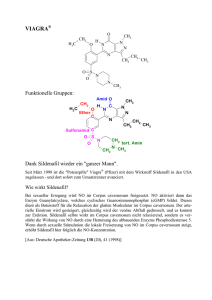
original PDF file
Werbung

Arbeiter-Samariter-Bund Regionalverband München/Oberbayern e.V. SEG-Behandlung ACS-Workshop 16.10.2015 Bernd Kaiser Material und Ergebnisse Gruppe 1 + 2 ACS-Workshop (Akutes Koronarsyndrom) Themen Gruppe 1 Anatomie Pathophysiologie Komplikationen Gruppe 2 Symptome Risikofaktoren Gruppe 3 Basismaßnahmen Diagnostischer Block Gruppe 4 Der BehandlungsAlgorithmus „akuter Inhalte Aufbau und Funktion des Herzens / Kranzgefäße Elektrische Reizleitung Aufbau des Gefäßsystems Arterien / Venen Ursachen eines MI / AP (stabil/instabil) Plaques, Ruptur, Gerinnung Kardialer Schock / Kardiales Lungenödem / Rhythmusstörungen Symptome, Unterschied Mann / Frau Unterschied MI / AP Spezielle Patientengruppen CoMorbidität Typische Risikofaktoren und deren CoMedikationen RR BZ SpO2 Immobilisierung / Lagerung Defibrillations-Bereitschaft EKG 3-Kanal, 12-Kanal Typische EKG-Bilder (STEMI) Vorstelle des Leitalgorith us „akuter Thorax-Sch erz“ Organisation: Notruf, NA, Transport-Ziel, Chest pain unit, Door to ballon time < 60 min Thorax-Schmerz“ Organisation *) Schema zur Vorstellung der Medikamente Wirkstoff / häufige Handelsnamen Anwendungsgebiete ACS-typische Dosierung, wie wird aufgezogen? Wirkmechanismus, was macht das Medikament? Gegenanzeigen Typische Wechselwirkungen Typische Nebenwirkungen Was tun bei Überdosierung? Medikamente *) Nitrolingual PDE-5Hemmer ASS Heparin Sauerstoff i.v. Zugang Morphin Vomex A Arbeiter-Samariter-Bund Regionalverband München/Oberbayern e.V. SEG-Behandlung ACS-Workshop 16.10.2015 Bernd Kaiser Gruppe 1 V. a. ACS – Algorithmus Glyceroltrinitrat-Spray Seite 7 Therapie des ACS: MONA Deutscher Berufsverband Rettungsdienst e.V. (DBRD) • • • • • • Algorithmus Morphin + Antiemetika Angepasste O2-Gabe Algorithmus Glyceroltrinitrat-Spray Algorithmus ASS + Heparin Algorithmus Sedation Monitoring + Defibrillationsbereitschaft Lagerung nach Kreislaufsituation, RR-Messung, EKG (*siehe Bemerkung), SpO2 Viagra®, Cialis®, Levitra®, Revatio®/48 h? JA KEIN Glyceroltrinitrat-Spray NEIN JA RRsys < 100 mmHg? KEIN Glyceroltrinitrat-Spray NEIN 0,4 - 1,2 mg Glyceroltrinitrat-Spray nach RR dosiert sublingual i.v.-Zugang engmaschige RR-Kontrolle, i.v.-Zugang ➡ Besserung der Beschwerden nach 3 - 5 Minuten? JA NEIN JA RRsys < 100 mmHg? KEIN Glyceroltrinitrat-Spray NEIN 0,4 - 0,8 mg Glyceroltrinitrat-Spray nach RR dosiert sublingual * V. a. STEMI-ACS mit inferioren Infarktzeichen: Bei ST-Streckenhebungen in den Extremitäten-Ableitungen II, III und aVF: Besonders bei Bradykardie vorsichtige Nitratgabe mit 0,4 mg je Einzelgabe, dann weiter dosiert nach Blutdruck! ESC Leitlinie STEMI 2012; ESC Leitlinie NSTEMI 2013; ERC-Leitlinie 2010; Nitrolingual-Fachinfo Nitrolingual® Pumpspray Bernd Kaiser, ASB-Langenau Name Nitrolingual® Pumpspray Wirkstoff • Glyceroltrinitrat („Nitroglycerin“) Substanz- • Koronar-Vasodilatator klasse • Nitroverbindungen Darrei- • Spray zur Anwendung in der Mundhöhle chungsform • Klare farblose Lösung in einer Glasflasche mit durchsichtiger roter Kunststoffummantelung Wirkstärke Nitrolingual® zugel. Indikationen (RD-relevant) • Arteriosklerose Koronare Herzkrankheit ACS Blutgefäß • Akuter Angina pectoris Anfall • Zur vorbeugenden Einnahme unmittelbar vor körperlichen Belastungen oder anderen Situationen, die erfahrungsgemäß A. pectoris Anfälle auslösen können • Akuter Myokardinfarkt • Akute Linksherzinsuffizienz ( 0,4mg / Sprühstoß kardiales Lungenödem) Blutfluß Plaque IschämieSchmerz durch O2-Mangel Nitrolingual® Indikationen Angina pectoris ACS Algorithmus ACS (Akutes Coronar Syndrom) Myokardinfarkt unmittelbare Symptomlinderung nitro-restistent nitro-sensitiv Nitrolingual® Indikationen Linksherzinsuffizienz Nitrolingual® Dosierung akute Angina pectoris R L↓ R↓ L 1 – 3 Sprühstöße: akute Linksherzinsuffizienz akuter Myokardinfarkt Wiederholung nach 10 min. bei gleicher Dosis möglich Unter Kontrolle der Kreislaufverhältnisse! D.h.: Infusion liegt und läuft RR sys > 100 mmHg kein Viagra®, Cialis® Levitra® i.d. letzten 48h Nitrolingual® Anwendung Nitrolingual® Wirkung am Gefäß • Unter die Zunge sprühen (s.l.) • Nicht inhaliert • Blutdrucksenkung! • Mit 30 Sek. Abstand sprühen • Entspannung der glatten Gefäßmuskulatur • Bei erstem Einsatz in die • Blut sammelt sich in den Venen Zunahme der venösen Kapazität („pooling“) Luft sprühen (Kammer füllen) • Entspannung der großen herznahen Arterien • Flasche senkrecht mit dem Sprühkopf nach oben halten • Sprühkopf nahe an den Mund heranzubringen Nitrolingual® Wirkort am Gefäß Nitrolingual® Wirkung am Herz Kapillare Arteriole • Venöser Rückstrom Venole Nitro Nitro • Herzkammervolumen • Herzkammer Füllungsdrücke Vene Arterie • Vorlast & Nachlast • Bedarf: Energie Intima (Endotel) Media (dicke glatte Muskulatur) Adventitia (elast. Kollagenfasern) & Sauerstoff • Erweiterung der Koronargefäße • Koronare Durchblutung Media (dünne glatte Muskulatur) höhere koronare Belastungstoleranz Nitrolingual® Wirkung am Herz Nitrolingual® Wirkung an der glatten Muskulatur Entspannung der gl. Muskulatur von: • Cranialen Gefäßen Perfusionsdruck Herz-Zeit-Volumen Nachlast Vorlast Nitro • Bronchien Nitro • Ableitenden Harnwegen *) • Gallenblase / Gallengang *) Vasotonus Vasotonus • Speiseröhre (Ösophagus) • Dünn- und Dickdarm Füllung Nitrolingual® Gegenanzeigen • Hypotonie • Sphinkter Peripherer Widerstand *) off label use: Gallenkolik, Harnleiterkolik Nitrolingual® Versteckte Risiken Gleicher Wirkstoff verschiedene Namen!!! RR sys. < 100 mmHg • kardiogener Schock, akutes Kreislaufversagen, Kreislaufkollaps • Behandlung mit Viagra® (Sildenafil), Cialis® (Tadalafil), Levitra® PDE-5 Hemmer Pulmonale arterieller Hypertonie (PAH) Erektile Dysfunktion (ED) (Vardenafil) gegen Erektionsstörungen, Pulmonale Arterielle Sildenafil Revatio® Viagra® -/- Vardenafil -/- Levitra® -/- Tadalafil Adcirca® Cialis® Cialis® Hypertonie, Benige Prostatahyperplasie (BPH) auch nicht bei akuten pectanginöse Beschwerden. • Allergien gegen Glyceroltrinitrat, anderen Nitratverbindungen, Pfefferminzöl ( allergisches Asthma)! Wirkstoff Benige Prostata Hyperplasie (BPH) Nitrolingual® Nebenwirkungen Nitrolingual® Nebenwirkungen Sehr häufig (≥1/10): Gelegentlich (≥1/1.000 bis <1/100) : • Kopfschmerzen (,,Nitratkopfschmerzen“) • starker Blutdruckabfall (Vorsicht: Wirkung schlecht steuerbar) Häufig (≥1/100 bis <1/10): • starker Blutdruckabfall und/oder orthostatische Hypotension • reflektorischen Erhöhung der Pulsfrequenz • Benommenheit sowie einem Schwindel- und Schwächegefühl mit Verstärkung der Angina pectoris Symptomatik • Kollapszustände, mit bradykarden Herzrhythmusstörungen und Synkopen • Übelkeit und Erbrechen • Allergien / Hautreaktionen, geschwollene Zunge • Brennen im Mund Nitrolingual® Wechselwirkungen Alkoholgehalt (87 Vol.%) Nitrolingual® Kommunikation mit Patient Ziele und Nutzen der Therapie erklären: • Verstärkung der Wirkung von Blutdrucksenkern • Bei gleichzeitiger Anwendung von Heparin kommt es zu einer Wirkungsabschwächung von Heparin. • Vorsicht - starker Blutdruckabfall • Scherzreduktion / Symptomreduktion Begleitmedikation erfragen: • RR-Medikam., Viagra®, Cialis®, Levitra® auch bei Frauen! Info über mögliche Nebenwirkungen / Rückmeldung: bei gleichzeitiger Anwendung von Präparaten zur Behandlung von • Schwindel und Benommenheit oder Kopfschmerzen Potenzstörungen / Erektionsstörungen (erektilen Dysfunktion) • starker Blutdruckabfall, Erhöhung der Pulsfrequenz (z.B. Viagra®, Cialis®, Levitra®), Lungenhochdruck und • Schwächegefühl, Übelkeit, Erbrechen, Durchfall Benigne (gutartige) Prostatahyperplasie (BPH). Anwendung des Sprays: • Sprühstöße alle 30 Sekunden, • Atem anhalten, nicht inhalieren! Nitrolingual® Überdosierung - ein Thema??? Ja, weil: • Standard-Medikation im Rettungsdienst • oft Bedarfsmedikation von Patienten Nitrolingual® Maßnahmen bei Überdosierung Monitoring: Maßnahmen: • • • • • Notarzt • Horizontal-Lagerung oder Schocklage • Sauerstoff-Gabe • Volumen-Gabe Puls Blutdruck Sauerstoffsättigung EKG • Intoxikation (Kinder!)…„Oma nimmt ja auch immer das Pfefferminz-Spray“ • An verschiedene Darreichungsformen denken! Pflaster Infusion (Ampullen) Zerbeißkapseln Nitrolingual® beim ASB-Langenau Medikamentenschrank Spray Quellen: • Rettungsdienst heute; D. Kühn, J. Luxem, K. Runggaldier; 4. Auflage • Nationale Versorgungs Leitlinie „Chronische KHK - Modul Medikamentöse Therapie“; 2. Auflage; Dezember 2011 • Fachinformation Nitrolingual® Pumpspray, Pohl Boskamp, Stand August 2011 • www.nitrolingual.de; Letzter Aufruf: 15.04.2013 • Arzneimittel pocket 2011; A. Ruß; Verlag Börm Bruckmeier Notfallrucksack / Ampullarium Trotz sorgfältiger Überprüfung sind die hier aufgeführten Hinweise, Dosierungsangaben ohne Gewähr. Der Autor übernimmt keine Haftung. Jeder Benutzer ist angehalten, durch Prüfung von Fachinformation / Beipackzettel der verwendeten Medikamente und Rücksprache mit einem Arzt festzustellen, ob die Empfehlungen für Anwendung und Dosierungen korrekt sind. 7/83-1 im Handschuhfach Nitrolingual® Wirkung im Detail… • Glycerintrinitrat wird in Stickstoffmonoxid (NO) verstoffwechselt • Calcium-Einstrom lässt glatte Muskelzellen der Gefäße kontrahieren • Hohe Stickstoffmonoxid (NO)-Konzentrationen glatte Muskeln entspannen • Wenig NO hoher Ca++ Einstrom verringert Ca++ -Einstrom Blutdruck sinkt starke Muskelkontraktion • Glyceroltrinitrat spaltet Stickstoffmonoxid (NO) ab • Calcium-Einstrom sinkt Nitrolingual® Leitlinien und Evidence based Med. Gesellschaft Leitlinie Deutschen Gesellschaft für Kardiologie - Herz- und Kreislaufforschung Infarkt-bedingter kardiogener Schock - Diagnose, Monitoring und Therapie (Gültig bis 5/2015) Indikation: Lungenstauung/ödem, Dyspnoe RR syst. > 90 mm Hg Akutes Koronarsyndrom mit persistierender ST- Streckenhebung (STEMI) (Update 2011) Geringgradige bis schwere Herzinsuffizienz ( IC) Prophylaktische Behandlung in der Akutphase (IIb A) Akutes Koronarsyndrom ohne ST-Hebung (NSTE-ACS) (Update 2012) Orale Nitrate zur symptomatischen Behandlung der Angina (IC) Chronische KHK - Modul Medikamentöse Therapie (Gültig bis 12/2015) Nitrate zum Kupieren von stabilen Angina pectoris Anfällen (symptomatische Behandlung). (soll) Nationale Versorgungs Leitlinien Nitrolingual- Empfehlung Keine Belege für eine Reduktion klin. Endpunkte (kardiovaskuläre Morbidität und Mortalität) Muskulatur erschlafft hoher Blutdruck hohe NO-Konzentrationen Blutdruck fällt Leitlinien-Info Fachinformation Nitrolingual® Pumpspray 1. BEZEICHNUNG DES ARZNEIMITTELS Nitrolingual® Pumpspray 0,4 mg/Sprühstoß, Spray zur Anwendung in der Mundhöhle Glyceroltrinitrat 2. QUALITATIVE UND QUANTITATIVE ZUSAMMENSETZUNG Wirkstoff: 1 Sprühstoß enthält 0,4 mg Glyceroltrinitrat Sonstige Bestandteile mit bekannter Wirkung: Pfefferminzöl und Ethanol. Vollständige Auflistung der sonstigen Bestandteile, siehe Abschnitt 6.1. 3. DARREICHUNGSFORM Spray zur Anwendung in der Mundhöhle 4. KLINISCHE ANGABEN 4.1 Anwendungsgebiete – Behandlung des akuten Angina pectoris Anfalls – Zur vorbeugenden Einnahme unmittelbar vor körperlichen Belastungen oder anderen Situationen, die erfahrungsgemäß Angina pectoris Anfälle auslösen können (Prophylaxe der Angina pectoris) – Akuter Myokardinfarkt – Akute Linksherzinsuffizienz – Katheterinduzierte Koronarspasmen bei Koronarangiographie 4.2 Dosierung, Art und Dauer der Anwendung Wenn sich die Symptome nach der maximal empfohlenen Dosierung nicht gebessert haben, sollte der Patient umgehend ärztlichen Rat einholen. Die Patienten sollten darauf hingewiesen werden, sich bei der sublingualen Anwendung von Glyceroltrinitrat hinzusetzen, wann immer dies möglich ist. Dezember 2013 MA 175/9 Dosierung Soweit nicht anders verordnet, werden bei Beginn eines Angina pectoris Anfalls oder unmittelbar vor Belastungen, die erfahrungsgemäß zur Auslösung eines Angina pectoris Anfalls führen können, je nach Schweregrad 1 – 3 Sprühstöße Nitrolingual® Pumpspray (entsprechend 0,4 – 1,2 mg Glyceroltrinitrat) zugeführt. Bei akuter Linksherzinsuffizienz und bei akutem Myokardinfarkt werden unter Kontrolle der Kreislaufverhältnisse (systolischer Blutdruck höher als 100 mmHg) je nach Schweregrad 1 – 3 Sprühstöße Nitrolingual® Pumpspray (entsprechend 0,4 – 1,2 mg Glyceroltrinitrat) gegeben. Bei Nichtansprechen kann nach 10 Minuten die Behandlung mit der gleichen Dosis wiederholt werden. Vorbeugend vor der Koronarangiographie werden 1 – 2 Sprühstöße Nitrolingual® Pumpspray (entsprechend 0,4 mg – 0,8 mg Glyceroltrinitrat) gegeben. Eine zusätzliche antianginöse Therapie mit Arzneimitteln, die keine Nitratverbindungen enthalten, sollte für das nitratfreie Intervall in Betracht gezogen werden. 011675-3410 Ältere Menschen Während der Verwendung von Nitraten bei älteren Menschen können Hypotonie und Synkopen ein Problem darstellen. Eine Dosisanpassung ist jedoch nicht erforderlich. Kinder und Jugendliche Es liegen keine Daten über die Verwendung von Glyceroltrinitrat bei Kindern vor. Art der Anwendung Zur Anwendung jeweils die Schutzkappe senkrecht nach oben abziehen. Um die Handhabung des Nitrolingual® Pumpsprays kennenzulernen und um beim ersten Einsatz die Dosierkammer vollständig aufzufüllen, wird das Ventil zunächst betätigt und der Inhalt in die Luft gesprüht (Sprühkopf zügig und vollständig durchdrücken und dann wieder loslassen). Dies kann auch erforderlich sein, wenn der Spray längere Zeit nicht benutzt wurde. Jetzt ist der Spray funktionsbereit. Den Spray vor der Anwendung nicht schütteln. Beim Sprühen die Flasche senkrecht mit dem Sprühkopf nach oben halten. Die Öffnung im Sprühkopf ist möglichst nahe an den Mund heranzubringen. Da sich die Öffnung leicht tasten lässt, dient sie auch als sichere Orientierungshilfe bei nächtlichen Anwendungen. Die Sprühstöße werden in Abständen von etwa 30 Sekunden in die Mundhöhle (bevorzugt unter die Zunge) gesprüht. Während des Sprühstoßes den Atem anhalten und nicht inhalieren. Durch das transparente Behältnis erhält man einen ständigen Überblick über den Verbrauch. Der Flascheninhalt kann nur soweit abgesprüht werden, wie die Öffnung am unteren Ende des Steigrohres vollständig in die Flüssigkeit eintaucht. Wie bei allen anderen Sprays auch, läßt sich der Bodenrest nicht mehr absprühen. Dies wird bei der Befüllung der Flasche berücksichtigt, so dass die deklarierte Menge entnommen werden kann. Dauer der Anwendung Über die Dauer der Anwendung entscheidet der behandelnde Arzt. 4.3 Gegenanzeigen Nitrolingual® Pumpspray darf nicht angewendet werden bei: – Überempfindlichkeit gegen Glyceroltrinitrat, andere Nitratverbindungen, Pfefferminzöl oder einen der in Abschnitt 6.1 genannten sonstigen Bestandteile – akutem Kreislaufversagen (Schock, Kreislaufkollaps, hypovolämischer Schock) – ausgeprägter Hypotonie (systolischer Blutdruck weniger als 90 mmHg) – kardiogenem Schock, sofern nicht durch intraaortale Gegenpulsation oder positiv inotrope Pharmaka ein ausreichend hoher linksventrikulärer, enddiastolischer Druck gewährleistet ist – hypertropher obstruktiver Kardiomyopathie, da es die Obstruktion während der Austreibungsphase verstärken kann – Erkrankungen, die mit einem erhöhten intrakraniellen Druck einhergehen, wie z. B. zerebrale Blutungen oder Schädelhirntrauma (bisher wurde nur bei hochdosierter i. v. Gabe von Glyceroltrinitrat eine Drucksteigerung beobachtet) – schwerer Anämie – Einnahme von Phosphodiesterase-5Hemmern zur Behandlung der erektilen Dysfunktion oder der pulmonalen arteriellen Hypertonie, da durch diese der blutdrucksenkende Effekt von Nitrolingual® Pumpspray erheblich verstärkt werden kann. Beachten Sie bei Patienten mit koronarer Herzkrankheit daher die Anwendungsbeschränkungen von solchen Arzneimitteln. Nitrolingual® Pumpspray darf auch dann nicht angewendet werden, wenn Patienten, die Phosphodiesterase-5-Hemmer zur Behandlung der erektilen Dysfunktion oder der pulmonalen arteriellen Hypertonie eingenommen haben, akute pectanginöse Beschwerden entwickeln. 4.4 Besondere Warnhinweise und Vorsichtsmaßnahmen für die Anwendung Glyceroltrinitrat darf nur mit Vorsicht angewendet werden bei: – konstriktiver Perikarditis und Perikardtamponade – niedrigen Füllungsdrücken z. B. bei akutem Herzinfarkt, eingeschränkter Funktion der linken Herzkammer (Linksherzinsuffizienz). Eine Blutdrucksenkung unter 90 mmHg systolisch sollte vermieden werden – Aorten- und/oder schwerer Mitralklappenstenose – Neigung zu orthostatischen Kreislaufregulationsstörungen – Patienten mit zerebrovaskulären Erkrankungen, da deren Symptome durch Hypotonie ausgelöst werden können. Wie bei allen glyceroltrinitrathaltigen Präparaten sollte der Gebrauch von Nitrolingual® Pumpspray bei Patienten mit beginnendem Glaukom vermieden werden. Durch die Auswirkungen einer Tachykardie und eines verringerten diastolischen Drucks könnte die Anwendung von Glyceroltrinitrat theoretisch die myokardiale Blutversorgung bei Patienten mit linksventrikulärer Hypertrophie und assoziierter Aortenklappenstenose gefährden. Ausführliche hämodynamische Studien mit einer kleinen Zahl an Patienten mit Aortenklappenstenose mit und ohne begleitende signifikante koronare Herzkrankheit haben in liegender Position keine negativen Auswirkungen von sublingualem Glyceryltrinitrat gezeigt. Dennoch ist bei der Behandlung von ambulanten Patienten mit einer Kombination von Angina pectoris und mittelschwerer bis schwerer Aortenklappenstenose Vorsicht geboten. Dieses Arzneimittel enthält 82 Vol. % Ethanol (Alkohol). Aufgrund des Alkoholgehaltes kann die Anwendung des Sprays zu einem brennenden Gefühl in der Mundhöhle führen. 1 Fachinformation Nitrolingual® Pumpspray 4.5 Wechselwirkungen mit anderen Arzneimitteln und sonstige Wechselwirkungen Folgende Wechselwirkungen dieses Arzneimittels müssen beachtet werden: Die gleichzeitige Einnahme von anderen Vasodilatatoren, anderen Antihypertensiva (z. B. Beta-Rezeptorenblocker, Kalziumantagonisten, ACE-Hemmer, Diuretika), Neuroleptika oder trizyklischen Antidepressiva, Alkohol und Sapropterin kann die blutdrucksenkende Wirkung von Nitrolingual® Pumpspray verstärken. N-Acetylcystein kann die gefäßerweiternde Wirkung von Glyceroltrinitrat verstärken. Bei zusätzlicher Einnahme von Phosphodiesterase-5-Hemmern zur Behandlung der erektilen Dysfunktion oder der pulmonalen arteriellen Hypertonie zu einer bestehenden Nitrattherapie (wie zum Beispiel mit Nitrolingual® Pumpspray) kann es zu einer erheblichen Verstärkung des blutdrucksenkenden Effektes kommen. Daher dürfen Patienten mit koronarer Herzkrankheit Phosphodiesterase-5-Hemmer zur Behandlung der erektilen Dysfunktion oder der pulmonalen arteriellen Hypertonie nicht einnehmen (siehe Abschnitt 4.3 Gegenanzeigen). Bei mit organischen Nitratverbindungen, z. B. Isosorbiddinitrat, Isosorbid-5-Mononitrat, vorbehandelten Patienten kann eine höhere Dosierung von Glyceroltrinitrat zur Erzielung der gewünschten hämodynamischen Wirkung erforderlich sein. Nitrolingual® Pumpspray kann bei gleichzeitiger Anwendung von Dihydroergotamin (DHE) zum Anstieg des DHE-Spiegels führen und damit dessen blutdrucksteigernde Wirkung verstärken. Bei gleichzeitiger Anwendung von Heparin und Nitrolingual® Pumpspray kommt es zu einer Wirkungsabschwächung von Heparin. Unter engmaschigen Kontrollen der Blutgerinnungsparameter ist die Heparindosis entsprechend anzupassen. Nach Absetzen von Glyceroltrinitrat kann es zu einer deutlich verminderten Blutgerinnung (sprunghafter Anstieg der PTT) kommen, so dass eine Reduktion der Heparindosis erforderlich sein kann. 4.7 Auswirkungen auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen Dieses Arzneimittel kann auch bei bestimmungsgemäßem Gebrauch das Reaktionsvermögen so weit verändern, dass die Fähigkeit zur aktiven Teilnahme am Straßenverkehr oder zum Bedienen von Maschinen oder zum Arbeiten ohne sicheren Halt beeinträchtigt wird. Es wird dem Patienten empfohlen nach der Anwendung des Sprays mindestens fünf Minuten zu warten, bevor er fährt oder Maschinen bedient. Wenn ein Patient sich schwach, schwindelig oder unwohl fühlt, sollte er warten, bis er sich besser fühlt. Dies gilt in verstärktem Maße bei Behandlungsbeginn, Dosiserhöhung und Präparatewechsel sowie im Zusammenwirken mit Alkohol. 4.8 Nebenwirkungen Bei Behandlungsbeginn können Nitrat-induzierte Kopfschmerzen sehr häufig auftreten, die erfahrungsgemäß bei weiterer Einnahme abklingen (Insgesamt: häufige Frequenz). Ebenso werden häufig bei der Erstanwendung, aber auch bei einer Dosiserhöhung, ein Abfall des Blutdrucks und/oder orthostatische Hypotension beobachtet, die mit einer reflektorischen Erhöhung der Pulsfrequenz (Tachykardie), Benommenheit sowie einem Schwindel- und Schwächegefühl einhergehen können. Gelegentlich treten Kollapszustände, mitunter mit bradykarden Herzrhythmusstörungen und Synkopen, auf. Tierstudien zeigten keine schädlichen Auswirkungen auf die Fertilität. Bei den Häufigkeitsangaben zu Nebenwirkungen werden folgende Kategorien zugrunde gelegt: Sehr häufig (≥ 1/10) Häufig (≥ 1/100 bis < 1/10) Gelegentlich (≥ 1/1.000 bis < 1/100) Selten (≥ 1/10.000 bis < 1/1.000) Sehr selten (< 1/10.000) Nicht bekannt (Häufigkeit auf Grundlage der verfügbaren Daten nicht abschätzbar) Schwangerschaft Erkrankungen des Immunsystems 4.6 Fertilität, Schwangerschaft und Stillzeit Fertilität Für Glyceroltrinitrat liegen keine klinischen Daten über Schwangere vor. Tierversuche ergaben keine Hinweise auf direkte oder indirekte schädigende Wirkungen im Hinblick auf Schwangerschaft, embryonale/fetale Entwicklung, Entbindung oder postnatale Entwicklung. In der Schwangerschaft sollte Glyceroltrinitrat aus Gründen besonderer Vorsicht nur nach sorgfältiger Nutzen-Risiko-Abwägung angewendet werden. Gelegentlich: Überempfindlichkeitsreaktionen Stillzeit Häufig: Tachykardie Gelegentlich: Verstärkung der Angina pectoris Symptome, Bradykardie Es ist unbekannt, ob Glyceroltrinitrat oder seine Metaboliten in die menschliche Mut2 termilch übergehen. Ein Risiko für das zu stillende Kind kann nicht ausgeschlossen werden. Die Entscheidung, das Stillen oder die Therapie mit Glyceroltrinitrat zu beenden, muss eine genaue Nutzen-RisikoAbwägung beinhalten, welche den Vorteil des Stillens des Kindes und den Vorteil der Arzneimitteltherapie für die Frau beinhaltet. Psychiatrische Erkrankungen Sehr selten: Unruhe Erkrankungen des Nervensystems Häufig: Kopfschmerzen, Schwindel und Benommenheit. Gelegentlich: Synkope Sehr selten: zerebrale Ischämie Herzerkrankungen: Gefäßerkrankungen Häufig: orthostatische Hypotension Gelegentlich: flüchtige Hautrötungen (Flush), Kreislaufkollaps Erkrankungen der Atemwege, des Brustraums und Mediastinums Sehr selten: Beeinträchtigung der Atmung Erkrankungen des Gastrointestinaltrakts Gelegentlich: Übelkeit, Erbrechen Nicht bekannt: geschwollene Zunge** Erkrankungen der Haut und des Unterhautzellgewebes Gelegentlich: allergische Dermatitis** Sehr selten: exfoliative Dermatitis Allgemeine Erkrankungen und Beschwerden am Verabreichungsort Häufig: Asthenie Nicht bekannt: Toleranzentwicklung* Untersuchungen Häufig: Abfall des Blutdrucks * Eine Toleranzentwicklung sowie das Auftreten einer Kreuztoleranz gegenüber anderen Nitratverbindungen wurden beschrieben. Zur Vermeidung einer Wirkungsabschwächung oder eines Wirkungsverlustes sollten hohe kontinuierliche Dosierungen vermieden werden. ** Gelegentlich können Überempfindlichkeitsreaktionen auftreten, welche sich z. B. als allergische Dermatitis oder in einzelnen Fällen als geschwollene Zunge zeigen. Bei entsprechend sensibilisierten Patienten können durch Pfefferminzöl Überempfindlichkeitsreaktionen (einschließlich Atemnot) ausgelöst werden. Hinweis: Bei der Gabe von Nitrolingual® Pumpspray kann, bedingt durch eine relative Umverteilung des Blutflusses in hypoventilierte Alveolargebiete, eine vorübergehende Hypoxämie auftreten und bei Patienten mit koronarer Herzkrankheit eine myokardiale Hypoxie auslösen. Meldung des Verdachts auf Nebenwirkungen Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von großer Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdachtsfall einer Nebenwirkung dem Bundesinstitut für Arzneimittel und Medizinprodukte, Abt. Pharmakovigilanz, KurtGeorg-Kiesinger Allee 3, D-53175 Bonn, Website: http://www.bfarm.de anzuzeigen. 4.9 Überdosierung Symptome einer Überdosierung Anzeichen und Symptome einer Überdosierung sind im Allgemeinen ähnlich den beschriebenen Nebenwirkungen: z. B. können Blutdruckabfall mit orthostatischen Regulationsstörungen, reflektorische Tachykardie und Kopfschmerzen, Schwächegefühl, Schwindel, Benommenheit, Flush, Übelkeit, Erbrechen und Durchfall auftreten. Bei hohen Dosen (größer 20 mg/kg Körpergewicht) ist infolge des beim GTN-Abbau entstehenden Nitrit-Ions mit Methämoglo011675-3410 Fachinformation Nitrolingual® Pumpspray binbildung, Zyanose, Atemnot und Tachypnoe zu rechnen. Bei sehr hohen Dosen kann es zur Erhöhung des intrakraniellen Druckes mit cerebralen Symptomen kommen. Bei chronischer Überdosierung wurden erhöhte Methämoglobinspiegel gemessen, deren klinische Relevanz umstritten ist. Therapiemaßnahmen bei Überdosierung Bei einer Überdosierung sollte der klinische Status des Patienten einschließlich der Vitalfunktion und des mentalen Status überwacht werden. Therapeutische Maßnahmen zur Unterstützung des kardiovaskulären Systems und der Atemwege sind entsprechend den klinischen Erfordernissen und, wenn vorhanden, den nationalen Empfehlungen durchzuführen. Im Falle einer milden Hypotonie kann das Hochlegen der Beine des Patienten und/ oder Absenken des Kopfes wirksam sein. Eine arterielle Blutgasanalyse (BGA) sollte bestimmt werden. Bei Azidose oder klinischen Zeichen einer Zyanose muss eine schwere Methämoglobinämie angenommen werden. Neben einer Sauerstoffbehandlung sollte Methylenblau i. v. (1 bis 2 mg/kg Körpergewicht) über 5 Minuten gegeben werden; es sei denn, der Patient leidet unter einem G-6-PD-Mangel. 5. PHARMAKOLOGISCHE EIGENSCHAFTEN 5.1 Pharmakodynamische Eigenschaften Pharmakotherapeutische Gruppe: Koronar-Vasodilatatoren, Nitroverbindungen ATC-Code: C01 DA 02 Wirkmechanismus Glyceroltrinitrat wirkt direkt relaxierend auf die glatte Gefäßmuskulatur und führt zu einer Vasodilatation. Die postkapillären Kapazitätsgefäße und die großen Arterien – insbesondere die noch reagiblen Teile von Koronararterien – sind hierbei stärker betroffen als die Widerstandsgefäße. Die Vasodilatation in der Strombahn führt zur Zunahme der venösen Kapazität („pooling“), der Rückstrom zum Herzen wird vermindert, Ventrikelvolumina und Füllungsdrücke sinken („preload“-Senkung). Dezember 2013 MA 175/9 Verkleinerter Ventrikelradius und verminderte systolische Wandspannung senken den myokardialen Energie- bzw. O2-Bedarf. Die Abnahme der kardialen Füllungsdrücke begünstigt die Perfusion ischämiegefährdeter, subendokardialer Wandschichten, regionale Wandbewegung und Schlagvolumen können verbessert werden. Die Dilatation der großen herznahen Arterien führt zu einer Abnahme sowohl des systemischen („afterload“-Senkung) als auch des pulmonalen Auswurfwiderstandes. Glyceroltrinitrat bewirkt eine Relaxation der Bronchialmuskulatur, der ableitenden Harnwege, der Muskulatur der Gallenblase, des 011675-3410 Gallenganges sowie des Ösophagus, des Dünn- und Dickdarmes einschließlich der Sphinkteren. Auf molekularer Ebene wirken die Nitrate sehr wahrscheinlich über die Bildung von Stickoxid (NO) und zyklischem Guanosylmonophosphat (cGMP), das als Mediator der Relaxation gilt. 5.2 Pharmakokinetische Eigenschaften GTN wird intestinal vollständig absorbiert, unterliegt aber einem extensiven hepatischen First-pass-Metabolismus sowie einer Spontanhydrolyse im Blut. Außerdem erfolgt eine hohe Erythrozytenbindung sowie eine Anreicherung in der Gefäßwand. Bei sublingualer Applikation wird Glyceroltrinitrat aus der Mundhöhle rasch resorbiert. Der First-pass-Effekt von GTN wird nach sublingualer und topischer Applikation in unterschiedlichem Ausmaß beobachtet. So beträgt die absolute Bioverfügbarkeit bei sublingualer Gabe ca. 39 % und nach topischer Anwendung als Pflaster ca. 55 %. Die Plasmaproteinbindung beträgt ca. 60 %. Die Eliminationshalbwertszeit für GTN ist kurz. Nach sublingualer Gabe werden Werte von 2,5 – 4,4 min, nach intravenöser Gabe von 2 – 2,5 min angegeben. Der GTN-Abbau, der in der Leber, aber auch in vielen anderen Zellen, z. B. in den Erythrozyten, erfolgt, beinhaltet die Abspaltung einer oder mehrerer Nitratgruppen. Neben der Verstoffwechselung des GTN findet eine renale Elimination der Metaboliten statt. Therapeutischer Blutspiegelbereich: 0,1 ng/ml – 3 (– 5) ng/ml. Plasmaspiegel: Nach sublingualer Applikation wurden große intra- und interindividuelle Schwankungen der Plasmaspiegel beobachtet. Für eine sublinguale Dosis von 0,4 mg betragen die Cmax-Werte 1,9 ± 1,6 ng/ml (Variationskoeffizient 87 %) und die tmax-Werte 5 ± 2 min (Bereich 2 – 10 min). Toleranz: Trotz gleich bleibender Dosierung und bei konstanten Nitratspiegeln wurde ein Nachlassen der Wirksamkeit beobachtet. Eine bestehende Toleranz klingt nach Absetzen der Therapie innerhalb von 24 Stunden ab. Bei entsprechend intermittierender Verabreichung wurde keine Toleranzentwicklung beobachtet. 5.3 Präklinische Daten zur Sicherheit Die Prüfung von Glyceroltrinitrat in Zellkulturen und im Tierversuch zeigte keine für den therapeutischen Dosisbereich relevanten mutagenen oder kanzerogenen Wirkungen. Reproduktionsstudien an Tieren liegen mit intravenöser, intraperitonealer und dermaler Gabe vor. In Studien zur Embryotoxizität und Fertilität ergaben sich bis in einen für die Elterntiere toxischen Dosisbereich keine Hinweise auf eine Beeinflussung des Embryos oder auf Fertilitätsstörungen. Insbesondere fanden sich keine Anhaltspunkte für teratogene Eigenschaften. Dosen oberhalb von 1 mg/kg/Tag (i.p.) und 28 mg/kg/Tag (dermal) zeigten fetotoxische Wirkungen (verminderte Geburtsgewichte) nach Anwendung während der Fetalentwicklung bei trächtigen Ratten. Untersuchungen zur Bestimmung der Wirkstoffkonzentrationen in der Muttermilch sind nicht bekannt. 6. PHARMAZEUTISCHE ANGABEN 6.1 Liste der sonstigen Bestandteile Glyceroltri(octanoat, decanoat, succinat) (50 : 35 : 15), Ethanol, Pfefferminzöl 6.2 Inkompatibilitäten Nicht zutreffend. 6.3 Dauer der Haltbarkeit 2 Jahre 6.4 Besondere Vorsichtsmaßnahmen für die Aufbewahrung Nicht über 25 °C lagern. 6.5 Art und Inhalt des Behältnisses OP 1 Flasche mit 16,7 ml (14,2 g) Spray zur Anwendung in der Mundhöhle AP 10 × 1 Flasche mit 16,7 ml (14,2 g) Spray zur Anwendung in der Mundhöhle OP 1 Flasche mit 20,9 ml (17,8 g) Spray zur Anwendung in der Mundhöhle AP 10 × 1 Flasche mit 20,9 ml (17,8 g) Spray zur Anwendung in der Mundhöhle Es werden möglicherweise nicht alle Packungsgrößen in den Verkehr gebracht. 6.6 Besondere Vorsichtsmaßnahmen für die Beseitigung und sonstige Hinweise zur Handhabung Nicht in Flammen oder auf glühende Körper sprühen. Nicht verwendetes Arzneimittel oder Abfallmaterial ist entsprechend den nationalen Anforderungen zu beseitigen. 7. INHABER DER ZULASSUNG G. POHL-BOSKAMP GmbH & Co. KG Kieler Straße 11 25551 Hohenlockstedt Telefon: (0 48 26) 59-0 Telefax: (0 48 26) 59-109 Internet: www.pohl-boskamp.de E-Mail: [email protected] 8. ZULASSUNGSNUMMER(N) 6008131.00.00 9. DATUM DER ERTEILUNG DER ZULASSUNG/VERLÄNGERUNG DER ZULASSUNG 08.10.1996/01.07.2002 3 Fachinformation Nitrolingual® Pumpspray 10. STAND DER INFORMATION Dezember 2013 11. VERKAUFSABGRENZUNG Verschreibungspflichtig Zentrale Anforderung an: Rote Liste Service GmbH Fachinfo-Service Mainzer Landstraße 55 60329 Frankfurt 4 011675-3410 Fachinformation CIALIS® 1. BEZEICHNUNG DES ARZNEIMITTELS CIALIS® 5 mg Filmtabletten CIALIS® 10 mg Filmtabletten CIALIS® 20 mg Filmtabletten 2. QUALITATIVE UND QUANTITATIVE ZUSAMMENSETZUNG CIALIS 5 mg Filmtabletten Jede Tablette enthält 5 mg Tadalafil. Sonstiger Bestandteil mit bekannter Wirkung: Jede Filmtablette enthält 121 mg Lactose (als Monohydrat). CIALIS 10 mg Filmtabletten Jede Tablette enthält 10 mg Tadalafil. Sonstiger Bestandteil mit bekannter Wirkung: Jede Filmtablette enthält 170 mg Lactose (als Monohydrat). störung liegen keine Daten zur Anwendung von Dosen höher als 10 mg Tadalafil vor. Tadalafil 10 und 20 mg Filmtabletten sind vorgesehen zur Einnahme vor einer erwarteten sexuellen Aktivität. Die tägliche Einnahme über einen längeren Zeitraum wird nicht empfohlen. Die einmal tägliche Anwendung von CIALIS zur Behandlung einer erektilen Dysfunktion und eines benignen Prostatasyndroms wurde bei Patienten mit Leberfunktionsstörung nicht untersucht; deshalb sollte vor einer Verordnung der verschreibende Arzt eine sorgfältige, individuelle Nutzen-Risiko Abwägung durchführen. (Siehe Abschnitte 4.4. und 5.2). Bei Patienten, die eine sehr häufige Anwendung von CIALIS erwarten (z. B. mindestens 2-mal pro Woche), kann unter Berücksichtigung der Patientenpräferenz und der Beurteilung durch den Arzt eine tägliche Anwendung von CIALIS in den niedrigsten Dosierungen als angemessen erachtet werden. Männer mit Diabetes Bei Diabetikern sind Dosisanpassungen nicht erforderlich. Die empfohlene Dosis für diese Patienten ist einmal täglich 5 mg jeweils zur etwa gleichen Tageszeit. Basierend auf der individuellen Verträglichkeit kann die Dosis auf einmal täglich 2,5 mg herabgesetzt werden. Kinder und Jugendliche CIALIS 20 mg Filmtabletten Jede Tablette enthält 20 mg Tadalafil. Sonstiger Bestandteil mit bekannter Wirkung: Jede Filmtablette enthält 233 mg Lactose (als Monohydrat). Die Angemessenheit der täglichen Dosierung sollte bei kontinuierlicher Anwendung in regelmäßigen Abständen überprüft werden. Art und Dauer der Anwendung CIALIS ist verfügbar als 2,5 mg*), 5 mg, 10 mg und 20 mg Filmtabletten zum Einnehmen. Vollständige Auflistung der sonstigen Bestandteile, siehe Abschnitt 6.1. Die empfohlene Dosis beträgt 5 mg und sollte etwa zur gleichen Zeit, unabhängig von den Mahlzeiten, täglich eingenommen werden. Für erwachsene Männer, die sowohl wegen eines benignen Prostatasyndroms als auch auf Grund einer erektilen Dysfunktion behandelt werden, beträgt die empfohlene Dosis ebenfalls 5 mg und sollte etwa zur gleichen Zeit täglich eingenommen werden. Patienten, die Tadalafil 5 mg zur Behandlung des benignen Prostatasyndroms nicht vertragen, sollten eine alternative Behandlung in Erwägung ziehen, da die Wirksamkeit von Tadalafil 2,5 mg zur Behandlung des benignen Prostatasyndroms nicht gezeigt wurde. 3. DARREICHUNGSFORM Filmtablette (Tablette). CIALIS 5 mg Filmtabletten: Hellgelbe und mandelförmige Tablette mit der Markierung „C 5“ auf einer Seite. CIALIS 10 mg Filmtabletten: Hellgelbe und mandelförmige Tablette mit der Markierung „C 10“ auf einer Seite. CIALIS 20 mg Filmtabletten: Gelbe und mandelförmige Tablette mit der Markierung „C 20“ auf einer Seite. Benignes Prostatasyndrom bei erwachsenen Männern 4. KLINISCHE ANGABEN Besondere Patientengruppen 4.1 Anwendungsgebiete Eine Dosisanpassung ist bei älteren Patienten nicht erforderlich. CIALIS 5 mg, 10 mg und 20 mg Filmtabletten: Zur Behandlung der erektilen Dysfunktion bei erwachsenen Männern. Tadalafil zur Behandlung einer erektilen Dysfunktion kann nur wirken, wenn eine sexuelle Stimulation vorliegt. CIALIS ist nicht angezeigt zur Anwendung bei Frauen. Zusätzlich CIALIS 5 mg Filmtabletten: Zur Behandlung des benignen Prostatasyndroms bei erwachsenen Männern. 