N-Methylcarbamoylphosphate, I. Synthese
Werbung

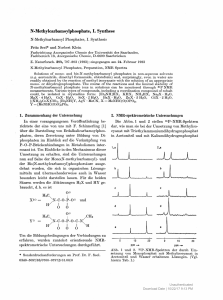
N-Methylcarbamoylphosphate, I. Synthese N-Methylcarbamoyl Phosphates, I. Synthesis Fritz Seel* und Norbert Klein Fachrichtung Anorganische Chemie der Universität des Saarlandes, Fachbereich 13, Anorganische Chemie, D-6600 Saarbrücken Z. Naturforsch. 38 b, 797-803 (1983); eingegangen am 24. Februar 1983 N-Methylcarbamoyl Phosphates, Preparation, NMR Spectra Solutions of mono- and bis-N-methylcarbamoyl phosphates in non-aqueous solvents [e.g. acetonitrile, dimethyl formamide, chloroform) and, surprisingly, even in water are readily obtained by the reaction of methyl isocyanate with the solution of an appropriate mono- or dihydrogenphosphate. The course of the reactions and the limited stability of N-methylcarbamoyl phosphate ions in solutions can be monitored through 31 P NMR measurements. Various types of compounds, including a coordination compound of cobalt could.be isolated in crystalline form: [Et 3 NH]HX, K H X , N H 4 H X , Na 2 X • H 2 0 , MgX • 4 H 2 0 , CaX • H 2 0 , SrX • 2 H a O, BaX • H 2 0 , Z n X • 5 H 2 0 , CdX • 2 H 2 0 , [(NH3)5COX]C104, [EtsNHjY, AgY • MeCN, X = M e N H C ( 0 ) 0 P 0 3 , Y = [MeNHC( 0 ) 0] 2 P0 2 . 1. Zusammenhang der Untersuchung 2. NMR-spektrometrische Untersuchungen In einer vorangegangenen Veröffentlichung berichtete der eine von uns mit F. Schinnerling [1] über die Darstellung von Erdalkalicarbamoylphosphaten, deren Zersetzung unter Bildung v o n Diphosphaten im Hinblick auf die Verknüpfung von P-O-P-Brückenbindungen in Metabolismen interessant ist. Um Einblicke in den Mechanismus dieser Umsetzung zu erhalten, sind die Untersuchungen nun auf Salze der Mono (N-methylcarbamoyl)- und der Bis(N-methylcarbamoyl)phosphorsäure ausgedehnt worden, die sich in organischen Lösungsmitteln und überraschenderweise auch in Wasser besonders leicht darstellen lassen. Für die beiden Säuren werden die Abkürzungen H 2 X und H Y gebraucht, d.h. es ist Die A b b n . 1 und 2 stellen 3 1 P-NMR-Spektren dar, wie man sie bei der Umsetzung von Methylisocyanat mit Triethylammoniumdihydrogenphosphat in Acetonitril und mit Kaliumdihydrogenphosphat X = 1 1 ^N-C-O-P-O© II II H' 0 0 09 | H3CL Y® = J 2,a 0© H3CX 2e 1 ,a und /CH3 ;N-C-O-P-O-C-N( H 1 0 Ji 0 JI 0 X H U m die Bildungsbedingungen der Verbindungen zu erfahren, wurden zunächst orientierende N M R spektrometrische Untersuchungen durchgeführt. * Sonderdruckanforderungen an Prof. Dr. F. Seel. 0340-5087/83/0700-0797/$ 01.00/0 10 ppm 20 - 10 20 ppm Abb. 1 und 2. 31 P-NMR-Spektren der durch Umsetzung von Monophosphat mit Methylisocyanat in Acetonitril und Wasser erhaltenen Lösungen. (Vgl. hierzu Tab. I.) Dieses Werk wurde im Jahr 2013 vom Verlag Zeitschrift für Naturforschung in Zusammenarbeit mit der Max-Planck-Gesellschaft zur Förderung der Wissenschaften e.V. digitalisiert und unter folgender Lizenz veröffentlicht: Creative Commons Namensnennung-Keine Bearbeitung 3.0 Deutschland Lizenz. This work has been digitalized and published in 2013 by Verlag Zeitschrift für Naturforschung in cooperation with the Max Planck Society for the Advancement of Science under a Creative Commons Attribution-NoDerivs 3.0 Germany License. Zum 01.01.2015 ist eine Anpassung der Lizenzbedingungen (Entfall der Creative Commons Lizenzbedingung „Keine Bearbeitung“) beabsichtigt, um eine Nachnutzung auch im Rahmen zukünftiger wissenschaftlicher Nutzungsformen zu ermöglichen. On 01.01.2015 it is planned to change the License Conditions (the removal of the Creative Commons License condition “no derivative works”). This is to allow reuse in the area of future scientific usage. 