Institut für MTA-Ausbildung am Klinikum Osnabrück - mtaschule
Werbung

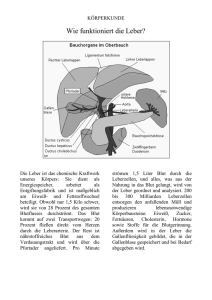
Institut für MTA-Ausbildung am Klinikum Osnabrück Fachgebiet Klinische Chemie (Dr. Krämer) Leber 1. Allgemeine Grundlagen 1.1 Hauptaufgaben der Leber: Kohlenhydratstoffwechsel: Glukosehomöostase, Glykogensynthese, Glykogenolyse, Glukoneogenese, Pentosephosphatzyklus, Metabolisierung von Zuckern (Glukose, Fruktose, Galaktose, Sorbit) Proteinstoffwechsel: Synthese der überwiegenden Anzahl von Plasmaproteinen (nicht der Immunglobuline) und der hepatogenen Gerinnungsfaktoren (II, V, VII, X, XI, XII, XIII), Katabolismus von Plasmaproteinen, Harnstoff-, Harnsäure und Kreatinsynthese, Aminosäuremetabolisierung Lipid- und Lipoproteinstoffwechsel : Synthese von Fettsäuren, Triglyceriden, Phospholipiden, Gallensäuren, Cholesterin, Ketonkörpern, Lipoproteinen, ß-Oxidation der Fettsäuren Biotransformation und Entgiftung : Metabolische Aktivierung und Deaktivierung endeogener und exogener Substanzen durch Oxidation, Reduktion und Hydrolyse, mit nachfolgender Glucuronidierung, Sulfatierung, Methylierung, Azetylierung oder Glycinierung. Dadurch Herstellung der Wasserlöslichkeit und Ausscheidung über die Nieren oder die Gallenflüssigkeit. Speicherung der fettlöslichen Vitamine : Gespeichert können in der Leber die Vitamine E, D, E und K. Vitamin K ist für die Prothrombinsynthese notwendig. Marcumar (Phenprocumon) ist ein Vitamin-K-Antagonist, senkt den Prothrombinspiegel und hemmt damit die Blutgerinnung. Die Hemmung wird erfaßt über die Bestimmung der Thromboplastinzeit nach Quick bzw. über die Bestimmung der INR. 1.2 Leberschädigungen und ihre Kenngrößen Die Leber ist aufgrund ihres hohen Differenzierungsgrades ein recht empfindliches Organ. Vier hauptsächliche Schädigungsprozesse führen zu Reaktionen der Leber. Diese Prozesse sind: Leberzelluntergang (Zellnekrose), metabolische Insuffizienz, Cholestase, Fibrose -2Durch Bestimmung geeigneter Laborparameter kann man Rückschlüsse auf eine der genannten Schädigungsursachen ziehen. 1.2.1 Leberzelluntergang (Zellnekrose) verursacht durch: Beeinträchtigung des Energiestoffwechsels der Mitochondrien mit ATP-Defizit. Es kommt zur Freisetzung lysosomaler Enzyme und toxischer Produkte durch aktivierte Makrophagen und als Folge davon zur Schädigung der Zellmembran. Schädigung der Zelle -> erhöhte Permeabilität der Plasmamembran -> Austritt von Zellinhaltsstoffen => Grundlage der Enzymdiagnostik von Lebererkrankungen. Höhe des Aktivitätsanstieges der Zellenzyme steht in Beziehung zu Umfang und Aktivität des Zellschadens. Parameter: Aspartataminotransferase (ASAT, GOT) gehört wie auch Alaninaminotransferase (ALAT, GPT) zur Gruppe der Transaminasen, katalysiert Transfer von Aminogruppen von Aspartat auf Ketoglutarat unter Bildung von Oxalacetat. Weite Verbreitung in Körpergeweben, besonders aber in der Leber vorhanden. Jedoch nicht leberspezifisch, vergleichsweise hohe Aktivitäten auch in Myokard, Skelettmuskel und Niere. Parameter: Alaninaminotransferase (ALAT, GPT) Transfer von Aminogruppen von Alanin auf Ketoglutarat unter Bildung von Pyruvat Spezifische Aktivität in der Leber am höchsten, daher Anstieg im Serum weitgehend spezifisch für Leberzellschädigung Transaminasen sind bis zu gewissem Grad bei allen Lebererkrankungen erhöht, Ausmaß des Schadens bestimmt die Höhe der Aktivität im Plasma/Serum. Parameter: Glutamatdehydrogenase (GLDH) weitgehend leberspezifisches Enzym, ausschließlich intramitochondrial lokalisiert, vorwiegend in azinusnahen Leberzellen . Erhöhte Aktivitäten bei tiefgehenden Leberschäden. Dient der vorläufigen Ammoniakelimination durch Übertragung des NH3 auf Ketoglutarat Parameter: Gamma-Glutamyl-Transferase (Gamma-GT) Weitläufig vorkommend. Leberaktivität aber bestimmend für Serumspiegel. Aktivitätserhöhung der Gamma-GT ist ein sehr sensitiver Indikator für eine hepatobiliäre Erkrankung. Höchste Spiegel bei alkoholtoxischer Hepatitis und bei Cholestase (s.u.). -3Leichte Leberzellschäden => Anstieg der Gamma-GT und der zytoplasmatischen Enzyme GPT(ALAT) und GOT(ASAT) (Quotient GOT/GPT <1) Schwere Leberzellschäden => zusätzlich weiterer Anstieg mitochondrialer Enzyme (GLDH, teilweise GOT). Quotient GOT/GPT steigt