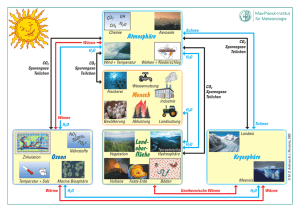
Infomaterial zur Vorlesung - Technische Universität Braunschweig
Werbung

Physikalische Chemie für Studierende im Nebenfach (Lehramt, Biologie, Geoökologie, Pharmazie) Vorlesung SoSe 2015 (1413003) Stand: Juli 2015 Prof. Dr. Sigurd Bauerecker, Institut für Physikalische und Theoretische Chemie, Hans-Sommer-Str. 10, Ruf 0531/391-5336 [email protected], http://www.tu-braunschweig.de/pci/forschung/bauerecker Vorlesung, 2 SWS, deutsch, jedes SoSe, 8:00 bis 9:30, Hörsaal SN 20.2 Seite 1, Bauerecker, PC für Nebenfächler Klausuren • Geoökologie und CuV Lehramt: Audimax, 05.08.2015, 07:30 Uhr 2stündig, Vorlesungsstoff, Wiederholungsklaursur: Audimax, 17.02.2016, ab 7:30 Uhr, Dauer: je 90 min. • Biologie: Audimax, 05.08.2015, 07:30 Uhr Vorlesungsstoff, Wiederholungsklaursur: Audimax, 17.02.2016, , ab 7:30 Uhr, Dauer: je 120 min. • Pharmazie: nach dem 4. Semester im Rahmen des Ersten Staatsexamens Bedingungen der Klausur: Namen, Matrikelnr., Fachbenennung eintragen; keine Hilfsmitteln, nur Kugelschreiber, Papier wird gestellt; Namen, Seitenzahl auf jedes Blatt Papier; Freitext-Klausur; gleiche Klausur für alle, aber Biologie 30 min länger -> ein bis zwei Aufgaben mehr; ♦ Vorlesung ist Grundlage für PC-Praktikum Seite 2, Bauerecker, PC für Nebenfächler Literatur & Lehrmaterial Software: SimChemistry für Windows • Molekulardynamik-Software zu Ausbildungszwecken kostenfrei herunterladbar: www.simchemistry.co.uk Nachschlagewerke • Chempagedia Physikalische Chemie: www.chemgapedia.de • IUPAC Gold Book - aktuelles Kompendium chemischer Terminologie: goldbook.iupac.org • CRC Handbook of Chemistry and Physics, umfangreiche Tabellen mit Stoffeigenschaften von chem. Verbindungen, CRC Press, erscheint jährlich neu, aktuell: 95. Aufl. (2013/14) • H. Kuchling: Taschenbuch der Physik. Carl Hanser Verlag, 20. Auflage, 2010, ca. 711 S. • Vorlesung C. Maul: Phys. Chemie f. Nebenfächler: www.tu-braunschweig.de/pci/research/maul/people/hcm/vl Lehrbücher Chemie-Nebenfach • G. Adam, P. Läuger, G. Stark: Physikalische Chemie u. Biophysik*. Springer, 5. Aufl., 2009, 642 S. • C. Czeslik, H. Seemann, R. Winter: Basiswissen Physikalische Chemie*. Teubner, 4. Aufl., 2010, 396 S. • P. Atkins, J. Paula: Kurzlehrbuch Physikalische Chemie. Wiley – VCH, 4. Auflage, 2008, 1154 S. Lehrbücher Chemie-Hauptfach • P. Atkins, J. Paula: Atkins: Physikalische Chemie. Wiley – VCH, 5. Auflage, 2008, 1316 S. • G. Wedler, H.-J. Freund: Lehrbuch der Physikalischen Chemie. Wiley – VCH, 6. Aufl., 2012, 1386 S. *verfügbar im IP-Adressbereich der TUBS, auch über VPN Seite 3, Bauerecker, PC für Nebenfächler Einordnung der Physikalischen Chemie Physik Chemische Physik Physikalische Chemie Chemie Biophysik Biophysikalische Chemie Molekularbiologie, Biochemie, Physiolog. Chemie Biologie Physikalische Grundpfeiler der PC ♦ Thermodynamik (auch irreversible) Seite 4, Bauerecker, PC für Nebenfächler ♦ Quantenmechanik Warum PC? • Thermodynamik: Energieerzeugung technisch, Kraftwerke, Motoren, Kühl- und Klimaanlagen … GM Hydrogen4 Solvis GmbH • Thermodynamik: Energieerzeugung biologisch, Stoffwechsel Organismus u. Zelle, Nahrungs- und Atmungsketten, Löslichkeiten … Seite 5, Bauerecker, PC für Nebenfächler Warum PC? • Kinetik und Transport: Reaktionsgeschwindigkeiten, Katalyse, Enzymkinetik, Atmung u. Blutkreislauf, Wirkstofftransport, Explosion Stoffkreisläufe Pflanze Quelle: Digitales Nachschlagewerk über Bäume Seite 6, Bauerecker, PC für Nebenfächler Explosion Natrium in Wasser P.E. Mason, F. Uhlig, V. Vaněk, T. Buttersack, S. Bauerecker, Jungwirth, Nature Chemistry (2015) Bereiche der Physikalischen Chemie ⇒ Statistische Theorie (Deutung makroskopischer Erscheinungen auf molekularer Ebene), wird hier überwiegend nur behandelt in: ⇒ Kinetische Gastheorie (Gaseigenschaften, ideales Gas, Van-der Waals Gas, …) ⇒ Chemische Thermodynamik (Energie, Entropie, Hauptsätze, Reinstoffe, Mischungen, Lösungen, Ein- und Mehrphasensysteme, …) ⇒ Transportprozesse (Diffusion, Wärmetransport, irreversible Prozesse, laminare und turbulente Strömungen, …) ⇒ Chemische Reaktionen (Gleichgewichte, Reaktionskinetik, Geschwindigkeit, …) ⇒ Grenzflächenerscheinungen (Oberflächenspannung, Kapillarität, selektiver Stofftransport, Osmose, Monolayer, Lipiddoppelschicht, …) ⇒ Spektroskopie (Wechselwirkung Licht mit Materie, …) ⇒ Elektrochemie (Elektrochemische Zellen, Elektrolyte, Redoxreaktion, Brennstoffzelle, Membranpotential, Nervenerregung, Photosynthese, …) ⇒ Aufbau der Materie (Atome, Moleküle, Gase, Aerosole, Flüssigkeiten, Festkörper, Plasma, …), kaum hier behandelt Seite 7, Bauerecker, PC für Nebenfächler Mit auf den Weg Biologen, Pharmazeuten brauchen fundierte Kenntnisse in Physikalischer Chemie. Biologie ist eine exakte Naturwissenschaft. Die Physikalische Chemie beschäftigt sich mit den Änderungen der physikalisch chemischen Eigenschaften von Materie Seite 8, Bauerecker, PC für Nebenfächler Wichtige Begriffe der Physikalischen Chemie 1 System - Komplexes Gebilde, kann feste, flüssige Stoffe, Gase enthalten - Teil der Welt, vom Rest der Welt (Umgebung) abgegrenzt - Materieaustausch Energieaustausch mit Umgebung möglich: nein nein abgeschlossenes System nein ja geschlossenes System ja ja offenes System - Homogenes System: einphasig (Wasser, Salzkristall, Luft, Salzlösung) - Heterogenes System: mehrphasig (Milch, Sand, Rauch, Nebel) Epot q Wärmezufuhr Umgebung Ekin U W Arbeitsleistung am System System Wärmeabgabe -q -W U innere Energie Seite 9, Bauerecker, PC für Nebenfächler Arbeitsleistung vom System Wichtige Begriffe der Physikalischen Chemie 2 Zustandsgröße Die Systemeigenschaften sind messbar und beschreiben den Zustand des Systems (Beschreibung Aggregatzustand z.B. über Kompressibilität). Das System ist im Gleichgewicht, wenn es seinen Zustand nicht selbsttätig zu verändern sucht. Die Gleichgewichtsbedingungen sind reproduzierbar, durch Zustandsgrößen (Zustandsvariable) charakterisierbar und hängen nicht von der Vorgeschichte des Systems ab. • Intensive Zustandsgrößen: sie sind unabhängig von der Stoffmenge der Phase (Druck, Temperatur, Konzentration). Sie sind nicht additiv. • Extensive Zustandsgrößen: sie sind abhängig von der Stoffmenge der Phase (Masse, Volumen). Sie sind additiv. Zustandsfunktion Die einzelnen Zustandsgrößen eines Systems sind durch die Zustandsgleichung (Zustandsfunktion) miteinander verknüpft (Zustandsgleichung ideales Gas: pV = NkT). Seite 10, Bauerecker, PC für Nebenfächler Wichtige Begriffe der Physikalischen Chemie 3 Phase Bis in molekulare Bereiche physikalisch homogener Teil eines Systems. Bildquelle: Chemgapedia 1 Komponente: 2 Phasen: Seite 11, Bauerecker, PC für Nebenfächler Wasser flüssig + fest 3 Komponenten: 2 Phasen: Wasser, Ether, Farbstoff wässrige + organische Temperatur Definitionen Zusammenhang zwischen absoluter thermodynamischer Temperatur T in K und Celsius-Temperatur θ in °C T / K = 273,15 + θ / °C Absoluter Nullpunkt von T Bildquelle: Vorlesung C. Maul 2014 Historisch geht die Definition der Celsius-Temperatur aus der Schmelz- und Siedetemperatur des Wassers (0 und 100 °C) hervor. Seite 12, Bauerecker, PC für Nebenfächler Das Ideale Gas Gase zeigen das einfachste Verhalten der möglichen Aggregatzustände. Das Gas wird umso einfacher beschreibbar (idealer), je niedriger der Druck und je höher die Temperatur ist. Die den Gaszustand beschreibenden physikalischen Größen Druck p in Pa, Volumen V in m3, Temperatur T in K und Molekülzahldichte n = N/V in m-3 sind abhängig voneinander und bilden eine Zustandsgleichung, die N ⋅ k ⋅T = n ⋅ k ⋅T V Ideale Gasgleichung: p= Ideale Gasgleichung (für 1 Mol): N mol ⋅ R ⋅T = c ⋅ R ⋅T p= V Teilchengesamtzahl Gasmasse Konzentration Molzahl, Stoffmenge Molmasse N m = Nmol⋅M c = Nmol/V Nmol M BOLTZMANN-Konstante k = 1,381⋅ 10-23 J⋅K-1, Allgem. Gaskonstante R = 8,3143 J⋅K-1⋅mol-1, in kg in mol⋅m-3, in mol, in kg⋅mol-1, ist gewissermaßen die Gaskonstante pro Teilchen, auch molare oder universelle Gaskonstante, ist für alle Gase gleich, AVOGADRO-Konstante NA= R / k = N / Nmol = 6,022 ⋅ 1023 mol-1, Teilchenzahl pro Stoffmenge; damit hat 1 mol 6,022 ⋅ 1023 Teilchen. Seite 13, Bauerecker, PC für Nebenfächler Norm- und Standard-Bedingungen International Union of Pure and Applied Chemistry Standardzustand nach IUPAC T°= 298,15 K = 25°C p° = 100 kPa = 1000 mbar Normzustand nach DIN 1343 Tn = 273,15 K = 0°C pn = 101.325 Pa = 1013,25 mbar Achtung: es werden auch andere Standards verwendet (z.B. früher 1013 mbar). Molares Volumen oder Molvolumen (Volumen von 1 mol ideales Gas) Im Standardzustand: Im Normzustand: Vm° = 24,79 L/mol Vm,n = 22,414 L/mol Seite 14, Bauerecker, PC für Nebenfächler 3dimensionales pVT-Diagramm ideales Gas N p = ⋅ k ⋅T V Seite 15, Bauerecker, PC für Nebenfächler Quelle: Vorlesung C. Maul 2014 Ideale Gasgleichung Anwendungen des idealen Gasgesetzes • Gasvolumetrische Umrechnungen (z.B. in Analytik) • Umrechnung auf Normalbedingungen p = 1,013 bar, 0 °C (z.B. Erdgas) • Molmassenbestimmung • Gasthermometer • CO2-Gehalt im Blut nach VAN SLYKE und NEILL H+ + HCO3- ⇔ H2O + CO2↑ • Physiologische Umsetzung unter Beteiligung von Gasen (z.B. in WARBURG-Apparaturen, Gärungsreaktionen) Seite 16, Bauerecker, PC für Nebenfächler Temperaturmessung, Gasthermometer • Flüssigkeitsthermometer • Gasthermometer • Widerstandsthermometer • Thermoelemente • Strahlungsthermometer Bildquelle: UNC College of Arts and Sciences - Gasvolumen wird konstant gehalten über Hg-Niveau - aus Höhendifferenz h wird Druck p bestimmt - über ideales Gasgesetz ergibt sich T (V und N konstant) Seite 17, Bauerecker, PC für Nebenfächler Kinetische Behandlung des idealen Gases I y (Beispiel für deduktive Behandlung phys.-chem. Vorgänge) Modell des idealen Gases: - Moleküldurchmesser klein gegen mittleren Molekülabstand - keine Kraftwirkungen zwischen Molekülen - geradlinig fortschreitende Bewegung, auch bei Stößen (Wand, Moleküle), klass. Gesetze Mechanik - n Teilchenzahldichte, m Molekülmasse. A vx x z In Zeitdifferenz dt kommen mit Fläche A genau ½·n·vx·dt·A Moleküle zum Stoss. Jedes überträgt den Impuls 2·m ·vx auf die Fläche. Alle zusammen daher den Impuls dB = Σ ½ ·n ·vx ·dt · A · 2·m ·vx = Σ n · m · vx2 · dt · A 2 Einführung des mittl. Geschwindigkeitsquadrats v x , Aufsummierung: ∆B = n · m · v x2 · ∆t · A Druck p ist definiert als p = F /A = 1/A · ∆B/∆t = n · m · v x2 Drei Raumrichtungen gleichwertig, Vekorbetrachtung (räumlicher Satz d. PYTHAGORAS): v 2 = v x2 + v y2 + v z2 ⇒ v x2 = v y2 = v z2 = 13 v 2 ⇒ Anderseits gilt nach id. Gasgesetz p = n ⋅ k ⋅ T ⇒ Seite 18, Bauerecker, PC für Nebenfächler p = 13 ⋅ n ⋅ m ⋅ v 2 v2 = 3⋅ k ⋅T m Ableitung des Drucks Ableitung mittl. quadratische Geschwindigkeit Beispiele f. mittl. quadratische Geschwindigkeit Mittl. quadratische Geschwindigkeit Seite 19, Bauerecker, PC für Nebenfächler v2 = 3⋅ k ⋅T m Wasserstoff H2 1888 m/s Sauerstoff O2 479 m/s Quecksilber Hg 189 m/s Anwendungen der Gaskinetik Die Diffusionsgeschwindigkeiten u1, u2 zweier verschiedener Gase bei gleichen Bedingungen (p,T) verhalten sich umgekehrt wie die Wurzeln aus den Quotienten der molekularen Massen und Molmassen: u1 m2 = = u2 m1 M2 M1 Dies ist wichtig für: • Molekulardestillation (Vitamine, Lipide, Aromen, …) • Hochvakuumsublimation (Reinstoffe, Arzneimittel, Pheromone, …) • Gefriertrocknung (biolog. Präparate, Kaffee*, Tee, Gemüse, …) • Sprühtrocknung (Kaffee, Milch, Tee, Extrakte, Waschmittel, Babynahrung, Instantprodukte allgemein, …) *Welthandelsgut Nr. 2 Seite 20, Bauerecker, PC für Nebenfächler Kinetische Behandlung des idealen Gases II Temperatur wird als kinetische Energie der Moleküle interpretiert: Mittlere kin. Energie eines Moleküls: Ekin = 12 m ⋅ v 2 = 32 k ⋅ T ⇒ nur Funktion von T! Also, Verknüpfung der mechanischen Größe Energie mit der Temperatur! ⇒ Bewegung d. Moleküle. Ekin setzt sich aus 3 Komponenten zusammen: Ekin = 12 m ⋅ v x2 + 12 m ⋅ v y2 + 12 m ⋅ v z2 = 32 k ⋅ T ⇒ E x = E y = E z = 12 k ⋅ T 3 Translationsfreiheitsgrade werden als Möglichkeiten der Moleküle bezeichnet, sich in 3 Raumrichtungen geradlinig-fortschreitend zu bewegen. Je Translationsfreiheitsgrad entfällt die Energie: 12 k ⋅ T Gleichverteilungssatz (CLAUSIUS, MAXWELL) oder Äquipartitionsprinzip: Auf jeden Freiheitsgrad eines Moleküls entfällt im Mittel die gleiche Energie Die Anzahl der Freiheitsgrade f eines Systems ergibt sich aus der Anzahl der Koordinaten, durch die der Bewegungszustand eindeutig bestimmt ist. Beispiel zweiatomiges Molekül: 3 Translationen, 2 Rotationen, 1 Schwingung. Seite 21, Bauerecker, PC für Nebenfächler 1 2 k ⋅T Daltonsches Gesetz Der Gesamtdruck des Gasgemisches ist gleich der Summe der Partialdrücke, d.h., Summe der Drücke der einzelnen Gase. Die Dampfbildung über einer verdunstenden Flüssigkeit wird durch Anwesenheit anderer Dämpfe/Gase nicht beeinflusst. Der entstehende Dampfdruck heißt Partialdruck. Seite 22, Bauerecker, PC für Nebenfächler Bsp. Gaseigenschaften Physikalische Eigenschaften Wasserstoff und Methan [Cer08, Roe11, Ull00] Größe Molare Masse Spezielle Gaskonstante Siedetemperatur Schmelztemperatur Krit. Temperatur Krit. Druck Krit. Dichte Dichte (20°C, 101325 Pa) Löslichkeit in Wasser (20°C) Viskosität, dynamische (20°C) Heizwert Symbol M Ri Tb Tm Tkr pkr Brennwert Ho Zündbereich in Luft Detonationsgrenzen Detonationsgeschwindigkeit Zündtemperatur in Luft Minimale Zündenergie Lamin. Brenngeschwindigkeit Diffusionskoeffizient in Luft Kohäsionsdruck (vdW-Konst.) Kovolumen (vdW-Konst.) Seite 23, Bauerecker, PC für Nebenfächler ρkr ρ η Hu Tz D a b Einheit kg/kmol J/kg/K °C °C °C MPa kg/m3 kg/m3=g/L mL/L kg/m/s MJ/Nm3 MJ/kg MJ/Nm3 MJ/kg kWh/kg Vol% Vol% km/s °C mJ m/s cm2/s Pa*m6/mol2 m3/mol H2 2,016 4124 -252,77 -259,20 -240 1,315 31 0,084 20 8,8*10-6 10,78 120 12,75 141,9 39,4 4 – 75 18,3 – 59 1,5 – 2,2 530 0,017 270 – 346 0,61 0,0247 2,66*10-5 CH4 16,043 518 -161,48 -182,47 -82,6 4,595 162,2 0,71 35 11,0*10-6 35,89 50 39,83 55,5 15,4 4,4 – 16,5 6,3 – 13,5 1,4 – 1,6 645 0,28 37 – 43 0,15 0,225 4,28*10-5 3,6 MJ = 1 kWh BOLTZMANNsche Energieverteilung BOLTZMANN-Verteilung (kanonische Verteilung), folgt aus klassischer Statistik = MaxwellBoltzmann-Statistik, ist Grenzfall der Quantenstatistik. Sie folgt aus dem Prinzip, dass der wahrscheinlichste Zustand angenommen wird, also der, der unter Berücksichtigung von Randbedingungen die meisten Realisationsmöglichkeiten hat. Sie beschreibt damit die Energieverteilung von N Teilchen mit der Gesamtenergie E, die die r Energiewerte E0 bis Er-1 annehmen können. Also jeder Energiewert Ei bekommt Ni Teilchen zugeordnet. Randbedingungen: - Teilchen sind unterscheidbar (nummeriert vorstellbar), - Teilchen sind unabhängig (keine wechselseitige Beeinflussung), - jedes Teilchen nimmt genau einen Energiewert an. E N − kTi Ni = ⋅ e Z BOLTZMANN-Verteilung Die Teilchen verteilen sich bezüglich der Energie pro Raumkoordinate in einer e-Funktion! Für Interessierte: Z = ∑je r −1 − Ej kT = konst. E = ∑ N i Ei = konst. i =0 Quelle: Vorlesung K.