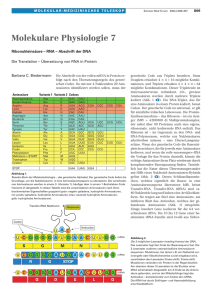
Dokument_1
Werbung

Versuche zum ribosomalen Einbau unnatürlicher Aminosäuren in Proteine und Untersuchung von Struktur-Funktionsbeziehungen in thermophilen ATP-Synthasen mittels Spin-markierter Nukleotide Vom Fachbereich Chemie der Technischen Universität Kaiserslautern zur Verleihung des akademischen Grades Doktor der Naturwissenschaften genehmigte DISSERTATION (D 386) vorgelegt von Dipl.-Chem. Dirk Mannweiler Betreuer: Prof. Dr. Wolfgang E. Trommer Tag der wissenschaftlichen Aussprache: 3. Juli 2006 Kaiserslautern 2006 Tag der wissenschaftlichen Aussprache: 03. Juli 2006 Promotionskommission: Vorsitzender: Prof. Dr. S. Ernst 1. Berichterstatter: Prof. Dr. W. E. Trommer 2. Berichterstatter: Prof. Dr. Dr. D. Schrenk Die vorliegende Arbeit entstand in der Zeit von Juni 2000 bis Dezember 2005 an der Technischen Universität Kaiserslautern. Herrn Prof. Dr. Trommer danke ich für den mir gewährten Freiraum bei der Gestaltung des Themas, die Diskussionsbereitschaft bei fachlichen Problemen sowie die Unterstützung bei der Durchführung der Arbeit Frau Prof. Dr. Vogel danke ich für die Unterstützung bei den Untersuchungen an der β-Untereinheit der FOF1-ATP-Synthase des thermophilen Bacillus PS3 Der Wissenschaftler ist verantwortlich für die Erklärung seines Wissens, nicht aber dafür, wie dieses Wissen angewendet wird. Edward Teller Inhaltsverzeichnis 1. Einleitung ........................................................................................................ 1 1.1 1.1.1 1.1.2 1.1.3 1.1.4 1.1.5 1.2 1.3 1.3.1 1.3.2 1.4 1.4.1 1.4.2 1.5 2. Translation in Prokaryonten ............................................................................ 1 Transfer-RNA (tRNA) ...................................................................................... 1 Bildung der Aminoacyl-tRNA........................................................................... 3 Wobble Theorie............................................................................................... 4 Das Ribosom .................................................................................................. 5 Mechanismus der Proteinsynthese ................................................................. 6 Ortsspezifischer Einbau unnatürlicher Aminosäuren in Proteine..................... 7 FOF1-ATP-Synthase ...................................................................................... 10 Die F1-ATPase .............................................................................................. 11 Katalytischer Mechanismus .......................................................................... 11 ESR-Spektroskopie....................................................................................... 14 Grundlagen ................................................................................................... 14 Spin-Label..................................................................................................... 20 Matrix-unterstützte Laser-Desorptions/Ionisations-Massenspektrometrie (MALDI-MS) .................................................................................................. 21 Problemstellung ............................................................................................ 22 3. Eigene Ergebnisse und Diskussion............................................................... 24 3.1 Synthese einer amber Suppressor tRNA mit Spin-markierter Aminosäure ... 24 3.1.1 Synthese des Cyanomethylesters der Aminosäure HO3007 ........................ 25 3.1.2 Acylierung der aktivierten Spin-Label Aminosäure an das Dinukleotid pdCpA.................................................................................. 26 3.1.3 Entfernung der tBoc-Schutzgruppe am N-Terminus der Spin-Label Aminosäure........................................................................... 27 3.1.4 Herstellung und Reinigung der verkürzten tRNA .......................................... 29 3.1.5. Ligation der verkürzten Suppressor tRNA mit dem aminoacylierten Dinukleotid dpCpA ........................................................................................ 32 3.2 Konstruktion des Expressionsvektors pIVEX2.3MCSMtj1pN-term................ 33 3.2.1 Das Mtj1p-Protein ......................................................................................... 33 3.2.2 Herstellung eines N-terminalen Mtj1p-Fragments......................................... 34 3.3 Ortsspezifischer Einbau einer Spin-markierten Aminosäure in den N-Terminus des Mtj1p-Proteins ........................................................................................ 37 3.4 Gentechnische Gewinnung und Reinigung der β-Untereinheit der FOF1-ATP-Synthase des thermophilen Bacillus PS3............................... 40 3.5 ESR-Untersuchungen der β-Untereinheit der FOF1-ATP Synthase des thermophilen Bacillus PS3 mit 2-N3-2’,3’-SL-ATP ......................................... 43 3.5 Massenspektrometrische Untersuchungen an der β-Untereinheit der FOF1-ATP-Synthase des thermophilen Bacillus PS3.....................................45 Inhaltsverzeichnis 4 Zusammenfassung .......................................................................................51 5 Material und Methoden ................................................................................. 54 5.1 Material ......................................................................................................... 54 5.1.1 E.coli Stämme............................................................................................... 54 5.1.2 Plasmide ....................................................................................................... 54 5.1.3 Enzyme und Marker ...................................................................................... 54 5.1.4 Medien .......................................................................................................... 55 5.1.5 Chemikalien .................................................................................................. 55 5.1.6 Sonstige Materialien...................................................................................... 56 5.1.7 Geräte ........................................................................................................... 57 5.2 Aufzucht und Lagerung von E.coli................................................................. 58 5.2.1 Autoklavieren ................................................................................................ 58 5.2.2 Flüssigmedien ............................................................................................... 58 5.2.3 Festmedien ................................................................................................... 59 5.2.4 Kulturbedingungen ........................................................................................ 59 5.2.5 Wachstumsmessungen................................................................................. 60 5.3 Allgemeine Arbeitsmethoden ........................................................................ 60 5.3.1 Herstellung kompetenter Zellen .................................................................... 60 5.3.2 Transformation.............................................................................................. 61 5.3.3 Plasmid-Minipräparation ............................................................................... 61 5.3.4 Restriktionsverdau ........................................................................................ 61 5.3.5 Agarose-Gelelektrophorese .......................................................................... 62 5.3.6 DNA-Größenstandards ................................................................................. 63 5.3.7 Konzentrationsbestimmung von Plasmid-DNA ............................................. 63 5.3.8 Isolierung von DNA aus Agarosegelen ......................................................... 64 5.3.9 Reinigung von PCR-Produkten ..................................................................... 64 5.3.10 Ligation von DNA-Fragmenten...................................................................... 64 5.3.11 Rekonstitution und Lagerung von Trypsin Gold ............................................ 64 5.4 Konstruktion des Vektors pIVEX2.3MCSMtj1pN-term................................... 65 5.4.1 Konstruktion des N-terminalen Mtj1p-Fragments mittels PCR ...................... 65 5.4.2 Linearisierung von pIVEX2.3MCS................................................................. 67 5.4.3 Ligation des PCR-Produkts mit linearisiertem Vektor pIVEX2.3MCS............ 68 5.5 Isolierung der β-Untereinheit der FoF1-ATP-Synthase des thermophilen Bacillus PS3 aus E.coli ................................................................................. 69 5.5.1 Transformation von pUCβ in E.coli................................................................ 69 5.5.2 Expression der β-Untereinheit in E.coli JM109 ............................................. 69 5.5.3 Isolation der β-Untereinheit aus E.coli JM109............................................... 70 5.5.4 Photoaffinitätsmarkierung der β-Untereinheit der FoF1-ATP-Synthase des thermophilen Bacillus PS3 ............................................................................ 72 5.6 ESR-Studien an der β-Untereinheit der FOF1-ATP-Synthase des thermophilen Bakteriums PS3 ............................................................................................ 73 5.6.1 Geräteeinstellungen ...................................................................................... 73 5.6.2 Vorbereitung des Proteins für die ESR-Messungen...................................... 73 5.7 Massenspektrometrische Untersuchungen von Peptiden und Proteinen ...... 74 5.7.1 Tryptischer Verdau von Proteinen im SDS-Gel............................................. 74 5.7.2 Vorbereitung von Peptiden und Proteinen für MALDI-MS............................. 75 5.7.3 Geräteeinstellungen ...................................................................................... 76 5.7.4 Kalibrierung................................................................................................... 77 5.8 Synthesen ..................................................................................................... 78 5.8.1 Cyanomethylester von HO3007 .................................................................... 78 Inhaltsverzeichnis 5.8.2 Acylierung von dpCpA mit HO3007............................................................... 78 5.8.3 Entschützen des acylierten Dinukleotids....................................................... 79 6 Abkürzungen................................................................................................. 80 7 Literatur......................................................................................................... 81 1 Einleitung 1 1. Einleitung 1.1 Translation in Prokaryonten Der Mechanismus der Proteinsynthese wird als Translation bezeichnet, da er das vierbuchstabige Alphabet der Nukleinsäuren in das völlig andere der Proteine übersetzt. Der komplexe Prozess der Translation erfordert ein Zusammenspiel von mehr als 100 Makromolekülen zusätzlich zu tRNA, mRNA und Ribosomen. In diesem Abschnitt wird die Proteinsynthese in E.coli beschrieben, da diese viele allgemeingültige Prinzipien aufweist. 1.1.1 Transfer-RNA (tRNA) Die tRNA fungiert als ein Adaptermolekül. Sie erkennt sowohl das zur Anknüpfung der richtigen Aminosäure erforderliche Enzym als auch das Codon auf der mRNA. Die erstmalige Bestimmung der Basensequenz der tRNA gelang Robert Holley (Holley 1965). Das Molekül besteht aus einem einzelnen Strang von 76 Ribonukleotiden. Am 5'-Ende ist es phosphoryliert, das 3'-Ende trägt eine freie Hydroxylgruppe. Charakteristisch ist der hohe Gehalt an seltenen Basen wie Inosin, Pseudouridin, Ribothymin und methylierten Derivaten von Guanosin und Inosin. Die 3'-OH Gruppe des Adenosinrestes am 3'-Ende ist die Aminosäurebindungsstelle. In der Mitte des Moleküls befindet sich das Anticodon. Es ist komplementär zu einem bestimmten Codon auf der mRNA (s. Abbildung 1). So ist die Sequenz GAA komplementär zu UUC, einem Codon für Phenylalanin. 1 Einleitung 2 Abbildung 1: Basensequenz der Hefe-Alanyl-tRNA (L.Stryer; Biochemie) Alle bekannten tRNA-Moleküle können in der Form eines Kleeblattes beschrieben werden, in dem circa 50% der Basen gepaart sind. Dieses immer gleiche Strukturmerkmal ermöglicht den tRNA's auf fast gleiche Weise mit Ribosomen, mRNA's und Elongationsfaktoren zu reagieren. So müssen alle tRNA Moleküle in die A-, P- und E-Stellen der Ribosomen passen, um mit dem enzymatischen Zentrum, welches die Knüpfung der Peptidbindung katalysiert, in Wechselwirkung treten zu können. Die Aufklärung der Raumstruktur der tRNA erfolgte durch die Arbeitsgruppen von A. Rich und A. Klug (Suddath 1974, Robertus 1974). Ihre voneinander unabhängigen Arbeiten lieferten eine Vielzahl von Informationen. Das Molekül besitzt zwei doppelhelikale Abschnitte. Diese Abschnitte stehen senkrecht aufeinander, dadurch entsteht eine L-Form. In den nichthelikalen Abschnitten sind die meisten Basen an ungewöhnlichen Wasserstoffbrücken beteiligt. Diese Wechselwirkungen finden zwischen Basen, die nicht komplementär sind, dem Ribose-Phosphat-Rückgrat und einigen verschiedenen Abschnitten des Rückgrats selbst statt. Basen sowie zwischen 1 Einleitung 3 Die Enden des L werden von der Aminosäurenbindungsstelle (CCA-Ende) und der Anticodonschleife gebildet. Das bedeutet, dass die Aminosäure in der AminoacyltRNA weit vom Anticodon entfernt ist. (s. Abbildung 2) Abbildung 2: Skelettmodell der Hefe-Phenylalanyl-tRNA (Sussmann 1978) 1.1.2 Bildung der Aminoacyl-tRNA Die Bildung einer Peptidbindung zwischen zwei Aminosäuren ist thermodynamisch ungünstig. Die Überwindung dieser Barriere erfolgt durch die Aktivierung der Carboxylgruppe der Aminosäuren. Es handelt sich um Aminosäureester, bei denen die Carboxylgruppe einer Aminosäure mit der 2' bzw. 3'-Hydroxylgruppe der Ribose am CCA-Ende der tRNA verknüpft ist. Diese Verbindungen werden als AminoacyltRNA bezeichnet. Die Aminoacyl-tRNA’s sind auch für den Transport der Aminosäuren zum Ribosom zuständig. Da die Aminosäuren die Codons der mRNA nicht ablesen können, werden sie von spezifischen tRNA's, die die Codons der mRNA erkennen können, zu den Ribosomen transportiert. Die Aktivierung und Bindung der Aminosäuren wird von spezifischen AminoacyltRNA-Synthetasen katalysiert (Hoagland 1957). 