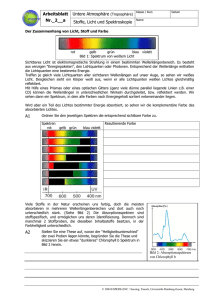
Protokoll Versuch 1-5
Werbung

Biochemie Hauptfach Sommersemester 2008 Protokoll Versuch 1-5 Melanie Thompson Inhalt I. Versuch: Proteinbestimmung ............................................................................... 2 II. Versuch: Reinigung von Fusionsproteinen ......................................................... 39 III. Versuch: Metabolitbestimmung....................................................................... 55 IV. Versuch: Derepressionskinetik ....................................................................... 68 V. Versuch: Analyse von Polysomen-Profilen ........................................................ 81 VI. Quellen ........................................................................................................... 88 VII. Anhang ........................................................................................................... 88 1 I. Versuch: Proteinbestimmung Einleitung Um den Proteingehalt einer Lösung quantitativ zu bestimmen, und damit Aussagen über die Konzentration zu liefern, gibt es verschiedene Methoden, die sich grob in zwei Kategorien einordnen lassen: spektroskopische und kolorimetrische Methoden. Spektroskopische Bestimmungsmethoden basieren im Prinzip auf der Absorption bestimmter Aminosäuren, Seitenketten oder Bindungen eines Peptids bei definierten Wellenlängen. So absorbieren die aromatischen Aminosäuren Tryptophan, Phenylalanin und Tyrosin bei einer Wellenlänge von 280nm. Peptidbindungen, also die Bindungen, die in einem Protein die einzelnen Aminosäuren verknüpfen, absorbieren wiederum bei 205nm. Die Ermittlung der Anzahl aromatischer Aminosäuren oder Peptidbindungen eines Proteins kann der Aufklärung unbekannter Proteine dienen. In beiden Fällen muss jedoch der Gehalt des Proteins an aromatischen Aminosäuren, bzw. die Größe des Protein und die damit vorhandene Anzahl an Peptidbindungen bekannt sein, um eine quantitative Bestimmung des Proteingehalts einer Lösung eines bekannten Proteins möglich zu machen. Vorteil der Spektroskopischen Konzentrationsbestimmung einer Proteinlösung ist, dass diese unter nativen Bedingungen stattfindet, die Zielproteine also nicht denaturiert werden (müssen). Nachteile spektroskopischer Methoden, sind zum einen die geringe Sensitivität, da z.B. nur die aromatischen Aminosäuren eines Peptides erfasst werden, zum anderen auch die Beeinflussung des Ergebnisses durch den pH-Wert der Lösung, das Lösungsmittel selbst, aber auch Seitenketten innerhalb des Peptides sowie weitere nicht-Protein-Substanzen. Außerdem, muss eine Proteinlösung in hochreiner Form vorliegen, da das Messergebnis sonst nicht aussagekräftig ist. 2 Kolorimetrische oder auch photometrische Proteinbestimmungsmethoden liegen die Reaktionen farbstoffbildender Reagenzien mit funktionellen Gruppen von Proteinen zugrunde. Anhand dieser Reaktion ist eine direkte Wechselbeziehung zwischen Farbintensität der Proteinlösung und Konzentration der reagierenden Gruppen gegeben. Die exakte Farbintensität kann dann mittels eines Photometers bei der für den jeweiligen Test spezifischen Wellenlänge bestimmt werden. Die wichtigsten vier Assays sind: Biuret-Assay, BCA-Assay, Bradford-Assay und Lowry-Assay. Die Biuret-Reaktion fußt auf der Reaktion zwischen Cu2+-Ionen und den Peptidbindungen eines Proteins sowie den TyrosinAbbildung Reagenz 1: Biuret- Resten in alkalischer Lösung zu einem rot-violetten-Farbkomplex. Diese Reaktion wurde das erste Mal zwischen Cu2+-Ionen und dem Biuret-Reagenz (Abb. 1) beobachtet, welches ebenfalls eine Peptidbindung enthält und namensgebend für diese Reaktion war. Die Absorption wird nach erfolgter Reaktion bei 546 nm gemessen. Diese Methode ist schnell, jedoch weniger genau und kann durch verschiedene Substanzen (z.B. den Puffer Tris) gestört werden. Das Lowry-Assay ist eine Erweiterung der Biuret-Reaktion, hierbei findet ein weiteres, das sog. Folin-Ciocalteu-Phenol-Reagenz, Anwendung. Dieses wird durch die während der Biuret-Reaktion im Farbkomplex gebildeten Cu2+-Ionen reduziert, woraufhin eine intensiv blaue Färbung entsteht. Die Intensität der Färbung wird im Anschluss bei 540nm gemessen. Die Lowry-Methode ist deutlich genauer als die Biuret-Methode, wird jedoch durch die gleichen Substanzen bedeutend stärker gestört. Bei der BCA-Methode werden Cu2+-Ionen durch Proteine bei alkalischen pH-Werten zu Cu+-Ionen reduziert. Diese komplexieren wiederum mit der hier Abbildung 2: Bicinchoninsäure (BCA) verwendeten Bicinchoninsäure (kurz: BCA, Abb. 2). Der Farbumschlag von grün auf violett macht die Reaktion deutlich, anschließend die bei Extinktion 562nm wird bestimmt. Vorteile dieser Methode ist die variable Abbildung 3: Coomassie-Brillantblau 3 Sensitivität durch Temperaturanpassung, die einfache Durchführung und die relative Stabilität des Farbkomplexes. Das BCA-Assay kann durch komplexbildende Reagenzien gestört werden. Bei der Proteinbestimmung nach Bradford findet der blaue Säurefarbstoff Coomassie-Brillantblau (Abb. 3) Anwendung. Dieses Reagenz bindet an Proteine, woraufhin sich das Absorptionsmaximum im Komplex von 464nm auf 595nm verschiebt. Dieses Assay hat die höchste Sensitivität bei gleicher simpler Durchführung. Da der Test jedoch im sauren Milieu stattfindet, fallen viele Proteine aus, zudem findet die Wechselwirkung des Farbkomplexes mit kationischen und nicht-polaren, hydrophoben Seitenketten statt, enthält ein Protein wenig dieser Seitenketten, kann es zu Ungenauigkeiten bei der Konzentrationsberechnung kommen. Entscheidend für diese kolorimetrischen Bestimmungsmethoden ist es, dass parallel zur Bestimmung des Proteins unbekannter Konzentration eine Eichgerade mit einem Protein1 bekannter, verschiedener Konzentrationen erstellt werden muss, um anhand dieser die Konzentration des isolierten Proteins ablesen zu können. Ziel dieses Versuches ist es, die oben genannten unterschiedlichen kolorimetrischen Methoden auf ihre Genauigkeit und Vergleichbarkeit zu untersuchen. Material und Methoden Siehe Skript. Die Verdünnungen des Rinderserum Albumins sowie des Ovalbumins wurden bei allen Assays mit deionisiertem Wasser angesetzt. Ergebnisse 1 Hierfür wird meist Rinderserum Albumin (BSA) oder Ovalbumin eingesetzt. Es können auch verschiedene Konzentrationen des Zielproteins Anwendung finden, so dieses bereits in reiner Form vorliegt. 4 Biuret-Assay Zunächst wurde mit Hilfe von Rinderserum Albumin in unterschiedlichen Konzentrationen (0-4mg im Test) in Doppelbestimmung eine Eichgerade erstellt. Tabelle 1: Biuret - Doppelbestimmung der Extinktion bei 546nm von Rinderserum Albumin (BSA) in verschiedenen Konzentrationen zur Erstellung einer Eichgeraden mg BSA E1 E2 E 0 0 0 0 0,5 0,141 0,095 0,118 1 0,198 0,214 0,206 1,5 0,323 0,307 0,315 2 0,383 0,377 0,38 2,5 0,484 0,484 0,484 3 0,574 0,574 0,574 3,5 0,675 0,699 0,687 4 0,729 0,743 0,736 Werden diese Werte (Tab. 1) nun graphisch aufgetragen und über eine Regressionsgerade verbunden, so fungiert diese Gerade als Eichgerade zur Proteinbestimmung einer Proteinlösung unbekannter Konzentration. 5 Abbildung 4: Biuret - Eichgerade erstellt mit Rinderserum Albumin Die Gerade muss nun so gezogen werden, dass sie den Nullpunkt schneidet. Abbildung 5: Biuret - Eichgerade mit Rinderserum Albumin, Schnittpunkt der Gerade muss im Nullpunkt liegen Eine BSA-Lösung unbekannter Konzentration wurde nun in verschiedenen Verdünnungen dem Biuret-Assay unterzogen. Die Messung fand erneut in Doppelbestimmung statt. 6 Tabelle 2: Biuret - Extinktion der BSA-Lösung unbekannter Konzentration in Doppelbestimmung bei 546nm Verdünnung unverdünnt 1:1 1:2 1:3 1:10 1:100 E1 0,737 0,438 0,296 0,234 0,099 0,024 E2 0,735 0,447 0,3 0,237 0,096 0,026 Mittelwert 0,736 0,4425 0,298 0,2355 0,0975 0,025 Diese Werte (Tab. 2) können nun entweder anhand der Eichgerade abgelesen oder mit Hilfe der Geradengleichung berechnet werden. Exemplarisch sei dies für die 1:1-Verdünnung gezeigt: Eichgerade ohne 0-Schnittpunkt umgeformt lautet die Gleichung wie folgt: 200 µl der 1:1-Verdünnung entsprechen einer BSA-Konzentration von 2,295 mg, da es sich aber um eine Verdünnung handelt, muss diese herausgerechnet werden: Damit entsprechen 200 µl der unbekannten BSA-Lösung einer BSA-Konzentration von 4,59 mg. Da jedoch nur 200 µl eingesetzt wurden, muss dieser Wert nun noch auf 1 ml hochgerechnet werden. 7 Da die Eichgerade in der Größenordnung mg BSA angesetzt wurde, handelt es sich bei diesem Wert bereits um die Endkonzentration in mg/ml. Diese Rechnung wird nun für alle Proben anhand der Eichgerade mit und ohne 0Schnittpunkt durchgeführt, und mit den handschriftlich abgelesenen Ergebnissen verglichen: Tabelle 3: Biuret - Konzentration der unbekannten BSA-Probe im Mittel anhand verschiedener Eichgeraden Verdünnung handschriftliche Eichgerade Eichgerade (Abb. 4) Eichgerade Schnittpunkt 0 (Abb. 5) 1:1 22,75 23,74 22,946 1:2 23,25 23,895 22,703 1:3 24 25,1 23,514 1:10 26,25 25,45 21,487 Mittelwert 24,0625 mg/ml 24,546 mg/ml 22,66 mg/ml Die unverdünnte sowie die 1:100 verdünnte unbekannte BSA-Probe wurden nicht mit in die Rechnungen einbezogen, da sie außerhalb der Eichgerade lagen. Anhand der Eichgeraden wurde ermittelt, dass die unbekannte BSA-Probe eine Konzentration von 24,546 mg/ml nach Eichgerade (Abb. 4), 22,66 mg/ml nach der Eichgerade mit Schnittpunkt Null (Abb. 5) bzw. 24,0625 mg/ml nach der handschriftlichen Eichgerade hat. In oben beschriebener Weise wird der Vorgang mit einer Ovalbumin-Probe unbekannter Konzentration wiederholt, wobei auch hier zunächst eine Eichgerade mit Hilfe von Ovalbumin unterschiedlicher Konzentrationen (0-4mg im Test) erstellt wurde. 8 Tabelle 4: Biuret - Doppelbestimmung der Extinktion bei 546nm von Ovalbumin in verschiedenen Konzentrationen zur Erstellung einer Eichgeraden mg E1 Ovalbumin E2 E 0 0 0 0 0,5 0,124 0,125 0,1245 1 0,235 0,242 0,2385 1,5 0,36 0,358 0,359 2 0,466 0,468 0,467 2,5 0,532 0,56 0,546 3 0,658 0,638 0,648 3,5 0,716 0,714 0,715 4 0,757 0,763 0,76 Anhand dieser Werte (Tab. 4) wurde im nächsten Schritt eine Eichgerade erzeugt: 9 Abbildung 6: Biuret - Eichgerade erstellt mit Ovalbumin Wird diese Gerade durch den Nullpunkt gesetzt, ergibt sich folgendes Bild: Abbildung 7: Biuret - Eichgerade erstellt mit Ovalbumin, Schnittpunkt der Geraden im Nullpunkt Im nächsten Schritt wurde eine unbekannte Ovalbumin-Lösung in verschiedenen Verdünnungen dem Biuret-Assay unterzogen. Die Messungen fanden in Doppelbestimmung statt. 10 Tabelle 5: Biuret - Extinktion der Ovalbumin-Lösung unbekannter Konzentration in Doppelbestimmung bei 546nm Verdünnung unverdünnt 1:1 1:2 1:3 1:10 1:100 E1 0,776 0,555 0,375 0,284 0,116 0,016 E2 0,778 0,549 0,379 0,297 0,113 0,014 Mittelwert 0,777 0,552 0,377 0,2905 0,1145 0,015 In oben beschriebener Weise wird auch für die unbekannte Ovalbumin-Lösung die Konzentration berechnet, und erneut mit den handschriftlich abgelesenen Werten verglichen. Tabelle 6: Biuret - Konzentration der unbekannten Ovalbumin-Probe im Mittel anhand verschiedener Eichgeraden Verdünnung handschriftliche Eichgerade Eichgerade (Abb. 6) Eichgerade Schnittpunkt 0 (Abb. 7) 1:1 24,5 28,2 26,53 1:2 26,625 28,695 26,19 1:3 27 29,3 25,96 1:10 27,5 27,65 19,3 Mittelwert 26,41 mg/ml 28,461 mg/ml 24,5 mg/ml Die Werte der unverdünnten bzw. der 1:100 verdünnten unbekannten OvalbuminProbe wurden nicht berücksichtig, da diese außerhalb der Eichgeraden lagen. Für die unbekannte Ovalbumin-Probe ergibt sich eine Konzentration von 28,461 mg/ml für die eine Eichgerade (Abb. 6), 24,5 mg/ml für die Eichgerade mit Schnittpunkt Null (Abb. 7), bzw. 26.41 mg/ml für die handschriftliche Eichgerade. 11 BCA-Assay Zunächst wurde für das BCA-Assay eine Eichgerade mit Hilfe bekannter Rinderserum Albumin Konzentrationen (0-100µg im Test) in Doppelbestimmung erstellt. Tabelle 7: BCA - Doppelbestimmung der Extinktion bei 562nm von Rinderserum Albumin (BSA) in verschiedenen Konzentrationen zur Erstellung einer Eichgeraden µg BSA E1 E2 E 0 0 0 0 10 0,207 0,159 0,183 20 0,338 0,312 0,325 30 0,458 0,481 0,4695 40 0,563 0,553 0,558 50 0,677 0,698 0,6875 60 0,827 0,893 0,86 70 1,043 0,975 1,009 80 1,174 1,077 1,1255 90 1,28 1,25 1,265 100 1,391 1,447 1,419 Diese Werte (Tab. 6) graphisch aufgetragen, ergibt die BSA-Eichgerade für das BCA-Assay. 12 Abbildung 8: BCA - Eichgerade erstellt mit BSA Der Schnittpunkt dieser Geraden muss anschließend durch den Nullpunkt gelegt werden. Abbildung 9: BCA - Eichgerade erstellt mit BSA, Schnittpunkt der Geraden im Nullpunkt Das BCA-Assay fand nun bei einer BSA-Lösung unbekannter Konzentration Anwendung, wobei die Messungen erneut in Doppelbestimmung stattfanden. 