren Kombination sehr begrenzt. Entsprechend lassen sich Mausinzuchtlinien mit PA alleine vor einer Infektion mit der kapseldefizienten SSLV zu 100 % schützen [95], aber nicht gegen eine Infektion mit einem voll virulentem Stamm von B. anthracis. Die hier durchgeführte Einzelvakzinierung mit rPA83 diente als Vergleichsgruppe für die Kombinationen. Um deren Schutzwirkung entsprechend abzugrenzen, war der Versuchsaufbau und die Infektionsdosis entsprechend so gewählt, dass Unterscheidungen zwischen den einzelnen Gruppen möglich waren. Das verwendet Lipopeptid als Adjuvans [23] war ebenso bereits in einer vorhergehenden Studie im Vergleich zu RIBI und Alhydrogel im NMRI-Mausmodell erprobt und in vielen Belangen als Zusatz für rPA83 für gleichwertig bzw. besser befunden worden [24]. Die Neuerungen der Protein-basierenden Vakzinierungsversuche umfassten die Erprobung eines Kapselkonjugats und rBclA einzeln, sowie Kombinationsversuche mit rPA83, rBclA, FIS und dem Kapselkonjugat. Der Zusatz des Kapselkonjugats hatte keinen nachweisbaren Einfluss auf die Schutzwirkung Bisherige Versuche mit allein auf PA basierenden Vakzineformulationen zeigten in Mausinzuchtlinien vorwiegend eine hohe Schutzwirkung bei der Verwendung kapseldefizienter Infektionsstämme, wie der SSLV und dem STI-1. Ohne die Anwesenheit der Kapsel sind die germinierten, vegetativen Bakterienkörper durch das Wirts-Immunsystem leichter zu detektieren und zu beseitigen [275], sodass ein nur auf einer Antitoxinwirkung beruhender Schutz bereits ausreichend sein kann. Da dies unter natürlichen Bedingungen nicht vorkommt und außerhalb des Inzuchtmodells keine Gültigkeit besitzt, ist eine zusätzliche Wirkung gegen die Kapsel wün- 83 4. Ergebnisse und Diskussion schenswert. In ihrer Eigenschaft als äußerste Schicht der vegetativen Zelle verdeckt die wenig immunogene Kapsel andere immunogene Antigene und vermindert die Detektion und Phagozytose des Erregers durch das Immunsystem des Wirts [242][163][208]. Die Immunogenität der Kapsel kann durch eine Konjugation an andere immunogene Proteine verändert werden. Immunisierungsversuche mit natürlichen oder künstlichen Kapselpräparaten konjugiert an BSA [50] [273], PA [256][105], OMPC (Outer Membran Protein Complex von Neisseria meningitidis serotyp B) [158][49] oder KLH (keyhole limpet hemocyanin) [344] resultierten teilweise in Abhängigkeit von der Länge der PGA und der Konjugationsart [273][174] in stark erhöhten (>104) kapselspezifischen Antikörpertitern. Diese waren im Falle eines Kapselkonjugats mit OMPC im BALB/c Mausmodell bei einer s. c. Infektion mit 20000 Sporen eines Ames Stammes auch ohne Zugabe weiterer Antigene zu 100 % protektiv, während die unkonjugierte Kapsel nur 30 % Schutzwirkung zeigte [158]. Der verbesserte immunogene Effekt trat entsprechend nicht auf, wenn die Konjugationspartner nur in Mischung verabreicht wurden. Tendenziell waren Kapselmultimere mit mindestens vier Glutaminsäureresten nötig, um ein funktionelles Konjugat zu erstellen [344], wobei Dekamere Optimallänge zu haben schienen [273]. In Anlehnung an diese erfolgversprechenden Experimente und zur Minimierung der Bestandteile einer zukünftigen NLV sollte ein Kapselkonjugat erstellt werden, das mit einem Lipopeptid als Adjuvans verknüpft ist. Das resultierende Kapsel-Nonamer Pam 3CysAQEKEAKSELDYD-(isoE9)-NH2 beinhaltete Bestandteile des B. cereus spore germination protein (GerD) und wurde von EMC-Microcollections (Herrn Dr. Wiesmüller) zur Verfügung gestellt. Die Testung erfolgte einzeln (NC), in Kombination mit rPA83 (NCP) oder Lipopeptid, rBclA, rPA83 (LBCP), sowie Lipopeptid, rBclA, rPA83 und FIS (LBCOP). Entgegen den Erwartungen konnte das Kapselkonjugat ohne das zusätzliche Lipopeptid keine signifikanten Titer erzeugen und induzierte auch bei Zusatz des Lipopeptids nur sehr niedrige Antikörpertiter. Zudem ist davon auszugehen, dass das Konjugat keinerlei Einfluss auf die Schutzwirkung hatte. Zwar konnte in der Gruppe NCP mit 70 % Überleben eine weitaus höhere Schutzrate erzeugt werden als mit PA allein (LP – 10 %), was auf den ersten Blick für eine Wirksamkeit des Konjugats spricht. Allerdings gilt es hier zu beachten, dass Gruppe LP mit ~50LD50 und NCP mit ~25LD50 infiziert wurden. Da frühere Versuche dieser Arbeitsgruppe bei einer Infektion mit ~21LD50 für eine mit Lipopeptid und rPA83 immunisierte Gruppe 80 % Überleben ergaben [24], kann davon ausgegangen werden, dass die Schutzwirkung in NCP einer rPA83-Immunisierung ohne Lipopeptid entspricht. Dies wird auch durch die Titerwerte von Gruppe NCP unterstützt, die für rPA83 unter denen von Gruppe LP bleiben. Des Weiteren zeigte das Kapselkonjugat allein keinerlei Wirkung (NC) und trug auch als Bestandteil der Kombinationsvakzine nichts bei, da die Gruppen LBP und LBCP, die sich nur im Zusatz des Konjugats unterschieden, in allen Belangen gleiche Werte aufwiesen. Die Erhöhung der anti-Kapsel IgG-Titer für die 84 4. Ergebnisse und Diskussion Gruppen LBCP und LBCOP auf ~2000 könnte dem zusätzlich verabreichten Lipopeptid zuzuschreiben sein, ist aber für die Erwartungen, die sich aus den Vorarbeiten an das Kapselkonjugat ergeben haben, viel zu niedrig. Einen Erklärungsversuch für den Fehlschlag dieses Ansatzes bietet ein 2013 veröffentlichtes Manuskript, welches Arbeiten zum gleichen Rumpfgerüst des hier verwendeten Kapselkonjugats beschreibt [306]. Darin wird die Wirksamkeit von Pam3Cys-AQEKEAKSELDYD, dem hier innerhalb des Kapselkonjugats verwendeten Epitops, als LPS-Toleranz (Lipopolysaccharid) erzeugendes Antiallergikum erörtert. Der beschriebene immunsupprimierende Effekt dieses Epitops war zuvor nicht bekannt und könnte die Unwirksamkeit des hier verwendeten Kapselkonjugats erklären. Damit wäre zudem das Prinzip der Erhöhung der Immunogenität des Kapselkonstrukts durch Konjugation an ein immunogenes Peptid unterwandert und dessen Unwirksamkeit begründet. Das bedeutet allerdings keinesfalls eine generelle Untauglichkeit von Lipopeptiden für ein Kapselkonjugat. Ebenfalls sollte angemerkt werden, dass das als generelles Adjuvans verwendete Lipopeptid in diesen Versuchen eine Mischung aus einem TLR2/1 Aktivator und einem promiskuitiven TH2-Epitop darstellte und nicht von den Ergebnissen des Kapselkonjugats betroffen ist. Auf Grundlage dieser Ergebnisse und der Annahme, dass ebenso die bei Gruppe LBCP und LBCOP bemerkten Verkapselungen nach der Vakzinierung auf das Kapselkonjugat zurückzuführen sind, wurde von einer Verwendung in weiteren Versuchen abgesehen. BclA und PA wirken einander unterstützend Sowohl rBclA als auch rPA83 erzeugten ohne den Zusatz weiterer Antigene signifikant hohe Antikörpertiter mit starker IgG1-Gewichtung. Dabei waren beide ähnlich immunogen mit Titerwerten, welche die der Kombinationsvakzinen übertrafen. Die Immunogenität beider Antigene ist hinlänglich bekannt, wobei BclA als immunodominantes Oberflächenprotein der Spore in Anwendung als Vakzine [310][296] im Vergleich zu PA und dessen geschichtlicher Relevanz für die Vakzineentwicklung [347] noch relativ unerforscht ist. In den wenigen Vergleichsstudien zur Immunisierung mit BclA als rekombinantes Protein oder DNA-Vakzine [125] konnten in Kaninchen, Meerschweinchen und verschiedenen Mauslinien [39][195][61] ebenfalls hohe Antikörpertiter gegen BclA erzeugt werden, die allerdings ebenfalls nur einen Schutz von < 20 % vermittelten. Versuche zur Kombination von BclA und PA wurden vor allem in Hinblick auf die Verbesserung der Schutzwirkung von suboptimalen Konzentrationen von PA durchgeführt und vermittelten den Eindruck eines antagonistischen Effekts von BclA auf die anti-PA Titer [39]. Eine Trennung der Antigene in einem Prime/Boost Ansatz nivellierte den Effekt und resultierte in 85 4. Ergebnisse und Diskussion kompletten Schutzraten bei einer Infektion von A/J-Mäusen mit Sporen der SSLV [39]. Spätere Versuche zeigten allerdings, dass der störende Effekt von BclA nur bei suboptimalen Konzentrationen von PA und während der frühen Impfphase auftritt, sodass eine Kombination beider Antigene ohne nachteilige Reaktionen problemlos möglich ist [125][61]. Dies konnte in den hier durchgeführten Versuchen bestätigt werden, wobei eine Schutzrate von bis zu 70 % nach einer Infektion mit voll virulentem B. anthracis für die Kombination beider Antigene erreicht wurde. Der offensichtlich synergetische Effekt beider Antigene in Kombination, wurde allerdings nicht in den gemessenen Antikörper- oder TNA-Titern reflektiert, die in der Kombination sogar eher niedriger ausfielen. Mit einem ähnlichen Ergebnis konfrontiert, folgerten Brahmbhat et al. (2007) [39] aufgrund weiterführender Experimente, dass der Zusatz von BclA zu einer PAbasierenden Vakzine die Schutzrate über die Erzeugung sporenopsonierender und germinationshemmender Antikörper erhöht. Überdies belegten weitere Studien die Fähigkeit von anti-PA Antikörpern zu Detektion von Sporen [352][62]. Dies beruht vermutlich auf einer Einlagerung von vegetativen Bestandteilen in den Sporenmantel bei der Sporulation und betrifft nicht nur PA [352][332]. Durch die Opsonierung der Sporen mittels anti-PA Antikörpern kann es zu einer Hemmung der Germination und einer Erhöhung der Phagozytose dieser durch Makrophagen kommen [352][62]. Schlussfolgernd könnte also die partielle Schutzrate von rPA83 und rBclA in Kombination sowohl ein Produkt des erzeugten anti-Sporeneffekts, als auch der und der Antitoxineigenschaften der Antikörper sein. Tabelle 34: Korrelation der Proteingruppen rPA83 rBclA rPA83 IgG vs IgG1 IgG vs IgG1 IgG vs TNA NCP +0,14 - +0,36 LP +0,28 - -0,05 Gruppe LB - +0,92 - LBP +0,81 +0,95 +0,85 LBCP +0,65 +0,85 +0,59 LBCOP +0,82 +0,85 +0,65 Angegeben sind die r s-Werte der Proteinbasierenden Mausgruppen; Korrelationen sind um so stärker je näher der rs-Wert an ±1 gelangt; signifikante Werte sind fett gedruckt Ein detaillierterer Blick auf die gemessenen Titer beider Antigene in allen relevanten Protein-basierenden Gruppen und deren Korrelationen, lässt weitaus mehr Schlüsse zur Synergie zu als die unprozessierten Rohdaten erahnen lassen. So scheint rBclA die Gewichtung der Antikörpertiter gegen rPA83 in Richtung IgG1 zu verschieben und hat eventuell auch Einfluss auf deren Korrelation zur Toxinneutralisationsfähigkeit. In Tabelle 34 sind zur Erläuterung noch einmal alle betreffenden Gruppen nebst ihrer r sWerte aufgeführt. Betrachtet man hierbei Gruppe NCP, in Anbetracht der bereits erläuterten Wirkungslosigkeit des Kapselkonjugats ebenfalls als reine rPA83 vakzinierte Gruppe, wird der Unterschied zwischen den Einzelvakzinierungen mit rPA83 und den Kombinationsvakzinen mit rBclA deutlich. Während rBclA allein und in Kombination eine nahezu gleiche und starke IgG:IgG1-Korrelation aufweist, wird diese für rPA83 erst in Kombination mit rBclA erzeugt. Im Weiteren schlägt sich dies auch in der Korrelation der anti-rPA83 IgG-Titer mit den TNATitern nieder. 86 4. Ergebnisse und Diskussion Die Anwesenheit von BclA in einer Kombinationsvakzine könnte also einen nivellierenden Einfluss auf die Richtung der Antikörpersubklassenentwicklung von PA und deren toxinneutralisierende Eigenschaften haben ohne dabei die Titerhöhe zu beeinflussen. Eine Wirkung dieser Art ist bisher nirgends dokumentiert. Umgekehrt scheint rPA83 keinen Einfluss auf rBclA zu haben. Zusatz von FIS bietet potentiell höheren Schutz, der nicht zwingend allein auf BclA beruht Anschließend an die vorhergehenden Betrachtungen zur Wirksamkeit von BclA schien ein Zusatz von FIS zu rPA83 und rBclA innerhalb der serologischen Parameter keinen weiteren Einfluss zu haben. Nahezu alle gemessenen Antikörpertiter der Gruppen LBP, LBCP und LBCOP waren identisch, was bei der Gegenüberstellung der IgG Titer aller Proteingruppen in Kapitel 4.1.4 , Abbildung 33 gut zu sehen ist. Ein ähnlich detaillierterer Blick auf die Subklassen wie bei BclA ist für FIS nicht möglich, da für FIS nur Messungen auf IgG-Titer durchgeführt und abgesehen von Gruppe LBCOP nur die vereinigten Mischseren der Gruppen getestet wurden. Eine nachweisbare starke Korrelation zwischen anti-FIS und anti-rBclA IgG-Titern ergab sich daher nur für Gruppe LBCOP. Da die Antikörpertiter gegen FIS und rBclA auch in den anderen Gruppen, die keine FIS erhalten hatten eine ähnliche Höhe aufwiesen, ist davon auszugehen, dass die über FIS generierten Antikörper mehrheitlich gegen BclA gerichtet waren. Die Möglichkeit anti-rBclA Antikörper mit FIS als Antigenkomponente zu detektieren spricht auch für die Funktionalität der Epitope des rekombinant in E. coli gebildeten BclA und auch für die Funktionalität der intrazellulär exprimierten BclAD1D3 DNA-Konstrukte. Im Umkehrschluss verdeutlicht dies, dass die FIS-Präparation intakte BclA-Filamente enthielt. Die fehlende Glykosylierung des rekombinant hergestellten BclA schien überdies keinen merklichen Einfluss zu haben, obwohl für den in die Glykosilierung involvierten Zucker bereits eine immunogene Wirkung beschrieben ist [214]. Mittels der vorhandenen, serologischen Werte ließ sich allerdings keine Aussage über den Beitrag von FIS zur Vakzineformulation treffen. Einzig der vollständige Schutz gegen eine Infektion mit ~50LD50 Sporen eines voll virulentem Stammes (Ames), der nur über den Zusatz von FIS erreicht wurde, spricht für deren zusätzlichen positiven Einfluss. Damit entsprechen die erzielten Ergebnisse den Vorarbeiten von Brossier et al. (2002) [42] und Gauthier et al. (2009) [106], denen es gelungen war, Mäuse und Meerschweinchen mit einer Vakzinekomposition aus PA und FIS gegen eine Infektion mit einem voll virulenten Stamm zu schützen (bis 30LD50). Beide erreichten 100 % Schutzraten, wobei PA und FIS allein unter gleichen Bedingungen keinen bis geringen (< 25 %) Schutz boten. 87 4. Ergebnisse und Diskussion In diesem Zusammenhang stellt sich die Frage nach dem Wert von BclA, da der Zusatz von FIS für einen Schutz ausreichend erscheint und über FIS generierte Antikörper auch gegen BclA gerichtet sind. Entsprechend könnten diese Antikörper die gleiche nivellierende Funktion wie reines BclA übernehmen. Hintergrund für den Einsatz von rBclA zusätzlich zu FIS war die Befürchtung, dass funktionale BclA-Epitope größtenteils durch die FIS-Präparation verloren gegangen sein könnten und FIS als undefinierte Mischung bestenfalls nur Adjuvanscharakter haben könnte. Neuere Erkenntnisse zeigen noch einen weiteren Grund für die Beibehaltung beider Antigene in einer Kombinationsvakzine und erklären die mögliche Zusatzwirkung von FIS. Nachdem Cybulsky et al. (2008) [64] 10 weitere immunogene, sporenspezifische Antigene fanden, verdichteten sich die Belege, wonach BclA die Erkennung anderer Antigene auf der Spore behindert. Untermauert wurde dies durch die Arbeiten von Côte et al. (2012) [61], die Kombinationen von suboptimalen Mengen von PA mit BclA und anderen Sporenantigenen testeten. Während der alleinige Zusatz von BclA und anderer Sporenantigene zu PA nur maximal 40 % der Tiere schützte, konnte eine Kombination aus PA, BclA und der Sporenantigen ExsFA und p5303 einen kompletten Schutz vermitteln. Die Detektion von Antikörpern gegen ExsFA und p5303 war hierbei mit BclA-defizienten Sporen wesentlich besser möglich, wodurch die überdeckenden Eigenschaften von BclA nochmals herausgestellt wurden. Schlussendlich zeigten die Ergebnisse von Vergis et al. (2013) [336] mit FIS eines B. cereus eine bessere Schutzwirkung von BclA-defizientem FIS im Vergleich zu BclA-tragenden Sporen und bestätigte das Vorhandensein von mindestens 11 weiteren immunogenen, sporenspezifischen Antigenen. Damit kann davon ausgegangen werden, dass die immunogene Wirkung von FIS nicht allein und potentiell nicht einmal hauptsächlich auf BclA beruht. Darüber hinaus scheint BclA weitere sporenspezifische Antigene zu überdecken und könnte als Bestandteil von Sporen einen negativen Einfluss auf der Immunogenität und Schutzwirkung haben. Die beobachtete zusätzliche Schutzwirkung von FIS in den hier gezeigten Versuchen könnte daher auf weiteren sporenspezifischen Antigenen beruhen. Da rBclA erheblich zur Schutzwirkung von rPA83 beitrug, sollte es aber weiterhin Bestandteil zukünftiger Versuche bleiben. Um beiden Erkenntnissen gerecht zu werden, wäre eine Vakzine bestehend aus BclA-defizienten FIS, rPA83 und rBclA ein sinnvoller Kompromiss, der sowohl BclA als auch die Gesamtheit an immunogenen Sporenantigenen berücksichtigt. Die Veröffentlichungen und die damit einhergehenden Erkenntnisse sind erst während oder nach den hier ebenfalls durchgeführten Ziegenversuchen erschienen, daher wurden auch für diese BclA-tragende FIS verwendet. 88 4. Ergebnisse und Diskussion 4.2.2 DNA-Vakzinen Wie unter 4.1.4 zusammengefasst, erzeugten die DNA-Vektoren generell signifikant niedrigere Titer gegen rPA83, aber äquivalent hohe anti-rBclA Titer wie die Proteinvakzinen. Bei vergleichsweise verminderter Infektionsdosis waren alle antigentragenden Vektoren partiell protektiv, konnten aber, analog zu den Proteinvakzinen in Kombination einen nahezu vollständigen Schutz vermitteln. Die eingesetzten Konstrukte umfassten die antigenkodierenden Vektoren pDNAVaccUltra1-TPA-LFD1PAD4-LAMP1, 7382-TPA-LFD1PAD4-mIPS-1, pDNAVaccUltra1-TPA-PA83-LAMP1, pDNAVaccUltra1-TPA-BclAD1D3-LAMP1, NTC- pDNAVacc- Ultra4-BclA-Ubiquitin und den Adjuvansvektor pDNAVaccUltra5-CIITA. In dieser Komposition waren alle Vektoren bisher unerprobt. Vektoren mit Toxinkomponenten zeigten eine geringere Immunogenität als vergleichbare Proteinvakzinen Die Grundlage für die Erstellung und Tests der Vektoren mit Toxinkomponenten bestand in einer früheren Studie, deren Zielsetzung es war, ein atoxisches Fusionsprotein des Letaltoxins zu erstellen, das vornehmlich neutralisierende Titer erzeugt [11]. Resultat war das in dieser Studie ebenfalls verwendete LFD1PAD4, welches die PA-Bindestelle von LF [6] und die Rezeptorbindestelle von PA miteinander [287] vereint. Eingesetzt als Protein war es damit möglich, A/JMäuse gegen eine i. p. Infektion mit ~105 Sporen der SSLV zu schützen [11]. Zuvor war dies ebenfalls für die Einzelkomponenten PAD4 und LFD1 im BALB/c bzw. A/J Mausmodell etabliert worden [243][95][307], wobei für beide Antigene eine starke IgG1-Gewichtung ungeachtet der Darreichungsform aufgefallen war. Um die Wirksamkeit von LFD1PAD4 innerhalb einer DNA-Vakzine in einem Versuchsaufbau mit einem voll virulenten Infektionsmodell zu testen, wurde es in zwei verschiedenen Vektoren im Vergleich zu einen Vektor mit PA in voller Länge getestet. Diese umfassten zum einen den von Dr. L. Baillie zur Verfügung gestellten Vektor NTC7382-TPA-LFD1PAD4-mIPS-1 und zum anderen, in Anlehnung an die Arbeiten von Midha et al. (2009) [211], pDNAVaccUltra1TPA-LFD1PAD4-LAMP1 und respektive pDNAVaccUltra1-TPA-PA83-LAMP1. Die integrierten Signalsequenzen TPA, LAMP1 und das für die BclA-Vektoren zum Einsatz gekommene Ubiquitin dienten zur gerichteten Expression der eingefassten Proteinsequenzen. Die Funktionalität solcher Sequenzen war bereits in anderen Arbeiten erprobt worden [211][124][210]. Alle drei getesteten Vektoren zeigten eine signifikante Schutzrate (30 – 40 %) gegenüber einer Infektion mit Ames-Sporen (~25LD50), wobei die gemessenen anti-rPA83 und TNA-Titer einen mindestens zehnfach niedrigeren Wert als die analogen Proteingruppen aufwiesen. Dies war auch schon in vorhergehenden Versuchen mit PA83 in einem sekretierenden und endosomal verankernden Vektor aufgefallen [125], der bei gleicher Infektionsdosis im gleichen Tier89 4. Ergebnisse und Diskussion modell ebenfalls eine ähnliche Schutzrate (30 %) zeigte. Da dies für Vektoren mit LFD1PAD4, PA83 (diese Arbeit [125]) und PAD4 [24] gleichermaßen zutraf, ist davon auszugehen, dass die serologischen Unterschiede zwischen der Protein- und DNA-Vakzine nicht auf die Verkürzung des Fusionsproteins auf einzelne Domänen zurückzuführen sind. Die generelle Funktion der Vektoren oder die Form der DNA-Vakzinierung als Ursache für die Diskrepanz anzunehmen, scheint aber ebenfalls unplausibel, da die gleichen Vektoren mit integrierter Sequenz von BclAD1D3 äquivalente Titer gegenüber der Proteinapplikation erzeugen konnten. Zudem erreichten die von Midha et al. (2009) [211] getesteten gleichartigen Vektoren mit PA63 äquivalente Titer im Vergleich zu PA63 und PA83 als Protein. Unterscheidungsmerkmal war hier allerdings die Form der Applikation als i. m. Injektion, sowie die applizierte Menge von 100 µg pro Dosis. Ebenso könnte das verwendete Tiermodell einen Einfluss haben, da Tests mit PA und dessen Domänen in verschiedenen Mausmodellen unterschiedliche Immunogenität aufwiesen [1]. Eine generelle Erhöhung der DNA-Menge für die applizierten Toxin-Vektoren wäre also möglicherweise zuträglich für eine verbesserte Titerentwicklung gewesen, was aber nicht zwingend gleichbedeutend mit einer besseren Schutzwirkung sein muss. Die internen Unterschiede der Toxinvektoren sind vermutlich dosisbedingt Unterschiede zwischen den getesteten DNA-Vektoren betrafen die Antikörpertiter gegen rPA83, welche für Gruppe TA (pDNAVaccUltra1-TPA-LFD1PAD4-LAMP1) im Gegensatz zu Gruppe NS (NTC7382-TPA-LFD1PAD4-mIPS-1) und TH (pDNAVaccUltra1-TPA-PA83-LAMP1) nur geringfügig erhöht waren. Gegen rLF waren vergleichsweise niedrigere Titer in Gruppe TA als für Gruppe NS gemessen worden. Abgesehen von der Höhe der gemessenen Titer, war das Antikörperspektrum von TA und NS gleich, wobei die gebildeten anti-rLF IgG-Titer signifikant höher als die anti-rPA83 IgG-Titer waren (p ≤ 0,035). Damit scheint die verminderte Immunogenität von pDNAVaccUltra1-TPA-LFD1PAD4-LAMP1 den Vektor, bzw. die Vakzinekomposition insgesamt zu betreffen. Abgesehen vom schlichten Unterschied der Menge an applizierter antigenkodierender DNA der Gruppen NS und TA, die für Gruppe NS aufgrund der Abwesenheit eines weiteren Vektors doppelt so hoch war wie für Gruppe TA sind auch funktionelle Unterschiede denkbar. Hierbei könnte eine Zurückhaltung des Fusionsproteins im Lysosom durch LAMP1 nachteilig sein, da für eine umfangreiche immunogene Wirkung von PA dessen Sekretion und Rezeptorbindung [75][43] essentiell wichtig sind. Eine Verminderung dieses Prozesses durch lysosomal gerichtete Expression könnte somit negative Auswirkung auf die Immunogenität des Vektors gehabt haben. Da PA83 und BclAD1D3 im gleichen Vektor funktional waren und ähnliche Studien mit Kombinationen aus sekretierenden und intrazellulär einschleusenden Vektoren [211][125] keine nachteiligen Wirkungen aufzeigten, ist der Konzentrations- 90 4. Ergebnisse und Diskussion unterschied zwischen Gruppe NS und TA als wahrscheinliche Ursache für die dokumentierten Unterschiede anzunehmen. Da diese Konzentrationsunterschiede auch Gruppe TH betraf und diese äquivalente anti-rPA83 Titer wie Gruppe NS vorwies, ist davon auszugehen, dass das Vorhandensein von PA in voller Länge bei dieser Gruppe die Konzentrationsunterschiede ausgeglichen haben könnte. Trotz der geringen immunogenen Wirkung des Vektors überlebten in Gruppe TA 40 % der Tiere, wobei nur die Überlebenden einen anti-rPA83 IgG-Titer vor der Infektion zeigten. Daher war für diese Gruppe eine signifikante Korrelation der anti-rPA83 und anti-rLF IgG-Titer zum Überleben vorhanden. Die Arbeiten von Brossier et al. (2000) [43] deuteten diesbezüglich an, dass innerhalb einer Lebendvakzine marginale anti-PA Titer die Schutzwirkung von LF erhöhen können, wohingegen bei gänzlicher Abwesenheit von PA kein Schutz durch LF erzielt wurde. Eine ähnliche Beziehung könnte für das Fusionsprotein in beiden Vektoren ebenfalls zutreffen, wenngleich entsprechende Korrelationen aufgrund der klaren serologischen Unterscheidbarkeit zwischen Lebenden und Verstorbenen im Bezug auf die Antikörpertiter vor der Infektion nur für Gruppe TA zu sehen sind. Diese Unterschiede könnten allerdings ebenfalls bei NTC7382-TPALFD1PAD4-mIPS-1 vorhanden, aber durch quantitativ dominantere, für den Schutz aber unerhebliche Antikörperfraktionen überdeckt worden sein. DNA-Vektoren mit BclAD1D3 stimulierten eine sterile Immunität bei 50 % Schutzrate Die verkürzte Sequenz von BclAD1D3 wurde in zwei verschiedenen Vakzinevektoren angewendet. Zum einen flankiert von TPA und LAMP1 (Gruppe TK) und zum anderen mit Ubiquitin (Gruppe TM) als Signalsequenz. Beide Konstrukte erzeugten hohe Antikörpertiter gegen rBclA, welche denen vergleichbarer Proteinanwendungen entsprachen. Einen signifikanten Unterschied zwischen den Konstrukten gab es nicht. Beide zeigten, wie auch die Toxinvektoren, eine hohen IgG1-Dominanz, sodass vektorspezifische Unterschiede bzw. Veränderungen in der Subklassengewichtung durch unterschiedliche Signalsequenzen nicht erkennbar waren. Dies kann auf die Art der Applikation mittels GeneGun zurückzuführen sein, die als Anwendung selbst die Subklassengewichtung stark in Richtung TH2 (IgG1) beeinflusst [91][350]. Damit traf die durch Midha et al. (2009) [211] beschriebene unterschiedliche Immunogenität der beiden Vektoren nicht auf die hier verwendeten Konstrukte zu. Dort zeigte der Vektor pPA63-Ubiquitin im Vergleich zu pTPA-PA63-Lamp1 deutlich niedrigere Titer mit einer stärkeren IgG2a-Gewichtung. Allerdings waren diese Ergebnisse wahrscheinlich nicht durch die Art der Applikation beeinflusst, da dort eine i. m. Injektion vorgenommen wurde. Die im Rahmen dieser Arbeit erzielten Ergebnisse für BclAD1D3 entsprachen bezüglich der erreichten Antikörpertiter und deren Gewichtung einer früheren Studie mit ähnlichem 91 4. Ergebnisse und Diskussion Versuchsaufbau, bei dem die gesamte Sequenz von BclA in einem sekretierenden Vektor erprobt wurde [125]. Die Menge an antigenkodierender DNA war doppelt so hoch wie in den in dieser Arbeit präsentierten Gruppen, erzielte aber keine höheren Titer und erzeugte nur Schutzraten von 20 % bei einer äquivalenten Infektion. Damit übertrafen beide hier getesteten Vektoren mit 50 % Überlebenden die bisher getesteten rein auf BclA basierenden DNA- und Proteinvakzine [39][61], was darauf hindeutet, dass die Deletion der CLR, wie von Liu et al. (2007) [195] beabsichtigt, einen signifikanten Effekt auf die Immunogenität von BclA hatte. Dieser Effekt könnte auch darauf zurückzuführen sein, dass BclAD1D3 durch das Fehlen der CLR nicht trimerisiert [254][32] und so eventuell weitere Epitope zugänglich sind. Ebenso ist davon auszugehen, dass die hier applizierte Menge an antigentragender DNA für eine maximale immunogene Wirkung von BclA ausreichend ist, da auch in anderen Studien mit höheren Konzentrationen keine höheren Titer erzeugt wurden [125]. Von besonderer Bedeutung ist, dass messbare anti-rPA83 IgG-Titer in den Seren aller überlebenden Tiere nach der Infektion abwesend waren. Dies deutet darauf hin, dass die durch die Impfung erzeugte Immunität zur Erkennung und Beseitigung der initialen Sporenlast der Infektion ausreichte, ohne eine wesentliche Etablierung der Infektion und damit einhergehender Germination und Ausschüttung von Toxinen zuzulassen. Die kombinierte DNA-Vakzine TKS vermittelte nahezu vollständigen Schutz gegen eine voll virulente Infektion mit B. anthracis Um herauszufinden, ob eine Kombination aus Toxin- und Sporenantigenen auf Ebene der DNA-Vakzine einen ähnlichen additiven Effekt wie bei den Proteinvakzinen aufweist, sollten die erfolgversprechendsten Vektoren gemeinsam appliziert werden. Als Toxinvektor wurde NTC7382-TPA-LFD1PAD4-mIPS-1 der Gruppe NS ausgesucht. Die Verwendung von pDNAVaccUltra1-TPA-LFD1PAD4-LAMP1 (Gruppe TA) wurde nicht in Erwägung gezogen, da ein rein sekretierender Vektor in Kombination mit einem zusätzlichen, vektorinternen Adjuvans entsprechend der zuvor erörterten Punkte vorteilhafter schien. Beide Vektoren für BclAD1D3 waren gleichermaßen geeignet. Da eine eventuelle negative Wirkung von Ubiquitin [211] auf den Toxinvektor vermieden werden sollte, bekam das Vektorkonstrukt VaccUltra1-TPA-BclAD1D3-LAMP1 (Gruppe TK) den Vorzug. Unter Zusatz des CIITA-kodierenden Vektors als Adjuvans stellte diese Kombinationsgruppe (TKS) nicht nur eine Vakzinekombination, bestehend aus den Antigenen BclA, LF und PA dar, sondern beinhaltete ebenso zwei funktionale Adjuvanzien. Zum besseren Vergleich der Vakzinekombination waren die Konzentrationen der einzelnen Vektoren so gewählt, dass sie denen der Einzelapplikationen entsprachen. 92 4. Ergebnisse und Diskussion Die entsprechend vakzinierte Gruppe TKS zeigte eine Schutzrate von 90 %, wobei das einzige verstorbene Tiere eine verlängerte Überlebenszeit aufwies. Dieser Anstieg der Schutzrate im Vergleich zu den Einzelapplikationen spiegelte sich auch in einer Erhöhung der anti-PA83 IgGTiter wider, obgleich diese weiterhin nicht das Maß der Proteinvakzinen erreichten. Interessanterweise ging diese Erhöhung nicht mit gesteigerten TNA-Titern einher, welche tendenziell sogar niedriger waren. Daher ist davon auszugehen, dass die zusätzlich gebildeten anti-rPA83 Antikörper eher markierende oder germinationshemmende Eigenschaften aufwiesen. Die Erhöhung der Immunogenität gegen PA könnte mit der Anwesenheit sowohl von CIITA als auch von BclAD1D3 in Zusammenhang stehen. Ein entsprechender positiver Einfluss von BclA auf antirPA83 Titer wurde bereits in einer anderen Proteinstudie bemerkt [61]. Bei genauerem Blick auf die Subklassenkorrelationen der Antikörper fällt auf, dass bezüglich der anti-rPA83 Titer nur Gruppe NS mit rs = +0,19 eine ebenso niedrige Korrelation der IgG- und IgG1-Titer zeigte wie die Proteinvakzinen ohne BclA. Dies war allerdings bei den anderen Toxinvektoren aus Gruppe TA und TH mit rs ≥ +0,85 und der Kombinationsgruppe TKS mit rs = +0,77 nicht der Fall. Die entsprechende Verschiebung muss also eher auf die Anwesenheit des zusätzlichen Adjuvans CIITA zurückzuführen sein, statt wie bei den Proteinen auf BclA, und könnte ebenfalls ein Grund für das veränderte Verhältnis von TNA zu anti-rPA83 IgG-Titern in der Kombination sein. Die initial niedrige Korrelation der IgG-/IgG1-Titer der NS Gruppe wiederum könnte mit dem Vektor zusammenhängen. Dieser ist im Gegensatz zu den anderen eingesetzten Vektoren CpG optimiert und sollte somit neben einer verbesserten Antigenpräsentation und der Auslösung der Produktion von Zytokinen und Chemokinen vornehmlich TH1-Zellen aktivieren [173][13][169]. Allerdings sind kaum IgG2a-Titer für Gruppe NS gemessen worden, welche für eine TH1-Antwort sprächen. Sowohl für BclA als auch für LF waren bei allen Einzelanwendungen der Vektoren starke Korrelationen zwischen IgG und IgG1 vorhanden (rs ≥ +0,91). Die Kombinationsgruppe TKS zeigte für BclA eine drastisch verringerte Korrelation zwischen IgG und IgG1 (r s = +0,61) und wies generell deutlich verringerte anti-rBclA Titer auf, die in der Höhe den Antikörpertitern gegen die Toxine entsprachen. Da diesbezüglich in der Kombination nur der Vektor NTC7382TPA-LFD1PAD4-mIPS-1 eine Neuerung darstellt, könnte der beschriebene Effekt der CpGMotive auch hierbei eine Rolle spielen. Eine weitere Begründung für die signifikant niedrigeren Titer gegen rBclA der Gruppe TKS könnte in der Anwendung von Mischpatronen bei der Applikation gelegen haben. Zur Ermöglichung eines vollständigen Zusammenwirkens aller Vektoren sollte die Mischung der Vektoren sicherstellen, dass über den GenGun-Beschuss applizierte Goldpartikel möglichst Kopien aller Vektoren enthalten. Damit sollte gewährleistet werden, dass möglichst alle Vektoren gleichzeitig in einer Zelle vorhanden sind. Bei einer Verteilung der Vektoren auf verschiedene Patronen 93 4. Ergebnisse und Diskussion wäre dies eher unwahrscheinlich gewesen. Das Zusammenwirken mehrerer antigentragender Vektoren in einer Zelle könnte aber auch nachteilig sein, da sie, wie schon im Bezug auf Gruppe NS angedeutet, gegenläufige stimulatorische Signale auslösen könnten. Außerdem wäre, bedingt durch die Kapazität einer Zelle zur Antigenpräsentation, eine Konkurrenz der Antigene möglich. Im Falle einer kapazitätsbedingten Konkurrenz müssten beide Antigene eine gleichstarke Reaktion hervorrufen, wenn keines der Antigene bevorzugt präsentiert wird und eine Absättigung der Antigenpräsentation erreicht ist. Die Ergebnisse von Gruppe TKS sprächen für ein solches Szenario, da die Antikörpertiter gegen rPA83, rLF und rBclA, im Gegensatz zu den Einzelapplikationen der Vektoren, eine ähnliche Höhe haben. Diese These unterstützend zeigten die Versuche von Hahn et al. (2006) [125][24], dass bei einer separaten Applikation der Vektoren für BclA und PA Antikörpertiter erzeugt werden konnten, die denen der Einzelapplikationen derselben Vektoren glichen. Die anti-rLF IgG-Titer der Kombinationsvakzine waren indes unverändert im Vergleich zur Einzelapplikation. Die einzige feststellbare Veränderung war ein Anstieg der Korrelation zwischen TNA-Titern und IgG- bzw. IgG1-Titern (rs ≥ +0,85). Denkbar wäre also, dass die neutralisierenden Eigenschaften der gebildeten Antikörper der Kombinationsvakzine aufgrund der erläuterten Verschiebungen eher auf LF, denn PA zurückzuführen sind, zumal die steigenden anti-rPA83 Titer die TNA-Titer nicht weiter erhöht haben. Generell waren die individuellen Titer in der Gruppe TKS weitaus weniger gestreut, was vermutlich auf die erhöhte Gesamtmenge an applizierter DNA zurückzuführen ist. Ähnliche Ergebnisse erzielten Hahn et al. (2004) [124], wo eine starke Streuung bei niedrigen DNAMengen (~1 µg) durch Applikation von ~5 µg DNA vermindert werden konnte. Die Vereinheitlichung der Titerwerte könnten aber auch auf die Zusammenwirkung der Vektoren und/oder Antigene zurückzuführen sein. Zusammenfassung der Mausversuche Die Erprobung von Vakzinekomponenten im NMRI-Mausmodell für eine NLV ergab sowohl für die DNA- als auch die Proteinvakzinen Kompositionen, deren Immunogenität und Schutzwirkung eine Erprobung im Ziegenmodell rechtfertigten. Dabei wurde nachgewiesen, dass das Kapselkonjugat nichts zur Schutzwirkung beitrug, was vermutlich auf ein immunsuppressives Epitop im Lipopeptid zurückzuführen war. Daher wurde ein potentieller Einsatz innerhalb der Kombinationsvakzine mit diesem Konstrukt nicht weiter verfolgt. Ein äquivalenter Ansatz mit einem anderen integrierten Epitop im Lipopeptid, im Speziellen sogar das in dieser Studie als eigenständiges Adjuvans verwendete Lipopeptid, wären aber für zukünftige Studien denkbar. 94 4. Ergebnisse und Diskussion PA und BclA wirkten einander unterstützend und erzeugten in Kombination eine Schutzwirkung, die höher als die der Einzelanwendung beider Antigene zusammengenommen war. Dieser Schutz wurde durch den Zusatz von FIS komplettiert, wobei das in den FIS vorhandene BclA wahrscheinlich mögliche weitere immunogene Antigene verdeckte. Daher wäre eine FIS Präparation mit einem BclA-defizienten Stamm unter separater Zugabe von rBclA bzw. rBclAD1D3 und rPA83 bzw. einem Kapselkonjugat mit PA eine erfolgversprechende Kombinationsvakzine für zukünftige Anwendungen. Für die nachfolgenden Ziegenversuche konnten diese Erkenntnisse noch nicht umgesetzt werden, sodass je eine Kombination aus rPA83, rBclA und Lipopeptid sowohl mit als auch ohne Zusatz von FIS im Vergleich zur SSLV getestet wurde. Im Vergleich zu den Proteinvakzinen und den auf BclA basierenden Vektoren zeigten die Toxinvektoren generell ca. zehnfach niedrigere Antikörpertiter. Trotzdem konnten durch den Einsatz des Fusionsproteins im Verhältnis zu den anti-Toxin Antikörpertitern ähnliche TNATiter wie in den Proteinvakzinen erzeugt werden. Die Verwendung von BclAD1D3 stimulierte eine sterile Immunität bei 50 % Schutzrate, was alle bisherigen Anwendungen dieser Art übertraf. Durch Kombination der Toxin- und BclA Vektoren konnte auch hier ein nahezu vollständiger Schutz erzeugt werden, wenngleich die Infektionsdosis im Vergleich zur Proteinstudie halbiert war. Auf Grundlage der hier erzielten Ergebnisse wären potentiell alle getesteten Vektoren für einen Einsatz in einer Kombinationsvakzine denkbar, wobei für einen auf PA basierenden Vektor generell ein rein sekretorisches Signal vermutlich vorteilhafter gewesen wäre. Ebenso könnte eine getrennte, aber gleichzeitige Applikation der antigentragenden Vektoren, verteilt auf verschiedene Patronen, sowie eine Erhöhung der Konzentration zur zukünftigen Verbesserung der Wirkung in Erwägung gezogen werden. Für die weiterführenden Tests im Ziegenmodell wurde eine Vektorkombination analog zur Mausgruppe TKS i. m. in weitaus höheren Konzentrationen angewendet. 95 4. Ergebnisse und Diskussion 4.3 Testung von Vakzinekomponenten in Burenziegen Die Vakzinierungs- und Infektionsversuche in Burenziegen fanden in Südafrika unter Kooperation mit der Universität Pretoria und SANParks statt. Entsprechend der Vorversuche im NMRIMausmodell wurden die in Tabelle 15 aufgeführten Gruppen immunisiert. Der Versuchsaufbau umfasste drei Vergleichsgruppen, die mit der SSLV immunisiert wurden (Gruppe 2 und 3 a/b), drei naive Kontrollgruppen (Gruppe 1, NK und LK), zwei Protein-basierende Vakzinegruppen (Gruppe 4 und 6) und eine DNA-basierende Vakzinegruppe (Gruppe 5). Ein separater Wiederholungsversuch der Gruppe 4 und 6, inklusive einer weiteren naiven Kontrollgruppe schloss sich in Zusammenarbeit mit der Kafkas Universität (Türkei) an. Da die Details zur Versuchsdurchführung leicht von den in Südafrika durchgeführten Versuchen abweichen, werden die Wiederholungsversuche im Anschluss gesondert behandelt (siehe Kapitel 4.3.8). Zur serologischen Evaluation der Vakzinen wurden Antikörpertiter gegen rPA83, rBclA, rLF, FIS und eine vegetative Antigenpräparation mittels ELISA bestimmt. Soweit nicht anders vermerkt, wurden nur Titerbestimmungen zur IgG Subklasse vorgenommen. Zur qualitativen Bewertung der Toxin-Antikörpertiter ist außerdem der Neutralisationstiter mittels TNA bestimmt worden. Im Kontrast zu den Mausversuchen wurde auch die Titerentwicklung über die Zeit berücksichtigt. Details zu den entsprechenden Probenahmen finden sich in Tabelle 53 und 54 im Anhang und entsprechend bei den Beschreibungen der Gruppen. Generell wurden alle Individuen mindestens vor dem Beginn der Immunisierung, vor jeder weiteren Immunisierung sowie vor und nach der Infektion für die Untersuchung der Serokonversion geblutet. Neben der Testung der Vakzinekomponenten war der Versuchsaufbau in Südafrika zudem darauf ausgelegt, die Pathogenität des zur Infektion verwendeten Feldisolats SD20 [181] zu verifizieren, sowie dessen minimale letale Dosis (MLD) in Burenziegen einzugrenzen. Zudem war eine detaillierte Betrachtung der Titerentwicklung der SSLV-immunisierten Tiere im Verlauf eines Jahres nebst Revakzinierung von Interesse, da Daten dieser Art für Großtiere und Feldversuche nur bedingt vorhanden oder veraltet sind. Diesbezüglich war auch die Durchführung der Infektion außerhalb der Rahmenbedingungen eines Labors und die Diagnostik auf Grundlage der Interpretation von Verhalten, Temperatur und systemischer Bakterienlast bisher unerprobt und barg für zukünftige Versuche entsprechend verwertbare Ergebnisse. Alle folgenden Daten und Angaben beziehen sich zunächst nur auf die Versuche in Südafrika. 4.3.1 Gesundheitszustand der Ziegen vor der Infektion Nach dem Transport der Ziegen zum Infektionsort, wurde ihr Zustand genau untersucht. Wie in Tabelle 35 ersichtlich wiesen einige Tiere teils durch den Transport (Husten und Schnupfen), teils durch die vorhergehende Haltung (kleinere bilaterale Abszesse) bedingte Krankheitssym96 4. Ergebnisse und Diskussion ptome auf. Einige der Symptome blieben auch nach der Akklimatisierung und während des gesamten Versuchs erhalten und könnten das Ergebnis beeinflusst haben. Die bemerkten Abszesse bei einigen Ziegen waren bereits zu Beginn der Haltung immer wieder aufgetreten und erfahrungsbedingt mit einer persistierende Infektion von Corynebacterium pseudotuberculosis in Verbindung gebracht worden. Ein nachträglicher Test exemplarischer Seren auf Pseudotuberkulose (pTBC) Antikörper (siehe Anhang Tabelle 59) durch Dr. Sting (CVUA Fellbach) bestätigte diese Vermutung insofern, als ein Großteil der Tiere positive Testergebnisse zeigte. Die Beeinträchtigung der Ziegen durch die genannten Krankheitssymptome schien jedoch gering. Auffällig war ebenfalls die gemessene Temperatur der Ziegen, die individuell stark schwankte und bereits vor der Infektion Werte von 37,6 – 40,2 °C annehmen konnte. Aufgrund des begrenzten Platzangebots an der Ver- Tabelle 35: Gesundheitszustand nach Transport suchsanlage wurde die Infektion in zwei Etappen Ziegen Nr. Gruppe durchgeführt. Für die erste Etappe wurden Gruppe 4 II NK 37,6 gesund VI NK 39,0 gesund und 6 sowie Gruppe NK und LK direkt von Pretoria 8202 LK 39,0 leichter Husten zur 8203 LK 38,6 leichter Husten 8185 1 38,9 gesund 8186 1 n.v. verstarb an der Herzwasserkrankheit 8187 1 39,9 leichter Husten 8210 2 38,8 gesund erhofften Verbesserung des Gesundheitszustands nach 8211 2 39,0 gesund 8212 2 39,0 leichter Husten dem Transport wurden die Versuchstiere der zweiten 8213 2 39,0 gesund 8214 2 38,4 leichter Husten Etappe (Gruppen 1 – 3) einige Wochen früher in den 8172 3a 37,7 gesund 8173 3a 40,2 leichter Husten Nationalpark transportiert und in der Nähe der Ver- 8174b 3b n.v. verstarb an der Herzwasserkrankheit suchsanlage versorgt. Die endgültige Überführung zur 8175 3b 37,9 gesund 8176 3a 38,3 leichter Husten Versuchsanlage geschah 14 Tage vor der Infektion. 8178 3a 38,2 gesund 8179 3b 38,8 infiziert mit der Herzwasserkrankheit 8180 3a n.v. gesund 8181 3b 39,0 gesund 8182 3b 38,6 gesund und läufig 8188 4 39,9 leichter Husten raturschwankungen. Zudem ergab sich durch die ver- 8190 4 39,4 tränende Augen 8192 4 39,3 gesund längerte Haltung in diesem Gebiet ein erhöhtes Risiko 8198 4 38,9 gesund 8199 4 39,8 bilaterale Abszesse (Wange) 8191 6 39,7 tränende Augen Etappe zum Verlust von drei Versuchstieren führte. 