über 1 Eigenschaften einiger Leberenzyme Aspartat-Amino-Transferase (GOT, ASAT) 80% mitochondrial, 20% zytoplasmatisch, Halbwertszeit ca. 17 Tage Alanin-Amino-Transferase (GPT, ALAT) 15% mitochondrial, 85% zytoplasmatisch Halbwertszeit 47 ± 10 Tage Glutamatdehydrogenase (GlDH): mitochondriales Enzym, Halbwertszeit ca. 18 Tage Lactat-Dehydrogenase-Isoenzym 5 (LDH 5) zytoplasmatisches Enzym, Halbwertszeit 10 ± 2 Tage Erkrankungen mit Erhöhung der Serumaktivitäten von Transaminasen und GLDH: Stark erhöhte Aktivitäten bei: Akuter Hepatitis, akuter toxischer Leberschädigung, akuter Leberstauung (z.B. bei Rechtsherzinsuffizienz), Verschlußikterus, biliärer Zirrhose, Lebermetastatsen Mäßige Aktivitätserhöhungen bei: Myokardinfarkt, Traumen, postoperativ, Muskeldystrophie, akuter Pankreatitis, Lungenembolie, Hirn- und Niereninfarkt, Mononucleosis infectiosa, Leberzirrhose, chronisch aktiver Hepatitis, Stauungsleber, Leberzirrhose, alkoholischer Fettleber, schwerer Ketoazidose bei Diabetesentgleisung. Geringe Aktivitätserhöhungen bei: Leberzirrhose (niedriger Aktivitätsgrad), Myokarditis, Mononucleosis infectiosa (in Abheilung) Myokardinfarkt (Spätphase), Pankreatitis, Lebertumoren/-metastasen -4- 1.2.2 Metabolische Insuffizienz und Einschränkung der Biotransformation Leber synthetisiert und sezerniert nahezu alle Plasmaproteine, ist Ort der Biotransformation vieler exogener (z.B. Pharmaka, Toxine) und endogener Substanzen (z.B. Ammoniak) Einschränkungen dieser Funktionalität => veränderte Plasmakonzentrationen der Syntheseprodukte bzw. Metabolite, allerdings erst bei ausgedehnterem Parenchymschaden. Geringere Schädigungen dann nur durch Belastungsteste nachweisbar. Konzentrationsabnahme von Syntheseprodukten : Leber ist vorherrschendes Organ der Plasmaproteinsynthese, daher ist die Plasmaproteinkonzentration ein Maß der Lebersyntheseleistung. Konzentrationsabfälle treten aber nicht in der Frühphase von Lebererkrankungen auf, da die Halbwertszeit der Proteine ziemlich lang ist: Albumin (20 Tage), Präalbumin (2 Tage) Bei Coeruloplasmin, Transferrin, und Gerinnungsfaktoren (Fibrinogen) sieht das anders aus. Diese Parameter können sich innerhalb von Stunden nach Eintritt einer Leberschädigung verändern (z.B. Knollenblätterpilzvergiftung). Allerdings ist Proteinkonzentration im Serum auch vom Sekretionsvermögen und -weg, von Elimination und Abbau in der Leber abhängig, nicht allein von der Syntheseleistung. Hinweis: Serumglobuline, d.h. Alpha-, Beta- und Serumimmun-Globuline (Gamma) sind bei chronischen Leberschäden nichtselektiv erhöht. Lipidstoffwechsel : Im Zusammenhang mit akuten und chronischen Lebererkrankungen kommt es zu qualitativen und quantitativen Verschiebungen des Lipoproteinspektrums (Dyslipoproteinämie). Deren Bedeutung für die Diagnostik der akuten Leberinsuffizienz ist gering. Fetteinlagerungen kommen insbesondere bei Diabetiker sowie bei erhöhtem Alkoholkonsum vor. Kohlenhydratstoffwechsel : Fehlregulationen der Blutglukosekonzentration können vorkommen durch Erschöpfung der Glykogenreserven in der geschädigten Leber sowie durch Störungen des Monosaccharidmetabolismus bei fortgeschrittenem Leberparenchymverlust. Konzentrationsabnahme von Sekretionsenzymen: Cholinesterase: Im Blut gibt es verschiedene Cholinesterasen (Abbau von Acylcholin -> Cholin) Nach Substratspezifität unterscheidet man zwei Gruppen:echte, substratspezifische Cholinesterasen (Hirn, Muskel, Erythrozyten und Thrombozyten) - spielen wichtige Rolle bei Erregungsübertragung in Synapsen, sind unbedeutend für Diagnostik der Leberinsuffizienz Ferner gibt es substratunspezifische Cholinesterasen (18 genet. Varianten), die Acetylcholin, aber auch andere Substrate (z.B. Butyrylthiocholin, Succinylcholin) spalten können. Diese Enzyme werden in der Leber synthetisiert und sind damit Marker der Syntheseleistung. -5Gemessen wird stellvertretend für alle die Acetylthiocholinesterase (ACHES bzw. einfach ChE) Es gibt Spezifische genetische Varianten mit verminderter Fähigkeit zum Abbau des Muskelrelaxans Succinycholin (Narkosezwischenfälle) können auftreten. Daher wird vor Allgemeinnarkosen mit Anwendung von Succinylcholin die Aktivität der ChE bestimmt. Verminderte Biotransformationsleistung : Das mikrosomales