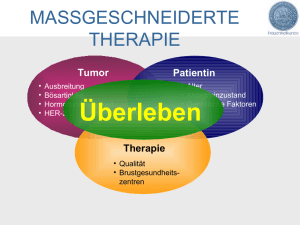
Koexpression von VEGF (Vascular Endothelial Growth Factor), c
Werbung
, c")
Aus dem Institut für Pathologie an den Berufsgenossenschaftlichen Kliniken Bergmannsheil Universitätsklinik der Ruhr-Universität Bochum Direktor: Prof. Dr. med. K.-M. Müller Koexpression von VEGF (Vascular Endothelial Growth Factor), c-Met - Rezeptor und HGF/SF (Hepatocyte Growth Factor / Scatter Factor) in Pleurametastasen Inaugural-Dissertation zur Erlangung des Doktorgrades der Medizin einer Hohen Medizinischen Fakultät der Ruhr-Universität Bochum vorgelegt von Ramin Naim aus Teheran (Iran) 2000 Dekan: Referent: Korreferent: Prof. Dr. med. G. Muhr Prof. Dr. med. K.-M. Müller Prof. Dr. med. G. Schulze-Werninghaus Tag der mündlichen Prüfung: 24. April, 2001 gewidmet meinen eltern Abstract Naim Ramin Koexpression von VEGF (Vascular Endothelial Growth Factor), c-Met - Rezeptor und HGF/SF (Hepatocyte Growth Factor / Scatter Factor) in Pleurametastasen Ziel dieser Untersuchung war die Charakterisierung angiogenetisch wirksamer Proteine in Pleurametastasen. Es wurde Biopsiegut sekundärer Pleuratumoren von 71 Patienten aus dem Einsendegut des Institutes für Pathologie an den Berufsgenossenschaftlichen Kliniken „Bergmannsheil“ immunhistochemisch hinsichtlich der Expression von HGF/SF (hepatischer Wachstumsfaktor bzw. Scatter Faktor; HGF und SF bezeichnen dasselbe Protein), c-Met (Rezeptor des HGF/SF) und VEGF/VPF (vaskulärer endothelialer Wachstumsfaktor, auch VPF: Vascular Permeability Factor genannt) analysiert. Die in-situ Hybridisierung wurde zum Nachweis von mRNA Trankripten von c-Met und VEGF eingesetzt. Eine deutliche intrazytoplasmatische Expression, teils Überexpression von VEGF, c-Met und HGF/SF konnte bei 83% bis 94% der pleuralen Metastasen von Mamma-, Lungen-, Gastrointestinaltrakt- und Urogenitaltrakt-Karzinomen sowie verschiedenen anderen Tumoren nachgewiesen werden. Eine Synthese von VEGF und c-Met war in Tumorzellen zu belegen mit Hilfe der in-situ Hybridisierung. Es lag eine statistisch signifikante Koexpression (p<0,05) von VEGF mit c-Met und HGF/SF im Tumorgewebe vor. Der Tyrosinkinaserezeptor c-Met (Rezeptor des hepatischen Wachstumsfaktors) bildet zusammen mit dem zugehörigen Liganden HGF/SF ein parakrines Signalsystem. Dieses System ist in den Prozeß der Angiogenese eingebunden, wobei HGF/SF nach Synthese in mesenchymalen Zellen eine angiogenetische Wirkung z.B. auf Gefäßendothelzellen und Gefäßwandmyozyten ausübt. Die Befunde belegen die Bedeutung des vaskulären endothelialen Wachstumsfaktors (VEGF), des hepatischen Wachstumsfaktors (HGF/SF) und des c-Met in Tumoren im Zusammenspiel bezüglich ihrer Wirksamkeit nachgemessenen Koexpression bei der kann Angiogenese. eine Unter Berücksichtigung autokrine/parakrine Stimulation der dieser angiogenetisch wirksamen Systeme diskutiert werden. Eine gleichzeitige Überexpression bzw. ein synergistischer Effekt dieser Proteine einschließlich VEGF ist über das Plasminogen-Urokinase-System möglich. Insbesondere unter Berücksichtigung der Wirkung von VEGF auf eine Steigerung der Gefäßpermeabilität können die hier erhobenen Untersuchungsergebnisse eine Grundlage möglicher zukünftiger palliativer Therapiestrategien mit Pleuraergüssen darstellen. antiangiogenetische n Substanzen bei refraktären malignen 1 EINLEITUNG ___________________________________________________________________5 1.1 DIE CHRONIK DER A NGIOGENESE _________________________________________________ 5 1.2 M ETASTASIERUNG ____________________________________________________________ 6 1.3 DIE PLEURAKARZINOSE ________________________________________________________ 9 1.4 ZIEL DER DURCHGEFÜHRTEN UNTERSUCHUNGEN ____________________________________ 10 2 ANATOMIE UND PHYSIOLOGIE DER PLEURA_______________________________________11 2.1 DIE A NATOMIE______________________________________________________________ 11 2.2 PLEURA VISCERALIS __________________________________________________________ 11 2.3 PLEURA PARIETALIS __________________________________________________________ 12 2.4 DIE BLUTGEFÄßE ____________________________________________________________ 13 2.5 DIE LYMPHGEFÄßE ___________________________________________________________ 13 2.6 DIE PLEURAFLÜSSIGKEIT ______________________________________________________ 14 2.7 PHYSIKALISCHE PHÄNOMENE IN DER PLEURAHÖHLE _________________________________ 14 2.8 DIE FUNKTIONEN DER PLEURA __________________________________________________ 15 2.9 STÖRUNG DES FLÜSSIGKEITSHAUSHALTES DER PLEURAHÖHLE __________________________ 15 2.10 DIE LYMPHANGIOSIS CARCINOMATOSA _________________________________________ 16 2.11 DIE M ORPHOLOGIE DER PLEURAKARZINOSE _____________________________________ 17 2.12 DAS A USBREITUNGSVERHALTEN SEKUNDÄRER PLEURATUMOREN _____________________ 18 2.13 DIE PLEURABIOPSIE ________________________________________________________ 18 3 VEGF ________________________________________________________________________19 3.1 EINFÜHRUNG IN DAS VEGF / VPF - SYSTEM UND IHRE REZEPTOREN ______________________ 19 3.2 DIE STRUKTUR VON VEGF _____________________________________________________ 20 3.3 DIE EXPRESSION DER VEGF-PROTEINE ____________________________________________ 20 3.4 3.5 3.3.1 VEGF in der Embryogenese ________________________________________________20 3.3.2 VEGF im adulten Gewebe _________________________________________________21 3.3.3 VEGF-Expression in pathologischen Veränderungen - insbesondere im Tumorgewebe ___22 3.3.4 Expression von VEGF in Tumorzellen, die von Endothelzellen abstammen ____________22 3.3.5 Ein Einblick auf die VEGF Genregulation_____________________________________23 DIE FUNKTION VON VEGF IN GEFÄßENDOTHELIEN ___________________________________ 24 3.4.1 Proliferationsstimulation und tubuläre Formation von Endothelzellen _______________24 3.4.2 Permeabilitätssteigerung an Gefäßen ________________________________________24 3.4.3 Andere biologische Aktivitäten des VEGF auf Endothelzellen ______________________24 3.4.4 VEGF-Effekte auf andere Zellen als Endothelien ________________________________25 DIE REGULATION DES TUMORWACHSTUMS DURCH SUPPRESSION DES VEGF-SYTEMS _________ 25 3.5.1 Suppression der Angiogenese ______________________________________________26 3.5.2 Die Kontrolle über Aszites-Bildung und Metastasierung __________________________26 1 4 SCATTER-FACTOR UND C-MET __________________________________________________28 4.1 SCATTER FACTOR____________________________________________________________ 28 4.1.1 Molekularbiologische Eigenschaften ________________________________________28 4.1.2 Die biologische Aktivität von SF ____________________________________________29 4.2 C-M ET ____________________________________________________________________ 30 4.3 DIE A KTIVITÄT VON SF AUF BLUTGEFÄßE IN VIVO UND IN VITRO _______________________ 30 4.3.1 4.3.1.1 Die Endothelzelle als SF-Produzent _________________________________________ 31 4.3.1.2 Die Endothelzelle als Zie lzelle des SF________________________________________ 31 4.3.2 4.4 5 Die Gefäßendothelzellen __________________________________________________30 DIE GEFÄßMUSKELZELLEN (SMC) UND DIE PERIZYTEN _____________________________ 32 4.3.2.1 SMC als SF-Produzent __________________________________________________ 32 4.3.2.2 SMC als Zielzellen des SF ________________________________________________ 33 SF IM TUMORGEWEBE _________________________________________________________ 33 4.4.1 Die Expression von SF und c-Met im Tumorgewebe ______________________________33 4.4.2 Die Herkunft des Tumor-SF ________________________________________________34 4.4.3 SF und Tumorangiogenese ________________________________________________34 MATERIAL UND METHODEN_____________________________________________________35 5.1 SEKUNDÄRE PLEURATUMOREN _________________________________________________ 35 5.2 EINTEILUNG DER PLEURATUMOREN ______________________________________________ 35 5.3 DIE IMMUNHISTOCHEMIE - DIE APAAP-M ETHODE __________________________________ 36 5.4 M ETHODEN-BESCHREIBUNG ____________________________________________________ 36 5.5 DIE A USWERTUNG ___________________________________________________________ 37 Tabelle 1: Immunreactive Score: SI- und PP-Einteilung _______________________________ 38 5.6 STATISTIK _________________________________________________________________ 38 5.7 DIE IN-SITU HYBRIDISIERUNG ___________________________________________________ 39 6 5.7.1 Definition _____________________________________________________________39 5.7.2 Die Fluoreszenz-in-situ-Hybridisierung (FISH) _________________________________39 ERGEBNISSE__________________________________________________________________41 6.1.1 Biopsien: Patientenzahl, Geschlechts- und Altersverteilung _______________________41 6.1.2 Verteilungsverhältnis der sekundären Pleuratumoren ____________________________44 Tabelle 2: Verteilung der einzelnen Biopsien auf Primärtumoren _________________________ 45 Zusammenfassung der wesentlichen Befunde (Teil I): __________________________________ 46 6.2 M AKROSKOPISCHE UND MIKROSKOPISCHE BEFUNDE _________________________________ 46 6.2.1 Ausgesuchte sekundäre Pleuratumoren mit differenzial-diagnostischen Kriterien _______46 6.2.1.1 Primärtumor: Lunge _____________________________________________________ 46 6.2.1.2 Primärtumor: Gastrointestinaltrakt __________________________________________ 47 6.2.1.3 Primärtumor: Mammakarzinom _____________________________________________ 47 2 6.2.1.4 Primärtumor: Nierenzellkarzinom ___________________________________________ 47 6.2.1.5 Die Lymphangiosis carcinomatosa _________________________________________ 48 Zusammenfassung der wesentlichen Befunde (Teil II): _________________________________ 48 6.3 DIE IMMUNHISTOCHEMISCHEN ERGEBNISSE ________________________________________ 49 6.3.1 SF, c-Met und VEGF _____________________________________________________49 6.3.1.1 Primärtumor: Lunge _____________________________________________________ 49 Tabelle 3: Ergebnisse der Immunhistochemie - Lunge ________________________________ 51 Zusammenfassung ____________________________________________________________ 52 6.3.1.2 Primärtumor: Gastrointestinaltrakt __________________________________________ 54 Tabelle 4: Ergebnisse der Immunhistochemie - Gastrointestinaltrakt _____________________ 56 Zusammenfassung ____________________________________________________________ 57 6.3.1.3 Primärtumor: Mamma____________________________________________________ 59 Tabelle 5: Ergebnisse der Immunhistochemie - Mamma _______________________________ 61 Zusammenfassung ____________________________________________________________ 61 6.3.1.4 Primärtumor: Urogenitaltrakt ______________________________________________ 63 Tabelle 6: Ergebnisse der Immunhistochemie - Urogenitaltrakt _________________________ 65 Zusammenfassung ____________________________________________________________ 65 6.3.1.5 Primäres Karzinom unbekannter Lokalisation __________________________________ 67 Tabelle 7: Ergebnisse der Immunhistochemie - Primäres Karzinom unbekannter Lokalisation ___ 69 Zusammenfassung ____________________________________________________________ 69 6.3.1.6 Verschiedene sekundäre Pleuratumoren______________________________________ 71 Pankreaskarzinom_____________________________________________________________ 71 Schilddrüsenkarzinom__________________________________________________________ 71 Kehlkopfkarzinom_____________________________________________________________ 72 Leiomyosarkom ______________________________________________________________ 72 Ewing-Sarkom _______________________________________________________________ 72 Tabelle 8: Ergebnisse der Immunhistochemie - Verschiedene Primärtumoren _______________ 73 6.3.1.7 Statistik der Immunhistochemie und Zusammenfassung der Ergebnisse______________ 74 Tabelle 9: Der exakte Test von R. A. Fisher in Übersicht ______________________________ 75 6.4 ERGEBNISSE DER IN-SITU HYBRIDISIERUNG _________________________________________ 76 6.5 ZUSAMMENFASSUNG DER ERGEBNISSE _____________________________________________ 78 7 DISKUSSION__________________________________________________________________80 7.1 M ETHODEN ________________________________________________________________ 80 7.1.1 Immunhistochemische Färbeverfahren________________________________________80 7.1.2 In-situ Hybridisierung ____________________________________________________80 7.1.2.1 7.2 Fluoreszenz-in-situ Hybridisierung (FISH)____________________________________ 81 BEFUNDE __________________________________________________________________ 82 7.2.1 Häufigkeit _____________________________________________________________82 3 7.2.2 Verteilung _____________________________________________________________82 Tabelle 10: Vergleich eigener Resultate zu den Primärtumoren als Ursache pleuraler Metastasen mit Angaben in der Literatur : ____________________________________________________ 84 7.3 7.2.3 Klinik pleuraler Metastasen _______________________________________________85 7.2.4 Lokalisation des malignen Ergusses _________________________________________86 7.2.5 Techniken zur Entnahme von Biopsien _______________________________________86 7.2.6 Die Thorakoskopie ______________________________________________________87 7.2.6.1 Die historische Entwicklung der Thorakoskopie _______________________________ 87 7.2.6.2 Die diagnostischen Vorteile der internistischen Thorakoskopie beim Pleuraerguß_______ 88 7.2.6.4 Therapeutische Vorteile der internistischen Thorakoskopie beim Pleuraerguß _________ 88 7.2.7 Morphologische Diagnostik _______________________________________________88 7.2.8 Charakterisierung pleuraler Metastasen ______________________________________89 DIE BEHANDLUNGSMÖGLICHKEITEN PLEURALER TUMOREN ___________________________ 90 7.3.1 Anti-VEGF-Therapie _____________________________________________________92 7.3.1.1 Konstruktion von VEGF-Rezeptor-Antagonisten _______________________________ 93 7.3.1.2 Bifunktionale VEGF-Toxine _______________________________________________ 93 7.3.1.3 Die Infektion der VEGF-exprimierenden Endothelzellen __________________________ 93 7.3.1.4 Angriff auf Rezeptorebene _______________________________________________ 93 7.3.2 Anti-SF- und Anti-c-Met-Therapie ___________________________________________94 8 ZUSAMMENFASSUNG __________________________________________________________95 9 LITERATUR___________________________________________________________________98 4 1 EINLEITUNG 1.1 DIE CHRONIK DER ANGIOGENESE Der Prozeß der Angiogenese, das Wachstum der Blutgefäße, ist ein wichtiger Vorgang während der Reproduktion, Entwicklung und Wundheilung. Während dieser Vorgänge unterliegt die Angiogenese einer sehr hochentwickelten und bis heute noch nicht vollständig nachvollziehbaren Regulation (Folkman 1991 und 1990, Hudlika 1986). Seit langem beschäftigen sich viele Forscher mit den Blutgefäßen und dem Phänomen der Gefäßbildung. 1616 stellte William Harvey (1578 - 1657) die These auf, daß bestimmte kommunizierende Poren zwischen Venen und Arterien existieren (communicating „pores“) (Abb. 1, 2). René Descartes (1596 - 1650) bezog sich in seinem Aufsatz „Le Discours de la Méthode“ 1637 auf dieselbe Entdeckung, er schrieb: „petits passages aux extrémités des artères par oú le sang qu`elles reçoivent du coer entre les petites branches des veines“ (Sherrington 1929). Marcello Malpighi (1628 - 1694) entdeckte, daß die Verbindungen durch mikroskopisch kleine Röhrchen, den Kapillaren, gebildet wurden (Malpighi 1686 - tom I). 11 Jahre später teilte er diese Neuentdeckung der Royal Society in London mit, wobei er die Kapillaren in einem Hühnerembryo beschrieb (Malpighi 1686 - tom II). Antony van Leeuwenhoek (1632 - 1723) entdeckte auch in anderen Geweben Kapillarstrukturen. Stephen Hales (1677 - 1761) erkannte, daß die „kleinen kapillaren Gefäße“ dilatierten oder sich zusammenzogen, wenn sie mit bestimmten Chemikalien behandelt wurden (Hales 1733). August Krogh (1874 - 1949), der an der Yale Universität tätig war, bestätigte die Feststellungen von Charles-Marie-Benjamin Rouget (1824 - 1904). Dieser hatte ein Netzwerk von verästelten, kontraktilen Zellen, die mit den Kapillaren in Verbindung standen, entdeckt - heute bekannt als Rouget Zellen oder Perizyten (Rouget 1873 und 1879, Krogh 1922). 5 § Abb. 1 Darstellung der äußeren Venen des Blutkreislaufs (nach William Harvey). § Abb. 2 Der englische Leibarzt William Harvey entwickelte das Konzept eines Blutkreislaufs, in welchem das Herz als zentraler Antrieb arbeitet. In mit Stichen bebilderten Werken korrigierte er zahlreiche Irrtümer, beispielsweise daß das Herz an der Verdauung teilnehme (Quelle: Hudlika 1986). 5a Arthur Tremain Hertig beschrieb 1935 die Organisation der Blutgefäße in der Plazenta und verwies darauf, daß neben den Endothelzellen die Perizyten ebenfalls in die Angiogenese einbezogen sind (Hertig 1935). John Hunter (1728 - 1793) beschrieb die Angiogenese in seinem Buch „Treatise on the blood, inflammation and gun shot wounds“, das nach seinem Tod 1794 erschien. Theodor Billroth veröffentlichte 1856 in Berlin seine Untersuchungen und Beobachtungen über die Entwicklung der Blutgefäße (Quelle: Georg Reimer, Berlin, 1856, 81 pp. + V plates) (Abb. 3). Das „Wie?“ der Angiogenese wurde schon im späten 19. Jahrhundert bis in das frühe 20. Jahrhundert ausführlich beschrieben. Die Forschung über das „Warum?“ der Angiogenese sowohl im physiologischen als auch im pathophysiologischen Prozeß (Beispiel: während der Wundheilung oder im Malignomen) begann in den frühen 70iger Jahren mit den Arbeiten von Judah Folkman (Folkman 1971). Er sah, daß Tumorzellen , die in isolierte Organe implantiert wurden, nicht größer als 1 bis 2 mm wurden. Ebenso blieb die Vaskularisation aus. Aufgrund dieser Tatsache stellte er die Hypothese auf, daß das Tumorwachstum von einer neuinduzierten Gefäßbildung abhängig sei (Folkman 1963 und 1971). Heute geht man in der Krebsforschung davon aus, daß der Angiogenese in der Tumorprogression und -metastasierung eine wesentliche Bedeutung zukommt. 1.2 METASTASIERUNG Der Begriff „Metastase“ stammt aus dem Griechischen und kann als „Veränderung“ oder „Wanderung“ übersetzt werden. In der medizinischen Anwendung wird mit „Metastase“ die Verschleppung einer Krankheit von einer Körperstelle an eine andere bezeichnet. Im engeren Sinne werden unter dem Begriff „Metastasierung“ all diejenigen Prozesse zusammengefaßt, die an der Verschleppung bösartiger Zellen oder infektiöser Keime beteiligt sind und ihr An- und Weiterwachsen in entfernten Körperregionen realisieren (Walther 1948). 6 § Abb. 3 Theodor Billroth Die Entwicklung der Blutgefäße (Quelle: Georg Reimer, Berlin, 1856, 81 pp. + V plates). 