Softwarewerkzeuge der Bioinformatik WS06/07 Übungen 3: multiple
Werbung

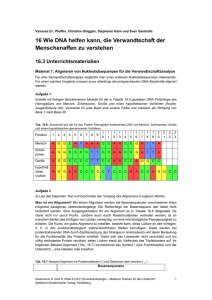
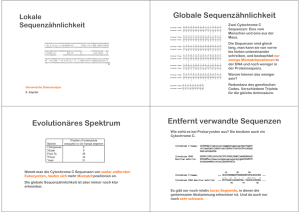
Softwarewerkzeuge der Bioinformatik WS06/07 Übungen 3: multiple Alignments, CLUSTALW 1. Einfaches multiples Sequenzalignment (a) Gehe zum ClustalW-Webinterface von EBI bei http://www.ebi.ac.uk/clustalw/. Schaue über die Seite und mache Dich mit den Funktionen und Informationen vertraut. Erklärungen zu den Feldern und Optionen von ClustalW gibt es im Anhang und im Menu auf der linken Seite (ClustalW Help). (b) Unter http://www.ebi.ac.uk/clustalw/alignment.txt findest Du eine Ansammlung aus Sequenzen im Multi-Fasta-Format. Kopiere diese Beispielsequenzen in das dafür vorgesehene Feld und starte das multiple Sequenzalignment mit den Standard-Parametern. (c) Mache Dich mit der Ausgabe vertraut. Mit Show Colors kannst Du die Aminosäuren gemäß dem ClustalW-Farbcode anzeigen lassen. Bestimme in JalView die Konsensussequenz und lasse die Konservierung berechnen. Wozu ist der Rollbalken Conservation Colour Increment nützlich? 2. Datenbanksuchen und multiple Sequenzalignments (a) Um welches Protein handelt es sich bei FOSB_MOUSE? (Entry name bei SRS, http://srs.ebi.ac.uk) Schaue dir das Alignment aus Aufgabe 1 noch mal an. Wo würdest Du funktionelle Domänen erwarten? Und liegen dort tatsächlich welche vor? Vergleiche mit dem Eintrag für FOSB_MOUSE in der Datenbank PRINTS und überprüfe, ob eines der aufgelisteten Motive tatsächlich konserviert ist. (b) Finde den Swiss-Prot-Eintrag von P00042 (Accession Number). Suche alle Proteine, die in derselben PFAMA-Familie wie P00042 sind und lasse Dir ihre Sequenzen im Fasta-Format anzeigen (Tip: Display Options – Show 500 results). Führe aus dieser Liste von Sequenzen ein Alignment in ClustalW durch und versuche mit Hilfe von JalView hochkonservierte Bereiche zu finden. Du wirst feststellen, daß manche Untergruppen zusätzliche konservierte Bereiche haben, die in anderen Sequenzen nicht auftreten. Vergleiche Deine potentiellen Domänen mit den Einträgen in der Datenbank PRINTS. 3. Phylogenetischer Baum (a) Suche in der Protein-Datenbank bei NCBI (http://www.ncbi.nlm.nih.gov/) folgende Sequenzen des Cathepsin L: gi|55958173 (Mensch), gi|27806673 (Rind), gi|15027272 (Schwein), gi|10185020 (Hund), gi|4887002 (Maus), gi|28194647 (Goldhamster) und gi|1752664 (Zebrafisch). (Tip: Du kannst alle 7 Proteinsequenzen gleichzeitig suchen, indem Du sie mit OR verknüpfst. Wähle dann Display FASTA und send to Text.) Erstelle ein multiples Alignment mit ClustalW. Betrachte das Alignment: Ist überall Konservierung zu erkennen? Welche Spezies unterscheidet sich auffällig von den anderen und sollte daher in einem phylogenetischen Baum die Outgroup sein? (b) Lasse Dir mit View Alignment File das Alignment anzeigen und kopiere es in das Sequenzen-Feld eines neuen ClustalW-Jobs, um eine Distanzmatrix und einen richtigen phylogentischen Baum zu erstellen. Hier kommen die Optionen der Rubrik PHYLOGENETIC TREE ins Spiel. Wähle TREE TYPE nj für die Methode neighbor joining, CORRECT DIST. on für eine Korrektur der Distanzen und IGNORE GAPS on, um alle Positionen, die Gaps enthalten, auszuschließen. Vergleiche das Ergebnis mit dem guide tree. Anhang: Erklärungen • Man kann das Alignment interaktiv durchführen oder sich die URL für das Ergebnis per E-Mail zuschicken lassen. (Empfehlenswert bei sehr großen Jobs, aber Achtung: kurze Speicherzeit!) • ALIGNMENT: Hier kann man die Stringenz des paarweisen Alignments einstellen, mit der der guide tree erstellt wird. Bei einer sehr großen Zahl von Sequenzen macht es mehr Sinn, fast auszuwählen, um schneller an die Ausgabe heranzukommen. Da dieser Schritt aber der • • • • • • wichtigste