13 Tabelle 8: BCA - Extinktion der BSA-Lösung unbekannter Konzentration in Doppelbestimmung bei 562nm Verdünnung unverdünnt 1:10 1:20 1:40 1:100 1:1000 E1 2,929 1,390 0,647 0,437 0,185 0,036 E2 2,967 1,464 0,454 0,564 0,225 0,043 Mittelwert 2,95 1,427 0,5505 0,5005 0,205 0,0395 Diese Werte (Tab. 8) können nun entweder anhand der Eichgerade abgelesen oder mit Hilfe der Geradengleichung berechnet werden. Exemplarisch sei dies für die 1:20-Verdünnung gezeigt: Eichgerade ohne 0-Schnittpunkt umgeformt lautet die Gleichung wie folgt: 50 µl der 1:20-Verdünnung entsprechen einer BSA-Konzentration von 40,19 µg, da es sich aber um eine Verdünnung handelt, muss diese herausgerechnet werden: Damit entsprechen 50 µl der unbekannten BSA-Lösung einer BSA-Konzentration von 803,85 µg. Da jedoch nur 50 µl eingesetzt wurden, muss dieser Wert nun noch auf 1 ml hochgerechnet werden: 14 Da die Eichgerade in der Größenordnung µg BSA angesetzt wurde, muss dieser Wert noch auf mg/ml umgerechnet werden: Diese Rechnung wird nun für alle Proben anhand der Eichgerade mit und ohne 0Schnittpunkt durchgeführt, und mit den handschriftlich abgelesenen Werten verglichen, was zu folgendem Ergebnis führt: Tabelle 9: BCA - Konzentration der unbekannten BSA-Probe im Mittel anhand verschiedener Eichgeraden Verdünnung handschriftliche Eichgerade Eichgerade (Abb. 8) Eichgerade Schnittpunkt 0 (Abb. 9) 1:20 14 16,9272 16,077 1:40 27,2 30,7776 29,08 1:100 27 31,482 27,23 Mittelwert 22,733 mg/ml 26,3956 mg/ml 24,129 mg/ml Die unverdünnte, sowie die 1:10 und 1:1000 Verdünnungen wurden in der Berechnung nicht berücksichtigt, da sie zum einen außerhalb der Eichgeraden lagen (gilt für unverdünnte sowie 1:10 verdünnte BSA-Lösung) bzw. sehr abweichende Ergebnisse zeigten (bei 1:1000 Verdünnung wäre im Mittel eine BSA-Konzentration von 58,31 mg/ml berechnet worden, die sich zu sehr von den restlichen Werten unterscheidet). Für die unbekannte BSA-Probe ergibt sich im Mittel also eine BSAKonzentration von 26,3956 mg/ml BSA für die Eichgerade (Abb. 8), 24,129 mg/ml für die Eichgerade mit Schnittpunkt Null (Abb. 9) bzw. 22.733 mg/ml für die handschriftliche Eichgerade. 15 In oben beschriebener Weise wird das BCA-Assay mit einer Ovalbumin-Probe unbekannter Konzentration wiederholt. Auch hierbei wurde zunächst eine Eichgerade mit Hilfe von Ovalbumin unterschiedlicher Konzentrationen (0-100 µg im Test) erstellt. Tabelle 10: BCA - Doppelbestimmung der Extinktion bei 562nm von Ovalbumin in verschiedenen Konzentrationen zur Erstellung einer Eichgeraden µg E1 Ovalbumin E2 E 0 0 0 0 10 0,182 0,152 0,167 20 0,325 0,296 0,3105 30 0,439 0,377 0,408 40 0,502 0,528 0,515 50 0,639 0,591 0,615 60 0,722 0,707 0,7145 70 0,876 0,836 0,856 80 0,972 0,965 0,9685 90 1,03 1,063 1,0465 100 1,247 1,307 1,277 Anhand dieser Werte (Tab. 10) wurde eine Eichgerade für das BCA-Assay erstellt: 16 Abbildung 10: BCA - Eichgerade erstellt mit Ovalbumin Diese Eichgerade muss anschließend noch durch den Nullpunkt gelegt werden: Abbildung 11: BCA - Eichgerade erstellt mit Ovalbumin, Schnittpunkt der Gerade im Nullpunkt Eine Ovalbumin-Lösung unbekannter Konzentration wurde anschließend in verschiedenen Verdünnungen dem BCA-Assay unterzogen. Die Messungen wurden in Doppelbestimmungen vollzogen. 17 Tabelle 11: BCA - Extinktion der Ovalbumin-Lösung unbekannter Konzentration in Doppelbestimmung bei 562nm Verdünnung unverdünnt 1:10 1:20 1:40 1:100 1:1000 E1 2,863 1,465 0,861 0,739 0,220 0,028 E2 2,920 1,605 0,654 0,697 0,226 0,040 Mittelwert 2,8915 1,535 0,7575 0,718 0,223 0,034 Die Konzentration der unbekannten Ovalbumin-Lösung wird in oben genannter Weise berechnet, und mit den handschriftlich abgelesenen Werten verglichen. Tabelle 12: BCA - Konzentration der unbekannten Ovalbumin-Probe im Mittel anhand verschiedener Eichgeraden Verdünnung handschriftliche Eichgerade Eichgerade (Abb. 10) Eichgerade Schnittpunkt 0 (Abb. 11) 1:20 23,8 27,5316 26,273 1:40 45,6 52,1904 49,67 1:100 35 40,476 34,182 Mittelwert 34,8 mg/ml 40,066 mg/ml 36,71 mg/ml Die unverdünnte, sowie die 1:10 und 1:1000 Verdünnungen wurden in der Berechnung der Konzentration nicht berücksichtigt. Zum einen lagen diese Werte außerhalb der Eichgeraden (gilt für unverdünnte sowie 1:10 verdünnte OvalbuminLösung), zum anderen zeigten sie sehr abweichende Ergebnisse (bei 1:1000 Verdünnung wäre im Mittel eine Ovalbumin-Konzentration von 58,89 mg/ml berechnet worden, die sich zu sehr von den restlichen Werten unterscheidet). Für die unbekannte Ovalbumin-Probe ergibt sich damit im Mittel eine Ovalbumin18 Konzentration von 40,066 mg/ml Ovalbumin für die Eichgerade (Abb. 10), 36,71 mg/ml für die Eichgerade mit Schnittpunkt Null (Abb. 11) bzw. 34,8 mg/ml für die handschriftliche Eichgerade. Lowry-Assay Auch beim Lowry-Assay wurde zunächst mit Hilfe bekannter BSA-Konzentrationen (0-80 µg im Test) in Doppelbestimmung eine Eichgerade erstellt: Tabelle 13: Lowry - Doppelbestimmung der Extinktion bei 540nm von Rinderserum Albumin in verschiedenen Konzentrationen zur Erstellung einer Eichgeraden µg BSA E1 E2 E 0 0 0 0 10 0,085 0,077 0,081 20 0,185 0,238 0,2115 30 0,274 0,333 0,3035 40 0,405 0,388 0,3965 50 0,505 0,473 0,489 60 0,538 0,555 0,5465 70 0,698 0,654 0,676 80 0,733 0,759 0,746 Anhand dieser Werte (Tab. 13) wurde dann die Eichgerade erstellt: 19 Abbildung 12: Lowry - Eichgerade erstellt mit Rinderserum Albumin Diese Eichgerade muss anschließend noch durch den Nullpunkt gelegt werden: Abbildung 13: Lowry - Eichgerade erstellt mit Rinderserum Albumin, Schnittpunkt der Geraden im Nullpunkt Verschiedene Verdünnungen einer unbekannten BSA-Lösung wurden anschließend dem Lowry-Assay unterzogen. Die durchgeführten Messungen wurden in Doppelbestimmung erstellt. 20 Tabelle 14: Lowry - Extinktion der BSA-Lösung unbekannter Konzentration in Doppelbestimmung bei 540nm Verdünnung 1:100 1:200 1:400 1:1000 E1 0,410 0,193 0,116 0,047 E2 0,393 0,157 0,094 0,033 Mittelwert 0,4015 0,175 0,105 0,04 Diese Werte (Tab. 14) können nun entweder anhand der Eichgerade abgelesen oder mit Hilfe der Geradengleichung berechnet werden. Exemplarisch sei dies für die 1:200-Verdünnung gezeigt: Eichgerade ohne 0-Schnittpunkt umgeformt lautet die Gleichung wie folgt: Damit entsprechen 200 µl der 1:200-Verdünnung einer BSA-Konzentration von 18,56 µg, da es sich aber um eine Verdünnung handelt, muss diese herausgerechnet werden: Damit entsprechen 200 µl der unbekannten BSA-Lösung einer BSA-Konzentration von 3711,1 µg. Da jedoch nur 200 µl eingesetzt wurden, muss dieser Wert nun noch auf 1 ml hochgerechnet werden: 21 Da die Eichgerade in der Größenordnung µg BSA angesetzt wurde, muss dieser Wert noch auf mg/ml umgerechnet werden: Diese Rechnung wird nun für alle Proben anhand der Eichgerade mit und ohne 0Schnittpunkt durchgeführt und anschließend mit den handschriftlich abgelesenen Werten verglichen. Tabelle 15: Lowry - Konzentration der unbekannten BSA-Probe im Mittel anhand verschiedener Eichgeraden Verdünnung handschriftliche Eichgerade Eichgerade (Abb. 12) Eichgerade Schnittpunkt 0 (Abb. 13) 1:100 22,25 22,3015 21,86 1:200 18 19,436 18,56 1:400 21 23,318 21,56 1:1000 20 22,18 17,78 Mittelwert 20,3125 mg/ml 21,8089 mg/ml 19,94 mg/ml Die unbekannte BSA-Lösung hat damit im Mittel eine Konzentration von 21,8089 mg/ml BSA für die Eichgerade (Abb. 12), 19,94 mg/ml für die Eichgerade, die durch den Nullpunkt verläuft (Abb. 13) bzw. 20,3125 mg/ml für die handschriftliche Eichgerade. 22 Auch für die unbekannte Ovalbumin-Lösung wurde zunächst eine Eichgerade anhand einer Ovalbumin-Lösung bekannter Konzentration (0-80 µg im Test) erstellt. Tabelle 16: Lowry - Doppelbestimmung der Extinktion bei 540nm von Ovalbumin in verschiedenen Konzentrationen zur Erstellung einer Eichgeraden µg E1 Ovalbumin E2 E 0 0 0 0 10 0,112 0,186 0,149 20 0,375 0,213 0,294 30 0,52 0,426 0,473 40 0,332 0,495 0,4135 50 0,604 0,439 0,5215 60 0,746 0,675 0,7105 70 0,771 0,758 0,7645 80 0,82 0,815 0,8175 Anhand dieser Werte (Tab. 16) wurde anschließend eine Eichgerade erstellt: 23 Abbildung 14: Lowry - Eichgerade erstellt mit Ovalbumin Diese Eichgerade (Abb. 14) wurde anschließend so gelegt, dass sie den Nullpunkt schneidet. Abbildung 15: Lowry - Eichgerade erstellt mit Ovalbumin, Schnittpunkt der Geraden im Nullpunkt Im nächsten Schritt wurde eine unbekannte Ovalbumin-Lösung in verschiedenen Verdünnungen dem Lowry-Assay unterzogen. Die Messung fand in Doppelbestimmung statt. 24 Tabelle 17: Lowry - Extinktion der Ovalbumin-Lösung unbekannter Konzentration in Doppelbestimmung bei 540nm Verdünnung 1:100 1:200 1:400 1:1000 E1 0,444 0,166 0,154 0,057 E2 0,501 0,174 0,126 0,060 Mittelwert 0,4725 0,17 0,14 0,0585 Die Ovalbumin-Konzentration dieser Proben wird wie in oben beschriebener Weise ermittelt. Anschließend werden die so berechneten mit den handschriftlich abgelesenen Werten verglichen. Tabelle 18: Lowry - Konzentration der unbekannten Ovalbumin-Probe im Mittel anhand verschiedener Eichgeraden Verdünnung handschriftliche Eichgerade Eichgerade (Abb. 14) Eichgerade Schnittpunkt 0 (Abb. 15) 1:100 20,75 23,595 20,625 1:200 14,5 16,94 11 1:400 24 27,88 16 1:1000 37,5 28,95 - Mittelwert 24,1875 mg/ml 24,341 mg/ml 15,875 mg/ml Damit hat die unbekannte Ovalbumin-Lösung im Mittel eine Konzentration von 24,341 mg/ml Ovalbumin anhand der Eichgerade (Abb. 14), 15,875 mg/ml anhand der Eichgerade, die den Nullpunkt schneidet (Abb. 15) bzw. 24,1875 mg/ml für die handschriftliche Eichgerade. 25 Bradford Auch das Bradford-Assay verlangt zunächst das Erstellen einer Eichgerade. Diese wurde mit Hilfe einer BSA-Lösung bekannter Konzentration (0-10 µg im Test) in Doppelbestimmung erzeugt. Tabelle 19: Bradford - Doppelbestimmung der Extinktion bei 595 nm von Rinderserum Albumin in verschiedenen Konzentrationen zur Erstellung einer Eichgeraden µg BSA E1 E2 E 0 0 0 0 1 0,063 0,092 0,0775 2 0,148 0,174 0,161 3 0,264 0,23 0,247 4 0,297 0,36 0,3285 5 0,351 0,355 0,353 6 0,375 0,415 0,395 7 0,439 0,439 0,439 8 0,495 0,487 0,491 9 0,535 0,521 0,528 10 0,55 0,572 0,561 Anhand dieser Werte (Tab. 19) wurde dann die Eichgerade erstellt: 26 Abbildung 16: Bradford - Eichgerade erstellt mit Rinderserum Albumin Diese Gerade wurde anschließend durch den Nullpunkt gelegt, was zu folgendem Ergebnis führte: Abbildung 17: Bradford - Eichgerade erstellt mit Rinderserum Albumin, Schnittpunkt der Geraden im Nullpunkt Eine unbekannte BSA-Lösung wurde anschließend in verschiedenen Verdünnungen dem Bradford-Assay unterzogen. Die durchgeführten Messungen wurden in Doppelbestimmung erstellt. 27 Tabelle 20: Bradford - Extinktion der BSA-Lösung unbekannter Konzentration in Doppelbestimmung bei 595 nm Verdünnung 1:200 1:400 1:1000 1:10000 E1 0,435 0,275 0,152 0,015 E2 0,421 0,279 0,148 0,011 Mittelwert 0,428 0,277 0,15 0,013 Diese Werte (Tab. 20) können nun entweder anhand der Eichgerade abgelesen oder mit Hilfe der Geradengleichung berechnet werden. Exemplarisch sei dies für die 1:400-Verdünnung gezeigt: Eichgerade ohne 0-Schnittpunkt umgeformt lautet die Gleichung wie folgt: Damit entsprechen 100 µl der 1:400-Verdünnung einer BSA-Konzentration von 4,13 µg, da es sich aber um eine Verdünnung handelt, muss diese herausgerechnet werden: Damit entsprechen 100 µl der unbekannten BSA-Lösung einer BSA-Konzentration von 1650,91 µg. Da jedoch nur 100 µl eingesetzt wurden, muss dieser Wert nun noch auf 1 ml hochgerechnet werden: 28 Da die Eichgerade in der Größenordnung µg BSA angesetzt wurde, muss dieser Wert noch auf mg/ml umgerechnet werden: Diese Rechnung wird nun für alle Proben anhand der Eichgerade mit und ohne 0Schnittpunkt durchgeführt, die Ergebnisse werden anschließend mit den handschriftlich abgelesenen Werten verglichen: Tabelle 21: Bradford - Konzentration der unbekannten BSA-Probe im Mittel anhand verschiedener Eichgeraden Verdünnung handschriftliche Eichgerade Eichgerade (Abb. 16) Eichgerade Schnittpunkt 0 (Abb. 17) 1:200 13,2 15,464 13,75 1:400 17,2 19,9456 16,51 1:1000 23 26,773 18,2 1:10000 10 18,64 - Mittelwert 15,85 mg/ml 20,20565 mg/ml 16,15 mg/ml Damit hat die unbekannte BSA-Lösung im Mittel eine Konzentration von 20,20565 mg/ml BSA für die Eichgerade (Abb. 16), 16,15 mg/ml BSA für die Eichgerade, die den Nullpunkt schneidet (Abb. 17) bzw. 15,85 mg/ml für die handschriftliche Eichgerade. 