4.2 Dosierung und Art der Anwendung August 2015 Die maximale Einnahmehäufigkeit ist einmal täglich. Ältere Männer Männer mit Nierenfunktionsstörung Bei Patienten mit leichter bis mäßiger Nierenfunktionsstörung ist eine Dosisanpassung nicht erforderlich. Bei Patienten mit schwerer Nierenfunktionsstörung ist 10 mg die empfohlene Maximaldosis für die Anwendung nach Bedarf. Die einmal tägliche Anwendung von 2,5 mg bzw. 5 mg Tadalafil zur Behandlung einer erektilen Dysfunktion oder eines benignen Prostatasyndroms, wird bei Patienten mit schwerer Nierenfunktionsstörung nicht empfohlen. (Siehe Abschnitte 4.4 und 5.2). Dosierung Männer mit Leberfunktionsstörung Erektile Dysfunktion bei erwachsenen Männern Zur Behandlung einer erektilen Dysfunktion mit CIALIS nach Bedarf beträgt die empfohlene Dosis 10 mg. Diese wird vor einer erwarteten sexuellen Aktivität und unabhängig von den Mahlzeiten eingenommen. Es existieren begrenzt klinische Daten zur Unbedenklichkeit von CIALIS bei Patienten mit schwerer Leberfunktionsstörung (ChildPugh Klasse C). Vor der Verordnung sollte der verschreibende Arzt eine sorgfältige, individuelle Nutzen-Risiko Abwägung durchführen. Bei Patienten mit Leberfunktions- Im Allgemeinen beträgt die empfohlene Dosis 10 mg. Diese wird vor einer erwarteten sexuellen Aktivität und unabhängig von den Mahlzeiten eingenommen. Bei Patienten, bei denen sich nach Einnahme von 10 mg Tadalafil keine entsprechende Wirkung zeigt, können 20 mg versucht werden. Die Einnahme sollte mindestens 30 Minuten vor einer sexuellen Aktivität erfolgen. 008049-15484 Es gibt im Anwendungsgebiet zur Behandlung der erektilen Dysfunktion keinen relevanten Nutzen von CIALIS bei Kindern und Jugendlichen. 4.3 Gegenanzeigen Überempfindlichkeit gegen den Wirkstoff oder einen der in Abschnitt 6.1 genannten sonstigen Bestandteile. In klinischen Studien wurde gezeigt, dass Tadalafil die blutdrucksenkende Wirkung von Nitraten verstärkt. Dies wird auf eine gemeinsame Wirkung von Nitraten und Tadalafil auf den Stickstoffmonoxid/cGMPStoffwechsel zurückgeführt. Daher ist die Anwendung von CIALIS bei Patienten kontraindiziert, die organische Nitrate in jeglicher Form einnehmen (siehe Abschnitt 4.5). Männer mit kardialen Erkrankungen, denen von sexueller Aktivität abgeraten wird, dürfen CIALIS nicht verwenden. Ärzte müssen das potentielle kardiale Risiko einer sexuellen Aktivität bei Patienten mit einer vorbestehenden kardiovaskulären Erkrankung berücksichtigen. Die folgenden Patientengruppen mit HerzKreislauf-Erkrankung waren in klinische Studien nicht eingeschlossen und daher ist die Anwendung von Tadalafil kontraindiziert: – Patienten mit Herzinfarkt während der vorangegangenen 90 Tage, – Patienten mit instabiler Angina pectoris oder einer Angina pectoris, die während einer sexuellen Aktivität auftrat, – Patienten mit Herzinsuffizienz Schweregrad II oder höher nach New York Heart Association (NYHA) während der letzten 6 Monate, – Patienten mit unkontrollierten Arrhythmien, Hypotonie (< 90/50 mm Hg) oder unkontrollierter Hypertonie, – Patienten mit einem Schlaganfall während der vorangegangenen 6 Monate. Bei Patienten, die aufgrund einer nicht arteriitischen anterioren ischämischen Optikusneuropathie (NAION) ihre Sehkraft auf einem Auge verloren haben, ist CIALIS kontraindiziert, unabhängig davon, ob der Sehverlust mit einer vorherigen Einnahme eines PDE5-Hemmers in Zusammenhang stand oder nicht (siehe Abschnitt 4.4). 1 Fachinformation CIALIS® Die Begleittherapie von PDE5-Hemmern, inklusive Tadalafil, mit Guanylatcyclase-Stimulatoren wie Riociguat ist kontraindiziert, da es möglicherweise zu einer symptomatischen Hypotonie kommen kann (siehe Abschnitt 4.5). 4.4 Besondere Warnhinweise und Vorsichtsmaßnahmen für die Anwendung Vor der Behandlung mit CIALIS Eine Anamnese und eine körperliche Untersuchung sollten durchgeführt werden, um eine erektile Dysfunktion oder eine benigne Prostatahyperplasie zu diagnostizieren und potentielle zugrunde liegende Ursachen zu bestimmen, bevor eine medikamentöse Behandlung in Betracht gezogen wird. Vor Beginn jedweder Behandlung der erektilen Dysfunktion sollte der Arzt den kardiovaskulären Status des Patienten erheben, da mit sexueller Aktivität ein gewisses kardiales Risiko verbunden ist. Tadalafil hat gefäßerweiternde Eigenschaften, die eine leichte und vorübergehende Blutdrucksenkung bewirken (siehe Abschnitt 5.1) und dadurch den blutdrucksenkenden Effekt von Nitraten verstärken (siehe Abschnitt 4.3). Die Abklärung einer erektilen Dysfunktion sollte die Bestimmung möglicher zugrundeliegender Ursachen einschließen. Nach einer entsprechenden ärztlichen Diagnose ist eine angemessene Behandlung festzulegen. Es ist nicht bekannt, ob CIALIS bei Patienten, bei denen eine Operation im Bereich des Beckens oder eine radikale Prostatektomie in nicht nervenerhaltender Technik vorgenommen wurde, wirksam ist. Bevor mit der Einnahme von Tadalafil zur Behandlung eines benignen Prostatasyndroms (CIALIS 5 mg) begonnen wird, müssen die Patienten sorgfältig untersucht werden, um ein Prostatakarzinom auszuschließen und um ihren kardiovaskulären Zustand zu überprüfen (siehe Abschnitt 4.3). Herz-Kreislauf Schwerwiegende kardiovaskuläre Ereignisse, einschließlich Myokardinfarkt, plötzlicher Herztod, instabile Angina pectoris, ventrikuläre Arrhythmien, Schlaganfall, vorübergehende ischämische Attacken, Brustschmerz, Palpitation und Tachykardie wurden aus klinischen Studien und/oder spontan nach Markteinführung berichtet. Bei den meisten Patienten, von denen diese Ereignisse berichtet wurden, waren vorbestehende kardiovaskuläre Risikofaktoren bekannt. Allerdings ist es nicht möglich, mit Gewissheit festzustellen, ob diese Ereignisse in ursächlichem Zusammenhang mit diesen Risikofaktoren, mit CIALIS, mit der sexuellen Aktivität oder einer Kombination dieser oder anderer Faktoren stehen. Bei Patienten, die gleichzeitig blutdrucksenkende Arzneimittel erhalten, kann Tadalafil eine Blutdrucksenkung induzieren. Wenn eine tägliche Anwendung von Tadalafil (CIALIS 2,5 mg oder 5 mg) begonnen wird, müssen entsprechende klinische Überlegungen bezüglich einer möglichen Dosisanpassung der antihypertensiven Therapie angestellt werden. 2 Bei Patienten, die Alpha1-Blocker einnehmen, kann die gleichzeitige Einnahme von CIALIS zu symptomatischer Hypotonie führen (siehe Abschnitt 4.5). Die Kombination von Tadalafil und Doxazosin wird nicht empfohlen. Visus Sehstörungen und Fälle von NAION sind in Zusammenhang mit der Einnahme von CIALIS und anderen PDE5-Hemmern berichtet worden. Die Patienten müssen darüber aufgeklärt werden, dass sie im Falle einer plötzlichen Sehstörung CIALIS absetzen und sofort einen Arzt aufsuchen sollen (siehe Abschnitt 4.3). Nieren- und Leberfunktionsstörung Aufgrund erhöhter Bioverfügbarkeit (AUC) von Tadalafil, begrenzter klinischer Erfahrung und der fehlenden Möglichkeit die Clearance durch Dialyse zu beeinflussen, wird die einmal tägliche Gabe von CIALIS (CIALIS 5 mg) bei Patienten mit schwerer Niereninsuffizienz nicht empfohlen. Es existieren begrenzt klinische Daten zur Unbedenklichkeit von CIALIS bei Verabreichung einer Einzeldosis bei Patienten mit schwerer Leberinsuffizienz (Child-Pugh Klasse C). Die einmal tägliche Anwendung von Tadalafil zur Behandlung einer erektilen Dysfunktion (CIALIS 2,5 mg oder 5 mg) oder eines benignen Prostatasyndroms (CIALIS 5 mg) wurde bei Patienten mit Leberinsuffizienz nicht untersucht. Deshalb sollte vor einer Verordnung von CIALIS der verschreibende Arzt eine sorgfältige, individuelle Nutzen-Risiko Abwägung durchführen. Priapismus und anatomische Deformation des Penis Patienten mit Erektionen, die länger als 4 Stunden dauern, sollten angewiesen werden, dringend ärztliche Hilfe in Anspruch zu nehmen. Wird Priapismus nicht sofort behandelt, können Schädigungen des Penisgewebes und ein dauerhafter Potenzverlust die Folge sein. CIALIS darf bei Patienten mit anatomischer Deformation des Penis (z. B. Deviation, Fibrose im Bereich der Corpora Cavernosa oder Induratio penis plastica) oder bei Patienten mit für Priapismus prädisponierenden Erkrankungen (z. B. Sichelzellenanämie, multiples Myelom oder Leukämie) nur mit Vorsicht angewendet werden. Anwendung mit CYP3A4-Inhibitoren Wenn CIALIS an Patienten verschrieben wird, die potente CYP3A4-Inhibitoren (Ritonavir, Saquinavir, Ketoconazol, Itraconazol und Erythromycin) einnehmen, ist Vorsicht geboten, da eine erhöhte Tadalafil-Exposition (AUC) bei Kombination dieser Arzneimittel beobachtet wurde (siehe Abschnitt 4.5). CIALIS und andere Behandlungsmethoden der erektilen Dysfunktion Unbedenklichkeit und Wirksamkeit einer Kombination von CIALIS mit anderen PDE5Inhibitoren oder anderen Behandlungsmethoden der erektilen Dysfunktion wurden nicht untersucht. Informieren Sie Ihre Patienten, dass sie CIALIS nicht in solchen Kombinationen einnehmen dürfen. Lactose CIALIS enthält Lactose. Patienten mit der seltenen hereditären Galactose-Intoleranz, Lactase-Mangel oder Glucose-GalactoseMalabsorption sollten dieses Arzneimittel nicht einnehmen. 4.5 Wechselwirkungen mit anderen Arzneimitteln und sonstige Wechselwirkungen Wie im Folgenden erläutert, wurden Interaktionsstudien mit 10 mg und/oder 20 mg Tadalafil durchgeführt. Bezüglich solcher Interaktionsstudien, bei denen nur die 10 mg Tadalafil Dosierung verwendet wurde, können klinisch relevante Wechselwirkungen bei höheren Dosierungen nicht völlig ausgeschlossen werden. Wirkungen anderer Substanzen auf Tadalafil Cytochrom P450 Inhibitoren Tadalafil wird hauptsächlich durch CYP3A4 abgebaut. Ein selektiver CYP3A4-Inhibitor, Ketoconazol (200 mg täglich), erhöhte die AUC von Tadalafil (10 mg) auf das Doppelte und cmax um 15 % im Vergleich zu den AUC und cmax-Werten bei alleiniger Gabe von Tadalafil. Ketoconazol (400 mg täglich) erhöhte die AUC von Tadalafil (20 mg) um das 4-fache und cmax um 22 %. Der ProteaseInhibitor Ritonavir, ein Inhibitor von CYP3A4, CYP2C9, CYP2C19 und CYP2D6, erhöhte bei einer Tagesdosis von 2 × 200 mg die AUC von Tadalafil (20 mg) auf das Doppelte bei gleichzeitig unveränderter cmax. Obwohl die spezifischen Wechselwirkungen nicht untersucht wurden, sollten andere Protease-Inhibitoren wie Saquinavir, und andere CYP3A4-Inhibitoren wie Erythromycin, Clarithromycin, Itraconazol und Grapefruitsaft mit Vorsicht gleichzeitig gegeben werden, da zu erwarten ist, dass sie den Tadalafil-Plasmaspiegel erhöhen (siehe Abschnitt 4.4). Folglich könnte sich die Häufigkeit der Nebenwirkungen, die in Abschnitt 4.8 aufgeführt sind, möglicherweise erhöhen. Transportmoleküle Die Rolle von Transportmolekülen (z. B. p-Glycoprotein) bei der Verteilung von Tadalafil ist nicht bekannt. Daher besteht die Möglichkeit von Arzneimittelwechselwirkungen, die durch Inhibierung von Transportmolekülen hervorgerufen werden. Cytochrom P450 Induktoren Ein CYP3A4-Induktor, Rifampicin, reduzierte die AUC von Tadalafil um 88 %, verglichen mit den AUC-Werten bei alleiniger Gabe von Tadalafil (10 mg). Diese reduzierte Exposition lässt erwarten, dass die Wirksamkeit von Tadalafil vermindert wird, der Umfang der Wirksamkeitsminderung ist nicht bekannt. Andere CYP3A4-Induktoren wie Phenobarbital, Phenytoin und Carbamazepin verringern ebenfalls die Plasma-Konzentration von Tadalafil. Wirkungen von Tadalafil auf andere Arzneimittel Nitrate In klinischen Studien wurde gezeigt, dass Tadalafil (5, 10 und 20 mg) die blutdrucksenkende Wirkung von Nitraten verstärkt. Daher ist die Gabe von CIALIS an Patienten kontraindiziert, die organische Nitrate in jeglicher Form einnehmen (siehe Abschnitt 4.3). 008049-15484 Fachinformation CIALIS® Entsprechend den Ergebnissen einer klinischen Studie, in der 150 Probanden 7 Tage lang eine tägliche Dosis von 20 mg Tadalafil und 0,4 mg Nitroglycerin sublingual zu verschiedenen Zeitpunkten erhielten, dauerte diese Wechselwirkung mehr als 24 Stunden an und war 48 Stunden nach der letzten Tadalafil-Gabe nicht mehr nachweisbar. Falls die Gabe von Nitraten in einer lebensbedrohlichen Situation für medizinisch erforderlich gehalten wird, sollten daher bei einem Patienten, der CIALIS (2,5 mg – 20 mg) erhält, mindestens 48 Stunden seit der letzten CIALIS-Einnahme verstrichen sein, bevor eine Nitrat-Gabe in Betracht gezogen wird. Nitrate sollten in diesen Situationen nur unter enger ärztlicher Überwachung mit einer angemessenen hämodynamischen Kontrolle gegeben werden. druck war die Blutdrucksenkung größer, obwohl dies in der Mehrheit der Fälle nicht mit Symptomen einer Hypotonie einherging. Bei Patienten, die gleichzeitig blutdrucksenkende Arzneimittel erhalten, können 20 mg Tadalafil eine Blutdrucksenkung hervorrufen, die (mit der Ausnahme von AlphaBlockern – siehe oben) im Allgemeinen geringfügig und wahrscheinlich nicht klinisch relevant ist. Die Analyse klinischer Daten aus Phase III Studien zeigte keine Unterschiede der unerwünschten Ereignisse bei Patienten, die Tadalafil mit oder ohne antihypertensive Arzneimittel einnahmen. Jedoch sollten Patienten, wenn sie mit Antihypertensiva behandelt werden, entsprechende ärztliche Hinweise über eine mögliche Blutdrucksenkung erhalten. Antihypertensiva (einschließlich Calciumkanal-Blocker) Präklinische Studien zeigten einen additiven Effekt auf die Senkung des systemischen Blutdrucks, wenn PDE5-Inhibitoren mit Riociguat kombiniert wurden. In klinischen Studien zeigte sich, dass Riociguat den hypotensiven Effekt von PDE5-Hemmern verstärkt. Es gab keinen Hinweis auf einen positiven klinischen Effekt dieser Kombination in der untersuchten Studienpopulation. Die gleichzeitige Verwendung von Riociguat zusammen mit PDE5-Hemmern, inklusive Tadalafil, ist kontraindiziert (siehe Abschnitt 4.3). August 2015 Die gleichzeitige Einnahme von Doxazosin (4 bzw. 8 mg täglich) und Tadalafil (5 mg tägliche Dosis bzw. 20 mg als einzelne Dosis) erhöht die blutdrucksenkende Wirkung dieses Alpha-Blockers in erheblicher Weise. Dieser Effekt hält mindestens 12 Stunden an und Symptome, einschließlich einer Synkope, können auftreten. Daher wird diese Kombination nicht empfohlen (siehe Abschnitt 4.4). Aus Wechselwirkungsstudien mit einer begrenzten Anzahl an gesunden Probanden wurde diese Wirkung bei Alfuzosin und Tamsulosin nicht berichtet. Dennoch sollte bei Patienten, die mit Alpha-Blockern behandelt werden, besondere Vorsicht gelten, wenn Tadalafil eingesetzt wird, dies gilt insbesondere für die Behandlung von älteren Menschen. Die Behandlungen sollten mit einer Minimaldosis begonnen und stufenweise angepasst werden. In klinisch-pharmakologischen Studien wurde untersucht, welches Potential Tadalafil besitzt, die blutdrucksenkende Wirkung antihypertensiver Arzneimittel zu verstärken. Wichtige Substanzklassen antihypertensiver Arzneimittel wurden untersucht, einschließlich Calciumkanal-Blockern (Amlodipin), Angiotensin Converting Enzym (ACE) Hemmern (Enalapril), Beta Rezeptoren Blockern (Metoprolol), Thiazid-Diuretika (Bendrofluazide) und Angiotensin II Rezeptor Blockern (verschiedene Arten und Dosierungen, allein oder in Kombination mit Thiaziden, Calcium-Kanalblockern, Beta-Blockern und/ oder Alpha-Blockern). Tadalafil (10 mg, außer in Studien mit Angiotensin II Rezeptorenblockern und Amlodipin, in denen eine 20 mg Dosis gegeben wurde) zeigte keine klinisch signifikante Wechselwirkung mit einer dieser Substanzklassen. In einer weiteren klinisch-pharmakologischen Studie wurde Tadalafil (20 mg) in Kombination mit bis zu 4 Klassen von Antihypertensiva untersucht. Bei Studienteilnehmern, die verschiedene Antihypertensiva einnahmen, schienen die ambulant gemessenen Blutdruck-Veränderungen im Zusammenhang mit der Blutdruck-Einstellung zu stehen. So war bei Studienteilnehmern, deren Blutdruck gut eingestellt war, die Senkung minimal und ähnlich der, die bei gesunden Probanden beobachtet wurde. Bei Studienteilnehmern mit schlecht eingestelltem Blut008049-15484 Riociguat 5-Alpha-Reduktase-Inhibitoren In einer klinischen Studie zur Behandlung der BPH Symptome wurde die gleichzeitige Einnahme von Tadalafil 5 mg und Finasterid 5 mg verglichen mit der gleichzeitigen Einnahme von Placebo und Finasterid 5 mg, dabei traten keine neuen Nebenwirkungen auf. Allerdings wurde keine formale Arzneimittelwechselwirkungsstudie zur Untersuchung der Effekte von Tadalafil und 5-AlphaReduktase-Inhibitoren (5-ARIs) durchgeführt, daher sollte man bei der Kombination von Tadalafil und 5-ARIs vorsichtig sein. CYP1A2 Substrate (z. B. Theophyllin) In einer klinisch-pharmakologischen Studie zeigte sich bei der Anwendung von 10 mg Tadalafil mit Theophyllin (einem nichtselektiven Phosphodiesterase-Hemmer) keine pharmakokinetische Wechselwirkung. Die einzige pharmakodynamische Wirkung war eine geringfügige Erhöhung der Herzfrequenz (um 3,5 Schläge pro Minute). Obwohl dieser Effekt geringfügig ist und in dieser Studie klinisch nicht signifikant war, sollte er bei gemeinsamer Anwendung dieser Arzneimittel berücksichtigt werden. Ethinylestradiol und Terbutalin Tadalafil zeigte eine Erhöhung der oralen Bioverfügbarkeit von Ethinylestradiol; eine ähnliche Erhöhung kann bei der oralen Anwendung von Terbutalin erwartet werden, obwohl die klinischen Auswirkungen unbekannt sind. Alkohol Alkohol-Konzentrationen (mittlere maximale Blutkonzentration 0,8 ‰) wurden durch gleichzeitige Gabe von Tadalafil (10 mg oder 20 mg) nicht beeinflusst. Auch wurde 3 Stunden nach gleichzeitiger Verabreichung von Alkohol keine Veränderung der Tadalafil-Konzentration beobachtet. Der Alkohol wurde dabei so verabreicht, dass die Alkohol-Absorption maximiert war (keine Nahrungsaufnahme seit dem vorangegangenen Abend bis 2 Stunden nach der Alkohol-Gabe). Tadalafil (20 mg) verstärkte nicht den durch Alkoholkonsum (0,7 g/kg oder etwa 180 ml 40%iger Alkohol [Wodka] bei einem Mann mit 80 kg Körpergewicht) verursachten mittleren Blutdruckabfall, aber bei einigen Probanden wurde Schwindel nach dem Aufrichten und orthostatische Hypotonie beobachtet. Bei Verabreichung von Tadalafil mit geringeren Alkoholmengen (0,6 g/kg) wurde keine Hypotonie beobachtet und Schwindel kam ähnlich häufig vor wie bei alleinigem Alkoholkonsum. Tadalafil (10 mg) verstärkte nicht die Alkoholwirkung auf kognitive Funktionen. Arzneimittel, die durch Cytochrom P450 metabolisiert werden Es ist nicht zu erwarten, dass Tadalafil eine klinisch signifikante Hemmung oder Verstärkung der Clearance solcher Arzneimittel bewirkt, die durch CYP450-Isoformen metabolisiert werden. Studien haben bestätigt, dass Tadalafil CYP450-Isoformen, einschließlich CYP3A4, CYP1A2, CYP2D6, CYP2E1, CYP2C9 und CYP2C19, weder inhibiert noch induziert. CYP2C9 Substrate (z. B. R-Warfarin) Tadalafil (10 mg und 20 mg) hatte weder eine klinisch signifikante Wirkung auf die Bioverfügbarkeit (AUC) von S-Warfarin oder R-Warfarin (CYP2C9 Substrat), noch hatte Tadalafil einen Einfluss auf eine mittels Warfarin eingestellte Prothrombin-Zeit. Acetylsalicylsäure Tadalafil (10 mg und 20 mg) hatte keinen Einfluss auf die durch Acetylsalicylsäure verlängerte Blutungszeit. Antidiabetika Spezifische Wechselwirkungsstudien mit Antidiabetika wurden nicht durchgeführt. 4.6 Fertilität, Schwangerschaft und Stillzeit CIALIS ist nicht zur Anwendung bei Frauen indiziert. Schwangerschaft Es gibt begrenzte Daten zur Anwendung von Tadalafil bei schwangeren Frauen. Tierexperimentelle Studien lassen nicht auf direkte oder indirekte schädliche Auswirkungen auf Schwangerschaft, embryonale/ fetale Entwicklung, Geburt oder postnatale Entwicklung schließen (siehe Abschnitt 5.3). Als Vorsichtsmaßnahme sollte vorzugsweise auf die Anwendung von CIALIS während der Schwangerschaft verzichtet werden. Stillzeit Verfügbare pharmakodynamische/toxikologische Daten zeigen eine Exkretion von Tadalafil in die Milch von Tieren. Ein Risiko für den Säugling kann nicht ausgeschlossen werden. CIALIS sollte während der Stillzeit nicht eingenommen werden. Fertilität Bei Hunden wurden Effekte beobachtet, die möglicherweise auf eine Beeinträchtigung der Fertilität hindeuten. Zwei sich daran anschließende klinische Studien zeigen, dass dieser Effekt beim Menschen unwahrscheinlich ist, obwohl bei einigen Männern 3 Fachinformation CIALIS® eine Abnahme der Spermienkonzentration beobachtet wurde (siehe Abschnitte 5.1 und 5.3). 4.7 Auswirkungen auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen CIALIS hat einen zu vernachlässigenden Einfluss auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen. Obwohl in klinischen Studien über Schwindel ähnlich häufig unter Placebo und Tadalafil berichtet wurde, sollten Patienten wissen, wie sie auf CIALIS reagieren, bevor sie Auto fahren oder Maschinen bedienen. 4.8 Nebenwirkungen Zusammenfassung des Sicherheitsprofils Die am häufigsten berichteten Nebenwirkungen bei Patienten, die CIALIS zur Behandlung einer erektilen Dysfunktion oder eines benignen Prostatasyndroms einnah- Sehr häufig men, waren Kopfschmerzen, Dyspepsie, Rückenschmerzen und Myalgie, wobei die Anzahl der berichteten Nebenwirkungen mit der verabreichten CIALIS-Dosis zunahm. Die berichteten unerwünschten Reaktionen waren vorübergehend und im Allgemeinen leicht bis mäßig. Die Mehrzahl der berichteten Kopfschmerzen, die bei der täglichen Anwendung von CIALIS berichtet wurden, trat meist innerhalb von 10 bis 30 Tagen nach Behandlungsbeginn auf. Tabellarische Zusammenfassung der Nebenwirkungen Die unten aufgeführte Tabelle beinhaltet die Nebenwirkungen aus spontan berichteten Fällen und placebo-kontrollierten klinischen Studien (mit insgesamt 7116 CIALIS-Patienten und 3718 Placebo-Patienten) zur Behandlung der erektilen Dysfunktion nach Bedarf bzw. in der täglichen Dosierung und zur Behandlung des benignen Prostatasyndroms in der täglichen Dosierung. Häufig Häufigkeitsangaben gemäß Konvention: sehr häufig (≥ 1/10), häufig (≥ 1/100 bis < 1/10), gelegentlich (≥ 1/1.000 bis < 1/100), selten (≥ 1/10.000 bis < 1/1.000), sehr selten (< 1/10.000) und nicht bekannt (Häufigkeit auf Grundlage der verfügbaren Daten nicht abschätzbar). Beschreibung spezieller Nebenwirkungen Verglichen mit Placebo gab es in der Gruppe, die einmal täglich mit Tadalafil behandelt wurde, eine etwas höhere Inzidenz von EKG Abnormalitäten, in erster Linie Sinusbradykardie. Die meisten dieser EKGUnregelmäßigkeiten standen nicht im Zusammenhang mit unerwünschten Reaktionen. Andere besondere Patientengruppen Daten zu Patienten über 65 Jahren, die in klinischen Studien Tadalafil entweder zur Behandlung der erektilen Dysfunktion oder zur Behandlung des benignen Prostatasyndroms erhalten haben, sind begrenzt. In Gelegentlich Selten Überempfindlichkeitsreaktionen Angioödem2 Schwindel Schlaganfall1 (einschließlich hämorrhagische Ereignisse), Synkope, vorübergehende ischämische Attacken1, Migräne2, Krampfanfälle, vorübergehende Amnesie (Gedächtnisstörung) Erkrankungen des Immunsystems Erkrankungen des Nervensystems Kopfschmerz Augenerkrankungen Verschwommenes Sehen, Empfin- Gesichtsfeldausfall, Schwellung der Augenlider, dungen, die als Augenschmerzen Bindehautrötung, nicht arteriitische anteriore ischämische Optikusneuropathie (NAION)2, beschrieben wurden Augenvenenverschluss2 Erkrankungen des Ohrs und des Labyrinths Tinnitus Plötzliche Schwerhörigkeit oder Taubheit Tachykardie, Palpitationen Myokardinfarkt, instabile Angina pectoris2, ventrikuläre Arrhythmien2 Herzerkrankungen1 Gefäßerkrankungen Hautrötung Hypotonie3, Hypertonie Erkrankungen der Atemwege, des Brustraums und Mediastinums Verstopfte Nase Atemnot, Epistaxis (Nasenbluten) Erkrankungen des Gastrointestinaltrakts Dyspepsie, gastroösophagealer Reflux Abdominale Schmerzen Erkrankungen der Haut und des Unterhautzellgewebes Hautausschlag, Hyperhidrosis (Schwitzen) Urtikaria, Stevens-Johnson Syndrom2, exfoliative Dermatitis2 Sklelettmuskulatur-, Bindegewebs- und Knochenerkrankungen Rückenschmerzen, Muskelschmerzen, Schmerzen in den Extremitäten Erkrankungen der Nieren und Harnwege Hämaturie Erkrankungen der Geschlechtsorgane und der Brustdrüse Penishämorrhagie, Hämatospermie Lang andauernde Erektionen, Priapismus2 Allgemeine Erkrankungen und Beschwerden am Verabreichungsort Brustschmerz1 (1) (2) (3) 4 Gesichtsödem2, plötzlicher Herztod1,2 Bei den meisten Patienten waren vorbestehende kardiovaskuläre Risikofaktoren bekannt (siehe Abschnitt 4.4). Nach Markteinführung berichtete Nebenwirkungen, die nicht in placebo-kontrollierten Studien beobachtet wurden. Vor allem wurde dies berichtet, wenn Tadalafil von Patienten eingenommen wurde, die bereits mit Antihypertensiva behandelt wurden. 008049-15484 Fachinformation CIALIS® klinischen Studien mit Tadalafil 5 mg in der einmal täglichen Anwendung zur Behandlung des benignen Prostatasyndroms wurden Schwindel und Diarrhö bei Patienten über 75 Jahren häufiger berichtet. Meldung des Verdachts auf Nebenwirkungen Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von großer Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdachtsfall einer Nebenwirkung dem Bundesinstitut für Arzneimittel und Medizinprodukte, Abt. Pharmakovigilanz, KurtGeorg-Kiesinger-Allee 3, D-53175 Bonn, Website: www.bfarm.de anzuzeigen. 4.9 Überdosierung Einzeldosen bis zu 500 mg wurden an gesunden Probanden und Mehrfachdosen bis zu 100 mg täglich an Patienten gegeben. Die unerwünschten Ereignisse waren denen vergleichbar, die bei niedrigeren Dosen gesehen werden. Im Fall einer Überdosierung sollten je nach Bedarf die üblichen unterstützenden Maßnahmen ergriffen werden. Hämodialyse trägt nur unerheblich zur Tadalafil-Elimination bei. 5. PHARMAKOLOGISCHE EIGENSCHAFTEN 5.1 Pharmakodynamische Eigenschaften Pharmakotherapeutische Gruppe: Urologika, Arzneimittel zur Behandlung der erektilen Dysfunktion, ATC-Code: G04BE 08. Wirkmechanismus Erektile Dysfunktion Tadalafil ist ein selektiver, reversibler Hemmstoff der zyklischen Guanosin Monophosphat (cGMP)-spezifischen Phosphodiesterase Typ 5 (PDE5). Wenn eine sexuelle Stimulation die lokale Ausschüttung von Stickstoffoxid verursacht, erzeugt Tadalafil durch die Hemmung der PDE5 erhöhte cGMPSpiegel im Corpus Cavernosum. Dadurch ergibt sich eine Entspannung der glatten Muskulatur und Blut fließt in das Penisgewebe, wodurch eine Erektion hervorgerufen wird. In der Behandlung der erektilen Dysfunktion zeigt Tadalafil ohne sexuelle Stimulation keine Wirkung. Benignes Prostatasyndrom August 2015 Die Wirkung einer PDE5-Hemmung auf die cGMP-Konzentration im Corpus Cavernosum wird auch in der glatten Muskulatur der Prostata, der Blase und den versorgenden Blutgefäßen beobachtet. Die daraus resultierende vaskuläre Entspannung erhöht die Durchblutung, dies könnte der Wirkmechanismus sein, der zu einer Reduktion des benignen Prostatasyndroms führt. Diese vaskulären Effekte könnten durch eine Aktivitätshemmung der afferenten Nerven der Blase und durch eine Entspannung der glatten Muskulatur der Prostata und der Blase unterstützt werden. Pharmakodynamische Wirkungen In vitro Studien haben gezeigt, dass Tadalafil ein selektiver PDE5-Inhibitor ist. PDE5 ist ein Enzym, das sich in der glatten Muskulatur des Corpus Cavernosum, in der 008049-15484 glatten Muskulatur der Gefäße und inneren Organe, im Skelettmuskel, in den Thrombozyten, in der Niere, Lunge und im Kleinhirn findet. Die Tadalafil-Wirkung ist auf PDE5 deutlich stärker als auf andere Phosphodiesterasen. Tadalafil wirkt mehr als 10.000-fach stärker auf PDE5 als auf PDE 1, PDE 2 und PDE 4, Enzyme, die im Herz, im Hirn, in den Blutgefäßen, der Leber und weiteren Organen vorkommen. Tadalafil wirkt mehr als 10.000-fach stärker auf PDE5 als auf PDE 3, ein Enzym, das im Herz und in Blutgefäßen vorkommt. Die im Vergleich zu PDE 3 höhere Selektivität für PDE5 ist von Bedeutung, da das Enzym PDE 3 die Kontraktionsfähigkeit des Herzens mit beeinflusst. Zusätzlich ist die Tadalafil-Wirkung auf PDE5 etwa 700-fach stärker als auf PDE 6, ein Enzym, das in der Retina gefunden wird und für die Phototransduktion verantwortlich ist. Tadalafil wirkt ebenfalls mehr als 10.000-fach stärker auf PDE5 als auf PDE 7 bis PDE 10. Klinische Wirksamkeit und Sicherheit Bei gesunden Probanden verursachte Tadalafil verglichen mit Placebo keine signifikanten Veränderungen des systolischen und diastolischen Blutdrucks im Liegen (mittlere maximale Abnahme 1,6 bzw. 0,8 mm Hg) sowie des systolischen und diastolischen Blutdrucks im Stehen (mittlere maximale Abnahme von 0,2 bzw. 4,6 mm Hg) und keine signifikante Änderung der Pulsfrequenz. In einer Studie zur Untersuchung der Wirkung von Tadalafil auf die Sehfähigkeit wurde mit dem Farnsworth Munsell 100-hue Test keine Beeinträchtigung der Farbunterscheidung (blau/grün) festgestellt. Dieses Ergebnis steht im Einklang mit der geringen Affinität des Tadalafils für PDE 6 verglichen mit PDE5. In allen klinischen Studien waren Berichte über Änderungen des Farbsehens selten (< 0,1 %). Bei Männern wurden drei Studien durchgeführt, um den möglichen Effekt von CIALIS 10 mg (eine 6-monatige Studie) und 20 mg (eine 6-monatige und eine 9-monatige Studie) bei einer täglichen Einnahme auf die Spermatogenese zu untersuchen. In zwei dieser Studien wurden eine Abnahme der Spermienzahl und der -konzentration im Zusammenhang mit der Tadalafil-Behandlung beobachtet, die wahrscheinlich nicht klinisch relevant sind. Diese Effekte standen nicht im Zusammenhang mit der Veränderung anderer Werte, wie z. B.: Motalität, Morphologie und FSH (follikelstimulierendes Hormon). Erektile Dysfunktion (Bedarfs- oder Konstanztherapie) Zu CIALIS nach Bedarf wurden drei klinische Studien mit 1054 Patienten unter häuslichen Bedingungen durchgeführt, um die Wirkdauer zu bestimmen. Tadalafil zeigte eine statistisch signifikante Verbesserung der erektilen Funktion. Es befähigte zu erfolgreichem Geschlechtsverkehr bis zu 36 Stunden nach der Einnahme, ebenso wie es den Patienten im Vergleich zu Placebo ermöglichte, bereits 16 Minuten nach der Einnahme eine Erektion zu bekommen und für einen erfolgreichen Verkehr aufrecht zu erhalten. In einer 12-wöchigen Studie, die mit 186 Patienten (142 Tadalafil, 44 Placebo) mit erektiler Dysfunktion bei Rückenmarksverletzung durchgeführt wurde, verbesserte Tadalafil signifikant die erektile Funktion. Die Einnahme von Tadalafil 10 oder 20 mg (flexible Dosierung, nach Bedarf) erhöhte die erfolgreichen Versuche pro Patient im Mittel auf 48 % im Vergleich zu 17 % unter Placebo. Zur Evaluierung der einmal täglichen Einnahme von Tadalafil wurden anfänglich 3 klinische Studien in den Dosierungen 2,5 mg, 5 mg und 10 mg an 853 Patienten durchgeführt. Die Patienten unterschiedlicher ethnischer Zugehörigkeit im Alter (von 21 – 82 Jahren) litten unter erektiler Dysfunktion unterschiedlicher Ätiologie und Ausprägung (leicht, mäßig, schwer). In den beiden primären Wirksamkeitsstudien in der Gesamtpopulation betrug die mittlere Rate erfolgreicher Versuche von Geschlechtsverkehr pro Person 57 und 67 % mit CIALIS 5 mg und 50 % mit CIALIS 2,5 mg im Vergleich zu 31 und 37 % mit Placebo. In der Studie bei Patienten mit erektiler Dysfunktion als Folge eines Diabetes betrug die mittlere Rate erfolgreicher Versuche Geschlechtsverkehr auszuüben, pro Person 41 und 46 % mit CIALIS 5 mg bzw. 2,5 mg im Vergleich zu 28 % mit Placebo. Die meisten Patienten in diesen 3 Studien hatten auf eine vorangegangene PDE5-InhibitorTherapie nach Bedarf angesprochen. In einer weiteren Studie wurden 217 Patienten, die noch keine PDE5-Inhibitoren erhalten hatten, auf CIALIS 5 mg einmal täglich bzw. Placebo randomisiert. Die mittlere Rate erfolgreicher Versuche von Geschlechtsverkehr betrug pro Person 68 % für CIALISPatienten im Vergleich zu 52 % für PlaceboPatienten. Tadalafil wurde in 16 klinischen Studien in Dosierungen zwischen 2 und 100 mg an 3250 Patienten untersucht. Die Patienten litten unter erektiler Dysfunktion unterschiedlicher Ausprägung (leicht, mäßig, schwer), Ätiologie, Alter (von 21 – 86 Jahre) und ethnischer Zugehörigkeit. Die meisten Patienten berichteten über eine schon mindestens ein Jahr andauernde erektile Dysfunktion. In den primären Wirksamkeitsstudien in der Gesamtpopulation berichteten 81 % der Patienten über eine verbesserte Erektion unter CIALIS im Vergleich zu 35 %unter Placebo. Ebenso berichteten Patienten mit erektiler Dysfunktion aller Schweregrade über eine Verbesserung der Erektion nach der Einnahme von CIALIS (86 %bei geringer, 83 % bei mittlerer und 72 %bei schwerer erektiler Dysfunktion im Vergleich zu 45 %, 42 % und 19 % bei Placebo). Versuche, Geschlechtsverkehr auszuüben, waren in den primären Wirksamkeitsstudien bei 75 % der mit CIALIS behandelten Patienten erfolgreich, verglichen mit 32 % unter Placebo. Benignes Prostatasyndrom CIALIS wurde bei Männern mit einem benignen Prostatasyndrom in 4 klinischen Studien über einen Zeitraum von 12 Wochen mit über 1500 Patienten untersucht. In diesen vier Studien war die Verbesserung des Gesamt IPSS (International Prostate Symptom Score)mit CIALIS 5 mg – 4,8; – 5,6; – 6,1 und – 6.3 im Vergleich zu Placebo mit 5 Fachinformation CIALIS® – 2,2; – 3,6; – 3,8 und – 4,2. Die Verbesserung des Gesamt IPSS (International Prostate Symptom Score) trat frühestens nach 1 Woche auf. In der Studie, die auch Tamsulosin 0,4 mg als einen aktiven Vergleichsarm führte, war die Verbesserung der Gesamtpunktzahl des IPSS (International Prostate Symptom Score) mit CIALIS 5 mg, Tamsulosin und Placebo jeweils entsprechend: – 6,3; – 5,7 und – 4,2. Eine dieser Studien untersuchte die Verbesserung der erektilen Dysfunktion und des benignen Prostatasyndroms bei Patienten mit beiden Erkrankungen. In dieser Studie wurde die Verbesserung anhand des erektilen Dysfunktionsbereichs des IIEF (International Index of Erectile Function) und des Gesamt IPSS (International Prostate Symptom Score) beurteilt. Der IIEF und der IPSS waren 6,5 und – 6,1 mit CIALIS5 mg im Vergleich zu 1,8 und – 3,8 bei Placebo. Mit CIALIS 5 mg lag der durchschnittliche prozentuale Anteil von erfolgreich durchgeführten Geschlechtsverkehrversuchen bei 71,9 % im Vergleich zu 48,3 % mit Placebo. Die Verbesserung, die im Gesamt IPSS nach 12 Wochen gesehen wurde, konnte in der open-label Verlängerung einer dieser Studien, in der die Behandlung mit CIALIS 5 mg bis zu einem Jahr fortgeführt wurde; über diesen Zeitraum erhalten bleiben. Kinder und Jugendliche Die Europäische Arzneimittel-Agentur hat eine Freistellung von der Verpflichtung zur Vorlage von Ergebnissen zu Studien in allen pädiatrischen Altersklassen in der Behandlung der erektilen Dysfunktion gewährt (siehe Abschnitt 4.2 bzgl. Informationen zur Anwendung bei Kindern und Jugendlichen). 