1 Einleitung 4 Im ersten Schritt erfolgt die Bildung eines Aminoacyladenylats aus der Aminosäure und ATP, es entsteht ein Aminoacyl-AMP. Im zweiten Schritt erfolgt die Übertragung der Aminoacylgruppe des Aminoacyl-AMP auf die tRNA. Es entsteht eine AminoacyltRNA. Die Aktivierung und die Übertragung für eine bestimmte Aminosäure werden von ein und derselben Aminoacyl-tRNA-Synthetase katalysiert. Die Erkennung des richtigen tRNA-Partners erfolgt bei einigen Synthetasen vor allem durch das jeweilige Anticodon. Bei anderen tRNAs erfolgt die Identifikation nach mehreren Kriterien. Beispielsweise erkennt die Glutaminyl-tRNA-Synthetase sowohl das Anticodon als auch den Akzeptorstamm der tRNA(Gln). 1.1.3 Wobble Theorie Nach einer einfachen Hypothese müsste jede Base eines Codons eine Watson-Crick Basenpaarung mit einer komplementären Base des Anticodons eingehen, vorausgesetzt, dass ein bestimmtes Anticodon nur ein Codon erkennt. Tatsächlich erkennen einige reine tRNA's mehr als ein Codon. Zum Beispiel bindet die Hefe-Alanyl-tRNA an die drei Codons GCU, GCC und GCA. Die Codons unterscheiden sich nur in der dritten Base. Das bedeutet, dass die Erkennung der dritten Base des Codons manchmal weniger genau erfolgt als die der ersten beiden. Die Degeneration des genetischen Codes deutet auch darauf hin. XYU und XYC codieren immer dieselbe Aminosäure, während das bei XYA und XYG meistens der Fall ist. Setzt man einige sterische Freiheiten bei der Paarung der dritten Base voraus, dann erscheinen folgende Kombinationen (engl.: wobble) (Tabelle 1). plausibel, wenn die Base "wackelt" 1 Einleitung 5 erste Base dritte Base des Anticodons des Anticodons C G A U U A oder G G U oder G I U,C oder A Tabelle 1: Erlaubte Paarungen der dritten Base des Codons nach der WobbleTheorie 1.1.4 Das Ribosom Das Ribosom ist der Ort der Proteinbiosynthese. Hier erfolgt die Translation der mRNA-Sequenz in die Proteinsequenz. Ein E.coli Ribosom ist ein Ribonukleinpartikel mit einer Masse von ca. 2.700 kd, einem Durchmesser von etwa 20 nm und einem Sedimentationskoeffizienten von 70 S. Dieser kann in eine kleine (30 S) und in eine große Untereinheit (50 S) dissoziieren. Die Untereinheiten können in 55 verschiedene Proteine und drei verschiedene RNA's aufgespalten werden (s. Abbildung 3). Abbildung 3: Aufspaltung eines Ribosoms in verschiedene Proteine und RNA's (L. Stryer Biochemie) Etwa zwei Drittel der Masse eines Ribosoms wird von RNA ausgemacht. Die drei RNA's (5 S, 16 S und 23 S) sind für die Funktion und Architektur des Ribosoms 1 Einleitung wichtig. Die 6 ribosomalen RNA's haben eine steuernde Funktion bei der Proteinbiosynthese. Zum Beispiel kommt durch Spaltung einer einzigen Bindung in der 16 S-rRNA die Proteinsynthese zum Erliegen. Außerdem wird die Startstelle auf der mRNA von einer Sequenz der 16 S-rRNA ausgewählt. 1.1.5 Mechanismus der Proteinsynthese Durch 3H Markierungsexperimente konnte H. Dintzis (Dintzis 1961) zeigen, dass das Kettenwachstum bei der Peptidsynthese vom Amino- zum Carboxylende verläuft. Im Gegenzug wird die mRNA in 5’ Æ 3’-Richtung übersetzt. Die Translation läuft zeitgleich zur Transcription ab, da die mRNA in E.coli ebenfalls in 5’ Æ 3’-Richtung synthetisiert wird. Schon kurz nach der Entstehung tritt das 5’Ende der mRNA mit dem Ribosom in Wechselwirkung. Mehrere Ribosomen können ein mRNA-Molekül gleichzeitig übersetzen. Eine Gruppe von Ribosomen die ein mRNA-Molekül übersetzt, bezeichnet man als Polyribosom oder Polysom (s. Abbildung 4). Abbildung 4: Darstellung eines Polyribosoms (L. Stryer Biochemie) Die Translation beginnt meist mehr als 25 Nukleotide vom 5’-Ende der mRNA entfernt. Außerdem sind mRNA's bei Prokaryonten oft polycistronisch, das heißt sie codieren für mehr als eine Peptidkette. 1 Einleitung 7 1.2 Ortsspezifischer Einbau unnatürlicher Aminosäuren in Proteine Es sind verschiedene Methoden entwickelt worden, bei denen, unter Ausnutzung der biosynthetischen Maschinerie der Zelle, unnatürliche Aminosäuren in Proteine inkorporiert werden. Diese Methoden nutzen die Tatsache aus, dass die Erkennung von Anticodon und Codon zwischen mRNA und tRNA nahezu unabhängig von der Struktur der Aminosäure ist, die mit dem 3’-Ende des Akzeptorstamms der tRNA verknüpft ist. Diese Adaptor Hypothese (Crick 1958) konnte von Chapeville (Chapeville1962) unter Beweis gestellt werden. Die cysteinyl-tRNACys wurde durch Reduktion mit H2/Ni in alanyl-tRNAcys umgewandelt. Mit dieser tRNA konnte in vitro sehr effizient Alanin, unter Erkennung des Codons für Cystein (UGU), in Polypeptide eingebaut werden. Die chemische Behandlung von tRNAs führte zu einer ganzen Reihe von Derivaten einfacher Aminosäuren (Johnson 1976). Der Nachteil dieser Methoden liegt darin, dass die modifizierten Aminosäuren an allen Stellen eingebaut werden, die vom Anticodon der modifizierten tRNA vorgegeben sind. Außerdem erfolgt der Einbau der Aminosäurenderivate in Konkurrenz mit den natürlichen Aminosäuren. Diese Faktoren führen zu einem heterogenen Produkt. Die Konsequenz daraus war die Entwicklung von semisynthetischen Methoden, bei denen verkürzte tRNA’s (fehlender CA-Schwanz am 3’-Ende) enzymatisch mit chemisch aminoacylierten Dinukleotiden ligiert werden. Zum Beispiel entwickelten Hecht und Mitarbeiter (Heckler 1984) einen Ansatz, bei dem das Dinukleotid pCpA chemisch mit Nα-geschützten Aminosäuren acyliert wird. Anschließend erfolgt die Ligation mit einer verkürzten tRNA. Auch diese Methode hat entscheidende Nachteile. Die Ausbeute der Aminoacylierung ist unzureichend und das Entfernen der N-terminalen Schutzgruppe führt zur Hydrolyse der Aminoacylesterbindung. Eine generelle Methode zur ortsspezifischen Einführung von künstlichen Aminosäuren in Peptide wurde 1989 von Schultz und Mitarbeitern vorgestellt (Noren 1989). Dieser Ansatz nutzt die Degeneration der drei Stopp-Codons UAA, UAG und UGA (sog. Nonsense Codons) aus. Diese Codons codieren keine Aminosäuren sondern bewirken die Termination der Polypeptidsynthese durch Bindung der release Faktoren (Crick 1961, Brenner 1965). Da nur ein Stopp-Codon für die Beendung der Proteinsynthese benötigt wird, stehen zwei Codons zur Verfügung, die spezifisch für unnatürliche Aminosäuren kodieren könnten (s. Abbildung 5). 1 Einleitung 8 Abbildung 5: Biosynthetische Methode zum ortsspezifischen Einbau unnatürlicher Aminosäuren in Proteine in vitro. Uaa = unnatürliche Aminosäure (Schultz 2005) Es konnte nachgewiesen werden, dass tRNA’s, die das Stopp-Codon erkennen (amber-Suppressor tRNA’s), natürliche Aminosäuren effizient in Proteine inkorporieren (Miller 1977, Bossi 1980). Durch Experimente mit [3H]Phe und α-Hydroxysäuren wurde demonstriert, dass die gewünschte Aminosäure in vitro selektiv an der Position eingebaut wird, die von dem Codon UAG vorgegeben ist (Noren 1989, Ellman 1992a). Für eine hohe Effizienz muss die verwendete tRNA orthogonal zu den Aminoacyltranferasen des Organismus sein, aus dem das in vitro Translationssystem gewonnen wird. Das bedeutet, dass die tRNA kein Substrat der anwesenden Aminoacyltransferasen sein darf, da es sonst zum proof-reading (Deacylierung und Reacylierung mit der natürlichen Aminosäure) kommt. Daher wurde eine orthogonale amber-Suppressor tRNA aus der Hefe tRNAPhe für den Einsatz in E.coli in vitro Translationssystemen modifiziert (Ellman 1992b). 1 Einleitung 9 Die Nukleotide 34-37 der Anticodonschleife wurden durch 5’-CUAA-3’ ersetzt. Mit Hilfe der runoff Transkription steht die Suppressor tRNA in hohen Ausbeuten zur Verfügung (Milligan 1987, Noren 1990). Eine Verbesserung der Methode von Hecht (Heckler 1984) ermöglicht die effiziente Aminoacylierung orthogonaler tRNA. Dabei monoacylieren Cyanomethylester Nα-geschützter Aminosäuren selektiv die 2’,3’-Hydroxy-Gruppe des Dinukleotides pCpA. Die Schutzgruppen können von der aminoacyl-tRNA (nach der Ligation) unter milden Bedingungen abgespalten werden (Ellman 1992a). Dieses Aminoacylierungsprotokoll ist relativ unkompliziert und verspricht hohe Ausbeuten sowohl bei der Aminoacylierung als auch bei der Ligation. Nach dieser Methode wurden eine ganze Reihe von unnatürlichen Aminosäuren ortsspezifisch in Proteine eingebaut (Ellman 1992b). 1 Einleitung 10 1.3 FOF1-ATP-Synthase Die FOF1-ATP-Synthase ist an den Plasmamembranen von Bakterien, der inneren Mitochondrienmembran und den Thylakoidmembranen der Chloroplasten lokalisiert. Sie katalysiert die Synthese von ATP aus ADP und Pi bei der oxidativen bzw. Photophosphorylierung. Die dazu notwendige Energie liefert ein Protonengradient, der durch Elektronentransportprozesse aufgebaut wird. Der FOF1-ATP-Synthase Komplex besteht aus zwei oligomeren Einheiten, FO und F1 (s. Abbildung 6). Abbildung 6: Cartoonhafte Darstellung der Raumstruktur der FoF1-ATP-Synthase Der FO-Teil ist ein hydrophober membranintegraler Proteinkomplex, der den Protonenkanal bildet. Der F1-Teil ist dagegen ein hydrophiler Proteinkomplex und trägt die Bindungsstellen für die Synthese und Hydrolyse von Nukleotiden. Trennt man FO von F1, so verliert F1 die Fähigkeit ATP zu synthetisieren, da die Triebkraft des Protonengradienten fehlt. Daher bezeichnet man diesen Teil oft als F1-ATPase. 1 Einleitung 11 1.3.1 Die F1-ATPase Die F1-ATPase aus Bakterien, Mitochondrien und Chloroplasten setzt sich aus fünf verschiedenen Untereinheiten der Stöchiometrie α3β3γδε zusammen. Die drei α- und die drei β-Untereinheiten sind alternierend um eine sechszählige Pseudosymmetrieachse angeordnet (s. Abbildung 6). In der Symmetrieachse befindet sich die γ-Untereinheit, die zum Teil aus dem α3β3 Hexamer herausragt und einen Stiel bildet, der die F1- und FO-Domänen des ATP Synthase-Komplexes miteinander verbindet. Alle F1-ATPasen besitzen insgesamt sechs Nukleotidbindungsstellen. Drei Bindungsstellen sind katalytisch aktiv und werden deshalb als katalytische Bindungsstellen bezeichnet. Die Nukleotidbindungsstellen befinden sich an den Grenzflächen zwischen den α- und β-Untereinheiten. Dabei werden die katalytischen Bindungsstellen größtenteils von den β-Untereinheiten die nichtkatalytischen Bindungsstellen von den α-Untereinheiten gebildet. 1.3.2 Katalytischer Mechanismus Die F1-ATPase arbeitet nach dem von Boyer formulierten Binding Change Mechanismus (Boyer 1973). Anhand der Kristallstruktur von mitochondrialer F1 konnten Walker und Mitarbeiter diesen Mechanismus weitgehend bestätigen. Für ihre herausragenden Leistungen zur Erforschung der F1-ATPase erhielten Boyer und Walker 1997 je zur Hälfte den Nobelpreis für Chemie. Drei Merkmale charakterisieren den Binding Change Mechanismus: 1. Die drei katalytischen Bindungsstellen sind im Prinzip identisch, jedoch sind sie zu keinem Zeitpunkt der Katalyse strukturell und funktionell äquivalent. 2. Die Energie des Protonengradienten dient nicht zur Synthese von ATP, sondern zur Freisetzung von ATP aus der Bindungstasche, in der es spontan aus ADP und Pi gebildet wird. Die Bindungstasche geht dabei von der tight in die loose Konformation über. 1 Einleitung 12 3. Die drei Prozesse Produktfreisetzung Substratbindung, laufen zeitgleich an Katalyse und verschiedenen Bindungsstellen ab. Die Bindungsstellen sind um 120° versetzt angeordnet und wechseln periodisch zwischen drei verschiedenen Konformationen. Nach ihrer unterschiedlich starken Affinität für Nukleotide werden die Konformere als O (open), T (tight) oder L (loose) bezeichnet. Durch Energiezufuhr wird die Konformationsänderung induziert und somit die Affinität der Bindungsstelle für Nukleotide verändert. Das erste katalytische Zentrum beginnt in der unbesetzten O-Form, das zweite in der katalytisch inaktiven L-Form und das dritte in der T-Form, die das Substrat fest bindet und katalytisch aktiv ist. Wenn man ein Enzymmolekül betrachtet, welches ATP im T-Zentrum gebunden hat, so werden ADP und Pi an das L-Zentrum binden. Die Energie aus dem Protonenfluss wandelt das T- in ein O-Zentrum, das L- in ein T-Zentrum und das O- in ein L-Zentrum um. Diese Umwandlungen ermöglichen die Freisetzung von ATP aus dem neuen OZentrum und die Synthese von ATP aus ADP und Pi im neuen T-Zentrum. Danach kann der Zyklus von Neuem beginnen (s. Abbildung 7). Abbildung 7: Binding Change Mechanismus der FOF1ATP-Synthase (L. Stryer Biochemie) Eine Weiterführung des Modells ergab, dass die Konformationsänderungen der Bindungsstellen durch die Rotation einer asymmetrischen Gruppe von Untereinheiten, relativ zum Rest des Enzyms, verursacht werden. Durch Experimente der Arbeitsgruppe von Yoshida konnte dieses Modell bestätigt werden (Noji 1997). Es gelang eine direkte Beobachtung der Rotation. Dazu wurden fluoreszenzmarkierte Actinfilamente mit der γ-Untereinheit verknüpft und das α3β3-Hexamer mit Histidin-Tags an einer nickelbeschichteten Glasplatte immobilisiert. Unter Hydrolysebedingungen konnte die Rotation direkt unter dem Fluoreszenzmikroskop beobachtet werden (s. Abbildung 8). 1 Einleitung Abbildung 8: Das zur Beobachtung der Rotation der γ-Untereinheit der F1-ATPase benutzte System (Noji 1997) 13 1 Einleitung 14 1.4 ESR-Spektroskopie Die Elektronenspinresonanz- (ESR-)Spektroskopie ist eine „schnelle“ Messmethode, deren Zeitfenster, in dem dynamische Prozesse beobachtet werden können, bei ca. 10-11 bis 10-7 Sekunden liegt. Bei der speziellen Methode der Sättigungstransfer-ESR erreicht man sogar bis 10-3 Sekunden. Ebenso wie die Kernspinresonanz ist auch die ESR eine Absorptionsmethode, das heißt, das zu untersuchende Molekül nimmt Energie in Form von elektromagnetischer Strahlung auf. Die zu untersuchende Substanz muss dazu paramagnetische Gruppen enthalten. In der Biochemie sind im Wesentlichen drei Arten von Interesse: a) Übergangsmetalle, z. B. in Proteinen und Enzymen b) freie Radikale, z. B. als Zwischenstufe bei der lichtinduzierten Reaktion der Photosynthese sowie Coenzyme wie Flavine und Semichinone (z. B. Ubichinon und Plastochinon) c) Spinsonden, das sind stabile organische Radikale, die meist an Biomoleküle gebunden und in das zu untersuchende System inkorporiert werden. 