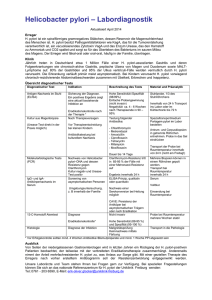
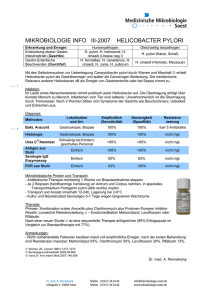
ZIP - FreiDok plus
Werbung

Aus der Sektion Experimentelle Anästhesiologie der Anästhesiologischen Universitätsklinik und dem Institut für Medizinische Mikrobiologie und Hygiene der Albert-Ludwigs-Universität Freiburg i.Br. Helicobacter pylori aktiviert den Transkriptionsfaktor AP-1 über Mitogen-aktivierte Proteinkaskaden und induziert die Protoonkogene c-fos und c-jun INAUGURAL DISSERTATION zur Erlangung des Medizinischen Doktorgrades der Medizinischen Fakultät der Albert-Ludwigs-Universtität Freiburg i. Br. vorgelegt 2003 von Tobias Meyer-ter-Vehn geboren in München Freiburg i.Br. Dekan : Prof. Dr. M. Schumacher 1. Gutachter : Frau Prof. Dr. rer. nat. H.L. Pahl 2. Gutachter : Prof. Dr. Dr. hc. H.E. Blum Promotionsjahr 2003 Meiner Familie gewidmet Der Mensch muß bei dem Glauben verharren, daß das Unbegreifliche begreiflich sei, er würde sonst nicht forschen. J.W. Goethe Teile dieser Arbeit wurden veröffentlicht : Meyer-ter-Vehn T, Covacci A, Kist M, Pahl HL (2000) Helicobacter pylori activates MAP Kinase cascades and induces expression of the proto-oncogenes c-fos and c-jun. J. Biol. Chem. 275 : 16064-16072. Meyer-ter-Vehn T, Fleckenstein M, Diehl S, Pahl HL (2002) Helicobacter pylori stimulates VEGF secretion from gastric epithelial cells through MAP kinase activation. submitted. Vorträge : Helicobacter pylori aktiviert MAP Kaskaden und induziert die Expression der ProtoOncogene c-fos und c-jun. 54. Jahrestagung der DGVS, 22.-25.September 1999, Leipzig Inhaltsverzeichnis 1 EINLEITUNG ....................................................................................................... 1 1.1 Helicobacter pylori (H. pylori)............................................................................................................... 1 1.2 Entwicklung der H.pylori Forschung.................................................................................................... 1 1.3 Epidemiologie .......................................................................................................................................... 2 1.4 Pathogentitätsfaktoren von H.pylori..................................................................................................... 2 1.4.1 Urease ............................................................................................................................................... 3 1.4.2 Motilität ............................................................................................................................................ 3 1.4.3 Adhäsion........................................................................................................................................... 4 1.4.4 Zytotoxin VacA ................................................................................................................................ 4 1.4.5 Zytotoxin assoziiertes Protein CagA ................................................................................................ 5 1.4.6 Pathogenitätsinsel CagI..................................................................................................................... 6 1.4.7 Molekularer Mimikry ....................................................................................................................... 6 1.5 H.pylori assoziierte Erkrankungen ....................................................................................................... 7 1.5.1 MALT Lymphom ............................................................................................................................. 8 1.5.2 Adenocarcinom des Magens............................................................................................................. 8 1.6 Reaktionen des Magenepithels auf die H.pylori Infektion .................................................................. 9 1.6.1 Interleukin 8 in der H.pylori Infektion.............................................................................................. 9 1.7 Der Transkriptionsfaktor AP-1 ........................................................................................................... 10 1.7.1 Die Komponenten des AP-1 Transkriptionsfaktors ........................................................................ 11 1.7.2 Transkriptionelle Aktivität.............................................................................................................. 11 1.8 Die MAP Kinase Kaskaden.................................................................................................................. 12 1.8.1 Allgemeine Wirkung von MAPK Kaskaden .................................................................................. 14 1.8.2 ERK-Signalkaskade ........................................................................................................................ 14 1.8.2.1 Zellproliferation........................................................................................................................ 15 1.8.2.2 Weitere Effekte der ERK-Kaskade........................................................................................... 15 1.8.3 JNK-Signalkaskade......................................................................................................................... 15 1.8.4 MAPK und Apoptose ..................................................................................................................... 16 1.9 Regulation von c-fos und c-jun ............................................................................................................ 16 1.9.1 Induktion der c-fos Expression ....................................................................................................... 16 1.9.2 Regulation von c-Jun ...................................................................................................................... 17 1.9.3 Bedeutung von c-Fos und c-Jun in der Onkogenese ....................................................................... 17 1.10 2 Zielsetzung der Arbeit .......................................................................................................................... 18 MATERIAL ........................................................................................................ 19 2.1 Geräte..................................................................................................................................................... 19 2.2 Chemikalien........................................................................................................................................... 20 2.3 Zellkulturmaterial................................................................................................................................. 21 2.4 Synthetische Oligonukleotide............................................................................................................... 22 2.5 Plasmide................................................................................................................................................. 22 2.6 Zellinien ................................................................................................................................................. 22 2.7 Zelltransfektion ..................................................................................................................................... 23 2.8 Zytokine & Inhibitoren ........................................................................................................................ 23 2.9 Antikörper ............................................................................................................................................. 23 3 METHODEN ...................................................................................................... 25 3.1 Prokaryontische Zellkultur.................................................................................................................. 25 3.1.1 Kultivierung von H.pylori............................................................................................................... 25 3.1.1.1 Anzucht auf Nährböden............................................................................................................ 25 3.1.1.2 Anzucht in Flüssigmedium ....................................................................................................... 26 3.1.1.3 Langzeitkonservierung ............................................................................................................. 27 3.1.2 Spezifiaktion von H.pylori.............................................................................................................. 27 3.1.3 Vorbereiten der Bakterien für ein Stimulationversuch ................................................................... 28 3.2 Eukaryontische Zellkultur ................................................................................................................... 28 3.2.1 Passage der Zellen .......................................................................................................................... 29 3.2.1.1 Adhärente Zellen ...................................................................................................................... 29 3.2.1.2 Nicht adhärente Zellen.............................................................................................................. 29 3.2.2 Langzeitkonservierung.................................................................................................................... 30 3.2.3 Bestimmung der Zellanzahl ............................................................................................................ 30 3.2.4 Ausplattieren der Zellen.................................................................................................................. 30 3.3 Molekularbiologische Methoden ......................................................................................................... 31 3.3.1 Plasmide.......................................................................................................................................... 31 3.3.1.1 Plasmidpräparation ................................................................................................................... 31 3.3.2 DNA-Quantifizierung ..................................................................................................................... 32 3.3.3 DNA – Agarosegelelektrophorese .................................................................................................. 32 3.3.4 Herstellung von cDNA Sonden ...................................................................................................... 32 3.3.5 Markieren der cDNA Sonden mit α-(32P)-dCTP ............................................................................ 33 3.3.6 Northern Blot.................................................................................................................................. 33 3.3.6.1 RNA-Präparation ...................................................................................................................... 33 3.3.6.2 Gelelektrophorese der RNA ..................................................................................................... 34 3.3.6.3 Nothern Blot ............................................................................................................................. 34 3.3.6.4 Hybridisierung .......................................................................................................................... 35 3.3.7 Transfektion.................................................................................................................................... 35 3.3.7.1 Vorbereiten der Zellen.............................................................................................................. 36 3.3.7.2 Zellernte.................................................................................................................................... 36 3.3.7.3 Bestimmung der Luciferase Aktivität....................................................................................... 36 3.4 Proteinbiochemische Methoden........................................................................................................... 37 3.4.1 Stimulation von Magenepithelzellen durch H.pylori ...................................................................... 37 3.4.2 Gewinnung der Gesamtzellextrakte................................................................................................ 37 3.4.3 Proteinbestimmung nach Bradford ................................................................................................. 38 3.4.4 Gelretardierungsassay / EMSA....................................................................................................... 39 3.4.4.1 Markieren von Oligonukleotiden mit γ-(32P)-dATP ................................................................. 39 3.4.4.2 EMSA Mix ............................................................................................................................... 40 3.4.4.3 Supershift.................................................................................................................................. 41 3.4.4.4 Kompetitionsassay.................................................................................................................... 41 3.4.5 Westernblott / Immunoblott............................................................................................................ 41 3.4.5.1 SDS-PolyAcrylamid-Gel-Elektrophorese (PAGE)................................................................... 42 3.4.5.2 Blotten ...................................................................................................................................... 43 3.4.5.3 Immundetektion........................................................................................................................ 44 3.4.5.4 Stripping ................................................................................................................................... 45 3.4.6 MAP Kinase Assay......................................................................................................................... 45 3.4.6.1 Immunpräzipitation .................................................................................................................. 45 3.4.6.2 Kinase Assay ............................................................................................................................ 46 4 4.1 ERGEBNISSE ................................................................................................... 47 Etablierung des AP-1 EMSA ............................................................................................................... 47 4.2 H.pylori induziert AP-1 in AGS Zellen ............................................................................................... 53 4.2.1 Identifikation der AP-Untereinheiten ............................................................................................. 53 4.2.2 Kinetik der AP-1 Aktivierung......................................................................................................... 55 4.2.3. Aktivierung von AP-1 durch Mutanten des H.pylori Stamm G27.................................................. 56 4.3 H.pylori aktiviert die ERK/MAP Kinase Kaskade ............................................................................ 57 4.3.1 Kinetik der ERK Aktivierung ......................................................................................................... 62 4.4 H.pylori aktiviert JNK.......................................................................................................................... 63 4.5 H.pylori induziert die Transkription von c-fos................................................................................... 64 4.6 H.pylori bewirkt c-Jun Phosphorelierung .......................................................................................... 67 4.7 Etablierung von Reportergen Versuchen ........................................................................................... 69 4.7.1 Etablierung der Transfektion von AGS-Zellen............................................................................... 69 4.7.2 Serumdeprivation............................................................................................................................ 72 4.8 H.pylori induziert AP-1 und SRF abhängige Genexpression............................................................ 75 4.9 H.pylori induziert keine MRE abhängige Genexpression ................................................................. 76 4.10 H.pylori und Cyclin D1 Expression...................................................................................................... 77 4.11 VEGF und H.pylori............................................................................................................................... 80 4.11.1 Regulation der Angiogenese durch VEGF...................................................................................... 80 4.11.2 H.pylori induziert die Transkription von VEGF in AGS Zellen..................................................... 81 5 DISKUSSION .................................................................................................... 83 5.1 H.pylori aktiviert den Transkriptionsfaktor AP-1 via MAPK Kaskaden ....................................... 83 5.1.1 ERK Signaltransduktionsweg ......................................................................................................... 83 5.1.1.1 H.pylori aktiviert MEK1/2 und ERK1/2................................................................................... 84 5.1.1.2 ERK Kaskade und Neoplastische Transformation.................................................................... 84 5.1.1.3 ERK-Signalkaskade als Regulator des Zellzyklus.................................................................... 85 5.1.2 JNK Signaltransduktionsweg.......................................................................................................... 88 5.1.2.1 H.pylori aktiviert JNK .............................................................................................................. 88 5.1.3 MAPK Kaskaden in der Interaktion zwischen Wirt und Erreger.................................................... 89 5.2 Welcher Bestandteil von H.pylori aktiviert die MAPK-Kaskaden und AP-1 ? .............................. 90 5.3 H.pylori aktiviert VEGF....................................................................................................................... 92 6 ZUSAMMENFASSUNG..................................................................................... 93 7 REFERENZEN .................................................................................................. 94 -1- 1. Einleitung H.pylori ist ein gramnegatives, spiralförmiges, 2-5 µm großes Stäbchenbakterium, das 1 Einleitung 1.1 Helicobacter pylori (H. pylori) unipolar 2-6 unbescheidete Flagellen mit terminalen Auftreibungen besitzt [Goodwin et al., 1990]. Seinen Namen verdankt es zum einen seiner spiralförmigen Morphologie (Helix), andererseits seiner präferentiellen Lokalisation im Pylorus des Magens. H.pylori besiedelt die Magenschleimhaut des Menschen und lebt in engem Kontakt mit dem Magenepithel, wo es durch die physiologische Schleimschicht vor dem sauren Milieu des Magens geschützt ist. Die Kolonisation ist wirtsspezifisch – der Mensch ist somit nach heutigem Wissensstand das einzige Reservoir der Spezies. Biochemisch zeichnet sich H.pylori primär durch seine Ureaseaktivtät aus, die ihm das Überleben im sauren Milieu des Magens ermöglicht. Weitere enzymatische Charakteristika sind die Produktion von Zytochromoxidase und γ-Glutamyl-Aminopeptidase, die in Verbindung mit der strikt mikroaerophilen Lebensweise der Identifikation des Erregers dienen. Darüberhinaus besitzt es alkalische Phosphatase Aktivität, kann H2S bilden und wird durch Cephalotin, nicht jedoch Nalixidinsäure im Wachstum gehemmt. Katalase und Superoxiddismutase (SOD) dienen der Inaktivierung von Sauerstoffradikalen. H.pylori kann weder Nitrate reduzieren noch Hippurate hydrolisieren oder Kohlenhydrate metabolisieren. 1.2 Entwicklung der H.pylori Forschung Im Jahre 1874 stellte Boetcher als erster die Hypothese auf, das chronische Magenulkus könne durch eine Infektion verursacht sein. Bizzozero berichtete 1893 über spiralförmige Organismen in Magenschleimhautbiopsien von Hunden [Bizzozero 1893]. Konjetzny beschäftigte sich mit dem Zusammenhang zwischen Ulkus und der chronischen Gastritis, in der er die Ursache für das Magenulkus sah. Erst die durch die chronische Gastritis geschädigte Magenschleimhaut ermögliche eine bakterielle Besiedelung, die die Gastritis verstärke und zum Ulkus führen kann [Konjetzny 1923]. Eine erhöhte Ureaseaktivität stellten Luck & Seth 1924 bei Patienten mit Magenbeschwerden fest. Die erste nähere Beschreibung von H.pylori gelang Steer & Collin-Jones elektronenmikroskopisch an endoskopisch entnommenen Magenbiopsien. Nachdem Marshall 1983 die erste Kultivierung von H.pylori -2- 1. Einleitung gelungen war, zeigte er 1985 im Selbstversuch die Kausalität zwischen Infektion und Magenbeschwerden. Anfangs wurde der Keim der Gattung Campylobacter zugeordnet. Hiervon unterscheidet er sich jedoch durch seine Begeißelung, die starke Ureaseaktivität, den niedrigeren GC-Gehalt seiner DNA (37%) [Kist 1994] und bzgl. der 16S rRNA-Sequenz, so daß das ursprünglich der Gattung Campylobacter zugeordnete Bakterium in Helicobacter pylori umbenannt wurde [Goodwin, 1989]. Die Entschlüsselung des Genoms von H.pylori Stamm 26695 gelang Tomb und seinen Mitarbeitern. Ein weiterer Helicobacter pylori Stamm – J99 – wurde von der Firma „Genome Therapeutics“ vollständig sequenziert, so daß Genomvergleiche zweier unterschiedlicher H.pylori Stämme ermöglicht wurden. 1.3 Epidemiologie H.pylori ist ein weltweit verbreiteter Keim. Während in den Industrieländern ca. 30%-50% der Bevölkerung infiziert sind [Pounder, 1995], liegt die Prävalenz in den Entwicklungsländern deutlich höher, so z.B. in Korea im Kindesalter bei mehr als 50% und bei Erwachsenen bis zu 90 % [Baik, 1990]. Eindeutige Risikofaktoren sind ein niedriger Hygienestandard und niedrige sozioökonomische Verhältnisse. 1.4 Pathogenitätsfaktoren von H.pylori Die Fähigkeit eines Krankheitserregers, Barrieren des Wirtes zu überwinden und somit eine ökologische Nische des Wirtsorganismus zu besiedeln, werden als Pathogenitätsfaktoren bezeichnet. Tabelle 1 gibt eine Übersicht über die Pathogenitätsfaktoren von H.pylori: Virulenzfaktor Beteiligte Gene Funktion Urease ureA, ureB Harnstoffspaltung ureI,ureE,ureF Akzessorische Gene ureG, ureH, hspA Beweglichkeit nixA Transport von Nickel flaA, flaB Flagellenfilament flgE Flagellen-Hakenprotein flbA Koordination der Flagellensynthese fliI Transport der Strukturproteine -3- 1. Einleitung Zytotoxin VacA Destruktion von Wirtszellen Immunstimulation Cag –Gene Induktion von Wirtsimmunantwort Superoxiddismutase SodB Inaktivierung von Sauerstoffradikalen Katalase KatA Mimikry GalE Synthese von Lewis x- & y- identischen Fucosyltransferase Oligosaccharidseitenketten im LPS alpA/B, babA-2 Bindung an Wirtszellen Adhäsion Tabelle 1 : Übersicht über wichtige Pathogenitätsfaktoren von H.pylori aus S.Bereswill, M.Kist : Mikrobiologie 7. Jg 1997 :131, sowie Habilitationsschrift von S.Bereswill: S.9 1.4.1 Urease H.pylori zeichnet sich insbesondere durch die Produktion großer Mengen an Urease aus, die Harnstoff in Kohlendioxid und Ammoniak spaltet. Die Ammoniak Moleküle können die Magensäure neutralisieren und so H.pylori die Kolonisation des Magens ermöglichen [Goodwin, 1986]. Desweiteren dient das gebildete Ammoniak dem Bakterium als Stickstoffquelle [Williams, 1996]. Das native Enzym ist ein 550 kDa Proteinkomplex und besteht aus 6 Dimeren der zwei Untereinheiten UreA und UreB. Für die enzymatische Aktivität ist Nickel als Kofaktor essentiell. Die Urease ist sowohl im Zytosol als auch an der Zelloberfläche lokalisiert, wo sie als immunogenes Protein die Migration von Monozyten und Granulozyten stimuliert [Mai, 1991]. Mutanten, die in den Genen für UreA und UreB inaktiviert wurden, konnten weder im Tierversuch die Magenschleimhaut kolonisieren [Eaton, 91], noch bei sauren pH-Verhältnissen überleben [Segal, 1992]. 1.4.2 Motilität Die unipolaren Flagellen, die aus den Proteinen FlaA und FlaB aufgebaut sind, verleihen H.pylori eine hohe Motilität und ermöglichen damit ein Durchdringen der Mukusschicht sowie eine Kolonisation des Magens. Flagellenlose Mutanten sind im Tierversuch nicht in der Lage, den Magen zu besiedeln [Eaton, 1992]. Somit stellt die Beweglichkeit sicherlich einen wichtigen Pathogenitätsfaktor dar. -41.4.3 1. Einleitung Adhäsion Nachdem die chemischen (Magensäure) und physikalischen Barrieren (Mukusschicht) überwunden sind, adhäriert H.pylori an das Magenepithel. Dabei bindet H.pylori v.a. an Polysaccharidstrukturen der Magenepithelzellen, so z.B. an das fibrilläre N- Acetylbeuraminyllactose bindende Hämaglutinin [Evans, 1988], sowie an ein 19,6 kD Adhäsin [Doig, 1992] und an Glycerollipidstrukturen [Lingwood, 1992]. Desweiteren adhäriert H.pylori mit dem BabA-2 Adhäsin an das Blutgruppenmarkerantigen Lewis b, das neben Erythrozyten auch auf Magenepithelzellen exprimiert wird [Ilver, 1998 & Boren, 1993]. Das Lewis b Antigen wird nur von Menschen der Blutgruppe 0 exprimiert – diese weisen eine erhöhte Inzidenz für Ulcera und Magenkarzinome auf. Die Mutation einer weiteren Gruppe von bakteriellen Adhäsinen - AlpA und AlpB - führt zum Verlust der Bindungsfähigkeit [Odenbreit, 1999]. Diese Adhäsine sind wahrscheinlich vom Lewis-b Antigen Rezeptor unabhängig. 