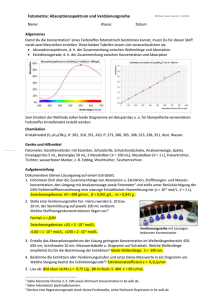
pδv
Werbung

2.
Energie und Temperatur
2.1 Ideales Gasgesetz
Das Volumen eines Gases erhöht sich bei konstantem Druck mit der
Temperatur und der Anzahl der Gasteilchen. Zunehmender Druck führt
bei konstanter Temperatur zur Verringerung des Gasvolumens.
Gesetz von Gay-Lussac
Volumen und Temperatur bei konstantem Druck und Stoffmenge sind
zueinander proportional: V ~ T
Das heißt:
V
= const.
T
Gesetz von Boyle-Marriot
Volumen und Druck bei konstanter Temperatur und Stoffmenge sind
€
umgekehrt
proportional: V ~ 1/p
Das heißt:
p ⋅V = const.
Avogadrosches Gesetz
Weiter gilt, dass Volumen und Stoffmenge bei konstantem Druck und
€
Temperatur
einander proportional sind: V ~ n
V
= const.
n
Gesetz von Jacques Alexandre Cesar Charles
Ebenso sind Druck und Temperatur bei konstanter Stoffmenge und Vo€ zueinander proportional: p ~ T
lumen
p
= const.
T
€
Aus diesen vier experimentell gefundenen Gesetzmäßigkeiten kann das
allgemeine Gasgesetz für ideale Gase aufgestellt werden:
p ⋅V = n ⋅ R ⋅T
Die Proportionalitätskonstante ist:
R = 8.314 J/mol.K = allgemeine Gaskonstante
k =€1.38.10-23 J/K = Boltzmankonstante
R = k . NA
Mit NA = Avogadrokonstante = Anzahl der Atome oder Moleküle in einem
Mol des jeweiligen Materials. NA = 6.022⋅1023 mol-1
Davon zu unterscheiden: die Loschmidtzahl NL = Anzahl der Atome oder
Moleküle pro Volumeneinheit eines idealen Gases unter Normalbedingungen NL = 2.686.1025 m-3.
In der älteren Literatur werden Avogadrozahl und Loschmidtzahl zum Teil
(fälschlicherweise) synonym verwendet.
Sie hängen über folgende Beziehung zusammen:
NL =
NA
Vm0
mit dem molaren Volumen bei Normalbedingungen
€
Beispiel:
Ein Gas hat bei einem Druck von 75 kPa ein Volumen von 360 ml. Wie ist
sein Volumen bei 100 kPa (bei gleicher Temperatur)?
Lösung:
pV
75 kPa ⋅ 360 ml
p1 ⋅ V1 = p2 ⋅ V2 → V2 = 1 1 =
= 270 ml
p2
100 kPa
€
Beispiel:
Ein Airbag wird durch Zersetzung von Natriumazid aufgeblasen:
2 NaN3(s) → 2 Na (l) + 3 N2 (g)
Die Treibladung enthält 120 g NaN3; es entsteht N2 bei 25°C und p = 125
kPa (leichter Überdruck). Wie groß ist das Volumen des Prallsackes?
Lösung:
V (N2 ) =
n(N2 ) ⋅ R ⋅T
p
3
n(N2 ) = n(NaN3 )
2
€
€
n(NaN3 ) =
120g
= 1,85mol
(23,0 + 3 ⋅14,0)g / mol
N(N2)=2,77mol
€
V (N2 ) =
2,77 mol ⋅ 8,31J ⋅ 298 K
mol ⋅ K ⋅125 kPa
V (N2 ) = 54,9 l
€
2.2
Drücke in einer Mischung idealer Gase
€
p = pA + pB + pC + . . .
Gesamtdruck ist Summe der
Partialdrücke der Gase A, B, C…
1 l Gas A bei 20 kPa
1 l Gas B bei 40 kPa
1 l Gas (A + B) bei 60 kPa
Stoffmengenanteil (früher auch Molenbruch genannt) von A
nA
xA + x B = 1
n A + nB
nA
pA =
⋅ p = xA ⋅ p
Partialdruck des Gases A
n A + nB
xA =
€
€
Beispiel:
Ein Gemisch von 40,0 g O2 und 40,0 g He hat einen Druck von 90,0 kPa.
Wie groß ist der O2-Partialdruck?
Lösung:
40,0 g
nO2 =
= 1,25 mo l
32,0 g / mol
nHe =
€
xO2 =
€
pO2 =
€
40,0 g
= 10,0 mo l
4,0 g / mol
nO2
nO2 + nHe
1,25 mol
⋅ 90 kPa
10 mol + 1,25 mol
= xO2 ⋅ 90kPa
€
= 10,0 kPa
€
Vorlesungsversuch 2a: Gesetz von Gay-Lussac
Prinzip: Das Gesetz idealer Gase (Teilchen ohne Wechselwirkung und
Eigenvolumen) wird durch Messung von Gasvolumen V, Druck p, Temperatur T und Teilchenzahl n bestimmt: p ⋅ V = n ⋅ R ⋅ T
Messmethode: Das Luftvolumen in einer Gasspritze mit kalibriertem
Volumen wird bei konstantem Druck (Atmosphärendruck) mit einem
Wasserbad erwärmt und die Zunahme des Gasvolumens an Hand der
Kolbenverschiebung gemessen.
Empirisch: Volumen ~ Temperatur (V ~ T)
Temperatur ϑ in °C (Eis: 0 °C; siedendes Wasser: 100°C) nach Celsius.
Extrapolation der V(ϑ)-Kurve führt zu einem Volumen von Null bei einer
Temperatur von -273,15°C.