1.4.1 Grundlagen 1.4.1.1 Die Resonanzbedingung Das Elektron besitzt einen Eigendrehimpuls, den so genannten Spin. Der Spin wird durch die Quantenzahl S = ½ charakterisiert, seine z-Komponente durch die Quantenzahl ms = + ½ oder ms = - ½ . Der Spin des Elektrons bewirkt sein magnetisches Moment µE. Dessen z-Komponente µ Ez darf nur die zwei Werte annehmen, die den Spinquantenzahlen ms = + ½ und ms = - ½ entsprechen. Die Beziehung zwischen µ Ez und ms lautet: µ Ez = -ms⋅gE⋅µB (1) 1 Einleitung 15 Dabei ist µB das Bohrsche Magneton und gE eine dimensionslose Zahl, die für ein freies Elektron 2,0023 beträgt. Der gE Wert von Elektronen in Atomen oder Molekülen kann von dem des freien Elektrons verschieden sein. Identifiziert man die Richtung der z-Komponente des magnetischen Moments mit der eines äußeren Magnetfeldes, so ist µ Ez für das Verhalten des Elektrons im Magnetfeld verantwortlich. Ohne ein äußeres Magnetfeld sind die Spinzustände ms = + ½ und ms = - ½ entartet. Wird aber ein Magnetfeld der Stärke H in z-Richtung angelegt, tritt eine Wechselwirkung zwischen diesem Feld und dem magnetischen Moment des Elektrons auf. Dadurch wird die Entartung der beiden Spinzustände aufgehoben. Diese Aufspaltung ist als Zeeman-Effekt bekannt. Für die Wechselwirkungsenergie gilt die folgende Formel: E = µ Ez ⋅H = (ms⋅gE⋅µB)⋅H (2) Der Energieunterschied zwischen den beiden bevorzugten Positionen beträgt E2 – E1 = gE⋅µB⋅H. (3) Das bedeutet, dass der Abstand der beiden Zeeman Energieniveaus von der Stärke des Magnetfeldes H abhängig ist (Abbildung 9). 1 Einleitung 16 Abbildung 9: Oben: Aufspaltung der Spinzustände eines Elektrons in einem äußeren Magnetfeld der Stärke H. Mitte: ESR-Signal bei konstanter Frequenz ν und variabler Stärke H des Feldes. Unten: Ableitung der Absorptionsintensität nach der Feldstärke H als Funktion von H. (F. Gerson, Hochauflösende ESR-Spektroskopie) Übergänge zwischen E1 und E2, bei denen das Elektron seinen Spinzustand ändert, können mit einer elektromagnetischen Strahlung der Resonanzfrequenz ν angeregt werden. Diese Frequenz muss der Resonanzbedingung hν = gE⋅µB⋅H (h = Planck’sches Wirkungsquantum) (4) gehorchen. Damit hängt die Resonanzfrequenz von der Feldstärke H ab. Der Proportionalitätsfaktor zwischen ν und H ist das so genannte gyromagnetische Verhältnis γE: γE = ν gE ⋅ µ B = H h (5) 1 Einleitung 17 1.4.1.2 Hyperfeinstruktur Ein gut aufgelöstes ESR-Signal eines Radikals in Lösung kann aus mehreren hundert Linien bestehen. Diese Aufspaltung, die man als Hyperfeinstruktur bezeichnet, ist unabhängig von der Stärke H des äußeren Magnetfeldes. Sie entsteht durch die Wechselwirkung zwischen dem ungepaarten Elektron und magnetischen Kernen im Radikal. Magnetische Kerne sind Kerne, deren Spinquantenzahl I von Null verschieden ist (1H, 14N). Die Kernspinmomente der Atome, in deren Umgebung sich das ungepaarte Elektron befindet, können selbst 2I + 1 Einstellungen im Magnetfeld einnehmen. Des Weiteren erzeugen sie ein schwaches lokales Magnetfeld, welches das äußere Magnetfeld verstärkt oder abschwächt. Das Ein-Linien-Signal des Elektrons erhält also eine Hyperfeinstruktur von 2I + 1 Linien für die Wechselwirkung mit jedem einzelnen Kern (Abbildung 10). Abbildung 10: Aufspaltung eines ESR-Signals durch Hyperfein-Wechselwirkung des Elektrons mit einem Kern der Spinquantenzahl I = ½ (links) oder I = 1 (rechts) (F. Gerson, Hochauflösende ESR-Spektroskopie) 1.4.1.3 Anisotropie Das Spektrum eines Nitroxidradikals in Lösung zeigt im Idealfall drei äquidistante Linien gleicher Intensität. Untersucht man jedoch einen Einkristall, so beobachtet man eine starke Abhängigkeit von der Richtung des Magnetfeldes relativ zur Orientierung der Nitroxidgruppe. Dieser Effekt ist in Abbildung 11 dargestellt. Deutlich erkennbar sind stark veränderte Werte der Kopplungskonstanten a und kleine Abweichungen des g-Faktors. 1 Einleitung 18 Im Falle des isotropen Spektrums gilt aiso = 1/3 (axx + ayy + azz) (6) giso = 1/3 (gxx + gyy + gzz) (7) sowie Betrachtet man keinen Einkristall, sondern eine Probe mit vielen unterschiedlich orientierten Kristallen, so erhält man ein Pulverspektrum. Dieses bildet die Summe aus den drei Extremen sowie allen statistisch gewichteten dazwischen liegenden Orientierungen. Abbildung 11: Orientierungsabhängigkeit des ESR-Spektrums eines Nitroxidradikals (H-J. Galla, Spektroskopische Methoden in der Biochemie) Obwohl auch im isotropen Spektrum statistisch orientierte Moleküle vorliegen, unterscheiden sich die Spektrenformen durch die unterschiedliche Rotationsgeschwindigkeit der Radikale stark. Anisotrope Effekte werden durch ungehinderte Rotation (τ = 1/3 (axx + ayy + azz) < 10-11 s) ausgemittelt. Einschränkungen der Rotation, die zu einer Erhöhung der Rotationskorrelationszeit τ führen, bewirken eine Verbreiterung der Linien. Dieser Effekt ist beim Hochfeldsignal am größten. Prozesse, die die Beweglichkeit einschränken, können die Kopplung an 1 Einleitung 19 ein Makromolekül oder die Erhöhung der Viskosität des Lösungsmittels sein. Der Effekt der Lösungsmittelviskosität ist in Abbildung 12 dargestellt. Abbildung 12: ESR-Spektrum eines Nitroxidradikals in Lösungen mit steigender Viskosität (H-J. Galla, Spektroskopische Methoden in der Biochemie) Im schwach immobilisierten Bereich ist eine Abnahme der Intensität der feldhöchsten Hyperfeinlinie zu beobachten. Im mittleren und stark immobilisierten Bereich kommt es zur Linienverbreiterung. Aus diesem Effekt ergibt sich die Anwendung der ESRSpektroskopie in der Biochemie. Bindet man Spin-Label an ein Makromolekül, kann man durch Ligandenbindung induzierte Konformationsänderungen untersuchen, da sich dabei der Immobilisierungsgrad des Labels und somit auch das ESR-Spektrum verändern. Die Verknüpfung des Labels mit Liganden dient hauptsächlich zur Untersuchung der Bindung dieser Substanz an ihre Rezeptoren. Durch Komplexierung des gelabelten Liganden mit dem entsprechenden Makromolekül wird das Radikal weitgehend immobilisiert. Die Rotationskorrelationszeiten liegen in der Regel nur wenig unter denen des Biopolymers. Das ESR-Spektrum zeigt dann praktisch keine Absorption im Bereich des Hochfeldpeaks des freien Labels. Die Intensitätsabnahme dieses Peaks ist ein direktes Maß für den gebundenen Anteil des Spin-Label-Liganden. Die verbliebene Absorption des Peaks liefert die absolute Konzentration des freien Liganden. 1 Einleitung 20 1.4.2 Spin-Label Stabile Nitroxide wurden 1968 von Hamilton und McConnell (Hamilton 1968) unter der Bezeichnung Spin-Label erstmals zur Verwendung als Reportergruppe beschrieben. Im Unterschied zu reaktiven Zwischenstufen sind diese Radikale langlebig und durch sterische Abschirmung, wie im klassischen Beispiel des Ditert.butyl-nitroxids, weitgehend vor chemischen Angriffen geschützt. Die Abwesenheit von α-H-Atomen verhindert auch intramolekulare Zersetzungs-reaktionen. Obwohl sie kinetisch stabilisiert sind, reagieren diese Substanzen mit einigen Reduktionsmitteln, Organometallverbindungen sowie unter sauren Bedingungen unter Verlust der Nitroxidgruppe. Trotzdem ist vor allem wegen der bahnbrechenden Arbeiten von Rozantsev (Rozantsev 1970) eine Vielzahl von Reaktionen bekannt, bei denen die paramagnetische Gruppe erhalten bleibt. Während die Piperidin- (TEMPO-) und Pyrrolinradikale als fertige Fragmente vorliegen, müssen Oxazolidin- (DOXYL-) Gruppen in der Regel am Zielmolekül aufgebaut werden. Zum Einbau von Nitroxiden wurden zahlreiche reaktive Gruppen eingesetzt, die den Spin-Label kovalent an Proteine binden können. Eine vergleichende Übersicht findet man bei Esmann et al (Esmann 1993). R R O R1 N . O TEMPO N . O Pyrrolin-SL N . R2 R2 R1 N . O O DOXYL Azethoxyl Abbildung 13: Verschiedene als Spin-Label eingesetzte Nitroxide 1 Einleitung 21 1.5 Matrix-unterstützte Laser-Desorptions/Ionisations-Massenspektrometrie (MALDI-MS) In einem Massenspektrometer werden gasförmige Ionen erzeugt und nach ihrem Masse/Ladungs-Verhältnis aufgetrennt und detektiert. Mit der Laser-Desorptions/Ionisations-Massenspektrometrie können Molekulargewichtsbestimmungen von Peptiden und Proteinen durchgeführt werden. Die MALDI-Massenspektrometrie zeichnet sich durch eine hohe Nachweis- empfindlichkeit aus. Es können Molekülmassen von bis zu mehreren kDa erfasst werden. (Hillenkamp 1991) Zur Analyse wird die Probe mit einer definierten Matrix co-kristallisiert. Die Probe wird im Hochvakuum des Massenspektrometers einem kurzwelligen Laserimpuls von wenigen Nanosekunden ausgesetzt. Die Wahl der Matrix richtet sich nach ihrem UVAbsorptionsmaximum, das in den Bereich der Wellenlänge des verwendeten Lasers fallen muss. Die Matrixmoleküle absorbieren die Energie des Lasers, die für die Desorption der Ionen notwendig ist. Hierbei relaxiert die in den Matrixmolekülen gespeicherte elektrostatische Anregungsenergie in das Gitter des Festkörpers. Dies führt zu einer Störung und Aufweitung der Gitterstruktur. Dadurch werden Matrix- und Probenmoleküle freigesetzt, die in die Gasphase übertreten und durch den Laserimpuls ionisiert werden. Die ionisierten Moleküle werden mit Hilfe eines elektrostatischen Feldes in Richtung des Analysators beschleunigt. Die Massenbestimmung erfolgt über die Erfassung der Zeit, die zwischen der Desorption der Ionen bis zum Eintreffen am Detektor vergeht. Nach der Beschleunigung der Ionen auf mehrere keV kinetische Energie durchlaufen diese eine feldfreie Driftstrecke von 0,5 – 2m Länge. Bei gleicher kinetischer Energie haben Ionen unterschiedlicher Masse damit unterschiedliche Flugzeiten. Leichte Ionen durchlaufen die Driftstrecke in kürzerer Zeit als schwere Ionen. Durch Vergleich mit bekannten Referenzmassen ergibt sich aus der gemessenen Flugzeit die Masse des Probenmoleküls. Die Methode der Flugzeitmessung wird in Anlehnung an die englische Bezeichnung als TOF (time of flight) bezeichnet. 2 Problemstellung 22 2. Problemstellung Im ersten Teil der Arbeit soll mit der amber-Suppressor-Technik, die von Schultz entwickelt wurde (Schultz 2005) eine geeignete Spin-markierte Aminosäure ortsspezifisch in ein Protein eingebaut werden. Der Einbau der Aminosäure soll in der Transmembranregion eines C-terminalen Fragments des humanen Mtj1pProteins erfolgen. Ziel dieses Ansatzes ist es zu untersuchen, ob es tatsächlich gelingt den Einbau der Spin-markierten Aminosäure mittels ESR-Spektroskopie nachzuweisen. Bei Erfolg des Experiments erwartet man ein typisches ESRSpektrum eines stark immobilisierten Spin-Labels (s. Abbildung 12). Diese Untersuchungen sollen den Weg zum mehrfachen Einbau von Spin-LabelAminosäuren in Proteine ebnen, um später Spin-Spin-Abstände in Proteinen bestimmen zu können. Mittels Photoaffinitätsmarkierug der F1-ATPase aus Rinderherzen mit dem Substratanalogon Verdau konnte [β,γ-32P]-2-N3-2’,3’-SL-ATP P. Vogel (Vogel 1992) und anschließendem zeigen, dass das für tryptischen Struktur- Funktionsuntersuchungen eingesetzte Substratanalogon trotz seiner Modifikationen an Tyr-345 der β-Untereinheit der FOF1-ATP-Synthase bindet. Damit war der Beweis erbracht, dass das Substratanalogon in der gleichen Weise wie das natürliche Substrat ATP an die F1-ATPase bindet, da sich Tyr-345 in der katalytischen Bindungstasche der β-Untereinheit befindet. In späteren Arbeiten (Burgard 2003) wurde gezeigt, dass auch die isolierte β-Untereinheit der FOF1-ATP-Synthase des thermophilen Baciilus PS3 kovalent mit dem Substratanalogon 2-N3-2’,3’-SL-ATP markiert werden kann. Allerdings konnte nicht geklärt werden, ob das Substratanalogon tatsächlich an die katalytische Bindungsstelle bindet. Im zweiten Teil dieser Arbeit soll daher überprüft werden, ob das Substratanalogon 2-N3-2’,3’-SL-ATP auch in der isolierten β-Untereinheit der FOF1-ATP-Synthase des thermophilen Bacillus PS3 an die katalytische Bindungsstelle bindet. Dazu soll die β-Untereinheit der FOF1-ATP-Synthase des thermophilen Bacillus PS3 mit dem 2 Problemstellung 23 Substratanalogon 2-N3-2’,3’-SL-ATP kovalent markiert und darauffolgend tryptisch verdaut werden. Durch die Untersuchung der entstandenen Peptidfragmente mit MALDI-TOF Massenspektrometrie soll anschließend das Peptidfragment identifiziert werden, das mit 2-N3-2’,3’-SL-ATP modifiziert wurde. Mit Datenbankanalyse kann dann geklärt werden, ob das Substratanalogon tatsächlich an das Tyrosin bindet, das sich in der katalytischen Bindungsstelle befindet. Ist dies tatsächlich der Fall, wäre der Beweis erbracht, dass die strukturellen Modifikationen des Substratanalogons auch bei der isolierten β-Untereinheit die Substratbindung nicht beeinflussen und es daher für weitergehende Untersuchungen als Reportermolekül geeignet ist. 3 Eigene Ergebnisse und Diskussion 24 3. Eigene Ergebnisse und Diskussion 3.1 Synthese einer amber Suppressor tRNA mit Spin-markierter Aminosäure Ziel dieses Teils der Arbeit war es, eine amber-Suppressor tRNA herzustellen, an deren Akzeptorstamm eine Spin-markierte Aminosäure acyliert ist. Mit dieser tRNA sollen in Peptide Spin-markierte Aminosäuren ortsspezifisch eingebaut werden. Dazu verwendet man eine messenger-RNA, die eine oder mehrere amber-Mutationen trägt. Wird das amber-Codon von der Suppressor tRNA erkannt, erfolgt statt des Abbruchs der Peptidsynthese der Einbau der Spin-markierten Aminosäure in das Polypeptid. Zur Synthese wurde folgende Strategie angewendet. Zuerst wurde das Dinukleotid dpCpA mit der Aminosäure acyliert und anschließend das modifizierte Dinukleotid mit einer verkürzten tRNA (tRNA-CA) zur vollständigen tRNA ligiert. Bei der Wahl der Spin-markierten Aminosäure muss darauf geachtet werden, dass der Spin-Label während des Syntheseprozesses nicht abgespalten wird. Damit schieden die weit verbreiteten Spin-markierten Cysteine aus, bei denen SH-reaktive Spin-Label an die Thiolgruppe des Cysteins unter Ausbildung einer Schwefelbrücke gebunden sind. Grund ist die Schwefel-Schwefel Einfachbindung, die chemisch unbeständig ist und leicht gespalten werden kann. Daher wurde eine Spin-Label Aminosäure gesucht, deren Label mit einer Kohlenstoff-Kohlenstoff Bindung an die Aminosäuren-Seitenkette gekoppelt ist. Eine Aminosäure, die diesen Anforderungen genügt, wurde freundlicherweise von Prof. K. Hideg (Universität Pécs, Ungarn) zur Verfügung gestellt. Die Aminosäure ist ein Derivat des Alanins, dessen Seitenkette mit dem Spinlabel 2,2,5,5-Tetramethylpyrrolin-nitroxid modifiziert ist. Des Weiteren ist der N-Terminus der Aminosäure mit der tBoc-Gruppe geschützt (s. Abbildung 14). Im weiteren Verlauf der Arbeit wird diese Aminosäure als HO3007 bezeichnet. 