29 Um die Konzentration einer unbekannten Ovalbumin-Lösung anhand des BradfordAssays zu bestimmen, muss auch hier zunächst eine Eichgerade erstellt werden. Hierzu wird eine Ovalbumin-Lösung bekannter Konzentration (0-10 µg im Test) dem Bradford-Assay in Doppelbestimmung unterzogen. Tabelle 22: Bradford - Doppelbestimmung der Extinktion bei 595 nm von Ovalbumin in verschiedenen Konzentrationen zur Erstellung einer Eichgeraden µg E1 Ovalbumin E2 E 0 0 0 0 1 0,048 0,047 0,0475 2 0,095 0,072 0,0835 3 0,127 0,11 0,1185 4 0,159 0,157 0,158 5 0,184 0,181 0,1825 6 0,213 0,211 0,212 7 0,238 0,228 0,233 8 0,279 0,253 0,266 9 0,313 0,298 0,3055 10 0,345 0,332 0,3385 Anhand dieser Werte (Tab. 22) wurde nun die graphische Auftragung in Form einer Eichgerade erzeugt: 30 Abbildung 18: Bradford - Eichgerade erstellt mit Ovalbumin Diese Gerade (Abb. 18) wurde anschließend so gelegt, dass sie den Nullpunkt schneidet: Abbildung 19: Bradford - Eichgerade erstellt mit Ovalbumin, Schnittpunkt der Geraden im Nullpunkt Im Anschluss wurde eine Ovalbumin-Lösung unbekannter Konzentration dem Bradford-Assay in verschiedenen Verdünnungen unterzogen. Die Messung erfolge in Doppelbestimmung. 31 Tabelle 23: Bradford - Extinktion der Ovalbumin-Lösung unbekannter Konzentration in Doppelbestimmung bei 595 nm Verdünnung 1:200 1:400 1:1000 1:10000 E1 0,296 0,194 0,099 0,025 E2 0,288 0,190 0,094 0,022 Mittelwert 0,292 0,192 0,0965 0,0235 Die Konzentration der unbekannten Ovalbumin-Lösung wurde anschließend in oben beschriebener Weise ermittelt, und mit den handschriftlich abgelesenen Werten verglichen. Tabelle 24: Bradford - Konzentration der unbekannten Ovalbumin-Probe im Mittel anhand verschiedener Eichgeraden Verdünnung handschriftliche Eichgerade Eichgerade (Abb. 18) Eichgerade Schnittpunkt 0 (Abb. 19) 1:200 16,5 18,22 17,31 1:400 21,4 23,94 22,125 1:1000 27 30 25,47 1:10000 55 72 26,56 Mittelwert 29,975 mg/ml 36,04 mg/ml 22,87 mg/ml Die unbekannte Ovalbumin-Probe hat damit im Mittel eine Ovalbumin-Konzentration von 36,04 mg/ml für die Eichgerade (Abb. 18), 22,87 mg/ml für die Eichgerade, die den Nullpunkt schneidet (Abb. 19) bzw. 29,975 mg/ml für die handschriftliche Eichgerade. 32 Diskussion Ziel dieses Versuches war es, verschiedene Proteinbestimmungsmethoden kennen zu lernen, und in ihrer Genauigkeit zu vergleichen. Um letzteres bewerkstelligen zu können, muss jedoch die gesuchte Proteinkonzentration der unbekannten BSA- bzw. Ovalbumin-Lösung gegeben sein, diese Betrug: Zunächst werden die vier verschiedenen Assays anhand der mit verschiedenen Eichgeraden ermittelten Werte verglichen: Handschriftliche Eichgerade (Siehe Anhang) Tabelle 25: Mittelwerte der Proteinkonzentration, Abweichung in mg/ml bzw. prozentuale Abweichung der mit der handschriftlichen Eichgerade erstellten Werte Assay Protein Konzentration [mg/ml] Abweichung [mg/ml] prozentuale Abweichung Biuret BSA 24,0625 4,5625 23,4 % Ovalbumin 26,41 1,59 5,679 % BSA 22,733 3,23 16,58 % Ovalbumin 34,8 6,8 24,29 % BCA prozentuale Abweichung d. Assays 14,5395 % 20,435 % 33 Lowry Bradford BSA 20,3125 0,8125 4,166 % Ovalbumin 24,1875 3,8125 13,616 % BSA 15,85 3,65 18,72 % Ovalbumin 29,975 1,975 7,054 % durchschnittliche Abweichung der mit der 3,304 handschriftlichen Eichgerade berechneten Werte 8,891 % 12,887 % 14,188 % So betrachtet zeigte bei der handschriftlich erstellten Eichgeraden das Lowry-Assay das beste Ergebnis, mit einer durchschnittlichen prozentualen Abweichung der realen Ergebnisse von 8,891 % (Tab. 25). Werden die reinen Proteinkonzentrationen betrachtet, so zeigt das Lowry-Assay bei der unbekannten BSA-Probe das beste Ergebnis mit 20,3125 mg/ml (real: 19,5 mg/ml) und nur einer 4,2 % Abweichung. Bei der unbekannten Ovalbumin-Probe zeigte das Bradford-Assay mit 29,975 mg/ml und einer 7,1 %igen Abweichung das beste Ergebnis (real: 28 mg/ml). Eichgerade ohne Schnittpunkt im Nullpunkt Tabelle 26: Mittelwerte der Proteinkonzentration, Abweichung in mg/ml bzw. prozentuale Abweichung der mit der Eichgerade erstellten Werte Assay Protein Konzentration [mg/ml] Abweichung [mg/ml] prozentuale Abweichung Biuret BSA 24,546 5,046 25,88 % Ovalbumin 28,461 0,461 1,65 % BSA 26,3956 6,8956 35,362 % BCA prozentuale Abweichung d. Assays 13,765 % 34 Lowry Bradford Ovalbumin 40,066 12,066 43,093 % BSA 21,809 2,309 11,84 % Ovalbumin 24,341 3,659 13,07 % BSA 20,20565 0,70565 3,619% Ovalbumin 36,04 8,04 28,71 % durchschnittliche Abweichung der mit der 4,898 Eichgerade berechneten Werte 39,2275 % 12,455 % 16,17 % 20,403 % Bei der Betrachtung der Werte, die mit der Eichgeraden ohne Nullschnittpunkt erzeugt wurden (Tab. 26) ergibt sich ein ähnliches Bild, wie bei der handschriftlichen Eichgeraden. Auch hier schneidet das Lowry-Assay mit einer prozentualen Abweichung von 12,5 % der realen Ergebnisse am besten ab. Werden wieder die reinen Proteinkonzentrationen betrachtet, so zeigt jedoch bei der unbekannten BSAProbe das Bradford-Assay das beste Ergebnis mit 20,21 mg/ml (real: 19,5 mg/ml) und damit einer prozentualen Abweichung von 3,6 %. Bei der unbekannten Ovalbumin-Probe zeigt das Biuret-Assay das beste Ergebnis mit 28,461 mg/ml (real: 28 mg/ml) und damit einer prozentualen Abweichung von 1,65 %, das zweit beste Ergebnis überhaupt. Eichgerade mit Schnittpunkt im Nullpunkt Tabelle 27: Mittelwerte der Proteinkonzentration, Abweichung in mg/ml bzw. prozentuale Abweichung der mit der Eichgerade, die den Nullpunkt schneidet, erstellten Werte Assay Protein Konzentration [mg/ml] Abweichung [mg/ml] prozentuale Abweichung Biuret BSA 22,66 3,16 16,21 prozentuale Abweichung d. Assays 35 Ovalbumin 24,5 3,5 12,5 BSA 24,13 4,63 23,74 Ovalbumin 36,71 8,71 31,11 BSA 19,94 0,44 2,26 Ovalbumin 15,88 12,12 43,29 BSA 16,15 3,35 17,18 Ovalbumin 22,87 5,13 18,32 durchschnittliche Abweichung der mit der 5,13 Eichgerade - Schnittpunkt Nullpunkt berechneten Werte 20,58 BCA Lowry Bradford 14,35 27,425 22,78 17,75 Bei der Betrachtung der Werte, die mit der Eichgeraden erstellt worden sind, die den Nullpunkt schneidet, ist das Biuret-Assay das am besten abschneidende Proteinbestimmungsverfahren mit 14,36 % Abweichung der Werte von den realen Ergebnissen. Auch bei der unbekannten Ovalbumin-Probe zeigt das Biuret-Assay mit 24,5 mg/ml (real: 28 mg/ml) und einer Abweichung von 12,5 % das zutreffendste Ergebnis. Bei der unbekannten BSA-Probe zeigt das Lowry-Assay eindeutig das beste Ergebnis mit 19,94 mg/ml (real: 19,5 mg/ml) und einer Abweichung von 2,26 %. Alles in allem weisen alle Assay ganz zutreffende Ergebnisse vor mit totalen prozentualen Abweichungen um die 12-20 %. Diese Abweichungen rühren wahrscheinlich von Pipetierfehlern her, und liegen nicht im Assay selbst begründet. Die Vorteile des Biuret-Assay liegen ihn seiner guten Reproduzierbarkeit sowie seiner Unempfindlichkeit, weshalb er für eine zügige Proteinbestimmung im Labor eingesetzt wird (sog. Mikrobiuret). Seine Nachweisgrenze liegt jedoch nur bei 1-10 µg Protein/ml, was ihn nur für größere Proteinmengen empfiehlt. 36 Das BCA-Assay zeichnet sich durch seine einfache Durchführung und die Stabilität des Farbkomplexes aus. Auch die Nachweisgrenze mit 0,1-1 µg Protein/ml macht ihn zu einer geeigneten Proteinbestimmungsmethode. Einzig die schlechte Reproduzierbarkeit ist als Nachteil zu nennen. Das Lowry-Assay weist, im Vergleich zum Biuret- bzw. BCA-Assay, eine zusätzliche Farbreaktion auf, was seine Sensitivität erhöht. Seine Nachweisgrenze liegt bei 0,1-1 µg Protein/ml, was ihn auch für geringere Proteinmengen qualifiziert. Die Nachteile des Lowry-Assay wiederum sind die Instabilität des Farbkomplexes sowie die Störung durch etliche, im Labor gängige Substanzen, darunter EDTA, SDS, Triton-X, Tris, Ammoniumsulfat usw. Das Bradford-Assay zeichnet sich durch seine leichte Durchführbarkeit, sowie seine Toleranz gegenüber Reduktionsmittel aus. Es besitzt die kleinste Nachweisgrenze mit 0,05-0,5 µg Protein/ml und eignet sich daher auch für sehr kleine Proteinmengen. Als Nachteil wäre die schlechte Reproduzierbarkeit zu nennen, die sehr stark vom Inkubationszeitraum abhängt. Als weiteres Unterscheidungsmerkmal des Bradford-Assays zu den anderen Assays wäre zu nennen, dass der Farbstoff hier, im Gegensatz zum Biuret-, BCA- oder Lowry-Assay nicht an die Peptidbindungen eines Proteins komplexiert, sondern sich an kationische sowie unpolare, hydrophobe Seitenketten anlagert. Dies kann als Voraber auch als Nachteil gesehen werden. Da die kolorimetrischen Proteinbestimmungsmethoden unter denaturierenden Bedingungen stattfinden, besteht die Möglichkeit, dass eigentlich vorhandene Peptidbindung durch die Auffaltung der Proteine gespalten, und damit auch nicht durch die hier angreifenden Färbereaktionen erkannt werden, was sich als Nachteil für das Biuret-, BCA- sowie Lowry-Assay erweisen könnte. Besitzt jedoch ein Protein sehr viele kationische sowie unpolare, hydrophobe Seitenketten, so könnte dies das Bradford-Assay in seiner Genauigkeit beeinflussen. Dies ist auch der Grund, warum sich beim Bradford-Assay die Eichgerade für BSA sowie die Eichgerade für Ovalbumin so stark in ihrer Steigung unterscheiden, was im direkten Vergleich deutlich wird: 37 Abbildung 20: Bradford - Vergleich der Eichgerade von BSA und Ovalbumin, Absorption bei 595 nm Vor allem die positiv geladene Aminosäure Arginin sorgt hier für die unterschiedlichen Wechselwirkungen. Ovalbumin besitzt 15, BSA jedoch 26 dieser Aminosäure-Reste pro Polypeptid. Die Wechselwirkungen des Coomassie- Farbkomplexes mit Arginin sind also im Rinderserum Albumin fast zweimal so stark vertreten wie im Ovalbumin. Dies sorgt für die stärkere Steigung der BSA-Eichgerade im Verhältnis zur Ovalbumin-Eichgerade und zeigt auf diesem Wege, dass es nicht ganz unwichtig ist, welches Protein zur Erstellung der Eichgerade eingesetzt wird. Welche Proteinbestimmungsmethode im Endeffekt eingesetzt wird, hängt schlussendlich von der Durchführbarkeit sowie der gewünschten Genauigkeit ab. Eindeutige Präferenzen gibt dieser Versuch nicht wieder. 38 II. Versuch: Reinigung von Fusionsproteinen Einleitung Bevor ein Protein eines bestimmten Organismus untersucht werden kann, geht bei intrazellulären, also cytosolischen, Proteinen zunächst der Zellaufschluss und die Proteinaufreinigung voran. Der Aufschluss von Zellen kann auf verschieden Art und Weise erfolgen, die in zwei Kategorien gegliedert werden können: chemische, enzymatische und mechanische Zellaufschlussmethoden. Zu den chemischen Zellaufschlussmethoden gehören das Versetzen und Inkubieren mit Toluol oder EDTA, was für das Auflösen der äußeren Zellmembran sorgt, und tiefer gelegene Zellwände dann für den enzymatischen Aufschluss zugänglich macht. Zellen bzw. deren Zellwände werden auf dem enzymatischen Weg mit Hilfe von Lysozym oder Zymolyase abgebaut. Auf mechanischem Wege werden Zellen mittels Ultraschall aufgeschlossen. Auch der Aufschluss mit Hilfe der French Press, einer Apparatur, die für einen plötzlichen Druckabfall sorgt, und die Zellen so zerreißen lässt, ist eine der möglichen mechanischen Zellaufschlussmethoden. Bei Hefezellen wird auch der Zellaufschluss mittels Glasperlen angewandt, hierbei wird eine Zellsuspension mit sehr kleinen Glaskügelchen versetzt, welche dann durch sehr starkes Vermischen die Zellen aufschließen. Wichtig beim Zellaufschluss ist es, einen Proteasehemmer wie Phenylmethylsulfonylfluorid (PMSF) oder Benzamidin der Zellsuspension hinzuzufügen, da ansonsten die intrazellulär enthaltenen Proteasen die Zielproteine nach dem Aufschluss irreversibel proteolytisch spalten. Das Zielprotein liegt nun in dem Gemisch aus Chemikalien, Enzymen, Zelltrümmern und Lösungsmittel vor, darum müssen als nächstes Aufreinigungsschritte erfolgen, die das gewünschte Protein von den restlichen Bestandteilen und auch anderen unerwünschten Proteinen trennen. Es gibt viele verschiedene Methoden zur Proteinaufreinigung, genannt sei hier nur die Aufreinigung mittels einer Gelmatrix. Hierbei wird die Proteinlösung auf eine Affinitäts-Gelmatrix gegeben, wobei die 39 Proteine über meist ionische Wechselwirkungen an die Partikel der Gelmatrix binden. Durch Elution mit Hilfe eines Salzgradienten werden die Proteine kompetetiv durch die Ionen des Salzes verdrängt, wobei das Eluat in Fraktionen gesammelt wird. Anschließend können diese Proteinlösungen verschiedensten Tests unterzogen werden, u.a. Aktivitätstests bei Enzymen oder Elektronenüberträgern von Enzymkomplexen. Dieser Ablauf wird häufig bei der Isolation bereits bekannter Proteine angewandt, wobei die resultierenden Eluat-Fraktionen meist keine hohe Reinheit und damit auch keine hohe Konzentration des Zielproteins vorweisen. Bei der Charakterisierung unbekannter Proteinen ist es jedoch von großer Wichtigkeit das Protein in ausreichender Reinheit und Menge vorliegen zu haben. Um eine hinreichende Menge an Protein mit hoher Reinheit zu erzielen, wird die Bildung sog. Fusionsproteine heterolog in einem Modellorganismus vorgezogen. Bei einem Fusionsprotein handelt es sich um das Zielprotein ausgestattet mit einer am N- oder C-Terminus addierten kurzen Sequenz, einem sog. ‚Tag‘. Die Anheftung des Tags, meist eines Poly-Histidin-Schwanzes mit mind. sechs Histidinen, erfolgt auf genetischem Wege, indem ein Fusionsgen2 erzeugt wird. Dieses Fusionsgen wird heterolog in einen Modellorganismus exprimiert, wobei es sich bei dem Modell meist um Escherichia coli, Hefe- oder Insektenzellen handelt. Die Expression kann hierbei ebenfalls kontrolliert werden, indem vor das Fusionsprotein der Promotor und der Operator des lac-Operons gesetzt wird. Das lac-Operon codiert unter anderem für die β-Galactosidase, einem Enzym, das in der Lage ist, glykosidische Bindungen zu spalten. Der Operator des lac-Operons wird bei Fehlen von Substrat durch den lac-Repressor blockiert, sodass der Promotor nicht durch die RNA-Polymerase angesteuert werden kann. Auf diesem Weg findet nur bei Anwesenheit von Substrat eine Genexpression statt. Ein möglicher Induktor stellt hierbei IPTG (Isopropyl-β-Dthiogalactopyranosid) dar. Dieser nicht verwertbare Induktor bindet den lacRepressor, sodass dieser nicht mit dem lac-Operator wechselwirken kann, der Promotor wird frei zugänglich und die Genexpression wird induziert. Auch die das 2 Dieses Fusionsgen ist eine Fusion aus dem das Zielprotein exprimierenden Gen und einer Sequenz aus Basenpaaren, die für die Aminosäure Histidin codieren. Aus diesem Fusionsgen wird das Zielprotein mit vorangeschalteter oder angehängtem Histidin-Tag gebildet. 