798 F. Seel-N. Klein • N-Methylcarbamoylphosphate bei quantitativer Umsetzung der Isocyanatkomponente erwartet werden kann, d.h. es gilt für die Anteile a an X und Y , wenn n die eingesetzten Stoffmengen sind: a ( X ) + 2 a ( Y ) = w(MeNCO)/w(P). (Vgl. Tab. I, Vers, l a - g . ) Die Beobachtung, daß die Konzentration von Y bei der Umsetzung von H3PO4, E t 3 N und MeNCO im Molverhältnis 1 : 1 : 1 nach Erreichen eines Maximums wieder abnimmt und die v o n X zunimmt (vgl. Vers. 1, c und f ) ist auf die Reaktion von Y~ mit noch nicht umgesetztem Dihydrogenphosphat zurückzuführen, die sich an (1) und (2) anschließt: PH 10 8 6 4 0 5 10 15 ml —» 20 Abb. 3. pH-Änderungen bei Titrationen von Monokalium- bzw. Dinatrium-N-methylcarbamoylphosphat mit 0,1-molarer Natronlauge (a) bzw. Salzsäure (b) bei 20 °C. bzw. Kaliummonohydrogenphosphat in Wasser erhalten kann. Die Einwirkung von MeNCO auf Dihydrogenphosphate ergibt zwei neue Signale mit Hochfeldverschiebung, wobei ein Überschuß an MeNCO die Substanz begünstigt, deren 3 1 P - N M R Signal v o m Phosphatsignal weiter entfernt ist. Die nachfolgend beschriebenen Präparationen ergaben eindeutig, daß es sich bei den entstehenden Teilchen um H X - bzw. X 2 - und Y " handelt. In Acetonitril verhalten sich die Signalflächen bei der Umsetzung von Dihydrogenphosphat bereits nach kurzer Reaktionszeit ( < 2 0 min) so, wie auf Grund der Gleichungen PO4H2- + MeNCO -> X H - (1) X H - + MeNCO (2) und Versuche in MeCN n(H 3 P0 4 )/mmol n(NEt3)/mmol n(MeNCO)/mmol t/min a(P04)[%] a(X) [%] a(Y) [%] Versuche in H2O n(K 2 HP0 4 )/mmol n(K 2 HP0 4 )/mmol n(MeNCO)/ mmol t/min a(P0 4 ) [ % ] a(X) [%] a(Y) [%] - * Y~ 1, a b c d e 25 25 16 20 40 50 10 25 25 25 20 20 60 20 25 25 32 20 7 53 40 25 25 40 20 b c d e 20 20 20 20 - - - - 25 25 8 20 70 30 - 2, a 20 20 34 32 34 20 240 30 62 8 40 20 - 30 70 40 240 14 80 6 Y - + P 0 4 H 2 - -> 2 X H - . Die Inhibierung der Bildung von Y~ in einer Lösung v o n [Et3NH]H2P04 durch überschüssiges Amin (Vers. 1, c und g) erklärt eine, zumindest teilweise, durch eine Tieffeldverschiebung des 3 1 P - N M R Signals angedeutete, Deprotonierung des Anions H X - . Bemerkenswert ist, daß präparativ isoliertes [EtaNHJHX in Acetonitril-Lösung in geringem Maße (d.h. zu etwa 5 % ) im Sinne einer Umkehrung v o n (3) disproportioniert. Gleichzeitig bildet sich langsam Polyphosphat. In Wasser setzt sich MeNCO auch mit Dihydrogenphosphaten sehr rasch, d . h . innerhalb von 10 bis 20 min, bei äquimolarem Ansatz, quantitativ zu N-Methylcarbamoylphosphaten um, wie die Gültigkeit der für einen vollständigen Umsatz des Isocyanats abgeleiteten Beziehung zeigt (vgl. Vers. 2, a). I m Laufe der Zeit (vgl. Vers. 2, b) wird jedoch a ( X ) + 2 a ( Y ) kleiner als w(MeNCO)/n(P), wobei f - 33 67 _ 20 32 20 - 100 - (3) 25 25 25 240 10 80 10 g 25 50 25 20 7 88 5 Tab. I. Ergebnisse der Umsetzungen von Triethylammonium- und Kaliumhydrogenphosphaten mit Methylisocyanat in Acetonitril bzw. Wasser. Lösungsmittel volumen 10 ml. a = Stoffmengenanteile in Mol-%, t = Reaktionszeit. 