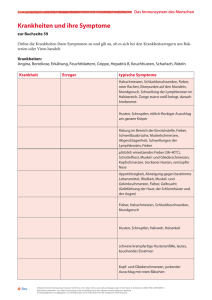
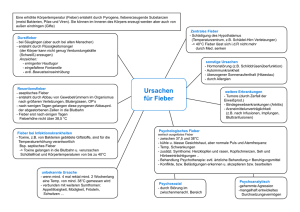
Kinder 10. Semester - Skript - mein
Werbung

Pädiatrie-Skript für das 10. Semester basiert auf ein Skript von Gerald Prechtl aktualisiert und erweitert von Karl Quint bearbeitet von Cornelia Reichardt im SS 2004 / SS 2006 6. Juli 2004 1 KINDERPRÜFUNG Neugeborenenperiode: - Frühgeborenes allgemein Typische Früh- und Mangelgeborenen-Komplikationen Wet-lung Syndrom Surfactantmangel ARDS Bronchopulmonale Dysplasie PDA Retinopathia prematurorum Periventrikuläre Leukomalazie Intrauterine Hypoxie NEC Aspiration Apnoe Fehlender Schluckreflex Hirnblutung Hyperbilirubinämie Hypothermie Hypoglykämie Hypocalzämie Infektgefährdet - Ösophagusatresie (S. 83) (140ff) Einteilung nach Vogt (IIIB am häufigsten, untere Fistel mit oberem Blindsack) 1:2500 Geburten K: Hydramnion, Zyanoseanfälle, Hustenattacken, schaumiges Sekret D: Sondierung mit kontrastdichter Sonde im Ösophagussack, Radiologie T: Bis zur OP Absaugkatheter im Ösophagusblindsack, Operation innerhalb 24h (Anastomose und Fistelverschluss) Kompl.: Aspiration, Magensaft über distale Fistel in Lunge DD: Aspiration, Sectio, Choanalatresie, Achalasie, Stenosen - Duodenalatresie (S. 86) (140ff) 1:3000, Assoziation mit anderen Fehlbildungen, v.a. Trisomie21 K: Hydramnion, Ileus, frühes Erbrechen (evtl gallig), Unterbauch eingefallen D: Rö im Hängen: doubble-bubble (Doppelspiegel: Magen und Duodenum) T: Magensonde (Dauersog); frühzeitige OP DD: Hiatushernie, Stenosen, Salzverlustsyndrom, zerebrales Erbrechen - Analatresie () (105) 1:3000 – 1:5000; 40% Kombination mit anderen Fehlbildungen (vor allem urogenital) Mit/ohne Fisteln: ♂: Blase, Urethra, Perineum; ♀: Vagina, Vestibulum, Blase, Perineum Hohe/intermediäre/untere Atresien (tiefe A. bessere Prognose wegen Kontinenz nach OP) K: sichtbare Atresie, Fistel mit Stuhlentleerung; wenn keine Fistel: keine Stuhl, aber Erbrechen, aufgetriebenes Abdomen D: Röntgen nach 24 Stunden in Bauchlage / Kopftieflage (zum Sichtbarmachen des Endes des Darms im Verhältnis zur markierten Analrinne) DD: Mekoniompfropfsyndrom, Ganglienzellanomalien (Stuhl gestaut in Darm) T: OP (direkte Anastomose) P: gut (evtl. Kontinenzprobleme bei hohen Atresien weil keine sensible Innervation) 2 - M. Hirschsprung (S. 253) Mangel an Ganglienzellen in Darmwand, 1:5000 (Jungen 3-4x häufiger) K: Subileussymptomatik, aufgetriebens Abdomen, leere, enge Ampulle D: rektale Untersuchung, Kontrasteinlauf , evtl. Histologie T: Operation (entfernen des aganglionären Segmentes) DD: Ileus, Rektumatresie, Chagas - Cholestase – siehe Gastroenterologie - Fazialisparese – siehe Neuropädiatrie - Atemnotsyndrom (S. 98) Veraschiedene Ursachen : Surfactantmangel (v.a. bei Frühgeborenen <32 SSW), Pneumonie, Aspiration, Fehlbildungen… K: Tachypnoe, inspirat. Einziehungen (nach Schwere jeweils Jugulum, dann substernal, am schwersten intercostal), exspirat. Stöhnen („Knorksen“), Nasenflügeln, Zyanose D: BGA (globale/partiale resp. Insuff.), Radiologie (fleckig, streifige Veränderungen, Bronchopneumogram, Herz unscharf begrenzt) T: CPAP/Intubation (Problem: bronchopulmonale Dysplasie, Hirnblutungen…), endotracheale Surfactant-Substitution Prophylaxe : Frühgeburt meiden, Dexametason 24-72h präpartal, Lecithin-Sphingomyelin-Ratio >2,0 reife Lunge - Aspirationspneumonie / neonatale Pneumonie () (128) Beruht auf Aspiration von infiziertem Fruchtwasser vor oder Vaginalsekret während er Geburt; Mögliches Vorkommen bei: vorzeitigem Blasensprung übelriechendem Fruchtwasser („Amnioninfektionssyndrom“) Fieber der Mutter und Besiedlung der mütterlichen Vagina mit hochpathogenen Bakterien (Streptokokken Gruppe B, E. coli). Bei Listeria monocytogenes Infektion (transplazentar, vaginal), hier v.a. schwere Sepsis (Neugeborenensepsis) Aspiration von Mekonium (Nährboden für Pneumonie (Staph. Aureus abszedierende Pneumonie)) Chlamydia trachomatis: nach 5-10 Tagen Konjunktivitis und 2-3 Wochen schwere interstitielle Pneumonie (Husten, Tachypnoe, Eosinophilie) Mykoplasmen, Ureaplasma, RS-Viren: interstitielle Pneumonien Pseudomonas, Pilze bei intubierten NG K: Atemnot, Fieber, eitriges Trachealsekret; evtl. Sepsiszeichen; frühe Sepsis des NG: an pulmonale Infektion denken (allgemein frühe Sepsis durch Keime der Vagina)! Sepsiszeichen: Irritabilität, Lethargie, Tachypnoe, Temperaturschwankungen, Perfusionsstörungen (Cutis marmorata und verlängerte Reperfusionszeit), Hypotension, blaß-graues Hautkolorit!; auch wenig Zeichen möglich und dennoch Sepsis); Labor: Leukozytose mit unreifen Granulozyten oder Leukopenie mit absoluter Neutropenie; auch normale Leukozytenzahlen möglich; septischer Schock: Thrombozytopenie, DIC D: Thorax-Rö, Mikroskopie und Mikrobiologie des Trachealsekrets T: wie Sepsis: nach Entnahme der Kulturen (Blut, Abstriche, Liquor, Urin) kalkülierte Antibiotikatherapie (Betalactamantibiotikum mit Aminoglykosid oder Cephalosporin); nach Ergebnis der MiBi gezielt. Verdacht auf Chlamydien, Mykoplasmen oder Ureaplasma: Erythromycin Bei RS-Viren bzw. Nachweis ihres Antigens: Ribavirin inhalativ 3 - NEC (S. 124) (168) Nekrot. Entzündung der Darmwand bei Fühgeborenen (ausgetragene Neugeborene selten betroffen); evtl. durch Minderperfusion (Schock, PDA, Hypoglykämie, Hypoxämie...); v.a. dist. Ileum & proximaler Colon Häufigste Ursache für akutes Abdomen beim Frühchen (2.-4. Lebenswoche) K: geblähtes Abdomen, Rückstau von Mageninhalt & Erbrechen, dann Blässe, Apathie, zunehmender Verfall; später Abwehrspannung und fehlende DG ( Peritonitis nach Perforation), Sepsis (Schock, Verbrauchskoagulopathie, DIC); manchmal gleich Sepsis als erstes Zeichen Spätsymptome: Schmerzen, Flankenrötung D: klinische Überwachung, CRP, Blutbild (Leukozytose und Linksverschiebung), Blutkultur, Radiologie – Pneumatosis intestinalis, nach Perforation: freie Luft unter Zwerchfell (Pneumoperitoneum), Sonographie – verdickte Darmwände T: parenterale Ernährung, nasogastr. Magensonde, Ursachenbeseitigung (PDA-Ligatur, Transfusion); bei Sepsis Antibiose, bei Perforation OP mit Resektion des nekrot. Darms P: Sepsis als Prognosefaktor, Letalität 5-10% Genetik: - Prader-Willi (S. 51, 133) (76) 1:15000, i.d.R. sporadisch, Mikrodeletion 15q12 paternal, maternales imprinting K: Kleinwuchs, Muskelhypotonie, zuerst Fütterungsschwierigkeiten, später Hyperphagie, Adipositas, kleine Hände und Füße, Hypogenitalismus, Minderbegabung (IQ 40-60) D: FISH - Trisomie 21 (S. 138) (66) 1:700, Risiko steigt mit Alter der Mutter (35 Jahre: 3/1.000, 45 Jahre: 3/100) verschiedene Formen: freie Trisomie (95%): Translokations-Tris. (2,5%): meist mit Chr. 13, 14, 21; hohes Wiederholungrisiko, bei balancierter Translokation 21/21 nur kranke Kinder! – gleiche Klinik wie freie Trisomie) Mosaik-Trisomie (2,5%; je nach trisom. Zellanteil Down-Syndrom bis hin zu normalem Phänotyp und Intellekt) K: Brachycephalus, nach oben schräge Lidachsen, Epikanthus, kleine Nase, verdickte Zunge (hängt meistens aus Mund heraus, weil zu groß), kleine dysplastische Ohren, kurzer Nacken mit Hautfalten, Vierfingerfurche, Sandalenlücke, muskuläre Hypotonie, Gelenke überstreckbar, Fehlbildungen innerer Organe (Fallot-Tetralogie; AV-Kanal, ASD, VSD, Duodenal-/Analatresie, Pancreas anulare, M.Hirschsprung), Becken charakteristisch (weit ausladende niedrige Darmbeischaufeln, horizontale Hüftgelenkspfannen mit abgeflachten Acetabulum- und Ileumwinkel), Hypothyreose, 10-20x erhötes Leukämierisiko (ALL), geistige Retardierung (IQ 50 und kleiner); Minderwuchs D: Chromosomenanalyse, Suche nach Begleitfehlbildungen T: symptomatisch bei Organfehlbildungen, Physio-, Ergo-, Sprachtherapie, Förderung, medikamentös bei Hypothyreose P: Lebenserwartung (wenn herzgesund) ca. 50% der Normalbevölkerung - Andere numerische Aberrationen, Trisomie 18 (Edwards-Syndrom) und Trisomie 13 (PätauSyndrom) ( ) (66) Fast alle anderen numerischen Aberrationen führen zu spontanaborten, nur bei Trisomie 13 und Trisomie 18 wird ein kleiner Teil der Kinder lebend geboren. Bei beiden Trisomien sterben ca. 90% der Kinder im ersten LJ 4 Stoffwechsel: - Diabetes mellitus (S. 154) (257 ff) Verschied. Typen IDDM (DM Typ I), NIDDM (DM Typ II), Gestationsdiabetes, MODY IDDM – 16/100.000 zwei Gipfel 4.-6. und 9.-15. LJ K: o langsame Entwicklung: Polydipsie/-urie (das meistens nur bei Diabetes mellitus und Diabetes insipidus!), gelegentlich Enuresis, Gewichstabnahme, Müdigkeit und Leistungsschwäche o rasche Entwicklung: Übelkeit, Erbrechen, Azetongeruch der Atemluft, Kussmaulatmung (metab. Azidose), Kopfschmerzen, Exsikkosezeichen, Adbominalbeschwerden („Pseudoappendizitis“); schließlich Bewusstseinsstörungen bis zum tiefen Koma D: Blutzucker, Glukosurie (sichern Diagnose); HbA1c, diabet. Keto-Azidose (BZ > 300 mg/dl, pH < 7.3, BE < -15 mÄqu/l); OGT (1,75g/kg Oligosaccharide Nüchternwert <110mg/dl, nach 60min <160mg/dl, nach 120min <140mg/dl) T: allg.: Blutzuckereinstellung (Diät, Insulin, körperl. Aktivität, Stoffwechselkontrollen); Initialtherapie, je nach Schweregrad der Stoffwechselentgleisung: o wenn keine Ketoazidose, kein Erbrechen, bewusstseinsklar: perorale Hydratation (Tee, Mineralwasser, etc.) und Alt-Insullin in häufigen Einzeldosen bis BZ normal; fettfreie Diät bis keine Ketonurie mehr o diabetische Ketoazidose (mit/ohne Koma) ist lebensbedrohlich (Dehydratation, Elektrolytstörungen, Hyperglykämie, Azidose): Flüssigkeitssubstitution (Defizit + Erhaltungsbedarf = 1500 ml/m² KO + 5-10% des KG): 50% innerhalb 8-12h, Rest bis 36h; bei Schock (RR, Puls , Zenralisation, Oligo-/Anurie) innerhalb 1-2h ausreichend substituieren. Elektrolytsubstitution (Na, K, PO4,). V.a. Kalium sinkt während Therapie (Insulin Gluc + K in Zelle hinein), häufig kontrollieren und als Kaliumphosphat geben). Alt-Insulin bis BZ 250 mg/dl, dann Insulin reduzieren und Gluc-Elektrolyt-Gemisch. BZAbfall max. 100 mg/dl pro Stunde. Bikarbonat-Gabe bei pH > 7.15 nicht nötig!!!; darunter Ausgleich des Basendefizits um ca. 1/3 pro 2 Stunden. o Langzeitbehandlung: 3 Säulen: Insulin, Diät, Stoffwechselkontrolle Insulin Alt-Insulin (=reguläres I., Normalinsulin): WiDauer/Wi-Maximum: 6h / 2-4h Verzögerungsinsulin: - / 36 h Mischinsulin (Alt- und VZ-Insulin) - / 8-12 h Verteilung der Injektionen auf 3-5 / Tag (gleichm. BZ-Spiegel, keine Spitzen) Konventionelle Basis-Bolus-Therapie: morgens, abends Depotinsulin, Bedarfsinsulin nach BZ Intensivierte Therapie: auf Mahlzeiten bezogene Injektionen Bei Erkrankungen ca. 20% mehr Insulin nötig. Diät Rasch resorbierbare KH ganz vermeiden KH:Fett:EW: 55% : 30% : 15% Ca. 6-7 Mahlzeiten pro Tag (gleichförmige Resorption der KH) „strenge Diät“: gesamte Nahrungszufuhr (KH, Fette, EW) in Menge und Verteilung festgelegt „kalorienfixierte Diät“: Menge von Fett und EW berechnet, aber nur die erteilung KH für jede Mahlzeit festgelegt Stoffwechselkontrolle BZ-Bestimmung, Glucosurie-Bestimmung: regelmäßig vom Patienten; Ziel: Normoglykämie; HbA1C (7 ± 1%); regelmäßige und wiederkehrende Schulung K: Hypoglykämie (treten schnell ein; Bewusstlosigkeit, Krämpfe, feuchte, schweissige Haut) Glukose (z.B. Saft, Traubenzucker und Brot); Hyperglykämie / Coma diabeticum (treten über Stunden/Tage auf; s. oben) 5 Weitere Kompl.: diabet. Nephropathie ( NI), Hypertonie, Retinopathie, Neuropathie, Arteriosklerose, etc. P: bei guter Stoffwechselführung ist Lebenserwartung und Lebensqualität nicht nennenswert eingeschränkt - Nesidioblastose (S. 161) Sporadische primäre Inselzellhypertrophie: Hypoglykämie der in den ersten Lebenstagen (möglicherweise Inselzelladenome) K: Hypoglykämie ohne Ketoazidose T: Glukoseinfusion, Diazoxid, bei Therapieresistenz 7/8-Resektion des Pankreas - Hyperammonämie (OCT Mangel) (S. 190) (266) Seltene Störung der Amoniakentgiftung (x-chromosomal, alle anderen bekanten Störungen der Amoniakentgiftung autosomal rezessiv) Typ I: Carbamylphosphat-Synthetase-Mangel (autosomal rezessiv) Typ II: Ornithintranscarbamylase-Mangel (OCT-Mangel) (x-chromosomal) D: Hauptkriterium: mehr oder weniger starke Ammoniakerhöhung im Blut (bei Bestimmung der Zwischenprodukte des HS-Zyklus genaue funktionelle Lokalisation des Defekts möglich); Amminosäuren im Blut/Urin bestimmen (bei jedem psychomotorischen Entwicklungsrückstand) K: schwache Erhöhung: Erbrechen, Trinkunlust, rezidivierende Bewusstseinsstörungen, neurologische Symptome (Muskelhypotonie, Hyperreflexie), Hepatomegalie, psychomot. Entwicklungsrückstand starke Erhöhung: zerebrale Symptomatik: Krämpfe (bei fast allen Stoffwechselkrankheiten!!), Muskelhypotonie, Koma und Tod chronisch: mentale Retardierung, Mikrozephalie, Spastizität T: Stop der Proteinzufuhr, hochkalorische Infusion, L-Karnitin, evtl. Dialyse Endokrinologie : - Pubertas praecox vera (S . 213) (255) Pubertätsentwicklung bei Knaben vor dem 9. LJ, bei Mädchen vor ca. 8. LJ durch hypothalamische/hypophysäre Aktivierung in physiologischer Weise; 1:10000-1:5000 (Mädchen 5x häufiger) Voraussetzung für eine Pubertas praecox ist das Auftreten pubertärer GnRH Spiegel (physiologische Stimulation) im Ggs. zur Pseudopubertas praecox, bei der die Pubertätsmerkmale ohne initialen GnRH Anstieg auftreten (Pubertätseinleitung unphysiologisch durch anderweitige Stimulation als durch GnRH; GnRH ist dabei supprimiert un dim GnRH-Test lassen sich FSH/LH nicht stimulieren) Ursachen: idiopatisch, symptomatisch (Hirntumor, AGS, Hirnmissbildungen, Neurofibromatose, tuberöse Sklerose, unbehandelte Hypothyreose, Traumen, Hydrozephalus); Jungs: meistens ZNSTumore, dann idiopathisch; Mädels: meistens idiopathisch, dann ZNS-Tumore! K: Pubertätsentwicklung in richtiger Reihenfolge vor dem 6.