5.2 Pharmakokinetische Eigenschaften Resorption Tadalafil wird nach oraler Gabe gut resorbiert und die mittlere maximale Plasmakonzentration (cmax) wird meist 2 Stunden nach Einnahme erreicht. Die absolute Bioverfügbarkeit von Tadalafil nach oraler Gabe wurde nicht ermittelt. Rate und Ausmaß der Tadalafil-Resorption werden durch Nahrungsmittel nicht beeinflusst, daher kann CIALIS unabhängig von den Mahlzeiten eingenommen werden. Der Zeitpunkt der Einnahme (morgens oder abends) hat keine klinisch relevanten Auswirkungen auf Geschwindigkeit und Ausmaß der Resorption. Verteilung Das mittlere Verteilungsvolumen beträgt etwa 63 l, was darauf hindeutet, dass Tadalafil im Gewebe verteilt wird. In therapeutischen Konzentrationen beträgt die Plasmaproteinbindung von Tadalafil 94 %. Die Proteinbindung wird durch eine gestörte Nierenfunktion nicht beeinträchtigt. Weniger als 0,0005 % der eingenommenen Dosis fand sich im Samen von gesunden Probanden. Biotransformation Tadalafil wird hauptsächlich durch die Cytochrom P450 (CYP) 3A4 Isoform metabolisiert. Der zirkulierende Hauptmetabolit ist das Methylcatecholglucuronid. Dieser Meta6 bolit ist auf PDE5 mindestens 13.000-fach weniger wirksam als Tadalafil. Eine klinische Wirkung des Metaboliten ist bei den ermittelten Konzentrationen daher nicht zu erwarten. Elimination Bei gesunden Probanden beträgt die mittlere Clearance für Tadalafil nach oraler Gabe 2,5 l/h und die mittlere Halbwertszeit 17,5 Stunden. Tadalafil wird hauptsächlich in Form inaktiver Metaboliten ausgeschieden, vorwiegend über die Faeces (etwa 61 % der Dosis) und zu einem geringeren Teil über den Urin (etwa 36 % der Dosis). Linearität/Nicht-Linearität Die Pharmakokinetik von Tadalafil ist bei gesunden Probanden im Hinblick auf Zeit und Dosis linear. Über den Dosisbereich von 2,5 bis 20 mg steigt die Exposition (AUC) proportional mit der Dosis. Eine Steady State Plasmakonzentration wird bei einmal täglicher Gabe innerhalb von 5 Tagen erreicht. Die mit dem Populationsansatz bestimmte Pharmakokinetik bei Patienten mit erektiler Dysfunktion ist der bei Personen ohne erektile Dysfunktion vergleichbar. Besondere Patientengruppen Ältere Männer Gesunde ältere Männer (65 Jahre oder älter) zeigten nach oraler Gabe von Tadalafil eine niedrigere Clearance, was zu einer 25 % höheren Bioverfügbarkeit (AUC) im Verhältnis zu gesunden Probanden im Alter zwischen 19 bis 45 Jahren führte. Dieser Effekt des Alters ist klinisch nicht signifikant und erfordert keine Dosisanpassung. Nierenfunktionsstörung In klinisch-pharmakologischen Studien, in denen Einzeldosen Tadalafil (5 mg bis 20 mg) verabreicht wurden, war bei Männern mit leichter (Kreatinin Clearance 51 bis 80 ml/ min) oder mäßiger (Kreatinin Clearance 31 bis 50 ml/min) Nierenfunktionsstörung sowie bei Dialyse-Patienten mit terminalem Nierenversagen die Bioverfügbarkeit (AUC) von Tadalafil ungefähr verdoppelt. Cmax war bei dialysepflichtigen Patienten gegenüber dem bei gesunden Männern gemessenen Wert um 41 % erhöht. Hämodialyse trägt nur unerheblich zur Tadalafil-Elimination bei. Leberfunktionsstörung Die Bioverfügbarkeit von Tadalafil (AUC) bei Männern mit leichter und mäßiger Leberfunktionsstörung (Child-Pugh Class A und B) ist mit der bei gesunden Männern vergleichbar, wenn eine 10 mg Dosis gegeben wird. Für Patienten mit Leberfunktionsstörung liegen keine Daten über die Gabe höherer Dosen als 10 mg Tadalafil vor. Es existieren nur begrenzt klinische Daten zur Unbedenklichkeit von CIALIS bei Patienten mit schwerer Leberinsuffizienz (Child-Pugh Klasse C). Die einmal tägliche Anwendung von Tadalafil wurde bei Patienten mit Leberfunktionsstörung nicht untersucht. Daher sollte vor der Verordnung von CIALIS in der einmal täglichen Dosierung oder auch in der Bedarfstherapie der verschreibende Arzt eine sorgfältige, individuelle NutzenRisiko Abwägung durchführen. Diabetiker Die Bioverfügbarkeit (AUC) von Tadalafil war bei Diabetikern etwa 19 %niedriger, als der AUC-Wert von gesunden Probanden. Dieser Unterschied in der Bioverfügbarkeit erfordert keine Dosisanpassung. 5.3 Präklinische Daten zur Sicherheit Basierend auf den konventionellen Studien zur Sicherheitspharmakologie, Toxizität bei wiederholter Gabe, Reproduktionstoxizität, Genotoxizität und zum kanzerogenen Potential lassen die präklinischen Daten keine besonderen Gefahren für den Menschen erkennen. Bei Ratten oder Mäusen, die bis zu 1000 mg/kg Tadalafil täglich erhielten, gab es keinen Hinweis auf Teratogenität, Embryotoxizität oder Fetotoxizität. Bei einer prä- und postnatalen Entwicklungsstudie an Ratten war die höchste Dosis, bei der keine toxikologischen Effekte beobachtet wurden, 30 mg/kg/Tag. Bei trächtigen Ratten war die AUC für die berechnete ungebundene Substanz bei dieser Dosis etwa 18-mal höher als die AUC beim Menschen bei einer 20 mg Dosis. Die Fertilität bei männlichen und weiblichen Ratten wurde nicht beeinträchtigt. Bei Hunden, denen Tadalafil 6 bis 12 Monate lang täglich in Dosierungen von 25 mg/kg/Tag und mehr gegeben wurde (und die dadurch einer zumindest 3-mal höheren Menge [Faktor 3,7 bis 18,6] ausgesetzt waren als Menschen nach einer 20 mg Einzeldosis), wurde eine Rückbildung des Epithels der Tubuli Seminiferi beobachtet, die zu einer Abnahme der Spermatogenese bei einigen Hunden führte. Siehe auch Abschnitt 5.1. 6. PHARMAZEUTISCHE ANGABEN 6.1 Liste der sonstigen Bestandteile Tablettenkern: Lactose-Monohydrat, Croscarmellose-Natrium, Hyprolose, mikrokristalline Cellulose, Natriumdodecylsulfat, Magnesiumstearat. Filmüberzug: Lactose-Monohydrat, Hypromellose, Triacetin, Titandioxid (E 171), Eisen (III)-hydroxid-oxid × H2O (E 172), Talkum. 6.2 Inkompatibilitäten Nicht zutreffend. 6.3 Dauer der Haltbarkeit 3 Jahre. 6.4 Besondere Vorsichtsmaßnahmen für die Aufbewahrung In der Originalverpackung aufbewahren, um den Inhalt vor Feuchtigkeit zu schützen. CIALIS 5 mg: Nicht über 25 °C lagern. CIALIS 10/20 mg: Nicht über 30 °C lagern. 008049-15484 Fachinformation CIALIS® 6.5 Art und Inhalt des Behältnisses Aluminium/PVC/PE/PCTFE Blisterpackungen in Faltschachteln CIALIS 5 mg: 14, 28 oder 84 Filmtabletten CIALIS 10 mg: 4 Filmtabletten CIALIS 20 mg: 2, 4, 8, 10 oder 12 Filmtabletten Es werden möglicherweise nicht alle Packungsgrößen in den Verkehr gebracht. *) CIALIS® 2,5 mg wird derzeit in Deutschland nicht vermarktet. 13. KONTAKTADRESSE IN DEUTSCHLAND Lilly Deutschland GmbH Werner-Reimers-Str. 2-4 61352 Bad Homburg Medizinische Information: Tel. 06172 273-2222 www.cialis.de 6.6 Besondere Vorsichtsmaßnahmen für die Beseitigung Keine besonderen Anforderungen. 7. INHABER DER ZULASSUNG Eli Lilly Nederland B.V. Grootslag 1-5 NL-3991 RA Houten Niederlande 8. ZULASSUNGSNUMMER(N) CIALIS® 5 mg, 14 Filmtabletten: EU/1/02/237/007 CIALIS® 5 mg, 28 Filmtabletten: EU/1/02/237/008 CIALIS® 5 mg, 84 Filmtabletten: EU/1/02/237/010 CIALIS® 10 mg, 4 Filmtabletten: EU/1/02/237/001 CIALIS® 20 mg, 2 Filmtabletten: EU/1/02/237/002 CIALIS® 20 mg, 4 Filmtabletten: EU/1/02/237/003 CIALIS® 20 mg, 8 Filmtabletten: EU/1/02/237/004 CIALIS® 20 mg, 10 Filmtabletten: EU/1/02/237/009 CIALIS® 20 mg, 12 Filmtabletten: EU/1/02/237/005 9. DATUM DER ERTEILUNG DER ZULASSUNG/VERLÄNGERUNG DER ZULASSUNG Datum der Erteilung der Zulassung: 12. November 2002 Datum der letzten Verlängerung der Zulassung:12. November 2012 10. STAND DER INFORMATION August 2015 Ausführliche Informationen zu diesem Arzneimittel sind auf den Internetseiten der Europäischen Arzneimittel-Agentur http://www.ema.europa.eu/ verfügbar. 11. VERSCHREIBUNGSSTATUS/ APOTHEKENPFLICHT August 2015 Verschreibungspflichtig 12. PACKUNGSGRÖSSEN in Deutschland Blisterpackungen in Faltschachteln CIALIS® 5 mg: 14, 28, 84 Filmtabletten CIALIS® 10 mg: 4 Filmtabletten CIALIS® 20 mg: 4, 8, 12 Filmtabletten 008049-15484 Zentrale Anforderung an: Rote Liste Service GmbH Fachinfo-Service Mainzer Landstraße 55 60329 Frankfurt 7 Fachinformation Levitra® 20 mg Filmtabletten 1. BEZEICHNUNG DES ARZNEIMITTELS Levitra® 20 mg Filmtabletten 2. QUALITATIVE UND QUANTITATIVE ZUSAMMENSETZUNG Jede Tablette enthält 20 mg Vardenafil (als Hydrochlorid). Vollständige Auflistung der sonstigen Bestandteile, siehe Abschnitt 6.1. 3. DARREICHUNGSFORM Filmtablette. Orangefarbene runde Tabletten, die auf einer Seite mit dem „Bayer-Kreuz“ und auf der anderen Seite mit „20“ gekennzeichnet sind. 4. KLINISCHE ANGABEN 4.1 Anwendungsgebiete Behandlung der erektilen Dysfunktion bei erwachsenen Männern. Erektile Dysfunktion ist die Unfähigkeit, eine für einen befriedigenden Geschlechtsverkehr ausreichende Erektion des Penis zu erreichen oder aufrechtzuerhalten. Damit Levitra wirken kann, ist eine sexuelle Stimulation erforderlich. 4.2 Dosierung und Art der Anwendung Dosierung Anwendung bei erwachsenen Männern Die empfohlene Dosis beträgt 10 mg, sie ist bei Bedarf ungefähr 25 bis 60 Minuten vor dem Geschlechtsverkehr einzunehmen. Entsprechend der Wirksamkeit und Verträglichkeit kann die Dosis auf 20 mg erhöht oder auf 5 mg verringert werden. Die empfohlene Maximaldosis beträgt 20 mg. Die Einnahme darf nicht häufiger als einmal täglich erfolgen. Levitra kann mit oder ohne Nahrung eingenommen werden. Der Wirkungseintritt kann bei gleichzeitiger Einnahme einer fettreichen Mahlzeit verzögert sein (siehe Abschnitt 5.2). Spezielle Patientengruppen Ältere Menschen (≥ 65 Jahre) Bei älteren Patienten ist keine Dosisanpassung erforderlich. Jedoch sollte bei ihnen eine Dosissteigerung bis zu einer Maximaldosis von 20 mg sorgfältig unter Berücksichtigung der individuellen Verträglichkeit abgewogen werden (siehe Abschnitte 4.4 und 4.8). Eingeschränkte Leberfunktion April 2014 DE/20 Die empfohlene Anfangsdosis für Patienten mit leichter und mäßig eingeschränkter Leberfunktion (Child-Pugh A-B) beträgt 5 mg. Entsprechend der Verträglichkeit und Wirksamkeit kann die Dosis anschließend erhöht werden. Die empfohlene Maximaldosis bei Patienten mit mäßig eingeschränkter Leberfunktion (Child-Pugh B) beträgt 10 mg (siehe Abschnitte 4.3 und 5.2). Eingeschränkte Nierenfunktion Bei Patienten mit einer leichten bis mäßigen Einschränkung der Nierenfunktion ist keine Dosisanpassung erforderlich. Bei Patienten mit stark eingeschränkter Nierenfunktion (Kreatinin-Clearance < 30 ml/min) beträgt die empfohlene An002574-4967 fangsdosis 5 mg. Entsprechend der Verträglichkeit und Wirksamkeit kann die Dosis auf 10 mg und dann auf 20 mg erhöht werden. Kinder und Jugendliche Levitra ist nicht für die Anwendung bei Personen unter 18 Jahren angezeigt. Für Levitra gibt es keine Indikation für die Anwendung bei Kindern. Anwendung bei Patienten, die andere Arzneimittel anwenden Gleichzeitige Anwendung von CYP3A4Inhibitoren Bei gleichzeitiger Anwendung von CYP3A4Inhibitoren wie Erythromycin oder Clarithromycin sollte die Vardenafil-Dosis 5 mg nicht überschreiten (siehe Abschnitt 4.5). Art der Anwendung Zum Einnehmen. 4.3 Gegenanzeigen Überempfindlichkeit gegen den Wirkstoff oder einen der in Abschnitt 6.1 genannten sonstigen Bestandteile. Die gleichzeitige Anwendung von Vardenafil und Nitraten oder Stickstoffmonoxid-Donatoren (wie Amylnitrit) in jeder Form ist kontraindiziert (siehe Abschnitte 4.5 und 5.1). Bei Patienten, die aufgrund einer nichtarteriitischen anterioren ischämischen Optikusneuropathie (NAION) ihre Sehkraft auf einem Auge verloren haben, ist Levitra kontraindiziert, unabhängig davon, ob der Sehverlust mit einer vorherigen Einnahme eines Phosphodiesterase-5 (PDE5)-Hemmers in Zusammenhang stand oder nicht (siehe Abschnitt 4.4). Arzneimittel zur Behandlung der erektilen Dysfunktion dürfen generell nicht von Männern angewendet werden, denen von sexuellen Aktivitäten abzuraten ist (z. B. Patienten mit schweren Herz-Kreislauf-Erkrankungen wie instabiler Angina pectoris oder schwerer Herzinsuffizienz [New York Heart Association III oder IV]). Bei folgenden Untergruppen von Patienten wurde die Sicherheit von Vardenafil nicht untersucht, daher ist die Anwendung kontraindiziert, bis weitere Daten vorliegen: – Patienten mit schwerer Leberinsuffizienz (Child-Pugh C), – im Endstadium einer Nierenfunktionsstörung mit Dialysepflicht, – Patienten mit Hypotonie (Blutdruck < 90/50 mmHg), – mit kürzlich erlittenem Schlaganfall oder Herzinfarkt (innerhalb der letzten 6 Monate), – mit instabiler Angina pectoris sowie mit bekannten erblich bedingten degenerativen Retinaerkrankungen wie Retinitis pigmentosa. Bei Männern über 75 Jahre ist die gleichzeitige Anwendung von Vardenafil mit den starken CYP3A4-Inhibitoren Itraconazol und Ketoconazol (orale Darreichungsform) kontraindiziert. Die gleichzeitige Anwendung von Vardenafil mit HIV Protease-Inhibitoren wie zum Beispiel Ritonavir und Indinavir ist kontraindiziert, weil sie sehr starke CYP3A4-Inhibitoren sind (siehe Abschnitt 4.5). 4.4 Besondere Warnhinweise und Vorsichtsmaßnahmen für die Anwendung Bevor eine medikamentöse Therapie in Betracht gezogen wird, muss die Diagnose einer erektilen Dysfunktion mittels Anamnese und körperlicher Untersuchung gestellt und zugrunde liegende Ursachen ermittelt werden. Vor dem Beginn jeglicher Behandlung einer erektilen Dysfunktion müssen die behandelnden Ärzte den kardiovaskulären Status ihrer Patienten berücksichtigen, da mit sexueller Aktivität ein gewisses kardiales Risiko einhergeht (siehe Abschnitt 4.3). Vardenafil besitzt vasodilatatorische Eigenschaften, die zu leichten und vorübergehenden Blutdrucksenkungen führen (siehe Abschnitt 5.1). Patienten mit einer Obstruktion der linksventrikulären Ausstrombahn, zum Beispiel Aortenstenose und idiopathischer hypertrophischer Subaortenstenose, können empfindlich gegenüber gefäßerweiternden Substanzen einschließlich PDE5-Inhibitoren reagieren. Die Behandlung einer erektilen Dysfunktion mit Arzneimitteln muss dann mit Vorsicht erfolgen, wenn anatomische Missbildungen des Penis (wie Angulation, Fibrose im Bereich der Corpora cavernosa oder die Peyronie-Krankheit) vorliegen, sowie bei Patienten mit für Priapismus prädisponierenden Erkrankungen (wie Sichelzellenanämie, Plasmozytom, Leukämie). Es liegen keine Studien zur Unbedenklichkeit und Wirksamkeit von Levitra Filmtabletten in Kombination mit Levitra Schmelztabletten oder anderen Behandlungen einer erektilen Dysfunktion vor. Die Anwendung solcher Kombinationen wird daher nicht empfohlen. Die Verträglichkeit der Maximaldosis von 20 mg kann bei älteren Patienten (≥ 65 Jahre) geringer sein (siehe Abschnitte 4.2 und 4.8). Gleichzeitige Anwendung von AlphaRezeptorenblockern Die gleichzeitige Anwendung von AlphaRezeptorenblockern und Vardenafil kann bei einigen Patienten zu einer unerwünscht starken Blutdrucksenkung führen, da beide Arzneimittel gefäßerweiternd wirken. Eine Begleitbehandlung mit Vardenafil sollte nur begonnen werden, wenn der Patient stabil auf die Alpha-Rezeptorenblocker-Therapie eingestellt ist. Bei den Patienten, die stabil auf Alpha-Rezeptorenblocker-Therapie eingestellt sind, sollte Vardenafil mit der niedrigsten empfohlenen Anfangsdosis von 5 mg Filmtabletten begonnen werden. Vardenafil kann zu jeder Zeit mit Tamsulosin oder mit Alfuzosin angewandt werden. Bei anderen Alpha-Rezeptorenblockern sollte ein zeitlicher Abstand der Gabe berücksichtigt werden, wenn Vardenafil als Begleitbehandlung verordnet wird (siehe Abschnitt 4.5). Bei den Patienten, die schon eine optimierte Dosis Vardenafil einnehmen, sollte die Alpha-Rezeptorenblocker-Therapie mit der niedrigsten Dosis begonnen werden. Die schrittweise Erhöhung der Alpha-Rezeptorenblocker-Dosis kann bei Patienten, die Vardenafil einnehmen, mit einer weiteren Blutdrucksenkung einhergehen. 1 Fachinformation Levitra® 20 mg Filmtabletten Gleichzeitige Anwendung von CYP3A4Inhibitoren Die gleichzeitige Anwendung von Vardenafil mit starken CYP3A4-Inhibitoren wie zum Beispiel Itraconazol und Ketoconazol (orale Darreichungsform) sollte vermieden werden, da bei Kombination dieser Arzneimittel sehr hohe Plasmakonzentrationen von Vardenafil erreicht werden (siehe Abschnitte 4.5 und 4.3). Bei gleichzeitiger Anwendung von moderaten CYP3A4-Inhibitoren wie Erythromycin und Clarithromycin kann eine Dosisanpassung von Vardenafil notwendig sein (siehe Abschnitte 4.5 und 4.2). Bei gleichzeitiger Einnahme von Grapefruit oder Grapefruitsaft ist ein Anstieg der Vardenafil-Plasmakonzentrationen zu erwarten. Daher muss diese Kombination vermieden werden (siehe Abschnitt 4.5). Wirkung auf das QTc Intervall Orale Einmalgaben von 10 mg und 80 mg Vardenafil führten im Mittel zu Verlängerungen des QTc Intervalls um 8 msec resp. 10 msec. Einmalgaben von 10 mg Vardenafil, die gleichzeitig mit 400 mg Gatifloxacin, einem Wirkstoff mit vergleichbarer Wirkung auf QT, eingenommen wurden, führten zu einer additiven Wirkung auf QTc von 4 msec im Vergleich zu den Wirkungen der einzelnen Wirkstoffe. Die klinische Relevanz dieser QT-Änderungen ist unbekannt (siehe Abschnitt 5.1). Die klinische Relevanz dieses Befundes ist unbekannt und kann nicht für alle Patienten unter allen Bedingungen verallgemeinert werden, da dies von den individuellen Risikofaktoren sowie der Empfindlichkeit des einzelnen Patienten zu einem gegebenen Zeitpunkt abhängt. Arzneimittel, die das QTc Intervall verlängern können, einschließlich Vardenafil, sollten bei Patienten mit relevanten Risikofaktoren, zum Beispiel Hypokaliämie, angeborener QT Verlängerung, gleichzeitiger Anwendung von Antiarrhythmika der Klasse IA (z. B. Chinidin, Procainamid) oder der Klasse III (z. B. Amiodaron, Sotalol) möglichst vermieden werden. Wirkung auf das Sehvermögen Sehstörungen und Fälle von nichtarteriitischer ischämischer Optikusneuropathie (NAION) sind in Zusammenhang mit der Einnahme von Levitra und anderen PDE5Hemmern beobachtet worden. Die Patienten müssen darüber aufgeklärt werden, dass sie im Falle einer plötzlichen Sehstörung Levitra absetzen und sofort einen Arzt aufsuchen sollen (siehe Abschnitt 4.3). Wirkung auf Blutungen In vitro Studien an menschlichen Thrombozyten haben Hinweise darauf erbracht, dass Vardenafil keine eigenständigen antiaggregatorischen Eigenschaften zeigt, aber bei hohen Konzentrationen (oberhalb des therapeutischen Bereichs) die antiaggregatorische Wirkung des StickstoffmonoxidDonators Nitroprussid-Natrium verstärkt. Beim Menschen hat weder die alleinige Anwendung von Vardenafil noch die Kombination mit Acetylsalicylsäure einen Effekt auf die Blutungszeit (siehe Abschnitt 4.5). Es liegen keine Daten über die Unbedenklichkeit der Verabreichung von Vardenafil an 2 Patienten mit Blutungsstörungen oder aktiven peptischen Ulzera vor. Daher sollte Vardenafil von diesen Patienten nur nach sorgfältiger Nutzen-Risiko-Abwägung angewendet werden. 4.5 Wechselwirkungen mit anderen Arzneimitteln und sonstige Wechselwirkungen Wirkungen Vardenafil anderer Arzneimittel auf In vitro Studien Vardenafil wird hauptsächlich in der Leber durch das Cytochrom P450 (CYP) Isoenzym 3A4 mit geringer Beteiligung der CYP3A5- und CYP2C-Isoenzyme metabolisiert. Inhibitoren dieser Isoenzyme können daher die Vardenafil-Clearance vermindern. In vivo Studien Die gleichzeitige Anwendung des HIV Protease-Hemmstoffs Indinavir (3-mal täglich 800 mg), eines starken CYP3A4-Inhibitors, mit Vardenafil (10 mg Filmtablette) bewirkte eine 16-fache Steigerung der VardenafilAUC und eine 7-fache Steigerung der Vardenafil-Cmax. Nach 24 Stunden waren die Vardenafil-Plasmaspiegel wieder auf ungefähr 4 % der maximalen Vardenafil-Plasmaspiegel (Cmax) gesunken. Die gleichzeitige Anwendung von Ritonavir (2-mal täglich 600 mg) und Vardenafil 5 mg führte zu einem 13-fachen Anstieg der Vardenafil-Cmax und einer 49-fachen Steigerung der Vardenafil-AUC0 – 24. Diese Wechselwirkung ist eine Folge der Blockierung der hepatischen Metabolisierung von Vardenafil durch Ritonavir, einen hochpotenten CYP3A4-Inhibitor, der gleichzeitig CYP2C9 hemmt. Ritonavir verlängert die Halbwertszeit von Vardenafil signifikant auf 25,7 Stunden (siehe Abschnitt 4.3). Die gleichzeitige Anwendung von Ketoconazol (200 mg), einem starken CYP3A4-Inhibitor mit Vardenafil (5 mg) bewirkte eine 10-fache Steigerung der Vardenafil-AUC und eine 4-fache Steigerung der VardenafilCmax (siehe Abschnitt 4.4). Obwohl spezifische Interaktionsstudien nicht durchgeführt wurden, ist zu erwarten, dass die gleichzeitige Anwendung anderer starker CYP3A4-Inhibitoren (wie Itraconazol) Vardenafil-Plasmaspiegel in vergleichbarer Höhe bewirken wie unter Anwendung mit Ketoconazol. Die gleichzeitige Anwendung von Vardenafil mit starken CYP3A4-Inhibitoren wie zum Beispiel Itraconazol und Ketoconazol (orale Anwendung) sollte vermieden werden (siehe Abschnitte 4.3 und 4.4). Bei Männern über 75 Jahre ist die gleichzeitige Anwendung von Vardenafil mit Itraconazol oder Ketoconazol kontraindiziert (siehe Abschnitt 4.3). Die gleichzeitige Anwendung von Erythromycin (3-mal täglich 500 mg), einem CYP3A4-Inhibitor, und Vardenafil (5 mg) bewirkte eine 4-fache Steigerung der Vardenafil-AUC und eine 3-fache Steigerung der Vardenafil-Cmax. Obwohl eine spezifische Interaktionsstudie nicht durchgeführt wurde, ist zu erwarten, dass die gleichzeitige Anwendung mit Clarithromycin zu vergleichbaren Wirkungen auf Vardenafil-AUC und Cmax führen würde. Bei gleichzeitiger Anwendung mit einem moderaten CYP3A4Inhibitor wie Erythromycin oder Clarithromycin kann eine Anpassung der VardenafilDosis notwendig sein (siehe Abschnitt 4.2 und Abschnitt 4.4). Cimetidin (2-mal täglich 400 mg), ein unspezifischer CytochromP450-Inhibitor, zeigte bei gleichzeitiger Anwendung von Vardenafil (20 mg) bei gesunden Probanden keinen Effekt auf die Vardenafil-AUC und -Cmax. Grapefruitsaft kann als schwacher Hemmstoff des CYP3A4-Stoffwechsels in der Darmwand eine geringe Steigerung des Vardenafil-Plasmaspiegels bewirken (siehe Abschnitt 4.4). Die Pharmakokinetik von Vardenafil (20 mg) wurde durch die gleichzeitige Anwendung des H2-Antagonisten Ranitidin (2-mal täglich 150 mg), Digoxin, Warfarin, Glibenclamid, Alkohol (mittlerer maximaler Blut-Alkohol-Spiegel von 73 mg/dl) oder die Einmalgabe von Antacida (Magnesiumhydroxid/Aluminiumhydroxid) nicht beeinflusst. Obwohl spezifische Interaktionsstudien nicht für alle Arzneimittel durchgeführt wurden, erbrachte die Analyse pharmakokinetischer Daten keine Veränderungen der pharmakokinetischen Parameter von Vardenafil bei gleichzeitiger Anwendung von folgenden Arzneimitteln: Acetylsalicylsäure, ACE-Hemmern, Betablockern, schwachen CYP3A4Inhibitoren, Diuretika und Arzneimitteln zur Behandlung von Diabetes (Sulfonylharnstoff und Metformin). Wirkungen von Vardenafil auf andere Arzneimittel Es liegen keine Daten zu Wechselwirkungen von Vardenafil mit nicht-spezifischen Phosphodiesterasehemmern wie Theophyllin oder Dipyridamol vor. In vivo Studien In einer Studie an 18 gesunden männlichen Probanden wurde keine Verstärkung des blutdrucksenkenden Effektes von sublingual gegebenem Nitroglycerin (0,4 mg) bei Einnahme von Vardenafil (10 mg) in unterschiedlichen Abständen (1 – 24 Stunden) vor der Anwendung von Nitroglycerin beobachtet. Vardenafil 20 mg Filmtablette verstärkte bei gesunden Probanden mittleren Alters die blutdrucksenkende Wirkung von sublingual gegebenem Nitroglycerin (0,4 mg), wenn dieses 1 Stunde und 4 Stunden nach Einnahme von Vardenafil angewandt wurde. Wenn Nitroglycerin 24 Stunden nach einer Einmaldosis von 20 mg Vardenafil Filmtablette verabreicht wurde, war kein Effekt auf den Blutdruck zu beobachten. Es liegen jedoch keine Informationen über eine mögliche Verstärkung des blutdrucksenkenden Effektes von Nitraten und Vardenafil bei Patienten vor, daher ist die gleichzeitige Anwendung kontraindiziert (siehe Abschnitt 4.3). Nicorandil ist ein Wirkstoff, der gleichzeitig als Kaliumkanalöffner und als Nitrat wirkt. Auf Grund der Nitratkomponente besteht die Möglichkeit einer schwerwiegenden Wechselwirkung mit Vardenafil. Da eine Alpha-Rezeptorenblocker-Monotherapie zu einer ausgeprägten Blutdrucksenkung, speziell zu orthostatischer Hypotonie und Synkope, führen kann, wurden Interaktionsstudien mit Vardenafil durchge002574-4967 Fachinformation Levitra® 20 mg Filmtabletten führt. In zwei Interaktionsstudien mit gesunden Probanden, bei denen normale Blutdruckwerte vorlagen, wurde nach forcierter Titration auf hohe Dosen des Alpha-Rezeptorenblockers Tamsulosin oder Terazosin bei gleichzeitiger Verabreichung mit Vardenafil von einer signifikanten Anzahl der Probanden über Hypotonie (in einigen Fällen über eine symptomatische Hypotonie) berichtet. Bei mit Terazosin behandelten Probanden wurde eine Hypotonie häufiger bei gleichzeitiger Gabe von Vardenafil und Terazosin beobachtet, als wenn ein Zeitintervall von 6 Stunden zwischen der Gabe lag. Auf Basis der Ergebnisse von mit Vardenafil durchgeführten Interaktionsstudien bei Patienten mit benigner Prostatahyperplasie (BPH) und stabiler Tamsulosin-, Terazosinoder Alfuzosin-Therapie ergibt sich: • Wenn Vardenafil (Filmtabletten) in einer Dosierung von 5, 10 oder 20 mg im Rahmen einer stabilen Tamsulosin-Therapie angewandt wurde, trat keine symptomatische Blutdrucksenkung auf, obwohl 3/21 mit Tamsulosin behandelten Patienten vorübergehend einen systolischen Blutdruck (im Stehen) von kleiner als 85 mmHg aufwiesen. • Wenn Vardenafil 5 mg (Filmtabletten) gleichzeitig mit Terazosin 5 oder 10 mg gegeben wurde, kam es bei einem von 21 Patienten zu einer symptomatischen orthostatischen Hypotonie. Es wurde keine Hypotonie beobachtet, wenn die Gabe von Vardenafil 5 mg und Terazosin in einem zeitlichen Abstand von 6 Stunden erfolgte. • Wenn Vardenafil (Filmtabletten) in einer Dosierung von 5 oder 10 mg im Rahmen einer stabilen Alfuzosin-Therapie angewendet wurde, trat keine symptomatische Blutdrucksenkung gegenüber Placebo auf. April 2014 DE/20 Deshalb sollte eine Begleitbehandlung nur begonnen werden, wenn der Patient stabil auf die Alpha-Rezeptorenblocker-Therapie eingestellt ist. Bei den Patienten, die stabil auf Alpha-Rezeptorenblocker-Therapie eingestellt sind, sollte Vardenafil mit der niedrigsten empfohlenen Anfangsdosis von 5 mg begonnen werden. Levitra kann zu jeder Zeit mit Tamsulosin oder mit Alfuzosin angewandt werden. Bei anderen AlphaRezeptorenblockern sollte ein zeitlicher Abstand der Gabe berücksichtigt werden, wenn Vardenafil als Begleitbehandlung verordnet wird (siehe Abschnitt 4.4). Bei gleichzeitiger Anwendung von Vardenafil (20 mg Filmtabletten) mit Warfarin (25 mg), das durch CYP2C9 verstoffwechselt wird, oder Digoxin (0,375 mg) konnten keine signifikanten Wechselwirkungen gezeigt werden. Die relative Bioverfügbarkeit von Glibenclamid (3,5 mg) wurde durch die gleichzeitige Anwendung von Vardenafil (20 mg) nicht beeinflusst. In einer gezielten Studie erhielten Hypertoniker Vardenafil (20 mg) zusammen mit verzögert freisetzendem Nifedipin (30 mg oder 60 mg). Es zeigte sich eine zusätzliche Senkung des Blutdruckes in Rückenlage um systolisch 6 mmHg und diastolisch um 5 mmHg, begleitet von einem Herzfrequenzanstieg um 4 Schläge/min. 002574-4967 Bei gleichzeitiger Einnahme von Vardenafil (20 mg Filmtabletten) und Alkohol (mittlerer maximaler Blut-Alkohol-Spiegel von 73 mg/dl) wurden die Wirkung von Alkohol auf Blutdruck und Herzfrequenz nicht verstärkt und die pharmakokinetischen Eigenschaften von Vardenafil nicht verändert. Die durch Acetylsalicylsäure (2-mal täglich 81 mg) bewirkte Verlängerung der Blutungszeit wurde durch Vardenafil (10 mg) nicht gesteigert. 4.6 Fertilität, Schwangerschaft und Stillzeit Für die Behandlung von Frauen ist Levitra nicht indiziert. Es gibt keine Studien mit Vardenafil bei Schwangeren. Es liegen keine Fertilitätsdaten vor. 4.7 Auswirkungen auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen Es wurden keine Studien zu den Auswirkungen auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen durchgeführt. Da in klinischen Studien mit Vardenafil über Schwindel und Sehstörungen berichtet wurde, müssen die Patienten darauf achten, wie sie auf die Einnahme von Levitra reagieren, bevor sie Auto fahren oder Maschinen bedienen. 4.8 Nebenwirkungen Die in klinischen Studien zu Levitra Filmtabletten oder Vardenafil 10 mg Schmelztabletten berichteten Nebenwirkungen waren überwiegend vorübergehender und leichter bis mäßiger Natur. Die häufigste, bei ≥ 10 % der Patienten aufgetretene Nebenwirkung ist Kopfschmerzen. Die Nebenwirkungen sind gemäß MedDRA Häufigkeitskonvention aufgeführt: sehr häufig (≥ 1/10), häufig (≥ 1/100, < 1/10), gelegentlich (≥ 1/1.000, < 1/100), selten (≥ 1/10.000, < 1/1.000) und nicht bekannt (Häufigkeit auf Grundlage der verfügbaren Daten nicht abschätzbar). Innerhalb jeder Häufigkeitsgruppe werden die Nebenwirkungen nach abnehmendem Schweregrad angegeben. Folgende Nebenwirkungen wurden beobachtet: Siehe Tabelle auf Seite 4 Penisblutung, Hämatospermie und Hämaturie wurden bei der Anwendung aller PDE5Hemmer einschließlich Vardenafil in klinischen Studien und spontanen Post-Marketing-Berichten gemeldet. Mit der Dosierung von 20 mg Levitra Filmtabletten traten bei älteren Patienten (≥ 65 Jahre) häufiger Kopfschmerzen (16,2 % im Vergleich zu 11,8 %) und Schwindel (3,7 % im Vergleich zu 0,7 %) auf als bei jüngeren Patienten (< 65 Jahre). Im Allgemeinen hat sich gezeigt, dass die Inzidenz von Nebenwirkungen (hauptsächlich „Schwindel“) bei Patienten mit bekannter Hypertonie etwas höher ist. Post-Marketing-Berichte über ein anderes Arzneimittel dieser Wirkstoffklasse Gefäßerkrankungen Schwerwiegende kardiovaskuläre Reaktionen, einschließlich Hirnblutung, plötzlicher Herztod, transitorische ischämische Attacke, instabile Angina pectoris und ventrikuläre Arrhythmie wurden nach der Markteinführung im zeitlichen Zusammenhang mit dem Gebrauch eines anderen Arzneimittels dieser Wirkstoffklasse berichtet. Meldung des Verdachts auf Nebenwirkungen Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von großer Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdachtsfall einer Nebenwirkung dem Bundesinstitut für Arzneimittel und Medizinprodukte, Abt. Pharmakovigilanz, KurtGeorg-Kiesinger-Allee 3, D-53175 Bonn, Website: http://www.bfarm.de, anzuzeigen. 4.9 Überdosierung In Studien an gesunden Probanden wurden Einmaldosen bis zu einschließlich 80 mg Vardenafil (Filmtabletten) pro Tag ohne schwerwiegende Nebenwirkungen vertragen. Bei Einnahme von Vardenafil in höherer Dosis und häufiger als empfohlen (2-mal täglich 40 mg Filmtabletten) wurden starke Rückenschmerzen berichtet. Dies war nicht verbunden mit toxischen Wirkungen auf Muskeln und Nervensystem. In Fällen einer Überdosierung sollten je nach Bedarf die üblichen unterstützenden Maßnahmen eingeleitet werden. Da Vardenafil in hohem Maße an Plasmaproteine gebunden ist und nicht wesentlich renal eliminiert wird, ist durch eine Dialyse keine Beschleunigung der Clearance zu erwarten. 5. PHARMAKOLOGISCHE EIGENSCHAFTEN 5.1 Pharmakodynamische Eigenschaften Pharmakotherapeutische Gruppe: Urologika, Mittel bei erektiler Dysfunktion, ATC-Code: G04BE09. Vardenafil ist eine orale Behandlung zur Verbesserung der erektilen Funktion bei Männern mit erektiler Dysfunktion. Liegt eine sexuelle Stimulation vor, wird die gestörte Erektionsfähigkeit durch eine Steigerung des Bluteinstroms in den Penis wiederhergestellt. Die Erektion des Penis basiert auf einem haemodynamischen Prozess. Während der sexuellen Stimulation erfolgt die Freisetzung von Stickstoffmonoxid (NO), das das Enzym Guanylatcyclase aktiviert, was zu erhöhten Spiegeln an zyklischem Guanosinmonophosphat (cGMP) im Corpus cavernosum führt. Daraus resultiert eine Relaxation der glatten Muskulatur, wodurch ein gesteigerter Bluteinstrom in den Penis ermöglicht wird. Der cGMP-Spiegel wird über die Syntheserate durch Guanylatcyclase und über die Abbaurate durch cGMP3 Fachinformation Levitra® 20 mg Filmtabletten Systemorganklasse Sehr häufig Häufig Gelegentlich Selten Nicht bekannt (≥ 1/10) (≥ 1/100 und < 1/10) (≥ 1/1.000 und < 1/100) (≥ 1/10.000 und < 1/1.000) (Häufigkeit auf Grundlage der verfügbaren Daten nicht abschätzbar) Infektionen und parasitäre Erkrankungen Konjunktivitis Erkrankungen des Immunsystems Allergische Ödeme und Angioödem Allergische Reaktionen Psychiatrische Erkrankungen Schlafstörungen Angstgefühle Somnolenz, Parästhesien und Dysästhesien Synkope, cerebrale Krämpfe, Amnesie Augenerkrankungen visuelle Störungen, Okuläre Hyperämie, Farbensehen, Augenschmerzen und Augenbeschwerden, Photophobie Anstieg des Augeninnendrucks, Vermehrte Tränenbildung Erkrankungen des Ohrs und des Labyrinths Tinnitus, Schwindel Herzerkrankungen Palpitationen, Tachykardie Erkrankungen des Nervensystems Kopfschmerzen Schwindel Gefässerkrankungen Flush Erkrankungen der Atemwege, des Brustraums und Mediastinums Verstopfte Nase Dyspnoe, Nasennebenhöhlenentzündung Erkrankungen des Gastrointestinaltraktes Dyspepsie Gastroösophageale Refluxerkrankung, Gastritis, Gastrointestinale und abdominelle Schmerzen, Diarrhö, Erbrechen, Übelkeit, Mundtrockenheit plötzliche Schwerhörigkeit oder Taubheit Myokardinfarkt, Ventrikuläre Tachyarrythmien, Angina pectoris Hypertonie, Hypotonie Nasenbluten Leber- und Gallenerkrankungen Transaminasenanstieg Anstieg des Gammaglytamyl-transpeptidase-Wertes Erkrankungen der Haut und des Unterhautzellgewebes Erythem, Exanthem Photosensibilität Skelettmuskulatur-, Bindegewebs- und Knochenerkrankungen Rückenschmerzen Anstieg der Kreatinphosphokinase Muskelschmerzen Verstärkter Muskeltonus und Krämpfe Erkrankungen der Nieren und Harnwege 4 nicht-arteriitische anteriore ischämische Optikusneuropathie, Störungen des Visus Hämaturie Erkrankungen der Geschlechtsorgane und der Brustdrüse Anstieg von Erektionen Priapismus Allgemeine Erkrankungen und Beschwerden am Verabreichungsort Unwohlsein Brustschmerzen Penisblutung, Hämatospermie 002574-4967 Fachinformation Levitra® 20 mg Filmtabletten hydrolysierende Phosphodiesterasen (PDEs) geregelt. Vardenafil ist ein starker und selektiver Hemmstoff der cGMP-spezifischen Phosphodiesterase Typ 5 (PDE5), der wichtigsten PDE im menschlichen Corpus cavernosum. Vardenafil verstärkt den Effekt des endogenen Stickstoffmonoxids im Corpus cavernosum beträchtlich, indem es PDE5 hemmt. Wenn als Reaktion auf sexuelle Stimulation Stickstoffmonoxid freigesetzt wird, bewirkt die PDE5-Hemmung durch Vardenafil erhöhte cGMP-Spiegel im Corpus cavernosum. Daher ist eine sexuelle Stimulation nötig, damit Vardenafil den beabsichtigten günstigen therapeutischen Effekt entwickeln kann. In vitro Studien zeigten, dass Vardenafil stärker auf PDE5 wirkt als auf andere bekannte Phosphodiesterasen (> 15-fach gegenüber PDE6, > 130-fach gegenüber PDE1, > 300-fach gegenüber PDE11 und > 1.000-fach gegenüber PDE2, PDE3, PDE4, PDE7, PDE8, PDE9 und PDE10). Eine Untersuchung mittels Penis-Plethysmographie (RigiScan) zeigte, dass bereits 15 Minuten nach Einnahme von 20 mg Vardenafil bei einigen Patienten für einen Geschlechtsverkehr ausreichende Erektionen (60 %ige Rigidität an der Peniswurzel, gemessen mit RigiScan) erreicht wurden. Im Allgemeinen trat eine signifikante Wirkung von Vardenafil im Vergleich zu Placebo innerhalb von 25 Minuten nach Einnahme ein. April 2014 DE/20 Vardenafil bewirkt eine leichte und vorübergehende Blutdrucksenkung, die in der Mehrzahl der Fälle keine klinischen relevanten Wirkungen zur Folge hat. Im Vergleich zu Placebo betrug die mittlere maximale systolische Blutdrucksenkung in Rückenlage nach Gabe von 20 mg Vardenafil 6,9 mmHg und nach Gabe von 40 mg Vardenafil 4,3 mmHg. Diese Effekte spiegeln die vasodilatatorischen Wirkungen von PDE5-Inhibitoren wider und werden wahrscheinlich durch erhöhte cGMP-Spiegel in den glatten Gefäßmuskelzellen verursacht. Orale Einmalsowie Mehrfachgaben von bis zu 40 mg Vardenafil führten bei männlichen Probanden nicht zu klinisch relevanten EKG-Veränderungen. In einer doppelblinden, randomisierten Crossover-Studie mit 59 gesunden Männern wurde die Wirkung einer Einmalgabe von Vardenafil (10 mg und 80 mg), Sildenafil (50 mg und 400 mg) und Placebo auf das QT Intervall untersucht. Moxifloxacin (400 mg) wurde als aktive Kontrolle eingesetzt. Die Wirkung auf das QT Intervall wurde 1 Stunde nach der Einnahme bestimmt (mittlere tmax für Vardenafil). Primäres Ziel dieser Studie war es, einen Effekt einer Einzelgabe von 80 mg Vardenafil auf das QT Intervall von mehr als 10 msec im Vergleich zu Placebo auszuschließen (d. h. das Fehlen eines Effektes zu zeigen); bestimmt wurde die Änderung des mit der Fridericia-Korrekturformel (QTcF = QT/ RR1/3) ermittelten Wertes 1 Stunde nach Einnahme im Vergleich zum Ausgangswert. Die Ergebnisse zeigten 1 Stunde nach Einnahme von 10 mg bzw. 80 mg Vardenafil im Vergleich zu Placebo eine QTc-Verlänge002574-4967 rung (Fridericia) um 8 msec (90 % KI: 6 – 9) bzw. 10 msec (90 % KI: 8 – 11) und eine QTci-Verlängerung um 4 msec (90 % KI: 3 – 6) bzw. 6 msec (90 % KI: 4 – 7). Zum Zeitpunkt tmax lag ausschließlich der mittlere QTcF-Wert nach Gabe von 80 mg Vardenafil außerhalb