-H. Gericke Seite 24, Bauerecker, PC für Nebenfächler ist die Zustandssumme, ist die Gesamtenergie. Ableitungen aus der BOLTZMANN-Verteilung Aus der BOLTZMANN-Verteilung lassen sich etliche wichtige Gesetze der PC ableiten. Beispiele: • Verteilung der kinetischen Energie der Teilchen pro Raumkoordinate (e-Funktion, siehe oben) • Verteilung der kinetischen Energie der Teilchen f(E) • Verteilung der Geschwindigkeit (Impuls) der Teilchen f(v) • Luftdruckverteilung in der Erdatmosphäre p(h) • Gleichgewichtskonzentration in chemischen Reaktionen c(G) h Höhe über Normal, G Gibbs-Energie, freie Enthalpie Seite 25, Bauerecker, PC für Nebenfächler Barometrische Höhenformel Ableitung aus der BOLTZMANN-Verteilung BOLTZMANN-Verteilung/Prinzip: Die N Gasteilchen in der Atmosphäre ordnen sich bezüglich ihrer potentiellen Energie so an, dass es einem Maximum der Realisationsmöglichkeiten entspricht, d.h. in einer e-Funktion. Randbedingungen: Luftmoleküle im Schwerefeld der Erde haben alle gleiche Teilchenmasse m. Temperatur T sei konstant, g Erdbeschleunigung, h Höhe über Meeresniveau. Potentielle Energie eines Luftteilchens: Dann ergibt sich für die Teilchenverteilung: Mit Idealem Gasgesetz: E(h) = m⋅g⋅h, N0 = N(h=0), N ( h) = N 0 ⋅ e p ( h ) = p0 ⋅ e − − E (h) kT = N0 ⋅ e − m⋅ g ⋅ h kT p0 = p(h=0) h m⋅ g ⋅ h kT In welcher Höhe halbiert sich der Druck? Quelle: Imagico.de m⋅ g ⋅h = ln 0,5 = -ln2 kT 8,3 ⋅ 300 kT RT = ⋅ ln 2 = ⋅ ln 2 = ⋅ 0.693 m = 6075 m 0,029 ⋅ 9,81 m⋅ g M ⋅g p (h) = 0,5 ⋅ p0 ⇒ h1/ 2 m⋅ g ⋅ h − kT ⇒ e = 0,5 ⇒ − Quelle: Vorlesung C. Maul Seite 26, Bauerecker, PC für Nebenfächler Verteilung kin. Energie ideales Gas Bei der Berechnung der Verteilung der gesamten kinetischen Energie müssen alle drei Raumrichtungen berücksichtigt werden. Jedes Teilchen kann jeweils drei von einander unabhängige Energiezustände für die x, y, und z Richtungen einnehmen (sogenannte Entartung), wodurch bei der Verteilung ein statistischer Faktor hinzu kommt. Daher steigt die Verteilung zunächst bei kleinen Energien mit einer Wurzelfunktion. Bei großen Energien dominiert die e-Funktion, so dass die Verteilung entsprechend fällt. Verteilung der kinetischen Energie 2 f (E) = π 1 RT 3/ 2 ⋅ E ⋅e − E RT BOLTZMANN-Faktor f(E) 3/2 RT enspricht 300 K Normierungs-Faktor Statistischer Faktor 6/2 RT enspricht 600 K Der Normierungs-Faktor wird so gewählt, dass alle Teilchen über alle Energien erfasst werden, also dass gilt: T = 300 K ∞ 0 2000 4000 6000 Energie (1 mol) E / J Seite 27, Bauerecker, PC für Nebenfächler 8000 10000 ∫ f (E ) = 1 0 Geschwindigkeitsverteilung id. Gas Die Geschwindigkeitsverteilung für ideale Gasteilchen lässt sich aus deren Verteilung der kinetischen Energie ableiten. Dabei geht man von der Teilchenzahl in Abhängigkeit von der Energie dN(E) = f(E) ⋅ dE aus, ersetzt in dieser Gleichung E und dE nach den Gleichungen E = ½ mv2 und dE = m⋅v⋅dv (aus Ableitung der vorhergehenden Gleichung) und erhält die Gleichung dN(v) = f(v) ⋅dv und damit f(v). MAXWELL-BOLTZMANNsche Geschwindigkeitsverteilung f(v) 2 m f (v ) = ⋅ π 2π ⋅ kT T = 300 K 0 200 400 600 800 1000 Geschwindigkeit Luftmoleküle v (m/s) Seite 28, Bauerecker, PC für Nebenfächler 1200 3/ 2 ⋅v ⋅e 2 − m 2 ⋅v 2⋅kT Geschwindigkeitsverteilung id. Gas II MAXWELL-BOLTZMANNsche Geschwindigkeitsverteilung 2 m f (v ) = ⋅ π 2π ⋅ kT 3/ 2 ⋅ v2 ⋅ e − 3 charakteristische Geschwindigkeiten • Wahrscheinlichste Geschwindigkeit (Maximalwert der Verteilung), Ableitung verschwindet: m 2 ⋅v 2⋅kT 2kT m vˆ = f ′(v) = 0 • Mittlere Geschwindigkeit (arithmetisches Mittel) ∞ v = ∫ v ⋅ f (v)dv 0 f(v) 8kT v= π ⋅m T = 300 K • Mittlere quadratische Geschwindigkeit (mittl. „energetische“ Geschwindigkeit) 3kT v = m 2 0 200 400 600 800 1000 Geschwindigkeit Luftmoleküle v (m/s) Seite 29, Bauerecker, PC für Nebenfächler 1200 ∞ v = ∫ v 2 f (v)dv 2 0 Geschwindigkeitsverteilung id. Gas III Temperaturabhängigkeit der MAXWELL-BOLTZMANNschen Geschwindigkeitsverteilung. Mit zunehmender Temperatur steigt der Anteil der hochenergetischen, hochreaktiven Moleküle im sogenannten „BOLTZMANNSchwanz“. Aus der Formel erkennt man, dass eine Verringerung der Molekülmasse exakt die gleiche Wirkung auf die Verteilung hat, wie eine Erhöhung der Temperatur. Bsp.: Luft, N2 2 f (v ) = π m ⋅ kT π 2 ⋅ 3/ 2 ⋅v ⋅e 2 − m 2 ⋅v 2⋅kT BOLTZMANN-Schwanz, energiereiche Teilchen sorgen für Reaktionen, Sublimation von Schnee/Eis, Verdunstung von Wasser, … Bildquelle: Vorlesung C. Maul 2014 Seite 30, Bauerecker, PC für Nebenfächler Mittlere freie Weglänge von Gasteilchen Mittlerer Abstand zwischen 2 Gasteilchen (ideales Gas) bei Standardbedingungen: V kT 3 3 r= = N p 3,5 nm −23 J 1 , 38 10 ⋅ K ⋅ 298,15 K =3 = 3,5 nm 105 Pa ⇒ Mittlere Gasdichte (Luft, N2) ist etwa 1000 mal geringer als die der dichtesten Kugelpackung. ⇒ Flüssige Luft ist grob 1000 mal dichter als Luft bei Standardbedingungen. Mittlere freie Weglänge bei Stoss zwischen zwei Teilchen: Stickstoff (N2) bei Standardbedingungen: V kT l= = 2 π 2 ⋅d ⋅ N π 2 ⋅d2 ⋅ p Seite 31, Bauerecker, PC für Nebenfächler Bildquelle: Chemgapedia ≈ 75 nm, Luft bei Standardbedingungen Stoßzahlen von Gasteilchen Stoßzahl für 1 Teilchen (ideales Gas). Stöße mit Stoßpartnern pro Zeit: Stoßzahl z = N2 bei Standardbedingungen mittlere Teilchengeschwindigkeit v 400 m/s = = 5,3 ⋅109 s -1 −9 mittlere freie Weglänge l 75 ⋅10 m Mittlere Stoßzeit ∆tS zwischen 2 Stößen (N2, Standardbedingungen): ∆t S = 1 = 1,88 ⋅10-10 s = 188 ps z Stoßzahl insgesamt (ideales Gas, in Volumen V). Faktor ½, damit Stöße nicht doppelt gezählt werden. Ohne Ableitung: Gesamtzahl der Stöße Z = 1 N 1 v N 1 v ⋅π 2 ⋅ d 2 ⋅ N 2 = 2⋅ ⋅ = 2⋅ 2⋅z⋅ V l V V2 Stoßzahlen sind wichtige Kenngrößen für Transportphänomene und Reaktionsgeschwindigkeiten. Seite 32, Bauerecker, PC für Nebenfächler Druck- und Vakuumtechnik Bezeichnung Bereich (mbar) Anwendungen, Bedeutung Ultrahochvakuum < 10-7 Wissenschaftliche Forschung, Weltraumsimulation Hochvakuum 10-7 – 10-4 Molekulardestillation, Hochvakuumsublimation, Hochvakuum-Schmelzen und Gießen Feinvakuum 10-4 – 1 Gefriertrocknung, Sublimative Reinigung, Glühlampenherstellung Grobvakuum 1 – 103 Konservierung („Einkochen“), Vakuumverpackung, Destillation unter vermind. Druck, Himalaja (8,85 km ⇒ 320 mbar) Normaldruck um 103 Luftdruck auf Meereshöhe, Sprühtrocknung Hochdruck 104 – 106 Synthesen (z.B. NH3), Hochdruckgasextraktion (z.B. Entkoffeinierung, Aromen), Untertagespeicherung von Gasen Höchstdruck 106 – 109 Geologische Prozesse, Diamantsynthese Seite 33, Bauerecker, PC für Nebenfächler Vakuumpumpe Quelle: GUT Speicherung von Wind- und Sonnenenergie Unvorteilhaft ist unregelmäßiges Windenergieangebot. Lösungsansätze: ⇒ Umwandlung in Wasserstoff über Elektrolyse ⇒ Speicherung über Druckluft, z.B. in Kavernen, siehe Bild unten ⇒ Pumpspeicherkraftwerke Seite 34, Bauerecker, PC für Nebenfächler Reale Gase: Lennard-Jones-Potenzial Lennard-Jones-Potential A B Φ (r ) = − 6 + 12 r r Potenzial Φ Reale Gasteilchen (Atome, Moleküle, Ionen) können sich in Abhängigkeit vom Abstand anziehen und abstoßen. Die potenzielle Energie zweier kugelförmiger Teilchen kann durch ein Teilchen-Wechselwirkungs-Potenzial ausgedrückt werden, z.B. durch das bekannte Für r > r0 überwiegt Anziehung, für r < r0 überwiegt (starke) Abstoßung, B r 12 0 bei r = rmin sind die beiden Teilchen im Potentialminimum, also in der Gleichgewichtslage (z.B. bei T = 0 K). r0 / pm He N2 Ar 258 368 342 Angström: 1 Å = 100 pm Seite 35, Bauerecker, PC für Nebenfächler − r0 Beispiele für r0 = r (Φ=0) Abstoßungsterm rmin A r6 Anziehungsterm Abstand der Teilchenzentren r Reale Gase VAN-DER-WAALS-Gleichung (1873): (p + a/V2) (V – b) = RT pV – Diagramm Bereich ideales Gas, Verflüssigung unmöglich, Isothermen ≈ Hyperbeln Anziehungsterm (Kohäsionsdruck, Berücksichtigt Anziehung der Teilchen) Kritischer Punkt mit (Sattelpunkt) dp d 2 p = =0 dV dV 2 Gas Zweiphasengebiet Flüssigkeit Phasenübergang Seite 36, Bauerecker, PC für Nebenfächler Abstoßungsterm (Kovolumen, berücksichtigt effektives Eigenvolumen der Teilchen) gleichgroße Flächen, Verlauf physikalisch unsinnig a, b, Van-der-Waals-Konstanten, müssen empirisch bestimmt werden, z.B. aus kritischen Daten : Tkr= 8a / 27bR, pkr = a / 27b2, Vkr = 3b. Realgasfaktor Das Realgasverhalten kann beliebig genau genähert werden durch eine Reihenentwicklung des Realgasfaktors Z als Virialgleichung mit tabellierten T-abhängigen Virialkoeffizienten: pVmol B(T ) C (T ) D(T ) = Z =1+ + 2 + 3 + ... RT Vmol Vmol Vmol Mit dem Realgasfaktor im Normzustand Zn definiert man die Kompressibilitätszahl K: Z K= Zn [Cer08] Verschiedene Gase bei 0°C, zunächst < 1, dann steigend Seite 37, Bauerecker, PC für Nebenfächler Erdgas bei verschiedenen Temperaturen Anwendung: Energiedichte Wasserstoff u. Methan Van der Waals Bildquelle: Eichelseder 2010 700 bar • Große Unterschiede bis Faktor 2 je nach Modell bei großen Drücken! • Beispiel Druckspeicher H2 bei 700 bar ⇒ Realgasfaktor bis 3. Virialkoeffizient notwendig! • Methan/Erdgas hat grob dreifache Energiedichte als Flüssigkeit und Druckgas Seite 38, Bauerecker, PC für Nebenfächler Volumenarbeit ideales Gas Hohm Das ideale Gas dehnt sich gegen äußeren Druck p im Zylinder gegen einen beweglichen Kolben mit der Fläche A aus. Die Kraft F ergibt sich aus dem Druck: dl dV F = p ⋅ A = p ⋅ A⋅ = p ⋅ dl dl Das System (Gas) leistet Arbeit an der Umgebung: δW = − F ⋅ dl = − p ⋅ dV = − Seite 39, Bauerecker, PC für Nebenfächler n ⋅ R ⋅T dV V Hohm Die vom Gas geleistete Arbeit ist (graue Fläche): V2 nRT dV = −nRT ln ∆W = ∫ − V V1 V1 V2 ∆W ist wegabhängig, siehe rotumspannte Fläche. Daher ist W keine Zustandsfunktion! Bsp.: Kompressionsarbeit H2 GM Hydrogen4 • Die Kompressionsarbeit ist proportional ln(p2/p1). • Bezogen auf den Energieinhalt ist sie im Idealfall für CH4 (Erdgas) etwa 3,3mal geringer. • Ein höherer Druck als 700 bar wird gebraucht, um H2 in den Drucktank einzufüllen. • Der H2 muss gegebenenfalls noch gekühlt werden, weil a) das Gas im Tank wieder komprimiert wird und sich dabei erwärmt, b) sich H2 beim Entspannen erwärmt (Joule-Thomson-Effekt, oberhalb Inversionstemperatur!). Die letzten beiden Punkte sind in der Tabelle noch nicht berücksichtigt! Gaskonstante R = 8,3143 J/K/mol, Molmasse 0,002 kg, 1 kg H2 entspricht 500 mol, Energieinhalt (Heizwert Hu) H2 = 120 MJ/kg, 293 K. Tabelle: Kompressionsarbeit für Wasserstoff Verdichtung Faktor ln(p2/p1) Anwendung /bar 1 –> 25 25 3,2 Rohrnetz 1 –> 700 700 6,6 Drucktank 25 –> 700 28 3,3 Drucktank/Rohrnetz Arbeit/MJ 1kg H2 3,90 8,04 4,02 in % H2-Energieinhalt ideal real 3,3 6,6 6,7 13,4 3,4 6,8 Wasserstofftransport in Druckbehältern bei 700 bar. Verdichtungsverluste als Anteil der H2-Energie betragen etwa 13 % (Anfangsdruck 2 bar, 4stufige Kompression), 7 % (Anfangsdruck 28 bar, 2stufige Kompression, relevant bei vorhandenem H2-Pipeline-System!) [Wue07, Bos09]. (Die reale Verdichtungsarbeit ist etwa doppelt so groß, weil noch der Wirkungsgrad des Verdichters mit etwa 50 % zu berücksichtigen ist [Eic10]). Seite 40, Bauerecker, PC für Nebenfächler Für Interessierte: Joule-Thomson-Effekt Joule-Thomson-Effekt: Gase in Behältern unter erhöhtem Druck kühlen sich beim Ausströmen ab, wenn die Gastemperatur unterhalb der Inversionstemperatur liegt (irreversible adiabatische Expansion realer Gase über Drossel). Dabei wird nahezu keine äußere Arbeit geleistet und wegen hoher Geschwindigkeit keine Wärme mit der Umgebung ausgetauscht, aber innere Arbeit gegen zwischenmolekulare Kräfte. Nutzung z.B. zur Luftverflüssigung, Linde 1876. [Her12] Bildquelle: Wikipedia Richtung und Stärke der Temperaturänderung bei isenthalper Zustandsänderung wird durch den Joule-ThomsonKoeffizient µ beschrieben: ∂T ∆T µ = = ∆p H ∂p H µ ist abhängig von Gasart, Anfangsdruck und Anfangstemperatur. µ > 0, Anfangstemperatur < Inversionstemperatur Abkühlung: µ < 0, Anfangstemperatur > Inversionstemperatur Erwärmung: Inversionstemperatur Van-der-Waals-Gas (nur Maximalwert, da auch vom Druck abhängig!): Ti ≈ 2a = 6,75 ⋅ Tk R ⋅b Das sind mit den Parametern a, b für H2 223 K. Inversionstemperatur Wasserstoff (1013 mbar) [Kuc07]: 193,15 K. Seite 41, Bauerecker, PC für Nebenfächler Joule-Thomson-Inversionskurve für H2 (para) mit Dampfdruckkurve [Ull00] µ<0 µ>0 Dynamische Viskosität Modell zur dynamischen Viskosität: Eine ebene Platte wird mit der Kraft K und der Geschwindigkeit v auf einer Flüssigkeitsschicht der Dicke x bewegt. Gase sind näherungsweise inkompressibel, wenn ihre Strömungsgeschwindigkeit höchstens 1/3 der Schallgeschwindigkeit beträgt [Her07] Reibungsgesetz nach Newton (1687) mit der dynamischen Viskosität η (Zähigkeit) mit der Einheit [η]= N·s·m-2 = Pa·s als Proportionalitätskonstante für ein Newtonsches Fluid: K =η ⋅ A ⋅ Seite 42, Bauerecker, PC für Nebenfächler dv dx Zahlenwerte dynamische Viskosität Substanz Wasser Blut Glykol Glykol/Wasser (50/50) Ethylether Glycerin n-Heptan n-Nonan n-Tetradekan n-Hexadekan Luft Wasserstoff-Gas Helium-Gas Neon-Gas T / °C 0 20 40 100 37 0 20 0 20 20 0 20 20 20 20 20 0 40 0 0 0 η / 10-3 Pa·s 1,789 1,005 0,653 0,282 3 bis 25! 