6a Die Metastasierung ist ein eindeutiges Zeichen der Malignität eines Tumorgewebes und ein ausgesprochen selektiver Vorgang, denn von den Millionen Zellen, die von einem Tumorgewebe abwandern, führen nur wenige zu einer Metastase. Besonders diese Tumormetastasen prägen in der Finalphase den Krankheitsverlauf eines malignen Tumorleidens. Das Zusammenspiel der an der Metastasierung beteiligten Faktoren wird von bestimmten Genen kontrolliert. Bei der Metastasierung spielen folgende Faktoren eine entscheidende Rolle: Kohäsionsverlust: Verliert die Zelle diejenigen Genprodukte, die die Zelladhäsionsmoleküle (auch Integrine genannt) exprimieren, so resultiert der Koadhäsionsverlust des Zellverbandes. Zellmotilität : Die Bildung von Motilitätsfaktoren führt bei den Tumorzellen zu einer auto- und parakrinen Wirkung, die eine erhöhte Motilität zur Folge hat. Intra-, Extravasation : Die Tumorzellen können Proteasen freisetzen, mit deren Hilfe sie sich aus dem ursprünglichen Zellverband lösen und so ins Gewebe und in Gefäße einbrechen können. Fremdverkennung : Bleiben sie nach dem Einbruch in Gefäße in der Strombahn haften, können sie sich vor dem Immunsystem schützen. Sie vermindern die eigene HLAExpression und umgeben sich mit einer Fibrinummantelung. Absiedelung : Ob Tumorzellen sich in ein bestimmtes Organ absiedeln können, hängt von den ortsspezifischen Anhaftungs- und Wachstumsbedingungen ab. Die Oberfläche der Tumorzelle spielt ebenso eine wichtige Rolle (Schmähl 1981, Selberg 1982). Man differenziert je nach der Struktur, innerhalb derer die Metastasierung erfolgt: 7 A) Lymphogene Metastasierung : Die Verschleppung der Tumorzellen durch Lymphgefäße; vermehren sie sich in diesen Gefäßen, so entsteht eine Lymphangiosis carcinomatosa, die dann auch pathologisch-anatomisch nachweisbar ist (Kap. 2.10). B) Hämatogene Metastasierung : Die Verschleppung auf dem Blutweg; es gibt vier Grundtypen der hämatogenen Metastasierung: § Lungentyp: Die Krebszellen stammen primär aus der Lunge und bewegen sich über die Lungenvene zum linken Herzen und von dort aus in den großen Kreislauf. § Lebertyp: Der Tumor hat sich primär in der Leber manifestiert und die Tumorzellen werden über die Lebervene forttransportiert. § Kavatyp: Der Primärtumor sitzt im Abflußgebiet der Hohlvene (Beispiel: Nieren- oder Knochenkrebs). § Pfortadertyp: Fast bei allen Darmtumoren findet man diesen Typ; die Tumorzellen gelangen über die Pfortader in die Leber, können dort in die Lebervene einbrechen und als Lebertyp-Metastasierung fortwandern. § vertebralvenöser Typ: Frühzeitig metastatische Tumoraus-breitung über das paravertebrale Venengeflecht. C) Kavitäre Metastasierung : Tumorzellen brechen in die Peritoneal- oder Pleurahöhle, in den Liquorraum oder in die Sehnenscheide ein. D) Kanalikuläre Metastasierung : Diese seltene Metastasierungsform liegt vor, wenn sie innerhalb eines epithelial ausgekleideten kanalikulären Systems stattfindet (Grundmann 1981 und 1994). 8 1.3 Die DIE PLEURAKARZINOSE Pleurakarzinose stellt als Manifestation sekundärer Pleuratumoren eine Veränderung der Pleurahöhle dar, die nicht nur aus klinischer Sicht, sondern auch aus pathologisch-anatomischer Sicht in der Diagnostik bisweilen erhebliche Probleme bereitet hat (Abb. 4a, 4b). Klinisch manifestieren sich die Pleurakarzinosen in Form von Pleuraergüssen, die selten einmalig sind und oft rezidivieren. Der Pathologe ist gefordert, aus Ergusspunktaten oder aus wenigen Millimeter großen Biopsaten eine zuverlässige Diagnose mit Hinweisen auf den meist klinisch noch nicht manifesten Primärtumor zu stellen. Le Roux meinte, daß die meisten pleuralen Tumoren Metastasen eines primären Lungentumors wären. Von 3000 Patienten mit Bronchialkarzinomen aus einer Studie von 1962 hatten 220 (7%) Pleurametastasen - während von 3500 Patienten mit Mammakarzinomen lediglich 27 (0,8%) Pleurametastasen aufwiesen (Le Roux 1985). Reshad (Reshad 1985) fand heraus, daß unter 200 malignen Pleuraergüssen 123 (60%) primäre Lungentumoren, 72 (35%) andere Primärtumoren überwiegend der Mamma vorhanden waren; 5 hatten primäre Mesotheliome. Untersuchungen haben bei malignem Pleuraerguß herausgestellt, daß die sekundären malignen Pleuratumoren als Ursache überwiegen. Geschätzt wurden Zahlen von 15000 bis 30000 sekundären Pleurakarzinosen jährlich in den alten Bundesländern der Bundesrepublik Deutschland (Kreuser 1984). In Wien wurden in 5 Jahren 427 „Pleurakarzinosen“ diagnostiziert, wovon nur 13 primäre Mesotheliome, 222 sekundäre Pleurakarzinosen bei Bronchialkarzinom und 132 sekundäre Pleurakarzinosen bei Mammakarzinom waren. Das hieße, daß 30% aller Pleurakarzinosen auf ein Mammakarzinom und 50% auf ein Bronchialkarzinom zurückzuführen wären (Titscher 1981). Die Unterscheidung zwischen der Manifestation eines primären oder eines sekundären Pleuratumors kann große versicherungsmedizinische Konsequenzen haben, da das Pleuramesotheliom nicht nur zu den berufsbedingten Erkrankungen (BK-Nr. 4105: durch Asbest verursachtes Mesotheliom des Rippenfells, Bauchfells oder Perikards) 9 Abb. 4a Makroskopischer Aspekt bei pleuraler Metastasierung (E.-Nr. 228/96) Metastase in der Pleura mediastinalis bei primärem Adenokarzinom im Lungenoberlappen (männlich, 69 Jahre): neugebildete, auf den Tumor zulaufende Gefäßsprossen. Abb. 4b Makroskopischer Aspekt bei pleuraler Metastasierung (E.-Nr. 28/96) Makroskopischer Aspekt einer knotigen Pleurametastasierung mit deutlich erkennbaren neugebildeten Gefäßsprossen in radiärer Ausrichtung auf die Tumorknoten, daneben pleurale Mischstaubablagerungen bei Mischstaubpneumokoniose (Bergmann, 58 Jahre, Primärtumor der Lunge). 9a zählt, sondern mit 50% gewissermaßen die Liste der Berufskrankheiten anführt (Müller 1989). 1.4 ZIEL DER DURCHGEFÜHRTEN UNTERSUCHUNGEN In der vorliegenden Untersuchung wird die Expression dreier Proteine, Scatter Factor (SF) bzw. Hepatocyte Growth Factor (HGF), das c-Met und der Vascular Endothelial Growth Factor (VEGF), mit wesentlicher Bedeutung in der Angiogenese in sekundären Pleuratumoren analysiert im Hinblick auf eine mögliche Assoziation dieser untereinander. Um zusätzliche Information über die Herkunft von c-Met und VEGF zu bekommen, wird auch eine in-situ Hybridisierung vorgenommen. 10 2 ANATOMIE UND PHYSIOLOGIE DER PLEURA 2.1 DIE ANATOMIE Zahlreiche pulmonale und extrapulmonale Erkrankungen betreffen die Pleura. Der Pleuraerguß ist ein häufiges Symptom und Beschwerdebild, welches ein gezieltes diagnostisches Vorgehen erfordert. Der Pleuraspalt ist der Raum zwischen parietaler und viszeraler Pleura, der 10 bis 20 µm weit (Agostoni 1969) ist und eine 0,1 bis 0,2 ml klare, farblose Pleuraflüssigkeit pro Kilogramm Körpergewicht enthält. Der Proteingehalt beträgt weniger als 1,5 g/dl (Yamada 1933, Agostoni 1972). Es finden sich pro Mikroliter ca. 1500 bis 4500 Zellen, vorwiegend Monozyten, wenige Makrophagen und Lymphozyten, vereinzelt auch Granulozyten, aber keine Erythrozyten (Stauffer 1978, Yamada 1933). Die gesamte Lunge sowie die Interlobärspalten werden vom viszeralen Blatt der Pleura bedeckt. Mit dem parietalen Blatt werden die Thoraxinnenwand, das Mediastinum und das Diaphragma ausgekleidet. Die viszerale Pleura schlägt an beiden Hili um und geht in das parietale Blatt über (v. Hayek 1960) (Abb. 5). 2.2 PLEURA VISCERALIS Die Pleura visceralis ist physiologisch 100 - 200 µm dick und kann histologisch in 5 Schichten differenziert werden: § eine Mesothelschicht § eine submesotheliale Bindegewebsschicht § eine elastische Schicht § eine relativ lockere subpleurale Bindegewebsschicht sowie § eine fibroelastische Schicht (v. Hayek 1960, Mariassay 1983). 11 Abb. 5 Anatomie und Topographie der Pleura (Quelle: Atlas der Anatomie des Menschen - Sobotta, U&S). 11a Die zur Pleurahöhle gerichtete Seite wird von einer einreihigen pleomorphen Mesothelschicht, die ca. 4 bis 8 µm hoch ist, ausgekleidet. Diese besitzt Mikrovilli (ca. 300 pro µm² Zelle), welche einen Durchmesser von ca. 0,1 µm und eine Länge bis zu 3 µm haben. Die Zellen sind alle untereinander durch Desmosomen verbunden und sind in der Lage, sich bei jeder Atembewegung zu verformen (Müller 1983 und 1994, Agostoni 1969, Cooray 1949). Bei Änderungen des Milieus sowie Verletzungen und entzündlichen Vorgängen können die Mesothelien aktiv reagieren. Durch Migration und Mitose werden Defekte wiederhergestellt. Dabei ist bemerkenswert, daß sie als freischwimmende Zellen in der Pleurahöhle ihre Gestalt ändern in rund-ovale, zum Teil zusammengezogene Zellen (Efrati und Nir 1976). Ob die reparativen Vorgänge jedoch von den Zellen der Subserosa als Mesothelersatz ausgehen, ist noch ungeklärt (Brockmann und Müller 1991). Es folgt zum Lungengewebe hin eine äußere elastische Grenzlamelle, eine dreischichtige, gefäßführende elastische Faserschicht (Tela subserosa) und eine innere elastische Grenzlamelle, die etwas straffer entwickelt ist. Von dieser elastischen Grenzlamelle besteht ein direkter Übergang zum elastischen und kollagenen Gewebe der angrenzenden Alveolar- und Lobularsepten. 2.3 PLEURA PARIETALIS Die Pleura parietalis hat wie die viszerale Pleura eine gleichartige Deckzellschicht, die einer Basalmembran aufliegt; dieser Schicht folgt eine hauptsächlich kollagene Zwischenschicht, in der vereinzelt elastische Fasern zu finden sind. Im Bereich der Zwischenrippenräumen ist in dieser Zwischenschicht Fett eingelagert. Über elastische Fasern besitzt die Pleura parietalis einen direkten Übergang in die Fascia endothoracica. 12 2.4 DIE BLUTGEFÄßE Die Bindegewebsschicht enthält Blutgefäße, Nerven und Lymphgefäße. Noch ist nicht sicher bekannt, welche Blutgefäße die Pleura viszeralis mit Blut versorgen; man nimmt an, daß sie von den Bronchialarterien versorgt werden (McLaughlin 1961, Miller 1947). Die parietale Pleura jedoch wird im Bereich der Rippen von den Interkostalarterien, im apikalen Bereich von Ästen der A. subclavia und im Mediastinum von Ästen der Bronchialarterien sowie der A. mammaria versorgt (Müller 1983 und 1994, Testut 1930). 2.5 DIE LYMPHGEFÄßE Das Lymphsystem der Lunge befindet sich im subpleuralen Bindegewebe und besteht aus einem oberflächlichem Plexus, das Netzstrukturen bildet (Müller 1983 und 1994). Es ist im Bereich der Lungenunterlappen besonders ausgeprägt. Ein tiefer Plexus umschließt die Bronchioli und Blutgefäße (Grant 1974, Miller 1947, Naigaishi 1972). Die subpleuralen Lymphgefäße drainieren in die größeren zwischen Lungensegmenten und Lungenlappen lokalisierten Gefäße (Naigaishi 1972). Für das Verständnis der Pathogenese von Pleuraergüssen ist die Lymphdrainage des Pleuraraumes sehr bedeutsam. Der Abfluß der Lymphe beginnt in den sogenannten Stomata, ca. 2 bis 6 µm große Öffnungen, hauptsächlich im Bereich des unteren Mediastinums, der parietalen infrakostalen Pleura und des Diaphragmas lokalisiert (Miller 1947, Wang 1975). Der Pleuraraum wird durch diese Stromata mit den Lymphgefäßen durch erweiterte Lymphräume, den sogenannten Lakunen, verbunden. An den Enden der Lakunen befindet sich ein ventilartiger Mechanismus, der atemsynchron öffnet und schließt, wodurch die Lymphe in die Lymphgefäße gelangt (Courtice 1954, Wang 1975). Der größte Abschnitt der Lungenunterlappen drainiert in die hinteren mediastinalen, die viszerale Pleura in die mittleren mediastinalen Lymphknoten. Die Lymphdrainage der vorderen Thoraxwandpleura und des anterioren Diaphragmas erfolgt über die 13 sternalen Lymphknoten, der mittlere Anteil der diaphragmalen Pleura über die mittleren, der hintere über die posterioren mediastinalen Lymphknoten. Die Lymphe der parietalen Pleura drainiert in die interkostalen Lymphknoten (Marsac 1982). Durch strukturelle und funktionale Untersuchungen fand man heraus, daß große Partikel und Zellen, die sich in der Pleurahöhle befinden, hauptsächlich durch lymphatische Drainage nur an einigen Stellen der parietalen Pleura wegbefördert werden (Wang 1975). 2.6 DIE PLEURAFLÜSSIGKEIT Die Pleurahöhle enthält eine proteinreiche Flüssigkeit, die ständig vom Mesothel der Pleura parietalis und visceralis, das makroskopisch als spiegelnd glatt imponiert, sezerniert wird. Diese Flüssigkeit überzieht den kapillären Spalt der Pleurahöhle und ermöglicht so die Verschiebbarkeit der Lunge gegenüber der Pleura mit einer gleichzeitig wirksamen adhäsiven Kraft. Der Sekretion der Pleuraflüssigkeit ist die ständige Resorption durch die Pleura parietalis und visceralis entgegengerichtet. Der Erhalt des kapillären Spaltes hängt vom Gleichgewicht der Produktion und Resorption der Pleuraflüssigkeit ab (Bakhle 1977). 2.7 PHYSIKALISCHE PHÄNOMENE IN DER PLEURAHÖHLE Das Mesothel besitzt eine sehr lockere Interzellularverbindung, wodurch es für Flüssigkeit leicht durchlässig ist. Besonders im Bereich der Pleura mediastinalis und diaphragmatica weicht das Mesothel sogar zu Netzen mit weiten Maschen auseinander, so daß das darunterliegende Bindegewebe mit dem Pleuraspalt direkt in Verbindung steht. Dies ist ein wichtiger Unterschied zwischen dem Pleuramesothel und den Alveolarepithelien der Interalveolarsepten (Bucher 1980). Der interpleurale Druck ist ein Unterdruck, der bei Ruheatmung in mittlerer Höhe der Pleurahöhlen zwischen -0,4 bis -0,5 kPa ( ca. -4 bis -5 cm H2O) beträgt und generell bei tiefer Inspiration höher wird (Piiper 1975). 14 2.8 DIE FUNKTIONEN DER PLEURA Die Pleura besitzt drei wichtige Funktionen: 1. Da innerhalb der Pleura ein atemabhängiger Unterdruck herrscht, bleibt hierdurch die Lunge entfaltet. 2. Die Pleura hat ebenso eine Stützfunktion gegenüber dem elastisch aufgespannten alveolären Lungengewebe 3. Eine optimale Beweglichkeit der Lunge wird durch die Pleuraflüssigkeit erreicht (Rennard 1984, Lee und Olak 1994). 2.9 STÖRUNG DES FLÜSSIGKEITSHAUSHALTES DER PLEURAHÖHLE Folgende Faktoren beeinflussen den Flüssigkeitshaushalt der Pleura: 1. Filtrationsdruck der in der Pleura parietalis lokalisierten Kapillaren 2. Der kolloidosmotische Druck 3. Der intrapleurale Unterdruck 4. Sekretionsdruck durch die Mesothelien 5. Resorption der Pleuraflüssigkeit durch das Lymphsystem Die unter Punkt 1, 3 und 4 aufgeführten Zustände führen zum erhöhtem Einstrom von Flüssigkeit in die Pleurahöhle. Diesem Phänomen stehen der kolloidosmotische Druck und insbesondere das reich ausgebildete Lymphgefäßsystem unter der parietalen und viszeralen Pleura entgegen, das ebenso unter dem intrapleuralen Unterdruck steht und mit den Atembewegungen rhythmisch schwankt. Dieses führt dazu, daß die proteinreiche Flüssigkeit besonders leicht von den Lymphgefäßen aufgenommen und durch die Klappen geleitet wird, und 15 am Ende ins Blutgefäßsystem zurückgeführt wird. Es sind ebenso besondere netzartige Öffnungen des Pleuramesothels ausgebildet, durch die in den Pleuraspalt eingedrungene Zellen oder Partikel abtransportiert werden. Eine Verlegung dieser Lymphdrainage durch massive Metastasierung tritt im Rahmen der Pleurakarzinose auf, die zu einem Pleuraerguß führen kann. Dieser Pleuraerguß ist ein Exsudat (Abb. 6). Störungen, die zum Pleuraerguß führen, entstehen durch: 1. Erhöhung des kapillären Drucks im Körper- und Lungenkreislauf 2. Absenken des Proteingehalts des Blutes (und somit auch des kolloidosmotischen Drucks) 3. Obstruktion der Lymphdrainage durch Tumormassen (Földi 1983, Guyton 1981). 2.10 DIE LYMPHANGIOSIS CARCINOMATOSA Dieser Ausdruck beschreibt den Befall von Lymphgefäßen durch Tumorzellen. Makroskopisch ist diese deutlich sichtbar als glasige Verdickung besonders der um die sekundären Lobuli angeordneten Lymphgefäße, die auf der Lungenoberfläche ein weiß prominentes Netz bilden. In der mikroskopischen Darstellung kann man dieses Phänomen als ausgeweitete Lymphgefäße mit bis zu mehreren 100 Zellen im Querschnitt enthaltenen Tumorzellsträngen erkennen. Dabei kann hierdurch eine reaktive Fibrose (breite Züge von Fibroblasten und Kollagenfasern) entlang der Tumorzellen entstehen, so daß das morphologische Bild hierdurch stärker als durch die Tumorzellen selbst beeinflußt werden kann (Müller 1983, Chrétien und Jaubert 1985). 16 Abb. 6 Flußdiagramm für das diagnostische Vorgehen beim Pleuraerguß (Loddenkämper `82). 16a 2.11 DIE MORPHOLOGIE DER PLEURAKARZINOSE Die Karzinose kann pleural oder bronchial, maximal aber broncho-pleuro-pulmonal sein. Auch im letzteren kann der Prozeß auf den Thorax beschränkt bleiben, und es muß noch keine Generalisation vorliegen. Einzelne größer als miliare Lungenherde sind als Metastasen, d.h. hämatogen und nicht als Teil der lymphogenen Aussaat anzusehen. Die Pleurakarzinose kann längere Zeit die einzige Tumormanifestation sein. Makroskopisch findet man eine fein- oder grobknotige, herdförmig oder diffuse Manifestation. Manchmal ist die Pleura nicht verdickt, sondern hauchdünn (2 bis 3 mm dick), sulzig, höckerig und selten auch schwartenähnlich. Im letzteren Fall ist eine Verwechslung mit einem Mesotheliom möglich. Mikroskopisch : Chrétien (1985) beschreibt drei unterschiedliche Typen der Krebszellentwicklung als pleurale Antwort beim metastatischen Tumor: § endolymphatisch § nodulär und § papillär. Beim endolymphatischen Typ weist die Pleuraoberfläche ein normales Mesothel auf. Wo nun der Zeitpunkt des Auftritts des Exsudates liegt und ebenso welche Beziehung es zu den Metastasen besitzt, ist weiterhin spekulativ. Mögliche Pleurareaktionen können sich in der Pleurabiopsie (bei Thorakoskopie, deutlich bei Nadelpunktion) zeigen: § papilläre Proliferation von Mesothelien § fibroblastische Proliferation und § exfoliierte maligne Mesothelien. 17 2.12 DAS AUSBREITUNGSVERHALTEN SEKUNDÄRER PLEURATUMOREN Häufig geht die initiale Metastasierung sekundärer Pleuratumoren über die Pleura visceralis vonstatten. Die Sekundärmanifestation der Mammatumoren erfolgt meist auf direktem Wege durch die Brustwand. Die im Bauchraum befindlichen Tumoren metastasieren über die paraaortalen und parakavalen Lymphbahnen. Ein direkter Weg transdiaphragmal ist ebenso möglich (Dail 1994). 2.13 DIE PLEURABIOPSIE Sie ist bei allen Ergüssen unklarer Ätiologie indiziert. Als Kontraindikation sind hämorrhagische Diathese und eine geringe Ergussmenge zu erwähnen. Die Sensitivität bei der Pleurakarzinose beträgt 57% (Tomlinson 1987). Um die Treffsicherheit zu erhöhen, sollten mindestens vier Biopsien pro Untersuchung entnommen werden (Mungall 1982). 18 3 3.1 VEGF EINFÜHRUNG IN DAS VEGF / VPF - SYSTEM UND IHRE REZEPTOREN Die Neovaskularisation beinhaltet die Begriffe Vaskulogenese und Angiogenese. Die Vaskulogenese ist die Blutgefäßbildung von endothelialen Vorstufen im Mesenchym während der Embryonalentwicklung. Die Angiogenese ist die Ausbildung von neuen Gefäßästen aus schon vorbestehenden kleinen Gefäßen. Die Gefäßbildung ist nicht nur für die embryonale Entwicklung der Vertebraten von Bedeutung, sondern auch unter physiologischen Bedingungen - bei der „schnellen“ vorübergehenden Angiogenese (Beispiel: während der Bildung des Corpus Luteum). Sie tritt aber auch unter pathologischen Bedingungen auf, wie beispielsweise bei entzündlichen Reaktionen, bei der Wundheilung und beim wachsenden Tumor in vivo. Besonders bei der Krebsentwicklung spielt die Angiogenese eine sehr wichtige Rolle (Shibuya 1995): Tumor-Angiogenese: bei soliden Tumoren größer als 3 mm Durchmesser erhalten diese Sauerstoff und Nährstoffe durch neugebildete Gefäße (Folkman 1990). Permeabilität von Gefäßen: sobald sich Metastasen in der abdominalen (Mediastinum) oder aber thorakalen (Pleura) Körperhöhle entwickeln, scheint eine abnormale Steigerung der Gefäßpermeabilität hier statt zu finden, was zur Aszitesbildung im Mediastinum und Pleuraerguß führt. 19 3.2 DIE STRUKTUR VON VEGF Der vaskulär endotheliale Wachstumsfaktor VEGF/VPF wurde ursprünglich von Senger als ein gefäßpermeabilitätsreguliernder Faktor isoliert. Sie fanden einen heparinbindenden Faktor mit einem Molekulargewicht von 40 bis 45 kDa. Es bestand aus einem Dimer von zwei 20-23 kDa schweren Untereinheiten. Dieser Faktor wurde VPF (Vascular Permeability Factor) genannt, da er bei einer intrakutanen Injektion die Gefäßpermeabilität steigern konnte (Senger 1983) (Abb. 7). Unabhängig von diesen fand eine andere Arbeitsgruppe (Ferrara und Henzel 1989), einen den Endothelzellen spezifischen Wachstumsfaktor in konditioniertem Medium von Nebennierenmarkzellen. Dieser Faktor, den sie VEGF nannten, bewirkte bei Anwendung auf andere Endothelzelltypen einen Zellzahlanstieg. VEGF konnte jedoch nicht zur Proliferation von Fibroblasten oder Epithelzellen führen. Molekulargenetische Versuche haben gezeigt, daß VPF und VEGF das gleiche Proteine darstellen (Leung 1989, Keck 1989, Ferrara 1991 und 1992). Im Folgenden wird zur Vereinfachung der vaskulär endotheliale Wachstumsfaktor als VEGF bezeichnet. 3.3 DIE EXPRESSION DER VEGF-PROTEINE 3.3.1 VEGF in der Embryogenese In den frühen Stadien der Embryogenese (8. Tag) wird VEGF mRNA in relativ hoher Konzentration in den Trophoblasten Riesenzellen der Plazenta gefunden, jedoch in niedrigerer Konzentration im Embryo. Nun steigt am 11. Tag der Embryogenese die VEGF mRNA-Konzentration in der thorakalen Region des Embryos, wo die sehr frühen Stadien der Herzentwicklung stattfinden, und erreicht hier in den nächsten Tagen (11. bis 14. Tag) seine höchste Konzentration (Jackman 1993, Shifren 1994). Zwischen dem 14. und 16. Tag sinkt die Konzentration der VEGF mRNA in der kardialen Region, steigt jedoch im Lungenbereich, Gehirn und Nierenmark an. Es 20 wurde festgestellt, daß die relativ höhere Konzentration an VEGF mRNA in der Lunge, Niere, Herz und Gehirn auch noch nach der Geburt vorhanden waren (Jakeman 1993). Am 15. Tag wurde in dem den Nierenglomerulus umgebendem Epithel ebenso eine starke VEGF mRNA-Expression festgestellt (Breier 1992). 