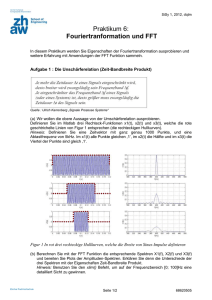
Exakte Massen der wichtigsten Isotope
Werbung

MS 1/24 König Induktiver und mesomerer Effekt Die Entstehung der Peaks eines Massenspektrums hängt im wesentlichen von drei Faktoren ab: (1) der Stärke der jeweiligen Bindung, (2) der induktive Effekt und (3) der mesomere Effekt. Eine typische kovalente Bindung weist eine Bindungsenergie von ca. 10 bis 12 eV auf. Beschießt man nun - wie in der Elektronenstoßionisation - die Moleküle mit 70 eV, so kann man primär annehmen, dass die Stoßenergie eines einzelnen Elektrons ausreichen müsste mehrere Bindungen in Folge zu spalten. Generell weiß man, dass Fragmente die durch Spaltung schwacher Bindungen entstehen bevorzugt auftreten. Bei genauerer Analyse eines Massenspektrums stellt man allerdings fest, dass viele Bindungen wenig oder gar nicht brechen, obwohl deren Bindungsenergie weit unter der Stoßenergie liegt. Für dieses Verhalten sind Faktoren verantwortlich, die sich aus der Elektronenverteilung im Molekül ableiten, und die dazu führen, dass positive Ionen stabilisiert werden. Induktiver Effekt: Falls die Nachbaratome eines positiv geladenen Atoms Elektronenspender sind können sie die positive Ladung teilweise neutralisieren und damit stabilisieren. Dieser Effekt wird "induktiver Effekt" genannt. Gruppen die Elektronen spenden, werden als +I-Gruppen bezeichnet, Gruppen die Elektronen abziehen als -I-Gruppen. +I-Gruppen stabilisieren ein positives Ion, -I-Gruppen destabilisieren es. Die am meisten verbreitete +I-Gruppe ist die Alkyl-Gruppe. Das erklärt auch die Tatsache dass das t-Butyl-Carbokation ausgesprochen stabil ist. Der induktive Effekt wirkt nur über maximal zwei Bindungen, ist als lokal sehr begrenzt. -I Gruppen +I Gruppen -NO2 -CHO -RC=CR2 -CH3 -COR -CH2R -COOH -F -CHR2 -COOR -Cl -SO2OH -CR3 -OH -Br -SH -OR -I -SR C6H5 -CH=CH2 -NH2 Mesomerer Effekt: Der mesomere Effekt tritt dann auf, wenn es im Molekül konjugierte Doppelbindungen gibt. Da die pi-Elektronen in konjugierten Systemen sehr leicht beweglich sind, können diese die positive Ladung über das gesamte konjugierte System verteilen ("delokalisieren"), was erheblich zur Stabilisierung eines positiven Ions beiträgt. Außerdem können Resonanzstrukturen (Strukturen, bei denen die Ladung zwischen den Atomen verschoben wird) zusätzlich zur Stabilisierung beitragen. Man nennt die Kombination von delokalisierter Ladung in konjugierten pi-Systemen und Resonanzstrukturen, den "mesomeren Effekt". Analog zum induktiven Effekt werden die Gruppen als +M- und -M-Gruppen bezeichnet. Eine +M-Gruppe stabilisiert eine positive Ladung, eine -M-Gruppe destabilisiert sie. MS 2/24 +M, -I Gruppen -M, -I Gruppen König +M, +I Gruppen -F -SH -NO2 -CONH2 CH3 -Cl -SR -SO2R -CH2R -BR -NH2 -CHO -CF3 -CHR2 -I -NHR -COR -CCl3 -CR3 -OH -NR2 -COOH -OR -NHCOR -COOR -OCOR -C6H5 -CH=CH2 -CH=CR2 Treten in einem Molekül sowohl der induktive als auch der mesomere Effekt auf, so überwiegt immer der mesomere Effekt. Interpretation: Das Massenspektrum Das Massenspektrum wird üblicherweise als Strichspektrum dargestellt, wobei die einzelnen Peaks jeweils auf ganzzahligen Massen aufgetragen werden. Den intensivsten Peak bezeichnet man als Basispeak, dessen Höhe wird willkürlich mit 100 angesetzt, alle anderen Peaks werden relativ dazu dargestellt. Dadurch erreicht man eine gut vergleichbare Darstellung der Spektren. Der Basispeak ist nur bei sehr stabilen Verbindungen (wie z. B. polyzyklische Aromaten) gleichzeitig auch der Molekülpeak. Verbindungen die weniger stabil sind, haben oft nur einen kleinen Molekülpeak, bei manchen Substanzklassen kann der Molekülpeak überhaupt fehlen. Da die meisten natürlich vorkommenden Elemente aus einem Isotopengemisch bestehen, findet man neben jedem Fragmentpeak die jeweiligen Isotopenpeaks. Aus dem Intensitätsverhältnis der Isotopenpeaks kann man Rückschlüsse auf die Summenformel einer Verbindung ziehen. Wichtig für die organische Massenspektrometrie sind folgende Isotope: C-12 (98.9 %), C-13 (1.1 %), Br-79 (50.7 %), Br-81 (49.3 %), Cl-35 (75.8 %) und Cl-37 (24.2 %) und S-32 (95.0 %) und S-34 (4.2 %). Die Elemente Fluor und Phosphor sind monoisotop. MS 3/24 König Neben den Isotopenpeaks gilt es noch eine Besonderheit bezüglich Wasserstoff zu berücksichtigen: Da der Wasserstoff einen hohen Massendefekt aufweist, weicht die Massenzahl von H-1 deutlich von der ganzzahligen Masse ab (1.0078 Dalton). Die vergleichsweise große Zahl an