40 Fusionsprotein tragende Zellen müssen oben genannten Zellaufschlussverfahren unterzogen werden. Die Aufreinigung eines Fusionsproteins kann ebenfalls über eine Affinitäts-Gelmatrix erfolgen, ist jedoch, im Gegensatz zur Aufreinigung von Proteinen ohne Tag, wesentlich vereinfacht. Zur Isolierung des gewünschten Proteins kann eine Nickelchelat-Affinitäts-Gelmatrix, oder jedes beliebige zweiwertige Ion, genutzt werden. Das Nickel-Ion (Ni2+) ist über vier Koordinationsstellen mit der Säule koordiniert. Die daraus resultierenden zwei freien Koordinationsstellen binden an den Poly-Histidin-Tag und „fangen“ so das Zielprotein aus dem Proteingemisch ab. Eluiert wird auch hier über einen Salzgradienten durch kompetetive Verdrängung des HisTags (und damit des Zielproteins) durch die Ionen des Salzes. Da ein Tag bei strukturellen Analysen störend wirkt und auch die biologische Funktion des Zielproteins beeinträchtigen könnte, ist es im letzten Schritt entscheidend den Tag vom nativen Protein zu entfernen. Um dies zu erreichen wird auf genetischem Wege neben dem Tag auch eine zusätzliche proteolytische Spaltstelle eingefügt. Durch die Zugabe einer geeigneten Protease3 kann so der Tag durch proteolytische Spaltung entfernt werden. Es gibt hier verschiedenste Kombinationsmöglichkeiten, hier findet die sog. TEV-Protease und ihre spezifische Schnittstelle, die Aminosäureabfolge ENLYFQ G/S, Anwendung. Diese Protease stammt aus dem Taback Mosaik Virus (Tobacco Etch Virus), was die Chancen erhöht, dass mögliche weitere Spaltstellen rein zufällig vorkommen, und damit spezifisch nur das Fusionsprotein vom Tag getrennt wird. Material und Methoden Siehe Skript, Änderungen: 3 Die proteolytische Spaltstelle besteht auf genetischer Ebene aus einer Sequenz, auf Proteinebene aus einer spezifischen Abfolge von Aminosäuren, an der eine jeweilige Protease spezifisch schneidet. Auf diesem Wege kann gezielt gespalten werden, wobei so auch verhindert wird, dass zelleigene Proteasen den Tag ungewollter Weise entfernen. 41 Die 50ml Medium wurden mit jeweils 500 µl der Antibiotika Ampicillin und Kanamycin sowie 500 µl Vorkultur versetzt. (S. 14) Nach Induktion der Expression wurde die Kultur für 3h bei 37°C unter ständigem Schütteln inkubiert. (S. 14) Auf das SDS-Gel wurden nur 14,4 µl der t0 (=P1) sowie der t3 (=P2) Probe aufgeladen, da von der t1 (=P2) Probe zu wenig Material entnommen wurde (444 µl statt 616 µl) Bei der TEV-Protease Kinetik wurden 6 µl Dialysat eingesetzt. (S. 21) Ergebnisse Über Nacht wurden Escherichia coli Transformanten bei 37°C unter ständigem Schütteln mit einem Expressionsplasmid inkubiert. Dieses Plasmid trug das Gen für ein SpaI Fusionsprotein mit einem His-Tag am N-Terminus, welches über eine TEV Spaltstelle fusioniert war. Gleichzeitig wies dieses Plasmid zwei AntibiotikaResistenz-Kassetten auf, zum einen die Resistenz gegen Ampizilin und die gegen Kanamycin um eine gezielte Selektion der transformierten Zellen zu ermöglichen. Am nächsten Morgen wurden 500 µl der Vorkultur in 50 ml frisches Medium überimpft und um mögliches Wachstum von nicht-Transformanten zu vermeiden mit Antibiotika versetzt. Anschließend wurde die erste Probe (= t0 bzw. P1) entnommen und das Zellsediment nach erfolgter Zentrifugation mit SDS-Probenpuffer versetzt und eingefroren. Anschließend wurde durch Zugabe von 1mM IPTG die Expression des SpaI-Gens induziert, wobei nach einer (= t1 bzw. P2) bzw. drei (= t3 bzw. P3) weiteren Stunden Proben entnommen wurden. Nach der letzten Probeentnahme (= t 3) wurde die verbleibende Kultur zentrifugiert und das Zellsediment in 1 ml Lysis-Puffer überführt. Die Zellen wurden anschließend aufgeschlossen, wobei nach erfolgter Zentrifugation vom Lysat (= P4) bzw. vom Sediment (= P5) die nächsten Proben entnommen wurden. Der restliche Überstand des Zellextraktes wurde anschließend über eine Nickel-Affinitäts-Gelmatrix gegeben, wobei vom Durchfluss (= P6), von der ersten Waschfraktion (= P7), sowie der ersten (= P8) und zweiten (= P9) Elutionsfraktion Proben entnommen wurden. Die erste und zweite Elutionsfraktion wurden nach der Probeentnahme vereinigt und über Nacht bei 4°C in Dialyse-Puffer dialysiert. Von diesem Dialysat wurde am nächsten Morgen ebenfalls eine Probe 42 entnommen (= P10). Alle Proben wurden anschließend fünf Minuten bei 96°C denaturiert, schnell auf Eis abgekühlt und auf ein SDS-Gel geladen um den Reinigungsverlauf bzw. -erfolg sichtbar zu machen. Abbildung 21: Verlauf der Reinigung des Fusionsproteins spaI, Legende: M = Marker, P = Probe Das Gelbild (Abb. 21) zeigt den Verlauf und Erfolg der Reinigung des Fusionsproteins SpaI. Das Gel wurde von links nach rechts wie folgt beladen: Marker, t0-Probe, t1-Probe, t3-Probe, Lysat, Sediment, Durchfluss, 1.Waschfraktion, 1.Elutionsfraktion, 2.Elutionsfraktion, Dialysat. Zu erkennen ist, dass nach jedem Reinigungsschritt Banden verschwinden und nur eine Bande zwischen 20 und 15 kDa erhalten bleibt. Das verbleibende Dialysat wurde nach erfolgter Probeentnahme einer Proteinbestimmung nach Bradford unterzogen. Hierzu wurde zunächst mit Hilfe von Rinderserum Albumin in Doppelbestimmung eine Eichgerade erstellt, anhand derer im Anschluss daran die Proteinkonzentration des Dialysats berechnet wurde. Tabelle 28: Bradford - Doppelbestimmung der Extinktion verschiedener BSA-Konzentrationen bei 595 nm zur Erstellung einer Eichgeraden µg BSA E1 E2 E 0 0 0 0 43 1 0 0,028 0,014 2 0,092 0,118 0,105 5 0,248 0,297 0,2725 10 0,379 0,478 0,4285 20 0,833 0,821 0,827 Anhand dieser Werte (Tab. 28) wurde anschließend graphisch die Eichgerade wiedergegeben. Abbildung 22: Bradford Eichgerade erstellt mit Rinderserum Albumin Anhand der Geradengleichung kann nun die Proteinkonzentration berechnet werden, diese lautet wie folgt: 44 Umgeformt ergibt sie sich zu: Der Term y stellt hierbei die Extinktion des Dialysates dar, diese wurde ebenfalls in Doppelbestimmung anhand zweier Konzentrationen ermittelt: Tabelle 29: Extinktion des Dialysates Probe 2 µl 5 µl E1 0,134 0,302 E2 0,134 0,301 E 0,134 0,3015 Die Berechnung der Proteinkonzentration geschah dabei exemplarisch an der 2 µl Probe wie folgt: Da in den Test jedoch nur 100 µl eingingen, muss dieser Wert auf 1 ml hochgerechnet werden: Es wurden jedoch nur 2 µl in den Test eingesetzt, was einer 1:50 Verdünnung entspricht, die anschließend noch herausgerechnet werden muss: 45 Da die Eichgerade jedoch im Maßstab µg angesetzt wurde, muss dieser Wert nun noch in mg überführt werden: Die Proteinkonzentration der 2 µl Probe entspricht also 1,595 mg/ml. Die Berechnung für die 5 µl Probe erfolgt in gleicher Weise, nur, dass es sich hierbei um eine 1:20 Verdünnung handelt; ihre Proteinkonzentration entspricht 1,436 mg/ml. Damit hat das Dialysat im Mittel eine Proteinkonzentration von 1,5155 mg/ml. Die Bestimmung der Proteinkonzentration kann weiterhin über die Ermittlung der Extinktion bei 280 nm erfolgen. Hierzu wurde die Extinktion bei 280 nm einer 1:100 Verdünnung des Dialysates ermittelt. Diese betrug 0,032, wobei das Dialysat unverdünnt so auf eine Extinktion von 3,2 kommt. Mit Hilfe des Lambert-Beer‘schenGesetzes kann so die Konzentration berechnet werden, hierzu muss nur der Extinktionskoeffizient (hier: 17420 1/(cm∙M)) sowie das Molekulargewicht (hier: 18886,8 g/mol) bekannt sein. Das Lambert-Beer’sche-Gesetz lautet: umgeformt: Um nun auf eine Proteinkonzentration in mg/ml zu kommen, muss dieses Ergebnis mit dem Molekulargewicht multipliziert werden: Auf diesem Weg hat das Dialysat eine berechnete Proteinkonzentration von 3,47 mg/ml. 46 Im zweiten Teil des Versuches wurde die Entfernung des His-Tags des Fusionsproteins durch die TEV-Protease in Form einer Kinetik untersucht. Hierzu wurden 3 verschiedene Ansätze hergestellt, Ansatz 1 ohne TEV-Protease, Ansatz 213 mit dem Fusionsprotein und der TEV-Protease, sowie Ansatz 14 ohne Fusionsprotein. Diese Ansätze wurden bei Raumtemperatur über Nacht inkubiert. Ansatz 1 und 14 fungierten hierbei als Kontrolle. Aus dem Ansatz 2-13 wurden in folgenden Zeitabständen Proben entnommen: 10, 20, 30, 40, 50 Minuten, sowie 1, 2, 3, 4, 5, 6 Stunden, und am nächsten Morgen, diese wurden fortlaufend durchnummeriert. Die Proben 1-14 enthielten damit folgende Kombination (Legende: FP = Fusionsprotein mit Tag, TEVP = TEV-Protease): Tabelle 30: Zusammensetzung der Proben 1-14 Probe 1 Inhalt FP 2 Entnahme ü.N. 10‘ 3 4 5 6 7 8 9 10 11 12 FP + TEVP 20‘ 30‘ 40‘ 50‘ 1h 2h 13 14 TEVP 3h 4h 5h 6h ü.N. ü.N. Die Proben (Tab. 30) wurden anschließend auf ein Tris-Tricin-Gel aufgetragen, um den Verlauf der proteolytischen Spaltung, die sog. TEV-Kinetik, optisch sichtbar zu machen. 47 Abbildung 23: Tris-Tricin-Gel der TEV-Kinetik Proben 1-14, M = Marker Das Tris-Tricin-Gelbild (Abb. 23) zeigt den Verlauf der proteolytischen Spaltung des Fusionsproteins. Als oberste Bande, zwischen 30 und 25 kDa, ist die TEV-Protease zu erkennen, die zweite Bande bei ca. 20 kDa zeigt das Fusionsprotein, die dritte Bande zwischen 20 und 15 kDa markiert das SpaI-Protein ohne Tag. Zu erkennen ist die Abnahme des Fusionsproteins und die stätige Zunahme des SpaI-Proteins ohne Tag. Die Probe 1 als erste Kontrolle sollte eigentlich das SpaI-Protein ohne TEVProtease zeigen, was offensichtlich nicht der Fall ist. Auch Probe 1 zeigt die oberste Bande, die TEV-Protease mit gespaltenem Fusionsprotein SpaI. Die Probe 14 als zweite Kontrolle sollte die TEV-Protease allein zeigen, die jedoch augenscheinlich nicht zu erkennen ist. Diskussion Um ein Protein z.B. zu charakterisierungszwecken in ausreichender Menge und Reinheit zu isolieren, wird die Methode der Erzeugung von Fusionsproteinen mit gleichzeitiger Überexpression in einem Modellorganismus angewandt. Ein 48 Fusionsprotein wird erzeugt, indem auf genetischem Wege ein sog. Tag, meist ein terminaler Poly-Histidin-Schwanz, an das Zielprotein angeheftet wird. Nebst diesem Tag wird der Promotorbereich des für das Zielprotein codierenden Gens durch den Promotor und den Operator des lac-Operons ersetzt, um eine gezielte Induktion und Überexpression des Zielgens zu ermöglichen. Die Induktion bzw. Überexpression wird meist durch den nicht-verwertbaren Induktor IPTG bewerkstelligt, da dieser nicht von der Zelle verstoffwechselt werden und damit eine konstante Induktion ermöglichen kann. Den Erfolg dieser Methoden zeigt auch der Reinigungsverlauf des hier isolierten Fusionsprotein SpaI (Abb. 21), dass mit einer Größe von ca. 19 kDa zwischen der 15 und 20 kDa Bande zu erkennen ist. Bevor der Reinigungsverlauf näher begutachtet werden kann, muss zunächst überprüft werden, ob die Induktion mittels IPTG funktioniert hat. Hierzu Fallen die Proben P1-P3 in nähere Betrachtung. Wird die Bande des SpaI-Proteins bei ca. 19 kDa isoliert beurteilt, so ist eindeutig eine Zunahme der Intensität zu beobachten. Dies lässt jedoch für sich noch nicht den Schluss der Induktion bzw. Überexpression zu, sondern könnte einfach nur auf die stärkere Beladung des Gels bezüglich der Proben P2 und P3 hindeuten. Neben der Bande des Zielproteins SpaI müssen also die restlichen Banden ebenfalls in