der in der Studie definierten Grenzen (im Mittel 10 msec, 90 % KI: 8 – 11). Nach Anwendung der individuellen Korrekturformel lag kein Wert außerhalb der Grenzen. In einer separaten, nach Markteinführung durchgeführten Studie an 44 gesunden Personen, wurden Einmalgaben von 10 mg Vardenafil oder 50 mg Sildenafil zusammen mit 400 mg Gatifloxacin, einem Präparat mit vergleichbarer Wirkung auf die QT-Zeit, eingenommen. Beide Präparate, Vardenafil und Sildenafil, zeigten einen Anstieg des Fridericia-Effekts auf QTc von 4 msec (Vardenafil) und 5 msec (Sildenafil) im Vergleich zu den Wirkungen dieser Präparate alleine. Die klinische Relevanz dieser Änderungen der QT-Zeit ist unbekannt. Weitere Informationen zu klinischen Studien mit Vardenafil 10 mg Schmelztabletten Die Wirksamkeit und Sicherheit von Vardenafil 10 mg Schmelztabletten wurde separat in zwei Studien zu 701 randomisierten Patienten mit erektiler Dysfunktion, die bis zu 12 Wochen lang behandelt wurden, bei einem breiten Patientenspektrum nachgewiesen. Die Verteilung der Patienten auf vorab definierte Untergruppen umfasste ältere Patienten (51 %) sowie Patienten mit einem Diabetes mellitus (29 %), einer Dyslipidämie (39 %) oder einer Hypertonie (40 %) in der Vorgeschichte. In den gepoolten Daten der beiden Studien zu Vardenafil 10 mg Schmelztabletten waren die Punktwerte in der IIEF-EF-Domäne unter Vardenafil 10 mg Schmelztabletten signifikant höher als unter Placebo. In den klinischen Studien gelang bei 71 % aller Koitus-Versuche eine erfolgreiche Penetration gegenüber 44 % aller Versuche in der Placebogruppe. Diese Ergebnisse spiegelten sich auch in den Untergruppen, bei älteren Patienten (65 % aller Koitus-Versuche mit erfolgreicher Penetration), bei Patienten mit Diabetes mellitus in der Vorgeschichte (63 %), bei Patienten mit Dyslipidämie in der Vorgeschichte (66 %) und bei Patienten mit Hypertonie wider (70 %). In Bezug auf eine Aufrechterhaltung der Erektion waren unter Vardenafil 10 mg Schmelztabletten etwa 63 % aller angegebenen Koitus-Versuche erfolgreich im Vergleich zu etwa 26 % aller Versuche in der Placebokontrollgruppe. In den vorab definierten Untergruppen waren 57 % (ältere Patienten), 56 % (Patienten mit Diabetes mellitus in der Vorgeschichte), 59 % (Patienten mit Dyslipidämie in der Vorgeschichte) und 60 % (Patienten mit Hypertonie in der Vorgeschichte) aller genannten Koitus-Versuche unter Vardenafil 10 mg Schmelztabletten im Hinblick auf eine Aufrechterhaltung der Erektion erfolgreich. Weitere Informationen über klinische Studien Vardenafil wurde in klinischen Studien mehr als 17.000 Männern mit erektiler Dysfunktion (ED) im Alter von 18 – 89 Jahren, von denen viele multiple Begleiterkrankungen aufwiesen, verabreicht. Mehr als 2.500 Patienten wurden 6 Monate oder länger mit Vardenafil behandelt. Von diesen wurden 900 Patienten ein Jahr oder länger therapiert. Die folgenden Patientengruppen waren vertreten: Ältere (22 %), Patienten mit Bluthochdruck (35 %), mit Diabetes mellitus (29 %), mit koronarer Herzkrankheit und anderen Herz-Kreislauf-Erkrankungen (7 %), mit chronischen Lungenerkrankungen (5 %), mit Hyperlipidämie (22 %), mit Depressionen (5 %) und mit radikaler Prostatektomie (9 %). Die folgenden Gruppen waren unterrepräsentiert: Ältere über 75 Jahre (2,4 %) und Patienten mit bestimmten Herz-KreislaufErkrankungen (siehe Abschnitt 4.3). Es wurden keine Studien durchgeführt an Patienten mit ZNS-Erkrankungen (ausgenommen Rückenmarkverletzungen), an Patienten mit stark eingeschränkter Nieren- und Leberfunktion, an Patienten nach Beckenoperation (ausgenommen nervenerhaltender Prostatektomie) oder Beckentrauma oder nach Bestrahlungstherapie, an Patienten mit einem verminderten sexuellem Bedürfnis oder an Patienten mit anatomischen Missbildungen des Penis. In allen relevanten Studien führte die Behandlung mit Vardenafil (Filmtabletten) im Vergleich zur Behandlung mit Placebo zu einer Verbesserung der erektilen Funktion. Bei der kleinen Anzahl von Patienten, die bis zu vier bis fünf Stunden nach der Einnahme versuchten, den Geschlechtsverkehr zu vollziehen, war die Erfolgsrate für die Penetration und das Aufrechterhalten der Erektion im Vergleich zu Placebo durchweg größer. In Studien mit festgelegter Dosierung (Filmtabletten) und einer Dauer von 3 Monaten in einer breiten Population von Männern mit erektiler Dysfunktion berichteten 68 % der mit 5 mg behandelten Männer, 76 % der mit 10 mg behandelten Männer und 80 % der mit 20 mg behandelten Männer über eine erfolgreiche Penetration (SEP 2) verglichen mit 49 % unter Placebogabe. In dieser breiten Population wurde die Fähigkeit zum Aufrechterhalten der Erektion (SEP 3) mit 53 % (5 mg), 63 % (10 mg) und 65 % (20 mg) im Vergleich zu 29 % unter Placebo angegeben. Anhand gepoolter Daten der größeren Wirksamkeitsstudien war der Anteil an Patienten, die nach Anwendung von Vardenafil über eine erfolgreiche Penetration berichteten, wie folgt: Männer mit psychogener erektiler Dysfunktion (77 – 87 %), mit gemischter Ätiologie der erektilen Dysfunktion (69 – 83 %), mit organisch bedingter erektiler Dysfunktion (64 – 75 %), Ältere (52 – 75 %), Männer mit koronarer Herzkrankheit (70 – 73 %), mit Hyperlipidämie (62 – 73 %), mit chronischen Lungenerkrankungen (74 – 78 %), mit Depressionen (59 – 69 %) und Patienten, die gleichzeitig mit Antihypertensiva behandelt wurden (62 – 73 %). In einer klinischen Studie bei Patienten mit Diabetes mellitus verbesserte Vardenafil bei einer Dosierung von 10 mg und 20 mg die erektile Funktion, die Fähigkeit zu einer für die Vollendung des Geschlechtsaktes aus5 Fachinformation Levitra® 20 mg Filmtabletten reichend lang anhaltenden Erektion sowie die Penis-Rigidität im Vergleich zu Placebo signifikant. Die Ansprechrate für die Fähigkeit, eine Erektion zu erlangen und aufrechtzuerhalten, betrug bei Patienten nach dreimonatiger Behandlungsdauer für die 10 mg Vardenafil-Dosis 61 % bzw. 49 % und für die 20 mg Vardenafil-Dosis 64 % bzw. 54 %, verglichen mit 36 % bzw. 23 % unter Placebo. In einer klinischen Studie mit Patienten nach einer Prostatektomie verbesserte Vardenafil bei einer Dosierung von 10 mg und 20 mg die erektile Funktion, die Fähigkeit zu einer für die Vollendung des Geschlechtsaktes ausreichend lang anhaltenden Erektion sowie die Penis-Rigidität im Vergleich zu Placebo signifikant. Die Ansprechrate für die Fähigkeit, eine Erektion zu erlangen und aufrechtzuerhalten, betrug bei Patienten nach dreimonatiger Behandlungsdauer für die 10 mg Vardenafil-Dosis 47 % bzw. 37 % und für die 20 mg Vardenafil-Dosis 48 % bzw. 34 %, verglichen mit 22 % bzw. 10 % unter Placebo. In einer klinischen Studie mit flexibler Dosierung bei Patienten mit Rückenmarkverletzungen verbesserte Vardenafil die erektile Funktion, die Fähigkeit zu einer für die Vollendung des Geschlechtsaktes ausreichend lang anhaltenden Erektion sowie die PenisRigidität im Vergleich zu Placebo signifikant. Die Zahl der Patienten, die eine normale erektile Funktion (IIEF-Domain-Score > 26) wieder erlangten, betrug 53 % nach Vardenafil-Behandlung, verglichen mit 9 % unter Placebo. Die Ansprechrate für die Fähigkeit, eine Erektion zu erlangen und aufrechtzuerhalten, betrug bei Patienten nach dreimonatiger Behandlungsdauer 76 % bzw. 59 % unter Vardenafil, verglichen mit 41 % bzw. 22 % unter Placebo; diese Ergebnisse waren klinisch und statistisch signifikant (p< 0,001). Die Unbedenklichkeit und Wirksamkeit von Vardenafil wurde in Langzeitstudien bestätigt. Kinder und Jugendliche Die Europäische Arzneimittel-Agentur hat eine Freistellung von der Verpflichtung zur Vorlage von Ergebnissen zu Studien in allen pädiatrischen Altersklassen in der Behandlung der erektilen Dysfunktion gewährt (siehe Abschnitt 4.2 bzgl. Informationen zur Anwendung bei Kindern und Jugendlichen). 5.2 Pharmakokinetische Eigenschaften Bioäquivalenzstudien zeigten, dass Vardenafil 10 mg Schmelztabletten und Vardenafil 10 mg Filmtabletten nicht bioäquivalent sind; daher sollte die Formulierung als Schmelztablette nicht als Äquivalent zu Vardenafil 10 mg Filmtabletten eingesetzt werden. Resorption Vardenafil in Vardenafil Filmtabletten wird schnell resorbiert – bei einigen Männern wurden bereits 15 Minuten nach Einnahme maximale Plasmaspiegel beobachtet. In 90 % der Fälle werden jedoch nach oraler Gabe im nüchternen Zustand maximale Plasmaspiegel innerhalb von 30 bis 120 Minuten (Mittel: 60 Minuten) erreicht. Die mitt6 lere absolute orale Bioverfügbarkeit beträgt 15 %. Nach oraler Einnahme von Vardenafil nehmen AUC und Cmax fast dosisproportional über den empfohlenen Dosisbereich (5 bis 20 mg) zu. Bei Einnahme von Vardenafil Filmtabletten zusammen mit einer sehr fettreichen Mahlzeit (Fettgehalt: 57 %) ist die Resorptionsgeschwindigkeit reduziert, die tmax verlängert sich im Mittel um 1 Stunde und Cmax ist im Mittel um 20 % verringert. Die VardenafilAUC wird nicht beeinflusst. Im Vergleich zur Nüchterneinnahme bleiben bei Einnahme von Vardenafil nach einer Mahlzeit mit 30 % Fettgehalt Resorptionsrate und Resorptionsausmaß von Vardenafil (tmax, Cmax und AUC) unverändert. Vardenafil wird nach Gabe von Vardenafil 10 mg Schmelztabletten ohne Wasser schnell resorbiert. Die mediane Zeit bis zum Erreichen von Cmax variierte zwischen 45 und 90 Minuten und fiel gegenüber den Filmtabletten ähnlich oder leicht verzögert (um 8 bis 45 Minuten) aus. Die mittlere Vardenafil-AUC war unter den 10 mg Schmelztabletten wegen der lokalen Resorption einer geringen Arzneimittelmenge in der Mundhöhle gegenüber den Filmtabletten um 21 bis 29 % (Patienten mit erektiler Dysfunktion mittleren und höheren Alters) oder um 44 % (junge gesunde Probanden) erhöht. Es war kein einheitlicher Unterschied in der mittleren Cmax zwischen den Schmelztabletten und den Filmtabletten erkennbar. Die Einnahme von Vardenafil 10 mg Schmelztabletten zu einer Mahlzeit mit hohem Fettgehalt hatte keinen Einfluss auf die AUC und tmax von Vardenafil, während die Cmax von Vardenafil nach Nahrungsaufnahme um 35 % verringert war. Auf der Grundlage dieser Ergebnisse können Vardenafil 10 mg Schmelztabletten mit oder ohne Nahrung eingenommen werden. Wenn Vardenafil 10 mg Schmelztabletten mit Wasser eingenommen werden, ist die AUC um 29 % reduziert, die Cmax bleibt unverändert und die mediane tmax ist gegenüber der Einnahme ohne Wasser um 60 Minuten verkürzt. Vardenafil 10 mg Schmelztabletten müssen ohne Flüssigkeit eingenommen werden. Verteilung Das mittlere Verteilungsvolumen von Vardenafil im Steady State beträgt 208 l, was auf eine Verteilung in die Gewebe hinweist. Vardenafil und sein wichtigster im Blutkreislauf zirkulierender Metabolit (M1) sind beide stark an Plasmaproteine gebunden (zu rund 95 % für Vardenafil oder M1). Die Proteinbindung ist sowohl für Vardenafil als auch für M1 unabhängig von der Gesamtkonzentration des Arzneimittels. Bei gesunden Probanden wurden 90 Minuten nach Anwendung von Vardenafil weniger als 0,00012 % der verabreichten Menge im Sperma gefunden. Biotransformation Vardenafil in Filmtabletten wird überwiegend in der Leber durch das Cytochrom P450 (CYP) Isoenzym 3A4 sowie anteilig durch die CYP3A5- und CYP2C-Isoenzyme metabolisiert. Der beim Menschen wichtigste zirkulierende Metabolit (M1) resultiert aus Deethylierung von Vardenafil und unterliegt einer weiteren Metabolisierung mit einer Plasmahalbwertszeit von etwa 4 Stunden. Anteile von M1 finden sich als Glukuronid im Blutkreislauf. Das Profil der Phosphodiesterase-Selektivität des Metaboliten M1 ist ähnlich dem von Vardenafil und weist in vitro eine Hemmwirkung für PDE5 auf, die rund 28 % derjenigen von Vardenafil beträgt und zu etwa 7 % zur Wirksamkeit beiträgt. Die mittlere terminale Halbwertzeit von Vardenafil betrug bei mit Vardenafil 10 mg Schmelztabletten behandelten Patienten 4 bis 6 Stunden. Die Eliminationshalbwertzeit des Metaboliten M1 beträgt 3 bis 5 Stunden, vergleichbar mit der Muttersubstanz. Elimination Die Gesamtkörperclearance von Vardenafil beträgt 56 l/h mit einer daraus resultierenden terminalen Halbwertszeit von etwa 4 bis 5 Stunden. Nach oraler Anwendung wird Vardenafil nach Metabolisierung überwiegend über die Fäzes (rund 91 – 95 % der verabreichten Dosis) und in geringerem Maß renal (rund 2 – 6 % der verabreichten Dosis) ausgeschieden. Pharmakokinetik bei speziellen Patientengruppen Ältere Patienten Gesunde ältere Probanden (65 Jahre oder älter) zeigten im Vergleich zu jüngeren Probanden (18 – 45 Jahre) eine herabgesetzte hepatische Vardenafil-Clearance. Ältere Männer zeigten bei der Einnahme von Vardenafil Filmtabletten durchschnittlich eine um 52 % höhere AUC und eine um 34 % höhere Cmax als jüngere Männer (siehe Abschnitt 4.2). Bei älteren Patienten (65 Jahre oder älter) fielen die AUC und Cmax von Vardenafil nach Einnahme von Vardenafil Schmelztabletten um 31 bis 39 % bzw. um 16 bis 21 % höher aus als bei Patienten im Alter von 45 Jahren und darunter. Nach einmal täglicher Einnahme von Vardenafil 10 mg Schmelztabletten über 10 Tage wurde bei Patienten im Alter von 45 Jahren und darunter oder 65 Jahren und darüber keine Akkumulation von Vardenafil im Plasma beobachtet. Eingeschränkte Nierenfunktion Bei Probanden mit leichter bis mäßiger Nierenfunktionsstörung (Kreatinin-Clearance 30 – 80 ml/min) war die Pharmakokinetik von Vardenafil vergleichbar mit der Kontrollgruppe aus Patienten mit normaler Nierenfunktion. Bei Probanden mit schwerer Nierenfunktionsstörung (Kreatinin-Clearance < 30 ml/min) waren verglichen mit Probanden mit normaler Nierenfunktion die mittlere AUC um 21 % erhöht und die mittlere Cmax um 23 % erniedrigt. Zwischen KreatininClearance und Vardenafil-Exposition (AUC und Cmax) wurde keine statistisch signifikante Korrelation beobachtet (siehe Abschnitt 4.2). Die Pharmakokinetik von Vardenafil bei dialysepflichtigen Patienten wurde nicht untersucht (siehe Abschnitt 4.3). 002574-4967 Fachinformation Levitra® 20 mg Filmtabletten Eingeschränkte Leberfunktion Bei Patienten mit leichter bis mäßiger Leberfunktionsstörung (Child-Pugh A und B) war die Clearance von Vardenafil proportional zum Grad der Leberfunktionsstörung herabgesetzt. Im Vergleich mit gesunden Probanden waren bei Patienten mit leichter Leberfunktionsstörung (Child-Pugh A) die mittlere AUC und Cmax um 17 % bzw. 22 % erhöht. Bei Patienten mit mäßigen Leberfunktionsstörungen (Child-Pugh B) waren im Vergleich mit gesunden Probanden mittlere AUC und Cmax um 160 % bzw. 133 % erhöht (siehe Abschnitt 4.2). Die Pharmakokinetik von Vardenafil bei Patienten mit schwerer Leberfunktionsstörung (Child-Pugh C) wurde nicht untersucht (siehe Abschnitt 4.3). 9. DATUM DER ERTEILUNG DER ZULASSUNG/VERLÄNGERUNG DER ZULASSUNG Datum der Erteilung der Zulassung 06. März 2003 Datum der letzten Verlängerung der Zulassung 06. März 2008 10. STAND DER INFORMATION April 2014 Ausführliche Informationen zu diesem Arzneimittel sind auf den Internetseiten der Europäischen Arzneimittel Agentur http://www.ema.europa.eu verfügbar. 5.3 Präklinische Daten zur Sicherheit Basierend auf den konventionellen Studien zur Sicherheitspharmakologie, Toxizität bei wiederholter Gabe, Reproduktionstoxizität, Genotoxizität und zum kanzerogenen Potenzial lassen die präklinischen Daten keine besonderen Gefahren für den Menschen erkennen. 6. PHARMAZEUTISCHE ANGABEN 6.1 Liste der sonstigen Bestandteile 11. VERKAUFSABGRENZUNG Verschreibungspflichtig Lokaler Ansprechpartner: Jenapharm GmbH & Co. KG Otto-Schott-Str. 15 07745 Jena Telefon: 03641-64 88 88 Telefax: 03641-64 88 89 e-Mail: [email protected] Tablettenkern: Crospovidon. Magnesiumstearat. Mikrokristalline Cellulose. Hochdisperses Siliciumdioxid. Filmüberzug: Macrogol 400. Hypromellose. Titandioxid (E 171). Eisenoxidhydrat (E 172). Eisen(III)-oxid (E 172). 6.2 Inkompatibilitäten Nicht zutreffend. 6.3 Dauer der Haltbarkeit 3 Jahre 6.4 Besondere Vorsichtsmaßnahmen für die Aufbewahrung Für dieses Arzneimittel sind keine besonderen Lagerungsbedingungen erforderlich. 6.5 Art und Inhalt des Behältnisses PP/Aluminiumfolie–Blister in Packungen zu 2, 4, 8, 12 und 20 Tabletten. Es werden möglicherweise nicht alle Packungsgrößen in den Verkehr gebracht. 6.6 Besondere Vorsichtsmaßnahmen für die Beseitigung April 2014 DE/20 Keine besonderen Anforderungen für die Beseitigung. 7. INHABER DER ZULASSUNG Bayer Pharma AG 13342 Berlin Deutschland 8. ZULASSUNGSNUMMER(N) EU/1/03/248/009-012, 023 002574-4967 Zentrale Anforderung an: Rote Liste Service GmbH Fachinfo-Service Mainzer Landstraße 55 60329 Frankfurt 7 Fachinformation (Zusammenfassung der Merkmale des Arzneimittels) VIAGRA ® 1. BEZEICHNUNG DER ARZNEIMITTEL VIAGRA ® 25 mg, Filmtabletten VIAGRA ® 50 mg, Filmtabletten VIAGRA ® 100 mg, Filmtabletten 2. QUALITATIVE UND QUANTITATIVE ZUSAMMENSETZUNG Jede Tablette enthält Sildenafilcitrat entsprechend 25 mg/50 mg/100 mg Sildenafil. Sonstiger Bestandteil mit bekannter Wirkung: Viagra 25 mg Filmtablette: Jede Tablette enthält 0,834 mg Lactose (als Monohydrat). Viagra 50 mg Filmtablette: Jede Tablette enthält 1,667 mg Lactose (als Monohydrat). Viagra 100 mg Filmtablette: Jede Tablette enthält 3,334 mg Lactose (als Monohydrat). Vollständige Auflistung der sonstigen Bestandteile siehe Abschnitt 6.1. 3. DARREICHUNGSFORM Filmtablette Viagra 25 mg Filmtablette: Blaue abgerundete rautenförmige Tabletten, auf einer Seite mit „PFIZER“ und auf der anderen mit „VGR 25“ gekennzeichnet Viagra 50 mg Filmtablette: Blaue abgerundete rautenförmige Tabletten, auf einer Seite mit „PFIZER“ und auf der anderen mit „VGR 50“ gekennzeichnet Viagra 100 mg Filmtablette: Blaue abgerundete rautenförmige Tabletten, auf einer Seite mit „PFIZER“ und auf der anderen mit „VGR 100“ gekennzeichnet 4. KLINISCHE ANGABEN 4.1 Anwendungsgebiete VIAGRA wird zur Behandlung von erwachsenen Männern mit erektiler Dysfunktion angewendet. Das ist die Unfähigkeit, eine für einen befriedigenden Geschlechtsverkehr ausreichende Erektion zu erreichen oder aufrechtzuerhalten. VIAGRA kann nur wirken, wenn eine sexuelle Stimulation vorliegt. 4.2 Dosierung und Art der Anwendung Dosierung Juni 2015 spcde-5v28vgr-ft-0 Anwendung bei Erwachsenen Die empfohlene Dosis beträgt 50 mg, sie ist ungefähr 1 Stunde vor dem Geschlechtsverkehr einzunehmen. Entsprechend der Wirksamkeit und Verträglichkeit kann die Dosis auf 100 mg erhöht oder auf 25 mg verringert werden. Die empfohlene Maximaldosis beträgt 100 mg. Die Einnahme darf nicht häufiger als einmal täglich erfolgen. Wenn VIAGRA zusammen mit Nahrung eingenommen wird, kann der Wirkungseintritt im Vergleich zur Nüchterneinnahme verzögert sein (siehe Abschnitt 5.2). Spezielle Patientengruppen Ältere Patienten Dosisanpassungen bei älteren Patienten (≥ 65 Jahre) sind nicht erforderlich. 003927-14827 Patienten mit Nierenfunktionsstörung Die Dosierungsempfehlungen unter „Anwendung bei Erwachsenen“ gelten auch für Patienten mit leichter bis mäßiger Niereninsuffizienz (Kreatinin-Clearance = 30 bis 80 ml/min). Da bei Patienten mit schwerer Niereninsuffizienz (Kreatinin-Clearance <30 ml/min) die Sildenafil-Clearance vermindert ist, sollte eine Dosierung von 25 mg erwogen werden. Entsprechend der Wirksamkeit und Verträglichkeit kann die Dosis bei Bedarf schrittweise auf 50 mg bis 100 mg erhöht werden. Patienten mit Leberfunktionseinschränkung Da bei Patienten mit Leberinsuffizienz (z. B. Leberzirrhose) die Sildenafil-Clearance vermindert ist, sollte eine Dosis von 25 mg erwogen werden. Entsprechend Wirksamkeit und Verträglichkeit kann die Dosis bei Bedarf schrittweise auf 50 mg bis 100 mg erhöht werden. Kinder und Jugendliche VIAGRA ist nicht für die Anwendung bei Patienten unter 18 Jahren angezeigt. Anwendung bei Patienten, die andere Arzneimittel einnehmen Mit Ausnahme von Ritonavir, für das eine gleichzeitige Gabe von Sildenafil nicht angezeigt ist (siehe Abschnitt 4.4), sollte bei erstmaliger Anwendung bei Patienten, die eine begleitende Behandlung mit CYP3A4-Hemmstoffen erhalten, eine Dosis von 25 mg erwogen werden (siehe Abschnitt 4.5). Um die Möglichkeit einer orthostatischen Hypotonie bei Patienten, die mit Alphablockern behandelt werden, möglichst gering zu halten, sollte die Alphablocker-Therapie vor Beginn der Sildenafil-Behandlung stabil eingestellt sein. Darüber hinaus sollte eine Initialdosis von 25 mg Sildenafil in Erwägung gezogen werden (siehe Abschnitte 4.4 und 4.5). Art der Anwendung Zum Einnehmen. 4.3 Gegenanzeigen Überempfindlichkeit gegen den Wirkstoff oder einen der in Abschnitt 6.1 genannten sonstigen Bestandteile Entsprechend seiner pharmakologischen Wirkung auf den Stickstoffmonoxid-zyklisches-Guanosinmonophosphat (cGMP)Stoffwechsel (siehe Abschnitt 5.1) konnte gezeigt werden, dass Sildenafil den blutdrucksenkenden Effekt von Nitraten verstärkt. Daher ist die gleichzeitige Gabe mit Stickstoffmonoxid-Donatoren (wie beispielsweise Amylnitrit) oder jeglichen Nitraten kontraindiziert. Mittel zur Behandlung der erektilen Dysfunktion wie auch Sildenafil sind bei Patienten, denen von sexueller Aktivität abzuraten ist (z. B. Patienten mit schweren Herz-KreislaufErkrankungen wie instabiler Angina pectoris oder schwerer Herzinsuffizienz), nicht anzuwenden. Bei Patienten, die aufgrund einer nicht arteriitischen anterioren ischämischen Optikus- neuropathie (NAION) ihre Sehkraft auf einem Auge verloren haben, ist Sildenafil kontraindiziert, unabhängig davon, ob der Sehverlust mit einer vorherigen Einnahme eines PDE5Hemmers in Zusammenhang stand oder nicht (siehe Abschnitt 4.4). Bei folgenden Patienten wurde die Sicherheit von Sildenafil nicht untersucht, daher ist die Anwendung kontraindiziert: schwere Leberinsuffizienz, Hypotonie (Blutdruck <90/50 mmHg), Patienten mit kürzlich erlittenem Schlaganfall oder Herzinfarkt sowie mit bekannter erblich bedingter degenerativer Retinaerkrankung wie beispielsweise Retinitis pigmentosa (eine Minderheit dieser Patienten hat eine genetisch bedingte Störung der retinalen Phosphodiesterasen). 4.4 Besondere Warnhinweise und Vorsichtsmaßnahmen für die Anwendung Bevor eine medikamentöse Therapie in Betracht gezogen wird, sollte die Diagnose einer erektilen Dysfunktion gestellt und zugrunde liegende Ursachen mittels Anamnese und körperlicher Untersuchung ermittelt werden. Kardiovaskuläre Risikofaktoren Vor dem Beginn jeglicher Behandlung einer erektilen Dysfunktion sollten die behandelnden Ärzte den kardiovaskulären Status ihrer Patienten berücksichtigen, da mit sexueller Aktivität ein gewisses kardiales Risiko einhergeht. Aufgrund seiner vasodilatatorischen Eigenschaften bewirkt Sildenafil eine leichte und vorübergehende Blutdrucksenkung (siehe Abschnitt 5.1). Vor der Verordnung von Sildenafil sollen Ärzte sorgfältig erwägen, ob Patienten mit bestimmten Grunderkrankungen durch diese gefäßerweiternde Wirkung beeinträchtigt werden könnten, insbesondere in Kombination mit sexueller Aktivität. Zu Patienten mit erhöhter Empfindlichkeit gegenüber gefäßerweiternden Substanzen gehören Patienten mit einer Obstruktion des linksventrikulären Ausflusstrakts (z. B. Aortenstenose, hypertroph-obstruktive Kardiomyopathie) oder Patienten mit dem seltenen Syndrom der Multisystematrophie, das sich in einer schweren Störung der autonomen Blutdruckkontrolle manifestiert. VIAGRA potenziert die blutdrucksenkende Wirkung von Nitraten (siehe Abschnitt 4.3). Schwerwiegende kardiovaskuläre Ereignisse, einschließlich Herzinfarkt, instabile Angina pectoris, plötzlicher Herztod, ventrikuläre Arrhythmie, zerebrovaskuläre Blutung, transitorische ischämische Attacke, Hypertonie und Hypotonie, wurden nach der Markteinführung im zeitlichen Zusammenhang mit dem Gebrauch von VIAGRA gemeldet. Die meisten dieser Patienten, aber nicht alle, hatten vorbestehende kardiovaskuläre Risikofaktoren. Für viele Ereignisse wurde gemeldet, dass sie während oder kurz nach dem Geschlechtsverkehr auftraten, und für einige wenige, dass sie kurz nach dem Gebrauch von VIAGRA ohne sexuelle Aktivität auftraten. Es ist unmöglich zu entscheiden, ob diese Ereignisse direkt mit diesen Faktoren oder mit anderen Faktoren zusammenhängen. 1 Fachinformation (Zusammenfassung der Merkmale des Arzneimittels) VIAGRA ® Priapismus Die Behandlung einer erektilen Dysfunktion, auch mit Sildenafil, sollte dann mit Vorsicht erfolgen, wenn anatomische Penismissbildungen wie Angulation, Fibrose im Bereich der Corpora cavernosa oder die PeyronieKrankheit vorliegen, sowie bei Patienten mit für Priapismus prädisponierenden Erkrankungen (wie Sichelzellanämie, Plasmozytom, Leukämie). In der Post-Marketing-Überwachung wurde unter Sildenafil-Behandlung von verlängerten Erektionen und Priapismus berichtet. Im Fall einer länger als 4 Stunden andauernden Erektion sollte der Patient sofort medizinische Hilfe aufsuchen. Wenn ein Priapismus nicht sofort behandelt wird, kann dies zu Gewebeschädigung im Penis und dauerhaftem Potenzverlust führen. Gleichzeitige Anwendung von anderen PDE5-Hemmern oder anderen Behandlungsmethoden für die erektile Dysfunktion Es liegen keine Studien zur Unbedenklichkeit und Wirksamkeit von Sildenafil in Kombination mit anderen PDE5-Hemmern oder mit anderen Sildenafil-haltigen Arzneimitteln (REVATIO) zur Behandlung einer pulmonalen arteriellen Hypertonie (PAH) oder mit anderen Behandlungen einer erektilen Dysfunktion vor. Die Anwendung solcher Kombinationen wird daher nicht empfohlen. Auswirkungen auf das Sehvermögen Fälle von Sehstörungen sind in Zusammenhang mit der Einnahme von Sildenafil und anderen PDE5-Hemmern spontan berichtet worden (siehe Abschnitt 4.8). Fälle von nicht arteriitischer anteriorer ischämischer Optikusneuropathie (NAION), einer seltenen Erkrankung, sind in Zusammenhang mit der Einnahme von Sildenafil und anderen PDE5-Hemmern berichtet worden, sowohl spontan erfasst als auch in einer Anwendungsbeobachtung (siehe Abschnitt 4.8). Patienten müssen darüber aufgeklärt werden, dass sie beim Auftreten einer plötzlichen Sehstörung jeglicher Art VIAGRA absetzen und sofort einen Arzt aufsuchen sollen (siehe Abschnitt 4.3). Gleichzeitige Anwendung von Ritonavir Eine gleichzeitige Gabe von Sildenafil und Ritonavir wird nicht empfohlen (siehe Abschnitt 4.5). Gleichzeitige Anwendung von Alphablockern Wenn Patienten unter Alphablocker-Therapie Sildenafil erhalten, ist Vorsicht geboten, da eine gleichzeitige Anwendung bei einigen wenigen empfindlichen Personen zu symptomatischer Hypotonie führen kann (siehe Abschnitt 4.5). Am wahrscheinlichsten tritt diese innerhalb von 4 Stunden nach Einnahme von Sildenafil auf. Um die Möglichkeit einer orthostatischen Hypotonie möglichst gering zu halten, sollten Patienten, die mit Alphablockern behandelt werden, vor Beginn der Behandlung mit Sildenafil hämodynamisch stabil eingestellt sein. Eine Initialdosis von 25 mg Sildenafil sollte in Erwägung gezogen werden (siehe Abschnitt 4.2). Darüber hinaus sollten Ärzte die Patienten darüber aufklären, wie sie sich 2 beim Auftreten von Symptomen einer orthostatischen Hypotonie verhalten sollen. Auswirkungen auf die Blutgerinnung Studien an menschlichen Thrombozyten haben Hinweise darauf erbracht, dass Sildenafil die anti-aggregatorische Wirkung von Nitroprussid-Natrium in vitro verstärkt. Es liegen keine Daten über die Unbedenklichkeit der Anwendung von Sildenafil an Patienten mit Blutungsstörungen oder aktiven peptischen Ulzera vor. Daher sollte die Gabe von Sildenafil an diese Patienten nur nach sorgfältiger Nutzen-Risiko-Abwägung erfolgen. Der Filmüberzug der Tablette enthält Lactose. Männer mit der seltenen hereditären Galactose-Intoleranz, Lactase-Mangel oder Glucose-Galactose-Malabsorption sollten VIAGRA nicht einnehmen. Frauen Für die Behandlung von Frauen ist VIAGRA nicht indiziert. 4.5 Wechselwirkungen mit anderen Arzneimitteln und sonstige Wechselwirkungen Wirkungen anderer Arzneimittel auf Sildenafil In-vitro-Studien Der Sildenafil-Metabolismus wird grundsätzlich durch die Cytochrom-P450 (CYP)-Isoenzyme 3A4 (Hauptweg) und 2C9 (Nebenweg) vermittelt. Die Sildenafil-Clearance kann folglich durch Inhibitoren dieser Isoenzyme herabgesetzt und durch Induktoren dieser Isoenzyme gesteigert sein. In-vivo-Studien Die Analyse pharmakokinetischer Daten aus den durchgeführten klinischen Studien wies auf eine reduzierte Sildenafil-Clearance bei gleichzeitiger Gabe von CYP3A4-Inhibitoren (wie Ketoconazol, Erythromycin, Cimetidin) hin. Obwohl bei den Patienten, die gleichzeitig CYP3A4-Inhibitoren erhielten, keine Zunahme von unerwünschten Ereignissen beobachtet wurde, sollte hier eine Anfangsdosis von 25 mg in Erwägung gezogen werden. Die gleichzeitige Gabe des HIV-ProteaseHemmstoffs Ritonavir im Steady State (zweimal täglich 500 mg), der ein hochpotenter P450-Hemmstoff ist, und Sildenafil (100-mgEinzeldosis) bewirkte eine 300%ige (4-fache) Steigerung der Sildenafil-Cmax und eine 1 000%ige (11-fache) Steigerung der Sildenafil-Plasma-AUC. Nach 24 Stunden betrugen die Sildenafil-Plasmaspiegel noch etwa 200 ng/ml im Vergleich zu 5 ng/ml, wenn Sildenafil alleine gegeben wurde. Dies entspricht den ausgeprägten Effekten von Ritonavir auf ein breites Spektrum von P450Substraten. Sildenafil hatte keine Auswirkungen auf die Pharmakokinetik von Ritonavir. Aufgrund dieser pharmakokinetischen Ergebnisse ist von der gleichzeitigen Gabe von Sildenafil und Ritonavir abzuraten (siehe Abschnitt 4.4), und in jedem Fall sollte die maximale Sildenafil-Dosis unter keinen Umständen 25 mg innerhalb 48 Stunden überschreiten. Die gleichzeitige Gabe des HIV-ProteaseHemmstoffs Saquinavir, eines CYP3A4- Hemmstoffs, im Steady State (dreimal täglich 1 200 mg) und von Sildenafil (100-mgEinzeldosis) bewirkte eine 140%ige Steigerung der Sildenafil-Cmax und eine 210%ige Steigerung der Sildenafil-Plasma-AUC. Sildenafil hatte keine Auswirkungen auf die Pharmakokinetik von Saquinavir (siehe Abschnitt 4.2). Bei stärkeren CYP3A4-Hemmstoffen wie Ketoconazol und Itraconazol dürften größere Effekte zu erwarten sein. Bei Gabe einer Einzeldosis von 100 mg Sildenafil mit Erythromycin, einem mäßigen CYP3A4-Hemmstoff, im Steady State (zweimal täglich 500 mg für 5 Tage) wurde die systemische Sildenafil-Exposition (AUC) um 182 % gesteigert. Bei gesunden männlichen Probanden konnte ein Einfluss von Azithromycin (500 mg täglich über 3 Tage) auf die AUC, Cmax, tmax, Eliminationsrate oder die sich daraus ergebende Halbwertszeit von Sildenafil oder seinem Hauptmetaboliten nicht nachgewiesen werden. Cimetidin (800 mg), ein Cytochrom-P450-Hemmstoff und unspezifischer CYP3A4-Hemmstoff, bewirkte eine 56%ige Steigerung der Sildenafil-Plasmaspiegel, wenn es gesunden Probanden gleichzeitig mit Sildenafil (50 mg) gegeben wurde. Grapefruitsaft ist ein schwacher Hemmstoff des CYP3A4-Stoffwechsels in der Darmwand und kann eine geringe Steigerung der Sildenafil-Plasmaspiegel bewirken. Durch die Einmalgabe eines Antazidums (Magnesiumhydroxid/Aluminiumhydroxid) wurde die Bioverfügbarkeit von Sildenafil nicht beeinflusst. Obwohl spezifische Interaktionsstudien nicht für alle Arzneimittel durchgeführt wurden, erbrachte die Analyse pharmakokinetischer Daten aus den durchgeführten klinischen Studien keine Veränderungen der pharmakokinetischen Parameter von Sildenafil bei gleichzeitiger Behandlung mit CYP2C9-Inhibitoren (wie Tolbutamid, Warfarin, Phenytoin), CYP2D6-Inhibitoren (wie selektiven Serotonin-Wiederaufnahme-Hemmern, trizyklischen Antidepressiva), Thiazidund ähnlichen Diuretika, Schleifen- und kaliumsparenden Diuretika, ACE-Hemmern, Calciumantagonisten, Betablockern oder Substanzen, die den CYP450-Stoffwechsel induzieren (wie Rifampicin, Barbiturate). In einer Studie an gesunden männlichen Probanden führte die gleichzeitige Anwendung des Endothelin-Antagonisten Bosentan (einem Induktor von CYP3A4 [mäßig], CYP2C9 und möglicherweise auch von CYP2C19) im Steady State (zweimal täglich 125 mg) zusammen mit Sildenafil im Steady State (dreimal täglich 80 mg) zu einer Verringerung der AUC und der Cmax von Sildenafil um 62,6 % bzw. 55,4 %. Daher wird bei gleichzeitiger Anwendung von starken CYP3A4-Induktoren wie Rifampicin erwartet, dass sie eine größere Abnahme der Plasmakonzentration von Sildenafil verursachen. Nicorandil ist ein Wirkstoff, der gleichzeitig als Kaliumkanalöffner und als Nitrat wirkt. Aufgrund der Nitratkomponente besteht die Möglichkeit, dass er zu einer schwerwiegenden Wechselwirkung mit Sildenafil führt. 003927-14827 Fachinformation (Zusammenfassung der Merkmale des Arzneimittels) VIAGRA ® Wirkungen von Sildenafil auf andere Arzneimittel In-vitro-Studien Sildenafil ist ein schwacher Inhibitor der Cytochrom-P450-Isoenzyme 1A2, 2C9, 2C19, 2D6, 2E1 und 3A4 (IC50 >150 µM). Angesichts der maximalen Plasmaspiegel von Sildenafil nach empfohlener Dosierung von etwa 1 µM erscheint es unwahrscheinlich, dass VIAGRA die Clearance von Substraten dieser Isoenzyme verändert. Es liegen keine Daten hinsichtlich Wechselwirkungen zwischen Sildenafil und unspezifischen Phosphodiesteraseinhibitoren wie Theophyllin oder Dipyridamol vor. In-vivo-Studien Entsprechend seiner pharmakologischen Wirkung auf den Stickstoffmonoxid-cGMPStoffwechsel (siehe Abschnitt 5.1) konnte gezeigt werden, dass Sildenafil den blutdrucksenkenden Effekt von Nitraten verstärkt. Daher ist die gleichzeitige Gabe mit Stickstoffmonoxid-Donatoren oder jeglichen Nitraten kontraindiziert (siehe Abschnitt 4.3). Wenn Patienten unter Alphablocker-Therapie gleichzeitig Sildenafil erhalten, kann dies bei einigen wenigen empfindlichen Personen zu symptomatischer Hypotonie führen. Am wahrscheinlichsten tritt diese innerhalb von 4 Stunden nach Einnahme von Sildenafil auf (siehe Abschnitte 4.2 und 4.4). In drei spezifischen Interaktionsstudien wurde der Alphablocker Doxazosin (4 mg und 8 mg) zusammen mit Sildenafil (25 mg, 50 mg oder 100 mg) an stabil eingestellte Doxazosin-Patienten mit benigner Prostatahyperplasie (BPH) gegeben. Bei dieser Studienpopulation wurde ein zusätzlicher mittlerer Blutdruckabfall von jeweils 7/7 mmHg, 9/5 mmHg und 8/4 mmHg im Liegen und 6/6 mmHg, 11/4 mmHg und 4/5 mmHg im Stehen beobachtet. Bei gleichzeitiger Gabe von Sildenafil an Patienten mit stabil eingestellter Doxazosin-Dosis gab es gelegentlich Berichte über eine symptomatische orthostatische Hypotonie. Gemeldet wurden dabei Schwindelgefühl und Benommenheit, jedoch keine Synkope. Bei gleichzeitiger Gabe von Sildenafil (50 mg) wurden keine signifikanten Wechselwirkungen mit Tolbutamid (250 mg) oder mit Warfarin (40 mg) gezeigt, die beide durch CYP2C9 verstoffwechselt werden. Die durch Acetylsalicylsäure (150 mg) bewirkte Verlängerung der Blutungszeit wurde durch Sildenafil (50 mg) nicht gesteigert. Juni 2015 spcde-5v28vgr-ft-0 Die blutdrucksenkende Wirkung von Alkohol (maximale Blutalkoholspiegel im Mittel 80 mg/dl) wurde bei gesunden Probanden durch Sildenafil (50 mg) nicht verstärkt. Bei gepoolter Analyse der Patientengruppe, die eine antihypertensive Medikation mit folgenden Substanzklassen: Diuretika, Betarezeptorenblocker, ACE-Hemmer, Angiotensin-II-Antagonisten, andere Antihypertensiva (direkte Vasodilatatoren und zentral wirksame Antihypertensiva), Ganglienblocker, Calciumantagonisten und Alpharezeptorenblocker erhielten, ergab sich kein Unterschied des Nebenwirkungsprofils zwischen Patienten, die zusätzlich Sildenafil, und Patienten, die zusätzlich Placebo erhielten. In einer gezielten Interaktionsstudie erhielten Hyper003927-14827 toniker Sildenafil (100 mg) zusammen mit Amlodipin. Es zeigte sich eine zusätzliche Senkung des Blutdrucks im Liegen um systolisch 8 mmHg und diastolisch um 7 mmHg. Das Ausmaß dieser zusätzlichen Blutdrucksenkung war ähnlich der Blutdrucksenkung, die beobachtet wurde nach alleiniger Gabe von Sildenafil an gesunde Probanden (siehe Abschnitt 5.1). Sildenafil (100 mg) beeinflusste nicht die Steady-State-Pharmakokinetik der HIV-Protease-Hemmstoffe Saquinavir und Ritonavir, die beide CYP3A4-Substrate sind. In gesunden männlichen Probanden führte Sildenafil im Steady State (80 mg dreimal täglich) zu einer Erhöhung der AUC von Bosentan um 49,8 % und zu einer Erhöhung der Cmax von Bosentan um 42 % (Bosentan: 125 mg zweimal täglich). 