-8./9. LJ D: LH, FSH (pulsatile Sekretion) im LH-RH-(=GnRH-)Test stimulierbar T: LH-RH-Analoga (= GnRH-Analoga) bis ca. 10-11 Jahren Verlangsamung der Skelettreifung und Erreichen einer normalen Endgröße, spontane pubertäre Entwicklung nach Therapieende verläuft normal - Kleinwuchs / Minderwuchs (S. 224) Körpergröße unter 3. Perzentile, proportiniert/unproportioniert, primär/sekundär, viele verschiedene Ursachen (mittlere Zielgröße: [(GrVater + GrMutter) / 2] +/- 6.5 cm (+ für Jungen, für Mädchen). Ursachen: 6 1. Konstitutionelle Entwicklungsverzögerung, fam. Minderwuchs; Merkmale: - Normale Körperproportionen - Keine organischen und hormonellen Abweichungen - Knochenalter = Lebensalter - Pubertät tritt zeitgemäß ein 2. Mangel an Aufbaustoffen Hypokalorischer M. Psychosozialer M. Intstinaler M. 3. Hormonale und neurale Störungen Hypothalamus und Hypophyse: z.B. Wachstumshormonmangel; echte Pub. praecox Thyreoidea: Hypothyreose Pankreas: schlecht kontroll. Diabetes mellitus NN: Cushing-Syndrom, AGS (Minderwuchs nach Epiphysenfugenschluss) Gonaden: Pseudopubertas praecox bei Gonadentumoren 4. Nichthormonale Störungen Renaler M.: Mißbildungen, Fanoni-Syndr., chron. Nephritis, renaler Diab. insip. Intestinaler M.: Zöliakie, Pankreasfibrose, Megacolon Hepatischer M.: Glykogenose, Zirrhose Anoxämischer M.: Vitium cordis congenitum, Bronchiektasen, chron. Anämie Rachitischer M.: Vit. D Mangel und Vit. D resistente Rachitis Lipoidosen: M. Gaucher, M. Nieman-Pick, Hand-Schüller-Christian, PfaundlerHurler 5. Mangelhafte Wachstumspotenz der Knochen Primordialer M. und Progerie Kongenitale Skelettstörungen: Chondrodystrophie, Dysostosen (D. multiplex Pfaundler-Hurler, D. enchondralis Morquio, D. cleidocranialis usw.) Erworbene Skelettstörungen: Rachitis, Hyperparathyreoidismus Kongenitale Dysmorphiesyndrome: Turner-Syndrom, Pseudohypoparathyreoidismus, autosomale Chromosomen-Störungen (Down-Syndrom), Prader-Willi-Syndrom D: Grundsätzlich: Elterngröße, Geburtsmaße! – dann: Perzentilenkurven, Zielgröße, Pubertätsstadien, Proportionen, chron. Erkrankungen?, Medikamente?, Labor (Glukose, Leber, Schilddrüse, Entzündungsparamter, Cortisol, Wachstumshomone – Stimulationstests), Urin (pH, Glukose, Eiweiß), Knochenalter (ist Standard bei Minderwuchs!; durch Rö linke Hand bestimmt); bei dysproportionierten Minderwuchs: Rö des Skeletts: Becken, WS; Chromosomenanalyse bei ungeklärtem Minderwuchs WH-Mangel kann ausgeschlossen werden wenn IGF-1 (Somatomedin C/insulin like growth factor) bzw. IGFBP-3 (IGF-Binding Protein) normal T: je nach Ursache, GH-Mangel: Hormonsubstitution Gastroenterologie: - Stomatitis (S. 235) (294, 592) Entzündung der Mundschleimhaut; siehe unten + Steven-Johnson-Syndrom Viral: o Gingivostomatitis herpetica (Stomatitis aphthosa, Mundfäule): Erstinfektion mit Herpesvirus; weiße Ulcera mit weiß-gelblichen Pseudomembranen bedeckt; Lokalisation: meist Gingiva, Lippen, Wangenschleimhaut; K: starke Schmerzen bei Nahrungsaufnahme, allg. Abgeschlagenheit, Fieber, submandibul. Lymphadenitis, Foetor ex ore; P: Verlauf meistens selbstlimitiert/abortiv o Herpangina: Coxsackie-A-Virus; Vorschulkinder im Sommer/Herbst (z.T. kleine Epidemien); K: polötzlich hohes Fieber (um 40°C) und starkes Krankheitsgefühl, dann Halsschmerzen und Auftreten von 2-3 mm großen Bläschen an Gaumenbögen und (weichen 7 - - & harten) Gaumen. Nach Ulzeration Fibrinbelag mit rotem Randsaum. P: Symptome und Beschwerden klingen in wenigen Tagen ab; häufig abortiver Verlauf o Varizellenstomatitis: aphtenähnliche flache Ulzera mit hyperämischem Hof v.a. in hintere Abschnitte der Mundhöhle o Infektiöse Mononukleose: (EBV-Viren aus der Herpesvirengruppe): Neben Monozytenangina oft Petechien im Bereich des weichen Gaumens, selten auch Gingivitis Therapie der viralen S.: meist symptomatisch: Mundpflege, Mundspülung und Gurgeln mit Kamillen-Lösung, Salbei-Lösung, Panthenol-Lösung und Sulcrabest (Schleimhautprotektivum); bei starken Schmerzen auch lokalanästhetische Mittel Bakteriell: o Pyogene Gingivitis: chronisch bei ausgedehnter Karies und ungenügender Mundpflege oder schlechter Abwehrlage des Körpers; ganze Mundschleimhaut kann betroffen sein Therapie: Mundpflege, Gebisssanierung, Mundspülungen; Vitaminpräparate. Anästhesie der Mundschleimhaut vor dem Essen; chronische Mundatmung kann Zustand verschlimmern; ist zu verhindern (Adenektomie, Tonsillektomie, Sinusitistherapie) o Gingivostomatitis ulcerosa, Stomatitis ulcerogangränosa, Gingivostomatitis ulceromembranacea (Angina Plaut Vincent): Heranwachsende und Jugendliche; durch schlechte Mundhygiene oder konsummierende Erkrankungen; Ulzera der Gingiva und Mundschleimhaut, mit Pseudomembranen bedeckt (bei Angina Plaut-Vincenti (akute nekrotisierende Gingivostomatitis) zusätzlich oberer Tonsillenpol); lokale oder allgemeine Beschwerden, auch symptomarm; Spirillen und fusiforme Stäbchen besiedeln die Ulzera Therapie: der evtl. bestehenden Grundleiden, Mundspülungen mit Dreierlösung (Benzocain+Panthenol+Perkamillon) oder verdünnter Wasserstoffperoxidlösung; Oberflächenanästhesie und Ätzen mit Silbernitratlösung; bei ausgedehntem Befall Antibiotika Mykotisch o Candidiasis, Soor: Candida albicans; weiße, mit Spatel abstreifbare flächige oder punktförmige Beläge; auf Mundschleimhaut oder weichem Gaumen, teilweise bis in Ösophagus; bes. bei schwächenden Allgemeinerkrankungen oder längerer antibiotischer Therapie; beim NG durch Besiedlung der mütterlichen Scheide Therapie: lokal antimykotisch; bei ausgedehntem Soor und schlechter Abwehrlage systemische Antimykose (z.B. Fluconazol) (Kinder > 16 J.) ; Kinder < 16 nur wenn keine therpeut. Alternative; Säuglinge: kontraindiziert K: allg: Fieber, Entzündung, geschwollene LK´s, Bläschen D: Klinik, evtl. Erregernachweis, Soor-Abstrich T: Grundkrankheit (Soor: Nystatin, Stevens-Johnson: Prednison), Mundspülungen - Laugenverätzungen (S.242) Koliquationsnekrose (gefährlicher als Koagulationsnekrose), Perforationsgefahr K: Verätzungen der Mundschleimhaut, retrosternale Schmerzen; Kind schreit, spuckt, speichelt, hustet 3 Grade der Verätzung: Grad I: nur Erythem und/oder Ödem der Schleimhaut Grad II: Schleimhautläsionen mit Ulzerationen und Nekrosen, nicht gesamte Zirkumferenz Grad III: Schleimhautläsionen mit Ulzerationen und Nekrosen, gesamte Zirkumferenz T: kein Erbrechen auslösen, mit Wasser spülen; Ösophago-Gastro-Duodenoskopie in Vollnarkose o Grad I: stationär bis Brei und klare Flüsigkeit vertragen; Antibiotika und Nachuntersuchungen nicht nötig o Grad II & III: komplette Nahrungskarenz (parenterale Ernährung), Breitbandantibiotikum, analgetische Therapie obligat; Prednisolon empfohlen; alle paar Monate Kontrolle (Strikturen) Allgemein, unabh. vom Grad: bei Kehlkopfschwellungen etc. Kortikosteroide; Ranitidin o.ä. gegen Säurereflux 8 K: früh: Perforation (Mediastinitis, Fisteln zur Trachea oder Aorta, Aspiration, Schock; spät: narbige Stenose ( Risiko für Ösophagus-Ca bei Narben), Bradyösophagus und sek. Hiatushernie und Sphinkterinsuff. - Akute Gastroenteritis (S. 263) (162) Infektiöse Enterokolitis durch Viren (90% Rota, Adeno, Echo, Entero...) Bakterien (Salmonella, E. coli, Campylobacter...) Protozoen (Entamoeba histolytika...) Enteropathogene Keime Enterotoxine Zerstörung der Mukosabarriere oder Störung der normalen Darmwandfunktion EPEC & ETEC: wäßrige Durchfälle EIEC: Durchfall + Dysenterie (blutige Durchfälle) EHEC: wäßrige, teilweise blutige Durchfälle & Hämolytisch Urämisches Syndrom (HUS) mögl. chron. Durchfälle (sekretorische und osmotische Diarrhoe, siehe entsprechendes Kapitel), Dystrophie (Nahrungsaufnahme gestört, Hyperperistaltik), Immunreaktionen (Kuhmilchintoleranz...) K: meist unabh. von Ursache: meist vorher Verhaltensändrung des Kindes; Blässe, Bauchschmerzen, Inappetenz, Erbrechen, Diarrhoe; bei chron. Geschehen: Exikose, Azidose, seltener Lidschlag, halonierte Augen bei Exsikkose, schwacher Saugreflex T: symptomatisch – Flüssigkeitssubstitution & Elektrolytausgleich sehr wichtig; dann Realimentation; evtl. Antiemetika, Antibiotika (gezielt nach Erregernachweis) (Trimethoprim/Sulfamethoxazol, Ampicillin, verschiedene Cephalosporine; auch Colistin und Polymyxin B) Rehydratation: in den ersten 6-12 h: geschätzte verlorene Flüssigkeitsmenge geben (Glucose-SalzTee oder kommerzielle Rehydratationslösungen) – oft ca. 50-100 ml/kg KG restliche 12-18 Stunden: täglichen Basisbedarf (Erhaltungsbedarf) 100 – 120 ml/kg/ KG am Tag ca. 4 ml/kg/ KG in der Stunde - kleine Einzelportionen da sonst leicht Erbrechen ältere Säuglinge leichte Kost möglich nach ca. 4 -6 Tagen sollte schon gewohnte Normalkost möglich sein Infusion: wenn wiederholtes Erbrechen oder keine Gewichtszunahme bei oraler Rehydratation Colitis ulcerosa (S. 271) (205) Chron. Entzündung von Mucosa/Submukosa, auf Kolon beschränkt, kontinuierlicher Befall; Kryptenabszesse, Pseudopolypen, “starres Rohr”, toxisches Megakolon, Perforation, Karzinomrisiko 2-4/100.000, davon 1/3 vor 18. LJ; Durchschnittsalter bei Diagnose: 12 J. K: Tenesmen, blutige Durchfälle/Blutauflagerungen auf Stuhl (in 100%), extraintest. Manifestationen (Sklerosiernede Cholangitis, Erythema nodosum...) (Fieber, Gewichtsverlust, Wachstumsverzögerung wie beim M. Crohn eher selten!) D: Klinik, Koloskopie (erosive oder ulzerative Entzündung, meist Rektosigmoid), Histologie, RöDoppelkontrastdarstellung; Lab: Eisenmangelanämie, Thrombozytose, leicht BSG; Sono: verdickte Colonwand (> 3-4 mm) T: 5-Aminosalizylsäure, Predinisolon bei schwerem Befall, Immunsuppressiva, Proktokolektomie Kompl.: schwere Blutungen, Infektionen, paralytischer Ileus bei toxischem Megacolon; bei chronischer Pankolitis (> 10 Jahre) Gefahr der Karzinomentstehung 9 DD: M. Crohn, akute Enterokolitis, Purpura Schönlein-Henoch, Pseudomembranöse Kolitis, Vaskulitiden mit enteraler Manifestation, P: Heilungen möglich; manchmal Proktocolektomie die einzige Möglichkeit (ist dann auch kurativ); nach > 10 Jahren Erkrankung Ca-Risiko erhöht - M. Crohn (S. 273) (200) Granulomatöse Entzündung aller Darmschichten (bis Mesenterialansatz), gesamter GI-Trakt, segmentaler Befall; Fistelneigung, Stenosen, Ulzerationen, entzündliche Konglomerattumoren 6-10/100.000 durchschnittliche Manifestation 10.-12. LJ; Durschschnittsalter bei ED: 11 Jahre K: erst oft monate- bis jahrelange Phase mit uncharakter. Symptomen: Bauchschmerzen, Appetitlosigkeit, Wachstumsverzögerung (Größe weniger als Gewicht), Durchfall; Anale Läsionen und Blut im Stuhl; extraintest. Manifestationen: Uveitis, Arthritis, Erythema nodosum, Pyoderma gangraenosum, Psoriasis D: Labor (Anämie, Leukos & Thrombos , α2-Globulin , Vit. B12 und FS; Entzündungsparameter Aktivitätsgrad; aber klinisch-chemisch nicht zu beweisen), Endoskopie, Sono (Wandverdickung), Biopsie (Beweis), Radiologie (Stenosen, Fisteln, Pflastersteinrelief, Wandstarre) Kompl.: Wachstumsretardierung (häufigste K.), Fisteln (perianale, rektovaginale, enteroenterale), Abszesse, Perforationen, Stenosen; Osteopathie, Nephrolithiasis T: 5-ASA, Salazosulfapyridin, Immunsuppression & Steroide (Prednisolon, Azathioprin) erst in zweiter Linie, Metronidazol bei Fisteln, evtl. OP (Stenosen, Fisteln; aber keine Heilung dadurch) DD: jRA, M. Bechterew, Psoriasis, endokrine Pubertas tarda, Anorexia nervosa, intestinale Tuberkulose, Lymphadenitis mesenterica, Colitis ulcerosa V/P: Sono zur Verlaufsbeurteilung, (Endosk. und Radiol. wichtig bei Indikationsstellung für OP); im Kindesalter meist akuter als beim Erwachsenen; nach Pubertät ruhigerer Verlauf; je nach Kompl. Lebensqualität eingeschränkt - Cholestase (S. 280) (220) Störung von Bildung, Sekretion und Transport der Galle mit Anhäufung von Gallenbestandteilen im Blut und Defizit im Darm Einteilung: obstruktiv (intra- oder extrahepatisch) o Cholelithiasis o Kompression der Gallenwege o Cholangitis / Cholangiolitis o Pankreaserkrankungen o Gallengangsatresie / -hypoplasie / -dysplasie o Choledochuszyste hepatozellulär o Hepatitis (viral / Medikamentös) o Toxisch (Medikamente, Enterotoxine, Pilzgifte) o Angeborene Stoffwechselerkrankungen Fruktoseintoleranz Galaktosämie Tyrosinose α1-Antitrypsinmangel Mukoviszidose Morbus Wilson auch: Rotor-Syndrom (chronische Hyperbilirubinämie ohne Hämolyse, selten), DubinJohnson-Syndrom (v.a. konj. Hyperbilirubinämie ohne Hämolyse, selten) Gallestau mit Zerstörung von Hepatozyten, Zirrhose K: Ikterus der Haut und Skleren, Braunfärbung des Urins, variabel entfärbte Stühle (acholische Stühle), Juckreiz, Leberfunktionsstörungen 10 D: Bilirubin (direkt, indirekt), Gallensäuren, alk. Phosphatase, γ-GT (empflindlichster Cholestaseparameter), Leberfunktion, Stoffwechselscreening, Sono, Biopsie, Lebersequenzszinti T: je nach Grunderkrankung, Diät bei Stoffwechseldefekten, OP bei extrahepat. Abflussstörung, Leber-TX, symptomatisch: Urocholsäure, Antihistaminika, Cholestyramin (gegen GS-bedingten Pruritus), Substitution der fettl. Vitamine! DD: Glukuronidierungsstörungen, hämolytische Phänomene - Mukoviszidose (S. 284) (176) Aut. rez. Erkrankung mit Dysfunktion der exokrinen schleimproduzierenden Drüsen (sondern hochvisköses eiweißhaltiges Sekret ab, das sie Ausführungsgänge verstopft und sekundär zu progredienten zystisch-fibrotischen Gewebsveränderungen führt; Defekt in Cl-- und Na+-TransportKanälen); 1:3000 Neugeborene (häufigste Erbkrankheit der weißen Rasse) Folgen: Pankreas: Fibrose und zunehmende exokrine Pankreasinsuff. mit Maldigestion Lunge: Einengung und Obstruktion der Bronchioli, manchmal auch der Bronchi; obstruktives Emphysem; chronische Bronchitis, Bronchopneumonie, Bronchiektasie Die anderen rein serösen Drüsen zeigen nur eine abnorme Zusammensetzung ihres Sekrets: Schweißtest (Na+ und Cl- Gehalt pathologisch) K: im Frühstadium sehr variabel; Mekoniumileus des NG (in 10%); intestinale Sx und Lungenveränderungen; exokrine Pankreasinsuff. (hartnäckige Durchfälle, fettig glänzender und übelriechender Stuhl) Wachstumsverzögerung, Hypoproteinämie, Anämie; dünne Extremitäten, aufgetriebenes Abdomen pulmonale Komplikationen: anfangs chronische Bronchitis und rezidiv. Bronchopneumonien, später schweres obstr. Emphysem und Cor pulmonale; dann Dyspnoe, Zyanose, eitriges zähes Sputum; Faßthorax, Kyphose der BWS, Trommelschlegelfinger und Uhrglasnägel (alles sekundär); Salzhunger (wegen des hohen Verlusts) und chronische Pan-Sinusitis D: Schweißtest (Na+ u/o Cl- Konz. im Schweiß); Gen-Analyse (ΔF 508 Mutation in 70%, viele andere auch); Screening durch Mekoniumtest ( Albuminkonz.); IRT-Test (immunreaktives Trypsin im Blut; Werte bei NG mit CF); Thorax-Rö (typische Veränderungen: Faßthorax, Emphysem, Bronchiektasien); Chymotrypsin und Pankreas-Elastase im Stuhl, Pankreasenzyme im Duodenalsaft; Pränatale Diagnose durch Chorionzottenbiopsie (8.-12.- SSW; DNA-Analyse, wenn bereits ein CF Kind in der Familie; sog. „Indexfall“) K.: intestinale Obstruktionen, Invagination, Rektumprolaps, biliäre Abflußbehinderung ( Leberzirrhose); Hypovitaminosen (ADEK); Pneumothorax; Sterilität bei männlichen Patienten DD: Malabsorptions- und Magdigestionssyndrome anderer Ursache, chron. Lungenerkrankungen anderer Genese; Shwachmann-Syndrom: exokrine Pankreasinsuff., Neutropenie, Minderwuchs, gelegentlich metaphysäre Dysostosen T: keine kausale Tx; symptomatisch: hochkalorische Diät, Vitaminsubstitution (ADEK), reichlich Kochsalz, Pankreasenzyme zu den Mahlzeiten, Physiotherapie (Atemgymnastik), Mukolytika, Antibiotika; körperliches Training; später Sauerstoffgabe und Cor pulmonale behandeln; Präfinalstadium: Herz-Lungen-Transplantation P: bestimmt durch Lungen-Koplikation und Cor pulmonale, 80% erreichen Erw.-Alter Luftwege: - Croup-Syndrom () (426) Kleinkindtypische Manifestation der Laryngitis und Tracheitis, die aufgrund der kleinen anatomischen Verhältnisse schell zur Obstruktion führt. K: inspiratorischer Stridor und Dyspnoe, je nach Ursache auch bellender laryngotrachealer Husten (bei Pseudocroup) Folgende 4 Krankheitsbilder: o subglottische stenosierende Laryngitis: Infektcroup (früher: Pseudocroup) o supraglottische stenosierende Laryngitis: (akute phlegmonöse) Epiglottitis 11 o Kehlkopfdiphterie: „echter“ Croup o eitrige Tracheobronchitis DD (inspiratorischer Stridor): obstruierende Tonsillitis; wenn afebril: Fremdkörperspiration, Glottisödem (Nahrungsmittelallergie, Insektenstich), Laryngospasmus - Subglottische Laryngitis / Pseudocroup / Infektcroup (S. 301) (304,426) Stenosierende subglottische Laryngitis durch RS-, Parainfluenza-, Influenzaviren v.a. zwischen 1.