Falk Gastro-Kolleg Leber und Gallenwege
Werbung

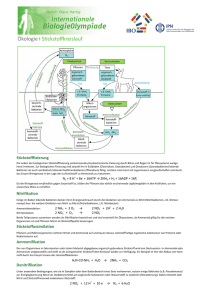
Ammoniakentgiftung in der Leber Blut vom Darm Blut zum Herz Ammoniak NAGS + Falk Gastro-Kolleg Leber und Gallenwege Ammoniak Harnstoffzyklus Harnstoff GlutaminSynthetase Glutamin Hyperammonämie bei Kindern und Erwachsenen: Einteilung und Krankheitsbilder Prof. Dr. Johannes Häberle Abteilung Stoffwechselkrankheiten Kinderspital Zürich Steinwiesstr. 7 0 Zürich Schweiz Zusammenfassung Die Hyperammonämie ist ein nicht-spezifischer Laborbefund, der als Surrogatmarker eine Störung im Stickstoffmetabolismus anzeigt und bei einer Reihe von Krankheitsbildern primär oder sekundär auftreten kann. Weil Ammoniak ab einer bestimmten Konzentration neurotoxisch ist, sind Zustände mit vermehrter Produktion oder eingeschränkter Entgiftung von Ammoniak stets als Notfall anzusehen, sofern klinische Symptome vorliegen. Ammoniak wird im Harnstoffzyklus in der Leber entgiftet. Bei einem angeborenen Defekt des Harnstoffzyklus (entweder eines der beteiligten Enzyme oder eines Transporters) resultiert eine primäre Hyperammonämie. Ist hingegen die Funktion des Harnstoffzyklus durch toxische Metabolite oder durch Substratmangel beeinträchtigt, wird die Situation als sekundäre Hyperammonämie bezeichnet. Diese Unterscheidung hat ihre Bedeutung nicht nur für das Verständnis der Pathophysiologie, sondern besitzt auch Relevanz für das diagnostische und therapeutische Vorgehen. Unabhängig von der Ätiologie führt eine Hyperammonämie zu irreversiblen Schäden des Gehirns, wenn keine rechtzeitige und effektive Behandlung erfolgt. Daher besitzen ein frühzeitiges Erkennen und die sofortige Einleitung der spezifischen Maßnahmen größte Bedeutung. Die wichtigsten prognostischen Faktoren sind, unabhängig von der zugrunde liegenden Ursache, die Dauer einer hyperammonämischen Bewusstseinsstörung und das Ausmaß der Hyperammonämie. Diese Übersicht beschreibt die biochemischen Hintergründe der primären und sekundären Hyperammonämie und gibt einen Überblick über die verschiedenen Aspekte im Management betroffener Patienten. Fragebeantwortung unter www.falkfoundation.de Falk Gastro-Kolleg Titelbild: Schematische Darstellung der Entgiftung von Ammoniak in 2 Schritten in den periportalen Hepatozyten (Harnstoffzyklus) sowie in den perivenösen Hepatozyten (Glutamin-Synthetase) 1 Schlüsselwörter Hyperammonämie | Harnstoffzyklusstörung | Organoazidopathie | Glutamin-Synthetase | N-Acetylglutamat-Synthase | hyperammonämisches Koma | Stoffwechselentgleisung | Bewusstseinsstörung Hyperammonämie bei Kindern und Erwachsenen: Einteilung und Krankheitsbilder 1. Einführung Die Messung der Konzentration von Ammoniak im Plasma gehört zu den notwendigen Notfalllaboruntersuchungen bei einem Patienten mit ungeklärter Bewusstseinsstörung [1]. Ist der Ammoniakwert im Plasma erhöht, ist dies zwar ein unspezifischer Befund, aber dennoch aufgrund der möglichen Neurotoxizität von großer Bedeutung für den Patienten. Die Hyperammonämie ist ein Indikator für eine Vielzahl von verschiedenen vererbten oder erworbenen Krankheitszuständen [2]. Hyperammonämie ist definiert als ein Plasma-Ammoniakwert > 50 µmol/L (> 100 µmol/L bei Neugebo­ renen) und sollte, sofern gleichzeitig klinische Symptome bestehen, stets als Notfall betrachtet werden. Eine Hyperammonämie kann bestehen i) bei einer erhöhten Produktion von Ammoniak (mögliche Ursachen: z. B. intestinale bakterielle Überwucherung, neurogene Blase, Medikamente, Infektionen oder andere katabole Zustände mit endogenem Abbau von Eiweiß) oder ii) bei verminderter Ammoniakentgiftung (mögliche Ursachen: z. B. verminderte Harnstoffzyklusfunktion, intrahepatische portosystemische Shunts oder eine unzureichende Aktivität der Glutamin-Synthetase) [2–3]. Die Entgiftung von Ammoniak erfolgt überwiegend durch den Harnstoffzyklus in den periportalen Hepatozyten [4]. Die Funktion des Harnstoffzyklus kann direkt durch einen Mangel an einem der beteiligten Enzyme und Transporter betroffen sein; die daraus resultierende Hyperammonämie wird als primäre Hyperammonämie bezeichnet [2]. Außerdem kann die Funktion des Harnstoffzyklus beeinträchtigt sein, wenn bestimmte inhibierende Stoffwechselprodukte anfallen oder wenn ein Mangel an Intermediärsubstraten besteht; ein solcher Zustand wird als sekundäre Hyperammonämie bezeichnet [2]. Neben dem Harnstoffzyklus dient das Enzym Glutamin-Synthetase (GS) der vollständigen Entgiftung von Ammoniak [5]. Dies ist vor allem bei erworbenen Leber­ erkrankungen von Bedeutung und beispielsweise die Ursache der Hyperammonämie bei Leberzirrhose. Die Notwendigkeit einer normalen Funktion der GS für eine vollständige Ammoniakentgiftung wurde kürzlich bei Patienten mit angeborenem GSDefekt belegt [6]. Die Einteilung in eine primäre oder sekundäre Hyperammonämie ist hilfreich zum Verständnis der zugrunde liegenden Pathophysiologie, aber auch für die Anpassung der diagnostischen und therapeutischen Strategien. Allerdings kann es bei beiden Formen der Hyperammonämie zu einer Enzephalopathie und irreversiblen Schädigung des Gehirns kommen, sofern die Behandlung nicht rechtzeitig und konsequent durchgeführt wird [7]. Das frühzeitige Erkennen einer Hyperammonämie und die sofortige Einleitung der spezifischen Behandlung sind von größter Bedeutung für die Prognose des Patienten [8]. Die wichtigsten prognostischen Faktoren sind, unabhängig von der zugrunde liegenden Ursache, die Dauer einer hyperammonämischen Bewusstseinsstörung und die maximale Ammoniakkonzentration. Neben einer hyperammonämischen Enzephalopathie gehen vor allem bei Erwachsenen mit erworbener Lebererkrankung manche Krankheitsbilder mit nur geringer Erhöhung der PlasmaAmmoniakwerte (im Bereich um 100 µmol/L) einher, die klinische Relevanz dieser Situation ist jedoch noch nicht vollständig verstanden. P Eine Hyperammonämie kann zu Enzephalopathie führen und ist stets ein Notfall. 