PowerPoint-Präsentation - Universität des Saarlandes
Werbung

Der Bauplan des Menschen
Effiziente Algorithmen
zur Berechnung
von Sequenz-Alignments
Zentrum für Bioinformatik
der Universität des Saarlandes
WS 2001/2002
Der Bauplan des Menschen
• Sequenz-Alignments ist eines der ältesten Forschungsgebiete der Bioinformatik.
„ ... the wholy grail of Bioinformatics... „ (Dan Gusfield)
• Sequenz-Alignments werden in vielen Bereichen der Bioinformatik verwendet:
1.
2.
3.
4.
5.
6.
Sequenzierung
Suche nach Genen
Bestimmung der Funktion von Genen und Proteinen
Bestimmung der Struktur von Proteinen
Berechnung von „Evolutionary Trees“
Design von Proteinen
Der Bauplan des Menschen
Kopien des Gens
Nanopartikel
Lektine
Lektine docken an gewissen Zuckermolekülen (GlcNAG) auf der Zelloberfläche an.
Dieser Vorgang ermöglicht die Aufnahme der Partikel in die Zelle.
Zellkern
Zelle
• Mutiertes Gen „verursacht“ Krankheit.
• Mögliche Behandlung: Gentherapie
d.h. das Einschleusen einer „normalen“
Kopie des Gens in den Zellkern.
mutiertes Gen
Der Bauplan des Menschen
VAIAEQCGRQAGGKLCPNNLCCSQWGWCGSTDEYCSPDHNCQSNCK
TAHAQRCGEQGSNMECPNNLCCSQYGYCGMGGDYCGKGCQNGACYT
ATNAQTCGKQNDGMICPHNLCCSQFGYCGLGRDYCGTGCQSGACCS
LVSAQRCGSQGGGGTCPALWCCSIWGWCGDSEPYCGRTCENKCWS
TAQAQRCGEQGSNMECPNNLCCSQYGYCGMGGDYCGKGCQNGACWT
TAQAQRCGEQGSNMECPNNLCCSQYGYCGMGGDYCGKGCQNGACWT
QRCGEQGSGMECPNNLCCSQYGYCGMGGDYCGKGCQNGACWT
TVKSQNCGCAPNLCCSQFGYCGSTDAYCGTGCRSGPCRS
SAEQCGRQAGDALCPGGLCCSSYGWCGTTVDYCGIGCQSQCDG
PAAAQNCGCQPNFCCSKFGYCGTTDAYCGDGCQSGPCRS
PAAAQNCGCQPNVCCSKFGYCGTTDEYCGDGCQSGPCRS
SAEQCGQQAGDALCPGGLCCSSYGWCGTTADYCGDGCQSQCDG
SAEQCGRQAGDALCPGGLCCSFYGWCGTTVDYCGDGCQSQCDG
EQCGRQAGGKLCPNNLCCSQYGWCGSSDDYCSPSKNCQSNCK
Hevein
Sequenz
Struktur
Design von Lektinen mit optimalen Bindungseigenschaften für GlcNAG.
Hevein ist das kleinste Lektin (Ausgangssubstanz).
Datenbanksuche mit Hevein liefert eine Reihe weiterer Lektine.
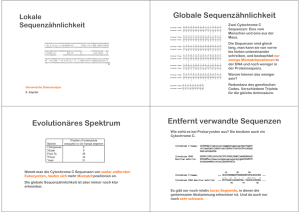
Sequenz-Alignments der gefundenen Lektine zeigt Gemeinsamkeiten
und Unterschiede der Sequenzen (in den Spalten des Alignments).
Funktion
Der Bauplan des Menschen
VAIAEQCGRQAGGKLC-PNNLCCSQWGWCGSTDEYCSPDHNCQSN-CKTAHAQRCGEQGSNMEC-PNNLCCSQYGYCGMGGDYCGKG--CQNGACYT
ATNAQTCGKQNDGMIC-PHNLCCSQFGYCGLGRDYCGTG--CQSGACCS
LVSAQRCGSQGGGGTC-PALWCCSIWGWCGDSEPYCGRT--CENK-CWS
TAQAQRCGEQGSNMEC-PNNLCCSQYGYCGMGGDYCGKG--CQNGACWT
TAQAQRCGEQGSNMEC-PNNLCCSQYGYCGMGGDYCGKG--CQNGACWT
----QRCGEQGSGMEC-PNNLCCSQYGYCGMGGDYCGKG--CQNGACWT
TVKSQNCG-------CAP-NLCCSQFGYCGSTDAYCGTG--CRSGPCRS
SAE--QCGRQAGDALC-PGGLCCSSYGWCGTTVDYCGIG--CQSQ-CDG
PAAAQNCG-------CQP-NFCCSKFGYCGTTDAYCGDG--CQSGPCRS
PAAAQNCG-------CQP-NVCCSKFGYCGTTDEYCGDG--CQSGPCRS
SAE--QCGQQAGDALC-PGGLCCSSYGWCGTTADYCGDG--CQSQ-CDG
SAE--QCGRQAGDALC-PGGLCCSFYGWCGTTVDYCGDG--CQSQ-CDG
----EQCGRQAGGKLC-PNNLCCSQYGWCGSSDDYCSPSKNCQSN-CK-
Hevein
Sequenz
Struktur
Design von Lektinen mit optimalen Bindungseigenschaften für GlcNAG.