8194 6 39,0 gesund 8195 6 39,4 gesund Die Infektionsgefahr für die Herzwasserkrankheit 8200 6 38,7 gesund 8201 6 39,3 gesund Versuchsanlage im Krüger Nationalpark (~600 km) verbracht und für 5 – 14 Tage akklimatisiert (Details siehe Tabelle 53 Anhang). Zwecks einer Trotz der umfangreich gestatteten Akklimatisierung an das Gebiet das Krüger Nationalparks zeigten auch diese Tiere die beschriebenen Symptome und Tempe- für eine Herzwassererkrankung, welche in der zweiten bestand dabei nur für die Zwischenhaltung im Nationalpark für den Zeitraum zwischen Transport und Infektion, da dort Zugang zu natürlichem Bewuchs vorhanden war. Die Stallungen für Immunisierung b b Temperatura Zustand nach Transport a rektal gemessen am Tag nach dem Transport b wurde nicht infiziert Gesundheitszustand und rektale Temperatur (°C) der Versuchstiere einen Tag nach dem Transport zur Versuchsanlage; An der Herzwasserkrankheit erkrankte Tiere (in grau) verstarben oder wurden behandelt und vom Infektionsversuch ausgeschlossen; Gruppe 5 ist nicht aufgeführt, da diese nicht infiziert wurde; n.v. nicht vorhanden 97 4. Ergebnisse und Diskussion und Infektion waren betoniert und entsprechend so eingezäunt, dass ein Kontakt zur Bepflanzung nicht möglich war. 4.3.2 Überprüfung der Infektiösität der Sporen im BALB/c Mausmodell Vor dem Beginn der Infektionsversuche an Ziegen mit dem vorbereiteten Feldisolat SD20 musste dessen Virulenz zweifelsfrei festgestellt werden. Dieser war zuvor bereits durch E. Lekota [181] mikrobiologisch und genetisch verifiziert und als voll virulent klassifiziert worden. Zur Bestätigung der Virulenz wurden 8 Wochen alte weibliche BALB/c-Mäuse in zwei Gruppen zu je 3 Tieren mit 500 und 1000 Sporen i. p. mit dem Feldisolat infiziert. Entsprechend der eingesetzten Konzentration sind die Mausgruppen mit M500 und M1000 bezeichnet worden. Einen Tag nach der Infektion waren in Gruppe M500 1 Tier und in M1000 2 Tiere verstorben. Alle übrigen Tiere verstarben bis zum 2. Tag nach der Infektion. Zur Verifizierung der Todesursache wurde allen Tieren durch Herzpunktion Blut entnommen und die Anwesenheit des Erregers mikroskopisch bzw. mittels Ausstrich auf Blutplatten nachgewiesen. 4.3.3 Kontrollgruppen Für den gesamten Versuchsaufbau wurden drei verschiedene Kontrollgruppen mitgeführt. Diese umfassten Gruppe 1 mit 3 Tieren, sowie LK und NK mit je 2 Tieren. Gruppe 1 erhielt einmalig eine Injektion mit 1 ml PBS, dem Verdünnungsmedium der SSLV und wurde während der kompletten Versuchsdauer zusammen mit den SSLV immunisierten Ziegen der Gruppe 3 gehalten und in gleichem Maße beprobt und analysiert, um einen permanenten serologischen Vergleich zu erhalten. Auf Gruppe 1 wird bis auf die für die Infektion relevanten Daten entsprechend in Kapitel 4.3.5.1 ausführlich im Vergleich mit den SSLV immunisierten Gruppen 2 und 3 a/b eingegangen. Die Ziegen der Gruppe LK wurden je einmal s. c. mit je 500 µg Lipopeptid paramunisiert, um etwaige negative Effekte des Adjuvans in Ziegen abzuklären und eine eventuell auftretende Schutzwirkung des Lipopeptids abschätzen zu können. Nebenwirkungen aufgrund der Applikation des Lipopeptids sind weder bei dieser Gruppe noch bei allen weiteren Gruppen, die damit immunisiert wurden, aufgetreten. Bei der Gruppe NK handelte es sich um komplett ungeimpfte Tiere, die zur Verifikation der Virulenz des eingesetzten Feldisolats SD20 in Ziegen und der Abschätzung der zu verwendenden Infektionsdosis dienten. Zum Abgleich zu den eigentlichen Testgruppen wurden alle genannten Kontrollgruppen auf IgG-Antikörper gegen rBclA, rPA83, FIS und vegetatives Antigen sowie TNA-Titer getestet. 98 4. Ergebnisse und Diskussion Die in Tabelle 36 dargestellten Titer-Werte stammen von vereinigten Blutseren der jeweiligen Gruppen, welche direkt vor der Infektion entnommen wurde. Für alle gemessenen Antigene wurden in allen Kontrollgruppen niedrige unspezifische IgG-Titer detektiert. Diese waren in schwankender Höhe bereits vor dem Beginn der Immunisierung und für den gesamten Versuchszeitraum für diese Gruppen und die Tabelle 36: Kontrollgruppen (Ziege) Gruppe rBclA rPA83 TNA FIS 1 162 800 <50 1550 Vegetativ 231 NK 247 271 <50 600 141 LK 247 235 <50 537 141 ELISA IgG- und TNA-Titer der Kontrollgruppen der Ziegenversuche vor der Infektion als vereinigte Seren gemessen oder als gemittelter Wert der Individualmessungen; Titer-Werte ergeben sich als reziproker Wert der letzten Verdünnungsstufe mit einer OD404nm ≥ 0,1. Negativseren der Impfgruppen messbar. Dies betraf auch die nur für die DNA-Vakzinen ermittelten anti-rLF IgG-Titer. Unter Einbeziehung aller gemessenen naiven Seren ergaben sich Schwankungsbreiten von <100 – 2539 für anti-rPA83 IgG, 400 – 1056 für anti-rLF IgG, 162 – 2122 für anti-rBclA IgG, 575 – 1856 für anti-FIS IgG und 158 – 300 für anti-vegetativ IgG. Die gemessenen unspezifischen Hintergrundtiter bei nicht immunisierten Tieren schwankten individuell stark und waren auch im zeitlichen Verlauf nicht einheitlich (siehe hierzu besonders Gruppe 1, Kapitel 4.3.5.1). Daher wurde bei den Einzeldarstellungen jeder Gruppe von einer Normalisierung gegen die gemessenen Werte vor der Immunisierung abgesehen, um die Schwankungen in die gruppeninterne Statistik mit einzubeziehen. Beim Vergleich der Gruppen untereinander wurden die individuellen Titerdaten gegen ihren jeweiligen Titerwert in Woche 0 normalisiert, um eine bessere Vergleichbarkeit zu gewährleisten. 4.3.4 Ermittlung der MLD Zur Eingrenzung der MLD des Feldisolats SD20 und Tabelle 37: Bestimmung der MLD der endgültigen Feststellung der Virulenz des Stammes Gruppe im Ziegenmodell wurde eine Infektion der Kontroll- 1 gruppen mit unterschiedlichen Sporenkonzentrationen Nr. ren, welche s. c. in den Hinterlauf unter Fixierung der Tiere appliziert wurden. Entsprechend der Vorgehensweise innerhalb der Mausversuche waren auch hier überzählig abgefüllte Infektionsdosen zur Bestimmung 8185 8187 Infektionsdosis Überlebenszeit (Sporen) (in h) 36 M NK durchgeführt. Das Infektionsvolumen betrug hierbei 1 ml mit avisierten Dosen von 50, 200 und 1000 Spo- Ziegen II VI 8202 8203 M 83,3 70,3 172 M LK 57,3 66,3 – 67,5 57,5 – 63,5 61,9 – 65,5 844 37,8 63,5 50,7 Überlebenszeit der Kontrollgruppen der Ziegenversuche nach Infektion mit 36, 172 und respektive 844 Sporen des voll virulenten Feldisolats SD20; Der Todeszeitpunkt der NK konnte nicht genau bestimmt werden und ist entsprechend als Zeitfenster angegeben; M – Mittelwert der tatsächlich applizierten Menge an Sporen auszuzählen (Kapitel 3.2.6) und ergaben durchschnittlich 36, 172 und 844 Sporen pro Dosis. Die Ergebnisse der Infektion sind in Tabelle 37 dargestellt. Durch die haltungsbedingte Exponiertheit der Versuchstiere war es möglich, den Zeitpunkt des Todes in den meisten Fällen 99 4. Ergebnisse und Diskussion genau zu bestimmen. Für die Gruppe NK war dies nicht der Fall, sodass entsprechend ein Zeitfenster angegeben wurde. Wenngleich anhand der überlebten Stunden eine Tendenz zu einer verkürzten Überlebenszeit bei steigender Sporenkonzentration erkennbar ist, so unterschieden sich die Überlebensdaten der Kontrollgruppen vermutlich aufgrund der kleinen Gruppengröße nicht signifikant voneinander (siehe Kapitel 4.3.11). Da auch eine Dosis von 36 Sporen spätestens 4 Tage nach der Infektion zum Tode führte, ist davon auszugehen, dass die MLD für diesen Stamm in diesem Tiermodel unter 36 Sporen liegt. Für alle weiteren Versuche durfte die Höhe der Infektionsdosis nicht zu niedrig angesetzt werden, um eine aussagekräftige Diskriminierung zwischen Impf- und Kontrollgruppen und einen schnellen Krankheitsfortschritt (< 3 Tage) in nicht geschützten Tieren zu bewirken. Eine zu stark erhöhte Dosis könnte ebenfalls negative Auswirkungen haben, da die Unterscheidbarkeit der Wirksamkeit verschiedener Impfgruppen beeinträchtigt sein könnte. Daher wurde für alle weiteren Gruppen eine Infektionsdosis von ~850 Sporen ausgewählt. 4.3.5 Einzelbetrachtungen der Immunisierungen von Burenziegen mit der SSLV (Sterne Sporen Lebendvakzine) Die Zielsetzung der Studie war es, alternative Impfkomponenten zur veterinärmedizinisch eingesetzten SSLV zu testen. Daher wurden mehrere Ziegengruppen mit der SSLV immunisiert, um sowohl vergleichende serologische Daten zu erheben als auch einen direkten Vergleich in Bezug auf die Schutzwirkung gegenüber einer tödlichen Infektion zu haben. Hierfür wurden drei Gruppen (Gruppe 2, 3a und 3b) verwendet. Für einen fortdauernden Impfschutz muss die Immunisierung mit der SSLV jährlich aufgefrischt werden, wobei ein Impfschutz bereits 2 – 4 Wochen nach der initialen Immunisierung bestehen soll (Angaben des Herstellers OBP). Aus diesem Grund war die schützende Wirkung der SSLV direkt nach der Vakzinierung, nach dem Verlauf von einem Jahr und direkt nach einer Auffrischungsimpfung von Interesse. Dementsprechend wurden 10 naive Tiere mit der SSLV immunisiert und im Verlauf eines Jahres monatlich geblutet (Gruppe 3). 58 Wochen nach der Erstimmunisierung wurden fünf dieser Tiere zufällig ausgewählt und erneut mit der SSLV immunisiert, sodass sich eine Teilung von Gruppe 3 in 3a (ohne Auffrischungsimpfung) und 3b (mit Auffrischungsimpfung) ergab. Zeitgleich mit der Auffrischungsimpfung wurde eine weitere naive Gruppe von 5 Ziegen ebenfalls mit der SSLV geimpft (Gruppe 2). Zum Zeitpunkt der Infektion waren drei verschieden SSLV-Gruppen vorhanden, welche einmal 62 Wochen zuvor (Gruppe 3a), zweimal 62 Wochen und 4 Wochen zuvor (Gruppe 3b) und einmal 6 Wochen zuvor (Gruppe 2) geimpft worden waren. Bei allen serologischen Betrachtungen wurden die 100 4. Ergebnisse und Diskussion Gruppen 3a und b bis zur 57. Woche nach der ersten Impfung gemeinschaftlich als Gruppe 3 ausgewertet. 4.3.5.1 Ziegengruppe 1 (PBS) Wie zuvor erwähnt (Kapitel 4.3.3), wurden die Ziegen dieser Gruppe zum andauernden serologischen Vergleich zusammen mit Gruppe 3 über den Verlauf von einem Jahr gehalten und jeweils im gleichen Maße beprobt und behandelt. Zum Zeitpunkt der ersten Impfung von Gruppe 3 erhielt Gruppe 1 einmalig eine Injektion s. c. mit PBS. Serologisch dient diese Gruppe dem Negativabgleich zum Rest der Impfgruppen. Da die Messzeitpunkte eng mit den Daten von Gruppe 3 verknüpft sind, werden die Titerdaten (Tabelle 38) hierzu vergleichend in den Abbildungen zu Gruppe 3 dargestellt. Von den 3 Ziegen der Gruppe 1 verstarb Ziege Nr. 8186 zwischen Woche 57 und 62 an der Herzwasserkrankheit und fehlte daher im Infektionsversuch. Die serologischen Daten dieser Ziege wurden bis einschließlich Woche 57 in alle Betrachtungen mit einbezogen. Die übrigen 2 Tiere wurden 62 Wochen nach Beginn des Versuchs mit 36 Sporen des voll virulenten Feldisolats SD20 infiziert. Beide Tiere verstarben innerhalb von 4 Tagen, wobei nur eine der Ziegen vor dem Tod (9 h) eine erhöhte Temperatur zeigte, welche bis zum Tod erhalten blieb und die andere ohne Symptome verstarb. Tabelle 38: Ziegengruppe 1 Antigen / Titer rPA83 Ziegen Nr. W ochen nach 1. Immunisierung 0 4 8 12 16 20 24 28 32 37 48 53 57 62b 8185 758 1058 688 736 353 318 194 223 464 336 360 624 <100 659 64c - 8186a 593 683 752 656 471 318 265 282 360 296 672 896 188 - - 8187 703 2529 736 560 353 259 212 205 360 376 360 368 156 941 - M 685 1423 725 651 392 298 224 237 395 336 464 629 115 800 - S 84 976 33 88 68 34 37 40 60 40 180 264 101 199 - <50 <50 <50 <50 <50 <50 <50 <50 <50 <50 <50 <50 <50 <50 - 236 189 298 298 267 307 280 342 334 246 196 185 165 162 - 1225 1525 1450 1400 1150 1050 1300 1175 1100 1300 1350 1856 1425 1550 - 262 188 188 158 158 158 184 175 156 150 162 285 215 231 - 8185 TNA 8186a 8187 8185 rBclA 8186a 8187 8185 FIS 8186a 8187 8185 Veg. 8186a 8187 a starb zwischen Woche 57 und 62 an der Herzwasserkrankheit und wurde entsprechend nicht infiziert b Probenzeitpunkt direkt vor der Infektion c Probenzeitpunkt nach Beendigung der Infektion ELISA IgG- und TNA-Titer der Ziegengruppe 1 im Verlauf von 64 Wochen gemessen. Titerwerte ergeben sich als reziproker Wert der letzten Verdünnungsstufe mit einer OD 404nm ≥ 0,1. Ausgegraut dargestellt sind alle Tiere die nicht infiziert werden konnten. An der Infektion Verstorbene sind entsprechend orange unterlegt; Detaillierte Todesdaten in hpi sind Tabelle 37 zu entnehmen; M – arithmetischer Mittelwert; S – Standardabweichung 101 4. Ergebnisse und Diskussion Wie in Tabelle 38 aufgeführt, konnten selbst bei nicht immunisierten Tieren über die Dauer von einem Jahr unspezifische IgG-Titer gegen rPA83, rBclA, FIS und vegetatives Vollantigen nachgewiesen werden. Dabei waren die gemessenen Titer, außer für rPA83, bei allen Tests als vereinigte Mischprobe der Gruppe gemessen worden. Für den TNA waren keine Titer oberhalb der Nachweisgrenze messbar. Signifikanzbetrachtungen der gruppeninternen Schwankungen für die anti-rPA83 IgG-Titer zeigten für Woche 4 bis 16, 58, 53 und 62 keine signifikanten Unterschiede zu Woche 0 (p ≥ 0,078), weshalb alle diese Titer als unspezifisch betrachtet wurden. Für alle weiteren Messzeitpunkte, waren die unspezifischen anti-rPA83 IgG-Titer niedriger als die in Woche 0 gemessenen (p ≤ 0,031). 4.3.5.2 Ziegengruppe 2 (SSLV) Eine Gruppe von 5 Ziegen wurde einmal mit der SSLV immunisiert und 6 Wochen später mit 850 Sporen des Tabelle 39: Ziegengruppe 2 Antigen / Titer voll virulenten Feldisolats SD20 infiziert. Es überlebten 3 von 5 Tieren, wobei die Beobachtungszeit nach der Infektion 2 Wochen betrug. Die Probennahmen zur rPA83 serologischen Auswertung umfassten das Negativserum, entnommen vor der Immunisierung (Woche 0), sowie Serum vor (Woche 6) und nach der Infektion W ochen nach 1. Immunisierung Tod in 0 6a 8b dpi 8210 471 9035 90353 - 8211 541 1600 - 7,6 8212 800 2653 256000 - 8213 612 776 - 3,0 8214 1634 5271 49694 - M 812 3867 132016 5,3 S 476 3,3 Ziegen Nr. 3348 109281 8210 112 1643 - 8211 <50 - 7,6 <50 6873 - <50 - 3,0 <50 4848 - - 4455 5,3 3,3 8212 TNA <50 8213 (Woche 8). Alle gemessenen Titer sind in Tabelle 39 8214 M - aufgeführt. Die Negativseren zeigten für alle betrachte- S - - 2637 8210 258 1050 - ten Antigene unspezifische Hintergrundtiter entspre- 8211 235 - 7,6 258 10541 - 305 - 3,0 chend der beschriebenen Kontrollgruppen. Durch die 8212 Veg. 8214 Immunisierung mit der SSLV konnten die anti-rPA83 IgG-Titer tendenziell leicht erhöht werden, wobei der Anstieg nicht signifikant (p = 0,110) und auch nicht für alle Tiere sichtbar war. So zeigten die Ziegen Nr. 8211, 8212 und 8213 6 Wochen nach der Immunisierung Titerwerte (Abbildung 35), die im Bereich der Schwankungsbreite der naiven Seren lagen. Bei der nachfolgenden Infektion verstarben die 2 Tiere mit den nied- 300 8213 M S 800 894 - - 371 4162 5,3 - 241 5525 3,3 rBclA Pool 173 173 196 - FIS Pool 400 2600 5000 - a Probenzeitpunkt direkt vor der Infektion b Probenzeitpunkt nach Beendigung der Infektion ELISA IgG- und TNA-Titer der Ziegengruppe 2 im Verlauf von 8 Wochen gemessen, sowie Eintritt des Todes in dpi. Titerwerte ergeben sich als reziproker Wert der letzten Verdünnungsstufe mit einer OD404nm ≥ 0,1. Die Überwachung des Überlebens erfolgte bis 14 dpi. Überlebende sind entsprechend grün hinterlegt, Verstorbene orange; M – arithmetischer Mittelwert; S – Standardabweichung rigsten anti-rPA83 IgG-Titern, entsprechend war eine Korrelation der Titer mit der Überlebenszeit vorhanden (rs = +0,9). Alle weiteren gemessenen Titer vor der Infektion, davon rBclA und FIS als vereinigte Mischprobe der Gruppe gemessen, 102 4. Ergebnisse und Diskussion zeigten keine bzw. nur leichte Veränderungen im Vergleich zu vor der Immunisierung (p ≥ 0,374, Abbildung 36). Eine sichtbare weitere Erhöhung der gemessenen Titer, exklusive der anti-rBclA IgG Titer war erst nach dem Überleben der Infektion zu sehen. Aufgrund der geringen Gruppengröße waren diese zwar nicht signifikant (p ≥ 0,1), aber deutlich verschieden zu den zuvor gemessenen Werten. Dies war besonders für die anti-rPA83 IgG- und TNA-Titer auf- Abbildung 35: Ziegengruppe 2 (anti-rPA83 IgG-Titer) fällig, die 10 – 100fach höhere Werte als vor Halblogarithmische Auftragung der anti-rPA83 IgG-Titer; Mittelwerte sind durch gepunktete Linien miteinander verknüpft; Grüne Pfeile deuten den Zeitpunkte der Immunisierung, schwarze Pfeile geben den Zeitpunkt der Infektion an. Die y-Achse ist logarithmisch dargestellt. der Infektion aufwiesen. Die IgG-Titer gegen das vegetative Antigen waren durch die Imp- fung unverändert, zeigten aber abhängig vom Individuum deutliche Erhöhungen durch die Infektion. Die gemessenen anti-FIS IgG-Titer waren sowohl durch die Immunisierung als auch durch die Infektion leicht erhöht, wobei die Messung einer vereinigten Mischprobe der Gruppenseren diesbezüglich fehleranfällig ist, da eine Mittlung aus zunächst fünf und nach der Infektion drei Individuen vorliegt. Daher könnte es sein, dass die sichtbare Erhöhung der Titer nach der Infektion nur durch den Wegfall der möglicherweise niedrigeren Titer der Verstorbenen zustande kommt. Eine merkliche Reaktion gegen rBclA konnte weder Abbildung 36: Ziegengruppe 2 (anti-FIS, -Vegetativ, -rBclA und TNA-Titer) durch die Immunisierung, noch durch die In- Lineare Darstellung der Antikörpertiter; Mittelwerte sind durch gepunktete Linien miteinander verknüpft; Grüne Pfeile deuten den Zeitpunkte der Immunisierung, schwarze Pfeile geben den Zeitpunkt der Infektion an. fektion hervorgerufen werden, was aufgrund der zumindest leicht erhöhten anti-FIS IgGTiter bemerkenswert ist. 103 4. Ergebnisse und Diskussion 4.3.5.3 Ziegengruppe 3a/b (SSLV) Eine Gruppe von 10 Ziegen wurde mit der SSLV immunisiert, im Verlauf von einem Jahr ca. alle 4 Wochen geblutet und das so gewonnene Serum auf Antikörper- und TNA-Titer untersucht. In der 58. Woche nach der ersten Impfung wurden 5 der 10 Tiere zufällig ausgewählt und erneut mit der SSLV immunisiert (3b). Die restlichen 5 Tiere blieben ohne Revakzinierung (3a). Für die grafische Darstellung und die gemittelten Werte wurden daher Gruppe 3a und b bis zur Trennung durch die Revakzinierung von 3b gesamtheitlich als Gruppe 3 betrachtet. Zur besseren Übersicht wird Gruppe 3a in den Abbildungen farblich wie zuvor die Gesamtgruppe 3 in Blautönen fortgeführt und 3b nach der Trennung in Rottönen. Bis zur Trennung waren die Mitglieder von Gruppe 3a und b für alle individuell gemessenen Antikörpertiter serologisch nicht voneinander zu unterscheiden (p ≥ 0,283). Zusätzlich zu Gruppe 3 sind in den Abbildungen auch die Daten von Gruppe 1 (PBS) für Vergleichszwecke abgebildet. Während der Akklimatisierungsphase im Krügerpark infizierten sich 2 Ziegen der Gruppe 3b (Nr. 8174 und 8179) mit der Herzwasserkrankheit, woran Nr. 8174 verstarb und Nr. 8179 erfolgreich behandelt werden konnte. Beide schieden zur Verwendung bei der Infektion aus, wurden aber serologisch in die Betrachtungen integriert. Die Infektion der restlichen 8 Tiere erfolgte in Woche 62 mit 850 Sporen des voll virulenten Feldisolats SD20, wobei die Tiere für weitere 2 Wochen auf den Ausgang der Infektion überprüft wurden. Die Infektion überlebten 4 von 5 Tieren der Gruppe 3a, wobei das verstorbene Tier eine verlängerte Überlebenszeit von 10 Tagen aufwies. Aus der revakzinierten Gruppe 3b überlebten drei von drei der infizierten Ziegen. Anti-rPA83 IgG-Titer und TNA-Titer In Tabelle 40 sind die gemessenen anti-rPA83 IgG-Titer von Gruppe 3 im Verlauf eines Jahres aufgeführt. Durch die erste Impfung konnte nach 4 Wochen bei allen Individuen ein deutlicher, signifikanter Anstieg (p ≤ 0,002) der anti-rPA83 IgG-Titer im Vergleich zu den Seren vor der Immunisierung gemessen werden. In Abbildung 37 ist gut zu sehen, dass zu diesem Zeitpunkt (Woche 4) die Einzelwerte sehr stark voneinander abwichen und somit kein einheitlicher IgGTiter gegen rPA83 vorhanden war. Da für den Zeitraum zwischen Impfung und Woche 4 keine weitere Probennahme erfolgte, ist zudem unklar, ob der sichtbare Höhepunkt in Woche 4 tatsächlich den Spitzentiter angibt oder bereits abfallende Titerwerte beinhaltet. Zumindest fügt sich ein weiter sinkender Trend für die anti-rPA83 IgG-Titer der Wochen 8, 12 und 16 an, die sich jeweils signifikant vom vorhergehenden Messzeitpunkt unterscheiden (p ≤ 0,04). Diese verbleiben ab Woche 16 auf einem relativ konstanten, im Vergleich zu Gruppe 1 leicht erhöhtem Niveau und verändern sich erst mit der nächsten Immunisierung bzw. der Infektion. 104 4. Ergebnisse und Diskussion Tabelle 40: Ziegengruppe 3a/b (anti-rPA83 IgG-Titer) Ziegen Nr. 3a 3b W ochen nach 1. Immunisierung 0 4 8 12 16 20 24 28 32 37 48 53 57c 62d 64e 8172 1379 24361 8704 5632 2918 2729 1882 2259 3200 3200 3200 4992 3712 5271 7529 8173 551 82580 14848 6400 6965 7529 2776 3765 3800 3200 3200 4992 1536 5271 87341 8176 965 44593 15872 5632 5271 5835 2071 4141 6400 6144 10338 4224 7138 15812 24847 8178 620 44593 7680 3200 2541 1741 671 1035 992 928 800 1056 800 1412 96376 8180 510 15277 4864 3200 1788 1600 1247 1882 1600 1856 1984 1856 1984 2653 - 8174a 303 51200 8704 5632 4518 4800 2588 3059 4736 7168 9088 6154 5046 - - 8175 1158 51200 11264 4480 3765 2729 2682 1412 1536 1856 2880 3008 2880 39153 24847 8179b 882 21058 6400 5632 4518 2729 2588 1318 2176 3200 2880 4224 3968 20329 - 8181 482 35509 7680 5120 2918 2729 2447 5365 2176 2436 2880 3200 800 39153 24847 8182 303 135432 23040 12288 4518 4800 3953 2635 4992 5632 5292 2560 800 M 715 50580 10906 5722 3972 3722 2291 2687 3161 3562 4254 3627 2866 S 365 35479 5527 2544 1514 1934 899 1400 1778 2065 a starb zwischen Woche 57 und 62 an der Herzwasserkrankheit und wurde entsprechend nicht infiziert b erkrankte zwischen Woche 62 und 64 an der Herzwasserkrankheit und wurde entsprechend nicht infiziert c Probenzeitpunkt vor der 2. Immunisierung von 3b in Woche 58 d Probenzeitpunkt vor der Infektion e Probenzeitpunkt nach Beendigung der Infektion 3099 1573 2116 31624 12800 6084 (3a) 54023 (3a) 32565 (3b) 20831 (3b) 5691 (3a) 44410 (3a) 8896 (3b) 6955 (3b) ELISA IgG-Titer der Ziegengruppe 3a/b im Verlauf von 64 Wochen gemessen; Titerwerte ergeben sich als reziproker Wert der letzten Verdünnungsstufe mit einer OD404nm ≥ 0,1. Ausgegraut dargestellt sind alle Tiere, die nicht infiziert werden konnten; Die Überwachung des Überlebens erfolgte bis 14 dpi. Überlebende sind entsprechend grün hinterlegt, Verstorbene orange; M – arithmetischer Mittelwert; S – Standardabweichung Abbildung 37: Ziegengruppe 3a/b und 1 (anti-rPA83 IgG-Titer) Halblogarithmische Darstellung der anti-rPA83 IgG-Titer (Tabelle 40) von Gruppe 3 a/b und 1 im Verlauf eines Jahres; Bis zur revakzinierung von Gruppe 3b wurde Gruppe 3a/b gesamtheitlich betrachtet (Gruppe 3, Blautöne); Nach der Splittung in Woche 57 wird Gruppe 3a in Blautönen weitergeführt und ab diesem Zeitpunkt entsprechend in Rottönen die revakzinierte Gruppe 3b. Die Rohdaten für Gruppe 1 (grau) sind Tabelle 38 zu entnehmen; Für jeden Messzeitpunkt sind die Einzelwerte aller Individuen angegeben, sowie durch einen horizontalen Strich, deren Mittelwerte, die durch gepunktete Linien miteinander verknüpft sind; Grüne Pfeile deuten den Zeitpunkte der Immunisierung, schwarze Pfeile geben den Zeitpunkt der Infektion an. Die y-Achse ist logarithmisch aufgeteilt. Werten unterhalb der Nachweisgrenze wurde ein Wert von 100, für eine bessere Darstellung zugewiesen. 105 4. Ergebnisse und Diskussion Die zweite Immunisierung von Gruppe 3b (rot), 58 Wochen nach der ersten Immunisierung, erzeugte in allen Tieren eine signifikante Erhöhung (p = 0,006) der anti-rPA83 IgG-Titer. Diese entsprachen der Höhe nach dem Titer nach der ersten Impfung in Woche 4 (p = 0,347) und stiegen durch die nachfolgende Infektion nicht weiter an, sondern fielen signifikant ab (p = 0,009). Für die nicht revakzinierte Gruppe 3a (blau) blieben die Titer-Werte bis Woche 62 konstant und wurden erst durch die überlebte Infektion auf ein ähnliches Niveau wie das von Gruppe 3b vor der Infektion bzw. das in Woche 4 erreichte Niveau angehoben (p ≥ 0,272). Da Ziege Nr. 8172 von Gruppe 3a durch die überlebte Infektion kaum erhöhte Titer im Vergleich zum vorhergehenden Zeitpunk aufwies, waren die Titer von Gruppe 3a in Woche 64 nicht signifikant gegenüber Woche 0 (p = 0,098) erhöht. Vergleicht man die individuellen Titer nach der ersten und zweiten Impfung fällt auf, dass die beobachtete starke Streuung der erzeugten Titer nach der zweiten Immunisierung (Gruppe 3b, rot) geringer ausfällt. Im Gegensatz dazu sind die Titer von Gruppe 3a (blau) nach einer überlebten Infektion wieder stark gestreut. Ebenfalls ungewöhnlich sind die Titer-Werte der Ziegen Nr. 8172 und 8173 62 und 64 Wochen nach der Immunisierung. Obwohl beide vor der Infektion den gleichen Ausgangstiter aufwiesen und diese auch überlebten, kam es bei Nr. 8173 zu einem erheblichen Anstieg der anti-rPA83 IgG-Titer durch die Infektion und bei Nr. 8172 nicht. In Tabelle 41 sind die gemessenen TNA-Titer aufgeführt. Aufgrund der niedrigen anti-rPA83 IgG-Titer wurden alle TNA-Titer der Wochen 0 bis 57 als vereinigte Mischprobe der Gruppe bestimmt, da Titer über der Nachweisgrenze nicht zu erwarten waren. In Abbildung 38 sind die aufgeführten Titer noch einmal grafisch gegen die gemessenen Titer von Gruppe 1 dargestellt. In Woche 4 nach der ersten Impfung konnte nur ein leicht erhöhter TNA-Titer gemessen werden, dieser war in Anbetracht der Höhe der generierten anti-rPA83 IgG-Titer (~50000) vergleichsweise gering. Im nachfolgenden Zeitraum bis einschließlich zur 32. Woche waren keine TNA-Titer messbar (<50). Erstaunlicherweise konnten ab Woche 37 bis zur Revakzinierung bzw. Infektion wieder fortdauernd leicht erhöhte TNA-Titer (>100) gemessen werden, obwohl die anti-rPA83 IgG-Titer für diesen Zeitraum konstant und niedrig waren (~3500). Durch die zweite Immunisierung in Woche 58 konnten bei Gruppe 3b (Abbildung 38, rot) signifikant erhöhte TNA-Titer erzeugt werden (p = 0,002), welche tendenziell höher als die TNA-Titer der ersten Immunisierung waren. In einem ähnlichen Maße waren die TNA-Titer von Gruppe 3a (blau) nach der Infektion erhöht. Somit wurden sowohl bei der zweiten Immunisierung von Gruppe 3b, als auch der Infektion von Gruppe 3a ähnliche anti-rPA83 IgG-Titer wie bei der ersten Immunisierung erzeugt (~30000 – 50000), der TNA-Titer war aber im Vergleich um ein ~Zehnfaches erhöht. 106 4. Ergebnisse und Diskussion Tabelle 41: Ziegengruppe 3a/b (TNA-Titer) Ziegen Nr. W ochen nach 1. Immunisierung 62d 64e 149 149 8173 74 2042 8176 285,5 348 8178 <50 2839 86,5 - 0 4 8 12 16 20 24 28 32 37 48 53 57c 8172 3a 8180 8174a <50 248 <50 <50 <50 <50 <50 <50 <50 100 149 100 100 8175 3b - - 1488 597 8179b 546 - 8181 1860 896 8182 M S - - - - - - - - - - - a starb zwischen Woche 57 und 62 an der Herzwasserkrankheit und wurde entsprechend nicht infiziert b erkrankte zwischen Woche 62 und 64 an der Herzwasserkrankheit und wurde entsprechend nicht infiziert c Probenzeitpunkt vor der 2. Immunisierung von 3b in Woche 58 d Probenzeitpunkt vor der Infektion e Probenzeitpunkt nach Beendigung der Infektion - - 1265 398 119 (3a) 1345 (3a) 1290 (3b) 630 (3b) 107 (3a) 1309 (3a) 553 (3b) 251 (3b) TNA-Titer der Ziegengruppe 3a/b im Verlauf von 64 Wochen gemessen; TNA-Titerwerte ergeben sich als reziproker Wert der Verdünnungsstufe bei der 50 % Neutralisation gemessen wurden. Ausgegraut dargestellt sind alle Tiere, die nicht infiziert werden konnten; Die Überwachung des Überlebens erfolgte bis 14 dpi. Überlebende sind entsprechend grün hinterlegt, Verstorbene orange; M – arithmetischer Mittelwert; S – Standardabweichung Abbildung 38: Ziegengruppe 3a/b und 1 (TNA-Titer) Lineare Darstellung der TNA-Titer (Tabelle 41) von Gruppe 3a/b und 1 im Verlauf eines Jahres; Bis zur revakzinierung von Gruppe 3b wurde Gruppe 3a/b gesamtheitlich betrachtet (Gruppe 3, Blautöne); Nach der Splittung in Woche 57 wird Gruppe 3a in Blautönen weitergeführt und ab diesem Zeitpunkt entsprechend in Rottönen die revakzinierte Gruppe 3b. Die Rohdaten für Gruppe 1 (grau) sind Tabelle 38 zu entnehmen; Für einige Messzeitpunkte sind die Einzelwerte aller Individuen angegeben, sowie durch einen horizontalen Strich, deren Mittelwerte, die durch gepunktete Linien miteinander verknüpft sind; Grüne Pfeile deuten den Zeitpunkte der Immunisierung, schwarze Pfeile geben den Zeitpunkt der Infektion an. Zur besseren Darstellung wurde die y-Achse mit einer Unterbrechung im Bereich 2100 – 2800 versehen. 107 4. Ergebnisse und Diskussion Analog zu den anti-rPA83 IgG-Titern konnten durch die Infektion die TNA-Titer von Gruppe 3b nicht weiter erhöht werden. Eine signifikante Korrelation der anti-rPA83 IgG-Titer zu den TNA-Titern war nicht vorhanden, da die Signifikanzschwelle aufgrund der Gruppengröße sehr hoch war. Dennoch war eine Korrelation in Tendenz für beide Gruppen vor der Infektion erkennbar (rs ≥ +0,825). Anti-rBclA und anti-FIS IgG-Titer In Tabelle 42 sind die gemessenen anti-rBclA und anti-FIS IgG-Titer aufgeführt, deren grafische Darstellung sich in Abbildung 39 im Vergleich zu Gruppe 1 befindet. Alle Titer wurden als vereinigte Mischprobe der Gruppe gemessen. Sowohl FIS als auch rBclA sind Sporenantigene, wobei im vorliegenden Fall BclA auch Bestandteil der FIS-Präparation war, da die dazu verwendeten Sporen BclA exprimierten. Da auch die Immunisierung mit der SSLV auf Sporen basiert, war daher eine immunogene Reaktion auf beide Antigene zu erwarten. Tabelle 42: Ziegengruppe 3a/b (anti-rBclA und -FIS IgG-Titer) W ochen nach 1. Immunisierung Antigen rBclA FIS 0 3a 3b 3a 3b 298 1400 4 307 13600 8 347 9813 12 400 5801 16 667 4906 20 702 5409 a Probenzeitpunkt vor der 2. Immunisierung von 3b in Woche 58 b Probenzeitpunkt vor der Infektion c Probenzeitpunkt nach Beendigung der Infektion 24 471 5180 28 533 3200 32 533 2742 37 277 2971 48 331 5638 53 154 3200 57a 62b 64c 647 752 188 200 223 200 2628 2742 4400 3961 32426 20000 ELISA IgG-Titer der Ziegengruppe 3a/b im Verlauf von 64 Wochen gemessen; Titerwerte ergeben sich als reziproker Wert der letzten Verdünnungsstufe mit einer OD 404nm ≥ 0,1. Die Überwachung des Überlebens erfolgte bis 14 dpi. Angegeben sind Werte von vereinigten Mischproben der Gruppe. Wie zu erkennen ist, konnten weder durch die Impfungen, noch die Infektion, die ebenfalls mit Sporen stattgefunden hat, erhöhte Titer gegen rBclA erzeugt werden. Somit blieben die IgGTiter gegen rBclA für den gesamten Versuchszeitraum konstant auf dem Niveau von Gruppe 1 (Abbildung 39, links) und den Werten, die vor der Immunisierung gemessen wurden. Im Vergleich dazu konnten durch die erste Immunisierung hohe anti-FIS IgG Titer erzeugt werden. Diese verhielten sich im weiteren Verlauf ähnlich den gemessenen Titern gegen rPA83 und fielen schrittweise nach 8, 12 und 16 Wochen auf einen niedrigen Wert. Ab Woche 16 verblieben die Werte auf einem konstanten Niveau, das höher lag als die Werte von vor der Immunisierung und der Kontrollgruppe im gesamten Jahresverlauf (Gruppe 1, grau). Wie in Abbildung 39 (rechts, rot) gut zu sehen ist, konnte die zweite Immunisierung von Gruppe 3b in Woche 58 den IgG-Titer gegen FIS noch weiter erhöhen, womit er ca. dreimal höher lag als der Titer nach der ersten Immunisierung. Wie auch schon für die anti-rPA83 IgG-Titer bemerkt, wurden die anti-FIS IgG-Titer bei Gruppe 3b durch die Infektion nicht weiter erhöht, sondern fielen in Woche 64 auf ein Niveau 108 4. Ergebnisse und Diskussion ab, das dem der ersten Immunisierung entsprach. Auffallend ist hierbei, dass in Gruppe 3a nach dem Überleben der Infektion kein bzw. nur ein marginal erhöhter anti-FIS IgG-Titer messbar war. Da im Gegensatz zu rBclA eine Veränderung der anti-FIS IgG-Titer durch die Immunisierungen detektierbar war, muss die Immunreaktion gegen FIS auf anderen Antigenen als BclA beruhen. Abbildung 39: Ziegengruppe 3a/b und 1 (anti-rBclA und -FIS IgG-Titer) Lineare Darstellung der anti-rBclA (A) und -FIS IgG-Titer (B) (Tabelle 42) von Gruppe 3a/b und 1 im Verlauf eines Jahres; Bis zur revakzinierung von Gruppe 3b wurde Gruppe 3a/b gesamtheitlich betrachtet (Gruppe 3, Blautöne); Nach der Splittung in Woche 57 wird Gruppe 3a in Blautönen weitergeführt und ab diesem Zeitpunkt entsprechend in Rottönen die revakzinierte Gruppe 3b. Die Rohdaten für Gruppe 1 (grau) sind Tabelle 38 zu entnehmen; Für jeden Messzeitpunkt sind Messwerte aus vereinigten Mischproben der Gruppe als horizontaler Striche angegeben und durch gepunktete Linien miteinander verknüpft; Grüne Pfeile deuten den Zeitpunkte der Immunisierung, schwarze Pfeile geben den Zeitpunkt der Infektion an. Anti-vegetativ IgG-Titer Das vegetative Antigen stellt ein undefiniertes Zelllysat eines unbekapselten, toxindefizienten Stammes von B. anthracis dar. Entsprechend kann damit eine Immunantwort gegen die vegetative Form des Bakteriums unabhängig von den Toxinen und der Kapsel detektiert werden. In Tabelle 43 und Abbildung 40 sind die gemessenen IgG-Titer von Gruppe 3a/b gegen das vegetatives Antigen aufgeführt. Durch die erste Immunisierung mit der SSLV konnten die IgG-Titer im Vergleich zur Negativkontrolle nur leicht erhöht werden, wobei das so entstandene Niveau bis zur nächsten Impfung erhalten blieb. Durch eine weitere Immunisierung mit der SSLV in Woche 58 (Gruppe 3b) konnten ca. zehnfach höhere IgG-Titer gegen vegetatives Antigen erzeugt werden. Diese wurden durch die nachfolgende Infektion in Woche 62 nicht weiter erhöht (Gruppe 3b). Erstaunlicherweise zeigte Gruppe 3a ebenso wie für die anti-FIS IgG-Titer durch die überlebte Infektion keine bzw. nur marginal erhöhte Titer gegen das vegetatives Antigen. 109 4. Ergebnisse und Diskussion Tabelle 43: Ziegengruppe 3a/b (anti-vegetativ IgG-Titer) Ziegen Nr. W ochen nach 1. Immunisierung 62d 64e 1500 941 8173 682 1411 8176 1317 941 8178 564 5082 447 - 0 4 8 12 16 20 24 28 32 37 48 53 57c 8172 3a 8180 8174a 215 1323 1046 1046 1046 1600 1600 1600 1600 1600 1969 1785 1508 8175 3b - - 7529 5553 8179b 8282 - 8181 10917 8658 8182 M S - - - - - - - - - - - a starb zwischen Woche 57 und 62 an der Herzwasserkrankheit und wurde entsprechend nicht infiziert b erkrankte zwischen Woche 62 und 64 an der Herzwasserkrankheit und wurde entsprechend nicht infiziert c Probenzeitpunkt vor der 2. Immunisierung von 3b in Woche 58 d Probenzeitpunkt vor der Infektion e Probenzeitpunkt nach Beendigung der Infektion - - 21083 10164 902 (3a) 2094 (3a) 11953 (3b) 8125 (3b) 474 (3a) 2004 (3a) 6258 (3b) 2351 (3b) Anti-vegetativ IgG-Titer der Ziegengruppe 3a/b im Verlauf von 64 Wochen gemessen; Titerwerte ergeben sich als reziproker Wert der letzten Verdünnungsstufe mit einer OD 404nm ≥ 0,1. Ausgegraut dargestellt sind alle Tiere, die nicht infiziert werden konnten; Die Überwachung des Überlebens erfolgte bis 14 dpi. Überlebende sind entsprechend grün hinterlegt, Verstorbene orange; M – arithmetischer Mittelwert; S – Standardabweichung Abbildung 40: Ziegengruppe 3a/b (anti-vegetativ IgG-Titer) Lineare Darstellung der anti-vegetativ IgG-Titer (Tabelle 43) von Gruppe 3a/b und 1 im Verlauf eines Jahres; Bis zur revakzinierung von Gruppe 3b wurde Gruppe 3a/b gesamtheitlich betrachtet (Gruppe 3, Blautöne); Nach der Splittung in Woche 57 wird Gruppe 3a in Blautönen weitergeführt und ab diesem Zeitpunkt entsprechend in Rottönen die revakzinierte Gruppe 3b. Die Rohdaten für Gruppe 1 (grau) sind Tabelle 38 zu entnehmen; Für einige Messzeitpunkte sind die Einzelwerte aller Individuen angegeben, sowie durch einen horizontalen Strich, deren Mittelwerte, die durch gepunktete Linien miteinander verknüpft sind; Grüne Pfeile deuten den Zeitpunkte der Immunisierung, schwarze Pfeile geben den Zeitpunkt der Infektion an. Zur besseren Darstellung wurde die y-Achse mit einer Unterbrechung im Bereich 12500 – 19500 versehen. 110 4. Ergebnisse und Diskussion 4.3.6 Zusammenfassung der SSLV-Gruppen Für Gruppe 3a/b erzeugte die erste Impfung nach 4 Wochen äquivalent hohe anti-rPA83 IgGTiter (p = 0,501) wie die zweite Impfung (Gruppe 3b, Woche 62). Die Antikörpertiter gegen FIS und das vegetative Antigen sowie die TNA-Titer waren nach der zweiten Immunisierung aber tendenziell höher als nach der ersten, sodass die Auffrischungsimpfung nach einem Jahr eine bessere immunogene Wirkung erzielte. Die Reaktion auf die Infektion war in Gruppe 3a und 3b ebenfalls unterschiedlich. Während sämtliche Titerwerte der frisch revakzinierten Gruppe 3b nach der Infektion (Woche 64) konstant blieben oder abfielen, waren für die Mehrheit der Überlebenden von Gruppe 3a im Vergleich zu den vor der Infektion gemessenen Werten die anti-rPA83 IgG- und TNA-Titer tendenziell erhöht. Alle weiteren Titer verblieben auf dem Niveau von vor der Infektion. Betrachtet man im Fall von Gruppe 3a die überlebte Infektion als zweite Immunisierung mit einer Lebendvakzine und vergleicht die immunologische Reaktion darauf mit einer zweiten SSLV Immunisierung (Gruppe 3b), ergeben sich ebenfalls Unterschiede. Die anti-rPA83 IgGund TNA-Titer bei Gruppe 3b nach der Revakzinierung (Woche 62) und Gruppe 3a nach der Infektion (Woche 64) stiegen für beide Gruppen in gleicher Weise an (p ≥ 0,380), wohingegen eine Erhöhung der anti-FIS und anti-vegetativ IgG Titer nur bei der Revakzinierung mit der SSLV (Gruppe 3b, Woche 62) zu sehen ist. Damit unterscheiden sich in ihrer Immunogenität sowohl die Erst- und Zweitimmunisierung mit der SSLV, als auch die Infektion mit einem voll virulentem Stamm voneinander. Gruppe 2 erhielt die gleiche Impfung wie Gruppe 3 und sollte sich daher von Woche 0 bis 6 serologisch analog zu dieser verhalten. Tatsächlich lagen die gemessenen Antikörpertiter für Gruppe 2 weit unter denen von Gruppe 3, wobei einzelne Tiere von Gruppe 2 Antikörpertiter aufwiesen, die denen der Kontrollgruppen glichen. Leider war es nicht möglich, Gruppe 2 analog zu Gruppe 3 in Woche 4 zu beproben, um die Seren für diesen Zeitpunkt direkt zu vergleichen. Da der Titerverlauf von Gruppe 3 eine rapide Abnahme der Antikörpertiter ab Woche 4 angibt, könnte der um 2 Wochen verspätete Probenzeitpunkt von Gruppe 2 als Begründung für die niedrigeren Titer angesehen werden. Ein Vergleich der anti-rPA83 IgG-Titer von Gruppe 2 (Woche 6) mit den in Woche 4 und 8 gemessenen Titern der Gruppe 3 ergibt allerdings für beide Zeitpunkte signifikant niedrigere Titer (p ≤ 0,022) für Gruppe 2. Eine ähnliche Tendenz war auch für alle weiteren Antikörpertiter dieser Gruppen zu diesen Zeitpunkten sichtbar. Damit sind die generell niedrigeren Titer von Gruppe 2 nicht auf den späteren Probenzeitpunkt zurückzuführen, wonach die Unterschiede zwischen Gruppe 2 und 3 nach der ersten Immunisierung andere Ursachen haben müssen. 111 4. Ergebnisse und Diskussion 4.3.7 Einzelbetrachtungen der Immunisierungen von Burenziegen mit NLV Der SSLV standen die Gruppen zur Testung der NLV gegenüber, die sich aus den Ergebnissen der Mausversuche ergaben. Die Ziegengruppen 4 und 6 wurden entsprechend dreimal im Abstand von 3 Wochen mit rPA83, rBclA und Lipopeptid bzw. rPA83, rBclA, FIS und Lipopeptid immunisiert und 4 Wochen nach der letzten Immunisierung s. c. mit 844 Sporen des voll virulenten Feldisolats SD20 infiziert. Ebenso wurde an Ziegengruppe 5 die analoge Kombination aus DNA-Vektoren der Mausgruppe TKS getestet, die ebenfalls dreimal im Abstand von 3 Wochen appliziert wurde. Da für diese Gruppe schon im Verlauf der Immunisierung kaum erhöhte Titer festgestellt werden konnten, wurde 9 Wochen nach der letzten Impfung eine Auffrischungsimpfung in Form des eingesetzten Proteingemischs von Gruppe 6 appliziert. Da auch diese keine zufriedenstellenden Titer erzeugte, wurde von einer Infektion von Gruppe 5 zum Schutz der Tiere gänzlich abgesehen. 4.3.7.1 Ziegengruppe 4 (rPA83, rBclA und Lipopeptid) Eine Gruppe von 5 Ziegen wurde dreimal im Abstand von 3 Wochen mit rPA83 (25 µg), rBclA (25 µg) und Lipopeptid (500 µg) s. c. immunisiert und 4 Wochen nach der letzten Immunisierung s. c. mit 844 Sporen des voll virulenten Feldisolats SD20 infiziert. Die Applikation der einzelnen Impfkomponenten erfolgte nicht wie geplant in Mischung, sondern jeweils einzeln in einem Volumen von 1 ml in das gleiche Areal. Serumentnahmen fanden vor dem Beginn der Immunisierung (Woche 0), nach jeder Immunisierung (Woche 3, 5 und 8) und direkt vor der Infektion (Woche 10) statt. Die serologische Auswertung umfasste die Ermittlung von Antikörpertitern gegen rPA83, rBclA, FIS und vegetativem Antigen, sowie TNA-Titern (Tabelle 44). Wie schon zuvor beobachtet, waren auch bei diesen Individuen bereits vor dem Start der Immunisierungen niedrige unspezifische Titer gegen die jeweiligen Antigene messbar. Besonders auffällig war hierbei Ziege Nr. 8188, die mit Abstand den höchsten gemessenen, unspezifischen Hintergrundtiter gegen rBclA aufwies und auch im weiteren Verlauf der Immunisierung beibehielt. Für alle anderen Mitglieder dieser Gruppe änderten sich die anti-rBclA IgG-Titer durch die Immunisierung ebenfalls kaum merklich (Abbildung 41, rechts), sodass ein signifikanter Anstieg der anti-rBclA IgG-Titer nicht ersichtlich war (p ≥ 0,104). Korrelierend zu diesen Ergebnissen konnten auch mit FIS nur marginal erhöhte anti-rBclA Titer detektiert werden, die sich weitestgehend im Rahmen der zuvor gemessenen unspezifischen Hintergrundtiter bewegten. Die Messung der anti-vegetativen Antikörper ergab ebenfalls keine Veränderung, was zu erwarten war, da die Impfung keine Antigene gegen den vegetativen Zellkörper enthielt. Einzig der anti-rPA83 IgG-Titer war bei einigen Tieren nach der ersten Immunisierung (Woche 3) ten- 112 4. Ergebnisse und Diskussion denziell und nach der zweiten Immunisierung (Woche 5) signifikant (p = 0,017) erhöht (Abbil- Tabelle 44: Ziegengruppe 4 Antigen / Titer dung 41, links). Die dritte Immunisierung (Woche 8) schien die Titer gegen rPA83 insgesamt nicht weiter rPA83 zu erhöhen, sondern nivellierte lediglich die Titer zuvor wenig reaktiver Tiere auf ein einheitlicheres Niveau. Bis zur Infektion in Woche 10 blieben die anti-rPA83 IgG-Titer signifikant erhöht (p ≤ 0,042), TNA obgleich sie im Vergleich zu Woche 5 und 8 signifikant abgefallen waren (p ≤ 0,034) und in der Höhe den gemessenen Werten von Woche 3 entsprachen (p = 0,704). Aufgrund der niedrigen anti-rPA83 IgG- rBclA Titer war ein TNA-Titer über der Nachweisgrenze (<50) nicht zu erwarten und konnte entsprechend mit einer Ausnahme auch nicht detektiert werden. Daher wurde von einer graphischen Darstellung dieser Titer und der ebenfalls kaum veränderlichen Titer gegen FIS und das vegetative Antigen abgesehen. Durch die Ziegen Nr. W ochen nach 1. Immunisierung 0 3 5 8 10a Tod in 8188 557 800 2571 2571 1371 3,2 8190 371 4457 5600 5028 2914 3,5 8192 400 1114 2857 1828 942 2,2 8198 285 700 1600 4800 1600 3,2 dpi 8199 700 3314 5828 6857 4571 3,2 M 463 2077 3691 4217 2280 3,0 S 165 1707 1906 2024 1477 0,5 8188 <50 <50 <50 <50 <50 3,2 8190 <50 <50 50 <50 <50 3,5 8192 <50 <50 <50 <50 <50 2,2 8198 <50 <50 <50 <50 <50 3,2 8199 <50 <50 <50 <50 <50 3,2 M - - - - - 3,0 S - - - - - 0,5 8188 2122 1795 3200 2448 1387 3,2 8190 318 612 1387 865 481 3,5 8192 465 726 465 726 661 2,2 8198 346 587 400 726 677 3,2 8199 628 800 800 930 563 3,2 M 776 904 1250 1139 754 3,0 0,5 S 762 506 1158 737 363 veg Pool 123 100 150 135 150 - FIS Pool 575 713 675 1150 1150 - a Probenzeitpunkt vor der Infektion ELISA IgG und TNA-Titer der Ziegengruppe 4 im Verlauf von 10 Wochen gemessen, sowie überlebte Zeit in dpi; Titerwerte ergeben sich als reziproker Wert der letzten Verdünnungsstufe mit einer OD404nm ≥ 0,1. Verstorbene sind orange hinterlegt; M – arithmetischer Mittelwert; S – Standardabweichung Infektion verstarben alle Tiere in einem relativ ähnlichem Zeitrahmen bis zum 3. Tag, wobei alle Tiere vor ihrem Tod eine erhöhte Temperatur aufwiesen. Korrelationen zwischen den gemessenen Titern und der überlebten Zeit waren nicht vorhanden (rs ≤ +0,5). Abbildung 41: Ziegengruppe 4 (anti-rPA83 und -rBclA IgG-Titer) Lineare Darstellung der anti-rPA83 IgG-Titer (A) und anti-rBclA IgG-Titer (B) von Gruppe 4 (Tabelle 44) im Versuchsverlauf; Für jeden Messzeitpunkt sind die Einzelwerte aller Individuen angegeben, sowie durch einen horizontalen Strich, deren Mittelwerte, die durch gepunktete Linien miteinander verknüpft sind; Blaue Pfeile deuten den Zeitpunkte der Immunisierung mit der Proteinkombination an, schwarze entsprechend den der Infektion. 