2 Dieser Übersichtsbeitrag beschreibt die verschiedenen Formen der Hyperammonämie, deren biochemische, genetische und pathophysiologische Grundlagen, die häufigsten Krankheitsbilder sowie die wesentlichen Aspekte des diagnostischen und therapeu­ tischen Managements betroffener Patienten. 2. Grundlagen der primären Hyperammonämie Der Harnstoffzyklus ist im Säugetierorganismus der Hauptweg für die Entgiftung von Stickstoff, sofern dieser nicht als Bestandteil von Aminosäuren für die Herstellung von zum Beispiel Strukturproteinen, Enzymen oder Hormonen verwendet werden konnte. Der vollständige Harnstoffzyklus ist nur in der Leber und dort nur in periportalen Hepatozyten exprimiert [4]. Dies ist verständlich, weil diese Zellen als erste in Kontakt mit Pfortaderblut aus dem Darm kommen, welches eine Ammoniakkonzentration von 100–300 µmol/L aufweist. Im ersten Schritt des Harnstoffzyklus werden Ammo­ niak und Bikarbonat durch das Enzym Carbamoylphosphat-Synthetase 1 (CPS1, OMIM *608307) zu Carbamoylphosphat umgewandelt. Dieser erste Schritt und auch der folgende, katalysiert durch die Ornithin-Transcarbamylase (OTC, OMIM *300461), ist im Mitochondrium lokalisiert. Dagegen erfolgen die 3 nachstehenden enzymatischen Reaktionen (katalysiert durch die Argininosuccinat-Synthetase [ASS], OMIM *603470; die Argininosuccinat-Lyase [ASL], OMIM *608310; die Arginase 1 [ARG1], OMIM *608313) im Cytosol. Zusätzlich werden für die Harnstoffzyklusfunktion 2 Transporter benötigt, der Ornithin-Transporter ORNT1 (OMIM *603861) und der Aspartat-Glutamat-Antiporter Citrin (OMIM *603859) [9] (Abb. 1). Abb. 1 Blut aus der Pfortader NH3 + HCO3- Glutamin GLNase Citrullin GDH ASS ORNT1 Argininosuccinat Citrullin Glutamat NAGS Aspartat Citrin Acetyl-CoA N-AcetylL-glutamat + CPS1 OTC Carbamoylphosphat + Ornithin Harnstoffzyklus ASL Arginin ARG1 ORNT1 Ornithin Mitochondrium Cytosol Harnstoff Carbamoylphosphat Orotsäure Urin ARG1 = Arginase 1; ASL = Argininosuccinat-Lyase; ASS = Argininosuccinat-Synthetase; CPS1 = Carbamoylphosphat-Synthetase 1; GDH = Glutamat-Dehydrogenase; GLNase = Glutaminase; NAGS = N-Acetylglutamat-Synthase; ORNT1 = Ornithin-Citrullin-Antiporter; OTC = Ornithin-Transcarbamylase Enzyme und Transporter des Harnstoffzyklus in Mitochondrium und Cytosol der periportalen Hepatozyten Für die oben genannten enzymatischen Reaktionen wie auch für die beiden Transporter existieren angeborene Krankheiten durch Defekte der jeweiligen Gene. Des Weiteren benötigt der Harnstoffzyklus die Aktivierung durch N-Acetylglutamat (NAG), welches durch die N-Acetylglutamat-Synthase (NAGS, OMIM *608300) in Mitochondrien 3 bereitgestellt wird und obligater Aktivator der CPS1 ist (s. Abb. 1). Auch die NAGS kann selten von angeborenen Defekten betroffen sein (NAGS-Mangel, OMIM #237310), häufiger jedoch beeinträchtigen diverse Metabolite die enzymatische Aktivität der NAGS und führen dann sekundär zur Hyperammonämie (s. unten) [2] (Tab. 1). Formen der Hyperammonämie, deren wesentliche Diagnostik, Leitmetabolite und Pathomechanismen Primäre Hyperammonämie Enzymdefekte des Harnstoffzyklus Krankheit Wesentliche Diagnostik Leitmetabolite Pathomechanismus N-Acetylglutamat-Synthase (NAGS)-Mangel Aminosäuren im Plasma Glutamin im Plasma erhöht, Orotsäure im Urin normal Enzymatischer Block im Harnstoffzyklus CarbamoylphosphatSynthetase-1 (CPS1)-Mangel Aminosäuren im Plasma Glutamin im Plasma erhöht, Orotsäure im Urin normal Enzymatischer Block im Harnstoffzyklus Ornithin-Transcarbamylase (OTC)-Mangel Aminosäuren im Plasma, Orotsäure im Urin Glutamin im Plasma erhöht, Orotsäure im Urin erhöht Enzymatischer Block im Harnstoffzyklus Argininosuccinat-Synthetase (ASS)-Mangel Aminosäuren im Plasma, Orotsäure im Urin Citrullin im Plasma erhöht, Orotsäure im Urin erhöht Enzymatischer Block im Harnstoffzyklus Argininosuccinat-Lyase (ASL)-Mangel Aminosäuren im Plasma, Orotsäure im Urin Argininosuccinat in Plasma und Urin vorhanden, Orotsäure im Urin erhöht Enzymatischer Block im Harnstoffzyklus Arginase-1 (ARG1)-Mangel Aminosäuren im Plasma Arginin im Plasma erhöht Enzymatischer Block im Harnstoffzyklus Tab. 1 P Bei primärer Hyperammonämie besteht ein angebore­ ner Defekt der Enzyme oder Transporter des Harnstoffzyklus, wäh­rend bei sekun­­­därer Hyperammo­nämie die Funktion des Harnstoffzyklus indirekt betroffen ist. Transporterdefekte des Harnstoffzyklus HyperammonämieHyperornithinämieHomocitrullinurie-Syndrom Aminosäuren in Plasma und Urin Ornithin im Plasma erhöht, Homocitrullin im Urin erhöht Mangel an mitochondrialem Ornithin (Substrat der OTC) Citrullinämie Typ II Aminosäuren im Plasma Citrullin im Plasma erhöht Mangel an Aspartat (Substrat der ASS) Sekundäre Hyperammonämie mit Hemmung des Harnstoffzyklus Organoazidopathien Krankheit Wesentliche Diagnostik Leitmetabolite Pathomechanismus Methylmalonazidurie (MMA) Organische Säuren im Urin, Acylcarnitine im Blut Methylmalonsäure und Methylcitrat im Urin erhöht, Proprionylcarnitin im Blut erhöht Hemmung der NAGS durch Metabolite der MMA Propionazidämie (PA) Organische Säuren im Urin, Acylcarnitine im Blut 3-Hydroxy-Propionsäure und Methylcitrat im Urin erhöht, Proprionylcarnitin im Blut erhöht Hemmung der NAGS durch Metabolite der PA Isovalerianazidämie (IVA) Organische Säuren im Urin, Acylcarnitine im Blut Isovalerylglycin und 3-Hydroxy-Isovaleriansäure im Urin erhöht, Isovaleryl­ carnitin im Blut erhöht Hemmung der NAGS durch Metabolite der IVA Sekundäre Hyperammonämie mit funktioneller Insuffizienz des Harnstoffzyklus Fettsäurenoxidationsdefekte und Defekte des Carnitinzyklus Krankheit Wesentliche Diagnostik Leitmetabolite Pathomechanismus Mittelkettiger Acyl-CoADehydrogenase-Mangel Acylcarnitine im Blut, organische Säuren im Urin Erhöhtes Octanoylcarnitin im Blut, vermehrt Hexanoyl­ glycin und Dicarbonsäuren im Urin Mangel an Acetyl-CoA, Acylierung der CPS1 Multipler Acyl-CoADehydrogenase-Mangel Acylcarnitine im Blut, organische Säuren im Urin Erhöhte mittel- und lang­ kettige Acylcarnitine im Blut, vermehrt Ethylmalonsäure und Dicarbonsäuren im Urin Mangel an Acetyl-CoA, Acylierung der CPS1 Carnitin-Palmitoyltransferase-II-Mangel Acylcarnitine im Blut Erhöhte langkettige Acylcarnitine im Blut Mangel an Acetyl-CoA, Hemmung der CPS1 4 Carnitin-AcylcarnitinTranslo­k ase-Mangel Acylcarnitine im Blut Tiefes Carnitin im Blut, erhöhte langkettige Acylcarnitine im Blut Mangel an Acetyl-CoA, Hemmung der CPS1 Substratmangel Lysinurische Proteinintoleranz Aminosäuren im Urin Vermehrte Ausscheidung von Lysin, Arginin und Ornithin Mangel der HarnstoffzyklusSubstrate Arginin und Ornithin Pyrrolin-5-CarboxylatSynthetase-Mangel Aminosäuren im Plasma Tiefe Konzentrationen von Prolin, Arginin, Citrullin und Ornithin im Plasma Mangel der HarnstoffzyklusSubstrate Arginin und Ornithin Pyruvatcarboxylase-Mangel Aminosäuren im Plasma, Laktat im Blut Erhöhtes Citrullin und Alanin, Laktat erhöht Mangel an Aspartat (Substrat der ASS) Ornithin-AminotransferaseMangel Aminosäuren im Plasma Ornithin im Plasma erhöht Mangel an Ornithin (Substrat der OTC) Carbonic-Anhydrase-V-Mangel Laktat im Blut Laktat erhöht Fehlendes Bikarbonat (Substrat der CPS1) Andere HyperinsulinismusHyperammonämie-Syndrom Glukose im Blut Hypoglykämie Erhöhte Produktion von Ammoniak, verminderte Verfügbarkeit von Glutamat (nicht nachgewiesen) Mitochondriale Krankheiten Laktat im Blut Laktat erhöht Eingeschränkte ATP-Produktion Glutamin-Synthetase (GS)-Mangel Aminosäuren in Plasma und Urin Glutamin erniedrigt Unzureichende Ammoniak­ entgiftung 2.1 Enzymatische Defekte des Harnstoffzyklus Sämtliche klassische Harnstoffzyklusdefekte (engl. urea cycle disorders, UCD), d. h. die Enzymdefekte der NAGS, CPS1, OTC, ASS, ASL oder ARG1 können zu lebensbedrohlicher Hyperammonämie bei Patienten aller Altersgruppen führen (s. Tab. 1) [9–10]. Die genannten Enzymdefekte werden autosomal-rezessiv vererbt, außer der Mangel an OTC (OMIM #311250), welcher einen X-chromosomalen Erbgang aufweist. Die kumulative Inzidenz der Harnstoffzyklusdefekte beträgt nach neu erhobenen Zahlen etwa 1:35.000 [11]. Nach einem kurzen unauffälligen Intervall erkrankt etwa die Hälfte der Patienten um den 3. Lebenstag mit rasch progredienter hyperammonämischer Enzephalopathie. Ohne frühzeitige Diagnosestellung und Behandlung besteht eine hohe Sterblichkeitsrate aufgrund eines irreversiblen Hirnödems. Aber auch bei aggressiver Therapie ist ein relevanter Teil der Patienten von den neurologischen Folgen einer neonatalen hyperammonämischen Enzephalopathie betroffen. Die wesentlichen Komplikationen sind Krampfanfälle sowie geistige oder psychomotorische Retardierung. Dies unterstreicht die Bedeutung der möglichst frühzeitigen Behandlung betroffener Neugeborener, gilt jedoch auch für die Erstmanifestation in jeder anderen Altersgruppe. Bemerkenswert ist weiter, dass viele der Neugeborenen mit UCD zunächst für einige Zeit unter der Verdachtsdiagnose einer bakteriellen Sepsis behandelt werden, ehe in zweiter Linie ein Stoffwechseldefekt differenzialdiagnostisch erwogen wird; dies führt oft zu relevanter Verzögerung und damit schlechter Prognose. Im Verlauf sind katabole Situationen vor allem bei (ansonsten banalen) viralen Infektionen, Fieber oder durch längeres Fasten die Hauptauslöser für wiederkehrende Stoffwechselentgleisungen. Für die neurologische Prognose sind die Dauer eines hyperammonämischen Komas und das Ausmaß der Ammoniakerhöhung die relevantesten Einflussgrößen [12–13]. Leider ist die Prognose von UCD-Patienten oft nach wie vor schlecht, weil die Möglichkeit einer endogenen Intoxikation durch Ammoniak bei differenzialdiagnostischen Überlegungen vielfach nur in zweiter Linie erwogen wird [14–15]. Die Entwicklung eines Hirnödems stellt die bedeutendste Komplikation einer Harnstoffzyklusstörung dar. Während viele der neurologischen Folgeerscheinungen auf die neurotoxische Wirkung von Ammoniak zurückzuführen sind, existieren weitere schädigende Metabolite. Etwa kann ein Mangel an Arginin oder von dessen Metaboliten ebenfalls das Gehirn schädigen [16–17]. Bei Patienten mit ASL-Mangel (OMIM #207900) treten neurokognitive Beeinträchtigungen auch ohne hyperammonämische Episoden P Harnstoffzyklusdefekte sind die häufigste Ursache einer (primären) Hyperammonämie; sie können in jedem Lebensalter erstmals auftreten. 5 auf; hier dient als spezifische pathophysiologische Erklärung, dass bei ASL-Mangel die Verfügbarkeit von Stickstoffmonoxid durch Beeinträchtigung eines zytosolischen Multienzymkomplexes eingeschränkt ist. Ein Sonderfall sind OTC-Patientinnen, die in Abhängigkeit vom Muster der X-Inaktivierung typische neurokognitive Defizite aufweisen, selbst wenn manche Patientinnen lebenslang klinisch asymptomatisch sind [18]. 