Skript
Werbung

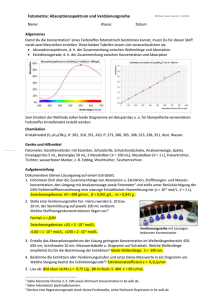
Physikalische Chemie 2005 T Fos Physikalische Chemie 2 0. SYMBOLE UND FORMELN .................................................................................................................... 1 0.1 0.2 0.3 1. LISTE WICHTIGER SYMBOLE ...................................................................................................................... 1 AUSWAHL VON FORMELN.......................................................................................................................... 2 LITERATURANGABEN ................................................................................................................................ 2 EINFÜHRUNG............................................................................................................................................ 3 1.1 GESCHICHTLICHE ENTWICKLUNG .............................................................................................................. 3 1.2 GEBIETE DER PHYSIKALISCHEN CHEMIE ................................................................................................... 4 1.3 PHASENDIAGRAMME, PHASENÜBERGÄNGE, WÄRMEKAPAZITÄT (WIEDERHOLUNG)................................. 6 1.3.1 Berechnung der Wärmemenge für Temperaturänderungen innerhalb einer Phase...................... 10 1.3.2 Phasenübergänge in Einstoffsystemen .......................................................................................... 13 1.3.3 Phasenübergang Flüssigkeit / Gas............................................................................................... 16 1.3.4 Phasenübergang Feststoff / Flüssigkeit........................................................................................ 23 1.3.5 Phasenübergang Feststoff / Gas................................................................................................... 24 1.3.6 Erwärmung und Phasenübergang................................................................................................. 25 0. Symbole und Formeln 0.1 Liste wichtiger Symbole b = Molalität [mol kg-1] c = Stoffmengenkonzentration [mol L-1] cp = spezifische Wärmekapazität bei konstantem Druck [J g-1 K-1] cV = spezifische Wärmekapazität bei konstantem Volumen [J g-1 K-1] Cp = molare Wärmekapazität bei konstantem Druck [J mol-1 K-1] CV = molare Wärmekapazität bei konstantem Volumen [J mol-1 K-1] ES = molale ebbuloskopische Konstante [K kg mol-1] F = Freiheitsgrad G = freie Enthalpie [J mol-1] H = Enthalpie [J mol-1] i = van´t Hoffscher Faktor K = Komponente Kf = molale kryoskopische Konstante [K kg mol-1] m = Masse [g, kg] M = molare Masse g mol-1 n = Stoffmenge [mol] N = Teilchenzahl NA = Avogadrozahl p = Druck [bar, Pa, atm, Torr] P = Leistung [W,J/s] P = Phase Q = Wärmemenge [J] R = Gaskonstante 8,314 J mol-1 K-1 R = Anzahl der Beziehungen unter den Teilchen S = Entropie [J mol-1 K-1] S = Teilchenzahl t = Zeit [s, min, h, etc] T = Temperatur [K] U = innere Energie [J mol-1] V = Volumen [m3, L, cm3] x = Stoffmengenanteil allgemein oder Stoffmengenanteil in der Flüssigphase y = Stoffmengenanteil in der Gasphase α = Dissoziationsgrad β = Massenkonzentration [g / L] ϑ = Temperatur [0C] µ = chemische Potential J mol-1 ω = Massenanteil ϕ = Volumenanteil σ = Volumenkonzentration Π = Osmotischer Druck [Pa, bar etc.] ν = Mögliche Teilchenzahl Seite 1 Physikalische Chemie 2005 0.2 T Fos Auswahl von Formeln Stoffmenge ni = Stoffmengenanteil xi = mi Mi ni = ni n Gesamt K = S−R Q = m ⋅ c p ⋅ ∆T Phasen, Komponenten etcWärmemenge N NA F=K−P+2 Q = n ⋅ C p ⋅ ∆T p⋅V = n ⋅R ⋅T H m , Verd. S dp = = m , Verd. Clausisus-Clapeyronsche-Gleichung dT T(Vm , Dampf − Vm , Flüssigkeit ) ∆Vm , Verd. Ideales Gasgesetz ⎛ T *T ⎞ p H m , Verd. = R * ⎜⎜ 2 1 ⎟⎟ * ln 2 p1 ⎝ T2 − T1 ⎠ B ϑ+C G = H − T *S Q P= t p A = x A * p 0, A log p = A − Antoine – Gleichung Gibbs-Helmholtz-Gleichung Leistung real Raoult ideal Siedepunktserhöhung ′ ∆TSiedepunkt = Es * c = Es * b Molmassenbestimmung M2 = Gefrierpunktserniedrigung ′ ∆TGefrierpunkt = K f * c = K f * b ∆T = p A = γ i * x A * p0, A R * T 2 * n2 * M 1 H m,Verd . * m1 Es * m2 R * T 2 * M 1 m2 = * ∆T * m1 H m ,Verd . ∆T * m1 M2 = K f Kf = m2 m1 * ∆T Osmotischer Druck Π = i * cB * R * T van´t Hoffscher Faktor i = (1 − α ) + ν * α Seite 2 Π=− R * T * ln xLm Vm , Lm R * T 2 * M1 H m , Sch Physikalische Chemie 2005 0.3 T Fos Literaturangaben Einführung in das chemische Rechnen Hübschmann/Links 10. Auflage 2000 Verlag Handwerk und Technik Hamburg ISBN 3.582.01231.X 30,20 € Arbeitsbuch Physikalische Chemie Atkins, Trapp 3. Auflage 2001 VCH Weinheim 42,90 € Basiswissen Physikalische Chemie Claus Czeslik, Heiko