Augenschein genommen werden. Gut ist dies bei den Banden zwischen 40 und 50 kDa zu erkennen. Wo die SpaI-Bande stetig an Intensität zunimmt, bleiben die Banden zwischen 40 und 50 kDa von ihrer Intensität her konstant. Der Erfolg der Induktion konnte hiermit gezeigt werden, nun muss im nächsten Schritt der Reinigungsverlauf auf sein Gelingen hin untersucht werden. Hierzu werden die einzelnen Reinigungsschritte, beginnend bei P4 betrachtet. P4 stellt das Lysat nach Aufschluss der Zellen dar. Hierbei ist wichtig zu beachten, ob das Zielprotein noch in ausreichender Menge vorhanden ist, da das restliche Lysat für die weitere Reinigung eingesetzt wird. Sollte die das Zielprotein charakterisierende Bande stark an Intensität abnehmen, muss die Probe P5 bzw. das abzentrifugierte Sediment näher betrachtet werden. Ein Phänomen des Modellorganismus E. coli ist es, Fremdproteine in sog. Inclusion Bodies einzuschließen. Diese Einschlusskörperchen sind dann so schwer, dass sie bei der Zentrifugation sedimentieren und im Überstand des Lysates (also P4) nicht mehr anzutreffen sind, weshalb ein großer Teil des Proteins dann während der Reinigung verschwinden würde. Dies ist der Fall, wenn im Sediment (P5) die Proteinkonzentration des Zielproteins, bzw. die Intensität der das Zielprotein charakterisierenden Bande stark zunehmen würde. Im direkten 49 Vergleich der SpaI-Bande des Lysats (P4) mit der des Sediments (P5) ist zu erkennen, dass im Sediment durchaus Protein verloren gegangen ist, die Intensität der SpaI-Bande des Sediments die Intensität der SpaI-Bande des Lysats jedoch nicht übersteigt, also noch ausreichend Protein für den weiteren Reinigungsverlauf und eine anschließende Charakterisierung vorhanden ist. Im nächsten Schritt der Reinigung wurde das Lysat auf die Nickel-Affinitäts-Gelmatrix gegeben. Hier sollte der His-Tag des Zielproteins an die zweiwertigen Nickel-Ionen binden, damit dieses durch die Matrix zurückgehalten wird. Die Betrachtung des Durchflusses (P6) zeigt jedoch, dass auch hier eine nicht unwesentliche Menge des Zielproteins vorhanden ist. Das Protein hat also nicht vollständig gebunden und wurde mit einem Teil der verbleibenden, nicht-bindenden Proteine eluiert. Es gibt in erster Linie zwei Gründe, weshalb ein Teil des Zielproteins nicht an die Säule gebunden hat: Zum einen könnte die Kapazität der Gelmatrix überschritten worden sein, zum anderen könnte der spontane Abbau des His-Tags für das Durchfließen des Zielproteins verantwortlich sein. Eine bestimmte Menge der Nickel-Affinitäts-Gelmatrix weist eine ebenso definierte Anzahl an Koordinationsstellen auf, an die z.B. der His-Tag eines Fusionsproteins binden kann. Wird die Anzahl der Koordinationsstellen der Matrix durch die Anzahl der binde-fähigen His-Tags überschritten, so bezeichnet man dies als Überschreitung der Kapazität der Matrix. Die Matrix wäre nicht in der Lage mehr His-Tags und damit mehr Protein binden zu können, und das überschüssige Protein passiert die Matrix ohne an ihr haften zu bleiben. Der nächste zu betrachtende Reinigungsschritt war das Waschen der Gelmatrix um unspezifisch gebundene Proteine zu entfernen (P7). Durch das Erhöhen der Imidazol-Konzentration im Wasch-Puffer sollen unspezifisch gebundene Proteine kompetitiv von den Koordinationsstellen der Matrix verdrängt werden. Wie gut an den verschiedenen Banden der Probe der ersten Waschfraktion (P7) zu erkennen ist, ist dies auch der Fall. Dennoch wird auch ein Teil des Zielproteins während des Waschens von der Matrix verdrängt, sodass auch hier kleinere Verluste auftreten. Die Eluat-Fraktionen (P8 und P9) zeigen dann den schlussendlichen Reinigungserfolg. Hier ist deutlich zu erkennen, dass sich kein weiteres, oder höchsten sehr kleine Mengen an FremdProtein in den Eluaten befinden. Das Zielprotein SpaI konnte erfolgreich isoliert und aufgereinigt werden. Die Probe des Dialysats (P10) zeigt das Eluat nach erfolgter Dialyse über Nacht. Durch das Dialysieren wird der Salzgehalt im das Zielprotein umgebenden Medium verringert, was jedoch zu einer Verdünnung des Zielproteins 50 führt. Die Tris-Tricin-Gel-Bande des Dialysats (P10) zeigt jedoch, dass noch eine ausreichende Menge an Fusionsprotein vorhanden ist, um damit weiterarbeiten zu können. Um für die TEV-Kinetik eine ausreichende Menge an Fusionsprotein einsetzen zu können, wurde die Proteinkonzentration des Dialysats berechnet. Diese Betrug ca. 1,52 mg Fusionsprotein/ml bei der Proteinbestimmung nach Bradford und 3,47 mg Fusionsprotein/ml bei der Bestimmung der Extinktion bei 280 nm. Dieser doch sehr starke Unterschied kommt zustande, weil bei der Ermittlung der Extinktion eine leichte Überbestimmung stattfindet, da bei 280 nm alle Proteine absorbieren und wahrscheinlich noch mit leichten Verunreinigungen zu rechnen ist. Die Proteinkonzentration von 1,52 mg/ml ist damit eher anzunehmen. Die dargestellte TEV-Kinetik zeigt sehr anschaulich die im Reaktionsgemisch ablaufende proteolytische Spaltung des Fusionsproteins bei Raumtemperatur (Abb. 23). Die erste Spur (Probe 2) zeigt die TEV-Protease (oberste Bande bei 28,75 kDa), das Fusionsprotein SpaI mit His-Tag (zweite Bande bei ca. 19 kDa) sowie das SpaIProtein mit bereits abgespaltenem His-Tag (dritte Bande bei 18,89 kDa) nach 10 Minuten Reaktionsverlauf. Im weiteren zeitlichen Ablauf nimmt das Fusionsprotein (zweite Bande von oben) fortwährend ab, und das SpaI-Protein ohne His-Tag stetig zu (dritte Bande), bis nach ca. einer Stunde (Probe 7) kaum noch etwas an ungespaltenem SpaI mit His-Tag vorhanden ist. Zwei Stunden nach Start der Reaktion ist kein ungespaltenes Fusionsprotein mehr vorhanden, und auch die Intensität der Bande, die das gespaltene SpaI-Protein ohne His-Tag charakterisiert, nimmt nicht weiter zu. Es liegt also nahe, dass nach ca. 1-2 Stunden die Reaktion bereits vollständig abgelaufen ist. Die Probe 13 stellt die Kontrolle der Stabilität der TEV-Protease dar, weshalb sie nach Inkubation über Nacht entnommen wurde. Die oberste Bande bei ca. 28 kDa ist nun nur noch sehr fein zu erkennen, was auf den Abbau der TEV-Protease unter Raumtemperaturbedingungen hindeutet. Da die für die TEV-Protease charakteristischen Banden der Proben 11 und 12 nach fünf bzw. sechs Stunden Inkubation bereits sehr an Intensität verloren haben, wird der Abbau hier schon begonnen haben. Die Stabilität der TEV-Protease ist also bei Raumtemperatur nach spätestens 6 Stunden nicht mehr gegeben. Die Proben 1 und 14 sollten eigentlich Kontrollen für die TEV-Kinetik darstellen, darum sollte Probe 1 ohne TEV-Protease und Probe 14 ohne Fusionsprotein über Nacht inkubiert werden. 51 Das Tris-Tricin-Gelbild (Abb. 23) weist jedoch zum einen das Fehlen der TEVProtease in Reaktionsansatz 14, als auch das Vorhandensein der TEV-Protease in Reaktionsansatz 1 auf. Dieses Erscheinungsbild lässt sich auf einen Pipetierfehler hin zurückführen. Die Spur der Probe 1 könnte doppelt beladen worden sein, also sowohl mit Probe 1, als auch mit Probe 14, was für die Intensität der Bande der TEVProtease sprechen würde. Warscheinlicher aber wurde die TEV-Protease anstatt in Reaktionsansatz 14 in Reaktionsansatz 1 gegeben, was für den Ablauf der Reaktion spricht, da das Gel ja unter denaturierenden Bedingungen gestartet wurde. Einwandfreie Kontrollen hätten eher wie auf diesem Gelbild aussehen sollen: Abbildung 24: TEV-Kinetik bei Raumtemperatur Die hier (Abb. 24) dargestellte Kinetik fand ebenfalls bei Raumtemperatur statt. Die Kontrolle 1 zeigt deutlich nur das Fusionsprotein. Die darunterliegende Bande, die charakteristisch für das gespaltene SpaI ist, rührt von spontaner Abspaltung des HisTags her. Kontrolle 14 zeigt deutlich die TEV-Protease ohne Zugabe des Fusionsproteins. Die Spaltung des Fusionsproteins zum SpaI-Protein ohne His-Tag dauerte bei dieser Kinetik etwas länger als bei der zuvor dargestellten (Abb. 23). Hier (Abb. 24) war nach ca. 5-6 Stunden die Reaktion durch vollständige Spaltung des Fusionsproteins beendet. 52 Deutlich länger dauert die Spaltung des Fusionsproteins unter Bedingungen bei 4°C, wie hier dargestellt: Abbildung 25: TEV-Kinetik bei 4°C im Kühlraum Hier (Abb. 25) ist deutlich zu erkennen, dass die Spaltung des Fusionsproteins selbst nach Inkubation über Nacht (Probe 13) noch nicht vollkommen beendet ist. Die Abnahme des Fusionsproteins bzw. die Zunahme des SpaI-Proteins ohne His-Tag vollzieht sich zwar auch stetig, jedoch deutlich langsamer als bei Inkubation bei Raumtemperatur. Dieses Phänomen, dass bei höheren Temperaturen eine stärkere Umsetzung des Ausgangsstoffes (in diesem Fall dem Fusionsprotein) beobachtet werden kann, kann mit der sog. RGT-Regel (Reaktionsgeschwindigkeits-Temperatur-Regel) erklärt werden. Die RGT-Regel besagt, dass sich die Reaktionsgeschwindigkeit einer enzymatischen Reaktion verdoppelt bei Erhöhung der Temperatur um 10°C. Diese Regel lässt sich leicht anhand der hier erstellten Kinetik nachvollziehen. Bei der Kinetik bei 4°C (Abb. 25) ist das Fusionsprotein nach 24 Stunden (Probe 13) beinah vollständig umgesetzt. Die vollständige Umsetzung des Fusionsproteins bei Raumtemperatur erfolgte bei der zugehörigen Kinetik (Abb. 24) nach ca. 5-6 Stunden. Da die Temperatur von 4°C auf Raumtemperatur um 20°C erhöht wurde, müsste sich die Reaktionsgeschwindigkeit ungefähr vervierfachen, was sie demnach auch tut. Einzig problematisch an diesem Vorgang ist, dass die Stabilität eines 53 Enzyms bei höherer Temperatur eher nachlässt als bei tieferen, wobei letzteres zu kosten der Reaktionsgeschwindigkeit gehen würde. 54 III. Versuch: Metabolitbestimmung Einleitung Vitale Zellen betreiben Stoffwechselvorgänge, wobei bestimmte Substrate verbraucht, und daraus synthetisierte Endprodukte abgegeben werden. Bei der Metabolitbestimmung wird die Abnahme eines Substrates verfolgt und in Korrelation zur Zunahme eines daraus resultierenden Produktes gesetzt. In diesem Versuch wird der Stoffwechsel der Bäckerhefe Saccharomyces cerevisiae untersucht. Dieser weißt eine Besonderheit auf: Ist S. cerevisiae hohen Glucose-Konzentrationen ausgesetzt, wird der respirative Stoffwechsel reprimiert und die Bäckerhefe zeigt auch unter aeroben Bedingungen Gärungsvorgänge. Dieses Phänomen wird als Crabtree-Effekt bezeichnet. Hierbei wird ein Molekül Glucose zunächst zu zwei Molekülen Pyruvat umgesetzt, welches anschließend fast vollständig zu zwei Molekülen Ethanol fermentiert wird. Dieses Phänomen geht mit der Repression aller Enzyme einher, die für die Respiration und die Gluconeogenese benötigt werden und wird als GlucoseRepression bezeichnet. Ziel ist es, den Verbrauch von Glucose und die Bildung von Ethanol zeitlich während der Kultivierung zu bestimmen, dieses geschieht mit Hilfe der sog. Endwertmethode. Hierbei werden zu verschiedenen Zeitpunkten während des Wachstums Proben entnommen und diese auf zwei Aliquots aufgeteilt. Eine Testreihe wird auf Glucose-Abnahme, die andere auf Ethanol-Zunahme hin untersucht. Es wird jeweils eine enzymatische Reaktion gestartet, die entweder Glucose oder Ethanol als Ausgangssubstrat benötigt und ein Produkt erzeugt, dessen Zunahme photometrisch quantifiziert werden kann. Die Extinktion wird dabei vor und nach der Zugabe des Enzyms gemessen um eine Extinktionsänderung zu verfolgen, wobei anhand dessen die jeweilige Konzentration an Glucose oder Ethanol mit Hilfe des Lambert-Beer’schen Gesetzes (a) berechnet werden kann. (a) 55 Material und Methoden Siehe Skript, Änderungen: Ein 1L Erlenmeyer-Kolben mit 100 ml SCD-Medium wurde mit 200 µl stationärer Vorkultur angeimpft. (S. 24) Ergebnisse Zunächst wurde ein 1L Erlenmeyer-Kolben mit 100 ml SCD-Medium mit 200 µl stationärer Vorkultur des S. cerevisiae-Stammes CEN.PK122 steril beimpft. Nach Inkubation über Nacht bei 30°C auf dem Schüttler wurde die optische Dichte bei 600 nm bestimmt, diese betrug OD600 = 4,8. Die benötigte Zellmenge, damit 20ml frisches SCD-Medium nach beimpfen eine OD600 von ungefähr 3 hat war 12,5ml. Diese Zellmenge wurde von der Vorkultur abgenommen, zentrifugiert, gewaschen und anschließend in das neue Medium überführt. Anschließend wurde direkt die t 0-Probe entnommen und der Kolben schüttelnd bei 30°C im Wasserbad inkubiert. Nun wurde alle 30 Minuten eine weitere Probe entnommen bis zur finalen Probe nach 5 Stunden Inkubation. Während der Probeentnahme wurde die optische Dichte der Kultur verfolgt, um diese in Korrelation zum Glucose-Verbrauch bzw. zur Ethanol-Zunahme zu setzen. So konnte zunächst eine Wachstumskurve des S. cerevisiae-Stammes CEN.PK122 erstellt werden: Tabelle 31: Bestimmung der OD600 des Saccharomyces cerevisiae-Stammes CEN.PK122 über einen Zeitraum