113 4. Ergebnisse und Diskussion 4.3.7.2 Ziegengruppe 5 (DNA-Vakzine) Eine Gruppe von 5 Ziegen wurde dreimal im Abstand von 3 Wochen mit einer Mischung aus je 1 mg NTC7382-TPA-LFD1PAD4-mIPS-1, Tabelle 45: Ziegengruppe 5 Antigen / Titer pDNAVaccUltra1- TPA-BclAD1D3-LAMP1 und pDNAVaccUltra5-CIITA in einem Endvolumen von 1 ml s. c. (erste Immunisie- 8205 rPA83 rung) bzw. i. c. (zweite und dritte Immunisierung) geimpft. Zur Erhöhung der Immunreaktion wurde 9 Wochen nach der letzten Immunisierung mit der rBclA DNA-Mischung eine weitere Immunisierung mit Proteinkomponenten vorgenommen. Diese beinhaltete rPA83 (25 µg), rBclA (25 µg), FIS (10 Sporen) und 8 Lipopeptid (500 µg) und wurde analog zu den Protein- rLF gruppen 4 und 6 s. c. appliziert. Die serologische Auswertung umfasste die Detektion von IgG Antikörper gegen rPA83, rBclA, FIS, rLF und vegetatives Antigen, sowie TNA-Titer. Die Probennahmen erfolgten vor dem Beginn der Immunisierung (Woche 0) und nach jeder DNA-Vakzinierung (Woche 3, 6 und 9) sowie vor (Woche 15) und 2 Wochen nach (Woche 17) dem Proteinboost (Tabelle 45). Wie schon für die anderen Zie- Ziegen Nr. W ochen nach 1. Immunisierung 0 3 6 9a 15b 17c 1262 1732 1356 1354 1323 1969 8206 985 1323 769 923 769 1415 8207 1908 1732 1415 1785 892 1846 8208 1169 892 831 1354 1323 2831 8209 723 662 631 769 677 1077 1828 M 1209 1268 1000 1237 997 S 441 485 360 402 307 663 8205 246 446 831 923 677 1538 8206 315 600 477 708 400 1385 8207 446 1015 677 692 354 1569 8208 754 754 738 862 400 4062 8209 492 1077 708 646 331 1723 M 451 778 686 766 432 2055 S 196 268 130 119 140 1128 8205 1056 1280 1152 1536 800 1664 8206 592 800 720 1280 768 1664 8207 864 1792 1216 1376 960 1920 8208 528 688 800 1440 1184 3456 8209 400 1120 768 1088 640 4864 M 688 1136 931 1344 870 2714 1415 S 267 437 234 171 209 TNA Pool <50 <50 <50 <50 <50 <50 FIS Pool 1050 1050 1200 950 800 1900 veg Pool 182 182 170 152 282 282 a 3 Wochen nach letzter DNA Impfung b 9 Wochen nach letzter DNA Impfung und direkt vor dem Proteinboost c 2 Wochen nach dem Proteinboost ELISA IgG und TNA-Titer der Ziegengruppe 5 im Verlauf von 17 Wochen gemessen; Titerwerte ergeben sich als reziproker Wert der letzten Verdünnungsstufe mit einer OD404nm ≥ 0,1; Verstorbene orange; M – arithmetischer Mittelwert; S – Standardabweichung gengruppen bemerkt, waren für alle Negativseren (Woche 0) unspezifische Hintergrundtiter detektierbar. Durch die 3fach-Immunisierung mit der DNA-Vakzine konnten die anti-rPA83 IgG-Titer nicht signifikant erhöht werden (p ≥ 0,107). Der Proteinboost in Woche 15 erzeugte zwar eine leichte Erhöhung der Antikörpertiter gegen rPA83, aber auch diese Erhöhung war nicht signifikant (Abbildung 42; p = 0,099). Die Titerentwicklung gegen rLF verlief ähnlich, wobei durch die niedrigeren unspezifischen Hintergrundtiter und die teilweise geringere Streuung der Messwerte die Titer ab Woche 3 signifikant erhöht waren (Abbildung 42; p ≤ 0,047). Der Proteinboost erzeugte hier ebenfalls, deutlich höher als bei rPA83, einen Titeranstieg, der mit p = 0,0498 sogar signifikant war. Die Erhöhung des anti-rLF IgG-Titers durch den Proteinboost war unerwartet, da nur rPA83, rBclA und FIS verabreicht wurden. Möglicher Hintergrund könnte eine Kreuzreaktion mit rPA83 sein, die bereits in anderen Studien bemerkt wurde [11][193], womit die sichtbaren Titer keine „echten“ Titer gegen rLF darstellen würden. Eine entsprechende Korrelation der anti-rPA83 und -rLF IgG-Titer für Woche 17 bestand jedoch nicht (rs = 0,175). Entsprechend der niedrigen Antitoxin-Titer waren zu keinem Zeitpunkt TNA-Titer über 114 4. Ergebnisse und Diskussion der Nachweisgrenze (<50) messbar (Abbildung 43, rechts). Titer gegen das vegetative Antigen waren erwartungsgemäß ebenfalls, abgesehen von den unspezifischen Hintergrundtitern, nicht messbar. Abbildung 42: Ziegengruppe 5 (anti-rPA83 und -rLF IgG-Titer) Lineare Darstellung der anti-rPA83 IgG-Titer (A) und anti-rLF IgG-Titer (B) von Gruppe 5 (Tabelle 45) im Versuchsverlauf; Für jeden Messzeitpunkt sind die Einzelwerte aller Individuen angegeben, sowie durch einen horizontalen Strich, deren Mittelwerte, die durch gepunktete Linien miteinander verknüpft sind; Rote Pfeile deuten den Zeitpunkte der Immunisierung mit der DNA-Vakzine an und blau entsprechend den der Immunisierung mit der Proteinkombination Auch die Antikörpertiter gegen rBclA und FIS (Abbildung 43) zeigten einen ähnlichen Verlauf wie für rPA83 und rLF. Wie bei rLF waren auch bei den anti-rBclA IgG-Titern die Werte zu einigen Zeitpunkten signifikant erhöht (Woche 3 und 9; p ≤ 0,043). Der Proteinboost bewirkte jedoch eine deutlichere Erhöhung der Antikörpertiter gegen die Spore als die DNA-Vakzinierungen, sodass die anti-rBclA IgG-Titer in Woche 17 signifikant erhöht waren (p = 0,02). Abbildung 43: Ziegengruppe 5 (anti-rBclA, -FIS, -vegetativ IgG- und TNA-Titer) Lineare Darstellung der anti-rBclA IgG-Titer (A) und anti-FIS, -vegetativ IgG- und TNA- Titer (B) von Gruppe 5 (Tabelle 45) im Versuchsverlauf; Für jeden Messzeitpunkt sind individuelle Einzelwerte oder Mittelwerte angegeben; Mittelwerte sind durch gepunktete Linien miteinander verknüpft; Rote Pfeile deuten den Zeitpunkte der Immunisierung mit der DNA-Vakzine an und blaue entsprechend den der Immunisierung mit der Proteinkombination 115 4. Ergebnisse und Diskussion Generell sind die durch die DNA-Vakzinierung erzeugten Titerveränderungen marginal und bewegen sich in einem Bereich, der auch bei unspezifischen Hintergrundtitern der Seren von nicht immunisierten Tieren (siehe Kapitel 4.3.3) gefunden wurde. Es kann davon ausgegangen werden, dass die DNA-Vakzinierung generell keine Titererhöhung erzeugte, die von den berichteten Schwankungen innerhalb naiver Tiere zu unterscheiden gewesen wären. Der nachträgliche Proteinboost erhöhte die Antikörpertiter nur in dem Umfang, wie es der Erstimmunisierung der Gruppe 6 entsprach (p ≥ 0,254), die die gleiche Vakzineformulation erhalten hatte. Daher ist ein humorales Priming durch die vorherige Gabe der DNA-Vakzine, das sich in Memoryreaktion gegen die Proteinantigene hätte äußern können, unwahrscheinlich. Aufgrund der geringen Immunogenität der DNA-Vakzine wurde, zur Vermeidung von unnötigem Leid, von einer Infektion abgesehen. 4.3.7.3 Ziegengruppe 6 (rPA83, rBclA, FIS und Lipopeptid) Eine Gruppe von 5 Ziegen wurde dreimal im Abstand von 3 Wochen mit rPA83 (25 µg), Tabelle 46: Ziegengruppe 6 Antigen / Titer Ziegen Nr. W ochen nach 1. Immunisierung Tod in 0 3 5 8 10a 12b dpi rBclA (25 µg), FIS (10 Sporen) und Lipopep- 8191 514 657 10285 6171 3200 - 4,1 8194 385 600 4000 2857 1485 - 2,6 tid (500 µg) s. c. immunisiert und 4 Wochen 8195 214 2285 17371 12342 5600 - 4,6 8200 685 771 3200 4228 1600 1314 - nach der letzten Immunisierung mit 844 Spo- 8201 685 2171 1942 5028 285 - 3,8 M 497 1297 7360 6125 2434 - 3,8 ren des voll virulentem Feldisolats SD20 infi- S 202 853 6456 3679 2051 - 0,8 ziert. Die Applikation der einzelnen Impfkom- 8191 <50 <50 99 <50 <50 - 4,1 8194 <50 <50 50 <50 <50 - 2,6 8195 <50 50 149 74 74 - 4,6 8200 <50 <50 <50 <50 <50 <50 - 8201 <50 <50 <50 <50 <50 - 3,8 8 ponenten erfolgte nicht wie geplant in rPA83 TNA Mischung, sondern jeweils einzeln in einem Volumen von 1 ml in das gleiche Areal. Die (Woche 12) statt. Die serologischen Tests umfassten die Detektion von IgG Antikörpern gegen rPA83, rBclA, FIS und vegetatives Antigen, sowie TNA-Titer (Tabelle 46). Wie schon zuvor beobachtet, waren auch - 60 - - - 3,8 - 65 - - - 0,8 281 759 3853 3853 1551 - 4,1 8194 387 1355 1795 1453 742 - 2,6 8195 185 2644 2906 1795 1077 - 4,6 8200 342 783 1600 1093 604 522 - 8201 661 628 383 1730 144 - 3,8 M 371 1234 2107 1985 824 - 3,8 SE 179 837 1324 1080 527 - 0,8 FIS Pool 625 6800 14400 20000 16800 12400 - veg Pool 170 200 200 282 247 1694 - rBclA Immunisierung (Woche 3, 5 und 8), sowie vor (Woche 10) und 2 Wochen nach der Infektion - 8191 Serumentnahme fand vor dem Beginn der Immunisierung (Woche 0) und nach jeder M S a Probenzeitpunkt vor der Infektion b Probezeitpunkt nach Beendigung der Infektion ELISA IgG und TNA-Titer der Ziegengruppe 6 im Verlauf von 12 Wochen gemessen, sowie überlebte Zeit in dpi; Beobachtungen zum Überleben wurden bis 8 dpi durchgeführt; Titerwerte ergeben sich als reziproker Wert der letzten Verdünnungsstufe mit einer OD404nm ≥ 0,1. Verstorbene sind orange hinterlegt, Überlebende entsprechend grün; M – arithmetischer Mittelwert; S – Standardabweichung bei dieser Gruppe bereits vor dem Start der Immunisierung niedrige unspezifische Titer gegen die jeweiligen Antigene messbar. Durch die Immunisierung konnten die anti-rPA83 IgG-Titer bereits ab der zweiten Immunisierung tendenziell erhöht werden (Abbildung 44). Dieser 116 4. Ergebnisse und Diskussion Anstieg war aber durch die starken Unterschiede zwischen den einzelnen Tieren nicht signifikant (p = 0,081). Obwohl die Titer gegen rPA83 nach der dritten Immunisierung (Woche 8) auf dem Niveau von Woche 5 verblieben (p = 0,464), waren sie im Vergleich zu Woche 0 signifikant erhöht (p = 0,03), da die individuellen Werte weniger streuten. Entsprechend der niedrigen anti-rPA83 IgG-Titer waren TNA-Titer nur marginal (<150) und nicht für alle Tiere messbar. Auch die anti-rBclA IgG-Titer in Woche 8 zeigten eine signifikante Erhöhung gegenüber den Werten von Woche 0 (p = 0,034). Dennoch lagen die individuellen Werte für rBclA Abbildung 44: Ziegengruppe 6 (anti-rPA83 IgG-Titer) Lineare Darstellung der anti-rPA83 IgG-Titer von Gruppe 6 (Tabelle 46) im Versuchsverlauf; Für jeden Messzeitpunkt sind die Einzelwerte aller Individuen angegeben, sowie durch einen horizontalen Strich, deren Mittelwerte, die durch gepunktete Linien miteinander verknüpft sind; Blaue Pfeile deuten den Zeitpunkte der Immunisierung an und schwarz entsprechend den Zeitpunkt der Infektion. Zur besseren Darstellung wurde die y-Achse mit einer Unterbrechung im Bereich 13000 – 17000 versehen. mehrheitlich in einem Bereich, der den unspezifisch gemessenen Hintergrundtitern entsprach (Abbildung 45). Daher ist davon auszugehen, dass auch in dieser Gruppe rBclA nur marginal immunogen war. Sowohl für rBclA, als auch für rPA83 fielen die Antikörpertiter in Woche 10, wie auch bei Gruppe 4, im Vergleich zu Woche 8 rapide ab (p ≤ 0,027). Abbildung 45: Ziegengruppe 6 (ELISA- und TNA-Titer) Lineare Darstellung der anti-rBclA IgG-Titer (A) und anti-FIS, -vegetativ und TNA-Titer (B)von Gruppe 6 (Tabelle 46). im Versuchsverlauf; Für jeden Messzeitpunkt sind durch einen horizontalen Strich Mittelwerte angegeben und durch gepunktete Linien miteinander verknüpft; Für Woche 12 sind die Einzeltiter der Überlebenden verzeichnet; Blaue Pfeile deuten den Zeitpunkte der Immunisierung an und schwarz entsprechend den Zeitpunkt der Infektion Durch die Immunisierung mit FIS konnte ein sehr hoher Titer anti-FIS Titer erzeugt werden. Dieser stieg, im Gegensatz zu den anderen gemessenen Antikörpertitern kontinuierlich mit jeder Immunisierung weiter an und fiel nach der Beendigung des Impfplans nur langsam wieder 117 4. Ergebnisse und Diskussion ab (Abbildung 45). Ein erhöhter Titer gegen das vegetative Antigen konnte auch bei dieser Gruppe durch die Immunisierung erwartungsgemäß nicht nachgewiesen werden. Die Infektion überlebte nur 1 von 5 Tieren, wobei eine leicht erhöhte Überlebenszeit der verstorbenen Tiere erkennbar war. Die überlebende Ziege zeigte nach der Infektion für alle gemessenen Antigene, exklusive des vegetativen Antigens, nahezu unveränderte Antikörpertiter. Der Titer gegen das vegetative Antigen schien marginal erhöht. Korrelationen der gemessenen Titer mit dem Überleben waren nicht vorhanden. Alle verstorbenen Ziegen hatten vor dem Tod eine erhöhte Temperatur, die Überlebende zu keinem Zeitpunkt. 4.3.8 Versuchswiederholung der Gruppen 4 und 6 (Türkei) Aufgrund des beschriebenen Fehlers bei der Immunisierung der Gruppen 4 und 6 und der dort beobachteten niedrigen Schutzraten, wurde eine Wiederholung dieser Gruppen angestrebt. Die generelle Durchführung der Wiederholung geschah analog zu den bereits beschriebenen Arbeitsschritten mit leichten, erfahrungsbedingten Änderungen (siehe hierzu auch 3.4.3). Diese betrafen die Messung der Temperatur, welche zweimal täglich regulär durchgeführt wurde (zuvor ein Mal) und die Erhöhung der Fütterungsintervalle (dreimal täglich, statt zuvor zweimal). Zudem waren die Tiere während der Infektion, durch die ausschließliche Haltung innerhalb eines Gebäudes, weniger stark äußeren Temperaturschwankungen ausgesetzt und auch nachts in Intervallen von 3 – 6 h gut beobachtbar. Ein Transport zwischen Immunisierung und Infektion entfiel. Ausschlaggebend für eine frequentere Kontrolle des Zustands der Tiere sowie eine Blutabnahme zur Abklärung von Bakterien im Blut war wie zuvor eine Temperatur von 40 °C und höher. Eine weitere Veränderung betraf die serologische Auswertung. Zur Verminderung der unspezifischen Hintergrundtiter wurde die Blocklösung im ELISA mit 10 % Milchpulver statt 10 % FKS verwendet. Wie in Tabelle 47 aufgeführt, konnten durch die Verwendung von Milchpulver die unspezifischen Hintergrundtiter gegen alle Antigene deutlich vermindert werden, sodass sich eine niedrigere Schwankungsbreite bis zu Titerwerten von ca. 200 ergab. In Einzelfällen, bei den Negativseren der Gruppen 4 (W) und 6 (W), waren aber auch höhere unspezifische Hintergrundtiter möglich. Daher ist für die Einzelbetrachtungen dieser Gruppen ebenfalls keine Normalisierung angewendet worden. Beim Vergleich der Gruppen untereinander wurde mit normalisierten Werten gearbeitet. Zur Verbesserung der Wirksamkeit wurde, neben der korrekten Applikation als Mischung, die Konzentration der Antigene rPA83 und rBclA von 25 µg auf 75 µg pro Dosis erhöht. Darüber hinaus beinhalteten die vakzinierten Gruppen 4 (W) und 6 (W) 10 statt 5 Tiere, um potentielle Verluste durch Umwelteinflüsse ausgleichen zu können und bessere Vergleichswerte zu erhalten. Da in der Versuchswiederholung ein anderer Infektionsstamm verwendet wurde, war 118 4. Ergebnisse und Diskussion zudem eine weitere unvakzinierte Kontrollgruppe (NK (W)) bestehend aus 5 Tieren Bestandteil des Wiederholungsversuchs. Die Impfung der Tiere erfolge dreimal im Abstand von 3 Wochen. Blutentnahmen wurden vor dem Beginn der Vakzinierung vorgenommen (Woche 0), vor jeder weiteren Vakzinierung (Woche 3 und 6), direkt vor der Infektion (Woche 10 bzw. 11) und nach der Beendigung des Infektionsversuchs (Woche 13). 4.3.8.1 Negativkontrolle (Wiederholung) Eine Gruppe von 5 Ziegen wurde nicht vakziniert und während des gesamten Versuchsverlaufs mit den Ziegen von Gruppe 4 (W) und 6 (W) gehalten und beprobt. Da für die Versuchswiederholungen ein anderer Infektionsstamm verwendet wurde, stellt diese Gruppe die negativen Vergleichswerte für die Serologie und Schutzraten der Wiederholungen dar. Von den fünf ursprünglich eingesetzten Ziegen dieser Gruppe verstarb Ziege Nr. 96132 zwischen Woche 6 und 9 an einer Infektion. Die serologischen Daten dieser Ziege wurden bis einschließlich Woche 6 in alle Betrachtungen mit einbezogen. Tabelle 47: Negativkontrolle (W) – Ziege (Türkei) Antigen / Titer rPA83 TNA Veg Ziegen Nr. W ochen nach 1. Immunisierungc 0 3 6 11d dpi Ziegen Nr. W ochen nach 1. Immunisierungc 0 3 6 11d 97014 <100 <100 <100 <100 dpi 2,5 97014 <100 <100 <100 118 97015 <100 <100 <100 2,5 <100 4,7 97015 102 104 142 136 96132a <100 <100 4,7 <100 - - 96132a 179 289 477 - 96163b <100 - <100 <100 <100 2,3 96163b 120 <100 101 155 2,3 kkb <100 <100 <100 <100 2,9 kkb 116 <100 <100 115 2,9 M - - - - 3 M 103 79 144 131 3 S - - - - 1 S 65 126 196 18 1 97014 <50 <50 <50 <50 2,5 97014 160 143 138 171 2,5 4,7 Tod in Antigen / Titer rBclA Tod in 97015 <50 <50 <50 <50 4,7 97015 182 188 326 338 96132a <50 <50 <50 - - 96132a 132 137 161 - - 96163b <50 <50 <50 <50 2,3 96163b 131 151 139 <100 2,3 kkb <50 <50 <50 <50 2,9 kkb 155 114 209 239 2,9 M - - - - 3 M 152 147 195 187 3 S - - - - 1 S 21 27 79 142 1 97014 <100 <100 <100 <100 2,5 a starb zwischen Woche 6 und 9 durch eine Infektion 97015 FIS <100 <100 <100 <100 4,7 b Infektion bereits in Woche 10 a 96132 <100 <100 <100 - - c bezogen auf Gruppe 4(W) und 6(W) 96163b <100 <100 <100 <100 2,3 d Probenzeitpunkt vor der Infektion kkb <100 <100 <100 <100 2,9 M - - - - 3 S - - - - 1 ELISA IgG- und TNA-Titer der Gruppe NK (W) im Verlauf von 13 Wochen gemessen, sowie Eintritt des Todes in dpi. Titerwerte ergeben sich als reziproker Wert der letzten Verdünnungsstufe mit einer OD 404nm ≥ 0,1. Die Überwachung des Überlebens erfolgte bis 14 dpi. Überlebende sind entsprechend grün hinterlegt, Verstorbene orange; Ausgegraut dargestellt (Hintergrund weiß) sind Tiere die vorzeitig aus dem Versuch ausgeschieden sind; M – arithmetischer Mittelwert; S – Standardabweichung Zur Verifizierung der Pathogenität des verwendeten Feldisolats K-136 in Ziegen wurden vor der Durchführung des Infektionsversuchs 2 der verbliebenen 4 Tiere dieser Gruppe zufällig ausgewählt (96163 und kesik kulak), abgetrennt und bereits in Woche 10 mit einer Dosis von 1027 Sporen infiziert (Tabelle 16). Die Infektion der restlichen 2 Tiere dieser Gruppe erfolgte in 119 4. Ergebnisse und Diskussion Woche 11 mit derselben Sporensuspension, zusammen mit allen Tieren von Gruppe 4 (W) und 6 (W), mit 918 Sporen. Die Schwankung der Infektionsdosis ergab sich aus den unterschiedlichen Auszählungen der jeweiligen überzähligen Infektionsdosen, wobei beide Infektionen mit der gleichen Sporensuspension durchgeführt wurden. Alle Tiere verstarben innerhalb von 5 Tagen, wobei nur zwei der Tiere vor dem Tod eine kurzzeitige Erhöhung der Temperatur (<10h vor dem Tod) zeigten, die bis zum Tod erhalten blieb. Keiner der gemessenen Titer (Tabelle 47) zeigte eine signifikante Veränderung während des Versuchszeitraums (p ≥ 0,098), daher wurde davon abgesehen die Werte grafisch darzustellen. 4.3.8.2 Ziegengruppe 4 (Wiederholung) Eine Gruppe von 10 Ziegen wurde dreimal im Abstand von 3 Wochen mit einer Mischung aus rPA83 (75 µg), rBclA (75 µg) und Lipopeptid (500 µg) s. c. immunisiert. Während der Immunisierungsphase fielen 2 Tiere (Nr. 97011 und Eks) zwischen Woche 3 und 6, bedingt durch den gewährten Freigang im Außengehege, wildernden Straßenhunden zum Opfer. Die serologischen Daten dieser Tiere wurden bis zu deren Ausscheiden aus dem Versuch in alle Betrachtungen einbezogen. Die Infektion der verbliebenen 8 Tiere erfolgte s. c. 5 Wochen nach der letzten Immunisierung in Woche 11 mit einer Dosis von 918 Sporen des voll virulenten Feldisolats K136. Die Infektion überlebten 4 von 8 Tieren, wobei bei den verstorbenen eine verlängerte Überlebenszeit von durchschnittlich 5 Tagen gemessen wurde. Bei zwei der als „verstorben“ gewerteten Tiere wurden frühzeitig Bakterien im Blut nachgewiesen und aufgrund des guten Gesundheitszustands der Tiere eine Behandlung mit Penicillin/Streptomycin begonnen. Der „Todeszeitpunkt“ dieser Tiere wurde daher artifiziell festgelegt und stellt den Zeitpunkt des positiven Befunds dar. Beide Tiere (Nr. 99834 und 627398) blieben am Leben und konnten ebenfalls nach der Beendigung des Versuchs beprobt werden. Die Daten für diesen Zeitpunkt sind zwar in den Tabellen und Darstellungen angegeben, flossen aber nicht in die Berechnungen und statistischen Kalkulationen für Woche 13 mit ein. Alle verstorbenen Tiere zeigten an mindestens einem Probenzeitpunkt vor dem Tod eine erhöhte Temperatur von >40 °C, bei zwei von vier Überlebenden war dies ebenfalls der Fall. In Tabelle 48 sind die serologischen Daten der Gruppe aufgelistet. Auch in dieser Gruppen waren für den Zeitpunkt vor dem Beginn der Immunisierung (Woche 0) teilweise unspezifische Hintergrundtiter für die einzelnen Antigene messbar. 120 4. Ergebnisse und Diskussion Tabelle 48: Ziegengruppe 4 (W) – ELISA- und TNA-Titer Antigen / Titer rPA83 3 6 11c 13d dpi 97011a 105 642 - - - 97013 <100 528 2836 2628 99348b 134 427 5415 580315 213 391 15903 580319 105 368 2872 Antigen / Titer W ochen nach 1. Immunisierung Tod in Ziegen Nr. 0 3 6 11c 13d - 97011a <100 250 - - - - 5,8 97013 <100 197 267 111 2921 1244641 8,7 99348b 101 105 157 175 192 23386 282292 - 580315 164 200 297 126 120 - 2415 - 2,7 580319 <100 128 179 171 - 2,7 627398b 103 <100 225 124 228 2,8 627399 369 624 701 382 366 - dpi 5,8 8,7 627398b <100 773 4704 3006 800475 2,8 627399 <100 157 2371 972 762 - 728761 <100 597 6600 11190 658286 - 728761 <100 <100 159 <100 <100 - Eksa 119 813 - - - - Eksa <100 179 - - - - Gk <100 1354 12719 5022 1404 - Gk <100 110 186 <100 <100 - M 68 605 6678 6443 235686 5,0 M 74 179 271 136 121 5,0 2,9 rBclA 77 329 4998 7527 311363 2,9 S 120 177 181 120 173 97011a <50 <50 - - - - 97011a 144 206 - - - - 97013 <50 <50 <50 <50 - 5,8 97013 164 177 316 191 - 5,8 99348b <50 <50 74,8 <50 10461 8,7 99348b 115 <100 170 262 190 8,7 580315 <50 <50 223,6 50 1315 - 580315 305 237 536 730 430 - 580319 <50 <50 74,8 <50 - 2,7 580319 180 172 265 319 - 2,7 627398b <50 <50 50 <50 9465 2,8 627398b 103 123 269 267 189 2,8 627399 <50 <50 <50 <50 <50 - 627399 198 194 326 267 197 - 728761 <50 <50 198,8 50 3225 - 728761 <100 <100 171 158 129 - Eksa <50 <50 - - - - Eksa 150 129 - - - - Gk <50 74,8 273,24 <50 <50 - Gk 258 350 568 391 286 - M - - 112 13 1135 5,0 M 162 159 328 323 261 5,0 S 84 105 150 179 130 2,9 S Veg Tod in 0 S TNA W ochen nach 1. Immunisierung Ziegen Nr. FIS - - 105 23 1525 2,9 97011a <100 <100 - - - - a b starb zwischen Woche 3 und 6 durch Wildhunde 97013 <100 <100 <100 <100 - 5,8 99348b <100 <100 <100 <100 110 8,7 580315 <100 <100 <100 <100 <100 - 580319 <100 <100 <100 <100 - 2,7 c Probenzeitpunkt vor der Infektion 627398b <100 <100 <100 <100 <100 2,8 d Probenzeitpunkt nach Beendigung der Infektion 627399 <100 <100 <100 101 <100 - 728761 <100 <100 <100 <100 <100 Eksa <100 <100 - - - - Gk <100 <100 <100 <100 <100 - M - - - - - 5,0 S - - - - - 2,9 wurde nach der Detektion von Bakterien im Blut mit Antibiotika behandelt und überlebte; zählt zu Verstorbenen; Zeitpunkt des Todes ist Zeitpunkt der Antibiotikagabe ELISA IgG- und TNA-Titer der Ziegengruppe 4 (W) im Verlauf von 13 Wochen gemessen, sowie Eintritt des Todes in dpi. Titerwerte ergeben sich als reziproker Wert der letzten Verdünnungsstufe mit einer OD 404nm ≥ 0,1. Die Überwachung des Überlebens erfolgte bis 14 dpi. Überlebende sind entsprechend grün hinterlegt, Verstorbene orange; Ausgegraut dargestellt sind Tiere die vorzeitig aus dem Versuch ausgeschieden sind; M – arithmetischer Mittelwert; S – Standardabweichung Durch die Immunisierung konnten die Antikörpertiter gegen rPA83 bereits nach der ersten Immunisierung (Woche 3) signifikant erhöht werden (p = 0,001). Diese stiegen durch die zweite Immunisierung weiter an (p = 0,007), sodass für den Probenzeitpunkt in Woche 6 ebenfalls signifikant erhöhte und stark mit den anti-rPA83 IgG-Titern korrelierende TNA-Titer messbar waren (p = 0,02; rs = +0,927; Abbildung 46). Für den Messzeitpunkt nach der dritten Immunisierung in Woche 11 waren die Titer gegen rPA83 zwar weiterhin signifikant erhöht (p = 0,046), jedoch nicht unterschiedlich zu den in Woche 6 gemessenen Titern (p = 0,064). Für die Mehrheit der Tiere waren keine nachweisbaren TNA-Titer für Woche 3 und 11 messbar. Das heißt die nach der zweiten Immunisierung messbaren TNA-Titer fielen nach der dritten Immunisierung wieder unter den Wert von 1 : 50 (Abbildung 46B). Durch die überlebte Infektion kam es für einige Tiere, insbesondere den behandelten Tieren, die nachweislich Bakterien im Blut aufgewiesen hatten, zu einem extremen Anstieg der anti-rPA83 IgG-Titer und damit korrelierend 121 4. Ergebnisse und Diskussion den TNA-Titern (rs = +0,95). Für zwei der überlebenden Tiere war keine Titererhöhung für rPA83 feststellbar, weshalb die gemessenen Werte für TNA und rPA83 in Woche 13 insgesamt aufgrund der hohen Streuung keine Signifikanz aufwiesen. Alle Tiere die eine Temperatur von >40 °C und höher aufwiesen, zeigten auch eine deutliche Titererhöhung gegen rPA83 von Woche 11 auf Woche 13. Abbildung 46: Ziegengruppe 4 (W) – anti-rPA83 und TNA-Titer Halblogarithmische Darstellung der anti-rPA83 IgG-Titer (A, links) und TNA-Titer (B, rechts) von Gruppe 4 (W) (Tabelle 48) im Versuchsverlauf; Für jeden Messzeitpunkt sind die Einzelwerte aller Individuen angegeben, sowie durch einen horizontalen Strich, deren Mittelwerte, die durch gepunktete Linien miteinander verknüpft sind; Blaue Pfeile deuten den Zeitpunkte der Immunisierung mit der Proteinkombination an, schwarze entsprechend den der Infektion; Mit * gekennzeichnete Zeitpunkte zeigen signifikant erhöhte Titer (p ≤ 0,05) im Vergleich zu Woche 0; Wie in Tabelle 48 aufgeführt, konnten gegen das vegetative Antigen im Verlauf der Immunisierung erwartungsgemäß keine signifikanten Titer gemessen werden. Dies änderte sich auch nach der überlebten Infektion nicht. Die Titer gegen rBclA nach der ersten und zweiten und für FIS nach der zweiten und dritten Immunisierung waren signifikant erhöht (p ≤ 0,02; Abbildung 47). Allerdings bewegten sich die Titer für beide Antigene in einem Bereich knapp oberhalb der unspezifischen Hin- Abbildung 47: Ziegengruppe 4 (W) – anti-rBclA IgG-Titer tergrundtiter, weswegen diese Titererhöhung Halblogarithmische Darstellung der anti-rBclA IgG-Titer von Gruppe 4 (W) (Tabelle 48) im Versuchsverlauf; Für jeden Messzeitpunkt sind die Einzelwerte aller Individuen angegeben, sowie durch einen horizontalen Strich, deren Mittelwerte, die durch gepunktete Linien miteinander verknüpft sind; Blaue Pfeile deuten den Zeitpunkte der Immunisierung mit der Proteinkombination an, schwarze entsprechend den der Infektion; Mit * gekennzeichnete Zeitpunkte zeigen signifikant erhöhte Titer (p ≤ 0,05) im Vergleich zu Woche 0; wenig aussagekräftig ist. Eine Korrelation der rBclA und FIS-Titer bestand nur für Woche 3 und 6 (rs ≥ +0,685). Nach der Infektion konnte auch für die anti-rBclA und -FIS IgG-Titer kei- ne Veränderung festgestellt werden. Keiner der gemessenen signifikant erhöhten Titer (rPA83 122 4. Ergebnisse und Diskussion und TNA eingeschlossen) korrelierte mit der überlebten Zeit (r s ≤ +0,49). Dies galt für alle Messzeitpunkte. 4.3.8.3 Ziegengruppe 6 (Wiederholung) Eine Gruppe von 10 Ziegen wurde dreimal im Abstand von 3 Wochen mit einer Mischung aus rPA83 (75 µg), rBclA (75 µg), FIS (108 Sporen) und Lipopeptid (500 µg) s. c. immunisiert und 5 Wochen nach der letzten Immunisierung (Woche 11) mit einer Dosis von 918 Sporen des voll virulenten Feldisolats K-136 infiziert. Die Infektion überlebten 8 von 10 Tieren, wobei eines der verstorbenen Tiere eine verlängerte Überlebenszeit von 5 Tagen aufwies. Die serologischen Tests umfassten die Detektion von IgG Antikörper gegen rPA83, rBclA, FIS und vegetatives Antigen, sowie TNA-Titer (Tabelle 49). Tabelle 49: Ziegengruppe 6 (W) – ELISA- und TNA-Titer Antigen / Titer rPA83 TNA Ziegen Nr. Tod in Tod in 0 3 6 11a 13b dpi - 96139 120 147 1093 380 517 - 1194 - 96157 <100 191 779 398 182 - 3591 283759 - 97162 <100 148 805 252 164 - 25111 20003 32358 - 580316 132 329 768 668 259 - 2335 50260 18230 - 5,1 627395 <100 166 1226 478 - 5,1 <100 143 2886 1494 - 2,5 627396 <100 159 649 286 - 2,5 198 740 8533 5194 2816 - 728758 <100 270 567 217 175 - 729277 140 345 17898 4413 134279 - 729277 <100 187 1248 456 216 - 729278 167 3541 1575 5566 34909 - 729278 100 151 177 256 137 - 729284 163 768 11387 6083 1479612 - 729284 <100 <100 305 159 <100 - M 232 3360 14018 6820 257901 3,8 M 35 175 762 355 206 3,8 S 235 6787 14706 6709 502403 1,8 S 57 86 360 152 146 1,8 96139 <50 <50 <50 <50 670 - 96139 172 1386 5221 4596 5030 - 96157 <50 <50 124 <50 <50 - 96157 121 1708 10150 5295 3612 - 97162 <50 75 199 <50 3473 - 97162 153 4595 6074 5230 5911 - 580316 <50 323 596 248 99 - 580316 184 4250 16053 11707 6378 - 627395 <50 50 373 50 - 5,1 627395 <100 10818 24160 12676 - 5,1 627396 <100 1492 3135 869 - 2,5 728758 159 737 7428 4802 3071 - 6 11a 13b dpi 96139 122 188 3017 903 94279 96157 104 1422 6471 2721 97162 110 1681 13044 580316 561 22434 627395 754 627396 728758 Antigen / Titer W ochen nach 1. Immunisierung Ziegen Nr. 3 rBclA 627396 <50 <50 50 <50 - 2,5 728758 <50 <50 50 <50 <50 - 729277 <50 <50 100 <50 3299 - 729277 <100 2253 2428 1834 3045 - 729278 <50 149 <50 75 322 - 729278 175 2319 2123 3681 1728 - 729284 <50 <50 50 75 4168 - 729284 <100 2627 3924 3869 1979 - - 60 154 45 1504 3,8 M 96 3218 8070 5456 3844 3,8 S 85 2936 7057 3834 1746 1,8 M S Veg W ochen nach 1. Immunisierung 0 FIS - 105 191 78 1804 1,8 96139 <100 <100 <100 <100 163 - a Probenzeitpunkt vor der Infektion 96157 <100 <100 <100 <100 <100 - b Probenzeitpunkt nach Beendigung der Infektion 97162 <100 <100 <100 <100 186 - 580316 <100 <100 <100 <100 <100 - 627395 <100 <100 <100 <100 - 5,1 627396 <100 <100 <100 <100 - 2,5 728758 <100 <100 <100 <100 <100 - 729277 <100 <100 <100 <100 <100 - 729278 <100 <100 <100 <100 <100 - 729284 <100 <100 <100 <100 176 - M - - - - 66 3,8 S - - - - 91 1,8 ELISA IgG- und TNA-Titer der Ziegengruppe 6 (W) im Verlauf von 13 Wochen gemessen, sowie Eintritt des Todes in dpi. Titerwerte ergeben sich als reziproker Wert der letzten Verdünnungsstufe mit einer OD 404nm ≥ 0,1. Die Überwachung des Überlebens erfolgte bis 14 dpi. Überlebende sind entsprechend grün hinterlegt, Verstorbene orange; M – arithmetischer Mittelwert; S – Standardabweichung 123 4. Ergebnisse und Diskussion Wie schon zuvor beobachtet, waren auch bei dieser Gruppe bereits vor dem Start der Immunisierung niedrige unspezifische Hintergrundtiter gegen die jeweiligen Antigene messbar. Die anti-rPA83 IgG-Titer konnten bereits durch die erste Immunisierung bei einigen Tieren erhöht werden. Da dies nicht auf alle Tiere zutraf und die Werte für diesen Zeitpunkt (Woche 3) entsprechend stark streuten, war diese Erhöhung nicht signifikant. Dies änderte sich erst mit der zweiten Immunisierung, die eine signifikante Titererhöhung bei allen Tieren bewirkte (p = 0,015; Abbildung 48). Die TNA-Titer waren ebenfalls nach der zweiten Immunisierung (Woche 6) signifikant erhöht (p = 0,031) und korrelierten für alle Messzeitpunkte nach der ersten Immunisierung stark mit den anti-rPA83 IgG-Titern (rs ≥ +0,833). Die Titerwerte für rPA83 IgG und TNA in Woche 11, nach der dritten Immunisierung fielen im Vergleich zu Woche 6 signifikant ab (p ≤ 0,047). Allerdings lagen die Werte für rPA83 noch immer in einem Bereich, der signifikant höher lag, als die gemessenen Titer vor der Immunisierung (p = 0,011). Durch die überlebte Infektion kam es, wie auch schon in Gruppe 4 (W) beobachtet, zu einem extremen Anstieg der anti-rPA83 IgG- und TNA-Titer in ca. der Hälfte der Tiere. Die restlichen Tiere zeigten nur leicht ansteigende, stagnierende oder sogar fallende Titer. Daher besteht für diesen Messzeitpunkt keine Signifikanz. Auch bei dieser Gruppe ging eine deutliche Erhöhung der anti-rPA83 IgG-Titer nach der Infektion tendenziell mit dem Auftreten von Fieber einher. Ähnlich wie bei Gruppe 4 (W) waren auch hier für den gesamten Messzeitraum keine signifikanten anti-vegetativ IgG-Titer feststellbar. Abbildung 48: Ziegengruppe 6 (W) – anti-rPA83 IgG- und TNA-Titer Halblogarithmische Darstellung der anti-rPA83 IgG- (A) und TNA-Titer (B) von Gruppe 6 (W) (Tabelle 49) im Versuchsverlauf; Für jeden Messzeitpunkt sind die Einzelwerte aller Individuen angegeben, sowie durch einen horizontalen Strich, deren Mittelwerte, die durch gepunktete Linien miteinander verknüpft sind; Blaue Pfeile deuten den Zeitpunkte der Immunisierung mit der Proteinkombination an, schwarze entsprechend den der Infektion; Mit * gekennzeichnete Zeitpunkte zeigen signifikant erhöhte Titer (p ≤ 0,05) im Vergleich zu Woche 0; Die in Abbildung 49 dargestellten anti-FIS und -rBclA IgG-Titer waren bereits nach der ersten Immunisierung signifikant erhöht (p ≤ 0,008), wobei die gemessen Titer gegen rBclA (< 1300) verglichen mit den anti-FIS IgG-Titern (< 25000) sehr gering waren. Eine weitere Erhöhung der 124 4. Ergebnisse und Diskussion Antikörpertiter nach der zweiten Immunisierung war für beide Antigene nachweisbar (p ≤ 0,012), nach der dritten Immunisierung jedoch nicht. Generell sanken die Titerwerte ab Woche 6 kontinuierlich ab und wurden auch durch die überlebte Infektion nicht weiter erhöht. Eine signifikante Korrelation der anti-rBclA und -FIS IgG Titer bestand für den Zeitraum der Immunisierung (rs ≤ +0,41) und nach der Infektion (rs = +0,619) nicht. Erstaunlicherweise war auch eine teils starke Korrelation der anti-FIS IgG-Titer sowohl mit den anti-rPA83 IgG-Titern, als auch den TNA-Titern für Woche 3 und 6 vorhanden (+0,600 ≤ rs ≥ +0,724). Weitere Korrelationen, die Überlebenszeit eingeschlossen, konnten nicht festgestellt werden. Abbildung 49: Ziegengruppe 6 (W) – anti-rBclA und -FIS IgG-Titer Halblogarithmische Darstellung der anti-FIS (A) und -rBclA IgG-Titer (B) von Gruppe 6 (W) (Tabelle 49) im Versuchsverlauf; Für jeden Messzeitpunkt sind die Einzelwerte aller Individuen angegeben, sowie durch einen horizontalen Strich, deren Mittelwerte, die durch gepunktete Linien miteinander verknüpft sind; Blaue Pfeile deuten den Zeitpunkte der Immunisierung mit der Proteinkombination an, schwarze entsprechend den der Infektion; Mit * gekennzeichnete Zeitpunkte zeigen signifikant erhöhte Titer (p ≤ 0,05) im Vergleich zu Woche 0; 4.3.9 Zusammenfassung der Ergebnisse der NLV Die Antikörpertiter der Ziegenversuche mit NLV in Südafrika (Gruppe 4 – 6) waren generell sehr niedrig und durch die individuell stark schwankenden, unspezifischen Hintergrundtiter schwer interpretierbar. Die Schwankungsbreite der Titer lag je nach Antigen bei <100 – 2600. Durch den Austausch von FKS mit Milchpulver in der Blocklösung des ELISA konnte die unspezifischen Hintergrundtiter in den Wiederholungsversuchen verringert werden (<100 – 800). Für eine bessere Vergleichbarkeit der Gruppen untereinander wurden daher die individuellen Titerwerte durch Abzug der gemessenen Titer aus Woche 0 normalisiert. Die entsprechenden Gegenüberstellungen und Signifikanzberechnungen beziehen sich daher auf normalisierte Werte. Da die Probennahmezeitpunkte der verschiedenen Gruppen unterschiedlich waren, wurden für die vergleichende Darstellung in Abbildung 50 die Probenzeitpunkte aus- 125 4. Ergebnisse und Diskussion gewählt, die einander am besten entsprachen. So ist anzumerken, dass für Gruppe 4 und 6 in Woche 8, 2 Wochen nach der letzten Immunisierung, zwar die höchsten Titerwerte gemessen wurden, aber der sinnvollere Vergleich zu den Wiederholungsgruppen bei den Werten aus Woche 10 besteht. Entsprechend beinhaltet Abbildung 50 die normalisierten Werte des Messpunkts vor der Infektion von Woche 62 für Gruppe 1, Woche 10 für Gruppe 4 und 6, Woche 17 für Gruppe 5 und Woche 11 für Gruppe 4 (W), 6 (W) und NK (W). Abbildung 50: IgG- und TNA-Titer der NLV (Ziege) Logarithmische Darstellung der ELISA-Titer (IgG) und TNA-Titer der Seren aller Ziegengruppen direkt vor der Infektion; Die Standardabweichung ist über den Balken als positiver Wert angegeben (bei Messwerten aus vereinigten Mischproben der Gruppe entsprechend nicht); Werten unterhalb der Nachweisgrenze wurde für eine bessere Darstellung ein Wert von 10 zugewiesen; BclAD1D3 bezieht sich auf den Vektor mit flankierenden Signalsequenzen von TPA und LAMP1; Die graue Trennlinie auf Höhe von 100 soll zur ungefähren Abschätzung des möglichen unspezifischen Hintergrunds der ELISA-Titer dienen Die DNA-Vakzinierung von Ziegen mit der erfolgversprechenden Kombination an Vektoren schien nur eine schwache Immunität zu erzeugen. So waren zwar teils leicht erhöhte Titer (≤ 1000, nach Normalisierung) gegen die eingesetzten Antigene vorhanden, diese waren aber nicht signifikant höher als die Titer der Kontrollgruppen (p ≥ 0,057). Der nachträglich verabreichte Proteinboost mit rPA83 (25 µg), rBclA (25 µg), FIS (108 Sporen) und Lipopeptid erzeugte zwar eine deutliche Erhöhung der Antikörpertiter gegen rLF, rPA83 und rBclA, dieser Anstieg entsprach aber für rPA83 und rBclA der gemessenen Titererhöhung von Gruppe 6 und, rBclA ausgenommen, von Gruppe 6 (W) nach der ersten Immunisierung (p ≥ 0,071). Da im 126 4. Ergebnisse und Diskussion Wiederholungsversuch generell niedrigere anti-rBclA IgG-Titer gemessen wurden, erzeugte der Proteinboost in Gruppe 5 signifikant höhere Titer gegen rBclA als in Gruppe 6 (W) (p ≤ 0,021). Damit schien die DNA-Vakzinierung selbst keinen nachweisbaren Effekt zu haben und war auch in Kombination mit einem Proteinboost nicht besser als die analoge Proteinapplikation allein. Beim Vergleich der Gruppen 4 und 6 waren über den gesamten Immunisierungsverlauf keine Unterschiede für die anti-rPA83, -rBclA IgG- und TNA-Titer erkennbar (p ≥ 0,084). Einzige Ausnahme bildeten die anti-rBclA IgG-Titer in Woche 8, die für Gruppe 6 signifikant höher lagen (p = 0,04). In den abgebildeten Werten ist dieser Unterschied tendenziell noch sichtbar, war aber für Woche 11 nicht mehr signifikant. Da für diese Gruppen die anti-FIS IgG-Titer nur für eine vereinigte Mischprobe der Gruppe erhoben wurden, ließ sich keine Aussage über die statistische Signifikanz der Unterschiede tätigen. Allerdings lagen die Werte für Gruppe 6 weit über denen von Gruppe 4, sodass diese das einzige nachweisbare Unterscheidungsmerkmal der beiden Gruppen darstellten. Dies wurde auch in den Wiederholungsversuchen untermauert und statistisch belegt. Während für die anti-rPA83 IgG- und TNA-Titer ebenfalls keine Unterschiede zwischen Gruppe 4 (W) und 6 (W) festgestellt werden konnten (p ≥ 0,142), waren die Antikörpertiter für rBclA und FIS in Gruppe 6 (W) für nahezu alle Probenpunkte signifikant höher als für Gruppe 4 (W) (p ≤ 0,007). Ebenso waren für Gruppe 6 (W) vermehrt TNA-Titer messbar, welche neben den anti-rPA83 IgG-Titern auch mit den anti-FIS IgG-Titern korrelierten (rs ≥ +0,6). Der Vergleich der Gruppen 4 und 6 mit den Wiederholungsversuchen zum Zeitpunkt vor der Infektion (Woche 10 bzw. 11; Abbildung 50) zeigte für keines der Antigene signifikante Unterschiede in der Titerhöhe (p ≥ 0,142). Bei dreifach erhöhter Dosis und Applikation der Vakzinekomponenten in Mischung bei Gruppe 4 (W) und 6 (W) war das unerwartet, könnten aber auch auf die starke Streuung der individuellen Titer zurückzuführen sein. Dies trifft besonders auf die Antikörpertiter gegen rPA83 zu, da diese in den Wiederholungen tendenziell erhöht waren und somit von der Konzentrationserhöhung und/oder Applikation als Mischung profitiert haben könnten. Die anti-FIS IgG-Titer schienen sich in beiden Versuchsdurchführungen ähnlich zu verhalten. Da die Konzentration von FIS in beiden Ansätzen mit 10 8 Sporen unverändert blieb, ist davon auszugehen, dass eine kombinierte Applikation keinen Einfluss auf die immunogene Wirkung hatte. Einzig die anti-rBclA IgG-Titer schienen in der Wiederholung vermindert zu sein, bewegten sich aber generell bei allen Gruppen in einem niedrigen Bereich. Obwohl es keinen signifikanten Unterschied der Antikörpertiter zwischen den Gruppen 4 und 6 und deren Wiederholungen gab, zeigten die Wiederholungen tendenziell bessere Antikörpertiter und höhere Schutzraten. Daher werden im Weiteren nur Gruppe 4 (W) und 6 (W) in der serologischen Gegenüberstellung mit den Lebendvakzine berücksichtigt. 127 4. Ergebnisse und Diskussion 4.3.10 Interner Vergleich der Titerdaten der Ziegenversuche In den vorhergehenden Unterkapiteln wurden bereits die mit der SSLV immunisierten Gruppen und die NLV-Gruppen untereinander kurz zusammengefasst dargestellt und verglichen. Abschließend sollen die serologischen Daten beider Versuchszweige miteinander verglichen werden. Da sich die Gruppen untereinander stark in der Anzahl der Impfungen, Probennahmen und dem Zeitpunkt der Infektion unterscheiden (siehe Tabelle 53 und 54 Anhang), ist ein direkter Vergleich schwierig. Serologisch gesehen, wurden die Gruppen teils gewollt, teils bedingt durch den Versuchsaufbau nicht im selben Zeitrahmen infiziert. Daher wurden für die vereinfachte Darstellung (Abbildung 51) die normalisierten Titerwerte der Zeitpunkte direkt nach dem Abschluss des jeweiligen Impfplans ausgewählt. Dies entspricht den Messwerten von Woche 62 für Gruppe 1 und Woche 4 für 3a, Woche 6 für Gruppe 2, Woche 11 für Gruppe 4 (W) und 6 (W), Woche 17 (nach dem Proteinboost) für Gruppe 5, sowie Woche 62 für Gruppe 3 b. Abbildung 51: Gesamtübersicht IgG- und TNA-Titer (Ziege) Logarithmische Darstellung der IgG-Titer und TNA-Titer der Seren aller Ziegengruppen direkt nach der Beendigung des jeweiligen Impfplans; Die Standardabweichung ist über den Balken als positiver Wert angegeben (bei Messwerten aus vereinigten Mischproben entsprechend nicht); Werten unterhalb der Nachweisgrenze wurde für eine bessere Darstellung ein Wert von 10 zugewiesen; BclAD1D3 bezieht sich auf den Vektor mit flankierenden Signalsequenzen von TPA und LAMP1; Die graue Trennlinie auf Höhe von 100 soll zur ungefähren Abschätzung des möglichen unspezifischen Hintergrunds der ELISA-Titer dienen In der Abbildung wird zunächst nochmal der Unterschied zwischen Gruppe 2 und 3a nach der Immunisierung mit der SSLV deutlich. Für den betrachteten Zeitpunkt sollten beide Gruppen 128 4. Ergebnisse und Diskussion äquivalente Titer zeigen, da sie beide 4 bzw. 6 Wochen zuvor einmalig mit der SSLV immunisiert wurden. Der Zeitunterschied der Messungen in Form von 2 Wochen spielte dabei keine Rolle, da auch die Werte von Gruppe 3a nach 8 Wochen deutlich über denen von Gruppe 2 lagen (p ≤ 0,022). Als funktionell schlechteste der SSLV-immunisierten Gruppen war Gruppe 2 damit serologisch nicht von Gruppe 4 (W) und 6 (W) zu unterscheiden (p ≥ 0,16) für rPA83, TNA und vegetatives Antigen). Das galt ebenso in Tendenz für die anti-FIS IgG-Titer im Bezug zu Gruppe 6 (W), die generell eine sehr hohe Ähnlichkeit im Titerspektrum mit Gruppe 2 aufwies. Diese Ähnlichkeit war auch mit Gruppe 3a und 3b vorhanden, allerdings zeigten beide signifikant höhere (p ≤ 0,001) anti-rPA83, TNA- und anti-vegetative IgG-Titer als Gruppe 6 (W). Die deutlichsten Unterschiede bestanden dabei für die TNA-Titer und anti-vegetativen IgGTiter, die bei den NLV nur knapp oberhalb der Nachweisgrenze messbar waren. Die Antikörperspektren für FIS und rPA83 waren jedoch sehr ähnlich zwischen den SSLV und Gruppe 6 (W). Beim direkten Vergleich der ersten und zweiten SSLV Immunisierung von Gruppe 3 wird deutlich, dass die anti-rPA83 IgG-Titer in beiden Gruppen relativ gleich ausfallen, während die TNA-Titer und die Titer gegen das vegetative Antigen und FIS durch die zweite Immunisierung merklich gestiegen sind. Interessant ist diesbezüglich auch, dass das in Gruppe 6 (W) zugesetzte FIS offenbar ähnlich hohe Titer erzeugen kann wie die lebensfähigen Sporen der SSLV. Zudem scheint die Wirkungen der FIS mehrheitlich nicht auf den Anteil an BclA zurückzuführen zu sein, da eine äquivalente Reaktion gegen BclA ausblieb. Dies ist besonders bei den Gruppen 4, 4 (W) und 5 erkennbar, denen nur BclA verabreicht wurde und deren anti-FIS und -rBclA IgG-Titer eine ähnliche Höhe haben. Im Bezug auf BclA gab es auch einen merklichen Unterschied zwischen den SSLV- und NLV-immunisierten Gruppen. 