Seemann, Roland Winter 1. Auflage 2001 Teubner 39,90 € Elektrochemie Hamann, Vielstich 1998 VCH Weinheim 62,90 € Kurzlehrbuch Physikalische Chemie Peter W. Atkins 3. Auflage 2001 VCH Weinheim 52,90 € Lehrbuch der Physikalischen Chemie Gerd Wedler 5. Auflage 2004 VCH Weinheim 79,00 € Physikalische Chemie Peter W. Atkins 3. Auflage 75 € 3. Auflage 2001 VCH Weinheim Physikalische Chemie und Biophysik Adam, Läuger, Stark 4. Auflage 2003 Springer 39,95 € Reaction Kinetics Pilling, Seakins 1. Auflage 1995 Oxford Science 45,49 € Thermodynamik Lüdecke, Lüdecke 1. Auflage 2000 Springer 49,95 € Seite 2 Physikalische Chemie 2005 1. Einführung 1.1 Geschichtliche Entwicklung T Fos Die Chemie wird in weitere Teilgebiete unterteilt. Eine übliche Unterteilung ist Anorganische Chemie, Organische Chemie, Physikalische Chemie und Technische Chemie. Die konsequente Anwendung physikalischer Methoden auf die Erforschung chemischer Vorgänge begann Ende des 18. Jahrhunderts, doch erst Ende des 19. Jahrhunderts hatte sich die Physikalische Chemie als eigenes Wissensgebiet herausgebildet. Hier eine Liste von bedeutenden Enddeckungen aus der Zeit der Bildung der Physikalischen Chemie als eigenständiges Teilgebiet der Chemie 1664, 1676 1774 1801 1801 1802, 1807 1803 1811 1824 1834 1833 1837 1840 1847 1850 1850, 1851 1853, 1859 1856,1857 1860 1860 1867 1873 1877 1877 1886 1887 1889 Gasgesetz nach Boyle und Mariotte p1 ⋅ v1 = p 2 ⋅ v 2 = konst. Lavoisier Konstanz der Gesamtmasse bei chemischen Umsetzungen Berthollet Chemische Gleichgewicht Dalton Partialdruckgesetz Gay-Lussacsche Gesetze Gasvolumen f (T) Henrysche Gesetz Löslichkeit von Gasen in Flüssigkeiten Avogadro Gleich viele Moleküle eines idealen Gases bei gleicher Temperatur nehmen das gleiche Volumen ein Carnot Kreisprozess Clapeyron Temperaturabhängigkeit des Dampfdruckes Faraday Gesetze bei der Elektrolyse Daniell Erste galvanische Kette mit konstanter EMK Hess-scher Satz Reaktionsenthalpie einer Reaktion ist unabhängig vom Reaktionsweg Helmholtz 1. Hauptsatz Wilhelmy Reaktionsgeschwindigkeit, Rohrzuckerinversion Clausius, Kelvin 2. Hauptsatz Hittorf Überführung gelöster Salze beim Stromdurchgang durch eine elektrolytische Zelle Krönig, Clausius Kinetische Gastheorie, mittlere freie Weglänge Maxwell Geschwindigkeitsverteilung in Gasen Kirchhoff, Bunsen Spektroskopie Guldberg, Waage Massenwirkungsgesetz Kohlrausch Unabhängige Wanderung der Ionen Pfeffer Osmotischer Druck Boltzmann Energieverteilung (Entropie und Zustandwahrscheinlichkeit bei Gasen Raoult Dampfdruckerniedrigung bei Lösungen Arrhenius Dissoziation gelöster Elektrolyte Arrhenius Aktivierungsenergie, Seite 3 Physikalische Chemie 2005 1891 Nernst 1895 1897 1900 1902 1905 1906 1911 1913 1922 1923 1925 1926 1927 1918, 1919 1927 Röntgen Bodenstein Planck Gibbs Einstein Nernstsches Wärmetheorem Rutherford Bohr Compton Debye-Hückel Heisenberg, Born, Jordan Schrödinger Heisenberg Born, Landé Heitler, London 1930 London 1935 Eyring 1.2 T Fos Temperaturabhängigkeit von Reaktionsgeschwindigkeitskonstanten Verteilungssatz ( Löslichkeit einer Substanz in verschiedenen Lösungsmitteln Röntgenstrahlung Gasreaktionen Schwarzer Strahler Statistische Thermodynamik Äquivalenz von Energie und Masse 3. Hauptsatz Versuche mit Alphateilchen Theorie des Atombaus Comptoneffekt Theorie für starke Elektrolyte Quantenmechanik Theorie der Wellenmechanik Unschärfenrelation Gitterenergie polarer Feststoffe Berechnung unpolarer chemischer Bindungen Theorie der waalsschen Wechselwirkungen Theorie des Übergangszustandes Gebiete der Physikalischen Chemie Die Unterteilung der Physikalischen Chemie in weitere Teilgebiete ist je nach zur Hand genommenem Lehrbuch unterschiedlich. Hier eine Einteilung aus dem Buch Basiswissen Physikalische Chemie von Czeslik, Seemann und Winter Aggregatzustände Ideale Gase Reale Gase Flüssigkeiten Kristalline Festkörper Thermodynamik Erster Hauptsatz der Thermodynamik Zweiter Hauptsatz der Thermodynamik Mischungen Chemische Gleichgewichte Phasendiagramme Aufbau der Materie Grenzen der klassischen Physik Einführung in die Quantenmechanik Mikroskopische Teilchen in Bewegung Atome Moleküle Photoelektronenspektroskopie Seite 4 Physikalische Chemie 2005 T Statistische Thermodynamik Isolierte Systeme Geschlossene Systeme Offene Systeme Anwendung: Ideale Gase Das Äquipartitionstheorem Anwendung: Wärmekapazität kristalliner Festkörper Grenzflächenerscheinungen Einleitung Die Oberflächenspannung Gleichgewichtsbedingungen für gekrümmte Oberflächen Thermodynamische Oberflächengrößen Oberflächenerscheinungen von Mischungen Gasadsorption an Festkörperoberflächen Elektrochemie Ionentransport in