3.3.2 VEGF im adulten Gewebe In zahlreichen maturen Geweben konnten hohe VEGF-Konzentrationen nachgewiesen werden, so z.B. im Alveolarepithel der Lunge, in Nierenglomeruli, in Herzmyozyten und ebenfalls im Hypophysengewebe (Brown 1992, Ferrara 1991, Berse 1992, Ilijima 1993). Da in diesen Geweben das Endothel sich in einem Wachstumsstillstand befindet, stellt sich hier die Frage, welche biologische Rolle VEGF in diesen Geweben spielt. Eine mögliche Antwort wäre, daß insbesondere in der Niere und der Lunge durch VEGFProduktion die Permeabilität und Integrität dieser Zellen aufrecht erhalten wird. Eine spontane Neovaskularisation taucht während der Bildung des Corpus luteum, dem Wachstum des Uterusendometriums und während der Plazentaentwicklung auf (Sharkey 1993, Dissen 1994). Man konnte während all dieser Prozesse eine hohe VEGFmRNA-Konzentration sowie eine erhöhte VEGF-Proteinsynthese § in Cumulus oopherus Zellen (Phillips 1990) § in Luteal Zellen des Ovars (Ravindranath 1992) § und in Endometriumzellen des Uterus (Shweiki 1993) feststellen, was die klare Beziehung von VEGF zur Angiogenese deutlich macht. Weitere Experimente haben gezeigt, daß die Induktion von VEGF-mRNA durch Östrogene im Rattenuterus eine enge Korrelation zur Steigerung der Kapillarpermeabilität und Angiogenese in diesem Gewebe besitzt (Cullinan-Bove und Koos 1993). Dabei exprimierten Granulosazellen hormonabhängig VEGF-mRNA (Garrido 1993, Yan 1993). 21 § Abb. 7 VEGF Dreidimensional-molekulare Struktur des VEGF. Disulfidbrückenbindungen: gelb Polypeptidketten: grün Rezeptorbindungsstellen: rot und blau (Quelle: Oefner 1992) 21a 3.3.3 VEGF-Expression in pathologischen Veränderungen - insbesondere im Tumorgewebe Die Angiogenese findet in der Regel in späten Stadien der Wundheilung in Form von Granulationsgewebe statt. Beispielsweise haben Keratinozyten in der Nähe von Wundheilungsbereichen eine hohe Expressionsrate an VEGF-mRNA (Brown 1992). In der Ovulation-Induktionsthrapie haben weibliche Patienten gelegentlich ein durch das Ovar verursachtes Hyperstimulations-Syndrom, wodurch proteinreicher Aszites durch dort befindliche Gefäße freigesetzt wird, aber auch zum Pleura- und Perikarderguss führen kann. Mc Clure (1994) zeigte, daß die Flüssigkeit eine sehr hohe VEGF-Konzentration besitzt, was impliziert, daß dies auf eine mögliche hormonelle Stimulation durch VEGF zurückgeführt werden kann. Eine abnorme Angiogenese § in der Retina von Diabetes mellitus Patienten (Aiello 1994) § im rheumatoiden Synovial-Gewebe (Miller 1994), § im vergrößerten Schilddrüsengewebe der Patienten mit Morbus Basedow (Fava 1994) konnte dadurch charakterisiert werden, daß sie mit einer ebenso abnorm hohen VEGFExpression korreliert. Insgesamt scheint bei der pathologischen Angiogenese auch das Rezeptorsystem des VEGF (flt-1, KDR etc.) eine wichtige Rolle zu spielen. In vielen Tumorzelllinien wurde beobachtet, daß sie gegenüber dem umgebenden normalen Gewebe eine erhöhte VEGF-mRNA-Konzentration besitzen (Tolnay 1997). 3.3.4 Expression von VEGF in Tumorzellen, die von Endothelzellen abstammen VEGF wirkt hauptsächlich auf Endothelzellen. Ebenso taucht VEGF in Tumorgewebe in einer erhöhten Konzentration auf. Somit erscheint es nicht abwegig, daß VEGF - und 22 ebenso seine Rezeptoren - in diesen Zellen möglicherweise als Onkogene aktiviert werden könnten. Morii fand heraus, daß die Konzentration von VEGF-mRNA im menschlichen GehirnHämangioblastom relativ hoch ist (Morii 1993), was eine gewisse autokrine Stimulation aufzeigt; dieses Phänomen ist jedoch mit Hilfe der Proteinstufen noch nicht vollständig aufgeklärt. Auf der anderen Seite hat man beim murinen Angiosarkom, welches durch chemische Karzinogene induziert wurde, sehr geringe Mengen an VEGF und seinen Rezeptoren festgestellt (Rosen 1990). Somit scheint die Karzinogenese endothelialer Tumoren nicht hauptsächlich auf die Wirkung von VEGF zu basieren. 3.3.5 Ein Einblick auf die VEGF Genregulation Sehr viele verschiedene Substanzen können zu einer „up“-Regulation des VEGF-Gens führen (Beispiel: cAMP, Östrogene, TGF-alpha, Prostaglandine, mutiertes p53-Gen) (Claffey 1992, Kieser 1994, Garrido 1993, Yan 1993, Pertovaara 1994, Harada 1994). Ebenso können hypoxische Zustände zu einer Induktion des VEGF-Gens führen, welches Ladoux und Frelin am Beispiel von Kardiomyozyten gezeigt haben (1993). Claffey postulierte, daß Bedingungen, die generell zu einer Umdifferenzierung einer Zelle führen, mit einer erhöhten VEGF-Genaktivität einhergehen (1992). Für die eigentliche Tumorentstehung erscheint das Phänomen der „up“-Regulation des VEGF-Gens bei Hypoxie sehr bedeutsam, denn erst wenn Tumorzellen Anschluß an das Gefäßsystem bekommen - also aus dem hypoxischen Zustand gleiten - können sie sich „explosionsartig“ vermehren. Diesen Anschluß erreichen die Tumorzellen wahrscheinlich durch die Freisetzung von VEGF, wodurch sie die Angiogenese einleiten. Aber nicht nur aus der Sicht der Tumorangiogenese ist dieser Aspekt wichtig. Vielmehr ist dieses Phänomen eine wichtige biologische Antwort auf die niedrige Sauerstoffbedingung in verschiedenen Zellen des höher entwickelten Organismus. 23 3.4 DIE FUNKTION VON VEGF IN GEFÄßENDOTHELIEN 3.4.1 Proliferationsstimulation und tubuläre Formation von Endothelzellen VEGF besitzt die Fähigkeit, bei allen bisher bekannten Endothelzellen zum Zellwachstum zu führen (Ferrara und Henzel 1989). Man hat ebenso festgestellt, daß Lebersinusoidalzellen stark VEGF-abhängig sind. Das impliziert, daß VEGF nicht nur als Wachstumsfaktor wirkt, sondern auch eine sehr wichtige physiologische Bedeutung für einige Endothelzellen besitzt (Yamane 1994). In vitro bewirkte VEGF bei Endothelzellen eine tubuläre Formation (Connolly 1989, Goto 1993). Obwohl dieser Prozeß sehr komplex ist, vermutet man, daß VEGF einen direkten Faktor der Angiogenese darstellt, sowohl in vivo als auch in vitro. 3.4.2 Permeabilitätssteigerung an Gefäßen VEGF verursacht eine Permeabilitätssteigerung an den Gefäßendothelien (Senger 1983). Die effektive Konzentration von VEGF ist bis zu 1000 Mal stärker als die von Histamin und Bradykinin (Connolly 1989 und 1991). Man nimmt an, daß die Permeabilitätssteigerung über eine Signaltransduktion, durch aktivierte VEGF-Rezeptoren initiiert, an das Zytoskelettsystem weitergegeben wird, wobei die Transzytose und/oder die Auflockerung des Zellverbandes der Endothelzellen die Folgeerscheinung darstellt (Collins 1993). Der Mechanismus ist jedoch noch nicht vollständig aufgeklärt. Es ist aber möglich, durch die Koinjektion von VEGF-Antikörper die VEGFPermeabilitätssteigerung aufzuheben (Sioussat 1993). 3.4.3 Andere biologische Aktivitäten des VEGF auf Endothelzellen Der erste Schritt der Angiogenese ist die Auflockerung der interzellulären Matrix durch Proteasen. In HUVEC-haltigem Gewebe (Human Umbilical Vein Endothelial Cells) konnte beispielsweise beobachtet werden, daß VEGF sowohl auf mRNA- als auch auf 24 der Proteinstufe die Kollagenasegene stimuliert (Unemori 1992). Ebenso wurde beobachtet, daß bei bestimmten Endothelzellen der Plasminogen-Aktivator (PA) und der PA-Inhibitor auf VEGF-Stimulus „up-reguliert“ wurde (Pepper 1994). Dies wurde jedoch in HUVEC nicht beobachtet (Bikfalvi 1991). Die unterschiedliche Antwort der Endothelien ist noch nicht aufgeklärt. Ku und Mitarbeiter (1993) berichteten, daß durch VEGF-Stimulation die Endothelien der Herzkranzgefäße mit einer Stickstoffoxid (NO)-Freisetzung reagierten (NO ist synonym zu EDRF = Endothelium Derived Relaxation Factor), was zur Koronargefäßdilation führte. Es konnte ebenso gezeigt werden, daß sowohl glatte Muskelzellen als auch Kardiomyozyten bei einer Hypoxie mit der Expression von VEGF mRNA reagieren (Ladoux und Frelin 1993). So ist anzunehmen, daß die durch VEGF induzierte NO-Bildung für die Aufrechterhaltung der kontinuierlichen Sauerstoffversorgung des Herzens obligat ist und somit eine physiologische Reaktion des Organismus darstellt. 3.4.4 Obwohl VEGF-Effekte auf andere Zellen als Endothelien sich die Wachstums- und Permeabilitätsstimulation des VEGF auf Endothelzellen beschränkt, hat man dennoch eine in seltenen Fällen auftauchende Migrationsstimulation auf menschliche Monozyten festgestellt (Clauss 1990). Zusätzlich wurde die Steigerung des prokoagulanten Gewebsfaktors und ebenso der erhöhte Einstrom von Kalziumionen in Monozyten nach VEGF-Bahandlung registriert (Clauss 1990). VEGF induziert ebenfalls die Differenzierung von Osteoblasten, was die Möglichkeit einer Beteiligung von VEGF beim Knochenumbau zeigt (Midy und Plouet 1994). 3.5 DIE REGULATION DES TUMORWACHSTUMS DURCH SUPPRESSION DES VEGF-SYTEMS 25 3.5.1 Suppression der Angiogenese Insgesamt ergib sich bisher zusammenfassend, daß § das schnelle Wachstum des Tumorgewebes in vivo erst durch Anschluß an das Gefäßsystem durch Angiogenese initiiert wird, § das VEGF-System eine wichtige Rolle bei der Stimulation der TumorAngiogenese spielt. Es muß daher möglich sein, durch artifizielle Suppression des VEGF-Systems die Wachstumsrate des Tumors in vivo reduzieren zu können. Dabei könnten verschiedene Stufen in der Signalkette des VEGF und seinem Rezeptorsystem Ansatzpunkte einer Blockade darstellen durch § Chemikalien, § Antikörper oder § andere Peptide. Kim und Mitarbeiter (1992 und 1993) haben gezeigt, daß es möglich ist, durch monoklonale Antikörper gegen das N- oder C-terminale Ende des VEGF-Moleküls eine Suppression der VEGF-Aktivität zu erreichen. Zudem können diese Antikörper eine Herabsetzung des Tumorwachstums in Mäusen verursachen. Es wurden ähnliche Ergebnisse auch durch den Einsatz von polyklonalen Antikörpern berichtet (Kondo 1993). Eine andere Strategie ist die Rezeptormodulation des VEGF, wodurch die Wirkung durch einen Bruch in der Signalkette nicht zur Geltung kommt (Kendall und Thomas 1993). 3.5.2 Die Kontrolle über Aszites-Bildung und Metastasierung VEGF wurde ursprünglich von Senger und Mitarbeiter (1983) in von Tumoren gebildeter Aszites-Flüssigkeit nachgewiesen. Es ist aber immernoch nicht geklärt, in welchem Ausmaß VEGF für die Aszitesbildung verantwortlich ist. Der Beitrag des 26 VEGF für die Aszites- oder Pleuraergußbildung ist bei jedem Patienten unterschiedlich. Wenn es jedoch gelingen würde, die Menge an VEGF anzugeben, die bei den verschiedenen Patienten zur abnormen Gefäßpermeabilität führt, so könnte die Behandlung der Patienten durch Blockade des VEGF- und des VEGF-Rezeptor-Systems durchaus einen Ansatzpunkt darstellen. 27 4 SCATTER-FACTOR UND C-MET 4.1 SCATTER FACTOR 4.1.1 Molekularbiologische Eigenschaften Scatter Factor (SF) ist ein Protein, das vom Mesenchym synthetisiert werden soll (nach neueren Artikeln auch in epithelialen Tumoren) und die Fähigkeit besitzt, Epithelzellverbände zu dissoziieren („to scatter“) (Stoker 1987, Rosen 1989). SF ist identisch mit dem Hepatocyte Growth Factor (HGF) (Weidner 1991, Bhargava 1992), der mitogene Wirkung auf Rattenhepatozyten besitzt und dessen Funktion ursprünglich als hepatotroph bei der Leberreparatur definiert wurde (Miyazawa 1989, Nakamura 1989). Im folgenden wird zur Vereinfachung von Scatter Factor (SF) gesprochen, wobei damit auch HGF gemeint ist. SF ist ein heparinbindendes Glykoprotein, das aus einer 60 kDa α-Kette und einer 30 kDa β-Kette besteht (Gherardi 1989, Rosen 1990, Weidner 1990). Die α-Kette besitzt am N-terminalen Ende eine „Haarnadel“-ähnliche Schleife und vier „Kringel“-ähnliche Komplexe (Disulfid-Brücken Bindungen, wodurch die ProteinLiganden-Bindung zustande kommt). Die β-Kette ist bezüglich der Aminosäuresequenz mit Serinproteasen verwandt. SF und das Proenzym Plasminogen besitzen eine 38 prozentige Homologie in der Aminosäure-Sequenz, jedoch besitzt SF keine Proteaseaktivität aufgrund des Unterschiedes von zwei essentiellen Aminosäuren an der katalytischen Triade der βKette (Nakamura 1989, Rosen 1990). Nach seiner intrazellulären Transkription und Translation wird SF - mit einer Signalsequenz versehen - nach außen abgegeben und durch den SF-Aktivator, eine Serin-Protease ähnlich dem Hagemann-Faktor XII, physiologisch in seine aktive Form übergeführt (Miyazawa 1993). Der SF-Aktivator wird in einer zymogenen Form gebildet und nach einer Gewebsverletzung durch eine proteolytische Kaskade aktiviert (Miyazawa 1994). Auch Plasminogen-Aktivatoren (tPA und uPA) können Pro-SF 28 spalten und aktivieren. Dies geschieht aber nur in einer sehr hohen Konzentration gegenüber dem physiologischen Wert (Naldini 1992). 4.1.2 Die biologische Aktivität von SF SF beeinflußt über die c-Met-Tyrosinkinase: § die Motogenese, § die Mitogenese, § die Morphogenese von bestimmten Zellen und § die Angiogenese (Weidner 1993, Zhu 1994). Zusätzlich zum „Auflösen“ von Epithelverbänden kann SF bei isolierten Epithelzellen eine ungerichtete Migration durch die extrazelluläre Matrix sowie eine Chemotaxis bewirken (Rosen 1990, Li 1994). SF erleichtert die Migration durch Verstärkung der Transkription und Proteinexpression von uPA und uPA-Rezeptoren (uPAR), die sich an der Zelloberfläche befinden (Pepper 1992, Grant 1993, Rosen 1994). Die an der Oberfläche gebundenen uPA sind katalytisch aktiv und könnten so einen Weg für hier migrierende Zellen durch Auflösung der extrazellulären Matrix „frei“ machen (Saksela und Rifkin 1988). Offensichtlich kann SF in diesen Funktionen eine Tumorinvasion und -metastasierung erleichtern. Wodurch dieser gesamte Mechanismus - durch SF initiiert in Gang gebracht wird, ist noch unklar. SF wirkt auf vielen Zelltypen mitogen, insbesondere auf Epithel- und Gefäßendothelzellen und Melanozyten (Kan 1991, Rubin 1991, Halaban 1992). SF ist ebenfalls ein sehr potenter morphogener Faktor. Er induziert bei Epithelzellen, die in Kollagen Typ I-Gel inkubiert wurden, ein Netzwerk von verzweigenden Tubuli mit konsequenter apikal-basolateraler Polarität (Montesano 1991, Santos 1992). Ähnlich induziert SF bei Epithelzellen der Mamma die Ausbildung einer milchgangsähnlichen Struktur (Tsarfaty 1992) (Abb. 8, 9). 29 Insgesamt kann SF verschiedene Differenzierungsvorgänge bei Zellen initiieren, die insbesondere vom Zelltypus abhängig sind. 4.2 C-MET Der Rezeptor des SF , das c-Met, ist ein Proteinprodukt des Proto-Onkogens c-met (Bottaro 1991), das als transmembranöse Tyrosin-Kinase (TK) insbesondere durch Epithelien und Fibroblasten exprimiert wird (Gonzatti-Haces 1988). Der c-Met-Rezeptor ist ein Glykoprotein mit einem Molekulargewicht von 190 kDa, das aus einer 50 kDa αKette besteht und sich an der Zellwandoberfläche befindet, sowie aus einer 145 kDa βKette mit transmembranöser Lokalisation. Die extrazelluläre Bindungsstelle, die transmembranöse sowie intrazelluläre Kinase und der nicht-katalytische Phosphatrezeptor befinden sich auf der β-Kette (Ponzetto 1994). Ursprünglich wurde das Proto-Onkogen c-Met in der Zellinie eines chemisch behandelten menschlichen Osteosarkoms entdeckt (Cooper 1984). c-Met, der SFRezeptor, wird hauptsächlich in epithelialen Zellen gebildet. Obwohl auch mesenchymale Zellen diesen Rezeptor exprimieren, wird c-Met hier in einem viel niedrigeren Umfang produziert (Faletto 1993, Sonnenberg 1993). Untersuchungen haben gezeigt, daß c-Met in vielen verschiedenen menschlichen Karzinomen und Sarkomen überexprimiert wird (Di Renzo 1991, Rong 1993). Es wurde auch festgestellt, daß die Tyrosinkinase-Aktivität mit dem Ausmaß einer malignen Zelltransformation eng assoziiert ist (Ottenhoff-Kalf 1992). Ferner wurde gezeigt, daß die erhöhte Expression von c-Met bei den Fibroblasten als Resultat eine erhöhte Invasivität aufwies, sowohl in vivo als auch in vitro (Giordano 1993, Rong 1994). 4.3 DIE AKTIVITÄT VON SF AUF BLUTGEFÄßE IN VIVO UND IN VITRO 4.3.1 Die Gefäßendothelzellen 30 4.3.1.1 Die Endothelzelle als SF-Produzent Die Gefäßendothelzellen § produzieren SF und § können Zielobjekte von SF sein. Die Endothelien produzieren in vitro und in vivo physiologisch geringe Mengen an SF (Stoker 1987, Rosen 1989, Matsumoto 1993). Es wurde auch festgestellt, daß nach Leberschädigung sowohl die Lebersinusoid-Endothelien als auch die Alveolarendothelzellen der Lunge SF produzieren. Dieses konnte man durch in-situ Hybridisierung bestätigen (Yanagita 1992, Noji 1990). Hieraus kann man folgern, daß durch bestimmte Stimuli (Beispiel: Leberschädigung) Endothelzellen zur Produktion von SF angeregt werden. Es ist möglich, daß parakrine oder endokrine Signale, die im Zusammenhang mit einer Noxe auftreten, für die Konzentrationserhöhung des SF - von Endothelzellen produziert - verantwortlich sind. 4.3.1.2 Die Endothelzelle als Zielzelle des SF In der frühen Phase der Angiogenese löst sich in vivo die Basalmembran durch den Einfluß von Endothelzellen an bestimmten Stellen auf. Die Endothelien wandern ins Interstitium in Richtung eines angiogenetischen Stimulus und formen einen kapillären Ast (Folkman 1985). Die verzweigenden Endothelzellen, die sich in der wandernden Spitze befinden, proliferieren. Hierbei organisieren sich die verzweigenden Endothelzellen in ein anastomosierendes Netzwerk von Kapillargängen. In der Schlußphase der angiogenetischen Reaktion kommt es zur Adhäsion der neugebildeten glatten Muskelzellen, der Perizyten und der Basalmembran (Folkman 1985, AntonellìOrlidge 1989). Die Stimulation der Endothelzellen zur Motilität, Proliferation und Bildung kapillargangsähnlicher Formation in vitro korrelieren mit der Angiogenese in vivo (Folkman 1985). 31 § Abb. 8 Mikroskopisch sichtbarer “Scatter-Effekt” Die Beimischung von Scatter Factor (SF / HGF) in eine Fibroblasten-Kultur führt zum “Auseinanderdriften” der Fibroblasten und zur mitotischen Teilung (Quelle: Bussolino 1992). § Abb. 9 Wirkspektrum des hepatischen Wachstumsfaktors (HGF/SF) (Quelle: Polverini 1995). 31a Die Endothelien kleiner und großer Gefäße exprimieren c-Met und können so durch SF stimuliert werden (Rosen 1990 und 1991, Bussolino 1992, Grant 1993, Naidu 1994). SF wirkt chemotaktisch auf Endothelzellen und beeinflußt die Motilität (Rosen, 1990 und 1991). Rosen hat in einer Versuchsreihe gezeigt, daß Endothelzellen auf einen SFStimulus hin durch ein Matrixgel auf einen porösen Filter (stellt die Basalmembran dar) penetrieren (Rosen 1991). SF beeinflußt ebenso die DNA-Synthese und die Proliferation von bestimmten Endothelzelltypen (Rubin 1991, Morimoto 1991). Die kapilläre Tubenformation scheint eine von der Motilität und Proliferation unabhängige Variable zu sein (Grant 1989). Sobald Endothelzellen auf der Oberfläche eines als Basalmembran funktionierenden Matrixgels aufgebracht werden, stellen sie DNA-Synthese und Proliferation ein. Es vollzieht sich eine zytoplasmatische „Verlängerung“, die mit dem Beginn einer kapillärtubulären Formation einhergeht. Diese kapillär-tubuläre Formation konnte bei Gehirnarterien-Endothelzellen und HUVEC (Human Umbilical Vein Endothelial Cell) durch SF bis zu einer zehnfach höheren Geschwindigkeit beschleunigt werden (Rosen 1991, Grant 1993). 4.3.2 DIE GEFÄßMUSKELZELLEN (SMC) UND DIE PERIZYTEN 4.3.2.1 SMC als SF-Produzent In vitro produzieren die Kalbsaorten-, die menschlichen Iliakalarterien- und die Rattenarterien-SMC SF in einer vergleichsweise hohen Konzentration wie die menschlichen Lungen-Fibroblasten (64 bis 128 Einheiten / 1000 000 Zellen / 48 h) (Rosen 1989 und 1990). Die biologischen und chemischen Eigenschaften des von den SMC produzierten SF sind denen des von den Fibroblasten produzierten SF sehr ähnlich und es ist anzunehmen, daß beide Proteine identisch sind (Rosen 1990). 