Neue Temperaturskala (Kelvin): T=0K bei V=0ml ->T/K = ϑ/°C + 273,15
Ergebnis: V1=50ml, ϑ1=20,8°C [T=294K] V1/T1=0,170ml/K
Steigung aus Abbildung ΔV/ΔT=0,135ml/K
Vorlesungsversuch 2b: Gesetz von Boyle-Marriot
Prinzip: Gasförmiger Stickstoff wird mit einem Kolben in einem Zylinder
zusammengedrückt und der Druck als Funktion des Volumens bei verschiedenen Temperaturen (p-V Isotherme) gemessen.
Messmethode: Eine thermostatisierte, druckfeste Glaskapillare mit Volumenskala wird evakuiert, mit N2 auf Atmosphärendruck gefüllt und geschlossen. Die Messkapillare ist auf eine mit Quecksilber gefüllte, stählerne Druckkammer montiert. Mit einem Handrad wird über einen Spindeltrieb ein Kolben in der Druckkammer bewegt und über den Kolbenhub
das Gasvolumen verkleinert. Der Gasdruck in der Kapillare wird von einem Manometer angezeigt.
Empirisch: Volumen ~ 1/Druck (V ~ 1/p)
Ergebnisbeispiel: N2
V/cm3
4,0
3,8
3,6
3,4
3,2
p/105Pa
10,1
10,5
10,9
11,5
12,1
40°C
Vorlesungsversuch 2c: Gesetz von Jacques Alexandre Cesar
Charles
Prinzip: Gasförmiger Stickstoff wird mit einem Kolben in einem Zylinder
zusammengedrückt und der Druck als Funktion der Temperatur bei konstantem Volumen gemessen.
Messmethode: Eine thermostatisierte, druckfeste Glaskapillare mit Volumenskala wird evakuiert, mit N2 auf Atmosphärendruck gefüllt und geschlossen. Die Messkapillare ist auf eine mit Quecksilber gefüllte, stählerne Druckkammer montiert. Die Messkapillare wird auf eine bestimmte
Temperatur thermostatisiert. Mit einem Handrad wird über einen Spindeltrieb ein Kolben in der Druckkammer bewegt und über den Kolbenhub
das Gasvolumen auf einen bestimmten Wert eingestellt. Der dazugehörige Gasdruck in der Kapillare wird am Manometer abgelesen. Danach
werden weitere Temperaturen an der Kapillare eingestellt und jeweils der
Druck beim gleichen Volumen bestimmt.
Empirisch: Temperatur ~ Druck (T~ p)
Ergebnisbeispiel: N2
T/K
303
213
323
333
p/105Pa
31,5
33.2
35,0
36,7
(V=2 ml)
Ergebnisbeispiel: Welchen Wert hat die Proportionalitätskonstante
(”allgemeine Gaskonstante” R) dieser Gleichung?
p.V = R.n.T, also R = p.V/n.T
R = (1,01325·105 N/m2 · 53.65·10-6m3) / (53.65ml/22414ml)mol · 293K
= 8,315 J/mol K
Literatur:
R = 8,3145 J/mol K
2.3 Interpretation von Druck und Temperatur
Die Aufschläge der Teilchen auf die Gefäßwände verursachen den Druck
des Gases.
Mit zunehmender Geschwindigkeit bzw. kinetischer Energie der Teilchen
steigt bei konstantem Volumen der Druck und unter Berücksichtigung der
idealen Gasgleichung die Temperatur. Es gilt (ohne Ableitung):
Ekin,x=1/2 RT
Mittlere kinetische Energie von 1 mol Teilchen pro
Bewegungs-Freiheitsgrad(Flug in x-Richtung)
Bei 3 Freiheitsgraden der Bewegung (in x-, y- und z- Richtung) gilt dann:
EKin=3/2 RT pro Mol (oder 3/2 kT pro Molekül)
Die Temperatur ist also der mittleren kinetischen Energie der Teilchen
eines Gases proportional. Wärme ist eine Art der (unkoordinierten) Bewegung. Mechanische Energie wird sichtbar, wenn die Bewegung der
Teilchen koordiniert in eine Richtung erfolgt.
Beispiel:
Wie hoch ist die mittlere kinetische Energie und die mittlere Geschwindigkeit eines mol H2-Gases bei Normbedingungen?
Lösung:
3
3
EKin = ⋅ R ⋅T = ⋅ 8,31Pa ⋅ m3 ⋅ mol −1 K −1 ⋅ 273K
2
2
€
Pa ⋅ m3
kJ
= 3403
= 3,4
mol
mol
3
1
RT = mN A < v 2 >
2
€ 2
v = < v2 > =
€
NA = 6.022⋅1023 Teilchen/Mol
3RT
M
2,016g / mol
Mittlere Geschwindigkeit eines Teilchens
€
0°C
→
€
=
(
)
3 ⋅ 8,314 ⋅10 3 g ⋅ m2 / s 2 ⋅ mol ⋅ K ⋅ 273K
2,016g / mol
= 1,84 ⋅ 103 m/s
100°C
→
= 2,15 ⋅ 103 m/s
Grahamsches-Effusionsgesetz
2 Gase A, B haben also die gleiche mittlere kinetische Energie, wenn sie
die gleiche Temperatur haben:
1
1
2
2
mA < v A >= mB < vB >
2
2
€
€
vA
MB
Diffusion durch Loch in ein Vakuum (”Effusion”)
=
vB
MA
Trennung eines Gasgemisches nach den Massen auf Grund der unterschiedlichen Diffusionsgeschwindigkeiten durch ein Loch oder eine
Membran (z.B. Urantrennung)
Beispiel:
Welche Molmasse hat ein Gas X, das 0,876 mal so schnell wie N2(g) effundiert?
Lösung:
vX
0.876
28g / mol
=
=
MX = 36,5 g/mol
vN2
1
MX
€
2.4
Geschwindigkeitsverteilung und mittlere Geschwindigkeit
In einem Gas haben die Moleküle unterschiedliche Geschwindigkeiten.