1.4.4 Zytotoxin VacA Das Zytotoxin wird zunächst als 140 kDa Protein exprimiert, dessen Mittelteil sich nach Abspalten von N- und C-terminalen Enden außerhalb der Zelle in zwei 37 und 58 kDa Untereinheiten teilt, und sich dann zu einem 600 kDa Multimer zusammenlagert. Das vakuolisierende Zytotoxin bewirkt eine charakteristische Vakuolenbildung in der Wirtszelle. Dies führt zu Schleimhautläsionen und bis zur Nekrose des Epithels. Das Gen für das Toxin, vacA, weist einen starken Polymorphismus auf, ist aber bei allen H.pylori Stämmen vorhanden, wohingegen nur 50% aller H.pylori Stämme auch ein funktionelles Toxin sezernieren. Dabei lassen sich klar Subtypen in der N-terminalen Gensequenz (s1, s2) und im mittleren Bereich (m1, m2) abgrenzen. Stämme mit dem Genotyp s1 weisen eine stärkere Toxizität des gebildeten Proteins auf und korrelieren mehr mit einer Ulkuserkrankung, während der Genotyp s2 eher mit einer Gastritis assoziiert ist. Die Sekretion des VacA Toxins zeigt eine starke Korrelation mit der Expression des „Cytotoxin associated Gene“ CagA [Covacci, 1993], wobei dies durch die Nähe der beiden Gene und nicht durch eine kausale Verknüpfung verursacht wird, da eine selektive Deletion des CagA Gens nicht zu einer Reduktion der VacA Sekretion führt [Akopyants, 1998]. -51.4.5 1. Einleitung Zytotoxin assoziiertes Protein CagA Das 120-140 kDa große, stark immunogene CagA Protein wird von ca. 70-80% der europäischen H.pylori Stämme exprimiert. Zuerst wurde die lokale IgA und systemische IgG Antikörperantwort gegen CagA in Seren von Patienten 1988 von Apel beschrieben, sowie die Infiltration der Magenmukosa mit polymorphkernigen Granulozyten. Die Klonierung und molekulare Charakterisierung gelang Covacci 1993. Die Präsenz des Gens korreliert eng mit dem Auftreten von H.pylori assoziierten Folgeerkrankungen - dementsprechend unterscheidet man pathogene Typ I H.pylori Stämme, die CagA exprimieren, und weniger pathogene Typ II H.pylori Stämme, die CagA nicht produzieren. Hierbei liegt, anders als bei VacA, immer ein Verlust des cagA Gens vor. Cover konnte zeigen, daß das Ulcus duodeni zu 100% mit einer Infektion durch cagA+ Stämme einhergeht, wohingegen bei einer blanden Gastritis nur in 60,8% cagA+ Stämme gefunden wurden. Im Vergleich mit cagA- Infektionen wiesen Patienten, die mit einem cagA+ Stamm infiziert waren, eine erhöhte Inzidenz von Adenokarzinom im distalen Abschnitt des Magens sowie MALT-Lymphomen des Magens auf [Blaser 1995, Rudi 1997, Eck 1997]. Um die H.pylori Pathogenese in vitro zu studieren, wurden Bakterien mit Magenepithelzellinien kokultiviert. cagA+ Stämme, nicht aber cagAführten zu einer Sekretion des Chemokins Interleukin 8 [Crabtree 94]. Diese Ergebnisse bekräftigten frühere Beobachtungen, daß Magenbiopsien von Patienten mit einer H.pylori Infektion gegenüber nichtinfizierten Patienten deutlich erhöhte IL-8 Spiegel, aber auch TNFα und IL-6 Spiegel aufweisen [Crabtree 1991, 1993; Gionchetti 1994]. In vitro Versuche mit H.pylori Mutanten, bei denen selektiv das cagA Gen oder das vacA Gen deletiert wurde, zeigten jedoch keine Verminderung der IL-8 Sekretion in Vergleich zum Wildstamm [Crabtree 95, Shamra 95]. Cag stellt somit ein Marker für erhöhte Virulenz dar, ist jedoch nicht direkt in die Induktion von IL-8 involviert. Einen besseren Einblick in die CagAassoziierte Virulenz gewährte die Entdeckung der 40 kb großen Pathogenitätsinsel (PAI) in direkter Nachbarschaft zum cagA Gen [Censini 1996]. Einige Gruppen konnten kürzlich zeigen, daß H.pylori das CagA Protein über einen Typ IV Sekretionsmechanismus in die Wirtszelle transferiert und es dort zu einer Phosphorelierung von CagA kommt, was wiederum zu Veränderungen des Tyrosinphosphorelierungszustandes von Zellproteinen führt [Odenbreit, 2000; Stein, 2000]. -61.4.6 1. Einleitung Pathogenitätsinsel CagI Da CagA offenbar nicht essentiell für die Sekretion von IL-8 ist, suchten Tummuru et al. nach anderen IL-8 induzierenden Genen und fanden stromaufwärts von cagA die Gene picA und picB (permits induction of cytokines). Eine ausführlichere Untersuchung der benachbarten DNA-Sequenz ergab, daß ein 40 Kb großer heterologer DNA-Abschnitt insgesamt für über 25 Proteine kodiert (u.a. picA und picB), sowie charakteristische Eigenschaften einer Pathogenitätsinsel (PAI) aufweist [Censini 1996]. Die Proteine der PAI wurden fortlaufend mit cagA, cagB, … benannt, die Pathogenitätsinsel selbst als cag Region. Die cag Region ist durch eine Insertion eines transposablen Elementes (IS605) in zwei Abschnitte – cagI und cagII – unterteilt. Daß die PAI immer am Ende des Glutamtat-Razemase Gens inseriert ist, weist auf ein Rekombinationsereignis als Ursprung hin. H.pylori Stämme, die die PAI in ihrem Genom enthalten (cagA+/vacA+), werden als pathogene Typ I Stämme klassifiziert; Stämme, die nicht über diesen Genabschnitt verfügen, als Typ II Stämme, die als weniger pathogen gelten. Die genaue Funktion der in der PAI kodierten Gene ist noch Gegenstand der aktuellen Forschung; Homologievergleiche zu anderen bakteriellen Genen ergaben u.a. eine Ähnlichkeit des cagE Gens zu dem Toxintransportprotein PtlC von Bordetella pertussis und dem virulenzassoziierten VirB4 Protein von Agrobacterium tumefaciens auf. Es wird daher vermutet, die in der cag Region liegenden Gene könnten für einen membrangebundenen Komplex kodieren, der als Sekretionsapparat fungiert [Censini 1996, Covacci 1997]. 1.4.7 Molekularer Mimikry Als ein weiterer Pathogenitätsfaktor von H.pylori wird ein Autoimmunprozess diskutiert: bei bis zu 50% der Patienten mit einer chronischen H.pylori Infektion lassen sich Antikörper gegen Magengewebe nachweisen [Negrini, 1991 & Faller G, 1997]. Da das bakterielle Lipopolysaccharid (LPS) von mehr als 85 % der H.pylori Stämme Seitenketten besitzt, die mit den Blutgruppenantigenen Lewis x und Lewis y des Menschen identisch sind [Appelmelk, 1997], wurde eine Kreuzreaktivität zwischen dem H.pylori LPS und den Lewis x/y Antigenen, die auf dem Magenepithel exprimiert werden, diskutiert. Im Tierversuch generierten mit H.pylori infizierte Mäuse Antikörper, die mit menschlicher Magenschleimhaut reagierten [Appelmelk,1996]. Neuere Arbeiten bezweifeln wieder einen -7- 1. Einleitung direkt kausalen Zusammenhang zwischen dem Lewis-Polymorphismus und dem Auftreten antigastraler Antikörper [Faller, 1998]. 1.5 H.pylori assoziierte Erkrankungen Die H.pylori-Infektion kann sich vielfältig präsentieren. Während viele Infizierte asymptomatisch bleiben oder eine geringgradige Oberflächengastritits entwickeln, bildet sich bei anderen Individuen eine antrumbetonte Gastritis oder eine Pangastritis aus, kommt es zum Ulcus duodeni oder seltener Ulcus ventriculi. Auch neoplastische Erkrankungen wie das MALT-Lymphom und selten das Adenokarzinom des Magens sind mit der H.pylori Infektion assoziiert. Die Gastritis stellt dabei die Vorstufe für Ulcus ventriculi und duodeni sowie für die Entwicklung von Neoplasien dar [Blaser 1992]. Die vielfältige Präsentation dieser Infektion ist zum einen auf die große Variabilität des Erregers zurückzuführen, zum anderen auf die unterschiedliche Immunsituation und genetische Prädisposition des Infizierten. Die häufigste Manifestation der H.pylori Infektion ist die chronische Gastritis vom Typ B (bakteriell-infektiös). Es handelt sich um eine oft asymptomatisch verlaufende Oberflächengastritits, die jedoch in eine chronisch-atrophische Gastritis übergehen kann. Histologisch ist sie gekennzeichnet durch Infiltration der Magenmukosa mit neutrophilen Granulozyten, Lymphozyten und Plasmazellen, Erosionen, Ersatz des Oberflächenepithels durch Regenerationsepithel, intestinale Metaplasie, Depletion der Schleimschicht bis hin zur Atrophie der Magenmukosa [Leung, 1992]. Die Entzündung ist meist verstärkt im Antrumbereich; als Erklärung wird angenommen, daß die Parietalzellen des Korpus durch Säureproduktion die Kolonisation für H.pylori erschweren. Die Prävalenz einer H.pylori Infektion bei Ulcus duodeni beträgt 95 %, bei Ulcus ventriculi 70%. Das Risiko ein Ulcus duodeni zu entwickeln wird durch Infektion mit H.pylori um das 20-fache gesteigert [Stolte, 1992]. Pathophysiologisch werden zwei Varianten unterschieden : zum einen führt eine Infektion des Korpusbereiches zu einer Reduktion der Säuresekretion, was einer Ausbreitung der Infektion auf den ganzen Magen Vorschub leistet. Es kann zu atrophischen Veränderungen und dem Ulcus ventriculi kommen. Ein Fortbestehen der Atrophie wird als neoplastische Prädisposition gewertet [Blaser, 1992]. Eine prädominante Infektion des Antrums führt zu einer vermehrten Aktivierung der Gastrinsekretion mit Gastrinämie und konsekutiv zu einer Hyperchlorhydrie, was wiederum zum Ulcus duodeni prädisponiert. Für die verstärkte Gastrinsekretion durch H.pylori gibt es -8- 1. Einleitung mehrere Theorien : das von H.pylori produzierte Ammonium erhöht den pH Wert und unterbricht somit die negative Rückkopplung der Säureproduktion auf die Gastrinproduktion in den G-Zellen des Antrum. Auch bewirkt das von H.pylori produzierte N-α-Methylhistamin eine ähnlich deutliche Stimulierung der Säurebildung in den Parietalzellen [Beales, 1997]. Desweiteren stimuliert H.pylori in vitro auch die Synthese der Histidin Decarboxylase, die für die Synthese von Histamin ein Schlüsselenzym darstellt [Wessler, 2000]. Histamin wiederum stellt einen wichtigen aktivierenden Stimulus für die Magensäureproduktion dar. 1.5.1 MALT Lymphom Die chronische Infektion mit H.pylori führt durch die Dauerstimulation des lokalen Immunsystems zur Ausbildung von Mukosa-assoziiertem lymphoidem Gewebe (MALT), bestehend aus reaktiven T-Zellen, Plasmazellen und anderen B-Zellen – histologisch gleicht es Lymphfollikeln aus Lymphknoten [Parsonnet, 1994]. Die MALT Lymphome, die man an der Stelle der H.pylori Infektion, v.a. im Antrum, findet, resultieren aus der klonalen Expansion von genetisch mutierten B-Zellen [Wotherspoon, 91]. Dabei ist der Übergang von H.pylori Gastritis mit lymphoiden Infiltraten zum frühen Magen MALT-Lymphom oft fließend und initial oft schwer zu unterscheiden. Die Eradikation von H.pylori führt zur Regression niedrigmaligner MALT-Lymphome des Magens [Wotherspoon, 1993 & Stolte, 1992]. 1.5.2 Adenocarcinom des Magens Obwohl die Prävalenz des Magenkarzinoms zurückgeht, ist es weltweit die zweithäufigste bösartige Neubildung nach dem Lungenkrebs [Neugut, 1996]. Mehrere epidemiologische Studien zeigten, daß eine chronische Infektion mit H. pylori das Risiko für die Entwicklung eines Adenokarzinoms des Magens um das 6-fache erhöht [Parsonnet, 1994; Eurogast Study Group, 1995]. Deshalb klassifizierte die IARC/WHO 1994 H.pylori als Klasse I Karzinogen. Neuere Untersuchungen am Tiermodell ergaben, daß die chronische Infektion mit H.pylori neoplastische Veränderungen im Pylorusbereich induzieren kann. Nach 62 Wochen H.pylori Infektion entwickelten 10 von 27 (= 37%) mongolischen Meerschweinchen ein Adenocarcinom des Magens, wohingegen keines der Kontrolltiere (n=30) ein Malignom aufwies [Watanabe, 1998; Honda, 1998; Sawada 1998]. Die pathologischen Veränderungen -9- 1. Einleitung schritten von einer regenerativen Hyperplasie der Magenschleimhaut (nach 6 Wochen) über die Entstehung einer intestinalen Metaplasie (nach 26 Wochen) und von hyperplastischen Polypen (nach 52 Wochen), bis hin zum Auftreten eines Adenokarzinoms (nach 62 Wochen) bei einem Drittel der Tiere [Watanabe, 1998]. Alle diese Veränderungen werden auch bei der H.pylori Infektion des Menschen beobachtet [Leung, 1992]. Der molekulare Mechanismus jedoch, der zur malignen Transformation durch H.pylori führt, ist noch nicht aufgeklärt. Einige Studien zeigten eine erhöhte Zellproliferation in Magenbiopsien von Patienten mit H.pylori Infektion verglichen mit Patienten ohne Infektion [Peek, 1997; Fraser, 1994; Bechi, 1996; Lynch, 1995; Harvard, 1996]. Für die Erhaltung der Gewebeintegrität ist bei einer erhöhten Zellproduktion auch ein erhöhter Zellabgang notwendig [Hall, 1994]. Studien, die sich mit der Apoptose bei H.pylori Infektion beschäftigten, fielen unterschiedlich aus : einige Forschergruppen berichten über eine erhöhte Apoptoserate der Magenmukosa bei Infektion mit cagA+ H.pylori [Moss, 2001; Yamaguchi, 2000; Jang, 2000], und spekulieren, daß die erhöhte Zellproliferation der Magenschleimhaut als Antwort auf die gesteigerte Apoptoserate zu verstehen ist. Andere Untersuchungen erbrachten eine gesteigerte Proliferation mit abgeschwächter Apoptoserate des Magenepithels bei Infektion mit cagA+ H.pylori Stämmen [Peek, 1997, Rokkas, 1999]. Eine erhöhte Proliferationsrate mit einer erniedrigten Apoptoserate könnte zu unkontrolliertem Zellwachstum führen [Peek 1997]. Einigkeit herrscht darüber, daß cagA+ H.pylori in vitro bei der Kokultivation mit AGS Zellen in diesen Apoptose induziert [Wagner, 1997; Rudi, 1998; Peek, 1999]. Erhöhter Zellumsatz und chronische Schädigung des Magenepithels sowie die Entwicklung einer atrophischen Gastritis werden als Ursachen für die maligne Transformation diskutiert. 1.6 1.6.1 Reaktionen des Magenepithels auf die H.pylori Infektion Interleukin 8 in der H.pylori Infektion Die durch H.pylori verursachte chronische Gastritis vom Typ B ist gekennzeichnet durch Infiltration der Magenmukosa mit neutrophilen Granulozyten, Lymphozyten und Plasmazellen. Diese Histologie und das Vorhandensein von sowohl akuten wie chronischen entzündlichen Komponenten legt die Präsenz eines löslichen Mediators nahe, der diese Zellen anlocken kann. Das CXC-Chemokin Interleukin 8 ist ein starker chemotaktischer Faktor und Aktivator von neutrophilen Granulozyten und Lymphozyten. Gastrointestinales Epithel sekretiert IL-8 bei Kontakt mit pathogenen Bakterien [Eckmann, 1993]. Die - 10 - 1. Einleitung Magenschleimhaut zeigt bei H.pylori Infektion eine verstärkte Interleukin 8 Expression [Gionchetti, 1994; Crabtree, 1995b]. Die Kokultivation von AGS Zellen mit cagA+ Stämmen, nicht aber cagA- führten zu einer Sekretion des Chemokins Interleukin 8 [Crabtree, 94; Shamra 95], wobei cagA deletierte Mutanten nach wie vor IL-8 Sekretion induzieren können [Crabtree 95; Shamra 95], cagA also nur ein Marker für diese Fähigkeit und nicht Verursacher hierfür ist. Ein Modell für die durch H.pylori verursachte Gastritis ist also, daß das Bakterium die Sekretion von IL-8 aus der Magenmukosa bewirkt, was zur Infiltration der Magenschleimhaut mit Entzündungszellen führt. Wie kommt es nun zu der Interleukin 8 Induktion ? Der humane IL-8 Promoter enthält Bindungsstellen für NF-κB (-80 bis –70), NF-IL6 (-94 bis –81), sowie für AP-1 (-126 bis – 120) (Abb. 1). Abb 1: Bindungsstellen für Transkriptionsfaktoren im humanen IL-8 Promoter (aus Peek, 2001) Reportergenversuche mit dem humanen IL-8 Promoter, in den die jeweiligen Bindungsstellen für NF-κB, NF-IL6 und AP-1 mutiert wurden, ergaben folgendes : NF-κB ist unabdingbar für die Sekretion von IL-8, die Mutierung der AP-1 Bindungsstelle führt zu einer Reduktion der Reporteraktivität auf 30%, wohingegen NF-IL6 keinen wesentlichen Beitrag zur Aktivierung des IL-8 Promoters durch H. pylori zu leisten scheint [Aihara, 1997]. A. Münzenmaier konnte 1997 zeigen, daß die Kokultivation von AGS Zellen mit cagA+ H.pylori Stämmen, nicht aber mit cagA- zur Aktivierung von NF-κB führt, und dieses mit der Sekretion von IL-8 korreliert. Dabei sind die meisten Gene der Pathogenitätsinsel notwendig für die NF-κB Induktion, nicht jedoch cagA, cagF und cagN [Glocker, 1999]. Desweiteren führt die Stimulation von AGS-Zellen mit cagA+ H.pylori Stämmen zu Neuordnung des Zytoskeletts sowie Phosphorelierung eines 145kDa Protein an einer TyrosinSeitenkette [Segal, 1997], welches später als von H.pylori in die Wirtszelle transloziertes cagA identifiziert wurde [Odenbreit, 2000; Stein, 2000]. 1.7 Der Transkriptionsfaktor AP-1 Der Transkriptionsfaktor AP-1 besitzt eine Schlüsselstelle in der Regulierung von Zellproliferation und maligner Transformation, sowie in der Zelldifferenzierung [Karin, - 11 - 1. Einleitung 1995]. Zuerst wurde er als Transkriptionsfaktor beschrieben, der an ein essentielles cisElement des humanen Metallotheonin IIA Promoters bindet [Lee, 1987a]. Kurze Zeit später wurde AP-1 auch als Transkriptionsfaktor für die Tetra Phorbol Acetat (TPA) - induzierte Expression von etlichen zellulären und viralen Genen beschrieben, so z.B. humane Kollagenase, Stremolysin, Interleukin 2, oder das virale SV40 Protein [Angel, 1989]. TPA ist ein hoch potenter Tumorpromoter und aktiviert die Proteinkinase C [Nishizuka, 1984]. Dabei bindet AP-1 an die palindromische DNA-Sequenz 5‘-TGA G/C TCA-3‘, die auch TPA Responsive Element (TRE) genannt wird [Angel 1989; Lee, 1987b]. Weitere Aktivatoren des AP-1 Transkriptionsfaktors sind neben TPA u.a. Wachstumsfaktoren, T-Zell-Aktivatoren, Neurotransmitter und UV-Bestrahlung [Angel, 1991]. 1.7.1 Die Komponenten des AP-1 Transkriptionsfaktors AP-1 besteht aus Homo- oder Heterodimeren der Onkogenfamilien Jun (c-Jun, JunB, JunD), Fos (c-Fos, FosB, Fra1, Fra2) und ATF (ATF2, ATF3/LRF1, B-ATF), die alle der bZIP (basic region leucine zipper) Familie von DNA-bindenden Proteinen angehören [zur Übersicht: Angel, 1991; Karin, 1995; Karin 1997]. Jun Proteine bilden sehr stabile Heterodimere mit Proteinen aus der Fos- und ATF-Familie, können aber auch Jun-Jun Homodimere bilden. Dabei binden Jun-Fos und Jun-Jun Komplexe präferentiell an das TPA-Responsive Element, während Jun-ATF und ATF-Homodimere eine hohe Affinität zu dem cAMP Responsive Element (CRE) haben. Die Klasse der bZIP Proteine, die mit Jun und Fos Proteinen Heterodimere bilden kann, wächst laufend – so kamen u.a. die Maf-Familie und das Neural Retina Specific Gene Product Nrl neu zu der Klasse der möglichen Dimerisierungspartner von Jun und Fos hinzu [Karin, 1997]. Dabei interagieren die Leucin Zipper Regionen, die meist Cterminal lokalisiert sind, durch hydrophobische Wechselwirkung und führen zur Dimerisierung, wohingegen die N-terminale Region für die sequenz-spezifische DNABindung verantwortlich ist [Glover, 1995]. Die DNA-bindenden Regionen weisen dabei eine hohe Homologie zu dem Hefe-Transkriptionsfaktor GCN-4 [Angel, 1991] auf. 1.7.2 Transkriptionelle Aktivität Die transkriptionelle Aktivität von AP-1 in ruhenden Zellen ist gering, meist liegen Jun-Jun Homodimere mit einer schwachen Transaktivierungspotenz vor. Im Gegensatz zu den Transkriptionsfaktoren NF-κB und den Glucocorticoid Rezeptoren ist aber meist eine basale - 12 - 1. Einleitung Transaktivierung vorhanden; es reguliert also sowohl basale wie induzierbare Transkription. Insbesondere proliferierende Zellinien, die in serumhaltigem Medium gehalten werden, weisen eine deutliche basale AP-1 Aktivität auf. Die meisten Gene, die für AP-1 Komponenten kodieren, verhalten sich wie „immediate early genes“, d.h. Gene, deren Transkription nach Stimulation schnell aktiviert wird, unabhängig von de-novo Proteinsynthese [Karin, 1995]. AP-1 Aktivität wird sowohl transkriptionell als auch posttranskriptionell reguliert. 1.8 Die MAP Kinase Kaskaden Eine wesentliche Rolle spielen dabei die sogenannten Mitogen Activated Protein (MAP) Kinase Kaskaden. Diese Signaltransduktionswege bestehen aus drei sequentiell agierenden Proteinkinasen : eine MAP Kinase Kinase Kinase (MAPKKK) phosphoreliert und aktiviert eine MAP Kinase Kinase (MAPKK), die wiederum ihrerseits eine MAP Kinase (MAPK) phosphoreliert und damit aktiviert. Die aktivierte MAP Kinase aktiviert andere Effektor Proteinkinasen, MAPK-activated protein kinases (MAPKAPK), oder transloziert in den Zellkern, um dort Transkriptionsfaktoren zu phosphorelieren und damit die Transkription eines Gens zu induzieren. Dieses Prinzip einer dreistufigen Proteinkinase Kaskade ist in der Evolution stark konserviert. Vieles über die Regulation von MAPK Kaskaden wurde an den homologen Enzymen der Hefe entdeckt : So aktiviert in der Hefe ein Phermon über die Bindung an seinen Rezeptor ein membranständiges G-Protein und induziert damit die Anlagerung des G-Protein bindenden Proteins STE20. Das aktivierte STE 20 bindet dann die MAPKKK STE11 (entspricht in humanen Zellen c-Raf), wodurch STE11 in Membrannähe lokalisiert und durch membranständige Kinasen aktiviert wird. Die Serin/Threonin Kinase STE11 ihrerseits aktiviert die Threonin/Tyrosin MAPK Kinase STE7 (entspricht in humanen Zellen MEK1/2), die wiederum durch Phosphorelierung des Thr-X-Tyr Motiv die MAPK KSS/Fus3 (entspricht in humanen Zellen ERK1/2) aktiviert. Diese aktivierte Kinase transloziert in den Zellkern und reguliert durch Phosphorelierung die Aktivität von Transkriptionsfaktoren und somit die Genexpression, was schließlich zur Zellteilung oder Differenzierung der Hefezellen führt. - 13 - Abb. 2 : 1. Einleitung Übersicht über MAPK in der Hefe (aus Minden, 1997) Säugetiere exprimieren mindestens vier verschiedene MAPK-Kaskaden, wobei die ERK1/2Signalkaskade, die JNK/SAPK-Kaskade und die p38 Kaskade besser untersucht sind, wohingegen über die Regulation und physiologische Bedeutung der ERK5-Signalkaskade noch nicht viel bekannt ist. Die einzelnen Signalkaskaden weisen dabei eine gewisse Beeinflussung untereinander, den sogenannten „Cross-Talk“, auf. So aktiviert z.B. die MAPKK MKK4 sowohl JNK als auch p38. Verschiedene Mechanismen gewährleisten die Spezifität der einzelnen Kaskaden. Zum einen wurden „Gerüst“-Proteine JIP1 und MP1 entdeckt, die beteiligte Proteinkinasen – JNK1/2 / MKK7 / MLK1 bzw. ERK1/2 / MEK-1 / c-Raf1 - in räumliche Nähe bringen. Desweiteren besitzen die Kinasen spezifische Bindungsdomänen, so daß sie jeweils mit den passenden Upstream- oder Downstream- Kinasen interagieren. Darüberhinaus besitzen die Kinasen Substratspezifität : so phosphoreliert MEK1/2 spezifisch das TEY Motiv von ERK1/2, während MKK4 bzw. MKK3/6 spezifisch für die Motive TPY von JNK bzw. TGY von p38 sind. - 14 - Abb.3 : 1.8.1 1. Einleitung Übersicht der verschiedenen MAPK Kaskaden des Menschen (aus Whitmarsh, 1996) Allgemeine Wirkung von MAPK Kaskaden Die drei MAP Kinasen ERK, JNK und p38 können ETS Transkriptionsfaktoren phosphorelieren, so insbesondere Elk-1 [Zink, 1995; Rinegaud, 1996], der an die SRE Bindungsstelle im c-fos Promoter bindet, und so die c-fos Expression reguliert [Hipskind, 1991]. 1.8.2 ERK-Signalkaskade Die ERK-Signalkaskade wird am stärksten durch Mitogene stimuliert. Als Paradebeispiel dient der Epidermal Growth Factor (EGF), der an seinen Rezeptor EGF-R bindet und somit die Rezeptor Tyrosin Kinase (RTK) von EGF-R aktiviert. Die Autophosphorelierung des Rezeptors bewirkt das Anlagern der Adapter Proteine Grb2 und Sos; letzteres wiederum katalysiert den Austausch von GDP zu GTP an dem kleinen GTP bindenden Protein ras. Anschließend erfolgt die Aktivierung der ras / c-Raf / MEK1/2 / ERK1/2 - 15 - 1. Einleitung Signaltransduktionswege analog zur Aktivierung in der Hefe (s.o.). Ein weiterer Induktor der ERK-Signalkaskade ist Tetraphorbolacetat (TPA), das als Diacylglycerol (DAG) Analogon Proteinkinase C aktiviert, die wiederum über ras-abhängige Signalwege die ERK-Kaskade anschaltet. 1.8.2.1 Zellproliferation Mitogene bewirken die Zellteilung, und so ist eine Hauptaufgabe von ERK die Regulierung der Zellproliferation. Zum einen stimuliert ERK die DNA Synthese durch Phosphorelierung der Carbamyl Phosphatase Synthetase II, ein Schlüsselenzym in der Pyrimidin Synthese [Graves, 2000]. Indirekt stimuliert ERK die Zellproliferation über den Transkriptionsfaktor AP-1, der zu einer Cyclin D1 Induktion führt [Lavoie, 1996]. Treinieset et al. berichten, daß dies allein für die Induktion von DNA-Synthese nicht hinreichend ist, sondern zusätzlich der Aktivierung von Phosphatidyl-3-OH Kinase bedarf, die wiederum autokrin durch AP-1 induzierte Wachstumsfaktoren aktiviert werden kann [Treinies, 1999]. Ein weiteres Ziel von ERK ist die pp90 Ribosomal S6 Kinase (RSK), die durch Inaktivierung von MYT1, einer zellzyklusinhibitorischen Kinase, die Zellteilung fördert [Palmer, 1998]. 1.8.2.2 Weitere Effekte der ERK-Kaskade Während eine transiente Aktivierung von ERK in PC12 Zellen zur Zellproliferation führt, bewirkt eine anhaltende Aktivierung der ERK-Kaskade mit Akkumulation von aktivierten ERK im Zellkern eine Differenzierung dieser Zellen [Marshall, 1995]. Auch scheint ERK an der Regulierung der Eicosanoide beteiligt zu sein, da es zytosolische Phospholipase A2 aktivieren kann und so zur Freisetzung von Arachnoidonsäure beiträgt [Lin, 1993]. 1.8.3 JNK-Signalkaskade Jun N-Terminal Kinase (JNK) wurde als Kinase kloniert, die den Transkriptionsfaktor Jun NTerminal an den Seitenketten Ser-63 und Ser-73 phosphoreliert und damit seine transkriptionelle Aktivität entscheidend erhöht [Hibi, 1993]. Der JNK Signaltransduktionsweg wird v.a. durch zellulären Streß aktiviert, so z.B. UV-Bestrahlung, Hitzeschock und die proinflammatorischen Zytokine IL-1 und TNF-α [Karin, 1995; Minden 1997]. Desweiteren - 16 - 1. Einleitung kann JNK auch durch die Wachstumsfaktoren EGF und NGF aktiviert werden; aktiviertes ras potenziert die UV-induzierte JNK-Aktivierung. Der Signalweg wird über Rac und Cdc42 vermittelt, zwei Mitglieder der Rho-Familie, die zu den kleinen GTP-bindenden Proteinen gehört. Aktiviertes Cdc42 bindet an PAK65 und aktiviert diese damit. PAK65 wiederum vermag die MAPKKK MEKK-1 zu stimulieren. Es folgt die bekannte dreistufige MAPKKaskade MEKK1 - JNKK/MKK4 – JNK. Neben Jun phosphoreliert JNK auch die Transkriptionsfaktoren Elk-1 und ATF-2 [Zinck, 1995]. 1.8.4 MAPK und Apoptose Als Faustregel gilt, daß ERK mit Zellüberleben assoziiert ist, während JNK und p38 eher zur Induktion der Apoptose führen [Xia, 1995]. Natürlich gibt es hierzu Ausnahmen, und die tatsächliche Rolle der verschiedenen MAPK in vivo bezüglich Zellüberleben ist stark zelltypspezifisch. Längere JNK-Aktivierung führt insbesondere in bestimmten neuronalen Zellen zu Apoptose [Xia, 1995]. Auch ist JNK notwendig für die UV-induzierte Apoptose in Fibroblasten [Tournier, 2000]. 1.9 1.9.1 Regulation von c-fos und c-jun Induktion der c-fos Expression Der c-fos Promoter besitzt drei Hauptbindungsstellen für Transkriptionsfaktoren : das SerumResponse Element (SRE), das cAMP-Response Element (CRE) und das sis-induzierbare Element (SIE) (Abb.4) [Whitmarsh, 1996]. Hier soll nur auf das SRE Element eingegangen werden, das im Rahmen dieser Arbeit näher untersucht wurde. Das SRE Element bindet den dimeren Serum Response Factor sowie einen Ternary Complex Factor (TCF). TCF gehören zu der Familie von DNA-bindenden Proteinen mit einer ETSDomäne, wie z.B. Elk-1 und SAP1. Wie weiter oben erwähnt, kann Elk-1 von allen drei MAP Kinasen phosphoreliert und damit aktiviert werden. - 17 - Abb. 4 : 1.9.2 1. Einleitung Regulation des c-fos und c-jun Promoters durch MAPK (aus Minden, 1997) Regulation von c-Jun Der Transkriptionsfaktor Jun wird sowohl transkriptionell als auch posttranskriptionell reguliert. Der Promoter von c-Jun enthält zwei TPA-Responsive Elemente (TRE), an die bevorzugt c-Jun/c-Fos oder c-Jun/ATF-2 Heterodimere und c-Jun/c-Jun Homodimere binden (Abb.4). In ruhenden Zellen liegen einige Jun-Jun Homodimere mit schwacher transkriptioneller Aktivität an dem TRE. Neu gebildetes c-Fos bildet mit dem in ruhenden Zellen präexistierenden c-Jun c-Fos/c-Jun Heterodimere mit einer deutlich erhöhten Transaktivierungspotenz. Durch positive Rückkopplung an das TRE des c-jun Promoters führt dies zu einer verstärkten c-Jun Expression. Posttranskriptionell wird c-Jun an den Seitenketten Ser-63 und Ser-73 von JNK phosphoreliert und so seine transkriptionelle Aktivität entscheidend angehoben [Hibi, 1993; Dérijard, 1994]. 