4.6 Fertilität, Schwangerschaft und Stillzeit Für die Behandlung von Frauen ist VIAGRA nicht indiziert. Es liegen keine geeigneten und gut kontrollierten Studien mit schwangeren oder stillenden Frauen vor. In Reproduktionsstudien bei Ratten und Kaninchen wurden nach oraler SildenafilApplikation keine relevanten unerwünschten Wirkungen festgestellt. Bei einmaliger oraler Gabe von 100 mg Sildenafil an gesunde Probanden waren keine Effekte auf die Motilität oder die Morphologie der Spermien festzustellen (siehe Abschnitt 5.1). 4.7 Auswirkungen auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen Es wurden keine Studien zu den Auswirkungen auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen durchgeführt. Da in klinischen Studien mit Sildenafil über Schwindel und Sehstörungen berichtet wurde, sollen die Patienten darauf achten, wie sie auf die Einnahme von VIAGRA reagieren, bevor sie Auto fahren oder Maschinen bedienen. 4.8 Nebenwirkungen Zusammenfassung des Sicherheitsprofils Das Sicherheitsprofil von VIAGRA beruht auf 9.570 Patienten aus 74 doppelblinden placebokontrollierten klinischen Studien. Die am häufigsten beschriebenen Nebenwirkungen bei den mit Sildenafil behandelten Patienten in klinischen Studien waren Kopfschmerzen, Flush, Dyspepsie, verstopfte Nase, Schwindel, Übelkeit, Hitzewallung, Sehstörungen, Zyanopsie und verschwommenes Sehen. Aus der Post-Marketing-Überwachung liegen gesammelte Berichte über Nebenwirkungen für einen geschätzten Zeitraum von >10 Jahren vor. Da nicht alle Nebenwirkungen an den Inhaber der Zulassung gemeldet und so in der Sicherheitsdatenbank erfasst werden, können die Häufigkeiten für diese Nebenwirkungen nicht zuverlässig bestimmt werden. Tabellarische Auflistung der Nebenwirkungen In der nachstehenden Tabelle werden alle medizinisch relevanten Nebenwirkungen, die in klinischen Studien mit einer höheren Inzidenz als mit Placebo beschrieben wurden, nach Systemorganklassen und Häufigkeit (sehr häufig [≥ 1/10], häufig [≥ 1/100, <1/10], gelegentlich [≥ 1/1.000, <1/100], selten [≥ 1/10.000, <1/1.000]) angeführt. Innerhalb jeder Häufigkeitsgruppe werden die Nebenwirkungen nach abnehmendem Schweregrad angegeben. Meldung des Verdachts auf Nebenwirkungen Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von großer Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-RisikoVerhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdachtsfall einer Nebenwirkung über das aufgeführte nationale Meldesystem anzuzeigen. Deutschland Bundesinstitut für Arzneimittel und Medizinprodukte Abt. Pharmakovigilanz Kurt-Georg-Kiesinger-Allee 3 D-53175 Bonn Website: www.bfarm.de Österreich Bundesamt für Sicherheit im Gesundheitswesen Traisengasse 5 1200 WIEN ÖSTERREICH Fax: + 43 (0) 50 555 36207 Website: http://www.basg.gv.at/ 4.9 Überdosierung In Studien erhielten gesunde Probanden Einzeldosen bis zu 800 mg. Die hierbei beobachteten Nebenwirkungen waren ähnlich wie die bei niedrigeren Dosen, lediglich Inzidenz und Schweregrad waren erhöht. Dosen von 200 mg führten nicht zu einer stärkeren Wirksamkeit, jedoch zu einem Anstieg der Inzidenz von Nebenwirkungen (Kopfschmerz, Flush, Schwindel, Dyspepsie, Verstopfung der Nase, Sehstörungen). In Fällen einer Überdosierung sollten je nach Bedarf die üblichen unterstützenden Maßnahmen eingeleitet werden. Da Sildenafil in hohem Maße an Plasmaproteine gebunden ist und renal nicht eliminiert wird, ist durch eine Dialyse keine Beschleunigung der Clearance zu erwarten. 5. PHARMAKOLOGISCHE EIGENSCHAFTEN 5.1 Pharmakodynamische Eigenschaften Pharmakotherapeutische Gruppe: Urologika; Arzneimittel zur Behandlung der erektilen Dysfunktion ATC-Code: G04B E03 Wirkmechanismus Sildenafil stellt eine orale Behandlung der erektilen Dysfunktion dar. Auf natürliche Weise, d. h. durch sexuelle Stimulation, wird die gestörte Erektionsfähigkeit durch eine Steigerung des Bluteinstroms in den Penis wiederhergestellt. 3 Fachinformation (Zusammenfassung der Merkmale des Arzneimittels) VIAGRA ® Tabelle 1: Medizinisch relevante Nebenwirkungen, die in kontrollierten klinischen Studien mit einer höheren Inzidenz als unter Placebo beschrieben wurden, und medizinisch relevante Nebenwirkungen, die aus der Überwachung nach Marktzulassung berichtet wurden. Systemorganklasse Sehr häufig (≥ 1/10) Häufig (≥ 1/100, <1/10) Infektionen und parasitäre Erkrankungen Gelegentlich (≥ 1/1.000, <1/100) Selten (≥ 1/10.000, <1/1.000) Rhinitis Erkrankungen des Immunsystems Überempfindlichkeit Erkrankungen des Nervensystems Kopfschmerzen Schwindel Somnolenz, Hypästhesie Schlaganfall, Transitorische ischämische Attacke, Krampfanfall,* Rezidivierende Krampfanfälle,* Synkope Augenerkrankungen Veränderungen des Farb- Tränenflussstörungen***, Nicht arteriitische anteriore sehens**, Sehstörungen, Augenschmerzen, Photo- ischämische Optikusneuverschwommenes Sehen phobie, Photopsie, Okulä- ropathie (NAION),* Retire Hyperämie, Visuelles naler Gefässverschluss,* Netzhautblutung, ArterioLeuchten, Konjunktivitis sklerotische Retinopathie, Erkrankung der Retina, Glaukom, Gesichtsfelddefekt, Doppeltsehen, Sehschärfe vermindert, Myopie, Asthenopie, Mouches volantes, Iriserkrankung, Mydriasis, Farbsäume, Augenödem, Schwellung des Auges, Augenerkrankung, Bindehauthyperämie, Augenreizung, Anomale Sinnesempfindung des Auges, Augenlidödem, Skleraverfärbung Erkrankungen des Ohrs und des Labyrinths Vertigo, Tinnitus Taubheit Herzerkrankungen Tachykardie, Palpitationen Plötzlicher Herztod,* Herzinfarkt, Ventrikuläre Arrhythmie,* Vorhofflimmern, instabile Angina pectoris Gefässerkrankungen Flush, Hitzewallung Hypertonie, Hypotonie Erkrankungen der Atemwege, des Brustraums und Mediastinums Verstopfte Nase Epistaxis, Sinus-Sekretstauung Engegefühl des Halses, Nasenödeme, trockene Nasenschleimhaut Erkrankungen des Gastrointestinaltrakts Übelkeit, Dyspepsie Gastroösophageale Refluxerkrankung, Erbrechen, Schmerzen im Oberbauch, Trockener Mund Orale Hypästhesia Erkrankungen der Haut und des Unterhautzellgewebes Ausschlag Syndrom Stevens-Johnson,* toxische epidermale Nekrolyse* Skelettmuskulatur-, Bindegewebsund Knochenerkrankungen Myalgie, Schmerzen in den Extremitäten Erkrankungen der Nieren und Harnwege Hämaturie Erkrankungen der Geschlechtsorgane und der Brustdrüse Penisblutung Priapismus,* Hämatospermie, prolongierte Erektion Allgemeine Erkrankungen und Beschwerden am Verabreichungsort Brustschmerzen, Müdigkeit, Wärmegefühl Untersuchungen erhöhte Herzfrequenz Reizbarkeit * Wurde nur während der Überwachung nach Markteinführung beschrieben ** Veränderungen des Farbsehens: Chloropsie, Chromatopsie, Zyanopsie, Erythropsie und Xanthopsie *** Tränenflussstörungen: Trockenes Auge, Erkrankungen des Tränenapparates, Tränensekretion verstärkt 4 003927-14827 Fachinformation (Zusammenfassung der Merkmale des Arzneimittels) VIAGRA ® Der für die Erektion des Penis verantwortliche physiologische Mechanismus schließt die Freisetzung von Stickstoffmonoxid (NO) im Corpus cavernosum während der sexuellen Stimulation ein. Das Stickstoffmonoxid aktiviert das Enzym Guanylatcyclase, was zu erhöhten Spiegeln an zyklischem Guanosinmonophosphat (cGMP) führt. Hierdurch kommt es zu einer Relaxation der glatten Muskulatur im Corpus cavernosum, was den Bluteinstrom ermöglicht. Sildenafil ist ein wirksamer und selektiver Hemmstoff der cGMP-spezifischen Phosphodiesterase Typ 5 (PDE5) im Corpus cavernosum, wo sie für den Abbau von cGMP verantwortlich ist. Sildenafil wirkt peripher auf Erektionen. Sildenafil übt keinen direkten relaxierenden Effekt auf isoliertes menschliches Corpus-cavernosum-Gewebe aus, es verstärkt jedoch die relaxierende Wirkung von NO auf dieses Gewebe. Wenn unter sexueller Stimulation die Aktivierung des NO/cGMP-Stoffwechselweges stattfindet, bewirkt die PDE5-Hemmung durch Sildenafil erhöhte cGMP-Spiegel im Corpus cavernosum. Daher ist eine sexuelle Stimulation nötig, damit Sildenafil den beabsichtigten günstigen pharmakologischen Effekt entwickeln kann. Pharmakodynamische Wirkungen In-vitro -Studien zeigten, dass Sildenafil für die PDE5, die am Erektionsprozess beteiligt ist, selektiv ist. Es wirkt stärker auf PDE5 als auf andere bekannte PhosphodiesteraseIsoenzyme. Gegenüber der PDE6, die an dem Phototransduktionsprozess in der Retina beteiligt ist, hat Sildenafil eine 10-fach höhere Selektivität. Bei den maximalen empfohlenen Dosen zeigt sich eine 80-fach höhere Selektivität gegenüber PDE1 und eine über 700-fach höhere Selektivität gegenüber PDE2, 3, 4, 7, 8, 9, 10 und 11. Sildenafil hat insbesondere eine mehr als 4 000-fach höhere Selektivität für PDE5 im Vergleich zu PDE3, dem an der Steuerung der kardialen Kontraktilität beteiligten, cAMP-spezifischen Phosphodiesterase-Isoenzym. Juni 2015 spcde-5v28vgr-ft-0 Klinische Wirksamkeit und Sicherheit In zwei klinischen Studien wurde gezielt überprüft, innerhalb welchen Zeitfensters Sildenafil auf sexuelle Stimulation eine Erektion auslösen kann. Eine Untersuchung mittels Penisplethysmographie (RigiScan) bei nüchternen Patienten zeigte, dass bei den Patienten, die eine 60%ige Rigidität des Penis (die einen Geschlechtsverkehr ermöglicht) unter Sildenafil erreichten, im Mittel innerhalb von 25 Minuten (Bereich: 12 bis 37 Minuten) die Wirkung eintrat. In einer weiteren RigiScan-Untersuchung konnte Sildenafil noch 4 bis 5 Stunden nach oraler Einnahme auf sexuelle Stimulation eine Erektion auslösen. Sildenafil bewirkt eine geringe und vorübergehende Reduktion des Blutdrucks, die in den meisten Fällen keine klinisch relevanten Erscheinungen zur Folge hat. Im Mittel betrugen die maximalen Blutdrucksenkungen im Liegen nach Einnahme von 100 mg Sildenafil systolisch 8,4 mmHg, diastolisch 5,5 mmHg. Diese Blutdrucksenkung spiegelt den vasodilatatorischen Effekt von Sildenafil wider, möglicherweise aufgrund erhöhter cGMP-Spiegel in der glatten Gefäßmusku003927-14827 latur. Orale Einzeldosen von bis zu 100 mg Sildenafil zeigten bei gesunden Probanden keine klinisch relevanten EKG-Veränderungen. In einer Studie zu den hämodynamischen Effekten einer oralen Einmalgabe von 100 mg Sildenafil bei 14 Patienten mit schwerer (>70%ige Stenose mindestens einer Koronararterie) koronarer Herzkrankheit (KHK) nahmen der mittlere systolische und der mittlere diastolische Blutdruck in Ruhe gegenüber dem Ausgangswert um 7 % bzw. 6 % ab. Der mittlere pulmonale systolische Blutdruck nahm um 9 % ab. Sildenafil beeinflusste weder das Herzminutenvolumen noch beeinträchtigte es die Durchblutung in den stenosierten Koronararterien. In einer doppelblinden placebokontrollierten Belastungsstudie wurden 144 Patienten mit erektiler Dysfunktion und stabiler chronischer Angina pectoris untersucht, die regelmäßig antianginöse Medikation (außer Nitraten) erhielten. Unter Sildenafil traten im Vergleich zu Placebo keine klinisch relevanten Unterschiede in der Zeit bis zum Auftreten einer zum Abbruch zwingenden Angina auf. Leichte und vorübergehende Veränderungen des Farbsehens (Blau/Grün) wurden bei einigen Studienteilnehmern durch den Farnsworth-Munsell-100-Farben-Test 1 Stunde nach Einnahme von 100 mg beobachtet, 2 Stunden nach Einnahme waren diese Veränderungen nicht mehr nachweisbar. Der vermutete Mechanismus für diese Veränderung des Farbsehens bezieht sich auf die Hemmung der PDE 6, die bei dem Phototransduktionsprozess der Retina eine Rolle spielt. Sildenafil übt keinen Einfluss auf die Sehschärfe oder das Kontrastsehen aus. In einer kleinen placebokontrollierten Untersuchung bei neun Patienten mit dokumentierter altersbedingter Makuladegeneration im Frühstadium zeigte Sildenafil als 100-mg-Einmaldosis in den durchgeführten Sehtests (Sehschärfe, Amsler-Gitter, Lichtertest, Humphrey-Perimeter und PhotostressTest) keine signifikanten Veränderungen. Bei einmaliger oraler Gabe von 100 mg Sildenafil an gesunde Probanden wurden keine Effekte auf Motilität oder Morphologie der Spermien festgestellt (siehe Abschnitt 4.6). Weitere Informationen über klinische Studien Sildenafil wurde in klinischen Studien an mehr als 8 000 Patienten im Alter von 19 bis 87 Jahren gegeben, wobei folgende Patientengruppen vertreten waren: ältere Patienten (19,9 %), Patienten mit Hypertonie (30,9 %), Diabetes mellitus (20,3 %), ischämischer Herzkrankheit (5,8 %), Hyperlipidämie (19,8 %), Rückenmarkverletzungen (0,6 %), Depressionen (5,2 %), transurethraler Prostataresektion (3,7 %) und radikaler Prostatektomie (3,3 %). Folgende Patientengruppen waren nur unzureichend vertreten oder wurden aus den Studien ausgeschlossen: Patienten nach Operationen im kleinen Becken, nach Radiatio, mit schweren Nieren- oder Leberfunktionsstörungen sowie mit bestimmten Herz-Kreislauf-Erkrankungen (siehe Abschnitt 4.3). In Studien mit festgelegter Dosis berichteten 62 % (25 mg), 74 % (50 mg) und 82 % (100 mg) der Patienten über eine Verbesserung ihrer Erektion gegenüber 25 % unter Placebo. In kontrollierten klinischen Studien war die Sildenafil-bedingte Abbruchrate niedrig und mit der unter Placebo vergleichbar. In allen klinischen Studien lag die Rate der Patienten, die über eine Verbesserung unter Sildenafil berichteten, je nach Patientengruppe bei: psychogene erektile Dysfunktion (84 %), gemischte erektile Dysfunktion (77 %), organisch bedingte erektile Dysfunktion (68 %), ältere Patienten (67 %), Diabetes mellitus (59 %), koronare Herzkrankheit (69 %), Hypertonie (68 %), transurethrale Prostataresektion (61 %), radikale Prostatektomie (43 %), Rückenmarkverletzungen (83 %), Depressionen (75 %). Die Unbedenklichkeit und Wirksamkeit von Sildenafil blieben in den Langzeitstudien erhalten. Kinder und Jugendliche Die Europäische Arzneimittel-Agentur hat für VIAGRA eine Freistellung von der Verpflichtung zur Vorlage von Ergebnissen zu Studien in allen pädiatrischen Altersklassen bei erektiler Dysfunktion gewährt (siehe Abschnitt 4.2 bzgl. Informationen zur Anwendung bei Kindern und Jugendlichen). 5.2 Pharmakokinetische Eigenschaften Resorption Sildenafil wird schnell resorbiert. Die maximalen beobachteten Plasmaspiegel werden innerhalb von 30 bis 120 Minuten (Mittel: 60 Minuten) nach oraler Gabe im nüchternen Zustand erreicht. Die mittlere absolute orale Bioverfügbarkeit beträgt 41 % (Streubreite: 25 bis 63 %). Nach oraler Einnahme von Sildenafil nehmen AUC und Cmax dosisproportional über den empfohlenen Dosisbereich (25 bis 100 mg) zu. Bei Einnahme von Sildenafil zusammen mit einer Mahlzeit ist die Resorptionsrate reduziert, die tmax verzögert sich um 60 Minuten, während die Cmax im Mittel um 29 % verringert ist. Verteilung Das mittlere Verteilungsvolumen von Sildenafil im Steady State beträgt 105 l, was auf eine Verteilung in die Gewebe hinweist. Nach einer oralen Einmalgabe von 100 mg beträgt die mittlere maximale Gesamtplasmakonzentration von Sildenafil ca. 440 ng/ml (CV 40 %). Da Sildenafil (und sein wichtigster im Blutkreislauf zirkulierender, N-desmethylierter Metabolit) zu 96 % an Plasmaproteine gebunden ist, ergibt sich hieraus eine mittlere maximale freie Sildenafil-Plasmakonzentration von 18 ng/ml (38 nM). Die Proteinbindung ist unabhängig von der Gesamtkonzentration des Arzneimittels. Bei gesunden Probanden wurden 90 Minuten nach Gabe von Sildenafil (100-mg-Einzeldosis) weniger als 0,0002 % (im Mittel 188 ng) der gegebenen Menge im Ejakulat gefunden. Biotransformation Sildenafil wird überwiegend hepatisch durch die mikrosomalen Isoenzyme CYP3A4 (Hauptweg) und CYP2C9 (Nebenweg) metabolisiert. Der wichtigste zirkulierende Metabolit resultiert aus N-Demethylierung 5 Fachinformation (Zusammenfassung der Merkmale des Arzneimittels) VIAGRA ® von Sildenafil. Das Profil der Phosphodiesterase-Selektivität dieses Metaboliten ist ähnlich dem von Sildenafil und weist in vitro eine Hemmwirkung für PDE5 auf, die rund 50 % derjenigen der Stammsubstanz beträgt. Die Plasmaspiegel dieses Metaboliten betragen rund 40 % der für Sildenafil beobachteten Werte. Der N-Desmethyl-Metabolit wird weiter verstoffwechselt, die terminale Halbwertszeit beträgt rund 4 Stunden. 6. PHARMAZEUTISCHE ANGABEN Elimination Die gesamte Clearance von Sildenafil beträgt 41 l/h mit einer daraus resultierenden terminalen Halbwertszeit von 3 bis 5 Stunden. Nach oraler oder intravenöser Gabe wird Sildenafil nach Metabolisierung hauptsächlich über die Fäzes (rund 80 % der gegebenen oralen Dosis) und in geringerem Maße über den Urin (rund 13 % der gegebenen oralen Dosis) ausgeschieden. Filmüberzug: Hypromellose Titandioxid (E 171) Lactose-Monohydrat Triacetin Indigocarmin, Aluminiumsalz (E 132) Pharmakokinetik bei speziellen Patientengruppen Ältere Patienten Gesunde ältere Probanden (65 Jahre oder älter) zeigten eine herabgesetzte SildenafilClearance, wobei die Plasmaspiegel von Sildenafil und des aktiven N-DesmethylMetaboliten ungefähr 90 % höher lagen als bei jüngeren gesunden Probanden (18 bis 45 Jahre). Aufgrund der altersabhängigen Veränderung der Plasmaproteinbindung lag der entsprechende Anstieg der Plasmaspiegel von freiem Sildenafil bei rund 40 %. Nierenfunktionsstörungen Bei Probanden mit leichter bis mäßiger Nierenfunktionsstörung (Kreatinin-Clearance = 30 bis 80 ml/min) war die Pharmakokinetik nach einer oralen Sildenafil-Einzeldosis von 50 mg unverändert. Die mittleren Werte für AUC und Cmax des N-DesmethylMetaboliten stiegen um bis zu 126 % bzw. bis zu 73 % im Vergleich zu Probanden gleichen Alters mit nicht eingeschränkter Nierenfunktion. Aufgrund der hohen interindividuellen Variabilität waren diese Unterschiede nicht statistisch signifikant. Bei Probanden mit schwerer Nierenfunktionsstörung (Kreatinin-Clearance <30 ml/min) war die Clearance von Sildenafil herabgesetzt und resultierte in Erhöhungen von AUC (100 %) und Cmax (88 %) im Vergleich zu Probanden gleichen Alters mit nicht eingeschränkter Nierenfunktion. Zusätzlich waren die AUC (200 %) und Cmax (79 %) des N-Desmethyl-Metaboliten signifikant erhöht. Leberfunktionsstörungen Bei Probanden mit leichter bis mäßiger Leberzirrhose (Child-Pugh-Klassen A und B) war die Clearance von Sildenafil herabgesetzt, was zu Erhöhungen von AUC (84 %) und Cmax (47 %) führte, im Vergleich zu Probanden gleichen Alters mit nicht eingeschränkter Leberfunktion. Die Pharmakokinetik von Sildenafil bei Patienten mit schwerer Leberfunktionsstörung wurde nicht untersucht. 5.3 Präklinische Daten zur Sicherheit Basierend auf den konventionellen Studien zur Sicherheitspharmakologie, Toxizität bei wiederholter Gabe, Reproduktions- und Ent6 wicklungstoxizität, Genotoxizität und zum kanzerogenen Potenzial lassen die präklinischen Daten keine besonderen Gefahren für den Menschen erkennen. 6.1 Liste der sonstigen Bestandteile Tablettenkern: Mikrokristalline Cellulose Calciumhydrogenphosphat Croscarmellose-Natrium Magnesiumstearat 6.2 Inkompatibilitäten Nicht zutreffend. 6.3 Dauer der Haltbarkeit 5 Jahre 6.4 Besondere Vorsichtsmaßnahmen für die Aufbewahrung Nicht über 30 °C lagern. In der Originalverpackung aufbewahren, um den Inhalt vor Feuchtigkeit zu schützen. 6.5 Art und Inhalt des Behältnisses PVC/Aluminium-Blisterpackungen in Umkartons zu 2, 4, 8, 12 oder 24 Tabletten oder als zusätzlich hitzeversiegelte Blisterkarten. Es werden möglicherweise nicht alle Packungsgrößen in den Verkehr gebracht. 6.6 Besondere Vorsichtsmaßnahmen für die Beseitigung und sonstige Hinweise zur Handhabung Keine besonderen Anforderungen 7. INHABER DER ZULASSUNG Pfizer Limited Sandwich Kent CT13 9NJ Vereinigtes Königreich 8. ZULASSUNGSNUMMERN EU/1/98/077/002 – 004 EU/1/98/077/006 – 008 EU/1/98/077/010 – 012 EU/1/98/077/013 EU/1/98/077/014 EU/1/98/077/015 EU/1/98/077/016 – 019 EU/1/98/077/024 EU/1/98/077/025 Ausführliche Informationen zu diesem Arzneimittel sind auf den Internetseiten der Europäischen Arzneimittel-Agentur http://www.ema.europa.eu verfügbar. Verkaufsabgrenzung in Deutschland Verschreibungspflichtig. Rezeptpflicht/Apothekenpflicht in Österreich Rezept- und apothekenpflichtig. Packungsgrößen in Deutschland VIAGRA 25 mg: Packung mit 4 Filmtabletten VIAGRA 50 mg: Packung mit 4 Filmtabletten Packung mit 12 Filmtabletten VIAGRA 100 mg: Packung mit 4 Filmtabletten Packung mit 12 Filmtabletten Packungsgrößen in Österreich VIAGRA 25 mg: Packung mit 4 Filmtabletten VIAGRA 50 mg: Packung mit 4 Filmtabletten Packung mit 12 Filmtabletten VIAGRA 100 mg: Packung mit 4 Filmtabletten Packung mit 12 Filmtabletten Repräsentant in Deutschland PFIZER PHARMA GmbH Linkstr. 10 10785 Berlin Tel.: 030 550055-51000 Fax: 030 550054-10000 Mitvertrieb: PHARMACIA GmbH Linkstr. 10 10785 Berlin Tel.: 030 550055-51000 Fax: 030 550054-10000 Repräsentant in Österreich Pfizer Corporation Austria Ges.m.b.H. Floridsdorfer Hauptstraße 1 A-1210 Wien Tel.: +43 (0)1 521 15-0 9. DATUM DER ERTEILUNG DER ZULASSUNG/VERLÄNGERUNG DER ZULASSUNG Datum der Erteilung der Zulassung: 14. September 1998 Datum der letzten Verlängerung der Zulassung: 14. September 2008 Zentrale Anforderung an: Rote Liste Service GmbH Fachinfo-Service 10. STAND DER INFORMATION Juni 2015 Mainzer Landstraße 55 60329 Frankfurt 003927-14827 Fachinformation (Zusammenfassung der Merkmale des Arzneimittels/SPC) Revatio ® 20 mg Filmtabletten 1. BEZEICHNUNG DES ARZNEIMITTELS Revatio ® 20 mg Filmtabletten 2. QUALITATIVE UND QUANTITATIVE ZUSAMMENSETZUNG Jede Filmtablette enthält 20 mg Sildenafil (als Citrat). Sonstige Bestandteile mit bekannter Wirkung: Jede Tablette enthält auch 0,7 mg Lactose. Vollständige Auflistung der sonstigen Bestandteile siehe Abschnitt 6.1. 3. DARREICHUNGSFORM Filmtablette Weiße, runde, bikonvexe Filmtabletten mit der Kennzeichnung „PFIZER“ auf der einen und „RVT 20“ auf der anderen Seite 4. KLINISCHE ANGABEN 4.1 Anwendungsgebiete Erwachsene Behandlung von erwachsenen Patienten mit pulmonaler arterieller Hypertonie (PAH) der WHO-Funktionsklassen II und III zur Verbesserung der körperlichen Leistungsfähigkeit. Die Wirksamkeit konnte nachgewiesen werden bei primärer PAH und bei pulmonaler Hypertonie in Verbindung mit einer Bindegewebskrankheit. Kinder und Jugendliche Behandlung von pädiatrischen Patienten im Alter von 1 bis 17 Jahren mit pulmonaler arterieller Hypertonie. Die Wirksamkeit konnte anhand der Verbesserung der körperlichen Belastbarkeit oder der pulmonalen Hämodynamik nachgewiesen werden bei primärer PAH und bei pulmonaler Hypertonie in Verbindung mit angeborenen Herzerkrankungen (siehe Abschnitt 5.1). 4.2 Dosierung und Art der Anwendung Die Behandlung sollte nur durch einen Arzt eingeleitet und überwacht werden, der Erfahrung mit der Behandlung von PAH hat. Im Falle einer klinischen Verschlechterung trotz einer Behandlung mit Revatio sollten andere Formen der Behandlung in Erwägung gezogen werden. Dosierung April 2015 spcde-5v31ro-ft-20 Erwachsene Die empfohlene Dosierung beträgt 20 mg dreimal täglich. Der Arzt sollte den Patienten, der eine Einnahme von Revatio vergessen hat, anhalten, so bald wie möglich eine Dosis einzunehmen und dann mit der normalen Dosierung fortzufahren. Zum Ausgleichen einer vergessenen Einnahme sollten die Patienten keine doppelte Dosis einnehmen. Kinder und Jugendliche (1 bis 17 Jahre) Bei pädiatrischen Patienten im Alter von 1 Jahr bis 17 Jahren beträgt die empfohlene Dosierung bei einem Körpergewicht ≤ 20 kg 10 mg (1 ml zubereitete Suspension) dreimal täglich und bei einem Körpergewicht >20 kg 20 mg (2 ml zubereitete Suspension oder 1 Tablette) dreimal täglich. Höhere als die empfohlenen Dosen sollten bei pädiatri009389-12885 schen Patienten mit PAH nicht angewendet werden (siehe Abschnitte 4.4 und 5.1). Hinweise zur Zubereitung des Arzneimittels vor der Anwendung siehe Abschnitt 6.6. Patienten, die zusätzlich weitere Arzneimittel anwenden Generell sollte jede Dosisanpassung nur nach einer sorgfältigen Nutzen-Risiko-Abschätzung vorgenommen werden. Wenn Sildenafil Patienten verabreicht wird, die bereits CYP3A4-Hemmer wie Erythromycin oder Saquinavir erhalten, sollte eine Dosisreduktion auf zweimal täglich 20 mg erwogen werden. Bei gleichzeitiger Gabe mit stärkeren CYP3A4-Hemmern wie Clarithromycin, Telithromycin und Nefazodon wird eine Dosisreduktion auf einmal täglich 20 mg empfohlen. Zur gleichzeitigen Anwendung von Sildenafil mit den stärksten CYP3A4-Hemmern siehe Abschnitt 4.3. Bei gleichzeitiger Gabe mit CYP3A4-Induktoren kann eine Dosisanpassung für Sildenafil notwendig werden (siehe Abschnitt 4.5). Spezielle Patientenpopulationen Ältere Patienten (≥ 65 Jahre) Bei älteren Patienten ist keine Dosisanpassung erforderlich. Die anhand der 6-Minuten-Gehstrecke gemessene klinische Wirksamkeit kann bei älteren Patienten verringert sein. Eingeschränkte Nierenfunktion Bei Patienten mit eingeschränkter Nierenfunktion einschließlich solchen mit schwerer Niereninsuffizienz (Kreatinin-Clearance <30 ml/min) ist keine initiale Dosisanpassung erforderlich. Nur wenn die Therapie nicht gut vertragen wird, sollte nach einer sorgfältigen Nutzen-Risiko-Bewertung eine Dosisreduktion auf 20 mg zweimal täglich erwogen werden. Eingeschränkte Leberfunktion Bei Patienten mit eingeschränkter Leberfunktion (Child-Pugh-Klassen A und B) ist keine initiale Dosisanpassung erforderlich. Nur wenn die Therapie nicht gut vertragen wird, sollte nach einer sorgfältigen NutzenRisiko-Bewertung eine Dosisreduktion auf 20 mg zweimal täglich erwogen werden. Bei Patienten mit schwerer Einschränkung der Leberfunktion (Child-Pugh-Klasse C) ist Revatio kontraindiziert (siehe Abschnitt 4.3). Kinder und Jugendliche Die Sicherheit und Wirksamkeit von Revatio bei Kindern unter 1 Jahr ist nicht erwiesen. Es liegen keine Daten vor. Absetzen der Behandlung Anhand bisheriger, limitierter Daten ist anzunehmen, dass ein plötzliches Absetzen von Revatio keinen Rebound-Effekt mit einer Verschlechterung der PAH verursacht. Allerdings sollte zur Vermeidung einer möglichen und plötzlichen klinischen Verschlechterung während des Absetzens eine allmähliche Dosisreduktion in Erwägung gezogen werden. Während des Absetzens wird eine engmaschigere Überwachung empfohlen. Art der Anwendung Revatio ist nur zur oralen Anwendung bestimmt. Die Tabletten sollten in Abständen von etwa 6 bis 8 Stunden mit oder unabhängig von einer Mahlzeit eingenommen werden. Zubereitete Suspension zum Einnehmen: Schütteln Sie die zubereitete Suspension zum Einnehmen sorgfältig für mindestens 10 Sekunden, bevor Sie die benötigte Dosis der Flasche entnehmen. 4.3 Gegenanzeigen Überempfindlichkeit gegen den Wirkstoff oder einen der in Abschnitt 6.1 genannten sonstigen Bestandteile Gleichzeitige Gabe mit NO-Donatoren (wie Amylnitrit) oder Nitraten jeglicher Form aufgrund der hypotensiven Effekte von Nitraten (siehe Abschnitt 5.1) Kombination mit den stärksten CYP3A4Hemmern (z. B. Ketoconazol, Itraconazol, Ritonavir) (siehe Abschnitt 4.5) Patienten, die aufgrund einer nicht arteriitischen anterioren ischämischen Optikusneuropathie (NAION) ihre Sehkraft auf einem Auge verloren haben, unabhängig davon, ob der Sehverlust mit einer vorherigen Einnahme eines PDE5-Hemmers in Zusammenhang stand oder nicht (siehe Abschnitt 4.4) Die Sicherheit von Sildenafil wurde bei folgenden Patientengruppen nicht untersucht und seine Anwendung bei diesen Gruppen ist daher kontraindiziert: – schwere Einschränkung der Leberfunktion – kürzlich zurückliegender Schlaganfall oder Herzinfarkt – ausgeprägte Hypotonie (Blutdruck <90/ 50 mmHg) bei Behandlungsbeginn 4.4 Besondere Warnhinweise und Vorsichtsmaßnahmen für die Anwendung Die Wirksamkeit von Revatio bei Patienten mit schwerer PAH (Funktionsklasse IV) wurde bisher nicht untersucht. Falls sich der klinische Zustand verschlechtert, sollten Therapien in Erwägung gezogen werden, die für ein schweres Stadium der Krankheit empfohlen werden (z. B. Epoprostenol) (siehe Abschnitt 4.2). Das Nutzen-Risiko-Profil von Sildenafil bei Patienten mit PAH der WHO-Funktionsklasse I wurde nicht untersucht. Studien mit Sildenafil wurden bei PAH in Verbindung mit primären (idiopathischen) Bindegewebskrankheiten und mit angeborenen Herzerkrankungen durchgeführt (siehe Abschnitt 5.1). Die Anwendung von Sildenafil bei anderen Formen der PAH wird nicht empfohlen. In der pädiatrischen Langzeit-Anschlussstudie wurde bei Patienten, die höhere als die empfohlenen Dosen erhielten, eine Zunahme von Todesfällen beobachtet. Deshalb sollten bei pädiatrischen Patienten mit PAH höhere als die empfohlenen Dosen nicht angewendet werden (siehe auch Abschnitte 4.2 und 5.1). Retinitis pigmentosa Die Sicherheit von Sildenafil wurde bei Patienten mit bekannter erblich bedingter degenerativer Retinaerkrankung wie beispielsweise Retinitis pigmentosa (eine Minderheit dieser Patienten hat eine genetisch bedingte Störung der retinalen Phosphodiesterasen) nicht untersucht und seine Anwendung kann daher nicht empfohlen werden. 1 Fachinformation (Zusammenfassung der Merkmale des Arzneimittels/SPC) Revatio ® 20 mg Filmtabletten Gefäßerweiternde Wirkung Bei der Verordnung von Sildenafil sollen Ärzte sorgfältig abwägen, ob Patienten mit bestimmten Grunderkrankungen durch die leichte bis mäßige gefäßerweiternde Wirkung von Sildenafil beeinträchtigt werden könnten. Hierzu zählen beispielsweise Patienten mit Hypotonie, solche mit Dehydratation, Patienten mit einer schweren Obstruktion des linksventrikulären Ausflusstrakts oder schwerer Einschränkung der autonomen Blutdruckkontrolle (siehe Abschnitt 4.4). Kardiovaskuläre Risikofaktoren Nach der Markteinführung von Sildenafil zur Behandlung der erektilen Dysfunktion wurden schwerwiegende kardiovaskuläre Ereignisse, einschließlich Herzinfarkt, instabiler Angina pectoris, plötzlichen Herztodes, ventrikulärer Arrhythmie, zerebrovaskulärer Blutung, transitorischer ischämischer Attacke, Hypertonie und Hypotonie im zeitlichen Zusammenhang mit der Anwendung von Sildenafil berichtet. Die meisten dieser Patienten hatten vorbestehende kardiovaskuläre Risikofaktoren. Für viele Ereignisse wurde berichtet, dass sie während oder kurz nach dem Geschlechtsverkehr auftraten, und für einige wenige, dass sie kurz nach der Anwendung von Sildenafil ohne sexuelle Aktivität auftraten. Es ist nicht möglich zu beurteilen, ob diese Ereignisse direkt mit den kardiovaskulären Risikofaktoren oder mit anderen Faktoren in Zusammenhang stehen. Priapismus Eine Anwendung von Sildenafil bei Patienten mit anatomischen Penismissbildungen (wie etwa Angulation, Fibrose im Bereich der Corpora cavernosa oder die Peyronie-Krankheit) sowie bei Patienten mit für Priapismus prädisponierenden Erkrankungen (wie Sichelzellenanämie, Plasmozytom, Leukämie) sollte mit entsprechender Vorsicht erfolgen. Nach der Markteinführung wurden verlängerte Erektionen und Priapismus unter Sildenafil-Behandlung berichtet. Falls eine Erektion länger als 4 Stunden anhält, sollte der Patient sofort medizinische Hilfe suchen. Wird ein Priapismus nicht sofort behandelt, kann dies zu Gewebeschädigung im Penis und dauerhaftem Potenzverlust führen (siehe Abschnitt 4.8). Vaso-okklusive Krise bei Patienten mit Sichelzellenanämie Bei Patienten mit pulmonaler Hypertonie nach einer Sichelzellenanämie sollte Sildenafil nicht angewendet werden. In einer klinischen Studie wurden vaso-okklusive Krisen, die zu einer Krankenhauseinweisung führten, bei den Patienten, die Revatio erhielten, häufiger berichtet als unter Placebo, was zu einem vorzeitigen Abbruch dieser Studie führte. Sehstörungen In Zusammenhang mit der Einnahme von Sildenafil und anderen PDE5-Hemmern sind Fälle von Sehstörungen spontan berichtet worden. Fälle von nicht arteriitischer anteriorer ischämischer Optikusneuropathie, einer seltenen Erkrankung, sind in Zusammenhang mit der Einnahme von Sildenafil und anderen PDE5-Hemmern spontan und in 2 einer Beobachtungsstudie berichtet worden (siehe Abschnitt 4.8). Im Falle jeglicher plötzlicher Sehstörungen sollte die Behandlung sofort abgebrochen und eine alternative Therapie in Betracht gezogen werden (siehe Abschnitt 4.3). Alphablocker Wenn Patienten unter Alphablocker-Therapie Sildenafil erhalten, ist Vorsicht geboten, da eine gleichzeitige Anwendung bei empfindlichen Personen zu symptomatischer Hypotonie führen kann (siehe Abschnitt 4.5). Um die Möglichkeit einer orthostatischen Hypotonie möglichst gering zu halten, sollten Patienten, die mit Alphablockern behandelt werden, vor Beginn der Behandlung mit Sildenafil hämodynamisch stabil eingestellt sein. Ärzte sollten die Patienten darüber aufklären, wie sie sich beim Auftreten von Symptomen einer orthostatischen Hypotonie verhalten sollen. Blutungsstörungen Studien an menschlichen Thrombozyten erbrachten Hinweise, dass Sildenafil die antiaggregatorische Wirkung von NitroprussidNatrium in vitro verstärkt. Es liegen keine Daten über die Unbedenklichkeit der Anwendung von Sildenafil an Patienten mit Blutungsstörungen oder aktiven peptischen Ulzera vor. Daher sollte die Gabe von Sildenafil bei diesen Patienten nur nach sorgfältiger Nutzen-Risiko-Abwägung erfolgen. Vitamin-K-Antagonisten Bei Patienten mit PAH, insbesondere bei einer PAH in Verbindung mit einer Bindegewebskrankheit, besteht bei Beginn einer Therapie mit Sildenafil unter laufender Behandlung mit einem Vitamin-K-Antagonisten möglicherweise ein erhöhtes Blutungsrisiko. Venenverschlusskrankheit Für die Anwendung von Sildenafil bei Patienten mit pulmonaler Hypertonie in Verbindung mit einer pulmonalen Venenverschlusskrankheit liegen bisher keine Daten vor. Allerdings wurden für die Anwendung von Vasodilatatoren (hauptsächlich Prostacyclin) bei solchen Patienten Fälle von lebensbedrohlichen Lungenödemen beschrieben. Sollten daher bei der Anwendung von Sildenafil bei Patienten mit pulmonaler Hypertonie Zeichen eines Lungenödems auftreten, ist an die Möglichkeit einer Venenverschlusskrankheit zu denken. Galactose-Intoleranz Der Filmüberzug der Tabletten enthält Lactose-Monohydrat. Patienten mit der seltenen hereditären Galactose-Intoleranz, LactaseMangel oder Glucose-Galactose-Malabsorption sollten Revatio nicht einnehmen. Anwendung von Sildenafil zusammen mit Bosentan Die Wirksamkeit von Sildenafil bei Patienten, die gleichzeitig Bosentan erhalten, wurde nicht abschließend nachgewiesen (siehe Abschnitte 4.5 und 5.1). Gleichzeitige Gabe von anderen PDE5Hemmern Die Sicherheit und Wirksamkeit von Sildenafil in Kombination mit anderen PDE5Hemmern, einschließlich Viagra, ist in PAHPatienten nicht untersucht worden. Die Anwendung in dieser Kombination wird nicht empfohlen (siehe Abschnitt 4.5). 