Molekulare Systeme 1 - Institut für Medizinische Informatik, Statistik
Werbung

Zellen
Molekulare Systeme 1
• Prokaryonte Zellen haben
keinen separaten Zellkern
(Bakterien, BlaugrünAlgen)
• Eukaryonte Zellen haben
einen durch eine Membran
vom Zytoplasma abgegrenzten Zellkern (echte
Algen, Pilze, Protozoen,
usw.)
Dr. Jochen Forberg
Institut für Medizinische Informatik,
Statistik und Epidemiologie
Zelluläre Substanzen
Proteine
Wasser
Kleine Ionen (Na+, K+, OH–, usw.)
Stoffwechselmoleküle (Kohlehydrate,
Lipide, Nucleotide, usw.)
Proteine
Nukleotide und Nukleinsäuren
• Ein Protein ist eine Kette einfacherer Moleküle
und zwar von Aminosäuren.
• In einem Protein sind die Aminosäuren durch
Peptidbindungen aneinander gekettet. Aus diesem
Grund heißen sie auch Polypeptidketten oder kurz
Polypeptide.
• Größe: kleinste weniger als 100 Aminosäuren
typisch ca. 300 Aminosäuren
größte mehr als 5000 Aminosäuren
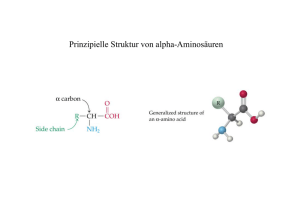
Aminosäuren
Beispiele für Aminosäuren
Jede Aminosäure hat ein zentrales Kohlenstoffatom Cα .
An das Cα – Atom ist ein
Wasserstoffatom H, eine
Aminogruppe NH2 , eine
Carboxylgruppe COOH und
eine Seitenkette gebunden.
Die verschiedenen Aminosäuren unterscheiden sich in
dieser Seitenkette.
CH3
HO
Seitenkette
H2N
C"
H
CH3
COOH
H2N
C"
CH
COOH
H2N
C"
H
H
Alanin
Threonin
COOH
1
Peptidbindung
Tabelle der 20 natürlichen Aminosäuren
A
C
D
E
F
G
H
I
K
L
Ala
Cys
Asp
Glu
Phe
Gly
His
Ile
Lys
Leu
Alanin
Cystein
Asparaginsäure
Glutaminsäure
Phenylalanin
Glycin
Histidin
Isoleucin
Lysin
Leucin
M
N
P
Q
R
S
T
V
W
Y
Met
Asn
Pro
Gln
Arg
Ser
Thr
Val
Trp
Tyr
Methionin
Asparagin
Prolin
Glutamin
Arginin
Serin
Threonin
Valin
Tryptophan
Tyrosin
H
N
C
C
H
O
H
OH + H
H
R1
N
C
C
H
O
N
C
C
H
O
H
R2
N
C
C
H
O
Peptidbindung
Tryptophansynthetase A aus E. coli
10
As3
ÂÅÅÅÅÅÅÅÅÅÅÅÅÅÅÅÅÅÅÅÅÄÅÅÅÅÅÅÅÅÅÅÅÅÅÅÅÅÅÅÅÅÃ
H R3
As1
As2
ÂÅÅÅÅÅÅÅÅÅÅÅÅÅÅÅÅÅÅÅÅÄÅÅÅÅÅÅÅÅÅÅÅÅÅÅÅÅÅÅÅÅÃ ÂÅÅÅÅÅÅÅÅÅÅÅÅÅÅÅÅÅÅÅÅÄÅÅÅÅÅÅÅÅÅÅÅÅÅÅÅÅÅÅÅÅÃ
H R1
H R2
OH + H
H
R3
N
C
C
H
O
N
C
C
H
O
OH
OH + 2(H2O)
Peptidbindung
Rinderinsulin
20
Met-Glu-Arg-Tyr-Glu-Ser-Leu-Phe-Ala-Gln-Leu-Lys-Glu-Arg-Lys-Glu-Gly-Ala-Phe-Val30
40
50
60
Pro-Phe-Val-Thr-Leu-Gly-Asp-Pro-Gly- Ile -Glu-Gln-Ser-Leu-Lys-Ile - Ile -Asp-Thr-LeuIle - Glu-Ala-Gly-Ala-Asp-Ala-Leu-Glu-Leu-Gly- Ile -Pro-Phe-Ser-Asp-Pro-Leu-Ala-Asp70