Der Mittelwert dieser Geschwindigkeiten verschiebt sich mit zunehmender Temperatur zu höheren Werten und liegt für Moleküle kleiner Masse
höher als für schwere Moleküle. Da die Fläche unter der Verteilung die
Gesamtzahl der Moleküle angibt und diese konstant bleibt, wird die Verteilung mit zunehmender Temperatur und abnehmender Masse flacher.
Vorlesungsversuch 3: Maxwell-Boltzmann Geschwindigkeitsverteilung
Prinzip: Atome und Moleküle haben eine Verteilung der Größe ihrer
Geschwindigkeiten, die von ihrer Masse und der Temperatur abhängt.
Die Geschwindigkeitsverteilung ist auf die statistisch erfolgenden Stösse
der Teilchen untereinander zurückzuführen, die durch Rütteln von vielen
kleinen Glasbällen simuliert werden kann. Ihre Geschwindigkeiten werden durch ihre Fluglängen bestimmt.
Messmethode: 400 Glasbälle werden mit 50 Hz (Stroboskopmessung)
durch einen Motor geschüttelt und durch eine Öffnung in einen Behälter
mit 24 ringförmig angeordneten Auffangrillen zunehmenden Durchmessers geschleudert. Die Anzahl der Glasbälle in jeder der Rillen wird durch
Auswiegen bestimmt (die durchschnittliche Masse einer Glaskugel wurde
vorher durch Auszählen und Wiegen ermittelt).
Aus der Flugstrecke s (Abstand Austrittsöffnung-Auffangrille) und der
Höhe h der Austrittsöffnung über der Rillenebene wird die Kugelgeschwindigkeit v bestimmt. Es gilt nämlich
shor=v t
svert=h=½ g t2
Flugzeit t=√2h/g
v=shor√g/2h
h=8cm g=9,81ms-2 (Erdbeschleunigung)
Die Rillenbreite Δs=1cm entspricht einer
Δv=0,078 ms-1. ∑Nv=400.
Ergebnisbeispiel:
shor[cm]
v[ms-1]
0
0
0,5
0,039
1,5
0,117
2,5
0,196
3,5
0,274
usw.: Tabelle mit 24 Zeilen…
Nv
0
16
19
20
21
Geschwindigkeitsbreite
Nv/∑Nv Δv
0
0,5
0,6
0,65
0,67
Der theoretische Ausdruck für die Maxwell-Boltzmannverteilung ist
dN/N=√2/π⋅(m/kT)3/2⋅v2⋅exp(-m⋅v2/2kT) ⋅dv
Die graphische Auftragung führt zu einem ähnlichen Kurvenverlauf wie im
Glaskugelversuch: Anstieg der Kurve mit v2 (parabelförmig) und glockenförmiger Verlauf (Gausskurve exp(-ax2)) bei größerem v.
Beispiel:
Berechnen Sie für Wasserstoffatome bei 300K den Anteil ΔN/N≅dN/N an
Atomen im Geschwindigkeitsintervall 1000-1100 ms-1.
v=1050ms-1 Δv=100ms-1 T=300K m=1.67262158⋅10-27kg (H-Atom)
Boltzmannkonstante k=1.3806503⋅10-23JK-1; Anteil ΔN/N=0,018 (1,8%)
2.5
Innere Energie und Enthalpie
Wärme (eine Form der Energie mit der Einheit [J]) fließt vom Körper mit
höherer Temperatur zu dem Körper mit niedrigerer Temperatur, denn
System und Umgebung streben maximale Unordnung (Gleichverteilung)
an.
Wärme in atomaren Gasen: Bewegungsenergie der Teilchen;
Wärme im Festkörper: Schwingungsenergie der Teilchen;
Wärme in Flüssigkeiten: beides.
2.5.1 Erster Hauptsatz: Erhaltung der Energie
1. Hauptsatz der Thermodynamik:
”Die Energie eines isolierten Systems bleibt konstant.”
ΔU = Q +W
€
2.5.2
ΔU = Änderung der inneren Energie
Q = Wärmeaustausch
W = Arbeit
(z.B. differentielle Volumenarbeit –pdV oder
elektrochemische Arbeit)
Reversible und irreversible Prozesse
Thermodynamische Prozesse wie Wärmeaustausch und Arbeit können
isotherm, isochor oder isobar ablaufen (Temperatur, Volumen oder Druck
sind konstant).
Thermodynamische Prozesse können reversibel oder irreversibel ablaufen.
reversibel:
ständig im Gleichgewicht
Beispiel:
Fahrradpumpe → langsame Kompression, dann innerer Druck ≙ äußerer Druck. Wenn bei Kompression der innere Druck (Gasdruck) steigt,
muss auch mehr äußerer Druck für die Kompression aufgewandt werden.
Im Gleichgewicht kann dann für den äußeren Druck p der innere Druck
des Gases p = nRT/V in dW = -pdV eingesetzt werden:
dW = -(nRT/V)dV. Nach Integration folgt die
reversible isotherme Volumenarbeit
Bei Expansion ist VB > VA , so dass W < 0 ist: Das System (Gas) gibt
Volumenarbeit nach außen ab (negatives Vorzeichen) und erhält die
Energie dafür aus einem Wärmereservoir. Dabei ändert sich die innere
Energie des Gases nicht (isothermer Prozess).
Bei Kompression ist VB < VA , so dass W > 0 ist: Am Gas wird Volumenarbeit geleistet, wobei die dabei zugeführte Energie an das Wärmereservoir abgegeben wird. Wiederum ändert sich die innere Energie des
Gases nicht (isothermer Prozess).
Beispiel:
Wie groß ist die Arbeit für schnelles, irreversibles und für langsames,
reversibles Expandieren von 1 mol Gasvolumen von 1l auf 2l bei Atmosphärendruck?
Lösung:
Irreversibel: ΔWirr = -pE⋅ΔV = 1,013⋅105N/m2 ⋅ 10-3m3 = 101 Nm
ΔWirr = -101 J.