62 22 10 4,4 0,243 12110 1499 0,409 0,711 2,18 3,34 0,0171 0,0190 0,00835 0,0186 0,0297 Quelle: Adam, Läuger, Stark 2002 Seite 43, Bauerecker, PC für Nebenfächler Beachte: • Viskosität η nimmt bei Flüssigkeiten mit steigender Temperatur sehr stark ab: η ≈ A eB/T A, B empirische Konstanten • Bei Gasen nimmt Viskosität η mit steigender Temperatur zu! Bsp. Kinematische Viskosität T-abhängige Viskosität von Glykol-Wasser-Gemischen Kinematische Viskosität ν in mm2/s ist Quotient aus dynamischer Viskosität η und Dichte ρ : ν=η/ρ Quelle: Viessmann Handbuch Solarthermie Anwendung z.B. in Solarthermie (Frage welches Wasser/Glykol-Gemisch liegt vor?) Quelle: Baukataloge.ch Seite 44, Bauerecker, PC für Nebenfächler Laminare Rohrströmng, Gesetz v. HAGEN-POISEUILLE In laminaren Rohrströmungen (z.B. in Niederdruckgasnetzen) haben die einzelnen Fluidschichten unterschiedliche Geschwindigkeiten v(r): null an der Wandung und maximal auf der Rohrachse. Dazwischen bildet sich eine parabolische Geschwindigkeitsverteilung aus, mit ∆p Druckdifferenz zwischen den Rohrenden, l Rohrlänge, r Radius der bewegten Zylinderschale, R Rohrdurchmesser: v(r ) = ∆p ⋅ (R 2 − r 2 ) 4 ⋅ l ⋅η Berechnung des Gesamtvolumen V, das den Zylinder (Rohr) pro Zeiteinheit t durchströmt. Durch die Zylinderschale zwischen r und r + dr strömt (R 2 − r 2 ) dV = 2π ⋅ r ⋅ dr ⋅ t ⋅ v = 2π ⋅ r ⋅ dr ⋅ t ⋅ ∆p 4 ⋅ l ⋅η R π ⋅ t ⋅ ∆p R 2 2 V = ∫ dV = ⋅ ∫ (R − r )rdr ⋅ ⋅ l 2 η 0 0 ⇒ Gesetz von HAGEN-POISEULLE π ⋅ t ⋅ ∆p ⋅ R 4 V= 8 ⋅η ⋅ l Seite 45, Bauerecker, PC für Nebenfächler Folgerungen: • Transportierte Fluidvolumen steigt mit 4. Potenz des Rohrradius! • Verdopplung Rohrradius bewirkt 16fachen (!) Volumentransport ⇒ über Rohrradius lässt sich das Strömungsvolumen wesentlich effektiver als über den Druck vergrößern. • Anwendung Biologie: Variation des Kapillarradius mit glatter Gefäßmuskulatur erlaubt eine effektive Regulation der Durchblutung von tierischem Gewebe. • Gilt nur für laminare Strömungen, also für Re < 2320. Reynoldszahl Charakterisierung von Fluidströmungen über Reynolds-Zahl Re (d Hauptabmessung des Körpers, z.B. Rohrdurchmesser, ρ Massendichte des Fluids, v Relativgeschwindigkeit Körper/Medium weit vom Körper entfernt, η dynamische Viskosität): Re = d ⋅ ρ ⋅v laminar turbulent η Unter der kritischen Reynoldszahl Rekrit = 2320 ist die Rohrströmung laminar [Cer08]. Darüber kann sie zunächst noch laminar sein (bis etwa 20000 [Kuc07]), ist aber so instabil, dass bei kleinster Störung bleibende Turbulenz eintritt . Reynoldsches Ähnlichkeitsgesetz: Strömungsbilder mit gleicher Reynoldszahl ähneln sich und lassen sich aufeinander übertragen. Beispiel 1: das Strömungsbild bleibt gleich bei Verdopplung des Rohrdurchmessers und Halbierung der Strömungsgeschwindigkeit. Beispiel 2: im Windkanal können 10fach kleinere Modelle bei 10fach höherer Strömungsgeschwindigkeit getestet werden. Seite 46, Bauerecker, PC für Nebenfächler Für Interessierte: Beispiele Reynoldszahl Beispiel Ideales Gas: Für das ideale Gas gilt ρ ∝ p / T, η ∝ T1/2 und η(p) = konst., η ist also unabhängig vom Druck. Damit ergibt sich: d ⋅ p ⋅v Re ∝ 3 / 2 T Damit lassen sich Druck-Temperaturpaare mit gleicher Reynoldszahl und gleichem Strömungsbild aufstellen, wie z.B. (1013 mbar, 293 K), (200 mbar, 100 K). Beispiel Gasherd [Cer08]: Durch Herdanschlussleitung mit Rohrinnendurchmesser von 16 mm (DN15) fließen bei 20°C etwa 0,9 m3/h Erdgas. Die Strömungsgeschwindigkeit ergibt sich aus dem Volumenstrom pro Rohrquerschnitt, η = 11*10-6 kg/m/s ⇒ Re = 0,016 * 0,71 * 0,9 / 3600 / 0,0162 / π *4 / 11*10-6 = 1285 < 2320. Die Strömung ist also laminar. Beispiel Wasserstoff: Gleicher Energiefluss bei 3 m3/h. ⇒ Re = 0,016 * 0,084 * 3 / 3600 / 0,0162 / π *4 / 8,8*10-6 = 1583 < 2320. Die Reynoldszahl ist für H2 um den Faktor 1,23 größer und immer noch laminar. Beispiel maximaler laminarer Wasserstoff-Energietransport: Rohrinnendurchmesser 0,4 m, p = 30 bar = 3 MPa, ideales Gas angenommen, T = 20°C, ⇒ v = 2320*8,8*10-6/0,4/(30*0,084) m/s = 0,02 m/s. ⇒ Volumenstrom = Rohrquerschnitt * v = 0,0025 m3/s ⇒ Energiefluss = Volumenstrom * Heizwert * Druck = 0,0025 * 10,78 * 30 MW = 0,8 MW. Der Energiefluss ist proportional zum Rohrdurchmesser und unabhängig vom Druck. Seite 47, Bauerecker, PC für Nebenfächler Für Interessierte: Rohrreibungsdruckverluste Rohrreibungsdruckverluste bei inkompressiblen Medien Druckverluste bei Rohrströmungen werden durch folgenden allgemeingültige Ansatz (für laminare und turbulente Strömungen) beschrieben: Gleichung von Darcy ∆p λ k l r, d ρ v ∆p = λ l ρ 2 v r 4 Druckabfall durch Rohrreibung Rohrreibungszahl Rohrrauigkeit Rohrlänge Rohrinnenradius (Durchmesser) Dichte Mittlere Geschwindigkeit Pa m m m kg/m3 m/s Laminare Strömung: λ = 64/Re ⇒ Hagen-Poiseuillesches Gesetz! Turbulente Strömung: - hydraulisch glatte Rohre, k = 0 ⇒ λ hängt nur von Re ab - hydraulisch rauen Rohren ⇒ λ hängt nur von d und k ab - Übergangsbereich zw. rau und glatt ⇒ λ hängt von Re, d und k ab Daher wird λ durch verschiedene Gleichungen beschrieben. Einfacher handhabbar ist das Rohrreibungsdiagramm, aus dem man λ entnehmen kann. Seite 48, Bauerecker, PC für Nebenfächler Für Interessierte: Rohrreibungsdruckverluste kompressible Medien Im Vergleich zu Flüssigkeiten sind Gase kompressibel (aber ≈ inkompressibel bei Strömungsgeschwindigkeit < 1/3 Schallgeschwindigkeit!), ⇒ Ausdehnung bei sinkendem Druck ⇒ Berechnung komplizierter Effizienter Transport im Hochdruckrohrnetz ⇒ Hoher Druck (30 – 120 bar), hohe Strömungsgeschwindigkeit v12 l 2 ⇒ Turbulente Rohrströmungen mit Re > 2320 p2 = p1 − p1 ⇒ Näherungen: Vernachlässigung kin. Energie, T = konst ⇒ V1 d ⇒ Enddruck ist Wurzelfunktion der Rohrlänge [Glu88] λ λ l d Enddruck Anfangsdruck Anfangsgeschwindigkeit Spez. Anfangsvolumen Rohrreibungszahl Rohrlänge Rohrinnendurchmesser Pa Pa m/s m3/kg m m Druckverlust Pipeline 1000 MW 30 bar Anfangsdruck Rohrdurchmesser D = 0,6 m, Temperatur 15°C, Massestrom: 8,33 kg/s (H2), 21,3 kg/s (CH4) 35 30 25 Rohrdruck / bar p2 p1 v1 V1 Wasserstoff D = 0,6 m Methan D = 0,6 m Methan D = 0,516 m 20 15 10 Vergleich der Druckverluste von Pipelines in Abhängigkeit von der Rohrlänge bei 15°C mit Transportkapazität 1000 MW, berechnet nach obiger Gleichung bei einem Anfangsdruck von 30 bar. Das entspricht einem Massestrom von 8,33 kg/s für Wasserstoff und 21,3 kg/s für Methan. Dabei schneidet Wasserstoff etwas schlechter ab als Methan: bei gleichem Rohrinnendurchmesser von 0,6 m kommt man bei H2 auf 370 km, bei CH4 auf knapp 800 km. Allerdings braucht man bei H2 mit 0,6 m gegenüber CH4 mit 0,516 m nur einen etwas größeren Rohrdurchmesser, um das gleiche Ergebnis zu erzielen. Seite 49, Bauerecker, PC für Nebenfächler 5 0 0 100 200 300 400 500 Rohrlänge / km 600 700 800 900 Für Interessierte: Berechnung Rohrdruckverluste v12 l p2 = p − p1λ V1 d 2 1 Seite 50, Bauerecker, PC für Nebenfächler Zum Vertrautmachen: Druckverlust-Online-Rechner [Sch11] http://www.druckverlust.de Beispiel Erdgaspipeline OPAL Ostsee-Pipeline-Anbindungs-Leitung: • 470 km lang • zwischen Lubmin und Südsachsen/ Tschechien • verschweißt aus 26000 Elementen • je 18 m lang, 2,2 cm stark, 15 t schwer • 1 Mrd. Euro Baukosten • Innendurchmesser 1,4 m • Anfangsdruck 84 bar (bis 120 bar) • Massenstrom 1200 kg/s CH4 ⇒ 60 Gigawatt an Transportleistung ( ≈ elektr. Durchschnittsleistung von Deutschland!) Seite 51, Bauerecker, PC für Nebenfächler Anwendung: H2-Pipeline-Transportverluste in Brennwert% von H2: Quelle: Ulf Bossel Leipzig 4.11.2009 Plausibler durchschnittlicher Transportweg in Europa bei lokaler H2Produktion, z.B. aus Photovoltaik, Wind oder Biomasse: 100 km ⇒ Minimale H2-Transportverluste Seite 52, Bauerecker, PC für Nebenfächler Anwendung: Energietransport Vergleich Stromnetz mit Gasnetz • Verteilungskosten Strom: 5 – 12 ct/kWh • Verteilungskosten Gas: 0,7 ct/kWh (Rohrmiete Erdgas 0,2 ct/kWh) • Grenzübergangspreis Erdgas (Polen/D, 2014): 2,5 ct/kWh! • Energieverteilung über Gas-Rohrnetz etwa 10 mal effizienter/günstiger als über elektrisches Stromnetz (Gesamtkosten) • Beispiel: Vergleich Hochspannungs-Überlandleitung (220 – 380 kV, Mast 60 m hoch) mit Rohrleitung (Erdgas oder H2, 40 cm Durchmesser): beide transportieren die Leistung von je 600 MW. ⇒ Strom wird als Energieträger überschätzt ⇒ Gasnetz hat Puffer- und Speicher-Option ⇒ Moderne Stromübertragung (HGÜ)? Quellen: [Tet08, Agf00, Har94] Seite 53, Bauerecker, PC für Nebenfächler Anwendung: Wasserstoff-Netze Wasserstofftransport in Rohrleitungssystemen (sehr hohe Sicherheitsstandards [Gei04]): • Deutschland/Ruhrgebiet seit 1938, Air Liquide, 240 km, bis 30 bar, bislang keine nennenswerten Unfälle. • Deutschland/Leuna, Linde AG, 100 km, 24 bar. • Nordfrankreich/Belgien seit 1966, Air Liquide, 400 km, 65 – 100 bar. • USA seit ca. 1955, Systeme mit insgesamt 720 km. • Stadtgas seit 150 Jahren Vision Wasserstoffenergienetz (Rifkin, 2002): „Hydrogen Energy Web“ (HEW) in Analogie zum „World Wide Web“ (WWW). Quelle: BMW http://www.bmw.dk/dk/da/insights/technology/cleanenergy/_sh ared/pdf/cleanenergy_map.pdf?download=true Seite 54, Bauerecker, PC für Nebenfächler Thermodynamisches System Bei chemischen Reaktionen werden nicht nur Stoffe umgewandelt, sondern auch Energien: • freigesetzt (exotherme Vorgänge), • aufgenommen (endotherme Vorgänge). Für chemische Reaktionen gilt die Erhaltung der Masse (nicht für Kernchemie: E = mc2). Epot Wärmezufuhr q Wärmeabgabe -q Ekin U System w -w Arbeitsleistung am System Arbeitsleistung vom System Vorzeichenfestlegung: „immer vom System aus gesehen“. Die Arbeit w kann sein: • Volumenarbeit (mechanische Arbeit), • elektrische Arbeit, • Oberflächenarbeit, ... . Die Gesamtenergie E des Systems setzt sich zusammen aus potentieller Energie Epot und kinetischer Energie Ekin des gesamten Systems (z.B., wenn es sich bewegt) und der inneren Energie U (alle restl. Energiebeiträge; U hier bitte von elektrischer Spannung unterscheiden!): E = E pot + Ekin + U Seite 55, Bauerecker, PC für Nebenfächler Erster Hauptsatz der Thermodynamik Nach MAYER 1842 und JOULE 1849 Erster Hauptsatz: Die innere Energie U eines abgeschlossenen Systems vergrößert sich durch Zufuhr von Wärme q oder Arbeit w und bleibt ansonsten konstant: dU = δq + δw Erster Hauptsatz, alternative Formulierung: Ein Perpetuum Mobile erster Art* ist unmöglich *eine Maschine, die in der Bilanz nichts anderes bewirkt, als Wärme in Arbeit umzuwandeln ⇒ Ist Erhaltungssatz und Erfahrungssatz Seite 56, Bauerecker, PC für Nebenfächler Innere Energie und Enthalpie Da Energie nicht verloren gehen kann, verändern zu- oder ab-geführte Energien die Innere Energie U eines Systems: ∆U = U 2 − U1 = q + w Oder differenziell ausgedrückt, d.h. für sehr kleine Änderungen (Symbol δ kennzeichnet mathematisch „nicht wohldefinierte“ Größe): dU = δ q + δ w U ist unabhängig davon, auf welchem Weg der Zustand des Systems erreicht wurde, nur Anfangs- und Endzustand sind wichtig! U ist eine extensive (von der Stoffmenge abhängige) Zustandsgröße. Meistens möchte man Systemeigenschaften nicht als Funktion von T und V, sondern von T und p beschreiben. Grund: Chemische Prozesse laufen meist bei konstantem Normaldruck ab ⇒ nur noch T ist variable Systemgröße. Darum wird eine weitere Zustandsgröße Enthalpie H zweckmäßig definiert als Enthalpie H ist um Volumenarbeit H = U + p ⋅V ergänzte innere Energie U Es ergibt sich: dU = δq − pdV (falls nur Volumenarbeit) dH = dU + pdV + Vdp (Produktregel), dH = δq + Vdp Also gilt für Einfache Systeme: Zusammengesetzte Systeme: Seite 57, Bauerecker, PC für Nebenfächler für p = konst ⇒ dp = 0 ⇒ dH = dU + pdV U = U(T,V) U = U(T,V,n1,n2,...) H = H(T,p) H = H(T,p,n1,n2,...) Wärmekapazitäten cV und cp Unter der Wärmekapazität C in J/K eines Körpers versteht man das Verhältnis von zugeführter Wärmemenge q zu erzielter Temperaturänderung ∆T: q q C= ∆T dU = δq − pdV V konst., dV = 0 ⇓ ∂U δq = = cV dT V = konst . ∂T V -q U System V, T, p dH = δq + Vdp p konst., dp = 0 ⇓ ∂H δq = = cp dT p = konst . ∂T p Dies sind die Definitionen der stoffmengenbezogenen Wärmekapazitäten (Molwärmen) bei konstantem Volumen cV und bei konstantem Druck cp. Beide werden durch Kalorimetrie gemessen. Seite 58, Bauerecker, PC für Nebenfächler w -w Kalorimetrie Messung von: 1. Wärmekapazitäten (T ändert sich, äußere Bedingungen und stoffliche Beschaffenheit bleiben konstant). 2. Wärmetönungen (T konst., Änderung der stofflichen Beschaffenheit beim Aggregatzustand, bei chemischen Reaktionen, ...). 3. Nichtthermische Vorgänge (radioaktive Umwandlungen, Strahlungsmessungen, ...). Beispiel: Messung der molaren Wärmekapazität cp,m Kalorimeter Definierte elektr. Energie R⋅I2⋅∆t (Strom I, Widerstand R, Zeitraum ∆t) erwärmt Probe um ∆T; cp,m wird bestimmt aus: ∆H = c p ,m ⋅ n ⋅ ∆T = RI 2 ∆t =q Seite 59, Bauerecker, PC für Nebenfächler H = H(T), p = const. Wärmekapazitäten Gase Ableitung aus Definitionen von cp und cV unter Anwendung des Id. Gasgesetzes: c p − cV = N mol ⋅ R C p − CV = R für 1 mol 1 mol = 6,022⋅1023 Teilchen 1 u = 1,66054⋅10-27 kg Die innere Energie U des idealen Gases ist nur durch die kinetische Energie der Moleküle bestimmt für 1 mol. (Wärmeenergie) und hängt damit nur von T ab: U = Ekin = 3/2 R⋅T Pro Freiheitsgrad kann das Gas ½ R pro mol Teilchen (oder ½ k pro Teilchen) aufnehmen, siehe auch Gleichverteilungssatz. Bei einatomigen Gasen gilt: CV = 3/2 R und Cp = 5/2 R, Seite 60, Bauerecker, PC für Nebenfächler Wärmekapazität mehratomiger Gase Zweiatomige Gase (O2, N2, H2, NO, HCl, …) Freiheitsgrade 3 Translation 2 Rotation (1 Schwingung, eingefroren bei tiefen T ⇒ Quantentheorie) Cp = CV + R = 3/2 R + 2/2 R + R = 7/2 R HantelModell Mehratomige Gase Lineare Moleküle (CO2, N2O, C2H2, …) 3 2 3N – 5 Translation Rotation Schwingung (2fach) Freiheitsgrade Gewinkelte Moleküle (H2O, SO2, NH3, …) 3 3 3N – 6 Translation Rotation Schwingung (2fach) Aufteilung der 3N Bewegungskoordinaten (= Freiheitsgrade): 3 für Schwerpunktstranslation, 3 für Schwerpunktsrotation (2 lineares Molekül!), Rest für relative Positionen der Atome im Molekül. Die Schwingungen sind wiederum erst bei rel. hohen T angeregt. Seite 61, Bauerecker, PC für Nebenfächler Wärmekapzität komplexe Moleküle Komplexe Moleküle haben sehr viele Schwingungsfreiheitsgrade! 