Hevein ist das kleinste Lektin (Ausgangssubstanz).
Datenbanksuche mit Hevein liefert eine Reihe weiterer Lektine.
Sequenz-Alignments der gefundenen Lektine zeigt Gemeinsamkeiten
und Unterschiede der Sequenzen (in den Spalten des Alignments).
Beispiel: Die gelben Balken zeigen die acht Cysteine, die vier Schwefelbrücken bilden
[ein essentielles Strukturelement aller hier gezeigten Lektine].
Funktion
Der Bauplan des Menschen
s1
s2
S ={A,C,G,U}
S‘ ={A,C,G,U,-}
AUGAACGC
AUAUCGGCC
AUGAACG--C
AU-AUCGGCC
Definition:
Gegeben k Sequenzen S={ s1 , ... , sk } über einem Alphabet .
Ein Sequenz-Alignment von S ist eine Menge von k Sequenzen AS = {as1 , ... , ask }
über dem erweiterten Alphabet ‘ = {-} mit den folgenden Eigenschaften:
(1) Alle Sequenzen in AS besitzen die gleiche Länge.
(2) Entfernt man die Lückensymbole {-} aus asi, so erhält man die Sequenz si (für alle i).
Der Bauplan des Menschen
s1
s2
S ={A,C,G,U}
S‘ ={A,C,G,U,-}
AUGAACGC
AUAUCGGCC
AUGAACG--C
AU-AUCGGCC
Substitution
Deletion
Insertion
Biologische Interpretation von Alignments:
Indels
Annahme: Die Ähnlichkeit von DNA-Sequenzen kann als ein Hinweis auf eine gemeinsame
evolutionäre Herkunft (gemeinsamen Vorfahren) gedeutet werden. Im Laufe
der Evolution kam es zu Mutationen, die die Unterschiede im Erbgut der
verschiedenen Spezies hervorgerufen haben.
Fragen: Wieviele Mutationen sind erforderlich, um eine Sequenz in eine andere
Sequenz zu überführen? Wie wahrscheinlich sind diese Mutationen?
Wie oft kommen solche Mutationen (Transformationen) in einem bestimmten Zeitraum im Durchschnitt vor?
Der Bauplan des Menschen
• Für die Bewertung von Alignments stehen viele unterschiedliche Funktionen zur Verfügung.
• Die meisten Bewertungsfunktionen kann man zwei Klassen zuordnen:
1. Abstandsfunktionen
2. Ähnlichkeitsfunktionen
• Die einfachen Bewertungsfunktionen bewerten jede Spalte eines Alignments.
• Die Summe der Spaltenwerte ergibt dann die Gesamtbewertung eines Alignments.
• Beispiel mit einer einfachen Abstandsfunktion:
AC G U -
S
P
AUGAACGC
AUAUCGGCC
AS
AP
AUGAACG--C
AU-AUCGGCC
A 0
1
1
1
1
C 1
0
1
1
1
0 0 10 1 0 0 1 1 0
G 1
1
0
1
1
=4
U 1
1
1
0
1
-
1
1
1
0
1
d ( AS , AP)
d (as , ap )
jSpalten
j
j
Der Bauplan des Menschen
Paarweises Sequenz-Alignment-Problem:
Gegeben zwei Sequenzen S und P über einem Alphabet S und eine Bewertungsfunktion d.
Berechne ein bezüglich d optimales paarweises Alignment Aopt[S,P] von S und P.
• Beispiel mit einer einfachen Abstandsfunktion:
Aopt [ S , P ] min [ AS , AP ] d ( AS , AP) min [ AS , AP ]
AC G U -
S
P
AUGAACGC
AUAUCGGCC
d (as , ap )
j
jSpalten
AS
AP
j
AUGAACG--C
AU-AUCGGCC
A 0
1
1
1
1
C 1
0
1
1
1
0 0 10 1 0 0 1 1 0
G 1
1
0
1
1
=4
U 1
1
1
0
1
-
1
1
1
0
1
d ( AS , AP)
d (as , ap )
j
j ( Spalten)
j
Der Bauplan des Menschen
Paarweises Sequenz-Alignment-Problem:
Gegeben zwei Sequenzen S und P über einem Alphabet S und eine Bewertungsfunktion d.
Berechne ein bezüglich d optimales paarweises Alignment Aopt[S,P] von S und P.