Elektrolytlösungen Thermodynamische Eigenschaften von Ionen in Lösungen Aktivitätskoeffizienten in Elektrolytlösungen Elektrochemische Thermodynamik Technisch wichtige Zellen Elektrolyse und Potentiale von Zellen unter Belastung Reaktionskinetik Grundbegriffe und Messmethoden Einfache Geschwindigkeitsgesetze (Formalkinetik) Bestimmung der Geschwindigkeitsgleichung Temperaturabhängigkeit der Geschwindigkeitsgleichung Komplexe Reaktionen Theorie der Elementarreaktionen Reaktionen in Lösungen, Reaktionen von Ionen Molekülspektroskopie Elektrische Eigenschaften der Materie Prinzipien der Spektroskopie Reine Rotationsspektren Schwingungsspektroskopie RAMAN-Spektroskopie Elektronenschwingungsspektren von Molekülen NMR-Spektroskopie Elektronen-Spin-Resonanz (ESR) Seite 5 Fos Physikalische Chemie 2005 1.3 T Fos Phasendiagramme, Phasenübergänge, Wärmekapazität (Wiederholung) Die Phasen (fest, flüssig, gasförmig) lassen sich anschaulich in Phasendiagrammen darstellen. Einfache Diagramme sind z.B. p,T, p,V oder T,V Diagramme. Diagramm 1-1 Dreidimensionale Diagramme sind komplexer und bedürfen mehr Erläuterung Es scheint uns einfach selbstverständlich, dass z.B. Wasser als eine Komponente anzusehen ist, die in den drei Phasen fest, flüssig und gasförmig auftreten kann. Diagramm 1-2 Seite 6 Physikalische Chemie 2005 T Fos Die Begriffe Phasen und Komponenten sollen hier etwas genauer beleuchtet werden Phase: Eine Phase ist bezüglich ihrer chemischen Zusammensetzung und ihres physikalischen Zustandes durch und durch gleichförmig Ein homogener Bereich in einem heterogen System wird als Phase bezeichnet; d.h. nun aber nicht, dass eine Phase nicht aus mehreren Komponenten bestehen kann. Beispiele für Phasen: Gas oder Gasmischung Eine Flüssigkeit Eine Flüssigkeit mit einem gelösten Gas Eine Flüssigkeit mit zwei vollkommen miteinander mischbaren Flüssigkeiten * Eine Flüssigkeit mit gelösten festen Stoffen Ein Kristall * Flüssigkeitsmischungen bei denen eine Komponente in deutlichem Überschuss vorliegt werden als Lösungen bezeichnet. Beispiele: Eis ob als Block oder in kleinen Stücken stellt eine Phase dar. Eine Eis - Wasser - Mischung (z.B. Schneematsch) besteht jedoch aus zwei Phasen, fest und flüssig. Eine Legierung aus 2 Metallen kann sowohl ein Einphasensystem als auch ein Zweiphasensystem darstellen, je nachdem ob die 2 Metalle mischbar oder nicht mischbar sind. Man spricht von e i n e r P h a s e, wenn diese im m o l e k u l a r e n B e r e i c h gleichförmig ist. So ist z.B. eine Dispersion aus Feststoffen und Flüssigkeit im makroskopischen Bereich gleichförmig, aber im molekularen Bereich heterogen. An diesem Beispiel kann man schon erahnen, dass es auch Grenzfälle geben wird, die nicht eindeutig zuordenbar sind. Seite 7 Physikalische Chemie 2005 T Fos Komponente: Eine chemische Verbindung oder ein chemisches Element stellt eine Komponente dar; so ist z.B. Wasser ein Einkomponentensystem K = 1. Betrachten wir eine Mischung aus Ethanol und Wasser so haben wir ein Zweikomponentensystem. Nun ist die Anzahl der Komponenten aber nicht einfach die Anzahl der chemischen Bestandteile, sondern es gibt hier eine Einschränkung. Die Anzahl der Komponenten ist die Anzahl der voneinander unabhängigen chemischen Bestandteile. Die Anzahl der Komponenten gibt an, wie viele unabhängige Substanzen man mindestens braucht, um die Zusammensetzung in allen Phasen des Systems zu beschreiben. 1. Beispiele: NaCl in Wasser Zunächst würde man vielleicht spontan sagen wir haben 2 Komponenten, nämlich NaCl und Wasser. Wir müssen die Sache jedoch etwas genauer betrachten und ermitteln wie viele Teilchenarten wir haben. Wassermoleküle, Natriumionen und Chloridionen Teilchenzahl, S = 3 Die einzelnen Teilchen sind jedoch nicht unabhängig voneinander. Es muss gelten, dass die Lösung elektrisch neutral ist und somit die Anzahl der Natriumionen gleich der der Chloridionen ist. K=S-R (K = Komponente, S = Teilchenzahl, R = Beziehungen unter Teilchen,) 2. Beispiel Ein System bestehend aus H2O, H2 und O2 bei Raumtemperatur stellt 3 Komponenten dar. Bei sehr hohen Temperatur können diese Stoffe über die thermische von → Wasser, jedoch ein 1 Komponentensystem sein. H 2 O ← H 2 + 0,5 O 2 Seite 8 Physikalische Chemie 2005 T Fos Von Gibbs (ca. 1875) stammt nun das Phasengesetz, welches den Zusammenhang von Phasen, Komponenten und Freiheitsgraden aufzeigt. F = K – P +2 F= P= K= Anzahl von Varianz, Freiheitsgrad Anzahl der intensiven (mengenunabhängigen) Zustandsgrößen, die unabhängig voneinander variiert werden können ohne dass die Anzahl Phasen sich verändert. z.B. Druck, Temperatur, Konzentration Anzahl der Phasen (z.B. feste, flüssig, gasförmig) Komponenten (Anzahl der voneinander unabhängigen chemischen Substanzen) Bei Einkomponentensystemen gibt es drei Möglichkeiten: a) Eine Phase P = 1, eine Komponente K = 1 z.B. ein Gas F = 1 - 1 +2 = 2 d.h. es können die Zustandsgrößen Druck und Temperatur in weiten Bereichen unabhängig voneinander variiert werden ( ohne dass z.B. eine flüssige - oder feste Phase auftritt). b) Zwei Phasen P = 2, eine Komponente K = 1, z.B. Wasser/Wasserdampf F=1-2+2=1 Geben wir z.B. die Temperatur mit 100°C vor so ist für ein Gleichgewicht der Druck automatisch mit dem Atmosphärendruck festgelegt. Senken wir die Temperatur auf 20°C haben wir im Gleichgewicht eine Druck von 23,4 mbar ( 25°C 32 mbar) ( Druck oder Temperatur ist variabel). c) Drei Phasen P = 3, eine Komponente K = 1 z.B. Eis, Wasser, Wasserdampf F=1-3+2=0 Wir befinden uns am Tripelpunkt. Der Druck ist mit 0,00611 bar und die Temperatur mit 273,16 K festgelegt. Das Phasengesetz lässt sich nun aber auch auf Mehrkomponentensysteme oder auch auf chemische Reaktionen anwenden. So erhält man z.B. bei einem Zweikomponentensystem mit nur einer Phase drei Freiheitsgrade F=K-P+2 =2-1+2=3 Seite 9 Physikalische Chemie 2005 T Fos Die vollständige Darstellung aller Zustände wird somit dreidimensional p x T In der Praxis wird meist eine intensive Variable konstant gehalten (z. B. Druck p oder Temperatur T) und die andere gegen die Zusammensetzung x variiert (zweidimensionale Darstellung). z.B. Dampfdruckdiagramm p = f (x), T = const. oder Siedediagramm T = f (x), p = const. T= const. p x Bei Systemen mit mehr als 2 Komponenten kann man angeben, dass die Zahl der maximalen Freiheitsgrade ≥ 4 wird. F = 3-1 +2 = 4 F= 4-1 +2 = 5 Die Darstellung in der Zeichenebene wird schwieriger. Mehrere intensive Variable müssen gleichzeitig konstant gehalten werden. C Hält man z.B. in einem Dreikomponentensystem p und T konstant kann man Dreieckskoordinaten für die Darstellung wählen. A B 1.3.1 Berechnung der Wärmemenge für Temperaturänderungen innerhalb einer Phase Wollen wir eine Substanz erwärmen so führen wir Energie in Form von Wärme zu. Für die quantitative Berechnung benötigen wir die Wärmekapazität. Ist diese auf die Masse bezogen so sprechen wir von spezifischer Wärmekapazität c, ist sie auf die Stoffmenge bezogen von der molaren Wärmekapazität C. Weiterhin muss beachtet werden, ob dies bei konstantem Volumen (cV bzw. CV ) oder bei konstantem Druck (cp bzw. Cp ). Bei Festkörpern und Flüssigkeiten wird diese Unterscheidung meist vernachlässigt, da der Unterschied hier sehr gering ist ( Volumenarbeit und Änderung der inneren Energie). Seite 10 Physikalische Chemie 2005 T Fos Im Falle des idealen Gases gilt C p − C V = R Beispiele: a) Für flüssiges Wasser findet man eine molare Wärmekapazität Cp von 75,291 J K-1 mol-1 Wie groß ist die spezifische Wärmekapazität ? Welche Energie wird benötigt um 1 kg Wasser von 250C auf 800C zu erwärmen ? cp = Cp M = 75,291 J ⋅ mol K ⋅ mol 18,015 g Q = m ⋅ c p ⋅ ∆T = 1 ⋅ 4,179 ⋅ 55 = 4,179 J kJ kcal = 4,179 =1 K⋅g K ⋅ kg K ⋅ kg kg ⋅ kJ ⋅ K = 229,86 kJ K ⋅ kg Einige Wärmekapazitäten zum Vergleich: Stoff Heptan (l) Ethanol(l) Aceton(l) Aluminium(s) Eisen(s) Sauerstoff(g) Wasserstoff(g) Wasser(l) Wasser(g) Cp [J/mol*K] 224,3 111,46 124,7 24,35 25,10 29,355 28,824 75,291 33,58 cp [ kJ/kg*K] 2,238 2,419 2,147 0,902 0,449 0,917 14,298 4,179 1,864 Tabelle 1-1 b) 10 Liter Wasserdampf bei Standarddruck (der Einfachheit halber als ideales Gas betrachten) werden von 1000C auf 1500C bei konstantem Volumen erwärmt. Welche Wärmemenge ist notwendig ? Welche Wärmemenge ist für 1 kg Wasserdampf notwendig ? p⋅V = n ⋅R ⋅T V = 10 Liter V = const. p ⋅ V 1,013 ⋅ 10 N ⋅ 10 ⋅ 10−3 m3 ⋅ mol ⋅ K Nm ⋅ mol n= = = 0,327 = 0,327 mol 2 R ⋅T J m ⋅ 8,314J ⋅ 373K 5 Q = n ⋅ C V ⋅ ∆T Q = 0,327mol ⋅ 25,27 C V = C p − R = 33,58 J 8,314J J − = 25,27 mol ⋅ K mol ⋅ K mol ⋅ K J ⋅ 50K = 0,413kJ mol ⋅ K Q = m ⋅ c V ⋅ ∆T = 1kg ⋅ 1,403 kJ ⋅ 50K = 70,15kJ kg ⋅ K Seite 11 Physikalische Chemie 2005 T Übungen: Welche Wärmemenge wird benötigt um einen Aluminiumblock von 10 kg von Raumtemperatur auf 300°C zu erwärmen ? 2480,5kJ Welche Wärmemenge muss abgeführt werden um 5 kg Wasser von 100°C auf Raumtemperatur abzukühlen? 1567,1kJ Seite 12 Fos Physikalische Chemie 2005 T Fos 1.3.2 Phasenübergänge in Einstoffsystemen Schmelzen, Sieden, Sublimieren, Erstarren, Kondensieren, Desublimieren sind Beispiele von Phasenübergängen. Betrachten wir den thermodynamischen Gleichgewichtszustand in einem Einstoffsystem mit mehr als einer Phase so muss gelten: TPhase1 = TPhase2 pPhase1 = pPhase2 Auch das chemische Potential µ muss gleich sein, das heißt die Freie Enthalpie G für diese Komponente muss gleich sein: µPhase1 = µPhase2 Anmerkung: G kann gleich µ gesetzt werden wenn nur eine Komponente vorhanden ist. ⎛ ∂G ⎞ ⎟⎟ µi = ⎜⎜ ⎝ ∂n i ⎠ p , T , n j ...