799 F. Seel-N. Klein • N-Methylcarbamoylphosphate gleichzeitig a ( X ) anwächst. Dies deutet auf eine von X Carbamoylierung v o n Phosphat durch Y~ entspre- zwischen 15 und 18 p p m (bez. auf 85-proz. H3PO4 chend (3) und eine Hydrolyse v o n Y ~ : extern). Die A r t des Kations und die Ionenstärke Y - + H 2 0 -> X H - + H O • CO • N H M e - > X 2 - + C 0 2 + MeNH 3 +. (4) Bei der Umsetzung v o n Monohydrogenphosphat in wäßriger Lösung entsteht auch bei Einsatz eines Überschusses an MeNCO kein Y _ (vgl. Vers. 2, e) weil das aus HPO4 2 "" gebildete X 2 - nicht mit dem I s o c y a n a t weiterreagieren kann. (Gibt m a n zu einer wäßrigen Natrium-, Kalium- oder Ammonium - monohydrogenphosphatlösung einen Überschuß an M e N C O und schüttelt kräftig, so wird das H y d r o genphosphation quantitativ in X 2 - übergeführt.) I m Hinblick darauf, daß die Umsetzung von Phosphat mit Cyanat in wäßriger Lösung selbst unter optimalen pH-Bedingungen nur zu einem Gleichgewicht führt, das kaum mehr als 5 0 % Carbam o y l p h o s p h a t liefert, ist es zunächst erstaunlich, d a ß die Umsetzung v o n phosphat ergibt und die v o n sogar Y wäßriger Lösungen üben einen geringen Einfluß aus. Sehr stark ändert sich d( 3 1 P), wenn X 2 ~ Ligand des C o b a l t ( I I I ) - I o n s ist. Die Koordinationsverschiebung des 3 1 P-Signals so groß ( ~ — 6 p p m ) ist jedoch nicht ganz wie bei anderen Phosphato-cobalt(III)- K o m p l e x e n (7 bis 11 p p m ) [2], D a t e n der ! H - N M R - und der 1 3 C-NMR-Spektren der Lösungen v o n [ E t 3 N H ] H X und [ E t 3 N H ] Y in CDC1 3 sind in der T a b . I I I zusammengestellt. Ergänzend hierzu sei mitgeteilt, daß die K o p p l u n g der an C und N gebundenen Protonen der C H 3 N H Gruppen an der Aufspaltung der entsprechenden Signale erkennbar ist ( J = 4,7 Hz). Ein Zusatz v o n nur 2 % Methanol hebt diese auf. Die Signale der schnell austauschenden Protonen der P O H - G r u p p e v o n H X - und der Et 3 NH+-Ionen sind stark verbreitert. Monohydrogenphosphat mit Methylisocyanat 1 0 0 % Mono(N-methylcarbamoyl)phosphat liegen zwischen 1 und 7 p p m , die v o n darüber DihydrogenBis(N- Bereits Allen, Richelson und Jones [3] versuchten methylcarbamoyl)phosphat. Die Erklärung hierfür 1966 das Triethylammoniumsalz [ E t 3 N H ] H X nach ist, daß Methylisocyanat im Gegensatz zur Cyan- einer v o n Cramer und Winter [4] zur Darstellung säure nicht protolytisch desaktiviert werden kann. N-substituierter Carbamoylphosphate entwickelten Die NMR-spektrometrischen B e f u n d e ergaben die M e t h o d e zu erhalten. D a es ihnen nicht gelang, das Grundlagen für die nachfolgend beschriebenen Prä- Salz aus einer Lösung in Acetonitril auszukristalli- parationen. sieren, fällten sie das Dilithiumsalz L i 2 X • H 2 0 aus. 31P-NMR-Daten hinausgehend 3. Darstellung von Mono (N-methylcarbamoyl) phosphaten v o n Lösungen präpara- t i v isolierter N-Methylcarbamoylphosphate sind in Porter, M o d e be u n d Stark [5] wiesen nach, daß das der T a b . I I zusammengestellt. Protonen bewirken P r o d u k t reichlich Monophosphat enthielt. (Dies ist keine Aufspaltung der 31 P-Signale. Deren Lage hängt verständlich, weil sich [ E t 3 N H ] H 2 P 0 4 und MeNCO im Falle v o n X ebenso wie bei PO4 v o m Grad der in äquimolarem Verhältnis nicht quantitativ Protonierung des Teilchens ab, und zwar in der [Et3NH]HX Weise, daß diese das Signal zu hohem Feld ver- gelang es ihnen durch schiebt. Die chemischen Verschiebungen der Signale graphie Li2X • H2O mit einer Reinheit v o n 9 0 % zu umsetzen.) In umständlicher zu Weise Ionenaustauschchromato- Tab. II. 