32 4.3.2.2 SMC als Zielzellen des SF Die Psoriasis vulgaris (Schuppenflechte) ist eine chronische Hautkrankheit, die durch die Proliferation von epidermalen Keratinozyten und Neovaskularisation im Stratum papillare gekennzeichnet ist. Die Zellen der Gefäßwände (Perizyten, Endothelzellen) sind im „psoriatischem Plaque“ immunhistochemisch positiv für das c-Met-Protein (Grant 1993). Demnach können diese Zelltypen durch SF stimuliert werden. Die SMC in Tumormikrogefäßen exprimieren SF. Die neugebildeten SMC und die Perizyten (die auch als SMC der Mikrogefäße bezeichnet werden) sind die essentiellen Komponenten der Angiogenese. Sie dienen dazu, die neugebildeten Gefäße zu stabilisieren und bilden den Abschluß eines angiogenetischen Prozesses (AntonellìOrlidge 1989). SF reguliert dementsprechend auch die Abschlußphase der Angiogenese. Die Fähigkeit von SMC, SF zu produzieren und ebenso die Zielzelle darzustellen, zeigt, daß SF eine autokrine Funktion für diesen Zelltyp besitzt. Die SMC ruhen in vivo, können aber durch eine Gefäßverletzung stimuliert werden. Dieses konnte durch die „phänotypische Modulation“, die bei der Exzision von in vitro befindlichen Gefäß-SMC auftritt, gezeigt werden (Campbell und Chamley-Campbell 1981). Die phänotypische Modulation ist charakterisiert durch die Transition einer „ruhigen“ kontraktilen SMC in eine aktive, synthetisierende und proliferative Variante. Man hat aber auch gemessen, daß nicht-stimulierte SMC signifikante Mengen an SF in vivo produzierten. Weitere Untersuchungen haben gezeigt, daß SF eine mögliche parakrine (SMC auf Endothelzelle) und autokrine (SMC auf SMC) Funktion - assoziiert mit einer Gefäßverletzung – besitzt (Rosen und Goldberg 1997). 4.4 SF IM TUMORGEWEBE 4.4.1 Die Expression von SF und c-Met im Tumorgewebe Die SF- und c-Met – Produktion wird sowohl im physiologischen als auch in pathologischen Prozessen „up“- und „down“-reguliert (Sonnenberg 1993, Matsumoto 33 und Nakamura 1993, Joannidis 1994). SF wird ebenso in verschiedenen Tumorgeweben (Beispiel: Mammakarzinom, Blasenkarzinom u.a.) überproduziert (Rosen 1994, Yamashita 1994, Joseph 1995). Möglicherweise führen spezifische SF-induzierende Proteine zur Überproduktion von SF und c-Met, die im Tumorgewebe in einer unphysiologisch hohen Konzentration vorliegen (Li 1996). 4.4.2 Die Herkunft des Tumor-SF Mesenchymale Zellen sind die Zelltypen mit der höchsten SF-Produktion in vitro einschließlich Fibroblasten, glatte Gefäßmuskelzellen, Gliazellen, Makrophagen, Endothelzellen und ebenso T-Lymphozyten (Stoker 1987, Rosen 1989 und 1994, Noji 1990, Shiota 1992, Yanagita 1992, Naidu 1994). Viele dieser Zellen sind im Tumorgewebe präsent und tragen ebenso zu einer Überproduktion des SF bei. Li meinte, daß durch die Interaktion zwischen Stroma und Epithel, Fibroblasten SF sezernieren und so Epithelzellen in vielen Organen (Haut, Gefäße u.a.) in ihrer Entwicklung regulieren (Li 1996). Durch viele Untersuchungen wurde deutlich, daß die SF-Konzentration im malignen Gewebe einen wichtigen prognostischen Faktor darstellt (Yamashita 1994, Weidner 1991, Bosari, 1992, Toi 1993). 4.4.3 SF und Tumorangiogenese Man geht davon aus, daß der Gehalt an SF im Tumor und die Gefäßdichte im Rahmen einer Tumorangiogenese eine nicht zu vernachlässigende prognostische Aussagekraft bei Brustkarzinomen besitzt (Weidner 1991, Bosari 1992, Toi 1993). Eine Korrelation zwischen SF und der Gefäßdichte wäre hierbei vorhanden. In der Angiogenese müssen jedoch auch weitere Proteine bzw. Faktoren, berücksichtigt werden (Beispiel: VEGF, TGF, bFGF u. a.). Es ist möglich, daß SF mit anderen angiogenetischen Faktoren eine additive oder synergistische Interaktion eingeht, wie beispielsweise mit VEGF. Der angiogenetische Phänotyp eines Tumors wird durch die Balance der „pro“- und „anti“angiogenetischen Faktoren determiniert und ebenso seine Tendenz zu wachsen. 34 5 MATERIAL UND METHODEN 5.1 SEKUNDÄRE PLEURATUMOREN Die in dieser Untersuchung ausgewerteten Pleurakarzinosen bzw. -metastasen sind in einem Zeitraum von 3 Jahren (1994 bis 1997) gesammelt worden. Es handelt sich um Pleurabiopsien, die durch bronchoskopischen, perkutanen, thorakoskopischen oder thorakotomischen Eingriff als PE`s (PE: Probeexzision, operative Gewebeentnahme für diagnostische Zwecke als Form der Biopsie) in das Institut für Pathologie an den Berufsgenossenschaftlichen Kliniken „Bergmannsheil Bochum“ gelangt sind. Anhand konventioneller Schnittpräparate in HE- (Hämatoxylin-Eosin) oder EvG-Färbung (Elastika van Gieson) wurde die Diagnose gestellt. Unter Berücksichtigung klinischer Angaben konnten die Absiedlungen verschiedenen Primärtumoren zugeordnet werden. Von diesen Patienten lagen nicht nur archivierte histologische Befunde vor, sondern auch Angaben zu personenbezogenen Daten. 5.2 EINTEILUNG DER PLEURATUMOREN Insgesamt wurden 82 sekundäre Pleuratumoren ausgewertet. Da im Besitz des Instituts ebenso 16 Primärtumor-Biopsien vorhanden waren, wurden diese als Vergleich zu den sekundären Tumoren in die Auswertung miteinbezogen. Zusätzlich wurden Pleurabiopsien untersucht, die bei langjährigem krankheitsfreien Verlauf als sogenannte „dormant cells“ klassifiziert wurden. Dabei ergab sich folgende Verteilung (Pleurakarzinosen nach Primärtumoren geordnet): § 16 Mammakarzinome § 21 Lungenkarzinome § 18 Karzinome des Gastrointestinaltraktes § 9 Karzinome des Urogenitaltraktes 35 § § 5.3 verschiedene Primärtumoren: - Pankreaskarzinom - Kehlkopfkarzinom - Ewing-Sarkom - Leiomyosarkom - Schildrüsenkarzinom 9 ungeklärte Primärtumoren. DIE IMMUNHISTOCHEMIE - DIE APAAP-METHODE Diese Technik beruht auf einem indirekten Nachweis (des zu untersuchenden Proteins), der durch einen Brücken-Antikörper, der den Primär-Antikörper mit den APAAPKomplex verbindet, erreicht wird. Der Komplex besteht aus einem Antikörper und Enzym (Alkalische Phosphatase), gegen das der Antikörper gerichtet ist. Man kann vereinfacht von einem Enzym-Antienzym Komplex sprechen. 5.4 METHODEN -BESCHREIBUNG 1. 3-4 µm dicke Schnittpräparate formalinfixierten und in Paraffin eingebetteten Gewebes werden auf einen speziellen Kapillarspalt-Objektträger der Firma DAKO aufgezogen; anschließend werden die Präparate über Nacht bei 58° C im Wärmeschrank getrocknet. 2. Die Schnittpräparate werden in Xylol entparaffiniert und in einer absteigenden Alkoholreihe rehydriert, anschließend in Tris-Puffer (pH = 7,6) für 10 Minuten gespült. 3. Die Vorbehandlung der Schnittpräparate erfolgt in Abhängigkeit vom verwendeten Primär-Antikörper. 4. Der Primär-Antikörper wird in der jeweiligen Gebrauchsverdünnung für 25 Minuten bei Raumtemperatur inkubiert; die Antikörperverdünnung erfolgt mit Hilfe eines Diluents (Firma DAKO). Die Präparate werden im Puffer (Puffer-Kit 36 K 5006, Firma DAKO) gespült. Bei polyklonalen Antikörpern wird anschließend „mausifiziert“, d.h. es wird mit MAR (Maus anti-Rabbit-Serum, 1:150 in Diluent verdünnt, Firma DAKO) für 25 Minuten bei Raumtemperatur inkubiert. 5. Die Präparate werden gespült und anschließend für 25 Minuten bei Raumtemperatur mit einem Sekundär-Antikörper (= LINK, APAAP-Kit K 5000, Firma DAKO) inkubiert; beim Brücken-Antikörper handelt es sich um Kaninchen anti-Maus Immunglobuline aller Isotypen. 6. Nach Spülung mit Puffer wird für weitere 25 Minuten bei Raumtemperatur mit einem APAAP-Komplex (APAAP-Kit K 500, Firma DAKO) inkubiert. Der APAAP- Komplex besteht aus monoklonalen Maus-Immunglobulin G1Antikörpern, welche spezifisch an die alkalische Phosphatase (aus KälberMukosa gewonnen) gebunden ist. 7. Zur Verstärkung der Reaktion werden nach Spülung mit Puffer wiederholt LINK und APAAP-Komplex für jeweils 10 Minuten Raumtemperatur aufgegeben. 8. Zur Farbentwicklung wird ebenfalls der APAAP-Kit benutzt; das Chromogen wird maximal 10 Minuten vor Gebrauch angesetzt. Der verwendete Farbstoff ist Fast-Red. Die endogene Alkalische Phosphatase wird dabei durch Zugabe von Levamisol (0,2 mmol, ebenfalls im APAAP-Kit vorhanden) geblockt. Die Inkubationszeit beträgt 4 x 5 Minuten, wobei zwischendurch immer wieder mit Puffer gespült wird. 9. Die Gegenfärbung erfolgt in Mayers-Hämatoxylin (Firma DAKO) für 1 Minute. Anschließend werden die Präparate erst in Puffer, dann in Aqua dest. gebläut. 10. Die Schnittpräparate werden in aufsteigender Alkoholreihe dehydriert und über Xylol mit Eukitt eingedeckt. 5.5 DIE AUSWERTUNG Damit die immunhistochemischen Untersuchungsergebnisse einheitlicher und somit vergleichbarer waren, wurde auf der Consensus-Tagung in Mainz 1986 der sogenannte 37 „Immunreaktive Score (IRS)“ ermittelt. Ursprünglich war diese Methode für den immunhistochemischen Nachweis von Östrogen- und Progesteronrezeptoren im Mammakarzinomgewebe entwickelt worden (Remmele und Stegner 1987). Anhand dieser Methode untersucht man die Färbeintensität (SI) und den Prozentsatz positiver Zellen (PP) im Gewebepräparat. Wir haben diese Methode in einer abgewandelten Form übernommen. 1. Die PP-Einteilung wurde modifiziert, d.h. die ursprüngliche Unterscheidung zwischen <10% und < 50% positive Zellen wurde in der Auswertung nicht eingehalten, da dieses unerheblich auf das Ergebnis war. 2. Wir haben den immunreaktive Score nicht berechnet und haben die einzelnen angiogenetischen Faktoren sowohl in der Färbeintensität (SI) als auch in der PPEinteilung gesondert betrachtet und anhand des Fisher-Tests eine Korrelation untersucht. Tabelle 1: Immunreactive Score: SI- und PP-Einteilung SI-Einteilung PP-Einteilung 0 = keine Farbreaktion 0 = keine positiven Zellen 1 = schwache Farbreaktion 1 = < 50 % positive Zellen 2 = mäßige Farbreaktion 2 = 50 - 80 % positive Zellen 3 = starke Farbreaktion 3 = > 80 % positive Zellen Die Auswertung erfolgt mit Hilfe eines Lichtmikroskopes, wobei bei verschiedenen Vergrößerungen (x250, x400) die Färbeintensität und der prozentuelle Anteil positiver Zellen ermittelt wird. 5.6 STATISTIK 38 Die histomorphologischen Befunde der sekundären Pleuratumoren wurden nach Methoden der deskriptiven Statistik ausgewertet. Unterstützend wurde hierzu der „exakte Test von R. A. Fisher“ (Sachs 1984) hinzugezogen. Das Signifikanzniveau wurde bei p=0,05 festgesetzt. Es wurde die SAS-Software als Rechenprogramm im Windows-NT-System benutzt. 5.7 DIE IN-SITU HYBRIDISIERUNG 5.7.1 Definition Die in-situ Hybridisierung gehört zu den molekularbiologischen experimentellen Techniken. Dabei kommt es zur Bindung von in der Zelle befindlichen mRNA an einsträngige komplementäre cDNA, die man in die Zelle eingebracht hat, mit Ausbildung stabiler Hybridmoleküle. 5.7.2 Die Fluoreszenz-in-situ-Hybridisierung (FISH) In dieser Arbeit wurde die Fluoreszenz-in-situ-Hybridisierung (FISH) - Technik für cMet und VEGF angewandt. Dabei geht man folgenderweise vor: 1. Insertion des Gens in das E. coli Genom 2. Vervielfältigung dessen durch E. coli 3. Ausschneiden des Inserts (cDNA) 4. Markierung des Inserts mit Digoxigenin 5. Erhitzen auf 95° C (um DNA-Einzelstränge zu produzieren) 6. Dann sofortiges Abkühlen im 4° C - Bad (damit wieder kein Doppelstrang entsteht) 7. Nun Behandlung der immunhistochemisch positiven Zellen mit dem Einzelstrang-DNA-Substrat (bei 42° C) 39 8. Inkubation mit Digoxigenin-Antikörpern und Fluorescein Hat die Zelle nun c-Met- oder VEGF-Proteine produziert, so muß dort auch mRNA des c-Met- oder VEGF-Gens vorhanden sein, so daß durch die Inkubation mit homologer cDNA eine Markierung der mRNA durchgeführt werden kann. Das Ergebnis wird durch die Reaktion mit Fluorescein sichtbar. 40 6 ERGEBNISSE 6.1.1 Biopsien: Patientenzahl, Geschlechts- und Altersverteilung Im Institut der Pathologie an den Berufsgenossenschaftlichen Kliniken Bergmannsheil werden Biopsien der Pleura nicht nur unter wissenschaftlichen, sondern auch unter gutachterlichen Gesichtspunkten untersucht. Hierbei handelt es sich besonders um die Klärung von Berufskrankheiten (BK). Die Begutachtung kann große versicherungsrechtliche Folgen haben. Insbesondere bei der Unterscheidung eines primären von einem sekundären Pleuratumor, wobei bei letzterem der Primärtumor klinisch zu diesem Zeitpunkt noch stumm sein kann. So ergab sich, daß im bei einigen Pleurakarzinosen kein Primärtumor bestimmt werden konnte. 1. Biopsiegut von 71 männlichen und weiblichen Patienten wurde untersucht (von einigen Patienten wurden mehrere Biopsien aus verschiedenen Bereichen der Pleura entnommen; daher insgesamt 82 Biopsien). 2. Das Durchschnittsalter aller Patienten lag zum Zeitpunkt der Biopsieentnahme bei 65 ± 8 Jahren (Median: 67 Jahre). 3. Im Kollektiv befanden sich 41 männliche Patienten. Das Alter lag bei durchschnittlich 63,5 ± 9 Jahren (Median: 67 Jahre). Der älteste Patient war 77 und der Jüngste 25 Jahre alt. Der Altersgipfel lag im männlichen Kollektiv bei 72 Jahren. 4. Die Zahl der weiblichen Patienten betrug 30. Das Durchschnittsalter lag bei 66 ± 6,5 Jahren (Median: 67 Jahre). Die älteste Patientin war 91, die jüngste 46 Jahre alt; der Altersgipfel lag bei 70 Jahren. Es zeigt sich somit, daß die männlichen Patienten im Durchschnitt 3 Jahre jünger waren als die weiblichen Patienten zum Zeitpunkt der Biopsieentnahme. Es gab bei beiden 41 keinen Unterschied im Altersgipfel. Bezüglich des Medians ist auch kein Unterschied zu erkennen. 42 DIAGRAMM 1: ABSOLUTE ALTERSVERTEILUNG: MÄNNER / FRAUEN: 86-95 Altersabschnitt 76-85 66-75 56-65 46-55 36-45 26-35 16-25 0 5 10 15 20 25 Anzahl der Patienten Männer Frauen DIAGRAMM 2: PROZENTUALES ALTERSVERHÄLTNIS ZWISCHEN MÄNNER UND FRAUEN: 96-105 86-95 Altersabschnitt 76-85 66-75 56-65 46-55 36-45 26-35 16-25 0% 10% 20% 30% 40% 50% 60% 70% 80% 90% 100% Prozentuales Verhältnis Männer Frauen 43 6.1.2 Verteilungsverhältnis der sekundären Pleuratumoren Die am häufigsten in die Pleura metastasierten Tumoren im Kollektiv waren die Lungentumoren. Mit 21 primär in der Lunge lokalisierten Karzinomen stellt dieses Kollektiv 26 Prozent der metastasierenden Primärtumoren. Die häufigsten Lungentumoren waren insbesondere die kleinzellig differenzierten Bronchialkarzinome. Unter den Tumoren waren auch häufig die Adenokarzinome vertreten. Nur wenige plattenepitheliale Lungentumoren hatten sich in der Pleura manifestiert. Zu den zweithäufigsten pleurapenetrierenden primären Karzinomen zählten die gastrointestinalen Tumoren und die Mammatumoren. Die im Gastrointestinaltrakt lokalisierten Tumoren befanden sich hauptsächlich im oberen Bereich, nämlich im Ösophagus und besonders häufig im Magen, daneben auch im Kolon. Insgesamt waren im Kollektiv 22 Prozent gastrointestinale Tumoren und 20 Prozent Mammakarzinome vorhanden, die in der Pleura zu einer Sekundärmanifestation geführt haben. Im Kollektiv waren 11 Prozent primär im Urogenitaltrakt lokalisierte Karzinome vorhanden, wobei bei den Nierenmalignomen besonders das hellzellige Karzinom dominierte (60%). Ein Zehntel der Fälle zeigten pleurale Metastasen bei Pankreas-, Kehlkopf- und Schildrüsenkarzinomen sowie bei Ewing- und Leiomyosarkomen. Diese Fallzahlen waren allerdings sehr niedrig. Bei ca. 11 Prozent der Fälle konnte trotz zusätzlicher Immunhistochemie der Primärtumor nicht ermittelt werden. Diese waren ausschließlich Adenokarzinome. Gesamtzahl: 82 sekundäre Pleuratumoren (Pleurakarzinosen) 44 Tabelle 2: Verteilung der einzelnen Biopsien auf Primärtumoren Sekundäre Pleuratumore Frauen Männer Gesamtanteil Lunge 8 16 26% Gastrointestinaltrakt 7 11 23% Mamma 15 0 20% Urogenitaltrakt 3 6 11% Verschiedene 0 7 9% Ungeklärte 1 8 11% Summe 34 48 100 DIAGRAMM 3: PROZENTUALVERHÄLTNIS DER SEKUNDÄREN PLEURATUMOREN: Verschiedene 9% Ungeklärte 11% Lunge 26% Urogen.-Trakt 11% Mamma 20% GI-Trakt 23% 45 Zusammenfassung der wesentlichen Befunde (Teil I): § Frauen waren bei der Biopsieentnahme durchschnittlich 3 Jahre älter. § Das Durchschnittsalter lag bei beiden Geschlechtern bei 65 Jahren. § Die Männer waren mit 41 Patienten in der Mehrheit gegenüber 30 weiblichen Patienten. § Der Altersgipfel war bei beiden ähnlich, nämlich zwischen 70 und 72 Jahren. § Die Pleurakarzinosen hatten ihren Ursprung hauptsächlich in der Lunge, Gastrointestinaltrakt und Mamma. § Histologisch war die häufigste Diagnose das Adenokarzinom. 6.2 MAKROSKOPISCHE UND MIKROSKOPISCHE BEFUNDE Die Unterscheidung sekundärer Pleuratumoren von primären Pleuratumoren, d.h. dem Mesotheliom, bereitet dem Pathologen nicht selten Schwierigkeiten, da diese oft nicht nur durch mikroskopische, sondern auch durch ähnliche makroskopische Befunde imponieren. Es werden bei der Obduktion meist knotige Tumorherde auch mit Infiltration des Lungengewebes beobachtet, was nicht selten zu einer Kompressionsatelektase im angrenzenden Lungengewebe führt. Die Patienten klagen dabei über Luftnot und geringeres Atemvolumen. 6.2.1 Ausgesuchte sekundäre Pleuratumoren mit differenzial- diagnostischen Kriterien 6.2.1.1 Primärtumor: Lunge Diese waren meist Adenokarzinome. In zahlreichen Präparaten waren heterogene Differenzierungen vorhanden. Die entdifferenzierten Karzinosen waren oftmals kaum 46 vom Mesotheliom zu unterscheiden und Klarheit konnte nur durch zusätzliche immunhistochemische Reaktion geschaffen werden. 6.2.1.2 Primärtumor: Gastrointestinaltrakt Auch diese Karzinosen waren zumeist von adenoidem Charakter. Die ösophagalen und gastralen Karzinome sowie die Kolonkarzinome waren die häufigsten Vertreter. Die Pleurakarzinosen mit gastrointestinalem Primärtumor waren meist mit klinisch manifesten Primärtumor assoziiert. Hierdurch wurde die Diagnose des Pleuratumors erleichtert. Die Tumoren metastasierten im relativ fortgeschrittenen Stadium in die Pleura. Im histologischen Präparat zeigte sich die Tendenz einer Karzinose zu einem fokal nekrotischen Zerfall. 6.2.1.3 Primärtumor: Mammakarzinom Da bei naher Lage der Mamma zur Pleura die kavitäre Form der Metastasierung vorliegen kann, geht das Wachstum der Metastase oft von der Pleura parietalis aus. Die Metastasen hatten in der Pleura meist eine polymorphe Struktur, teils durch solides, teils durch tubuläres Wachstum gekennzeichnet. Ein weiteres Charakteristikum war das multifokal ödematös aufgelockerte Bindegewebe mit zum Teil starken Fibrosierungsprozessen. Häufig waren Primärmalignom und seine Pleurakarzinose uniform bezüglich des Differenzierungsgrades. 6.2.1.4 Primärtumor: Nierenzellkarzinom Auch hier bestand eine relativ vermehrte Fibrosierung einhergehend mit einem Stromaödem. Angrenzend befand sich ein hochdifferenziertes Karzinom, das durch ein papilläres Wachstumsmuster mit epitheloidem Charakter imponierte. Das umgebende 47 Stroma war häufig gefäßreich. Bei den Primärtumoren handelte es sich meist um hellzellige Karzinome. 6.2.1.5 Die Lymphangiosis carcinomatosa Da das bioptische Material häufig zu klein für die sichere Diagnose einer Lymphangiosis carcinomatosa war, konnte diese nicht immer mit Sicherheit diagnostiziert werden. Jedoch fanden sich besonders bei den Mammakarzinomen im Bereich der parietalen Pleura und bei den Lungentumoren eine Lymphangiosis carcinomatosa. Zusammenfassung der wesentlichen Befunde (Teil II): § Adenokarzinome stellen den größten Anteil der hier untersuchten Pleurakarzinosen. § Bei allen Präparaten zeigten sich reaktive Veränderungen. § Es bestand meist eine chronische Begleitentzündung in Form einer (fibrosierenden) Pleuritis. § Das Ausmaß der Entzündung korrelierte mit dem Ausmaß der Tumorzellnekrosen. Die Entzündung war dann randständig betont und mit einem gemischtzelligem Infiltrat gekennzeichnet. § Bei Karzinosen gastrointestinaler Tumoren war häufig ein Primärtumor klinisch gesichert mit Metastasierung im fortgeschrittenem Stadium. § Metastasen der Nierenzellkarzinome waren häufiger höher differenziert. 48 6.3 DIE IMMUNHISTOCHEMISCHEN ERGEBNISSE 6.3.1 SF, c-Met und VEGF Durch die Immunhistochemie markiert, ließen sich die Zellen, beziehungsweise die Strukturen, die SF und c-Met oder VEGF enthielten, durch eine tiefpurpurne Färbung (Fast Red) darstellen. In wenigen Fällen konnte aufgrund einer Überfärbung kein zuverlässiges Urteil gefällt werden. 6.3.1.1 Primärtumor: Lunge Im Kollektiv waren 24 Lungenbiopsien von 8 weiblichen und 16 männlichen Patienten vorhanden. VEGF und SF zeigten in 20 Fällen eine positive Reaktion und in 4 Fällen keine. c-Met besaß nur in 2 Fällen keine erkennbare Reaktion (Abb. 10). a) VEGF: Es fanden sich 6 schwache, 10 mäßige und 4 starke Reaktionen. 4 reagierten nicht. Demgegenüber hatten 2 Biopsien unter 50%, 9 Biopsien 50 bis 80% und ebenso 8 Biopsien über 80% positiv reagierende Zellen. Dabei war auffällig, daß bei einer mäßigen Farbreaktion (SI = 2) über 50% Tumorzellen eines Präparates eine Reaktion zeigten. Dies war auch bei den zwei stark reaktiven Präparaten (SI = 3) ähnlich. Es bestand somit eine Korrelation zwischen der Farbintensität (SI) und dem Prozentsatz positiver Zellen (PP). Dies ließ sich auch anhand des „exakten Tests“ von R. A. Fisher belegen: Bei einem Signifikanzniveau von p=0,05 konnte man durch den Test ein p < 0,001 ermitteln, was auf eine sichere Korrelation hinwies. Die im Gewebe am stärksten gefärbten Zellen waren eindeutig die Tumorzellen. Im Gegensatz zur c-Met-Immunhistochemie war das Bindegewebe in den meisten Fällen nicht reaktiv oder sehr schwach reaktiv. Ebenso verhielt es sich bei den Fibroblasten (Abb. 11). 