80
90
100
Gly-Pro-Thr-Ile -Gln-Asn-Ala-Thr-Leu-Arg-Ala-Phe-Ala- Ala- Gly-Val -Thr-Pro-Ala-GlnCys-Phe-Glu-Met-Leu-Ala-Leu-Ile-Arg-Gln-Lys-His-Pro-Thr- Ile -Pro- Ile -Gly-Leu-Leu110
120
130
140
S
A- Kette
S
Gly-Ile-Val-Glu-Gln-Cys-Cys-Ala-Ser-Val-Cys-Ser-Leu-Tyr-Gln-Leu-Glu-Asn-Tyr-Cys-Asn
5
Met-Tyr-Ala-Asn-Leu-Val-Phe-Asn-Lys-Gly-Ile-Asp-Glu-Phe-Tyr-Ala-Gln-Cys-Glu-Lys-
10
S
15
S
21
Val-Gly-Val -Asp-Ser-Val-Leu-Val -Ala-Asp-Val-Pro-Val-Gln-Glu-Ser-Ala-Pro-Phe-Arg150
160
Gln-Ala-Ala-Leu-Arg -His-Asn-Val-Ala-Pro -Ile-Phe -Ile-Cys-Pro-Pro-Asn-Ala-Asp-Asp170
180
Asp-Leu-Leu-Arg-Gln-Ile-Ala-Ser-Tyr -Gly-Arg-Gly -Tyr-Tyr-Tyr-Leu-Leu-Ser-Arg -Ala190
200
Gly -Val-Thr-Gly-Ala-Glu-Asn-Arg-Ala -Ala-Leu-Pro-Leu-Asn-His-Leu-Val-Ala-Lys-Leu210
220
230
240
250
260
Lys-Glu-Tyr-Asn-Ala-Ala-Pro-Pro-Leu-Gln-Gly-Phe-Gly -Ile -Ser-Ala-Pro-Asp-Gln-Val-
S
S
B- Kette
Phe-Val-Asn-Gln-His-Leu-Cys-Gly-Ser-His-Leu-Val-Glu-Ala-Leu-Tyr-Leu-Val-Cys-Gly-Glu5
15
10
20
Arg-Gly-Phe-Phe-Tyr-Thr-Pro-Lys-Ala
25
30
Lys-Ala-Ala -Ile -Asp-Ala-Gly-Ala -Ala-Gly-Ala - Ile -Ser-Gly-Ser-Ala- Ile -Val -Lys- IleIle -Glu-Gln-His-Asn- Ile -Glu-Pro-Glu-Lys-Met-Leu-Ala-Ala-Leu-Lys-Val-Phe-Val-Gln268
Pro-Met-Lys-Ala-Ala-Thr-Arg-Ser
Struktur von Proteinen
Primär-, Sekundär-, Tertiär- und Quartärstruktur
Enzyme
2
Sichelzellhämoglobin
Sichelzellanämie
Nukleinsäuren
Ribose und Desoxyribose
Ähnlich den Proteinen sind die Aminosäuren
Ketten aus einfacheren Molekülen. Diese
Bausteine heißen Nukleotide und bestehen aus
Phosphaten, Zucker und Pyrimidin- oder PurinBasen.
Lebende Organismen enthalten zwei Arten von
Nukleinsäuren: Ribonukleinsäure (RNA) mit
Ribose als Zuckerrest und Desoxyribonukleinsäure (DNA) mit Desoxyribose als Zuckerrest.
NH2
N
H
C
N
C
C
HN
CH
N
O
H
Adenin (A)
H2N
C
C
N
N
CH3
C
CH
H
Thymin (T)
O
HN
C
C
O
NH2
C
C
N
N
CH
N
Guanin (G)
O
H
Purine
C
Cytosin (C)
C
N
H
HO 5' H
OH
O
4'
H
H
H
3'
2'
HO
OH
HO 5' H
OH
O
1'
4'
H
H
Ribose
H
H
3'
2'
HO
H
1'
H
2'-Desoxyribose
(Base + Zucker)
O
N
H
Nukleosid
Nukleotidbasen
C
H
CH
HN
CH
C
O
Uracil (U)
C
N
H
CH
CH
Ein Nukleosid ist die
Verbindung eines
Zuckerrestes (Ribose oder
Desoxyribose) und einer
Nukleotidbase. Die
Verbindung erfolgt zwischen
dem C-Atom in Position 1
des Zuckers und einem NAtom der Base (N-glykosidische Bindung).