1.9.3 Bedeutung von c-Fos und c-Jun in der Onkogenese Unkontrollierte Expression von c-Fos und c-Jun kann zur neoplastischen Transformation führen [Curran, 1984; Miller 1984]. Die viralen Äquivalente v-Fos und v-Jun werden durch - 18 - 1. Einleitung die murinen FBJ und FBR Viren transduziert, die ein Osteosarkom induzieren können [Curran, 1982a&b]. Transgene Mäuse, die c-Fos überexprimieren, entwickeln Osteo- und Chondrosarkome. Saez konnte zeigen, daß c-Fos notwendig für die maligne Progression von Hauttumoren ist. Die Behandlung von kultivierten Fibroblasten mit c-fos antisense RNA oder die Injektion von Antikörpern gegen c-Fos stoppte das Zellwachstum [Raibowol, 1988; Holt, 1986]. Fibroblasten, die von c-jun -/- Knockout Mäusen gewonnen wurden, wiesen Störungen in der Proliferation auf [Johnson, 1993], was eine Beteiligung von c-Jun an der Zellproliferation in vivo nahe legt. 1.10 Zielsetzung der Arbeit Da AP-1 eine solche Schlüsselposition in der Regulation der Zellproliferation besitzt, und die Infektion mit H.pylori Hyperproliferation der Magenschleimhaut induziert (s. oben), sowie manchmal zum Magenadenokarzinom führt, sollte in dieser Arbeit die Beteiligung des Transkriptionsfaktors AP-1 bei der H.pylori Infektion in einem in vitro System mit der Magenepithelzellinie AGS untersucht werden. Desweiteren sollte geklärt werden, ob H.pylori AP-1 aktivieren kann, und so synergistisch mit der Induktion von NF-?B die Interleukin-8 Expression induzieren kann. Zuerst wurde ein DNA-Bindungsassay für AP-1 in Form eines Gelretardierungsassays etabliert. Daraufhin sollten die Signalwege, insbesondere die MAP Kinase Kaskaden, durch die H.pylori AP-1 aktiviert, näher betrachtet werden. Hierzu wurden mehrere Ansätze gewählt, so u.a. der Einsatz von spezifischen Inhibitoren, in vitro Kinaseassays und Westernblots. Da die Schwere der H.pylori Infektion und das Risiko Magenkrebs zu entwickeln von dem einzelnen Erregerstamm abhängt und mit dem Vorhandensein der Pathogenitätsinsel cag korreliert, sollte die Abhängigkeit der AP-1 Induktion von der Präsenz der einzelnen Gene der Pathogenitätsinsel näher untersucht werden. Auch sollte die transkriptionelle Aktivität der induzierten AP-1 Komplexe im Reportergenassay gemessen werden. Ein weiterer Teil der Arbeit beschäftigte sich mit der Suche nach weiteren durch H.pylori aktivierten Zielgenen neben IL-8, die durch die Aktivierung der MAPK Kaskaden und die Induktion von AP-1 beeinflußt werden. Hierbei wurden das den Zellzyklus regulierende Gen Cyclin D1, sowie das in der Heilung der Ulcera wichtige Angiogenese Gen VEGF näher betrachtet. - 19 - 2. Material 2 Material 2.1 Geräte 1.5 ml Reaktionsgefäße Eppendorf, Hamburg Falcon 15ml Reaktionsgefäß Becton Dickinson / UK Falcon 50ml Reaktionsgefäß Becton Dickinson / UK Filme, X-OMAT TM AR. # 853 2665 Kodak, Rochester/ USA Filmentwicklermaschine, Curix 60 Agfa-Geveart, Leverkusen Geiger-Zählrohr WellhöferDosimetrie, Schwarzenbruck Gelkammern Thoma, Freiburg Geltrockner Bio-Rad, München Hybond-N Nylon-Membran Amersham Life Science Immobilon-P Transfer-Membran Millipore, Bedford/Massachusetts Laminar Flow Sterilbank Heraeus, Stuttgart Magnetrührer IKA-Labortechnik, Staufen Millex GP 0,22 µm Filter Unit #P25390 Millipore, Bedford, MA Microbank Container ProLab Diagnostik, Augsburg Mikroskop Hund, Wetzlar Mikrospin G-25, #27-5325-01 Amersham Pharmacia Biotech Mikrospin G-200, #27-5120-01 Amersham Pharmacia Biotech Photometer Pharmacia, Freiburg Power-supply Roth, Karlsruhe Pipettenspitzen Roth, Karlsruhe Qiagen Plasmid Maxi Kit, #12162 Qiagen, Hilden Rotator Wheels PSI (pool of scientific instruments), Laudenbach Semi-dry-Elektroblotter Owl scientific Inc. Woburn, USA Spektrophotometer Beckmann, Palo Alto, USA DU 640 Thermomixer Eppendorf, Hamburg Waagen Mettler-Toledo, Gießen Whatman 3 mm Papier Schneider&Düll, Dassel Vortexer Bender&Hobein, Zürich/ Schweiz - 20 Zellinkubator Zentrifugen 2.2 Heraeus, Stuttgart Biofuge fresco Heraeus, Stuttgart Biofuge 13 Heraeus, Stuttgart Megafuge 1.0 Heraeus, Stuttgart Beckmann J2-21 Beckmann, Palo Alto / USA Chemikalien Aprotinin Sigma, Deisenhofen Acrylamid-Lösung, 30% (w/v) Roth, Karlsruhe Polyacrylamid; 0,8% (w/v) Bisacrylamid Roth, Karlsruhe Ammoniumperoxodisulfat (APS), Merk, Darmstadt # 529K791001 [γ-32P]-ATP, 3000Ci/mmol Amersham Pharmacia Biotech Bactor Dextrose Bactor Peptamin Bactor Trypton Bactor Yeast Extrakt Betamerkaptoethanol Difco Difco Difco Difco Roth, Karlsruhe Borsäure Roth, Karlsruhe Bradford Protein Assay, # 500-0006 Bio-Rad, München Bromphenolblau Roth, Karlsruhe Bovines Serum Albumin Sigma, Deisenhofen CL4B-Sepharose, # 17-0780-01 Amersham Parmacia Biotech Color Marker – Wide Range, (C3437) Sigma, Deisenhofen Dextran T500 Roth, Karlsruhe DMSO Roth, Karlsruhe DTT (1,4-Dithiothreitol), #6908.2 Roth, Karlsruhe ECL-Detektion Pack Amersham Pharmacia Biotech EDTA Merck, Darmstadt EGTA Roth, Karlsruhe Ethanol Roth, Karlsruhe ExpressHyb Hybridization Solution, #8015 Clontech, Heidelberg Ficoll, 17-1440-03 Amersham Pharmacia Biotech Fleischextrakt Merck, Darmstadt Glycerol Roth, Karlsruhe 2. Material - 21 HCl Merck, Darmstadt Hefeextrakt Heparin-Na, Braun multi, 10 000I.E/ml Difco Braun Melsungen AG, Melsungen HEPES Roth, Karlsruhe I-Block, # AI 300 Tropix, Bedford/Massachusetts Isopropanol Roth, Karlsruhe L-Cyteiniumchloridmonohydrat Merck Leupeptin L-2023 Sigma, Deisenhofen Methanol Roth, Karlsruhe MOPS Sigma, Deisenhofen MgCl2 Roth, Karlsruhe NaCl Roth, Karlsruhe NaF Sigma, Deisenhofen Na-Desoxycholat Roth, Karlsruhe Na3VO4 Sigma, Deisenhofen NP-40 Sigma, Deisenhofen PBS Sigma, Deisenhofen Pepstatin A P-5318 Sigma, Deisenhofen Peptone tryptisch aus Casein Merck PMSF (Phenylmethylsulfonylfluorid), Sigma, Deisenhofen #P-7626 Prime It II Random Labeling Kit, #300385 Stratagene, Heidelberg Skim Milk Powder, #70166 Fluka BioChemika TEMED, T´,T´,T´,T-Tetramethylendiamin Sigma, Deisenhofen # T-8133 T4-PNK, # M0201S New England Biolabs, Frankfurt TWEEN 20 Roth, Karlsruhe 2.3 Zellkulturmaterial BSA, Bovine Serum Albumin Fraction Sigma, Deisenhofen V 96%, # A-9647 DMEM,Dulbecco´s Modifiziertes Eagle Medium, # 41966-029, Lot # 3056458 Gibco BRL, Karlsruhe 2. Material - 22 Fötales Kälberserum (FCS), # F-7524; 2. Material Sigma, Deisenhofen Lot # 64H3386 L-Glutaminsäure, #11048-048 Gibco BRL, Karlsruhe HAM’s F-12 Medium, #12-615 BioWhittaker, Verviers/Belgien Lot #OMB0155 Penicillin/ Streptomyzin Sigma, Deisenhofen RPMI 1640 ohne Glu, # 31870-025, Gibco BRL, Karlsruhe Lot # 3056474 Trypsin/ EDTA Gibco BRL, Karlsruhe Zellkulturschalen Becton Dickinson / UK 2.4 Synthetische Oligonukleotide AP-1 Consesus Oligonucleotide, #E3201, Promega, Mannheim Lot # 81221 NF-κB Oligonucleotide, #E3291, Lot# 80223 Promega, Mannheim Sequenzen : AP-1 : 5‘ - CGC TTG ATG AGT CAG CCG GAA – 3‘ NF-κB : 5‘ – AGT TGA GGG GAC TTTCCC AGG C – 3‘ Poly (d(i-c)) stammte von Roche Diagnostics GmbH, Mannheim (1219847) verwendet in folgender Konzentration: 50 A 260 Units in 2,5 ml ddH2O aufgelöst. 2.5 Plasmide Mercury Pathway Profiling System, #K2049-1 Clontech, Heidelberg 2.6 Zellinien AGS : Zellinie aus einem Resektat von einem Adenokarzinom des Magens, Patient hat keine vorherige Chemotherapie erhalten; ATCC #CRL 1739 - 23 KATO III : Zellinie von einem Magenkarzinom im metastitierten Zustand; ATCC #HTB103 NIH 3T3 : 2.7 murine, embryonal Fibroblasten ; ATCC #CRL-1658 Zelltransfektion DOTAP Tranfection Agent, #1811177 Boehringer Mannheim FuGENE 6 Transfection Reagent, #1814443 Boehringer Mannheim Superfect Transfection Agent, #301305 Qiagen, Hilden 2.8 Zytokine & Inhibitoren PD 98059, #513000 Calbiochem, Bad Soden U 0126, #V1121, Lot # 95665 Promega, Mannheim TNF- α TPA 2.9 2. Material Antikörper c-Fos, rabbit polyclonal #052x Santa Cruz Biotechnology c-Fos all, rabbit polyclonal #253x Santa Cruz Biotechnology Fra-1, rabbit polyclonal #048x Santa Cruz Biotechnology Fra-2, rabbit polyclonal #171x Santa Cruz Biotechnology c-Jun, rabbit polyclonal #1694x Santa Cruz Biotechnology c-Jun all, rabbit polyclonal #044x Santa Cruz Biotechnology Jun B, rabbit polyclonal #073x Santa Cruz Biotechnology Jun D, rabbit polyclonal #074x Santa Cruz Biotechnology JNK1, rabbit poyclonal #571x Santa Cruz Biotechnology ERK 1/2, rabbit poyclonal #V1141 Promega, Mannheim Active MAPK, rabbit poyclonal #V8031 Promega, Mannheim Active JNK, rabbit poyclonal #V7931 Promega, Mannheim Cyclin D1, mouse IgG2a #66271A PharMingen, Hamburg PhosphoPlus c-Jun (Ser63), c-Jun (Ser 73) New England Biolabs, Schwalbach - 24 Antibody Kit , #9260 MEK 1/2 Kinase Assay Kit, #9830 New England Biolabs, Schwalbach p44/42 MAP Kinase Assay Kit, #9800 New England Biolabs, Schwalbach Anti-mouse-Antibody, # NA 931 Amersham Pharmacia Biotech Anti-rabbit-Antibody, # NA 934 Amersham Pharmacia Biotech 2. Material - 25 - 3. Methoden 3 Methoden 3.1 Prokaryontische Zellkultur Die Kultivierung sowie Spezifikation von Helicobacter pylori wurde an Sterilbänken bei Laminarfluß und in Inkubationsschränken unter S2 Sicherheitsbedingungen durchgeführt. Alle verwendeten Arbeitsmaterialien, Medien und Puffer waren autoklaviert oder steril filtriert. Folgende H.pylori Stämme wurden untersucht : Stamm Herkunft Referenz G27 & Mutanten klinisches Isolat Censini et al ,96 NTCT Referenzstämme Warren et al., 83 11638, ATCC 43405 klinische Isolate von einer aktiven chron. Gastritis 151, 2012 klinisches Isolat Apel et al., 82; Odenbreit et al., 96 69A, 2074 klinisches Isolat Labor Kist, Freiburg Tabelle 2 : Quellennachweis der H.pylori Stämme 3.1.1 Kultivierung von H.pylori 3.1.1.1 Anzucht auf Nährböden Die Kultivierung auf Nährplatten, insbesondere zur Überprüfung auf Kontaminationen, geschah auf HCB-Platten. Hierfür wurde mit einer ausgeglühten Impföse etwas Material von tiefgefrorenen Bakterien, Flüssigkultur oder von einer anderen Agarplatte abgenommen und auf der HCB-Platte im Dreiösenausstrich verteilt. Die Inkubation der HCB-Platten erfolgte in speziellen Anaerobiertöpfen unter mikroaerophilen Bedingungen (5% O2, 10% CO2 und 85% N2) bei 37°C und einem Unterdruck von 100 mbar. Erste Kolonien wuchsen innerhalb von 23 Tagen. HCB-Platte : 10,0 g Peptone tryptisch aus Casein (Merck) - 26 5,0 g 2,0 g 5,0 g 0,3 g 18,0 g 2,0 g 0,5 ml 10 ml ad 1 l 3. Methoden NaCl (Roth) Fleischextrakt (Merck) Hefeextrakt (Difco) L-Cysteiniumchloridmonohydrat (Merck) Bactor Agar (Difco) Glukose Vitamin K3 Hämin ddH2O pH7,4 3.1.1.2 Anzucht in Flüssigmedium Die Kultivierung von H.pylori in Flüssigmedium bietet einige Vorteile: (1) Die Kultivierungszeiten, bis die Bakteriensuspension gerade eben dicht zu werden beginnt – dann weisen die Bakterien die höchste Motilität auf und eignen sich für Stimulationsversuche am besten – ist bei Verwendung von Vorkulturen relativ gut vorhersagbar. So gewonnene Bakterienkulturen weisen hohe Motilität auf und eignen sich am besten für Stimulationsexperimente. (2) Gegenüber der Kultivierung auf Nährböden können leichter größere Mengen Bakterien produziert werden. Erkauft werden diese Vorteile durch eine höhere Anfälligkeit für Kontaminationen; diese werden insbesondere nicht so leicht entdeckt wie bei der Anzucht auf Nährböden. Insofern muß sehr genau auf steriles Arbeiten geachtet werden, und begleitend Kontrollen (Mikroskopie und Kontrollkulturen auf Nährplatten) durchgeführt werden. Für eine Vorkultur wurde 15ml Brucella Medium in eine 75ml-Kulturflasche mit Belüftungskappe und integriertem Sterilfilter gegeben und mit der ausgeglühten Impföse die Bakterien inokuliert. Um eine Kontamination der Belüftungskappe zu vermeiden, wurden die Kulturflaschen in Schräglage in einen Anaerobiertopf gelegt und unter mikroaerophilen Bedingungen bei 37°C auf einem Schüttler (100 U/min) für 1-2 Tage inkubiert. Die dichte Kultur wurde unter dem Phasenkontrastmikroskop kontrolliert. Sofern hierbei eine frische Kultur herauskam (Bakterien beweglich, wenig Kokken), wurden 100-150µl Bakteriensuspension in eine neue Kulturflasche mit 15ml Brucellamedium gegeben und unter denselben Bedingungen (s.o.) für 14-16 Stunden kultiviert. - 27 Brucella Medium : 10 g 10 g 10 g 1g 5g 0,1 g ad 1l 3. Methoden Bactor Dextrose (Difco) Bactor Trypton (Difco) Bactor Peptamin (Difco) Bactor Yeast Extrakt (Difco) NaCl (Roth) Natriumbisulfat (Roth) ddH2O Das Medium wurde autoklaviert und mit sterilfiltriertem, 10% fetalem Kälberserum (FCS) supplementiert. Das Serum mußte vor Gebrauch 30 min auf 56°C erwärmt werden, um Komponenten des Komplementsystems zu inaktivieren, und wurde anschließend durch einen Millex-GP 0,22 µm Filter (Millipore) sterilfiltriert. 3.1.1.3 Langzeitkonservierung Die Langzeitkultivierung erfolgte mittels Kryokonservierungsröhrchen namens Mikrobank der Firma Prolab Diagnostik. 15 ml einer dichten Bakterienkultur wurden abzentrifugiert (4000rpm, 5 min, Megafuge 1.0), das Pellet in dem Konservierungspuffer resuspendiert, und die Konservierungskügelchen aus dem Röhrchen für 5 min mit dieser Suspension inkubiert. Anschließend wurde die Suspension wieder abgesaugt und das Röhrchen mit den Konservierungskügelchen bei -70°C gelagert. Alternativ wurde einer dichten Bakterienkultur 20 vol % Glycerin zugegeben und in einem Konservierungsröhrchen bei -70°C gelagert. 3.1.2 Spezifikation von H.pylori Die Identifikation von H.pylori erfolgte durch Kontrollkulturen auf HCB-Platten sowie durch Bestimmung der Katalase-, Oxidase- und Ureaseaktivität. Durch Phasenkontrastmikroskopie konnte eine orientierende Spezifikation einzelner H.pylori Stämme erbracht werden - die verschiedenen Stämme besitzen eine unterschiedliche Morphologie - und ebenfalls orientierend die Kultur auf Kontamination mit anderen Bakterien geprüft werden. Desweiteren wurde jede für Stimulationsexperimente eingesetzte H.pylori Kultur mikroskopisch auf die Vitalität der Bakterien, sowie die Kulturphase – spiralförmig (frühe Kulturphase) oder kokkoid (späte Kulturphase) – überprüft. Nur spiralförmige Bakterien der frühen Kulturphase, die zudem eine deutlich höhere Motilität aufwiesen als Bakterien aus älteren Kulturen, wurden für die Stimulation von AGS-Zellen benutzt. Ein weiteres - 28 - 3. Methoden Phänomen und auch Problem der späteren Kulturphase bestand darin, daß sich die Bakterien zu Klumpen zusammenlagerten. Hierfür war besonders H.pylori G27 und bestimmte seiner isogenen Mutanten anfällig. Durch das Zusammenkleben der Bakterien wurde zum einen die photometrische Dichtebestimmung der Bakterien unvalide – größere Bakterienaggregate haben ein anderes Streuverhalten als eine dichte homogene Suspension einzelner Bakterien. Zum anderen zeigten die verklumpten Bakterien eine deutlich geringere Motilität und bewirkten nur eine schwache NF-κB bzw. AP-1 Stimulation. 3.1.3 Vorbereiten der Bakterien für einen Stimulationsversuch Bakterien, die in serumhaltigen Stimulationsversuchen, die sich Flüssigmedium um Teile des angezogen MAP wurden, Kinase / mußten c-Fos / vor AP-1 Signaltransduktionsweges sowie AP-1 abhängige Genexpression drehten, von dem Serum gereinigt werden, da dieses sonst mögliche Effekte von H.pylori überdecken würde. Hierfür wurde eine entsprechende Menge Bakterienkultur aus der frühen Wachstumsphase (meist 15ml) in ein Falcon gegeben, herunterzentrifugiert (4000rpm, 5 min / Megafuge 1.0), in 1ml unsupplementiertes (kein Antibiotikum, kein Serum) HAM’s F-12 Medium resuspendiert (bei Stimulation von AGS-Zellen, ansonsten entsprechend anderes Medium) und in ein 1,5ml Eppendorfgefäß transferiert. Nach zwei weiteren Waschschritten (Sedimentation bei 6000 rpm / 5 min in einer Tischzentrifuge (Biofuge 13) und Resuspension in serumfreiem Medium, wurde die optische Dichte einer 1:20 in Medium verdünnten Bakteriensuspension bei λ=695nm bestimmt und auf die unverdünnte Kultur hochgerechnet. Durch den Verdünnungsschritt wurde meist der lineare Meßbereich des Photometers (OD zwischen 0,05 und 0,3) erreicht. Da bei der Sedimentation während den verschiedenen Waschschritten die Bakterien gerne zur Klumpenbildung neigten, wurden die Bakterien nach den Waschschritten mehrmals durch eine sehr dünne Nadel (26 Gauche) in eine Spritze gesaugt – durch die auftretenden Scherkräfte konnten die Komplexe meist wieder vollständig aufgelöst werden (mikroskopische Kontrolle). 3.2 Eukaryontische Zellkultur Alle Arbeiten mit eukaryontischen Zelllinien inklusive der Stimulationsversuche von den Zellen mit H.pylori Stämmen erfolgte in Sterilbänken unter Laminarfluß in einem separaten - 29 - 3. Methoden Zellkulturlabor mit S2 Sicherheitsbedingungen. Eingesetzte Materialien wurden vor Gebrauch desinfiziert (Medien, Lösungen) oder waren steril verpackt bzw. autoklaviert. Die verwendeten Zellinien und ihre Medien sind in folgender Tabelle zusammengestellt : Cell Line Bezug Ursprung Medium AGS ATCC Adenokarzinom des HAM’s F-12 + 10 %FCS + 50µg/ml Penicillin/ CRL 1739 Magens Streptomycin + Glutamin ATCC Adenokarzinom des RPMI 1640 + 10 %FCS + 50µg/ml Penicillin/ HTB 103 Magens Streptomycin + Glutamin ATCC Murine, embyonale RPMI 1640 + 10 %FCS + 50µg/ml Penicillin/ CRL-1658 Fibroblasten Streptomycin + Glutamin KATO III NIH 3T3 Tabelle 3 : Quellennachweis der Zellinien Die Kulturflaschen sowie Zellkulturschalen wurden in einem Brutschrank bei 37°C, 5% CO2 Atmosphäre inkubiert. 3.2.1 Passage der Zellen 3.2.1.1 Adhärente Zellen Das Teilen der AGS-Zellen erfolgte nach Möglichkeit jeden 2.Tag, um die Zellen in der logarithmischen Wachstumsphase zu halten und so optimale Bedingungen für Stimulationsversuche zu gewährleisten. Nach einem Vorwaschschritt mit 2ml 1mM Trypsin/EDTA wurde die Kulturflasche mit 5ml 1mM Trypsin/EDTA für 4 - 6 min bei 37°C inkubiert. Danach wurde das Trypsin durch Zugabe von 10ml Mediums/ 10% FCS inaktiviert und die gelösten Zellen in einem Verhältnis 1:8 -1:10 weiterkultiviert. Die übrigen Zellen wurden entweder verworfen oder für Stimulationsversuche ausplattiert (s.u.) . 3.2.1.2 Nicht adhärente Zellen Die Zellen wurden bei einer Zelldichte von ca. 106 pro ml Medium geteilt. - 30 3.2.2 3. Methoden Langzeitkonservierung 5 - 10 106 Zellen in Suspension wurden in ein 15ml Falcon Röhrchen transferiert und bei 1000 U/min 5 min abzentrifugiert. Das Zellpellet wurde in 1,5ml 90% FCS / 10% DMSO resuspendiert und in ein Kryoröhrchen überführt. Das Einfrieren erfolgte langsam durch Lagerung in Styropor bei -80°C über 3-4 Tage und zur Langzeitkonservierung dann in flüssigem Stickstoff. Das Auftauen erfolgte bei Raumtemperatur und Kultivierung in 25ml Medium mit Mediumwechsel nach 1-2 Tagen. 3.2.3 Bestimmung der Zellanzahl 10µl der Zellsuspension wurden in der Neubauer Zählkammer ausgezählt und die Zellzahl auf die Gesamtsuspension hochgerechnet. 3.2.4 Ausplattieren der Zellen Bei Versuchen, bei denen Serum im Medium nicht mit den zu messenden Variabeln interferierte (u.a. Stimulation von NF-κB), wurden ca. 5 105 Zellen in 5ml Serum supplementiertem Medium – im folgenden SER+ genannt – in eine Kulturschale (Durchmesser 6cm) gegeben und über Nacht bei 37°C inkubiert. Bei Versuchen, bei denen das supplementierte Serum mit den interessierenden Variablen interferiert – so z.B. aktiviert Serum den Transkriptionsfaktor AP-1 und die MAPKSignaltransduktionswege und führt zu hoher Hintergrundaktivität – sowie bei Versuchen, bei denen sich die Zellen zur guten Stimulierbarkeit in der Ruhephase G0 befinden müssen, wurden die Zellen zuerst von Serum gereinigt. Hierzu wurden die Zellen 2x 5 min bei 1000 U/min sedimentiert und in 10ml serumfreiem – im folgenden SER∅ genannt – Medium resuspendiert. Anschließend wurden 8 – 12 105 Zellen in 5ml Medium SER∅ in eine 6cm Kulturschale gegeben, durch Schwenken der Kulturschale möglichst homogen verteilt und für 8-14h im Brutschrank inkubiert. Bei Versuchen mit Kulturschalen anderer Größe wurden folgenden Zellmengen verwendet : - 31 Größe SER∅ Ansatz 5 SER+ Ansatz 5 10 5 3. Methoden Verwendungszweck 6cm 8 -12 10 EMSA 10cm 2-3 106 Westernblot 14cm 6-8 106 Northernblot Tabelle 4 : Zellkulturschalgrößen 3.3 Molekularbiologische Methoden 3.3.1 Plasmide Die verwendeten cDNA-Sonden für Northernblot wurden freundlicherweise von folgenden Kollegen bereitgestellt : c-fos 600bp Exon 4 in pBFH 480, Dr. Judith Müller (ZKF, Universität Freiburg) Schnittstelle : HindIII,EcoRI VEGF Cyclin D1 600 bp Dr. J. Finkenzeller (Klinik für Tumorbiologie, Schnittstelle : EcoRI, BamH1 Freiburg) 900bp in pCMX Dr. Mattiaz Humar (ZKF, Universität Freiburg) Schnittstelle : EcoRI, BamH1 β-Actin 1200 bp Prof. Dr. Heike Pahl (ZKF, Universität Freiburg) Schnittstelle : PstI Tabelle 5 : Quellennachweis der verwendeten Plasmide Die Reportergenkonstrukte pTal, pAP-1, pNF-κB, pSRE stammen aus dem „Mercury Pathway Profiling Set“ (Clontech). pKoll-Luc, ein Luciferase Reporterkonstrukt mit einer AP-1 binding site aus dem Kollagen Promoter, wurde von Dr. Rupec (Universitätshautklinik, LMU München) zur Verfügung gestellt. 3.3.1.1 Plasmidpräparation Die Plasmidpräparation erfolgte säulenchromatographisch mittels der Plasmid Maxi Kits von Qiagen – die Plasmidausbeute betrug durchschnittlich 500-800 µg. - 32 3.3.2 3. Methoden DNA-Quantifizierung Die Konzentration von Nukleinsäure kann photometrisch bestimmt werden, da Nukleinsäuren bei λ=260 nm ein Absorptionsmaximum aufweisen, wohingegen dieses bei Aminosäuren/Proteinen eher bei λ=280 nm liegt. Die Konzentration ergibt sich aus der Formel: c(µg/µl) = E260 nm * Verdünnungsfaktor * const Verdünnungsfaktor : i.a. wurde die DNA bzw. RNA 1:80 in ddH2O verdünnt const : 50 für DNA / 40 für RNA Der Quotient E260 nm/ E280 nm ist ein Maß für die Reinheit der DNA bzw. RNA; ein Wert von 1,8 oder höher entspricht ausreichender DNA-Reinheit. 3.3.3 DNA – Agarosegelelektrophorese Für die Auftrennung von DNA wurde ein 0,8 bzw. 1 % TAE-Agarose Gel verwendet: 0,8 % / 1,0 % TAE –Gel : 0,4 / 0,5 g 50 ml 1 µl Agarose TAE – Puffer Ethidiumbromid (1 µg/µl) Für das Gel wurde die Agarose und der TAE-Puffer in einen Erlenmeierkolben gegeben, in der Mikrowelle kurz zum Kochen gebracht, bis auf ca. 60°C abkühlen gelassen und dann das Ethidiumbromid hinzugegeben. Die DNA-Proben wurden mit 6x Lade Puffer (0,2 % Bromphenolblau, 0,2 % Xylencyanol, 30 % Glycerin) geladen, und die Elektrophorese erfolgte bei 70 V / 25mA. Das Ergebnis wurde unter UV-Durchleuchtung mit Hilfe des EagleEye Apparates (Stratagene) dokumentiert. 3.3.4 Zuerst Herstellung von cDNA Sonden wurde die cDNA Sonde aus 3µg des entsprechenden Plasmids durch Restriktionsverdau geschnitten – die Reaktionsbedingungen wurde nach den Empfehlungen - 33 - 3. Methoden der Hersteller der Restriktionsenzyme gewählt. Daraufhin wurden die Reaktionsprodukte – das Insert, der linearisierte Vektor und unverdautes Plasmid - in einem lowmelt Agarose Gel aufgetrennt und das Insert unter einem λ=365 nm UV-Schirm ausgeschnitten. Die ausgeschnittene Bande wurde gewogen, in ein Eppendorfreaktionsgefäß transferiert, mit 2 Volumen ddH2O resuspendiert und 10 min bei 65°C erhitzt. Aufbewahrt wurden die lowmelt Sonden bei -20°C. 3.3.5 Markieren der cDNA Sonden mit α-(32P)-dCTP Hierzu wurde der „Prime-It-II Labeling Kit“ (Stratgene) verwendet : 50ng Insert in lowmelt Agarose (ca. 10-20 µl) wurden auf 23µl mit ddH2O verdünnt, 10 µl Random Primers hinzugegeben, 5 min auf 95°C erhitzt und dann kurz runterzentrifugiert. Nach Zugabe von 10µl 5x dCTP Puffer, 5µl α-(32P)-dCTP (3000 Ci/ml) und 1µl Klenow Exo – Polymerase, wurde der Ansatz durch Antippen an das Eppendorfreaktionsgefäß kurz gemischt, kurz abzentrifugiert und bei 37°C für 30 min inkubiert. Danach wurde das Reaktionsprodukt mittels einer G-200 SpinColumn aufgereinigt und die Aktivität der markierten Sonde in einem β-Counter gemessen (durchschnittlich 106 cpm). 3.3.6 Northern Blot 3.3.6.1 RNA-Präparation Die Gesamtzell-RNA wurde durch saure Phenol Extraktion (Trizol, Life Technologies) gewonnen. Die Zellen einer 14cm∅ Kulturschale wurden nach dem Stimulationsversuch in 5 ml 1xPBS geerntet, in ein 15ml Falcon Röhrchen transferiert und abzentrifugiert. Das Zellpellet wurde nun in 5ml Trizol resuspendiert, 1ml Chloroform hinzugefügt, kräftig gemischt, in ein Corex Röhrchen überführt und 3 min bei Raumtemperatur stehengelassen. Durch eine 15 min Zentrifugation (10000rpm, 4°C) wurde das Gemisch in eine obere wässrige Phase (beinhaltet die RNA), eine mittlere weiße Phase (beinhaltet das Protein) und eine untere rote PhenolChloroform Phase (beinhaltet die DNA) getrennt. Die obere Phase wird in ein neues CorexRöhrchen transferiert und die RNA mit 1 Volumen (ca. 2.5ml) Isopropanol gefällt. Nach Zugabe des Isopropanols erfolgte eine 10 min Inkubation bei Raumtemperatur und 10 min - 34 - 3. Methoden Zentrifugation (10000rpm, 4°C). Das RNA-Pellet wurde in 2,5ml Ethanol durch vortexen gelöst, nochmals zentrifugiert (5 min, 8000 rpm, 4°C) und luftgetrocknet. Die RNA wurde in 20 µl RNAse freiem DEPC-ddH2O (Wasser mit 0,1 % Diethylpyrocarbonat, ein RNAse Inhibitor) aufgenommen und bei -20°C aufbewahrt. 3.3.6.2 Gelelektrophorese der RNA Zur Auftrennung der RNA wurde ein 1,0 % Agarose Gel in 1xMOPS-Puffer verwendet. Zur Linearisierung der RNA wurde 0,6% Formaldehyd beigesetzt. Jeweils 10µg der RNA-Proben (5-15µl) wurden in ein Eppendorfreaktionsgefäß gegeben, in einer Vakuumzentrifuge auf 4µl konzentriert und mit 15,5 µl Probenpuffer und 5 µl 6x Ladepuffer auf das Gel geladen. Die Elektrophorese erfolgte in 1x MOPS mit 0,6 % Formaldehyd (37%) für 1-2h bei 30mA. 1x MOPS : 20 mM 5 mM 1 mM MOPS NaOAc EDTA 6x Ladepuffer : 50 % 0,4 % 0,4 % 1 mM Glycerin Bromphenolblau Xylenblau EDTA Probenpuffer : 20 µl 35 µl 100 µl 845 µl 10x MOPS Formaldehyd (37%) Formamid (100%) DEPC - ddH2O 3.3.6.3 Nothern Blot Für den Transfer der RNA vom Gel auf eine Nitrozellulose Membran Hybond-N wurde das TurboBlotter System (Schleicher & Schuell) verwendet, das nach dem Neutralblot Prinzip arbeitet. Das Gel wurde für 10 min in DEPC – ddH2O gewaschen, die Nitrozellulose Membran 1 min in ddH2O und 5 min in 20x SSC inkubiert. Der Aufbau erfolgte nach Empfehlungen des Herstellers; im wesentlichen wurde das Gel auf die Membran gelegt, die wiederum auf einem - 35 - 3. Methoden Stoß von Filterpapier platziert war. Nachdem nun auf das Gel ein größeres Filterpapier aufgelegt wurde, das Verbindung zu einem Transferpuffer – Reservoir (20x SSC) hatte, ergab sich ein Fluß von Ionen durch das Gel in die Membran, der die RNA auf die Membran „blottete“. Als Transferpuffer diente 20x SSC. Nach mindestens 12h Inkubation wurde die Membran 5 min in 2x SSC-Puffer gewaschen und durch UV-Bestrahlung an die Membran fixiert. 20x SSC –Puffer : 3M 300 mM NaCl NaCitrat 3.3.6.4 Hybridisierung Die Membran mit der fixierten RNA wurde 1 Stunde in 10ml Express-Hyb Lösung (ClonTech) vorinkubiert. Die radioaktiv markierte DNA-Sonde wurde zur Trennung in ssDNA für 10 min auf 95°C erhitzt, kurz abzentrifugiert, auf Eis gekühlt und dem Hybridisierungspuffer beigegeben. Die Hybridisierung erfolgte mindestens 2h bei 68°C. Daraufhin wurde der Blot dreimal für 10 min bei Raumtemperatur in 2x SSC mit 0,05 % SDS und in einem zweiten stringenterem Waschschritt zweimal für 20 min bei 50°C in 0,1x SSC mit 0,1% SDS inkubiert. Der Blot wurde bei -80°C für eine geeignete Zeit autoradiographiert. 3.3.7 Transfektion Bei der Transfektion werden DNA-Plasmide in eukaryontische Zellen eingeschleust. In dieser Arbeit wurden drei kommerzielle Transfektionskits auf liposomaler Basis – DOTAP (Boehringer Mannheim), Fugene 6 (Boehringer Mannheim) und SuperFect (Qiagen) - für die Eignung zur Transfektion der Magenepithelzell-Linie AGS getestet. Fugene 6 erwies sich dabei als die effizienteste Methode, die darüber hinaus auch die am wenigsten zytotoxische war. Im weiteren soll nur die zum Schluß für die Reportergenversuche verwendete Methode mit Fugene 6 näher beschrieben werden – alle drei Methoden wurde entsprechend den Empfehlungen des Herstellers durchgeführt. Zur Optimierung der Transfektionseffizienz wurden Parameter wie die eingesetzte Menge an DNA bzw. Transfektionsagens variiert (siehe auch Etablierung der Transfektion von AGS-Zellen). - 36 - 3. Methoden 3.3.7.1 Vorbereiten der Zellen AGS-Zellen in der logarithmischen Wachstumsphase wurden in 2ml HAM´s F-12 / 10% Serum in einer Dichte von 4-6*104 in eine 6-well Platte ausplattiert. Nach 30-36 Stunden Kultivierung im Brutschrank war eine 40-60% Konfluenz des Zellrasens erreicht und somit ein geeigneter Zeitpunkt für die Transfektion der Zellen. Zu 97 µl HAM´s F-12 Medium, dem kein Serum oder Antibiotikum zugefügt war, wurden 3 µl Fu6 Reagenz pipettiert, wobei das Transfektionsagens die Wand des Eppendorfgefäßes nicht berühren durfte, und für 5-10 min bei Raumtemperatur inkubiert. In ein zweites Eppendorfgefäß wurde 2µg DNA des Reporterkonstrukts, sowie 50ng eines tk-Renilla Luciferase Konstrukts als Standardisierungsvektor gegeben. Dieses Gemisch wurde nun tropfenweise dem ersten Eppendorfgefäß mit dem in Medium verdünnten Transfektionsreagens hinzugegeben. Zur Ausbildung von DNA-Liposomen Komplexen wurde der Ansatz 15 min bei Raumtemperatur stehengelassen und dann tropfenweise den Zellkulturschalen hinzugefügt. Nach 8h Inkubation im Brutschrank wurden optional die Zellen für 6h gehungert, indem das serumhaltige Medium durch serumfreies Medium ersetzt wurde. Anschließend erfolgte die Stimulation der Zellen für 8 Stunden und daraufhin die Zellernte. 