3 Eigene Ergebnisse und Diskussion 25 COOH NH C O N O O . Abbildung 14: Spin-markierte Aminosäure HO3007 Bevor die Kopplung der Aminosäure mit dem Dinukleotid erfolgen kann, muss die Aminosäure am Carboxy-Ende aktiviert werden. 3.1.1 Synthese des Cyanomethylesters der Aminosäure HO3007 Zur Aktivierung der Carboxylgruppe wurde die literaturbekannte Methode der Umsetzung mit Chloracetonitril gewählt (Schwyzer 1955). Dabei entsteht unter Freisetzung von HCl der entsprechende Cyanomethylester (s. Abbildung 15). O C COOH O CH2CN ClCH2CN NH Boc NH Boc NEt3 / -NEt3.HCl N N . . O O Abbildung 15: Schematische Darstellung der Synthese des Cyanomethylesters Dazu wird die Spin-Label Aminosäure auf Eis in Gegenwart von Triethylamin mit einem Äquivalent Chloracetonitril versetzt. Anschließend lässt man die Reaktionslösung über Nacht auftauen. Zur Aufarbeitung werden die entstandenen Triethylammoniumsalze durch Zentrifugation entfernt und mit Triethylamin extrahiert. Aus dem Lyophilisat der Triethylammoniumphasen erhält man ein Reinprodukt. Die Reinheitskontrolle erfolgt mit RP18 DC-Platten und 30%igem, wässrigem Acetonitril als Laufmittel. 3 Eigene Ergebnisse und Diskussion 26 3.1.2 Acylierung der aktivierten Spin-Label Aminosäure an das Dinukleotid pdCpA Der Reaktionsverlauf ist in Abbildung 16 schematisch dargestellt. Außerdem zeigt die Abbildung den Vorteil der Verwendung des DNA/RNA Hybrid-Dinukleotids pdCpA anstelle des reinen RNA Dinukleotids pCpA. Aufgrund des Fehlens der 2‘-OH Gruppe am Cytidylrest kann die Aminosäure nur an die 2‘-OH bzw. 3’-OH Gruppe der entsprechenden Ribose binden. Eine fehlerhafte Verknüpfung wird somit ausgeschlossen. Außerdem wird die biologische Aktivität einer tRNA, die am CCA-Ende dieses Hybrid-Dinukleotid trägt, nicht beeinträchtigt (Schultz 2005). O NH2 H N C C O NH O CH2 CN NH2 C N O O - O O P O O N O O - N NH2 . - O N O O P O O N NH2 O - N N N DMF, NEt3 O - O P N O N - O O O O P N O N O O OH OH O O H R C O C H NH O C O Abbildung 16: Schematische Darstellung des Reaktionsverlaufs zur Synthese des Spin-markierten Dinukleotids Für die Synthese löst man in der Kälte die aktivierte Spin-Label Aminosäure in trockenem, frisch destilliertem DMF und einem leichten Überschuss an Triethylamin. Dazu gibt man einen leichten Überschuss des in DMF gelösten Dinukleotids. Anschließend taut das Reaktionsgemisch über Nacht auf. Zur Aufarbeitung wird die Reaktionslösung mit Ethylacetat verdünnt und lyophilisiert. Die anschließende Reinigung erfolgt mittels Reversed Phase HPLC. Zur Identifikation Reaktionsprodukts werden alle Fraktionen ESR-spektroskopisch untersucht. Man erhält das Spin-markierte Dinukleotid (pdCpA-SLAA) als Reinsubstanz. des 3 Eigene Ergebnisse und Diskussion 27 Die Fraktion, die das Spin-markierte Dinukleotid enthält, zeigt das typische ESR-Spektrum eines leicht immobilisierten Spin-Labels mit einem Hochfeld zu Mittelfeld Verhältnis von 0,83 (s. Abbildung 17). Abbildung 17: ESR-Spektrum des Spin-markierten Dinukleotids 3.1.3 Entfernung der tBoc-Schutzgruppe am N-Terminus der Spin-Label Aminosäure Bevor das Spin-markierte Dinukleotid mit einer verkürzten tRNA ligiert werden kann, muss die tBoc-Schutzgruppe am N-Terminus der Spin-Label Aminosäure entfernt werden, da sonst nach dem Einbau dieser Aminosäure in das Protein die Proteinbiosynthese abbrechen würde. Die häufigste Methode für die Abspaltung dieser Schutzgruppe ist die Behandlung mit Trifluoressigsäure (TFA). Dazu löst man das Dinukleotid in DMF und versetzt mit wenig wasserfreier TFA. Anschließend wird das Lösungsmittel im Vakuum entfernt und der Rückstand erneut in DMF aufgenommen. Die oft beobachtete irreversible Zerstörung des Spin-Labels in der Gegenwart von TFA tritt hier nicht auf. 3 Eigene Ergebnisse und Diskussion 28 Zwar sinkt die Radikalkonzentration direkt nach der Zugabe von TFA ab, doch kann eine fast vollständige Reoxidierung des Radikals beobachtet werden, wie die nachstehenden Abbildungen eindrucksvoll beweisen. 10.000 8.000 6.000 Intensität 4.000 2.000 0 -2.000 -4.000 -6.000 -8.000 -10.000 3.400 3.420 3.440 3.460 3.480 3.500 magnetic field [G] Abbildung 18: ESR-Spektrum des Spin-markierten Dinukleotids vor der Zugabe von Trifluoressigsäure 3.000 2.000 Intensität 1.000 0 -1.000 -2.000 -3.000 3.400 3.420 3.440 3.460 3.480 3.500 magnetic field [G] Abbildung 19: ESR-Spektrum der selben Probe direkt nach der Zugabe von TFA 3 Eigene Ergebnisse und Diskussion 29 10.000 8.000 6.000 Intensität 4.000 2.000 0 -2.000 -4.000 -6.000 -8.000 3.400 3.420 3.440 3.460 3.480 3.500 magnetic field [G] Abbildung 20: ESR-Spektrum der obigen Probe 40 min nach TFA Zugabe. Wie man durch Vergleich der Abbildungen Abbildung 19 und Abbildung 20 erkennt, steigt die Radikalkonzentration nach 40 min auf etwa 90% des Ausgangswertes an. Das aminoacylierte Dinukleotid ist entschützt und ESR-aktiv. Im nächsten Schritt kann es mit einer verkürzten tRNA ligiert werden. 3.1.4 Herstellung und Reinigung der verkürzten tRNA Zur Darstellung der verkürzten tRNA wurde die Methode der runoff Transkription verwendet. Eine linearisierte DNA-Matrize wurde mit Hilfe von RNA Polymerase in die komplementäre RNA transcribiert. Als DNA-Matrize diente das Plasmid pStR1 (Graf 1998). Die cDNA des aus pUC19 gewonnenen Plasmids codiert in der multiple cloning site stromabwärts zum T7 Promotor für eine am 3’-Ende verkürzte amberSuppressor tRNA (s. Abbildung 21). Am Ende der tRNA-Sequenz befindet sich eine FokI Restriktionsschnittstelle. Die Sequenz der Suppressor tRNA entspricht, mit Ausnahme des Anticodons, der E.coli tRNAGly. 3 Eigene Ergebnisse und Diskussion 30 Das Plasmid pStR1 wurde freundlicherweise von Prof. J. Brunner von der Eidgenössischen Technischen Hochschule Zürich (Schweiz) zur Verfügung gestellt. Abbildung 21.: Plasmidkarte von pStR1 mit den wichtigsten Restriktionsschnittstellen 3.1.4.1 Linearisierung von pStR1 Für die Linearisierung wird das Plasmid im Reaktionspuffer gelöst und entsprechend der Plasmidmenge das Restriktionsenzym FokI zugegeben. Der anschließende Verdau erfolgt bei 37°C. Am Ende des Verdaus wird FokI inaktiviert. Um die Vollständigkeit des Verdaus zu garantieren, wurde ein Teil des Ansatzes auf ein Agarosegel aufgetragen und elektrophoretisch getrennt. Als Marker diente λ-DNA/HindIII. Wie aus Abbildung 21 ersichtlich ist, trägt pStR1 sechs Schnittstellen für FokI. Dadurch können sechs Fragmente mit Längen von 108, 181, 244, 287 643, und 1.309 Basenpaaren entstehen. Das aus 1.309 bp bestehende Fragment trägt die Sequenz der tRNA. Im Agarosegel findet man allerdings nur eine Bande, die dem Fragment von 1.309 bp Größe entspricht (s. Abbildung 22). Unverdautes Plasmid wurde nicht dedektiert. Die anderen Fragmente wandern aufgrund ihrer geringen 3 Eigene Ergebnisse und Diskussion 31 Größe bei der Elektrophorese so schnell, dass sie im Gel nicht mehr detektiert werden können. 1 2 3 Abbildung 22: Gelelektrophorese des mit FokI verdauten Plasmids pStR1 Bahn 1: λ-HindIII-Marker, Bahn 2: pStR1 unverdaut Bahn 3: pStR1 verdaut mit FokI 3.1.4.2 Runoff Transkription Die runoff Transkription wurde mit dem T7-MEGAshortscript Kit der Firma Ambion durchgeführt. Das linearisierte Plasmid pStR1, die vier Nukleotidtriphosphate (ATP, CTP, GTP, UTP) und die T7 RNA-Polymerase wurden nach Herstellerangaben im Reaktionspuffer vermischt und je nach eingesetzter DNA Menge für zwei bis vier Stunden bei 37°C inkubiert. Anschließend erfolgte in der Kälte die Fällung der tRNA mit Ethanol. Das weiße tRNA Pellet wurde getrocknet und in TE-Puffer aufgenommen. Die so erhaltene verkürzte Suppressor tRNA kann bis zur Verwendung bei –78°C gelagert werden. 3 Eigene Ergebnisse und Diskussion 32 3.1.5. Ligation der verkürzten Suppressor tRNA mit dem aminoacylierten Dinukleotid dpCpA Zur Ligation der verkürzten Suppressor tRNA mit dem Dinukleotid wurden die beiden Substanzen und die T4 RNA Polymerase nach Herstellerangaben im Reaktionspuffer gemischt und inkubiert. Nach der Reaktion wurde das RNA-Pellet mit Ethanol ausgefällt und in TE-Puffer resolubilisiert. Das Reaktionsprodukt ist eine vollständige aminoacylierte Suppressor tRNA die es ermöglicht, mittels amber-Suppression Aminosäure in ein Protein einzuführen. ortsspezifisch eine Spin-markierte 3 Eigene Ergebnisse und Diskussion 33 3.2 Konstruktion des Expressionsvektors pIVEX2.3MCSMtj1pN-term 3.2.1 Das Mtj1p-Protein Das Mtj1p-Protein ist das Analogon von Sec63p in Hefe (Dudek 2002). Es wechselwirkt mit dem Sec62p-Komplex und dem Binding Protein (BiP). Mtj1p kommt in Pankreas Mikrosomen in niedrigerer Konzentration als Sec63p vor, besitzt aber eine höhere Affinität zu BiP. Außer einer lumelnalen J-Domäne besitzt das Protein eine einzelne TransmembranDomäne und eine cytosolische Domäne. Der enge Kontakt der cytosolischen Domäne mit den translatierenden Ribosomen ermöglicht dem Protein die Modulation der Translation. Das Gen des Mtj1p-Proteins wurde in die multiple cloning site (MCS) des pIVEX2.4MCS Expressionsvektors (s. Abbildung 23) einkloniert und freundlicherweise von Prof. Zimmermann (Universitätsklinikum Homburg) zur Verfügung gestellt. Abbildung 23: Restriktionskarte des Plasmids pIVEX2.3MCS (Roche Applied Science) 3 Eigene Ergebnisse und Diskussion 34 3.2.2 Herstellung eines N-terminalen Mtj1p-Fragments Um eine hohe Effizienz in der in vitro Translation von Mtj1p zu erzielen, sollte ein N-terminales Fragment des Proteins erzeugt werden, welches eine amber-Mutation (TAG) anstelle von Phe113 (TAC) besitzt. Unter Verwendung der Oligonukleotidprimer amber1 und amber2 sowie des Vektors pIVEX2.3MCSMtj1p wurde mittels PCR ein PCR-Produkt erzeugt, dass N-terminal eine NdeI-Schnittstelle und C-Terminal eine XhoI-Schnittstelle besitzt. Die beiden verwendeten Oligonukleotidprimer haben die folgende Sequenz: amber1 5’- GGG AAT TTC ATA TGT GGG AGA GCG GAG ACC TG-3’ amber2 5’- CCG CTC GAG GTA GAT GGA CCA AAC CAC AGC ATA GTG ACC CAC TGT GAG AAT AAT CTA CAA GAG-3’ Das PCR-Produkt codiert für ein 137 Aminosäuren langes N-terminales Fragment des Mtj1p-Proteins. An Position 113 befindet sich anstatt der Aminosäure Phenylalanin die amber-Mutation. Nach Aufreinigung des PCR-Produkts mittels des QIAquick PCR Purification Kits erfolgte der Restriktionsverdau mit den Restriktionsenzymen NdeI und XhoI. Das entstandene 429 bp große Fragment wurde im Anschluss durch Agarosegelelektrophorese von Fremd-DNA befreit und mit Hilfe des QIAEX II Gel Extraction Kits aus dem Agarosegel isoliert. Gleichzeitig wurde der pIVEX2.3MCSVektor ebenfalls mit NdeI und XhoI verdaut, durch Agarosegelelektrophorese gereinigt und aus dem Gel extrahiert. Anschließend erfolgte die Ligation des 429 bp Oligonukleotids (Mtj1pN-term) mit dem 3534 bp großen linearisierten pIVEX2.3MCS mit T4 DNA Ligase. Das Ligationsprodukt ist der 3963 bp große Vektor pIVEX2.3MCSMtj1pN-term. Nach Inaktivierung der Ligase erfolgte die Transformation der Ligationslösung in kompetente E.coli DH5α. Der erzeugte Vektor pIVEXMtj1N-term codiert für ein 137 Aminosäuren langes N-terminales Fragment des Mtj1p-Proteins. Zusätzlich befindet sich in der Transmembranregion anstelle der Aminosäure Phe113 eine amber Mutation. Das für 3 Eigene Ergebnisse und Diskussion 35 das Mtj1p-Fragment codierende Gen wurde so in den pIVEX2.3MCS-Vektor einkloniert, dass das Transcript C-terminal einen 6-fachen Histidin-Tag (His-Tag) trägt, der eine Aufreinigung des Expressionsprodukts mittels Nickel- Affinitätschromatographie ermöglicht. Um den Erfolg der Klonierung zu verifizieren, wurde der Vektor mit den Restriktionsenzymen XbaI und PstI einem Doppelverdau unterworfen. Für beide Restriktionsenzyme liegt auf dem Vektor jeweils nur eine Schnittstelle vor. Bei richtiger Orientierung des Inserts (Mtj1pN-term) erhält man zwei Fragmente von 139 und 3.824 bp Größe. Bei falscher Orientierung des Inserts erhält man dagegen Fragmente von 350 und 3.613 bp Größe. Nach Hitzeinaktivierung der Restriktionsenzyme wurde der Verdau mittels Agarosegelelektrophorese analysiert. Das in Abbildung 24 gezeigte Agarosegel veranschaulicht den Erfolg der Ligation. In den Bahnen 1 und 2 ist die Ligation gelungen. Man erkennt eine Bande nahe 4000 bp. Diese repräsentiert das Fragment von 3824 bp Größe. Das andere zu erwartende Fragment mit einer Größe von 139 bp kann nicht detektiert werden. Die "Gesamtmasse“ der DNA dieses Fragments ist zu gering um detektiert zu werden. In den Bahnen 3 und 4 sieht man eine fehlgeschlagene Ligation des linearisierten Vektors pIVEX2.3MCS mit dem Fragment Mtj1pN-term. In diesem Fall wurde das Insert nicht in den Vektor eingebaut. Man erkennt jeweils eine Bande zwischen 3000 und 4000 bp, die dem linearisierten Vektor entspricht. Die Bande zwischen 400 und 500 bp repräsentiert das Insert. 1 2 3 4 5 Abbildung 24: Bahnen 1 und 2 gelungene Ligation, Bahnen 3 und 4 fehlgeschlagene Ligation, Bahn 5: 2-Log Ladder Marker Mit dem neu konstruierten Vektor pIVEX2.3MCSMtj1pN-term ist man in der Lage, ortsspezifisch eine künstliche Aminosäure in das N-terminale Fragment des Mtj1p- 3 Eigene Ergebnisse und Diskussion 36 Proteins einzubauen. Mit einer entsprechenden aminoacylierten amber Supressor tRNA kann eine unnatürliche Aminosäure an Position 113 des Proteinfragments eingebaut werden. Außerdem ist das Proteinfragment aufgrund seiner geringen Größe gut in in vitro Translationssystemen zu handhaben. Zur Plasmidamplifikation wurde der Verktor in kompetente E.coli DH5α transformiert und in Glycerinkultur gelagert. 