Wasserstoffatomen in organischen Molekülen, führt daher zu einer Verschiebung der Fragmente zu höheren Massen. Spezielle Korrekturen bei der Erfassung der Spektren sorgen aber dafür, dass die Massen immer bei der richtigen ganzzahligen Masse erscheinen. Informationsgehalt von Massenspektren Bei konstanter Elektronenenergie (typ. 70 eV) ist das Fragmentierungsmuster bei Elektronenstoßionisierung gut reproduzierbar und kann einerseits zur Bibliothekssuche, andererseits auch zur (automatischen) Spektreninterpretation herangezogen werden. Ziel der Interpretation ist zum einen die Substanz zu identifizieren, und zum anderen Vorgänge beim Zerfall des Moleküls zu verstehen. Für die klassische Analytik ist der erste Anwendungsfall der wichtigste. Betrachtet man ein Massenspektrum, so kann man zwei Typen von Informationen herauslesen: zum einen Information die sich aus der Form des Massenspektrums ergibt, und zum anderen Detailinformationen aus den einzelnen Peaks und deren Abständen zueinander. Wichtige Peaks in einem Massenspektrum Die Peaks in einem Massenspektrum sind normalerweise auf den höchsten Peak normiert, so dass dieser 100% erhält. Dieser Peak wird auch Basispeak (engl. base peak) genannt (m/e = 91 in obiger Abbildung). Ein weiterer wichtiger Peak ist der Molekülpeak, der dem Molekulargewicht der Substanz entspricht (m/e = 126 im Beispiel oben). Allerdings ist der Molekülpeak nicht bei allen Substanzen sichtbar. Bei leicht zerfallenden Stoffen kann der Molekülpeak eventuell zur Gänze verschwinden. Die meisten der Peaks haben einen oder mehrere Isotopenpeaks, die meist von 13C, 34S, oder Chlor- und Bromisotopen herrühren. MS 4/24 König Die weiteren Fragmentpeaks können durch Abspaltungen von ungeladenen Molekülbruchstücken aus dem ionisierten Molekül oder einem ionisierten Bruchstück erklärt werden. Interpretation von Massenspektren Zur Interpretation von Massenspektren wird man neben der automatischen Spektrensuche immer auch einen Blick auf das jeweilige Spektrum werfen. Für eine erste Interpretation wird man folgende Vorgangsweise wählen: 1. Allgemeine Übersicht zur Spektrenform Die Form des Spektrums gibt Auskunft über die Stabilität des Moleküls. Stabile Moleküle weisen wenige kleine Peaks bei niedrigen Massen (< 70 Da) auf, und nur wenige, dafür aber große Peaks bei hohen Massen. Für stabile Moleküle ist das Molekülion immer zu sehen. Andererseits weisen Molküle, die leicht fragmentieren eine typische Form auf, bei der die höchsten Peaks im Bereich unter 70 Da auftreten. 2. Molekülion identifizieren Kennt man das Molekulargewicht der Substanz, sollte es nicht schwer sein, das Molekülion zu finden. Allerdings gibt es Substanzen die keine oder nur sehr kleine Molekülionen erzeugen (wie z.B. Alkohole). Falls das Molekülion nicht identifiziert werden kann, hilft entweder die Aufnahme des EI-Spektrums bei reduzierter Elektronenenergie oder der Einsatz chemischer Ionisierung weiter. Ist das Molekülion bekannt, so sollte man folgende Überprüfungen durchführen: Gibt es Peaks unterhalb des Molekülions im Abstand kleiner als 14? Falls es sich bei der Substanz um eine Reinsubstanz handelt dürfen keine Peaks zwischen M1 und M-13 auftreten (ausgenommen Isotopenpeaks). Ist das doch der Fall, so ist die Probe entweder verunreinigt oder das vermeintliche Molekülion ist doch nicht das Molekülion. Versuchen Sie auch das Molekülion über metastabile Ionen abzusichern. Ist die Masse des Molekülions gerade oder ungerade? Eine Substanz mit einer geraden Zahl von N-Atomen muss eine geradzahlige Masse des Molekülions aufweisen. 3. Allgemeine Hintergrundionen MS 5/24 König Peaks bei 18 (Wasser), 28 (O2), 32 (N2), 40 (Ar) und 44 (CO2 ) sind praktisch immer vorhanden, zusätzlich können Peaks von Lösungsmitteln, von Weichmachern, oder GCSepten verusacht werden. 4. Augenfällige Isotopenmuster Chlor- und Brom-Verbindungen, aber auch viele metallorganische Verbindungen weisen ein ausgeprägtes Isotopenmuster auf, das Rückschlüsse auf die Struktur der Substanz zulässt. 