2.2 Transporterdefekte des Harnstoffzyklus Die beiden Transporter des Harnstoffzyklus, ORNT1 und Citrin, führen, sofern defekt, in unterschiedlicher Weise zu Hyperammonämie [19]. Das Hyperammonämie-Hyperorni­ thinämie-Homocitrullinurie (HHH)-Syndrom (OMIM #238970), verursacht durch einen Defekt des Ornithin-Transporters ORNT1, hat viele Gemeinsamkeiten mit den klassischen Harnstoffzyklusstörungen. So sind Neugeborene ebenfalls von einer hyperammonämischen Stoffwechselentgleisung bereits wenige Tage nach der Geburt betroffen. Im Gegensatz dazu ist der Citrin-Mangel (Synonym: Citrullinämie Typ II) klinisch davon sehr verschieden. Patienten können hier als Neugeborene oder Säuglinge/Kleinkinder eine cholestatische Lebererkrankung entwickeln, welche meist ohne Hyperammonämie abläuft (OMIM #605814). Dagegen sind erwachsene Patienten mit einem Defekt im gleichen Transporter klinisch in anderer Weise betroffen: Es bestehen ein chronisches Hirnödem sowie neurologische Symptome wie Verwirrtheit oder gestörtes Bewusstsein (OMIM #603471). P Transporterdefekte des Harnstoff­ zyklus sind eine seltene Ursache einer (primären) Hyperammonämie. 3. Grundlagen der sekundären Hyperammonämie Wie oben bereits erwähnt, kann die Funktion des Harnstoffzyklus auf verschiedene Weise sekundär beeinträchtigt sein. Durch Anstau von Metaboliten aus anderen Stoffwechselwegen kann der Harnstoffzyklus inhibiert werden; dies ist am häufigsten der Fall bei einer Krankheit aus der Gruppe der Organoazidopathien. Beispiele sind hier vor allem Stoffwechseldefekte im Abbau verzweigtkettiger Aminosäuren, etwa die Methylmalonazidurie bei Defekt der Methylmalonyl-CoA-Mutase (MMA, OMIM #251000), die Propionazidurie bei Defekt einer der Untereinheiten der Propionyl-CoA-Carboxylase (PA, OMIM #606054) oder die Isovalerianazidurie bei Defekt der Isovaleryl-CoA-Dehydrogenase (IVA, OMIM #243500) [2]. Bei all diesen angeborenen Stoffwechselkrankheiten kommt es zu einer Hemmung der Funktion des Harnstoffzyklus und dadurch zu einer sekundären Hyperammonämie. In gleicher Weise können Medikamente den Harnstoffzyklus hemmen, etwa Valproinsäure durch direkte Hemmung der NAGS durch den Metaboliten Valproyl-CoA [20]. Außer einer Hemmung des Harnstoffzyklus kann dessen Funktion auch durch Substratmangel beeinträchtigt werden; beispielsweise besteht eine verringerte Verfügbarkeit der Aminosäuren Arginin, Citrullin und Ornithin bei der lysinurischen Proteinintoleranz (LPI) bei Defekt des kationischen Aminosäuren-Transporters (OMIM #222700) oder bei Pyrrolin-5-Carboxylat-Synthetase-Mangel (P5CSD, OMIM #219150) [21–22]. Zudem führen alle Zustände, bei denen vermindert das Substrat der NAGS, Acetyl-CoA, bereitgestellt wird, zu einer beeinträchtigten Produktion von NAG und dadurch zu verminderter Aktivierung des Harnstoffzyklus. Dies ist etwa der Fall bei Fettsäurenoxidationsstörungen (z. B. mittelkettiger Acyl-CoA-Dehydrogenase-Mangel [MCADD], OMIM #201450; multipler Acyl-CoA-Dehydrogenase-Mangel [MADD], OMIM #231680) sowie bei De­ fekten des Carnitinzyklus (z. B. die neonatale Form des Carnitin-Palmitoyltransferase-IIMangels, neonataler CPT-II-Mangel, OMIM #608836) [23] (s. Tab. 1). P Bei Hemmung der Enzymfunktion oder bei Substratmangel kann der Harnstoffzyklus sekundär betroffen sein; dieser Situation können verschiedene angeborene Stoffwechselkrankheiten zugrunde liegen. 3.1 Sekundäre Hyperammonämie aufgrund einer Hemmung des Harnstoffzyklus Organoazidopathien Organoazidopathien sind eine Gruppe angeborener Enzymdefekte im Abbau verzweigtkettiger Aminosäuren. Die Symptome dieser Krankheiten sind vorwiegend neurologisch, zudem bestehen verschiedenste Komplikationen mehrerer Organe (einschl. Gehirn, Nieren, Herz, Knochen und Bauchspeicheldrüse). Ähnlich den Harnstoffzyklusstörungen sind aus klinischer Sicht Neugeborene erneut am meisten betroffen, weil diese ebenfalls von einer schwerwiegenden Hyperammonämie mit hoher 6 Morbidität und Mortalität bedroht sein können. Auch Organoazidopathien können sich in jedem Lebensalter erstmals manifestieren. Die Höhe des Ammoniakwerts erlaubt keine Unterscheidung zu den primären Störungen des Harnstoffzyklus. Eher kann die Konzentration von Glutamin im Blut zur Unterscheidung dienen, weil diese Aminosäure bei Organoazidopathien oft geringer oder gar nicht erhöht ist. Bei Organoazidopathien ist die Ammoniakkonzentration sekundär erhöht, weil Metabolite wie Methylmalonsäure, Methylzitronensäure oder Propionsäure zu einer Hemmung der NAGS- und CPS1-Funktion führen. Aus diesem Grund wird der Einsatz von N-Carbamylglutamat, einem chemischen NAG-Analogon, für die Behandlung der akuten Hyperammonämie bei den häufigsten Organoazidopathien MMA, PA und IVA empfohlen (und ist für diese Indikation auch durch die EMA zugelassen) [24–25]. Valproat Valproinsäure ist eine Carbonsäure (C8-verzweigtkettige Fettsäure) und wird seit 1967 als Medikament für die Behandlung von epileptischen Anfällen, später auch für die Behandlung von psychiatrischen Störungen und Migräne, verwendet. Während dieser mehr als 40 Jahre wurden wiederholt unerwünschte Ereignisse berichtet, am relevantesten ist hier sicherlich das akute Leberversagen zu nennen. Als ursächlich werden eine Hemmung der Fettsäurenoxidation oder des Abbaus von Carnitin und AcetylCoA oder eine noch nicht näher bekannte mitochondriale Störung diskutiert. Immer wieder wurde das Auftreten einer Hyperammonämie unter Valproatbehandlung beobachtet. Vor Kurzem wurde nachgewiesen, dass die Metabolite der Valproin­ säure, etwa Valproyl-CoA, eine direkte hemmende Wirkung auf die Aktivität der NAGS haben und dies zu einer geringeren Aktivierung der CPS1 und somit des Harnstoff­ zyklus führt [20]. Diese Hemmung ist reversibel, sodass nach Absetzen von Valproat eine normale NAGS-Funktion wiederkehrt und die Hyperammonämie verschwindet. Bis dies tatsächlich eintritt, kann als Überbrückung die Gabe von N-Carbamylglutamat erwogen werden. P Organoazidopathien sind die häufigste Ursache einer sekundären Hyperammonämie; sie können in jedem Lebensalter erstmals auftreten und sind vom Ausmaß der Hyperammonämie nicht von den primären Störungen zu unterscheiden. P Valproat führt zur Hemmung der N-Acetylglutamat-Synthase und damit sekundär zu Hyperammonämie. 