1.0 Nucleophile Addition an das ungesättigte Carbonyl
Werbung

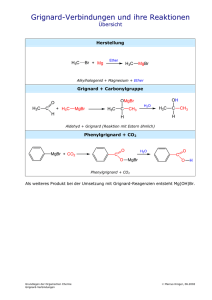
Skript zum 5. OC-Kolloquium Seite 1 von 39 1.0 Nucleophile Addition an das ungesättigte Carbonyl-CAtom: 1.1 Allgemeines In einer Carbonylgruppe sind wie in der olefinischen C=C_Doppelbindung zwei Atome, hier jedoch ein Kohlennstoffatom und ein Sauerstoffatom, über eine σ- und eine π-Bindung aneinander gebunden. Aufgrund des großen Elektronegativitätsunterschieds zwischen Kohlenstoff und Sauerstoff ist die Carbonylgruppe jedoch extrem polarisiert, was durch folgende Grenzformeln beschrieben werden kann. R2 δ+ C R2 δ− O (-) O (+) C R1 R1 π-Bindung O σ-Bindung C Im Prinzip handelt es sich bei den Reaktionen der Carbonylgruppe um ähnliche Reaktionen wie bei Additionen an C=C-Doppelbindungen. Eine riesige Zahl synthetisch wertvoller Reaktionen von und mit Carbonylverbindungen sowie bei C=CDoppelbindungen nicht zu beobachtende Folgereaktionen der Additionsprodukte rechtfertigt aber, die Reaktionen der Carbonylgruppe separat zu behandeln. Wegen ihrer Polarität reagiert eine Carbonylgruppe mit Elektrophilen und Nucleophilen an verschiedenen Enden. Dabei kommt dem Angriff eines Nucleophils an das positiv polarisierte Carbonyl-C-Atom die größere Bedeutung zu. Elektrophile dagegen suchen das elektronenreiche Ende, greifen also das Carbonyl-O-Atom an. Wichtig ist, dass sie dadurch einen Angriff eines Nucleophils erleichtern können, denn durch ihren Angriff erhöhen sie die Elektrophilie des Carbonyl-C-Atoms. Dies gilt für Protonen sowie für Lewis-Säuren. E E + O O O E(+) + C Angriff eines Nucleophils wird erleichtert O H + O H O + C Nu (-) Nu (-) O + (-) BF3 BF3 O + C Nu (-) Die Vielzahl der Reaktionen von Carbonylverbindungen lässt sich in zweifacher Weise unterteilen: nach der Art der Nucleophile und nach der Art der Skript zum 5. OC-Kolloquium Seite 2 von 39 Carbonalverbindungen. Je nach Nucleophil können C-Hetereoatom- C-C- oder C-HBindungen gebildet werden. Die Carbonylverbindungen kann man unterteilen nach der Anzahl der Bindungen zu Hetereoatomen: Aldehyde und Ketone (2-Bindungen), Carbonsäure-Derivate (3 Bindungen) und Kohlensäure-Derivate (4 Bindungen). Gegenüber verschiedenen Nucleophilen verhalten sich Carbonylverbindungen und Carbonsäure-Derivate recht unterschiedlich. Grund hierfür ist die unterschiedliche Elektrophilie der Carbonyl-C-Atome, die durch die beiden Substituenten R1 und R2 stark beeinflusst wird. δ− O C δ+ R1 R2 Elektronenschiebende Gruppen wie +M- und +I-Substituenten erhöhen die Elektronendichte am C-Atom und erniedrigen daher die Reaktivität der Csarbonylgruppe, während elektronenziehende Gruppen –M- und –I-Substituenten die elektronendichte erniedrigen und zu schnelleren Reaktionen führen. Die unreaktivsten Carbonsäure-Derivate sind daher die Carbonsäure selbst, Amide sowie Carbonsäure-Anionen, während Säurehalogenide und –anhydride zu den reaktivsten Carbonsäure-Derivaten gehören. O Cl R OH R O NR'2 R R' R O O OR' O H R R' O R O R O O O O R Bezüglich ihrer Reaktivität gegenüber Nucleophilen liegen Aldehyde und Ketone in der Mitte, aber auch hier lässt sich aufgrund der mesomeren und induktiven, aber auch sterischen Effekten eine abgestufte Reaktivität beobachten. O O O O O Cl3C H H H Me H But- O Me Ph Ketonen und Carbonsäurederivaten H O Ph Ph Me Me Skript zum 5. OC-Kolloquium stereochemische Aspekte der Carbonyladdition Eigenschaften der Carbonylgruppe: Acetale Ketale Thioacetale Thioketale Seite 3 von 39 Skript zum 5. OC-Kolloquium Hydratisierung Paraformaldehyd Methaldehyd Seite 4 von 39 Skript zum 5. OC-Kolloquium Seite 5 von 39 Addition von HCN Durch Addition von Blausäure an Carbonylverbindungen erhält man Cyanhydrine. R1 OH R1 O R1 C O + langsam CN- + H(+) C R2 CN R2 CN R2 C Cyanhydrin R1 = R2 = Alkyl R1 = H, R2 = Alkyl, Aryl Das stark basische Cyanidion lagert sich im ersten Schritt langsam nucleophil an den Carbonylkohlenstoff an. Das primäre Anlagerungsprodukt wird dann in einer Gleichgewichtsreaktion durch Protonen unter Ausbildung des Cyanhydrins abgefangen. Die praktische Durchführung der Cyanhydrinreaktion erfolgt so, dass man zu einem aus Carbonylverbindung und wässrige NaCN-Lösung bestehenden Gemisch langsam wässrige verdünnte Mineralsäure gibt. Zu stark saure Lösungen müssen vermieden werden, da dadurch das Gleichgewicht der Blausäure auf die Seite der undissozierten Säure verschoben wird und somit nur wenig CN--Ionen zur Verfügung stehen. Zu stark basisch ist ebenfalls ungünstig, weil das CN- dann nicht mehr als freies Ion vorliegt. Cyanhydrine von Aldehyden sind stabiler als die von Ketonen und somit auch besser darstellbar. Dafür sind vor allem sterische und elektronische Faktoren verantwortlich. Alternativ besteht auch die Möglichkeit der Herstellung von Cyanhydrinen aus den Hydrogensulfit-Addukten durch Austauschreaktion: C O + HSO3(-)Na(+) SO3 (-) Na(+) C + NaCN C CN + Na2SO3 OH OH Hierbei wirkt das