5. Versuchen Sie anhand der 13C-Satelliten die Zahl der C-Atome im Molekülion abzuschätzen 13C kommt mit 1.1 % in der Natur vor. Es lässt sich leicht zeigen, dass man die Zahl der C-Atome aus den 13C-Satelliten abschätzen kann. Dazu berechnet man das Verhältnis der Intensitäten des 13C-Satelliten zur Intensität des zugehörigen 12C-Peaks und dividiert dieses Verhältnis durch 0.01. Molekülionen Je nach Art der Ionisierung treten Molekülionen, das sind Ionen die durch Ionisation des Moleküls ohne weiteren Zerfall entstehen, mehr oder weniger häufig auf. Bei der Elektronenstoßionisation mit 70 eV, die die bei weitem gängigste Ionisationsmethode darstellt, hängt die Bildung von Molekülionen von der Stabilität der jeweiligen Moleküle bzw. Molekülionen ab. Große Molekülpeaks treten auf, wenn der unimolekulare Zerfall des Molekülions energiereich ist (> 4 eV). So können bei sehr stabilen Molekülionen mehr als 90% des gesamten Ionenstroms aus den Molekülionen stammen. Aromatische Verbindungen z. B. zeigen einen ausgeprägten Molekülpeak, während tertiäre Alkohole oder langkettige Aliphaten meist überhaupt keinen aufweisen. starker Molekülpeak, wenig Fragmente mittlerer Molekülpeak MS 6/24 König kein Molekülpeak, viele Fragmente Generell ist die Entscheidung ob der höchste im Spektrum vorhandene Peak der Molekülpeak ist, nicht einfach und meist nur mit Hilfe zusätzlicher Experimente oder durch veränderte Messbedingungen zu treffen. Eine einfache Möglichkeit bei Elektronenstoßspektren besteht darin, die Elektronenenergie von 70 eV auf ca. 10 eV zu senken. Durch die verringerte Elektronenenergie werden labile Moleküle weniger leicht fragmentiert, wodurch im Spektrum bei niedrigerer Energie der Molekülpeak höher wird (oder überhaupt erst sichtbar) und die Fragmentionen zurückgedrängt werden. Ist eine Änderung der experimentellen Parameter nicht möglich, so bieten sich ein paar Regeln an mit denen man zumindest einschränken kann, ob der jeweilig höchste Peak das Molekulargewicht widerspiegelt. Ganz besonders wichtig für diese Entscheidung sind Fragmente innerhalb der ersten 14 Masseneinheiten unterhalb des höchsten Peaks. Treten in diesem Bereich Peaks auf, so ist davon auszugehen, dass der Molekülpeak nicht sichtbar ist, da für Verbindungen, die nur C, H, O, und N enthalten, keine Neutralteilchenabspaltung mit der Masse 5 bis 13 gibt ("unmögliche Massendifferenzen"). Auch der Verlust von 3 bis 5 H-Atomen wird praktisch nie beobachtet (meist nur durch Dehydrierung im Einlasssystem). Hinweis: Das Molekulargewicht von Verbindungen die nur C, H und O und keine oder eine gerade Zahl von N-Atomen enthalten, ist immer geradzahlig. Molekülpeaks von Substanzen aus den Elementen C, H, N und O sind immer ungeradzahlig wenn die Zahl der Stickstoffatome ungerade ist. Isotopenpeaks Bei genauerer Betrachtung der Massenspektren zeigt sich, dass fast alle Peaks in Massenspektren von Kohlenstoffverbindungen einen kleineren Satellitenpeak bei der nächsthöheren Masse aufweisen. Grund dafür ist das C-13 Isotop, das mit einer Häufigkeit von etwa 1.1 % natürlich vorkommt. Dadurch ergibt sich für ein Fragment mit n Kohlenstoffatomen eine Intensität von n * 1.1 % bei der nächst-höheren Masse. Die MS 7/24 König Intensität eines Satellitenpeaks ist proportional zur Wahrscheinlichkeit, dass das jeweilige Fragment ein C13-Atom enthält. Von den für organische Substanzen wichtigen Elementen sind Fluor, Jod, und Phosphor monoisotop, alle anderen weisen mehr oder weniger stark ausgeprägte Isotopenmuster auf (siehe auch Isotopentabelle). Speziell chlorierte und bromierte Verbindungen weisen ein sehr charakteristisches Muster auf, das leicht erkannt werden kann und sich auch zur automatischen Auswertung gut eignet. Die folgende Abbildung zeigt die Isotopenmuster der wichtigsten Chlor/Brom-Kombinationen. Isotopenmuster verschiedener Br/Cl Kombinationen Exakte Massen der wichtigsten Isotope Die folgende Tabelle zeigt die exakten Massen der wichtigsten Isotope. Die relative Häufigkeit ist auf 100 normiert (das häufigste Isotop eines Elements wird gleich 100 gesetzt). Um die tatsächlichen Häufigkeiten zu ermitteln, müssen diese Zahlen jeweils durch die Summe der angegebenen Häufigkeiten eines Elements dividiert werden Element Symbol Exakte Masse (Da) Rel. Häufigkeit Wasserstoff 1H 1.007825037 2 Deuterium H or D 2.014101787 12 Kohlenstoff-12 C 12.00000 Kohlenstoff-13 13C 13.003354 14 Stickstoff-14 N 14.003074 15 Stickstoff-15 N 15.00011 100.0 0.015 100.0 1.11223 100.0 0.36734 MS 8/24 Sauerstoff-16 Sauerstoff-17 Sauerstoff-18 Fluor Natrium Silizium-28 Silizium-29 Silizium-30 Phosphor Schwefel-32 Schwefel-33 Schwefel-34 Schwefel-36 Chlor-35 Chlor-37 16O 17O 18O 19F 23Na 28Si 29Si 30Si 31P 32S 33S 34S 36S 35Cl 37Cl 15.99491464 16.9991306 17.99915939 18.998405 22.9897697 27.9769284 28.9764964 29.9737717 30.9737634 31.972074 32.9707 33.96938 35.96676 34.968854 36.965896 