Pyrimidine
3
Nukleotid
(Base + Zucker + Phosphat)
Ein Nukleotid ist die
Verbindung eines
Zuckerrestes (Ribose
oder Desoxyribose) mit
einer Nukleotidbase
(Pyrimidin- oder PurinBase) mit einer
Phosphatgruppe
Nukleotid-Kette
Da in einer Nukleotidkette die
Hydroxy-Gruppe an der
Position 5' des Zuckerrestes
des einen Nukleotids über eine
Phosphordiesterbrücke mit der
Hydroxy-Gruppe des nächsten
Zuckerrestes in Position 3'
verbunden ist, besitzt die
Nukleotidkette eine Polarität.
Nach Konvention wird die
Sequenz von Nukleotidbasen
in der Richtung von 5' nach 3'
angegeben.
DNA
Die Sequenz der
Nukleotidbasen des
einen Stranges der
DNA entspricht
komplementär der
Basensequenz des
anderen Stranges in 3'
nach 5' Richtung.
Nukleotid-Kette
Eine Nukleotidkette entsteht
durch Verbindung der
Hydroxy-Gruppe eines
Zuckers über eine Phosphatverbindung mit dem nächsten
Zuckerrest. Die über die
Phosphat-Gruppen miteinander verbundenen Zuckerreste bilden den invariablen
Teil der Kette. Variabel ist
die Sequenz der Nukleotidbasen A, T oder U, C und G.
DNA
DNA ist ein Doppelstrang
zweier Ketten von
Nukleotiden mit Desoxyribose als Zucker. Dabei
liegen sich immer zwei
Nukleotidbasen gegenüber
und zwar ist stets ein Purin
(Adenin oder Guanin) mit
einem Pyrimidin (Thymin
oder Cytosin) gepaart.
DNA
James D. Watson und
Francis H. Crick
erkannten 1953, dass
DNA aus einer Doppelhelix bestehen muss.
Diese Struktur erklärt die
beiden wichtigen
funktionellen Aspekte Replikation und genetische
Informationsübertragung.
4
RNA
RNA ist ebenfalls eine Kette von
Nukleotiden. Es bestehen folgende
Unterschiede zur DNA:
– Ribose statt Desoxyribose als Zucker
– Uracil (U) statt Thymidin (T) als Base
– RNA ist ein einfacher Strang
Replikation
Da die sich in der
Doppelhelix
gegenüber liegenden
Nukleotidketten strikt
komplementär sind,
kann nach Öffnung
jede als Vorlage
(Templat) für die
Bildung (Replikation)
einer neuen dienen.
Genetischer Code
Genetische Information
Nukleotidbase
• Die genetische Information besteht in der Abfolge
von Nukleotidbasen. Die Sequenz von jeweils drei
Basen bedeutet ein Codewort (Codon) für eine
Aminosäure.
• Die Folge von Codons ergibt die
Aminosäuresequenz eines Polypeptides.
• Ein Gen kann als Abschnitt in der DNA definiert
werden, der für die Bildung eines Polypeptides
verantwortlich ist. Ein oder mehrere Polypeptide
bilden ein Protein.
Genetische
Informationsübertragung
• Die Abfolge der
Basenpaare in der DNA
wird zunächst in ein die
Information übertragendes
Botenmolekül aus RNA
(Messenger-RNA,
mRNA) übertragen
(Transkription).
• Dieses dient anschließend
als Vorlage für die
Produktion des
Polypeptids (Translation).