4.3.11 Todesdatenübersicht der Ziegenversuche Alle Versuchstiere wurden nach dem Tod bzw. der Euthanasie via Blutausstrich auf die Anwesenheit von B. anthracis überprüft. Für keines der überlebenden Tiere waren Erreger im Blut nachweisbar, sodass davon ausgegangen werden kann, dass diese Tiere die Infektion klären konnten. Bei allen verstorbenen Tieren war der Erreger nachweisbar. Für die Betrachtung des Ausgangs der Infektion wurden die Sterbedaten im log-rank-Test miteinander verglichen, wobei als ausschlaggebende Intervalle volle überlebte Tage galten. Die Überlebenszeit ergab sich aus dem Zeitintervall vom Beginn der Infektion bis zu dem Zeitpunkt an dem der Tod des Tieres festgestellt wurde bzw. es euthanasiert oder behandelt wurde. Da in den Versuchswiederholungen der Gruppen 4 (W) und 6 (W) ein anderer Infektionsstamm als für die restlichen Gruppen angewendet wurde, beziehen sich die angegebenen Vergleiche zur Negativkontrolle auf die 129 4. Ergebnisse und Diskussion Gruppe NK (W). Alle anderen Gruppen wurden gegen die Negativgruppen LK, NK und 1 auf Signifikanz geprüft, welche als Kontrollgruppen für die Versuche in Südafrika mitgeführt wurden. Abbildung 52: Überlebensdaten Ziegengruppen (Vergleich der Wiederholungen) Prozentuale Auftragung der Überlebenden über die Zeit in dpi bei einer Infektionsdosis von 36 Sporen für Gruppe 1; 172 Sporen für Gruppe NK, 844 für Gruppe LK, Gruppe 4 und 6 und 918 bzw. 1027 Sporen für die Wiederholungsversuche; Angegebene farblich angepasste p-Werte beziehen sich auf die jeweilige farblich passende Gruppe im Vergleich zu den jeweiligen Kontrollgruppen; p-Werte in schwarz sind Vergleich zwischen den entsprechend gekennzeichneten Gruppen In Abbildung 52 sind zunächst Gruppe 4 und 6 gegen die Wiederholungsversuche, inklusive sämtlicher Kontrollgruppen aufgetragen. Visuell und auch statistisch belegt waren alle vier Kontrollgruppen (Gruppe 1, NK, LK und NK (W)) nicht voneinander verschieden (p ≥ 0,317), obgleich sie jeweils mit einer unterschiedlichen Dosis (36, 172 und 844 Sporen) oder einem anderen Isolat infiziert worden waren. Da in Gruppe 4 keines der 5 Tiere überlebte, lag hier dieselbe Schutzrate vor, wie in den Kontrollgruppen. Ähnlich verhielt es sich für Gruppe 6 bei der 1 von 5 Tieren die Infektion überlebte, allerdings war die Schutzrate aufgrund der teilweise verlängerten Überlebenszeit nur im Vergleich zu Gruppe NK nicht signifikant verschieden.Verglichen zur Gruppe 1 und LK war für Gruppe 6 die Schutzwirkung signifikant (p ≤ 0,039). In den Wiederholungsversuchen mit dreifach höherer Dosis von rPA83 und rBclA und einer verbesserten Applikation, waren die Schutzraten von Gruppe 4 (W) (50 %) und von Gruppe 6 (W) (80 %) signifikant. Der Unterschied zwischen beiden Gruppen war aber aufgrund der verlängerten Überlebenszeit einiger Tiere aus Gruppe 4 (W) nicht signifikant. Der direkte Vergleich bei- 130 4. Ergebnisse und Diskussion der Vakzinegruppen mit ihren Wiederholungsversuchen ergab eine deutliche signifikante Erhöhung der Schutzraten (p ≤ 0,023). Es ist davon auszugehen, dass die Erhöhung der Dosis oder/und die verbesserte Applikation für die deutlich höheren Schutzraten verantwortlich ist. In den nachfolgenden Vergleichen zu den SSLV-immunisierten Gruppen wurden nur die Wiederholungsversuche betrachtet. Abbildung 53: Gesamtübersicht der Überlebensdaten der Ziegengruppen Prozentuale Auftragung der Überlebenden über die Zeit in dpi bei einer Infektionsdosis von 36 Sporen für Gruppe 1; 172 Sporen für Gruppe NK, 844 für Gruppe LK, 850 Sporen für die SSLV-immunisierten Gruppen (2 und 3a/b) und 918 bzw. 1027 Sporen für die Wiederholungsversuche; Angegebene farblich angepasste p-Werte beziehen sich auf die jeweilige farblich passende Gruppe im Vergleich zu den jeweiligen Kontrollgruppen; In Abbildung 53 sind die Sterbedaten der NLV gegen die SSLV-immunisierten Ziegengruppen aufgetragen. Innerhalb der mit der SSLV immunisierten Gruppen 2, 3a und 3b gab es keinen signifikanten Unterschied im Überleben (p ≥ 0,246), was auch an der verringerten Tierzahl aufgrund der Infektionen mit Ehrlichia ruminantium (Herzwasserkrankheit) für Gruppe 3 a und b gelegen haben könnte. Es überlebten 3 von 5 Tieren in Gruppe 2, vier von fünf in Gruppe 3a und drei von drei in Gruppe 3b. Die Vakzinegruppen 4 (W) und 6 (W) waren im Vergleich zu allen SSLV-immunisierten Gruppen äquivalent geschützt (p ≥ 0,165). 131 4. Ergebnisse und Diskussion 4.3.12 Diagnoseprozeduren Feldversuche dieser Art in Großtieren, außerhalb einer umfassend kontrollierbaren Umwelt, sind selten bzw. in neueren Studien kaum vorhanden. Daher sind die diagnostisch erhobenen Daten bezüglich der Temperatur, dem Verhalten und der detektierbaren Bakterienlast aller untersuchten Gruppen sowie deren Implikationen für zukünftige Versuche im Folgenden dargelegt. Eine entsprechende Übersicht der Rohdaten findet sich im Anhang (Tabellen 56 und 57). Bereits während der Versuchsdurchführung in Südafrika wurden initial festgelegte Handlungskaskaden anhand der gemachten Erfahrungen minimal abgeändert, um eine bessere Praktikabilität und Prognose vor Ort zu gewährleisten. Bei der Wiederholung der NLV mit Proteinkomponenten kamen diese Verbesserungen ebenfalls zum Einsatz. Dazu zählte die Erhöhung der Frequenz der Beobachtungsintervalle, der Fütterungen und der Temperaturmessung sowie die bessere Durchführbarkeit der Blutentnahme zu diagnostischen Zwecken mittels Insulinspritzen. Zur Beurteilung des Verlaufes der Infektion und zur Prävention von unnötigem Leid wurden die Tiere regelmäßig auf abnormale Temperatur und auffälliges Verhalten überprüft. Die Temperatur wurde rektal gemessen, wobei die Normalwerte zwischen 38,5 °C und 40,5 °C angenommen wurden. Bei auffälligem Verhalten und ab einer Temperatur von 40,0 °C musste, wenn möglich, eine Blutentnahme zum Bakteriennachweis über einen Blutausstrich erfolgen. Bei einem positiven Befund war das entsprechende Tier mit Penicillin/Streptomycin zu behandeln oder entsprechend des Gesundheitszustands mit Pentobarbital zu euthanasieren. Bei einem negativen Befund war das Tier genauer zu beobachten und in regelmäßigen Abständen erneut Temperatur und Blut zu überprüfen. Für die Beurteilung des Verhaltens galt lebhaftes, aufmerksames und neugieriges Verhalten als normal. Die Fressgewohnheiten und Interaktionen mit den Pflegern und Artgenossen sowie die Körperhaltung und der Zustand des Fells waren weitere Kriterien. 4.3.12.1 Temperatur Bereits zu Beginn des Infektionsversuchs in Südafrika, im Zuge der Eingangsuntersuchung im Anschluss an den Transport, war die enorme Schwankungsbreite der individuellen Temperaturen mit 37,6 – 40,2 °C (Kapitel 4.3.1) auffällig. Dies trat auch direkt vor und während der Infektion auf und war vermutlich auch auf den mit dem Transport und der Infektion verbundenen Stress zurückzuführen, zumal sich die Temperaturen am Folgetag der Infektion mit 36,5 – 39,3 °C bei deutlich niedrigeren Temperaturen einpegelten. Außerdem waren die Tiere durch die Haltung in einem Außengehege mit Überdachung den Temperaturschwankungen (15 – 35 °C) der Umgebung permanent ausgesetzt. Wichtig hierbei ist, dass die Temperatur nie bei allen Tieren gleich stark schwankte, sodass theoretisch auch individuelle Faktoren wie Alter, 132 4. Ergebnisse und Diskussion Gewicht, Geschlecht, Brunst, persistierende Infektionen oder die Stellung in der Gruppe eine Rolle spielen könnten. Die Versuchswiederholungen in der Türkei beinhalteten keinen Transport zwischen Immunisierung und Infektion und fanden im Zeitraum der Infektion ausschließlich innerhalb eines abgeschlossenen Gebäudes mit konstanter Außentemperatur (20 – 22 °C) statt. Daher waren die individuellen Temperaturschwankungen vor (38,1 – 40,1 °C) und innerhalb der Infektionszeit geringer, sodass eine bessere Einschätzung des Gesundheitszustands über die Temperatur möglich war. Trotzdem waren auch hier bereits vor der Infektion, wie auch bei den Versuchen in Südafrika, 2 Tiere mit Temperaturen über 40 °C vorhanden. Da diese Temperaturerhöhung nicht auf die Infektion zurückzuführen sein konnten und diese sich in den meisten Fällen nach der Infektion wieder verringerten, wurden erhöhte Temperaturen bis 3 hpi nicht in den Betrachtungen berücksichtigt. Die Festlegung der Temperaturschwelle von 40,0 °C als Indikator für weitere diagnostische Maßnahmen stellt eine artifizielle Grenze dar, deren Sinnhaftigkeit im Zuge der erzielten Ergebnisse erörtert werden sollte. Aufgrund der Gruppenstärke und den äußeren Gegebenheiten in Südafrika wurde die Temperatur der Ziegen nur einmal täglich morgens nach der Reinigung des Geheges und vor der Fütterung durchgeführt. Weitere Messungen wurden individuell entsprechend des Zustandes der Tiere durchgeführt. Daher sind die Zusammenhänge zwischen erhöhter Temperatur (>40,0 °C) und dem Infektionsstatus der Tiere mit einer Ungenauigkeit (ca. 24 h) behaftet. Die Wiederholungsversuche in der Türkei erlaubten eine reguläre Messung der Temperatur aller Tiere zweimal täglich, sodass die Veränderungen der Temperatur über die Zeit dort besser dokumentiert sind. Die Ergebnisse der Versuche in Südafrika und der Türkei werden daher zunächst getrennt analysiert. Von den in Südafrika insgesamt 29 infizierten Tieren überlebten 11 den Versuch. In Abbildung 54 sind die Temperaturspektren jedes Tiers für den Infektionszeitraum, mit maximal und minimal gemessener sowie durchschnittlicher Temperatur, aufgeführt. Es ist zu sehen, dass drei der 11 überlebenden Tiere dabei zu mindestens einem Messzeitpunkt eine erhöhte Temperatur aufwiesen. Von den 18 verstorbenen Tieren zeigten 13 eine Abbildung 54: Temperaturspektrum Ziegenversuch (Südafrika) Gemessene Temperaturen der einzelnen Versuchstiere für den Zeitraum von 3 hpi bis zum Tod bzw. Ende des Versuchs; Angegeben sind jeweils die Durchschnittstemperatur (Symbol) und die maximal bzw. minimal gemessene Temperatur jedes infizierten Versuchstiers; Die Gruppenzugehörigkeit ist durch entsprechende Kürzel gekennzeichnet (L – Lipopeptidkontrolle; N – Negativkontrolle). Die Gruppen sind in Überlebende und Verstorbene unterteilt und anhand aufsteigender Maximaltemperatur sortiert. 133 4. Ergebnisse und Diskussion erhöhte Temperatur, welche teilweise auch bis zum Tod erhalten blieb. Die niedrigsten gemessenen Temperaturen bei überlebenden und verstorbenen Ziegen unterschieden sich mit 36,2 °C und 36,3 °C kaum. Die höchste gemessene Temperatur bei den überlebenden Ziegen betrug 40,5 °C, während bei den Verstorbenen Temperaturen bis 41,6 °C messbar waren. Somit verstarben alle Tiere, die eine Temperatur von mindestens 40,6 °C aufwiesen im weiteren Verlauf an der Infektion. Von den in der Türkei infizierten 22 Tieren überlebten 12 den Versuch. Die gemessenen Temperaturen dieser Tiere sind analog zu den Versuchen in Südafrika in Abbildung 55 dargestellt. Die Hälfte der Überlebenden und acht der 10 verstorbenen Tiere hatte dabei zu mindestens einem Zeitpunkt vor dem Tod eine erhöhte Temperatur. Die niedrigsten gemessenen Temperaturen waren mit 37,6 °C bei den Überlebenden und 38,0 °C bei den Verstorbenen kaum unterschiedlich, lagen aber bedeutend höher als die Messungen in Südafrika. Auch die maximal gemessene Höchsttemperaturen war mit 41,6 °C bei VerAbbildung 55: Temperaturspektrum Ziegenversuch (Türkei) Gemessene Temperaturen der einzelnen Versuchstiere für den Zeitraum von 3 hpi bis zum Tod bzw. Ende des Versuchs; Angegeben sind jeweils die Durchschnittstemperatur (Symbol) und die maximal bzw. minimal gemessene Temperatur jedes infizierten Versuchstiers; Die Gruppenzugehörigkeit ist durch entsprechende Kürzel gekennzeichnet (N – Negativkontrolle (W); 4 – Gruppe 4 (W); 6 – Gruppe 6 (W)). Rot markiert sind die Tiere, die nach dem Nachweis von Bakterien im Blut behandelt wurden. Die nach der Behandlung gemessenen Temperaturen wurden nicht berücksichtigt. Die Gruppen sind in Überlebende und Verstorbene unterteilt und anhand aufsteigender Maximaltemperatur sortiert. storbenen und Überlebenden gleich, wohingegen es bei den Versuchen in Südafrika einen Unterschied gegeben hatte. Da bei den Versuchen in Südafrika aber nur drei überlebende Tiere überhaupt eine Temperaturerhöhung zeigten, ist davon auszugehen, dass der dort notierte Unterschied zufällig war und die umfangreicheren Daten der Nachfolgeversuche ein realistischeres Bild ergeben. Die deutlich höher ausfallenden Minimaltemperaturen der Versuchstiere in der Türkei sind vermutlich ein weiterer Indikator für die geringeren Schwankungsbreiten der Temperatur der einzelnen Tiere durch die haltungsbedingten, konstanteren Umgebungstemperaturen. Betrachtet man die Versuche in Südafrika und der Türkei zusammengenommen, fällt auf, dass vor allem naive Tiere, die paramunisiert wurden oder keine Impfung erhalten hatten, mehrheitlich ohne messbares Fieber verstarben. Dies betraf insgesamt sechs der 10 naiven Tiere. Die restlichen 4 Tiere wiesen zumindest kurzzeitig (1 – 7 h) Temperaturen von 40 °C und höher auf. Da die regulären Temperaturmessintervalle 12 – 24 h betrugen, könnte es möglich sein, dass auch die unauffälligen Tiere kurzzeitig Fieber aufwiesen, dies aber nicht detektiert wurde. 134 4. Ergebnisse und Diskussion Darüber hinaus schienen nicht nur versterbende Tiere zeitweilig ein Fieber aufzuweisen, da auch neun der überlebenden Tiere eine erhöhte Temperatur aufwiesen, die nicht von der der später versterbenden zu unterscheiden waren. Damit gab es keine eindeutige Temperaturschwelle. Betrachtet man allerdings die Veränderung des Anteils an versterbenden Tieren mit Erhöhung der maximal gemessenen Temperatur (Abbildung 56), lässt sich ein optimaler Schwellenwert besser abschätzen. Insgesamt beinhalteten die Versuche 51 Tiere, von denen 28 durch die Infektion verstarben, was einen Anteil von 55 % ausmacht. Mit steigender maximal gemessener Temperatur erhöht sich der Anteil versterbender Tiere an der Gesamtzahl an Tieren die ebenfalls mindestens eine solche Temperatur aufwiesen. Die entsprechende Kurve (Abbildung 56, dunkelblaue Kurve) stellt somit den prozentualen Anteil richtiger Vorhersagen für den Tod eines Tieres auf Grundlage einer bestimmten Temperaturschwelle dar. Die niedrigste gemessenen Höchsttemperatur aller involvierten Tiere betrug 38,7 °C. Würde man diese Temperatur als Schwellenwert verwenden, wären 100 % der später versterbenden Tiere erfasst (Abbildung 56, blaue Kurve), der Anteil an richtig diagnostizierten Tieren läge aber nur bei 55 % und würde alle 51 Tiere mit einbeziehen. Damit hätte ein Schwellenwert bei dieser Temperatur keine Funktionalität als praktikable Diagnosehilfe. Eine Verbesserung ergibt sich, wenn der Schwellenwert so gewählt wird, dass möglichst viele richtige Vorhersagen getroffen werden, bei möglichst geringer Zahl an falsch Negativen. Wie in Abbildung 56 zu sehen ist, sinkt der Anteil versterbender Tiere an deren Gesamtzahl bereits zwischen 38,9 und 39,8 °C auf 75 % ab. Für einen diagnostischen Schwellenwert sind die Temperaturen zwischen 37,4 – 39,6 °C aber ungeeignet, weil sie der natürlichen Schwankungsbreite nicht infizierter Tiere entsprechen (Tabelle 55, Anhang). Daher kommt erst ein Schwellenwert ab 39,6 °C in Frage. Für die Höchsttemperaturen zwischen 39,8 und 40,4 °C bleibt der Anteil verstorbener Tiere an deren Gesamtzahl konstant (hellblaue Kurve), während der Anteil richtiger Vorhersagen weiter zunimmt (dunkelblaue Kurve). Das bedeutet, dass in diesem Bereich nur falsch positive Tiere aus dem Anteil ausgeschlossen werden und somit die Temperatur von 40,4 °C einen optimalen Schwellenwert darstellt, der eine korrekte Vorhersage für 75 % der verstorbenen Tiere ermöglicht. Dies unterstützend zeigt der weitere Verlauf der Kurven, dass die Anzahl Verstorbener an der Gesamtzahl Verstorbener, die diese Temperatur mindestens aufwiesen, ab 40,4 °C rapide abnimmt und gleichzeitig die Wahrscheinlichkeit für eine richtige Aussage nur langsam weiter ansteigt (dunkelblau). Die Fieberdauer im Temperaturbereich zwischen 40,0 und 40,8 °C ist zudem mit <7 h sehr kurz (Abbildung 56, Balken), was die Detektionsfähigkeit einschränkt. Ebenso ist das Auftreten eines kurzen Fiebers bei einem später versterbenden Tier erstmals bei 40,4 °C vermerkt, während bei niedrigeren Maxi135 4. Ergebnisse und Diskussion maltemperaturen nur falsch positive Tiere Fieber zeigten. Länger andauerndes Fieber über teils mehrere Tage ist sowohl bei Überlebenden, als auch bei Verstorbenen erst ab einem Temperaturbereich von 40,8 – 41,6 °C aufgetreten. Für eine Verwendung der Temperatur als diagnostisches Mittel sollte daher weiterhin eine Temperatur von 40,0 °C und höher als Indikator für Fieber angenommen werden. Weiterführende diagnostische Mittel, wie eine Blutentnahme zur Detektion von Bakterien, sollten aber erst ab einer Temperatur von 40,4 °C vorgenommen werden. Frequentere Kontrollen auf Temperatur und Verhalten wären aber weiterhin ab 40,0 °C angeraten. Abbildung 56: Temperaturverhältnisse Die Kurvenverläufe (y-Achse links) geben den prozentualen Anteil verstorbener Ziegen an der Gesamtzahl an Ziegen (schwarz) bzw. der Gesamtzahl an verstorbenen Ziegen (blau) an, die eine Höchsttemperatur von mindestens der an der xAchse angegebenen Temperatur entsprach. Es wurden alle 51 Tiere der Infektionsversuche in Südafrika und der Türkei berücksichtigt. Das integrierte Balkendiagramm (y-Achse rechts) stellt die Dauer des Fiebers (≥ 40,0 °C) von Tieren mit der angegeben Höchsttemperatur dar (Anzahl der Tiere farblich gekennzeichnet über den entsprechenden Balken), unterteilt in Überlebende (grün) und Verstorbene (rot). Für eine verbesserte Darstellung wurde eine Maximaldauer von 48 h als Obergrenze festgesetzt, sodass alle Balken auf dieser Höhe einer Fieberdauer von ≥ 48 h entsprechen. Da 90 % der Tiere (32,1 % der Gesamtzahl Verstorbener) mit einer Höchsttemperatur von ≥ 41,4 °C im weiteren Verlauf verstarben, könnte diese Temperatur als zusätzlicher Schwellenwert dienen. Zur Vermeidung von Leid könnten Tiere mit dieser Temperatur auch ohne den Nachweis von Bakterien im Blut als verstorben gewertet und entsprechend frühzeitig behandelt oder euthanasiert werden. Außerhalb eines Infektionsversuchs wäre eine Behandlung von Verdachtsfällen auf Grundlage der rektal gemessenen Temperatur ab 40,4 °C angeraten. Allerdings ist zu beachten, dass naive Tiere tendenziell ohne detektierbares Fieber verstarben bzw. nur kurz vor dem Tod ein Fieber zeigten und die hier betrachteten Tiere zum Teil eine partielle Immunität aufwiesen. 136 4. Ergebnisse und Diskussion 4.3.12.2 Zusammenhänge von Fieber, Antikörpertitern und Schutz Die Entwicklung einer Vorhersage der Schutzrate auf Grundlage der immunologischen Daten ohne die Notwendigkeit eines Infektionsversuchs ist häufiger Bestandteil der Vakzineentwicklung. Daher werden im Folgenden die Zusammenhänge zwischen der beobachteten Schutzrate, den gemessenen anti-rPA83 IgG-Titern Tabelle 50: Zusammenhänge von Fieber, Antikörpertitern und Schutz (Ziegen) Gruppe Ziegen anti-rPA83 Fieber Nr. IgG-Titerb [h] G1 8185 <100 7 G6 8201 <100 9 LK 8202 <100 und der Entwicklung von Fieber nach der Infektion näher betrach- LK 8203 <100 NK II <100 tet. In Tabelle 50 sind alle 51 Tiere der Ziegenversuche aus Süd- NK VI <100 NK (W) 96163 <100 NK (W) 97014 <100 afrika und der Türkei in aufsteigender Reihenfolge ihrer normalisierten anti-rPA83 IgG-Titer aufgeführt. Überlebende sind dabei grün hinterlegt und Verstorbene orange. Die Fieberdauer ist ebenfalls vermerkt. Dabei war der zeitliche Abstand zwischen dem ersten und letzten Auftreten einer kontinuierlich erhöhten Temperatur ausschlaggebend. Daher stellt der angegebene Zeitraum eine Mini- NK (W) 97015 <100 NK (W) kk <100 G2 8213 164 3 6 7 G1 8187 238 G4 8192 542 (1) G6 (W) 96139 781 9 G3a 8178 792 (1) G4 8188 814 (1) G6 8200 915 G4 (W) 627399 972 malangabe dar, wobei nochmals anzumerken ist, dass die reguläre G2 8211 1059 72 G6 8194 1100 (1) Frequenz der Temperaturmessung unauffälliger Tiere bei 12 – 24 h G4 8198 1315 (1) G6 (W) 627396 1494 32 lag, sodass die tatsächliche Fieberdauer entsprechend länger sein G2 8212 1853 (1) G3a 8180 2143 95 konnte. Für einige Tiere war eine erhöhte Temperatur an nur G4 (W) 580319 2310 8 G4 8190 2543 9 einem Messzeitpunkt feststellbar, diese sind in der Tabelle mit G6 (W) 96157 2617 G4 (W) 97013 2628 39 G6 8191 2686 26 G4 (W) 99348a 2787 48 G4 (W) 627398a 3006 15 G6 (W) 97162 3481 41 einer artifiziellen Fieberdauer von 1 h angegeben. Aufgrund der beschriebenen Ungenauigkeit der Temperaturerfassung, bedingt durch die Messintervalle, besteht daher generell die Möglichkeit, dass auch Tiere ohne ein detektierbares Fieber ebenfalls eine kurzzeitige Temperaturerhöhung aufwiesen. G2 8214 3637 G4 8199 3871 G3a 8172 3892 G6 (W) 729277 4273 120 G3a 8173 4720 (1) Anhand der sortierten Daten in Tabelle 50 ist ersichtlich, dass G6 (W) 728758 4996 G4 (W) Gk 5022 alle Tiere bis zu einem anti-rPA83 IgG-Titer von 542 vor der G6 8195 5386 G6 (W) 729278 5399 Infektion mit teils nur kurzzeitig gemessenem Fieber (< 10 h) aus- G6 (W) 729284 5920 G2 8210 8564 nahmslos verstorben sind. Dies betraf insgesamt 46,5 % aller ver- G4 (W) 728761 11190 G3a 8176 14847 storbenen Tiere. Ab dem genannten Antikörpertiter zeigten zudem G6 (W) 627395 17476 G6 (W) 580316 19442 alle später verstorbenen Tiere an mindestens einem Messzeitpunkt G4 (W) 580315 23173 G3b 8182 31321 ein detektierbares Fieber. Generell wiesen Tiere mit anti-rPA83 IgG-Titern unter 972 oder 2617, wenn nur die Überlebenden betrachtet werden, kein oder nur kurzzeitig ein Fieber (< 10 h) auf. Bei Titerwerten zwischen 781 und 3006 war keine eindeutige Tendenz zu erkennen, da sowohl 26 % der Überlebenden als auch 43 % der Verstorbenen Antikörpertiter in dieser Höhe aufwiesen. (1) G3b 8175 37995 G3b 8181 38671 a wurde behandelt und überlebte b direkt vor der Infektion 35 48 24 71 (1) Auftragung aller 51 infizierten Tiere in aufsteigender Reihenfolge ihrer antirPA83 IgG-Titer; Tieren bei denen nur eine einzige Messung eine Temperatur von ≥ 40,0 °C ergab wurde eine Fieberdauer von (1) h zugeschrieben; 137 4. Ergebnisse und Diskussion Ab einem Antikörpertiter von 3481 überlebte die Mehrheit (85 %) der Tiere die Infektion, wobei 74 % aller Überlebenden und 11 % der Verstorbenen mindestens diesen Titer aufwiesen. Auffällig ist hierbei, dass die beiden antibiotisch behandelten Tiere bezüglich ihrer Antikörpertiter direkt an diesem Schwellenwert angrenzen, was eventuell den Erfolg der Behandlung bedingt hat. Einen merklichen Unterschied zwischen Verstorbenen und Überlebenden bezüglich der Fieberdauer gab es nicht, da unabhängig vom Ausgang der Infektion kurzzeitiges Fieber bis teils tagelang anhaltende Fieberperioden möglich waren. Ähnliche Zusammenhänge zwischen den gemessenen anti-rPA83 IgG-Titern und dem beobachteten Schutz ergaben sich für die analoge Gegenüberstellung aller Einzeltiter der Mausgruppen (siehe Anhang, Tabelle 58), die mit ~1000 Sporen infiziert wurden. Alle Tiere mit einem Antikörpertiter von ~800 und niedriger verstarben ausnahmslos, was insgesamt 33 % der Verstorbenen Tiere betraf. Ab einem Antikörpertiter von ~30000 überlebten 75 % aller Tiere, wobei 69 % aller Überlebenden und 25 % aller Verstorbenen mindestens diesen Titer aufwiesen. Der dazwischenliegende Wertebereich beinhaltete (nahezu identisch zu den Ergebnissen aus den Ziegenversuchen) 42 % der Verstorbenen und 26 % der Überlebenden. Die Gegenüberstellung der Mausgruppen, deren Infektionsdosis ~2000 Sporen betrug, zeigte keine entsprechenden Schwellenwerte. In den Korrelationsberechnungen innerhalb der einzelnen Gruppen waren, bis auf zwei Ausnahmen, keine signifikanten Korrelationen zwischen Antikörpertitern und der überlebten Zeit nachweisbar. Aufgrund der sichtbaren Schwellenwerte unter Berücksichtigung aller Individuen wurden gruppenübergreifende Korrelationsberechnungen durchgeführt. Einbezogen wurden alle Messwerte, für die Einzelmessungen gegen das jeweilige Antigen vorhanden waren, unterteilt in Ziegen- und Mausversuche, sowie im Falle der Mausversuche nach unterschiedlichen Infektionsdosen (~1000 und ~2000 Sporen). Korrespondierend zu den zuvor beschriebenen Schwellenwerten in Ziegen ergab sich eine signifikante Korrelation der anti-rPA83 IgG-Titer zur überlebten Zeit (rs = +0,606). Ähnlich verhielt es sich für FIS (rs = +0,562) und das vegetative Antigen (rs = +0,528). Für die TNAund anti-rBclA IgG-Titer waren keine signifikanten Korrelationen messbar (r s ≤ +0,192). Damit schienen in Ziegen bei einer Infektionsdosis von ~1000 Sporen vor allem die Antikörpertiter gegen die Spore (exklusive BclA), den vegetativen Zellkörper und PA für das Überleben von Bedeutung zu sein, wobei nur für rPA83 eindeutige Schwellenwerte sichtbar waren. Die Vergleiche der Mausversuche beschränkten sich, aufgrund der vorhandenen Daten, auf rLF, rBclA, rPA83, TNA und die Kapsel. Bei einer Infektionsdosis von ~1000 Sporen korrelierten die anti rPA83 IgG-Titer ebenfalls signifikant mit der überlebten Zeit (r s = +0,507). Eine weitaus schwächere signifikante Korrelation ergab sich für die anti-rLF IgG- und TNA-Titer (rs ≤ +0,334). Für rBclA und die Kapsel waren keine signifikanten Korrelationen sichtbar 138 4. Ergebnisse und Diskussion (rs ≤ ±0,188). Die Vergleiche innerhalb der Mausgruppen die mit ~2000 Sporen infiziert wurden, ergaben für die anti-rBclA IgG- und anti-Kapsel IgG-Titer (rs ≤ ±0,213), sowie TNA-Titer (rs = +0,313) ähnliche Werte, wohingegen die anti-rPA83 IgG-Titer mit r s = -0,406 zwar signifikant, aber entgegengesetzt zur überlebten Zeit korrelierten. Daher scheint auch in AuszuchtMäusen ein Überleben mit den anti-rPA83 IgG-Titern in Zusammenhang zu stehen, wobei deren Relevanz für das Überleben abhängig von der Infektionsdosis variiert. In Tabelle 51 sind die Titerdaten für rPA83 von vor und nach der Infektion für alle Überlebenden, inklusive der behandelten Ziegen dargestellt. Bei Tabelle 51: Zusammenhang zwischen Antikörpertiter und der Entwicklung von Fieber Gruppe Ziegen anti-rPA83 IgG-Titer Fieberdauer Nr. vor Infektion nach Infektion G6 8200 915 629 0 der Betrachtung der Daten war aufgefallen, dass 4 (W) 627399 972 762 0 größtenteils nur diejenigen Tiere die ein nachweis- 6 (W) 96157 2617 1090 0 4 (W) Gk 5022 1404 0 6 (W) 728758 4996 2618 0 G3a 8172 3892 6150 0 G3b 8182 31321 12497 0 G3b 8175 37995 23689 0 Diese Tiere sind entsprechend gelb hinterlegt. Tie- G3a 8176 14847 23882 0 G3b 8181 38671 24365 0 re, deren anti-rPA83 IgG-Titer nach der Infektion 6 (W) 580316 19442 31797 0 6 (W) 729278 5399 34742 0 den Titern vor der Infektion entsprachen oder sogar G2 8214 3637 48060 0 G3a 8173 4720 86790 (1) sanken, hatten hingegen kein nachweisbares Fieber G2 8210 8564 89882 0 6 (W) 96139 781 94157 9 (blau). Dies galt ohne Ausnahme. Die Sortierung G3a 8178 792 95756 (1) 6 (W) 729277 4273 134139 120 der Daten in aufsteigender Höhe der anti-rPA83 G2 8212 1853 255200 (1) IgG-Titer nach der Infektion (Tabelle 51) zeigt 4 (W) 580315 23173 282079 (1) 6 (W) 97162 3481 283649 41 4 (W) 728761 11190 658286 24 4 (W) 627398a 3006 800475 15 4 (W) 99348a 2787 1244507 48 6 (W) 729284 5920 1479449 48 bares Fieber hatten, eine deutliche Erhöhung der anti-rPA83 IgG-Titer nach der Infektion aufwiesen. deutlich die Abhängigkeit der Titer nach der Infektion zu einer Entwicklung von Fieber. Dabei sind die vermeintlichen 3 Ausnahmen (weiß) im Übergangsbereich angesiedelt, sodass bei diesen entweder eine Titererhöhung ohne Fieber stattgefunden haben muss oder die Detektion des Fiebers bei diesen Tieren durch die geringe Dauer des Fiebers nicht funktioniert hat. Dafür spricht auch, mit Aus- a [h] zählte zu den Verstorbenen, wurde aber behandelt und überlebte Aufgelistet sind alle 23 Überlebenden der Ziegenversuche inklusive der zwei Individuen die durch eine Behandlung gerettet werden konnten. Die Reihenfolge ergibt sich aus den aufsteigend sortierten anti-rPA83 IgG-Titern nach der Infektion; Blau hinterlegt sind alle Tiere bei denen die Titer nach der Infektion ähnlich hoch wie vor der Infektion lagen oder absanken und bei denen kein Fieber gemessen werden konnte; Gelb hinterlegt sind alle Tiere deren Titer nach der Infektion deutlich anstieg und die ein Fieber aufwiesen; Weiß hinterlegt sind alle Tiere die ebenfalls einen Titeranstieg zeigten, aber kein Fieber aufwiesen. nahme von Ziege Nr. 729277, die eine Fieberdauer von 120 h aufwies, die geringe Fieberdauer im Titerbereich nach der Infektion von ~34000 – ~280000. Eine Abhängigkeit der Fieberentwicklung bzw. Titererhöhung nach der Infektion zu den Titern vor der Infektion scheint dabei nicht zu existieren, da Tiere mit ähnlichen Titern vor der Infektion in beiden Kategorien zu finden sind. Auch ein Zusammenhang zur Stärke der Titererhöhung ist nicht ersichtlich. 139 4. Ergebnisse und Diskussion 4.3.12.3 Verhalten Die Beurteilung des Verhaltens und des Zustandes der Tiere war subjektiv und damit ungenau, da Stress und Wetterverhältnisse sowie die Variation in den Möglichkeiten zur Beobachtung die Ergebnisse beeinflussten. Hierbei gilt es vor allem zu beachten, dass zwischen 19:00 – 06:00 Uhr unter den Bedingungen der Versuchsanlage im Krüger Nationalpark keine ununterbrochene Beobachtung möglich war. Tiere, die in diesem Zeitraum starke Symptome entwickelten, machten aber für gewöhnlich auf sich aufmerksam und wurden entsprechend versorgt und beobachtet. Der Versuchsaufbau in der Türkei erlaubte auch Beobachtungen zwischen 19:00 – 06:00, was bei auffälligen Tieren in Intervallen von 3 Stunden wahrgenommen wurde. Allerdings konnten außerhalb dieser Intervalle, aufgrund der Entfernung zu den Schlafplätzen der Helfer, kranke Tier nicht auf sich aufmerksam machen. Tagsüber wurden die Tiere im Zuge der regelmäßigen Temperaturmessung genau überprüft und im Tagesverlauf ununterbrochen beobachtet. Bei den Fütterungen konnten Agilität, Appetit und Durchsetzungsvermögen beim Ringen um Futterplätze eingeschätzt werden, welche im Gesamtbild den stärksten, früh sichtbaren Indikator für eine Veränderung des Verhaltens ergaben. In einigen Fällen war auch auffällig, dass die Tiere zwar Appetit hatten, jedoch das Fassen und Kauen der Nahrung schwerzufallen schien. Deutlicher auffallende Verhaltensweisen waren erst später erkennbar und beinhalteten das stoische Herumstehen mit gekreuzten Beinen und gesenktem Kopf und die Abtrennung von der Gruppe. Selbst wenn es zuvor keine Anzeichen einer Infektion gab, waren ab ca. eine Stunde vor dem Tod die Symptome und Verhaltensauffälligkeiten, wie lautes Stöhnen, Gleichgewichtsstörungen, Orientierungslosigkeit und Krämpfe, unmissverständlich. Aufgrund der unterschiedlichen Versuchsbedingungen werden die Versuche in Südafrika und der Türkei zunächst getrennt beschrieben. Unter Ausschluss der Symptome, die erst kurz vor dem Tod (<1 h) erkennbar waren, zeigten in Südafrika sieben der überlebenden 11 und 11 der 18 verstorbenen Tiere auffälliges Verhalten. In der ursprünglichen Versuchsplanung sollten Temperatur und Verhalten Aufschluss über den Gesundheitszustand bezüglich der Infektion geben. Es wurde davon ausgegangen, dass nicht erkrankte Tiere weder auffälliges Verhalten noch eine erhöhte Temperatur haben. In der praktischen Durchführung zeigte sich aber, dass nur drei der 11 überlebenden Tiere dieser Annahme entsprachen. Die restlichen überlebenden Tiere zeigten vordergründig auffälliges Verhalten, was im Falle des zusätzlichen Auftretens von hoher Temperatur gleichzeitig oder zuerst bemerkbar war. Für die Beurteilung kam erschwerend hinzu, dass auch fünf der 18 verstorbenen Tiere im Bezug auf Temperatur und Verhalten vollkommen unauffällig waren. Die restlichen Tiere, die der Infektion erlagen, waren mehrheitlich sowohl verhaltensauffällig als auch fiebrig, wobei die erhöhte Temperatur erstes und/oder hauptsächliches Merkmal war. 140 4. Ergebnisse und Diskussion Bei der Versuchsdurchführung in der Türkei zeigten sieben der 10 verstorbenen Tiere auffälliges Verhalten, was sich mehrheitlich in Appetitlosigkeit äußerte. Mit einer Ausnahme traten Verhaltensauffälligkeiten erst nach der Detektion einer erhöhten Temperatur auf, wobei bei sieben der verstorbenen Tiere beides vorhanden war. Zwei Tiere verstarben ohne jegliche frühzeitig (<1 h vor dem Tod) erkennbare Symptomatik. Von den überlebenden Tieren zeigte kein Tier auffälliges Verhalten und 6 der 12 Tiere hatten eine erhöhte Temperatur. Damit war die Beurteilung von Temperatur und Verhalten in den Wiederholungsversuchen eindeutiger, was zum einen an der frequenteren Fütterung und Temperaturmessung gelegen haben könnte aber auch an der Haltung der Tiere in einem abgeschlossenen Gebäude. Unabhängig von der stark schwankenden Außentemperatur war innerhalb des Gebäudes eine konstante Temperatur von 20 – 22 °C gegeben. Bei den Versuchen in Südafrika war nur ein Außengehege mit Überdachung vorhanden, das zwar Schutz vor Regen und direkter Sonneneinstrahlung bot, aber die Tiere den Temperaturschwankungen der Umwelt aussetzte. Dies hatte möglicherweise zu erhöhtem Stress geführt, der sich in teils unspezifischem, auffälligem Verhalten äußerte und die Aussagekraft von Verhaltensauffälligkeiten als diagnostisches Mittel beeinträchtigt haben könnte. Da generell eine Temperaturerhöhung mehrheitlich das eindeutigere und im Versuchsaufbau in der Türkei auch das erste detektierbare Symptom darstellte, sollte die Priorität bei der Diagnostik auf der Temperaturmessung liegen und die Veränderung des Verhaltens nur unterstützende Funktion haben. Die Relevanz der Verhaltensbeobachtungen ist dabei aber umso höher, je reduzierter die regelmäßigen Temperaturerhebungen ausfallen. 4.3.12.4 Detektion von Bakterien im Blut Auffälligkeiten bei Temperatur und Verhalten galten als Indikatoren für ein Fortschreiten der Infektion, deren Bestätigung über einen Blutausstrich und visuellen Nachweis von B. anthracis nötig war. Ein entsprechend positiver Befund hierbei galt als ausreichendes Anzeichen für einen im weiteren zum Tode führenden Krankheitsverlauf und hatte die Euthanasie des betreffenden Tieres zur Vermeidung von Leid zur Folge. Zu Beginn der Versuche war die Möglichkeit der Blutentnahme durch die Verwendung von entsprechenden für Ziegen vorgesehenen Kanülen limitiert, da es nicht praktikabel war mit diesen, mehrmals täglich an ähnlichen Stellen Blut abzunehmen. Zusätzlich erschwert wurde die Diagnose durch die nicht vorhandenen Daten zum Zeitpunkt des Auftretens von B. anthracis im Blutkreislauf bei Ziegen. Somit war die Detektionsrate von Bakterien im Blut vor dem Tod der Tiere begrenzt und größtenteils aufgrund der geringen Frequenz der Probennahme dem Zufall überlassen. Anlässlich der Unzulänglichkeit dieser Methode wurde gegen Ende der Versuchsdurchführung in Südafrika eine Blutentnahme an der Halsvene mittels Insulinspritzen 141 4. Ergebnisse und Diskussion erprobt. Mit diesen war es möglich minimal invasiv mehrmals täglich sehr kleine Mengen (<100 µl) Blut zu entnehmen. Zur Gewinnung besserer Daten bezüglich des Auftretens von Bakterien im Blut von Ziegen wurden die Ziegen der Gruppe LK nach der Infektion im Abstand von 3 – 6 h auf die beschriebene Art geblutet und auf Bakterien untersucht. Ebenso fand diese verbesserte Blutentnahme zur Detektion von Bakterien im Blut in der Gesamtheit der Ziegenversuche in der Türkei Anwendung. Zusammengenommen ergaben die gesammelten Daten beider Versuche (Tabelle 52) ein deutlicheres Bild. Der frühest mögliche Zeitpunkt ab dem Bakterien Tabelle 52: Detektion von Bakterien im Blut Ziegen Gruppe Nr. II NK VI NK 97014 NK (W) 97015 96163 letzter negativer erster positiver Blutabstrich Blutabstrich [h vor Tod] [h vor Tod] 8,2 – 10 2,8 – 4 6 n.v. * n.v. n.v. * NK (W) n.v. n.v. * NK (W) 5,75 1 kk NK (W) 9,25 – 10,25 5,25 – 6,25 8202 LK 4,83 1,83 8203 LK 5,5 0 8185 1 9,3 2,3 8187 1 35,3 0 8211 2 14,67 0 8213 2 n.v. 0 8180 3a 25,2 1,2 8188 4 n.v. 0,58 8190 4 n.v. 0 8192 4 n.v. 0,33 8198 4 n.v. 0 8199 4 n.v. 0,25 627398 4 (W) 580319 4 (W) 97013 4 (W) 99348 4 (W) 8191 6 n.v. 0 8194 6 n.v. n.v. * 8195 6 12,75 0 8201 6 n.v. 0 627396 6 (W) 4 – 8,75 n.v. * 627395 6 (W) 7,5 n.v. * wurde behandelt und überlebte 9,25 – 13,25 n.v. * 52 25,5 wurde behandelt und überlebte * Verifikation von Bakterien im Blut erst nach dem Tod Ergebnisse der Blutausstriche aller an B. anthracis verstorbenen Ziegen aus den Versuchen in Südafrika und der Türkei. Zeitangaben beziehen sich auf Stunden vor dem Todeszeitpunkt und sind als Zeitintervall angegeben, wenn der Zeitpunkt nicht eindeutig bestimmt werden konnte; Die aussagekräftigsten Ergebnisse sind fett hervorgehoben; n.v. - nicht vorhanden; 0 – positiver Blutausstrich zum Zeitpunkt des Todes im Blut nachgewiesen wurden, lag bei 25,5 h vor dem Tod. Allerdings war dies verglichen mit den restlichen Ergebnissen, die zudem mehrheitlich von naiven Tieren stammten, vermutlich eine Ausnahme. Außerdem beinhaltete der Blutausstrich dieser Ziege nur zwei vereinzelte Bakterien, sodass ein eindeutig positiver Befund erst später durch Kultivierung des Bluts auf CBA erbracht wurde. Daher ist davon auszugehen, dass bei der hier verwendeten Art der Detektion mittels mikroskopischer Beurteilung eines Blutausstrichs ein positiver Befund regulär erst bei weiterem Fortschreiten des Krankheitsverlaufs möglich ist. Unter Ausschluss dieses Einzelfalls lag die frühste Detektion von Bakterien im Blut bei 2,8 bis 5,25 h, wobei beide Daten durch den nicht eindeutig festgestellten Todeszeitpunkt ungenau waren. Allerdings werden diese Werte durch weitere Ergebnisse gestützt, bei denen in anderen Tieren frühstens 2,3 h bzw. 1,83 h vor dem Tod Bakterien nachgewiesen werden konnten. Ebenso lagen die Zeitpunkte für einen negativen Abstrich bei frühstens 4,83, 5,5, 5,75 oder 6,0 h oder vor dem Tod. Dabei war die Anzahl der gefunde- nen Bakterien 1 – 3 h vor dem Tod zunächst gering (< 10) und bestand mehrheitlich aus Einzelzellen oder kurzen Ketten. Probennahmen zum Zeitpunkt des Todes enthielten hohe Konzentrationen, bestehend aus zum Teil langen Bakterienketten mit bis zu 40 Bakterien. Auf Grundlage dieser Ergebnisse kann vorläufig postuliert werden, dass wenigstens 2 – 5 Stunden vor dem Tod einer Ziege Bakterien im Blut nachweisbar waren, während 4 – 6 Stunden vor dem Tod die Diagnose noch negativ ausfallen konnte. Dies bedeutet, dass bei einer Detekti142 4. Ergebnisse und Diskussion on von Bakterien im Blut einer infizierten Ziege in einem Zeitraum von 2 – 5 h mit deren Tod zu rechnen ist, wohingegen ein negatives Resultat nur Prognosen für die nächsten 4 – 6 Stunden zulässt. Daraus ergeben sich Beobachtungs- und Kontrollintervalle mit denkbaren initialen Zeitfenstern von maximal 6 h bzw. 3 h bei auffälligen Tieren. 