-3. LJ, auftreten Nachts im Herbst, Spätwinter K: Stadium 1: bellender Husten (Krupphusten) oder Schluckbeschwerden; Heiserkeit 2: inspirat. Stridor, Einziehungen im Jugulum, Epigastrium 3: Stridor, intercost. Einziehungen, Atemnot, Tachykardie, Hautblässe, Angst, Unruhe 4: Stridor, höchste Atemnot, Zyanose, Sopor, Puls klein, frequent, max. Einziehungen D: Klinik: bellender Husten, inspir. Stridor, T: Stadium I: Fiebersenkung (Wadenwickel, Paracetamol/ASS); kühle feuchte Luft (manche reagieren darauf mit Symptomverschlechterung!; Aufsicht!); evtl. Sekretolyse; Corticoide in hoher Dosis (Prednis(ol)on; Solu-Decortin H (SDH)); leichte Sedierung Stadium II: stationär: Adrenalin-Inhalation + evtl. Sauerstoff Stadium III & IV: Intensivstation mit Intubationsbereitschaft; H2O und E-lyte; Azidoseausgleich; Antibiotikum (wg. möglicher maligner Laryngotracheitis, Epiglottitis) (Amoxi 50 mg/kg p.o., Ampi 100 mg/kg i.v., Cefotaxim 150 mg/kg i.v.)) DD: akute Epiglottititis - Zwerchfellhernie (S. 306) (129, 131) Meist posterolateral li mit Verlagerung von Abdominalorganen in den Thorax 1/2.000, Mädchen 2x häufiger; links 4-8x häufiger als rechts K: meist sofort nach Geburt Atemnot, niedrige O2-Sättigung, fehlendes Atemgeräusch, Darmgeräusche über Thorax; seltener entwickelt sich die ZFH erst in den ersten Tagen p.p. D: Röntgen (Bauchorgane im Thorax, Mediastinalverlagerung); DG über Thorax T: KEINE Maskenbeatmung (Luft geht in Magen und Darm), sofortige Intubation, Magensonde (Luft aus Magen herausbekommen und Verdrängung der Lunge vermindern), OP K: Lungenhypoplasie, Letalität bis 50% - Obstrukt. Bronchitis (S. 309) (436) Virale (RSV, Adeno-Viren, Rhino-Viren) Infektion der Atemwege mit Schleimhautödem / Hypersekretion bei Säuglingen und Kleinkindern K: expiratorisches Giemen, Brummen, hypersonorer Klopfschall, evtl. Atemnot und respiratorische Insuffizienz D: Giemen, Brummen, Röntgen (Überblähung/Zwerchfelltiefstand, verstärkte Bronchialzeichnung) T: symptomatisch: Sekretolyse (oral, inhal.), β2-Mimetikum, Parasympatholytikum, Theophyllin, evtl. Kortikoide; akute Exazerbationen: Prednison 3-5 Tage in rasch abfallender Dosis (1-2 mg/kg tgl. initial); bei Infektion: Antibiotika 10-14 Tage; primäre Antibiose nicht sinnvoll da viral! DD: Asthma (bei >3 Episoden), Fremdkörperaspiration (akutes Geschehen), laryngotrachela Obstruktion (bellender Husten und inspir. Dyspnoe) Im akuten Stadium folgenermaßen vorgehen: - inhalativ Sotalol und/oder Atrovent - O2-Gabe und feuchte kalte Luft - Absaugen wenn sehr verschleimt - Blutabnahme: Hb, Hkt, BB, Na+, K+, Cl- (evtl.) Fieber senken (Paracetamol, Ibuprofen) - Bei behinderter Nasenatmung: Nasivin, Otriven - Bei ungenügendem Ansprechen: Theophyllin i.v. P: Übergang in Asthma bei entspechender atopischer Disposition (IgE oder Familienanamnese) 12 - Asthma (S. 311) (439) Überempfindlichkeit der Bronchialschleimhaut mit anfallsweiser (reversibler) Atemwegsobstruktion Inzidenz 10%, im Kindesalter vorwiegend allergische Ursache Allergen Mastzelldegranulation Histamin/Entzündungsmediatoren Sofortreaktion (Bronchospasmus, Schleimhaut-Ödem, Dyskrinie/Hypersekretion), Spätreaktion (Entzündung durch Granulozyten) K: Anfall: akut einsetzende Atemnot, Angstgefühl, Schweißausbruch; verlängertes Expirium und Einsatz der Atemhilfsmuskulatur. Giemen, Pfeifen und Brummen bei der Auskultation, hypersonorer Klopfschall (Überblähung); bei hochgradiger Obstruktion „stumme Lunge“ (stark verminderte Ventilation) D: Klinik, Anamnese (Anfallshäufigkeit, Auslösefaktoren), LuFu (erhöhter Widerstand in den Atemwegen und Reversibilität der Obstruktion (im ggs. zu COPD)), Röntgen, Prick-Test/RAST, Provokationstest (siehe auch Allergiediagnostik weiter unten); bei Infektion Abklärung dieser (Art der Infektion und Lokalisation (Sinusitis, Bronchopneumonie)) Allergiediagnostik bei Asthma bronchiale 1. Anamnese (saisonal, perennial usw., Auslöser) 2. Hauttest auf Allergene 3. ggf. IgE im Serum quantitativ 4. ggf. allergenspezifische IgE-AK (z.B. mittels Radioallergosorbent-Test (RAST) 5. Nachweis der akuten Pathogenität eines Allergens durch bronchialen Provokationstest T: 3 Ziele: Dämpfen der bronchialen Hyperreagibilität, Prophylaxe & Senken der Anfallshäufigkeit und Vorbeugen von Spätschäden, z.B. Cor pulmonale) Prophylaxe: Dämpfen der Entzündung: Vorbeugen eines Anfalls Nedocromil, Chromoglycinsäure (DNCG) Glokokortikoide (lokal/systemisch) und Antileukotriene β2-Sympathomimetikum, Theophyllin, Anticholinergikum Stufentherapie: allgemein: Allergenkarenz, meiden unspezifischer Reize (Rauch & Staub, keine Tiere); ggf. Hyposensibilisierung I. (<6 Anfälle/Jahr) inhalativ β2-Mimetikum bei Bedarf II. (>6 Anfälle/Jahr, 1x/Woche, 2x/Monat nachts) DNCG oder Nedocromil zur Prophylaxe, bei Bedarf β2-Mimetikum; wenn nicht genug: inhalative Steroide III. (>1x/Woche, >2x/Monat nachts) inhalative Steroide + langwirksames inhal. β2-Mimetikum/Theophyllin; evtl. Anticholinergika (Atropin); bei Bedarf: kurzwirksames β2-Mimetikum, notfalls Prednis(ol)on oral; möglichst alternierend IV. (ständig Symptome) inhalative, orale Steroide + β2Mimetikum/Theophyllin; bei Bedarf: kurzwirksames β2-Mimetikum, notfalls Prednis(ol)on oral; möglichst alternierend Vorgehen beim schweren Asthmaanfall: (O2 B2 Pred Theo) o geeignete Körperposition mit Entlastung des Brustkorbs; O2 Gabe o schnellwirksames β2-Mimetikum inhalativ über „Inhalationshilfe“/Vernebler (Albuterol, Terbutalin, Fenoterol, etc., kein Salmeterol – zu langsamer Wirkungseintritt) o Prednison 6-8 mg/kg i.v. (nicht inhalativ) o Theophyllin langsam 5-6 mg/kg i.v. o evtl. Terbutalin s.c. o wenn kein genügender Erfolg: Intensivtherapie (Sedierung (Ketamin, Propofol), Intubation und Beatmung) DD: Fremdkörperaspiratin, CF, obstruktive Bronchitis - Pneumonien (S. 317) (442 ff) 13 Entzündungen des Lungenparenchyms, verschl. Einteilungen nach Ätiologie (bakteriell/viral), Morphologie (Lobär-, Broncho-, interstitielle, atypische Pneumonie, eosinophile Pneumonie, primär abszedierende Pneumonie, Aspirationspneumonie), Klinik, Alter: Neugeborene: B-Streptokokken, gram.neg. Keime, Staph. aureus Säuglinge: Viren, gram.neg. Keime, Pneumokokken, Staph. aureus Kleinkinder: Viren, Pneumokokken, Haemophilus Schulkinder: Mykoplasmen/Chlamydien, Viren, Pneumokokken K: Allgemeinzustand , Fieber, Husten, Dyspnoe (mit Nasenflügelatmung), Tachypnoe, periorale Zyanose, atemabhängige Schmerzen, geblähtes Abdomen, Bauschmerzen; sehr diskreter Befund bei hilusnahen Prozessen (zentrale Pneumonie) D: Klinik, Auskultation, Röntgen, Astrup (resp. Insuff); diff. BB (Ätiologie); wichtig: Impfstatus, Tuberkulintestung; Mikrobiologie T: Maßnahmen, je nach Schwere der Erkrankung, z.B.: Bettruhe, ausreichende Flüssigkeitszufuhr, Expektorantien, Frischluft, leichte physiotherapeutische Maßnahmen, Antipyretika, O2-Gabe bei Hypoxie; Antitussivum bei trockenem Reizhusten und (schmerzhafter) Begleitpleuritis; Rückenlagerung bei Säuglingen (wegen der physiologischen Bauchatmung, die bei Bauchlagerung erschwert wird; schlechte Atmung – schlechte Heilung!) o viral: meist nur symptomatisch; wenn Ätiologie unklar oder erneuter Fieberanstieg: Antibiotika (evtl. bakterielle Superinfektion) o bakteriell: Antibiose nach erwartetem Keim; später Modifizieren nach Antibiogramm o Neugeborene: Ampicillin+Aminoglykosid oder + Cephalosporin 3. Gen. o Säuglinge (2-12 Wochen): Ampicillin+Aminoglycosid+Oxacillin o Kleinkinder (2-6 J): Cephalosporin 2. Generation o Schulkinder: Makrolide, >9 Jahre Doxycyclin o Mykoplasmen/Chlamydien: Erythromycin Tetrazyklin bei älteren Kindern o Pneumozystis carinii bei Zytostatika-Therapie oder AIDS Cotrimoxazol hochdosiert; dennoch hohe Letalität o Bei Säuglingen <6 Monate i.v., darüber in der Regel orale Therapie 7-10 Tage - TBC (S. 656) Chron. Granulomatöse Entzündung durch säurefeste Stäbchen (M. tuberculosis), Tröpfcheninfektion, Hauptmanifestation Lunge; Primärkomplex aus Lungenrundherd und LK kann Jahre latent persistieren und bei Immunsuppression erneut exazerbieren/kanalikulär, hämatogen streuen Weltweit eine der häufigsten Todesursachen, in Deutschland v.a. ausländische Kinder K: Primär-Tbc: Infektion, dann Inkubationszeit 2-8 Wochen, dann Bildung des Primärkomplexes (fast immer nur allg. Sx); nur Konversion nach tuberkulinpositiv einziger Hinweis unspez. Symptome: Appetitlosigkeit, Gewichtsverlust, Nachtschweiß, Müdigkeit, Fieber, evtl. Erythema nodosum; für 1-3 Wochen nach Infektion; NB: Husten ist bei Kinder-Tbc selten selten ausgedehnte Entzündungsreaktion um Primärherd (pneumonische oder obstruktive Erscheinung) Kleinkinder: unklares Fieber, Appetitlosigkeit, Abgschlagenheit, Gewichtsverlust Dann oft Ausheilung mit/ohne Verkalkung LK-Tbc (cervicale, indolente LK-Schwellung) – hierzulande selten Pleuritis tubercul. (Erguss, Husten, Schmerzen) Miliartuberkulose mit Organbeteiligung Hämtogene Streuung aus dem Primärkomplex 14 3-6 Monate nach Primärinfektion zu befürchten bei Säuglingen und Kleinkindern Fieber, allg. Krankheitszeichen Betroffene Organe: Lunge, Auge, Leber, Gehirn; Knochen, Gelenke, Otitis media; Nieren- und Haut-Tbc allerdings erst nach Jahren Meningitis tuberculosa V.a. bei Kleinkindern Oft nach miliärer Streuung; oft letal Stimmungs- und Wesensveränderungen, Irritabilität oder Apathie; Kopfschmerzen, Erbrechen Nach 1-3 Wochen: Fieber, Meningismus, Krämpfe, Hirnnervenausfälle, Hirnödem, Bewusstseinsstörung, Tremor, Athetose, Opisthotonus Schließlich Koma und Tod (wenn keine Behandlung) In fortgeschrittenem Stadium hohe Letalität; oft bleiben neurologische Ausfälle Liquorbefund: Pleozytose (vorw. Lymphozyten), Eiweiß , Glucose ; Nachweis der Bakterien im Spinngewebsgerinnsel des abgestandenen Liquors abdominelle, Urogenital-, Skelett-Tbc D: Anamnese, Hauttest intracutan – nach Mendel-Mantoux mit gereinigtem Tuberkulin; intrakutane Injektion von Tuberkulin, ablesen nach 72h positiv bei Induration >10mm (>5mm bei Risikogruppe); Röntgen (Lungenrundherd, Verkalkungen), Lumbalpunktion bei ZNSBeteiligung, Urin („sterile“ Leukozyturie); Mikrobiologie: Anzucht aus Sputum; Bactec; PCR T: immer Kombinationstherapie in der Regel ambulant, bei offener Tbc, schlechtem AZ und resistenten Keimen auch stationär Pulmonale primäre Manifestation ohne Komplikation: Therapiedauer 6-9 Monate 2 Monate Isoniazid+Rifampicin+Pyrazinamid tgl., danach 4-7 Monate INH+RMP tgl.; alternativ Streptomycin (SM), Ethambutol Pulmonale primäre Menifestation mit Komplikation: 9 Monate Meningitis tuberculosa: 2-3 Mon. INH+RMP+PZA+ SM tgl; dann 10 Mon. INH + RMP P: Expositionsprophylaxe (Isolation bei offener Tbc, bis 3 Sputumkontrollen negativ ausfallen) Chemoprophylaxe INH über 3 Monate nach Kontakt mit offener Tbc INH über 9-12 Monate bei Serokonversion ohne Klinik BCG-Impfung nicht mehr empfohlen - Pleuritis (S. 326) Meist Übergreifen von benachbarter Lunge, entzündliches Exsudat; Pleuritis: sicca, serofibrinosa oder purulenta K: atemabhängige, stechende Schmerzen, evtl. Husten, Fieber, Verminderung der Vitalkapazität (Erguss, Verschwartung) D: Pleurareiben, Klopfschalldämpfung, abgeschwächtes Atemgeräusch, Röntgen, evtl. Punktion T: Antibiose, Drainage, selten Dekortikation DD: Transudat durch Herzinsuffizienz, Hypoproteinämie..., hämorrhagischer Erguss bei Metastasen/malignen Tumoren der Pleura - Akute otitis media und Akute Mastoiditis () (430) Akute Om: v.a. Säuglinge und Kleinkinder, später wird die Erkrankung seltener; viral (Rhinopharyngitis-Viren & Masern-Virus) und meistens bakterielle Superinfektion; auch direkt bakteriell: Pneumokokken, Haemophilus influenzae, Streptokokken, Staphylokokken; meistens tubogen Viral: einseitig, blande; bakteriell: beidseitig, heftiger K: plötzlich starke Ohrenschmerzen, hohes Fieber, ans-Ohr-Fassen, Tragusdruckschmerz; Fazialisparese bei Otitis media acutissima. D: Otoskopisch: gefäßinjiziertes gerötetes Trommelfell ohne Lichtreflex; bei Grippeototitis: hämorrhagische Blasen; bei Perforation schneller Schmerzrückgang 15 - Säuglinge: kaum stürmische Allgemeinsymptome: Trinkschwäche, Unruhe, evtl. Durchfall immer Ohr untersuchen! Wenn nach 2 Wochen nicht besser Verdacht auf akute Mastoiditis. K: Senkung der hinteren Gehörgangswand und Schmerzen und Rötung über Warzenfortsatz. D: Rö nach Schüller, BB, (CT ) Kompl: Einbruch ins Innenohr, Sinus transversus, Schädelinnere. Säuglinge entwickeln leicht chronische Mastoiditis. DD: Otitis externa, Gehörgangsfurunkel mit Lymphadenitis (LK hinter Ohr) T: Amoxicillin oder Erythromycin für 10 Tage schon bei Verdacht auf bakterielle Otitis media, abschwellende Nasentropfen (Tubenbelüftung), b.B. systemische Analgetika; Parazentese bei Innenohrdruck und Fazialisparese. Mastoiditis: operative Eröffnung + Antibiotika P: bei richtiger Therapie nach 1-3 Tage Beschwerdefreiheit und evtl. für einige Zeit Serotympanon Prophylaxe: beseitigen einer behinderten Nasenatmung und Tubenbelüfungsstörungen Bei Kindern mit entsprechender Disposition: chronische Otitis media K: Schleimhauteiterung mit zentraler Trommelfellperforation oder Knocheneiterung mit randständiger Perforation, Cholesteatom, fötide Ohrsekretion und weitere otogene oder endokranielle Komplikationen. Schalleitungsschwerhörigkeit. T: OP: Tympanoplastik evtl. mit Gehörknöchelchenrekonstruktion Herzkreislauf : - PDA (S. 333) (466) 10% der angeborenen Herzfehler, v.a. bei Frühgeborenen (Hypoxämie, hohe Prostaglandine; je Gewicht, desto eher PDA), links-rechts-Shunt, Volumenbelastungvon Lunge/li Ventrikel K: kleiner PDA: kaum Sx; keine Zyanose oder Ermüdung; evtl. Pulse leicht betont HG: Lokomotiv-/Maschinengeräusch (hochfrequentes kontinuierliches Crescendo-Decrescendo-Geräusch mit p.m. über 2. ICR parasternal li.; systolisch (und diastolisch) maximale Lautstärke ca. bei 2. HT) großer PDA: Zeichen der Herzinsuffizienz (Dyspnoe, rasche Ermüdbarkeit, vermehrtes Schwitzen), Gedeihstörungen, wenig Appetit; pulsus celer et altus, periphere Minderversorgung HG: Maschinengeräusch wie beim kleinen PDA; systolisch und immer auch diastolisch; diastolische Komponente verschwindet bei pulmonaler Drucksteigerung (dann reicht der Druck in der Diastole nicht mehr aus um Blut in die A. pulmonalus zu pumpen); Herzspitzenstoß vermehrt tastbar; evtl. Palmarpulse tastbar (Pulse in den Handflächen); gespaltener 2. HT mit Pulmonalis-Akzentuierung D: syst. Crescendo-, diast. Decrescendogeräusch 2-.ICR links, Schwirren, Linkshypertrophie in EKG, Röntgen, Echo T: Verschluss medikamentös (Indometacin), interventionell (Coils), operativ - VSD (S. 338) (462) Häufigster angeborener Herzfehler 30-35%, oft mit weiteren Herzfehlbildungen kombiniert Links-rechts-Shunt mit Entwicklung einer pulmonalen Hypertonie K: kleiner VSD kleiner li-re Shunt, normaler Pulmonalisdruck; Kinder beschwerdefrei lautes Systolikum mit p.m. li. Parasternal 3.-4. ICR; 3-4/6 Tx: häufig Spontanverschlüsse großer VSD ohne Eisenmenger-Reaktion 16 großer li-re Shunt, erhöhter Pulmonalisdruck; schon früh (Säugling) Gedeihstörungen, Trinkschwierigkeiten, vermehrtes Schwitzen, vermehrte Infektanfälligkeit; 2-3/6 Systolikum 3.-4. ICR, betonter 2. HAT EKG: bi- oder re.