3.2 Sekundäre Hyperammonämie aufgrund einer funktionellen Störung des Harnstoffzyklus Defekte der Fettsäurenoxidation Patienten mit Störungen der mitochondrialen Fettsäurenoxidation können sich in jedem Alter und mit großer klinischer Variabilität präsentieren. Die gefürchtetsten klinischen Zeichen bei schwerer Krankheit sind ein hypoketotisch-hypoglykämisches Koma und Leberversagen sowie Kardiomyopathie und Rhabdomyolyse. Als Labor­ befunde während einer Entgleisung werden eine hypoketotische Hypoglykämie, Laktatazidose und Hyperammonämie gefunden. Als Ursache der Hyperammonämie bei Fettsäurenoxidationsdefekten wird ein Mangel an Acetyl-CoA als Folge der Blockierung des Abbaus der Acylcarnitine angenommen [26]. Dadurch steht ein Substrat der NAGS nicht ausreichend zur Verfügung. Zudem wurde eine Hemmung der Aktivität der CPS1 durch langkettige Fettsäuren-Acyl-CoA, insbesondere Palmitoyl-CoA, beschrieben. Diese Hemmung der CPS1, bedingt durch Acylierung, ist Bestandteil der Stickstoff-sparenden Strategie des Organismus bei Hunger, kann jedoch im Fall von vor allem (sehr) langkettigen Acyl-CoA-Oxidationsdefekten fatal sein. Defekte des Carnitinzyklus und Mangel an Carnitin Defekte des Carnitinzyklus stellen eine Gruppe erblicher Krankheiten dar, bei denen der Transport von Carnitin-gebundenen langkettigen Fettsäuren aus dem Cytosol in das Mitochondrium gestört ist. Dadurch ist die Oxidation langkettiger Fettsäuren beeinträchtigt, sodass toxische Metabolite akkumulieren, welche zum Teil den Harnstoffzyklus inhibieren können. So sind für alle Defekte des Carnitinzyklus hyperammonämische Entgleisungen beschrieben, die mitunter sehr ausgeprägt verlaufen können (z. B. bei einem Defekt der Carnitin-Acylcarnitin-Translokase, CACT-Mangel, OMIM #212138). Zudem sind auch bei alimentärem Carnitin-Mangel hyperammonämische Enzephalopathien beschrieben. P Störungen der mitochondrialen Fettsäurenoxidation und des Carnitin­ zyklus führen sekundär zu Hyper­ ammonämie, weil toxische Metabolite akkumulieren oder Acetyl-CoA fehlt. 7 Lysinurische Proteinintoleranz Die lysinurische Proteinintoleranz (LPI) ist eine autosomal-rezessiv vererbte Störung der Reabsorption der dibasischen Aminosäuren Lysin, Arginin und Ornithin an der basolateralen Membran in Darm und Nieren [21]. Als Ergebnis des renalen Verlusts dieser Aminosäuren besteht eine funktionelle Insuffizienz des Harnstoffzyklus durch Mangel an Intermediärmetaboliten. Dabei kann eine hyperammonämische Enzephalopathie auftreten, wobei diese nicht im Vordergrund der LPI steht, welche als Multiorgankrankheit (mit u. a. Gedeihstörung, interstitieller Pneumonie, chronischer Niereninsuffizienz, Osteopenie und immunologischer Dysfunktion) verstanden werden sollte. Ursächlich ist ein Defekt im SLC7A7-Gen, welcher sehr selten außerhalb von Süditalien oder dem Norden Japans auftritt. Pyrrolin-5-Carboxylat-Synthetase-Mangel Der Pyrrolin-5-Carboxylat-Synthetase-Mangel (P5CSD) ist autosomal-rezessiv vererbt und führt ebenfalls zu einem Mangel an Substraten des Harnstoffzyklus und damit funktioneller Harnstoffzyklus-Insuffizienz. Eine metabolische Besonderheit sind die erniedrigten Konzentrationen der Aminosäuren Prolin, Ornithin und Arginin als Folge der gestörten Umwandlung von Glutamat zu Δ1-Pyrrolin-5-Carboxylat, einer Vorstufe des Prolins [22]. Die Hyperammonämie bei P5CSD ist typischerweise präprandial am ausgeprägtesten und unterscheidet sich damit von anderen hier vorgestellten Störungen. Allerdings ist P5CSD eine sehr seltene und bislang nur im Kindesalter beschriebene Entität (weitere Symptome sind angeborene Katarakt, Cutis laxa, Hyper­ mobilität der Gelenke und geistige Behinderung). P Die lysinurische Proteinintoleranz und der Pyrrolin-5-Carboxylat-Synthe­ tase-Mangel führen sekundär zu Hyperammonämie, weil Intermediär­ metabolite des Harnstoffzyklus fehlen. 4. Weitere angeborene Krankheitsbilder mit Hyperammonämie Hyperinsulinismus-Hyperammonämie (HIHA)-Syndrom Das HIHA-Syndrom (OMIM #606762) entsteht bei „gain of function“-Mutationen im Gen der mitochondrialen Glutamat-Dehydrogenase 1 (GDH1), GLUD1 [27]. Dieses Gen wird durch Guanosin-5‘-Triphosphat (GTP) reguliert, welches die Enzymaktivität in­hibiert. Diese Inhibition geht bei bestimmten Mutationen verloren, sodass durch die GDH1Überaktivität vermehrt α-Ketoglutarat (stimuliert die Insulinsekretion aus β-Zellen des Pankreas) und Ammoniak entstehen. Allerdings ist die Hyperammonämie bei einigen Patienten mit HIHA-Syndrom nicht besonders ausgeprägt (Werte um 100 µmol/L) und erfordert dann möglicherweise keine Therapie. Störungen der Atmungskette, ATP-Mangel und Pyruvat-Dehydrogenase-Mangel Störungen der Atmungskette umfassen eine große und sehr heterogene Gruppe angeborener Defekte des Energiestoffwechsels. Häufig resultiert ein ATP-Mangel durch Beeinträchtigung der mitochondrialen oxidativen Phosphorylierung. Es wird vermutet, dass der ATP-Mangel den Harnstoffzyklus beeinträchtigt, weil einige der daran beteiligten Enzyme (CPS1 und ASS) ATP-abhängig sind. So wird bei einigen Patienten mit Störungen der Atmungskettenfunktion eine zum Teil ausgeprägte Hyperammonämie beobachtet, wobei dies bislang nur für wenige der bekannten Defekte beschrieben ist. Ein Beispiel ist der Defekt des TMEM70-Gens (mitochondrialer Komplex-VMangel, OMIM #614052), welches