• Beispiel mit einer einfachen Ähnlichkeitsfunktion:
Aopt [ S , P ] max [ AS , AP ] d ( AS , AP) max [ AS , AP ]
A
C
G
U
-
A
1
-1
-1
-1
-1
C
-1
1
-1
-1
-1
G
-1
-1
1
-1
-1
U
-1
-1
-1
1
-1
-
-1
-1
-1
-1
1
S
P
AUGAACGC
AUAUCGGCC
d (as , ap )
j
jSpalten
AS
AP
j
AUGAACG--C
AU-AUCGGCC
1 1-1 1 -11 1 -1-11
=6
d ( AS , AP)
d (as , ap )
jSpalten
j
j
Der Bauplan des Menschen
Gegeben zwei Sequenzen S und P über einem Alphabet S und eine Bewertungsfunktion d.
Berechne ein bezüglich d optimales paarweises Alignment Aopt[S,P] mit Wert d(Sn,Pm)
von S und P, wobei S die Länge n und P die Länge m hat..
Si = Präfix der Länge i von S
Pj = Präfix der Länge j von P
d(Si ,Pj ) = Wert des opt. Alignments von Si und Pj
Idee: Betrachte die Möglichkeiten für die letzte Spalte
und leite hieraus eine Rekursionformel ab.
AC G U A 0
1
1
1
1
C 1
0
1
1
1
G 1
1
0
1
1
U 1
1
1
0
1
-
1
1
1
0
1
Si
Pj
• Beispiel für Distanzfunktion d
AUGAACGC
AUAUCGGCC
d(Si-1 ,Pj )
d(Si-1 ,Pj-1 )
d(Si ,Pj-1 )
C
C
C
C
d(Si-1 ,Pj-1 ) + d(si,pj)
d(Si ,Pj ) = min
d(Si-1 ,Pj ) + d(si,-)
d(Si ,Pj-1 ) + d(-,pj)
Der Bauplan des Menschen
Gegeben zwei Sequenzen S und P über einem Alphabet S und eine Bewertungsfunktion d.
Berechne ein bezüglich d optimales paarweises Alignment Aopt[S,P] mit Wert d(Sn,Pm)
von S und P, wobei S die Länge n und P die Länge m hat..
Fülle Matrix Aji mit
aji = d(Si ,Pj ) für alle i{0, ... ,n}
für alle j{0, ... ,m}
S0 S1 S2 S3 S4 S5 S6 S7 S8
A U G A A C G C
0 1 2 3 4 5 6 7 8
P0
P1 A 1 0
P2 U 2 1
P3 A 3 2
P4 U 4 3
P5 C 5 4
P6 G 6 5
P7 G 7 6
P8 C 8 7
P9 C 9 8
1 2 3 4 5 6 7
0 1 2 3 4 5 6
1 1
1
2 3
AC G U A 0
1
1
1
1
C 1
0
1
1
1
G 1
1
0
1
1
U 1
1
1
0
1
-
1
1
1
0
1
4 5
2 2 2 2 3 4 5
3 3 3 3 2 3 4
4 3 4 4 3 2 3
5 4 4 5 4 3 3
6 5 5 5 5 4 3
7 6 6 6 5 5 4
AUGAAC-G - C
AU-AUCGG C C
d(Si ,P0 ) = i
d(S0 ,Pj ) = j
d(Si-1 ,Pj-1 ) + d(si,pj)
d(Si ,Pj ) = min
d(Si-1 ,Pj ) + d(si,-)
d(Si ,Pj-1 ) + d(-,pj)
Der Bauplan des Menschen
Gegeben zwei Sequenzen S und P über einem Alphabet S und eine Bewertungsfunktion d.
Berechne ein bezüglich d optimales paarweises Alignment Aopt[S,P] mit Wert d(Sn,Pm)
von S und P, wobei S die Länge n und P die Länge m hat..
Fülle Matrix Aji mit
aji = d(Si ,Pj ) für alle i{0, ... ,n}
für alle j{0, ... ,m}
S0 S1 S2 S3 S4 S5 S6 S7 S8
A U G A A C G C
0 1 2 3 4 5 6 7 8
P0
P1 A 1 0
P2 U 2 1
P3 A 3 2
P4 U 4 3
P5 C 5 4
P6 G 6 5
P7 G 7 6
P8 C 8 7
P9 C 9 8
1 2 3 4 5 6 7
0 1 2 3 4 5 6
1 1
1
2 3
4 5
2 2 2 2 3 4 5
3 3 3 3 2 3 4
4 3 4 4 3 2 3
5 4 4 5 4 3 3
6 5 5 5 5 4 3
7 6 6 6 5 5 4
AUGAAC-G - C
AU-AUCGG C C
(1) Intitialisierung (der Ränder der Matrix).
(2) Berechnung der Matrixeinträge mittels Rekursion.
(3) Backtracking: Berechne ein optimales Alignment.
Laufzeit: O(mn). Speicherplatzbedarf: O(mn)
d(Si ,P0 ) = i
d(S0 ,Pj ) = j
d(Si-1 ,Pj-1 ) + d(si,pj)
d(Si ,Pj ) = min
d(Si-1 ,Pj ) + d(si,-)
d(Si ,Pj-1 ) + d(-,pj)
Der Bauplan des Menschen
Zunächst hat sich die Bioinformatik mit globalen Alignments beschäftigt,
d.h., man hat versucht, die Sequenzen vollständig miteinander zu vergleichen.