( j≠ i ) Hat z.B. Dampf bei einem bestimmten Druck und bei einer bestimmten Temperatur ein kleineres chemisches Potential als die entsprechende Flüssigkeit, so hat die Flüssigkeit das bestreben zu verdampfen. Die Thermodynamik macht jedoch keine Aussage über die Geschwindigkeit mit der diese Vorgänge ablaufen. Bei Gasen, Flüssigkeiten und Lösungen stellen sich die Gleichgewichte meist schnell ein. Bei Festkörpern kann eine thermodynamische Instabilität eingefroren sein. µ C , Graphit < µ C, Diamant ∆ B G Θm = 0 ∆B = Bildungs- kJ kJ für Graphit und ∆ B G Θm = 2,9 für Diamant mol mol m = molar Θ = Standardwerte bei 25 0C, 1,013 bar Seite 13 Physikalische Chemie 2005 T Fos Für ein Phasengleichgewicht zwischen Gas und Flüssigkeit, sowie für ein Phasengleichgewicht zwischen Flüssigkeit und Feststoff gilt: µ Gas = µ Flüssigkeit Die Richtung der Temperatur - und Druckabhängigkeit kann man aus der Thermodynamik ableiten: und µ Flüssigkeit = µ Feststoff chemisches Potential, µ ⎛ dµ ⎞ ⎜ ⎟ = −S und aus dµ = −S * dT ⎝ dT ⎠ P ergibt sich, dass das chemische Potential µ mit der Temperatur abnimmt. ⎛ dµ ⎞ ⎜⎜ ⎟⎟ = V und aus dµ = V * dp ⎝ dp ⎠T hoher Druck Festkörper Flüssigkeit niedriger Druck Schmelz punkt erhöht Siedepunkt erhöht Gas Temperatur, ϑ Diagramm 1-3 ergibt sich, dass das chemische Potential µ mit dem Druck zunimmt. Bei der Umwandlung von Flüssigkeit in Dampf bei konstanter Temperatur wird Energie verbraucht. Diese in Form von Wärme zugeführte Energie wird in erster Linie zur Überwindung der Anziehungskräfte zwischen den Molekülen der Flüssigkeit verbraucht (innerer Energie U). Als Zweitens ist jedoch das Volumen des Dampfes meist um drei Größenordnungen über der der Flüssigkeit, so dass auch eine Volumenarbeit (p . dV) zu leisten ist. dH Verdampfung = dU Verdampfung + p ⋅ dV Bei der Verdampfung von Wasser bei 100°C und Normaldruck wird z.B. 92,5% zur Erhöhung der inneren Energie benötigt und nur 7,5% wird für die Volumenarbeit gebraucht. Die Größe der Verdampfungsenthalpie HVerd. ist nun abhängig von der Temperatur bei der diese Verdampfung erfolgt. Seite 14 Physikalische Chemie 2005 T Fos E. Clapeyron fand 1834 eine Beziehung die diesen Zusammenhang gut beschreibt. Die thermodynamische Begründung wurde 1850 von R. Clausius geliefert. H m , Verd. S dp = = m , Verd. dT T(Vm , Dampf − Vm , Flüssigkeit ) ∆Vm , Verd. H m , Verd. = H m , Gas − H m , Flüssigkeit Sm , Verd. = Sm , Gas − Sm , Flüssigkeit Für Wasser ergeben sich bei 25°C und 1013 mbar kJ kJ ∆ B H Θm,Gas = −241,82 ∆ B H Θm,Flüssigkeit = −285,83 mol mol J J SΘm , Gas = 188,83 SΘm , Flüssigkeit = 69,91 mol * K mol * K Die Clausius-Clapeyronsche Beziehung kann aus der Thermodynamik abgeleitet werden. Sie zeigt, dass die für die Verdampfung aufgebrachte Energie sich in der Unordnung der Entropie wiederfindet. Die Clausius-Clapeyronsche Beziehung kann jedoch allgemein angewendet werden, das heißt nicht nur für den Übergang Flüssigkeit - Gas sondern auch für Fest - Flüssig oder allgemein Phase 1 - Phase 2 Die Clausius-Clapeyronsche Gleichung stellt einen Bezug zwischen der Temperaturabhängigkeit des Druckes mit der Enthalpie her. Es kann sich hierbei sowohl um Schmelz- , Verdampfungs- , Sublimations- oder Modifikationsänderungen handeln. Seite 15 Physikalische Chemie 2005 T Fos 1.3.3 Phasenübergang Flüssigkeit / Gas Für den Phasenübergang Flüssigkeit / Gas ergibt sich die Clausius-Clapeyronsche Gleichung in der molaren Form zu: H m , Verd. dp = dT T(Vm , Dampf − Vm , Flüssigkeit ) Man kann die Gleichung einerseits dazu benutzen, den Sättigungsdampfdruck bei bekannter Verdampfungsenthalpie und bei bekannten Flüssigkeits- und Dampfdichten zu berechnen. Andererseits lässt sich bei Kenntnis des Dampfdruckverhaltens und der Dichten die Verdampfungsenthalpie berechnen. Bei genauer Betrachtung ergeben sich allerdings einige Probleme weil die Verdampfungsenthalpie ebenfalls temperaturabhängig ist. dH m, Verd. = C p, Gas − C p, Fl. dT und CP = a + b * T + c T2 Cp = molare Wärmekapazität bei konstantem Druck des Gases bzw. der Flüssigkeit die Abhängigkeit des Dampfdruckes von der Temperatur ergibt dann selbst in differentieller Form einen komplexen Ausdruck. Für die Ermittlung des Dampfdruckes in Abhängigkeit von der Temperatur stehen uns eine ganze Reihe von Möglichkeiten offen: a) b) Anwendung der Clausius-Clapeyronsche Gleichung für kleine Druck- und Temperaturdifferenzen. Die Differentiale d werden dann einfach durch Differenzen ∆ ersetzt. Vereinfachung