3 1 P-NMR-Daten von N-Methylcarbamoylphosphaten (positive (5-Werte bedeuten Hochfeldverschiebung bez. auf 85-proz. H 3 P04). Lösemittel: [Et3NH]HX KHX NH4HX Na2X • H 2 0 [(NH 3 ) 5 COX]C10 4 [Et 3 NH]Y KY A g Y • MeCN H20 — MeOH MeC0 2 H DMSO NC5H5 6,8 6,0 6,5 L6 4,3 16,0 MeCN 16,2 18,0 17,8 18,0 16,4 15,1 800 F. Seel-N. Klein • N-Methylcarbamoylphosphate Tab. III. iH- und isC-NMR-Daten von [Et 3 NH]HX und [Et 3 NH]Y. (Lösemittel: CDC13, 6 in ppm, bezogen auf TMS). Daten des Et 3 NH+-Ions: Anion <5h(CH3) <5H(CH2) <5H(NH, OH) öc (CH3) <5C(CH2) XHY- 3,14 3,14 9,7 10,5 8,58 8,58 45,9 46,0 Daten der MeNHCO-Gruppe: Anion <5H (CH3) <5H(NH) <5C(CH3) (5C (CO) XHY- 6,9 6,9 27,6 27,6 153,1 152,0 1,31 1,33 2,76 2,77 präparieren. Tatsächlich ist [ E t 3 N H ] H X recht einfach in reiner Form zu erhalten: Man stellt zunächst eine Lösung von Triethylammoniumdihydrogenphosphat in Acetonitril her und setzt diese mit Methylisocyanat in äquimolarer Menge um. Das Produkt kristallisiert bei Wahl geeigneter Konzentrationsverhältnisse aus der Lösung beim Stehen im Kühlschrank aus. In analoger Weise lassen sich weitere Salze isolieren. So erhält man die Zinkverbindung, wenn man Phosphorsäure, Triethylamin und Methylisocyanat im Mol Verhältnis 1 : 2 : 1 , 6 in Wasser zu X 2 ~ umsetzt und eine wäßrige Zinknitratlösung zufügt. Bei Zusatz von Acetonitril fällt Z n X • 5 H 2 0 in der Kälte aus. Auf die gleiche Weise läßt sich auch C d X • 2 H 2 0 darstellen. Versuche zur Isolierung der Kalium- und der Ammoniumsalze von H 2 X ergaben, daß nur die Hydrogensalze K H X und N H 4 H X gut kristallisieren. Es erwies sich für die Darstellung dieser Substanzen als vorteilhaft, Monohydrogen-, Dihydrogenphosphat und Methylisocyanat im Molverhältnis 1 : 1 : 3 umzusetzen und die Verbindungen durch Zugabe von Methanol bei 0 °C zur Kristallisation zu bringen. Bei dem Versuch, in analoger Weise das Natriumhydrogensalz zu isolieren, kristallisierte wider Erwarten das Dinatriumsalz N a 2 X • H 2 0 aus. Zu seiner Darstellung setzt man deshalb besser Dinatriumhydrogenphosphat ein. Die Kalium- und Ammonium Verbindungen, K H X und N H 4 H X , konnten auch aus dem entsprechenden Monohydrogenphosphat erhalten werden, wenn man nach der Carbamoylierung soviel Essigsäure zufügte, wie notwendig war, um X 2 - zu H X - zu protonieren. Auch die komplexgebundene Hydrogenphosphatgruppe kann mittels Methylisocyanats leicht carbamoyliert werden. So gelang die Darstellung des Pentaammin(N-methylcarbamoyl)phosphatocobalt(IH)perchlorats [(NH 3 ) 5 CoX]C10 4 durch Umsetzen des Neutralkomplexes [(NH 3 ) 5 CoP0 4 ] • 2 H 2 0 mit MeNCO nach Zusatz von Essigsäure und Natriumperchlorat. Interessant ist auch die Möglichkeit, in einem Anionenaustauscher gebundenes Hydrogenphosphat durch Zugabe von MeNCO zur Suspension des Harzes in Wasser und kräftiges Schütteln quantitativ zu carbamoylieren. Die Erdalkalisalze der Säure H 2 X können nicht ebenso wie die Kalium-, Ammonium- und Natriumverbindungen durch Carbamoylieren der entsprechenden Phosphate hergestellt werden. Die Synthese dieser Substanzen gelingt jedoch leicht, wenn man von einer wäßrigen Lösung des Salzes K 2 X ausgeht. Bei Zugabe einer wäßrigen Erdalkalisalzlösung kristallisieren M g X - 4 H 2 0 , CaX H 2 0 , S r X - 2 H a O und B a X H 2 0 nach einer gewissen Zeit aus. Die Reindarstellung der Salze ermöglichte die Ermittlung der zweiten Dissoziationskonstante der Säure X H 2 durch potentiometrische Titration wäßriger Lösungen von primären und sekundären NMethylcarbamoylphosphaten mit Natronlauge bzw. Salzsäure. (Vgl. Abb. 3.) Der pK s -Wert von H X ~ ist bei einer Ionenstärke von 0,02 mol/1 5,3. (Der zweite Säureexponent der nicht substituierten Carbamoylphosphorsäure ist 4,9 [3].) 