3N – 6 (Normal-)Schwingungen bei N Atomen. Quelle: Wikipedia, engl. Ausgabe Seite 62, Bauerecker, PC für Nebenfächler Wärmekapazität Festkörper 1 Gittermodell: Atome „Federn“ U = Ekin + Epot , für 1 Mol ⇒ U = 3⋅ ½ RT + 3⋅ ½ RT = 3RT (Gleichverteilungssatz, s.o.), d.h. auf jedes Atom kommen 3 „kinetische“ und 3 „potentielle“ Freiheitsgrade. ∂U -1 -1 CV = = 3R ≈ 25 J ⋅ K ⋅ mol ∂T V Dieses Ergebnis wird bei einatomigen Festkörpern, z.B. Metallen, bei Raumtemperatur experimentell bestätigt (DULONG-PETIT-Regel). Seite 63, Bauerecker, PC für Nebenfächler Wärmekapazität Festkörper 2 Das Experiment zeigt jedoch eine T-Abhängigkeit von Cv! Die Schwingungsfreiheitsgrade „frieren ein“ (Begründung durch Quantenstatistik). Dies gilt besonders für leichte Metalle. Raumtemperatur ⇒ Regel von DULONG-PETIT 3R Für Flüssigkeiten und Festkörper ist die Differenz Cp – Cv klein wegen i.a. geringer thermischer Ausdehnung (⇒ geringe Volumenarbeit). Quelle: Wedler, Lehrbuch der Physik. Chemie Seite 64, Bauerecker, PC für Nebenfächler Wärmekapazität Flüssigkeiten In Flüssigkeiten können die Moleküle sowohl kinetische (Translation, Rotation, Vibration) als auch potentielle Energie aufnehmen (→ intermolekulare Wechselwirkungen). Daher ist Cp von Flüssigkeiten i.a. höher als bei Festkörpern und Gasen! Beispiel: Wasser hat eine etwa zweifach höhere Wärmekapazität als (Wasser-)Eis. Darstellung der T-Abhängigkeit von Cp z.B. als Polynom: Cp = a + bT + cT2 , a, b, c gasspez. Konstanten. Beispiel spezifische Wärmekapazität cp • Wassereis (0 °C) 2,1 kJ/(kg⋅K) • Wasser (0 °C) 4,2 kJ/(kg⋅K) Seite 65, Bauerecker, PC für Nebenfächler Prozessrealisierung Prozess Bedingung für System Mathem. Formulierung isotherm Temperatur konstant T konst., dT = 0 isochor Volumen konstant V konst., dV = 0 isobar Druck konstant p konst., dp = 0 adiabatisch Kein Wärmeaustausch mit Umgebung dq = 0 isenthalpisch Enthalpie konstant H konst., dH = 0 isoster Molzahlen Einzelkomponenten konstant ni konst., dni = 0 reversibel Ständig im Gleichgewicht S konst., dS = 0 irreversibel Nicht immer im Gleichgewicht S wächst, dS > 0 Seite 66, Bauerecker, PC für Nebenfächler Phasenumwandlungs-, Reaktions-, u. Bildungs-Enthalpien Enthalpie Symbol Phasenumwandlungsenthalpien: Prozess, Beispiel Abkürzung Latente Wärme, Wärmezufuhr bewirkt keine T-Erhöhung, T = const. s solid, l liquid, g gaseous Verdampfungsenthalpie ∆vapHθ H2O (l) → H2O (g) vap vaporisation Schmelzenthalpie ∆fusHθ H2O (s) → H2O (l) fus fusion Sublimationsenthalpie ∆subHθ H2O (s) → H2O (g) sub sublimation Umwandlungsenthalpie ∆trHθ Graphit → Diamant (Fest-fest-Umwandlung) tr transition Reaktionsenthalpie ∆rHθ H2 (g) + ½ O2 (g) → H2O (l) ∆rHθ < 0 exotherme, ∆rHθ > 0 endotherme Reaktion r reaction Verbrennungsenthalpie ∆cHθ C + O2 → CO2 c combustion Hydratationsenthalpie ∆hydrHθ Anlagerung Wassermoleküle an gelöste/dispergierte Stoffe hydr hydration Bildungsenthalpie ∆fHθ für Bildungsreaktion für 1 Mol der Verbindungen, aus den bei 25°C thermodyn. stabilsten Modifikationen ihrer Elemente f formation ∆Hθ molare Standardenthalpien (bei 1 bar = 105 Pa) Seite 67, Bauerecker, PC für Nebenfächler Anwendung Latentwärmespeicher ... nutzen Umwandlungsenthalpien von Stoffen (Schmelz-, Lösungs-, Adsorptionswärme → (Phasenwechselmaterialien, engl. PCM). Für Speicher-temperaturen zwischen 0 und 150°C eignen sich u.a. Wasser, Paraffine, Salzhydrate, Zuckeralkohole. Quelle: BINE Quelle: PowerTank Beispiel Paraffin-Speicher: Eigenschaften Paraffin: Wachsartiges Gemisch aus Alkanen (CnH2n+2 , n = 18 – 32 oder größer), Dichte 720 – 780 kg/m3, Schmelzwärme 200 – 240 kJ/kg, Schmelztemperatur 45 – 80°C, Preis 2008 ca. 1 (1,2) €/kg [Eci08]. Annahme: Heizölpreis 0,6 (0,9) €/l entspricht 0,714 €/kg bei Dichte von 0,84 kg/l. Für gut isoliertes Einfamilienhaus braucht man ca. 1000 kg Erdöl (ca. 4,3 x 1010 J; 714 €). Für Vollversorgung mit Solarenergie in D braucht man ca. 20 t Paraffin-Speicher (20000 €), mit Volumen von π/4⋅3,62⋅2 m3 = 20,4 m3 als Rundtank. Ohne Zinsen und Zusatzinvestition für Solaranlage und Speichersystem braucht man also etwa 30 Jahre, um allein den Paraffinpreis wieder hereinzuholen (10 Füllungszyklen pro Jahr angenommen). Paraffinspeicher auf dem Markt → z.B. Fa. PowerTank. Gegenüber Wasserwärmespeichern haben sie einen etwa 3 x geringeren Raumbedarf, kosten aber etwa 4 x so viel. Überlegung: Kombination mit Warmwasserspeicher (Patronen passiv integrieren), optional? Seite 68, Bauerecker, PC für Nebenfächler Brennwert und Heizwert Ermittlung des Energieinhalts einer Probe durch Verbrennungskalorimetrie. Wichtig für Lebens- und Genussmittel sowie für Brennstoffe. Man unterscheidet Heizwert Hu in J⋅g-1 Brennwert Ho in J⋅g-1 → H2O ist dampfförmig → H2O ist flüssig Ho ist um Verdampfungsenthalpie des entstehenden (oder kondensierenden) Wassers größer als Hu ⇒ Brennwertkessel. Seite 69, Bauerecker, PC für Nebenfächler Bsp. Brennwerttechnik Abluft ca. 80 - 150°C Altes Prinzip Neue Brennwerttechnik (ab 1985) Ventilator, Schornstein fällt weg Abluft nur 30°C ! Zuluft Quelle: Veritherm Quelle: Wikipedia CH4 + 2 O2 → CO2 + 2 H2O Kondensationsenthalpie H2O Brennwert Ho > Heizwert Hu Ho = 1,11 Hu (Erdgas) Ho = 1,05 Hu (Öl) Einsparung 4 Großkraftwerke in D ! Richard Vetter Preisträger Dieselmedaille Seite 70, Bauerecker, PC für Nebenfächler Brennwert Physikalischer Brennwert von • Kohlehydraten • Proteine • Fette • Alkohol Ho ≈ 16 MJ/kg Ho ≈ 16 MJ/kg Ho ≈ 38 MJ/kg Ho ≈ 29 MJ/kg • Wasserstoff • Methan • Öl Ho = 1,18 Hu = 141,9 MJ/kg (H2-Verbrennung): Ho = 1,11 Hu = 55,5 MJ/kg (Erdgasverbrennung): Ho = 1,05 Hu = 42,7 MJ/kg 2 H2 + O2 → 2 H2O CH4 + 2 O2 → CO2 + 2 H2O Der physiologische Brennwert ist natürlich kleiner, vor allem bei Proteinen → unvollständiger Abbau im Organismus, nicht zu CO2 (g), H2O (lq), N2 (g)! 1 kcal = 4.187 kJ Energiebedarf des Menschen, täglich: EMensch ≈ 2000 kcal = 8400 kJ = 8,4 MJ → entspricht 526 g Kohlehydrate/Proteine Durchschnittsleistung Mensch: PMensch = EMensch / d = 8,4 MJ / (3600*24 s) ≈ 100 W → entspricht Abwärme Seite 71, Bauerecker, PC für Nebenfächler Satz von HESS Kirchhoffsches Gesetz der Temperatur- HESS‘scher Satz: Die Reaktionsenthalpie einer gege- abhängigkeit der Reaktionsenthalpie: benen Reaktion ist unter gleichen Reaktionsbedingungen eine konstante Größe. Dies gilt unabhängig davon, in wie vielen Schritten und über welche Zwischenstufen ein Reaktionsprodukt gewonnen wird. Thermodynamische Grundlage ist die Wegunabhängigkeit von ∆H. T2 ∆ R H T2 − ∆ R H T1 = ∫ ∆c p ⋅ dT T1 Für temperaturkonstante Wärmekapazität: ∆ R H T2 − ∆ R H T1 = ∆c p ⋅ (T2 − T1 ) Anwendungsbeispiel 1: Anwendungsbeispiel 2: Bestimmung der Bildungsenthalpie von CO: ∆cHθ (Graphit) = -393 kJ·mol-1 C + O2 CO2 + ½ O2 ∆cHθ (CO) = -283 kJ·mol-1 CO ∆fHθ (CO) = (-393 – (-283)) kJ·mol-1 = -110 kJ·mol-1 Seite 72, Bauerecker, PC für Nebenfächler ∆fHθ(H2O, 373K) -242 373 K H2, ½ O2 + ½ O2 373 K H2O(g) (∆cp(H2(g))+ ½ ∆cp(O2(g))) • 75 K 3,2 298 K H2, ½ O2 ∆vapHθ 40,6 Werte in kJ/mol -285,8 ∆fHθ(H2O, 298K) 373 K H2O(l) 5,6 298 K H2O(l) ∆cp(H2O(l)) • 75 K Überleitung zum 2. Hauptsatz Bisher wurden behandelt: energetische Umsetzungen (z.B. Phasenumwandlungen, chem. Reaktionen) Im Folgenden befassen wir uns mit: Ablaufrichtung und Gleichgewichtslage von Reaktionen. Ansicht früher: Reaktionen verlaufen umso besser und vollständiger, je exothermer sie sind (BERTHELOT-Prinzip). Dies ist nicht richtig, denn z.B. verlaufen endotherm (unter Abkühlung, dem System Wärme entziehend): NH4Cl(s) + H2O(l) → NH4+(aq) + Cl- (aq) N2(g) + 2 O2(g) → 2 NO2(g) ⇒ Die Affinität (chem. Triebkraft) einer chemischen Reaktion muss noch durch eine andere Größe bestimmt sein als allein durch den Wert einer exothermen Reaktionsenthalpie! Seite 73, Bauerecker, PC für Nebenfächler Irreversible (spontan ablaufende) Prozesse 1 • Temperaturausgleich durch Wärmeleitung, z.B heiße Tasse Kaffee • Konzentrationsausgleich durch Diffusion, z.B. Tropfen Tinte in Wasser • Ausströmen eines Gases ins Vakuum (Effusion) • Auflösen von Zucker in Kaffee (Lösung) • Springende Kugel/Ball verliert kinetische Energie (Wärmeumwandlung) • Unordnung im Zimmer nimmt zu (Entropiezuwachs) Quelle: Wikipedia, engl. Ausgabe Seite 74, Bauerecker, PC für Nebenfächler Irreversible Prozesse 2 Solche Prozesse verlaufen zeitlich offenbar spontan in einer Richtung. Man nennt sie irreversibel. Der Ablauf in umgekehrter Richtung ist nicht spontan. Er kostet Energie. Arbeit muss geleistet werden, um den Anfangszustand wieder zu erreichen. Extreme Beispiele für irreversible Reaktionen sind: Explosionen, Verbrennungen. Was passiert hier? Kann es sein, dass die Energie im betrachteten System ein Minimum annimmt? Nein, denn solche Vorgänge laufen auch in abgeschlossenen Systemen ab. Dabei erfolgt nach dem 1. HS keine Änderung der Gesamtenergie. Es scheint eher mit der Verteilung der Energie zusammenzuhängen, (z.B. T-Ausgleich, Verteilung id. Gas). • Irreversible (spontan ablaufende) Prozesse hängen mit der Verteilung der Energie und der Materie zusammen. • Irreversible Prozesse verlaufen vom Zustand geringerer zum Zustand größerer Wahrscheinlichkeit. Beispiel: Ein Gas kann sich auf einer Seite des Raumes befinden (vor allem wenn wenige Moleküle da sind). Dies ist aber sehr unwahrscheinlich. geringe hohe Wahrscheinlichkeit Seite 75, Bauerecker, PC für Nebenfächler Reversible Prozesse Neben den häufigeren irreversiblen Prozessen gibt es auch seltenere reversible (oder zumindest angenähert reversible) Prozesse. Diese bewegen sich meist sehr langsam von Gleichgewichtszustand zu Gleichgewichtszustand, quasi unendlich langsam. Bei der Umkehr reversibler Prozesse bleibt keine dauernde Veränderung zurück. Ist die vollständige Umwandlung von Wärme in Arbeit möglich? Eine solche Maschine wäre ein Perpetuum Mobile 2. Art. Die Erfahrung zeigt: dies ist nicht möglich. Die Umkehrung ist jedoch möglich, sogar die Regel: Umwandlung Energie in Wärme, z.B. bei elektrischer Heizung, Bremsvorgang Auto, ... . Beispiele: • Kompression eines Gases (Luftpumpe) • Laden/Entladen eines Akkus (Auto) • elektrischer Schwingkreis ohne el. Widerstand (reversibel und schnell ablaufend!) • Schwingendes Pendel im Vakuum (reversibel und schnell ablaufend!) Ist eine BELOUSOV-ZHABOTINSKY-Reaktion ein reversibler Prozess? Quelle: Wikipedia Seite 76, Bauerecker, PC für Nebenfächler Wärmekraftmaschine Wir betrachten eine Wärmekraftmaschine (Dampfmaschine, Automotor, Kohle-, Kern-Kraftwerk, ...). Sie wird zwischen zwei Wärmereservoirs mit hoher und mit geringer Temperatur betrieben, indem die Wärmemenge Q2 aufgenommen und eine kleinere Wärmemenge Q1 (Verlustwärme) plus einer (erwünschten) Arbeit W abgegeben wird. Man definiert den Wirkungsgrad η der Maschine als −W q η= = 1+ 1 q2 q2 (1) T1 T2 (2) Dabei werden vier reversible Teilprozesse am idealen Gas (jeweils adiabatische und isotherme Expansionen und Kompressionen) unter Berücksichtigung des 2. Hauptsatzes durchgeführt (CARNOT 1824). Seite 77, Bauerecker, PC für Nebenfächler Q2 W Wärmekraftmaschine Q1 Aus dem Gedankenexperiment des Carnotschen Kreisprozesses ergibt sich der maximale Wirkungsgrad eines reversiblen Vorgangs zu: η max = 1 − Wärmereservoir T2 > T1 Wärmereservoir T1 Die Entropie Obige Gleichungen (1) und (2) für ηmax liefern: Wärmereservoir T 2 > T1 q1 q2 + =0 T1 T2 Q2 q selbst ist keine Zustandsfunktion aber q/T ! Daher führte CLAUSIUS (1850) eine neue Zustandsgröße Entropie S ein, deren Änderung definiert wurde als δq dS = rev , T ∆q ∆S = rev T W Wärmekraftmaschine Q1 Wärmereservoir T1 In abgeschlossenen Systemen kann diese Größe nie abnehmen: dS = 0 für reversible Vorgänge dS > 0 für irreversible Vorgänge Anschaulich: Entropie ist Maß für Qualität von Wärme. Wärme mit höherer Temperatur (also geringerer Entropie) ist „wertvoller“, weil sie mehr Arbeit verrichten kann. Beispiel 1: 1000 J Wärme bei 1000 K können bei Abkühlung auf 300 K maximal 700 J Arbeit verrichten. 1000 J Wärme bei 500 K aber nur 400 J. (Bitte selbst nachvollziehen). Seite 78, Bauerecker, PC für Nebenfächler Beispiel CARNOT-Maschine Beispiel 2 Kohlekraftwerk: Maximaler Wirkungsgrad für Arbeitsmedium Wasser bei kritischer Temperatur (647 K) und Kühlung durch Umgebung wie Luft oder Fluss (293 K): Prinzipskizze Verbrennungsmotor η = (647 – 293 / 647) = 0,547 ≈ 55 %. Der reale Wirkungsgrad ist niedriger aufgrund zusätzlicher Verluste (Reibung, nichtideale Isolierung, ...). Folgerung: • Wärme kann durch periodische Vorgänge nicht vollständig in Arbeit umgewandelt werden (nur bis CARNOT-Wirkungsgrad). • Das Konzept der Carnot-Maschine mit dem maximalen Carnot-Wirkungsgrad ist äquivalent zur Aussage des 2. Hauptsatzes der Thermodynamik. Wäre nämlich ein höherer Wirkungsgrad als ηmax möglich, so könnte man mit Hilfe der obigen Wärmekraftmaschine und einer umgekehrt arbeitenden Wärmekraftmaschine Wärme von einem tieferen auf ein höheres Temperaturniveau transformieren, ohne dass sich sonst etwas am Gesamtsystem ändern würde. Umgekehrt gäbe es bei Nichtgültigkeit des 2. HS keine theoretische Beschränkung im Wirkungsgrad von Wärmekraftmaschinen. Seite 79, Bauerecker, PC für Nebenfächler Zweiter Hauptsatz der Thermodynamik 1 ⇒ Erfahrungssatz Zweiter Hauptsatz: Alle irreversiblen Prozesse in einem abgeschlossenen System vergrößern die Entropie: ∆S > 0 Unter Arbeits-Aufwendung kann in Teilsystemen ∆S < 0 gelten. Zweiter Hauptsatz alternativ: Es gibt kein Perpetuum Mobile zweiter Art, also keine Maschine die ausschließlich Arbeit unter Abkühlung eines Wärmereservoirs liefert. Seite 80, Bauerecker, PC für Nebenfächler Zweiter Hauptsatz der Thermodynamik 2 Weitere Formulierungen: • Alle spontan im abgeschlossenen System ablaufenden Vorgänge produzieren Entropie (erniedrigen den Grad der Ordnung). • Die Entropie des Universums nimmt zu. • Für jeden homogenen Bereich (System) existiert eine extensive Zustandsfunktion, die Entropie S des Bereichs (Systems), mit folgenden Eigenschaften: A) Für kleinste (infinitesimale) Zustandsänderungen gilt: TdS = dU + pdV, (U, V, unabhängige Zustandsvariablen, konstante Stoffmengen). B) Die Entropieänderung dS des Bereichs (Systems) kann immer in zwei Anteile zerlegt werden: dS = deS + diS, (Indices: e für Austausch mit Umgebung (exchange), i für inneres System). Es gilt: deS = 0 deS = dQ/T diS = 0 diS > 0 Seite 81, Bauerecker, PC für Nebenfächler für thermisch isoliertes System, für geschlossenes System (thermisch leitende Wände), für reversible Zustandsänderung, für irreversible Zustandsänderung, diS < 0 ist stets unmöglich. Thermodynamische Maschinen Weitere thermodynamische Maschinen sind die Wärmepumpe und die Kältemaschine: Hier wird der Maschine die Arbeit W zugeführt und damit die Wärme Qzu der Temperatur T1 auf ein höheres Temperaturniveau T2 transformiert, so dass die Maschine die Wärme Qab = Qzu + W bei T2 abgibt. Ein Kühlschrank ist z.B. eine solche Maschine. Umgebung T2 > T1 Wärmereservoir T2 > T1 Qab Qzu Wärmekraftmaschine W Kältemaschine Wirkungsgrad: η= W Qzu = T2 − T1 T2 Carnot-Rechtsprozess Bsp.: Stirlingmotor T2 = 1200 K, T1 = 300 K ηid = 0,75 ηreal= 0,3 bis 0,4 Seite 82, Bauerecker, PC für Nebenfächler Qab W Wärmepumpe