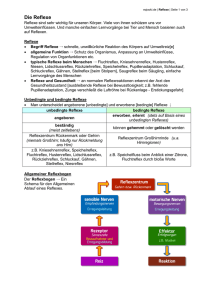
Synthese und Charakterisierung von neuen und
Werbung

Synthese und Charakterisierung von neuen und bekannten Hartstoffen und die technische Relevanz von Härteangaben - Einkristallzüchtung und Darstellung von Sinterkörpern - INAUGURAL-DISSERTATION ZUR ERLANGUNG DES AKADEMISCHEN GRADES DOKTOR DER NATURWISSENSCHAFTEN DER FAKULTÄT FÜR CHEMIE UND PHARMAZIE DER ALBERT – LUDWIGS – UNIVERSITÄT FREIBURG IM BREISGAU vorgelegt von Dipl. Chem. Dominik Kotzott aus Tarnowitz (OS) - 2009 - Vorsitzender des Promotionsausschusses : Prof. Dr. R. Schubert Dekan: Prof. Dr. H. Hillebrecht Leiter der Arbeit: Prof. Dr. H. Hillebrecht Referent: Prof. Dr. H. Hillebrecht Referentin: Prof. Dr.-Ing. C. Röhr Tag der Einreichung: 17.04.2009 Tag der Promotion: 07.05.2009 Tag der Verkündigung des Prüfungsergebnisses: 07.05.2009 Publikationen Folgende Teile der Arbeit wurden veröffentlicht: Publikationen J. Etzkorn, M. Ade, D. Kotzott, M. Kleczek, H. Hillebrecht „Ti2GaC, Ti4GaC3 and Cr2GaC – synthesis, crystal growth and structure analysis of Ga-containing MAX-phases Mn+1GaCn with M = Ti, Cr and n = 1, 3“ Akzeptiert am 3. Januar 2009, bei J. Solid State Chem., (in Druck) D. Kotzott, M. Ade, H. Hillebrecht „Singel crystal studies on Co-containing, τ-Borides Co23-xMxB6 (M = Al, Ga, Sn, Ti, V, Ir) and the boron rich τ-Boride Co12,3Ir8,9B10,5“ Akzeptiert am 18. Oktober 2008, bei J. Solid State Chem., (in Druck) D. Kotzott, M. Ade, H. Hillebrecht „Einkristalluntersuchungen an Co- haltigen τ-Boriden in den Systemen Co/M’/B (M’ = Al, Ga, In, Sn, Ir)“ Z. Kristallogr. Suppl. 21 (2007) 079-54. D. Kotzott, M Ade, H. Hillebrecht „Synthesis and Crystal Structures of alpha- and beta-Modifications of Cr2IrB2 containing B4-Chains, τ-Borides Cr23-xIrxB6 and Cr2B“ J. Solid State Sciences 10 (2008) 291-302. Tagungsbeiträge (I) Vorträge H. Hillebrecht, D. Kotzott „Ternary carbides Ti3MC2 (M = Al, Si, Ga, Ge)-Synthesis, crystal growth, solid solutions and micro-hardness measurements“ 17. DGK (Deutsche Gesellschaft für Kristallographie) 9. - 12. März 2009 Hannover. D. Kotzott, H. Hillebrecht „Mikrohärtemessung nach Vickers und Knoop an Hartstoffen“ 17. FMF (Freiburger Material Forschung) Kolloquium 16. - 17. Oktober 2008, Hornberg. D. Kotzott, H. Hillebrecht „Synthesen mit dem Puls-Plasma-Ofen SPS“ 16. FMF (Freiburger Material Forschung) Kolloquium, 15. - 16. Oktober 2007, Rust. Publikationen D. Kotzott, H Hillebrecht „τ-Boride - Ternäre Boride von Übergangs- und Hauptgruppenmetallen mit Kobalt als Hauptkomponente“ Seminar für Festkörperchemie, 15. - 18. Juni 2006, Hirschegg, Österreich. (II) Posterbeiträge D. Kotzott, M. Ade, H. Hillebrecht „Single crystal studies on Co-containing τ-Borides“ 16th ISBB 2008 (International Symposium on Boron, Borides) and Related Materials, 7. – 12. September 2008, Matsue, Shimane, Japan. D. Kotzott, H. Hillebrecht „Vickers and Knoop Micro Hardness Test of Single Crystals of Borides“ 16th ISBB 2008 (International Symposium on Boron, Borides) and Related Materials. 7. – 12. September 2008, Matsue, Shimane, Japan. N. Vojteer, D. Kotzott, S. Haseloff, M. Schroeder, H. Hillebrecht „B6P – sometimes electron precise“ 16th ISBB 2008 (International Symposium on Boron, Borides) and Related Materials. 7. – 12. September 2008, Matsue, Shimane, Japan. N. Vojteer, J. Stauffer, D. Kotzott, S. Haseloff, H. Hillebrecht „LiB12PC: the first Boron Rich Metal Boride with Phosphorus: Synthesis, Crystal, Structure, Hardness, EDX, Spectroscopic Investigations“ 16th ISBB 2008 (International Symposium on Boron, Borides) and Related Materials. 7. – 12. September 2008, Matsue, Shimane, Japan. D. Kotzott, H. Hillebrecht „Vickers and Knoop Micro Hardness Test of Single Crystals of Hard Materials“ Tag der Forschung der Fakultät für Chemie, Pharmazie und Geowissenschaften, 4 Juli 2008, Freiburg i. Br.. M. Kleczek, D. Kotzott, H. Hillebrecht „Single Crystals analysis of H-Phase Compounds and the Substitution of Special Atoms“ Tag der Forschung der Fakultät für Chemie, Pharmazie und Geowissenschaften, 6 Juli 2008, Freiburg i. Br.. D. Kotzott, M. Ade, H. Hillebrecht „Einkristalluntersuchungen an Co-haltigen τ-Boride in den Systemen Co/M‘/B (M‘ = Al, Ga, In, Sn, Ir“ 15. DGK (Deutsche Gesellschaft für Kristallographie), 5. – 9. März 2007, Bremen. Für meine Familie Die vorliegende Arbeit entstand in der Zeit von April 2006 bis März 2009 am Institut für Anorganische und Analytische Chemie der Albert-Ludwigs-Universität Freiburg im Breisgau unter der Leitung von Herrn Prof. Dr. Harald Hillebrecht Ihm danke ich besonders für die interessante und spannende Aufgabenstellung, für das mir entgegengebrachte Vertrauen, für die Unterstützung und immerwährende Diskussionsbereitschaft bei der Anfertigung dieser Arbeit. Frau Prof. Dr.-Ing. Caroline Röhr danke ich für Übernahme des Korreferats und ihre stete Hilfsbereitschaft. Weiterhin möcht ich mich bei allen ehemaligen und aktuellen Mitgliedern des Arbeitskreises herzlich für die überaus angenehme Arbeitsatmosphäre bedanken – bei Dr. Martin Ade, Anita Becherer, Klaus Bickel, Sonia Bimmler, Lisa Boschmann, Stefanie Haseloff, Dr. Henning Höppe, Karolina Kazmierczak, Katrin Keller, Monika Kleczek, Stefan Lösel, Dr. Thilo Ludwig, Alexis Pediaditakis, Hasiba Richter, Dr. Heinz Rotter, Kai Schillinger, Melanie Schroeder und Dr. Natascha Vojteer. Meinem Vater Heinrich Kotzott danke ich besonders für die Hilfe und Diskussionsbereitschaft bei meinen ersten Versuchen der Präparation und der Härtemessung an Einkristallen. Außerdem danke ich meinen Mitarbeitern Monika Kleczek und Kai Schillinger, die auf dem Teilgebiet der MAX-Phasen eine große Hilfe waren, und allen ehemaligen und aktuellen Mitarbeitern des Arbeitskreises Röhr für die Hilfsbereitschaft. Meinen Eltern Boguslawa und Heinrich Kotzott gebührt Dank und Respekt für ihre Stärke, sämtliche Sorgen von mir fernzuhalten, damit ich nun hier stehe. Ich danke meine Bruder Leszek Kotzott für Beistand und Unterstützung in allen Situationen. Und für die regelmäßigen Ablenkungen und die wichtigen Pausen in den entscheidenden letzten Monaten möchte ich mich ganz besonders bei Nora Borst, Thomas Cuber, Fabian Zehender, Andrea Kinzler, Stephan Wießler und Nikola Schwarzer und allen meinen Freunden bedanken. Inhaltsverzeichnis Verzeichnis der verwendeten Symbole _____________________________17 Verzeichnis der verwendeten Abkürzungen _________________________19 1 Thema ____________________________________________________21 1.1 Einleitung _____________________________________________________________ 21 1.2 Zielsetzung und Synthesemethoden ________________________________________ 22 1.2.1 1.2.2 1.2.3 2 Ziele ______________________________________________________________________ 22 Charakterisierung ____________________________________________________________ 23 Synthesemethoden____________________________________________________________ 23 Methoden__________________________________________________25 2.1 Härtemessung__________________________________________________________ 25 2.1.1 Einleitung __________________________________________________________________ 2.1.2 Die verschiedenen Messverfahren________________________________________________ 2.1.3 Die physikalische Einheit bei der Härteprüfung _____________________________________ 2.1.4 Zusammenhang von Härte und Probeneigenschaften _________________________________ 2.1.5 Härteprüfung nach Brinell______________________________________________________ 2.1.5.1 Fehlerquellen bei der Härtemessung nach Brinell _______________________________ 2.1.6 Härteprüfung nach Vickers _____________________________________________________ 2.1.6.1 Fehlerquellen bei der Härtemessung nach Vickers ______________________________ 2.1.7 Härteprüfung nach Knoop ______________________________________________________ 2.1.8 Mikrohärtemessung: Vickers vs. Knoop ___________________________________________ 2.1.9 Umwerten von Härtewerten ____________________________________________________ 25 26 28 29 30 31 33 36 38 41 42 2.2 Hilfsmetallbadtechnik ___________________________________________________ 44 2.3 Hochtemperatursynthesen _______________________________________________ 45 2.4 Die Sintermethoden _____________________________________________________ 46 2.5 SPS-Anlage „Dr. SINTER-LAB™“ ________________________________________ 51 am Institut für Anorganische und Analytische Chemie ______________________________ 51 2.6 3 Reaktionen im Pulsplasmaofen____________________________________________ 52 MAX-Phasen_______________________________________________54 3.1 Vorkommen der MAX-Phasen ____________________________________________ 54 3.2 Struktur der Komplexcarbide ____________________________________________ 56 3.3 Eigenschaften der MAX-Phasen___________________________________________ 60 3.3.1 3.3.2 3.3.3 3.4 Chemische Reaktivität_________________________________________________________ 60 Mechanische Eigenschaften ____________________________________________________ 61 Elektrische und thermische Leitfähigkeit __________________________________________ 61 Das System Titan-Aluminium-Kohlenstoff __________________________________ 63 3.4.1 Die binären Randsysteme um das System Titan-Aluminium-Kohlenstoff _________________ 3.4.2 Das ternäre System Titan-Aluminium-Kohlenstoff___________________________________ 3.4.3 Arbeiten in einer Aluminium-Schmelze ___________________________________________ 3.4.3.1 Präparative Untersuchung im System Titan-Aluminium-Kohlenstoff________________ 3.4.3.2 Synthese der MAX-Phase im SPS-Ofen ______________________________________ 3.4.3.3 EDX - Untersuchung an Einkristallen der 312-Phase ____________________________ 3.4.3.4 Röntgenographische Untersuchung an Einkristallen der 312-Phase _________________ 3.4.3.5 Strukturdiskussion von Ti3AlC1,82N0,18 _______________________________________ 3.4.3.6 EDX - Untersuchung an Einkristallen der 211-Phase ____________________________ 3.4.3.7 Röntgenographische Untersuchung an Einkristallen der 211-Phase _________________ 63 64 65 65 68 71 73 76 77 78 - 11 - Inhaltsverzeichnis 3.4.3.8 Strukturdiskussion von Ti2AlC0,28N0,72 _______________________________________ 81 3.4.4 Arbeiten in einer Aluminium/Zinn-Schmelze _______________________________________ 82 3.4.4.1 Die binären Systeme Aluminium-Zinn und Titan-Zinn___________________________ 82 3.4.4.2 Präparative Untersuchung im System Titan-Aluminium/Zinn-Kohlenstoff ___________ 83 3.4.4.3 Synthese der 312-Phase im SPS-Ofen ________________________________________ 85 3.4.4.4 EDX - Untersuchung an Einkristallen ________________________________________ 86 3.4.4.5 Röntgenographische Untersuchung an Einkristallen der 211-Phase _________________ 89 3.4.4.6 Allgemeine Strukturdiskussion _____________________________________________ 94 3.4.4.7 Substitutionsmöglichkeiten im System Titan-Aluminium/Zinn-Kohlenstoff/Stickstoff __ 95 3.4.4.8 EDX - Untersuchung an Einkristallen ________________________________________ 96 3.4.4.9 Röntgenographische Untersuchung an Einkristallen der 413-Phase _________________ 98 3.4.4.10 Strukturdiskussion ______________________________________________________ 102 3.4.4.11 EDX - Untersuchung an Einkristallen der 211-Phase ___________________________ 103 3.4.4.12 Röntgenographische Untersuchung an Einkristallen der 211-Phase ________________ 104 3.4.4.13 Strukturdiskussion ______________________________________________________ 106 3.4.4.14 Synthese im SPS-Ofen___________________________________________________ 108 3.4.5 Arbeiten in einer Aluminium/Gallium-Schmelze ___________________________________ 111 3.4.5.1 Die binären Systeme Aluminium-Gallium und Titan-Gallium ____________________ 111 3.4.5.2 Präparative Untersuchung im System Ti-Al/Ga-C _____________________________ 112 3.4.5.3 Härtemessung an der 312-Phase (Ti3GaC2) ___________________________________ 114 3.4.5.4 EDX - Untersuchung an Einkristallen der 312-Phase ___________________________ 115 3.4.5.5 Röntgenographische Untersuchung an Einkristallen der 312-Phase ________________ 116 3.4.5.6 Strukturdiskussion ______________________________________________________ 119 3.4.5.7 Röntgenographische Untersuchung an Einkristallen der 211-Phase ________________ 120 3.4.5.8 Strukturdiskussion ______________________________________________________ 122 3.4.6 Arbeiten in einer Aluminium/Indium-Schmelze ____________________________________ 124 3.4.6.1 Die binären Randsysteme Al-In und Ti-In sowie In-C __________________________ 124 3.4.6.2 Präparative Untersuchung im System Titan-(Aluminium/Indium)-Kohlenstoff _______ 125 3.4.6.3 Röntgenographische Untersuchung an Einkristallen der 211-Phase ________________ 126 3.4.6.4 Strukturdiskussion ______________________________________________________ 128 3.4.7 Arbeiten in einer Aluminium/Germanium-Schmelze ________________________________ 130 3.4.8 Die binären Systeme _________________________________________________________ 130 3.4.8.1 Präparative Untersuchung im System Titan-Germanium-Kohlenstoff ______________ 131 3.4.8.2 Röntgenographische Untersuchung an Einkristallen ____________________________ 132 3.4.8.3 Strukturdiskussion ______________________________________________________ 135 3.4.8.4 Härtemessung _________________________________________________________ 135 3.4.8.5 Synthese im Pulsplasma-Ofen _____________________________________________ 136 3.5 Das System Vanadium-Aluminium-Kohlenstoff_____________________________ 138 3.5.1 Die binären Randsysteme um das System V-Al-C __________________________________ 3.5.2 Das ternäre System Titan-Aluminium-Kohlenstoff__________________________________ 3.5.3 Arbeiten in einer Aluminium/Zinn-Schmelze ______________________________________ 3.5.3.1 Präparative Untersuchung im System Vanadium-(Aluminium/Zinn)-Kohlenstoff _____ 3.5.3.2 Röntgenographische Untersuchung an Einkristallen der 211-Phase ________________ 3.5.3.3 Strukturdiskussion ______________________________________________________ 3.5.3.4 Röntgenographische Untersuchung an Einkristallen der 531-Phase ________________ 3.5.3.5 Strukturdiskussion ______________________________________________________ 3.5.4 Arbeiten in einer Aluminium/Gallium-Schmelze ___________________________________ 3.5.4.1 Präparative Untersuchung im System Vanadium-(Aluminium/Gallium)-Kohlenstoff __ 3.5.4.2 Röntgenographische Untersuchung an Einkristallen der 211-Phase ________________ 3.5.4.3 Strukturdiskussion ______________________________________________________ 3.5.5 Arbeiten in einer Aluminium/Indium-Schmelze ____________________________________ 3.5.5.1 Präparative Untersuchung im System Vanadium-(Aluminium/Indium)-Kohlenstoff ___ 3.5.5.2 Röntgenographische Untersuchung an Einkristallen der 211-Phase ________________ 3.5.5.3 Strukturdiskussion ______________________________________________________ 3.5.5.4 Weitere Einkristalle _____________________________________________________ 3.5.5.5 Strukturdiskussion ______________________________________________________ 3.5.6 Weitere Untersuchungen ______________________________________________________ 3.5.6.1 Untersuchungen mit Aluminium/Eisen ______________________________________ 3.5.6.2 Untersuchungen mit Aluminium/Kobalt _____________________________________ 3.5.6.3 Untersuchungen mit Aluminium/Nickel _____________________________________ 3.5.6.4 Untersuchungen mit Zinn/Chrom __________________________________________ 138 138 139 140 141 143 145 148 151 151 152 155 156 156 157 160 161 163 165 165 166 167 168 - 12 - Inhaltsverzeichnis 3.5.6.5 3.5.6.6 3.6 3.6.1 3.6.2 3.7 3.7.1 3.7.2 3.7.3 3.7.4 4 Untersuchungen mit Aluminium/Silizium ____________________________________ 169 Untersuchungen mit Chrom/Zinn __________________________________________ 170 Weitere Ergebnisse aus dem Pulsplasma-Ofen ______________________________ 171 Darstellung von Ti3SiC2 im Pulsplasma Ofen ______________________________________ 171 Darstellung von Ta4AlC3 und Ta2AlC im Pulsplasma Ofen ___________________________ 172 h-Carbide im Pulsplasma Ofen___________________________________________ 174 Darstellung von Ta3Al2CoC und Ta3Al2CoN im Pulsplasma Ofen______________________ Härteprüfung _______________________________________________________________ Darstellung von Nb3Al2CoC im Pulsplasma Ofen __________________________________ Strukturdiskussion___________________________________________________________ 174 176 177 178 τ-Boride __________________________________________________181 4.1 4.1.1 4.1.2 4.1.3 4.1.4 4.2 Literaturübersicht _____________________________________________________ 181 Phasenbreite und Charakterisierung der τ-Phase____________________________________ Der Cr23C6-Strukturtyp _______________________________________________________ Sonderverhalten in der τ-Phase _________________________________________________ Das System Kobalt-Bor_______________________________________________________ Das System Kobalt-Iridium-Bor__________________________________________ 192 4.2.1 Die Phasenbreite des ternären Systems Kobalt-Iridium-Bor___________________________ 4.2.2 Präparative Untersuchung im System Kobalt-Iridium-Bor ____________________________ 4.2.3 EDX-Untersuchung__________________________________________________________ 4.2.4 Röntgenographische Untersuchung an Einkristallen_________________________________ 4.2.4.1 Untersuchungen an borarmen Einkristallen___________________________________ 4.2.4.2 Strukturdiskussion ______________________________________________________ 4.2.4.3 Untersuchungen an borreichen Einkristallen __________________________________ 4.2.5 Strukturdiskussion___________________________________________________________ 4.3 4.3.1 4.3.2 4.3.3 4.3.4 4.4 4.4.1 4.4.2 4.4.3 4.5 4.5.1 4.5.2 4.5.3 4.5.4 4.5.5 4.6 4.6.1 4.6.2 4.6.3 4.7 4.7.1 4.7.2 4.7.3 4.7.4 4.7.5 4.8 4.8.1 4.8.2 4.8.3 182 185 188 189 192 192 193 194 194 197 198 201 Das System Kobalt-Gallium-Bor _________________________________________ 203 Präparative Untersuchung im System Kobalt-Gallium-Bor ___________________________ Härtemessung an Einkristallen _________________________________________________ Röntgenographische Untersuchung an Einkristallen_________________________________ Strukturdiskussion___________________________________________________________ 203 205 205 208 Das System Kobalt-Aluminium-Bor_______________________________________ 209 Präparative Untersuchung im System Kobalt-Aluminium-Bor_________________________ 210 Röntgenographische Untersuchung an Einkristallen_________________________________ 211 Strukturdiskussion___________________________________________________________ 214 Das System Kobalt-Zinn-Bor ____________________________________________ 215 Der kobaltreiche Bereich des ternären Systems Kobalt-Zinn-Bor ______________________ Präparative Untersuchung im System Kobalt-Zinn-Bor ______________________________ EDX-Untersuchung__________________________________________________________ Röntgenographische Untersuchung an Einkristallen_________________________________ Strukturdiskussion___________________________________________________________ 215 215 217 218 221 Das System Kobalt-Titan-Bor____________________________________________ 222 Präparative Untersuchung im System Kobalt-Titan-Bor______________________________ 222 Röntgenographische Untersuchung an Einkristallen_________________________________ 224 Strukturdiskussion___________________________________________________________ 227 Das System Kobalt-Vanadium-Bor _______________________________________ 228 Der kobaltreiche Bereich des ternären Systems Kobalt-Vanadium-Bor __________________ Präparative Untersuchung im System Kobalt-Vanadium-Bor__________________________ Härtemessung an Einkristallen _________________________________________________ Röntgenographische Untersuchung an Einkristallen_________________________________ Strukturdiskussion___________________________________________________________ 228 228 230 230 233 Das System Kobalt-Wolfram-Bor_________________________________________ 234 Präparative Untersuchung im System Kobalt-Wolfram-Bor___________________________ 234 Härtemessung an Einkristallen _________________________________________________ 235 Röntgenographische Untersuchung an Einkristallen_________________________________ 236 - 13 - Inhaltsverzeichnis 4.8.4 4.9 Strukturdiskussion___________________________________________________________ 239 Das System Kobalt-Mangan-Bor _________________________________________ 240 4.9.1 4.9.2 4.9.3 4.10 Präparative Untersuchung im System Kobalt-Mangan-Bor ___________________________ 240 Röntgenographische Untersuchung an Einkristallen_________________________________ 241 Strukturdiskussion___________________________________________________________ 244 Das System Kobalt-Molybdän-Bor________________________________________ 245 4.10.1 4.10.2 4.10.3 4.11 Das System Kobalt-Magnesium-Bor ______________________________________ 250 4.11.1 5 5.1.1 5.1.2 5.1.3 5.2 5.2.1 5.2.2 5.2.3 5.2.4 7 8 Cr2ZrB6 ______________________________________________________________ 253 Präparative Untersuchungen ___________________________________________________ 253 Röntgenographische Untersuchungen an Einkristallen _______________________________ 255 Strukturdiskussion___________________________________________________________ 259 Cr2AlB2 ______________________________________________________________ 261 Präparative Untersuchungen ___________________________________________________ EDX-Untersuchungen ________________________________________________________ Röntgenographische Untersuchungen an Einkristallen _______________________________ Strukturdiskussion___________________________________________________________ 261 262 263 266 κ-Carbide ________________________________________________267 6.1 Präparative Untersuchungen ____________________________________________ 267 6.2 EDX- Untersuchung____________________________________________________ 268 6.3 Härtemessung an Einkristallen___________________________________________ 269 6.4 Röntgenographische Untersuchung an Einkristallen _________________________ 270 6.5 Strukturdiskussion_____________________________________________________ 273 Weiter Untersuchungen zur Härte ____________________________274 7.1 Boride _______________________________________________________________ 274 7.2 Boridcarbide__________________________________________________________ 279 7.3 Boridnitride __________________________________________________________ 280 7.4 Diskussion der Härteergebnisse __________________________________________ 281 Experimenteller Teil________________________________________284 8.1 8.1.1 8.1.2 8.1.3 8.2 8.2.1 8.2.2 8.2.3 8.2.4 8.2.5 9 Präparative Untersuchung im System Kobalt-Magnesium-Bor ______________________ 250 Ternäre Boride ____________________________________________253 5.1 6 Präparative Untersuchung im System Kobalt-Molybdän-Bor _______________________ 245 Röntgenographische Untersuchung an Einkristallen ______________________________ 246 Strukturdiskussion ________________________________________________________ 249 Allgemeine Angaben zu den Synthesen ____________________________________ 284 Spezifikation der verwendeten Chemikalien_______________________________________ 284 Synthesen in Hochtemperaturöfen ______________________________________________ 286 Aufarbeitung der Proben ______________________________________________________ 287 Untersuchungsmethoden________________________________________________ 287 Röntgenbeugung an Pulvern ___________________________________________________ Röntgenbeugung an Einkristallen _______________________________________________ EDX – Untersuchungen an Einkristallen _________________________________________ Einbetten von Einkristallen ____________________________________________________ Härtemessung ______________________________________________________________ 287 288 288 288 289 Verwendete Programme ____________________________________290 - 14 - Inhaltsverzeichnis 10 Zusammenfassung und Ausblick _____________________________291 11 Literaturverzeichnis ________________________________________296 - 15 - - 16 - Verzeichnis der wichtigsten Abkürzungen und Symbole Verzeichnis der verwendeten Symbole Symbol a, b, c, aw,bw d Gitterparameter Å (=100pm) Parameter des Gewichtschemas w des Programms SHELXL[199] Atomabstand Å (=100pm) Berechneter Strukturfaktor Fc Fc Dimension 2 Quadrat des berechneten Strukturfaktors Fo Beobachteter Strukturfaktor Fo2 Quadrat des beobachteten Strukturfaktors h, k, l Miller’sche Indizes I Intensität M Molare Masse g·mol-1 n Stoffmenge Mol N Anzahl p Druck GPa rh Aufheizgeschwindigkeit (heating rate) K·h-1 rc Abheizgeschwindigkeit (cooling rate) K·h-1 R1(F) Zuverlässigkeitsfaktor (verfeinert gegen Fo) Rint Interner Standard s.o.f. Besetzungsfaktoren (site occupation factor) S(F2) goodness-of-fit (verfeinert gegen Fo2) T Transmissionsgrad K oder °C T Temperatur K oder °C Ttrans Phasenübergangstemperatur K oder °C Tmax. Maximaltemperatur K oder °C t Zeit t(Tmax.) Haltezeit bei Maximaltemperatur d U Auslenkungsparameter Å2 (=104 pm2) V Volumen der Elementareinheit Å3 (=106 pm3) Tc Curie-Temperatur K h (Stunden); min. (Minuten) - 17 - Verzeichnis der wichtigsten Abkürzungen und Symbole Symbol w wR2(F2) Dimension Wichtungsschema des Programms SHELXL[199] w-1 = σ2F02 + (aw * P)2 + bw * P mit P = (F02 + 2 FC2) / 3 Gewichteter Zuverlässigkeitsfaktor (verfeinert gegen Fo2) x, y, z, Atomkoordinaten x Homogenitätsbereich Z Anzahl der Formeleinheiten pro Elementarzelle CN Koordinationszahl (coordination number) HV Härte nach Vickers HK Härte nach Knoop HB Härte nach Brinell HRC / A* Härte nach Rockwell F Kraft (Force) A Fläche d Durchmesser mm D Kugeldurchmesser mm α, β, γ Winkel einer Elementarzelle ° α, β Winkel der Eindringkörper °, ` α- Tieftemperaturmodifikation β- Hochtemperaturmodifikation Extinktionskorrekturfaktor des Programms ε SHELXL[199] θ Röntgenbeugungswinkel ° λ Wellenlänge Å (=100pm) µ Absorptionskoeffizient mm-1 ρcalc Berechnete röntgenographische Dichte g·cm-3 σ Standardabweichung φ Diffraktometerwinkel ° ω Diffraktometerwinkel ° Ð Winkel Ø Durchmesser mm * HRC bzw. HRA sind die wichtigsten Härteverfahren nach Rockwell - 18 - Verzeichnis der wichtigsten Abkürzungen und Symbole Verzeichnis der verwendeten Abkürzungen Abkürzungen e- Elektron EZ Elementarzelle EK Einkristall RG Raumgruppe Gl. Gleichung k. A. keine Angabe HT- Hochtemperatur (high temperature) LT- Tieftemperatur (low temperature) M Übergangsmetall M’ Übergangsmetall hier τ-Stabilisator PSE Periodensystem der Elemente Ref. Referenz PXRD powder X-ray diffraction SXRD single-crystal X-ray diffraction REM Rasterelektronenemikroskop EDX energiedispersive Röntgenspektroskopie WDX wellenlängendispersive Röntgenspektroskopie SPS Spark Plasma Sintering DIN Deutsche Industrie Norm ISO Internationale Organisation für Standardisierung At.-% Atomprozent - 19 - - 20 - 1 Thema 1.1 Einleitung Die heutige Materialforschung befasst sich vertieft mit der Darstellung und Untersuchung von Verbindungen die aus Mehrkomponenten aufgebaut sind[1]. Ziel ist es dabei, die Werkstoffen, welche aus mehreren Komponenten bestehen, durch die Vereinigung von den charakteristischen Eigenschaften der einzelnen Stoffe zu kombinieren. Je nach Einsatzgebiet müssen die neuen Multifunktionswerkstoffe neben einer hohen Verschleißfestigkeit und Härte auch chemische und thermische Widerstandsfähigkeit aufweisen, sowie leicht und billig zugänglich sein. Zu den wichtigsten Werkstoffen in der Stahl- und Metallindustrie gehören Carbide, Nitride sowie Boride der Übergangsmetalle. Ihre mechanischen Eigenschaften sind durch den Zusatz von weiteren Legierungsbestandteilen gut steuerbar. Je nach Legierungselement werden Härte, Festigkeit, Streckgrenze, Dehnung, Kerbschlagzähigkeit, Elastizität, Wärmefestigkeit, Verschleißfestigkeit, Zerspanbarkeit, Korrosionsbeständigkeit sowie magnetische Eigenschaften beeinflusst[2]. Durch eine gut gewählte Kombination der Legierungselemente kann die Bildung von intermetallischen Phasen dazu beitragen, dass unerwünschte Eigenschaften minimiert oder sogar vollständig beseitigt werden. Nachdem lange Zeit versucht worden ist, den Verschleiß durch Veränderungen der Werkstoffeigenschaften zu vermindern, geht man in der letzten Zeit zunehmend zur Anwendung verschleißhemmender Schichten über. Das ternäre Borid Ni21M2B6 mit der Cr23C6 Struktur hat sich bereits aufgrund seiner Reibungs- und Verschleißresistenz für eine Oberflächenbeschichtung von Eisenwerkstoffen in der Industrie durchgesetzt[3,4]. Die τBoride der Zusammensetzung Co23-xMxB6 (M = ÜM) zeigen neben der hohen Härte[126] auch noch ferro- bzw. paramagnetische Eigenschaften[5,6]. Die MAX-Phasen1 überzeugen durch ihre Vielfalt an Eigenschaften und repräsentieren typische Hybridmaterialen. Hier sind die Eigenschaften von Keramiken und Metallen kombiniert. Somit zeigt diese Verbindungsklasse der MAX-Phasen Eigenschaften, wie einen typischen metallischen (elektrische und thermische Leitfähigkeit, duktil und elastisch auch bei hohen Temperaturen) und keramischen Charakter (hohe Härte, geringe Dichte, Oxidationsbeständigkeit und thermische Stabilität) in einer einzigen Verbindung vereinen. 1 MAX-Phasen sind Verbindungen mit der allgemeinen Zusammensetzung von Mn+1AXn (n=1, 2, 3) Thema In der Industrie kommt schon seit Jahren die Härteprüfung an Werkstoffe zum Einsatz, um die Eigenschaften wie Härte, Zugfestigkeit, Elastizitätsmodul und den Verschleiß mit Hilfe einer “zerstörungsfreien Methode“ zu ermitteln. Die Härteprüfung lässt sich als ein bequemes “Screening“ der mechanischen Eigenschaften bezeichnen. Vorteile dieser Analysemethode sind eine einfache und schnelle Durchführung. Durch Umwerten der gewonnenen Werte lassen sich Größen erhalten, die sonst nur durch aufwendige Experimente unter Zerstörung der Probe zugänglich wären. 1.2 Zielsetzung und Synthesemethoden 1.2.1 Ziele Das Ziel dieser Arbeit war die Synthese und eine möglichst umfangreiche Charakterisierung von MAX-Phasen und τ-Boriden sowie die Inbetriebnahme des neuen SPSOfens am Institut der Anorganischen und Analytischen Chemie in Freiburg. Zudem sollte ein Mikrohärtetester für Härtemessungen an Superhartstoffen betriebsam gemacht werden. In beiden Verbindungsklassen stand die Züchtung von Einkristallen hoher Güte und Qualität bei Hochtemperatursynthese im Vordergrund wie auch die Darstellung eines Sinterkörpers mit der Pulsplasmatechnik. Die Synthesebedingungen wurden in dieser Arbeit so optimiert, dass die erhaltenen Einkristalle für eine Härtemessung geeignet waren. Bei den MAX-Phasen mit der Zusammensetzung Mn+1AXn (n = 1, 2, 3) wurden die Einkristalle aus einer Aluminium/M (M = Ga, Ge, In, Sn) Schmelze bei Temperaturen von 1350°C bis 1650°C gezüchtet und charakterisiert. Aus den gewonnenen Erkenntnissen der Hochtemperatursynthese sollte eine Reproduktion der MAX-Phasen in Form eines Sinterkörpers nach einer neuen Synthesemethode im Pulsplasmaofen (SPS) erfolgen. Des Weiteren sollten Carbide aus den Verbindungsklassen der h-Carbide mit der Zusammensetzung M3Al2CoC (mit M = Ta und Nb) sowie κ-Carbide der Zusammensetzung M6-xM’xC (x = 2 – 4; M = W; M’ = Al, Co) im Pulsplasma Ofen synthetisiert werden. Bei der Untersuchung der τ-Boriden2 stand Kobalt als Hauptkomponente im Vordergrund. Als τ-Stabilisator3 wurde Iridium, Gallium, Aluminium, Zinn, Titan, Vanadium, Wolfram, Mangan, Molybdän und Magnesium eingesetzt. Hier wurden geeignete Einkristalle aus der Schmelze bei einer Temperatur von 1250°C bis 1500°C für eine detaillierte 2 Bei den τ-Boriden handelt es sich um Boride mit der allgemeinen Summenformel von M23-xM’xB6+y. Bei dem so genannten τ-Stabilisator handelt es sich um eine Hauptgruppen-, Seltenerd- oder frühem Übergangsmetall, dass auf einer speziellen Lage in der M23-xM’xB6+y -Struktur eingebaut wird. 3 - 22 - Thema Strukturaufklärung gezüchtet. Neben der ausführlichen Strukturanalyse der erhaltenen Verbindung wurde zudem der Homogenitätsbereich bezüglich des Borgehalts für die untersuchten Phasen geklärt. Frühere Untersuchungen haben gezeigt, dass eine Analyse von Einkristallen essentielll ist. 1.2.2 Charakterisierung Für eine detaillierte und vollständige Charakterisierung der dargestellten Verbindungen standen Analysetechniken wie Röntgenbeugung am Pulver (PXRD) und Einkristall (SXRD), energiedispersiver Röntgenemmissionsspektroskopie (EDX) sowie Härteprüfung (nach Vickers (HV) und Knoop (HK)) und eine Gefügeuntersuchung an Einkristallen und Sinterkörpern zur Verfügung. 1.2.3 Synthesemethoden Synthesen aus der Schmelze wurden im Silitrohrofen (bis 1450°C), im Korundrohrofen (bis 1650°C) sowie im Graphitofen (bis 1650°C) aus den Elementen in Korundtiegeln unter Argonschutzgas durchgeführt. Für die Darstellung von Sinterkörpern wurde ein Pulsplasmaofen (bis 2000°C) eingesetzt. Hier wurden die Elemente in einer Graphitmatrize unter Druck und Vakuum bzw. Argonschutzgas umgesetzt. - 23 - - 24 - 2 Methoden 2.1 Härtemessung 2.1.1 Einleitung Mit dem Fachbegriff Härte assoziieren wir heute eine Vorstellung über die Verwendungsfähigkeit eines Gegenstandes. Mit einem harten Gegenstand verbinden wir Eigenschaften wie Verschleißfestigkeit oder die Möglichkeit andere Gegenstände zu verformen. Eine allgemeine Formulierung für die Härte, die in der gesamten Technik Gültigkeit besitzt, lautet: „Härte“ ist der mechanische Widerstand, den ein Körper dem Eindringen eines anderen Körpers entgegensetzt. Diese Formulierung ist jedoch keineswegs eindeutig, da keine Angaben gemacht werden, in welcher Form der Körper eindringt. Die Erkenntnis „harte Stoffe deformieren weiche“, hat Friedrich Mohs schon Anfang des 18. Jahrhunderts gemacht. Somit war seine Überlegung der Grundstein für viele Methoden der Härteprüfung, um Gegenstände, Materialien und Werkstoffe nach ihrer Deformationsfähigkeit einzuteilen. Der Mineraloge Mohs ritzte verschiedene Minerale gegeneinander und ordnete sie nach ihrer Härte. Die Zuordnung der Härte wurde auf einer einheitslosen Skala die von dem Mineral Talk mit der Härte von 1 bis zu dem Edelstein Diamant der Härte 10 reichte, eingeteilt. Die sogenannte Mohs-Skala, die heute noch Anwendung findet, ist in Abbildung 2.1.1 dargestellt. Die dargestellte Mohs-Skala mit allen dazugehörigen Mineralien ist hier mit Vickers-Skala gegenüber gestellt, die heute einen größeren Stellenwert in der Industrie besitzt. Abbildung 2.1.1: Vergleich der Mohs-Skala (mit den dazugehörigen Mineralien) mit der Vickers-Skala In der heutigen Zeit ist die Mohs-Skala zu ungenau, in der Werkstoffkunde sind absolute Werte von Nöten. Jedoch ist die damals entwickelte Härte-Skala nach Mohs immer noch ein guter Anhaltspunkt für die Einteilung von Werkstoffen und Keramiken und somit ein sehr guter Vergleichswert. - 25 - Methoden - Härtemessung In der Werkstoffprüfung werden heute vor allem Prüfverfahren eingesetzt, welche Eindringhärten messen. So kommen genormte Prüfkörper zum Einsatz, die unter festgelegten Bedingungen in der zu prüfenden Probe einen bleibenden Eindruck hinterlassen. Im Anschluss wird die Oberfläche oder die Tiefe des bleibenden Eindruckes gemessen. Grundsätzlich unterscheidet man heute die Härteprüfverfahren nach der Art, wie die Belastung auf den Prüfkörper aufgebaut wird. Man spricht von statischen oder dynamischen Härteprüfverfahren. Die dynamischen Prüfverfahren bringen die Belastung des zu prüfenden Teiles schlagartig auf. Bei den statischen Verfahren ist die Belastung gleichbleibend oder allmählich zunehmend. 