4.5 Wechselwirkungen mit anderen Arzneimitteln und sonstige Wechselwirkungen Wirkungen anderer Arzneimittel auf Sildenafil In-vitro -Studien Der Sildenafil-Metabolismus wird grundsätzlich durch die Cytochrom-P450 (CYP)Isoenzyme 3A4 (Hauptweg) und 2C9 (Nebenweg) vermittelt. Die Sildenafil-Clearance kann folglich durch Inhibitoren dieser Isoenzyme herabgesetzt und durch Induktoren dieser Enzyme erhöht sein. Zu Dosisempfehlungen siehe Abschnitte 4.2 und 4.3. In-vivo -Studien Die gleichzeitige Gabe von oral appliziertem Sildenafil und intravenös appliziertem Epoprostenol wurde untersucht (siehe Abschnitte 4.8 und 5.1). Die Wirksamkeit und Sicherheit bei gleichzeitiger Gabe von Sildenafil mit anderen Behandlungen der PAH (z. B. Ambrisentan, Iloprost) wurden in kontrollierten klinischen Studien nicht untersucht. Daher ist im Falle gleichzeitiger Gabe Vorsicht geboten. Die Sicherheit und Wirksamkeit von Sildenafil bei gleichzeitiger Gabe mit anderen PDE5-Inhibitoren wurden bei Patienten mit PAH nicht untersucht (siehe Abschnitt 4.4). Eine populationspharmakokinetische Analyse der Daten für alle Patientengruppen in klinischen Studien bei PAH zeigte eine reduzierte Sildenafil-Clearance und/oder eine erhöhte orale Bioverfügbarkeit von Sildenafil bei gemeinsamer Anwendung mit CYP3A4Substraten und mit der Kombination von CYP3A4-Substraten und Betablockern. Diese waren die einzigen Faktoren mit einem statistisch signifikanten Einfluss auf die Pharmakokinetik von Sildenafil bei Patienten mit PAH. Die Plasma-AUC von Sildenafil war bei Patienten mit CYP3A4-Substraten und CYP3A4-Substraten plus Betablockern um 43 % bzw. 66 % höher als bei Patienten, die keine solchen Arzneimittel erhielten. Die PlasmaAUC von Sildenafil war bei einer Dosis von 80 mg dreimal täglich um das 5-Fache höher als bei einer Dosis von 20 mg dreimal täglich. Dieser Konzentrationsbereich entspricht der Erhöhung der Sildenafil-Exposition, die bei speziell konzipierten Interaktionsstudien mit CYP3A4-Hemmern (mit Ausnahme der stärksten CYP3A4-Hemmer wie Ketoconazol, Itraconazol, Ritonavir) beobachtet wurden. CYP3A4-Induktoren dürften einen erheblichen Einfluss auf die Pharmakokinetik von Sildenafil bei Patienten mit PAH haben. Dies konnte in einer In-vivo -Interaktionsstudie mit dem CYP3A4-Induktor Bosentan bestätigt werden. Bei gesunden Freiwilligen führte die gleichzeitige Anwendung von zweimal täglich 125 mg Bosentan (einem mäßigen Induktor von CYP3A4, CYP2C9 und möglicherweise auch von CYP2C19) mit dreimal täglich 80 mg Sildenafil (im Steady State) über 6 Tage zu einer Verringerung der AUC von Sildenafil um 63 %. Bei einer populationspharmakokinetischen Analyse von Sildenafil-Daten erwachsener PAH-Patienten in klinischen Studien, darunter eine 12-wöchige Studie zur Beurteilung der Wirksamkeit und 009389-12885 Fachinformation (Zusammenfassung der Merkmale des Arzneimittels/SPC) Revatio ® 20 mg Filmtabletten Sicherheit von 20 mg Sildenafil oral dreimal täglich zusätzlich zu einer stabilen Dosis von Bosentan (62,5 mg bis 125 mg zweimal täglich), zeigte sich eine Verminderung der Sildenafil-Exposition unter der gleichzeitigen Gabe von Bosentan, ähnlich wie sie bei gesunden Freiwilligen beobachtet wurde (siehe Abschnitte 4.4 und 5.1). Bei Patienten, die gleichzeitig starke CYP3A4-Induktoren wie Carbamazepin, Phenytoin, Phenobarbital, Johanniskraut und Rifampicin anwenden, muss die Wirksamkeit von Sildenafil genau überwacht werden. Die gleichzeitige Anwendung des HIVProtease-Hemmers Ritonavir, eines hochpotenten P450-Hemmstoffs, im Steady State (zweimal täglich 500 mg) mit Sildenafil (100-mg-Einzeldosis) bewirkte eine 300%ige (4-fache) Steigerung der Cmax von Sildenafil und eine 1.000%ige (11-fache) Steigerung der Plasma-AUC von Sildenafil. Nach 24 Stunden betrugen die SildenafilPlasmaspiegel noch immer etwa 200 ng/ml im Vergleich zu etwa 5 ng/ml nach alleiniger Gabe von Sildenafil. Dies entspricht den ausgeprägten Effekten von Ritonavir auf ein breites Spektrum von P450-Substraten. Aufgrund dieser pharmakokinetischen Ergebnisse ist die gleichzeitige Einnahme von Sildenafil und Ritonavir bei Patienten mit PAH kontraindiziert (siehe Abschnitt 4.3). April 2015 spcde-5v31ro-ft-20 Die gleichzeitige Anwendung des HIV-Protease-Hemmers Saquinavir, eines CYP3A4Hemmstoffs, im Steady State (dreimal täglich 1.200 mg) mit Sildenafil (100-mg-Einzeldosis) bewirkte eine 140%ige Steigerung der Cmax von Sildenafil und eine 210%ige Steigerung der Plasma-AUC von Sildenafil. Sildenafil hatte keine Auswirkungen auf die Pharmakokinetik von Saquinavir. Zu Dosisempfehlungen siehe Abschnitt 4.2. Bei Anwendung einer Einzeldosis von 100 mg Sildenafil gemeinsam mit Erythromycin, einem mäßigen CYP3A4-Hemmstoff, im Steady State (zweimal täglich 500 mg für 5 Tage) erhöhte sich die systemische Sildenafil-Exposition (AUC) um 182 %. Zu Dosisempfehlungen siehe Abschnitt 4.2. Bei gesunden männlichen Freiwilligen konnte kein Einfluss von Azithromycin (500 mg täglich über 3 Tage) auf die AUC, Cmax, tmax, Eliminationsrate oder auf die sich daraus ergebende Halbwertszeit von Sildenafil oder auf seinen Hauptmetaboliten im Kreislauf festgestellt werden. Eine Dosisanpassung ist nicht notwendig. Cimetidin (800 mg), ein Cytochrom-P450-Hemmstoff und ein unspezifischer CYP3A4-Hemmstoff, bewirkte eine 56%ige Steigerung der Sildenafil-Plasmaspiegel, wenn es gesunden Freiwilligen gleichzeitig mit Sildenafil (50 mg) gegeben wurde. Eine Dosisanpassung ist nicht notwendig. Bei den stärksten CYP3A4-Hemmern wie beispielsweise Ketoconazol oder Itraconazol dürften ähnliche Effekte wie bei Ritonavir zu erwarten sein (siehe Abschnitt 4.3). Bei CYP3A4-Hemmern wie z. B. Clarithromycin, Telithromycin und Nefazodon wird erwartet, dass der Effekt zwischen dem von Ritonavir und dem von CYP3A4-Hemmern wie z. B. Saquinavir oder Erythromycin liegt – man vermutet eine 7-fach höhere Exposition. Bei 009389-12885 CYP3A4-Hemmern werden daher Dosisanpassungen empfohlen (siehe Abschnitt 4.2). Eine populationspharmakokinetische Analyse der Daten für alle Patientengruppen mit PAH lässt vermuten, dass die gemeinsame Anwendung von Betablockern mit CYP3A4-Substraten zu einer zusätzlichen Erhöhung der Plasma-AUC von Sildenafil im Vergleich zu einer alleinigen Anwendung des CYP3A4-Substrats führen könnte. Grapefruitsaft ist ein schwacher Hemmstoff des CYP3A4-Stoffwechsels in der Darmwand und kann eine geringe Steigerung der Sildenafil-Plasmaspiegel bewirken. Eine Dosisanpassung ist nicht notwendig, die gleichzeitige Einnahme von Sildenafil mit Grapefruitsaft wird jedoch nicht empfohlen. Durch die Einmalgabe eines Antazidums (Magnesiumhydroxid/Aluminiumhydroxid) wurde die Bioverfügbarkeit von Sildenafil nicht beeinflusst. Die gleichzeitige Anwendung von oralen Kontrazeptiva (Ethinylestradiol 30 µg und Levonorgestrel 150 µg) hatte keinen Einfluss auf die Pharmakokinetik von Sildenafil. Nicorandil ist ein Wirkstoff, der gleichzeitig die Kaliumkanäle aktiviert und als Nitrat wirkt. Aufgrund der Nitratkomponente besteht die Möglichkeit einer schwerwiegenden Wechselwirkung mit Sildenafil (siehe Abschnitt 4.3). Wirkungen von Sildenafil auf andere Arzneimittel In-vitro -Studien Sildenafil ist ein schwacher Inhibitor der Cytochrom-P450-Isoenzyme 1A2, 2C9, 2C19, 2D6, 2E1 und 3A4 (IC50 >150 µM). Es liegen keine Daten hinsichtlich Wechselwirkungen zwischen Sildenafil und unspezifischen Phosphodiesteraseinhibitoren wie Theophyllin oder Dipyridamol vor. In-vivo -Studien Bei gleichzeitiger Anwendung von Sildenafil (50 mg) zeigten sich keine signifikanten Wechselwirkungen mit Tolbutamid (250 mg) oder mit Warfarin (40 mg), die beide durch CYP2C9 verstoffwechselt werden. Sildenafil hatte keine signifikante Wirkung auf die Plasma-AUC von Atorvastatin (AUC um 11 % erhöht), was vermuten lässt, dass Sildenafil keinen klinisch relevanten Effekt auf CYP3A4 hat. Es wurden keine Wechselwirkungen zwischen Sildenafil (100-mg-Einzeldosis) und Acenocoumarol beobachtet. Die durch Acetylsalicylsäure (150 mg) bewirkte Verlängerung der Blutungszeit wurde durch Sildenafil (50 mg) nicht gesteigert. Die blutdrucksenkende Wirkung von Alkohol (maximale Blutalkoholspiegel im Mittel 80 mg/dl) wurde bei gesunden Probanden durch Sildenafil (50 mg) nicht verstärkt. In einer Studie an gesunden Freiwilligen führte Sildenafil im Steady State (80 mg dreimal täglich) zu einer Erhöhung der AUC von Bosentan (125 mg zweimal täglich) um 50 %. Eine populationspharmakokinetische Analyse der Daten aus einer Studie mit erwachsenen PAH-Patienten mit einer bestehenden Bosentan-Therapie (62,5 mg bis 125 mg zweimal täglich) ergab bei gleichzeitiger Gabe von Sildenafil im Steady State (20 mg dreimal täglich) eine Erhöhung (20 % [KI 95 %: 9,8 bis 30,8]) der AUC von Bosentan, die geringer war als die bei gesunden Freiwilligen mit gleichzeitiger Gabe von 80 mg Sildenafil dreimal täglich (siehe Abschnitte 4.4 und 5.1). In einer Interaktionsstudie erhielten Hypertoniker Sildenafil (100 mg) zusammen mit Amlodipin. Es zeigte sich eine zusätzliche Senkung des Blutdrucks im Liegen um 8 mmHg systolisch und um 7 mmHg diastolisch. Das Ausmaß dieser zusätzlichen Blutdrucksenkung war ähnlich der Blutdrucksenkung, die nach alleiniger Anwendung von Sildenafil an gesunden Probanden beobachtet wurde. In drei spezifischen Arzneimittelinteraktionsstudien wurden der Alphablocker Doxazosin (4 mg und 8 mg) und Sildenafil (25 mg, 50 mg oder 100 mg) gemeinsam an Patienten mit benigner Prostatahyperplasie (BPH) angewendet, die eine Therapie mit Doxazosin in stabiler Dosis erhielten. Bei diesen Studienpopulationen zeigten sich mittlere zusätzliche Senkungen des systolischen und diastolischen Blutdrucks im Liegen um 7/7 mmHg, 9/5 mmHg bzw. 8/4 mmHg und mittlere zusätzliche Senkungen des systolischen und diastolischen Blutdrucks im Stehen um 6/6 mmHg, 11/4 mmHg bzw. 4/5 mmHg. Bei gleichzeitiger Anwendung von Sildenafil und Doxazosin an Patienten mit stabil eingestellter Doxazosin-Dosis gab es gelegentlich Berichte über eine symptomatische orthostatische Hypotonie. Gemeldet wurden dabei Schwindel und Benommenheit, jedoch keine Synkope. Eine gleichzeitige Anwendung von Sildenafil bei Patienten mit Alphablocker-Therapie kann bei empfindlichen Personen zu orthostatischer Hypotonie führen (siehe Abschnitt 4.4). Sildenafil (100-mg-Einzeldosis) hatte keinen Einfluss auf die Steady-State-Pharmakokinetik des HIV-Protease-Hemmstoffs Saquinavir, der ein CYP3A4-Substrat-Hemmer ist. Entsprechend seiner pharmakologischen Wirkung auf den Stickstoffmonoxid-cGMPStoffwechsel (siehe Abschnitt 5.1) konnte gezeigt werden, dass Sildenafil den blutdrucksenkenden Effekt von Nitraten verstärkt. Daher ist die gleichzeitige Gabe mit Stickstoffmonoxid-Donatoren oder jeglichen Nitraten kontraindiziert (siehe Abschnitt 4.3). Sildenafil hatte keinen klinisch relevanten Einfluss auf die Plasmaspiegel von oralen Kontrazeptiva (Ethinylestradiol 30 µg und Levonorgestrel 150 µg). Kinder und Jugendliche Interaktionsstudien wurden nur bei Erwachsenen durchgeführt. 4.6 Fertilität, Schwangerschaft und Stillzeit Frauen im gebärfähigen Alter und Kontrazeption bei Männern und Frauen Wegen fehlender Daten zu den Auswirkungen von Revatio bei Schwangeren wird Revatio bei Frauen im gebärfähigen Alter nur dann empfohlen, wenn gleichzeitig ein wirksamer Empfängnisschutz angewandt wird. 3 Fachinformation (Zusammenfassung der Merkmale des Arzneimittels/SPC) Revatio ® 20 mg Filmtabletten Schwangerschaft Es liegen keine Daten für die Anwendung von Sildenafil bei schwangeren Frauen vor. Tierstudien zeigen keine direkt oder indirekt schädlichen Wirkungen in Bezug auf Schwangerschaft und embryonale/fetale Entwicklung. Studien an Tieren zeigten eine Toxizität hinsichtlich der postnatalen Entwicklung (siehe Abschnitt 5.3). Aufgrund fehlender Daten sollte Revatio bei schwangeren Frauen nicht angewendet werden, es sei denn, eine Anwendung ist dringend erforderlich. Stillzeit Es ist nicht bekannt, ob Sildenafil in die Muttermilch übertritt. Revatio sollte nicht von stillenden Müttern eingenommen werden. Fertilität Auf der Basis von Standarduntersuchungen zur Fertilität zeigten die präklinischen Daten keine besonderen Risiken für den Menschen (siehe Abschnitt 5.3). 4.7 Auswirkungen auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen Revatio hat geringen Einfluss auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen. Da in klinischen Studien mit Sildenafil über Schwindel und Sehstörungen berichtet wurde, sollen die Patienten darauf achten, wie sie auf die Einnahme von Revatio reagieren, bevor sie ein Fahrzeug lenken oder Maschinen bedienen. 4.8 Nebenwirkungen Zusammenfassung des Nebenwirkungsprofils In der placebokontrollierten Zulassungsstudie mit Revatio bei pulmonaler arterieller Hypertonie wurden insgesamt 207 Patienten auf Revatio in einer Tagesdosis von 20 mg, 40 mg oder 80 mg dreimal täglich randomisiert und 70 Patienten auf Placebo. Die Behandlungsdauer betrug 12 Wochen. Bei den mit 20 mg, 40 mg oder 80 mg Sildenafil dreimal täglich behandelten Patienten betrug die Gesamtabbruchrate 2,9 %, 3,0 % oder 8,5 % im Vergleich zu 2,9 % unter Placebo. Von den 277 Patienten, die in der Zulassungsstudie behandelt wurden, wurden 259 Patienten in eine Langzeit-Fortsetzungsstudie aufgenommen, in der Dosen von bis zu 80 mg dreimal täglich (das 4-Fache der empfohlenen Dosis von 20 mg dreimal täglich) gegeben wurden. Nach 3 Jahren erhielten noch 87 % der 183 Patienten unter Studienmedikation 80 mg Revatio dreimal täglich. In einer placebokontrollierten Studie mit Revatio als Begleitmedikation zu intravenös verabreichtem Epoprostenol bei pulmonaler arterieller Hypertonie wurden insgesamt 134 Patienten mit Epoprostenol und Revatio (mit einer fixen Dosissteigerung von anfangs 20 mg, dann 40 mg und schließlich 80 mg jeweils dreimal täglich, entsprechend der Verträglichkeit) sowie 131 Patienten mit Epoprostenol und Placebo behandelt. Die Behandlungsdauer betrug 16 Wochen. Die Häufigkeit von Therapieabbrüchen aufgrund unerwünschter Arzneimittelwirkungen lag 4 insgesamt unter Sildenafil/Epoprostenol bei 5,2 %, im Vergleich zu 10,7 % unter Placebo/Epoprostenol. Zu den bis dahin nicht berichteten Nebenwirkungen, die in der Sildenafil/Epoprostenol-Gruppe häufiger auftraten als mit Placebo/Epoprostenol, zählten: okulare Hyperämie, verschwommenes Sehen, Nasenschleimhautschwellung, nächtliche Schweißausbrüche, Rückenschmerzen und Mundtrockenheit. Bekannte Nebenwirkungen wie Kopfschmerzen, Erytheme, Gliederschmerzen und Ödeme wurden häufiger bei mit Sildenafil/Epoprostenol behandelten als bei mit Placebo/ Epoprostenol behandelten Patienten beobachtet. Von den Patienten, die diese initiale Studie abschlossen, wurden 242 Patienten in eine Langzeit-Fortsetzungsstudie aufgenommen. Dabei wurden Dosen bis zu 80 mg dreimal täglich gegeben, und nach 3 Jahren erhielten noch 68 % der 133 Patienten unter Studienmedikation 80 mg Revatio dreimal täglich. In den beiden placebokontrollierten Studien waren die Nebenwirkungen im Allgemeinen leichter bis mäßiger Art. Die am häufigsten beschriebenen Nebenwirkungen (Häufigkeit: 10 % oder größer) mit Revatio im Vergleich zu Placebo waren Kopfschmerzen, Flush, Dyspepsie, Durchfall und Gliederschmerzen. Tabelle der Nebenwirkungen Die Tabelle zeigt die Nebenwirkungen, die bei mindestens 1 % der mit Revatio behandelten Patienten und häufiger (Unterschied >1 %) als unter Placebo auftraten (Datenbasis ist die Zulassungsstudie zu Revatio bzw. eine gemeinsame Auswertung der beiden placebokontrollierten Studien zu PAH mit Dosierungen von 20, 40 oder 80 mg Sildenafil dreimal täglich). Die Nebenwirkungen sind nach Organsystem und Häufigkeit gegliedert: sehr häufig (≥ 1/10), häufig (≥ 1/100, <1/10), gelegentlich (≥ 1/1.000, <1/100) und nicht bekannt (Häufigkeit auf Grundlage der verfügbaren Daten nicht abschätzbar). Innerhalb jeder Häufigkeitsgruppe werden die Nebenwirkungen nach abnehmendem Schweregrad angegeben. Die Meldungen nach Markteinführung sind kursiv angegeben. Kinder und Jugendliche In der placebokontrollierten Revatio-Studie bei Patienten mit pulmonaler arterieller Hypertonie im Alter von 1 bis 17 Jahren wurden insgesamt 174 Patienten dreimal täglich mit niedrigen Dosen von Revatio (10 mg bei Patienten >20 kg; kein Patient ≤ 20 kg erhielt diese niedrige Dosis), mittleren Dosen (10 mg bei Patienten ≥ 8 bis 20 kg; 20 mg bei Patienten ≥ 20 bis 45 kg; 40 mg bei Patienten >45 kg) oder hohen Dosen (20 mg bei Patienten ≥ 8 bis 20 kg; 40 mg bei Patienten ≥ 20 bis 45 kg; 80 mg bei Patienten >45 kg) behandelt und 60 Patienten erhielten Placebo. Das in der Studie bei Kindern beobachtete Nebenwirkungsprofil entsprach im Allgemeinen dem bei Erwachsenen (siehe Tabelle). Die häufigsten Nebenwirkungen (mit einer Häufigkeit von ≥ 1 %) die bei mit Revatio behandelten Patienten (alle Dosierungen) auftraten, mit einer Häufigkeit von >1 % gegenüber der Placebo-Gruppe, waren Fieber, Infektionen der oberen Atemwege (je 11,5 %), Erbrechen (10,9 %), vermehrte Erektionen (einschließlich spontaner Erektion des Penis bei männlichen Individuen) (9,0 %), Übelkeit, Bronchitis (je 4,6 %), Pharyngitis (4,0 %), Rhinorrhö (3,4 %) und Pneumonie, Rhinitis (je 2,9 %). Von den 234 pädiatrischen Patienten, die in der placebokontrollierten Kurzzeit-Studie behandelt wurden, haben 220 Patienten an der Langzeit-Anschlussstudie teilgenommen. Teilnehmer, die eine aktive SildenafilTherapie erhalten hatten, haben die gleichen Dosierungsschemata fortgeführt, während die Teilnehmer aus der Placebo-Gruppe der Kurzzeit-Studie randomisiert einer Sildenafil-Behandlung zugeordnet wurden. Die häufigsten Nebenwirkungen, die während der gesamten Dauer der Kurzzeit- und Langzeit-Studie berichtet wurden, waren im Allgemeinen den Nebenwirkungen ähnlich, die in der Kurzzeit-Studie beobachtet wurden. Nebenwirkungen, die bei >10 % der 229 mit Sildenafil behandelten Patienten (kombinierte Dosisgruppe, einschließlich 9 Patienten, die die Langzeit-Studie nicht fortgesetzt haben) auftraten, waren Infektion der oberen Atemwege (31 %), Kopfschmerz (26 %), Erbrechen (22 %), Bronchitis (20 %), Pharyngitis (18 %), Fieber (17 %), Diarrhoe (15 %) und Grippe, Epistaxis (jeweils 12 %). Der Schweregrad der meisten dieser Nebenwirkungen wurde als leicht bis mäßig eingestuft. Bei 94 (41 %) der 229 Patienten, die Sildenafil erhielten, wurden schwerwiegende Nebenwirkungen berichtet. Von diesen 94 Patienten, die eine schwerwiegende Nebenwirkung gemeldet haben, waren 14/55 (25,5 %) in der Gruppe mit der geringen Sildenafil-Dosierung, 35/74 (47,3 %) in der Gruppe mit der mittleren Sildenafil-Dosierung und 45/100 (45 %) in der Gruppe mit der hohen Sildenafil-Dosierung. Die häufigsten schwerwiegenden Nebenwirkungen, die mit einer Häufigkeit von ≥ 1 % bei SidenafilPatienten (kombinierte Dosierungen) auftraten, waren Pneumonie (7,4 %), Herzversagen, pulmonare Hypertonie (je 5,2 %), Entzündungen der oberen Atemwege (3,1 %), Versagen der rechten Herzkammer, Gastroenteritis (je 2,6 %), Synkope, Bronchitis, Bronchopneumonie, pulmonale arterielle Hypertonie (je 2,2 %), Brustschmerzen, Karies (je 1,7 %) und kardiogener Schock, virale Gastroenteritis, Harnwegsinfektionen (je 1,3 %). Die folgenden schwerwiegenden Nebenwirkungen wurden als behandlungsbedingt bewertet, Enterokolitis, Konvulsion, Hypersensitivität, Stridor, Hypoxie, neurosensorische Taubheit und ventrikuläre Arrhythmie. Meldung des Verdachts auf Nebenwirkungen Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von großer Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdachtsfall einer Nebenwirkung dem Bundesinstitut für Arzneimittel und Me009389-12885 Fachinformation (Zusammenfassung der Merkmale des Arzneimittels/SPC) Revatio ® 20 mg Filmtabletten 5. PHARMAKOLOGISCHE EIGENSCHAFTEN Systemorganklassen Nebenwirkung gemäß MedDRA (V. 14.0) Infektionen und parasitäre Erkrankungen Häufig 5.1 Pharmakodynamische Eigenschaften Cellulitis, Grippe, Bronchitis, Sinusitis, Rhinitis, Gastroenteritis Erkrankungen des Blutes und des Lymphsystems Häufig Anämie Stoffwechsel- und Ernährungsstörungen Häufig Flüssigkeitsretention Psychiatrische Erkrankungen Häufig Schlaflosigkeit, Angst Erkrankungen des Nervensystems Sehr häufig Kopfschmerzen Häufig Migräne, Tremor, Parästhesie, Brennen, Hypästhesie Augenerkrankungen Häufig Retinablutungen, Sehstörungen, verschwommenes Sehen, Photophobie, Chromopsie, Zyanopsie, Augenreizungen, okuläre Hyperämie Gelegentlich verminderte Sehschärfe, Doppeltsehen, Fremdkörpergefühl im Auge Nicht bekannt nicht arteriitische anteriore ischämische Optikusneuropathie (NAION) *, Verschluss von Netzhautgefäßen *, Gesichtsfelddefekte * Erkrankungen des Ohrs und des Labyrinths Häufig Vertigo Nicht bekannt plötzlicher Hörverlust Gefäßerkrankungen Sehr häufig Flush Hypotonie Erkrankungen der Atemwege, des Brustraums und Mediastinums Nicht bekannt Häufig Nasenbluten, Husten, Nasenschleimhautschwellung Erkrankungen des Gastrointestinaltrakts Sehr häufig Durchfall, Dyspepsie Häufig Gastritis, gastroösophagealer Reflux, Hämorrhoiden, abdominelles Spannungsgefühl, Mundtrockenheit Erkrankungen der Haut und des Unterhautzellgewebes Häufig Alopezie, Erythem, nächtliche Schweißausbrüche Nicht bekannt Ausschlag Skelettmuskulatur- und Bindegewebserkrankungen Sehr häufig Gliederschmerzen Häufig Myalgie, Rückenschmerzen Erkrankungen der Nieren und Harnwege Gelegentlich Hämaturie Erkrankungen der Geschlechtsorgane und der Brustdrüse Gelegentlich Penisblutung, Hämatospermie, Gynäkomastie Nicht bekannt Priapismus, vermehrte Erektionen Allgemeine Erkrankungen und Beschwerden am Verabreichungsort Häufig Fieber April 2015 spcde-5v31ro-ft-20 * Diese unerwünschten Ereignisse wurden bei Patienten, die PDE5-Hemmer zur Behandlung der erektilen Dysfunktion einnahmen, berichtet. dizinprodukte, Abt. Pharmakovigilanz, KurtGeorg-Kiesinger-Allee 3, D-53175 Bonn, Website: www.bfarm.de anzuzeigen. 4.9 Überdosierung In Studien erhielten gesunde Probanden Einzeldosen bis zu 800 mg. Die hierbei beobachteten Nebenwirkungen waren ähnlich wie die bei niedrigeren Dosen, lediglich Inzidenz und Schweregrad waren erhöht. Bei Einzeldosen von 200 mg war die Inzi009389-12885 denz der Nebenwirkungen (Kopfschmerz, Flush, Schwindel, Dyspepsie, Verstopfung der Nase und Sehstörungen) erhöht. In Fällen einer Überdosierung sollten je nach Bedarf die üblichen unterstützenden Maßnahmen eingeleitet werden. Da Sildenafil in hohem Maße an Plasmaproteine gebunden ist und renal nicht eliminiert wird, ist durch eine Dialyse keine Beschleunigung der Clearance zu erwarten. Pharmakotherapeutische Gruppe: Urologika, Arzneimittel zur Behandlung der erektilen Dysfunktion ATC-Code: G04BE03* * In der amtlichen deutschen Fassung des ATC-Codes wird Revatio wie folgt klassifiziert: Andere Antihypertonika (C02KX06) Wirkmechanismus Sildenafil ist ein wirksamer und selektiver Hemmstoff der für zyklisches Guanosinmonophosphat (cGMP) spezifischen Phosphodiesterase Typ 5 (PDE5), dem Enzym, das für den Abbau von cGMP verantwortlich ist. Abgesehen von seinem Vorliegen im Corpus cavernosum des Penis findet sich PDE5 auch in den Lungengefäßen. Sildenafil erhöht somit cGMP innerhalb der glatten Muskelzellen der Lungengefäße und führt so zu einer Entspannung. Bei Patienten mit PAH kann dies zu einer selektiven Vasodilatation im pulmonalen Gefäßsystem und – in geringerem Ausmaß – zu einer Vasodilatation im systemischen Kreislauf führen. Pharmakodynamische Wirkungen In-vitro -Studien zeigten, dass Sildenafil für PDE5 selektiv ist. Es wirkt stärker auf PDE5 als auf andere bekannte Phosphodiesterasen. Die Selektivität von Sildenafil für PDE5 ist um das 10-Fache höher als für PDE6, die am Phototransduktionsprozess in der Retina beteiligt ist. Es zeigte sich eine 80-fach höhere Selektivität für PDE5 als für PDE1 und eine um mehr als das 700-Fache höhere Selektivität für PDE5 als für PDE2, 3, 4, 7, 8, 9, 10 und 11. Insbesondere hat Sildenafil eine mehr als 4000-fach höhere Selektivität für PDE5 im Vergleich zu PDE3, dem an der Steuerung der kardialen Kontraktilität beteiligten cAMP-spezifischen Phosphodiesterase-Isoenzym. Sildenafil bewirkt eine geringe und vorübergehende Reduktion des Blutdrucks, die in den meisten Fällen keine klinisch relevanten Effekte zur Folge hat. Nach Langzeitapplikation von 80 mg dreimal täglich an Patienten mit systemischer Hypertonie war die mittlere Veränderung des systolischen und des diastolischen Blutdrucks eine Senkung von 9,4 mmHg bzw. 9,1 mmHg gegenüber dem Ausgangswert. Nach Langzeitapplikation von 80 mg dreimal täglich an Patienten mit PAH wurden geringere Effekte auf die Blutdrucksenkung beobachtet (eine Senkung von jeweils 2 mmHg systolisch und diastolisch). Bei der empfohlenen Dosierung von dreimal täglich 20 mg wurde keine Senkung des systolischen oder diastolischen Blutdrucks beobachtet. Die einmalige orale Gabe von bis zu 100 mg Sildenafil ergab bei gesunden Freiwilligen keine Auswirkung auf das EKG. In der Langzeitanwendung von dreimal täglich 80 mg Sildenafil bei Patienten mit PAH wurden keine signifikanten Veränderungen des EKG beobachtet. In einer Studie zu den hämodynamischen Effekten einer oralen Einmalgabe von 100 mg Sildenafil bei 14 Patienten mit 5 Fachinformation (Zusammenfassung der Merkmale des Arzneimittels/SPC) Revatio ® 20 mg Filmtabletten schwerer (>70%ige Stenose mindestens einer Koronararterie) koronarer Herzkrankheit (KHK) nahmen der mittlere systolische und der mittlere diastolische Blutdruck in Ruhe im Vergleich zum Ausgangswert um 7 % bzw. 6 % ab. Der mittlere pulmonale systolische Blutdruck nahm um 9 % ab. Sildenafil beeinflusste weder das Herzminutenvolumen, noch beeinträchtigte es die Durchblutung in den stenosierten Koronararterien. Leichte und vorübergehende Veränderungen des Farbsehens (Blau/Grün) wurden bei einigen Studienteilnehmern durch den Farnsworth-Munsell-100-Farben-Test 1 Stunde nach Einnahme von 100 mg beobachtet, 2 Stunden nach Einnahme waren diese Veränderungen nicht mehr nachweisbar. Der vermutete Mechanismus für diese Veränderung des Farbsehens bezieht sich auf die Hemmung der PDE6, die bei dem Phototransduktionsprozess der Retina eine Rolle spielt. Sildenafil übt keinen Einfluss auf die Sehschärfe oder das Kontrastsehen aus. In einer kleinen, placebokontrollierten Untersuchung bei 9 Patienten mit dokumentierter altersbedingter Makuladegeneration im Frühstadium zeigte Sildenafil als 100-mg-Einmaldosis in den durchgeführten Sehtests (Sehschärfe, Amsler-Gitter, Lichtertest, Humphrey-Perimeter und PhotostressTest) keine signifikanten Veränderungen. Klinische Wirksamkeit und Sicherheit Wirksamkeit bei erwachsenen Patienten mit PAH Es wurde eine randomisierte, doppelblinde, placebokontrollierte Studie bei 278 Patienten mit primärer PAH, PAH in Verbindung mit einer Bindegewebskrankheit und PAH nach chirurgischer Korrektur eines angeborenen Herzfehlers durchgeführt. Die Patienten wurden für eine von 4 Behandlungsgruppen randomisiert: Placebo, Sildenafil 20 mg, Sildenafil 40 mg oder Sildenafil 80 mg, je dreimal täglich. Von den 278 randomisierten Patienten erhielten 277 zumindest 1 Dosis der Studienmedikation. Die Studienpopulation umfasste 68 (25 %) Männer und 209 (75 %) Frauen mit einem mittleren Alter von 49 Jahren (Altersbereich: 18 bis 81 Jahre) und einer 6-Minuten-Gehstrecke zwischen 100 und 450 Meter (Mittelwert: 344 Meter) bei Studienbeginn. 175 Patienten (63 %) hatten eine Diagnose mit primärer pulmonaler Hypertonie, 84 (30 %) eine Diagnose mit PAH in Verbindung mit einer Bindegewebskrankheit und 18 (7 %) eine Diagnose mit PAH nach einer chirurgischen Korrektur eines angeborenen Herzfehlers. Die meisten Patienten gehörten bei Studienbeginn in die WHO-Funktionsklasse II (107/277; 39 %) oder III (160/277; 58 %) und wiesen eine durchschnittliche 6-MinutenGehstrecke von 378 Meter bzw. 326 Meter auf, weniger Patienten in die Funktionsklasse I (1/277; 0,4 %) oder IV (9/277; 3 %). Patienten mit einer linksventrikulären Auswurffraktion <45 % oder mit linksventrikulärer Verkürzungsfraktion <0,2 % waren von einer Teilnahme ausgeschlossen. Sildenafil (oder Placebo) wurde zusätzlich zur bestehenden Therapie der Patienten verabreicht, die eine Kombination von Antikoagulanzien, Digoxin, Calciumantagonisten, 6 Diuretika oder Sauerstoff umfassen konnte. Die Anwendung von Prostacyclin, Prostacyclinanaloga oder Endothelinantagonisten als Zusatzbehandlung war ebenso wenig gestattet wie eine Argininsupplementation. Patienten, die zuvor auf eine Therapie mit Bosentan nicht angesprochen hatten, waren von einer Teilnahme an der Studie ausgeschlossen. Der primäre Endpunkt für die Wirksamkeit war die Veränderung der 6-Minuten-Gehstrecke in Woche 12 gegenüber dem Ausgangswert. Für alle drei Sildenafil-DosisGruppen zeigte sich im Vergleich zu den Patienten mit Placebo eine statistisch signifikante Erhöhung der 6-Minuten-Gehstrecke. Die relative Erhöhung der 6-Minuten-Gehstrecke gegenüber Placebo betrug 45 Meter (p <0,0001), 46 Meter (p <0,0001) bzw. 50 Meter (p <0,001) für Sildenafil 20 mg, 40 mg bzw. 80 mg dreimal täglich. Es gab keinen signifikanten Unterschied in der Wirkung zwischen den einzelnen Dosen von Sildenafil. Bei Patienten mit einem Ausgangwert der 6-Minuten-Gehstrecke unter 325 Meter wurde eine verbesserte Wirksamkeit bei den höheren Dosen beobachtet (die Verbesserung gegenüber Placebo betrug 58 Meter, 65 Meter bzw. 87 Meter für 20 mg, 40 mg bzw. 80 mg dreimal täglich). Unter Berücksichtigung der WHO-Funktionsklassen konnte in der 20-mg-DosisGruppe eine statistisch signifikante Erhöhung der 6-Minuten-Gehstrecke beobachtet werden: Für die Funktionsklassen II und III wurden gegenüber Placebo Erhöhungen um 49 Meter (p = 0,0007) und 45 Meter (p = 0,0031) gemessen. Die Verbesserung der 6-Minuten-Gehstrecke war bereits nach 4 Wochen Behandlung eindeutig feststellbar und konnte auch über 8 und 12 Wochen aufrechterhalten werden. Die Therapieeffekte waren bei den verschiedenen Subgruppen vergleichbar, wobei die Subgruppen nach der Ätiologie (primäre PAH und pulmonale Hypertonie in Verbindung mit einer Bindegewebskrankheit), den verschiedenen WHO-Funktionsklassen, Geschlecht, Rasse, den geographischen Regionen, dem mittleren Pulmonalarteriendruck und dem pulmonalen Gefäßwiderstand definiert waren. Bei allen Dosierungen von Sildenafil zeigten die Patienten eine statistisch signifikante Reduktion des mittleren Pulmonalarteriendrucks (mPAP) und des pulmonalen Gefäßwiderstands (PVR) im Vergleich zu denen mit Placebo. Die placebokorrigierten Behandlungseffekte auf den mPAP betrugen – 2,7 mmHg (p = 0,04), – 3,0 mmHg (p = 0,01) bzw. – 5,1 mmHg (p <0,0001) für dreimal täglich 20 mg, 40 mg bzw. 80 mg Sildenafil. Die gegenüber Placebo relativen Behandlungseffekte auf den PVR betrugen – 178 dyn · sec/cm5 (p = 0,0051), (p = 0,0017) bzw. – 195 dyn · sec/cm5 – 320 dyn · sec/cm5 (p <0,0001) für dreimal täglich 20 mg, 40 mg bzw. 80 mg Sildenafil. Nach 12 Wochen mit dreimal täglich 20 mg, 40 mg bzw. 80 mg Sildenafil war die prozentuale Senkung des PVR proportional größer (11,2 %, 12,9 % bzw. 23,3 %) als die Reduktion für den systemischen Gefäßwiderstand (7,2 %, 5,9 % bzw. 14,4 %). Der Ein- fluss von Sildenafil auf die Mortalität ist nicht bekannt. Bei allen Sildenafil-Dosierungen ergab sich in Woche 12 bei einem größeren Prozentsatz der Patienten (nämlich bei 28 %, 36 % bzw. 42 % der Personen unter dreimal täglich 20 mg, 40 mg bzw. 80 mg) eine Verbesserung um mindestens 1 WHO-Funktionsklasse im Vergleich zu 7 % unter Placebo. Die jeweilige Odds-Ratio betrug 2,92 (p = 0,0087), 4,32 (p = 0,0004) bzw. 5,75 (p <0,0001). Langzeit-Überlebensdaten bei nicht vorbehandelten Patienten Die Patienten der Zulassungsstudie konnten als Fortsetzung an einer offenen Langzeitstudie teilnehmen. Nach 3 Jahren erhielten 87 % der Patienten eine Dosierung von dreimal täglich 80 mg. In der Zulassungsstudie wurden insgesamt 207 Patienten mit Revatio behandelt, und ihre Langzeit-Überlebensrate wurde über mindestens 3 Jahre verfolgt. In dieser Patientenpopulation betrug die Kaplan-Meier-Schätzung für die 1-Jahres-, 2-Jahres- und 3-Jahres-Überlebensrate 96 %, 91 % und 82 %. Bei Patienten mit einer WHO-Funktionsklasse II zu Studienbeginn betrug die Überlebensrate nach 1, 2 und 3 Jahren 99 %, 91 % und 84 % und bei Patienten mit einer WHO-Funktionsklasse III zu Studienbeginn 94 %, 90 % und 81 %. Wirksamkeit bei erwachsenen Patienten mit PAH (bei Kombination mit Epoprostenol) Es wurde eine randomisierte, placebokontrollierte Doppelblindstudie mit 267 PAH-Patienten durchgeführt, die mit intravenös verabreichtem Epoprostenol eingestellt waren. Die Studienpopulation bestand sowohl aus Patienten mit primärer pulmonaler arterieller Hypertonie (212/267, 79 %) als auch aus Patienten mit PAH in Verbindung mit einer Bindegewebskrankheit (55/267, 21 %). Die meisten Patienten entfielen zu Studienbeginn auf die WHO-Funktionsklassen II (68/267, 26 %) und III (175/267, 66 %), weniger Patienten waren der Funktionsklasse I (3/267, 1 %) oder IV (16/267, 6 %) zugeordnet, und bei einigen Patienten (5/267, 2 %) war die WHO-Funktionsklasse unbekannt. Die Patienten wurden in zwei Gruppen randomisiert: intravenös verabreichtes Epoprostenol plus Placebo oder plus Sildenafil (bei einer fixen Dosissteigerung von anfangs 20 mg, dann 40 mg und schließlich 80 mg jeweils dreimal täglich, entsprechend der Verträglichkeit). Der primäre Endpunkt für die Wirksamkeit war die Veränderung der 6-Minuten-Gehstrecke in Woche 16 gegenüber dem Ausgangswert. Mit Sildenafil zeigte sich im Vergleich zu Placebo eine statistisch signifikante Erhöhung der 6-Minuten-Gehstrecke. Die mittlere placebokorrigierte Verlängerung der Gehstrecke unter Sildenafil betrug 26 Meter (95 %-KI: 10,8 bis 41,2; p = 0,0009). Bei Patienten mit einer Gehstrecke von ≥ 325 Metern zu Studienbeginn war der Behandlungseffekt 38,4 Meter zugunsten von Sildenafil; bei Patienten mit einer Gehstrecke von <325 Metern zu Studienbeginn war der Behandlungseffekt 2,3 Meter zugunsten von Placebo. Bei Patienten mit primärer PAH war 009389-12885 Fachinformation (Zusammenfassung der Merkmale des Arzneimittels/SPC) Revatio ® 20 mg Filmtabletten der Behandlungseffekt 31,1 Meter im Vergleich zu 7,7 Metern bei Patienten mit PAH in Verbindung mit einer Bindegewebskrankheit. Aufgrund der geringen Fallzahlen in den einzelnen randomisierten Untergruppen könnten diese Unterschiede auch zufällig sein. Im Vergleich zu Placebo wurde bei den Patienten unter Sildenafil eine statistisch signifikante Senkung des mittleren Pulmonalarteriendrucks (mPAP) erreicht. Dabei war Sildenafil überlegen mit einer mittleren placebokorrigierten Senkung von – 3,9 mmHg (95 %-KI: – 5,7 bis – 2,1; p = 0,00003). Ein sekundärer Endpunkt war die Zeit bis zum Eintreten einer klinischen Verschlechterung, die als die Zeitspanne von der Randomisierung bis zum ersten Auftreten eines die Erkrankung verschlechternden Ereignisses definiert war (Tod, Lungentransplantation, Beginn einer Bosentan-Therapie oder eine klinische Verschlechterung, die eine Veränderung der Epoprostenol-Therapie notwendig machte). Im Vergleich zu Placebo führte die Behandlung mit Sildenafil zu einer signifikanten Verlängerung der Zeit bis zum Eintreten einer klinischen Verschlechterung der PAH (p = 0,0074). In der Placebo-Gruppe kam es bei 23 Personen zum Auftreten von die Erkrankung verschlechternden Ereignissen (17,6 %) im Vergleich zu 8 Personen in der Sildenafil-Gruppe (6,0 %). April 2015 spcde-5v31ro-ft-20 Langzeit-Überlebensdaten aus der Studie bei bestehender Epoprostenol-Therapie Die an der Studie mit bestehender Epoprostenol-Therapie eingeschlossenen Patienten konnten als Fortsetzung an einer offenen Langzeitstudie teilnehmen. Nach 3 Jahren erhielten 68 % der Patienten eine Dosierung von dreimal täglich 80 mg. Zu Studienbeginn wurden insgesamt 134 Patienten mit Revatio behandelt und ihre Langzeit-Überlebensdaten wurden über mindestens 3 Jahre verfolgt. In dieser Patientenpopulation betrugen die Kaplan-Meier-Schätzwerte der 1-, 2- und 3-Jahres-Überlebensrate 92 %, 81 % und 74 %. Wirksamkeit und Sicherheit bei erwachsenen Patienten mit PAH (bei gleichzeitiger Anwendung von Bosentan) Es wurde eine randomisierte, placebokontrollierte Doppelblindstudie mit 103 klinisch stabilen PAH-Patienten (WHO FK II und III) durchgeführt, die seit mindestens 3 Monaten eine Bosentan-Therapie erhalten hatten. Darunter waren Patienten mit primärer PAH und solche mit PAH in Verbindung mit einer Bindegewebskrankheit. Die Patienten wurden randomisiert einer Behandlung mit Placebo oder Sildenafil (20 mg dreimal täglich) in Kombination mit Bosentan (62,5 bis 125 mg zweimal täglich) zugeordnet. Der primäre Endpunkt für die Wirksamkeit war eine Veränderung der 6-Minuten-Gehstrecke in Woche 12 gegenüber dem Ausgangswert. Die Ergebnisse zeigen, dass kein signifikanter Unterschied in der mittleren Veränderung der 6-Minuten-Gehstrecke gegenüber dem Ausgangswert zwischen Sildenafil 20 mg dreimal täglich (13,62 Meter [KI 95 %: – 3,89 bis +31,12]) und Placebo (14,08 Meter [KI 95 %: – 1,78 bis +29,95]) zu beobachten ist. 009389-12885 Unterschiede bei der 6-Minuten-Gehstrecke wurden beobachtet zwischen Patienten mit primärer PAH und PAH in Verbindung mit einer Bindegewebskrankheit. Bei Patienten mit primärer PAH (67 Teilnehmer) betrug die mittlere Veränderung gegenüber dem Ausgangswert 26,39 Meter (KI 95 %: 10,70 bis 42,08) in der Sildenafil-Gruppe und 11,84 Meter (KI 95 %: – 8,83 bis 32,52) in der Placebo-Gruppe. Bei Patienten mit PAH in Verbindung mit einer Bindegewebskrankheit (36 Teilnehmer) betrug die mittlere Veränderung gegenüber dem Ausgangswert jedoch – 18,32 Meter (KI 95 %: – 65,66 bis 29,02) in der Sildenafil-Gruppe und 17,50 Meter (KI 95 %: – 9,41 bis 44,41) in der Placebo-Gruppe. Insgesamt waren die Nebenwirkungen in den beiden Behandlungsgruppen (Sildenafil plus Bosentan gegenüber Bosentan alleine) grundsätzlich ähnlich und entsprachen dem bekannten Sicherheitsprofil von Sildenafil als Monotherapie (siehe Abschnitte 4.4 und 4.5). Kinder und Jugendliche In einer randomisierten, doppelblinden, placebokontrollierten, parallelen Multizenterstudie mit verschiedenen Dosierungen wurden insgesamt 234 Personen im Alter von 1 bis 17 Jahren behandelt. Die Teilnehmer (38 % männlich und 62 % weiblich) hatten ein Körpergewicht ≥ 8 kg und litten zu 33 % an primärer pulmonaler Hypertonie (PPH) oder einer PAH in Verbindung mit angeborenen Herzerkrankungen (systemischpulmonale Shunts 37 %, chirurgische Reposition 30 %). 63 der 234 Patienten (27 %) in dieser Studie waren jünger als 7 Jahre (niedrige Sildenafil-Dosis: n = 2; mittlere Sildenafil-Dosis: n = 17; hohe SildenafilDosis: n = 28; Placebo: n = 16), und 171 der 234 Patienten (73 %) waren 7 Jahre oder älter (niedrige Sildenafil-Dosis: n = 40; mittlere Sildenafil-Dosis: n = 38; hohe Sildenafil-Dosis: n = 49; Placebo: n = 44). Die meisten Personen waren als Ausgangwert in der WHO-Funktionsklasse I (75/234, 32 %) oder II (120/234, 51 %). Weniger Patienten waren Funktionsklasse III (35/234, 15 %) oder IV (1/234, 0,4 %). Bei einigen wenigen Patienten (3/234, 1,3 %) war die WHO-Funktionsklasse nicht bekannt. Die Patienten waren nicht mit einer spezifischen PAH-Therapie vorbehandelt und die Anwendung von Prostacyclin, Prostacyclinanaloga sowie EndothelinrezeptorAntagonisten waren in der Studie nicht erlaubt, ebenso wenig wie Argininsupplementierung, Nitrate, Alphablocker und starke CYP450-3A4-Hemmer. Das primäre Ziel der Studie war, die Wirksamkeit von oralem Sildenafil auf die Verbesserung der körperlichen Belastbarkeit bei Kindern in der Dauertherapie über 16 Wochen anhand des CardiopulmonaryExercise-Tests (CPET) bei den Teilnehmern, die von ihrer Entwicklung her dazu imstande waren (n = 115) zu prüfen. Die sekundären Endpunkte schlossen u. a. ein hämodynamisches Monitoring, Erfassung der Symptome, die WHO-Funktionsklasse, Veränderung der Begleitmedikation und die Erfassung der Lebensqualität mit ein. Die Teilnehmer wurden entweder einer der drei Sildenafil-Gruppen zugeteilt (niedrige [10 mg], mittlere [10 bis 40 mg] oder hohe [20 bis 80 mg] Revatio-Dosen dreimal täglich) oder auf die Placebo-Gruppe. Die innerhalb einer Gruppe tatsächlich gegebene Dosis orientierte sich am Körpergewicht (siehe Abschnitt 4.8). Der Anteil der Teilnehmer, die zu Beginn eine unterstützende Therapie (Antikoagulanzien, Digoxin, Calciumkanalblocker, Diuretika und/oder Sauerstoff) erhielten, war in der kombinierten SildenafilGruppe (47,7 %) und in der Placebo-Gruppe (41,7 %) vergleichbar. Der primäre Endpunkt war die durch CPET in den kombinierten Sildenafil-Gruppen erhobene, placebokorrigierte prozentuale Veränderung des max. VO2 in Woche 16 gegenüber dem Ausgangswert (siehe Tabelle). Insgesamt waren 106 von den 234 Teilnehmern (45 %) mittels CPET auswertbar. Es handelte sich hierbei um die Kinder, die 7 Jahre und älter und von ihrer Entwicklung her imstande waren, den Test durchzuführen. Bei den Kindern unter 7 Jahren (kombinierte Sildenafil-Gruppe: n = 47; PlaceboGruppe: n = 16) konnten nur die sekundären Endpunkte erhoben werden. Die durchschnittlichen Ausgangswerte für die max. Sauerstoffaufnahme waren innerhalb der Sildenafil-Gruppen vergleichbar (17,37 bis 18,03 ml/kg/min) und in der Placebo-Gruppe geringfügig höher (20,02 ml/kg/min). Die Ergebnisse der Gesamtauswertung (kombinierte Dosis-Gruppe vs. Placebo) unterschieden sich nicht signifikant (p = 0,056; siehe Tabelle). Zwischen der mittleren Sildenafil-Dosis und Placebo betrug der berechnete Unterschied 11,33 % (95 %-KI: 1,72 bis 20,94; siehe Tabelle 2). Dosisabhängige Verbesserungen wurden beim pulmonalen Gefäßwiderstands-Index (PVRI) und dem durchschnittlichen pulmonalen arteriellen Druck (mPAP) beobachtet. Mit – 18 % (95 %-KI: 2 % bis 32 %) und – 27 % (95 %-KI: 14 % bis 39 %) gegenüber der Placebo-Gruppe ergab sich in den Tabelle 2: Placebokorrigierte prozentuale Veränderung des Peak-VO2 gegenüber dem Ausgangswert in den aktiven Behandlungsgruppen. Behandlungsgruppe Niedrige Dosis (n = 24) Mittlere Dosis (n = 26) Hohe Dosis (n = 27) Kombinierte Dosen (n = 77) Berechneter Unterschied 95 %-Konfidenzintervall 3,81 – 6,11 bis 13,73 11,33 1,72 bis 20,94 7,98 – 1,64 bis 17,60 7,71 (p = 0,056) – 0,19 bis 15,60 Placebo-Gruppe: n = 29 Schätzwerte beruhend auf ANCOVA mit den Kovariablen Ausgangswert der max. VO2, Ätiologie und Gewichtsgruppe 7 Fachinformation (Zusammenfassung der Merkmale des Arzneimittels/SPC) Revatio ® 20 mg Filmtabletten Sildenafil-Gruppen mit mittlerer und hoher Dosis eine Verringerung des PVRI. Die Gruppe mit der niedrigen Dosis zeigte keine signifikanten Unterschiede gegenüber der Placebo-Gruppe (Unterschied: 2 %). Mit – 3,5 mmHg (95 %-KI: – 8,9 bis 1,9) und – 7,3 mmHg (95 %-KI: – 12,4 bis – 2,1) gegenüber dem Ausgangswert ergab sich in den Sildenafil-Gruppen mit mittlerer und hoher Dosis eine Veränderung des mPAP im Vergleich zur Placebo-Gruppe. Die Gruppe mit der niedrigen Dosis zeigte nur kleine Unterschiede gegenüber Placebo (Unterschied: 1,6 mmHg). Alle drei Sildenafil-Gruppen zeigten gegenüber Placebo eine Verbesserung des Herzindex von 10 %, 4 % und 15 % jeweils für die Gruppe mit niedriger, mittlerer und hoher Dosis. Gegenüber Placebo zeigte sich lediglich bei den Teilnehmern mit der hohen SildenafilDosis eine signifikante Verbesserung der Funktionsklasse. Im Vergleich zu Placebo betrug die Odds-Ratio in den SildenafilGruppen mit niedriger, mittlerer und hoher Dosis 0,6 (95 %-KI: 0,18 bis 2,01), 2,25 (95 %-KI: 0,75 bis 6,69) und 4,52 (95 %-KI: 1,56 bis 13,10). Daten der Langzeit-Anschlussstudie Von den 234 pädiatrischen Patienten, die in der placebokontrollierten Kurzzeit-Studie behandelt wurden, haben 220 Patienten an der Langzeit-Anschlussstudie teilgenommen. Dabei wurden Teilnehmer aus der Placebo-Gruppe der Kurzzeit-Studie randomisiert einer Sildenafil-Behandlung zugeordnet; Patienten mit einem Gewicht ≤ 20 kg wurden in die Gruppen mit mittlerer oder hoher Dosis (1 : 1) aufgenommen, während Patienten mit einem Gewicht >20 kg in die niedrige, mittlere oder hohe Dosisgruppe (1 : 1 : 1) aufgenommen wurden. Von den insgesamt 229 Patienten, die Sildenafil erhielten, waren 55, 74 und 100 Patienten in den Gruppen mit niedriger, mittlerer bzw. hoher Dosis. Die Gesamtbehandlungsdauer während der Kurzzeit- und Langzeit-Studien, beginnend mit der Doppelverblindung für die einzelnen Patienten, lag zwischen 3 und 3.129 Tagen. In der Gruppe mit Sildenafil-Behandlung betrug die mittlere Dauer der Sildenafil-Behandlung 1.696 Tage (darin nicht enthalten die 5 Patienten, die in der doppelblinden Phase Placebo erhielten und nicht in der Langzeit-Anschlussstudie behandelt wurden). In den Gruppen mit niedriger, mittlerer und hoher Dosis betrugen die KaplanMeier-Schätzwerte der 3-Jahres-Überlebensrate für die Patienten mit einem Gewicht >20 kg bei Studienbeginn 94 %, 93 % und 85 %. In den Gruppen mit mittlerer und hoher Dosis betrugen die Schätzwerte der Überlebensrate für die Patienten mit einem Gewicht ≤ 20 kg bei Studienbeginn 94 % und 93 % (siehe Abschnitte 4.4 und 4.8). Während der Studiendurchführung wurden insgesamt 42 Todesfälle gemeldet, entweder im Verlauf der Behandlung oder während der Nachbeobachtung des Überlebens. 37 Todesfälle traten auf, bevor das Datenüberwachungskomitees (Data Monitoring Committee) die Entscheidung gefällt hatte, die Dosis der Patienten auf eine nied8 rigere Dosierung zu reduzieren. Diese Entscheidung basierte auf einem beobachteten Ungleichgewicht der Sterblichkeit mit zunehmenden Sildenafil-Dosen. Von diesen 37 Todesfällen betrug die Anzahl (%) in der Gruppe mit niedriger Sildenafil-Dosis 5/55 (9,1 %), mit mittlerer Sildenafil-Dosis 10/74 (13,5 %) und mit hoher Sildenafil-Dosis 22/100 (22 %). Im Anschluss wurden 5 weitere Todesfälle gemeldet. Die Todesursachen wurden mit der PAH in Verbindung gebracht. Höhere als die empfohlenen Dosen dürfen bei pädiatrischen Patienten mit PAH nicht angewendet werden (siehe Abschnitte 4.2 und 4.4). 1 Jahr nach Beginn der placebokontrollierten Studie wurde die max. VO2 bestimmt. Von den mit Sildenafil behandelten Personen, die von ihrer Entwicklung her imstande waren, den CPET durchzuführen, zeigte sich bei 59/114 Personen (52 %) gegenüber dem Zeitpunkt zu Beginn der Sildenafil-Behandlung keinerlei Verschlechterung der max. VO2. In ähnlicher Weise hatte sich die WHO-Funktionsklasse bei 191 von 229 Personen (83 %), die Sildenafil erhalten hatten, bei der Beurteilung nach 1 Jahr unverändert erhalten oder verbessert. Die Europäische Arzneimittel-Agentur hat für Revatio eine Zurückstellung von der Verpflichtung zur Vorlage von Ergebnissen zu Studien bei Neugeborenen mit pulmonaler arterieller Hypertonie gewährt (siehe Abschnitt 4.2 bzgl. Informationen zur Anwendung bei Kindern und Jugendlichen). 5.2 Pharmakokinetische Eigenschaften Resorption Sildenafil wird rasch resorbiert. Die maximalen Plasmaspiegel werden innerhalb von 30 bis 120 Minuten (Median: 60 Minuten) nach oraler Gabe im nüchternen Zustand erreicht. Die mittlere absolute orale Bioverfügbarkeit beträgt 41 % (Streubreite: 25 bis 63 %). Nach dreimal täglicher oraler Einnahme von Sildenafil nehmen AUC und Cmax dosisproportional über den Dosisbereich von 20 bis 40 mg zu. Nach oralen Dosen von 80 mg dreimal täglich wurde ein höherer als dosisproportionaler Anstieg der Plasmaspiegel von Sildenafil beobachtet. Bei Patienten mit PAH war die orale Bioverfügbarkeit von Sildenafil nach einer Dosis von 80 mg dreimal täglich durchschnittlich 43 % (90%-KI: 27 % bis 60 %) höher als mit den niedrigeren Dosen. Bei Einnahme von Sildenafil zusammen mit einer Mahlzeit ist die Resorptionsrate reduziert, die tmax verzögert sich im Mittel um 60 Minuten, während die Cmax im Mittel um 29 % verringert ist; allerdings war das Ausmaß der Resorption nicht signifikant beeinträchtigt (AUC verringerte sich um 11 %). Verteilung Das mittlere Verteilungsvolumen im Steady State (Vss) von Sildenafil beträgt 105 l, was auf eine Verteilung in die Gewebe hinweist. Nach oraler Gabe einer 20-mg-Dosis dreimal täglich beträgt die mittlere maximale Gesamtplasmakonzentration von Sildenafil ca. 113 ng/ml. Sildenafil und sein wichtigster im Blutkreislauf zirkulierender, N-demethylierter Metabolit sind zu etwa 96 % an Plas- maproteine gebunden. Die Proteinbindung ist unabhängig von der Gesamtkonzentration des Arzneimittels. Biotransformation Sildenafil wird überwiegend hepatisch durch die mikrosomalen Isoenzyme CYP3A4 (Hauptweg) und CYP2C9 (Nebenweg) metabolisiert. Der wichtigste zirkulierende Metabolit resultiert aus der N-Demethylierung von Sildenafil. Das Profil der Phosphodiesterase-Selektivität dieses Metaboliten ist ähnlich jenem von Sildenafil, und er zeigt eine In-vitro -Hemmwirkung für PDE5, die rund 50 % derjenigen der Stammsubstanz beträgt. Der N-Demethyl-Metabolit wird weiter verstoffwechselt, die terminale Halbwertszeit beträgt rund 4 Stunden. Bei Patienten mit PAH betragen die Plasmaspiegel des N-Demethyl-Metaboliten nach einer Gabe von 20 mg dreimal täglich etwa 72 % jener von Sildenafil (was einem Beitrag von 36 % zu den pharmakologischen Wirkungen von Sildenafil entspricht). Der weitere Effekt auf die Wirksamkeit ist nicht bekannt. Elimination Die gesamte Clearance von Sildenafil beträgt 41 l/h mit einer daraus resultierenden terminalen Halbwertszeit von 3 bis 5 Stunden. Nach oraler oder intravenöser Applikation wird Sildenafil nach Metabolisierung hauptsächlich über die Fäzes (rund 80 % der verabreichten oralen Dosis) und in geringerem Maße renal (rund 13 % der verabreichten oralen Dosis) ausgeschieden. Pharmakokinetik bei speziellen Patientengruppen Ältere Patienten Gesunde ältere Freiwillige (65 Jahre oder älter) zeigten eine herabgesetzte SildenafilClearance, wobei die Plasmaspiegel von Sildenafil und des aktiven N-Demethyl-Metaboliten ungefähr 90 % höher lagen als bei jüngeren gesunden Freiwilligen (18 bis 45 Jahre). Aufgrund der altersabhängigen Unterschiede in der Plasmaproteinbindung lag der entsprechende Anstieg der Plasmaspiegel von freiem Sildenafil bei rund 40 %. Patienten mit Nierenfunktionsstörungen Bei Probanden mit leichter bis mäßiger Nierenfunktionsstörung (Kreatinin-Clearance = 30 bis 80 ml/min) war die Pharmakokinetik nach einer oralen Sildenafil-Einzeldosis von 50 mg unverändert. Bei Probanden mit schwerer Nierenfunktionsstörung (KreatininClearance <30 ml/min) war die Clearance von Sildenafil herabgesetzt und resultierte in Erhöhungen von AUC und Cmax um 100 % bzw. 88 % im Vergleich zu Probanden gleichen Alters ohne eingeschränkte Nierenfunktion. Zusätzlich waren die AUC (200 %) und Cmax (79 %) des N-DemethylMetaboliten bei Patienten mit schwerer Nierenfunktionsstörung im Vergleich zu Probanden mit normaler Nierenfunktion signifikant erhöht. Patienten mit Leberfunktionsstörungen Bei Probanden mit leichter bis mäßiger Leberzirrhose (Child-Pugh-Klassen A und B) war die Clearance von Sildenafil herabgesetzt, was zu Erhöhungen von AUC (85 %) und Cmax (47 %) führte, im Vergleich zu Probanden gleichen Alters ohne einge009389-12885 Fachinformation (Zusammenfassung der Merkmale des Arzneimittels/SPC) Revatio ® 20 mg Filmtabletten schränkte Leberfunktion. Zusätzlich waren AUC und Cmax für den N-Demethyl-Metaboliten bei Probanden mit Leberzirrhose im Vergleich zu Probanden mit normaler Leberfunktion signifikant um 154 % bzw. 87 % erhöht. Die Pharmakokinetik von Sildenafil bei Patienten mit schwerer Leberfunktionsstörung wurde nicht untersucht. Pharmakokinetik bei verschiedenen Patientengruppen Bei Patienten mit PAH waren die durchschnittlichen Plasmaspiegel im Steady State über den untersuchten Dosisbereich von 20 bis 80 mg dreimal täglich um 20 bis 50 % höher als bei gesunden Freiwilligen. Es zeigte sich eine Verdoppelung der Cmin im Vergleich zu gesunden Freiwilligen. Diese beiden Befunde lassen eine geringere Clearance und/oder eine höhere orale Bioverfügbarkeit von Sildenafil bei Patienten mit PAH im Vergleich zu gesunden Freiwilligen vermuten. Kinder und Jugendliche Die Analyse des pharmakokinetischen Profils von Sildenafil bei den in klinische Studien mit Kindern eingeschlossenen Patienten zeigte, dass das Körpergewicht eine gute Vorhersage der Arzneimittelexposition bei Kindern erlaubt. Bei einem Körpergewicht von 10 bis 70 kg beträgt die Plasmahalbwertzeit von Sildenafil 4,2 bis 4,4 Stunden und zeigt dabei keine Unterschiede, die man als klinisch relevant einstufen könnte. Bei Patienten mit einem Gewicht von 70, 20 und 10 kg betrug die Cmax nach einer oralen Einzeldosis von 20 mg Sildenafil 49, 104 und 165 ng/ml. Bei Patienten mit einem Gewicht von 70, 20 und 10 kg betrug die Cmax nach einer oralen Einzeldosis von 10 mg Sildenafil 24, 53 und 85 ng/ml. Die tmax betrug etwa 1 Stunde und war nahezu unabhängig vom Körpergewicht. 5.3 Präklinische Daten zur Sicherheit April 2015 spcde-5v31ro-ft-20 Basierend auf den konventionellen Studien zur Sicherheitspharmakologie, Toxizität bei wiederholter Gabe, Genotoxizität und zum kanzerogenen Potenzial sowie zur Reproduktions- und zur Entwicklungstoxizität lassen die präklinischen Daten keine besonderen Gefahren für den Menschen erkennen. Bei Jungtieren von Ratten, die prä- und postnatal mit 60 mg/kg Sildenafil behandelt worden waren, zeigten sich bei einer Exposition, die etwa dem 50-Fachen der erwarteten Exposition beim Menschen bei einer Dosis von 20 mg dreimal täglich entsprach, eine verminderte Wurfgröße, ein geringeres Gewicht der Jungtiere an Tag 1 und ein vermindertes Überleben bis zum Tag 4. Effekte in präklinischen Studien wurden bei einer Exposition beobachtet, die so weit über der maximalen Dosis beim Menschen lagen, dass sie als für den klinischen Einsatz nicht relevant erachtet wurden. Im Tiermodell wurden bei klinisch relevanten Konzentrationen keine Nebenwirkungen mit möglichen Auswirkungen auf die klinische Anwendung beobachtet, die nicht ebenfalls in klinischen Studien aufgetreten sind. 009389-12885 6. PHARMAZEUTISCHE ANGABEN 6.1 Liste der sonstigen Bestandteile Tablettenkern: mikrokristalline Cellulose Calciumhydrogenphosphat Croscarmellose-Natrium Magnesiumstearat Filmüberzug: Hypromellose Titandioxid (E 171) Lactose-Monohydrat Triacetin 5. 6. 7. 6.2 Inkompatibilitäten Nicht zutreffend. 6.3 Dauer der Haltbarkeit 8. Tabletten: 5 Jahre Zubereitete Suspension zum Einnehmen: stabil für 28 Tage bei Aufbewahrung in einem Kühlschrank (2 °C bis 8 °C) 6.4 Besondere Vorsichtsmaßnahmen für die Aufbewahrung Tabletten: Nicht über 30 °C lagern. In der Originalverpackung aufbewahren, um den Inhalt vor Feuchtigkeit zu schützen. 9. 10. 11. Zur Aufbewahrung der zubereiteten Suspension zum Einnehmen siehe Abschnitt 6.3. 6.5 Art und Inhalt des Behältnisses PVC/Aluminium-Blister mit 90 Filmtabletten Packung mit 90 Tabletten in einer Faltschachtel 12. PVC/Aluminium-Blister mit 300 Filmtabletten Packung mit 300 Tabletten in einer Faltschachtel Es werden möglicherweise nicht alle Packungsgrößen in den Verkehr gebracht. 6.6 Besondere Vorsichtsmaßnahmen für die Beseitigung und sonstige Hinweise zur Handhabung Keine besonderen Anforderungen für die Beseitigung. 13. Anwendung bei Kindern und Jugendlichen Siehe Abschnitt 4.2 Herstellung einer fallweise zubereiteten Suspension zum Einnehmen aus Revatio 20 mg Filmtabletten (Endkonzentration: 10 mg/ml) Mit der Herstellung einer Suspension zum Einnehmen durch einen Apotheker nach dieser Anleitung wird einem Patienten genügend Arzneimittel für eine 28-tägige (Dosierung: 1 ml; Dosis: 10 mg) oder eine 14-tägige (Dosierung: 2 ml; Dosis: 20 mg) Behandlung zur Verfügung stehen. Zur Herstellung einer Revatio-Suspension zum Einnehmen aus Revatio 20 mg Filmtabletten werden Ora-Sweet und Ora-Plus als Verdünnungsmittel benutzt. Herstellungsanleitung für Apotheker 1. Stellen Sie sicher, dass sich Ora-Sweet und Ora-Plus auf Raumtemperatur erwärmt haben. 2. Zählen Sie 62 Revatio 20 mg Filmtabletten ab. 3. Zerreiben Sie diese 62 Tabletten, immer 2 bis 10 Tabletten gleichzeitig, mittels Mörser und Stößel zu einem feinen Pulver. 4. Messen Sie 30 ml Ora-Plus ab (eine trübe, weiße Flüssigkeit) und warten 14. Sie ab, bis sich alle Luftblasen aufgelöst haben. Geben Sie einen Teil des Ora-Plus aus Schritt 4 in den Mörser (üblicherweise 15 bis 20 ml) und vermischen Sie es zu einem dicken homogenen Brei. Bei Bedarf können Sie mehr Ora-Plus aus Schritt 4 hinzugeben. Geben Sie den Brei in eine Braunglasoder HDPE(hochdichtes Polyäthylen)Flasche mit einem Volumen von mindestens 150 ml. Spülen Sie Mörser und Stößel mit dem restlichen Ora-Plus aus Schritt 4 ab und schütten Sie die Flüssigkeit ebenfalls in die Flasche, um eine vollständige Überführung des Breis zu gewährleisten. Messen Sie 90 ml Ora-Sweet ab (eine klare, pinkfarbene Flüssigkeit) und warten Sie ab, bis sich alle Luftblasen aufgelöst haben. Geben Sie etwa die Hälfte des OraSweet aus Schritt 8 in die Flasche mit der oben hergestellten Formulierung. Verschließen Sie die Flasche und schütteln Sie sie kräftig für mindestens 30 Sekunden. Geben Sie das restliche Ora-Sweet aus Schritt 8 in die Flasche und schütteln Sie sie wieder kräftig für mindestens 30 Sekunden, um eine homogene Suspension zum Einnehmen zu erhalten. Kleben Sie ein zusätzliches Etikett auf die Flasche mit dem Warnhinweis „Patienten mit der seltenen hereditären Fructose-Intoleranz, Glucose-Galactose-Malabsorption oder SaccharaseIsomaltase-Mangel sollten dieses Arzneimittel nicht einnehmen.“ Auf dem Aufkleber sollte ebenfalls stehen „Vor jeder Einnahme mindestens 10 Sekunden kräftig schütteln“. Geben Sie weiterhin den Namen des Patienten, die Dosierungsanleitung, das Verfalldatum und die Bezeichnung des Arzneimittels an. Informieren Sie die Person, die das zubereitete Produkt verabreichen wird, darüber, dass nach Beendigung der Therapie verbliebene Restmengen entsorgt werden müssen. Dies kann entweder über den Aufkleber auf der Flasche oder durch einen zusätzlichen Hinweis auf dem Etikett für die Anleitung für die Apotheken erfolgen. Geben Sie entsprechend den Aufbewahrungshinweisen (siehe Abschnitt 6.3) das Verfalldatum auf dem Etikett an. Überlegen Sie gegebenenfalls, die Suspension zum Einnehmen mit einer zur Dosierung geeigneten Applikationsspritze für Zubereitungen zum Einnehmen mit Graduierung zur Abmessung des benötigten Volumens an Suspension zum Einnehmen abzugeben. Markieren Sie, wenn möglich, für jeden Patienten die Graduierung für die jeweilige Dosis auf der Applikationsspritze für Zubereitungen zum Einnehmen (1 ml oder 2 ml). Besondere Aufbewahrungshinweise Bei Aufbewahrung der mit Ora-Sweet/OraPlus als Verdünnungsmittel zubereiteten Suspension zum Einnehmen in einem Kühlschrank (bei 2 °C bis 8 °C) und in Braun9 Fachinformation (Zusammenfassung der Merkmale des Arzneimittels/SPC) Revatio ® 20 mg Filmtabletten glas- und HDPE-Flaschen konnte eine Stabilität von 28 Tagen nachgewiesen werden. Mit anderen Vehikeln oder anderen Flaschentypen wurden keine Stabilitätsuntersuchungen durchgeführt. Verdünnungsmittel Ora-Plus (eine trübe, weiße Flüssigkeit) enthält gereinigtes Wasser, mikrokristalline Cellulose, Natriumcarboxymethylcellulose, Xanthangummi, Carrageen (enthält Calciumsulfat und Trinatriumphosphat), Natriumphosphat, Citronensäure, Entschäumungsemulsion und ist konserviert mit Methylparaben und Kaliumsorbat. Ora-Sweet (eine klare, pinkfarbene Flüssigkeit) enthält gereinigtes Wasser, Sucrose, Glycerin, Sorbitol, Citronensäure, Natriumphosphat, Geschmacksstoffe und ist konserviert mit Methylparaben und Kaliumsorbat. 7. INHABER DER ZULASSUNG Pfizer Limited Sandwich Kent CT13 9NJ Vereinigtes Königreich 8. ZULASSUNGSNUMMER EU/1/05/318/001 EU/1/05/318/004 9. DATUM DER ERTEILUNG DER ZULASSUNG/VERLÄNGERUNG DER ZULASSUNG Datum der Erteilung der Zulassung: 28. Oktober 2005 Datum der letzten Verlängerung der Zulassung: 23. September 2010 10. STAND DER INFORMATION April 2015 Ausführliche Informationen zu diesem Arzneimittel sind auf den Internetseiten der Europäischen Arzneimittel-Agentur http://www.ema.europa.eu/ema/ verfügbar. Verkaufsabgrenzung Verschreibungspflichtig. Packungsgrößen in Deutschland Packung mit 90 Filmtabletten N 2 Packung mit 300 Filmtabletten N 3 Repräsentant in Deutschland PFIZER PHARMA GmbH Linkstr. 10 10785 Berlin Tel.: 030 550055-51000 Fax: 030 550054-10000 Zentrale Anforderung an: Rote Liste Service GmbH Fachinfo-Service Mainzer Landstraße 55 60329 Frankfurt 10 009389-12885 Arbeiter-Samariter-Bund Regionalverband München/Oberbayern e.V. SEG-Behandlung ACS-Workshop 16.10.2015 Bernd Kaiser Gruppe 2 Nitrolingual® Indikationen ACS (Akutes Coronar Syndrom) Angina pectoris Myokardinfarkt unmittelbare Symptomlinderung nitro-restistent nitro-sensitiv V. a. ACS – Algorithmus Acetylsalicylsäure + Heparin Seite 8 Therapie des ACS: MONA Deutscher Berufsverband Rettungsdienst e.V. (DBRD) • • • • • • ➡ Algorithmus Morphin + Antiemetika Angepasste O2-Gabe Algorithmus Glyceroltrinitrat-Spray Algorithmus ASS + Heparin Algorithmus Sedation Monitoring + Defibrillationsbereitschaft Hinweis für aktive Blutung? JA KEINE ASS + Heparin-Gabe JA KEINE ASS + Heparin-Gabe NEIN Hinweise auf frischen Schlaganfall? NEIN • • • • Hinweise für Aortendissektion? Schmerzen zwischen den Schulterblättern Reissender Schmerzcharakter RR-Differenz von mehr als 20 mmHg zwischen beiden Armen Bekanntes thorakales Aortenaneuyrsma Medikation mit oralen Antikoagulantien? JA JA KEINE ASS + Heparin-Gabe KEINE ASS + Heparin-Gabe Allergie gegen ASS? JA KEINE ASS-Gabe Allergie gegen Heparin? JA KEINE Heparin-Gabe NEIN NEIN Acetylsalicylsäure 300 mg i.v. 70 - 100 I.E. / kg KG bis max. 5000 I.E. Heparin i.v. ggf. andere Antikoagulantien nach lokalem Protokoll ESC Leitlinie STEMI 2012; ESC Leitlinie NSTEMI 2013; ERC-Leitlinie 2010 FACHINFORMATION Aspirin® i. v. 500 mg 1. BEZEICHNUNG DES ARZNEIMITTELS Aspirin® i. v. 500 mg, Pulver und Lösungsmittel zur Herstellung einer Injektions- oder Infusionslösung Kinder und Jugendliche Anwendungsgebiet Tagesdosen bei Kindern und Jugendlichen Rekonstituierte Lösung in ml (bzw. mg Acetylsalicylsäure) 2. QUALITATIVE UND QUANTITATIVE ZUSAMMENSETZUNG 1 Durchstechflasche mit Pulver zur Herstellung einer Injektions- oder Infusionslösung enthält 1000 mg D,L-Lysinacetylsalicylat ∙ Glycin, entsprechend 500 mg Acetylsalicylsäure. 1 ml rekonstituierte Lösung enthält 100 mg Acetylsalicylsäure. Vollständige Auflistung der sonstigen Bestandteile, siehe Abschnitt 6.1. 3. DARREICHUNGSFORM Pulver und Lösungsmittel zur Herstellung einer Injektions- oder Infusionslösung. Die Farbe des Pulvers ist weiß. Die rekonstituierte Lösung hat einen pHWert von 4,5 bis 6,5. 4. KLINISCHE ANGABEN 4.1 Anwendungsgebiete – Akute mäßig starke bis starke Schmerzen (falls eine orale Anwendung nicht angezeigt ist), – akute Behandlung der Kopfschmerzphase von Migräneanfällen mit oder ohne Aura, – Fieber (wenn eine sofortige Temperatursenkung erforderlich und eine orale Anwendung nicht angezeigt ist). Bitte beachten Sie die Angaben für Kinder und Jugendliche (siehe Abschnitt 4.4). 4.2 Dosierung und Art der Anwendung Dosierung Erwachsene Soweit nicht anders verordnet, werden folgende Richtdosen empfohlen: Anwendungsgebiete Einzeldosen bei Erwachsenen Anzahl Durchstechflaschen (bzw. mg Acetylsalicylsäure) Akute mäßig starke bis starke Schmerzen Mai 2014 DE/12 Fieber Akute Behandlung der Kopfschmerzphase von Migräneanfällen mit oder ohne Aura 009217-5043 1–2 (500 – 1000 mg Acetylsalicylsäure) Bei Bedarf ist eine wiederholte Anwendung möglich; maximale Tagesdosis: 10 Durchstechflaschen (5000 mg Acetylsalicylsäure). 2 (1000 mg Acetylsalicylsäure) Fieber 0,1 – 0,25 ml/kg KG/Tag (10 – 25 mg Acetylsalicylsäure/kg KG/Tag), verteilt auf 2 – 3 Gaben im Abstand von 4 – 8 Stunden je nach individueller Dosis Art der Anwendung Intravenöse Anwendung. Die Injektionslösung muss stets frisch hergestellt und gleich nach der Herstellung verwendet werden. Nur klare Lösungen ohne sichtbare Partikel dürfen verabreicht werden. Die Lösung ist nur für den einmaligen Gebrauch bestimmt. Nicht verwendete Lösung muss verworfen werden. Die Lösung wird langsam intravenös injiziert. Sie kann auch einer Kurzinfusion beigemischt (siehe Abschnitt 6.6) oder langsam in den Infusionsschlauch nahe der Kanüle einer bereits laufenden Infusion injiziert werden. Dauer der Anwendung Aspirin i. v. 500 mg ist für eine Dauerbehandlung nicht vorgesehen. Die Dauer der Anwendung bestimmt der behandelnde Arzt. 4.3 Gegenanzeigen – Überempfindlichkeit gegen den Wirkstoff oder andere Salicylate oder einen der in Abschnitt 6.1 genannten sonstigen Bestandteile. – Asthmaanfälle in der Vergangenheit, die durch die Verabreichung von Salicylaten oder Substanzen mit ähnlicher Wirkung, insbesondere nichtsteroidalen Antiphlogistika, ausgelöst wurden. – Akute gastrointestinale Ulcera. – Hämorrhagische Diathese. – Leber- und Nierenversagen. – Schwere, nicht eingestellte Herzinsuffizienz. – Kombination mit Methotrexat in einer Dosierung von 15 mg oder mehr pro Woche (siehe Abschnitt 4.5). – Letztes Trimenon der Schwangerschaft (siehe Abschnitt 4.6). 4.4 Besondere Warnhinweise und Vorsichtsmaßnahmen für die Anwendung – bei Überempfindlichkeit gegen andere Analgetika/Antiphlogistika/Antirheumatika oder andere allergene Stoffe (siehe Abschnitt 4.3); – bei Bestehen von Allergien (z. B. mit Hautreaktionen, Juckreiz, Nesselfieber), Asthma, Heuschnupfen, Nasenschleimhautschwellungen (Nasenpolypen) oder chronischen Atemwegserkrankungen; – bei gleichzeitiger Therapie mit Antikoagulantien; – bei gastrointestinalen Ulcera oder Blutungen in der Vorgeschichte; – bei Patienten mit eingeschränkter Leberfunktion; – bei Patienten mit eingeschränkter Nierenfunktion oder mit verminderter kardiovaskulärer Durchblutung (z. B. renale Gefäßerkrankung, kongestive Herzinsuffizienz, Volumenverlust, größere Operationen, Sepsis oder schwere Blutungsereignisse): Acetylsalicylsäure kann das Risiko einer Nierenfunktionsstörung und eines akuten Nierenversagens weiter erhöhen; – vor Operationen (auch bei kleineren Eingriffen wie z. B. Zahnextraktionen): es kann zu verstärkter Blutungsneigung kommen; – bei Patienten mit schwerem Glukose-6Phosphat-Dehydrogenasemangel: Acetylsalicylsäure kann eine Hämolyse oder eine hämolytische Anämie induzieren. Das Risiko einer Hämolyse kann durch Faktoren wie z. B. hohe Dosierung, Fieber oder akute Infektionen erhöht werden. Bei Operationen, bei denen intraoperativ eine absolute Blutstillung erforderlich ist, sollte Aspirin i. v. 500 mg möglichst nicht präoperativ gegeben werden. Acetylsalicylsäure vermindert in niedriger Dosierung die Harnsäureausscheidung. Bei Patienten, die bereits zu geringer Harnausscheidung neigen, kann dies unter Umständen einen Gichtanfall auslösen. Kinder und Jugendliche Aspirin i. v. 500 mg soll bei Kindern und Jugendlichen mit fieberhaften Erkrankungen nur auf ärztliche Anweisung und nur dann angewendet werden, wenn andere Maßnahmen nicht wirken. Sollte es bei diesen Erkrankungen zu lang anhaltendem Erbrechen kommen, so kann dies ein Zeichen des Reye-Syndroms, einer sehr seltenen, aber lebensbedrohlichen Krankheit sein, die unbedingt sofortiger ärztlicher Behandlung bedarf. 4.5 Wechselwirkungen mit anderen Arzneimitteln und sonstige Wechselwirkungen Verstärkung der Wirkung bis hin zu erhöhtem Nebenwirkungsrisiko – Antikoagulanzien/Thrombolytika: Acetylsalicylsäure kann das Blutungsrisiko erhöhen, wenn es vor einer ThrombolyseTherapie eingenommen wurde. Daher muss bei Patienten, bei denen eine Thrombolysebehandlung durchgeführt werden soll, auf Zeichen äußerer oder innerer Blutung aufmerksam geachtet werden; – Thrombozytenaggregationshemmer, z. B. Ticlopidin, Clopidogrel: erhöhtes Risiko für Blutungen; – Andere nichtsteroidale Analgetika/Antiphlogistika (in Dosierungen ab 3 g Acetylsalicylsäure pro Tag und mehr): erhöhtes Risiko für gastrointestinale Ulcera und Blutungen; – Systemische Glucokortikoide (mit Ausnahme von Hydrocortison als Ersatztherapie bei Morbus Addison): erhöhtes Risiko für gastrointestinale Nebenwirkungen; – Alkohol: erhöhtes Risiko für gastrointestinale Ulcera und Blutungen; – Digoxin: Erhöhung der Plasmakonzentration; 1 FACHINFORMATION Aspirin® i. v. 500 mg – Antidiabetika: der Blutzuckerspiegel kann sinken; – Methotrexat: Verminderung der Ausscheidung und Verdrängung aus der Plasmaproteinbindung durch Salicylate; – Valproinsäure: Verdrängung aus der Plasmaproteinbindung durch Salicylate; – Selektive-Serotonin-WiederaufnahmeHemmer (SSRIs): erhöhtes Risiko für gastrointestinale Blutungen aufgrund synergistischer Effekte. Abschwächung der Wirkung – Diuretika (in Dosierungen ab 3 g Acetylsalicylsäure pro Tag und mehr); – ACE-Hemmer (in Dosierungen ab 3 g Acetylsalicylsäure pro Tag und mehr); – Urikosurika (z. B. Probenecid, Benzbromaron). 4.6 Fertilität, Schwangerschaft und Stillzeit Fertilität Es gibt Hinweise, dass Arzneistoffe, die die Cyclooxygenase/Prostaglandinsynthese hemmen, durch einen Effekt auf die Ovulation eine Beeinträchtigung der weiblichen Fertilität hervorrufen können. Dieser Effekt ist nach Absetzen der Behandlung reversibel. Schwangerschaft Eine Hemmung der Prostaglandinsynthese kann die Schwangerschaft und/oder die embryonale/fetale Entwicklung ungünstig beeinflussen. Aufgrund von Daten aus epidemiologischen Studien ergeben sich Bedenken hinsichtlich eines erhöhten Risikos für Fehlgeburten und für Fehlbildungen nach der Einnahme von Prostaglandinsynthesehemmern in der Frühschwangerschaft. Es wird angenommen, dass dieses Risiko mit der Dosis und Anwendungsdauer zunimmt. Die verfügbaren epidemiologischen Daten für Acetylsalicylsäure deuten auf ein erhöhtes Risiko von Gastroschisis hin. Tierexperimentelle Studien zeigten Reproduktionstoxizität (siehe Abschnitt 5.3). Während des 1. und 2. Trimenons der Schwangerschaft darf Acetylsalicylsäure nicht gegeben werden, außer dies ist eindeutig notwendig. Frauen mit Kinderwunsch oder Frauen im 1. und 2. Trimenon der Schwangerschaft sollten bei Einnahme acetylsalicylsäurehaltiger Arzneimittel die Dosis niedrig und die Behandlung so kurz wie möglich halten. Während des 3. Trimenons der Schwangerschaft ist eine Exposition mit Prostaglandinsynthese-Hemmstoffen mit folgenden Risiken verbunden: Beim Feten – Kardiopulmonale Toxizität (mit vorzeitigem Verschluss des Ductus arteriosus und pulmonaler Hypertonie); – Renale Dysfunktion, die bis zum Nierenversagen mit Oligohydramniosis fortschreiten kann. Bei der Mutter und beim Kind am Ende der Schwangerschaft – Mögliche Verlängerung der Blutungszeit, ein anti-aggregatorischer Effekt, der auch schon bei sehr geringer Dosierung auftreten kann; – Hemmung der Wehentätigkeit, die zu einer verzögerten oder verlängerten Geburt führen kann. 2 Folglich ist Aspirin i. v. 500 mg während des 3. Trimenons der Schwangerschaft kontraindiziert (siehe Abschnitt 4.3). Stillzeit Der Wirkstoff Acetylsalicylsäure und seine Abbauprodukte gehen in geringen Mengen in die Muttermilch über. Da nachteilige Wirkungen auf den Säugling bisher nicht bekannt geworden sind, ist bei gelegentlicher Anwendung der empfohlenen Dosis eine Unterbrechung des Stillens nicht erforderlich. Bei längerer Anwendung bzw. Einnahme höherer Dosen sollte dennoch abgestillt werden. 4.7 Auswirkungen auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen Aspirin i. v. 500 mg hat keinen Einfluss auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen. 4.8 Nebenwirkungen Die Aufzählung der folgenden unerwünschten Wirkungen umfasst alle bekannt gewordenen Nebenwirkungen unter der Behandlung mit Acetylsalicylsäure, auch solche unter hochdosierter Langzeittherapie bei Rheumapatienten. Die Häufigkeitsangaben, die über Einzelfälle hinausgehen, beziehen sich auf die kurzzeitige Anwendung bis zu Tagesdosen von maximal 3 g Acetylsalicylsäure. Bei den Häufigkeitsangaben zu Nebenwirkungen werden folgende Kategorien zugrunde gelegt: Sehr häufig (≥ 1/10) Häufig (≥ 1/100 bis < 1/10) Gelegentlich (≥ 1/1.000 bis < 1/100) Selten (≥ 1/10.000 bis < 1/1.000) Sehr selten (< 1/10.000) Nicht bekannt (Häufigkeit auf Grundlage der verfügbaren Daten nicht abschätzbar) Erkrankungen des Blutes und des Lymphsystems Selten bis sehr selten sind auch schwerwiegende Blutungen wie z. B. cerebrale Blutungen, besonders bei Patienten mit nicht eingestelltem Bluthochdruck und/oder gleichzeitiger Behandlung mit Antikoagulantien berichtet worden, die in Einzelfällen möglicherweise lebensbedrohlich sein können. Hämolyse und hämolytische Anämie wurden bei Patienten mit schwerem Glukose-6-Phosphat-Dehydrogenasemangel berichtet. Blutungen wie z. B. Nasenbluten, Zahnfleischbluten, Hautblutungen oder Blutungen des Urogenitaltraktes mit einer möglichen Verlängerung der Blutungszeit (siehe Abschnitt 4.4). Diese Wirkung kann über 4 bis 8 Tage nach der Einnahme anhalten. Erkrankungen des Gastrointestinaltrakts Häufig Gastrointestinale Beschwerden wie Sodbrennen, Übelkeit, Erbrechen, Bauchschmerzen. Selten Gastrointestinale Ulcera, die sehr selten zur Perforation führen können. Gastrointestinale Blutungen, die sehr selten zu einer Eisenmangelanämie führen können. Gastrointestinale Entzündungen. Erkrankungen des Nervensystems Kopfschmerzen, Schwindel, gestörtes Hörvermögen, Ohrensausen (Tinnitus) und mentale Verwirrung können Anzeichen einer Überdosierung sein (siehe Abschnitt 4.9). Erkrankungen der Haut und des Unterhautzellgewebes Gelegentlich Hautreaktionen (sehr selten bis hin zu Erythema exsudativum multiforme) Erkrankungen des Immunsystems Sehr selten Überempfindlichkeitsreaktionen der Haut, des Respirationstrakts, des Gastrointestinaltrakts und des kardiovaskulären Systems, vor allem bei Asthmatikern. Symptome können sein: Blutdruckabfall, Anfälle von Atemnot, Rhinitis, verstopfte Nase, anaphylaktischer Schock oder Quincke-Ödeme. Leber- und Gallenerkrankungen Sehr selten Erhöhungen der Leberwerte wurden beobachtet. Erkrankungen der Nieren und Harnwege Nierenfunktionsstörungen und akutes Nierenversagen wurden berichtet. Allgemeine Erkrankungen und Beschwerden am Verabreichungsort Reaktionen an der Einstichstelle (z. B. Schmerzen, Rötung). Meldung des Verdachts auf Nebenwirkungen Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von großer Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdachtsfall einer Nebenwirkung dem Bundesinstitut für Arzneimittel und Medizinprodukte, Abt. Pharmakovigilanz, KurtGeorg-Kiesinger-Allee 3, D-53175 Bonn, Website: http://www.bfarm.de anzuzeigen. 4.9 Überdosierung Mit einer Intoxikation muss bei älteren Personen und vor allem bei Kleinkindern gerechnet werden (therapeutische Überdosierung oder versehentliche Intoxikationen können bei ihnen tödlich wirken). Symptomatologie Mäßige Intoxikation Tinnitus, Hörstörungen, Schwitzen, Übelkeit, Erbrechen, Kopfschmerzen und Schwindel werden in allen Fällen von Überdosierung festgestellt und können durch Reduzierung der Dosierung abgestellt werden. Schwere Intoxikation: Fieber, rische Koma, sagen, Hyperventilation, Ketose, respiratoAlkalose, metabolische Azidose, kardiovaskulärer Schock, Atemverschwere Hypoglykämie. Notfallbehandlung – Sofortige Einweisung in die Fachabteilung des Krankenhauses; – Magenspülung und Verabreichung von Aktivkohle, Kontrolle des Säure-BasenGleichgewichts; – Alkalische Diurese, um einen Urin-pHWert zwischen 7,5 und 8 zu erreichen; 009217-5043 FACHINFORMATION Aspirin® i. v. 500 mg eine gesteigerte alkalische Diurese muss berücksichtigt werden, wenn die Plasmasalicylatkonzentration bei Erwachsenen größer als 500 mg/l (3,6 mmol/l) oder bei Kindern größer als 300 mg/l (2,2 mmol/l) ist; – Möglichkeit der Hämodialyse bei schwerer Intoxikation; – Flüssigkeitsverlust muss ersetzt werden; – Symptomatische Behandlung. 5. PHARMAKOLOGISCHE EIGENSCHAFTEN 5.1 Pharmakodynamische Eigenschaften Pharmakotherapeutische Gruppe: andere Analgetika und Antipyretika, Salicylsäure und Derivate; ATC-Code: N02BA01. Acetylsalicylsäure gehört zur Gruppe der säurebildenden nichtsteroidalen Antiphlogistika mit analgetischen, antipyretischen und antiphlogistischen Eigenschaften. Ihr Wirkungsmechanismus beruht auf der irreversiblen Hemmung von Cyclo-Oxygenase-Enzymen, die an der Prostaglandinsynthese beteiligt sind. Nach intravenöser Applikation einer wässrigen Lösung einer Lysin-AcetylsalicylsäureVerbindung (Aspirin i. v. 500 mg) ist die analgetische Wirkung wesentlich stärker als die nach oraler Gabe von Acetylsalicylsäure. Das Wirkungsspektrum entspricht jedoch qualitativ dem der Acetylsalicylsäure. Acetylsalicylsäure hemmt außerdem die Thrombozytenaggregation, da sie die Synthese von Thromboxan A2 in den Thrombozyten blockiert. Aus diesem Grund werden bei verschiedenen kardiovaskulären Indikationen (z. B. instabile Angina Pectoris, akuter Myokardinfarkt) Dosierungen von 75 bis 300 mg täglich (oral) eingesetzt. Mai 2014 DE/12 5.2 Pharmakokinetische Eigenschaften Nach intravenöser Gabe von Acetylsalicylsäure erfolgt durch die hohe Invasionsgeschwindigkeit in das Gewebe ein schneller Wirkungseintritt. Acetylsalicylsäure wird rasch in ihren aktiven Hauptmetaboliten Salicylsäure umgewandelt. Nach Injektion von 1 Durchstechflasche Aspirin i. v. 500 mg (entsprechend 500 mg Acetylsalicylsäure) werden die maximalen Plasmaspiegel von Salicylsäure nach ca. 30 Minuten erreicht. Sowohl Acetylsalicylsäure als auch Salicylsäure werden weitgehend an Plasmaproteine gebunden und schnell in alle Teile des Körpers verteilt. Salicylsäure tritt in die Muttermilch über und ist plazentagängig. Salicylsäure wird vor allem durch Metabolisierung in der Leber eliminiert; die Metaboliten sind Salicylursäure, Salicylphenolglucuronid, Salicylacylglucuronid, Gentisinsäure und Gentisursäure. Die Eliminationskinetik von Salicylsäure ist dosisabhängig, da der Metabolismus durch die Kapazität der Leberenzyme begrenzt wird. Die Eliminationshalbwertzeit variiert daher und liegt nach niedrigen Dosen zwischen 2 bis 3 Stunden, während sie nach hohen Dosen bis zu etwa 15 Stunden beträgt. Salicylsäure und ihre Metaboliten werden überwiegend über die Nieren ausgeschieden. 009217-5043 5.3 Präklinische Daten zur Sicherheit Das präklinische Sicherheitsprofil von Acetylsalicylsäure ist gut dokumentiert. Salicylate haben in tierexperimentellen Untersuchungen außer Nierenschäden bei hohen Dosierungen keine weiteren Organschädigungen gezeigt. Acetylsalicylsäure wurde ausführlich in vitro und in vivo bezüglich mutagener Wirkungen untersucht. Die Gesamtheit der Befunde ergibt keine relevanten Verdachtsmomente für eine mutagene Wirkung. Gleiches gilt für Untersuchungen zur Kanzerogenität. Salicylate haben in Tierversuchen an mehreren Tierspezies teratogene Wirkungen gezeigt (z. B. kardiale Missbildungen, skelettale Missbildungen und Bauchwanddefekte). Implantationsstörungen, embryo- und fetotoxische Wirkungen sowie Störungen der Lernfähigkeit bei Nachkommen nach pränataler Exposition sind beschrieben worden. 6. PHARMAZEUTISCHE ANGABEN 6.1 Liste der sonstigen Bestandteile Lösungsmittel: Wasser für Injektionszwecke 6.2 Inkompatibilitäten Das Arzneimittel darf, außer mit den unter Abschnitt 6.6 aufgeführten, nicht mit anderen Arzneimitteln gemischt werden. 6.3 Dauer der Haltbarkeit 18 Monate Haltbarkeit nach Rekonstitution/Verdünnung Die gebrauchsfertige Lösung kann für 2 Stunden bei 25 °C gelagert werden. Aus mikrobiologischen Aspekten ist eine sofortige Verwendung des rekonstituierten Arzneimittels zu empfehlen. 6.4 Besondere Vorsichtsmaßnahmen für die Aufbewahrung Nicht über 25 ºC lagern. Aufbewahrungsbedingungen nach Rekonstitution, siehe Abschnitt 6.3. 6.6 Besondere Vorsichtsmaßnahmen für die Beseitigung und sonstige Hinweise zur Handhabung Keine besonderen Anforderungen für die Beseitigung. Bitte das Produkt unmittelbar nach Zugabe des Lösungsmittels bis zur vollständigen Auflösung des Pulvers schütteln. Für die folgenden Infusionslösungen wurde Kompatibilität mit Aspirin i. v. 500 mg nachgewiesen – Natriumchlorid 9 mg/ml (0,9 %), – Glucose 50 mg/ml (5 %), – Glucose 100 mg/ml (10 %), – Ringer-Lösung, – Ringer-Lactat-Lösung. Wird das Arzneimittel einer Kurzinfusion beigemischt, soll eine Durchstechflasche Aspirin i. v. 500 mg auf nicht mehr als 250 ml Lösung zur Infusion hinzugefügt werden. Die rekonstituierte Lösung ist nur für den einmaligen Gebrauch und nicht verwendete Lösung muss verworfen werden. 7. INHABER DER ZULASSUNG Bayer Vital GmbH 51368 Leverkusen Tel.: 0214/30-5 13 48 Fax: 0214/30-5 16 03 E-Mail: [email protected] 8. ZULASSUNGSNUMMER(N) 54243.00.00 9. DATUM DER ERTEILUNG DER ZULASSUNG/VERLÄNGERUNG DER ZULASSUNG Datum der Erteilung der Zulassung: 06. Juli 2005 Datum der letzten Verlängerung der Zulassung: 30. November 2010 10. STAND DER INFORMATION Mai 2014 11. VERKAUFSABGRENZUNG Verschreibungspflichtig 6.5 Art und Inhalt des Behältnisses Pulver 8 ml Typ 3 Klarglas-Durchstechflaschen mit blauem Chlorbutyl-Stopfen, AluminiumBördelkappe und Plastik-Schutzkappe Lösungsmittel 5 ml Typ 1 Klarglas-Ampullen Packungsgrößen Packung mit 5 Durchstechflaschen zu 1 g Pulver und 5 Ampullen zu 5 ml Lösungsmittel. Packung (gebündelt) mit 20 (4 × 5) Durchstechflaschen zu 1 g Pulver und 20 (4 × 5) Ampullen zu 5 ml Lösungsmittel. Zentrale Anforderung an: Klinikpackung (gebündelt) mit 25 (5 × 5) Durchstechflaschen zu 1 g Pulver und 25 (5 × 5) Ampullen zu 5 ml Lösungsmittel. Rote Liste Service GmbH Es werden möglicherweise nicht alle Packungsgrößen in den Verkehr gebracht. Mainzer Landstraße 55 Fachinfo-Service 60329 Frankfurt 3 Fachinformation Heparin-Natrium-5 000-ratiopharm® 1. BEZEICHNUNG DES ARZNEIMITTELS Heparin-Natrium-5 000-ratiopharm ® (Ampullen/Fertigspritzen) Injektionslösung 2. QUALITATIVE UND QUANTITATIVE ZUSAMMENSETZUNG Jede Ampulle/Fertigspritze zu 0,2 ml Injektionslösung enthält 5.000 I. E. Heparin-Natrium (aus Schweinedarm-Mukosa). Vollständige Auflistung der sonstigen Bestandteile siehe Abschnitt 6.1. 3. DARREICHUNGSFORM Injektionslösung Klare, farblose bis schwach gelbliche Lösung. 4. KLINISCHE ANGABEN 4.1 Anwendungsgebiete – Prophylaxe von thromboembolischen Erkrankungen – im Rahmen der Behandlung von venösen und arteriellen thromboembolischen Erkrankungen (einschließlich der Frühbehandlung des Herzinfarktes sowie der instabilen Angina pectoris) – zur Antikoagulation bei Behandlung oder Operation mit extrakorporalem Kreislauf (z. B. Herz-Lungen-Maschine, Hämodialyse) Hinweis: Heparin-Natrium-5 000-ratiopharm ® Fertigspritzen sind nur bei der Prophylaxe von thromboembolischen Erkrankungen anzuwenden. 4.2 Dosierung und Art der Anwendung Dosierung Heparin-Natrium muss individuell dosiert werden. Die Dosierung ist abhängig von den Gerinnungswerten (siehe Abschnitt 4.4), Art und Verlauf der Erkrankung, Ansprechen des Patienten, Nebenwirkungen, Gewicht und Alter des Patienten. Zu berücksichtigen sind die unterschiedliche Heparin-Empfindlichkeit und eine mögliche Änderung der Heparin-Toleranz im Therapieverlauf. Thromboembolieprophylaxe („low-dose“-Behandlung) Zur Thromboembolieprophylaxe empfiehlt sich die subkutane Injektion. Die Fertigspritze Heparin-Natrium-5 000ratiopharm ® enthält 5.000 I. E. Heparin-Natrium und ist daher nur für die subkutane Gabe dieser Einzeldosen geeignet. Allgemeine Dosierungsempfehlung für die Thromboembolieprophylaxe: – Prä- und postoperative Thrombo- Januar 2013 embolieprophylaxe Präoperativ 5.000 – 7.500 I. E. subkutan ca. 2 h vor der Operation. Postoperativ in Abhängigkeit vom Thromboserisiko in der Regel 5.000 I. E. subkutan alle 8 – 12 h oder 7.500 I. E. subkutan alle 12 h bis zur Mobilisierung des Patienten oder bis zur ausreichenden Wirkung von Vitamin-K-Antagonisten. Laborkontrollen (Gerinnungswerte) zur Dosisanpassung können in Einzelfällen erforderlich sein. Heparin-Natrium sollte über mindestens 48 h gegeben werden. – Prophylaxe in der nicht-operativen – Als Begleittherapie bei Thrombolyse Medizin (z. B. bei längerer Bettlägerigkeit, erhöhter Thromboseneigung des Patienten, Erkrankungen mit erhöhtem Thromboserisiko). In Abhängigkeit vom Thromboserisiko in der Regel 5.000 I. E. subkutan alle 8 – 12 h oder 7.500 I. E. subkutan alle 12 h. Die Dosierung muss dem Thromboserisiko und dem Aktivitätsgrad des Gerinnungssystems angepasst werden und lässt sich durch Gerinnungskontrollen festlegen. Im Rahmen der Therapie von venösen und arteriellen thromboembolischen Erkrankungen Bei bestehenden Gerinnseln in Blutgefäßen empfiehlt sich die kontinuierliche intravenöse Verabreichung. Dosierung bei Erwachsenen Im Allgemeinen einleitend 5.000 I. E. Heparin-Natrium als Bolus intravenös, gefolgt von einer fortlaufenden Infusion mit 1.000 I. E. Heparin-Natrium pro Stunde mittels Perfusor. Dosierung bei Kindern Initial 50 I. E./kg Körpergewicht, anschließend 20 I. E./kg Körpergewicht pro Stunde. Ist eine intravenöse Dauerinfusion nicht möglich, kann alternativ auf eine subkutane Therapie (verteilt auf 2 – 3 Einzeldosen) unter engmaschiger Therapiekontrolle ausgewichen werden (z. B. 10.000 – 12.500 I. E. Heparin-Natrium alle 12 h). mit fibrinspezifischen Thrombolytika (z. B. r-tPA) zur Behandlung des akuten Myokardinfarkts Initial 5.000 I. E. Heparin-Natrium intravenös als Bolus, gefolgt von einer intravenösen Infusion von 1.000 I. E. pro Stunde. Die Infusion sollte gemäß den aPTT-Werten auf eine Verlängerung des Ausgangswertes um das 1,5 – 2,5-fache eingestellt werden. Heparin-Natrium sollte über 48 h gegeben werden. Bei Thrombolyse mit nicht fibrinspezifischen Thrombolytika (z. B. Streptokinase) können auch subkutan 12.500 I. E. Heparin-Natrium alle 12 h gegeben werden, beginnend 4 h nach Thrombolyse. Die genaue Dosierung der Heparin-Begleittherapie richtet sich nach der Art des Thrombolytikums und ist entsprechend den Angaben zu den einzelnen Thrombolytika vorzunehmen. Auf eine genaue Kontrolle des Gerinnungsstatus ist in jedem Fall zu achten! Antikoagulation bei Behandlung oder Operation mit extrakorporalem Kreislauf Hämodialyse Individuelle Dosierung je nach Ausfall der Gerinnungsbestimmungen und dem Maschinentyp. Herz-Lungen-Maschine Die Dosierung ist abhängig vom Typ der Herz-Lungen-Maschine und der Dauer der Operation und ist individuell zu handhaben. Eine engmaschige Therapiekontrolle unter Bestimmung der Gerinnungswerte ist in jedem Fall unbedingt erforderlich. Therapieüberwachung und Dosisanpassung erfolgen im Allgemeinen anhand der aktivierten partiellen Thromboplastinzeit (aPTT), die um das 1,5 – 2,5-fache der Norm erhöht sein soll. Empfohlen werden Kontrollen der aPTT bei kontinuierlicher intravenöser HeparinGabe 1 – 2 h, 6 h, 12 h und 24 h nach Therapiebeginn und bei subkutaner Applikation 6 h nach Verabreichung der 2. Dosis. (Ampullen) Zur subkutanen und intravenösen Injektion oder zur intravenösen Infusion nach Verdünnen. – Behandlung von venösen Thrombo- Handhabung der Ampullen embolien Einleitend sollten 5.000 I. E. Heparin-Natrium intravenös als Bolus appliziert werden, gefolgt von einer intravenösen Infusion von in der Regel 1.000 I. E. Heparin-Natrium pro Stunde. Die Dosierung sollte entsprechend der aPTT-Werte adjustiert werden, wobei eine Verlängerung der aPTT auf das 1,5 – 2,5-fache des Ausgangswertes erreicht werden sollte (möglichst innerhalb der ersten 24 h). Die Behandlung sollte über mindestens 4 Tage erfolgen bzw. sollte so lange fortgesetzt werden bis eine ausreichende orale Antikoagulation erreicht worden ist. – Im Rahmen der Behandlung der instabilen Angina pectoris oder des Non-Q-Wave-Myokardinfarkts Im Allgemeinen 5.000 I. E. Heparin-Natrium intravenös als Bolus, gefolgt von einer kontinuierlichen Infusion mit 1.000 I. E. pro Stunde. Die Dosis richtet sich nach der aPTT, die auf das 1,5 – 2,5-fache des Normalwertes verlängert sein sollte. Art der Anwendung Heparin-Natrium-5 000-ratiopharm ® (Fertigspritzen) Zur subkutanen Injektion. Heparin-Natrium-5 000-ratiopharm ® Nach Aufziehen des Ampulleninhaltes in eine Spritze wird eine frische, feine Injektionsnadel auf die Spritze aufgesetzt. Die subkutane Injektion der Lösung erfolgt wie unten unter „Verabreichung der subkutanen Injektion“ beschrieben. Hinweis: Wegen der erforderlichen Dosierungsgenauigkeit bitte ausschließlich sehr fein graduierte, totraumfreie Einmalspritzen verwenden! Handhabung der Fertigspritzen Schutzkappe von der Fertigspritze parallel zur Spritzenachse entfernen. Luftblase nicht entfernen (sie sorgt für eine vollständige Entleerung der Fertigspritze). Verabreichung der subkutanen Injektion Der Einstich soll mit einer feinen Injektionsnadel senkrecht zur Körperachse in eine abgehobene Bauchfalte oder an der Vorderseite des Oberschenkels erfolgen, die Injektion ist streng subkutan vorzunehmen. Ein an der Injektionsnadel haftender Tropfen ist 1 Fachinformation Heparin-Natrium-5 000-ratiopharm® vor der Injektion zu entfernen, da ein Einbringen von Heparin-Natrium in den Stichkanal zu einem oberflächlichen Bluterguss bzw. in seltenen Fällen zu einer lokalen allergischen Reizung führen kann. Hinweis: Zur Verminderung von Lymphabflussstörungen sollte Heparin-Natrium-5 000ratiopharm ® bei Patienten mit operativer Ausräumung von Lymphknoten im Abdominalbereich/Urogenitalbereich am Oberarm appliziert werden. Hinweis: Da Heparin durch Plättchenbestandteile (PF4) gebunden und dadurch die Wirkung neutralisiert wird, sollte das für die Gerinnungsuntersuchung entnommene und mit Citrat versetzte Blut möglichst bald nach der Abnahme zur Trennung von Blutzellen und Blutplasma zentrifugiert und dekantiert werden. Über die Dauer der Anwendung entscheidet der behandelnde Arzt. Regelmäßige Kontrollen der aktivierten partiellen Thromboplastinzeit (aPTT) sowie der Thrombozytenwerte sind bei der HeparinTherapie erforderlich. 4.3 Gegenanzeigen Heparin-Natrium-ratiopharm ® darf nicht angewendet werden bei: – Überempfindlichkeit gegen den Wirkstoff Heparin oder einen der in Abschnitt 6.1 genannten sonstigen Bestandteile – aktueller oder aus der Anamnese bekannter allergisch bedingter Thrombozytopenie (Typ II) durch Heparin – Erkrankungen, die mit einer hämorrhagischen Diathese einhergehen, z. B. Thrombozytopenien, Koagulopathien, schweren Leber-, Nieren- oder Bauchspeicheldrüsenerkrankungen – Erkrankungen, bei denen der Verdacht einer Läsion des Gefäßsystems besteht, z. B. Ulzera im Magen- und/oder Darmbereich, Hypertonie (>105 mmHg diastolisch), Hirnblutung, Traumata oder chirurgische Eingriffe am Zentralnervensystem, Augenoperationen, Retinopathien, Glaskörperblutungen, Hirnarterienaneurysma, infektiöse Endokarditis – Abortus imminens – Spinalanästhesie, Periduralanästhesie, Lumbalpunktion – Organläsionen, die mit Blutungsneigung einhergehen. 4.4 Besondere Warnhinweise und Vorsichtsmaßnahmen für die Anwendung Heparin-Natrium-ratiopharm ® sollte nicht angewendet werden bei: – Verdacht auf Malignom mit Blutungsneigung – Nieren- und Harnleitersteinen – chronischem Alkoholismus Eine besonders sorgfältige ärztliche Überwachung ist erforderlich: – während der Schwangerschaft, insbesondere bei längerer Anwendung (siehe Abschnitt 4.6) – bei älteren Patienten, insbesondere bei Frauen – bei gleichzeitiger Behandlung mit Fibrinolytika oder oralen Antikoagulanzien, 2 mit Thrombozytenaggregationshemmern (wie z. B. Acetylsalicylsäure, Ticlopidin, Clopidogrel) und/oder Glykoprotein-IIb/ IIIa-Rezeptorantagonisten – bei gleichzeitiger Anwendung von Arzneimitteln, die den Serum-Kaliumspiegel erhöhen. Die Serum-Kaliumspiegel sollten bei entsprechenden Risikopatienten (z. B. aufgrund von Diabetes mellitus, Einschränkung der Nierenfunktion oder Einnahme von Arzneimitteln, die den SerumKaliumspiegel erhöhen) kontrolliert werden. Während der Behandlung mit Heparin-Natrium sind i.m.-Injektionen wegen der Gefahr von Hämatomen zu vermeiden. Treten unter Heparin-Gabe thromboembolische Komplikationen auf, muss differenzialdiagnostisch an eine Heparin-induzierte Thrombozytopenie Typ II gedacht und die Thrombozytenzahl kontrolliert werden. Bei Säuglingen, Kindern und Patienten mit Nieren- und/oder Leberinsuffizienz ist eine sorgfältige Überwachung und Kontrolle der Gerinnungswerte erforderlich; dies gilt auch für die Thromboembolieprophylaxe („lowdose“-Behandlung). Patienten unter Heparin-Therapie (über 22.500 I. E./Tag) sollten keiner Verletzungsgefahr ausgesetzt werden. Heparin kann die Menstruationsblutung verstärken und verlängern. Bei ungewöhnlich starken oder azyklischen Blutungen sollte eine behandlungsbedürftige organische Ursache durch eine ergänzende gynäkologische Untersuchung ausgeschlossen werden. Für unfraktionierte wie auch für fraktionierte, niedermolekulare Heparine wurde, insbesondere bei intravenöser Applikation bzw. bei Applikation höherer Dosen als zur „lowdose“-Thromboembolieprophylaxe empfohlen (über 15.000 I. E. unfraktioniertes Heparin pro Tag s.c. hinaus), im zeitlichen Zusammenhang mit einer Spinal- oder Periduralanästhesie in Einzelfällen über das Auftreten von spinalen und periduralen Hämatomen berichtet. Diese können zu neurologischen Komplikationen unterschiedlicher Ausprägung bis hin zu lang dauernder oder permanenter Paralyse führen. Heparin-Natriumratiopharm ® soll deshalb bei geplanten oder bereits durchgeführten rückenmarksnahen Anästhesieverfahren erst nach eingehender individueller Nutzen-Risiko-Abwägung eingesetzt werden. Entsprechend einer Empfehlung der Deutschen Gesellschaft für Anästhesie und Intensivmedizin soll zur Sicherheit ein punktionsfreies Intervall von 4 Stunden zwischen letzter Gabe von Heparin-Natriumratiopharm ® in prophylaktischer Dosierung („low-dose“) und der Neuanlage bzw. dem Entfernen eines Spinal-/Periduralkatheters eingehalten werden. Danach soll eine erneute Gabe von niedrig dosiertem HeparinNatrium-ratiopharm ® frühestens nach 1 Stunde erfolgen. Die Patienten sind nach Anwendung eines rückenmarksnahen Anästhesieverfahrens sorgfältig neurologisch zu überwachen, wobei insbesondere auf persistierende sensorische oder motorische Ausfälle zu achten ist. Bei klinischem Verdacht auf ein rückenmarksnahes Hämatom sind unverzüglich geeignete diagnostische und/oder therapeutische Maßnahmen einzuleiten. Hinweise zu labordiagnostischen Untersuchungen: Kontrollen der Thrombozytenzahl sollen erfolgen: – vor Beginn der Heparin-Gabe – am 1. Tag nach Beginn der Heparin-Gabe – anschließend während der ersten 3 Wochen regelmäßig alle 3 – 4 Tage – am Ende der Heparin-Therapie Heparin kann die Ergebnisse zahlreicher Laboruntersuchungen verfälschen, z. B. die Blutsenkungsgeschwindigkeit, Erythrozyten-Resistenz und Komplementbindungstests. Heparin kann die Prothrombinzeit beeinflussen; dies ist bei der Einstellung auf Cumarin-Derivate zu beachten. Unter Heparin-Therapie können die Ergebnisse von Schilddrüsenfunktionsuntersuchungen verfälscht werden (z. B. fälschlich hohe T3- und T4-Spiegelmessungen). 4.5 Wechselwirkungen mit anderen Arzneimitteln und sonstige Wechselwirkungen Thrombozytenaggregationshemmer (Acetylsalicylsäure, Ticlopidin, Clopidogrel, Dipyridamol in hohen Dosen), Fibrinolytika, andere Antikoagulanzien (Cumarin-Derivate), nicht-steroidale Antiphlogistika (Phenylbutazon, Indometacin, Sulfinpyrazon), Glykoprotein-IIb/IIIa-Rezeptorantagonisten, Penicillin in hohen Dosen, Dextrane: Klinisch bedeutsame Wirkungsverstärkung und erhöhte Blutungsgefahr. Zytostatika Wirkungsverstärkung von Heparin; Doxorubicin schwächt die Wirkung wahrscheinlich ab. Nitroglycerin, intravenös appliziert Unter intravenöser Gabe von Nitroglycerin kann es zu einer klinisch bedeutsamen Wirkungsabschwächung von Heparin kommen. Nach Absetzen von Nitroglycerin kann es zu einem sprunghaften Anstieg der aPTT kommen. Engmaschige Kontrollen der aPTT und eine Dosisanpassung von Heparin sind bei gleichzeitiger Infusion von Nitroglycerin notwendig. Ascorbinsäure, Digitalis, Tetracycline, Nikotinabusus Hemmung der Heparin-Wirkung. An Plasmaproteine gebundene Arzneimittel (z. B. Propranolol) Wirkungsverstärkung durch Verdrängung aus der Plasma-Eiweißbindung. Arzneimittel, die den Serum-Kaliumspiegel erhöhen Arzneimittel, die den Serum-Kaliumspiegel erhöhen, dürfen nur unter besonders sorgfältiger medizinischer Überwachung gleichzeitig mit Heparin-Natrium-ratiopharm ® angewendet werden. Fachinformation Heparin-Natrium-5 000-ratiopharm® Basische Arzneimittel (trizyklische Psychopharmaka, Antihistaminika und Chinin) Gegenseitige Wirkungsabschwächung durch Salzbildung mit Heparin. Selten: 4.6 Fertilität, Schwangerschaft und Stillzeit Schwangerschaft Heparin ist nicht plazentagängig. Es gibt bisher keine Berichte darüber, dass die Anwendung von Heparin in der Schwangerschaft zu Missbildungen führt. Tierexperimentelle Untersuchungen haben ebenfalls keine Hinweise auf fruchtschädigende Einflüsse ergeben (siehe 5.3). Es liegen jedoch Berichte über ein erhöhtes Risiko von Aborten und Frühgeburten vor. Behandlungs- oder krankheitsbedingte Komplikationen sind bei Schwangeren nicht auszuschließen. Tägliche hochdosierte Heparin-Gaben über einen Zeitraum von mehr als 3 Monaten können bei Schwangeren das Osteoporoserisiko erhöhen. Unter der Geburt ist die epidurale Anästhesie bei Schwangeren, die mit Antikoagulanzien behandelt werden, kontraindiziert. Ebenso ist die gerinnungshemmende Therapie bei Blutungsneigung, wie z. B. bei Abortus imminens, kontraindiziert (siehe 4.3). Stillzeit Heparin geht nicht in die Muttermilch über. Tägliche hochdosierte Heparin-Gaben über einen Zeitraum von mehr als 3 Monaten können bei stillenden Frauen das Osteoporoserisiko erhöhen. 4.7 Auswirkungen auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen Es sind keine Auswirkungen auf die Verkehrstüchtigkeit und das Bedienen von Maschinen bekannt. 4.8 Nebenwirkungen Unter der Therapie mit Heparin-Natriumratiopharm ® können die nachfolgend genannten Nebenwirkungen auftreten. Bei den Häufigkeitsangaben zu Nebenwirkungen werden folgende Kategorien zugrunde gelegt: sehr häufig häufig gelegentlich selten sehr selten nicht bekannt ≥ 1/10 ≥ 1/100 bis <1/10 Erkrankungen des Immunsystems Gelegentlich: Allergische Reaktionen mit Symptomen wie: Übelkeit, Kopfschmerzen, Temperaturanstieg, Gliederschmerzen, Urtikaria, Erbrechen, Pruritus, Dyspnoe, Bronchospasmen und Blutdruckabfall. Lokale und generalisierte Hypersensibilität, einschließlich Angioödem, reversible Alopezie, Hautnekrosen. Sehr selten: Auftreten eines anaphylaktischen Schocks, insbesondere bei sensibilisierten Patienten, die früher bereits Heparin erhalten haben. Calcinosis am Injektionsort tritt sehr selten, hauptsächlich bei Patienten mit schwerem Nierenversagen, auf. Endokrine Erkrankungen Selten: Hypoaldosteronismus, verbunden mit Hyperkaliämie und metabolischer Azidose, besonders bei Patienten mit Einschränkung der Nierenfunktion und Diabetes mellitus. ≥ 1/1.000 bis <1/100 Erkrankungen des Nervensystems ≥ 1/10.000 bis <1/1.000 In seltenen Fällen wurde bei der Verwendung von Heparin-Natrium im Zusammenhang mit einer Spinal- oder Epiduralanästhesie oder postoperativen Verweilkathetern über spinale und epidurale Hämatome berichtet. Diese Ereignisse haben zu neurologischen Komplikationen unterschiedlicher Ausprägung wie zum Beispiel langdauernder oder permanenter Paralyse geführt (siehe auch 4.4) <1/10.000 Häufigkeit auf Grundlage der verfügbaren Daten nicht abschätzbar Erkrankungen des Blutes und des Lymphsystems Januar 2013 pervermittlung (Thrombozytenzahl: 100.000 – 150.000/µl), ohne Thrombose. Heparin-induzierte, antikörpervermittelte Thrombozytopenie Typ II (Thrombozytenzahl: <100.000/µl oder einem schnellen Abfall der Thrombozytenzahl auf <50 % des Ausgangswertes), mit arteriellen und venösen Thrombosen oder Embolien, Verbrauchskoagulopathie, evtl. Hautnekrosen an der Injektionsstelle, Petechien, Purpura und Melaena. Bei nicht Sensibilisierten beginnt der Thrombozytenabfall in der Regel 6 – 14 Tage nach Behandlungsbeginn, bei Sensibilisierten unter Umständen innerhalb von Stunden. Dabei kann die blutgerinnungshemmende Wirkung des Heparins vermindert sein (Heparin-Toleranz). Sehr häufig: In Abhängigkeit von der Heparin-Dosierung vermehrtes Auftreten von Blutungen, insbesondere aus Haut, Schleimhäuten, Wunden, Gastrointestinal- und Urogenitaltrakt. Häufig: Zu Beginn der Behandlung, Heparin-induzierte Thrombozytopenie Typ I ohne Antikör- Gefäßerkrankungen Sehr selten: Vasospasmen Leber- und Gallenerkrankungen Sehr häufig: klinisch nicht bedeutsamer, in der Regel reversibler Anstieg der Serum-Transaminasen (GOT, GPT), GammaGlutamyl-Transpeptidase (Gamma-GT) sowie LDH und Lipase. Erkrankungen der Geschlechtsorgane Sehr selten: Priapismus Skelettmuskulatur-, Bindegewebs- und Knochenerkrankungen Nach längerer Anwendung (Monate) kann sich, vor allem bei Verwendung höherer Dosierungen und insbesondere bei dazu disponierten Patienten, eine Osteoporose entwickeln. Allgemeine Erkrankungen und Beschwerden am Verabreichungsort Häufig kommt es an der Injektionsstelle zu lokalen Gewebsreaktionen (Verhärtungen, Rötungen, Verfärbungen und kleineren Hämatomen). 4.9 Überdosierung a) Symptome einer Überdosierung Blutungen zumeist aus Haut und Schleimhäuten, Wunden, Gastrointestinal- und Urogenitaltrakt (Epistaxis [Nasenbluten], Hämaturie, Melaena, Hämatome, Petechien). Blutdruckabfall, Abfall des Hämatokrits oder andere Symptome können Zeichen einer okkulten Blutung sein. b) Therapie einer Überdosierung Leichte Blutungen Gegebenenfalls Reduzierung der HeparinDosis. Mäßig, vital nicht bedrohliche Blutungen Unterbrechen der Heparin-Therapie. Ernstere, vital bedrohliche Blutungen Aufhebung der Heparin-Wirkung mit Protamin nach Ausschluss anderer Blutungsursachen (z. B. Verbrauchskoagulopathie, Faktorenmangel). Protamin soll nur bei lebensbedrohlichen Blutungen verabreicht werden, da bei vollständiger Neutralisierung des Heparins ein erhöhtes Risiko für thromboembolische Komplikationen besteht. Der Patient muss unter intensivmedizinischen Bedingungen überwacht und weiterversorgt werden. Das Antidot Protamin ist ein argininreiches Protein, das üblicherweise als Chlorid oder Sulfat verwendet wird. Als Regel gilt, dass 1 mg Protamin die Wirkung von ca. 100 I. E. Heparin neutralisiert (1 I. E. Protamin neutralisiert 1 I. E. Heparin). Für die Therapie muss die Halbwertszeit des Heparins und die Applikationsart berücksichtigt werden, d. h. – 90 min nach intravenöser Heparin-Applikation sollen nur 50 % der errechneten Protamin-Menge gegeben werden – 3 h nach der intravenösen Verabreichung nur 25 % Bei Übertitrierung kann Protamin über verschiedene Mechanismen selbst eine verstärkte Blutungsneigung verursachen. Bei zu rascher i.v.-Injektion von Protamin kann es zu Blutdruckabfall, Bradykardie, Dyspnoe und Beklemmungsgefühl kommen. Protamin wird rascher aus dem Blut eliminiert als Heparin. Die Wirkung der Neutralisation muss daher durch regelmäßige Bestimmungen der aktivierten partiellen Thromboplastinzeit (aPTT) überprüft werden. Heparin ist nicht dialysierbar. 3 Fachinformation Heparin-Natrium-5 000-ratiopharm® 5. PHARMAKOLOGISCHE EIGENSCHAFTEN 5.3 Präklinische Daten zur Sicherheit 5.1 Pharmakodynamische Eigenschaften Pharmakotherapeutische thrombotische Mittel ATC-Code: B01AB01 Gruppe: Anti- Heparin ist ein Mucopolysaccharid-Polyschwefelsäure-Ester und besteht aus Glukosamin-N-Schwefelsäure und Schwefelsäure-Estern der Glukuronsäure, die glykosidisch miteinander verknüpft sind. Heparin bildet aufgrund seiner starken negativen Ladung mit bestimmten Proteinen Komplexe und verändert so deren biologische Eigenschaften. Dies trifft vor allem für das Antithrombin III (ATIII) zu, das durch seine Komplexbildung mit Heparin eine Aktivitätssteigerung etwa um das 700-fache erfährt. Aktiviertes ATIII bewirkt eine Hemmung von Serinproteasen, zu denen auch die Gerinnungsfaktoren XIIa, XIa, Xa, VIIa und IIa gehören. Dabei werden Faktor VIIa relativ schwach und Faktor IIa (Thrombin) besonders stark durch Heparin-ATIII-Komplex inhibiert. Bereits niedrige Heparin-Dosen beschleunigen die Inhibition von ATIII gegenüber Faktor IIa (Thrombin) und Faktor Xa. So erklärt sich die prophylaktische Wirkung von niedrig dosiertem („low-dose“) Heparin zur Vorbeugung von thromboembolischen Erkrankungen. Die gerinnungshemmende Wirkung hängt vor allem von der verfügbaren Menge von ATIII und der Fibrinogenkonzentration ab; bestimmte Thrombozyteninhaltsstoffe (Plättchenfaktor 4) neutralisieren ebenfalls die Heparin-Wirkung. Hohe Heparin-Dosen inaktivieren zusätzlich im Überschuss gebildetes Thrombin und verhindern so die Entstehung von Fibrin aus Fibrinogen. Heparin beeinflusst auch die Thrombozytenfunktionen. 5.2 Pharmakokinetische Eigenschaften Heparin kann subkutan oder intravenös verabreicht werden. Wegen seiner Molekülgröße und negativen Oberflächenladung wird Heparin vom Darm nicht resorbiert, eine inhalative Aufnahme ist möglich. Die Wirkung von Heparin setzt nach intravenöser Gabe sofort ein, nach subkutaner Injektion innerhalb von 20 – 30 min. Die Bioverfügbarkeit nach subkutaner Anwendung ist individuell verschieden. Nach subkutaner Applikation von 2-mal täglich 5.000 I. E. Heparin wurden Plasmaspiegel zwischen 0,02 I. E./ ml und 0,8 I. E./ml gemessen. Die interindividuelle Halbwertszeit ist sehr variabel, die mittlere Halbwertszeit wird mit 90 – 120 min angegeben und ist abhängig von der Dosis und der Funktion von Leber und Nieren sowie der Komorbidität. Heparin wird in hohem Maße an Plasmaproteine gebunden (LDL, Globuline [insbesondere ATIII] und Fibrinogen), das Verteilungsvolumen bei Erwachsenen wird mit ca. 0,07 l/kg angegeben. Nach parenteraler Gabe wird Heparin aus dem Blut durch Aufnahme in das retikuloendotheliale System, durch Spaltung in der Leber (Heparinasen) und durch Ausscheidung über den Urin überwiegend als depolymerisiertes, inaktiviertes Heparin eliminiert. Die Heparin-Ausscheidung erfolgt sowohl durch glomeruläre Filtration als auch durch tubuläre Sekretion. 4 In tierexperimentellen Untersuchungen traten nur Effekte (Osteoporose und Blutungen) auf, die bereits unter Abschnitt 4.8 beschrieben sind. Aus In-vitro- und In-vivoUntersuchungen auf genotoxische Wirkungen haben sich keine Hinweise auf ein mutagenes Potenzial ergeben. Untersuchungen zum tumorerzeugenden Potenzial wurden nicht durchgeführt. Tierexperimentelle Untersuchungen haben keine Hinweise auf fruchtschädigende Einflüsse ergeben. 6. PHARMAZEUTISCHE ANGABEN 6.1 Liste der sonstigen Bestandteile Wasser für Injektionszwecke, zur pH-Einstellung: Natriumhydroxid, Schwefelsäure 96%. 6.2 Inkompatibilitäten Wegen der Gefahr physikalisch-chemischer Inkompatibilitäten darf Heparin nicht zusammen mit anderen Arzneimitteln in einer Spritze aufgezogen oder in einer Infusion verabreicht werden. 6.3 Dauer der Haltbarkeit Ampullen 5 Jahre Fertigspritzen 3 Jahre 6.4 Besondere Vorsichtsmaßnahmen für die Aufbewahrung Die Injektionslösung kann sich nach längerer Lagerung dunkler färben, die therapeutische Wirkung wird dadurch nicht beeinträchtigt. 6.5 Art und Inhalt des Behältnisses Packung mit 5 Ampullen zu 0,2 ml Injektionslösung Packung mit 20 Ampullen zu 0,2 ml Injektionslösung Packung mit 10 Fertigspritzen zu 0,2 ml Injektionslösung 6.6 Besondere Vorsichtsmaßnahmen für die Beseitigung und sonstige Hinweise zur Handhabung Keine besonderen Anforderungen. 7. INHABER DER ZULASSUNG ratiopharm GmbH Graf-Arco-Str. 3 89079 Ulm 8. ZULASSUNGSNUMMER 5394.00.00 9. DATUM DER ERTEILUNG DER ZULASSUNG/VERLÄNGERUNG DER ZULASSUNG Datum der Erteilung der Zulassung: 1. Oktober 1984 Datum der Verlängerung der Zulassung: 25. August 2005 10. STAND DER INFORMATION Januar 2013 11. VERKAUFSABGRENZUNG Verschreibungspflichtig Zentrale Anforderung an: Rote Liste Service GmbH | FachInfo-Service Postfach 11 01 71 10831 Berlin |