Qzu Qab Wärmereservoir T1 Heizung T2 > T1 Qzu Kühlraum T1 Umgebung T1 Leistungszahl: Leistungszahl: ε KM = Qzu T1 = W T2 − T1 Carnot-Linksprozess Bsp.: Kühlschrank T2 = 310 K, T1 = 270 K εid = 6,75 W ε WP = Qab W = T2 T2 − T1 Carnot-Linksprozess Bsp.: Erdwärmepumpe T2 = 313 K, T1 = 273 K εid = 7,8 εreal = 3 – 5 Arbeitsanteil: 20 bis 33 %! Beispiel Wärmepumpe generell Die Wärmepumpe ist eine Maschine, die mit elektrischer/mechanischer Pumpe Heizwärme aus einer Niedertemperatur-Wärmequelle zur Raumheizung/Warmwasserbereitung erzeugt. Je geringer die Differenz zwischen beiden Temperaturniveaus, desto effizienter arbeitet die Wärmepumpe (⇒ Leistungszahldefinition oben). Beispiel: Zieltemperatur = 40°C, Wärmequellentemperatur = 0 °C ⇒ maximale theoretische Leistungszahl = 7,8 (realistisch nur ε = 3 – 5!). Verstärkter Einsatz wegen steigender Öl- und Gaspreise in den letzten Jahren. Mögliche Wärmequellen: • Grundwasser (Wasser/Wasser-Wärmetauscher) ⇒ Vorteil: rel. hohe WQ-Temperatur, Nachteil: Unsicherheit, weil offenes System • Erdreich mit Erdkollektoren (Sole/WasserWärmetauscher) ⇒ robustes System, Gartenboden kühlt etwas aus • Erdreich mit Erdsonden (Sole/Wasser-Wärmetauscher) ⇒ robustes System • Umgebungsluft (Luft/Wasser-Wärmetauscher) ⇒ preisgünstig, aber bei T << 0°C nicht so effizient • Solarkollektoren einbeziehen ⇒ Niedertemperaturbereich (20 – 60°C) und Überschusswärme kann in Boden und Eis-Speichern gespeichert werden. Seite 83, Bauerecker, PC für Nebenfächler Quelle: Engstfeld Beispiel Wärmepumpe Funktionsprinzip Kompressionswärmepumpe: ein Kältemittel mit niedrigem Siedepunkt wird bei tiefen Temperaturen unter Nutzung der Erdwärme verdampft/erwärmt, mit einem Kompressor verdichtet und damit auf die höhere Zieltemperatur gebracht. Auf diesem Temperaturniveau wird die Nutzwärme über einen weiteren Wärmetauscher abgeführt, wobei sich das Kältemittel verflüssigt und die Kondensationswärme frei wird. Über ein Expansionsventil wird das Kältemittel entspannt und zum Sieden gebracht, die Temperatur erniedrigt sich deutlich, so dass der Kreislauf geschlossen ist. T2 = 313 K Bildquelle: Wikipedia Quelle: K+W Vlasak GmbH, www.k-w-info.de Seite 84, Bauerecker, PC für Nebenfächler T1 = 273 K Anwendung: Effizienzvergleich Raumwärmeerzeugung 70 % der Nutzenergie ist Wärme (50 % Raumwärme, 20 % Prozesswärme)! Daher hat WärmepumpenTechnik große Bedeutung ⇒ riesiges Energieeinsparpotential, das mit Wärmepumpen realisiert werden kann: Verluste: Primärenergieaufbereitung / Stromerzeugung / Stromtransport Gesamtwirkungsgrad zur Erzeugung von Raumwärme ohne Wärmepumpe: mit elektr. Strom mit Öl, Gas (Brennwertkessel) mit Öl, Gas (Diesel- oder Stirlingmotor) mit Wärmepumpe: mit elektr. Strom mit Gas (Gas - oder Stirlingmotor) mit Wasserstoff (Brennstoffzelle) 0,9 x 0,4 x 0,9 x 1 = 0,32 0,9 x 0,95 = 0,86 0,9 x (0,25 + 0,75) El. Strom 0,9 x 0,4 x 0,9 x 4 = 1,3 0,9 x (0,25 x 4 + 0,75) = 1,6 0,8 x (0,6 x 4 + 0,4) = 2,2 Effizienzsteigerung durch solargetriebene Erdwärmespeicher: 20 – 50 %? Seite 85, Bauerecker, PC für Nebenfächler Berechnung der Entropie 1. Berechnung Entropieänderung für GGszustände einfach, z.B. Phasenumwandlungen. Da q = ∆ trs H ⇒ ∆ trs S = ∆ H q = trs Ttrs Ttrs 2. Verknüpfung 1. und 2. Hauptsatz 1. Hauptsatz: δq = dU + pdV dS = 2. Hauptsatz: Beide H.-Sätze: dU + pdV cv dT + pdV = dS = T T ⇒ dU = TdS − pdV S = S(T, V) S(T, p) wird durch Integration berechnet Seite 86, Bauerecker, PC für Nebenfächler δq = dH − Vdp δq T dH − Vdp c p dT − Vdp dS = = T T ⇒ dH = TdS + Vdp S = S(T, p) Wichtigere Darstellung! T -Abhängigkeit der Entropie S eines Stoffes S(T, p) wird durch Integration berechnet: S = S0 + T2 ∫ c p (T )dT T T1→0 Integrationskonstante („Nullpunktsentropie“) Vdp T p1 −∫ Glied klein, außer für Gase hinzu kommen ev. noch Terme ∆ H Schema T-Abhängigkeit Entropie S p2 trs ∆vapS Ttrs vgl. 1. ∆fusS g ∆trsS ∆trsS S0 s, I s, II s, III 0 lq Ttrs Seite 87, Bauerecker, PC für Nebenfächler Aussagen über die Integrationskonstante S0 ? Ttrs Tfus Tvap T2 T 3. Hauptsatz der Thermodynamik Dieser ist wie der 1. und 2. Hauptsatzes ein Erfahrungssatz (NERNST, PLANCK): Die Entropien aller ideal kristallinen Substanzen besitzen bei T = 0 den gleichen Wert. Dieser Wert wird gleich null gesetzt: S(T = 0) = 0. • in Einklang mit der Anschauung versteht man die Entropie als Maß für die Unordnung. • dieser Satz gilt für ideale Kristalle unterschiedlicher Modifikationen (z.B. monokliner oder rhombischer Schwefel) • dieser Satz gilt nicht für amorphe Substanzen! Mit dem 3. Hauptsatz kann man S = S(T, p) explizit berechnen. Für viele Stoffe sind die Entropie-Werte tabelliert, vor allem für Standardbedingungen: ° (Molare Standard- S SATP = standard ambient temperature and pressure: 298,15 K und 105 Pa = 1 bar Seite 88, Bauerecker, PC für Nebenfächler Entropie) Freie Enthalpie → Triebkraft spontaner Vorgänge in geschlossenen Systemen Abgeschlossene Systeme: dS ≥ 0 (2. Hauptsatz) Geschlossene Systeme: Gibt es hier ein ähnliches Kriterium? → wichtig, weil häufig auftretend, z.B. Reaktionsgefäß Wir betrachten ein geschlossenes System zusammen mit seiner Umgebung als abgeschlossenes System: geschlossenes System diS Umgebung T = konst. deS Umgebung wird als unendlich großes abgeschlossenes System betrachtet ⇒ T = konst. dS = diS + deS ≥ 0, diS ≥ 0 (= für reversible, > für irreversible Vorgänge) deS = δq/T diS = dS − δq/T ≥ 0, isobarer Prozess, p = konst. ⇒ δq = dH diS = dS − dH/T ≥ 0 (Multiplikation mit −1) ⇒ dH − TdS ≤ 0 gilt für geschlossene Systeme Seite 89, Bauerecker, PC für Nebenfächler Freie Enthalpie - Definition Definition Freie (GIBBSsche) Enthalpie G nach J. W. GIBBS: G = H −T ⋅ S Für isobare Prozesse (p = konst.) gilt: dH – TdS ≤ 0 Also: ∆G = ∆H − T∆S ≤ 0 dG = dH − TdS ≤ 0 (siehe Vorfolie, beachte auch: T = konst. ). GIBBS-HELMHOLTZ-Gleichung Wir haben damit eine wichtige Beziehung für freiwillig ablaufende (= spontane) isotherme und isobare Vorgänge in geschlossenen Systemen: G strebt einem Minimum zu. Vorgänge laufen nur spontan ab, wenn ∆G < 0. ⇒ Aussage über die Triebkraft einer Reaktion (eines Prozesses). Seite 90, Bauerecker, PC für Nebenfächler Differenzial der Freien Enthalpie … Wir leiten Ausdruck für dG ab: G = H − T⋅S dG = dH − T⋅dS − S⋅dT (Produktregel) dH = δq + V⋅dp (s.o.) und dS = δq/T (m. Umgebung ausgetauschte Entropie) eingesetzt, ergibt: verschwindet bei p = konst. und T = konst. ! dG = V⋅dp − S⋅dT … und Beispiele für chemische Gleichgewichte • chemische Reaktion • Koexistenz verschiedener Phasen (z.B. flüssig/fest) • Lösungsgleichgewichte •… Seite 91, Bauerecker, PC für Nebenfächler Bildquelle: Reininger Freie Reaktionsenthalpie ∆rG Freiwilliger Ablauf chemischer Reaktionen bei ∆rG = ∆r H − T ⋅ ∆r S < 0 ∆ rH ∆ rS Folgerung über Freiwilligkeit des Vorgangs: a) <0 >0 Bei jeder Temperatur möglich, ∆rG stets < 0 b) >0 <0 Bei keiner Temperatur möglich, ∆rG stets > 0 c) <0 <0 Bei niedriger Temperatur begünstigt, da dort ∆rG < 0 wahrscheinlicher d) >0 >0 Bei hoher Temperatur begünstigt, da dort ∆rG < 0 wahrscheinlicher Vorgänge vom Typ c) Vorgänge vom Typ d) Vorgänge mit ∆G stets < 0 Vorgänge mit ∆G stets > 0 Seite 92, Bauerecker, PC für Nebenfächler heißen enthalpiegetrieben. heißen entropiegetrieben. heißen exergonisch. heißen endergonisch. Beispiel Anwendung Freie Enthalpie Bildquelle: superlehrer.de Beispielreaktion: Oxidation von Glukose (Verbrennung) C6H12O6(s) + 6 O2(g) → 6 CO2(g) + 6 H2O(l) ∆ r G ° = ∆ r H ° − T ⋅ ∆ r S ° = (−2794 − 298 ⋅ 0,262) kJ/mol = − 2872 kJ/mol Berechnet über Freie Standardbildungsenthalpien: ∆ r G ° = [6 ⋅ (−394) + 6 ⋅ (−237) − (−911) − 6 ⋅ 0] kJ/mol = − 2875 kJ/mol Seite 93, Bauerecker, PC für Nebenfächler ∆rG bei chemischer Reaktion Änderung der Freien Enthalpie bei chemischer Reaktion schematisch: A+B ⇌ C+D Rückreaktion läuft auch immer ab! Edukte Produkte Bildquelle: pixabay G spontan ∆rG < 0 nur A+B Seite 94, Bauerecker, PC für Nebenfächler spontan Warum verbrennt Glukose nicht spontan in Sauerstoffatmosphäre, wie es nach obiger Betrachtung sein müsste? ⇒ Kinetische Hemmung liegt vor ⇒ Energiebarriere ∆rG = 0 nur C+D Man muss also unterscheiden: - thermodynamische Betrachtung - kinetische Betrachtung Gleichgewicht, thermodynamische Triebkraft verschwindet, → dynamisches Gleichgewicht aus Hin- und Rückreaktion Freie Energie Definition der Freien (HELMHOLTZ) Energie F : F =U −T ⋅ S ∆F = ∆U − T∆S ≤ 0 dF = dU − TdS ≤ 0 dF = dU − T⋅dS − S⋅dT (Produktregel), dU = δq + p⋅dV (s.o.) und dS = δq/T eingesetzt, ergibt: dF = p ⋅ dV − S ⋅ dT Wir haben damit eine weitere wichtige Beziehung für freiwillig ablaufende (= spontane) isotherme und isochore Vorgänge: F strebt einem Minimum zu. Vorgänge laufen nur spontan ab, wenn ∆F < 0. Anwendbar bei V = konst. und T = konst. Seite 95, Bauerecker, PC für Nebenfächler Übersicht Thermodynamische Potentiale Innere Energie Freie Energie Freie Enthalpie Enthalpie dU = TdS – pdV dF = – SdT – pdV dG = – SdT + Vdp dH = TdS + Vdp GUGGENHEIM Quadrat Beispiel dU = – pdV + TdS Natürliche Variable Merkschema: Unheimlich viele Forscher trinken gern Pils hinterm Schreibtisch Das Potential (hier U) wird ausgewählt, benachbart stehen die zugehörigen Differenziale (hier dS und dV) ohne Vorzeichen. An den Ecken gegenüber stehen die zugehörigen Koeffizienten (hier – p und T) mit Vorzeichen. Die natürlichen Variablen des thermodynamischen Potentials U sind S und V. Für energetische Betrachtungen/Rechnungen wählt man das Potential dem vorliegenden System entsprechend so, dass die Differentiale Null sind, z.B. bei abgeschlossenen Systemen das Potential U (keine Wärmeaustausch, keine Volumenänderung. Seite 96, Bauerecker, PC für Nebenfächler Aggregatzustände und Phasenumwandlungen Festkörper α umwandeln Festkörper β Seite 97, Bauerecker, PC für Nebenfächler DICHTE Gläser (amorphe Stoffe) desublimieren Flüssigkeiten sublimieren ENERGIEINHALT Gase (Dämpfe) Phasengleichgewichte eines Reinstoffes Schmelzkurve Dampfdruckkurve Gibbssche Phasenregel: F=K–P+2 F Anzahl der Freiheitsgrade des Systems (Zahl der Zustandsvariablen die unabhängig von der Zahl der Phasen geändert werden können). K Anzahl der Komponenten, P Anzahl der Phasen, Phasenregel gilt im Gleichgewicht. Beispiele: 1) Auf Phasengrenzlinie: P = 2, K = 1 ⇒ F = 1. Man kann sich auf Linie bewegen ⇒ 1 Freiheitsgrad. Sublimationskurve 2) Auf Tripelpunkt: P = 3, K = 1 ⇒ F = 0. Man ist auf Punkt beschränkt ⇒ 0 Freiheitsgrade. 3) Innerhalb einer Phase: P = 1, K = 1 ⇒ F = 2. Man kann sich in p-T-Fläche des Phasengebietes bewegen. Phasengleichgewichte eines Reinstoffes werden im p-T-Phasendiagramm dargestellt: • gibt stabilste Phasen an; • Phasengrenzlinien: benachbarte Phasen stehen mitein-ander im GG (Dampfdruck-, Sublimations-, Schmelz-Kurve); • Dampf/Flüssigkeit stehen im dynamischen Gleichgewicht; • kritischer Punkt: Oberhalb Tkrit keine Verflüssigung möglich; • Tripelpunkt: alle drei Phasen sind im GG. Seite 98, Bauerecker, PC für Nebenfächler CLAUSIUS-CLAPEYRON-Gleichung Wir betrachten ein geschlossenes System aus einer Komponente, die in zwei Phasen vorliegt (gekennzeichnet durch '' und '). Phasengleichgewicht bedeutet, dass die Freie Umwandlungsenthalpie null ist (sonst würde sich die eine Phase weiter in die andere umwandeln): G'' – G' = ∆trG = 0 ⇒ dG'' = dG' mit dG = Vdp – SdT folgt V''dp – S''dT = V'dp – S'dT, (V'' – V') dp = (S'' – S') dT ⇒ (dp/dT)koex = (S'' – S') / (V'' – V'), mit S'' – S' = ∆ trS = ∆ trH / T und V'' – V' = ∆ trV folgt ∆ tr H dp = dT koex T ⋅ ∆ trV Seite 99, Bauerecker, PC für Nebenfächler CLAUSIUS – CLAPEYRONsche Gleichung Folgerungen aus CLAUSIUS-CLAPEYRONscher .. … Gleichung: ∆ H dp = tr dT koex T ⋅ ∆ trV 1. Steigung (dp/dT)koex der Phasengrenzlinie lässt sich durch rechten Teil der Gleichung bestimmen. 2. Übergang (l) → (g): ∆vapH > 0 (Verdampfung erfordert Energiezufuhr) ∆vapV > 0 (Molvolumen Gasphase 1000 x größer als in fl. Phase) ⇒ dp/dT > 0 3. Übergang (s) → (g): ∆subH > 0 (Sublimation erfordert Energiezufuhr) ∆subV > 0 (Molvolumen Gasphase 1000 x größer als in fester Phase) ⇒ dp/dT > 0 4. Übergang (s) → (l): ∆fusH > 0 (Schmelzen erfordert Energiezufuhr) aber! ∆fusV ≈ 0 (Molvolumina flüssige und feste Phase unterscheiden sich sehr gering) ⇒ dp/dT ist groß, aber Vorzeichen ist a priori nicht bestimmbar ⇒ siehe H2O, hier ∆fusV < 0 ⇒ negative Steigung im Phasendiagramm Seite 100, Bauerecker, PC für Nebenfächler T - Abhängigkeit des Dampfdrucks (einer Flüssigkeit) Für Übergang (l) → (g) gilt ∆vapV = V(g) – V(l) ≈ V(g), mit id. Gasgesetz V = N/p ⋅RT in CLAUSIUS-CLAPEYRONsche Gleichung eingesetzt: dp p ⋅ ∆ vap H = dT n ⋅ RT 2 (DGL lösbar durch Trennung der Variablen) Integration der Gleichung liefert T- Abhängigkeit des Dampfdruckes: p(T ) = p0 (T0 ) ⋅ e ∆vap H 1 1 (T −T ) n⋅R 0 (nimmt exponentiell mit 1/T zu) T0, p0 sind Referenztemperatur und –druck, d.h., wenn ein Punkt auf Dampfdruckkurve bekannt ist, lässt sich diese mit dieser Gleichung berechnen. Entsprechende Gleichungen gelten für Übergang (s) → (g). Hier muss ∆vapV durch ∆subV = ∆vapV + ∆fusV ersetzt werden. ⇒ Sublimationskurve hat größere Steigung als Dampfdruckkurve. Seite 101, Bauerecker, PC für Nebenfächler Flüssigkeiten metallische (geschmolzene Metalle) polare (auch geschmolzene Salze) unpolare Hohe elektr. Leitfähigkeit mäßige Leitfähigkeit ⇒ teilweise Ionisation ⇒ vollständige Ionisation in Rumpfionen und Elektronengas, metallischer Glanz Sehr geringe elektrische Leitfähigkeit („Isolatoren“) ⇒ Keine Ionisation Na, Hg, Al, Au H2O, HNO3, BiCl3, CH3OH, ... CCl4, C6H6, C6H12, C6H14, ... Stark assoziiert (Zusammenlagerung) Gering assoziiert → Viskosität, Strömungseigenschaften siehe oben! Seite 102, Bauerecker, PC für Nebenfächler Wasser - Struktur Wasser spielt für Leben und Klima auf der Erde eine herausragende Rolle. Es hat einige „anomale“ Eigenschaften. Die erhöhte Verdampfungsentropie deutet z.B. auf einen erhöhten Ordnungszustand in der flüssigen Phase hin → lokale Cluster aus Molekülen, über H-Brücken gebunden. Lewis-Strukturmodell Gittermodell von Eis mit Wasserstoffbrückenbindungen Seite 103, Bauerecker, PC für Nebenfächler Wassermolekül Struktur des Wassermoleküls Wasser – Besondere Eigenschaften GrotthußMechanismus Bildquelle: K.-D. Keller Wikimedia Eigenschaft Im Vergleich Bedeutung / Auswirkung Wärmekapazität Extrem hoch, nur fl. NH3 höher Temp.-Ausgleich, Dämpfung v. Temp.Schwankungen Schmelzenthalpie Hoch Temp.