der Clausius-Clapeyronsche Gleichung und anschließende Integration c) Eine Reihe einfacher und komplexer empirischer Ansätze d) Berechnung mit Hilfe thermodynamischer Größen Seite 16 Physikalische Chemie 2005 a) T Fos Beispiel: Berechnung der Verdampfungsenthalpie von Wasser Für kleine Bereiche d durch ∆ ersetzt. H m,Verd. ∆p = ∆T T(Vm,Dampf − Vm,Flüssigkeit ) p1 = 900 mbar bei ϑ1 = 96,71°C p2 = 1200 mbar bei ϑ2 = 104,81°C Vm,Wasserdampf = 30,147 Liter * mol-1 bei ϑ = 100°C Vm,fl. Wasser = 0,018 Liter * mol-1 bei ϑ = 100°C Werte: gesucht ist die molare Verdampfungsenthalpie bei 100°C ∆p * T * (Vm , Dampf − Vm , Flüssigkeit ) ∆T 30000 N * 373,15K * 30,129L 30000 N * 373,15K * 30,129L * 1m3 41639 Nm = = = m 2 * 8,1K * mol m 2 * 8,1K * mol * 1000L mol kJ 41639 Nm = = 41,64 mol mol H m , Verd. = H m , Verd. H m , Verd. Der berechnete Wert liegt nahe bei dem Literaturwert von 40,71 kJ/mol b) Vereinfachung der Clausius-Clapeyronsche Gleichung und anschließende Integration Vor der Integration werden 3 Vereinfachungen / Annahmen eingeführt: Vm,Fl. wird vernachlässigt weil Vm,Fl. oft nur ca. 1/1000 des Gasvolumens Vm,Gas ist. Für Vm,Gas wird die Gültigkeit des idealen Gasgesetzes angenommen (z.B. ergibt sich für p*V Wasserdampf bei 100°C ein Fehler von ca. 1,5% ⇒ z id. = 1 und z real = = 0,985 mit R *T z = Kompressibilitätsfaktor Die Verdampfungsenthalpie wird als temperaturunabhängig angenommen. H m , Verd. ≠ f (T ) Bei Anwendung der Gleichung ist zu überprüfen ob diese drei Vereinfachungen auch vertretbar sind. Seite 17 Physikalische Chemie 2005 T Fos Ableitung: H m , Verd. dp = dT T * Vm , Gas Vm , Gas = dp H m , Verd. * p = dT T2 * R R *T p T p2 dp H m , Verd. dT = * 2 p R T dp H m , Verd. 2 dT ∫ p = R * T∫ T 2 p1 1 Ergibt: ln p 2 H m , Verd. ⎛ 1 1 ⎞ * ⎜⎜ − ⎟⎟ = p1 R ⎝ T1 T2 ⎠ oder einfacher ln p 2 H m , Verd. ⎛ T2 − T1 ⎞ ⎟⎟ * ⎜⎜ = p1 R ⎝ T2 * T1 ⎠ Die Gleichung kann nun nach p, T oder Hm,Verd. aufgelöst werden. ⎛ H m ,Verd. ⎛ T2 − T1 ⎞ ⎞ ⎜ ⎟⎟ *⎜⎜ ⎜ R T2 *T1 ⎟⎠ ⎟⎠ ⎝ ⎝ p 2 = p1 * e T1 = oder ⎛ T *T ⎞ p H m , Verd. = R * ⎜⎜ 2 1 ⎟⎟ * ln 2 p1 ⎝ T2 − T1 ⎠ 1 ⎛ p 2 H m , Verd. ⎞ R ⎜⎜ ln + ⎟⎟ * ⎝ p1 R * T2 ⎠ H m , Verd. Beispiel: p 2 = p1 Berechnung der Dampfdrucke von Wasser bei verschiedenen Temperaturen mit Hilfe der soeben abgeleiteten Beziehung . Die Verdampfungsenthalpie wird im betrachteten Bereich als konstant angenommen. ⎛ H m,Verd . ⎛ T2 − T1 ⎞ ⎞ ⎜ ⎟⎟ *⎜⎜ ⎜ R T2 *T1 ⎟⎠ ⎟⎠ ⎝ ⎝ *e Hm,Verd.(H2O, 60,9°C) = 42,47 kJ/mol R = 8,314 J K-1 mol-1 p1(H2O, 60,9°C) = 207,58 mbar Hm,Verd. / R = 5108,3 K Temperatur 0 C K 0 20 40 60 60,9 80 100 120 273,15 293,15 313,15 333,15 334,05 353,15 373,15 393,15 T1 = 334,05 K gemessene - Werte - berechnetet p [mbar] p [mbar] % d. gem. Wertes 6,10 6,86 112,5 23,4 24,6 105,1 73,8 74,8 101,4 199 199,2 100,1 207,6 473 474,7 100,3 1013 1030,6 101,7 ? Seite 18 Physikalische Chemie 2005 T Fos Tabelle 1-2 Übungen: Welchen Dampfdruck berechnet sich für Wasser von 120°C? 2,068 Bar Literatur Handbook of Chemistry and Physics 1,985 Bar Bei kleinen Temperatur- oder Druckunterschieden im Vergleich zum gemessenen Ausgangswert kann die Verdampfungsenthalpie als konstant angesehen werden (wenn man weit genug vom kritischen Punkt entfernt ist). Trägt man hier ln p gegen 1/T ( oder log p gegen 1/T ) auf so erhält man eine Gerade deren Steigung - Hm,Verd. / R oder (- Hm,Verd. / 2,303 R) beträgt. c) Einfache und komplexe empirischer Ansätze Eine der einfachsten empirischen Gleichungen hat die Form (Augustsche Gleichung): ln p = A − B T B= H m ,Verd . R A und B sind stoffspezifische Konstanten, die mit Hilfe experimenteller Dampfdruckdaten bestimmt werden können. Für p = 1 kann die Gleichung nach A aufgelöst werden. p=1 ⇒ ln p = 0 0 = A− B T A= B H m ,Verd . = T R *T Die Gleichung ist für kleinere Temperaturbereiche erstaunlich genau. Ursache ist ein ähnlicher Temperaturgang der Verdampfungsenthalpie Hm,Verd. und des Kompressibilitätsfaktors z, der indirekt enthalten ist. Ist die Gleichung zu ungenau kann sie durch weitere Konstanten erweitert werden: B B + C * ln T oder log p = A − + C * log T T T Kirchhoffsche Dampfdruck-Gleichung bzw. Dupre´-Rankine-Gleichung ln p = A − Wenn es sich um Berechnungen in der Nähe der kritischen Temperatur handelt: B + C * ln T + D * T 6 Das Problem liegt dann in der exakten Ermittlung der T Koeffizienten A, B, C und D (Tabellenwerke wie D´Ans-Lax oder Landolt-Börnstein). ln p = A − Wohl die bekannteste empirische Gleichung ist die Antoine - Gleichung mit p in [kPa] log p = A − B ϑ+C Seite 19 Physikalische Chemie 2005 T Fos Relativ genau, indirekt ist die Temperaturabhängigkeit der Verdampfungsenthalpie berücksichtigt. Substanz Antoinekonstanten