gebildete NaHSO3 als Säure. Die synthetische Bedeutung der Cyanhydrin-Reaktion liegt darin, dass man durch saures Verseifen zu αHydroxycarbonsäuren gelangen kann: + C OH R1 OH R1 2 H2O + H(+) C + NH4(+) COOH R2 CN R2 Ein Beispiel für die technische Anwendung der Cyanhydrin-Reaktion ist die Herstellung von Methacrylsäure (Plexiglas). Strecker-Synthese Diese von Strecker bereits 1850 gefundene Reaktion ist die amonoanaloge Cyanhydrin-Reaktion; denn die zur C=O-Gruppe isoeletronische Gruppe im Ammonosystem ist die Imin-Gruppe C=NH, die ebenfalls nucleophil von CNangegriffen werden kann und zu Aminonitrilen führt: H3C C H O NH3 - H2O H3C C H NH HCN H3C H C NH2 CN 2 H2O/ H(+) H3C H C NH2 COOH Skript zum 5. OC-Kolloquium Seite 6 von 39 Die Aminosäuren liegen am isoelektrischen Punkt in einer Zwitterion-Form vor, der so genannten Betainform: COOH H COOH NH2 H2N H2N H CH3 CH3 "D" "L" H COOH H HOOC NH2 CH3 CH3 "R" "S" + H3N D,L-Nomenklatur (Fischer-Konvention) R, S- Nomenklatur nach Cahn, Ingold, Prelog O H C O CH3 Diese Zwitter-Ionen sind im Allgemeinen kristalline, hochschmelzende und gut wasserlösliche Verbindungen. Addition von Ammoniak und Aminen (Imine, Schiff’sche Basen, Aminale) Allgemeines: Grundsätzlich reagieren Basen mit Carbonylverbindungen nach folgendem Schema: konjugierte Base HB + C Protonenübertragung + HB O O C B C OH N C OH (im Sinne einer SäureBase-Reaktion) konjugierte Säure R1 R1 NH R2 + C O H N + R2 R1 C O R2 Halbaminal Skript zum 5. OC-Kolloquium Seite 7 von 39 Die Halbaminale sind ebenso wie die Halbacetale sehr unbeständig und nicht isolierbar. Je nachdem, ob es sich um primäre Amine (R1 = H, R2 = Alkyl, Aryl) oder um sekundäre Amine (R1 = R2 = Alkyl, Aryl) handelt, ergeben sich versiedne Möglichkeiten zur Weiterreaktion: Die Addition primärer Amine an Carbonylverbindungen Die gebildeten Halbaminale reagieren nach folgendem Mechanismus weiter: R1 N H C OH H(+) R1 N H C OH2 - H2 O + R1 C+ N H R1 N C H Carbimmoniumion - H(+) R1 N C Schiff'sche Base Das durch Protonierung und Abspaltung von Wasser gebildete Carbimonium-Ion (= Carbeniumimmonium-Ion) spaltet als konjugierte Säure einer N-Base leicht ein Proton ab, und man erhält für R1 = H sogenannte Imine, für R1 = Alkyl, Aryl sogenannte Azomethine, die auch Schiff’sche Basen genannt werden. Die gut kristallisierenden Produkte aus den Kondensationsreaktionen werden zur Reinigung und in der Analytik zur Identifizierung von Carbonylverbindungen verwendet. primäres Amin Derivat H2N OH (Hydroxylamin) NOH C H2N NH2 (Oxim) (Hydrazin) C N NH2 (Hydrazon) H2N N H C6H5 (Phenylhydrazin) C N N H C6H5 (Phenylhydrazon) H2N N H NO2 O 2N C C NH2 NO2 O2N (2,4- Dinitrophenylhydrazon) O O N H N H (2,4- Dinitrophenylhydrazin) H2N N C (Semicarbazid) N N H (Semicarbazon) C NH2 Skript zum 5. OC-Kolloquium Seite 8 von 39 Addition sekundärer Amine an Carbonylverbindungen Werden sekundäre Amine an Carbonylverbindungen addiert, so gibt es verschiedene Möglichkeiten zur Weiterreaktion. a) Enaminbildung: R1 N R2 + OH2 C C R1 R1 - H2O N R2 H + N C+ C C C R2 H H Carbimonium-Ion - H(+) R1 R1 N C R2 (+) N C C (-) R2 C Enamin (mesomeriestabilisiert) Hier wäre die Abspaltung eines R+ energetisch zu ungünstig; daher stabilisiert sich das Carbeniumion durch Abspaltung eines H+ vom β-C-Atom unter Bildung von Enaminen. Die Enaminbildung findet im allgemeinen nicht mit primären Aminen statt. Ausnahme findet man jedoch, wenn die Enamin-Doppelbindung durch Konjugation weiter stabilisiert werden kann. O H3C H3C O C C H2 C +NH3 C OR NH2 -H2O H2N C + C HOOC CH3 C H HOOC H "E" "Z" β-Aminocrotonsäureester Zu den meisten genutzten sekundären Aminen für die Enaminbildung gehören Pyrolidin, Morpholin, Piperidin. Beispiel: O O O N (+) N O + H N H (-) Skript zum 5. OC-Kolloquium Seite 9 von 39 Aufgrund der Elektronenverteilung im Enamin ist eine elektrophile Substitution in αStellung leicht möglich. Anschließende Spaltung des Enamin ins Ausgangsketon liefert das auf anderem Weg kaum zugängliche α-substituierte Keton. b) Vollaminalbildung Ist am β-C-Atom kein abspaltbarer Wasserstoff vorhanden, so ist keine Stabilisierung durch Enaminbildung möglich. In diesem Falle findet eine Vollaminalbildung statt: R1 R1 (+) N C (+) N R2 R2 C C N R2 (+) R1 NH C R2 C R1 N R2 R1 NHR2 C R1 -H(+) R1 C N Vollaminal R2 C Beispiel: + H2C O +H(+) -H2O N N H Piperidin C H2 N Aminal des Formaldehyds Formaldehyd Ein Vollaminal mit Adamantanstruktur liegt im Urotropin (Hexamethylentetramin) vor: N 6 CH2O + 4 NH3 -6H 2O N N N Enamine (Reaktionen der Enamine) Oxime Mehrere Aminderivate kondensieren mit Aldehyden und Ketonen zu Iminen, die gut kristallisieren und oft scharfe Schmelzpunkte haben. Zum Beispiel kondensieren Hydroxylamin, H2NOH, in Form seines Hydrochlorids zu Oximen. Skript zum 5. OC-Kolloquium Seite 10 von 39 O CH3(CH2)5 CH CH3(CH2)5 H(+) -H2O + H3(+)NOHCl(-) NOH C H Heptanal-Oxim HydroxylaminHydrochlorid Hydrazone Hydrazin, H2NNH2, und einige