Uracil
(U)
Cytosin
(C)
Adenin
(A)
Guanin
(G)
Dritte
Zweite
Erste
Uracil (U)
Cytosin (C)
Adenin (A)
Guanin (G)
F Phenylalanin (Phe)
S Serin (Ser)
Y Tyrosin (Tyr)
C Cystein (Cys)
F Phenylalanin (Phe)
S Serin (Ser)
Y Tyrosin (Tyr)
C Cystein (Cys)
C
L Leucin (Leu)
S Serin (Ser)
Stop-Codon
Stop-Codon
A
L Leucin (Leu)
S Serin (Ser)
Stop-Codon
W Tryptophan (Trp)
G
L Leucin (Leu)
P Prolin (Pro)
H Histidin (His)
R Arginin (Arg)
L Leucin (Leu)
P Prolin (Pro)
H Histidin (His)
R Arginin (Arg)
L Leucin (Leu)
P Prolin (Pro)
Q Glutamin (Gln)
R Arginin (Arg)
A
L Leucin (Leu)
P Prolin (Pro)
Q Glutamin (Gln)
R Arginin (Arg)
G
I Isoleucin (Ile)
T Threonin (Thr)
N Asparagin (Asn)
S Serin (Ser)
U
I Isoleucin (Ile)
T Threonin (Thr)
N Asparagin (Asn)
S Serin (Ser)
C
I Isoleucin (Ile)
Start (Methionin)
T Threonin (Thr)
K Lysin (Lys)
R Arginin (Arg)
A
T Threonin (Thr)
K Lysin (Lys)
R Arginin (Arg)
G
V Valin (Val)
A Alanin (Ala)
D Asparaginsäure (Asp)
G Glycin (Gly)
U
V Valin (Val)
A Alanin (Ala)
D Asparaginsäure (Asp)
G Glycin (Gly)
C
V Valin (Val)
A Alanin (Ala)
E Glutminsäure (Glu)
G Glycin (Gly)
A
V Valin (Val)
A Alanin (Ala)
E Glutminsäure (Glu)
G Glycin (Gly)
G
U
U
C
Transkription
Die Codierung für ein Polypeptid ist immer nur in einem
Strang festgelegt (Sinnstrang, coding strand). Als Vorlage
für die Transkription in mRNA dient die komplementäre
Sequenz des Gegenstranges (template strand). Dieser wird
in 3´- nach 5´-Richtung gelesen. Die RNA-Synthese
erfolgt in 5´- nach 3´-Richtung.
5
Translation
Genexpression bei Eukaryonten
Die Translation erfolgt in den Ribosomen unter
Zwischenschaltung einer weiteren Klasse von RNA, der
Transfer-RNA (tRNA). Für jede Aminosäure gibt es eine
eigene tRNA mit einem Bereich, der komplementär zum
Codon der mRNA ist (Anticodon).
DNA
primäres
RNATranskript
Transkription
RNA-Processing
reife mRNA
Aminosäurekette
Transport ins Cytoplasma
mRNA
Translation
Ribosom
RNA-Prozessierung
DNA
Intron 1
Intron 2
Exon 2
Exon 1
Exon 3
Primäres Transkript
mRNA
Promotorsequenzen
'
Gen(e)
35-Region
Pribnow-Box
(' 10-Region)
• Die Region eines Gens auf der DNA, die die
Erkennungssignale für den Start der Transkription
beinhalten, bezeichnet man als Promotor.
• Eine dissoziierbare Untereinheit der RNAPolymerase, der sogenannte Sigma-Faktor, sorgt
dafür, dass die RNA-Polymerase die
Promotorregionen erkennt und an sie binden kann.
• Damit eine Transkription erfolgen kann, müssen
bestimmte Bedingungen im Promotorbereich
erfüllt sein.