A B Wasser 7,19621 1730,63 Methanol 7,20587 1582,27 Ethanol 7,23710 1592,86 Aceton 6,24204 1210,59 1-Butanol 6,96290 1558,19 Heptan 6,01876 1264,37 Gültigkeitsbereich in °C C von bis 233,426 1 100 239,726 15 84 226,184 20 93 229,664 -13 55 196,881 -1 118 216,640 -3 127 Tabelle 1-3 Beispiel H2O Temperatur in °C Druck in Druck in log p kPa mbar 1 -0,18620492 0,651321 6,51 5 -0,06235241 0,86625867 8,66 10 0,08673936 1,22106662 12,21 15 0,22982967 1,69757773 16,98 20 0,36727374 2,32955915 23,30 25 0,49939931 3,15790679 31,58 30 0,62650921 4,23164485 42,32 35 0,74888374 5,60897798 56,09 40 0,86678266 7,35838759 73,58 45 0,9804471 9,55976253 95,60 50 1,09010117 12,3055541 123,06 55 1,19595344 15,7019444 157,02 60 1,29819824 19,8700169 198,70 65 1,3970169 24,946918 249,47 70 1,4925788 31,0869992 310,87 75 1,58504233 38,4629269 384,63 80 1,67455577 47,2667526 472,67 90 1,84527965 70,0292779 700,29 95 1,92674291 84,4778603 844,78 100 2,00576294 101,33581 1013,36 Tabelle 1-4 Seite 20 Physikalische Chemie 2005 T Fos Eine weitere Methode zur Berechnung von Dampfdrucken beruht auf dem Zusammenhang zwischen p Gm,Verd . = − R * T * ln Θ = H m ,Verd . − T * S m ,Verd . p Mit Hilfe von Tabellenwerten für die Standardenthalpie, Standardentropie und Molwärme lässt sich der Dampfdruck ermitteln. Beispiel Wasser bei Standardbedingungen (25°C) H m ,Verd . = H mΘ, H 2O ( g ) − H mΘ, H 2O ( l ) S m,Verd . = S mΘ, H 2O ( g ) − S mΘ, H 2O ( l ) H m ,Verd . = −241,82 − (−285,83) = 44,01 Eingesetz für GmΘ,Verd . = 44010 kJ mol S m,Verd . = 188,8 − 69,9 = 118,9 GmΘ,Verd . = H mΘ,Verd . − T * S mΘ,Verd . J J J − 298,15K * 118,9 = 8560 mol mol * K mol Θ Und weiter für den Druck : ln ln G p = − m,Verd . Θ p R *T p 8560 J * mol * K =− = −3,4533 Θ p mol * 8,314 J * 298,15 K p = p Θ * e −3, 4533 = 1013mbar * 0,0316 = 32,0mbar Seite 21 ergibt sich : J mol * K Physikalische Chemie 2005 T Fos Möchte man den Dampfdruck nicht bei Standardbedingung, sondern bei einer anderen Temperatur ermitteln sind verschiedene Umrechnungen notwendig Beispiel Wasser bei 100°C = 373,15 K H m, H 2O(l)373K = H Θ m, H 2O(l) + C p ⋅ ∆T H m, H 2O(g )373K = H Θ m, H 2O(g ) + C p ⋅ ∆T = −285,83 J kJ + 75,29 ⋅ 75K mol ⋅ K mol = −241,82 J kJ + 33,58 ⋅ 75K mol ⋅ K mol = −285,83 kJ kJ + 5,647 mol mol = −241,82 kJ kJ + 2,519 mol mol = −280,183 kJ mol = −239,301 kJ mol H m, Verd. = H m, H 2O(g )373K − H m, H 2O(l)373K = −239,301 − (−280,183) = 40,882 T2 Sm, H 2O(l)373K = SΘ m, H 2O(l) + C p ⋅ ln T1 373,15 K J J J = 69,91 + 75,29 ⋅ ln = 86,80 mol ⋅ K mol ⋅ K 298,15 K mol ⋅ K T2 Sm, H 2O(g )373K = SΘ m, H 2O(g ) + C p ⋅ ln T1 = 188,83 373,15 K J J J + 33,58 ⋅ ln = 196,36 mol ⋅ K mol ⋅ K 298,15 K mol ⋅ K S m,Verd . = S m ,H 2O ( g ) 373 K − S m, H 2O ( l ) 373 K = 196,36 − 86,80 = 109,56 Eingesetz für G m, Verd. = 40882 G m, Verd. = H m, Verd. − T ⋅ Sm, Verd. J J J − 373,15K ⋅ 109,56 = −0,314 mol mol ⋅ K mol Und weiter für den Druck ln p pΘ =− J mol ⋅ K G p ln Θ = − m, Verd. R ⋅T p − 0,314J ⋅ mol ⋅ K = 1,01 ⋅10 − 4 mol ⋅ 8,314J ⋅ T ⋅ 373,15K p = pΘ ⋅ e0,000101 = 1013mbar ⋅ 1,000101 = 1013mbar Seite 22 ergibt sich: kJ mol Physikalische Chemie 2005 T Fos 1.3.4 Phasenübergang Feststoff / Flüssigkeit Auch für den Phasenübergang fest / flüssig kann man analoge Betrachtungen wie beim Übergang flüssig / gasförmig durchführen. Die Clausius-Clapeyronsche - Gleichung erhält dann die Form: H m , Schm. dp = dT T (Vm , Flüssigkeit − Vm , Feststoff ) Im Falle des Schmelzens oder des Erstarrens einer Substanz ist die Änderung des Volumens und damit auch des Molvolumens gering. Deshalb gilt näherungsweise: ⇒ H m, Schm. = U m , Schm. + p * ∆Vm H m, Schm. ≈ U m, Schm. Normalerweise ist das Molvolumen eines Festkörpers kleiner als das Molvolumen der entsprechenden Flüssigkeit. Eine Ausnahme bildet hier wieder Wasser. Die Änderungen sind in beiden Fällen sehr klein, so dass als Folgerung die Kurve dp/dT sehr steil ist. Für die üblichen Bereiche (nicht für die Hochdrucktechnik) kann man die Schmelzenthalpie und die auftretenden Volumenänderungen als temperatur- und druckunabhängig betrachten. Man kann deshalb meistens anstelle der Differentiale mit Differenzen arbeiten. H m, Schm. ∆p = ∆T T * ∆Vm, Schm. oder p2 = p1 + H m , Schm. * (T2 − T1 ) T1 * ∆Vm , Schm. oder T2 = T1 + T1 * ∆Vm, Schm. * ( p2 − p1 ) H m, Schm. Will man die Gleichung etwas exakter angehen ist die Gleichung natürlich zu integrieren: p2 H m, Schm. T2 ∫ dp = ∆Vm, Schm. * ∫ p1 T1 dT T H ⎛T ⎞ p2 = p1 + m, Schm. * ln⎜⎜ 2 ⎟⎟ ∆Vm, Schm. ⎝ T1 ⎠ man erhält dann: ( p 2 − p1 )* ∆V m , Schm . oder T2 = T1 * e Seite 23 H m , Schm . Physikalische Chemie 2005 T Fos Beispiel einer Berechnung mit beiden Varianten: Wie ändert sich die Schmelztemperatur von Eis, wenn es unter einem Druck von 100 bar steht (z.B. Gletscher). Daten: Schmelzenthalpie Hm,Schm. bei 0°C = 6008 J/mol spezifische Schmelzenthalpie hSchm.