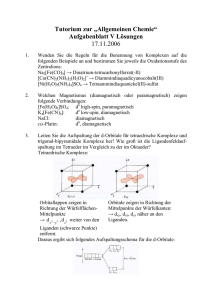
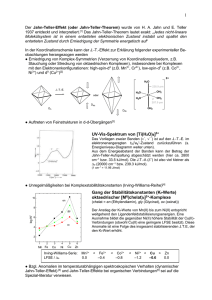
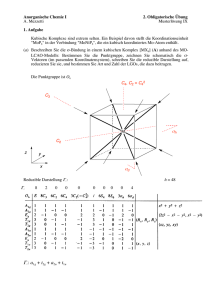
in D o
Werbung

Ligandenfeld-Theorie Einleitung & Abgrenzung Energieaufspaltung der d-Orbitale im Ligandenfeld Experimentelle Ermittlung der Energieaufspaltung Do Faktoren die Do beeinflussen High-Spin / Low-Spin-Komplexe Ligandenfeldstabilisierungsenergie Vorteile & Schwächen der Theorie Prof. Dr. Berthold Kersting, E-mail: [email protected] Skript: www.uni-leipzig.de/~bkerst/~lehre, password: LFSE 1. Einleitung & Abgrenzung Bindungsmodelle für Übergangsmetallverbindungen a) b) c) d) Lewis-Säure/Lewis-Base Wechselwirkung (Sidgwick, 1923) Valenzbindungs-Theorie (Pauling, Sidgwick, 1930) Ligandenfeld-Theorie (Bethe, van Vleck, ab ~ 1930) Molekülorbital-Theorie Komplex: [Fe(H2O)6]2+ 3d 4p 4s 4d L 2 d sp oktaedrisch Komplex: [Fe(CN)6] L 3 2- 2- L Fe L L L d2sp3 oktaedrisch Die ersten beiden Theorien können die Anzahl der ungepaarten Elektronen bei manchen Komplexen nicht erklären (high-spin/low-spin), machen keine Aussagen zu den Farben oder der Reaktivität von Komplexen. 2. Energieaufspaltung der d-Orbitale im Ligandenfeld Annahme der Ligandenfeldtheorie: ● abstoßende elektrostatische Wechselwirkungen zwischen Elektronen der Liganden u. Metallionen ● L und M = Punktladungen ● Ligandenfeld = gesamtes elektrisches Feld, das von den Liganden ausgeht Ergebnis: ● Änderung der energetischen Lage der fünf d-Orbitale in einem Ligandenfeld z y x dz2 dx2-y2 dxy dxz eg-Orbitale (Orbitallappen auf den Achsen) t2g-Orbitale (Orbitallappen zwischen den Achsen) dzy Beispiel: Oktaedrischer Komplex Oktaedrisches Ligandenfeld ● Liganden weisen direkt auf die dz2- und dx2-y2-Orbitale (starke elektrost. WW) ● Liganden liegen zwischen den dxy, dxz, dyz-Orbitalen (geringere elektrost. WW) Ligand dxz dx2-y2 stärkere Abstoßung Anhebung der Energie geringere Abstoßung Absenkung der Energie Ergebnis: ● Zwei Sätze von d-Orbitalen unterschiedlicher Energie (t2g, eg) Beispiel: Oktaedrischer Komplex Resultat ● zwei Sätze von d-Orbitalen: t2g- und eg-Orbitale*; ● eg-Orbitale haben die höhere Energie ● Bezeichnung der Energiedifferenz: Do (oder 10Dq) Oktaederfeldaufspaltung Do Do = 84 – 480 kJ/mol Energie dz2, d x2-y2 eg 0,6 Do Do 0,4 Do xy, xz, yz freies Ion Ion im sphärischen Kristallfeld t2g Ion im oktaedrischen Kristallfeld *eg, t2g: Mullikensymbole, Symmetrieverhalten (-typ) der d-Orbitale Beispiel: Tetraedrischer Komplex Orbitalenergie eg Do t2 0.6 Do 0.4 Dt Dt 0.4 Do 