fremdstoffmetabolisierendes Enzymsystem (Cytochrom P450) metabolisiert viele exogene und endogene Substrate (mehr als 200). Wesentliche Funktion: Transformation lipidlöslicher Stoffe in wasserlösliche => somit durch Galle und Urin ausscheidbar. Außerdem "Giftungsprozesse", z.B. Aktivierung von Pharmaka und Bildung zytotoxischer Stoffe, und Entgifungsprozesse (Pharmaka, Alkohol) Es stehen diverse direkte und indirekte Methoden zur Einschätzung der Biotransformation zur Verfügung, nur kurz: direkt: Bestimmungen der Aktivität von Biotransformationsenzymen in der Leberbiopsie indirekt: Ermittlung von Halbwertszeiten bestimmter Substanzen (z.B. Pharmaka, radioaktiv markierte Stoffe, z.B. Aminopyrin) Veränderungen im Ammoniumstoffwechsel : Die Leber hat herausragende Bedeutung im Ammoniumstoffwechsel. Zum einen werden über die Pfortader aus dem Intestinaltrakt große Ammoniummengen herangeführt, zum anderen ist die Leber der nahezu ausschließliche Ort der Harnstoffsynthese. Hyperammonämie (erhöhter Ammoniakspiegel im Blut) durch verminderte Harnstoffproduktion durch Leberzellinsuffizienz (Parenchymschädigung) oder Parenchymrarefizierung (Zirrhose), Umgehung der Leber durch portocavale Shunts (operativ bei ausgeprägten Ösophagusvarizen), metabolische Alkalose mit Hyperkaliämie mit Verschiebung des Dissoziationsgleichgewichts zugunsten freien Ammoniaks, ferner durch proteinkatabole Zustände 1.2.3 Einschränkung der Exkretionsfunktion (Cholestase) Bildung und Abfluss der Gallenflüssigkeit zur Elimination endogener (Bilirubin, Gallensäuren, Cholesterin, Hormonmetabolite) und exogener (manche Pharmaka, Kontrastmittel) Substanzen und zur Förderung der Fettverdauung. Cholestase = mehr oder minder ausgeprägte Reduktion des Galleflusses. Klinisch: als Ikterus, in schweren Fällen mit den Folgen der verminderten Absorption fettlöslicher Vitamine (A,D,E und K). -6- Klinisch-chemisch finden sich Konzentrationszunahme bestimmter Galleexkretionsprodukte im Serum und Urin (direktes Bilirubin) sowie charakteristische Enzymveränderungen. Cholestase kann mechanisch durch Obstruktion extrahepatischer Gallenwege (Steine, Tumoren, Strikturen) oder intrahepatischer Gallenwege (Tumoren, Zysten, Cholangitis, Gallenwegsatresie) oder funktionell durch Störungen des Exkretionsmechanismus (somit immer intrahepatisch und damit durch eine Erkrankung der Leber selbst) entstehen. Wichtigste Folgen der Cholestase sind: Retention von Bilirubin und Gallensäuren. Parameter: Bilirubin Bilirubin = lipophiles, toxisches Produkt des Abbaus von Hämoproteinen (Hämoglobin, Myoglobin) Es wird im Plasma an Albumin gebunden transportiert, von der Leber aufgenommen, mit Glucuronsäure verestert und mit der Galle ausgeschieden. Im Gastrointestinaltrakt dekonjugiert, z.T. im Rahmen des enterohepatischen Kreislaufs reabsorbiert, der überwiegende Teil wird jedoch zu Urobilinogen, das in geringem Umfang reabsorbiert und über den Urin ausgeschieden, zum größten Teil als Sterkobilinogen mit dem Stuhl ausgeschieden wird. Im Labor Bestimmung von Gesamt-Bilirubin und konjugiertem (= direktem) Bilirubin (indirektes Bilirubin = Differenz Gesamt-Bili minus Bili direkt). Störungen des Bilirubinstoffwechsels sind auf allen Stufen möglich und erschinen klinisch als Ikterus (zunächst der Skleren). Man unterscheidet: Prähepatischer Ikterus: bei übermäßigem Anfall von Bilirubin (Überschreiten der Konjugationskapazität) infolge vermehrten Erythrozytenabbaus (Hämolyse, Hämatomabbau, ineffektive Hämatopoese wie perniziöse Anämie, Thalassämie etc.) Intrahepatischer Ikterus: Störung der Zellfunktion: Störung der Bilirubinaufnahme in die Leberzelle (Gilbert-Syndrom), Störungen der Bilirubinkonjugation (angeboren, physiologischer postnataler Glucuronyltransferase-Mangel, Hämochromatose, Zirrhose, alkohol. Fettleber, akute Hepatitis) oder Störungen der Exkretion konjugierten Bilirubins (viele Leberzellschäden, Steroide, Kontrazeptiva, genetische Störungen) Posthepatischer Ikterus: bei Verschluß extrahepatischer Gallenwege durch Gallensteine, Tumoren, Verwachsungen -7Einteilung und Ursachen der Hyperbilirubinämien: Vorwiegend konjugiertes ("direktes") Bilirubin im Serum vermehrt bei Einschränkung der biliären Exkretion : verursacht durch Leberschäden (Hepatitis, Leberverfettung, Zirrhose Medikamente (Kontrazeptiva, Methyltestosteron, u.a.) Postoperative Cholestase (Operationskomplikation bei Baucheingriffen) Familiäre Cholestase (Erbanlage) Evtl. bei Schwangerschaft (im Rahmen einer Gestose) Obstruktion extrahepatischer Gallenwege Konkremente Tumoren Strikturen Atresie Gesteigerte Bilirubinproduktion Hämolytische Anämie Shunt-Hyperbilirubinämie bei ineffektiver Erythropoese (z.B. Perniziosa) Abbau extravasaler Blutmassen (Hämorrhagien) Vorwiegend unkonjugiertes ("indirektes") Bilirubin im Serum erhöht bei: Einschränkung der hepatozellulären Bilirubinaufnahme: z. B. durch - Gilbert-Meulengracht-Syndrom (Icterus juvenilis intermittens) (hierbei ist die Aktivität der UDP-Glucuronyltransferase, die in der Leber Bilirubin mit Glucuronsäure zum Bilirubindiglucuronid verknüpft, auf etwa 30% der Norm vermindert). - Ikterus neonatorum (gesteigerter Abbau von fetalem Hämoglobin bei Neugeborenen) - Medikamente (Chloramphenicol, Novobiocin) - Crigler-Najjar-Syndrom (Molekulardefekt der Glucuronyltransferase, erblich, sehr selten) Gesteigerte Bilirubinproduktion, z. B. bei - Hämolytischer Anämie - Hyperbilirubinämie bei ineffektiver Erythropoese (z.B. Perniziosa) - Abbau extravasaler Blutmassen (Hämatome) Parameter: Gallensäuren Gallensäuren werden fast ausschließlich in der Leber synthetisiert, über die Galle ausgeschieden, stehen im oberen Intestinaltrakt zur Fettverdauung zur Verfügung, werden im terminalen Ileum zu 95% reabsorbiert und über das Portalvenenblut der Leber wieder zugeführt. Beeinträchtigte Leberfunktion (nahezu alle akuten und chronischen Lebererkrankungen) führt zur erhöhten Gallensäurekonzentration im Serum und erhöhter Ausscheidung im Urin. Enzymaktivitäten im Serum bei Cholestase / Galleabflußstörungen Als sogenannte Cholestase-anzeigende Enzyme gelten die alkalische Phosphatase (AP), die Gamma-Glutamyltransferase (Gamma-GT), die Leucinarylamidase (LAP) und evtl. die 5'-Nucleotidase. -8Parameter: Alkalische Phosphatase (AP) AP-Anstiege im Serum sind nicht zwingend Cholestase-spezifisch, da gewebsspezifische Isoenzyme auch in Knochen, Darm, Niere, Lunge und Plazenta vorhanden sind. Die Gesamtaktivität ist überwiegend von Leber- und Knochenisoenzymen getragen. Im letzten Trimenon der Schwangerschaft steigt das plazentare Isoenzym - daher deutlich erhöhte Gesamtaktivitäten bei Schwangeren. Bei Cholestase kommt es zu einer starken Konzentrationserhöhung des membranständigen Anteils des hepatischen Isoenzyms der AP durch 1.) Syntheseinduktion 2.) partielle Membranzerstörung durch intrazelluläre Gallensäuren. Diese Mechanismen sind unabhängig von der Ursache der Cholestase, so dass aus einem AP-Anstieg keine Schlussfolgerung über die Ursache der Cholestase gezogen werden kann. Erfahrungsgemäß ist der AP-Anstieg bei extrahepatischer Obstruktion jedoch höher. Differentialdiagnose zwischen hepatischer und ossärer Herkunft eines AP-Anstieges ist durch Bestimmung des Isoenzyms Knochen-AP möglich. Erhöhte Aktivitäten der AP findet man bei Lebererkrankungen mit extrahepatische Cholestase hepatozellulärer Schädigung Cholangitis, Cholangiolitis Lebertumoren Osteopathien mit erhöhter Osteoblastenaktivität Hyperparathyreoidismus Osteomalazie Morbus Paget Knochenmetastasen Multoples Myelom bei Frakturheilung bei nephrogener Rachitis im 3. Trimenon der Schwangerschaft Erniedrigte Werte der AP im Serum findet man bei: Erblicher AP-Defizienz (Mangel oder Molekulardefekt) Hypothyreose Achondroplasie Schwerer Anämie Parameter: Gamma-Glutamyltransferase (Gamma-GT) Nahezu ubiquitäres Vorkommen. Vergleichsweise geringe Aktivität in der Leber (Niere 25x mehr, Pankreas doppelt so viel), jedoch wegen Membranfixierung in diesen Organen ist der Zustand der Leber bestimmend für Serumspiegel. -9Aktivitätserhöhung der Gamma-GT = sehr sensitiver Indikator für eine hepatobiliäre Erkrankung. Anstiege finden sich jedoch auch bei 80% der mit Antikonvulsiva und Sedativa behandelten Patienten. Bei 70% der Alkoholiker ohne manifeste Lebererkrankungen finden sich durch Abstinenz reversible Gamma-GT-Anstiege. Diese kommen durch Enzyminduktion (Mehrsynthese von Enzymprotein) zustande. Erkrankungen mit Veränderung der Gamma-GT im Serum: Normalaktivitäten