3 Eigene Ergebnisse und Diskussion 37 3.3 Ortsspezifischer Einbau einer Spin-markierten Aminosäure in den N-Terminus des Mtj1p-Proteins Der Einbau der entschützten, Spin-markierten Aminosäure HO1007 (s. Abbildung 14) an Position 113 der Aminosäurensequenz des N-terminalen Fragments des Mtj1p-Proteins soll nach der Methode der amber Suppression erfolgen (s. 1.2). Für diese in vitro Methode benötigt man den Expressionsvektor pIVEX2.3MCSMtj1pN-term (s. 3.2.2.) und eine entsprechende amber-Suppressor tRNA (s. 3.1.5). Der Expressionsvektor codiert für ein N-terminales Fragment des Mtj1p Proteins und trägt an Position 113 der Aminosäurensequenz die amber-Mutation. Zwei reguläre Stopp-Codons am Ende des Gens sorgen für die Termination der Translation und für die Dissoziation des synthetisierten Peptids vom Ribosom. Die amber-Suppressor tRNA trägt am Akzeptor-Stamm die entschützte Aminosäure HO3007 und erkennt mit ihrem Anticodon das Basentriplett des amber-Codons. Zur in vitro Translation wurde das RTS 100 E.coli HY Kit der Fa. Roche Diagnostics verwendet. Mit Hilfe dieses Kits kann der N-Terminus des Mtj1p-Proteins exprimiert werden. Nach erfolgter in vitro Translation wurde der Gesamtansatz bis zur weiteren Verwendung bei -80°C gelagert. Bei erfolgreichem Verlauf des Experiments sollte an Position 113 des Proteins, mit Hilfe der amber-Suppressor tRNA, die Spin-markierte Aminosäure eingebaut werden. Wird die Spin-Label Aminosäure eingebaut, erwartet man bei der Untersuchung des Protein-Fragments im ESR-Spektrometer, wenn es in nativer Faltung vorliegt, das typische Signal eines stark immobilisierten Spin-Labels. Das vom Translationsansatz gemessene ESR-Spektrum zeigte kaum freie SpinLabel Anteile (s. Abbildung 25). Im Gegensatz dazu traten die Anteile von gebundenem Spin-Label mit einer Aufspaltung von 62,8 Gauß deutlich hervor. Der geringe Anteil von freiem Spin-Label deutet darauf hin, dass fast die gesamte amber-Suppressor tRNA während der in vitro Tranlation verbraucht wurde und dass die Suppressor tRNA nicht oder kaum deacyliert wurde. Im Fall der Deacylierung würde man im ESR-Spektrum Anteile von freiem Spin-Label beobachten, die von der abgespaltenen Spin-Label Aminosäure HO3007 stammen. Außerdem kann ein freies 3 Eigene Ergebnisse und Diskussion 38 Spin-Label Signal von ungefaltetem Protein verursacht werden, in das die Spin-Label Aminosäure inkorporiert wurde. In diesem Fall können keine höheren Proteinstrukturen die Beweglichkeit des Spin-Labels einschränken. Es können daher drei Effekte gleichzeitig für Signale von freiem Spin-Label verantwortlich sein, die hier aber nur in untergeordnetem Maße auftraten. Zu welchem Anteil jede dieser Möglichkeiten an der Entstehung des schwachen Signals des freien Spin-Label beteiligt waren, konnte anhand des ESR-Spektrums nicht geklärt werden. 25.000 20.000 15.000 10.000 5.000 0 -5.000 -10.000 -15.000 62,8 G -20.000 3.400 3.420 3.440 3.460 3.480 3.500 Magnetfeld [G] Abbildung 25: ESR-Spektrum des N-Terminus des Mtj1p-Proteins Die hohen gebundenen Anteile im ESR-Spektrum zeigen, dass die Aminosäure HO1007 tatsächlich in das Proteinfragment inkorporiert wurde und dass die SpinLabel Seitenkette der Aminosäure stark immobilisiert ist. Das bedeutet, dass das Mtj1p Fragment in nativer Faltung vorliegt und damit die Beweglichkeit des SpinLabels stark eingeschränkt ist. Die zusammenfassende Betrachtung dieser Ergebnisse zeigt, dass es gelungen ist die Spin-Label Aminosäure HO3007 ortsspezifisch an Position 113 der Aminosäurensequenz des nativ gefalteten N-terminalen Fragments des Mtj1pProteins einzubauen. 3 Eigene Ergebnisse und Diskussion 39 Damit konnte nachgewiesen werden, dass mit der amber-Suppressor Technik SpinLabel Aminosäuren ortsspezifisch in Proteine eingebaut und ESR-spektroskopisch detektiert werden können. 3 Eigene Ergebnisse und Diskussion 40 3.4 Gentechnische Gewinnung und Reinigung der β-Untereinheit der FOF1-ATP Synthase des thermophilen Bacillus PS3 Die β-Untereinheit der FOF1-ATP Synthase des thermophilen Bacillus PS3 sollte mit Hinsicht auf hohe Reinheit gentechnisch aus E.coli gewonnen werden. Das für die β-Untereinheit codierende Plasmid pUC118β wurde freundlicherweise von T. Hisabori (Chemical Resources Laboratory, Tokyo Institute of Technology, Yokohama, Japan) zur Verfügung gestellt. Die Aufzucht der mit pUC118β transformierten E.coli erfolgte mit einigen Modifikationen in Anlehnung an eine Veröffentlichung von Ohtsubo (Ohtsubo 1987). Die Aufreinigung des Zellysates fand in zwei Schritten mit Ionenaustausch- und Gelchromatographie statt. Laut SDS-PAGE erhält man die β-Untereinheit als nahezu reines Protein. Zur weiteren Kontrolle wurde die entsprechende Bande des SDS-Gels tryptisch verdaut und im MALDI-TOF Massenspektrometer analysiert. Durch Analyse des gemessenen Massenspektrums mit der MASCOT-Datenbank stellte sich heraus, dass das augenscheinlich reine Protein eine Mixtur von Tryptophanase aus E.coli und der β-Untereinheit ist. Der Treffer der Datenbank hat einen Score (Punktzahl) von 211 (s. Abbildung 26). Bei einem Score von über 67 ist das Ergebnis signifikant. Alle anderen signifikanten Treffer beinhalten nur die E.coli Tryptophanase. Abbildung 26: Trefferdiagramm der MASCOT-Datenbank für die Mixtur aus Tryptophanase und β-Untereinheit In der untersuchten Bande wurde die Tryptophanase mit einer Sequenzabdeckung von 43,9% und die β-Untereinheit mit einer Sequenzabdeckung von 37,0% gefunden. 3 Eigene Ergebnisse und Diskussion 41 Das Molekulargewicht der Tryptophanase ist mit 52.789 Da dem der β-Untereinheit (51.938 Da) sehr ähnlich. Daher verhalten sich beide Proteine im SDS-Gel gleich und erscheinen als eine Bande. Aufgrund dieses Ergebnisses wurde die Aufreinigung leicht modifiziert. Anstelle eines linearen Kochsalzgradienten bei der Ionenaustauschchromatographie wurde ein Stufengradient angelegt (s. 5.5.3). Anschließend wurde mit SDS-PAGE in zwei Elutionsbanden Protein mit einem Molekulargewicht von ca. 52.000 Da nachgewiesen. Mittels MALDI-TOF Massenspektrometrie der tryptisch verdauten Banden und anschließender Datenbankanalyse konnte verifiziert werden, dass sich in den Banden jeweils nur die Tryptophanase oder die β-Untereinheit befindet. Die Tryptophanase konnte mit einem maximalen Score von 94 und einer Sequenzabdeckung von 49,7% gefunden werden, die β-Untereinheit entsprechend mit einem maximalen Score 138 und einer Sequenzabdeckung von 41,4% (s. Abbildung 27 und Abbildung 28). Abbildung 27: Trefferdiagramm der MASCOT-Datenbank für die Tryptophanase Abbildung 28: Trefferdiagramm der MASCOT-Datenbank für die β-Untereinheit 3 Eigene Ergebnisse und Diskussion 42 Die beiden Proteine konnten durch die Modifikation bei der Durchführung der Ionenaustauschchromatographie eindeutig separiert werden. Nach der gelchromatographischen Reinigung der β-Untereinheit konnte die Reinheit nochmals mit SDS-PAGE und MALDI-TOF Massenspektrometrie bestätigt werden. Bis auf nicht signifikante Verunreinigungen im höher- und niedermolekularen Bereich ist das Protein sauber (s. Abbildung 38 im experimentellen Teil). Das Protein wurde bis zur weiteren Verwendung bei -80°C in Elutionspuffer gelagert. 3 Eigene Ergebnisse und Diskussion 43 3.5 ESR-Untersuchungen der β-Untereinheit der FOF1-ATP-Synthase des thermophilen Bacillus PS3 mit 2-N3-2’,3’-SL-ATP NH2 N O O O -O P O P O P O OOO- N N H N3 N O H H H OH O O N O Abbildung 29: 2-N3-2’,3’-SL-ATP Das ESR-Spektrum der β-Untereinheit nach Inkubation mit dem Substratanalogon 2-N3-SL-ATP (s. Abbildung 29) in magnesiumhaltigem Puffer (SHPMg) ist in Abbildung 30 zu sehen. Neben den Signalen für freie Spin-Label Radikale erkennt man im Hoch- und Tieffeldbereich zwei zusätzliche Signale mit einer Aufspaltung von 52 G (s. Pfeile in Abbildung 30). Dieses Signalpaar resultiert aus hochimmobilisierten, enzymgebundenen Spin-Label Radikalen. Die beobachtete Aufspaltung entspricht praktisch den von S. Burgard bestimmten Werten von 53 – 54 G (Burgard 1995). Das Substratanalogon bindet an eine Bindungsstelle der β-Untereinheit und kann anschließend durch Bestrahlung mit UV-Licht kovalent mit dem Protein verknüpft werden. 3 Eigene Ergebnisse und Diskussion 44 8.000 6.000 4.000 2.000 0 -2.000 -4.000 -6.000 3.400 3.420 3.440 3.460 3.480 3.500 Magnetfeld [G] Abbildung 30: ESR-Spektrum der β-Untereinheit in magnesiumhaltigem Puffer inkubiert mit 2-N3-2’,3’-SL-ATP 3 Eigene Ergebnisse und Diskussion 45 3.5 Massenspektrometrische Untersuchungen an der β-Untereinheit der FOF1-ATP-Synthase des thermophilen Bacillus PS3 Unter UV-Bestrahlung kann das Substratanalogon 2-N3-2’,3’-SL-ATP mit der Seitenkette der Aminosäure massenspektrometrische Tyrosin Untersuchungen stabile soll Addukte überprüft bilden. werden, Durch ob das Substratanalogon der FOF1-ATP-Synthase kovalent an Tyr-341 in der katalytischen Bindungsstelle der isolierten β-Untereinheit des thermophilen Bacillus PS3 bindet. Anhand der Ergebnisse soll bewiesen werden, dass das Substratanalogon in der gleichen Weise an die β-Untereinheit bindet wie in der vollständigen F1-ATPase. Aufbauend auf die Ergebnisse von P. Vogel (Vogel 1992) und S. Burgard (Burgard 2003) könnte so gezeigt werden, dass die Modifikationen des Substratanalogons 2-N3-2’,3’-SL-ATP im Vergleich zum natürlichen Substrat ATP auch in der isolierten β-Untereinheit der F1-ATPase keinen Einfluss auf das Bindungsverhalten haben. Nach Inkubation der β-Untereinheit mit 2-N3-2’,3’-SL-ATP in magnesiumhaltigem Puffer wird der Enzymsubstratkomplex mit UV-Licht bestrahlt, um gebundenes Substrat kovalent mit dem Enzym zu verknüpfen. Nach Aufreinigung des Proteins mittels SDS-PAGE erfolgte ein Verdau mit Trypsin. Die auf diese Weise erhaltenen Peptidfragmente wurden im MALDI-TOF-Massenspektrometer vermessen und mittels Datenbankanalyse identifiziert. Intens. [a.u.] 3 Eigene Ergebnisse und Diskussion 46 x10 4 1471.629 2.5 2140.082 2268.995 1921.883 1781.797 2.0 2307.224 1605.636 1.5 1262.442 1.0 63.216 0.5 2018.971 1000.441 481.493 2751.444 1130.538 710.204 2433.224 2968.504 103.314 0.0 500 1000 1500 2000 2500 3000 m/z Abbildung 31: Typisches Massenspektrum der tryptisch verdauten β-Untereinheit Bei erfolgreichem kovalenten Einbau des Substratanalogons sollten Anteile des Peptidfragmentes, das Tyr-341 beinhaltet, zu höheren Massen verschoben sein. Ein entsprechendes Massenspektrum zeigt Abbildung 31. Durch Analyse der Fragmente mit der MASCOT-Datenbank konnte die β-Untereinheit mit einem Score von 81 (s. Abbildung 32) und einer Sequenzabdeckung von 78,9 % gefunden werden. Abbildung 32: Trefferdiagramm der MASCOT Datenbank für die kovalent modifizierte β-Untereinheit 3 Eigene Ergebnisse und Diskussion 47 Ebenfalls wurde ein Fragment identifiziert, welches β-Tyr341 als einziges Tyrosin beinhaltet. Dieses Fragment hat ein Molekulargewicht von 1.890,85 und tritt mit einer relativen Intensität von 21,8 % auf. Ausgehend davon wurde nach dem entsprechenden modifizierten Peptid gesucht. Eine Markierung mit dem ATP-Derivat konnte nicht gefunden werden. Allerdings kann ATP unter den Bedingungen im Massenspektrometer Phosphatgruppen abspalten. Einen ersten Hinweis darauf lieferte in diesem Fall die Software Bio Tools der Fa. Bruker, die nach dem Einlesen von Massenspektren, unabhängig vom eingestellten Detektionsbereich, nach abgespaltenen Phosphatgruppen sucht. Wurden Phosphatgruppen detektiert, erscheint auf dem Bildschirm ein kleines Ausgabefenster (s. Pfeil in Abbildung 33). Dabei spielt es keine Rolle, dass wie hier der Detektionsbereich von m/z = 500 bis m/z = 3.500 eingestellt ist. Die Software analysiert auch den Bereich m/z < 500, obwohl in der Steuersoftware des Spektrometers die Option Matrix-Suppression aktiviert wurde. Das bedeutet, dass der Messbereich m/z < 400 abgeschnitten wird. Der Nachweis von abgespaltenen Phosphatgruppen ist eindeutig. Eine Kontaminaton der Probe kann ausgeschlossen werden, da jede Probe vor der Applikation auf ein MALDI-Target mit ZipTip®-Pipettenspitzen entsalzt wurde. 3 Eigene Ergebnisse und Diskussion 48 Abbildung 33: Ausgabefenster der Software Bio Tools Den Ergebnissen der Software Bio Tools entsprechend konnte die Modifizierung mit dem ADP-Derivat erfolgreich nachgewiesen werden. Gemäß eines berechneten Molekulargewichts von 2.497,25 wurde experimentell ein schwacher Peak mit einem Molekulargewicht von 2.497,252 gefunden (s. Abbildung 34). Damit konnte eindeutig bestätigt werden, dass 2-N3-2’,3’-SL-ATP in das Protein eingebaut wird und dass eine Phosphatgruppe des Substratanalogons abgespalten wird. 3 Eigene Ergebnisse und Diskussion 49 Intensität [au] 120000 60000 m / z = 2497,252 0 2300 2350 2400 2450 m/z Abbildung 34: Ausschnitt aus dem Massenspektrum 2500 3 Eigene Ergebnisse und Diskussion 50 Einen weiteren Beweis für den kovalenten Einbau konnte durch das Massespektrum des unverdauten Enzyms erbracht werden (s. Abbildung 35). 7500 β-Untereinheit nicht modifiziert β-Untereinheit modifiziert 7000 6500 6000 5500 5000 4500 4000 3500 3000 2500 2000 1500 1000 500 0 20000 40000 60000 80000 100000 120000 140000 m/z Abbildung 35: Massenspektren der unverdauten modifizierten und nicht modifizierten β-Untereinheit Im Vergleich zum unmodifizierten Protein weist das modifizierte Protein ein deutliches Peak-tailing zu höheren Massen auf (s. Abbildung 35). Dies ist ein Zeichen kovalenter Bindung niedermolekularer Substanzen an das Protein. Die Proben wurden vor der Messung mit ZipTip®-Pipettenspitzen entsalzt, da Ionen wie Na+ ebenfalls Peak-tailing verursachen. Anhand dieser Ergebnisse konnte eindeutig nachgewiesen werden, dass das Substratanalogon kovalent an Tyr-341 der β-Untereinheit bindet. Das bedeutet, dass das Substratanalogon auch in der isolierten β-Untereinheit an die katalytische Bindungsstelle bindet. 