König 100.0 0.03809 0.20048 100.0 100.0 100.0 5.0634 3.3612 100.0 100.0 0.78931 4.43065 0.02105 100.0 31.97836 Berechnung der Elementarzusammensetzung Die Elementarzusammensetzung einer Verbindung (bzw. des jeweiligen Fragments) lässt sich berechnen, falls man von den jeweiligen Peaks nicht nur die Nominalmasse kennt, sondern die exakte Masse mit einer Mindestauflösung von 5000 (besser 10000). Da die einzelnen Isotopen keine ganzzahligen Massen aufweisen, lassen sich durch einfache Kombination alle möglichen Summenformeln für einen gegebenen Peak berechnen. Allerdings kann diese Berechnung bereits bei mittleren Massenzahlen (ab 200 Dalton) ziemlich langwierig sein, da sich eine astronomische Zahl von Möglichkeiten ergibt. Um die Berechnung dennoch durchführen zu können, muss man einige Einschränkungen vorgeben (z.B. die Minimal- und die Maximalzahl der beteiligten Atome). Außerdem sollte der Algorithmus auch noch chemisch sinnlose Kombinationen von vornherein ausschließen. Als Beispiel sei die Berechnung aller möglichen Summenformeln für die Masse 252.09 0.02 gezeigt, wobei folgende Einschränkungen gelten: C = 6..21, H = 0..52, N = 0..4, O = 0..4. Die angegebene Toleranz entspricht einer Auflösung von 6000. verschiedene Summenformeln mit exakter Molekülmasse Summenformel Masse C10H12N4O4 C11H14N3O4 C14H10N3O2 C14H12N4O1 C15H12N2O2 C16H12O3 C16H14NO2 C19H10N C20H12 252.085788 252.098364 252.0772656 252.101088 252.0898416 252.0785952 252.1024176 252.0813192 252.0938952 MS 9/24 König Fragmentierungsregeln Die folgenden algemeinen Regeln zur Fragmentierung sollen einen kurzen Überblick zu den wichtigsten Fragmentierungsmechanismen geben. Stevenson-Regel Unmittelbar nach der Ionisation eines Moleküls in der Ionenquelle, versucht dieses möglichst viel Energie abzugeben. Das geht am einfachsten durch Abspaltung eines möglichst großen Radikals (je größer das Radikal, desto mehr Schwingungsfreiheitsgrade hat es, und um so mehr Energie kann es übernehmen). Dies hat Stevenson in den frühen 50er Jahren des 20. Jahrhunderts erkannt, weshalb folgende Regel auch Stevenson-Regel genannt wird: Bei der Fragmentierung spaltet sich immer das größte Radikal ab. Das restliche Fragment bildet meist den Basispeak. Diese Regel kann sehr schön zum Beispiel bei den Isomeren des Butanols verfolgt werden: 1-Butanol hat als Basispeak die Masse 31 (Abspaltung des C3H7-Radikals), 2Butanol die Masse 45 (Abspaltung der Ethylgruppe) und t-Butanol den Basispeak 59 (Abspaltung eines CH3-Radikals). McLafferty-Umlagerung Die McLafferty-Umlagerung ist eine allgemeine Umlagerungsreaktion, die in vielen Varianten von Carbonyl-Verbindungen abläuft und zu charakteristischen Fragmentpeaks führt. Dabei wandert in einer konzertierten Bindungsumlagerung ein -Wasserstoffatom auf den Sauerstoff der Carbonylverbindung. Dadurch entstehen zwei Fragmente: Ein meist neutrales Alken und der geladene Rest. Mechanismus der Mc Lafferty Umlagerung Die Gruppen R1 bis R6 sind beliebige Gruppen, die Atome A, B, und D können eine beliebige Kombination von C, O oder S sein. Der Rest R ist eine für Carbonylverbindungen typische Gruppe, wie z.B. H für Aldehyde, oder CH3O für Methylester. Die folgende Tabelle gibt einen Überblick zu den wichtigsten Substituenten R und den daraus entstehenden Peaks. MS 10/24 König Verbindungstyp Substituent R McLafferty-Peak Aldehyd Methylketon Amid Karbonsäure Ethylketon Methylester Propylketon Ethylester Phenylketon Phenylester H CH3 NH2 OH C2H5 OCH3 C3H7 OC2H5 C6H5 OC6H5 44 48 59 60 72 74 86, 58 (zweifache Umlagerung!) 88 120 136 Da die eliminierte Gruppe ein neutrales Molekül ist, muss in CHO-Verbindungen die Masse des McLafferty-Peaks geradzahlig sein (bei sonst ungeradzahligen Fragmenten). Für Verbindungen mit einer ungeraden Zahl an Stickstoffatomen gilt das umgekehrte. Dadurch sind McLafferty-Umlagerungen gut im Spektrum zu erkennen (Beispiel 2Octanon). Umlagerungen Neben dem einfachen Bindungsbruch kommen auch Umlagerungen von Atomen oder funktionellen Gruppen vor, die sowohl durch einen Bindungsbruch als auch durch Knüpfen einer neuen Bindung erzeugt werden. In den meisten Fällen handelt es sich bei den Umlagerungen um Wasserstoffwanderungen, gelegentlich kommt es auch zu Wanderungen eines Methyl- oder Phenylrestes. Außerdem können durch Umlagerungen auch Teile einer Struktur eliminiert werden, wodurch es zu einer Skelettumlagerung kommt. Wasserstoffumlagerung Die wichtigste Wasserstoffumlagerung ist die