3.3.7.2 Zellernte Die Zellen wurden einmal mit 1x PBS gewaschen und anschließend in der Kulturschale mit 200 µl 1x PLB (Passive Lysis Puffer / Promega) für 2 min lysiert. Die Zellen wurden mittels eines Gummischabers gesammelt, mit der Pipette durch Auf- und Abpipettieren weiter homogenisiert und der Zelldebris durch Zentrifugation (13000rpm, 5 min, 4°C) abgetrennt. Der Überstand wurde bei -80°C aufbewahrt. 3.3.7.3 Bestimmung der Luciferase Aktivität Die Luciferase Aktivität wurde an einem LB 96P Luminometer (EG&G Bertold) mit 100 µl Festinjektoren und unter Verwendung des Dual Luciferase Reporter Assay Systems (Promega) bestimmt. Hierfür wurden 25 µl des Proteinextraktes in eine spezielle 96Wellplatte pipettiert und die Luciferase Aktivität nach Zugabe von 100 µl LARII-Puffers (Promega) über ein 10 s-Intervall im Luminometer quantifiziert. In einem zweiten Schritt - 37 - 3. Methoden wurde durch Zugabe von 2 µl Renilla-Luceferin, das in 98 µl Stop&Glow Puffer gelöst war, die erste Chemolumineszens Reaktion beendet und eine zweite, die durch die kotransfizierte Renilla-Luciferase initiiert wurde, gestartet. Wieder erfolgte die Messung über ein 10 sIntervall. Die zweite Messung diente als Abgleich für die erste Messung, um Schwankungen der Transfektionseffizienz auszugleichen. 3.4 3.4.1 Proteinbiochemische Methoden Stimulation von Magenepithelzellen durch H.pylori Für einen Stimulationsversuch wurde 1ml Bakterien Suspension mit einer optischen Dichte von 1,0 bei λ=695nm (entspricht 108 Bakterien) pro 5 ml Medium zu den ausplattierten Zellen gegeben und für die angegebene Zeit im Brutschrank kokultiviert. 3.4.2 Gewinnung der Gesamtzellextrakte Adhärente Zellen wurden zuerst 1-2 mal mit 4°C 1x PBS gewaschen und anschließend in 5ml selbigen Puffers mit einem Gummispatel abgekratzt, in ein 15ml Falcon transferiert, bei 1000 U/min 5 min abzentrifugiert und in 1ml 1x PBS resuspendiert. Dieses wurde in ein 1,5ml Eppendorfröhrchen pipettiert, kurz sedimentiert (13.000 U/min, 15 sec), in 4 Volumen Totex Puffer (=50µl) resuspendiert und 30 min auf Eis inkubiert. Zum Schluß wurde der Zelldebris 5 min bei 13000 U/min und 4°C abzentrifugiert und der Überstand in ein neues Eppendorfröhrchen pipettiert. Für die Langzeitkonservierung wurde der Extrakt im flüssigen Stickstoff schockgefroren und bei -80°C aufbewahrt. Für die Gewinnung von Proteinextrakten für MAP Kinase Assays oder auch Western Blots wurden alternativ zu oben beschriebener Methode die Zellen zweimal mit 4°C 1x PBS gewaschen und anschließend direkt in der Kulturschale mit 200µl NEB Lysis Puffer lysiert. Nach 5-10 min Inkubation auf Eis erfolgte das Abkratzen mit dem Gummispatel; die Zellsuspension wurde dann in ein Eppendorfröhrchen transferiert. Die Proteinextrakte wurden 4mal für 5 sec im Ultraschallbad homogenisiert und dann der Zelldebris analog oben entfernt. - 38 Totex 3. Methoden NEB Lysis Puffer 20 mM HEPES (pH 7,9) 20 mM Tris (pH 7.5) 350 mM NaCl 150 mM NaCl 1% NP-40 1% Triton X-100 0,1 mM EDTA 1mM EDTA 0,5 mM EGTA 1 mM EGTA 1mM MgCl2 1 mM Glycerolphosphate 1mM Na3VO4 2.5 mM NaPyrophosphat 1µg/ml Leupeptin 1mM PMSF ** * 5mM Na3VO4 30 mM NaPyrophosphat * 1% Aprotinin ** ** 1mM PMSF 5mM DTT ** Tabelle 6 : Zelllysispuffer * wurde bei Zellextrakten als Phosphatase Inhibitoren hinzugefügt Na3VO4 Zubereitung (0,2 M Stock) : [nach Gordon 1991] Mw Na3VO4 : 184 g/mol • 0,37 g Na3VO4 in 10ml lösen • pH auf 10.0 mit 37 % HCl titrieren (Cave bei Erreichen des Zielbereiches) • 1 Stunde bei 100° C kochen – Farbe schlägt von orange auf durchsichtig • bei 4° C aufbewahren (verwendbar für ca. 1-2 Wochen) ** Verbindung nicht stabil - wurde kurz vor dem Versuch dem Puffer hinzugefügt 3.4.3 Proteinbestimmung nach Bradford Zur Bestimmung der Proteinkonzentration wurde 10µl Proteinextrakt zu 1ml Bioradreagenz gegeben, das zuvor 5-fach verdünnt in 250 mM TRIS Puffer wurde, und nach 5 min Inkubation bei Raumtemperatur die Lichtabsorption bei λ=595 nm gemessen. Der Proteingehalt wurde mittels Standardverdünnungsreihe ermittelt, wofür in Lysis Puffer verdünntes BSA verwendet wurde. - 39 3.4.4 3. Methoden Gelretardierungsassay / EMSA Der Gelretardierungsassay oder Electrophoretic Mobility Shift Assay dient dem Nachweis von Protein-DNA Wechselwirkung, wie sie u.a. physiologisch bei der Interaktion von Transkriptionsfaktoren mit Promoter und Enhancer DNA-Sequenzen stattfindet. Hierzu wird ein Proteinextrakt – entweder Gesamtproteinextrakt oder auch Kernextrakt – mit einem radioaktiv markierten Oligonukleotid inkubiert, welches die Konsensusbindungssequenz des zu untersuchenden Transkriptionsfaktors beinhaltet. Um unspezifische Bindungen zu vermeiden, wird BSA zum Blockieren von proteinbindenden DNA-Sequenzen neben der markierten Sonde und Poly [dIdC] zum Abfangen von unspezifisch DNA-bindenden Proteinen hinzugefügt. Zur Detektion der gebildeten DNA-Protein Komplexe wird das Reaktionsprodukt in der Polyacrylamid Gelelktrophorese (PAGE) unter nativen nicht-denaturierenden Bedingungen aufgetrennt und durch Autoradiographie oder im Phosphoimager visualisiert. 3.4.4.1 Markieren von Oligonukleotiden mit γ-(32P)-dATP Die für den EMSA verwendete Sonde wurde mittels einer T4-Polynukleotidkinase radioaktiv markiert: Reaktionsansatz : 37 µl 5 µl 1 µl 5µl 1,5µl ddH2O 10x PNKinase Puffer (NEB) AP-1/ NF-κB Oligonukleotid (1.75 pmol/µl) (Promega) γ-(32P)-dATP (3000 Ci/mmol) T4-Polynukleotidkinase Der Reaktionsansatz wurde bei 37°C 30 min inkubiert und anschließend mittels einer G-25 SpinColumn aufgereinigt. Zur Vorbereitung hierfür wurde die Säule eine Minute bei 3000 rpm in der Tischzentrifuge (Biofuge 13) abzentrifugiert, um die in der Säule befindliche Flüssigkeit zu entfernen. Anschließend wurde das Reaktionsprodukt auf die Säule aufgebracht und für 2 min bei 3000 rpm zentrifugiert. Die so gewonnenen radioaktiv markierten Oligonukleotide wurden bei 4°C gelagert und waren für ca. 10 Tage verwendbar. - 40 - 3. Methoden 3.4.4.2 EMSA Mix Zur Bildung der DNA-Protein Komplexe wurden 5µl des Proteinextraktes (ca. 10-20 µg) mit 15µl EMSA Bindungsmix 25 min bei Raumtemperatur inkubiert. Für die Analyse wurde ein 4% natives Polyacrylamid Gel verwendet, die Elektrophorese wurde bei 210V in 0,5x TBE Puffer durchgeführt. Die Gele wurden auf 3MM Filterpapier für 35 min bei 80°C vakuumgetrocknet und dann autoradiographiert. EMSA Mix : 2 µl 2 µl 2 µl 4 µl 2 µl 2 µl 1 µl ddH2O MgCl2 (50mM) Puffer D+ 5x Ficoll Puffer BSA (10 µg/µl) Poly [dIdC] (1 µg/µl) γ-(32P) markerites Oligo Bei NF-κB wird kein MgCl2 benötigt und diese 2 µl durch ddH2O ersetzt. Puffer D+ : 20 mM 100 mM 0,25 % 20 % 0,5 mM 2mM HEPES (pH 7,9) KCl NP-40 Glycerol EDTA DTT 5x Ficoll Puffer : 100 mM 300 mM 20 % 10 mM 0,1 mM HEPES (pH 7,9) KCl Ficoll 400 DTT PMSF 10x TBE Puffer : 890 mM 890 mM 20 mM TRIS Borsäure EDTA 4 % PA-Gel : 60 ml 10 ml 3,5 ml 400 µl ddH2O Acrylamid (30%) 10x TBE APS (10%) - 41 40 µl 3. Methoden TEMED (100%) 3.4.4.3 Supershift Der Supershift dient der weiteren Spezifikation des zu untersuchenden DNA-bindenden Proteins – bindet ein hinzugegebener Antikörper spezifisch an den DNA-Protein Komplex, hat dieser größere Komplex eine langsamere Laufgeschwindigkeit in der PAGE - so wird die Bande nach oben „geshiftet“. Ist der Antikörper gegen die DNA-bindende Domäne des Transkriptionsfaktors gerichtet, kann es zur vollständigen Aufhebung der DNA-Protein Interaktion kommen, und die Bande im EMSA verschwinden. Zur Durchführung wurden zu den 15 µl EMSA Mix 2µl Antikörper und 3 µl 1:2 verdünnter Proteinextrakt gegeben. 3.4.4.4 Kompetitionsassay Dieser Assay dient dem Nachweis der Spezifität der Bindung des Transkriptionsfaktors an das markierte Oligonukleotid. Hierbei wird dem Bindungsmix 1µl 100x konzentriertes (1,75 pmol/µl) nicht radioaktiv markiertes Oligonukleotid hinzugegeben. Handelt es sich um das gleiche Oligonukleotid wie das radioaktiv markierte, sollte die spezifische Bande im EMSA durch kompetitive Hemmung verschwinden. Wird hingegen ein anderes Oligonukleotid als das zu untersuchende im Überschuß hinzugefügt, sollte bei spezifischer Bindung des Transkriptionsfaktors an seine Consensus-Sequenz-Oligonukleotid keine kompetitive Hemmung stattfinden. 3.4.5 Westernblot / Immunoblot Der Westernblot stellt eine Kombination dar aus der (1) Polyacrylamid Gel Elektrophorese (PAGE) zum Auftrennen von Proteinextrakten meist unter denaturierenden Bedingungen (SDS-PAGE) und nach Blotten der Proteine aus dem Gel auf eine Membran - z.B. Nitrocellulose (NC) oder Polyvinylidene difluorid (PVDF) Membran - (2) der Immundetektion einer speziellen Proteinbande mit Antikörpern gegen das zu untersuchende Protein. Die Immundetektion erfolgte in 2 Stufen : ein erster Antikörper erkennt das Protein, der zweite Antikörper, der gegen den ersten - 42 - 3. Methoden Antikörper gerichtet ist, ist mit einem Marker bestückt – in dieser Arbeit wurden Zweitantikörper verwendet, die mit Horseradish Peroxidase (HRP) markiert waren, und so mittels Chemolumineszens detektiert werden konnten. 3.4.5.1 SDS-PolyAcrylamid-Gel-Elektrophorese (PAGE) Für die lineare Auftrennung der Proteine entsprechend ihrer Größe in der Gelelektrophorese ist es zunächst wichtig, diese zu linearisieren und die Ladungen der Seitenketten, die mit der in der Gelelektrophorese angelegten Spannung interferieren, zu egalisieren. Nach Bestimmung des Proteingehaltes der Gesamtzellextrakte wurden hierfür gleiche Mengen (ca. 30µg) in einem Laemli Sample Puffer 5 min bei 95°C denaturiert und auf einem 10% SDSPolyacrylamidgel bei 30 mA für 1-1,5 h elektrophoretisch aufgetrennt. Das eingesetzte SDSPolyacrylamidgel unterteilte sich in ein oberes Sammelgel (1/3) und ein unteres Trenngel (2/3). Zuerst wurde das Trenngel in die Gelkammer gegossen. Nachdem es nach ca. 30 min auspolymerisiert war – der obere Rand wurde mit ddH2O bedeckt und so vor Austrocknung geschützt – wurde das Wasser abgesaugt und das Sammelgel in die verbleibende Kammer gegossen. Im Sammelgel wird das in die Ladetasche gegebene Proteinextrakt auf eine dünne Bande fokussiert, im Trenngel erfolgt die Auftrennung der Proteine entsprechend ihrer Größe. Trenngel : 3,3 ml 2 ml 2,7 ml 50 µl 4 µl ddH2O 4x lower buffer Acrylamid (30%) APS (10%) TEMED (100%) Sammelgel : 2,5 ml ddH2O 1 ml 4x upper buffer 0,5 ml Acrylamid (30%) 40 µl APS (10%) 4 µl TEMED (100%) lower buffer : 500 mM 0,4 % TRIS-HCl pH 6,8 SDS upper buffer : 1500 mM 0,4 % TRIS-HCl pH 6,8 SDS - 43 5x SDS running : buffer 125 mM 1000 mM 0,5 % TRIS Glycin SDS 4x Laemli buffer : 200 mM 40 % 8% 400 mM 0,4 % TRIS Glycerol SDS DTT Bromphenolblau 3. Methoden 3.4.5.2 Blotten Danach wurden die Proteine mit einem Semi-dry-Elektroblotter bei 0.8 mA/cm2 für 1h auf eine Immobilon-P PVDF Membran transferiert. Hierbei wurde folgender Aufbau gewählt : Kathode 6 Stück 3MM Filterpapier äquilibriert in Kathodenpuffer Gel Membran 2 Stück 3MM Filterpapier äquilibriert in Anodenpuffer 2 4 Stück 3MM Filterpapier äquilibriert in Anodenpuffer 1 Anode Anodenpuffer 1 : 300 mM 10 % TRIS Methanol Anodenpuffer 2 : 25 mM 10 % TRIS Methanol Kathodenpuffer : 25 mM 40 mM 10 % TRIS 6-amino-Hexansäure Methanol - 44 - 3. Methoden Die Membran wurde zuerst 15 sec in Methanol quellen gelassen und dann nacheinander 2 min in ddH2O und 5 min in Anodenpuffer 2 inkubiert. 3.4.5.3 Immundetektion Zur Vermeidung unspezifischer Bindungen wurde die Membran in 1x TBS mit 5% BSA ( bei Antikörpern von NEB oder Santacruz) bzw. 0,2% I-Block (bei phosphospezifischen Antikörpern von Promega) für 1 Stunde bei Raumtemperatur oder bei 4°C über Nacht inkubiert. Nach Austausch des Blocking Puffers wurde der 1. Antikörper in einer Verdünnung 1:1000 – 1:5000 (den Angaben des Herstellers folgend / siehe Tabelle 7) dazugegeben und für 1 Stunde bei Raumtemperatur oder bei 4°C über Nacht inkubiert. Antikörper Hersteller Verdünnung c-Fos, rabbit polyclonal #052x Santa Cruz 1:1000 PhosphoPlus c-Jun (Ser63), c-Jun (Ser 73) NEB 1:1000 JNK1, rabbit poyclonal #571x Santa Cruz 1:250 ERK 1/2, rabbit poyclonal #V1141 Promega 1:5000 Active MAPK, rabbit poyclonal #V8031 Promega 1:5000 Active JNK, rabbit poyclonal #V7931 Promega 1:5000 Cyclin D1, mouse IgG2a #66271A PharMingen 1:500 Anti-mouse-Antibody, # NA 931 Amersham 1:1000 Anti-rabbit-Antibody, # NA 934 Amersham 1:1000 Antibody Kit , #9260 Tabelle 7 : Verdünnung der Antikörper Nach dreimaligen Waschen der Membran in 1x TBS für 5 min wurde gebundener 1.Antikörper mit einem 2.Antikörper (anti-Mouse IgG oder anti-Rabbit IgG), an den eine Peroxidase gekoppelt ist, dekoriert (Verdünnung 1:1000 in Milk Powder Blocking Puffer – Inkubation 45 min bei Raumtemperatur). Wieder folgte ein dreifacher Waschschritt in 1xTBS und dann die Detektion durch Chemolumineszens. Hierbei wird Luminol unter alkalischen Pufferbedingungen durch die an den Zweitantikörper gekoppelte Peroxidase und Wasserstoffperoxid oxidiert und dabei das Luminol in einen - 45 - 3. Methoden angeregten Zustand versetzt. Bei Übergang in den Grundzustand wird ein Lichtquant emittiert, welches auf einem Röntgenfilm detektiert werden kann. Zur Durchführung wurde die Membran für 1 min in eine ECL-Chemolumineszenz Reagenz inkubiert und die nun fluoreszierenden Immunkomplexe auf XAR-5 Filmen detektiert. 3.4.5.4 Stripping Um einen Blot ein zweites Mal mit Antikörpern zu dekorieren, wurden die gebundenen Antikörperkomplexe durch dreimaliges Waschen in Stripping Buffer bei 50°C gelöst. Anschließend wurde die Membran wieder in Blocking Puffer inkubiert und dann neu mit Antkörpern dekoriert. Stripping Buffer : 3.4.6 62,5 mM 100 mM 2% TRIS β-Mercaptoethanol SDS MAP Kinase Assay Der Kinase Assay ist eine Methode, die Aktivität einzelner Kinasen zu bestimmen. Die entsprechende Kinase wird zuerst mittels eines an Sepharose gekoppelten Antikörpers immunpräzipitiert, die daraufhin in vitro nach Zugabe von ATP ein entsprechendes Substrat für eine bestimmte Zeit phosphoreliert. Als Substrat diente das rekombinant hergestellte physiologische Substrat. Anschließend wurde der Phosphorelierungsgrad des Substrats (Maß für die Aktivität der Kinase) mittels eines phosphospezifischen Antikörpers im Westernblot detektiert. 3.4.6.1 Immunpräzipitation Zu 200µg Gesamtzellextrakten, die mit dem NEB Lysis Puffer hergestellt wurden, wurde 15 µl Sepharose-immobilisierter phosphospezifischer Antikörper gegen MEK1/2 oder ERK1/2 gegeben, über Nacht bei 4°C auf der Rotationsschaukel inkubiert, und dann für zwei Minuten bei 2000 U/min abzentrifugiert. Die Sepharose gekoppelten Antikörper wurden nun zweimal in 500 µl NEB Lysis Puffer und danach zweimal in 500 µl NEB Kinase Puffer gewaschen. - 46 Kinase Puffer : 25 mM 10 mM 0.1 mM 5 mM 2mM 3. Methoden TRIS MgCl2 Na3VO4 β-Glycerolphosphat DTT 3.4.6.2 Kinase Assay Das Präzipitat wurde in 50 µl Kinase Puffer resuspendiert und mit 200 µM ATP und 2 µg Substrat – entweder rekombinantes ERK2 oder Elk-1-Gluthtation-S-Transferase Fusionsprotein – supplementiert. Die Kinase Reaktion wurde 30 min bei 37°C ausgeführt. Das Reaktionsprodukt wurde in der SDS-PAGE aufgetrennt und der Phosphorelierungsgrad des Substrats durch Westernblot mit phosphospezifischen Antikörpern detektiert. - 47 - 4. Ergebnisse 4 Ergebnisse 4.1 Etablierung des AP-1 EMSA Der Gelretardierungsassay oder Electrophoretic Mobility Shift Assay dient dem Nachweis von Protein-DNA Wechselwirkung, wie sie u.a. physiologisch bei der Interaktion von Transkriptionsfaktoren mit Promoter und Enhancer DNA Sequenzen stattfindet. Hierzu wird ein Proteinextrakt – entweder Gesamtproteinextrakt oder auch Kernextrakt – mit einem radioaktiv markierten Oligonukleotid inkubiert, welches die Konsensusbindungssequenz des zu untersuchenden Transkriptionsfaktors beinhaltet. Um unspezifische Bindungen zu vermeiden, wird BSA zum Blockieren von Protein-bindenden DNA-Sequenzen neben der markierten Sonde und Poly [dIdC] zum Abfangen von unspezifisch DNA-bindenden Proteinen hinzugefügt. Zur Detektion der gebildeten DNA-Protein Komplexe wird das Reaktionsprodukt in der Polyacrylamid Gelelktrophorese (PAGE) unter nativen nicht-denaturierenden Bedingungen aufgetrennt und durch Autoradiographie oder im Phosphoimager visualisiert. Ein zentraler Teil dieser Arbeit beschäftigt sich mit den Reaktionen der MagenepithelWirtszelle nach Kokultivation mit H.pylori und dies im besonderen bezogen auf den Transkriptionsfaktor AP-1 und die zu seiner Aktivierung führenden „Mitogen Activated Protein Kinases“ (MAPK) Signaltransduktionswege. Wir interessierten uns für die Frage, ob H.pylori AP-1 in Magenepithelzellen aktiviert. Bei der Etablierung eines AP-1 EMSA ergaben sich folgende Schwierigkeiten : (1) AP-1 und die es aktivierenden MAPK Signaltransduktionswege werden durch Serum aktiviert. In proliferierenden Zellen/Zellinien, insbesondere in Tumorzellinien, führt dies zu konstitutiv aktiven AP-1 und MAP-Kinasen, so daß eine mögliche zusätzliche Aktivierung durch H.pylori verdeckt wird. Auch die Bakteriensuspensionen, die in serumhaltigem Medium kultiviert werden, können das Ergebnis verfälschen : war es nun das Bakterium oder nur das im Medium enthaltende Serum, das zu einer Aktivierung von AP-1 führte ? (2) AP-1 wird sowohl transkriptionell als auch posttranskriptionell reguliert (s. Einleitung 1.9) – bei letzterem kann die DNA-Bindungsaffinität von AP-1 durch Abspalten von - 48 - 4. Ergebnisse Phosphatgruppen an Thr231, Ser243, Ser249 durch unspezifische Phosphatasen erhöht werden, und so im EMSA zu hohen Hintergrundwerten führen. Ad (1) Zuerst sollte mit Hilfe des EMSA untersucht werden, ob H.pylori in der Lage ist, den Transkriptionsfaktor AP-1 zu aktivieren. Bei den ersten Versuchen erfolgten keine Waschschritte zur Vermeidung von Störeffekten durch Serum : die Magenepithelzellinien AGS und KATO III wurden in serumhaltigen Medium ausplattiert und die Bakterien in 12 TP A 20 1 15 uc ell a1 0m uc in ell a1 h G2 7 Br Br H. pylori Stamm : - serumhaltigem Brucella Medium zur Infektion hinzugefügt. Nach 1h Kokultivierung der Exp 8 : Stimulation von in Serum kultivierten (10% FCS) AGS Zellen mit verschiedenen H.pylori Stämmen, die ebenfalls in Serumhaltigen Medium kultiviert wurden. AGS Zellen in der logarithmischen Wachstumsphase wurden mit verschiedenen cagA+ H.pylori Stämmen (G27, 151) und cagA- (2012), sowie Bakteriummedium (Brucella) für 1h kokultiviert. Als Negativkontrolle dienten unstimulierte Zellen, als Positivkontrolle TPA (100ng/µl). Gesamtzellextrakte wurden im EMSA auf DNA-Bindeaktivität von AP-1 untersucht. Das ausgefüllte Dreieck zeigt die Position von spezifischen AP1/DNA Komplexen an, das leere Dreieck markiert ungebundenes Oligonukleotid. 1 2 3 4 5 6 7 Zellen mit verschiedenen H.pylori Stämmen wurden Gesamtzellproteinextrakte erstellt und auf DNA-Protein Bindungen mittels EMSA untersucht. Abb. Exp 8 demonstriert das wesentliche Problem : sämtlichen Linien, auch die als Nullkontrolle dienenden unstimulierten Zellen, wiesen eine deutliche AP-1 Bande auf. Auch zu hohe Zelldichten kann Streß für die Zelle bedeuten und so zur Aktivierung von AP-1 führen (Daten nicht gezeigt). - 49 - 15 1 NT AT CC TP A - 15 1 NT CT AT CC TP A AGS Serum 10% - H. pylori Stamm : CT AGS Serum 0% 4. Ergebnisse 1 2 3 4 7 8 5 6 9 10 Exp 23 : Vergleich von für 12h Serum gehungerten AGS Zellen mit nicht gehungerten Zellen bzgl. der AP-1 Stimulierbarkeit durch H.pylori. AGS Zellen in der logarithmischen Wachstumsphase wurden (1) entweder 2x in serumfreien Medium gewaschen, dann in diesem ausplattiert und für 12h serumdepriviert oder (2) ohne Waschschritt in serumhaltigen Medium ausplattiert. Nach 12 h wurden die Zellen für 1h mit verschiedenen cagA+ H.pylori Stämmen stimuliert, und anschließend die Zellextrakte im EMSA auf aktiviertes AP-1 untersucht. Um eine niedrigere AP-1 Hintergrundsaktivität zu erreichen, wurde versucht, die Zellen mittels 12h Kultivierung in HAM’s F-12 SER∅ Medium in die Ruhephase G0 zu bringen. Abb. Exp. 23 stellt die Ergebnisse von gehungerten Zellen (Spur 1-5) und in Serum kultivierten Zellen (Spur 6-10) gegenüber – die serumverarmten Zellen weisen eine deutlich niedrigere AP-1 Aktivität in der Nullkontrolle auf. Auch das als Nullkontrolle mitgeführte Bakterienmedium Brucella wies regelmäßig eine hohe AP-1 Aktivität auf. Dies legte den Schluß nahe, daß gar nicht H.pylori als vielmehr das serumenthaltende Nährmedium für die AP-1 Aktivierung verantwortlich war (Abb. Exp. 29). 4. Ergebnisse 1xP BS 1m FC l S( 100 %) Br 1 uc( 10% 00µl FC S) 1m l Stimulus : - - 50 - Exp. 29 : AP-1 Induktion von 12h gehungerten AGSZellen durch serumhaltiges Bakterienmedium oder FCS. Für 12h serumdeprivierte Zellen wurden für 1h mit 1xPBS, 100 µl FCS oder 1ml Bakterienmedium (10% Serum) inkubiert. Dann wurden die Zellextrakte im EMSA auf aktiviertes AP-1 untersucht. 1 2 3 4 Nun wurden die Bakterien zuerst immer 3x in HAM’s F-12 SER∅ Medium gewaschen, bevor sie für Stimulationsversuche eingesetzt wurden. Nach Einführen dieser Waschschritte sowohl für die Bakterien als auch für die Zellen konnte die Hintergrundsaktivität auf ein vertretbares Maß reduziert werden (Abb. Exp 34). Daß nun tatsächlich H.pylori für die Stimulation von AP-1 verantwortlich ist, zeigt sich am besten daran, daß der cag A- H.pylori Stamm 2012 nicht in der Lage ist, AP-1 zu aktivieren (Spur 3). Würde die Aktivierung auf mögliche Kontamination mit Serum zurückzuführen sein, sollte C AT C CT NT 20 12 1 51 H. pylori Stamm : - man erwarten, daß alle mit Bakterien stimulierten Zellen AP-1 Aktivität aufweisen. Exp. 34 : AP-1 Induktion in 12h gehungerten AGS-Zellen durch H.pylori in serumfreiem Medium. Für 12h serumdeprivierte Zellen wurden für 1h mit verschienen H.pylori Stämmen stimuliert. Dabei waren die in serumhaligen Medium kultivierten Bakterien vor dem Stimulationsexperiment 3x in serumfreiem Medium gewaschen worden. Zellextrakte wurden im EMSA auf aktiviertes AP-1 untersucht. 1 2 3 4 5 - 51 - 4. Ergebnisse Da nach zu langer Kultivierung der Zellen in serumfreiem Medium die Zellen in Apoptose gehen und zuvor der Entzug von Überlebenssignalen in Form von Serumfaktoren als externer Streß eine Aktivierung von AP-1 zu Folge hat, ist das Zeitfenster, indem die Zellen langsam in die G0-Ruhephase übergehen, aber noch nicht in „Apoptose Streß“ geraten, begrenzt. Die beste Stimulierbarkeit ergab sich nach 10-12h (Spur 1-3 und 4-6) (Abb. Exp 33), was sich in weiteren Experimenten bestätigte. Werden die Zellen länger gehungert, steigt zum einen die Hintergrund AP-1 Aktivität an (Spur 10), gravierender noch aber nimmt die Stimulierbarkeit der Zellen ab (Spur 7&8 bzw. 10&11). 11 12 7 9 - 8 TP A 7 G2 6 16 TP A 5 7 7 4 TP A - 3 14 G2 1 - 2 7 TP A - H. pylori Stamm : 12 G2 11:30 G2 Hungern [h] : Exp 33 : Einfluß der Länge der Hungerzeit auf die AP-1 Induzierbarkeit in AGS-Zellen. AGS Zellen wurden für unterschiedliche Zeiten Serum verarmt und anschließend für 1h mit H.pylori G27 oder TPA (100ng/ml) stimuliert. Zellextrakte wurden im EMSA auf aktiviertes AP-1 untersucht. Ad (2) Eine weitere Möglichkeit, die zu hohen Hintergrundaktivitäten von AP-1 im EMSA führen kann, wird durch die posttranslationale Regulierung von AP-1 bedingt. Wird AP-1 an den Stellen Thr231, Ser243 und Ser249 durch Phosphatasen, die sich in jeder Zelle befinden und bei der Erstellung der Zellextrakte ihre Wirkung an den AP-1 Molekülen entfalten können, dephosphoreliert, erhöht sich hierdurch die DNA-Bindungsaffinität von AP-1 [Boyle, 1991]. Prinzipiell gibt es zwei Möglichkeiten, die Phosphatasen zu inhibieren : Man kühlt die Zellkulturschalen direkt nach Beendigung des Stimulationversuches für die Zeit der Proteinextraktgewinnung auf Eis, und senkt so die enzymatische Aktivität der Phosphatasen. Alternativ kann man Phosphatase Inhibitoren (NaF, Na Pyrophosphat, Na Vanadat) dem Lysis-Puffer hinzufügen. - 52 - 4. Ergebnisse Das Ernten und die Herstellung der Zellextrakte erfolgte von nun immer auf Eis. Verschiedene Phosphatase Inhibitoren wurden bei Zellextrakten von unstimulierten Zellen versus TPA-stimulierten Zellen ausgetestet (Abb. Exp 46). Bei AGS Zellen konnten die besten Resultate mit Hinzufügen von 5mM Na3VO4 und 50mM NaF zum Lysis-Puffer erzielt werden (Spur 7-10). Bei den KATO III Zellen ergaben sich auch nach Hinzugabe der Phosphatase Inhibitoren oder Veränderung der Hungerzeiten (s.o.) keine wirklich zufriedenstellende Ergebnisse (Daten nicht gezeigt). Dies mag unter anderem dadurch erklärt werden, daß viele Tumorzellinien eine konstitutiv aktivierende ras-Mutation besitzen, die zu permanent aktiven MAP-Kinasen und AP-1 führt, unabhängig, ob die Zellen vorher gehungert wurden oder nicht. Aus diesem Grunde wurde für die weiteren Experimente v.a. die AGS- 4 35 50 0 mM mM N Na aC l F i 35 5 m 0 mM M Na NaC l 3 VO 35 30 0 mM mM N Na aC l PP TP A 5 TP A - TP A - 4 - 3 TP A 2 - NF To -κB tex 1 TP A - Lysis Buffer : H. pylori Stamm : 27 0m MN aC l Zellinie verwendet. 6 7 8 10 9 Exp 46 : Einfluß des Lysispuffer auf die DNA-Bindungsaktivität von aktivierten AP1Molekülen. Serumgehungerte AGS Zellen wurden entweder unbehandelt gelassen oder für 1h mit TPA stimuliert (100ng/ml). Anschließend wurden die Zellen auf Eis geerntet, Zellextrakte mit verschiedenen Lysispuffer erstellt und dann im EMSA auf aktiviertes AP-1 untersucht. Desweiteren wurde auch ein Lysis-Puffer der Firma NEB zur Herstellung von Proteinextrakten für MAP-Kinase Assays, der auch Phosphatase Inhibitoren enthält, auf die Tauglichkeit für EMSA getestet. Für AP-1 EMSA ergaben sich im Vergleich zu dem eigenen Lysis Puffer mindestens gleichwertige Ergebnisse (Abb. Exp 62), bei den NF-κB EMSA waren die Ergebnisse mit NEB Lysis-Puffer in punkto Auflösen der einzelnen DNA-Protein Komplexe in der PAGE sogar besser (Daten nicht gezeigt). Da die mit NEB Lysis-Puffer erstellten Proteinextrakte sowohl für EMSA, als auch für MAP-Kinase Assays und Western Blots benutzt werden konnten, wurde dieser fortan eingesetzt. 4 5 6 7 TP A 3 G2 7 TP A 2 - 1 G2 H. pylori Stamm : - Lysis Buffer : 4. Ergebnisse NE B NF To -κB te x - 53 - Exp.: 62 : Vergleich von NF-κB Lysis-Puffer mit Lysis-Puffer von NEB. Serumgehungerte AGS Zellen wurden unbehandelt gelassen oder für 1h mit H.pylori G27 oder TPA (100ng/ml) stimuliert. Anschließend wurden die Zellen auf Eis geerntet, Zellextrakte mit verschiedenen Lysis-Puffer erstellt und dann im EMSA auf aktiviertes AP-1 untersucht. 4.2 H.pylori induziert AP-1 in AGS Zellen Als erstes sollte untersucht werden, ob H.pylori den Transkriptionsfaktor AP-1 in AGS-Zellen aktivieren kann. Die Kokultivierung von AGS Zellen mit verschiedenen H.pylori Stämmen für 1h führt – nach Herstellen der Zellextrakte und Analyse im EMSA - zum Erscheinen neuer DNA-Protein Komplexe (Abb. Exp 96), die durch einen Kompetitionsassay als AP-1 identifiziert wurden (Abb. Exp 37, Spur 11,12). Dabei variiert die Fähigkeit, AP-1 Komplexe zu induzieren, unter den verschiedenen H.pylori : während die Stämme G27 und ATCC 43405 deutliche DNA-Protein-Banden (Spur 2,5) erzeugten, zeigten Stamm 151 und NTCT 11638 mittlere Aktivierung von AP-1 (Spur 3,4). Der cag A- Stamm 2012, der somit als apathogen eingestuft wird, vermochte keine Aktivierung von AP-1 (Spur 6). Als Negativkontrolle wurden unstimulierte Zellen verwendet (Spur 1), zur Positivkontrolle TPA stimulierte Zellen (Spur 7). 