4 Zusammenfassung 51 4 Zusammenfassung Im ersten Teil dieser Arbeit sollte untersucht werden, ob Spin-Label Aminosäuren mit der Methode der amber-Suppression in Proteine inkorporiert werden können. Des Weiteren sollte der Einbau ESR-spektroskopisch bewiesen werden. Dazu wurde eine neue amber-Suppresor tRNA synthetisiert. Diese tRNA leitet sich von einer Hefe tRNA ab und ist somit orthogonal zu E.coli Aminoacylsynthetasen. Am Akzeptorstamm trägt die tRNA die entschützte Spin-Label-Aminosäure HO3007. Mit ihrem Anticodon erkennt die tRNA das amber Codon. Weiterhin wurde in den in vitro Expressionsvektor pIVEX2.3MCS das Gen für ein 137 Aminosäuren langes N-terminales Fragment des humanen Mtj1p-Proteins einkloniert. Das Gen wurde so mutiert, dass sich an der Position des Codons für die Aminosäure Phe113 (TAC) die amber-Mutation (TAG) befindet. Der neu synthetisierte Vektor trägt die Bezeichnung pIVEX2.3MCSMtj1pN-term. Der Vektor und die amber-Suppressor tRNA wurden in einem E.coli in vitro Translationssystem eingesetzt. Durch ESR-spektroskopische Untersuchung des Reaktionsansatzes konnte nachgewiesen werden, dass fast die gesamte eingesetzte tRNA verbraucht wurde. Außerdem kann anhand des hohen gebundenen Anteils im ESR-Spektrum darauf geschlossen werden, dass die Spin-Label Aminosäure in das nativ gefaltete Nterminale Fragment des Mtj1p-Proteins eingebaut wurde. Die Methode der amber-Suppression konnte erfolgreich auf die Spin-Label Aminosäure HO3007 übertragen werden. In Versuchen, die späteren Arbeiten vorbehalten bleiben, kann untersucht werden, ob durch den ortsspezifischen Einbau von zwei Spin-Label Aminosäuren Spin-Spin Abstände in nativ gefalteten Proteinen gemessen werden können. Durch geeignete Variation der Abstände der Spin-Label Aminosäuren in der Aminosäurensequenz könnten die verschiedenen gemessenen Spin-Spin Abstände zur Strukturaufklärung beitragen. So werden in α-helicalen Strukturen die Spin Abstände kleiner sein als in ungefalteten Aminosäureketten. Auf diese Weise könnte man zum Beispieldirekt während der Translation von 4 Zusammenfassung 52 sekretorischen Proteien nachweisen, ob bereits während der Translation die Faltung der Proteine erfolgt. Im zweiten Teil der vorliegenden Arbeit sollte nachgewiesen werden, dass das Substratanalogons 2-N3-2’,3’-SL-ATP auch in der isolierten β-Untereinheit der FOF1-ATP-Synthase des thermophilen Bacillus PS3 an die katalytische Bindungsstelle bindet und somit für weiteregehende Untersuchungen von StrukturFunktionsbeziehunhen als Reportermolekül geeignet ist. Bereits P. Vogel konnte zeigen, dass sich das Bindungsverhalten des Substratanalogons für die F1-ATPase aus Rinderherzen trotz der strukturellen Modifikationen nicht von dem des natürlichen Substrats ATP unterscheidet (Vogel 1992). Die F1-ATPase aus Rinderherz wurde dazu mit dem Substratanalogon [β,γ-32P]2-N3-2’,3’-SL-ATP kovalent markiert, tryptisch verdaut und die Peptidfragmente mit Reversed Phase HPLC identifiziert. Später zeigte S. Burgard (Burgard 2003), dass auch die isolierte β-Untereinheit des thermophilen Bacillus PS3 kovalent mit 2-N3-2’,3’-SL-ATP markiert werden kann. In der vorliegenden Arbeit wurde überprüft ob das Substratanalogon tatsächlich an die katalytische Bindungsstelle der isolierten β-Untereinheit der FOF1-ATP-Synthase des thermophilen Bacillus PS3 bindet. Dazu wurde die in der Literatur beschriebene Methode zur gentechnischen Gewinnung der β-Untereinheit vereinfacht und die Reinheit des Proteins mittels MALDI-TOF Massenspektrometrie und Datenbankanalyse bestätigt. Die isolierte Untereinheit wurde anschließend mit dem Substratanalogon 2-N3-2’,3’-SL-ATP inkubiert. Das danach gemessene ESR-Spektrum des EnzymSubstratkomplexes entspricht dem für die F1-ATPase (Burgard 1995). Nach der kovalenten Markierung der β-Untereinheit mit dem Substratanalogon wurde das Protein tryptisch verdaut. Die entstandenen Peptidfragmente wurden anschließend mittels MALDI-TOF Massenspektrometrie analysiert. Mit Hilfe der MASCOTDatenbank konnte die β-Untereinheit mit einer Sequenzabdeckung von 78,9 % identifiziert werden. Außerdem wurde ein Peptid gefunden, das Tyr 341 beinhaltet. Diese Aminosäure befindet sich in der katalytischen Bindungstasche der βUntereinheit. Das entsprechende Fragment hat ein Molekulargewicht von 1.890,85 und tritt mit einer relativen Intensität von 21,8% auf. Ausgehend von diesem Peptid konnte das entsprechende mit dem ATP-Analogon markierte Peptid nicht gefunden 4 Zusammenfassung 53 werden. Die Software BioTools der Fa. Bruker lieferte die Begründung. Bei der Auswertung der Massenspektren detektierte die Software Phosphorylierung, also abgespaltene Phosphatgruppen. Modifizierung mit dem Dieser ADP-Derivat Aussage entsprechend nachgewiesen werden. konnte Gemäß die eines berechneten Molekulargewichts von 2.497,25 konnte experimentell ein Peak mit einem Molekulargewicht von 2.497,252 gefunden werden. Das Substratanalogon hat zweifelsfrei kovalent an Tyr 341 der katalytischen Bindungsstelle gebunden. Einen weiteren Hinweis für die erfolgreiche kovalente Modifizierung der β-Untereinheit mit 2-N3-2’,3’-SL-ATP lieferte die Untersuchung des unverdauten Proteins im MALDI-TOF Massenspektrometer. Im Vergleich zum unmodifizierten Protein weist der Peak der modifizierten β-Untereinheit ein deutliches Peak-tailing zu höheren Massen auf. Da die Proben vor der Messung entsalzt wurden, spricht diese Beobachtung deutlich für die kovalente Modifizierung des Proteins. Die Ergebnisse dieses Teils der Arbeit beweisen, dass das Substratanalogon 2-N3-2’,3’-SL-ATP auch im Fall der isolierten β-Untereinheit an die katalytische Bindungsstelle bindet. Aufbauend auf die Arbeiten von P. Vogel (Vogel 1992) und S. Burgard (Burgard 2003) bedeutet das, dass die strukturellen Modifikationen des Substratanalogons im Vergleich zum natürlichen Substrat ATP auch in der isolierten β-Untereinheit keinen Einfluss auf das Bindungsverhalten an die katalytische Bindungsstelle haben. Das Substratanalogon 2-N3-2’,3’-SL-ATP kann daher für weitergehende Untersuchungen Reportermolekül verwendet werden. von Struktur-Funktionsbeziehungen als 5 Material und Methoden 54 5 Material und Methoden 5.1 Material 5.1.1 E.coli Stämme Stamm DH5α JM109 Genotyp Quelle F- (Φ80d∆(lacZ)M15) gyrA96 (Nalr) endA1 recA1 relA1 λ-thi-1 deoR glnV44 hsdR17 (rk-, mk-) ∆(lacZYA-argF)U169 F- traD36 proA+B+ lac/q ∆(lacZ)M15/ e14- (McrA-) ∆(lac-proAB) gyrA96 (Nalr) endA1 recA1 relA1 λthi-1 glnV44 hsdR17 (rk-, mk-) Woodcock et al. 1989 Yanisch-Perron et al, 1995. 5.1.2 Plasmide pStR1 pUCβ pIVEX2.3MCSMtj1p 5.1.3 Enzyme und Marker 2-Log DNA Ladder New England BioLabs Alkaline Phosphatase (CIP) New England BioLabs DpnI New England BioLabs FokI New England BioLabs λ-DNA/HindIII-Marker MBI-Fermentas Nde I New England BioLabs Pfu Turbo DNA Polymerase Stratagene 5 Material und Methoden 55 PstI New England BioLabs Roti®-Mark STANDARD Roth T4 DNA-Ligase New England BioLabs T4 RNA-Ligase New England BioLabs Trypsin Gold Promega XbaI New England BioLabs XhoI New England BioLabs 5.1.4 Medien Agar ICN Biochemicals Bacto-Trypton (-Pepton) Difco Hefeextrakt Fluka 5.1.5 Chemikalien Acrylamidlösung Roth Agarose Roth Ampicillin Sigma Borsäure MERCK Bromphenolblau Sigma Butanol Riedel-de Haen BSA Roche Diagnostics Chloracetonitril Fluka Coomassie Brilliant Blue R 250 Serva α-Cyno-4-hydroxyzimtsäure Sigma DEAE Sephacel Serva dpCpA Photo Probes N,N-Dimethylformamid MERCK EDTA Serva Ethanol Roth Ethidiumbromid USB Fractogel TSK HW-40 (S) MERCK Glycerin Caldic Deutschland 5 Material und Methoden 56 Glycin Serva IPTG Roth β-Mercaptoethanol Fluka Natriumazid MERCK Natriumchlorid MERCK Pefabloc SC (AEBSF) Roche Rubidiumchlorid Roche Schwefelsäure MERCK SDS Roche Sinapinsäure Sigma TEMED Sigma Tetrabutylammoniumacetat Fluka Trifluoressigsäure Riedel-de Haen Tris MERCK Alle nicht aufgeführten Chemikalien waren im Arbeitskreis vorhanden oder wurden über die Chemikalienausgabe bezogen. 5.1.6 Sonstige Materialien QIAEXII Gel Extraction Kit Qiagen QIAprep Spin Miniprep Kit Qiagen QIAquick PCR Purification Kit Qiagen RTS 100 E.coli HY Kit Roche Diagnostics Starter Kit for MALDI-TOF-MS Bruker Daltonics T7 - MEGAshortscript Kit Ambion 5 Material und Methoden 57 5.1.7 Geräte Gerät Bezeichnung Hersteller Autoklav Dampfkochtopf Sitram Brutschrank S40 Ü Memmert Elektrophoresezubehör Mini Protean 3 Kammern BIO-RAD Lichtbank REX Sofortbildkamera DS 34 Polaroid Fraktionssammler 2111 MULTIRAC LKB BROMMA Gefriertrocknung LYOVAC GT 2 Leybold Heraeus Geltrockner 2003 Slab Gel Dryer LKB HPLC System Gold Beckman HPLC-Säulen Supersphere 100 RP18 GROM PARTISIL 10 SAX Whatman innova 4000 New Brunswick Scientific AQUATRON INFORS Amicon Ultra (10000 MWCO) Amicon Inkubatoren Konzentratoren VIVASPIN 2 30,000 MWCO RC VIVASCIENCE Küvetten Plastibrand Brand Suprasil Hellma Kryostat UKT Edmund Bühler Leistungssteller voltron messner emtronic MALDI-Target MTP Anchor Chip var/384 Bruker Daltonics Massenspektrometer ultraflex II TOF/TOF Bruker Daltonics Mixer Mixer 5432 Eppendorf pH-Meter pH 521 WTW Spektralphotometer DU-640 Beckman Rotoren JA-10, JA-14, JA-20 Beckman Schreiber 2210 LKB BROMMA Spannungsgeräte Power Pac 3000 BIO-RAD DA 150/0,5 GB zentro-elektrik Thermocycler PCR Sprint Thermo Hybaid Thermostat ULTRATEMP 2000 F30 julabo 5 Material und Methoden 58 Ultraschall-Zelldisruptor SONIFIER B-12 BRANSON UV-Monitor 2138 UVICORD S LKB BROMMA UV-Transilluminator TFX-20M Waagen Zentrifugen Labortechnik Fröbel Feinwaage 1204 M Sartorius 1601 MP8-1 Sartorius 5415 C Eppendorf J2-21 Beckman UNIVERSAL Hettich 5.2 Aufzucht und Lagerung von E.coli 5.2.1 Autoklavieren Die Flüssigmedien werden in einem Dampfkochtopf bei 121°C und 1,5 bar für zwanzig Minuten autoklaviert. 5.2.2 Flüssigmedien 2xYT-Medium: 16 g Bacto-Trypton 10 g Hefeextrakt 5 g NaCl pH 7,5 mit 1 M NaOH; ad 1000 ml mit H2Obidest LB-Medium: 10 g Bacto-Trypton 5 g Hefeextrakt 10 g NaCl pH 7,5 mit 1 M NaOH; ad 1000 ml mit H2Obidest 5 Material und Methoden 59 Ampicillin, IPTG, Pefabloc SC Ampicillin-Stocklösung: 50 mg/ml in H2Obidest IPTG-Stocklösung: 1 mol/l in H2Obidest Pefablock-Stocklösung 80 mg/ml in H2Obidest Die Stocklösungen werden durch einen Membranfilter mit 0,45 µm Porenweite steril filtriert und bei –20°C gelagert. Da Ampicillin hitzeempfindlich ist, darf es dem Medium erst zugegeben werden, wenn dessen Temperatur unter 50°C gesunken ist. Die Konzentration des Ampicillins im Flüssigmedium beträgt üblicherweise 150 µg/ml. 5.2.3 Festmedien Zum Anfertigen der Agarplatten wird dem Flüssigmedium 1,5 % Agar zugegeben. Anschließend wird autoklaviert und nach dem Abkühlen auf unter 50 °C Ampicillinlösung bis zur Endkonzentration von 50 µg/ml zugesetzt. 5.2.4 Kulturbedingungen 5.2.4.1 Festmedien Aus einer Glycerinkultur (Kap. 5.4.2.3) wird ein Verdünnungsausstrich auf einer Agarplatte angefertigt und über Nacht bei 37°C im Brutschrank kultiviert. Beimpfte Platten können eine Woche bei 4°C, unbeimpfte Platten maximal zwei Monate bei 4°C gelagert werden. 5.2.4.2 Flüssigmedium Es wird eine einzelne Kolonie von einer frischen plattierten Agar-Platte in 15 ml des gewünschten Mediums überführt. Die Inkubation der Kulturen erfolgt über Nacht bei 37°C und 200 bis 300 rpm im Luft- oder Wasserbadschüttler. Ausgehend von dieser Übernachtkultur kann eine größere Kultur angeimpft werden. 5 Material und Methoden 60 5.2.4.3 Glycerinkulturen 500 bis 750 µl einer Übernachtkultur, die sich noch im exponentiellen Wachstum befindet, werden in einem sterilen Kryoröhrchen mit demselben Volumen 80%igem sterilen Glycerin vermischt und im Aceton/Trockeneis-Bad schockgefroren. Die Glycerinkulturen werden bei –80°C gelagert. Jede Glycerinkultur wird höchstens zweimal für einen Verdünnungsausstrich verwendet. Zur Kultivierung von Einzelkolonien wird mit einer sterilen Impföse ein kleiner Teil von der Oberfläche der tiefgefrorenen Glycerinkultur abgeschabt und ausgestrichen. 5.2.5 Wachstumsmessungen Die Bestimmung der Bakterienzahl erfolgt indirekt durch eine Trübungsmessung der Bakteriensuspension. Das Messprinzip beruht auf der teilweisen Streuung sichtbaren Lichts beim Durchtritt durch die Bakteriensuspension. Die Streuung hängt von der Form, Größe und Zahl der Bakterien ab (Süßmut et al., 1987). Die Lichtschwächung wird durch einen Vergleich mit unbeimpftem Medium (Nullwert, Blank) bei 600 nm bestimmt, so lässt sich als Maß für das Bakterienwachstum die optische Dichte (OD) einführen. Bei einer OD über 0,3 muss die Kultur vor der Messung verdünnt werden, da nur bis zu diesem Wert eine lineare Abhängigkeit zwischen der Trübung der Flüssigkultur und der Zellzahl besteht. 5.3 Allgemeine Arbeitsmethoden 5.3.1 Herstellung kompetenter Zellen Um die Transformation eines Plasmids in eine E.coli-Zelle zu ermöglichen, benötigt man kompetente Zellen. Dabei ist eine sehr hohe Transformationseffizienz erwünscht, welche man in Gegenwart von Rubidiumchlorid (Hanahan, 1983) erreicht. Je nach verwendetem Plasmid bzw. E.coli Stamm können mit dieser Art kompetenter Zellen über 107 Transformanden pro µg eingesetzter DNA erhalten werden. 5 Material und Methoden 61 5.3.2 Transformation Die kompetenten Zellen werden auf Eis aufgetaut und mit der gewünschten Menge an Plasmid-DNA (1-2 µl, ca. 10ng) versetzt. Anschließend wird eine Stunde auf Eis inkubiert. Nach der Inkubation werden die Zellen für 3 Minuten bei 42°C einem Hitzeschock ausgesetzt und danach sofort auf Eis gestellt. Die Bakteriensuspension wird dann in ein steriles Reagenzglas mit 1 ml LB-Medium überführt und für eine Stunde bei 37°C und 150 rpm im Luftschüttler inkubiert. Von der Suspension werden je 100 µl mit einem Trigalski-Spatel auf Agarplatten ausgestrichen, die das entsprechende Antibiotikum enthalten und über Nacht im Brutschrank bei 37°C inkubiert. 5.3.3 Plasmid-Minipräparation Die Isolierung von Plasmid DNA erfolgt mit dem kommerziellen System QIAprep Spin Miniprep der Fa. Qiagen. Das Verfahren basiert auf der Bindung von PlasmidDNA an eine Silica-Matrix. Die Isolierung erfolgt aus 3 ml einer Übernachtkultur von E.coli-Zellen nach dem Protokoll des Herstellers. Die isolierte und gereinigte DNA wird mit 50 µl EB-Puffer (1 mM Tris⋅HCl pH 8,5) oder H2Obidest von der Säule eluiert. 