anhang i zusammenfassung der merkmale des
Werbung

ANHANG I
ZUSAMMENFASSUNG DER MERKMALE DES ARZNEIMITTELS
1
1.
BEZEICHNUNG DES ARZNEIMITTELS
Pradaxa 75 mg Hartkapseln
2.
QUALITATIVE UND QUANTITATIVE ZUSAMMENSETZUNG
Jede Hartkapsel enthält 75 mg Dabigatranetexilat (als Mesilat).
Sonstige Bestandteile: Jede Hartkapsel enthält 2 Mikrogramm Gelborange S (E 110).
Die vollständige Auflistung der sonstigen Bestandteile siehe Abschnitt 6.1.
3.
DARREICHUNGSFORM
Hartkapsel
Bedruckte Kapseln, bestehend aus einer hellblauen, undurchsichtigen Kappe und einem cremefarbenen,
undurchsichtigen Korpus der Größe 2, gefüllt mit gelblichen Pellets. Auf der Kappe ist das Firmenlogo von
Boehringer Ingelheim, auf dem Korpus „R75“ aufgedruckt.
4.
KLINISCHE ANGABEN
4.1
Anwendungsgebiete
Primärprävention von venösen thromboembolischen Ereignissen bei erwachsenen Patienten nach elektivem
chirurgischen Hüft- oder Kniegelenksersatz.
4.2
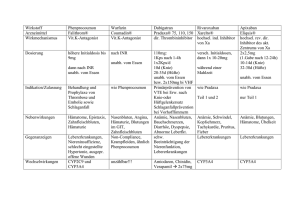
Dosierung, Art und Dauer der Anwendung
Prävention einer venösen Thromboembolie (VTE) bei Patienten nach elektivem chirurgischen
Kniegelenksersatz:
Die empfohlene Dosis von Pradaxa ist 220 mg einmal täglich, eingenommen als 2 Kapseln zu 110 mg. Die
Behandlung sollte postoperativ innerhalb von 1-4 Stunden mit 1 Kapsel oral eingeleitet und anschließend mit
2 Kapseln einmal täglich über insgesamt 10 Tage fortgesetzt werden.
Prävention einer venösen Thromboembolie (VTE) nach elektivem chirurgischen Hüftgelenksersatz:
Die empfohlene Dosis von Pradaxa ist 220 mg einmal täglich, eingenommen als 2 Kapseln zu 110 mg. Die
Behandlung sollte postoperativ innerhalb von 1-4 Stunden mit 1 Kapsel oral eingeleitet und anschließend mit
2 Kapseln einmal täglich über insgesamt 28-35 Tage fortgesetzt werden.
Für beide chirurgische Eingriffe gilt: Bei nicht gesicherter Hämostase ist die Einleitung der Behandlung
aufzuschieben. Wird die Behandlung nicht am Tag des chirurgischen Eingriffs begonnen, sollte der
Therapiebeginn mit 2 Kapseln einmal täglich erfolgen.
Besondere Patientengruppen:
Eingeschränkte Nierenfunktion:
Die Behandlung von Patienten mit schwerer Beeinträchtigung der Nierenfunktion (Kreatinin Clearance
< 30 ml/min) mit Pradaxa ist kontraindiziert (siehe Abschnitt 4.3).
2
Bei Patienten mit mittelgradiger Beeinträchtigung der Nierenfunktion (Kreatinin Clearance 30-50 ml/min)
liegen nur begrenzte klinische Erfahrungen vor. Diese Patienten sollten mit Vorsicht behandelt werden. Die
empfohlene Dosis ist 150 mg einmal täglich, eingenommen als 2 Kapseln zu 75 mg (siehe Abschnitte 4.4
und 5.1).
Nach chirurgischem Kniegelenkersatz sollte die Behandlung postoperativ innerhalb von 1-4 Stunden mit
1 Kapsel eingeleitet und anschließend mit 2 Kapseln einmal täglich über insgesamt 10 Tage fortgesetzt
werden.
Nach chirurgischem Hüftgelenkersatz sollte die Behandlung postoperativ innerhalb von 1-4 Stunden mit
1 Kapsel eingeleitet und anschließend mit 2 Kapseln einmal täglich über insgesamt 28-35 Tage fortgesetzt
werden.
Ältere Personen:
Bei älteren Patienten (über 75 Jahren) liegen nur begrenzte klinische Erfahrungen vor. Diese Patienten
sollten mit Vorsicht behandelt werden. Die empfohlene Dosis ist 150 mg einmal täglich, eingenommen als
2 Kapseln zu 75 mg (siehe Abschnitte 4.4 und 5.1).
Nach chirurgischem Kniegelenkersatz sollte die Behandlung postoperativ innerhalb von 1-4 Stunden mit
1 Kapsel eingeleitet und anschließend mit 2 Kapseln einmal täglich über insgesamt 10 Tage fortgesetzt
werden.
Nach chirurgischem Hüftgelenkersatz sollte die Behandlung postoperativ innerhalb von 1-4 Stunden mit
1 Kapsel eingeleitet und anschließend mit 2 Kapseln einmal täglich über insgesamt 28-35 Tage fortgesetzt
werden.
Beeinträchtigte Leberfunktion:
Patienten mit Erhöhung der Leberenzym-Werte über das 2fache des oberen Grenzwertes des Normbereichs
waren von den klinischen Prüfungen ausgeschlossen. Die Anwendung von Pradaxa bei dieser
Patientengruppe wird daher nicht empfohlen (siehe Abschnitte 4.4 und 5.2). Der ALT-Wert sollte im
Rahmen der präoperativen Untersuchungen routinemäßig gemessen werden (siehe Abschnitt 4.4).
Körpergewicht:
Zur Anwendung der empfohlenen Dosierung bei Patienten mit einem Körpergewicht < 50 kg bzw. > 110 kg
liegen nur sehr begrenzte klinische Erfahrungen vor. Angesichts der verfügbaren klinischen und kinetischen
Daten ist eine Dosisanpassung nicht erforderlich (siehe Abschnitt 5.2); eine engmaschige klinische
Überwachung wird jedoch empfohlen (siehe Abschnitt 4.4).
Patienten mit erhöhtem postoperativen Blutungsrisiko:
Patienten mit erhöhtem Blutungsrisiko oder Patienten mit Risiko einer übermäßigen Exposition,
insbesondere Patienten mit mittelgradiger Beeinträchtigung der Nierenfunktion (Kreatinin Clearance
30-50 ml/min) sollten mit Vorsicht behandelt werden (siehe Abschnitt 4.4 und 5.1).
Kinder und Jugendliche:
Es gibt keine Erfahrungen bei Kindern und Jugendlichen. Pradaxa wird nicht empfohlen für die Anwendung
bei Patienten unter 18 Jahren aufgrund des Fehlens von Daten zur Unbedenklichkeit und Wirksamkeit.
Gleichzeitige Anwendung von Pradaxa und Amiodaron:
Bei Patienten die gleichzeitig Dabigatranetexilat und Amiodaron erhalten, sollte die Tagesdosis auf 150 mg
Pradaxa verringert werden (siehe Abschnitt 4.5).
3
Umstellung von Pradaxa auf ein parenterales Antikoagulans:
Es wird empfohlen nach der letzten Dosis 24 Stunden zu warten, bevor von Pradaxa auf ein parenterales
Antikoagulans umgestellt wird (siehe Abschnitt 4.5).
Umstellung von der Behandlung mit einem parenteralen Antikoagulans auf Pradaxa:
Es liegen keine Daten vor. Daher wird nicht empfohlen mit der Anwendung von Pradaxa vor Fälligkeit der
nächsten ursprünglich planmäßigen Gabe des parenteralen Antikoagulans zu beginnen (siehe Abschnitt 4.5).
Pradaxa sollte unzerkaut mit Wasser zu oder unabhängig von den Mahlzeiten geschluckt werden.
4.3
•
•
•
•
•
•
•
4.4
Gegenanzeigen
Überempfindlichkeit gegen den Wirkstoff oder gegen einen der sonstigen Bestandteile
Patienten mit schwerer Beeinträchtigung der Nierenfunktion (Kreatinin-Clearance < 30 ml/min)
Akute, klinisch relevante Blutung
Organschäden, die das Blutungsrisiko erhöhen
Spontane oder pharmakologisch bedingte Einschränkung der Hämostase
Beeinträchtigung der Leberfunktion oder Lebererkrankung, die Auswirkungen auf das Überleben
erwarten lässt
Gleichzeitige Behandlung mit Chinidin (siehe Abschnitt 4.5)
Besondere Warnhinweise und Vorsichtsmaßnahmen für die Anwendung
Beeinträchtigte Leberfunktion:
Patienten mit Erhöhung der Leberenzym-Werte über das 2fache des oberen Grenzwertes des Normbereichs
waren von den kontrollierten klinischen Prüfungen ausgeschlossen. Die Anwendung von Pradaxa bei dieser
Patientengruppe wird daher nicht empfohlen. Der ALT-Wert sollte im Rahmen der präoperativen
Untersuchungen routinemäßig gemessen werden.
Blutungsrisiko:
Eine engmaschige klinische Überwachung (Kontrolle auf Anzeichen für eine Blutung oder Anämie) wird
über den gesamten Behandlungszeitraum hinweg empfohlen, insbesondere in folgenden Situationen, in
denen ein erhöhtes Blutungsrisiko bestehen könnte: Erkrankungen, die mit erhöhtem Blutungsrisiko
einhergehen, wie angeborene oder erworbene Gerinnungsstörungen, Thrombozytopenie oder funktionelle
Thrombozytendefekte, aktive ulzerierende gastrointestinale Erkrankung, kürzlich durchgeführte Biopsie oder
aufgetretenes schweres Trauma, kürzlich aufgetretene intrakranielle Blutung oder kürzlich erfolgter
chirurgischer Eingriff an Hirn, Rückenmark oder Augen, bakterielle Endokarditis.
Patienten mit mittelgradiger Beeinträchtigung der Nierenfunktion sind einer erhöhten Exposition gegenüber
Dabigatran ausgesetzt. Für Patienten mit einem Körpergewicht < 50 kg sowie älteren Patienten liegen
begrenzte Daten vor (siehe Abschnitte 4.2 und 5.2). In diesen Situationen sollte Pradaxa mit Vorsicht
angewendet werden und eine engmaschige klinische Überwachung (Kontrolle auf Anzeichen für eine
Blutung oder Anämie) ist über den gesamten Behandlungszeitraum hinweg erforderlich (siehe
Abschnitt 4.2).
Beim Auftreten schwerer Blutungen ist die Behandlung abzusetzen und die Blutungsquelle zu ermitteln
(siehe Abschnitt 4.9).
Die gleichzeitige Verabreichung von möglicherweise das Blutungsrisiko erhöhenden Wirkstoffen und
Pradaxa sollte unterbleiben oder mit Vorsicht erfolgen (siehe Abschnitt 4.5).
4
Patienten mit hohem operativen Mortalitätsrisiko und mit spezifischen Risikofaktoren für
thromboembolische Ereignisse:
Bei diesen Patienten liegen begrenzte Daten zur Wirksamkeit und Sicherheit für Dabigatran vor. Sie sollten
daher mit Vorsicht behandelt werden.
Spinalanästhesie/Epiduralanästhesie/Lumbalpunktion:
Bei Patienten nach einem größeren orthopädischen Eingriff können bei gleichzeitiger Anwendung von
Dabigatran und einer Spinal- oder Epiduralanästhesie oder Spinalpunktion Epidural- oder Spinalhämatome,
die zu einer lang andauernden oder permanenten Paralyse führen, nicht ausgeschlossen werden. Das Risiko
für diese seltenen Ereignisse kann bei postoperativem Einsatz von liegendem Epiduralkathether oder
gleichzeitigem Einsatz anderer Arzneimittel, die die Hämostase beeinflussen, höher sein.
Die Anwendung von Pradaxa wird daher bei Patienten, die sich einer Anästhesie mit postoperativ liegendem
Epiduralkatheter unterziehen müssen, nicht empfohlen.
Nach dem Entfernen des Katheters sollte bis zur Verabreichung der ersten Pradaxa-Dosis ein Abstand von
mindestens 2 Stunden liegen. Bei diesen Patienten sind häufige Kontrollen hinsichtlich neurologischen
Anzeichen und Symptomen erforderlich.
Operationen nach Hüftfraktur:
Bei Patienten, die sich einer Operation nach Hüftfraktur unterziehen müssen, liegen keine klinischen Daten
zur Anwendung von Pradaxa vor. Die Anwendung wird daher nicht empfohlen.
Farbstoffe:
Pradaxa Hartkapseln enthalten den Farbstoff Gelborange S (E 110), der allergische Reaktionen hervorrufen
kann.
4.5
Wechselwirkungen mit anderen Arzneimitteln und sonstige Wechselwirkungen
Wechselwirkungsstudien wurden nur bei Erwachsenen durchgeführt.
Antikoagulanzien und Thrombozytenaggregationshemmer:
Die gleichzeitige Anwendung folgender Arzneimittel mit Pradaxa wird nicht empfohlen: unfraktionierte
Heparine und Heparinderivate, niedermolekulare Heparine (NMH), Fondaparinux, Desirudin,
thrombolytische Wirkstoffe, GPIIb/IIIa-Rezeptor-Antagonisten, Clopidogrel, Ticlopidin, Dextran,
Sulfinpyrazon und Vitamin-K-Antagonisten. Es sollte jedoch beachtet werden, dass unfraktioniertes Heparin
in Dosen gegeben werden kann, die notwendig sind, um die Durchgängigkeit eines zentralvenösen- oder
arteriellen Katheters zu erhalten (siehe Abschnitte 4.2 und 4.4).
Wechselwirkungen von Dabigatranetexilat und Stoffwechselprofil von Dabigatran:
Dabigatranetexilat und Dabigatran werden nicht über das Zytochrom-P450-System abgebaut und zeigten in
vitro keine Wirkung auf menschliche Zytochrom-P450-Enzyme. Daher sind für Dabigatran keine
diesbezüglichen Wechselwirkungen mit anderen Arzneimitteln zu erwarten.
NSARs: Bei der gleichzeitigen Verabreichung von Pradaxa und Diclofenac blieb die Plasmaexposition beider
Arzneimittel unverändert, was auf fehlende pharmakokinetische Wechselwirkungen zwischen
Dabigatranetexilat und Diclofenac schließen lässt. Dennoch werden aufgrund des Blutungsrisikos,
insbesondere bei NSAR mit einer Eliminationshalbwertszeit > 12 Stunden, engmaschige Kontrollen
hinsichtlich Anzeichen für eine Blutung empfohlen (siehe Abschnitt 4.4).
5
Wechselwirkungen mit Transportern:
Amiodaron: Amiodaron ist ein Inhibitor des Effluxtransporters P-Glykoprotein und Dabigatranetexilat ist ein
Substrat dieses Transporters. Bei gleichzeitiger Verabreichung von Pradaxa mit Amiodaron waren Ausmaß
und Rate der Resorption von Amiodaron und seines aktiven Metaboliten DEA im Wesentlichen unverändert.
AUC und Cmax von Dabigatran waren um ca. 60 % bzw. 50 % erhöht. Der Mechanismus dieser
Wechselwirkung ist nicht vollständig geklärt. Unter Berücksichtigung der langen Halbwertszeit von
Amiodaron besteht die Möglichkeit einer Arzneimittelwechselwirkung unter Umständen auch über Wochen
nach Absetzen von Amiodaron.
Bei Patienten, die gleichzeitig Dabigatranetexilat und Amiodaron erhalten, sollte die Tagesdosis auf 150 mg
Pradaxa reduziert werden (siehe Abschnitt 4.2).
P-Glykoproteininhibitoren:
Vorsicht ist bei starken P-Glykoproteininhibitoren wie Verapamil, Clarithromycin und andere geboten. Der
P-Glykoproteininhibitor Chinidin ist kontraindiziert (siehe Abschnitt 4.3).