4.2.1 Identifikation der AP-Untereinheiten - 4. Ergebnisse NT CT AT 1163 CC 8 43 40 20 12 5 TP A H.pylori Strain : G2 7 15 1 - 54 - Exp. 96 : AP-1 Induktion in AGS Zellen durch verschiedene H.pylori Stämme 1 2Zellen 3 wurden 4 5für 6 7 verschienen cagA+ H.pylori Stämmen (G27, 151, Für 12h serumdeprivierte 1h mit NTCT,ATCC) und cagA Stämmen (2012) kokultiviert. Zellextrakte wurden im EMSA auf aktiviertes AP-1 untersucht. AP-1 tritt als Homo- oder Heterodimer auf, wobei sich die Subeinheiten aus den Portoonkogenfamilien Fos (c-Fos, FosB, Fra-1, Fra-2), Jun (c-Jun, JunB, JunD) und ATF (ATF2, ATF3, B-ATF) rekrutieren. Die Zusammensetzung des Homo- oder Heterodimeres beeinflußt die DNA-Affinität und Spezifität sowie die Transaktivierungsfähigkeit. Um die genaue Zusammensetzung des durch H.pylori induzierten AP-1 zu bestimmen, wurde eine Supershift Analyse durchgeführt. Der Antikörper α-c-Fos all, der allgemein Proteine der FosFamilie erkennt, und der Antikörper α-c-Fos führten zu einem Shift der Bande (Abb. Exp37, Spur 3,4), die Antikörper α-Fra1 und α-Fra2 hatten keinen Einfluß auf den DNA-Protein Komplex (Abb. Exp37, Spur 5,6). Analog zeigten die Antikörper α-c-Jun und α-c-Jun all (erkennt alle Jun-Proteine) Interaktion mit dem AP-1 Komplex (Abb. Exp37, Spur 7,8), nicht jedoch die Antikörper gegen JunB und JunD (Abb. Exp37, Spur 9,10). Zusammengefaßt besteht der durch H.pylori G27 induzierte AP-1 Komplex v.a. aus einem Heterodimer von cJun und c-Fos. Antibody : - - Fr a- Competitor : - - - - - - - - - - H.pylori G27 : - + + + + + + + + + 1 2 3 4 5 6 7 8 9 10 - - AP -1 NF -κB 2 c-J un a ll c-J un Ju nB Ju nD 4. Ergebnisse Fr a- 1 s c-F o c-F o sa ll - 55 - + + 11 12 Exp 37 : Supershift und Kompetitionsanalyse von H.pylori aktivierten AP-1 AGS Zelle wurden für 1h mit H.pylori G27 kokultiviert und Zellextrakte im EMSA auf AP-1 Aktivität untersucht. Spur 1, unbehandelte Zellen; Spur 2, mit G27 stimulierte Zellen; Spur 3-10, Zellextrakat von Spur 2 wurden mit angegebenem Antikörper oder nicht markierten Oligonukleotid (100x Überschuß) inkubiert. Das ausgefüllte Dreieck markiert die Position des AP-1/DNA Komplexes, während der gefüllte Kreis die Lage der Antikörper/AP-1/DNA Komplexe markiert. Das mittlere Dreieck kennzeichnet ungebundenes Oligonukleotid. 4.2.2 Kinetik der AP-1 Aktivierung Neben der Zusammensetzung des AP-1 Dimers ist auch der zeitliche Verlauf entscheidend für die physiologische Wirkung – siehe auch Einleitung (1.8.2). Die AP-1 Aktivierung erreichte nach 90-120 min ihr Maximum (Abb. EXP115, Spur 5,6) und dauerte über mehrere Stunden an (Abb. EXP 92). H.pylori bewirkt also eine anhaltende AP-1 Aktivierung mit einem Maximum nach 90-120 min. 4. Ergebnisse 45 60 90 15 30 60 90 90 Zeit (h) : 1 2 3 4 5 6 7 8 9 10 11 G2 G2 G2 TP A TP A 30 7 20 1 15 7 H. pylori : 0 H. pylori : Zeit (min) : 2 7-∆ G - 56 - 0 2 4 6 2 4 12 13 14 15 16 17 Exp. 115 : Kinetik der AP-1 Stimulation durch verschiedene H.pylori Stämme AGS Zellen wurden unbehandelt gelassen (Spur 1) oder für die angegebenen Zeiten mit H.pylori G27 (Spur 2-6, 12-15) bzw. der isogenen Mutante G27-∆G (Spur 7-9), H.pylori 2012 (Spur 10) oder TPA (100ng/ml) (Spur 11, 16,17) kokultiviert. Zellextrakte wurden im EMSA auf aktiviertes AP-1 untersucht. 4.2.3 Aktivierung von AP-1 durch Mutanten des H.pylori Stamm G27 In früheren Arbeiten aus unserem Labor konnte A. Münzenmeier und E. Glocker zeigen, daß für die Aktivierung des Transkriptionsfaktor NF-κB durch H.pylori bakterielle Gene notwendig sind, die auf der Pathogenitätsinsel cag I lokalisiert sind. Wie in der Einleitung beschrieben, kodieren diese Gene für einen Typ IV Sekretionsapparat, der entweder selbst durch Zelloberflächenkontakt für die NF-κB Aktivierung verantwortlich ist, oder für die Sekretion eines NF-κB induzierendem Agens. Wir interessierten uns nun für die Gene, die für die AP-1 Aktivierung notwendig sind. Hierfür wurden 12 isogene Mutanten des Stammes G27, jede in einem Gen der Pathogenitätsinsel verändert, im EMSA auf ihre Fähigkeit getestet, AP-1 zu induzieren. Mutationen in cagF und cagN veränderten nicht die Fähigkeit von H.pylori G27 AP-1 zu aktivieren (Abb. EXP 65, Spur 4,13,14), wohingegen H.pylori G27 mit Mutationen in cagE, cagG, cagH, cagI, cagL und cagM AP-1 nicht über Hintergrundniveau zu aktivieren vermochten (Abb. EXP 65, Spur 3,5-12). Interessanterweise stimmen diese Ergebnisse exakt mit denen für die Aktivierung von NF-κB überein [Glocker, 1998]; dieses Ergebnis spricht dafür, daß dieselben Pathogenitätsgene für die Aktivierung von NF-κB und AP-1 verantwortlich sind, obwohl diese Transkriptionsfaktoren über ganz - 57 - 4. Ergebnisse unterschiedliche intrazelluläre Signaltransduktionswege reguliert werden. AP-1 kann durch 1 2 3 4 5 6 7 8 G2 7-∆ L G2 7-∆ M G2 7-∆ M G2 7-∆ M G2 7-∆ N G2 7-∆ N 20 12 20 74 - G2 7-∆ E G2 7-∆ F G2 7-∆ G G2 7-∆ H G2 7-∆ H G2 7-∆ I H.pylori Strain : G2 7 eine Vielzahl von Signalwegen aktiviert werden, u.a. durch MAP Kinase Kaskaden. 9 10 11 12 13 14 15 16 Exp 65 : AP-1 Stimulation durch H.pylori mit Mutationen in der cag Pathogenitätsinsel AGS Zelle wurden für 1h mit H.pylori G27 (Spur 2) und isogenen Mutanten (Spur 3-14), sowie den cagA- Stämmen 2012 & 2074 kokultiviert. Kontroll-Zellen wurden unbehandelt gelassen (Spur 1). Zellextrakte wurden im EMSA auf aktiviertes AP-1 untersucht. 4.3 H.pylori aktiviert die ERK/MAP Kinase Kaskade Verschiedene MAP-Kinase Kaskaden, u.a. der ERK, der SAPK/JNK und der p38 Signaltransduktionsweg, spielen in der Regulation von AP-1 eine wichtige Rolle. Um die Signalkaskaden, durch die H.pylori AP-1 aktiviert, näher zu untersuchen, kann man sich verschiedener Methoden bedienen. Ein indirekter Nachweis ist durch spezifische Inhibition einer bestimmten Kinase möglich, entweder durch einen spezifischen chemischen Inhibitor oder genetischen Knock-out bzw. Transfektion der entsprechenden isogen dominant negativen Kinase. Der chemische Inhibitor beherbergt immer das Problem der Spezifität : mit welcher Konzentration kann man spezifisch die zu untersuchende Kinase möglichst - 58 - 4. Ergebnisse vollständig inhibieren, ohne andere Enzyme zu beeinflussen ? Für die MEK1/2-Kinase existieren zwei in der Fachliteratur anerkannte Inhibitoren, PD 98059 und U0126 [Dudley, 1995; Favata 1998]. Zellen mit einem Knock-out für die betreffende Kinase sind ein ideales System, aber wegen den oft letalen oder proliferationsinhibierenden Auswirkungen des Knock-out schwer zu realisieren. Die Überexpression einer dominant negativen Mutante der zu untersuchenden Kinase ist eine oft verwendete Methode für die Untersuchung von Signaltransduktionswegen. Jedoch bedeutet die unphysiologisch starke Expression der dominant negativen Mutante eine Veränderung zum physiologischen Ruhezustand der Zelle, so daß gewonnene Ergebnisse unter dieser Veränderung zu sehen sind. In dieser Arbeit wurden die chemischen Inhibitoren von MEK1/2, PD 98059 und U0126, verwendet. AGS-Zellen wurden 30 min vor der Stimulation durch H.pylori und TPA als Positivkontrolle mit unterschiedlichen Konzentration der Inhibitoren behandelt. Beide Inhibitoren führten bei Konzentrationen von 10 µM (PD 98059) bzw. 1 µM (U0126) zu einer PD 98059 Inhibitor : Concentration (µM) : - - 0.1 1 10 50 H.pylori G27 : - + + + + + 1 2 3 4 5 6 Inhibitor : U0126 Concentration (µM) : - - 0.1 1 10 - H.pylori G27 : - + + + + - 1 - 10 - TPA : - - - - - + + + 1 2 3 4 5 6 7 8 Exp 49 : Effekt von MAP Kinase Inhibitoren auf die Stimulation von AP-1 durch H.pylori AGS Zellen wurden 30 min vor Stimulationbeginn mit PD98059 oder U0126 in den angegebenen Konzentrationen inkubiert. Daraufhin wurden sie für 1h mit H.pylori G27 oder TPA kokultiviert. Zellextrakte wurden im EMSA auf aktiviertes AP-1 untersucht. - 59 - 4. Ergebnisse vollständigen Inhibition der AP-1 Aktivierung durch H.pylori G27 (Abb. Exp 49,79). Dies bedeutet unter der Prämisse der Spezifität (s.o.) eine notwendige Beteiligung des MEK1 – ERK 1/2 Signaltransduktionsweges an der Aktivierung von AP-1 durch H.pylori. Der direkte Nachweis, ob H.pylori eine bestimmte Signalkaskade aktiviert, ist u.a. durch einen Kinase Assay möglich. Hierfür wurden AGS-Zellen mit H.pylori G27 bzw. H.pylori G27 und 2012, sowie TPA als Positivkontrolle, stimuliert und Zellextrakte erstellt. Aktivierte MEK1/2 wurde mit einem phosphospezifischen Antikörper gegen phospho-Ser-217 und phospho-Ser-221 immunpräzipitiert – dies sind die Regulationsstellen für MEK1, eine Phosphorelierung entspricht der Aktivierung des Enzyms. Anschließend wurde die Phosphorelierungs/Kinase Aktivität des Immunpräzipitats in vitro mit ERK2 als Substrat getestet und der Phosphorelierungsgrad von ERK2 dann im Westernblot mit einem H. - H. - Treatment : py lor py i G27 TP lori G A 27 phosphospezifischen Antikörper gegen phospho-Thr-202 und phospho-Tyr-204 gemessen. ERK P ERK 1 2 4 5 6 Exp 71 : H.pylori induziert MEK1/2 in AGS Zellen AGS Zellen wurden unbehandelt gelassen (Spur 2,3) oder für 1h mit H.pylori G27 (Spur 4,5) oder TPA (100ng/ml) (Spur 6) stimuliert. Anschließend wurden die Zellextrakte mit einem Antikörper gegen aktivierte MEK1/2 Kinase immunpräzipitiert. Spur 1, Größenmarker. (Oben) Die präzipitierte Kinase wurde in einem in vitro Kinase Assay auf ihre Aktivität überprüft, wobei rekombinantes ERK-2 als Substrat diente. Daraufhin wurde der Phosphorelierungsgrad des Substrates im Westernblot mit einem phosphospezifischen Antikörper gegen ERK-2 detektiert. (Unten) Der Westernblot wurde gestrippt und mit einem Antikörper gegen ERK-2 dekoriert. - 60 - 4. Ergebnisse Mit H.pylori G27 kokultivierte Zellen wiesen eine deutliche Phosphorelierung von ERK2 auf (Abb. Exp71, Spur 3), was auf eine Aktivierung von MEK1/2 durch H.pylori G27 hinweist. Als Kontrolle, daß in jedem Ansatz gleiche Mengen an ERK2 verwendet wurden, wurde der Westernblot gestrippt und mit einem Antikörper gegen ERK2 neu dekoriert – in allen Spuren waren gleiche Mengen ERK2 vorhanden (Abb. Exp71). Die Phosphorelierung von ERK durch MEK1 führt zu einer Aktivierung dieser Kinase, die wiederum ihrerseits den Transkriptionsfaktor Elk-1 phosphoreliert und damit aktiviert. Es steht zu erwarten, daß die Aktivierung von MEK1 durch H.pylori G27 konsekutiv zu einer Aktivierung von ERK1/2 führt. Dies wurde nun in einem weiteren Kinase-Assay für ERK1/2 H. H. - Treatment : py lor py i G2 7 TP lori 2 A 01 2 überprüft. Die Immunpräzipitation erfolgte mit einem phosphospezifischen Antikörper gegen Elk-1 P Elk-1 1 2 3 4 5 Exp 76 : H.pylori induziert ERK in AGS Zellen AGS Zellen wurden unbehandelt gelassen (Spur 2) oder für 1h mit H.pylori G27 (Spur 3) bzw. 2012 (Spur 4) oder TPA (100ng/ml) (Spur 5) stimuliert. Anschließend wurden die Zellextrakte mit einem Antikörper gegen aktivierte ERK1/2 Kinase immunpräzipitiert. (Oben) Die präzipitierte Kinase wurde in einem in vitro Kinase Assay auf ihre Aktivität überprüft, wobei rekombinantes Elk-1 als Substrat diente. Daraufhin wurde der Phosphorelierungsgrad des Substrartes im Westernblot mit einem phosphospezifischen Antikörper gegen Elk-1 detektiert. (Unten) Der Westernblot wurde gestript und mit einem Antikörper gegen Elk-1 dekoriert. - 61 - 4. Ergebnisse ERK (Thr-202 und Tyr-204), welches die Regulationsstellen von ERK1/2 sind. Als Substrat diente das physiologische Substrat Elk-1, dessen Phosphorelierungsgrad mit einem phosphospezifischen Antikörper (Ser-383) im Western Blot detektiert wurde. Während H.pylori G27 eine starke Phosphorelierung von Elk-1 bewirkte, was einer starken Aktivierung von ERK1/2 entspricht, führte der cag A- H.pylori Stamm 2012 nur zu einer sehr schwachen Elk-1 Phosphorelierung / ERK1/2 Aktivierung (Abb. Exp76). Auch dieser Westernblot wurde gestrippt und zur Kontrolle mit einem Antikörper gegen Elk-1 dekoriert, was gleiche Mengen Substrat in allen Ansätzen ergab (Abb. Exp76). Für die Frage, ob H.pylori auch in vivo zu einer Phosphorelierung und Aktivierung von Elk-1 führt, wurden die Zell-Extrakte aus dem letzten Versuch im Westernblot mittels eines phosphospezifischen Antikörpers gegen Elk-1 getestet. Wiederum zeigte sich, daß H.pylori G27, nicht aber der cag A- Stamm 2012 eine Phosphorelierung und damit Aktivierung von Elk-1 vermag (Abb. Exp 82). Der Nachweis, daß in allen Ansätzen gleiche Mengen an Elk-1 vorhanden waren, konnte aufgrund der physiologisch extrem geringen Konzentration von Elk1 sowie der hierfür nicht ausreichend hohen Sensitivität des „normalen“, nicht phosphospezifischen Antikörpers gegen Elk-1 nicht direkt erbracht werden, eine Färbung des ri 2 py lo TP A - H. Treatment : H. py lo ri G 27 01 2 Blots in Poinceau-Rot zeigte jedoch gleiche Mengen an Gesamtprotein in allen Spuren. Elk-1 P 1 2 3 4 5 Exp. 82 : H.pylori bewirkt Phosphorelierung von Elk-1 AGS Zellen wurden unbehandelt gelassen (Spur 2) oder für 1h mit H.pylori G27 (Spur 3) bzw. 2012 (Spur 4) oder TPA (100ng/ml) (Spur 5) stimuliert. Spur 1, Größenmarker. Zellextrakte wurden im Westernblot mit einem Antikörper analysiert, der spezifisch phosphoreliertes Elk-1 erkennt. - 62 4.3.1 4. Ergebnisse Kinetik der ERK Aktivierung Da die Aktivität der MAP Kinasen exklusiv über die Phosphorelierung des Thr-X-Tyr Motivs reguliert wird, kann der Aktivitätszustand der MAP Kinasen durch entsprechende phosphospezifische Antikörper bestimmt werden. Dies bedeutet im Vergleich zum Kinase Assay eine weniger komplexe Methode. Wir interessierten uns nun für den zeitlichen Verlauf der durch H.pylori bewirkten ERK-Aktivierung. Hierzu wurden Proteinextrakte, mit denen auch der zeitliche Verlauf für die AP-1 Aktivierung untersucht wurde (Abb. Exp 115), im Westernblot mit einem Antikörper gegen das Motiv pTyr-E-pThr von aktivem ERK untersucht. Die höchste Aktivität von ERK wurde nach 30-45 min erreicht (Abb. Exp 115 III; Exp 115-3 : H.pylori bewirkt ERK Phosphorelierung in AGS Zellen. AGS Zellen wurden unbehandelt gelassen (Spur 2) oder für die angegebenen Zeiten mit H.pylori G27 (Spur 3-7) bzw. der isogenen Mutante G27-∆G (Spur 8-10), H.pylori 2012 (Spur 11) oder TPA (100ng/ml) (Spur 12) kokultiviert. Spur 1, Größenmarker. Zellextrakte wurden im Westernblot mit einem Antikörper analysiert, der spezifisch phosphoreliertes ERK1/2 erkennt. Der Blot wurde gestrippt und mit einem nicht phosphospezifischen Antikörper gegen ERK1/2 redekoriert. - 63 - 4. Ergebnisse Spur 4,5). Der cagA- H.pylori Stamm 2012 sowie die isogene Mutante G27 ∆G vermochten ERK nicht zu aktivieren (Abb. Exp 115 III; Spur 8-10,11). Für den Nachweis, daß sich in jeder Spur gleiche Mengen an ERK befanden, der Unterschied der Banden also tatsächlich verschiedene Phosphorelierungs/Aktivierungslevels repräsentierte, wurde der Blot gestrippt und mit einem Antikörper, der sowohl phosphoreliertes wie unphosphoreliertes ERK erkennt dekoriert. 4.4 H.pylori aktiviert JNK Als nächstes stellten wir uns die Frage, ob H.pylori auch JNK aktivieren kann, die gleich wie ERK den Transkriptionsfaktor Elk-1 zu phosphorelieren vermag, darüber hinaus aber auch die Transaktivierungspotenz von c-Jun durch Phosphorelierung entscheidend erhöhen kann, und über weitere Effektoren eine zentrale Rolle in der Signaltransduktion von extrazellulären Streß wie UV-Bestrahlung und präinflammatorischen Zytokinen spielt (s.Einleitung 1.8.3). AGS Zellen wurden für die angegebenen Zeiten mit dem cagA+ H.pylori Stamm G27, dem cagA- H.pylori Stamm 2012 sowie der isogenen G27-Mutante G27-∆G kokultiviert und Proteinextrakte angefertigt. Diese wurden im Westernblot mit einem Antikörper, der spezifisch das pThr-E-pTyr der aktiven JNK erkennt, analysiert. H.pylori stimuliert JNK; dabei wurde das Maximum nach 30 min erreicht (Abb. Exp 115IV; Spur 4). Der cagAStamm 2012 sowie die Mutante G27-∆G hingegen bewirkten keine Aktivierung von JNK (Abb. Exp 115IV; Spur 8-10,11). Um sicherzustellen, daß in allen Spuren gleiche Mengen an JNK vorhanden waren, wurde der Blot gestrippt und mit einem nicht phosphospezifischen Antikörper gegen JNK dekoriert. - 64 - 4. Ergebnisse Exp 115-4 : H.pylori bewirkt JNK Phosphorelierung in AGS Zellen. AGS Zellen wurden unbehandelt gelassen (Spur 2) oder für die angegebenen Zeiten mit H.pylori G27 (Spur 3-7) bzw. der isogenen Mutante G27-∆G (Spur 8-10), H.pylori 2012 (Spur 11) oder TPA (100ng/ml) (Spur 12) kokultiviert. Spur 1, Größenmarker. Zellextrakte wurden im Westernblot mit einem Antikörper analysiert, der spezifisch phosphoreliertes JNK1/2 erkennt. Der Blot wurde gestript und mit einem nicht phosphospezifischen Antikörper gegen JNK1/2 redekoriert. 4.5 H.pylori induziert die Transkription von c-fos Der c-fos Promoter enthält mehrere Kontrollelemente, u.a. ein „Serum Responsive Element“ (SRE). Die Aktivierung der MAPK/ERK führt zu Phosphorelierung des Transkriptionsfaktors Elk-1, der dann als Teil des Tertianary Complex Factor (TCF) zusammen mit dem Serum Response Factor (SRF) an das SRE im c-fos Promoter binden kann. Dies führt zu einer Induktion der c-fos Transkription, und somit zu einem Ansteigen der c-fos mRNA. Wir interessierten uns deshalb für die Frage, ob Kokultivation von AGS-Zellen mit H.pylori zu einer Zunahme der c-fos mRNA führt. Hierzu wurden AGS-Zellen eine Stunde mit H.pylori - 65 - 4. Ergebnisse G27 oder mit TPA (100 ng/ml) stimuliert, Gesamt-RNA gewonnen und jeweils 10µg RNA im Northernblot mit einer c-fos cDNA Sonde untersucht. Sowohl H.pylori als auch TPA bewirkten einen deutlichen Anstieg von c-fos RNA, die bei unstimulierten Zellen nicht detektierbar war (Abb. Exp 68). Zum Nachweis, daß in allen Spuren gleiche Mengen RNA vorlagen, wurde der Blot gestrippt und mit einer Sonde für das Haushaltsgen β-Actin - - H. Treatment : py lor TP iG A 27 hybridisiert. 28S c-fos 18S actin 1 2 3 4 Exp 68 : H.pylori induziert c-fos mRNA Expression in AGS Zellen AGS Zellen wurden unbehandelt gelassen (Spur 1,2) oder für 1h mit H.pylori G27 (Spur 3) oder TPA (100ng/ml) (Spur 4) inkubiert. Gesamt-RNA-Extrakte wurden angefertigt, und jeweils 10 µg im Northernblot mit einer 500bp Sonde für c-fos untersucht (oben). Anschließend wurde der Blot gestript, und mit einer 1200 bp Sonde für β-Actin rehybridisiert (unten). Die Positionen der 28S und 18S rRNA sind gekennzeichnet. Für die Frage, ob die erhöhte c-fos RNA Menge auch zu einer verstärkten c-Fos Expression mit erhöhter c-Fos Proteinkonzentration führt, wurden wieder AGS-Zellen für eine Stunde (A) mit H.pylori G27 inkubiert, wobei in einem Ansatz die AGS Zellen 30 min vor Stimulation mit dem MEK1/2 Inhibitor PD98059 behandelt wurden, oder (B) mit H.pylori G27 und H.pylori 2012 kokultiviert. Zur Positivkontrolle wurden die Zellen mit TPA stimuliert. Proteinextrakte wurden angefertigt und im Westernblot auf c-Fos Protein - 66 - 4. Ergebnisse untersucht. Stimulation von AGS-Zellen mit H.pylori G27 bewirkte eine deutliche c-Fos Expression (Abb. Exp75B, Spur 3) gegenüber ruhenden Zellen (Abb. Exp75B, Spur 2), wohingegen H.pylori 2012, der keine AP-1 Aktivierung im EMSA zeigte (Abb. Exp96, Spur - H. PD 98059 : - - + - Treatment : - H. p H. ylor py i G TP lori 27 20 A 12 Treatment : H. py lor py i G2 l 7 TP ori G A 27 6), auch keine c-Fos Transkription induzierte (Abb. Exp75B, Spur 4). c-Fos 1 2 3 4 c-Fos 5 Actin 1 2 3 4 5 Exp 75 : Effekt von H.pylori induziert c-Fos Proteinlevel in AGS Zellen (A) AGS Zellen wurden unbehandelt gelassen (Spur 2) oder für 1h mit H.pylori G27 (Spur 3,4) oder TPA (100ng/ml) (Spur 5) stimuliert. AGS Zellen in Spur 4 wurden 30 min vor Stimulation mit 50µM PD98059 behandelt. Zellextrakte wurden im Westernblot mit einem Antikörper gegen cFos analysiert. (B) AGS Zellen wurden unbehandelt gelassen (Spur 2) oder für 1h mit H.pylori G27 (Spur 3) bzw. 2012 (Spur 4) oder TPA (100ng/ml) (Spur 5) stimuliert. Spur 1, Größenmarker. Zellextrakte wurden im Westernblot mit einem Antikörper gegen c-Fos analysiert. Der Blot wurde gestript und mit einem Antikörper gegen β-Actin redekoriert. Der Inhibitor PD 98059 hob die Stimulation von c-Fos durch H.pylori vollständig auf (Abb. Exp75A, Spur 4), was konsistent mit den früheren Ergebnissen ist, daß der MEK1/ERKSignaltransduktionsweg notwendig für die AP-1 Induktion durch H.pylori ist. Zur Kontrolle, daß in jeder Spur gleiche Mengen an Protein geladen wurden, wurde der Blot gestrippt und mit einem Antikörper gegen β-Actin dekoriert. Analog der Kinetik für AP-1 im EMSA (Abb. Exp 115 & Exp 92), sollte nun mit denselben Proteinextrakten der zeitliche Verlauf der c-Fos Proteinlevel im Westernblot studiert werden. c-Fos Protein erscheint zuerst nach 45 min Stimulation mit H.pylori (Abb. Exp 115, Spur 5) und erreicht seine maximale Konzentration gleich wie die AP-1 Bindungsaktivität im EMSA nach 90-120 min (Abb. Exp 115, Spur 6,7). 7 G2 H. pylori : Zeit (min) : 4. Ergebnisse TP A 20 12 G2 7 -∆ G - 67 - 0 15 30 45 60 90 90 90 90 c-Fos 2 3 4 5 6 7 8 9 10 Stimulus : Zeit (h) : 0 2 4 Se G2 7 ru m 10 % 1 6 8 2 4 6 8 c-Fos 1 2 3 4 5 6 7 8 9 10 Exp. 115 : Kinetik der c-fos Expression durch verschiedene H.pylori Stämme (A) AGS Zellen wurden unbehandelt gelassen (Spur 2) oder für die angegebenen Zeiten mit H.pylori G27 (Spur 3-7) bzw. der isogenen Mutante G27-∆G (Spur 10), H.pylori 2012 (Spur 9) oder TPA (100ng/ml) (Spur 8) kokultiviert. Zellextrakte wurden im Westernblot mit einem Antikörper gegen c-Fos analysiert. (B) Für die Langzeitkinetik wurden AGS Zellen unbehandelt gelassen (Spur 2) oder für die angegebenen Zeiten mit H.pylori G27 (Spur 3-6) oder in serumhaltigen Medium (10%) (Spur 710) inkubiert. Zellextrakte wurden im Westernblot mit einem Antikörper gegen c-Fos analysiert. 4.6 H.pylori bewirkt c-Jun Phosphorelierung Wie oben gezeigt wurde, besteht der durch H.pylori induzierte AP-1 Komplex aus c-Fos und c-Jun (Abb. Exp37). c-Jun wird sowohl transkriptionell wie posttranslational durch Phosphorelierung reguliert (siehe Einleitung 1.9.2). Wir gingen nun der Frage nach, auf welche Weise H.pylori c-Jun aktiviert. Hierfür wurden AGS-Zellen für 15, 30, 60 und 90 min - 68 - 4. Ergebnisse mit H.pylori G27 sowie für 90 min mit H.pylori 2012 und TPA (Positivkontrolle) inkubiert. Die Proteinextrakte wurden im Westernblot mit einem phosphospezifischen Antikörper für cJun (Ser-73) auf Phosphorelierung an dieser Stelle getestet. Inkubation von AGS-Zellen mit H.pylori G27 für 60 min, stärker nach 90 min bewirkte eine solche Phosphorelierung, wohingegen 90 min Inkubation mit H.pylori 2012 keine Phosphorelierung bewirkte. Anschließend wurde der Blot gestrippt, und mit einem Antikörper gegen c-Jun (NEB) neu dekoriert. Alle Proben zeigten eine vergleichbare Menge an c-Jun (Daten nicht gezeigt), was darauf hinweist, daß H.pylori c-Jun durch Phosphorelierung und nicht transkriptionell aktiviert. Exp. 116 : H.pylori bewirkt c-Jun Phosphorelierung in AGS Zellen. AGS Zellen wurden unbehandelt gelassen (Spur 2) oder für die angegebenen Zeiten mit H.pylori G27 (Spur 1-5) bzw 2012 (Spur 6) oder TPA (100ng/ml) (Spur 7) kokultiviert. Zellextrakte wurden im Westernblot mit einem Antikörper analysiert, der spezifisch an Ser-73 phosphoreliertes c-Jun bindet. - 69 - 4.7 4. Ergebnisse Etablierung von Reportergen Versuchen Nachdem im EMSA das Binden des Transkriptionsfaktors AP-1 an die Consensussequenz nachgewiesen wurde, sollte nun die physiologische Wirkung dieser DNA-Protein Interaktion mittels Reportergenversuche näher untersucht werden. Bei einem Reportergenkonstrukt handelt es sich um ein Plasmid, das ein Reportergen – in dieser Arbeit wurde hierfür das Luciferase-Gen gewählt – unter einem künstlichen Promoter beinhaltet. Je nach Versuch besitzt dieser Promoter eine oder mehrere Bindungsstellen für den zu untersuchenden Transkriptionsfaktor. Alternativ kann auch der Originalpromoter eines Genes oder Teile davon verwendet werden, um die Regulation der Expression dieses Gens zu untersuchen. 4.7.1 Etablierung der Transfektion von AGS-Zellen Bei der Transfektion werden DNA-Plasmide in eukaryontische Zellen eingeschleust. Hierfür stehen eine Vielzahl von beschriebenen Methoden zur Verfügung : (1) Elektroporation (2) Calciumphosphat (3) Liposomale Transfektion (4) Virale / Retrovirale Transfektion In dieser Arbeit wurden drei kommerzielle Transfektionskits auf liposomaler Basis – DOTAP (Boehringer Mannheim), Fugene 6 (Boehringer Mannheim) und SuperFect (Qiagen) - für die Eignung zur Transfektion der Magenepithelzellinie AGS getestet. Folgende Parameter, die die Transfektionseffizienz beeinflussen, wurden optimiert : • Menge der zu transfizierenden DNA : 0,1 – 3 µg • Verhältnis DNA : Transfektionsagens Desweiteren sind auch die Zeitpunkte, wann die Zellen transfiziert werden, wann stimuliert wird und wann geerntet, wichtig für den Ausgang des Experimentes. Ein typischer Zeitplan sah wie folgt aus : - 70 - 4. Ergebnisse Ausplattieren der Zellen 30 -36 Stunden Transfektion 8-10 Stunden Serum Deprivation (optional) 6-10 Stunden Stimulation 8 Stunden Zellernte Abb. : Zeitplan zur Transfektion von AGS-Zellen Als erstes wurden die 3 liposomalen Transfektionsagentien miteinander verglichen. Hierzu wurden 5*104 AGS Zellen in 2ml HAM’s F-12 SER+ für 24h kultiviert und dann mit einem CMV-Luciferase Konstrukt transfiziert – dabei wurden folgende Menge an DNA und Transfektionsagens benutzt : Methode DNA [µg] Ratio DNA:Agens max. RLU Superfect 1-4 1:1 – 1:10 350000 DOPTAP 1-3 1:5 – 1:10 900000 Fugene 6 0,5 – 3 2:3 – 1:4 4000000 Nach 15h Transfektionsdauer wurden die Zellen geerntet und die Luciferase Aktivität, welche unter identischen Bedingungen ein Maß für die Transfektionseffizienz des jeweiligen Agens darstellt, bestimmt. Für die Superfect Methode nahmen die RLU mit steigender Menge an DNA zu, das beste Verhältnis von DNA zu Agens lag bei 1:1 – 1:2 . Anzumerken ist auch, daß bei größeren Mengen Superfect (ab 10µl) die Zellen sichtlich in Mitleidenschaft gezogen wurden und eine Großteil der Zellen Apoptose begangen. - 71 - 4. Ergebnisse DOTAP zeigte bei kleineren DNA-Mengen (1 µg) gute Ergebnisse, das beste Verhältnis von DNA zu Agens betrug 1:7,5. Auch hier wirkte das Agens bei größeren Mengen (ab 15 µl) zytotoxisch. Mit Fugene 6 ließen sich die besten Ergebnisse erzielen. Besonders geeignet erwies sich ein Verhältnis von 2 µg DNA zu 3 µl Fugene 6. Darüberhinaus war Fugene 6 auch am wenigsten zytotoxisch. Superfect 400000 350000 RLU 300000 250000 RLU 200000 150000 100000 50000 0 1 2 3 4 5 6 7 8 9 10 1:1 1:1 1:1 1:2 1:2 1:2 1:5 1:5 1:5 1:10 11 12 1:10 - 72 - 4. Ergebnisse DOTAP 1000000 800000 600000 400000 RLU 200000 0 1 2 3 4 5 6 1:5 1:75 1:10 1:5 1:7,5 1:10 Fugene 6 5000000 RLU 4000000 3000000 RLU 2000000 1000000 0 1 2 3 4 5 6 2 2 2 3 3 3 Exp86: Transfektion eines pCMV-Luc in AGS Zellen mittels verschiedener liposomaler Transfektionskits (A) AGS Zellen wurden mit den angegebenen Mengen an pCMV-Luc Plasmids und Superfect Transfektionsagenz (Qiagen) transient transfiziert. Nach 2 Stunden Inkubation der AGS Zellen mit den DNA-Liposom Komplexen wurde das Medium ausgetauscht. Nach weiteren 13h wurden die Zellen geerntet und die Zellextrakte auf die Luciferase Aktivität unter Verwendung des Luciferase ReportergenAssay Systems (Promega) mit einem Luminometer (EG&G Bertold) gemessen. (B) Analog (A), jedoch wurde das DOTAP Transfektionsagnz (Boehringer/ Mannheim) verwendet. Es erfolgte kein Mediumwechsel. Die Zellen wurden 15h nach Transfektion geerntet und auf Luciferase Aktivität untersucht. (C) Analog (B), jedoch wurde das Fugene6 Transfektionsagens (Boehringer/ Mannheim) verwendet. 