seiner Derivate kondensieren zu Hydrazonen. Erfolgt die Reaktion an beiden Enden des Hydrazinmoleküls, werden Azine gebildet. NH2 N O H3C C CH3 + H2N NH2 C -H2O H3C Hydrazin CH3 Propanon (Aceton)-Hydrazon C6H5 H C O C6H5 NH CH + H2N NH2 N -H2O C H5C6 H Mannich-Reaktion Die Umsetzung eines primären oder sekundären Amins mit einem Aldehyd, i.a. Formaldehyd, und einer C,H-aciden Verbindung bezeichnet man als MannichKondensation. Hierbei findet wie allgemein bei Aminen zunächst eine Addition des Amins an die Carbonylverbindung mit nachfolgender Ausbildung des mesomeriestabilisierten Carbimmonium-Ions statt: R1 NH + R2 R3 R1 O R4 R3 + NH R2 Protonenübertragung O R4 R3, R4 meist H R3 R1 OH N R2 R4 + H +/ -H 2O R3 R1 N R2 + R3 R1 + C N R4 R2 R4 Carbimmoniumion Die Carbimmonium-Ionen stellen gleichfalls auch ammonoanaloge Carbonylverbindungen dar, die sich in einer normalen sauer katalysierten Skript zum 5. OC-Kolloquium Seite 11 von 39 Aldolreaktion mit dem Enol einer C,H-aciden Verbindung zu Mannichbasen umsetzen lassen: R3 R1 N C R1 + R2 N R4 OH R3 + + R2 für R3, R4 = H CH2 R R4 Carbimmoniumion OH R1 N C H2 R2 C H2 C (+) R N R2 C H2 C H2 C UmprotonierungR1 R R2 N C H2 C H2 C (+) NH C H2 O C H2 C R Salz der Mannichbase O R1 -H+ (+)OH R1 R R2 Mannich-Base = β-Aminoketon Cannizarro-Reaktion Bei der Cannizzaro-Reaktion handelt es sich um eine Disproportionierung von aromatischen und nicht enolisierbaren aliphatischen Aldehyden unter der Einwirkung von Alkali- oder Erdalkalihydroxiden zu Carbonsäuren und Alkoholen durch HydridTransfer: R H + R + OH- O langsam + H R O H O R O + H R O H O R OH R OH H OH H O Die Kinetik dieser Reaktion ist 2. Ordnung. Die Kinetik und vor allem D2O-Untersuchungen machen einen Mechanismus über das Dianion wahrscheinlich. R O OH H O + OH- R H + OH-H2O R H O O In diesem Falle wird das Salz der Säure direkt gebildet, und das Proton kommt vom Solvens. Benzoin-Addition Skript zum 5. OC-Kolloquium H Seite 12 von 39 OH HO O Umpolung C + CN(-) O CN Ar Ar Ar H Ar Aldol-Addition und Aldol-Kondensation Carbanionen von Carbonylverbindungen addieren sich ebenfalls an die Carbonylgruppe. Bei der Aldoladdition kann man sowohl mit Basen als auch mit Säuren die Reaktion katalysiert. Basenkatalyse: R H3C O + B C R (-) CH2 C H3C R O Carbonylkomponente H3C O H2C C (-) O Base = Carbanion CH-Acide Verbindung (Methylenkomponente) R R C R C H2 C O O (+) + BH -B + (+) BH konjugierte Säure der Base R H3C C C H2 C O OH Aldol (=Aldehydalkohol) β-Hydroxyketon Im ersten geschwindigkeitsbestimmenden Schritt löst die Base ein Proton von der CH-aciden Methylenkomponente unter Ausbildung eines mesomeriestabilisierten Carbanions, das dann an die Carbonylkomponente addiert wird. Dieses intermediär gebildete Ion wird dann durch Protonierung durch die konjugierte Base der Säure stabilisiert. Säurekatalyse: R H3C C O + H(+) H3C C (+) R R R OH H3C C (+) OH -H(+) H2C C OH Im Anschluss daran führt dann die Addition des Enols an das Carbenium-Ion (= protonierte Carbonylverbindung) zu Aldolen: Skript zum 5. OC-Kolloquium R R C (+) H3C Seite 13 von 39 + H2C R Aldoladdition C H3C OH Retro-Aldoladdition OH R - H(+) H3C C C R C H2 (+) OH C OH R C H2 OH C O Aldol Im sauren Milieu sind die rch Addition gebildeten Aldole nicht stabil, sondern durch Protonierung der Hydroxylgruppe und Wasserabspaltung findet eine AldolKondensation statt: R H3C C R R C H2 C + O H(+) H3C R - H3C C R C H C C H2 C R O - H2O H3C C (+) OH (+) 2 OH H(+) C R R C H2 C O O Ist R = H (Acetaldehyd als Carbonylverbindung), dann wird Crotonaldehyd gebildet, und zwar das thermodynamisch stabilere trans-Isomere. Gemischte Aldolkondensation und gezielte Aldolkondensation nach G. Wittig Setzt man zwei verschiedene Carbonylverbindungen bei der Aldolkondensation ein, so hängt die Tatsache, welche der Carbonylverbindungen die Methylenkomponente und welche die Carbonylkomponente darstellt, im wesentlichen von der Carbonylaktivität der jeweiligen Verbindung ab. Da die Unterschiede häufig jedoch nur ggraduell sind, ist ein eindeutiger Reaktionsablauf im allgemeinen nicht gegeben. Problemlos verläuft die gemischte Aldolkondensation nur dann, wenn eine Carbonylverbindung keinen α-ständigen Wasserstoff besitzt, z.B. beim Benzaldehyd: C6H5 O O CH + H3C C OH C6H5 C6H5 C H O C H2 C C6H5 O -H2O C6H5CH C H C C6H5 Als CH-acide Verbindung ist auch Nitromethan geeignet, das hier nur als Methylenkomponente fungieren kann: Skript zum 5. OC-Kolloquium O H3C + B N Seite 14 von 39 - BH(+) CH2 O O O H2C N N O O So reagiert Nitromethan durch successiven Ersatz aller Wasserstoffatome mit Formaldehyd zu Trihydroxymethylnitromethan. Auch Cyclopentadien bzw. Alkine können als Methylenkomponente verwendet werden. Verbidnungen, die auf dem Wege einer gemischtten Aldolkondensation nicht zu erhalten sind, wie z.B. β,β-Dimethylacrolein aus Aceton und Acetaldehyd, lassen sich auf folgende Weise durch eine gezielte Aldolkondensation nach