Reversibel: ΔWrev = -nRTln(VE/VA)
ΔWrev = -1729J
Pfeilrichtung: Expansion
Die Volumenarbeit ist jeweils die Fläche unter der reversiblen und irreversiblen Isotherme: WABrev hat größere Fläche, also
.
Bei reversibler isothermer Expansion wird mehr Wärme aus der Umgebung aufgenommen als bei irreversibler Expansion.
2.5.3
Wärmekapazität
Wenn man nur Volumenarbeit zulässt, gilt für die Änderung der inneren
Energie eines geschlossenen Systems ΔU=Q – pΔV.
Bei konstantem Volumen (Autoklav) gilt ΔV=0 und damit ΔU=Q, d.h. die
ausgetauschte Wärmemenge unter isochoren Bedingungen entspricht
der Änderung der inneren Energie des Systems.
Die Wärmemenge, um die Temperatur eines Körpers mit Masse m um
1°C zu erhöhen, ist die
Wärmekapazität C
Die durch die Masse m dividierte Wärmekapazität ist die spezifische
Wärmekapazität c [J/kg.K]=C/m
Beispiel:
Wasser hat eine spezifische Wärmekapazität c von 4,184 Jg-1K-1. Wie
groß ist die Wärmekapazität von 125 g Wasser?
Lösung:
C = 125 g · 4,184 Jg-1K-1 = 523
Beispiel:
Wie groß ist die Wärmemenge, um 125 g Wasser von 20,0°C auf 25,0°C
zu erwärmen?
Lösung:
J
Q = C ⋅ ΔT = 523 ⋅ (298,15 K − 293,15 K ) = 2615 J = 2,615 kJ
K
5K
Ein Mikrowellenofen mit 300 Watt = 300 J/s Leistung braucht dazu 9 s
[ohne Berücksichtigung von Verlusten].
€ 1 W = 1 V · 1 A (300 W = 220 V · 1,4 A)
Die durch die Stoffmenge n dividierte Wärmekapazität ist die molare
Wärmekapazität Cm [J/mol K]=C/n.
Ein Kalorimeter misst die Wärme, die bei chemischen Reaktionen freigesetzt oder aufgenommen werden.
Cgesamt = CWasser + CGerät → Eichung durch Verbrennung einer
Substanz mit bekannter Wärmemenge.
→ oder: Berechnung CWasser, CGerät aus
Temperaturerhöhung nach Zufuhr einer
bekannten Wärmemenge durch elektrische
Heizung.
Q = Cgesamt (T2 – T1)
Beispiel:
Im Bombenkalorimeter wird Traubenzucker verbrannt. Wieviel Wärme
wird pro Mol Traubenzucker frei?
C6H12O6(s) + 6 O2 (g) → 6 CO2 (g) + 6 H2O (l)
T1 = 19,00°C; T2 = 25,50°C bei 3,00 g Traubenzucker/1,20 kg
Wasser-Füllung
CGerät = 2,21 kJ/K
Lösung:
Cgesamt = 1,20 kg ⋅ 4,184 kJkg-1K-1 + 2,21 kJK-1 = 7,23 kJK-1
Q = 7,23 kJ ⋅ K-1 ⋅ (25,5 – 19,0)K = 47,0 kJ
Bei 1 mol Traubenzucker
Zu erwarten ist die folgende Temperaturabhängigkeit der Wärmekapazität bei konstantem Volumen cv für ein ideales Gas aus 2-atomigen Molekülen:
Die Energie pro Freiheitsgrad ist ½ RT (für 1 Mol Gas)
Die Wärmekapazität pro Freiheitsgrad ist dann ½ R.
Bei 3 Freiheitsgraden für Translation, 2 für Rotation und 2 für Schwingung
(potentielle und kinetische Energie) folgt für das 2-atomige Molekül
cv=7/2R.
Die Molwärmen (molare Wärmekapazitäten) von Flüssigkeiten cv,fl sind
i.a. größer als bei Gasen.
z.B. flüssiges Wasser bei 100°C: cv,fl
Wasserdampf bei 100°C:
=
cv,Gas =
Die Ursache liegt in der größeren Zahl von inneren Freiheitsgraden, in die
Energie deponiert werden kann (z.B. intermolekulare Schwingungen in
der Flüssigkeit).
Die Wärmekapazität nimmt bei Flüssigkeiten mit steigender Temperatur
zu (zunehmende Anregung von Freiheitsgraden).
Für die Molwärme von Festkörpern gilt die Dulong-Petitsche Regel
(1819):
cv,solid = 3 R = 25 J/K ⋅ mol
konstant
N ⋅ 3 Gitterschwingungen bei N-Atomen,
k Wärmekapazität pro Schwingungfreiheitsgrad,
2 Freiheitsgrade pro Schwingung (kinetische und potentielle Energie)
→
cv (298 K)
Al
23,8
Cu 23,4 €
Pb 24,7
Bi
25,9
1
N ⋅ 3 ⋅ k ⋅ 2 = 3R
2
bei Nichtmetallen Ausnahmen:
Graphit:
Silizium:
Bor:
8,4 J/K ⋅ mol
19,7 J/K ⋅ mol
11,7 J/K ⋅ mol
Bei höheren Temperaturen nähert sich cv dem Wert von 25 J/K ⋅ mol an.
Bei kleineren Temperaturen gilt cv = a T3 , da nicht alle Gitterschwingungen des Festkörpers angeregt werden können (nur die niederfrequenten
Schwingungen) und ihre Anregung stark mit der Temperatur ansteigt.
Bei T = 0K ist cv = 0 J/mol.K.
2.5.4
Isobarer Wärmeaustausch und Enthalpie
Zustandsfunktionen hängen nur vom gegenwärtigen Zustand des Systems ab, sie sind unabhängig davon, wie dieser Zustand erreicht wurde.