McLafferty-Umlagerung. Dabei wandert der Wasserstoff am -Atom und gleichzeitig wird die Bindung zwischen und -Atom gespalten. Die McLafferty-Umlagerung führt sehr oft zu intensiven Peaks im Massenspektrum, wobei die positive Ladung auf beiden Bruchstücken etwa gleichwahrscheinlich auftreten kann (womit die beiden Fragmentpeaks auch etwa dieselbe Intensität aufweisen). MS 11/24 König Skelettumlagerung Ca. 10% der organischen Verbindungen zeigen eine Umlagerung des Skeletts durch Eliminierung eines besonders stabilen Neutralteilchens (z.B. CO, CO2, N2, NO, SO2, HCN). Dabei geht der Fragmentierung des Moleküls eine Umlagerung voraus. So verliert z.B. 2-Nitrotoluol eine OH-Gruppe und eine CO-Gruppe, die ja als solche gar nicht im Molekül vorhanden sind. Typische Fragmentionen (Masse 26-60) m/z Formel 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 C2H2+ CN+ C2H3+ C2H4+ CO+ N2+ C2H5+ CHO+ CH2=NH2+ NO+ CH2=OH+ CH3NH2+ O2+ SH+ CH5O+ H2S+ Cl+ HCl+ Cl+ HCl+ C3H3+ Ar C3H4+ C3H5+ C3H6+ CH2=CO+ Verbindungen ungesättigte Kohlenwasserstoffe, aromatische Verbindungen aromatische Nitrile ungesättigte Kohlenwasserstoffe Ethylverbindungen Carbonylverbindungen Azoverbindungen Kohlenwasserstoffe (Ethylverbindungen) Aldehyde Primäre Amine Nitro-Verbindungen, Nitrosamine Methoxy-Verbindungen, primäre Alkohole N-Methylamine Sauerstoff, Luft, zyklische Peroxide Thiole, Isothiocyanate (H2O+CH3)+ Thiole Chlorverbindungen, auf Isotopenverhältnis achten (35Cl:37Cl = 3:1) Chlorverbindungen, auf Isotopenverhältnis achten (35Cl:37Cl = 3:1) Chlorverbindungen, auf Isotopenverhältnis achten (35Cl:37Cl = 3:1) Chlorverbindungen, auf Isotopenverhältnis achten (35Cl:37Cl = 3:1) aromatische Kohlenwasserstoffe Argon aus der Luft Kohlenwasserstoffe ungesättigte Kohlenwasserstoffe Kohlenwasserstoffe Acetate, Acetylverbindungen MS 43 44 45 46 47 49 50 51 55 56 57 58 59 59 60 12/24 C3H7+ CH3CO+ CO2+ CH3CH=NH2+ CH2=CH(OH)+ H2N=CO+ CH3CH=OH+ CH3CH2O+ COOH+ NO2+ PO CH2=SH+ König Kohlenwasserstoffe Methylketone Kohlendioxid aus der Luft, Karbonate, Anhydride aliphatische Amine Aldehyde mit Wasserstoffatomen in -Position primäre Amine sekundäre Alkohole Ethoxyverbindungen Karbonsäuren Nitroverbindungen Phosphorylverbindungen primäre Thiole Chlormethylverbindungen - auf Isotopenverhältnis achten (35Cl:37Cl CH2Cl+ = 3:1) C4H2+ aromatische Verbindungen Chlormethylverbindungen - auf Isotopenverhältnis achten (35Cl:37Cl CH2Cl+ = 3:1) + C4H3 aromatische Verbindungen + C4H7 ungesättigte Kohlenwasserstoffe, zyklische Ketone (5-, 6-Ring) C4H8+ Kohlenwasserstoffe + C4H9 Kohlenwasserstoffe + CH3CH2CO Ethylketone + CH2=C(OH)CH3 Methylketone mit -Wasserstoffatomen (CH3)2COH+ Methylester + CH2=C(OH)NH2 primäre Amide CH2=C(OH)OH+ Essigsäureester, aliphatische Karbonsäuren mit -Wasserstoff Typische Fragmentionen (Masse 61-150) m/z Formel Verbindungen 61 CH3COOH2+ S2+ 64 SO2+ 65 C5H5+ 66 C5H6+ 68 C4H4N+ 69 C5H9+ 70 C5H10+ 71 C3H7CO+ 72 CH2=C(OH)C2H5+ 72 C3H7CH=NH2+ C3H7OCH3+ (CH3)3Si+ 73 CO2C2H5+ CH2=CHC(OH)=OH+ 74 CH2=C(OH)OCH3+ 75 (CH3)3Si=OH+ Essigsäureester, nicht aber Methylazetat Disulfide Sulfone, Sulfonate Benzyl- und Toluyl-Verbindungen, Phenole, Aniline aromatische Verbindungen Pyrrole ungesättigte Kohlenwasserstoffe Kohlenwasserstoffe Propylketone Ethylketone mit -Wasserstoffatomen Amine Ether Trimethylsilyl-Derivate Ethylester aliphatische Säuren Methylester mit -Wasserstatomoffen MS 75 76 77 78 79 13/24 C2H5CO(OH2)+ C6H4+ C6H5+ C6H6+ C6H7+ 79Br+ 80 79BrH+ 81 81Br+ 82 83 85 86 86 88 89 90 91 92 93 94 95 81BrH+ C5H6N+ C4H3S+ C6H13+ CH2=C(OH)C3H7+ C4H9CH=NH2+ C3H7COOH+ C4H9S+ C4H9O2+ C7H6+ C7H7+ (TropyliumIon) C6H6N+ 79BrCH + 2 C6H5O+ C7H9+ C6H6O+ 81BrCH + 2 C7H11+ Propionate Benzolderivate (mono- oder di-substituiert) Benzolderivate Benzolderivate mit- oder -Wasserstoffatomen Benzolderivate Bromverbindungen - auf Isotopenverhältnis achten (79Br:81Br = 1:1) Bromverbindungen - auf Isotopenverhältnis achten (79Br:81Br = 1:1) Bromverbindungen - auf Isotopenverhältnis achten (79Br:81Br = 1:1) Bromverbindungen - auf Isotopenverhältnis achten (79Br:81Br = 1:1) Methylpyrrole monosubstituierte Thiophene Hexyl-Verbindungen Propylketone mit -Wasserstoffen Amine Butanoate Sulfide Glykolether, Diole disubstituierte Aromaten Alkylbenzol Monoalkylpyridine Br-CH2-R - auf Isotopenverhältnis achten (79Br:81Br = 1:1) Phenole, Nitrobenzole Mono- und Sesquiterpene C6H5O-R (R > Methyl) Br-CH2-R - auf Isotopenverhältnis achten (79Br:81Br = 