Punkt im Alignment ist, sollte sonst immer ein volles Alignment (full) ausgeführt werden, wobei jede Sequenz gegen jede andere vollständig ausgerichtet wird. Die Optionen aus der 2. Reihe (KTUP, WINDOW LENGTH, SCORE TYPE, ...) beziehen sich alle auf das schnelle BLAST-ähnliche Alignment (fast) zum Erstellen des guide tree. MATRIX: Hier kann man die Austauschmatrix wählen. Default ist Gonnet 250. GAP OPEN: Die Bestrafung zum erstmaligen Einfügen eines Gaps (Insertion oder Deletion). END GAP: Die Bestrafung zum Schließen eines Gaps. GAP EXTENSION: Ist aufzubringen um eine Lücke um eine Position zu verlängern. GAP DISTANCES: Dieser Wert definiert den Strafwert für das Trennen zweier Gap-Bereiche OUTPUT • Default ist Alignment mit Positionsnummern (aln w/numbers). Alignments im Format phylip dienen als Eingabe für das Programmpaket PHYLIP, das phylogenetische Bäume nach verschiedenen Methoden erstellt (http://evolution.genetics.washington.edu/phylip.html). PHYLOGENETIC TREE • Durch Wahl von dist bei TREE TYPE erhält man die Distanzmatrix angezeigt, nj konstruiert einen phylogenetischen Baum nach der neighbor joining-Methode. • CORRECT DIST. on führt eine Korrektur der Distanzen nach Motoo Kimura (1983) durch. Hintergrund ist, daß die Zahl der beobachteten Unterschiede (D) die Zahl der tatsächlichen Mutationen unterschätzt: es kann mehr als eine Mutation pro Residue aufgetreten sein. Daher wird die Zahl der Mutationen geschätzt durch die Formel K = - ln (1 - D - D²/5). Dies bewirkt, daß Zweige gestreckt werden. • IGNORE GAPS on betrachtet zur Erstellung der Distanzmatrix nur solche Positionen, an denen keine Sequenz ein gap hat, vergleicht also nur Aminosäuren untereinander. Vorteil: die Bewertung von gaps gegen Aminosäuren ist nicht eindeutig, also werden solche Teile des Alignments einfach ignoriert. Nachteil: das Alignment wird kürzer, d.h. es gibt weniger Positionen, um die Distanz zu errechnen; außerdem entsprechen gaps Insertionen/Deletionen, die durchaus etwas über die Evolution aussagen. Ausgabe • Die erste Sektion der ClustalW-Ausgabeseite beinhaltet eine Übersicht von der Suche und direkten Links zu den entsprechenden Ergebnissen. Zusätzlich erlaubt das Java-Applet JalView eine schöne graphische Darstellung des Alignments. Was diese Anwendung aber am meisten auszeichnet, ist die Möglichkeit, das Alignment manuell zu bearbeiten. • In der Sektion Scores Table ist eine Übersicht von den Alignments aufgezeigt. Alle paarweisen Alignmentkombinationen werden aufgezeigt und nach Verwandtschaft gewertet (Achtung, hier handelt es sich noch um Ähnlichkeits-Scores und nicht um Distanz-Scores). Unter den ausgerichteten Proteinsequenzen findet man eine Zeile mit ".", ":" und "*". "." = Spalte/Position im Alignment besitzt ungleiche, aber ähnliche Aminosäuren ":" = Spalte/Position im Alignment markiert hoch konservierte Bereiche "*" = Spalte/Position im Alignment zeigt identische Aminosäuren in allen Sequenzen • Die letzte Sektion ist der Guide Tree, nach dem die Sequenzen ausgerichtet wurden. Dieser (und auch ein echter phylogenetischer Baum) wird zunächst als Cladogramm dargestellt, in dem alle Zweige gleich lang sind. Show as Phylogram Tree wandelt ihn so um, daß die Länge der Zweige der Distanz entspricht, d.h. dem Prozentsatz der Mutationen, die seit der Aufspaltung der beiden Spezies an der Gabelung (die dem gemeinsamen Vorfahren enspricht) ereignet haben. Show Distances zeigt diese Entfernung als Zahl an. Anmerkung: SRS hat auch ein ClustalW-Modul. Hier wird per default ein schnelles Alignment durchgeführt es gibt keine Gonnet-Matrix und keine Baumdarstellung. Unter Display Option ClustalwAli findet man ebenfalls ein JalView-Applet, allerdings in einer älteren Version.