2.1.2 Die verschiedenen Messverfahren Wie schon oben erwähnt, wurde die erste Unterteilung und Klassifizierung von Mineralien durch den Mineralogen Mohs im Jahre 1811 vorgenommen. Hier wurde mit dem Ritzhärteprüfverfahen nach dem Prinzip „Harte Stoffe ritzen weiche Stoffe“ eine Einteilung in zehn Stufen vorgenommen. Brinell stellte auf der Weltausstellung in Paris das Kugeldruckverfahren zur Bestimmung der Härte vor[7]. Dieses noch heute beliebte und angewandte Härteprüfverfahren war der Auslöser für viele weitere Härtemessungsmethoden, die sich vom Brinellprüfverfahren ableiten lassen. Ludwik präsentierte nur kurz darauf den Kegeldruckversuch und im etwa gleichen Zeitraum wurde das Rücksprungverfahren nach Shore veröffentlicht. Im Jahre 1922 fand auch das Härteprüfverfahren nach Rockwell Einzug in die Industrie. Nur drei Jahre später wurde die Vickers- Pyramide vorgestellt und als Universalhärteprüfverfahren etabliert. Für die Bestimmung der Härte an Einkristallen und Untersuchung der Gefügebestandteile sind die Eindring- Härteprüfverfahren von Interesse. Da Härtewerte über 9 Mohs nur sehr schlecht oder gar nicht dokumentiert sind. Aus diesem Grund werden in dieser Arbeit nur die Eindring- Härteprüfverfahren nach Vickers und Knoop angewendet und detailliert vorgestellt. Abbildung 2.2 zeigt die heutigen Härteprüfverfahren unterteilt nach den statischen und dynamischen Methoden. Rot unterlegt sind die Verfahren, die für eine Härteprüfung an Einkristallen und sehr dünnen Schichten geeignet sind. - 26 - Methoden - Härtemessung Härteprüfverfahren Statische Härteprüfverfahren Verfahren mit Messungen unter Prüfkraft Universalhärte HU # Verfahren mit Umwertung Verfahren mit Messung nach Wegnahme der Prüfkraft Verfahren mit Energiemessung Equotip# # UCI HBT# mit # gekennzeichnet sind Härteprüfverfahren die nicht genormt sind Dynamische Härteprüfverfahren Härtewert definiert als Quotient aus Prüfkraft und Eindruckoberfläche Härtewert definiert als Quotient aus Prüfkraft und Projektionsfläche des Eindrucks Härtewert definiert mit Hilfe der Eindringtiefe Brinell Vickers (Makro, Kleinlast und Mikro) Knoop Rockwell A, B, C, D, E, F, G, H, K Rockwell 15 N, 30 N, 45 N Rockwell 15 T, 30 T, 45 T Rockwell Bm, Fm, 30Tm Rockwell HR 10/3000, HR 10/1000, HR 5/750 HR 5/250 Abbildung 2.1.2 Flussdiagramm und Übersicht der Härteprüfverfahren - 27 - Methoden - Härtemessung 2.1.3 Die physikalische Einheit bei der Härteprüfung Die in der Härteprüfung angegebenen Einheiten sind durch die DIN-Normen vorgeschrieben und leiten sich aus den einzelnen Härteprüfverfahren ab. 1 N = 1 kg · 1 m/s2 Formel 2-1 Die Definition eines Newtons ist somit die Kraft, die eine Masse von 1 Kilogramm mit der Geschwindigkeit von 1m/s2 beschleunigt. Oft gebräuchlich für die Kraft ist auch das Kilopond kp, definiert als die Kraft, die eine Masse von 1 Kilogramm mit der Erdschwerebeschleunigung von 9,80620 m/s2 (auf dem 45. Breitengrad) beschleunigt. 1 kp = 1 kg · 9,80665 m/s2 Formel 2-2 1 kp = 9,80665 N Formel 2-3 1 N = 0,101972 kp Formel 2-4 somit Für die Härteprüfung von Werkstoffen (hier nicht Kunststoffe) wurde zur zahlenmäßigen Aufhebung der Umrechnung der Prüfkraft in Newton ein Korrekturfaktor von 0,102 nach Gl. 2.4 eingeführt. Somit wurde die Härte H in die Gl. 2.5 zusammengefasst. H = 0,102 ⋅ F A Formel 2-5 F = Force (Kraft) A = Area (Fläche) Die Angabe der Härte wird als dimensionslose Zahl angegeben. Abbildung 2.1.3 zeigt eine Übersicht der wichtigsten Härteprüfmethoden und einen Vergleich der Härteskalen. Die Härteskala nach Vickers ist diejenige, die als Universelle Härteskala beschrieben wird, da sie das ganze Spekturum der bekannten Stoffe abdeckt. - 28 - Methoden - Härtemessung Brinell HB 75 Rockwell HRF 300 85 Rockwell HRB 630 115 50 100 Rockwell HRC 20 Rockwell HRA 70 65 Vickers HV 100 94 200 400 600 800 1000 1400 2000 10000 8 Topas 9 Korund Mohshärte 1 Speck- 2 Gips 3 Calcit 4 Flußspat stein 5 Apatit 6 Feldspat 7 Quarz Abbildung 2.1.3: Härteskalen Übersicht. 2.1.4 Zusammenhang von Härte und Probeneigenschaften Tatsache ist, die Härte beschreibt die Verformbarkeit eins Werkstoffs. Jedoch kann diese abhängig von der Probenzusammensetzung die Verformbarkeit einer ganzen Probe umfassen oder auch nur in unterschiedlicher Weise ortsabhängig in einer Probe geschehen. Somit sind Werkstoffe in unterschiedliche Gruppen aufzuteilen. Eine erste wichtige Unterteilung ist in amorphe und kristalline Werkstoffe vorzunehmen. Bei amorphen Werkstoffen hängen die Eigenschaften im Wesentlichen vom Molekülaufbau, der Molekülstruktur im Werkstoff, sowie von der Menge und Art etwaiger Fremdbeimischungen ab. Bei den kristallinen Werkstoffen sind die Eigenschaften der Werkstoffe hauptsächlich vom Aufbau der Elementarzelle und somit von den Atomen auf ihren festen Kristallgitterpositionen und den dazugehörigen Bindungen abhängig. Je nach Bindungsart ist der Beitrag zur Härte des gesamten Werkstoffs größer oder geringer. Bei mikrokristallinen Stoffen ist die Verteilung der Kristalle und ihrer Lagen statistisch in alle Richtungen gleich häufig verteilt. Somit sind die Werkstoffeigenschaften von kristallinen Stoffen in der Regel richtungsunabhängig. Bearbeitungsmethoden wie Walzen oder Gießen - 29 - 10 Methoden - Härtemessung können Einfluss auf die Mikrostruktur nehmen und die Kristalle in eine Vorzugsrichtung orientieren. In solchem Fall verleiten wir dem Werkstoff richtungsabhängige Werkstoffeigenschaften. Kristalline Werkstoffe können je nach Zusammensetzung und Größe der beteiligten Atome oder Fehlbesetzungen, Lehrstellen sowie Mischbesetzungen aufweisen, dieses können die Ursachen für eine Abnahme der Härte sein. 2.1.5 Härteprüfung nach Brinell Die Bestimmung der Brinell-Härte[8] HWB (HB alte Schreibweise) wurde im Jahr 1900 von Johan August entwickelt[9] und war lange Zeit die einzige Härteprüfung, die eindeutig reproduzierbare Messergebnisse lieferte[9]. Bei diesem heute genormten Verfahren (DIN EN ISO 6506-1:2005) wird eine gehärtete Stahl- oder Hartmetallkugel vom Durchmesser D (1 / 2,5 / 5 oder 10 mm) mit der Last F in das Werkstück eingedrückt. Mit der gewählten Prüflast F wird für eine definierte Einwirkdauer (10 -15 oder max. 30 Sekunden) in die zu untersuchende Probe belastet. Der Härtewert nach Brinell ergibt sich als Quotient aus der Prüflast F zur Oberfläche A(Kugelkalotte) des hinterlassenen Eindrucks (Kugelkalotte). Die Probendicke soll mindestens das Zehnfache der Eindringtiefe betragen, damit der Eindruckdurchmesser zwischen dem 0,25- und dem 0,6fachen des Kugeldurchmessers liegt. Für die Ermittlung des Durchmessers d werden zwei Abmessungen d1 und d2 vorgenommen, diese werden anschließend zu d gemittelt[9,10]. Abbildung 2.1.4: Härteprüfung nach Brinell Abbildung 2.1.5: Brinell Eindruck in der Aufsicht - 30 - Methoden - Härtemessung Die Brinell-Härte (HBW) wird für eine idealisierte Eindruckgeometrie mit Hilfe des Eindruckdurchmessers berechnet, gemäß der Formel: HBW ( S ) = k⋅F A( Kugelkalotte ) = 0,102 ⋅ F 0,102 ⋅ 2 ⋅ F = A( Kugelkalotte ) π ⋅ D D − D 2 − d 2 [ ( )] Formel 2-6 Der Härtekennwert wird in der folgenden Form angegeben: 5 Kennzeichnung für Die Härte nach Brinell Kugeldurchmesser in mm / 62,5 / 30 Einwirkdauer der Prüfkraft in Sekunden HBW Prüfkraft F in kp* 185 Härtewert Tabelle 2.1.5.1: Angabe des Härtewertes nach Brinell (*3000kp = 29430N) 2.1.5.1 Fehlerquellen bei der Härtemessung nach Brinell Werkstoffbedingte Fehler Eine der größten Fehlerquellen ist sicherlich auf die elastische Rückstellung zurückzuführen. Der Krümmungsradius des rückverformten Eindrucks kann das Dreifache des verwendeten Kugeldurchmessers betragen[10]. Das zweite Problem bringt der kaltverfestigte Zustand mit sich. Dieser führt nicht nur zu einer Veränderung der plastischen Fließvorgänge in der Probe, sondern auch zu einer Wallbildung um die Eindruckränder. Die Folge ist das Einsinken des gesamten Abdrucks unter das Niveau der Bezugsfläche. Somit führt dieses zu einem größeren Härtewert. Von geringerer Bedeutung aber nicht immer vernachlässigbar spielen Korngröße, inhomogene Härteverteilung und das Fließvermögen eine Rolle. In der Tabelle 2.1.5.1 sind die wichtigsten Merkmale für die Härteprüfung nach Brinell zusammengefasst. - 31 - Methoden - Härtemessung Tabelle 2.1.5.2: Härtemessung nach Brinell Anwendungsbereiche Für alle metallischen Werkstoffe bis zu einer Härte von 650 HBW Eindringkörper Kugel (D = 1, 2, 2,5, 5 oder 10 mm) HBW Hartmetallkugel (E-Modul ≥ 600.000 N/mm2) HBS gehärteter Stahl ( ≥ Härte – 850 HV 10) Prüfkraft Abhängig von Kugeldurchmesser Aufbringzeit 2 bis 8 sec. stoß- und schwingungsfrei Einwirkdauer 10 – 15 sec. (max. 30 sec.) Probenfläche Sauber und frei von fremden Materialien Probengröße Probenoberfläche 3 · Eindruckdurchmesser Probenmindestdicke 10faches der Eindringtiefe der Prüfkugel Abstand zweier Mindestens das Vierfache des Kugeldurchmesser (4 · d) bei Stahl, benachbarter Grauguss, Kupfer und Kupferlegierungen Eindrücke Mindestens das Sechsfache des Kugeldurchmesser (6 · d) bei Leichtmetallen, Blei, Zinn und deren Legierungen Abstand des Mindestens 2 · d für Stahl, Grauguss, Kupfer und Kupferlegierungen und Eindrucks vom bei Leichtmetallen, Blei, Zinn und deren Legierungen sollte das Dreifache Probenrand des Kugeldurchmessers berücksichtigt werden. Messung des Es werden zwei Diagonalen d1 und d2 vermessen und anschließend einer zu Eindrucks d gemittelt (Abweichung der zwei Diagonalen sollte nicht mehr als 5% betragen) Definition des HBS bzw. HBW = 0,102 ⋅ Härtewertes ( 2⋅F π ⋅ D ⋅ D − D2 − d 2 ) D = Kugeldurchmesser d = Eindruckdurchmesser Kurzzeichen 380 HB S(W) 10 / 470 / 15 Einwirkdauer in sec. Kennziffer für die Prüfkraft Kugeldurchmesser D Art des Prüfkörpers und Kennbuchstaben für die BrinellHärte S = Stahlkugel W = Hartmetallkugel Härtewert - 32 - Methoden - Härtemessung 2.1.6 Härteprüfung nach Vickers Im Jahre 1925 entwickelten Smith und Sandland die Vickers-Pyramide[11] und benannten diese nach der britischen Flugzeugbaufirma Vickers[12]. Die Einführung eines Prüfkörpers aus einem Diamanten mit spezieller Geometrie sollte die Hauptnachteile des Brinell-Verfahren vermeiden (die wechselnde Geometrie des Eindrucks und die mögliche Verformung der Stahl- bzw. Hartmetallkugel). Der Prüfkörper besteht aus einer regelmäßigen Diamantpyramide mit quadratischer Grundfläche und einem Flächenwinkel von 136(±2)°. Der spezielle Winkel von 136° beruht auf der Erfahrung, dass der Eindrucksdurchmesser beim Brinell-Verfahren üblicherweise zwischen 0,25- und 0,5fachen der Kugeldurchmesser beträgt. Somit resultiert ein mittleres Durchmesserverhältnis von 0,375. Anlegen von Tangenten an den Berührungskreis ergibt ein Öffnungswinkel von 136°[9,10]. Die Abbildung 2.1.6 zeigt die Beziehung zwischen dem Prüfverfahren nach Vickers und Brinell. Abbildung 2.1.6: Darstellung der Ableitung der Vickers- Messmethode aus der Brinell- Messmethode Das Vickers-Verfahren ist das Zweite von den drei genormten Härteprüfverfahren (DIN 50 133)[8]. Das Vickers-Härteprüfverfahren ist eine Weiterentwicklung Härteprüfverfahrens. Somit sind Durchführung und Auswertung sehr ähnlich. - 33 - des Brinell- Methoden - Härtemessung Zur Bestimmung der Härte nach Vickers wird die Prüflast F stoßfrei in ca. 10 Sekunden aufgebracht und für eine definierte Einwirkdauer üblicherweise 10 -15 Sekunden, gehalten. Längere Haltezeiten müssen in der Härtekennzahl angegeben werden. Abbildung 2.1.7: Härteprüfung nach Vickers Abbildung 2.1.8: Vickers- Abdruck in der Aufsicht Für die Ermittlung der Härtekennziffer wird die Prüfkraft auf die Kontaktoberfläche des verbleibenden Eindruckes bezogen. Somit ist die Härte wieder definiert als Quotient aus der Prüfkraft und dem bleibenden Eindruck nach Wegnahme der Kraft. HV = 0,102 ⋅ F A Formel 2-7 Der bleibende Abdruck ist eine geometrisch getreue Abbildung des Eindringkörpers. Somit lässt sich die Fläche aus den beiden Diagonalen ermitteln. A= d2 d2 = 136° 1,854 2 ⋅ sin 2 mit d= d1 + d 2 2 Beim Einsetzen der Gleichung 2-8 in die Gleichung 2-7 folgt ein neuer Vorfaktor - 34 - Formel 2-8 Methoden - Härtemessung HV = 0,120 ⋅ 1,854 ⋅ F 0,1891 ⋅ F = d2 d2 Formel 2-9 Sollten sich die beiden vermessenen Diagonalen bei isotropen Materialien um mehr als 5% unterscheiden, ist die Messung zu wiederholen. Immer häufiger wird die Vickershärte in GPa (Gigapascal) angegeben. GPa ist eine Druckangabe und steht somit im Zusammenhang mit der Kraft pro Fläche. p= N kg Pa = m 2 ≡ m ⋅ s 2 F A Formel 2-10 Für eine Angabe in Gigapascal [GPa] muss die Umrechnung der Einheiten erfolgen, somit wird die Prüfkraft in kg und die hinterlassenen Diagonalen in mm in die Gleichung 1-10 eingesetzt und die Prüflast wird in kg angegeben. Nach Umrechung der Einheiten lässt sich die Vickershärte als GPa angeben. HV = 0,018191 ⋅ F d2 (GPa) Formel 2-11 Der Härtewert wird angegeben gemäß: Kennzeichnung für Die Härte nach Vickers 5 / 30 Einwirkdauer der Prüfkraft in Sekunden HV Prüfkraft in N 145 Härtewert Tabelle 2.1.6.1: Angabe des Härtewertes nach Vickers Der wesentliche Vorteil der Vickershärteprüfung gegenüber der Brinellhärteprüfung ist die universelle Anwendbarkeit. Die spezielle Pyramidengeometrie sorgt dafür, dass die Eindringtiefe nur 1/7 der Diagonallänge beträgt. Somit eignet sich die Härteprüfung nach Vickers bei Variation der Prüflast für einen weiten Bereich der an Werkstoffwerten und sogar - 35 - Methoden - Härtemessung zur lokalen Charakterisierung einzelner Gefügebestandteile und Einkristalle. Die Möglichkeit zur Variation der Prüflast unterteilt das Vickers-Verfahren in drei Bereiche[9]. 1. Makro-Härtemessung: Prüfkraft > 49,03 N 2. Kleinlast-Härteprüfung: Prüfkraft 3. Mikro-Härteprüfung: Prüfkraft < 1,961N 1,961 N bis 49,03 N Bei der Härtebestimmung im Kleinlastbereich und insbesondere im Mikrobereich steigt die Vernachlässigbarkeit der Prüflast. Mit abnehmender Prüflast nimmt die Härte zu, verantwortlich für diesen Effekt ist die Kaltverfestigung in der Randzone. 2.1.6.1 Fehlerquellen bei der Härtemessung nach Vickers Da sich das Vickers-Härteprüfverfahren vom Brinell-Härteprüfverfahren ableitet, entsprechen die Fellerquellen denen der Brinell- Härtemessung. Es kommt zu einer Wallbildung und zu einem Einsinken der Eindrücke bei nichtverfestigten Proben. Zusätzlich können konkave oder konvexe Eindruckkanten beobachtet werden. Eine Fehlerquelle, die das Vickers- Härteprüfverfahren mit sich bringt, liegt in der möglichen Formungenauigkeit der Diamantpyramide, was sowohl den Flächenwinkel als auch den Schliff betreffen kann. - 36 - Methoden - Härtemessung Tabelle 2.1.6.2: Härteprüfung nach Vickers (HV) DIN EN ISO 6507-1. Anwendungsbereiche Alle Metalle sowie spröde Materialen wie Keramiken, Sinterwerkstoffe und Einkristalle. Auch Einsatz für Makro- und Mikrohärtemessungen. Eindringkörper quadratische Pyramide aus Diamant Flächenwinkel α = 136° ±30’ Kantenwinkel β = 148° ±15’ Eindringtiefe 1/7 der Länge der längeren Diagonalen Prüfkraft Makrobereich F ≥ 49,03 N Kleinlastbereich F = 49,03 N bis 1,961 N Mikrobereich F ≥ 1,961 N Aufbringzeit 2 bis 8 sec. stoß- und schwingungsfrei Einwirkdauer 10 bis 15 sec. Probenfläche Sauber und frei von fremden Materialien, blank und glatt Abstand zweier Mindestens das Dreifache der Diagonalen bei Stahl, Keramik und benachbarter Hartstoffen. Eindrücke Mindestens das Sechsfache der Diagonalen bei Leichtmetallen, Blei, Zinn und deren Legierungen Abstand des Eindrucks 2,5·d für Stahl, Keramik und Hartstoffe und 3·d für Leichtmetalle, vom Probenrand Zinn, Blei, und Kunststoffe Messung des Die Messung erfolgt nach Entlastung. Die beiden Eindruckdiagonalen Eindrucks des bleibenden Eindrucks d1 und d2 werden vermessen und das arithmetische Mittel d wird gebildet. Definition des Härtewertes Kurzzeichen HV = 0,1891 ⋅ F d2 bzw. HV = 0,018191 F (GPa) d2 1900 HV 1 / 20 Einwirkdauer in sec. Kennziffer für die Prüfkraft 1 entspricht 9,807 N Kennbuchstaben für die Vickershärte Härtewert - 37 - Methoden - Härtemessung 2.1.7 Härteprüfung nach Knoop Die nicht genormte Härteprüfung nach Knoop[13] wurde 1939 von dem Physiker und Ingenieur Frederick Knoop vorgestellt[14,15]. Das Härtemessverfahren nach Knoop stellt ein Sonderverfahren dar, abgeleitet von der Härteprüfung nach Vickers. Auch hier wird der Härtewert als Quotient aus der Prüfkraft und der Projektionsfläche des bleibenden Eindrucks definiert. Diese Härteverfahren unterscheiden sich im Wesentlichen nur in der Form des Eindringkörpers. Die in der Vickershärteprüfung gleichseitige Diamantenspitze besitzt in der Knoophärteprüfung eine rhombische Form. Der Längskantenwinkel beträgt 172°30’ und der Querkantenwinkel 130°. Der verbleibende Abdruck ist sehr flach und seine Tiefe beträgt gerade nur 1/30 der langen Diagonalen. Die besondere Geometrie des Eindringkörpers erlaubt Messungen an dünnen Schichten und ist somit hervorragend geeignet für Untersuchungen von Gefügebestandteilen und Einkristallen. Die Geometrie des Prüfkörpers vermindert die Bruchgefahr und stellt dadurch die genauste Messmethode bei der Untersuchung von spröden Materialen da[14,16]. Das Diagonalenverhältnis beträgt 7,11:1, dieser Charakter erlaubt das Platzieren viele Eindrücke nebeneinander ohne den Mindestabstand vom 2.5fachen der Diagonalenlänge einhalten zu müssen. Anders als bei Brinell- und Vickers-Verfahren wird für die Ermittlung der Härtekennziffer nur die lange Diagonale vermessen. Abbildung 2.1.9 zeigt einen Knoop- Abdruck und die zu vermessene Diagonale. 512 HK1 Abbildung 2.1.9: Resultierender Knoop- Abdruck und Messstellen Die Knoopkennziffer HK (oder auch KHN = KNOOP Hardness Number) wird aus der Projektionsfläche und somit aus der längeren Diagonalen d1 ermittelt. Die Projektionsfläche lässt sich somit zusammenfassen: - 38 - Methoden - Härtemessung A= tan β 2 tan 2 ⋅ d 2 = 0,07028 ⋅ d 2 mit α = 172° 30’ und β = 130° 0’ Formel 2-12 α 2 Für die Ermittlung der Knoophärte muss der Faktor von 0,102 für die Einführung des Newtons berücksichtigt werden. HK = 0,102 ⋅ F F = 1,451 2 2 0.07028d d Formel 2-13 Tabelle 2.1.7.1: Abkürzungen und Prüfkräfte bei der Härteprüfung nach Knoop Kurzzeichen Prüfkraft [N] Kurzzeichen Prüfkraft [N] HK 0,01 0,09807 HK 0,3 2,942 HK 0,02 0,1961 HK 0,5 4,903 HK 0,25 0,2451 HK 1 9,807 HK 0,05 0,4903 HK 0,1 0,9807 HK 0,2 1,961 In der Tabelle 2.1.7.1 sind die Kurzzeichen und die dazugehörigen Prüfkräfte nach ISONorm zusammengestellt. Das Knoop-Härtemässverfahren ist genau wie die anderen bis jetzt vorgestellten Härteprüfverfahren abhängig von der Prüflast. Die Diagonalen werden jedoch unterschiedlich stark von der elastischen Rückstellung beeinflusst. Die kurze Diagonale unterliegt der elastischen Rückstellung beträchtlich. Während die lange Diagonale, wenn überhaupt, nur einer sehr kleinen Längenänderung unterworfen wird. Somit lassen sich zudem wichtige Informationen über das elastische Verhalten des zu untersuchenden Materials gewinnen, wenn die kurze Diagonale betrachtet wird. Das Härteprüfverfahren nach Knoop ist unter anderem auch dadurch beliebt, weil es die Möglichkeit zur Erkennung der Werkstoffanisotropie bietet! Wie auch beim Vickers-Messverfahren wird die Probenoberfläche nur in einem sehr geringen Ausmaß beschädigt. Einer der größten Fehler bei der speziellen Härteprüfung entsteht durch Nichterkennung der Werkstoffanisotropie. - 39 - Methoden - Härtemessung Tabelle 2.1.7.2: Charakterisierung der Knoophärteprüfung Anwendungsbereiche Spröde Materialen wie Keramiken, Sinterwerkstoffen und Einkristallen ebenfalls für alle Metalle. Bei der Härtebestimmung von Schichtsystemen, ist dies die genausteMessmethode. Eindringkörper rhombische Pyramide aus Diamant Längskantenwinkel α = 172° 30’ Querkantenwinkel β = 130° 0’ Diagonalenverhältnis d1 : d2 = 7,11 : 1 Eindringtiefe 1/30 der Länge der längeren Diagonalen Prüfkraft von 0,09807 N (HK 0,01) bis 9,807 N (HK 1) Aufbringzeit 2 bis 10 sec. stoß- und schwingungsfrei Einwirkdauer 10 bis 15 sec. Probenfläche Sauber und frei von fremden Materialien Abstand zweier Mindestens das Dreifache der kürzeren Diagonalen (3·d2) bei Stahl, benachbarter Keramik und Hartstoffen Eindrücke Für Leichtmetalle und Kunststoffe sollte ein Mindestabstand vom Sechsfachen der kürzeren Diagonalen (6·d2) eingehalten werden Abstand des Eindrucks 2,5·d2 für Stahl, Keramik und Hartstoffe vom Probenrand und 3·d2 für Leichtmetalle, Zinn, Blei und Kunststoffe Messung des Es wird nur die lange Diagonale d1 vermessen. Eindrucks Definition des Härtewertes Kurzzeichen HK = 0,102 ⋅ F F = 1,451 2 2 0.07028 d d 1208 HK 0,2/10 Einwirkdauer in sec. Kennziffer für die Prüfkraft 0,2 entspricht 1,961 N Kennbuchstaben für die Knoophärte Härtewert - 40 - Methoden - Härtemessung 2.1.8 Mikrohärtemessung: Vickers vs. Knoop Vergleicht man die Abdrücke, die durch die Eindringkörper hinterlassen werden, lassen sich folgende Aussagen machen: • Der Vickers-Eindringkörper dringt etwa doppelt so tief ein wie der Knoop Eindringkörper • Der Vickers-Eindringkörper hinterlässt nur etwa 1/3 der langen Diagonalen des Knoop-Eindringkörpers • Die Vickershärteprüfung ist im Allgemeinen weniger beeinflussbar durch die Oberfläche als die Knoophärteprüfung • Die Vickershärteprüfung zeigt größere Empfindlichkeit gegenüber Messfehlern als die Knoophärteprüfung • Die Vickershärteprüfung eignet sich besser für abgerundete Oberflächen als die Knoophärteprüfung • Die Knoophärteprüfung eignet sich besser für Schichtsysteme und kleine längliche Bereiche als die Vickershärteprüfung • Die Knoophärteprüfung eignet sich besser für sehr harte und spröde Werkstoffe und sehr dünne Prüfflächen Abbildung 2.1.11: Härteprüfung nach Vickers Abbildung 2.1.10: Härteprüfung nach Knoop Abbildung 2.1.10 und 2.1.11 zeigen die Geometrie der Prüfkörper und die hinterbliebenen Abdrücke nach Wegnahme der Prüflast. - 41 - Methoden - Härtemessung Kleinlast- und Mikrohärteprüfungen werden im industriellen Bereich überwiegend nach der Vickersprüfmethode durchgeführt. Zunehmend gewinnt das Knoop-Prüfverfahren an Bedeutung. Will man beide Verfahren miteinander vergleichen, so spielt die Eindringtiefe die größte Rolle. Die Eindringtiefe bei Verwendung des Knoopdiamanten ist 50% geringer als bei der Vickerspyramide bei Verwendung gleicher Last[17,18]. Nach umfangreichen Untersuchungen konnte die Beziehung von HV 0,2 ↔ HK 0,5 und HV 0,3 ↔ HK 1,0 aufgestellt werden. 2.1.9 Umwerten von Härtewerten Die Praxis verlangt oft die gemessenen Werte aus der Vickershärteprüfung als eine Angabe in SI-Einheiten oder GPa bzw. MPa. Jedoch erhält man die Vickershärte als eine einheitslose Angabe (ohne die Einheit von kp/mm2). Der erhaltene Härtewert ist bei der Vickershärteprüfung eine Funktion der verwendeten Prüflast. Diese Tatsache kann zur Verwirrung führen. Je nach Einsatzgebiet ist die Variation der Prüfkraft von 1 kp bis 100 kp möglich. Für die Umwertung der Vickershärtezahl ist es notwendig, die Prüflast von Kilopond [kp] in Newton [N] und die Fläche von mm2 in m2 umzuwandeln. Das resultierende Ergebnis ist die Einheit in Pascal [Pa]. Das Umwerten ist in der Praxis von großer Notwendigkeit, denn nur so gelangt man zur aussagekräftigen Zugfestigkeit. Um aus der Härteangabe des (HV) zu Megapascal (MPa) zu gelangen, muss mit dem Faktor 9,807 multipliziert werden. Die Umwertung von HV in GPa erfolgt analog durch die Multiplikation mit dem Faktor 0,009807. Die Tabelle 2.1.9.1 soll ein Gefühl für die Handhabung der Härtewerte vermitteln. Tabelle 2.1.9.1: Zusammenhang von HV, MPa und GPa Nr. HV MPa aus (HV) GPa aus (HV) Beispiel 1 100 980,7 0,98 CaF2 2 500 4904 4,90 Ca[(F,Cl,OH)(PO4)3] 3 1000 9807 9,81 SiO2 4 2000 1,96x10-4 19,61 α-Al2O3 5 3000 2,92x10-4 29,42 B4C 6 5000 4,90x10-4 49,04 c-BN 7500 -4 73,55 C (Diamant) 7 7,36x10 - 42 - Methoden - Härtemessung Die Abbildung 2.1.12 zeigt die Stellung der unterschiedlichen Hartstoffe. R. Riedel[19] präsentiert in der Gegenüberstellung von unterschiedlichen Stoffklassen die härtesten Konkurrenten beim Wetteifern um das härteste Material. Der Diamant ist mit 7575 kp/mm2 immer noch ungeschlagen als der härteste Werkstoff, der uns bekannt ist. Das kubische Bornitrid besetzt den zweiten Platz mit einer Härte von 4700kp/mm2. In jüngerer Zeit wurden für Übergangsmetallbordide der 5d-Metall (ReB2[20], OsB2[21]) ebenfalls außergewöhnliche hohe Härtewerte bestimmt. Abbildung 2.1.12: Gegenüberstellung der Mikrohärte einiger bekannter Verbindungen/Elemente[19] - 43 - Methoden - Hilfsmetallbadtechnik 2.2 Hilfsmetallbadtechnik Bei der Hilfsmetallbadtechnik handelt es sich um ein präparatives Verfahren, bei dem die Synthese in einer Metallschmelze durchgeführt wird. Das Metall fungiert hier als gemeinsames Fluxmittel für die eingesetzten Edukte. Das flüssige Metall kann, muss aber nicht, an der Reaktion beteiligt sein. Arbeiten in einem flüssigen Medium bringt viele Vorteile, wie eine Erhöhung der Reaktivität der Edukte, schnellere Diffusionsprozesse und die Minimierung der inneren Spannungen der entstehenden Produkte. Ein weiterer Vorteil ist die Möglichkeit die Reaktionstemperatur gegenüber den klassischen Festkörperreaktionen deutlich zu senken. Bei der Züchtung von Einkristallen ist die Metallschmelze eine sehr beliebte Methode um Einkristalle guter Güte und hoher Qualität zu züchten. Metalle mit vergleichsweise niedrigen Schmelzpunkten und weitern Flüssigkeitsbereichen bei einem möglichst geringen Dampfdruck kommen für eine Anwendung als Hilfsmetallbad in Frage. Zudem sollte eine gute Löslichkeit der Edukte im ausgewählten Reaktionsmedium vorliegen. Außerdem darf das Hilfsmetall mit dem Tiegel und der Reaktionskammer nicht in Reaktion treten. Als Lösungsmittel sollte sich das Medium inert gegenüber den Reaktanden verhalten, mit der Ausnahme, bei der das Reaktionsmedium im Überschuss als Edukt eingesetzt wird. Falls möglich sollte die Löslichkeit mit der Temperatur kontinuierlich abnehmen, um hochwertige Einkristalle guter Quantität zu erhalten. Für eine erfolgreiche Isolierung von den aus der Schmelze gewonnen Einkristallen sollte sich das Hilfsmetall relativ schnell und schonend abtrennen lassen. Anlässlich der geforderten Eigenschaften für das Arbeiten in einer Metallschmelze ist die Auswahl eingeschränkt. Die Metalle, die dafür in Frage kommen, befinden sich überwiegend in der Gruppe der Hauptgruppenmetalle IIIa (Al, Ga, In, Tl). Als ein weiteres Metall hat sich Zinn aus der IVa Gruppe als Hilfsmetall bewährt. Auch ein Gemisch aus zwei (oder mehreren) Metallen ist denkbar. So lässt sich das Angebot der zu Verfügung stehenden Lösungsmittel beträchtlich erweitern. Hierfür sollten die Phasendiagramme der eingesetzten Systeme herangezogen werden. Leider besteht da die Gefahr der Bildung weitere Phasen. Somit ist genaue Kenntnis der Systeme notwendig, um das Spektrum an unerwünschten Nebenprodukten zu reduzieren. - 44 - Methoden - Hochtemperatursynthesen 2.3 Hochtemperatursynthesen Die Edukte wurden zunächst in einem Achat-Mörser homogenisiert und mit Hilfe einer Handpresse zu Tabletten verdichtet. Die Reaktionsmischung wurde in Korundtiegeln platziert, diese in Korundschiffchen und diese in der Mitte eines Rohrofens gestellt. Die geschlossene Anlage wurde dreimal mit dem Schutzgas gespült. Unter einem schwachen Schutzgasstrom wurde auf Reaktionstemperatur erwärmt. Die Umsetzungen erfolgten bei Maximaltemperaturen zwischen 950 und 1650°C. Ein Zr-Getter diente zur Vermeidung von unerwünschtwen Nebenprodukten, unterbildung von ZrO2 bzw. ZrN. Abbildung 2.3.1 zeigt einen schematischen Aufbau des Laborrohrhochtemperaturofens (genauere Angaben zu den verwendeten Öfen sind dem Kapitel 8 zu entnehmen). Die exakten Temperaturprogramme sind mit Aufheiz- und Abkühlraten sowie Haltezeiten bei den jeweiligen untersuchten Systemen angegeben. Korundrohr Ar-Schutzgas Zufuhr Ofengehäuse Zr-Getter Korundtiegel mit Probe im Korundschiffchen Hahn Gasflansch Ar-Schutzgas Abluft Zr-Getter Gasflansch Hahn und Blasenzähler Abbildung 2.3.1: Schematische Darstellung des Hochtemperaturreaktionsofens Nach der Beendigung der Reaktion wurde das Korundschiffchen aus dem Hochtemperaturofen entnommen und je nach Bedarf (beim Arbeiten in einer Schmelze) der Schmelzregulus bis zu mehreren Tagen mit Säure4 behandelt. Der Rückstand wurde mit einer Saugflache und geeigneter Fritte abgesaugt und mehrmals mit dest. Wasser gewaschen und getrocknet. Jeder Rückstand wurde röntgenographisch auf die gebildeten Phasen hin untersucht. Unter dem Lichtmikroskop wurden, wenn vorhanden, Einkristalle für eine röntgenographische Untersuchung an den Einkristallen sowie auch für eine qualitative 4 Die verwendeten Säuren oder Säuregemische wie auch ihre Konzentration stehen im präparativen Teil des entsprechenden Kapitels. - 45 - Methoden - Hochtemperatursynthesen Elementaranalyse mittels energiedispersiver Röntgenemmissionsspektroskopie (EDX) isoliert. Die untersuchten Einkristalle wurden dann, wenn die geforderte Größe erfüllt war, weiter für eine Härtebestimmung verwendet. 2.4 Die Sintermethoden Die Abkürzung SPS steht für Spark Plasma Sintering und darunter ist ein neu entwickeltes Heißpress-Sinterverfahren zu verstehen. Mit Hilfe von gepulstem Strom wird Gleichstrom durch eine Graphitmatrize geleitet, wobei es von innen heraus zu einer hohen Erwärmung kommt. Dieses neue Sinterverfahren erlaubt Heiz- und Kühlraten von 1000°C / min. Herkömmliche Heißpress-Sintermethoden (HIP) erwärmen die Probe von außen. Abbildung 2.4.1: Probe im SPS-Ofen Sintern Unter Sintern versteht man in der technischen Terminologie die Verdichtung und Verfestigung eines Pulvers bei einer gleichzeitigen Wärmebehandlung unterhalb des „Schmelzpunktes“ der Pulverprobe. Die Verdichtung der Probe führt zur einen gleichmäßigen Schwindung. Das Sinterprodukt ist ein homogener und rissfreier Werkstoff (sogenannter Grünling oder Halbzeug) mit feinkristallinem Gefüge (Abbildung 2.4.2). Die treibende Kraft des Prozesses ist das Bestreben des Systems seine Ober- und Grenzflächenenergie durch Kornwachstum und Porenschrumpfung zu reduzieren. Abbildung 2.4.2: Sinterprobe aus Al2O3 Bei den zahlreichen Sintermethoden gibt es zwei Hauptprozesse, die sich nach der Reihenfolge der Druck- und Temperaturbehandlung unterscheiden. Abbildung 2.4.3 gibt eine Übersicht über die herkömmlichen Sinterprozesse. Beim Sinterprozess kommt es kontinuierlich zur einen Veränderung der Mikrostruktur. Diese Veränderung lässt sich in drei Sinterstadien beschreiben. In der Abbildung 2.4.4 wird der Sinterprozess am Beispiel von Al2O3 diskutiert. - 46 - Methoden – Pulsplasma Sinter (SPS) Abbildung 2.4.3: Übersicht der Sintermethoden Für einen gut gesinterten Körper ist eine homogene Pulvermischung sowie möglichst kleine Partikel notwendig. Je kleiner die Partikel sind, desto größer ist der Volumenverlust, folglich erreicht man eine größere Verdichtung des Sinterkörpers nach dem Sintern. Für ein homogenes Gefüge wie in der Abbildung 2.4.2 dargestellt ist es notwendig, die Probe nahe (jedoch unter) dem Schmelzpunkt der verwendeten Edukte zu tempern. Lange Haltezeiten bei der Sintertemperatur fördern den Sinterprozess. I. Anfangsstadium II. Hauptstadium III. Endstadium Beginn der Halsbildung Halswachstum Plastische Verformung Es kommt zur einen Umordnung In diesem Bereich kommt es Dichtes Versintern der Probe. der Partikel. An den Berührungs- zum Kornwachstum kommt zum stellen bilden Wachstum der Hälse. sich Material- Partikel sind dichter gepackt. Die Stillstand brücken, sogenannte „Hälse“. Zahl der Poren nimmt stark ab. Verunreinigungen und Fremd- Beginn von Oberflächen- Somit kommt es zu einer starken einschlüssen. diffusion. Schrumpfung der Probe. I. II. III. Abbildung 2.4.4: Mechanismus des Sinterprozesses - 47 - aufgrund von Methoden – Pulsplasma Sinter (SPS) Der klassische Festphasen-Sinterprozess verläuft nach den Regeln einer Festphasenreaktion. Hier ist die Reaktionsgeschwindigkeit direkt proportional zu der Diffusion. Die Kontaktflächen der Partikel lassen sich durch die Anlegung eines Drucks von außen erhöhen, dieses wirkt reaktionsbeschleunigend für den Diffusionsprozess. Die klassischen Sinterprozesse haben gemeinsam, dass die Erwärmung der Probe von außen erfolgt, etwa durch Widerstandsöfen oder mit Hilfe von Mikrowellen- oder Laserstrahlung. Der SPS-Prozess hat unter den Sinterverfahren eine Sonderstellung. Hier wird die Probe durch den elektrischen Stromfluss direkt von innen erwärmt. Pulsplasma Sintern (SPS Prozess) Die neue Sintermethode mittels gepulsten Gleichstroms wurde bereits in den sechziger Jahren in Japan entwickelt[22,23]. Wie auch bei dem klassischen Heißpress-Sintern wird die Probe auch beim SPS-Prozess axial unter Druck in einer Graphitmatrize umgesetzt. Die Produktpalette der nach diesem Verfahrenen dargestellten Produkte ist enorm. Das Pulsplasma-Sintern erlaubten die Synthesen von Keramikstoffen aller Art (Oxide, Nitride, Carbide und Boride). Aber auch Metalle wie Eisen, Kupfer, Aluminium, Gold und Nickel wurden mit Hilfen der gepulsten Sintertechnik in Form von Grünlingen dargestellt[24]. Der schematische Aufbau eines SPS-Ofens ist in der Abbildung 2.4.5 abgebildet, die wichtigsten Ofenelemente sind hier abgebildet und beschrieben. Die Reaktion wird in der wassergekühlten Vakuumkammer mit Überwachungsfenster durchgeführt. In der Vakuumkammer befindet sich eine vertikale einarmige Presse. Das Herzstück des Ofens ist der leistungsstarke Gleichstromgenerator. Die Synthese wird zwischen den Elektroden in einer Graphitmatrize durchgeführt. Je nach Generator und Matrizendurchmesser kann eine Temperatur von bis zu 2500°C erreicht werden. - 48 - Methoden – Pulsplasma Sinter (SPS) Überwachung - z-Position - Kammerdruck - Temperatur - Spannung - Stromstärke - Kühlung - Schutzgas/Luft Überwachungsfenster obere Elektrode Pulver o Graphitstempel Sintermatrize SPS DC-Puls* Generator SPS Steuereinheit untere Elektrode Kammerdrucksteuerung VakuumSinterkammer (Wassergekühlt) Abbildung 2.4.5: Schematische Darstellung des Pulsplasma Sinter Ofens (SPS) (*DC -Direct Current) Der SPS-Prozess, der noch nicht vollständig geklärt ist, wird über eine Funkenentladung zwischen den Partikeln, die durch den gepulsten Strom entstehen, angenommen. Die Funkenentladung verursacht lokale Temperaturen von bis zu 10.000°C. Der Pulsplasma-Sintermechanismus wird über fünf Zwischenschritten beschrieben[23]. Die einzelnen Stadien sind in der Abbildung 2.3.1 dargestellt. Die Pulverprobe befindet sich analog dem Heißpress-Verfahren in einer Graphitmatrize. Durch einen Graphitstempel wird ein axialer Druck von mehreren Tonnen eingestellt. Ströme (gepulste Ströme im Millisekundenbereich) von mehreren tausend Ampère werden dann durch die Probe geleitet. Je nach Materialeigenschaften, metallischer Leiter oder Isolator/Halbleiter, wird der elektrische Strom durch die Probe oder durch die Graphitmatrize geleitet. Abbildung 2.4.6 zeigt den Temperaturgradienten bei einer isolierenden Probe (A) und einer leitfähigen Probe (B)[25]. - 49 - Methoden – Pulsplasma Sinter (SPS) (A) Isolierendes Material (B) Leitfähiges Material Abbildung 2.4.6: Unterschied zwischen isolierendem (A) und leitfähigen (B) Material An den Kontaktflächen und zwischen den Lücken der Partikel kommt es zu einer Funkenentladung. Folge ist die Entwicklung von begrenzter, sehr hoher Temperatur. Oxidationsschichten, wenn vorhanden, werden auf diese Art gesprengt. Die entstandene Wärme (bis zu 10.000°C) führt zum Aufschmelzen und Verdampfen der Partikeloberfläche. Auf diese Weise wird ein sehr schneller Materialtransport ermöglicht. Es kommt zur Ausbildung von Sinterhälsen zwischen benachbarten Partikeln. Bildungsmechanismus der Pulsplasmahälse nach einem Gleichstrom-Impuls (I) Anfangsstadium der Funkenentladung nach ON-OFF Impuls (II) Entstehung von Plasma (IV) Reaktionsphase zwischen den Partikeln (III) Verdampfung und Aufschmelzen der Partikel (V) Ausbildung von Sinterhälsen Abbildung 2.4.7: Möglicher Mechanismus des Pulsplasma-Sinterns[26]. - 50 - Methoden – Pulsplasma Sinter (SPS) 2.5 SPS-Anlage „Dr. SINTER-LAB™“ am Institut für Anorganische und Analytische Chemie Am Institut für Anorganische und Analytische Chemie in Freiburg i. Br. steht der Pulsplasma Ofen „Dr. SINTER-LAB™ - SPARK PLASMA SINTERING SYSTEM SPS-515S“. Hier handelt es sich um einen Sinterplasmaofen im Labormaßstab mit einer einachsigen vertikalen hydraulischen Presse, die Drücke bis zu 50 kN (die Einstellung des Sinterdrucks ist von dem Durchmesser der verwendeten Graphitmatrize abhängig) erzeugen kann. Für diesen Ofentyp sind Matrizen aus Graphit mit einem Reaktionsdurchmesser von 10 bis 20 Millimeter vorgesehen. Der leistungsstarke Gleichstromgenerator arbeitet mit einer durchschnittlichen Stromstärke von 1500 A bei einer Spannung von bis zu 20V. Dieses ermöglicht eine Arbeitstemperatur von bis zu 2000°C. Bei der Vakuumkammer handelt es sich um eine von außen mit Wasser (18°C) gekühlte doppelschichtige Reaktionskammer aus Edelstahl, die auf einen Druck von 6,0 Pa (4,5·10-2 Torr) evakuiert werden kann. Die Reaktionskammer besitzt zwei Fenster, das eine dient der Temperaturüberwachung durch das Pyrometer und das zweite Fenster bietet einen Einblick in das Reaktionsgeschehen für eine visuelle Überwachung. 