1:1) Mono- und Sesquiterpene 96 C5H4NO+ 97 C5H5S+ 99 C7H15+ 103 C6H5CH=CH+ Methylthiophene Heptylverbindungen Zimtsäure 111 C5H3OS+ 127 I+ 127 C10H7+ 128 HI+ König Iodverbindungen Naphthylverbindungen Iodverbindungen MS 14/24 149 König Dialkylphthalate (Weichmacher) Ionenserien Aliphatische Moleküle mit verschiedenen funktionellen Gruppen ergeben charakteristische Ionenserien, die immer einen Abstand von 14 Massen aufweisen, jedoch bei unterschiedlichen Startmassen beginnen. Dies erklärt sich aus der Spaltung jeweils benachbarter C-C-Bindungen, so dass die Fragmente sich um jeweils eine CH2Gruppe (14 Massen) unterscheiden. Die unterschiedliche Startmasse hängt natürlich von der kleinsten Einheit mit der jeweiligen funktionellen Gruppe ab. Die folgende Tabelle enthält die wichtigsten Ionenserien: funktionelle Gruppe Startmasse Ionenserie Kohlenwasserstoff 29 (C2H5+) 29, 43, 57, 71, 85, 99, 113, ... Äther, Alkohol 31 (H2C=O+H) 31, 45, 59, 73, 87, 101, .... Keton 43 (H2C-CO+) Amin 30 (H2C=N+H2) 30, 44, 58, 72, 86, 100, .... 43, 57, 71, 85, 99, 113, .... Das folgende Beispiel zeigt die charakteristische Serie eines Alkohols (der Molekülpeak ist nicht sichtbar): Neutralteilchenabspaltung m Neutralteilchen typische Verbindungen 1 H 2 H2 15 CH3 16 17 Aldehyde, Acetale, Alkine, Aryl-CH3, =N-CH3 kondensierte aromatische Ringsysteme Acetale, Methylderivate, (CH3)3SiO-Derivate Aromatische Nitroverbindungen, aromatische Amide Karboxylsäuren MS 18 H2O 20 HF 26 C2H2 27 28 29 30 31 32 33 34 35 Cl 36 HCl 40 H2C=C=N| 41 43 45 46 47 48 55 57 79 79Br 81 81Br 91 127 I 15/24 König geradkettige Aldehyde, primäre Alkohole Fluoralkane aromatische Kohlenwasserstoffe aromatische Amine, N-Heterozyklen Phenole, Aldehyde, Quinone aromatische Aldehyde, aliphatische Nitrile aromatische Nitroverbindungen aromatische Methylether Methoxy-Derivate o-Methylbenzoate kurze primäre Alkohole, Isothiocyanate Thiole sekundäre und tertiäre Chloralkane n-Chloralkane aliphatische Nitrile, Dinitrile Propylester Propyl-Verbindungen, aliphatische Nitrile, t-Amide Karboxylsäuren Nitroverbindungen Phosphorylverbindungen Sulfoxide Butylester Ethyl- und Butylketone Bromverbindungen (Isotopenverhältnis beachten - 79Br:81Br = 1:1) Bromverbindungen (Isotopenverhältnis beachten - 79Br:81Br = 1:1) Benzyl- und Tolylverbindungen Iodverbindungen Spektroskopische Datenbanken Die folgende Tabelle enthält eine alphabetische Liste der spektroskopischen Datenbanken mit den jeweiligen Links zu den Webseiten der Anbieter. Die Angaben zu den Datenbanken stammen von den Anbietern. Produkt/Datenbank Kurzbeschreibung AAFS MSDC Database Ca. 1600 Massenspektren ACD/CNMR DB ACD/HNMR DB 3.0 ACD 19F NMR Prediction and Database ACD 31P NMR Prediction and Database Aldrich FT-IR Vapor Phase Library 13C-NMR-Spektren von 58000 Strukturen; enthält Referenzen auf die Originalspektren, Summenformel, Molekulargewicht und IUPAC-Name. 1H-NMR, 300000 experimentell ermittelte chemische Verschiebungen, 50000 Kopplungskonstanten für ca. 50000 Strukturen. Die ACD Vorhersagealgorithmen basieren auf einer internen Datenbank von über 11000 Strukturen und den zugehörigen 19F-Verschiebungen. Die ACD Vorhersagealgorithmen basieren auf einer internen Datenbank von über 9000 Strukturen und den zugehörigen 31P-Verschiebungen. 5,010 FTIR-Spektren; enthält Gasphasen-Spektren die von Aldrich über einen GC gewonnen wurden um chromatographisch reine Proben sicher zu stellen. MS 16/24 König Aldrich FT-IR Condensed Phase Library 18,500 FTIR Spektren; diese Sammlung enthält die wichtigsten Substanzen aus dem Aldrich Catalog Handbook of Fine Chemicals. Diese Spektrenbibliothek ist die größte Sammlung von FTIR-Spektren. Aldrich FT-Raman Condensed Phase Library 14,033 FT-Raman Spektren der wichtigsten Laborchemikalien AntiBase BMRB Biological NMR Data CSEARCH David Sullivan FT-IR Library EPA FT-IR Reference Spectra EPA FT-IR Vapor Phase Library AntiBase enthält verschiedene chemisch-physikalische Daten inklusive UV-, 13C-NMR-, IR- and Massenspektren für ca. 20000 Substanzen aus dem Bereich der Mikrobiologie. Die BMRB Datenbank enthält mehr als 100000 chemische Verschiebungen von Proteinen und Peptiden. Die Datenbank ist in der Publikation "A Relational Database for Sequence-Specific Protein NMR Data", B.R. Seavey, E.A. Farr, W.M. Westler, and J.L. Markley, J. Biomol. NMR 1, 217-236 (1991) beschrieben. 