Beispiel: Innere Energie, Enthalpie
Wegfunktionen sind abhängig von dem Weg, wie der Zustand erreicht
wurde. Beispiel: Wärme, Arbeit
Enthalpie
H = U + pV
Zustandsfunktion
ΔH = ΔU + pΔV + VΔp = δQ – pΔV + pΔV + VΔp
isobar:
Δp = 0
→
ΔΗ = δQ
Bei konstantem Druck (z.B. chemische Reaktionen bei Normaldruck) ist
Δp=0 und damit ΔH = δQ, d.h. die ausgetauschte Wärmemenge unter
isobaren Bedingungen entspricht der Änderung der Enthalpie des Systems (griechisch: en = darin, thalpos = Wärme).
Standard-Bildungsenthalpie ΔHf0:
ΔH-Wert, der zur Bildung von 1 mol reiner Substanz aus den reinen
Elementen in ihrer stabilsten Form unter Standard-Bedingungen (Normdruck = 1 atm = 101,325 kPa; Standardtemperatur: meist 25°C) gehört.
Messung von ΔH mit dem Bombenkalorimeter, siehe Kapitel 2.5.3.
Beispiel:
ΔHf0 (CH4) = - 74,9 kJ/mol
f
0
= Formation
= Normbedingungen
Die Reaktionsenthalpien können aus den tabellierten ΔHf0 nach dem Satz
von Hess berechnet werden:
ΔHR0 =
®
®
ΔHf0 (Produkte) -
ΔHf0 (Edukte)
ΔHf0 mit Koeffizienten (Zahl der Mole) aus Reaktionsgleichung multiplizieren
ΔHf0 (Elemente in stabilster Form) = 0
Beispiel:
2NH3 (g) + 3Cl2 (g) → N2 (g) + 6HCl (g)
−46,19
0
0
−92,3
€
0
ΔHR = ?
tabellierte Werte
ΔHR0 = {(6 · [- 92,3]) – (2 · [- 46,19])}kJ/mol = - 461,4 kJ/mol werden bei
dem angegebenen molaren Stoffumsatz frei.
Beispiel:
Die Bildung von Methan aus den Elementen in stabilster Form
C (Graphit) + 2 H2 (g) → CH4 (g)
ΔHR =ΔHf0=?
bzw. die Umkehrreaktion läuft im Bombenkalorimeter nicht direkt ab.
Deshalb wird ΔHR über eine ausgewählte Folge von Reaktionen mit
meßbarer Reaktionsenthalpie bestimmt.
1) C (Graphit) + O2 (g) → CO2 (g)
ΔHR = -393,5 kJ/mol
2) 2 H2 (g) + O2 (g)
→ 2 H2O (l)
ΔHR = -571,8 kJ/mol
3) CO2 (g) + 2 H2O (l) → CH4 (g) + 2 O2 (g) ΔHR = +890,4 kJ/mol
Gl. 1 + Gl. 2 + Gl. 3 =
C + O2 + 2 H2 + O2 + CO2 +2 H2O → CO2 +2 H2O + CH4 + 2 O2
C + 2 H2 → CH4
ΔHR = -393,5 kJ/mol - 571,8 kJ/mol + 890,4 kJ/mol = - 74,9 kJ/mol
Die Bildungsenthalpie von CH4 erhält man also indirekt aus den im
Bombenkalorimeter oder mit anderen Methoden gemessenen Reaktionsenthalpien ausgewählter Reaktionen.
2.6 Wärmeleitfähigkeit
Die Erwärmung von Gas, Flüssigkeit oder Festkörper (Wärmeaustausch)
dauert eine gewisse Zeit, die u.a. von der Wärmeleitfähigkeit der Substanz, der Querschnittsfläche des Wärmeüberträgers und der Temperaturdifferenz zwischen warmer und kalter Stelle abhängt.
Die Wärme fließt dabei immer von der wärmeren zur kälteren Seite, also
in Richtung niedrigerer Temperatur. Zum Transport der Energie durch
Wärmeleitung ist ein Medium erforderlich, da sich der Energieaustausch
durch Schwingung der Festkörperatome und Stoß nur zwischen unmittelbar benachbarten Teilchen vollzieht.
2.7 Reale Gase und Dampfkondensation
Bisher haben wir Gase als Ansammlung punktförmiger Atome oder Moleküle ohne Eigenvolumen und ohne Wechselwirkung aufgefasst (ideales
Gas).
Tatsächlich kondensieren reale Gase aber bei niedrigen Temperaturen
und hohen Drücken spontan, d.h. die Teilchen üben bei geringem Abstand eine anziehende Wechselwirkung aufeinander aus. Das Kondensat
hat ein (relativ zum Gas kleines) Volumen, d.h. die Gasmoleküle haben
ein endliches Eigenvolumen und sind keine Punkte. Das Kondensat lässt
sich nur bei sehr hohen Drücken weiter komprimieren, weil sich bei den
noch kleineren Teilchenabständen einer Flüssigkeit die Teilchen abstoßen: abstoßendes Wechselwirkungspotential.
Oberhalb einer kritischen Temperatur Tc lässt sich das Gas auch bei extremer Verkleinerung des Volumens und entsprechendem Druckanstieg
nicht mehr verflüssigen – es existiert keine Phasengrenze flüssig-gasförmig mehr.
Am kritischen Punkt Tc,pc,Vc existiert diese Phasengrenze gerade nicht
mehr. Jenseits des kritischen Punktes sind die Abstände zwischen den
Molekülen im Dampf genau so groß wie in der Flüssigkeit – es liegt ein
überkritischer Zustand vor ohne Unterschied zwischen Dampf- und Flüssigkeitsdichte.
Die Verdampfungswärme ist knapp unterhalb des kritischen Punktes sehr
gering, so dass die Substanz dauernd zwischen Gas- und Flüssigzustand
wechselt (siehe Schlierenbildung in Vorlesungsversuch 5).