143 4. Ergebnisse und Diskussion 4.4 Diskussion Ziegenversuche 4.4.1 Versuchsdurchführung und Diagnoseprozeduren Ein nicht zu vernachlässigender Faktor bei Versuchen dieser Art in Großtieren außerhalb einer Laborumgebung ist der Mangel an verifizierbaren Daten zu den Rahmenbedingungen der Versuchsdurchführung. Vor allem neuere Daten zu Infektionsversuchen in Tiermodellen wie Ziege, Schaf und Rind sind, durch den damit verknüpften hohen Aufwand und die einzuhaltenden Sicherheitsrichtlinien, rar. Die Möglichkeit, solche Versuche unter Feldbedingungen in Endemiegebieten durchzuführen, war daher sowohl einzigartig, als auch im Bezug auf die sonst gesetzten Maßstäbe und Vereinheitlichungen eines Laborumfelds schwierig. Daher sollen zunächst die gewonnenen Daten zur Durchführung und Diagnose anhand der vorhandenen Literatur ausgewertet werden, um zukünftigen Vorhaben dieser Art eine bessere Ausgangslage zu ermöglichen. Das Auftreten von Fieber steht in Zusammenhang mit den Antikörpertitern gegen rPA83 Die Ermittlung der rektalen Temperatur war im Zuge des Infektionsversuchs eines der wichtigsten Mittel zur Früherkennung einer voranschreitenden Infektion und ausschlaggebend für weitere diagnostische Maßnahmen. Daher war eine Abgrenzung zwischen normalen Körpertemperaturen und erhöhten Temperaturen notwendig. Im Zuge der Vorbereitung der Versuche in Südafrika wurde daher die Temperatur der Ziegen aus Gruppe 1 bei den monatlichen Probenentnahmen bestimmt. Entsprechend der Messungen schwankten die Normaltemperaturen sehr stark und erreichten Werte von 37,4 – 39,6 °C (Tabelle 55, Anhang), was auch während der Infektion auffällig war. Zur Unterscheidung eines Fiebers war in früheren Studien von Schlingman et al. (1956) und Jackson et al. (1957) [272][150] mit Großtieren wie Rind und Schaf eine Temperatur von 39,4 °C und höher angenommen worden, welche entsprechend der gemessenen Normalwerte für diesen Versuch auf 40,0 °C angehoben wurden. In den Experimenten von Schlingman und Jackson nahm die Schutzwirkung der applizierten Vakzinen mit fortschreitender Entfernung vom Zeitpunkt der Vakzinierung ab und damit einhergehend die Inzidenz von Fieber sowohl in Überlebenden wie Verstorbenen mit verlängerter Überlebenszeit zu. Diese erhöhten Temperaturen konnten bei Rindern und Schafen generell im Zeitraum von 1 – 9 Tagen nach der Infektion auftreten und hielten bei Schafen 1 – 3 Tage an [272][150]. Die höchste Inzidenz an Fieber trat am 3. Tag nach der Infektion auf [150]. Ein ähnlicher Verlauf wurde auch für Schweine beschrieben [250]. Diese Beobachtungen konnten auch in den hier beschriebenen Versuchen an Ziegen bestätigt werden. Der Zeitraum des Auftretens erhöhter Temperaturen lag bei 2 – 10 Tagen nach der 144 4. Ergebnisse und Diskussion Infektion, wobei die Mehrheit der Tiere mit erhöhter Temperatur von Tag 2 – 4 auftraten und Tag 3 mit Abstand die höchste Rate an auffälligen Temperatur aufwies. Die Fieberdauer betrug 1 – 5 Tage. Ausgehend von den gemessenen serologischen Werten vor und nach der Infektion lassen sich in Bezug auf die gemessenen Temperaturen zudem einige Tendenzen feststellen. So verstarben in den vorliegenden Versuchen, wie in Abbildung 54 und 55 zu sehen ist, insgesamt 7 Versuchstiere ohne ein detektierbares Fieber (≥ 40,0 °C). Sechs dieser Tiere stammten aus den Kontrollgruppen, die keine funktionelle Vakzinierung erhalten hatten und alle 7 Tiere zeigten vor der Infektion einen unspezifischen anti-rPA83 IgG-Titer von ≤ 238. Ähnliche Versuche in Stieren zeigten ebenfalls, dass ein Großteil der Tiere, die ohne ein messbares Fieber verstarben, zur Gruppe der unvakzinierten naiven Tiere gehörten [150]. Da auch sechs weitere verstorbene Tiere mit einem ähnlichen anti-rPA83 IgG-Titer (< ~550) nur kurzzeitig auftretendes Fieber (< 10) aufwiesen, ist bei den praktizierten Messintervallen nicht auszuschließen, dass auch unauffällige Tiere eine transiente Temperaturerhöhung aufwiesen, die nicht detektiert wurde. Kein Tier mit einem anti-rPA83 IgG-Titer vor der Infektion von 550 und niedriger überlebte und keines dieser Tiere zeigte eine Temperaturerhöhung, die länger als 10 h detektierbar war. Ebenfalls keine Temperaturerhöhung zeigten Überlebende mit anti-rPA83 IgG-Titern vor der Infektion von > ~30000, was ausschließlich auf die frisch revakzinierten Tiere der SSLV-immunisierten Gruppe 3b zutraf. Auch bei den Versuchen von Schlingman et al. (1956) und Jackson et al. (1957) [272][150] mit AVP und Lebendvakzinen war auffällig, dass Tiere mit einem frischen Impfschutz durch eine Infektion keinerlei Temperaturerhöhung erfuhren. Diese abwesende Temperaturerhöhung ging zudem in der hier beschriebenen Versuchsgruppe 3b auch mit einer Verminderung der anti-rPA83 IgG-Titer nach der Infektion einher. Es ist daher denkbar, dass eine ausreichende Höhe an anti-rPA83 IgG-Titern im Falle einer Infektion den Progress der Infektion und damit einhergehend die Entwicklung von Fieber unterbindet und somit zu einer „sterilen Immunität“ [49] führt. Dies würde über die schnelle Klärung der Antigene durch die vorhandenen Antikörper eine weitere spezifische Immunreaktion auf diese verhindern und so konstante oder sinkenden Antikörpertiter nach der Infektion zur Folge haben. Entsprechend müsste bei Überlebenden ein konstanter Antikörpertiter vor und nach der Infektion mit der Abwesenheit von Fieber einhergehen. Dies konnte in dieser Arbeit bestätigt werden, da alle 11 Tiere mit konstanten oder fallenden Antikörpertitern gegen rPA83 auch kein detektierbares Fieber aufwiesen. Umgekehrt wiesen auch 11 der 14 überlebenden Tiere mit einer starken Erhöhung der Antikörpertiter gegen rPA83 nach der Infektion ein detektierbares Fieber auf. Dabei war ab einer Titerhöhe von ~90000 nach der Infektion in allen Fällen Fieber nachweisbar, während dies bei Titerhöhen bis ~32000 nie der Fall war. 145 4. Ergebnisse und Diskussion Die Titerhöhe vor der Infektion schien für eine Vorhersage der Entwicklung von Fieber nicht aussagekräftig zu sein. Dies bestätigt die zuvor festgestellte Tendenz, wonach sowohl Tiere mit besonders niedrigen (≤ 238), also auch besonders hohen (> ~30000) anti-rPA83 IgG-Titern vor der Infektion kein oder nur kurzzeitiges Fieber entwickelten. In beiden Fällen ist es vermutlich nicht zu einer systemischen Reaktion auf die Infektion gekommen, wobei die Ursache dafür jeweils eine andere war. Tiere mit besonders niedrigen Titern starben an der Infektion bevor sich ein Fieber entwickeln konnte, während Tiere mit besonders hohen Titern vermutlich erst gar keine systemische Infektion entwickelten. Titerwerte zwischen diesen Extremen bieten mitunter nur einen partiellen Schutz, wodurch ein ambivalentes Krankheitsbild entsteht. Damit scheint der anti-rPA83 IgG-Titer vor der Infektion keinen linearen Bezug zur Entwicklung von Fieber zu haben, wohl aber zum Verlauf der Infektion. Der Unterschied zwischen den anti-rPA83 IgG-Titern vor und nach der Infektion könnte zu einer quantifizierbaren Auswertung der Schutzrate hinzugezogen werden, da dieser in Zusammenhang mit einer Fieberentwicklung und damit dem Fortschreiten der Infektion steht. Somit wäre eine Unterscheidung bei Überlebenden zu einer sterilen Immunität ohne eine Temperaturund Titerentwicklung durch die Infektion für eine generell bessere Beurteilung von Vakzinen sinnvoll. Daher sollte in Vakzinestudien bei Überlebenden generell auch das Blut nach der Infektion auf Titerveränderungen untersucht werden. Entsprechend konstante oder abfallende Titer im Vergleich zu vor der Infektion würden demnach auf eine sterile Infektion hindeuten, qualitative Unterscheidungen bei den Überlebenden ermöglichen und so potentiell eine tiefere Diskriminierung zwischen ähnlich schützenden Ansätzen erlauben. Die Höhe der anti-rPA83 IgG-Titer vor der Infektion ist in Ziegen und Auszucht-Mäusen prädiktiv für das Überleben, aber möglicherweise abhängig von der Infektionsdosis Bei der Beurteilung neuer Vakzinekomponenten gilt letztendlich die Schutzwirkung vor einem letalen Infekt mit einem voll virulentem Stamm als ausschlaggebendes Kriterium. Besonders im Bezug auf Humanvakzinen und der Vermeidung von Tierversuchen im Sinne des Tierschutzes ist dies aber nicht immer möglich oder praktikabel. Daher wird seitdem es möglich ist die Serokonversion durch verschiedene Antigene mittels ELISA und TNA zu messen, versucht, diese mit eine Prognose zur Überlebenswahrscheinlichkeit zu verknüpfen. Korrelationen der gemessenen Antikörpertiter zum Überleben geben aber bisher kein schlüssiges Bild, sodass es besonders auch im Hinblick auf die Antitoxin-Titer namhafte Verfechter für deren Korrelation zum Überleben (auszugsweise: [211][102][307][20][240]) und auch dagegen (auszugsweise: [124] [95][148][326][61]) gibt. Hauptgründe für die widersprüchlichen Berichte sind die fehlenden Richtlinien zum Zeitpunkt der Probennahme und die Variationen in den Beurteilungsmaßstäben. Wie besonders bei der Titerentwicklung der SSLV über ein Jahr ersichtlich ist, können 146 4. Ergebnisse und Diskussion Korrelationsversuche zu verschiedenen Probenzeitpunkten gänzlich andere Ergebnisse liefern. Überdies beeinflussen auch Faktoren wie die Infektionsdosis, Infektionsart oder der eingesetzte Stamm sowie das Tiermodell die Ergebnisse [192][89][1], sodass die Vergleichbarkeit zwischen den Arbeiten kaum gegeben ist. Im Zuge der hier vorliegenden Arbeit wurde dennoch nach Korrelationen zwischen den Titern vor der Infektion und der überlebten Zeit in Stunden mittels des Spearman Rangkorrelationskoeffizienten gesucht. Entsprechende Vergleiche innerhalb einzelner Gruppen waren aufgrund der teilweise geringen Gruppengröße nicht für alle Gruppen möglich bzw. resultierten in sehr hohen Signifikanzschwellen. Die Mausversuche mit einbegriffen, ergaben sich nur signifikante Korrelationen für die zwei Gruppen, deren Immunogenität weniger gut anmutete, was natürlich auch dadurch bedingt ist, dass Korrelationsberechnungen bei vollständiger Schutzwirkung nicht möglich sind. Dies betraf Gruppe 2 der Ziegenversuche und Gruppe TA der Mausversuche, bei denen die anti-rPA83 IgG-Titer (und für TA auch die anti-rLF IgG-Titer) mit der überlebten Zeit korrelierten (rs = +0,9). Beide Gruppen zeigten durch die jeweilige Immunisierung nur sehr niedrige Antikörpertiter und in beiden Gruppen verstarben die Tiere mit den niedrigsten bzw. nicht nachweisbaren anti-rPA83 IgG-Titern. Möglicherweise sind daher die für den Schutz relevanten Antikörperfraktionen nicht zwingend gegen die immunogensten Epitope gerichtet. Für einen weiteren Versuch, Zusammenhänge zwischen den anti-rPA83 IgG-Titern und dem Überleben aufzudecken, wurden alle Versuchstiere der Ziegenversuche anhand ihrer anti-rPA83 IgG-Titer direkt vor der Infektion sortiert. Dabei ergab sich, dass Tiere mit einem anti-rPA83 IgG-Titern von ~550 und weniger nicht mehr ausreichend geschützt waren und ausnahmslos verstarben. Dies betraf 46,5 % aller verstorbenen Tiere (25 % aller Tiere insgesamt) und diese blieben tendenziell symptomlos mit einem möglicherweise äußerst kleinem Zeitfenster (< 10 h) in dem eine erhöhte Temperatur messbar gewesen wären. Alle verstorbenen Ziegen mit einem Ausgangstiter von ~550 und höher wiesen eine detektierbare Temperaturerhöhung auf. In einem Bereich zwischen ~550 bis ~3400 war keine eindeutige Tendenz zu erkennen, da sowohl 26 % der Überlebenden als auch 43 % der Verstorbenen Antikörpertiter in dieser Höhe aufwiesen. Ab einem Titerwert von 3400 überlebten 85 % aller Tiere, wobei eine eindeutige Prognose für einen vollständigen Schutz erst ab einem Titer von 20000 möglich war. Die gleiche Analyse von Mausgruppen mit einer Infektionsdosis von ~1000 Sporen ergab eine Überlebensrate von 75% ab einem Schwellenwert von 30000, wobei eine vergleichbare Tendenz bei einer Infektionsdosis von ~2000 Sporen nicht vorhanden war. Analoge Beobachtungen gaben Minimaltiter gegen rPA83 von 1132 für eine Schutzrate von 90 % in Kaninchen [102] und 2500 – 4200 für 100 % Schutzraten in Meerschweinchen an [255]. Der postulierte Schwellenwert von 3400 für 147 4. Ergebnisse und Diskussion Ziegen bei einer Schutzrate von 85 % läge damit in einem ähnlichen Bereich, wäre aber nicht absolut. Für alle weiteren Antigene waren keine Schwellenwerte erkennbar. Um die Zusammenhänge zwischen den gemessenen Antikörpertitern und der überlebten Zeit statistisch zu belegen, wurden gruppenübergreifende Korrelationsberechnungen durchgeführt. Es ergaben sich korrespondierend zu den beschriebenen Schwellenwerten für die Ziegen- und Mausgruppen, die eine Infektionsdosis von ~1000 Sporen erhielten, signifikante Korrelationen der anti-rPA83 IgG-Titer vor der Infektion zur überlebten Zeit (r s ≥ +0,507), wohingegen die Mausgruppen, die eine Infektionsdosis von ~2000 Sporen erhielten eine gegenläufige Korrelation aufwiesen (rs = -0,406). Daher scheint in Ziegen und Auszucht-Mäusen ein Überleben mit den anti-rPA83 IgG-Titern in Zusammenhang zu stehen, wobei deren Relevanz für das Überleben in Auszucht-Mäusen abhängig von der Infektionsdosis sein kann. Die MLD für das Feldisolat SD20 ist kleiner als 36 Sporen in Ziegen Für die Infektion der Ziegen in Südafrika wurden Sporen des bis dato unerprobten Feldisolats SD20 verwendet. Dieser war 2001 aus dem Ohr eines an Milzbrand verendetem Schafes im Gebiet von Standerton, Provinz Mpumalanga, Südafrika isoliert und als voll virulent typisiert [181] worden. Die Testung des Stammes vor Ort mit 500 und 1000 Sporen in BALB/c-Mäusen bestätigte dessen Virulenz. Für den Einsatz in der Ziege fehlten direkte Vergleichsangaben zur einzusetzenden Dosis, sodass vor der eigentlichen Infektion verschiedene Konzentrationen getestet wurden. Andere Infektionsversuche in sensiblen Tierarten stellten hierbei eine Orientierungshilfe. So gaben Schlingman et al. (1956) [272] eine LD50 (s. c.) von 100 Sporen für den Stamm V770-2-P in Schafen und de Vos et al. (1996) [66] eine MLD von < 100 für den Stamm 17JB in Impalas an. In Anlehnung an diese Ergebnisse sollten 1000, 200 und 50 Sporen des SD20 Stammes s. c. in naiven Ziegen erprobt werde. Die Auszählung der Infektionsdosen ergab eine tatsächlich applizierte Dosis von 844, 172 und 36, wobei alle Dosen mit korrelierender Überlebenszeit tödlich wirkten. Da selbst die geringste Dosis einen Tod innerhalb von 4 Tagen zur Folge hatte, ist davon auszugehen, dass die MLD dieses Stammes in der Ziege < 36 Sporen für eine s. c. Applikation beträgt. Beim Nachweis von Bakterien im Blut infizierter Ziegen ist mit einem Tod binnen 2 – 5 h zu rechnen Die Einwanderung von B. anthracis in den Blutkreislauf und die dortige Replikation wird als finales Stadium der Infektion angesehen, weswegen ein Nachweis von Bakterien im Blut ein sicheres Zeichen für eine letale Infektion darstellt. Abhängig von der Spezies, dem eingesetzten Stamm, dessen Dosis und der Infektionsroute [189][44][89][357] kann der Zeitpunkt des Auf- 148 4. Ergebnisse und Diskussion tretens von Bakterien und deren Verdopplungszeit variieren. Dabei ist der Nachweis von Bakterien nicht generell prädiktiv für einen tödlichen Verlauf, da auch Einzelfälle bekannt sind, bei denen sich Infizierte sogar ohne Behandlung voll erholten [89][264][156][272]. Ausschlaggebend für eine Remission ist aber die Konzentration der vorhandenen Bakterien [293]. Dennoch wurde aus technischen und ethischen Gründen in dieser Studie die Detektion von Bakterien im Blut als Kriterium für eine therapeutische Intervention angenommen. Ähnliche Studien dieser Art ergaben eine frühest mögliche Detektion von 5 – 24 Stunden vor dem Tod für Kaninchen und Meerschweinchen [174], bei einem Detektionslimit von 100 cfu/ml, sowie 24 – 46 Stunden für Rinder [272][150], 12 – 16 Stunden für Schafe [272] und 11,5 – 12 Stunden für Rhesusaffen [166][188]. Beim Vergleich dieser Daten ist zu beachten, dass das Detektionslimit bedingt durch die Art der Blutabnahme beeinflusst werden kann. Überdies kann auch ein etwaiger bestehender Impfschutz das Ergebnis beeinflusst haben. Für die aus dieser Arbeit vorliegenden Ergebnisse kann das Detektionslimit nur mit einem Schätzwert von 100 cfu/ml angegeben werden, da bei jeder Probennahme nur jeweils 1 – 2 Objektträger mit je einem Tropfen Blut (~10 µl) komplett begutachtet wurden. Die unter diesen Voraussetzungen frühste Entdeckung von Bakterien im Blut lag bei 2 – 5 Stunden vor dem Tod, wobei bei einzelnen Tieren 4 – 6 Stunden vor dem Tod noch keine Bakterien nachweisbar waren. Damit war mit dem Tod von unbehandelten Ziegen nach Detektion von Bakterien im Blut innerhalb der nächsten 2 – 5 Stunden zu rechnen. Die Mehrheit der gesammelten Messdaten zu diesem Thema wurde in den Kontrollgruppen erhoben, wodurch etwaige Verfälschungen durch einen bestehenden Impfschutz weniger ins Gewicht fallen. Im Vergleich liegt die Detektionsfähigkeit von Bakterien im Blut infizierter Ziegen weit unter der anderer Großtiere, wobei die größte Ähnlichkeit mit Schafen [272] und möglicherweise mit Schweinen besteht, da letztere terminal sehr niedrige Bakterienkonzentrationen aufzuweisen scheinen [250]. Alle genannten Ergebnisse könnten zusätzlich auch durch die Art der Blutentnahme über große Blutgefäße beeinflusst worden sein, wobei gerade bei den zitierten Großtierversuchen diesbezüglich keine Angaben gemacht wurden. Daher ist davon auszugehen, dass die Beprobung dort auf ähnlich Weise stattgefunden hat. Unter der Voraussetzung der Praktikabilität könnte eine Blutentnahme aus kleinvolumigeren Gefäßen eine bessere Detektion ermöglichen, da dort Bakterien in größeren Konzentrationen vorhanden sein könnten. Die hohen unspezifischen Hintergrundtiter bei Ziegenseren im ELISA lassen sich durch den Austausch von FKS mit Magermilchpulver stark reduzieren Im Vergleich zu den Mausversuchen zeichneten sich die serologischen Analysen der Ziegenseren mittels ELISA durch starke unspezifische Hintergrundtiter (< ~2500) gegen verschiedene Antigene, insbesondere rPA83, rBclA und FIS aus. Diese schienen individuell unterschiedlich 149 4. Ergebnisse und Diskussion auszufallen, sowohl in den Kontrollgruppen, als auch in den Seren der Vakzinegruppen vor der Immunisierung und waren auch über die Zeit (Gruppe 1) Schwankungen unterlegen. Ähnlich hohe Titer bei naiven Tieren waren auch in den Arbeiten von Hahn et al. (2006) [123] in Schafen bezüglich rPA83 festgestellt worden, die dort in einer Serumverdünnung von im Durchschnitt bis zu ~1:2000 nachweisbar waren [123]. Auch für Seren humanen Ursprungs, von Rhesusaffen und sogar diversen Mausmodellen ist dieses Phänomen bekannt und betrifft neben PA auch LF und EF [40][49][75]. Damit ist davon auszugehen, dass es sich um ein generelles Phänomen handelt, das vermutlich auch mehr oder weniger stark von der individuellen Zusammensetzung der Seren abhängt. Der Austausch von FKS gegen Magermilchpulver als Bestandteil des Blockpuffers im ELISA bewirkte eine deutliche Verminderung der unspezifischen Hintergrundtiter auf < ~200. Dies wurde ausschließlich bei den Seren der Wiederholungsversuche (Türkei) angewendet, weswegen die Vergleiche der Ziegengruppen untereinander mit normalisierten Titerwerten durchgeführt wurden. Die Höhe der unspezifischen Hintergrundtiter war nur im Bezug auf die anti-rBclA Titer problematisch, da die eventuell sichtbaren Titererhöhungen in einem Bereich lagen, der den unspezifischen Titern entsprach. Für FIS, rPA83 und das vegetative Antigen waren spezifische Titererhöhungen aufgrund der Titerhöhe deutlicher unterscheidbar. Allerdings waren auch bei diesen Antigenen aufgrund der unspezifischen Hintergrundtiter marginale Titerveränderungen im Verlauf der Immunisierung weniger aussagekräftig. Adverse Reaktionen von Ziegen auf die Immunisierung mit der SSLV beruhen möglicherweise auf parallel vorhandenen Infektionen Während bereits M. Sterne selbst Zweifel an der Sicherheit seiner Vakzine in Ziegen äußerte [300] und die generelle Empfehlung zur Anwendung der SSLV in diesen Tieren eine PräInokulation, vorzugsweise in der Schwanzregion mit einem Viertel der eigentlichen Dosis, vorsieht [357], sind publizierte Fallbeschreibungen zu dieser Thematik kaum vorhanden. Zwar wird in vielen Publikationen die Sensitivität von Ziegen und Lamas aufgegriffen und deren Neigung zu negativen Reaktionen auf die Vakzine postuliert [326], dennoch beruhen die meisten auf einer einzigen echten Referenz. Dieser von Cartwright et al. (1987) [48] berichtete Fall betraf drei 3 Monate alte Lama-Kälber, die durch die Immunisierung mit der SSLV erkrankten und von denen zwei im weiteren Verlauf verstarben. Dabei zeigte keines der älteren Tiere eine ähnliche Reaktion. Weitere Berichte dieser Art auch in anderen Tiermodellen wie Ziegen sind nur mündlich überliefert oder beruhen auf Erfahrungswerten. Hinlänglich nachgewiesen und publiziert, ist die Restvirulenz der SSLV indes nur in verschiedenen sensitiven Mausinzuchtmodellen und Meerschweinchen [354][353][192][148]. 150 4. Ergebnisse und Diskussion Ebenso gibt der Hersteller (OBP – Onderstepoort Biological Products) der hier verwendeten Vakzine (SSLV, Stamm 34F2) bezüglich einer Verwendung in Ziegen an, dass es regional zu Schockreaktionen aufgrund der Immunisierung kommen kann. Zudem sollte von einer Applikation wie üblich in der Halsreaktion aufgrund von möglichen Schwellungen abgesehen werden, sodass eine Sicherheit der Vakzine nicht in allen Tieren gewährleistet werden kann. Die Grundlage dieser Empfehlung wird allerdings nicht erläutert. Entgegen der generellen Einschätzung und den vom Hersteller geäußerten Bedenken verlief die Immunisierung von Gruppe 2 und 3 a/b mit der SSLV vollkommen nebenwirkungsfrei, wobei zudem auf eine Prä-Inokulation verzichtet und entsprechend sofort eine reguläre Dosis verabreicht wurde. Todesfälle aufgrund der Immunisierung traten ebenfalls nicht auf. Damit schließen sich die Ergebnisse diesbezüglich an die von Shakya et al. (2007) [280] gemachten Erfahrungen an, die ebenfalls keinerlei adversen Reaktionen der SSLV in Ziegen feststellen konnten. Einzig die aufgetretenen generellen gesundheitlichen Probleme sowie die Verluste durch parallel auftretende Infektionen könnten einen Anhaltspunkt bieten. Ähnliche Vakzinierungsversuche in Ziegen, anderen Großtieren und Wildtieren waren vermutlich eher, wie die in dieser Arbeit beschriebenen Versuche, als Feldversuche ausgelegt und auch eine reguläre Impfung findet zumeist in Tieren statt, deren Gesundheitszustand nicht genau erfasst ist. Daher scheint es möglich, dass adverse Effekte oder gar Verluste von Tieren durch die Immunisierung, gerade auch bei jungen Tieren, auf mögliche Zusatzbelastungen durch Nebeninfektionen oder einen generell schlechten Zustand zurückzuführen sind. Die hier verwendeten Ziegen zeigten mehrheitlich positive Reaktionen im Test auf Pseudotuberkulose, welche oft vor allem junge Tiere befällt, latent verläuft und erst bei älteren Tieren durch Stress z. B. beim Transport oder einer Impfung zum Ausbruch kommt [55]. Ebenso erkrankten und verstarben mehrere Tiere vor der Infektion durch Freilandhaltung an der Herzwasserkrankheit. Wäre die Ausprägung besonders der Herzwasserkrankheit direkt nach der Immunisierung oder Infektion aufgetreten, hätte man die Todesursache ausschließlich auf B. anthracis zurückgeführt und nicht auf eine Zusammenwirkung mit einem relevanten Parallelinfekt. Die beobachteten Krankheitsfälle waren in dieser Arbeit eindeutig auf die kurzzeitige Haltung in einem Freilandgehege zurückzuführen. Während der Haltung in betonierten Ställen traten keine Fälle auf. Verbesserung der Diagnoseprozeduren Zusammenfassend war die Beobachtung von Verhalten und Temperatur abhängig vom Versuchsaufbau kein vollends verlässlicher Indikator zur Beurteilung des Infektionsverlaufs. Dies war auch darauf zurückzuführen, dass die Temperatur nur punktuell gemessen werden konnte 151 4. Ergebnisse und Diskussion und Verhaltensauffälligkeiten vornehmlich nachts nicht oder schwer bemerkbar waren und zudem durch äußere Faktoren beeinflusst werden konnten. Generell ist daher eine kontinuierliche Messung der Temperatur über einen Chip bei dieser Art von Experiment anzuraten, da so eventuelle kurze durch Stress oder andere Einflüsse induzierte Temperaturerhöhungen besser als solche erkannt werden können und sich so ein schlüssigeres Beurteilungskonzept erstellen lässt. Muss die Temperatur manuell erhoben werden, so sollte dies mehrmals täglich entsprechend der Praktikabilität in Abständen von 6 – 12 h (optimal 8 h) geschehen. Hierbei gewinnt die zusätzliche Beurteilung des Verhaltens um so mehr an Bedeutung je weniger frequent eine reguläre Temperaturmessung möglich ist. Zur Verbesserung der Aussagekraft von auffälligem Verhalten ist die Haltung innerhalb eines abgeschlossenen Gebäudes anzuraten, um Stress durch Umwelteinflüsse zu minimieren. Generell sollte darauf geachtet werden, die Betreuung und Untersuchung der Tiere mit möglichst großer Routine durchzuführen, um Stress zu vermindern. Zudem sind regelmäßige Fütterungen, mehrmals täglich mit Heu oder Laub zur Einschätzung des Appetits, des Durchsetzungsvermögens und der Stellung in der Gruppe hilfreich. Beides wird von Ziegen gerne angenommen und deckt besser als pelletiertes Futter Probleme beim Zerkauen der Nahrung auf. Beim Auftreten von Verhaltensauffälligkeiten sollte die Temperatur der jeweiligen Tiere auch außerhalb der normalen Messintervalle ermittelt werden. Der Schwellenwert für die Indikation von Fieber liegt bei 40,0 °C. Dabei sollte bei ausreichender Versuchsgruppengröße in Erwägung gezogen werden, Tiere nicht zu infizieren, die vor der Applikation der Infektionsdosis eine Temperatur von 40,0 °C und höher aufweisen. Mit einer infektionsbedingten Erhöhung der Temperatur ist zwischen 2 und 10 Tagen nach der Infektion zu rechnen, wobei zwischen Tag 2 und 5 die Inzidenz am höchsten ist. Tiere mit einer Temperatur von < 40,0 °C oder/und auffälligem Verhalten nach der Infektion sollten bei jeder regulären Überprüfung genau auf eine Veränderung ihres Zustands überprüft werden. Weiterführende diagnostische Maßnahmen (Blutausstrich) und eine häufigere Überprüfung der entsprechenden Tiere (alle 2 – 5 h) sind erst ab einer Temperatur von 40,4 °C sinnvoll und sollten so lange in dieser Frequenz weitergeführt werden, bis sich der Zustand verbessert (< 40,0 °C) oder Bakterien im Blut nachweisbar sind. Entsprechend der Anzahl an gefundenen Bakterien und des Gesamtzustands des Tiers kann bei einer Detektion von Bakterien im Blut statt einer Euthanasie ebenfalls eine antibiotische Behandlung in Erwägung gezogen werden. Als weiteres frühzeitiges Abbruchkriterium könnte eine Temperaturerhöhung auf 41,4 °C verwendet werden, da die Ergebnisse dieser Arbeit ergaben, dass 90 % der Tiere mit einer Höchsttemperatur von ≥ 41,4 °C im weiteren Verlauf verstarben. Tiere mit einer solchen Temperatur könnten auch ohne den Nachweis von Bakterien im Blut behandelt oder euthanasiert werden, wenn es die Zielsetzung des Versuchs erlaubt. 152 4. Ergebnisse und Diskussion Als weiteres diagnostisches Mittel könnten, sofern es durchführbar ist, die anti-rPA83 IgGTiter der Seren direkt vor der Infektion ermittelt werden. In den Versuchen dieser Arbeit wiesen 46,5 % der verstorbenen Tiere einen anti-rPA83 IgG-Titer von < ~550 auf und verstarben symptomlos oder mit kurz andauerndem Fieber (< 10 h). Daher waren diese Tiere anhand der Beobachtung von Temperatur und Verhalten nur schwer korrekt zu diagnostizieren. Mit entsprechender Kenntnis der anti-rPA83 IgG-Titerwerte von vor der Infektion könnten Tiere dieser Kategorie verstärkt beobachtet werden oder von der Infektion ausgeschlossen werden, wenn es der Versuchsaufbau zulässt. Ein zusätzliches Hilfsmittel vor allem zur Unterscheidung von infektionsbedingt auffälligem Verhalten wäre die Inklusion von 1 – 2 nicht infizierten Tieren in die Kohorte, anhand derer sich ein normales Verhalten abschätzen lässt. Dies ist besonders von Vorteil, wenn äußere Stressfaktoren nicht vermieden werden können. Die Anwendung der genannten Erkenntnisse aus dieser Studie können dazu beitragen, innerhalb ähnlicher Versuche einen irreversiblen Krankheitsverlauf frühzeitig zu erkennen und durch Behandlung oder Euthanasie das Leid der Tiere und das Kontaminationsrisiko zu vermindern. 4.4.2 Impfversuche mit NLV Während die Ergebnisse zum diagnostischen Prozedere nur Beiwerk der eigentlichen Versuche waren, lag das Hauptaugenmerk auf der Testung von NLV im Vergleich zur gängigen SSLV. Geeignete Kandidaten und deren Kombinationen waren zuvor im NMRI-Mausmodell etabliert worden. Während die DNA-basierenden Vakzinen in Ziegen eine geringere Wirksamkeit aufwiesen, konnten die Protein-basierenden Vakzinen im Vergleich zur Immunisierung mit der SSLV einen äquivalenten Schutz erzeugen, waren aber weniger immunogen. Daher sollen im Folgenden die Ergebnisse anhand der vorhandenen Literatur verglichen und Gründe für die unterschiedliche Wirksamkeit, sowie Verbesserungsmöglichkeiten erörtert werden. Die DNA-Vakzine Kombination war in dieser Form weitestgehend wirkungslos Die Ergebnisse der DNA-Vakzinierung der Ziegen belegten, dass keine der drei Impfungen eine merkliche Titererhöhung bewirkte. Ebenso schien auch kein Potential als vorbereitende Vakzinierung für einen Proteinboost vorhanden zu sein, da auch eine entsprechende Boosterdosis keine besseren Ergebnisse brachte als die vergleichbare Proteinimmunisierung alleine. Die fehlende Wirkung der DNA-Vakzinierung war vor allem im Bezug auf die gute, teils zu den Proteinen äquivalente Wirkung der Vektoren im NMRI-Mausmodell erstaunlich. Zu beachten ist hierbei allerdings die unterschiedliche Applikation der Vakzine in den Maus- und Ziegenversuchen, wenngleich bereits in Schafen gezeigt wurde, dass generell eine i. m. Applikation von DNA153 4. Ergebnisse und Diskussion Vakzinen wirkungsvoll sein kann. So konnten Hahn et al. (2006) [123] erfolgreich mit einer ähnlichen DNA-Vakzine anti-rPA83 IgG-Titer (~104) in Merino-Schafen erzeugen. Abgesehen von der Verwendung einer anderen Wirtsspezies und anderer Vektoren lag ein großer Unterschied in der Verwendung eines anderen Adjuvans (Vaxfektin), das gerade bei i. m. Immunisierungen mit DNA eine Rolle spielen kann. Die verstärkende Wirkung von Vaxfektin war auch innerhalb der Versuche von Hahn et al. (2006) [123] nachgewiesen, da eine Immunisierung ohne das Adjuvans im Vergleich ebenfalls kaum eine Wirkung der Vektoren zur Folge hatte. Die in der vorliegenden Arbeit verwendeten Adjuvanzien mIPS-1 und CIITA sind zudem stark auf eine intrazelluläre Wirkung angewiesen, welche durch eine Injektion nur über aktive Aufnahme der betreffenden Zellen gewährleistet werden kann. Dabei ist die generelle Aufnahme von extrazellulärer DNA durch APCs ohnehin sehr ineffektiv [262]. Darüber hinaus ist besonders CIITA auf eine gemeinsame Wirkung mit den applizierten Antigenvektoren angewiesen, was ebenfalls außerhalb einer GeneGun-Anwendung weitestgehend dem Zufall überlassen ist. Ein weiterer Faktor könnte die Herkunft der genannten Sequenzen selbst sein. Sowohl mIPS-1 als auch CIITA waren murinen Ursprungs und könnten in anderen Tiermodellen eine andere Wirkung haben. Aus den Versuchen von Hahn et al. (2006) [123] schließend, könnte eine erneute Anwendung der Vektoren NTC7382-TPA-LFD1PAD4-mIPS-1, pDNAVaccUltra1-TPA-BclAD1D3LAMP1 und pDNAVaccUltra5-CIITA mit anderen Adjuvanzien, beispielsweise Vaxfektin gänzlich andere Ergebnisse erzielen. Ebenso wären alternative Applikationsarten, die ein intrazelluläres Eintreffen der Vektoren sicherstellen, wie z. B. Elektroporation, Tätowierung oder Mikropartikelbeschuss [262] für das zwingende Zusammenwirken mehrere Vektoren vorteilhafter. FIS als Bestandteil einer Proteinvakzine erhöht die schützende Wirkung von rPA83 Trotz der veränderten Dosis und Applikation unterschieden sich die Wiederholungsversuche von den Vorversuchen in der Höhe der gebildeten Antikörpertiter nicht signifikant, was wahrscheinlich auf die in beiden Versuchsdurchgängen beobachtete starke Streuung der Titerwerte innerhalb der Gruppen zurückzuführen ist. Die anti-rPA83 IgG-Titer in den Wiederholungsversuchen lagen tendenziell höher und es wurde eine signifikant bessere Schutzwirkung erzielt. Inwiefern neben der Erhöhung der Dosis auch die gemeinsame Applikation der Antigene als Mischung einen Einfluss hatte, ist dabei nicht abzusehen, da die Titerentwicklung in beiden Szenarien ähnlich verlief. So scheint bei allen getesteten Gruppen eine dritte Immunisierung die erzeugten Antikörpertiter nicht weiter zu erhöhen. Die unerwartet niedrige Immunogenität von rBclA in Ziegen betraf alle Gruppen gleichermaßen und wird in späteren Abschnitten diskutiert. 154 4. Ergebnisse und Diskussion Eine Veränderung der anti-rPA83 IgG-Titer durch den Zusatz von FIS war weder bei den Vorversuchen, noch bei den Wiederholungsversuchen vorhanden. Einzig die TNA-Titer schienen durch die Kombination mit FIS tendenziell erhöht und wiesen im Falle von Gruppe 6 (W) eine Korrelation mit den anti-FIS IgG-Titern auf. Ebenso lagen die Schutzraten bei Zusatz von FIS mit 20 % bei Gruppe 6 und 80 % bei Gruppe 6 (W) höher als ohne FIS (respektive 0 % und 50 %). Die verbesserte Wirkung der Kombinationsvakzine beruhte also entweder auf der Erhöhung der gebildeten neutralisierenden Antikörper gegen rPA83, bedingt durch die Zugabe von FIS oder/und auf den durch Fis generierten anti-Spore Antikörpern. Damit konnte die zuvor in Mäusen nachgewiesene, verbesserte Schutzwirkung von Vakzinekombinationen mit FIS und PA [42][106], die ebenfalls bereits in den hier beschriebenen Vorversuchen in Mäusen gezeigt wurde auch in Ziegen nachgewiesen werden. Im Vergleich zur Vakzinierung mit der SSLV (diese Studie) und der von Hahn et al. (2006) getesteten Kombination aus rPA83 und Alhydrogel in Schafen [123] waren die anti-rPA83 IgGTiter der NLV insgesamt weitaus niedriger. Nur vereinzelte Tiere wiesen Titerwerte (> 104) auf, die mit den genannten Versuchen vergleichbar wären. Allerdings war die erzielte Schutzrate von 80 %, mit einer Kombination von rPA83, rBclA, Lipopeptid und FIS vergleichbar mit den von Jackson et al. (1957) und Schlingman et al. (1956) erreichten Schutzraten von 100 % durch AVP in Stieren und Schafen [150][272] und den in dieser Studie durchgeführten Versuchen mit der SSLV. Tendenziell scheint die Wirksamkeit der Kombination von rPA83, (rBclA), Lipopeptid und FIS allerdings nicht vollständig ausgeschöpft worden zu sein, da die Titer innerhalb der Gruppe unabhängig von der Applikationsart stark schwankten. Eine weitere Erhöhung der Antigendosis, FIS eingeschlossen, oder/und die Verwendung eines anderen Adjuvans [123] könnte die Schwankungen reduzieren und für ein höheres, einheitlicheres Titerbild sorgen, was sich entsprechend auch positiv auf die Schutzrate auswirken könnte. 4.4.3 Impfversuche mit der SSLV Während eine oder mehrere verstärkende Folgeimmunisierungen zur Gewährleistung eines vollständigen Schutzes Bestandteil der meisten Impfpläne sind, wird für die SSLV ein Impfschutz ab der ersten Immunisierung angenommen. Die maximale Schutzdauer wird für jede Immunisierung mit einem Jahr angegeben, sodass Auffrischungen jährlich notwendig sind. Inwiefern die Aufrechterhaltung dieses Impfplans nach der ersten Auffrischungsimpfung notwendig ist oder ein den Schutz beeinflussender Unterschied zwischen der ersten und allen weiteren Impfung besteht, ist jedoch nicht schlüssig geklärt. Initiale Arbeiten zu diesem Thema zur Zeit der Testung und Einführung der Vakzine befassten sich bei der Einschätzung der Wirksam- 155 4. Ergebnisse und Diskussion keit hauptsächlich mit der klinischen Reaktion der Tiere und der praktischen Schutzwirkung. Inzwischen sind detailliertere Untersuchung zur Grundlage einer Schutzwirkung möglich. Daher waren die Daten zur Titerentwicklung der SSLV, neben der Funktion als Referenz, insbesondere mit Abdeckung der hier getesteten Antigene von generellem Interesse. Der Erfolg einer Erstimmunisierung mit der SSLV kann stark variieren Während neuere Studien zur Wirksamkeit der SSLV in Großtieren kaum vorhanden sind, finden sich zahlreiche Studien in Mäusen, Kaninchen und Meerschweinchen, in denen die Vakzinierung mit der SSLV vergleichend zu anderen Ansätzen erprobt wird. Leider beschränkt sich der serologischen Anteil dieser Studien oft auf die Antikörpertiter gegen die Toxine, insbesondere PA, und erstreckt sich zudem meist nicht über längere Zeiträume. Wurde eine Immunisierung mit der SSLV in diesen Studien mehrfach vorgenommen, so geschah dies in Angleichung an die Impfpläne der zu vergleichenden Ansätze im Abstand von 2 – 4 Wochen, was nicht der Anwendungsrealität der SSLV entspricht. Daher sind reale Vergleichsstudien, wie die in dieser Arbeit präsentierte, mit einem über PA hinausgehenden Titerspektrum nahezu nicht vorhanden. Einzig die Arbeiten von Shakya et al. (2007) [280] bieten eine direkte Vergleichsmöglichkeit zu den Versuchen dieser Arbeit. Dort sollte die Möglichkeit einer oralen Immunisierung mit der SSLV im Vergleich zur regulären Impfung mit der SSLV in Ziegen getestet werden. Analog zu Gruppe 3 wurden durch die initiale Impfung in Woche 3 hohe, aber auch stark streuende anti-rPA83 Titer (~13000) gemessen. Diese sanken vergleichsweise schnell in Woche 6 auf ein stabiles, niedriges Niveau (~1500), welches sich bis Woche 9 nicht weiter änderte. Vergleichend dazu war die Immunisierung von Gruppe 3 langlebiger und auch der Initialtiter in Woche 4 (~50000) sehr viel höher. Zwar fielen auch hier die Titer danach rapide ab, pegelten sich aber erst ab Woche 12 – 16 auf ein unveränderliches Niveau (~3000) ein. Vergleicht man die Daten von Shakya et al. (2007) [280] mit der weniger gut verlaufenen Immunisierung von Gruppe 2 ergibt sich eine größere Überschneidung, da diese 6 Wochen nach der Immunisierung ebenfalls bereits sehr niedrige Titer aufwies (~3000). Aufgrund der fehlenden Messung für Woche 3 kann allerdings nicht gesagt werden, ob diese zu einem früheren Zeitpunkt ebenfalls entsprechend höhere Titer gehabt hätte. Dennoch zeigt dies, sowie der Bericht von Salmon et al. (1992) [266] deutlich, dass eine solche Variation in der Immunreaktion auf einen initiale Immunisierung mit der SSLV kein Einzelfall sein muss. Auch bei der Anwendung in Pferden ist bekannt, dass sich die erzeugte Immunität bei einer initialen Immunisierung mit der SSLV nur sehr langsam entwickelt [228]. Darüber hinaus ist auch eine unterschiedliche Wirksamkeit der Vakzinierung durch Chargenunterschiede denkbar. Damit ist fraglich ob eine Schutzwirkung der SSLV ab der ersten Immunisierung umfassend für alle Tiere gegeben ist, zumal sogar ein vollständiges Ausbleiben 156 4. Ergebnisse und Diskussion einer detektierbaren Reaktion, einhergehend mit dem Verlust der Schutzwirkung, wie in 2 Tieren von Gruppe 2 beobachtet wurde, möglich ist. Die Versuche von Shakya et al. (2007) [280] sahen weiterhin eine Reimmunisierung mit der SSLV in Woche 9 vor, die eine Erhöhung der Titer gegen PA bereits nach einer Woche auf ein weitaus höheres Niveau (~40000) als das der initialen Impfung bewirkte. Dieses Niveau blieb noch mindestens drei weitere Wochen erhalten und entsprach von der Höhe eher den in unseren Versuchen gemessenen Titern für Gruppe 3, sowohl nach der ersten Immunisierung, als auch nach der Revakzinierung von Gruppe 3b. Damit scheint der grobe Verlauf der Immunisierungen sehr ähnlich zu sein, in den Details sind aber durchaus Unterschiede erkennbar, wie sie auch analog zu den Ergebnissen von Gruppe 3 und 2 vorlagen. Die unspezifische phagozytotische Wirkung von Ziegenseren könnte die Wirkung der Immunisierung mit der SSLV beeinflussen Neben der Bedeutung für die Schutzwirkung einer Erstimmunisierung mit der SSLV werfen die Ergebnisse auch die Frage nach der Ursache der ausbleibenden Wirkung der SSLV in einzelnen Tieren auf. Wie zuvor erwähnt, scheint die zwischen Gruppe 3a und 2 bezüglich der Erstimmunisierung notierte Diskrepanz keine Ausnahme zu sein, da auch Beispiele in anderen Arbeiten zu finden sind, bei denen die erste Immunisierung mit der SSLV kaum erhöhte anti-rPA83 Titer erzeugte [280][192][144][84]. Erstaunlicherweise waren die Unterschiede in den Überlebensraten zu Gruppen, die mit hohen anti-rPA83 Titern reagierten nur gering und auch in den hier beschriebenen Versuchen überlebten noch 60 % (Gruppe 2) trotz kaum erhöhter Titer. Dies ist ein deutlicher Hinweis darauf, dass die Schutzwirkung durch die SSLV vermutlich durch zusätzliche Faktoren, als nur die anti-rPA83 IgG-Titer klassifiziert werden sollte. Allerdings geben diesbezüglich für Gruppe 2 die gemessenen Titer für FIS, TNA, BclA und das vegetative Antigen nach der Immunisierung keine weiteren Hinweise, da keiner der Titer eine Veränderung aufwies. Einzig die gemessenen anti-FIS IgG-Titer waren leicht erhöht, lagen aber ebenfalls weit unter den Werten von Gruppe 3 für Woche 4 und 8. Neben potentiellen Unterschieden in der Wirksamkeit verschiedener Vakzinechargen könnte die von Welkos et al. (2001) [352] beschriebe Wirkung von naiven Tierseren auf Sporen einen Hinweis auf die Ursache für die unterschiedlichen Reaktionen auf die Vakzinierung bieten. Demnach scheinen besonders bei Ziegenseren eine hohe unspezifische phagozytosefördernde Wirkung zu haben, die zu einer vermehrten Aufnahme der Sporen durch murine Makrophagen führt. Diese Eigenschaft, sofern sie auch auf andere Makrophagen zutrifft, könnte einen Einfluss auf die individuelle Wirksamkeit der SSLV haben. 