G. Wittig herstellen: CH2OH CH2OH C NO2 CH2OH Trihydroxymethylnitromethan Dabei wird die Komponente mit höher Carbonylaktivität durch Überführung in die Schiff’sche Base zur Methylenkomponente. Die Schiff’sche Base wird durch Lithiumdiisopropylamid in das Carbanion überführt, das sich dann an eine weitere Carbonylkomponente addieren kann. Tollens-Kondensation Einen Mehrfach-Ersatz aciden Wasserstoffs in der Aldolreaktion findet man in der sogenannten Tollens-Reaktion, bei der Tollens-Reaktion handelt es sich um eine gemischte Aldolkondensation mit nachfolgender gekreutzter Cannizarro-Reaktion. Bruttoreaktion: CH2OH 4 H2C O + H3C C H O NaOH CH2OH C CH2OH + HCO2Na CH2OH Pentaerythrit Primär reagieren alle drei aciden H-Atom der Methylgruppe des Acetaldehyds in einer normalen Aldoladdition. Dieses primäre Reaktionsprodukt ist unter den Reaktionsbedingungen(starke Basen) nicht stabil, sondern es reagiert mit einem weiteren Molekül Formaldehyd unter Disprotortionierung in der sogenannten gekreutzen Cannizzaro-Reaktion. Skript zum 5. OC-Kolloquium H C O Seite 15 von 39 + OH(-) C H CH2OH OH H O + CH2OH C H C C H2 CH2OH O CH2OH OH CH2OH CH2OH C H O + H C O O Protonentransfer Pentaerythrit + R C O Formiat Die CH2OH-Gruppen werden von OH- wegen der Neopentylstellung nicht angegriffen. Schließlich erfolgt Protonentransfer unter Ausbildung der Salze der Ameisensäure (Formiate), da die Ameisensäuure die stärkere Säure ist. Aldolkondensation mit anderen CH-aciden Verbindungen (aliphatische Nitroverbindungen) Knoevenagel-Reaktion Bei dieser Reaktion handelt es sich um einen Spezialfall der Aldolkondensation, wobei Methylenkomponenten mit besonders hoher CH-Acidität verwendet werden. Z1 R1 Z1 R1 Base C O + H2C C C - H2 O Z2 R2 Z2 R2 Das entstehende Reaktionswasser wird azeotrop entfernt. Im allgemeinen reagieren Aldehyde besser als Ketone. Durch Variation der Reste Z lassen sich eine Vielzahl von Verbindungen einsetzen, z.B. Malonester, Cyanessigester, Acetessigester, Nitromethan, Dipenylmethan, Malodinitril und S-haltige Verbindungen. Als Basen werden verwendet: Piperidin, NEt3, KOH, NaOH (wässrig oder alkoholisc), KNH2, RO-, CH3CO2- und NH3. Michael-Addition O C O C C + C C O O C C C C C H2O -OH(-) O O C C C C C H Reaktionen OH-, NH- und CH-acider Verbindungen mit a,b ungesättigten Aldehyden, Ketonen, Estern und anderen aktivierter C-C-Doppelbindungen Cyanethylierung Skript zum 5. OC-Kolloquium Seite 16 von 39 Olefinierungsreaktionen (Wittig- und Horner-Emmons-Reaktion) Kondensation von Phosphoniu-Yliden mit Crbonyverbindungen: Wittig-Reaktion Ein Ylid ist eine Verbindung, für den sich eine Grenzstruktur formulieren lässt, bei welcher zwei benachbarte Atome mit vollem Oktett entgegengesetzte Ladungen aufweisen. Ylide sind relativ stabil, falls man das Oktenn aufweiten und die entsprechende YlenForm schreiben kann (also ab 3. Periode: P-Ylide, S-Ylide, aber nicht N-Ylide) Betain-Struktur Nucleophiles Zentrum Me Me P CH2 Me Me Me P CH2 Me Ph Ph P CH2 Ph Ph Ph P CH2 Ph Wichtiges Phoshonium-Ylid Stammsubstanz der PhosphoniumYlide en = Doppelbindung yl = Kovalenzbindung, id = Ionenbindung Darstellung von Phosphonium-Yliden: Ph3P R H2C X Ph3P R Ph3P R n-BuLi RCH2PPh3 X -LiX - BuH Skript zum 5. OC-Kolloquium Seite 17 von 39 Bei einer Wittig-Reaktion werden Ylide eingesetzt: Li PPh3 Hal H R2 O H R1 Li Hal kcis O PPh3 kDrift PPh3 O R1 H H Oxaphosphetane R2 R1 R2 R1 O=PPh3 O PPh3 ktrans R1 R2 R2 R1 R2 Li Hal Li O H R1 PPh3 Hal R2 H cis-selektive Wittig-Reaktionen: Unterbindung des stereochemischen Drift bei Wittig-Reaktionen labiler Ylide durch Anwendung sogenannter „salzfreier-Bedingungen“ (Basen: NaNH2, KOtert-Bu, KHMDS). Wichtig: salzfrei-Wittig-Reaktionen labiler Ylide stellen eine stereoselektive Synthese von cis-Olefinen dar. R O NaNH2 R Ar3P CH2 Me Ar3P CH2 Me Br THF Me % cis Olefin für ... ... R = Pent Ph Pr Pr Ar = Ph 96 90 92 84 Ar = o-Tolyl 98 97 96 95 Skript zum 5. OC-Kolloquium Seite 18 von 39 P-Ylide reagieren mit C=O-Doppelbindungen umso rascher, je elektrophiler sie sind: Aldehyde lassen sich sogar in Gegenwart von Ketonen olefenieren (Chemoselektivität). O Ph3P C Hex H2 Br O Oct KOtertBu O Ph3P C Hex H THF Hex Oct Verwendet man labile P-Ylide unter nicht-salzfrei Bedingungen, so erhält man ein cis/trans-Produktgemisch. Ein Beispiel dafür ist die Heterolyse von Oxaphophetanen: O Ph3P CH2 (dargestellt aus Br Ph3PHCH3 nBuLi) R Ph3P CMe2 O R (dargestellt aus Ph3PHCHMe2 I nBuLi) Eine trans-selektive Form der Wittig-Reaktion stellt die Schlosser-Variante. O PPh3 R1 R2 O ktrans R1 H H PPh3 kcis R2 kDrift O PPh3 R1 Li Hal Li PPh3 Hal R2 H O H R1 R2 Li Hal PhLi Li O H R1 PPh3 Hal R2 PhLi Li PPh3 Hal H R2 O H R1 A B C Lithiobetain Oxidoylid Lithiobetain HCl oder t-BuOH KOtert-Bu (- LiOtert-Bu) PPh3 O R2 R1 D R2 O PPh3 R1 E R2 R1 O=PPh3 Skript zum 5. OC-Kolloquium Seite 19 von 39 Wittig-Horner-Olefenierung Die Wittig-Horner-Reaktion ist eine C,C-verknüpfende