Dies macht nur dann Sinn, wenn
(a) die Sequenzen sehr ähnlich und
(b) fast gleich lang sind.
Sind die Sequenzen jedoch unterschiedlich lang, so sucht man nach lokalen Alignments.
(a)
Falls zum Beispiel eine kleine Sequenz in einer großen
Sequenz versteckt ist (eine ähnliche oder identische Teilsequenz in der Großen versteckt ist).
Beispiel: Suche Verwandte eines Genes in einem langen,
neu sequenzierten DNA-Molekül.
(b)
Falls zwei Sequenzen nur an den Enden „überlappen“.
Beispiel: Fragmente (reads) bei der Sequenzierung.
(c)
Falls eine Sequenz in Teilstücken in der anderen auftaucht.
Beispiel: Suche mit EST oder Protein nach Exons in DNA.
Der Bauplan des Menschen
Bei lokalen Alignments werden die überstehenden Endstücke und die leeren Mittelstücke
nicht bewertet:
d(Si ,P0 ) = 0
d(Si-1 ,Pj-1 ) + d(si,pj)
d(Si ,Pj ) = max
d(Si-1 ,Pj ) + d(si,-)
d(S0 ,Pj ) = 0
d(Si ,Pj-1 ) + d(-,pj)
0
Als Bewertungsfunktionen werden in der Regel Ähnlichkeitsfunktionen verwendet.
A
C
G
U
-
A
1
-1
-1
-1
-1
C
-1
1
-1
-1
-1
G
-1
-1
1
-1
-1
U
-1
-1
-1
1
-1
-
-1
-1
-1
-1
1
Der Bauplan des Menschen
Bei lokalen Alignments werden die überstehenden Endstücke und die leeren Mittelstücke
nicht bewertet:
d(Si-1 ,Pj-1 ) + d(si,pj)
d(Si ,P0 ) = 0
d(Si ,Pj ) = max
d(Si-1 ,Pj ) + d(si,-)
d(S0 ,Pj ) = 0
d(Si ,Pj-1 ) + d(-,pj)
0
Als Bewertungsfunktionen werden in der Regel Ähnlichkeitsfunktionen verwendet.
S 0 S1 S2 S3 S4 S5 S6 S7 S 8
P0
0
P1 G 0
P2 A 0
P3 C 0
P4 C 0
A
0
0
1
0
0
U
0
0
0
0
0
G
0
1
0
0
0
A
0
0
2
1
0
A
0
0
1
1
0
C
0
0
0
2
2
C
0
0
0
1
3
C
0
0
0
0
2
A
C
G
U
-
A
1
-1
-1
-1
-1
C
-1
1
-1
-1
-1
G
-1
-1
1
-1
-1
U
-1
-1
-1
1
-1
-
-1
-1
-1
-1
1
Man beachte, dass bei der Berechnung von lokalen Alignments der beste Wert
irgendwo in der Matrix erscheinen kann (modifiziertes Back-Tracing).
Der Bauplan des Menschen
Ein Lücke (Gap) ist eine maximale Aufeinanderfolge von (mindestens einem) Lückensymbolen (-):
ATACGCGGTTGAT
ATA---GGT-GCT
Lücken
Die Bewertungsfunktionen können um einen additiven Term für Lücken erweitert werden.
S
P
AS
AP
AUGAACGC
AUAUCGGCC
AUGAACG--C
AU-AUCGGCC
Affine Gap-Kostenfunktion:
d ( AS , AP)
d (as , ap ) w
jSpalten
j
j
gLücken
s
wv * | g |
Konvexe Gap-Kostenfunktion:
d ( AS , AP)
d (as , ap ) w
jSpalten
j
j
gLücken
s
wv * log(| g |)
Der Bauplan des Menschen
Affine Gap-Kostenfunktion:
d ( AS , AP)
d (as , ap ) w
jSpalten
j
j
gLücken
s
wv * | g |
Falls die Bewertungsfunktion eine affine Gap-Kostenfunktion beinhaltet, benötigt man vier Matrizen
der Größe [n+1,m+1], um ein optimales paarweises Alignment von zwei Sequenzen S und P mit den
Längen n und m zu berechnen.
d (Si , Pj ) = Wert eines (maximalen) optimalen Alignments der Präfixe Si und Pj
d (Si , Pj ) = max { E(Si , Pj ) , F (Si , Pj ) , H (Si , Pj ) } , wobei
si
E (Si , Pj ) = Wert des max. Alignments von Si und Pj, das mit der Spalte p endet.
j
F (Si , Pj ) = Wert des max. Alignment von Si und Pj, das mit der Spalte _ endet.
pj
H (Si , Pj ) = Wert des max. Alignment von Si und Pj, das mit der Spalte si endet.