4. Darstellung von Salzen der Bis (N-methylcarbamoyl)phosphorsäure Zunächst wurde versucht, entsprechend der Vorschrift, die Cramer und Winter [4] für die Darstellung N-substituierter Bis-carbamoyl phosphate ange- 801 F. Seel-N. Klein • N-Methylcarbamoylphosphate geben haben, die Kristallisation v o n [Et3NH]Y durch Zugabe v o n Ether auszulösen. Als vorteilhafter erwies es sich, die Umsetzung in Chloroform durchzuführen. Aus der so erhaltenen Lösung scheiden sich nach Zugabe v o n A c e t o n bei 10 °C nach 2 h schöne Kristalle aus. Bei Zugabe der entsprechenden Thiocyanat- Lösungen zu einer Lösung v o n [ E t 3 N H ] Y in MeCN fielen die Natrium-, Kalium-, A m m o n i u m - , Magnesium- und Calciumsalze, N a Y , K Y , N H 4 Y , M g Y 2 und C a Y 2 - allerdings nicht in völlig reiner F o r m aus. ( P M R - und IR-spektroskopisch ließ sich nachweisen, daß die Substanzen infolge Mitfällung stets Triethylammoniumthiocyanat enthielten.) K Y und N H 4 Y konnten aus einer Mischung v o n D i m e t h y l f o r m a m i d und Aceton umkristallisiert werden. Es gelang auch, diese Verbindungen in D M F A herzustellen und anschließend durch Zugabe v o n A c e t o n auszukristallisieren. Die Elementaranalyse lieferte für N H 4 Y befriedigende, f ü r K Y aber keine reproduzierbaren Ergebnisse. Das Silbersalz v o n HY konnte als Solvat A g Y • MeCN rein synthetisiert werden, indem zu einer Lösung v o n [ E t 3 N H ] Y in Acetonitril ebenfalls in MeCN gelöstes Silbernitrat gegeben wurde. Sämtliche Salze der Mono- und Bis(N-methyl- carbamoyl)phosphorsäuren sind bei R a u m t e m p e r a tur mehr oder weniger unbeständig. So zersetzen sich [ E t 3 N H ] H X und [ E t 3 N H ] Y im Verlaufe eines Tages. Die Präparate müssen deshalb im Tiefkühlschrank a u f b e w a h r t werden. 5. Experimenteller Teil A. Präparative Vorschriften Triethylammonium-mono(N-methylcarbamoyl hydrogenphosphat )- I n einem 100 ml fassenden R u n d k o l b e n wurden 100 m m o l (9,8 g) kristalline Phosphorsäure in 10 ml Acetonitril suspendiert und unter kräftigem Rühren langsam mit 100 m m o l (14 ml) wasserfreiem Triethylamin versetzt. N a c h 2 min war eine klare Lösung v o n Triethylammoniumdihvdrogenphosphat in Acetonitril entstanden. I n diese wurden nach dem Abkühlen auf R T (noch b e v o r das Salz auskristallisierte) 100 m m o l (6,2 ml) Methvlisocyanat unter Rühren eingetropft. Bei — 3 0 °C kristallisierte das Endprodukt nach etwa 6 h aus. (Die Kristallisation kann auch verzögert sein.) N a c h dem Stehen über Nacht wurde es abgesaugt u n d mit kaltem Acetonitril nachgewaschen. Die Ausbeute an [ E t 3 N H ] H X betrug 11,5 g ( = 4 5 % bez. auf die eingesetzte Phosphorsäure). Kalium- und Ammonium-mono( N-methylcarbamoyl Jhydrogenphosphat Z u deren Darstellung wurden in einem 250 ml fassenden R u n d k o l b e n mit Magnetrührstäbchen u n d P o l y p r o p y l e n s t o p f e n 50 m m o l KH 2 P(>4 bzw. N H 4 H 2 P 0 4 und 50 m m o l K 2 H P 0 4 bzw. ( N H 4 ) 2 H P 0 4 in 25 ml Wasser gelöst und mit 145 m m o l (9 ml) Methylisocyanat versetzt. I n dem danach verschlossenen Gefäß wurde die Mischung in einem gut ziehenden A b z u g ( M A K v o n MeNCO = 0,02 p p m b z w . 