von 5 Stunden bei 30°C auf dem Schüttelwasserbad Probe [t = ] Verdünnung OD600 1 OD600 2 OD600 0 1:100 0,039 0,039 3,9 56 0,5 1:100 0,037 0,036 3,65 1 1:100 0,049 0,049 4,9 1,5 1:100 0,056 0,056 5,6 2 1:100 0,071 0,073 7,2 2,5 1:100 0,075 0,081 7,8 3 1:100 0,096 0,077 8,65 3,5 1:100 0,09 0,087 8,85 4 1:100 0,085 0,082 8,35 4,5 1:100 0,082 0,074 7,8 5 1:100 0,085 0,076 8,1 Die Wachstumskurve sieht hierbei wie folgt aus: 57 Abbildung 26: Wachstumskurve des Saccharomyces cerevisiae-Stammes CEN.PK122 über einen Zeitraum von 5 Stunden bei 30°C auf dem Schüttelwasserbad Zur Bestimmung der Glucose- bzw. Ethanol-Konzentrationen während der verschiedenen Zeitpunkte wurden die Proben zunächst in Einfachbestimmung gemessen, um anhand dieser Werte geeignete Verdünnung abzuschätzen. Die Bestimmung der Glucose-Konzentration erfolgte mit Hilfe einer chemischen Reaktion, die auf der Umsetzung von Glucose durch das Enzym Glucose-Oxidase zu Glukonsäure und Wasserstoffperoxid beruht. Das entstehende Wasserstoffperoxid wird anschließend durch das Enzym Peroxidase zu Wasser umgesetzt. Bei diesem letzten Reaktionsschritt wird das farblose ABTS in die grüne, oxidierte Form überführt, deren Absorption bei 420nm gemessen wird. Ist also Glucose in der Probe vorhanden, wird dies durch einen grünen Farbumschlag sichtbar. Für die Konzentration der Glucose im Medium konnte folgendes berechnet werden: Tabelle 32: Glucose-Konzentration des Mediums im Verlauf von 5 Stunden GlucoseKonzentration Probe [t = ] Verdünnung E1 E2 E [mg/ml] [mM] 58 0 1:100 0,654 0,67 0,662 7,71 42,83 0,5 1:100 0,528 0,533 0,5305 6,18 34,33 1 1:100 0,381 0,416 0,3985 4,64 25,78 1,5 1:100 0,31 0,333 0,3215 3,74 20,8 2 1:10 1,559 1,582 1,5705 1,83 10,16 2,5 1:10 0,523 0,527 0,525 0,61 3,4 3 1:10 0,073 0,075 0,074 0,09 0,48 3,5 1:10 0,017 0,017 0,017 0,02 0,11 4 1:10 0,017 0,018 0,0175 0,02 0,11 4,5 1:10 0,018 0,018 0,018 0,02 0,12 5 1:10 0,018 0,018 0,018 0,02 0,12 Standard 1:100 0,789 0,774 0,7815 9,1 50,56 SCDMedium 1:100 (0,103) 0,748 0,4255 8,71 48,39 Die Berechnung der Glucose-Konzentration erfolgte dabei, exemplarisch gezeigt an der Probe t0, wie folgt: EProbe = Mittelwert der Extinktion der Probe (bei t0 = 0,662) F = Verdünnungsfaktor (bei t0 = 100) 59 EStandard = Mittelwert der Extinktion des mit-getesteten Standards (hier: 0,7815) Für t0 sähe die Berechnung also wie folgt aus: Die Glucose-Konzentration in mM wird anschließend wie folgt berechnet: Für t0 würde die Berechnung der Glucose-Konzentration in mM wie folgt aussehen: Die Betrachtung der Entwicklung der Glucose-Konzentration zeigt, dass die GlucoseKonzentration im Medium von der Probe t0 zur Probe t3 stetig abnimmt, bis sie bei t3,5 vollständig verbraucht ist und nicht weiter sinkt bis t5. Die Ethanol-Konzentration wird auf einem ähnlichen Weg nachgewiesen, hierbei wird Ethanol durch das Enzym Alkoholdehydrogenase zu Acetaldehyd überführt. Dies geschieht unter der Umwandlung eines Reduktionsäquivalents NAD+ zu NADH+H+. Die NADH-Bildung wird bei 366 nm gemessen. Ist also Ethanol im Medium vorhanden nimmt die durch die Reaktion gebildete Menge an NADH zu. Die EthanolKonzentration wird mit Hilfe der Extinktion vor und nach Start der Reaktion ermittelt. Für die Ethanol-Konzentration im Medium konnte folgendes berechnet werden: Tabelle 33: Ethanol-Konzentration des Mediums im Verlauf von 5 Stunden Probe EthanolKonzentration 60 [t = ] Verd. E1.1 E1.2 E1 E2.1 E2.2 E2 0 1:5 0,034 0,032 0,033 0,179 0,178 0,1785 0,104 2,249 0,5 1:50 0,039 0,036 0,0375 0,142 0,138 0,14 15,84 1 1:50 0,041 0,040 0,0405 0,223 0,222 0,2225 1,3 28,13 1,5 1:50 0,064 0,038 0,051 0,316 0,311 0,3135 1,87 40,57 2 1:100 0,038 0,034 0,036 0,239 0,243 0,241 2,92 63,36 2,5 1:100 0,039 0,185 0,112 0,278 0,279 0,2785 2,37 51,46 3 1:100 0,065 0,038 0,0515 0,305 0,303 0,304 3,6 78,05 3,5 1:100 0,04 0,04 0,04 0,298 0,287 0,2925 3,6 78,05 4 1:100 0,044 0,039 0,0415 0,306 0,292 0,299 3,67 79,59 4,5 1:100 0,044 0,039 0,0415 0,292 0,285 0,2885 3,52 76,35 5 1:100 0,04 0,035 0,0375 0,268 0,273 0,2705 3,32 72,02 Leerwert 1:100 0,04 0,043 0,0415 0,063 0,065 0,064 0,32 6,95 SCD 1:100 0,04 0,04 0,04 0,062 0,0625 0,32 6,95 0,063 [mg/ml] 0,73 [mM] Die Berechnung der Ethanol-Konzentration in mM, exemplarisch an der Probe t0, erfolgte so: E = E2-E1 der jeweiligen Probe (bei t0 also. 0,1785-0,033 = 0,1455) F = Verdünnungsfaktor (bei t0 = 50) 61 V = Testvolumen (also 1020 µl) v = Volumen der Probe (also 100 µl) d = Schichtdicke der Küvette in cm (also 1) 366 (NADH) = 3,3 ∙ 10-6 cm2 mol-1 oder 3300 M-1cm-1 Für t0 lautet diese Rechnung also: Die Berechnung der Ethanol-Konzentration in mg/ml für t0 lautet wie folgt: Die Begutachtung der ermittelten Ethanol-Konzentration zeigt, dass die EthanolKonzentration im Medium bis zum Zeitpunkt t4 kontinuierlich steigt, und dann zum Zeitpunkt t5 hin wieder etwas abfällt. Werden die Glucose- und Ethanol-Konzentrationen in mg/ml in Korrelation zum Wachstum der Zellen gesetzt, kann folgender Graph erstellt werden: 62 Abbildung 27: Metabolitbestimmung, Glucose- und Ethanol-Konzentrationen in mg/ml Dieser Graph (Abb. 27) zeigt das Wachstum der S. cerevisiae-Stamm CEN.PK122 Kultur in Korrelation zu den Konzentrationen der Metabolite. Die Abnahme der Glucose, sowie die Zunahme des Ethanols sind deutlich zu erkennen. Werden die Glucose- bzw. Ethanol-Konzentrationen in mM aufgetragen, sieht dies wie folgt aus: 63 Abbildung 28: Metabolitbestimmung, Glucose- und Ethanol-Konzentrationen in mM Die graphische Darstellung (Abb. 28) der Glucose- bzw. Ethanol-Konzentration in mM zeigt ebenfalls die Abnahme der Glucose und die Zunahme des Ethanols, hier jedoch direkt vergleichbar. Ein Molekül Glucose wird zu zwei Molekülen Ethanol umgesetzt, was diese Abbildung anschaulich wiedergibt. Der Ausgangswert der Glucose lag ungefähr bei 40 mM, rein rechnerisch sollten hieraus ca. 80 mM Ethanol gebildet werden. Dieser theoretische Wert wird durch den gemessenen Wert von 79,59 mM Glucose (Tab. 33, Abb. 28) bestätigt. Diese theoretische Überlegung kann nun für alle Werte umgesetzt und anschließend mit den real gemessenen Werten überprüft werden: Tabelle 34: Überprüfung der theoretischen mit den tatsächlichen Werten Probe Glucose [mM] [t = ] Differenz [mM] theoretisch gebildeter Ethanol [mM] theoretische Gesamtmenge Ethanol tatsächlich gebildeter Ethanol [mM] 0 40,24 2,249 0,5 34,44 5,8 11,6 11,6 15,84 1 25,78 8,66 17,32 28,92 28,13 64 1,5 20,8 4,98 9,96 38,88 40,57 2 10,16 10,64 21,28 60,16 63,36 2,5 3,4 6,76 13,52 73,68 51,46 3 0,48 2,92 5,84 79,52 78,05 3,5 0,11 0,37 0,74 80,26 78,05 4 0,11 0 0 80,26 79,59 4,5 0,12 0 0 80,26 76,35 5 0,12 0 0 80,26 72,02 Graphisch aufgetragen, sieht dies wie folgt aus: Abbildung 29: Graphische Auftragung der theoretischen und tatsächlichen Werte, Legende: ZE = Zeiteinheit, hier: 0,5 Std 65 Die Graphik (Abb. 29) zeigt, dass sich die theoretische und die tatsächliche EthanolKonzentration im Medium in mM sehr stark ähneln. Die theoretische Erwartungshaltung konnte also praktisch bestätigt werden. Diskussion Dieser Versuch zeigt anschaulich, was mit im Medium enthaltenen Metaboliten während des Wachstums einer Zellkultur passiert. Gleichzeitigt zeigt er auch die Stoffwechselaktivitäten von Zellen während des Wachstums sehr deutlich. Wird das Wachstum der S. cerevisiae Stamm CEN.PK122 Kultur in Verbindung mit dem Glucose-Abbau bzw. dem Ethanol-Zuwachs betrachtet (Abb. 28) so lassen sich daraus einige Schlussfolgerungen ziehen: Der Glucose-Verbrauch begann direkt nach überimpfen der Vorkultur in frisches Medium. Die Abnahme der GlucoseKonzentration vollzog sich relativ zügig, sodass bereits nach 3-3 ½ Stunden die Vorhandene Glucose im Medium verbraucht war. Zum gleichen Zeitpunkt zeigte die Kultur kein nennenswertes Wachstum mehr. Dies lässt den Schluss zu, dass die Kultur stationär wird, sobald alle Glucose verbraucht ist. Diese Schlussfolgerung ist logisch, wenn in Betracht gezogen wird, dass der für den Vorgang der GlucoseFermentation vorhandene Enzymsatz nach Verbrauch aller im Medium vorhandener Glucose zunächst auf ein anderes Substrat umgestellt werden muss. Die EthanolKonzentration steigt mit jedem verbrauchten Molekül Glucose selbst um zwei Moleküle an, was sich ebenfalls in der Graphik (Abb. 28) wiederfindet. Die Kultur startet mit ca. 40 mM Glucose und verstoffwechselt diese vollständig; die Kultur startet ebenfalls mit einem Minimum an Ethanol, welcher sich bis auf maximal ca. 80 mM anreichert. In das Medium wurde zu Beginn des Versuches kein Ethanol hinzugefügt, da sich die Bildung dieses Substrates zwar schnell, aber nicht so schnell vollzieht, ist der Anfangswert auf Messschwankungen zurückzuführen. Diese Schwankungen lassen sich vor allem beim Leerwert bzw. beim reinen SCD-Medium wiederfinden, so weisen beide dieselbe Ethanolkonzentration von knapp 7 mM auf. Dieser Wert ist als Abweichung zu betrachten und muss bei der Betrachtung der Messwerte berücksichtigt werden. Der Einbruch der Ethanolkonzentration bei t 2,5 lässt sich auf eine nicht-einwandfreie Doppelbestimmung zurückführen und sollte vernachlässigt werden (Tab. 33). Wird die tatsächlich gebildete Ethanolkonzentration 66 in mM mit der theoretisch gebildeten Ethanolkonzentration verglichen, so ist eine signifikante Abweichung nur zum Ende hin zu erkennen (Abb. 29). Dass die Ethanolkonzentration zum Ende hin abfällt, lässt sich z.B. durch bereits erfolgtes Umschalten des Enzymsatzes auf Ethanolverbrauch der Zellen erklären. Da ab dem Zeitpunkt t3,5 alle im Medium vorhandene Glucose verbraucht ist, kann kein weiterer Ethanol dem Medium zugeführt werden. Die die Ethanolkonzentration beschreibende Kurve sollte eigentlich konstant bleiben, so wie in der Theorie dargestellt (Abb. 29). Tatsächlich fällt die Konzentration zum Ende hin jedoch ab, was dafür sprechen könnte, dass die Zellen ihren Enzymsatz bereits umgestellt haben, und nun Ethanol als Kohlenstoffquelle nutzen (Abb. 28). Was dafür sprechen würde ist, dass die Zellen nach einem kurzen Wachstumseinbruch zum Zeitpunkt t 4,5 erneut Wachstum zum Zeitpunkt t5 zeigen (Abb. 28). Dies würde jedoch bedeuten, dass die Zellen ihre gesamte Enzymausstattung innerhalb von ca. 1-1 ½ Stunden umgestellt haben und nun in der Lage sind, Ethanol zu verstoffwechseln. Ob dies der Fall ist, kann nicht mit Sicherheit gesagt werden, jedoch sprechen genannte Gründe dafür. Würde dieser Fall außer Acht gelassen werden, so lässt sich die sinkende Ethanolkonzentration auf Mess- bzw. Pipetierfehler hin zurückführen, die natürlich nicht ausgeschlossen werden können. 67 IV. Versuch: Derepressionskinetik Einleitung Der Stoffwechsel der Hefe Saccharomyces cerevisiae hat die Besonderheit, dass unter Anwesenheit hoher Konzentrationen an Glucose zunächst nur diese verbraucht wird. Gene für Enzyme die mögliche weitere Substrate4 umsetzen, werden reprimiert. Dieses Phänomen bezeichnet man als Glucoserepression, diese tritt auch auf, wenn das Medium ein Gemisch aus verschiedenen Zuckern ist. Ist die im Medium vorhandene Glucose verbraucht, kommt es zur Umschaltung des genetischen Apparates und Enzyme zum Abbau weiterer Metabolite werden gebildet. Damit werden die vorher reprimierten Gene nun exprimiert, was wiederum als Derepression bezeichnet wird. In diesem Versuch wird gezielt ein Gen auf Glucoserepression hin untersucht. Dieses Gen codiert für die Fructose-1,6-bisphosphatase (FBPase), ein Enzym der Gluconeogenese das Fructose-1,6-Bisphosphat zu Fructose-6-Phosphat umsetzt. Um eine mögliche Repression mit nachfolgender Derepression anschaulich zu machen, wird eine Reportergenfusion erzeugt. Hierbei wird der Promotor der FBPase mit dem Strukturgen lacZ, welches für die bakterielle β-Galactosidase codiert, fusioniert und über ein Plasmid in die Zelle gebracht. Wird der Promotor der FBPase nun abgelesen, wird das Enzym β-Galactosidase gebildet, wird der Promotor reprimiert, ist keine oder nur sehr wenig β-Galactosidase in der Zelle zu finden. Die βGalactosidase hat die Fähigkeit glykosidische Bindungen zu spalten. Die Besonderheit ist, dass dieses Enzym keine sehr hohe Substratspezifität aufweist, und damit auch z.B. die glykosidische Bindung des angebotenen Substrates ortho-Nitrophenol- Abbildung 30: orthoNitrophenol-Galactosid (oNPG) Abbildung 31: Gelbgefärbtes Monomer: ortho4 S. cerevisiae kann auf verschiedenen Zuckern wie Raffinose, Galaktose oder Saccharose, aber auch Nitrophenol auf Substanzen mit kürzeren C-Ketten wie Ethanol, Glycerin, Laktat oder Acetat wachsen. 