4.7.2 Serumdeprivation Da Serum AP-1 aktiviert und Zellen in der logarithmischen Wachstumsphase konstitutiv aktives AP-1 aufweisen (vgl. Abschnitt über Etablierung des AP-1 EMSA), sollte nun der - 73 - 4. Ergebnisse Frage nachgegangen werden, ob sich das Verhältnis der Luciferase Aktivität von unstimulierten Zellen zu TPA-stimulierten Zellen dadurch verbessern ließe, in dem man vor einer 4-8 stündigen Stimulation eine Hungerphase von 6h einlegen würde, um so die Zellen in die Ruhephase G0 zu bringen. Subkonfluent gewachsene Zellen wurden mit 2 µg von dem AP-1 Reportergenkonstrukt pAP1 oder dem Kollagenreporterkonstrukt pKoll-Luc, das eine AP-1 Bindungsstelle beinhaltet, mit dem Fugene Reagens transfiziert. Nach 15 h Transfektion wurden die Zellen entweder für 6h gehungert (Mediumwechsel mit serumfreien Medium) oder ohne Hungerphase für 4, 6 und 8 h mit 100 ng/ml TPA stimuliert. Die größten Unterschiede bzgl. der normalisierten RLU (relative Luciferase Einheiten) zwischen unstimulierten und stimulierten Zellen wurden bei 6h Hungern und 8h Stimulation erreicht. Die Normalisierung erfolgte durch 50ng kotransfizierten Renilla-Luciferase-Normalisierungsvektor. AP-1 Induktion nach Serum Deprivation 25,00 20,00 15,00 Luc/ Ren 10,00 5,00 0,00 Stimulation [h] : 0 Stimulus : ø 4 4 ø TPA 6 ø Reporter : Hungern [h] : 6 TPA 8 ø 8 0 TPA ø 4 ø 4 TPA 6 6 ø TPA 8 ø 8 TPA pAP-1 0 6 ø 6 8 TPA ø 8 TPA pKoll-Luc 6 Exp97: AP-1 Stimulation im Reportergenversuch in AGS Zellen nach Serum Deprivation Ein subkonfluente Monolayer von AGS Zellen wurde mit 2 µl pAP-1 Reporterplasmid (Spur 1-14) bzw. pKoll-Luc Reporterplasmid (Spur 15-18) und 3µl Fu6 Transfektionsagenz transient transfiziert. Als Normierung wurden 50ng ptk-Renilla kotransfiziert. Nach 15h Transfektion wurden die Zellen entweder sofort mit TPA für die angegebene Zeit stimuliert (Spur 1-7) oder vor Stimulation für 6h serumdepriviert (Spur 8-18) und dann stimuliert. Anschließend wurden die Zellen geerntet und Zellextrakte auf Luciferaseaktivität untersucht. Zur Kontrolle, ob die verstärkte RLU-Antwort tatsächlich auf die AP-1 Bindestelle vor dem tk-Promoter in dem pAP-1 Vektor zurückzuführen ist, es sich also um eine AP-1 induzierte Antwort handelt, wurde neben dem pAP-1 Plasmid auch das Mutter-Plasmid pTal, das nur den tk-Promoter vor dem Luciferase Gen beinhaltet, sowie das Reporterkonstrukt pSRE, welches Serum Responsive Elements als Enhancer besitzt, transfiziert. Der Zeitplan war folgender : Tansfektionszeit 10h, Hungerphase 6h, Stimulation 8h. - 74 - 4. Ergebnisse Auch bei den mit dem Basisvektor pTal transfizierten Zellen wiesen die TPA-stimulierten Zellen eine 3fach erhöhte Luciferase Aktivität auf; verglichen mit den Induktionsraten der pAP-1 oder pSRE transfizierten Zellen, die bei 8-10 lagen, lagen sie aber deutlich niedriger. Es handelt sich also zumindest zu einem wesentlichen Teil um AP-1 und SR-Faktor vermittelte Prozesse. H.pylori ist somit in der Lage, physiologisch aktives AP-1 und SRF in ähnlicher Stärke zu induzieren wie TPA. G27 induziert pAP-1 und pSER 12 10 8 Induction 6 4 2 0 Stimulus : ø Reporter : TPA pTal ø G27 pAP-1 TPA ø G27 TPA pSER Exp98 : H.pylori induziert AP-1 abhängige Luciferase Konstrukte AGS Zellen wurden mit 2µl Reporterkonstrukt ohne TF-Bindungsstellen (pTal – Spur 1-3), AP-1 Bindungsstellen (pAP-1) (Spur 4-6) oder SRF-Bindungsstellen sowie 3 µl Fu6 transient transfiziert. Als Normierung wurden 50ng ptk-Renilla kotransfiziert. Nach 15h Transfektion wurden die Zellen für 6h serumdepriviert und anschließend für 8h unbehandelt gelassen oder mit H.pylori G27 oder TPA stimuliert. Zellextrakte wurden auf Luciferaseaktivität untersucht. Die gemessenen RLU (Relative Light Units) wurden mit den RLU-Werten der Renilla Luciferase normiert. Die unstimulierten Kontrolen wurde der Wert 1,0 zugewiesen und die stimulierten Werte jeweils auf diesen bezogen. In weiteren Versuchen wurde versucht, durch andere Wahl der Transfektion- / Hunger- / Stimulations-Zeiten die Aktivierung des Basiskonstrukts pTal zu minimieren, aber es wurden hierdurch auch die Induktionsraten der Plasmide pAP-1 und pSRE verändert (Daten nicht gezeigt). - 75 - 4.8 4. Ergebnisse H.pylori induziert AP-1 und SRF abhängige Genexpression AGS-Zellen wurden mit dem Basisvektor pTal sowie den AP-1 bzw. SRF getriebenen Luciferase-Konstrukten pAP-1 und pSRE transfiziert und mit dem cag A+ H.pylori Stamm G27 sowie dem cag A- H.pylori Stamm 2012 für 8 Stunden stimuliert. Als Negativkontrolle wurden die Zellen unstimuliert gelassen, als Positivkontrolle diente TPA. Sämtliche Stimuli führten zu einer unspezifischen Aktivierung des Basisvektors pTal um den Faktor 2-3. Der cag A+ H.pylori Stamm G27, nicht jedoch der CagA - H.pylori Stamm 2012 vermochte eine deutliche AP-1 bzw. SRF abhängige Antwort um den Faktor 5 bzw. 7 zu induzieren. Dies ist konsistent mit den Ergebnissen für Aktivierung der MEK1-ERK1/2-Elk-1 Signalkaskade, welche zur Induktion von SRF und konsekutiv von AP-1 führt, durch H.pylori G27, nicht jedoch durch H.pylori 2012. Der Promoter von c-fos besitzt eine SRF-Bindungsstelle – und, wie oben gezeigt wurde, führt die Stimulation mit H.pylori G27, nicht jedoch durch H.pylori 2012 zur Expression von c-fos. So fügen sich die Ergebnisse dieses Reportergenversuches nahtlos in die früher erhobenen Ergebnisse und untermauern diese somit bezüglich ihrer physiologischen Relevanz – es wird nicht nur Elk-1 phosphoreliert, es ist auch in der Lage, SRF-abhängige Genexpression zu induzieren. Indutkion von pAP-1 und pSER durch G27 und 2012 8,00 7,00 6,00 5,00 4,00 Induction 3,00 2,00 1,00 0,00 Stimulus : Reporter : ø G27 2012 TPA pTal ø G27 2012 TPA pAP-1 ø G27 2012 TPA pSER Exp110 : Abhängigkeit des cag Status von H.pylori auf die Induktion AP-1 abhängiger Luciferase Konstrukte Transfektion und Stimulation erfolgte analog zu Exp. 98. Zusätzlich wurde hier der cagA- H.pylori Stamm 2012 auf die Induktion AP-1 abhängiger Luciferase Konstrukte untersucht. - 76 4.9 4. Ergebnisse H.pylori induziert keine MRE abhängige Genexpression Die Exposition mit Metallen, wie z.B. Zink, bedeutet für die meisten Zellen eine Stresssituation und führt oft zur Aktivierung einer Metall-Responsive-Element (MRE) induzierten Genexpression, u.a. auch von Metalloproteinasen. Da die chronische Infektion mit H.pylori auch eine andauernde Stressituation darstellt, untersuchten wir, ob die Stimulation von AGS-Zellen mit H.pylori G27 eine MRE-abhängige Genexpression bzw. eine Aktivierung des Promoter der mMTE bewirken kann. Hierzu wurden AGS Zellen mit den Reportergenkonstrukten p4xMRE und pmMTE transfiziert und für 4 Stunden mit H.pylori G27 kokultiviert; zur Positivkontrolle wurden die Zellen mit 200 µM ZnSO4 stimuliert. Die mit ZnSO4 behandelten Zellen wiesen eine sehr deutliche Aktivierung beider Reporterplasmide auf, wohingegen bei den mit H.pylori G27 stimulierten Zellen sich kein Unterschied zu den unstimulierten Zellen ergab. H.pylori G27 führt demnach nicht zu der Induktion einer MRE-abhängigen Genexpression – die Infektion mit H.pylori scheint demnach molekularpathologisch nicht mit der Stressituation durch Metallexposition vergleichbar zu sein. G27 bewirkt keine MRE Antwort 12,00 10,00 8,00 6,00 Induction 4,00 2,00 0,00 ø G27 ZnSO4 ø G27 ZnSO4 Exp117 : Effekt von H.pylori G27 auf Induktion MRE-abhängiger Genexpression AGS Zellen wurden mit 2µl Reporterkonstrukt pmMTE (Spur 1-3), oder Reporterkonstrukt p4xMRE (Spur 4-6) sowie 3 µl Fu6 transient transfiziert. Als Normierung wurden 50ng ptk-Renilla kotransfiziert. Nach 6h Transfektion wurden die Zellen für 4h unbehandelt gelassen oder mit H.pylori G27 oder 200 µM ZnSO4 stimuliert. Zellextrakte wurden auf Luciferaseaktivität untersucht. Die gemessenen RLU (Relative Light Units) wurden mit den RLU-Werten der Renilla Luciferase normiert. Die unstimulierten Kontrollen wurde der Wert 1,0 zugewiesen und die stimulierten Werte jeweils auf diesen bezogen. - 77 - 4. Ergebnisse 4.10 H.pylori und Cyclin D1 Expression Unkontrollierte Aktivierung von MAP Kinase Kaskaden und AP-1, welche beide wesentliche Bestandteile der Signaltransduktion für Zellteilung und -proliferation sind, kann offensichtlich zur Transformation und zur unkontrollierten Proliferation von Zellen führen. Physiologischerweise wird die MAP Kinase ERK durch ein Mitogen aktiviert, z.B. das Binden von EGF an seinen Rezeptor EGF-R mit konsekutiver Rezeptorphosphorelierung und Aktivierung der Raf-MEK1-ERK1 Kaskade (s. Einleitung), welches konsekutiv zur AP-1 Aktivierung führt. Die Progression durch den Zellzyklus von G1 zur S Phase und damit Übergang zur Zellteilung wird durch D-Typ Cycline reguliert, welche Komplexe mit „Cyclin Dependent Kinase“ CDK 4 & 6 bilden. Diese Cyclin-D-CDK Komplexe wiederum phosphorelieren daraufhin das Rb-Protein, wodurch der Rb-gebundener Transkriptionsfaktor E2-F freigesetzt wird. Daraufhin initiiert E2-F die Transkription von Genen für die Synthese von DNA und bewegt die Zelle so in die S-Phase. D-Cycline ihrerseits werden wiederum durch AP-1 und die ERK/MAPK Signalwege reguliert – u.a. besitzt der Cyclin D1-Promoter eine AP-1 Bindungsstelle [Watanabe, 1996] und ein konstitutiv aktiver ERK/MAPK Signalweg führt zu G1/S Übergang im Zellzyklus [Lavoie, 1996]. Da eine chronische Infektion mit H.pylori zu einer Hyperproliferation des Magenepithels und seltener auch zu einem Adenokarzinom des Magens führt (siehe auch Einleitung), interessierten wir uns für die Frage, ob H.pylori auch in der AGS-Zellinie zu einer Aktivierung von D-Cyclinen und Hyperproliferation der Zellen führt. Hierzu wurden für 12h gehungerte AGS-Zellen für eine Stunde mit H.pylori G27 und TPA (100ng/ml) stimuliert, Gesamt-RNA Extrakte hergestellt, und diese im Northernblot (Abb. Exp 68) mit einer Sonde für Cyclin D1 hybridisiert : die in unstimulierten Zellen deutlich nachweisbare Cyclin D1 Bande wurde nach Stimulation mit H.pylori G27 oder TPA deutlich schwächer. Darüberhinaus wurde auch RNA aus AGS-Zellen, die durch eine 12h Hungerperiode in die G0-Phase gebracht wurden, mit AGS-Zellen in der früh logarithmischen Wachstumsphase verglichen. Die proliferierenden Zellen wiesen eine deutlich stärkere Cyclin D1-Bande auf (Abb. Exp 68). 4. Ergebnisse gG row Sta r ve th P ha d s Lo 7 TP A G2 - Stimilus : - e - 78 - cyclin D1 1 2 3 4 5 6 Exp 68cyc : H.pylori induziert cyclin D1 mRNA Expression in AGS Zellen AGS Zellen wurden unbehandelt gelassen (Spur 1,2) oder für 1h mit H.pylori G27 (Spur 3) oder TPA (100ng/ml) (Spur 4) inkubiert. Gesamt-RNA-Extrakte wurden angefertigt. Desweiteren wurde RNA von für 12h serumdeprivierten AGS Zellen (Spur 6) bzw. AGS-Zellen in der frühen logarithmischen Wachstumsphase (Spur5) gewonnen. Jeweils 10 µg RNA wurden im Northernblot mit einer 900bp Sonde für cyclin D1 untersucht. Die Positionen der 28S und 18S rRNA sind gekennzeichnet. Um sich die Cyclin D Expression auch auf Proteinebene anzuschauen, wurden für 12 h komplett serumdeprivierte AGS Zellen für 2, 4, 6 und 8 Stunden mit H.pylori kokultiviert, sowie als Positivkontrolle mit frischem 10% serumhaltigen Medium inkubiert und anschließend Proteinextrakte hergestellt. Bei H.pylori stimulierten Zellen sank das in ruhenden Zellen schwach detektierbare Cyclin D1 (Abb. Exp 92, Spur 1) unter die Nachweisgrenze (Abb. Exp 92, Spur 2-5), wohingegen die serumbehandelten Zellen einen deutlichen Anstieg der Cyclin D1 Konzentration mit Maximum nach 6 Stunden aufwiesen (Abb. Exp 92, Spur 7-10). Se r 7 G2 Stimilus : Zeit (h) : 0 2 4 6 4. Ergebnisse um - 79 - 8 2 4 6 8 Cyclin D1 1 2 3 4 5 6 7 8 9 10 Exp 92cyc : H.pylori bewirkt Cyclin D1 Expression in AGS Zellen AGS Zellen wurden unbehandelt gelassen (Spur 2) oder für die angegebenen Zeiten mit H.pylori G27 (Spur 3-6) oder in serumhaltigen Medium (10%) (Spur 7-10) inkubiert. Zellextrakte wurden im Westernblot mit einem Antikörper gegen Cyclin D1 analysiert. Zusammenfassend läßt sich sagen, daß die Kokultivation von AGS-Zellen mit H.pylori G27 zu einer Herunterregulierung von cyclin D1 sowohl auf mRNA als auch auf Proteinebene führt und somit die Zellen eher in einen G0-Arrest versetzt werden als zur Proliferation angeregt zu werden. - 80 - 4. Ergebnisse 4.11 VEGF und H.pylori Sowohl das Ulcus ventriculi et duodeni, als auch das Magenkarzinom werden von Gefäßneubildung begleitet. Bei ersterem wird vermutet, daß die Neovaskularisation den Heilungsprozeß unterstützt, in der Karzinogenese ist die Gefäßbildung essentiell für die Nährstoffversorgung des wachsenden Tumors. Im Randgebiet von Magenulcera bei H.pylori infizierten Patienten fanden sich erhöhte VEGF-Level [Takahashi, 1997] – die Autoren vermuteten, daß dies im Zusammenhang mit der Wundheilung des Ulcus zu sehen ist, wie VEGF allgemein in der Proliferationsphase der Wundheilung eine wichtige Rolle spielt. 4.11.1 Regulation der Angiogenese durch VEGF Die Angiogenese ist ein eng kontrollierter Prozeß, der von pro-angiogenetischen – u.a. Vascular Endothelial Growth Factor – und anti-angiogenetischen Peptiden, wie z.B. Angiostatin, reguliert wird. VEGF besitzt eine zentrale Rolle sowohl bei der Gefäßneubildung als auch als Mitogen für endotheliale Zellen [Leung, 1989]. VEGF Expression wird durch eine Vielzahl von Stimuli induziert. Präinflammatorische Zytokine, wie TNF-α, IL-1, IL-6 und IL-8, sowie die Wachstumsfaktoren bFGF, EGF, TGF-α & β [Gille, 1997; Enholm, 1997; Cohen 1996] führen über die Aktivierung der Protoonkogene ras & c-Raf sowie die konsekutive Aktivierung der ERK-MAPK-Kaskade zur Aktivierung der Transkriptionsfaktoren AP-2 und SP-1, die an den Promoter von VEGF binden und die Expression starten [Milani, 1998 ; Rak, 1995; Grugel, 1995]. Ein weiterer wichtiger Stimulus für die VEGF Synthese ist Hypoxie, die sowohl durch transkriptionelle wie posttranskriptionelle Mechanismen zur verstärkten Expression von VEGF führt - zum einen durch die Aktivierung der Transkriptionsfaktoren HIF-1 und AP-1 [Forsythe, 1996], zum anderen durch Stabilisierung der VEGF mRNA [Stein, 1995]. Kürzlich fanden Pages et. al, daß auch die Aktivierung der MAPK JNK und p38 die VEGF mRNA stabilisiert [Pages, 2000]. - 81 - 4. Ergebnisse 4.11.2 H.pylori induziert die Transkription von VEGF in AGS Zellen Da VEGF in der Pathogenese der Ulcuserkrankung wie auch der Karzinogenese beteiligt ist, und H.pylori die MAPK ERK und JNK aktivieren kann, stellten wir uns die Frage, ob die verstärkte VEGF-Expression in Magenulcera direkt von H.pylori stimuliert wird, oder eher als Reaktion des Körpers auf die Entzündung zu sehen ist. Hierzu wurden für 12h gehungerte AGS-Zellen für 2, 4 und 6 Stunden mit H.pylori G27 Exp94 : H.pylori induziert VEGF mRNA Expression in AGS Zellen. AGS Zellen wurden unbehandelt gelassen (Spur 2) oder für die angegebenen Zeiten mit H.pylori G27 (Spur 3-6) stimuliert. Gesamt-RNA-Extrakte wurden angefertigt, und jeweils 10 µg im Northernblot mit einer 600bp Sonde für VEGF untersucht. (Unten) Ethidiumbromid gefärbtes RNAGel. - 82 - 4. Ergebnisse stimuliert, Gesamt-RNA-Extrakte hergestellt, und diese im Northernblot (Abb. Exp 68) mit einer Sonde für VEGF hybridisiert : in unstimulierten Zellen ließ sich keine VEGF-RNA nachweisen, wohingegen nach 4 Stunden Stimulation eine deutliche Bande sichtbar wurde, die nach 6 Stunden noch stärker wurde (Abb. Exp94, Spur 3,4). Die Kokultivation von AGSZellen mit H.pylori führt also zu einer deutlichen Induktion der VEGF-Expression. Es konnte also gezeigt werden, daß H.pylori direkt die Expression von VEGF-mRNA induziert. Spätere Versuche von cand.med. M. Fleckenstein ergaben weiterhin, daß die Stimulation von AGS-Zellen mit H.pylori Stämmen auch die verstärkte Sekretion von VEGF bewirkt (unveröffentlichte Daten). - 83 - 5. Diskussion 5 Diskussion In dieser Arbeit konnte erstmals die Aktivierung des Transkriptionsfaktors AP-1 durch das gramnegative Bakterium H.pylori in AGS Zellen gezeigt werden. Dabei korrelierte die Aktivierung von AP-1 mit dem Vorhandensein des virulenzassoziierten cagA Gens und dem Vorhandensein der Pathogenitätsinsel cag, wenngleich sich nicht alle Gene dieses 40 kbp DNA-Abschnittes als notwendig erwiesen. 5.1 H.pylori aktiviert den Transkriptionsfaktor AP-1 via MAPK Kaskaden Wir konnten in dieser Arbeit zeigen, daß die Kokultivierung von AGS-Zellen mit H.pylori Stämmen, die eine intakte Pathogenitätsinsel cag besitzen, den Transkriptionsfaktor AP-1 induzieren können, wobei erste DNA Bindeaktivität von AP-1 im EMSA nach ca. 45 min nachweisbar wird und das Maximum nach ca. 2 Stunden erreicht wird. Für die Regulation von AP-1 sind die MAPK Signaltransduktionswege von entscheidender Bedeutung. In Zellen von Säugetieren sind mittlerweile vier verschiedene MAPK-Kaskaden beschreiben : die ERK1/2Kaskade, die JNK-Kaskade, der p38 Signalweg und die ERK5 Kaskade. 5.1.1 ERK Signaltransduktionsweg Die ERK Signalkaskade ist oft an der Signalvermittelung von Wachstumsfaktoren und konsekutiv an der Regulation der Zellproliferation beteiligt. Nach Binden des Wachstumsfaktors – u.a. EGF, NGF, VEGF, HGF – an seinen Rezeptor kommt es zu einer Autophosphorelierung desselbigen und einer konsekutiven Anlagerung der Adapterproteine Grb2 und Sos. Sos bewirkt einen Austausch von GDP gegen GTP an dem kleinen GTP bindendem Protein ras und aktiviert es damit. Aktiviertes ras führt u.a. zur Bindung und Aktivierung der MAPKKK c-Raf1 und weiter zu Aktivierung der MEK1/2-ERK1/2-Elk1 Signalkaskade. Mutationen von ras spielen in der Karzinogenese eine wesentliche Rolle – viele epitheliale und hämatopoetische Tumoren weisen ein durch Mutation konstitutiv aktives ras Protein auf [Bos, 1989]. Die für die Funktionalität von ras wichtigen Farnesylgruppen sind Angriffspunkt der als Anti-Krebs Medikamente getesteten Farnesyltransferaseinhibitoren, die, indem sie das Übertragen von Farnesylgruppen auf ras verhindern, dessen Funktion hemmen und somit die ungehemmte Zellteilung bremsen sollen [Oliff, A; 1999]. - 84 - 5. Diskussion 5.1.1.1 H.pylori aktiviert MEK1/2 und ERK1/2 Einer der Downstream-Effektoren von ras ist MEK1/2. In unseren Experimenten konnten wir zeigen, daß auch H.pylori G27 die MAPKK MEK1/2 stimuliert (Exp 71). Einen indirekten Beweis für die Beteiligung von MEK1/2 an der H.pylori induzierten Aktivierung von AP-1 wurde durch Versuche mit selektiven Inhibitoren für MEK1/2, PD98059 und U0126, erbracht (Exp49). Die Mechanismen, durch die H.pylori eine Aktivierung von MEK1/2 bewirkt, sind noch nicht bekannt. Zur Zeit werden in unserem Labor Versuche durchgeführt, die klären sollen, ob H.pylori auch in der Lage ist, ras oder c-Raf zu aktivieren, oder ob die H.pylori induzierte Aktivierung von MEK1/2 mit der konsekutiven Stimulation von AP-1 über andere, noch unbekannte Signalwege verläuft. Wessler et al. berichten, daß cagA+ H.pylori Stämme den Histidin Decarboxylase Promoter transaktivieren, und konnten durch Kotransfektion von dominant negativen Mutanten von Signalmediatoren zeigen, daß hierbei ERK und c-Raf eine Rolle spielen, nicht aber ras und MEKK-1 [Wessler, 2000]. Bezüglich der Aktivierung des Histidin Decarboxylase Promoters sind also c-Raf und ERK an der Signaltransduktion beteiligt, nicht aber ras und MEKK-1. Im Analogieschluß könnte man folgern, daß ras und MEKK-1 wohl auch nicht in der H.pylori induzierten, MEK1/2-ERK-Elk1 vermittelten AP-1 Aktivierung beteiligt sind. Da die Signaltransduktion aber selten rein linear erfolgt, sondern oft mehrere Kaskaden gleichzeitig aktiviert werden, die sich wiederum untereinander beeinflussen, sollte man hier den Versuch mit einem AP-1 getriebenen Reporterkonstrukt als Auslese wiederholen. Das physiologische Substrat von MEK1/2 ist die MAPK ERK. Kokultivation von AGS Zellen mit H.pylori G27 bewirkt eine Aktivierung von ERK1/2, wie wir durch Messung der Aktivität der immunpräzipitierten ERK1/2 in vitro, als auch im Westernblot mit für aktive MAPK ERK1/2 spezifische Antikörper herausfanden. Weiterhin stimulierte H.pylori die Induktion durch einen MEK1/2 vermittelten Signalweg, die Expression des Protoonkogens c-Fos (Exp 75) und die Phosphorelierung von c-Jun an der Seitenkette Ser-73 (Exp 116), wodurch dessen transkriptionelle Aktivität potenziert wird [Hibi 1993]. 5.1.1.2 ERK Kaskade und Neoplastische Transformation - 85 - 5. Diskussion Die chronische Infektion mit H.pylori ist mit einem 6-fach erhöhten Risiko für die Entwicklung eines Magenadenokarzinoms assoziiert [Parsonnet, 1994; Eurogast Study Group, 1995]. Magenbiopsien von Patienten mit einer H.pylori Infektion wiesen im Vergleich zu einer uninfizierten Kontrollgruppe Hyperproliferationen der Magenschleimhaut auf [Peek, 1997; Fraser, 1994; Bechi, 1996; Lynch, 1995; Harvard, 1996]. Beide Beobachtungen waren mit der Präsenz des virulenzassoziierten cagA Gens sowie dem Vorhandensein der Pathogenitätsinsel cag korreliert. Mutationen, die eine konstitutive Aktivierung der ERK-Kaskade bewirken, sind oft mit transformierenden Eigenschaften verbunden. So bewirkt die Expression von durch Mutation konstitutiv aktivem MEK1/2 in Säugetierzellen Fokusformation dieser Zellen und Wachstum in Softagar, sowie Tumorinduktion in Nacktmäusen [Mansour, 1994]. Die Aktivierung von ERK1/2 ist sowohl notwendig, wie auch hinreichend für die Transformation von NIH 3T3 Zellen [Cowely, 1994]. Auch die konstitutive Aktivierung von c-Fos und c-Jun kann Tumorwachstum vorantreiben. Die Überexpression von c-Fos in transgenen Mäusen kann zur Entwicklung von Osteo- und Chondrosarkomen führen [Rüther, 1987]. Die Transformation von Zellen durch c-Fos Überexpression ist dabei nicht abhängig vom Zellzyklus; vielmehr scheint die deutliche Verzögerung, mit der die Transformation nach Beginn der Expression von c-Fos auftritt, darauf hinzuweisen, daß vor der Transformation c-Fos Zielgene akkumulieren müssen [Miao, 1994]. Die deregulierte c-Jun Expression bewirkt die Transformation von Fibroblasten der Ratte [Curran, 1982 ; Schuermann, 1989]. Die in dieser Arbeit vorgestellten Ergebnisse deuten daraufhin, daß die konstitutive Aktivierung von MAP Kinasen und der Protoonkogene c-Fos und c-Jun durch eine Infektion mit H.pylori die Magenschleimhaut zu einer Zellproliferation anregen könnte, sowie eine neoplastische Transformation der Magenmukosa bewirken, die in der Entwicklung eines Adenokarzinoms gipfeln könnte. 5.1.1.3 ERK-Signalkaskade als Regulator des Zellzyklus Mitogene Stimuli wie z.B. Wachstumsfaktoren oder Immunstimulation von B- und T-Zellen durch Aktivierung ihrer Rezeptoren können ruhende Zellen aus der G0 Phase (Ruhephase) in den Zellzyklus bringen und die DNA-Synthese stimulieren. Der Zellzyklus teilt sich in die 4 Phasen G1 (Ruhephase), S (Synthesephase), G2 (Intermediärphase) und M (Mitosephase). Der durch Mitogene ausgelöste Übergang von der G1 Phase in die S Phase wird über Cycline der Klasse D vermittelt [Matsushime, 1991; Übersicht : Sherr, 1995], die mit den Cyclin - 86 - 5. Diskussion Dependent Kinases (CDK) CDK4 und CDK6 Komplexe bilden und diese dadurch aktivieren. Die Proteine p21cip und p27 kip , die an den Cyclin-CDK Komplex binden können und dessen katalytische Aktivität inhibieren, agieren als Gegenspieler zu den Cyclinen. Dabei ist das Verhältnis zwischen D-Typ Cyclinen, die in der G1 Phase stetig zunehmen, und den Inhibitoren entscheidend : übersteigt die Anzahl der Cyclin-CDK Komplexe die Anzahl der inhibitorischen Proteine, kommt es zur Phosphorelierung von Rb und damit zum Übergang in die S-Phase. Dementsprechend führt die Überexpression von cyclin D1 in Fibroblasten zur Beschleunigung der G1 Phase, wohingegen die Mikroinjektion von Antikörpern gegen Cyclin D1 diese Zellen in der G1 Phase blockiert [Quelle, 1993]. Die Cyclin D-CDK Komplexe können das Retinoblastom (Rb) Protein phosphorelieren und dadurch die Bindung zwischen dem Rb-Protein und E2F, einem für die DNA-Synthese essentieller Transkriptionsfaktor, lösen. Das freigesetzte E2F wandert in den Zellkern, stimuliert die DNA-Synthese und bringt die Zelle damit in die S-Phase. Auf ähnliche Weise bewirken auch Tumorviren die Transformation : das E1A Gen (Adenovirus), das SV40 T Antigen (Simvian Virus 40) und das TAX Gen (HTLV I) binden Rb Protein mit hoher Affinität, so daß Komplexe von Rb Protein mit E2F gespalten werden und freies E2F entsteht [Jansen Dürr, 1996]. Konstitutive Aktivierung von ERK führt zur Induktion von D-Typ Cyclinen [Lavoie, 1996] der cyclin D1 Promoter besitzt sowohl eine AP-1 wie eine Ets Bindungsstelle [Watanabe, 1996]. Aktives ERK kann auch über andere Mechanismen den Zellzyklus vorantreiben : ERK kann in vitro den Zellzyklus Inhibitor p27kip phosphorelieren, so daß dieser von dem Cyclin D-CDK Komplex gelöst wird und verstärkt der Ubiquitin vermittelten Degradation ausgesetzt ist. Zu starke Aktivierung der MEK-ERK Signalkaskade durch ein konstitutiv hochaktives cRaf Konstrukt kann jedoch auch die Zellproliferation inhibieren [Woods, 1997]. Hierbei bewirkt das überexprimierte c-Raf eine starke Expression des Zellzyklusinhibitors p21cip, was in einem Proliferationsarrest resultiert. Wie in der Einleitung (1.8.2) bereits angesprochen, ist sowohl die Dauer als auch die Intensität der Aktivierung der MEK-ERK Kaskade für die physiologische Wirkung – Proliferation oder Wachstumshalt / Differenzierung – entscheidend. Eine dritte proliferationsfördernde Wirkung von ERK besteht in der Phosphorelierung (und Aktivierung) der Carbomyl Phosphatase Synthetase II, ein Schlüsselenzym in der Pyrimidin Synthese [Graves, 2000]. Es liegt nahe zu vermuten, daß H.pylori über eine Aktivierung der MEK-ERK Kaskade und Expression der Protoonkogene c-Fos und c-Jun eine Induktion von Cyclin D1 bewirken könnte und so die Hyperproliferation in den Magenschleimhautbiopsien von H.pylori - 87 - 5. Diskussion infizierten Patienten zu erklären sei. Die Kokultivation von AGS Zellen mit H.pylori zeigte jedoch sowohl auf mRNA Ebene (Exp 68cyc) als auch auf Protein Ebene (Exp 92_cyc), daß H.pylori in AGS Zellen zu einer Reduktion von Cyclin D1 führt. Hierfür gibt es mehrere Erklärungsmöglichkeiten : (1) Die Inkubation der AGS-Zellen mit H.pylori bedeutet für diese ein Stressituation, in der es biologisch nicht sinnvoll erscheint, in Mitose zu gehen. Dies deckt sich auch mit morphologischen Beobachtungen, daß die AGS-Zellen bei Kokultivation mit H.pylori G27 ähnlich wie bei Stimulation mit TPA Philopoden ausbilden, wohingegen sie bei Proliferation wie unter Standardbedingungen in Kultur eher eine rundliche Form aufweisen und sich in Nestern anhäufen. (2) Bei der AGS-Zellinie handelt es sich um eine Zellinie, die aus einem Adenokarzinom des Magens gewonnen wurde. Diese Zellen proliferieren unter Zugabe von Wachstumsfaktoren in Form von Serum von sich aus. Dennoch können die Zellen durch Serumdeprivation in einen Wachstumsarrest gebracht werden, und somit den physiologischen Verhältnissen der Magenmukosa angenähert werden. Dieses System stellt somit ein nützliches und in der Fachliteratur oft beschriebenes Modell für die Magenschleimhaut dar, ist aber gerade bei Fragenstellungen bezüglich der Proliferation auch begrenzt und nicht vollständig mit der normalen Magenmukosa des Menschen gleichzusetzen. Weitere Versuche sollten mit Primärkulturen menschlicher Magenmukosa durchgeführt werden, die bei Aspekten der Proliferation wohl ein besseres Modell sind. (3) Die Kokultivierung von H.pylori mit AGS-Zellen bewirkt neben einer Aktivierung der ERK-Signalkaskade auch die Aktivierung der p38-Signalkaskade (nicht gezeigte Daten), die sonst v.a. durch präinflammatorische Zytokine, Endotoxine und osmotischen Streß angeschaltet wird. Aktives p38 führt im Gegensatz zu ERK zu einer Herunterregulierung von Cyclin D1 [Lavoie, 1996]. In dem verwendeten in vitro Modell mit AGS-Zellen scheint dieser Effekt durch aktiviertes p38 den von aktivierten ERK1/2 zu überwiegen. Shirin et al. berichtet im Einklang mit unseren Daten über einen Zellzyklus-Stop durch H.pylori in AGS Zellen und eine dosisabhängige Induktion von Apoptose in AGS Zellen bei Kokultivation mit H.pylori. Die Langzeitinfektion von AGS-Zellen führte jedoch zur Selektion von Apoptose resistenten AGS Zellen, die den Zellzyklusinhibitor p27kip1 deutlich schwächer als normal exprimieren [Shirin, 2000]. - 88 - 5. Diskussion Im Tiermodell konnte gezeigt werden, daß mit H.hepaticus infizierte Mäuse hepatozelluläre Tumore entwickeln, die eine erhöhte Expression von Cyclin D1, Cdk4 und c-Myc sowie ein hyperphosphoreliertes Rb-Protein aufweisen [Ramljak, 1998]. Die Wirkung von H.pylori auf den Zellzyklus scheint also stark von dem zellulären Hintergrund und dem verwendeten Modell – in vitro oder in vivo – abzuhängen. Weiterführend wären sicher immunhistochemische Untersuchungen und in situ Hybridisierungen mit Cyclin D1 spezifischen Sonden an Magenbiopsien von H.pylori infizierten Patienten und einer Kontrollgruppe. Hierbei ergibt sich allerdings wieder das Problem, daß sich der Verlauf der H.pylori Infektion sehr verschieden gestalten kann und stark von Faktoren des Wirtes – aktueller Gesundheitszustand, genetische Prädisposition – abhängt. 