Olefinsynthese aus einem Alkyldiphenylphosphanoxid R1-CH2-P(=O)Ph2 und einem Aldehyd R2-CHO. Man kann die Reaktion einstufig oder zweistufig führen. Der Vorteil der zweistufigen Reaktionsführung ist das der Protonierungsschritt involviert ist und die phospholierten Alkohole leicht trennbar sind. Bei der einstufigen Form erhält man ein 1:1-Gemisch der diastereomeren Alkoholaten. Die entstandenen Olefine können meist nur schwer getrennt werden. O O PPh2 PPh2 nBuLi PPh2 -BuH H R1 O R1 Li R1 Zugabe von R2 Li B A O O O PPh2 PPh2 R2 R1 HCl R2 R1 OH OH 50 : 50 bis 10:90 syn-C O O PPh2 PPh2 R2 R1 O anti-C R2 R1 R1 R2 cis-E O O K trans-G Li Otert-Bu K Ph2P O K O O PPh2 PPh2 R2 O R2 R1 O K K syn-F R1 R1 R2 anti-D Otert-Bu R1 O Ph2P O Li Otert-Bu K via cis-G trans-E Ph2P O syn-D Li Otert-Bu K via trans-G R2 R1 O cis-G R2 K anti-F Skript zum 5. OC-Kolloquium Seite 20 von 39 Wittig-Wadsworth-Emmons-Reaktion (HWE-Reaktion) Die HWE-Reaktion stellt eine C=C-Doppelbindung bildende Kondensation zwischen β-Ketooder dem Li-, Naoder K-Salz eines β(Alkoxycarbonyl)phosphonsäuredialkylesters und einem Aldehyd dar. Die Vorteile dieser Reaktion ist die Bildung eines wasserlöslichen PhosphorsäuredialkylesterAnions, und die hohe trans-Selektivität. Mechanismus: O O (EtO)2P CH2 O R2 in situ: n-BuLi oder NaH oder KOtertBu O O (EtO)2P CH O R2 O (EtO)2P C H M O O R2 O (EtO)2HP C H M O O R2 M O ? R1 O O P(OEt)2 O O M P(OEt)2 R1 O O R2 R1 O O R2 B (racemisch) (racemisch) M O O O P(OEt)2 R1 P(OEt)2 O O R2 R1 O O R2 D E (racemisch) (racemisch) Leuckart-Wallach-Reaktion R1 O C A O O R2 O O P(OEt)2 Skript zum 5. OC-Kolloquium Seite 21 von 39 Eigenschaften von Carbonsäureestern, Amiden, Nitrilen und Anhydriden Ester Ester besitzen die allgemeine Formel RC=O-OR’. Sie sind die wichtigsten Carbonsäure-Derivate. Sie entstehen durch die Reaktion von Carbonsäuren mit Alkoholen. Gibt man eine Carbonsäure sowie in einen Alkohol in einem Reaktionsgefäß zusammen, so wird man keine Reaktion feststellen können. Erst nach der Zugabe von katalytischen Mengen einer starken Säure wie Schwefelsäure findet eine Reaktion unter Wasserabspaltung statt. Eine derartige Reaktion wird als Vereseterung bezeicchnet. Veresterung Bei einer Veresterung handelt es sich um eine Gleichgewichtsreaktion deren Lage durch die Konzentration der Edukte sowie der Produkte bestimmt werden kann. Eine Veresterung verläuft über eine säurekatalysierte Additions-Eliminierungsreaktion. Mechanismus Schritt 1: Protonierung der Carboxylgruppe H O O + H(+) H H + O O + O R C -H(+) H O R H O R + O H O H R H Dihydroxycarbeniumion Schritt 2: Angriff durch den Alkohol O + O H3C O R H H H OH R H O H3C O O -H(+) + H R + H(+) H3C H O Schritt 3: Wasserabspaltung H H O O R O H + H(+) H3C O O -H(+) + H(+) R O CH3 H + R O -H(+) H3C H O H + H O -H2O +H2O R H3C O O + R C H3C O Skript zum 5. OC-Kolloquium Seite 22 von 39 Lactone Eine Veresterung kann auch intramolekular verlaufen, falls ein Molekül sowohl eine Alkoholfunktion sowie eine Carboxylgruppe trägt. Man erhält einen cyclischen Ester, welcher als Lacton bezeichnet wird: Beispiel: O HOCH2CH2CH2CH2COOH O H2SO4, H2O + H2O Neben der Säurekatalysierten Veresterung können Carbonsäuren auch über andere Reaktionen in Ester überführt werden. Zwei besonders wichtige Wege sind die nucleophile Substitution von Halogenalkanen mit Carboxylat-Ionen und die Bildung von Methylestern durch Reaktion von Carbonsäuren mit Diazomethan, CH2N2. Verseifung Die Umkehrung der Veresterung wird als Esterhydrolyse bezeichnet. Diese verläuft nachdem gleichen Mechanismus wie die Esterbildung. Dies ist möglich, da alle Reaktionsschritte reversibel sind: O O H3C COH + CH3OH H2SO4, ∆ H3C COCH3 + H2O Eine irreversible Methode Ester zu spalten ist die Verseifung hierbei wird der Ester mit einer starken Base versetzt und man erhält den entsprechenden Alkohol sowie das Carboxylat der Carbonsäure. Skript zum 5. OC-Kolloquium Seite 23 von 39 Amide Eine weitere wichtige Gruppe von Carbbonsäurederivate sind die Amide, welche die allgemeine Formel RC=ONR2’ aufweisen. Da Stickstoff nicht so elektronegativ wie Sauerstoff ist, sind Amine nucleophiler und basischer als Alkohole, und sie können auf beide Arten mit Carbonsäuren reagieren. Gibt man eine Säure und ein Amin zusammen, bildet sich sofort das Ammoniumsalz. O O (+) NH4 R C O CO H R + NH 3 Die Salzbildung ist beim Erhitzen reversibel, dabei läuft ein langsamer, aber thermodynamisch günstiger Prozess, bei dem das Stickstoffatom das CarbonylKohlenstoffatom angreift, ab. Eine Additions-Eliminierungs-Reaktion führt zu einem Amid, in dem NR2 die OH-Gruppe ersetzt hat. OH R O O O R + NH3 OH + NH R Protonenübertragung 3 O + OH2 + H2O R NH2 NH2 Man bezeichnet diese Reaktion als Ammonolyse. Man kann aber Amide auch durch die Reaktion von Carbonsäuren mit Aminen gewinnen. Die Amidbildung ist reversibel. Beim Behandeln von Amiden mit heißer, basischer oder saurer wässriger Lösung gewinnt man die