_
Der Bauplan des Menschen
Affine Gap-Kostenfunktion:
d ( AS , AP)
d (as , ap ) w
jSpalten
j
j
gLücken
s
wv * | g |
Falls die Bewertungsfunktion eine affine Gap-Kostenfunktion beinhaltet, benötigt man vier Matrizen
der Größe [n+1,m+1], um ein optimales paarweises Alignment von zwei Sequenzen S und P mit den
Längen n und m zu berechnen.
Der folgende Algorithmus für affine Gap-Kosten hat Laufzeit und Speicherplatzbedarf O(nm).
d (Si , Pj ) = Wert eines (maximalen) optimalen Alignments der Präfixe Si und Pj
d (Si , Pj ) = max { E(Si , Pj ) , F (Si , Pj ) , H (Si , Pj ) } , wobei
si
E (Si , Pj ) = Wert
max.
d (Sdes
+ d(si , p j) von Si und Pj, das mit der Spalte p endet.
i-1 , P
j-1 ) Alignments
j
F (Si , Pj ) = Wert
desdmax.
das
max{
(Si , Alignment
Pj-1 ) + ws von
+ wvS,i und
F(SiP,j,Pj-1
) +mit
wvder
} Spalte _ endet.
pj
H (Si , Pj ) = Wert
desdmax.
mit
max{
(Si-1Alignment
, Pj ) + ws von
+ wvS,i und
H(SPi-1j, , das
Pj) +
wvder
} Spalte si endet.
_
Der Bauplan des Menschen
mandrill
presbytis_francoisi
crab_eating_macaque
green_monkey
br._capped_capuchin
chimpanzee
orangutan
gorilla
human
bovine
sheep
mule_deer
rat
golden_hamster
mouse
TTTTKGENFTETDVKMMERVVEQMCITQYEKESQAYYQ__RGSSMVLFSS
TTTTKGENFTETDVKMMERVVEQMCITQYEKESQAYYQ--RGSSMVFFSS
TTTTKGENFTETDVKMMERVVEQMCITQYEKESQAYYQ--RGSSMVLFSS
TTTTKGENFTETDVKMMERVVEQMCITQYEKESQAYYQ--RGSSMVLFSS
TTTTKGENFTETDVKMMERVVEQMCITQYERESQAYYQ--RGSSMVLFSS
TTTTKGENFTETDVKMMERVVEQMCITQYERESQAYYQ--RGSSMVLFSS
TTTTKGENFTETDVKMMERVVEQMCITQYERESQAYYQ--RGSSMVLFSS
TTTTKGENFTETDVKMMERVVEQMCITQYERESQAYYQ--RGSSMVLFSS
TTTTKGENFTETDVKMMERVVEQMCITQYERESQAYYQ--RGSSMVLFSS
TTTTKGENFTETDIKMMERVVEQMCITQYQRESQAYYQ--RGASVILFSS
TTTTKGENFTETDIKIMERVVEQMCITQYQRESQAYYQ--RGASVILFSS
TTTTKGENFTETDIKMMERVVEQMCITQYQRESQAYYQ--RGASVILFSS
TTTTKGENFTETDVKMMERVVEQMCVTQYQKESQAYYDG-RRSSAVLFSS
TTTTKGENFTETDIKIMERVVEQMCTTQYQKESQAYYDG-RRSSAVLFSS
TTTTKGENFTETDVKMMERVVEQMCVTQYQKESQAYYDGRRSSSTVLFSS
Das obige multiple Alignment zeigt einen Abschnitt eines multiplen Alignments von Prionen
Proteinen verschiedener Spezies. Die Prionen gelten als Verursacher von BSE, Scrapie und der
Jacob-Kreutzfeld-Erkrankung.
Der Bauplan des Menschen
Multiples Sequenzalignment-Problem:
Gegeben k Sequenzen über einem Alphabet S und eine Bewertungsfunktion d.
Berechne ein optimales multiples Sequenz-Alignment der k Sequenzen.
Wir betrachten der Einfachheit halber im folgenden nur den Fall k = 3,
d.h., gegeben drei Sequenzen S, P, Q der Längen n, m , l.
AC G U Sum-of-Pairs-Problem (SP):
A 0
1
1
1
1
• Die Bewertungsfunktion d sei eine Distanz- oder Ähnlichkeitsfunktion.
C 1
0
1
1
1
• Die Bewertung einer Spalte eines multiplen Alignments [AS,AP,AQ]
ist gleich der Summe der Abstände (Ähnlichkeiten) aller Paare von
Buchstaben in dieser Spalte.
• Die Summe der Spalten liefert die Gesamtbewertung (plus eventuell
Gap-Kosten).
G 1
1
0
1
1
U 1
1
1
0
1
-
1
1
1
0
d ( AS , AP, AQ )
1
d (as , ap , aq ) d (as , ap ) d (as , aq ) d (ap , aq )
jSpalten
j
j
j
jSpalten
j
j
j
j
j
j
Der Bauplan des Menschen
Sum-of-Pairs-Problem (SP):
Ein optimales SP-Alignment kann man mittels Dynamischer Programmierung (DP) berechnen.