0,05 m g / m 3 ! ) kräftig geschüttelt, wobei durch vorsichtiges L ü f t e n des Stopfens für die Entspannung des entstehenden Kohlendioxidüberdruckes gesorgt wurde. (Die Mischung erwärmte sich u m etwa 10 °C). N a c h 15 min langem Rühren wurde auf etwa 10 °C abgekühlt und die Lösung mit 150 ml Methanol versetzt. I m Eisbad begann die Kristallisation nach 2 bis 3 h. Nach 24 h wurde das P r o d u k t abgesaugt, zweimal mit je 20 ml Methanol gewaschen und in einem Exsikkator durch Evakuieren v o n anhaftendem Lösungsmittel befreit. Die auf eingesetztes Phosphat bezogene Primärausbeute v o n 5 0 % d . T h . (9,75 g K H X bzw. 8,5 g N H 4 H X ) erhöhte sich auf 9 0 % , wenn die Mutterlauge mit 2,85 ml Eisessig sowie weiteren 50 ml Methanol versetzt und zwei Tage lang im Tiefkühlschrank bei — 2 0 °C a u f b e w a h r t wurde. Man kann auch bei sonst gleich bleibender Arbeitsweise v o n 100 m m o l der Monohydrogenphosphate allein ausgehen, m u ß dann aber v o r d e m Methanolzusatz 50 m m o l (2,9 ml) Eisessig zugeben. Dinatrium-mono(N-methylcarbamoyl )phosphat Zunächst wurden wie bei der Darstellung v o n K H X und N H 4 H X beschrieben worden ist, 25 ml einer 1-molaren Lösung v o n N a 2 H P 0 4 mit 40 m m o l (2,5 ml) M e N C O umgesetzt. Z u der entstandenen L ö s u n g wurden bei 0 °C in 25-ml-Portionen 200 ml Methanol zugefügt. N a c h 24 h langem Stehen im Kühlschrank wurde abgesaugt und weiterhin, wie bereits angegeben, verfahren. Primärausbeute 4,6 g ( 8 5 % ) , nach Tiefkühlung der Mutterlauge 5,4 g N a 2 X • H 2 0 (99%). Magnesium-monofN-methylcarbamoyl )phosphat I n einem 100-ml-Rundkolben wurden 5 ml einer 2-molaren L ö s u n g v o n K 2 H P 0 4 mit 16 m m o l (1 ml) M e N C O in der beschriebenen Weise umgesetzt. Z u der entstandenen Lösung v o n 10 m m o l K 2 X wurden unter R ü h r e n eine Lösung v o n 12 m m o l (2,5 g) Mg(SCN) 2 • 4 H 2 0 in 5 ml Wasser und anschließend 10 ml Acetonitril hinzugegeben. (Durch Zusatz v o n Methanol oder Ethanol konnte kein kristallines P r o d u k t erhalten werden.) Nach dem Einstellen des K o l b e n s in Eiswasser kristallisierte das P r o d u k t bald aus u n d konnte nach einem T a g abfiltriert, mit Methanol u n d A c e t o n gewaschen und durch A b kondensieren der Lösungsmittel getrocknet werden. A u s b e u t e : 2,1 g M g X • 4 H 2 0 ( 8 4 % ) . 802 Calciummono( F. Seel-N. Klein • N-Methylcarbamoylphosphate dung mit Ethanol konnten größere Kristalle erhalten werden. Strontium- und BariumN-methylcarbamoyl)phosphat Die Calciumverbindung schied sich als feinkristalliner Niederschlag nach wenigen Minuten aus, wenn zu 10 ml einer 1-molaren Lösung v o n K 2 X unter Rühren tropfenweise eine Lösung 11 m m o l (2,6 g) C a ( N 0 3 ) 2 • 4 H 2 0 in 20 ml Wasser zugegeben wurde. Ausbeute nach dem Waschen mit kaltem Wasser, Methanol und A c e t o n : 8 0 % (1,7 g) Subst. Die Strontium- und die Barium Verbindung m u ß t e n aus verdünnteren Lösungen ausgefällt werden, u m filtrierbare Produkte zu erhalten. Die in 10 ml W a s ser hergestellte Lösung v o n 10 m m o l K 2 X wurde deshalb zunächst mit 40 ml Wasser verdünnt u n d erst danach mit den Lösungen v o n 11 m m o l (2,9 g) SrCl 2 • 6 H 2 0 bzw. 11 m m o l BaCl 2 • 2 H 2 0 (2,7 g) in 40 ml H 2 0 gefällt. S r X • 2 H 2 0 kristallisierte bei 0 °C sehr feinkörnig in kurzer Zeit aus u n d war schwer abzusaugen, B