5.3.4 Restriktionsverdau Für den Restriktionsverdau von DNA werden die Herstellervorgaben befolgt und die mitgelieferten 10x Reaktionspuffer verwendet. Der Ansatz variiert abhängig vom Volumen und der Konzentration der verwendeten DNA-Lösung. Ein Standardansatz setzt sich wie folgt zusammen: 2 µl 10x Puffer 2-3 µl DNA 1 µl Enzym (5-10 U/µl) ad 20 µl H2Obidest Der Verdau wird bei der empfohlenen Temperatur (i. A. 37°C) durchgeführt. Die anschließende Inaktivierung Herstellerangaben. der Restriktionsenzyme erfolgt nach den 5 Material und Methoden 62 5.3.5 Agarose-Gelelektrophorese Das wichtigste Verfahren zur Auftrennung und Identifikation von DNA-Molekülen ist die Elektrophorese in Agarosegelen. Als Trennmedium dient eine Agarose-Matrix. Die Geschwindigkeit mit der sich DNA im elektrischen Feld auf die Anode zubewegt, ist von der Struktur, Größe und Mobilität der DNA abhängig. So bewegt sich lineare, doppelsträngige DNA mit einer Geschwindigkeit, die umgekehrt proportional zum Logarithmus ihres Molekulargewichts ist, durch das Agarosegel. Die Mobilität der DNA ist von der Konzentration der Agarose abhängig. Im Allgemeinen verwendet man 0,9%-ige Agarosegele. Durchführung: Elektrophoresepuffer (TBE-Puffer) Loading Dye 0,089 M Tris 0,25% Bromphenolblau 0,089 M H3BO3 0,25% Xylenblau 0,002 M EDTA 30% Glycerin in H2Obidest pH 8,3 mit H3BO3 50 mM EDTA Die entsprechende Menge Agarose wird in 70 ml TBE-Puffer aufgeschmolzen. Nach dem Abkühlen auf 50°C erfolgt die Zugabe von 3,5 ml Ethidiumbromid-Stammlösung (10 mg/ml). Die Lösung wird in eine entsprechende Form gegossen und mit einem Kamm, der die Probentaschen ausbildet, versehen. Nach dem Erstarren des Agarosegels wird der Kamm entfernt und das Gel in der Elektrophoresekammer positioniert. Die vorbereiteten DNA-Proben werden in die Geltaschen pipettiert. Anschließend wird die Elektrophorese bei konstant 100 V für 1,5 bis 2 Stunden durchgeführt. Die Detektion der DNA erfolgt mit Hilfe des Ethidiumbromids, welches in dopplesträngige DNA-Moleküle intercaliert und somit die DNA unter UV-Licht sichtbar macht. 5 Material und Methoden 63 Probenvorbereitung: Unverdaute Plasmide und DNA-Marker Lösungen werden mit TE-Puffer auf 20 µl aufgefüllt und mit 7µl Loading Dye versetzt. Zu den PCR-Produkten und den verdauten DNA-Ansätzen werden direkt 7 µl Loading Dye zugegeben. Nach fünfminütiger Inkubation bei 65°C werden die Proben bis zum Beladen des Gels auf Eis gelagert. 5.3.6 DNA-Größenstandards Zur Abschätzung der Größe der verdauten und unverdauten DNA-Fragmente werden λ-DNA/HindIII-Marker sowie 2-Log DNA Ladder Marker benutzt. Die Größe der Fragmente ist in Basenpaaren (bp) angegeben λ-DNA/HindIII 23.130, 9.416, 6.557, 4.361, 2.322, 2.027, 564, 125 2-Log DNA Ladder 10.000, 8.000, 6.000, 5.000, 4.000, 3.000, 2.000, 1.500, 1.200, 1.000, 900, 800, 600, 500, 400, 300, 200, 100 5.3.7 Konzentrationsbestimmung von Plasmid-DNA Die DNA besitzt bei 260 nm ein Absorptionsmaximum. Aufgrund dessen kann die Konzentration von Plasmidlösungen unter Verwendung der folgenden Gleichung bestimmt werden (Sambrook et al., 1989): 1 OD260 = 50 µg/ml ds DNA Die Messungen erfolgen in Quartzküvetten mit einer Schichtdicke von 1 cm. Das Absorptionsspektrum wird im Bereich von 230 – 350 nm aufgenommen. 5 Material und Methoden 64 5.3.8 Isolierung von DNA aus Agarosegelen Für die Isolierung von DNA-Fragmenten aus Agarosegelen wurde das QIAEX®II Gel Extraction Kit der Fa. Qiagen benutzt. Die Durchführung erfolgte nach dem Protokoll des Herstellers. 5.3.9 Reinigung von PCR-Produkten Die Reinigung von PCR-Produkten erfolgte mit dem QIAquick PCR Purification Kit der Fa. Qiagen nach dem Protokoll des Herstellers. Die Elution der DNA erfolgte mit 50 µl sterilem Wasser. 5.3.10 Ligation von DNA-Fragmenten Die Ligation von zwei DNA-Fragmenten erfolgte ausschließlich mit der T4-DNALigase, einem Enzym, das die Bildung einer Phosphordiester-Bindung zwischen der 5’-Phosphat- und der 3’-Hydroxylgruppe in doppelsträngiger DNA katalysiert. Die Ligase besitzt die Fähigkeit, sowohl sticky-ends als auch blunt-ends zu ligieren. Ein typischer Ligationsansatz bestand aus den zu ligierenden Fragmenten, die aus Agarosegelen isoliert wurden (s. 5.3.8). Die DNA-Lösung wurde mit Wasser auf 17 µl aufgefüllt. Der Ansatz wurde mit 2µ 10x Ligasepuffer und 1µl T4-DNA-Ligase komplettiert. Die genauen Reaktionsbedingungen sind der entsprechenden Versuchs- beschreibung zu entnehmen. 5.3.11 Rekonstitution und Lagerung von Trypsin Gold 100 µg des lyophilisierten Enzympulvers werden nach Herstellerangaben in 100 µl 50 mM Essigsäure rekonstituiert. Die Enzymlösung wird in 5 µl Portionen aufgeteilt und kann so bei –78°C gelagert werden. Zum Verdau von Proteinen wird die Trypsinkonzentration mit 40 mM Ammoniumhydrogencarbonat-Lösung in 10%igem Acetonitril auf 20 µg/ml eingestellt. Dieser Verdaupuffer kann bei –20°C gelagert werden. 5 Material und Methoden 65 5.4 Konstruktion des Vektors pIVEX2.3MCSMtj1pN-term Die nachfolgenden Arbeitschritte erfolgten ausschließlich im pIVEX2.3MCS-Vektor. 5.4.1 Konstruktion des N-terminalen Mtj1p-Fragments mittels PCR Bei der Polymerase-Kettenreaktion (PCR) katalysiert eine DNA-Polymerase die Verlängerung eines DNA-Einzelstranges in Anwesenheit des komplementären Stranges. Der Strang kann durch Hinzufügen komplementärer Basen so lange ergänzt werden, bis beide Stränge gleichlang sind und ein normales doppelsträngiges DNA-Molekül bilden. Die Polymerase-Kettenreaktion läuft nur dann, wenn dem zu kopierenden Strang eine kurze doppelsträngige DNA-Sequenz, die sogenannte Primer-Sequenz, vorausgeht. Das PCR-Verfahren besteht im Wesentlichen aus zwei Schritten, die so lange wiederholt werden, bis ausreichend viele Kopien vorhanden sind. Im ersten Schritt wird die vorliegende doppelsträngige DNA durch vorsichtiges Erhitzen getrennt. Im zweiten Schritt koppelt man die Einzelstränge mit dem Primer und ergänzt die komplementären Basen mit der DNA-Polymerase (Lenhof 1997). Das Design des Plasmids pIVEX2.3MCSMtj1pN-term wurde so gewählt, dass das Codon für die Aminsäure Phenylalanin (TTC) in der Transmembranregion des Proteins Mtj1p durch ein amber-Codon (TAG) ersetzt wird. Der zur PCR verwendete Vektor pIVEX2.3MCSMtj1p wird nach der Qiagen Methode (s. 5.3.3) isoliert und gereinigt. Zur Durchführung der PCR verwendeten Lösungen wurden gemäß Tabelle 2 in 200 µl PCR-Reaktionsgefäße pipettiert. Das Temperatur- und Zeitprofil der PCR ist in Tabelle 3 dargestellt 5 Material und Methoden 66 10x Reaction Buffer 10 µl dNTP-Mix (je 2 mM) 2,5 µl pIVEX2.3MCSMtj1p (50 ng/µl) 4 µl Primer amber1 (750 ng/µl) 2 µl Primer amber1 (750 ng/µl) 2 µl DMSO 2 µl H2Obidest 77,5 µl Pfu Turbo Polymerase (2,5 U/ml) 2 µl Tabelle 2: Pipettierschema für die PCR Segment Temperatur [°C] Zeit [Minuten] Zyklenzahl 1 95 3 1 95 0,5 60 0,75 72 1 72 10 4 ∞ 2 3 30 1 Tabelle 3: Zusammenfassung der PCR-Parameter zur Konstruktion des PCRProdukts Die PCR-Ansätze werden vor und nach der PCR auf Eis gelagert. Nach Beendigung der PCR wird ein Aliquot der amplifizierten Probe auf ein Agarosegel aufgetragen (s. 5.3.5), um den Erfolg des Experiments zu überprüfen. Bei erfolgreicher PCR wird das PCR Produkt mittels des QIAquick PCR Purification Kits von Kontaminationen befreit (s. 5.3.9) und mit den Restriktionsenzymen NdeI und XhoI nach folgendem Schema verdaut: 5 Material und Methoden 67 PCR-Produkt 30 µl Buffer NEB 2 4 µl BSA (1mg/ml) 4 µl NdeI 1 µl XhoI 1 µl Tabelle 4: Pipettierschema für den Verdau des PCR-Fragments Die Reaktion erfolgt über Nacht bei 37°C und wird anschließend durch zwanzigminütige Inkubation bei 65°C gestoppt. Man erhält das 429 bp lange PCR-Produkt Mtj1pN-term. 5.4.2 Linearisierung von pIVEX2.3MCS Die Linearisierung des Vektors erfolgt ebenfalls mit den Restriktionsenzymen NdeI und XhoI. Es wurde folgendes Pipettierschema angewendet: Vektor (1mg/ml) 10 µl Buffer NEB 2 4 µl BSA (1mg/ml) 4 µl NdeI 1 µl XhoI 1 µl H2Obidest 20 µl Tabelle 5: Pipettierschema für den Verdau von pIVEXMCS2.3 Die Reaktion erfolgt ebenfalls bei 37°C über Nacht mit anschließender Hitzeinaktivierung der Enzyme bei 65°C. Der auf diese Weise erzeugte linearisierte Vektor ist 3.534 bp lang. 5 Material und Methoden 68 5.4.3 Ligation des PCR-Produkts mit linearisiertem Vektor pIVEX2.3MCS Für die Ligation werden die Fragmente in Ligase Puffer gelöst mit T4-DNA-Ligase versetzt und homogenisiert. Die Inkubation erfolgt über Nacht bei 25 °C. Für den Ansatz wurde folgendes Pipettierschema benutzt: 10x Ligase Buffer 1 µl PCR-Produkt 2 µl lineariserter Vektor 1 µl H2Obidest 4 µl T4 DNA Ligase 1 µl Tabelle 6: Pipettierschema für die Ligation Nach thermischer Inaktivierung der Ligase bei 65°C für 20 min wird der Ligationsansatz in kompetente E.coli DH5α transformiert. Der erzeugte Expressionsvektor pIVEX2.3MCSMtj1pN-term ist 3963 bp lang. Der Kontrollverdau des neuen Vektors mit NdeI und XbaI erfolgt über Nacht bei 37°C nach dem in Tabelle 7 angegebenen Pipettierschema. Vektor (1mg/ml) 10 µl Buffer NEB 3 4 µl BSA (1mg/ml) 4 µl XbaI 1 µl PstI 1 µl H2Obidest 20 µl Tabelle 7: Pipettierschema für den Kontrollverdau Nach erfolgtem Verdau werden die Restriktionsenzyme bei 65°C hitzeinaktiviert. 5 Material und Methoden 69 5.5 Isolierung der β-Untereinheit der FoF1-ATP-Synthase des thermophilen Bacillus PS3 aus E.coli 5.5.1 Transformation von pUCβ in E.coli Das Plasmid pUCβ, welches sich von pUC118 ableitet und für die β-Untereinheit der FOF1-ATP-Synthase des thermophilen Bacillus PS3 codiert, wurde in kompetente E.coli DH5α (zur Plasmidamplifikation) und in E.coli JM109 (zur Expression) transformiert. Von beiden transformierten Kulturen wurden Glycerinkulturen angelegt und bei –80 °C gelagert. 5.5.2 Expression der β-Untereinheit in E.coli JM109 Aus einer Glycerinkultur wird ein Verdünnungsausstrich auf einer LBAmp-Agarplatte angefertigt und bei 37°C über Nacht inkubiert. Am nächsten Tag impft man 15 ml 2xYT Nährmedium (mit 50 µg Ampicillin pro ml) mit einer Einzelkolonie E.coli an. Anschließend wird über Nacht bei 37°C und 300 min-1 im Wasserbadschüttler inkubiert. Mit der so erhaltenen Startkultur werden 500 ml 2xYT Nährmedium (100 µg/ml Amp.) angeimpft. Es wird bei 37°C und 200 min-1 im Luftschüttler inkubiert. Von Zeit zu Zeit wird die Zelldichte photometrisch gemessen. Die optische Dichte bei 600 nm ist bis zu einem Wert von 0,3 proportional zur Zelldichte. Bei höheren Zelldichten muss die Lösung entsprechend verdünnt werden. Bei einer OD600 von 0,5 bis 0,7 werden die Zellen durch Zugabe einer 1 molaren IPTG-Lösung zu einer Endkonzentration von 1mM induziert. Man lässt dann die Zellen noch bis zu einer OD600 von ca. 1,5 wachsen und erntet ab. Dazu stoppt man das Zellwachstum zuerst auf Eis ab und zentrifugiert dann die Zellsuspension bei 8.700 g bei 4 °C für 20 min. ab. Der Überstand wird verworfen und das Zellpellet in Lysepuffer (50 mM Tris⋅H2SO4 pH 8,0) resuspendiert. Alternativ kann das Zellpellet bis zur weiteren Verwendung bei –80 °C gelagert werden. 5 Material und Methoden 70 5.5.3 Isolation der β-Untereinheit aus E.coli JM109 Die im vorherigen Schritt erhaltene Zellsuspension wird mittels eines UltraschallZelldisruptors bei aufgeschlossen. 70% Leistungsabgabe Unmittelbar nach dem 10 mal 10 Zellaufschluss Sekunden wird 1 auf Eis mg/ml des Proteaseinhibitors Pefabloc SC zugegeben. Danach wird der Zellaufschluss bei 22.000 g bei 4°C für 20 min abzentrifugiert. Der Überstand wird zur Fraktionierung auf eine Ionenaustauschersäule (DEAE Sephacel 1,6x65 cm) appliziert, die in Lysepuffer equillibriert ist. Dann wird die Säule mit ca. 150 ml Lysepuffer gewaschen. Anschließend wird ein stufenweisen Natriumchloridgradient (100 mmolar 200ml, 200 mmolar 200 ml, 400 mmolar 200 ml, 600 mmolar 400 ml) angelegt. Verfolgt man die Absorption der einzelnen Fraktionen bei 280 nm, erhält man das in Abbildung 36 dargestellte Elutionsdiagramm. 6 1,2 1,0 1 E280 0,8 0,6 2 0,4 3 4 0,2 5 7 0,0 0 20 40 60 80 100 120 140 160 180 Fraktionen Abbildung 36: Elutionsdiagramm zur Reinigung der β-Untereinheit an einer DEAESephacel Säule 5 Material und Methoden 71 Mit Hilfe der Proteinbestimmung nach Bradford kann in den Fraktionen 3; 4 und 5 Protein nachgewiesen werden. Zur Charakterisierung werden Proben der Fraktionen auf ein SDS-Gel appliziert und im elektrischen Feld aufgetrennt. Die β-Untereinheit findet sich in den Fraktionen des Peaks 5, ist aber mit niedermolekularen Proteinen verunreinigt. Zur weiteren Aufreinigung werden die entsprechenden Fraktionen gesammelt und mittels einer Amicon 8050 Zelle mit YM30 Ultrafiltrationsmembran bis auf ca. 5 ml aufkonzentriert. Danach wird das Konzentrat auf eine Gelfiltrationssäule (Fractogel TSK HW-40 (S) 16 x 1,5 cm) appliziert. Die Säule ist in in 50 mM Tris⋅H2SO4 pH 8,0/10% Glycerin equillibriert. Mit dem gleichen Puffersystem erfolgt die Elution. Durch Verfolgung der Absorption bei 280 nm erhält man das in Abbildung 37 wiedergegebene Elutionsdiagramm. 1,4 1 1,2 E280 1,0 0,8 0,6 4 0,4 2 3 0,2 0,0 0 5 10 15 20 Fraktionen Abbildung 37: Elutionsdiagramm zur Reinigung der β-Untereinheit an einer Fractogel TSK HW-40 (S)-Säule 5 Material und Methoden 72 Mit Hilfe der Proteinbestimmung nach Bradford und SDS-PAGE wird die β-Untereinheit in Peak 1 identifiziert (s. Abbildung 38). 1 2 Abbildung 38: SDS-Gel der β-Untereinheit nach Gelfiltration Bahn 1: Marker, Bahn 2: β-Untereinheit 5.5.4 Photoaffinitätsmarkierung der β-Untereinheit der FoF1-ATPSynthase des thermophilen Bacillus PS3 Die Photoaffinitätsmarkierung wird mit dem Cofaktor-Analogon 2-N3-2’,3’-SL-ATP durchgeführt. Die Durchführung erfolgt in Anlehnung an eine Publikation von P. Vogel (Vogel 1992). Das Protein liegt nach der Isolierung in einem glycerinhaltigen Puffer vor. Durch mehrmaliges Verdünnen mit dem entsprechenden Puffer und anschließendem Aufkonzentrieren mittels VIVASPIN 2 30,000 MWCO RC wird das Enzym umgepuffert. Es wird der folgende Puffer verwendet: SHPMg: 150 mM Sucrose 10 mM Hepes 10 mM KH2PO4 1mM MgSO4 pH 8,0 5 Material und Methoden 73 Anschließend wird die Proteinlösung auf eine Konzentration von 50 µM eingestellt. Zum Markieren der katalytischen Bindungsstellen wird die Proteinlösung mit 150 µM 2-N3-2’,3’-SL-ATP 90 Minuten bei Raumtemperatur inkubiert. Danach wird die Probe 4 mal 1 Minute bei 254 nm mit einer Handlampe der Firma Camag bestrahlt. Der Abstand Lampe – Probe beträgt dabei 1 cm. Zwischen den Bestrahlungen werden Abstände von 30 Minuten eingehalten, um das Azido-Tetrazol-Gleichgewicht wieder herzustellen. Der nichtgebundene Anteil an Spin-markiertem Nukleotid wird durch mehrmaliges starkes Verdünnen der Proteinlösung mit SHPMg-Puffer und anschließendem Aufkonzentrieren mittels VIVASPIN 2 30,000 MWCO RC entfernt. Für die ESR-Messungen wird die Probe auf 40 bis 60 µM aufkonzentriert. 