Carbonsäure und Amine wieder zurück. O CH3CH2CH2COOH + (CH3)2NH 155C CH3CH2CH2 CN(CH3)2 + H2O N,N-Dimethylbutanamid Die entstehende Bindung wird als Peptidbindung bezeichnet. Setzt man beispielsweise Aminosäuren ein so kann man Polypeptide synthetisieren. Aminosäuren enthalten sowohl eine Aminfunktion als auch eine Carboxylgruppe, somit gibt es pro Molekül zwei „aktive“ Gruppen und es kann zu einer Polykondensation kommen. Lactame Setzt man hingegen größere Moleküle mit beiden funktionellen Gruppen ein, so bilden sich intramolekular, cyclische Amide und man erhält die so genannten Lactame. O (+) HN H C H2 C H2 C H2 (-) COO H2NCH2CH2CH2COOH ∆ NH + H2O Skript zum 5. OC-Kolloquium Seite 24 von 39 Verseifung Man kann amide analog zu Estern in Gegenwart von starken Basen verseifen. Die Reaktion verläuft prinzipiell über den gleichen Mechanismus. Nitrile Eine besondere Klasse von Carbonsäure-Derivaten stellen die Nitrile mit der allgemeinen Formel RC≡N, dar. Der Kohlenstoff liegt bei Nitrilen in der gleichen Oxidationsstufe wie in einer Carboxylgruppe vor. Außerdem lassen sich Nitrile leicht in andere Derivate der Carbonsäuren überführen. Die elektronenziehende Kraft des Stickstoffs der Nitrilgruppe lässt sich über eine dipolare Resonanzformel darstellen: R C N R C + N Das freie Elektronenpaar am Stickstoffatom macht die Nitrilfunktion leicht basisch, das Stickstoffatom kann also ebenso wie das Carbonylsauerstoffatom der Carbonsäure ein Proton addieren. Im VVergleich zu einem Imin oder Amin wird das sp-hybridisierte Nitrilsystem jedoch weitaus weniger leicht protoniert. Basizität des freien Elektronenpaars am Stickstoff R R R C N R R R N C N R sp3 sp2 sp Herstellung siehe Cyanhydrin Bildung Verseifung Nitrile lassen sich säure- oder basenkatalysiert zu Carbonsäuren hydrolisieren. Mechanismus der säurekatalysierten Hydrolyse von Nitrilen NH R C N +H(+) -H(+) R C NH -H(+) +H(+) +H(+) C R -H(+) OH R + N + R H C + NH2 NH2 C C OH R N -H2O NH2 -H(+) C +H(+) OH + +H2O H R O +H(+) C R +H2O OH2 + RCOOH + NH4+ Skript zum 5. OC-Kolloquium Seite 25 von 39 Mechanismus der basenkatalysierten Hydrolyse von Nitrilen H N R C N + OH(-) C N N O R NH2 +H2O H + OH(-) +H(+) C C C O R H H -H2O + OH(-) C OH R R N +H2O O R O RCOOH + NH4+ Anhydride Carbonsäureanhydride gehören zu den reaktivsten Carbonsäurederivaten. Herstellung Carbonsäureanhydride können durch die Reaktion von Alkanoylhalogeniden mit einem Carboxylatsalz gebildet werden. O O O CH3CH2CH2COONa + H3C H3C CCl C H2 C H2 CO CCH3 + NaCl "gemischtes" Anhydrid Wie aus dem Namen schon ersichtlich ist leiten sich die Anhydride von der Carbonsäure unter Abspaltung eines Wassermoleküls ab. Daher ist ebenso eine Darstellung durch thermische Dehydratisierung möglich. Geht man dabei von einer Dicarbonsäure aus, so erhält man ein cyclische Anhydrid. O C COOH H2C H2C H2C 573K O H2C + H2O C COOH O Dutandisäureanhydrid Butandisäure Desweiteren kann man die Anhydride durch die Reaktion eines Ketens mit der entsprechenden Carbonsäure darstellen: Darstellung des Ketens: O R C H2 CCl N(CH2CH3)3 RCH C O + HN(+)(CH2CH3)3Cl(-) O R H C Br CBr Zn RCH C O + ZnBr2 Skript zum 5. OC-Kolloquium Seite 26 von 39 Darstellung des Anhydrids O O RCH C Keten O + H3C H3C COH Essigsäure O CO CCH3 Essigsäureanhydrid Alkoholyse Versetzt man ein Carbonsäureanhydrid mit einem Alkohol, so tritt ein AdditionsEliminierungs-Mechanismus ein und man erhält die entsprechenden Carbonsäureester: O O O O CH3OH, 372K HO CCH2C H2 COCH3 O Ammonolyse Durch das versetzen eines Anhydrids mit Ammoniak erhält man ein Ammoniumsalz sowie eine Aminogruppe. O O CH3 O H3C 2 NH3 NH4+ O CCH2CCH2CNH2 O H3C O Herstellung von Carbonsäurederivaten Halbester CH3 Skript zum 5. OC-Kolloquium Carbaminsäure Harnstoff Phosgen Urethane Isocyanate Seite 27 von 39 Skript zum 5. OC-Kolloquium Seite 28 von 39 Claisen-Dieckmann-Kondensation (mit Oxalsäure- und Ameisensäureester) Bei dieser Synthese werden als Carbonylkomponente Carbonsäureester und als Methylenkomponente ebenfalls Carbonsäureester oder Ketone oder Nitrile eingesetzt. Die Claisen’sche Esterkondensation und die Diekmannkondensation unterscheiden sich wie folgt: O R C C (-) + C Carbanion O intermolekular OR' O C O Ester C R + R C b-Dicarbonylverbindung O Alkoholat Diekmann-Kondensation O intramolekular C OR' C C (-) C O + RO(-) Allgemeiner Mechanismus: Die Esterkondensationen beginnen mit der Bildung von Carbanionen: Wegen der Enolatbildung sind stöchiometrische Mengen Base notwendig: H C + C R (-) C O + C R OH O O Der Nachweis für das intermediär Auftreten von Carbanionen gelingt einmal durch Markierungversuche mit C2H5-OD als Base, (-) C + C D ROD C + RO(-) C O O weiterhin durch Racemisierung in der α-Position zur Estergruppe von chialen Estern: R1 H + R2 COOR R O R1 (-) C R2 OR C O H R1 COOR R2 +ROH Das durch die Base aus der CH-aciden Komponente gebildete Carbanion greift dann eine Carbonylverbindung an: Skript zum 