-ventrikuläre Hypertrophie Rö-Thorax: ausgeprägte Kardiomegalie, Lungenüberflutung Tx: OP-Korrektur im 1. LJ; bei multiplen VSD’s (swiss cheese): Bändelung der A. pulmonalis (künstliche Stenosierung), OP dann später Großer VSD mit Eisenmenger-Reaktion Ausgeprägte Widerstandserhöhung im kleinen Kreislauf mit Shuntumkehr (reli) Subjektiv Besserung des klin. Zustandes (Abnahme des li-re Shunts); progrediente Zyanose Systolikum verschwindet; 2. HT knallend EKG: massive RVH Rö: Hiluszeichnung stark vermehrt, periphere Zeichnung stark vermindert („Hilusamputation“) Tx: OP kontraindiziert; Lebenserwartung: 20-30 Jahre D: Systolikum 3.-4. ICR li parasternal, EKG (Rechtsherzbelastung bei pulmonaler Hypertonie), Röntgen (Kardiomegalie, pulmonale Hyperämie), Echo T: ( allg) Endokarditisprophylaxe, operativer Verschluss - Pulmonalstenose (S. 341) (457) Subvalvulär, supravalvulär, valvulär – wie bei der Aortenstenose auch (Verwachsungen, Verklebungen, Dysplasien der Klappen, zu enger Ring); poststenotische Dilatation der A. pulmonalis Rechtsventrikuläre Hypertrophie, evtl. gleichzeitiger rechts-links-Shunt K: selten Klinik bei milden Stenosen; stärkere Stenosen: Belastungsdyspnoe; hochgradige Stenosen: schon im Säuglingsalter akute Rechtsherzdekompensation D: raues Systolikum 2.ICR li parasternal, Fortleitung zum Hals, li. Axilla oder Rücken; systolischer Klick (je schwerer die Stenose desto näher am 1. HT); gespaltener 2. HT ( verlängerte Ejektionszeit im pulmonalen System; je schwerer die Stenose desto gespaltener); EKG (Rechtstyp), Echo, Herzkatheter; Röntgen (Pulmonalisbogen poststenotisch dilatiert) T: Ballondilatation bei valvulären Stenosen, OP (plumpe, dysplastische Klappen) P: auch ohne Therapie Überleben bis ins Erwachsenenalter (bei kleiner Stenose symptomfrei), dann meistens einsetzende Symptome ( Ermüdbarkeit, Symptome der Rechtsherzinsuffizienz) - Aortenstenose () (458) Valvuläre Stenose; hypertrophierte linke Kammer, poststenotischer Druck in Aorta und Koronarien (bei zunehmener Hypertrophie des li. Herzens: Koronarinsuffizienz) Supravalvuläre Stenose meistens bei Williams-Beuren-Syndrom (Gnomgesicht, Zahnanomalien, psychomotorische Retardierung) Subvalvuläre Stenose (Subaortenstenose): membranöse oder fibromuskuläre subvalv. Stenosen, subvalv. Tunnelstenosen; auch bei hypertrophischer Kardiomyopathie, K: >70% beschwerdefrei, nur Geräusch über Aortenklappe; später Belastungsdyspnoe, Blässe, stenokardische Beschweden; bei Säuglingen mit kritischer Aost: manifeste Herzinsuff. hebender Herzspitzenstoß Schwirren im Jugulum und über Karotiden Systolisches Geräusch rechts parasternal 2. ICR mit Fortleitung in die Karotiden; vom 1. HT abgesetzt und beginnt kurz danach mit einem Klick EKG: linksventrikuläre Hypertrophie D: RR-Differenz, Systolikum zw. Schulterblättern, Echo (Druckgradient, Domstellung der valv. Aost), Katheter (Druckgradient, Differenzierung der einzelnen Formen) 17 T: Verbot von Leistungssport (auch bei geringem Schweregrad); Ballonvalvuloplastik oder OP (risikoreiche OP) - Aortenisthmusstenose / Coarctatio aortae (S. 351) (460) 2 Formen: Präduktal: bei Neugeborenen/Säuglingen, untere Körperhälfte wird über Ductus arteriosus versorgt, in 60% Kombination mit anderen Fehlern Postduktal: adulte Form 4-5% aller Herzfehler, häufig bei Ullrich-Turner –Syndrom K: Präduktal: K: Dyspnoe und Tachypnoe in den ersten Lebenswochen; Trinkschwierigkeiten, Ödeme, Lebervergrößerung Zeichen der kradialen Dekompensation (praktisch wie Vorwärtspumpversagen wegen der Enge); Blaßgraues Hautkolorit und Zyanose der unteren Körperhälfte; bei starker Stenose ist Durchblutung der unteren Körperhälfte vom offenen Ductus arteriosus abhängig: erstmal offenhalten (mit Prostaglandinen) D: Systolikum am ganzen linken Sternalrand (sofern noch nicht dekompensiert); RR-Differenz obere und untere Extremitäten; EKG: RVH; Rö: Kardiomegalie mit Stauungszeichen/Lungenüberflutung; Doppler-Echo: Flussbeschleunigung im Stenosenbreich; Herzkatheter: obligat, da Druckverhältnisse bekannt sein müssen für OP-Entscheidung und –methodik T: OP; Therapie der Herzinsuffizienz P: Letalität zw: 4 - 10 % Postduktal: K: erst normale Entwicklung; dann Kopfschmerzen, Schwindel, Nasenbluten (hoher RR obere Körperpartie), kalte Füße und Wadenschmerzen (niedriger RR untere Körperhälfte); wenn’s schlimm läuft: Apoplex; Versorgung der unteren Körperhälfte über die Ausbildung von Kollateralkreisläufen über Rippenarterien (Rö: Rippenusuren durch Pulsationen der Arterien) oder A. mammaria interna D: fehlende Femoralispulse; Systolikum am rechten Sternalrand und zwischen Schulterblättern (Fortleitung in Richtung Aorta desc.); Echo; Angio (Darstellung des Defekts und der Kollateralen); Herzkatheter (Druckmessungen) T: OP (je früher desto rückbildungsfähiger der RR in den oberen Körperteilen); bei Rezidiven Ballondilatation D: (allg.) RR-Differenz, Systolikum zw. Schulterblättern, Echo, MRT, Katheter T: (allg.) medikamentös Ductus offen halten, OP - Hypoplastisches Linksherzsyndrom (HLHS) (S. 354) (472) Hochgradige Stenose/Atresie der Aortenklappe oder hochgradige Stenose/Atresie der Mitralklappe = HLHS; linker Ventrikel sehr klein; Aortendurchmesser bis zum Ductus nur im mm-Bereich; Blut zum linken Herzen geht durch Vorhof-Links-Rechts-Shunt (wenn vorhanden) ins rechte Herz und von da über Ductus arteriosus in die unteren Körperpartien; Koronarien retrograd vom Ductus Aorta ascendens durchblutet; häufigste kardiale Todesursache im Säuglingsalter Kinder nur durch weitere Fehlbildungen (Shunt, PDA) lebensfähig K: Neugeborene zunächst o.k., dann am 4-7 Tag dekompensieren rasch kardialer Schock (Ductus geht zu?); blaßgraues zyanotisches Hautkolorit, ausgeprägte Tachy-Dyspnoe; Hepatosplenomegalie (infolge Volumenüberlastung des re. Herzens Stauung im großen Kreislauf); Azidose; D: EKG: Rechtstyp mit kleiner R-Amplitude links präkordial; Rö-Thorax: Kardiomegalie und Lungenstauung; Echo: Dilatierter rechter Ventrikel und A. pulmonalis, linker Ventrikel oft nur noch kleiner Hohlraum, Aorta ascendens extrem schmal; Injektion von KM in A. axillaris retrograder Fluß zur Aortenklappe (keine Vorwärtsbewegung des Blutes in diesem Segment) 18 T: OP in drei Schritten, Herz-TX, ansonsten versterben 95% in den ersten 10 Tagen - Trikuspidalatresie () (472) Es fehlt die direkte Verbindung zwischen re. Vorhof und re. Kammer, wobei meistens keine Klappe angelegt ist. Verschiedene Varianten: immer mit ASD (nötig zum Überleben); rechter Ventrikel hypoplastisch; andere Gegebenheiten können sein: Stellung der großen Arterien normal/pathol., großer/kleiner VSD, Pulmonalstenose/-atresie, PDA. Lungendurchblutung kann , oder sein K: sehr variabel mit/ohne Zyanose; entsprechende HG bei Pulonalstenose, VSD oder PDA D: EKG: pathognomonisch: Linkstyp, P cardiale, LVH; Echo: rudimentäre re. und dilatierte li. Kammer, rel. einfache Diagnosestellung mit Echo (auch Erfassen weiterer Anomalien: PDA, VSD, Fluss über rechtsventrikuläre Ausflussbahn, etc.) T: PGE1 um Ductus offen zu halten wenn Lungendurchblutung ductus-abh. Ist; Korrektur von / Lungendurchblutung; OP nach Fontan: Schaffung zweier getrennter Kreisläufe bei Fehlen eines funktionstühtigen re. Ventrikels: weitere Einzelheiten kompliziert :o) P: schwer zu beherrschende Herzrhythmusstörungen, Folgen der Einflußstauung (Ödeme, Hepatomegalie, Aszites, Pleura- und Perikardergüsse) - Fallot-Tetralogie (S. 243) (468) 1. Pulmonalstenose 2. Rechtsherzhypertrophie (als Folge der rechtsventrikul. Obstruktion und des VSD) 3. großer subaortaler VSD 4. Überreitende Aorta (durch Dextro- und Anteroposition) (bekommt Blut aus beiden Ventr.) Häufigster zyanotischer Herzfehler; verminderte Lungendurchblutung durch Pulmonalstenose und (je nach Ausmaß der Stenose) resultierendem li-re- / re-li-Shunt (Zyanose)! K: im ersten Lebensmonat unauffällig; dann Belastungs- und später Ruhezyanose; schlechtes Gedeihen; im 2. LJ hypoxämische Anfälle: durch Schreien/Aufregung/Weinen (‚Luftmangel’) zunehmende Zyanose (Teufelskreis) bis zur Bewusstlosigkeit/Krampfanfällen/Paresen; später körperliche Leistungsfähigkeit stark eingeschränkt; evtl. Trommelschlegelfinger/Uhrglasnägel; Polyglobulie Hochstellung: Druck im systemischen Kreislauf, re-li-Shunt nimmt ab und Lungendurchblutung wird besser. D: lautes, scharfes Systolikum 3.-4. ICR links parasternal (Pulmonalklappenregion); schwacher 2. HAT; Rö: Herzspitze vermehrt gerundet und angehoben; Echo: dilatierte Aorta, reitend über gut sichtbaren VSD; Herzkatheter T: Unterbrechung des hypoxämischen Anfalls durch Morphium oder Sedierung (Diazepam) + O2Gabe; Propranolol (allg. β-Blocker) prophylaktisch; OP-Korrektur früh und dann im Alter von 3-6 Jahren - Supravent. Tachykardien (S. 377) (480 ff) Sinustachykardie, -bradykardie und resp. Sinusarrhythmie keine eigentlichen Rhythmusstörungen, sondern Normvarianten Mögliche Ursachen: Fieber, Anämie, HI, Perikarderguss, Medikamente Respiratorische Arrhythmie: Inspiration Zu-, Expiration Abnahme T: in der Regel nicht notwendig; evtl. Katheterablation der akzessorischen Leitungsbahn(en) Tachykardieanfall: SVT vom Reentrytyp im Kindesalter die häufigste akut zu behandelnde HRS Säugling: Pressen eines mit Eiswasser gefüllten Beutels für 15-30 s aufs Gesicht Älteres Kind: 1-2 Gläser eiskalten Sprudel schnell trinken oder Vagusstimulation (Vasalva) oder Karotissinusmassage (vom Arzt) Wenn erfolglos (bei allen Altersstufen) wahlweise o Adenosin-Bolus-Injektion o Elektrokonversion 19 o i.v. Propafenon (kein Verapamil im NG und Säugl.-Alter (UAW!) Dauerbehandlung schon nach 1. Tachykardieanfall, da > 90% Rezidiv (Digitalis, Propafenon; Sotalol, Amiodaron)) – nach 1 Jahr Auslassversuch - Extrasystolen T: im allgemeinen keine Tx nötig; nur wenn polytop/polymorph, R-auf-T-Phänomen (schnell einfallende ES), unter Belastung zunehmende ES ( Gefahr der ventrikul. Tachykardie oder des Kammerflimmers) - Ventrikuläre Tachykardien (480ff) Im Kindesalter seltener als SVT. Bei Myokarditis, Digitalisintoxikation, angeborene Herzfehler, nach Herz-OP’s, Herztumoren; bei völlig gesunden Kindern T: allgemein bedarf die VT einer Dauertherapie wenn bedrohlicher Zustand: sofortige Elektroversion (wenn keine Digitalisintox.); wenn EV nicht möglich: Lidocain als Bolus und Dauerinfusion Dauertherapie: v.a. bei Herzfehlern: Propafenon, Flecainid, Mexiletin, β-Blocker; wenn kein Erfolg: Sotalol, Amiodaron Keine Therapie, wenn: akzelerierter idioventr. Rhythmus (HF <150/min) oder bei kurzen Salven einer monomorphen extrasystolischen Tachykardie - Long QT-Syndrom (S. 384) Ursachen : angeboren – erworben (Elyt-Störungen, Medikamente, Hypothyreose…) Angeborene Formen: Romano-Ward-Syndrom, Jervell/Lange-Nielsen-Syndrom verlängerte QT-Zeit mit rezidivierenden Synkopen durch ventrikuläre Tachykardien K: unerwartete Synkopen, Krampfanfälle, oft nach Kontakt mit kaltem Wasser D: QT-Verlängerung T: b-Blocker zur Tachykardie-Prophylaxe, darunter evtl. Schrittmacher notwendig Urogenital : - Akute Glomerolunephritis (S. 394) Verschiedene Einteilungen nach Ätiologie, Klinik, Histologie.... Seltene angeborene Glomerulopathien, erworbene Glomerulopathien – Immunkomplexnephritis, Basalmembran-Ak-Nephritis Unterscheidung nephritisches/nephrotisches Syndrom Nephritisches Syndrom: Hämaturie, mäßige Proteinurie, Zylindrurie; (Oligurie, Hypertonus, Ödeme) Nephrotisches Syndrom: klinischer Symptomenkomplex gekennzeichnet durch großen und selektiven (<70 kDa) renalen Eiweißverlust; Folge: Hypo- und Dysproteinämie (Hypalbuminämie, von α2- und β-Mikroglobulin). Weitere Symptome durch Eiweißverlust: Hyperlipidämie, Ödeme, Oligurie. Ursachen nicht einheitlich, sondern verschiedene Prozesse: minimal change GN, fokal-segmentale GN, etc. Therapeutische Unterteilung in: (klar muss man das nicht alles wissen :o) – auch die Therapie ist etwas zu umfangreich geworden, aber für später kann’s nicht schaden kausal zu beeinflussende nephrotische Syndome Lupusnephritis Toxoplasmosenephritis Koagulase-negative Staphylokokken-Nephritis (Shuntnephritis) Intoxikationen (Penicillamin, Schwermetalle, u.a.) Allergisch bedingte nephrotische Syndome (Bienenstich, Inhalationsallergene) 20 Malignom mit nephrotischem Syndom (Lymphome, Leukämien, Lymphogranulomatose) steroidsensible nephrotische Syndome Lipoidnephrose (minimale glomeruläre Läsionen) Nephrotisches Syndrom bei fokaler globaler Glomerulosklerose steroidresistente nephrotische Syndome angeboren I. kongenitales NS vom finnischen Typ II. diffuse mesangiale Sklerose III. NS bei angeborenen Nephropathien (Lowe-, Alport-, Drash-Syndrom) Idiopathisch I. Fokale segmentale Glomerulosklerose II. Mesangioproliferative GN III. Membranoproliferative GN IV. Membranöse GN V. Chronische GN Symptomatisch I. Lupus erythematodes II. Purpura Schönlein-Henoch III. Panarteriitis nodosa, Hepatitis-B-assoziierte GN IV. Malarianephritis V. Diabetes mellitus VI. Amyloidose VII. Nierenvenenthrombose K: Ödeme, Ergüsse, Oligurie, Neigung zu Thrombosen (AT III-Mangel) Kompl.: lange bestehendes nephrotisches Syndrom: Ödeme, Hypoproteinämie, Hypovolämie, Hyperkoagulabilität, trophische Störungen, Infektionsneigung (Ig Verlust); unter langer Steroidtherapie zusätzlich: Elektrolytstörungen, Hypertonie, zerebrale und psychische Symptome, Katarakt, Minderwuchs u.a. D: Urin (Proteinurie, Zylinder), Blut (Hypalbuminämie, Hyperlipid-/cholesterinämie), Histologie T: kausal + symptomatisch; Steroide (wenn sensibel); Zytostatika (Cyclophosphamid, Chlorambucil, Cyclosporin A) Symptomatische Therapie: Ödeme: leichte: keine Behandlung nötig; schwere (Aszites, Pleuraergüsse): Diuretika (Furosemid + Hydrochlorothiazid+zusätzlich Kalzium) Hypoproteinämie, Hypovolämie: nur in Notfällen, da nur kurzfristig wirksam ( renale Elimination nach Gabe) und Belastung durch NaCl Hyperlipidämie: Diät; Sitosterol Verminderte Infektresistenz/Infektionen: Prophylaxe ( Hygiene), keine AntibiotikaProphylaxe; bei akuten Infektionen Gabe von Antibiotika und γ-Globulinen (werden auch wieder ausgeschieden durch die nephrotische Niere) Elektrolytstörungen ( Steroide, Diuretika und salzarme Kost): NaCl und K-reiche Diät trotz Ödeme; Infusion mit Ringer-Lactat und 20%ige Humanalbuminlösung wenn Hypovolämie Thrombosen ( Hyperfibrinogenämie, AT-III , Störung Fibrinolyse, Thrombozytenaggregabilität): Heparin (PTT aufs Doppelte der Norm), Cumarine, ASS; bei frischer arterieller Thrombose: Urokinase; alte Thrombosen: chirurgisch Poststreptokokken-Nephritis: akute, exsudativ-proliferative Immunkomplex-GN, 1-3 Wochen nach A-Streptokokken-Infekt; v.a. Kinder zw. 2-12 Jahren K: Hämaturie, Oligurie, Ödeme, Hypertension, Allgemeinsymptome (Fieber, Erbrechen, Kopfschmerzen) D: Urin (Hämaturie, Zylinder, Proteinurie), Blut (ASL, C3 vermindert, BSG, Retentionswerte) 21 T: (gegen Erreger und gegen Komplikationen; renale Prozesse können nicht direkt beeinflusst werden): Bettruhe bis keine Hämaturie, bis Blutdruck normal und keine Ödeme Diät: kochsalzarm (Hypertonus, Ödeme), Eiweiß (bei Oligurie), Flüssigkeitsrestriktion nach Harnvolumen des Vortags + 500 ml/m² KOF (Perspiratio); regelmäßige Gewichtskontrolle Penicillin oral 20.000 – 40.000 IE/kg/Tag für 10 d (Streptokokken, sonst nach Antibiogramm), Furosemid, evtl. Herdsanierung (Tonsillektomie im infektfreien Intervall mit Penicillinschutz) K: Niereninsuffizienz: Hypertonus: Flüssigkeits- und Salzbilanzierung, evtl. Furosemid Herzinsuffizienz: Digitalis Ödeme: Furosemid; bei Lungenödem mit Tachypnoe und Herzinsuff.: Dialyse Enzephalopathie mit zerebralen Krampfanfällen: Diazepam langsam i.v., Phenobarbital Rasch progressive Glomerulonephritis / Perakute GN: stürmisch verlaufendes Krankheitsbild; rasch einsetzende Oligurie und Anurie; nach bakteriellen Infektionen, bei Systemerkrankungen oder idiopathisch; schwere nekrotisierende und proliferative Veränderungen, Exsudationen von Fibrinogen und Fibrin mit Kapselproliferationen und Halbmondbildung an fast allen Glomeruli K: Hämaturie, Oligurie, Ödeme; wie akute postrept. GN D: wie akute poststrept. GN, nur raschere Progression T: keine kausale Therapie: wie akutes Nierenversagen: Flüssigkeitsüberwachung, Kaliumrestriktion, eiweißarme Diät, Peritoneal- und Hämodialyse; evtl. Plasmapherese; Immunsuppression: Wirkung noch nicht gesichert: Corticosteroide evtl. in Kombination mit Cyclophosphamid; neueste Studien: Leukapherese (Leukos aus Blut raus erstmal): besser als nur medikamentös P: nach akutem Stadium kann sich Nierenfunktion wieder normalisieren; meist bleibt eine chhronische GN mit Übergang in chronische Niereninsuffizienz nach mehreren Jahren IgA-Nephritis (M. Berger): Immunkomplex-GN mit mesangialen Ablagerungen durch IgAÜberproduktion bei Immunregulationsstörung, meist spätes Schulalter und Mädchen K: persist. Mikro- mit bei Infekten rezid. Makrohämaturie; in schweren Fällen: große Proteinurie, Hypertonie, progred. Niereninsuff., IgA Ablagerungen in Basalmembran D: Immunfluoreszenz T: (keine kausale Tx bekannt, aber) Infekttherapie (bessert die Nieren-Symptome); bei schweren Verläufen: Prednison, Azathioprin; hochdosierte Immunglobuline, ACE-Hemmer P: günstig; bleibende Mikrohämaturie; leichte Formen heilen innerh. 