a X • H2O fiel sofort aus. Ausbeuten: S r X • 2 HsO 1,7 g ( 6 1 % , B a X • H 2 0 2,9 g (95%). Zink- und phosphat Cadmium-mono(N-methylcarbamoyl)- Zunächst wurde in einem 100-ml-Rundkolben eine Lösung v o n [ E t 3 N H ] 2 H P 0 4 aus 10 m m o l (0,98 g) kristalliner Monophosphorsäure und 20 m m o l (2,8 ml) Triethylamin in 10 ml Wasser hergestellt. N a c h d e m Abkühlen wurden 16 m m o l (1 ml) MeNCO zugefügt und wie beschrieben umgesetzt. Danach wurde eine Lösung v o n 13 mmol (3,4 g) Z n ( N 0 3 ) 2 • 4 H 2 0 bzw. 11 m m o l (3,4 g) C d ( N 0 3 ) 2 • 4 H 2 0 in 10 ml Wasser zugegeben und schließlich wurden etwa 8 ml A c e t o nitril bis zur schwachen Trübung zugetropft. Ü b e r Nacht bildete sich im Eisschrank ein feinkristalliner Niederschlag, der abfiltriert, gewaschen und durch Abkondensieren des anhaftenden Lösungsmittels getrocknet werden konnte. Ausbeuten: Z n X • 5 H2O 8 1 % (2,5 g), C d X • 2 H 2 0 8 0 % (2,4 g) bez. auf H3PO4. Pentaamin-mono( phosphatocobalt N-methylcarbamoyl (111)-perchlorat )- 1 m m o l (0,27 g) Pentaamminphosphatocobalt( I I I ) - D i h y d r a t wurde in 5 ml Wasser suspendiert und mit 2 Tropfen Eisessig in Lösung gebracht. N a c h Zugabe v o n 1,4 m m o l (0,2 g) NaC10 4 • H 2 0 und 1,6 m m o l MeNCO wurde die zunächst entstandene Emulsion 15 min lang kräftig geschüttelt. Schließlich wurde die gebildete Lösung auf 10 °C abgekühlt und mit 5 ml Ethanol versetzt. Bei weiterem Abkühlen fiel nach wenigen Minuten ein feinkristalliner, rotvioletter Niederschlag aus. N a c h 2 h wurde nochmals 5 ml Ethanol zugegeben, nach 4 h wurde abgesaugt und mehrmals mit Ethanol gewaschen. Lösungsmittelreste wurden durch Evakuieren entfernt. Ausbeute: 0,22 g ( 5 5 % ) [(NH 3 ) 4 COX]C10 4 . Durch Überschichten einer konzentrierten wäßrigen Lösung der K o m p l e x v e r b i n - Triethylammonium-bis( phosphat N-methylcarbamoyl )- I n der gleichen Weise wie bei der Darstellung des Monocarbamoylphosphats beschrieben, wurden 100 m m o l H 3 P 0 4 mit 100 m m o l E t 3 N in 10 ml Chloroform umgesetzt. Es bildete sich eine sirupöse Masse, die sich nur wenig löste. Zu dieser wurden 210 m m o l Methylisocyanat (13 ml) unter Rühren tropfenweise zugegeben. Nach d e m Abkühlen auf 10 °C wurde mit 10 ml A c e t o n verdünnt. Nach etwa 2 h waren schöne, jedoch an der L u f t sehr zerfließ liehe Kristalle entstanden, die nach 12 h abgesaugt, mit kaltem wasserfreiem Chloroform nachgewaschen u n d durch Evakuieren im Exsikkator v o n restlichem Lösungsmittel befreit Avurden. Die Ausbeute an [ E t 3 N H ] Y betrug 25,5 g, d . h . 8 1 % bez. auf die eingesetzte Phosphorsäure. Kalium-bis( N-methylcarbamoyl) phosphat Zur Darstellung der Verbindung in Dimethylf o r m a m i d wurden 10 m m o l (0,98 g) H 3 P 0 4 in 20 ml D M F gelöst und mit 10 m m o l (1,4 ml) Triethylamin sowie 3,2 m m o l (2 ml) MeNCO versetzt. Nach 30 min wurde eine Lösung v o n 1,1 m m o l (1,1 g) K S C N in D M F zugegeben und mit 20 ml A c e t o n verdünnt. N a c h 1 h begann sich ein fest an der Gefäßwand haftendes Kristallisat zu bilden. Nach 12 h wurde abgesaugt und das P r o d u k t zweimal mit je 10 ml A c e t o n , das 0,1 ml MeNCO enthielt, gewaschen und i m Vakuumexsikkator getrocknet. Ausbeute an K Y : 2,2 g = 8 8 % bez. auf eingesetzte Phosphorsäure. Silber-bis( N-methylcarbamoyl )phosphat 10 m m o l (0,98 g) H 3 P 0 4 wurden in 20 ml MeCN suspendiert und nach Zugabe v o n 10 mmol (1,4 ml) N