für ein Protein der ATP-Synthase kodiert; Patienten zeigen bereits als Neugeborene eine schwere Hyperammonämie mit Laktatazidose. Defekte der Pyruvat-Dehydrogenase (PDH) umfassen eine Gruppe von Störungen einer der Komponenten des PDH-Multienzymkomplexes. Patienten zeigen oft eine globale Retardierung, Fehlbildungen des Gehirns sowie eine Laktatazidose. In einzelnen Fällen wurde zudem eine Hyperammonämie beschrieben, welche möglicherweise ebenfalls durch eine verminderte Funktion der CPS1 bei ATP-Mangel bedingt ist. Hyperammonämie bei Defekt der Glutamin-Synthetase In der Literatur sind einzelne Patienten mit einem angeborenen Defekt der GlutaminSynthetase (GS) und daraus resultierendem systemischem Glutamin-Mangel beschrieben. Alle Patienten sind bereits als Neugeborene mit schwerer Enzephalopathie aufgefallen [6, 28]. Neben der sehr niedrigen Konzentration von Glutamin in Blut, Urin und Liquor bestand eine Hyperammonämie mit Ammoniakwerten zwischen 100 und 200 µmol/L. Dies illustriert perfekt die Notwendigkeit der GS für eine vollständige Ammoniakentgiftung im Säugetierorganismus und beim Menschen. 8 Schließlich gibt es auch Patienten, bei denen ein erworbener GS-Mangel sowie eine Hyperammonämie nach orthotoper Lungen- oder Knochenmarktransplantation bestehen. Deren Ursache ist nicht vollständig geklärt, vermutet wird eine deutlich reduzierte GS-Expression und -Aktivität. 5. G rundlagen nicht-metabolischer oder erworbener Ursachen einer Hyperammonämie Neben den oben genannten Situationen gibt es weitere, in denen selten auch eine Hyperammonämie resultieren kann: bei Überwucherung mit Urease-bildenden Bakte­ rien in Blase und Harnwegen (v. a. bei Infektionen der Harnwege und bei einer neuro­ genen Blase) oder im Darm (v. a. bei verminderter Motilität oder Dilatation intestinaler Segmente), bei Verwendung von Medikamenten wie L-Asparaginase (Hydrolyse von Asparagin zu Asparaginsäure und Ammoniak) oder von Chemotherapeutika oder bei hoch dosierten Aminosäureninfusionen im Rahmen einer parenteralen Ernährung. Zudem können vaskuläre Besonderheiten, vor allem bei Vorliegen eines portokavalen Shunts, zu einer verminderten Ammoniakentgiftung führen [29]. Dies kann angeboren der Fall sein, noch häufiger jedoch erworben im Rahmen einer Leberzirrhose (Tab. 2). Erworbene Hyperammonämie-Formen Situation/Erkrankung Pathomechanismus Akute oder chronische Leberinsuffizienz Reduzierte Kapazität des Harnstoffzyklus, verminderte Funktion der Glutamin-Synthetase aufgrund einer Hemmung durch Tyrosin-Nitrierung, intrahepatische portosystemische Shunts Malformation von Gefäßen Intrahepatische portosystemische Shunts Valproatbehandlung Hemmung der N-Acetylglutamat-Synthase durch Metabolite des Valproats, Carnitin-Mangel L-Asparaginase-Behandlung Erhöhte Produktion von Ammoniak durch Hydrolyse von Asparagin Urease-produzierende Organismen Erhöhte Produktion von Ammoniak in den Harnwegen und im Darm Totale parenterale Ernährung (TPN) Unausgewogene TPN-Zusammensetzung mit relativem Arginin-Mangel Ernährungsbedingter Carnitin-Mangel Beeinträchtigung der Fettsäurenoxidation und dadurch Mangel an Acetyl-CoA Zystoskopie mit Glycinhaltigen Lösungen Erhöhte Produktion von Ammoniak bei Stickstoffüberladung Status nach Lungen- oder Knochenmarktransplantation Reduzierte Expression und Aktivität der Glutamin-Synthetase unbekannter Ursache Transiente Hyperammonämie des Neugeborenen Intrahepatische portosystemische Shunts (nicht nachgewiesen) Akute oder chronische Leberinsuffizienz Neben den vielfältigen angeborenen Defekten, welche mit Hyperammonämie einhergehen können, kann auch eine erworbene Leberinsuffizienz die Entgiftung von Ammoniak betreffen. Dies ist etwa der Fall bei einer Leberzirrhose, bei der zum einen der normale Blutfluss durch die Leber beeinträchtigt ist und zum anderen die Anzahl der funktionsfähigen Hepatozyten unter einen Schwellenwert für ausreichende Ammoniakentgiftung vermindert sein kann. Im fortgeschrittenen Stadium kann eine hepatische Enzephalopathie mit ausgeprägter Hyperammonämie resultieren. Tab. 2 P Mehrere Medikamente sowie portokavale Shunts bei Malformationen von Gefäßen können zu Hyperammo­n­ ämie führen. P Erworbene Lebererkrankungen gehen oft mit Hyperammonämie einher, unter anderem, weil die Entgiftungs­ leistung der Leber für Ammoniak nicht mehr ausreichend ist. 9 Ein weiterer Faktor bei Lebererkrankungen ist der erworbene Defekt der GlutaminSynthetase (GS) durch Tyrosin-Nitrierung [30]. Als deren Folge ist der notwendige zweite Schritt der Ammoniakentgiftung neben dem Harnstoffzyklus, die Reaktion der GS, beeinträchtigt. Hyperammonämie bei gesteigerter Produktion von Ammoniak Bei Infektionen der Harnwege mit Urease-produzierenden Organismen (z. B. Proteus mirabilis und einige Klebsiella-Arten) kann durch Spaltung von Harnstoff eine Hyperammonämie resultieren. Dies kann auch bei Patienten mit Ureter-Dilatation, neuro­ gener Blasenstörung und Restharn oder bei solchen mit eingeschränkter Motilität und Dilatation des Kolons der Fall sein, wobei jeweils eine bakterielle Überwucherung Ursache des vermehrten Anfalls von Ammoniak ist. Unter diesen Bedingungen kann die Konzentration von Ammoniak im Plasma (nach Erfahrung des Autors) Werte von > 300 µmol/L erreichen und damit Auslöser einer hyperammonämischen Enzephalopathie sein. Weiterhin können einige Medikamente zu einer erhöhten Produktion von Ammoniak führen; dies sind Chemotherapeutika (Auslöser eines ausgeprägten Katabolismus), L-Asparaginase sowie pegylierte Asparaginase (dieses bakteriell gewonnene Enzym katalysiert die Hydrolyse von Asparagin in Asparaginsäure und Ammoniak) oder