Die Rekursionsgleichung leitet man aus einer Fallunterscheidung für die letzte Spalte ab (siehe Übung).
Falls alle k Sequenzen die Länge n haben, so ist die Laufzeit und der Speicherplatzbedarf
O(nk).
DP funktioniert nur für sehr kleine k (< 10).
Für k > 4 und entsprechende Längen n findet man nur dann ein optimales Alignment, wenn man
den Suchraum verkleinern kann (Branch-and-Bound).
Die Berechnung eines optimalen Alignments entspricht der Suche nach einem kürzesten (oder
längsten) Pfad in einem Alignment-Graphen (gerichteter Graph mit Quelle und Senke).
Suchraum verkleinern!
Nur den „interessanten“
Teilgraphen durchmustern!
Branch-and-Bound!
Die Kanten des Graphen entsprechen
den möglichen Spalten des Alignments.
Das Gewicht einer Kante ist gleich
der Bewertung der zugehörigen Spalte.
Der Bauplan des Menschen
Sum-of-Pairs-Problem (SP):
Betrachte ein optimales SP-Alignment Aopt[S,P,Q] von S, P und Q.
Sei d(Sn , Pm , Ql ) der Wert des optimalen SP-Alignments.
AATGCGTCTC
AA-GCGTATC
AA-GCGT-TG
AATGCGTCTC
AA-GCGTATC
AATGCGTCTC
AA-GCGTATC
I(Sn , Pm)
AA-GCGTATC
AA-GCGT-TG
AAGCGTATC
AAGCGT-TG
I(Sn , Ql)
AATGCGTCTC
AA-GCGT-TG
AATGCGTCTC
AA-GCGT-TG
I(Pm , Ql)
Betrachte alle durch das optimale Alignment implizierten paarweisen Alignments.
Die implizierten paarweisen Alignments erhält man, indem man die Spalten ohne
Informationen (Doppelminus => Bewertung 0) streicht.
Es gilt (d Ähnlichkeitsfunktion):
d(Sn , Pm , Ql ) = d(I(Sn , Pm )) + d(I(Sn , Ql )) + d(I( Pm , Ql ))
d(Sn , Pm )
+ d(Sn , Ql )
+ d( Pm , Ql )
Der Bauplan des Menschen
Sum-of-Pairs-Problem (SP):
Die Summe der optimalen paarweisen Alignments ist eine obere Schranke OS für den Wert des
optimalen SP-Alignments:
d(Sn , Pm , Ql ) OS = d(Sn , Pm ) + d(Sn , Ql ) + d( Pm , Ql )
Sei d(Si , Pj , Qr ) der Wert des optimalen SP-Alignments für die Präfixe Si , Pj , Qr .
Es gilt (d Ähnlichkeitsfunktion) für beliebige Präfixe Si , Pj , Qr :
d(Si , Pj , Qr ) d(Si , Pj ) + d(Si , Qr ) + d( Pj , Qr)
Bezeichnung:
Seien RSi der Suffix [si+1si+2....sn]
von S
RPj der Suffix [pj+1pj+2....pm] von P
RQr der Suffix [qr+1qr+2....ql] von Q
Es gilt natürlich auch für alle Suffixe RSi , RPj , RQr :
d(RSi , RPj , RQr ) d(RSi , RPj ) + d(RSi , RQr ) + d( RPj , RQr)
Der Bauplan des Menschen
Branch-and-Bound:
Sei d(Si , Pj , Qr ) der Wert des optimalen SP-Alignments für die Präfixe Si , Pj , Qr .
Dieser Wert wird im Alignment-Graphen (bzw. der Matrix) in dem zugehörigen Knoten gespeichert.
Wurde dieser Wert gerade berechnet, so kann man die Frage stellen, ob dieser Knoten überhaupt
auf dem optimalen Pfad liegen kann, d.h., ob man die Kanten, die von diesem Knoten ausgehen,
betrachten muss.
Nehmen wir einmal an, dass wir eine untere Schranke US für den Wert des optimalen SP-Alignments
von S, P ,Q kennen.
Ist
d(Si , Pj , Qr ) + d(RSi , RPj , RQr ) US
so kann der (d(Si , Pj , Qr ) ) Knoten nicht auf einem optimalen Pfad liegen.
Aus
d(RSi , RPj , RQr ) d(RSi , RPj ) + d(RSi , RQr ) + d( RPj , RQr)
Ist
d(Si , Pj , Qr ) + d(RSi , RPj ) + d(RSi , RQr ) + d( RPj , RQr) US
so kann der (d(Si , Pj , Qr ) ) Knoten nicht auf einem optimalen Pfad liegen.
folgt:
Der Bauplan des Menschen
Branch-and-Bound:
Um die unten angegebene Bedingung zur Reduzierung des Suchraums einsetzen zu können,
benötigt man
(1) die Werte aller optimalen paarweisen Alignments d(RSi , RPj ), d(RSi , RQr ), d( RPj , RQr).