5.6 ESR-Studien an der β-Untereinheit der FoF1-ATP-Synthase des thermophilen Bakteriums PS3 5.6.1 Geräteeinstellungen Die ESR-Messungen erfolgten mit einem ELEXSYS E580 der Fa. Bruker im X-Band bei 9,6 GHz. Die Temperatur wurde mit einer Temperiereinheit auf 293 K eingestellt. Die Proben wurden mittels einer Quarzkapillare im Hohlraumresonator positioniert. Die Modulationsfrequenz betrug 100 kHz. Die Mikrowellenleistung wurde zwischen 12,2 und 24,5 mW variiert. Die Modulation des Messsignals lag bei 1G. 5.6.2 Vorbereitung des Proteins für die ESR-Messungen Vor Beginn der Messungen muss das Protein in der gewünschten Konzentration in den entsprechenden Messpuffer überführt werden. Dies geschieht entsprechenden in diesem Puffer und Fall durch mehrmaliges anschließendem Verdünnen Aufkonzentrieren mit mit Microkonzentrator (VIVASPIN 2 30,000 MWCO RC der Fa.VIVASCIENCE). dem einem 5 Material und Methoden 74 5.7 Massenspektrometrische Untersuchungen von Peptiden und Proteinen 5.7.1 Tryptischer Verdau von Proteinen im SDS-Gel Alle Arbeitsschritte wurden steril durchgeführt, um Kontaminationen mit Keratin zu verhindern. 01 Zu untersuchende Bande aus dem SDS-Gel ausschneiden und in Stücke von ca. 1x1 mm zerkleinern 02 100 µl 50 mM Ammoniumhydrogencarbonat-Lösung hinzufügen, vortexen und 5 min inkubieren 03 Zentrifugieren und Überstand entfernen 04 50 µl 10 mM DDT-Lösung hinzufügen, vortexen und 30 min bei 56°C inkubieren 05 Zentrifugieren und Überstand entfernen 06 50 µl Acetonitril hinzufügen und 10 min inkubieren 07 Zentrifugieren und Überstand entfernen 08 50 µl Iodacetamid (55 mM in 50 mM Ammoniumhydrogencarbonat-Lösung) hinzufügen und 20 min inkubieren 09 Zentrifugieren und Überstand entfernen 10 100 µl 50 mM Ammoniumhydrogencarbonat-Lösung hinzufügen, vortexen und 10 min inkubieren 11 Zentrifugieren und Überstand entfernen 12 100 µl Acetonitril hinzufügen und 10 min inkubieren 13 Zentrifugieren und Überstand entfernen 14 Gel-Stücke 20 min im Vakuum trocknen 15 Gelstücke in 20 µl Verdau-Puffer (s. 5.3.11) rehydratisieren und 45 min bei 4°C inkubieren. Falls die Gelstücke die gesamten Lösung absorbieren, etwas VerdauPuffer hinzufügen 16 Verdau bei 37°C über Nacht 17 Hinzufügen von 10 µl 5 mM Trifluoressigsäure 18 Extraktion der Peptide aus den Gelstücken durch heftiges Schütteln für 45 min 5 Material und Methoden 75 5.7.2 Vorbereitung von Peptiden und Proteinen für MALDI-MS Vor der Applikation auf ein MALDI-Target müssen die Peptide entsalzt und aufkonzentriert werden. Dazu verwendet man C18-ZipTip®-Pipettenspitzen der Fa. Millipore. Die Peptidlösung soll einen pH-Wert von 2 – 4 haben, damit die Peptide vollständig an die C18-Matrix der Pipettenspitzen binden. Sollte dies nicht der Fall sein, werden einige µl 5 M Trifluoressigsäure der Peptidlösung hinzugefügt und der pH-Wert erneut überprüft. Folgende Lösungen werden benötigt: Befeuchtungslösung: 50%iges Acetonitril Equillibrierlösung: 0,1%ige Trifluoressigsäure Waschlösung: 0,1%ige Trifluoressigsäure Elutionslösung: HCCA gesättigt in 0,1%ige Trifluoressigsäure und 50%iges Acetonitril gesättigt mit HCCA Die Durchführung erfolgt nach dem Protokoll des Herstellers. Die Elution der Peptide von der C18-Matrix der Pipettenspitzen erfolgt mit 2,5 µl Elutionslösung. Das Eluat wird direkt im Anschluss auf ein MALDI-Target präpariert. Nach dem vollständigen Verdampfen des Lösungsmittels kann die Probe im Massenspektrometer analysiert werden. Im Fall von intakten Proteinen wird analog verfahren. Allerdings findet in der Elutionslösung Sinapinsäure anstelle von HCCA Verwendung. 5 Material und Methoden 76 5.7.3 Geräteeinstellungen 5.7.3.1 Peptide Peptide werden im Reflektor Modus als positive Ionen gemessen. Desweiteren finden folgende Geräteparameter Anwendung. Ionen Quelle 1 25,00 kV Ionen Quelle 2 22,35 kV Linse 6,00 kV Reflektor 26,3 kV Reflektor 2 14,8 kV Laser Frequenz 20 Hz Messbereich 500 – 3200 Da Die Energie des Lasers wird bei jeder Messung manuell an die jeweilige Probe angepasst. 5.7.3.2 Proteine Intakte Proteine werden ebenfalls als positive Ionen gemessen. Im Gegensatz zu den Peptiden erfolgt die Detektion im linearen Modus. Die Geräteparameter werden wie folgt eingestellt: Ionen Quelle 1 25,00 kV Ionen Quelle 2 22,90 kV Linse 6,25 kV Laser Frequenz 20 Hz Meßbereich 2000 – 150000 Da Analog zu Peptiden erfolgt die Anpassung der Laserenergie ebenfalls manuell. In beiden Fällen werden für ein Spektrum 1000 – 2000 Shots aufsummiert. 5 Material und Methoden 77 5.7.4 Kalibrierung Zur Kalibrierung des Massenspektrometers wird für Peptide der Peptide calibration standard der Fa. Bruker verwendet. Im Fall von Proteinen findet der Protein calibration standard II, ebenfalls von der Fa. Bruker, Anwendung. Die Standards werden nach der dried droplet Methode auf ein MALDI-Target appliziert. Das heisst, zuerst werden 0,8 µl Matrixlösung (Elutionslösung, s. 5.7.2) auf das Target gebracht Herstellerangaben und bereiteten getrocknet. Danach Standardlösung werden auf Anschließend lässt man das Lösungsmittel verdampfen. den 0,8 µl Matrixfleck der nach pipettiert. 5 Material und Methoden 78 5.8 Synthesen 5.8.1 Cyanomethylester von HO3007 2,3 mg (7,02.10-6 mol) der Spin-Label Aminosäure HO3007 werden in 20 µl Triethylamin gelöst und auf 0°C abgekühlt. Anschließend werden 0,9 µl (14,04.10-6 mol, zweifacher Überschuss) Chloracetonitril unter Rühren hinzugefügt. Dann lässt man die Lösung auftauen und über Nacht bei Umgebungstemperatur rühren. Am nächsten Tag wird der entstandene Brei mit 250 µl Ethylacetat versetzt. Die ausgefallenen Triethylammoniumsalze werden abzentrifugiert und der Überstand mit je 100 µl 0,5 N HCl, 0,5 N KHCO3 und Wasser gewaschen. Die organische Phase wird im Anschluss auf -78°C abgekühlt und lyophilisiert. Man erhält das Produkt als gelben Feststoff. Laut DC auf Kieselgel 60 ist das Produkt frei von Verunreinigungen. Die Analyse des Produkts und des Edukts mittels DC (Kieselgel 60) liefert folgende Ergebnisse: Rf = 0,6 (CH2Cl2 94%, MeOH 6%) Produkt Rf = 0,33 (CH2Cl2 94%, MeOH 6%) Edukt 5.8.2 Acylierung von dpCpA mit HO3007 2,4 mg des Ammoniumsalzes des Dinukleotids dpCpA werden mit 200 µl 0,1 M Tetrabutylammoniumacetatlösung versetzt und lyophilisiert. Man erhält das in DMF lösliche Tetrabutylammoniumsalz des Dinukleotids. Das erhaltene dpCpA Pulver wird in 300 µl frisch destilliertem DMF aufgenommen. Der Cyanomethylester von HO3007 wird in 200µl DMF aufgenommen. Beide Lösungen werden vereint. Nach 2h rühren wird die Reaktionslösung über Nacht lyophilisiert. Das Produktgemisch wird in 50 µl Portionen mittels einer C18-RP-HPLCSäule gereinigt. Die Säule ist in 0,1 M Ammoniumhydrogencarbonat-Lösung (pH 4,5) equillibriert. Nach dem Auftragen der Probe auf die Säule wird ein linearer Gradient aus 0,1 M NH4OAc-Lösung und Acetonitril angelegt. Bei einem Laufmittelgemisch von 40%/60% wird der Gradient angehalten und mit diesem Gemisch weiter eluiert. 5 Material und Methoden 79 Alle Fraktionen werden im ESR-Spektrometer untersucht. Die das Produkt enthaltenden Fraktionen werden gesammelt und lyophilisiert. 5.8.3 Entschützen des acylierten Dinukleotids Zum Entfernen der tBoc-Schutzgruppe von der an das Dinukleotid acylierten SpinLabel Aminosäure HO3007 wird das Dinukleotid in 500 µl frisch destilliertem DMF aufgenommen und mit einem Tropfen wasserfreier Trifluoressigsäure versetzt. Zur Reoxidation des Spin-Labels rührt man die Lösung eine Stunde unter Luftzufuhr und entfernt anschließend das Lösungsmittel im Vakuum. 6 Abkürzungen 6 Abkürzungen ADP Adenosindiphosphat AMP Adenosinmonophosphat ATP Adenosintriphosphat BiP Binding Protein bp Basenpaare Da Dalton DC Dünnschichtchromatographie DDT 1,4-Dithiothreitol DMF N,N-Dimethylformamid DNA Desoxyribonukleinsäure E.coli Escherichia coli ESR Elektronenspinresonanz G Gauß HCCA α-Cyano-4-hydroxyzimtsäure HPLC high performance liquid chromatography IPTG Isopropyl-Thio-β-D-Galactopyranosid MALDI Matrix assisted laser desorption/ionisation mRNAmessenger-Ribonukleinsäure MS Massenspektrometrie PAGE Polyacrylamidgelelektrophorese PCR polymerase chain reaction Pi anorganisches Phosphat RNA Ribonukleinsäure RP Reversed Phase SA Sinapinsäure SDS Natriumdodecylsulfat SL Spin-Label TEMED Tetramethylethylendiamin TFA Trifluoressigsäure TOF time of flight tRNA transfer-Ribonukleinsäure 80 7 Literatur 81 7 Literatur Bossi, L., Roth, J. R. (1980) Nature 286 123 Boyer, P. D., Cross, R. L., Momsen, W. (1973) Proc. Nat. Acad. Sci. USA 70 2837-9 Brenner, S., Stretton, A. O., Kaplan, S. (1965) Nature 206 994 Burgard, S. (1995) Dissertation, 54 Burgard, S., Harada, M., Kagawa, Y., Trommer, W. E., Vogel, P. D. (2003) Cell. Biochem. Biophys. 39 175-181 Chapeville, F., Lipmann, F., von Ehrenstein, G., Weisblum, B., Ray Jr., W. J., Benzer, S. (1962) Proc. Nat. Acad. Sci. USA 48 1086 Crick, F. H. C. (1958) Symp. Soc. Exp. Biol. 12 38 Crick, F. H. C., Barret, L., Brenner, S., Watts-Tobin, R., (1961) Nature 192 1227 Dintzis, H. M. (1961) Proc Natl. Acad. Sci. U S A (1961) Mar 15 47 247-61 Ellman, J. A., Mendel, D., Schultz, P. G. (1992a) Science 255 197 Ellman, J. A., Mendel, D., Anthony-Cahill, S., Noren, C. J., Schultz, P. G. (1992b) Methods Enzymol. 202 301 Esmann, M., Sar, P. C., Hideg, K., Marsh, D., (1993) Anal. Biochem. 213 336 - 348, Graf, R., Brunner, J., Dobberstein, B., Martoglio, B. (1998) CELL BIOLOGY: A LABORATORY HANDBOOK, Second Edition Vol. 4 495-501 Hamilton, C. L., McConnell, H. M., (1968) Structural Chemistry and Molecular Biology, (Rich, A., Davidson, n., eds.), p. 115, Freeman, San Francisco, Hanahan D., (1983) J Mol Biol 166 558-580 Heckler, T. G., Chang, L. H., Zama, Y, Naka, T., Chorghade, M. S., Hecht, S. M., (1984) Biochemistry 23 1468 Hillenkamp, F., Karas, M., Beavis, R. C., Chait, B. T (1991) Anal. Chem. 63 1193 Hoagland, M. B., Zamecnik, P. C., Stephenson, M.L. (1957) Biochim Biophys Acta. Apr 24(1) 215-6 Holley, (1965) Science Mar 19 147 1462-5 Holley, (1965) J Biol Chem. May 240 2122-8 Johnson, A. E., Woodward, W. R., Herbert, E., Menninger, J. R., (1976) Biochemistry 15 569 Miller, J. H., Ganem, D., Lu, P., Schmitz, A. (1977) J. Mol. Biol. 109 275 7 Literatur Milligan, J. F., Groebe, D. R., Witherell, G. W., Uhlenbeck, O. C. (1987) Nucleic Acid Res. 15 8783 Noji, H., Yasuda, R., Yoshid, M., Kinosita, K. (1997) Nature 386 299 Noren, C. J., Anthony-Cahill, S. J., Griffith, M C., Schultz, P. G., (1989) Science 27 182 Noren, C. J., Anthony-Cahill, S. J., Suich, D. J., Noren, K. A., Griffith, M. C., Schultz, P. G. (1990) Nucleic Acids Res 18 83 Ohtsubo, M., Yoshida, M., Ohta, S., Kagawa, Y., Yohda, M., Date, T. (1987) Biochem. Biophys. Res. Comm. 146 705-710 Robertus, J. D., Ladner. J. E., Finch. J. T., Rhodes D., Brown R. S., Clark B. F., Klug A. (1974) Nature Aug 16; 250 (467) 546-51 Rozantsev, E. G. (1970) Free Nitroxyl Radicals, Plenum, New York Sambrook, J., Fritsch, E. F., Maniatis, T. (1989) Molecular Cloning: A Laboratory Manual (Bdd. 1-3), Cold Spring Laboratory Press Schultz, P. G., Wang, L. (2005) Angew. Chem Int. Ed. 44 34-66 Schwyzer, R., Iselin, B., Feurer, M. (1955) Helv. Chim. Acta 38 69 Stratagene (1997) QuickChange Site-Directed Mutagenesis Suddath, F. L., Quigley, G. J., McPherson, A., Sneden, D., Kim, J. J., Kim, S. H., Rich, A. (1974) Nature. Mar 1 248 (443):20-4 Sussmann, J. L.; Holbrook, S.R.; Warrant, R.W.; Curch, G.M.; Kim, S.H. (1978) J. Mol. Biol. 123 607 Süßmut, R., Eberspächer, J., Haag, R., Springer, W (1987) Biochemischmikrobiologisches Praktikum, Georg-Thieme Verlag, Stuttgart Woodcock, D.M., Crowther, P.J., Doherty, J., Jefferson, S., De Cruz, E., NoyerWeidner, M., Smith, S.S., Michael, M.Z., Graham, M.W. (1999) Nucleic Acids Res. 17 3469-78 Vogel, P. D., Nett, J. H., Sauer, H. E., Schmadel, K., Cross, R. L., Trommer, W. E. (1992) J Biol Chem. June 267 11982-86 Yanish-Perron, C., Vieira, J., Messing, J. (1985) Gene 33 103-19 82 Danksagung Mein Dank gilt allen, die zum Gelingen dieser Arbeit beigetragen haben. Herrn Prof. Dr. J. Brunner danke ich für das großzügige Überlassen der Plasmids pStR1. Herrn Prof. R. Zimmermann danke ich für die Überlassung des Expressionsvektors pIVEX2.3MCSMtj1p. Herrn Prof. K. Hideg danke ich für die großzügige zur Verfügungstellung der Aminosäure HO3007. Herrn Dr. M. Jung und Herrn Dr. P. Maurer danke ich für die Hilfe bei der Erarbeitung der Klonierungsstrategien und der in vitro Translation. Frau Prof. P. Vogel und Herrn Dr. T. Hisabori danke ich für das großzügige Überlassen des Plasmids pUCβ. Weiterhin danke ich meinen Kollegen im Arbeitskreis Prof. Dr. W. E. Trommer für die freundliche Atmosphäre und die vielen anregenden Gespräche. In besonderem Maß möchte ich mich bei meinen Eltern bedanken, mit deren Unterstützung das Studium und diese Arbeit erst möglich wurden. Lebenslauf Name: Dirk Mannweiler Geburtsdatum: 12. Februar 1974 Familienstand: ledig Staatsangehörigkeit: deutsch Schulausbildung: 1980 – 1990 14. Polytechnische Oberschule in Halle/S 1990 – 1992 Torgymnasium in Halle/S (Abschluss: Abitur) Studium Oktober 1993 – Dezember 1999 Studium der Chemie an der Universität Kaiserslautern Mai 1999 – Dezember 1999 Diplomarbeit im Arbeitskreis von Herrn Prof. W. E. Trommer mit dem Thema: Kinetische Untersuchungen an der Glucose-Dye- Oxidoreduktase Promotionsstudium: Im Arbeitskreis von Herrn Prof. W. E. Trommer Juni 2000 – Dezember 2005 an der Universität Kaiserslautern