P-Glykoproteininduktoren:
Stark wirksame P-Glykoproteininduktoren wie Rifampicin oder Johanniskraut (Hypericum perforatum)
können die systemische Exposition gegenüber Dabigatran verringern. Vorsicht ist bei der gleichzeitigen
Verabreichung dieser Arzneimittel angebracht.
Digoxin: Bei gleichzeitiger Verabreichung von Pradaxa und Digoxin wurden in einer Studie an 24 gesunden
Probanden keine Veränderungen der Digoxin- und keine klinisch relevanten Veränderungen der DabigatranExposition beobachtet.
Magensaft-pH:
Pantoprazol: Bei gleichzeitiger Verabreichung von Pradaxa und Pantoprazol wurde für Dabigatran eine
Verringerung der AUC um ca. 30 % beobachtet. Bei gleichzeitiger Verabreichung von Pantoprazol sowie
anderen Protonenpumpen-Hemmern und Pradaxa wurde im Rahmen klinischer Prüfungen kein Effekt im
Hinblick auf Blutungen oder Wirksamkeit festgestellt.
Ranitidin: Die gleichzeitige Verabreichung von Ranitidin und Pradaxa zeigte keine klinisch relevante
Wirkung auf das Ausmaß der Resorption von Dabigatran.
4.6
Schwangerschaft und Stillzeit
Schwangerschaft:
Es liegen keine hinreichenden Daten für die Anwendung von Pradaxa bei Schwangeren vor.
Tierexperimentelle Studien haben eine Reproduktionstoxizität gezeigt (siehe Abschnitt 5.3). Das potentielle
Risiko für den Menschen ist nicht bekannt.
Frauen in gebärfähigem Alter sollten während der Behandlung mit Dabigatranetexilat eine Schwangerschaft
vermeiden. Pradaxa sollte nicht während der Schwangerschaft angewendet werden, es sei denn, dies ist
unbedingt erforderlich.
Stillzeit:
Es liegen keine klinischen Daten über die Wirkung von Dabigatran auf Säuglinge während der Stillzeit vor.
Das Stillen sollte während der Behandlung mit Pradaxa unterbrochen werden.
4.7
Auswirkungen auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen
6
Es wurden keine Studien zu den Auswirkungen auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen
von Maschinen durchgeführt.
4.8
Nebenwirkungen
Insgesamt wurden im Rahmen von 4 aktiv kontrollierten VTE-Präventionsstudien 10.084 Patienten mit
mindestens einer Dosis des Arzneimittels behandelt. Von diesen wurden 5.419 Patienten mit 150 mg oder
220 mg Pradaxa täglich behandelt, während 389 Patienten Dosen unter 150 mg täglich und 1.168 Patienten
Dosen über 220 mg täglich erhielten.
Die am häufigsten berichteten Nebenwirkungen sind Blutungen, die bei etwa 14 % der Patienten auftraten;
die Häufigkeit von schweren Blutungen (einschließlich Blutungen an der Wundstelle) ist geringer als 2 %.
Tabelle 1 zeigt Dosis-bezogen die Anzahl der Patienten (%) mit Blutungsereignissen, die während der
Behandlungsphase in den beiden pivotalen Studien zur VTE-Prävention aufgetreten sind.
Tabelle 1: Blutungen (schwere Blutungen/Blutungen insgesamt) in den pivotalen Studien zum Hüft- und
Kniegelenksersatz
Anzahl Behandelte
Schwere Blutungen
Blutungen insgesamt
Dabigatranetexilat
150 mg
N (%)
1.866 (100,0)
24 (1,3)
258 (13,8)
Dabigatranetexilat
220 mg
N (%)
1.825 (100,0)
33
(1,8)
251 (13,8)
Enoxaparin
N (%)
1.848 (100,0)
27
(1,5)
247 (13,4)
Tabelle 2 zeigt die Nebenwirkungen geordnet nach System-Organ-Klassen und Häufigkeit gemäß folgender
Einteilung: Sehr häufig (≥ 1/10), häufig (≥ 1/100, < 1/10), gelegentlich (≥ 1/1.000, < 1/100), selten
(≥ 1/10.000, < 1/1.000), sehr selten (< 1/10.000).
Systemorganklasse /
Bevorzugter Begriff
Dabigatranetexilat
220 mg
N (%)
2.682 (100)
Dabigatranetexilat
150 mg
N (%)
2.737 (100)
Anzahl der behandelten
Patienten
Erkrankungen des Blutes und des Lymphsystems
Häufig
Anämie
110 (4,0)
117
Gelegentlich
Thrombozytopenie
5 (0,2)
2
Gefäßerkrankungen
Häufig
Hämatom
38 (1,4)
37
Traumatisches Hämatom
37 (1,4)
41
Wundblutung
35 (1,3)
28
Gelegentlich
Blutung
5 (0,2)
18
Erkrankungen der Atemwege, des Brustraums und Mediastinums
Gelegentlich
Nasenbluten
19 (0,7)
15
Erkrankungen des Gastrointestinaltrakts
Häufig
Gastrointestinale Blutung
33 (1,2)
17
Gelegentlich
Rektale Blutung
12 (0,4)
15
7
Enoxaparin
N (%)
3.108
(100)
(4,4)
141
(4,5)
(0,1)
5
(0,2)
(1,4)
(1,5)
(1,0)
55
51
31
(1,8)
(1,6)
(1,0)
(0,7)
21
(0,7)
(0,6)
13
(0,4)
(0,6)
20
(0,6)
(0,6)
5
(0,2)
Dabigatranetexilat
Dabigatranetexilat
220 mg
150 mg
N (%)
N (%)
Hämorrhoidale Blutung
4 (0,2)
8 (0,3)
Leber- und Gallenerkrankungen
Gelegentlich
ALT erhöht
18 (0,7)
7 (0,3)
AST erhöht
9 (0,3)
5 (0,2)
Abnorme Leberfunktion /
6 (0,2)
10 (0,4)
Abnormer Leberfunktionstest
Leberenzyme, erhöht
4 (0,2)
5 (0,2)
Hyperbilirubinämie
4 (0,1)
3 (0,1)
Transaminasen, erhöht
0 (0,0)
2 (0,1)
Erkrankungen der Haut und des Unterhautzellgewebes
Häufig
Hautblutung
45 (1,6)
57 (2,1)
Skelettmuskulatur-, Bindegewebs- und Knochenerkrankungen
Gelegentlich
Hämarthros
9 (0,3)
7 (0,3)
Erkrankungen der Nieren und Harnwege
Häufig
Hämaturie
38 (1,4)
33 (1,4)
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort
Gelegentlich
Blutung an der Injektionsstelle
21 (0,8)
19 (0,7)
Blutige Absonderung
2 (0,1)
6 (0,2)
Blutung an der Eintrittsstelle
2 (0,1)
1 (0,0)
des Katheters
Untersuchungen
Häufig
Hämoglobin vermindert
45 (1,6)
35 (1,3)
Gelegentlich
Hämatokrit vermindert
0 (0,0)
6 (0,2)
Verletzung, Vergiftung und durch Eingriffe bedingte Komplikationen
Häufig
Wundsekretion
130 (4,8)
130 (4,9)
Anämie, postoperativ
99 (3,6)
87 (3,2)
Hämatom, postoperativ
66 (2,4)
45 (1,7)
Blutung, postoperativ
37 (1,4)
54 (2,0)
Absonderung, postoperativ
31 (1,1)
34 (1,3)
Chirurgische und medizinische Eingriffe
Gelegentlich
Drainage, postoperativ
11 (0,4)
13 (0,5)
Wunddrainage
1 (0,0)
4 (0,2)
Systemorganklasse /
Bevorzugter Begriff
Enoxaparin
N (%)
2
(0,1)
28
15
7
(0,9)
(0,5)
(0,2)
11
4
1
(0,4)
(0,1)
(0,0)
61
(2,0)
17
(0,6)
25
(0,8)
27
6
7
(0,9)
(0,2)
(0,2)
74
(2,4)
4
(0,1)
93
120
78
56
31
(3,0)
(3,7)
(2,5)
(1,8)
(1,0)
16
2
(0,5)
(0,1)
Neben den berichteten ALT-Werten wurden folgende laborchemische Daten in den Phase-3-Studien ermittelt
(siehe Tabelle 3).
Tabelle 3: ALT-Werte
Gesamthäufigkeit von ALTErhöhungen auf das 3fache der
Dabigatranetexilat
150 mg
N (%)
68 (2,5)
Dabigatranetexilat
220 mg
N (%)
58 (2,2)
8
Enoxaparin
N (%)
95
(3,5)
oberen Grenze des
Normbereichs
4.9
Überdosierung
Es gibt kein Antidot gegen Dabigatran. In höheren als den empfohlenen Dosierungen führt
Dabigatranetexilat zu einem erhöhten Blutungsrisiko. Im Falle hämorrhagischer Komplikationen ist die
Behandlung abzubrechen und die Blutungsquelle festzustellen. Da Dabigatran überwiegend renal
ausgeschieden wird, ist eine ausreichende Diurese sicherzustellen. Die Einleitung einer entsprechenden
Behandlung, z. B. eine chirurgische Hämostase oder der Transfusion gefrorenen Frischplasmas, ist in
Erwägung zu ziehen.
Dabigatran ist dialysefähig; es liegen keine klinischen Erfahrungen vor, die den Nutzen dieses Ansatzes in
klinischen Prüfungen zeigen.
5.
PHARMAKOLOGISCHE EIGENSCHAFTEN
5.1
Pharmakodynamische Eigenschaften
Pharmakotherapeutische Gruppe: Direkter Thrombin-Hemmer, ATC-Code: B01AE07
Dabigatranetexilat ist ein kleinmolekulares Prodrug, das keine pharmakologische Aktivität aufweist. Nach
oraler Verabreichung wird Dabigatranetexilat rasch resorbiert und mittels Esterase-katalysierter Hydrolyse
im Plasma und in der Leber in Dabigatran umgewandelt. Dabigatran ist ein stark wirksamer, kompetitiver,
reversibler direkter Thrombin-Hemmer und das wichtigste Wirkprinzip im Plasma.
Da Thrombin (Serinprotease) in der Gerinnungskaskade die Umwandlung von Fibrinogen zu Fibrin bewirkt,
verhindert seine Hemmung folglich die Thrombusentstehung. Darüber hinaus hemmt Dabigatran sowohl
freies als auch fibringebundenes Thrombin und die thrombininduzierte Thrombozytenaggregation.
Im Rahmen tierexperimenteller In-vivo- und Ex-vivo-Studien wurden die antithrombotische Wirksamkeit und
die antikoagulierende Wirkung von Dabigatran nach intravenöser Verabreichung sowie von
Dabigatranetexilat nach oraler Verabreichung in verschiedenen Thrombose-Tiermodellen nachgewiesen.
Es besteht eine eindeutige Korrelation zwischen der Dabigatran-Plasmakonzentration und dem Grad der
antikoagulierenden Wirkung basierend auf Phase II Studien.
Im Steady state (nach 3 Tagen) liegt die maximale Dabigatran-Plasmakonzentration 2-4 Stunden nach
Verabreichung von 220 mg Dabigatranetexilat ungefähr bei 270 ng/ml, mit einem erwarteten
Konzentrationsbereich von 80-460 ng/ml. Die am Ende des Dosierungsintervalls (24 Stunden nach der
letzten 220 mg Dabigatran-Dosis) gemessene minimale Dabigatran-Plasmakonzentration wird bei ungefähr
40 ng/ml mit einem Konzentrationsbereich von 10-90 ng/ml erwartet.
Ethnische Herkunft:
Mehr als 99 % der Daten zur Wirksamkeit und Sicherheit wurden an Personen mit weißer Hautfarbe
erhoben.
Klinische Studien zur Prävention einer venösen Thromboembolie (VTE) nach Implantation von
Endoprothesen in großen Gelenken:
In 2 großen randomisierten, doppelblinden Parallelgruppenstudien mit Dosisbestätigung erhielten Patienten,
die sich einem elektiven größeren orthopädischen Eingriff (chirurgischer Kniegelenks- bzw.
Hüftgelenksersatz) unterzogen, bei sichergestellter Hämostase innerhalb von 1-4 Stunden nach dem Eingriff
75 mg oder 110 mg Pradaxa sowie anschließend 150 mg oder 220 mg täglich oder aber 40 mg Enoxaparin
9
am Tag vor dem Eingriff sowie im Anschluss täglich. In der RE-MODEL-Studie (Kniegelenksersatz)
erfolgte die Behandlung über 6-10 Tage, in der RE-NOVATE-Studie (Hüftgelenksersatz) über 28-35 Tage.
Die Gesamtzahl der behandelten Patienten betrug 2.076 (Knie) bzw. 3.494 (Hüfte).
Primärer Endpunkt in beiden Studien war der kombinierte Endpunkt aus Gesamtzahl der VTE-Ereignisse
(einschließlich Pulmonalembolie, proximaler und distaler tiefer Venenthrombose, sowohl symptomatische
als auch asymptomatische, festgestellt mittels routinemäßig durchgeführter Phlebografie) und Mortalität
jeglicher Ursache dar. Ein sekundärer Endpunkt, der als klinisch relevanter betrachtet wird, war der
kombinierte Endpunkt aus schwerer VTE (einschließlich Pulmonalembolie und proximaler tiefer
Venenthrombose, sowohl symptomatische als auch asymptomatische, festgestellt mittels routinemäßig
durchgeführter Phlebografie) und VTE-bezogener Mortalität.
Die Ergebnisse beider Studien zeigten, dass der antithrombotische Effekt von 220 mg und 150 mg Pradaxa
im Vergleich zu Enoxaparin bezüglich Gesamtzahl der VTE-Ereignisse und Mortalität jeglicher Ursache
statistisch nicht unterlegen war. Das Risiko einer schweren VTE und VTE-bezogenen Mortalität war für die
150 mg Dosis geringfügig höher als für Enoxaparin (Tabelle 4). Bessere Ergebnisse wurden für die 220 mg
Dosis beobachtet: Das Risiko einer schweren VTE war geringfügig niedriger als für Enoxaparin (Tabelle 4).
Die klinischen Studien wurden in einer Patientenpopulation mit einem durchschnittlichen Alter > 65 Jahren
durchgeführt.
Es bestanden keine Unterschiede in den klinischen Studien der Phase 3 bezüglich Daten zu Wirksamkeit und
Sicherheit zwischen Männern und Frauen.
In der untersuchten Patientenpopulation von RE-MODEL und RE-NOVATE (5.539 behandelte Patienten)
lagen folgende Begleiterkrankungen vor: Hypertonie 51 %, Diabetes und koronare Herzkrankheit je 9 %. Bei
20 % der Patienten war anamnestisch eine venöse Insuffizienz bekannt. Keine dieser Erkrankungen zeigte
einen Einfluss auf die Wirkung von Dabigatran bezüglich VTE-Prävention oder Blutungsraten.
Die Daten für den Endpunkt „schwere VTE und VTE-bezogene Mortalität“ waren hinsichtlich des primären
Wirksamkeits-Endpunktes homogen und sind in Tabelle 4 dargestellt.
Die Daten für die Endpunkte „Gesamtzahl der VTE-Ereignisse und Mortalität jeglicher Ursache“ sind in
Tabelle 5 dargestellt.
Die Daten für den Endpunkt „schwere Blutung“ sind in Tabelle 6 enthalten.