finden sich (evtl trotz vorliegender Störung) bei Schwangerschaft, Knochenwachstum, Knochenerkrankungen, Muskelerkrankungen, Nierenerkrankungen, chronisch persistierender inaktiver Hepatitis Mäßig erhöhte Werte bei Unkomplizierte Virushepatitis, alkoholischer Fettleber, mäßigem Alkoholismus Leberstauung bei Rechtsherzinsuffizienz Deutliche erhöhte Werte bei Chronisch aktiver Hepatitis, allkoholtoxischer Hepatitis, alkoholtoxischer Zirrhose primär biliärer Zirrhose, primären und sekundären Lebertumoren, akuter und chronischer Pankreatitis, Langzeittherapie mit Antikonvulsiva u. Sedativa, Starke Erhöhungen finden sich bei: Verschlußikterus, cholestatischen Formen der akuten Hepatitis, toxischen Leberschäden Parameter Leucinarylamidase (LAP) wird relativ selten bestimmt, hat allerdings höhere Leberspezifität. Wird genutzt zur Differentialdiagnose unklarer, solitärer AP-Erhöhungen. AP hoch, LAP normal -> ossäre Ursache. Die 5'-Nucleotidase ist der LAP in der Wertigkeit vergleichbar. 1.2.4 Zunahme des intrahepatischen Bindegewebes (Fibroplasie) Fibrose, d.h die Konzentrationszunahme und Umverteilung der interzellulären Bindegewebsproteine, hat große Bedeutung bei der Entstehung einer chronischen Schädigungen der Leber bis hin zur Zirrhose mit möglicher Konsequenz des Pfortaderhochdruckes und der Ausbildung von Ösophagusvarizen. Klinisch-chemische Parameter zur Erfassung der fibrotischen Transformation sind wenig gebräuchlich, da es ihnen an Organ- und Krankheitsspezifität fehlt. Man wählt für die Diagnostik vorzugweise die Leberbiopsie mit anschließender histologischer Untersuchung. -102. Spezielle klinisch-chemische Diagnostik wichtiger Lebererkrankungen 2.0 Ich will wissen: Liegt eine Lebererkrankung vor? Ich bestimme: ALAT oder Gamma-GT: Sensitivität von 80% ALAT + Gamma-GT + Cholinesterase: Sensitivität von 95% ALAT + Gamma-GT + AP + Cholinesterase: Sensitivität von 97% Quotient ASAT/ALAT: < 1 Entzündungstyp (z.B. Hepatitis) > 1 Nekrosetyp (z.B. Alkohol) 2.1 Akute Virushepatitis 2.1 Ich will wissen: Liegt eine Hepatitis vor? Vorab: Das Hepatitis-Virus an sich ist nicht zytopathogen, sondern ruft erst in Kombination mit der zellulären Immunantwort eine Hepatitis hervor. Obligat zu untersuchen: Leberzellschaden- und Cholestaseparameter (Transaminasen, AP, Gamma-GT, Bilirubin) Fakultativ zu untersuchen: Syntheseparameter (ChE) Eine enzymchemische Differenzierung der Hepatitistypen ist nicht möglich. Hier hilft nur die Serologie weiter. Als Suchtests sind zu empfehlen: HAV-IgG und HAV-IgM (für Hepatitis A) => Hepatitis-A-Diagnostik HBs-AG, Anti-HBs und Anti-HBc (für Hepatitis-B) => Hepatitis-B-Diagnostik HCV-Antikörper (für Hepatitis C) => Hepatitis-C-Diagnostik Hepatitis-A-Diagnostik: Norm: Test Norm HAV-IgG negativ nach Impfung: positiv HAV-IgM negativ HAV-Antigen negativ -11Bewertung: Test Material Anti-HAV-IgG-ELISA Serum Anti-HAV-IgM-ELISA Serum HAV-Antigen-ELISA Stuhl Bewertung Antikörper von Beginn der Krankheitssymptome an nachweisbar Antikörper persistiert meist lebenslang (Parameter für Durchseuchungsrate) nach Impfung positiv, bei Nachweis von IgG-Antikörpern ist Immunität anzunehmen. Marker für Erkrankung (frisch/zurückliegend) oder Impfung beweisend für eine frische Infektion Antikörper von Beginn der Krankheitssymptome an 3-6 Monate lang nachweisbar nur zur Abklärung einer frischen Infektion in der Inkubationsphase sinnvoll 1-3 Wochen vor bis 3-6 Wochen nach Erkrankungsbeginn nachweisbar Hepatitis- B-Diagnostik Norm: Test Norm HBcAntikörper HBc-IgMAntikörper Antikörpernachweis HBeAntikörper Antigennachweis negativ negativ negativ HBsAntikörper ungeimpft: 0-10 U/l nach Impfung: >10 U/l HBsAntigen HBeAntigen negativ HBV-PCR negativ negativ Bewertung: Test HBs-AntigenELISA* Material Serum Bewertung Nachweis von Virus-Hüllprotein (Surface = Oberfläche) Parameter für Infektiosität: PCR positiv Æ hohe Infektiosität PCR negativ Æ geringe Infektiosität Bei Persistenz >6 Monate liegt per definitionem eine chronische Infektion vor. 5-10 % der HBV-Infektionen sind HBs-Antigen negativ! zusammen mit Anti-HBc Parameter für ausgeheilte Infektion ohne vorhandenes Anti-HBc meist Parameter für Impfschutz: Anti-HBsELISA Anti-HBcELISA Anti-HBc-IgMELISA Serum kein ausreichender