-Ausgleich, langsames Gefrieren Verdampfungsenthalpie Extrem hoch Atmosphär. Wärmetransport, Schwitzen, etc. Thermische Ausdehnung anormal Dichtemaximum (3,98 °C), Temp.-Konvektion, Eis schwimmt oben Oberflächenspannung Extrem hoch Membranen, Tropfenbildung, Tensidwirkungen Transparenz Relativ groß Absorption IR und UV, kaum im Sichtbaren (Photosynthese, Lebensvorgänge) Viskosität Relativ klein Blutkreislauf (Kapillaren) Wärmeleitfähigkeit Extrem hoch Energieaustausch durch Wärmeleitung groß (Zelle!) Dielektrizitätskonstante Sehr hoch Solvatisierung von Ionen Dissoziation Sehr gering Bei H+ und HO- hohe Ionenbeweglichkeit durch besonderen Transportmechanismus (GROTTHUSS-) Lösungsfähigkeit außergew. hoch für verschied. Stoffe Elektrolyte, polare Nichtelektrolyte, Blutplasma, Bioflüssigkeiten Seite 104, Bauerecker, PC für Nebenfächler Phasendiagramm Wasser Tripelpunkt T: 0,01°C ; 6,1 hPa negative Steigung! Bildquelle: Chemgapedia Seite 105, Bauerecker, PC für Nebenfächler Kritischer Punkt K: 374 °C; 220 bar = 22 MPa Festkörper kristalline amorphe Metalle, Salze, Minerale, Halbleiter, org. Stoffe regelmäßige Anordnung der Bausteine Scharfe Schmelztemperatur, wenn rein: Tfus Gläser, Bitumen, Si, Ge, Kunststoffe, Zucker regellose Anordnung der Bausteine Erweichungsintervall, Glastemperatur: Tg Strukturaufklärung durch Beugung und Interferenz v. Röntgenstrahlen (v. LAUE 1912, BRAGG 1916), Elektronen und Neutronen. Informationen aus Reflexen: Lage ⇒ Abstand Gitterebenen Intensität ⇒ Atomgewicht Breite ⇒ Partikelgröße Seite 106, Bauerecker, PC für Nebenfächler Gittertypen: 1. Atomgitter 2. Ionengitter 3. Molekülgitter Bildquelle: Wikipedia Mischungen/Lösungen (Mehrkomponentensysteme) Wir sehen Lösung und Mischung als gleichbedeutend an und betrachten sie allgemein als Mehrkomponentensysteme. Einteilung 1. Beide reine Stoffe sind Flüssigkeiten a) vollständig mischbar (Ethanol/H2O, CCl4/Benzol, …) b) unvollständig mischbar (Ether/H2O, CS2/Methanol, Nikotin/H2O → Mischungslücke 2. Flüssigkeit und Festkörper a) gesättigte Lösungen (ohne und mit Bodenkörper) b) ungesättigte Lösungen b1) Stoff 2 löst sich als Molekül: H2O/Rohrzucker H2O/Harnstoff b2) Stoff 2 dissoziiert: H2O/Salze H2O/Säuren 3. Flüssigkeit und Gas a) keine Reaktion mit Lösemittel: H2O/O2, H2O/CH4 b) Reaktion mit Lösemittel: H2O/CO2, H2O/NH3 Seite 107, Bauerecker, PC für Nebenfächler System Wasser/Nikotin T/°C Bildquelle: Vorlesung Gericke Konzentrationsmaße (Mengenanteile) … einer chemischen Komponente in einer Mischung. xi = Ni NA Ni NA ni = n ∑i ∑ = Ni ∑ Ni xi = Stoffmengenanteil (Molenbruch) einer Komponente i ni = Stoffmenge einer Komponente i Ni = Teilchenanzahl einer Komponente i NA = Avogadro-Konstante = 6,022 ⋅ 1023 mol-1 Ni/NA = Molzahl einer Komponente i = Stoffmenge Hier bevorzugt verwendet. mi/V = Massenkonzentration (Partikeldichte) einer Komponente i Vi/V = Volumenkonzentration einer Komponente i Vi/ΣVi = Volumengehalt (Volumenbruch) einer Komponente i mi/Σmi = Massengehalt (Massenbruch) einer Komponente i ci = ni/V = Stoffmengenkonzentration (Molarität) yi = Ni/(NA·m) = Stoffmenge pro Masse (Molalität) 100·Vi/VLsg Für zwei Komponenten (binäre Mischung): x1 = n1 n1 + n2 x2 = 1 − x1 = Seite 108, Bauerecker, PC für Nebenfächler n2 n1 + n2 = Volumenprozent einer Komponente i Ideale Mischungen Extensive Zustandsgrößen addieren sich aus denen der reinen Komponenten (Ausnahme Entropie). Bsp. Volumen ideale Mischung (m für Mischung): V = n1V1 + n2V2 + n3V3 + ... Vmol = x1Vmol ,1 + x2Vmol , 2 + x3Vmol ,3 + ... U = n1U1 + n2U 2 + n3U 3 + ... U mol = x1U mol ,1 + x2U mol , 2 + x3U mol ,3 + ... ... ... • Entspricht „naiver“ Erwartung. • Zwischenmolekulare Wechselwirkungen zw. allen Komponenten sind gleich groß. • Eigenschaftsänderungen hängen bei Konzentrationsänderung nur von dieser Konzentrationsänderung ab, nicht jedoch von den Eigenschaften der gelösten Stoffe. • Gilt annähernd für gleichartige Stoffe (z.B. Gasmischungen). • Ideale Mischungen sind die Ausnahme, nicht die Regel! 26,7 cm3 bei verdünnter Lsg. (n1 groß) Dies gilt nicht für reale Mischungen! 31,3 cm3 bei gesättigter Lsg. (n1 klein). Bsp.: Zugabe 1 mol KCl zu n1 mol H2O ⇒ Volumenzunahme d. Lsg. Bei realen Mischungen sind die extensiven Größen nicht additiv aus denen der reinen Komponenten zu errechnen. Hier gilt: V = n1V1 + n2 V2 + n3V3 + ..., mit ∂V Vi = ∂n1 p ,T ,ni ist eine Funktion von den anderen Molzahlen, Druck und Temperatur. Seite 109, Bauerecker, PC für Nebenfächler als partielles molares Volumen. Dieses Partielle molare Größe Bsp.: Molvolumen ist Funktion mehrerer Veränderlicher (z.B. p,T,ni): Partielles Molvolumen der Komponenten i: V = f ( p, T , ni ) ∂V Vi = ∂ni p ,T ,ni • Partielle Ableitung von V nach Variable ni • unter Konstanthaltung aller anderer Variablen (p,T, ni ≠ nk) • partielle Ableitung ist gekennzeichnet durch geschwungenes ∂ Beispiele partieller Ableitungen: g = g ( x, y, z,♥) = 2 x + 3 yz + ♥ 3 2 Seite 110, Bauerecker, PC für Nebenfächler ∂V ∂ni p ,T , ni manchmal auch so geschrieben! ∂g ∂ 2 x + 3 yz 3 +♥2 = 2 = ∂x ∂x ∂g ∂ 2 x + 3 yz 3 +♥2 = 3z 3 = ∂y ∂y ∂g ∂ 2 x + 3 yz 3 +♥2 = 9 yz 2 = ∂z ∂z ∂g ∂ 2 x + 3 yz 3 +♥2 = 2♥ = ∂♥ ∂♥ ( ) ( ) ( ) ( = ) Reale Mischungen Beispiel: Mischung Ethanol – Wasser Bsp.: bei einem Anteil von etwa 10 % hat das Ethanol in der Mischung nur ein molares (partielles) Volumen von ca. 52 anstelle der „idealen“ 58,3 mL/mol (gestrichelte Linie). Partielles Molvolumen von Ethanol in der Mischung: ∂V/ ∂nE Molvolumen der Mischung: VMischung(xE) Partielles Molvolumen von Wasser in der Mischung: ∂V/ ∂nW Bildquelle: Vorlesung Maul Molvolumen der realen Mischung: ∂V ∂V + xW VMischung = xE ∂nE p ,T ,nW ∂nW 1 = xE + xW Seite 111, Bauerecker, PC für Nebenfächler p ,T ,nE Chemisches Potential Das Chemische Potenzial µi eines Stoffes i in einer Mischphase (Gasmischung, Lösung, chem. Reaktionssystem, …) ist eine sehr wichtige Größe in der Thermodynamik (siehe auch oben). Definition: ∂G µi = ∂ni p ,T ,n j Anschaulich: µi beschreibt also die Fähigkeit einer kleinen Stoffmenge ∂ni der Substanz i, die dem System zugegeben wird, an diesem System Arbeit zu verrichten, die bei konstanter Temperatur T, konstantem Druck p und konstanten Stoffmengen nj der anderen Substanzen j als kleine Änderung der Freien Enthalpie ∂G auftritt. Erweiterung des Differentials der Freien Enthaltpie (s.o.) um die Wirkung von Materieaustausch des Systems mit der Umgebung durch die einzelnen Komponenten-Stoffmengen n1, n2, n3, …, zur Gibbschen Fundamentalgleichung: dG = − SdT + Vdp + ∑ µi dni Wenn p und T konstant, ergibt sich die gesamte Freie Enthalpie des Systems zu Seite 112, Bauerecker, PC für Nebenfächler G = ∑ ni µi Chemisches Potential und Aktivität Das chemische Potential lässt sich in zwei Anteile aufspalten: µi(p,T) = µi°(p=105Pa, T) + RT⋅lnai chem. Standardpotential Aktivität mit ai = fi ⋅ xi Anders geschrieben: µi = µi° + RT⋅lnxi + RT⋅lnfi id. Anteil (id. Gas, id. Lösung) Bei ideal verdünnter Lösung gilt: fi ≈ 1 ⇒ ai ≈ xi Seite 113, Bauerecker, PC für Nebenfächler Zusatzanteil / Exzessanteil für reales Verhalten Aktivitätskoeffizient berücksichtigt Abweichung vom id. Verhalten HENRYsches Gesetz Der Dampfdruck pi einer flüchtigen gelösten Substanz ist proportional zu ihrem mit HENRY-Konstante KH Stoffmengenanteil xi in der Lösung: pi = KHxi (gilt insbesondere für ideal verdünnte Lösungen) Gasgemisch mit Komponente i Flüssigkeit mit gelöstem Gas i HENRY-Konstante einiger Gase in Wasser bei 25°C, KH / mbar Methan, CH4 Kohlendioxid, CO2 Wasserstoff, H2 Stickstoff, N2 Sauerstoff, O2 Seite 114, Bauerecker, PC für Nebenfächler 105 4,19 x 1,67 x 106 7,12 x 107 8,68 x 107 4,40 x 107 Physiologische Bedeutung: A) In Luft ist bei 25°C die 30fach größere Menge O2 als in gleichem Volumen in Wasser! ⇒ Lungenatmung ist gegenüber Kiemenatmung im Vorteil! B) T-Abhängigkeit der Gaslöslichkeit: Löslichkeit von O2, N2, CO2 nimmt bei TAnstieg von 0 auf 30°C auf die Hälfte ab! ⇒ vitales Meeresleben in Polarregionen (auch wg. Durchmischung!). Kolligative Eigenschaften von Lösungen Dies sind Eigenschaften verdünnter Lösungen, die praktisch nicht von der Natur der beteiligten Stoffe, sondern ausschließlich von deren Konzentrationen abhängen. Alle vier kolligativen Effekte haben unterschiedliche Ausprägung. Vergleiche die Zahlenwerte (Bsp.: einmolare wässrige Lösung, 25°C): Kolligativer Effekt Symbol Zahlenwert / Ausprägung 1. Dampfdruckerniedrigung ∆p/p0 -0,02 2. Siedepunktserhöhung ∆Tvap 0,51 K 3. Gefrierpunktserniedrigung ∆Tfus -1,9 K 4. Osmotischer Druck π 25 bar (!) Seite 115, Bauerecker, PC für Nebenfächler Dampfdruckerniedrigung Wir betrachten eine verdünnte Lösung einer nicht-flüchtigen Substanz 2 in einem flüchtigen Lösungsmittel 1 mit p0 als Dampfdruck des reinen Lösungsmittels. RAOULTsches Gesetz (1890): Der Dampfdruck p des Lösungsmittels nimmt proportional mit seiner Stoffmengenkonzentration x1 ab: p = x1 ⋅ p0 Mit ∆p = p0 – p und x2 = 1 – x1 gilt: Die relative Dampfdruckerniedrigung ∆p/p0 ist gleich dem Molenbruch der gelösten Substanz: ∆p/p0 = x2 p p0 RAOULTsches Gesetz wahrer Verlauf 0,5 Seite 116, Bauerecker, PC für Nebenfächler x1 1 Siedepunktserhöhung u. Gefrierpunktserniedrigung Wir betrachten beide Phänomene schematisch am p-T-Phasendiagramm (s. auch oben): p Schmelzdruckkurven Dampfdruckkurven 1013 mbar Bsp. H2O (mit negativ steigender Schmelzdruckkurve) reines Lösungsmittel Lösung, z.B. von Zucker ∆Tfus ∆Tvap T Dampfdruckkurve und Schmelzdruckkurve der Lösung verlaufen unterhalb der entsprechenden Kurven des reinen Lösungsmittels. Aus den Gleichungen von RAOULT und CLAUSIUS-CLAPEYRON folgt durch Gleichsetzen (über ∆p/p0): RT 2 (Siedepunktserhöhung) ∆Tvap = x2 ⋅ ∆ vap H ∆T fus Seite 117, Bauerecker, PC für Nebenfächler RT 2 = x2 ⋅ ∆ fus H (Gefrierpunktserniedrigung) Beispiele Gefrierpunktserniedrigung 1. Winterdienst-Salzstreuung. Es kommt nur auf die Zahl der gelösten Teilchen an. Also ließe sich auch Zucker streuen. 2. Winterharte Pflanzen. Umwandlung von Stärke (Polysaccharid) in Glucose (Monosaccharid) erhöht Zahl gelöster Teilchen ⇒ Gefrierpunktsabsenkung. Seite 118, Bauerecker, PC für Nebenfächler Osmotischer Druck Osmotische Erscheinungen bilden eine bestimmte Klasse von Transporterscheinungen an Membranen. Osmose ist ein wichtiges Phänomen in der Biologie. Semipermeable Membran: Ideale Membran, ist durchlässig für eine Komponente (meist Lösungsmittel, z.B. H2O) und undurchlässig für andere Komponente(n) (z.B. geladene Salzionen wie Na+, Mg2+, Cl-). Geeignete technische Membranen sind in Praxis realisiert ⇒ z.B. bei Meerwasserentsalzung. Modellaufbau Lösungsmittel, Druck: p Lösungsmittel plus weitere Komponente, Druck: p + π Bildquelle: Chemgapedia semipermeable Membran Lösung (L) und Lösungsmittel (LM) (bzw. 2 Lösungen unterschiedl. Konzentration) sind durch semipermeable Membran getrennt ⇒ LM wandert vom Ort höheren chem. Potentials zum Ort niedrigeren chem. Potentials in L. π = osmotischer Druck a) Gleichgewicht (kein LM-Fluss): b) Osmose (LM fließt zur L und verdünnt diese: 0 < π < osmotischer Druck π > osmotischer Druck c) Umkehrosmose, Hyperfiltration (LM wird aus L gepresst, ⇒ L konzentrierter): Seite 119, Bauerecker, PC für Nebenfächler Osmose: VAN‘T HOFFsches Gesetz Für verdünnte Lösungen gilt für den osmotischen Druck: π = c ⋅ R ⋅T VANT‘HOFFsche Gleichung (1886) Die Gleichung ist formal analog zur idealen Gasgleichung: c = Nmol /V ist Konzentration des gelösten Stoffes. p= N mol RT = cRT V Lösungen von gleichem osmotischen Druck heißen isotonisch. Sie sind wichtig für • Augen-, Nasen-Tropfen, • Injektions- und Infusionslösungen, • Blutplasma-Ersatzlösungen (physiolog. Kochsalzlösung), • Turgor von Gewebs- und Pflanzenzellen (Zellflüssigkeitsdruck, → Spannungszustand, • Isotonische Getränke für Leistungssportler. Seite 120, Bauerecker, PC für Nebenfächler Umkehrosmose: Anwendungen 1. Reinwasserspender Bildquelle: Siemens 2. Großtechnische Meerwasserentsalzung Bildquelle: Taprogge Seite 121, Bauerecker, PC für Nebenfächler Bildquelle: Siemens Kolligative Eigenschaften: Anwendungen Die Gesetze für die vier kolligativen Eigenschaften von Lösungen sind anwendbar zur Bestimmung von: • „Osmolarität“ der Lösung (Gesamtkonzentration der gelösten Stoffe), • Aktivitätskoeffizient des Lösungsmittels und des gelösten Stoffes, • Dissoziationsgrad des Gelösten, • Relative Molekülmasse des Gelösten (hochmolekulare Stoffe) Seite 122, Bauerecker, PC für Nebenfächler Chemische Gleichgewichte Wir betrachten eine chemische Reaktion, deren stöchiometrische Gleichung gegeben sei durch: Hinreaktion Edukte Produkte νAA + νBB ⇌ νCC + νDD Rückreaktion νi sind stöchiome- Bildquelle: SchulLV trische Koeffizienten Unabhängig davon, ob die Reaktion mit den Produkten oder Edukten startet, stellt sich im thermodynamischen Gleichgewicht (GG) ein eindeutig bestimmter dynamischer Gleichgewichtszustand ein. Beispiel Ammoniaksynthese: 3 H2 + N2 ⇌ 2 NH3 mit νA = 3, νB = 1, νC = 2 Seite 123, Bauerecker, PC für Nebenfächler VAN‘T HOFFsche Reaktionsisotherme Wir betrachten die Freie Reaktionsenthalpie ∆rG und gehen vom totalen Differenzial von G(T,p,ni) bei isothermen und isobaren Bedingungen (dT = 0, dp = 0) aus, s.o.: =0 =0 d r G = ∑ µi dni [+ Vdp − SdT ] Bezogen auf Reaktionsgleichung, mit dni ersetzt durch νi i ∆ r G = ∑ν i µi i ∆ r G = ∑ν i µi + RT ∑ν i ln ai i Die µi sind im allgemeinsten Fall gegeben durch (s.o.): µi = µi° + R⋅T ⋅ lnai , womit i ∆rG° = Freie Standardreaktionsenthalpie Anwendung Logarithmusgesetze ∆ r G = ∆ r G + RT ⋅ ln ∏ aνi i VAN‘T HOFFsche Reaktionsisotherme i aνCC ⋅ aνDD ∆ r G = ∆ r G + RT ⋅ ln ν A ν B a A ⋅ aB Seite 124, Bauerecker, PC für Nebenfächler für die Reaktion νAA + νBB ⇌ νCC + νDD Massenwirkungsgesetz Im Gleichgewichtsfall gilt: mit K = ∏ aνi i ∆rG = 0 = ∆rG° + RT⋅lnK Massenwirkungsgesetz (fundamental wichtig!), i aνCC ⋅ aνDD K = νA νB a A ⋅ aB Man erhält für die Reaktion νAA + νBB ⇌ νCC + νDD, K ist thermodynamische Gleichgewichtskonstante d. Reaktion. ∆rG ln K = − RT (fundamental wichtig!) Man kann damit aus bekannten Freien Standardreaktionsenthalpien die Gleichgewichtskonstante einer chemischen Reaktion berechnen! [NH3] andere Schreibweise f. Obiges Beispiel 3 H2 + N2 ↔ 2 NH3 liefert Konzentration oder Molenbruch! [ NH 3 ]2 a( NH 3 ) 2 x( NH 3 ) 2 K= ≈ = a(H 2 ) 3 ⋅ a( N 2 ) x(H 2 ) 3 ⋅ x( N 