Vorlesungsversuch 5: Reale Gasgleichung und kritischer Punkt
Prinzip: Gasförmiges SF6 wird mit einem Kolben in einem Zylinder zusammengedrückt und der Druck als Funktion des Volumens bei verschiedenen Temperaturen (p-V Isotherme) gemessen. Oberhalb der kritischen Temperatur Tc lässt sich das Gas auch bei den höchsten Drücken
nicht mehr verflüssigen. Wenn unterhalb Tc das Gas kondensiert, steigt
der Druck mit abnehmendem Volumen nicht mehr an, bis aller Dampf
kondensiert ist (Plateaugebiet). Am kritischen Punkt Tc,pc,Vc ist das Plateau gerade nicht mehr zu erkennen. Aus den kritischen Daten lassen
sich die Konstanten der van der Waals Gleichung für reale Gase und die
Parameter des SF6-Wechselwirkungspotentials berechnen.
Messmethode: Eine mit einem Wasserumwälzer thermostatisierte,
hochdruckfeste Glaskapillare mit Volumenskala wird evakuiert und dann
mit SF6 auf Atmosphärendruck gefüllt und geschlossen. Die Messkapillare ist auf eine mit Quecksilber gefüllte, stählerne Druckkammer montiert. Mit einem Handrad wird über einen Spindeltrieb ein Kolben in der
Druckkammer bewegt und über den Kolbenhub das SF6-Gasvolumen
verkleinert (hydraulischer Druck: Beispiel p=F/A = 10kg⋅9,81ms-2/1mm2 =
0,98 MPa ≈ 100bar). Der Gasdruck in der Kapillare wird von einem Federmanometer angezeigt.
Ergebnisbeispiel: SF6
V/cm3
4,0
3,8
3,6
3,4
3,2
3
p/105Pa
10,1
10,5
10,9
11,5
12,1
usw. bis 0,2cm und 5MPa
20°C
p/105Pa
10,3
10,8
11,3
11,9
12,5
30°C
p/105Pa
10,6
11,1
11,7
12,3
13,0
40°C
Zur Beschreibung des realen Verhaltens von Gas: Van der Waals Gleichung:
p
⋅
Vm
= R ⋅T
a
p +
⋅ (Vm − b) = R ⋅T
2
Vm
a = Konstante für Binnendruck durch Molekülanziehung. Der Binnendruck
ist a/V2. b = Eigenvolumen der Moleküle
€
Die van der Waals Gleichung
beschreibt den Druckverlauf überhalb
der kritischen Temperatur richtig.
Je höher die Temperatur, um so
idealer das Verhalten.
Die Parameter a und b der van der Waals Gleichung hängen von den
kritischen Parametern ab:
a = 9/8 ⋅ RTcVc = 9/8 ⋅ 8,314Jmol-1K-1 ⋅ 319K ⋅ 2,62⋅10-4m3mol-1 =
0,78Jm3mol-2 = 0,78m6Pa mol-2
b = 1/3 ⋅ Vc = 1/3 ⋅ 2,62⋅10-4m3mol-1 = 8,73⋅10-5m3mol-1
Vc = 3/8 ⋅ RTc/pc = 3/8 ⋅8,314Jmol-1K-1 ⋅ 319K/3,8⋅106Pa = 2,62⋅10-4m3mol-1
Korrektur des idealen Gasgesetzes für 1 mol Gas mit V = 1m3 bei 300K:
pideal = RT/V = 8,314Jmol-1K-1 ⋅ 300K/1m3 = 2494Pa = 25mbar
Binnendruck a/V2 = 0,78m6Pa mol-2/1m6 mol-2 = 0.78Pa → minimal bei
kleinen Drücken/großen Volumina.
Bei Kompression auf V = 1l = 10-3m3 ist pideal = 25bar und der Binnendruck
a/V2 = 0,78Mpa = 7,8bar
Auch bei Kompression auf V = 10-3m3 bleibt die Korrektur des idealen
Gasgesetzes durch das Eigenvolumen b klein.
Die molekularen Wechselwirkungen des realen Gases lassen sich formal
auch durch eine Expansion der idealen Gasgleichung mit einer Potenzreihe des Drucks mit den Virialkoeffizienten B(T), C(T) usw. beschreiben.
Virialgleichung: pV=RT + B(T)p + C(T)p2 + …… Bei Vernachlässigung
der quadratischen und höheren Terme erhält man durch Koeffizientenvergleich mit der Van der Waals Gleichung:
B(T)=b – a/(RT)= 8,73⋅10-5m3mol-1 - 0,78m6Pa mol-2/(8,314Jmol-1K-1⋅T[K])
B(300K)= - 2,25⋅10-4m3mol-1
Aus den kritischen Daten lassen sich auch die Wechselwirkungspotentiale von Molekülen bestimmen:
E(r) = 4ε[(r0/r)12 - (r0/r)6] = ε[(rm/r)12 - 2(rm/r)6]
Lennard-Jones-Potential
1.Term: Pauli-Abstoßung, wenn sich Elektronen gleichen Spins soweit
nähern, dass die Orbitale übereinander geschoben werden (bedingt das
Eigenvolumen).
2.Term: Anziehung der Moleküle bei größeren Abständen r durch (induzierte) Dipol-Dipol Wechselwirkung (bedingt den Binnendruck).
r0: Abstand der Nullstelle des LJ-Potentials E(r= r0)=0, wo sich Anziehung
und Abstoßung gerade aufheben.
rm: Abstand des Energieminimums vom Ursprung.
ε: ”Tiefe” der Potentialmulde, die durch Anziehung und Abstoßung entsteht.
2.8
Adiabatische Prozesse
Adiabatische Prozesse: kein Wärmeaustausch mit der Umgebung
δQ = 0
ΔU = -pΔV
Wenn System von Umgebung isoliert ist oder Prozesse zu schnell für
Wärmeaustausch ablaufen.