Impfschutz, Auffrischung empfohlen >100 U/l ausreichender Impfschutz ≤100 U/l Serum Parameter für eine Infektion (frisch/chronisch/ausgeheilt) Nachweis 1-≥ Wochen später als HBs-Antigen langjährige (bis lebenslange) Persistenz nach Impfung nicht positiv Serum Parameter für eine akute Infektion Nachweis häufig schon vor Auftreten des HBs-Ag Persistenz bis zu 12 Monaten PCR EDTA(PolymeraseBlut kettenreaktion) Die DNA des Virus wird vermehrt und nachgewiesen (höchste Empfindlichkeit schon bei geringster Virämie!). Marker für die Infektiosität des Patienten. Hepatitis-C-Diagnostik Norm: Test Antikörpernachweise Antigennachweise Norm HCV-Antikörper negativ HCV-Immunoblot keine Banden nachweisbar HCV-PCR negativ Bewertung: Test HCV-ELISA Material Serum Bewertung sensibler Suchtest zur Erkennung von Antikörpern gegen das Hepatitis C-Virus Antikörperbildung setzt frühestens nach 4-6 Wochen (meist 2-6 Monaten!) ein. Aufgrund möglicher falsch positiver Befunde sollte ein positives Ergebnis mit einer spezifischen Methode (z.B. Immunoblot, siehe unten) kontrolliert werden! Ursachen für falsch-positive Befunde: Paraproteinämie Autoantikörper EBV-Infektionen andere Ursachen für erhöhte IgG-Konzentrationen Der Test kann nicht zwischen einer akuten, chronischen (infektiösen) oder ausgeheilten (nicht mehr infektiösen) Erkrankung unterscheiden! HCVImmunoblot Serum HCV-PCR EDTA(PolymeraseBlut kettenreaktion) spezifischer Bestätigungstest zur Abklärung eines positiven ELISABefundes (siehe oben) Durch die Interpretation des Bandenmusters (Nachweis spezifischer Antikörper) ist eine Differentialdiagnose zwischen Erkrankung und unspezifischer Reaktion möglich: Bei Nachweis einer spezifischen Bande ist das Ergebnis fraglich (Kontrolle empfohlen) Bei Nachweis von mehr als einer spezifischen Bande ist das Ergebnis positiv! Der Test kann nicht zwischen einer akuten, chronischen (infektiösen) oder ausgeheilten (nicht mehr infektiösen) Erkrankung unterscheiden! Indikationen: Antikörper-positive Patienten (ELISA und/oder Blot) DD: akute oder ausgeheilte Hepatitis? frische Infektion vor dem Auftreten von Antikörpern (ELISA und/oder Blot) Diagnose/Verlauf einer chronischer Infektion mit fehlendem Antikörpernachweis (ELISA und/oder Blot) unklarer serologischer Befund des ELISA und/oder Blots Bei der PCR (Polymerasekettenreaktion, Virusdirektnachweis) wird das Erbgut des Virus (RNA) vermehrt und danach nachgewiesen. Derzeit die einzige verfügbare Methode um eine Aktivität und Infektiosität einer Hepatitis C-Erkrankung nachzuweisen. Bei typischen akuten Hepatitis- Verläufen rasche Anstiege von ASAT und ALAT, mit proportional höherem ALAT-Anstieg (im Gegensatz zu Alkoholtoxikose) und nur mäßiger GLDH-Erhöhung bei leichter Zellschädigung und relativ stärkeren Anstiegen von ASAT und GLDH bei nekrotisierenden Verlaufsformen. Innerhalb weniger Tage Maximum des Bilirubinanstieges (fehlt allerdings bei anikterischen Formen). Der Anteil des konjugierten Bilirubins beträgt etwa 60-80%. Frühzeitig Urobilinogenurie. Die Parameter der Syntheseleistung ändern sich bei normal verlaufender Virushepatitis kaum, bei fulminanten Formen sind rascher Abfall von Cholinesterase und Gerinnungsparametern prognostisch ungünstig. 2.2 Ich will wissen: Liegt eine chronische Hepatitis vor? Im Vergleich zur Akutform, bei der Leberzellnekrosen im Vordergrund stehen, dominieren bei der chronischen Hepatitis entzündlich-mesenchymale Reaktionen. Nach Histologie und klinischem Verlauf kann man eine chronisch-persistierende (milde, nicht progredient) und eine chronisch-aktive Form (mit Zeichen der fortschreitenden Parenchymdestruktion) unterscheiden. Hier brauche ich also besonders eine Histologie und nicht nur die Klinische Chemie. Ich untersuche wie unter 2.2.1 und 2.2.2 im folgenden ausgeführt: 2.2.1 Chronisch persistierende Hepatitis Obligat zu untersuchen: Leberzellschadenparameter (meist nur geringe Aktivitätserhöhungen) Hepatitis-Serologie, IgM-Anstieg (Elektrophorese) Fakultativ zu untersuchen: Cholestaseparameter, Histologie entscheidend. -142.2.2 Chronisch aggressive Hepatitis Obligat: Leberzellschadenparameter, Cholestaseparameter, Hepatitis-Serologie, IgG stark angestiegen (Elektrophorese) Fakultativ: Syntheseparameter Histologie entscheidend. 