157 4. Ergebnisse und Diskussion Der Höchstpunkt der anti-rPA83 IgG-Titer ist 1 – 2 Wochen nach der Immunisierung zu erwarten Im Hinblick auf die erst spät erfolgte Beprobung von Gruppe 2 in Woche 6 stellt sich die Frage, ob die gemessenen Titer für Gruppe 3 in Woche 4 noch dem Höhepunkt entsprachen oder diese dort schon im Absinken begriffen waren. Die bereits eine Woche nach der Revakzinierung deutlich erhöhten Titer bei Shakya et al. (2007) [280] würden für Letzteres sprechen, wenn auch klar ist, dass eine Auffrischungsimpfung anders verläuft als eine Erstimmunisierung. Andere Studien mit der SSLV, vornehmlich in Meerschweinchen, machen teils nur sehr ungenaue Angaben bezüglich des Zeitpunkts der Blutentnahme, wobei dieser oft mit „nach der Immunisierung“ angegeben wird und daher davon auszugehen ist, dass die Probennahme entsprechend zeitnah nach der Immunisierung erfolgte. Die angegebenen Titer gegen PA umfassten dabei, auch in Abhängigkeit der applizierten Sporenzahl bei einer einmaligen Immunisierung, 200 – 6000 [192][148][144][329], was eher den von Shakya et al. (2007) [280] angegebenen Werten bzw. Gruppe 2 entspricht. Ein von Cohen et al. (2000) [58] durchgeführter Versuch mit einem SSLV-ähnlichen EF- und LF-defizientem Stamm ergab einen Höchsttiter gegen PA erst 8 Wochen nach der Immunisierung, wohingegen Versuche mit azellulärem PA bereits nach 2 Wochen Höchstwerte erreichten [365] [191]. Damit bleibt der genaue Zeitpunkt der höchsten Immunreaktion weiterhin ungenau, wobei dieser, aufgrund den Ergebnissen von Shakya und der rasanten Titerentwicklung 2 Wochen nach der Infektion für Gruppe 2 und 3 a, vermutlich eher innerhalb der ersten 2 Wochen nach der Immunisierung zu erwarten ist. Eine Vakzinierung mit der SSLV von Tieren mit erhöhten Antikörpertitern (gegen PA) ist wirkungslos Der danach folgende rasante Abfall der Titer scheint ebenfalls bereits in anderen Applikationen beobachtet worden zu sein [280][365][191] und kann eventuell in Abhängigkeit der initial erzeugten Titer wie in unserem Fall bis zu 16 Wochen nach der Immunisierung andauern, bevor ein unveränderliches Niveau erreicht ist. Dies kann insbesondere im Zuge einer rasch folgenden Revakzinierung besonders ins Gewicht fallen. Betrachtet man die überlebte Infektion mit dem voll virulenten Stamm SD20 als weitere Immunisierung mit einer Lebendvakzine, so konnte durch diese in der frisch revakzinierten Gruppe 3b keiner der gemessenen Titer weiter erhöht werden. Ähnlich verhielt es sich auch für ein Individuum von Gruppe 3a (8176), welches vor der Infektion noch leicht erhöhte anti-rPA83 IgG-Titer aufwies. In beiden Fällen könnten die erhöhten Antikörpertiter die Infektion vollständig abgefangen haben, sodass diese keine weitere Immunreaktion auslösen konnte [82][284]. Setzt man also eine gute Wirksamkeit der Erstimmunisierung voraus, könnte eine zu dicht darauf folgende zweite Immunisierung wirkungslos verpuffen. Teilergebnisse von Ivins et al. (1990) [148] unterstützen diese Annahme, wonach 158 4. Ergebnisse und Diskussion einmalige Immunisierungen mit der SSLV in Meerschweinchen äquivalent hohe anti-rPA83 Titer erzeugte wie zwei Immunisierungen im Abstand von 4 Wochen. Überdies ergaben die zusammengefassten Ergebnisse anderer Studien kaum Unterschiede in der Titerhöhe bei ein und mehrfach in rascher Folge immunisierten Meerschweinchen, wenn die Erstimmunisierung Wirkung zeigte [192][144][84]. Eine gedoppelte Erstimmunisierung könnte die sporadischen Immunisierungsausfälle mit der SSLV beheben Zusammengenommen haben die beschriebenen Ergebnisse der Versuche mit der SSLV Implikationen für die Praxis. So muss davon ausgegangen werden, dass eine Erstimmunisierung, eventuell auch chargenabhängig, mit der SSLV nicht allen Tieren einen Schutz vermittelt und eine umfassende Schutzwirkung für alle immunisierten Tiere erst frühestens ab der zweiten Immunisierung gegeben ist. Daher scheint es unplausibel eine zweite Immunisierung erst nach 9 – 12 Monaten durchzuführen. In Anbetracht des zuvor beschriebenen Zeitfensters 12 – 16 Wochen in dem nicht nur die anti-rPA83 IgG-Titer, sondern auch die anti-FIS IgG-Titer zwar fallend, aber immer noch leicht erhöht sind, würde sich eine zweite Impfung nach 16 Wochen anbieten. Dadurch wäre eine erneute immunogene Reaktion ohne Verluste durch bestehende Antikörpertiter wahrscheinlich. Für einen ungewissen Impfschutz ist allerdings auch eine Wartezeit von 4 Monaten recht lang. Daher wäre auch eine doppelte initiale Impfung im Abstand von 1 bis 2 Wochen möglich. Davon könnten Tiere profitieren bei denen die Erstimmunisierung nicht angegangen ist. Dieser Ansatz wird auch indirekt durch die zuvor genannten Arbeiten unterstützt. Diese führten Fallbeispiele mit sehr niedrigen Titerentwicklungen bei einmal immunisierten Tieren auf, aber niemals für schnell aufeinander folgende, mehrfach Immunisierte und dies galt auch für Tests mit niedrigeren Sporenkonzentrationen. Eine mindestens doppelte, rasch aufeinander folgende Immunisierung scheint das sporadische Fehlschlagen der Erstimmunisierung zu beheben. Eine entsprechende Empfehlung dieser Art der Erstimmunisierung ist für Pferde bereits vorhanden [228]. Überdies würde es, abgesehen von der verminderten Konzentration der Prä-Inokulation, exakt den Vorgaben der WHO zur Immunisierung von Ziegen entsprechen [357], die aber nicht aufgrund der verbesserten Wirksamkeit, sondern zur Verminderung von adversen Reaktionen auf die Vakzine postuliert wurde. Die Auffrischungsimpfung sollte 3 – 4 Monate nach der Erstimmunisierung erfolgen Unabhängig davon, ob in einem veränderten Impfplan mit initial zwei dicht aufeinanderfolgenden Impfungen oder nur einer Erstimmunisierung operiert wird, wäre aber dennoch die Applikation der ersten Folgeimpfung 3 – 4 Monate nach der letzten Impfung sinnvoll, da diese fakti- 159 4. Ergebnisse und Diskussion sche Auffrischungsimpfung einen zusätzlichen Effekt zu haben scheint. Vergleicht man die Titerwerte der ersten und zweiten Immunisierung von Gruppe 3b, scheinen die anti-rPA83 IgGTiter ähnlich und für die zweite Immunisierung sogar leicht niedriger zu sein. Dies könnte aber auch darauf zurückzuführen sein, dass sie, wie bei Shakya et al. (2007) [280] dargestellt, früher ihren Höhepunkt erreicht haben und in Woche 4 nach der Impfung bereits wieder abfallen. Davon abgesehen sind die anti-FIS und anti-vegetativ IgG-Titer für die erste Immunisierung deutlich geringer ausgefallen als für die zweite, sodass diese drei- bzw. zehnfach höhere Werte zeigte. Zudem scheint auch der Anteil an neutralisierenden Antikörpern im Vergleich zur Erstimmunisierung, trotz ähnlich hoher anti-rPA83 Titer, zugenommen zu haben. Prozentual machten die TNA-Titer nach der ersten Immunisierung nur 0,5 % der anti-rPA83 IgG-Titer aus, nach der 2. Immunisierung bzw. überlebten Infektion betrugen diese 2 – 4 %. Hierbei ist es allerdings anzumerken, dass bereits ab Woche 37 und für alle folgenden Zeitpunkte TNA-Titer knapp oberhalb der Nachweisgrenze messbar waren, obwohl die anti-rPA83 IgG-Titer unverändert blieben. Da diese Erhöhung nicht mit den Titern gegen rPA83 einherging, wäre auch eine Beteiligung der hier nicht gemessenen anti-LF Antikörpertiter möglich, die nachweislich ebenfalls den TNA beeinflussen können [199][43] und im Zuge einer Immunisierung mit der SSLV ebenfalls produziert werden [192]. Zusammenfassend sprechen die Ergebnisse dieser Arbeit dafür, dass eine zweite Immunisierung nach einem Jahr ein deutlich anderes Titerspektrum erzeugt als die Erstimmunisierung. So werden neben einer Auffrischung der Immunreaktion gegen rPA83 durch die zweite Immunisierung auch verstärkt Antikörper gegen die Spore und den vegetativen Zellkörper gebildet. Eine jährliche Auffrischung der Impfung mit der SSLV könnte generell obsolet sein oder durch eine Änderung des Impfplans obsolet werden Abschließend ist auch die Dauer des Impfschutzes durch die Vakzinierung mit der SSLV und die damit einhergehende Notwendigkeit jährlicher Auffrischungen zu erörtern. Langzeitstudien vor allem mit den Humanvakzinen AVA und AVP zeigen deutlich, dass zumindest deren auf PA basierende immunogene Wirkung bis zu 2 Jahre anhält [241][367], während für die SSLV und SSLV-ähnliche Lebendvakzinen eine Mindestwirkzeit von nur 9 – 12 Monaten angegeben wird [357][228][58]. Ausgehend von den hier erläuterten Ergebnissen wäre eine solche Begrenzung der Wirkdauer für die SSLV nicht anzunehmen. Zunächst scheinen die nach Woche 16 stabil, leicht erhöhten Titer gegen rPA83 und FIS bis zur Revakzinierung oder respektive Infektion auf einem einheitlichen Niveau zu verbleiben (p ≥ 0,114). Daher gibt es keinen Grund anzunehmen, dass sich dies nach einem Jahr ändert, zumal die Titer im Bezug auf PA auch in Woche 62 immer noch signifikant höher waren als die vor dem Beginn der Immunisierung gemessenen (p = 0,006). Zudem waren vier von fünf der 160 4. Ergebnisse und Diskussion nicht revakzinierten Tiere (Gruppe 3a) auch 62 Wochen nach der Erstimmunisierung geschützt, was zudem sogar eine marginal bessere Schutzrate erzeugte als die frisch immunisierte Gruppe 2 mit drei von fünf Überlebenden. Hinzu kommt, dass die einzige verstorbene Ziege aus Gruppe 3a nicht nur eine verlängerte Überlebenszeit zeigte, sondern nach der ersten Immunisierung (Woche 4) mit Abstand die niedrigsten anti-rPA83 IgG-Titer der Gruppe 3 aufwies und somit womöglich eher Ähnlichkeit mit den Tieren und der beschriebenen Problematik von Gruppe 2 hatte. Legt man die zuvor erörterten Theorien zugrunde, hätte gerade diese von einer „doppelten“ Erstimmunisierung profitiert und wäre vermutlich wie alle anderen auch nach einem Jahr nicht verstorben. Auf Grundlage dieser Ergebnisse ist daher nicht davon auszugehen, dass der Impfschutz auf ein Jahr begrenzt ist und noch weniger, wenn der Erfolg einer Erstimmunisierung gewährleistet und durch eine zweite Impfung 3 – 4 Monate später aufgefrischt ist. Um dies zu untermauern, müssten langandauernde Impfstudien über mehrere Jahre durchgeführt werden. Eine Begrenzung des Impfplans auf maximal 2 – 3 Immunisierungen innerhalb von einem halben Jahr ohne weitere Auffrischungen wäre aber eine deutliche Verbesserung zum jetzigen Szenario. 4.4.4 Generelle Anmerkungen zu den Ergebnissen Neben den diagnostischen Ergebnissen und den Vergleichen der Versuche untereinander und zur vorhandenen Literatur warfen die Versuche generelle Fragen auf bzw. gaben weitere Denkanstöße zu bereits bestehenden Fragestellungen. Diese sollen im Folgenden beleuchtet werden. BclA ist in Ziegen nicht bzw. wenig immunogen Zur Unterstützung und Verbesserung der Wirksamkeit von rPA83 wurde bei den Maus- und Ziegenversuchen das Sporenantigen rBclA eingesetzt in der Hoffnung, den Vakzinen neben einer Antitoxinwirkung auch eine Antisporenwirkung hinzuzufügen. Die Eignung von rBclA für diese Aufgabe schien dabei klar, da es als äußerster Bestandteil der Spore zu deren immunogensten Antigenen gehört [310][296] und bereits in anderen Versuchen die Schutzwirkung von PA verbessern konnte [39][125]. Diese Eigenschaft wurde auch in den Mausversuchen dieser Arbeit bestätigt, wobei rBclA nicht nur äquivalent hohe Antikörpertiter wie rPA83 erzeugte, sondern innerhalb einer DNA-Vakzine auch eine eigenständige Schutzwirkung zu haben schien. Auch in anderen Tierarten wie Kaninchen, Rind, Schaf und Rhesusaffe [195][64][264] war die Immunogenität von BclA bereits erwiesen, sodass es keinen Anlass gab, an der analogen Wirkung in Ziegen zu zweifeln. Daher war die Abwesenheit einer deutlichen Titerentwicklung gegen rBclA in den getesteten Ziegen höchst erstaunlich. Durch die SSLV konnten keine signifikanten Titer gegen rBclA erzeugt werden und auch die NLV mit rBclA und FIS erzeugten teil161 4. Ergebnisse und Diskussion weise zwar signifikante, aber nur marginal erhöhte Antikörpertiter. Dabei schienen die Gruppen mit FIS und BclAD1D3 als DNA-Vektor im Vergleich etwas besser zu funktionieren als rBclA. Als mögliche Ursache im Bezug auf FIS könnte hier die fehlende Glykosylierung von rBclA [296] in Betracht gezogen und damit einhergehend die verringerte Stabilität des Trimers [254] herausgestellt werden, wobei mögliche Folgen unklar sind. Des Weiteren sind fehlerhafte Messungen diesbezüglich auszuschließen, da die gleichen Präparationen im Mausmodell und im ELISA funktional waren und auch über den ELISA mit FIS, welches nachweislich ebenfalls BclA enthielt (siehe Kapitel 4.1.1.5, 4.1.2.2 und 4.1.2.3), keine erhöhten Titer in Gruppe 4 und 4 (W) (erhielt rBclA aber keine FIS) detektiert werden konnten. Die immunogene und möglicherweise auch Schutz vermittelnde Wirkung von FIS beruht nicht auf BclA Überraschend in diesem Zusammenhang war die hohe Immunogenität von FIS bzw. der lebenden Sporen der SSLV, war man doch davon ausgegangen, dass ein Großteil von deren Immunogenität auf BclA zurückzuführen sei. So zeigten die mit der SSLV immunisierten Gruppen und die zusätzlich mit FIS immunisierte Gruppe 6 Antikörpertiter gegen FIS, die denen gegen rPA83 entsprachen. Ein ähnliches Bild hatte sich bereits für die Mausversuche abgezeichnet, war aber hauptsächlich dem vorhanden BclA auf der FIS Präparation zugeschrieben worden. Zur Aufklärung dieses Missverhältnisses gereichen neuere Studien, welche die Funktion von BclA in einem etwas anderen Licht als bisher darstellen. So revidieren die Arbeiten von Cybulsky et al. (2008) [64], Côte et al. (2012) [61] und Vergis et al. (2013) [336] nicht nur das Alleinstellungsmerkmal von BclA als einzigem relevanten Immunogen der Spore, sondern definieren dessen Rolle sogar eher als maskierenden Täuschkörper mit der Funktion der Überdeckung anderer möglicherweise viel relevanterer Antigene der Spore. Unter diesem Gesichtspunkt scheint die geringe immunogene Reaktion von Ziegen auf BclA in Kombination mit einer hohen Immunogenität von FIS bzw. der lebenden Sporen der SSLV merkwürdig. Wie exemplarisch innerhalb der Mausversuche (siehe Kapitel 4.1.1.5, 4.1.2.2 und 4.1.2.3) gezeigt wurde, war die Detektion von Antikörpern gegen rBclA und BclAD1D3 mittels FIS möglich. Daher ist davon auszugehen, dass die BclA-Filamente sowohl auf FIS, als auch auf den Sporen der SSLV noch weitestgehend intakt sind. Trotzdem erzeugten FIS und die SSLV in Ziegen hohe Titer gegen FIS, die nicht auf eine immunogene Wirkung von BclA zurückzuführen waren und trotz der Anwesenheit von BclA auf den Sporen gebildet wurden. Auf Grundlage der Beobachtung der überdeckenden Eigenschaften von BclA auf andere Sporenantigen in vorhergehenden Studien [64][61][336] sind mehrere Erklärungen denkbar, die die hier präsentierten Ergebnisse mit einbeziehen. 162 4. Ergebnisse und Diskussion Zunächst könnte das Potential BclAs zur Überdeckung anderer Antigene ein Faktor der Immunogenität selbst sein, sodass eine verstärkte Immunreaktion auf BclA eine vergleichbare Reaktion auf konkurrierende Antigene vermindert. Ist die Immunogenität von BclA herabgesetzt, wie es bei Ziegen zu sein scheint, bestünde dieser Effekt nicht und eine Reaktion auf weitere Sporenantigene auch bei Anwesenheit von BclA wäre ungehindert möglich. Unter diesen Umständen hätte eine Sporenpräparation ohne BclA eine ähnliche immunogene Wirkung wie mit BclA. Eine weitere mögliche Erklärung, die für die tatsächliche mechanische Überdeckung anderer Antigene durch BclA sprechen würde, ist die Beschädigung des Exosporiums oder der BclAFilamente. Sowohl FIS, als auch die SSLV sind Produkte eines präparativen Protokolls mit mehreren Reinigungsschritten. Dabei könnte das Exosporium und dessen BclA-Filamente teilweise beschädigt worden sein, womit der Zugang zu weiteren Sporenantigenen, trotz Anwesenheit von BclA ermöglicht wäre. Die in Ziegen gemessenen anti-FIS IgG-Titer müssten demnach durch die komplette Entfernung des BclAs von der Sporenoberfläche weiter erhöhbar sein. Eine Bestätigung dieses Szenario hätte allerdings weitreichende Implikationen für die Vakzineproduktion und die Vergleichbarkeit verschiedener Arbeiten mit Lebendvakzinen, FIS und sogar Infektionsstämmen. Sogar die in den vorherigen Kapiteln bemerkte unterschiedliche Wirksamkeit der SSLV könnte durchaus auf entsprechende Schwankungen im Präparationsprotokoll zurückzuführen sein. Ein weiterer Erklärungsversuch bezieht die Länge der BclA-Filamente mit ein. Bedingt durch die variable Länge der CLR variiert auch die Länge der BclA-Filamente stammspezifisch [296][310][254]. Bisher hat man diesem Umstand wenig Bedeutung beigemessen, da die gänzliche Abwesenheit von BclA keinen nachweislichen Einfluss auf die Resistenz und Virulenz von B anthracis Sporen zu haben schien [296][310] [33]. Trotzdem könnte durch die Länge der Filamente, die Zugänglichkeit weiterer Antigene unterschiedlich stark beeinträchtigt sein. Auch die Struktur der BclA-Filamente, mit einem haarähnlichem Mittelteil und bulbärem Ende (Abbildung 57, [310]), könnte dabei von Bedeutung sein und zur Abschirmung der Sporenoberfläche beitragen. In diesem Zusammenhang zeichnet sich gerade der für die SSLV verwendete und innerhalb dieser Arbeit auch zur Erstellung von Elektronenmikroskopische Aufnahme eines BclA Filaments nach Färbung und Immunogold Markierung aus [310]; der Maßstab entspricht 100 nm Abbildung 57: BclA-Filament FIS benutzte Stamm 34F2 durch die längsten bislang getesteten BclA-Filamente aus [311]. Inwiefern diese Eigenschaft die besondere Eignung als Vakzinestamm beeinflusst haben könn163 4. Ergebnisse und Diskussion te, ist allerdings unklar. Ebenso geben die vorangegangenen Arbeiten über die Wirkung von BclA als Täuschkörper wenig Aufschluss über den potentiellen Anteil der Länge der BclA-Filamente an der immunogenen Wirkung der SSLV-Sporen, da diese mit Sporen von B. anthracis Ames [61] oder B. cereus [336] durchgeführt wurden. Wie auch immer die tatsächlichen Begebenheiten sein mögen, die Grundlage der Immunogenität von FIS in Ziegen scheint nicht auf BclA zu beruhen, wobei die fehlende Immunogenität von BclA in diesem Tiermodell für sich genommen Grundlage neuer Experimente sein sollte. Möglicherweise ist auch die beschriebene Sensitivität dieser und anderer sensibler Spezies auf diesem Merkmal begründet. Trotzdem sollte die Relevanz von BclA als Antigen innerhalb einer universell einsetzbaren NLV erhalten bleiben. Die Versuche in NMRI-Mäusen haben deutlich gezeigt, dass eine additive Schutzwirkung durch den Zusatz von BclA ausgehen kann und dass diese durch FIS noch verstärkt wird. Allerdings sollte in Anbetracht der überdeckenden Eigenschaften von BclA bei einen Zusatz von lebenden oder inaktivierten Sporen die Verwendung BclA-defizienter Stämme in Erwägung gezogen werden. Davon abgesehen, sollten die Auswirkungen unterschiedlicher Präparationsprotokolle für lebende und inaktivierte Sporen sowie die Verwendung unterschiedlicher Stämme eingehend untersucht werden. Nicht nur die unterschiedliche Länge von BclA oder dessen Konzentration auf der Oberfläche könnten eine größere Relevanz haben als bisher angenommen, sondern auch die von Welkos et al. (2001) [352] beschriebene, möglicherweise auch präparationsbedingte Einlagerung von vegetativen Bestandteilen in die Sporenoberfläche. Eine überlebte Infektion erzeugt ein anderes Titerspektrum als eine Impfung mit der SSLV Neben den bereits beschriebenen Unterschieden für die erste und zweite Immunisierung mit der SSLV waren auch serologische Unterschiede zur überlebten Infektion mit dem voll virulenten Stamm SD20 vorhanden. Während die Auffrischungsimpfung mit der SSLV (Gruppe 3b) bei gleichen anti-rPA83 IgG-Titern in höheren TNA, anti-FIS und anti-vegetativ IgG-Titern resultierte, erzeugte die überlebte Infektion bei Gruppe 3a nur äquivalent hohe anti-rPA83 IgG-Titer und erhöhte TNA-Titer. Verstärkte Reaktionen gegen FIS und das vegetative Antigen blieben aus. Die Reaktionen auf das Toxin schien äquivalent zu einer zweiten Immunisierung mit der SSLV zu sein. Da eine Reaktion auf das Toxin erfolgte, ist davon auszugehen, dass eine Germination nebst Ausschüttung von Toxinen in den meisten infizierten Tieren, abgesehen von der frisch revakzinierten Gruppe 3b, stattgefunden haben muss. Um so verwunderlicher ist die schwache oder gänzlich fehlende Reaktion gegen die Spore und den vegetativen Zellkörper. Die einfachste generelle Erklärung zu diesem Unterschied wäre schlicht die unterschiedliche Konzentration der Sporen. Während sowohl die SSLV als auch FIS mit einer Dosis von >107 Sporen 164 4. Ergebnisse und Diskussion appliziert wurde, betrug die Infektionsdosis nur ~900 Sporen. Daher wäre es möglich, dass die immunogene Reaktion auf die zellulären Komponenten, besonders die der Sporenantigene konzentrationsbedingt weitaus geringer ausgefallen ist, sodass sie weniger deutlich sichtbar ist. Davon abgesehen, scheint als Begründung für die mangelnde Reaktion auf den vegetativen Zellkörper die Bildung einer Kapsel durch den Infektionsstamm naheliegend. Diese ist nur wenig immunogen, inhibiert die Phagozytose der vegetativen Zelle und verhindert die Erkennung darunter liegender Antigene, sowie die Opsonierung durch das Komplementsystem und Antikörper [163]. Da der zur Immunisierung verwendete SSLV-Stamm aufgrund des Verlusts des entsprechenden Plasmids keine Kapsel ausbildet, war im Gegensatz zu Infektion eine Reaktion auf den vegetativen Zellkörper bei der Immunisierung uneingeschränkt möglich. BclA könnte im Ziegenmodell die gleiche Funktion für die Spore einnehmen wie die Kapsel für den vegetativen Zellkörper Lässt man die konzentrationsbedingte Begründung für die Unterschiede außer acht, war die mangelnde Reaktion auf die Spore zwar unerwartet, aber im Zuge der zuvor erläuterten eventuellen Unterschiede verschiedener Stämme oder/und Präparationen bezüglich der BclA-Filamente plausibel. Sollten strukturelle Unterschiede die Detektionsfähigkeit anderer sporenspezifischer Antigene als BclA bedingen und im schlechtesten Falle gänzlich verhindern, so könnte die Immunogenität der Spore gänzlich von einer Reaktion auf BclA abhängig sein. Da im vorliegendem Fall BclA kaum eine immunogene Wirkung erzeugte, ist die Spore, wie auch der durch die Kapsel geschützte vegetative Zellkörper, immunologisch inert, wodurch einzig die Antitoxinwirkung für einen Schutz verbleibt. Sollte dies wirklich innerhalb des Ziegenmodells Bestand haben, erklärt dies eventuell deren Sensitivität gegenüber dem Erreger, da innerhalb einer natürlichen Infektion weder die Spore, noch die vegetative Zelle spezifisch erkennbar wären und deren Beseitigung hauptsächlich auf unspezifischer Phagozytose und der Antitoxinwirkung beruhen würde. Diesen Gedanken weiterführend, stellt sich die Frage, inwiefern die Einbeziehung immunogener Antigene gegen die Spore und das vegetative Antigen sinnvoll ist, wenn diese nicht zur Beseitigung einer virulenten Infektion beitragen. Sinnvoller erscheint die Erhöhung der Immunogenität der Kapsel und BclA, um eine bessere Detektion der des Bakterienkörpers zu ermöglichen, was beispielsweise mittels Konjugation dieser an PA erreicht werden könnte. 165 5. Konklusion und Ausblick 5 Konklusion und Ausblick Die Ergebnisse dieser Arbeit führten neben der Evaluation neuer Ansätze für eine NLV und deren Abgleich mit der Sterne Sporen Lebendvakzine (SSLV) zu neuen Erkenntnissen für die Diagnostik und die Auswertung ähnlicher Versuche. Besonders zukünftige Versuche in Ziegen und anderen Großtieren könnten durch die beschriebenen Verbesserungsvorschläge reibungsloser ablaufen und eine frühzeitigere Diagnose ermöglichen. So konnte das Auftreten von Bakterien im Blut bei Ziegen in einem Zeitraum von 2 – 5 h vor dem Tod nachgewiesen und, durch eine praktikablere Blutentnahme mittels Insulinspritze, als geeignetes Diagnosemittel zum Nachweis einer terminalen Infektion etabliert werden. In Kombination mit der ermittelten Temperaturschwelle von 40,4 °C und optimalen Beobachtungsintervallen sollte eine frühzeitige Diagnose, nebst aussichtsreicher Behandlung, von 75 % der später versterbenden Tiere möglich sein. Überdies konnte ein Zusammenhang zwischen der Entwicklung von Fieber (≥ 40,0 °C) und den nach der Infektion gemessenen anti-rPA83 IgG-Titern nachgewiesen werden. Demnach ging eine deutliche Erhöhung der anti-rPA83 IgG-Titer nach der Infektion mehrheitlich mit dem Auftreten von Fieber einher. Tiere bei denen kein Fieber detektierbar war, zeigten ausnahmslos keine Titererhöhung gegen rPA83. Unter der Annahme, dass bei einer Infektion ohne Titer- und Temperaturerhöhung diese schon frühzeitig geklärt werden konnte, lassen sich anhand der Veränderungen der Messwerte Aussagen über die Qualität einer Schutzwirkung treffen. Eine detaillierte serologische Analyse der Seren nach der Infektion wird bisher nicht regulär durchgeführt bzw. ist selten Bestandteil von Publikationen, würde aber eine tiefere Diskriminierung der Ergebnisse eines Infektionsversuchs erlauben, die bisher nur das Überleben und die überlebte Zeit bei Verstorbenen in die Auswertung mit einbezieht. Ob dieser Zusammenhang zwischen erhöhter Temperatur, Titerentwicklung und Verlauf der Infektion auch in anderen Spezies als der Ziege Gültigkeit hat, muss aber noch gezeigt werden. Beim Nachweis eines generell gültigen Zusammenhangs könnte die Titerveränderung durch die Infektion auch Rückschlüsse auf die Entwicklung von Fieber erlauben, wenn es spezies- oder situationsbedingt nicht gemessen werden kann. Für die Antikörpertiter gegen rPA83 vor der Infektion waren Schwellenwerte erkennbar, die bei Ziegen und Auszucht-Mäusen mit einer Infektionsdosis von ~1000 Sporen eine Aussage zum Ausgang der Infektion erlaubten. Nahezu 50 % aller später verstorbenen Ziegen wiesen einen anti-rPA83 IgG-Titer von < ~550 auf und zeigten maximal eine kurzzeitige Erhöhung der Temperatur (< 10 h). Alle Ziegen mit höheren Antikörpertitern als 550, die im weiteren Verlauf starben, wiesen eine unterschiedlich lang anhaltende, erhöhte Temperatur auf. Erst ab einem anti-rPA83 IgG-Titer von ~3400 überlebten 85 % der infizierten Ziegen, wobei eine eindeutige 166 5. Konklusion und Ausblick Zuordnung zu den Überlebenden erst auf Tiere mit einem Titer von mindesten ~20000 zutraf. Im Mausmodell war ein Schwellenwert von ~30000 prädiktiv für 75 % der Überlebenden, wobei Tiere mit Titerwerten unterhalb von 800 ausnahmslos verstarben. Damit könnten die anti-rPA83 IgG-Titer vor der Infektion als prädiktive Diagnosehilfe bei der Infektion verwendet werden und im Falle der Ziegen zusammen mit der erörterten Temperaturschwelle eine nahezu vollständige Erfassung aller gefährdeten Tiere ermöglichen. Eine statistisch belegbare Korrelation der Antikörpertiter innerhalb der einzelnen Gruppen ergab sich aber nur bei Maus- und Ziegengruppen, die insgesamt sehr niedrige Antikörpertiter aufwiesen. Erst bei einer gruppenübergreifenden Betrachtung der Titerwerte, unterteilt nach Spezies und Infektionsdosis ergaben sich aussagekräftige Korrelationen. Damit scheinen für Ziegen die Antikörpertiter gegen die Spore (exklusive BclA), den vegetativen Zellkörper und PA für das Überleben von Bedeutung zu sein. In Auszucht-Mäusen war eine Korrelation, in Abhängigkeit von der Infektionsdosis nur für die anti-rPA83 IgG-Titer vorhanden, wobei Berechnungen für FIS und das vegetative Antigen aufgrund fehlender Daten nicht durchführbar waren. Neben der Analyse der Seren nach der Infektion und dem Hinzuziehen der Titerdaten vor der Infektion für eine bessere Diagnose, sind weitere Tests zur Verbesserung der Auswertung denkbar. Während die Immunreaktion auf die Toxinkomponenten mittels ELISA und TNA quantitativ und qualitativ bestimmt wurde, war für die anti-Sporen Antikörper keine qualitative Einschätzung möglich. Dies könnte durch Messungen der Fähigkeit der anti-Sporen Antikörper zur Opsonierung von Sporen und deren Einfluss auf die Phagozytoserate von Sporen durch Makrophagen erweitert werden [352][351][79][64]. Zudem wäre eine Erfassung der gebildeten Gedächtniszellen für die Einschätzung der Dauer der Immunität und der entsprechenden Prognosen zur Schützbarkeit sinnvoll. Zur Erprobung neuer Ansätze für eine NLV wurden in Mäusen und Ziegen antigenkodierenden DNA-Vektoren angewendet. Dabei bot die Kombination der erfolgversprechendsten DNAVektoren in Mäusen mit 90 % Überlebenden einen äquivalenten Schutz wie eine analoge Proteinkombination und erzeugte bessere Resultate als ähnliche Ansätze mit anderen Vektorkonstrukten. Trotzdem lagen die gegen die Toxine gebildeten Antikörpertiter der DNA-Vakzinen in Mäusen weit unter den für die Proteingruppen gemessenen. Für eine weitere Erhöhung der Immunogenität und Schutzwirkung könnte daher eine separate Applikation der antigentragenden Vektoren und eine rein sekretorische Expression der Toxine förderlich sein. Die Immunogenität von BclA ohne die CLR innerhalb eines DNA-Vektors war von rekombinantem BclA nicht zu unterscheiden. Sowohl innerhalb der DNA-Vektoren als auch in Proteinform konnten hohe Antikörpertiter gegen BclA in Mäusen erzeugt werden, die innerhalb der 167 5. Konklusion und Ausblick DNA-Vektoren bei niedrigerer Infektionsdosis ohne weitere Antigene sogar partiell protektiv waren (50 %). Da die Überlebenden dieser Gruppen nach der Infektion keinen erhöhten antirPA83 IgG-Titer aufwiesen, ist zudem davon auszugehen, dass die durch BclA vermittelte Schutzwirkung auf einer sterilen Immunität beruhte. Die Übertragung der erfolgversprechenden Ergebnisse der Mausversuche mit DNA-Vakzinen auf Ziegen war nicht möglich. Dieselbe Vektorkombination, die in den Mausversuchen eine Schutzrate von 90 % erzielte, hatte eine kaum nachweisbare immunogene Wirkung in Ziegen. Auch eine zusätzliche Immunisierung mit den entsprechenden Proteingegenstücken ergab keine Ergebnisse, die auf eine Zusatzwirkung der DNA-Vakzinen schließen ließen. Daher wurde von eine Infektion gänzlich abgesehen. Da die Immunisierung der Ziegen i. m. erfolgte und nicht mittels GeneGun, wie in den Mausversuchen, kann der verminderte Effekt durch die Applikationsart zustande gekommen sein. Zwar wurde innerhalb der Ziegen mit einer höheren Dosis gearbeitet, diese schien aber den verminderten Effekt durch die veränderte Applikationsart nicht aufzuwiegen. Neben einer weiteren Erhöhung der Dosis für eine bessere Wirksamkeit und der Applikation der Vektoren durch GeneGun oder Elektroporation ist auch der Einsatz weiterer Adjuvanzien denkbar, die die Wirksamkeit von DNA-Vakzinen bei Injektionen unterstützen. Ausgehend von den Ergebnissen der Mausversuche und ähnlichen Studien in Großtieren [123] war die niedrige Wirksamkeit der DNA-Vektoren im Ziegenmodell vermutlich nicht auf eine mangelnde Funktionalität der Vektoren zurückzuführen, sondern auf die Art und Form der Applikation. Zur vollständigen Klärung der Anwendbarkeit der DNA-Vektoren in Ziegen wären daher weiterführende Versuche mit entsprechenden Verbesserungen in der Vakzinekomposition und Applikation notwendig. Die Erprobung neuer Vakzinekompositionen auf Proteinbasis ergab in Mäusen eine synergetische Wirkung von rBclA und rPA83 mit einer Schutzrate von 70 %, die durch den Zusatz von FIS zu einem vollständigen Schutz komplettiert werden konnte. Mit 80 % Überlebenden konnte die gleiche Komposition, bestehend aus Lipopeptid, rPA83, rBclA und FIS auch in Ziegen eine ähnliche Schutzrate erzeugen. Damit war die zuvor in mehreren Studien beschriebene Wirksamkeit einer Kombination von PA mit Sporenantigenen auch im NMRI-Mausmodell und Ziegen bestätigt. Allerdings scheint die immunogene Wirkung von FIS nicht wie bisher angenommen, ausschließlich auf BclA zurückzuführen zu sein und ist mitunter sogar negativ durch BclA beeinflusst oder zumindest gänzlich unabhängig davon. Hintergrund für diese Annahme war die unerwartet geringe bis nicht vorhandene Immunogenität von BclA im Ziegenmodell in Kombination mit einer hohen Immunogenität von FIS. Das im Mausversuch innerhalb der DNA-Vakzine und als Protein applizierte hoch immunogene BclA erzeugte in Ziegen kaum Antikörpertiter. Durch verschiedene Teilergebnisse war die 168 5. Konklusion und Ausblick Anwesenheit von BclA auf FIS und dessen Detektionsfähigkeit durch anti-rBclA Antikörper bereits nachgewiesen. Da auch eine Kombination von rPA83, rBclA und FIS kaum Antikörpertiter gegen rBclA erzeugte, war auch für natürliches, auf den FIS befindliches BclA keine nachweisliche Wirkung in Ziegen zu finden. In bisherigen Studien mit verschiedenen Mausinzuchtlinien, Kaninchen und Rhesusaffen war die Immunogenität von BclA unstrittig [310][296][39] [125][195][64][264], sodass die beobachtete Wirkung in Ziegen diese Spezies herausstellt und womöglich eine Grundlage für neue Erkenntnisse zu deren Sensibilität bietet. Im Licht dieser Erkenntnis war die hohe immunogene Wirkung von FIS in Ziegen umso erstaunlicher. Neuere Studien hatten BclA-Filamenten auf Sporen überdeckende Eigenschaften zugeschrieben, die die Immunogenität anderer, darunter befindlicher Antigene einschränkt [64] [61][336]. Daher war bei einer abwesenden Reaktion auf BclA nicht mit einer starken immunogenen Wirkung durch FIS zu rechnen. Eine Erklärung, die beiden Erkenntnissen gerecht wird, wäre eine präparationsbedingte Beschädigung der BclA-Filamente oder des Exosporiums in FIS-Präparaten, die zur Freilegung sonst verdeckter Antigene führt. Dies hätte entsprechende Konsequenzen für alle Präparationsprotokolle mit Sporen, da kleine unprotokollierte Eigenschaften, wie das Alter der Kultur, die Anzahl der Waschschritte, die Lagerung und Handhabung einen verstärkten Einfluss auf die Wirksamkeit des Präparats haben könnten. Dies beträfe neben FIS Präparationen auch Sporensuspensionen zur Infektion und Immunisierung und könnte mitunter auftretende Fluktuationen der Schutzrate, Infektiösität oder Wirkung erklären. Alternativ wäre auch eine Überdeckung der BclA-Filamente denkbar, die möglicherweise abhängig von der Länge und Dichte der Filamente einen flexiblen Zugang zu den darunter liegenden Antigenen erlaubt. Da die Länge der BclA-Filamente stammspezifisch variiert, ergäben sich entsprechende graduelle Unterschiede, die eventuell besonderen Einfluss auf Spezies haben könnten, die nicht auf BclA reagieren. In diesem Zusammenhang ist zu erwähnen, dass sich gerade die SSLV, die auch zur Produktion von FIS in dieser Arbeit verwendet wurde, durch die längsten bisher vermessenen BclA-Filamente auszeichnet [311], was dessen besondere Eignung als Lebendvakzine beeinflusst haben könnte. Eine genaue Aufklärung dieser Mechanismen und deren Implikationen für die Vergleichbarkeit von Studien mit verschiedenen Sporenpräparaten sollte Bestandteil zukünftiger Studien sein. Ungeachtet der Hintergründe der Reaktion von Ziegen auf BclA und FIS konnte sowohl in Ziegen als auch in Mäusen gezeigt werden, dass die zusätzliche Schutzwirkung von FIS nicht allein und nicht hauptsächlich auf BclA beruht. In Kombination mit den überdeckenden Eigenschaften BclAs und der vorhandenen Immunogenität anderer sporenspezifischer Antigene scheint daher die Verwendung von FIS ohne BclA für weitere Vakzinekompositionen sinnvoller. Zur Erhöhung der Immunogenität von BclA in Ziegen und eventuell ähnlich reagierenden Tieren wären zudem Konjugationsversuche in Anlehnung an die Konjugationsversuche der Kapsel 169 5. Konklusion und Ausblick denkbar. Ein genereller Zusatz von rekombinantem BclA zu einer universell einsetzbaren Vakzineformulation hätte aber auch ohne dessen Verbesserung weiterhin Bestand, da es in diversen Spezies hoch immunogen ist und in diesen zur Schutzwirkung beitragen kann. Ebenso sollten weiterhin Untersuchungen zur Konjugation der Kapsel und der Integration dieses Antigens in eine NLV unternommen werden. Die Dysfunktionalität des hier getesteten Kapselkonjugats war vermutlich einem Epitop des konjugierten Lipopeptids geschuldet. In Anlehnung an andere Studien [256][273][50] wäre daher eine Konjugation der Kapsel, und weiterführend auch BclA, an PA eine sinnvolle Alternative. Im Zusammenhang mit der beobachteten, extrem unterschiedlichen Wirkung von BclA in NMRI-Mäusen und Ziegen sollte die Wahl des Tiermodells zur Testung von Vakzinekomponenten kritisch betrachtet werden. Die Verwendung von Mausinzuchtlinien zur Testung einer breiten Auswahl an potentiellen Vakzinekomponenten hat neben den praktischen Gründen auch den Vorteil, diese, aufgrund der genetischen Ähnlichkeit, in einer Gruppe möglichst gleichförmig reagierender Individuen zu erproben. Dennoch ist hinlänglich bekannt, dass die Sensitivität gegenüber einer Infektion mit B. anthracis und entsprechend die Schützbarkeit speziesbedingt stark variiert [354][353]. Zudem konnte in dieser Arbeit gezeigt werden, dass auch in der Reaktion auf ein Antigen starke Unterschiede auftreten können. Entsprechend sind eindeutige Schlussfolgerungen aus Vorversuchen mit anderen Spezies als der Zielspezies bedenklich. Die Entscheidung über den Ein- oder Ausschluss eines Antigens als potentielle Komponente einer Vakzineformulation sollte daher nicht auf Grundlage von Vorversuchen in Spezies getroffen werden, die aufgrund der Durchführbarkeit der Versuche ausgewählt wurden. Eine solche Auswahl könnte zur Exklusion für die Zielspezies wichtiger Antigene führen, weil diese in den Vorversuchen mit anderen Spezies unreaktiv waren und vice versa. Entsprechend sollten vorbereitende Versuche in anderen Spezies nur verwendet werden, um graduelle Unterschiede ähnlicher Formulationen oder Kombinationen von Komponenten, deren Wirksamkeit in der Zielspezies belegt ist, zu evaluieren. Zusammenfassend war die Testung der NLV, bestehend aus rPA83, rBclA, FIS und Lipopeptid mit einer Schutzrate von 80 % sehr erfolgversprechend, kann aber noch weiter verbessert werden. Hintergrund für Testung neuer NLV waren die bekannten Defizite der SSLV als favorisierte Veterinärvakzine gegen B. anthracis. Neben der Unvereinbarkeit einer antibiotischen Behandlung mit der Applikation einer wirksamen Lebendvakzine stand vor allem die beschriebene Restvirulenz in sensiblen Tierarten wie Ziegen und Lamas im Fokus. Dabei beruhen die meisten Beschreibungen zur Restvirulenz der SSLV auf Erfahrungswerten und sind, mit wenigen Ausnahmen, nicht publiziert. In den hier durchgeführten Versuchsgruppen mit der SSLV traten 170 5. Konklusion und Ausblick keine adversen Effekte auf. Allerdings kam es durch verschiedene Nebeninfektionen zu Tierverlusten, was unter Umständen auch der Immunisierung mit der SSLV hätte zugeschrieben werden können. Daher wäre es prinzipiell möglich, dass die beschriebene Restvirulenz der SSLV eher durch latente Nebeninfektionen oder einen generell schlechten Gesundheitszustand der Tiere bedingt wird, als durch die Immunisierung selbst. Generell blieb bei allen Immunisierungen mit der SSLV und auch bei den Tieren, die eine Infektion mit voll virulenten Sporen überlebten, analog zu den NLV, eine Reaktion gegen rBclA aus. Damit konnten neben inaktivierten Sporen und rekombinantem BclA auch mittels intakter Sporen keine nennenswerten anti-rBclA Antikörpertiter erzeugt werden. Analog zu den Erfahrungen mit FIS konnte auch die SSLV deutliche Antikörpertiter gegen FIS erzeugen, wobei dies nach einer zweiten Immunisierung noch gesteigert wurde. Erstaunlicherweise hatte die Infektion mit einem voll virulenten Stamm keinen Effekt auf die anti-FIS Antikörpertiter. Dies könnte auf die in der Infektion verwendete, verhältnismäßig geringe Sporenzahl (~10 3) im Vergleich zur SSLV (107) zurückzuführen sein oder mit den zuvor beschriebenen Ursachen durch die Anwesenheit von BclA begründet werden. Für einen Teil der insgesamt 15 Tiere, die mit der SSLV immunisiert wurden, zeigte die Erstimmunisierung nur einen geringen bzw. keinen nachweisbaren Effekt, was sich teilweise auch in einer verminderten Schutzrate widerspiegelte. Auch innerhalb anderer Studien und bei der Immunisierung von anderen Spezies wurde eine individuell verminderte Wirksamkeit der SSLV ohne Angabe von Gründen bereits beschrieben [266][280][228]. Daher scheint zumindest für die betreffenden Spezies eine Veränderung des Impfplans, der erst nach einem Jahr eine weitere Immunisierung vorsieht, sinnvoll. Demnach sollte eine gedoppelte Erstimmunisierung gefährdeter Tiere in einem Abstand von 1 – 2 Wochen vorgenommen werden, um langsam reagierenden oder unreaktiven Tieren bis zur Revakzinierung ebenfalls einen Schutz zu vermitteln. Entsprechend der vorhandenen Richtlinien [357] könnte die erste der beiden Immunisierungen auch nur mit einer verminderten Dosis vorgenommen werden, um adversen Reaktionen in sensitiven Tierarten vorzubeugen. Auch die potentielle Schutzdauer der SSLV von 9 – 12 Monaten und die damit verbundene Notwendigkeit von jährlichen Auffrischungen wurde in dieser Studie näher betrachtet. Durch eine funktionierende Erstimmunisierung konnte in den meisten Tieren ein hoher anti-rPA83 IgG-Titer und TNA-Titer erzeugt werden, der sich im Verlauf der folgenden 3 – 4 Monate auf ein Niveau absenkte, das signifikant höher als das naiver Tiere lag. Dieses Niveau blieb auch bis zur Revakzinierung bzw. Infektion nach einem Jahr erhalten. Darüber hinaus waren Tiere ohne Revakzinierung auch nach einem Jahr noch signifikant geschützt. Die Annahme, dass die Schutzdauer nach dem Ablauf eines Jahres rapide abnimmt, konnte jedoch nicht gestützt werden. Die beobachteten Einzelfälle, bei denen ein Schutz nach einem Jahr nicht mehr gegeben 171 5. Konklusion und Ausblick war, könnten zudem gerade diejenigen Tiere betreffen, die wenig oder gar nicht auf die Erstimmunisierung reagiert haben. Damit wäre deren Schützbarkeit, wie in Gruppe 2 deutlich gezeigt wurde, bereits direkt nach der ersten Immunisierung fraglich und hätte nichts mit der Schutzdauer einer sonst immunogene Impfung mit der SSLV zu tun. Wie lange daher die Schutzwirkung der Erstimmunisierung anhält, wenn diese immunogen war, und inwiefern die Schutzdauer bei einer Revakzinierung denselben Verlauf zeigt, bleibt offen. Nachweislich hatten die nach einem Jahr revakzinierten Tiere ein anderes Antikörpertiterspektrum als nach der Erstimmunisierung. So waren zwar die anti-rPA83 IgG-Titer im gleichen Maße erhöht wie bei der Erstimmunisierung, zeigten aber deutlich höhere neutralisierende Eigenschaften. Zudem waren nach der Revakzinierung weitaus höhere anti-FIS und -vegetativ IgG-Titer messbar, die nach der Erstimmunisierung nicht oder kaum vorhanden waren. Aufgrund des deutlichen Unterschieds beider Vakzinierungen könnte potentiell eine stärkere oder/und länger andauernde Schutzwirkung nach der Revakzinierung vorliegen. Sollte dies in weiteren Studien bestätigt werden, wäre an dieser Stelle ebenfalls über eine Veränderung des bisherigen Impfplans nachzudenken, da die zusätzliche Wirkung der Revakzinierung in gefährdeten Tieren möglichst früh anzustreben wäre. Entsprechend der hier gezeigten Wirkungslosigkeit einer Impfung mit lebenden Sporen bei noch erhöhten Antikörpertitern, wie bei der Infektion der frisch revakzinierten Gruppe 3b gesehen, sollte eine Revakzinierung frühstens 3 – 4 Monate nach der Erstimmunisierung erfolgen. Ab diesem Zeitpunkt waren die Antikörpertiter der immunisierten Tiere auf einem leicht erhöhten, unveränderlichem Niveau. Auf Grundlage der aufgeführten Ergebnisse wäre zusammenfassend in weiterführenden Versuchen ein verkürzter Impfplan für die SSLV mit einer gedoppelten Erstimmunisierung und einer Revakzinierung nach 3 – 4Monaten in Ziegen oder andern sensitiven Spezies auszutesten. Zudem sollte hierbei auch die absolute bzw. maximale Dauer der Schutzwirkung nach der Revakzinierung festgestellt werden, da Grund zur Annahme besteht, dass weitere Immunisierungen obsolet sein könnten wenn Erstimmunisierung und Revakzinierung funktional waren. Bei einer Bestätigung dieser Annahme würde eine entsprechende Anpassung des Impfplans die Handhabung und Anwendbarkeit der SSLV verbessern und potentielle Verluste durch nachlassende oder unvollständige Immunität von Einzeltieren vermindern. Zudem sollte auch im Hinblick auf die beschriebene Unwirksamkeit von BclA die Anwendung einer BclA-defizienten SSLV erprobt werden, da die beschriebenen Vakzinierungsausfälle oder/und die Restvirulenz potentiell auch auf die Anwesenheit von BclA auf Sporen zurückzuführen sein könnten. Abschließend könnte die in dieser Arbeit erfolgreich erprobte Protein-basierende NLV oder darauf aufbauende Nachfolger als alternative Immunisierungen innerhalb des Impfplans der SSLV erprobt werden. Unter Bedingungen, die den Einsatz der SSLV nicht zulassen, beispielsweise bei einer bestehenden antibiotischen Behandlung oder bei Bedenken zum Einsatz der SSLV in 172 5. Konklusion und Ausblick sensitiven Tierarten, könnte die Erstimmunisierung mit der Protein-basierenden NLV vorgenommen werden. Bei der Anwendung einer gedoppelten Erstimmunisierung wäre sogar generell in sensitiven Tierarten oder Tieren mit ungewissem Gesundheitszustand eine vorbereitende Impfung mit der Protein-basierenden NLV denkbar, die dann nach 1 – 2 Wochen mit der Injektion der SSLV komplettiert wird. Weiterführende Revakzinierungen könnten je nach Bedarf mit einer der beiden Vakzinen fortgeführt werden. Bei einem entsprechenden Nachweis der Funktionalität einer solchen kooperativen Anwendung könnten die positiven Aspekte beider Immunisierungen genutzt werden, ohne in Konkurrenz zueinander zu stehen. 