Bei infinitesimal kleinen Änderungen gilt δQ = 0 und dU = -pdV. Die
Energie für die Gasexpansion kann bei der adiabatischen Expansion
wegen δQ = 0 nur aus der inneren Energie des Gases kommen, also dU
= cvdT.
cv dT = −
€
cv
1
1
dT = − nR dV
T
V
cv ∫
€
€
nRT
dV
V
1
1
dT = − nR ∫ dV
T
V
reversible Expansion: äußerer Druck
p = innerer Druck nRT/V
Variablenseparation
Unbestimmtes Integral:
cv lnT = − nRlnV + const
Integrationskonstante: const
lnT = − (nR / cv )lnV + const ′
const’ = const/cv
€
€
€
lnT +
e
nR
lnV = const ′
cv
lnT +
nR
lnV
cv
e lnT ⋅ e
€
€
€
nR
lnV
cv
= e const ′
= e const ′
T ⋅V nR / cv = const ′′
e
const’
= const’’
Mit cp - cv = nR folgt nR/cv = cp-cv/cv = γ-1 mit γ = cp/cv, also
T ⋅V γ −1 = const ′′
€
Für ein Gas folgt dann für die Temperaturen TA und TB und die Volumina
VA und VB
TA ⋅VA
γ −1
= TB ⋅VB
γ −1
Mit T = pV/nR folgt
€
p AVA ⋅VA
γ
€
γ −1
= pBVB ⋅VB
p AVA = pB ⋅VB
γ −1
und daraus die Poissonsche Gleichung:
γ
€
Die Adiabate liegt unterhalb der Isothermen im pV Diagramm, denn wegen cp = cV+nR ist cp > cv, also γ = cp/cv > 1, so dass die Adiabate mit einer
höheren Potenz als die Isotherme abfällt.
Physikalische Ursache: Bei der adiabatischer Expansion sinkt die Gastemperatur und somit auch der Druck stärker als bei der isothermen Expansion (gleichbleibende Gastemperatur).
2.9
Adiabatische Expansion realer Gase (Joule-Thomson Effekt)
Der Joule-Thomson Effekt: Temperaturabnahme eines realen Gases bei
schneller Druckabnahme, also ΔT/Δp>0.
Ziehen sich die Gasmoleküle an, so muss bei der Expansion Arbeit gegen diese Anziehung geleistet werden. Die hierfür aufzubringende Energie wird der kinetischen Energie des Gases entnommen ⇒ Abkühlung.
Stossen sich die Gasmoleküle ab, so wird bei der Expansion kinetische
Energie frei und die Temperatur erhöht sich.
Es gibt für jedes Gas eine Temperatur unterhalb derer sich die Teilchen
anziehen und oberhalb derer sich abstoßen: Inversionstemperatur.
Oberhalb dieser Temperatur erwärmt sich ein Gas bei der Expansion,
unterhalb kühlt es sich ab.
Die Inversionstemperatur für Luft beträgt 659 K, die für H2 202 K und die
für He 35 K.
Beispiel:
Unterhalb der Inversionstemperatur von 866K für N2 ist (ΔT/Δp) > 0 bei
einem zur Verflüssigung nötigen Mindestdruck von 3,39 MPa.
(ΔT/Δp) = 0,25 K bar-1 für N2.
Technische Anwendung: Luftverflüssigung nach Linde.
Vorlesungsversuch 6: Joule Thomson Effekt
Prinzip: Wenn ein unter Druck stehendes reales Gas durch eine Düse in
einen Niederdruckbereich schnell (ohne Wärmeaustausch mit der Umgebung) entspannt wird, kühlt sich das Gas dabei ab (adiabatische Expansion; Sprühdoseneffekt). Bei der Expansion erfolgt das energie-aufwändige Trennen der attraktiven intermolekularen Wechselwirkungen auf Kosten der inneren Energie des Gases und seiner Translationsenergie: Abkühlung. Bei verschiedenen Expansionsdrücken p wird die
Abkühlung ΔT gemessen und der molekülspezifische Joule-Thomson
Koeffizient µ als Steigung ΔT/Δp bestimmt.
Messmethode: Der Stahlzylinder der Joule-Thomson-Apparatur wird mit
100kPa (1bar) CO2 -Überdruck gegen Umgebungsdruck gefüllt , durch
einen Luftwärmetauscher auf Raumtemperatur gebracht, dann durch
Öffnen eines Drosselventils (Fritte) auf Umgebungsdruck entspannt und
dabei die Temperaturdifferenz vor und hinter dem Drosselventil nach 1
Minute Druckentspannung (Gleichgewichtseinstellung!) gemessen. Diese
Prozedur wird für eine Reihe sukkzessiv abnehmender Drucke wiederholt.
Ergebnisbeispiele: µCO2=(ΔT/Δp)adiab=1,084K/bar
Literatur
µCO2=1,16K/bar
µN2 =0,253K/bar
µN2=0,25K/bar
Positiver Joule-Thomson-Koeffizient: Bei Druckerniedrigung Δp<0 kühlt
sich das Gas ab ΔT<0, so daß µ>0 ist. Oberhalb der so genannten Inversionstemperatur (z.B. 866K für N2) wird µN2<0, also führt Druckerniedrigung dann zur Temperaturerhöhung.
Berechnung von µ aus van der Waals Konstanten und Wärmekapazität:
µ =(ΔT/Δp)=1/cp⋅(2a/RT – b)
CO2: a=3,60Pa m6/mol2 b=42,7cm3/mol cp= 366,1J/molK
µCO2=0,795K/bar
2.9.1 Der Temperaturgradient in der Troposphäre
Die Temperatur der Troposphäre nimmt mit der Höhe nahezu linear ab
Nach dem 1. Hauptsatz der Thermodynamik gilt:
dU = δQ − pdV
Für adiabatische Änderungen gilt:
€
dU = −pdV
Die Luft der Atmosphäre kann in guter Näherung als ideales Gas betrachtet werden für das die innere Energie nur von der Temperatur ab€
hängt:
dU = cv dT
cv ist die Molwärme bei konstantem Volumen.