Leseraster
Initiationsstelle
ACCCCAGGCTTTACACTTTATGCTTCCGGCTCGTATGTTGTGTGGAATTGTGAGCGG
lac1
CCATCGAATGGCGCAAAACCTTTCGCGGTATGGCATGATAGCGCCCGGAAGAGAGTC
gal P2
ATTTATTCCATGTCACACTTTTCGCATCTTTGTTATGCTATGGTTATTTCATACCAT
araB,A,D GGATCCTACCTGACGCTTTTTATCGCAACTCTCTACTGTTTCTCCATACCCGTTTTT
araC
GCCGTGATTATAGACACTTTTGTTACGCGTTTTTGTCATGGCTTTGGTCCCGCTTTG
trp
AAATGAGCTGTTGACAATTAATCATCGAACTAGTTAACTAGTACGCAAGTTCACGTA
bioA
TTCCAAAACGTGTTTTTTGTTGTTAATTCGGTGTAGACTTGTAAACCTAAATCTTTT
bioB
CATAATCGACTTGTAAACCAAATTGAAAAGATTTAGGTTTACAAGTCTACACCGAAT
tRNATyr
CAACGTAACACTTTACAGCGGCGCGTCATTTGATATGATGCGCCCCGCTTCCCGATA
rrn D1
CAAAAAAATACTTGTGCAAAAAATTGGGATCCCTATAATGCGCCTCCGTTGAGACGA
rrn E1
CAATTTTTCTATTGCGGCCTGCGGAGAACTCCCTATAATGCGCCTCCATCGACACGG
rrn A2
AAAATAAATGCTTGACTCTGTAGCGGGAAGGCGTATTATGCACACCCCGCGCCGCTG
lac
Konsensussequenz:
Initiation der Transkription
TGTTGACA----- 11-15 bp-----TATAAT--- 5-8 bp--- Initiationsstelle
TAATCGAATGGGC
TAA TCG AAT GGG C
T AAT CGA ATG GGC
TA ATC GAA TGG GC
6
Sequenzvergleiche
Typische Fragestellungen
• Der Vergleich verschiedener Basen- oder
Aminosäurensequenzen ist eine elementare
Aufgabe der Bioinformatik.
• Wozu werden solche Sequenzvergleiche
durchgeführt?
• Welche Algorithmen benutzt man hierfür?
1. Wir haben zwei Sequenzen über dem gleichen
Alphabet, beide von etwa der gleichen Länge
(Tausende von Zeichen). Wir wissen, dass die
Sequenzen im wesentlichen gleich sind und nur
an wenigen isolierten Stellen Differenzen durch
Insertion oder Deletion oder Substitution von
Zeichen auftreten. Wir wünschen die Stellen, an
denen diese Differenzen auftreten, zu finden.
=> Suche nach Polymorphismen,
Mutationssuche
Typische Fragestellungen
Typische Fragestellungen
2. Wir haben zwei Sequenzen über dem
gleichen Alphabet von einigen 100
Zeichen Länge. Wir wollen wissen, ob die
eine Sequenz eine Präfixsequenz hat, die
Suffixsequenz der anderen Sequenz ist.
Wenn die Antwort ja ist, so soll diese
Überlappungssequenz angegeben werden.
=> Fragmentmontage beim Sequenzieren
3. Wir haben zwei Sequenzen über dem gleichen
Alphabet von einigen 100 Zeichen Länge. Gibt
es Substrings in den beiden Sequenzen, die
einander gleich sind oder wenigstens einander
sehr ähnlich sind?
=> Identifikation von Bindungsstellen in
Promotorbereichen oder von Bindungsdomänen bei Proteinen
Typische Fragestellungen
Sequenzvergleich
4. Wir haben viele Sequenzen über dem
gleichen Alphabet von einigen 100
Zeichen Länge. Gibt es Substrings in
diesen Sequenzen, die einander gleich
sind oder wenigstens einander sehr
ähnlich sind?
=> Suche nach evolutionär konservierten
Sequenzen
Aus informatischer Sicht handelt es sich um den
Vergleich von Strings über einem bestimmten
Alphabet von Zeichen
- Vergleich von Nukleotidsequenzen:
Alphabet bestehend aus den 4 Zeichen
für die 4 Nukleotidbasen
- Vergleich von Aminosäuresequenzen:
Alphabet bestehend aus den 20 Zeichen
für die 20 Aminosäuren
7
Ähnlichkeit und Alignment
• Ähnlichkeit von zwei Sequenzen ist ein
Maß dafür, wie gut die Übereinstimmung
dieser Sequenzen ist.
• Ein Alignment ist die Anordnung einer
Sequenz über einer anderen, um die
Korrespondenz zwischen ähnlichen Zeichen
oder Substrings zu erkennen.