0.6 Dt e t2g Oktaederfeld Tetraederfeld Resultat ● wiederum zwei Sätze von d-Orbitalen: t2- (dxy, dxz, dyz) und e-Orbitale (dz2, dx2-y2); ● t2-Orbitale haben die höhere Energie ● Bezeichnung der Energiedifferenz: Dt (Dt = 4/9 Do ~ ½ Do) 3. Experimentelle Ermittlung der Energieaufspaltung Do ● Farbe von Komplexen beruht auf Elektronenübergängen t2g -> eg, ● Energie der Elektronenübergänge entspricht dem Wert von Do ● UV/Vis-Spektroskopie zur Messung von Elektronenübergängen Beispiel: [Ti(OH2)6]3+ absorbiert blaues Licht der Wellenlänge 490 nm h Do t2g1eg0 = 490 nm DE h h t2g0eg1 c DE D0 = 240 kJ/mol 4. Faktoren, die Do beeinflussen a) Die Oxidationsstufe des Metallions Do nimmt mit steigender Oxidationsstufe des Metallions zu: [CoII(NH3)6]2+ [CoIII(NH3)6]3+ Do = 980 nm = 122 kJ/mol Do = 437 nm = 273 kJ/mol b) Die Art des Metallions Do nimmt innerhalb einer Gruppe von oben (3d-Elemente) nach unten (5d-Elemente) zu: 3d < 4d <5d => 4d- und 5d-Elemente liegen fast immer als low-spin-Komplexe vor [CoIII(NH3)6]3+ [RhIII(NH3)6]3+ [IrIII(NH3)6]3+ Do = 437 nm = 122 kJ/mol Do = 293 nm = 408 kJ/mol Do = 242 nm = 493 kJ/mol Zahlenwerte aus L.H. Gade, Koordinationschemie, Wiley-VCH, 1999, S. 201. Verwendete Umrechnungsfaktoren: 1 cm1 = 11.96 J/mol , [nm] = 107 / [cm-1] Faktoren, die Do beeinflussen c) Die Koordinationszahl [NiCl6]4- Do = 7700 cm-1 [NiCl4]2- Dt = 3850 cm-1 Do = -4/9 Dt Orbitalenergie eg Do t2 0.6 Do 0.4 Dt 0.4 Do Dt 0.6 Dt e t2g Oktaederfeld Tetraederfeld Konsequenz: tetraedrische Komplexe fast immer high-spin Faktoren, die Do beeinflussen d) Die Liganden Es wurde gefunden, daß der Wert von Do in der folgenden Reihe von links nach rechts zunimmt Spektrochemische Reihe: I- < Br- < S2- < SCN- < Cl- < N3- < F- < OH- < O2- < OH2 < NCS- < NH3 ~ py < en < bpy < NO2- < CH3- < CN- < CO Konsequenz: Low-spin- und high-spin-Konfigurationen möglich bei d4 bis d7 Konfiguration 5. High-Spin- / Low-Spin-Komplexe Betrag von Do ist maßgeblich für die Elektronenkonfiguration von Metallkomplexen ● bei d1-d3 sowie d8-d10 nur eine Möglichkeit ● bei d4-d7 zwei Möglichkeiten: high-spin und low-spin Komplexe P, die Spinpaarungsenergie, besitzt einen Wert von ca. 200 kJ/mol Low-spin-Komplex High-spin-Komplex dz2, dx2-y2 P P = Spinpaarungsenergie dz2, dx2-y2 Do < P Do > P dxy, dxz, dyz dxy, dxz, dyz Faustregel: Starke Liganden machen Low-Spin-Komplexe, schwache Liganden ergeben High-Spin-Komplexe 6. Ligandenfeldstabilisierungsenergie (LFSE) ● LFSE = Energiegewinn einer dn-Konfiguration