173 6 Anhang 6.1 Sequenzen BclAD1D3 gcatttgaccctaatcttgtaggacctacattaccaccgataccaccatttacccttcctaccggaccatccggactaggacttccagcagg actatatgcatttaactccggtgggatttctttagatttaggaattaatgatccagtaccatttaatactgttggatctcagtttggtacagcaattt ctcaattagatgctgatactttcgtaattagtgaaactggattctataaaattactgttatcgctaatactgcaacagcaagtgtattaggaggtc ttacaatccaagtgaatggagtacctgtaccaggtactggatcaagtttgatttcactcggagcacctatcgttattcaagcaattacgcaaat tacgacaactccatcattagttgaagtaattgttacagggcttggactatcactagctcttggcacgagtgcatccattattattgaaaaagttg cttaa PA83 gaagtaaagcaagagaaccgtctgctgaacgaatctgaatccagctctcagggcctgcttggttactatttctctgacctgaacttccaagca ccgatggttgtaaccagctctaccactggcgatctgtccatcccgtctagtgaacttgagaacattccaagcgagaaccagtatttccagtct gcaatctggtccggttttatcaaagtcaagaaatctgatgaatacacgtttgccacctctgctgataaccacgtaaccatgtgggttgacgatc aggaagtgatcaacaaagcatccaactccaacaaaattcgtctggaaaaaggccgtctgtatcagatcaagattcagtaccaacgcgagaa cccgactgaaaaaggcctggactttaaactgtattggactgattctcagaacaagaaagaagtgatcagctctgacaatctgcaactgccgg aattgaaacagaaaagctccaactctcgtaagaaacgttccaccagcgctggcccgaccgtaccagatcgcgacaacgatggtattccgg actctctggaagttgaaggctacacggttgatgtaaagaacaaacgtaccttccttagtccgtggatctccaatattcacgagaagaaaggtc tgaccaaatacaaatccagtccggaaaaatggtccactgcatctgatccgtactctgactttgagaaagtgaccggtcgtatcgacaagaac gtctctccggaagcacgccatccactggttgctgcgtatccgatcgtacatgttgacatggaaaacatcattttgtccaagaacgaagacca gtccactcagaacactgactctgaaactcgtaccatctccaagaacacctccacgtctcgtactcacaccagtgaagtacatggtaacgctg aagtacacgcctctttctttgacatcggcggctctgttagcgctggcttctccaactctaattcttctactgttgccattgatcactctctgagtct ggctggcgaacgtacctgggcagagaccatgggtcttaacactgctgataccgcgcgtctgaatgctaacattcgctacgtcaacactggt acggcaccgatctacaacgtactgccaaccaccagcctggttctgggtaagaaccagactcttgcgaccatcaaagccaaagagaacca actgtctcagattctggcaccgaataactactatccttccaagaacctggctccgatcgcactgaacgcacaggatgacttctcttccactcc gatcaccatgaactacaaccagttcctggaacttgagaagaccaaacagctgcgtcttgacactgaccaagtgtacggtaacatcgcgacc tacaactttgagaacggtcgcgtccgcgttgacacaggctctaattggtctgaagtactgcctcagattcaggaaaccaccgctcgtatcatc ttcaacggtaaagacctgaacctggttgaacgtcgtattgctgctgtgaacccgtctgatccattagagaccaccaaaccggatatgactctg aaagaagccctgaagatcgcctttggcttcaacgagccgaacggtaatcttcagtaccaaggtaaagacatcactgaatttgacttcaacttt gatcagcagacctctcagaatatcaagaaccaactggctgagctgaacgcgaccaatatctatacggtactcgacaagatcaaactgaacg cgaaaatgaacattctgattcgcgacaaacgtttccactacgatcgtaataacatcgctgttggcgctgatcaatctgttgtgaaagaagcgc atcgcgaagtcatcaactccagcaccgaaggcctgcttctgaacatcgacaaagacattcgtaagatcctgtctggttacattgttgagatcg aagacaccgaaggcctgaaagaagtgatcaatgatcgttacgacatgctgaacatcagctctctgcgtcaagatggtaagacgttcattga cttcaagaaatacaacgacaaacttccgctgtatatctctaatccgaactacaaagtgaacgtttacgctgttaccaaagagaacaccatcat caatccatctgagaacggcgatacctctaccaacggtatcaagaagattctgatcttctccaagaaaggttacgagatcggttaa I 6.2 Versuchsdurchführung Abbildung 58: GeneGun Patronenverschuss Probeverschuss der für die GeneGun Immunisierung verwendeten Patronen der Mausgruppen auf Filterpapier II Tabelle 53: Probennahmepunkte Ziegenversuch (Südafrika) Gruppe Immunisierunga 1 2 3 4 Transport b Probennahmen (Blut) a naive 1 2 3 4 5 6 7 8 9 11 12 13 14 15 Infektiona 1 0,0 - - - 2,0 0,0 3,6 7,6 11,6 15,6 19,6 23,6 27,6 31,6 36,7 47,6 52,6 56,9 61,6 - 2 0,0 - - - 2,0 0,0 6,0 8,0c - - - - - - - - - - - - 61,7 6,1 3a 0,0 - - - 2,0 0,0 3,6 7,6 11,6 15,6 19,6 23,6 27,6 31,6 36,7 47,6 52,6 56,9 61,6 63,6c 61,7 3b 0,0 58,4 - - 2,0 0,0 3,6 7,6 11,6 15,6 19,6 23,6 27,6 31,6 36,7 47,6 52,6 56,9 61,6 63,6c 61,7 4 0,0 3,0 6,0 - 1,3 0,0 2,9 4,9 8,1 10,3 - - - - - - - - - - 10,4 5 0,0 3,0 6,0 14,7 - 0,0 3,0 6,0 9,0 14,7 16,7 - - - - - - - - - - 6 0,0 3,0 6,0 - 1,3 0,0 2,9 4,9 8,1 10,3 11,6c - - - - - - - - - 10,4 LK 0,0 - - - 2,0 11,3 - - - - - - - - - - - - - - 11,3 NK - - - - 0,7 9,9 - - - - - - - - - - - - - - 9,9 a in Wochen nach erster Immunisierung (für die Negativgruppe gilt die Immunisierung von Gruppe 3a/b) b in Wochen vor Infektion c Blutprobennahme nach Überleben der Infektion Immunisierungszeitpunkte, Blutungen, Transport zum Infektionsort und Infektionszeitpunkt aller Ziegengruppen (Südafrika); Probennahme 10 war geplant konnte aber nicht durchgeführt werden Tabelle 54: Probennahmepunkte Ziegenversuch (Türkei - Wiederholungen) Gruppe Immunisierunga 1 2 Probennahmen (Blut) a 3 naive 1 2 3 4c Infektiona 4 (W) 0,0 3,3 6,0 0,0 3,3 6,0 11,0 13,1 11,0 6 (W) 0,0 3,3 6,0 0,0 3,3 6,0 11,0 13,1 11,0 - - - 0,0 3,3 6,0 10,4 (11,0) b - 10,4 (11,0) b NK (W) a in Wochen nach erster Immunisierung (für die Negativgruppe gilt die Erstimmunisierung von Gruppe 4 und 6) b Zeitpunkt leicht unterschiedlich, da in 2 Etappen infiziert wurde c Blutprobennahme nach Überleben der Infektion Immunisierungszeitpunkte, Blutungen und Infektionszeitpunkt aller Ziegengruppen (Türkei) Tabelle 55: exemplarische Temperaturerhebung in naiven Ziegen (Südafrika) Ziegen Nr. 8185 W ochen nach Erstimmunisierung 3 5 7 9 11 13 15 17 19 21 23 25 27 29 39,1 39,5 39,2 39,2 39,5 39,6 39,5 39,5 39,2 39,2 38,4 39,1 39,3 38,8 8186 39,5 39,5 39,3 39,4 39,4 39,5 39,3 38,7 38,7 38,7 38,1 38,5 39,3 38,7 8187 39,3 38,8 38,9 39,2 39,2 39,3 39,1 39,2 39,1 39,1 37,4 38,3 39,4 38,6 rektale Temperaturen der Gruppe 1, in Wochen nach der Erstimmunisierung mit PBS Tabelle 56: Ziegenversuche (Südafrika), diagnostische Daten hpi NK LK G1 G2 G3a G3b G4 G6 0 3 II 38,6 38,9 VI 38,0 38,8 7 8202 37,7 8203 37,5 9 17 38,5 38,0 19 38,7 21 37,5 37,6 38,4 24 30 33 34 36 38 40 37,7 38,0 39,3 39,5 39,4 36,5 38,2 38,7 38,2 38,0 42 43 45 46 48 50 51 52 53 54 55 57 37,8 37,7 38,6 38,6 37,2 38,1 37,7 39,1 38,6 38,4 37,2 37,2 37,3 39,2 40,5 58 63 64 65 36,3 T 37,3 66 67 68 69 71 72 40,0 8185 39,0 40,1 40,6 8187 38,9 38,8 38,8 8210 38,5 38,0 38,3 8211 38,7 38,2 38,4 8212 38,8 40,4 37,9 8213 39,0 37,6 8214 39,3 39,4 38,2 8172 38,7 38,4 38,5 8173 38,5 40,1 39,8 8176 38,6 37,8 38,2 8178 38,8 38,0 40,5 8180 38,5 38,7 38,6 8175 38,5 38,0 38,1 8181 39,0 38,1 38,1 8182 39,0 38,1 38,6 8188 39,2 38,0 41,6 39,4 8190 39,5 38,1 8192 39,0 38,1 41,6 41,4 8198 39,4 38,1 A 40,4 8199 40,8 37,6 A 40,6 8191 38,8 38,1 8194 39,4 38,0 A 41,5 8195 39,3 37,6 8200 39,3 37,2 8201 40,4 37,8 40,4 39,7 37,7 40,1 39,3 41,5 40,8 41,1 40,1 37,6 37,7 A Temperatur- und Verhaltensdaten aller Versuchstiere während der Infektion; Angegebene Zeitintervalle der Messpunkte sind in Stunden nach Infektion (hpi) aufgeführt; Verstorbene Individuen sind mit orange unterlegt, Überlebende entsprechend mit grün; Gelb unterlegt sind Zeitpunkte für positive Blutausstriche vor dem Eintreten des Todes (grau hinterlegt); Rot und dick hervorgehoben sind Zeitpunkte an denen erstmals eine Veränderung des Verhaltens (gewöhnlich Verlust des Appetits) festgestellt wurden; NK – Negativkontrolle; LK – Lipopeptidkontrolle; G – Gruppe; T – teilnahmslos; A – Appetitverlust Tabelle 56: Ziegenversuche (Südafrika), diagnostische Daten Part 2 (76 – 336 hpi) hpi NK LK G1 G2 76 77 83 84 93 96 98 103 111 120 129 132 144 150 153 168 183 192 216 240 37,6 37,9 241 264 288 312 336 38,4 38,4 38,6 38,4 38,7 II VI 8202 8203 8185 8187 8210 38,7 38,0 38,6 38,3 8211 41,2 39,6 41,4 41,4 8212 38,6 38,3 38,8 38,6 37,7 37,4 38,0 38,8 38,8 38,0 38,0 8214 38,6 38,0 38,5 38,5 37,4 37,4 38,6 38,2 38,3 38,0 38,0 8172 38,4 38,0 38,5 38,4 38,2 37,7 38,3 37,7 38,9 38,2 37,2 8173 38,6 37,8 40,0 38,4 37,5 37,3 37,7 37,7 37,8 36,2 36,9 8176 38,1 38,8 38,5 38,2 37,8 38,2 37,9 38,5 37,6 37,5 37,4 8178 39,2 37,7 38,1 38,4 38,0 37,8 37,5 38,5 38,6 38,0 38,0 8180 39,0 39,3 40,6 41,5 40,4 40,2 40,4 8175 38,4 37,9 39,0 38,1 37,3 37,3 37,9 38,0 38,6 37,4 37,5 8181 38,3 38,7 38,2 38,2 38,5 37,2 38,0 39,1 38,4 38,4 37,2 8182 38,0 38,3 38,2 38,3 37,5 37,7 37,8 38,5 37,9 37,3 38,0 8213 G3a G3b 8188 8190 G4 8192 8198 8199 8191 40,8 8194 G6 8195 8200 40,4 38,2 36,6 39,0 38,2 38,7 38,0 8201 Temperatur- und Verhaltensdaten aller Versuchstiere während der Infektion; Angegebene Zeitintervalle der Messpunkte sind in Stunden nach Infektion (hpi) aufgeführt; Verstorbene Individuen sind mit orange unterlegt, Überlebende entsprechend mit grün; Gelb unterlegt sind Zeitpunkte für positive Blutausstriche vor dem Eintreten des Todes (grau hinterlegt); Rot und dick hervorgehoben sind Zeitpunkte an denen erstmals eine Veränderung des Verhaltens (gewöhnlich Verlust des Appetits) festgestellt wurden; NK – Negativkontrolle; LK – Lipopeptidkontrolle; G – Gruppe; T – teilnahmslos; A – Appetitverlust Tabelle 57: Ziegenversuche (Türkei), diagnostische Daten hpi NK (W) G4 (W) G6 (W) 0 16 96163 39,5 39,6 19 114 122 138 162 170 186 189 194 210 218 234 258 282 306 330 354 97014 38,8 38,9 38,9 97015 39,5 39,5 39,4 kk 40,0 97013 38,9 38,7 39,2 39,1 99348 38,9 38,4 39,5 39,5 41,1 41,4 A 39,7 38,2 39,0 580315 38,7 38,1 38,8 39,0 39,2 38,9 38,1 41,2 41,5 A 41,3 B 41,5 41,3 39,9 39,5 38,6 38,5 38,9 38,6 38,2 38,7 38,3 38,5 39,0 40,0 38,3 38,6 38,5 38,3 580319 39,1 38,6 41,3 40,9 A 38,3 627398 38,9 38,1 39,2 41,2 A 627399 39,6 39,4 39,3 39,1 38,8 38,2 40,2 39,3 39,0 36,1 38,3 38,9 38,6 38,5 38,5 38,7 38,4 38,5 39,5 39,0 38,7 38,9 38,8 38,4 39,0 38,4 39,1 39,3 39,0 39,8 728761 38,1 38,0 38,3 38,9 38,2 38,6 39,3 B 37,9 38,4 41,0 41,0 39,8 38,4 38,3 38,3 38,4 37,9 38,4 38,0 Gk 39,0 38,1 38,0 38,8 38,2 B 38,7 38,5 38,2 37,9 37,8 37,8 38,2 38,3 38,4 37,9 38,2 38,4 38,5 96139 39,4 38,4 39,0 38,8 B 41,3 40,5 39,9 38,7 39,4 38,4 38,0 38,4 38,6 38,6 37,6 38,1 38,7 38,2 96157 39,1 39,1 39,3 B 39,0 39,1 39,0 38,4 38,7 38,7 38,7 38,4 38,6 38,3 39,1 38,7 38,3 38,8 38,6 97162 39,7 39,3 B 41,1 41,5 41,6 41,2 40,7 39,4 39,4 39,2 39,2 39,2 39,0 39,3 39,0 38,9 39,0 38,8 580316 39,1 B 38,9 38,7 38,8 37,9 38,7 38,0 38,0 38,0 38,3 38,2 38,9 38,8 38,9 39,1 38,4 38,7 38,7 627395 B 38,6 39,0 40,9 41,5 41,6 41,5 41,3 41,5 627396 40,1 40,1 41,1 41,0 728758 38,9 39,1 39,5 39,3 38,7 38,9 38,6 38,7 38,6 38,6 38,4 39,0 39,0 38,8 38,8 38,7 38,5 39,1 B 729278 38,6 38,7 39,1 39,0 38,3 39,1 39,0 39,0 38,3 38,4 38,4 38,7 38,5 38,9 38,6 38,6 38,8 39,1 B 729277 39,0 38,4 39,1 39,3 38,5 39,1 39,1 38,9 38,6 38,3 39,2 40,4 40,8 40,0 40,0 40,6 40,3 39,8 B 729284 39,7 39,2 39,1 39,5 39,0 39,1 39,4 38,7 41,1 40,8 40,1 39,8 39,1 38,7 38,2 38,4 38,5 38,3 B 39,1 25 40 39,5 39,1 38,0 43 39,0 49 51 54 40,5 39,7 40,5 55 60 64 40,2 40,7 66 67 69 70 75 88 38,9 38,9 38,9 41,2 39,5 39,0 38,6 140 359 39,3 39,4 39,1 39,7 38,9 39,8 41,1 41,0 B B Temperatur- und Verhaltensdaten aller Versuchstiere während der Infektion; Angegebene Zeitintervalle der Messpunkte sind in Stunden nach Infektion (hpi) aufgeführt; Verstorbene Individuen sind mit orange unterlegt oder rot, wenn diese behandelt wurden und überlebten, Überlebende entsprechend mit grün; Gelb unterlegt sind Zeitpunkte für positive Blutausstriche vor dem Eintreten des Todes (grau hinterlegt); Rot und dick hervorgehoben sind Zeitpunkte an denen erstmals eine Veränderung des Verhaltens (gewöhnlich Verlust des Appetits) festgestellt wurden; N – Negativkontrolle; G – Gruppe; A – Appetitverlust; B (blau) – Behandlung Tabelle 58: Zusammenhänge von Antikörpertitern und Schutz (Mäuse) Infektionsdosis: ~1000 Sporen Gruppe TA Maus anti-rPA83 Nr. IgG-Titera 12 0 Infektionsdosis: ~2000 Sporen Gruppe LBP Maus anti-rPA83 Nr. IgG-Titera 23 143515 TA 13 0 LBCOP 54 172408 TA 14 0 LBCP 27 180363 TA 15 0 LBCP 29 180363 TA 20 0 LBP 24 213333 TA 11 0 LBCOP 56 214204 NS 8 800 LBCP 30 226909 TH 27 800 LBCOP 48 256000 TA 16 1600 LBCP 32 308363 TA 18 2096 LBP 18 310303 NS 5 2610 LBP 22 310303 TH 30 4342 LBCP 31 349090 TA 19 4855 LBP 19 364606 NS 9 4884 LBCOP 49 381387 NS 2 6400 LBCOP 51 381387 NS 10 10778 LBCP 26 407272 TA 17 12800 LBP 25 411151 TH 23 12800 LBCOP 53 412734 TKS 274 15872 LP 41 414254 NS 4 16168 LP 38 432872 TH 24 19657 LBCOP 50 433632 TH 25 21028 LBCP 33 442181 NS 7 21557 LP 37 442181 NS 1 21557 LBCOP 47 454530 TKS 277 27136 LBCP 28 471272 TH 28 28342 LBP 20 473212 TKS 276 29696 LBP 16 496484 TKS 287 32768 LP 43 512000 TH 22 37485 LP 44 512000 NS 6 45810 LBCOP 52 512000 TH 21 51200 LBP 17 589575 TH 26 56685 LP 40 623709 TKS 292 58122 LP 42 623709 TKS 286 76800 LP 39 660945 TKS 275 81920 LP 36 660945 TKS 288 81920 LBCP 34 663272 TKS 281 82286 LP 45 735418 TH 29 84114 LBCP 35 768000 NS 3 94315 LBP 21 837818 46 1191183 TKS 289 113633 LBCOP NCP 59 122181 a NCP 60 197818 NCP 65 256000 NCP 58 256000 NCP 62 256000 NCP 57 350000 NCP 64 350000 NCP 61 453818 NCP 63 512000 NCP 66 512000 direkt vor der Infektion Auftragung aller 90 infizierten Tiere mit Einzelmessungen für die anti-rPA83 IgG Titer, unterteilt nach Infektionsdosis in aufsteigender Reihenfolge ihrer anti-rPA83 IgG-Titer VII Tabelle 59: Test auf pTBC Ziegen Nr. Gruppe Probenzeitpunkt II NK vor Infektion + VI NK vor Infektion + 8202 LK vor Infektion + 8203 LK vor Infektion - 8185 1 Negativserum - 8186a 1 Negativserum + 8187 1 Negativserum + 8172 3a Negativserum + 8173 3a Negativserum + 8174a 3b Negativserum + 8175 3b Negativserum + 8176 3a Negativserum + 8178 3a Negativserum ? 8179a 3b Negativserum ? 8180 3a Negativserum + 8181 3b Negativserum + 8182 3b Negativserum + Negativserum + vor Infektion + Negativserum - vor Infektion - Negativserum - vor Infektion - Negativserum - vor Infektion - 8188 8190 8192 4 4 4 8198 4 8199 4 8191 6 8194 6 8195 6 8200 6 8201 6 a pTBC-Test Negativserum - vor Infektion + Negativserum - vor Infektion - Negativserum ? vor Infektion + Negativserum + vor Infektion + Negativserum - vor Infektion ? Negativserum - vor Infektion - waren mit der Herzwasserkrankheit infiziert exemplarischer Test der Seren Vor der Impfung und/oder vor der Infektion auf pTBC; +/- geben an ob der Test positiv ausgefallen ist, bei ? war der Test inkonklusiv; alle nicht aufgeführten Seren wurden nicht getestet VIII Literaturverzeichnis [1] Abboud N, Casadevall A: Immunogenicity of Bacillus anthracis Protective Antigen Domains and Efficacy of Elicited Antibody Responses Depend on Host Genetic Background. Clin Vaccine Immunol 2008; 15:1115-23 [2] Akira S, Sato S: Toll-like Receptors and Their Signaling Mechanisms. Scand J Infect Dis 2003; 35:555-62 [3] Allison AC, Byars NE: An adjuvant formulation that selectively elicits the formation of antibodies of protective isotypes and of cell mediated immunity. J Immunol Methods 1986; 95:157-68 [4] Anderson AO, Reynolds JA: Adjuvant effects of the lipid amine CP-20,961. J Reticuloendothel Soc 1979; 26:667-80 [5] Aronson AI, Fitz-James P: Structure and morphogenesis of the bacterial spore coat. Bacteriol Rev 1976; 40:360-402 [6] Arora N, Leppla SH: Residues 1-254 of Anthrax Toxin Lethal Factor Are Sufficient to Cause Cellular Uptake of Fused Polypeptides. J Bacteriol Chem 1993; 268:3334-41 [7] Bail O: Untersuchungen über natürliche und künstliche Milzbrandimmunität. X Die künstliche Immunität des Kaninchens. Centralbl Bakt Jena Abt I 1904; 36:366-72 [8] Baillie L: The development of new vaccines against Bacillus anthracis. Appl Microbiol 2001; 91[4]:609-13 [9] Baillie L, Hebdon R, Flick-Smith H, Williamson D: Characterisation of the immune response to the UK human anthrax vaccine. FEMS Immunol Med Mic 2003; 36:83-6 [10] Baillie L, Read TD: Bacillus anthracis, a bug with attitude!. Curr Opin Microbiol 2001; 4[1]:78-81 [11] Baillie LW, Huwar TB, Moore S, Mellado-Sanchez G, Rodriguez L, Neeson BN et al: An anthrax subunit vaccine candidate based on protective regions of Bacillus anthracis protective antigen and lethal factor. Vaccine 2010; 28:6740-8 [12] Ballachanda ND, Singer SS: CIITA and its dual roles in MHC gene transcription. Front Immun 2013; 4:1-6 [13] Ballas ZK, Rasmussen WL, Krieg AM: Induction of NK activity in murine and human cells by CpG motifs in oligodeoxynucleotides and bacterial DNA. J Immunol 1996; 157:1840-5 [14] Barnes JM: The development of Anthrax following the administration of spores by inhalation. Br J Exp Pathol 1947; 28:385-94 [15] Bast E: Mikrobiologische Methoden 3. Edition. Spektrum Akademischer Verlag 2014. [16] Basu S, Kang TJ, Chen WH, Fenton MJ, Baillie L, Hibbs S, Cross AS: Role of Bacillus anthracis Spore Structures in Macrophage Cytokine Responses. Infect Immun 2007; 75:2351-8 [17] Beall FA, Taylor MJ, Thorne CB: Rapid lethal effect in rats of a third component found upon fractionating the toxin of Bacillus anthracis. J Bacteriol 1962; 83:1274-80 [18] Beaman TC, Pankratz HS, Gerhardt P: Ultrastructure of the exosporium and underlying inclusions in spores of Bacillus megaterium strains. J Bacteriol 1972; 109:1198-209 [19] Beaumont G: Anthrax in a Scottish intravenous drug user. J Forensic Leg Med 2010; 17:443-5 [20] Beedham RJ, Turnbull PCB, Williamson ED: Passive transfer of protection against Bacillus anthracis infection in a murine model. Vaccine 2001; 19:4409-16 [21] Bell JH: On anthrax and anthracaemia in wool sorters, heifers and sheep. Br Med J 1880; 2:656 [22] Belton FC, Strange RE: Studies on a protective antigen produced in vitro from Bacillus anthracis: medium and methods of production. Br J Exp Pathol 1954; 35:144-152 [23] Bessler WG, Heinevetter L, Wiesmüller KH, Jung G, Baier W, Huber M, Lorenz AR, Esche UV, Mittenbühler K, Hoffmann P: Bacterial cell wall components as IX immunomodulators--I. Lipopeptides as adjuvants for parenteral and oral immunization. Int J Immunopharmacol 1997; 19:547-50 [24] Beyer W: Entwicklung und präklinische Testung rekombinanter Vakzinekandidaten gegen die Infektion mit Bacillus anthracis. Habilitationsschrift Universität Hohenheim 2005 [25] Beyer W, Glöckner P, Otto J, Böhm R.: A nested PCR method for the detection of Bacillus anthracis in environmental samples collected from former tannery sites. Microbiol Res 1995; 150:179-86 [26] Bird AP: CpG islands as gene markers in the vertebrate nucleus. Trends Genet 1987; 3:342-7 [27] Blancou J: History of the surveillance and control of transmissible animal diseases. Office International des Èpizooties, Paris 2003. [28] Böhm R: Resistance, survival, sterilization and desinfection of spores of Bacillus anthracis. Salisbury Ned Bull 1990; 68:99-101 [29] Boor AK: An antigen prepared in vitro effective for immunisation against anthrax. I. Preparation and evaluation of the crude protective antigen. J Infect Dis 1955; 97:194-202 [30] Boor AK, Tresselt HB: An antigen prepared in vitro effective for immunisation against anthrax. II. Concentration of the protective antigen by chemical fractionation. J Infect Dis 1955; 97:194-202 [31] Boyden ED, Dietrich WF: Nalp1b controls mouse macrophage susceptibility to anthrax lethal toxin. Nat Genet 2006; 38:240-244 [32] Boydston JA, Chen P, Steichen CT, Turnbough CL: Orientation within the exosporium and structural stability of the collagen-like glycoprotein BclA of Bacillus anthracis. J Bacteriol 2005; 187:5310-7 [33] Bozue J, Cote CK, Moody KL, Welos SL: Fully virulent Bacillus anthracis does not require the immunodominant protein BclA for pathogenesis. Infect Immun 2007; 75:508-11 [34] Bozue J, Moody KL, Cote CK, Stiles BG, Friedlander AM, Welkos SL, Hale ML: Bacillus anthracis Spores of the bclA Mutant Exhibit Increased Adherence to Epithelial Cells, Fibroblasts, and Endothelial Cells but Not to Macrophages. Infect Immun 2007; 75:4498-505 [35] Bozue JA, Parthasarathy N, Phillips LR, Cote CK, Fellows PF, Mendelson I, Shafferman A, Friedlander AM: Construction of a rhamnose mutation in Bacillus anthracis affects adherence to macrophages but not virulence in guinea pigs. Microb Pathogenesis 2005; 39:1-12 [36] Brachman PS, Gold H, Plotkin SA, Fekety FR, Werrin M, Ingraham NR: Field evaluation of a human anthrax vaccine. Am J Public Health 1962; 52:632-45 [37] Bradley KA, Mogridge J, Mourez M, Collier RJ, Young JAT: Identification of the cellular receptor for anthrax toxin. Nature 2001; 414:225-9 [38] Bragg TS, Robertson DL: Nucleotide sequence and analysis of the lethal factor gene (lef) from Bacillus anthacis . Gene 1989; 81:45-54 [39] Brahmbhatt TN, Darnell SC, Carvalho HM, Sanz P, Kang TJ, Bull RL, Rasmussen SB et al: Recombinant exosporium protein BclA of bacillus anthracis is effective as a booster for mice primed with suboptimal amounts of protective antigen. Infect Immun 2007; 75:5240-7 [40] Brenneman KE, Doganay M, Akmal A, Goldman S, Galloway DR, Mateczun AJ, Cross AS, Baillie LW: The early humoral immune response to Bacillus anthracis toxins in patients infected with cutaneous anthrax. FEMS Immunol Med Microbiol 2011; 62:164-72 [41] Brewer CR, McCullough WG et al: Studies on the nutritional requirements of Bacillus anthracis. Arch Biochem 1946; 10:65-75 [42] Brossier F, Levy M, Mock M: Anthrax Spores Make an Essential Contribution to Vaccine Efficacy. Infect Immun 2002; 70:661-4 [43] Brossier F, Weber-Levy M, Mock M, Sirard J-C: Role of Toxin Functional Domains in Anthrax Pathogenesis. Infect Immun 2000; 68:1781-6 X [44]: Bryden HB, de Vos V, Anthrax in the Kruger National Park, South Africa: The effect of different wild animal'sera on the in vitro germination of Bacillus anthracis spores. 1998 [45] Buchan S, Gronevik E, Mathiesen I, King CA, Stevenson FK, Rice J: Electroporation as a ‘prime/boost’ strategy for naked DNA vaccination against a tumor antigen. J Immunol 2005; 15:6292-8 [46] Buwitt-Beckmann U, Heine H, Wiesmüller KH, Jung G, Brock R, Ulmer AJ: Lipopeptide structure determines TLR2 dependent cell activation level. FEBS J 2005; 272:6354-64 [47] Cano RJ, Borucki MK: Revival and identification of bacterial spores in 25- to 40million-year-old Domican amber. Science 1995; 268:1060-4 [48] Cartwright ME, McChesney AE, Jones RL: Vaccination-related anthrax in three llamas. J Am Vet Med Assoc 1987; 191:715-6 [49] Chabot DJ, Joyce J, Caulfield M, Cook J, Hepler R, Wang S, Vietri NJ, Ruthel G, Shoop W, Pitt L, Leffel E, Ribot W, Friedlander AM: Efficacy of a capsule conjugate vaccine against inhalational anthrax in rabbits and monkeys. Vaccine 2012; 30:846-52 [50] Chabot DJ, Scorpio A, Tobery SA, Little SF, Norris SL, Friedlander AM: Anthrax capsule vaccine protects against experimental infection. Vaccine 2004; 23:43-7 [51] Chada VG, Sanstad EA, Wang R, Driks A: Morphogenesis of Bacillus spore surfaces. J Bacteriol 2003; 185:6255-61 [52] Chavarría-Smith J, Vance RE: Direct proteolytic cleavage of NLRP1B is necessary and sufficient for inflammasome activation by anthrax lethal factor. PLoS Pathog 2013; 9:e1003452 [53] Chen JW, Murphy TL, Willingham MC, Pastan I, August JT: Identification of two lysosomal membrane glycoproteins. J Cell Biol 1985; 101:85-95 [54] Chopra AP, Boone SA, Liang X, Duesberry NS: Anthrax Lethal Factor Proteolysis and Inactivation of MAPK Kinase. J Bacteriol Chem 2003; 278:9402-6 [55] Clarke CJ: The pathology and pathogenesis of paratuberculosis in ruminants and other species. J Comp Pathol 1997; 116:217-61 [56] Cleret A, Quesnel-Hellmann A: Lung dendritic cells rapidly mediate anthrax spore entry through the pulmonary route. J Immunol 2007; 178:7994-8001 [57] Cohen AD, Boyer JD, Weiner DB: Modulating the immune response to genetic immunization. FASEB J 1998; 12:1611-26 [58] Cohen S, Mendelson I, Altboum Z, Kobiler D, Elhanany E, Bino T, Leitner M, Inbar I, Rosenberg H, Gozes Y, Barak R, Fisher M, Kronman C, Velan B, Shafferman A: Attenuated Nontoxinogenic and Nonencapsulated Recombinant Bacillus anthracis Spore Vaccines Protect against Anthrax. Infect Immun 2000; 68:4549-58 [59] Coker PR, Smith KL, Fellows PF, Rybachuck G, Kousoulas KG, Hugh-Jones ME: Bacillus anthracis Virulence in Guinea Pigs Vaccinated with Anthrax Vaccine Adsorbed Is Linked to Plasmid Quantities and Clonality. J Clin Microbiol 2003; 411212-8 [60] Collier RJ: Mechanism of membrane translocation by anthrax toxin: insertion and pore formation by protective antigen. J Appl Microbiol 1999; 87:283 [61] Cote CK, Kaatz L, Reinhardt J, Bozue J, Tobery SA, Bassett AD et al: Characterization of a multi-component anthrax vaccine designed to target the initial stages of infection as well as toxaemia. J Med Microbiol 2012; 61:1380-92 [62] Cote CK, Rossi CA, Kang AS, Morrow PR, Lee JS, Welkos SL: The detection of protective antigen (PA) associated with spores of Bacillus anthracis and the effects of anti-PA antibodies on spore germination and macrophage interactions. Microb Pathogenesis 2005; 38:209-25 [63] Cromartie WJ, Watson DW, Bloom WL, Heckly RI: Studies on infection with Bacillus anthracis. I. The immunological and tissue damaging properties of extracts prepared from lesions of Bacillus anthracis infection. J Infect Dis 1947; 80:14-27 XI [64] Cybulski RJ Jr, Sanz P, McDaniel D, Darnell S, Bull RL, O’Brien AD: Recombinant Bacillus anthracis spore proteins enhance protection of mice primed with suboptimal amounts of protective antigen. Vaccine 2008; 26:4927-39 [65] Davies DG: The influence of temperature and humidity on spore formation and germination in Bacillus anthracis. J Hyg 1960; 58:177-186 [66] De Vos V, Scheepers GJ: Remote Mass Vaccination of Large Free-Ranging Wild animals for Anthrax using Sterne Spore Vaccine. Salisbury Med Bull 1996; 87:116-120 [67] De Vos V, Turnbull PCB: Anthrax. In: Coetzer JAW, Trustin RC: Infectious Diseases of Livestock, Vol. 3. Oxford University Press Southern Africa 2004, 1788-1818. [68] Defoort JP, Nardelli B, Huang W, Ho DD, Tam JP: Macromolecular assemblage in the design of a synthetic AIDS vaccine. Proc Natl Acad Sci USA 1992; 89:3879-83 [69] Delogu G, Howard A, Collins FM, Morris SL: DNA Vaccination against Tuberculosis: Expression of a Ubiquitin-Conjugated Tuberculosis Protein Enhances Antimycobacterial Immunity. Infect Immun 2000; 68:3097-102 [70] Deres K, Schild H, Wiesmüller K-H, Jung G, Rammensee H-G: In vivo priming of virus specific cyto-toxic T lymphocytes with synthetic lipopeptide vaccine. Nature 1989; 342:561-4 [71] Dixon TC, Meselson M, Guillemin J, Hanna PC: Anthrax. N Engl J Med 1999; 341:815–26 [72] Driks A: The Bacillus subtilis spore coat. Microbiol Mol Biol Rev 1999; 63:1-20 [73] Driks A: The Bacillus anthracis spore. Mol Aspects Med 2009; 30:368-73 [74] Driks A: Maximum shields: the armor plating of the bacterial spore. Trends Microbiol 2002; 10:251-5 [75] Ducle H, Hong HA, Atkins HS, Flick-Smith HC, Durrani Z, Riipkema S, Titball RW, Cutting SM: Immunization against anthrax using Bacillus subtilis spores expressing the anthrax protective antigen. Vaccine 2007; 25:346-55 [76] Duesbery NS, Webb CP, Leppla SH, Gordon VM, Klimpel KR, Copeland TD, Ahn NG, Oskarsson MK, Fukasawa K, Paull KD, Woude GFV: Proteolytic Inactivation of MAP-KinaseKinase by Anthrax Lethal Factor. Science 1998; 280:734-7 [77] Duguid JP: The demonstration of bacterial capsules and slime. J Pathol Bacteriol 1951; 63:673-685 [78] Dyer CM, Zhan Y, Brady JL, Carbone FR, Smyth MJ, Lew AM: Unexpectedly, induction of cytotoxic T-lymphocytes enhances the humoral response after DNA immunization. Blood 2004; 103:3073-5 [79] Enkhtuya J, Kawamoto K, Kobayashi Y, Uchida I, Neeraj R, Makino S: Significant passive protective effect against anthrax by antibody to Bacillus anthracis inactivated spores that lack two virulence plasmids. Microbiology 2006; 152:3103-10 [80] Escuyer V, Collier RJ: Anthrax Protective Antigen Interacts with a Specific Rezeptor on the Surface of CHO-K1 Cells. Infect Immun 1991; 59:3381-6 [81] Etienne-Toumelin I, Sirard J-C, Duflot E, Mock M, Fouet A: Characterization of the Bacillus anthracis S-Layer: Cloning and Sequencing of the Structural Gene. J Bacteriol 1995; 177:614-620 [82] Eyles JE, Butcher WA, Titball RW, Hill J: Concomitant administration of Yersinia pestis specific monoclonal antibodies with plague vaccine has a detrimental effect on vaccine mediated immunity. Vaccine 2007; 25:7301-6 [83] Ezzel JW, Abshire TG: Serum protease cleavage of Bacillus anthracis protective antigen. J Gen Microbiol 1992; 138:543-9 [84] Ezzel JW, Abshire TG: Immunological Analysis of Cell-Associated Antigens of Bacillus anthracis. Infect Immun 1988; 56:349-56 XII [85] Ezzel JW, Abshire TG, Panchal R, Chabot D, Bavari S, Leffel EK, Purcell B, Friedlander AM, Ribot WJ: Association of Bacillus anthracis capsule with lethal toxin during experimental infection. Infect Immun 2009; 77:749-55 [86] Ezzell JW: Anthrax - Pasteur to the Present. CMNEEJ 1988; 10:113-20 [87] Farchaus JW, Ribot WJ, Downs MB, Ezzell JW: Purification and characterization of the major surface array protein from the avirulent Bacillus anthracis Delta Sterne-1. J Bacteriol 1995; 177:2481-9 [88] Fasanella A, Galante D, Garofolo G, Jones MH: Anthrax undervalued zoonosis. Vet Microbiol 2010; 140:318-31 [89] Fellows PF, Linscott MK, Ivins BE, Pitt MLM, Rossi CA, Gibbs PH, Friedlander AM: Efficacy of a human anthrax vaccine in guinea pigs, rabbits, and rhesus macaques against challenge by Bacillus anthracis isolates of diverse geographical origin. Vaccine 2001; 19:32417 [90] Fellows PF, Linscott MK, Lettle SF, Gibbs P, Ivins BE: Anthrax vaccine efficacy in golden Syrian hamsters. Vaccine 2002; 20:1421-4 [91] Feltquate DM, Heaney S, Webster RG, Robinson HL: Different T helper cell types and antibody isotypes generated by saline and gene gun DNA immunization. J Immunol 1997; 158:2278-84 [92] Fernelius AL: Comparison of two heat-shock methods and a phenol treatment method for determining germination rates of spores of Bacillus anthracis. J Bacteriol 1960; 79:755-6 [93] Firoved AM, Miller GF, Moayeri M, Kakkar R, Shen Y, Wiggins JF, McNally EM, Tang WJ, Leppla SH: Bacillus anthracis Edema Toxin Causes Extensive Tissue Lesions and Rapid Lethality in Mice. Am J Pathol 2005; 167:1309-20 [94] Fisher N, Carr KA, Giebel JD, Hanna PC: Anthrax Spore Germination. In: Bergman NH: Bacillus Anthracis and Anthrax. Wiley-Blackwell 2011, 39-54. [95] Flick-Smith HC, Walker NJ, Gibson P, Bullifent H, Hayward S, Miller J et al: A Recombinant Carboxy-Terminal Domain of the Protective Antigen of Bacillus anthracis Protects Mice against Anthrax Infection. Infect Immun 2002; 70:1653-6 [96] Fouet A: The surface of Bacillus anthracis. Mol Aspects Med 2009; 30:374-85 [97] Fouet A, Mock M: Differential influence of the two Bacillus anthracis plasmids on regulation of virulence gene expression. Infect Immun 1996; 64:4928-32 [98] Friedlander AM: Macrophages Are Sensitive to Anthrax Lethal Toxin through an Aciddependent Process. J Bacteriol Chem 1986; 261:7123-6 [99] Friedlander AM, Bhatnagar R, Leppla SH, Johnson L, Singh Y: Characterization of Macrophage Sensitivity and Resistance to Anthrax Lethal Toxin. Infect Immun 1993; 61:245-52 [100] Friedlander AM, Welkos SL, Ivins BE: Anthrax Vaccines. In: Anthrax. Current topics in microbiology and immunology. Springer Verlag 2002, 33-60 [101] Fynan EF, Webster RG, Fuller DH, Haynes JR, Santoro JC, Robinson HL: DNA vaccines: protective immunizations by parenteral, mucosal and gene-gun inoculations. P Natl Acad Sci USA 1993; 90:11478-82 [102] Galloway D, Liner A, Legutki J, Mateczun A, Barnewall R, Estep J: Genetic immunization against anthrax. Vaccine 2004; 22:1604-8 [103] Galloway DR, Baillie L: DNA vaccines against anthrax. Expert Opin Biol Ther 2004; 4:1661-7 [104] Garrod LP: The sensitivity of Bacillus anthracis to antibiotics. Antibiotics Chemother 1952; 2:689-92 [105] Garufi G, Wang Y-T, Oh S-Y, Maier H, Missiakas DM, Schneewind O: Sortaseconjugation generates a capsule vaccine that protects guinea pigs against Bacillus anthracis. Vaccine 2012; 30:3435-44 XIII [106] Gauthier YP, Tournier J, Paucod J, Corre J, Mock M, Goossens PL, Vidal DR: Efficacy of a Vaccine Based on Protective Antigen and Killed Spores against Experimental Inhalational Anthrax. Infect Immun 2009; 77:1197-207 [107] Gerber LD, Kodukula K, Udenfriend S: Phosphatidylinositol glycan (PI-G) anchored membrane proteins. Amino acid requirements adjacent to the site of cleavage and PI-G attachment in the COOH-terminal signal peptide. J Biol Chem 1992; 267:12168-73 [108] Gerhardt P: Cytology of Bacillus anthracis. Fed Proc 1967; 26:1504-17 [109] Ghielmetti M, Reschner A, Zwicker M, Padovan E: Synthetic bacterial lipopeptide analogs: structural requirements for adjuvanticity. Immunbiol 2005; 210:211-5 [110] Gill DM: Bacterial Toxins and Cell Membranes. Academic Press, New York 1978. [111] Giorno R, Bozue J, Cote C, Wenzel T, Moody KS, Mallozzi M, Ryan M, Wang R, Zielke R, Maddock JR, Friedlander A, Welkos S, Driks A: Morphogenesis of the Bacillus anthracis Spore. J Bacteriol 2007; 189:691-705 [112] Gladstone GP: Immunity to anthrax. Protective antigen present in cell-free culture filtrates. Br J Exp Pathol 1946; 27:394 [113] Goldberg HA, Warner KJ: The staining of acidic proteins on polyacrylamide gels: enhanced sensitivity and stability of "Stains-all" staining in combination with silver nitrate. Anal Biochem 1997; 251:227-33 [114] Gómez-Miguel MJ, Moriyón I, Alonso-Urmeneta B, Riezu-Boj JI, Díaz R: Serological response to the outer membrane lipoprotein in animal brucellosis. Infect Immun 1988; 56:716-8 [115] Gordon VM, Leppla SH, Hewlett EL: Inhibitors of receptor-mediated endocytosis block the entry of Bacillus anthracis adenylate cyclase toxin but not that of Bordetella pertussis adenylate cyclase toxin. Infect Immun 1988; 56:1066-9 [116] Grabenstein JD: Countering Anthrax: Vaccines and Immunoglobulins. Clin Infect Dis 2008; 46:129-36 [117] Green BD, Battisti L, Koehler TM, Thorne CB, Ivins BE: Demonstration of a Capsule Plasmid in Bacillus anthracis. Infect Immun 1985; 49:291-297 [118] Gu M-L, Leppla SH, Klinman DM: Protection against anthrax toxin by vaccination with a DNA plasmid encoding anthrax protective antigen. Vaccine 1999; 17:340-4 [119] Guidi-Rontani C, Levy M, Ohayon H, Mock M: Fate of germinated Bacillus anthracis spores in primary murine macrophages.. Mol Microbiol 2001; 42:931-8 [120] Guidi-Rontani C, Weber-Levy M, Labruyère E, Mock M: Germination of Bacillus anthracis spores within alveolar macrophages. Mol Microbiol 1999; 31:9-17 [121] Guinebretière MH, Auger S, Galleron N, Contzen M, De Sarrau B, De Buyser ML et al: Bacillus cytotoxicus sp. nov. is a novel thermotolerant species of the Bacillus cereus Group occasionally associated with food poisoning. Int J Syst Evol Mivrobiol 2013; 63:31-40 [122] Gupta RK, Siber GR: Adjuvants for human vaccines--current status, problems and future prospects. Vaccine 1995; 13:1263-76 [123] Hahn UK, Aichler M, Boehm R, Beyer W: Comparison of the immunological memory after DNA vaccination and protein vaccination against anthrax in sheep. Vaccine 2006; 24:4595-7 [124] Hahn UK, Alex M, Czerny CP, Böhm R, Beyer W: Protection of mice against challenge with Bacillus anthracis STI spores after DNA vaccination. Int J Med Microbiol 2004; 294:3544 [125] Hahn UK, Boehm R, Beyer W: DNA vaccination against anthrax in mice—combination of anti-spore and anti-toxin components. Vaccine 2006; 24:4569-71 [126] Hambleton P, Carman JA, Melling J: Anthrax: the disease in relation to vaccines. Vaccine 1984; 2:125-32 [127] Hammamieh R, Ribot WJ, Abshire TG, Jett M, Ezzell J: Activity of the Bacillus anthracis 20 kDa protective antigen component. BMC Infect Dis 2008; 8:124 XIV [128] Hanby WE, Rydon HN: The capsular Substance of Bacillus anthracis. Biochem J 1946; 40[2]:297-309 [129] Hanna PC, Acosta D, Collier RJ: On the role of macrophages in anthrax. Proc Natl Acad Sci USA 1993; 90:10198-201 [130] Hanson JF, Taft SC, Weiss AA: Neutralizing Antibodies and Persistence of Immunity following Anthrax Vaccination. Clin Vaccine Immunol 2006; 13:208-13 [131] Hantke K, Braun V: Covalent binding of lipid to protein. Diglyceride and amide-linked fatty acid at the N-terminal end of the murein-lipoprotein of the Escherichia coli outer membrane. Eur J Biochem 1973; 34:284-96 [132] Heath WR, Carbonne FR: Cross-presentation in viral immunity and self-tolerance. Nat Rev Immunol 2001; 1:126-34 [133] Heine H, Lien E: Toll-like receptors and their function in innate and adaptive immunity. Int Arch Allergy Immunol 2003; 130:180-92 [134] Hellmich KA, Levinsohn JL, Fattah R, Newman ZL, Maier N, Sastalla I et al: Anthrax lethal factor cleaves mouse nlrp1b in both toxin-sensitive and toxin-resistant macrophages. PLoS One 2012; 7:e49741 [135] Hemmi H, Takeuchi O, Kawai T, Kaisho T, Sato S, Sanjo H, Matsumoto M, Hoshino K, Wagner H, Takeda K, Akira S: A Toll-like receptor recognizes bacterial DNA. Nature 2000; 408:740-5 [136] Henderson I, Duggleby CJ, Turnbull PCB: Differentiation of Bacillus anthracis from other B. cereus group bacteria with PCR. Int J Syst Bacteriol 1994; 44:99-105 [137] Henriques AO,Moran CP Jr: Structure, Assembly, and Function of the Spore Surface Layers. Annu Rev Microbiol 2007; 61:555-88 [138] Hermanson G, Whitlow V, Parker S, Tonsky K, Rusalov D, Ferrari M, Lalor P, Komai M, Mere R, Bell M, Brenneman K, Mateczun A, Evans T, Kaslow D, Galloway D, Hobart P: A cationic lipid-formulated plasmid DNA vaccine confers sustained antibody-mediated protection against aerosolized anthrax spores. PNAS 2004; 101:13601-6 [139] Hills GM: Chemical Factors in the Germination of Spore-bearing Aerobes. The Effects of Amino-acids on the Germination of Bacillus anthracis, with some Observations on the Relation of Optical Form to Biological Activity. Biochem J 1949; 45:363-370 [140] Hoffmaster AR, Ravel J, Rasko DA, Chapman GD, Chute MD, Marston CK, De BK, Sacchi CT, Fitzgerald C, Mayer LW, Maiden MC, Priest FG, Barker M, Jiang L, Cer RZ, Rilstone J, Peterson SN, Weyant RS, Galloway DR, Read TD, Popovic T, Fraser CM: Identification of anthrax toxin genes in a Bacillus cereus associated with an illness resembling inhalation anthrax. Proc Natl Acad Sci USA 2004; 101:8449-54 [141] Holt SC, Leadbetter ER: Comparative ultrastructure of selected aerobic spore-forming bacteria: a freeze-etching study. Bacteriol Rev 1969; 33:346-378 [142] Hugh-Jones ME, De Vos V: Anthrax and Wildlife. Rev Sci Tech Off Int Epiz 2002; 21:359-83 [143] Ivanovics G, Bruckner V: Chemische und immunologische Studien über den Mechanismus der Milzbrandinfecktion und Immunitat, die chemische Struktur der Kapselsubstanz des Milzbrandbazillus und der serolisch identischen spezifichen Substanz des Bacillus mesentericus. Z Immun Forsch 1937; 90:304-18 [144] Ivins BE, Ezzell JW Jr, Jemski J, Hedlund KW, Ristroph JD, Leppla SH: Immunization Studies with Attenuated Strains of Bacillus anthracis. Infect Immun 1986; 52:454-8 [145] Ivins BE, Fellows P, Pitt L, Estep J, Farchaus J, Friedlander A, Gibbs P: Experimental anthrax vaccines; efficacy of adjuvants combined with PA against an aerosol Bacillus anthracis spore challenge in guinea pigs. Vaccine 1995; 13:1779-84 [146] Ivins BE, Fellows PF, Nelson GO: Efficacy of a standard human anthrax vaccine against Bacillus anthracis spore challenge in guinea-pigs. Vaccine 1994; 12:872-4 XV [147] Ivins BE, Welkos SL: Recent Advances in the Development of an Improved, Human Anthrax Vaccine. Eur J Epidemiol 1988; 4:12-19 [148] Ivins BE, Welkos SL, Knudson GB, Little SF: Immunization against anthrax with aromatic compound-dependent (Aro-) mutants of Bacillus anthracis and with recombinant strains of Bacillus subtilis that produce anthrax protective antigen. Infect Immun 1990; 58:3038 [149] Ivins BE, Welkos SL, Little SF, Crumrine MH, Nelson GO: Immunization against Anthrax with Bacillus anthracis Protective Antigen Combined with Adjuvants. Infect Immun 1992; 60:662-8 [150] Jackson FC, Wright GG, Armstrong J: Immunization of cattle against experimental anthrax with alum-precipitated protective antigen or spore vaccine. Am J Vet Res 1957; 18:7717 [151] Janeway CA: Immunologie. Spektrum Heidelberg Berlin 2001. [152] Jang J, Cho M, Chun J-H, Cho M-H, Jungchan P, Oh H-B, Yoo C-K, Rhie G-E: The Poly-y-D-Glutamic Acid Capsule of Bacillus anthracis enhances lethal toxin activity. Infect Immun 2011; 79:3846-54 [153] Jansohn M, Rothämel S: Gentechnische Methoden 5. Edition. Spektrum Akademischer Verlag 2011. [154] Jelacic TM, Chabot DJ, Bozue JA, Tobery SA, West MW, Moody K et al: Exposure to Bacillus anthracis capsule in supression of human monocyte derived dendritic cells. Infect Immun 2014; 82:3405-16 [155] Jernigan DB et al: Investigation of Bioterrorism-related Anthrax, United States, 2001: Epidemiologic Findings. Emerg Infect Dis 2001; 8:1019-28 [156] Jernigan DB, Raghunathan PL, Bell BP, Brechner R, Bresnitz EA, Buttler JC et al: Investigation of bioterrorism-related anthrax, United States, 2001: epidemiologic findings. Emerg Infect Dis 2002; 10:1019-28 [157] Johnson AD, Spero L: Comparison of Growth and Toxin Production in Two Vaccine Strains of Bacillus anthracis. Appl Environ Microbiol 1981; 41:1479-81 [158] Joyce J, Cook J, Chabot D, Hepler R, Shoop W, Xu Qiuwei, Stambaugh T, AsteAmezaga M, Wang S, Indrawati L, Bruner M, Friedlander A, Keller P, Caulfield M: Immunogenicity and protective efficacy of Bacillus anthracis poly-gamma-D-glutamic acid capsule covalently coupled to a protein carrier using a novel triazine-based conjugation strategy. J Biol Chem 2006; 281:4831-43 [159] Kawai T, Akira S: The roles of TLRs, RLRs and NLRs in pathogen recognition. Int Immunol 2009; 21:317-37 [160] Kawai T, Takahashi K, Sato S, Coban C, Kumar H, Kato H: IPS-1, an adaptor triggering RIG-I- and MDA5-mediated type 1 interferon induction. Nat Immunol 2005; 6:981-8 [161] Kensil CR, Newman MJ, Coughlin RT, Soltysik S, Bedore D, Recchia J, Wu J, Marciani DJ: The use of Stimulon adjuvant to boost vaccine response. Vacc Res 1993; 2:273-81 [162] Kensil CR, Patel U, Lennick M, Marciani D: Separation and characterization of saponins with adjuvant activity from Quillaja saponaria molina cortex. J Immunol 1991; 146:431-7 [163] Keppie J, Harris-Smith W, Smith H: The chemical basis of the virulence of Bacillus anthracis. IX. Its Aggressins and their mode of action. Brit J Ex Pathol 1963; 44:446-53 [164] Kim D, Hoory T, Monie A, Ting JP-Y, Hung C-F, Wu T-C: Enhancement of DNA vaccine potency through co-administration of CIITA DNA with DNA vaccines via gene gun. J Immunol 2008; 180:7019-27 [165] Klee SR, Muhsin O, Appel B, Boesch C, Ellerbrook H: Characterization of Bacillus anthracis-like bacteria isolated from wild great apes from Cote d'Ivoire and Cameroon. J Bacteriol 2006; 188:5333-44 XVI [166] Klein F et al: Anthrax toxin: causative agent in the death of rhesus monkeys. Science 1962; 138:1331-3 [167] Klimpel KR, Arora N, Leppla SH: Anthrax toxin lethal factor contains a zinc metalloprotease consensus sequence which is required for lethal toxin activity. Mol Microbiol 1994; 13:1093-100 [168] Klimpel KR, Molloy SS, Thomas G, Leppla SH: Anthrax toxin protective antigen is activated by a cell surface protease with the sequence specificity and catalytic properties of furin. Proc Natl Acad Sci USA 1992; 89:10277-81 [169] Klinman DM, Yi AK, Beaucage SL, Conover J, Krieg AM: CpG motifs expressed by bacterial DNA rapidly induce lymphocytes to secrete IL-6, IL-12 and IFN. Proc Natl Acad Sci USA 1996; 93:2897-83 [170] Knudson GB: Treatment of anthrax in man: history and current concepts. Milit Med 1986; 151:71-7 [171] Koch R: Untersuchungen über Bakterien. V. Die Aetiologie der Milzbrand Krankheit, begradet auf Entwicklungsgeschichte des Bacillus anthracis. Beiträge zur Biologie der Pflanze 1876; 2:277-310 [172] Kozuka S, Tochikubo K: Properties and origin of filamentous appendages on spores of Bacillus cereus. Microbiol Immunol 1985; 29:21-37 [173] Krieg AM, Yi A-K, Matson S, Waldschmidt TJ, Bishop GA, Teasdale R, Koretzky GA, Klinman DM: CpG motifs in bacterial DNA trigger direct B-cell activation. Nature 1995; 374:546-9 [174] Kubler-Kielb J, Liu T-Y, Mocca C, Majadly F, Robbins JB, Schneerson R: Additional Conjugation Methods and Immunogenicity of Bacillus anthracis Poly-y-D-Glutamic Acid– Protein Conjugates. Infect Immun 2006; 74:4744-9 [175] Kumar A, Kanungo R, Bhattacharya S, Badrinath S, Dutta TK, Swaminathan RP: Human anthrax in India: urgent need for effective prevention. J Commun Dis 2000; 32:240-6 [176] Labruyère E, Mock M, Ladant D, Michelson S, Gilles A-M, Laoide B, Bârzu O: Characterization of ATP and Calmodulin-Binding Properties of aTruncated Form of Bacillus anthracis Adenylate Cyclase. Biochemistry 1990; 29:4922-8 [177] Labruyère E, Mock M, Surewicz WK, Mantsch HH, Rose T, Munier H, Sarfati RS, Bârzu: Structural and ligand-binding properties of a truncated form of Bacillus anthracis adenylate cyclase and of a catalytically inactive variant in