2. 1. 3. Abbildung 0.1: Vakuumkammer (offen) mit einer Probe: 1. Pyrometer, 2. Reaktionskammer, 3. Thermoelement - 51 - Methoden – Pulsplasma Sinter (SPS) SPS-Anlage der Universität Freiburg 1. Pyrometer 5. Temperaturüberwachung (Einsatz ab einer Temperatur von 600°C) 4. 2. Vakuumkammer (offen) mit Probe 3. Thermoelement Platin-Rhodium (Einsatz nur bei Temperaturen bis ca.1100°C) 4. Vakuumkammer (geschlossen) Überwachungsfenster aus Quarz 5. Steuerpult der SPS-Anlage 6. Programmsteuerung, Druck, Stromstärke, Kühlung, z-Achse, Vakuumpumpe 6. Steuerung für die Vakuumkammer Luft / Schutzgas (Ar) Abbildung 0.2 SPS-Anlage: 4. Vakuumkammer, 5. Steuereinheit 2.6 Reaktionen im Pulsplasmaofen Für die Synthese in einem Pulsplasmaofen ist nur wenig Vorarbeit zu leisten. Die Edukte werden in Pulverform in entsprechenden Mengen eingewogen und gut in einem Achat-Mörser homogenisiert. Die Reaktionsmatrize besteht aus Graphit, diese wird im Inneren mit einer dünnen Graphitfolie ausgekleidet. Auch die Stirnflächen der Stempel werden mit einer dünnen Graphitfolie ausgelegt. Diese soll die Matrize vor Beschädigungen schützen, zudem wird das Entnehmen des Produkts erleichtert. Das Befüllen der Matrize ist in Abbildung 2.6.1 schematisch dargestellt. Die Unterseite der Graphitmatrize wird verschlossen und von oben mit der Reaktionsmischung befüllt. Mit dem oberen Graphitstempel wird die Matrize verschlossen. Die verschlossene Graphitmatrize wird in der Mitte der Reaktionskammer ausgerichtet. Die Reaktionstemperatur kann auf zwei unterschiedlichen Wegen gesteuert werden, entweder über ein Thermoelement in der Reaktionskammer oder von außen über ein Pyrometer. Für eine Temperatursteuerung über ein Thermoelement ist in der Mitte der Graphitmatrize eine Bohrung vorgelegt. Bei einer Temperatursteuerung über das Pyrometer (ab einer Temperatur von 600°C) wird das Pyrometer auf die Mitte der Matrize durch das - 52 - Methoden – Pulsplasma Sinter (SPS) Überwachungsfenster (Fenster sollte vor jedem Gebrauch auf Sauberkeit geprüft werden) ausgerichtet. Probe Graphitfolie Verdichtete Probe unterer Graphitstempel Abstandsplatte aus Graphit Eingang für das Thermoelement Geschlossene Graphitmatrize Abbildung 2.6.1: Darstellung der Auskleidung und Befüllung der Graphitmatrize für eine Synthese im SPS - Ofen Anschließend wird die Reaktionskammer geschlossen. Auf die Probe wird der gewünschte Reaktionsdruck angelegt (abhängig vom verwendeten Matrizendurchmesser). Die Anlage wird darauf ca. 20 Minuten evakuiert bzw. mit Argon gespült. Die Umsetzungen erfolgten im Temperaturbereich von 900 bis 1600°C (TMAX = 2100°C) und Reaktionszeiten von 3 bis 40 Minuten. Die exakten Temperaturprogramme sind mit Aufheiz- und Abkühlraten sowie Haltezeiten bei den jeweiligen untersuchten Systemen angegeben. Nach dem Abkühlen der Reaktionsmischung (in der Regel ca. 45 Minuten) wurde die Tablette aus der Graphitmatrize entnommen und mit Hilfe von Schleifpapier (mittlere Körnung von 220µm, anschließend feine Körnung von 120 µm) die überschüssige Graphitfolie, die noch am Probenkörper haftete, entfernt. Nach dem vollständigen Befreien der Tablette vom Graphitmaterial wurde die Tablette in einem Wolframcarbidmörser zertrümmert und fein gemörsert. Der Rückstand wurde mittels einer röntgenographischen Untersuchung am Pulver auf die gebildeten Phasen hin analysiert. - 53 - MAX - Phasen 3 MAX-Phasen Die MAX-Phasen (Mn+1AXn Phasen) sind ternäre Phasen, die keramische und metallische Eigenschaften in einer Verbindungsklasse vereinen. Die ersten Untersuchungen liegen nun ein halbes Jahrhundert zurück[27,36]. Die Studien in den sechziger Jahren von Nowotny et al. präsentierten mehr als fünfzig der bis heute bekannten MAX-Phasen, bei denen eine Vielzahl von Kombinationsmöglichkeiten der frühen Übergangsmetalle (M) mit einigen der Hauptgruppenelemente, meist aus den Gruppen IIIa und IVa und den Nichtmetallatomen Kohlenstoff oder Stickstoff (Abbildung 3.1.1), gegeben ist. Heute spricht man von Nano-Komposit-Materialien[28], wenn über MAX-Phasen gesprochen wird. Die systematischen Untersuchungen dieser ternären Phasen zeigen die Besonderheit von guter Oxidationsbeständigheit und hoher Härte bei einem beträchtlichen Young-Modul sowie einer guten Beständigkeit bei hohen Temperaturen bei gleichzeitig guter elektrischer und thermischer Leitfähigkeit[59]. Die meisten Arbeiten zu MAX-Phasen beziehen sich auf das Titan-SiliziumKohlenstoffsystem mit der Verbindung Ti3SiC2. Hier wurden die Synthesebedingungen, Phasenbreite wie auch die Eigenschaften eingehend untersucht. Gegenstand der aktuellen Forschung sollen aufgrund ihrer Möglichkeiten der Ausbildung von unterschiedlicher Phasenvarianten die Systeme Titan-Aluminium/M-Kohlenstoff (M = Sn, Ga, In und Ge) wie auch Vanadium-Aluminium/M-Kohlenstoff (M = Sn, Ga und In) sein. In der vorliegenden Arbeit soll der Bereich der Primärkristallisation wie auch das geeignete Fluxmittel für die Züchtung von Einkristallen aus der Schmelze erforscht werden, um Einkristalle geeigneter Größe für die Härtemessung sowie eine vollständige Charakterisirung (PXRD, SXRD und EDX) zu synthetisieren. Als eine weitere Synthesemethode soll die Reproduzierbarkeit der erhaltenen Phase in einem Funkenpulsplasmasinterofen durchgeführt werden. 3.1 Vorkommen der MAX-Phasen Abbildung 3.1.1 und 3.2 gibt eine Übersicht über die im Cr2AlC-Typ[33] kristallisierenden Verbindungen. Die meisten bekannten Vertreter sind Carbide des Typs M2AX[59]. Als Hauptkomponente M treten bevorzugt die frühen Übergangsmetalle aus der Gruppen IVb und Vb. Als A-Bestandteil wurden überwiegend die Hauptgruppenelemente aus den Gruppen IIIa - 54 - MAX - Phasen und IVa eingesetzt. Als C-Komponente wurde zum aktuellen Zeitpunkt nur Kohlenstoff und Stickstoff beobachtet. Bezüglich der Struktur (siehe Kapitel 3.2) zeigen sich abgeleitet aus der M2AX (211) Struktur zwei weitere Varianten M3AX2 (312) und M4AX3 (413). Fast alle Verbindungen der bekannten MAX-Phasen sind der 211-Variante zuzuordnen. Nur fünf Verbindungen sind bislang mit der 312-Variante dargestellt und charakterisiert worden (Ti3AlC2[29], Ti3GeC2[30], Ti3SiC2[45], Ta3AlC2[203] und Ti3SnC2[31])5. In der 413-Variante wurden nur drei Verbindungen in der Literatur beschrieben (Ti4AlN3[32], Ta4AlC3[203] und V4AlC3-x[203]). Ia IIa IIIb IVb Vb VIb VIIb VIIIb Ib IIb IIIa IVa Va VIa VIIa VIIIa H He Li Be B C N O F Ne Na Mg Al Si P S Cl Ar K Ca Sc Ti V Cr Mn Fe Co Ni Cu Zn Ga Ge As Se Br Kr Rb Sr Y Zr Nb Mo Tc Ru Rh Pd Ag Cd In Sn Sb Te I Xe Cs Ba La L Hf Ta W Re Os Ir Pt Au Hg Tl Pb Bi Po At Rn Fr Ra Ac A Rf Db Sg Bh Hs Mt Ds Rg L = Lanthanoiden A = Actinoiden Abbildung 3.1.1: In der Literatur beschriebene Phasen mit Cr2AlC-, Ti3SiC2- und Ti4AlN3-Struktur. Aus den Abbildungen 2.1.9.1 und 2.1.9.2 sind die drei bekannten Prototypen der MAXPhasen mit ihren charakteristischen Gitterkonstanten und den zugehörigen Modellen abzuleiten. Prototyp: 2 1 1 - M2A1X1 Cr2AlC - Typ[33] (über 100 Verbindungen) P63/mmc (194); Prototyp: 3 1 2 - M3A1X2 Ti3SiC2 – Typ[34] (genau 5 Verbindungen)5 P63/mmc (194); Prototyp: 4 1 3 - M4A1X3 a = 2,854 Å; c = 12,82 Å a = 3,068 Å; c = 17,669 Å Se4CsYb3 – Typ[35] (genau 3 Verbindungen) P63/mmc (194); a = 4,0578 Å; c = 31,303 Å Abbildung 3.1.2: Varianten der Mn+1AXn – Phasen, Prototyp Betrachtung 5 Im Rahmen dieser Arbeit konnte zum ersten Mal die 312-Variante aus dem System Titan-Gallium-Kohlenstoff dargestellt werden. Diese bereichert die 312-Familie um eine weitere Phase mit der stöchiometrischen Zusammensetzung Ti3GaC2. - 55 - MAX - Phasen 3.2 Struktur der Komplexcarbide Die MAX-Phasen kristallisieren hexagonal im Cr2AlC-Typ[36] mit der Raumgruppe P63/mmc (Nr. 194) mit zwei Formeleinheiten. Abbildung 3.2.1, .2 und .3 zeigt die drei möglichen Varianten. Die 211-Phase, wird auch als H-Phase bezeichnet, die 312-Phase und die 413-Phase sind weitere Varianten. Das Herleiten der Strukturen der 312- bzw. 413- Phasen lässt sich durch einen weiteren Einbau von MX-Einheiten erklären. Ausgehend von der 211-Phase, die sich aus den Elementen oder aus den MX und MA Bausteinen darstellen lässt, geht es über einen Stufenmechanismus bis zur nachgewiesenen 413-Phase. Jedoch konnte dieser Mechanismus bislang nicht an allen bekannten Verbindungen des Cr2AlC-Typ[36] nachgewiesen werden. Vielmehr gibt es bislang nur die oben genannten vier Verbindungen der 312-Phase (Ti3AlC2[37], Ti3GeC2[38], Ti3SiC2[45] und Ta3AlC2[203]) sowie nur drei Verbindungen der 413Phase (Ti4AlN3[39], Ta4AlC3[203] und V4AlC3[203]), bei denen ein Einbau einer MX-Einheit erfolgreich durchgeführt wurde. Die Reaktionsgleichung (Formel 3-1) zeigt den zusammenhang der M3AX2 und M4AX3 Produkte ausgehend von der M2AX-Phase. + MX + MX M 2 AX → M 3 AX 2 → M 4 AX 3 Formel 3-1 Diese formale Betrachtungsweise erlaubt die Abschätzung der Gitterkonstanten in cRichtung sowie die Vorhersage für mögliche Strukturen, die sich durch Addition von weiteren Bauelementen in Richtung [001] ergeben. Folglich sind Phasen der Zusammensetzung M5AX4 oder M6AX5 plausibel und denkbar. Es steigt aber die Schwierigkeit, die Phasen zu charakterisieren + MX + MX + MX M 4 AX 3 → M 5 AX 4 → M 6 AX 5 → ....... Formel 3-2 In der 211-Phasen bilden M- und A-Atome eine hexagonale dichteste Packung, wobei auf zwei M-Schichten eine A-Schicht folgt. In den von den M-Atomen gebildeten Oktaederlücken befinden sich ausschließlich Kohlenstoff- oder Stickstoffatome. Abbildung 3.2.1 repräsentiert die H-Phase mit Blick entlang [100]. - 56 - MAX - Phasen Die Metallatome besetzen die Punktlagen 4f und 2d Die Position 2a wird ausschließlich von Kohlenstoff besetzt. Tabelle 2.1.9.1 gibt die Besetzungsfaktoren der Ti2AlC Verbindung wieder[40]. Tabelle 2.1.9.1: Ortskoordinaten und Besetzungsfaktoren in der Ti2AlC-Struktur. Atom Wyckoff Symmetrie Ti 4f Al C Position x y 3m 1 2 2d -6m2 1 2a -3m 0 3 3 2 3 3 0 z 7/12 1 4 0 In der Abbildung 3.2.1 (rechts) ist die abfolge der einzelnen Schichten wiedergegeben. Die mit Kohlenstoff besetzten Oktaeder sind zu Schichten über Flächen miteinander verknüpft. Diese Ti6C-Oktaederschichten lassen sich als Ti2C-Bausteine visualisieren, die durch eine Aluminiumschicht getrennt sind. Ti2AlC – Stapelfolge Abbildung 3.2.1: Kristallstruktur von der H-Phase (auch als 211-Phase bekannt) am Beispiel von Ti2AlC (links). Die Stapelfolge der Metallschichten ist im Bild rechts dargestellt. Bei der 312-Phase handelt es sich gelegentlich um eine weitere Variante zu Ti2AlC. Bei diesem Aufbau werden die Aluminiumschichten durch drei Titanschichten getrennt. Die Oktaederlücken sind auch hier mit Kohlenstoffatomen vollständig besetzt. Die Oktaeder sind miteinander über Kanten verknüpft. So kommt es zur Ausbildung von Ti3C2-Bausteinen die - 57 - MAX - Phasen periodisch durch eine Aluminiumschicht getrennt werden. Die Besetzungsfaktoren (Tabelle 2.1.9.2) sowie die Struktur werden in der Abbildung 3.2.2 dargestellt. Tabelle 2.1.9.2: Ortskoordinaten und Besetzungsfaktoren in der Ti3AlC2-Struktur. Atom Wyckoff Symmetrie Ti 1 2a -3m. Ti 2 2f 3m Al 2b -6m2 C 4f 3m Position x y z 0 0 0 2 1 3 1/8 0 0 1 1 2 3 3 3 4 1/16 In der 312-Phase wird nicht mehr wie bei der 211-Phase (oben) eine einfache Sequenz der hexagonalen dichtesten Kugelpackung der Metallatome (hdp ABAB) versinnlicht, sondern eine, die sich als (hhhc)2, nach Jagodzinski mit der Stapelsequenz ABABCBCB beschreiben lässt. Ti3AlC2 – Stapelfolge Abbildung 3.2.2: Kristallstruktur von der 312-Phase am Beispiel von Ti3AlC2 (links). Die Stapelfolge der Metallschichten ist im Bild rechts dargestellt. - 58 - MAX - Phasen Der Aufbau der 413-Phase verläuft analog dem oben diskutierten Bauprinzip. Vier Titanlagen, deren Oktaederlücken mit Stickstoffatomen besetzt sind, werden zu einem Ti4N3Bausteine über gemeinsame Flächen miteinander verknüpft. Diese Ti4N3-Sichten werden durch Aluminiumschichten getrennt. Abbildung 3.2.3 gibt den Aufbau der Schichten in den 413-Phasen wieder. Tabelle 2.1.9.3: Ortskoordinaten und Besetzungsfaktoren in der Ti4AlN3-Struktur. Atom Wyckoff Position Symmetrie x y 2 Ti 1 4f 3m 1 Ti 2 4e 3m Al 2b N1 N1 z 3 ~1/20 0 0 ~3/20 -6m2 1 2 4f 3m 1 2a -3m 0 3 3 3 2 3 1 4 3 ~1/10 0 0 Die Stapelsequenz in der 413-Phase lässt sich als ABABCACACB beschreiben (nach Jagodzinski (hhhcc)2). Ti4AlN3 – Stapelfolge Abbildung 3.2.3: Kristallstruktur von der 413-Phase am Beispiel von Ti4AlN3 (links). Die Stapelfolge der Metallschichten ist im Bild rechts dargestellt. - 59 - MAX - Phasen 3.3 Eigenschaften der MAX-Phasen 3.3.1 Chemische Reaktivität Um das Verhalten gegenüber Säuren und Basen von MAX-Phasen zu studieren, wurde Ti3SiC2 in einer Versuchsdauer von 6 Monaten einer Behandlung durch konzentrierte Schwefelsäure, Salpetersäure, Salzsäure sowie Fluorwasserstoffsäure unterzogen. Das Ergebnis war überzeugend. Eine Behandlung mit konz. H2SO4 zeigt keine angegriffene Oberfläche und auch das Wiegen nach 6 Monaten keinen Gewichtsverlust, somit ist Ti3SiC2 gegen konz. H2SO4 vollständig resistent. Mit der verd. H2SO4 konnte eine geringfügige Gewichtsabweichung beobachtet werde, die den Rückschluss von einer Abtragung von < 1 µm/Jahr erlaubt. Eine größere Säurenwirkung konnte mit verd. HCl (< 2 µm/Jahr) und verd. HF (< 5 µm/Jahr) beobachtet werden. Bei einer Einwirkung von NaOH über den Zeitraum von 6 Monaten konnte keine Gewichts- und Oberflächenveränderung vermerkt werden. Basierend auf den gesammelten Daten aus dieser Studie lässt sich sagen, dass Ti3SiC2 säureund basenresistent ist. Die Übergangsmetallcarbide und -nitride sind an Luft in Temperaturbereichen ab 500°C bis 800°C meist schon vollständig oxidiert. Untersuchungen von Ti3SiC2 zeigten eine gute Oxidationsresistenz bei hoch Temperaturen[41,42,43]. Eine merkliche Oxidation setzte erst ab einer Temperatur von 900°C ein. Eine Erhöhung der Temperatur führt zu einer TiO2 Geschwindigkeitsbeschleunigung der Oxidation[44] nach Gl. 3-3. SiO2 + +TiO2 Ti3SiC2 + 5 O2 → 3 TiO2 + SiO2 + CO Formel 3-3 Der Querschnitt einer oxidierten Ti3SiC2-Probe ist in Abbildung 3.3.1 dargestellt. Ti3SiC2 Abbildung 3.3.1: Querschnitt einer Ti3SiC2-Probe nach sechsmaligem Aufheizen von RT auf 1100°C für 12 Stunden an der Luft[59]. Bei der Probe Ti3SiC2 zeigt sich nach einer thermischen Behandlung an Luft eine 20 µm dicke Schicht an reinem TiO2 und einer darunter liegenden Schicht an SiO2 neben TiO2. - 60 - MAX - Phasen 3.3.2 Mechanische Eigenschaften Vergleicht man die Mn+1AXn Phasen mit den zugehörigen MX Phasen erscheinen diese relativ weich[45]. Härtemessungen nach Knoop an Schichten, die durch ein CVD Verfahren gewonnen wurden, zeigten eine Knoophärte von 3-4 GPa gemessen parallel zu basalen Schicht und eine erstaunlich hohe Knoophärte senkrecht zu dieser von 12-15 GPa[45]. Weitere Untersuchungen der Härte an Sinterproben von MAX-Phasen wuden von Pampuch et al.[46] durchgeführt und zeigten ein Vickershärte im Bereich von 5 ± 1 GPa. 3.3.3 Elektrische und thermische Leitfähigkeit Alle untersuchten Verbindungen aus der Familie der MAX-Phasen zeigten eine gute elektrische Leitfähigkeit. Die Leitfähigkeit lag bei den untersuchten Verbindungen im Bereich von 0,07 µΩ·m (Ti2SnC) bis 2,7 µΩ·m (Ti4AlN2,9)[47,48,49,50;51,52]. Untersuchungen der thermischen Leitfähigkeit wurden von Barsoum et al. im Bereich bis 1300°C durchgeführt. Untersucht wurden die Verbindungen Ti3SiC2, Nb2SnC, Ti4AlN3 und Ti2AlC. Das Resultat war beeindruckend, die thermische Leitfähigkeit lag in der Größenordnung von Titan. Das Ergebnis ist in Abbildung 3.3.2 und der Tabelle 3.3.3.1 zu entnehmen. Tabelle 3.3.3.1: Ergebnis der Messung von thermischer Leitfähigkeit von ausgewählten MAX-Phasen bei 300K und 1300K[59]. Verbindung Ti3SiC2[53] 34 33 Nb2SnC[54] 17,5 30,5 12 20 Ti2AlC 46 36 TiC[57] 14,4 33,4 TiN[58] 27,4 45,3 Ti4AlN3 [55] [56] Abbildung 3.3.2: thermische Leitfähigkeit der ternären Verbindung, sowie die des reinen Titans[59]. Leitfähigkeit [W/m·K] 300K 1300K Die MAX-Phasen schmelzen nicht, sie zeigen eine thermische Zersetzung nach folgender Reaktionsgleichung (Formel 2-1): - 61 - MAX - Phasen M n+1 AX n → M n+1 X n + A Formel 3-4 Die Zersetzungstemperatur der untersuchten MAX-Phasen liegt im Bereich von 850°C beim Nitrid Cr2GaN, bis über 2300°C im Fall von Ti3SiC2[59]. Somit vereinen die MAX-Phasen Eigenschaften wie gute Bearbeitbarkeit und gleichzeitig hohe Festigkeit bis zu hohen Temperaturen in einem Stoff. Zugleich bleiben sie gute elektrische und thermische Leiter. Zudem sind sie beständig gegenüber starken Oxidationsmitteln. Abbildung 3.3.3: Übersicht der Eigenschaften von MAX-Phasen Die Eigenschaften der MAX-Phasen lassen Einsatzgebiete im Hoch-temperaturbereich zu. Somit stellen die MAX-Phasen eine potentielle Anwendungsmöglichkeit als Wärmeüberträger oder Wärmetauscher (sogenannte „heat exchangers“), die die Wärme bzw. thermische Energie von einem Stoffstrom auf einen anderen übertragen, durch die Befähigung der thermischen Leitfähigkeit bei hohen Temperaturen. Ti3SiC2 kann aufgrund seiner extremen Temperaturbeständigkeit mit einer Zersetzungstemperatur oberhalb von 2200 °C auch bei Wärmeüberträgern eingesetzt werden, deren Materialtemperaturen oberhalb der Einsatzgrenze der Metalle liegen. Ti3SiC2 gehört zu den neu entwickelten HochtemperaturWärmeüberträgern[60]. - 62 - 3.4 Das System Titan-Aluminium-Kohlenstoff 3.4.1 Die binären Randsysteme um das System Titan-Aluminium-Kohlenstoff Das binäre Phasendiagramm von Titan und Kohlenstoff (Abbildung 3.4.1) zeigt eine nicht stöchiometrische Phase TiCx[61] (weitere bekannte Phasen stellen Defektvarianten da) mit einem erstaunlich großen Homogenitätsbereich (x = 0,52 - 0,98) und eine zweite stöchiometrische Verbindung Ti2C, die sich jedoch unterhalb von 1900°C zersetzt[62]. TiCx kristallisiert im Natriumchlorid-Typ in der Raumgruppe Fm 3 m. TiCx (bei ca. 45 At.-% Kohlenstoff) schmilzt kongruent bei einer Temperatur von 3067 ± 15°C. Abbildung 3.4.1: Das binäre Phasendiagramm des Systems Titan-Kohlenstoff[175] Abbildung 3.4.2: Das binäre Phasendiagramm des Systems Aluminium-Kohlenstoff[175] Das Phasendiagramm von Aluminium-Kohlenstoff offenbart nur eine einzige Verbindung Al4C3. Diese kristallisiert stöchiometrisch in der Raumgruppe R 3 m mit den Gitterkonstanten von a = 3,3350 Å und c = 3,3350 Å[63]. Al4C3 beginnt erst ab einer Temperatur von 2500°C sich zu zersetzen. Das binäre System Titan-Aluminium zeigt zahlreiche Phasen, diese bilden sich im Homogenitätsbereich von TiAl3 über TiAl2 bis Ti3Al[64,65]. TiAl3 schmilzt peritektisch bei einer Temperatur von ca. 1350 ± 15°C. An diesem Punkt liegt auch das Maximum das Lösungsvermögen vom Titan in Aluminium. Abbildung 3.4.3: Das binäre Phasendiagramm des Systems Aluminium-Titan[175] - 63 - MAX – Phase – Das System Ti-Al-C 3.4.2 Das ternäre System Titan-Aluminium-Kohlenstoff Das System Titan-Aluminium-Kohlenstoff wurde bereits bei Temperaturen von 850°C, 1000°C und 1250°C auf die Phasenbreiten hin untersucht[66]. Aus zahlreichen Proben konnte mit Hilfe von unterschiedlichen Probezusammensetzungen das ternäre Phasendiagramm erstellt werden. Es konnten unter den durchgeführten Versuchsbedingungen zwei ternäre Verbindungen im gegebenen Dreistoffsystem erfasst werden, Ti3AlC (P-Phase ein PerowskitCarbid, in der RG: Pm 3 m[67]) und Ti2AlC (H-Phase ein Cr2AlC-Typ in der RG: P63/mmc[33]). In der Abbildung 3.4.4 sind die zwei ternären Verbindungen gekennzeichnet (Ti3AlC, blaugestrichelt und Ti2AlC rot) und auch der Bereich in dem die ternären Verbindungen kristallisieren. Für die H-Phase existiert nur ein sehr schmaler Bereich in der titanreichen Ecke in dem nicht das thermodynamische Gleichgewicht der binären Phasen dominiert. Für eine phasenreine und einkristalline Darstellung der H-Phasen und den noch nicht in diesem Diagramm erfassten 312- und der eventuellen 413-Phasen steht nur ein sehr schmaler Bereich zur Verfügung. Die hohe thermische Stabilität des TiC1-x sowie des TiAl3 sind das Hauptproblem in der Synthese der H-Phase im System Ti-Al-C. Diese binären Verbindungen kristallisieren und konkurrieren mit der H-Phase in der titanreichen Ecke beim Arbeiten in einer reinen Aluminiumschmelze. . Abbildung 3.4.4: Das ternäre Phasendiagramm Titan-Aluminium-Kohlenstoff. Abgebildet ist ein isothermer Schnitt bei einer Temperatur von 1000°C. - 64 - MAX – Phase – Das System Ti-Al-C 3.4.3 Arbeiten in einer Aluminium-Schmelze Aus dem in der Abbildung 3.4.4 gezeigten ternären Phasendiagram geht hervor, dass eine Züchtung von Einkristallen aus einer reinen Aluminiumschmelze nicht ganz trivial ist. Um das Kristallisieren der binären Produkte zu verhindern, wurde eine Reaktionstemperatur von 1600°C ausgewählt. Diese liegt deutlich über der Zersetzungstemperatur von den intermetallischen Phasen. Das thermische Gleichgewicht liegt somit vollständig auf der Seite der MAX-Phase. 3.4.3.1 Präparative Untersuchung im System Titan-Aluminium-Kohlenstoff Für eine erfolgreiche Züchtung von Einkristallen hoher Qualität wurde der Einfluss der Ausgangsmaterialien getestet. Die erste Versuchsreihe, ausgehend von den Elementen6, wurde in der Konzentration der Elemente mehrfach variiert (Tabelle 3.4.3.1). Ein sehr langsames Abkühlen 5°C ( 1600°C → 1500°C ) der Schmelze im angegeben Temperaturbereich sollte zum Wachstum von Einkristallen der MAX-Phasen führen, sowie die primäre Kristallisation von anderen Nebenprodukten unterdrücken. Die Elemente wurden in den unten angegebenen Verhältnissen in einem Achat-Mörser verrieben und zu einer Tablette gepresst. Die Umsetzung erfolgte in einem Korundtiegel unter einer Ar-Schutzgasatmosphäre. Dabei wurde folgendes Temperaturprogramm verwendet: Temperaturprogramm: 150°C/h RT -5°C/h -20°C/h 1600°C 1500°C (20h halten) (2h halten) -100°C/h 1000°C RT (1h halten) Der gewonnene Schmelzregulus wurde über Nacht in verd. Salzsäure eingelegt. Nach dem aAuflösen der Schmelzrückstände wurde das kristalline Produkt (hexagonale Plättchen) gewaschen und abgesaugt. Die Phasenanalyse zeigte in allen Ansätzen das deutliche Auftreten der 312-Phase, in einigen Fällen (Probe 2, 3 und 5 aus der Tabelle 3.4.3.1) sogar phasenrein. Die Indizierung der Reflexe zeigte eine hexagonale Zelle mit den charakteristischen Gitterkonstanten in der Raumgruppe P63/mmc (alle Gitterkonstanten sowie die Volumina sind in der Tabelle Tabelle 3.4.3.1 angegeben). Das Vorkommen der 211-Phase konnten nur in einem Ansatz in Spuren beobachtet werden (Probe 1, eine Indizierung der 6 Als Kohlenstoffquelle wurde „carbon black“ eingesetzt, die Metalle wurden vor dem Einsatz röntgenographisch auf die Reinheit geprüft - 65 - MAX – Phase – Das System Ti-Al-C Reflexe wurde auf Grund der schwachen Intensität der Reflexe nicht durchgeführt). Als weiteres Nebenprodukt (Spuren) fielen auch die binären Verbindungen TiC und TiB2 an. Das Erscheinen von Titanborid lässt sich nur durch eine Verunreinigung erklären. TiB2 ist thermodynamisch sehr stabil. Ist Bor in der Probe anwesend, kommt es auch sehr rasch zur Reaktion mit Titan. Das Bor ist vermutlich als Verunreinigung in geringen Mengen in den Reaktionstiegel eingebracht worden. Tabelle 3.4.3.1: Proben im System Titan-Aluminium-Kohlenstoff und ihre pulverdiffraktometrische Charakterisierung. Nr. Zusammensetzung Gehalt [At.-%] Ti Al C 1 Ti6Al15C1 27,3 68,2 4,5 2 Ti6Al15C1 27,3 68,2 4,5 3 Ti5,9Al15,3C1 26,6 68,9 4,5 4 Ti5,8Al15,7C1,1 25,7 69,5 4,8 5 Ti5,9Al15,3C0,8 26,8 69,5 3,6 6 Ti6Al20C1,5 21,8 72,7 5,5 Gitterkonstante [Å] weitere kristalline MAX-Phase Phasen a = 3,0768(13); c = 18,557(8) V=152,14(7) a =3,0786(17); c = 18,629(1) V=152,73(13) a = 3,093(3); c = 18,754(4) V=155,3(3) a = 3,0879(19); c = 18,679(2) V=154,24(12) a = 3,0778(7); c = 18,722(16) V=153,59(11) a = 3,082(3); c = 18,824(5) V=154,7(3) Ti2AlC in Spuren Phasenrein Phasenrein > TiC Phasenrein TiC, >> TiB2 In der zweiten Versuchsreihe (Tabelle 3.4.3.2) sollte die 312-Phase reproduziert werden, wobei der Einfluss der Art der Edukte überprüft wurden. So wurde hier Titancarbid als Titanund Kohlenstoffquelle eingesetzt. In den Proben 1 bis 6 aus Tabelle 3.4.3.2 (Aluminiumgehalt variierte von 13,9-32,6 At.-%) konnte die 312-Phase als kristallines Hauptprodukt mit einem geringem Anteil von kristallinem TiC dargestellt werden. Nur die Probe Nummer 7, bei der ein deutlicher Überschuss an Aluminium eingesetzt wurde, zeigt die 312-Phase nur als kristallines Nebenprodukt, hier dominierte das binäre Titancarbid. Die Umsetzung der Proben (Präparation sowie Temperaturprogramm) erfolgte wie oben beschrieben. In Tabelle 3.4.3.2 ist neben der Einwaage auch das Ergebnis der röntgenographischen Phasenanalyse aufgeführt. Das Pulverdiffraktogramm ist mit den charakteristischen Reflexen der 312-Phase mit den Gitterkonstanten von a = 3,073(1) Å und c = 18,532(8) Å in der - 66 - MAX – Phase – Das System Ti-Al-C Raumgruppe: P 63/mmc in der Abbildung 3.4.5 dargestellt. Nur ein beobachteter Reflex im 43,302° 2θ –Bereich konnte nicht zugeordnet werden (blau markiert). Erwähnenswert ist die Kristallform der Produkte aus beiden Versuchsreihen. Aus dem Syntheseweg, ausgehend vom TiC (zweite Synthesestrategie), konnten Einkristalle einer Kantenlänge von 0,01 mm und einer Dicke von 0,003 mm erhalten werden. Die erste Synthese führte zu einer deutlich besseren Kristallisation der MAX-Phase, die gewonnenen, metallisch glänzenden, hexagonalen Einkristalle fielen mit einer durchschnittlichen Kantenlänge von ca. 0,03 mm und einer Dicke von 0,01 mm an. Der Vergleich der beiden Kohlenstoffquellen zeigt, dass „carbon black“ eine bessere Kristallform und größere Einkristalle unter gleichen Synthesebedienungen hervorbringt. Tabelle 3.4.3.2: Proben im System Titan-Aluminium-Kohlenstoff und ihre pulverdiffraktometrische Charakterisierung. Fettgedruckt sind die Hauptprodukte. Nr. Zusammensetzung Gehalt [At.-%] TiC Ti Al 1 Ti3AlC2 61,5 24,6 13,9 2 Ti3Al1.1C2 60,7 24,3 15,0 3 Ti3Al1.2C2 59,9 23,9 16,2 4 Ti3Al1.1C1,8 55,5 29,5 15,2 5 Ti3Al2C2 54,1 21,6 24,3 6 Ti3Al3C2 48,2 19,3 32,6 7 Ti3Al10C2 27,4 10,9 61,7 Gitterkonstante [Å] weitere kristalline der MAX-Phase Phasen a = 3,0796(15); c = 18,559(8) V=152,42(9) a= 3,0746(12); c = 18,584(9) V=152,13(8) a = 3,0748(10); c = 18,589(7) V=152,20(6) a =3,0730(13); c = 18,532(8) V=151,56(8) a = 3,0919(17); c = 18,710(2) V=154,90(15) a = 3,086(2); c = 18,691(16) V=155,33(15) TiC* > TiC > TiC > TiC Phasenrein > TiC > TiC > Ti3AlC2 Exemplarisch für die 312-Phase aus dem System Titan-Aluminium-Kohlenstoff ist das Pulverdiffraktogramm der Verbindung Ti3AlC2 in der Abbildung 3.4.4 dargestellt. Die Indizierung der 21 stärksten Reflexe konnte zu der Verfeinerung einer Zelle mit Gitterkonstanten von a = 3,0730(13) Å, c = 18,532(8) Å und einem Volumen von V=151,56(8) Å3 in der Raumgruppe P63/mmc führen. - 67 - MAX – Phase – Das System Ti-Al-C 170000 160000 150000 140000 130000 120000 Lin (Counts) 110000 100000 90000 80000 70000 60000 50000 40000 30000 20000 10000 0 6 10 20 30 40 50 60 70 80 90 2-Theta - Scale Abbildung 3.4.5: Pulverdiffraktogramm von Ti3AlC2. Der blaumarkierte Reflex konnte keiner bekannten Phase zu geordnet werden. 3.4.3.2 Synthese der MAX-Phase im SPS-Ofen Bei den Synthesen im Pulsplasma-Ofen wurden mehrere Strategien erprobt. Nicht jede führte zum gewünschten Erfolg. In mehreren Versuchsserien wurden die Edukte und die Temperaturprogramme variiert. Aus technischen Gründen ist das Arbeiten in einer Schmelze bei der Benutzung des SPS-Ofens unmöglich. Aus diesem Grunde wurde die Zusammensetzung weitgehend konstant gehalten. Verändert wurden die Edukte wie auch die Reaktionszeit und -temperatur. Die Versuche wurden bei einer Maximaltemperatur im Bereich von 1100 bis 1350°C und bei Synthesezeiten von 3 bis 25 Minuten durchgeführt. Bei der Herstellung der 312-Phase im System Titan-Aluminium-Kohlenstoff wurden folgende Edukte eingesetzt: Als Titanquelle wurde elementares Titan wie auch Titancarbid herangezogen (analog der Hochtemperatur Synthese). Für das Aluminium wurde elementares Aluminium wie auch Aluminiumcarbid (Al4C3) eingesetzt. Der Kohlenstoff wurde entweder über das Aluminium in Form von Al4C3 oder über das Titancarbid (TiC) mit eingebracht oder elementar als carbon black oder Graphit zugeführt. In den meisten Ansätzen bildete sich TiC als Hauptprodukt. Oft war die MAX-Phase nachweisbar, jedoch nur als Nebenprodukt. Das beste Ergebnis für die 312-Phase konnten aus dem Ansatz mit der Zusammensetzung Ti3Al1,1C2 0,243 g (4,056 mmol) Titancarbid, 0,097 g (2,025 mmol) Titan und 0,06 g (2,223 mmol) Aluminium gewonnen werden. - 68 - MAX – Phase – Das System Ti-Al-C Die Pulver wurden homogenisiert und in einer Graphitmatrize (Durchmesser von 10 mm) ausgekleidet mit Graphitfolie bei einer Temperatur von 1300°C für 10 Minuten umgesetzt. Temperaturprogramm: 2 min. RT 2 min. 600°C 2 min 800°C 2 min 1100°C 45 min. 1300 RT (10 min. halten) Die Reaktion wurde unter einem Vakuum von etwa 0,12 mbar durchgeführt. Der Sinterprozess wurde unter einem Druck von 3,9 kN durchgeführt. Dabei konnte eine Schrumpfung der Probe während des Sinterns von ungefähr 6% beobachtet werden. Nach dem Abkühlen der Probe wurde der Sinterkörper aus der Matrize entnommen, das Graphit auf der Tablette wurde mit Hilfe eines Schleifpapiers (120 µm) entfernt. Nach vollständigem Beseitigen der Graphitrückstände wurde die Tablette in einem Wolframcarbidmörser zertrümmert und röntgenographisch auf die gebildeten Phasen hin analysiert. Die Auswertung des aufgenommenen Pulverdiffraktogramms (Abbildung 3.4.6) zeigt die Bildung der Ti3AlC2 Phase als eindeutiges Hauptprodukt und ein Nebenprodukt, das sich anhand der charakteristischen Reflexe als TiC identifizieren lassen konnte (in der Abbildung blau markiert). 