auch Glycin-haltige Spüllösungen während Zystoskopie-Untersuchungen (vermehrter Anfall von Stickstoff bzw. Ammoniak). Auch bei Diuretika und Antiepileptika sind Hyper­ ammonämien als Nebenwirkung beschrieben. Daneben kann eine totale parenterale Ernährung, sofern diese (bei relativem Mangel an Arginin) hohe Konzentrationen an Eiweiß enthält, zu Dysfunktion des Harnstoff­ zyklus und Hyperammonämie führen. Interessanterweise wurde diese Komplikation vor allem in den 1980er- und 1990er-Jahren berichtet, während möglicherweise nach Verbesserung der Zusammensetzung der verwendeten Lösungen keine weiteren Berichte in der Literatur nach 2001 existieren. PVerschiedene seltene Situationen können zu vermehrter Produktion von Ammoniak führen; dazu zählen Ureaseproduzierende Bakterien sowie mehrere Medikamente. Hyperammonämie bei intrahepatischen portosystemischen Shunts Vaskuläre Malformationen können zu einer Verbindung von Pfortader und Vena cava inferior und damit zur Umleitung von Blut aus dem Darm unter Umgehung der Leber direkt in den Körperkreislauf führen. Das Ausmaß der Hyperammonämie liegt im Bereich der Ammoniakkonzentration in der Portalvene (100–300 µmol/L). 6. Fazit Eine Hyperammonämie kann sehr verschiedene Ursachen haben. Im Kindesalter sind darunter vorwiegend angeborene Stoffwechselkrankheiten, wenngleich diese sich grundsätzlich in jedem Alter manifestieren können. Im Erwachsenenalter spielen eher erworbene Erkrankungen der Leber eine Rolle. Das wichtigste klinische Zeichen einer Hyperammonämie ist die (nicht anderweitig erklärte) Bewusstseinsstörung. Für das Verständnis der Situation und das diagnostische und therapeutische Management ist die Zuordnung der vorliegenden Störung zu primärer oder sekundärer Hyperammo­n­ ämie hilfreich. In jedem Fall ist ein unverzügliches Handeln dringend anzuraten, sofern eine symptomatische Hyperammonämie, also eine Enzephalopathie, besteht. Im Vordergrund steht die möglichst rasche Entgiftung von Ammoniak. Die Prognose ist von einem sofortigen und konsequenten Therapiebeginn abhängig. Für die Details des diagnostischen und therapeutischen Prozederes wird auf die verfügbaren Leitlinien verwiesen [8, 31]. Danksagung und Hinweis für den Leser Die Arbeiten des Autors zu Harnstoffzyklusdefekten und Hyperammonämie werden vom Schweizerischen Nationalfonds unterstützt (Projekt-Nr. 310030_127184). Dieser Artikel basiert auf einer kürzlich veröffentlichten Übersichtsarbeit des Autors [2], die Struktur des Manuskripts sowie der Text wurden jedoch mit Blick auf die erwartete Leserschaft stellenweise sehr stark verändert. 10 Zu empfehlende Literatur Literatur 1 Häberle J. Clinical practice: the management of hyperammonemia. Eur J Pediatr. 2011;170(1):21–34. 2 Häberle J. Clinical and biochemical aspects of primary and secondary hyperammonemic disorders. Arch Biochem Biophys. 2013;536(2):101–8. 3 Bachmann C. Mechanisms of hyperammonemia. Clin Chem Lab Med. 2002;40(7):653–62. 4 Häussinger D. Nitrogen metabolism in liver: structural and functional organization and physiological relevance. Biochem J. 1990;267(2):281–90. 5 Häussinger D, Sies H, Gerok W. Functional hepatocyte heterogeneity in ammonia metabolism. The intercellular glutamine cycle. J Hepatol. 1985;1(1):3–14. 6 Häberle J, Shahbeck N, Ibrahim K, Hoffmann GF, Ben-Omran T. Natural course of glutamine synthetase deficiency in a 3 year old patient. Mol Genet Metab. 2011;103(1):89–91. 7 Walker V. Ammonia toxicity and its prevention in inherited defects of the urea cycle. Diabetes Obes Metab. 2009;11(9):823–35. 8 Häberle J, Boddaert N, Burlina A, Chakrapani A, Dixon M, Huemer M, et al. Suggested guidelines for the diagnosis and management of urea cycle disorders. Orphanet J Rare Dis. 2012;7:32. 9 Brusilow S, Horwich A. Urea cycle enzymes. In: Scriver C, et al. (eds): The metabolic & molecular bases of inherited disease. McGraw-Hill, New York 2001:1909–63. 10 Brusilow SW, Maestri NE. Urea cycle disorders: diagnosis, pathophysiology, and therapy. Adv Pediatr. 1996;43:127–70. 11 Summar ML, Koelker S, Freedenberg D, Le Mons C, Häberle J, Lee HS, et al. The incidence of urea cycle disorders. Mol Genet Metab. 2013;110(1–2):179–80. 12 Picca S, Dionisi-Vici C, Abeni D, Pastore A, Rizzo C, Orzalesi M, et al. Extracorporeal dialysis in neonatal hyperammonemia: modalities and prognostic indicators. Pediatr Nephrol. 2001;16(11):862–7. 13 Msall M, Batshaw ML, Suss R, Brusilow SW, Mellits ED. Neurologic outcome in children with inborn errors of urea synthesis. Outcome of urea-cycle enzymopathies. N Engl J Med. 1984;310(23):1500–5. 11 14 Kido J, Nakamura K, Mitsubuchi H, Ohura T, Takayanagi M, Matsuo M, et al. Long-term outcome and intervention of urea cycle disorders in Japan. J Inherit Metab Dis. 2012;35(5):777–85. Literatur 15 Bachmann C. Long-term outcome of patients with urea cycle disorders and the question of neonatal screening. Eur J Pediatr. 2003;162 Suppl 1:S29–33. 16 Braissant O, McLin VA, Cudalbu C. Ammonia toxicity to the brain. J Inherit Metab Dis. 2013;36(4):595–612. 17 Braissant O. Current concepts in the pathogenesis of urea cycle disorders. Mol Genet Metab. 2010;100 Suppl 1:S3–S12. 18 Gropman AL, Summar M, Leonard JV. Neurological implications of urea cycle disorders. J Inherit Metab Dis. 2007;30(6):865–79. 19 Palmieri F. Diseases caused by defects of mitochondrial carriers: a review. Biochim Biophys Acta. 2008;1777(7–8):564–78. 20 Aires CC, van Cruchten A, Ijlst L, de Almeida IT, Duran M, Wanders RJ, et al. New insights on the mechanisms of valproate-induced hyperammonemia: inhibition of hepatic N-acetylglutamate synthase activity by valproyl-CoA. J Hepatol. 