68 Galactopyranosid (oNPG, Abb. 6) in der Lage ist zu spalten. Das resultierende Monomer ortho-Nitrophenol weist eine gelbe Färbung auf, die anhand der Messung der optischen Dichte quantifiziert werden kann. Dies lässt Rückschlüsse auf die Promotoraktivität und damit auf die Repression oder Derepression der FBPase zu. Um eine Derepressionskinetik beobachten zu können, werden die Zellen zunächst in Glucose-haltigem Medium angezogen und anschließend auf Glucose-freies Medium überimpft. Es werden zu verschiedenen Zeiten Proben genommen und diese dem sog. β-Galactosidase-Test unterzogen. Da bei diesem Test nicht die FBPase selbst, sondern eine künstliche βGalactosidase-Fusion bzw. deren Genprodukt die β-Galactosidase selbst untersucht wird, muss im letzten Schritt noch das Vorhandensein der FBPase ermittelt werden. Dies geschieht mit Hilfe eines sog. Western Blots, bei der das Vorhandensein der FBPase immunologisch nachgewiesen wird. Hierzu werden die Proteine zunächst auf einem SDS-Polyacrylamid-Gel aufgetrennt und anschließend auf eine Membran transferiert. Diese Membran wird mit spezifischen Antikörpern gegen die FBPase inkubiert und die Antigen-Antikörper-Bindung (und damit die FBPase) mittels einer Farbreaktion sichtbar gemacht. Die Ergebnisse des β-Galactosidase-Tests sollten mit den Ergebnissen des Western Blots korrelieren. Material und Methoden Siehe Skript, Änderungen: Bei der Chloroform-Methanol-Fällung wurden 70µg Protein bei jeder Probe gefällt. (S. 37) Es wurde KPP-Puffer hinzugegeben, bis das Gesamtvolumen 150 µl betrug. (S. 37) Ergebnisse 69 Zunächst wurde eine Saccharomyces cerevisiae Kultur, Stamm CEN.PK115, mit dem Plasmid pJS151 transformiert und anschließend in einer SCD 4-ura Vorkultur angezogen. Diese Vorkultur wurde anschließend auf 200 ml frisches SCD4-uraMedium überimpft und über Nacht inkubiert. Am nächsten Morgen wurde die optische Dichte bei 600 nm der Kultur bestimmt, diese betrug OD 600 = 3,59 und konnte so weiter verwendet werden. Die Zellen wurden anschließend durch Zentrifugation geerntet, gewaschen, nach erneutem Zentrifugieren mit SCE3-uraMedium resuspendiert und anschließend in 200 ml selbiges überführt. Nun wurden über einen Zeitraum von acht Stunden hinweg stündlich zwei Mal ca. 5ml entnommen, daraus die optische Dichte bei 600 nm bestimmt, sowie die Zellen abzentrifugiert, mit eiskalten KPP-Puffer gewaschen, erneut zentrifugiert, geerntet und eingefroren. Durch das stündliche Bestimmen der optischen Dichte, konnte auf diesem Weg eine Wachstumskurve erzeugt werden: Tabelle 35: Verlauf der optischen Dichte der S. cerevisiae Kultur Stamm CEN.PK115 über einen Zeitraum von acht Stunden bei 30°C auf dem Schüttler Probe [t = ] Uhrzeit/Std Verdünnung OD600 t.1 OD600 t.2 OD600 0 8.34Uhr /0 1:10 0,275 0,274 2,745 1 9.35Uhr/1 1:20 0,150 0,148 2,98 2 10.34Uhr/2 1:20 0,151 0,148 2,99 3 11.35Uhr/3 1:20 0,153 0,169 3,22 4 12.34Uhr/4 1:20 0,166 0,160 3,26 5 13.33Uhr/5 1:20 0,165 0,170 3,35 6 14.33Uhr/6 1:20 0,180 0,188 3,68 7 15.34Uhr/7 1:20 0,191 0,192 3,83 8 16.35Uhr/8 1:20 0,20 0,197 3,97 70 Die graphische Auftragung dieser Werte (Tab. 35) führt zu folgender Wachstumskurve: Abbildung 32: Wachstumskurve des S. cerevisiae Stammes CEN.PK115 über einen Zeitraum von acht Stunden bei 30°C auf dem Schüttler Als nächstes wurden die eingefrorenen Proben (gekennzeichnet mit t.1 und t.2) mit 500 µl eiskalten KPP-Puffer und ca. 0,5 ml Glasperlen versetzt. Dieses Gemisch wurde anschließend für ca. 1 ½ Minuten intensiv geschüttelt, dann mit 1 ml eiskaltem KPP-Puffer versetzt und erneut ca. 3 Sekunden intensiv gemischt. Dieser Rohextrakt wurde anschließend 10 Minuten bei 4000 upm und 4°C zentrifugiert um Zelltrümmer und nicht-aufgeschlossene Zellen abzutrennen. Nach dem Zentrifugieren wurde der Überstand knapp über den Glasperlen abgenommen und in ein neues Reagenzglas überführt. Anschließend erfolgte die Proteinbestimmung nach Mikrobiuret. Hierzu wurden je 100 µl Probe mit 1 ml KPP-Puffer und 500 µl Mikrobiuret-Reagenz versetzt. Dieser Ansatz wurde dann jeweils ca. 5 Sekunden intensiv gemischt und im nächsten Schritt sofort die Extinktion bei 290 nm gegen einen Puffer-Leerwert bestimmt. Alle Messungen wurden in Doppelbestimmung durchgeführt. Die Proteinkonzentration der einzelnen Proben lautete wie folgt: 71 Tabelle 36: Proteinkonzentrationen der Proben 0.1-8.2, Doppelbestimmung und Einzelwerte siehe Anhang Probe [t = ] Proteinkonzentration [mg/ml] 0.1 0,760727 0.2 0,729081 1.1 0,872461 1.2 0,848269 2.1 0,856374 2.2 0,868937 3.1 0,912290 3.2 0,796727 4.1 0,937784 4.2 0,857238 5.1 0,743232 5.2 0,894873 6.1 0,910688 6.2 0,951769 7.1 0,896495 7.2 0,801819 8.1 0,998625 8.2 1,017903 72 Der Vergleich zweier Proben derselben Stunde gibt Auskunft über den Erfolg des Zellaufschlusses. So variiert die Proteinkonzentration der einzelnen Proben zueinander zwischen 1,5-20,5 % und im Durchschnitt um 8,88 %. Als nächstes wurde der β-Galactosidase-Test durchgeführt. Hierbei gibt die Spaltung des angebotenen Substrates oNPG und die daraufhin erfolgende Gelbfärbung Auskunft über die Aktivität des Fructose-1,6-bisphosphatase-Promotors und dabei schlussendlich über die theoretische Aktivität der Fructose-1,6-bisphosphatase. Die Durchführung war dabei folgende: Zunächst wurden die Proben 0.1-8.2 jeweils in Doppelbestimmung vorbereitet und gekennzeichnet mit A und B (also: 0.1 A, 0.1 B, 0.2 A, 0.2 B usw.). In die Reagenzgläser kamen nun 50 µl der jeweiligen Probe, 1ml Z-Puffer sowie 100 µl 2-Mercaptoethanol. Die Ansätze wurden anschließend gemischt und bei 30°C für ca. 10 Minuten vortemperiert. Als Referenz diente der im Test verwendete Z-Puffer. Der Test wurde gestartet, indem in konstanten Zeitintervallen je 250 µl oNPG-Lösung zum jeweiligen Ansatz hinzupipetiert und intensiv gemischt wurde. Die Reaktion wurde bei ausreichender jedoch nicht übermäßiger Gelbfärbung durch die Zugabe von je 500 µl 1M Na 2CO3-Lösung in den bereits zuvor angewandten Zeitintervallen abgestoppt. Die Reaktion dauerte je Probe ca. 10 Minuten und 52 Sekunden (bzw. 10,867 Minuten). Nach Ablauf der Reaktion wurde die Extinktion der Proben bei 600 nm gemessen. Die Extinktion der Proben sah dabei wie folgt aus: Tabelle 37: Extinktion der Proben 0.1-8.2 nach erfolgter β-Galactosidase-Reaktion Probe [t = ] Verdünnung Extinktion Extinktion unverdünnt 0.1 / 0,009892 <- 0.2 / 0,011087 <- 1.1 / 0,007805 <- 1.2 / 0,007693 <- 2.1 / 0,027845 <- 73 2.2 / 0,029044 <- 3.1 / 0,079560 <- 3.2 / 0,085417 <- 4.1 / 0,306348 <- 4.2 / 0,294861 <- 5.1 1:5 0,140283 0,701415 5.2 1:5 0,160208 0,80104 6.1 1:10 0,118205 1,18205 6.2 1:10 0,130208 1,30208 7.1 1:10 0,135293 1,35293 7.2 1:10 0,125456 1,25456 8.1 1:10 0,184882 1,84882 8.2 1:10 0,187464 1,87464 Anhand dieser Werte kann nun die Aktivität nach Miller der einzelnen Proben berechnet werden. Die Berechnung erfolgte anhand des Lambert-Beer’schen Gesetzes: VA = Volumenaktivität E = Extinktion bei 420 nm 74 V = Volumen des gesamten Testansatzes (hier: 1150 µl) t = Dauer der Reaktion vom Start durch oNPG bis zum Stopp durch Na 2CO3 (hier: 10,867 min) = Extinktionskoeffizient des Spaltprodukts ortho-Nitrophenol: 4500 M-1cm-1 d = Schichtdicke der Küvette (hier: 1 cm) v = Volumen der Probe (hier: 50 µl) Exemplarisch sei die Rechnung für die Probe t = 8.2 gezeigt: Beim β-Galactosidase-Test ist das Unit jedoch definiert mit 1 nMol Substratumsatz/min, was der Einheit [nmol/min∙ml] entspricht. Es muss damit folgende Umformung vorgenommen werden: Die Probe t = 8.2 besitzt damit eine Volumenaktivität von 881,705 nmol/min∙ml oder 881,705 Miller-Units/ml. Mit Hilfe dieses Wertes kann nun die spezifische Aktivität anhand folgender Formel berechnet werden: 75 Die Proteinkonzentrationen der einzelnen Proben wurden zuvor anhand des Mikrobiuret-Assays bestimmt (Tab. 36). Die Proteinkonzentration der Probe t = 8.2 betrug 1,017903 mg/ml, damit ergibt sich folgende spezifische Aktivität: Damit ergibt sich für die Probe t = 8.2 eine spezifische Aktivität von 866,1975 MillerUnits/ml oder 866,1975 nmol/min∙mg. Zusammengefasst weisen die Proben folgende Aktivitäten auf: Tabelle 38: Aktivität der Proben 0.1-8.2 nach Miller Probe Ansatz VA [Unit/ml VA min] VA/cprot [Unit/mg] 0 .1 4,653 6,1165 .2 5,215 .1 3,671 .2 3,618 .1 13,096 .2 13,66 .1 37,42 .2 40,174 1 2 3 4,934 7,1528 VA/cprot 6,63465 4,2076 3,6445 4,2652 4,2364 15,2924 13,378 15,7204 15,5064 41,0177 38,797 50,424 45,7209 76 4 5 6 7 8 .1 144,086 .2 138,683 .1 329,899 .2 376,7558 .1 555,9574 .2 612,4115 .1 636,328 .2 590,0612 .1 869,562 .2 881,705 153,645 141,3845 161,7789 157,71195 443,8708 353,3274 421,0159 432,4434 610,4807 584,1845 643,4455 626,9631 709,7953 613,1946 735,90324 722,8493 870,7588 875,6335 866,1975 868,4782 Werden diese Werte graphisch aufgetragen und in Korrelation zum Wachstum der. S. cerevisiae Kultur gesetzt, ergibt sich folgendes Bild: 77 Abbildung 33: Aktivität nach Miller in Korrelation zum Wachstum der S. cerevisiae Kultur Die graphische Auftragung (Abb. 33) zeigt, dass die Aktivität des FBPase-Promotors zunächst noch sehr schwach ist, jedoch innerhalb der ersten drei Stunden langsam, ab da an sehr stark ansteigt. Abgesehen vom Wert der ersten Stunde verdreifacht sich die Aktivität nach Miller zu jedem Zeitpunkt innerhalb der ersten fünf Stunden. Danach steigt die Aktivität um ungefähr das 1 - 1 ½ fache zu jedem Zeitpunkt an. Im letzten Schritt wurde ein SDS-Gel hergestellt. Von jeder Probe wurde hier jeweils diejenige aufgetragen, die die höchste Proteinkonzentration zeigt (also 0.1, 1.1, 2.2 usw. Tab. 36). Dieses Gel wurde anschließend genutzt, um einen Western Blot zu erstellen. Hierbei werden die Proteine von einem SDS-Gel über das Anlegen von Spannung auf eine Membran übertragen. Diese Membran wird anschließend mit einem Antikörper gegen das jeweilige Protein inkubiert. Zuletzt wird die Membran mit gebundenem erstem Antikörper mit einem weiteren Antikörper inkubiert, der wiederum gegen den ersten Antikörper gerichtet ist. Der zweite Antikörper ist Teil einer Reaktion, die die Bindung beider Antikörper sichtbar macht. Dies könnte entweder durch die Bestrahlung mit Röntgenstrahlen aber auch durch eine gewöhnliche Farbreaktion besorgt werden. In diesem Fall wurde die Membran zur Indikatorreaktion in einem Färbelösungsmix geschwenkt, was folgendes Bild ergab: 78 Abbildung 34: Western Blot, Legende: M = Marker in [kDa], 0-8 = jeweilige Probe Auch der Western Blot zeigt deutlich die Zunahme der Intensität, hier jedoch direkt die der Fructose-1,6-Bisphosphatase (FBPase), die ein Molekulargewicht von 38,1 kDa aufweist. Die Intensität der Bande nimmt nach ca. zwei Stunden etwas, ab drei Stunden deutlich zu. Dies korreliert mit den im β-Galactosidase-Tests gewonnenen Ergebnissen, dass die Aktivität der FBPase zunächst nur langsam, dann jedoch sehr deutlich ansteigt. Diskussion Ziel dieses Versuches war es zu zeigen, dass bestimmte Enzyme unter Wachstumsbedingungen bei Anwesenheit von Glucose reprimiert sind, und diese bei Veränderung der Bedingungen auf glucosefreies Wachstum eine Derepression erfahren, also dann exprimiert werden. Betrachtet wurde hierbei die Fructose-1,6Bisphosphatase, ein Enzym der Gluconeogenese. Beim Überimpfen des S. cerevisiae Stammes CEN.PK115 von Glucose-haltigem SCD4-, auf Glucose-freies SCE3-Medium wurde innerhalb weniger Stunden der Enzymsatz der Vorkultur, eingestellt auf Glucose-Repression, so umgestellt, dass nun vorher reprimierte Enzyme exprimiert werden, also eine Derepression stattgefunden hat. Gezeigt wurde dies u.a. durch die Reportergenfusion des FBPase Promotors mit der β- 79 Galactosidase und anschließender Aktivitätsbestimmung letzterer (Tab. 38, Abb. 33). Dieser Nachweis ist als eher indirekt zu betrachten, da er Auskunft über die Promotoraktivität des FBPase-Promotors zulässt, jedoch nicht die FBPase selbst betrachtet. Zu sehen ist dennoch, dass die Aktivität nach Miller der β-Galactosidase, einhergehend mit der Aktivität des FBPase-Promotors, nach ca. zwei Stunden leicht anstieg, und ab ca. drei Stunden kontinuierlich zunahm. Dies lässt den Rückschluss zu, dass nach ca. 1-2 Stunden die Repression des FBPase-Promotors bereits zu großen Teilen aufgehoben wurde, dass Enzym also nun wieder exprimiert wird. Auch die Betrachtung des Western Blots lässt ähnliche Schlüsse zu (Abb. 34). Hierbei wurde mit spezifischen Antikörpern gegen die FBPase gearbeitet. Dies lässt direkte Schlussfolgerungen der Derepression auf die FBPase im Bezug auf die Konzentration dieses Enzymes zu. Zu erkennen ist, dass auch hier das Vorhandensein des Enzyms innerhalb der ersten Stunde mehr oder minder konstant geblieben ist. Nach einer weiteren Stunde (also insgesamt zwei Stunden) fand bereits eine sichtbare Mengenzunahme der FBPase statt, die ab der dritten Stunde kontinuierlich erfolgte. Die FBPase besitzt ein Molekulargewicht von 38,1 kDa, ist also ungefähr auf der Höhe der 37 kDa-Bande zu erkennen. Die Bande um 150 kDa zeigt das Fusionsprotein aus 14 Aminosäuren der FBPase sowie der βGalactosidase mit ca. 120 kDa, wobei der hier eingesetzte Antikörper die kurze FBPase-Sequenz erkennt. Die Banden unter 25 kDa zeigen Abbauprodukte der FBPase, da sie in der gleichen Weise an Intensität zunehmen, wie die FBPase selbst. Die verbleibenden Banden sind unspezifische Wechselwirkungen der Antikörper mit den Proteinen, und von daher zu vernachlässigen. Alles in allem zeigt dieser Versuch sehr anschaulich den Ablauf und auch die zeitliche Dauer des Umstellens eines Enzymsatzes. Was dieser Versuch nicht vermag ist zu vermitteln, was für ein Aufwand diese Umgestaltung für die Zellen bedeuten. Die Umstellung eines ganzen Enzymsatzes auf neue Umweltbedingungen ist eine energetische Anstrengung, die eine Zelle nicht leichtfertig vollziehen sollte. Vielleicht ist ein Grund, warum diese Modifikation einige Zeit benötigt, nicht nur der zeitlichen Dauer der Neu-Synthese von Enzymen zuzuschreiben, sondern auch dem Durchlaufen verschiedener Kontrollsequenzen, ob sich die Neu-Synthese oder gar die vollständige Umstellung des Enzymsatzes wirklich lohnt. 