Softwarewerkzeuge der Bioinformatik WS05/06
Werbung

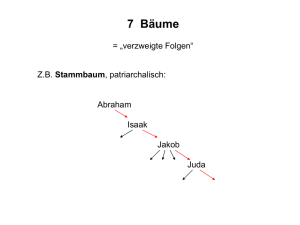
Softwarewerkzeuge der Bioinformatik WS05/06 - Übungen #4 Phylogenetische Bäume mit ClustalW (http://www.ebi.ac.uk/clustalw/) A) Guide tree ist nicht gleich phylogenetischer Baum 1. Finde unter Benutzung von SRS (http://www.srs.ebi.ac.uk) die Proteinsequenzen der Acetylcholinesterase von Mensch, Maus, Ratte, Rind, Katze, Kaninchen und Huhn. (Tip: SRS Feld Species) Führe ein multiples Alignment mit ClustalW durch. Betrachte das Alignment: Ist überall Konservierung zu erkennen? Welche Spezies unterscheidet sich auffällig von den anderen und sollte daher die outgroup sein? Notiere den guide tree. Speichere das Alignment-File (.aln) und benutze es als Eingabe für die folgende Aufgabe. 2. Zur Erstellung eines phylogenetischen Baums muß ClustalW ein multiples Alignment vorgegeben werden, aus dem eine Distanzmatrix erstellt wird. Hier kommen die Optionen der Rubrik PHYLOGENETIC TREE ins Spiel. Durch Wahl von dist bei TREE TYPE erhält man die Distanzmatrix angezeigt. CORRECT DIST. on führt eine Korrektur der Distanzen nach Motoo Kimura durch. Hintergrund ist, daß die Zahl der beobachteten Unterschiede (D) die Zahl der tatsächlichen Mutationen unterschätzt: es kann mehr als eine Mutation pro Residue aufgetreten sein. Daher wird die Zahl der Mutationen geschätzt durch die Formel K = - ln (1 – D – (D² /5)). Dies bewirkt, daß Zweige gestreckt werden. IGNORE GAPS on betrachtet zur Erstellung der Distanzmatrix nur solche Positionen, an denen keine Sequenz ein gap hat, vergleicht also nur Aminosäuren untereinander. Vorteil: die Bewertung von gaps gegen Aminosäuren ist nicht eindeutig, also werden solche Teile des Alignments einfach ignoriert. Nachteile: das Alignment wird kürzer, d.h. es gibt weniger Positionen, um die Distanz zu errechnen; außerdem entsprechen gaps Insertionen/Deletionen, die durchaus etwas über die Evolution aussagen. Der konstruierte phylogenetische Baum wird zunächst als Cladogramm dargestellt, in dem alle Zweige gleich lang sind. Show as Phylogram Tree wandelt ihn so um, daß die Länge der Zweige der Distanz entspricht, d.h. dem Prozentsatz der Mutationen, die seit der Aufspaltung der beiden Spezies an der Gabelung (die dem gemeinsamen Vorfahren enspricht) ereignet haben. Show Distances zeigt diese Entfernung als Zahl an. Experimentiere mit verschiedenen Kombinationen von CORRECT DIST. und IGNORE GAPS off/on. Worin unterscheiden sich die phylogenetischen Bäume vom guide tree und untereinander und warum? Betrachte auch die Distanzmatrizen. B) Konstruktion eines "Tree of Life" Erstelle ein Alignment aller Cytochrome C Sequenzen, die in der UniprotKB/Swiss-Prot Datenbank erhältlich sind (EntryName CYC_ - es sollten 115 Einträge sein) und konstruiere daraus einen phylogenetischen Baum. Welche biologischen Gruppen (Pflanzen, Pilze, Tiere, ...) kannst Du wiederfinden? (Tip: SRS Feld Species) Einige Abkürzungen: ALLMI - Alligator mississippiensis, Alligator APIME – Apis mellifera, Honigbiene ASPNG - Aspergillus niger - Schimmelpilz CAEEL – Caenorhabditis elegans, Fadenwurm (Nematode) EUGGR – Euglena gracilis, Grünalge GINBI – Ginkgo biloba, Ginkgobaum HELAS – Helix aspersa, Schnecke MACGI – Macropus giganteus, Känguruh PONPY - Pongo pygmaeus, Orang-Utan RANCA - Rana catesbeiana, Ochsenfrosch SAMNI – Smbucus nigra, Holunderbaum SCHGR - Schistocerca gregaria, Wüstenheuschrecke SCHPO – Schizosaccharomyces pombe, Spalthefe SPINOL – Spinacia oleracea, Spinat TETPY - Tetrahymena pyriformis, Wimpertierchen USTSP - Ustilago sphaerogena, Beulenbrandpilz XENTR – Xenopus tropicalis, Krallenfrosch