5. OC-Kolloquium O R1 H R1 C + RO(-) C (-) CH COOR R2 COOR R2 (-) CH + OR R3 + ROH R2 O O R1 Seite 29 von 39 O R3 H C R1 R2 O R3 OR R2 R1 O -RO(-) + RO(-) R1 O H C R3 R2 + RO(-) R2 O -ROH M R3 O Von größerer Bedeutung ist die Tatsache, dass es sich bei allen Schritten dieser Reaktion um Gleichgewichte handelt, der letzte Schritt der Enolatbildung jedoch die Reaktion irreversibel in Richtung der Produkte verschiebt. Durch die Enolatbildung wird wieder freiwerdendes Alkoholat verbraucht, das ist auch der Grund dafür, dass stöchiometrisches Mengen Alkoholat benötigt werden. Durch die verwendeten M+Ionen wird die Reaktion unterschiedlich schnell bbeschleunigt. Sie steigt in der Reihe Li < Na < K << Cs. Dies zeigt, dass das Metallatom als Koordinationszentrum wirkt und mit steigendem Atomradius immer besser in der Lage ist, die Reaktionspartner in eine für die Reaktion günstige Lage zu bringen. Die präparative Bedeutung dieser Reaktion liegt in ihrer großen Anwendungsbreite: mit dem Rest R1 der Carbonylkomponente R1 = H (Ameisensäure) = Alkyl (Carbonsäureester) = OR (Kohlensäureester) mit dem Rest R2 der Methylenkomponente R2 = Alkyl, Aryl durch Variation des Restes R3 der Methylengruppe R3 = Alkyl, Aryl (Ketone) R3 = OR (Carbonsäureester) -CO-R = CN (Carbonsäurenitrile werden eingeführt Formylgruppen Acylgruppen Acyloxygruppen werden eingeführt Substituenten in α-Stellung erhält man folgende Produkte β-Diketone β-Ketoester β-Ketonitrile Skript zum 5. OC-Kolloquium Seite 30 von 39 Diekmann-Reaktion R1 R2 Cyclische β-Dicarbonylverbindung α,ϖ -Dicarbonsäureester Bei Variation der Reste R und verschiedenen Kombinationen ist somit eine große Zahl von Verbindungen zugänglich. Wie bei der gemischten Aldolreaktion ist es jedoch auch bei der gemischten Esterkondensation wichtig, dass einer der Reaktionspartner eindeutig als Carbonylkomponente reagiert. Andernfalls erhält man Produktgemische. Beispiele: I. Carbonylkomponente und Methylenkomponente identisch Darstellung von Acetessigester H3C + CO2R R (-) CH2 O O RO C + (-) CH2 CO2R + ROH O CO2R RO CH3 Säure-Base-Gleichgewicht O C H2 C AdditionsphaseGleichgewicht C OR CH3 CH3 -R O O H3C O OR C C H2 b-Diketo-Form des Acetessigesters +R O O + ROH C RO O Enolat-Form des Acetessigesters Durch Enolat-Bildung wird die Reaktion ganz zur Seite des Reaktionproduktes ( des Enolats der β-Dicarbonylverbindung) gezogen Bei Raumtemperatur liegt der Acetessigester in einem Gleichhgewicht von 92,5 % βDiketo-Form und 7,5% Enolform vor: CH3 O H RO O O O C H3C C H2 C OR β-Diketo-Form Enol-Form (7%) Der Nachweis von enolisierbaren β-Dicarbonylverbindungen gelingt mit wässrigethanolischer FeCl3-Lösung durch Bildung von tief-violett gefärbten ChelatKomplexen unter der Voraussetzung, dass der Enolgehalt mindestens 1-2% beträgt: Skript zum 5. OC-Kolloquium Seite 31 von 39 O O Fe H O O O β-Diketon II. O Fe3+ Metall-Chelat cis-Enol "H-Brücke" Carbonylkomponente und Methylkomponente verschieden Darstellung von Acetylaceton: Die Durchführung erfolgt so, dass man 0,5 mol Keton mit 1 mol Ester in Ether mit pulverisiertem Natrium in Gegenwart von katalytischen Mengen C2H5OH behandelt: O O RO C + CH2 O C CH3 RO O C H2 C C CH3 CH3 CH3 O -RO(-) H3C C +ROH CH3 O C H2 C O +RO(-) CH3 -ROH H3C Keto-Form (15%) O Enolat-Form (85%) Hierbei handelt es sich um eine gemischte Aldolkondensation, in der beide Komponenten sowohl die Methylen- als auch die Carbonylkomponente darstellen können. Aufgrund der unterschiedlichen Carbonylaktivität und der Reaktionsdurchführung (Ester-Überschuss) lässt sich die Reaktion im oben dargestellten Sinne lenken. III. Ethylcarbonat als Carbonylkomponente Ester als Methylenkomponente + C EtO O O O (-) CH OEt C EtO OR O OEt C +RO(-) OR CO2R O C R OEt C (-) C R R EtO OR C O O H C C C H C OEt R R -EtO(-) C O kaum Enol O C OR Skript zum 5. OC-Kolloquium Seite 32 von 39 Man erhält substituierte Malonester. In einer Variante arbeitet man mit Ketonen als Methylenkomponente und erhält β-Ketoester: O OEt C O H C C R R Dies erfolgt vor allem dann, wenn der Ester zu stark zur Selbstkondensation neigt. IV. Oxalester als Carbonylkomponente Ester als Methylenkomponente RO O O C C O O OR (-) CH + RO OR C O H C C OR R C R O RO C O O O H C C -CO C OR R V. RO C ∆ Fe-Pulver u. B(OH)3 O OR C O H C C OR substituierter Malonester R Ameisensäure als Carbonylkomponente Ester als Methylenkomponente H O O O OR C + (-) CH H OR C O H C H C C OR OR R R -RO(-) C O O H C OR C Formylessigester R VI. Diekmann-Kondensation Cyclische intramolekulare Esterkondensation COOR COOR +RO(-) O -RO(-) O OR OR COOR +RO(-)Me(+) Me(+) + ROH O COOR Me = Metall O Skript zum 5. OC-Kolloquium Seite 33 von 39 Der mittlere Ringbereich wird durch diese Reaktion jedoch nicht abgedeckt. Die transannularen Wechselwirkungen sind thermodynamisch ungünstige Faktoren zur Einstellung einer Gleichgewichtsreaktion. Eigenschaften und Reaktionen der entsprechenden β-Ketoester und βDiketone (Acetessigestersynthese, Ketospaltung, Säurespaltung, ChelatKomplex) I. Alkylierung von β-Dicarbonylverbindungen Beim Acetessigester tritt wie bei allen β-Dicarbonylverbindungen Keto-EnolTautomerie auf: COOR COOR COOR + RO(-) O -RO(-) O OR OR O COOR +RO(-)Me(+) Me(+) + ROH O Aufgrund dieser Keto-Enol-Tautomerie sind die zur Carboxyl-Funktion α-ständigen HAtome acid und lassen sich durch Basen leicht als Protonen abspalten. Dabei entsteht dann das mesomeriestabilisierte, ambidente Anion des Acetessigesters: CH3 H H O RO (-) -H(+) H RO O CH3 CH3 H O O O RO O Beide Zentren können nucleophile Substitutionsreaktionen eingehen; es handelt sich also wieder um ein ambidentes Ion. Für dieses Ion gelten nun auch die bereits besprochenen Voraussetzungen: CH3 H OCH2OCH3 SN1 H CH3 (-) RO CH3 O O +CH3OCH2Cl RO O H O RO O SN2 +CH3I CH3 H3C CH3 OH H3C O H RO O RO O Skript zum 5. OC-Kolloquium Seite 34 von 39 Ein weiters Beispiel für derartige Reaktionen stellt das Cyclohexadion-1,3 dar: O O O O C +Base -HB(+) OH O O O trans-fixiertes Enol hohes Gewicht geringeres Gewicht Nach der Abspaltung eines Protons erhält man ein mesomeristabilisiertes Anion. Die mesomere Grenzform, bei der die negative Ladung am Sauerstoff sitzt, entspricht dabei dem elektronegativsten Ende (SN1), diejenige, bei der die negative Ladung am Kohlenstoff sitzt, entspricht dem polarisierbaren Ende (SN2). Bei nucleophilen Substitutionen unter verschiedenen Bedingungen erhält man folgende Reaktionen: O O C O O + (CH3O)SO2 in NaOH/ H2O + CH3I in CH3OH SN2 SN1 O O O CH3 CH3 H OMe O OH II. Esterspaltung (Retro-Claisen-Reaktion) Die Esterspaltung gelingt mit überschüssigem wasserfreiem Alkoholat. Man muss jedoch die gleiche Alkoholkomponente einsetzen, die auch der Ester enthält, da sonst durch Umesterung Estergemische gebildet werden. O O R C C H2 CO2R R C OR RO(-) O C H2 CO2R H3C + (-) CH2 OR H2O/ H(+) Ester CO2R Skript zum 5. OC-Kolloquium III. Seite 35 von 39 Säurespaltung von β-Ketoestern mit konz. Alkali O R C R O O C H OH(-)-Überschuss R C OR O H C C O OH R OH(-) O R + C (-) CH O + H2O CO2 H2C -OH(-) (-) CO2 R R Diese Reaktion hat jedoch präparativ nur geringe Bedeutung, da substituierte Essigester auf dem Wege einer Malonestersynthese leichter zugänglich sind. IV. Ketospaltung von β-Ketoestern mit verd. Alkali oder verd. Säure O R C O H C O OH(-) R O C -CO2 O R O O O C C OR R R H C (-) CH R C H C R R H2 O R C H2 C R Reaktionen von Carbonyl- und Carboxylverbindungen mit GrignardReagenzien Bei Grignard-Verbindungen handelt es sich um metallorganische Verbindungen mit Magnesium. R-Hal + Mg → R-Mg-Hal Grignard-Verbindungen werden von Lösungsmittel (meistens Ether) stabilisiert. R R' R' O Mg O R' R' R Die Struktur von Grignard-Verbindungen ist noch nicht in allen Einzeheiten geklärt. Sie hängt von der Konzentration und vom Lösungsmittel ab, wobei das sogenannte Schlenk-Gleichgewicht eine Rolle spielt. Die Addition von Grignard-Reagenzien an Carbonalverbindungen besitzt große präparativer Bedeutung zur Darstellung von Alkoholen (Carbinolen): Formaldehyd Aldehyde Ketone CO2 → → → → primäre Alkohole sekundäre Alkohole tertiäre Alkohole Carbonsäuren Skript zum 5. OC-Kolloquium Seite 36 von 39 a) Reaktionen mit Aldehyden und Ketonen Mechanismus: R R R' 2 R'MgX Mg C C O R O R R' X R' R H2O C R OH Mg X An der Reaktion sind im allgemeinen 2 Mol Reagens pro mol Carbonylverbindung beteiligt, wobei ein sechsgleidriger cyclischer Übergangszustand gebildet wird. b) Reaktion mit Carbonsäurederivaten Carbonsäurederivate (z.B. Ester) reagieren zunächst analog zu den Aldehyden und Ketonen: R2O O R R 2 R1MgX C R1 C " O OR2 " Mg R1 Mg X RO R1 C R OMgX X O R1 RO R C R C R1 + OMgX R2 OMgX Man erhält primär Salze von Halbacetalen, die jedoch instabil sind und in Ketone zerfallen. Die Ketone reagieren dann wie oben beschrieben zu tertiären Alkoholen weiter. Herstellung von primären, sekundären und tertiären Alkoholen und von Carbonsäure Reaktionen von Carbonylund Carboxylierungverbindungen mit Organozinkverbindungen (Reformatzky-Reaktion) Die Reformatsky-Reaktion ist eine Grignard-analoge Reaktion von α-Chlor- oder Bromessig- bzw. Propionsäureestern mit Aldehyden oder Ketonen unter Bildung von β-Hydroxycarbonsäuren, aus denen man durch H2O-Abspaltung α,β-ungesättigte Ester erhalten kann. 1. Zn/ Ether H C H3C OH 3 (+) C O + H2C H3C +H(+)/ -H2O CO2R C H3C C H3C Cl H3C 2. H /H2O H C C H2 CO2R β,β-Dimethylacrylsäureester CO2R Es bildet sich intermediär eine zinkorganische Verbindung. Skript zum 5. OC-Kolloquium 2.0 Identifizierung organischer Substanzen Vorproben Seite 37 von 39 Skript zum 5. OC-Kolloquium Prüfung auf funktionelle Gruppen Seite 38 von 39 Skript zum 5. OC-Kolloquium Seite 39 von 39 Darstellung geeigneter Derrivate (Reagenzien, Reaktionsgleichungen)

![6.3.1 1-Oxa-spiro[2.5]octan - Institut für Organische Chemie](http://s1.studylibde.com/store/data/001356875_1-96e669e5c88ad586db9f9f199d424d05-300x300.png)