Beispiel globales Alignment
s: GATCGGAATAG
t: GACGGATTAG
s’:GATCGGAATAG
t’:GA-CGGATTAG
Score: +1 match
-1 mismatch
-2 gap
sim(s,t) = 9∗(+1) + 1∗ (-1) + 1∗ (-2) = 6
Globales Alignment zweier
Sequenzen
1. Einfügen von Leerstellen (gaps), so dass beide
Sequenzen danach gleiche Länge haben und
möglichst gut übereinstimmen.
2. Übereinanderlegen der durch Leerstellen
erweiterten Sequenzen, so dass eine
Korrespondenz zwischen den Zeichen und
Leerstellen der einen Sequenz und den Zeichen
und Leerstellen der anderen Sequenz entsteht.
Alignment zweier Sequenzen
Ähnlichkeit zweier Sequenzen:
1. Spaltenweiser Score = g falls Alignment Zeichen-gap
p(i,j) falls Alignment s[i] mit t[j]
2.
Totaler Score =
∑ Spaltenweiser Score
Spalten
Optimales Alignment zweier Sequenzen:
Alignment so, dass totaler Score maximal möglichen
Wert annimmt!
Globales Alignment zweier
Sequenzen
3. Es wird zusätzlich vereinbart, dass keine
Leerstelle in der einen Sequenz mit einer
Leerstelle in der anderen Sequenz
korrespondieren darf.
4. Leerstellen dürfen sowohl am Anfang als auch
am Ende einer Sequenz eingefügt werden.
Zahl der Alignments
Zwischen zwei Sequenzen der Länge n sind
2n (2n )! 2 2 n
~
=
2
πn
n (n!)
globale Alignments möglich.
Systematisches Aufzählen ist also nicht
vertretbar.
8
Dynamische Programmierung
(Needleman – Wunsch – Algorithmus)
Schrittweise Erzeugung des Alignments:
Welche Möglichkeiten gibt es, ein Alignment des
Präfixes s[1..i-1] der Sequenz s bis zum Zeichen
i-1 mit dem Präfix t[1..j-1] von t bis zum Zeichen
j-1 fortzusetzen?
i-1,j-1
1. Ordne dem t[j] eine Lücke zu.
2. Ordne dem s[i] das t[j] zu.
3. Ordne dem s[i] eine Lücke zu.
Weitere Möglichkeiten gibt es nicht, da
eine Zuordnung von zwei Lücken
ausgeschlossen wurde.
i-1,j-1
i-1,j
2.
i,j-1
Needleman – Wunsch - Algorithmus
1.
3.
2.
i,j
1. Align s[1..i] mit t[1..j-1] und match ein gap mit t[j]
2. Align s[1..i-1] mit t[1..j-1] und match s[i] mit t[j]
3. Align s[1..i-1] mit t[1..j] und match t[j] mit einem gap
i-1,j
i,j-1
1.
3.
i,j
sim( s[1.. i ], t[1.. j − 1]) − g
sim( s[1.. i ], t[1.. j ]) = max sim( s[1.. i − 1], t[1.. j − 1]) + p(i , j )
sim( s[1.. i − 1], t[1.. j ]) − g
t
Beispiel:
0
A
G
C
1
2
3
0
• Globales Alignment von s = AAAC mit t = AGC
• Ähnlichkeitsscore:
g = -2
p(i,j) = +1 , falls s[i] = t[j]
p(i,j) = -1 , falls s[i] ≠ t[j]
A
1
A
2
A
3
C
4
s
9
t
t
A
G
C
0
1
2
3
0
0
-2
-4
-6
A
1
-2
A
2
-4
A
G
0
1
2
3
0
0
-2
-4
-6
A
1
-2
A
2
-4
+1
s
C
-2
-2
s
A
3
-6
A
3
-6
C
4
-8
C
4
-8
t
t
A
G
C
0
1
2
3
0
0
-2
-4
-6
A
1
-2
1
A
2
-4
A
G
0
1
2
3
0
0
-2
-4
-6
A
1
-2
1
A
2
-4
-1
s
C
-2
-2
s
A
3
-6
A
3
-6
C
4
-8
C
4
-8
t
A
t
A
G
C
0
1
2
3
0
0
-2
-4
-6
1
-2
1
-1
A
A
G
0
1
2
3
0
0
-2
-4
-6
1
-2
1
-1
+1
A
2
-4
s
A
2
-4
C
-2
-2
s
A
3
-6
A
3
-6
C
4
-8
C
4
-8
10
t
t
A
G
C
0
1
2
3
0
0
-2
-4
-6
A
1
-2
1
-1
A
2
-4
-1
s
A
G
0
1
2
C
3
0
0
-2
-4
-6
A
1
-2
1
-1
-3
A
2
-4
-1
0
-2
s
A
3
-6
A
3
-6
-3
-2
-1
C
4
-8
C
4
-8
-5
-4
-1
t
t
A
G
C
0
1
2
3
0
0
-2
-4
-6
A
1
-2
1
-1
-3
A
2
-4
-1
0
-2
s
A
G
0
1
2
C
3
0
0
-2
-4
-6
A
1
-2
1
-1
-3
A
2
-4
-1
0
-2
s
A
3
-6
-3
-2
-1
A
3
-6
-3
-2
-1
C
4
-8
-1
-4
-1
C
4
-8
-1
-4
-1
s:
C
s:
AC
t:
C
t:
-C
t
t
A
G
C
0
1
2
3
0
0
-2
-4
-6
A
1
-2
1
-1
-3
A
2
-4
-1
0
-2
s
A
G
0
1
2
C
3
0
0
-2
-4
-6
A
1
-2
1
-1
-3
A
2
-4
-1
0
-2
s
A
3
-6
-3
-2
-1
A
3
-6
-3
-2
-1
C
4
-8
-1
-4
-1
C
4
-8
-1
-4
-1
s:
AAC
s: A A A C
t:
G -C
t: A G - C
11
Rekursiver Algorithmus zur Bestimmung des Alignments
Needleman – Wunsch - Algorithmus
Algorithm Similarity
input: sequences s and t
output: similarity between s and t
m:=|s|
n:=|t|
for i:=0 to m do a[i,0]:=i*g
for j:=0 to n do a[0,j]:=j*g
for i:=0 to m do
for j:=0 to n do a[i,j]:= max(a[i,j-1]+g,
a[i-1,j-1]+p(i,j),
a[i-1,j]+g)
return a[m,n]
Algorithm Align
input: indices i,j, parameter len, Array a given by Similarity
output: alignment in align_s,align_t, and length in len
if i=0 and j=0 then
len:=0
else if i>0 and a[i,j]=a[i-1,j]+g then
Align(i-1,j,len)
len:=len+1
align_s[len]:=s[i]
align_t[len]:= else if i>0 and j>0 and a[i,j]=a[i-1,j-1]+g then
Align(i-1,j-1,len)
len:=len+1
align_s[len]:=s[i]
align_t[len]:=t[j]
else //has to be j>0 and a[i,j]=a[i,j-1]+g
Align(i,j-1,len)
len:=len+1
align_s[len]:= align_t[len]:=t[j]
align_s und align_t sind global zu Align
max( |s|, |t|) ≤ len ≤ m + n
Vergleich von Proteinsequenzen
BLOSUM50 Substitutionsmatrix
A
R
N
D
C
Q
E
G
H
I
L
K
M
F
P
S
T
W
Y
V
A
R
N
D
C
Q
E
G
H
I
L
K M
F
P
S
T W
Y
V
5
-2
-1
-2
-1
-1
-1
0
-2
-1
-2
-1
-1
-3
-1
1
0
-3
-2
0
-2
7
-1
-2
-4
1
0
-3
0
-4
-3
3
-2
-3
-3
-1
-1
-3
-1
-3
-1
-1
7
2
-2
0
0
0
1
-3
-4
0
-2
-4
-2
1
0
-4
-2
-3
-2
-2
2
8
-4
0
2
-1
-1
-4
-4
-1
-4
-5
-1
0
-1
-5
-3
-4
-1
-4
-2
-4
13
-3
-3
-3
-3
-2
-2
-3
-2
-2
-4
-1
-1
-5
-3
-1
-1
1
0
0
-3
7
2
-2
1
-3
-2
2
0
-4
-1
0
-1
-1
-1
-3
-1
0
0
2
-3
2
6
-3
0