Tabelle 4: Analyse schwerer VTE und VTE-bezogener Mortalität während der Behandlungsphase in den
Studien RE-MODEL und RE-NOVATE zu orthopädischen Eingriffen
Studie
RE-NOVATE (Hüfte)
N
Inzidenz (%)
Risikoverhältnis zu
Enoxaparin
95 %-Konfidenzintervall
RE-MODEL (Knie)
N
Inzidenz (%)
Risikoverhältnis zu
Enoxaparin
95 %-Konfidenzintervall
Dabigatranetexilat
220 mg
Dabigatranetexilat
150 mg
909
28 (3,1)
888
38 (4,3)
0,78
1,09
0,48; 1,27
0,70; 1,70
506
13 (2,6)
527
20 (3,8)
0,73
1,08
0,36; 1,47
0,58; 2,01
10
Enoxaparin
40 mg
917
36 (3,9)
511
18 (3,5)
Tabelle 5: Analyse der Gesamtzahl der VTE-Ereignisse und Mortalität jeglicher Ursache während der
Behandlungsphase in den Studien RE-MODEL und RE-NOVATE zu orthopädischen Eingriffen
Studie
RE-NOVATE (Hüfte)
N
Inzidenz (%)
Risikoverhältnis zu
Enoxaparin
95 %-Konfidenzintervall
RE-MODEL (Knie)
N
Inzidenzen (%)
Risikoverhältnis zu
Enoxaparin
95 %-Konfidenzintervall
Dabigatranetexilat
220 mg
Dabigatranetexilate
150 mg
880
53 (6,0)
0,9
874
75 (8,6)
1,28
(0,63; 1,29)
(0,93; 1,78)
503
183 (36,4)
0,97
526
213 (40,5)
1,07
(0,82; 1,13)
(0,92; 1,25)
Enoxaparin
40 mg
897
60 (6,7)
512
193 (37,7)
Tabelle 6: Ereignisse schwerer Blutungen in der RE-MODEL- und der RE-NOVATE-Studie nach
Behandlungsgruppe
Studie
RE-NOVATE (Hüfte)
Anzahl der Patienten N
Anzahl der ESB N (%)
RE-MODEL (Knie)
Anzahl der Patienten N
Anzahl der ESB N (%)
5.2
Dabigatranetexilat
220 mg
Dabigatranetexilat
150 mg
Enoxaparin
40 mg
1.146
23 (2,0)
1.163
15 (1,3)
1.154
18 (1,8)
679
10 (1,5)
703
9 (1,3)
694
9 (1,3)
Pharmakokinetische Eigenschaften
Nach oraler Verabreichung wird Dabigatranetexilat rasch und vollständig in Dabigatran, die aktive Form im
Plasma, umgewandelt. Die Aufspaltung des Prodrugs Dabigatranetexilat durch Esterase-katalysierte
Hydrolyse in den aktiven Wirkstoff Dabigatran stellt den vorherrschenden Stoffwechselvorgang dar. Die
absolute Bioverfügbarkeit von Dabigatran nach oraler Verabreichung von Pradaxa lag etwa bei 6,5 %.
Nach oraler Verabreichung von Pradaxa an gesunde Probanden ist das pharmakokinetische Profil von
Dabigatran durch einen raschen Anstieg der Plasmakonzentration gekennzeichnet, wobei Cmax innerhalb von
0,5 und 2,0 Stunden nach der Verabreichung erreicht wird.
Resorption:
Eine Studie zur Beurteilung der postoperativen Resorption von Dabigatranetexilat, 1-3 Stunden nach der
Operation verabreicht, ergab im Vergleich zu gesunden Probanden eine relativ langsame Resorption mit
einem ebenmäßigen Plasmakonzentrationszeitprofil ohne hohe maximalen Plasmakonzentrationen. Die
maximalen Plasmakonzentrationen werden 6 Stunden nach der Verabreichung im postoperativen Intervall
erreicht; dies ist auf von der oralen Formulierung des Arzneimittels unabhängige Faktoren wie Anästhesie,
gastrointestinale Parese und Auswirkungen der Operation zurückzuführen. In einer weiteren Studie wurde
nachgewiesen, dass eine langsame und verzögerte Resorption normalerweise nur am Tag des Eingriffs selbst
vorliegt. An den folgenden Tagen wird Dabigatran rasch resorbiert, mit maximalen Plasmakonzentrationen
2 Stunden nach Verabreichung.
Mahlzeiten beeinflussen die Bioverfügbarkeit von Dabigatranetexilat nicht, verzögern jedoch die Zeit bis zur
maximalen Plasmakonzentration um 2 Stunden.
11
Verteilung:
Eine niedrige (34-35 %) konzentrationsunabhängige Bindung von Dabigatran an menschliche
Plasmaproteine wurde beobachtet. Das Verteilungsvolumen von Dabigatran in Höhe von 60-70 l übersteigt
das Volumen des Körperwassers, was auf eine mäßige Verteilung von Dabigatran ins Gewebe schließen
lässt.
Cmax und die AUC waren dosisproportional. Die Plasmakonzentration von Dabigatran sank biexponentiell
mit einer mittleren terminalen Halbwertszeit von 12-14 Stunden bei gesunden Probanden und 14-17 Stunden
bei Patienten, die sich einem größeren orthopädischen Eingriff unterzogen. Die Halbwertszeit war
dosisunabhängig.
Metabolismus und Elimination:
Metabolismus und Ausscheidung von Dabigatran wurden nach einmaliger intravenöser Verabreichung von
radioaktiv markiertem Dabigatran bei gesunden männlichen Probanden untersucht. Nach intravenöser
Verabreichung wurde die von Dabigatran ausgehende Radioaktivität hauptsächlich über den Urin eliminiert
(85 %). Insgesamt 6 % der verabreichten Dosis wurden über die Faeces ausgeschieden. Die
Rückgewinnungsrate der Gesamtradioaktivität betrug 168 Stunden nach Verabreichung 88-94 % der
verabreichten Dosis.
Durch Konjugation entstehen aus Dabigatran pharmakologisch wirksame Acylglucuronide. Es liegen vier
Positionsisomere (und zwar 1-O-, 2-O-, 3-O- und 4-O-Acylglucuronid) vor, von denen jedes weniger als
10 % des Gesamtdabigatrans im Plasma ausmacht. Spuren anderer Metaboliten waren lediglich bei
Verwendung hoch empfindlicher Analysemethoden nachweisbar. Dabigatran wird hauptsächlich in
unveränderter Form über den Urin ausgeschieden. Die Rate entspricht mit ca. 100 ml/min der glomerulären
Filtrationsrate.
Besondere Patientengruppen:
Niereninsuffizienz:
Die Dabigatran-Exposition (AUC) nach oraler Verabreichung von Pradaxa beträgt bei Probanden mit
mittelgradiger Niereninsuffizienz (Kreatinin-Clearance 30-50 ml/min) etwa das 2,7fache verglichen mit der
Exposition bei Probanden ohne Niereninsuffizienz.
Bei einer geringen Zahl von Probanden mit schwerer Niereninsuffizienz (Kreatinin-Clearance 10-30 ml/min)
war die Dabigatran-Exposition (AUC) etwa sechsmal höher und die Halbwertszeit etwa zweimal länger als
bei Patienten ohne Niereninsuffizienz (siehe Abschnitte 4.2, 4.3 und 4.4).
Ältere Patienten:
Spezielle Studien zur Pharmakokinetik mit älteren Probanden ergaben eine Steigerung der AUC von
40-60 % und eine Erhöhung von Cmax um mehr als 25 % im Vergleich zu jungen Probanden. Im Rahmen
populationsbasierter pharmakokinetischer Studien wurde die Pharmakokinetik von Dabigatran nach
wiederholter Gabe bei Patienten (bis 88 Jahre) untersucht. Der beobachtete Anstieg der DabigatranExposition korrelierte mit der altersbedingten Abnahme der Kreatinin-Clearance (siehe Abschnitte 4.2 und
4.4).
Leberinsuffizienz:
Bei 12 Probanden mit mittelgradiger Leberinsuffizienz (Child-Pugh B) wurde im Vergleich zu 12 Kontrollen
keine Veränderung der Dabigatran-Exposition festgestellt (siehe Abschnitte 4.2 und 4.4).
Körpergewicht:
In populationsbasierten Studien zur Pharmakokinetik wurden die pharmakokinetischen Eigenschaften von
Dabigatran bei Patienten mit einem Körpergewicht von 48 bis 120 kg beurteilt. Das Körpergewicht hatte nur
einen geringen Einfluss auf die Plasma-Clearance von Dabigatran, was zu einer höheren Exposition von
Patienten mit niedrigem Körpergewicht führte (siehe Abschnitte 4.2 und 4.4).
12
Geschlecht:
Die Wirkstoffexposition ist bei weiblichen Patienten um etwa 40 bis 50 % höher als bei männlichen
Patienten. Eine Dosisanpassung wird nicht empfohlen.
Ethnische Herkunft:
Die Pharmakokinetik von Dabigatran wurde bei Probanden mit weißer Hautfarbe und japanischen Probanden
nach einmaliger und mehrfacher Verabreichung untersucht. Die ethnische Herkunft hat keinen klinisch
relevanten Einfluss auf die Pharmakokinetik von Dabigatran. Für Patienten mit dunkler Hautfarbe liegen
keine Daten zur Pharmakokinetik vor.
Pharmakokinetische Wechselwirkungen:
In-vitro-Wechselwirkungsstudien ergaben keine Inhibition oder Induktion der wichtigsten Isoenzyme von
Cytochrom P450. Dies wurde im Rahmen von In-vivo-Studien mit gesunden Probanden bestätigt, bei denen
keine Wechselwirkungen zwischen Dabigatran und den folgenden Wirkstoffen auftraten: Atorvastatin
(CYP3A4), Digoxin (P-Glykoprotein-Transporterwechselwirkung) und Diclofenac (CYP2C9).
Bei Anwendung von Amiodaron war die Dabigatran-Exposition bei gesunden Probanden um 60 % erhöht.
5.3
Präklinische Daten zur Sicherheit
Basierend auf den konventionellen Studien zur Sicherheitspharmakologie, Toxizität bei wiederholter Gabe
und Genotoxizität lassen die präklinischen Daten keine besonderen Gefahren für den Menschen erkennen.
Die in den Studien zur Toxizität bei wiederholter Gabe beobachteten Effekte waren auf die übersteigerte
pharmakodynamische Wirkung von Dabigatran zurückzuführen.
Bei 70 mg/kg (entsprechend dem 5fachen der Plasmaexposition bei Patienten) wurde eine Wirkung auf die
weibliche Fertilität in Form einer Abnahme der Implantationen und eines Anstiegs der
Präimplantationsverluste beobachtet. Bei Verabreichung maternaltoxischer Dosen (entsprechend dem 5 bis
10fachen der Plasmaexposition bei Patienten) an Ratten und Kaninchen war eine Verminderung des
Körpergewichts und der Lebensfähigkeit der Föten, einhergehend mit einem Anstieg fötaler Missbildungen,
zu verzeichnen. In der Prä-/Postnatalstudie wurde bei maternaltoxischen Dosen (entsprechend einer 4fach
höheren Plasmaexposition, als sie bei Patienten zu beobachten ist) eine Zunahme der fötalen Mortalität
festgestellt.
Die Karzinogenitätsstudien mit Dabigatran sind noch nicht abgeschlossen.
6.
PHARMAZEUTISCHE ANGABEN
6.1
Liste der sonstigen Bestandteile
Kapselfüllung:
• Weinsäure
• Arabisches Gummi
• Hypromellose
• Dimeticon 350
• Talkum
• Hyprolose
Kapselhülle:
• Carrageenan
• Kaliumchlorid
• Titandioxid
• Indigokarmin (E 132)
• Gelborange S (E 110)
• Hypromellose
13
•
Gereinigtes Wasser
Schwarze Druckfarbe:
• Schellack
• Butan-1-ol
• 2-Propanol
• Ethanol vergällt (mit Aceton, Methanol und Acetylacetat)
• Eisen(II,III)-oxid (E 172)
• Gereinigtes Wasser
• Propylenglycol
6.2
Inkompatibilitäten
Nicht zutreffend.
6.3
Dauer der Haltbarkeit
Blisterpackung und Flasche: 2 Jahre
Nach dem ersten Öffnen der Flasche ist das Arzneimittel innerhalb von 30 Tagen zu verbrauchen.
6.4
Besondere Vorsichtsmaßnahmen für die Aufbewahrung
Blisterpackung:
In der Originalverpackung aufbewahren, um den Inhalt vor Feuchtigkeit zu schützen.
Flasche:
In der Originalverpackung aufbewahren, um den Inhalt vor Feuchtigkeit zu schützen. Die Flasche fest
verschlossen halten.
6.5
Art und Inhalt des Behältnisses
Faltschachteln mit 1, 3 oder 6 Blisterpackung-Folienstreifen (10 x 1, 30 x 1 oder 60 x 1 Hartkapsel) in
perforierten Blisterpackungen zur Abgabe von Einzeldosen aus beschichtetem Aluminium. Die AluminiumBlisterpackungen zur Abgabe von Einzeldosen sind mit Polyvinylchlorid-vinylacetat-copolymer-acrylat
(PVACAC) und Polyvinylchlorid (PVC) beschichtet.
Polypropylen-Flasche mit Schraubdeckel mit 60 Hartkapseln.
Es werden möglicherweise nicht alle Packungsgrößen in den Verkehr gebracht.
6.6
Besondere Vorsichtsmaßnahmen für die Beseitigung und sonstige Hinweise zur Handhabung
Bei der Entnahme der PradaxaKapseln aus der Blisterpackung sollten die folgenden Anweisungen beachtet
werden:
• Um die Hartkapseln zu entnehmen, sollte die rückseitige Folie der Karte der Blisterpackung
abgezogen werden.
• Die Hartkapseln sollten nicht durch die Folie der Blisterpackung gedrückt werden.
• Die Folie der Blisterpackung sollte erst dann abgezogen werden, wenn eine Hartkapsel benötigt wird.
Bei der Entnahme einer Hartkapsel aus der Flasche sollten die folgenden Anweisungen beachtet werden:
• Zum Öffnen den Deckel drücken und drehen.
14
Nicht verwendetes Arzneimittel oder Abfallmaterial ist entsprechend den nationalen Anforderungen zu
entsorgen.
7.
INHABER DER ZULASSUNG
Boehringer Ingelheim International GmbH
D-55216 Ingelheim am Rhein
Deutschland
8.
ZULASSUNGSNUMMER(N)
9.
DATUM DER ERTEILUNG DER ZULASSUNG / VERLÄNGERUNG DER ZULASSUNG
{TT Monat JJJJ}
10.
STAND DER INFORMATION
{MM/JJJJ}
Ausführliche Informationen zu diesem Arzneimittel sind auf der Website der Europäischen ArzneimittelAgentur (EMEA) http://www.emea.europa.eu/ verfügbar.
15
1.
BEZEICHNUNG DES ARZNEIMITTELS
Pradaxa 110 mg Hartkapseln
2.
QUALITATIVE UND QUANTITATIVE ZUSAMMENSETZUNG
Jede Hartkapsel enthält 110 mg Dabigatranetexilat (als Mesilat).
Sonstige Bestandteile: Jede Hartkapsel enthält 3 Mikrogramm Gelborange S (E 110).
Die vollständige Auflistung der sonstigen Bestandteile siehe Abschnitt 6.1.
3.
DARREICHUNGSFORM
Hartkapsel
Bedruckte Kapseln, bestehend aus einer hellblauen, undurchsichtigen Kappe und einem cremefarbenen,
undurchsichtigen Korpus der Größe 1, gefüllt mit gelblichen Pellets. Auf der Kappe ist das Firmenlogo von
Boehringer Ingelheim, auf dem Korpus „R110“ aufgedruckt.
4.
KLINISCHE ANGABEN
4.1
Anwendungsgebiete
Primärprävention von venösen thromboembolischen Ereignissen bei erwachsenen Patienten nach elektivem
chirurgischen Hüft- oder Kniegelenksersatz.