[Ordne die Buchstaben von hinten nach vorne und berechne die paarweisen Alignments.]
(2) eine „gute“ untere Schranke US für den Wert des optimalen SP-Alignments.
[Zur Berechnung der unteren Schranke benötigt man gute Heuristiken.]
Ist
d(Si , Pj , Qr ) + d(RSi , RPj ) + d(RSi , RQr ) + d( RPj , RQr) US
so kann der (d(Si , Pj , Qr ) ) Knoten nicht auf einem optimalen Pfad liegen.
Der Bauplan des Menschen
Heuristiken zur Berechnung von multiplen Sequenz-Alignments:
Eine ganze Reihe von Heuristiken basieren auf der folgenden
Idee: Man kombiniere optimale paarweise Alignments zu multiplen Alignments.
Frage (1): Welche paarweisen Alignments lassen sich kombinieren (sind kompatibel)?
Frage (2): Wie kann man paarweise Alignments kombinieren ?
Satz [Feng & Doolitle (1987)]:
Gegeben ein Baum, in dem jeder Knoten eine Sequenz repräsentiert und jede Kante ein
optimales paarweises Alignment der beiden Sequenzen repräsentiert, die in den Knoten
der Kante gespeichert sind. Dann gibt es ein multiples Alignment aller vom Baum repräsentierten Sequenzen, das zu den paarweisen Alignments der Kanten kompatibel ist.
Kompatibel bedeutet hier genauer: Für jedes Paar von Sequenzen, das im Baum durch
eine Kante repräsentiert wird, ist das vom multiplen Alignment induzierte paarweise
Alignment gleich dem paarweisen Alignment, das durch die Baumkante repräsentiert wird.
Der Bauplan des Menschen
Heuristiken zur Berechnung von multiplen Sequenz-Alignments:
A G-T
A GCT
ACT
AGT
A-C T
AG- T
A-CT
AG-T
AGC T
AGCT
AGG
AGCT
AGG
AGCT
AGC
AGCT
AGAGGT
A-C
A G G -T
Greedy-Ansatz:
A G G CT
AGGT
AGG-T
AGGCT
AGGCT
• E sei die Kantenmenge.
• MA eine (zunächst leere) Alignment-Menge.
• (Si , Sj) seien die Sequenzen der Kante e.
• ae sei das paarweise Alignment der Kante e.
• beste Kante = optimaler Alignment-Wert.
-T
CT
-T
-T
-T
=
A
A
A
A
A
G
G
G
G
-
G
G
C
C
-T
CT
-T
-T
-T
Solange E noch nicht leer ist,
a. Wähle die beste Kante e=(Si , Sj) in E.
b. Sind Si und Sj Teil von Alignments in MA,
dann kombiniere mittels ae (ersetze die zwei
alten durch das neue kombinierte Alignment).
c. Ist Si oder Sj Teil eines Alignments in MA,
dann kombiniere die andere Sequenz mit dem
entspr. Alignment in MA.
d. Ist weder Si noch Sj in MA vertreten, dann füge
ae zu MA hinzu.
e. Entferne e=(Si , Sj) aus E
Der Bauplan des Menschen
Heuristiken zur Berechnung von multiplen Sequenz-Alignments:
AGT
AG-T
AGGT
ACT
A-CT
AGGT
AGCT
AGGT
AGCT
AGGT
AGG-T
AGGCT
AGGCT
Wir nehmen o.B.d.A. an, dass die
Metrik d ein Maß für die Zahl der
Transformationen (Mutationen) ist,
um einen String S in einen String P
zu überführen.
Sterne (Stars) sind eine besondere Form von Bäumen
mit einem zentralen Knoten, dem Zentrum, und sonst
nur Blätter.
Gegeben einen Stern G. Ein Alignment AG heißt
Star-Alignment von G, falls das Alignment zu den
optimalen paarweisen Alignments der Kanten des Sterns
kompatibel ist.
Pevzner und Gusfield haben Approximationsverfahren
für das Sum-of-Pairs-Problem mit einer Metrik d als
Bewertungsfunktion (Distanzfunktion) entwickelt.
Für drei beliebige Sequenzen S, P, Q gilt:
d(S,Q) d(S,P) + d(P,Q) Dreiecksungleichung.
Der Bauplan des Menschen
Heuristiken zur Berechnung von multiplen Sequenz-Alignments:
AGT
ACT
Sei G=(E,V) ein ungerichteter Graph mit gewichteten Kanten.
Den kürzesten Pfad zwischen zwei Knoten v und w bezeichnen
AG-T
AGGT
A-CT
AGGT
wir mit sp(v,w).
Die Kommunikationskosten c(e) einer Kante e definieren wir als
die Zahl der kürzesten Pfade sp(v,w), die diese Kante benutzen.
AGCT
AGGT
AGG-T
AGGCT
AGCT
AGGT
In einem Stern mit k Knoten hat jede Kante e Kommunikationskosten
c(e) = k-1.