2 ) [H 2 ]3 ⋅ [ N 2 ] unter Verwendung der bei Gasen guten Näherung ai ≈ xi. Einsetzen von ∆rG° = -32,9 kJ⋅mol-1 bei 25°C liefert K = 5,8 x 105 ⇒ x(H2) = 0,046, x(N2) = 0,0154, x(NH3) = 0,938, wobei Σxi = 1 gelten muß! Seite 125, Bauerecker, PC für Nebenfächler Chemische Reaktionskinetik Chemische Thermodynamik macht Aussagen zu: - Gleichgewichten, - Ablaufrichtung und - Energetik von chem. Reaktionen - keine Aussage zur Geschwindigkeit - ohne Begriff „Zeit“ Der Reaktionsmechanismus chem. Reaktionen besteht i.d.R. aus zahlreichen, experimentell schwer erfassbaren Zwischenstufen (Elementarreaktionen). Chemische Kinetik ist Lehre der Dynamik molekularer u. zellulärer Prozesse, sie behandelt: - Transportvorgänge im System (Makrokinetik) - Geschwindigkeit und - Mechanismus von Reaktionen (Mikrokinetik) Homogenkinetik Reaktion verläuft vollständig in einer Phase Heterogenkinetik Reaktion verläuft an Phasengrenzflächen -Historisches: Anscheinend erstes Wissen zu Reaktionskinetik bei Bierbrauern, Weingärtnern, Metallurgen - Stoffwechselkinetik (z.B. Diagnostik Organfunktion) - Pharmakokinetik Seite 126, Bauerecker, PC für Nebenfächler Transportvorgänge Konvektion „erzwungen“ - Schütteln - Rühren - Ultraschall - Umschwenken - ... Diffusion (molekularer Transport) „frei“ durch Unterschiede in - Temperatur - Dichte - Oberflächenspannung A c2 A c1 1. FICKsches Gesetz: dN/dt = – D · A · dc/dx STOKES-EINSTEIN-Gleichung D = kB T / (6 π ηr) Zähigkeit Molekülradius Seite 127, Bauerecker, PC für Nebenfächler Stoffmengenfluss Fläche Konzentrationsgradient (-gefälle) Diffusionskoeffizient D ist Maß für Diffusionsgeschwindigkeit [D] = m2 s-1 Reaktions-Geschwindigkeit und -Ordnung Wir betrachten ein geschlossenes Reaktionsgefäß mit Volumen V, in dem die Reaktion A + B ⇌ C + D abläuft. Dann gilt wegen Stöchiometrie (Lehre von den chemischen Umsetzungen) für die Reaktionsgeschwindigkeit: v=− dc dc A dc dc =− B = C = D dt dt dt dt (Negative Vorzeichen, weil A, B verschwinden, wenn C, D zunehmen). v hängt von allen Konzentrationen cA, cB, cC, cD ab. Ansatz für diesen Zusammenhang: (Geschwindigkeitsgleichung) v = k(T) ⋅ [A]a⋅[B]b⋅[C]c⋅[D]d Exponenten werden experimentell bestimmt, können nicht abgeleitet werden. Konzentrationen ci oder auch Stoffmengenanteile xi der Reaktanden Geschwindigkeits-Konstante (besser: -Koeffizient) Definition: Reaktionsordnung = Summe aller Exponenten = a + b + c + d muss nicht unbedingt eine ganze Zahl sein! Seite 128, Bauerecker, PC für Nebenfächler Beispiele Reaktionsordnung • Reaktion 1. Ordnung (Zerfall Distickstoffpentoxid): 2 N2O5 → 4 NO2 + O2 v = k⋅[N2O5] (Exponent 1, unabhängig v. anderen Konz.) • Reaktion 2. Ordnung (Zerfall Stickstoffdioxid, T > 150°C): 2 NO2 → 2 NO + O2 v = k⋅[NO2]2 (Exponent 2, unabhängig v. anderen Konz.) • Reaktion 3. Ordnung (Oxidation Stickstoffmonoxid): 2 NO + O2 → 2 NO2 v = k⋅[NO]2⋅[O2] (Exponent 3, unabhängig v. NO2 Konz.) • Reaktion 0. Ordnung: selten, Konzentrationen haben keinen Einfluss auf Reaktionsgeschwindigkeit (z.B. Zersetzung von Gasen an Oberflächen): 2 N2O Au→ 2 N2 + O2 • Reaktion „Pseudo“ 1. Ordnung: A+B → C v = k1⋅[A]⋅[B] = k2⋅[B] = konstant, da im Überschuss vorhanden Seite 129, Bauerecker, PC für Nebenfächler Wasser: Autoprotolyse – das Ionenprodukt KW Einige wenige Wassermoleküle dissoziieren (→ el. Leitfähigkeit), Dissoziationsgleichgewicht: OxoniumIon HydroxidIon 2 H2O ⇌ H3O+ + OHUnter Anwendung des Massenwirkungsgesetzes und weil der Stoffmengenanteil des Wassers nahezu unverändert bleibt ([H2O] ≈ 1) ergibt sich für die das Ionenprodukt des Wassers (Gleichgewichtskonstante): = 1 ⋅ 10-14 mol2⋅L-2 (bei 25°C, ist T-abhängig!). KW = [H3O+]⋅[OH-] / [H2O]2 ≈ [H3O+]⋅[OH-] Bei Autoprotolyse werden gleiche Mengen Oxonium (Hydronium)- und Hydroxid-Ionen gebildet ⇒ [H O ] = [OH ] = + 3 - 10 -14 mol2 -7 mol 10 = L2 L Der pH-Wert ist der negative dekadische Logarithmus der Oxonium-Ionen-Konzentration: pH = − lg[H3O+] Seite 130, Bauerecker, PC für Nebenfächler ⇒ pH(H2O, 25°C) = 7 Dissoziation von Säuren – die Säurekonstante KA Definition nach BRØNSTED: Säuren sind Protonendonatoren, Basen sind Protonenakzeptoren. Wasser ist ein Ampholyt, d.h. sowohl Säure (H2O ⇌ H+ + OH-) als auch Base (H2O + H+ ⇌ H3O+). Die Stärke einer Säure HA drückt sich durch die Säurekonstante KA bei ihrer Reaktion mit Wasser aus, wiederum unter Berücksichtiung, dass [H2O] ≈ 1: KA [H O ]⋅ [A ] ≈ [H O ]⋅ [A ] = KB [HB ]⋅ [OH ] ≈ [HB ]⋅ [OH ] = + HA + H2O ⇌ H3O+ + AEntsprechend gilt für eine Base B: [HA]⋅ [H 2O] + B + H2O ⇌ HB+ + OH- + - 3 - [B]⋅ [H 2O] 3 - [HA] + - [B] Wiederum wird der negative dekadische Logarithmus verwendet, um die Stärke der Säure oder Base auszudrücken : pKA = − lg(KA) ⇒ Säure pKB = − lg(KB) Seite 131, Bauerecker, PC für Nebenfächler ⇒ Base Starke und schwache Säuren Starke Säuren sind in der Regel vollständig dissoziiert ⇒ Dissoziationsgrad αstark ≈ 1 Schwache Säuren sind in der Regel unvollständig dissoziiert ⇒ Dissoziationsgrad αschwach < 1 pKA Eigenschaft Beispiel <0 sehr stark HCl, H2SO4, HNO3 0–4 stark HCOOH, H3PO4 4–8 mittelstark CH3COOH, H2CO3, H2S 8 – 12 schwach HCN, NH4+, HCO3- > 12 sehr schwach NH3 Quelle: Vorlesung Maul Seite 132, Bauerecker, PC für Nebenfächler Molekularität und Aktivierungsenergie Die Molekularität einer chem. Reaktion gibt die notwendige (Stoß-)Partnerzahl bei Elementarreaktionen an. • Unimolekulare (monomolekulare) Reaktionen, Zerfallsreaktionen, sind immer Reaktion 1. Ordnung. Bsp. 1: Zerfall von Ozon O3 → O2 + O Bsp. 2: Radioaktiver Zerfall • Bimolekular sind die weitaus meisten Elementarreaktionen, sind immer Reaktionen 2. Ordnung. Bsp.: Bildung von Ozon O2 + O → O3 oder • Trimolekular, drei Stoßpartner sind notwendig, selten. Bsp.: Bildung des Sauerstoffmoleküls O + O + O2 → 2 O 2 Seite 133, Bauerecker, PC für Nebenfächler Vor der Reaktion müssen Partner i.d.R. energetisch angeregt werden, d.h. es bildet sich ein aktivierter Komplex Geschwindigkeitsgesetze einfacher Reaktionen Ziel: Berechnung der Zeitabhängigkeit der Konzentration bei Reaktionen aus Differenzialgleichungen (DGL). Reaktion 1. Ordnung v=− dc = k ⋅ c = k[A] dt Integration der DGL, mit c = c(A), c0 = c(A, t=0) als Konzentration des Edukts A bei t und t = 0. dc − = k ⋅ dt c c ln = −kt c0 c t dc ∫c c = −k ∫0 dt 0 ⇒ c(t ) = c0 ⋅ e − kt c/c0 1 - Exponentieller Abfall der Konzentration von A k nimmt zu Günstige Form zur Auswertung ⇒ Gerade mit Steigung –k Halbwertszeit t1/2 → Nach t1/2 ist nur noch die halbe Stoffmenge vorhanden: c(t1/2) = ½ c0 → Einsetzen in ⇒ ln2 = k⋅ t1/2 ⇒ t1/2 = ln2/k ⇒ t1/2 ist unabhängig von c0! Seite 134, Bauerecker, PC für Nebenfächler t Geschwindigkeitsgesetze einfacher Reaktionen 2 Reaktion 2. Ordnung v=− dc = k ⋅ c 2 = k[A]2 , s.o. dt Integration der DGL, mit c = c(A), c0 = c(A, t=0) wie oben, Trennung der Variablen. c t dc − 2 = k ⋅ dt c dc ∫c c 2 = −k ∫0 dt 0 1 1 − = kt c c0 c0 c(t ) = 1 + k ⋅ c0 ⋅ t ⇒ Günstige Form zur Auswertung ⇒ Gerade mit Steigung k Halbwertszeit t1/2 → c(t1/2) = ½ c0 → Einsetzen in ⇒ t1/2 ist abhängig von c0! Seite 135, Bauerecker, PC für Nebenfächler c/c0 1 - Hyperbolischer Abfall der Konzentration von A k nimmt zu t ⇒ 2/c0 – 1/c0 = k⋅ t1/2 ⇒ t1/2 = 1/(k⋅ c0) T-Abhängigkeit der Reaktionsgeschwindigkeit Faustregel: Temperaturerhöhung um 10 K führt zur Verdopplung der Reaktionsgeschwindigkeit ARRHENIUS-Gleichung (empirisch): Logarithmierte Auftragung: ln k = ln A − Ea / RT lnA k (T ) = A e − Ea / RT A Präexponentieller Faktor Ea Aktivierungsenergie Seite 136, Bauerecker, PC für Nebenfächler Steigung EA Enzymkatalysierte Reaktion Enzyme → hochwirksame, hochspezifische Biokatalysatoren (von lebenden Zellen gebildeter Eiweißkörper) Katalysator → Reaktionsbeschleuniger Substrat S lagert sich spezifisch und reversibel an aktives Zentrum im Enzym E an. Komplex ES reagiert zu Produkt P ab, E wird zurückgebildet: k1 k2 E Enzym + S Substrat ⇌ k-1 ES Komplex ⇀ P + E Produkt Enzym GG liegt weit auf rechter Seite Geschieht die Bildung von ES schnell, der Zerfall von ES jedoch langsamer = geschwindigkeitsbestimmender Schritt, so gilt: vP = d [P] = k2 [ES] dt Die Komplexkonzentration [ES] lässt sich mit Hilfe der Enzymgesamtkonzentration [E]0 und der Substratkonzentration [S] ausdrücken: [ES] = [E]0 [S] K M + [S] MICHAELIS-MENTEN-Gleichung MICHAELIS-Konstante KM Seite 137, Bauerecker, PC für Nebenfächler MICHAELIS-MENTEN-Gleichung vP = k2 [ES] = k2 [E]0 [S] K M + [S] MICHAELIS-MENTEN-Gleichung (Schreibweise 1, s.o.) Es gibt 2 Grenzfälle: a) [S] << KM ⇒ vP = k2 [E]0⋅[S] / KM Reaktion 1. Ordnung b) [S] >> KM ⇒ vP = vmax = k2 [E]0 Reaktion 0. Ordnung konstant Damit kann man die MM-Glg. auch so schreiben: vP vmax vP = ½ vmax vmax ⋅ [S] K M + [S] MICHAELIS-MENTEN-Gleichung (Schreibweise 2) KM Seite 138, Bauerecker, PC für Nebenfächler [S] Grenzflächenerscheinungen Mehrphasensysteme haben Phasengrenzen. An Grenzflächen sind physik.-chem. Eigenschaften anders als im Inneren der Phase. Der Grenzflächeneinfluss nimmt bei fein verteilten Stoffen mit ihrem Verhältnis von Grenzfläche A zu Volumen V zu. Beispiele: a) Kugelförmige Dispersion (Mehrphasensystem): b) Einfacher kubischer Kristall: N = 6⋅1023 Teilchen groß A 4π ⋅ r 2 3 = = 3 4 V r 3π ⋅r xOberfl. = 7⋅10-8 ⇒ geringer Oberflächenanteil ((42 ⋅ 2 ⋅ 106)2 ⋅ 6)/(6 ⋅ 1023) 0,5 nm Teilchendurchmesser 42 mm N = 109 Teilchen klein 500 nm Seite 139, Bauerecker, PC für Nebenfächler xOberfl. = 0,01 = 1% ⇒ größerer Oberflächenanteil Große relative Oberflächen haben: • Tröpfchen in Emulsionen (z.B. Milch) • Aerosole, Sprays, Rauche • Zellen (Oberfläche = zelluläre Membran) • poröse, gefurchte Teilchen Aerosolpartikal in der Umwelt UV VIS IR Öl/Dieselrauch Wolkentröpfchen Mineralstaub Pollen Meersalzkerne Algen Atome Metallstäube Bakterien Viren Moleküle 0,1 1 Seite 140, Bauerecker, PC für Nebenfächler Proteine 10 100 1000 104 105 in nm Beispiele häufige Aerosole in der Umwelt Wolken: Wassertröpfchen- oder Eispartikel-Aerosol Vulkane: Staub- und Säure-Aerosole Meeresbrandung, Gischt: Salzwassertröpfchen- und Salzkern-Aerosole Seite 141, Bauerecker, PC für Nebenfächler Phänomen Leben Das Phänomen Leben beruht wesentlich auf speziellen Grenzflächenvorgängen. Seite 142, Bauerecker, PC für Nebenfächler Oberflächenspannung von Flüssigkeiten Flüssigkeit zeigt Bestreben, einen Zustand minimaler Energie einzunehmen ⇒ möglichst kleine Grenzfläche ⇒ Kugelform Isotherme Vergrößerung eines Tropfens: δw = pi⋅dV = σ ⋅dA dV = d(4/3⋅ π ⋅r3) = 4π⋅r2⋅dr dA = d(4π ⋅r2) = 8π⋅rdr ⇒ pi = 2 σ / r Bildquelle: Backhaus Uni Duesseldorf YOUNG-LAPLACE-Gleichung → „Luftballoneffekt“ Bildquelle: Chemgapedia Kräfte in Flüssigkeit an Oberfläche und im Inneren anziehende Kräfte (molekulare WWen) Seite 143, Bauerecker, PC für Nebenfächler σ Grenzflächenspannung (ist Kraft) [σ] = N⋅m-1 pi resultierende Kraft zieht nach innen Messmethoden der Grenzflächenspannung Messmethoden der Grenzflächenspannung • kapillare Steighöhe • Tropfenmasse • maximaler Blasendruck • Ringmethode • Plattenmethode (WILHELMY) Bsp.: Kapillare Steighöhe r Benetzbarkeit der Kapillarwand bewirkt Kapillarkraft: - Kapillarszension (Flkt. steigt auf, z.B. Wasser) - Kapillardepression (Flkt. wird unter Oberfläche gedrückt, z.B. Quecksilber) Kapillare Kontaktwinkel = Randwinkel θ h Steighöhe Hier halten sich im Gleichgewichtsfall die Gewichtskraft der Flüssigkeitssäule und die durch die Oberflächenspannung bewirkte „Zugkraft“ die Waage: π r2g h ∆ρ = 2π r σ cosθ Dichtedifferenz zw. Flkt. u. Dampf Erdbeschleunigung ⇒ σ= Seite 144, Bauerecker, PC für Nebenfächler g ⋅ h ⋅ r ⋅ ∆ρ 2 cosθ Grenzflächenspannung Anwendung des Kapillaritätsverhaltens Flüssigkeit bei 20°C Oberflächenspannung in mN/m Ethanol 22,6 Wasser 80°C 62,6 Wasser 50°C 67,9 Wasser 20°C 72,8 Quecksilber 476 • Messung σ • Safttransport in Pflanzen • Blutkreislauf • Transport Wasser in Böden (Poren) Temperaturabhängigkeit Grenzflächenspannung und Mischbarkeit • σ nimmt für alle Flüssigkeiten annähernd linear mit T ab • σ = 0 bei T = TK • σ12 = 0 ⇒ zwei Flüssigkeiten sind vollständig mischbar Seite 145, Bauerecker, PC für Nebenfächler Kapillaraktive und –inaktive Stoffe Kapillarinaktive Stoffe streben volle Solvatisierung an und reichern sich in der Grenzfläche ab: z.B. Salze, starke Säuren, Polysacharide, kleine Aminosäuren, Zucker, mehrwertige Alkohole. ⇒ erhöhen σ (max. 15%). Kapillaraktive Stoffe („Tenside“) werden angereichert in der Grenzfläche. Moleküle sind amphiphil, d.h. hydrophil und lipophil. ⇒ erniedrigen σ (teilweise sehr stark). Zusammenlagerungsformen amphiphiler Substanzen Monolayer Bilayer PhospholipidMolekül hydrophober, unpolarer Fettsäureschwanz hydrophiler, polarer Kopf Bildquelle: Encyclopedia Britannica Micelle (Kugel-, Stäbchen-, Scheibchen-) Seite 146, Bauerecker, PC für Nebenfächler Elektromagnetisches Spektrum Sonne als Schwarzkörperstrahler mit Oberflächentemperatur 6000 °C Intensitätsmaximum Sonnenlicht grün (530 nm): • Blätter sind grün ⇒ grünes Licht wird absorbiert, d.h. Komplementärfarbe des Restlichts ist wieder grün. • Augen sind am empfindlichsten im grün ⇒ Naturvölker haben viele verschiedene Namen für grün. Seite 147, Bauerecker, PC für Nebenfächler Energiespektrum Sonnenstrahlung Spektrale Strahlungsleistungsdichte in W/(m2µm) Das Energieverteilungsspektrum der Sonnenstrahlung (unten, Bildquelle [Kal06]) stimmt außerhalb der Atmosphäre gut mit dem Schwarzkörperspektrum bei 5700°C überein. Man erkennt, dass ein großer Teil der Strahlungsenergie von der Atmosphäre gestreut und absorbiert wird. Seite 148, Bauerecker, PC für Nebenfächler Globale Energiebilanz 30 % 100 % 70 % Quelle: [IPC07] Seite 149, Bauerecker, PC für Nebenfächler Lichtwechselwirkung mit Teilchen (Aerosol) Streuung Eingestrahlte Photonen Transmission Absorption Reflexion Seite 150, Bauerecker, PC für Nebenfächler Lumineszenz Einteilung Optische Spektroskopie Optische Spektroskopie Absorption MolekülAbsorption AtomAbsorption Körper (z.B. Blatt) nimmt Lichtenergie auf. Seite 151, Bauerecker, PC für Nebenfächler Reflexion Normale Reflexion Diffuse Reflexion Körper reflektiert Licht. nach W. Schmidt Streuung Raleigh Mie Fraunhofer RamanStreuung Körper (z.B. Blatt) streut (=verteilt) Licht. Lumineszenz Fluorreszenz Phosphoreszenz (lang anhaltend) Chemolumineszenz → Biolumineszenz (Glühwürmchen, Tiefseefische, Bakterien, …) Körper leuchtet selbst, ohne sehr heiß zu sein. Elektrochemie