which glutamine substitutes for lysine-346. Biochemistry 1991; 30:2619-24 [178] Laforce FM: Woolsorter's disease in England. Bull N Y Acad Med 1978; 54:956-63 [179] Lai E-M, Phadke ND, Kachman MT, Giorno R, Vazquez S, Vazquez JA, Maddock JR, Driks A: Proteomic analysis of the spore coats of Bacillus subtilis and Bacillus anthracis. J Bacteriol 2003; 185:1443-54 [180] Lee D-Y, Chun J-H, Ha H-J, Park J, Kim B-S, Oh H-B, Rhie G-E: Poly-gamma-dglutamic acid and protective antigen conjugate vaccines induce functional antibodies against the protective antigen and capsule of Bacillus anthracis in guinea-pigs and rabbits. FEMS Immunol Med Microbiol 2009; 57:165-72 [181] Lekota KE, Polyphasic and genome characterization of Bacillus species from anthrax outbreaks in animals in South Africa and Lesotho. Masterarbeit Universität Pretoria 2013 [182] Leppla SH: Bacillus anthracis calmodulin-dependent adenylate cyclase: chemical and enzymatic properties and interactions with eucaryotic cells. Adv Cyclic Nucleotide Protein Phosphorylation Res 1984; 17:189-98 [183] Leppla SH: Anthrax toxin edema factor: A bacterial adenylate cyclase that increases cyclic AMP concentrations in eukaryotic cells. P Natl Acad Sci USA 1982; 79:3162-6 XVII [184] Levinsohn JL, Newman ZL, Hellmich KA, Fattah R, Getz MA, Liu S et al: Anthrax lethal factor cleavage of Nlrp1 is required for activation of the inflammasome. PLoS Pathog 2012; 8:e1002638 [185] Lewis V, Green SA, Marsh M, Vihko P, Helenius A, Mellman I: Glycoproteins of the lysosomal membrane. J Cell Biol 1985; 100:1839-47 [186] Li Z, Howard A, Kelley C, Delogu G, Collins F, Morris S: Immunogenicity of DNA vaccines expressing tuberculosis proteins fused to tissue plasminogen activator signal sequences. Infect Immun 1999; 67:4780-6 [187] Lightfoot NF, Scott RJD, Turnbull PCB: Antimicrobial susceptibility of Bacillus anthracis. Salisbury Med Bull 1990; 68:95-8 [188] Lincoln RE et al.: Sucessful treatment of rhesus monkeys for septicemic anthrax. Antimicrob Agents Ch 1964; 4:759-63 [189] Lincoln RE, Walker JS, Klein F, Rosenwald AJ, Jones WI Jr: Value of field data for extrapolation in anthrax. Fed Proc 1967; 26:1558-62 [190] Lindeque PM, Turnbull PCB: Ecology and epidemiology of anthrax in the Etosha National Park, Namibia. Onderstepoort J Vet Res 1994; 61:71-83 [191] Little SF, Ivins BE, Webster WM, Fellows PF, Pitt ML, Norris SL et al.: Duration of protection of rabbits after vaccination with Bacillus anthracis recombinant protective antigen vaccine. Vaccine 2006; 24:2530-6 [192] Little SF, Knudson GB: Comparative Efficacy of Bacillus anthracis Live Spore Vaccine and Protective Antigen Vaccine against Anthrax in the Guinea Pig. Infect Immun 1986; 52:509512 [193] Little SF, Leppla SH, Friedlander AM: Production and characterization of monoclonal antibodies against the lethal factor component of Bacillus anthracis lethal toxin. Infect Immun 1990; 58:1606-13 [194] Little SF, Novak JM, Lowe JR, Leppla SH, Singh Y, Klimpel KR, Lidgerding BC, Friedlander AM: Characterization of lethal factor-binding and cell-receptor binding domains of protective antigen of Bacillus anthracis using monoclonal antibodies. Microbiology 1996; 142:707-15 [195] Liu C-Q, Nuttall SD, Tran H, Wilkins M, Streltsov VA, Alderton MR: Construction, Crystal Structure and Application of a Recombinant Protein That Lacks the Collagen-Like Region of BclA From Bacillus anthracis Spores. Biotechnol Bioeng 2007; 99:774-82 [196] Liu MA, McClements W, Ulmer JB, Shiver J, Donnelly J: Immunization of non-human primates with DNA vaccines. Vaccine 1997; 15:909-12 [197] Liu S, Moayeri M, Leppla SH: Anthrax lethal and edema toxins in anthrax pathogenesis. Trends in Microbiology 2014; 22:317-25 [198] Liu S, Zhang Y, Moayeri M, Liu Jie, Crown D, Fattah R: Key tissue targets responsible for anthrax toxin-induced-lethality. Nature 2013; 501:63-8 [199] Liu T-H, Oscherwitz J, Schnepp B, Jacobs J, Yu F, Cease KB, Johnson PR: Genetic Vaccines for Anthrax Based on Recombinant Adeno-associated Virus Vectors. Molecular Therapy 2009; 17:373-9 [200] Luo D, Saltzman WM: Synthetic DNA delivery systems. Nat Biotechnol 2000; 18:33-7 [201] Luxembourg A, Hannaman D, Nolan E, Ellefsen B, Nakamura G, Chau L, Tellez O, Little S, Bernard R: Potentiation of an anthrax DNA vaccine with electroporation. Vaccine 2008; 28:5216-22 [202] M'Fadyean J: A further note with regard to the staining rection of anthrax blood with methylene blue. J Comp Pathol 1903; 16:360-1 [203] Makino S, Watarai M, Cheun HI, Shirahata T, Uchida I: Effect of the Lower Molecular Capsule Released from the Cell Surface of Bacillus anthracis on the Pathogenesis of Anthrax. J Infect Dis 2002; 186:227-33 XVIII [204] Maldonado-Arocho FJ, Fulcher JA, Lee B, Bradley KA: Anthrax oedema toxin induces anthrax toxin receptor expression in monocyte-derived cells. Mol Microbiol 2006; 2:324-37 [205] Malyala P, Singh M: Endotoxin limits in formulations for preclinical research. J Pharm Sci 2008; 97:2041-4 [206] Martin BK, Chin KC, Olsen JC, Skinner CA, Dey A, Ozato K, Ting JP: Induction of MHC class I expression by the MHC class II transactivator CIITA. Immunity 1997; 6:591-600 [207] Martinon F, Tschopp J: Inflammatory caspases and inflammasomes: master switches of inflammation. Cell Death Differ 2007; 14:10-22 [208] Mesnage S, Tosi-Couture E, Gounon P, Mock M, Fouet A: The Capsule and S-Layer: Two Independent and Yet Compatible Macromolecular Structures in Bacillus anthracis. J Bacteriol 1998; 180:52-8 [209] Mesnage S, Tosi-Couture E, Mock M, Gounon P, Fouet A: Molecular characterization of the Bacillus anthracis main S-layer component: evidence that it is the major cell-associated antigen . Mol Microbiol 1997; 23:1147-55 [210] Midha S, Bhatnagar R: Genetic immunization with GPI-anchored anthrax protective antigen raises combined CD1d- and MHC II-restricted antibody responses by natural killer T cell-mediated help. Vaccine 2009; 27:1700-9 [211] Midha S, Bhatnagar R: Anthrax protective antigen administered by DNA vaccination to distinct subcellular locations potentiates humoral and cellular immune responses. Eur J Immunol 2009; 39:159-177 [212] Mignot T, Mesnage S, Coututre-Tosi E, Mock M, Fouet A: Developmental switch of Slayer protein synthesis in Bacillus anthracis. Mol Microbiol 2002; 43:1615-27 [213] Mikesell P, Ivins BE, Ristroph JD, Dreier TM: Evidence for Plasmid-Mediated Toxin Production in Bacillus anthracis. Infect Immun 1983; 39:371-6 [214] Milhomme O, Köhler SM, Ropartz D, Lesur D, Pilard S, Djedaini-Pilard F, Beyer W, Grandjean C: Synthesis and immunochemical evaluation of a non-methylated disaccharide analogue of the anthrax tetrasaccharide. Org Biomol Chem 2012; 10:8524-32 [215] Miller CJ, Elliot JL, Collier RJ: Anthrax protective antigen: prepore-to-pore conversion. Biochemistry 1999; 38:10432-41 [216] Milne JC, Furlong D, Hanna PC, Wall JS, Collier RJ: Anthrax Protective Antigen Forms Oligomers during Intoxication of Mammalian Cells. J Bacteriol Chem 1994; 269:2060712 [217] Minett FC: Sporulation and viability of B. anthracis in relation to environmental temperature and humidity. J Comp Pathol 1950; 60:161-76 [218] Moayeri M, Crown D, Newman ZL, Okugawa S, Eckhaus M, Cataisson C et al: Inflammasome sensor Nlrp1b-dependent resistance to anthrax is mediated by caspase-1, IL-1 signaling and neutrophil recruitment. PLoS Pathog 2010; 6:e1001222 [219] Moayeri M, Leppla SH: The roles of anthrax toxin in pathogenesis. Curr Opin Microbiol 2004; 7:19-24 [220] Moayeri M, Leppla SH: Abthrax Toxins. In: Bergman NH: Bacillus anthracis und Anthrax. Wiley-Blackwell 2011, 121-56. [221] Moayeri M, Martinez NW, Wiggins J, Young HA, Leppla SH: Mouse susceptibility to anthrax lethal toxin is influenced by genetic factors in addition to those controlling macrophage sensitivity. Infect Immun 2004; 72:4439-47 [222] Mogridge J, Cunningham K, Collier RJ: Stoichiometry of anthrax toxin complexes. Biochem 2002; 41:1079-82 [223] Morens DM: Characterizing a “New” Disease: Epizootic and Epidemic Anthrax, 1769– 1780. Am J Public Health 2003; 93:886-93 [224] Murrell WG: Chemical composition of spores and spore structure. In: Gould GW, Hurst A: The Bacterial Spore. Academic Press, London 1969, 215. XIX [225] Nguyen DT, Witte de L, Ludlow M, Yuksel S, Wiesmüller K-H, Geijtenbeek TBH, Osterhaus ADME, Swart de RL: The synthetic bacterial lipopeptide Pam3CSK4 modulates respiratory syncytial virus infection independent of TLR activation. PLoS Pathog 2010; 6:1-13 [226] Nicholson WL, Munakata N, Horneck G, Melosh HJ, Setlow P: Resistance of Bacillus endospores to extreme terrestrial and extraterrestrial environments. Microbiol Mol Biol Rev 2000; 64:548-72 [227] Novak JM, Stein M-P, Little SF, Leppla SH, Friedlander AM: Functional Characterization of Protease-treated Bacillus anthracis Protective Antigen. J Bacteriol Chem 1992; 267:17186-93 [228] OIE: Anthrax. In: World Organisation for Animal Health: Manual of Diagnostic Tests and Vaccines for Terrestrial Animals 2014. 2014, . [229] Okusawa T, Fujita M, Nakamura J, Into T, Yasuda M, Yoshimuta A, Hara Y, Hasebe A, Golenbock DT, Morita M, Kuroki Y, Ogawa T, Shibata K: Relationship between Structures and Biological Activities of Mycoplasmal Diacylated Lipopeptides and Their Recognition by TollLike Receptors 2 and 6. Infect Immun 2004; 72:1657-65 [230] Otten GR, Schaefer M, Doe B, Liu H, zur Megede J, Donnelly J, Rabussay D, Barnett S, Ulmer JB: Potent immunogenicity of an HIV-1 gag–pol fusion DNA vaccine delivered by in vivo electroporation. Vaccine 2006; 24:4503-9 [231] Owen MP, Schauwers W, Hugh-Jones ME, Kiernan JA, Turnbull PCB, Beyer W: A simple, reliable M'Fadyean stain for visualizing the Bacillus anthracis capsule. J Microbiol Meth 2013; 92:264-9 [232] Paccani SR, Tonello F, Ghittoni R, Natale M, Muraro L, D'Elios MM, Tang WJ, Montecucco C, Baldari CT: Anthrax toxins suppress T lymphocyte activation by disrupting antigen receptor signaling. J Exp Med 2005; 201:325-31 [233] Pannifer AD, Wong TY, Schwarzenbacher R, Renatus M, Petosa C, Bienkowska J, Lacy DB, Collier RJ, Park S, Leppla SH, Hanna P, Liddington RC: Crystal structure of the anthrax lethal factor. Nature 2001; 414:229-33 [234] Pardoll DM, Beckerleg AM: Exposing the immunology of naked DNA vaccines. Immunity 1995; 3:165-9 [235] Park JM, Greter FR, Li ZW, Karin M: Macrophage apoptosis by anthrax lethal factor through p38 MAP kinase inhibition. Science 2002; 297[5589]:2048-51 [236] Parry JM, Turnbull PCB, Gibson JR: Wolfe Medical Atlases. Wolfe Medical Publications 1983. [237] Pasteur L: De l'atténuation des virus et de leur retour à la virulence. CR Acad Sci Agric Bulg 1881; 92:429-35 [238] Patra G, Sylvestre P, Ramisse V, Thérasse J, Guesdon JL: Isolation of a specific chromosomic DNA sequence of Bacillus anthracis and its possible use in diagnosis. FEMS Immunol Med Microbiol 1996; 15:223-31 [239] Petosa C, Collier RJ, Klimpel KR, Leppla SH, Liddington RC: Crystal structure of the anthrax toxin protective antigen. Nature 1997; 385:833-8 [240] Pitt MLM, Little SF, Ivins BE, Fellows P, Barth J, Hewetson J et al: In vitro correlate of immunity in a rabbit model of inhalational anthrax. Vaccine 2001; 19:4768-73 [241] Pittman PR, Fisher D, Quinn X, Schmader T, Barrera-Oro JG: Effect of delayed anthrax vaccine dose on Bacillus anthracis protective antigen IgG response and lethal toxin neutralization activity. Vaccine 2014; 31:5000-14 [242] Preisz H: Experimentelle Studien über Virulenz, Empfänglichkeit und Immunität beim Milzbrand. Z Immunitätsforschung 1909; 5:341-452 [243] Price BM, Liner AL, Park S, Leppla SH, Mateczun A, Galloway DR: Protection against Anthrax Lethal Toxin Challenge by Genetic Immunization with a Plasmid Encoding the Lethal Factor Protein. Infect Immun 2001; 69:4509-15 XX [244] Puziss M, Wright GG: Studies on immunity in anthrax. X. Gel-adsorbed protective antigen for immunization of man. J Bacteriol 1962; 85:230-6 [245] Rasko DA: Bacillus anthracis Plasmids: Species Definition or Niche Adaptions?. In: Bergman NH: Bacillus anthracis and Anthrax. Wiley-Blackwell 2011, 89-106. [246] Raz E, Tighe H, Sato Y, Corr M, Dudler JA, Roman M, Swain SL, Spiegelberg HL, Carson DA: Preferential induction of a Th1 immune response and inhibition of specific IgE antibody formation by plasmid DNA immunization. Proc Natl Acad Sci USA 1996; 93:5141-5 [247] Reason D, Liberato J, Sun J, Keitel W, Zhou J: Frequency and Domain Specificity of Toxin-Neutralizing Paratopes in the Human Antibody Response to Anthrax Vaccine Adsorbed. Infect Immun 2009; 77:2030-5 [248] Reason DC, Ullal A, Liberato J, Sun J, Keitel W, Zhou J: Domain specificity of the human antibody response to Bacillus anthracis protective antigen. Vaccine 2008; 26:4041-7 [249] Record BR, Wallis RG: Physicochemical Examination of Polyglutamic Acid from Bacillus anthracis Grown in vivo. Biochem J 1956; 63:453-4 [250] Redmond C et al: Experimentally assessed public health risks associated with pigs from farms experiencing anthrax. Veterinary Record 1997; 141:244-7 [251] Redmond C, Baillie LW, Hibbs S, Moir AJ, Moir A: Identification of proteins in the exosporium of Bacillus anthracis. Microbiology 2004; 150:355-63 [252] Redmond C, Pearce MJ, Manchee RJ, Berdal BP: Deadly relic of the Great War. Nature 1998; 393:747-8 [253] Reith W, LeibundGut-Landmann S, Waldburger JM: Regulation of MHC class II gene expression by the class II transactivator. Nature 2005; 5:793-806 [254] Réty S, Salamitou S, Garcia-Verdugo I, Hulmes DJS, Le Hégarat F, Chaby R, LewitBentley A: The Crystal Structure of the Bacillus anthracis Spore Surface Protein BclA Shows Remarkable Similarity to Mammalian Proteins. J Biol Chem 2005; 280:43073-8 [255] Reuveny S, White MD, Adar YY, Kafri Y, Altboum Z, Gozes Y et al: Search for Correlates of Protective Immunity Conferred by Anthrax Vaccine. Infect Imnun 2001; 69:288893 [256] Rhie G-E, Roehrl MH, Mourez M, Collier RJ, Mekalanos JJ, Wang JY: A dually active anthrax vaccine that confers protection against both bacilli and toxins. PNAS 2003; 100:1092530 [257] Ribi E, Cantrell J, Takayama K: A new immunomodulator with potential clinical applications: monophosphoryl lipid A, a detoxified endotoxin. Clin Immunol 1985; 6:33-6 [258] Ringertz SH, Hoiby EA, Jendenius M, Maehlen J, Caugant DA, Myklebust A, Fossum K: Injectional anthrax in a heroin skin popper. Lancet 2000; 356:1574-5 [259] Robertson DL, Tippetts MT, Leppla SH: Nucleotide sequence of the Bacillus anthracis edema factor gene (cya): a calmodulin-dependent adenylate cyclase. Gene 1988; 73:363-71 [260] Rodriguez F, An LL, Harkins S, Zhang J, Yokoyama M, Widera G, Fuller JT, Kinkaid C, Campbell IA, Whitton L: DNA Immunization with Minigenes: Low Frequency of Memory Cytotoxic T Lymphocytes and Inefficient Antiviral Protection Are Rectified by Ubiquitination. J Virol 1998; 72:5174-81 [261] Ross JM: The pathogenesis of Anthrax following the administration of spores by the respiratory route. Pathology 1957; 73:485-94 [262] Rottinghaus ST, Poland GA, Jacobson RM, Barr LJ, Roy MJ: Hepatitis B DNA vaccine inducesprotective antibody responses in human non-responders to conventional vaccination. Vaccine 2003; 21:4604-8 [263] Rouse RJ, Nair SK, Lydy SL, Bowen JC, Rouse BT: Induction in vitro of primary cytotoxic T-lymphocyte responses with DNA encoding herpes simplex virus proteins. J Virol 1994; 68:5685-9 XXI [264] Saile E, Boons G-J, Buskas T, Carlson RW, Kannenberg EL, Barr JR, Boyer AE, Gallegos-Candela M, Quinn CP: Antibody Responses to a Spore Carbohydrate Antigen as a Marker of Nonfatal Inhalation Anthrax in Rhesus Macaques. Clin Vaccine Immunol 2011; 18:743-8 [265] Saile E, Koehler TM: Bacillus anthracis Multiplication, Persistence, and Genetic Exchange in the Rhizosphere of Grass Plants. Appl Env Microbiol 2006; 72:3168-74 [266] Salmon DD, Ferrier GR: Post vaccination occurrence of anthrax in cattle. Vet Rec 1992; 130:140-1 [267] Sára M, Sleytr UB: S-Layer Proteins. J Bacteriol 2000; 182:859-68 [268] Sarkar D, Desalle R, Fisher PB: Evolution of MDA-5/RIG-I-dependent innate immunity: independent evolution by domain grafting. Proc Natl Acad Sci USA 2008; 105:17040-5 [269] Sastalla I, Tang S, Crown D, Liu S, Eckhaus MA, Hewlett K: Anthrax edema toxin impairs clearance in mice. Infect Immun 2012; 80:529-38 [270] Schild H, Deres K, Wiesmüller KH, Jung G, Rammensee HG: Efficiency of peptides and lipopeptides for in vivo priming of virus-specific cytotoxic T cells. Eur J Immunol 1991; 21:2649-54 [271] Schlecht S, Wiesmüller K-H, Jung G, Bessler WG: Lipopeptide als natürliche Adjuvantien für Impfstoffe aus Gram-negativen Bakterien. Naturwissenschaften 1993; 80:9-17 [272] Schlingman AS, Devlin HB, Wright GG, Maine RJ, Manning MC: Immunizing activity of alum-precipitated protective antigen of Bacillus anthracis in cattle, sheep, and swine. Am J Vet Res 1956 1956; 17:256-61 [273] Schneerson R, Kubler-Kielb J, Liu T-Y, Dai Z-D, Leppla SH, Yergey A, Backlund P, Shiloach J, Majadly F, Robbins JB: Poly(gamma-d-glutamic acid) protein conjugates induce IgG antibodies in mice to the capsule of Bacillus anthracis a potential addition to the anthrax vaccine. PNAS 2003; 100:8945-50 [274] Scobie HM, Rainey GJ, Bradley KA, Young JA: Human capillary morphogenesis protein 2 functions as an anthrax toxin receptor. Proc Natl Acad Sci USA 2003; 100:5170-4 [275] Scorpio A, Chabot DJ, Day WA, O'Brian DK, Vietri NJ, Itoh Y, Mohamadzadeh M, Friedlander AM: Poly-γ-glutamate capsule-degrading enzyme treatment enhances phagocytosis and killing of encapsulated Bacillus anthracis. Antimicrob Agents Ch 2007; 51:215-22 [276] Seth RB, Sun L, Ea CK, Chen ZJ: Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-κB and IRF 3. Cell 2005; 122:66982 [277] Setlow P: Mechanisms for the prevention of damage to DNA in spores of Bacillus species. Annu Rev Microbiol 1995; 49:29-54 [278] Setlow P: I will survive: DNA protection in bacterial spores. Trends Microbiol 2007; 15[4]:172-80 [279] Severson KM, Mallozzi M, Bozue J, Welkos SL, Cote CK, Knight KL, Driks A: Roles of the Bacillus anthracis sporeprotein ExsK in exosporium maturation and germination. J Bacteriol 2009; 191:7587-96 [280] Shakya KP, Hugh-Jones ME, Elzer PH: Evaluation of immune response to orally administered Sterne strain 34F2 anthrax vaccine. Vaccine 2007; 25:5374-7 [281] Shen Y, Lee YS, Soelaiman S, Bergson P, Lu D, Chen A: Physiological calcium concentrations regulate calmodulin binding and catalysis of adenylyl cyclase exotoxins. EMBO J 2002; 24:6721-32 [282] Shlyakhov EN, Rubinstein E: Human live anthrax vaccine in the former USSR. Vaccine 1994; 12:727-30 [283] Shoop WL, Xiong Y, Wiltsie J, Woods A, Guo J, Pivnichny JV, Felcetto T, Michael BF, Bansal A, Cummings RT, Cunningham BR, Friedlander AM, Douglas CM, Patel SB, XXII Wisniewski D, Scapin G, Salowe SP, Zaller DM et al.: Anthrax lethal factor inhibition. Proc Natl Acad Sci USA 2005; 102:7958-63 [284]: Siegrist C: Vaccine immunology. In: Plotkin SA, Orenstein WA, Offit PA: Vaccines. Saunders Elsevier Philadelphia 2008 [285] Singh Y, Klimpel KR, Arora N, Sharma M, Leppla SH: The chymotrypsin-sensitive site, FFD315, in anthrax toxin protective antigen is required for translocation of lethal factor. J Biol Chem 1994; 269:29039-46 [286] Singh Y, Klimpel KR, Goel S, Swain PK, Leppla SH: Oligomerization of Anthrax Toxin Protective Antigen and Binding of Lethal Factor during Endocytic Uptake into Mammalian Cells. Infect Immun 1999; 67:1853-9 [287] Singh Y, Klimpel KR, Quinn CP, Chaudhary VK, Leppla SH: The Carboxyl-terminal End of Protective Antigen Is Required for Receptor Binding and AnthraxToxin Activity. J Bacteriol Chem 1991; 266:15493-7 [288] Sirard JC, Mock M, Fouet A: The three Bacillus anthracis toxin genes are coordinately regulated by bicarbonate and temperature. J Bacteriol 1994; 176:5188-92 [289] Sjöstedt A, Eriksson U, Ramisse V, Garrigue H: Detection of Bacillus anthracis spores in soil by PCR. FEMS Microbiol Ecol 1997; 23:159-68 [290] Sleytr UB, Messner P: Crystalline Surface Layers on Bacteria. Ann Rev Microbiol 1983; 37:311-39 [291] Sleytr UB, Messner P, Pum D, Sára M: Crystalline bacterial cell surface layers. Mol Microbiol 1993; 10:911-6 [292] Smith H, Gallop RC: The chemical basis of the virulence of Bacillus anthracis. VI. An extracellular immunising aggressin isolated from exudates of infected guinea-pigs. Brit J Exp Pathol 1956; 37:144-55 [293] Smith H, Keppie J: Observations on experimental anthrax: Demonstration of a specific lethal factor produced in vivo by bacillus anthracis. Nature 1954; 173:869-70 [294] Stanley JL, Smith H: The Three Factors of Anthrax Toxin: Their Immunogenicity and Lack of Demonstrable Enzymic Activity. J gen Microbiol 1963; 31:329-37 [295] Stanley JL, Smith H: Purification of Factor I and Recognition of a Third Factor of the Anthrax Toxin . J Gen Microbiol 1961; 26:49-66 [296] Steichen C, Chen P, Kearney JF, Turnbough Jr C: Identification of the immunodominant protein and other proteins of the Bacillus anthracis exosporium. J Bacteriol 2003; 185:1903-10 [297] Steichen CT, Kearney JF, Turnbough CL: Characterization of the exosporium basal layer protein BxpB of Bacillus anthracis. J Bacteriol 2005; 187:5868-76 [298] Stepanov AV, Marinin LI, Pomerantsev AP, Staritsin NA: Development of novel vaccines against anthrax in man. J Bacteriol 2006; 44:155-160 [299] Sterne M: The use of saponin spore vaccine for inoculation against anthrax in South Africa. Onderstepoort J Vet Sci Ani In 1939; 12:279-302 [300] Sterne M: The use of Anthrax Vaccines prepared from avirulent (unencapsulated) variants of Bacillus anthracis. Onderstepoort J Vet Sci An Ind 1939; 13:307-312 [301] Sterne M: The immunization of laboratory animals against anthrax. Onderstepoort J Vet Sci An Ind 1939; 13:315-7 [302] Sterne M: The effects of different carbon dioxide concentrations on the growth of virulent anthrax strains. Pathogenicity and immunity tests on guinea-pigs and sheep with anthrax variants derived from virulent strains. Onderstepoort J Vet Sci An Ind 1937; 9:49-67 [303] Sterne M: Avirulent anthrax vaccine. Onderstepoort J Vet Sci An Ind 1946; 21:41-3 [304] Sterne M, Nichol J, Lambrechts MC: The effect of large scale active immunization against anthrax. J S African Vet Med Assoc 1942; 13:53-63 XXIII [305] Sterne M, Robinason EM: The preparation of anthrax spore vaccines (for cattle and sheep) in South Africa. Onderstepoort J Vet Sci Ani In 1939; 12:9-12 [306] Stiehm M, Peters K, Wiesmüller KH, Bufe A, Peters M: A novel synthertic lipopeptide is allergy-protective by the induction of LPS-tolerance. Clin Exp Allergy 2013; 43:785-97 [307] Stokes MGM, Titball RW, Neeson B, Galen JE, Walker NJ, Stagg AJ et al: Oral Administration of a Salmonella enterica-Based Vaccine Expressing Bacillus anthracis Protective Antigen Confers Protection against Aerosolized B. anthracis. Infect Immun 2007; 75:1827-34 [308] Sussman AS, Halvorson HO: Spores: Their dormancy and Germination. Harper & Row New York 1966 [309] Sylvestre P, Couture-Tosi E, Mock M: Contribution of ExsFA and ExsFB proteins to the localization of BclA on the spore surface and to the stability of the Bacillus anthracis exosporium. J Bacteriol 2005; 187:5122-8 [310] Sylvestre P, Couture-Tosi E, Mock M: A collagen-like surface glycoprotein is a structural component of the Bacillus anthracis exosporium. Mol Microbiol 2002; 45:169-78 [311] Sylvestre P, Couture-Tosi E, Mock Michèle: Polymorphism in the Collagen-Like Region of the Bacillus anthracis BclA Protein Leads to Variation in Exosporium Filament Length. J Bacteriol 2003; 185:1555-63 [312] Takeuchi O, Akira S: Toll-like receptors; their physiological role and signal transduction system. Int Immunol 2001; 1:625-35 [313] Tang DC, DeVit M, Johnston SA: Genetic immunization is a simple method for eliciting an immune response. Nature 1992; 356:152-4 [314] Terra JK, Cote CK, France B, Jenkins AL, Bozue JA, Welkos SL et al.: Cutting edge: resistance to Bacillus anthracis infection mediated by a lethal toxin sensitive allele of Nalp1b/Nlrp1b. J Immunol 2010; 184:17-20 [315] Thompson BM, Waller LN, Fox KF, Fox A, Stewart GC: The BclB glycoprotein of Bacillus anthracisis involved in exosporium integrity. J Bacteriol 2007; 189:6704-13 [316] Thorne CB, Molnar DM, Strange RE: Production of toxin in vitro by Bacillus anthracis and its separation into two components. J Bacteriol 1960; 79:450-75 [317] Tigertt WD: William Smith Greenfield, MD, FRCP, Professor Superintendent, The Brown Animal Sanatory Institution (1876-81). Concerning the priority due to him for the production of the first vaccine against anthrax. J Hyg Camb 1980; 415-20 [318] Ting J P-Y, Trowsdale J: Genetic Control of MHC Class II Expression. Cell 2002; 109:21-33 [319] Tomcsik J, Szongott H: Über ein spezifisches Protein der Kapsel des Milzbrandbazillus. Z Immun Forsch 1933; 78:86-99 [320] Tresselt HB, Boor AK: An antigen prepared in vitro effective for immunisation against anthrax III. Immunisation of monkeys against anthrax. J Infect Bacteriol 1955; 97:203-6 [321] Turell MJ, Knudson GB: Mechanical transmission of Bacillus anthracis by stable flies (Stomoxys calcitrans) and mosquitoes (Aedes aegypti and Aedes taeniorhynchus). Infect Immun 1987; 55:1859-61 [322] Turnbull PCB: Guidelines for the Surveillance and Control of Anthrax in Human and Animals, 3rd edition. World Health Organziation Press 1998. [323] Turnbull PCB: Introduction: Anthrax History, Disease and Ecology. In: Koehler TM: Anthrax. Current topics in microbiology and immunology. Berlin Heidelberg: Springer-Verlag 2002, 1-19. [324] Turnbull PCB: Definitive identification of Bacillus anthracis—a review. J Appl Microbiol 1999; 87:237-40 [325] Turnbull PCB: Current status of immunization against anthrax: old vaccines maybe here to stay for a while. Opin Infect Dis 2000; 13:113-120 XXIV [326] Turnbull PCB: Anthrax vaccines: past, present and future. Vaccine 1991; 9:533-9 [327] Turnbull PCB: Anthrax is alive and well. PHLS Microbiol Dig 1996; 9:103-6 [328] Turnbull PCB, Broster MG, Carman A, Manchee RJ, Melling J: Development of Antibodies to Protective Antigen and Lethal Factor Components of Anthrax Toxin in Humans and Guinea Pigs and Their Relevance to Protective Immunity. Infect Immun 1986; 52:356-63 [329] Turnbull PCB, Leppla SH, Broster MG, Quinn CP, Melling J: Antibodies to anthrax toxin in humans and guinea pigs and their relevance to protective immunity. Med Microbiol Immunol 1988; 177:293-303 [330] Turnbull PCB, Quinn CP, Hewson R, Stockbridge MC, Melling J: Protection conferred by microbially-supplemented UK and purified PA vaccines. Salisbury Med Bull 1990; 68:89-91 [331] Uchida I, Sekizaki T, Hashimoto K, Terakado N: Association of the Encapsulation of Bacillus anthracis with a 60 Megadalton Plasmid. J Gen Microbiol 1985; 131:363-7 [332] Uchida M, Harada T, Enkhtuya J, Kusumoto A, Kobayashi Y, Chiba S, Shyaka A, Kawamoto K: Protective effect of Bacillus anthracis surface protein EA1 against anthrax in mice. Biochem Bioph Res Co 2012; 421:323-8 [333] Ulmer JB, Wahren B ,Liu MA: Gene-based vaccines recent technical and clinical advances. Trends Mol Med 2006; 12:216-22 [334] Van Ness GB: Ecology of Antrax. Science 1971; 172:1303-7 [335] Varughese M, Teixeira AV, Liu S, Leppla SH: Identification of a Receptor-Binding Region within Domain 4 of the Protective Antigen Component of Anthrax Toxin. Infect Immun 1999; 67:1860-5 [336] Vergis JM, Cote CK, Bozue J, Alem F, Ventura CL, Welkos SL, O'Brian AD: Immunization of Mice with Formalin-Inactivated Spores from Avirulent Bacillus cereus Strains Provides Significant Protection from Challenge with Bacillus anthracis Ames. Clin Vacc Immunol 2013; 20:56-65 [337] Vitale G, Bernardi L, Napolitani G, Mock M, Montecucco C: Susceptibility of mitogenactivated protein kinase kinase family members to proteolysis by anthrax lethal factor. Biochem J 2000; 352:739-45 [338] Vitale G, Pellizzari R, Recchi C, Napolitani G, Mock M, Montecucco C: Anthrax lethal factor cleaves the N-terminus of MAPKKs and induces tyrosine/threonine phosphorylation of MAPKs in cultured macropahges. Biochem Biophys Res Commun 1998; 248:706-11 [339] Vreeland RH, Rosenzweig WD, Powers DW: Isolation of a 250 million-year-old halotolerant bacterium from a primary salt crystal. Nature 2000; 407:897-900 [340] Waley SG: The structure of bacterial polyglutamic acid. J chem Soc 1955; 0:517-22 [341] Waller LN, Stump MJ, Fox KF, Harley WM, Fox A, Steward GC, Shahgholi M: Identification of a Second Collagen-Like Glycoprotein Produced by Bacillus anthracis and Demonstration of Associated Spore-Specific Sugars. J Bacteriol 2005; 187:4592-7 [342] Walz A, Mujer CV, Connolly JP, Alefantis T, Chafin R, Dake C, Whittington J, Kumar SP, Khan AS, DelVecchio VG: Bacillus anthracis secretome time course under host-simulated conditions and identification of immunogenic proteins. Prot Sci 2007; 5[1]:11 [343] Wang D, Fang L, Li T, Luo R, Xie L, Jiang Y, Chen H, Xiao S: Molecular cloning and functional characterization of porcine IFN-beta promoter stimulator 1 (IPS-1). Vet Immunol Immunopathol 2008; 125:344-53 [344] Wang TT, Fellows PF, Leighton TJ, Lucas AH: Induction of opsonic antibodies to the gamma-D-glutamic acid capsule of Bacillus anthracis by immunization with a synthetic peptide-carrier protein conjugate. FEMS Immunol Med Microbiol 2004; 40:231-7 [345] Wang TT, Lucas AH: The capsule of Bacillus anthracis behaves as a thymusindependent type 2 antigen. Infect Immun 2004; 72:5460-3 [346] Warth AD, Ohye DF, Murrell WG: The composition and structure of bacterial spores. J Cell Biol 1963; 16:579-92 XXV [347] Watson DW, Cromartie WJ, Bloom WL, Kegels G, Heckly RJ: Studies on infection with Bacillus anthracis III. Chemical and immunological properties of the protective antigen in crude extracts of skin lesions of B. anthracis. J Infect Dis 1947; 80:1-13 [348] Webster A: Inhibiting effects of antibiotics on anthrax vaccination. Aust Vet J 1973; 49:545 [349] Weiss R, Leitner WW, Scheiblhofer S, Chen D, Bernhaupt A, Mostböck S, Thalhamer J, Lyon JA: Genetic vaccination against malaria infection by intradermal and epidermal injections of a plasmid containing the gene encoding the Plasmodium berghei circumsporozoite protein. Infect Immun 2000; 56:5914-9 [350] Weiss R, Scheiblhofer S, Freund J, Ferreira F, Livey I, Thalhamer J: Gene gun bombardment with gold particles displays a particular Th2-promoting signal that overrules the Th1-inducing effect of immunostimulatory CpG motifs in DNA vaccines. Vaccine 2002; 20:3148-54 [351] Welkos S, Friedlander A, Weeks S, Little S, Mendelson I: In-vitro characterisation of the phagocytosis and fate of anthrax spores in macrophages and the effects of anti-PA antibody. J Med Microbiol 2002; 51:821-31 [352] Welkos S, Little S, Friedlander A, Fritz D, Fellows P: The role of antibodies to Bacillus anthracis and anthrax toxin components in inhibiting the early stages of infection by anthrax spores. Microbiol 2001; 147:1677-85 [353] Welkos SL, Friedlander AM: Comparative safety and efficacy against Bacillus anthracis of protective antigen and live vaccines in mice. Microb Pathogenesis 1988; 5:127-39 [354] Welkos SL, Keener TJ, Gibbs PH: Differences in susceptibility of inbred mice to Bacillus anthracis. Infect Immun 1986; 51:795-800 [355] Welkos SL, Lowe JR, Eden-McCutchan F, Vodkin M, Leppla SH, Schmidt JJ: Sequence and analysis of the DNA encoding protective antigen of Bacillus anthracis. Gene 1988; 69:287-300 [356] Wheelis M: First shots fired in biological warfare. Nature 1998; 395:213 [357] WHO: Anthrax in humans and animals - 4th ed.. World Health Organziation Press 2008 [358] Wiesmüller K-H, Fleckenstein B, Jung G: Peptide Vaccines and Peptide Libraries. Biol Chem 2001; 382:571-579 [359] Wiesmüller KH, Bessler WG, Jung G: Solid phase peptide synthesis of lipopeptide vaccines eliciting epitope-specific B-, T-helper and T-killer cell response. Int J Pept Protein Res 1992; 40:255-60 [360] Williams JA, Carnes AE, Hodgson CP: Plasmid DNA Vaccine vector design: impact on efficacy, safety and upstream production. Biotechnol Adv 2009; 27:353-370 [361] Williams JA, Luke J, Johnson L, Hodgson C: pDNAVACCultra vector family: high throughput intracellular targeting DNA vaccine plasmids. Vaccine 2006; 24:4671-6 [362] Williams MA, Fukuda M: Accumulation of membrane glycoproteins in lysosomes requires a tyrosine residue at a particular position in the cytoplasmic tail. J Cell Biol 1990; 111:955-66 [363] Williams RC, Rees ML, Jacobs MF, Práqai Z, Thwaite JE, Baillie LW, Emmerson PT, Harwood CR: Production of Bacillus anthracis protective antigen is dependent on the extracellular chaperone, PrsA. J Biol Chem 2003; 278:18056-62 [364] Williamson ED, Beedham RJ, Bennett AM, Perkins SD, Miller J, Baillie LW: Presentation of protective antigen to the mouse immune system: immune sequelae. J Appl Microbiol 1999; 87:315-7 [365] Williamson ED, Hodgson I, Walker NJ, Topping AW, Duchars MG, Mott JM et al.: Immunogenicity of Recombinant Protective Antigen and Efficacy against Aerosol Challenge with Anthrax. Infect Immun 2005; 73:5978-87 XXVI [366] Wright GG, Hedberg MA, Slein JB: Studies on immunity in anthrax III. Elaboration of protective antigen in a chemically defined, non-protein medium. J Immunol 1954; 72:263-9 [367] Wright JG, Plikaytis BD, Rose CE, Parker SC, Babcock J, Keitel W et al: Effect of reduced dose schedules and intramuscular injection of anthrax vaccine adsorbed on immunological response and safety profile: A randomized trial. Vaccine 2014; 32:1019-28 [368] Wu TC, Guarnieri FG, Staveley-O'Carroll KF, Viscidi RP, Levitsky HI, Hedrick L, Cho KR, August JT, Pardoll DM: Engineering an intracellular pathway for major histocompatibility complex class II presentation of antigens. Proc Natl Acad Sci USA 1995; 92:11671-5 [369] Xia ZG, Storm DR: A-type ATP binding consensus sequences are critical for the catalytic activity of the calmodulin-sensitive adenylyl cyclase from Bacillus anthracis. J Biol Chem 1990; 265:6517-20 [370] Young JAT, Collier RJ: Anthrax Toxin: Receptor Binding, Internalization, Pore Formation, and Translocation. Annu Rev Biochem 2007; 76:243-65 [371] Yu HB, Finlay BB: The caspase-1 inflammasome: apilot of innate immune responses. Cell Host Microbe 2008; 4:198-208 [372] Zhongming L, Howard A, Kelley C, Delogu G, Collins F, Morris S: Immunogenicity of DNA Vaccines Expressing Tuberculosis Proteins Fused to Tissue Plasminogen Activator Signal Sequences. Infect Immun 1999; 67:4780-6 XXVII Eidesstattliche Versicherung gemäß § 7 Absatz 7 der Promotionsordnung der Universität Hohenheim zum Dr. rer. nat. 1. Bei der eingereichten Dissertation zum Thema „ Entwicklung und Testung neuer DNA- und Protein-basierter Multikomponentenvakzinen, sowie regulatorischer Adjuvanzien gegen eine Infektion mit B. anthracis in Auszucht-Mäusen und Ziegen“ handelt es sich um eine eigenständig erbrachte Leistung. 2. Ich habe nur die angegebenen Quellen und Hilfsmittel benutzt und mich keiner unzuläs- sigen Hilfe Dritter bedient. Insbesondere habe ich wörtlich oder sinngemäß aus anderen Werken übernommene Inhalte als solche kenntlich gemacht. 3. Ich habe nicht die Hilfe einer kommerziellen Promotionsvermittlung oder -beratung in Anspruch genommen. 4. Die Bedeutung der eidesstattlichen Versicherung und der strafrechtlichen Folgen einer unrichtigen oder unvollständigen eidesstattlichen Versicherung sind mir bekannt. Die Richtigkeit der vorstehenden Erklärung bestätige ich: Ich versichere an Eides statt, dass ich nach bestem Wissen die reine Wahrheit erklärt und nichts verschwiegen habe. ------------------------------- ----------------------------------- Ort und Datum Unterschrift XXVIII Lebenslauf Name: Susanne Melanie Köhler Geboren: 18.04.1981 in Eisenach Eltern: Barbara und Andreas Köhler SCHULAUSBILDUNG Realschulabschluss: 1987 – 1997 Schule: Wartburgschule (Eisenach) Note: 1,1 Auslandsaufenthalt (USA): 1998 – 1999 Schule: Shell-Rock High School (Waverly, Iowa) Abitur: 1998 – 2001 Schule: Ernst-Abbe-Gymnasium (Eisenach) Note: 1,3 STUDIUM Georg-August-Universität Göttingen von 2001 bis 2007 Hauptfach: Mikrobiologie; Nebenfächer: Immunologie und Biochemie Abschluss als Diplom-Biologin mit einer Arbeit zum Thema: „Quantitative Bestimmung von Enten Hepatitis B Virus Ausbreitung und Titer im Verlauf von in vitro Infektionen“ Abschlussnote: „Gut“ (1,5) Universität Hohenheim von 2009 bis 2014 Projektarbeit und Dissertation zum Thema: „Entwicklung und Testung neuer DNA- und Protein-basierter Multikomponentenvakzinen, sowie regulatorischer Adjuvanzien gegen eine Infektion mit B. anthracis in Auszucht-Mäusen und Ziegen“ WEITERBILDUNG GV-SOLAS zertifizierter Kurs zur Versuchstierkunde (B) XXIX PUBLIKATIONEN „Synthesis and immunochemical evaluation of a nonmethylated disaccharide analogue of the anthrax tetrasaccharide“ [Milhomme et al. 2012] „BclA and toxin antigens augment each other to protect NMRI mice from lethal Bacillus anthracis challenge“ [Koehler et al. 2015 – eingereicht] „Novel furin inhibitors with potent anti-infectious activity“ [Hardes et al. 2015 - eingereicht] VORTRÄGE UND POSTERPRÄSENTATIONEN „Vaccination of NMRI mice against Bacillus anthracis using DNA-vectors and novel adjuvants as an approach to a new acellular vaccine-combination.“ BACT conference 2013 und Nationales Symposium für Zoonoseforschung Berlin 2013 [Koehler et al. 2013] „Recombinant acellular vaccines tested in goats for immunogenicity and protectivity.“ BACT conference 2013 und DFG [Koehler et al. 2013] „Vaccination of NMRI mice against Bacillus anthracis using DNA and protein based acellular vaccine candidates and novel adjuvants.“ BACT conference 2011 [Koehler et al. 2011] „Highly potent inhibitors of the proprotein convertase furin as potential drugs for the treatment of viral and bacterial infectious diseases.“ DPHG Annual Meeting 2013 [Steinmetzer et al. 2013] „Immunogenicity and protective efficacy of the Sterne 34F2 live spore anthrax vaccine in goats.“ BACT conference 2013 [Ndumnego et al. 2013] „Quantitative anti-anthrax IgG ELISA correlates with the anthrax toxin neutralization assay in goats.“ BACT conference 2013 [Ndumnego et al. 2013] ------------------------------- ----------------------------------- Ort und Datum Unterschrift XXX Danksagung Eine Arbeit wie diese ist ein langwieriger Prozess, dessen Realisierung neben der eigenen Kraft, Initiative und Ausdauer vor allem von den Menschen im persönlichen und beruflichen Umfeld abhängt. Dabei ist nicht von Bedeutung welcher Beitrag, wann geleistet wurde, wie einschneidend dieser war oder gar ob der entscheidende Einfluss direkt mit dieser Arbeit verknüpft ist. Die Tatsache dass ich jetzt, hier an diesem Punkt in meinem Leben das Privileg habe die Doktorwürde zu erreichen, ist allen Menschen geschuldet die mich seit meiner Geburt unterstützt, begleitet, gefördert und beflügelt haben. Und so gilt mein Dank zuoberst und zuallererst meiner Mutter, Barbara Köhler, die immer an mich geglaubt und nie etwas unversucht gelassen hat, mir dabei zu helfen meine Träume zu verwirklichen, selbst dann wenn diese schwer nachvollziehbar, utopisch oder schmerzhaft für sie waren. Ich danke dir dafür von Herzen und mit Stolz. In meiner Kindheit und Jugend haben mir die wenigsten Leute Wertschätzung entgegen gebracht sodass ich oft als beliebtes Negativbeispiel - „Werde bloß nie wie Susanne!“ - bei der Erziehung gleichaltriger herhalten musste. Glücklicherweise konnte ich in dieser Zeit auf Gabi und Jürgen Meißner zählen, die mich bedingungslos und vorurteilsfrei an ihrem Familienleben teilhaben ließen und mir wie einer eigenen Tochter Geborgenheit und Unterstützung gewährten. Im Bezug auf meine akademische Laufbahn möchte ich mich besonders bei Jürgen bedanken. Ich war damals nur eine mittelmäßige Schülerin. Erst durch deine Hilfe, mir mittels Übung die Angst vor den topografischen Tests in den Geografiestunden zu nehmen, habe ich überhaupt begriffen was es bedeutet zu lernen und erst danach avancierte ich zu einer Musterschülerin. Ich bin mir daher sicher, dass Du, Deine Frau und auch eure Kinder einen entscheidenden Einfluss auf meinen Werdegang hatten und dafür bin ich sehr dankbar. Aus dieser für mich sehr schwierigen Zeit und darüber hinaus möchte ich mich auch bei meiner Freundin Franziska Groß bedanken, die mit Freude meine Fantasien, Spinnereien und Ideen teilte und damals schon meine Andersartigkeit zu schätzen wusste. Nach ihr kamen noch viele weitere Weggefährten die mich durch ihren Einfluss und ihre Freundschaft auch über diese Arbeit hinaus geprägt haben, ihnen möchte ich an dieser Stelle ebenfalls danken: Martin Baumgartl, Annika Benner, Elisabeth Blaschke, Rene Eling, Holger Engelhardt, Dora Finkeisen, Daniel Graeber, Dieter Graeber, Irina Graeber, Hannlore Goerz, Matthias Goerz, Ayesha Hassim, Nicole Hesse, Jan Hönneman, Philipp Klemm, Jörg Lüdke, Dirk Mathes, Marion Mikoteit, Eric Morfa-Morales, Kathy Olson, Lynn Olson, Fabian Patzke, Tobias Peter, Sandra Seelke, Britta von Terzi und Arne Walter XXXI Für die Ermöglichung dieser Arbeit und der damit verbundenen Abenteuer, Eindrücke und Erfahrungen möchte ich mich vor allem bei Herrn PD Dr. med. vet. habil. Wolfgang Beyer bedanken. Sie waren mir in den 6 Jahren unserer Zusammenarbeit eine große Stütze, bereichernder Lehrer und beflügelnder Diskussionspartner. Überdies erlaubte mir die Arbeit in ihrer Arbeitsgruppe, unterstützt durch Elisabeth Blaschke, Karen Hills und Sabine Hoche, eine freie Entfaltung in einem familiären Umfeld, dass sich auch auf unsere Projektpartner in der Türkei und Südafrika erstreckte. Besonders möchte ich hierbei Dr. Fatih Büyük, Dr. Özgür Ҫelebi, At Decker, Schalk van Dyk, Dr. Henriette van Heerden, Okechukwu Ndumnego, Prof. Dr. Mitat Şahin, Dr. Louis van Schalkwyk, Dr. Peter Turnbull und deren Mitarbeiter herausstellen, deren Hilfe und Ratschläge oft über das Notwendige und Machbare hinausgehend, bei der Realisation der Versuche in Südafrika und der Türkei essentiell waren. Zudem danke ich Frau Prof. Dr. rer. nat. Ute Mackenstedt für die Übernahme der Betreuung meiner Dissertation. Allen weiteren Mitarbeitern des ehemaligen Instituts für Umwelt und Tierhygiene möchte ich ebenfalls für ihre Hilfe und Unterstützung bei dieser Arbeit, aber auch für ihre Freundschaft und Anteilnahme meinen Dank aussprechen und dazu anhalten weiterhin mit Güte und Hilfsbereitschaft jungen Wissenschaftlern zur Seite zu stehen. Abschließend ein großes DANKE auch Daniel Goerz, für deinen Beistand, deine Bienchen, deinen Humor, deine Freundschaft und deine Liebe. XXXII