13C 80000 Spektren 31P 6000 Spektren 15N 5000 Spektren 19F 5000 Spektren 11B 9000 Spektren 17O 5000 Spektren Ca. 100 FT-IR Spektren zum freien Download. Ca. 350 FT-IR Referenzspektren von ungefähr 100 gefährlichen Stoffen die zur Luftverschmutzung beitragen. Sammlung von 3297 FT-IR Spektren. Ca. 50 FT-IR Spektren von Fasern. Diese Spektren wurden für eine FBI- FBI FT-IR Fibers Library Projekt zur Identifizierung von Fasern mittels FTIR gemessen (Spectrochimica Acta, V.46, p.1513, 1991) FDM FTIR Spectra of Polymers and Polymer Additives FDM FTIR Spectra of Surfactants FDM FTIR Spectra of Organic Compounds FDM FTIR Spectra of Minerals and Inorganic Compounds FDM FTIR Spectra of Drugs / Canadian Forensic FTIR Spectra FDM VP FTIR Spectra of Organic Compounds 580 Spektren 430 Spektren 950 Spektren 310 Spektren 3750 Spektren 5220 Spektren Frei zugängliche online-Datenbank mit ca. 6,000 IR-, MS-, UVVIS-, NIR- Galactic Spectra Online und NMR-Spektren. Die Spektren können in SPC-Format vom Server geladen werden. JINNO Drug Database Ca. 80 UV/VIS Spektren von Drogen JINNO PAH Data Base Ca. 50 UV/VIS-Spektren von PAHs (Polycyclic Aromatic Hydrocarbons) JICST Mass Spectral Database Ca. 800 Messenspekren, online verfügbar MassLib MassLib kann mit einer Reihe verschiedener Spektrenbibliotheken ausgestattet werden: MS 17/24 König - NIST (>100.000 Spektren) - Wiley Registry (>220.000 Spektren) - geo- und petrochemische Verbindungen (1.100 spectra) - Pharmazeutika und Metaboliten (2.200 spectra) - Androstane (2.000 spectra) IR spectra von über 5000 Verbindungen Massen Spektren von über 10000 Verbindungen UV/VIS Spektren für mehr als 2000 Verbindungen NIST WebBook NIST/EPA/NIH Mass 130000 Massenspektren von mehr als 100000 Verbindungen Spectral Library Public database of NMR ca. 30 13C und 1H Spektren spectra Sadtler Condensed enthält IR-Spektre von mehr als 75000 Verbindungen Phase IR Standards Database Sadtler Vapor Phase IR 9,200 IR-Spektren reiner Verbindungen in der Gasphase (bei 25 bis 300°C) Standards SDBS SpecInfo SpecLib USCA UV/VIS spectra Wiley AccessPak Integrierte Spektrendatenbank für organische Substanzen: MS (ca. 19,000 Spektren), 13C NMR (ca 10,200 Spektren), 1H NMR (ca 11,500 Spektren), ESR (ca 1,300 Spektren), IR (ca 47,000 Spektren), Raman (ca 3,500 Spektren) Integrierte Spektrendatenbank für organische Substanzen: 13C-NMR (200000 Spektren), 1H-NMR (4000 Spektren), 15N (1000 Spektren), 17O (800 Spektren), 19F (23500 Spektren), 31P (16000 Spektren), MS (360000 Spektren), IR (24000 Spektren), NIR (4000 Spektren) SpecLib ist eine Spektrenbibliothek als Ergänzung zu SpecTool (550 MS-, >550 H-NMR-, >550 C-NMR-, >440 IR-, >1,000 UV/VIS-Spektren). ca. 40 UV/VIS Spektren von Farbstoffen und Indikatoren 275000 Massenspektren auf CDROM Werkzeuge zur Bearbeitung spektroskopischer Daten Werkzeuge zur Bearbeitung von Spektren gibt es sehr viele; ihre Funktionalität reicht von einfachen Konversionsprogrammen bis zu vollständigen Interpretationssystemen. In der folgenden Liste sind die wichtigsten Werkzeuge zusammengefasst. Produktname / Link ACD NMR Viewer BenchTop/PBM Chemscape Chime Software CSEARCH Kurzbeschreibung Viewer für NMR-Daten. Mit diesem Programm können verschiedene Formate aus dem Bereich der NMR-Spektroskopie geladen und dargestellt werden (JCMP/DX, Varian FIDs und Phasenfiles, Bruker FIDs, NUTs, Galactic SPC Format und ACS/ESP Dateien. BenchTop/PBM ist ein Suchsystem für die Massenspektrometrie mit ca. 275,000 Referenzspektren. Ein frei verfügbarer Viewer für Molekülstrukturen, der auch Spektren im JCMP/DX Format darstellen kann. Online-NMR-Suche basierend auf HOSE-Codes (W. Bremser, Anal. Chim. Acta, 103(1978), 355-365). Es stehen folgende Spektrenbibliotheken zur Verfügung. 13C 80000 Spektren 31P 6000 Spektren 15N 5000 Spektren 19F 5000 Spektren 11B 9000 Spektren MS 18/24 König O17 5000 Spektren FDM Electronic Handbook of FTIR Spectra gNMR Galactic Spectra Online FTIR Spektren und entsprechende Software zum Zugriff über Netzwerk. NMR-Simulation von Molkülen mit bis zu 12 Spinsystemen. Spectra Online ist eine kostenlos über das Internet suchbare Datenbank mit 6,000 Spektren (IR, MS, NMR (proton, carbon and other nuclei), UV/VIS und NIR). Generische Spektroskopie-Software mit vielen eingebauten Algorithmen. GRAMS/32 Instrument Data File Import und Konvertierungsfilter für die wichtigsten Datenformate. Compatibility Suchsoftware für IR-Spektren IR SearchMaster Isotopensimulator für Windows IsoPro MassLib Molecular Weight Calculator MS Lab MSLIB MassLib ist das