4.2
Dosierung, Art und Dauer der Anwendung
Prävention einer venösen Thromboembolie (VTE) bei Patienten nach elektivem chirurgischen
Kniegelenksersatz:
Die empfohlene Dosis von Pradaxa ist 220 mg einmal täglich, eingenommen als 2 Kapseln zu 110 mg. Die
Behandlung sollte postoperativ innerhalb von 1-4 Stunden mit 1 Kapsel oral eingeleitet und anschließend mit
2 Kapseln einmal täglich über insgesamt 10 Tage fortgesetzt werden.
Prävention einer venösen Thromboembolie (VTE) nach elektivem chirurgischen Hüftgelenksersatz:
Die empfohlene Dosis von Pradaxa ist 220 mg einmal täglich, eingenommen als 2 Kapseln zu 110 mg. Die
Behandlung sollte postoperativ innerhalb von 1-4 Stunden mit 1 Kapsel oral eingeleitet und anschließend mit
2 Kapseln einmal täglich über insgesamt 28-35 Tage fortgesetzt werden.
Für beide chirurgische Eingriffe gilt: Bei nicht gesicherter Hämostase ist die Einleitung der Behandlung
aufzuschieben. Wird die Behandlung nicht am Tag des chirurgischen Eingriffs begonnen, sollte der
Therapiebeginn mit 2 Kapseln einmal täglich erfolgen.
Besondere Patientengruppen:
Eingeschränkte Nierenfunktion:
Die Behandlung von Patienten mit schwerer Beeinträchtigung der Nierenfunktion (Kreatinin Clearance
< 30 ml/min) mit Pradaxa ist kontraindiziert (siehe Abschnitt 4.3).
16
Bei Patienten mit mittelgradiger Beeinträchtigung der Nierenfunktion (Kreatinin Clearance 30-50 ml/min)
liegen nur begrenzte klinische Erfahrungen vor. Diese Patienten sollten mit Vorsicht behandelt werden. Die
empfohlene Dosis ist 150 mg einmal täglich, eingenommen als 2 Kapseln zu 75 mg (siehe Abschnitte 4.4
und 5.1).
Nach chirurgischem Kniegelenkersatz sollte die Behandlung postoperativ innerhalb von 1-4 Stunden mit
1 Kapsel eingeleitet und anschließend mit 2 Kapseln einmal täglich über insgesamt 10 Tage fortgesetzt
werden.
Nach chirurgischem Hüftgelenkersatz sollte die Behandlung postoperativ innerhalb von 1-4 Stunden mit
1 Kapsel eingeleitet und anschließend mit 2 Kapseln einmal täglich über insgesamt 28-35 Tage fortgesetzt
werden.
Ältere Personen:
Bei älteren Patienten (> 75 Jahren) liegen nur begrenzte klinische Erfahrungen vor. Diese Patienten sollten
mit Vorsicht behandelt werden. Die empfohlene Dosis ist 150 mg einmal täglich, eingenommen als
2 Kapseln zu 75 mg) (siehe Abschnitte 4.4 und 5.1).
Nach chirurgischem Kniegelenkersatz sollte die Behandlung postoperativ innerhalb von 1-4 Stunden mit
1 Kapsel eingeleitet und anschließend mit 2 Kapseln einmal täglich über insgesamt 10 Tage fortgesetzt
werden.
Nach chirurgischem Hüftgelenkersatz sollte die Behandlung postoperativ innerhalb von 1-4 Stunden mit
1 Kapsel eingeleitet und anschließend mit 2 Kapseln einmal täglich über insgesamt 28-35 Tage fortgesetzt
werden.
Beeinträchtigte Leberfunktion:
Patienten mit Erhöhung der Leberenzym-Werte über das 2fache des oberen Grenzwertes des Normbereichs
waren von den klinischen Prüfungen ausgeschlossen. Die Anwendung von Pradaxa bei dieser
Patientengruppe wird daher nicht empfohlen (siehe Abschnitte 4.4 und 5.2). Der ALT-Wert sollte im
Rahmen der präoperativen Untersuchungen routinemäßig gemessen werden (siehe Abschnitt 4.4).
Körpergewicht:
Zur Anwendung der empfohlenen Dosierung bei Patienten mit einem Körpergewicht < 50 kg bzw. > 110 kg
liegen nur sehr begrenzte klinische Erfahrungen vor. Angesichts der verfügbaren klinischen und kinetischen
Daten ist eine Dosisanpassung nicht erforderlich (siehe Abschnitt 5.2); eine engmaschige klinische
Überwachung wird jedoch empfohlen (siehe Abschnitt 4.4).
Patienten mit erhöhtem postoperativen Blutungsrisiko:
Patienten mit erhöhtem Blutungsrisiko oder Patienten mit Risiko einer übermäßigen Exposition,
insbesondere Patienten mit mittelgradiger Beeinträchtigung der Nierenfunktion (Kreatinin Clearance
30-50 ml/min) sollten mit Vorsicht behandelt werden (siehe Abschnitte 4.4 und 5.1).
Kinder und Jugendliche:
Es gibt keine Erfahrungen bei Kindern und Jugendlichen. Pradaxa wird nicht empfohlen für die Anwendung
bei Patienten unter 18 Jahren aufgrund des Fehlens von Daten zur Unbedenklichkeit und Wirksamkeit.
Gleichzeitige Anwendung von Pradaxa und Amiodaron:
Bei Patienten die gleichzeitig Dabigatranetexilat und Amiodaron erhalten, sollte die Tagesdosis auf 150 mg
Pradaxa verringert werden (siehe Abschnitt 4.5).
17
Umstellung von Pradaxa auf ein parenterales Antikoagulans:
Es wird empfohlen nach der letzten Dosis 24 Stunden zu warten, bevor von Pradaxa auf ein parenterales
Antikoagulans umgestellt wird (siehe Abschnitt 4.5).
Umstellung von der Behandlung mit einem parenteralen Antikoagulans auf Pradaxa:
Es liegen keine Daten vor. Daher wird nicht empfohlen mit der Anwendung von Pradaxa vor Fälligkeit der
nächsten ursprünglich planmäßigen Gabe des parenteralen Antikoagulans zu beginnen (siehe Abschnitt 4.5).
Pradaxa sollte unzerkaut mit Wasser zu oder unabhängig von den Mahlzeiten geschluckt werden.
4.3
•
•
•
•
•
•
•
4.4
Gegenanzeigen
Überempfindlichkeit gegen den Wirkstoff oder gegen einen der sonstigen Bestandteile
Patienten mit schwerer Beeinträchtigung der Nierenfunktion (Kreatinin-Clearance < 30 ml/min)
Akute, klinisch relevante Blutung
Organschäden, die das Blutungsrisiko erhöhen
Spontane oder pharmakologisch bedingte Einschränkung der Hämostase
Beeinträchtigung der Leberfunktion oder Lebererkrankung, die Auswirkungen auf das Überleben
erwarten lässt
Gleichzeitige Behandlung mit Chinidin (siehe Abschnitt 4.5)
Besondere Warnhinweise und Vorsichtsmaßnahmen für die Anwendung
Beeinträchtigte Leberfunktion:
Patienten mit Erhöhung der Leberenzym-Werte über das 2fache des oberen Grenzwertes des Normbereichs
waren von den kontrollierten klinischen Prüfungen ausgeschlossen. Die Anwendung von Pradaxa bei dieser
Patientengruppe wird daher nicht empfohlen. Der ALT-Wert sollte im Rahmen der präoperativen
Untersuchungen routinemäßig gemessen werden.
Blutungsrisiko:
Eine engmaschige klinische Überwachung (Kontrolle auf Anzeichen für eine Blutung oder Anämie) wird
über den gesamten Behandlungszeitraum hinweg empfohlen, insbesondere in folgenden Situationen, in
denen ein erhöhtes Blutungsrisiko bestehen könnte: Erkrankungen, die mit erhöhtem Blutungsrisiko
einhergehen, wie angeborene oder erworbene Gerinnungsstörungen, Thrombozytopenie oder funktionelle
Thrombozytendefekte, aktive ulzerierende gastrointestinale Erkrankung, kürzlich durchgeführte Biopsie oder
aufgetretenes schweres Trauma, kürzlich aufgetretene intrakranielle Blutung oder kürzlich erfolgter
chirurgischer Eingriff an Hirn, Rückenmark oder Augen, bakterielle Endokarditis.
Patienten mit mittelgradiger Beeinträchtigung der Nierenfunktion sind einer erhöhten Exposition gegenüber
Dabigatran ausgesetzt. Für Patienten mit einem Körpergewicht < 50 kg sowie älteren Patienten liegen
begrenzte Daten vor (siehe Abschnitte 4.2 und 5.2). In diesen Situationen sollte Pradaxa mit Vorsicht
angewendet werden und eine engmaschige klinische Überwachung (Kontrolle auf Anzeichen für eine
Blutung oder Anämie) ist über den gesamten Behandlungszeitraum hinweg erforderlich (siehe
Abschnitt 4.2).
Beim Auftreten schwerer Blutungen ist die Behandlung abzusetzen und die Blutungsquelle zu ermitteln
(siehe Abschnitt 4.9).
Die gleichzeitige Verabreichung von möglicherweise das Blutungsrisiko erhöhenden Wirkstoffen und
Pradaxa sollte unterbleiben oder mit Vorsicht erfolgen (siehe Abschnitt 4.5).
18
Patienten mit hohem operativen Mortalitätsrisiko und mit spezifischen Risikofaktoren für
thromboembolische Ereignisse:
Bei diesen Patienten liegen begrenzte Daten zur Wirksamkeit und Sicherheit für Dabigatran vor. Sie sollten
daher mit Vorsicht behandelt werden.
Spinalanästhesie/Epiduralanästhesie/Lumbalpunktion:
Bei Patienten nach einem größeren orthopädischen Eingriff können bei gleichzeitiger Anwendung von
Dabigatran und einer Spinal- oder Epiduralanästhesie oder Spinalpunktion Epidural- oder Spinalhämatome,
die zu einer lang andauernden oder permanenten Paralyse führen, nicht ausgeschlossen werden. Das Risiko
für diese seltenen Ereignisse kann bei postoperativem Einsatz von liegendem Epiduralkathether oder
gleichzeitigem Einsatz anderer Arzneimittel, die die Hämostase beeinflussen, höher sein.
Die Anwendung von Pradaxa wird daher bei Patienten, die sich einer Anästhesie mit postoperativ liegendem
Epiduralkatheter unterziehen müssen, nicht empfohlen.
Nach dem Entfernen des Katheters sollte bis zur Verabreichung der ersten Pradaxa-Dosis ein Abstand von
mindestens 2 Stunden liegen. Bei diesen Patienten sind häufige Kontrollen hinsichtlich neurologischen
Anzeichen und Symptomen erforderlich.
Operationen nach Hüftfraktur:
Bei Patienten, die sich einer Operation nach Hüftfraktur unterziehen müssen, liegen keine klinischen Daten
zur Anwendung von Pradaxa vor. Die Anwendung wird daher nicht empfohlen.
Farbstoffe:
Pradaxa Hartkapseln enthalten den Farbstoff Gelborange S (E 110), der allergische Reaktionen hervorrufen
kann.
4.5
Wechselwirkungen mit anderen Arzneimitteln und sonstige Wechselwirkungen
Wechselwirkungsstudien wurden nur bei Erwachsenen durchgeführt.
Antikoagulanzien und Thrombozytenaggregationshemmer:
Die gleichzeitige Anwendung folgender Arzneimittel mit Pradaxa wird nicht empfohlen: unfraktionierte
Heparine und Heparinderivate, niedermolekulare Heparine (NMH), Fondaparinux, Desirudin,
thrombolytische Wirkstoffe, GPIIb/IIIa-Rezeptor-Antagonisten, Clopidogrel, Ticlopidin, Dextran,
Sulfinpyrazon und Vitamin-K-Antagonisten. Es sollte jedoch beachtet werden, dass unfraktioniertes Heparin
in Dosen gegeben werden kann, die notwendig sind, um die Durchgängigkeit eines zentralvenösen- oder
arteriellen Katheters zu erhalten (siehe Abschnitte 4.2 und 4.4).
Wechselwirkungen von Dabigatranetexilat und Stoffwechselprofil von Dabigatran:
Dabigatranetexilat und Dabigatran werden nicht über das Zytochrom-P450-System abgebaut und zeigten in
vitro keine Wirkung auf menschliche Zytochrom-P450-Enzyme. Daher sind für Dabigatran keine
diesbezüglichen Wechselwirkungen mit anderen Arzneimitteln zu erwarten.
NSARs: Bei der gleichzeitigen Verabreichung von Pradaxa und Diclofenac blieb die Plasmaexposition beider
Arzneimittel unverändert, was auf fehlende pharmakokinetische Wechselwirkungen zwischen
Dabigatranetexilat und Diclofenac schließen lässt. Dennoch werden aufgrund des Blutungsrisikos,
insbesondere bei NSAR mit einer Eliminationshalbwertszeit > 12 Stunden, engmaschige Kontrollen
hinsichtlich Anzeichen für eine Blutung empfohlen (siehe Abschnitt 4.4).
19
Wechselwirkungen mit Transportern:
Amiodaron: Amiodaron ist ein Inhibitor des Effluxtransporters P-Glykoprotein und Dabigatranetexilat ist ein
Substrat dieses Transporters. Bei gleichzeitiger Verabreichung von Pradaxa mit Amiodaron waren Ausmaß
und Rate der Resorption von Amiodaron und seines aktiven Metaboliten DEA im Wesentlichen unverändert.
AUC und Cmax von Dabigatran waren um ca. 60 % bzw. 50 % erhöht. Der Mechanismus dieser
Wechselwirkung ist nicht vollständig geklärt. Unter Berücksichtigung der langen Halbwertszeit von
Amiodaron besteht die Möglichkeit einer Arzneimittelwechselwirkung unter Umständen auch über Wochen
nach Absetzen von Amiodaron.
Bei Patienten, die gleichzeitig Dabigatranetexilat und Amiodaron erhalten sollte die Dosierung auf 150 mg
Pradaxa täglich reduziert werden (siehe Abschnitt 4.2).
P-Glykoproteininhibitoren:
Vorsicht ist bei starken P-Glykoproteininhibitoren wie Verapamil, Clarithromycin und andere geboten. Der
P-Glykoproteininhibitor Chinidin ist kontraindiziert (siehe Abschnitt 4.3).
P-Glykoproteininduktoren:
Stark wirksame P-Glykoproteininduktoren wie Rifampicin oder Johanniskraut (Hypericum perforatum)
können die systemische Exposition gegenüber Dabigatran verringern. Vorsicht ist bei der gleichzeitigen
Verabreichung dieser Arzneimittel angebracht.
Digoxin: Bei gleichzeitiger Verabreichung von Pradaxa und Digoxin wurden in einer Studie an 24 gesunden
Probanden keine Veränderungen der Digoxin- und keine klinisch relevanten Veränderungen der DabigatranExposition beobachtet.
Magensaft-pH:
Pantoprazol: Bei gleichzeitiger Verabreichung von Pradaxa und Pantoprazol wurde für Dabigatran eine
Verringerung der AUC um ca. 30 % beobachtet. Bei gleichzeitiger Verabreichung von Pantoprazol sowie
anderen Protonenpumpen-Hemmern und Pradaxa wurde im Rahmen klinischer Prüfungen kein Effekt im
Hinblick auf Blutungen oder Wirksamkeit festgestellt.
Ranitidin: Die gleichzeitige Verabreichung von Ranitidin und Pradaxa zeigte keine klinisch relevante
Wirkung auf das Ausmaß der Resorption von Dabigatran.
4.6
Schwangerschaft und Stillzeit
Schwangerschaft:
Es liegen keine hinreichenden Daten für die Anwendung von Pradaxa bei Schwangeren vor.
Tierexperimentelle Studien haben eine Reproduktionstoxizität gezeigt (siehe Abschnitt 5.3). Das potentielle
Risiko für den Menschen ist nicht bekannt.
Frauen in gebärfähigem Alter sollten während der Behandlung mit Dabigatranetexilat eine Schwangerschaft
vermeiden. Pradaxa sollte nicht während der Schwangerschaft angewendet werden, es sei denn, dies ist
unbedingt erforderlich.
Stillzeit:
Es liegen keine klinischen Daten über die Wirkung von Dabigatran auf Säuglinge während der Stillzeit vor.
Das Stillen sollte während der Behandlung mit Pradaxa unterbrochen werden.