49 b) c-Met: Hier fanden sich 11 stark und 10 mäßig reagierende Präparate. Ein Präparat war schwach reaktiv. Bei diesem waren weniger als 50% der Zellen positiv. Da bei der c-Met Färbung 19 Präparate einen positiv gefärbten Tumorzellanteil von über 80% besaßen, kann man daraus schließen, daß auch hier eine Korrelation zwischen SI und PP vorhanden ist. Dieses wird durch den Fisher-Test bestätigt; p ist wieder kleiner 0,001. Die Tumorzellen besaßen die stärkste Farbreaktion. Sie imponierten häufig durch tubuläre Anordnung und durch fokale Färbereaktionen. Ähnlich verhielten sich die in der Nähe befindlichen Endothelien der kleinen Gefäße. Das Bindegewebe zeigte häufig eine mäßige Farbreaktion; die Fibroblasten waren fokal intensiv reaktiv, blieben insgesamt in ihrer Reaktionsstärke hinter den Tumorzellen. c) SF: Bei der SF-Immunhistochemie zeigte sich größtenteils dasselbe Bild wie bei der c-MetFärbung. Der Fisher-Test zeigte hier ein p < 0,001. Jedoch waren Biopsiepräparate vorhanden, bei denen sich fokal keine Tumorzellreaktion zeigte. Hier waren die Fibroblasten insgesamt besser reaktiv als das Bindegewebe. In entzündlichen Gebieten wiesen die Lymphozyten generell keine Reaktion mit den SF-Antikörpern auf. 50 Tabelle 3: Ergebnisse der Immunhistochemie - Lunge Färbeintensität (SI) Prozentsatz Primärtumor: Positiver Zellen (PP) in % Lunge VEGF c-Met SF VEGF c-Met SF 1 mäßig mäßig stark > 80 > 80 > 80 2 mäßig mäßig stark > 80 > 80 > 80 3 mäßig mäßig schwach 50 - 80 > 80 50 - 80 4 keine stark keine - > 80 - 5 schwach mäßig mäßig 50 - 80 > 80 > 80 6 schwach mäßig mäßig 50 - 80 > 80 > 80 7 mäßig stark stark > 80 > 80 > 80 8 keine keine keine - - - 9 keine stark mäßig - > 80 > 80 10 schwach stark mäßig 50 - 80 > 80 > 80 11 mäßig mäßig stark - > 80 > 80 12 stark mäßig stark > 80 > 80 50 - 80 13 keine stark stark - > 80 50 - 80 14 mäßig mäßig mäßig < 50 > 80 > 80 15 schwach schwach keine < 50 < 50 - 16 mäßig stark mäßig 50 - 80 > 80 < 50 17 schwach keine keine 50 - 80 - - 18 mäßig stark stark > 80 > 80 > 80 19 stark stark mäßig > 80 > 80 > 80 20 mäßig mäßig schwach 50 - 80 < 50 50 - 80 21 stark stark stark > 80 > 80 > 80 22 stark stark mäßig > 80 > 80 < 50 23 mäßig stark stark 50 - 80 > 80 > 80 24 schwach mäßig stark 50 - 80 50 - 80 > 80 51 Pleurametastasierung bei bekanntem Lungentumor (E.-Nr. 27968/96) § Abb. 10 EvG; x20 Disseminiert gelagerte Tumorzellverbände mit kräftiger desmoplastischer Reaktion (breite Kollagenfaserbündel). § Abb. 11 VEGF - Reaktion; x20 Einzeln gelegene Gruppen von Tumorzellen mit starker feingranulärer intrazytoplasmatischer Expression. Geringgradige positive Markierung auch benachbarter Stromazellen. 51a Zusammenfassung Bei einem Signifikanzniveau von p=0,05 war eine deutliche Korrelation zwischen der Färbeintensität (SI) und dem Prozentsatz positiver Zellen (PP) bei allen drei immunhistochemischen Färbungen vorhanden, dabei war p<0,001. Tumorzellen waren immer stärker gefärbt als das umliegende Gewebe. 52 DIAGRAMM 4: SI & PP LUNGE Färbeintensität (SI): 12 Anzahl 10 8 6 4 2 0 0 1 2 3 SI-Grad VEGF c-Met SF Prozentsatz positiver Zellen (PP): 20 Anzahl 15 10 5 0 0% < 50% 50-80% >80% Positive Zellen VEGF c-Met SF 53 6.3.1.2 Primärtumor: Gastrointestinaltrakt Es befanden sich 18 Biopsiefälle aus dem Gastrointestinaltrakt im Kollektiv, wobei 7 von weiblichen und 11 von männlichen Patienten stammen (Abb. 12). a) VEGF: Bei diesem Faktor reagierten 8 (44%) stark, 4 (22%) mäßig und 3 (17%) schwach. Weitere 3 (17%) Fälle zeigten keine Reaktion. Ungefähr 78% der Präparate war so gefärbt, daß mehr als 50% - bei 8 sogar mehr als 80% - der Tumorzellen immunhistochemisch positiv reagierten. Hier war auch nach dem Fisher-Test eine enge Korrelation zwischen SI und PP vorhanden. Auffallend war die häufige Nichtreaktion des Bindegewebes. Hingegen zeigten die Fibroblasten und Myozyten eine mäßige Reaktion. Tumorzellen sind nach wie vor die am stärksten reaktiven Zelleinheiten (Abb. 18, 19, 20, 21). b) c-Met: Hier waren 2 Biopsien nicht reaktiv. 5 waren schwach, 5 mäßig und 6 stark reagierend. Auch hier war die Mehrzahl der Präparate, 8 (44%), dadurch gekennzeichnet, daß mehr als 80% der Tumorzellen eine positive Reaktion aufwiesen. Mikroskopisch waren die Endothelzellen sehr gut immunhistochemisch gefärbt. Eine Korrelation zwischen SI und PP war vorhanden. Wie bei den Lungenbiopsien waren auch hier die Tumorzellen generell stärker reaktiv als das umliegende Gewebe. Auffällig war, daß die Fibroblasten eine fast ebenso gute immunhistochemische Reaktion aufwiesen. Jedoch war bei einigen Präparaten fokal keine Reaktion gegen jene vorhanden. Das Bindegewebe und die glatte Muskulatur waren in variabler Form manchmal gut und manchmal kaum reaktiv. Im Gegensatz zur SF-Immunreaktion der Lunge wiesen die Lymphozyten hier in den entzündlichen Gebieten der Biopsie eine relativ gute Farbreaktion auf (Abb. 13, 16, 17). 54 c) SF: Beim SF zeigte die Mehrheit der Biopsien (11 Fälle = 61%) eine mäßige Reaktion. 6 (33%) waren stark reagierend, 1 Präparat zeigte keine immunhistochemische Färbung. Bei 7 Präparaten besaßen weniger als 50% der Karzinosezellen eine Farbreaktion, bei 11 reagierten mehr als 50% der Tumorzellen mit einer Färbung. Die Tumorzellen waren hier die reaktivsten Zellen. Das Bindegewebe und die Fibroblasten waren selten gefärbt. Glatte Muskelzellen der Darmwand waren häufig reaktiv. In wenigen Präparaten befindliche lymphozytäre Infiltrate waren nur fokal schwach farbreaktiv (Abb. 14, 15). 55 Tabelle 4: Ergebnisse der Immunhistochemie - Gastrointestinaltrakt FÄRBEINTENSITÄT (SI) Primärtumor: Prozentsatz Positiver Zellen (PP) in % Gastrointestinal-trakt VEGF c-Met SF VEGF c-Met SF 1 keine schwach keine - < 50 - 2 mäßig schwach mäßig > 80 < 50 50 - 80 3 stark keine mäßig 50 - 80 - 50 - 80 4 stark mäßig mäßig 50 - 80 50 - 80 50 - 80 5 stark mäßig mäßig > 80 50 - 80 50 - 80 6 keine mäßig stark - 50 - 80 > 80 7 mäßig mäßig mäßig > 80 > 80 < 50 8 mäßig mäßig mäßig > 80 > 80 < 50 9 schwach schwach mäßig 50 - 80 50 - 80 < 50 10 schwach schwach mäßig 50 - 80 > 80 < 50 11 mäßig schwach mäßig 50 - 80 > 80 < 50 12 keine keine mäßig - - < 50 13 stark stark stark > 80 > 80 > 80 14 stark stark stark > 80 > 80 > 80 15 stark stark stark > 80 > 80 > 80 16 schwach stark stark < 50 50 - 80 > 80 17 stark stark stark > 80 > 80 > 80 18 stark stark mäßig 50 - 80 50 - 80 > 80 56 Zusammenfassung Eine deutliche Korrelation war auch hier zwischen SI und PP vorhanden (p < 0,05). Die meisten Präparate reagierten gut, 10% reagierten nicht. Insgesamt gab es keine Korrelation zwischen dem Geschlecht und dem SI- oder PPGrad. 57 Pleurametastasierung bei primärem Karzinom im Gastrointestinaltrakt (E.-Nr. 1915/96) § Abb. 12 HE; x10 Desmoplastische Reaktion in Nähe der Tumorabsiedlung. § Abb. 13 c-Met - Reaktion; x20 Mittelstarke c-Met - Expression der Tumorzellen; fehlende Reaktion in Stroma. 57a Pleurametastasierung bei primärem Karzinom im Gastrointestinaltrakt (E.-Nr. 19548/96) § Abb. 14 Scatter Factor (SF) - Reaktion; x20 Strangartige Tumorzellverbände mit deutlich positiver SF-Expression. § Abb. 15 Scatter Factor (SF) - Reaktion; x40 Feingranuläre zytoplasmatisch nachweisbare SF-Expression. 57b Pleurametastasierung bei Gallengangskarzinom im Ductus Choledochus (E.-Nr. 19548/96) § Abb. 16 c-Met - Reaktion; x40 Starke c-Met - Immunoreaktivität der Tumorzellen intrazytoplasmatisch mit umgebender desmoplastischer Reaktion. § Abb. 17 c-Met - In-situ-Hybridisierung; x625 Punktförmiger Nachweis der c-Met - mRNA - Transkripte in den Tumorzellen (FISH). 57c Pleurametastasierung bei primärem Karzinom im Gastrointestinaltrakt (E.-Nr. 1537/96) § Abb. 18 VEGF - Reaktion; x40 § Abb. 19 VEGF - In-situ-Hybridisierung; x625 Vergleich zwischen der VEGF-Immunoreaktion und der VEGF - in-situ-Hybridisierung. Deutlicher Nachweis der mRNA-Transkripte für VEGF in komprimiert gelagerten Tumorzellen korreliert mit der mittelstarken VEGF-Immunoreaktion. 57d Pleurametastasierung bei primärem Karzinom im Gastrointestinaltrakt (E.-Nr. 1537/96) § Abb. 20 VEGF - Reaktion; x40 § Abb. 21 VEGF - In-situ-Hybridisierung; x625 Mittelstarke VEGF-Immunoreaktion und Fluoreszenz-in-situ-Hybridisierung (FISH) mit Nachweis von VEGF-mRNA. Zentral traubenförmig gelagerte Tumorzellen im ausgeweiteter Gefäßstruktur mit starker VEGF-Syntheses. 57e DIAGRAMM 5: SI & PP GASTROINTESTINALTRAKT Färbeintensität (SI): 12 Anzahl 10 8 6 4 2 0 0 1 2 3 SI-Grad VEGF c-Met SF Prozentsatz positiver Zellen (PP): 8 Anzahl 7 6 5 4 3 2 1 0 0% < 50% 50-80% >80% Positive Zellen VEGF c-Met SF 58 6.3.1.3 Primärtumor: Mamma Im Kollektiv befanden sich 15 Pleurakarzinose-Biopsien primärer Mammatumoren von insgesamt 12 Patientinnen. Die jüngste Patientin war 56, die älteste 71 Jahre alt (Abb. 23). a) VEGF: 9 Fälle (60%) zeigten eine gute, jeweils 2 (13%) eine starke und 2 schwache Reaktion. 2 waren nicht reaktiv. 6 Präparate (40%) waren durch Tumorzellen gekennzeichnet, von denen mehr als 80% eine positive Farbreaktion aufwiesen. Bei weiteren 6 waren 50 bis 80% der Tumorzellen positiv. Es war also eine enge Korrelation zwischen SI und PP vorhanden. In dieser immunhistochemischen Reaktion dominierten wiederum die Tumorzellen. Das Bindegewebe war häufig nicht bzw. fokal sehr schwach reaktiv. Die Fibroblasten wiesen ein inhomogenes Bild auf - teilweise reagierten sie mäßig bis sehr schwach (Abb. 22, 24, 28, 29). b) c-Met: Es zeigten 11 Präparate (73%) eine mäßige, 2 eine starke und weitere 2 eine schwache Reaktion. Weiterhin waren 10 (67%) Biopsien mit Tumorzellen charakterisiert, von denen über 80% immunhistochemisch positiv reagierten. Tatsächlich war auch hier eine Reaktionsdominanz der Tumorzellen zu sehen. Generell wies das Bindegewebe keine Farbreaktion auf, die Fibroblasten waren teilweise schwach bis sehr gut gefärbt (Abb. 25, 26). c) SF: Bei der Reaktion gegen SF waren 8 Biopsien (53%) stark positiv. 6 (40%) waren mäßig, und eine Biopsie schwach in der Reaktion. 6 Präparate besaßen Tumorzellen, von 59 denen über 80% positiv reaktiv waren. Weitere 6 Präparate zeigten Tumorzellen, von denen zwischen 50 und 80% der Zellen eine positive Reaktion aufwiesen. Insgesamt waren die Tumorzellen im Vergleich zum umliegenden Gewebe sehr stark reaktiv. Die Fibroblasten waren eher mäßig bis gut, und das Bindegewebe kaum farbreaktiv (Abb. 27). 60 Tabelle 5: Ergebnisse der Immunhistochemie - Mamma FÄRBEINTENSITÄT (SI) Primärtumor: Prozentsatz Positiver Zellen (PP) in % Mamma VEGF c-Met SF VEGF c-Met SF 1 mäßig mäßig stark > 80 > 80 50 - 80 2 mäßig schwach mäßig 50 - 80 < 50 50 - 80 3 keine mäßig stark - > 80 > 80 4 schwach mäßig stark > 80 > 80 > 80 5 keine mäßig schwach - < 50 50 - 80 6 schwach schwach mäßig < 50 > 80 50 - 80 7 stark stark mäßig > 80 > 80 > 80 8 mäßig mäßig stark 50 - 80 > 80 > 80 9 mäßig mäßig stark > 80 > 80 50 - 80 10 mäßig mäßig stark > 80 > 80 < 50 11 stark stark stark 50 - 80 > 80 > 80 12 mäßig mäßig mäßig 50 - 80 50 - 80 < 50 13 mäßig mäßig mäßig 50 - 80 50 - 80 < 50 14 mäßig mäßig mäßig 50 - 80 50 - 80 50 - 80 15 mäßig mäßig stark > 80 > 80 > 80 Zusammenfassung Eine mäßige Korrelation war zwischen SI und PP vorhanden (p < 0,05). Bei dieser Organgruppe war eine relativ mäßige Reaktion typisch. Umliegende Gewebe war zum Teil stärker reagierend als Tumorzellen selbst (Beispiel: cMet - Antikörperreaktion bei Fibroblasten). 61 Pleurametastasierung eines Mammakarzinoms - (E.-Nr. 2354/96) § Abb. 22 VEGF - Reaktion; x40 Pleurametastasierung bei primärem Karzinom in der Mamma (E.-Nr. 10914/96) § Abb. 23 HE; x10 Metastasierende Tumorzellverbände der Pleura bei invasivem duktalen Mammakarzinom. 61a Pleurametastasierung bei primärem Karzinom in der Mamma (E.-Nr. 16362/96) § Abb. 24 VEGF - Reaktion; x20 § Abb. 25 c-Met - Reaktion; x40 Mittelstark bis mäßige VEGF- und c-Met - Expression innerhalb der Tumorzellkonglomerate. 61b Pleurametastasierung bei primärem Karzinom in der Mamma (E.-Nr. 15521/96) § Abb. 26 c-Met - Reaktion; x20 § Abb. 27 Scatter Factor (SF) - Reaktion; x40 Deutliche immunhistochemische intrazytoplasmatische Expression von c-Met, schwache positive Reaktion von Bindegewebszellen. Ebenso eine deutliche Scatter Factor Expression der Tumorzellen. 61c Pleurametastasierung eines Mammakarzinoms - (E.-Nr. 2354/96) § Abb. 28 VEGF - Reaktion; x40 § Abb. 29 VEGF - Reaktion; x20 Starke VEGF-Expression der Tumorzellen eines primären Mammakarzinoms bei lymphangtischer Ausbreitung in der Pleura. Befund sogenannter “dormant cells”: Die Resektion des Tumors fand 1985 statt, d.h. ca. 11 Jahre vor klinischer Manifestation der Pleurakarzinose. In Tumornähe gelegene stimulierte Mesothelzellen zeigen eine deutliche VEGF-Expression bei malignem Pleuraerguß. 61d DIAGRAMM 6: SI & PP MAMMA Färbeintensität (SI): 12 10 Anzahl 8 6 4 2 0 0 1 2 3 SI-Grad VEGF c-Met SF Anzahl Prozentsatz positiver Zellen (PP): 10 9 8 7 6 5 4 3 2 1 0 0% < 50% 50-80% >80% Positive Zellen VEGF c-Met SF 62 6.3.1.4 Primärtumor: Urogenitaltrakt Die meisten dieser Präparate stellten Absiedlungen hellzelliger Nierenkarzinome dar, daneben Metastasen eines Ovarialkarzinoms und eines Urothelkarzinoms (Abb. 30, 31). In dieser Versuchsreihe zeigten sich die intensivsten c-Met-Reaktionen. a) VEGF: Bei dieser immunhistochemischen Färbung reagierten 2 (22%) stark positiv, 4 (44%) mäßig positiv und 1 (11%) Präparat schwach positiv. Zwei Biopsien waren nicht reaktiv. Nur bei 4 bioptischen Fällen reagierten mehr als 80% der Zellen mit einer positiven immunhistochemischen Färbung. Hier waren die Tumorzellen die reaktivsten Zellen. Das Bindegewebe und ebenso die Fibroblasten waren relativ reaktionsarm. Beim Ovarialkarzinom waren alle Zellen des Präparates reaktionsarm. Das Bindegewebe und die Fibroblasten der Metastase des Harnblasentumors zeigten keine Reaktion. b) c-Met: Es waren hier 4 (44%) stark, 3 (33%) mäßig und 2 (22%) Biopsien schwach reaktiv. Bei 7 (78%) Präparaten besaßen mehr als 80% der Zellen eine immunhistochemisch positive Färbung! Sämtliche Gewebsstrukturen waren gut reagierend gegenüber der c-Met-Färbung. Zum Teil fanden hier die in der gesamten Untersuchung stärksten Reaktionen der Strukturen statt. Unter den Zellen dominierten auch hier die Tumorzellen; die anderen Strukturen (Myozyten, Fibroblasten und Lymphozyten) wiesen häufig eine relativ gute Farbreaktion auf. c) SF: 63 Ein ähnliches Bild bot sich auch bei der SF-Immunhistochemie. 6 (67%) Biopsien hatten eine stark positive Reaktion, eine Biopsie war mäßig reaktiv. Weitere 2 waren nicht reaktiv. Ebenso waren hier bei 5 (56%) Präparaten über 80% der Zellen reaktiv bezüglich der Immunhistochemie. 64 Tabelle 6: Ergebnisse der Immunhistochemie - Urogenitaltrakt FÄRBEINTENSITÄT (SI) Primärtumor: Prozentsatz Positiver Zellen (PP) in % Urogenitaltrakt VEGF c-Met SF VEGF c-Met SF 1 keine mäßig keine - > 80 - 2 keine schwach keine - > 80 - 3 schwach stark mäßig < 50 > 80 < 50 4 mäßig stark stark > 80 > 80 50 - 80 5 mäßig stark stark > 80 > 80 > 80 6 mäßig schwach stark > 80 < 50 > 80 7 stark mäßig stark 50 - 80 > 80 > 80 8 mäßig mäßig stark < 50 < 50 > 80 9 stark stark stark > 80 > 80 > 80 Zusammenfassung Insgesamt ist eine gute Korrelation zwischen SI und PP vorhanden (p < 0,05). Die Urogenitaltrakt-Gruppe zeigt generell eine gute c-Met- und SF-Reaktion. Die VEGF-Reaktion ist schwächer ausgeprägt als bei den bisher besprochenen BiopsieGruppen. Diese Tumorzellen haben die stärkste Reaktion. 65 Pleurametastasierung bei primärem Urothelkarzinom (E.-Nr. 28106/95) § Abb. 30 HE; x10 Hochdifferenzierte urothel-ähnliche Tumorformationen bei pleuraler Metastasierung. § Abb. 31 HE; x4 Inselartige Lagerung von Tumorzellen mit Bindegewebsreaktion, Brenner-Tumor ähnliches Bild. 65a DIAGRAMM 7: SI & PP UROGENITALTRAKT Färbeintensität (SI): 6 5 Anzahl 4 3 2 1 0 0 1 2 3 SI-Grad VEGF c-Met SF Prozentsatz positiver Zellen (PP): 7 6 Anzahl 5 4 3 2 1 0 0% < 50% 50-80% >80% Positive Zellen VEGF c-Met SF 66 6.3.1.5 Primäres Karzinom unbekannter Lokalisation Bei diesen Präparaten handelt es sich um Fälle, bei denen ein Mesotheliom anhand eingehender Histologie und Immunhistochemie sicher ausgeschlossen werden konnte, jedoch eine sichere und eindeutige Diagnose hinsichtlich der Einordnung bei einem anderen Orts lokalisierten Primärtumors nicht möglich war. Im Kollektiv waren insgesamt 9 Biopsien vorhanden (8 männlich). a) VEGF: Bei der VEGF-Immunhistochemie waren 4 (44%) stark, 3 (33%) mäßig und eine Biopsie schwach positiv. Eine Biopsie zeigte keine Reaktion. Bei 6 (67%) Präparaten waren über 80% der Zellen positiv reaktiv, bei 2 (22%) zeigten weniger als 50% der Zellen eine Reaktion. Da die Präparate unterschiedlicher Herkunft waren, zeigten sie eine unterschiedliche Bindegewebs- und Zellreaktion. Manche waren stark positiv, manche aber auch schwach bis nicht reaktiv. Hinsichtlich der Tumorzellreaktion wiesen die Biopsiepräparate eine durchgehend gute Reaktion auf, was die Dominanz der Tumorzellen bezüglich der Konzentration an VEGF unterstreicht. b) c-Met: Hier fanden sich 6 (67%) stark reagierende Biopsien. Jeweils eine Biopsie reagierte schwach, eine mäßig und eine zeigte keine Reaktion. 5 Präparate besaßen Tumorzellen, von denen über 80% immunhistochemisch reaktiv waren. Wie bei der VEGF-Immunhistochemie zeigten sich auch hier unterschiedlich starke Reaktionen des umliegenden Gewebes. Insgesamt war eine relativ gute c-Met-Färbung nicht nur der Tumorzellen - zu sehen (Abb. 32, 33). c) SF: 67 Bei der PP-Auswertung zeigten die Präparate in der SF-Immunhistochemie dieselben Resultate wie beim c-Met. Jedoch lag der Anteil der stark reagierenden Präparate bei 4 (44%). 3 Biopsien waren mäßig, eine schwach reaktiv und eine reagierte nicht. 68 Tabelle 7: Ergebnisse der Immunhistochemie - Primäres Karzinom unbekannter Lokalisation FÄRBEINTENSITÄT (SI) Positiver Zellen (PP) in % PRIMÄRTUMOR: Primäres Prozentsatz Karzinom unbekannter Lokalisation VEGF c-Met SF VEGF c-Met SF 1 stark stark mäßig > 80 > 80 < 50 2 schwach stark mäßig > 80 > 80 > 80 3 stark mäßig mäßig > 80 50 - 80 > 80 4 mäßig stark stark < 50 50 - 80 > 80 5 keine schwach keine - < 50 - 6 stark stark stark > 80 > 80 > 80 7 mäßig keine stark > 80 - > 80 8 stark stark stark > 80 > 80 > 80 9 mäßig stark schwach < 50 > 80 50 - 80 Zusammenfassung Eine enge Korrelation ist zwischen SI und PP vorhanden (p < 0,05). Jedoch ist insgesamt eine gute immunhistochemische Reaktion aller Biopsien hinsichtlich der Tumorzellen kennzeichnend; 11% sind nicht reaktiv. Das Gewebe besitzt aufgrund unterschiedlicher Herkunft der ungeklärten Tumoren kein einheitliches Reaktionsverhalten gegenüber den Antikörpern. 69 DIAGRAMM 8: SI & PP PRIMÄRES KARZINOM UNBEKANNTER LOKALISATION Färbeintensität (SI): 6 5 Anzahl 4 3 2 1 0 0 1 2 3 SI-Grad VEGF c-Met SF Prozentsatz positiver Zellen (PP): 6 Anzahl 5 4 3 2 1 0 0% < 50% 50-80% >80% PositiveZellen VEGF c-Met SF 70 Pleurametastasierung bei Primärtumor ungeklärter Lokalisation (E.-Nr. 26214/96) § Abb. 32 c-Met - Reaktion; x20 Immunhistochemisch deutlich positive Reaktion der Tumorzellen gegen c-Met. § Abb. 33 c-Met - In-situ-Hybridisierung; x625 Gleichzeitiger Nachweis einer c-Met - Synthese bei positiver c-Met-FISH im Tumorgewebe. 70a 6.3.1.6 Verschiedene sekundäre Pleuratumoren Im Kollektiv waren § 3 Pankreaskarzinome § 1 Schildrüsenkarzinom § 1 Kehlkopfkarzinom § 1 Leiomyosarkom sowie § 1 Ewing-Sarkom vorhanden. Pankreaskarzinom Bei dieser Organgruppe waren von 3 Biopsien bei der • VEGF-Färbung 2 mäßig und eine schwach reaktiv • c-Met-Färbung 2 stark und eine mäßig reaktiv • SF-Färbung 2 schwach und eine mäßig reaktiv. Bei der Mehrzahl der immunhistochemisch gefärbten Präparate zeigten über 80% der Tumorzellen eine positive Reaktion. Sie waren sehr reaktiv. Das Bindegewebe zeigte ebenso eine gute Farbreaktion, die Fibroblasten sind teilweise sehr reaktiv. Insgesamt sind alle Zellen reaktiv. Eine Korrelation zwischen SI und PP war vorhanden (p < 0,05). Schilddrüsenkarzinom Bei dieser Karzinose zeigten bei der VEGF-Immunhistochemie die Tumorzellen und das umliegende Gewebe keine Reaktion; jedoch waren bei der Reaktion gegen c-Met und SF alle Zellen gleichermaßen stark positiv. Eine Korrelation zwischen SI und PP war vorhanden (p < 0,05). 