5.1.2 JNK Signaltransduktionsweg Die JNK Signalkaskade ist mit der Signalweiterleitung durch extrazelluläre Stressoren assoziiert; so wird sie u.a. durch UV-Bestrahlung, Hitzeschock und inflammatorische Zytokine wie IL-1 und TNF-α aktiviert [Minden, 1997]. Die Aktivierung kann dabei durch verschiedene Upstream-Mediatoren erfolgen, die auf der JNKK1/2 (auch als MKK4/7 bezeichnet) konvergieren, die die Phosphorelierung und Aktivierung von JNK bewirkt. JNK kann Transkriptionsfaktoren der Jun Familie, so u.a. c-Jun an den Seitenketten Ser-63 und Ser-73 phosphorelieren und damit dessen transkriptionelle Aktivität entscheidend verstärken. Darüberhinaus vermag es auch weitere Transkriptionsfaktoren zu aktivieren. So kann JNK Elk1 phosphorelieren und ist so auch an der c-fos Induktion durch proinflammatorische Zytokine und UV-Bestrahlung beteiligt [Minden, 1997]. Ein weiterer Transkriptionsfaktor, der von JNK durch Phosphorelierung aktiviert werden kann, ist ATF-2. Dieser wiederum bildet mit c-Jun Heterodimere, bindet an das TRE-Motiv im c-jun Promoter und stimuliert so die c-Jun Expression [van Dam, 1993]. In vivo ist JNK und c-Jun mit der Apoptose-Induktion verbunden. So führt die anhaltende Aktivierung von JNK-Aktivierung in neuronalen Zellen zum programmierten Zelltod [Xia, 1995]. Auch sind JNK1 und 2 notwendig für die Apoptose Induktion nach UV-Bestrahlung [Tournier, 2000]. 5.1.2.1 H.pylori aktiviert JNK - 89 - 5. Diskussion Die Kokultivation von AGS mit dem cagA+ H.pylori Stamm G27, nicht jedoch seine isogene Mutante G27-∆G oder der cagA- Stamm 2012 bewirken eine Phosphorelierung von JNK1/2, wobei das Maximum nach 30-45 min erreicht wird und sich dann langsam wieder zurück bildet. Diese Ergebnisse stehen im Einklang mit Resultaten aus der Arbeitsgruppe von M.Naumann, der durch Reportengenversuche mit einem AP-1 getriebenen Promoter und Kotransfektion einer dominant negativen JNKK1 auch die Notwendigkeit der JNKSignalkaskade für die Aktivierung von AP-1 belegen konnte [Naumann, 1999]. Desweiteren konnte er ebenfalls durch Kotransfektion dominant negativer Mutanten zeigen, daß die kleinen G-Proteine Rac und Cdc42, die seit längerem als Upstream-Mediatoren des JNKSignalweges beschrieben sind [Minden, 1997], auch für die AP-1 Induktion durch H.pylori notwendig sind. Die Aktivierung von JNK könnte neben der Aktivierung von AP-1 und Expression AP-1 abhängiger Gene auch mitverantwortlich für die erhöhte Apoptoserate des Magenepithels bei einer H.pylori Infektion sein. 5.1.3 MAPK Kaskaden in der Interaktion zwischen Wirt und Erreger Die ERK-Kaskade wird oft im Zusammenhang mit Proliferation und Differenzierung von Zellen beschrieben, so daß man sie nicht primär mit der Pathogenese einer bakteriellen Infektion in Verbindung bringen würde. Anders dagegen der JNK Signalweg, der zuerst als Mediator als Reaktion auf UV-Bestrahlung entdeckt wurde [Hibi, 1993], heute aber allgemeiner als Signalweg für extrazellulären Streß wie Hitzeschock oder auch als Entzündungsmediator eingeordnet wird. Beide Signalwege bewirken die Aktivierung des Transkriptionsfaktors AP-1 und so zum einem die Expression von Genen, die für Zellproliferation und Differenzierung wichtig sind, zum anderen - insbesondere in Verbindung mit dem Transkriptionsfaktor NF-κB – die Expression von proinflammatorischen Zytokinen wie IL-8, IL-6, TNF-α, GRO-α und MIP. Desweiteren kann JNK zur Induktion der Cyclooxygenase-2 führen, die als Schlüsselenzym für die Produktion der Prostaglandine in der Entzündungsvermittelung eine kritische Position einnimmt [Xie, 1995]. In dieser Arbeit konnte gezeigt werden, daß H.pylori sowohl die ERK als auch die JNK Signalkaskade aktiviert. Auch das intrazelluläre Bakterium Neisseria gonorrhoeae aktiviert den JNK Signalweg [Naumann, 1998]. Doch nicht nur eine Aktivierung von MAPK-Kaskaden und der Transkriptionsfaktoren NFκB und AP-1 mit einer konsekutiven Entzündungsantwort konnten in der Molekularpathogenese von bakteriellen Infektionen beobachtet werden; auch das Gegenteil, - 90 - 5. Diskussion die Herunterregulierung dieser Signalmediatoren und somit ein Entkommen von der Immunantwort des Wirtes wurde beschrieben. So führt die Infektion von Makrophagen mit Yersinia enterocolitica Stämmen, die das Yersinia Virulenzplasmid besitzen, zu einer deutlichen Inhibierung der MAPK ERK, JNK und p38, sowie einer Inaktivierung des Transkriptionsfaktors NF-κB, was zu einer deutlichen Supprimierung der TNF-α vermittelten Immunantwort führt [Ruckdeschel, 1997]. Dabei interagiert das auf dem Virulenzplasmid kodierte Gen YopJ, das durch einen Typ III Sekretionsapparat in die Wirtszelle geschleust wird, mit der MAPKK-Protein Superfamilie sowie dem auch dieser Gruppe zugeordneten IKK-β, die eine zentrale Rolle in der Aktivierung von NF-κB spielt. Durch das Binden an die MAPKK Proteine wird deren Aktivierungsdomäne blockiert, so daß verschiedene Signalwege (u.a. die MAPK-Kaskaden und der NF-κB Transduktionsweg), die entscheidend an der Initiierung der Immunantwort des Wirtes beteiligt sind, gestört werden [Orth, 1999]. Auch das Lipid Lipoarabinomannan aus der Hülle von Mycobacterium tuberculosis kann die Dephosphorelierung von Tyrosinseitenketten und die Inhibierung der MAPK ERK 2 bewirken [Knutson, 1998]. Neben diesen Pathomechanismen, die primär mit der Akutreaktion des Wirtes auf das Pathogen assoziiert sind, könnte die Aktivierung des ERK-Signalweges durch eine chronische Infektion mit H.pylori zu einer Hyperproliferation der Magenschleimhaut führen. Dies wäre im Gegensatz zu ersteren ein Effektormechanismus für einen länger andauernden Prozeß und somit eine neue Facette der MAPK Kaskaden in der Molekularpathogenese von Infektionen. 5.2 Welcher Bestandteil von H.pylori aktiviert die MAPK-Kaskaden und AP-1 ? Ein wesentliches Ergebnis dieser Arbeit stellt sicherlich die Beobachtung dar, daß Wildtyp Klasse I und cagA+ H.pylori Stämme, wie G27, 151, NTCT 11638 oder ATCC 43405, in der Lage sind AP-1 zu aktivieren. Im Gegensatz dazu fehlt H.pylori Stämmen, die die Pathogenitätsinsel nicht besitzen, so z.B. der cagA- Stamm 2012, oder isogene Stämme von H.pylori G27 mit Mutationen in der Pathogenitätsinsel cag, diese Eigenschaft. Dabei läßt sich dieser Unterschied auf vielen Ebenen feststellen : der cagA+ Stamm G27 aktiviert die MAPK ERK und JNK, nicht jedoch cagA- Stamm 2012, oder die G27 Mutante ∆G (Exp 115 III & IV). Gleiches gilt für die Expression von c-Fos (Exp 75), AP-1 Bindungsaktivität im EMSA (Exp 96II), sowie AP-1 Funktionalität im Reportergenversuch (Exp 110). Da die verschiedenen Bakterienstämme unter identischen Bedingungen kultiviert wurden, kann ausgeschlossen werden, daß es sich bei der Aktivierung der Signalkaskaden um ein Artefakt - 91 - 5. Diskussion handelt, die Aktivierung also gar nicht auf H.pylori zurückzuführen sei, sondern auf äußere Parameter wie z.B. das Kulturmedium der Bakterien. Dabei ist anzumerken, daß auch cagAStämme – u.a. 2012 – manchmal zur diskreten Induktion von AP-1 bzw. leichten Expression von c-Fos (Exp 115 IV) im Vergleich zu unstimulierten Zellen führten. Auch die Interleukin 8 Sekretion von Magenepithelzellen kann bei Stimulation mit cagA+ H.pylori Isolaten beträchtlich schwanken, und selbst cagA- Isolate können zu einem bestimmten Anteil Interleukin 8 Sekretion induzieren [Sharma, 1995]. Diese Beobachtungen lassen vermuten, daß neben cagA und den Genen der Pathogenitätsinsel weitere bakterielle Determinanten existieren, die die Antwort der Wirtszelle beeinflussen können; so z.B. die genetische Zusammensetzung der cag Insel, die eine große Heterogenität aufweist oder auch Gene außerhalb der Pathogenitätsinsel cag, wie z.B. das kürzlich entdeckte „outer membrane protein“ oipA von H.pylori, daß synergistisch mit dem cagE Gen die IL-8 aus Magenepithelzellen fördert [Yamaoka, 2000]. Von den isogenen Mutanten des Stammes G27 konnten nur die Mutanten ∆F und ∆N ΑP-1 Bindungsaktivität im EMSA induzieren. Interessanterweise sind dies die gleichen Mutanten, die auch NF-κB zu aktivieren vermochten [Glocker, 1998]. Diese Ergebnisse legen nahe, daß der gleiche bakterielle Proteinkomplex zur Aktivierung von NF-κB und AP-1 führt. Die Pathogenitätsinsel cag besteht aus 31 Genen, die wahrscheinlich für einen Typ IV Sekretionsapparat kodieren [Censini, 1996]. Kürzlich konnte von einigen Gruppen gezeigt werden, daß H.pylori das CagA Protein durch diesen Typ IV Sekretionsapparat in AGS Zellen transferieren kann, und CagA dann in der Wirtszelle phosphoreliert wird [Odenbreit, 2000; Stein, 2000]. Ein ähnlicher Mechanismus ist für das Tir-Protein von enteropathogenen Escherichia coli (EPEC) beschrieben, das mit einer Typ III Sekretionsmaschine in Epithelzellen geschleust wird, und dort phosphoreliert wird. Aktiviertes Tir bewirkt eine Kondensation von Actin und dient als Rezeptor für das bakterielle Protein Intimin und ist so an der Adhäsion von EPEC an Epithelzellen beteiligt [Kenny, 1997; Rosenshine, 1992]. Jedoch besitzen CagA und Tir keine Ähnlichkeit bzgl. ihrer Aminosäurensequenz und weisen auch strukturell wenig Ähnlichkeit auf [Kenny, 1997]. Die Zytoskelett Neuordnung und Actin Polymerisation [Segal, 1996], die H.pylori in Magenepithelzellen verursacht, könnte durch das translozierte CagA Protein vermittelt werden. Die Kondensation von Actinfilamenten benötigt aktiviertes Arp2/3 Protein, das über Proteine der WASP-Familie (Wildrich Albert Syndrom Protein) mit den Proteinen der Rho Familie - u.a. Cdc 42 – interagiert. Phosphoreliertes CagA könnte WASP nachahmen, WASP - 92 - 5. Diskussion durch spezifische kleine Rho-GTPasen rekrutieren oder über andere Mechanismen das Rearrangement des Zytoskeletts bewirken [Stein, 2000]. Da CagA weder direkt für die Interleukin 8 Sekretion notwendig ist [Carbtree 95, Shamra 95], noch für die Aktivierung von NF-κB [Sharma, 1998], existiert anscheinend ein Unterschied zwischen Rearrangement des Zytoskeletts und der präinflammatorischen, NF-κB abhängigen Entzündungsantwort, die H.pylori in AGS auslöst. Die Notwendigkeit von cagA für die Aktivierung von AP-1 bleibt zu untersuchen. 5.3 H.pylori aktiviert VEGF Ein pathophysiologisches Charakteristikum für die Entwicklung des Ulcus ventriculi et duodeni und des Magenkarzinoms ist die Neubildung von Gefäßen. Erhöhte VEGF- Spiegel konnten am Rand von Ulcera bei Patienten mit H.pylori Infektion festgestellt werden [Takahashi, 1997]. In dieser Arbeit konnte gezeigt werden, daß H.pylori direkt die Transkription von VEGF-mRNA in AGS stimuliert. Einerseits könnte das erhöhte VEGF in Zusammenhang mit der Wundheilung der Ulcera gesehen werden. Andererseits könnte VEGF auch in einer autokrinen Schleife proliferationsfördernd wirken : Während in normaler Magenschleimhaut kein VEGF vorhanden ist, konnten die VEGF Rezeptoren Flt-1 und KDR immunhistochemisch nachgewiesen werden [Takahashi, 1997]. Die Stimulation der VEGF Rezeptoren aktiviert MAPK Signalwege, die wiederum zur Zellproliferation führen können [Guo, 1995]. Desweiteren bewirkt VEGF die Expression des antiapoptotischen Proteins Bcl-2 [Nor, 1999]. Interessanterweise findet sich auch in Dysplasien des Magen-Darm-Traktes eine verstärkte Bcl-2 Expression [Cho, 1998]. Sowohl die Hyperproliferation als auch die Entwicklung von apoptoseresistenten Zellen sind im Zusammenhang mit der chronischen H.pylori Infektion beschrieben worden [Shirin,2000]. - 93 - 6. Zusammenfassung 6 Zusammenfassung In der vorliegenden Arbeit wurden die Aktivierung des Transkriptionsfaktors AP-1 durch H.pylori Stämme sowie die beteiligten Signaltransduktionswege näher untersucht. Desweiteren wurde die Induktion zweier putativer AP-1 abhängiger Gene, Cyclin D1 und VEGF, analysiert. Die Versuche wurden durch Kokultivation von H.pylori mit der Magenepithelzellinie AGS als ein Modellsystem für die H.pylori Infektion des Magens durchgeführt. Es konnte erstmals gezeigt werden, daß H.pylori Stämme, die die Pathogenitätsinsel cag besitzen, den Transkriptionsfaktor AP-1 induzieren können, der durch Supershiftanalyse als Heterodimer der Proteine c-Fos und c-Jun identifiziert werden konnte. Die Transaktivierungsaktivität des induzierten AP-1 Komplexes konnte im Reportergenassay nachgewiesen werden. Die durch Signaltransduktion der H.pylori verursachten AP-1 Stimulation erfolgt dabei über die Aktivierung von MEK-1/2 und ERK1/2 sowie der Phosphorelierung des Transkriptionsfaktors Elk-1 und resultiert in einer Neusynthese des Protoonkogens c-Fos auf mRNA und Proteinebene. Auch vermag H.pylori in AGS Zellen die MAPK JNK zu aktivieren und konsekutiv die Transaktivierungskapazität von c-Jun durch Phosphorelierung zu erhöhen. Als weiteres wichtiges Ergebnis ist festzuhalten, daß jeweils nur H.pylori Stämme, die eine intakte Pathogenitätsinsel cag besitzen (G27, 151, 69A, NTCT 11638, ATCC 43405), zu einer Stimulation der Signalkaskaden und Aktivierung des Transkriptionsfaktors AP-1 führten, wohingegen H.pylori Stämme ohne Pathogenitätsinsel (Stamm 2012, 2074) oder isogene Mutanten des H.pylori Stammes G27 mit Mutation der Gene cagE, cagG, cagH, cagI, cagL, cagM im Vergleich zu Hintergrundsaktivität keine oder nur geringfügige Aktivierung der Signalkaskaden verursachen. Die gleichen Gene werden auch für die Aktivierung des Transkriptionsfaktors NF-κB benötigt, was dafür spricht, daß der bakterielle Virulenzkomplex, der für die Stimulation der Transkriptionsfaktoren AP-1 und NF-κB verantwortlich ist, identisch ist. Zusammenfassend vermag H.pylori neben der bekannten Stimulation des Transkriptionsfaktors NF-κB auch den Transkriptionsfaktor AP-1, sowie die ERK und JNK MAPK Signalkaskade zu aktivieren. Hierdurch kann nicht nur die akute Entzündungsreaktion des Magens bei einer H.pylori Infektion auf einer molekularen Ebene besser erklärt werden, sondern es können auch Phänomene der chronischen Infektion mit H.pylori wie die Hyperproliferation der Magenschleimhaut sowie die neoplastische Transformation besser verstanden werden. - 94 - 7. Referenzen 7 Referenzen Akopyants NS. Clifton SW. Kersulyte D. Crabtree JE. Youree BE. Reece CA. Bukanov NO. Drazek ES. Roe BA. Berg DE. (1998) Analyses of the cag pathogenicity island of Helicobacter pylori. Molecular Microbiology. 28(1) : 37-53 Aihara M, Tsuchimoto D, Takizawa H, Azuma A, Wakebe H, Ohmoto Y, Imagawa K, Kikuchi M, Mukaida N, and Matsushima K. (1997) Mechanisms involved in Helicobacter pylori-induced interleukin-8 production by a gastric cancer cell line, MKN45. Infect Immun 65: 3218–3224,. Alm R.A., Ling L.S., Moir D.T., King B.L., Brown E.D. et al. (1999) Genomic sequence comparison of two unrelated isolates of the gastric pathogen Helicobacter pylori. Nature 397 :176-180 Angel P. Imagawa M. Chiu R. Stein B. Imbra RJ. Rahmsdorf HJ. Jonat C. Herrlich P. Karin M. (1987) Phorbol ester-inducible genes contain a common cis element recognized by a TPAmodulated trans-acting factor. Cell. 49(6) : 729-39, Angel P., Karin M. (1991) The role of Jun, Fos and the AP-1 complex in cell-proliferation and transformation. Biochem Biophys Acta 1072 : 129-157 Apel, I., Jacobs, E., Kist, M. and Bredt, W. (1988). Antibody response of patients against a 120 kDa ssurface protein of Campylobacter pylori. Zbl. Bakt. Hyg. 268, 271-276 Appelmelk BJ. Simoons-Smit I. Negrini R. Moran AP. Aspinall GO. Forte JG. De Vries T. Quan H. Verboom T. Maaskant JJ. Ghiara P. Kuipers EJ. Bloemena E. Tadema TM. Townsend RR. Tyagarajan K. Crothers JM. Monteiro MA. Savio A. De Graaff J. (1996) Potential role of molecular mimicry between Helicobacter pylori lipopolysaccharide and host Lewis blood group antigens in autoimmunity. Infection & Immunity. 64(6) : 2031-40 Appelmelk BJ. Negrini R. Moran AP. Kuipers EJ. Molecular mimicry between Helicobacter pylori and the host. (1997) Trends in Microbiology. 5(2) : 70-3 Bauerfeind P. Garner RM. Mobley LT. (1996) Allelic exchange mutagenesis of nixA in Helicobacter pylori results in reduced nickel transport and urease activity. Infection & Immunity. 64(7) : 2877-80 Beales IL. Calam J. (1997) Effect of N alpha-methyl-histamine on acid secretion in isolated cultured rabbit parietal cells: implications for Helicobacter pylori associated gastritis and gastric physiology. Gut. 40(1) : 14-9 Bechi, P., Balzi, M., Becciolini, A., Maugeri, A., Raggi, C.C., Amarosi, A. and al., e. (1996). Helicobacter pylori and cell proliferation of the gastric mucosa: possible implications for gastric carcinogenesis. Am. J. Gastroenterol. 91, 271-276 Blaser, M.J. (1992) Perspectives on the pathogensis of Helicobacter pylori infections. Helicobactewr pylori and gastroduodenal disease,eds. Rathbone B.J. & Heathley R.V. (Oxford, Blackwell Scientific Publication) - 95 - 7. Referenzen Blaser, M.J., Perez-Perez, G.I., Kleanthous, H., Cover, T.L., Peek, R.M., Chyou, P.H., Stemmermann, G.N. and Nomura, A. (1995). Infection with Helicobacter pylori strains possesing CagA associated with an increased risk of developing adenocarcinoma of the stomach. Cancer Res 55, 2111-2115 Bizzozero G. (1893) Über die schlauchförmigenDrüsen des Magendarmkanals und die Beziehungen ihres Epithels zu dem Oberflächenepithel der Schleimhaut. Arch. Mikr. Anat. 42 : 82-154 Boren T. Falk P. Roth KA. Larson G. Normark S. Attachment of Helicobacter pylori to human gastric epithelium mediated by blood group antigens.(1993) Science. 262(5141) : 1892-5 Bos JL (1989). ras oncogenes in human cancer: a review. Cancer Research 49(17) : 4682-9, Boyle WJ, Smeal T, Deifze LHK, Angel P, Woodgett JR, Karin M, Hunter T (1991) : Activation of Protein Kinase C Decreases Phosphorylation of c-Jun at Sites that negatively regulate its DNA-Binding Activity. Cell 1991, 64 ;573-584 Censini S. Lange C. Xiang Z. Crabtree JE. Ghiara P. Borodovsky M. Rappuoli R. Covacci A. cag, a pathogenicity island of Helicobacter pylori, encodes type I-specific and diseaseassociated virulence factors. (1996) Proceedings of the National Academy of Sciences of the United States of America. 93(25) : 14648-53 Censini Stefano, Stein Markus and Covacci Antonello (2001) Cellular responses induced after contact with Helicobacter pylori. Curr Opin in Microbiol 4 : 41–46 Chang L. Karin M. (2001) Mammalian MAP kinase signalling cascades. Nature 410(6824) : 37-40, Cho, J.H. and Kim, W.H. (1998) Altered topographic expression of p21WAF1/CIP1/SDI1, bcl2 and p53 during gastric carcinogenesis. Pathology, Research & Practice 194, 309-317. Claeys D. Faller G. Appelmelk BJ. Negrini R. Kirchner T. (1998) The gastric H+,K+-ATPase is a major autoantigen in chronic Helicobacter pylori gastritis with body mucosa atrophy. Gastroenterology. 115(2) :340-7 Cohen, T., Nahari, D., Cerem, L.W., Neufeld, G. and Levi, B.Z. (1996). Interleukin 6 induces the expression of vascular endothelial growth factor. Journal of Biological Chemistry 271, 736-741. Covacci, A., Censini, S., Bugnoli, M., Petracca, R., Burroni, D., Macchia, G., Massone, A., Papini, E., Xiang, Z., Figura, N. and Rappuoli, R. (1993). Molecular characterization of the 128-kDa immunodominant antigen of Helicobacter pylori associated with cytotoxicity and duodenal ulcer. Proc. Natl. Acad. Sci. USA 90, 5791-5795 Covacci, A., Falkow, S., Berg, D.E. and Rappuoli, R. (1997). Did the inheritance of a pathogenicity island modify the virulence of Helicobacter pylori. Trends Microbiol. 5, 205208. - 96 - 7. Referenzen Cowley, S., Paterson, H., Kemp, P. and Marshall, C.J. (1994). Activation of MAP kinase is necessary and sufficient for PC12 differentiation and for transformation of NIH3T3 cells. Cell 77, 841-850 Crabtree JE. Shallcross TM. Heatley RV. Wyatt JI. (1991) Mucosal tumour necrosis factor alpha and interleukin-6 in patients with Helicobacter pylori associated gastritis. Gut. 32(12) : 1473-7 Crabtree JE. Peichl P. Wyatt JI. Stachl U. Lindley IJ.(1993) Gastric interleukin-8 and IgA IL8 autoantibodies in Helicobacter pylori infection. Scandinavian Journal of Immunology. 37(1) : 65-70 Crabtree, J.E., Farmery, S.M., Lindley, I.J.D., Figura, N., Peichl, P. and Tompkins, D.S. (1994). CagA cytotoxic strains of Helicobacter pylori and interleukin-8 in gastric epithelial cell lines. J. Clin. Pathol. 47, 945-950 Crabtree, J.E., Xiang, Z., Lindley, I.J.D., Tompkins, D.S., Rappuoli, R. and Covacci, A. (1995a). Induction of interleukin-8 secretion from gastric epithelial cells by a cagA negative isogenic mutant of Helicobacter pylori. J. Clin. Pathol. 48, 967-969 Crabtree JE, Wyatt JI, Trejdosiewicz LK, Peichl P, Ni-cholsH, Ramsay N, Primrose JN, and Lindley IJ (1995b) Interleukin-8 expression in Helicobacter pylori infected, normal, andneoplastic gastroduodenal mucosa. J Clin Pathol 47: 61–66, Curran, T., Miller, A.D., Zukas, L. and Verma, I.M. (1984). Viral and cellular fos proteins: a comparative analysis. Cell 36, 259-268. Curran, T. and Teich, N.M. (1982a). Candidate product of the FBJ murine osteosarcoma virus oncogene: characterization of a 55,000 dalton phophoprotein. J. Virol. 42, 114-122. Curran, T. and Teich, N.M. (1982b). Identification of 39,000 dalton protein in cells transformed by FBJ murine osteosarcoma virus. Virology 116, 221-235 Dérijard, B., Hibi, M., Wu, I.-H., Barrett, T., Su, B., Deng, T., Karin, M. and Davis, R.J. (1994). JNK1: a protein kinase stimulated by UV light and Ha-Ras that binds and phosphorylates the c-Jun activation domain. Cell 76, 1025-1037 Doig P. Austin JW. Kostrzynska M. Trust TJ. (1992) Production of a conserved adhesin by the human gastroduodenal pathogen Helicobacter pylori. Journal of Bacteriology. 174(8) : 2539-47 Dudley, D.T., Pang, L., Decker, S.J., Bridges, A.J. and Saltiel, A.R. (1995). A synthetic inhibitor of the mitogen-activated protein kinase cascade. Proc. Natl. Acad. Sci. USA 92, 7686-7689. Eaton KA. Brooks CL. Morgan DR. Krakowka S. (1991) Essential role of urease in pathogenesis of gastritis induced by Helicobacter pylori in gnotobiotic piglets. Infection & Immunity. 59(7) : 2470-5 Eaton KA. Morgan DR. Krakowka S. (1992) Motility as a factor in the colonisation of gnotobiotic piglets by Helicobacter pylori. Journal of Medical Microbiology. 37(2) : 123-7 - 97 - 7. Referenzen Eck, M., Schmaußer, B., Haas, R., Greiner, A., Czub, S. and Müller-Hermelink, H.K. (1997). MALT-type lymphoma of the stomach is associated with Helicobacter pylori strains expressing the cagA protein. Gastroenterology 112, 1482-1486. Eckmann L, Kagnoff MF, and Fierer J (1993) . Epithelial cells secrete the chemokine interleukin-8 in response to bacterial entry. Infect Immun 61: 4569–4574, English J. et al (1999). New insights into the control of MAP kinase pathways . Exp. Cell Res. 253, 255-270 Enholm, B., Paavonen, K., Ristimaki, A., Kumar, V., Gunji, Y., Klefstrom, J., Kivinen, L., Laiho, M., Olofsson, B., Joukov, V., Eriksson, U. and Alitalo, K. (1997). Comparison of VEGF, VEGF-B, VEGF-C and Ang-1 mRNA regulation by serum, growth factors, oncoproteins and hypoxia. Oncogene 14, 2475-2483. Eurogast Study Group. (1993) Lancet 341, 1359-1362 Evans DG. Evans DJ Jr. Moulds JJ. Graham DY (1988) N-acetylneuraminyllactose-binding fibrillar hemagglutinin of Campylobacter pylori: a putative colonization factor antigen. Infection & Immunity. 56(11) : 2896-906 Favata, M.F., Horiuchi, K.Y., Manos, E.J. and al., e. (1998). Identification of a novel inhibitor of mitogen-activated protein kinase kinase. J. Biol. Chem. 273, 18623-18632. Faller G. Steininger H. Kranzlein J. Maul H. Kerkau T. Hensen J. Hahn EG. Kirchner T. (1997) Antigastric autoantibodies in Helicobacter pylori infection: implications of histological and clinical parameters of gastritis. Gut. 41(5) : 619-23 Forsythe, J.A., Jiang, B.H., Iyer, N.V., Agani, F., Leung, S.W., Koos, R.D. and Semenza, G.L. (1996). Activation of vascular endothelial growth factor gene transcription by hypoxiainducible factor 1. Molecular & Cellular Biology 16, 4604-4613. Fraser, A.G., Sim, R., Sankey, E.A., Dhiillon, A.P. and Pounder, R.E. (1994). Effect of eradication of Helicobacter pylori on gatric epithelial cell proliferation. Aliment. Pharmacol. Ther. 8, 167-173 Gille, J., Swerlick, R.A. and Caughman, S.W. (1997). Transforming growth factor-alphainduced transcriptional activation of the vascular permeability factor (VPF/VEGF) gene requires AP-2-dependent DNA binding and transactivation. EMBO Journal 16, 750-759 Gionchetti, P., Vaira, D., Campieri, M., Holton, J., Menegatti, M., Belluzzi, A., Bertinelli, E., Ferretti, M., Brignola, C., Migliolo, M. and Barbara, L. (1994). Enhanced mucosal interleukin-6 and -8 in Helicobacter pylori-positive dyspeptic patients. Am. J. Gastroent. 89, 883-887. Glocker, E., Lange, C., Covacci, A., Bereswill, S., Kist, M. and Pahl, H.L. (1998). Genes encoded in the Cag-pathogenicity island ofHelicobacter pylori are required for NF-kB activation. Infect. Immun. 66, 2346-2348. - 98 - 7. Referenzen Glover JN. Harrison SC. (1995) Crystal structure of the heterodimeric bZIP transcription factor c-Fos-c-Jun bound to DNA. Nature. 373(6511) : 257-61 Goodwin C.S, Armstrong J.A, Chilvers T., Peters M., Collins M.D., Sly L., McConell W., Harper W.E.S.(1989) Transfer of Camylobacter pylori and Camylobacter mutuluae to Helicobacter gen. nov. and H.pyloricomb.nov. as H. mustulae comb.nov. Int J. System. Bacteriol. 