4.7
Auswirkungen auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen
20
Es wurden keine Studien zu den Auswirkungen auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen
von Maschinen durchgeführt.
4.8
Nebenwirkungen
Insgesamt wurden im Rahmen von 4 aktiv kontrollierten VTE-Präventionsstudien 10.084 Patienten mit
mindestens einer Dosis des Arzneimittels behandelt. Von diesen wurden 5.419 Patienten mit 150 mg oder
220 mg Pradaxa täglich behandelt, während 389 Patienten Dosen unter 150 mg täglich und 1.168 Patienten
Dosen über 220 mg täglich erhielten.
Die am häufigsten berichteten Nebenwirkungen sind Blutungen, die bei etwa 14 % der Patienten auftraten;
die Häufigkeit von schweren Blutungen (einschließlich Blutungen an der Wundstelle) ist geringer als 2 %.
Tabelle 1 zeigt Dosis-bezogen die Anzahl der Patienten (%) mit Blutungsereignissen, während der
Behandlungsphase in den beiden pivotalen Studien zur VTE-Prävention aufgetreten sind.
Tabelle 1: Blutungen (schwere Blutungen/Blutungen insgesamt) in den pivotalen Studien zum Hüft- und
Kniegelenksersatz
Anzahl Behandelte
Schwere Blutungen
Blutungen insgesamt
Dabigatranetexilat
150 mg
N (%)
1.866 (100,0)
24 (1,3)
258 (13,8)
Dabigatranetexilat
220 mg
N (%)
1.825 (100,0)
33
(1,8)
251 (13,8)
Enoxaparin
N (%)
1.848 (100,0)
27
(1,5)
247 (13,4)
Tabelle 2 zeigt die Nebenwirkungen geordnet nach System-Organ-Klassen und Häufigkeit gemäß folgender
Einteilung: Sehr häufig (≥ 1/10), häufig (≥ 1/100 < 1/10), gelegentlich (≥ 1/1.000 < 1/100), selten
(≥ 1/10.000 < 1/1.000), sehr selten (< 1/10.000).
Systemorganklasse /
Bevorzugter Begriff
Dabigatranetexilat
220 mg
N (%)
2.682 (100)
Dabigatranetexilat
150 mg
N (%)
2.737 (100)
Anzahl der behandelten
Patienten
Erkrankungen des Blutes und des Lymphsystems
Häufig
Anämie
110 (4,0)
117
Gelegentlich
Thrombozytopenie
5 (0,2)
2
Gefäßerkrankungen
Häufig
Hämatom
38 (1,4)
37
Traumatisches Hämatom
37 (1,4)
41
Wundblutung
35 (1,3)
28
Gelegentlich
Blutung
5 (0,2)
18
Erkrankungen der Atemwege, des Brustraums und Mediastinums
Gelegentlich
Nasenbluten
19 (0,7)
15
Erkrankungen des Gastrointestinaltrakts
Häufig
Gastrointestinale Blutung
33 (1,2)
17
Gelegentlich
Rektale Blutung
12 (0,4)
15
21
Enoxaparin
N (%)
3.108
(100)
(4,4)
141
(4,5)
(0,1)
5
(0,2)
(1,4)
(1,5)
(1,0)
55
51
31
(1,8)
(1,6)
(1,0)
(0,7)
21
(0,7)
(0,6)
13
(0,4)
(0,6)
20
(0,6)
(0,6)
5
(0,2)
Dabigatranetexilat
Dabigatranetexilat
220 mg
150 mg
N (%)
N (%)
Hämorrhoidale Blutung
4 (0,2)
8 (0,3)
Leber- und Gallenerkrankungen
Gelegentlich
ALT erhöht
18 (0,7)
7 (0,3)
AST erhöht
9 (0,3)
5 (0,2)
Abnorme Leberfunktion /
6 (0,2)
10 (0,4)
Abnormer Leberfunktionstest
Leberenzyme, erhöht
4 (0,2)
5 (0,2)
Hyperbilirubinämie
4 (0,1)
3 (0,1)
Transaminasen, erhöht
0 (0,0)
2 (0,1)
Erkrankungen der Haut und des Unterhautzellgewebes
Häufig
Hautblutung
45 (1,6)
57 (2,1)
Skelettmuskulatur-, Bindegewebs- und Knochenerkrankungen
Gelegentlich
Hämarthros
9 (0,3)
7 (0,3)
Erkrankungen der Nieren und Harnwege
Häufig
Hämaturie
38 (1,4)
33 (1,4)
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort
Gelegentlich
Blutung an der Injektionsstelle
21 (0,8)
19 (0,7)
Blutige Absonderung
2 (0,1)
6 (0,2)
Blutung an der Eintrittsstelle
2 (0,1)
1 (0,0)
des Katheters
Untersuchungen
Häufig
Hämoglobin vermindert
45 (1,6)
35 (1,3)
Gelegentlich
Hämatokrit vermindert
0 (0,0)
6 (0,2)
Verletzung, Vergiftung und durch Eingriffe bedingte Komplikationen
Häufig
Wundsekretion
130 (4,8)
130 (4,9)
Anämie, postoperativ
99 (3,6)
87 (3,2)
Hämatom, postoperativ
66 (2,4)
45 (1,7)
Blutung, postoperativ
37 (1,4)
54 (2,0)
Absonderung, postoperativ
31 (1,1)
34 (1,3)
Chirurgische und medizinische Eingriffe
Gelegentlich
Drainage, postoperativ
11 (0,4)
13 (0,5)
Wunddrainage
1 (0,0)
4 (0,2)
Systemorganklasse /
Bevorzugter Begriff
Enoxaparin
N (%)
2
(0,1)
28
15
7
(0,9)
(0,5)
(0,2)
11
4
1
(0,4)
(0,1)
(0,0)
61
(2,0)
17
(0,6)
25
(0,8)
27
6
7
(0,9)
(0,2)
(0,2)
74
(2,4)
4
(0,1)
93
120
78
56
31
(3,0)
(3,7)
(2,5)
(1,8)
(1,0)
16
2
(0,5)
(0,1)
Neben den berichteten ALT-Werten wurden folgende laborchemische Daten in den Phase-3-Studien ermittelt
(siehe Tabelle 3).
Tabelle 3: ALT-Werte
Gesamthäufigkeit von ALTErhöhungen auf das 3fache der
Dabigatranetexilat
150 mg
N (%)
68 (2,5)
Dabigatranetexilat
220 mg
N (%)
58 (2,2)
22
Enoxaparin
N (%)
95
(3,5)
oberen Grenze des
Normbereichs
4.9
Überdosierung
Es gibt kein Antidot gegen Dabigatran. In höheren als den empfohlenen Dosierungen führt
Dabigatranetexilat zu einem erhöhten Blutungsrisiko. Im Falle hämorrhagischer Komplikationen ist die
Behandlung abzubrechen und die Blutungsquelle festzustellen. Da Dabigatran überwiegend renal
ausgeschieden wird, ist eine ausreichende Diurese sicherzustellen. Die Einleitung einer entsprechenden
Behandlung, z. B. eine chirurgische Hämostase oder der Transfusion gefrorenen Frischplasmas, ist in
Erwägung zu ziehen.
Dabigatran ist dialysefähig; es liegen keine klinischen Erfahrungen vor, die den Nutzen dieses Ansatzes in
klinischen Prüfungen zeigen.
5.
PHARMAKOLOGISCHE EIGENSCHAFTEN
5.1
Pharmakodynamische Eigenschaften
Pharmakotherapeutische Gruppe: Direkter Thrombin-Hemmer, ATC-Code: B01AE07
Dabigatranetexilat ist ein kleinmolekulares Prodrug, das keine pharmakologische Aktivität aufweist. Nach
oraler Verabreichung wird Dabigatranetexilat rasch resorbiert und mittels Esterase-katalysierter Hydrolyse
im Plasma und in der Leber in Dabigatran umgewandelt. Dabigatran ist ein stark wirksamer, kompetitiver,
reversibler direkter Thrombin-Hemmer und das wichtigste Wirkprinzip im Plasma.
Da Thrombin (Serinprotease) in der Gerinnungskaskade die Umwandlung von Fibrinogen zu Fibrin bewirkt,
verhindert seine Hemmung folglich die Thrombusentstehung. Darüber hinaus hemmt Dabigatran sowohl
freies als auch fibringebundenes Thrombin und die thrombininduzierte Thrombozytenaggregation.
Im Rahmen tierexperimenteller In-vivo- und Ex-vivo-Studien wurden die antithrombotische Wirksamkeit und
die antikoagulierende Wirkung von Dabigatran nach intravenöser Verabreichung sowie von
Dabigatranetexilat nach oraler Verabreichung in verschiedenen Thrombose-Tiermodellen nachgewiesen.
Es besteht eine eindeutige Korrelation zwischen der Dabigatran-Plasmakonzentration und dem Grad der
antikoagulierenden Wirkung basierend auf Phase II Studien.
Im Steady state (nach 3 Tagen) liegt die maximale Dabigatran-Plasmakonzentration 2-4 Stunden nach
Verabreichung von 220 mg Dabigatranetexilat ungefähr bei 270 ng/ml, mit einem erwarteten
Konzentrationsbereich von 80-460 ng/ml. Die am Ende des Dosierungsintervalls (24 Stunden nach der
letzten 220 mg Dabigatran-Dosis) gemessene minimale Dabigatran-Plasmakonzentration wird bei ungefähr
40 ng/ml mit einem Konzentrationsbereich von 10-90 ng/ml erwartet.
Ethnische Herkunft:
Mehr als 99 % der Daten zur Wirksamkeit und Sicherheit wurden an Personen mit weißer Hautfarbe
erhoben.
Klinische Studien zur Prävention einer venösen Thromboembolie (VTE) nach Implantation von
Endoprothesen in großen Gelenken:
In 2 großen randomisierten, doppelblinden Parallelgruppenstudien mit Dosisbestätigung erhielten Patienten,
die sich einem elektiven größeren orthopädischen Eingriff (chirurgischer Kniegelenks- bzw.
Hüftgelenksersatz) unterzogen, bei sichergestellter Hämostase innerhalb von 1-4 Stunden nach dem Eingriff
75 mg oder 110 mg Pradaxa sowie anschließend 150 mg oder 220 mg täglich oder aber 40 mg Enoxaparin
23
am Tag vor dem Eingriff sowie im Anschluss täglich. In der RE-MODEL-Studie (Kniegelenksersatz)
erfolgte die Behandlung über 6-10 Tage, in der RE-NOVATE-Studie (Hüftgelenksersatz) über 28-35 Tage.
Die Gesamtzahl der behandelten Patienten betrug 2.076 (Knie) bzw. 3.494 (Hüfte).
Primärer Endpunkt in beiden Studien war der kombinierte Endpunkt aus Gesamtzahl der VTE-Ereignisse
(einschließlich Pulmonalembolie, proximaler und distaler tiefer Venenthrombose, sowohl symptomatische
als auch asymptomatische, festgestellt mittels routinemäßig durchgeführter Phlebografie) und Mortalität
jeglicher Ursache dar. Ein sekundärer Endpunkt, der als klinisch relevanter betrachtet wird, war der
kombinierte Endpunkt aus schwerer VTE (einschließlich Pulmonalembolie und proximaler tiefer
Venenthrombose, sowohl symptomatische als auch asymptomatische, festgestellt mittels routinemäßig
durchgeführter Phlebografie) und VTE-bezogener Mortalität.
Die Ergebnisse beider Studien zeigten, dass der antithrombotische Effekt von 220 mg und 150 mg Pradaxa
im Vergleich zu Enoxaparin bezüglich Gesamtzahl der VTE-Ereignisse und Mortalität jeglicher Ursache
statistisch nicht unterlegen war. Das Risiko einer schweren VTE und VTE-bezogene Mortalität war für die
150 mg Dosis geringfügig höher als für Enoxaparin (Tabelle 4). Bessere Ergebnisse wurden für die 220 mg
Dosis beobachtet: Das Risiko einer schweren VTE war geringfügig niedriger als für Enoxaparin (Tabelle 4).
Die klinischen Studien wurden in einer Patientenpopulation mit einem durchschnittlichen Alter > 65 Jahren
durchgeführt.
Es bestanden keine Unterschiede in den klinischen Studien der Phase 3 bezüglich Daten zu Wirksamkeit und
Sicherheit zwischen Männern und Frauen.
In der untersuchten Patientenpopulation von RE-MODEL und RE-NOVATE (5.539 behandelte Patienten)
lagen folgende Begleiterkrankungen vor: Hypertonie 51 %, Diabetes und koronare Herzkrankheit je 9 %. Bei
20 % der Patienten war anamnestisch eine venöse Insuffizienz bekannt. Keine dieser Erkrankungen zeigte
einen Einfluss auf die Wirkung von Dabigatran bezüglich VTE-Prävention oder Blutungsraten.
Die Daten für den Endpunkt „schwere VTE und VTE-bezogene Mortalität“ waren hinsichtlich des primären
Wirksamkeits-Endpunktes homogen und sind in Tabelle 4 dargestellt.
Die Daten für die Endpunkte „Gesamtzahl der VTE-Ereignisse und Mortalität jeglicher Ursache“ sind in
Tabelle 5 dargestellt.
Die Daten für den Endpunkt „schwere Blutung“ sind in Tabelle 6 enthalten.
Tabelle 4: Analyse schwerer VTE und VTE-bezogener Mortalität während der Behandlungsphase in den
Studien RE-MODEL und RE-NOVATE zu orthopädischen Eingriffen
Studie
RE-NOVATE (Hüfte)
N
Inzidenz (%)
Risikoverhältnis zu
Enoxaparin
95 %-Konfidenzintervall
RE-MODEL (Knie)
N
Inzidenz (%)
Risikoverhältnis zu
Enoxaparin
95 %-Konfidenzintervall
Dabigatranetexilat
220 mg
Dabigatranetexilat
150 mg
909
28 (3,1)
888
38 (4,3)
0,78
1,09
0,48; 1,27
0,70; 1,70
506
13 (2,6)
527
20 (3,8)
0,73
1,08
0,36; 1,47
0,58; 2,01
24
Enoxaparin
40 mg
917
36 (3,9)
511
18 (3,5)
Tabelle 5: Analyse der Gesamtzahl der VTE-Ereignisse und Mortalität jeglicher Ursache während der
Behandlungsphase in den Studien RE-MODEL und RE-NOVATE zu orthopädischen Eingriffen
Studie
RE-NOVATE (Hüfte)
N
Inzidenz (%)
Risikoverhältnis zu
Enoxaparin
95 %-Konfidenzintervall
RE-MODEL (Knie)
N
Inzidenzen (%)
Risikoverhältnis zu
Enoxaparin
95 %-Konfidenzintervall
Dabigatranetexilat
220 mg
Dabigatranetexilate
150 mg
880
53 (6,0)
0,9
874
75 (8,6)
1,28
(0,63; 1,29)
(0,93; 1,78)
503
183 (36,4)
0,97
526
213 (40,5)
1,07
(0,82; 1,13)
(0,92; 1,25)
Enoxaparin
40 mg
897
60 (6,7)
512
193 (37,7)
Tabelle 6: Ereignisse schwerer Blutungen in der RE-MODEL- und der RE-NOVATE-Studie nach
Behandlungsgruppe
Studie
RE-NOVATE (Hüfte)
Anzahl der Patienten N
Anzahl der ESB N (%)
RE-MODEL (Knie)
Anzahl der Patienten N
Anzahl der ESB N (%)
5.2
Dabigatranetexilat
220 mg
Dabigatranetexilat
150 mg
Enoxaparin
40 mg
1.146
23 (2,0)
1.163
15 (1,3)
1.154
18 (1,8)
679
10 (1,5)
703
9 (1,3)
694
9 (1,3)
Pharmakokinetische Eigenschaften
Nach oraler Verabreichung wird Dabigatranetexilat rasch und vollständig in Dabigatran, die aktive Form im
Plasma, umgewandelt. Die Aufspaltung des Prodrugs Dabigatranetexilat durch Esterase-katalysierte
Hydrolyse in den aktiven Wirkstoff Dabigatran stellt den vorherrschenden Stoffwechselvorgang dar. Die
absolute Bioverfügbarkeit von Dabigatran nach oraler Verabreichung von Pradaxa lag etwa bei 6,5 %.
Nach oraler Verabreichung von Pradaxa an gesunde Probanden ist das pharmakokinetische Profil von
Dabigatran durch einen raschen Anstieg der Plasmakonzentration gekennzeichnet, wobei Cmax innerhalb von
0,5 und 2,0 Stunden nach der Verabreichung erreicht wird.
Resorption:
Eine Studie zur Beurteilung der postoperativen Resorption von Dabigatranetexilat, 1-3 Stunden nach der
Operation verabreicht, ergab im Vergleich zu gesunden Probanden eine relativ langsame Resorption mit
einem ebenmäßigen Plasmakonzentrationszeitprofil ohne hohe maximalen Plasmakonzentrationen. Die
maximalen Plasmakonzentrationen werden 6 Stunden nach der Verabreichung im postoperativen Intervall
erreicht; dies ist auf von der oralen Formulierung des Arzneimittels unabhängige Faktoren wie Anästhesie,
gastrointestinale Parese und Auswirkungen der Operation zurückzuführen. In einer weiteren Studie wurde
nachgewiesen, dass eine langsame und verzögerte Resorption normalerweise nur am Tag des Eingriffs selbst
vorliegt. An den folgenden Tagen wird Dabigatran rasch resorbiert, mit maximalen Plasmakonzentrationen
2 Stunden nach Verabreichung.
Mahlzeiten beeinflussen die Bioverfügbarkeit von Dabigatranetexilat nicht, verzögern jedoch die Zeit bis zur
maximalen Plasmakonzentration um 2 Stunden.
25
Verteilung:
Eine niedrige (34-35 %) konzentrationsunabhängige Bindung von Dabigatran an menschliche
Plasmaproteine wurde beobachtet. Das Verteilungsvolumen von Dabigatran in Höhe von 60-70 l übersteigt
das Volumen des Körperwassers, was auf eine mäßige Verteilung von Dabigatran ins Gewebe schließen
lässt.
Cmax und die AUC waren dosisproportional. Die Plasmakonzentration von Dabigatran sank biexponentiell
mit einer mittleren terminalen Halbwertszeit von 12-14 Stunden bei gesunden Probanden und 14-17 Stunden
bei Patienten, die sich einem größeren orthopädischen Eingriff unterzogen. Die Halbwertszeit war
dosisunabhängig.
Metabolismus und Elimination:
Metabolismus und Ausscheidung von Dabigatran wurden nach einmaliger intravenöser Verabreichung von
radioaktiv markiertem Dabigatran bei gesunden männlichen Probanden untersucht. Nach intravenöser
Verabreichung wurde die von Dabigatran ausgehende Radioaktivität hauptsächlich über den Urin eliminiert
(85 %). Insgesamt 6 % der verabreichten Dosis wurden über die Faeces ausgeschieden. Die
Rückgewinnungsrate der Gesamtradioaktivität betrug 168 Stunden nach Verabreichung 88-94 % der
verabreichten Dosis.
Durch Konjugation entstehen aus Dabigatran pharmakologisch wirksame Acylglucuronide. Es liegen vier
Positionsisomere (und zwar 1-O-, 2-O-, 3-O- und 4-O-Acylglucuronid) vor, von denen jedes weniger als
10 % des Gesamtdabigatrans im Plasma ausmacht. Spuren anderer Metaboliten waren lediglich bei
Verwendung hoch empfindlicher Analysemethoden nachweisbar. Dabigatran wird hauptsächlich in
unveränderter Form über den Urin ausgeschieden. Die Rate entspricht mit ca. 100 ml/min der glomerulären
Filtrationsrate.
Besondere Patientengruppen:
Niereninsuffizienz:
Die Dabigatran-Exposition (AUC) nach oraler Verabreichung von Pradaxa beträgt bei Probanden mit
mittelgradiger Niereninsuffizienz (Kreatinin-Clearance 30-50 ml/min) etwa das 2,7fache verglichen mit der
Exposition bei Probanden ohne Niereninsuffizienz.
Bei einer geringen Zahl von Probanden mit schwerer Niereninsuffizienz (Kreatinin-Clearance 10-30 ml/min)
war die Dabigatran-Exposition (AUC) etwa sechsmal höher und die Halbwertszeit etwa zweimal länger als
bei Patienten ohne Niereninsuffizienz (siehe Abschnitte 4.2, 4.3 und 4.4).
Ältere Patienten:
Spezielle Studien zur Pharmakokinetik mit älteren Probanden ergaben eine Steigerung der AUC von
40-60 % und eine Erhöhung von Cmax um mehr als 25 % im Vergleich zu jungen Probanden. Im Rahmen
populationsbasierter pharmakokinetischer Studien wurde die Pharmakokinetik von Dabigatran nach
wiederholter Gabe bei Patienten (bis 88 Jahre) untersucht. Der beobachtete Anstieg der DabigatranExposition korrelierte mit der altersbedingten Abnahme der Kreatinin-Clearance (siehe Abschnitte 4.2 und
4.4).
Leberinsuffizienz:
Bei 12 Probanden mit mittelgradiger Leberinsuffizienz (Child-Pugh B) wurde im Vergleich zu 12 Kontrollen
keine Veränderung der Dabigatran-Exposition festgestellt (siehe Abschnitte 4.2 und 4.4).
Körpergewicht:
In populationsbasierten Studien zur Pharmakokinetik wurden die pharmakokinetischen Eigenschaften von
Dabigatran bei Patienten mit einem Körpergewicht von 48 bis 120 kg beurteilt. Das Körpergewicht hatte nur
26
einen geringen Einfluss auf die Plasma-Clearance von Dabigatran, was zu einer höheren Exposition von
Patienten mit niedrigem Körpergewicht führte (siehe Abschnitte 4.2 und 4.4).
Geschlecht:
Die Wirkstoffexposition ist bei weiblichen Patienten um etwa 40 bis 50 % höher als bei männlichen
Patienten. Eine Dosisanpassung wird nicht empfohlen.
Ethnische Herkunft:
Die Pharmakokinetik von Dabigatran wurde bei Probanden mit weißer Hautfarbe und japanischen Probanden
nach einmaliger und mehrfacher Verabreichung untersucht. Die ethnische Herkunft hat keinen klinisch
relevanten Einfluss auf die Pharmakokinetik von Dabigatran. Für Patienten mit dunkler Hautfarbe liegen
keine Daten zur Pharmakokinetik vor.
Pharmakokinetische Wechselwirkungen:
In-vitro-Wechselwirkungsstudien ergaben keine Inhibition oder Induktion der wichtigsten Isoenzyme von
Cytochrom P450. Dies wurde im Rahmen von In-vivo-Studien mit gesunden Probanden bestätigt, bei denen
keine Wechselwirkungen zwischen Dabigatran und den folgenden Wirkstoffen auftraten: Atorvastatin
(CYP3A4), Digoxin (P-Glykoprotein-Transporterwechselwirkung) und Diclofenac (CYP2C9).
Bei Anwendung von Amiodaron war die Dabigatran-Exposition bei gesunden Probanden um 60 % erhöht.
5.3
Präklinische Daten zur Sicherheit
Basierend auf den konventionellen Studien zur Sicherheitspharmakologie, Toxizität bei wiederholter Gabe
und Genotoxizität lassen die präklinischen Daten keine besonderen Gefahren für den Menschen erkennen.
Die in den Studien zur Toxizität bei wiederholter Gabe beobachteten Effekte waren auf die übersteigerte
pharmakodynamische Wirkung von Dabigatran zurückzuführen.
Bei 70 mg/kg (entsprechend dem 5fachen der Plasmaexposition bei Patienten) wurde eine Wirkung auf die
weibliche Fertilität in Form einer Abnahme der Implantationen und eines Anstiegs der
Präimplantationsverluste beobachtet. Bei Verabreichung maternaltoxischer Dosen (entsprechend dem 5 bis
10fachen der Plasmaexposition bei Patienten) an Ratten und Kaninchen war eine Verminderung des
Körpergewichts und der Lebensfähigkeit der Föten, einhergehend mit einem Anstieg fötaler Missbildungen,
zu verzeichnen. In der Prä-/Postnatalstudie wurde bei maternaltoxischen Dosen (entsprechend einer 4fach
höheren Plasmaexposition, als sie bei Patienten zu beobachten ist) eine Zunahme der fötalen Mortalität
festgestellt.
Die Karzinogenitätsstudien mit Dabigatran sind noch nicht abgeschlossen.
6.
PHARMAZEUTISCHE ANGABEN
6.1
Liste der sonstigen Bestandteile
Kapselfüllung:
• Weinsäure
• Arabisches Gummi
• Hypromellose
• Dimeticon 350
• Talkum
• Hyprolose
Kapselhülle:
• Carrageenan
• Kaliumchlorid
27
•
•
•
•
•
Titandioxid
Indigokarmin (E 132)
Gelborange S (E 110)
Hypromellose
Gereinigtes Wasser
Schwarze Druckfarbe:
• Schellack
• Butan-1-ol
• 2-Propanol
• Ethanol vergällt (mit Aceton, Methanol und Acetylacetat)
• Eisen(II,III)-oxid (E 172)
• Gereinigtes Wasser
• Propylenglycol
6.2
Inkompatibilitäten
Nicht zutreffend.
6.3
Dauer der Haltbarkeit
Blisterpackung und Flasche: 2 Jahre
Nach dem ersten Öffnen der Flasche ist das Arzneimittel innerhalb von 30 Tagen zu verbrauchen.
6.4
Besondere Vorsichtsmaßnahmen für die Aufbewahrung
Blisterpackung:
In der Originalverpackung aufbewahren, um den Inhalt vor Feuchtigkeit zu schützen.
Flasche:
In der Originalverpackung aufbewahren, um den Inhalt vor Feuchtigkeit zu schützen. Die Flasche fest
verschlossen halten
6.5
Art und Inhalt des Behältnisses
Faltschachteln mit 1, 3 oder 6 Blisterpackung-Folienstreifen (10 x 1, 30 x 1 oder 60 x 1 Hartkapsel) in
perforierten Blisterpackungen zur Abgabe von Einzeldosen aus beschichtetem Aluminium. Die AluminiumBlisterpackungen zur Abgabe von Einzeldosen sind mit Polyvinylchlorid-vinylacetat-copolymer-acrylat
(PVACAC) und Polyvinylchlorid (PVC) beschichtet.
Polypropylen-Flasche mit Schraubdeckel mit 60 Hartkapseln.
Es werden möglicherweise nicht alle Packungsgrößen in den Verkehr gebracht.
6.6
Besondere Vorsichtsmaßnahmen für die Beseitigung und sonstige Hinweise zur Handhabung
Bei der Entnahme der Pradaxa-Kapseln aus der Blisterpackung sollten die folgenden Anweisungen beachtet
werden:
• Um die Hartkapseln zu entnehmen, sollte die rückseitige Folie der Karte der Blisterpackung
abgezogen werden.
• Die Hartkapseln sollten nicht durch die Folie der Blisterpackung gedrückt werden.
• Die Folie der Blisterpackung sollte erst dann abgezogen werden, wenn eine Hartkapsel benötigt wird.
28
Bei der Entnahme einer Hartkapsel aus der Flasche sollten die folgenden Anweisungen beachtet werden:
• Zum Öffnen den Deckel drücken und drehen.
Nicht verwendetes Arzneimittel oder Abfallmaterial ist entsprechend den nationalen Anforderungen zu
entsorgen.
7.
INHABER DER ZULASSUNG
Boehringer Ingelheim International GmbH
D-55216 Ingelheim am Rhein
Deutschland
8.
ZULASSUNGSNUMMER(N)
9.
DATUM DER ERTEILUNG DER ZULASSUNG / VERLÄNGERUNG DER ZULASSUNG
{TT Monat JJJJ}
10.
STAND DER INFORMATION
{MM/JJJJ}
Ausführliche Informationen zu diesem Arzneimittel sind auf der Website der Europäischen ArzneimittelAgentur (EMEA) http://www.emea.europa.eu/ verfügbar.
29
ANHANG II
A.
HERSTELLER DES WIRKSTOFFS UND INHABER DER
HERSTELLUNGSERLAUBNIS, DER FÜR DIE
CHARGENFREIGABE VERANTWORTLICH IST
B.
BEDINGUNGEN DER GENEHMIGUNG FÜR DAS
INVERKEHRBRINGEN
30
A.
HERSTELLER DES WIRKSTOFFS UND INHABER DER HERSTELLUNGSERLAUBNIS,
DER FÜR DIE CHARGENFREIGABE VERANTWORTLICH IST
Name und Anschrift des Herstellers, der für die Chargenfreigabe verantwortlich ist
Boehringer Ingelheim Pharma GmbH & Co. KG
Binger Strasse 173
D-55216 Ingelheim am Rhein
Deutschland
B.
BEDINGUNGEN DER GENEHMIGUNG FÜR DAS INVERKEHRBRINGEN
•
BEDINGUNGEN ODER EINSCHRÄNKUNGEN FÜR DIE ABGABE UND DEN GEBRAUCH,
DIE DEM INHABER DER GENEHMIGUNG FÜR DAS INVERKEHRBRINGEN
AUFERLEGT WERDEN
Arzneimittel, das der Verschreibungspflicht unterliegt.
•
BEDINGUNGEN ODER EINSCHRÄNKUNGEN HINSICHTLICH DER SICHEREN UND
WIRKSAMEN ANWENDUNG DES ARZNEIMITTELS
Nicht zutreffend.
•
SONSTIGE BEDINGUNGEN
Pharmakovigilanz-System
Der Inhaber der Genehmigung für das Inverkehrbringen muss sicherstellen, dass das PharmakovigilanzSystem wie in der im Modul 1.8.1 des Zulassungsantrags präsentierten Version 4.0 vom 30. Juli 2007
beschrieben, vorhanden und funktionsfähig ist, bevor und während das Arzneimittel in den Verkehr
gebracht wird.
Risiko Management Plan:
Der Inhaber der Genehmigung für das Inverkehrbringen verpflichtet sich, die im Pharmakovigilanz-Plan
beschriebenen Studien und zusätzlichen Pharmakovigilanz-Aktivitäten durchzuführen, wie sie in der
Version 1 vom 11. Januar 2007 des Risk Management Plans (RMP) akzeptiert wurden und im Modul
1.8.2 der Zulassungsdokumentation beschrieben sind, sowie alle nachfolgenden Aktualisierungen des
RMP, die durch das CHMP genehmigt werden.
Entsprechend der „CHMP Guideline on Risk Management Systems for medicinal products for human
use” sollte der aktualisierte RMP gleichzeitig mit dem nächsten Periodic Safety Update Report (PSUR)
eingereicht werden.
Zusätzlich sollte ein aktualisierter RMP eingereicht werden:
• wenn neue Informationen erhalten werden, die einen Einfluß auf die aktuelle Sicherheitsspezifikation,
Pharmakovigilanz-Plan oder Aktivitäten zur Risikominimierung haben
• innerhalb von 60 Tagen nach Erreichen eines wichtigen Meilensteines (in der Pharmakovigilanz oder
Risikominimierung)
• auf Anforderung der EMEA
31
ANHANG III
ETIKETTIERUNG UND PACKUNGSBEILAGE
32
A. ETIKETTIERUNG
33
ANGABEN AUF DER ÄUSSEREN UMHÜLLUNG
FALTSCHACHTEL FÜR BLISTERPACKUNG FÜR 75 mg
1.
BEZEICHNUNG DES ARZNEIMITTELS
Pradaxa 75 mg Hartkapseln
Dabigatranetexilat
2.
WIRKSTOFFE
Jede Hartkapsel enthält 75 mg Dabigatranetexilat (als Mesilat).
3.
SONSTIGE BESTANDTEILE
Enthält Gelborange S (E 110) (nähere Informationen siehe Packungsbeilage)
4.
DARREICHUNGSFORM UND INHALT
10 x 1 Hartkapsel
30 x 1 Hartkapsel
60 x 1 Hartkapsel
5.
HINWEISE ZUR UND ART(EN) DER ANWENDUNG
Zum Einnehmen.
Nicht kauen.
Packungsbeilage beachten.
6.
WARNHINWEIS, DASS DAS ARZNEIMITTEL FÜR KINDER UNERREICHBAR UND
NICHT SICHTBAR AUFZUBEWAHREN IST
Arzneimittel für Kinder unzugänglich aufbewahren.
7.
WEITERE WARNHINWEISE, FALLS ERFORDERLICH
8.
VERFALLDATUM
Verwendbar bis: MM JJJJ
9.
BESONDERE LAGERUNGSHINWEISE
In der Originalverpackung aufbewahren, um den Inhalt vor Feuchtigkeit zu schützen.
34
10.
GEGEBENENFALLS BESONDERE VORSICHTSMASSNAHMEN FÜR DIE BESEITIGUNG
VON NICHT VERWENDETEM ARZNEIMITTEL ODER DAVON STAMMENDEN
ABFALLMATERIALIEN
Nicht verwendetes Arzneimittel oder Abfallmaterial ist entsprechend den nationalen Anforderungen zu
entsorgen.
11.
NAME UND ANSCHRIFT DES PHARMAZEUTISCHEN UNTERNEHMERS
Boehringer Ingelheim International GmbH
Binger Str. 173
D-55216 Ingelheim am Rhein
Deutschland
12.
ZULASSUNGSNUMMER(N)
EU/0/00/000/000
EU/0/00/000/000
EU/0/00/000/000
13.
CHARGENBEZEICHNUNG
Ch.-B.:
14.
VERKAUFSABGRENZUNG
Verschreibungspflichtig
15.
HINWEISE FÜR DEN GEBRAUCH
16.
INFORMATION IN BRAILLE-SCHRIFT
Pradaxa 75 mg
35
ANGABEN AUF DER ÄUSSEREN UMHÜLLUNG
FALTSCHACHTEL FÜR BLISTERPACKUNG FÜR 110 mg
1.
BEZEICHNUNG DES ARZNEIMITTELS
Pradaxa 110 mg Hartkapseln
Dabigatranetexilat
2.
WIRKSTOFFE
Jede Hartkapsel enthält 110 mg Dabigatranetexilat (als Mesilat).
3.
SONSTIGE BESTANDTEILE
Enthält Gelborange S (E 110) (nähere Informationen siehe Packungsbeilage)
4.
DARREICHUNGSFORM UND INHALT
10 x 1 Hartkapsel
30 x 1 Hartkapsel
60 x 1 Hartkapsel
5.
HINWEISE ZUR UND ART(EN) DER ANWENDUNG
Zum Einnehmen.
Nicht kauen.
Packungsbeilage beachten.
6.
WARNHINWEIS, DASS DAS ARZNEIMITTEL FÜR KINDER UNERREICHBAR UND
NICHT SICHTBAR AUFZUBEWAHREN IST
Arzneimittel für Kinder unzugänglich aufbewahren.
7.
WEITERE WARNHINWEISE, FALLS ERFORDERLICH
8.
VERFALLDATUM
Verwendbar bis: MM JJJJ
9.
BESONDERE LAGERUNGSHINWEISE
In der Originalverpackung aufbewahren, um den Inhalt vor Feuchtigkeit zu schützen.
36
10.
GEGEBENENFALLS BESONDERE VORSICHTSMASSNAHMEN FÜR DIE BESEITIGUNG
VON NICHT VERWENDETEM ARZNEIMITTEL ODER DAVON STAMMENDEN
ABFALLMATERIALIEN
Nicht verwendetes Arzneimittel oder Abfallmaterial ist entsprechend den nationalen Anforderungen zu
entsorgen.
11.
NAME UND ANSCHRIFT DES PHARMAZEUTISCHEN UNTERNEHMERS
Boehringer Ingelheim International GmbH
Binger Str. 173
D-55216 Ingelheim am Rhein
Deutschland
12.
ZULASSUNGSNUMMER(N)
EU/0/00/000/000
EU/0/00/000/000
EU/0/00/000/000
13.
CHARGENBEZEICHNUNG
Ch.-B.:
14.
VERKAUFSABGRENZUNG
Verschreibungspflichtig
15.
HINWEISE FÜR DEN GEBRAUCH
16.
INFORMATION IN BRAILLE-SCHRIFT
Pradaxa 110 mg
37
MINDESTANGABEN AUF BLISTERPACKUNGEN ODER FOLIENSTREIFEN
BLISTER FÜR 75 mg
1.
BEZEICHNUNG DES ARZNEIMITTELS
Pradaxa 75 mg Hartkapseln
Dabigatranetexilat
2.
NAME DES PHARMAZEUTISCHEN UNTERNEHMERS
Boehringer Ingelheim (Logo)
3.
VERFALLDATUM
Verw. bis: MM JJJJ
4.
CHARGENBEZEICHNUNG
Ch.-B.:
5.
WEITERE ANGABEN
Abziehen
38
MINDESTANGABEN AUF BLISTERPACKUNGEN ODER FOLIENSTREIFEN
BLISTER FÜR 110 mg
1.
BEZEICHNUNG DES ARZNEIMITTELS
Pradaxa 110 mg Hartkapseln
Dabigatranetexilat
2.
NAME DES PHARMAZEUTISCHEN UNTERNEHMERS
Boehringer Ingelheim (Logo)
3.
VERFALLDATUM
Verw. bis: MM JJJJ
4.
CHARGENBEZEICHNUNG
Ch.-B.:
5.
WEITERE ANGABEN
Abziehen
39
ANGABEN AUF DER ÄUSSEREN UMHÜLLUNG UND DEM PRIMÄRPACKMITTEL
FALTSCHACHTEL UND FLASCHEN-ETIKETT FÜR 75 mg
1.
BEZEICHNUNG DES ARZNEIMITTELS
Pradaxa 75 mg Hartkapseln
Dabigatranetexilat
2.
WIRKSTOFFE
Jede Hartkapsel enthält 75 mg Dabigatranetexilat (als Mesilat).
3.
SONSTIGE BESTANDTEILE
Enthält Gelborange S (E 110) (nähere Informationen siehe Packungsbeilage)
4.
DARREICHUNGSFORM UND INHALT
60 Hartkapseln
5.
HINWEISE ZUR UND ART(EN) DER ANWENDUNG
Zum Einnehmen.
Nicht kauen.
Packungsbeilage beachten.
6.
WARNHINWEIS, DASS DAS ARZNEIMITTEL FÜR KINDER UNERREICHBAR UND
NICHT SICHTBAR AUFZUBEWAHREN IST
Arzneimittel für Kinder unzugänglich aufbewahren.
7.
WEITERE WARNHINWEISE, FALLS ERFORDERLICH
8.
VERFALLDATUM
Verwendbar bis MM JJJJ
Nach dem ersten Öffnen, das Arzneimittel innerhalb von 30 Tagen verbrauchen.
9.
BESONDERE LAGERUNGSHINWEISE
Die Flasche fest verschlossen halten. In der Originalverpackung aufbewahren, um den Inhalt vor
Feuchtigkeit zu schützen.
40
10.
GEGEBENENFALLS BESONDERE VORSICHTSMASSNAHMEN FÜR DIE BESEITIGUNG
VON NICHT VERWENDETEM ARZNEIMITTEL ODER DAVON STAMMENDEN
ABFALLMATERIALIEN
Nicht verwendetes Arzneimittel oder Abfallmaterial ist entsprechend den nationalen Anforderungen zu
entsorgen.
11.
NAME UND ANSCHRIFT DES PHARMAZEUTISCHEN UNTERNEHMERS
Boehringer Ingelheim International GmbH
Binger Str. 173
D-55216 Ingelheim am Rhein
Deutschland
12.
ZULASSUNGSNUMMER(N)
EU/0/00/000/000
13.
CHARGENBEZEICHNUNG
Ch.-B.:
14.
VERKAUFSABGRENZUNG
Verschreibungspflichtig
15.
HINWEISE FÜR DEN GEBRAUCH
16.
INFORMATION IN BRAILLE-SCHRIFT
Pradaxa 75 mg (nur auf der Faltschachtel, nicht auf dem Flaschen-Ettiket)
41
ANGABEN AUF DER ÄUSSEREN UMHÜLLUNG UND DEM PRIMÄRPACKMITTEL
FALTSCHACHTEL UND FLASCHEN-ETIKETT FÜR 110 mg
1.
BEZEICHNUNG DES ARZNEIMITTELS
Pradaxa 110 mg Hartkapseln
Dabigatranetexilat
2.
WIRKSTOFFE
Jede Hartkapsel enthält 110 mg Dabigatranetexilat (als Mesilat).
3.
SONSTIGE BESTANDTEILE
Enthält Gelborange S (E 110) (nähere Informationen siehe Packungsbeilage)
4.
DARREICHUNGSFORM UND INHALT
60 Hartkapseln
5.
HINWEISE ZUR UND ART(EN) DER ANWENDUNG
Zum Einnehmen.
Nicht kauen.
Packungsbeilage beachten.
6.
WARNHINWEIS, DASS DAS ARZNEIMITTEL FÜR KINDER UNERREICHBAR UND
NICHT SICHTBAR AUFZUBEWAHREN IST
Arzneimittel für Kinder unzugänglich aufbewahren.
7.
WEITERE WARNHINWEISE, FALLS ERFORDERLICH
8.
VERFALLDATUM
Verwendbar bis: MM JJJJ
Nach dem ersten Öffnen, das Arzneimittel innerhalb von 30 Tagen verbrauchen.
9.
BESONDERE LAGERUNGSHINWEISE
Die Flasche fest verschlossen halten. In der Originalverpackung aufbewahren, um den Inhalt vor
Feuchtigkeit zu schützen.
42
10.
GEGEBENENFALLS BESONDERE VORSICHTSMASSNAHMEN FÜR DIE BESEITIGUNG
VON NICHT VERWENDETEM ARZNEIMITTEL ODER DAVON STAMMENDEN
ABFALLMATERIALIEN
Nicht verwendetes Arzneimittel oder Abfallmaterial ist entsprechend den nationalen Anforderungen zu
entsorgen.
11.
NAME UND ANSCHRIFT DES PHARMAZEUTISCHEN UNTERNEHMERS
Boehringer Ingelheim International GmbH
Binger Str. 173
D-55216 Ingelheim am Rhein
Deutschland
12.
ZULASSUNGSNUMMER(N)
EU/0/00/000/000
13.
CHARGENBEZEICHNUNG
Ch.-B.:
14.
VERKAUFSABGRENZUNG
Verschreibungspflichtig
15.
HINWEISE FÜR DEN GEBRAUCH
16.
INFORMATION IN BRAILLE-SCHRIFT
Pradaxa 110 mg (nur auf der Faltschachtel, nicht auf dem Flaschen-Ettiket)
43
B. PACKUNGSBEILAGE
44
GEBRAUCHSINFORMATION: INFORMATION FÜR DEN ANWENDER
Pradaxa 75 mg - Hartkapseln
Pradaxa 110 mg - Hartkapseln
Dabigatranetexilat
Lesen Sie die gesamte Packungsbeilage sorgfältig durch, bevor Sie mit der Einnahme dieses
Arzneimittels beginnen.
-
Heben Sie die Packungsbeilage auf. Vielleicht möchten Sie diese später nochmals lesen.
Wenn Sie weitere Fragen haben, wenden Sie sich an Ihren Arzt oder Apotheker.
Dieses Arzneimittel wurde Ihnen persönlich verschrieben. Geben Sie es nicht an Dritte weiter. Es kann
anderen Menschen schaden, auch wenn diese dieselben Beschwerden haben wie Sie.
Wenn eine der aufgeführten Nebenwirkungen Sie erheblich beeinträchtigt oder Sie Nebenwirkungen
bemerken, die nicht in dieser Gebrauchsinformation angegeben sind, informieren Sie bitte Ihren Arzt
oder Apotheker.
Diese Packungsbeilage beinhaltet:
1.
Was ist Pradaxa und wofür wird es angewendet?
2.
Was müssen Sie vor der Einnahme von Pradaxa beachten?
3.
Wie ist Pradaxa einzunehmen?
4.
Welche Nebenwirkungen sind möglich?
5.
Wie ist Pradaxa aufzubewahren?
6.
Weitere Informationen
1.
WAS IST PRADAXA UND WOFÜR WIRD ES ANGEWENDET?
Was ist Pradaxa:
Pradaxa ist ein Arzneimittel, das zur Vorbeugung der Bildung von Blutgerinnseln eingesetzt wird. Es wirkt
über die Blockade einer körpereigenen Substanz, die an der Bildung von Blutgerinnseln beteiligt ist.
Wofür wird Pradaxa angewendet:
Pradaxa wird angewendet, um der Bildung von Blutgerinnseln in den Venen nach chirurgischem Knie- oder
Hüftgelenksersatz vorzubeugen.
2.
WAS MÜSSEN SIE VOR DER EINNAHME VON PRADAXA BEACHTEN?
Pradaxa darf NICHT eingenommen werden
-
wenn Sie überempfindlich (allergisch) gegen Dabigatranetexilat, Dabigatran oder einen der sonstigen
Bestandteile von Pradaxa sind.
wenn Ihre Nierenfunktion stark eingeschränkt ist.
wenn Sie gegenwärtig bluten
wenn Sie an einer Erkrankung eines Körperorgans leiden, die das Risiko einer schweren Blutung
erhöht.
wenn bei Ihnen eine erhöhte Blutungsneigung besteht. Diese kann angeboren sein, aus ungeklärter
Ursache auftreten oder durch andere Arzneimittel verursacht werden.
wenn Sie eine schwer eingeschränkte Leberfunktion oder eine möglicherweise lebensbedrohliche
Lebererkrankung haben.
wenn Sie Chinidin, ein Arzneimittel zur Behandlung von Herzrhythmusstörungen, einnehmen.
Besondere Vorsicht bei der Einnahme von Pradaxa ist erforderlich
45
Informieren Sie Ihren Arzt, wenn Sie an einer Störung oder Krankheit leiden oder gelitten haben,
insbesondere wenn diese in der folgenden Liste aufgeführt ist:
-
Wenn Sie an einer Lebererkrankung leiden, die mit einer Veränderung der Blutwerte einhergeht, wird
die Anwendung von Pradaxa nicht empfohlen.
-
Wenn Sie ein erhöhtes Blutungsrisiko haben, wie möglicherweise in den folgenden Situationen:
• wenn bei Ihnen im vergangenen Monat eine Biopsie (chirurgische Entnahme von Gewebe)
durchgeführt worden ist.
• wenn Sie eine schwere Verletzung erlitten haben (z.B. einen Knochenbruch, eine Kopfverletzung
oder jegliche Verletzung, die chirurgisch behandelt werden muss).
• wenn Sie eine Behandlung erhalten, die das Blutungsrisiko erhöhen könnte.
• wenn Sie entzündungshemmende Arzneimittel einnehmen.
• Wenn Sie an einer Herzentzündung (bakterielle Endokarditis) leiden.
• wenn Ihre Nierenfunktion mäßig beeinträchtigt ist.
• Pradaxa sollte nicht bei Kindern angewendet werden.
-
Wenn bei Ihnen ein Schlauch (Katheter), in den Rücken eingeführt wird:
Es besteht die Möglichkeit, dass ein Schlauch in Ihren Rücken eingeführt wird, z. B. für eine Narkose
oder zur Schmerzlinderung während oder nach der Operation. Falls Ihnen Pradaxa verabreicht wird,
nachdem der Katheter entfernt wurde, wird Ihr Arzt Sie regelmäßig untersuchen.