im Ligandenfeld ● jedes t2g-Elektron liefert –0.4 Do, jedes eg-Elektron benötigt +0.6 Do Ti3+ V3+ dz2, dx2-y2 dz2, dx2-y2 -0.4 Do dxy, dxz, dyz LFSE = -0.4 Do -0.4 Do dxy, dxz, dyz LFSE = 2x(-0.4) Do = -0.8 Do Gang der Ligandenfeldstabilisierungsenergie (LFSE) Beispiel d0, Ca2+ d1, Ti3+ d2, V3+ d3, Cr3+ d4, Mn3+ d5, Fe3+ d6, Co3+ d7, Co2+ d8, Ni2+ d9, Cu2+ d10, Zn2+ (t2g)0 (t2g)1 (t2g)2 (t2g)3 (t2g)3eg1 (t2g)3eg2 (t2g)4eg2 (t2g)5eg2 (t2g)6eg2 (t2g)6eg3 (t2g)6eg4 LFSE (in 0.0 0.4 0.8 1.2 0.6 0.0 0.4 0.8 1.2 0.6 0.0 Do) (t2g)4eg0 (t2g)5eg0 (t2g)6eg0 (t2g)6eg1 1.6 2.0 2.4 0.8 LFSE ist groß bei low-spin-Komplexen, maximal bei low-spin d6 (-2.4 Do) ● erklärt Beständigkeit von low-spin-Komplexen (z.B. Co3+-Komplexe) ● erklärt bevorzugte oktaedrische Koordination von Fe2+, Co3+-Komplexen ● erklärt erhöhte Reaktivität von Fe3+ (high-spin) bzw. Zn2+ (=> Spurenelemente) 7. Vorteile / Schwächen der Ligandenfeldtheorie Vorteile der LFT: ● Gutes, qualitatives Verständnis von Elektronischen Spektren, Farben ● Magnetische Eigenschaften und ● Strukturen (z.B. high-spin/low-spin) von Übergangsmetallkomplexen Schwächen der LFT-Theorie: ● Liganden = Punktladungen! ● keine Berücksichtigung kovalenter Bindungsanteile, ● keine Erklärung für Stellung der Liganden in der spektrochemischen Reihe Erklärbar ebenfalls ● Gang der Ionenradien ● Kinetische Stabilität /Labilität ● Verlauf der Hydratationsenthalpien ● Gitterenergien der Metalldihalogenide ● Verzerrungen von Idealgeometrien Bedeutung am Beispiel des Rubin-Lasers Rubin-Laser Termsymbol E (eV) Elektronenkonfiguration 4 4 T1g (t2g2eg1) 2 T2g Angeregte Zustände 1 (t2g eg ) 2 Al2O3·xCr2O3 539 nm 620 nm Eg (t2g3eg0) Rot-Emission (Phosphoreszenz) bei 627 nm = LASER Übergang 4 Grundzustand A2g (t2g3eg0) Absorptionen für rote (Komplementärfarbe) des Edelsteins verantwortlich Pigmente, Leuchtstoffe, Werkstoffwissenschaften (CD, DVD, LASER), Elektronikindustrie Ligandenanordnung in der spektrochemischen Reihe Die Ligandenanordnung in der spektrochemischen Reihe wird im Wesentlichen durch kovalente Metall-Ligand-p-Wechselwirkungen bestimmt p-Donor reiner -Donor schwacher p-Donor starker p-Akzeptor schwacher p-Akzeptor zunehmendes Do Spektrochemische Reihe: I- < Br- < S2- < SCN- < Cl- < N3- < F- < OH- < O2- < OH2 < NCS- < NH3 ~ py < en < bpy < NO2- < CH3- < CN- < CO MO-Theorie Molekülorbitale des Metallkomplexes Orbitale des Metallatoms 4p antibindende Orbitale 4s eg Do 3d t2g nichtbindende Orbitale Orbitale der Liganden bindende Orbitale