71 Kehlkopfkarzinom Hier war die Immunhistochemie bei allen Faktoren schwach reaktiv; jedoch zeigten über 80% der Tumorzellen eine Reaktion. Das Bindegewebe und die darin enthaltenen Strukturen waren reaktionsarm. Eine Korrelation zwischen SI und PP war nicht vorhanden (p < 0,05). Leiomyosarkom Die SF- und die VEGF-Immunhistochemie zeigten keine positive Reaktion. c-Met hingegen wies eine mäßige Farbreaktion fast aller Tumorzellen auf. Hier waren die Tumorzellen und die Fibroblasten gleichermaßen sehr reaktiv. Eine Korrelation zwischen SI und PP war nicht vorhanden (p < 0,05). Ewing-Sarkom Hier zeigte die SF-Färbung keine Reaktion; hingegen waren VEGF und c-Met jeweils mäßig positiv. Das Bindegewebe war sehr schwach, die Fibroblasten mäßig bis schwach reaktiv. Eine Korrelation zwischen SI und PP war vorhanden. 72 Tabelle 8: Ergebnisse der Immunhistochemie - Verschiedene Primärtumoren FÄRBEINTENSITÄT (SI) Primärtumor Prozentsatz Positiver Zellen (PP) in % VEGF c-Met SF VEGF c-Met SF Nr.1 mäßig stark schwach 50 - 80 > 80 < 50 Nr.2 schwach stark schwach < 50 > 80 < 50 Nr.3 mäßig mäßig mäßig > 80 > 80 > 80 II.Schilddrüsen- keine stark stark - > 80 50 - 80 III.Kehlkopf-karzinom schwach schwach schwach < 50 > 80 > 80 IV.Leiomyosarkom keine mäßig keine - > 80 - V.Ewing-Sarkom mäßig mäßig keine > 80 > 80 - I.Pankreas-karzinom karzinom 73 6.3.1.7 Statistik der Immunhistochemie und Zusammenfassung der Ergebnisse Als statistisches Hilfsmittel wurde für die Auswertung der verschiedenen Farbreaktionen der „exakte Test“ von R. A. Fisher benutzt. Dieser Test besitzt die Fähigkeit, mit einem relativ kleinen Versuchskollektiv (Bei dieser Untersuchung: 26 Biopsien von Lungenmetastasen) aussagefähiges und eindeutiges statistisches Ergebnis zu liefern. Als Alternative wäre der Chi-Quadrat-Test ebenso möglich gewesen; jedoch wäre die Validität aufgrund des kleinen Kollektives begrenzt. Ziel war es, die Beziehung zwischen der jeweiligen Farbreaktion (SI) der Immunhistochemie und dem Prozentsatz positiver Zellen (PP) zu ermitteln. Desweiteren sollte die Beziehung zwischen den angiogenetischen Faktoren untereinander anhand der Farbreaktion (SI) und dem Prozentsatz positiver Zellen (PP) festgestellt werden. Das Signifikanzniveau lag bei p = 0,05, d.h., mit einer 95 prozentigen Wahrscheinlichkeit kann davon ausgegangen werden, daß eine Beziehung oder eine Korrelation zwischen den untersuchten Größen vorhanden ist. Trifft dies zu, so muß p kleiner als 0,05 sein. Dabei ist es irrelevant, in wieweit p kleiner als 0,05 ist. Insgesamt wurde deutlich, daß bei allen drei angiogenetischen Faktoren eine klare Korrelation zwischen der Reaktivität (SI) und dem Prozentsatz positiver Zellen (PP) vorhanden war. Je stärker die Biopsiereaktion mit den Antikörpern war, desto mehr Zellen erschienen als tiefpurpurfarben. Dabei war p häufig kleiner als 0,001. Verglich man nun die Reaktivität (SI) der einzelnen angiogenetischen Faktoren miteinander, so ergab sich folgendes Ergebnis: VEGF-SI mit c-Met-SI => p < 0,01 VEGF-SI mit SF-SI => p < 0,001 74 c-Met-SI mit SF-SI => p < 0,05 Da das Signifikanzniveau p nie größer als 0,05 war, lag eine Korrelation zwischen den einzelnen Faktoren vor. Ein Vergleich der Prozentsätze positiver Zellen (PP) der einzelnen Faktoren ergab folgendes: VEGF-PP mit c-Met-PP => p = 0,085 VEGF-PP mit SF-PP => p< 0,01 c-Met-PP mit SF-PP => p < 0,05 Hier war eine Korrelation zwischen VEGF-PP und SF-PP sowie zwischen c-Met-PP und dem SF-PP zu erkennen. Zwischen VEGF-PP und c-Met-PP bestand lediglich eine Tendenz, jedoch keine signifikante Korrelation (p = 0,085). Bei Ausschluß der Gruppe variabler Primärtumoren (d.h. Pankreaskarzinom, Schildrüsenkarzinom, Kehlkopfkarzinom, Leiomyosarkom sowie Ewing-Sarkom - Kap. 6.3.1.6) in der Korrelationsbestimmung, war auch zwischen VEGF-PP und c-Met-PP eine signifikante Korrelation vorhanden (p < 0,05). Tabelle 9: Der exakte Test von R. A. Fisher in Übersicht VEGF c-Met SF VEGF - (SI): p<0,01 (SI): p<0,001 c-MET (PP): p=0,085 - (SI): p<0,05 SF (PP): p<0,01 (PP):p<0,05 - 75 6.4 ERGEBNISSE DER IN-SITU HYBRIDISIERUNG Die in-situ Hybridisierung wurde für c-Met und VEGF durchgeführt. Hierbei wurden jeweils 7 Präparate aus dem Gesamtkollektiv untersucht. Es wurden Präparate verwendet, die immunhistochemisch für c-Met und VEGF reaktiv waren. Anhand der in-situ Hybridisierung konnten die c-Met und VEGF kodierenden mRNA-Transkripte in Tumorzellen in unterschiedlicher Signalintensität semiquantitativ nachgewiesen werden, beurteilt die wurde (+ = schwach, ++ = mäßig, +++ = stark fluoreszierend). Eine Synthese von c-Met und VEGF in Tumorzellen konnte hierdurch bestätigt werden. Daneben fanden sich auch RNA-Transkript-Signale z.B. in Gefäßendothelzellen oder auch Perizyten und Stromazellen. Eine eigenständige Synthese in Tumorzellen war demnach hier auch zu sichern. Tabelle 10: In-situ Hybridisierung - Signalintensität Signalintensität Lunge E26214/96 Gastrointestinaltrakt E1537/96 Mamma E19737/94 ungeklärte Primärtumoren E19548/96 Urogenitaltrakt E16895/96 Gastrointestinaltrakt Präparat 12 Lunge Präparat 8 c-Met VEGF ++ ++ +++ +++ +++ +++ + + ++ ++ - - - 76 6.5 ZUSAMMENFASSUNG DER ERGEBNISSE § Das Signifikanzniveau wurde bei p = 0,05 angesetzt. § Es lag eine statistisch signifikante Korrelation zwischen den einzelnen angiogenetischen Faktoren (VEGF, c-Met und SF) bezüglich der Stärke der immunhistochemischen Reaktion vor. § Auch für die prozentuale Verteilung immunhistochemisch positiver Zellen ergab sich eine statistisch signifikante Korrelation, lediglich für die prozentuale Immunreaktivität hinsichtlich VEGF und c-Met zeigte sich zwar eine Tendenz der immunhistochemischen Korrelation, jedoch keine statistisch signifikante Korrelation. § Von 82 Pleurabiopsien zeigten 67 (83%) pleurale Metastasen eine positive immunhistochemische Reaktion gegen VEGF. § Die VEGF-Expression war intrazytoplasmatisch in den Tumorzellen lokalisiert. § In 76 (93%) der 82 Biopsien der Pleurametastasen war auch eine c-Met-Expression nachweisbar. § Bei 73 (89%) der 82 Fälle war eine positive Reaktion mit dem Antikörper gegen SF nachweisbar. § Eine Überexpression lag überwiegend in malignen Zellen vor, jedoch zeigten auch Endothelzellen benachbarter Gefäße eine Expression von c-Met (siehe Lunge). § Das Bindegewebe war hauptsächlich in der Lunge reaktiv für alle drei angiogenetischen Faktoren. Das Bindegewebe der Mammatumor-Biopsien war häufig reaktionslos. § Die Fibroblasten waren in Metastasen der Mammatumoren meistens genauso stark reaktiv wie die Tumorzellen selbst. Insgesamt verhielten sich die Fibroblasten relativ variabel gegenüber der immunhistochemischen Reaktionen (schwach bis sehr reaktiv). § In entzündlichen Gebieten waren die Lymphozyten häufig reaktiv. 78 § Die hier untersuchten Sarkome verhielten sich ähnlich: Das Leiomyosarkom war zwar - wie auch das Ewing-Sarkom - reaktiv gegen die c-Met - Immunhistochemie, jedoch fiel die Reaktion gegen SF und VEGF negativ aus. § Insgesamt können die obigen Ergebnisse mit vergleichbarer immunhistochemischer Expression von VEGF, c-Met und SF in Tumorzellen pleuraler Metastasen als Hinweis auf ein mögliches Zusammenspiel dieser angiogenetisch wirksamen Faktoren im Rahmen der Tumorangiogenese gelten. 79 7 7.1 DISKUSSION METHODEN Bei der Untersuchung des Kollektivs wurden die Verfahren der immunhistochemischen Färbung und der Fluoreszenz in-situ Hybridisierung (FISH) angewandt. 7.1.1 Immunhistochemische Färbeverfahren Das Probenkollektiv bestand aus 82 Biopsien pleuraler Metastasen von primär in anderen Organen lokalisierten Tumoren. Die immunhistochemischen Reaktionen wurden durch eine „Fast Red“-Reaktion farblich sichtbar gemacht und semiquantitativ ausgewertet. Dabei ist die Intensität der Färbung nur bedingt ein Anhaltspunkt für die Stärke der Immunreaktion. Die Auswertung wurde durch den (von uns modifizierten siehe Kap. 5.5) „Immunreactive Score (IRS)“ (nach Remmele und Stegner 1987) vorgenommen und um eine objektivere Evaluation zu erhalten, von zwei voneinander unabhängigen Beobachtern durchgeführt. Hierbei ergab sich keine signifikante Abweichung zwischen den beiden Auswertungen. Da die immunhistochemische Färbung maschinell und nicht manuell erfolgte, konnte eine höchstmögliche Standardisierung erreicht werden. Durch häufige Kontrollen wurden zufällige und systemische Fehler weitgehend eliminiert. 7.1.2 In-situ Hybridisierung Anhand der Fluoreszenz- bzw. nicht-radioaktiven in-situ Hybridisierung (FISH) war es möglich, die Synthese von den hier untersuchten angiogenetisch wirksamen Faktoren, endothelialer Wachstumsfaktor (VEGF) und c-Met (Rezeptor des HGF/SF), auf mRNA Ebene zu untersuchen. 80 Die Hybridisierung basiert auf der Fähigkeit, denaturierte (einzelsträngige) DNA mit entsprechenden komplementären Sequenzen unterhalb des Schmelzpunktes Tm zu renaturieren. In-situ Hybridisierungstechniken erlauben eine Detektion von spezifischen Nukleinsäuresequenzen in morphologisch erhaltenen Chromosomen, Zellen oder Gewebeschnitten. Wir haben letztere untersucht. Im Gegensatz zur Filterhybridisierung wird die Nukleinsäure nicht aus den Zellen extrahiert, sondern direkt in der intakten Zelle nachgewiesen (in-situ = in natürlicher Lage). Somit stellt die in-situ Hybridisierung ein Verfahren dar, Nukleinsäurestranges wodurch anhand der einer genetische chemischen Informationsgehalt „Kreuzung“ mit eines analogen Nukleinsäuren ermittelt werden kann. Dabei entstehen Hybridmoleküle, die durch fluoreszierende Stoffe optisch sichtbar gemacht werden können. 7.1.2.1 Fluoreszenz-in-situ Hybridisierung (FISH) Die Handhabung dieser Methode hat gegenüber der radioaktiven in-situ Hybridisierung Vorteile bezüglich einer ungefährlicheren Durchführung und einer besseren Qualität der Signale, was zur Verdrängung der radioaktiven in-situ Hybridisierung geführt hat. Die Signale können wie bei der immunhistochemischen Färbereaktion semiquantitativ ausgewertet werden. Insgesamt ist festzuhalten, daß anhand der immunhistochemischen Reaktionen die hier untersuchten angiogenetischen Faktoren in den pleuralen Metastasen qualitativ bestimmt werden konnten; eine quantitative Bestimmung ist nicht zulässig. Bei einer weiterführenden positiven Fluoreszenz in-situ Hybridisierung (FISH) war es möglich zu folgern, daß die Synthese der Faktoren – und nicht eine Speicherung - in den pleuralen Metastasen stattgefunden hat, da zu den Proteinen die jeweiligen mRNA erfaßt werden konnten. 81 7.2 BEFUNDE 7.2.1 Häufigkeit Im Untersuchungsgut, das aus 82 Pleurabiopsien von 41 männlichen und 30 weiblichen Patienten bestand, fanden sich am häufigsten pleurale Metastasen primärer Lungenkarzinome (26%). Pleurale Metastasen gastrointestinaler Tumoren waren mit 23%, die der Mammakarzinome mit 20% im Kollektiv vorhanden. Die Häufigkeitsverteilung im Probenkollektiv stimmte größtenteils mit dem Ergebnis von Chernow und Sahn (1977) überein. 7.2.2 Verteilung Mehr als 50% aller Pleurakarzinosen bei Frauen waren auf ein Mammakarzinom zurückzuführen. Farcchia zeigte anhand statistischer Erhebungen, daß ungefähr 50% aller Patientinnen mit Mammakarzinomen ein „malignes Pleuraexsudat“ entwickelten (Fracchia 1974). Bei Männern war eine deutliche Dominanz der primären Lungentumoren für pleurale Metastasen zu erkennen. Bei 39% der bioptisch gesicherten Pleurakarzinosen waren Neoplasien der Lungen die Primärtumoren. Mit 27% stellten gastrointestinale Tumoren die zweithäufigste Tumorengruppe für Pleurakarzinosen beim Mann dar. In ungefähr 20% der Fälle konnte der Primärtumor der gesicherten pleuralen Metastasierungen im männlichen Patientengut nicht abschließend diagnostiziert werden. Chrétien zeigte in seinem Untersuchungsgut, daß bei „jungen“ Patienten (bis 30. Lebensjahr) mit Pleuraergüssen ganz vorwiegend entzündliche Veränderungen als Ursache vorlagen, während bei den über 50 Jahre alten Patienten maligne Tumoren dominierten (Chrétien 1985). Das Durchschnittsalter lag bei den untersuchten Patienten zwischen 63 und 65 Jahren und ist mit dem Ergebnis von Chrétien vergleichbar. 82 Tabelle 11: Vergleich eigener Resultate zu den Primärtumoren als Ursache pleuraler Metastasen mit Angaben in der Literatur : Primärtumoren mit sekundären Pleuraergüssen Lunge Mamma GI- Urogenital- Trakt trakt Verschiedene Ungeklärt Mesotheliom 26% 20% 23% 11% 9% 11% * 36% 25% *) 5% 10% *) *) 13% 14% *) *) 5% 48% 5% Reshad 1985 60% 30% Eigene Resultate 1998 (n=82) Sahn 1988 (n=1783) Chrétien 1985 (n=163) 5% 5% (n=200) Hausheer 35% 23% Titscher 1981 52% 31% *) 6% 10% *) *) 1985 (n=811) 14% 3% (n=427) *)Diese Daten wurden entweder bei der Auswertung nicht berücksichtigt oder nicht ins Kollektiv aufgenommen. 84 7.2.3 Klinik pleuraler Metastasen Pleurale Metastasen manifestieren sich in der Initialphase klinisch meist als Pleuraerguß. Obwohl heute bei erheblich verbesserter Tumordiagnostik durch radiologische und klinisch-chemische Verfahren Neoplasien in frühen Entwicklungsstadien erkannt werden können, wird die Manifestation sekundärer Pleuratumoren oft erst über den Pleuraerguß erkannt. Radiologisch imponieren Pleuraergüsse bei Aufnahme im Stehen als Verschattungen der kostophrenischen Winkel. Im Liegen können Ergüsse übersehen werden. Bei Unsicherheit kann die diagnostische Klärung auch mittels Sonographie, Computertomographie, NMR und die Mikroskopie mit Probeentnahme erfolgen (Woodring 1984). Mattay fand heraus, daß bei 1868 untersuchten Patienten mit Pleuraergüssen 785 (42%) Folge bösartiger Tumoren waren (Mattay 1990). Chernow führte aus, daß eine kurative Therapie beim malignen Pleuraerguß in der Regel nicht mehr möglich ist (Chernow 1977). Klinische Symptome eines Pleuraergusses können sein: • Schmerzen • Lungenatelektasen • Belastungsdyspnoe • Kurzatmigkeit • Husten Der Pleuraerguß besitzt benigne und maligne Ursachen. In über 90% lassen sie sich auf folgende Erkrankungen zurückführen: Benigne Ursachen: • (Links-) Herzinsuffizienz • Herzoperationen (Durchgangssyndrom) • Leberzirrhose mit Aszites • Pneumonien • Lungenembolien 85 Maligne Ursachen: • Primäre und sekundäre Tumoren in der Pleura 7.2.4 Lokalisation des malignen Ergusses Amnamnestisch imponierte bei mehr als 50% der Patienten der Erguß einseitig, wobei der Erguß bei Mammakarzinomen am häufigsten war (in über 80% der Fälle). In der Literatur werden verschiedene Zahlen bezüglich der mit Mammakarzinomen assoziierten Ergüsse beschrieben: Raju fand in 83% der Fälle eine einseitige und in 9% einen kontralateralen Erguß (Raju 1970). Chrétien hingegen beziffert den einseitigen Erguß mit 50%, den kontralateralen mit 40% und den beidseitigen Erguß mit 10% (Chrétien 1985). Huzly führte 1991 aus, daß eine simultane Doppelseitigkeit sehr verdächtig auf ausgedehntere pleurale Dissemination des Primärtumors bzw. eine Generalisation sei. Dies sei Anlaß für ausgedehntere Untersuchungen einschließlich Knochenmarksanalyse der Beckenkammstanze, Szintigraphie des Skelettes, CT und NMR. Bei allen anderen Tumoren sei die Seitenlokalisation uninteressant. Er meinte weiterhin, daß eine pleurale Manifestation andernorts lokalisierter Primärtumoren in 2 bis 21% der Fälle auftrete (Huzly 1991). 7.2.5 § Techniken zur Entnahme von Biopsien Bei Pleuraergüssen unklarer Ätiologie muß die Klärung durch Analyse von Punktat oder Biopsiematerial erfolgen. Die Biopsieentnahme kann unterschiedlich durchgeführt werden. Dabei ist die Sensitivität unterschiedlich. § Bei einer (ersten) zytologischen Untersuchung der Biopsie beträgt die Sensitivität ungefähr 66% (Loddenkemper 1998). 86 § Die Nadelblindbiopsie besitzt bei sekundären malignen Pleuratumoren lediglich eine Sensitivität von ca. 57% (Tomlinson 1987). Sahn beziffert sie sogar mit nur 46% (1988). § Eine Kombination von Ergußzytologie und Nadelbiopsie sichert in 73% die Diagnose (Sahn 1988). § Mit thorakoskopisch gewonnenem Material wird eine Sensitivität bis zu 95% angegeben (Ganzemeier 1988). § Bei Kombination von thorakoskopischer Pleurabiopsie mit einer Ergußzytologie kann eine diagnostische Sensitivität von ebenfalls 95% erreicht werden (Ganzemeier 1988). 7.2.6 Die Thorakoskopie 7.2.6.1 Die historische Entwicklung der Thorakoskopie § 1910 Diagnostische Thorakoskopie (Jacobaeus) § 1915 Therapeutische Thorakoskopie (Jacobaeus) § 1937 Therapeutische Thorakoskopie beim Spontanpneumothorax (Sattler) § 1961 Operative Thorakoskopie zur Sympathektomie (Wittmoser) § 1972 Direkte Thorakoskopie (Maaßen) § 1982 Erweiterte Thorakoskopie § 1990 Interventionelle Thorakoskopie (Inderbitzi) § 1991 Thoracoscopic laser assisted lung resection (Landreneau) § 1992 Imaged thoracoscopic surgery (Lewis) § 1992 Minimal invasive Thoraxchirurgie (Loddenkemper 1998) 87 7.2.6.2 Die diagnostischen Vorteile der internistischen Thorakoskopie beim Pleuraerguß § Schnelle und sichere bioptische Diagnostik (incl. Tbc-Kultur und HormonrezeptorStatus). § Biopsien nicht nur von der Brustpleura, sondern auch von Zwerchfell, Lunge und Mediastinum. § Ausschluß einer malignen oder tuberkulösen Ätiologie mit hoher Sicherheit. § Goldstandard für wissenschaftliche Untersuchungen. (Loddenkemper 1998) 7.2.6.4 Therapeutische Vorteile der internistischen Thorakoskopie beim Pleuraerguß § Komplette sofortige Entfernung des Ergusses § Eröffnung von gekammerten Flüssigkeitshöhlen § Einschätzung des Reexpansionspotentials der Lunge § Talkpuderung zur Pleurodese (nicht-chirurgischer Goldstandard) § Früher Therapiebeginn (z.B. bei Tbc) (Loddenkemper 1998) 7.2.7 Morphologische Diagnostik Die beste Tumorsicherung (Goldstandard) wird durch morphologische Auswertungen von Biopsien unter Einsatz immunhistochemischer Zusatzuntersuchungen erzielt. Bei der Diagnose ist thorakoskopisch gewonnenes Material (4-5 mm im Durchmesser) der perthorakalen Nadelblindbiopsie (Ramel-Biopsie) deutlich überlegen. Seit 15 Jahren werden immunhistochemische Methoden bei der Diagnostik von malignen Pleuraergüssen eingesetzt (Müller 1998, Loy 1990). Hierbei werden mit Hilfe von monoklonalen Antikörpern maligne Zellen im Erguß durch Markierung z.B. von 88 Oberflächenantigenen nachgewiesen. Die Spezifität beträgt 100%, die Sensitivität schwankt zwischen 75 und 100% (Brockmann 1991, Loy 1990, Pavesi 1988). Mit gezieltem Einsatz immunhistochemischer Markerprofile kann die Dignitätsbestimmung und histogenetische Ableitung pleuraler Tumoren dem Kliniker wesentlich sicherer mitgeteilt werden. Mit Antikörpern, die gegen Zytokeratine, Vimentin, HEA, BMA 120, Leu-M-1 sowie Calretinin gerichtet sind, ist die versicherungsmedizinisch relevante Frage der Abgrenzung primärer Mesotheliome zu Pleurakarzinosen insbesondere primärer Adenokarzinome im Regelfall zuverlässig möglich (Müller 1998). Das Untersuchungsgut besitzt für den Pathologen die höchste diagnostische Wertigkeit. In dieser Arbeit war die Anzahl pleuraler Metastasen primärer Mammatumoren mit einem Anteil von 20% (15 Biopsien) gegenüber den Metastasen der gastrointestinalen Tumoren (23% mit 18 Biopsien) vergleichsweise unterrepräsentiert, wenn man davon ausgeht, daß gerade bei den Mammakarzinomen der Pleuraerguß unklarer Genese das Initialsymptom einer Pleurametastasierung darstellt (Pfitzner 1989). Die Pleurametastasen der Lungenkarzinome stellten im eigenen Untersuchungsgut mit 26% (24 Fälle) den größten Anteil an sekundären Pleurakarzinosen. Dabei betrug das Verhältnis der Männer zu den Frauen 2:1. Der in dieser Arbeit beschriebene Anteil unklarer maligner Ergüsse korrelierte mit 11% (9 Fälle) durchaus mit dem von Sahn gewonnenen Untersuchungsergebnis; er bezifferte sie mit 7% (Sahn 1988). 