200000 190000 180000 170000 160000 150000 140000 Lin (Counts) 130000 120000 110000 100000 90000 80000 70000 60000 50000 40000 30000 20000 10000 0 6 10 20 30 40 50 60 70 80 90 2-Theta - Scale Abbildung 3.4.6: Pulverdiffraktogramm von Ti3AlC2 synthetisiert im Pulsplasma Ofen. Die blaumarkierten Reflexe konnten TiC zu geordnet werden. - 69 - MAX – Phase – Das System Ti-Al-C Die Verfeinerung der Gitterkonstante in der Raumgruppe P 63/mmc ergab einen Wert von a = 3,0894(16) Å; c = 18,632 (6) Å und ein Volumen von V=154,0(1) Å3. Eine phasenreine Darstellung der 312-Phase konnte auch bei anderen Ansätzen nicht beobachtet werden. Untersuchungen zur Bildung der 312-Phase im SPS-Ofen zeigten unabhängig von den Edukten eine große Tendenz zur Entstehung von TiC. Der Einsatz von carbon black zeigte eine sehr große Reaktionsträgheit. Mit einer Kohlenstoffquelle konnte kaum eine Reaktion des Kohlenstoffs wahrgenommen werden. Selbst bei Reaktionszeiten von 25 Minuten (auch bei unterschiedlichen Reaktionstemperaturen) wurde bei der Phasenanalyse noch viel unumgesetzter Kohlenstoff detektiert. Bei tieferen Temperaturen (d.h. 1100 -1250°C) traten beide Phasen TiC und Ti3AlC2 nahezu im Verhältnis eins zu eins nebeneinander auf. Selbst ein kleiner Überschuss (Einwaage Ti3Al1,1C2) an Aluminium konnte das Gleichgewicht nicht auf die Seite von der 312-Phase verschieben. Bei einer höheren Menge an Aluminium wie oben angegeben, wurde das Aluminium aus der Probe verdampft. Dieses machte sich während der Synthese als ein metallischer Niederschlag auf den Kontrollfenstern bemerkbar. Nach dem Abkühlen konnten metallisch glänzende Perlen auf dem Ofenboden eingesammelt werden, die als reines Aluminium identifiziert werden konnten. Als Alternative wurde Aluminiumcarbid (Al4C3) herangezogen, jedoch zeigt das Arbeiten mit Al4C3 eine deutlich größere Tendenz zur Bildung der ternären Verbindung mit der Summenformel Ti3AlC, allerdings nicht phasenrein. Die Bildung con TiC so wie nicht abreagiertes Al4C3 waren als weitere Nebenprodukte deutlich im Diffraktogramm detektierbar. Als weitere Nebenprodukte wurde neben viel TiC und nicht umgesetzten Titan auch Ti2AlC wahrgenommen. Aus einem Ansatz mit identischer Einwage, wie schon oben die 312-Phase erhalten wurde, konnte die Bildung einer 211-Phase bei einer Temperatur von 1350°C beobachtet werden. Es wurde 0,242 g (4,039 mmol) Titancarbid, 0,098 g (2,046 mmol) Titan und 0,062 g (2,297 mmol) Aluminium eingewogen, gut vermischt und ungepresst in die Graphitmatrize (Durchmesser 10 mm) überführt. Die Synthese wurde unter einem Vakuum von 0,14 mbar durchgeführt. Beim Sinterprozess wurde ein Druck von 4,0 kN eingestellt. Temperaturprogramm: 2 min. RT 2 min. 600°C 2 min. 900°C 3 min. 1100°C 45 min. 1350°C (9 min. halten) - 70 - RT MAX – Phase – Das System Ti-Al-C 40000 Lin (Counts) 30000 20000 10000 0 6 10 20 30 40 50 60 70 80 90 100 2-Theta - Scale Abbildung 3.4.7: Pulverdiffraktogramm von Ti2AlC synthetisiert im Pulsplasma Ofen. Die grün markierten Reflexe konnten TiC zu geordnet werden. Die orange markierten Reflexe konnten keiner bekannten Verbindung zugeordnet werden. Über die röntgenographische Untersuchung am Pulver werden in der Probe zwei Phasen nachgewiesen. Das Hauptprodukt war die MAX-Phase einer 211-Variante mit Gitterkonstanten von a = 3,0640(23) Å; c = 13,705(12) Å und einem Volumen von V = 111,43(16) Å3. Die Verfeinerung zeigt eine sehr gute Übereinstimmung mit den in der Literatur erfassten Werten. Eine phasenreine Darstellung gelang auch bei der 211-Phase nicht. Als Nebenprodukt wurde in sehr hohen Anteilen TiC beobachtet. In der Abbildung 3.4.7 ist das Pulverdiagramm von Ti2AlC (blau) dargestellt. Die grünmarkierten Reflexe konnten der binären Phase TiC zugeordnet werden. Zwei Reflexe (orange markiert) konnten keiner bekannten Verbindung zugeordnet werden. 3.4.3.3 EDX - Untersuchung an Einkristallen der 312-Phase Aus dem Ansatz Nummer 1 mit der Ausgangszusammensetzung von Ti6Al15C1 (Tabelle 3.4.3.1) konnten hexagonale Plättchen, wie in den Abbildungen 3.16 und 3.17 gezeigt, isoliert werden. Diese wurden einer qualitativer Elementaranalyse mittels energiedispersiver Röntgenemmissionsspektroskopie (EDX) unterzogen. Im ersten Fall konnte die EDX-Untersuchung sicherstellen, dass es sich hier um eine 312Phase mit einem geringen Anteil an Stickstoff handelt (siehe das Spektrum in Abbildung 3.4.9 - 71 - MAX – Phase – Das System Ti-Al-C und Tabelle 3.4.3.3). Bei der Untersuchung des zweiten Einkristalls handelte es sich um eine 211-Phase (siehe Kapitel 3.4.3.6), die Kristalle konnten optisch nicht voneinander unterschieden werden. Abbildung 3.4.8: REM-Aufnahme der Probe Ti3AlC1,82N0,18 Abbildung 3.4.9: EDX-Spektrum der Probe Ti3AlC1,82N0,18. Das dargestellte Ergebnis aus der EDX-Untersuchung ist das Resultat aus acht Messstellen an der Kristalloberfläche. Das Verhältnis der Metallatome steht in sehr guter Übereinstimmung mit der theoretischen Summenformel von Ti3AlC2 (Ti/Al : 3/1). Der gefundene Stickstoff lässt sich als Verunreinigung in den Edukten als TiN erklären. Tabelle 3.4.3.3: Verhältnis der Metallatome in der Probe Ti3AlC1,82N0,18 Element Gewichts [%] Atomist [%] σ [%] Atomssoll [%] Ti K 72,48 48,33 0,72 50,0 Al K 13,61 16,29 0,16 16,6 CK 11,92 32,07 0,27 33,3-x NK 1,73 3,02 0,56 x Die erhaltenen Informationen aus der EDX-Untersuchung dienen jetzt als Anhaltspunkt für die Verfeinerung des mit dem IPDS II gemessenen Einkristalls (Kapitel 3.4.3.4). Der LαÜbergang des Titans, der Kα-Übergang des Stickstoffs und der Kα-Übergang des Kohlenstoffs liegen bei sehr ähnlichen Energien. Somit tritt eine Peaküberlappung wie in Abbildung 3.4.8 gezeigt ein, welche den Nachweis von Stickstoff neben dem Titan erschwert. - 72 - MAX – Phase – Das System Ti-Al-C 3.4.3.4 Röntgenographische Untersuchung an Einkristallen der 312-Phase Bei der röntgenographischen Untersuchung an Einkristallen konnten Gitterkonstanten von a = 3,0606(5)7 Å; c = 18,498(5) Å in P63/mmc bestimmt werden. Die Größe der Gitterkonstanten ist charakteristisch für die 312-MAX-Phasen. Aus den 3204 gesammelten Reflexen stand ein Datensatz von 136 unabhängigen Reflexen nach einer Mittelung in der Lauenklasse 6/mmm zu Verfügung. Als Startmodell für die Verfeinerung wurden die Lageparameter von Ti3SiC2 benutzt, wobei die Lage 2b vollständig mit Aluminium besetzt wurde. Nach der vollständigen Besetzung aller kristallographischen Lagen (2a(Ti), 4f(Ti), 2b(Al), 4f(C)) konvergierte die Verfeinerung problemlos zu guten Zuverlässigkeitswerten. Die gewonnenen Informationen aus der EDX zeigten einen Anteil an Stickstoff mit 3,0(6) At.-%. Die Besetzungsfaktoren der Nichtmetalle wurden aneinander gekoppelt, fixiert und erneut verfeinert. Die wechselweise Freigabe der Besetzungsfaktoren auf den Lagen 4f, 2a und 2b zeigte im Rahmen der Standardabweichungen keine signifikante Abweichung von der vollständigen Besetzung. Bei der Überprüfung der Aluminiumlage konnte keine Misch- oder Fehlbesetzung festgestellt werden. Die anisotrope Verfeinerung der 12 freien Parameter lieferte Zuverlässigkeitswerte von R1(F) = 0,0338 und wR2(I) = 0,0826 bei einer Zusammensetzung von Ti3AlC1,82N0,18. In den Tabelle 3.4.3.4, .5 und .6 sind alle Informationen aus der röntgenographischen Untersuchung aufgeführt. 7 Die etwas verkleinerte Gitterkonstante deutet auf den Einbau von Stickstoff hin. - 73 - MAX – Phase – Das System Ti-Al-C Tabelle 3.4.3.4: Angaben zur Strukturanalyse von Ti3AlC1,82N0,18. Summenformel Ti3AlC1,82N0,18 Kristallsystem hexagonal Raumgruppe P63/mmc – Nr. 194 a = 3,0606(5); c = 18,498(5) Gitterkonstante [Å] 3 Volumen der Elementarzelle [Å ] 150,06(6) Zahl der Formeleinheiten 2 3 röntgenogr. Dichte [g/cm ] 4,310 F(000) 182,1 Kristallform, -farbe metallisch glänzenden hexagonale Plättchen Diffraktometer Rotation STOE IPDS II (Mo-Kα-Strahlung; λ=0,71073 Å Graphitmonochromator 0° ≤ ω ≤ 180° ψ = 0°, 111; ∆ω = 2° Belichtungszeit [sec.] 300 Messtemperatur [K] Raumtemperatur [293(2)] Kristallgröße [mm] 0,1 × 0,1 × 0,02 Absorptionskoeffizient [mm-1] 7,77 Messbereich [°] 4°<2θ<70° -2 ≤ h ≤ 0, Indexbereich 0 ≤ k ≤ 4, 0 ≤ l ≤ 27 Zahl der gemessenen Reflexe 3204 Zahl der unabh. Reflexe mit I > 0 136 I > σ2(I): 103 freie Parameter 12 Korrekturen Lorentz, Polarisation Absorptionskorrektur numerical / equivalent method (XSHAPE[68]) Strukturlösung Isotypie Ti3SiC2[69] Strukturverfeinerung SHELXTL-97[199] Wichtungsschema w-1 = σ2F02 + (0,0610 * P)2 + 0,00 * P P = (F02 + 2 FC2) / 3 Goodness of fit 0,814 Extinktionskorrektur 0,027(11) Restelektronendichte [e-/Å3]: +1,14 / -0,74 - 0,22 e-/Å3 R-Werte für I > 0: Alle Reflexe R1(F) = 0,0338 wR2(I) = 0,0826 R1(F) = 0,0392 - 74 - MAX – Phase – Das System Ti-Al-C Tabelle 3.4.3.5: Ortskoordinaten, Besetzungsfaktoren (in [%]) und Auslenkungsparameter von Ti3AlC1,82N0,18. Atom Lage Ti 1 y ⅔ 2a Ti 2 x ⅓ z 0,62780(7) sof Ueq 100* 0,0108(4) * 0,0105(5) 0 1 ½ 100 Al 4f 2b 0 ¾ ¾ 100* 0,0149(7) C/N 4f ⅓ ⅔ 0,5695(3) 93/7# 0,014(1) * bei wechselweiser Freigabe der Besetzungsfaktoren konnte keine Abweichung von vollständigen Besetzung beobachtet werden # Besetzungsfaktoren und Temperaturfaktoren auf EDX-Ergebnis gekoppelt und fixiert Atom U11 U22 U33 U23 U13 U12 Ti 1 0,0098(4) 0,0098(4) 0,0131(5) 0 0 0,0051(2) Ti 2 0,0102(4) 0,0102(4) 0,0127(6) 0 0 0,0047(3) Al 0,0153(10) 0,0153(10) 0,0142(13) 0 0 0,0078(5) C/N 0,0123(16) 0,0123(16) 0,015(23) 0 0 0,006(8) Die folgende Tabelle zeigt benachbarte Atome, ihren charakteristischen Abstand in Å und die Koordinationszahl in der ermittelten Ti3AlC1,82N0,18-Struktur. Tabelle 3.4.3.6: Ausgewählte Abstände und Winkeln in der Ti3AlC1,82N0,18-Struktur Benachbarte Abstand Anzahl der Winkel Atome [Å] Bindungen Ti - C/N - Ti Ti 1 2,869(1) x6 Al 3,061(1) x6 Ti 1 - C/N 2,1861(4) x6 Ti 2 - C/N 2,069(3) x3 95,36(2)° Al 2,8691(10) x3 87,78(3)° Ti 1 2,9515(11) x3 Ti 2 3,061(1) x6 Ti 1 2,069(3) x3 Ti 2 2,186(4) x3 Al - C/N - - 75 - MAX – Phase – Das System Ti-Al-C 3.4.3.5 Strukturdiskussion von Ti3AlC1,82N0,18 Ti3AlC1,82N0,18 kristallisiert im Ti3SiC-Typ in der Raumgruppe P63/mmc mit den Gitterkonstanten von a = 3,0606(5) Å und c = 18,498(5) Å. Die etwas verkürzte a- und c-Achse lassen sich durch die Tatsache der Substitution von Kohlenstoff durch Stickstoff erklären (vgl. Lit. Ti3AlC2 a = 3,072 Å; c = 18,732 Å[29]), die anhand von EDX- Untersuchungen dargelegte werden konnte. Hier ist eine lineare Abhängigkeit der Gitterkonstanten von Stickstoff und Kohlenstoff nach der Vegard’schen Regel zu erkennen. Das Einschleppen von Stickstoff kann als Verunreinigung erklärt werden, die in Form von TiN bereits in den Pressling eingewogen wurde. Die Struktur (Abbildung 3.2.2) lässt sich als eine dichteste Kugelpackung nach Jagodzinski als (hhhc)2 mit einer Stapelsequenz von ABABCBCB beschreiben. Die Oktaederlücke zwischen den drei Titanatomschichten sind mit Kohlen-/Stickstoffatomen besetzt, so dass die Dopelschichten aus kantenverknüpften (X)Ti6-Oktaedern bestehen (X = C/N). Diese Ti3X2 (mit x = C/N) Stapelelemente werden jeweils von vollständig besetzten Aluminiumschichten getrennt. Die Koordinationszahl der Aluminiumatome in den Schichten ist jeweils zwölf. An dem Beitrag sind jeweils sechs Aluminiumatome und sechs Titanatome beteiligt. Dabei sind immer drei Titanatome der darüber bzw. darunter liegenden Schicht in einem Abstand von 2,869(1) Å beteiligt. Abbildung3.4.10: Koordinationspolyeder Die Aluminiumatome sind mit 3,061(1) Å etwas von Aluminium weiter voneinander entfernt. In der Ti3AlC1,82N0,18-Struktur befinden zwei unterscheidbare Titanatome (mit Ti1 und Ti2 gekennzeichnete). Ti1 befindet sich auf der kristallographischen Lage 2a, hier kommt es zur einer 18fachen Koordination durch Kohlen-/ Stickstoffatome (sechsfach) im Abstand von 2,186(4) Å sowie durch Titanatome aus der darüber, darunter und in der Ebenen liegenden Schicht (zwölffach) mit einem Abstand von je 3,069(1) Å. Es kommt zu einer kuboktaedrischen Abbildung 3.4.11: Koordination durch Ti 1, Ti 2 und C/N-Atome um das Ti 1. - 76 - Koordinationspolyeder von Ti 1 MAX – Phase – Das System Ti-Al-C Bei der Koordination von Ti2 sind alle Atomsorten beteiligt. Drei Aluminiumatome aus der Aluminiumschicht sind im Abstand von 2,869(1) Å um Ti 2 umgeben. Sechs Atome des gleichen Typs (Ti2) entlang der gleichen Schicht zeigen eine Distanz von 2,9515(11) Å. Abbildung 3.4.12: Koordinationspolyeder von Ti 2 Die sich in den Oktaedern befindenden Nichtmetallatome C/N koordinieren das Ti2 dreifach im Abstand von 2,069(3) Å. Weitere drei Metallatome aus der Ti1 - Schicht sind an der Umgebung von Ti2 im Abstand von 2,952(1) Å beteiligt. Die Lage 4f ist hauptsächlich durch das Kohlenstoffatom besetzt. Aus der EDX- Untersuchung ginge eine Besetzung von 10 At.-% mit Stickstoff auf der Nichtmetalllage hervor, welches anschließend bei der Strukturverfeinerung berücksichtigt wurde. Das Nichtmetallatom C/N 1 befindet sich im Zentrum des von den Titanatomen Ti1und Ti2- Atomen aufgebauten Oktaeders. Dabei ist das Ti 1 mit einem Abstand von 2,069(3) Å etwas näher als das Ti2 mit 2,186(4) Å. Somit liegt der Abstand von Ti2- C/N mit 2,186(4) Å sehr nahe am Abstand wie es in TiC (2,164 Å) zu beobachten ist. 3.4.3.6 EDX - Untersuchung an Einkristallen der 211-Phase Wie schon oben angedeutet konnten aus dem Ansatz Nummer 1 (Tabelle 3.4.3.1) auch Einkristalle isoliert werden, die der H-Phase zugeordnet werden konnten. Rein optisch ist eine Unterscheidung der 312 und 211- Phase nicht möglich. Eine röntgenographische Untersuchung auf dem Einkristalldiffraktometer und die dazugehörige qualitative Elementaranalyse bestätigte die Anwesenheit der 211-Phase mit einer Zusammensetzung von Ti2AlC0,28N0,72. Abbildung 3.4.13: REM-Aufnahme der Probe Ti2AlC0,28N0,72. Abbildung 3.4.14: Ti2AlC0,28N0,72. - 77 - Messspektrum der Probe MAX – Phase – Das System Ti-Al-C Das zusammengefasste Resultat aus vier Messstellen an den untersuchten Einkristallen ist in Tabelle 3.4.3.7 zusammengetragen. Auch hier zeigt sich der Stickstoff jedoch in einer viel größeren Präsenz als gedacht. Für die Nichtmetalllage 2a bedeutet das eine Besetzung von 72% mit Stickstoffatomen. Tabelle 3.4.3.7: Verhältnis der Metallatome in der Probe Ti2AlC0,28N0,72. Element Gewichts [%] Atomist [%] σ [%] Atomssoll [%] Ti K 68,71 46,21 1,2 50,0 Al K 19,01 22,73 0,61 25,0 CK 3,87 11,67 1,18 25,0-x NK 7,78 18,67 1,4 x 3.4.3.7 Röntgenographische Untersuchung an Einkristallen der 211-Phase Bei der röntgenographischen Untersuchung an einem isolierten Einkristall konnte durch die charakteristischen Gitterkonstanten auf das Auftreten von einer 211-Phase geschlossen werden. Die bestimmten Gitterkonstanten waren a = 3,0167(4) Å und c = 13,684(3) Å. Nach der Sammlung von Reflexintensitäten im Bereich von 2° < 2θ < 70° und der Mittelung in der Laueklasse 6/mmm gefolgt von einer Absorptionskorrektur, erhielt man einen Datensatz mit 92 unabhängigen Reflexen. Aufgrund der charakteristischen Gitterkonstanten und die sichere Wahrscheinlichkeit, dass es sich auch hier um eine 211-Phase handelt, wurde in der zentrosymmetrischen Raumgruppe P 63/mmc mit der Strukturanalyse begonnen. Die ermittelte Gitterkonstante a = 3,0167(4) Å liegt deutlich unter der in der Literatur bekannten stöchiometrischen Ti2AlC-Phase (mit a = 3,056 Å[40]), die vollständig mit Kohlenstoffatomen auf der Nichtmetalllage besetzt ist. Der Vergleich der Literaturdaten zeigt eine a-Gitterkonstante für Ti2AlN von a = 3,0099 Å[70], bei der eine reine Besetzung durch Stickstoffatome auf der 2a Lage vorliegt. Aus der EDX-Untersuchung war bekannt, dass der Stickstoffanteil in der Probe bei 18(1,5) At.-% liegt. Auch bei der Strukturverfeinerung lag das Startmodell einer bekannten H-Phase (Cr2AlC[33]) zugrunde. Die Verfeinerung der Nichtmetalle erfolgte hier durch die Kopplung der Besetzungs- und Temperaturfaktoren. Die anschließende, anisotrope Verfeinerung der 9 freien Parameter führte zur einer Struktur mit der Summenformel von Ti2AlC0,28N0,72 mit den Zuverlässigkeitswerten von R1(F) = 0,0235 und von wR2(I) = 0,0541. - 78 - MAX – Phase – Das System Ti-Al-C Tabelle 3.4.3.8: Angaben zur Strukturanalyse von Ti2AlC0,28N0,72. Summenformel Ti2AlC0,28N0,72 Kristallsystem hexagonal Raumgruppe P63/mmc a = 3,0167(4); c = 13,684(3) Gitterkonstante [Å] 3 Volumen der Elementarzelle [Å ] 107,8(4) Zahl der Formeleinheiten 2 3 röntgenogr. Dichte [g/cm ] 4,195 F(000) 127,4 Kristallform, -farbe metallisch glänzende, hexagonale Plättchen Diffraktometer Rotation STOE IPDS II (Mo-Kα-Strahlung; λ=0,71073 Å Graphitmonochromator 0° ≤ ω ≤ 180° ψ = 0°, 111; ∆ω = 2° Belichtungszeit [sec.] 300 Messtemperatur [K] Raumtemperatur [293(2)] Kristallgröße [mm] 0,08 × 0,08 × 0,01 Absorptionskoeffizient [mm-1] 7,36 Messbereich [°] 4°<2θ<70° -4 ≤ h ≤ 4; -4 ≤ k ≤ 4; Indexbereich -20 ≤ l ≤ 20 Zahl der gemessenen Reflexe 1762 Zahl der unabh. Reflexe mit I > 0 92 I > σ2(I): 68 freie Parameter 9 Korrekturen Lorentz, Polarisation Absorptionskorrektur numerical / equivalent method (XSHAPE[68]) Strukturlösung Isotypie Cr2AlC[33] Strukturverfeinerung SHELXTL-97[199] Wichtungsschema w-1 = σ2F02 + (0,346 * P)2 + 0,00 * P P = (F02 + 2 FC2) / 3 Goodness of fit 0,877 Extinktionskorrektur 0,069(15) Restelektronendichte [e-/Å3]: +0,80 / -0,67 - 0,15 e-/Å3 R-Werte für I > 0: Alle Reflexe R1(F) = 0,0235 wR2(I) = 0,0541 R1(F) = 0,0367 - 79 - MAX – Phase – Das System Ti-Al-C Auch hier wurden die Besetzungsfaktoren der Metalle wechselweise freigegeben, um auf mögliche Abweichungen von der vollständigen Besetzung zu prüfen. Weitere Einzelheiten der Strukturbestimmung sowie Besetzungsfaktoren, Temperatur-faktoren und Abstände der benachbarten Atome sind den Tabelle 3.4.3.8, 3.4.3.9 und 3.4.3.10 zu entnehmen. Tabelle 3.4.3.9: Ortskoordinaten, Besetzungsfaktoren (in [%]) und Auslenkungsparameter von Ti2AlC0,28N0,72. Atom Lage x y z sof Ueq ⅓ ⅔ 0,58497(7) 100 * 0,0059(3) Al 4f 2d ⅔ ⅓ ¾ 100 * 0,0116(6) C/N 2a 0 0 0 28/72 # 0,0086(12) Ti * bei wechselweiser Freigabe der Besetzungsfaktoren keine signifikante Restelektronendichte vorhanden # Besetzungsfaktoren und Temperaturfaktoren auf EDX-Ergebnis gekoppelt und fixiert Atom U11 U22 U33 U23 U13 U12 Ti 0,0055(4) 0,0055(4) 0,0068(4) 0 0 0,0027(2) Al 0,0108(8) 0,0108(8) 0,01318(9) 0 0 0,0054(4) C/N 0,0068(18) 0,0068(18) 0,011(2) 0 0 0,0034(9) Die folgende Tabelle zeigt benachbarte Atome, ihren charakteristischen Abstand und die Koordinationszahl. Tabelle 3.4.3.10: Ausgewählte Abstände und Winkeln in der Ti2AlC0,28N0,72-Struktur Benachbarte Abstand Anzahl der Winkel Atome [Å] Bindungen Ti - C/N - Ti Ti 2,8518(8) x6 Al 3,0167(4) x6 C/N 2,0941(6) x6 92,16(3)° Al 2,8518(8) x3 87,84(3)° Ti 2,9053(16) x3 Ti 3,0167(4) x3 Ti 2,0941(6) x6 Al - Ti - C/N - - 80 - MAX – Phase – Das System Ti-Al-C 3.4.3.8 Strukturdiskussion von Ti2AlC0,28N0,72 Ti2AlC0,28N0,72 kristallisiert im Cr2AlC-Typ somit in der Raumgruppe P63/mmc mit den Gitterkonstanten von a = 3,0167(4) Å und c = 13,684(3) Å. Die Struktur der 211-Phase wurde bereits im Kapitel 3.2 vorgestellt. Titan besetzt vollständig die Lage 4f und bildet die Oktaeder aus, in denen sich das Nichtmetallatom befindet. Jedes Nichtmetall liegt im Zentrum des Oktaeders im Abstand von 2,0941(6) Å. Aluminium belegt vollständig die Position 2d. Die Aluminiumatome bilden eine Trennschicht zwischen den kantenverknüpften Oktaedern. Die Nichtmetalllage ist durch eine Mischbesetztung von Kohlenstoffatomen und Stickstoffatomen geprägt. Mit 72 % Stickstoff auf der 2a Lage ist auch die Verkürzung der a-Gitterkonstanten plausibel. Die Veränderung der a-Gitterkonstanten ist in Formel 3-5 dargestellt. Der Einbau von Stickstoff in Ti2AlC macht sich überwiegend in der a-Gitterkonstanten bemerkbar. In c-Richtung kommt es nur zu einer sehr geringen Schrumpfung von ca. 0.5% somit von 13,684 Å in Ti2AlC auf 13,623 Å in Ti2AlN. Einbau von C-Atomen Ti2 AlN : a = 3,0099Å → Ti 2 AlC : a = 3,056Å - 81 - Formel 3-5 MAX – Phase – Das System Ti-(Al/Sn)-C 3.4.4 Arbeiten in einer Aluminium/Zinn-Schmelze Der Bereich für eine Darstellung von MAX-Phasen im System Titan-AluminiumKohlenstoff in einer reinen Aluminiumschmelze ist sehr schmal (siehe Kapitel 3.4.3). Bei Temperaturen von 1600°C konnten erfolgreich die zwei MAX-Phasen mit den Varianten 211 und 312 in einkristalliner Form gezüchtet werten, jedoch mit einer Mischbesetzung der Nichtmetalllage durch Kohlenstoff und Stickstoff. Eine Hilfsmetallbadtechnik hat viele Vorteile wie die Darstellung von großen und gut ausgebildeten Kristallen aus der Schmelze. Doch der größte Vorteil ergibt sich bei der Wahl des richtigen Lösungsmittelgemisches durch eine erhebliche Herabsenkung der Temperatur des Bereichs der homogenen Schmelze. Für eine weitere Darstellung von MAX-Phasen wurde hier als Lösungsmittelgemisch Aluminium mit Zinn ausgewählt. Das Phasendiagramm des ausgewählten Lösungsmittelgemisch (Abbildung 3.4.15) wird im nächsten Kapitel vorgestellt. Das Ziel der Versuchsserie ist zum einen die Kristallisationsbedingungen der MAX-Phasen in unterschiedlichen Schmelzen zu erforschen, sowie die Analyse des Substitutionsschemas der Hauptgruppenmetalle auf der speziellen Lage, d. h. hier der Austausch von Aluminium gegen Zinn gesteuert durch das Mischverhältnis der Schmelzzusammensetzung. Frühere Untersuchungen im quaternären System konnte die Entstehung der H-Phase mit einer vollständigen Besetzung durch Zinn auf der 2d Metalllage (Ti2SnC: a = 3,163 Å und c=13,670 Å [71] ) bestätigen. Auch von weiteren 211-Phasen, bei denen eine Mischbesetzung durch Aluminium und Zinn vorlag (Ti2(Al/Sn)C)[167], wurde berichtet. Die „höheren“ MAXPhasen mit der 312- und 413-Variante konnte erstmalig 1998 von Lückeroth beobachtet werden[72]. 3.4.4.1 Die binären Systeme Aluminium-Zinn und Titan-Zinn Das Phasendiagramm von Aluminium und Zinn ist ein einfaches eutektisches System mit der Unlöslichkeit der Metalle im festen Zustand und einer vollständigen Mischbarkeit der Schmelze. Somit liegt oberhalb von 660 ± 15°C eine homogene Schmelze der Metalle vor. Im Fall des Phasendiagramms von Titan und Zinn bilden sich die binären Phasen nur in der titanreichen Ecke des Zweistoffsystems. Die titanreichste Phase ist Ti3Sn kristallisiert im - 82 - MAX – Phase – Das System Ti-(Al/Sn)-C Ni3Sn-Typ, titanärmer sind die Phasen Ti2Sn (Ni2Sn), Ti5Sn3 (Mn5Si3-Typ) und α- sowie βTi6Sn5 (jeweils eigener Typ). Abbildung 3.4.15: Das binäre Phasendiagramm des Systems Aluminium-Zinn Abbildung 3.4.16: Das binäre Phasendiagramm des Systems Titan-Zinn Für das binäre System von Kohlenstoff und Zinn liegt kein Phasendiagramm vor, die Untersuchungen von diesem System zeigten ein Unlöslichkeit der beiden Elemente ineinander[73]. Untersuchungen in dem Dreistoffsystem Titan-Zinn-Kohlenstoff wurden bislang nur in der titanreichen Ecke durchgeführt. Im Laufe dieser Arbeit konnte Ti3SnC2 von der Arbeitsgruppe Dubois in Frankreich zum ersten Mal synthetisiert und mit Hilfe einer Rietveldverfeinerung charakterisiert werden (a = 3,1366 Å; c = 18,650 Å in P63/mmc)[74]. Die H-Phase ist bereits vollständig an den Einkristallen in früheren Arbeiten charakterisiert worden (Ti2SnC a = 3,1860 Å; c = 13,630 Å in P 63/mmc)[36]. 3.4.4.2 Präparative Untersuchung im System Titan-Aluminium/Zinn-Kohlenstoff In dieser Versuchsserie wurde der Titan- und Kohlenstoffgehalt konstant gehalten. Ausgehend von einem zinnarmen (somit aluminiumreichen) Ansatz bis zu einem nur mit Zinn eingewogenen Ansatz sollte das Substitutionsverhalten ausgiebig ergründet werden. Die Elemente wurden als Pulver eingesetzt, homogenisiert und kalt verpresst (Tabelle 3.4.4.1). Die erhaltenen Tabletten wurden in Korundtiegeln bei Temperaturen von 1500°C in einem Rohrofen unter Argon-Schutzgasatmosphäre umgesetzt. - 83 - MAX – Phase – Das System Ti-(Al/Sn)-C Temperaturprogramm: 150°C/h RT -5°C/h -20°C/h 1500°C 1400°C (20h halten) (2h halten) -100°C/h 1000°C RT (1h halten) Nach dem Abkühlen der Proben wurde das überschüssige Hilfsmetall mit verd. Salzsäure über die Nacht vollständig entfernt. Der Rückstand wurde abgesaugt und mehrmals mit Wasser gewaschen. In jedem Ansatz befanden sich metallisch glänzende, hexagonale Plättchen mit einer durchschnittlichen Kantenlänge von 0,03 mm und einer Dicke von 0,005 mm. Die Einwaage, das Ergebnis der Phasenanalyse der sechs Ansätze sowie die Verfeinerung der ermittelten Gitterkonstanten der MAX-Phasen sind in der Tabelle 3.4.4.1 zu finden. Nr. Gehalt [At.-%] Zusammensetzung Gitterkonstante [Å] Ti Al Sn C 1 Ti6Al10Sn5C1 27,3 45,5 22,7 4,5 2 Ti5,8Al9Sn6C1 26,6 41,3 27,5 4,6 3 Ti6Al7,5Sn7,5C1 27,3 34,1 34,1 4,5 4 Ti6Al5Sn10C1 27,3 22,7 45,5 4,5 5 Ti6Sn15C1 27,3 - 68,2 4,5 der MAX-Phase a = 3,1158(5); c = 13,716(4) V=115,32(4) a = 3,125(1); c = 13,726(4) V=116,06(5) a = 3,116(3); c = 13,724(8) V=115,43(9) a = 3,117(2); c = 13,733(7) V=115,51(9) a = 3,158(7); c = 13,701(3) V=118,17(4) weitere kristalline Phasen - - - - - Tabelle 3.4.4.1: Phasenanalyse der Proben aus dem Vierstoffsystem Titan- Aluminium/Zinn-Kohlenstoff. sowie die H-Phase mit den verfeinerten Gittekonstanten. In allen Ansätzen ist eine phasenreine Darstellung der 211-Phase gelungen. Das Ergebnis ist jedoch nur nicht zufriedenstellend. Die Betrachtung der ermittelten Gitterkonstanten und der Volumina zeigt nahezu einer identischen Größenordnung. Nur Probe Nummer 58 zeigt eine Übereinstimmung des Volumens und der Gitterkonstanten mit der aluminiumfreien Ti2SnC-Phase. Diese Tatsache lässt die Vermutung aufkommen, dass sich eine 8 Bei diesem Ansatz wurde ausschließlich in einer Zinnschmelze gearbeitet um eine Referenzsubstanz zu synthetisieren die den maximalen Gehalt an Zinn trägt. - 84 - MAX – Phase – Das System Ti-(Al/Sn)-C thermodynamische stabile Form der MAX-Phase durchgesetzt hat. Somit ist unabhängig vom Aluminium/Zinn-Verhältnis ein bestimmtes Al/Sn Verhältnis in der Struktur begünstigt. Zu erwarten wäre ausgehend von der Ti2AlC Verbindung eine Aufweitung der aGitterkonstanten von bis zu ∆ = 0,11 Å. Tabelle 3.4.4.2: Vergleich der Gitterkonstanten und Volumina Ti2AlC Substitution Ti2SnC gegen Sn a = 3,056 Å a = 3,163 Å c = 13,623 Å c = 13,670 Å Vol. = 110,18 Å3 Vol. = 118,44 Å3 3.4.4.3 Synthese der 312-Phase im SPS-Ofen Bei der Darstellung der von MAX-Phasen aus dem Dreistoffsystem Titan-Zinn-Kohlenstoff ist man wie üblich vorgegangen. Die Pulver wurden in stöchiometrischen Verhältnissen innig miteinander verrieben und in einer Graphitmatrize, die mit Graphitfolie ausgekleidet war, umgesetzt. Die Versuche wurden bei Temperaturen von 950 bis 1500°C umgesetzt. Die Reaktionszeit betrug dabei jeweils 5 Minuten. Alle Versuche wurden aus den Elementen durchgeführt, für Kohlenstoff wurde das Graphit eingewogen. Nur bei einer Probe bestehend aus 0,191 g (3,993 mmol) Titan, 0,189 g (1,592 mmol) Zinn und 0,019 g (1,598 mmol) Graphit konnte die erfolgreiche Bildung der 211-Phase beobachtet werden. Die Umsetzung erfolgte bei einer Temperatur von 1200°C. Die 211-Phase entstand als eines von vier Produkten. Als Hauptprodukt dominierte das Ti3Sn (orange). In der Abbildung 3.4.17 sind alle Phasen mit ihren charakteristischen Reflexen dargestellt (hellgrün Ti3Sn, blau Ti2SnC). Temperaturprogramm: 2 min. RT 2 min. 600°C 2 min 800°C 2 min 1000°C 45 min 1200 (5 min. halten) - 85 - RT MAX – Phase – Das System Ti-(Al/Sn)-C 40000 Lin (Counts) 30000 20000 10000 0 5 10 20 30 40 50 60 70 80 90 100 110 120 2-Theta - Scale Abbildung 3.4.17: Pulverdiffraktogramm von Ti2SnC (Nebenprodukt blau); synthetisiert im PulsplasmaOfen. Die grünen markierten Reflexe konnten Ti3Sn zugeordnet werden. Als weitere Phase wurden Ti8C5 (orange), Ti5Sn3 (dunkelrot) und Graphit (violett) beobachtet. Die Umsetzung der Sinterprobe erfolgte unter einem Vakuum von 0,13 mbar. Der Druck für die Sintersynthese wurde auf 4,2 kN eingestellt. Bei der Sinterung wurde ein Schwund der Probe von 9% aufgezeichnet. Nach der Abkühlung der Sinterprobe wurde der Pressling vom der Graphitfolie befreit und röntgenographisch analysiert. Die beobachtete Gitterkonstante der 211-Phase betrug a = 3,164(3) Å und c = 13,933(12) Å mit einem Volumen von V = 120,8(6) Å3. Die verfeinerten Gitterkonstanten finden sehr gut Übereinstimmung mit der Literatur. 3.4.4.4 EDX - Untersuchung an Einkristallen Aus jedem Ansatz wurde mindestens ein Einkristall entnommen und qualitativ auf die Elemente hin analysiert. Alle untersuchten Einkristalle waren hexagonale Plättchen, wie sie in den folgenden Abbildungen abgebildet sind. Jeder Kristall wurde an mehreren Stellen gemessen. Das dargestellte Ergebnis der folgenden EDX-Messungen ist jedes Mal ein Mittelwert aus mindestens fünf Messstellen. Bei den Untersuchungen der MAX-Phasen hat sich die EDX-Untersuchung als sehr nützliche, wenn nicht schon essentielle Analysemethode - 86 - MAX – Phase – Das System Ti-(Al/Sn)-C durchgesetzt. Das Ergebnis der Untersuchung an Einkristallen mittels energiedispersiver Röntgenemission ist in den folgenden Tabellen dargestellt. Einkristall aus der Probe Nummer 1 Tabelle 3.4.4.3: Ergebnis der EDX-Analyse: Das Verhältnis der Elemente als Durchschnittswert aus fünf Messungen am Einkristallen aus dem Ansatz Nummer 1. (rechts) REM-Bild des gemessenen Einkristalls. Element Gewichts [%] Atomist [%] σ [%] Atomssoll [%] Ti K 51,70 48,08 1,9 50,0 Al K 8,08 12,94 0,39 16,7 Sn K 31,82 12,01 0,45 8,3 CK 3,64 12,20 1,21 25,0-x NK 4,75 14,69 1,18 x Das eingewogene Metallverhältnis konnte die EDX-Untersuchung nicht bestätigen. Die EDX-Analyse zeigt ca. 13 At.-% Al anstatt der Erwarteten 16,7 At.-%. Zu alldem konnte ein beträchtlicher Anteil an Stickstoff detektiert werden und fällt mit 14,7 At.-% stark ins Gewicht. Der Stickstoffanteil in der Probe lässt sich als Verunreinigung erklären. Einkristall aus der Probe Nummer 2 Tabelle 3.4.4.4: Ergebnis der EDX-Analyse: Das Verhältnis der Elemente als Durchschnittswert aus sechs Messungen am Einkristallen aus dem Ansatz Nummer 2. (rechts) REM-Bild des gemessenen Einkristalls. Element Gewichts [%] Atomist [%] σ [%] Atomssoll [%] Ti K 48,15 47,18 0,23 50,0 Al K 5,34 9,93 0,64 15 Sn K 48,15 14,78 0,66 10 CK 5,82 22,37 0,77 25,0-x NK 1,57 5,75 0,45 x Auch der untersuchte Einkristall aus dem zweiten Ansatz zeigt eine große Diskrepanz bezüglich der erwartenden Werte. Das Metallverhältnis entspricht auch hier nicht der Einwaage. In etwas geringeren Anteilen, jedoch nicht vernachlässigbar klein, konnte der Stickstoff wieder einmal detektiert werden. - 87 - MAX – Phase – Das System Ti-(Al/Sn)-C Einkristall aus der Probe Nummer 4 Tabelle 3.4.4.5: Ergebnis der EDX-Analyse: Das Verhältnis der Elemente als Durchschnittswert aus acht Messungen an Einkristallen aus dem Ansatz Nummer 4. (rechts) REM-Bild des gemessenen Einkristalls. Element Gewichts [%] Atomist [%] σ [%] Atomssoll [%] Ti K 47,85 47,74 0,91 50,0 Al K 4,87 8,74 0,34 8,3 Sn K 39,99 16,34 0,44 16,7 CK 4,41 17,46 1,42 25,0-x NK 2,89 9,74 0,45 x Bei der Untersuchung des Einkristalls aus dem Ansatz Nummer 3 konnte zum ersten Mal im Rahmen der Standardabweichung eine Übereinstimmung des Metallverhältnisses mit der Schmelzzusammensetzung bestätigt werden. Stickstoff konnte auch hier in recht hohen Anteilen gefunden werden. Einkristall aus der Probe Nummer 5 Tabelle 3.4.4.6: Ergebnis der EDX-Analyse: Das Verhältnis der Elemente als Durchschnittswert aus fünf Messungen an Einkristallen aus dem Ansatz Nummer 5. (rechts) REM-Bild des gemessenen Einkristalls. Element Gewichts [%] Atomist [%] σ [%] Atomssoll [%] Ti K 41,00 46,22 0,73 50,0 Al K 0,87 1,57 0,15 - Sn K 51,48 23,53 0,34 25 CK 4,26 19,11 0,49 25,0-x NK 2,40 9,58 1,26 x Im letzten Ansatz der Versuchsreihe sollte ohne Aluminium gearbeitet werden. Die Untersuchung des gezüchteten Einkristalls aus diesem Ansatz brachte Spuren von Aluminium mit einem Anteil von 1,6(2) At.