2 Jahren aus, schwere Formen in 50% terminale Niereninsuffizienz DD: Purpura Schoenlein-Henoch - Glomerulonephritis bei Systemerkrankungen Schönlein Henoch Nephritis () (501) Bei Purpura Schönlein Henoch: anaphylaktoide Purpura, allergische Entzündung der Gefäßwand (Arteriolen und Kapillaren) bei Kleinkindern und Schulkindern K: streng symmetrisch angeordnetes hämorrhagisch-urtikarielles Ekzem, Arthralgien, periartikuläre Ödeme, Ödeme and Hand- und Fußrücken, kolikartige heftige Bauschmerzen (Darmwandödem), hämorrhagische Nephritis; schubartiger Verlauf möglich; keine allgemeine Blutungsneigung D: Klinik, Urin, Biopsie/Histologie T: keine kausale Tx; bei leichter Hämaturie und Proteinurie keine Behandlung nötig; Steroide in üblicher Dosierung nicht wirksam; Glucocorticoide bei abdominellen Sx; immunsuppressiv bei anhaltendem schubweisem Verlauf P: in Mehrzahl der Fälle günstig; in 5-10% chronische GN, die zur terminalen NI führen kann 22 Weitere Formen: Alport-Syndrom, GN bei Lupus, Goodpasture-Syndrom, Purpura... - VUR (S. 411) Ureterostiuminsuffizienz (angeboren/erworben) mit der Gefahr der Druckatrophie und rez. Infektionen 0,5-1%, 4x mehr Mädchen; Radiologisch 5 Grade (Füllung des Ureters bis massive Dilatation und Schlängelung) K: nicht durch Reflux, sondern durch Infekte D: MCU T: hohe Rückbildungstendenz, Grad I-III abwarten, Infektionsprophylaxe, bei höheren Graden und Hypertonie OP - Urethralklappen (S. 412) (488) Persistierende Schleimhautfalten der Urethra in der Pars prostatica, Abflusshindernis Balkenblase, Reflux, Hydronephrose, Niereninsuffizienz, Infekte K: Miktionsstörung, später: schlechtes Gedeihen, Erbrechen, Urosepsis, Anämie D: Sono, bds. Hydronephrose, große Harnblase T: suprapubische Harnableitung, ab 3.LM TUR, evtl. Antirefluxplastik - HI/Pyelonephritis (S. 414) Bakteriurie+Leukozyturie; Infektion hämatogen (Neugeborenen) oder durch Keimaszension Begünstigende Faktoren: Missbildungen, Abflussstörungen, Immunsuppression, hohe Virulenz Meist E.coli, daneben Proteus, Enterokokken, Pseudomonas... Zystourethritis – Pyelonephritis Sehr häufig! Säuglinge mehr Jungen, danach Mädchen 10:1 K: oft uncharakteristische (v.a. Säuglinge) Allgemeinsymptome, Dysurie, Pollakisurie, Enuresis Bei Pyelonephritis: Fieber, Flankenschmerz D: Urin (>5x104 Bakterien/ml, >50 Leukos/µl im Mittelstrahlurin); bei Pyelonephritis (Leukozytenzylinder, CRP, BSG) Sono, MCU (Säuglinge/Jungen, bei Mädchen nach Rezidiv) Ursachensuche T: Antibiose (7d Cotrimoxazol, danach nach Antibiogramm), evtl. OP, Reinfekt-Prophylaxe; bei Pyelonephritis hochdosierte Antibiose nach Antibiogramm - Nephroblastom (S. 506) Wilms-Tumor, maligner embryonaler Mischgewebs-Tumor (Adenomyosarkom); 1:100.000, 85% <6 Jahre, 10% mit weiteren Fehlbildungen, gute Prognose Stadien I-V (auf Niere beschränkt – Fernmetastasen – bilateral) Stadium I: intrarenal Stadium II: intrarenal und extrakapsulär, komplett resezierbar Stadium III: intraabdominelle Aussat Stadium IV: Metastasierung (meist pulmonal) Stadium V: bilateral (ca. 5%) Histologisch 3 Malignitätsgrade K: schmerzloser Abdominaltumor, unspez. Allgemeinsymptome, Zufallsbefund; Makrohämaturie (wenn ins Nierenkelchsystem eingebrochen) D: Sono, i.v. Pyelogramm; CT/MRT; Tumormarker negativ!; andere Laborparameter auch. T: präoperative Chemo und postoperative Radio-Chemotherapie, Operation, Radiatio P: Stadium I-III: gute Heilungschancen (> 90%), Stadium IV und V: 60% - Nierentransplantation (Herold 537) Voraussetzungen: Blutgruppenkompatibilität im A-B-0-System und fehlende KI: Malignome, Psychosen, chron. Infektionskrankheiten (bes.Tbc), fortgeschrittene Arteriosklerose, floride 23 Ulkuskrankheit, biologisches Alter >55-60 Jahre, zytotoxische Antikörper im Empfängerserum gegen Spenderlymphozyten! Vorlauf: Die Spendernieren werden unter Berücksichtigung von Histokompatibilität und Priorität vom Eurotransplantatzentrum (in Leiden/Holland) dem geeignetesten Empfänger vermittelt. Warme Ischämiezeit: max. tolerable Zeitspanne zwischen Kreislaufstillstand des Spenders und Nierenentnahme (15 Min); Kalte Ischämiezeit: max. tolerable Zeitspanne zwischen Nierenentnahme und Transplantation (36 Std). Die Nierentransplantation erfolgt extraperitoneal in die linke oder rechte Fossa iliaca. Wegen der Gefahr der Abstoßungraktionen müssen die Patienten immunsuppressiv behandelt werden T: langfristig mit Ciclosporin A, anfangs zusätzlich mit Kortikosteroiden, evtl auch Tacrolimus oder Mycophenolatmofetil Komplikationen nach Transplantation: o postoperativ: Nierenruptur, Blutungen oder Thrombose der Nierengefäße, Ureterleckage u.a. o Abstoßungsrektionen: perakut: infolge präformierter zytotoxischer Antikörper akut: innerhalb 8 Wo nach Transplantation chron.: nach 8 Wochen Folgen der Immunsuppressiven Therapie: Leukozytopenie, Thrombozytopenie, Immunschwäche, etc. Hämatologie / Onkologie: - Sichelzellanämie (S. 453) Häufigste Hämoglobinopathie, Punktmutation HbS (Aggregation, sichelförmige Verformung bei Hypoxie: Gefahr von Thrombembolien) K: Heterozygote erkranken seltener an Malaria und haben selten Symptome Homozygote bei Abfall des HbF Infarkte, Koliken, Krämpfe, Kopfschmerzen, Sehstörungen, Splenomegalie (später Schrumpfung durch multiple Infarkte) D: BB (Sichelzellen, evtl. nach Sauerstoff-Entzug), Hb-Elektrophorese, hämolyt. Anämie T: symptomatisch (Azidoseausgleich, Flüssigkeitszufuhr, Schmerztherapie), Hydroxyurea (erhöht HbF-Anteil); als kurativer Ansatz: KM-TX - Aplast. Anämie (S. 456) Panzytopenie mit Verminderung Zellreihen in KM und Blut Ursachen: idiopathisch, genetisch (Fanconi-Anämie, Shwachman-Syndrom), erworben (Medikamente, Strahlen, Infekte – häufig Virushepatitis) K: Blässe, Infektanfälligkeit, Blutungsneigung (Nase, Haut), bei genetischen Formen auch Missbildungen D: Retikulozyten <20.000/µl, Granulozyten <500/µl, Thrombos <20.000/µl, Markhypoplasie T: Absetzen auslösender Medikamente, Infektionsprophylaxe, Substitutionstherapie, kurativ: KMTX - Idiopathische Thrombozytopenie (S. 471) (350) Thrombos <100.000/µl; Immunreaktion nach Virusinfekt: Plättchen mit Ig oder mit komplementhaltigen Immunkomplexen beladen; Überlebenszeit der Plättchen verkürzt; v.a. Kinder <8 Jahre K: vorausgegangener Virusinfekt; guter AZ trotz ausgeprägter Petechien, Hämatome, Schleimhautblutungen D: BB T: Thrombozytenanstieg nach Tagen bis Wochen; symptomatisch: Druckverbände, Tamponaden; Kortikoide, Immunglobuline beschleunigen evtl. den Anstieg (nicht eindeutig bewiesen); PlättchenSubstitution bei Blutungen 24 Chronische ITP: wenn Thrombozytopenie mehr als 6-12 Monate andauert (10% aller ITP’s). Autoimmun-Raktion mit spezifischen Plättchen-AK T: Kortikosteroide, Immunglobuline, Anti-D-Immunglobuline; Splenektomie als ultima ratio - Purpura Schoenlein Henoch (S. 476) Generalisierte Entzündung der kleinen Gefäße häufig bei Klein-/Schulkindern, schubförmiger Verlauf K: Petechien, papulöse Veränderungen an Streckseiten der Extremitäten, Gelenkbeteiligung, Magen-Darm-Befall (Koliken, Blutungen), 1/3 Nierenbeteiligung (Hämaturie bei GN), selten Hirngefäße (Krämpfe, Paresen) D: Histologie T: hohe Spontanheilungsrate, evtl. Prednison - Leukämie (S. 486) Proliferation pathologischer Vorstufen weißer Blutzellen mit Verdrängung der normalen Hämatopoese; 4/100.000; Altersgipfel zwischen 3.-5. LJ Prädisponierend: chrom. Anomalien, Immundefekterkrankungen, Karzinogene 35-40% aller kindlichen Malignome, 80% ALL (allg. eher bei Kinder), 18% AML (allg. eher bei Erw.), 2% CML K: erst uncharakteristisch: Müdigkeit, Appetitlosigkeit, mäßiges Fieber, Infekte, Nasen/Schleimhautblutungen, Knochen-/Gelenkschmerzen, generalisierte LK-Schwellung, Leber/Milz vergrößert; spezifische Sx: Blässe und hämorrhagische Diathese (Petechien, Hämatome, Nasen/Zahnfleischblutungen) Befunde: v.a. Blässe; LK-Schwellung und Hepatosplenomegalie; manchmal Hyperplasie des Zahnfleisches (Leukämiezellinfiltrate); im BB Panzytopenie, Leukos können sein (bei 50% sogar unter 10.000/µl); D: BB, KM-Punktion (leukämische Blasten verdrängen Hämatopoese), Diff-BB (Hiatus leukämikus bei akuten Leukämien), MRT (Ausmaß der KM-Infiltration), Liquor-Punktion (immer durchzuführen! Meningiosis leukaemica), Sono, Chemie (Harnsäure, LDH, Kalium, Phosphat hoch, Kalzium niedrig) DD: aleukämische Formen manchmal zu verwechseln mit aplastischer Anämie (KM bei Leuk. voll und bei aplast. Anämie: leer!); lymphatische Organe bei Mononukleose auch vergrößert (Unterschied im BB: keine hämatopoetische Insuff.) T: allgemein: mehrere Phasen der Therapie: 1. Phase: Induktionstherapie (Senken der leukämischen Leukozyten) meistens schon durch Prednison und Vincristin (>90% Erfolg). Nennt man Remission.; 2. Phase: (Präventive) Behandlung des ZNS: intrathekale Gabe von Methotrexat (wenn nicht gemacht, nach einigen Monaten in 50% der Fälle ZNS-befall); 3. Phase: Dauertherapie (ca. 2 Jahe) mit Antimetaboliten (Mercaptopurin, Methotrexat). Wichtig: Erhaltung der Remission, da Rezidive (häufig im KM, ZNS, Hoden) die Prognose verschlechtern ALL: häufigste Form bei Kindern, >70% heilbar, meist B-Zell-ALL T: Chemotherapie (MTX, Steroide, Cyclophosphamid...), KM-TX AML: zweithäufigste Form, 8 Subtype T: Chemotherapie, ZNS-Bestrahlung, KM-TX P: schlechter als bei ALL CML: myelodysplastisches Syndrom, gesteigerte Hämatopoese in KM, Milz, Leber, stadienhafter Verlauf mit terminalem Blastenschub K: chron. Verlauf schleichend oder in Schüben; Hepato-Splenomegalie, Infektionen, Blutungen D: BB (meist Panmyelopathie: alle Reihen betroffen), KM-Ausstrich, alkalische Leukozytenphosphatase erniedrigt, Philadelphia-Chromosom T: Chemotherapie; kurativ nur KM-TX 25 - NHL (S. 495) (369) Maligne Proliferation von Lymphozyten (B: 70%, T: 20%, Null: 10%) im lymphatischen Gewebe; hohe Malignität bei Kindern; prädisponierend: Immundefektsyndrome, Immunsuppression, EBV (Burkitt-Lymphom), Translokationen [ t(2;8), t(8;14), t(8;22), ; Grenze zur ALL unscharf (Anteil lymphatischer Blasten im KM< 25% NHL; > 25% ALL); meist schnelles Wachstum 0,8/100.000 Kinder <15 Jahre; Jungen 2,8x häufiger Kiel-Klassifikation (histologisch) Stadieneinteilung: I – Einzellokalisation II – mehrere Herde auf einer Zwerchfellseite III – beide Zwerchfellseiten IV – ZNS-, KM-Befall K: kurze Anamnese (schnelle Proliferation): schmerzlose LK-Vergrößerung (derb, verbacken) (auch mediastinal/abdominell Atemprobleme / Appendizitis-ähnliche Symptome, Invagination), oft akutes Geschehen (Dyspnoe, akutes Abdomen, Querschnitt, Krämpfanfälle) D: Biopsie, Staging (Sono, Röntgen, MRT, Liquor); KM-Aspirat; BB, LDH, Harnstoff, Krea, Harnsäure, K+, Ca++; chirurgische Probe-Biopsie (sichert Diagnose) DD: Infektiös bedingte LK-Erkrankungen (Tbc, infektiöse Mononukleose, Toxoplasmose, Listeriose, Zytomegalie, HHV-6-(=Exanthema subitum) und HIV-Lymphadenopathie) und M. Hodgkin T: Polychemotherapie P: bei hochmalignen NHL Heilung möglich, bei niedrigmalignen nur Verlangsamung der Progression ( eher abwartendes Verhalten, Symptomatische Therapie) Heilungschancen: Stadium I & II: 95% – 100%; Stadium III & IV: 75% – 85% - Neuroblastom (S. 502) (382) Aus Zellen der Neuralleiste (Sympathikus, NNM), zweithäufigstes solides Malignom bei Kindern, v.a. bei Säuglingen; Infiltrativ, frühe Metastasen (meist schon bei Dx-Stellung) in Knochen, KM, entfernte LK, Leber, Haut (NICHT in Lunge); Besonderheiten: Aggressivität des Wachstums, Fähigkeit zur Ausreifung, sponatane Rückbildung Drei Differenzierungsgrade, 5 Stadien (auf Ursprungsorgan begrenzt Metastasen) K: Allgemeinsymptome: Fieber, Schmerzen, Abgeschlagenheit; typische Neuroblastom Symptome: Horner-Syndrom (Tumor im Ganglion stellatum) Querschnittssymptomatik (Einwachsen des Tumors) (aka Sanduhrtumor) Therapieresistenter Durchfall (VIP Produktion durch Neuroblastomzellen) Opsoclonus-Myoclonus-Ataxie-Syndrom (tanzende Augenbewegungen, Kloni, Ataxie) Monokel-/Brillenhämatom bei Schädelknocheninfiltration und Protrusio bulbi D: Katecholaminmetaboliten in Urin und Serum (Vanillinmandelsäure, Homovanillinsäure, Dopamin) Neurospezifische Enolase (NSE) im Serum Metajodbenzylguanidin-Szinti (123mIBG) (Tumor selbst); Technetium-Knochen-Szinti (Metastasen) CT/MRT/Sono T: nach Stadien, OP (I), OP+Chemo-/Strahlentherapie (II-III) P: Langzeitüberlebenschancen: Stadium 1-3 & 4S: 80%; Stadium 5: 30%; allgemein: 55% Screeninguntersuchung auf Katecholamine mit 12 Monaten zur Frühdiagnose (noch als Bundesweite Studie) - Ewing-Sarkom (S. 510) (386) Zweithäufigster Knochentumor, Jugendliche betroffen (5< Ewing-S. <30 LJ); frühe Metastasierung in Lunge und Knochen; Schwarze und gelbe Menschen fast nie betroffen L: Lokalisation v.a. Becken und Diaphysen von Femur, Tibia, Fibula (manchmal auch Wirbelkörper, Rippen & Scapula) 26 K: Schmerzen, Schwellungen, Bewegungseinschränkung, Schwäche, Fieber, Nachtschweiß, Jucken, Gewichtsabnahme D: Röntgen (Zwiebelschalen-Periost und mottenfraßähnliche Knochendestruktionen), CT/MRT (Ausdehnung in Weichteile und intramedullär; Metastasen), Histo, Staging (Szinti, Lungen-CT); Diagnosestellung allgemein schwierig bei Ewing-S. DD: Osteomyelitis, andere Knochentumoren, Metastasen T: Polychemo – OP – Bestrahlung – Polychemo (über 12 Monate) P: bei lokalisierten Tumoren 5-JÜR: 50-60%, bei Lungenmetastasen: 20-30%, bei Knochenmetastasen: infaust - Osteosarkom () (384) Primär maligner Tumor des Knochens, dessen Zellen Osteoid oder unreifen Knochen bilden; bevorzugt in Adoleszenz, Häufigkeitsgipfel zum Zeitpunkt des höchstes Wachstumstempos (J: 14.5 J; M: 13.5 J); L: typischerweise in Knochenmetaphyse, meist Kniegegend: distale Femur- oder prox. TibiaMetaphyse; andere Lok.: prox. Humerusmetaphyse, selten: distales Radius- und Tibiaende K: Schmerzen (v.a. nachts); später Schwellung mit örtlich begrenzter Hyperthermie D: Rö: Knochendestruktion (osteolytisch/osteosklerotisch), periostale Knochenneubildung; Rö-Th: Lungenmetastasen MRT: intramedulläre Ausdehnung und Weichteilanteil (häufig mehr als man auf Rö-Bild sieht) Tc-Szinti: hochgradige Anreicherung im Tumor; Abschätzung des Ansprechens auf Chemo-Tx (je mehr Tc Tumor speichert, desto Anspechen auf Chemo möglich) Histo (aber keine initiale Tumorentfernung!) T: präoperative Chemo (Tumorverkleinerung) OP postoperative Chemo: 60% Heilungsrate (OP alleine hat auch bei Amputation im Gesunden nur 20% Heilungsrate wegen präexistierender Mikro-Lungenmetastasen) Osteosarkom ist strahleninsensibel! DD: strahleninduzierte osteogene Sarkome (6-10 Jahre nach Radiotherapie) – v.a. wenn untypische Lokalisation und entsprechende Anamnese; Myositis ossificans, Osteomyelitis - Rhabdomyosarkom () (387) Maligner mesenchymaler Tumor mit (unreifen) Markern quergestreifter Muskulatur; zwei Altersgipfel (<5 Jahre: Hals, Kopf, Prostata, Blase, Vagina; Jugendliche 15-19 J: Urogenitaltrakt (testikulär, paratestikulär)); häufiger in Familien mit Hirntumoren und Mamma-Karzinomen L: überall, auch wo sonst keine quergestreifte Muskulatur zu finden ist Histologische Typen: embyronal, alveolär, botryoid (in Hohlräumen wachsend), pleomorph, undifferenziert; Metastasen bevorzugt: Lunge, Knochen und KM, LK K: typisch: schmerzlose Weichteilschwellung (v.a. nach Trauma als „Hämatom“, das sich aber nicht zurückbildet); weitere Sx von Lokalisation abhängig: Orbita: Bulbusverlagerung, Chemosis (=konjunktivale Schwellung) Kopf-Hals: verlegte Nasenatmung, Nasenbluten, Dysphagie, chronische Otorrhoe, Fazialisparesen, Reizhusten Urogenitaltumoren: Hämaturie, Harnverhalten, polypoide Vaginalextrusion, schmerzlose testikuläre Knoten D: Sono, CT, MRT: Sitz, Größe und Ausdehnung; Szinti: Skelettmetastasen; KM-Punktion: KMMetastasen ; Rö-Thorax: Lungenmetastasen; Histo: Probebiopsie immer! T: nur kleine Tumoren oprativ entfernen (R0); Chemo-Tx (Dauer je nach Stadium, Sitz, Ansprechen und Histologie), bei parameningealen Tumoren auch intrathekal; Radiotherapie P: lokalisierte Tumoren: Langzeitprognose von 60-70%; Stadium IV 20% Stadieneinteilung: I: lokalisiert, mirkroskopisch komplett reseziert, LK negativ II, A/B: lokalisiert, reseziert mit mikroskopischen Resten (mit/ohne regionale LK Beteiligung) 27 III: IV: - lokalisiert, makroskopische Reste, maligner Erguss Fernmetastasen ZNS-Tumoren () (379) Sehr heterogen was Lokalisation und Histologie angeht Infratentoriell: Mehrzahl aller ZNS-Tumoren haben infratentoriellen Ursprung zerebelläres Astrozytom Medulloblastom des Kleinhirns Hirnstammgliom Ependymom des 4. Ventrikels Supratentoriell: Astrozytom (im supratentoriellen Stammhirn) Ependymom Kraniopharyngeom Spinal: Astrozytom Myxopapilläre Ependymome (Cauda equina – Bereich) K: Obstruktion des Liquorweges und lokal durch herdförmige Läsion des Tumors im Hirngewebe; Allgemeinsymptome: Kopfschmerzen, morgendliches Nüchternerbrechen, Lethargie, kontinuierlicher Leistungsabfall in der Schule; bei Säuglingen Sx manchmal auch fehlend, da Innendruck durch Nahterweiterung der Schädeldecke kompensiert wird; ältere Kinder dagegen: Papillenödem bzw. Stauungspapille Sx nach Tumor-Lokalisation Ataxie: Kleinhirntumoren (Medulloblastom, Astrozytom cerebellär oder im Stammhirn) Bulbär-Sx: Hirnstamm-Tu endokrinologische Folgen (Wachstum, verzögerte Pubertät, Diabetes insipidus) und Gesischtsfeldeinschränkungen: Kraniopharyngeom epileptische Anfälle (Grand mal oder fokal): Neoplasien in den Hemisphären Querschnittssymptomatik: bei Cauda-Tumoren (Ependymom) oder Neuroblastom D: MRT, CT, Angio: Sitz, Ausbreitung, Größe EEG, evozierte Potentiale (je nach Lokalisation: VEP, SEP, MEP) Stereotaktische Biopsien T: je nach Histologie, Lokalisation; von Tumor zu Tumor sehr verschieden Astrozytom () (379) häufigster Hirntumor bei Kindern, Grad 1-4 (3+4: Glioblastom), Radikal-OP, sonst Radiatio; gute Prognose; zwei Arten: Kleinhirnastrozytom Häufigkeit bes. zwischen 5 – 15 Jahren; T: mikrochirurgische Entfernung (85% erfolgreich), Residuen lokal bestrahlen Supratentorielle Astrozytome Arten: pilozytisch, fibrillär, höhergradig (Grad 3 und 4: Glioblastom) T: radikale chirurgische Entfernung; lokale Strahlentherapie bei Residuen niedriggradiger A. und bei höhergradigen A. allgemein P: Glioblastom: 2% 5-JÜR; 80% pilozytische A. - Keimzelltumor (S. 513) Intra-/extragonadal (vorders Mediastinum, Sakrokokzygeum, intrakraniell) 0,5/100.000 Mädchen>Jungen; Germinom/Seminom/Dysgerminom – Dottersacktumor – Chorion-Ca – Teratom 28 K: abhängig von Lokalisation: schmerzlose Skrotalschwellung, endokrine Symptome, Bauchschmerzen, Dyspnoe, obere Einflussstauung, Störung der Blasen-/Mastdarmfunktion, Hirndruck, Sehstörungen D: CT/MRT, Tumormarker (AFP – Dottersacktumor, b-HCG – Chorion-Ca) T: nach Stadien: OP bei benignen Tumoren, bei malignen zusätzlich Chemo; Germinom/Seminom/Dysgerminom strahlensensibel P: bei gonadaler Lokalisation besser Immunologie: - DiGeorge (S. 539) 22q11 Deletion auf Chromosom 22: Fehlende Migration von Zellen der Neuralleiste in die 3. und 4. Schlundtasche T-Zell-Defekt, Thymushypo-/aplasie, Hypoparathyreodismus, Herzfehler, faziale Dysmorphie Assoziation mit Catch22 cardiac abnormality abnormal facies tiefe, dysplast. Ohren, Mikrognath., char.Lidachse Thymic hypoplasia Infektionen (Pilze, Viren) cleft palate hypocalcemia Tetanie 22nd chromosome K: makulopap. Exanthem, Tetanie, rezid. Pilz-/Virusinfekte, evtl. Symptome der Herzfehler, Facies (tiefe, dysplast. Ohren, antimongoloide Lidachse, Mikrognathie) D: Hypokalziämie, wenige T-Lymphos T. Ca-Substitution, Vit. D, Infekttherapie, bei schwerem T-Zell-Mangel KM-Transplantation P: T-Zell-Defekt regredient, bestimmend: Herzfehler, Hypoparathyreodismus; Lernschwierigkeiten - Velocardiofaziales Syndrom (VCF, Sphrintzen-Syndrom) Ähnlichkeit/Überlappung mit DiGeorge: Herzfehler, fasziale Dysmorphie; auch 22q11 Deletionen - Nahrumgsmittelallergie (S. 549) Gipfel um 6.LM; Typ1-Immunreaktion, im Kleinkindalter häufig Toleranzentwicklung mit Rückbildung der Symptome K: atopische Dermatitis, Urtikaria, Quinke-Ödeme, seltener GI-Symptome D: Anamnese, Hauttest, RAST T: Expositionsprophylaxe - JCA (S. 561) Chronische Arthritis (>3 Monate) mit Beteiligung von Knorpel und Kapsel-Bandapp. 5-6/100.000; 5 Formen: Polyarthritis seronegativ: Mädchen>Jungen, in jedem Alter, Allgemeinsymptome, symmetrischer Gelenkbefall, IgM-Rheumafaktor negativ Polyarthritis seropositiv: ab 8.LJ, Mädchen> Jungen, symmetrischer Gelenkbefall, Rheumaknoten, Rheumafaktor positiv Oligoarthritis Typ 1: frühkindliche Form, Mädchen>>Jungen, asymmetrischer Befall der großen Gelenke, Uveitis; häufig ANA´s Oligoarthritis Typ 2: HLA-B27-Assoziiert, Jungen>>Mädchen, meist Schulalter, asymmetrische Arthritis, Rückenschmerzen, Iridozyklitis System. Polyarthritis: Still-Syndrom, hohes Fieber, Exanthem, Lymphadenopathie, Hepatosplenomegalie, Polyserositis, Arthritis K: Fieber, Müdigkeit, Morgensteifigkeit, Gelenkbefall, Tenosynovitis, Bursitis, Wachstumsstörungen (Knochenreifung beschleunigt/verlangsamt) 29 D: Klinik, Alter, BKS/CRP, evtl. ANA´s , Rheumafaktor +/T: NSAR, Basistherapeutika (Gold, Chloroquin...), Glukokortikoide, Physiotherapie, OP (Synovektomie, Korrekturen, Gelenkersatz) Infektionen: - Dreitagefieber (S. 588) Exanthema subitum, Roseola infantum; HHV-6 Infektion; häufigste exanthematische Viruserkrankung; fast nur Kinder unter 3 Jahren K: IKZ 5-15 Tage, hohes, unklares Fieber bis 40°C, Kontinua für 3-5 Tage, häufig Fieberkrämpfe; bei kritischer Entfieberung makulopap. Exanthem (Rumpf, Arme, Beine, Hals) (auch Abortivverläufe häufig) D: Klinik, typisches BB (Granulozytopenie mit relativer Lymphozytose), evtl. Serologie T: symptomatisch Fiebersenkung, Diazepam rektal bei Fieberkrampf P: Fieberkrämpfe hinterlassen keine bleibenden Schäden - EBV (S. 600) Infektiöse Mononukleose, Pfeiffersches Drüsenfieber; EBV-Virus, Übertragung durch Speichel K: IKZ 10-50 Tage, Fieber (bis 3 Wochen), LK-Schwellung, Angina, Hepatosplenomegalie Bei Immunsupprimierten: schwere Verläufe, maligne Lymphome, Haarleukoplakie D: Klinik, BB (Leukozytose mit 90% mononukleären Zellen), Transaminasen, Serologie T: Symptomatisch (Antipyretika, Bettruhe), evtl. Steroide Kein Ampicillin ( Exanthem) K: Meningoenzephalitis, Hepatitis, Milzruptur, Anämie, Nephritis, Perimyokarditis - Varizellen (S. 612) Exanthematische und enanthematische Erstinfektion (Windpocken) mit Varicella-Zoster-Virus Zweitinfektion (Gürtelrose). Konnatale Varizellen sind definiert als Windpocken des Neugeborenen (in den ersten 10 Lebenstagen oder bei manifester Erkrankung der Mutter 4 Tage vor bis 2 Tage nach der Geburt); akute, hochkontagiöse (2 Tage vor bis 5 Tage nach Exanthem) Erkrankung mit generalisiertem, schubweisem vesikulärem Exanthem K: IKZ 14 – 16 Tage , Exanthem, versch. Stadien nebeneinander, rote Makulae, Bläschen, und Krusten Heubnersche Sternkarte (Sternenhimmelphänomen), Effloreszenzen treten zuerst am Stamm auf und greifen auf Mundhöhle, Gesicht und – ganz typisch – auf den behaarten Kopf über. D: Klinik, evtl. Serologie (Nachweis spezifischer Antikörper im Serum) DD: Scharlach (Exanthem tritt 2-3 Tage nach Fieberbeginn auf, kann aber auch flüchtig sein oder ganz fehlen), Röteln (Keine Bläschen), Masern (Exanthem beginnt hinter den Ohren und breitet sich von dort über den Stamm auf die Extremitäten) T: symptomatisch mit Juckreiz stillenden Mitteln (Schüttelmixturen, Antihistaminika), keine Salizylate ( Reye-Syndrom mit Übelkeit, Erbrechen, Kopfschmerzen, Erregbarkeit, Delirium und Koma), bei Risikopatienten Aciclovir K: bakterielle Sekundärinfektionen, Zerebellitis, Enzephalitis, Thrombozytopenie, Hepatitis, Arthritis Prophylaxe: Expositionsprophylaxe, passive/aktive Immunprophylaxe (Risikopatienten), Chemoprophylaxe Konnatales Varizellen-Syndrom: Embryopathie, Haut- Skelettdefekte, Augen-, ZNS-Anomalien Konnatale Varizellen: bei Erkrankung der Mutter kurz vor Geburt, Kind erkrankt am 10.-12. LT, schwere Verläufe mit Pneumonie/Enzephalitis möglich Zoster: Reaktivierung, Fieber, Abgeschlagenheit, neuralgische Schmerzen - CMV (S. 615) 30 CMV-Virus, v.a. konnatal und bei Immunsupprimierten; verursacht chronisch persistierende Infektion (Eulenaugenzellen :o) Infektionen meist asymptomatisch für Mutter und Kind.; Infektion des Embyros: Abort oder schwere zerebrale Schädigung; 0.1% der NG: gravierende zerebrale oder viszerale CMV-Infektion K: NG: Mikrozephalie, Hydrocephalus, lymphozytäre Meningitis, periventrikuläre Verkalkungen, NG-Krämpfe, Chorioretinitis; viszerale Sx: Hepatitis, Hepatosplenomegalie, Anämie, Thrombozytopenie, Sepsis; Geburtsgewicht Pränatal: (v.a. 1. Trimenon) geistige Retardierung, Schwerhörigkeit Perinatal: Immunkompetenten: meist asymptomatisch, sonst mononukleoseähnlich; harmlos D: Klinik + Serologie (spezifische IgM, Virusnachweis im Urin/Speichel), „Eulenaugenzellen“ T: Ganciclovir P: NG: schlecht, hohe Letalität; pränatal: geistige Retardierung - TORCH Merkhilfe für paä- und perinatale Infektionen: soll an Infektionen erinnern, die zur Beeinträchtigung des Embryos, des Feten oder des Neugeborenen führen können: T O R C H - = Toxoplasma gondii = other infectious microorganisms = Rubella virus = Cytomegalievirus (CMV) = Herpes simplex-Virus Meningitis (S. 621) Primäre Meningitis : hämatogen (meist viraler Infekt voraus), sekundäre Meningitis : direktes Übergreifen (Otitis media, ZNS-Eingriff…) Typische Erreger nach Alter: 1-3 Monate: B-Strept., E-coli, Listerien Säuglinge/Kleinkinder: Meningo-, pneumokokken, Haemophilus Schulkinder/Erwachsene: Meningo-, Pneumokokken, Borrelia K: Fieber, Erbrechen, Kopfschmerzen, Lichtscheu, meningitische Zeichen (ab Kleinkindalter); bei Säuglingen: Apathie, schrilles Schreien, vorgewölbte Fontanelle, Apnoe D: Lumbalpunktion (Mikrobiologie, Zellzahl, Eiweiß, Laktat, Zucker), Blutkultur, evtl. weitere Diagnostik bei Hirndruck, Rezidiven T: sofortige Antibiose! Kalkuliert mit Cefotaxim/Ceftriaxon, danach nach Antibiogram (Meningokokken/Pneumokokken – Penicillin) Supportive Maßnahmen: Fiebersenkung, Krampftherapie... Pneumokokken: gram+ Diplokokken, Splenektomie als Dispositionsfaktor, Penicillin (zunehmend Resistenzen in Süd-/Südosteuropa), Prognose schlechter, Impfung möglich Meningokokken: gram- Diplokokken, häufige Meningitiserreger, petechiale Blutungen, Waterhouse-Friedrichsen-Syndrom (Endotoxinschock mit DIC), bei adäquater Therapie gute Prognose, Rifampicinprophylaxe, Meldepflicht Weitere: Tbc-Meningitis, Borrelienmeningitis, virale Meningitis - Pertussis (S. 639) Keuchhusten, B. pertussis, hochkontagiös, Tröpfcheninfektion K: IKZ 1-2 Wochen, Krankheitsdauer 6-8 Wochen o Stadium catarrhale (1-2 Wochen): Husten, Schnupfen, Konjunktivitis, subfebrile Temp. o Stadium convulsivum (2-4 Wochen): Hustenattacken (nachts!), (Typisch sind mehrere stakkatoartige Hustenstöße gefolgt von einer laut hörbaren, tiefen Inspiration (Reprise), gewöhnlich wiederholen sich die sog. Hustenperoxysomen mehrmals, bis schließlich zäher, 31 glasiger Schleim ausgwürgt wird) , Zyanose, Einflussstauung, Apnoen, evtl. Petechien, Konjunktivalblutungen (HustenIntrathorakale Druckanstieg) o Stadium decrementi (1-2 Wochen): abklingende Hustenanfälle D: Klinik (charakteristische Hustenanfälle), Mikrobiologie, Serologie, BB (Leukozytose mit Lymphozytose) DD: Virale Bronchitis, Chlamydienpneumonie, zystische Fibrose oder Fremdkörperaspiration T: Prophylaxe Aktive immunisierung: Hitzeabgetöte toxinhaltige Bakterien (Ganzkeim) ab dem 3. Lebensmonat 3x im Abstand von 4 Wochen, dann mit 12-14 Mo. Erwachsenenimpfung alle 10 Jahre sinnvoll Pertussis azellulär: zellfreier impfstoff mit antigenen Komponenten, Impfung von älteren Kindern und Erwachsenen mit Pa möglich Keuchhusten-Immunglobulin: verleiht für 8-10 Tage Schutz, im Stadium catarrhale wirkt es verlaufsmildernd. Bei Säuglingen bzw. schweren Verläufen: stationäre Aufnahme. Eine durchgemachte Infektion hinterlässt keine lebenslange Immunität. Antibiose (Erythromycin), Salbutamol, evtl. Kortikoide - Borreliose (S. 650) B. burgdorferi, Übertragung durch Zecken, v.a. Sommer und Herbst K: IKZ ca.7 Tage Stadium 1 (Erythema migrans), evtl. Fieber, Müdigkeit Myalgien, Arthralgien Stadium 2 (frühe disseminierte