Unterscheiden wir noch zwischen dem kürzesten Pfad von
v nach w und von w nach v, so hat jede Kante e in einem Stern
mit k Knoten (k-Star) Kommunikationskosten.
AGGCT
c(e) = 2(k-1).
Seien {v1, ... , vk} die Knoten von G.
Sei C(G) = (cij) die k x k Matrix mit
• cij = c(e)
falls e=(vi , vj ) eine Kante von G ist, und
• cij = 0
sonst.
Der Bauplan des Menschen
Heuristiken zur Berechnung von multiplen Sequenz-Alignments:
AGT
ACT
Für ein vorgegebenes multiples Alignments A von k Sequenzen
definieren wir W(A) = (w(Aij)) als eine k x k Matrix mit den
AG-T
AGGT
A-CT
AGGT
Werten w(Aij), der vom Alignment A induzierten paarweisen
Alignments Aij.
AGCT
AGGT
AGCT
AGGT
Sei E = (ij) eine k x k Matrix mit 0 auf der Diagonale und 1 sonst.
Der Sum-of-Pairs-Wert eines Alignments A ist
AGG-T
AGGCT
W ( A) ij * w( Aij )
AGGCT
i, j
Den Sum-of-Pairs-Wert SP(G) eines zu einen Baum G=(V,E)
kompatiblen Alignments AG können wir wie folgt abschätzen:
?
SP(G ) C (G ) W ( AG ) cij * w(( AG )ij )
i, j
Der Bauplan des Menschen
Heuristiken zur Berechnung von multiplen Sequenz-Alignments:
ACT
AGT
Für jedes Alignment AG , das zu einem Baum G kompatibel ist,
und für jedes durch AG induzierte paarweise Alignment (AG)ij gilt:
A-CT
AGGT
AG-T
AGGT
w(( AG )ij )
AGCT
AGCT
AGGT
AGGT
AGGCT
AC G U A 0
1
1
1
1
C 1
0
1
1
1
G 1
1
0
1
1
U 1
1
1
0
1
-
1
1
1
0
1
e ( va ,vb )sp ( vi ,v j )
w(( A )
e ( va ,vb )sp( vi ,v j )
G ab
)
Beispiel (siehe Abbildung auf der rechten Seite):
2 1 2 3
Betrachten wir alle paarweisen Alignments (AG)ij und
summieren wir jeweils die Werte links und rechts in der
obigen Ungleichung, so erhalten wir die unten angegebene Formel:
AGG-T
AGGCT
AGG-T
AGGCT
AGC-T
AG--T
A-C-T
d ( e)
Den Sum-of-Pairs-Wert SP(G) eines zu einen Baum G=(V,E)
kompatiblen Alignments AG können wir wie folgt abschätzen:
?
SP(G ) C (G ) W ( AG ) cij * w(( AG )ij )
i, j
Der Bauplan des Menschen
Heuristiken zur Berechnung von multiplen Sequenz-Alignments:
Sei K eine Menge (Kollektion) von k-Stars in einem Graphen mit k Knoten.
K heißt balanciert, wenn es eine ganze Zahl r gibt, so dass
C (G) r
GK
Da C(G) nur für Kanten in G Einträge ungleich 0 hat und da ein zu G kompatibles Alignment AG
optimale paarweise Alignments für die Kanten von G induziert (garantiert), gilt für jedes beliebige
Alignment A:
C (G ) W ( AG ) C (G ) W ( A )
Lemma:
Ist K eine (mit Skalar) r-balancierte Menge von Stars, dann gilt
r
min (C (G ) W ( AG ))
min ( W ( A ))
GK
|K | A
Der Bauplan des Menschen
Lemma:
Ist K eine (mit Skalar) r balancierte Menge von Stars, dann gilt
r
min (C (G ) W ( AG ))
min ( W ( A ))
GK
|K | A
Beweis:
1
min (C (G ) W ( AG ))
(C (G ) W ( AG ))
GK
| K | GK
1
für jedes Alignment A
(C (G ) W ( A ))
| K | GK
r
( W ( A ))
|K |
Da diese Ungleichung für
jedes beliebige Alignment
gilt, gilt sie natürlich auch
für das optimale Alignment
min ( W ( A ))
A
Der Bauplan des Menschen
Der Star-Alignment-Algorithmus:
• Konstruiere eine balancierte Menge K von k-Stars.
• Berechne für jeden k-Star GK ein Star-Alignment AG.
• Bestimme den k-Star GBestK mit minimalem Wert (C(G) o W(AG)).
• Gib AG
zurück.
Best
Satz (Gusfield [1993])
Gegeben eine r-balancierte Menge K von k-Stars. Der obige Algorithmus liefert
eine Approximation des optimalen multiples Alignment mit einem garantierten
Approximationsfaktor von (2-(2/k)).
Beweis:
2(k 1)( k 1)
r | K |
k (k 1)
r
2
2
|K |
k
Die obige Aussage folgt jetzt aus dem vorhergehenden Lemma.