2011;55(2):426–34. 21 Ogier de Baulny H, Schiff M, Dionisi-Vici C. Lysinuric protein intolerance (LPI): a multi organ disease by far more complex than a classic urea cycle disorder. Mol Genet Metab. 2012;106(1):12–7. 22 Martinelli D, Häberle J, Rubio V, Giunta C, Hausser I, Carrozzo R, et al. Understanding pyrroline-5-carboxylate synthetase deficiency: clinical, molecular, functional, and expression studies, structure-based analysis, and novel therapy with arginine. J Inherit Metab Dis. 2012;35(5):761–76. 23 Mitchell GA, Gauthier N, Lesimple A, Wang SP, Mamer O, Qureshi I. Hereditary and acquired diseases of acyl-coenzyme A metabolism. Mol Genet Metab. 2008;94(1):4–15. 24 Ah Mew N, McCarter R, Daikhin Y, Nissim I, Yudkoff M, Tuchman M. N-carbamylglutamate augments ureagenesis and reduces ammonia and glutamine in propionic acidemia. Pediatrics. 2010;126(1):e208–14. 25 Häberle J. Carglumic acid for the treatment of N-acetylglutamate synthase deficiency and of acute hyperammonemia. Expert Rev Endocrinol Metab. 2012;7(2):263–71. 26 Corkey BE, Hale DE, Glennon MC, Kelley RI, Coates PM, Kilpatrick L, et al. Relationship between unusual hepatic acyl coenzyme A profiles and the pathogenesis of Reye syndrome. J Clin Invest. 1988;82(3):782–8. 12 27 Stanley CA, Lieu YK, Hsu BY, Burlina AB, Greenberg CR, Hopwood NJ, et al. Hyperinsulinism and hyperammonemia in infants with regulatory mutations of the glutamate dehydrogenase gene. N Engl J Med. 1998;338(19):1352–7. Literatur 28 Häberle J, Görg B, Rutsch F, Schmidt E, Toutain A, Benoist JF, et al. Congenital glutamine deficiency with glutamine synthetase mutations. N Engl J Med. 2005;353(18):1926–33. 29 Hoover W, Ackerman V, Schamberger M, Kumar M, Marshalleck F, Hoyer M. The congenital porto-caval fistula: a unique presentation and novel intervention. Pediatr Pulmonol. 2008;43(2):196–9. 30 Schliess F, Görg B, Fischer R, Desjardins P, Bidmon HJ, Herrmann A, et al. Ammonia induces MK-801-sensitive nitration and phosphorylation of protein tyrosine residues in rat astrocytes. FASEB J. 2002;16(7):739–41. 31 Deutsche Gesellschaft für Kinder- und Jugendmedizin (2012): Diagnostik und Therapie von Harnstoffzyklusstörungen, AWMF-Leitlinienregisternummer 027-006, http://www.awmf.org/leitlinien/detail/ll/027-006.html (gültig bis 30.04.2017). 13 Fragen zur Hyperammonämie Welche Antworten sind richtig? Frage 1: Als Ursache einer Hyperammonämie kommt folgende Erklärung nicht infrage: EE EE EE EE EE Vermehrte Produktion von Ammoniak Urease-spaltende Bakterien Überaktivität der Glutamin-Synthetase Transporterdefekte des Harnstoffzyklus Portosystemische intrahepatische Shunts Frage 2: Unter einer primären Hyperammonämie wird verstanden: EE EE EE EE EE Ein Anstieg des Ammoniaks vor dem Auftreten von Symptomen Der erstmalige Nachweis einer Hyperammonämie Ein Ammoniakanstieg ohne klinische Zeichen Ein Anstau des Ammoniaks vor der Leber Ein unmittelbarer Defekt des Harnstoffzyklus Frage 3: Als nicht-relevant für die Prognose bei Hyperammonämie wird angesehen: EE EE EE EE EE Falk Gastro-Kolleg Leber und Gallenwege Bitte beachten Sie: Bei der Beantwortung der Fragen ist immer nur 1 Antwort möglich. Die Beantwortung der Fragen und Erlangung des Fortbildungszertifikats ist nur online möglich. Bitte gehen Sie dazu auf unsere Homepage www.falkfoundation.de. Unter dem Menüpunkt Falk Gastro-Kolleg können Sie sich anmelden und die Fragen beantworten. Bitte diesen Fragebogen nicht per Post oder Fax schicken! Die Zeit eines hyperammonämischen Komas Das Ausmaß der Hyperammonämie Die Geschwindigkeit des Abfalls von Ammoniak Die maximale Ammoniakkonzentration Das Ausmaß eines etwaigen Hirnödems Frage 4: Eine sekundäre Hyperammonämie kann auftreten bei ... EE EE EE EE EE angeborener Störung eines der Enzyme des Harnstoffzyklus Fehlernährung mit Eiweißüberladung defektem mitochondrialen Ornithin-Transporter Fehlen von Substraten des Harnstoffzyklus raschem Anfall von Ammoniak als Folge einer Infektion Frage 5: Zu den erworbenen Ursachen einer Hyperammonämie gehören nicht: EE EE EE EE EE Vaskuläre Malformationen Impfungen Verminderte Darmmotilität Einnahme von Medikamenten (z. B. Valproat) Leberzirrhose Wichtig: Fragebeantwortung unter www.falkfoundation.de Falk Gastro-Kolleg 14 Frage 6: Die klinischen Zeichen einer Hyperammonämie ... Falk Gastro-Kolleg EE EE EE EE EE Leber und Gallenwege können bei Patienten jeden Alters erstmals auftreten sind spezifisch in jeder Altersgruppe betreffen die meisten Körperfunktionen erlauben oft eine Zuordnung zur zugrunde liegenden Diagnose sind vorwiegend gastrointestinal und hepatisch Frage 7: Verschiedene Medikamente können eine Hyperammonämie auslösen, dazu zählen nicht: EE EE EE EE EE Valproat Chemotherapeutika Diuretika Antiepileptika Antibiotika Frage 8: Eine akute Hyperammonämie mit Zeichen der Enzephalopathie ... EE erfordert Kontrollen des Ammoniakwerts bereits nach 1–2 Stunden EE ist bei Erwachsenen meist Folge einer spät diagnostizierten angeborenen Stoffwechselstörung EE sollte zunächst Anlass sein, ein Leberversagen auszuschließen EE ist stets als Notfall zu betrachten EE kann bei bakterieller Sepsis sekundär auftreten Frage 9: Die Entgiftung von Ammoniak ... EE EE EE EE EE erfolgt gleichermaßen in Leber und Nieren erfordert die vollständige Expression des Harnstoffzyklus setzt eine ubiquitäre Expression der Glutamin-Synthetase voraus erfolgt überwiegend mitochondrial unterliegt tageszeitlichen Schwankungen Frage 10: Zu den Symptomen einer Hyperammonämie zählen nicht: EE EE EE EE EE Die Entwicklung eines Hirnödems Episodisches Erbrechen Somnolenz Ataxie oder Tremor Rezidivierende Infekte 15