Infektion) Arthralgien, Myalgien, Hautveränderungen, ZNS (Fazialisparese, Menigitis, Enzephalitis, radikuläre Schmerzen), Myokarditis, Uveitis Stadium 3 (persitierende Infektion) periphere Neuropathie, progressive Enzephalomyelitis, LymeArthritis, Hautveränderungen D: Anamnese, Klinik (Erythema migrans), Serologie (bei Neuroborreliose aus Liquor) T: Stadium 1 (2) Doxycyclin (<10 J Amoxixillin) 2 Wochen (4-6 Wochen), Stadium 3 Ceftriaxon parenteral P: v.a. bei Neuroborreliose Rezidive und bleibende neurologische Schäden - Orbitaphlegmone: Akute Entzündung des Orbitainhaltes mit den Leitsymptomen Motilitätsstörungen und allgemeinem Krankheitsgefühl. Bei Kindern ist die Orbitaphlegmone die häufigste Ursache für einen Exophthalmus. Ä: meist eine aus der Umgebung fortgeleite Entzündung. Bei Kinder über 84 % sinugen, insbesondere von den Siebbeinzellen und der Sirnhöhle ausgehend. Bei Säuglingen können fortgeleitete Zahnkeimentzündungen ursächlich sein. Seltener: Gesichtsfurunkel, Erysipel, Hordeolum Panophthalmitis, Orbitaverletzungen und Sepsis K: erhebliches Krankheitsgefühl, manchmal mit Fieber und Schmerzen, die durch Augenbewegungen verstärkt werden. D: Exophthalmus mit Bindehaut- und Lidschwellung sowie die deutliche Einschränkung der Bulbusbeweglichkeit (eingemauerter Bulbus), Leukozytose mit erhöhter BSG DD: Rhabdomyosarkom T: stationär hochdosierten i.v. Antibiose (bei Kleinkindern Ceftriaxon, bei Schulkindern: Oxacillin + Cefuroxim), sowie Sanierung der NNH bei sinugener Ätiologie K: Destruktion des N. opticus, Sinus-cavernosus-Thrombose, Meningitis, Hirnabszeß oder Sepsis Neuropädiatrie: - Apnoen () (125) v.a. bei Neugeborene! Atemstillstände oder Atempausen, die mehr als 10 s. dauern. Atmung kann dabei normal/unregelmäßig sein; begleitet von Bradykardien (P: <100/min) und Verlust des Muskeltonus; periodische oder unregelmäßige Atmung und lange Atempausen charakteristisch für 32 Fetus; ein FG mit noch nicht ausgereiftem Atemzentrum wird mit diesem geboren und reagiert auf unterschiedliche Auslöser mit Apnoen (siehe Ursachen), v.a. Hypoxie: während der Geburt erspart er sich durch die Apnoe Energie (keine vergeblichen Atembewegungen); A. können innerhalb weniger Minuten zu erheblichem Hirnschaden/Tod führen. Obstruktiv (= mit vergeblichen Atembemühungen): bei Obstruktion der Atemwege Zentral (= ohne Atembemühungen): direkte Hemmung des Atemzentrums Gemischt: obstruktiv und zentral Reflektorisch (=Vagusreflex): auch ohne Atembemühung Periodische Atmung: Aufeinanderfolge von (mind. 3) Atempausen unterbrochen von jeweils (maximal 20 Sekunden anhaltenden oder 20) regulären Atemzügen Ursachen: Zentrale A. (Hemmung des Atemzentrums) 1. Unreife (unregelmäßige oder periodische Atmung) 2. Hypoxie, Acidose, Schock, zentrale Ischämie (beim Geburtsvorgang spart hier die Apnoe Energie) 3. Medikamente an Mutter oder Kind (Sedative, Morphin(derivate), Narkosen) 4. Hypoglykämie, Hypokalzämie, Hyponatriämie 5. Sepsis 6. Angeborene Herzfehler 7. Schädigung des Gehirns (postasphyktisch, Trauma, Blutung, Meningitis, Fehlbildung) 8. Krampfanfall 9. Temperaturschwankungen (Hyperthermie, Phototherapie) Obstruktive A. Verlegung der Atemwege durch Schleim (Schnupfen), Aspiration Angeborene Stenose, Atresie oder Kompression der Atemwege (Nase, Rachen, Larynx, Trachea) Zentral bedingt durch Tonusschwankungen der glossopharyngealen Muskulatur während des Schlafs Reflektorische A (Vagusreflex) durch Absaugen (Rachen, Magen, Larynx) durch gastroösophagealen Reflux D: jedes NG mit Risiko zu Apnoen und jedes FG (< 34. SSW) überwachen: EKG-Herz/Atemfrequenzmonitor, Pulsoxy, transkutaner arterieller pO2. P: auslösende Ursachen vermeiden (v.a. Temperaturschwankungen im Inkubator!), T: wenn (wie fast immer) im Schlaf: sanfte Stimulation und Wecken des Kindes; bei Hypoxie: Beatmung (hypoxische Atemdepression beseitigen); Intubation/Beatmung; bei Unreife des Atemzentrums ständige Stimulation („Wiegematratze“, CPAP) oder medikamentös (Theophyllin, Coffein); Heimmonitor für zu Hause - Arnold-Chiari / Dysrhaphische Fehlbildungen (S. 682) (521) Dysraphische Fehlbildungen entstehen durch den fehlenden Verschluss des Wirbelkanals, 1-3/1000 Geburten Verschiedene Schweregrade: Spina bifida occulta/aperta, Meningocele, Meningomyelocele, Cranium bifidum, Anenzephalie Arnold Chiari Malformation (Typ II in fast allen Patienten mit Myelomeningozele): Verlagerung der Medulla oblongata und des 4. Ventrikels nach kaudal ins Foramen magnum und den oberen Zervikalkanal (sekundär Hydrozephalus) Lange und schmale obere Medulla und unterer Ponsteil und Fortbestehen der embryonalen Knicks im Zervikalmark Herniation des Kleinhirnwurms durch das Foramen magnum Knochendefekte des Foramen magnum, Occiputs und der oberen Halswirbel Einteilung in Typ I, II und III: je nach Grad der kaudalen Verlagerung; dabei tritt ein Hydrozephalus obstructivus auf: 33 Typ I: Typ II: in später Kindheit: 1-3 Ventrikel vergrößert; Herniation der Kleinhirntonsillen 1-12 Monat: 1-3 Ventrikel , Herniation von Kleinhirnwurm, -tonsillen und 4. Ventrikel, kaudale Dystopie von Pons und Medulla ins Foramen magnum Typ III: 1-12 Monat: 1-3 Ventrikel , Herniation von Kleinhirnwurm, -tonsillen und 4. Ventrikel, kaudale Dystopie von Kleinhirnteilen und Medulla in zervikale Meningozele Folgen: multiple neurologische Ausfälle (allein durch die Chiari-Malformation): Hirnnervenausfälle, Hydrozephalus, spinale Hinterstrangsymptome, Pyramidenbahnzeichen, zerebelläre Ataxie, …) K: nach Schweregrad: Hautgrübchen bis Zele, schlaffe Parese, Sensibilitätsstörungen, Blasen/Mastdarmstörungen, Gelenkkontrakturen D: Klinik, Sono, MRT, Blasenfunktion; Pränatal: Amnion-AFP, Sono T: Defektdeckung, Shunt bei Hydrocephalus, interdisziplinäre Therapie (Physio-, Ergotherapie...) - Hydrozephalus (S. 689) Erweiterung der Liquorräume (Liquorzirkulation, -resorption, -bildung) Versch. Ursachen: Fehlbildungen, Tumoren, Entzündungen, Blutungen K: offene Schädelnähte: Kopfvergrößerung, Sonnenuntergangsphänomen, vorgewölbte Fontanelle, Blickparesen, schrilles Schreien Geschlossene Nähte: Hirndrucksteigerung mit Kopfschmerzen, Einklemmungzeichen (Bewusstlosigkeit, Streckkrämpfe...) D: Klinik, Sono, CT/MRT T: Ursache beseitigen, Liquorableitung P: relativ gut, abhängig von Komplikationen (Shuntinfektionen...) - Enzephalozele Herniation des Gehirns und/oder der Meningen durch einen Knochendefekt des Schädels (Cranium bifidum); Neuralrohr-Störung; je früher die Störung in der Entwicklung, desto schlimmer die Folgen (evtl. nur Meningozele wenn später) K: Größe: von ganz kleinem Liquorkissen bis größer als der Kopf selbst Inhalt: Meningen mit Liquor, auch Groß-/Kleinhirn oder dysplastisches neurales Gewebe Beschaffenheit: mit Haut überdeckt oder nur dünne Membran Sonstiges: evtl. längere Haare um die Zele; vaskuläre Abnormalitäten um oder auf der Zele Lokalisation: 75% occipital, frontal (nasal, nasofrontal, nasopharyngeal), parietal Assoziationen: mit anderen Fehlbildungen, teilweise im Rahmen von Syndromen D: v.a. nasale Zelen nicht sichtbar oder wenn mit Haut überdeckt Verwechslungsgefahr mit Polypen oder Tumoren. Sonst gut sichtbar. MRT/CT: genaue Darstellung der Struktur, Hydrocephalus, assoziierte Fehlbildungen T: i.d.R. operativ; Shunt bei Hydrocephalus Prog.: bei Zelen Infektanfäligkeit; abh. von den anderen Fehlbildungen; reine Meningozelen: bessere Prognose (wohl keine zu erwartenden motorischen oder psychischen Defekte); Meningomyelozelen (je nach Schwere): mentale Retardierung, Ataxie, Spastizität, Anfälle, Blindheit, Augenmotilitätsstörungen) - VP-Shunt-Revision Prinzip des VP-Shunts: dauerhafte Liquorableitung aus dem Ventrikelsystem über Schlauchverbindung vorzugsweise in die freie Bauchhöhle (Verntriculo-peritonealer Schunt, VPShunt) Ind: Hydrocephalus und klinische Symptomatik Verwedung von Shuntsystemen Rickhamkapsel (subkutan gelegenes Liquorreservoir, kann mit Butterfly-Nadel punktiert werden zur Liquordiagnostik und Bestimmng der sog. Steighöhe 34 magnetisch verstellbarem Ventil zur Regulation des Drucks für den Liquorabfluss hohe Druckstufe des Ventils=hoher Widerstand=geringe Liquorabflußmenge niedrige Druckstufe des Ventils=niedriger Widerstand=hohe Liquorabflußmenge Cave: bei NMR Untersuchung duch Magnetfeld des MR Gerätes mögliche Ventilverstellung Neu-Einstellung mit Spezialgerät (über megnetischem Feld) und dann radiologische Kontrolle. Bei bestimmten Indikationen Verikulostomie als Alternative zur Shuntanlage Überwachung der Shuntfunkion: Anamnese, klinische Untersuchung, Kopfumfangskurve, Untersuchungs des Angenhintergrundes, Druckmessung, bildgebende Diagnostik: Ultraschall (Darstellung des Vetrikelsystemes, Dopplersono), Röntgen (Schädel / Shuntverlauf, Druckzeichen: Impressiones digitatae, Sella, Diskonnektion) CT / MRT - Phakomatosen (S. 693) Neurokutane Syndrome, auto.-dom. Vererbung Neurofibromatose: häufigste Phakomatose, zwei Formen, Typ1: Café-au-lait-Flecken, multiple Neurinome, Neurofibrome, Astrozytome, Irishamartome Typ2 : bilat. Akustikusneurinome, Astrozytome Tuberöse Sklerose (Bourneville-Pringle): Adenoma sebaceum, Epilepsie, geistige Behinderung K: BNS-Anfälle, weiße Hautflecken, Adenoma sebaceum, Konen-Tumoren (Nagelfalzfibrome), intrazerebrale Verkalkungen, Astrozytome, Rhabdomyome T: nur symptomatisch Sturge-Weber-Syndrom: Naevus flammeus (Trigeminusast), Angiome in homolateraler Hirnhälfte Weitere Formen: v.Hippel-Lindau-, Louise-Bar-, Peutz-Jegers-Syndrom - MELAS (S. 702) Mitochondrial encephalomyopathy with lactat acidosis and strokelike episodes - Spinale Muskelatrophie (S. 866) Versch. Erkrankungen mit Untergang motorischer Vorderhornzellen Meist auto.-rez. Erbgang, Unterscheidung proximale/(distale) Formen Typ1 (Werdnig-Hoffmann), sehr früh, generalisierte proximale Muskelschwäche, Areflexie, Faszikulationen der Zunge, Glockenthorax (überwiegende Zwechfellatmung bei Schwäche der Interkostalmuskeln), Pneumonieneigung, selten werden Kinder älter als 2,5 Jahre Typ2, symmetrische proximale Muskelschwäche, Hypotonie, Hyporeflexie, feinschlägiger Fingertremor, Kinder lernen Sitzen, Lebenserwartung 3-20 Jahre Typ3 (Kugelberg-Welander), verzögertes Laufenlernen, Schwäche, Faszikulationen, Tremor, Kontrakturen; Manifestation ab 3.LJ, Lebenserwartung 40-60Jahre, teilweise normal D: Klinische Kriterien: symmetrische Schwäche proximal>distal, Beine>Arme, Rumpf-, Interkostalmuskulatur beteiligt EMG, Biopsie, Faszikulationen T: nur symptomatisch - Enzephalitis (S. 707) Entzündung des Hirnparenchymes, häufig in Kombination mit Meningitis, meist viral (Entero, HSV2, CMV, Rubella, VCV, FSME-V) 35 K: katarrhalische Vorgeschichte, Fieber, Kopfschmerz, Erbrechen, Bewusstseinsstörungen, Krämpfe, asymmerische schlaffe Paresen; symptomarmer Beginn ohne Fieber möglich; wenn praenatal E. (Röteln, CMV), dann postnatal Defekte (Mikrozephalus, Zerebralparesen, BNSAnfälle) D: Klinik + o BSG o CRP: hoch: bakteriell, niedrig: möglicherweise viral o BB: Leukozytose o EEG/ CT/MRT: Herdsymptomatik; Besonderheit bei SSPE: „Radermecker-Komplexe“: intermitierend generalisierte, synchrone Entladungen bei sonst normalem EEG) o LP: Zellzahl (/Leukozytose bei bakt. M.), Glucose (erniedrigt bei bakterieller M.), Eiweiß () o Serologie: DNS oder > 4-facher Anstieg spezifischer AK Komplik.: Hirnödem, Abszeßeinbruch ins Ventrikelsystem, Hirnstamminfektion T: bei V.a. Virus-Enzephalitis Virostatikum, bei bakterieller: liquorgängiges Antibiotikum (Cephalosporin, z.B. Spicef); bei akuter bakterieller Meningitis kann man vor Antibiotika-Gabe einmalig Corticosteroide geben, vermindert Hirndefekt nach Abtöten der Bakterien durch Makrophagen usw.; dennoch vorsichtig damit sein, es schächt ja das Immunsystem! Proph: MMR, FSME: Impfung! SSPE: Subakute, sklerosierende Panencephalitis. Lange IKZ; schleichend progrediente Krankheit durch Masernvirus (typisches EEG) mit Zeichen der Demenz. Dann Myoklonien und schubweiser Zerfall der Willkürmotorik. Im 3. Stadium: vegetative Symptomatik. Endstadium wie Dezerebrationsstarre. T: keine Herpesenzephalitis: hämorrhagisch-nekrotisierende Entzündung v.a. des Temporallappens; fokale neurologische Symptome, Partialanfälle D: EEG, CT/MRT, Liquor, PCR T: Aciclovir schon bei Verdacht (3x10mg/kgKG über 10 Tage) Parainfektiöse Enzephalitis: Immunreaktion nach Virusinfektion (Masern-, Pertussis-, Röteln-, Mumpsenzephalitis) - MS (S. 712) Bei Kindern selten, evtl. Autoimmunprozess nach Virusinfektion; disseminierte Entmarkunkungsherde im ZNS, schubförmiger Verlauf K: Sehstörungen (Retrobulbärneuritis), Ataxie D: Liquor (Oligoklonale Banden), visuell evozierte Potentiale, MRT, Ausschluss von Tumoren T: Kortikoide, Immunsuppressiva - Guillain-Barre (S. 715) Polyneuroradikulitis durch Immunprozess nach Infektionen K: Symmetrische, schlaffe, aufsteigende Paresen, Sensibilitätsstörungen, Areflexie D: Liquor (zytalbuminäre Dissoziation), ENG T: Plasmapherese, Immunglobuline K: Herzrhythmusstörungen, Atemlähmung - Fazialisparese (S. 716) Zentral – peripher (Stirnast beteiligt) Ursachen: idiopathisch, durch Borrelien, bei Otitis media, traumatisch, angeboren K: Ausfall der mimischen Muskulatur, Tränen-/Speichelträufeln, Geschmacksstörungen D: Liquor (Borrelien!), ENG T: bei Borreliose Cephalosporin, sonst symptomatisch (Kortikoide, Augenschutz) 36 - Schädelfraktur (S. 717) Schädel-Hirn-Trauma, Commotio (Bewusstlosigkeit <1h), Contusio (>1h), Compressio cerebri (als Folge von Blutungen) Relativ häufig – großer, exponierter Kopf; an Kindsmisshandlung denken K: Bewusstseinsstörung, neurolog. Herdsymptome, vegetative Symptome D: Unfallhergang, Vitalfunktionen, Neurologie, Begleitverletzungen, CT/MRT Komplikationen: Blutungen (Subduralhämatom – Schütteltrauma!), Ödeme, Infektionen T: Vitalfunktionen sichern, Schockbekämpfung, Hirndrucktherapie, Blutungen ausräumen Bei leichtem Trauma Überwachung für 24h - Migräne (S. 726) Funktionelle Störung der cerebralen Gefäße, in jedem Alter möglich K: plötzliche, halbseitig wechselde Kopfschmerzen, Lichtscheu, Übelkeit, Blässe, evtl. Flimmerskotome, Aura Komplizierte Migräne: zusätzlich neurologische Symptome Ophthalmoplegische Migräne: Augenmuskellähmungen Basilarismigräne: Schwindel, Übelkeit D: Tumoren/Gefäßveränderungen ausschließen, Anamnese, Klinik T: akut Paracetamol, Prophylaxe: b-Blocker, Ergotaminpräparate, Ca-Antagonisten, Magnesium P: 1/3 verschwinden mit Pubertät, 1/3 Besserung - Epilepsie (S. 727) Verminderte Krampfschwelle 10%, Gelegenheitskrämpfe 1,5%, Epilepsie (rezidiv. Anfälle) 0,5% der Bevölkerung; Primär generalisierte Anfälle: Grand mal (Bewußtlosigkeit, tonisch-klonischer Anfall, Terminalschlaf) Petit mal (teils altersgebunden) Fokale Anfälle : Herdanfälle (Aura, ohne/mit Bewusstseinstrübung) Sekundär generalisierte Anfälle fokaler Genese D: Anamnese, EEG, andere Ursachen ausschließen, ZNS-Fokussuche (CT/MRT, Liquor) T: akut: Diazepam, Phenobarbital, Phenytoin Prophylaxe: Altersgebundene Anfälle: Valproat, lokale Anfälle: Carbamazepin, generalisierte Anfälle: Primidon/Carbamazepin Altersgebunden fokale Anfälle: Rolando-Epilepsie: gutartig; häufigste