39 : 397-405 Goodwin C.S. & Armstrong J.A (1990) Microbiological aspects of Helicobacter pylori (Campylobactor pylori). Europ. j. Microbiol. Inf. Dis. 9 : 297-308 Gordon JA. (1991) Use of vanadate as protein-phosphotyrosine phosphatase inhibitor. Methods in Enzymology 201:477-82 Graves, L. M. et al (2000) Regulation of carbamoyl phosphate synthetase by MAP kinase. Nature 403, 328-332 Grugel, S., Finkenzeller, G., Weindel, K., Barleon, B. and Marme, D. (1995). Both v-Ha-Ras and v-Raf stimulate expression of the vascular endothelial growth factor in NIH 3T3 cells. Journal of Biological Chemistry 270, 25915-25919. Guo, D., Jia, Q., Song, H.Y., Warren, R.S. and Donner, D.B. (1995). Vascular endothelial cell growth factor promotes tyrosine phosphorylation of mediators of signal transduction that contain SH2 domains. Association with endothelial cell proliferation. Journal of Biological Chemistry 270, 6729-6733. Hall P. A., Coates P. J., Ansari B., Hopwood D. (1994) Regulation of cell number in the mammalian gastrointestinal tract: the importance of apoptosis.. J. Cell Sci., 107: 3569-3577 Havard TJ. Sarsfield P. Wotherspoon AC. Steer HW. (1996) Increased gastric epithelial cell proliferation in helicobacter pylori associated follicular gastritis. Journal of Clinical Pathology. 49(1) : 68-71 Hazell SL. Lee A. Brady L. Hennessy W. Campylobacter pyloridis and gastritis: association with intercellular spaces and adaptation to an environment of mucus as important factors in colonization of the gastric epithelium. (1986) Journal of Infectious Diseases. 153(4) : 658-63 Hibi M. Lin A. Smeal T. Minden A. Karin M (1993). Identification of an oncoprotein- and UV-responsive protein kinase that binds and potentiates the c-Jun activation domain. Genes & Development. 7(11) : 2135-48 Hipskind, R.A., Rao, V.N., Mueller, C.G., Reddy, E.S.P. and Nordheim, A. (1991). Etsrelated protein Elk-1 is homologous to the c-fos regulatory factor p62TCF. Nature 354, 531534 Hirata Y. Maeda S. Mitsuno W. Akanuma M. Yamaji Y. Ogura K. Yoshida H. Shiratori Y. Omata M. (2001) Helicobacter pylori activates the cyclin D1 gene through mitogen-activated protein kinase pathway in gastric cancer cells. Infection & Immunity. 69(6) : 3965-3971 - 99 - 7. Referenzen Holt, J.T., Venkat Gopal, T., Moulton, A.D. and Nienhuis, A.W. (1986). Inducible production of c-fos antisense RNA inhibits 3T3 cell proliferation. Proc. Natl. Acad. Sci. USA 83, 47944798. Honda, S., Fujioka, T., Tokieda, M., Satoh, R., Nishizono, A. and Nasu, M. (1998). Development of Helicobacter pylori-induced gastric carcinoma in mongolian gerbils. Cancer Res. 58, 4255-4259 Ilver D. Arnqvist A. Ogren J. Frick IM. Kersulyte D. Incecik ET. Berg DE. Covacci A. Engstrand L. Boren T. (1998) Helicobacter pylori adhesin binding fucosylated histo-blood group antigens revealed by retagging. Science. 279(5349) : 373-7 International Agency for Research on Cancer (1994) IARC Monographs on the evaluation of Carcinogenic Risks to Humans, Lyon,France, IARC Jang TJ. Kim JR. Proliferation and apoptosis in gastric antral epithelial cells of patients infected with Helicobacter pylori. (2000) Journal of Gastroenterology. 35(4) : 265-271 Jansen Dürr (1995) How viral oncogenes make the cell cycle. Trends in Genetics 12(7) : 270274 Johnson, R.S., Van Lingen, B., Papaioannou, V.E. and Spiegelmann, B.M. (1993). A null mutation at the c-jun locus causes embryonic lethality and retarded cell growth in culture. Genes Dev. 7, 1309-1317. Kawada M. Yamagoe S. Murakami Y. Suzuki K. Mizuno S. Uehara Y. (1997) Induction of p27(kip1) degradation and anchorage independence by ras through the map kinase signaling pathway. Oncogene. 15(6) : 629-637 Karin, M. (1995). The regulation of AP-1 activity by mitogen activated protein kinases. J. Biol. Chem. 270, 16483-16486 Karin M. Liu Zg. Zandi E. (1997) AP-1 function and regulation. Current Opinion in Cell Biology. 9(2) : 240-6 Kenny B. DeVinney R. Stein M. Reinscheid DJ. Frey EA. Finlay BB. (1997) Enteropathogenic E. coli (EPEC) transfers its receptor for intimate adherence into mammalian cells. Cell. 91(4):511-20, Kist M (1994) Die Gattung Streptobacillus, Campylobacter und Helicobacter. Lehrbuch der Medizinischen Mikrobiologie 7.Auflage, Gustav Fischer Verlag : 489-495 Knutson KL. Hmama Z. Herreravelit P. Rochford R. Reiner NE. (1998) Lipoarabinomannan of mycobacterium tuberculosis promotes protein tyrosine dephosphorylation and inhibition of mitogen-activated protein kinase in human mononuclear phagocytes. Journal of Biological Chemistry. 273(1) : 645-652 Konjetzny G:E., Henke F., Lubarsch O. (1928) Die Entzündung des Magens. Handbuch der speziellen pathologischen Anatomie und Histologie; Bd 4/2 Springerverlag, Berlin - 100 - 7. Referenzen Lavoie, J.N., I'Allemain, G., Brunet, A., Müller, R. and Pouyssegur, I. (1996). Cyclin D1 expression is regulated positively by the p42/p44MAPK and negatively by the p38/HOGMAPK pathway. J. Biol. Chem. 274, 20608-20616 Lee W. Haslinger A. Karin M. Tjian R. (1987b) Activation of transcription by two factors that bind promoter and enhancer sequences of the human metallothionein gene and SV40. Nature. 325(6102) : 368-72 Lee W. Mitchell P. Tjian R. (1987b) Purified transcription factor AP-1 interacts with TPAinducible enhancer elements. Cell. 49(6) : 741-52, Leung, D.W., Cachianes, G., Kuang, W.J., Goeddel, D.V. and Ferrara, N. (1989). Vascular endothelial growth factor is a secreted angiogenic mitogen. Science 246, 1306-1309 Leung KM. Hui PK. Chan WY. Thomas TM. (1992) Helicobacter pylori-related gastritis and gastric ulcer. A continuum of progressive epithelial degeneration. American Journal of Clinical Pathology. 98(6) : 569-74 Lin LL. Wartmann M. Lin AY. Knopf JL. Seth A. Davis RJ (1993) cPLA2 is phosphorylated and activated by MAP kinase. Cell. 72(2) : 269-78 Lingwood CA. Huesca M. Kuksis A. (1992) The glycerolipid receptor for Helicobacter pylori (and exoenzyme S) is phosphatidylethanolamine. Infection& Immunity. 60(6) : 2470-4 Luck J.M., Seth T.N. (1924) Gastric Urease. Biochem. J. 19 :357-365 Lynch DAF. Mapstone NP. Clarke AMT. Sobala GM. Jackson P. Morrison L. Dixon MF. Quirke P. Axon ATR. (1995) Cell proliferation in helicobacter pylori associated gastritis and the effect of eradication therapy. Gut. 36(3) : 346-350 Mai UE. Perez-Perez GI. Wahl LM. Wahl SM. Blaser MJ. Smith PD. (1991) Soluble surface proteins from Helicobacter pylori activate monocytes/macrophages by lipopolysaccharideindependent mechanism. Journal of Clinical Investigation. 87(3) : 894-900 Mansour, S.J., Matten, W.T., Hermann, A.S., Candia, J.M., Rong, S., Fukasawa, K., Van de Woude, G.F. and Ahn, N.G. (1994). Transformation of mammalian cells by constitutively active MAP kinase kinase. Science 265, 966-970. Marshall CJ. (1995) Specificity of receptor tyrosine kinase signaling - transient versus sustained extracellular signal-regulated kinase activation Cell. 80(2) : 179-185 Matsushime, H., Roussel, M.E., Ashmun, R.A. and Sherr, C.J. (1991). Colony stimulating factor 1 regulates novel cyclins during the G1 phase of the cell cycle. Cell 65, 701-713 Miao GG, Curran T (1994) Cell transformation by c-fos requires an extended period of expression and is independent of the cell cycle. Mol Cell Biol 14 : 4295-4310 Milanini, J., Vinals, F., Pouyssegur, J. and Pages, G. (1998). p42/p44 MAP kinase module plays a key role in the transcriptional regulation of the vascular endothelial growth factor gene in fibroblasts. Journal of Biological Chemistry 273, 18165-18172. - 101 - 7. Referenzen Miller, A.D., Curran, T. and Verma, I.M. (1984). c-fos protein can induce cellular transformation. a novel mechanism for activation of a cellular oncogene. Cell 36, 51-60 Minden, A. and Karin, M. (1997). Regulation and function of the JNK subgroup of MAP kinases. Biochem. Biophys. Acta 1333, F85-F194 Moss SF. Sordillo EM. Abdalla AM. Makarov V. Hanzely Z. Perez-Perez GI. Blaser MJ. Holt PR. Increased gastric epithelial cell apoptosis associated with colonization with cagA plus Helicobacter pylori strains. (2000) Cancer Research. 61(4) : 1406-1411 Münzenmaier, A., Lange, C., Glocker, E., Moran, A., Covacci, A., Bereswill, S., Baeuerle, P.A., Kist, M. and Pahl, H.L. (1997). A shed/secreted product of Helicobacter pylori is required for activation of the transcription factor NF-kB. J. Immunol. 159, 6140-6147. Naumann M. Rudel T. Wieland B. Bartsch C. Meyer TF. (1998) Coordinate activation of activator protein 1 and inflammatory cytokines in response to Neisseria gonorrhoeae epithelial cell contact involves stress response kinases. Journal of Experimental Medicine. 188(7) : 1277-86 Naumann M. Wessler S. Bartsch C. Wieland B. Covacci A. Haas R. Meyer TF. (1999) Activation of activator protein 1 and stress response kinases in epithelial cells colonized by Helicobacter pylori encoding the cag pathogenicity island. Journal of Biological Chemistry. 274(44) : 31655-62 Negrini R. Lisato L. Zanella I. Cavazzini L. Gullini S. Villanacci V. Poiesi C. (1991) Albertini A. Ghielmi S. Helicobacter pylori infection induces antibodies cross-reacting with human gastric mucosa. Gastroenterology. 101(2) : 437-45 Neugut, A.I., Hayek, M. and Howe, G. (1996). Epidemiology of gastric cancer. Sem. Onc. 23, 281-291 Nishizuka Y. (1984) The role of protein kinase C in cell surface signal transduction and tumour promotion. Nature. 308(5961) : 693-8 Nor, J.E., Christensen, J., Mooney, D.J. and Polverini, P.J. (1999). Vascular endothelial growth factor (VEGF)-mediated angiogenesis is associated with enhanced endothelial cell survival and induction of Bcl-2 expression. American Journal of Pathology 154, 375-384. Odenbreit S. Till M. Hofreuter D. Faller G. Haas R. (1999) Genetic and functional characterization of the alpAB gene locus essential for the adhesion of Helicobacter pylori to human gastric tissue. Molecular Microbiology. 31(5) : 1537-48 Odenbreit S. Puls J. Sedlmaier B. Gerland E. Fischer W. Haas R. (2000) Translocation of Helicobacter pylori CagA into gastric epithelial cells by type IV secretion. Science. 287(5457) : 1497-500 Oliff A (1999) Famesyltransferase inhibitors: targeting the molecular basis of cancer. Biochimica et Biophysica Acta - Reviews on Cancer. 1423(3) : C19-C30 - 102 - 7. Referenzen Orth K. Xu ZH. Mudgett MB. Bao ZQ. Palmer LE. Bliska JB. Mangel WF. Staskawicz B. Dixon JE. (2000) Disruption of signaling by Yersinia effector YopJ, a ubiquitin-like protein protease. Science. 290(5496):1594-1597 Pages, G., Berra, E., Milanini, J., Levy, A.P. and Pouyssegur, J. (2000). Stress-activated protein kinases (JNK and p38/HOG) are essential for vascular endothelial growth factor mRNA stability. Journal of Biological Chemistry 275, 26484-26491. Palmer, A., Gavin, A. C. & Nebreda, A. R. (1998).A link between MAP kinase and p34(cdc2)/cyclin B during oocyte maturation: p90(rsk) phosphorylates and inactivates the p34(cdc2) inhibitory kinase Myt1. EMBO J. 17, 5031-5047 Parsonnet, J., Friedman, G.D., Vanderstetten, D.P., Chang, Y., Vogelman, J.H., Orentreich, N. and Sibley, R.K. (1991). Helicobacter pylori infection and the risk of gastric carcinoma. N. Eng. J. Med. 325, 1127-1131 Parsonnet J. Hansen S. Rodriguez L. Gelb AB. Warnke RA. Jellum E. Orentreich N. Vogelman JH. Friedman GD.(1994) Helicobacter pylori infection and gastric lymphoma.. New England Journal of Medicine. 330(18) : 1267-71 Peek, R.M., Moss, S.F., Tham, K.T., Perez-Perez, G.I., Wang, S., Miller, G.G., Atherton, J.C., Holt, P.R. and Blaser, M.J. (1997). Helicobacter pylori cagA+ strains and dissociation of gastric epithelial cell proliferation from apoptosis. J. Natl. Cancer Inst. 89, 863-868 Peek RM. Blaser MJ. Mays DJ. Forsyth MH. Cover TL. Song SY. Krishna U. Pietenpol JA.(1999) Helicobacter pylori strain-specific genotypes and modulation of the gastric epithelial cell cycle. Cancer Research. 59(24) : 6124-6131 Peek RM (2001) Microbes and Microbial Toxins: Paradigms for Microbial-Mucosal Interactions IV. Helicobacter pylori strain-specific activation of signal transduction cascades related to gastric inflammation. Am J Physiol Gastrointest Liver Physiol 280: G525–G530 Pounder RE. Ng D. The prevalence of Helicobacter pylori infection in different countries (1995) Alimentary Pharmacology & Therapeutics. 9 Suppl 2 : 33-9 Quelle, D.F., Ashmun, R.A., Shurtleff, S.A., Kato, J., Bar-Sagi, D., Roussel, M.F. and Sherr, C.J. (1993). Overexpression of mouse D-type cyclins accelerates G1 phase in rodent fibroblasts. Genes. Dev. 7, 1559-1571. Raibowol, K.T., Vosatka, R.J., Ziff, E.B., Lamb, N.J. and Feramisco, J.R. (1988). Microinjection of fos-specific antibodies blocks DNA synthesis in fibroblast cells. Mol. Cell. Biol. 8, 1670-1676 Rak, J., Mitsuhashi, Y., Bayko, L., Filmus, J., Shirasawa, S., Sasazuki, T. and Kerbel, R.S. (1995). Mutant ras oncogenes upregulate VEGF/VPF expression: implications for induction and inhibition of tumor angiogenesis. Cancer Research 55, 4575-4580 Ramljak, D., Jones, A.B., Diwan, B.A., Perantoni, A.O., Hochadel, J.F. and Anderson, L.M. (1998). Epidermal growth factor and transforming growth factor-a-associated overexpression of cyclin D1, Cdk4 and c-Myc during hepatocarcinogenesis in Helicobacter hepaticusinfected A/JCr mice. Cancer Res. 58, 3590-3597. - 103 - 7. Referenzen Rinegaud, J., Whitmarsh, A.J., Barrett, T., Derijard, B. and Davis, R.J. (1996). MKK3- and MKK6-regulated gene expression is mediated by the p38 mitogen-activated protein kinase signal transduction pathway. Mol. Cell. Biol. 16, 1247-1255 Rokkas T. Ladas S. Liatsos C. Petridou E. Papatheodorou G. Theocharis S. Karameris A. Raptis S. (1999) Relationship of Helicobacter pylori CagA status to gastric cell proliferation and apoptosis. Digestive Diseases & Sciences. 44(3) : 487-493, Rosenshine I. Donnenberg MS. Kaper JB. Finlay BB. (1992) Signal transduction between enteropathogenic Escherichia coli (EPEC) and epithelial cells: EPEC induces tyrosine phosphorylation of host cell proteins to initiate cytoskeletal rearrangement and bacterial uptake. EMBO Journal. 11(10):3551-60 Ruckdeschel K. Machold J. Roggenkamp A. Schubert S. Pierre J. Zumbihl R. Liautard JP. Heesemann J. Rouot B. (1997) Yersinia enterocolitica promotes deactivation of macrophage mitogen-activated protein kinases extracellular signal-regulated kinase-1/2, p38, and c-Jun NH2-terminal kinase. Correlation with ist inhibitory effect on tumor necrosis factor-alpha production. Journal of Biological Chemistry. 272(25) : 15920-7 Rudi, J., Kolb, C., Maiwald, M., Zuna, I., von Herbay, A., Galle, P.R. and Strempel, W. (1997). Serum antibodies agains Helicobacter pylori proteins VacA and CagA are associated with increased risk for gastric adenocarcinoma. Dig. Dis. Sci. 42, 1652-1659 Rudi J. Kuck D. Strand S. Vonherbay A. Mariani SM. Krammer PH. Galle PR. Stremmel W. (1998) Involvement of the CD95 (apo-1/fas) receptor and ligand system in helicobacter pylori-induced gastric epithelial apoptosis. Journal of Clinical Investigation. 102(8) : 15061514 Saez, E., Rutberg, S.E., Mueller, E., Oppenheim, H., Smoluk, J., Yuspa, S.H. and Spiegelman, B.M. (1995). c-fos is required for malignant progression of skin tumors. Cell 82, 721-732 Sawada, Y., Sashio, H., Yamamoto, N., Hida, N., Akashi, H., Tonokatsu, Y., Sakagami, T., Fukuda, Y., Shimoyama, T., Nishigami, T. and Uematsu, K. (1998). Pathologic changes in the glandular stomach and duodenum in an H. pylori-infected mongolian gerbil model. J. Clin. Gastroent. 27, S141-S143 Schuermann, M., Neuberg, M., Hunter, J.B., Jenuwein, T., Rysek, R., Bravo, R. and Müller, R. (1989). The leucine repeat motif in Fos protein mediates complex formation with Jun/AP-1 and is required for transformation. Cell 56, 507-516. Segal ED. Shon J. Tompkins LS. (1992) Characterization of Helicobacter pylori urease mutants. Infection & Immunity. 60(5) : 1883-9 Segal ED. Falkow S. Tompkins LS. Helicobacter pylori attachment to gastric cells induces cytoskeletal rearrangements and tyrosine phosphorylation of host cell proteins. (1996) Proceedings of the National Academy of Sciences of the United States of America. 93(3) : 1259-64 - 104 - 7. Referenzen Segal ED. Lange C. Covacci A. Tompkins LS. Falkow S. Induction of host signal transduction pathways by Helicobacter pylori. (1997) Proceedings of the National Academy of Sciences of the United States of America. 94(14) : 7595-9 Sharma, S.A., Tumuru, M.R., Miller, G.G. and Blaser, M.J. (1995). Interleukin-8 response of gastric epithelial cell lines to Helicobacter pylori stimulation in vitro. Infect. Immun. 63, 1681-1687. Sharma SA, Tummuru MK, Blaser MJ, and Kerr LD (1998) Activation of IL-8 gene expression by Helicobacter pylori is regulated by transcription factor nuclear factor-kB in gastric epithelial cells. J Immunol 160 : 2401–2407 Sherr CJ. D-TYPE CYCLINS (1995) Trends in Biochemical Sciences. 20(5) : 187-190 Shirin H., Sordillo EM., Kolevska TK., Hibshoosh H., Kawabata Y., Oh SH. Kuebler JF., Delohery T., Weghorst CM., Weinstein IB., Moss SF (2000). Chronic Helicobacter pylori infection induces an apoptosis-resistant phenotype associated with decreased expression of p27(kip1). Infection & Immunity. 68(9) : 5321-8 Steer H.W., Coli_jones D.G.(1975) Mucosal changes in gastric ulceration and their response to carbenoxolon sodium. Gut 16 : 590-597 Stein, I., Neeman, M., Shweiki, D., Itin, A. and Keshet, E. (1995). Stabilization of vascular endothelial growth factor mRNA by hypoxia and hypoglycemia and coregulation with other ischemia-induced genes. Molecular & Cellular Biology 15, 5363-5368. Stein M. Rappuoli R. Covacci A. (2000) Tyrosine phosphorylation of the Helicobacter pylori CagA antigen after cag-driven host cell translocation. Proceedings of the National Academy of Sciences of the United States of America. 97(3) : 1263-8 Stolte M. H.pylori – Krankheit aus Sicht des Pathologen. (1992) Z. Gastroenterologie 30 :1022 Takahashi, M., Kawabe, T., Ogura, K., Maeda, S., Mikami, Y., Kaneko, N., Terano, A. and Omata, M. (1997). Expression of vascular endothelial growth factor at the human gastric ulcer margin and in cultured gastric fibroblasts: a new angiogenic factor for gastric ulcer healing. Biochemical & Biophysical Research Communications 234, 493-498. Treinies, I., Paterson, H. F., Hooper, S., Wilson, R. & Marshall, C. J (1999). Activated MEK stimulates expression of AP-1 components independently of phosphatidylinositol 3-kinase (PI3-kinase) but requires a PI3-kinase signal To stimulate DNA synthesis. Mol. Cell. Biol. 19, 321-329 Tomb J.F. (1996) The complete genome sequence of the gastrc pathogen Helicobacter pylori. Nature 388 : 539-547 Tournier, C. et al (2000) Requirement of JNK for stress-induced activation of the cytochrome c-mediated death pathway. Science 288 , 870-874. - 105 - 7. Referenzen Tummuru MK. Sharma SA. Blaser MJ. (1995) Helicobacter pylori picB, a homologue of the Bordetella pertussis toxin secretion protein, is required for induction of IL-8 in gastric epithelial cells. Molecular Microbiology. 18(5) : 867-76 van Dam H. Duyndam M. Rottier R. Bosch A. de Vries-Smits L. Herrlich P. Zantema A. Angel P. van der Eb AJ. (1993) Heterodimer formation of cJun and ATF-2 is responsible for induction of c-jun by the 243 amino acid adenovirus E1A protein. EMBO Journal. 12(2) : 479-87, Wagner S., Beil W., Westermann J., Logan R. P., Bock C. T., Trautwein C., Bleck J. S., Manns M. P.(1997) Regulation of gastric epithelial cell growth by Helicobacter pylori: evidence for a major role of apoptosis. Gastroenterology, 113: 1836-1847, 1997 Watanabe, G., Howe, A., Lee, R.J., Albanese, C., Shu, I.W., Karnezis, A., Zon, I., Kyriakis, J., Rundell, K. and Pestell, R.G. (1996). Induction of cyclin D1 by simian virus 40 small tumor antigen. Proc. Natl. Acad.Sci. USA 93, 12861-12866. Watanabe, T., Tada, M., Nagai, H., Sasaki, S. and Nakao, M. (1998). Helicobacter pylori infection induces gastric cancer in mongolian gerbils. Gastroenterology 115, 642-648 Wessler S. Hocker M. Fischer W. Wang TC. (2000) Rosewicz S. Haas R. Wiedenmann B. Meyer TF. Naumann M. Helicobacter pylori activates the histidine decarboxylase promoter through a mitogen-activated protein kinase pathway independent of pathogenicity islandencoded virulence factors. Journal of Biological Chemistry. 275(5) : 3629-36 Williams CL. Preston T. Hossack M. Slater C. McColl KE. (1996) Helicobacter pylori utilises urea for amino acid synthesis. FEMS Immunology & Medical Microbiology. 13(1) : 87-94 Woods D. Parry D. Cherwinski H. Bosch E. Lees E. Mcmahon M. (1997) Raf-induced proliferation or cell cycle arrest is determined by the level of raf activity with arrest mediated by p21(cip1) Molecular & Cellular Biology. 17(9) : 5598-5611 Wotherspoon AC. Ortiz-Hidalgo C. Falzon MR. Isaacson PG. (1991) Helicobacter pyloriassociated gastritis and primary B-cell gastric lymphoma. Lancet. 338(8776) : 1175-6 Wotherspoon AC. Doglioni C. Diss TC. Pan L. Moschini A. de Boni M. Isaacson PG. (1993) Regression of primary low-grade B-cell gastric lymphoma of mucosa-associated lymphoid tissue type after eradication of Helicobacter pylori. Lancet. 342(8871) : 575-7 Xia, Z., Dickens, M., Raingeaud, J., Davis, R. J. & Greenberg, M. E. (1995) Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science 270 , 1326-1331 Xie W. Herschman HR. (1995) V-src induces prostaglandin synthase 2 gene expression by activation of the c-Jun N-terminal kinase and the c-Jun transcription factor. Journal of Biological Chemistry. 270(46) : 27622-8 Yamaguchi T. Nakajima N. Kuwayama H. Ito Y. Iwasaki A. Arakawa Y. Gastric epithelial cell proliferation and apoptosis in Helicobacter pylori-infected mice. (2000) Alimentary Pharmacology & Therapeutics. 14(Suppl 1) : 68-73 - 106 - 7. Referenzen Yamaoka Y, Kwon DH, and Graham DY. A (2000) Mr 34,000 proin-flammatory outer membrane protein (oipA) of Helicobacter py-lori. Proc Natl Acad Sci USA 97: 7533–7538 Zinck, R., Cahill, M.A., Kracht, M., Sachsenmaier, C., Hipskind, R.A. and Nordheim, A. (1995). Protein synthesis inhibitors reveal differential regulation of mitogen-activated protein kinase and stress activated protein kinase pathways that converge on Elk-1. Mol. Cell. Biol. 15, 4930-4938 - 107 - Abkürzungen # °C λ µ Abb. AK AP-1 APS ATCC ATP bp bZIP bzw. BSA Cag cDNA Ci cpm CMV CTP ddH2O DEPC DMSO DNA DNAse DTT EDTA EPEC ERK EGF EGF-R EGTA EMSA FCS h GTP H.pylori HEPES HSV HTLV IL JNK Kb kD Lb m M MAPK Nummer Grad Celsius Wellenlänge Mikro Abbildung Antikörper Activator Protein 1 Ammoniumperoxidsulfat American Type Culture Collection Adenosintriphosphat Basenpaare Transkriptionsfaktoren mit einer β-Zipperdomäne beziehungsweise Bovines Serumalbumin Cytotoxin assoziiertes Gen komplementäre DNA Curie counts per minute Zytomegalievirus Cytosintriphosphat Bidestilliertes Wasser Diethylpyrocarbonat Dimethylsulfoxid Desoxyribonukleinsäure Desoxyribonuklease Dithiothreitol Ethylendiamintetraacetat Enteropathogene Escherichia coli Extracellular Regulated Kinase Epidermal Growth Factor Epidermal Growth Factor Rezeptor Ethylenglycol-bis(β-aminethylether) N,N,N‘,N‘-tetraacetat Elektrophoretic Mobility Shift Assay, Gelretentionsanalyse Fetales Kälberserum Stunden Guanisintriphosphat Helicobacter pylori N-2-Hydroxyethylpiperazin-N‘-2-Ethansulfonsäure Herpes Simplex Virus human T-cell leukenia virus Interleukin c-Jun N-terminal Kinase Kilobasen Kilo Dalton Lewis b Antigen Milli Molar Mitogen aktivierte Protein Kinase Abkürzungen - 108 MAPKK MAPKKK MCP-1 MHC min mRNA MOPS NF-κB NP-40 p38 p42 p44 PAGE PBS pH PMSF Poly(dIdC) RLU RNA RNAse RT rpm s S s.o. s.u. SDS Ser SRE SRF SV 40 TBE TEMED Thr tk TNF TPA TRE Tris Tyr U UV V VacA VEGF v/v WASP z.B. Mitogen aktivierte Protein Kinase Kinase Mitogen aktivierte Protein Kinase Kinase Kinase Monocyte Chemoattractant Protein 1 Major Histocompatibility Complex Minuten messanger RNA 3-N-Morpholinopropanesulfonsäure Nuclear Factor κB Nonidet-P-40 p38 MAP Kinase ERK 1 ERK 2 Polyacrylamid Gelelektorphorese Phosphat Buffered Saline Pondus hydrogenii Phenylmethylsulfonylfluorid Poly-deoxy-Inosin-desoxy-Cytidinsäure Relative Light Units Ribonukleinsäure Ribonuklease Raumtemperatur Rounds per minute / Umdrehungen Sekunden Svedberg Konstante siehe oben siehe unten Natriumdodecylsulfat Serin Serum Response Element Serum Response Factor simvian virus 40 Tris-Borat-EDTA Puffer N,N,N‘,N‘-Tetramethylethyldiamin Threonin Thymidinkinase Tumornekrosefaktor 12-O-Tetradecanoyl-phorbol-13-acetat TPA Response Element Trishydroxymethylaminomethan Tyrosin Umdrehungen Ultraviolett Volt Vakuolisierendes Cytotoxin A Vascular Endothelial Growth Factor volume per volume Wildrich Albert Syndrom Protein zum Beispiel Abkürzungen Danksagung Ganz herzlich möchte ich mich bei Frau Prof. Dr. Heike L. Pahl für die Überlassung des Themas, sowie die umfassende Betreuung, die gute Einarbeitung und die immer vorhandene Diskussionsbereitschaft während der gesamten Durchführung dieser Arbeit bedanken. Weiterhin möchte ich mich bei Prof. Dr. M. Kist und PD Dr. Stefan Bereswill, sowie dem gesamten Helicobacter pylori Labor für die gute Einarbeitung sowie die stets vorhandene Hilfsbereitschaft bei Problemen mit der Kultivierung von H.pylori bedanken. Für die gute Atmosphäre im Labor bedanke ich mich bei Alex, Alfonso, Christpoher, Erik, Guido, Sabine und Snezana, als Team der ersten Stunde (zumindest meiner Labortätigkeit), sowie später Stefan, Steffen, Gesa, Matijaz und meiner Nachfolgerin Monika, die mein Projekt mit großem Engagement weitergeführt hat. Mein Dank gilt auch unserem Nachbarlabor Schüle, insbesondere Philip und Judith, die mir oft methodisch weiterhalfen. Zum Schluß möchte ich meiner Familie danken, die mich über meine Studienzeit stets unterstützt hat und somit mein Studium und das Gelingen dieser Arbeit ermöglicht hat. Ehrenwörtliche Erklärung Hiermit erkläre ich ehrenwörtlich, daß ich die vorliegende Arbeit selbstständig angefertigt und keine anderen als die von mir angegebenen Quellen und Hilfsmittel benutzt habe. Weiterhin erkläre ich, daß diese Dissertation weder in gleicher Weise noch in anderer Form bereits in einem anderen Prüfungsverfahren vorgelegen hat. Auch habe ich keinen anderen Promotionsversuch unternommen. Freiburg, den 19.7.2002 Curriculum vitae Name : Geburtstag : Geburtsort : Vater : Mutter : Familienstand : Tobias Meyer-ter-Vehn 26.Dez.1972 München Herr Prof. Dr. Jürgen Meyer-ter-Vehn, Physiker am MPI für Quantenoptik, Garching Frau Dipl. Sozialwirtin Helga Meyer-ter-Vehn, Mitarbeiterin bei der Unternehmensberatung Roland Berger ledig Schulbildung 1979 - 1983 1983 - 1992 8.Juli 1992 Grundschule Garching Ost Werner Heisenberg Gymnasium Garching Allgemeine Hochschulreife Studium Nov1992 – Okt1994 Studium Physik an der TU München Okt 1994 Diplomvorprüfung in Physik an der TU München Okt1994 - Sep1996 Sep 1996 Sep 1997 Mär 2000 Doppelstudium Physik an der TU München und Medizin an der LMU München. Dabei Erwerb sämtlicher Scheine des Hauptstudiums Physik. Ärztliche Vorprüfung an der LMU I. Staatsexamen - Medizin an der LMU München II. Staatsexamen - Medizin an der ALU Freiburg Mai - Jul 2000 Aug - Okt 2000 Nov/Dez 2000 Jan/Feb 2001 PJ-Tertial Neurologie an dem UKL Freiburg PJ-Tertial Chirurgie am Kantonspital Chur / Schweiz PJ-Tertial Innere Medizin an der University of Toronto / Kanada PJ-Tertial Innere Medizin an der University of Birmingham / UK Mai 2001 III. Staatsexamen Medizin an der ALU Freiburg Beruf Seit Okt 2001 AiP in der Abteilung für Rheumatologie und Immunologie (Prof. Peter) am UKL Freiburg Promotion Mär1998 - Dez 1999 Doktorarbeit an der Klinik für Tumorbiologie - Sektion Experimentelle Anästhesiologie der Anästhesiologischen Universitätsklinik Freiburg Thema : Immunreaktion bei Helicobacter-pylori Gastritis Sep 1999 Teilnahme an der Jahrestagung 1999 der DGVS in Leipzig mit Vorstellung von eigenen Ergebnissen als Referat