-%. Das Aluminium kann bei der Herstellung der Tabletten mit eingeschleppt worden sein, da der gleiche Achat-Mörser wie auch die gleiche Tablettepresse für die Herstellung verwendet wurde. Auch bei dieser Probe konnte Stickstoff mit recht hohen Anteilen nachgewiesen werde. In allen untersuchten Einkristallen konnte Stickstoff als Verunreinigung detektiert werden, was die Interpretation der Versuchsserie erschwert (mehr dazu in Kapitel 3.4.4.6). Jedoch - 88 - MAX – Phase – Das System Ti-(Al/Sn)-C lässt sich jetzt schon sagen, dass die Steuerung der Zusammensetzung nicht so einfach über die Stöchiometrie der Schmelzzusammensetzung realisierbar ist. 3.4.4.5 Röntgenographische Untersuchung an Einkristallen der 211-Phase Einkristalle aus jedem Ansatz aus dem oben diskutierten quaternären System wurden unter einem Lichtmikroskop entnommen und auf dem IPDS II vermessen. Die sehr kleinen hexagonalen Plättchen hatten eine durchschnittliche Kantenlänge von 50 µm und ein Dicke von ca. 10 µm. Bei allen analysierten Einkristallen konnten Gitterkonstanten zugeordnet werden, die für die 211-Phasen repräsentativ sind. Es wurde eine Strukturverfeinerung in der zentrosymmetrischen Raumgruppe P63/mmc basierend auf den Atomkoordinaten von Cr2AlC[33] mit zufriedenstellenden Zuverlässigkeitswerten durchgeführt (Tabelle 3.4.4.7). Aus der im Vorfeld vorangegangen Charakterisierung (Phasenanalyse und EDX-Untersuchung) wurde eine Mischbesetzung von Aluminium mit Zinn auf der 2d Lage und Kohlenstoff mit Stickstoff auf der kristallographischen 2a Position angenommen. Die Besetzung wurde gemäß dem Resultat aus der EDX-Analyse angeglichen und bei wechselweiser Freigabe der Besetzungsfaktoren frei verfeinert. Die Freigabe der Besetzungsfaktoren der Titanposition auf der Lage 4f zeigte keine signifikante Abweichung von einer vollständigen geordneten Besetzung. Die Restelektronendichten lagen in einem sinnvollen Rahmen. Die Strukturverfeinerung aus den gesammelten Daten zeigt eine nicht steuerbare Substitution der Hauptgruppenmetalle. Eine genaue Gegenüberstellung der gesammelten Daten aus den fünf Einkristalluntersuchungen, sowie die Messparameter sind in der Tabelle 3.4.4.7 zu finden. Einen Überblick über die Ortskoordinaten, Besetzungsfaktoren und Auslenkparameter der verfeinerten Strukturen gibt Tabelle 3.4.4.8. Der Vergleich der ausgewählten Abstände mit den dazu gehörigen Koordinationszahlen wird in Tabelle 3.4.4.9 dargelegt. - 89 - MAX - Phase – Das System Ti-(A/Sn)l-C Tabelle 3.4.4.7: Angaben zur Strukturanalyse von den H-Phasen aus einer Aluminium/Zinn-Schmelze Ti2Al1-xSnxC1-yNy. Summenformel Ti2Al0,46Sn0,54C0,45N0,55 Ti2Al0,42Sn0,58C0,69N0,31 Ti2Al0,48Sn0,52C0,7N0,3 Kristallsystem hexagonal Raumgruppe P 63/m m c – Nr. 194 Gitterkonstante [Å] Volumen der Elementarzelle [Å3] Ti2Al0,33Sn0,67C0,33N0,67 Ti2Al0,06Sn0,94C0,67N0,33 a=3,0840(4); a=3,1141(4); a=3,0822(4); a=3,1041(4); a=3,1465(4); c=13,6822(27) c=13,6907(27) c=13,648(3) c=13,6595(27) c=13,663(4) 112,7(7) 115,13(10) 112,29(6) 114,05(5) 117,15(6) Z=2 Zahl der Formeleinheiten röntgenogr. Dichte [g/cm3] 5,464 5,461 5,397 5,743 6,271 F(000) 167,1 169,9 164,5 176,5 195,9 metallisch glänzende hexagonale Plättchen Kristallform, -farbe STOE IPDS II - (Mo-Kα-Strahlung; λ=0,71073 Å Graphitmonochromator Diffraktometer 0° ≤ ω ≤ 180° und ψ = 0°,111°; ∆ω = 2° Belichtungszeit [sec.] 300 Messtemperatur [K] Raumtemperatur [293(2)] Kristallgröße [mm] 0,06 × 0,06 × 0,012 0,03 × 0,03 × 0,01 0,04 × 0,04 × 0,01 0,04 × 0,04 × 0,01 0,04 × 0,03 × 0,01 Absorptionskoeffizient [mm-1] 12,67 12,88 12,50 12,10 16,15 Messbereich [°] 4° < 2θ <64,99° 4° < 2θ <64,99° 4° < 2θ <64,99° 4° < 2θ <64,99° 4° < 2θ <64,99° -4 ≤ h ≤ 4 -4 ≤ h ≤ 4 -4 ≤ h ≤ 4 -4 ≤ h ≤ 4 -4 ≤ h ≤ 4 -4 ≤ k ≤ 4 -4 ≤ k ≤ 4 -4 ≤ k ≤ 4 -4 ≤ k ≤ 4 -4 ≤ k ≤ 4 -20 ≤ l ≤ 20 -20 ≤ l ≤ 20 -20 ≤ l ≤ 20 -20 ≤ l ≤ 20 -20 ≤ l ≤ 20 Indexbereich - 90 - MAX - Phase – Das System Ti-(A/Sn)l-C 1693 1720 1401 2769 1426 103 105 102 102 104 I > σ2(I): 78 76 88 78 97 freie Parameter 8 9 10 9 9 Tmin. = 0,3116 Tmin. = 0,1307 Tmin. = 0,3693 Tmin. = 0,1466 Tmin. = 0,4049 Tmax. = 0,6724 Tmax. = 0,2578 Tmax. = 0,651 Tmax. = 0,2185 Tmax. = 0,6243 Zahl der gemessenen Reflexe Zahl der unabh. Reflexe mit I > 0 Transmission Lorentz, Polarisation Korrekturen numerical / equivalent method (XSHAPE[68]) Absorptionskorrektur Strukturlösung Isotypie Cr2AlC[33] Strukturverfeinerung SHELXTL-97[199] Wichtungsschema w-1 = σ2F02 + (a * P)2 + b * P a = 0,0368 a = 0,0452 a = 0,0599 a = 0,0170 a = 0,0 b = 0,0 b = 0,0 b = 0,2 b = 0,0 b = 0,37 Goodness of fit 1,041 0,847 1,173 0,805 1,032 Extinktionskorrektur 0,033(9) 0,031(8) 0,071(17) 0,0167(26) 0,0397(5) +0,64 / -0,78 +0,88 / -1,04 +0,84 / -0,87 +0,46 / -0,45 +0,68/ -1,29 0,20 e-/Å3 0,24 e-/Å3 0,25 e-/Å3 0,13 e-/Å3 R1(F) = 0,0193 R1(F) = 0,0282 R1(F) = 0,0328 R1(F) = 0,0141 R1(F) = ,0166 wR2(I) = 0,0556 wR2(I) = 0,0658 wR2(I) = 0,0891 wR2(I) = 0,0271 wR2(I) =0,0388 R1(F) = 0,0234 R1(F) = 0,0372 R1(F) = 0,0353 R1(F) = 0,0184 R1(F) = 0,0166 P= (F02 +2 FC2) /3 Restelektronendichte [e-/Å3]: 0,17 e-/Å3 R-Werte für I > 0: Alle Reflexe - 91 - MAX - Phase – Das System Ti-(A/Sn)l-C Tabelle 3.4.4.8: Ortskoordinaten, Besetzungsfaktoren (in [%]) und Auslenkungsparameter von den H-Phasen Ti2Al1-xSnxC1-yNy. # Besetzungsfaktoren und Temperaturfaktoren gekoppelt, *Der Besetzungsfaktor des Ti wurde wechselweise freigegeben, es zeigte sich keine signifikante Abweichung (σ= 2%) Summenformel Ti2Al0,46Sn0,54C0,45N0,55 Ti2Al0,42Sn0,58C0,69N0,31 Ti2Al0,48Sn0,52C0,7N0,3 Ti2Al0,33Sn0,67C0,33N0,67 Ti2Al0,06Sn0,94C0,67N0,33 Ti x ⅓ ⅓ ⅓ ⅓ ⅓ y ⅔ ⅔ ⅔ ⅔ ⅔ z 0,08333(4) 0,08286(7) 0,08334(4) 0,08288(5) 0,08198(6) sof 100% * 100% * 100% * 100% * 100% * Ueq. 0,0077(2) 0,0052(3) 0,0113(5) 0,0053(2) 0,0068(3) U11 = U22 0,0088(3) 0,0053(4) 0,0119(6) 0,0057(2) 0,0069(3) U33 0,0057(4) 0,0050(4) 0,0101(7) 0,0048(3) 0,0065(5) 0 0 0 0 0 0,0044(2) 0,0026(2) 0,0059(3) 0,0028(1) 0,0034(1) U23 = U13 U12 Al/Sn x ⅓ ⅓ ⅓ ⅓ ⅓ y ⅔ ⅔ ⅔ ⅔ ⅔ z ¾ ¾ ¾ ¾ ¾ 54,0% Al 1 / 46,0% Sn 1 # 41,4% Al 1 / 58,6% Sn 1 # 47,8% Al 1 / 52,2 % Sn 1 # 33,3% Al 1 / 66,7% Sn 1 # 93,5% Al 1 / 6,5% Sn 1 # sof Ueq. 0,0128(3) 0,0093(4) 0,0155(5) 0,0117(2) 0,0089(3) U11 = U22 0,0164(4) 0,0110(5) 0,0186(5) 0,0142(3) 0,0103(2) U33 0,0056(3) 0,0059(5) 0,0092(5) 0,0068(3) 0,0061(3) 0 0 0 0 0 0,0082(2) 0,0054(2) 0,0093(3) 0,0071(2) 0,0052(1) x 0 0 0 0 0 y 0 0 0 0 0 z 0 0 0 0 0 45,0% C 1 / 55,0% N 1 # 68,9% C 1 / 31,1% N 1 # 70,0% C 1 / 30,0% N 1 # 33,3% C 1 / 66,7% N 1 # 66,7% C 1 / 33,0% N 1 # 0,0100(11) 0,0069(16) 0,0125(11) 0,0105(10) 0,010(1) U23 = U13 U12 C/N 1 sof Ueq. - 92 - MAX - Phase – Das System Ti-(A/Sn)l-C Tabelle 3.4.4.9: Ausgewählte Abstände in den H-Phasen, gewonnen aus einer Aluminium/Zinn-Schmelze mit der Summenformel Ti2Al1-xSnxC1-yNy. Summenformel Ti2Al0,46Sn0,54C0,45N0,55 Ti2Al0,42Sn0,58C0,69N0,31 Ti2Al0,48Sn0,52C0,7N0,3 Ti2Al0,33Sn0,67C0,33N0,67 Ti2Al0,06Sn0,94C0,67N0,33 Ti - C/N 2,1143(4) x3 2,1259(6) x3 2,1119(4) x3 2,1198(4) x3 2,1342(5) x3 - Ti 2,8930(9) x3 2,8949(15) x3 2,8881(4) x3 2,8876(11) x3 2,8842(12) x3 - Al/Sn 2,8932(5) x3 2,9101(8) x3 2,8880(6) x3 2,9023(7) x3 2,9276(7) x3 - Ti 2,8932(5) x6 2,9101(8) x6 2,8880(6) x6 2,9023(7) x6 2,9276(7) x6 - Al/Sn 3,0840(4) x3 3,1141(4) x3 3,0822(4) x3 3,1041(4) x3 3,1465(4) x3 - Ti 2,1143(4) x6 2,1259(6) x6 2,1119(4) x6 2,1198(4) x6 2,1342(5) x6 Al/Sn 1 C/N1 - 93 - MAX - Phase – Das System Ti-(Al/Sn)-C 3.4.4.6 Allgemeine Strukturdiskussion Alle untersuchten Einkristalle kristallisieren im Cr2AlC-Typ. In der Versuchsdurchführung stand die Substitution von Aluminium gegen Zinn im Vordergrund. Hier sollte mit Hilfe der Einkristalle ein gesteuerter Austausch der Hauptgruppenmetalle gezeigt werden. Jeder der analysierten Einkristalle zeigte eine Substitution von Aluminium gegen Zinn. Zudem konnte mit Hilfe der EDX-Untersuchung auch der unkontrollierte Einbau von Stickstoff nachgewiesen werden. Diese Tatsache erschwert die Interpretation des Ergebnisses. In allen untersuchten Einkristallen war die Titanbesetzung eindeutig nur auf der Lage 4f. Hier bildet das frühe Übergangsmetall Titan die Oktaederschichten entlang der [001]-Ebene. Im Zentrum der über kantenverknüpften Oktaeder befindet sich das Nichtmetall statistisch verteilt über alle Lücken. Eine Freigabe des Besetzungsfaktors zeigte eine vollständige Besetzung der Position (Abweichungen lagen im Rahmen der Standardabweichung). Die Kantenlänge (dTi1-Ti1: 2,8842(12) – 2,8949(15) Å) sowie der Abstand (dTi1-C/N1: 2,1119(4) – 2,1342(5) Å) der Nichtmetallatome zu den Oktaederecken steht im engen Verhältnis zum Mischungsverhältnis von Stickstoff und Kohlenstoff. Bei der kristallographischen Lage 2d wurde bei allen Einkristallen eine Mischbesetzung von Aluminium und Zinn festgestellt, die jedoch nicht eindeutig über die Schmelzzusammensetzung zu steuern ist. Die Trennschicht zwischen den carbidischen Elementen besteht aus einer statistischen Verteilung von Aluminium- und Zinnatomen. Hier sind jeweils drei Titanatome aus der darüber liegenden und darunter liegenden Schicht mit sechs Hauptgruppenmetallen aus gleicher Schicht an der Koordination jedes Atoms auf der Lage 2d beteiligt. Als Resümee lässt sich sagen, dass die Steuerung des gezielten Einbaus von Hauptgruppenmetallen über die Schmelzzusammensetzung nicht erfolgreich zu lenken ist. Es zeigt sich die Tendenz, dass Aluminium und Zinn in etwa gleichen Anteilen eingebaut werde und somit unabhängig vom Verhältnis der Schmelzzusammensetzung sind. - 94 - MAX - Phase – Das System Ti-(Al/Sn)-C 3.4.4.7 Substitutionsmöglichkeiten im System Titan-Aluminium/ZinnKohlenstoff/Stickstoff Wie in der vorangegangenen Untersuchung zu sehen war, wurde in den Einkristallen unbeabsichtigt eine erhebliche Menge an Stickstoff eingebaut. In der nächsten Versuchsreihe sollte ein Überprüfung erfolgen, inwiefern sich der Einbau an Stickstoff steuern lässt. Auch hier sollten Einkristalle aus einer Aluminium- und einer Aluminium/Zinn-Schmelze gezüchtet werden. Der Stickstoff wurde durch gezieltes Einwiegen von Titannitrid in die Probe eingebracht. Als Kohlenstoffquelle wurde wie schon in anderen Proben „carbon black“ eingesetzt. Die Edukte wurden in entsprechenden Verhältnissen zu Tabletten verpresst und bei einer Temperatur von 1600°C zwanzig Stunden unter einer Ar-Strom umgesetzt. Temperaturprogramm: 150°C/h RT -5°C/h -20°C/h 1600°C 1500°C (20h halten) (2h halten) -100°C/h 1000°C RT (1h halten) Tabelle 3.4.4.10: Proben im System Titan-(Aluminium/Zinn)-Kohlenstoff/Stickstoff und ihre pulverdiffraktometrische Charakterisierung. Fettgedruckt sind die Hauptprodukte. * wurde wegen der geringen Intensität nicht zugeordnet. Nr. 1 Atomgehalt [At.-%] Zusammensetzung Ti Al Ti6Al15C0,5N0,5 27,3 68,2 Sn Gitterkonstante [Å] N C 2,3 2,3 a = 3,057(7); c = 18,66(3) - 2 Ti6Al15C0,3N0,7 27,3 68,2 - 1,4 3,2 3 Ti6Al15C0,7N0,3 27,3 68,2 - 3,2 1,4 Ti6Al7,5Sn7,5C0,5N0,5 27,3 34,1 34,1 2,3 2,3 Ti6Al10Sn5C0,5N0,5 27,3 45,5 22,7 2,3 2,3 Ti6Al5Sn10C0,5N0,5 27,3 22,7 45,5 2,3 2,3 4 5 6 der MAX-Phase V=151,0(4) - weitere kristalline Phasen AlN Ti3AlC2*, AlN, Ti3Al2, a = 3,061(4); c = 18,63(3) TIC, V=151,2(4) TiN, a = 3,061(2); c = 13,70(3) - V=111,2(1) a = 3,0820(8); c = 13,714(5) - V=112,81(5) a = 3,053(2); c = 13,722(6) - V=111,91(18) Der Schmelzregulus wurde über zwei Tage in verd. HCl eingelegt. Nach dem Absaugen und Waschen des Rückstandes waren nur wenige und sehr kleine Kristalle unter dem Lichtmikroskop zu erkennen. Das Produkt fiel überwiegend in Pulverform an. Anhand der - 95 - MAX - Phase – Das System Ti-(Al/Sn)-C pulverdiffraktometrischen Charakterisierung konnten die Phasen identifiziert und ihre Gittekonstanten problemlos verfeinert werde. Die zinnfreie Schmelze zeigte das Erscheinen der 312-Phase und eine Vielzahl an Nebenprodukten. In den Ansätzen bei denen mit einer Aluminium/Zinn-Schmelze gearbeitet wurde, konnten die 211-Phase als phasenreines Produkt erhalten werden. 3.4.4.8 EDX - Untersuchung an Einkristallen Aus dem Ansatz mit der Schmelzzusammensetzung von Ti6Al7,5Sn7,5C0,5N0,5 und Ti6Al5Sn10C0,5N0,5 wurden metallisch glänzende, hexagonale Einkristalle für eine Untersuchung mittels energiedispersiver Röntgenemission präpariert. Die Untersuchungen sollte das genaue Verhältnis der Besetzung der Nichtmetalllage und der Hauptgruppenmetalllage geben. Jeder Kristall wurde an sechs Stellen vermessen I) Einkristall, der aus einer Schmelzzusammensetzung von Ti6Al7,5Sn7,5C0,5N0,5 halten wurde. Abbildung 3.4.18 REM-Aufnahme der Probe Ti4Al0,84Sn0,16C0,2N2,8 Abbildung 3.4.19:Messspektrum der Probe Ti4Al0,84Sn0,16C0,2N2,8. Aus der Messung geht hervor, dass die Nichtmetallposition nahezu vollständig mit Stickstoff besetzt ist, nur in Spuren konnte der Kohlenstoff beobachtet werden (3(2) At.-%). Der Erwartungsgemäß hoher Anteil am frühen Übergangsmetall (49,3(5) At.-%, Ti) deutet auf eine vollständige Besetzung der kristallographischen Punktlagen 4f und 4e hin. Bei den Hauptgruppenmetallen konnte ein nahezu fünfmal höherer Anteil an Aluminium zu Zinn detektiert werden. Die aus der EDX-Untersuchung ermittelte Zusammensetzung, konnten - 96 - MAX - Phase – Das System Ti-(Al/Sn)-C einen Hinwies auf das Vorliegen eine 413-Phase geben. Die der PXRD-Untersuchung konnte diese nicht beobachtet werden. Tabelle 3.4.4.11: Verhältnis der Elementaranalyse an Einkristallen der Zusammensetzung Ti4Al0,84Sn0,16C0,2N2,8. Element Gewichts [%] Atomist [%] σ [%] Atomssoll [%] Ti K 69,85 49,3 0,5 50,0 Al K 8,47 10,44 0,3 12,5-x Sn K 6,71 2,0 0,5 x CK 1,29 3,4 2,3 37,5-y NK 13,69 34,97 1,1 y Laut der EDX-Untersuchung liegt hier ein Einkristall mit der ungefähren Zusammensetzung von Ti4Al0,84Sn0,16C0,2N2,8 vor. Die ermittelte Zusammensetzung ist nun ein wichtiger Richtwert für die folgende Strukturverfeinerung. II) Einkristall, der aus einer Schmelzzusammensetzung von Ti6Al5Sn10C0,5N0,5 halten wurde. Abbildung 3.4.20 REM-Aufnahme der Probe Ti4Al0,83Sn0,17C0,4N2,6 Abbildung 3.4.21:Messspektrum der Probe Ti4Al0,83Sn0,17C0,4N2,6 Der zweite Einkristall zeigt einen etwas größeren Gehalt an Kohlenstoff, hier konnten 4,9(7) At.-% nachgewiesen werden. Die Metalle waren in beinahe gleichen Verhältnissen wie zuvor beobachtet. Weitere Details zur Messung sind der Tabelle 3.4.4.13 zu entnehmen. Aus der qualitativen Analyse konnte hier eine Zusammensetzung von Ti4Al0,83Sn0,17C0,2N2,6 bestimmt werden. - 97 - MAX - Phase – Das System Ti-(Al/Sn)-C Tabelle 3.4.4.12: Verhältnis der Elementaranalyse an Einkristallen der Zusammensetzung Ti4Al0,83Sn0,17C0,4N2,6. Element Gewichts [%] Atomist [%] σ [%] Atomssoll [%] Ti K 68,09 47,9 1,0 50,0 Al K 8,02 10,0 0,4 12,5-x Sn K 6,84 2,0 0,3 x CK 1,78 4,9 0,7 37,5-y NK 15,27 35,2 0,8 y 3.4.4.9 Röntgenographische Untersuchung an Einkristallen der 413-Phase Nach der Hydrolyse der Proben konnten in den Ansätzen mit reiner Aluminiumschmelze nur wenige und sehr kleine Kristalle beobachtet werden. Das Produkt lag größtenteils als ein metallisch glänzendes Pulver vor. Aus den ersten und dritten Ansatz (Tabelle 3.4.4.10) konnten zwei geeignete Einkristalle für weitere Untersuchungen isoliert werden. Die Größenordnung der Gitterkonstanten der gefundenen Zellen deutete auf eine Variante der 413-Phase hin. Die Strukturverfeinerung wurde für beide Einkristalle in der Raumgruppe P63/mmc durchgeführt. Aus der EDX-Untersuchung ging hervor, dass nur ein sehr geringer Anteil an Kohlenstoff in der Probe vorhanden ist. Zudem hat die qualitative Elementanalyse das exakte Metallverhältnis preisgegeben. Somit wurde die Strukturverfeinerung mit Hilfe der EDX- Ergebnisse zur Zusammensetzungen von Ti4Al0,84Sn0,16C0,2N2,8 und Ti4Al0,83Sn0,17C0,4N2,6 verfeinert. Die Verfeinerung der freien Parameter konvergierte nach Einführung anisotroper Temperaturfaktoren zu Zuverlässigkeitswerten von R1(F) = 0,0306 wR2(I) = 0,0763 für den Einkristall aus den Ansatz Nummer 1. Der zweite untersuchte Einkristall, der aus der dritten Probe herrührte, ergeben Zuverlässigkeitswerte von R1(F) = 0,0278 und wR2(I) = 0,0777. Weitere Einzelheiten zur Strukturbestimmung sind in den Tabelle 3.4.4.13 und 3.4.4.21 aufgezeigt. - 98 - MAX - Phase – Das System Ti-(Al/Sn)-C Tabelle 3.4.4.13: Angaben zur Strukturanalyse von Ti4Al0,84Sn0,16C0,2N2,8 (links) und Ti4Al0,83Sn0,17C0,4N2,6 (rechts). Summenformel Ti4Al0,84Sn0,16C0,2N2,8 Ti4Al0,83Sn0,17C0,4N2,6 Kristallsystem hexagonal Raumgruppe Gitterkonstante [Å] P 63/m m c – Nr. 194 a = 3,0048(4); c = 23,497(5) a = 3,0036(5); c = 23,381(9) Volumen der Elementarzelle [Å3] 182,68(9) 183,67(3) Zahl der Formeleinheiten röntgenogr. Dichte [g/cm3] F(000) Z=2 4,821 4,988 255,1 Kristallform, -farbe 248,8 metallisch glänzend, hexagonale Plättchen STOE IPDS II (Mo-Kα-Strahlung; λ=0,71073 Å Diffraktometer Graphitmonochromator 0° ≤ ω ≤ 180° Rotation ψ = 0°, 111; ∆ω = 2° 300 Belichtungszeit [sec.] Messtemperatur [K] Kristallgröße [mm] Raumtemperatur [293(2)] 0,06 × 0,06 × 0,01 0,06 × 0,05 × 0,01 Absorptionskoeffizient [mm-1] 9,47 9,40 Messbereich [°] 4°<2θ<65° 4°<2θ<65° Zahl der gemessenen Reflexe -4 ≤ h ≤ 3, -3 ≤ k ≤ 4, -32 ≤ l ≤ 32 1997 -4 ≤ h ≤ 3, -3 ≤ k ≤ 4, -32 ≤ l ≤ 32 1310 Zahl der unabh. Reflexe mit I > 0 160 124 I > σ2(I): 123 102 freie Parameter 14 14 Indexbereich Lorentz, Polarisation Korrekturen numerical / equivalent method (XSHAPE[68]) Isotypie Cr2AlC[33] Absorptionskorrektur Strukturlösung SHELXTL-97[199] Strukturverfeinerung Wichtungsschema w-1 = σ2F02 + (a * P)2 + b * P P = (F02 + 2 FC2) / 3 Goodness of fit - 3 Restelektronendichte [e /Å ]: Alle Reflexe a = 0,0481 b = 0,162 b =0,3251 1,005 1,088 0,028(7) Extinktionskorrektur R-Werte für I > 0: a =0,0515 0,027(7) - +0,88 / -0,99 - 0,21 e /Å 3 +0,66 / -0,69 - 0,19 e-/Å3 R1(F) = 0,0306 R1(F) = 0,0278 wR2(I) = 0,0763 wR2(I) = 0,0777 R1(F) = 0,0361 R1(F) = 0,0322 - 99 - MAX - Phase – Das System Ti-(Al/Sn)-C Tabelle 3.4.4.14: Ortskoordinaten, Besetzungsfaktoren (in [%]) und Auslenkungsparameter von Ti4Al0,84Sn0,16C0,2N2,8 (links) und Ti4Al0,83Sn0,17C0,4N2,6 (rechts). Ti4Al0,84Sn0,16C0,2N2,8 Summenformel Ti4Al0,83Sn0,17C0,4N2,6 Ti 1 x y z sof Ueq. U11 = U22 U33 U23 = U13 U12 0 0 0,15420(4) 100% * 0,0092(2) 0,0085(4) 0,0108(5) 0 0,0042(2) 0 0 0,15405(4) 100% * 0,0090(5) 0,0077(6) 0,0771(7) 0 0,0038(3) x y z sof Ueq. U11 = U22 U33 U23 = U13 U12 ⅓ ⅔ 0,05353(4) 100% 0,0091(2) 0,0082(4) 0,0110(4) 0 0,0041(2) ⅓ ⅔ 0,05345(4) 100% 0,0090(5) 0,0081(6) 0,0109(7) 0 0,0041(3) x y z sof Ueq. U11 = U22 U33 U23 = U13 U12 ⅓ ⅔ ¼ 84% Al 1 / 16% Ti 1 # 0,145(4) 0,161(5) 0,0128(7) 0 0,0080(3) ⅓ ⅔ ¼ 85% Al 1 / 15% Sn 1 # 0,0164(6) 0,0174(7) 0,0143(9) 0 0,0087(4) x y z sof Ueq. 0 0 0 78% N 1 / 22% C 1# 0,0120(13) 0 0 0 60,0% N 1 / 40% C 1 # 0,0131(15) x y z sof Ueq. ⅔ ⅓ 0,1055(2) 100%# 0,0124(10) ⅔ ⅓ 0,1053(2) 100% # 0,0114(11) Ti 2 Al/ Sn N/C 1 N2 # Besetzungsfaktoren und Temperaturfaktoren auf EDX-Ergebnis gekoppelt und fixiert - 100 - MAX - Phase – Das System Ti-(Al/Sn)-C Tabelle 3.4.4.15: Ausgewählte Abstände in Ti4Al0,84Sn0,16C0,2N2,8 (links) und Ti4Al0,83Sn0,17C0,4N2,6 (rechts). Struktur. Ti4Al0,84Sn0,16C0,2N2,8 Summenformel Ti4Al0,83Sn0,17C0,4N2,6 Ti 1 2,074(3) x3 2,078(3) x3 2,8361(9) x3 2,8447(9) x3 - Ti 2 2,927(1) x3 2,9321(12) x3 -N2 2,121(3) x6 2,120(3) x6 2,1400(7) x3 2,1417(7) x3 - Ti 1 2,973(1) x3 2,9321(12) x3 - Ti 2 3,0036(4) x3 3,0048(4) x3 - Ti 1 2,8361(9) x6 2,8447(9) x6 - Al/Sn 3,0036(4) x6 3,0048(4) x6 - Ti 2 2,1400(6) x6 2,1417(7) x6 - Ti 1 2,074(3) x3 2,078(3) x3 - Ti 2 2,121(3) x3 2,120(3) x3 -N2 - Al/Sn Ti 2 - N/C 1 Al/Sn N/C 1 N2 Tabelle 3.4.4.16: Ausgewählte Winkel in Ti4Al0,84Sn0,16C0,2N2,8 (links) und Ti4Al0,83Sn0,17C0,4N2,6 (rechts). Struktur. Winkel Ti4Al0,84Sn0,16C0,2N2,8 Ti4Al0,83Sn0,17C0,4N2,6 Ti 2 – C/N 1 – Ti 2 90,88(4)° 90,86(3)° 90,38(9) 90,35(7)° 89,12(4)° 89,14(3)° Ti 1 – N 2 – Ti 1 92,68(18)° 92,81(15)° Ti 2 – N 2 – Ti 2 90,12(17)° 90,16(15)° 88,50(3) 88,50(3)° - 101 - MAX - Phase – Das System Ti-(Al/Sn)-C 3.4.4.10 Strukturdiskussion Ti4Al0,84Sn0,16C0,2N2,8 (a = 3,0036(5) Å; c = 23,381(9) Å) und Ti4Al0,83Sn0,17C0,4N2,6 (a = 3,0048(4) Å; c = 23,497(5) Å) kristallisieren im Se4CsYb3-Typ und der Raumgruppe P63/mmc. Bei dieser MAX-Phase handelt es sich um die 413-Variante. Die Sichten der Zusammensetzung Ti4N3 werden durch eine Metallschicht getrennt, welche aus einer Mischbesetzung von Aluminium und Zinn geprägt wird. Die Metallatome bilden einen dichteste Kugelpackung, welche als ABABCACACB (nach Jagodzinski (hhhcc)2) beschrieben werden kann. In der Kristallstruktur konnten zwei kristallographisch unterschiedliche Titanatome bestimmt werden. Die Ti1 besetzen vollständig die Lage 4f und sind von drei Ti2 (dTi1-Ti2: 2,928(1)-2,9323(12) Å) und von drei Al/Sn (dTi1-Al/Sn: 2,8357(9)-2,8444(8) Å) aus den darüber bzw. darunter liegenden Schichten und von sechs gleichen Ti 1- Atomen umgeben (in der Abbildung zwölffachen 3.4.22 grünes Koordination Polyeder). kommt Aus der es zur auf der antikuboktaederische Koordinationssphäre. Ebenfalls sind die Atome Al/Sn kristallographischen Lage 2c in einer antikuboktaederische Koordination eingeschlossen. An der Abbildung 3.4.22: Struktur von Ti4Ti0,57Al0,43N3 und die charakteristischen Polyeder. Koordination sind sechs Atome aus gleicher Schicht (dAl/Sn-Al1/Ti3: 3,0036(4)- 3,0048(4) Å) sowie jeweils drei Ti 1 - Atome (dTi1-Al1/Ti3: 2,8357(9)-2,8444(8) Å) aus der darüber und darunter liegenden Schicht beteiligt (blaues Polyeder in der Abbildung 3.4.22). Die Ti 2 Atome befindet sich auf der Lage 2d, d.h. in der Mitte der nitridischen Einheit (rotes Polyeder in der Abbildung 3.4.22) und wird von Ti 1 (dTi1-Ti2: 2,928(1)-2,9323(12) Å) und Ti 2 (dTi2-Ti2: 3,0036(4)3,0048(4) Å) Atomen kuboktaedrisch umgeben. Stickstoff besetzt ausschließlich die Oktaederlücken die von Titanatomen gebildet werden. Dabei müssen zwei Arten von Oktaedern unterschieden werden. Die TiN6-Oktaeder welche aus sechs Ti 2 bestehen besitzen jeweils einen Abstand von dTi2-N1: 2,1399(7)-2,1417(7) Å. Die TiN6-Oktaeder in der Mitte bestehen aus drei Ti 1 (dTi1-N2: 2,073(3)-2,078 (3) Å) und drei - 102 - MAX - Phase – Das System Ti-(Al/Sn)-C Ti 2 Atomen (dTi2-N2: 2,122(3)-2,121(3) Å). Der charakteristischen Abstand in TiN besitzt einen Titan-Stickstoffabstand von 2,166 Å, so spiegeln die Oktaeder in der Mitte der Ti4N3Einheit am besten die Bindungsverhältnisse von TiN wider. 3.4.4.11 EDX - Untersuchung an Einkristallen der 211-Phase Aus dem Ansatz Nummer 5 (Tabelle 3.4.4.10) konnte ein hexagonales, metallisch glänzendes Plättchen isoliert werden. Nach der röntgenographischen Untersuchung an isolierten Einkristallen wurde auch auf die Elemente hin analysiert um eine Sicherstellung des Stickstoffs zu machen. Bei dem untersuchten Einkristall handelte es sich eindeutig um eine 211 Phase. Genaueres zur Strukturbestimmung ist in der Tabelle 3.4.4.18 aufgelistet. Die EDX-Analyse sollte das Metallverhältnis und den Stickstoffanteil in der Probe preisgeben. Abbildung 3.4.23: REM-Aufnahme der Probe Ti2Sn0,52Al0,48C0,52N0,48 Abbildung 3.4.24: Messspektrum Ti2Sn0,52Al0,48C0,52N0,48. der Probe Die EDX-Untersuchung zeigte, dass der Stickstoff wie beabsichtigt mit 13±1 At.-% eingebaut wurde. Bei den Metallen konnte das Titan im Rahmen der Standardabweichung gute Übereinstimmung mit den erwarteten Werten liefern. Die Metalle Aluminium und Zinn konnten nahezu gleichen Verhältnissen detektiert werden. - 103 - MAX - Phase – Das System Ti-(Al/Sn)-C Tabelle 3.4.4.17: Verhältnis der Elementaranalyse an Einkristallen der Zusammensetzung. Ti2Al0,48Sn0,52C0,52N0,48. Element Gewichts [%] Atomist [%] σ [%] Atomssoll [%] Ti K 50,84 48,37 0,98 50,0 Al K 7,11 12,72 0,78 Sn K 34,56 12,70 0,78 CK 3,88 13,57 0,16 NK 3,56 12,63 1,74 25,0-x (16,7) x (8,3) 25,0-y (12,5) y (12,5) 3.4.4.12 Röntgenographische Untersuchung an Einkristallen der 211-Phase Mit dem IPDS II wurde der Kristall mit fünf Minuten pro Aufnahme vermessen. Eine anisotropen Verfeinerung der Temperaturfaktoren konvertierte zu Zuverlässigkeitswerten von R1(F) = 0,0160 und wR2(I) = 0,0305. Bei der Freigabe des Besetzungsfaktors der Titanposition konnte keine Abweichung von der vollständigen, geordneten Besetzung beobachtet werden. Die Metallposition der Hauptgruppenmetalle sowie die Nichtmetalle wurden aneinander gekoppelt und nach dem EDX- Ergebnis frei verfeinert. Die Restelektronendichten befanden sich in einem vernünftigen Rahmen (+0,38 und -0,34 e/Å3) - 104 - MAX - Phase – Das System Ti-(Al/Sn)-C Tabelle 3.4.4.18: Angaben zur Strukturanalyse von Ti2Al0,48Sn0,52C0,52N0,48. Summenformel Ti2Al0,48Sn0,52C0,52N0,48 Kristallsystem hexagonal Raumgruppe P 63/m m c – Nr. 194 a = 3,072(1); c = 13,657(6) Gitterkonstante [Å] 3 Volumen der Elementarzelle [Å ] 111,63(7) Zahl der Formeleinheiten 2 3 röntgenogr. Dichte [g/cm ] 5,458 F(000) 165,5 Kristallform, -farbe metallisch glänzende hexagonale Plättchen Diffraktometer Rotation STOE IPDS II (Mo-Kα-Strahlung; λ=0,71073 Å Graphitmonochromator 0° ≤ ω ≤ 180° ψ = 0°, 111; ∆ω = 2° Belichtungszeit [sec.] 300 Messtemperatur [K] Raumtemperatur [293(2)] Kristallgröße [mm] 0,04 × 0,04 × 0,01 Absorptionskoeffizient [mm-1] 16,74 Messbereich [°] 4° < 2θ < 65° -4 ≤ h ≤ 4, Indexbereich -4 ≤ k ≤ 4, -20 ≤ l ≤ 20 Zahl der gemessenen Reflexe 1498 Zahl der unabh. Reflexe mit I > 0 97 I > σ2(I): 77 freie Parameter 9 Korrekturen Lorentz, Polarisation Absorptionskorrektur numerical / equivalent method (XSHAPE[68]) Strukturlösung Isotypie Cr2AlC75 Strukturverfeinerung SHELXTL-97[199] Wichtungsschema w-1 = σ2F02 + (0,00 * P)2 + 0,3825 * P P = (F02 + 2 FC2) / 3 Goodness of fit 1,143 Extinktionskorrektur 0,027(3) Restelektronendichte [e-/Å3]: +0,38 / - 0,34; R-Werte für I > 0: Alle Reflexe 0,14 e-/Å3 R1(F) = 0,0160 wR2(I) = 0,0305 R1(F) = 0,0195 - 105 - MAX - Phase – Das System Ti-(Al/Sn)-C Tabelle 3.4.4.19: Ortskoordinaten, Besetzungsfaktoren (in [%]) und Auslenkungsparameter von Ti2Al0,48Sn0,52C0,52N0,48. Atom Lage x y ⅓ Al/Sn 4f 2d ⅓ ⅔ C/N 2a 0 0 Ti ⅔ z sof 0,08357(4) Ueq 100* 0,0046(2) ¾ # 48/52(2) 0,0121(3) 0 52/48(3)# 0,0065(9) * bei wechselweiser Freigabe der Besetzungsfaktoren keine signifikante Restelektronendichte vorhanden # Besetzungsfaktoren und Temperaturfaktoren auf EDX-Ergebnis gekoppelt und fixiert Atom U11 U22 U33 U23 U13 U12 Ti 0,0051(3) 0,0051(3) 0,037(3) 0 0 0,0027(1) Al/Sn 0,0149(9) 0,0149(9) 0,0066(3) 0 0 0,0074(2) Die folgende Tabelle zeigt benachbarte Atome, ihren charakteristischen Abstand und die Koordinationszahl. Tabelle 3.4.4.20: Ausgewählte Abstände und Winkeln in der Ti2Al0,48Sn0,52C0,52N0,48-Struktur. Anzahl der Winkel Bindungen Ti - C/N - Ti Benachbarte Atome Abstand [Å] Al/Sn- Ti 2,928(2) x6 Al/Sn 3,0723(4) x6 C/N 2,1093(4) x3 94,04(2)° Al/Sn 2,8831(6) x3 85,96(2)° Ti 3,0723(4) x6 Ti 2,1093(4) x6 Ti- C/N- 3.4.4.13 Strukturdiskussion Ti2Al0,48Sn0,52C0,52N0,48 kristallisiert im Cr2AlC-Typ in der Raumgruppe P63/mmc mit den Gitterkonstanten von a = 3,072(1) Å und c = 13,657(6) Å. - 106 - MAX - Phase – Das System Ti-(Al/Sn)-C Der untersuchte Einkristall zeigt eine Mischbesetzung auf der Nichtmetall- und Hauptgruppenmetalllage mit guter Übereinstimmung der Einwaage. Es konnte ein Verhältnis Al:Sn von 48% :52% und C:N von 52% : 48% laut EDX- und verfeinert werden. Titan zeigt auf der kristallographischen Lage 4f eine vollständige Besetzung und formt somit die Oktaeder die zu Schichten entlang der [110]-Eben unendlich sind (dTi1-Ti1: 2,928(2) Å). Die dabei entstandenen Oktaederlücken sind vollständig mit Kohlenstoff und Stickstoff besetzt (dTi 1-C/N 1: 2,1093(4) Å). Zwischen den Oktaederschichten befindet sich die Schicht der Hauptgruppenmetalle, hier sind die Titanatome mit 2,8831(6) Å von Al/Sn1 Atomen entfernt. Jedes Hauptgruppenmetall ist innerhalb der Schicht um 3,0723(4) Å von seinem Gleichen entfernt. - 107 - MAX - Phase – Das System Ti-(Al/Sn)-C 3.4.4.14 Synthese im SPS-Ofen Darstellung von Ti4AlN3 Für die Darstellung von Ti4AlN3 mit dem Pulsplasma-Ofens wurde 0,282 g (4,556 mmol) Titannitridpulver mit 0,073 g (1,524 mmol) Titan und 0,045 g (1,667 mmol) Aluminium im nahezu stöchiometrischen Verhältnis eingewogen. Die Synthese wurde bei Temperaturen von 1200 bis 1350°C durchgeführt, bei einer Reaktionszeit von 5 bis 15 Minuten. Das beste Ergebnis konnte bei einer Temperatur von 1350°C (15 min.) beobachtet werden. Temperaturprogramm: 2 min. RT 2 min. 600°C 2 min 800°C 2 min 1100°C 2 min 1350°C RT (15 min. halten) 300000 Lin (Counts) 200000 100000 0 6 10 20 30 40 50 60 70 80 90 100 110 2-Theta - Scale Abbildung 3.4.25: Pulverdiffraktogramm der MAX-Phasen Ti4AlN3 und Ti2AlN. Die blaumarkierte Reflexe konnte TiN zu geordnet werden. Eine phasenreine Darstellung von Ti4AlN3 (grün) im Pulsplasma Ofen ist nicht gelungen. Die 413-Phase erscheint neben der 211-Phase (rot) als Nebenprodukt ab einer Temperatur von 1350°C. Als weiters kristallines Nebenprodukt konnte binäres TiN (blau) identifiziert werden. Weitere Versuche mit längeren Reaktionszeiten sowie höheren Reaktionstemperaturen konnten die Bildung der 211-Phase sowie das Verhältnis der 211-Phase zu TiN nicht verändern. Die in der Abbildung 3.4.25 gezeigte 413-Phase kristallisiert in der Raumgruppe P 63/mmc mit den Gitterkonstanten a = 2,997(2) Å, c = 23,383(18) und einem Volumen von V = 181,90(14) Å3. Weitere Details zu Ti4AlN3 sind in der Tabelle 3.4.4.21 angegeben. - 108 - MAX - Phase – Das System Ti-(Al/Sn)-C Tabelle 3.4.4.21: Pulverdaten von Ti4AlN3 (P 63/mmc) - Ergebnis der Synthese im SPS-Ofen N 2θ h k l Ibeob dbeob dber 1 7,500 0 0 2 12,0 11,7779 11,7057 2 15,138 0 0 4 4,2 5,8480 5,5829 3 35,361 1 0 2 113,3 2,5363 2,5321 4 37,898 1 0 4 10,1 2,3721 2,3712 5 38,375 0 0 10 9,9 2,3437 2,3411 6 39,687 1 0 5 100,0 2,2692 2,2688 7 41,748 1 0 6 33,1 2,1618 2,1599 8 55,919 1 0 11 3,8 1,643 1,6453 9 59,199 1 0 12 6,9 1,5595 1,5591 10 61,861 1 1 0 79,0 1,4986 1,4974 11 72,786 2 0 0 5,0 1,2983 1,2968 12 73,079 2 0 1 4,9 1,2938 1,2948 13 75,287 1 1 10 7,3 1,2612 1,2614 Die ermittelten Gitterparameter aus dem Pulver der Sinterprobe stimmen gut mit den Daten überein, die aus Untersuchungen an den Einkristallen erhalten wurden. Darstellung von Ti2AlN Für die Synthese von Ti2AlN im Pulsplasma-Ofen wurde Titanntitridpulver mit Aluminiumpulver und Titanpulver umgesetzt. Titannitrid sollte als Stickstofflieferant bei der Reaktion fungieren. Das Aluminium wurde in einem kleinen Überschuss eingesetzt. Somit wurden 0,133 g (2,149 mmol) TiN mit 0,103 g (2,150 mmol) Ti und 0,064 g (2,372 mmol) Al gut miteinander vermischt und ungepresst in der Graphitmatrize bei einer Temperatur von 1350°C für 5 Minuten zur Reaktion gebracht. Das Sintern wurde unter Vakuum (0,11mbar) durchgeführt. Dabei wurde ein Sinterdruck von 4.3 kN eingestellt. Temperaturprogramm: 2 min. RT 2 min. 600°C 2 min 800°C 2 min 1100°C 45 min. 1350°C RT (5 min. halten) - 109 - MAX - Phase – Das System Ti-(Al/Sn)-C 280000 270000 260000 250000 240000 230000 220000 210000 200000 190000 180000 Lin (Counts) 170000 160000 150000 140000 130000 120000 110000 100000 90000 80000 70000 60000 50000 40000 30000 20000 10000 0 5 10 20 30 40 50 60 70 80 90 100 110 120 2-Theta - Scale Abbildung 3.4.26: Pulverdiffraktogramm von einer Ti2AlN - Phase (rot), nicht umgesetztes TiN (blau) und Spuren von Ti4AlN3 (grün). Ergebnis aus den Pulsplasma-Ofen. Eine röntgenographische Phasenanalyse der Probe zeigte das angestrebte Hauptprodukt Ti2AlN mit den charakteristischen Gitterkonstanten von a =2,9912(10) Å und c = 13,643(4) Å (Lit.: a =2,994 Å und b = 13,610 Å)[76] als Hauptprodukt. Neben der 211-Phase konnten noch TiN und wenig Ti4AlN3 gefunden werden. Tabelle 3.4.4.22: Pulverdaten von Ti2AlN - Ergebnis der Synthese im SPS-Ofen- N 2θ h k l Ibeob dbeob dber 1 12,903 0 0 2 26,5 6,8553 6,8214 2 34,552 1 0 0 15,8 2,5938 2,5905 3 35,213 1 0 1 5,9 2,5466 2,5450 4 39,990 1 0 3 100,0 2,2527 2,2509 5 43,810 1 0 4 5,5 2,0648 2,0629 6 53,562 1 0 6 17,6 1,7096 1,7089 7 72,177 1 0 9 11,2 1,3077 1,3083 8 72,981 2 0 0 2,8 1,2953 1,2952 9 76,163 1 1 6 14,9 1,2489 1,2495 10 102,990 2 0 9 3,6 0,9843 0,9847 11 104,703 1 0 13 3,4 0,9729 0,9727 12 107,161 2 1 3 6,5 0,9573 0,9572 - 110 - MAX - Phase – Das System Ti-(Al/Ga)-C 3.4.5 Arbeiten in einer Aluminium/Gallium-Schmelze Im System Titan-Aluminium-Kohlenstoff konnte von zahlreichen binären und nur zwei ternären Phasen berichtet werden. Bei den ternären Verbindungen handelt es sich um MAXPhasen der 211- und 413-Variante9. 3.4.5.1 Die binären Systeme Aluminium-Gallium und Titan-Gallium Das eutektische Phasendiagramm von Aluminium und Gallium zeigt eine vollständige Mischbarkeit der Metalle in der Schmelze, somit liegt hier oberhalb von 660 ± 15°C eine homogene Schmelze vor. Die maximale Löslichkeit von Gallium in Aluminium beträgt ungefähr 9 At.-%. Abbildung 3.4.27: Das binäre Phasendiagramm des Systems Aluminium-Gallium Abbildung 3.4.28 Das binäre Phasendiagramm des Systems Titan-Gallium Im etwas komplexeren binären System Titan-Gallium wurde in den letzten Jahren von immer mehr stöchiometrischen Verbindungen berichtet. Angefangen in der titanreichen Ecke mit Ti3Ga[77] (Ni3Sn-Typ) gefolgt von Ti2Ga[78] (Ni2In-Typ), Ti5Ga3[79] (α- und βModifikation), Ti5Ga4[80] (eigener Struktur-Typ) und TiGa[80] (AuCu-Typ) und bei den titanarmen Verbindungen gibt es Ti2Ga3[80] (eigener Struktur-Typ), TiGa2[81] (HfGa2-Typ) sowie TiGa3[82] (TiAl3-Struktur-Typ). Zudem ist eine Allotropie des Titans bekannt. α-Titan geht bei einer Temperatur von 882±15°C in die β-Modifikation über. Für die Untersuchungen des ternären (Ti-Ga-C) bzw. quaternären (Ti-Ga/Al-C) Systems gibt es kaum Literatur. Währen die H-Phase[202] bereits lange bekannt ist, wurde erst kürzlich 9 Bei der 413-Phase nur als Nitrid bekannt ist. - 111 - MAX - Phase – Das System Ti-(Al/Ga)-C von Etzkorn beschriebene 413-Phase mit der stöchiometrischen Zusammensetzung Ti4GaC3[202,203]. Binäre Verbindungen im Gallium-Kohlenstoff-System sind nicht bekannt. Im Rahmen dieser Arbeit konnte noch die fehlende 312-Phase mit der Zusammensetzung von Ti3GaC2 in einkristalliner Form zu ersten Mal dargestellt und untersucht werden. 3.4.5.2 Präparative Untersuchung im System Ti-Al/Ga-C Die oben diskutierten 211- und 413-Phasen wurde aus einer reinen Galliumschmelze gewonnen. In dieser Arbeit sollten unterschiedliche Hilfsmetallbäder auf den Einfluss der Züchtung von Einkristallen getestet werde. Für die Synthese wurden die Elemente (als Kohlenstoffquelle wurde das carbon black eingesetzt) in verschiedenen stöchiometrischen Verhältnissen homogenisiert und zu Tabletten gepresst. Diese wurden dann anschließend bei einer Reaktionstemperatur von 1600°C für 20 Stunden unter Argon-Schutzgas in Korundtiegeln umgesetzt. Nach dem Abkühlen wurde der entstandene Schmelzregulus mit verd. Salzsäure behandelt. Der Rückstand bestand aus vielen kleinen rundlichen Einkristallen. Diese wurden abgesaugt, mit Wasser mehrmals gewaschen und getrocknet. Alle Proben wurden einer röntgenographischen Phasenanalyse auf die entstandenen Produkte hin untersucht. Temperaturprogramm: 150°C/h RT Nr. -5°C/h -15°C/h 1600°C 1500°C (20h halten) (2h halten) Gehalt [At.-%] Zusammensetzung -100°C/h 1000°C RT (1h halten) Gitterkonstante [Å] Ti Al Ga C 1 Ti6Al10Ga5C1 27,3 45,5 22,7 4,5 2 Ti6Al7,5Ga7,5C1 27,3 34,1 34,1 4,5 3 Ti6Al5Ga10C1 27,3 22,7 45,5 4,5 4 Ti6Al3Ga12C1 27,3 13,6 54,5 4,5 der MAX-Phase a = 3,0828(1); c = 18,387(10) V=151,33(8) a = 3,0797(9); c = 13,410(4) V=110,15(4) a = 3,0762(9); c = 13,408(5) V=109,88(5) a = 3,078(2); c = 13,399(7) V=109,92(9) weitere kristalline Phasen >>Ti >>TiC - Ga3Ti Tabelle 3.4.5.1: Proben (Schmelzzusammensetzung) im System Titan-Gallium-Kohlenstoff und ihre pulverdiffraktometrische Charakterisierung. - 112 - MAX - Phase – Das System Ti-(Al/Ga)-C In dem galliumarmen Ansatz Tabelle 3.4.5.1 konnte eine Gitterkonstante von a = 3,0828(1) Å und c = 18,387(10) Å mit einem Volumen von V=151,33(8) Å3 in der Raumgruppe P63/mmc mit etwas Nebenprodukt an nicht umgesetztem Titan bestimmt werden. Die ermittelten Gitterkonstanten waren das erste Indiz für das Auftreten einer 312-Phase im TitanGallium-Kohlenstoff System. In Betrachtung der c-Gitterkonstanten konnte die 312-Phase (Ti3AlC2: a = 3,072 Å; c = 18,732 Å und V = 151,3 Å3)[83] mit einer reinen Besetzung mit Aluminium auf 2b ausgeschlossen werden. Abbildung 3.4.29: Ti3GaC2-Kristalle Die in der Abbildung 3.4.29 gezeigten Einkristalle der Ti3GaC2-Phase zeigen eine bis jetzt nicht erreichte Größe. Es konnte Einkristalle von bis zu 1,5 mm Kantenlänge isoliert werden. Die durchschnittliche Dicke der Einkristalle betrug 0,04-0,06 mm. In den anderen Ansätzen konnte Ti2GaC anhand der charakteristischen Gitterkonstanten eindeutig identifiziert werden. In allen Pulverdiffraktogrammen war Ti2GaC dominantes Hauptprodukt und ließ sich problemlos verfeinern. Die Gitterkonstante ist nahezu identisch in allen drei Ansätzen was für ein und die gleich thermodynamische Phase spricht. Ein Einbau von Aluminium auf der Galliumposition wurde durch die erhaltenen Dimension der Gitterkonstanten und das Ergebnis der EDX-Analyse ausgeschlossen. In der galliumreichsten Probe Nummer 4 hat sich als einziges Nebenprodukt Ga3Ti gebildet. - 113 - MAX - Phase – Das System Ti-(Al/Ga)-C 3.4.5.3 Härtemessung an der 312-Phase (Ti3GaC2) Aus dem Ansatz mit der Schmelzzusammensetzung Ti6Al10Ga5C wurden Einkristalle isoliert (hier handelt es sich um Einkristalle mit der Zusammensetzung Ti3GaC2), die für eine Härtemessung nach Vickers geeignet waren. Die Einkristalle konnten erstmalig parallel und senkrecht zur basalen Schicht vermessen werden. Es wurden insgesamt fünf Einkristalle der Härteprüfung unterzogen. Die Kristalle wurden in Epoxidharz eingebettet, geläppt und poliert, anschließend wurden diese an den entsprechenden Stellen vermessen. Auf Grund der sehr kleinen zur Verfügung stehenden Flächen konnte nur ein Abdruck pro Einkristall erfolgen, jedoch zeigte die Untersuchung an mehreren Kristallen keine signifikante Abweichung der Härtewerte. Für eine verlässliche Härte muss der Kristalle eine Mindestdicke vom dreifachem der Eindringtiefe mit sich bringen. Die Abbildung 3.4.30 zeigt einen Vickersabdruck senkrecht zur basalen Sicht. Abbildung 3.4.31 präsentiert die Aufsicht parallel zur basalen Schicht. Der grün markierte Bereich ist Epoxidharz, welches bei der Aufarbeitung weggesprungen ist. 20 µm Abbildung 3.4.30: Vickers Abdruck auf zur basalen Schicht (d1=24,19µm, d2=25,63µm: 301 HV 0,1). 