7.2.8 Charakterisierung pleuraler Metastasen Beurteilt man die pleuralen Metastasen verschiedener Primärtumoren, so ergeben sich folgende Beobachtungen: § Pleuraergüsse bei Primärtumoren der Lunge manifestierten sich anamnestisch häufig in frühen Stadien der Tumorenbildung - insbesondere bei den kleinzelligen Bronchialkarzinomen und bei den Adenokarzinomen. Daher muß von einer frühzeitigen Metastasierung in die Pleura ausgegangen werden. Meyer kam nach Untersuchungen von metastasierenden Bronchialkarzinomen zu einem ähnlichen 89 Resultat. Er fand heraus, daß diese Tumorgruppe frühzeitig eine Pleurakarzinose durch Tumorembolien in den Ästen der Pulmonalarterie bildet (Meyer 1966). § Hingegen war bei der Pleuramanifestation gastrointestinaler Tumoren (n=18), insbesondere der Magentumoren, anamnestisch eine multiple Metastasierung in verschiedenen Organen des Patienten vorausgegangen, so daß hier eine pleurale Metastasierung in einem relativ fortgeschrittenen Stadium diskutiert werden muß. § Die Mammatumoren waren im Probenkollektiv unterrepräsentiert, obgleich diese Tumorgruppe nach Fracchia eine der häufigsten Ursachen des Pleuraergusses darstellen (Fracchia 1974). § Unter den untersuchten Biopsien waren Adenokarzinome verschiedener Primärtumoren am stärksten vertreten. Nach Huzly sind Ausbreitungsweg und Stadium pleuraler Metastasierung vom Sitz des Primärtumors abhängig: Beim primär bronchialen und bronchopulmonalen Tumor verläuft der Weg vom Bronchus zum Lungenparenchym und von dort aus zur Pleura visceralis. Diese Tumoren metastasieren relativ früh über die Lymphbahnen. Bei einem extrapulmonalen Karzinom wie z.B. beim Magenkarzinom, findet eine Metastasierung nach dem Kavatyp statt; dabei können Lunge und Bronchus isoliert oder simultan oder sukzedan befallen werden. Im Anschluß kann auch eine Pleurabeteiligung erfolgen. Diese Manifestation ist in einem relativ fortgeschrittenen Stadium vorzufinden (Huzly 1991). Unsere Beobachtungen bestätigen die Befunde von Huzly. 7.3 DIE BEHANDLUNGSMÖGLICHKEITEN PLEURALER TUMOREN Kreuser und Huzly schätzen die Neuinzidenz pleuraler Metastasen in Deutschland auf 15000 bis 30000 pro Jahr (Kreuser 1984, Huzly 1991). Dabei ist nicht auszuschließen, 90 daß ein großer Teil kausal behandelbarer Fälle der Diagnostik entgeht. Isolierte große Pleuratumoren stellen eine dringliche Indikation zur notwendigen Operation dar. Diese können infolge schnellen Wachstums die Respiration des Patienten erheblich behindern und zusätzlich kann der enorme Eiweißverlust durch die Ergußbildung (Exsudat) erheblich sein und zu sekundären Erkrankungen (z.B. Hypalbuminämie mit Ödemen, Hyperlipoproteinämie, Hypercholesterinämie) führen. Hier entfällt die sogenannte Pleurodese. Die Pleurodese stellt im Idealfall eine Sklerosierung des Pleuraspaltes mittels Verklebung durch eine aseptische Entzündung dar, die z.B. durch Talkum erreicht werden kann. Pleurapunktionen als Entlastung sind nur kurzfristig wirksam (Huzly 1991). Ebenso zeigt eine allgemeine zytostatische Behandlung bei metastasierenden pleuralen Prozessen nur in wenigen Fällen ausreichende Wirkung, so daß eine systemische Therapie oft nur mit lokalen Maßnahmen zum Erfolg führt (Feiereis 1983). Die Bestimmung der Ursache eines Pleuraergusses sollte so gut wie möglich erfolgen. Hiervon hängt häufig das weitere Vorgehen des Klinikers ab. In den Frühstadien kann die Abgrenzung zwischen Pleuramesotheliom, pulmonalen und extrapulmonalen Karzinomen sehr schwierig sein (Battifora 1985). Auch im eigenen Probenkollektiv war es gelegentlich erst nach immunhistochemischen Untersuchungen möglich, Hinweise auf die Lokalisation des Primärtumors an den Kliniker weiterzuleiten. Fracchia meinte, daß bei weiblichen Patienten mit unklarem Erguß in ungefähr 50% der Fälle ein Mammakarzinom die Ursache darstellt (Fracchia 1974). Die sicherste Methode der Therapie der Pleuraexsudation als Folge metastatischer Prozesse ist laut (nicht-chirurgischer) Autoren die operative Pleurektomie. Jedoch wird auch erwähnt, daß die Pleurektomie ein Eingriff mit größerem Risiko für den Patienten darstellt als eine nicht-operative Methode. Von daher sollte versucht werden, initial die Drainage und die Pleurodese der operativen Form vorzuziehen. Versagen diese, so kann immer noch die operative Pleurektomie erfolgen. 91 7.3.1 Anti-VEGF-Therapie Die „Angriffszelle“ von VEGF ist das Endothel der Kapillaren. Die Expression von VEGF in normalen Geweben ist durchaus als physiologisch einzustufen und sogar notwendig. VEGF wird aber in Tumorzellen offensichtlich eindeutig vermehrt exprimiert, wie auch in eigenen Untersuchungen bestätigt werden konnte. Plate konnte anhand seiner Versuchsreihen bezüglich des hochaffinen Tyrosinrezeptors von VEGF, dem flt-1, zeigen, daß im Gegensatz zum normalen Hirnkapillarendothel dieser in Tumorkapillarendothelien von Hirntumoren stark exprimiert wurde (Plate 1992). Mit Eingriff in das VEGF-Rezeptorsystem könnte eine mögliche therapeutische Strategie erfolgreich sein. Millauer transfizierte normale Gefäßendothelzellen von Tumoren in vivo mit einem Retrovirus, das in seinem Genom die negative Mutation des VEGFRezeptors flt-1 enthielt. Es resultierte eine Suppression des Tumorwachstums in den Versuchstieren (Millauer 1994). Kim wies durch Gabe von monoklonalen Antikörpern, die spezifisch gegen VEGF gerichtet waren, eine therapeutische Wirksamkeit nach, wodurch das Tumorwachstum inhibiert wurde. Dabei stellte sich heraus, daß in Korrelation zum Ausmaß der VEGFSynthese in verschiedenen Tumoren eine stärkere Tumorinhibition mittels Antikörper meßbar war (Kim 1993). Gegenwärtig orientieren sich die therapeutischen Strategien an § den molekularen Mechanismen der VEGF-Synthese § der VEGF-Sekretion § der rezeptorvermittelten Signaltransduktion von VEGF an der Kapillarendothelzellen von Tumoren Dabei liegen folgende Möglichkeiten einer Tumorinhibition vor: 92 7.3.1.1 Konstruktion von VEGF-Rezeptor-Antagonisten Diese orientieren sich an den verschiedenen VEGF-Aminosäuresequenzen, die die Bindung von VEGF am Rezeptor kompetitiv hindern. 7.3.1.2 Bifunktionale VEGF-Toxine VEGF-Toxine sind Giftstoffe, die in der Lage sind, die Synthese von VEGF zu unterbinden. Sie können ebenso mittels Endozytose die Kapillarendothelzelle direkt schädigen (Stetler-Stevenson 1997). 7.3.1.3 Die Infektion der VEGF-exprimierenden Endothelzellen Man versucht die Endothelproliferation durch Generation rekombinanter Retroviren, die ausschließlich diese infizieren, zu unterbinden (Millauer1994). 7.3.1.4 Angriff auf Rezeptorebene Die VEGF-Rezeptor-Dimerisierung, die Tyrosinkinase-Inhibitoren und andere rezeptorinhibierende Konstruktionen erlauben einen Eingriff in die Signaltransduktion von VEGF (Rosen 1997). Insgesamt ergeben sich bei der Anti-VEGF-Therapie eine Vielzahl an Möglichkeiten, die sich derzeit in Anfangsstadien der Forschung befinden. 93 7.3.2 Anti-SF- und Anti-c-Met-Therapie Grundsätzlich gilt für die Anti-SF- und Anti-c-Met-Therapie (SF = hepatischer Wachstumsfaktor; c-Met = Rezeptor des SF) dasselbe wie für die Anti-VEGF-Therapie. Die Antikörpertherapie gegen SF oder anderseits die Blockade seines Rezeptors c-Met stellen Ansatzpunkte dar. Man nimmt an, so die Tumorproliferation und Neovaskularisation, die durch den hepatischen Wachstumsfakor SF und seinem Rezeptor c-Met positiv beeinflußt werden, regulieren zu können. Eine andere Strategie ist die Inhibition der SF-Synthese. Polverini meinte, daß möglicherweise auch entzündliche Krankheiten (Beispiel: Psoriasis) auf diese Weise therapiert werden könnten, da das SF - c-Met-System - wie in tumorösen Prozessen auch - eine erhöhte Expressionsdichte zeigt (Polverini 1995). Die Inhibition der Angiogenese als neues therapeutisches Prinzip in der Behandlung maligner Tumoren ist sicherlich zukunftsweisend und es ist zu erwarten, daß sie in die klinische Anwendung gelangen wird (Marné 1997). 94 8 ZUSAMMENFASSUNG Ziel dieser Untersuchung war die Charakterisierung der Koexpression angiogenetisch wirksamer Proteine in pleuralen Metastasen primär andernorts lokalisierter bösartiger Tumoren. Bei den angiogenetischen Faktoren handelt es sich um § HGF / SF = hepatischer Wachstumsfaktor bzw. Scatter Faktor; (HGF und SF bezeichnen dasselbe Protein) § c-Met = Rezeptor des HGF / SF § VEGF / VPF = vaskulärer endothelialer Wachstumsfaktor; auch VPF (Vascular Permeability Factor) genannt Geprüft wurden hierbei Hypothesen bezüglich charakteristischer Expressionsmuster und Koexpressionen der einzelnen angiogenetischen Faktoren. Es wurde Biopsiegut sekundärer Pleuratumoren von 71 Patienten aus dem Einsendegut des Institutes für Pathologie an den Berufsgenossenschaftlichen Kliniken „Bergmannsheil“ analysiert. Mit den Methoden der Immunhistochemie und in-situ Hybridisierung konnten folgende Resultate dokumentiert werden: 1. Eine deutliche intrazytoplasmatische Expression, teils Überexpression von VEGF, cMet und HGF/SF konnte beim Großteil der pleuralen Metastasen primär andernorts lokalisierter bösartiger Tumoren (Mamma, Lunge, Gastrointestinaltrakt, Urogenitaltrakt und verschiedene andere Tumoren, wie Pankreas-, Kehlkopf- und Schilddrüsenkarzinom) nachgewiesen werden. 2. Die Expression der Faktoren war nicht an die histogenetische Abstammung des Tumors gebunden. 95 3. Die Synthese von HGF/SF und c-Met fand überwiegend in Tumorzellen statt. Dies konnte durch die in-situ Hybridisierung bestätigt werden. Ebenso kam es in den Endothelzellen der Gefäße, die sich in Tumorzellnähe befanden, zu verstärkter Expression aller Faktoren. 4. Es lag eine statistisch signifikante Koexpression (p<0,05) von VEGF mit c-Met und HGF/SF durch das Tumorgewebe sowie durch die Endothelien der tumorgebundenen Gefäße vor. Daher kann hier eine autokrine Stimulation dieser angiogenetisch wirksamen Systeme diskutiert werden. In HGF/SF-positiven Zellen fand sich, vergleichsweise zu HGF/SF-negativen Befunden, immunhistochemisch eine vermehrte c-Met Expression (p<0,05). 5. Auf der anderen Seite war beim Vergleich der prozentual reaktiven Zellen (PP) lediglich eine Tendenz feststellen, jedoch keine signifikante Korrelation. Eine mögliche Erklärung ist der unterschiedliche Expressionsmuster der Tumorzellen verschiedener Abstammung. Unterschiedliche Tumortypen könnten im Rahmen der Angiogenese verschiedene für sie typische angiogenetische Proteine (hier HGF/SF und VEGF) synthetisieren, jedoch könnten ebenso unterschiedliche Tumoren durch die Expression eines Rezeptors (hier c-Met) durch verschiedene Liganden stimuliert werden. Daher liegt die Schlußfolgerung nahe, daß zwar die Färbeintensität (SI) der Tumorzellen bezüglich c-Met und VEGF korreliert (p<0,05), die prozentual reaktiven Tumorzellen (PP) jedoch nur eine Tendenz (p=0,085) aufzeigen. Folgende Schlußfolgerungen sind aus diesen Ergebnissen abzuleiten: 1. Anamnestisch war bei allen Patienten, deren Biopsien hier untersucht wurden, zum Zeitpunkt der Biopsieentnahme ein Pleuraerguß vorhanden. Die verstärkte VEGFExpression durch Tumorzellen beim Prozeß der pleuralen Metastasierung kann u.a. als eine Erklärungsmöglichkeit für die Entwicklung des Pleuraergusses bei der Tumoraussaat sein. VEGF, auch (VPF/Vascular Permeability Factor) genannt, 96 erhöht die Gefäßpermeabilität im und um das Tumorgewebe – z.T. durch eine entzündliche Begleitreaktion – hierdurch kommt es zum Pleuraerguß. 2. Die obigen Befunde belegen die Bedeutung des vaskulären endothelialen Wachstumsfaktors (VEGF), des hepatischen Wachstumsfaktors (HGF/SF) und des cMet in Tumoren im Zusammenspiel bezüglich ihrer Wirksamkeit bei der Angiogenese. Der Tyrosinkinaserezeptor c-Met (Rezeptor des hepatischen Wachstumsfaktors) bildet zusammen mit dem zugehörigen Liganden HGF/SF ein parakrines Signalsystem. Dieses System ist offensichtlich in den Prozeß der Angiogenese eingebunden, wobei gerade HGF/SF nach Synthese in mesenchymalen Zellen (Makrophagen, Mastzellen, aber auch Endothelzellen) eine angiogenetische Wirkung z.B. Insbesondere auf muß Gefäßendothelzellen der bevorzugte und Gefäßwandmyozyten autokrine Mechanismus ausübt. zwischen überexprimiertem c-Met und HGF/SF in malignen Tumoren bei Nachweis der Proteine in denselben Tumorzellen auch beim Prozeß der pleuralen Metastasierung angenommen werden. Eine gleichzeitige Synthesestimulation dieser unterschiedlichen drei Proteine ist zudem über das Plasminogen-Urokinase-System möglich (siehe 3.4.3 und 4.1.1). Zumindest scheint ein synergistischer Effekt in der Angiogenese im Prozeß der metastatischen Absiedelung maligner Tumorzellen in der Pleura wahrscheinlich. Die hier erhobenen Untersuchungsergebnisse können eine Grundlage möglicher zukünftiger palliativer Therapiestrategien bei malignen Pleuraergüssen darstellen. Die lokale Instillation anti-angiogenetisch wirksamer Substanzen z.B. im Rahmen der diagnostischen Pleurapunktion ist als therapeutischer Ansatz bei therapierefraktären schmerzhaften malignen Pleuraergüssen denkbar. Ob möglicherweise auch blockierende Antikörper gegen HGF/SF-produzierenden Tumorzellen in Zukunft eine therapeutische Bedeutung erlangen, steht zur Diskussion. 97 9 LITERATUR Adams, J. C.; Furlong, R. A.; Watt, F. M.(1991): Production of SF by ndk, a strain of epithelial cells, and inhibition of SF activity by suramin. J. Cell. Sci. 98: 385-394. Agostonie, E.; D`Angelo, E. (1969): Thikness and pressure of the pleural liquid at various heights and with various hydrothoraces. Respir. Physiol. 6: 330-342. Agostonie, E. (1972): Mechanics of the pleural space. Physiol. Rev. 52: 57-128. Aiello, L. P.; Avery, R. L.; Arrigg, P. G.; Keyt, B. A.; Jampel, H. D.; Ferrara, N. (1994): New Engl. J. Med. 331: 1480-1487. Anderson, C. B.; Phillpott, G. U.; Ferguson, T. B. (1974): The treatment of malignant pleural effusion. Cancer 41: 1188-1192. Annen, K.; Nojima, T.; Hata, Y. (1995/96): Analysis of the HGF receptor in regeneration and oncogenesis of the liver. Ge, Diagn. Pathol. 141: 179-186. Antonellí-Orlidge, A.; Smith, S. R.; D`Amore, P. A. (1989): Influence of pericytes on capillary endothelial cell growth. Rev. Resp. Dis. 140: 1129-1131. Bakhle, Y. S.; Vane, J. R. (1977): Metabolic functions of the lung. Dekker, M., New York. Ballard, S. G.; Ward, D. C. (1993): Fluorescence in-situ Hybridization (FISH) using digital imaging microscopy. J. Histochem. Cyt. 41(12): 1755-1759. Battifora, H.; Kopinski, M. I. (1985): Distinction of mesothelioma from adenocarcinoma. An immunohistochemical approach. Cancer 55: 1679-1685. 98 Berse, B.; Brown, L. F.; Van de Water, L.; Dvorak, H. F.; Senger, D. R. (1992): VEGF gene is expressed differetially in normal tissues, macrophages, and tumors. Mol. Biol. Cell 3: 211-220. Bhargava, M.; Joseph, A.; Rosen, E. M. (1992): Cell Growth Differen. 3: 11-20. Bikfalvi, A.; Sauzeau, C.; Moukadiri, H.; Maclouf, J.; Busso, N.; Bryckaert, M.; Plouet, J.; Tobelem, G. (1991): J. Cell. Physiol. 149: 50-59. Billroth, T. (1991): Untersuchungen über die Entwicklung der Blutgefäße. Georg Reimer, Berlin: pp. 81 + V plates. Billroth, T. (1910): „Briefe“. 8. Veränd. Aufl., Hannover/Leipzig, Hahnsche Buchhandlung: pp. 17-18. Bosari, S.; Lee, A. K.; DeLellis, R. A.; Silverman, M. L. (1992): Microvessel quantitation and prognosis in invasive breast carcinoma. Hum. Pathol. 23: 755-761. Bottaro, D. P.; Rubin, J. S.; Faletto, D. L.; Aaronsen, S. A. (1991): A broad-spectrum human lung fibroblast-derived mitogen is a variant of HGF. Science 251: 802-804. Breier, G.; Albrecht, U.; Sterrer, S.; Risau, W. (1994): VEGF and glioma angiogenesis: coodinate induction of VEGF receptors, distribution of VEGF protein and possible in vivo regulatory mechanisms. Development 114: 521-532. Boocock,C. A.; Charnock-Jones, D. S.; Sharkey, A. M.; McLaren, J.; Barker, P. J.; Wright, K. A.; Twentyman, P. R.; Smith, S. K. (1995): Expression of VEGF and ist recepetors flt and KDR in ovarian carcinomas. J. Natl. Cancer Inst. 87: 506-515. Brockmann, M.; Müller, K. M. (1991): Die pathologische Anatomie der primären und sekundären Pleuratumoren. In: Drings, P., Vogt-Moykopf (Hersg.) Thoraxtumoren. 99 Siagnostik - Staging - gegenwärtiges Thrapiekonzept. Springer-Verlag, Berlin, Heidelberg, New York: 271 - 284. Brown, L. F.; Yeo, K. T.; Berse, B. B. (1992): Expression of VEGF by epidermal keratinocytes during wound healing. J. Exp. Med. 176: 1375-1379. Brown, L. F.; Berse, B. B.; Jackman, R. W.; Tognazzi, K.; Manseau, E. J.; Dvorak, H. F.; Senger, D. R. (1993): Increased expression of VEGF and ist receptor in kidney and bladder carcinomas. Am. J. Pathol. 143: 1255-1262. Bucher, O., Wartenberg, H. (1989): Zytologie , Histologie und mikroskopische Anatomie des Menschen. 11. Aufl., Huber-Verlag, Bern Stuttgart, Toronto. Bussolino, F.; DiRenzo, M. F.; Comoglio, P. M. (1992): HGF is a potent angiogenic factor which stimulates endothelial cell motility and growth. J. Cell. Biol. 119: 629-641. Campbell, G. R.; Chamley-Campbell, J. H. (1981): Smooth muscle phenotypic modulation: role in atherogenesis. Med. Hypotheses 7: 729-735. Chernow, B.; Sahn, S. A. (1977): Carcinomatous involvement of the pleura: An analysis of 96 patients. Am. J. Med. 63: 695-702. Chrétien, J.; Jaubert, F. (1985): Pleural responses in malignant metastatic tumors. In: Chretien, J., Bignon, J., Hirsch, A.(eds). The pleura in health and disease. Dekker, M., New York: 489-505. Claffey, K. P.; Wilkison, W. O.; Spiegelman, B. M. (1992): VEGF. Regulation by cell differentiation and activated second messenger pathways. J. Biol. Chem. 267: 16317-16322. 100 Collins, P. D.; Connolly, D. T.; Williams, T. J. (1993): Charakerization of the increase in vascular permeability induced by VEGF in vivo. Br. J. Pharmacol. 109: 195-199. Connolly, D. T.; Heuvelman, D. M.; Nelson, R. (1989): Tumor VPF stimulates endothelial cell growth and angiogenesis. J. Clin. Invest. 84: 1470-1478. Cooper, C. S.; Park, M.; Blair, D. G.; Vande Woude, G. F. (1984): Molecular cloning of a new transforming gene from a chemically transformed human cell line. Nature 311: 29-34. Cooray, G. H.: Defense mechanism in the mediastinum with special reference to the mechanism of pleural absorptions. J. Pathol. Bacteriol. 6: 551-567. Courtice, F. C.; Simmonds, W. J. (1949): Physiological significance of lymph drainage of the serous cavities and lungs. Physiol. Rev. 34: 419-448 (1954). Cullinan-Bove, K.; Koos, R. D. (1993): VEGF expression in rat uterus: rapid stimulation by estrogen correlates with estroen-induced increases in uterine capillary permeability and growth. Endocrinology 133: 829-837. Dail, D. H. (1994): Metastasis to and from the lung. In: Dail, D. H., Hammer, S. P. (eds.). Pumonary Pathology (2nd edition). Springer-Verlag, Berlin, Heidelberg, New York: 1581-1615. DiRenzo, M. F.; Narsimhyn, R. P.; Olivero, M.; Comoglio, P. M. (1991): Expression of the met/HGF-receptor in normal and neoplastic human tissues. Oncogene 6: 11972003. Dissen, G. A.; Lara, H. E.; Fahrenbach, W. H. (1994): Immature rat ovaries become revascularized rapidly after autotranplatation and show a gonadotropin- 101 dependent increase in angiogenic factor gene expression. Endocrinology 134: 11461154. Dvorak, H. F.; Brown, l. F.; Detmar, M.; Dvorak, A. M. (1995): VEGF, microvacular hyperpermeability, and angiogenesis. Am. J. Pathol. 146: 1029-1039. Eagles, G.; Warn, A.; Ball, R. Y.; Warn, R. M. (1996): HGF/SF is present in most pleural effusion fluids from cancer patients. Br. J. Cancer 73: 377-381. Ebert, M.; Yokoyama, M.; Friess, H.; Büchler, M. W.; Kore, M. (1994): Coexpression of the c.met proto-oncogene and HGF in human pancreatic cancer. Cancer Res. 54: 5775-5778. Efrati, P.; Nir, E. (1976): Morphological and cytochemical investigation of human mesothelial cells from pleural and peritoneal effusions. A light and electron microscopic study. Isr. J. Med. Sci. 12: 662-673. Faletto, D. L.; Kaplan, D. R.; Halverson, D. O.; Rosen, E. M.; Vande Woude, G. F. (1993): Singnal transduction in c-Met mediated motogenesis. In I. D. Goldberg and E. M. Rosen (eds): HGF/SF and c-Met Receptor. Birkhäuser Verlag, Basel: 107-130. Fava, R. A.; Olsen, N. J.; Spencer-Green, G. (1994): J. Exp. Med. 180: 341-346. Feiereis, H.; Grewe, H. E.; Johanigmann, J.; Keiser, p.; Schmidt, M. A.; Siebert, W. (1983): Brustkrebs der Frau. Marseille, München: 197-212. Ferrara, N.; Henzel, W. J. (1989): Pituitary follicular cells secrete a novel heparinbinding growth factor specific for vascular endothelial cells. Biochem. Biophys. Res. Commun. 161: 851-858. Ferrara, N.; Houck, K. A.; Jakeman, L. B. (1991): The VEGF family of polypeptides. J. Cell Biochem. 47: 211-218. 