= 333 J /g oder 80 cal /g Molare Volumenänderung ∆Vm,Schm. = − 1,7 cm3/mol Eingesetzt in die Gleichung mit den Differenzen ergibt sich: 273,15K * −1,7 * 10 −6 m 3 * (1 * 10 7 − 1 * 10 5 ) N * mol T2 = 273,15K + mol * m 2 * 6008 Nm T2 = 273,15 K − 0,765 K = 272,385 K oder - 0,765°C Mit der integrierten Form erhält man: (1*107 −1*105 ) N*−1, 7*10 −6 m 3 *mol T2 = 273,15K * e mol*m 2 *6008 Nm T2 = 273,15K * e −0,002801 = 273,15K * 0,997202 T2 = 272,386 K oder - 0,764°C 1.3.5 Phasenübergang Feststoff / Gas Der Dampfdruck wird beim Gefrieren der Flüssigkeit natürlich nicht 0, sondern auch Feststoffe haben einen Dampfdruck der mit weiterer Temperaturabsenkung immer mehr abnimmt. (Verdunsten von Eis, Gefriertrocknung, Dampfdruck von Metallen, Sublimation von Iod). Auch hier können wir wieder die Clausius-Clapeyronsche Gleichung verwenden in der Form mit der Sublimationsenthalpie. H m, Sub lim ation dp = dT T (Vm,Gas − Vm, Feststoff ) Das Molvolumen des Feststoffes kann hier gegenüber dem Molvolumen des Gases wieder vernachlässigt werden. Für die weitere Betrachtung können die Überlegungen analog wie beim Übergang Flüssigkeit - Gas herangezogen werden. Für Vm,Gas wird wieder Vm , Gas = R *T , so dass wir analog Flüssigkeit - Gas in der p integrierten Form schreiben können: p2 = p1 * e ⎛ H m , Sub lim ation ⎛ T2 − T1 ⎜ *⎜⎜ ⎜ R ⎝ T2 *T1 ⎝ ⎞⎞ ⎟⎟ ⎟ ⎟ ⎠⎠ Schmelzenthalpie und Verdampfungsenthalpie lassen sich zur Sublimationsenthalpie addieren. H m, Sub lim ation = H m , Schmelzen + H m ,Verdampfung Seite 24 Physikalische Chemie 2005 T Fos 1.3.6 Erwärmung und Phasenübergang Phasenübergänge lassen sich in vielfältiger Weise beschreiben und berechnen. Um die Berechnungen einigermaßen einfach zu gestalten wird sehr häufig die Temperaturabhängigkeit der Phasenübergangsenthalpien vernachlässigt. Bei der konkreten Berechnung ist zu prüfen. welche der Formeln einsetzbar ist. Je mehr man sich der kritischen Temperatur nähert umso mehr Sorgfalt ist notwendig. Die Verdampfungsenthalpie wird mit steigender Temperatur geringer. Bei der kritischen Temperatur wird sie null. Wie aus den folgenden Stoffbeispielen zu ersehen ist, sinkt die Verdampfungsenthalpie bei großen Wechselwirkungskräften (Wasser) langsamer als bei geringeren Wechselwirkungskräften ( Kohlenwasserstoffe). Diagramm 1-4 Seite 25 Physikalische Chemie 2005 T Fos Kritische Daten TK, pK, Vmol,K einiger Gase Gas He Ne Ar Kr Xe TK [K] pK [bar] Vmol,K [cm3/mol] 5,3 2,29 57,7 44,5 26,2 41,6 151 48,6 75,2 210,6 54,9 92 290 58,7 120,2 H2 N2 O2 33,2 126,0 154,3 13,0 33,9 50,4 65,0 90,0 74,0 CO2 NH3 H2O n-C4H10 CH3COOH 304,2 405,5 647,1 426,0 594 74,0 113,0 220,6 42,6 58,0 95,6 72,3 45,0 214 171 Tabelle 1-5 Berechnung von Wärmemengen Wenn Substanzen erwärmt (oder abgekühlt) werden gilt für die dafür notwendige Wärmemenge Q = m * c * ∆T oder Q = n * C p * ∆T Q = Wärmemenge in Joule m = Masse in Gramm n = Stoffmenge in mol c = spezifische Wärmekapazität in J*g-1*K-1 Cp = molare Wärmekapazität bei konst. Druck in J*mol-1*K-1 ∆T = Temperaturdifferenz in Kelvin Beispiel: Ein Wasserkocher hat eine elektrische Leistung P von 1000 W (J/s). Welche Zeit benötigt man um 1Liter (1 kg ) Wasser von 20°C zum Kochen zu bringen ? Q = m * c * ∆T Q = 1000 g * 4,184 Pelektrisch = Q t J * 80 K = 334720 J = 334,7 kJ g*K P = Leistung in W oder kW t = Q Pelektrisch Seite 26 = 334720 J *1W * s = 335s = 5,6 min 1000W *1J Physikalische Chemie 2005 T Fos Werden Substanzen erwärmt (oder abgekühlt ) und es werden Phasenumwandlungen mit einbezogen müssen in die Formeln die Umwandlungsenthalpien mit einbezogen werden. Q = mFestst . * cFestst . * ∆T + m * hSchmelzen + mFl . * cFl . * ∆T + m * hverd . + mGas. * cGas. * ∆T oder Q = nFestst . * C p , Festst . * ∆T + n * H m, Schmelzen + nFl . * C p , Fl . * ∆T + n * H m , verd . + nGas. * C p ,Gas. * ∆T Beispiel: 100 mol(1,8kg) Eis von - 18°C werden so lange erwärmt bis daraus Dampf von 105°C entstanden ist. n = 100 mol Cp,Festst. = 37,7 J/mol*K Hm,Schm. = 6008 J/mol Q= Cp,Fl.. = 75,3 J/mol*K Hm,Verd. = 40656 J/mol Cp,Gas. = 33,58 J/mol*K 100mol * 37,7 J *18K 100mol * 6008 J 100mol * 75,3J *100 K 100mol * 40656 J 100mol * 33,58 J * 5K + + + + mol * K mol mol * K mol mol * K Q = 67,86kJ + 600,8kJ + 753kJ + 4065,6kJ + 16,79kJ = 5504,05kJ Diagramm 1-5 6000 Enthalpie [kJ] H = f(T) 5504,05 5487,26 5000 Erwärmung von Wasser von -18°C bis 105°C 4000 3000 2000 1421,66 668,66 1000 Temperatur [°C] 67,86 0 0 -20 0 20 40 Seite 27 60 80 100 120