80 V. Versuch: Analyse von Polysomen-Profilen Einleitung Damit aus einem Gen ein Protein wird müssen verschiedene Teilschritte ablaufen. Zunächst wird das Gen bzw. die DNA aus der es besteht, in mRNA überschrieben. Dieser Vorgang wird als Transkription bezeichnet und findet bei Eukaryoten im Kern, bei Prokaryoten im Cytoplasma statt. Diese mRNA (messenger RNA) wird nun im Cytoplasma in ein Protein übersetzt, wobei sie bei Eukaryoten dazu zunächst aus dem Kern exportiert werden muss. Im Cytoplasma wird die mRNA dem Vorgang der Translation unterzogen und so in ein Protein übersetzt. Dies geschieht mit Hilfe kleiner RNA-Synthesemaschinen, den sog. Ribosomen. Ein Ribosom besteht aus einer großen und einer kleinen Untereinheit. Diese Untereinheiten lagern sich am Startpunkt der Translation (bei Prokaryoten der sog. Shine-Dalgarno-Sequenz) auf der mRNA an und verknüpfen sequenzspezifisch die zugehörigen Aminosäuren zu einem Peptid. Ist die Translation beendet, die mRNA also vollständig in eine Kette aus Aminosäuren übersetzt, fallen die Ribosomen von der mRNA ab und zerfallen wieder in ihre Untereinheiten, bis sie die nächste Translationsrunde starten. Die Charakterisierung der Untereinheiten in „große“ und „kleine“ Untereinheit findet aufgrund ihres Sedimentationsverhaltens. Das Sedimentationsverhalten wird bestimmt durch den sog. Sedimentationskoeffizienten, dieser beschreibt den Quotienten aus der maximalen Sedimentationsgeschwindigkeit eines Teilchens in einer Zentrifuge geteilt durch die Stärke des Zentrifugalfelds. Die Maßeinheit des Sedimentationskoeffizienten ist Svedberg (S) benannt nach dem schwedischen Chemiker Theodor Svedberg. Die größe des Sedimentationskoeffizienten ist abhängig hauptsächlich von der Masse und Form des sedimentierenden Teilchens, wird jedoch auch durch die Wechselwirkung des Teilchens mit dem umgebenden Medium beeinflusst. Wird ein Medium bekannter Eigenschaften genutzt, kann der Sedimentationskoeffizient herangezogen werden um die Beschaffenheit eines Teilchens, allen voran seiner Masse zu bestimmen, wie im Falle der Ribosomen bzw. Ihrer Untereinheiten. So wird die kleine Ribosomen-Untereinheit als 30S (bei 81 Prokaryoten) bzw. 40S (bie Eukaryoten) Untereinheit, und die große als 50S (bei Prokaryoten) bzw. 60S (bei Eukaroten) bezeichnet. Da Svedberg-Einheiten nicht additiv sind, handelt es sich bei den zusammengelagerten Untereinheiten um 70S (bei Prokaryoten) bzw. 80S (bei Eukaryoten) Ribosomen. Durch Zentrifugation können jedoch nicht nur einzelne Untereinheiten oder ganze Ribosomen aufgetrennt werden, sondern auch sog. Polysomen. Als Polysom bezeichnet man eine Vielzahl von aktiven 70S (bei Prokaryoten) oder 80S (bei Eukaryoten) Ribosomen, die gemeinsam auf einer mRNA liegen. Der Vergleich von freien großen und kleinen Untereinheiten und Polysomen gibt Auskunft über mögliche Defekte bei der Reifung der Untereinheiten oder während der verschiedenen Stadien der Translation. Für gewöhnlich liegen große und kleine Untereinheiten in äquimolaren Konzentrationen vor, da diese in gleichen Mengen synthetisiert werden. Ist dies nicht der Fall kann es sich um einen Defekt während der Ribosomen-Biogenese handeln, wie es z.B. in der Saccharomyces cerevisiae-Nop14-Mutante der Fall ist. Diese Mutante weist einen Defekt im Nop14-Protein auf, welches verantwortlich für die Reifung und den Export der kleinen Ribosomen-Untereinheit aus dem Kern ist. Die Nop14-Mutante zeigt daher eine Verringerung der kleinen 40S-Untereinheit. Dies ist nur eine Möglichkeit die Methode der Zentrifugation zu nutzen. Es gibt verschiedenste Techniken der Zentrifugation, je nachdem, welches Ziel gewünscht ist. Allgemein dient die Zentrifugation der Sedimentation von Zellen, Zellorganellen und größeren Molekülen, und damit der Auftrennung nach Größe und Dichte der Teilchen unter Ausnutzung Zentrifugationsarten, der darunter: Zentrifugalkraft. die Es differentielle gibt verschiedenste Zentrifugation, die Dichtegradientenzentrifugation bzw. darunter die Zonenzentrifugation und die isopyknische Zentrifugation. Bei der differentiellen Zentrifugation wird die Zentrifugalbeschleunigung und/oder Zentrifugalzeit stufenweise erhöht, sodass ein Gemisch aus verschiedenen Substanzen in Fraktionen unterschiedlichen Sedimentationskoeffizienten aufgetrennt wird. Während eine durchschnittliche Zentrifuge nur verhältnismäßig kleine Beschleunigungen erreichen kann, kann eine Ultrazentrifuge bis zu 10 6g erzeugen, dies wiederum entspricht der 106-fachen Erdbeschleunigung. Der Rotor einer 82 solchen Zentrifuge bewegt sich daher im Vakuum, um auftretende Luftreibung und damit übermäßige Wärmebildung zu vermeiden. Bei der Ultrazentrifugation lassen sich verschiedene Rotoren einsetzen: Festwinkel und Ausschwingrotoren. Bei einem Festwinkelrotor bleibt, wie der Name erkennen lässt, der Winkel konstant. Das Sediment einer zentrifugierten Probe würde sich hier an der Seitenwand des Zentrifugenröhrchens befinden. Bei einem Ausschwingrotor ist dies nicht der Fall. Hierbei wird während der Zentrifugation das Zentrifugenröhrchen in die Waagerechte gebracht, sodass sich das Sediment der Probe anschließend am Boden des Röhrchens befindet. Eine Technik der Ultrazentrifugation ist die Dichtegradientenzentrifugation. Hierbei wird mit Hilfe eines Salz- oder Zucker-Gradienten die Auftrennung eines Proteinoder RNA/DNA-Gemisches in seine unterschiedlich dichten Bestandteile erzielt. Die Teilchen lagern sich während der Zentrifugation im Gradienten in den Bereich ein, der ihrer jeweiligen Dichte entspricht. Anschließend kann das Profil des gesamten Gradienten aufgenommen, oder aber die Isolierung von Teilchen bekannter Dichte vorgenommen werden. Es gibt zwei Arten der Dichtegradientenzentrifugation: Die Zonenzentrifugation und die isopyknische Zentrifugation. Bei der Zonenzentrifugation handelt es sich um eine Zentrifugationsart, bei der mit Hilfe eines linearen, stabilisierenden, sehr flachen Gradienten, z.B. aus Saccharose bestehend, Teilchen aufgrund ihrer Dichte aufgetrennt werden. Die Teilchen starten bei der Zentrifugation alle von einer schmalen Zone. Da kleinere Teilchen ein niedrigeres Trägheitsmoment aufweisen als große, beschleunigen diese schneller bei der Zentrifugation als größere Teilchen, werden jedoch durch den zunehmend dichter werdenden Gradienten abgebremst. Damit wird eine konstante Sedimentationsgeschwindigkeit aller Teilchen erreicht. So sedimentieren die Teilchen im Gradienten an der Stelle, die ihrer Dichte entspricht. Die Dichte des Gradienten nimmt von oben im Zentrifugenröhrchen, nach unten hin zu, weshalb kleinere leichte Teilchen oben zu finden sind, und größere schwerere unten. Wichtig bei der Zonenzentrifugation ist, dass die maximale Dichte des Gradienten kleiner sein muss als die niedrigste Dichte der Partikel. Die isopyknische Zentrifugation beschreibt eine Zentrifugations-Methode, bei der, im Gegensatz zur Zonenzentrifugation, die maximale Dichte des Gradientenmaterials 83 die Dichte der Teilchen übersteigt. Als Gradientenmaterial kann z.B. Cäsiumchlorid eingesetzt werden. Die Teilchen sollten alle die gleiche Größe aufweisen, weshalb sie dann aufgrund ihrer reinen Dichte aufgetrennt werden. Ziel dieses Versuches ist es durch Dichtegradientenzentrifugation ein PolysomenProfil eines Wildtyp-Stammes und einer Nop14-Mutante zu erstellen und mögliche Unterschiede z.B. in den Kozentrationen der einzelnen Untereinheiten oder aber der Menge an Ribosomen und Polysomen festzustellen. Material und Methoden Siehe Skript, Änderungen: Die optische Dichte der Wildtyp-Vorkultur betrug 20, die der Nop14-Mutante 13,5. Die 100ml YEPD-Kolben wurden nur mit 2,2ml Wildtyp-Vorkultur (statt 2,5ml) bzw. 3ml Nop14-Mutante-Vorkultur (statt 3,7ml) beimpft. (S. 45) Auf den Gradienten wurden 25 OD254-Einheiten aufgeladen. (S.46) Zentrifugation in Ultrazentrifuge mit dem Rotor SW40 bei 19.600rpm, 4°C über 16 Stunden. (S.46) Ergebnisse Nach Aufschluss der Zellen und Zentrifugation wurde folgendes Polysomenprofil der Nop14 Mutante aufgenommen: 84 Abbildung 35: Polysomenprofil der Nop14-Mutante nach erfolgter Zentrifugation 16Std 19.600rpm 4°C, Gradient: 20-50%ige Saccharose Der gezeigte Peak entspricht dem Cytoplasma der S. cerevisiae Zellen, außer diesem traten jedoch keine weiteren Peaks auf (Abb. 35). Die anschließende Nulllinie deutet auf das Fehlen weiterer Zellbestandteile v.a. der Ribosomen-Untereinheiten, der Ribosomen oder Polysomen hin. Da dieses Ergebnis sehr ungewöhnlich ist, wurde versucht durch Anpassen der Sensitivität an der Detektoreinheit ein besseres Ergebnis zu erzielen, was jedoch erfolglos blieb. Um eventuelle Gerätedefekte auszuschließen, wurde weiterhin ein leerer Gradient vermessen, dieser zeigte jedoch keinen Cytoplasma-Peak, was auch Funktionstüchtigkeit des Gerätes hindeutet. Auch das Vermessen eines KontrollProfils, das bereits einmal erfolgreich war, zeigte kein verwertbares Ergebnis. Ein älteres Profil zeigt, wie das Ergebnis hätte aussehen sollen: 85 Abbildung 36: Polysomenprofil des Wildtyps nach erfolgter Zentrifugation 18Std 18.500rpm 4°C, Gradient: 20-50%ige Saccharose Dieses Profil (Abb. 36) zeigt wie das obere (Abb. 35) bei ca. 2 Minuten den Cytoplasma-Peak. Nach ca. 3,8 Minuten ist ein erster kleinerer Peak zu sehen, der der kleinen 40S-Ribosomenuntereinheit entspricht. Der zweite kleinere Peak bei ca. 4,8-5 Minuten entspricht der größeren 60S-Ribosomenuntereinheit. Der folgende Peak bei ca. 5,4-5,6 Minuten entspricht den freien 80S Ribosomen und die Peaks ab ca. 7,4 Minuten den an der mRNA haftenden Polysomen. Diskussion Ribosomen, deren Untereinheiten sowie die an der mRNA haftenden Polysomen sind in jeder lebenden und wachsenden Zelle vertreten. Da Ribosomen essentiell für die Proteinbiosynthese und damit die Vitalität jeglicher Zellen sind, ist ihre Abwesenheit nur in toten Zellen zu beobachten, und auch dann nur bei vorheriger Ausschaltung aller für ihre Synthese bzw. Reifung zuständigen Gene. Das Fehlen jeglicher ribosomaler Bestandteile nicht nur in der in diesem Versuch prozessierten Nop14 Mutante, sondern ebenfalls im Wildtyp deutet daher auf einen elementaren Fehler bei der Versuchsdurchführung hin. Die einzig mögliche Erklärung wäre das zerstören des Gradienten, was schlussendlich auf die Bestandteile selbigen, und damit auf den verwendeten Puffer zurückzuführen wäre. Der für den Gradienten verwendete Puffer 86 bzw. dessen falscher pH-Wert hat vermutlich zur Zerstörung, bzw. zum Nicht-Aufbau des Gradienten geführt, weshalb die Auftrennung der Ribosomen, ihrer Untereinheiten sowie der Polysomen bei der Ultrazentrifugation nicht möglich war. Warum kein Pellet nach der Ultrazentrifugation zu erkennen war, ist nicht klar und spricht für die Vollständige Abwesenheit jeglicher ribosomaler Bestandteile. Ob eine ungewollte Zugabe von RNasen eventuell für die Zerstörung genannter Bestandteile gesorgt hat konnte nicht aufgeklärt werden. 87 VI. Quellen Skript „Biochemisches Praktikum im Hauptstudium" David Nelson, Michael Fox, Lehninger Biochemie, 3. Auflage, Springer Verlag, Berlin Heidelberg, 2001/2005 VII. Anhang Versuch 1: Handschriftliche Eichgeraden der verschiedenen Proteinbestimmungsmethoden Versuch 3: Originalmesswerte OD600, Glucose- und Ethanolbestimmung Versuch 4: Originalmesswerte OD600, Mikrobiuret, β-Galactosidase-Test 88