102 Ferrara, N.; Houck, K.; Jakeman, L.; Leung, D. W. (1992): Molecular and biological properties of the VEGF family of proteins. Endocrinol. Rev. 13: 18-32. Földi, M.; Casley-Smith, J. R. (1983): Lymphangiology. Schattauer, Stuttgart. Folkman, J. (1985): Tumor angiogenesis. Adv. Cancer Res. 43: 175-203. Folkman, J. (1991): Tumor Angiogenesis. In: Cancer Medicine. Ed. By J. F. Holland, 3rd edn, Lea & Febiger, Melbourne, Pennsylvania, USA. Folkman, J. (1990): „What is the evidence that tumors are angiogenesis dependant?“ J. Natl. Canc. Inst. 82(1): 4-6. Folkman, J.; Long, D.; Becker, F. (1963): Growth and Metastasis of tumor in organ culture. 16: 453-467. Folkman, J. (1971): Tumor Angiogenesis: therapeutic implications. N. Engl. J. Med. 285: 1182-1186. Fracchia, A. A. (1974): Intrapleural chemotherapy for effusion from metastatic breast carcinoma. Cancer 33: 196-229. Garrido, C.; Saule, S.; Gospodarowicz, D. (1993): Transcriptional regulation of VEGF gene expression in ovarian bovine granulosa cells. Growth Factors 8: 109-117. Ganzemeier, U.; Magnussen, H. (1988): Diagnostik und Differentialdiagnose von Pleuraergüssen. In: D. K. Hossfeld und U. Ganzemeier (Hersg.). Maligne Ergüsse. Pleura - Peritoneum - Perikard. Beitr. Onkol. 30: 31-42. 103 Gherardi, E.; Grey, J.; Stoker, M. (1989): Purification of SF, a fibroblast-derived basic protein that modulates epithelial interactions and movement. Proc. Natl. Acad. Sci. USA 86: 5844-5848. Gherardi, E.; Stoker, M. (1990): Hepatocytes and SF. Nature (Lond.) 346: 228. Gonzatti-Haces, M.; Seth, A.; Vande Woude, G. F. (1988): Proc. Natl. Acad. Sci. USA 85: 21-25. Goto, F.; Goto, K.; Weindel, K.; Folkman, J. (1993): Synergistic effects of VEGF and bFGF on the proliferation and cord formation of bovine capillary endothelial cells within collagen gels. Lab. Invest. 69: 508-517. Grant, D. S.; Kleinman, H. K.; Goldberg, I. D.; Rosen, E. M. (1993): SF induces blood vessel formation in vivo. Proc. Natl. Acad. Sci. USA 90: 1937-1941. Grant, T.; Levin, B. (1974): Lymphangiographic visualizationof pleural and pulmonary lymphatics in a patient without chylothorax. Radiology 113. 49-50. Grundmann, E. (1981): Das Wesen des malignen Wachstums. Klin. Wschr. 59:931. Grundmann, E. (1994): Allgemeine Pathologie. Gustav Fischer Verlag, Stuttgart, Jena, New York. Guyton, A. C. (1981): Textbook of medical physiology. 6. Aufl. Saunders-Company, Philadelphia. Guzman, J.; Bross, K. J.; Würtemberger, G.; Costable, U. (1989): Immunocytology in malignant pleural mesothelioma. Chest 95: 590-595. 104 Halaban, R.; Rubin, J.; Moellman, G. (1992): Met and HGF/SF signal transduction in normal melanocytes and melanoma cells. Oncogene 7: 2195-2206. Hales, S. (1733): Statical essays: containing haemastaticks: or, an account of some hydraulick and hydrostatical experiments made on the blood and blood-vessels of animals. London, vol. 2, XXII: 361 pp. Harada, S.; Nagy, J. A.; Sullivan, K. A.; Thomas, K. A. (1994): Induction of VEGF expression by prostaglandine E2 and E1 in osteoblasts. J. Clin. Invest. 93: 24902496. Harvey, P.; Warn, A.; Newman, P.; Perry, L. J.; Ball, R. Y.; Warn, R. M. (1996): Immunreactivity for HGF/SF and ist receptor, met, in human lung carcinomas and malignant mesotheliomas. J. Pathol. 180: 389-394. Hayek, v. H. (1960): The parietal pleura. The human lung. Haffner, New York. Hayek, v. H. (1960): The visceral pleura. The human lung. Haffner, New York. Hertig, A. T. (1935): Angiogenesis in the early human chorion and in the primary placenta of the macaque monkey. Contr. Embryol. 25(146): 39-81. Hickman, J. A; Jones, M. C. (1970): Treatment of neoplastic pleural effusion with local instillations of Quinacrin (Mepacrine) Hydrochlorid. Thorax 25: 226-229. Hudlicka, O. (1984): Growth of vessels - historical review. In: Prog. Appl. Mikrocirc. Vol. 4, Karger, Basel: pp. 1-8. Huzly, A.; Ortlieb, H. (1991): Sekundäre Pleuratumoren. Atemw.-Lungenkrkh. 17(6): 254-266. 105 Jakeman, L. B.; Armanini, M.; Phillips, H. S.; Ferrara, N. (1993): Developmental expression of binding sites and messenger ribonucleic acid for VEGF suggests a role for this protein in vasculogenesis and angiogenesis. Endocrinology 133: 848-859. Joannidis, M.; Spokes, K.; Nakamura, T.; Faletto, D.; Canley, L. G. (1994): Regional expression of HGF/c-Met in experimental renal hypertrophy and hyperplasia. Am. J. Physiol. 267: F231-F236. Joseph, A.; Weiss, G. H.; Rosen, E. M. (1995): Expression of SF in human bladder cancer. J. Natl. Cancer 87: 372-377. Kan, M.; Zhang, G. H.; Zarneger, R.; Stevens, J. L. (1991): HGF/hepatopoetin A stimulates the growth of rat kidney proximal tubule epithelial cells (RPTE), rat nonparenchymal liver cells, human melanoma cells, mouse keratinocytes and stimulates anchorage-independent growth of SV-40 transformed RPTE. Biochem. Biophys. Res. Commun. 174: 331-337. Kendall, R. L.; Thomas, K. A. (1993): Inhibition of VEGF activity by an endogenously encoded soluble receptor. Proc. Natl. Acad. Sci. USA 90: 10705-10709. Kieser, A.; Weich, H. A.; Brandner, G.; Marme, D.; Kolch, W. (1994): Mutant p53 potentiates protein kinase C induction of VEGF expression. Oncogene 9: 963-969. Kim, K. J.; Li, B.; Houck, K.; Winer, J.; Ferrara, N. (1992): The VEGF proteins: identification of biologically relevant regions by neutralizing monoclonal antibodies. Growth Factors 7: 53-64. Kim, K. J.; Li, B.; Winer, J.; Armanini, M.; Gillett, N.; Phillips, H. S.; Ferrara, N. (1993): Inhibition of VEGF-induced angiogenesis supresses tumor growth in vivo. Nature 362: 841-844. 106 Kondo, S.; Asano, M.; Suzuki, H. (1993): Significance of VEGF for solid tumor growth, and ist inhibition by antibodies. Biochem. Biophys. Res. Commun. 194: 1234-1241. Kortsik, C. S. F.; Kroehel, C.; Virchow, C.; Werner, P.; Matthys, H. (1993): Erkrankungen der Pleura. Atemw.-Lungenkrkh. 17(6): 252-259. Kreuser, E. D. (1984): Behandlung maligner Pleuraergüsse durch Fibrinklebung. Tumor Diagn. Ther. 2: 55-58. Ku, D. D.; Zaleski, J. K.; Liu, S.; Brock, T. A. (1993): VEGF induces ERDF-dependent relaxation in coronary arteries. Am. J. Physiol. 265(2 Pt 2): H586-H592. Kuhnen, C.; Tolnay, E.; Naim, R.; Müller, K. M. (1997): VEGF-overexpression in metastatic tumors to the pleura. J. Cancer Res. Clin. Oncol. 123 (Suppl. 1): S38. Kullmann, H. J.; Wiesner, B. (1991): Pleuratumoren. Atemweg.-Lungenkrkh. 17 (6): 251-253. Krogh, S. A. S. (1922): The anatomy and physiology of capillaries. Silliman Memorial Lectures, New Haven, Yale University Press, XVII: 276 pp. Ladoux, A.; Frelin, C. (1993): Hypoxia is a strong inducer of VEGF mRNA expression in the heart. Biochem. Biophys. Res. Commun. 195: 1005-1010. Lee, K. F.; Olak, J. (1994): Anatomy and physiology of pleural space. Ches. Surg. Clin. N. Am. 4 (3): 391-403. Leung, D. W.; Cachianes, G.; Kuang, W. J.; Goeddel, D. V.; Ferrara, N. (1989): VEGF is secreted angiogenic mitogen. Science 246: 1306-1309. 107 Li, Q.; Wenig, J.; Mohan, R.; Bennett, G. (1996): HGF and HGF receptor in lacrimal gland, tears and cornea. Invest. Ophthalm. 37(5): 727-739. Li, Y.; Bhargava, M. M.; Goldberg, I. D. (1994):Effect of HGF/SF and other growth factors on motility and morphology of non-tumorgenic and tumor cells. In Vitro Cell Dev. Biol. 30 A: 105-110. Loddenkemper, R. (1998): Klassische pneumologische Thorakoskopie. Atemw. Lungenkrkh. 24 (5): 220-223. Loddenkemper, R.; Grosser, H.; Gabler, A.; Mau, J.; Preussler, H.; Brand, H. J. (1983): Prospective evaluation of biopsy methodes in the diagnosis of malignant pleural effusion. Intrapatient comparison between pleural fluid cytology, blind needle biopsy and thoracoscopy. Am. Rev. Respir. Dis. 127 (Suppl. 4): 114. Loy, T. S.; Diaz-Ariaz, A. A.; Bickel, J. T. (1990): Value of BCA-225 in the cytologic diagnosis of malignant effusions: an immunocytochemical study of 197 cases. Modern Pathology 3: 294-297. Malpighi, M. (1686): idem. De Ovo Incubato. Opera tom I, London. Malpighi, M. (1686): De Pulmonibus. Epist II; Opera tom II, London. Mariassay, A. T.; Wheeldon, E. B. (1983): A pleura: combined light microscopic, scanning, and transmission electron microscopic study in the sheep. I. Normal pleura. Exp. Lung. Res., 4: 293-314. Marsac, J.; Frija, G.; Bismuth, V. (1982): Chylothorax et physiologie lymphatique de la plèvre. Rev. Fr. Mal. Respir. 10: 227-241. 108 Matsumoto, K.; Tajima, H.; Okazaki, R.; Nakamura, T. (1993): Heparin as an inducer of HGF. J. Biochem. (Tokyo) 114: 820-826. Mattay, R. A.; Coppage, L.; Shaw, C.; Filderman, A. E. (1990): Malignancies metastatic to the pleura. Invest. Radiol. 25: 601-619. McClure, N.; Healy, D. L.; Rogers, P. A. (1994): VEGF as capillary permeability agent in ovarian hyperstimulation syndrome. Lancet 344: 235-236. McLaughlin, R. F.; Tyler, W. S.; Canada, R. O. (1961).: A study of the subgross pulmonary anatomy in various animals. Am. J. Anat. 108: 149-165. Meyer, B. C. (1966): Metastatic carcinoma of the pleura. Thorax 21: 437-442. Midy, V.; Plouet, J. (1994): Vasculotropin/VEGF induces differentiation in cultured osteoblasts. Biochem. Biophys. Res. Commun. 199: 380-386. Millauer, B.; Shawver, L. K.; Plate, K. H.; Risau, W.; Ullrich, A. (1994): Glioblastoma growth inhibited in vivo by a dominant-negative Flk-1 mutant. Nature 367 (6463): 576-579. Miller, W. S. (1947): The lung. 2nd ed. Thomas, Springfield: 118. Miyazawa, K.; Tsubouchi, H.; Naka, D.; Kitamura, N. (1989): Proteolytic activation of HGF in response to tissue injury. J. Biol. Chem. 163: 967-973. Miyazawa, K.; Shimomura, T.; Kitamura, N. (1993): Activation of the zymogen of HGF activator by thrombin. J. Biol. Chem. 268: 10024-10028. Miyazawa, K.; Shimomura, T.; Kitamura, N. (1994): J. Biol. Chem. 269: 8966-8979. 109 Montesano, R.; Matsumoto, K.; Orci, L. (1991): Identification of fibroblast-derived epithelial morphogen as HGF. Cell 67: 901-908. Moores, D. W. O. (1994): Management of the malignant pleural effusion. In: L. P. Faber und V.W. Rusch (eds.). Chest surgery clinics of north america. Vol. 4, No. 3: 481496. Müller, K. M. (1983): Pathologie der Lunge. In: Doerr, W. und Seifert, G. (Hersg.). Spezielle pathologische Anatomie. Springer-Verlag, Berlin, Heidelberg, New York. Bd. 16: 1373-1398. Müller, K. M. (1989): Pleuramesotheliom / Mesotheliomregister. Z. Herz Thoray Gefäßchir. 3 (Suppl. I): 100-105. Müller, K. M.; Respondek, M.; Hesse, H. (1990): Die Metastasierung in Lunge und Pleura. Langenbecks Arch. Chir. - Verh. Dtsch. Ges. Chir. (Suppl. II): 767-774. Müller, K. M. (1994): Erkrankungen der Pleura - Pathologische Anatomie. In Nakhosteen, J. A., Inderbitzi, R. Atlas und Lehrbuch der thorakalen Endoskopie. Springer-Verlag, Berlin, Heidelberg. 325-326. Müller, K. M., Theile, A. (1994): Voraussetzungen zur Metastasierung des Bronchialkarzinoms. Langenbecks Arch. Chir. - Verh. Dtsch. Ges. Chir. (Suppl. II): 767-774. Mungall, I. P. F.; Cowen, P. N.; Cooke, N. T.; Roach, T. C.; Cooke, N. J. (1982): Multiple pleural biopsy with the Abrams needle. Thorax 35: 600-602. Naigaishi, C. (1972): Functional anatomy and histology of the lung. Univ. Park Press, Baltimore: 79-179. 110 Naldini, L.; Tamagnone, L.; Vigna, E.; Sachs, M.; Comoglio, P. M. (1992): Extracellular proteolytic cleavage by urokinase is required for activation of HGF. EMBO J. 11: 4825-4833. Nakamura, T.; Nishizawa, T.; Shimizu, S. (1989): Molecular cloning and expression of human HGF. Nature 342: 440-443. Natali, P. G.; Nicotra, M. R.; Cavaliere, R.; Bigotti, A.; Romano, G.; Temponi, M.; Ferrone, S. (1990): Differential expression of intracellular adhesion molecule I in primary and metastatic melanoma lesions. Cancer Res. 50: 1271-1278. Natali, P. G.; Prat, M.; Nicotra, M. R.; Comoglio, P. M.; DiRenzo, M. F. (1996): Overexpression of the met/HGF receptor in renal cell carcinomas. Int. J. Cancer (Pred. Oncol.) 69: 212-217. Orr, F. W.; Young, L.; King, G. M.; Adamson, I. Y. R. (1988): Preferential growth of metastatic tumors at the pleural surface of mouse lung. Clin. Expl. Metastasis 6: 221-232. Ottenhoff-Kalff, A.E.; Rijksen, G. (1992): Charakterization of protein tyrosine kinase from human breast cancer: involvement of the c-src oncogene product. Cancer Res. 52: 4773-4877. Pavesi, F.; Lotzniker, M.; Cremaschi, P.; Marbello, L.; Moratti, R. (1988): Detection of malignant pleural effusions by tumor marker evaluation. Eur. J. Cancer Clin. Oncol. 24: 1005-1011. Pepper, M. S.; Matsumoto, K.; Montesano, R. (1993): Upregulation of urokinase receptor expression on migrating endothelial cells. J. Biol. Chem. 267: 20493-20496. 111 Pfitzner, P. (1989): Growth patterns iof adenocarcinomas in effusion. MD-GBK 55: 6164. Piiper, J. (1975).: Physiologie der Atmung. In: Gauer, Kramer, Jung (Hersg.), Physiologie des Menschen, Bd 6, 2. Aufl., Urban & Schwarzenberg, München. Pisters, L. L.; Troncoso, P.; Zhau, H. E.; Chung, L. W. K. (1995): c-Met proto-onkogen expression in benign and malignant human prostate tissue. J. Urol. 154: 293-298. Plate, K. H.; Breier, G.; Weich, H. A.; Risau, W. (1992): Vascular endothelial growth factor is a potential tumor angiogenesis factor in human gliomas in vivo. Nature 359: 845-848. Polverini, P. J.; Nickoloff, B. J. (1995): The role of SF and the c-met proto-oncogene in angiogenetic responses. In I. D. Goldberg and E. M. Rosen (eds): HGF/SF and cMet Receptor. Birkhäuser Verlag, Basel: 51-67. Ponzetto, C.; Bardelli, A.; Zhen, Z.; Comoglio, P. M. (1994): A multifunctional docking site mediates signaling and transformation by the HGF/SF receptor family. Cell 77: 261-271. Prat, M.; Narsimhan, R. P.; Crepaldi, T.; Comoglio, P. M. (1991): The receptor encoded by the human c-Met oncogene is expressed in hepatocytes, epithelial cells and solid tumors. Int. J. Cancer 49: 323-328. Raju, R. N.; Kardinal, C. G. (1970): Pleural effusion in breast carcinoma. Cancer 26: 626-629. Rennard, S. I.; Jaurand, M. C.; Bignon, J.; Kawanami, O.; Ferrans, V. J.; Davidson, J.; Crystal, R. G. (1984): Role of pleural mesothelial cells in the production of the 112 submesothelial conective tissue matrix of the lung. Am. Rev. Respir. Dis. 130: 267274. Reshad, K. I.; Invi, K.; Takeuchi, Y. O.; Hitomi, S.; Takahashi, Y. (1985): Treatment of malignant pleural effusion. Chest 88: 393-397. Rong, S.; Jeffers, M.; Resau, J. H.; Vande Woude, G. F. (1993): Met-expression and sarcoma tumorigenicity. Cancer Res. 53: 5355-5360. Rosen, E. M.; Goldberg, I. D.; Vinter, D. W. (1989): Smooth muscle releases an epithelial cell SF which binds to heparin. In Vitro Cell Dev. Biol. 25: 163-173. Rosen E. M.; Meromski, L.; Setter, E.; Vinter, D. W.; Goldberg, I. D. (1990): Smooth muscle-derived factor stimulates mobility of human tumor cells. Invasion Metast. 10: 49-64. Rosen, E. M.; Jaken, S.; Carley, W.; Goldberg, I. D. (1991): Regulation of motility in bovine brain endothelial cells. J. Cell. Physiol. 146: 325-335. Rosen, E. M.; Knesel, J.; Goldberg, I. D.; Rockwell, S. (1994): SF modulates the metastatic phenotype of the EMT6 mouse mammary tumor. Int. J. Cancer 57: 706714. Rouget, C. M. B. (1879): Mémoire sur le dévélopment de la tunique contractile des vaisseaux. C. R. Acad. Sci. Paris. 1873; 79: 559 - 562. Idem. Sur la contractibilité des capillaires sanguins. C. R. Acad. Paris, 88: 916-918. Le Roux, B. T. (1962): Pleural Tumors. Thorax 2: 111-119. Rubin, J. S.; Chan, A. M. L.; Bottaro, D. P.; Aaronsen, S. A. (1991): Identification of a competetive HGF antagonist encoded by an alternative transcript. Proc. Natl. Acad. Sci. USA 88: 415-419. 113 Sachs, L. (1984): Angewandte Statistik. Anwendung statistischer Methoden. Springer Verlag, Berlin, Heidelberg, New York, Tokyo. Sahn, S. A. (1988): Malignant pleural effusions. In: A. Fishman: Pulmonary disease and disorders. 2nd ed. Mc Graw Hill, New York: 267-285. Santos, O. F. P.; Moura, L. A.; Rosen, E. M.; Nigram, S. K. (1993): Modulation of HGFinduced tubulogenesis and branching by multiple phosphorylation mechanism. Dev. Biol. 159: 538-545. Schmähl, D. (1981): Maligne Tumoren: Enstehung - Wachstum - Chemotherapie. 3. Aufl. Editio Cantor, Aulendorf. Schmidt, C.; Bladt, F.; Goedecke, S.; Brinkmann, V.; Gherardi, E.; Birchmeier, C. (1995): SF/HGF is essential for liver development. Nature 373: 699702. Selberg, W. (1983): Definition häufiger Begriffe in Zusammenhang mit der Präneoplasie. Pathologe 4: 174. Sherington, C. S. (1929): On capillary blood-flow. In: Mammalian Physiology. 2nd edn., Clarendon Press, London: 21. Shibuya, M. (1995): Role of VEGF-FLT receptor system in normal and tumor angiogenesis. Adv. Cancer Res. 67: 281-313. Shiota, G.; Rhoads, D. B.; Wang, T. C.; Nakamura, T.; Schmidt, E. V. (1992): HGF inhibits growth of hepatocellular carcinoma cells. Proc. Natl. Acad. Sci. USA 89: 373-377. 114 Sonnenberg, E.; Meyer, D.; Weidner, K. M.; Birchmeier, C. (1993): SF/HGF and ist receptor, the c-Met-tyrosine-kinase, can mediate a signal exchange btween mesenchym ena epithelia during mous development. J. Cell. Biol. 123: 223-235. Stauffer, J. L.; Potts, D. E.; Sahn, S. A. (1978): Cellular content of the normal rabbit pleural space. Acta Cytol. 22: 570-574. Stoker, M.; Gherardi, E.; Peryman, M.; Gray, J. (1987): SF is a fibroblast-derived modulator of epithelial motility. Nature 327: 238-242. Takahashi, Y.; Kitadai, Y.; Ellis, L. M. (1995): Expressionof VEGF and ist receptor, KDR, correlates with vascularity, metastasis, and proliferation of human colon cancer. Cancer Research 55: 3964-3968. Testut, L. (1930): Traite d´anatomie humaine. Tome 3, 8th ed. Doin, Paris: 1003. Titscher, R.; Boeck, D. (1981): Wandel der Therapie bei Pleurakarzinose in den letzten 20Jahren. Tagungsbericht 16. Tagung der Österreichischen Ges. für Lungenerkrankungen und Tbk.: 123-126. Tolnay, E.; Fisseler-Eckhoff, A.; Wulf, M.; Müller, K. M. (1997): VEGF-mRNA Expression in Plattenepithelkarzinomen der Lunge. Path. Res. Pract. 193 (2): 94. Tomlinson, J. R.; Sahn, S. A. (1987): Invasive procedures in the diagnosis of pleural disease. Semin. Respir. Med. 9: 30-36. Tsao, M. S.; Zhu, H.; Giaid, A.; Park, M. (1993): HGF/SF is an autocrine factor for human normal bronchial epithelial and lung carcinoma cells. Cell Growth Differen. 4: 571-579. 115 Tsafarty, I.; Resau, J. H.; Rulong, S.; Keydar, I.; Vande Woude, G. F. (1992): The met proto-oncogen mesenchymal to epithelial cell conversion. Science 257: 117-119. Unger, C.; Marmé, D. (1995): Gibt es alternative Therapieformen beim Mammakarzinom? Stand und Perspektiven für die Behandlung des metastasierten Mammakrzinoms. Schweizerische Rundschau für Medizin (Praxis) 84: 390-394. Wang, N. S. (1975): The preformed stoma connecting the pleural cavity and the lymphatics in the parietal pleura. Am. Rev. Respir. Dis. 111: 12-20. Walther, H. E. (1948): Krebsmetastasen. Benno Schwabe & Co Verlag, Basel. Warren, R. S.; Yan, H.; Matli, M. R.; Gillett, N. A.; Ferrara, N. (1995): Regulation by VEGF of human colon cancer tumorgenesis in a mous model of experimental liver metastasis. J. Clin. Invest. 95: 1789-1797. Weidner, K. M.; Behrens, J.; Vandekerckhove, J.; Birchmeier, W. (1990): SF: molecular characteristics and effect on the invasiveness of epithelial cells. J. Cell. Biol. 111: 2097-2108. Weidner, K. M.; Sachs, M.; Birchmeier, W. (1993): The met receptor tyrosine kinase transduces motility, proliferation and morphogenic signals of SF/HGF in epithelial cells. J. Cell. Biol. 121: 145-154. Weidner, N.; Semple, J. P.; Welch, W. R.; Folkman, J. (1991): Tumor angiogenesis and metastasis - correlation in invasive breast carcinoma. N. Engl. J. Med. 324: 1-8. Woodring, J. H. (1984): Recognition of pleural effusion on supine radiographs. How much fluid is required? Am. J. Roentgenol. 142: 59-64. 116 Yamada, S. (1933): Über die seröse Flüssigkeit in der Pleurahöhle des gesunden Menschen. Z. Ges. Exp. Med. 90: 342-348. Yamashita, J.; Ogawa, M.; Yamashita, S.; Nomura, K.; Sadahito, S. (1994): Immunoreactive HGF is a strong and indepentent predictor of recurrence and survival in human breast cancer. Cancer Res. 54: 1630-1633. Yanagita, K.; Nagaike, M.; Ishibashi, H.; Nakamura, T. (1992): Lung may have an endocrine function producing HGF in response to injury of distal organs. Biochem. Biophys. Res. Commun. 182: 802-809. Zhu, H.; Naujokas, M. A.; Park, M. (1994): Receptor chimeras indicate that the met tyrosine kinase mediates the motility and morphogenic responses of HGF/SF. Cell Growth Differen. 5: 359-366. 117