20 µm Abbildung 3.4.31: Vickers Abdruck senkrecht zur basalen Schicht (d1=17,49µm, d2=18,07µm: 598 HV 0,1). Aus der Härteuntersuchung an mehreren Einkristallen der Zusammensetzung Ti3GaC2 mit unterschiedlicher Ausrichtung der kristallographischen Ebenen konnte ein anisotropes Verhalten der Verbindung beobachtet werden. Die Messung parallel zur basalen Schicht konnte ein Mittelwert (aus je drei Abdrücken) von 308 HV 0,1 (~3 GPa) und senkrecht zur dieser 651 HV 0,1 (~6,4 GPa) wiedergeben. - 114 - MAX - Phase – Das System Ti-(Al/Ga)-C 3.4.5.4 EDX - Untersuchung an Einkristallen der 312-Phase Der isolierte Einkristall der aus der Schmelze mit der Zusammensetzung von Ti6Al10Ga5C1 erhalten wurde, konnte mit Hilfe einer qualitativen Elementaranalyse mittels energiedispersiver Röntgenemmissionsspektroskopie auf die Zusammensetzung untersucht werden. Die beobachtet Aufweitung der a-Gitterkonstanten am Pulver und Einkristall, war ein Indiz dafür das neben dem Gallium noch ein weiteres Metall anwesend war. Abbildung 3.4.32: REM-Aufnahme der Probe Ti3Ga0,85Sn0,15C2. Abbildung 3.4.33: Messspektrum der Probe Ti3Ga0,85Sn0,15C2. Tabelle 3.4.5.2 zeigt die detektierten Elemente und ihre Verhältnisse am untersuchten Einkristallen. Aus der Messung geht hervor, dass neben Gallium noch Zinn in einem Anteil von 2,5(2) At.-% Präsenz zeigt10. Somit konnte eine Zusammensetzung von Ti3Ga0,85Sn0,15C2 postuliert werden. Tabelle 3.4.5.2: Verhältnis der Metallatome in der Probe Ti3Ga0,85Sn0,15C2. Element Gewichts [%] Atomist [%] σ [%] Atomssoll [%] Ti K 59,56 51,0 2,8 50,0 Ga K 23,29 13,6 0,1 16,67-x Sn K 7,48 2,5 0,2 x CK 9,67 32,6 2,7 33,3 10 Bei der Überprüfung der Elemente auf ihre Reinheit, konnte bei den verwendeten Galliumstückchen eine geringe Menge an Zinn nachgewiesen werden. - 115 - MAX - Phase – Das System Ti-(Al/Ga)-C 3.4.5.5 Röntgenographische Untersuchung an Einkristallen der 312-Phase Bei der röntgenographischen Untersuchung eines aus dem galliumarmen Ansatz isolierten Einkristalls wurde Gitterkonstante von a = 3,0723(6) Å und c = 18,221(6) Å bestimmt. Aus den beobachteten 2688 Reflexen wurde nach der Mittelung in der Laueklasse 6/mmm ein Datensatz von 129 unabhängigen Reflexen erhalten. Die Strukturverfeinerung erfolgte in der Raumgruppe P63/mmc, als Startmodell dienten die Lageparameter von Ti3SiC2, wobei die Position 2b mit Gallium- und Zinnatomen entsprechend dem EDX-Ergebnis besetzt und fixiert wurden. Die anisotropen Verfeinerung der Metalllagen konvergierte zu Zuverlässigkeitswerten bei R1(F) = 0,0248 und wR2(I) = 0,0594. Bei der Freigabe des Besetzungsfaktors der Titanposition konnte keine Abweichung von der vollständigen, geordneten Besetzung beobachtet werden. Alle weiteren Details zur Strukturbestimmung sind in den Tabellen 3.4.5.3, 3.4.5.4. und 3.4.5.5 zusammengefasst. - 116 - MAX - Phase – Das System Ti-(Al/Ga)-C Tabelle 3.4.5.3: Angaben zur Strukturanalyse von Ti3Ga0,85Sn0,15C2. Summenformel Ti3Ga0,85Sn0,15C2 Kristallsystem hexagonal Raumgruppe P 63/m m c – Nr. 194 a = 3,0723(6); c=18,221(6) Gitterkonstante [Å] 3 Volumen der Elementarzelle [Å ] 148,94(8) Zahl der Formeleinheiten 2 3 röntgenogr. Dichte [g/cm ] 5,455 F(000) 223,7 Kristallform, -farbe metallisch glänzende, hexagonale Plättchen Diffraktometer Rotation STOE IPDS II (Mo-Kα-Strahlung; λ=0,71073 Å Graphitmonochromator 0° ≤ ω ≤ 180° ψ = 0°, 111; ∆ω = 2° Belichtungszeit [sec.] 300 Messtemperatur [K] Raumtemperatur [293(2)] Kristallgröße [mm] 0,09 × 0,08 × 0,02 Absorptionskoeffizient [mm-1] 16,28 Messbereich [°] 4°<2θ<70° -4 ≤ h ≤ 4, Indexbereich -4 ≤ k ≤ 4, -20 ≤ l ≤ 20 Zahl der gemessenen Reflexe 2688 Zahl der unabh. Reflexe mit I > 0 129 I > σ2(I): 97 freie Parameter 12 Korrekturen Lorentz, Polarisation Absorptionskorrektur numerical / equivalent method (XSHAPE[68]) Strukturlösung Isotypie Ti3SiC2[84] Strukturverfeinerung SHELXTL-97[199] Wichtungsschema w-1 = σ2F02 + (0,0362 * P)2 + 0,00 * P P = (F02 + 2 FC2) / 3 Goodness of fit 0,830 Extinktionskorrektur 0,0092(48) Restelektronendichte [e-/Å3]: +0,72 / -1,19; R-Werte für I > 0: Alle Reflexe 0,21 e-/Å3 R1(F) = 0,0248 wR2(I) = 0,0594 R1(F) = 0,0311 - 117 - MAX - Phase – Das System Ti-(Al/Ga)-C Tabelle 3.4.5.4: Ortskoordinaten, Besetzungsfaktoren (in [%]) und Auslenkungsparameter von Ti3Ga0,85Sn0,15C2. Atom Ti 1 Ti 2 Lage x y ⅔ 2a ⅓ z 0,62877(6) sof Ueq 100* 0,0037(3) * 0,0034(4) 0 1 ½ 100 Ga/Sn 4f 2b 0 ¾ ¾ 85/15# 0,0118(4) C 4f ⅓ ⅔ 0,5699(3) 100* 0,0047(11) * bei wechselweiser Freigabe der Besetzungsfaktoren keine signifikante Restelektronendichte vorhanden # Besetzungsfaktoren und Temperaturfaktoren auf EDX-Ergebnis gekoppelt und fixiert Atom U11 U22 U33 U23 U13 U12 Ti 1 0,0034(4) 0,0034(4) 0,0034(4) 0 0 0,0017(2) Ti 2 0,0031(5) 0,0031(5) 0,0041(6) 0 0 0,0015(3) Ga/Sn 0,0139(5) 0,0139(5) 0,0077(5) 0 0 0,0069(2) Die folgende Tabelle zeigt benachbarte Atome, ihren Charakteristischen Abstand und die Koordinationszahl. Tabelle 3.4.5.5: Ausgewählte Abstände und Winkeln in Ti3Ga0,85Sn0,15C2 Anzahl der Winkel Bindungen Ti - C - Ti Benachbarte Atome Abstand [Å] Ga/Sn - Ti 1 2,833(1) x6 Ga/Sn 3,0731(4) x6 C 2,184(3) x3 90,60(17)° Ti 1 2,942(1) x6 89,40(17)° C 2,073(2) x3 95,68(18)° Ga/Sn 2,833(1) x3 89,40(17)° Ti 2 2,942(1) x3 87,37(3)° Ti 1 3,0731(4) x6 Ti 1 2,073(3) x3 Ti 2 2,184(3) x3 Ti 2- Ti 1- C- - 118 - MAX - Phase – Das System Ti-(Al/Ga)-C 3.4.5.6 Strukturdiskussion Ti3Ga0,85Sn0,15C2 kristallisiert im Ti3SiC2-Typ in der Raumgruppe P 63/mmc mit den Gitterkonstanten von a = 3,0723(6) Å und c = 18,221(6) Å. Ti3GaC2 wurde im Rahmen dieser Arbeit zum ersten Mal dargestellt. Ti3Ga0,85Sn0,15C2 vervollständigt die Reihe der MAXPhase, somit präsentiert das Dreistoffsystem Ti-Ga-C zum ersten Mal eine komplette Reihe aller Varianten. Somit ist hier die Möglichkeit gegeben, die Winkel und Bindungsabstände in der Serie zu studieren. Die Struktur von Ti3GaC2 kristallisiert in der dichtesten Stapelfolge Kugelpackung von mit einer ABABCBCB. Die Oktaederlücken zwischen den Titanatome sind vollständig mit Kohlenstoff besetzt (dC-Ti2: 2,184(3) Å und dC-Ti1: 2,073(3) Å). Gallium bildet die Metallschicht zwischen den Doppelschichten von CTi6-Oktaedern (dTi1-Ti2: 2,942(1) Å). Die Titanatome der Sorte 1 sind von neun Titanatomen (6 x Ti1 dTi2-Ti2: 3,0731(4) Å und 3 Ti2dTi1-Ti2: 2,942(1) Å) und von drei Galliumatomen in Form eines Abbildung 3.4.34: Struktur von Ti3GaC2 Antikuboktaeders umgeben. Die Koordinationssphäre des Galliumatoms besteht aus jeweils sechs gleichen Atomen (Ga-Schicht dGa-Ga: 3,0731(4) Å) sowie dem Beitrag von sechs Titanatomen, jeweils drei aus der darüber bzw. darunter liegenden Schichten. Das Titan 2 in der mittleren Schicht ist gemäß der kubischen Sequenz kuboktaedrisch von zwölf Titanatomen koordiniert (dTi1-Ti2: 2,942(1) Å). Hier wird der Beitrag aus sechs Titananatomen der gleichen Schicht (Ti2) und jeweils von drei Titanatomen aus der darüber liegenden und darunter liegenden Schicht erbracht. Die Kohlenstoffatome besetzen alle gebildeten Oktaederlücken der Titanatome und bringen so noch einen Beitrag zu der Koordination von Titan 2 zwischen den Schichten. Die Abstände von Titan und Kohlenstoff liegen mit einem Abstand von 2,184(3) Å sehr nahe an den vom binärem TiC (dTi-C: 2,164 Å). - 119 - MAX - Phase – Das System Ti-(Al/Ga)-C 3.4.5.7 Röntgenographische Untersuchung an Einkristallen der 211-Phase Mit dem IPDS II wurden 1197 Reflexe gemessen. Nach einer Mittelung in der Laueklasse 6/mmm und den üblichen Korrekturen konnte ein Datensatz mit 93 unabhängigen Reflexen gebildet werden. Temperaturfaktoren Die Verfeinerung problemlos bei konvergierte nach Zuverlässigkeitswerten Einführung von anisotroper R1(F)=0,0165 und wR2(I)=0,0378. Bei einer wechselweisen Freigabe der Besetzungsfaktoren konnte keine signifikante Abweichung von der vollständigen Besetzung vernommen werden. Die ermittelten Restelektronendichten lagen mit 0,93 und -0,42 in einem normalen Rahmen. Der Vergleich der erhalten Daten mit der Literatur (Ti2GaC a = 3,0764(4) Å; c = 13,378(3) Å)[202] zeigte eine gute Übereinstimmung der Parameter. - 120 - MAX - Phase – Das System Ti-(Al/Ga)-C Tabelle 3.4.5.6: Angaben zur Strukturanalyse von Ti2GaC. Summenformel Ti2GaC Kristallsystem hexagonal Raumgruppe P 63/m m c – Nr. 194 a = 3,0627(9); c = 13,336(6) Gitterkonstante [Å] 3 Volumen der Elementarzelle [Å ] 108,33(8) Zahl der Formeleinheiten 2 3 röntgenogr. Dichte [g/cm ] 5,45 F(000) 162,3 Kristallform, -farbe metallisch glänzende, hexagonale Plättchen Diffraktometer Rotation STOE IPDS II (Mo-Kα-Strahlung; λ=0,71073 Å Graphitmonochromator 0° ≤ ω ≤ 180° ψ = 0°, 111; ∆ω = 2° Belichtungszeit [sec.] 300 Messtemperatur [K] Raumtemperatur [293(2)] Kristallgröße [mm] 0,1 × 0,1 × 0,01 Absorptionskoeffizient [mm-1] 19,08 Messbereich [°] 4°<2θ<65° -2 ≤ h ≤ 0, Indexbereich 0 ≤ k ≤ 4, 0 ≤ l ≤ 19 Zahl der gemessenen Reflexe 1197 Zahl der unabh. Reflexe mit I > 0 93 I > σ2(I): 75 freie Parameter 9 Korrekturen Lorentz, Polarisation Absorptionskorrektur numerical / equivalent method (XSHAPE[68]) Strukturlösung Isotypie Cr2AlC[71] Strukturverfeinerung SHELXTL-97[199] Wichtungsschema w-1 = σ2F02 + (0,285 * P)2 + 0,00 * P P = (F02 + 2 FC2) / 3 Goodness of fit 0,966 Extinktionskorrektur 0,118(9) Restelektronendichte [e-/Å3]: +0,93 / -0,42; R-Werte für I > 0: Alle Reflexe 0,16 e-/Å3 R1(F) = 0,0165 wR2(I) = 0,0378 R1(F) = 0,0169 - 121 - MAX - Phase – Das System Ti-(Al/Ga)-C Tabelle 3.4.5.7: Ortskoordinaten, Besetzungsfaktoren (in [%]) und Auslenkungsparameter von Ti2GaC. Atom Lage x y z sof Ueq ⅓ ⅔ 0,58555(3) 100* 0,0059(2) Ga 4f 2d ⅔ ⅓ ¾ 100* 0,0095(3) C 2a 0 0 0 100 0,0066(7) Ti * bei wechselweiser Freigabe der Besetzungsfaktoren konnte keine signifikante Restelektronendichte vorhanden Atom U11 U22 U33 U23 U13 U12 Ti 0,0047(3) 0,0047(3) 0,0059(3) 0 0 0,0023(1) Ga 0,0113(3) 0,0113(3) 0,0059(3) 0 0 0,0056(1) Die folgende Tabelle 3.4.5.8 zeigt benachbarte Atome, ihren charakteristischen Abstand und die Koordinationszahl. Tabelle 3.4.5.8: Ausgewählte Abstände und Winkeln in der Ti2GaC.-Struktur Anzahl der Winkel Bindungen Ti - C/N - Ti Benachbarte Atome Abstand [Å] Ga - Ti 2,8172(5) x6 Ga 3,0627(9) x6 C 2,1044(3) x6 93,39(1)° Ga 2,8172(5) x3 86,61(1)° Ti 2,8867(7) x3 Ti 3,0627(9) x6 Ti 2,1044(3) x6 Ti - C- 3.4.5.8 Strukturdiskussion Ti2GaC kristallisiert im Cr2AlC-Typ in der Raumgruppe P63/mmc mit den Gitterkonstanten a = 3,0627(9) Å und c = 13,336(6) Å. Die ermittelten Gitterkonstanten stehen in sehr guter Übereinstimmung mit der Literatur. Die H-Phase der Zusammensetzung Ti2GaC wurde aus einer Aluminium/Gallium-Schmelze gezüchtet, zeigt jedoch keine Anzeichen, dass Aluminium in die Struktur eingebaut wurde. Das Gallium besetzt vollständig die - 122 - MAX - Phase – Das System Ti-(Al/Ga)-C kristallographische Lage 2d, hier erfährt das Hauptgruppenmetall von sechs weiteren Galliumatomen und sechs Titanatomen eine zwölffach Koordination (dGa-Ga: 3,0627(3) Å und dTi1-Ga1:2,8172(5) Å). Die Titanatome bilden die Oktaeder mit einem Abstand zu nächstem Titanatome von 2,8867(7) Å. Die Oktaeder sind über gemeinsame Kanten unendlich zu Schichten verknüpft. In den Oktaederlücken befinden sich die Kohlenstoffatome mit einer sechsfach Koordination vom Titan (dC1-Ti1: 2,1044(3) Å). Diese sind etwas kürzer als in TiC (dC1-Ti1: 2,164 Å). - 123 - MAX - Phase – Das System Ti-(Al/In)-C 3.4.6 Arbeiten in einer Aluminium/Indium-Schmelze Das ternäre System Titan-Indium-Kohlenstoff ist nur wenig erforscht. Die durchgeführten Untersuchungen wurden überwiegend in der titanreichen Ecke unternommen. Es konnten nur zwei ternäre Verbindungen nachgewiesen werden, Nämlich Ti3InC[85] (CaTiO3-Typ AntiPerovskit in der RG: Pm 3 m; a = 4,199 Å) und eine MAX-Phase der Zusammensetzung Ti2InC[27] mit dem Gitterkonstanten a = 3,132 Å und c = 14,06 Å. 3.4.6.1 Die binären Randsysteme Al-In und Ti-In sowie In-C Das binäre System bestehend aus Aluminium und Indium bietet keine einzige Phase aus den bisherigen Untersuchungen[175]. Charakteristisch für dieses System ist die Mischungslückeder Schmelze unterhalb des Punktes mit 875 ± 5°C, und ein eutektischer Punkt bei 88,5± 1°C. Wie der Abbildung 3.4.36 zu entnehmen ist, wurde das Titan-Indium System noch nicht vollständig aufgeklärt. Jedoch wurde von einigen binären Phasen aus diesem System berichtet. Ausgehend von der indiumreichen Ecke kristallisiert In5Ti3 (Mg3Cd-Typ; RG: P 63/mmc)[86] bei 57,2 At.-% Indium schon bei einer Temperatur von 156 ± 2°C. In dem nicht aufgeklärten Bereich (hier als Fragezeichen markiert) wurde von drei Phasen der Zusammensetzung In4Ti3 (Hg5Mn2-Typ; P4/mbm)[87], InTi3 (Mg3Cd-Typ; RG: P63/mmc)[88] und In5Ti8 (eigener Struktur-Typ, RG: P-43m)[88] berichtet. Abbildung 3.4.35: Das binäre Phasendiagramm des Systems Aluminium-Indium[175]. Abbildung 3.4.36: Das binäre Phasendiagramm des Systems Titan-Indium[175]. Es gibt keine bekannten Phasen im binäres System aus Indium und Kohlenstoff. - 124 - MAX - Phase – Das System Ti-(Al/In)-C 3.4.6.2 Präparative Untersuchung im System Titan-(Aluminium/Indium)Kohlenstoff Die Synthese erfolgte aus den Elementen. Titan-, Aluminiumpulver und „carbon black“ wurden in entsprechenden Verhältnissen homogenisiert und in Tabletten gepresst. Indium wurde aus einem Indiumdraht in Stücke portioniert und auf der Tablette platziert. Das Aufschmelzen der Proben wurde bei einer Temperatur von 1600°C in einer ArgonAtmosphäre durchgeführt. Die Reaktionszeit betrug 25 Stunden, dieses ermöglichte eine qualitative Umsetzung der Edukte. Die Proben wurden sehr langsam (5°C/h) bis auf eine Temperatur von 1300°C abgekühlt, um ein möglichst gute Kristallisation zu ermöglichen. Die Zusammensetzung der Proben wurde durch die Veränderung der Schmelzzusammensetzung verändert. Somit ergaben sich vier Ansätze von indiumarm bis indiumreich. Nach dem Abkühlen wurde das überschüssige Hilfsmetall mit verd. Salzsäure gelöst. Der Rückstand bestehand aus hexagonalen, metallischen Plättchen, welche abgesaugt, mit Wasser mehrmals gewaschen und getrocknet wurden. Die pulverdiffraktometrische Charakterisierung der Produkte ermöglichte die Bestimmung der Phasen wie auch die Verfeinerung der Gitterkonstanten der gebildeten MAX-Phasen. Temperaturprogramm: 150°C/h RT -5°C/h -15°C/h 1300°C 800°C (25h halten) (1h halten) (1h halten) Tabelle 3.4.6.1: Proben im System pulverdiffraktometrische Charakterisierung. Nr. Ti Al In C 1 Ti6Al10In5C1 27,3 45,4 22,7 4,5 2 Ti6Al7,5In7,5C1 27,3 34,1 34,1 4,5 3 Ti6Al5In10C1 27,3 22,7 45,4 4,5 Ti6Al3In12C1 27,3 13,6 54,5 4,5 4 RT Titan-Aluminium/Indium-Kohlenstoff Gehalt [At.-%] Zusammensetzung -100°C/h 1600°C und ihre Gitterkonstante [Å] weitere kristalline der MAX-Phase Phasen a = 3,0714(8); c = 14,027(6) V=114,60(5) a =3,0661(12); c = 14,031(3) V=114,19(2) a =3,044(2); c = 13,854(1) V=111,17(11) a =3,0419(14); c = 13,958(3) V=111,85(16) - - - - - 125 - MAX - Phase – Das System Ti-(Al/In)-C Das Ergebnis der pulverdiffraktometrischen Analyse war phasenrein. In jedem der Ansätze konnte eine Gitterkonstante verfeinert werden, die für eine MAX-Phase mit der 211Stapelsequenz charakteristisch ist. In den indiumreichen Ansätzen konnte jedoch eine Diskrepanz des Volumens beobachtet werden. Bei der Betrachtung der Ansätze mit höherem Indiumgehalt zeigt sich eine Verkleinerung der Zelle. Eine Volumenbetrachtung von Ti2AlC Phase (V = 110,18 Å) und von Ti2InC Phase (V = 119,44 Å) lässt das Resümee zu, dass die entstandenen kristallinen 211-Phasen eine Mischbesetzung auf der Position der Hauptgruppenmetalle aufweisen könnten. 3.4.6.3 Röntgenographische Untersuchung an Einkristallen der 211-Phase Aus dem indiumärmsten Ansatz konnten geeignete Einkristalle für eine Strukturverfeinerung isoliert werde. Mit dem STOE-IPDS II wurde 1846 Reflexe gesammelt. Nach der Mittelung in der Laueklasse 6/mmm und den üblichen Korrekturen wurde ein Datensatz mit 98 unabhängigen Reflexen erhalten. Die Strukturverfeinerung wurde in der Raumgruppe P63/mmc mit den Atomkoordinaten von Ti2AlC begonnen. Die Strukturverfeinerung konvertierte problemlos zu guten Zuverlässigkeitswerten von R1(F) = 0,0159 und wR2(I) = 0,0287. Aus den ermittelten Gittekonstanten war anzunehmen, dass eine Mischbesetzung vorlag. Somit wurden die Temperatur- und Besetzungsfaktoren gekoppelt und frei verfeinert. Bei Freigabe des Besetzungsfaktors des Titanatoms auf der kristallographischen Lage 4f konnte keine signifikante Abweichung von der vollständigen Besetzung beobachtet werden. Eine Überprüfung auf das Vorhandensein einer Mischbesetzung durch Stickstoff und Kohlenstoff der Nichtmetallposition 2a wurde durch die Freigabe der Besetzungsparameter durchgeführt. Es zeigte sich eine Überbesetzung von 3%, was im Rahmen der Standardabweichung (2,7%) vereinbar ist. An dieser Stelle sollte darauf hingewiesen werden, dass die röntgenographische Erfassung von leichten Elementen wie Kohlenstoff und Stickstoff nur vergleichsweise ungenau möglich ist. Die Restelektronendichte verblieb in einem überzeugenden Rahmen. Die Tabelle 3.4.6.2, .42 und .43 fassen die Einzelheiten aus der Strukturanalyse sowie die Temperatur-, Besetzungsfaktoren und Atomkoordinaten mit den ausgewählten Abständen der benachbarten Atome zusammen. - 126 - MAX - Phase – Das System Ti-(Al/In)-C Tabelle 3.4.6.2: Angaben zur Strukturanalyse von Ti2Al0,85In0,15C. Summenformel Ti2Al0,85In0,15C Kristallsystem hexagonal Raumgruppe P63/mmc Gitterkonstante [Å] a = 3,0588(3); c = 13,852(2) Volumen der Elementarzelle [Å3] 112,24(3) Zahl der Formeleinheiten 2 röntgenogr. Dichte [g/cm3] 4,257 F(000) 133,5 Kristallform, -farbe metallisch glänzende, hexagonale Plättchen STOE IPDS II (Mo-Kα-Strahlung; λ=0,71073 Diffraktometer Å Graphitmonochromator 0° ≤ ω ≤ 180° Rotation ψ = 0°, 111; ∆ω = 2° Belichtungszeit [sec.] 300 Messtemperatur [K] Raumtemperatur [293(2)] 0,1 × 0,1 × 0,02 Kristallgröße [mm] -1 Absorptionskoeffizient [mm ] 8,02 Messbereich [°] 4°<2θ<65° -2 ≤ h ≤ 0, 0 ≤ k ≤ 4, Indexbereich 0 ≤ l ≤ 20 Zahl der gemessenen Reflexe 1846 Zahl der unabh. Reflexe mit I > 0 98 I > σ2(I): 58 freie Parameter 9 Korrekturen Lorentz, Polarisation Absorptionskorrektur numerical / equivalent method (XSHAPE[68]) Strukturlösung Isotypie Cr2AlC89 Strukturverfeinerung SHELXTL-97[199] w-1 = σ2F02 + (0,0179 * P)2 + 0,00 * P Wichtungsschema P = (F02 + 2 FC2) / 3 0,688 Goodness of fit 0,024(5) Extinktionskorrektur - 3 Restelektronendichte [e /Å ]: R-Werte für I > 0: Alle Reflexe +0,39 / -0,39; 0,08 e-/Å3 R1(F) = 0,0159 wR2(I) = 0,0287 R1(F) = 0,0243 - 127 - MAX - Phase – Das System Ti-(Al/In)-C Tabelle 3.4.6.3: Ortskoordinaten, Besetzungsfaktoren (in [%]) und Auslenkungsparameter von Ti2Al0,85In0,15C. Atom Lage x y z sof Ueq ⅓ ⅔ 0,58371(8) 100* 0,0094(2) Al/In 4f 2d ⅓ ⅔ ¾ 85(3)/15# 0,0192(5) C 2a 0 0 ½ 100* 0,012(1) Ti * bei wechselweiser Freigabe der Besetzungsfaktoren keine signifikante Restelektronendichte vorhanden # Besetzungsfaktoren und Temperaturfaktoren auf EDX-Ergebnis gekoppelt und fixiert Atom U11 U22 U33 U23 U13 U12 Ti 0,0079(3) 0,0079(3) 0,0125(4) 0 0 0,0039(1) Al/In 0,0156(6) 0,0156(6) 0,0263(9) 0 0 0,0078(4) Die folgende Tabelle zeigt benachbarte Atome, ihren charakteristischen Abstand in der typischen Koordination. Tabelle 3.4.6.4: Ausgewählte Abstände und Winkeln in der Ti2Al0,85In0,15C -Struktur Anzahl der Winkel Bindungen Ti - C - Ti Benachbarte Atome Abstand [Å] Al/In - Ti 2,9025(10) x6 Al/In 3,0588(4) x6 C 2,126(37 x6 92,76(3)° Al/In 2,9026(10) x3 87,24(3)° Ti 3,0588(4) x6 Ti 2,1126(7) x6 Ti - C - 3.4.6.4 Strukturdiskussion Ti2Al0,85In0,15C kristallisiert im Cr2AlC-Typ mit der Raumgruppe P 63/mmc mit den sehr kleinen Gitterkonstanten von a = 3,0588(3) Å und c = 13,852(2) Å. Die Gitterkonstanten von Ti2AlC sind in der c-Richtung deutlich kleiner als in der untersuchten Mischphase (a = 3,056 Å; c = 13,363 Å). Die Aufweitung der Zelle ist durch die Substitution von Aluminium gegen Indium plausibel. Außer durch die Mischbesetzung der - 128 - MAX - Phase – Das System Ti-(Al/In)-C Hauptgruppenmetalllage gibt es keine signifikante Abweichung von der charakteristischen HPhase. Das Titanatom formen die Oktaeder, der Abstand der Titanatome zueinander ist mit 3,0723(4) Å in einer bekannten Dimension wie schon in den anderen H-Phasen beobachtet. In den Oktaederlücken der Titandoppelschicht befinden sich Kohlenstoffatome (dTi-C: 2,1126(7) Å). Die Hauptgruppenmetalllage ist durch das Arbeiten in einer Aluminium/Indium-Schmelze mischbesetzt. Das Mischverhältnis beträgt 85 % Aluminium und 15 % Indium auf der vollständig besetzten Lage. Die Hauptgruppenmetalle bekommen die gewöhnliche Koordinationssphäre von sechs Gleichartigen Atomen innerhalb der Schicht (dAl/In-Al1/In: 3,0588(4) Å) sowie von den darüber und darunter liegenden Titanatomen (2 x 3 d Al/In-Ti: 2,9026(10) Å). - 129 - MAX - Phase – Das System Ti-(Al/Ge)-C 3.4.7 Arbeiten in einer Aluminium/Germanium-Schmelze Das Dreistoffsystem bestehend aus Titan, Germanium und Kohlenstoff wurde nicht hinreichend untersucht, um ein qualitatives Phasendiagramm zu erstellen. Die Untersuchungen wurden lediglich in der titanreichen Ecke auf die Anwesenheit der MAXPhasen hin durchgeführt. So konnten die MAX-Phase mit der 211- (Ti2GeC: a = 3,079 Å; c = 12,93 Å)[27] und der 312-Variante (Ti3GeC2: a = 3,077 Å; c = 17,67 Å)[90] in den sechziger Jahren erfolgreich dargestellt werden. 3.4.8 Die binären Systeme Bei dem Schmelzdiagramm des Systems Aluminium-Germanium handelt es sich um ein typisches Beispiel für unbegrenzte Mischbarkeit im flüssigen Zustand und begrenzte Mischbarkeit im festen Zustand. Das binäre Phasendiagramm der Metall Titan und Germanium ist etwas komplexer. Insgesamt sind es fünf binäre, isolierbare Phasen die die Metalle miteinander bilden. Ti3Ge (Ni3P-Strukturtyp, RG: I-4)[177] ist die titanreichste Phase, gefolgt von Ge3Ti5 (Mn5Si3-Strukturtyp, RG: P63/mcm)[177], Ge5Ti6 (V6Si5-Strukturtyp, RG: Ibam)[177], GeTi (TiSi-Strukturtyp, RG: Pmm2)[177] und Ge2Ti (TiSi2-Strukturtyp, RG: Fddd)[177]. Abbildung 3.4.37: Das binäre Phasendiagramm des Systems Aluminium-Germanium Abbildung 3.4.38: Das binäre Phasendiagramm des Systems Titan-Germanium Wie aus der Abbildung 3.4.39 zu entnehmen ist, gibt es noch keine bekannte Verbindung im binären System des Kohlenstoffs mit Germanium. - 130 - MAX - Phase – Das System Ti-(Al/Ge)-C Die vollständige Charakterisierung des binären Phasendiagramms von Germanium und Kohlenstoff ist noch nicht dokumentiert. Abbildung 3.4.39: Das binäre Phasendiagramm des Systems Kohlenstoff-Germanium 3.4.8.1 Präparative Untersuchung im System Titan-Germanium-Kohlenstoff Wie schon bei den zuvor gegangenen Untersuchungen wurde auch hier das Verhältnis der Schmelzlösung durchvariiert. Die Elemente wurden in den Verhältnissen, wie in der Tabelle 3.4.8.1 abgegeben, kalt zu Tabletten verpresst. Diese wurden anschließend für 30 Stunden bei 1500°C unter Argon-Schutzgas in Korundtiegeln zur Reaktion gebracht. Temperaturprogramm: 150°C/h RT -4°C/h -15°C/h -200°C/h 1500°C 1000°C 800°C (30h halten) (2h halten) (1h halten) RT Tabelle 3.4.8.1: Proben im System Titan-Germanium-Kohlenstoff und ihre pulverdiffraktometrische Charakterisierung. Fettgedruckt sind die Hauptprodukte. *Phase wurde auf Grund von geringer Intensität nicht Indiziert. Nr. Zusammensetzung Gehalt [At.-%] Ti Ge Al C Gitterkonstante [Å] weitere kristalline der MAX-Phase Phasen 1 Ti2Al5Ge10C1 11,1 27,8 55,6 5,6 - 2 Ti2Al10Ge5C1,5 10,8 54,1 27,0 7,1 - 3 Ti6Al10Ge5C1,5 26,7 44,4 22,2 6,7 - 4 Ti3Al5Ge10C2 15,0 25,0 50,0 10,0 - 5 Ti10Al10Ge5C2 37,0 37,0 18,5 7,5 a = 3,11(1); c=18,05(7) Ge, Al4C3 >>Ti3GeC2* Ge, Ti2Al5, C Al4C3, Ge, Ti5C3,75, C, TiC2 Ge, Ti2Al5, >Ti3GaC2* Ge > , Ti2Al5 V=151,5(1) 6 Ti6Al10Ge5C3 25,0 41,7 20,8 12,5 Ge, C, Ti8C5, - Al4C3, Ti3GeC2* - 131 - MAX - Phase – Das System Ti-(Al/Ge)-C Die Abkühlrate wurde auch hier sehr langsam gewählt, was für eine gute Kristallisation der Schmelze sorgen sollte. Nach dem Abkühlen wurde der Rückstand in verd. Salzsäure eine Woche lang eingelegt. Die säurestabilen Einkristalle wurden anschließend abgesaugt, mit Wasser gewaschen und Luft getrocknet. Die Überprüfung der gebildeten Phasen erfolgte durch ein röntgenographisches Beugungsexperiment am Pulver. Die MAX Phase mit der 312-Variante konnte zwar in drei Ansätzen von sechs beobachtet werden, jedoch nur in Spuren. In einem germaniumarmen Ansatz (Tabelle 3.4.8.1, Nummer 5) wurde die Gitterkonstante aus dem Pulver verfeinert (a = 3,11(1) Å; c = 18,05(7) Å und V=151,5(1) in P63/mmc). Eine phasereine Darstellung war nicht möglich, weitere Verbindungen wie die 211- oder 413-Phasen konnte nicht beobachtet werden. 3.4.8.2 Röntgenographische Untersuchung an Einkristallen Aus dem Ansatz mit der Schmelzzusammensetzung Ti10Al10Ge5C2 konnten Einkristalle von ausreichender Qualität für weitere Untersuchungen isoliert werden. - 132 - MAX - Phase – Das System Ti-(Al/Ge)-C Tabelle 3.4.8.2: Angaben zur Strukturanalyse von Ti3GeC2. Summenformel Ti3GeC2 Kristallsystem hexagonal Raumgruppe P63/mmc a = 3,0807(6); c = 17,779(7) Gitterkonstante [Å] 3 Volumen der Elementarzelle [Å ] 146,13(7) Zahl der Formeleinheiten 2 3 röntgenogr. Dichte [g/cm ] 5,438 F(000) 219,0 Kristallform, -farbe metallisch glänzendes Polyeder STOE IPDS II (Mo-Kα-Strahlung; λ=0,71073 Diffraktometer Å Graphitmonochromator 0° ≤ ω ≤ 180° Rotation ψ = 0°, 111; ∆ω = 2° 300 Belichtungszeit [sec.] 0,025 × 0,025 × 0,025 Kristallgröße [mm] -1 Absorptionskoeffizient [mm ] 17,53 Messbereich [°] 4°<2θ<70° -2 ≤ h ≤ 0, 0 ≤ k ≤ 4, Indexbereich 0 ≤ l ≤ 28 Zahl der gemessenen Reflexe 2595 Zahl der unabh. Reflexe mit I > 0 129 I > σ2(I): 101 freie Parameter 11 Korrekturen Lorentz, Polarisation Absorptionskorrektur numerical / equivalent method (XSHAPE[68]) Strukturlösung Isotypie Ti3SiC2[84] Strukturverfeinerung SHELXTL-97[199] w-1 = σ2F02 + (0,0338 * P)2 + 0,00 * P Wichtungsschema P = (F02 + 2 FC2) / 3 0,923 Goodness of fit 0,0242(5) Extinktionskorrektur - 3 Restelektronendichte [e /Å ]: R-Werte für I > 0: Alle Reflexe +1,46 / -0,64; 0,20 e-/Å3 R1(F) = 0,0213 wR2(I) = 0,0522 R1(F) = 0,0253 - 133 - MAX - Phase – Das System Ti-(Al/Ge)-C Tabelle 3.4.8.3: Ortskoordinaten, Besetzungsfaktoren (in [%]) und Auslenkungsparameter von Ti3GeC2. Atom Lage x y z sof Ueq Ti 1 2a 1 0 ½ 100* 0,0028(3) Ti 2 ⅔ ⅓ 0,63293(5) 100* 0,0031(3) Ge 4f 2b 1 0 ¾ 100* 0,0082(3) C 4f ⅔ ⅓ 0,42862(27) 100* 0,0041(9) * bei wechselweiser Freigabe der Besetzungsfaktoren keine signifikante Restelektronendichte vorhanden Atom U11 U22 U33 U23 U13 U12 Ti 1 0,0023(5) 0,0023(5) 0,0038(5) 0 0,0048(2) 0,0011(2) Ti 2 0,0025(3) 0,0025(3) 0,0044(4) 0 0,0052(3) 0,0012(1) Ge 0,0097(4) 0,097(4) 0,0051(4) 0 0 0,0048(2) C 0,0059(15) 0,0059(15) 0,00681(16) 0 0 0,0029(8) Die folgende Tabelle zeigt benachbarte Atome, ihren charakteristischen Abstand und die Koordinationszahl. Tabelle 3.4.8.4: Ausgewählte Abstände und Winkeln in der Ti3GeC2-Struktur Anzahl der Winkel Bindungen Ti - C - Ti Benachbarte Atome Abstand [Å] Ge - Ti 2 2,7369(8) x6 Ge 3,0812(4) x6 C 2,185(3) x6 89,54(2)° Ti 2 2,9567(9) x6 87,54(2)° C 2,088(3) x3 95,09(15)° Ge 2,7369(8) x3 90,32(14)° Ti 1 2,9567(9) x3 89,68(14)° Ti 2 3,0812(4) x6 Ti 2 2,088(3) x3 Ti 1 2,185(3) x3 Ti 1- Ti 2- C- - 134 - MAX - Phase – Das System Ti-(Al/Ge)-C 3.4.8.3 Strukturdiskussion Ti3GeC2 kristallisiert in Ti3SiC2-Typ in der Raumgruppe P63/mmc mit den Gitterkonstanten von a = 3,0807(6) Å und c = 17,779(7) Å. Die Gitterkonstanten liegen in einer guten Übereinstimmung mit der in der Literatur dokumentierten. Bisherige Vergleichsdaten wurden aus Beugungsexperimenten am Pulver erhalten. Wie bei allen MAX-Phasen der 312 Varianten ist auch hier eine dichteste Packung der Metalle mit der Stapelfolge von ABABCBCB gegeben. Die Titanatome besetzen die kristallographischen Punktlagen 4f und 2a und bilden hier die Doppelschicht an über die Kantenverknüpften Oktaedern, die in der [001]-Ebene unendlich ist. Die Oktaeder sind leicht verzerrt und besitzen ein Kantenlänge entlang der Schichten von 3,0812(4) Å und entlang der c-Achse eine Abstand von 2,9567(9) Å. Alle Oktaederlücken in der Doppelschicht sind mit Kohlenstoffen besetzt (3 x dTi1-C1: 2,088(3) Å und 3 x dTi2-C1:2,185(3) Å. Das Germaniumatom bildet die metallische Schicht zwischen den carbidischen Einheiten (Ti3C2) in der Struktur. Die Titanatome der äußeren Schichten befinden sich im Abstand von 2,7369(8) Å zu den Germaniumatomen und koordinieren somit mit jeweils drei Atomen aus der darüber und darunter liegenden Schicht sechsfach das Germaniumatom. Zudem besitzt jedes Germaniumatom entlang der [001]-Ebene sechs weiter Germaniumnachbaren in der Koordinationssphäre (dGe1-Ge1: 3,0807(6)Å). 3.4.8.4 Härtemessung Von den auf dem IPDS II untersuchten Einkristalle (Tabelle 3.4.8.1/Nr. 5) der Zusammensetzung Ti3GeC2 konnten zwei Einkristalle von ausreichender Größe für die Härteprüfung nach Vickers eingebettet werden. Die Einkristalle wurden so eingebettet das nur eine Härteprüfung parallel zur basalen Ebene möglich war. An den zwei Einkristallen konnten jeweils zwei Abdrücke vorgenommen werden. Abbildung 3.4.40 zeigt einen der untersuchten Einkristalle mit typischen Abdrücken der Vickerspyramide. - 135 - MAX - Phase – Das System Ti-(Al/Ge)-C (I) (II) (I) oben: d1=32,70µm, d2=30,68µm: 370 HV 0,2 (II) unten: d1=34,42µm, d2=32,30µm: 333 HV 0,2 Abbildung 3.4.40: Zwei Vickers Abdrücke Aus den vier Abdrücken konnte ein Mittelwert von 362 HV 0,2 gebildet werden. Somit konnte der Härtewert von 3,5 GPa für Ti3GeC2 bestimmt werden. Dieser Wert liegt in der Größenordnung, wie er schon an bekannten MAX-Phasen gemessen werden konnten[45,46]. 3.4.8.5 Synthese im Pulsplasma-Ofen Für die Synthese einer MAX-Phase des oben diskutierten Systems im Pulsplasma Ofen wurde im System ein Temperaturbereich zwischen 900°C bis 1100°C untersucht. Die Synthese wurde ausgehend von den Elementen durchgeführt. 0,213 g (4,447 mmol) Titan, 0,161 g (2,218 mmol) Germanium sowie 0,027 g (2,250mmol) Graphit wurden eingewogen und in einer Graphitmatrize mit einem Durchmesser von 10 mm unter einem Sinterdruck von 3,9 kN und einem Vakuum von 0,11 mbar für 5 Minuten11 bei einer Temperatur von 1000°C zur Reaktion gebracht. Dabei wurde folgendes Temperaturprogramm verwendet. Temperaturprogramm: 2 min. RT 2 min. 600°C 2 min 750°C 3 min 900°C 45 min. 1000 RT (5 min. halten) 11 Hier wurden Synthesen bei Reaktionszeiten von 2 bis 15 Minuten durchgeführt. Das in der Abbildung 3.4.41 dargestellte Pulverdiffraktogramm ist das Ergebnis bei einer Reaktionszeit von 5 Minuten, hier trat die MAXPhase am deutlichsten in Erscheinung. - 136 - MAX - Phase – Das System Ti-(Al/Ge)-C 24000 23000 22000 21000 20000 19000 18000 17000 16000 Lin (Counts) 15000 14000 13000 12000 11000 10000 9000 8000 7000 6000 5000 4000 3000 2000 1000 0 5 10 20 30 40 50 60 70 80 90 100 110 120 2-Theta - Scale Abbildung 3.4.41: Pulverdiffraktogramm von Ti2GeC (HP) grün. Die blauen Reflexe konnten Titan zugeordnet werden. Bei den rot markierten Reflexen handelt es sich um Ge3Ti5 (NP); die violetten Reflexe konnten elementarem Germanium zugeordnet werden. Der Sinterkörper wurde aus der Graphitmatrize entnommen, von der Graphitfolie befreit und zertrümmert. Durch eine röntgenographische Phasenanalyse konnten die gebildeten Phasen eindeutig identifiziert und die Gitterkonstante der MAX-Phase verfeinert werden. Bei dem Hauptprodukt handelt es sich um Ti2GeC mit den Zellenparametern von a = 3,092(4) Å und c = 12,946(14) Å in der Raumgruppe P63/mmc. In einem hohen Anteil konnten noch nicht umgesetztes Titan, Germanium und Ge3Ti5 beobachtet werden. Bei Versuchen die MAX-Phase bei Temperaturen unter 1000°C zu erhalten, konnte keine Bildung der MAXPhase beobachtet werden. Bei Temperaturen über 1000°C ist das Germanium aus der Matrize ausgelaufen. - 137 - MAX - Phase – Das System V-(Al/Sn)-C 3.5 Das System Vanadium-Aluminium-Kohlenstoff 3.5.1 Die binären Randsysteme um das System V-Al-C Das binäre System Vanadium-Kohlenstoff zeigt sechs verschiedene Carbide VC[91] (NaClTyp) V2C[92,93] kommt in zwei unterschiedlichen Modifikationen vor (Anti-PbO2-Typ und Defekt-NiAs-Typ). V4C3-x wie auch die V6C5[94] und V8C7[95] kristallisiert im Ti7S12-Typ, bei den letzten drei Carbiden handelt es sich um eine Tieftemperaturmodifikation von VC1-x bei denen die Fehlstellen im Kohlenstoffteilgitter ausgeordnet vorliegen. Abbildung 3.5.1: Das binäre Phasendiagramm des Systems Vanadium-Kohlenstoff[175] Abbildung 3.5.2: Das binäre Phasendiagramm des Systems Vanadium-Aluminium[175] Im System Vanadium-Aluminium konnten sieben Verbindungen beschrieben werden. V3Al (Cr3Si-Typ), V50Al50 (W-Typ), V5Al8 (γ-Messing-Typ), VAl3, (TiAl3-Typ), V4Al23. V7Al45 und Val10 (die drei zuletzt genannten Verbindungen kristallisieren alle als eigener Strukturtyp). Das Phasendiagramm von Aluminium-Kohlenstoff wurde bereits im Kapitel 3.4.1 diskutiert. Als einzige Verbindung konnte Al4C3 beschrieben werden. 3.5.2 Das ternäre System Titan-Aluminium-Kohlenstoff Das System Vanadium-Aluminium-Kohlenstoff wurde bereits von Schuster et al. bei einer Temperatur von 1000°C hinsichtlich des Gleichgewichtsphasen hin untersucht[96]. Das ternäre Phasendiagramm wurde auf der Basis dieser Untersuchungen aufgestellt. Nur von einer einzigen ternären Verbindung mit der Zusammensetzung von V2AlC [33] (Cr2AlC-Typ in der - 138 - MAX - Phase – Das System V-(Al/Sn)-C RG: P 63/mmc[33]) konnte zu dieser Zeit berichtet werden. Neue Untersuchungen bei Temperaturen von 1500°C ermöglichten die Darstellung von V4AlC3-x (V12Al3C8) aus den Elementen[202]. Neben den MAX-Phasen wurden noch zahlreiche intermetallische Verbindungen und Carbide von Aluminium und Vanadium beobachtet, die in Konkurrenz zur den MAX-Phasen kristallisieren. In der Abbildung 3.5.3 sind die Gebiete markiert in welchen die 211-Phase beobachtet wurden. Abbildung 3.5.3: Das ternäre Phasendiagram von Vanadium-Aluminium-Kohlenstoff. Abgebildet ist ein isothermer Schnitt bei einer Temperatur von 1000°C. Hervorgehoben ist die H-Phase. 