Form im Kindesalter (2-12 Jahre), aus dem Schlaf sensorische Missempfindungen im Kopfbereich, partielle oder generalisierte Anfälle, tonsichklonische krämpfe im Gesichtsbereich; EEG sharpe-wave-Muster, verschwinden oft in der Pubertät; manchmal persistieren des EEG-Befundes, aber Kinder anfallsfrei Altersgebundene Petit mal: Neugeborenenkrämpfe: uncharakteristische Symptome (Apnoen, Hypotonie, Myoklonie), Ursachen Hypokalziämie, Hypoglykämie, Infekte, Blutungen BNS: Säuglingsalter (1-3. LJ), Blitz-Nick-Salam, EEG Hypsarrhythmie (=unregelmäßige Frequenzfolge und Amplituden), Hirnschädigung als Ursache schlechte Prognose Myoklonisch-astatische Krämpfe: Lennox-Gastaut, Kleinkindalter, plötzlicher Tonusverlust mit Hinstürzen, Myoklonien Pyknoleptische Absencen: Schulalter, Mädchen, kurze Bewusstseinspausen, EEG 3/s-Spikewave, gute Prognose Impulsiv-Anfälle: myoklonisch-impulsives Petit mal, im Jugendalter, v.a. beim Aufwachen, Zuckungen, Aufwach-Grand mal, gute Prognose 37 Fieberkrämpfe / Infektkrämpfe: Alter 1,5-5 Jahre, rascher Fieberanstieg; keine (entzündliche) Mitbeteiligung des Gehirns!; Unterteilung in einfache und komplizierte Fieberkrämpfe Komplizierte FK: o Vorschädigungen des ZNS o Herdsymptome im Erscheinungsbild des Anfalls oder Herdsymptome nach dem Anfall im EEG o Neurologische Störungen nach dem Anfall o Konstant bleibende Veränderungen im EEG Wochen nach dem Anfall o > 15 min Dauer o Auftreten vor dem 1. und nach dem 4. Geburtstag o Mherere Fieberkrämpfe während eines Infekts o Mehr als insgesamt 4 Infektkrämpfe o Epilepsiebelastung der Familie einfache FK: ohne diese Erscheinungen T: Wichtig: Fiebersenken, sonst kann man schlecht Krämpfe beenden! Diazepam i.v. oder Rektiole (max. 20 mg/Tag) und prophylaktisch bis zur Entfieberung, Fieber senken (physikalisch, antipyretisch medikamentös); wenn nicht Anfall so zu beenden nacheinander versuchen (ab Phenobarbital-Narkose auf Intensiv): Phenytoin, Phenobarbital, Midazolam-Narkose, Phenobarbital-Narkose (selten nötig) Dosen Diazepam i.v. / Rektiole: 2-5 mg Säuglinge 5 mg bei KG < 15 kg 5-10 mg Kleinkinder 10 mg bei KG > 15 kg 10-15 mg Schulkinder - Zerebralparese (S. 733) (514) Little-Krankheit, Lähmungserscheinungen, Haltungs-/Bewegungsstörungen nach frühkindlichem (nicht progredientem) Hirnschaden (Blutungen, Hypoxie, Infektionen...); 3-4/1000 Neugeborene Charakteristika der neurologischen Symptomatik: o positive Pyramidenbahnzeichen o gesteigerte Muskeleigenreflexe o spastischer Muskeltonus o extrapyramidal-motorische Zeichen (verschieden ausgeprägt): Choreoathetose, Dystonie o manchmal auch zusätzlich: geistige Retardierung Sehstörung Sprachentwicklungsverzögerung Anfallsleiden Unterteilung: spastische Hemiparese: eine Körperhälfte; arm- oder beinbetont; meist gute geistige Entw. spastische Diplegie: beinbetonte (Arme weniger) spast. Parese; gute geistige Entwicklung spastische Tetraparese: schwere Lähmungen Arme und Beine; beeinträchtigte geist. Entw. Dyskinetische Syndrome: EPMS neben Spaszitität und Lähmungen (Basalganglien); geist. Entw. variabel (meist motorische Störungen und motorische Sprachstörungen) Ataktische Syndrome: Körperhaltungs- und Bewegungskontrolle mit Stand- und Gangataxie, Intentionstremor, Dysmetrie; zusätzlich Spastik, Dyskinesien, Lähmungen; ohne S & L: andere Fehlbildungen oder progerdiente Erkrankungen! K: Spastik, abnorme Reflexe, Dyskinesien, Ataxie; oft kombiniert mit Intelligenzminderung, Anfällen... Komplikationen: Deformierungen, Fehlstellungen, Kontrakturen D: neurologisches Syndrom oft erst im 2.LJ deutlich, vorher Enrwicklungsverzögerung T: Physiotherapie (Verhinderung von Folgeschäden (Kontrakturen); Förderung von vorhandenen Muskelfunktionen); orthopädische Versorgung; wenn nötig operative Korrekturen von Fehlstellungen 38 - ZNS-Tumoren (S. 518) Bei hereditären Syndromen (Phakomatosen, ...), nach Bestrahlung, Karzinogene (Nitrosamine...)... 2.häufigste Tumoren im Kindesalter 20% (häufigste solide Tumoren); versch. Formen: Astrozytome 50% Primitive neuroektodermale Tumoren 20% (Medulloblastom, Ependymoblastom...) Hirnstammtumoren 15% Kraniopharyngeom 8% Lokalisation bei Kindern überwiegend median/infratentoriell (Ventrikelbeteiligung Hydrozephalus) K: Hirndruckzeichen, fokal-neurologische Symptome D: Anamnese, MRT, Angiographie, wenn möglich Biopsie T: Radiatio, OP, evtl. Chemo Medulloblastom : Kleinhirnwurm, Liquorabtropfmetastasen, Radikal-OP + Nachbestrahlung, Chemo ; 5-JÜR 30-70% Astrozytom : häufigster Hirntumor bei Kindern, Grad 1-4, Radikal-OP, sonst Radiatio; gute Prognose Hirnstammtumoren: unterschiedliche Entitäten mit gemeinsamer infauster Prognose; Bestrahlung Kraniopharyngeom: aus Rathke-Tasche, benigne, intr-/suprasellär, Sehstörungen, endokrine Symptome, OP/Radiatio, Hormonsubstitution - Commotio cerebri / traumatische Schäden Commotio: Plötzlich einsetzender Bewusstseinsverlust mit Amnesie Contusio: fokale oder multifokale Hirnschädigung Compressio: intrakranielle raumfordernde Komplikationen (Ödem, Blutungen) Meist nicht scharf abzugrenzen (z.B. Commotio mit Contusio und lokalis. Ödem; nach Contusio tritt Compressio auf) D: Anamnese; Bildgebung wenn Komplikationen vermutet werden; stationäre Überwachung 48 h (hier in Erlangen) (Blutungen können mit einer Latenz bis zu 7 Tagen auftreten und sich auch schleichend entwickeln) T: keine nötig, evtl. Vorbeugen eines Hirnödems (keine Glucose-Infusionen: Gluc. wird aufgenommen, dann hat man freis Wasser das ins Gehirn diffundiert) Psychatrie: - Enuresis (S. 741) Einnässen nach Vollendung des 4.LJ; primär/sekundär (nach vorübergehender Trockenheit), Enuresis nocturna/diurna D: organische Ursachen ausschliessen (HI, neurolog. Störungen, Fehlbildungen...), Anamnese T: medikamentös Antidepressiva, ADH-Analoga, Verhaltenstherapie („Klingelhose“), Blasentraining - Anorexie / Anorexia nervosa (S. 766) V.a. bei Mädchen in der (Prä-)Pubertät, von psychischen Faktoren abhängige extreme Gewichtsabnahme bzw. Verweigerung der Nahrungsaufnahme, mit der Befürchtung, zu dick zu werden.; Altersgipfel 14.LJ, Prävalenz bis 1% Anfangs Dinge wie Überwachen der Kalorienmenge mit Kalorienplan usw.; Erbrechen, Laxantien. Bei fast allen übermäßiger Bewegungsdrang. Persönlichkeit: depressive Verstimmungen, Sthenizität, Leistungsehrgeiz, überdurchschnittliche Intelligenz. Zwänge und Rituale bezüglich des Essverhaltens. 39 K: Lanugobehaarung, Cutis marmorata, Harausfall, Speicheldrüsenschwellung, Gewichtsverlust, abnormes Essverhalten, sekundäre Amenorrhoe, BB-Veränderungen (leichte Panzytopenie), ElytStörungen, Labor (Transaminasen & Amylase , Eiweiß & Albumin ), Sport, Laxantienabusus; Störungen Hypothalamus-Hypophysen-Achse; CT: Pseudoatrophia cerebri… T: stationär/ambulant: Ziel: Gewichtsnormalisierung, durch verschiedene Methoden Patienten „dazu zu bringen“ P: Letalität 5-13%! - Misshandlung (S. 769) Schätzungen: 20.000-400.000 Fälle/Jahr in Deutschland K: unerklärliche Verletzungen (Subduralhämatom, unterschiedlich alte Hämatome, metaphysäre Absprengungen, alte Frakturen..), Ängstlichkeit, intellektuelle Beeinträchtigungen, Verhaltensauffälligkeiten Auffälligkeit bei Eltern (Erklärungsversuche, unkooperatives Verhalten...) T: individuell angepasst (Gespräch mit Eltern bis Entziehung des Sorgerechts) Haut: - Arzneimittelreaktionen (S. 808) Sehr vielgestaltig, allergisch/nichtallergisch, von anderen Hauterkrankungen oft schwer zu unterscheiden Exanthematisch: Typ4-Immunreaktion am 2.-3., bzw. 9.-18.Tag nach Arzneimitteleinnahme, Morphologie ähnlich den exanthematischen Kinderkrankheiten, selten Fieber, Allgemeinsymptome D: Medikamentenanamnese, selten Hauttests T: Medis absetzen, Antihistaminika, evtl. Kortikoide Nodös/Erythema nodosum: schmerzhafte Knotenbildung, Hautrötung an Unterschenkelstreckseiten; Ursachen: Tbc, Sarkoidose, M. Crohn, Arzneimittel D: Klinik, Tuberkulintest, Röntgen... T: Bettruhe, Steroidcremes, Salicylate (als Ursache ausschließen) Erytheme exudativum multiforme: akuter Verlauf, kokardenförmige Läsionen, Schleimhautblasen; Ursachen: Viren (HHV!), Streptokokken, Yersinien, Mykoplasmen, Medikamente K: symmetrisches, kokardenförmiges Exanthem, Blasen an Haut/Schleimhaut, Fieber Lyell-Syndrom als Maximalform der bullösen Arzneimittelreaktion („Syndrom der verbrühten Haut“) T: Steroidlotionen, antiseptische Lösungen, bei schwerem Verlauf Intensivtherapie, system. Steroide Lyell-Syndrom: Isolation, Intensivpflege, Flüssigkeits-/Eiweißzufuhr, Plasmapherese Letalität bis 30%, abhängig von Komplikationen Knochen: - Osteomyelitis (S. 834) Hämatogene Entzündung des Markraumes, nach Traumen, OP´s; v.a. Streptokokken und Staphylokokken Bei Säuglingen und Erwachsenen Übergreifen auf Gelenk (Pyarthros) möglich, bei Kindern verhindert das die Epiphysenfuge K: häufig zuerst Allgemeininfektion, danach hohes Fieber, Allgemeinsymptome 40 D: Sono (Gelenkerguss), Entzündungszeichen, MRT, Röntgen (erst nach 3 Wochen Veränderungen), Punktion zum Keimnachweis T: i.v.-Antibiose, Ruhigstellung, bei Gelenkbeteiligung Spül-Saug-Drainage Komplikationen: Zerstörung der Wachstumsfuge , Chronifizierung, Gelenkversteifung Chron. Osteomyelitis nach Traumen, Operationen, durch Tbc - Epiphysiolysis (S. 860) Abgleiten der Hüftkopfepiphyse im präpubertären Wachstumsschub, jungen 3x häufiger, Prädisposition: schnelles Wachstum, hormonelle Faktoren (Dystrophia adiposogenitalis, eunuchoider Hochwuchs); Verlauf akut oder chronisch K: Hüft-, Knieschmerzen, Beinverkürzung, Außenrotation; bei akutem Geschehen klinisches Bild wie Schenkelhalsfraktur D: Röntgen (Launsteinaufnahme in Flexin, Abduktion), Drehmann-Zeichen (Bein weicht bei Beugung mit Abduktion und Außenrotation aus) T: Reposition und Fixation Drogen / Substanzen: - Alkoholintoxikation () (643) Bier, Wein, „Kölnisch Wasser“, gelegentlich Alkoholverbände Kleinkinder ab 2 ‰ schnell einsetzendes Koma manchmal ohne Exzitationsstadium K: Lallen, Ataxie, Schläfrigkeit oder Erregung, Übelkeit/Erbrechen, Atemdepression, rasch fortschreitendes Koma, Krämpfe, Hirnödem, Hypoglykämie T: primäre Giftentfernung (Erbrechen), Warmhalten!, 10-40%ige Glucoselösung i.v., Salz-Wasserausgleich; evtl. Beatmung, Hirnödem und Pneumonieprophylaxe (Lagerung, Antibiotika); bei schwersten Vergiftungen Dialyse Leitsymptome: - Durchfall / Diarrhoe Unter Diarrhoe versteht man: o erhöhte Stuhlfrequenz (mehr als 3/Tag, Säuglinge mehr als 6/Tag sind auffällig) o vermehrtes Stuhlvolumen o erhöhter Wassergehalt des Stuhls Sehr breites Symptom, daher hier nur Versuch einer einfachen DD: o akute Diarrhoe infektiöse Enteritis (Fieber, Erbrechen, evtl. blutg. Stühle) Nahrungsmittelvergiftung (Erbrechen, gleiche Sx bei anderen bei gleichem Essen) Botulismus (Erbrechen, Übelkeit, Paresen, Sehstörungen) Pseduomembranöse Colitis (bltg/schleimg Stühle, Fieber, vorher Antibiotikagabe) Antibiotika assozz. Diarrhoe (Antibiotikatherapie, evtl. Erbrechen) o Chronische Diarrhoe im Säuglings- und Kleinkindesalter chr.D., nicht sekretorisch hohe Fruktose und Sorbitzufuhr Kuhmilchproteinintoleranz Zöliakie D. bei Störungen im Immunsystem Folatmalabsorption Enterokinasemangel Acrodermatitis enteropathica Transcobalamin-II-Defekt chr.D., sekretorisch Glucose-Galaktose-Malabsorption Saccharase-(Isomaltase-)Mangel 41 Familiäre Chloriddiarrhoe Natriumdiarrhoe tumorassoziierte Diarrhoe kongenitale Mirkovilus-Atrophie idiopathische Enteropathie mit/ohne Zottenatrophie Kurzdarmsyndrom, erworben; selten kongenital Syndrom der blinden Schlinge, bakterielle Kontamination primäre Gallensäure-Malabsorption chr.D., vorwiegend mit Steatorrhoe zystische Fibrose Shwachmann-Syndrom Abetalipoproteinämie Hypobetalipoproteinämie Wolman-Krankheit chr.D., häufig mit Zeichen einer Kolitis autoimmune Enterokolitis eosinophile Enteropathie M. Hirschsprung o Diarrhoe bei älteren Kindern D., nichtsekretorisch M. Crohn Dermatitis herpetiformis M. Whipple D., mit blutigen Stühlen Colitis ulcerosa - Synkope anfallsartige, kurz dauernde Bewusstlosigkeit infolge Minderduchblutung des Gehirns; im Unterschied zum epileptischen Anfall zeigt sie meist keine motorischesn Begleiterscheinungen; bei Kindern vor allem wegen gestörter Kreislaufregulation in Form einer vasovagaler Synkope oder einer orthostatischen Dysregulation Vasovagale Synkope: Assoziation zu Ereignissen, die initial zu einem verstärkten Sympathikotonus führen: Angst, Schmerz K: zunächst Schwindel, Gesichtsblässe und Herzklopfen evtl. Verwirrtheit, Schweißausbrüche, Übelkeit oder Hyperventilation Orthostatische Dysregulation: charakteristische Zusammenhang zu Lagewechsel; weitere Auslöser: Hitzestau mit Flüssigkeitsmangel oder Dehydratation K: orthostatischer Kollaps (Ohnmacht), Kopfschmerzen, Schwindel und Verwirrtheitzustände D: Anamnese, Blutdruckmessung, EKG, Pulsoxymetrie, Schellong-Test DD: obstruktive Herzerkrankungen, Ischämien, Hypoglykämie, Hypoxie oder Hyperventilation, neurologische und neuropsychiatrischen Ursachen T: Vasovagale S: nach Bewußtseinsverlust Patient flach lagern; werden präsynkopale Symptome rechtzeitig erkannt, kann das sofortige Hinlegen mit Hochlagerung der Beine den Bewußtseinsverlust vermeiden. medikamentös: ß-Blocker Orthostatische Dysregulation:Kreislauftraining, Gymnasik, Wechselduschen und das Vermeiden von langem Stehen (einfach aber oft effektiv), evtl. Etilefrin (Sympathomimietikum) - Splenomegalie bei akuten und chron. Infektionen, Speicherkrankheiten, malignes Geschehen o stark: M.Gaucher, chronische Myelose, Osteomyelofibrose o mäßig: chronische Lymphadenose, Haarzellenleukämie, Thalssamie, aktue Leukämien, hämolytische Anämien, M. Hodgkin, infekiöse Mononukease, NHL, Policythaemia vera, primäre Thrombozytose, thrombotisch-thrombozytoopenische Purpura 42 o leicht: megaloblastäre Anämie, Plasmazytom, Präleukämie Eine kongestive Splenomegalie findet sich häufig bei portaler Hypertension; auch: extrahepatischer Pfortader- oder Milzvenenverschluß Rückstau des venösen Blutes in die Milz Splenomegalie - VACTERL – Assoziation Häufige Kombination / Assoziation von Fehlbildungen. V vertebral defects A anal atresia C cardiac defekts T tracheo-esophageal fistula E esophageal atresia R renal defects (dysplasia) L limb defects (radial aplasia) - Psoriasisarthritis Gemeinsames Auftreten von: Arthritis + Psoriasis Daktylitis Nagelveränderungen (Tüpfelnägel, Hyperkeratosen, Oncholyse oder Ölfleck) Psoriasis bei erstgradigen Verwandten 15% der chron. Athritiden Psoriasis geht selten der Arthritis voraus, aber kann gleichzeitig oder soger Jahre später auftreten. K: Arthritis meist oligoartikulär, gelegentlich polyartikulär; typisch ist Arthritis in den Finger-bzw. Zehenmittel- oder -endgelenken sowie der Befall eines Fingerstrahls D: Klinik, allgemeine Entzündungszeichen oft diskret. HLA-B27 in 30-40%, ANA in 40-60% positiv. T: symptomatisch, medikamentöse Therapie: NSAR, Basismedikamente: Methotraxat, Chloroquin, u.a.; Glukokortikoide Abkürzungen: Sx: Symptom(e) Dx: Diagnose(n) NB: nota bene! (beachte!) Seitenzahlen: 1. Zahl weiß nicht welches Buch (von Gerhard) 2. Zahl: Niessen Pädiatrie 5. & 6. Auflage 43