2.3 Ich will wissen: Liegt eine medikamentös-toxische Leberschädigung vor? Durch ihre Aufgabe und Funktion wird die Leber zum Stoffwechselort einer Vielzahl exogener und endogener Toxine. Man unterscheidet dabei obligate Hepatotoxine (CCl4, Benzole, Aflatoxine) sowie fakultative Hepatotoxine (viele Medikamente, Alkohol). Häufig findet die "Giftung" einer Substanz auch erst in der Leber statt (z.B. CCl4 zum CCl3Radikal). Im Spektrum toxininduzierter Lebererkrankungen finden sich diffuse Leberzellnekrosen (Nekrosetyp), entzündliche Veränderungen (Hepatitistyp), Galleabflußstörungen (Cholestasetyp) sowie intrazelluläre Fettakkumulation (Steatosetyp). Die erhebliche Bandbreite klinisch-chemisch nachweisbarer Veränderungen richtet sich nach dem Schädigungstyp und kann von extremen Veränderungen bis zum völligen Fehlen von Auffälligkeiten reichen. 2.4 Ich will wissen: Liegt eine alkoholtoxische Leberschädigung vor? Die Leber nimmt eine dominierende Stellung unter den vom Alkoholabusus geschädigten Organen ein, jedoch führt chronischer Alkoholkonsum auch zu Schädigungen anderer, nicht am Ethanolstoffwechsel beteiligter Organe (ZNS, Herz, Gastrointestinaltrakt). Ethanol wird im oberen Dünndarm rasch absorbiert und in der Leber zu 90% metabolisiert, zunächst zu Acetaldehyd und dann weiter zum Acetyl-CoA, welches in den Citratcyclus oder Fettsäureaufbau bzw. in die Cholesterinsynthese eingeschleust wird. Der Hauptabbau erfolgt mittels der Alkoholdehydrogenase (ADH). Daneben erfolgt eine Oxidation im sog. Microsomal Ethanol Oxidizing System (MEOS), insbesondere bei Kampftrinkern. Der Abbau des toxischen Azetaldehyds erfolgt über die Aldehyddehydrogenase. Chronischer Alkoholabusus führt zur Induktion des MEOS-Enzymsystems, welches neben Alkohol auch Medikamente und Schadstoffe metabolisiert und teilweise aktiviert, was Interferenzen zwischen Alkohol und Medikamentenwirkung als auch die üble Beziehung zwischen Alkoholabusus und Tumorgenese (beschleunigte Prokanzerogenaktivierung) erklärt. (MEOS = microsomal ethanol oxidising system; im endoplasmatischen Reticulum). Ich untersuche: Gamma-GT, ALAT, ASAT, MCV, Ethylglucuronid im Urin und in Haarproben (!) sowie evtl. das CDT (carbohydrate deficient transferrin), Fettsäureethylester (in Haarproben). (Haarprobenuntersuchungen dienen dem Nachweis des Langzeitkonsums) Formen der alkoholischen Leberschädigung: Alkoholabusus löst ein breites Spektrum morphologischer, klinischer und klinisch-chemischer Veränderungen aus, drei typische Formen werden aufgeführt. -152.4.1 Fettleber Die Fettakkumulation (meist Triglyceride) ist eine frühe, noch reversible Form der alkoholischen Leberschädigung. Die klinische Symptomatik ist blande, und so auch die klinisch-chemischen Veränderungen. Typisch: mit fortschreitender Verfettung kommt es zur Freisetzung von GLDH, sowie einem Absinken des (ASAT+ALAT)/GLDH-Verhältnisses. Die Aktivitätserhöhung der Gamma-GT als Ausdruck einer alkoholinduzierten Synthesesteigerung und Membranfreisetzung ist oft der früheste Marker. 2.4.2 Alkoholische Hepatitis Oft ausgelöst durch einen akuten Exzess, kann es beim chronischen Alkoholabusus zu einer akut nekrotisierenden, entzündlich infiltrativen Leberschädigung kommen, die der akuten Virushepatitis ähnelt. Ein Anstieg der Parameter der Zelläsion und der Cholestase geht einher mit einer Abnahme der Syntheseparameter. 2.4.3 Ich will wissen: Liegt eine Leberzirrhose vor? Die Zirrhose ist die schwerste und irreversible Form der alkoholischen Leberschädigung. Ihre Entwicklung ist abhängig von den konsumierten Alkoholmengen. Jedoch nur 40% der schweren Alkoholiker erreichen dieses Stadium, so daß von genetischen und umweltbedingten Faktoren ausgegangen werden muß. Die Umbauprozesse der Leber führen zur zunehmenden Syntheseinsuffizienz (Serumproteine, Gerinnung), zur funktionellen Cholestase mit eingeschränkter Vitaminabsorption, verminderten Biotransformationsleistungen und steigenden Metabolit- und Ammoniakkonzentrationen. Zusammen mit der fibrotisch bedingten portalen Hypertension und der metabolischen Insuffizienz der Leber kommt es zu den klassischen Symptomen der dekompensierten Leberzirrhose. Ich untersuche: Cholinesterase, Albumin und den Quick-Wert / INR-Wert. (erniedrigte Werte) Die Leberenzyme ASAT, ALAT sowie γ-GT, Bilirubin und Ammoniak können erhöht sein.Die definitive Diagnose wird durch eine Leber-Biopsie gestellt. Eine Elektrophorese ist hilfreich. Bei einer Leberzirrhose kommt es durch die verminderte Biosynthese von Albumin zu dessen Abfall im Blutplasma. Die gleichzeitig verminderte Synthese der α1 und α2-Globuline macht sich weniger stark bemerkbar, weil auch ihr Abbau durch die Leber gestört ist. Eine Erhöhung – und zwar relativ wie absolut – findet man bei den β- und γ-Globulinen. Die Ursache dafür wird darin gesehen, dass sich durch die Zirrhose das Blut in der Pfortader zurück in die Milz staut. Dort erfolgt dann wegen der (ebenfalls staubedingten) erhöhten Bakterienzahl in der Vena splenica eine vermehrte Produktion von Antikörpern in den ansässigen Plasmazellen. Der Anstieg der β-Globuline ist dabei durch die in dieser Fraktion laufenden Immunglobuline A und M zu erklären. -162.5. Hepatozelluläres Karzinom Unter den klinisch-chemischen Kenngrößen ist hier das Alpha-Fetoprotein (AFP) zu nennen. Erst im fortgeschrittenen Stadium der Erkrankung finden sich die bereits genannten Veränderungen der Leberzellschädigung und Cholestase. 2.6 Hepatische Enzephalopathie Die Pathogenese des Coma hepaticum stützt sich auf die experimentell gut untermauerte Annahme, dass wegen einer verminderten Entgiftungsfunktion der Leber toxische Substanzen in den Liquorraum übertreten können und Wirkungen auf das ZNS ausüben können. Genannt seien die Faktoren Hyperammonämie (Glutaminbildung im Gehirn blockiert Neurotransmitter Glutamat, Hemmung des zerebralen Energiestoffwechsels und Ammoniakwirkung auf neuronale Membranen), kurzkettige Fettsäuren, Mercaptane, Phenole und ihre Derivate, die allesamt neurotoxisch wirken. Na denn Prost. Die Bestimmung von Ammoniak im venösen bzw. arteriellen Blut stellt die hauptsächliche diagnostische Methode zur Einschätzung einer hepatischen Enzephalopathie dar. 2.7 Cholezystolithiasis / Cholelithiasis Unkomplizierte Gallensteinleiden verursachen kaum klinische Symptome und auch keine Veränderungen klinisch-chemischer Parameter. Erst Komplikationen wie Verschluß von Gallenwegen oder Entzündung führen in Abhängigkeit von Schweregrad und Dauer des Prozesses zur Cholestase und ggf. zur Zellnekrose. Die Zusammensetzung von Gallensteinen (Cholesterin, Gallepigment, Kalksalze) kann infrarotspektrometrisch ermittelt werden. Kontrollfragen zu diesem Artikel: (1) Welcher Parameter weist am ehesten auf einen alkoholtoxischen Leberschaden hin? (2) Sie vermuten bei einem Patienten einen Leberschaden. Welche Parameter würden Sie zunächst untersuchen? Berücksichtigen Sie dabei die Grundsätze der Wirtschaftlichkeit und des sinnvollen Handelns. (3) Warum hat ein Patient, der z. B. das Antieptileptikum Phenytoin oder Carbamazepin regelmäßig einnimmt, eine erhöhte Gamma-GT-Aktivität im Serum? (4) Warum brauchen Alkoholiker im Rahmen von Operationen oft vermehrt Narkosemittel? (5) Sie messen bei einem Patienten eine hohe Gamma-GT-Aktivität. Er versichert glaubhaft, Antialkoholiker zu sein. Lügt er? Wie klärt man das Problem? (6) Sie finden folgende Befundkonstellation: GPT 78 GOT 64 Bili direkt 10,4 Bili gesamt 16,0, Stuhl hell, Urin dunkel. Was ist die wahrscheinliche Ursache? Warum finden Sie im Urin kein Urobilinogen? -17(7) Welche physiologische Aufgabe hat die Gallenflüssigkeit? (8) Unter welchen Umständen führt ein Gallensteinleiden zu klinisch-chemischen Auffälligkeiten? (9) Warum gibt es kurz nach Verzehr eines Knollenblätterpilzsüppchens mit Sahnehaube und frischen Gartenkräutern Blutungskomplikationen? (10) Welche Vitamine sind fettlöslich? (11) Welche Gerinnungsfaktoren werden in der Leber gebildet? (12) Welche elektrophoretischen veränderungen findet man bei einer Leberzirrhose? (13) Wann findet man bei einem Gallensteinleiden erhöhte Leberwerte? (13) Was ist die Meulengrachtsche Erkrankung? Ist sie schlimm? (14) Durch welche Substanzen wird ein hepatisches Koma bewirkt? (15) Wie heißt der Tumormarker für das hepatozelluläre Carcinom? (16) Welche Suchtests führe ich durch bei Verdacht auf Hepatitis A, B, C ? (17) Welches sind die typischen Befunde bei prä-, intra- und posthepatischem Ikterus?