Bei Einnahme von Pradaxa mit anderen Arzneimitteln
Bitte informieren Sie Ihren Arzt oder Apotheker, wenn Sie andere Arzneimittel einnehmen / anwenden bzw.
vor kurzem eingenommen / angewendet haben, auch wenn es sich um nicht verschreibungspflichtige
Arzneimittel handelt. Z.B.:
- blutverdünnende Arzneimittel (z.B. Warfarin, Heparin)
- nichtsteroidale Antirheumatika
- Johanniskraut, Rifampicin, Verapamil, Clarithromycin
- Amiodaron
Wenn Sie ein Amiodaron-enthaltendes Arzneimittel einnehmen, sollten Sie mit einer reduzierten Dosis von
150 mg Pradaxa behandelt werden
Bei Einnahme von Pradaxa zusammen mit Nahrungsmitteln und Getränken
Pradaxa kann zu oder unabhängig von den Mahlzeiten eingenommen werden.
Schwangerschaft und Stillzeit
Die Wirkungen von Pradaxa auf die Schwangerschaft und das ungeborene Kind sind nicht bekannt. Sie
sollten Pradaxa nicht einnehmen, wenn Sie schwanger sind, es sei denn, Ihr Arzt teilt Ihnen mit, dass dies
unbedenklich ist. Wenn Sie eine Frau im gebärfähigen Alter sind, sollten Sie vermeiden, schwanger zu
werden, während Sie Pradaxa einnehmen.
Sie sollten nicht stillen, während Sie Pradaxa einnehmen.
Verkehrstüchtigkeit und das Bedienen von Maschinen
Die Wirkung von Pradaxa auf die Verkehrstüchtigkeit und das Bedienen von Maschinen ist nicht bekannt.
Ihr Arzt wird Ihnen mitteilen, wann Sie wieder ein Fahrzeug führen dürfen.
Wichtige Informationen über bestimmte sonstige Bestandteile von Pradaxa
46
Pradaxa Hartkapseln enthalten den Farbstoff Gelborange S, der allergische Reaktionen hervorrufen kann.
3.
WIE IST PRADAXA EINZUNEHMEN?
Wenn Sie Pradaxa-Kapseln aus der Blisterpackung entnehmen, beachten Sie bitte folgende Anweisungen:
• entnehmen Sie die Kapseln, indem Sie die rückseitige Folie der Blisterkarte abziehen.
• drücken Sie die Kapseln nicht durch die Folie der Blisterpackung.
• ziehen Sie die Folie der Blisterpackung erst dann ab, wenn eine Sie eine Kapsel benötigen.
Wenn Sie Pradaxa-Kapseln aus der Flasche entnehmen, beachten Sie bitte folgende Anweisungen:
• zum Öffnen drücken und drehen.
Die übliche empfohlene Dosis von Pradaxa beträgt 220 mg einmal pro Tag (eingenommen als 2 Kapseln zu
110 mg).
Wenn Ihre Nierenfunktion um mehr als die Hälfte herabgesetzt ist oder wenn Sie älter als 75 Jahre sind
beträgt die empfohlene Dosis 150 mg einmal pro Tag (eingenommen als 2 Kapseln zu 75 mg).
Nach chirurgischem Kniegelenksersatz
Sie sollten die Behandlung mit Pradaxa innerhalb von 1-4 Stunden nach dem Ende des Eingriffs mit einer
einzelnen Kapsel beginnen. Anschließend sollten 2 Kapseln einmal pro Tag über insgesamt 10 Tage
eingenommen werden.
Nach chirurgischem Hüftgelenksersatz
Sie sollten die Behandlung mit Pradaxa innerhalb von 1-4 Stunden nach dem Ende des Eingriffs mit einer
einzelnen Kapsel beginnen. Anschließend sollten 2 Kapseln einmal pro Tag über insgesamt 28-35 Tage
eingenommen werden.
Nach beiden Operationen sollte die Behandlung nicht begonnen werden, wenn eine Blutung aus der
Operationsstelle vorliegt. Falls die Behandlung erst am Tag nach der Operation eingeleitet werden kann,
sollte diese mit 2 Kapseln einmal täglich begonnen werden.
Nehmen Sie Pradaxa immer genau nach Anweisung des Arztes ein. Bitte fragen Sie bei Ihrem Arzt nach,
wenn Sie sich nicht ganz sicher sind. Die Kapsel sollte mit etwas Wasser eingenommen werden. Kauen Sie
die Kapsel nicht.
Umstellung von einer Behandlung mit Pradaxa auf eine durch Injektion verabreichte
gerinnungshemmende Behandlung
Beginnen Sie eine Behandlung mit injizierbaren gerinnungshemmenden Arzneimitteln (z. B. Heparin) nicht
vor Ablauf von 24 Stunden nach der letzten Einnahme von Pradaxa.
Umstellung von einer durch Injektion verabreichten gerinnungshemmenden Behandlung auf eine
Behandlung mit Pradaxa
Beenden Sie die Injektionsbehandlung und beginnen Sie mit der Einnahme von Pradaxa zu dem Zeitpunkt,
an dem Ihre nächste Injektion fällig wäre.
Wenn Sie eine größere Menge von Pradaxa eingenommen haben, als Sie sollten
Wenn Sie eine größere Menge von Pradaxa eingenommen haben, als empfohlen, können Sie ein erhöhtes
Blutungsrisiko haben.
47
Ihr Arzt kann zur Abschätzung des Blutungsrisikos einen Bluttest durchführen. Verständigen Sie Ihren Arzt
so rasch wie möglich, wenn Sie mehr als die vorgeschriebene Dosis von Pradaxa eingenommen haben. Wenn
es zu einer Blutung kommt, kann eine chirurgische Behandlung oder die Verabreichung von
Bluttransfusionen erforderlich sein.
Wenn Sie die Einnahme von Pradaxa vergessen haben
Setzen Sie die Einnahme der restlichen Tagesdosen von Pradaxa zu derselben Zeit am nächsten Tag fort.
Nehmen Sie nicht die doppelte Dosis ein, wenn Sie die vorherige Einnahme vergessen haben.
Wenn Sie die Einnahme von Pradaxa abbrechen
Brechen Sie die Einnahme von Pradaxa nicht ohne vorherige Rücksprache mit Ihrem Arzt ab, da das Risiko
in einer Vene ein Blutgerinnsel zu entwickeln erhöht sein könnte, wenn Sie die Behandlung vorzeitig
abbrechen.
Wenn Sie weitere Fragen zur Anwendung des Arzneimittels haben, fragen Sie Ihren Arzt oder Apotheker.
4.
WELCHE NEBENWIRKUNGEN SIND MÖGLICH?
Wie alle Arzneimittel kann Pradaxa Nebenwirkungen haben, die aber nicht bei jedem auftreten müssen.
Da dieses Arzneimittel die Blutgerinnung beeinflusst, äußern sich die meisten Nebenwirkungen in Form von
Blutergüssen oder Blutungen.
Nachfolgend sind die möglichen Nebenwirkungen, entsprechend der Wahrscheinlichkeit ihres Auftretens,
aufgeführt:
Die folgenden häufigen und gelegentlichen Nebenwirkungen sind bei Pradaxa bekannt:
Häufige Nebenwirkungen (treten bei mehr als 1 bis 10 von 100 Anwendern auf):
- Abnahme der Anzahl roter Blutkörperchen im Blut
- Hämatombildung
- Blutung infolge einer Verletzung
- Abnahme der Menge an Hämoglobin im Blut (der Substanz in den roten Blutkörperchen)
- Wundsekretion (Austreten von Flüssigkeit aus der Operationswunde)
- Blutergussbildung nach einem chirurgischen Eingriff
- Blutung nach einem chirurgischen Eingriff
- Abnahme der Anzahl roter Blutkörperchen nach einem chirurgischen Eingriff
- Blutergussbildung infolge einer Verletzung
- Austreten einer geringen Menge an Flüssigkeit aus dem Hautschnitt, der für einen chirurgischen Eingriff
durchgeführt wurde
- durch Labortest nachweisbares Blut im Urin.
Gelegentliche Nebenwirkungen (bei 1 bis 10 von 1.000 Anwendern auf):
- Blutung
- Blutung in ein Gelenk
- Abnahme der Anzahl der Blutplättchen im Blut
- Nasenbluten
- Magen- oder Darmblutung
- Blutung aus Hämorrhoiden
- Blutung in den Mastdarm
- Blut im Urin, das diesen rosa oder rot färbt
- Unterhautblutung
- blutgefärbte Absonderung aus der Eintrittsstelle eines Katheters in eine Vene
- Blutung aus der Eintrittsstelle eines Katheters in eine Vene;
- Nachweis von Blut im Stuhl durch einen Labortest
- Absinken der Anzahl roter Blutkörperchen
48
-
Blutung an der Stelle des chirurgischen Schnitts
von der Norm abweichende Leberfunktionswerte.
Informieren Sie bitte Ihren Arzt oder Apotheker, wenn eine der aufgeführten Nebenwirkungen Sie erheblich
beeinträchtigt oder Sie Nebenwirkungen bemerken, die nicht in dieser Gebrauchsinformation angegeben
sind.
5.
WIE IST PRADAXA AUFZUBEWAHREN?
Arzneimittel für Kinder unzugänglich aufbewahren.
Sie dürfen Pradaxa nach dem auf dem Umkarton, der Blisterpackung oder der Flasche angegebenen
Verfalldatum nicht mehr anwenden. Das Verfalldatum bezieht sich auf den letzten Tag des Monats.
Blisterpackung:
Flasche:
In der Originalverpackung aufbewahren, um den Inhalt vor Feuchtigkeit zu schützen.
Nach dem ersten Öffnen das Arzneimittel innerhalb von 30 Tagen verbrauchen. Die
Flasche fest verschlossen halten. In der Originalverpackung aufbewahren, um den Inhalt
vor Feuchtigkeit zu schützen.
Das Arzneimittel darf nicht im Abwasser oder Haushaltsabfall entsorgt werden. Fragen Sie Ihren Apotheker,
wie das Arzneimittel zu entsorgen ist, wenn Sie es nicht mehr benötigen. Diese Maßnahme hilft die Umwelt
zu schützen.
6.
WEITERE INFORMATIONEN
Was Pradaxa enthält
Der Wirkstoff ist Dabigatran, das in Form von 75 mg oder 110 mg Dabigatranetexilat angewendet wird,
gegeben als Mesilat.
Die sonstigen Bestandteile sind Weinsäure, Arabisches Gummi, Hypromellose, Dimeticon 350, Talkum und
Hyprolose.
Die Kapselhülle enthält Carrageenan, Kaliumchlorid, Titandioxid, Indigokarmin (E 132), Gelborange S
(E 110), Hypromellose und Gereinigtes Wasser.
Die schwarze Druckfarbe enthält Schellack, Butan-1-ol, 2-Propanol, Ethanol vergällt (mit Aceton, Methanol
undAcetylacetat), Eisen(II,III)-oxid (E 172), Gereinigtes Wasser und Propylenglycol.
Wie Pradaxa aussieht und Inhalt der Packung
Pradaxa ist eine Hartkapsel.
Pradaxa 75 mg - Hartkapseln besitzen eine undurchsichtige, hellblaue Kappe und einen undurchsichtigen,
cremefarbenen Korpus. Auf der Kappe ist das Firmensymbol von Boehringer Ingelheim, auf dem Korpus der
Kapsel „R75“ aufgedruckt.
Pradaxa 110 mg - Hartkapseln besitzen eine undurchsichtige, hellblaue Kappe und einen undurchsichtigen,
cremefarbenen Korpus. Auf der Kappe ist das Firmensymbol von Boehringer Ingelheim, auf dem Korpus der
Kapsel „R110“ aufgedruckt.
Pradaxa 75 mg und 110 mg Hartkapseln sind in Packungen mit 10 x 1, 30 x 1 oder 60 x 1 Kapsel in
perforierten Blisterpackungen zur Abgabe von Einzeldosen aus Aluminium erhältlich.
49
Pradaxa 75 mg und 110 mg Hartkapseln sind außerdem in Polypropylen-(Kunststoff-) Flaschen mit
60 Hartkapseln erhältlich.
Es werden möglicherweise nicht alle Packungsgrößen in den Verkehr gebracht.
Pharmazeutischer Unternehmer
Boehringer Ingelheim International GmbH
Binger Straße 173
D-55216 Ingelheim am Rhein
Deutschland
Hersteller
Boehringer Ingelheim Pharma GmbH & Co. KG
Binger Straße 173
D-55216 Ingelheim am Rhein
Deutschland
Falls weitere Informationen über das Arzneimittel gewünscht werden, setzen Sie sich bitte mit dem örtlichen
Vertreter des pharmazeutischen Unternehmers in Verbindung.
50
België/Belgique/Belgien
S.C.S. Boehringer Ingelheim Comm.V.
Tél/Tel: +32 2 773 33 11
Luxembourg/Luxemburg
S.C.S. Boehringer Ingelheim Comm.V.
Tél/Tel: +32 2 773 33 11
България
Бьорингер Ингелхайм Фарма ГмбХ
Тел: +359 2 958 79 98
Magyarország
Boehringer Ingelheim Pharma Fióktelep
Tel: +36 1 224 7120
Česká republika
Boehringer Ingelheim spol. s r.o.
Tel: +420 234 655 111
Malta
Boehringer Ingelheim Ltd.
Tel: +44 1344 424 600
Danmark
Boehringer Ingelheim Danmark A/S
Tlf: +45 39 15 88 88
Nederland
Boehringer Ingelheim b.v.
Tel: +31 (0) 800 22 55 889
Deutschland
Boehringer Ingelheim Pharma GmbH & Co. KG
Tel: +49 (0) 800 77 90 900
Norge
Boehringer Ingelheim Norway KS
Tlf: +47 66 76 13 00
Eesti
Boehringer Ingelheim Pharma GmbH
Eesti Filiaal
Tel: +372 60 80 940
Österreich
Boehringer Ingelheim Austria GmbH
Tel: +43 1 80 105-0
Ελλάδα
Boehringer Ingelheim Ellas A.E.
Tηλ: +30 2 10 89 06 300
Polska
Boehringer Ingelheim Sp.zo.o.
Tel: +48 22 699 0 699
España
Boehringer Ingelheim España S.A.
Tel: +34 93 404 58 00
Portugal
Boehringer Ingelheim, Lda.
Tel: +351 21 313 53 00
France
Boehringer Ingelheim France S.A.S.
Tél: +33 3 26 50 45 33
România
Boehringer Ingelheim Pharma Ges mbH
Reprezentanţa din România
Tel: +40 21 330 99 63
Ireland
Boehringer Ingelheim Ireland Ltd.
Tel: +353 1 295 9620
Slovenija
Boehringer Ingelheim Pharma
Podružnica Ljubljana
Tel: +386 1 586 40 00
Ísland
Vistor hf.
Sími: +354 535 7000
Slovenská republika
Boehringer Ingelheim Pharma
organizačná zložka
Tel: +421 2 5810 1211
Italia
Boehringer Ingelheim Italia S.p.A.
Tel: +39 02 5355 1
Suomi/Finland
Boehringer Ingelheim Finland Ky
Puh/Tel: +358 10 3102 800
Κύπρος
Boehringer Ingelheim Ellas A.E.
Sverige
Boehringer Ingelheim AB
51
Tηλ: +30 2 10 89 06 300
Tel: +46 8 721 21 00
Latvija
Boehringer Ingelheim Pharma GmbH
Pārstāvniecība Latvijā
Tel: +371 7 240 068
United Kingdom
Boehringer Ingelheim Ltd.
Tel: +44 1344 424 600
Lietuva
Boehringer Ingelheim Pharma Ges mbH
Atstovybė Lietuvoje
Tel: +370 37 473922
Diese Gebrauchsinformation wurde zuletzt genehmigt im {MM/JJJJ}.
Ausführliche Informationen zu diesem Arzneimittel sind auf der Website der Europäischen ArzneimittelAgentur (EMEA) http://www.emea.europa.eu/ verfügbar.
52