3.5.3 Arbeiten in einer Aluminium/Zinn-Schmelze Die Phasendiagramme von Zinn-Kohlenstoff, Zinn-Aluminium wurden bereits in den Kapitel 3.4.4.1 diskutiert. Im binären System von Vanadium mit Zinn (Abbildung 3.5.4 - 139 - MAX - Phase – Das System V-(Al/Sn)-C Phasendiagramm) konnten nur zwei intermetallische Verbindungen beschrieben werden, nämlich Sn2V[97] (Mg2Cu-Typ) und SnV3[98] (Cr3Si-Typ). Abbildung 3.5.4: Das binäre Phasendiagramm des Systems Vanadium-Zinn[175] Der Einbau von Zinn in einer Vanadium- MAX-Phase konnte bis jetzt noch nicht beobachtet werden. Das Benutzen einer Zinn/Aluminium-Schmelze sollte dem Versuch dienen, eine mit Zinn dotierte MAX-Phase zu bilden. Die Untersuchungen wurden bei Temperaturen von 1600°C durchgeführt. 3.5.3.1 Präparative Untersuchung im System Vanadium-(Aluminium/Zinn)Kohlenstoff In der Versuchsreihe wurden unterschiedliche Mischungen der Hilfsmetalle untersucht. So wurde der Vanadium- und Kohlenstoffanteil konstant gehalten während das Mischverhältnis von der Schmelze variiert wurde. In der Tabelle 3.5.3.1 sind die Zusammensetzungen der Schmelze und das Ergebnis aus der pulverdiffraktometrischen Charakterisierung angegeben. Die Elemente wurden in den unten angegebenen Verhältnissen eingewogen, in einem Achat-Mörser innig verrieben und zu Tabletten mit Hilfe einer Handpresse verarbeitet. In Korundtiegeln wurden die Proben bei einer Temperatur von 1600°C für 25 Stunden unter Argonschutzgas zur Reaktion gebracht. Als Kohlenstoffquelle wurde auch hier „carbon black“ verwendet. Temperaturprogramm: 150°C/h RT -5°C/h -15°C/h -100°C/h 1600°C 1300°C 800°C (25h halten) (1h halten) (1h halten) RT - 140 - MAX - Phase – Das System V-(Al/Sn)-C Tabelle 3.5.3.1: Proben im Titan-Aluminium/Indium-Kohlenstoff pulverdiffraktometrische Charakterisierung. Nr. Gehalt [At.-%] Zusammensetzung V Al Sn C 1 V6Al10Sn5C1 27,3 45,5 22,7 4,5 2 V6Al7,5Sn7,5C1 27,3 34,1 34,1 4,5 3 V6Al5Sn10C1 27,3 22,7 45,5 4,5 4 V6Al3Sn12C1 27,3 13,6 54,5 4,5 System und ihre Gitterkonstante [Å] weitere kristalline der MAX-Phase Phasen a = 2,926(2); c = 13,281(16) V=98,42(14) a = 2,925(18); c = 13,289(18) V=98,49(18) a = 2,936(17); c = 13,196(3) V=98,51(7) - V6C5, V5Al3C V6C5, V3Al, V5Al3C V6C5, V5Al3C V2C, V6C5, >V2AlC Der gewonnene Schmelz Regulus wurde jeweils über Nacht mit verd. Salzsäure behandelt. Der Rückstand wurde abgesaugt, gewaschen und getrocknet. Unter einem Lichtmikroskop wurden metallisch glänzende Plättchen mit einer durchschnittlichen Kantenlänge von 50µm isoliert und auf einem Glasfaden für eine röntgenographische Analyse unterzogen. Die übrige Ansammlung von Kristallen wurde mit Hilfe eins Mörsers verrieben und einer Phasenanalyse unterzogen. In den ersten drei Ansätzen konnte die MAX-Phase mit der 211-Variante anhand der charakteristischen Gitterkonstanten eindeutig identifiziert werden (Lit. a = 2,9116 Å; c = 13,140(3) Å mit einem Volumen von V = 96,47 Å3 in P63/mmc)[33]. Es zeigt sich eine kleine Abweichung der ermittelten Parameter (Volumen und Gitterkonstante) aus den Pulverdaten mit dem der Literatur. So konnte eine Aufweitung der Zelle beobachtet werden, die sich durch ein geringes Maß an Substitution von Aluminium gegen Zinn erklären lässt. Somit lassen sich auch bei diesem System nach der Vegard’schen Regl eine lineare Abhängigkeit bezogen auch die Gitterkontante und das Volumen erkennen und folglich vorherzusagen. 3.5.3.2 Röntgenographische Untersuchung an Einkristallen der 211-Phase Mit dem IPDS II wurde ein Datensatz mit 2642 Reflexen gesammelt und auf der Basis einer H-Phase verfeinert. - 141 - MAX - Phase – Das System V-(Al/Sn)-C Tabelle 3.5.3.2: Angaben zur Strukturanalyse von V2Al0,92Sn0,08C2. Summenformel V2Al0,92Sn0,08C Kristallsystem hexagonal Raumgruppe P63/mmc Gitterkonstante [Å] a = 2,9201(20); c = 13,074(11) Volumen der Elementarzelle [Å3] 96,54(12) Zahl der Formeleinheiten 2 röntgenogr. Dichte [g/cm3] 5,019 F(000) 134,1 Kristallform, -farbe metallisch glänzende, hexagonale Plättchen STOE IPDS II (Mo-Kα-Strahlung; λ=0,71073 Diffraktometer Å Graphitmonochromator 0° ≤ ω ≤ 180° Rotation ψ = 0°, 111; ∆ω = 2° Belichtungszeit [sec.] 300 Messtemperatur [K] Raumtemperatur [293(2)] 0,03 × 0,03 × 0,01 Kristallgröße [mm] -1 Absorptionskoeffizient [mm ] 16,74 Messbereich [°] 4° < 2θ < 65° -2 ≤ h ≤ 0, Indexbereich 0 ≤ k ≤ 4, 0 ≤ l ≤ 19 Zahl der gemessenen Reflexe 2642 Zahl der unabh. Reflexe mit I > 0 90 I > σ2(I): 53 freie Parameter 10 Korrekturen Lorentz, Polarisation Absorptionskorrektur numerical / equivalent method (XSHAPE[68]) Strukturlösung Isotypie Cr2AlC[71 Strukturverfeinerung SHELXTL-97[199] Wichtungsschema w-1 = σ2F02 + (0,0297 * P)2 + 0,0 * P P = (F02 + 2 FC2) / 3 Goodness of fit 0,662 Extinktionskorrektur 0,045(14) Restelektronendichte [e-/Å3]: +0,39 / - 0,77; R-Werte für I > 0: Alle Reflexe ] 0,12 e-/Å3 R1(F) = 0,0177 wR2(I) = 0,0384 R1(F) = 0,0343 - 142 - MAX - Phase – Das System V-(Al/Sn)-C Tabelle 3.5.3.3: Ortskoordinaten, Besetzungsfaktoren (in [%]) und Auslenkungsparameter von V2Al0,92Sn0,08C2. Atom Lage x y z ⅔ sof ⅓ Al/Sn 4f 2d ⅓ ⅔ ¾ 92 / 8 C 2a 0 0 0 100* V 0,5849(1) 100* Ueq 0,0045(3) # 0,0040(6) 0,0058(12) * bei wechselweiser Freigabe der Besetzungsfaktoren keine signifikante Restelektronendichte vorhanden # Besetzungsfaktoren und Temperaturfaktoren gekoppelt und fixiert Atom U11 U22 U33 U23 U13 U12 V 0,0052(4) 0,0052(4) 0,031(4) 0 0 0,0026(2) Al/Sn 0,0060(7) 0,0060(7) 0,000(1) 0 0 0,0030(3) C 0,0068(14) 0,0068(14) 0,0038(19) 0 0 0,0034(7) Die folgende Tabelle zeigt benachbarte Atome, ihren charakteristischen Abstand und die Koordinationszahl. Tabelle 3.5.3.4: Ausgewählte Abstände und Winkeln in der V2Al0,92Sn0,08C2-Struktur Anzahl der Winkel Bindungen Ti - C - Ti Benachbarte Atome Abstand [Å] Al/Sn - V 2,7381(14) x6 Al/Sn 2,9201(4) x6 C 2,0191(10) x6 92,53(9)° Al/Sn 2,7381(14) x3 87,47(9)° V 2,789(3) x3 V 2,9201(4) x6 V 2,0191(10) x6 V- C- 3.5.3.3 Strukturdiskussion V2Al0,92Sn0,08C kristallisiert in der Raumgruppe P63/mmc mit den Gitterkonstanten von a = 2,920(2) Å und c = 13,074(11) Å. - 143 - MAX - Phase – Das System V-(Al/Sn)-C Die in der Literatur beschriebene Phase V2AlC besitzt Gitterkonstanten von a = 2,9116 Å und c = 13,127 Å. Die geringe Abweichung lässt sich durch die partielle Substitution von Aluminium gegen Zinn erklären. Vanadium besetzt vollständig die Punktlage 4f, hier werden die über Kanten miteinander verknüpfte Oktaeder ausgebildet mit einer Kantenlänge von 2,7892(2) Å. Im Zentrum jedes Oktaeders befindet sich ein Kohlenstoffatom und wird so von jeweils sechs Vanadiumatomen mit 2,0191(10) Å koordiniert. Die Oktaederlücken sind vollständig von Kohlenstoff besetzt. Die Lage des Hauptgruppenmetalls ist in einem Verhältnis von 92% Al : 8 % Sn durch Aluminium und Zinn mischbesetzt. Das Hauptgruppenmetall erfährt eine Koordination von sechs gleichen Atomen im Abstand von 2,9201(4) Å und von jeweils drei Vanadiumatomen aus der darüber und darunter liegenden Schicht (dAl/Sn–V = 2,7381(14) Å). - 144 - MAX - Phase – Das System V-(Al/Sn)-C 3.5.3.4 Röntgenographische Untersuchung an Einkristallen der 531-Phase Neben den metallischen Plättchen konnten im Ansatz 1 und 3 metallisch glänzende Nadeln unter dem Lichtmikroskop beobachtet werden. Über eine röntgenographische Untersuchung an Einkristallen konnten die isolierten Einkristalle als eine neue Phase mit der Zusammensetzung von V5Al3C1-x (x bis 0,07) charakterisiert werden. Mit dem IPDS II wurden zwei Einkristalle vermessen werden. Aus den gesammelten Daten konnte eine hexagonale Elementarzelle mit den Gitterkonstanten von a = 7,1438(18)Å; und c = 4,8692(10) Å für V5Al3C bzw. a = 7,1657(4) Å und c = 4,86461(2) Å für V5AlC0,93N0,07 ermittelt werden. Nach Mittelung in der Laueklasse 6/mmm stand ein Datensatz von je 152 bzw. 157 unabhängigen Reflexen zur Verfügung. Entsprechend den Werten der E-Statistik wurde die Strukturlösung in der Raumgruppe P63/mcm begonnen. Mit der direkten Methode wurde ein Strukturmodell erhalten mit den Atomkoordinaten von Ti5,71Al2,29C0,67[167]. Die anschließende Verfeinerung der 13 bzw. 14 freien Parameter konvergierte nach Einführung von anisotropen Temperaturfaktoren zu Zuverlässigkeitswerten von R1(F) = 0,0284 und wR2(I) = 0,0829 und für V5Al3C0,93N0,07 mit R1(F) = 0,0240 und wR2(I) = 0,0596. Bei der Freigabe der Besetzungsfaktoren der Metallposition konnte keine signifikante Abweichung von der geordneten, vollständigen Besetzung beobachtet werden. Abbildung 3.5.5: V5Al3C- Einkristall metallisch, glänzende Nadel - 145 - MAX - Phase – Das System V-(Al/Sn)-C Tabelle 3.5.3.5: Angaben zur Strukturanalyse von V5Al3C (links) und V5Al3C0,93N0,07 (rechts). Summenformel V5Al3C1 V5Al3C0,93N0,07 Kristallsystem hexagonal Raumgruppe P63/mcm Gitterkonstante [Å] 3 Volumen der Elementarzelle [Å ] a = 7,1438(18); c = 4,8692(10) a = 7,1657(4); c = 4,8646(2) 215,20(9) 216,322(19) Z=8 Zahl der Formeleinheiten 3 röntgenogr. Dichte [g/cm ] 5,365 5,340 F(000) 320,0 320,1 metallisch glänzende Nadeln Kristallform, -farbe STOE IPDS II (Mo-Kα-Strahlung; λ=0,71073 Å Diffraktometer Graphitmonochromator 0° ≤ ω ≤ 180° Rotation ψ = 0°, 111; ∆ω = 2° Belichtungszeit [sec.] 480 Messtemperatur [K] Raumtemperatur [293(2)] Kristallgröße [mm] 0,2 × 0,01 × 0,03 0,7 × 0,1 × 0,05 Absorptionskoeffizient [mm-1] 10,87 10,82 Messbereich [°] 4° < 2θ < 70° 4° < 2θ < 70° -11 ≤ h ≤ 11 -11 ≤ h ≤ 11 -11 ≤ k ≤ 11 -11 ≤ k ≤ 11 -6 ≤ l ≤ 7 -6 ≤ l ≤ 7 Indexbereich Zahl der gemessenen Reflexe 5313 5799 Zahl der unabh. Reflexe mit I > 0 152 157 I > σ2(I): 140 137 freie Parameter 13 14 Lorentz, Polarisation Korrekturen numerical / equivalent method (XSHAPE[68]) Absorptionskorrektur Isotypie Mn5Si3-Typ[99] Strukturlösung SHELXTL-97[199] Strukturverfeinerung Wichtungsschema w-1 = σ2F02 + (a * P)2 + b * P 2 2 P = (F0 + 2 FC ) / 3 Goodness of fit - 3 Restelektronendichte [e /Å ]: Alle Reflexe a = 0,0450 b = 0,02 b = 0,0 1,223 0,988 0,022(3) Extinktionskorrektur R-Werte für I > 0: a = 0,0604 +0,88 / -0,85; 0,0328(7) - 3 0,20 e /Å +1,00 / -0,98; R1(F) = 0,0284 R1(F) = 0,0240 wR2(I) = 0,0829 wR2(I) = 0,0596 R1(F) = 0,0311 R1(F) = 0,0280 0,18 e-/Å3 - 146 - MAX - Phase – Das System V-(Al/Sn)-C Tabelle 3.5.3.6: Ortskoordinaten, Besetzungsfaktoren (in [%]) und Auslenkungsparameter von V5Al3C (links) und V5Al3C0,93N0,07 (rechts). Summenformel V5Al3C1 V5Al3C0,93N0,07 V1 x y z sof Ueq. U11 U22 U33 U13 = U23 U12 0 0,2618(1) ¼ 100%* 0,00999(10) 0,0086(5) 0,0096(4) 0,0115(5) 0 0,0043(3) 0 0,2368(1) ¼ 100%* 0,0069(3) 0,0064(4) 0,0064(3) 0,0073(4) 0 0,0032(2) x y z sof Ueq. U11 U22 U33 U13 = U23 U12 ⅔ ⅓ 0 100% 0,0093(4) 0,0067(4) 0,0097(4) 0,0086(5) 0 0,0048(2) ⅔ ⅓ 0 100% 0,0062(3) 0,00736(4) 0,00736(4) 0,00367(4) 0 0,0037(2) x y z sof Ueq. U11 U22 U33 U13 = U23 U12 0,4070(2) 0 ¼ 100% 0,0017(4) 0,0038(8) 0,0053(6) 0,0094(7) 0 0,0019(4) 0,4010(2) 0 ¼ 100% 0,0033(3) 0,0021(4) 0,0015(6) 0,0061(6) 0 0,0008(3) x y z sof Ueq. 0 0 0 100% C 1 0,047(4) 0 0 0 92,8% C 1 / 7,2% N 1 # 0,044(4) V2 Al C/N * bei wechselweiser Freigabe der Besetzungsfaktoren keine signifikante Restelektronendichte vorhanden # Besetzungsfaktoren und Temperaturfaktoren gekoppelt und fixiert - 147 - MAX - Phase – Das System V-(Al/Sn)-C Die folgende Tabelle zeigt benachbarte Atome, ihren charakteristischen Abstand und die Koordinationszahl. Tabelle 3.5.3.7: Ausgewählte Abstände in der V5Al3C.-Struktur V5Al3C(links) und V5Al3C0,93N0,07 (rechts) Summenformel V5Al3C1 V5Al3C0,93N0,07 V1 - C/N 2,0923(8) x2 2,0876(7) x2 - Al 2,4938(14) x2 2,5022(12) x2 - Al 2,580(2) x1 2,5953(17) x1 - Al 2,6972(10) x2 2,7021(9) x2 -V 2,9475(18) x4 2,9388(7) x4 -V 2,4346(5) x2 2,4323(5) x2 - Al 2,4976(6) x6 2,5022(5) x6 -V 3,0384(7) x4 3,0521(6) x4 -V 2,4938(14) x2 2,5022(12) x2 -V 2,4976(6) x4 2,5022(5) x4 -V 2,580(2) x1 2,59535(17) x1 -V 2,6972(10) - Al 2,8178(17) -V 2,0923(9) x6 2,0876(7) x6 - C/N 2,4346(5) x2 2,432(1) x2 V2 Al x2 x2 2,7021(9) 2,8157(15) x2 x2 C/N 3.5.3.5 Strukturdiskussion V5Al3C1-x (x=0 - 0,7) kristallisiert in der Raumgruppe P63/mcm mit den Gitterkonstanten von a = 7,1438(18) Å und c = 4,8692(10) Å. Diese Kristallstruktur kann als eine Variante der Mn5Si3-Struktur betrachtet werden. In der Abbildung 3.5.6 und 3.57. sind die - 148 - MAX - Phase – Das System V-(Al/Sn)-C charakteristischen Oktaeder als Merkmal der M5M`3C Struktur dargestellt. Die CV6-Oktaeder sind flächenverknüpft in c-Richtung. Abbildung 3.5.7: Kristallstruktur von V5Al3C – Darstellung der CV6-Oktaeder in c-Richtung Abbildung 3.5.6:Kristallstruktur von V5Al3C – Blick entlang der CV6-Oktaeder in c-Richtung Vanadiumatome der Sorte 1 formen Oktaeder die über gemeinsame Flächen in c-Richtung zu Ketten miteinander verknüpft sind, die Kantenlänge beträgt 2,948(1) Å. Die Oktaederlücken, die dabei entstehen, sind mit Kohlenstoffatomen besetzt. Somit sind alle Kohlenstoffe sechsfach von Vanadiumatomen im Abstand von 2,0923(9) Å umgeben. Die Vanadiumatome der Sorte 2 besetzen die Zentren der verzerrten Oktaeder welche ausschließlich von den Aluminiumatomen geformt sind (dV2-Al1: 2,4976(6) Å). Über die Dreiecksflächen der verzerrten Aluminiumoktaeder sind jeweils sechs weitere Vanadiumatome 1 an der Koordination von V2 im Abstand von 3,0384(7) Å und zwei Vanadiumatome 1 (dV2-V1: 2,4346(5) Å x 2) beteiligt, Koordination der V2, Atome ist in der Abbildung 3.5.9 dargestellt. Die Koordinationssphäre von V 1 und der Aluminiumatome ist sehr Abbildung 3.5.8: Koordination des C-Atome unregelmäßig. Das Aluminiumatom ist in einer sehr stark verzerrten, dreifach überkappten, unregelmäßig oktaedrischen Koordinationssphäre eingeschlossen. An der Koordination sind neun Vanadiumatome und zwei Aluminiumatome beteiligt. Somit sind V1 und V2 Atome im Abstand von 2,494 (1) Å bis zum 2,697 (1) Å unregelmäßig um das Aluminiumatom koordiniert. Das Oktaeder wird von V1 Atomen 2 x 2,494(1) Å und V2 Atomen 4 x 2,4976(6) Å gebildet. Weitere drei Vanadiumatome der Sorte 1 koordinieren über die Flächen im - 149 - MAX - Phase – Das System V-(Al/Sn)-C Abstand von 2 x 2,580(2) Å und 1 x 2,697(1) Å. Weitere zwei Aluminiumatome befinden sich in der Koordinationssphäre im Abstand von 2,818(2) Å. Die Koordinationssphäre des Aluminiumatoms ist in der Abbildung 3.5.10 dargestellt. Abbildung 3.5.10: Koordination von Aluminium Abbildung 3.5.9: Koordination von V 2 Die Koordination von Vanadium 1 wird aus fünf Aluminiumatomen im Bereich vom 2,494(1) – 2,698 (1) Å sowie von sechs Vanadiumatomen im Abstand von 2,948(2) Å ausgebildet. Zwei Kohlenstoffatome koordinieren mit 2,0923(8) Å an das Vanadium 1 Atom. Abbildung 3.5.11: Koordination von V 1 Die Abstände der Vanadiumatome zu den Kohlenstoffatomen liegen in guter Übereinstimmung zu den des binärem VC (dV-C: 2,086 Å). Der Abstand der Kohlenstoffatome untereinander beträgt in VC d = 3,0411 Å und ist deutlich länger als der Abstand der Kohlenstoffatome in den Oktaederketten (dC-C: 2,4346(5) Å). - 150 - MAX - Phase – Das System V-(Al/Ga)-C 3.5.4 Arbeiten in einer Aluminium/Gallium-Schmelze Das leicht schmelzende Gallium wurde zu dem Dreistoffsystem als Hilfsmetall zugesetzt. Es existieren mit beiden Hauptgruppenmetallen MAX-Phasen, mit Gallium konnte bis jetzt nur die 211-Phase beobachtet werden. V2GaC kristallisiert mit sehr kleinen Gitterkonstanten von a = 2,938 Å und c = 12,840 Å[36], während mit Aluminium bereits von zwei Varianten berichtet wurde, genauer gesagt von der 211- (V2AlC Lit. a = 2,924 Å; c = 13,063 Å)[202] und 413- Varianten (erst seit kurzem bekannten V4AlC3-x Lit. a = 2,9301 Å; c = 22,746 Å)[202]. Abbildung 3.5.12: Das binäre Phasendiagramm des Systems Vanadium-Gallium[175] Vanadium bildet mit Gallium vier stabile intermetallische Phasen, das galliumreiche Ga41V8[100] kristallisiert im eigenem Strukturtyp-Typ, GaV3[101] (Cr3Si-Typ), Ga5V2[102] (Hg5Mn2-Typ) und Ga5V6[103] (Ti6Sn5-Typ). Außerdem wurden Phasen der Zusammensetzung Ga7V6 (Cu5Zn8-Typ) und Ga3V5 (Mn5Si3-Typ) beschrieben, die nicht im binären Phasendiagramm eingezeichnet sind. 3.5.4.1 Präparative Untersuchung im System Vanadium-(Aluminium/Gallium)Kohlenstoff Für eine gute Umsetzung wurden die Elemente Vanadium, Aluminium und Kohlenstoff (carbon black) in den unten angegebenen Verhältnissen kaltgepresst. Die Tabletten wurden in Korundtiegeln positioniert und mit dem zurechtgeschnittenen Galliumstückchen, das auf der Tablette platziert wurde, zur Reaktion gebracht. Die Synthese wurde bei einer Temperatur von 1600°C für 25h unter leichtem Argon-Schutzgasstrom durchgeführt. - 151 - MAX - Phase – Das System V-(Al/Ga)-C Temperaturprogramm: 150°C/h RT -5°C/h -15°C/h 1600°C 1300°C 800°C (25h halten) (1h halten) (1h halten) Tabelle 3.5.4.1: Proben im System pulverdiffraktometrische Charakterisierung. Nr. RT Titan-Aluminium/Indium-Kohlenstoff Gehalt [At.-%] Zusammensetzung -100°C/h und weitere Gitterkonstante [Å] V Al Ga C kristalline der MAX-Phase a = 3,005(6); c = 13,193(18) 1 V6Al10Ga5C1 27,3 45,5 22,7 4,5 2 V6Al7,5Ga7,5C1 27,3 34,1 34,1 4,5 3 V6Al5Ga10C1 27,3 22,7 45,5 4,5 - 4 V6Al3Ga12C1 27,3 13,6 54,5 4,5 - V=103,2(3) a = 3,009(5); c = 13,190(15) V=103,4(3) ihre Phasen V6C5 V6C5 CV, GaV2 V6C5, V8Ga4, Ga5V2 Der erhaltene Schmelzregulus wurde über einige Stunden mit verd. Salzsäure behandelt. Der säurestabile, kristalline Rückstand wurde abgesaugt, mit Wasser gewaschen und getrocknet. In den ersten zwei Probe der Tabelle 3.5.4.1 konnten unter einem Lichtmikroskop runde, metallisch glänzende Plättchen beobachtet werden. In den beiden anderen Ansätzen war das Produkt feinkristallin. Die Proben wurden pulverdiffraktometrisch auf die gebildeten Phasen hin analysiert und die Gitterkonstante der MAX-Phase verfeinert. Die 211-Phase konnte mit Gitterkonstanten von a = 3,00 Å und c = 13,19 Å in der Raumgruppe P63/mmc eindeutig verfeinert werden. 3.5.4.2 Röntgenographische Untersuchung an Einkristallen der 211-Phase Aus dem Ansatz mit der Zusammensetzung V6Al7,5Ga7,5C1 wurde ein geeigneter Kristall für eine röntgenographische Untersuchung isoliert. Die ermittelten Gitterkonstanten von a = 2,9284 (6) Å und c = 12,879(3) Å stimmen sehr gut mit den in der Literatur gefundenen Gitterparametern für eine H-Phase der Zusammensetzung V2GaC überein. So wurde nach der Mittelung und einer numerischen Absorptionskorrektur unmittelbar mit den Atomkoordinaten von Cr2AlC-Modell verfeinert. - 152 - MAX - Phase – Das System V-(Al/Ga)-C Tabelle 3.5.4.2: Angaben zur Strukturanalyse von V2GaC2. Summenformel V2GaC Kristallsystem hexagonal Raumgruppe P63/mmc Gitterkonstante [Å] a = 2,9284(6); c = 12,879(3) 3 Volumen der Elementarzelle [Å ] 95,65(3) Zahl der Formeleinheiten 2 3 röntgenogr. Dichte [g/cm ] 6,3751 F(000) 166,0 Kristallform, -farbe metallisch glänzende, hexagonale Plättchen Diffraktometer STOE IPDS II (Mo-Kα-Strahlung; λ=0,71073 Å Graphitmonochromator 0° ≤ ω ≤ 180° ψ = 0°, 111; ∆ω = 2° Rotation Belichtungszeit [sec.] 300 Messtemperatur [K] Raumtemperatur [293(2)] 0,02 × 0,02 × 0,005 Kristallgröße [mm] -1 Absorptionskoeffizient [mm ] 23,03 Messbereich [°] 4° < 2θ < 65° -2 ≤ h ≤ 0, Indexbereich 0 ≤ k ≤ 4, 0 ≤ l ≤ 18 Zahl der gemessenen Reflexe 2285 Zahl der unabh. Reflexe mit I > 0 87 I > σ2(I): 44 freie Parameter 9 Korrekturen Lorentz, Polarisation Absorptionskorrektur numerical / equivalent method (XSHAPE[68]) Strukturlösung Isotypie Cr2AlC104 Strukturverfeinerung SHELXTL-97[199] Wichtungsschema w-1 = σ2F02 + (0,000 * P)2 + 0,00 * P P = (F02 + 2 FC2) / 3 Goodness of fit 0,575 Extinktionskorrektur 0,0442(86) - 3 Restelektronendichte [e /Å ]: R-Werte für I > 0: Alle Reflexe +0,70 / - 0,90; 0,16 e-/Å3 R1(F) = 0,0230 wR2(I) = 0,0326 R1(F) = 0,0522 - 153 - MAX - Phase – Das System V-(Al/Ga)-C Mit 9 freien Parametern konvergierte die Verfeinerung bei Zuverlässigkeitswerten von R1(F) = 0,0230 und wR2(I) = 0,0326. Einzelheiten zur Strukturuntersuchung von V2GaC sowie die Besetzungs- und Temperaturfaktoren sind in folgenden Tabellen aufgeführt. Tabelle 3.5.4.3: Ortskoordinaten, Besetzungsfaktoren (in [%]) und Auslenkungsparameter von V2GaC2 Atom Lage x y z sof Ueq ⅔ ⅓ 0,58629(3) 100* 0,0042(5) Ga 4f 2d ⅓ ⅔ ¾ 100* 0,0067(7) C 2a 0 0 0 100* 0,0018(15) V * bei wechselweiser Freigabe der Besetzungsfaktoren keine signifikante Restelektronendichte vorhanden Atom U11 U22 U33 U23 U13 U12 V 0,0079(6) 0,0079(6) 0,0029(1) 0 0 0,0024(3) Ga 0,0093(9) 0,0093(9) 0,0020(12) 0 0 0,0046(5) Die folgende Tabelle zeigt benachbarte Atome, ihren charakteristischen Abstand und die Koordinationszahl. Tabelle 3.5.4.4: Ausgewählte Abstände und Winkeln in der V2GaC2-Struktur Anzahl der Winkel Bindungen Ti – C - Ti Benachbarte Atome Abstand [Å] Ga - V 2,703(3) x6 Ga 2,9284(4) x6 C 2,023(2) x3 92,72(11)° Ga 2,703(3) x3 87,28(11)° V 2,793(5) x3 V 2,9284(4) x6 V 2,023(2) x6 V- C- - 154 - MAX - Phase – Das System V-(Al/Ga)-C 3.5.4.3 Strukturdiskussion V2GaC kristallisiert im Cr2AlC-Typ mit der Raumgruppe P63/mmc mit den Gitterkonstanten von a = 2,9284(6) Å; c = 12,879(3) Å. Die Gitterkonstanten liefern eine nahezu perfekte Übereinstimmung mit der Literatur. Die Struktur der H-Phase wurde bereits ausführlichst besprochen. Die Lage des frühen Übergangsmetalls ist vollständig durch Vanadium besetzt. Die zwei übereinanderliegenden Schichten von Vanadium bilden Oktaeder mit einer Kantenlänge von 2,9284(6) Å. Die Oktaederlücken sind vollständig mit Kohlenstoff besetzt (dV1-C1 = 2,023(2) Å). Dieser Abstand ist ertwas kürzer als in dem binären VC (2,086 Å). Die Oktaederschichten werden alternierend durch Galliumschichten getrennt. Dabei besitzen die Galliumatome einen Abstand von 2,9284(4) Å zueinander. Der Abstand der Vanadiumatome zu der Schicht der Galliumatome beträgt 2,703(3) Å. - 155 - MAX - Phase – Das System V-(Al/In)-C 3.5.5 Arbeiten in einer Aluminium/Indium-Schmelze Als weiteres Hilfsmetall ist in dieser Arbeit Indium zum Einsatz gekommen. Die Kombination aus dem frühen Übergangsmetall Vanadium mit Indium in einer MAX-Phase ist in der Literatur nicht bekannt. Zur Darstellung der ternären Carbide wurde in diesem System die Schmelzzusammensetzung mehrfach variiert, Die Temperatur wurde dabei mit 1500°C konstant gehalten. Abbildung 3.5.13: Das binäre Phasendiagramm des Systems Vanadium-Indium[175] Es existiert nur eine einzige Verbindung im System Vanadium-Indium InV3[105] (Cr3SiTyp), und diese ist nur bei hohen Drücken zugänglich. Die anderen binären Phasendiagramme wurden bereits in den Kapiteln 3.4.6.1 beschrieben. 3.5.5.1 Präparative Untersuchung im System Vanadium-(Aluminium/Indium)Kohlenstoff Die Synthese erfolgte aus den Elementen. Hierzu wurden Vanadium, Aluminium und Kohlenstoff „carbon black“ homogenisiert und in Tabletten verpresst. Das Indium wurde in Form von keinen Kugeln (d = 1 mm) auf der Tablette platziert. Die Umsetzung wurde in Korundtiegel unter Schutzgasatmosphäre (Ar) durchgeführt. Für eine möglichst gute Kristallisation wurde sehr langsam abgekühlt (5°C/h). Dabei wurde folgendes Temperaturprogramm verwendet: - 156 - MAX - Phase – Das System V-(Al/In)-C Temperaturprogramm: 150°C/h RT -5°C/h -15°C/h -100°C/h 1500°C 1200°C 850°C (20h halten) (1h halten) (1h halten) RT Tabelle 3.5.5.1: Proben im System Vanadium-Aluminium/Indium-Kohlenstoff pulverdiffraktometrische Charakterisierung. Fettgedruckt sind die Hauptprodukte. Nr. Zusammensetzung Gehalt [At.-%] V Al In C 45,5 22,7 4,5 1 V6Al10In5C1 27,3 2 V6Al7,5In,5C1 27,3 34,1 34,1 4,5 3 V6Al5In10C1 27,3 22,7 45,5 4,5 4 V6Al3In12C1 27,3 13,6 54,5 4,5 und ihre Gitterkonstante [Å] weitere kristalline der MAX-Phase Phasen a = 2,9177(16); c = 13,249(10) V2C, VC5, Al3V, V=97,67(9) a = a=2,9227(23); c = 13,205(14) V=97,69(10) a = 2,91897(7); c=13,253(6) V=97,79(5) a = 2,925(4); c = 13,128(15) V=97,27(14) C Ti3Al, V2C, VC V8C7, V2C V8C7, V2C Nach dem Abkühlen wurde der erhaltene Schmelzregulus in verd. Salzsäure eingelegt. In Anschluss an die Hydrolyse wurde der Rückstand abgesaugt, mit Wasser gewaschen und getrocknet. Die gewonnenen Produkte wurden röntgenographisch auf die gebildeten Phasen hin analysiert. Nicht in einer der vier Ansätze ist es gelungen eine der MAX-Phase phasenrein darzustellen. In jedem Ansatz konnten charakteristische Reflexe für eine H-Phase beobachtet werden. Die beobachteten Reflexe der MAX-Phasen wurden verfeinert. Unabhängig von der Schmelzzusammensetzung bildete sich in allen Ansätzen eine Phase mit nahezu identischen Zellenparametern. Aus der Betrachtung der Gitterkonstanten ist auch hier eine deutliche Mischbesetzung anzunehmen. Die binären Carbide des Vanadiums waren sehr überlegen vertreten. Bei den aluminiumreichen Proben wurde auch die intermetallische Phase Ti3Al beobachtet. 3.5.5.2 Röntgenographische Untersuchung an Einkristallen der 211-Phase Aus dem Ansatz mit der Schmelzzusammensetzung von V6Al5In10C1 wurde ein geeigneter Einkristall auf dem IPDS II in einem Winkelbereich von 180° in Schritten von zwei Grad vermessen. - 157 - MAX - Phase – Das System V-(Al/In)-C Tabelle 3.5.5.2: Angaben zur Strukturanalyse von V2In0,63Al0,37C. Summenformel V2In0,63Al0,37C Kristallsystem hexagonal Raumgruppe P63/mmc Gitterkonstante [Å] a = 3,0620(3); c = 13,3408(16) Volumen der Elementarzelle [Å3] 108,33(2) Zahl der Formeleinheiten 2 3 röntgenogr. Dichte [g/cm ] F(000) 6,011 175,2 Kristallform, -farbe metallisch glänzende hexagonale Plättchen STOE IPDS II (Mo-Kα-Strahlung; Diffraktometer λ=0,71073 Å Graphitmonochromator 0° ≤ ω ≤ 180° Rotation ψ = 0°, 111; ∆ω = 2° Belichtungszeit [sec.] 900 Messtemperatur [K] Raumtemperatur [293(2)] 0,04 × 0,04 × 0,01 Kristallgröße [mm] -1 Absorptionskoeffizient [mm ] 14,85 Messbereich [°] 4° < 2θ < 65° -4 ≤ h ≤ 4, -4 ≤ k ≤ 4, Indexbereich -20 ≤ l ≤ 20 Zahl der gemessenen Reflexe 3556 Zahl der unabh. Reflexe mit I > 0 100 I > σ2(I): 97 freie Parameter 9 Korrekturen Lorentz, Polarisation Absorptionskorrektur numerical / equivalent method (XSHAPE[68]) Strukturlösung Isotypie Cr2AlC[75] Strukturverfeinerung SHELXTL-97[199] w-1 = σ2F02 + (0,0096 * P)2 + 0,191 * P Wichtungsschema Goodness of fit P = (F02 + 2 FC2) / 3 1,556 Extinktionskorrektur 0,0168(6) Restelektronendichte [e-/Å3]: +0,39 / - 0,50; R-Werte für I > 0: Alle Reflexe 0,12 e-/Å3 R1(F) = 0,0217 wR2(I) = 0,0489 R1(F) = 0,0201 - 158 - MAX - Phase – Das System V-(Al/In)-C Bei einer Belichtungszeit von 15 Minuten und einem Detektorabstand von 50 mm konnten 3556 Reflexe gesammelt werden. Nach einer Mittelung in der Laueklasse 6/mmm und einer numerischen Absorptionskorrektur lag ein Datensatz von 97 unabhängigen Reflexen vor. Auch hier wurde für die Verfeinerung das Modell mit den Atomkoordinaten von Cr2AlC herangezogen, da die Gitterkonstanten eindeutig auf eine H-Phase hindeuteten. Die deutliche Aufweitung in alle Richtung der Zelle war ein erstes Indiz für eine Substitution von Aluminium gegen Indium. Die Verfeinerung der kristallographischen Lage 2d wurde unter der Annahme der Mischbesetzung durchgeführt. Die Besetzungsfaktoren und Temperaturfaktoren wurden gekoppelt und frei verfeinert. Die anisotrope Verfeinerung der 9 freien Parameter lieferte Zuverlässigkeitswerte von R1(F) = 0,0217 und wR2(I) = 0,0489 bei einer Zusammensetzung von V2In0,63Al0,37C. Eine Freigabe der Besetzungsfaktoren der drei Lagen zeigte keine signifikante Abweichung von der Idealbesetzung. Die Restelektronendichten lagen in einem vernünftigen Rahmen mit 0,39 und -0,50 e/Å3. In den folgenden Tabellen sind weitere Einzelheiten der Strukturanalyse aufgeführt. Tabelle 3.5.5.3: Ortskoordinaten, Besetzungsfaktoren (in [%]) und Auslenkungsparameter von V2In0,63Al0,37C. Atom Lage x y z sof Ueq ⅔ ⅓ 0,58561(5) 100* 0,0056(3) In/Al 4f 2d ⅓ ⅔ ¾ 63(2)/37*# 0,0132(3) C 2a 0 0 0 100* 0,0046(9) V * bei wechselweiser Freigabe der Besetzungsfaktoren keine signifikante Restelektronendichte vorhanden # Besetzungsfaktoren und Temperaturfaktoren gekoppelt und fixiert Atom U11 U22 U33 U23 U13 U12 V 0,0053(3) 0,0053(3) 0,0063(4) 0 0 0,0026(2) In/Al 0,0148(3) 0,0148(3) 0,0099(4) 0 0 0,0074(1) C 0,0046(15) 0,0046(15) 0,0046(21) 0 0 0,0023(7) Die folgende Tabelle zeigt benachbarte Atome, ihren charakteristischen Abstand und die dazugehörige Koordinationszahl. - 159 - MAX - Phase – Das System V-(Al/In)-C Tabelle 3.5.5.4: Ausgewählte Abstände und Winkeln in der V2In0,63Al0,37C-Struktur Anzahl der Winkel Bindungen Ti - C - Ti Benachbarte Atome Abstand [Å] In/Al - V 2,8169(6) x6 In/Al 3,0620(4) x6 C 2,1047(4) x6 94,63(47)° In/Al 2,8170(6) x3 85,37(48)° V 2,8883(11) x3 V 3,0620(4) x6 V 2,1047(4) x6 V- C- 3.5.5.3 Strukturdiskussion V2In0,63Al0,37C kristallisiert im Cr2AlC-Typ in der Raumgruppe P 63/mmc mit den Gitterkonstanten von a = 3,0620(3) Å; c = 13,3408(16) Å V = 108,33(2) ų. V2In0,63Al0,37C ist die erste Vanadium MAX-Phase, bei der es gelungen ist, Indium einzubauen. Der Vergleich der Gitterkonstanten mit V2AlC (a = 2,9116 Å; c = 13,140 Å) zeigt eine deutliche Aufweitung in alle Richtungen. Dieses lässt sich mit dem Einbau von Indium erklären. MAX-Phasen mit Indium zeigen in der Regel eine starke Aufweitung der Gitterparameter. Eine Volumenbetrachtung der Bekannten carbidischen MAX-Phase (Ti2InC (V = 119,44 ų), Zr2InC (V = 144,65 ų), Hf2InC (V = 139,51 ų), Nb2InC (V = 125,21 ų)) zum Vergleich V2AlC (V = 96,47 ų) zeigt eine deutlichen Ausdehnung der Zelle. Die kristallographische Lage 4f ist vollständig mit Vanadiumatomen besetzt. Die Lage des Hauptgruppenmetalls 2b ist durch eine Mischbesetzung der Metalle Indium und Aluminium geprägt. Die Metalle der beiden Lagen bilden eine hexagonale dichteste Packung, wobei auf jede zwei Vanadiumlagen eine Lage mit der Mischbesetzung folgt. Die Vanadiumatome bilden die Oktaeder mit einer Kantenlänge von 2,8883(11) Å. Diese sind über gemeinsame Kanten miteinander verknüpft. Die Lücken der Oktaeder sind vollständig mit Kohlenstoffatomen besetzt, so erhält das Nichtmetall hier eine sechsfach Koordination im Abstand von 2,1047(4) Å). Prinzipiell besteht strukturell kein großer Unterschied zu den bis jetzt beschrieben 211-Phasen. - 160 - MAX - Phase – Das System V-(Al/In)-C 3.5.5.4 Weitere Einkristalle Bei der Untersuchung der erhaltenen Einkristalle aus dem indiumreichsten Ansatz mit der Schmelzzusammensetzung von V6Al3In12C1 ist ein Einkristall mit erstaunlich kleiner Gitterkonstante aufgefallen. Die auf den IPDS II beobachteten Gittekonstanten betrugen a = 2,9190(3) Å und in c- Richtung 12,989(12) Å. Ein gut ausgeprägter Einkristall mit diesen Gittekonstanten wurde auf dem IPDS II in einem Winkelbereich von 180° mit zwei Schritten und 15 Minuten pro Aufnahme mit einem Detektorabstand von 40 mm vermessen. Die kristallographischen Lagen 4f und 2d wurden jeweils vollständig mit Vanadiumatomen besetzt. Die Freigabe der Besetzungsfaktoren zeigte eine geordnete und vollständig besetzte Position. Die Abweichung lag unter einem Prozent. Eine Besetzung mit Indium- und Aluminiumatomen ist durchaus denkbar, wurde aber auf Grunde der sehr kleinen Gitterparametern ausgeschlossen. Der Einbau von Indium führt in der Regel zur Aufweitun der Zelle, wie schon im analogen System Ti-Al/In-C beobachtet wurde. Substitution Ti2AlC[40] von Al gegen In a = 3,056 Å Ti2InC[27] a = 3,132 Å c = 13,623 Å c = 14,060 Å 3 Vol. = 110,18 Å Vol. = 119,44 Å3 Abbildung 3.5.14: Vergleich der Gitterkonstanten und Volumina Bei dem Besetzungsfaktor des Kohlenstoffs zeigte sich ein Defizit von ca. 4%, was jedoch noch im Rahmen der Standardabweichung lag (4,1%). Eine Unterbesetzung des Kohlenstoffs ist durchaus denkbar. In der Strukturchemie der Übergangsmetallcarbide wurde dieses schon oft beobachtet. Die Restelektronendichte lag in einer guten Dimension und war zufriedenstellend (0,32 e-/Å3 und -0,39 e-/Å3). - 161 - MAX - Phase – Das System V-(Al/In)-C Tabelle 3.5.5.5: Angaben zur Strukturanalyse von V3C. Summenformel V3C Kristallsystem hexagonal Raumgruppe P63/mmc Gitterkonstante [Å] (V2VC) a = 2,9190(4); c = 12,989(3) 3 Volumen der Elementarzelle [Å ] 95,85(2) Zahl der Formeleinheiten 2 röntgenogr. Dichte [g/cm3] F(000) 5,7111 150,0 Kristallform, -farbe metallisch glänzende hexagonale Plättchen STOE IPDS II (Mo-Kα-Strahlung; λ=0,71073 Å Diffraktometer Graphitmonochromator 0° ≤ ω ≤ 180° Rotation ψ = 0°, 111; ∆ω = 2° Belichtungszeit [sec.] 900 Messtemperatur [K] Raumtemperatur [293(2)] 0,03 × 0,03 × 0,01 Kristallgröße [mm] -1 Absorptionskoeffizient [mm ] 13,80 Messbereich [°] 4° < 2θ < 65° -2 ≤ h ≤ 0, Indexbereich 0 ≤ k ≤ 4, 0 ≤ l ≤ 19 Zahl der gemessenen Reflexe 2353 Zahl der unabh. Reflexe mit I > 0 147 I > σ2(I): 90 freie Parameter 10 Korrekturen Lorentz, Polarisation Absorptionskorrektur numerical / equivalent method (XSHAPE[68]) Strukturlösung Isotypie Cr2AlC[71] Strukturverfeinerung SHELXTL-97[199] w-1 = σ2F02 + (0,0165 * P)2 + 0,00 * P Wichtungsschema Goodness of fit P = (F02 + 2 FC2) / 3 0,697 Extinktionskorrektur 0,167(012) Restelektronendichte [e-/Å3]: +0,32 / - 0,39; R-Werte für I > 0: Alle Reflexe 0,11 e-/Å3 R1(F) = 0,0129 wR2(I) = 0,0259 R1(F) = 0,0384 - 162 - MAX - Phase – Das System V-(Al/In)-C Tabelle 3.5.5.6: Ortskoordinaten, Besetzungsfaktoren (in [%]) und Auslenkungsparameter von V3C. Atom Lage x y z sof Ueq ⅔ ⅓ 0,58597(12) 100* 0,0046(3) V2 4f 2d ⅓ ⅔ ¾ 100* 0,0094(5) C 2a 0 0 0 100* 0,0061(9) V1 * bei wechselweiser Freigabe der Besetzungsfaktoren keine signifikante Restelektronendichte vorhanden Atom U11 U22 U33 U23 U13 U12 V1 0,0048(4) 0,0048(4) 0,0044(4) 0 0 0,0024(2) V2 0,0112(8) 0,0112(8) 0,0058(7) 0 0 0,0056(4) C 0,0070(13) 0,0070(13) 0,0044(2) 0 0 0,0035(7) Die folgende Tabelle zeigt benachbarte Atome, ihren charakteristischen Abstand und die dazugehörige Koordinationszahl. Tabelle 3.5.5.7: Ausgewählte Abstände und Winkeln in der V3C.