Die Elektrochemie verknüpft elektrische Phänomene (Strom, Spannung, …) mit chemischen Prozessen. Sie ist wichtig zum Verständnis zahlreicher biologischer, alltäglicher Erscheinungen, wie zum Beispiel : Membranpotential 1) Spannungspotentiale Nervenerregung 2) Transport von Ladungen Verrosten 3) Anwendungen 4) Übungsaufgaben Seite 152, Bauerecker, PC für Nebenfächler Elektrochemie – Transport von Ladungen Elektrolyte sind Stoffe, die in Wasser in frei bewegliche Ionen (Anionen, Kationen) dissoziieren. Geladene Teilchen (Ionen, Elektronen) tragen Vielfache der Elementarladung e0 = 1,602 · 10-19 C mit der Einheit 1 C = 1 Coulomb. Sie üben eine abstandsabhängige Kraftwirkung aufeinander aus: 1 q1 ⋅ q2 F= ⋅ 2 4πε r r q1, q2 ε = εr · ε0 ε0 εr COULOMB – Gesetz (Analogie Gravitationsgesetz) Teilchenabstand Ladungen der Teilchen (positiv oder negativ) Dieletrizitätskonstante Vakuum-Permittivität (= 8,854 · 10-12 A·s/(V·m)) relative Dielektrizitätskonstante (z.B. 79 für H2O und 2,2 für CCl4) Seite 153, Bauerecker, PC für Nebenfächler Elektrochemie – Transport von Ladungen Der Stromfluß I im Elektrolyt hängt mit der Spannung U über den Widerstand R zusammen. Im Idealfall gilt das Ohmsche Gesetz: U = R⋅I Empirisch findet man: (→ Elektrolysezelle) Elektrodenabstand R= ρ⋅ l A Spezifischer Widerstand κ = 1/ρ Λ = κ/c c spez. Leitfähigkeit, Einheit Siemens S = Ω-1·m-1 molare Leitfähigkeit, Einheit S·m2·mol-1 Konzentration Seite 154, Bauerecker, PC für Nebenfächler Querschnittsfläche der Zelle • Gelöste Elektrolyte : o starke Elektrolyte (z.B. NaCl, NaOH) o schwache Elektrolyte (z.B. Essigsäure) Dissoziationsgrad α= caq c0 o Biologische Elektrolyte : im Cytosol sind Na+, K+, Mg2+, Cl-, PO43-, HCO32- gelöst Seite 155, Bauerecker, PC für Nebenfächler Kathode • Frei bewegliche Elektronen in Metallen (sehr gute Leitfähigkeit) Anode Elektrochemie – Transport von Ladungen Elektrochemie – Transport von Ladungen Beschleunigung durch E-Feld FE Bremsende Reibungskraft FR → → FR = 6πriη ν i Elektrische Beweglichkeit: vD = Seite 156, Bauerecker, PC für Nebenfächler z ⋅e⋅ E 6π ⋅ ri ⋅η vD z ⋅e u= = E 6π ⋅ ri ⋅η elektrisches Feld E Ladungszahl z Elementarladung e Viskosität η(Wasser, 20 °C) Ionenradius r → FE = zi ⋅ e ⋅ E (Stokessches Gesetz) Driftgeschwindigkeit: Beispiel → 1000 V/m 1 1.609·10-19 C 10-3 kg/(ms) 10-9 m E e ri η elek. Feldstärke Elementarladung Radius Teilchen Viskosität (Tabellierte Materialkonstante) m vD = 8,5 ⋅10 s −6 m2 u = 8,5 ⋅10 V ⋅s −3 Elektrochemie – Transport von Ladungen Elektrische Leitfähigkeit κ= 1 ρ = F ⋅ c ⋅ (ν ⋅ z ⋅ u +ν ⋅ z ⋅ u ) + + + − − − Λ= κ c Kation Anion Unabhängige Ionenwanderung ! ρ c z u ν Λ F spezifischer Widerstand Konzentration Ladung der Ionen elektrische Beweglichkeit Geschwindigkeit des Ions molare Leitfähigkeit Faraday-konstante Seite 157, Bauerecker, PC für Nebenfächler Leiter Leitfähigkeit [1/(Ω·cm)] Au Graphit NaCl(flüssig) 1 M NaCl-Lösung 1 M HCl-Lösung 4,85 · 105 1.2 · 10-3 3,77 0,074 0,332 Elektrochemie – Transport von Ladungen • Anionen und Kationen leiten in der Regel unterschiedlich gut! Hittorfsche Überführungszahlen I+ = t+ I − I = t− I • Anteil am Gesamtstrom I • Experimentell durch Elektrolyse bestimmbar Seite 158, Bauerecker, PC für Nebenfächler + − t +t =1 Elektrochemie – Transport von Ladungen Elektrolyse - + +++++ Kathodenraum Anodenraum Mittelraum ----K A ++++ - ++++ M Diffusion und Rühreffekte vermeiden Bsp.: Kationen leiten 5 mal besser − ∆cKathodenraum t = + ∆c Anodenraum t M Seite 159, Bauerecker, PC für Nebenfächler K A Elektrochemie – Transport von Ladungen Kohlrausch‘sches Quadratwurzelgesetz Λc = Λ0 − k ⋅ c Ursache für Konzentrationsabhängigkeit: Coulomb Wechselwirkungen zwischen den Ionen bremsen die Beweglichlichkeit je geringer die Konzentration (= großer Abstand) desto geringer die Rolle der interionischen WW Was hat eine größere Beweglichkeit ein kleines oder ein großes Ion? Seite 160, Bauerecker, PC für Nebenfächler Elektrochemie – Transport von Ladungen • Debye-Hückel-Onsager-Theorie: Ionen besitzen eine Ionenwolke Je größer die Ladungsdichte des Ions, desto größer der Radius der Ionenwolke • Relaxationseffekt (Störung der Nahordnung durch EFeld Ionenwolke muss neu Aufgebaut werden Ion wandert dem Schwerpunkt der Wolke voraus Bremswirkung) • Elektrophoretischer Effekt (Erhöhung der Stokesschen Reibungskraft durch Ionenwolke) Seite 161, Bauerecker, PC für Nebenfächler Elektrochemie – Transport von Ladungen • H+ und OH- sind klein und leiten besonders gut Anderer Mechanismus der Leitung ! Nicht die Moleküle wandern sondern nur die Ladung (Dissoziation) GROTTHUß-Mechanismus GROTTHUßMechanismus Seite 162, Bauerecker, PC für Nebenfächler Elektrochemie – Transport von Ladungen Schwache Elektrolyte OSTWALDSCHES Verdünnungsgesetz α² c( K + ) ⋅ c( A− ) = ⋅ c0 Kd = c( KA) 1−α Λc =α Λ0 Λ2c Kd = ⋅c (Λ 0 − Λ c )Λ 0 W. F. Ostwald 1853-1932 Nobelpreis 1909 Kd α c(A-) c(K+) c(KA) c0 Λc Λ0 Dissoziationskonstante Dissoziationsgrad Konzentration Anionen Konzentration Kationen Konzentration des nicht dissoziierten Elektrolyts Einwaagekonzentration Äquivalenzleitfähigkeit Grenzleitfähigkeit (d.h. bei unendlicher Verdünnung) Durch Verdünnen der Lösung nimmt der Dissoziationsgrad α zu ! Seite 163, Bauerecker, PC für Nebenfächler Elektrochemie – Transport von Ladungen Zusammenfassung Elektrolyte • Starke Elektrolyte zeigen schwache Abhängigkeit der mol. Leitfähigkeit von Konzentration (Kohlrausch Quadratwurzelgesetz) interionische WW (Debye-Hückel-Theorie: Nahordnung von Zentralionen und Gegenionenwolken, Coulombkräfte) • Schwache Elektrolyte zeigen starke Abhängigkeit von mol. Leitfähigkeit von der Konzentration (Ostwaldsches Verdünnungsgesetz) abnehmender Dissoziationsgrad durch Konz.-Erhöhung Schwache : Ver-dünn-ungsgesetz Starke: Kohl-Rausch-mittel Seite 164, Bauerecker, PC für Nebenfächler Elektrochemie - Potential ~ Faraday-konst= 9,6· 104 C/mol µ i = µi + zFϕ Elektrochemisches Potential [J/mol] Elektrisches Potential [V=J/C] Chemisches Potential [J/mol] µi (T , p) = µi0 + RT ln(ai ) Aktivität • korrigiertes Konzentrationsmaß • Kolligative Eigenschaft (nur von Teilchenzahl abhängig) • Kationen und Anionen tragen zur Teilchenzahl bei • Experimentell bestimmbar p1 ai = f i ⋅ xi = * p1 p1 1 − * = xi p1 (Raoultsches Gesetz 1890) Dampfdruckerniedrigung Seite 165, Bauerecker, PC für Nebenfächler Elektrochemie – Potential Redox-Reaktion 2− Fe + Cu 2+ SO 4 ⇔ Fe 2+ + Cu + SO4 2− ΔG0= -150 kJ/mol < 0 GG auf Seite der Produkte läuft spontan ab Übertragung von Elektronen von Fe zu Cu Reaktionsschritte: 1) Oxidation von Eisen: Fe Fe2+ + 2e(Erhöhung der Ox zahl, Anode) 2) Reduktion von Kupfer: (Kathode) Cu2+ + 2e- Cu Ox+Red Form = Redoxpaar: z.B. Fe2+/Fe Seite 166, Bauerecker, PC für Nebenfächler Elektrochemie – Potential Elektrochemische Spannungsreihe E0 > 0 reduzierte Form bevorzugt Nernstsche Gleichung: aOx RT ln E = E0 + zF aRe d R: Gaskonst; T: Temperatur; F: Faradaykonst.; z: Ladungszahl; E: Potentialdifferenz; ΔG: freie Enthalpie; a: Aktivität Seite 167, Bauerecker, PC für Nebenfächler Reduzierende Wirkung nimmt zu ∆G 0 E0 = − zF Oxidierende Wirkung nimmt zu Standard-Redox-Potential E0 Halb-Reaktion E0 Elektrochemie – Potential Aufbau einer Galvani-Spannung + + + + + + + - + - + - + - Me - + - + - + - + - + + Me+ a) Übertritt von positiven Ionen in Lösung + + + + + + + Me + + + + + + + + + - - Me+ b) Übertritt von positiven Ionen aus Lösung • Aufbau einer Doppelschicht (Dicke ~ 1 nm) • elektrochemisches GG: • Galvani-Spannung nicht direkt messbar sondern nur Vergleichbar (Standard-WasserstoffElektrode) ! Seite 168, Bauerecker, PC für Nebenfächler Elektrochemie – Potential Galvanische Zelle • Zwei verbundene galvanische Halbzellen • Elektromotorische Kraft (EMK): ΔE = EReduktion - EOxidation Bsp.: ECu = E0Cu + EZn = E0Zn + RT 2F RT 2F ln aCu ln aZn 2+ 2+ E0Cu = 0.34 V E0Zn = −0.76 V (Daniell-Element) Δ E0 = E 0Cu−E0Zn = 0.34 V−(−0.76 V)=1.1 V Seite 169, Bauerecker, PC für Nebenfächler Elektrochemie – Potential Ag/AgCl-Bezugselektrode •„natürliche“ Bezugselektrode ist Standard-H2-Elektrode (negativ Handling) • Am häufigsten genutzte Referenzelektrode • Gebrauchsfertig kommerziell verfügbar • Standard-Elektrodenpotential: E0 Ag+/Ag = 0,8 V • Konstantes Potential • AgCl sehr schwer löslich KL= 2·10-10 mol²/L² • Ag + Cl- AgCl + e- • Alternative : Kalomelelktrode (Hg/Hg2Cl2) Seite 170, Bauerecker, PC für Nebenfächler Elektrochemie – Potential Lithium-Ionen-Akku CoO2 + Li+ + e- LiCoO2 LiCoO2 Seite 171, Bauerecker, PC für Nebenfächler Li-Graphit LiCn Cn + Li+ + e- 3,6 V Elektrochemie – Brennstoffzelle Brennstoffzelle • Katalytisch kontrollierte Knallgasreaktion (benötigt Platin teuer) • Anode: 2 H2 4 H+ + 4 e• Kathode: O2 + 4 H+ + 4 e- 2 H2O • Standardzellspannung E0 = 1,23 V • verschiedene Typen: • SOFC (Solid Oxid, 600-1000°C) • MCFC (Molten Carbonate, 600°C) • PEMFC (Polymerelektrolyt, < 200°C) Seite 172, Bauerecker, PC für Nebenfächler Elektrochemie – Brennstoffzelle Stacks von Brennstoffzellen Wirkungsgrad vs. Leistungsgrad optimiert 65 % Elektrischer Wirkungsgrad Seite 173, Bauerecker, PC für Nebenfächler Hohe Leistung P = U ·I Elektrochemie – Brennstoffzelle Elektroautos • Seit etwa 1830 (T. Davenport, R. Anderson, Flocken Elektrowagen) • ab 1910 Nischendasein (Verbrennungsmotor) 1) Wasserstoff-Tank + Brennstoffzelle • Schneller Aufladevorgang (5 min) • Hohe Energiedichte (33 kW/kg) • Wenige Tankstellen ( in DE < 20) • Teuer (z.B. Platin) • Serienfahrzeug: Toyota Mirai 2) Li-Ionen-Akkumulator • Hohes Gewicht geringe Reichweite • Sehr langer Aufladevorgang • Serienreife Modelle verschiedener Hersteller auf dem Markt (Renault ZOE, Nissan Leaf, Tesla S, BMW i3, Opel Ampera, VW e-Golf) Seite 174, Bauerecker, PC für Nebenfächler Toyota Mirai Nissan Leaf (meist verkauftes Elektroauto) Elektrochemie – Potential Konzentrationselement aOx RT E = E0 + ln zF aRe d a groß RT ∆E = ⋅ ln zF aklein (für kleine Konzentrationen kann mit c statt a gerechnet werden) a1 = 10 · a2 59 mV @ RT • Membranpotential • Lambda-Sonde Seite 175, Bauerecker, PC für Nebenfächler Oxidation Cu Cu2+ + 2e- Reduktion Cu2+ + 2e- Cu Elektrochemie – Potential Membranpotential • Eigenschaft aller Zellen • Besonders wichtig: Nerven-, Muskel-, Sinneszellen • Kommunikation innerhalb des Körpers und mit Umgebung • etwa 30 bis 100 mV Seite 176, Bauerecker, PC für Nebenfächler Elektrochemie – Potential Gibbs-Donnan-Gleichgewicht: • Lebende Zellen (und technische Systeme) • Semipermeable Membran ( z.B. Protein zu groß) • unterschiedliche K+, Na+, ClKonzentrationen • Summe der Ladungen ausgeglichen Zelle (innen) Goldmann-Gleichung: (vgl. Nernst-Gleichung) UM PNa ⋅ [ Na + ]a + PK ⋅ [ K + ]a + PCl ⋅ [Cl − ]i RT = ⋅ ln F PNa ⋅ [ Na + ]i + PK ⋅ [ K + ]i + PCl ⋅ [Cl − ]a Seite 177, Bauerecker, PC für Nebenfächler P = Permeabilität =(Diffusionskonst ./ Membrandicke) i = Innen a = außen Um = Membranpotential Elektrochemie – Potential Lambdasonde • Vergleicht Restsauerstoffgehalt im Abgas mit O2 in Atmosphäre Ziel: Schadstoffabgabe minimieren (NOx, CxHy, Ruß) • Zr-Membran ist bei hohen T ( ~650 °C) O2Durchlässig • O2 wird ionisiert 450 mV Seite 178, Bauerecker, PC für Nebenfächler Lambdasonde Volvo 240 Elektrochemie – Potential Korrosion von Eisen (Verrosten) • Redoxvorgänge im belüfteten Wassertropfen auf Eisen • Oxidation: Fe Fe2+ + e- (E0 = -0,44 V) • Reduktion: 0,5 O2 +H2O + 2e- 2 OH- (E0 = +0,4 V) • Aktivitäten der Reaktanten sind entscheidend (pH-Wert) • Gelöste Salze beschleunigen die Korrosion Seite 179, Bauerecker, PC für Nebenfächler Elektrochemie – Potential Korrosionsschutz durch Opferanode • Zusatz eines leichter oxidierbaren (unedel) Metalls • z.B. Zink oder Magnesium (Me/Me2+ < Fe/Fe2+) • Opferanode muss regelmäßig ersetzt werden • Anwendungen: Wasserspeicher, Bohrinseln, Öltanks, Schiffe, … Opferanode an Schiffsrumpf Seite 180, Bauerecker, PC für Nebenfächler Opferanoden aus Wasserspeicher Elektrochemie - Übungsaufgaben 1. Nimmt der Dissoziationsgrad beim Verdünnen einer Elektrolytlösung zu oder ab ? 2. Die Dissoziationskonstante Kd eines Elektrolyten sei 0,5 und die Anfangskonzentration c0 = 1mol/L. Auf welche Konzentration c1 muss Verdünnt werden, um einen 20% größeren Dissoziationsgrad α1 zu erhalten? 3. Ist ein ähnliches Verhalten auch für eine HCl-Lösung zu erwarten und warum? 4.a) Welche Potentialdifferenz liegt in einer Au/Al-Zelle vor? (die Aktivitäten seien jeweils 1) b) Notieren Sie die Redox-Gleichungen! c) Welches Metall wird oxidiert? 5. Was beschreibt das Raoulsche Gesetz ? Seite 181, Bauerecker, PC für Nebenfächler Verzeichnis der Formelzeichen a A b B E Ekin Epot c c cp cv d D F g GG h h H Ho Hu k, K kB K K K KH l I m M n N NA Nmol Kohäsionsdruck (VdW-Gas) Fläche Kovolumen (VdW-Gas) Impuls Energie kinetische Energie potentielle Energie Konzentration in mol/m3 Lichtgeschwindigkeit Wärmekapazität b. konst. Druck Wärmekapazität b. konst. Volumen Durchmesser, Dicke, Länge Diffusionskoeffizient Kraft Erdbeschleunigung Gleichgewicht Höhe Planck-Konstante Enthalpie Brennwert Heizwert Reaktionsgeschwindigkeitskonstante Boltzmannkonstante Gleichgewichtskonstante Kompressibilitätszahl Anzahl der Komponenten Henry-Konstante Länge, freie Weglänge Stromstärke Masse Teilchen, Körper Molare Masse Teilchenzahlvolumendichte Teilchenzahl Avogadrokonstante (Teilchen pro Mol) Molzahl Seite 183, Bauerecker, PC für Nebenfächler p p° pn P q q Q r R R S t T T° Tn u U v vx V Vm Vm° Vm,n w w, W W WW x xi z z Z Z Zn Z Druck Standarddruck Normaldruck Anzahl der Phasen Elektrische Ladung Wärme Wärme Radius, Abstand, Länge molare Gaskonstante, Radius Elektrischer Widerstand Entropie Zeit absolute Temperatur Standardtemperatur Normaltemperatur (Diffusions-)Geschwindigkeit innere Energie Geschwindigkeit Geschwindigkeitskomponente in x-Richung Volumen molares Volumen Vm° im Standardzustand Vm,n im Normalzustand Arbeit Wahrscheinlichkeit Arbeit Wechselwirkung Länge, Abstand Stoffmengenanteil, Molenbruch Stoßzahl Ladungszahl Anzahl, Gesamtstoßzahl Realgasfaktor Realgasfaktor im Normzustand Zustandssumme α ε ε η η λ ν ρ ρ σ σ θ µ Λ Π Dissoziationsgrad Leistungszahl Kältemaschine Dielektrizitätskonstante Viskosität thermodynamischer Wirkungsgrad Wellenlänge Frequenz Dichte spez. Widerstand Moleküldurchmesser Oberflächenspannung Celsius-Temperatur Joule-Thomson-Koeffizient Ionenleitfähigkeit Osmotischer Druck