E t 3 mit 24 m m o l (1,5 ml) MeNCO bei etwa 25 °C zu [ E t 3 N H ] Y umgesetzt. N a c h 30 min wurde eine Lösung v o n 10 m m o l (1,7 g) A g N 0 3 in 10 ml MeCN zugegeben, w r obei sich die Lösung trübte. Nach kurzer Zeit bildeten sich im Filtrat bei 0 °C gut ausgebildete Kristalle. N a c h 3 h wurde die Mutterlösung abdekantiert und dreimal mit je 20 ml MeCN -f0,5 ml MeNCO gewaschen. Die Ausbeute an A g Y • CH 3 CN betrug 2,6 g = 7 2 % bez. auf eingesetzte H 3 P 0 4 . I n der gleichen Weise lassen sich in MeCN durch Umsetzung v o n Natrium-, A m m o n i u m - , Magnesiumund Calciumthiocyanat (allerdings nicht völlig frei v o n Triethylammoniumthiocyanat) N a Y , N H 4 Y , MgY2 und C a Y 2 herstellen. B. Analyse der Produkte Ergebnisse v o n Elementaranalysen sind in der T a b . I V zusammengestellt. ( C , H , N : Ultramikro- 803 F . Seel-N. Klein • N-Methylcarbamoylphosphate Tab. I V . Analysenergebnisse in Gew.-%. R = Glührückstand, a = NH3, berechnete Werte in Klammern. Verbindung M Met. C H N 36,3(37,5) 12,2(12,4) 13,6(14,0) 11,0(11,1) 9,3 (9,6) 10,6(11,4) 8,7 (8,7) 7,7 (7,8) 7,8 (7,8) 8,1 (8,0) 6,1 (6,1) 37,7(38,3) 19,7(20,5) 22,6(19,2) 21,7(21,0) 20,3(20,0) 7,8(8,3) 2,5(2,6) 5,0(5,3) 2,7(2,8) 4,3(4,8) 2,6(2,9) 2,1(2,9) 1,8(2,0) 3,8(4,6) 2,3(2,7) 4,6(4,8) 7,6(7,7) 3,3(3,5) 3,8(3,2) 5,0(5,3) 3,0(3,1) 10,5(10,9) 7,2 (7,3) 16,3(16,3) 6,3 (6,5) 5,5 (5,6) 6,3 (6,6) 5,2 (5,1) 4,6 (4,5) 4,4 (4,5) 4,2 (4,6) 21,1(21,2) 12,9(13,4) 10,9(12,0) 11,3(11,2) 17,8(18,3) 11,7(11,7) P R 16,1(16,0) 18,0(18,0) 14,3(14,3) 12,2(12,4) 14,6(14,7) 11,2(11,2) 10,0(10,0) 10,2(10,0) 9,9(10,3) 7,7 (7,8) 58(61) 40(41) 62(61) 45(45) 60(60) 68(63) 74(73) 49(49) 66(66) 42(46) 12,7(13,2) 11,2(12,4) 13,3(13,5) 8,3 (8,6) 45(44) 45(47) 30(31) 51(52) H20 g/mol [EtsNHJHX KHX NH4HX Na2X • H 2 0 MgX • 4 H 2 0 CaX • H 2 0 SrX • 2 H 2 0 BaX • H20 ZnX • 5 H20 CdX • 2 H 2 0 [a 5 CoX]C10 4 [Et3NH]Y NaY KY NH4Y A g Y • CH 3 CN 256,2 193,1 172,1 217,0 249,4 211,1 276,7 308,4 308,5 301,5 396,6 313,3 234,1 250,2 229,1 360,0 9,6 (9,7) 18,7(19,0) 33,0(31,7) 43,5(44,5) 21,3(21,2) 37,3(37,3) 14,6(14,9) 29,8(30,0) m e t h o d e nach Walisch [6], P : turbidimetrisch nach Eibl u n d Lands [7], Mg, Ca, Sr, Ba, Z n , Cd, C o : E D T A - T i t r a t i o n , A g nach Volhard.) Abgesehen v o n den sehr schwer löslichen Verbindungen C a X • H2O, S r X • 2 H2O u n d B a X • H2O konnten Wassergehalte nach Karl Fischer bestimmt werden. Die bei 900 °C erhaltenen Glührückstände der Monocarba- [1] F. Seel und F. Schinnerling, Z. Naturforsch. 33b, 374 (1978). [2] F. Seel und G. Bohnstedt, Z. Anorg. Allg. Chemie 435, 257 (1977). [3] Ch. M. Allen (Jr.), E . Richelson und M. E . Jones, Current aspects of biochemical energetics, Herausgeber N. O. Kaplan und E . P. Kennedy, New York, Academic Press 1966, S. 401. 9,5 (8,3) 30,3(28,9) 28,5(29,1) 13,8(12,0) moylphosphate konnten als Diphosphate, die v o n K H X , N H 4 H X und der Biscarbamoylphosphate als Metaphosphate angesehen werden. D i e I R Spektren der Verbindungen sind erwartungsgemäß im Bereich v o n 300 bis 1800/cm sehr bandenreich und es lassen sich Schwingungen der O C N H - , P O H - , PO2- und P 0 3 - G r u p p e n identifizieren. [4] F. Cramer und M. Winter, Chem. Ber. 92, 2761 (1959). [5] R . W . Porter, M. O. Modebe und G. R . Stark, J. Biol. Chem. 244, 1846 (1969). [6] W . Walisch, Chem. Ber. 94, 2341 (1961). [7] H . Eibl und W . E. M. Lands, Anal. Biochem. 30, 51 (1969).