führende Werkzeug zur Spektrensuche in MSDatenbanken und zur Interpretation von Massenspektren. Berechnet das Molekulargewicht und die prozentuale Zusammensetzung chemischer Verbindungen. To assist in the structural understanding of glycoproteins and the interpretation of their complex mass spectra, we are developing a software package named “MS Lab” Ein MSDOS-basiertes Programm für die Verwaltung von Massenspektren (veraltet). NUTS NMR Toolbox zur Bearbeitung und zur Darstellung von NMR-Daten processing software Potsdam University ONLINE wizzard to support the interpretation of IR, MS, and NMR spectra Spectroscopic Tools SpecTool WSearch SpecTool is a Hypermedia application containing reference data, reference spectra and computational tools for the interpretation of Mass Spectra, NMRIR- and UV-spectra. This is a Windows program that can read KRATOS DS90 files, HP Chemstation files, Finnigan ITS40 files and Varian Saturn files. Spektrenbeispiele: MS-Spektrum: 1-Bromhexan MS 19/24 König Im Spektrum des 1-Bromhexans sieht man sehr schön die beiden Isotopenpeaks des Broms bei den Massen 107/109, 135/137 und 164/166. Der Molekülpeak bei m/z = 164 ist nur wenig ausgebildet, da das Molekül wenig beständig ist und durch Abspaltung eines Neutralteilchens in die zyklische Struktur bei Masse 135 übergeht. Als alternativer Fragmentierungsweg, der zu einem stabileren Fragment führt, ist die Abspaltung von Brom zu sehen, wodurch das Hexyl-Ion bei Masse 85 entsteht. Der Peak bei Masse 55 entsteht durch Abspaltung von HBr aus den Fragmenten 135 und 137. MS-Spektrum: Butanol-Isomere Ein schönes Beispiel für die Stevenson-Regel findet sich in den Fragmentierungsmustern der Isomere des Butanols wieder. Bei der -Spaltung wird generell jenes Radikal eliminiert, das die meisten Schwingungsfreiheitsgrade aufweist (also am größten ist). 1-Butanol MS 20/24 2-Butanol t-Butanol MS-Spektrum: 2-Oktanon König MS 21/24 König Die Fragmentierung von 2-Oktanon erfolgt entsprechend der McLafferty-Umlagerung. Dabei wird 1-Penten als Neutralteilchen abgespalten, wodurch der Peak bei m/z=58 (McLafferty-Peak) entsteht. Die Peaks bei 43, 71, 85 und 113 entstehen durch Spaltung der Alkylkette an der entsprechenden Stelle. MS-Spektrum: Benzol Die Fragmentierung von aromatischen Kohlenwasserstoffen ist generell geringfügig, da die Ringe sehr stabil sind und leicht Molekülionen bilden. Meist weisen daher Spektren aromatischer Kohlenwasserstoffe einen prominenten Molekülpeak auf, der oft auch der Basispeak ist. Wenn Aromaten fragmentieren, spalten sie meist C2H2 oder C3H3 ab, was zu charakteristischen Peaks bei den Massen 39 und 52 führt. MS 22/24 König MS-Spektrum: Benzylalkohol Aromatische Alkohole haben einen klar ausgeprägten Molekülpeak. Es tritt nicht wie bei aliphatischen Alkoholen -Spaltung (Stevenson-Regel ) auf, sondern bevorzugt die Bildung eines Ions aus der Tropylium-Ionen-Familie. Beim Benzylalkohol entsteht ein Hydroxytropylium-ion, das für den kräftigen M-1-Peak verantwortlich ist. MS Spektrum: Toluol Befindet sich an einem Benzolring eine Alkylkette, so tritt bevorzugt eine ß-Spaltung bezüglich des Ringes auf, wodurch sich das Tropylium-Ion (m/z = 91) bildet. Das Spektrum oben zeigt die Fragmentionen von Toluol; neben dem Molekülion (m/z = 92) entsteht ein sehr intensives Signal bei der Masse 91. MS 23/24 König Das Tropylium-Ion ist so stabil, dass z.B. die Fragmentierung eines Dimethylbenzols zu einem Methyl-Tropylium-Ion führt (m/z = 105). Entsprechend geringe Intensitäten weisen die restlichen Fragmente im Spektrum auf. MS-Spektrum: Methylbutyrat Die Fragmentionen des Methylbutyrats entstehen auf zwei für Ester typischen Wegen. Zum einen tritt normaler Bindungsbruch an der Karbonylgruppe auf (m/z = 43, 59, und 71), zum anderen tritt parallel dazu eine McLafferty-Umlagerung auf, die zum Ion mit der Masse 74 führt. Das Fragment aus der McLafferty-Umlagerung kann benutzt werden um auf den Typ des Alkohols im Ester Rückschlüsse zu ziehen. MS-Spektrum: Diethylamin Amine sind in ihrem Fragmentierungsverhalten den Alkoholen und Ethern vergleichbar. Das Immonium-Ion (=C=NH+) ist deutlich stabiler als das Oxonium-Ion, da der Stickstoff die positive Ladung besser stabilisieren kann. Dies macht sich dadurch bemerkbar, dass in Spektren von Aminen bevorzugt Fragmentpeaks mit geraden Massenzahlen auftreten MS 24/24 König (typische Reihe: 30, 44, 58, 72, 86, 100,...). Das Spektrum von Diethylamin macht dies deutlich: