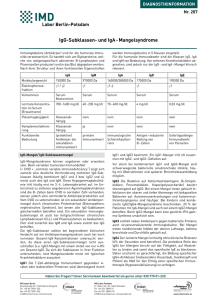
Häufigkeit und klinische Bedeutung von Defekten des
Werbung

Häufigkeit und klinische Bedeutung von Defekten des Immunglobulin G (IgG) und der IgG-Subklassen bei Patienten mit rheumatoider Arthritis Dissertation zur Erlangung des akademischen Grades doctor medicinae (Dr. med.) vorgelegt dem Rat der Medizinischen Fakultät der Friedrich-Schiller-Universität Jena von Sara Kästner geboren am 28.08.1988 in Heiligenstadt . Gutachter 1. Prof. Dr. rer. nat. Rolf Bräuer, Universitätsklinikum Jena 2. Prof. Dr. med. Peter Oelzner, Universitätsklinikum Jena 3. Prof. Dr. med. Hans-Hartmut Peter, Universitätsklinik Freiburg Tag der öffentlichen Verteidigung: 04. März 2014 Abkürzungsverzeichnis Abkürzungsverzeichnis ACPA Antikörper gegen citrullinierte Peptide/ Proteine ACR American College of Rheumatology AID Activation Induced Cytidine Deaminase AIDS Acquired Immunodeficiency Syndrome ANA antinukleäre Antikörper Anti-CCP-AK Anti-CCP-Antikörper APC Antigen-präsentierende Zellen BAFF B Cell Activating Factor BSG Blutsenkungsgeschwindigkeit CCP cyclisches citrulliniertes Peptid COX-2-Hemmer Cyclooxygenase-2-Hemmer CRP C-reaktives Protein CVID Common Variable Immunodeficiency DMARDs Disease-Modifying Anti-Rheumatic Drugs DNA Desoxyribonukleinsäure DR Klasse-2-Antigene ELISA Enzyme-linked Immunosorbent Assay EULAR European League Against Rheumatism Fab antigenbindendes Fragment eines Antikörpers Fc kristallines Fragment eines Antikörpers HLA Human Leukocyte Antigen Ig Immunglobulin IgA Immunglobulin A IgD Immunglobulin D IgE Immunglobulin E IgG Immunglobulin G IgM Immunglobulin M IL Interleukin IFN Interferon IU International Units kDa Kilodalton I Abkürzungsverzeichnis KI Konfidenzintervall MCP Metacarpophalangealgelenk MHC Major Histocompatibility Complex ml Milliliter mm Millimeter MMP Matrixmetalloproteinasen mRNA Messenger-Ribonukleinsäure NEMO NF-κB essential modulators NSAR nichtsteroidale Antirheumatika PAD Peptidylarginin-Deaminase PTPN Protein Tyrosine Phosphatase Non-receptor RA rheumatoide Arthritis RF Rheumafaktor RID Radioimmunodiffusion s.c. subkutan SD Standardabweichung SpA Spondylitis ankylosans TGF-β Transforming Growth Factor-β TNF-α Tumornekrosefaktor α UNG Uracil-DNA-Glycosylase VAS Visuelle Analogskala II Inhaltsverzeichnis Inhaltsverzeichnis Abkürzungsverzeichnis ............................................................................ I Inhaltsverzeichnis................................................................................... III 1 Zusammenfassung ............................................................................ 1 2 Einleitung........................................................................................... 3 2.1 Rheumatoide Arthritis ........................................................................................ 3 2.1.1 Epidemiologie.............................................................................................. 4 2.1.2 Klinik ........................................................................................................... 5 2.1.3 Diagnostik ................................................................................................... 6 2.1.4 Klassifikationskriterien ................................................................................. 7 2.1.5 Krankheitsaktivität ....................................................................................... 8 2.1.6 Ätiologie und Pathogenese ......................................................................... 9 2.1.7 Therapie .................................................................................................... 11 2.2 Antikörper ........................................................................................................ 14 2.2.1 Struktur und Funktion von IgG-Antikörpern ............................................... 14 2.2.2 Antikörperbildung ...................................................................................... 16 2.2.3 B-Zell-Aktivierung und Klassenwechsel .................................................... 18 2.3 Antikörpermangelsyndrome............................................................................. 22 2.3.1 X-chromosomale Agammaglobulinämie .................................................... 23 2.3.2 Autosomal rezessive Agammaglobulinämie .............................................. 24 2.3.3 Common Variable Immunodeficiency (CVID) ............................................ 24 2.3.4 Hyper-IgM-Syndrome ................................................................................ 25 2.3.5 Selektiver IgA-Mangel ............................................................................... 25 2.3.6 IgG-Subklassenmangel ............................................................................. 26 3 Ziel dieser Arbeit.............................................................................. 28 4 Material und Methoden .................................................................... 30 4.1 RA-Patienten ............................................................................................... 30 4.2 Geräte ......................................................................................................... 30 4.3 Material ........................................................................................................ 30 4.4 Methoden .................................................................................................... 31 4.4.1 Bestimmung der IgG-Subklassen.............................................................. 31 III Inhaltsverzeichnis 4.4.2 Bestimmung weiterer Laborwerte.............................................................. 32 4.4.3 Zugrunde liegende Normwerte .................................................................. 32 4.4.4 Bestimmung des Aktivitätsstatus............................................................... 33 4.4.5 Bestimmung der VAS ................................................................................ 34 4.5 Statistik ............................................................................................................ 34 5 Ergebnisse ...................................................................................... 35 5.1 Epidemiologische Daten des Patientenkollektivs ............................................ 35 5.2 Klinische Prozessaktivität und Laborergebnisse der Patientengruppe ............ 40 5.3 Konzentrationen von IgG und IgG-Subklassen bei RA-Patienten ................... 44 5.4 Regressionsanalysen aus den Patientendaten ............................................... 49 5.4.1 Regression mit den abhängigen Variablen IgM und IgA ........................... 50 5.4.2 Regression mit IgG als abhängige Variable .............................................. 51 5.4.3 Regression mit IgG1 als abhängige Variable ............................................ 53 5.4.4 Regression mit IgG2 als abhängige Variable ............................................ 56 5.4.5 Regression mit IgG3 als abhängige Variable ............................................ 58 5.4.6 Regression mit IgG4 als abhängige Variable ............................................ 58 5.4.7 Zusammenfassung der wichtigsten Ergebnisse aus den Regressionsanalysen ......................................................................................... 59 5.4.8 Regressionen aus allen Patientendaten .................................................... 59 6 Diskussion ....................................................................................... 62 6.1 Untersuchungen bei Gesunden ....................................................................... 63 6.2 Untersuchungen bei Patienten mit RA ............................................................ 66 6.2.1 Diskussion zu IgG, IgA und IgM ................................................................ 67 6.2.2 Diskussion zu IgG-Subklassen.................................................................. 71 6.2.3 Kombinierte Defekte .................................................................................. 73 6.2.4 Beziehungen der IgG-Subklassen zu anderen Parametern ...................... 73 6.2.5 IgG-Subklassenbestimmung bei anderen Erkrankungen .......................... 76 6.3 Limitationen ..................................................................................................... 78 6.4 Ausblick ........................................................................................................... 79 7 Schlussfolgerungen ......................................................................... 81 8 Literatur- und Quellenverzeichnis .................................................... 83 9 Anhang ............................................................................................ 92 IV 1 Zusammenfassung 1 Zusammenfassung Wissenschaftlicher Hintergrund und aktueller Forschungsstand Die Häufigkeit von Defekten der IgG-Subklassen bei Patienten mit rheumatoider Arthritis ist bis heute nicht bekannt. Man weiß außerdem wenig über die Bedeutung der IgG-Subklassen immunmodulierender besonders Medikamente im Zusammenhang wie Kortikosteroide, mit dem synthetische Einsatz sowie biologische DMARDs. Fragestellung Das Ziel dieser Arbeit ist die Bestimmung der IgG- und IgG-Subklassenkonzentrationen und die Häufigkeit von Defekten zu erfassen. Weiterhin stellt sich die Frage, ob und inwieweit die IgG-Subklassen von anderen Faktoren wie Alter, Geschlecht, Krankheitsaktivität, aber auch Einsatz von Medikamenten, die in das Immunsystem eingreifen, beeinflusst werden. Methodik Wir haben die Konzentrationen von IgG und IgG-Subklassen sowie die Häufigkeit von Defekten in Sera von 539 Patienten mit rheumatoider Arthritis bestimmt. Für die Messung der Immunglobuline nutzten wir die Nephelometrie. Um Zusammenhänge zwischen IgG und IgG-Subklassen und anderen Parametern zu untersuchen, analysierten wir Alter, Geschlecht, Raucherstatus, Krankheitsaktivität anhand des DAS 28, Erkrankungsdauer, Röntgenstatus, Medikation sowie Anzahl und Art von Infektionen bei den RA-Patienten. Weiterhin bestimmten wir die Laborwerte BSG, CRP, IgM, IgA, IgM-RF, IgA-RF, IgG-RF und Anti-CCP-AK. Die statistische Analyse erfolgte mit SPSS Version 20.0. Ergebnisse und Diskussion Wir erfassten in unseren Untersuchungen eine hohe Rate an Defekten sowohl im Bereich von IgG als auch bei den IgG-Subklassen. Die häufigsten Defekte traten mit 42,3% bei IgG1 auf. Das Risiko für IgG1- und IgG2-Defekte war für ältere Patienten höher. Der Raucherstatus sowie der Röntgenstatus zeigten Assoziationen zur Häufigkeit von IgG-Subklassendefekten. In der Regressionsanalyse zeigten sich 1 1 Zusammenfassung interessanterweise Abhängigkeiten zur Häufigkeit von Infektionen und Einnahme von Kortison, sowie synthetischen und biologischen DMARDs. Defekte des IgG waren mit vermehrtem Auftreten von Infektionen assoziiert. Schlussfolgerungen Da die Rate der IgG- und IgG-Subklassendefekte bei RA-Patienten unerwartet hoch ausfiel und die Medikamente einen Einfluss zu haben scheinen, empfehlen wir die Bestimmung zumindest der Konzentration von IgG, bei Risikopatienten mit häufigen und/ oder schweren Infektionen zudem die Bestimmung der Konzentration von IgGSubklassen. Unsere Ergebnisse lassen sich aufgrund fehlender Vergleichsstudien nur schwer einordnen. Es ist notwendig, weitere Untersuchungen in Form großer prospektiver Studien zu diesem Thema durchzuführen, um die Bedeutung dieser IgG-Subklassendefekte bei Patienten mit rheumatoider Arthritis besonders hinsichtlich der Infektanfälligkeit und der Therapiewirksamkeit besser einschätzen zu können. 2 2 Einleitung 2 Einleitung 2.1 Rheumatoide Arthritis Medizinhistoriker sind sich über die erstmalige Beschreibung der rheumatoiden Arthritis (RA) uneinig. Manche werten die Beschreibungen von Soranus aus dem zweiten Jahrhundert als erste Niederschrift, die zu einem Patienten mit RA existiert (Soranus 1950). Andere bezeichnen den französischen Chirurgen Landré-Beauvais als Erstbeschreiber der RA mit seiner Dissertation aus dem Jahre 1800, die unter starkem Einfluss seines Lehrers Philippe Pinel gestanden habe (Hansen 2009). Eingeführt wurde der Begriff der rheumatoiden Arthritis 1876 durch den Engländer Alfred Garrod. Die ersten Knochenfunde, die man mit der Erkrankung assoziiert, sind jedoch circa 6500 Jahre alt und stammen von Skeletten amerikanischer Ureinwohner (Rothschild et al. 1988). Die RA ist eine chronisch-entzündliche Systemerkrankung, die meist in einem progredienten Verlauf zu Synovialitis, Arthritis mit teils destruierenden Veränderungen der Gelenke, Bursitis und Tendovaginitis führt (Hettenkofer 2003). Fakultativ kann es zu extraartikulären Organmanifestationen kommen, die Herz, Lunge, Leber, Nieren, Augen und Gefäße betreffen können. Mit einer Prävalenz von über 1% handelt es sich um eine häufig auftretende Autoimmunerkrankung und um die häufigste entzündliche Arthritis, die somit große Auswirkungen auf Kosten im Gesundheitssystem, Arbeitsfähigkeit und Produktivität hat (Lee und Weinblatt 2001). Ein Problem stellt die zunehmende Funktionseinschränkung dar, die durch die schubförmig verlaufende Gelenkdestruktion entsteht. Hinzu kommen häufige Nebenwirkungen der antirheumatischen Therapie wie Magen- und Duodenalulzera sowie –blutungen durch NSAR, Infektionen unter Immunsuppressiva und Biologika, Osteoporose durch Langzeittherapie mit Kortikosteroiden u.a. Dadurch sind nach 10 Jahren Krankheitsdauer bis zu 50% der Patienten erwerbsunfähig (Herold 2010). Aus Studien geht hervor, dass die Mortalitätsrate bei der RA zweifach erhöht ist. Verantwortlich hierfür sind Infekte, Nieren- und Atemwegserkrankungen, gastrointestinale Blutungen und kardiovaskuläre Komplikationen, hinzu kommen Auswirkungen von Komorbiditäten. Diese wiederum sind einerseits verursacht durch 3 2 Einleitung die Grundkrankheit selbst, zusätzlich handelt es sich aber auch um Folgen der notwendigen therapeutischen Immunsuppression (Wolfe et al. 1994). Die Pathogenese der Erkrankung ist allerdings noch nicht ausreichend geklärt. Ein besseres Verständnis würde eine Optimierung von Therapieverfahren und somit eine langsamere Krankheitsprogression ermöglichen. Mit der Bestimmung zur Häufigkeit von Defekten des Immunglobulin G (IgG) und der IgG-Subklassen und folglich dem Erkennen eines möglichen Risikos könnte ein Beitrag zur Verbesserung der Sicherheit insbesondere unter aggressiven Therapieverfahren wie biologischen DMARDs erreicht werden. 2.1.1 Epidemiologie Die RA ist die häufigste entzündliche Gelenkerkrankung. Sie kommt bei ca. 1-2% der Bevölkerung vor, wobei die Prävalenz mit dem Alter zunimmt, bis auf 5% bei Frauen und 2% bei Männern über 55 Jahre. Frauen sind durchschnittlich viermal häufiger betroffen als Männer. Die Erkrankung kann in jedem Lebensalter auftreten, wobei der Erkrankungsgipfel im 4. Lebensjahrzehnt und die jährliche Inzidenz bei ca. 1% liegen (Lee und Weinblatt 2001, Classen et al. 2009). Es sind epidemiologische Unterschiede zwischen verschiedenen Bevölkerungsgruppen beschrieben, so liegen Prävalenz und Inzidenz der RA in Nordeuropa und Nordamerika höher als beispielsweise in Südeuropa (Tobon et al. 2010). Die Inzidenz wird mit 0,3% in China weitaus niedriger beschrieben als in Europa. Die Pima-Indianer in Nordamerika haben die höchste Inzidenz mit ca. 5% (Harris et al. 2005). Interessanterweise zeigt eine Studie Schwankungen in den Inzidenzen für Männer und Frauen im Verlaufe der Zeit (Gabriel et al. 1999). Diese Beobachtung unterstützt die Hypothese der Bedeutung der Interaktionen zwischen Mensch und Umwelt in der Pathogenese der RA. Die Prävalenz des Histokompatibilitätsantigens HLA-DR4, welches bei Trägern zur Ausprägung einer RA prädisponiert, beträgt 70% bei RA-Patienten im Gegensatz zu 28% in der Gesamtbevölkerung (Classen et al. 2009). 4 2 Einleitung 2.1.2 Klinik Bei der klinischen Untersuchung steht die Polyarthritis im Vordergrund. In Frühstadien finden sich noch vor den häufig asymmetrisch beginnenden Schwellungen der Gelenke mit Druck- und Spontanschmerz unspezifische Symptome, wie Fatigue, allgemeines Krankheitsgefühl oder diffuse muskuloskelettale Schmerzen. Der Erkrankungsbeginn verläuft häufig schleichend über Wochen bis Monate. Nur in 10-15% der Fälle findet sich ein akuter Beginn der Symptome innerhalb weniger Tage. Im Verlauf finden sich typischerweise symmetrische Gelenkbeschwerden. In fortgeschrittenen Stadien kommt es zu Deformierungen sowie Funktionseinschränkungen der proximalen Interphalangeal-, MCP- und Handgelenke. Die distalen Interphalangealgelenke sind bei der RA nur selten betroffen. Zunächst bestehen die Beschwerden vordergründig in kleineren Gelenken und nachfolgend zusätzlich in großen Gelenken, wie Ellenbogen-, Knie- und Schultergelenken. Oftmals geht ein allgemeines Krankheitsgefühl, verbunden mit subfebrilen Temperaturen, Gewichtsverlust und Morgensteifigkeit von mindestens 45 Minuten mit der Symptomatik einher. Weitere Symptome sind neben Synovialitis Bursitis und Tendovaginitis. Rheumaknoten finden sich in 20% der Fälle subkutan und in Sehnen, selten auch in viszeralen Organen wie z.B. Leber, Lunge, Herz und Gehirn. Bedingt durch die entzündliche Schwellung entwickeln sich häufig Nervenkompressionssyndrome wie das Karpaltunnelsyndrom. Es bilden sich Muskelatrophien in der Umgebung betroffener Gelenke heraus, die alltägliche Tätigkeiten wie Treppensteigen oder Türen öffnen erheblich erschweren können. Extraartikuläre Organmanifestationen können das hämatopoetische System in Form von Anämie sowie Augen, Haut, Herz, Lunge, Leber, Nieren und Gefäße betreffen. Depressionen und spezifische sowie unspezifische Angsterkrankungen sind ebenfalls mit der RA assoziiert (Bukhari et al. 2002, Harris et al. 2005, Herold 2010). Auch die Entwicklung von Malignomen ist mit der RA assoziiert, wird aber wahrscheinlich durch die medikamentösen Wirkungen verstärkt (Chakravarty und Genovese 2004). Schwerste arthritische Gelenkveränderungen werden heutzutage immer seltener beobachtet. Dies ist auf die sich stetig verbessernden therapeutischen Optionen zurückzuführen, mit denen man die Krankheitsaktivität und –progression stärker beeinflussen kann als noch in jüngster Vergangenheit. 5 2 Einleitung 2.1.3 Diagnostik Die Diagnosestellung sollte auf einer zielführenden Anamneseerhebung und effektiven klinischen Untersuchung, labordiagnostischen Tests sowie bildgebender Diagnostik basieren. RA-typische Zeichen sind symmetrische Gelenkschwellungen in multiplen Gelenken mit Druckschmerz sowie Überwärmung und Steifigkeit. Davon sind insbesondere die kleinen Gelenke der Hände und Füße betroffen, wobei die distalen Interphalangealgelenke häufig von den Manifestationen ausgespart sind. Subkutane Rheumaknoten befinden sich häufig an den Streckseiten der Gelenke. Bei der klinischen Untersuchung können zudem Lymphknotenschwellungen und subfebrile Temperaturen auffallen. Im Rahmen der Labordiagnostik einer RA werden unspezifische Entzündungszeichen wie Blutsenkungsgeschwindigkeit (BSG) und C-reaktives Protein (CRP) untersucht, die typischerweise erhöht sind und die Krankheitsaktivität gut widerspiegeln (Sokka und Pincus 2009). IgM-Rheumafaktoren (RF) werden bestimmt, bei denen es sich um Autoantikörper gegen körpereigenes Immunglobulin G (IgG) handelt. Die RF sind nicht spezifisch für die RA, sie kommen bei 65-80% der Patienten mit RA, aber auch häufig bei anderen rheumatischen Erkrankungen wie Kollagenosen und bei 5% der Gesunden vor. Im Gegensatz zu den RF mit einer Spezifität von 84-90% haben die Antikörper gegen cyclisch citrulliniertes Peptid (AntiCCP-AK) eine höhere Spezifität von über 95%, die Sensitivität liegt vergleichbar bei 64-76% (Nishimura et al. 2007, Whiting et al. 2010). Sowohl Anti-CCP-AK als auch IgM-RF können der Krankheitsmanifestation lange vorausgehen (Nielen et al. 2004). Bei gleichzeitigem Vorhandensein von RF und Anti-CCP-AK ist der Vorhersagewert für einen aggressiven Krankheitsverlauf höher. Antinukleäre Antikörper (ANA) sind in ca. 30% der Fälle positiv und deuten ebenfalls einen schwereren Verlauf an. In der Blutuntersuchung können Leukozytose bei normalem Differentialblutbild, Thrombozytose und leichte Anämie beobachtet werden. Neben der Bestimmung von Laborparametern ist die Röntgenuntersuchung der Hände und Vorfüße zur Erfassung von erosiven Veränderungen und Deformationen wichtiger Bestandteil sowohl der Primär- als auch der Verlaufsdiagnostik (Aletaha et al. 2010). Zu weiteren diagnostischen Maßnahmen zählen die Arthrosonografie einschließlich Powerdoppler und die Kontrastmittel-MRT, während die Szintigraphie mittels Technetium ihren Stellenwert weitgehend verloren hat. Mit diesen bildgebenden Verfahren ist zum Teil 6 eine frühere Erfassung von 2 Einleitung Gelenkveränderungen möglich (Ostergaard et al. 2008). Die Fluoreszenskontrastuntersuchung mit Indozyaningrün befindet sich noch in der Erprobung und dürfte das diagnostische Repertoire zukünftig bereichern (Fischer et al. 2010). Beweisende diagnostische Tests existieren für die RA nicht, die Gesamtheit der Untersuchungsbefunde muss zu einer Diagnose zusammengefügt werden. 2.1.4 Klassifikationskriterien Als diagnostische Charakteristika dienten die 1987 überarbeiteten Kriterien des American College of Rheumatology. Diese umfassen sieben klinische Kriterien: Morgensteifigkeit, Auftreten der Arthritis an drei oder mehr Gelenkbereichen, Befall von Finger- und Handgelenken, symmetrisches Befallsmuster, Rheumaknoten, Nachweis von Rheumafaktoren und typische röntgenologische Veränderungen. Diese Kriterien ermöglichten aber eine relativ späte diagnostische Sicherung der RA (Arnett et al. 1988). Tabelle 1: EULAR-/ ACR-Klassifikationskriterien der RA (2010) Gelenkverteilung (0-5) 1 großes Gelenk 2-10 große Gelenke 1-3 kleine Gelenke 4-10 kleine Gelenke >10 Gelenke (zumindest ein kleines Gelenk) Serologie (0-3) RF und Anti-CCP-AK negativ RF und/ oder Anti-CCP-AK niedrig positiv (bis 3x über Grenzwert) RF und/ oder Anti-CCP-AK hoch positiv (>3x über Grenzwert) Symptomdauer (0-1) <6 Wochen ≥6 Wochen Akutphase (0-1) CRP und BSG normal CRP und/ oder BSG erhöht 0 1 2 3 5 0 2 3 0 1 0 1 Die neuen EULAR-/ ACR-Klassifikationskriterien aus dem Jahre 2010 sollen eine frühere Diagnosestellung ermöglichen (Tab. 1). Voraussetzung für die Anwendung der tabellarisch gelisteten Kriterien ist das Vorliegen mindestens einer sicheren Synovialitis in einem Prädilektions-Gelenk bei fehlenden Hinweisen für eine andere Ursache wie Trauma oder andere entzündliche oder degenerative Gelenkaffektion. 7 2 Einleitung Wenn nach den Items in der Tabelle mindestens 6 Punkte erfüllt sind, kann die Erkrankung als RA klassifiziert werden. Es gilt jeweils nur der höchste Punktwert (Aletaha et al. 2010, Schneider et al. 2011). Ergänzende Erläuterungen zu Tab. 1: „Definition kleine Gelenke: Fingergrund- (MCP) und Fingermittelgelenke (PIP) 1–5; Zehengrundgelenke (MTP) 2–5, Großzehenmittelgelenke (IP 1) und Handgelenke. Ausgeschlossen von der Bewertung sind: Daumensattelgelenke (CMC 1), Großzehengrundgelenke (MTP 1), Finger- und Zehenendgelenke (DIP). Definition mittlerer und großer Gelenke: Schulter-, Ellenbogen-, Hüft-, Knie-, Sprunggelenke. Für die Bewertung eines Gelenkes als betroffenes Gelenk muss nicht die Eingangsdefinition einer definitiven Synovialitis erfüllt sein. Es wird jedes geschwollene oder druckschmerzhafte Gelenk der obigen Liste gewertet. Serologie: Rheumafaktor oder ACPA werden als hoch-positiv gewertet, wenn deren Wert über dem 3-fachen des oberen Normwertes liegt. Das Kriterium einer akut-PhaseReaktion ist erfüllt, wenn CRP oder BSG erhöht sind. Bei der BSG sind physiologisch erhöhte Werte (Alter, Geschlecht, Schwangerschaft) zu berücksichtigen und im Zweifelsfalle nicht zu werten. Definition Symptomdauer: bezieht sich auf das Gelenk, welches zum Zeitpunkt der Untersuchung nach Angabe des Patienten am längsten betroffen ist“ (Schneider et al. 2011). 2.1.5 Krankheitsaktivität Bei der Krankheitsaktivität unterscheidet man zwischen klinischer und paraklinischer Prozessaktivität. Bevorzugt eingesetzt wird der Disease Activity Score 28 (DAS 28). Die "28" bezieht sich auf die Anzahl der untersuchten Gelenke (Finger-, Hand-, Ellenbogen-, Schulter- und Kniegelenke). In die Berechnung fließen die Anzahl der geschwollenen und druckschmerzhaften Gelenke, die visuelle Analogskala (VAS) von 0-100 mm sowie ein laborchemisches Kriterium ein, entweder die Blutsenkungsgeschwindigkeit (BSG) oder das C-Reaktive Protein (CRP) (Prevoo et al. 1995). Zur Berechnung des DAS 28 und der Einteilung in Remission, niedrige sowie hohe Krankheitsaktivität siehe Kapitel 4.4.4. Neuere Ergebnisse mittels Powerdoppleruntersuchungen zeigen allerdings, dass die mittels DAS 28 bestimmte Remission teils inkomplett ist. Hier sind sicher weitere Untersuchungen zur Präzisierung notwendig (Balsa et al. 2010). 8 2 Einleitung 2.1.6 Ätiologie und Pathogenese Obwohl in den vergangenen Jahren signifikante Fortschritte im Verständnis der Entstehung der Erkrankung erzielt werden konnten, sind die Ursachen und die Krankheitsentwicklung der RA bis heute größtenteils unbekannt. Sowohl genetische als auch umweltbedingte Faktoren spielen eine wichtige Rolle in der komplexen Krankheitsentstehung. Zwillingsstudien zeigen, dass monozygote Zwillinge 8-mal häufiger an RA erkranken als dizygote, wenn deren Zwilling ebenfalls an RA erkrankt ist (Jarvinen und Aho 1994, Raychaudhuri 2010). Epitope sind Molekülabschnitte eines Gens, die eine spezifische Immunantwort auslösen können. Die Allele HLA-DRB1*01 und HLA-DRB1*04 tragen bei manchen Personen eine identische Aminosäuresequenz, die man Shared Epitope nennt. Menschen, bei denen das HLA-DRB1 mit Shared Epitope vorhanden ist, haben ein bis zu 10-mal höheres Risiko für eine erosive RA im Vergleich zu Personen ohne diese Sequenz (Huizinga et al. 2005). Weiterhin führt ein Polymorphismus im Protein Tyrosine Phosphatase Non-receptor Typ 22 (PTPN22-Gen) zu einem erhöhten RARisiko (Begovich et al. 2004). Eine defekte Galaktosylierung von Immunglobulinen des Isotyps G wird ebenso als Risikofaktor für das Auftreten einer RA diskutiert (Perdriger und Chales 1997). Diese tritt bereits vor dem Erkrankungsbeginn auf und betrifft die IgG-Subklassen IgG1, IgG2 und IgG4 der RF (Tsuchiya et al. 1994). Da Frauen signifikant häufiger an einer RA erkranken als Männer, hat man angenommen, dass es eine Verbindung zwischen der autoimmunologischen Reaktion und weiblichen Hormonen, wie Östrogenen, gibt. Im Allgemeinen stimulieren Östrogene das Immunsystem. Doch insbesondere bei einem Abfall der Östrogenkonzentrationen im weiblichen Organismus mit der Menopause steigt die Inzidenz für RA, weshalb man als weiteren Einflussfaktor die Konzentration weiblicher Sexualhormone anführt (Islander et al. 2011). Es wird angenommen, dass ein noch unbekanntes Antigen mit einem Tropismus für Gelenke über das Blut in die Synovialis gelangt. Dort wird es durch Antigenpräsentierende Zellen (APC) den T-Lymphozyten präsentiert. Die Antigenpräsentation erfolgt über das Zusammenspiel von HLA-Molekülen auf den APC und auf der Oberfläche von CD4+ T-Zellen. Das gehäufte Vorkommen von HLADRB1 bei RA lässt auf einen Zusammenhang zur Krankheitsentstehung schließen, jedoch findet sich das oben genannte Allel bei 30% der Bevölkerung, also auch bei 9 2 Einleitung gesunden Personen. Folglich erkranken längst nicht alle Individuen mit HLA-DRB1 an einer RA (Silman und Pearson 2002). Durch die Interaktion der APC mit T-Zellen kommt es zur Zellaktivierung und zur Freisetzung von Zytokinen, u.a. Interleukin (IL)-1, -6, -15 sowie Tumornekrosefaktor (TNF) α. Diese führen zur B-Zell-Proliferation und Antikörperbildung. Es kommt in der Folge zur Stimulation von synovialen Fibroblasten, welche u.a. Matrix- Metalloproteinasen (MMP) produzieren, und zur Osteoklastenaktivierung. Aus diesen immunologischen Vorgängen ergeben sich die typischen Veränderungen bei der RA: Es kommt durch überschießende Fibroblastenstimulation, durch Einwanderung von Makrophagen und Lymphozyten zur Verdickung der Synovialmembran und somit zur Pannusbildung im Gelenk. Durch die aktivierten Osteoklasten entstehen destruktive Knochenläsionen. Die Exsudation von Flüssigkeit ins Gelenk erfolgt aufgrund der erhöhten Kapillarpermeabilität. Klinisch manifestiert sich dies als Gelenkerguss. Die entzündlichen Prozesse können auch auf Sehnen und Ligamente übergreifen und führen letztendlich zu Schmerzen, Funktionseinschränkungen und über Zerstörungsprozesse zu Gelenkdeformierungen sowie -fehlstellungen. Neben der genetischen Disposition existieren Hinweise darauf, dass virale oder bakterielle Antigene, aber auch das Rauchen und Luftverschmutzung sowie physischer und psychischer Stress die Entstehung der RA triggern können (Klareskog et al. 2011, Hart et al. 2012). In der Studie von Klareskog (siehe Abb. 1) wird ein hypothetischer Weg zur Entwicklung der RA aufgezeigt: Durch langjähriges Rauchen wird die Peptidylarginin-Deaminase (PAD) aktiviert, die Proteine in der Lunge zitrulliniert. Die toxischen Produkte des Rauchens bei einem Träger des HLADRB1 Shared Epitope führen zur Antigenpräsentation von zitrullinierten Peptiden, für welche daraufhin B-Gedächtniszellen gebildet werden. Ein erneuter Trigger, wie beispielsweise ein Trauma oder eine Infektion, führen nun zur Manifestation der Krankheit. 10 2 Einleitung Abbildung 1. Pathophysiologie der RA. Modifiziert nach Klareskog et al. 2011. Hypothese der Krankheitsentstehung der CCP-positiven RA: (1) Langjähriges Rauchen induziert die Aktivierung von PAD-Enzymen, wodurch es zur vermehrten Zitrullinierung von Proteinen und Peptiden in der Lunge kommt. (2) Aktivierung Antigen-präsentierender Zellen durch toxische Komponenten des Rauchens führt zu vermehrter Aufnahme und Präsentation von zitrullinierten Peptiden im Zusammenhang mit HLA-DRB1 shared epitope und (3) CD4+ TZell-vermittelter Aktivierung spezifischer B-Zellen, die PTPN22 exprimieren; es folgen Proliferation, Differenzierung, Anti-CCP-AK-Produktion und (5) Bildung von Gedächtnis-BZellen, die spezifisch für zitrullinierte Peptide sind. Ein zweites Ereignis, z.B. Infektionen, Trauma, (6) führt zur Makrophagenaktivierung (7), Produktion proinflammatorischer Zytokine (8), vermehrter Antigenpräsentation, T- und B-Zell-Aktivierung und somit zu einem Circulus vitiosus, der die chronische Gelenkinflammation und RA-Entwicklung bedingt. CCP: cyclisches citrulliniertes Peptid, PAD: Peptidylarginin-Deaminase, PTPN: Protein Tyrosine Phosphatase Non-receptor. 2.1.7 Therapie Das Hauptziel der Behandlung ist die Minderung der klinischen und paraklinischen Prozessaktivität und damit eine Abbremsung oder Verhinderung der sonst progredient verlaufenden Gelenkdestruktionen. Zu Beginn kommen nichtsteroidale Antirheumatika (NSAR) unter Nutzung ihrer analgetischen und antiphlogistischen Eigenschaften zur Anwendung (Brooks und Day 1991). Sie wirken über die Hemmung des Enzyms Cyclooxygenase (COX), ein Schlüsselenzym in der Synthese entzündungsfördernder Prostaglandine. Die Aktivität von COX1 und COX2 wird gleichermaßen gehemmt. An diese komplexe Hemmung sind aber erhebliche unerwünschte Effekte gekoppelt, insbesondere die Schädigung der Magen- und 11 2 Einleitung Darmschleimhaut. Die Präparate sind nur eingeschränkt oder nicht anwendbar bei niereninsuffizienten und kardiovaskulär vorgeschädigten Patienten (Morris et al. 1991, Fries et al. 1993). Die Entwicklung der selektiven COX2-Hemmer stellte eine neue Etappe in der Therapie mittels Antirheumatika dar, da diese weniger unerwünschte gastrointestinale Wirkungen haben. Eine wesentliche Rolle spielt der Einsatz von Kortikosteroiden; diese beeinflussen die akute Entzündungsreaktion und wirken symptomlindernd, sie können sogar die Progression der Erkrankung mindern. Die Präparate haben aber dosisabhängig erhebliche unerwünschte Effekte. Sie stellen eine wichtige Behandlungsoption v.a. als Hochdosistherapie bei Schüben dar. Sie können insbesondere bei mon- und oligoartikulären Verläufen auch intraartikulär zur schnellen Suppression der lokalen Prozessaktivität eingesetzt werden (Laan et al. 1999, Bijlsma 2012). In den letzten Jahrzehnten ist es zu einer ständigen Verbesserung der Basistherapie mit Disease-Modifying Anti-Rheumatic Drugs (DMARDs) gekommen. Dabei wird zwischen synthetischen und biologischen DMARDs unterschieden. Entscheidend ist die frühzeitige Diagnosesicherung der Arthritis. Es gibt ein sogenanntes „window of opportunity“, während dieses Zeitraumes kann durch frühzeitigen Einsatz von synthetischen DMARDs, ggf. auch von biologischen Präparaten, das Auftreten von Gelenkzerstörungen vermieden oder zumindest vermindert werden. Die neuen 2010er Kriterien zur frühen Diagnosesicherung der RA sowie der Einsatz der modernen Bildgebung ermöglichen ein entsprechendes Vorgehen und damit eine Reduktion des Risikos einer radiologischen Progression (Bukhari et al. 2003). Mit Methotrexat (MTX), das gegenwärtig den Goldstandard darstellt, steht bereits seit Mitte der 1980er Jahre ein wichtiges Medikament zur Verfügung. Es handelt sich um einen synthetischen Folsäureantagonist, der kompetitiv und reversibel die Dihydrofolat-Reduktase hemmt. Der Wirkmechanismus ist allerdings nicht vollständig geklärt. Der Einsatz von MTX erfolgt als Monotherapie oder in Kombination mit anderen DMARDs, neuerdings auch mit biologischen. Durch diese Kombination kann die Effektivität deutlich gesteigert werden (Braun und Rau 2009). Eine weitere wichtige Substanz ist Leflunomid, (Dihydroorotat-Dehydrogenase) ein mit Hemmer der Pyrimidin-Synthese immunmodulatorischen und anti- inflammatorischen Eigenschaften (Katayama und Matsuno 2009). Sowohl bei MTX als auch bei Leflunomid tritt die Wirkung verzögert 4-12 Wochen nach Therapiebeginn ein. Beide sind für die Behandlung mittelschwerer bis schwerer 12 2 Einleitung Verlaufsformen zugelassen. Für letztere ist auch der Einsatz von Ciclosporin A möglich. Für leichtere Verläufe werden Sulfasalazin und Hydroxychloroquin eingesetzt. Goldpräparate wurden wegen ihres langsamen Wirkungseintritts und hohen Nebenwirkungsspektrums weitestgehend von neueren DMARDs verdrängt (Tony 2010, Lorenz 2012). Durch die seit 1998 erstmals mit dem Vertreter Infliximab zugelassenen Biologika hat sich die Behandlungssituation der RA enorm verbessert. Therapeutische Ansätze sind die Blockade des proinflammatorischen TNF-α mit den Vertretern Etanercept, Adalimumab, Infliximab, Golimumab und Certolizumab, die Inhibition von IL-6 durch Tocilizumab sowie die B-Zelldepletion durch den Anti-CD20-Antikörper Rituximab. Anakinra als IL1-Antagonist spielt in der Therapie der RA entgegen anfänglichen Erwartungen keine Rolle. Sämtliche Biologika greifen in bestimmte Teilschritte der Pathogenese ein, z.B. durch Beeinflussung von Zytokinen. Da die beeinflussten Mediatoren ebenfalls eine Rolle im Abwehrsystem spielen, kann allerdings die Immunbalance gestört werden. Essentiell ist eine frühzeitige Diagnosesicherung mit folgender früh einsetzender Behandlung und damit Schaffung der Voraussetzung für verminderte Funktionsverluste (Wiles et al. 2001). Aktuelle Studien belegen zudem ein signifikant besseres Outcome für Patienten unter Kombinationstherapie im Gegensatz zur Monotherapie (van der Kooij et al. 2009). Zu weiteren Behandlungsmöglichkeiten zählen chemische und Radiosynoviorthese sowie die operative Synovialektomie. Letztere hat allerdings durch die Entwicklung der modernen medikamentösen Therapieverfahren erheblich an Bedeutung verloren. Da die aktuell eingesetzten aggressiven Therapieverfahren neben ihrem therapeutischen Nutzen aufgrund ihrer unspezifischen Angriffspunkte auch die Immunhomöostase beeinflussen, besteht zumindest theoretisch das Risiko des vermehrten Auftretens von Infektionen und Malignomen. Umso wichtiger ist es, Patienten mit prävalenten Immundefekten zu erkennen und so vorbestehende Risiken in die Therapieentscheidung einzubeziehen. 13 2 Einleitung 2.2 Antikörper 2.2.1 Struktur und Funktion von IgG-Antikörpern Die Y-förmigen Monomere des Immunglobulin G haben eine relative Molekülmasse von 150kDa, die aus vier Polypeptidketten, zwei jeweils identischen schweren oder H-Ketten und zwei leichten oder L-Ketten aufgebaut sind. Somit besitzen die Polypeptide zwei gleiche Antigenbindungsstellen. Disulfidbrücken stellen die Verbindung zwischen den H-Ketten sowie auch zwischen H-und L-Kette her (siehe Abb. 2). Es existieren zwei Arten von leichten Ketten, die als Lambda-(λ-) und Kappa-(κ-)Kette bezeichnet werden. Jedoch kommen in einem Antikörper immer nur entweder λ- oder κ-Ketten vor. Die Klasse des Antikörpers wird durch die Struktur des konstanten Teils der schweren Kette festgelegt, diese bestimmt die Effektorfunktion des Immunglobulins. Im Falle des IgG handelt es sich bei dem zugehörigen Isotyp um eine Gamma-(γ-)Kette, bei IgM, IgD, IgA und IgE entsprechend jeweils um eine μ-, δ-, α- oder ε-Kette. Die Subklassen IgG1 bis IgG4 beinhalten schwere Ketten, die mit γ1 bis γ4 benannt sind. Die Antikörper setzen sich aus konstanten und variablen Regionen zusammen. Der variable Teil befindet sich im aminoterminalen Bereich der schweren und leichten Ketten und variiert zwischen einzelnen Antikörpern erheblich. Die konstante oder C-Domäne dagegen ist unverändert und wird vom Amino- zum Carboxylende mit CH1, CH2 usw. durchgezählt. Die Gelenkregion teilt einen Antikörper in zwei Fab-Fragmente, die für die Antigenbindung verantwortlich sind, und in ein Fc-Fragment, welches die Effektorfunktionen ausführt (Gemsa et al. 1997, Murphy et al. 2009). 14 2 Einleitung Abbildung 2. Antikörperaufbau. Modifiziert nach Lehninger Principles of Biochemistry (Nelson und Cox 2010). Fab: Antigen-bindendes Fragment, Fc: cristallines Fragment. Die IgG-Subklassen können auf drei verschiedene Weisen an der Immunabwehr beteiligt sein. Durch die Neutralisierung verhindern die Antikörper über das Binden an der Oberfläche des Antigens, dass sich Toxine und Bakterien auf der Zelloberfläche festsetzen. Diese Eigenschaft besitzen alle vier IgG-Subklassen in etwa gleichem Maße. Weiterhin fördern die Antikörper über ihre Bindung am Substrat dessen Phagozytose, auch Opsonierung genannt. Phagozyten können mit ihren FcRezeptoren an den Fc-Teil des Antikörpers binden und somit ihre Funktion ausüben. Hierfür sind hauptsächlich IgG1, in geringerem Maße auch IgG3 zuständig. Ein dritter Mechanismus stellt die durch Aktivierung von Komplement hervorgerufene Lyse von Bakterien sowie verstärkte Opsonierung dar. Antikörper binden mit ihrem Fc-Teil an C1q, ein Protein des Komplementsystems und sorgen darüber für die Aktivierung des klassischen Komplementweges. Hieran ist maßgeblich IgG3 beteiligt, auch IgG1 und selten IgG2 sind zur Komplementaktivierung in der Lage. Zu weiteren Eigenschaften der Subklassen siehe Tab. 2. 15 2 Einleitung Tabelle 2: Eigenschaften der IgG-Subklassen IgG1 Komplementaktivierung ++ Plazentagängigkeit ++ Serumhalbwertzeit (in Tagen) 21-23 Fc-Rezeptorbindung ++ Monozyten-Bindung ++ Mastzell-Bindung Neutrophilen-Bindung ++ Lymphozyten-Bindung ++ Tabelle modifiziert nach Hamilton (1987) IgG2 + + 21-23 + +/+/- IgG3 ++ ++ 7-8 + ++ ++ ++ IgG4 ++ 21-23 +/++ + + 2.2.2 Antikörperbildung Auf einem B-Lymphozyten befinden sich zahlreiche Kopien eines einzigen Antikörpers. Durch die große Anzahl der B-Zellen im Körper wird es möglich, auf viele verschiedene Antigene zu reagieren. Das Fab-Fragment des Antikörpers wird durch dessen variable Region gebildet, die ihre Vielfalt durch unterschiedliche Aminosäuresequenzen in diesem Bereich erhält. Das Antikörperrepertoire umfasst beim Menschen mindestens 1011 verschiedene Antikörpermoleküle. Doch wie kommt diese Vielfalt zustande? Nicht jede Variante der Aminosäureketten der variablen Region ist im Genom kodiert, stattdessen kommt es zur Genumlagerung, d.h. die Ketten werden in mehreren Gensegmenten kodiert und durch somatische Rekombination zu einer kompletten Sequenz zusammengefügt. Die Bildung der V-Region der leichten Kette erfolgt zunächst durch die Kodierung von zwei verschiedenen DNA-Abschnitten. Dabei handelt es sich um das V- und das J-Gen-Segment, die beide zu einem Exon zusammengelagert werden. Hierbei gelangt der für die C-Domäne kodierende DNAAbschnitt näher an die V- und J-Segmente. Nach der Transkription von DNA in RNA werden die V- und die J-Sequenz durch Spleißen miteinander verknüpft, wobei die dazwischenliegenden Introns entfernt werden. Die schwere Kette entsteht aus den drei Gensegmenten D, J und V. Der Vorgang ähnelt der Bildung der leichten Kette, nur werden hier zuerst D- und J-Gen-Segment verknüpft. Darauf folgt die Verbindung mit dem V-Gen-Segment und nach dem Spleißvorgang entsteht die Verknüpfung mit der C-Region (siehe Abb 3). Die genannten Gensegmente liegen in zahlreichen Kopien in der Keimbahn-DNA vor. Die große Variabilität wird durch zufällige Auswahl eines Gensegments von jedem Typ erzeugt. Für die leichte Kette existieren 40 VSegmente der κ-Kette bzw. 30 V-Segmente der λ-Kette und 5 bzw. 4 J-Segmente. Die schwere Kette wird aus einer Auswahl von 40 V-Segmenten, 25 D-Segmenten 16 2 Einleitung und 6 J-Segmenten gebildet. Diese kombinatorische Diversität, die aus der zufälligen Kombination jeweils eines der Segmente entsteht, sorgt für einen großen Teil der Antikörpervielfalt. Auch die Zusammenlagerung verschiedener schwerer und leichter Ketten zu einer variablen Region trägt zu der großen Anzahl unterschiedlicher Antikörper bei. Hinzu kommt das zufällige Anfügen oder Entfernen von Nukleotiden an den Verknüpfungsstellen der Gensegmente. Bis zu diesem Punkt haben die BZellen noch keinen Kontakt zu Antigenen. Zusammen mit der somatischen Hypermutation erhält man die oben genannten mehr als 10 11 unterschiedlichen Antikörper. Die somatische Hypermutation gehört wie auch die Genkonversion und der Klassenwechsel zur zweiten Phase der Diversifikation. Der Klassenwechsel verändert nur die konstante Region, während somatische Hypermutation und Genkonversion die variable Region betreffen. Bei der Hypermutation kommt es zu Punktmutationen in der variablen Region, wodurch sich die Affinität des Antikörpers ändern kann. Das für diesen Vorgang zuständige Enzym Activation Induced Cytidine Deaminase (AID) verursacht zufällige Veränderungen der DNA in aktivierten BZellen, wodurch das Risiko für fehlerhaft reparierte DNA und somit vermehrte Mutationen gesteigert wird. Lymphozyten, die ihr passendes Antigen optimal binden, überleben die Selektion und reifen zu antikörperproduzierenden Zellen heran. Jene, die das Antigen schwächer oder gar nicht binden können, gehen mittels Apoptose zugrunde. Dieser Vorgang, welcher in den sekundär lymphatischen Organen wieder und wieder durchlaufen wird, wird als Affinitätsreifung bezeichnet. Bei der Genkonversion werden infolge Punktmutation und Einzelstrangbrüchen Sequenzabschnitte durch Pseudogene ersetzt. Die so veränderten Sequenzen haben ebenfalls eine veränderte Affinität der Antikörper zufolge (Murphy et al. 2009, Vollmar et al. 2013). 17 2 Einleitung Abbildung 3. Somatische Rekombination von Immunglobulinketten. Modifiziert nach: http://commons.wikimedia. org/wiki/File: VDJ_recombination. png. Durch die Rearrangierung der Gene werden jeweils mehrere Gensegmente individuell zu einer mRNA zusammengefügt, die dann in Proteine translatiert wird. V: Gensegmente für variablen Abschnitt, D: diversity-Gensegmente, J: joining-Gensegmente, C: konstante Gensegmente. 2.2.3 B-Zell-Aktivierung und Klassenwechsel Naive B-Lymphozyten tragen als Antigenrezeptoren zuerst IgM und IgD auf ihrer Oberfläche. IgM stellt gleichzeitig den ersten Antikörper dar, der sezerniert wird. Mithilfe des Klassenwechsels (class switch) ist dieselbe Zelle in der Lage, in Folge eines Antigenkontakts den produzierten Immunglobulin-Isotyp zu ändern. Zugrunde liegen dem Klassenwechsel Zytokine, die vor allem von CD4 + T-Zellen sezerniert werden, die mit B-Zellen interagieren. Nach der B-Zell-Aktivierung durch die Bindung von Antigenen und der B-Zell-Rezeptor-Quervernetzung kommt es zur Wechselwirkung zwischen CD40 auf der B-Zelle und CD40L auf der aktivierten TZelle (Abb. 4). Ohne diese Oberflächenmoleküle und die Interaktion zwischen B- und T-Zellen ist ein Klassenwechsel nicht möglich. 18 2 Einleitung Abbildung 4. B-Zellaktivierung durch T-Helferzellen. Modifiziert nach Vollmar et al. (2013). Nach dem Erkennen eines Antigens durch BZellrezeptoren, Aufnahme und Prozessierung des Antigens als MHC-II/PeptidKomplex bindet die Th2-Zelle über ihren T-Zellrezeptor an den MHC-II-Komplex (1. Signal). Daraufhin kommt es zur Interaktion zwischen B- und T-Zelle über die Exprimierung des CD40-Liganden (CD40-L) auf der T-Zelloberfläche mit CD40 auf der B-Zelle (2. Signal). Es folgt die Freisetzung der Zytokine IL-4, IL-5 und IL-6. Diese fördern die Proliferation und Differenzierung der B-Zellen zu antikörperproduzierenden Plasmazellen und ruhenden Gedächtniszellen. Die Plasmazellklone produzieren monoklonale Antikörper, die der Antigenspezifität des B-Zellrezeptors der ursprünglichen B-Zelle gleichen. Tierexperimentelle Arbeiten an Mäusen zeigten, dass der Klassenwechsel nicht zufällig erfolgt, sondern unterschiedliche Zytokine den Wechsel zu den jeweiligen Isotypen auslösen. Bei der Maus werden im Gegensatz zum Menschen IgGSubklassen mit IgG1, IgG2a und IgG2b sowie IgG3 bezeichnet. Betrachtet man diese IgG-Subklassen, so wird deutlich, dass das von Th2-Zellen produzierte IL-4 v.a. für einen Klassenwechsel zu IgG1 und das von Th1-Zellen produzierte IFN-γ für die Produktion von IgG2a und IgG3 sorgt (Finkelman et al. 1990, Snapper et al. 19 2 Einleitung 1992). Finkelman et al. (1990) wie auch Kaplan et al. (2002) beschreiben eine Korrelation zwischen IgG2a und dem Erkrankungsbeginn sowie der Schwere der Arthritis im Mausmodell. T-bet, ein T-box-Transkriptionsfaktor, der für die Differenzierung von Th1-Zellen und IFN-γ benötigt wird, spielt auch eine wichtige Rolle beim Klassenwechsel der B-Zellen. Peng et al. (2002) zeigen am Mausmodell, dass T-bet für den selektiven Wechsel zu IgG2a essenziell ist, da bei T-bet negativen Mäusen kein IgG2a nachweisbar ist. Das von Th-Zellen und weiteren Zelltypen gebildete TGF-β führt zur Produktion von IgG2b (McIntyre et al. 1993). Das murine IgG1 entspricht beim Menschen am ehesten dem IgG4. Die Produktion dieser beiden Immunglobuline erfolgt jeweils durch die Sezernierung von IL-4 durch Th2-Zellen. TZellen vom Th1-Typ sezernieren IFN-γ, welches die Bildung von IgG1 und IgG3 beim Menschen bewirkt (Abb. 5). Durch diese IgG-Subklassen erfolgt eine Opsonierung und somit eine erleichterte Phagozytose, was wiederum die Th1-Antwort unterstützt (Vollmar et al. 2013). Der B Cell Activating Factor (BAFF) wird von Alsaleh et al. (2011) als weiterer Faktor für den Klassenwechsel von IgM zu IgG beschrieben. Dieses Zytokin ist ein wichtiger Überlebensfaktor für B-Zellen und wird von Monozyten, Neutrophilen und Makrophagen gebildet. BAFF liegt bei Patienten mit RA in erhöhten Konzentrationen vor und scheint die Bildung pathologischer Auto-AK zu unterstützen (Cheema et al. 2001). Es wurde gezeigt, dass Fibroblasten-ähnliche Synoviozyten, die eine vorherrschende Zellart in der hyperplastischen Synovialis von RA-Patienten darstellen, den Klassenwechsel unterstützend beeinflussen (Alsaleh et al. 2011). Die Umwandlung der schweren Kette der Immunglobuline wird durch CD40Lvermittelte Signale bewirkt und mithilfe verschiedener Zytokine reguliert. Die Zytokine verursachen den Klassenwechsel wahrscheinlich dadurch, dass sie die Transkription der zugehörigen switch-Region des Isotyps induzieren (Vollmar et al. 2013). 20 2 Einleitung Abbildung 5. Isotyp-Switching. Modifiziert nach Vollmar et al. (2013). B-Zellen, die durch Antigenkontakt aktiviert wurden, aber keinen T-Zellkontakt haben, differenzieren zu IgM-produzierenden Plasmazellen. Im Falle der Interaktion mit T-Helferzellen werden je nach T-Zellsubtyp verschiedenen Zytokine freigesetzt, die zum Klassenwechsel der produzierten Antikörper führen. Gesteuert wird der Vorgang durch sich wiederholende Bereiche auf der DNA, sogenannte switch-Regionen (S-Regionen). Die S-Regionen befinden sich zwischen VDJ-Sequenz und der für die konstante Domäne kodierenden C-Sequenz des μIsotyps sowie vor der C-Sequenz jedes anderen Isotyps mit Ausnahme des δ-Gens. Beim Klassenwechsel von IgM zu IgG kommt es zur DNA-Rekombination zwischen der Sμ-Region und der Sγ-Region, wobei die dazwischenliegende Cδ-Sequenz entfernt wird. Während der Transkription des Doppelstrangs kann das Enzym Activation Induced Cytidine Deaminase (AID) an die entwundene DNA binden und sorgt für zahlreiche Einzelstrangbrüche an Cytidinresten, wodurch es ebenfalls zu Doppelstrangbrüchen kommt. Reparaturmechanismen könnten daraufhin zur Zusammenführung der beiden switch-Regionen μ und γ führen. Nach dem Herausschneiden der dazwischen befindlichen DNA werden die beiden switch-Enden 21 2 Einleitung zu einem geschlossenen DNA-Strang zusammengefügt. Die Antigenspezifität bleibt hierbei unverändert, nur die Effektorfunktion ändert sich (Murphy et al. 2009). Von den aktivierten B-Zellen geht die Mehrzahl nach einer Infektion in Apoptose, wodurch die Immunreaktion limitiert werden kann. Auch die Antikörper selbst sind dazu fähig, ihre Produktion zu regulieren. Diesen Vorgang bezeichnet man als „Antikörper-Feedback“. IgG-Moleküle bilden hierbei Immunkomplexe mit dem entsprechenden Antigen und binden über den Fc-Rezeptor an B-Zellen. Das Antigen wird zudem von B-Zell-Rezeptoren gebunden, wodurch es zur intrazellulären Inhibierung der B-Zell-Aktivierung kommt. Über diese beiden Prozesse werden die Dauer und Stärke der Antikörperproduktion reguliert (Vollmar et al. 2013). 2.3 Antikörpermangelsyndrome Der erste Immundefekt bei einem Patient mit Agammaglobulinämie wurde 1952 von Bruton beschrieben. 1964 wurde durch Agarwal herausgefunden, dass das Immunglobulin G aus 4 Subklassen besteht. 4 Jahre später wurde erstmalig ein IgGSubklassenmangel bei einem Patienten mit gehäuften Infekten beschrieben (Terry et al. 1968). Hierauf folgten unzählige Einzelfallberichte und Untersuchungen zu Immundefekten. Mittlerweile sind zahlreiche Immundefekte bekannt, wobei in diesem Kapitel nur auf die wichtigsten humoralen Immundefekte eingegangen werden soll. Erkrankungen mit Antikörpermangel zählen zu den humoralen Immundefekten. Hierunter versteht man die Verminderung bzw. das Fehlen mindestens einer Immunglobulin-Klasse oder –subklasse. Häufig sind das humorale und das zelluläre Immunsystem betroffen, man spricht dann von kombinierten Immundefekten. Es werden Störungen der Antigen-unabhängigen B-Zell-Entwicklung im Knochenmark von Antigen-abhängigen Störungen der B-Zellreifung in der Peripherie unterschieden. Liegt ein Defekt im Knochenmark vor, kommt es nicht zur Ausreifung der Zellen und somit zum Fehlen reifer B-Zellen in Blut und lymphatischen Organen. Diese Patienten werden bereits im Kindesalter durch eine Agammaglobulinämie und häufige Infekte auffällig. Hierzu zählen die X-chromosomale Agammaglobulinämie und die autosomal rezessive Agammaglobulinämie. Zu den Syndromen mit antigenabhängiger Reifungsstörung der B-Zellen zählen u.a. der variable Immundefekt, IgGSubklassendefekte und selektive Antikörperdefekte sowie Erkrankungen, die mit einem Antikörpermangel einhergehen. Eine weitere Klassifikationsmöglichkeit von 22 2 Einleitung Antikörpermangelsyndromen besteht in der Einteilung in primäre (angeborene) und sekundäre (erworbene) Defekte. Das Acquired Immunodeficiency Syndrome (AIDS) ist die häufigste Ursache für sekundäre Immundefekte, weiterhin können diese iatrogen durch Medikamente hervorgerufen werden oder bei hämatologischen Grunderkrankungen bestehen. Leitsymptome aller Antikörpermangelsyndrome sind rezidivierende bakterielle Infektionen der Atemwege sowie des Gastrointestinaltraktes. Im Verlauf der Erkrankung nimmt häufig die Konzentration der Immunglobuline weiter ab, somit wird der Schweregrad der Infektionen noch verstärkt. Bei der Hälfte der Patienten mit langer Infektanamnese bilden sich schließlich Bronchiektasen, die zu einer schlechteren Prognose führen. 2.3.1 X-chromosomale Agammaglobulinämie Dieser Immundefekt, auch Morbus Bruton oder XLA für X-linked agammaglobulinemia genannt, geht mit rezidivierenden bakteriellen Infektionen schon ab dem Säuglingsalter von ca. einem Jahr einher, wenn der immunologische Nestschutz durch die mütterlichen Antikörper nicht mehr wirksam ist. Die Erkrankung wird durch eine Mutation im Bruton-Tyrosinkinase-Gen hervorgerufen, welches auf dem X-Chromosom lokalisiert ist. Es kommt zu einem Entwicklungsstopp der BLymphozyten auf der Stufe der Prä-B-Zellen. Die fehlenden reifen B-Zellen führen zu rezidivierenden Infektionen der Atemwege und durch septische Streuung zu schweren Infektionen wie Sepsis, Meningitiden und Osteomyelitiden. Typische Erreger sind Streptokokkus pneumoniae, Staphylokokkus aureus, Haemophilus influenzae und Pseudomonas. Es kommt zu Verminderungen aller Immunglobulinklassen und B-Zellen im Blut sowie in lymphatischen Geweben. Therapeutisch wird die fehlende B-Zell-Funktion mit der Generierung passiver Immunität durch lebenslange Substitution von Immunglobulinen verbessert. Somit können Anzahl und Schwere der Infektionen reduziert werden. Der frühzeitige Einsatz von Antibiotika ist entscheidend für eine suffiziente Infekttherapie. Die Wirksamkeit von Impfungen ist vermindert, Lebendimpfungen sind kontraindiziert und dürfen nur nach guter Nutzen-Risiko-Abwägung eingesetzt werden (Schmidt et al. 2011). 23 2 Einleitung 2.3.2 Autosomal rezessive Agammaglobulinämie Diese Form der Agammaglobulinämie ist unabhängig vom Bruton-Tyrosinkinase-Gen und wird autosomal rezessiv vererbt. Es handelt sich pathogenetisch ebenfalls um einen Reifungsstopp in der B-Zellentwicklung mit folgendem Funktionsverlust. Der Krankheitsphänotyp entspricht dem der X-chromosomalen Agammaglobulinämie, beginnt aber bereits mit drei Monaten. Labordiagnostisch finden sich eine ausgeprägte Hypogammaglobulinämie, keine B-Zellen und evtl. eine Neutropenie. Die Therapie besteht auch hier in der Substitution von Immunglobulinen. Allerdings ist die Prognose durch schwerere Verlaufsformen bei der rezessiven schlechter als bei der X-chromosomalen Agammaglobulinämie (Baumann et al. 2010). 2.3.3 Common Variable Immunodeficiency (CVID) Das variable Immundefektsyndrom (CVID) ist als primäre Antikörperdefizienz mit erniedrigten IgG- und IgA-Spiegeln, gehäuften bakteriellen Infektionen und vermindertem Impfansprechen definiert. IgM-Verminderungen können ebenfalls auftreten. Typische Krankheitssymptome sind rezidivierende bakterielle Infektionen der oberen und unteren Atemwege und des Gastrointestinaltraktes (Schmidt et al. 2011). Bei der CVID ist die Inzidenz lymphoproliferativer sowie granulomatöser Erkrankungen, von Autoimmunprozessen und gastrointestinalen Malignomen erhöht (Pfluger et al. 2000). Der Erkrankungsbeginn liegt in der zweiten bis dritten Lebensdekade. Die Diagnose wird mithilfe der Bestimmung von mindestens zwei Immunglobulinklassen zu mindestens zwei verschiedenen Zeitpunkten gestellt. Außerdem besteht ein reduzierter Antikörpertiter nach Impfungen. Es handelt sich um eine Ausschlussdiagnose, es müssen Urin-Status, Protein im Urin zum Ausschluss eines Eiweißverlustes, Erregernachweis und Ausschluss einer Leukämie oder eines Multiplen Myeloms erfolgen. Therapiert werden die Patienten mit Immunglobulinen und Antibiotika bei Infektionen bzw. prophylaktisch. Ein CVID kann mit IgG-Subklassendefekten assoziiert sein. Hierbei sind einige IgG-Subklassen häufiger betroffen als andere mit der Verteilung IgG4 > IgG2 > IgG1 > IgG3 (Wedgwood et al. 1984). 24 2 Einleitung 2.3.4 Hyper-IgM-Syndrome Bei diesen Syndromen handelt es sich um Defekte des ImmunglobulinKlassenwechsels, wodurch es zu erhöhten bis normalen IgM-Spiegeln bei niedrigen IgG-, IgA- und IgE-Spiegeln kommt. Defekte liegen in verschiedenen Genen vor, z.B. für CD40 und CD40-Ligand, die für die B-T-Zell-Kooperation essentiell sind, in B-Zellintrinsischen Defekten, wie NF-κB Essential Modulators (NEMO), Activation induced Cytidine Deaminase (AID), Uracil-DNA-Glykosylase (UNG) und anderen. Durch diese genetischen Defekte kommt es zu Störungen der B- und T-Zell-Interaktion und somit zu Defekten im Immunglobulin-Klassenwechsel und der somatischen Hypermutation. Dies führt zu fehlendem Klassenwechsel von IgM zu den anderen ImmunglobulinIsotypen mit Anhäufung des IgM sowie Verminderung der anderen Immunglobuline. Klinisch treten rezidivierende bakterielle Atemwegsinfekte, Infektionen mit opportunistischen Erregern wie Pneumocystis jirovecii und Kryptosporidien auf. Es kommt gehäuft zu oralen und rektalen Ulzerationen, Autoimmunerkrankungen wie hämolytischer Anämie, Arthritiden, Hepatitis, Neutro- und Thrombozytopenie. Die Prognose ist ungünstig, die Patienten versterben größtenteils im jugendlichen Alter. Hinzu kommt ein erhöhtes Malignomrisiko v.a. für Hodgkin- und Non-HodgkinLymphome, Pankreas-, Leber- und Gallengangskarzinome (Davies und Thrasher 2010). 2.3.5 Selektiver IgA-Mangel Der selektive IgA-Mangel ist der häufigste Immundefekt überhaupt. Er verläuft oft asymptomatisch. Die IgA-Spiegel sind vermindert oder nicht nachweisbar bei normalen IgG- und IgM-Spiegeln, normaler B- und T-Zellzahl und intakter Impfantwort. Bei oftmals gleichzeitig vorliegendem IgG-Subklassenmangel kommt es zu vermehrter Infektanfälligkeit. Es liegt eine erhöhte Assoziation zu Autoimmunerkrankungen und Allergien vor. Bei der RA tritt der IgA-Mangel ca. 10mal häufiger auf als in Kontrollkollektiven. Bei alleinigem IgA-Mangel ohne erhöhte Infektanfälligkeit besteht keine Indikation zur Immunglobulinsubstitution. Diese ist gegeben bei zusätzlichem IgG-Defekt und rezidivierenden respiratorischen Infektionen. 50% der Patienten entwickeln jedoch Antikörper gegen IgA, wodurch es bei Gabe von Immunglobulinen zu schweren anaphylaktischen Reaktionen kommen kann (Baumann et al. 2010, Schmidt et al. 2011). 25 2 Einleitung 2.3.6 IgG-Subklassenmangel Pathogenese und genetische Ursachen des IgG-Subklassenmangels sind bislang ungeklärt. Auch die Prävalenz ist nicht genau bekannt, geschätzt wird sie auf 1:10.000. Die tatsächliche Häufigkeit liegt vermutlich weit höher, da leichte Krankheitsverläufe gar nicht diagnostiziert werden. Klinisch können vermehrt Infektionen des Respirations- und Gastrointestinaltraktes beobachtet werden, entsprechend des jeweiligen Effektorprofils der defekten IgG-Subklassen (Jefferis und Kumararatne 1990). Neben einer oder mehreren IgG-Subklassen kann, muss aber nicht, auch die IgG-Gesamtkonzentration vermindert sein. Einen Mangel an IgG-Subklassen ohne verminderte IgG-Konzentration bezeichnet man als spezifischen Antikörperdefekt. Ein IgG1-Mangel kann mit schweren Infektionen bis hin zu septischen Verläufen einhergehen. Bei einem IgG2-Subklassendefekt kommt es häufig zu rezidivierenden Infekten mit bekapselten Bakterien, die Polysaccharide auf ihrer Oberfläche tragen, wie Streptococcus pneumoniae und Haemophilus influenzae. Schwere Infektionen sind bei reinem IgG2-Mangel selten. Es kann aber zu einer verminderten Impfantwort auf Polysaccharidimpfstoffe kommen. Daher sind bei solchen Patienten der Einsatz von konjugierten Impfstoffen sowie die nachträgliche Titerbestimmung ratsam. Der IgG2-Mangel tritt teils zusammen mit asthmatischen und chronisch obstruktiven Lungenerkrankungen auf. Ein IgG3Mangel kann zu viralen Infektionen der oberen Atemwege führen. IgG4 ist bei bis zu 40% der Bevölkerung als erniedrigt beschrieben, jedoch ohne Krankheitseffekte. Häufig treten Subklassendefekte in Kombination auf. Dabei treten v.a. kombinierte Defekte von IgG2 und IgG4 auf, auch Defekte des IgG1 kommen häufiger zusammen mit IgG3-Defekten vor (Schmidt et al. 2011). Bei asymptomatischen und leichten Krankheitsverläufen wird keine Dauertherapie mit Antibiotika empfohlen. Aber bei rezidivierenden Infektionen soll frühzeitig mit einer Antibiotika-Therapie begonnen werden. Bei der anfangs ungezielten Therapie sollten Antibiotika eingesetzt werden, die gegen bekapselte Bakterien wirksam sind. Bei rezidivierenden Infektionen und/ oder IgG1- oder IgG2-Mangel ist der Einsatz von Immunglobulinen indiziert (Baumann et al. 2010). Ein Übergang in ein CVID ist möglich. Eine Hypogammaglobulinämie und IgG-Subklassendefekte können auch bei schwerem kombiniertem Immundefekt auftreten. Weitere primäre Immundefekte, bei 26 2 Einleitung denen der AK-Mangel im Vordergrund steht, sind sehr selten und sollen deshalb hier nicht weiter ausgeführt werden. 27 3 Ziel dieser Arbeit 3 Ziel dieser Arbeit In den letzten Jahren haben sich die diagnostischen Möglichkeiten und die Therapieoptionen der RA entscheidend verbessert. Insbesondere aufgrund der aktuellen Entwicklung Therapieergebnisse der zu biologischen erreichen. Erstmals DMARDs gelingt sind es, verbesserte dem Ziel der Krankheitsremission näher zu kommen. Gleichzeitig bestehen aber auch Risiken beim Einsatz dieser Medikamente. Diese greifen nicht selektiv in die Pathogenese der RA ein, sondern beeinflussen das Immunsystem und die Immunhomöostase. Eine erhöhte Neigung zum Auftreten von Infektionen ist bereits allein durch die RA bekannt, welche durch Biologika noch verstärkt werden kann. Insbesondere ist ein intaktes Immunsystem der Patienten wichtig, da vorbestehende Immundefekte im humoralen und zellulären Bereich ein zusätzliches Risiko für das Auftreten von Infektionen hervorrufen können. Bislang sind die Defekte im Bereich der Immunglobuline, insbesondere des IgG und dessen Subklassen, nicht ausreichend untersucht. Ein intaktes IgG und dessen Subklassen sind jedoch wesentlich für die Abwehr von bakteriellen Infektionen. Das Ziel dieser Arbeit ist es, die Häufigkeit von IgG-Defekten an einer großen Fallzahl von RA-Patienten zu erfassen. Weiterhin soll die Häufigkeit der IgGSubklassendefekte in dieser Population beschrieben werden. Zusätzlich bestimmten wir die IgA- und IgM-Isotypen. Das Auftreten von Infekten, Nikotinkonsum, Anwendung von NSAR, Kortikosteroiden, synthetischen und biologischen DMARDs, RA-Krankheitsaktivität, Geschlechtsverteilung, RF, Anti-CCP-AK sowie deren Beziehung zum Auftreten von Immundefekten wurden untersucht, um mögliche korrelative Zusammenhänge zu beschreiben. Von diesen Untersuchungen erwarten wir ein besseres Verständnis der Bedeutung von Defekten der Immunglobuline für den Krankheitsverlauf, den Einfluss der Therapien bzw. der Wirksamkeit von Therapien. Die genauere Kenntnis des Immunstatus des einzelnen Patienten kann die Möglichkeit ergeben, ein individuelles Therapieschema erstellen zu können, das v.a. auf die Abwehrlage abgestimmt ist, um somit schwere Infektionen zu vermindern oder gar zu vermeiden. Insbesondere war der Zusammenhang von IgG-Subklassendefekten zur Infekthäufigkeit der RAPatienten von hohem Interesse. 28 3 Ziel dieser Arbeit Im Ergebnis der Arbeit erwarten wir ein besseres Verständnis der Bedeutung von Defekten der Immunglobuline auf den Krankheitsprozess und deren Einfluss auf den Therapieverlauf. Möglicherweise wird in Zukunft die genaue Kenntnis des Immunstatus des einzelnen Patienten das individuelle Therapieschema determinieren, um schwere Infektionen durch eine geschwächte Abwehrlage zu vermeiden. Sollten sich die Hinweise auf gehäufte Defekte im IgG-/IgG-Subklassenbereich durch unsere Untersuchungen verfestigen, wäre es empfehlenswert, die Sicherheitsuntersuchungen vor Einleitung einer Therapie von synthetischen und insbesondere biologischen DMARDs zu überprüfen. 29 4 Material und Methoden 4 Material und Methoden 4.1 RA-Patienten Untersucht wurden 539 Patienten mit der Diagnose rheumatoide Arthritis, die nach den ACR/ EULAR-Klassifikationskriterien von 2010 gestellt wurde (Aletaha 2010). Alle Patienten waren im Ambulanten Rheumazentrum in Erfurt in Betreuung. Die Patientendaten wurden einmalig im Rahmen der normalen Langzeitkontrolle erhoben. Die epidemiologischen Daten sind in Tab. 5 zusammengestellt. 4.2 Geräte Die Immunglobulin-Subklassenbestimmung wurde mittels Immunonephelometrie mit dem Beckman Immage 800 Analyser von Beckman Coulter (Birmingham, UK) im Labor des Ambulanten Rheumazentrums Erfurt durchgeführt. Die Konzentrationen der Immunglobulin-Isotypen G, A und M, des CRP und der IgM-RF wurden ebenfalls mit dem Beckman Immage 800 Analyser erhoben. Für die Erhebung von IgA-RF, IgG-RF und Anti-CCP-AK verwendeten wir den EUROIMMUN Analyzer 1 der EUROIMMUN AG Lübeck. 4.3 Material Für die Subklassenbestimmung wurden folgende Substanzen der Firma Bindingsite (Birmingham, UK) benötigt: 2,5 ml Human IgG1/2 Antiserum (Antiserum der jeweiligen Subklasse) Human IgG1/2 Beckman Immage Calibrator 1-6 (Kallibrator-Set, 6x1 ml der jeweiligen Subklasse) 1 ml Human IgG Subclass Low Control (Kontrolle, Low Level) 1 ml Human IgG Subclass High Control (Kontrolle, High Level) 3,4 ml Human IgG3/4 Latexreagenz der jeweiligen Subklasse 30 4 Material und Methoden 3,0 ml Reactions-Accelerator der jeweiligen Subklasse Human IgG3/4 Beckman Immage Calibrator 1-6 (Kalibrator-Set, 6x1ml der jeweiligen Subklasse) Beckman Immage Diluens1 (Phosphatpuffer) Beckman Immage Puffer1 (Phosphatpuffer mit Reaktionsbeschleuniger) 4.4 Methoden 4.4.1 Bestimmung der IgG-Subklassen Die Subklassen wurden mittels Nephelometrie bestimmt, hierbei kommt es zur Reaktion löslicher Antigene mit spezifischen Antiseren unter Bildung unlöslicher Antigen-Antikörper-Komplexe. Daraufhin wird ein Lichtstrahl durch die Suspension geführt, wobei ein Teil des Lichts gestreut wird. Die Intensität des Streulichts wird bei einer 90°-Ablenkung gemessen und mit entsprechend erstellter Kalibrationskurve wird die Konzentration des spezifischen Proteins, hier der IgG-Subklassen, ermittelt. Da IgG3 und IgG4 nicht genügend große Immunkomplexe mit den Antisera bilden und in dieser Form nicht gemessen werden könnten, werden die Antikörper an Latexpartikel geeigneter Größe gekoppelt. So kann der Antigen-Antikörper-Komplex ausreichend Licht streuen und das Antigen wird der Messung zugänglich. Das mit einem Serum-Gel-Röhrchen abgenommene Blut wurde 5min bei 3000 g zentrifugiert und das Serum in ein Sekundärröhrchen abgetrennt. Die Identifizierung erfolgte mittels Barcodierung. Die Proben wurden eingefroren, bis eine Serie von 40 Patientenproben vorhanden war. Zur Messung wurden die Proben am Immage 800 von Beckman eingerichtet und es wurde eine Kalibration durchgeführt. Anschließend wurden zwei Kontrollen mit hoher und niedriger Konzentration gemessen und dokumentiert. Lagen diese im vom Hersteller vorgegebenen Bereich, wurden die Patientenproben aufgetaut, mittels Vortexer gemischt und gemessen. Über den Barcode und die Kommunikation mit der elektronischen Labordatenverarbeitung erfolgte die Probenzuweisung durch das Gerät. Die Bestimmung der einzelnen IgGSubklassen erfolgte immer gleichzeitig. Die Ergebnisse wurden an den Computer übermittelt und auf Plausibilität technisch geprüft. Anschließend erfolgte eine medizinische Validierung durch den Laborarzt des Rheumazentrums Erfurt (Dr. Wolf). 31 4 Material und Methoden 4.4.2 Bestimmung weiterer Laborwerte Die Bestimmung von IgA, IgG, IgM, CRP, IgM-RF erfolgte ebenfalls durch Nephelometrie, Beckmann Immage 800 Analyser von Beckmann, Birmingham (s.o.). Die IgA-RF, Immunosorbent IgG-RF, Assay) Anti-CCP-AK Verfahren wurden und durch ELISA- (Enzyme-linked Auswertung mittels colorimetrischer Endpunktbestimmung analysiert (Gerät: EUROIMMUN Analyzer 1, EUROIMMUN AG Lübeck). Bestimmt wurden IgG gegen CCP, indem in einem ersten Analyseschritt die Reagenzgefäße mit verdünnten Patientenproben inkubiert wurden. Bei positiven Proben binden sich spezifische Antikörper der Klasse IgG, auch IgA und IgM an die jeweiligen Antigene. In einem zweiten Schritt kommt es durch Inkubation mit einem Enzym-markierten Anti-Human-IgG zur Darstellung dieser Antikörper. Hierdurch wird eine Farbreaktion katalysiert, die anschließend photometrisch gemessen wird. Entsprechend des oben dargestellten Vorgehens erfolgte die Bestimmung der RF, die bei positiven Proben an ihr jeweiliges Antigen binden, daraufhin wurde mit Enzym-markiertem Anti-Human-IgA bzw. –IgG inkubiert, wodurch eine Farbreaktion ausgelöst wird. Die Höhe des Titers wurde in IU/ml angegeben. Die Bestimmung der BSG erfolgte durch die Westergren-Methode, indem 1,6 ml Vollblut mit 0,4 ml 3,8%iger Natriumcitratlösung ungerinnbar gemacht wurden. Dieses wurde in ein senkrecht stehendes Röhrchen mit Millimeterskala gefüllt, auf der nach einer Stunde die Länge der zellfreien Säule bestimmt wurde. 4.4.3 Zugrunde liegende Normwerte Pathologisch verminderte Werte werden als Defekte bezeichnet. Sie werden als vermindert definiert, sobald sie den Normwert unterschreiten. Z.B. gelten im Falle des IgG1 alle Konzentrationen <4,90 g/l als Defekte und Konzentrationen >11,40 g/l als Erhöhungen. In Tab. 3 finden sich die in dieser Arbeit verwendeten Normwerte. 32 4 Material und Methoden Tabelle 3: Auflistung der verwendeten Normwerte Normwerte BSG 0-15 mm/h CRP 0-5 mg/l IgA 0,9-4,50 g/l IgG 8,00-18,00 g/l IgM 0,70-2,80 g/l IgG1 4,90-11,40 g/l IgG2 1,50-6,40 g/l IgG3 0,20-1,10 g/l IgG4 0,080-1,40 g/l RF IgM 0-20 IU/ml RF IgG 0-20 RU/ml RF IgA 0-20 RU/ml Anti-CCP-AK 0-5 RU/ml Normwerte des rheumatologisch-immunologischen Labors Erfurt, Dr. Wolf Für die binär logistische Regression wurde die abhängige Variable so eingeteilt, dass sich zwei Ausprägungen ergaben. Im Falle der Immunglobuline A, M und G sowie der IgG-Subklassen entsprach die Grenze der beiden Ausprägungen dem unteren Grenzwert der Norm. Dies ist in Tab. 4 gezeigt. Tabelle 4: Ausprägungen der abhängigen binären Variablen mit zugehörigen Laborgrenzwerten abhängige Ausprägung 1 Ausprägung 2 Variable kein Defekt Defekt IgA (g/l) > 0,90 ≤ 0,90 IgM (g/l) > 0,70 ≤ 0,70 IgG (g/l) > 8,00 ≤ 8,00 IgG1 (g/l) > 4,90 ≤ 4,90 IgG2 (g/l) > 1,50 ≤ 1,50 IgG3 (g/l) > 0,20 ≤ 0,20 IgG4 (g/l) > 0,08 ≤ 0,08 4.4.4 Bestimmung des Aktivitätsstatus Die Berechnung des Aktivitätsstatus mittels Disease Activity Score 28 (DAS 28) erfolgte nach folgenden Vorschriften: DAS 28 (BSG) = 0,56∙ +0,28∙ +0,70∙ln(BSG)+0,014∙VAS 33 4 Material und Methoden DAS 28 (CRP) = 0,56∙ +0,28∙ +0,36∙ln(CRP+1)+0,014∙VAS+0,96 Das mit einer dieser Formeln, abhängig von der Verwendung des Laborparameters, ermittelte Ergebnis kann Werte zwischen 0 und 10 annehmen, wobei ein Wert bis 3,2 eine geringe Krankheitsaktivität darstellt, Werte von 3,2 bis 5,1 entsprechen mittlerer und über 5,1 hoher Krankheitsaktivität. Von fehlender Krankheitsaktivität bzw. Remission spricht man bei Werten zwischen 0 und 2,6 (Wells et al. 2009). 4.4.5 Bestimmung der VAS Die visuelle Analogskala (VAS) stellt eine subjektive Methode zur Messung der Empfindungsstärke für Schmerz dar. Die Patienten wurden aufgefordert, ihre Schmerzempfindung auf einer Skala von 0 mm (entspricht keinen Schmerzen) bis 100 mm (entspricht stärksten vorstellbaren Schmerzen) einzutragen. 4.5 Statistik Zur statistischen Datenbearbeitung wurde SPSS 14.0 sowie SPSS 20.0 für Windows verwendet. Die grafische Aufarbeitung erfolgte mit Sigmaplot 12.0 und Origin 8.0. Tabellen wurden mit Excel erstellt. Die Beschreibung der epidemiologischen Daten erfolgte mit Hilfe eines zweiseitigen t-Tests und die Erhebung von Zusammenhängen wurde mittels binär logistischer Regression durchgeführt. Die Signifikanz der Korrelation für die verschiedenen Parameter wurde für p ≤ 0,05 definiert. 34 5 Ergebnisse 5 Ergebnisse 5.1 Epidemiologische Daten des Patientenkollektivs Das Alter der 539 untersuchten RA-Patienten betrug durchschnittlich 60,6 ± 12,6 Jahre (Mittelwert ± Standardabweichung), wobei der jüngste Patient 17 Jahre und der älteste Patient 92 Jahre alt waren (Abb. 6). Die Geschlechtsverteilung entsprach mit 383 (71,1%) weiblichen Patienten der zu erwartenden Häufigkeit bei RA. Abbildung 6. Altersvergleich unter den RA-Patienten (n.s. = nicht signifikant; n = 539, □ = Median,(-) = Mittelwert, untere und obere Boxbegrenzung = 25. und 75. Perzentile, x = Maxima/ Minima, Fehlerbalken = 1. und 99. Perzentile). Die weiteren epidemiologischen Daten zu Raucherstatus, Krankheitsdauer, Röntgenund Gelenkstatus sind in Tabelle 5 für das Gesamtkollektiv sowie getrennt für die Geschlechter zusammengefasst. 35 5 Ergebnisse Tabelle 5: Epidemiologische Daten der Patienten Gesamtgruppe % bezogen auf Gesamtgruppe Alter (in Jahren) Männer (♂) Frauen (♀) % bezogen auf alle % bezogen auf alle ♀ ♂ Vergleich ♀-♂ 60,6 ± 12,6 60,5 ± 12,2 60,8 ± 14 n.s. Anzahl der Raucher 115 (21,6 %) 72 (19,0 %) 43 (27,7 %) Ehemalige Raucher 19 (3,6 %) 6 (1,6 %) 13 (8,4 %) Anzahl Nichtraucher 399 (74,9 %) 300 (78,3 %) 99 (63,9 %) 9,2 ± 9,5 9,7 ± 9,7 8,1 ± 8,7 257 (51,1 %) 176 (49,2 %) 81 (55,9 %) 33 (6,6 %) 23 (6,4 %) 10 (6,9 %) 213 (42,3 %) 159 (44,4 %) 54 (37,2 %) Gelenkstatus: Anzahl DS 3,2 ± 4,0 3,3 ± 4,2 3 ± 3,3 n.s. Anzahl GS 3,0 ± 3,6 3,1 ± 3,8 2,7 ± 3,1 n.s. VAS (in mm) 21,3 ± 16 22,0 ± 16,5 19,7 ± 14,6 n.s. Rauchen Krankheitsdauer (in Jahren) p < 0,001 n.s. Röntgenstatus: Nicht erosiv Beginnend erosiv Erosiv n.s. Deskriptiver Vergleich mittels zweiseitigen t-Tests zur Beschreibung der epidemiologischen Verteilung. Angegeben sind Mittelwerte ± Standardabweichung und Häufigkeiten mit den zugehörigen Prozentangaben. (DS = druckschmerzhafte Gelenke, GS = geschwollene Gelenke, n.s. = nicht signifikant, p = Signifikanz) Unter den Patienten wurde der Raucherstatus erhoben. Hierbei fanden sich 399 Nichtraucher (74%), 115 (21,3%) Raucher und 19 (3,5%) ehemalige Raucher. Patienten, die seit mindestens 2 Jahren mit dem Rauchen aufgehört haben, fielen in diese Gruppe. Bei 6 Patienten (1,1%) gab es keine Angaben zum Raucherstatus. Hierbei fiel auf, dass unter den Patienten im prozentualen Vergleich signifikant mehr Männer als Frauen rauchten (Tab. 5). Die Krankheitsdauer ab Diagnosestellung betrug im Mittel 9,2 Jahre (SD = 9,5) mit einer Gesamtspanne von 0 bis 57 Jahren, wobei das Minimum der Krankheitsdauer 3 Monate betragen musste, um die Diagnose der RA stellen zu können. 55 Patienten wiesen zum Zeitpunkt unserer Studie eine frühe RA mit einer Krankheitsdauer unter 2 Jahren auf, die restlichen 484 hatten ein bereits fortgeschrittenes Stadium. Die Häufigkeiten der Röntgenstadien der Erkrankung waren im Bereich der Hände und Vorfüße wie folgt verteilt: 257 (47,7%) hatten keine erosiven Veränderungen, bei 36 5 Ergebnisse 213 (39,5%) erbrachte die Röntgendiagnostik erosive Befunde und bei 33 (6,1%) beginnende Erosionen. Bei 36 (6,7%) gab es keine aktuellen Röntgenbefunde. Für das Patientenkollektiv ergaben sich folgende Werte für den Gelenkstatus: die druckschmerzhaften Gelenke lagen im Mittelwert bei 3,2 (SD = 4,0). Der Mittelwert der geschwollenen Gelenke betrug 3 (SD = 3,6). Zur Beurteilung des Einflusses von Medikamenten auf die Konzentration von IgG und dessen Subklassen wurden die unter der Diagnose RA verordneten Medikamente betrachtet. Dazu zählten NSAR, COX-2-Hemmer, Glukokortikoide, DMARDs und Biologika. NSAR, welche die COX-1 und COX-2 hemmen, wurden zum Zeitpunkt unserer Messung von 245 Patienten (45,5%) eingenommen, 130 Patienten (24,1%) nahmen COX-2-Hemmer und 164 (30,4%) bekamen aktuell weder ein NSAR noch einen COX-2-Hemmer. Zu den Häufigkeiten der Einnahme von NSAR bzw. COXHemmern siehe Tab. 6. Tabelle 6: Häufigkeiten der Einnahme von NSAR und COX-2-Hemmern unter den RA-Patienten NSAR/ COX-2-Hemmer n=539 (100%) kein NSAR 164 (30,4%) Ibuprofen 68 (12,6%) Naproxen 5 (0,9%) Indometacin 8 (1,5%) Diclofenac 115 (21,3%) Acemetacin 22 (4,1%) Meloxicam 24 (4,5%) Piroxicam 2 (0,4%) Rantudil 1 (0,2%) Celecoxib 42 (7,8%) Etericoxib 88 (16,3%) Prednisolon, ein synthetisches Glukokortikoid, wurde von 365 Patienten (67,7%) eingenommen im Gegensatz zu 174 Patienten (32,3%) ohne dessen Einnahme. Von den 365 Patienten nahmen 29 (5,4%) bis 2,5 mg Prednisolon, 221 (41%) bis 5 mg, 43 (8%) bis 7,5 mg und 74 (13,7%) bekamen mehr als 7,5 mg. Eine Basistherapie mit DMARDs erhielten zum Zeitpunkt der Erhebung 414 Patienten (76,8%). Den restlichen 125 (23,2%) Patienten wurde keine Basistherapie verabreicht oder sie Unverträglichkeitsreaktionen mussten das absetzen. 37 Medikament Zu den auf Grund von Basistherapeutika der 5 Ergebnisse Patientengruppe gehörten MTX und Leflunomid, Chloroquin und Hydroxychloroquin, Sulfasalazin, Azathioprin und Gold-Präparate. MTX nahmen 327 der 539 Patienten ein, Leflunomid 79 Patienten, Sulfasalazin bekamen 23, Chloroquin 13, Hydroxychloroquin 10, Azathioprin 3 und das Gold-Präparat Tauredon 2 Patienten. In der Tab. 7 sind die Einnahmehäufigkeiten aller verordneten DMARDs dargestellt. 41 der 414 unter Basistherapie stehenden Patienten nahmen zwei DMARDs ein, ein Patient bekam drei DMARDs verschrieben. Tabelle 7: Einnahmehäufigkeiten der Basistherapie mit konventionellen DMARDs DMARDs Anzahl der Patienten (%) DMARDs aufgelistet nach Dosierung kein DMARD 125 (23,2) kein DMARD MTX 327 (60,7) MTX 5mg pro Woche 1 MTX 7,5mg pro Woche 26 MTX 10mg pro Woche 74 MTX 12,5mg pro Woche Leflunomid Sulfasalazin 79 (14,6) 23 (4,3) Anzahl der Patienten 125 2 MTX 15mg pro Woche 202 MTX 20mg pro Woche 16 MTX 22,5mg pro Woche 6 Leflunomid 10mg pro Tag 22 Leflunomid 20mg pro Tag 57 Sulfasalazin 1000mg pro Tag 7 Sulfasalazin 2x1000mg pro Tag 15 Sulfasalazin 3x1000mg pro Tag 1 Chloroquin 13 (2,4) Resochin 200mg pro Tag 13 Hydroxychloroquin 10 (1,8) Quensyl 250mg pro Tag 10 Azathioprin 3 (0,6) Azathioprin 2x50mg pro Tag 2 Azathioprin 3x50mg pro Tag 1 Tauredon 50mg alle 3-4 Wochen 2 Gold-Präparat 2 (0,4) Einem kleineren Teil der Patienten wurden Biologika verschrieben. Die Zahl der Patienten beläuft sich auf 107 (19,9%). Die Aufschlüsselung der biologischen DMARDs ist in Tab. 8 dargestellt. 17 der Patienten unter Tozilizumab erhielten 200mg, die anderen 12 Patienten bekamen 400mg. Etanercept wurde in 13 Fällen mit 50mg und in 8 Fällen mit 25mg verschrieben. Alle Patienten unter Certolizumab injizierten sich 200mg des Präparates im Abstand von 2 Wochen s.c.. 38 5 Ergebnisse Tabelle 8: Häufigkeiten von BiologikaEinnahmen unter den RA-Patienten Häufigkeiten der Biologika-Einnahmen n=539 (100%) kein Biologikum 432 (80,1%) Adalimumab 32 (5,9%) Tozilizumab 29 (5,4%) Etanercept 21 (3,9%) Certolizumab 15 (2,8%) Rituximab 7 (1,3%) Infliximab 1 (0,2%) Abatacept 1 (0,2%) Golimumab 1 (0,2%) Um den Einfluss von Wechselwirkungen unter den einzelnen RA-Medikamenten auf die Subklassenkonzentrationen zu erfassen, haben wir die Patienten nach der Anzahl eingenommener Medikamente eingeteilt. Dabei betrachteten wir lediglich Medikamente, die aufgrund der Diagnose rheumatoide Arthritis verordnet wurden. Unter den 539 Patienten befanden sich 9 (1,7%), die keine Medikamente einnahmen, 70 (13%) mit einem verordneten Mittel, 186 (34,5%) mit zwei, 230 Patienten (43,7%) mit drei, 41 (7,6%) mit vier Medikamenten und 3 Patienten (0,6%) bekamen 5 Medikamente. Die Probanden wurden außerdem zu Infekten und Infekthäufigkeit befragt. Von den 538 Befragten gaben 186 Infekte im vergangenen Jahr an. Nach Infekthäufigkeit wurden folgende Gruppen gebildet: selten mit bis zu zwei Infekten im letzten Jahr, häufig mit mehr als zwei auftretenden Infekten und persistierende Infekte. Wer weniger als zwei Infekte angab, wurde in die Gruppe keine Infekte eingeteilt. Dies betraf 352 (65,4%) der Befragten, seltene Infekte gaben 116 (21,6%) an und häufige Infekte 68 (12,6%). Persistierend traten diese bei zwei der Befragten (0,4%) auf. Die Häufigkeiten sowie Arten der retrospektiv erfassten Infektionen der Patienten sind in den Tab. 9 und 10 aufgelistet. Tabelle 9: Infekthäufigkeit der RA-Patienten Häufigkeiten n = 539 (100%) 353 (65,4) 117 (21,7) 67 (12,5) 2 (0,4) keine Infekte selten Infekte häufig Infekte persistierende Infekte (selten ≤ 2 Infekte pro Jahr, häufig > 2 Infekte pro Jahr) 39 5 Ergebnisse Tabelle 10: Arten der aufgetretenen Infektionen Infektion obere Atemwegsinfekte Harnwegsinfekte Herpes simplex Infektion Gastrointestinale Infekte Bronchitis Zystitis Infizierte Ulcera crura Pneumonie Augenentzündung Otitis externa Wundinfektion Tuberkulose Häufigkeiten n = 211 (100%) 122 (57,8) 34 (16,1) 13 (6,2) 10 (4,7) 7 (3,3) 6 (2,8) 4 (1,9) 4 (1,9) 4 (1,9) 3 (1,4) 2 (0,9) 2 (0,9) Der T-Test zwischen Männern und Frauen zeigte keine signifikanten Unterschiede bezüglich Medikamenteneinnahme und des Auftretens von Infektionen. 5.2 Klinische Prozessaktivität und Laborergebnisse der Patientengruppe Für die Berechnung des DAS 28 werden vier verschiedene Messgrößen benötigt, dazu gehören druckschmerzhafte und geschwollene Gelenke, VAS, wie oben schon beschrieben, und BSG. Für die Berechnung des DAS 28 mit CRP wird dieses anstelle der BSG verwendet. Die BSG lag im Mittel bei 24,8 ± 20,7 mm/h. Hierbei lagen 318 Proben (59%) im erhöhten Bereich. Der Mittelwert des CRP war 8,7 mg/l mit einer SD von 13,8. Von den gemessenen CRP-Werten fanden sich 226 (41,9%) oberhalb des Normbereiches. Die VAS von 0-100 mm betrug durchschnittlich 21,3 ± 16 mm. Aus diesen Werten lässt sich der DAS 28 auf zwei verschiedenen Wegen berechnen. Der Mittelwert für den DAS 28 mit BSG in der Berechnung ergab 3,4 bei einer SD von 1,3. Dieser Berechnung zufolge befanden sich 155 Patienten (28,8%) in Remission, 96 Patienten (17,8%) wiesen geringe, 230 Patienten (42,7%) mittlere und 58 Patienten (10,8%) hohe Krankheitsaktivität auf (Tab. 11). Die Berechnung des DAS 28 mit CRP ergab im Vergleich abweichende, deutlich niedrigere Werte. Der Mittelwert lag bei 3,0 bei einer SD von 1,2. Laut dieser Berechnung wiesen 205 40 5 Ergebnisse Patienten (38%) eine Remission auf, 112 (20,8%) geringe Krankheitsaktivität, 186 Patienten (34,5%) mittlere und 36 Patienten (6,7%) eine hohe Krankheitsaktivität auf. Tabelle 11: DAS 28 im Vergleich. Mittelwert mit Standardabweichung, Häufigkeiten und deren Prozente. n=539. Mittelwert ± SD DAS 28 mit BSG DAS 28 mit CRP 3,4 ± 1,3 3,0 ± 1,2 Anzahl an Patienten mit: Remission 155 (28,8%) 205 (38%) geringer Krankheitsaktivität 96 (17,8%) 112 (20,8%) mittlerer Krankheitsaktivität 230 (42,7%) 186 (34,5%) hoher Krankheitsaktivität 58 (10,8%) 36 (6,7%) Häufig wird noch die Berechnung des DAS 28 mithilfe der BSG durchgeführt, jedoch ist diese Methode anfälliger für Einflüsse, die nicht aus der Krankheitsaktivität resultieren, sondern andere Ursachen haben. So entsteht durch den Einfluss der unspezifischen BSG der Eindruck von höherer Krankheitsaktivität und weniger Remissionen, der bei der Verwendung des CRP nicht so ausfällt. Deshalb ist es sinnvoller, die Berechnung mit Einsatz des CRP durchzuführen. Zu den erhobenen Laborwerten zählten weiterhin die Immunglobulin-Isotypen G, A und M, wobei die Häufigkeiten von IgG und dessen Subklassen im nächsten Kapitel näher erläutert werden. Das IgA wurde bei den 539 Patientenproben mit einem Mittelwert von 2,5 ± 1,2 g/l gemessen (Tab. 12). Die im Folgenden beschriebenen Defekte lagen unter den Normwerten aus Tab. 3, die Erhöhungen über diesen Normwerten. Hierbei trat bei 31 Patienten (5,8%) ein IgA-Defekt auf und bei 28 Patienten (5,2%) lagen die Konzentrationen über dem Normwert. Die restlichen 480 (89,1%) wiesen Werte im Normbereich von 0,9-4,5 mg/l auf. Die IgM-Messungen ergaben einen Mittelwert von 1,2 ± 0,9 g/l (Tab. 12). 132 von 539 Proben (24,5%) zeigten IgM-Erniedrigungen neben 19 Proben (3,5%), die eine Erhöhung aufwiesen. Im Normbereich befanden sich 388 der Patientenproben (72%). Die IgMVerminderungen traten signifikant häufiger bei RA-Patienten mit positivem Raucherstatus auf. 41 5 Ergebnisse Tabelle 12: Laborergebnisse der RA-Patienten Gesamtgruppe Frauen (♀) Männer (♂) Vergleich ♀ ♂ BSG (mm/h) 24,8 ± 20,7 26,3 ± 20,7 21,0 ± 20,1 p < 0,01 CRP (mg/l) 8,7 ± 13,8 8,1 ± 11,9 10,1 ± 17,5 n.s. IgA (in g/l) 2,5 ± 1,2 2,5 ± 1,2 2,6 ± 1,1 n.s. IgM (in g/l) 1,2 ± 0,9 1,2 ± 0,7 1,,2 ± 1,2 n.s. (69,9) (69,1 ± 81,5) (71,7 ± 82,5) (n.s.) Anti-CCP-AK (in RU/ml)* DAS 28 mit BSG 3,4 ± 1,3 3,5 ± 1,3 3,2 ± 1,2 p < 0,05 DAS 28 mit CRP 3,0 ± 1,2 2,9 ± 1,2 3,1 ± 1,2 n.s. Deskriptiver Vergleich mittels zweiseitigen t-Tests. * Anti-CCP-Antikörper wurden im Labor bis 200 RU/ml bestimmt, darüberliegende Werte wurden mit >200 RU/ml angegeben. Die Konzentrationen der Immunglobulin-Isotypen G, A und M in absteigender Reihenfolge werden in Abb. 7 dargestellt. Abbildung 7. Konzentrationen von IgG, IgA und IgM im Vergleich. Mittelwerte ± SD der Ig-Konzentrationen der RA-Patientengruppe (n = 539). In Tab. 13 sind die auftretenden Kombinationen an Defekten der 3 ImmunglobulinIsotypen dargestellt. Die Hälfte der 28 Patienten mit IgA-Defekt wies gleichzeitig einen IgG-Defekt auf. Wir ermittelten 7 Patienten, die bei vorhandenem IgA-Defekt auch einen IgG2-Defekt hatten. 42 5 Ergebnisse Tabelle 13: Häufigkeiten von gleichzeitigem Auftreten von Immunglobulindefekten Kombination Patienten mit: Kombination aus IgG- und IgA-Defekt Kombination aus IgG- und IgM-Defekt Kombination aus IgA- und IgM-Defekt Defekt aller 3 Ig-Isotypen Anzahl der Patienten 16 (3,0 %) 9 (1,7 %) 14 (2,6 %) 9 (1,7 %) 16 Patienten fanden sich mit defektem IgG sowie IgA. Diese Befunde stellen Teilkriterien für ein CVID dar. 13 dieser Patienten erhielten eine Kortikosteroidtherapie, 14 eine Therapie mit konventionellen DMARDs, darunter MTX, Leflunomid, Sulfasalazin und Hydroxychloroquin, und jeweils ein Patient erhielt eine Biologika-Therapie mit Adalimumab bzw. Etanercept. Bei immerhin 9 RA-Patienten wurde eine Erniedrigung aller 3 Immunglobuline festgestellt. Die betroffenen 9 Patienten waren alle weiblich, der Mittelwert des Alters betrug 70,1 ± 9,2 Jahre. Die Mittelwerte der IgG-Subklassen dieser Patienten mit globalem Immunglobulinmangel lagen alle deutlich unter den Werten der anderen untersuchten RA-Patienten (Tab. 14). Tabelle 14: Vergleich der Mittelwerte der IgG-Subklassenkonzentrationen zwischen Patienten mit oder ohne globalem Immunglobulindefekt IgG, IgA, IgM RA-Patienten mit globalem Immunglobulindefekt 3,91 1,94 0,57 0,16 RA-Patienten ohne IgG1 (g/l) IgG2 (g/l) IgG3 (g/l) IgG4 (g/l) 5,61 3,25 0,69 0,44 Unter diesen 9 Patienten mit globaler Immunglobulinverminderung fanden sich 7-mal IgG1-Defekte, 3-mal IgG2-Defekte und 5-mal IgG4-Defekte. Jedoch traten bei den Patienten mit Globalinsuffizienz der Ig nicht mehr als 2 verminderte Subklassen auf. 7 dieser 9 Patienten standen unter Steroid-Medikation, 4-mal wurde MTX, 2-mal Sulfasalazin, einmal Leflunomid verschrieben und 2 Patienten waren ohne Basistherapie. Keiner der 9 Patienten stand unter Biologika-Therapie. 4 der Patienten klagten über Atemwegsinfekte, 3 davon über seltenes und einer über häufiges Infektgeschehen. Die zu den Diagnosekriterien der RA zählenden Rheumafaktoren wurden mit drei Untergruppen bestimmt. Zunächst wurde der am häufigsten in der täglichen Routine bestimmte IgM-RF betrachtet, bei welchem 330 der 539 Proben (61,2%) positiv 43 5 Ergebnisse ausfielen, also einen Wert über 20 IU/ml aufwiesen. Im Gegensatz zu der großen Anzahl von ca. 2/3 an Positivmessungen des IgM-RF zeigte der IgG-RF lediglich bei 27 Proben (5,0%) positive Werte. Die negativen Proben beliefen sich auf 511 (95,0%) von 538 gemessenen Blutproben. Der IgA-RF wurde ebenfalls in 538 Proben bestimmt, wovon 338 (62,8%) positiv ausfielen. Anti-CCP-AK wurden in 318 von 536 Patientenseren (59,3%) positiv gemessen, die restlichen 218 Proben (40,7%) waren negativ. Eine genaue Aussage über den Mittelwert war nicht möglich, da die Anti-CCP-Konzentration im Labor nur bis zu einem Wert von 200 RU/ml exakt bestimmt wurde. Höhere Werte sind mit >200 RU/ml angegeben. Daher wurden die Anti-CCP-positiven Seren zusätzlich unterteilt in niedrigtitrige Konzentrationen bis zum dreifachen des obersten Normwertes von 5 RU/ml, also 5-15 RU/ml und hochtitrige >15 RU/ml. Die Gruppe der niedrigtitrigen Anti-CCP-Werte beinhaltete 36 Proben (6,7%), jene der hochtitrigen Proben 282 (52,6%). 5.3 Konzentrationen von IgG und IgG-Subklassen bei RA-Patienten Tabelle 15: Serumkonzentrationen von IgG und IgG-Subklassen bei RA-Patienten (Mittelwert ± SD). Normwerte Gesamtgruppe Frauen (♀) Männer (♂) Vergleich ♀ ♂ IgG (g/l) 8,0-18,0 10,4 ± 2,8 10,3 ± 2,8 10,4 ± 2,8 n.s. IgG1 (g/l) 4,9-11,4 5,6 ± 2,3 5,6 ± 2,3 5,4 ± 2,3 n.s. IgG2 (g/l) 1,5-6,4 3,2 ± 1,5 3,2 ± 1,4 3,3 ± 1,6 n.s. IgG3 (g/l) 0,2-1,1 0,7 ± 0,4 0,7 ± 0,4 0,7 ± 0,4 n.s. IgG4 (g/l) 0,08-1,4 0,4 ± 0,5 0,4 ± 0,5 0,5 ± 0,4 p < 0,05 Die Werte für IgG sowie die IgG-Subklassen 1-4 sind bei 539 RA-Patienten bestimmt worden. Der Mittelwert aller Patienten des IgG lag bei 10,4 g/l (Tab. 15), die Standardabweichung betrug 2,8. Bei 78,5% der Patienten befand sich die IgGKonzentration im Normbereich. Bei 19,9% der Werte fiel ein Defekt auf. Bei den restlichen 1,7% ließ sich eine IgG-Erhöhung feststellen (Abb. 8 und Tab. 16). Betrachtet man die Subklassen des IgG, so fällt in der Gruppe des IgG1 mit 42,3%, bei 228 Patienten eine hohe Anzahl an verminderten Konzentrationen auf gegenüber 54,5% der Werte im Normbereich. Erhöhte Werte wiesen 3,2% der Patienten auf. Der Mittelwert des IgG1 betrug 5,6 ± 2,3 g/l. 44 5 Ergebnisse IgG2 wies von den Subklassen den höchsten Anteil an Werten im Normbereich mit 89,4% auf. Hier fanden sich bei 7,2% der Patienten ein Mangel und bei 3,3% ein Überschuss der Konzentrationen. Durchschnittlich errechnete sich für IgG2 ein Wert von 3,2 g/l mit einer SD von 1,5. Der Mittelwert des IgG3 betrug 0,7 ± 0,4 g/l. 83,5% der gemessenen IgG3Konzentrationen befanden sich in der Norm, 5,4% wichen nach unten ab und 11,1% nach oben. Beim IgG4 fanden sich bei einem Mittelwert von 0,4 ± 0,5 g/l 84,8% der Werte im Normbereich, 10,4% wiesen einen Defekt auf und 4,8% eine Erhöhung (zu den IgGSubklassen siehe Tab. 15, 16 und Abb. 8). Abbildung 8. IgG- und IgG-Subklassenverteilung bei RA-Patienten (n=539). Tabelle 16: Häufigkeitsverteilung von Normalbefund, Defekten und Erhöhungen des IgG und der IgG-Subklassen bei RA-Patienten IgG IgG1 Normalbefund 423 (78,5%) 294 (54,5%) Defekt 107 (19,9%) 9 (1,7%) Überschuss IgG2 n=539 (100%) IgG3 IgG4 482 (89,4%) 450 (83,5%) 457 (84,8%) 228 (42,3%) 39 (7,2%) 29 (5,4%) 56 (10,4%) 17 (3,2%) 18 (3,3%) 60 (11,1) 26 (4,8%) 45 5 Ergebnisse Unterschiede zwischen den Geschlechtern traten nur bei der Subklasse 4 des IgG auf, hierbei war die Konzentration bei den Frauen durchschnittlich niedriger mit einer Signifikanz von p < 0,05 (Abb. 9). Die anderen IgG-Subklassen unterschieden sich nicht signifikant zwischen den Geschlechtern. Die gemessenen Konzentrationen der IgG-Subklassen entsprachen der Reihenfolge der Subklassenbezeichnung. IgG1 war am stärksten vertreten, die nächsthöchste Konzentration lag bei IgG2 vor, gefolgt von IgG3 und IgG4, welche die niedrigsten Konzentrationen aufwiesen. Abbildung 9. Vergleich der IgG-Subklassen zwischen Frauen und Männern (* = p < 0,05; n = 539). Die Patienten wiesen teilweise einzeln auftretende, teilweise kombinierte Mängel an IgG und dessen Subklassen auf. In Abb. 10 wird die Häufigkeitsverteilung der Kombinationen sichtbar. Dabei traten Minderungen von IgG1 und IgG4 am häufigsten miteinander auf, gefolgt von der Kombination aus Mangel von IgG1 und IgG2. Bei 33 Patienten zeigte sich ein kombinierter IgG1- und IgG4-Defekt, wobei hiervon 24 Patienten unter Steroid-Therapie standen. Bei 28 dieser Patienten wurden DMARDs verschrieben, darunter 18-mal MTX, außerdem Leflunomid, Sulfasalazin, Chloroquin und Hydroxychloroquin. 2 der 33 Patienten bekamen ein Biologikum, 46 5 Ergebnisse Etanercept bzw. Adalimumab. Über Infekte berichteten 18 Patienten, davon 11 über seltene und 7 über gehäufte Infekte. 21 der 30 Patienten mit kombiniertem IgG1- und IgG2-Defekt nahmen Steroide ein, 26 Patienten standen unter Basistherapie mit MTX, Sulfasalazin oder Leflunomid. 5 der 30 Patienten erhielten Biologika, darunter waren 3 Patienten mit Tozilizumab, 1 Patient mit Adalimumab und 1 Patient mit Etanercept. 15 Patienten klagten über eine Infektneigung, 10 davon über seltene und 5 Patienten über häufige Infekte. Abbildung 10. Häufigkeiten von kombinierten IgG-Subklassendefekten. Parallel zu einem IgG-Mangel wiesen 98 Patienten (18,2 %) auch einen IgG1-Mangel auf, 30 Patienten (5,6 %) hatten zusätzlich zu einem IgG-Mangel einen IgG2-Mangel, lediglich 6 Patienten (1,1 %) hatten einen zusätzlichen IgG3-Mangel und 23 (4,3 %) einen IgG4-Mangel. 14 Patienten wiesen drei Mängel unter den Subklassen auf, darunter hatten die meisten eine Kombination aus erniedrigtem IgG1, 2 und 4 (Tab. 17). Bei einem Patienten waren alle Subklassen des IgG, IgG selbst und IgA erniedrigt, lediglich IgM lag im Normbereich. Da während der Laborbestimmungen auffiel, dass Patienten mit IgG im Normbereich nicht immer normwertige IgG-Subklassen aufwiesen, haben wir eine Übersicht zu 47 5 Ergebnisse allen Patienten mit IgG in normaler Konzentration und der entsprechenden Subklassenverteilung erstellt (Tab. 18). Tabelle 17: Häufigkeiten der Kombinationen von IgG-SubklassenMinderungen bei RA-Patienten kein kombinierter Mangel kombinierter Mangel IgG1 und 2 509 (94,4 %) 30 (5,6 %) IgG1 und 3 528 (98,0 %) 11 (2 %) IgG1 und 4 506 (93,9 %) 33 (6,1 %) IgG1 und IgG 441 (81,8 %) 98 (18,2 %) IgG2 und 3 532 (98,7 %) 7 (1,3 %) IgG2 und 4 529 (98,1 %) 10 (1,9 %) IgG2 und IgG 509 (94,4 %) 30 (5,6 %) IgG3 und 4 536 (99,4 %) 3 (0,6 %) IgG3 und IgG 533 (98,9 %) 6 (1,1 %) IgG4 und IgG 516 (95,7 %) 23 (4,3 %) IgG1,2,3 535 (99,3 %) 4 (0,4 %) IgG1,2,4 531 (98,2 %) 8 (1,5 %) IgG1,3,4 538 (99,4 %) 1 (0,2 %) IgG2,3,4 538 (99,4 %) 1 (0,2 %) IgG1,2,3,4 538 (99,4 %) 1 (0,2%) Tabelle 18: Patienten mit IgG im Normbereich und entsprechende IgG-Subklassenverteilung n = 432 (100%) IgG1 IgG2 Häufigkeit Prozent kein Defekt n = 302 69,9 Defekt n = 130 30,1 kein Defekt n = 423 97,9 n=9 2,1 n = 409 94,7 Defekt IgG3 IgG4 kein Defekt Defekt n = 23 5,3 kein Defekt n = 399 92,4 Defekt n = 33 7,6 48 5 Ergebnisse Diese Untersuchung zeigt, dass 30,1% der Patienten mit normaltitrigem IgG einen IgG1-Defekt aufwiesen, obwohl es sich beim IgG1 um die Subklasse mit der höchsten Konzentration handelt. Es darf folglich nicht bei normwertiger IgGKonzentration auf ebenfalls normale IgG-Subklassen geschlossen werden. Eine Bestimmung der Subklassen ist notwendig, um einzelne Defekte ausschließen zu können. 5.4 Regressionsanalysen aus den Patientendaten Weiterhin sollte die Abhängigkeit von IgM, IgA, IgG und IgG-Subklassen mit Einflussparametern wie Geschlecht, Alter, Entzündungs- und Laborwerten für die RA, Medikamenten sowie Infektionen verglichen werden. Dafür verwendeten wir die statistische Analysemethode der binär logistischen Regression. Mit dieser ist es möglich, Gruppenunterschiede zu erklären und gleichzeitig mehrere Variablen in ein Modell einfließen zu lassen. In unserer Arbeit nehmen diese Gruppen jeweils nur zwei Ausprägungen an, die Konzentration des jeweiligen Immunglobulins ist entweder oberhalb der unteren Normwertgrenze oder darunter, deshalb nennt man die Analyse binär (siehe Tab. 4). Diese Werte stellen die abhängige Variable dar, den so genannten Regressand. Dabei werden die Werte unter dem Normbereich mit „Defekt“ bezeichnet und die Werte darüber mit „kein Defekt“. Das Skalenniveau der unabhängigen Variablen, auch Regressoren genannt, hingegen ist beliebig. Das Konfidenzintervall (KI), auch als Vertrauensbereich bezeichnet, beschreibt die Präzision der Lageschätzung eines Parameters. Es beträgt in unserer Arbeit 95%, d.h. 95% der berechneten Konfidenzintervalle beinhalten den wahren Wert des zu untersuchenden Hintergrundes. Die Odds Ratio ist das Quotenverhältnis, das die Stärke eines Zusammenhangs von zwei Merkmalen beschreibt. Ist der Wert genau 1, liegt kein Unterschied zwischen den einzelnen Odds, den Wahrscheinlichkeiten, vor. Das bedeutet, es gibt keine Wahrscheinlichkeit für einen Zusammenhang. Je weiter der Wert von 1 abweicht, umso stärker stellt sich ein Zusammenhang dar. Dabei gilt für eine Odds Ratio > 1 ein direkter Bezug, in der Berechnung sind die Wahrscheinlichkeiten der ersten Gruppe größer, und für eine Odds Ratio < 1 ein entgegengesetzter Bezug, wobei die Wahrscheinlichkeiten der zweiten Gruppe größer sind. 49 5 Ergebnisse Zuerst folgen die Regressionen mit dem Einsatz ausgewählter unabhängiger Variablen in einzelne Modelle. Danach werden Gesamtregressionen beschrieben, bei denen alle erhobenen Variablen in die Modelle einfließen. 5.4.1 Regression mit den abhängigen Variablen IgM und IgA Die beiden Ausprägungen der abhängigen Variablen lauten jeweils „kein Defekt“ und „Defekt“. Zunächst wurden einzelne Regressionen berechnet (siehe Anhang Tab. 34 und 35). Hierbei zeigte IgM mit p < 0,001 in der statistischen Regression ein signifikant erhöhtes Risiko für Defekte bei älteren Patienten. Bei IgA hingegen bestanden gleiche Defekthäufigkeiten zwischen jüngeren und älteren Patienten. Männer wiesen bei der Berechnung der Regression zu IgA signifikant mehr Defekte im Vergleich zu Frauen auf. Beim IgM fällt dieser Befund mit p = 0,053 knapp nicht signifikant aus. Beide Variablen zeigen keine Abhängigkeiten zu Röntgenbefunden und Raucherstatus (p > 0,05). Mit längerer Krankheitsdauer hatten unsere untersuchten Patienten weniger IgM-Defekte. Dieser Befund ließ sich beim IgA nicht nachweisen. Zu den Parametern BSG, CRP, VAS, DAS 28 und Anti-CCP-AK wiesen beide Immunglobuline keine Signifikanzen auf (p > 0,05). Bei beiden ImmunglobulinIsotypen M und A zeigten sich signifikant abhängige Beziehungen zu den äquivalenten Rheumafaktor-Subtypen M und A, nicht aber zu den anderen RF. Die RF-negativen Patienten hatten jeweils ein höheres Risiko für Ig-Defekte. Die Immunglobuline wiesen auch untereinander Abhängigkeiten auf. Je höher die IgA- und IgG-Konzentrationen, desto weniger IgM-Defekte fanden sich bei den Messungen und je höher die IgG-Konzentration ist, desto weniger IgA-Defekte zeigten sich. Nur die IgG-Subklassen 1 und 2 zeigten Auffälligkeiten in Bezug auf die anderen Immunglobuline, nicht aber die IgG-Subklassen 3 und 4. Mit höheren IgG1Konzentrationen fanden sich weniger IgM-Defekte. So war es auch zwischen IgG2 und IgA, je höher die IgG2-Konzentrationen lagen, desto weniger IgA-Defekte zeigten sich. Zur Regressionsanalyse mit Medikamenten, Anzahl an Medikamenten und Infekten zeigten sich keine signifikanten Werte. Zu den Regressionsberechnungen von IgM und IgA siehe Tab. 34 und 35 im Anhang. 50 5 Ergebnisse 5.4.2 Regression mit IgG als abhängige Variable Zunächst soll als abhängige Variable das IgG mit den Ausprägungen „kein Defekt“ und „Defekt“ betrachtet werden. Generell gilt, dass die jeweils in einer Tabelle befindlichen unabhängigen Variablen in das jeweilige Regressionsmodell eingeflossen sind. Hierbei traten signifikante Abhängigkeiten zum Alter, Röntgenstadium, zu BSG, IgA, IgM, IgG1 und IgG2, zur Einnahme von Biologika und zu Infekten auf. Bei der Betrachtung des Alters, Geschlechts, Raucherstatus, Krankheitsdauer und des Röntgenstatus als Einflussfaktoren auf die IgG-Konzentration wiesen zwei Größen eine Abhängigkeit auf (Tab. 19). Das Alter zeigte bei der logistischen Regression einen p-Wert < 0,001 bei einer Odds Ratio von 1,041. Das bedeutet, pro Lebensjahr vergrößert sich das Risiko, einen IgG-Mangel zu bekommen, um 4,1%. Die weiteren Werte blieben ohne Auffälligkeiten, außer dem Röntgenstadium „beginnend erosiv“. Bei einer Signifikanz mit einem p-Wert von <0,05 zeigten Patienten in diesem Stadium eine um 77,7% niedrigere Ausprägung eines IgG-Defektes im Vergleich zum „nicht erosiven“ Stadium. Tabelle 19: Logistische Regression des IgG mit den Patientendaten Unabhängige Variable 95% Konfidenzintervall (KI) der Odds Ratio Unterer Wert Oberer Wert p-Wert Odds Ratio Alter (in Jahren) 0,000 1,041 1,019 1,063 Geschlecht männlich 0,781 1,073 0,651 1,768 Raucher 0,275 1,379 0,774 2,457 Ex-Raucher Krankheitsdauer (in Jahren) Röntgenstadium 0,635 0,732 0,201 2,660 0,525 0,992 0,967 1,017 erosiv 0,762 0,928 0,574 1,502 beginnend erosiv 0,046 0,223 0,051 0,974 Raucher-Status Bei der Betrachtung der Laborwerte (siehe Tab. 20) fiel eine Abhängigkeit zwischen IgG und BSG auf (p < 0,05). Bei höherer BSG fanden sich weniger IgG-Defekte. Zwischen IgG und RF sowie Anti-CCP-AK fanden sich keine Abhängigkeiten. 51 5 Ergebnisse Tabelle 20: Logistische Regression des IgG mit den Laborwerten p-Wert Odds Ratio BSG (in mm/h) 0,023 CRP (in mg/l) unabhängige Variable 95% KI der Odds Ratio Unterer Wert Oberer Wert 0,981 0,965 0,997 0,903 0,999 0,976 1,022 VAS 0,756 0,997 0,980 1,015 DAS 28 mit BSG 0,258 1,158 0,898 1,493 DAS 28 mit CRP 0,177 1,133 0,945 1,358 RF IgM (in IU/ml) 0,144 1,451 0,881 2,391 RF IgG (in RU/ml) 0,242 2,425 0,550 10,706 RF IgA (in RU/ml) 0,861 1,052 0,593 1,867 Anti-CCP-AK (in RU/ml) 0,208 0,998 0,994 1,001 Bei der Betrachtung der Immunglobuline A und M fielen deutliche Abhängigkeiten auf. Je höher die Werte für IgA und auch IgM sind, desto weniger IgG-Defekte waren nachweisbar, dieses Ergebnis änderte sich auch dann nicht, wenn andere Variablen in das Modell einbezogen wurden. Die beiden IgG-Subklassen mit den größten Konzentrationen IgG1 und IgG2 standen ebenfalls mit der Konzentration des IgG in Zusammenhang, je höher das IgG ausfiel, desto höher waren auch diese beiden Subklassen. Dieser Zusammenhang ließ sich bei den Subklassen 3 und 4 mit niedrigerer Konzentration nicht feststellen (Tab. 21). Tabelle 21: Logistische Regression des IgG mit den anderen Immunglobulinen p-Wert Odds Ratio IgA (in g/l) 0,000 IgM (in g/l) 95% KI der Odds Ratio Unterer Wert Oberer Wert 0,561 0,446 0,705 0,007 0,581 0,391 0,865 IgG1 (in g/l) 0,000 0,348 0,262 0,464 IgG2 (in g/l) 0,000 0,243 0,163 0,362 IgG3 (in g/l) 0,706 0,811 0,273 2,411 IgG4 (in g/l) 0,135 0,384 0,109 1,347 Betrachtet man die Abhängigkeit zwischen IgG und der Kortison- sowie BiologikaEinnahme, so wird deutlich, dass Patienten unter Kortison sowie auch unter Biologika weniger IgG-Defekte gegenüber Patienten ohne diese Behandlungen aufwiesen. Allerdings wurde dieses Ergebnis bei Einbeziehung von mehr unabhängigen Variablen in das Modell nicht signifikant. Das deutet darauf hin, dass dieser Effekt nur durch andere Einflüsse zustande kommt. Die Anzahl der Medikamente, die für die RA verschrieben wurden, hatte keinen signifikanten Einfluss auf die IgG52 5 Ergebnisse Konzentration. Dagegen wiesen Patienten mit Infekten eine höhere Rate an IgGDefekten im Gegensatz zu infektfreien Patienten auf (Tab. 22). Tabelle 22: Logistische Regression des IgG mit den Therapeutika sowie Infektionen Unabhängige p-Wert Odds Ratio NSAR 0,984 COX-Hemmer Variable 95%KI der Odds Ratio Unterer Wert Oberer Wert 1,005 0,611 1,652 0,484 0,810 0,448 1,463 Kortison 0,036 1,690 1,034 2,762 DMARD 0,354 1,287 0,754 2,197 Biologikum 0,031 0,470 0,237 0,932 Anzahl an Medikamenten 0,130 1,229 0,941 1,605 Infektionen 0,006 1,845 1,196 2,848 Um die Auswirkung der einzelnen Biologika zu beurteilen, haben wir diese Regressionsanalyse erneut für die einzelnen Medikamente durchgeführt. Als abhängige Variablen der einzelnen Berechnungen wurden jeweils IgG und dessen Subklassen verwendet, die 8 unterschiedlichen Biologika stellten die unabhängigen Variablen dar. Bei der Betrachtung des Einflusses der Biologika auf das IgG fiel ein signifikanter Zusammenhang zu Adalimumab, aber zu keinem anderen Biologikum auf (Anhang Tab. 38). Die 32 Patienten unter Adalimumab wiesen mit p < 0,05 eine geringere Rate von IgG-Defekten gegenüber Patienten ohne Biologikum auf. 5.4.3 Regression mit IgG1 als abhängige Variable Die Ergebnisse der logistischen Regression unter Betrachtung des IgG1 als abhängige Variable ähneln den Ergebnissen des IgG. Auch hier fand sich eine Abhängigkeit zum Alter, wobei die Rate für einen IgG1-Defekt ebenfalls mit höherem Alter stieg. Für Geschlecht, Raucherstatus sowie Krankheitsdauer ergaben sich keine signifikanten Zusammenhänge (jeweils p > 0,05). Zum Röntgenstatus lässt sich sagen, dass beim IgG1 nicht nur die Patienten mit beginnend erosiven Stadien, sondern auch jene mit erosiven Veränderungen weniger IgG1-Defekte aufwiesen als nicht erosive (Tab. 23). 53 5 Ergebnisse Tabelle 23: Regression des IgG1 mit Patientendaten p-Wert Odds Ratio Alter (in Jahren) 0,004 Geschlecht männlich 95% KI der Odds Ratio Unterer Wert Oberer Wert 1,024 1,007 1,040 0,398 1,193 0,792 1,796 Raucher 0,908 0,973 0,610 1,552 ehemalige Raucher 0,953 1,029 0,396 2,671 Krankheitsdauer (in Jahren) 0,420 0,992 0,971 1,012 erosiv 0,017 0,616 0,413 0,917 beginnend erosiv 0,022 0,386 0,171 0,873 Raucherstatus Röntgenstadium Bei der Betrachtung der Werte aus folgender Tabelle 24 fand sich wie schon zuvor eine Abhängigkeit von BSG und der abhängigen Variablen IgG1. Je höher die BSG ausfiel, desto geringer war die Anzahl an IgG1-Defekten. Die anderen Werte ergaben in der Regressionsanalyse keine signifikanten Abhängigkeiten. Die Ergebnisse der Regression des IgG1 zu den Immunglobulin-Isotypen sowie den IgG-Subklassen sind in Tab. 25 dargestellt. Es zeigte sich ein starker Zusammenhang zwischen IgG1 und IgG mit einem p-Wert <0,001, nicht aber zu den Immunglobulinen A und M. Tabelle 24: Regression des IgG1 mit Laborwerten 95% KI der Odds Ratio p-Wert Odds Ratio Unterer Wert Oberer Wert BSG (in mm/h) 0,000 0,977 0,965 0,990 CRP (in g/l) 0,591 1,005 0,988 1,022 VAS 0,287 1,008 0,993 1,023 DAS 28 mit BSG 0,772 ,970 0,790 1,191 DAS 28 mit CRP 0,742 1,026 0,883 1,192 RF IgM (in IU/ml) 0,313 1,239 0,817 1,878 RF IgG (in RU/ml) 0,199 1,822 0,730 4,550 RF IgA (in RU/ml) 0,368 1,248 0,771 2,020 Anti-CCP-AK (in RU/ml) 0,353 1,001 0,999 1,004 Starke Signifikanzen traten auch beim Einsetzen der weiteren IgG-Subklassen in das Modell in Bezug auf IgG1 auf. Die Wahrscheinlichkeit für einen IgG1-Mangel sank, je höher die anderen Subklassen ausfielen (Tab. 25). 54 5 Ergebnisse Tabelle 25: Regression des IgG1 mit den anderen Immunglobulinen 95% KI der Odds Ratio p-Wert Odds Ratio Unterer Wert Oberer Wert IgA (in g/l) 0,387 0,909 0,732 1,128 IgG (in g/l) 0,000 0,401 0,341 0,471 IgM (in g/l) 0,542 0,923 0,713 1,194 IgG2 (in g/l) 0,007 0,798 0,677 0,941 IgG3 (in g/l) 0,000 0,231 0,124 0,431 IgG4 (in g/l) 0,000 0,334 0,185 0,603 Unter Verwendung der Medikamente NSAR, COX-Hemmer, Kortison und DMARDs als unabhängige Variablen ergaben sich zwei signifikante Beobachtungen. Patienten, die NSAR, sowie auch Patienten, die COX-Hemmer einnahmen, hatten ein niedrigeres Risiko für IgG1-Defekte. Setzte man aber in das Regressionsmodell alle erhobenen Daten ein, so ergab sich kein signifikanter Unterschied zwischen den Gruppen „Patient mit NSAR“ bzw. „mit COX-Hemmer“ und „Patient ohne NSAR“/ „ohne COX-Hemmer“. Dies lässt einen Einfluss durch andere Variablen vermuten, der in der Gesamtregression berücksichtigt wurde und dadurch die Signifikanzen verschwanden. Patienten, die ein Biologikum einnahmen, hatten ein signifikant niedrigeres Risiko für IgG1-Subklassendefekte als Patienten ohne Biologikum. Diese Signifikanz errechnete sich unter Verwendung der unabhängigen Variablen Biologikum, Anzahl an Medikamenten und Infektionen, aber auch unter Verwendung aller erhobenen Daten im Regressionsmodell. Die Anzahl an unterschiedlichen Antirheumatika, auch das Auftreten von Infektionen, hatte keinen Einfluss auf das Vorkommen von IgGSubklassendefekten (Tab. 26). Tabelle 26: Regression des IgG1 mit den Therapeutika sowie Infekten p-Wert Odds Ratio NSAR 0,011 COX-Hemmer Kortison 95% KI der Odds Ratio Unterer Wert Oberer Wert 0,590 0,393 0,886 0,025 0,585 0,366 0,936 0,084 1,392 0,956 2,025 DMARD 0,856 1,039 0,685 1,576 Biologikum 0,005 0,475 0,283 0,798 Anzahl an Medikamenten 0,444 0,921 0,745 1,137 Infektionen 0,126 1,328 0,924 1,911 55 5 Ergebnisse Unter der Gabe von Adalimumab und Certolizumab zeigten sich in der Regressionsanalyse weniger IgG1-Subklassendefekte als unter anderen Medikamenten und im Vergleich zu Patienten ohne Biologikaeinnahme. Unter der Einnahme der Medikamente fanden sich mit p < 0,001 für Adalimumab und p < 0,05 für Certolizumab weniger IgG1-Subklassendefekte (Anhang Tab. 39). 5.4.4 Regression mit IgG2 als abhängige Variable Die einfließenden unabhängigen Variablen wurden wie auch schon in den vorherigen Berechnungen eingeteilt. Auch für das IgG2 bestand eine Abhängigkeit zum Alter und zur BSG (Tab. 27) im gleichen Sinne wie im Falle der schon zuvor beschriebenen Immunglobuline. Die IgG2-Defekte häuften sich im Alter und bei niedriger BSG. Zudem haben Raucher ein erhöhtes Risiko für einen IgG2-Defekt gegenüber Nichtrauchern. Der DAS 28 mit der BSG in der Berechnung zeigte ebenfalls eine Abhängigkeit. Je höher der Aktivitätsstatus in Form des DAS 28, desto höher war die Wahrscheinlichkeit für einen Defekt an IgG2. Hier schien die Abhängigkeit der BSG selbst eine große Rolle zu spielen, da in Bezug auf den DAS 28 mit CRP keine Abhängigkeit bestand. Tabelle 27: Regression des IgG2 mit den Patientendaten sowie Laborwerten p-Wert Odds Ratio 95% Konfidenzintervall der Odds Ratio Unterer Wert Oberer Wert Alter (in Jahren) 0,012 1,044 1,009 1,08 Geschlecht männlich 0,199 1,598 0,781 3,268 Raucher 0,009 2,935 1,308 6,589 ehemalige Raucher 0,814 0,777 0,095 6,364 Krankheitsdauer (in Jahren) 0,546 0,988 0,95 1,027 erosiv 0,308 1,462 0,704 3,038 beginnend erosiv 0,425 0,43 0,054 3,413 BSG (in mm/h) 0,005 0,957 0,928 0,987 CRP (in g/l) 0,736 0,992 0,946 1,04 VAS 0,117 0,976 0,947 1,006 DAS 28 mit BSG 0,004 1,79 1,2 2,672 DAS 28 mit CRP 0,194 1,199 0,912 1,578 RF IgM (in IU/ml) 0,439 1,364 0,622 2,993 RF IgG (in RU/ml) 0,688 1,531 0,192 12,229 RF IgA (in RU/ml) 0,143 0,509 0,206 1,257 Anti-CCP-AK (in RU/ml) 0,263 0,997 0,992 1,002 Raucherstatus Röntgenstadium 56 5 Ergebnisse Die Immunglobuline IgA und IgM wiesen keine signifikanten Abhängigkeiten zum IgG2 auf, dagegen aber das IgG selbst und auch die restlichen IgG-Subklassen. Das Risiko für IgG2-Defekte sank jeweils mit höheren Konzentrationen an IgG, IgG1, IgG3 und IgG4 (Tab. 28). Tabelle 28: Regression des IgG2 mit den anderen Immunglobulinen 95% KI der Odds Ratio p-Wert Odds Ratio IgA 0,737 IgG Unterer Wert Oberer Wert 0,947 0,69 1,300 0,000 0,513 0,413 0,637 IgM 0,409 0,779 0,430 1,409 IgG1 0,028 0,777 0,620 0,974 IgG3 0,001 0,074 0,017 0,323 IgG4 0,011 0,089 0,014 0,569 Eine Abhängigkeit zwischen Biologika und IgG2-Spiegeln ließ sich nicht nachweisen, im Gegensatz zum IgG1, bei dessen Berechnung ein Zusammenhang zur BiologikaEinnahme gefunden wurde. Es bestand in der Regressionsanalyse auch kein Zusammenhang zu Infektionen. Der Einfluss der Anzahl an Medikamenten, die von den Patienten eingenommen wurden, war mit einem p-Wert von 0,03 signifikant. Die Analyse ergab, dass mit steigender Medikamentenzahl auch mehr IgG2-Defekte auftraten. Patienten mit der Einnahme von COX-Hemmern zeigten eine Tendenz zu weniger IgG2-Subklassendefekten als Patienten, die keine COX-Hemmer einnahmen. Andersherum verhielt es sich bei der Betrachtung der DMARD-Therapie. Hierbei war das Risiko für einen IgG2-Defekt mit Therapie höher als für jene Patienten, die keine DMARDs erhielten (Tab. 29). Tabelle 29: Regression des IgG2 mit den Therapeutika sowie Infektionen p-Wert Odds Ratio NSAR 0,394 COX-Hemmer 95% KI der Odds Ratio Unterer Wert Oberer Wert 1,368 0,666 2,808 0,053 0,283 0,079 1,017 Kortison 0,880 1,057 0,517 2,158 DMARD 0,037 3,103 1,069 9,014 Biologikum 0,857 1,084 0,448 2,626 Anzahl an Medikamenten 0,030 2,073 1,074 4,0 Infektionen 0,401 1,193 0,79 1,8 57 5 Ergebnisse Ein signifikanter Einfluss der einzelnen Medikamente auf die IgG-Subklassen 2, 3 und 4 im Vergleich zu Patienten ohne Biologika konnte nicht nachgewiesen werden (Anhang Tab. 40-42). 5.4.5 Regression mit IgG3 als abhängige Variable Bei der Betrachtung des IgG3 traten zwei signifikante Abhängigkeiten auf. Einerseits in Bezug auf die Krankheitsdauer, mit steigender Krankheitsdauer stieg das Risiko auf einen IgG3-Defekt an. Andererseits bestand eine Abhängigkeit zur IgG2Konzentration. Je höher die IgG2-Konzentration war, desto weniger IgG3-Defekte konnte man erwarten (Tab. 30). Die restlichen Vergleiche blieben ohne signifikante Ergebnisse (siehe Anhang Tab. 36). Tabelle 30: Regression IgG3: signifikante Ergebnisse p-Wert Odds Ratio 95% Konfidenzintervall der Odds Ratio Unterer Wert Oberer Wert Krankheitsdauer (in Jahren) 0,047 1,035 1,0 1,072 IgG2 0,001 0,494 0,328 0,742 5.4.6 Regression mit IgG4 als abhängige Variable IgG4-Subklassendefekte zeigten eine Abhängigkeit zum IgA- und IgG-Serumspiegel, ebenso zu IgG2-Subklassendefekten. Dabei war die Richtung der Abhängigkeit, zu sehen am Vorzeichen der Odds Ratio, immer gleich. So sank das Risiko für einen IgG4-Defekt mit steigenden Konzentrationen der jeweiligen Immunglobuline. Patienten mit einer Basistherapie hatten ein höheres Risiko für IgG4-Defekte im Vergleich zu Patienten ohne Basistherapie (Tab. 31). Führte man aber die Regressionsberechnung der einzelnen DMARDs durch, fanden sich keine signifikanten Ergebnisse in Bezug auf die IgG4-Defekte. Dieser Effekt kam also nur bei gleichzeitigem Einfluss aller DMARDs in das Modell zustande, nicht jedoch bei der Einzelbetrachtung. Die weiter berechneten Regressionen blieben ohne signifikante Ergebnisse und finden sich im Anhang (Anhang Tab. 37). 58 5 Ergebnisse Tabelle 31: Regression IgG4: signifikante Ergebnisse und Immunglobuline p-Wert Odds Ratio 95% Konfidenzintervall der Odds Ratio Unterer Wert Oberer Wert IgA 0,014 0,688 0,512 0,926 IgG 0,001 0,796 0,698 0,908 IgM 0,403 0,836 0,549 1,272 IgG1 0,140 0,883 0,748 1,042 IgG2 0,000 0,493 0,363 0,671 IgG3 0,094 2,101 0,881 5,011 DMARD 0,034 2,451 1,070 5,616 5.4.7 Zusammenfassung der wichtigsten Ergebnisse aus den Regressionsanalysen Die oben aufgeführten signifikanten Ergebnisse aus der Berechnung mithilfe der binär logistischen Regression wurden in Tab. 32 zusammengestellt. Tabelle 32: Übersicht zu den Ergebnissen aus den Regressionsanalysen IgG-Defekte ↑ bei höherem Alter, Kortison-Einnahme, vermehrten Infektionen ↓ bei Biologikum-Einnahme ↓ bei beginnend erosivem Röntgenstadium ↓ bei hoher BSG, hohen Konzentrationen an IgA, IgM, IgG1, IgG2 IgG1-Defekte ↑ bei höherem Alter ↓ bei beginnend erosivem und erosivem Röntgenstadium ↓ bei hohen Konzentrationen an IgG, IgG2, IgG3, IgG4 ↓ bei Biologikum-, NSAR-, COX-Hemmer-Einnahme IgG2-Defekte ↑ bei höherem Alter, Rauchern ↑ bei höherer Medikamentenanzahl und DMARD-Einnahme ↑ bei hohem DAS 28 (mit BSG) ↓ bei hoher BSG, hohen Konzentrationen an IgG, IgG1, IgG3, IgG4 IgG3-Defekte ↑ bei längerer Erkrankungsdauer ↓ bei hohen Konzentrationen an IgG2 IgG4-Defekte ↑ bei DMARD-Einnahme ↓ hohen Konzentrationen an IgG, IgA, IgM, IgG2 ↑: nehmen zu/ sind gesteigert, ↓: nehmen ab/ sind vermindert. 5.4.8 Regressionen aus allen Patientendaten Zunächst wurden die Patientendaten der Regression in kleineren Gruppen unterzogen, da es so möglich ist, mehrere metrische Werte in die Regressionsanalyse einfließen zu lassen. Verwendet man viele Daten, so würden die Ergebnisse ungenauer werden. Zuletzt sollte aber der gegenseitige Einfluss aller erhobenen Faktoren betrachtet werden. Hierzu flossen als unabhängige Variablen alle Daten in das Modell ein, wurden aber bei metrischen Daten nur mit zwei 59 5 Ergebnisse Ausprägungen angegeben. Bei den zuvor beschriebenen Modellen war es möglich, dass sich gewisse Signifikanzen ergaben, die aber durch andere, nicht betrachtete Einflüsse entstanden sind. Oder aber es konnten Einflüsse verschleiert werden, die erst in einem Modell mit allen betrachteten Variablen erschlossen werden können. Aus diesen Gründen haben wir im letzten Teil der Ergebnisse die Regression mit allen vorhandenen Variablen durchgeführt. Die Ergebnisse aus diesem Teil der Regressionsanalyse unterschieden sich teilweise von den vorher berechneten Daten. Die Regression des IgM mit Einfluss aller Variablen erbrachte einen fortbestehenden Zusammenhang zum Alter. Das erhöhte Risiko für IgM-Defekte bei älteren Patienten blieb bestehen. Die anderen signifikanten Werte zeigten sich nun jedoch nicht mehr. Weniger IgM-Defekte waren mit erhöhten Konzentrationen an IgM-RF und Anti-CCPAK assoziiert (Anhang Tab. 43). Die schon zu Beginn beschriebene Abhängigkeit des IgA zum Geschlecht bestätigte sich ebenfalls in der Gesamtberechnung der Regression, doch die weiteren Werte aus den Einzelberechnungen waren nicht signifikant. Hinzu kam die Berechnung von weniger IgA-Defekten unter der Einnahme von NSAR sowie bei höheren IgM RF (Anhang Tab. 44). In der Gesamtanalyse des IgG zeigten sich die signifikanten Ergebnisse aus der Einzelberechnung im Zusammenhang mit dem Alter und der Kortison-Einnahme nicht mehr. Das erhöhte Risiko für IgG-Defekte bei häufiger auftretenden Infektionen ergab einen p-Wert von 0,07. Es zeigte sich folglich weiterhin eine Tendenz im Zusammenhang mit Infektionen, die aber nicht mehr signifikant war. Das verminderte Risiko an IgG-Defekten bei Biologikum-Einnahme ergab sich vermutlich durch andere Faktoren und war in der Gesamtregression nicht nachweisbar. Ebenso verhielt es sich in Bezug auf die Röntgenstadien. Ein signifikant vermindertes Risiko an Defekten des IgG bestand aber weiterhin mit steigenden Konzentrationen an IgG1 und IgG2 sowie grenzwertig signifikant mit steigenden Konzentrationen an IgA und Anti-CCP-AK (Anhang Tab. 45). Die Berechnung zum IgG1 ergab folgende Ergebnisse: Die als signifikant berechnete Abhängigkeit zum Alter bestand in der Gesamtregression nicht mehr. Dies lässt sich evtl. auf die Einflüsse des veränderten Immunsystems im Alter und auch die vermehrte Einnahme an Medikamenten zurückführen. Das sind Faktoren, die bei der Gesamtregression berücksichtigt werden, bei der einzelnen Regression aber nicht in das Modell eingeflossen sind. Es ergab sich ein erhöhtes Risiko für IgG1-Defekte bei 60 5 Ergebnisse höheren Titern an Anti-CCP-AK sowie hohen IgG2-Konzentrationen. Das niedrigere Risiko für Defekte des IgG1 blieb in der Gesamtregression bestehen für hohe Konzentrationen an IgG und IgG3 sowie für erosive Röntgenveränderungen. Die weiteren im ersten Modell errechneten Signifikanzen wurden in dieser Berechnung nicht bestätigt. Hierzu zählten das beginnend erosive Röngtenstadium, die Einnahme von COX-Hemmern sowie die NSAR-Einnahme (Anhang Tab 46). Bei der Betrachtung der Ergebnisse zum IgG2 ergab sich ein höheres Risiko für Defekte im IgG2-Bereich und weiterhin für Raucher und erhöhte Krankheitsaktivität in Form des DAS 28. Außerdem errechnete sich in der Regression mit allen Daten ein höheres Risiko für IgG2-Defekte bei männlichen im Gegensatz zu weiblichen RAPatienten und bei Patienten mit höheren IgG1-Konzentrationen. Die Zusammenhänge zu Alter, Anzahl an Medikamenten und DMARD-Einnahme, die sich in der vorherigen Regression berechneten, konnten nicht bestätigt werden. Das signifikant verminderte Risiko für IgG2-Defekte durch die BSG und die Konzentrationen von IgG und IgG3 blieb auch in dieser Regression bestehen (Anhang Tab. 47). Die Berechnung zum IgG3 zeigte, dass durch das Einsetzen aller Variablen in das Regressionsmodell sich die meisten vorher signifikanten Ergebnisse als nicht mehr signifikant darstellen ließen (Anhang Tab. 48). Hervorzuheben war das Resultat für Kortison. Die Patienten mit Kortisoneinnahme hatten ein geringeres Risiko für einen IgG3-Defekt verglichen mit Patienten ohne dieses Medikament (p = 0,06). Patienten mit höheren IgG2-Konzentrationen hatten ebenso ein niedrigeres Risiko für IgG3Defekte. Das Risiko für IgG4-Defekte stieg signifikant mit der Anzahl an eingenommenen Medikamenten sowie mit der Anzahl an Infektionen (Anhang Tab. 49). Dieser statistische Zusammenhang ergab sich aus der vorherigen Regression mit Daten aus einzelnen Gruppen nicht, sondern zeigt sich erst in der Gesamtregression. Dagegen sank das Risiko für IgG4-Defekte mit höheren Konzentrationen an IgA und IgG2, wie schon oben beschrieben (Tab. 32). Das höhere Risiko für IgG4-Defekte unter DMARD-Therapie konnte nicht bestätigt werden. 61 6 Diskussion 6 Diskussion Die Therapie der RA hat sich seit der Einführung der biologischen DMARDs deutlich verbessert. Dazu trägt besonders die Kombination biologischer und synthetischer DMARDs bei. Bewährt hat sich die gleichzeitige Applikation verschiedener Substanzen, wie z.B. die Gabe von TNF-α-Blockern und Methotrexat. Obwohl dadurch partielle oder auch komplette Remissionen erreichbar sind, kommt es auch zum Auftreten unerwünschter Wirkungen. So werden in Metaanalysen mit über 3000 eingeschlossenen Patienten signifikant häufiger schwere Infektionen, aber auch Malignome beobachtet (Bongartz et al. 2006, Atzeni et al. 2012). Die neuen Therapeutika greifen nicht kausal in die Krankheitsabläufe der RA ein. Sie beeinflussen pathogenetische Teilschritte im Bereich der humoralen und zellulären Abwehr. Dabei kann es neben den erwünschten Effekten zu einer Störung der Immunhomöostase kommen, verbunden mit Veränderungen von Interleukinen und Depletion von B-Zellen. Diese führt zur Reduktion der Immunglobuline und zur Störung der T-zellulären Abwehr. Zur Einschätzung des Nebenwirkungsprofils ist daher die Kenntnis des Immunstatus der Patienten von besonderer Bedeutung, um potentielle Interaktionen rechtzeitig berücksichtigen zu können. In der vorliegenden Arbeit werden Defekte des IgG und der IgG-Subklassen in den Mittelpunkt gestellt, weil diese möglicherweise mit vermehrten Malignomen, aber auch Infekten in Verbindung stehen können. Bereits 1970 konnte gezeigt werden, dass IgG-Subklassendefekte mit erhöhter Anfälligkeit für respiratorische Infekte assoziiert sind (Schur et al. 1970). Seitdem sind IgG-Subklassenbestimmungen bei Verdacht auf Immundefektsyndrome ein wichtiges diagnostisches Werkzeug. Daher begründet sich die Notwendigkeit, dem Auftreten solcher Defekte bei Eingriffen in das Immunsystem, z.B. durch Einsatz von synthetischen und biologischen DMARDs, mehr Aufmerksamkeit zu schenken. Angeregt wurde diese Fragestellung durch die klinische Beobachtung, dass in unserer Arbeitsgruppe bei zwei RA-Patienten unter TNF-Therapie Pneumonien auftraten, die auf Antibiotikagabe nicht ansprachen. Bei subnormalen IgGSerumspiegeln bestand ein deutlicher IgG-Subklassendefekt. Nach Substitution mit Immunglobulinen kam es bei diesen Patienten zur schnellen Abheilung. Diese Beobachtung veranlasste uns zur häufigeren Bestimmung der Immunglobulin- und 62 6 Diskussion IgG-Subklassenspiegel. Überraschenderweise weisen viele Patienten solche Defekte auf. Da sich in der Literatur kaum Untersuchungen zur Häufigkeit und Ausprägung der IgG-Defekte bei RA-Patienten finden, wurde die vorliegende Arbeit konzipiert, um ein größeres Kollektiv von RA-Patienten hinsichtlich des Auftretens dieser Defektsituation zu untersuchen. Die Hauptergebnisse unserer Arbeit betreffen die Anzahl an Defekten im Bereich von IgG sowie der IgG-Subklassen. Diese belaufen sich auf 20% für IgG und 42% für IgG1 mit den am häufigsten eruierten Defekten. Im Bereich von IgG2 und IgG3 finden sich weniger häufig Defekte mit einem Anteil von 7% bzw. 5%. IgG4 stellt mit 10% die IgG-Subklasse mit den zweithäufigsten Defekten dar. Im IgG3-Bereich finden sich außerdem mit 11% auffällig viele Erhöhungen. Die anderen Subklassen zeigen weit weniger Konzentrationserhöhungen. Da die vorliegende Arbeit einen Vergleich zu gesunden Probanden nicht beinhaltet, erfolgt zunächst die Betrachtung der IgG-Subklassen bei Kontrolluntersuchungen in der Literatur. 6.1 Untersuchungen bei Gesunden In der Literatur gibt es kaum Studien zur Häufigkeit von Defekten im IgG- und IgGSubklassenbereich bei Gesunden. Wenige kleine Vergleichsstudien zur Untersuchung an Gesunden existieren, spiegeln aber aufgrund der geringen Probandenzahlen nicht die Verhältnisse in der Gesamtbevölkerung wider. Die in der Literatur zu findenden Angaben beziehen sich oftmals nicht auf Gesunde, sondern betreffen Patienten, die andere Erkrankungen aufweisen. Die Firma Bindingsite ® trifft hierzu die Aussage, dass es in der Normalbevölkerung nur wenige dieser IgGSubklassendefekte gäbe und es weiterhin zu kostenaufwändig sei, eine umfassende Erhebung zu diesem Thema durchzuführen (persönliche Mitteilung der Firma Bindingsite®). Die Prävalenz des IgG-Subklassenmangels bei Gesunden ist also nicht genau bekannt. Schmidt et al. (2011) beschreiben eine Prävalenz von mindestens 1:10.000. Von diesen Autoren wird auch diskutiert, ob es bei einem solchen Mangel einen genetischen Hintergrund geben könne und ob dieser eine klinische Relevanz besitzt. Dazu sind jedoch keine sicheren Fakten bekannt (Ocampo und Peters 2013). Häufig würden im Falle des Auftretens die Defekte in Kombination von 2 Subklassenverminderungen auftreten, IgG2- mit einem IgG4- und 63 6 Diskussion IgG1- mit einem IgG3-Mangel. Klinische Symptome treten hauptsächlich assoziiert mit einem IgG2-Mangel auf, da IgG2 für die Abwehr von kapseltragenden Bakterien wie Streptococcus pneumoniae, Staphylococcus aureus und Haemophilus influenzae essenziell ist. Somit kommt es bei einem vorliegenden IgG2-Defekt zu einer erhöhten Suszeptibilität gegenüber Atemwegsinfekten. Auch findet sich eine verminderte Antwort auf Polysaccharidimpfstoffe (Schmidt et al. 2011). Dazu zählen Impfungen gegen Pneumokokken, Meningokokken und Haemophilus influenzae Typ B. Um bei Immunglobulinmangel einen ausreichenden Impferfolg erzielen zu können, sollte es zum Einsatz von Konjugatimpfstoffen kommen, die zusätzlich zu dem Polysaccharidanteil an Proteine gekoppelt sind. Bei Lebendimpfstoffen gilt erhöhte Vorsicht, da es nach der Impfung zu abgeschwächten oder gar fehlenden Immunreaktionen kommen kann, woraus ein mangelnder Impfschutz resultiert. Es treten aber auch mehr Nebenwirkungen auf als bei Impfungen mit Totimpfstoffen. Dennoch profitieren Patienten mit Teildefekten wie einem IgG-Subklassenmangel nachweislich und sollten auch mit Lebendvakzinen geimpft werden (MannhardtLaakmann und Wahn 2011). Nun stellt sich die Frage, was passiert, wenn diese IgG-Subklassendefekte durch eine Erkrankung wie die RA oder durch immunsuppressive Medikationen noch verstärkt werden. Zunächst betrachten wir die Studien zu IgG-Subklassenbestimmungen an Kontrollgruppen. Diese wurden in kleinem Umfang an gesunden Probanden durchgeführt (Tab. 33). Doch es ist zu beachten, dass die Konzentrationen der verschiedenen Studien mit unterschiedlichen Analysemethoden bestimmt wurden und daher die Mittelwerte voneinander abweichen (Shakib et al. 1975, Klein et al. 1985, Madassery et al. 1988). So wurden in früheren Untersuchungen die Radioimmunodiffusion mit monooder polyklonalen Antikörpern, ELISA-Techniken und die Nephelometrie verwendet. Die Nephelometrie stellt die schnellste und effektivste Methode zur Bestimmung der IgG-Subklassen dar. Sie bietet weiterhin geringere Variationen der Ergebnisse zwischen und innerhalb einzelner Radioimmunodiffusion (Vlug et al. 1994). 64 Laboratorien im Gegensatz zur 6 Diskussion Tabelle 33: Publikationen zu IgG-Subklassenkonzentrationen in Kontrollgruppen Autoren (Jahr) Leddy et al. (1970) Morrel und Skvaril (1971) van der Giessen et al. (1975) Shakib et al. (1975) Oxelius (1978) French und Harrison (1984) Madassery et al. (1988) Schauer et al. (2003) Proben 10 108 Methoden RIA (poly) RIA (poly) Konzentrationen in g/l IgG1 IgG2 IgG3 6,70 2,90 0,54 6,63 3,22 0,58 107 RID (poly) 7,10 3,80 0,65 0,60 111 20 RID (poly) RID (poly) 8,01 7,55 2,17 3,80 0,91 0,72 0,08 0,54 172 RID (mono) 5,91 3,04 0,61 0,24 100 68 PCFI (mono) Neph (poly) 6,10 5,00 3,00 3,00 0,56 0,64 0,23 0,35 IgG4 0,36 0,46 Modifiziert nach Schauer et al. 2003. poly = polyklonale Seren gegen IgG-Subklassen, mono = monoklonale Seren gegen IgG-Subklassen, RIA = Radioimmunassay, RID = radial immunodiffusion assay, PCFI = particle concentration fluorescence immunoassay, Neph = Nephelometrie. 2003 führten Schauer et al. eine Studie an 68 gesunden Personen unter Verwendung von Assays von Binding Site® durch, in welcher analog unserer Studie die Nephelometrie verwendet wurde. Die Werte der IgG-Subklassen 1-4 bei unseren RAPatienten liegen alle höher als die Mittelwerte der Studie von Schauer et al. (2003). Diese Beobachtung, dass IgG-Subklassen bei Patienten mit RA durchschnittlich höhere Konzentrationen aufweisen, wurde bereits durch eine andere Studie gemacht (Lin und Li 2010). Die meisten Autoren trafen jedoch keine Aussagen zur Häufigkeit von IgG-Subklassendefekten, daher ist es nicht möglich, die Defekte vergleichend zu betrachten. Bei der Mehrzahl der vorliegenden Untersuchungen mit kleineren Probandenzahlen handelt es sich um Querschnittuntersuchungen, Verlaufsuntersuchungen fehlen weitestgehend. So könnten asymptomatisch verlaufende subklinische Infektionen den Immunstatus zeitweise beeinflussen und in einmaligen Untersuchungen erfasst werden, die aber in weiteren Verlaufsuntersuchungen keine Relevanz mehr besitzen. Die Angaben vieler Arbeiten beziehen sich häufig nur auf die Konzentrationen der IgG-Subklassen, ohne die eigentliche Defektsituation anzugeben. Es existieren nur wenige Studien, die die Häufigkeit des Auftretens von Defekten beschreiben. So fanden z.B. Vlug et al. (1994) in einer der größten Untersuchungen zu IgGSubklassen an mehr als 2000 Probanden bei 9,8% IgG2-Defekte und bei 1,9% IgG2Erhöhungen. Allerdings wurde in dieser Studie nur die IgG2-Subklasse bestimmt. In der Studie von Lock und Unsworth (2003) mit 982 auf IgG-Subklassen untersuchten Kontrollen fanden sich bei einer Unterteilung in drei Altersgruppen 65 6 Diskussion geringe Unterschiede zwischen den Geschlechtern. Die Mediane von IgG und IgG4 fielen bei Männern höher aus als bei Frauen über 71 Jahren. Im Bezug auf das Alter beschrieb Lock eine Reduzierung der IgG-Konzentrationen bei über 70-Jährigen. Des Weiteren fanden sich in jener Studie reduzierte Konzentrationen von IgG2 bei älteren Patienten. Im Gegensatz dazu fanden Radl et al. (1975) in einer der wenigen Arbeiten bei Älteren, hier bei über 95-Jährigen, zu IgG-Subklassenkonzentrationen erhöhte IgG-Konzentrationen mit dem Alter sowie auch IgG1- und IgG3Konzentrationssteigerungen. Schauer et al. (2003) fanden keine Unterschiede zwischen den Geschlechtern. In Bezug auf Geschlechtsunterschiede bei gesunden Probanden fanden French und Harrison (1984) unter 172 Gesunden Auffälligkeiten bei IgG3 und IgG4. IgG3 wies in dieser Studie höhere Werte bei den Frauen auf (p = 0,034) und IgG4 war durchschnittlich bei den Männern höher (p < 0,001). In mehreren Studien zur Erfassung der Auswirkungen intensiven körperlichen Trainings auf das Immunsystem wurden bei Schwimmern auch die IgG- und IgGSubklassenkonzentration bestimmt. Diese fielen bei den Sportlern signifikant niedriger aus verglichen mit Kontrollen (Gleeson et al. 1996, Pyne und Gleeson 1998). Eine weitere Studie beschreibt Verminderungen der Immunglobulin-Isotypen G, A und M sowie des IgG2 bei Eliteschwimmern bei konstanten Konzentrationen von B- und T-Zellen (Gleeson et al. 1995). Aus den Ergebnissen der genannten Studien können allerdings keine genauen Rückschlüsse zur IgG- und IgG-Subklassenverteilung in der Normalbevölkerung gezogen werden. Wünschenswert wären multizentrische prospektive Studien, die IgG- sowie IgG-Subklassenkonzentrationen an einer großen Population gesunder Probanden mit Verlaufskontrollen untersuchen. 6.2 Untersuchungen bei Patienten mit RA Im Folgenden werden die im Rahmen unserer Arbeit untersuchten Konzentrationen der Immunglobuline sowie mögliche Einflussparameter diskutiert. 66 6 Diskussion 6.2.1 Diskussion zu IgG, IgA und IgM Bei den 539 untersuchten Proben von RA-Patienten wiesen die IgG-Konzentrationen im Mittel einen Wert von 10,4 g/l auf. Hierbei fanden sich bei 19,9% Defekte und bei 1,7% Konzentrationen über dem Normbereich. IgM lag bei den untersuchten RA-Patienten unserer Studie im Mittel bei 1,2 g/l. Bei 24,5% der Patienten lagen die erzielten Ergebnisse unter dem Normbereich und bei 3,5% darüber. Die IgA-Werte betrugen durchschnittlich 2,5 g/l und waren bei 5,8% defekt und bei 5,2% erhöht. Bemerkenswert waren die vielen Defekte in der Patientengruppe im IgG- und IgMBereich. Leider finden sich unseres Wissens keine vergleichbaren Messungen in anderen Studien zum Auftreten von Defekten, sodass sich dieser Befund schwer zu anderen Untersuchungen in Beziehung setzen lässt. Es existieren Arbeiten, die sich mit dem Verhalten der Immunglobuline bei Patienten mit RA auseinandersetzen. Veys und Claessens (1968) beschrieben im Vergleich zur Normalbevölkerung erhöhte IgG- und IgA-Werte bei RA-Patienten und gleiche IgM-Werte. Diese Ergebnisse wurden durch andere Studien bestätigt (Marcolongo et al. 1967), aber es gibt auch Studien mit anderen Ergebnissen zum Verhalten der Immunglobuline bei RA, bei denen die Isotypen der Immunglobuline A, G und M alle erhöht waren (Claman und Merrill 1966, Barden et al. 1967, Liu et al. 2003). In einer aktuellen Untersuchung von Neumann et al. (2013) wurde auch auf die Defekte der Immunglobuline bei RA-Patienten eingegangen. Die Autoren beschrieben ein gehäuftes Auftreten von bis zu 24,5% an IgG-Defekten. Diese korrelierten mit Medikamenteneinnahmen von Leflunomid und Rituximab, waren aber weder mit der Infekthäufigkeit, noch mit der Schwere von Infekten assoziiert. Eine große, mit gegensätzlichen Aussagen behaftete Arbeit stammt von Pappas et al. (2013) zu IgG-Defekten und deren Einflüsse. In dieser Studie fanden sich mit 6,6% IgG- und 5,5% IgM-Defekten weniger als in der Studie von Neumann et al. (2013) sowie in unserer Untersuchung. Pappas et al. (2013) beschrieben eine Häufung von Defekten im IgG-Bereich bei hoher Krankheitsaktivität der RA sowie unter Kortisoneinnahme. Eine finnische Untersuchung beschrieb das Verhalten der Immunglobuline G, A und M vor dem Auftreten der RA anhand einer knapp 20.000 Probanden umfassenden Studie, von denen 124 im Verlauf eine RA entwickelten (Aho et al. 1997). Es wurde hierbei ein signifikanter Zusammenhang 67 der IgG-Konzentrationen vor 6 Diskussion Krankheitsbeginn mit dem Risiko, eine RA zu entwickeln, beschrieben. In dieser Studie fanden sich, wie auch von Anti-CCP-AK bekannt, erhöhte IgG- Konzentrationen bereits mehr als 10 Jahre vor Ausbruch der Erkrankung. Dies könnte pathogenetisch wichtige immunologische Prozesse widerspiegeln, die sich vor der Krankheitsmanifestation im Körper abspielen. Interessanterweise bestand bei unseren Patienten eine Korrelation zwischen IgMDefekten sowie dem Raucher- und Röntgenstatus. Bei Patienten, die rauchten, traten tendenziell weniger IgM-Defekte auf. In einer Studie zu Parodontitis wurden die Immunglobuline und deren Subklassen in Abhängigkeit vom Rauchen untersucht (AlGhamdi und Anil 2007). Es handelt sich hierbei, wie auch bei der RA, um eine Erkrankung, bei welcher das Rauchen einen Risikofaktor für den Beginn sowie für die weitere Entwicklung der Krankheit darstellt. Dabei fanden sich bei Patienten signifikant niedrigere IgG- und IgA-Konzentrationen. Die Häufigkeit von Defekten wurde allerdings nicht beschrieben. Eine weitere Studie zu Immunglobulinen in der Normalbevölkerung in einer großen Population an Rauchern zeigte signifikant niedrigere Konzentrationen von IgG unter den Rauchern, verglichen mit Nichtrauchern, aber keine Unterschiede der IgA- und IgM-Konzentrationen (Gonzalez-Quintela et al. 2008). Roseman et al. (2012) beschrieben in einer Studie zum Effekt des Rauchens auf das Immunsystem bei RA-Patienten verminderte IgGWerte bei höheren Entzündungsmarkern. Im Zusammenhang mit dem Raucherstatus finden sich sowohl Arbeiten, die mit unseren Ergebnissen vergleichbar sind, als auch voneinander differierende Ergebnisse. Ein weiteres Ergebnis der vorgelegten Arbeit ist der Zusammenhang zwischen BSG und IgG- sowie IgM-Immunglobulinspiegeln. Je höher die BSG, desto weniger Immunglobulindefekte waren auch vorhanden. Da die BSG sehr stark von der Immunglobulinkonzentration abhängig ist, stellte sich die Frage, ob diese Korrelation einen kausalen Zusammenhang beschreibt oder ob eine niedrige BSG nicht durch niedrige IgG- und IgM-Spiegel entsteht. Die Titer von krankheitsbezogenen Antikörpern wie RF wurden bei Patienten mit niedrigen IgG-Spiegeln niedriger beschrieben als bei Patienten mit hohen IgGKonzentrationen (Liu et al. 2003). Allerdings fand sich in unserer Arbeit kein Zusammenhang von IgG-Konzentration zur Höhe der RF. Unsere Gesamtregression zu den Anti-CCP-AK ergab aber den gleichen Zusammenhang wie auch von Liu et al. (2003) beschrieben. 68 6 Diskussion IgG zeigte sich in unserer Arbeit in der Einzelregression signifikant abhängig vom Alter und Röntgenstatus. Mit zunehmendem Alter wiesen die Patienten ein höheres Risiko für IgG-Defekte auf. Die Funktion der B-Zellen scheint bei älteren Patienten beeinträchtigt zu sein, was sich in der Konzentration der Immunglobuline und der Defekte widerspiegelte. Diesen Vorgang des immunologischen Alterns und der damit verbundenen Immunmodulation nennt man Immunseneszens (McKenna et al. 2001). In zahlreichen Studien wurden die Veränderungen des Immunsystems im Alterungsprozess untersucht, die hauptsächlich die verminderte Effektivität der TZellen als Ursache der verschlechterten Abwehrlage im Alter ansehen. Im Laufe des Lebens häufen sich T-Zell-Klone und Gedächtnis- sowie Effektor-T-Zellen an, die die Anzahl an naiven T-Zellen ohne Antigen-Kontakt zurückdrängen (Fagnoni et al. 2000, Pawelec et al. 2002). Somit kommt es zu häufigeren Infektionen und schlechterer Infektbekämpfung. Listi et al. (2006) beschrieben die Rolle der B-ZellImmunseneszens mithilfe der Messungen von IgG und IgG-Subklassen sowie der BZellzahl an gesunden Probanden. Die Autoren fanden erhöhte Konzentrationen an IgA, IgG, IgG1 und IgG3 im Alter sowie eine verminderte Anzahl an naiven B-Zellen. Eine der größten Studien über IgG-Subklassen von Lock et al. (2003) beschreibt an über 1100 Patienten sinkende IgM- und IgG- sowie IgG2-Konzentrationen bei gleich bleibenden IgA-Werten bei über 60 Jährigen. In unserer Untersuchung fanden sich ebenso sinkende IgM-, IgG- und IgG2- sowie unveränderte IgA-Konzentrationen, aber auch IgG1-Verminderungen mit dem Altern. Ein wesentliches Ergebnis dieser Arbeit war der statistische Zusammenhang zwischen der Anzahl unserer Patienten mit Infekten und dem Vorkommen für IgGErniedrigungen. Dieser interessante Befund deckt sich mit unserer klinischen Beobachtung, die wir vor dieser Studie machten, bei der zwei Patienten mit verminderten IgG-Werten an schweren pulmonalen Infekten erkrankten. Auch in unserer großen Kohorte war das Risiko für Infekte bei Patienten mit IgG-Defekten höher als bei Patienten ohne diese Defekte. Nun stellte sich die Frage, wie ein Zusammenhang zwischen beiden Parametern begründet werden kann. Da IgG für die Immunabwehr essentiell ist und für Opsonierung, Neutralisation von Antigenen und deren Agglutination verantwortlich ist, können Erreger eher Infektionen auslösen, wenn diese immunologische Funktion vermindert ist. In unserer Regressionsanalyse konnte gezeigt werden, dass eine verminderte Konzentration an IgG mit einer 69 6 Diskussion erhöhten Anfälligkeit für Infekte einhergeht. Dies unterstreicht wiederum die hohe Bedeutung der Immunglobuline für die Infektabwehr. Interessanterweise fanden wir bei Patienten mit Biologika-Therapie signifikant erhöhte IgG-Konzentrationen. Bei einer genaueren Subanalyse der eingesetzten Biologika konnte ein signifikanter Zusammenhang zu Adalimumab gefunden werden, welches 32 Patienten erhielten. In diesen Fällen fanden sich signifikant erhöhte IgGWerte und somit ein geringeres Risiko für IgG-Defekte unter der Therapie mit einem Medikament, welches entscheidend in das Immunsystem eingreift und die Abwehr herabsetzt. Es handelt sich um einen Anti-TNF-α-Antikörper, unter dessen Wirkung durch die TNF-Neutralisierung Entzündungsmediatoren wie CRP und IL-6 entscheidend gesenkt werden. Womit lässt sich der IgG-Anstieg erklären? Eine wissenschaftliche Erklärung für dieses Phänomen liegt derzeit unseres Wissens noch nicht vor und es kann daher nur spekuliert werden. Möglicherweise sind Antikörper gegen das Medikament selbst dafür verantwortlich und führen so zu einer Abschwächung der Wirkung (neutralisierende AK). Ein solcher Mechanismus wurde bereits für IFN-β und Natalizumab in der Therapie der Multiplen Sklerose beschrieben (Deisenhammer 2009). Auch zu Adalimumab in der Therapie der RA existieren Studien zur Bildung neutralisierender AK gegen das Medikament und einer damit verbundenen Effektminderung (Bartelds et al. 2011). Da anzunehmen ist, dass Patienten, die Biologika einnehmen, eine aggressivere RA mit chronifiziertem Verlauf haben und deshalb diese Medikamente erhalten, könnte das höhere IgG auf erhöhte Autoantikörper zurückzuführen sein. Zusätzlich sei angemerkt, dass es bei chronifiziert verlaufenden RA-Erkrankungen auch zu einer Erhöhung der IgG-Spiegel kommen könnte. Bei den länger erkrankten Patienten mit erosiven Verläufen zeigten sich aber keine Abhängigkeiten zu Defekten des IgG. Im beginnend erosiven Stadium gab es weniger IgG-Defekte unter den RA-Patienten. Dies deutet auf den Einfluss des IgG auf den Krankheitsverlauf hin, aber auch auf Veränderungen des IgG in dessen Subklassen im Verlauf der Erkrankung. Jedoch könnte dieser Zusammenhang auch durch andere Faktoren beeinflusst worden sein, da er sich in der Gesamtregression als nicht mehr signifikant darstellte. Bei unserer kleinen mit Rituximab behandelten Patientengruppe fanden sich keine Auffälligkeiten beim IgG und den IgG-Subklassen. Im europäischen Konsensusbericht von Buch et al. (2011) wurden bei 5% der RA-Patienten vor 70 6 Diskussion Rituximab-Therapie erniedrigte IgG-Spiegel gefunden. Bei diesen Patienten kam es nach Therapie zu einem erhöhten Risiko für schwere Infektionen. Über die Vorwerte bei unseren Patienten lagen uns keine Informationen vor. In Langzeitsicherheitsanalysen von RA-Studien kam es bei 2% der Patienten zu Herpes zoster-Infektionen. Diese Rate ist vergleichbar mit den Komplikationen unter AntiTNFα-Therapie. In einer Studie zu Immunglobulinspiegeln unter Rituximab-Therapie wurden ebenfalls verstärkte IgG- und IgM-Defekte bei Patienten beschrieben, die bereits vor der Therapie niedrige IgG-Spiegel aufwiesen (De La Torre et al. 2012). Gottenberg et al (2010) beschrieben ein erhöhtes Risiko an schweren Infektionen bei Patienten mit Rituximab-Therapie. Das Infektionsrisiko war nochmal erhöht bei Patienten, die gleichzeitig Kortikosteroide erhielten. Die Autoren empfehlen eine Kontrolle des IgG vor Therapiebeginn und darauf folgend eine Nutzen-RisikoAbwägung in Bezug auf dieses Medikament bei Patienten mit niedrigen IgGSpiegeln. Die Auswirkung auf die IgG-Subklassen wurde nicht untersucht, fraglich bleibt, ob eine vorherige Bestimmung der IgG-Subklassen die Entscheidungsfindung vereinfachen würde. Unter Rituximab wurden zudem im Verlauf der Therapie gehäuft Verminderungen an IgM und IgA beobachtet, verglichen mit dem Zustand vor Rituximab-Gabe (van Vollenhoven et al. 2010). Auch Shaffu et al. (2013) beschrieben verminderte Konzentrationen von IgG, IgA und IgM in einer Untersuchung an 133 Patienten unter Rituximab. Bei der Untersuchung unserer RA-Patienten fielen 16 Patienten mit Defekten des IgG und IgA auf, bei 9 dieser Patienten war zusätzlich IgM defekt. Diese Defekte waren vorher nicht bekannt und wurden deshalb auch bei der Planung der bisherigen Therapie nicht beachtet. 13 dieser 16 Patienten standen unter KortikosteroidTherapie sowie 14 unter Basistherapie mit MTX, Sulfasalazin, Leflunomid oder Hydroxychloroquin mit potentiell erhöhtem Infektionsrisiko. Zwei der betroffenen Patienten wurden mit Adalimumab bzw. Etanercept therapiert. 6.2.2 Diskussion zu IgG-Subklassen Die IgG- und IgG-Subklassenwerte von 539 RA-Patienten sind in der vorliegenden Arbeit in den Tabellen 15 und 16 zusammengefasst. Sie unterschieden sich von den Ergebnissen anderer kleinerer Studien. Eine der ersten Arbeiten zur IgG-Subklassenverteilung stammt von Shakib et al. aus dem Jahre 1976. Hierbei wurden die Subklassen von 24 RA-Patienten mittels RID 71 6 Diskussion untersucht. Es fanden sich, verglichen mit 111 Normalsera, bei den Patienten ein höherer Mittelwert bei IgG1 und jeweils niedrigere Mittelwerte für IgG2 bis IgG4. Die Mittelwerte des IgG sowie der IgG-Subklassen aus der Arbeit von Shakib et al. (1976) wichen durchaus von unserer Studie ab. Dabei ist zu beachten, dass wir andere Messverfahren nutzten, eine größere Patientenanzahl hatten und die Patienten früher ein anderes Spektrum an Medikamenten bekamen, die evtl. die Subklassenkonzentrationen beeinflussten. Shakib et al. (1976) beschrieben erniedrigte IgG-Subklassenwerte bei 6,8% für IgG2, 0,7% für IgG3 und 0,3% für IgG4, lediglich für IgG1 wurde eine Erhöhung erwähnt. In unserer Studie wiesen immerhin 42,3% der Patienten eine IgG1-Verminderung auf, auch die weiteren Subklassen zeigten jeweils stärkere prozentuale Verminderungen (siehe Tab. 16). In der Studie von Lin und Li aus dem Jahre 2010 wurden Seren von 72 RA-Patienten und 45 Kontrollen Patientengruppe für mittels alle 4 Nephelometrie Subklassen untersucht. signifikant Es höhere wurden in der Konzentrationen beschrieben als in der Vergleichsgruppe. Zwischen aktiver und in Remission befindlicher Erkrankung fanden sich in dieser wie auch in unserer Arbeit keine Unterschiede. Eine aktuelle Untersuchung beschrieb höhere Konzentrationen von IgG, IgG1 und IgG3 bei RA-Patienten verglichen mit Kontrollwerten (Matt et al. 2013). Im Verlauf der Therapie der RA wurde ein Absinken der Antikörper beobachtet. Dies betraf die Konzentrationen von IgG, der IgG-Subklassen, RF und Anti-CCP-AK. Offen bleibt, ob und wie schnell sich die Immunglobuline nach Absetzen einer Therapie wieder erholen. In einer Arbeit aus dem Jahr 1986 fanden sich bei Juveniler Arthritis im Vergleich zu gesunden Kontrollen erhöhte Konzentrationen von IgG1, IgG2 sowie IgG3 bei äquivalentem IgG4 (Martini et al. 1986). Diese Werte wurden allerdings mittels RID erhoben und sind somit nur eingeschränkt vergleichbar mit unseren Ergebnissen. Die meisten Erhöhungen in unserer Arbeit lagen im IgG3-Bereich. Beschrieben wurde erhöhtes IgG3 bei Autoimmunerkrankungen, Virusinfekten und auf BakterienToxinen. Die wichtigste Eigenschaft der IgG3-Antikörper ist die Virusneutralisation. Ein Mangel geht mit rezidivierenden Infekten der oberen Atemwege, obstruktiven Lungenerkrankungen und Harnwegsinfekten einher (Oxelius et al. 1986). In Kombination mit einem IgG1-Mangel können v.a. T-Zell-abhängige Antigene schlechter abgewehrt werden. Die Folge sind schwere Infektionen. 72 6 Diskussion 6.2.3 Kombinierte Defekte In unserer Untersuchung traten nicht nur einzelne, sondern auch kombinierte IgGSubklassendefekte auf. Am häufigsten beobachteten wir diese bei IgG1 und IgG4, die zweithäufigste Kombination bestand aus IgG1- und IgG2-Defekten. Solche kombinierten Defekte wurden in den anderen Arbeiten zu IgG- Subklassenmessungen bei RA-Patienten nicht betrachtet. Allein die Defekte der Subklassen an sich werden nur selten von Autoren aufgegriffen. Ein größerer Teil unserer Patienten mit kombinierten IgG-Subklassendefekten bekam eine Basistherapie mit DMARDs sowie eine Therapie mit Kortikosteroiden. Ein kleinerer Teil erhielt auch Biologika. Hierbei stellt sich die Frage, ob es überhaupt vertretbar ist, ohne vorherigen Ausschluss solcher Immundefektsituationen eine immunsuppressive Therapie mit diesen Medikamenten durchzuführen. Es sollte gerade bei diesen Patienten die Empfehlung berücksichtigt werden, die Immunsituation weiter zu kontrollieren und eventuell bestehende Infektgeschehen genau zu erfragen. Sollte die Infektanamnese auffällig sein, muss die Therapie überdacht und ggf. mit einem Immunologen abgesprochen werden. 6.2.4 Beziehungen der IgG-Subklassen zu anderen Parametern Die Altersabhängigkeit in Bezug auf verstärkte Defektsituationen zeigte sich nicht nur wie oben dargestellt für IgG und IgM, sondern auch bei den Subklassen IgG1 und IgG2. Folglich ist erhöhte Vorsicht bei der Therapie gerade von älteren RAPatienten mit immunsuppressiven Medikamenten geboten, die auch noch Immundefekte aufwiesen. In den wenigen anderen Arbeiten zu IgG-Subklassen (Shakib und Stanworth 1976, Martini et al. 1986, Lin und Li 2010) wurden keine Angaben zu Altersabhängigkeiten gemacht. Bei der Verwendung der Gesamtregression stellten sich unsere Ergebnisse zum Alter lediglich im Falle des IgM signifikant abhängig dar. Geschlechtsunterschiede bezüglich der IgG-Subklassen fanden sich in unseren Untersuchungen lediglich beim IgG4, welches bei weiblichen Patienten niedrigere Konzentrationen aufwies als bei männlichen. In den Arbeiten von Shakib et al. (1976) und Lin und Li (2010) wurden eventuelle Geschlechtsunterschiede nicht beschrieben, Martini et al. (1986) fanden keine Unterschiede zwischen männlichen und weiblichen Patienten. In der Studie von Gunsolley et al. (1997) zu IgG- Subklassenbestimmungen bei Parodontitis-Patienten ergab die Untersuchung zu 73 6 Diskussion Geschlechtsunterschieden die gleichen Ergebnisse wie auch in unserer Studie. In unserer Gesamtregression stellte sich zudem ein höheres Risiko für IgG2-Defekte bei Männern heraus. Es fanden sich in unserer Untersuchung im beginnend erosiven sowie auch im erosiven Röntgenstadium weniger IgG1-Subklassendefekte im Vergleich zu Patienten mit nicht erosiven Veränderungen. Die anderen Subklassen des IgG zeigten hierbei keine Unterschiede. Die Wertigkeit dieser Befunde ist offen und muss in zukünftigen Untersuchungen detailliert analysiert werden. Wir beobachteten gehäuft IgG3-Defekte bei längerer Erkrankungsdauer der RAPatienten. Bei der Betrachtung der anderen Subklassen und auch in Bezug auf die Krankheitsaktivität fanden sich keine weiteren Unterschiede. Dies wird durch die Arbeit von Lin und Lee (2010) bestätigt, die ebenfalls keine Unterschiede bezüglich der IgG-Subklassen zwischen Patienten in Remission und mit aktiver Erkrankung gefunden haben. Auch hier ist noch keine sichere Interpretation der Ergebnisse möglich, eventuelle Einflüsse der RA hinsichtlich Prozessaktivität sowie Erkrankungsdauer und auch Therapie bleiben beim gegenwärtigen Kenntnisstand spekulativ. In zwei unabhängigen Arbeiten zu Immunglobulinen bei Parodontitis ergab die Bestimmung der IgG-Subklassen eine durchschnittlich niedrigere Konzentration des IgG2 unter den Rauchern (Gunsolley et al. 1997, Al-Ghamdi und Anil 2007). Diese Ergebnisse sind äquivalent unserem Ergebnis bei RA-Patienten, bei denen ebenfalls mehr IgG2-Defekte unter den Rauchern in der einzelnen sowie der Gesamtregression auftraten. Die anderen Subklassen blieben in den genannten Publikationen wie auch in unserer Arbeit ohne signifikante Unterschiede zwischen Rauchern und Nichtrauchern. Es könnte ein Zusammenhang zwischen dem Rauchen als ein Auslöser für die RA und den veränderten IgG-Subklassen bestehen. Jedoch können solche Annahmen lediglich als rein spekulativ gewertet werden. Eine weitere Arbeit zu IgG-Subklassen bei chronischer Bronchitis zeigte ebenfalls IgG2Erniedrigungen bei rauchenden Patienten, aber auch IgG-Verminderungen im Gegensatz zu Nichtrauchern (Qvarfordt et al. 2001). IgG2 ist v.a. für die Abwehr von bekapselten Bakterien wie Pneumokokken, Haemophilus und Meningokokken verantwortlich. Weiter müsste genauer untersucht werden, ob bei Rauchern vermehrt Infektionen mit den genannten Erregern vorkommen und ob diese unter Immunsuppressiva häufiger sind oder schwerer verlaufen. 74 6 Diskussion Einfluss der Medikation In unserer Untersuchung zeigte sich in den Einzelregressionen, je mehr Medikamente eingenommen wurden, desto höher ist das Risiko für IgG2-Defekte. Im Speziellen traten mehr IgG2-Defekte unter der Einnahme von DMARDs auf, aber in der Subanalyse zu den einzelnen DMARDs zeigten sich keine signifikanten Unterschiede. Die Anzahl der eingenommenen Medikamente ergab in Bezug zu den anderen IgG-Subklassen nur zu IgG4 signifikante Unterschiede. Hier stieg das Risiko für Defekte im IgG4-Bereich mit steigender Anzahl an Medikamenten signifikant an. Dieses Ergebnis zeigte sich in der Gesamtregression mit dem Einfluss aller erhobenen Daten. In anderen Studien zu RA wurden über einen möglichen Einfluss der Anzahl eingenommener Medikamente auf die IgG-Subklassen keine Aussagen gemacht. Patienten unter NSAR, Kortikosteroiden, COX-Hemmern und auch unter Biologika hatten signifikant weniger IgG-Defekte als Patienten ohne diese Medikamente. Auch IgG1-Defekte traten unter Biologika-Medikation seltener auf. In der Betrachtung der einzelnen Biologika zeigten sich diese Ergebnisse besonders bei Adalimumab und Certolizumab. Allerdings dürften die Patienten ohne Biologika-Therapie mildere Krankheitsverläufe aufweisen, da Biologika eher bei schwereren und aggressiveren Krankheitsverläufen eingesetzt werden. Damit erklärt sich möglicherweise das geringere Vorkommen von IgG- sowie IgG-Subklassendefekten bei Patienten mit Biologika-Therapie. Unsere Ergebnisse zur Häufigkeit an IgG-Defekten unter Biologika decken sich mit den Studien von Pappas et al. (2013) sowie Neumann et al. (2013), die ebenfalls, allerdings nur bei TNF-α-Blockern, weniger IgG-Defekte gefunden haben. Die IgG-Subklassen wurden in diesen Arbeiten nicht untersucht. Kiely et al. (2000) beschrieben in ihrer Untersuchung zur Auswirkung von Goldpräparaten auf die IgG-Subklassen signifikante Verminderungen an IgG1-IgG3 in einem Drittel der RA-Patienten, im Gegensatz zu Kontrollen, die eine RA hatten, aber keine Goldbehandlung erhielten. Bei diesen zeigten sich dennoch bei 8,5% IgGSubklassendefekte. Diese Arbeit stellt eine der wenigen Arbeiten zu IgGSubklassenbestimmungen im Zusammenhang mit dem Einsatz der Medikamente dar. Weiterhin monoklonalen liegen Arbeiten Anti-CD20-AK, zu vor. IgG-Subklassen Die Therapie und mit Rituximab, Rituximab einem führte in verschiedenen Studien zur Verminderung der Konzentration von IgG4. Somit könnte es zu einer Verbesserung IgG4-assoziierter 75 Erkrankungen wie 6 Diskussion Autoimmunpankreatitis und sklerosierende Cholangitis kommen (Khosroshahi et al. 2010, 2012). Spezifische Beobachtungen, die sich auf die RA beziehen, wurden jedoch bisher nicht publiziert. Infektrisiko bei Patienten mit RA Das Risiko für Infektionen bei Patienten mit RA ist per se schon erhöht (Doran et al. 2002a), verstärkt wird dieses noch durch die Einnahme immunmodulierender Medikamente. Besondere Vorsicht ist diesbezüglich bei TNFα-Blockern geboten, unter deren Einnahme ebenso vermehrte und v.a. schwere Infektionen beobachtet wurden (Harboe et al. 2012). Unsere Ergebnisse zeigten signifikant mehr Infekte bei Patienten mit IgG-Defekten. Dieser Zusammenhang bestand in der Gesamtregression für die Subklasse IgG4. Da die IgG-Subklassenkonzentrationen bei RA-Patienten laut unserer Studie verglichen mit anderen Arbeiten erhöht sind, scheinen die Subklassen selbst kein guter Prädiktor für infektgefährdete Patienten zu sein. Doch betrachtet man die Defektsituation im Zusammenhang mit anderen Faktoren, könnte es möglich sein, eine gute Aussagekraft für die Prognose von Infekten stellen zu können. Als starke Prädiktoren für Infekte bei der RA zählen höheres Alter, extraartikuläre Manifestationen der Erkrankung, Leukopenie und Steroidtherapie sowie Komorbiditäten wie chronische Lungenerkrankungen, Alkoholabhängigkeit und Diabetes mellitus (Doran et al. 2002b). In der Arbeit von Doran et al. (2002b) gelten Kortikosteroide als Risikofaktor für Infekte, die Therapie mit DMARDs hingegen nicht. Diese Risikofaktoren sollten vor Initiierung einer immunmodulierenden Therapie bekannt sein, um eine erhöhte Infektanfälligkeit berücksichtigen zu können. Aus der Kombination dieser Informationen über den jeweiligen Patienten könnte ein geeignetes Risikoprofil bezüglich der Infektanfälligkeit erstellt werden. Hierzu sind allerdings noch weitere vertiefende Studien notwendig. 6.2.5 IgG-Subklassenbestimmung bei anderen Erkrankungen Untersuchungen zu IgG-Subklassen sind zu mehreren Erkrankungen publiziert worden. Aus dem rheumatischen Formenkreis existieren Untersuchungen zu Patienten mit systemischem Lupus erythematodes. In einer dieser Arbeiten von Lin und Li (2009) wurden ähnlich der Ergebnisse einer späteren Arbeit derselben Forschergruppe zur RA (Lin und Li 2010) IgG-Subklassenerhöhungen im Bereich von IgG1, IgG2 und IgG3 beschrieben. IgG4 fiel auch hier normal aus, verglichen mit der 76 6 Diskussion Kontrollgruppe. Feng et al. (1994) beschrieben IgG-Subklassenverteilungen bei Kindern mit Eisenmangel. Es fanden sich hierbei vermehrt IgG-Subklassendefekte beim Auftreten eines Eisenmangels. Eine Untersuchung an Patienten, die an Sichelzellanämie leiden, zeigte erhöhte Konzentrationen an IgG, IgA, IgM, IgG1 und IgG3 im Vergleich zu gesunden Kontrollen (Hedo et al. 1993). Khosroshahi et al. (2010) beschrieben Verminderungen im IgG4-Bereich unter Rituximab-Therapie und damit einhergehender Besserung von mit IgG4 assoziierten systemischen Erkrankungen. Zu diesen zählen u.a. Autoimmunpankreatitis, retroperitoneale Fibrose und Riedel´s Thyreoiditis. Eine holländische Studie untersuchte Immunglobuline und Subklassen bei Patienten mit Parodontitis (Gunsolley et al. 1997). Hierbei traten höhere IgG1- und IgG2Konzentrationen bei den Patienten im Vergleich zu einer Kontrollgruppe auf. Unter den Patienten wurden weiter niedrigere IgG- und IgG2-Konzentrationen bei Rauchern beschrieben. Die Parodontitis ist ähnlich wie die RA eine chronische und in Schüben verlaufende Erkrankung, sodass man möglicherweise die immunologischen Veränderungen miteinander vergleichen kann. Neuerdings wird die Parodontitis auch als möglicher Auslöser oder einer unter mehreren Triggerfaktoren für die Initiierung der RA diskutiert. Diese Zusammenhänge stellen eventuell eine gewisse Vergleichbarkeit der beiden Erkrankungen sowie der immunologischen Prozesse dar. Es zeigen sich sehr ähnliche Ergebnisse bei der Subklassenbestimmung innerhalb beider Erkrankungen. Eine zum Sjögren-Syndrom publizierte Arbeit beschrieb erhöhte Konzentrationen von IgG1, IgG2 und IgG3 bei vermindertem IgG4 unter den 155 Patientensera verglichen mit Kontrollen (Liu und Li 2011). Diese Ergebnisse zu IgG-Subklassen- konzentrationen fanden sich auch bei Arbeiten von Lin et al. (2009, 2010) zur RA und zum systemischen Lupus erythematodes (s.o.). IgG-Subklassen gegen humanes und bakterielles Heat Shock Protein (HSP60) wurden bei Patienten mit Spondylitis ankylosans (SpA) bestimmt, da die Vermutung besteht, dass die rheumatische Erkrankung durch eine Kreuzreaktion auf humanes und bakterielles HSP60 getriggert wird (Carlsen et al. 2013). Erhöhte Konzentrationen an IgG1 und IgG3 fanden sich bei den SpA-Patienten verglichen zu gesunden Kontrollen. Es wurden aber keine Assoziationen zwischen humanen und bakteriellen IgG-Subklassen sowie zur Krankheitsaktivität registriert. 77 6 Diskussion In einer Studie aus dem Jahre 2013 wurde die IgG-Subklassendefektsituation bei Patienten mit chronisch-lymphatischer Leukämie beschrieben (Freeman et al. 2013). Bei 64,6% der Patienten wurden IgG-Subklassendefekte gemessen, wobei die einzelnen Subklassen IgG1, IgG2, IgG3 und IgG4 Defekte in Höhe von 28%, 19,3%, 52% und 22,7% aufwiesen. Infektionen waren in dieser Arbeit mit IgG3- und IgG4Defekten assoziiert. IgG4 zeigte sich auch in unserer Arbeit assoziiert zur Infekthäufigkeit. Unter den Patienten mit rekurrenten Infektionen wurden in 100% IgG-Subklassendefekte beschrieben, allerdings zeigten nicht alle Patienten mit Subklassendefekten vermehrt klinische Infektionen. Eine andere Untersuchung zu Patienten mit Non-Hodgkin-Lymphom ergab verminderte IgG- sowie IgG4Subklassenkonzentrationen im Vergleich zu Kontrollen (Biggar et al. 2009). IgG und dessen Subklassen wurden weiterhin bei Rauchern mit chronischer Bronchitis untersucht, mit dem Ergebnis, dass bei Rauchern IgG sowie auch IgG2 vermindert waren, verglichen mit gesunden Nichtrauchern. Diese Verhältnisse zwischen Rauchern und Nichtrauchern fanden wir auch in unserer Arbeit unter den RA-Patienten. In der Literatur wurden Subklassendefekte im IgG2 und IgG4 auch bei Patienten mit alkoholassoziierten Lebererkrankungen und damit einhergehenden vermehrten Infekten beschrieben (Spinozzi et al. 1992). 6.3 Limitationen Die Aussagen dieser Arbeit werden durch die Anzahl der Patienten begrenzt, die lediglich einen kleinen Teil der Gesamtbevölkerung darstellen. Es fehlt eine große Studie zu den IgG-Subklassenkonzentrationen bei gesunden Probanden, um die in dieser Arbeit erhobenen Ergebnisse besser vergleichen zu können. Auch sind keine Verlaufsstudien publiziert worden, weder zur Situation der IgG-Subklassen bei Gesunden noch bei RA-Patienten. Auch eine große Studie zu Normwerten liegt derzeit unseres Wissens nicht vor. Somit ist es nicht möglich, die auffälligen Ergebnisse unserer Studie mit anderen Aussagen und Erkenntnissen zu vergleichen. Das Wissen über IgG-Subklassen in Bezug zur RA, deren Krankheitsverlauf, der Dauer sowie zur Medikamenteneinnahme ist bisher noch sehr eingeschränkt und könnte möglicherweise in Zukunft verbessert werden. Weiterhin erfolgte die Erhebung der Patientendaten teils retrospektiv, die Aussagen zu den Infektionen innerhalb der letzten zwei Jahre könnten fehlerbehaftet sein, da 78 6 Diskussion nicht objektivierbar. Methodisch wäre eine prospektive, standardisierte Erhebung der Infektionen zu bevorzugen. Aussagen über den Immunstatus vor Krankheitsbeginn bzw. vor Therapiebeginn waren ebenfalls nicht möglich, somit kann nicht eindeutig festgestellt werden, inwieweit die Laborwerte im Zusammenhang mit der RA sowie der Therapie stehen. Abweichungen der Laborwerte könnten durch die verwendete Messmethode der Immunglobulinsubklassen entstanden sein, der Nephelometrie. Es kommt durch die RF zu erhöhter Absorption in RF-positiven Proben. Allerdings haben wir keine Unterschiede der IgG-Subklassenkonzentrationen zwischen RF-positiven und – negativen Patientenproben beobachtet. 6.4 Ausblick Mit umfangreicherem Wissen zum Immunstatus eines RA-Patienten kann es in Zukunft möglich sein, die Therapie besser auf den einzelnen abzustimmen und v.a. schwere Infektionserkrankungen durch immunmodulierende Medikamente zu verringern. Für diese Patienten kann es vorteilhaft sein, eine multimodale Therapie anzustreben, die nicht nur aus der Medikamenten-induzierten Modulation des Immunsystems besteht, sondern ebenfalls eine Stimulation des Immunsystems durch sportliche Aktivität, Entspannungsverfahren sowie Infektprävention einschließt. Aktuelle Untersuchungen von Andersson und Tracey (2012a) zeigen eine verminderte Funktion des Vagus-Nervs bei Patienten mit RA. Der Vagus ist eng verknüpft mit dem Immunsystem und besitzt immunregulatorische Funktionen. Bei einer Deprimierung scheint es jedoch zu einer Dysregulation des Immunsystems zu kommen, die autoimmunologische Erkrankungen wie die RA verschlimmern. Die Idee der Forschergruppe um Tracey ist es, die vagale Aktivität bei RA-Patienten zu erhöhen und so die Krankheitsaktivität zu vermindern. Dies kann in Form von sportlicher Aktivität umsetzbar sein oder durch die elektrische Stimulation des Nervus vagus über einen vagalen Schrittmacher, der wie ein Herzschrittmacher implantiert werden muss. Bisher ist diese neuartige Herangehensweise an die Behandlung jedoch nur an einem Patienten beschrieben (Andersson und Tracey 2012b). Falls sich dieses Therapiekonzept als Erfolg herausstellt, wäre es gerade für Patienten mit einem Immundefekt, die dennoch mit einem Arsenal an immunsupprimierenden Medikamenten therapiert werden, eine neue therapeutische Option. 79 6 Diskussion In weiteren Studien ist außerdem zu klären, wie diese enorme Anzahl an Immundefekten im IgG-Subklassenbereich bei RA-Patienten, die wir in unserer Arbeit gefunden haben, einzuschätzen ist, welche Risikofaktoren und Abhängigkeiten es gibt und wie die Situation in einer Kontrollgruppe aus Gesunden aussieht. 80 7 Schlussfolgerungen 7 Schlussfolgerungen Aufgrund der überraschenden Häufigkeit von Defekten im Bereich der Immunglobulin-Isotypen IgG, IgA und IgM sowie der IgG-Subklassen und der möglicherweise damit assoziierten Risiken im Sinne einer Infektionsgefährdung ergibt sich die Forderung, solche Defekte vor Therapiebeginn zu erfassen. In aktuellen Empfehlungen zur Zytokintherapie wird für Eingangs- und Verlaufsuntersuchungen bei Anti-TNFα- sowie bei Tozilizumab-Therapie die Bestimmung von Immunglobulinen und damit die mögliche Erfassung von Defekten bisher nicht gefordert (Naumann et al. 2013). Wir schlagen die Einbeziehung von insbesondere IgG-Bestimmungen, ggf. auch von IgG-Subklassen in diese Empfehlungen vor. Dies sollte auch bei anderen Biologika wie Rituximab und Adalimumab sowie für synthetische DMARDs gelten. Die Auswirkungen der in unseren Untersuchungen gefundenen häufigen Defekte unter therapeutischer Intervention mit Kortikosteroiden, konventionellen und biologischen DMARDs sind bisher nicht untersucht und somit nicht bekannt, es könnte jedoch ein erhebliches Gefährdungspotential bestehen. Weiterhin kann es bei Patienten mit IgG- und speziell mit IgG2-Subklassendefekten zu einer unzureichenden Impfantwort und damit zu einem nicht ausreichenden Impfschutz kommen, auch hier kann unter entsprechender Therapie eine verstärkte Gefährdung bestehen. Hier ergibt sich die Empfehlung, bei bestehenden IgG2Subklassendefekten den Impfeffekt durch Bestimmung der Impfantwort zu erfassen und damit mögliche non-Responder zu erkennen. Vor jeder Therapie mit immunmodulierenden Substanzen bei Patienten mit RA sollten die Immunglobulinisotypen IgG, IgA und IgM sowie die IgG-Subklassen bestimmt werden. Weiterhin sind prospektive Verlaufsuntersuchungen von RA- Patienten zu den Auswirkungen der beschriebenen Immundefekte wünschenswert, da bislang die möglichen Infektionsrisiken bei bestehenden Defekten nicht sicher abzuschätzen sind. Auch ist unter immunmodulierender Therapie das Auftreten solcher Defekte möglich. Ebenso könnten insbesondere IgG-Subklassendefekte nur passager auftreten. 81 7 Schlussfolgerungen Die vielen noch offenen Fragen sollten zukünftig in prospektiv angelegten Studien weiter untersucht und abgeklärt werden, um die Wirksamkeit der einzelnen Therapien weiter zu verbessern und individueller anpassen zu können. 82 8 Literatur- und Quellenverzeichnis 8 Literatur- und Quellenverzeichnis Agarwal SC. 1964. Nomenclature for human immunoglobulins. Bull World Health Organ, 30:447-450. Aho K, Heliovaara M, Knekt P, Reunanen A, Aromaa A, Leino A, Kurki P, Heikkila R, Palosuo T. 1997. Serum immunoglobulins and the risk of rheumatoid arthritis. Ann Rheum Dis, 56 (6):351-356. Al-Ghamdi HS, Anil S. 2007. Serum antibody levels in smoker and non-smoker Saudi subjects with chronic periodontitis. J Periodontol, 78 (6):1043-1050. Aletaha D, Neogi T, Silman AJ, Funovits J, Felson DT, Bingham CO, 3rd, Birnbaum NS, Burmester GR, Bykerk VP, Cohen MD, Combe B, Costenbader KH, Dougados M, Emery P, Ferraccioli G, Hazes JM, Hobbs K, Huizinga TW, Kavanaugh A, Kay J, Kvien TK, Laing T, Mease P, Menard HA, Moreland LW, Naden RL, Pincus T, Smolen JS, Stanislawska-Biernat E, Symmons D, Tak PP, Upchurch KS, Vencovsky J, Wolfe F, Hawker G. 2010. 2010 Rheumatoid arthritis classification criteria: an American College of Rheumatology/European League Against Rheumatism collaborative initiative. Arthritis Rheum, 62 (9):2569-2581. Alsaleh G, Francois A, Knapp AM, Schickel JN, Sibilia J, Pasquali JL, Gottenberg JE, Wachsmann D, Soulas-Sprauel P. 2011. Synovial fibroblasts promote immunoglobulin class switching by a mechanism involving BAFF. Eur J Immunol, 41 (7):2113-2122. Andersson U, Tracey KJ. 2012a. Neural reflexes in inflammation and immunity. J Exp Med, 209 (6):1057-1068. Andersson U, Tracey KJ. 2012b. A new approach to rheumatoid arthritis: treating inflammation with computerized nerve stimulation. Cerebrum, 2012:3. Arnett FC, Edworthy SM, Bloch DA, McShane DJ, Fries JF, Cooper NS, Healey LA, Kaplan SR, Liang MH, Luthra HS, Medsger TA, Mitchell DM, Neustadt DH, Pinals RS, Schaller JG, Sharp JT, Wilder RL, Hunder GG. 1988. The American Rheumatism Association 1987 revised criteria for the classification of rheumatoid arthritis. Arthritis Rheum, 31 (3):315-324. Atzeni F, Sarzi-Puttini P, Botsios C, Carletto A, Cipriani P, Favalli EG, Frati E, Foschi V, Gasparini S, Giardina A, Gremese E, Iannone F, Sebastiani M, Ziglioli T, Biasi D, Ferri C, Galeazzi M, Gerli R, Giacomelli R, Gorla R, Govoni M, Lapadula G, Marchesoni A, Salaffi F, Punzi L, Triolo G, Ferraccioli G. 2012. Long-term anti-TNF therapy and the risk of serious infections in a cohort of patients with rheumatoid arthritis: comparison of adalimumab, etanercept and infliximab in the GISEA registry. Autoimmun Rev, 12 (2):225-229. Balsa A, de Miguel E, Castillo C, Peiteado D, Martin-Mola E. 2010. Superiority of SDAI over DAS-28 in assessment of remission in rheumatoid arthritis patients using power Doppler ultrasonography as a gold standard. Rheumatology (Oxford), 49 (4):683-690. Barden J, Mullinax F, Waller M. 1967. Immunoglobulin levels in rheumatoid arthritis: Comparison with rheumatoid factor titers, clinical stage and disease duration. Arthritis Rheum, 10 (3):228-234. Bartelds GM, Krieckaert CL, Nurmohamed MT, van Schouwenburg PA, Lems WF, Twisk JW, Dijkmans BA, Aarden L, Wolbink GJ. 2011. Development of 83 8 Literatur- und Quellenverzeichnis antidrug antibodies against adalimumab and association with disease activity and treatment failure during long-term follow-up. JAMA, 305 (14):1460-1468. Baumann U, Belohradsky B, von Bernuth H, Friedrich W, Linde R, Niehues T, Renner E, Schöndorf I, Schulze I, Wahn V, Warnatz K. 2010. Primäre Immundefekte - Warnzeichen und Algorithmen zur Diagnosefindung. Bremen: Uni-Med-Verlag. Begovich AB, Carlton VE, Honigberg LA, Schrodi SJ, Chokkalingam AP, Alexander HC, Ardlie KG, Huang Q, Smith AM, Spoerke JM, Conn MT, Chang M, Chang SY, Saiki RK, Catanese JJ, Leong DU, Garcia VE, McAllister LB, Jeffery DA, Lee AT, Batliwalla F, Remmers E, Criswell LA, Seldin MF, Kastner DL, Amos CI, Sninsky JJ, Gregersen PK. 2004. A missense single-nucleotide polymorphism in a gene encoding a protein tyrosine phosphatase (PTPN22) is associated with rheumatoid arthritis. Am J Hum Genet, 75 (2):330-337. Biggar RJ, Christiansen M, Rostgaard K, Smedby KE, Adami HO, Glimelius B, Hjalgrim H, Melbye M. 2009. Immunoglobulin subclass levels in patients with non-Hodgkin lymphoma. Int J Cancer, 124 (11):2616-2620. Bijlsma JW. 2012. Disease control with glucocorticoid therapy in rheumatoid arthritis. Rheumatology (Oxford), 51 Suppl 4:iv9-13. Bongartz T, Sutton AJ, Sweeting MJ, Buchan I, Matteson EL, Montori V. 2006. AntiTNF antibody therapy in rheumatoid arthritis and the risk of serious infections and malignancies: systematic review and meta-analysis of rare harmful effects in randomized controlled trials. JAMA, 295 (19):2275-2285. Braun J, Rau R. 2009. An update on methotrexate. Curr Opin Rheumatol, 21 (3):216223. Brooks PM, Day RO. 1991. Nonsteroidal antiinflammatory drugs - differences and similarities. N Engl J Med, 324 (24):1716-1725. Bruton OC. 1952. Agammaglobulinemia. Pediatrics, 9 (6):722-728. Buch MH, Smolen JS, Betteridge N, Breedveld FC, Burmester G, Dörner T, Ferraccioli G, Gottenberg JE, Isaacs J, Kvien TK, Mariette X, Martin-Mola E, Pavelka K, Tak PP, van der Heijde D, van Vollenhoven RF, Emery P. 2011. Updated consensus statement on the use of rituximab in patients with rheumatoid arthritis. Ann Rheum Dis, 70 (6):909-920. Bukhari M, Lunt M, Harrison BJ, Scott DG, Symmons DP, Silman AJ. 2002. Erosions in inflammatory polyarthritis are symmetrical regardless of rheumatoid factor status: results from a primary care-based inception cohort of patients. Rheumatology (Oxford), 41 (3):246-252. Bukhari MA, Wiles NJ, Lunt M, Harrison BJ, Scott DG, Symmons DP, Silman AJ. 2003. Influence of disease-modifying therapy on radiographic outcome in inflammatory polyarthritis at five years: results from a large observational inception study. Arthritis Rheum, 48 (1):46-53. Carlsen TG, Hjelholt A, Jurik AG, Schiottz-Christensen B, Zejden A, Christiansen G, Deleuran B, Birkelund S. 2013. IgG subclass antibodies to human and bacterial HSP60 are not associated with disease activity and progression over time in axial spondyloarthritis. Arthritis Res Ther, 15 (3):R61. Chakravarty EF, Genovese MC. 2004. Associations between rheumatoid arthritis and malignancy. Rheum Dis Clin North Am, 30 (2):271-284. Cheema GS, Roschke V, Hilbert DM, Stohl W. 2001. Elevated serum B lymphocyte stimulator levels in patients with systemic immune-based rheumatic diseases. Arthritis Rheum, 44 (6):1313-1319. Claman HN, Merrill D. 1966. Serum immunoglobulins in rheumatoid arthritis. J Lab Clin Med, 67 (5):850-854. 84 8 Literatur- und Quellenverzeichnis Classen M, Diehl V, Kochsiek K, Halle, M, Böhm M, Wolff S. 2009. Innere Medizin. Urban & Fischer Verlag/Elsevier GmbH. Davies EG, Thrasher AJ. 2010. Update on the hyper immunoglobulin M syndromes. Br J Haematol, 149 (2):167-180. De La Torre I, Leandro MJ, Valor L, Becerra E, Edwards JC, Cambridge G. 2012. Total serum immunoglobulin levels in patients with RA after multiple B-cell depletion cycles based on rituximab: relationship with B-cell kinetics. Rheumatology (Oxford), 51 (5):833-840. Deisenhammer F. 2009. Neutralizing antibodies to interferon-beta and other immunological treatments for multiple sclerosis: prevalence and impact on outcomes. CNS Drugs, 23 (5):379-396. Doran MF, Crowson CS, Pond GR, O'Fallon WM, Gabriel SE. 2002a. Frequency of infection in patients with rheumatoid arthritis compared with controls: A population-based study. Arthritis Rheum, 46 (9):2287-2293. Doran MF, Crowson CS, Pond GR, O'Fallon WM, Gabriel SE. 2002b. Predictors of infection in rheumatoid arthritis. Arthritis Rheum, 46 (9):2294-2300. Fagnoni FF, Vescovini R, Passeri G, Bologna G, Pedrazzoni M, Lavagetto G, Casti A, Franceschi C, Passeri M, Sansoni P. 2000. Shortage of circulating naive CD8(+) T cells provides new insights on immunodeficiency in aging. Blood, 95 (9):2860-2868. Feng XB, Yang XQ, Shen J. 1994. Influence of iron deficiency on serum IgG subclass and pneumococcal polysaccharides specific IgG subclass antibodies. Chin Med J (Engl.), 107(11):813-816. Finkelman FD, Holmes J, Katona IM, Urban JF, Jr., Beckmann MP, Park LS, Schooley KA, Coffman RL, Mosmann TR, Paul WE. 1990. Lymphokine control of in vivo immunoglobulin isotype selection. Annu Rev Immunol, 8:303-333. Fischer T, Ebert B, Voigt J, Macdonald R, Schneider U, Thomas A, Hamm B, Hermann KG. 2010. Detection of rheumatoid arthritis using non-specific contrast enhanced fluorescence imaging. Acad Radiol, 17 (3):375-381. Freeman JA, Crassini KR, Best OG, Forsyth CJ, Mackinlay NJ, Han P, Stevenson W, Mulligan SP. 2013. Immunoglobulin G subclass deficiency and infection risk in 150 patients with chronic lymphocytic leukemia. Leuk Lymphoma, 54 (1):99104. French MA, Harrison G. 1984. Serum IgG subclass concentrations in healthy adults: a study using monoclonal antisera. Clin Exp Immunol, 56 (2):473-475. Fries JF, Williams CA, Ramey D, Bloch DA. 1993. The relative toxicity of diseasemodifying antirheumatic drugs. Arthritis Rheum, 36 (3):297-306. Gabriel SE, Crowson CS, O'Fallon WM. 1999. The epidemiology of rheumatoid arthritis in Rochester, Minnesota, 1955-1985. Arthritis Rheum, 42 (3):415-420. Garrod AB. 1876. A treatise on gout and rheumatic gout (rheumatoid arthritis). Longmans, Green. Gemsa D, Kalden JR, Resch K, Vorlaender KO. 1997. Immunologie: Grundlagen Klinik - Praxis. Thieme, Stuttgart. Gleeson M, McDonald WA, Cripps AW, Pyne DB, Clancy RL, Fricker PA. 1995. The effect on immunity of long-term intensive training in elite swimmers. Clin Exp Immunol, 102 (1):210-216. Gleeson M, Pyne DB, McDonald WA, Clancy RL, Cripps AW, Horn PL, Fricker PA. 1996. Pneumococcal antibody responses in elite swimmers. Clin Exp Immunol, 105 (2):238-244. Gonzalez-Quintela A, Alende R, Gude F, Campos J, Rey J, Meijide LM, FernandezMerino C, Vidal C. 2008. Serum levels of immunoglobulins (IgG, IgA, IgM) in a 85 8 Literatur- und Quellenverzeichnis general adult population and their relationship with alcohol consumption, smoking and common metabolic abnormalities. Clin Exp Immunol, 151 (1):4250. Gottenberg JE, Ravaud P, Bardin T, Cacoub P, Cantagrel A, Combe B, Dougados M, Flipo RM, Godeau B, Guillevin L, Le Loet X, Hachulla E, Schaeverbeke T, Sibilia J, Baron G, Mariette X. 2010. Risk factors for severe infections in patients with rheumatoid arthritis treated with rituximab in the autoimmunity and rituximab registry. Arthritis Rheum, 62 (9):2625-2632. Gunsolley JC, Pandey JP, Quinn SM, Tew J, Schenkein HA. 1997. The effect of race, smoking and immunoglobulin allotypes on IgG subclass concentrations. J Periodontal Res, 32 (4):381-387. Hamilton RG. 1987. Human IgG subclass measurements in the clinical laboratory. Clin Chem, 33 (10):1707-1725. Hansen SE. 2009. Did Philippe Pinel frame the concept of the disease rheumatoid arthritis in the year 1800? Scand J Rheumatol, 38 (6):488-491. Harboe E, Damas JK, Omdal R, Froland SS, Sjursen H. 2012. Risk of infection through use of selective immunomodulating drugs for rheumatoid arthritis. Tidsskr Nor Laegeforen, 132 (16):1867-1871. Harris ED, Ruddy S, Kelley WN. 2005. Kelley's textbook of rheumatology. Elsevier/Saunders. Hart JE, Kallberg H, Laden F, Bellander T, Costenbader KH, Holmqvist M, Klareskog L, Alfredsson L, Karlson EW. 2012. Ambient air pollution exposures and risk of rheumatoid arthritis: results from the Swedish EIRA case-control study. Ann Rheum Dis, 72 (6):888-894. Hedo C, Aken'Ova Y, Okpala I, Salimonu L. 1993. Serum immunoglobulin (G, A and M) classes and IgG subclasses in sickle cell anaemia. APMIS, 101 (5):353357. Herold G. 2010. Innere Medizin 2010: eine vorlesungsorientierte Darstellung. Selbstverl. Hettenkofer HJ. 2003. Rheumatologie: Diagnostik - Klinik - Therapie. Thieme, Stuttgart. Huizinga TW, Amos CI, van der Helm-van Mil AH, Chen W, van Gaalen FA, Jawaheer D, Schreuder GM, Wener M, Breedveld FC, Ahmad N, Lum RF, de Vries RR, Gregersen PK, Toes RE, Criswell LA. 2005. Refining the complex rheumatoid arthritis phenotype based on specificity of the HLA-DRB1 shared epitope for antibodies to citrullinated proteins. Arthritis Rheum, 52 (11):34333438. Islander U, Jochems C, Lagerquist MK, Forsblad-d'Elia H, Carlsten H. 2011. Estrogens in rheumatoid arthritis; the immune system and bone. Mol Cell Endocrinol, 335 (1):14-29. Jarvinen P, Aho K. 1994. Twin studies in rheumatic diseases. Semin Arthritis Rheum, 24 (1):19-28. Jefferis R, Kumararatne DS. 1990. Selective IgG subclass deficiency: quantification and clinical relevance. Clin Exp Immunol, 81 (3):357-367. Kaplan C, Valdez JC, Chandrasekaran R, Eibel H, Mikecz K, Glant TT, Finnegan A. 2002. Th1 and Th2 cytokines regulate proteoglycan-specific autoantibody isotypes and arthritis. Arthritis Res, 4 (1):54-58. Katayama K, Matsuno T. 2009. Long-term efficacy of leflunomide on disease activity and inhibition of joint damage: retrospective comparison with methotrexate for Japanese rheumatoid arthritis patients. Mod Rheumatol, 19 (5):513-521. 86 8 Literatur- und Quellenverzeichnis Khosroshahi A, Bloch DB, Deshpande V, Stone JH. 2010. Rituximab therapy leads to rapid decline of serum IgG4 levels and prompt clinical improvement in IgG4related systemic disease. Arthritis Rheum, 62 (6):1755-1762. Khosroshahi A, Carruthers MN, Deshpande V, Unizony S, Bloch DB, Stone JH. 2012. Rituximab for the treatment of IgG4-related disease: lessons from 10 consecutive patients. Medicine (Baltimore), 91 (1):57-66. Kiely PDW, Helbert MR, Miles J, Oliveira DBG. 2000. Immunosuppressant effect of gold on IgG subclasses and IgE; evidence for sparing of Th2 responses. Clin Exp Immunol, 120 (2):369-374. Klareskog L, Malmström V, Lundberg K, Padyukov L, Alfredsson L. 2011. Smoking, citrullination and genetic variability in the immunopathogenesis of rheumatoid arthritis. Semin Immunol, 23 (2):92-98. Klein F, Skvaril F, Vermeeren R, Vlug A, Duimel WJ. 1985. The quantification of human IgG subclasses in reference preparations. Clin Chim Acta, 150 (2):119127. Laan RF, Jansen TL, van Riel PL. 1999. Glucocorticosteroids in the management of rheumatoid arthritis. Rheumatology (Oxford), 38 (1):6-12. Leddy JP, Deitchman J, Bakemeier RF. 1970. IgG subclasses measurement of radioimmunoassay in normal and hypogammaglobulinaemic sera. Arthritis Rheum, 13:331-332. Lee DM, Weinblatt ME. 2001. Rheumatoid arthritis. Lancet, 358 (9285):903-911. Lin G, Li J. 2009. IgG subclass serum levels in systemic lupus erythematosus patients. Best Pract Res Clin Rheumatol, 28 (11):1315-1318. Lin G, Li J. 2010. Elevation of serum IgG subclass concentration in patients with rheumatoid arthritis. Rheumatol Int, 30 (6):837-840. ListÌ F, Candore G, Modica MA, Russo M, Lorenzo GD, Esposito-Pellitteri M, Colonna-Romano G, Aquino A, Bulati M, Lio D, Franceschi C, Caruso C. 2006. A study of serum immunoglobulin levels in elderly persons that provides new insights into B cell immunosenescence. Ann NY Acad Sci, 1089 (1):487495. Liu F, Wang Q, Qiu YR, Li MM. 2003. Analysis of the relation between serum immunoglobulin and auto-antibody levels in patients with rheumatoid arthritis. Di Yi Jun Yi Da Xue Xue Bao, 23 (5):472-473. Liu Y, Li J. 2011. Preferentially immunoglobulin (IgG) subclasses production in primary Sjogren's syndrome patients. Clin Chem Lab Med, Epub ahead of print. Lock RJ, Unsworth DJ. 2003. Immunoglobulins and immunoglobulin subclasses in the elderly. Ann Clin Biochem, 40 (Pt 2):143-148. Lorenz HM. 2012. Rheumatoid arthritis: diagnostics and therapy 2012. Orthopade, 41 (7):514-519. Madassery JV, Kwon OH, Lee SY, Nahm MH. 1988. IgG2 subclass deficiency: IgG subclass assays and IgG2 concentrations among 8015 blood donors. Clin Chem, 34 (7):1407-1413. Mannhardt-Laakmann W, Wahn V. 2011. Impfungen bei primären Immundefekten. Monatsschrift Kinderheilkunde, 159 (5):451-460. Marcolongo R, Jr., Carcassi A, Frullini F, Bianco G, Bravi A. 1967. Levels of serum immunoglobulins in patients with rheumatoid arthritis. Ann Rheum Dis, 26 (5):412-418. Martini A, Plebani A, Ravelli A, Avanzini MA, Jefferis R, Zonta L, Notarangelo LD, Ugazio AG, Burgio R. 1986. IgG subclass serum levels in juvenile chronic arthritis. Ann Rheum Dis, 45 (5):400-404. 87 8 Literatur- und Quellenverzeichnis Matt P, Lindqvist U, Rönnelid J, Kleinau S. 2013. Effects of anti-rheumatic treatment on antibody levels and monocytic Fc receptor status in early RA. Ann Rheum Dis, 72 (3): 816. McIntyre TM, Klinman DR, Rothman P, Lugo M, Dasch JR, Mond JJ, Snapper CM. 1993. Transforming growth factor beta 1 selectivity stimulates immunoglobulin G2b secretion by lipopolysaccharide-activated murine B cells. J Exp Med, 177 (4):1031-1037. McKenna RW, Washington LT, Aquino DB, Picker LJ, Kroft SH. 2001. Immunophenotypic analysis of hematogones (B-lymphocyte precursors) in 662 consecutive bone marrow specimens by 4-color flow cytometry. Blood, 98 (8):2498-2507. Morell A, Skavaril F. 1971. A modified radioimmunoassay for quantitative determination of IgG subclasses in man. Prot Biol Fluids, 19:533-540. Morris AJ, Madhok R, Sturrock RD, Capell HA, MacKenzie JF. 1991. Enteroscopic diagnosis of small bowel ulceration in patients receiving non-steroidal antiinflammatory drugs. Lancet, 337 (8740):520. Murphy KM, Travers P, Walport M, Seidler L, Hauaer-Siller I. 2009. Janeway Immunologie. Spektrum Akademischer Verlag. Naumann L, Feist E, Burmester GR. 2013. Antizytokintherapie. Internist, 54 (4):449463. Neumann C, Proft F, Hammitzsch A, Gruenke M, Schulze-Koops H. 2013. Hypogammaglobulinemia is a frequent finding in patients under immunosuppressive therapy but does not correlate with susceptibility to infections. Ann Rheum Dis, 72 (3):338. Nelson DL, Cox MM. 2010. Lehninger Principles of Biochemistry. W. H. Freeman. Nielen MM, van Schaardenburg D, Reesink HW, van de Stadt RJ, van der HorstBruinsma IE, de Koning MH, Habibuw MR, Vandenbroucke JP, Dijkmans BA. 2004. Specific autoantibodies precede the symptoms of rheumatoid arthritis: a study of serial measurements in blood donors. Arthritis Rheum, 50 (2):380386. Nishimura K, Sugiyama D, Kogata Y, Tsuji G, Nakazawa T, Kawano S, Saigo K, Morinobu A, Koshiba M, Kuntz KM, Kamae I, Kumagai S. 2007. Meta-analysis: diagnostic accuracy of anti-cyclic citrullinated peptide antibody and rheumatoid factor for rheumatoid arthritis. Ann Intern Med, 146 (11):797-808. Ocampo CJ, Peters AT. 2013. Antibody deficiency in chronic rhinosinusitis: epidemiology and burden of illness. Am J Rhinol Allergy, 27 (1):34-38. Ostergaard M, Pedersen SJ, Dohn UM. 2008. Imaging in rheumatoid arthritis--status and recent advances for magnetic resonance imaging, ultrasonography, computed tomography and conventional radiography. Best Pract Res Clin Rheumatol, 22 (6):1019-1044. Oxelius V. 1978. Crossed immunoelectrophoresis and electroimmunoassay of human IgG subclasses. Acta Pathol Microbiol Scand C, 86C (3):109-116. Oxelius VA, Hanson LA, Bjorkander J, Hammarstrom L, Sjoholm A. 1986. IgG3 deficiency: common in obstructive lung disease. Hereditary in families with immunodeficiency and autoimmune disease. Monogr Allergy, 20:106-115. Pappas DA, John A, Curtis JR, Kremer J, Reiss W, Shewade A, Silverman GJ, Greenberg J. 2013. Prevalence of low immunoglobulin levels and associations with rheumatoid arthritis factors. Ann Rheum Dis, 72 (3): 448. Pawelec G, Barnett Y, Forsey R, Frasca D, Globerson A, McLeod J, Caruso C, Franceschi C, Fulop T, Gupta S, Mariani E, Mocchegiani E, Solana R. 2002. T cells and aging, January 2002 update. Front Biosci, 7:d1056-1183. 88 8 Literatur- und Quellenverzeichnis Peng SL, Szabo SJ, Glimcher LH. 2002. T-bet regulates IgG class switching and pathogenic autoantibody production. Proc Natl Acad Sci U S A, 99 (8):55455550. Perdriger A, Chalès G. 1997. Influence of non HLA factors in rheumatoid arthritis: role of enzyme abnormalities in joint destruction. Rev Rhum Engl Ed, 64 (10):523-526. Pfluger H, Helbling A, Mordasini C, Pichler WJ. 2000. CVID (common variable immunodeficiency): heterogeneous clinical manifestation of the commonest symptomatic primary immunodeficiency disease. Schweiz Med Wochenschr, 130 (43):1590-1599. Prevoo ML, van 't Hof MA, Kuper HH, van Leeuwen MA, van de Putte LB, van Riel PL. 1995. Modified disease activity scores that include twenty-eight-joint counts. Development and validation in a prospective longitudinal study of patients with rheumatoid arthritis. Arthritis Rheum, 38 (1):44-48. Pyne DB, Gleeson M. 1998. Effects of intensive exercise training on immunity in athletes. Int J Sports Med, 19 Suppl 3:S183-191; discussion S191-194. Qvarfordt I, Riise GC, Andersson BA, Larsson S. 2001. IgG subclasses in smokers with chronic bronchitis and recurrent exacerbations. Thorax, 56 (6):445-449. Radl J, Sepers JM, Skvaril F, Morell A, Hijmans W. 1975. Immunoglobulin patterns in humans over 95 years of age. Clin Exp Immunol, 22 (1):84-90. Raychaudhuri S. 2010. Recent advances in the genetics of rheumatoid arthritis. Curr Opin Rheumatol, 22 (2):109-118. Roseman C, Truedsson L, Kapetanovic MC. 2012. The effect of smoking and alcohol consumption on markers of systemic inflammation, immunoglobulin levels and immune response following pneumococcal vaccination in patients with arthritis. Arthritis Res Ther, 14 (4):R170. Rothschild BM, Turner KR, DeLuca MA. 1988. Symmetrical erosive peripheral polyarthritis in the Late Archaic Period of Alabama. Science, 241 (4872):14981501. Schauer U, Stemberg F, Rieger CH, Borte M, Schubert S, Riedel F, Herz U, Renz H, Wick M, Carr-Smith HD, Bradwell AR, Herzog W. 2003. IgG subclass concentrations in certified reference material 470 and reference values for children and adults determined with the binding site reagents. Clin Chem, 49 (11):1924-1929. Schmidt RE, Baumann U, Boztug K, Hennig C, Stoll M, Witte T. 2011. Humorale Immundefekte. Primäre und sekundäre Immundefekte. Bremen: Uni-MedVerlag. Schneider M, Lelgemann M, Abholz HH, Blumenroth M, Flügge C, Gerken M, Jäniche H, Kunz R, Krüger K, Mau W, Specker C, Zellner M. 2011. Interdisziplinäre Leitlinie - Management der frühen rheumatoiden Arthritis. Springer-Verlag Berlin Heidelberg New York. Schur PH, Borel H, Gelfand EW, Alper CA, Rosen FS. 1970. Selective gamma-g globulin deficiencies in patients with recurrent pyogenic infections. N Engl J Med, 283 (12):631-634. Shaffu S, Royle JG, Neame R. 2013. Rituximab and immunoglobulin level monitoring in rheumatoid arthritis – a clinical audit. Ann Rheum Dis, 72 (3):888. Shakib F, Stanworth DR, Drew R, Catty D. 1975. A quantitative study of the distribution of IgG sub-classes in a group of normal human sera. J Immunol Methods, 8 (1-2):17-28. Shakib F, Stanworth DR. 1976. IgG subclass composition of rheumatoid arthritic sera and joint fluids. Ann Rheum Dis, 35 (3):263-266. 89 8 Literatur- und Quellenverzeichnis Silman AJ, Pearson JE. 2002. Epidemiology and genetics of rheumatoid arthritis. Arthritis Res, 4 Suppl 3:S265-272. Snapper CM, McIntyre TM, Mandler R, Pecanha LM, Finkelman FD, Lees A, Mond JJ. 1992. Induction of IgG3 secretion by interferon gamma: a model for T cellindependent class switching in response to T cell-independent type 2 antigens. J Exp Med, 175 (5):1367-1371. Sokka T, Pincus T. 2009. Erythrocyte sedimentation rate, C-reactive protein, or rheumatoid factor are normal at presentation in 35%-45% of patients with rheumatoid arthritis seen between 1980 and 2004: analyses from Finland and the United States. J Rheumatol, 36 (7):1387-1390. Soranus. 1950. On acute diseases and on chronic diseases. Translated into Latin by Caelius Aurelianus (5th century). English translation by Drabkin IE. Chicago, University of Chicago Press. Spinozzi F, Cimignoli E, Gerli R, Agea E, Bertotto A, Rondoni F, Grignani F. 1992. IgG subclass deficiency and sinopulmonary bacterial infections in patients with alcoholic liver disease. Arch Intern Med, 152 (1):99-104. Terry WD, Matthews BW, Davies DR. 1968. Crystallographic studies of a human immunoglobulin. Nature, 220 (5164):239-241. Tobon GJ, Youinou P, Saraux A. 2010. The environment, geo-epidemiology, and autoimmune disease: Rheumatoid arthritis. Autoimmun Rev, 9 (5):A288-292. Tony HP. 2010. Langzeittherapie der Rheumatoiden Arthrtitis. Bremen: Uni-MedVerlag. Tsuchiya N, Endo T, Shiota M, Kochibe N, Ito K, Kobata A. 1994. Distribution of glycosylation abnormality among serum IgG subclasses from patients with rheumatoid arthritis. Clin Immunol Immunopathol, 70 (1):47-50. van der Giessen M, Rossouw E, van Veen TA, van Loghem E, Zegers BJ, Sander PC. 1975. Quantification of IgG subclasses in sera of normal adults and healthy children between 4 and 12 years of age. Clin Exp Immunol, 21 (3):501-509. van der Kooij SM, de Vries-Bouwstra JK, Goekoop-Ruiterman YP, Ewals JA, Han KH, Hazes JM, Kerstens PJ, Peeters AJ, van Zeben D, Breedveld FC, Huizinga TW, Dijkmans BA, Allaart CF. 2009. Patient-reported outcomes in a randomized trial comparing four different treatment strategies in recent-onset rheumatoid arthritis. Arthritis Rheum, 61 (1):4-12. van Vollenhoven RF, Emery P, Bingham CO, 3rd, Keystone EC, Fleischmann R, Furst DE, Macey K, Sweetser M, Kelman A, Rao R. 2010. Longterm safety of patients receiving rituximab in rheumatoid arthritis clinical trials. J Rheumatol, 37 (3):558-567. Veys EM, Claessens HE. 1968. Serum levels of IgG, IgM, and IgA in rheumatoid arthritis. Ann Rheum Dis, 27 (5):431-440. Vlug A, Nieuwenhuys EJ, van Eijk RV, Geertzen HG, van Houte AJ. 1994. Nephelometric measurements of human IgG subclasses and their reference ranges. Ann Biol Clin (Paris), 52 (7-8):561-567. Vollmar A, Zündorf I, Dingermann T. 2013. Immunologie: Grundlagen und Wirkstoffe. Wiss.Verlag-Ges. Wedgwood R, Ochs H, Oxelius V. 1984. IgG subclass levels in the serum of patients with primary immunodeficiency. Am J Med, 76 (3, Part 1):219-231. Wells G, Becker JC, Teng J, Dougados M, Schiff M, Smolen J, Aletaha D, van Riel PL. 2009. Validation of the 28-joint Disease Activity Score (DAS28) and European League Against Rheumatism response criteria based on C-reactive protein against disease progression in patients with rheumatoid arthritis, and 90 8 Literatur- und Quellenverzeichnis comparison with the DAS28 based on erythrocyte sedimentation rate. Ann Rheum Dis, 68 (6):954-960. Whiting PF, Smidt N, Sterne JA, Harbord R, Burton A, Burke M, Beynon R, BenShlomo Y, Axford J, Dieppe P. 2010. Systematic review: accuracy of anticitrullinated peptide antibodies for diagnosing rheumatoid arthritis. Ann Intern Med, 152 (7):456-464. Wiles NJ, Lunt M, Barrett EM, Bukhari M, Silman AJ, Symmons DP, Dunn G. 2001. Reduced disability at five years with early treatment of inflammatory polyarthritis: results from a large observational cohort, using propensity models to adjust for disease severity. Arthritis Rheum, 44 (5):1033-1042. Wolfe F, Mitchell DM, Sibley JT, Fries JF, Bloch DA, Williams CA, Spitz PW, Haga M, Kleinheksel SM, Cathey MA. 1994. The mortality of rheumatoid arthritis. Arthritis Rheum, 37 (4):481-494. 91 9 Anhang 9 Anhang Regressionen in Gruppen Tabelle 34: Regressionsanalyse mit IgM als abhängige Variable p-Wert. Odds Ratio 95% Konfidenzintervall der Odds Ratio Unterer Wert Oberer Wert 0 1,039 1,019 1,06 0,053 1,586 0,994 2,532 Raucher 0,153 0,644 0,352 1,178 ehemalige Raucher 0,196 0,425 0,116 1,553 Krankheitsdauer (in Jahren) 0,018 0,967 0,941 0,994 erosiv 0,597 0,88 0,548 1,412 beginnend erosiv 0,524 1,314 0,567 3,044 BSG (in mm/h) 0,847 0,999 0,985 1,012 CRP (in g/l) 0,211 1,011 0,994 1,028 VAS 0,431 0,993 0,976 1,01 DAS 28 mit BSG 0,454 0,911 0,714 1,163 DAS 28 mit CRP 0,203 1,117 0,942 1,326 RF IgM (in IU/ml) 0,006 1,929 1,209 3,077 RF IgG (in RU/ml) 0,601 1,349 0,439 4,145 RF IgA (in RU/ml) 0,088 1,607 0,932 2,773 Anti-CCP-AK (in RU/ml) 0,46 0,999 0,995 1,002 IgA (in g/l) 0,038 0,821 0,682 0,989 IgG (in g/l) 0,003 0,885 0,816 0,959 IgG1 (in g/l) 0,042 0,893 0,8 0,996 IgG2 (in g/l) 0,103 0,866 0,729 1,029 IgG3 (in g/l) 0,482 0,796 0,422 1,502 IgG4 (in g/l) 0,39 1,212 0,782 1,88 Biologika 0,259 0,72 0,406 1,275 Anzahl an Medikamenten 0,393 0,901 0,71 1,144 Infektionen 0,607 0,898 0,594 1,355 Alter (in Jahren) Geschlecht männlich Raucherstatus Röntgenstadium 92 9 Anhang Tabelle 35: Regressionsanalyse mit IgA als abhängige Variable p-Wert. Odds Ratio 95% Konfidenzintervall der Odds Ratio Unterer Wert Oberer Wert Alter (in Jahren) 0,649 0,993 0,961 1,025 Geschlecht männlich 0,028 0,193 0,044 0,835 Raucher 0,795 1,138 0,429 3,017 ehemalige Raucher 0,998 0,000 0,000 Krankheitsdauer (in Jahren) 0,713 0,991 0,944 1,041 erosiv 0,659 0,828 0,358 1,915 beginnend erosiv 0,998 0,000 0,000 BSG (in mm/h) 0,464 0,988 0,957 1,020 CRP (in g/l) 0,257 0,963 0,902 1,028 VAS 0,690 1,006 0,977 1,036 DAS 28 mit BSG 0,366 1,234 0,782 1,948 DAS 28 mit CRP 0,188 1,235 0,902 1,693 RF IgM (in IU/ml) 0,296 1,607 0,660 3,909 RF IgG (in RU/ml) 0,998 RF IgA (in RU/ml) 0,001 10,680 2,557 44,608 Anti-CCP-AK (in RU/ml) 0,090 1,007 0,999 1,015 IgG (in g/l) 0,001 0,755 0,639 0,893 IgM (in g/l) 0,306 0,704 0,360 1,378 IgG1 (in g/l) 0,371 0,906 0,730 1,125 IgG2 (in g/l) 0,004 0,547 0,363 0,824 IgG3 (in g/l) 0,195 2,088 0,686 6,356 IgG4 (in g/l) 0,302 0,444 0,095 2,075 Biologika 0,693 0,818 0,302 2,214 Anzahl an Medikamenten 0,405 1,213 0,770 1,911 Infektionen 0,962 0,982 0,459 2,101 Raucherstatus Röntgenstadium 93 9 Anhang Tabelle 36: Regression mit IgG3 als abhängige Variable 95% Konfidenzintervall der Odds Ratio p-Wert. Odds Ratio Unterer Wert Oberer Wert Alter (in Jahren) 0,312 0,984 0,953 1,015 Geschlecht männlich 0,700 1,183 0,504 2,772 Raucher 0,476 0,686 0,244 1,931 ehemalige Raucher 0,395 1,996 0,407 9,795 Krankheitsdauer (in Jahren) 0,047 1,035 1,000 1,072 erosiv 0,682 1,191 0,516 2,750 beginnend erosiv 0,799 1,225 0,258 5,817 BSG (in mm/h) 0,643 1,006 0,982 1,029 CRP (in g/l) 0,419 1,012 0,983 1,041 VAS 0,349 0,983 0,949 1,019 DAS 28 mit BSG 0,969 0,991 0,627 1,567 DAS 28 mit CRP 0,880 1,025 0,746 1,408 RF IgM (in IU/ml) 0,301 1,592 0,659 3,848 RF IgG (in RU/ml) 0,998 RF IgA (in RU/ml) 0,733 0,841 0,311 2,275 Anti-CCP-AK (in RU/ml) 0,363 0,997 0,991 1,003 IgA 0,545 1,101 0,807 1,502 IgG 0,252 0,919 0,794 1,062 IgM 0,115 1,257 0,946 1,672 IgG1 0,351 1,081 0,918 1,272 IgG4 0,445 1,355 0,621 2,959 IgG2 0,001 0,494 0,328 0,742 NSAR 0,997 1,002 0,444 2,261 COX-Hemmer 0,108 0,344 0,094 1,264 Kortison 0,268 0,648 0,301 1,396 DMARD 0,194 2,058 0,693 6,111 Biologika 0,384 1,560 0,573 4,241 Anzahl an Medikamenten 0,406 1,383 0,643 2,975 Infektionen 0,930 1,021 0,640 1,630 Raucherstatus Röntgenstadium 94 9 Anhang Tabelle 37: Regression mit IgG4 als abhängige Variable p-Wert. 95% Konfidenzintervall der Odds Ratio Odds Ratio Unterer Wert Oberer Wert Alter (in Jahren) 0,226 1,016 0,99 1,042 Geschlecht männlich 0,164 0,593 0,284 1,239 Raucher 0,299 0,634 0,268 1,498 ehemalige Raucher 0,526 0,512 0,065 4,049 Krankheitsdauer (in Jahren) 0,538 0,99 0,957 1,023 erosiv 0,436 1,282 0,686 2,396 beginnend erosiv 0,524 0,615 0,138 2,745 BSG (in mm/h) 0,799 1,003 0,983 1,022 CRP (in g/l) 0,278 0,981 0,948 1,016 VAS 0,179 0,982 0,956 1,008 DAS 28 mit BSG 0,388 1,162 0,827 1,633 DAS 28 mit CRP 0,372 1,115 0,878 1,417 RF IgM (in IU/ml) 0,495 1,26 0,649 2,445 RF IgG (in RU/ml) 0,998 RF IgA (in RU/ml) 0,161 1,743 0,802 3,786 Anti-CCP-AK (in RU/ml) 0,977 1 0,995 1,005 IgA 0,014 0,688 0,512 0,926 IgG 0,001 0,796 0,698 0,908 IgM 0,403 0,836 0,549 1,272 ÍgG1 0,14 0,883 0,748 1,042 IgG2 0,000 0,493 0,363 0,671 IgG3 0,094 2,101 0,881 5,011 NSAR 0,229 1,488 0,779 2,844 COX-Hemmer 0,408 0,697 0,296 1,639 Kortison 0,26 1,443 0,762 2,734 DMARD 0,034 2,451 1,07 5,616 Biologika 0,071 0,445 0,185 1,072 Anzahl an Medikamenten 0,406 1,383 0,643 2,975 Infektionen 0,93 1,021 0,64 1,63 Raucherstatus Röntgenstadium 95 9 Anhang Tabelle 38: Regression mit IgG als abhängige Variable und der BiologikaEinnahme 95 % KI der Odds Ratio p-Wert Odds Ratio Unterer Wert Oberer Wert Adalimumab 0,036 0,118 0,016 0,873 Tozilizumab 0,447 1,389 0,596 3,236 Etanercept 0,787 0,858 0,282 2,611 Certolizumab 0,196 0,260 0,034 2,006 Rituximab 0,999 0,000 0,000 Infliximab 1,000 0,000 0,000 Abatacept 1,000 0,000 0,000 Golimumab 1,000 0,000 0,000 Referenzvariable: Patienten ohne Biologikum Tabelle 39: Regression mit IgG1 als abhängige Variable und der BiologikaEinnahme p-Wert Odds Ratio Adalimumab 0,001 Tozilizumab 0,798 Etanercept 95 % KI der Odds Ratio Unterer Wert Oberer Wert 0,169 0,058 0,490 1,103 0,520 2,341 0,266 0,591 0,234 1,493 Certolizumab 0,026 0,182 0,041 0,815 Rituximab 0,374 0,473 0,091 2,463 Infliximab 1,000 0,000 0,000 Abatacept 1,000 0,000 0,000 Golimumab 1,000 0,000 0,000 Referenzvariable: Patienten ohne Biologikum Tabelle 40: Regression mit IgG2 als abhängige Variable und der BiologikaEinnahme p-Wert Odds Ratio Adalimumab 0,397 Tozilizumab Etanercept 95 % KI der Odds Ratio Unterer Wert Oberer Wert 0,417 0,055 3,160 0,202 2,070 0,677 6,324 0,687 1,362 0,303 6,115 Certolizumab 0,940 0,924 0,118 7,260 Rituximab 0,999 0,000 0,000 Infliximab 1,000 0,000 0,000 Abatacept 1,000 0,000 0,000 Golimumab 1,000 0,000 0,000 Referenzvariable: Patienten ohne Biologikum 96 Anhang Tabelle 41: Regression mit IgG3 als abhängige Variable und der BiologikaEinnahme p-Wert Odds Ratio Adalimumab 0,243 Tozilizumab 95 % KI der Odds Ratio Unterer Wert Oberer Wert 2,131 0,598 7,593 0,582 1,526 0,339 6,872 Etanercept 0,978 1,030 0,132 8,064 Certolizumab 0,146 3,169 0,669 15,005 Rituximab 0,264 3,433 0,394 29,892 Infliximab 1,000 0,000 0,000 Abatacept 1,000 0,000 0,000 Golimumab 1,000 0,000 0,000 Referenzvariable: Patienten ohne Biologikum Tabelle 42: Regression mit IgG4 als abhängige Variable und der BiologikaEinnahme p-Wert Odds Ratio Adalimumab 0,187 Tozilizumab 0,223 Etanercept 95 % KI der Odds Ratio Unterer Wert Oberer Wert 0,258 0,034 1,933 0,286 0,038 2,148 0,272 1,882 0,608 5,826 Certolizumab 0,593 0,571 0,074 4,442 Rituximab 0,792 1,333 0,157 11,312 Infliximab 1,000 0,000 0,000 Abatacept 1,000 0,000 0,000 Golimumab 1,000 0,000 0,000 Referenzvariable: Patienten ohne Biologikum 97 9 Anhang Regressionen mit dem Einfluss aller erhobenen Variablen Tabelle 43: Gesamtregression mit IgM als abhängige Variable IgM 95% Konfidenzintervall der Odds Ratio p-Wert Odds Ratio Unterer Wert Oberer Wert Alter 0,001 1,036 1,014 1,059 Geschlecht 0,142 1,454 0,882 2,395 Raucher 0,532 0,814 0,427 1,552 Krankheitsdauer 0,092 0,975 0,947 1,004 Röntgenstadium erosiv 0,822 1,062 0,628 1,797 Röntgenstadium beginnend erosiv 0,475 1,419 0,543 3,708 BSG 0,425 1,007 0,991 1,023 CRP 0,486 1,007 0,988 1,026 VAS 0,201 0,987 0,968 1,007 DAS 28 0,659 0,941 0,719 1,232 IgA 0,166 0,858 0,691 1,066 IgG 0,298 0,927 0,805 1,069 IgG1 0,882 1,012 0,862 1,188 IgG2 0,525 0,932 0,751 1,157 IgG3 0,441 1,320 0,652 2,672 IgG4 0,292 1,307 0,794 2,153 RF IgM 0,002 0,462 0,284 0,750 Anti-CCP-AK 0,020 0,996 0,993 0,999 NSAR 0,640 0,853 0,439 1,658 Kortison 0,932 0,974 0,536 1,772 DMARD 0,748 1,120 0,561 2,234 Biologika 0,929 1,034 0,499 2,140 Anzahl an Medikamenten 0,664 0,838 0,377 1,862 Infektionen 0,578 1,150 0,704 1,878 98 9 Anhang Tabelle 44: Gesamtregression mit IgA als abhängige Variable IgA p-Wert Odds Ratio Alter 0,157 Geschlecht 95% Konfidenzintervall der Odds Ratio Unterer Wert Oberer Wert 0,971 0,932 1,011 0,015 0,147 0,031 0,690 Raucher 0,895 1,081 0,339 3,446 Krankheitsdauer 0,988 1,000 0,947 1,057 Röntgenstadium erosiv 0,974 1,017 0,357 2,897 Röntgenstadium beginnend erosiv 0,998 0,000 0,000 BSG 0,225 0,977 0,941 1,014 CRP 0,484 0,974 0,905 1,049 VAS 0,761 1,005 0,971 1,041 DAS 28 0,255 1,359 0,801 2,305 IgM 0,690 0,839 0,353 1,992 IgG 0,197 0,798 0,566 1,124 IgG1 0,393 1,179 0,808 1,721 IgG2 0,062 0,599 0,349 1,027 IgG3 0,119 2,848 0,764 10,610 IgG4 0,748 0,754 0,135 4,224 RF IgM 0,010 0,266 0,097 0,724 Anti-CCP-AK 0,491 0,998 0,991 1,004 NSAR 0,020 0,184 0,044 0,762 Kortison 0,798 1,175 0,343 4,020 DMARD 0,804 0,828 0,187 3,667 Biologika 0,456 0,575 0,134 2,464 Anzahl an Medikamenten 0,095 4,067 0,784 21,094 Infektionen 0,204 0,519 0,189 1,429 99 9 Anhang Tabelle 45: Gesamtregression mit IgG als abhängige Variable IgG p-Wert Odds Ratio Alter 0,511 Geschlecht 95% Konfidenzintervall der Odds Ratio Unterer Wert Oberer Wert 1,011 0,979 1,044 0,669 1,174 0,564 2,444 Raucher 0,789 1,126 0,472 2,690 Krankheitsdauer 0,982 1,000 0,963 1,038 Röntgenstadium erosiv 0,213 1,626 0,756 3,495 Röntgenstadium beginnend erosiv 0,091 0,220 0,038 1,273 BSG 0,354 1,012 0,987 1,038 CRP 0,577 0,991 0,960 1,023 VAS 0,869 1,002 0,976 1,029 DAS 28 0,598 1,108 0,758 1,619 IgA 0,059 0,727 0,522 1,013 IgM 0,455 0,873 0,612 1,246 IgG1 0,000 0,330 0,237 0,458 IgG2 0,000 0,267 0,172 0,415 IgG3 0,928 0,943 0,266 3,343 IgG4 0,286 0,472 0,119 1,875 RF IgM 0,415 0,750 0,375 1,498 Anti-CCP-AK 0,056 0,995 0,991 1,000 NSAR 0,363 0,619 0,221 1,739 Kortison 0,551 1,323 0,528 3,314 DMARD 0,869 0,918 0,331 2,544 Biologika 0,753 0,838 0,280 2,511 Anzahl an Medikamenten 0,425 1,624 0,494 5,342 Infektionen 0,077 1,845 0,936 3,636 100 9 Anhang Tabelle 46: Gesamtregression mit IgG1 als abhängige Variable 95% Konfidenzintervall der Odds Ratio IgG1 p-Wert Odds Ratio Unterer Wert Oberer Wert Alter 0,688 1,006 0,979 1,033 Geschlecht 0,717 1,131 0,582 2,197 Raucher 0,759 1,13 0,518 2,464 Krankheitsdauer 0,94 0,999 0,966 1,032 Röntgenstadium erosiv 0,053 0,516 0,263 1,009 Röntgenstadium beginnend erosiv 0,416 1,66 0,489 5,633 BSG 0,132 0,983 0,961 1,005 CRP 0,724 1,005 0,979 1,032 VAS 0,165 1,016 0,994 1,039 DAS 28 0,849 0,97 0,706 1,332 IgA 0,688 1,062 0,791 1,427 IgM 0,867 0,971 0,688 1,371 IgG 0,000 0,24 0,178 0,323 IgG2 0,000 2,676 1,887 3,795 IgG3 0,000 0,094 0,033 0,272 IgG4 0,129 0,523 0,226 1,209 RF IgM 0,823 1,074 0,575 2,006 Anti-CCP-AK 0,029 1,004 1,000 1,008 NSAR 0,108 0,501 0,216 1,163 Kortison 0,619 1,229 0,546 2,764 DMARD 0,317 0,65 0,28 1,512 Biologika 0,178 0,403 0,107 1,511 Anzahl an Medikamenten 0,404 0,648 0,233 1,797 Infektionen 0,505 0,806 0,428 1,519 101 9 Anhang Tabelle 47: Gesamtregression mit IgG2 als abhängige Variable IgG2 p-Wert Odds Ratio Alter 0,092 Geschlecht 95% Konfidenzintervall der Odds Ratio Unterer Wert Oberer Wert 1,039 0,994 1,087 0,038 2,697 1,054 6,898 Raucher 0,039 2,818 1,053 7,542 Krankheitsdauer 0,956 0,999 0,953 1,046 Röntgenstadium erosiv 0,332 1,648 0,600 4,523 Röntgenstadium beginnend erosiv 0,994 1,009 0,095 10,704 BSG 0,030 0,956 0,917 0,996 CRP 0,529 0,984 0,935 1,035 VAS 0,539 0,987 0,946 1,029 DAS 28 0,058 1,661 0,983 2,806 IgA 0,961 1,009 0,697 1,461 IgM 0,518 0,785 0,377 1,634 IgG 0,000 0,417 0,287 0,605 IgG1 0,007 1,751 1,165 2,634 IgG3 0,012 0,099 0,016 0,596 IgG4 0,138 0,184 0,020 1,719 RF IgM 0,438 1,465 0,558 3,846 Anti-CCP-AK 0,405 0,997 0,991 1,004 NSAR 0,585 0,692 0,185 2,594 Kortison 0,115 0,388 0,120 1,260 DMARD 0,497 1,678 0,376 7,482 Biologika 0,485 1,608 0,424 6,104 Anzahl an Medikamenten 0,622 1,502 0,298 7,570 Infektionen 0,202 1,793 0,731 4,399 102 9 Anhang Tabelle 48: Gesamtregression mit IgG3 als abhängige Variable IgG3 p-Wert Odds Ratio Alter 0,283 Geschlecht 95% Konfidenzintervall der Odds Ratio Unterer Wert Oberer Wert 0,980 0,945 1,017 0,424 1,469 0,572 3,774 Raucher 0,222 0,475 0,144 1,567 Krankheitsdauer 0,108 1,034 0,993 1,076 Röntgenstadium erosiv 0,951 1,031 0,387 2,747 Röntgenstadium beginnend erosiv 0,887 1,137 0,192 6,723 BSG 0,390 1,012 0,985 1,041 CRP 0,806 0,995 0,959 1,033 VAS 0,554 0,988 0,950 1,028 DAS 28 0,668 0,894 0,537 1,489 IgA 0,236 1,249 0,865 1,803 IgM 0,066 1,424 0,977 2,076 IgG 0,213 1,224 0,891 1,682 IgG1 0,538 0,899 0,639 1,263 IgG2 0,000 0,378 0,230 0,621 IgG4 0,516 1,342 0,553 3,258 RF IgM 0,063 0,414 0,163 1,048 Anti-CCP-AK 0,272 0,996 0,990 1,003 NSAR 0,312 0,555 0,178 1,737 Kortison 0,062 0,358 0,122 1,052 DMARD 0,295 2,098 0,524 8,407 Biologika 0,577 1,414 0,419 4,767 Anzahl an Medikamenten 0,374 1,955 0,446 8,564 Infektionen 0,271 1,629 0,683 3,882 103 9 Anhang Tabelle 49: Gesamtregression mit IgG4 als abhängige Variable IgG4 p-Wert Odds Ratio Alter 0,647 Geschlecht 95% Konfidenzintervall der Odds Ratio Unterer Wert Oberer Wert 1,007 0,977 1,039 0,241 0,619 0,278 1,381 Raucher 0,115 0,452 0,169 1,212 Krankheitsdauer 0,677 0,992 0,956 1,030 Röntgenstadium erosiv 0,187 1,656 0,783 3,500 Röntgenstadium beginnend erosiv 0,482 0,557 0,109 2,839 BSG 0,969 1,000 0,977 1,024 CRP 0,458 0,986 0,948 1,024 VAS 0,158 0,977 0,946 1,009 DAS 28 0,583 1,118 0,751 1,663 IgA 0,044 0,710 0,509 0,990 IgM 0,243 0,695 0,377 1,281 IgG 0,767 0,958 0,724 1,270 IgG1 0,792 0,960 0,708 1,301 IgG2 0,009 0,571 0,376 0,867 IgG3 0,060 2,580 0,959 6,940 RF IgM 0,659 0,852 0,418 1,737 Anti-CCP-AK 0,350 0,998 0,993 1,002 NSAR 0,447 0,668 0,236 1,890 Kortison 0,300 0,622 0,253 1,526 DMARD 0,808 0,869 0,278 2,713 Biologika 0,256 0,538 0,184 1,567 Anzahl an Medikamenten 0,027 4,055 1,175 13,992 Infektionen 0,025 2,161 1,103 4,233 104 9 Anhang Brief abstract for the American College of Rheumatology 2013: Deficiencies of IgG and its subclasses in patients suffering from rheumatoid arthritis Sara Kaestnera,b, Peter Kaestnera, Hans-Juergen Wolffa, Rolf Braeuerb Affiliation(s): a Ambulantes b Rheumazentrum Erfurt, Germany Institute of Pathology, University Hospital Jena, Germany Keywords: IgG-subclasses; rheumatoid arthritis; deficiency; humoral immune system; infections Abstract: Background: The occurrence and clinical significance of IgG subclass deficiencies in rheumatoid arthritis (RA) has not been determined in relation to immune modulatory treatment. The present study aimed therefore to determine IgG subclass deficiencies in patients with RA and the identification of putative contributing factors. Methods: We evaluated IgG subclass levels in blood samples of 539 patients with rheumatoid arthritis by an immunonephelometric assay. In addition, we determined age, gender, smoking habits, duration and activity of the disease (DAS 28), the degree erosions, number of infections as well as inflammatory markers to study an assumed relation to IgG levels. Results: According to laboratory standard values, we found IgG deficiencies in 19.9 % of patients with a mean value (± standard deviation) of 10.4 ± 2.8 g/l. Most frequent deficiencies of subclasses were observed for IgG1 (42.3 %), while less prominent for IgG2 (7.2 %), IgG3 (5.4 %) and IgG4 (10.4 %). Mean values of all patients of IgG-subclasses were 5.6 ± 2.3 g/l for IgG1, 3.2 ± 1.5 g/l for IgG2, 0.7 ± 0.4 g/l for IgG3 and 0.4 ± 0.5 g/l for IgG4. IgG1- and IgG2 deficiencies were more prevalent in older patients. In addition, smoking habits were closely associations with IgG2 deficiency while disease progression was related to IgG1 deficiency. A logistic 105 9 Anhang regression revealed that patients with IgG deficiency have a significant higher risk of infections. Patients presenting with Normal value [in %] Deficiency [in %] Increase [in %] IgG IgG1 IgG2 IgG3 IgG4 78,5 19,9 1,7 54,5 42,3 3,2 89,4 7,2 3,3 83,5 5,4 11,1 84,8 10,4 4,8 Conclusion: An unexpected high occurrence rate of IgG and IgG subclass deficiencies was observed in our patients. We suggest therefore that the assessment of IgG and IgG subclass status should prevail the application of immune modulating drugs to prevent a potential risk of infectious diseases. Prospective studies are needed to investigate both the influence of immune modulating drugs on IgG and IgG subclass levels and on the immune defense reaction. 106 9 Anhang Ehrenwörtliche Erklärung Hiermit erkläre ich, dass mir die Promotionsordnung der Medizinischen Fakultät der Friedrich-Schiller-Universität bekannt ist, ich die Dissertation selbst angefertigt habe und alle von mir benutzten Hilfsmittel, persönlichen Mitteilungen und Quellen in meiner Arbeit angegeben sind, mich folgende Personen bei der Auswahl und Auswertung des Materials sowie bei der Herstellung des Manuskripts unterstützt haben: Prof. Dr. rer. nat. Rolf Bräuer Dr. sc. med. Peter Kästner Dr. med. Rosemarie Kästner Dr. med. habil. H.- Jürgen Wolf Dr. rer. pol. Thomas Lehmann die Hilfe eines Promotionsberaters nicht in Anspruch genommen wurde und dass Dritte weder unmittelbar noch mittelbar geldwerte Leistungen von mir für Arbeiten erhalten haben, die im Zusammenhang mit dem Inhalt der vorgelegten Dissertation stehen, dass ich die Dissertation noch nicht als Prüfungsarbeit für eine staatliche oder andere wissenschaftliche Prüfung eingereicht habe und dass ich die gleiche, eine in wesentlichen Teilen ähnliche oder eine andere Abhandlung nicht bei einer anderen Hochschule als Dissertation eingereicht habe. Jena, den 07.08.2013 Unterschrift des Verfassers 107 9 Anhang Danksagung Mein Dank gilt meinem Doktorvater Prof. Dr. rer. nat. Rolf Bräuer aus dem Institut für Pathologie der Universität Jena für die sehr gute Beratung und Betreuung bei Konzeption sowie Erstellung der Arbeit, auch für die Unterstützung bei der Bewertung und Diskussion der Ergebnisse bedanke ich mich hiermit recht herzlich. Ebenso danke ich den Ärzten des Ambulanten Rheumazentrums Erfurt, insbesondere dem Leiter Dr. sc. med. Peter Kästner, dem Leiter des Labors Herrn Dr. med. habil. Jürgen Wolf, der mir bei labormedizinischen Fragen mit Rat und Tat geholfen hat und Frau Dr. med. Rosemarie Kästner sowie dem Mitarbeiter des statistischen Institutes IMSID der Universität Jena Dr. rer. pol. Thomas Lehmann, der mir in wissenschaftlichen Fragen zur Seite stand. Nicht zuletzt gilt mein sehr herzlicher Dank meinen Eltern und Freunden, die mich auf dem Weg zur Fertigstellung dieser Arbeit begleitet haben. Herzlichen Dank. 108 Anhang Curriculum Vitae Persönliche Daten Name Sara Kästner Anschrift Hinter der Kirche 7 07743 Jena, Deutschland Telefon 0049171/6825828 E-Mail [email protected] Staatsangehörigkeit Deutsch Geburtsdatum 28. August 1988 Geburtsort Heiligenstadt Familienstand ledig Ausbildung 1995 - 1997 Grundschule “Sahlenburg”, Cuxhaven 1997 - 1999 Grundschule “Heinrich Heine”, Uhlstädt 1999 - 2001 Gymnasium “Fridericianum-Gymnasium”, Rudolstadt 2001 - 2008 Sportgymnasium “GutsMuths-Gymnasium” Jena 2008 Abitur seit 2008 Medizinstudium an der Friedrich-Schiller-Universität, Jena 2010 1. Staatsexamen im Rahmen des Medizinstudiums 2011 - 2013 Bearbeitung der vorliegenden Doktorarbeit unter der Leitung von Prof. Dr. Rolf Bräuer am Institut für Pathologie der Universität Jena Fremdsprachen Englisch: fließend in Wort und Schrift Französisch: befriedigend in Wort und Schrift Datum Unterschrift Jena, den 03.08.2013 109