-4
-3
1
-2
-3
-1
-1
-1
-3
-2
-3
0
-3
0
-1
-3
-2
-3
8
-2
-4
-4
-2
-3
-4
-2
0
-2
-3
-3
-4
-2
0
1
-1
-3
1
0
-2
10
-4
-3
0
-1
-1
-2
-1
-2
-3
2
-4
-1
-4
-3
-4
-2
-3
-4
-4
-4
5
2
-3
2
0
-3
-3
-1
-3
-1
4
-2
-3
-4
-4
-2
-2
-3
-4
-3
2
5
-3
3
1
-4
-3
-1
-2
-1
1
-1
3
0
-1
-3
2
1
-2
0
-3
-3
6
-2
-4
-1
0
-1
-3
-2
-3
-3
-3
-4
-5
-2
-4
-3
-4
-1
0
1
-4
0
8
-4
-3
-2
1
4
-1
-1
-3
-2
-1
-4
-1
-1
-2
-2
-3
-4
-1
-3
-4
10
-1
-1
-4
-3
-3
1
-1
1
0
-1
0
-1
0
-1
-3
-3
0
-2
-3
-1
5
2
-4
-2
-2
0
-1
0
-1
-1
-1
-1
-2
-2
-1
-1
-1
-1
-2
-1
2
5
-3
-2
0
-2
-1
-2
-3
-3
-1
-2
-3
2
-1
-1
-2
0
4
-3
-2
-2
2
8
-1
0
-3
-3
-4
-1
-3
-3
-4
-4
4
1
-3
1
-1
-3
-2
0
-3
-1
5
-1
-2
-2
-4
-2
0
-2
-3
-1
2
3
-2
7
0
-3
-2
-1
-1
0
1
-3
-3
-4
-5
-5
-1
-3
-3
-3
-3
-2
-3
-1
1
-4
-4
-3
15
2
-3
Lokales Alignment von zwei Sequenzen
Suche nach Substrings, die mit hohem Score matchen.
Beispiel:
H E A G A W G H E E
P A W H E A E
Optimale lokale Übereinstimmung
G A W G H E E
P A W – H E A
Globales Alignment von Proteinsequenzen
0
H
E
A
G
A
W
G
H
E
E
-8
-16
-24
-32
-40
-48
-56
-64
-72
-80
P
-8
-2
-9
-17
-25
-33
-42
-49
-57
-65
-73
A
-16
-10
-3
-4
-12
-20
-28
-36
-44
-52
-60
W
-24
-18
-11
-6
-7
-15
-5
-13
-21
-29
-37
H
-32
-14
-18
-13
-8
-9
-13
-7
-3
-11
-19
E
-40
-22
-8
-16
-16
-9
-12
-15
-7
3
-5
A
-48
-30
-16
-3
-11
-11
-12
-12
-15
-5
2
E
-56
-38
-24
-11
-6
-12
-14
-15
-12
-9
1
H E A G AW G H E ! E
! ! P ! A W! H E A E
Multiples Alignment
Beispiel: Gesucht ist das optimale
Alignment folgender vier Sequenzen
M Q P I L L L
M L R L L
M K I L L L
M P P V L I L
Optimales Alignment
M Q P I L L L
M L R – L L –
M K – I L L L
M P P V L I L
Vergleich durch
sum-of-pairs Score.
Z.B. Score für die vierte Spalte
des obigen Alignments:
SP-Score(I,-,I,V) =
p(I,-) + p(I,I) + p(I,V) +
p(-,I) + p(-,V) + p(I,V)
12
Abbildungsnachweis
E.Passarge. Taschenatlas der Genetik. Stuttgart; New York: Thieme. 1994:
Folien 2, 17-24, 26, 29-31.
D.T.Suzuki, A.J.F.Griffiths, J.H.Miller, R.C.Lewontin. Genetik. Übersetzung herausgegeben
von S.Achten und P.Böhm. Weinheim: VCH, 1991:
Folien 8-10, 12-14, 32, 35.
J.Setubal and J.Meidanis. Introduction to computational molecular biology.
Boston: PWS Publishing Company, 1997:
Folien 11, 44, 53, 67, 68.
R.Durbin, S.Eddy, A.Krogh, G.Mitchison. Biological sequence analysis: Probalistic models
of proteins and nucleic acids. Cambridge University Press, 1998:
Folien 69, 70.
13