Mit der hydrostatischen Grundgleichung dp = −ρ ⋅ g ⋅ dh folgt:
€
Vdp = −M ⋅ g ⋅ dh
mit dem Molvolumen V und der €
Molmasse M.
€ Gasgesetz gilt:
Nach dem idealen
d(pV) =Vdp + pdV = RdT
Einsetzen in die umgeformte hydrostatische Grundgleichung:
€
RdT − pdV = −M ⋅ g ⋅ dh
Unter der Annahme, dass die Luft beim Aufsteigen adiabatisch expandiert, erhält man:
€
RdT + cv dT = −M ⋅ g ⋅ dh
Umformen:
€
€
(R + cv )dT = −M ⋅ g ⋅ dh
dT −M ⋅ g
=
dh R + cv
Mit g = 9.81 m/s2, M = 29 g/mol, R = 8.314 J/(mol.K) und cv = 20.8
J/(mol.K) erhält man:
€
dT −29 ⋅10 −3 ⋅ 9.81 kg ⋅ m⋅ mol ⋅ K ⋅ s 2
K
=
= −9.8 ⋅10 −3
dh
8.314 + 20.8 mol ⋅ s 2 ⋅ kg ⋅ m
m
Dieses bezeichnet man als den trockenadiabatischen Temperaturgradienten der Luft in der Stratosphäre.
€
Abbildung: Trockenadiabatischer Temperaturgradient in der Troposhäre
2.10 Die Entropie und der 2. Hauptsatz der Thermodynamik
Der 1. Hauptsatz der Thermodynamik beschreibt die Erhaltung der Energie. Er trifft aber keine Aussagen über die Richtung in der ein bestimmter Prozeß abläuft.
Gibt es seine Zustandsfunktion, die sich in Richtung eines spontan (freiwillig) ändernden Prozesses, in definierter Weise ändert?
Diese Zustandsfunktion muß dann im Gleichgewichtszustand einen Extremwert besitzen, der einem Zustand maximaler Stabilität entspricht.
Die gesuchte Zustandsfunktion ist die Entropie S, auch reduzierte Wärme
genannt:
dS =
δQrev
T
Die Gesamtentropie bleibt bei reversiblen Zustandsänderungen in abgeschlossenen Systemen konstant.
€
Bei allen irreversiblen Prozessen nimmt die Gesamtentropie eines Systems immer zu:
ΔSirrev > 0
Da alle in der Natur ablaufenden Prozesse irreversible sind strebt nach
Clausius die Entropie der Welt einem Maximum zu.
€
Der Begriff der Entropie hat durch die statistische Thermodynamik eine
anschauliche Bedeutung bekommen:
Der Zustand maximaler Stabilität ist identisch mit dem Zustand maximaler
Wahrscheinlichkeit. Diese Wahrscheinlichkeit wird dann maximal, wenn
die Anzahl der nichtidentischen Anordnungsmöglichkeiten maximal wird.
Wenn diese Anzahl groß wird, dann kann man das System als ungeordnet bezeichnen, die Entropie wird also mit der “Unordnung” eines Systems verknüpft.
Lokal kann die Entropie aber sehr wohl abnehmen. Bei einer spontanen
Kristallisation nimmt die Ordnung der kristallisierenden Substanz offensichtlich zu, also die Entropie ab. Durch die Erwärmung der Umgebung
durch die freiwerdende Kristallisationswärme, nimmt aber die Entropie in
der Umgebung starker zu, als sie im Kristall abnimmt. Die Gesamtentropie nimmt also auch hier zu.
Boltzmannsche Definition der Entropie:
S = k B ⋅ lnW
mit kB = Boltzmannkonstante und W = wahrscheinlichste Verteilung.
Der 2. Hauptsatz der €
Thermodynamik hat viele Beschreibungen. Die
häufigsten lauten:
• Bei irreversiblen Prozessen nimmt die Gesamtentropie eines Systems immer zu.
• Es ist unmöglich ein Perpetuum mobile 2. Art zu bauen.
• Es existiert kein Kreisprozess, dessen einzige Wirkung darin
besteht, Wärme von einem kälteren Reservoir zu einem wärmeren
Reservoir zu transportieren. (Clausius)
• Es existiert kein Kreisprozess, der eine Wärmemenge aus einem
Reservoir entnimmt und vollständig in Arbeit verwandelt. (Kelvin)
• dS =
€
δQ δWdiss,inn
+
T
T
δWdiss,inn ist hierbei die die innerhalb des Systems dissipierte Arbeit. Die ist immer positiv und erhöht die Entropie. Ein Beispiel ist
die nicht zu vermeidende Reibungswärme in realen Prozessen.
€
2.11 Die freie Energie und freie Enthalpie
Das Produkt TΔS stellt den nichtverwertbaren Anteil an isotherm erzeugter (Volumen)Arbeit aus innerer Energie dar. Diese wird in Form von
Abwärme an die Umgebung abgeführt. Die maximal mögliche Arbeit ist
dann:
€
ΔF = ΔU −TΔS
€
ΔF stellt eine weitere Zustandsgröße dar und wird als freie Energie
(Helmholtzenergie) bezeichnet.
€
Die freie Energie wird also bei isothermen Prozessen bei denen Volumenarbeit verrichtet wird verwendet.
Unter isobaren Bedingungen ist eine andere Zustandsfunktion von Belang, die freie Enthalpie (Gibbsenergie).
ΔG = ΔH −TΔS
2.12 Der dritte Hauptsatz der Thermodynamik
€
“Unerreichbarkeit des absoluten Nullpunkts”
lim ΔSkond = 0
T →0
€
Die drei Hauptsätze der Thermodynamik zusammen:
1. Unmöglichkeit eines Perpetuum mobiles 1. Art
2. Unmöglichkeit eines Perpetuum mobiles 2. Art
3. Unmöglichkeit der Erreichbarkeit des absoluten Nullpunkts