-Struktur Anzahl der Winkel Bindungen Ti - C - Ti Benachbarte Atome Abstand [Å] V2- V1 2,7165(12) x6 V2 2,9190(4) x6 C 2,0217(9) x6 94,6(5)° V2 2,7165(12) x3 85,4(5)° V1 2,7979(24) x3 V1 2,9190(4) x3 V1 2,1047(4) x6 V 1- C- 3.5.5.5 Strukturdiskussion V2VC kristallisiert im Cr2AlC-Typ mit der Raumgruppe P 63/mmc (Nr. 194) mit den sehr kleinen Gitterkonstanten von a = 2,9190(3) Å; c = 12,989(3) Å. Bei der Verfeinerung zeigte sich eine Besetzung beider Metalllagen ausschließlich durch das Vanadiumatom. Zum ersten Mal konnte an der hier diskutierten Struktur der Einbau vom - 163 - MAX - Phase – Das System V-(Al/In)-C frühen Übergangsmetall auf der 2b Position beobachtet werden. Die Koordinationssphäre der Atome ist wie schon an all den anderen H-Phasen zuvor beschrieben. Die Gitterkonstanten der verfeinerten 211-Phase besitzen die Größenordnung von V2GaC. Die Struktur lässt sich wie üblich als eine hexagonal dichteste Kugelpackung der Metallatome beschreiben. Hier sind immer zwei gleiche Vanadiumschichten auf einer weiteren kristallographisch Oktaederlücken der unterscheidbaren Vanadiumschicht der Vanadiumschicht Punktlagen 4f sind aufgebaut. Alle vollständig mit Kohlenstoffatomen besetzt (dV1-C:2,1047(4) Å). So ergeben sich CV6-Schichten die durch eine metallische Vanadiumschicht voneinander getrennt sind. Jedes Vanadiumatom auf der 2b Lage ist von sechs gleichen Vanadiumatomen (dV2V2:2,9190(4) Å) und jeweils von drei Vanadiumatomen aus der darüber bzw. darunter liegenden Schicht umgeben (dV2-V1:2,7165(12) Å). Das Vanadiumatom auf 4f ist dabei von sechs Vanadiumatomen der gleichen Schicht (dV1-V1:2,9190(4) Å), drei Vanadiumatomen aus der benachbarten Schicht (dV1-V1:2,7979(24) Å) und drei Vanadiumatomen von der 2b Schicht(dV1-V2:2,7165(12) Å). Somit zeigen die Metallatome ausnahmslos Antikuboktaeder die typischen Koordinationspolyeder gemäß der hexagonal dichtesten Kugelpackung. - 164 - MAX - Phase – Weitere Untersuchungen 3.5.6 Weitere Untersuchungen Im Rahmen dieser Arbeit wurden weitere Schmelzlösungen untersucht. Für die Untersuchungen des Systems Titan-Aluminium-Kohlenstoff wurden als Hilfsmetalle Eisen, Kobalt, Nickel und Chrom eingesetzt. Beim System Vanadium-Aluminium-Kohlenstoff wurden die Elemente Silizium, Chrom und Zinn zugegeben. Hier sollte auf mögliche Bildung von neuen MAX-Phasen eingegangen werden. 3.5.6.1 Untersuchungen mit Aluminium/Eisen Bei dem Zusatz von Eisen wurde wie üblich vorgegangen. Die Elemente wurden innig miteinander verrieben und mit einer Handpresse in Tabletten verdichtet. Diese wurden in Korundtiegeln bei einer Temperatur von 1600°C unter Argon umgesetzt. Temperaturprogramm: 150°C/h RT -5°C/h -20°C/h 1600°C 1500°C (20h halten) (2h halten) -100°C/h 1000°C RT (1h halten) Tabelle 3.5.6.1: Proben im System Titan-Aluminium/Eisen-Kohlenstoff und ihre pulverdiffraktometrische Charakterisierung. Fettgedruckt sind die Hauptprodukte. Nr. Gehalt [At.-%] Zusammensetzung Ti Al Fe C Gitterkonstante [Å] weitere kristalline der MAX-Phase Phasen a = 3,072(6); c = 13,87(3) TiC, TiN, 1 Ti6Al12Fe3C1 27,3 54,5 9,1 4,5 2 Ti6Al10Fe5C1 27,3 45,4 22,7 4,5 - TiC, TiN 3 Ti6Al5Fe10C1 27,3 22,7 45,4 4,5 - TiC, TiN 4 Ti6Fe15C1 27,3 - 68,2 4,5 - TiN, TiC 5 Ti6Al7,5Fe7,5C1 27,3 34,1 34,1 4,5 - TiC, TiN 6 Ti4Al3Fe12C1 20,0 15,0 70,0 5,0 - V=113,36(16) >>Ti2AlC TiC, TiN, Ti3Al, >Fe3C - 165 - MAX - Phase – Weitere Untersuchungen Nach der Aufarbeitung mit verd. Salzsäure wurden alle säurestabilen Produkte einer Phasenanalyse unterzogen. In allen Pulverdiffraktogrammen ging eindeutig Titancarbid bzw. Titannitrid12 als dominierende Hauptprodukte hervor. Als binäre Nebenphasen konnten das intermetallische Ti3Al und das Carbid Fe3C bestimmt werden. In einem eisenarmen Ansatz konnte die MAX-Phase mit der 211- Variante beobachtet werden. Nach Indizierung der charakteristischen Reflexe wurden Gitterkonstanten von a = 3,072(6) Å und c = 13,87(3) Å bestimmt. Aus diesem Ansatz konnte ein hexagonaler, metallisch glänzender Einkristall isoliert werden. Bei der Strukturverfeinerung und EDX-Untersuchung zeigte sich, dass es sich bei diesem Einkristall um einen eisenfreien Ti2AlC -Kristall handelt (a = 3,059(2) Å und c = 13,689(2) Å). 3.5.6.2 Untersuchungen mit Aluminium/Kobalt Weiter Untersuchungen wurden mit dem Zusatz von Kobalt als Hilfsmetall vorgenommen. Die Pulver wurden in den entsprechenden stöchiometrischen Verhältnissen eingewogen (Tabelle 3.5.6.2) und mit einer Handpresse zu Tabletten verarbeitet. Tabelle 3.5.6.2: Proben im System Titan-Aluminium/Kobalt-Kohlenstoff und ihre pulver- diffraktometrische Charakterisierung. Fettgedruckt sind die Hauptprodukte. Nr. Gehalt [At.-%] Zusammensetzung Ti Al Co C Gitterkonstante [Å] weitere kristalline der MAX-Phase Phasen a = 3,056(2); c = 18,596(6) TiC, TiCo 1 Ti6Al12Co3C1 27,3 54,5 20,7 4,5 2 Ti6Al10Co5C1 27,3 45,5 22,7 4,5 - TiC 3 Ti6Al5Co10C1 27,3 22,7 45,5 4,5 - TiC 4 Ti6Co15C1 27,3 - 68,2 4,5 - TiC 5 Ti6Al3Co12C1 27,3 20,7 54,5 4,5 a = 3,065(2); c=18,484(6) TiCo 6 Ti4Al12Co3C1 20,0 60,0 15,0 5,0 V=151,47(11) V=150,5(1) a = 3,068(4); c=18,63(4) V=151,91(18) > Ti3AlC2 > Ti3AlC2 TiC > Ti3AlC2 Als Tiegelmaterial wurde auch hier der Korund verwendet. Die Tabletten wurden in der Mitte des Ofens platziert und die Reaktion wurde bei einer Temperatur von 1600°C für 25 Stunden unter Argon-Schutzgas durchgeführt. Dabei wurde folgendes Temperaturprogramm verwendet. 12 Der Stickstoff kann durch das Titan als TiN mit eingeschleppt geworden sein. - 166 - MAX - Phase – Weitere Untersuchungen Temperaturprogramm: 150°C/h RT -5°C/h -15°C/h -100°C/h 1600°C 1300°C 800°C (25h halten) (1h halten) (1h halten) RT Der Schmelzregulus wurde mit Hilfe von verd. Salzsäure über Nacht entfernt. Der Rückstand bestand unabhängig von der Zusammensetzung der Schmelze nahezu immer aus TiC Kristallen. Bei den untersuchten Einkristallen die aus dem Rückstand isoliert werden konnten, wurden metallische Plättchen untersucht, die als Ti3AlC2 identifiziert werden konnten. Aus der EDX-Analyse der untersuchten Kristalle ging hervor, dass kein Einbau von Kobalt existierte. Es lagen ausschließlich Einkristalle von Typ Ti3AlC2-xNx vor. Die verfeinerten Gitterkonstanten der untersuchten Einkristalle sind etwas kleiner als die in der Literatur angegebenen. Eine Erklärung dafür ist eine Mischbesetzung von Kohlenstoff mit Stickstoff auf der Nichtmetalllage, was mit Hilfe der EDX-Analyse auch bestätigt werden konnte. 3.5.6.3 Untersuchungen mit Aluminium/Nickel Mit Nickel wurden weitere Versuche unternommen, MAX-Phasen aus der Schmelze zu gewinnen. Nach einer guten Verreibung der Elementgemische miteinander wurden diese in eine Tablette gepresst und unter Argon-Atmosphäre bei 1600°C in Korundtiegel umgesetzt. Tabelle 3.5.6.3: Proben im System Titan-Aluminium-Kohlenstoff und ihre pulverdiffraktometrische Charakterisierung. Fettgedruckt sind die Hauptprodukte. Nr. Gehalt [At.-%] Zusammensetzung Ti Al Ni C Gitterkonstante [Å] weitere kristalline der MAX-Phase Phasen 1 Ti6Al12Ni3C1 27,3 54,5 13,6 4,5 - TiC, NiTi 2 Ti6Al10Ni5C1 27,3 45,5 22,7 4,5 - Al4Ni3, TiC, TiN 3 Ti6Al5Ni10C1 27,3 22,7 45,5 4,5 - TiN, Ni2TiAl, TiC 4 Ti6Ni15C1 27,3 68,2 - 4,5 - NiTi, Ni3Ti, TiC 5 Ti6Al7,5Ni7,5C1 27,3 34,1 34,1 4,5 - NiAl, NiTi, TiC 6 Ti4Al3Ni12C1 20,0 15,0 60,0 5,0 - TiC, NiAl, Ti2AlNi - 167 - MAX - Phase – Weitere Untersuchungen Temperaturprogramm: 150°C/h RT -5°C/h -20°C/h 1600°C 1500°C (20h halten) (2h halten) -100°C/h 1000°C RT (1h halten) Über die röntgenographische Phasenanalyse konnte in keiner der Probe eine Spur von MAX-Phasen gefunden werden. In allen Ansätzen konnte kristallines Titancarbid wie auch einige intermetallische Phasen (NiTi, Al4Ni3, Ni2TiAl) gefunden werden. 3.5.6.4 Untersuchungen mit Zinn/Chrom Bei der Untersuchung des Einflusses von Chrom im System Titan-Zinn-Kohlenstoff wurden die Elemente in nahezu unterschiedlichen Verhältnissen verpresst und bei 1600°C umgesetzt. Die Synthese erfolgte 25 h unter Argon-Schutzgas und einem sehr langsamen Abkühlen. Der Schmelzregulus wurde unter Verwendung von verd. HCl von der Schmelze gelöst. Temperaturprogramm: 150°C/h RT -5°C/h -15°C/h -100°C/h 1600°C 1300°C 800°C (25h halten) (1h halten) (1h halten) RT Tabelle 3.5.6.4: Proben im System Titan-Zinn/Chrom-Kohlenstoff und ihre pulverdiffraktometrische Charakterisierung. Fettgedruckt sind die Hauptprodukte. Nr. Gehalt [At.-%] Zusammensetzung Ti Sn Cr C 1 Ti6Cr10Sn5C1 27,3 45,4 22,7 4,5 2 Ti6Cr7,5Sn7,5C1 27,3 34,1 34,1 4,5 3 Ti6Cr5Sn10C1 27,3 22,7 45,4 4,5 4 Ti6Cr3Sn12C1 27,3 13,6 54,5 4,5 Gitterkonstante [Å] weitere kristalline der MAX-Phase Phasen a = 3,1507(2); c = 13,710(6) V=117,86(6) a = 3,1638(12); c = 13,721(6) V=118,94(6) a = 3,1609(11); c = 13,710(5) V=118,64(6) a = 3,1588(11); c = 13,692(4) V=118,32(5) >TiC >TiC >TiC TiC - 168 - MAX - Phase – Weitere Untersuchungen Bei der Verwendung von Chrom als Zusatz im System Titan-Zinn-Kohlenstoff zeigte sich kein Einfluss auf die Produktverteilung in Abhängigkeit von der Schmelzzusammensetzung. Aus der zinnhaltigen Chromschmelze konnte in jedem Ansatz die Bildung der 211-Phase beobachtet werden. Bei einer röntgenographischen Analyse am Pulver zeigten sich immer geringe Anteile des binären TiC. Die Gitterkonstanten, die aus dem Pulver erhalten wurden, deuteten auf die Bildung von Ti2SnC hin. Dieses konnte mit Hilfe von Untersuchungen an den Einkristallen bestätigt werden. Die untersuchten Einkristalle wurden auch einer qualitativer Elementaranalyse mittels energiedispersiver Röntgenemmissionsspektroskopie unterzogen. Bei dieser Untersuchung zeigte sich, wie schon oft beobachtet, ein bedeutender Anteil an Stickstoff. Es konnte ein Stickstoffanteil von bis zu 15 At.-% an den Einkristallen detektiert werden. Der Einbau von Stickstoff liefert auch eine gute Erklärung für die etwas kleineren Gitterkonstanten, die aus den röntgenographischen Untersuchungen am Pulver erhalten wurden. Außer Titan und Zinn konnte kein weiteres Metall an Einkristallen nachgewiesen werden. 3.5.6.5 Untersuchungen mit Aluminium/Silizium Bei der Untersuchung im System Vanadium-Aluminium-Kohlenstoff wurde Silizium als Zusatz untergemischt. Die Zugabe an Silizium wurde mehrmals variiert, die Zusammensetzung der Elementgemische und das Ergebnis aus der Phasenanalyse ist in der Tabelle 3.5.6.5 angegeben. Die Umsetzung der handgepressten Tabletten erfolgte unter ArSchutzgas bei 1600°C. Tabelle 3.5.6.5: Proben im System Vanadium-Aluminium/Silizium-Kohlenstoff pulverdiffraktometrische Charakterisierung. Fettgedruckt sind die Hauptprodukte. Nr. Gehalt [At.-%] Zusammensetzung V Al Si C und ihre Gitterkonstante [Å] weitere kristalline der MAX-Phase Phasen V5Al8, V2C, V6C5, 1 V6Al10Si5C1 27,3 45,5 22,7 4,5 - 2 V6Al7,5Si7,5C1 27,3 34,1 34,1 4,5 - 3 V6Al5Si10C1 27,3 22,7 45,5 4,5 - Si2V, Si, SiC, V6C5 4 V6Al3Si12C1 27,3 13,6 54,5 4,5 - Si2V, Si, SiC SiC, Al4SiC4 Si2V, V6C5, V2C, Si - 169 - MAX - Phase – Weitere Untersuchungen Temperaturprogramm: 150°C/h RT -5°C/h -15°C/h -100°C/h 1600°C 1200°C 800°C (20h halten) (1h halten) (1h halten) RT Die Analyse der Syntheseprodukte ergab bei der siliziumarmen Probe die intermetallische Phase V5Al8 als Hauptprodukt. Bei den siliziumreicheren Phasen wurde als Hauptprodukt Si2V identifiziert. Keine der umgesetzten Proben zeigte Anzeichen auf die Bildung einer MAX-Phase. 3.5.6.6 Untersuchungen mit Chrom/Zinn Die Analysen des ternären Systems Vanadium-Zinn-Kohlenstoff mit dem Zusatz an Chrom sollte Erkenntnis im gegeben System zeigen, ob eine der MAX-Phasen bei ausgewählten Bedingungen zugänglich ist. Dazu wurden die Elemente in unterschiedlichen Verhältnissen gut homogenisiert und mit einer Handpresse in Form einer Tablette gebracht. Diese wurden anschließend in Korundtiegeln bei einer Temperatur von 1600°C für 25 h zur Reaktion gebracht. Temperaturprogramm: 150°C/h RT -5°C/h -15°C/h -100°C/h 1600°C 1300°C 800°C (25h halten) (1h halten) (1h halten) RT Tabelle 3.5.6.6: Proben im System Vanadium-Chrom/Zinn-Kohlenstoff und ihre pulverdiffraktometrische Charakterisierung. Nr. Gehalt [At.-%] Zusammensetzung V Cr Sn C 1 V6Cr10Sn5C1 27,3 45,5 22,7 4,5 2 V6Cr7,5Sn7,5C1 27,3 34,1 34,1 4,5 3 V6Cr5Sn10C1 27,3 22,7 45,5 4,5 4 V6Cr3Sn12C1 27,3 13,6 54,5 4,5 Gitterkonstante [Å] weitere kristalline der MAX-Phase Phasen Keine Ausbeute nach der Hydrolyse Tabelle 3.5.6.6 gibt eine Übersicht der Elementgemische. Bei Aufarbeiten des Schmelzregulus wurde durch die verd. Salzsäure der gesamte Rückstand aufgelöst. Alle bekannten MAX-Phasen gelten als säuerresistent, somit ist hier ausgeschlossen, dass unter den gebildeten Produkten eine MAX-Phase vorlag. - 170 - MAX - Phase – Ergebnisse aus dem Pulsplasma Ofen 3.6 Weitere Ergebnisse aus dem Pulsplasma-Ofen Im Rahmen dieser Arbeit wurden noch weitere Dreistoffsysteme wie das Titan-SiliziumKohlenstoff und das Tantal-Aluminium-Kohlenstoff System auf die Möglichkeiten der Darstellung von MAX-Phasen mit Hilfe der neuen Synthesemethode mittels gepulsten Strömen durchgeführt. Dabei wurde die Zusammensetzung, Temperatur und Reaktionszeit variiert. 3.6.1 Darstellung von Ti3SiC2 im Pulsplasma Ofen Für eine erfolgreiche Darstellung von Ti3SiC2 wurde ein Temperaturbereich von 1450°C bis 1600°C untersucht. Dabei wurden Reaktionszeiten von 3 Minuten bis zu 18 Minuten variiert. Jedoch konnten eine phasenreine Darstellung an Ti3SiC2 nicht erfolgreich durchgeführt werden. Es gelang jedoch, Ti3SiC2 als eindeutiges Hauptprodukt neben geringen Mengen an TiC und C herzustellen. 80000 70000 Lin (Counts) 60000 50000 40000 30000 20000 10000 0 6 10 20 30 40 50 60 70 80 90 2-Theta - Scale Abbildung 3.6.1: Pulverdiffraktogramm von Ti3SiC2 (HP). Die blauen Reflexe (NP) konnten Kohlenstoff zugeordnet werden. Bei den Grünmarkierten Reflexen handelt es sich um TiC. Bei der Darstellung von Ti3SiC2 wurden die Elemente 0,294 g (6,138 mmol) Titan, 0,057 g (2,029 mmol) Silizium und 0,049 g (4,083 mmol) Kohlenstoff in stöchiometrischen - 171 - MAX - Phase – Ergebnisse aus dem Pulsplasma Ofen Verhältnis eingewogen und gut homogenisiert. Das Pulvergemisch wurde in der Graphitmatrize bei einer Temperatur von 1600°C für 5 min. umgesetzt. Die Reaktion wurde im Vakuum (0,12 mbar) durchgeführt. Es wurde ein Sinterdruck von 4,2 kN für die Umsetzung eingestellt. Bei einer längeren Reaktionszeit (die Reaktionen wurden in Bereichen von 3 bis auf 18 Minuten durchgeführt) wurde die Bildung vom SiC beobachtet. Bei der Herabsetzung der Reaktionstemperatur wurde TiC gebildet. Bei Reaktionszeit unter 5 Minuten wurden überwiegend Edukte in der Phasenanalyse detektiert. Bei der Umsetzung der Probe im Temperaturbereich von 1600°C für 5 Minuten wurde das beste Ergebnis erzielt. Die Gitterkonstanten konnten auf a = 3,0785(16) Å und c = 17,745(8) Å in der Raumgruppe P63/mmc verfeinert werden. 3.6.2 Darstellung von Ta4AlC3 und Ta2AlC im Pulsplasma Ofen Neben den bisher beschriebenen Systemen mit Titan und Vanadium wurde in den letzten Jahren von MAX-Phasen mit Tantal als Hauptkomponente berichtet [202]. Die Untersuchung im System Tantal-Aluminium-Kohlenstoff wurde in Temperaturen von 1200°C bis 1600°C durchgeführt. Dabei wurden stöchiometrische Ansätze mit der Zielsetzung für die Bildung von MAX-Phasen aller Varianten eingewogen. Nahezu alle Proben wurden nicht nur bei unterschiedlichen Temperaturen sondern auch bei unterschiedlichen Reaktionszeiten umgesetzt. So wurden die Synthesezeiten von 3 bis 15 Minuten gewählt. Bei der Darstellung von Ta4AlC3 wurden die Elemente 0,460 g (2,555 mmol) Tantal, 0,017g (0,633 mmol) Aluminium und 0,023 g (1,908 mmol) Kohlenstoff in stöchiometrischen Verhältnis eingewogen und innig verrieben. Das Pulvergemisch wurde in einer Graphitmatrize mit Graphitfolie ausgekleidet bei einer Temperatur von 1300°C für 5 Minuten umgesetzt. Währen des Sinterns wurde ein Druck auf die Probe von 4,1 kN angelegt. Die Reaktion wurde in eine Vakuum von 0,14 mbar durchgeführt. Temperaturprogramm: 2 min. RT 2 min. 600°C 3 min. 900°C 3 min. 1200°C 2 min. 1300 RT (5 min. halten) - 172 - MAX - Phase – Ergebnisse aus dem Pulsplasma Ofen 240000 230000 220000 210000 200000 190000 180000 170000 160000 Lin (Counts) 150000 140000 130000 120000 110000 100000 90000 80000 70000 60000 50000 40000 30000 20000 10000 0 5 10 20 30 40 50 60 70 80 90 100 110 120 2-Theta - Scale Abbildung 3.6.2: Pulverdiffraktogramm von Pulverdiffraktogramm von Ta4AlC3 (grün NP) und Ta2AlC (rot NP). Die blauen Reflexe konnten TaC (HP) zugeordnet werden. Der erhaltenen Sinterkörper wurde im WC-Mörser zertrümmert und auf die gebildeten Phasen hin untersucht. Ganz klar wurde TaC als Hauptprodukt identifiziert. Bei den zwei Nebenprodukten, die entstanden sind, handelt es sich um MAX-Phasen. Die rotmarkieren Reflexe konnten Ta2AlC (RG: P63/mmc mit a = 3,081(2) Å und c = 13,939(18) Å) und die grünen Ta4AlC3 (RG: P63/mmc mit a = 3,101(3) Å und c = 24,00(4) Å) zugeordnet werden. Bei der Untersuchung von höheren Temperaturen konnte kein Hinweis auf die Bildung einer MAX-Phase gefunden werden. Die Veränderung der Stöchiometrie zu Bildung von 211bzw. 312-Phasen, um einer der Verbindungen abgesondert darzustellen, ist gescheitert. Die Produkte waren binäre Carbide (TaC, Ta2C) und eine intermetallische Phasen (Ta2Al). - 173 - MAX - Phase – h-Carbide im Pulsplasma Ofen 3.7 h-Carbide im Pulsplasma Ofen 3.7.1 Darstellung von Ta3Al2CoC und Ta3Al2CoN im Pulsplasma Ofen Bei der Untersuchung der MAX-Phasen des Tantals im SPS-Ofen konnte die Entstehung des h-Carbids mit der Summenformel Ta3Al2CoC[202,106] beobachtet werden. Bei der Reproduktion der h-Phase wurde versucht, diese phasenrein bei Temperaturen von 1400°C – 1500°C darzustellen. Dazu wurde 0,116 g (0,599 mmol) TaC, 0,2168 g (1,198 mmol) Ta, 0,033 g (1,197 mmol) Al und 0,035 g (0,599 mmol) Co in einem Achat-Mörser miteinander homogenisiert und in einer Graphitmatrize mit einer Graphitfolie ausgekleidet und bei einer Temperatur von 1500°C für 5 Minuten zu Reaktion gebracht. Bei einem Vakuum von 0,11 mbar und einem Sinterdruck von 4,5 kN konnte ein nahezu phasenreines Produkt erhalten werden. Temperaturprogramm: 2 min. RT 3 min. 600°C 3 min. 1000°C 4 min. 1300°C 2 min. 1500 RT (5 min. halten) 180000 170000 160000 150000 140000 130000 Lin (Counts) 120000 110000 100000 90000 80000 70000 60000 50000 40000 30000 20000 10000 0 6 10 20 30 40 50 60 70 80 90 2-Theta - Scale Abbildung 3.7.1: Pulverdiffraktogramm von Ta3Al2CoC (HP rot). Die blauen Reflexe (NP) konnten TaC und die grünmarkierten Reflexe konnten dem Ta2Al (NP) zugeordnet werden. - 174 - MAX - Phase – h-Carbide im Pulsplasma Ofen Nach einer Phasenanalyse konnte als Hauptprodukt h-Carbid das mit der Zusammensetzung von Ta3Al2CoC mit einer Gitterkonstanten von a = 11,5756(11) Å in der Raumgruppe Fd 3 m bestimmt werden. Als Nebenprodukte konnten das binäre TaC und das intermetallische Ta2Al beobachtet werden. Bei weiteren Versuchen wurde mit TaN gearbeitet. Hier wurde versucht, die h-Phase als Nitrid darzustellen. Dazu wurde 0,1164 g (0,603 mmol) TaN, 0,2162 g (1,201 mmol) Ta, 0,032 g (1,186 mmol) Al mit 0,035 g (0,598 mmol) Co bei einer Reaktion unter Vakuum (0,12mbar) bei einer Temperatur von 1500°C (5 Minuten) zur Reaktion gebracht. Der Sinterdruck wurde wie zuvor auf 4,5 kN eingestellt. Temperaturprogramm: 2 min. RT 3 min. 600°C 3 min. 1000°C 4 min. 1300°C 2 min. 1500 RT (5 min. halten) Nach einer Phasenanalyse des entstandenen Produkts zeigt sich eine quartären Verbindung mit deutlich kleiner Elementarzelle. Für das Hauptprodukt Ta3Al2CoN konnte eine Gitterkonstante von a = 11,395(3) Å in der Raumgruppe Fd 3 m bestimmt werden. Die Verkleinerung der Zellenparameter ist durch die Substitution von Kohlenstoff durch Stickstoff durchaus plausibel. Die h-Phase konnte als Hauptprodukt dargestellte werden, jedoch ist eine phasenreine Darstellung nicht gelungen. Neben TaN konnten auch zwei intermetallische Phasen beobachtet werden (Co2Al5 und Al31Ta19). Gitterkonstantenvergleich 11.6 Erhaltenen Gitterkonstanten Gitterkonstante [Å] 11.55 11.5 11.45 11.4 11.35 11.3 Ta3Al2CoC Ta3Al2CoC0.5N0.5 Ta3Al2CoN Verbindung Abbildung 3.7.2: Substitution von Stickstoff gegen Kohlenstoff - 175 - MAX - Phase – h-Carbide im Pulsplasma Ofen In einem dritten Versuch wurde die Zusammensetzung so gewählt, dass die Position des Kohlenstoffs bis zur Hälfte mit Stickstoff besetzt werden sollte. Somit wurde 0,073 g (0,374 mmol) TaN, 0,072 (0,373mmol) TaC, 0,271 g (1,506 mmol)Ta, 0,040 g (1,487 mmol) Al mit 0,044 g (0,747mmol) Co innig mit einander verreiben und im Pulsplasma-Ofen unter gleichen Konditionen wie die Proben zuvor umgesetzt. Das Ergebnis war eine h-Phase mit Gitterkonstanten von a = 11,563(3) Å in der Raumgruppe Fd 3 m, neben TiC, TiN, Co2Al5 und Al31Ta19. Die Gitterkonstante liegt sehr nahe an der vom h-Carbid, was dafür spricht dass nur ein geringer Anteil an Stickstoff gegen Kohlenstoff substituiert wurde. Eine Übersicht der Gitterparameter zu den Verbindungen gibt Abbildung 3.7.2. 3.7.2 Härteprüfung Nach erfolgreicher Reproduktion der h-Phasen wurden Sinterkörper von ausreichender Größe für eine Härtemessung nach Vickers und Knoop dargestellt. An jeder Probe wurden mehrere Messungen durchgeführt. Für die das h-Carbid mit der Zusammensetzung von Ta3Al2CoC konnte ein mittlere Härte von 1361 HV 0,2/10 (~13,2 GPa) und 1321 HK 0,2/10 bestimmt werden. Abbildung 3.7.3: Vickers -Abdruck Abbildung 3.7.4: Knoop- Abdruck (d1=16,18µm, d2=16,92µm: 1381 HV). (d1=46,27µm, 1355 HK). Für die das h-Carbid mit der Zusammensetzung von Ta3Al2CoN konnte ein mittlere Härte von 1390 HV 0,2/10 (~13,3 GPa) und 1224 HK 0,2/10 bestimmt werden. - 176 - MAX - Phase – h-Carbide im Pulsplasma Ofen Abbildung 3.7.6: Knoop- Abdruck Abbildung 3.7.5 Vickers- Abdruck (d1=47,60µm, 1281 HK). (d1=16,18µm, d2=17,17µm: 1360 HV). Für beide Sinterkörper zeigte sich nahezu die gleiche Härte von 13 GPa, diese liegt zwischen der von Mineralien Quarz (~11 GPa) und Topas (~14). 3.7.3 Darstellung von Nb3Al2CoC im Pulsplasma Ofen Nach der erfolgreichen Darstellung der oben diskutierten h-Phasen, wurden weitere Systeme auf die Existenz der h-Phase hin untersucht. Ein nahezu phasenreiner Sinterkörper von Nb3Al2CoC in Form einer schwarzen Tablette bildete sich in einem Ansatz der Zusammensetzung 0,129 (1,231 mmol) NbC, 0,2287 g (0,246 mmol) Nb, 0,070 g (2,583 mmol) Al und 0,073 g (1,238 mmol) Co. Es wurde folgendes Temperaturprogramm verwendet. Temperaturprogramm: 2 min. RT 3 min. 600°C 3 min. 1000°C 4 min. 1300°C 2 min. 1500 RT (5 min. halten) Für das Sintern im Vakuum (0,14 mbar) wurde ein Druck von 4,2 kN eingestellt. Die Synthese wurde in einer Graphitmatrize mit dem Durchmesser von 10 mm ausgekleidet mit Graphitfolie durchgeführt. - 177 - MAX - Phase – h-Carbide im Pulsplasma Ofen 120000 110000 100000 90000 Lin (Counts) 80000 70000 60000 50000 40000 30000 20000 10000 0 6 10 20 30 40 50 60 70 80 90 2-Theta - Scale Abbildung 3.7.7: Pulverdiffraktogramm von Nb3Al2CoC (HP rot). Die blauen Reflexe (NP) konnten NbC0,84 und die grünmarkierten Reflexe dem Co2B (NP) zugeordnet werden. Der Sinterkörper wurde nach dem Erkalten vollständig von der Graphitfolie befreit. Durch eine mechanische Zertrümmerung des Sinterkörpers im Wolfrahcarbidmörser konnte eine röntgenographische Phasenanalyse erfolgen. Als Hauptprodukt konnte eindeutig Nb3Al2CoC mit ein Gitterkonstanten von a = 11,6573(15) Å in der Raumgruppe Fd 3 m beobachtet werden. Als weitere Phasen konnten das binäre NbC0,84 und das Kobaltborid Co2B erfasst werden. Das Borid lässt sich nur als Produkt einer kleinen, mit eingebrachter Verunreinigung bei der Einwaage der Edukte erklären. 3.7.4 Strukturdiskussion Nb3Al2CoC (a = 11,6573(15) Å), Ta3Al2CoC (a = 11,5756(11) Å) und Ta3Al2CoN (a = 11,395(3) Å) kristallisieren im Ta3Co3C-Typ[106] in der Raumgruppe Fd 3 m. Die Metallatome besetzen die Punktlagen 48f und 32e und 16d, Die Position 16c wird ausschließlich von Kohlenstoff bzw. Stickstoff besetzt. Tabelle 3.7.4.1 gibt die Besetzungsfaktoren der Ta3Al2Co(C/N) Verbindung wieder[202]. Im Folgenden wird die Strukturdiskussion am Beispiel von Ta3Al2CoC exemplarisch durchgeführt. - 178 - MAX - Phase – h-Carbide im Pulsplasma Ofen Tabelle 3.7.4.1: : Ortskoordinaten und Besetzungsfaktoren in der Ta3Al2CoC-Struktur[203]. Atom Wyckoff Position Symmetrie x y z Ta 48f 2.mm 0,93669(6) ⅛ ⅛ Al 32e .3m 0,7075(4) x x Co 16d .3m 0 0 0 C (N) 16c .3m ½ ½ ½ In der Abbildung 3.7.8 ist die Kristallstruktur von Ta3Al2CoC dargestellt. Hervorgehoben sind die CTa6-Oktaeder, die in der Struktur über die gemeinsamen Spitzen verknüpft sind. In den Lücken zwischen den Oktaedern befinden sich Aluminium- und Kobaltatome. Abbildung 3.7.8: Kristallstruktur von Ta3Al2CoC Der Kohlenstoff ist oktaedrisch von sechs Tantalatomen umgeben. Der Kohlenstoff hat in seiner unmittelbaren Nähe keine weiteren Wechselwirkungen zu den anderen Metallen. Das Aluminiumatom bekommt eine Koordinationssphäre in Form eines verzerrten Ikosaeders. Daran sind sechs Tantal-, drei Aluminium- und drei Kobaltatome beteiligt. Die Umgebung des Kobaltatoms ist ebenfalls ein verzerrter Ikosaeders, jedoch nicht ganz so stark wie das des Aluminiums. Jeweils sechs Tantal und sechs Aluminiumatome koordinieren das Kobaltatom. Abbildung 3.7.9: Koordination von Al - 179 - MAX - Phase – h-Carbide im Pulsplasma Ofen Abbildung 3.7.10: Koordination von Co Abbildung 3.7.11: Koordination von Ta Die Umgebung des Tantalatoms ist über ein unregelmäßiges Polyeder mit der Koordinationszahl 16 charakterisiert. Hier sind acht Tantal-, vier Aluminium-, zwei Kobalt und zwei Kohlenstoffatome beteiligt. - 180 - 4 τ-Boride 4.1 Literaturübersicht Das Komplexcarbid mit der geordneten Cr23C6-Struktur wurde bei Untersuchungen von rostfreien Stählen zum ersten Mal beobachtet und genauer unter die Lupe genommen[107]. Die Kristallstruktur konnte im Jahre 1933 mit Hilfe von Pulverfilmaufnahmen von Westgren[108] aufgeklärt werden. Weitere Verbindungen des Cr23C6-Typs folgten noch im gleichen Jahr, mit dem kleinen Unterschied, dass es sich hier um ternäre Verbindungen handelt[109]. Die Arbeitsgruppe von Stadelmaier beobachtete den Cr23C6-Strukturtyp an zahlreichen Boriden[110,111,112,191], diese und wie auch die Arbeitskreise von Nowotny[113,114,163,179] und Kuz’ma[115,154] starteten eine systematische Untersuchungsreihe, gefolgt von röntgenographischen und metallographischen Analysemethoden, um die Existenz der mit τ gekennzeichneten Phase und deren Homogenitätsbereich zu erfassen. Bei den nahezu 90 Verbindungen, die inzwischen von der Literatur[116] erfasst wurden, handelt es sich überwiegend um Boride und Carbide. Jedoch sind auch wenige Boridcarbide[117,116,118], Phosphide[119,120], Silicide[121] und auch Germanide[121] bekannt. In der Tabelle 4.1.1.1 und Tabelle 4.1.1.2 sind die in der Literatur bekannten und untersuchten Phasen mit Cr23C6-Struktur zusammengefasst. Die meisten Untersuchungen der drei oben genannten Arbeitskreise beschränkten sich hauptsächlich auf die Bestimmung des Homogenitätsbereichs. Somit wurden selten Einkristalle untersucht, die eine Aussage über die Verteilung und das Verhältnis der beteiligten Metalle erlauben. In jüngerer Zeit wurde der Einbau von B4-Tetraedern auf einer der Metallpositionen nachgewiesen[167,174], die Erarbeitung solcher Erkenntnisse ist nur mit Hilfe von Einkristalldaten möglich. Als allgemeine Summenformel für die ternäre Verbindung hat sich die Schreibweise als M23-xMxB6+y etabliert. Wie in den folgenden Tabellen deutlich zu erkennen ist, ist die Mehrheitskomponente M vorwiegend Nickel oder Kobalt. etwas seltener ist auch Chrom zu finden. Als Minderheitskomponente oder auch τ-Stabilisator genannt, können Hauptgruppen-, Übergangs- und Seltenerdmetalle eingesetzt werden. Neben den zahlreichen ternären Verbindung des Cr23C6-Typs konnten drei binäre τ-Boride diskutiert werden Fe23B6[129,130], Co23B6[154,162,167] und Ni23B6[128]. Wobei nur Co23B6 als Einzige dieser Reihe mit Hilfe von Einkristalldaten aufgeklärt werden konnte. Die restlichen Verbindungen wurden als metastabil beschrieben. - 181 - τ-Boride - Literaturübersicht Von technischem Interesse sind die τ-Boride, die unterschiedliche Eigenschaften in einer Verbindung kombinieren, wie z.B. (Co,Fe)20Al3B6, welches als Borocube[122] bezeichnet wird. Hier wird die mechanische Härte mit den magnetischen Eigenschaften in einem Werkstoff verknüpft. Die Arbeitsgruppe von Buschow untersuchte die τ-Boride des Kobalts auf ihre ferromagnetischen Eigenschaften mit Tc zwischen 410 K und 740 K[123]. Eine weitere Anwendung der τ-Phase wurde bei der Beschichtung durch das Plasmaspritzen auf Stahlteilen diskutiert. Hier kommen mehrere τ-Boride in Frage mit der Zusammensetzung M21M’2B6 mit M = Fe, Ni, Co und M’ = Ti, Nb, W. Diese werden als verschleißfeste Schicht auf Fertigteile aufgebracht[124]. Hartstoffe auf der Basis solcher komplexer Systeme stellen ein großes Potential für die moderne Werkstoffentwicklung dar. Bei der Erforschung thermisch stabiler Legierungen zum Verschleißschutz wurde das τBorid mit der Zusammensetzung Ni20,7Ta2,3B6 auf seine Eigenschaften in einem Temperaturbereich von 850°C untersucht[125]. Weitere Untersuchungen zur Härte an τ-Boriden konnten im System Nickel-AluminiumBor und Nickel-Gallium-Bor durchgeführt werden[126]. Im Ni-Al-B System konnten nur borreiche Proben untersucht werden, für die Probe der Zusammensetzung Ni22AlB14 konnte ein Härte von 1100 HV 1 (~10,8 GPa) erhalten werden dierdamit vergleichbar mit Quarz (SiO2; HV=1103HV 1[127]) ist. Proben im Ni-Ga-B System lieferten Härtewerte im Bereich von 900 HV 1 (~8,8 GPa). Diese Werte, sind im Bereich von Nitrierstählen. Alle Härteuntersuchungen wurden jedoch nicht an Einkristallen sondern an Gussgefügen durchgeführt. 4.1.1 Phasenbreite und Charakterisierung der τ-Phase Wie in der Übersicht eindeutig zu erkennen ist, treten die meisten τ-Phasen mit einer geringen Phasenbreite auf. Ausnahmen sind Phasen, die ausschließlich aus den Elementen der 17. und 18. Gruppe aufgebaut sind Mn23-xRexB6 mit (x=4,5-15)[137] und Co23-xMnxB6 (x=311,5)[160] sind nur zwei von einigen Beispielen, bei der einen große Variation bezüglich des τStabilisators gegeben ist. Neben den reinen τ-Boriden und τ-Carbiden wurden auch τ-Borocarbide beschrieben wie Co23B4C2[117], bei denen eine Mischbesetzung der Nichtmetallposition durch Bor und Kohlenstoff auftritt. - 182 - τ-Boride - Literaturübersicht Tabelle 4.1.1.1: Übersicht der in der Literatur bekannten τ-Phasen Bekannte τ-Phasen mit der allgemeinen Summenformel M23-xM’xA6+y Boride mit M = Ni x a [Å] Boride mit M = Fe Ref. x k. A. Fe23-xRexB6 7 – 20 k. A. [155,191] Fe23-xIrxB6 7 – 13 k. A. [137,] Fe23-xMnxB6 9 – 18 k. A. [116] [135] 10,761 [128] Fe23B6 Ni20Li3B6 – 10,478 [132] 10,549 – 10,585 10,472 – 10,602 [133,191] 10,560 [155,191] Fe22NbB5 – 10,819 [136, 191] Fe21Nd2B6 – 10,0803 [134] Ref. – – 3,5 – 4,0 a [Å] [129,130] Ni23B6 Ni23-xMgxB6+y Carbide mit M = Cr Ni21Ca2B6+y – Ni20Sc3B6 – Ni20-xTixB6 2,5 – 3,0 Ni23-xZrxB6 1,5 – 4,5 Ni23-xHfxB6 2,2 – 2,7 Ni23-xVxB6 2,0 – 2,7 Ni20-xNbxB6 2,2 – 2,5 Ni20-xTaxB6 2,2 – 2,3 Ni23-xMnxB6 2,0 – 8,0 10,507 – 10,538 10,561 – 10,681 10,585 – 10,669 10,473 – 10,492 10,539 – 10,593 10,534 – 10,576 10,495 – 10,678 Ni23-xRexB6 10 – 12 11,250 [191] Ru23-xTaxB6 2–4 k. A. Ni18,5Fe4,5B6 – 10,501 [138] Ru23-xNbxB6 2–4 k. A. [116] [116,191] [191] Boride mit M = Cr, Mn, Ru [191] x a [Å] 11,202 [137,162] 11,175 – 11,193 [137] [191] Cr13Ir10B6+y – [191] Mn23-xRexB6 4,5 – 15 [116,191] Mn16Ir7B6 – Ref. [137] k. A. [139] [139] Cr23C6 x a [Å] – 10,6595 Cr23-xTixC6 0 – 1,2 Cr23-xHfxC6 0 – 0,3 Cr23-xVxC6 0 – 4,0 Cr23-xNbxC6 0 – 0,3 Cr23-xTaxC6 0 – 0,3 Cr21,7Co1,3C6 Cr23-xMoxC6 Cr21W2C6 Cr23-xMnxC6 Cr23-xRexC6 Cr23-xFexC6 Ref. [131,191] k. A. [116] k. A. [116] [116,191] 10,724 k. A. [116] k. A. [116] 0 – 1.7 10,637 [148] k. A. [116] 0 – 4,4 10,760 – 10,790 10,630 [109] 10,890 [116] 2,6 – 4,0 0 – 23 0 – 0,2 0 – 7,5 [116] 10,5966 – 10,6537 [116,131] [116,191] Cr17Fe5Mo2C6 – k. A. Cr16Fe6NiC6 – 10,636 [116] - 183 - τ-Boride - Literaturübersicht Ni23-xZnxB6+y 3–4 Ni23-xAlxB6+y 2,5 – 3,0 Ni23-xGaxB6 2,0 – 3,5 Ni23-xInxB6+y 1–2 Ni21,6Ge1,4B6 – Ni23-xSnxB6+y 1,7 – 2,0 Ni23-xSbxB6 1,9 – 2,0 Ni21Ce2B6 – Ni21U2B6 – 10,498 – 10,555 10,485 – 10,620 10,481 – 10,525 10,581 – 10,609 10,4890 [140,141,191] [145,191] Co23-xMnxByC6-y – 2,5 – 6 10,584 – 10,598 10,585 – 10,596 10,678 [146,191] Fe23ByC6-y – 1,3 – 3,6 Fe23-xMnxByC6-y 0 – 23 0–6 k. A. Fe23-xCrxByC6-y 0 – 23 1,5 k. A. 10,647 – 10,652 [115,149] – 0–4 k. A. [142,191] Boridcarbide mit M = Co, Fe, Mn, Cr [143,162,191] [144,162,167] [147] [148] x Co23B4C2 y 10,470 – Mn23ByC6-y Ni21Er2B6 – 10,640 [116] Mn23-xCrxByC6-y Ni21Tm2B6 – 10,633 [116] Cr23ByC6-y Ni21Yb2B6 – 10,636 Ni21Lu2B6 – 10,632 a [Å] k. A. 10,594 – 10,627 Carbide mit M = Mn Ref. [117] x a [Å] Ref. [116] [116] [116] Mn23-xMoxC6 0 – 0,06 10,595 – 10,729 k. A. [116,118] Mn23-xWxC6 0 – 0,06 k. A. Mn23C6-y – [116] [116] [116] [116,150] [116] 0 - 23 0-3 k. A. – 0 – 1,8 k. A. Carbide mit M = Fe [116] x Fe21Mo2C6 – Fe21W2C6 – a [Å] Ref. 10,510 – 10,560 10,533 – 10,540 [109,116] [109,116] [116] [116] Ni12,6Ir10,4B11,8 – 10,932 Phosphide mit M = Ni [162] x a [Å] Silicide mit M = Pd Ref. Ni20Mn3P6 – 11,0850 [151] Ni20Mg3P6 – 11,1130 [151] Ni21In2P6 – 11,1120 [153] Pd20Ce3Si6 x a [Å] – 12,1610 Ref. [152] Germanide mit M = Pd Pd20Ce3Ge6 x a [Å] – 12,4453 Ref. [152] - 184 - τ-Boride - Literaturübersicht Diese Arbeit soll sich mit der Untersuchung des Systems beschäftigen, in der Kobalt als Hauptkomponente dient. Die metallreichen Verbindungen des Kobalts mit der Cr23C6Struktur sind in der Tabelle 4.1.1.2 angegeben. Tabelle 4.1.1.2: Vorkommen und Homogenitätsbereich der in der Literatur erfassten Verbindungen, die mit Kobalt als Hauptkomponente in der Cr23C6-Struktur kristallisieren. Verbindung x Gitterkonstante [Å] Ref. Co23B6 - 10,4618 [162], [154] Co21Sc2B6 - 10,546 [155],[191] Co21U2B6 k. A. 10,575 – 10,620 [156] Co20Ti3B6 2,5 – 3,5 10,508 – 10,552 [191],[157] Co21Zr2B6 k. A. 10,582 – 10,597 [156] 10,574 [156],[158] Co23-xHfxB6 1,3 – 1,7 Co23-xVxB6 2–3 10,476 – 10,497 [191] Co21Nb2B6 1,7 – 2,0 10,520 – 10,574 [191],[159] Co21Ta2B6 1,0 – 2,2 10,549 [139,191] Co21Cr2B6 k. A. 10,471 [160,191] Co23-xMoxB6 1,2 – 15 10,500 – 10,505 [191] Co23-xWxB6 1,3 – 1,5 10,500 – 10,506 [191] Co23-xMnxB6 3,0 – 11,5 10,518 – 10,641 [160] Co23-xRexB6 k. A. 10,494 – 10,516 [161] Co23-xIrxB6+y k. A. 10,798 – 10,943 [162],[163] Co23-xAlxB6 2,8 – 3,0 10,480 – 10,520 [164],[165] Co23-xGaxB6 k. A. 10,520 [166] Co23-xInxB6 0,6 – 1,0 10,494 – 10,567 [167],[162] Co23-xGexB6 k. A. 10,499 [168] Co23-xSnxB6 1,9 – 2,0 10,601 [191], [169] Co23-xSbxB6 1,2 – 1,6 10,540 – 10,596 [191], [170] Co23-xMgxB6 k. A. 10,541 [191] Die Verbindungen Co15Ir8B6+y, Cr13Ir10B14, Ni12,6Ir10,4B11,8 und Ni22AlB14 fallen etwas aus der Reihe. Diese zeigen einen beträchtlichen Homogenitätsbereich bezüglich des Borgehaltes. Dieses überstöchiometrische Verhalten wird in Kapitel 4.1.3 vorgestellt. 4.1.2 Der Cr23C6-Strukturtyp Der Cr23C6-Strukturtyp ist der Prototyp für die so genannten τ-Phasen. Der Urtyp der hier diskutierten τ-Boriden kristallisiert kubisch in der Raumgruppe Fm 3 m (Nr. 225) mit einer - 185 - τ-Boride - Literaturübersicht Gitterkonstanten von a = 10,6595(4) Å[172]. Die Elementarzelle enthält 116 Atome verteilt auf fünf Punktlagen. In der Tabelle 4.1.2.1 ist die Besetzung der kristallographischen Lagen wie auch die Atompositionen wiedergegeben. Tabelle 4.1.2.1:Atomparameter in der Cr23C6-Struktur. Die Idealwerte wurden über das Hartkugelmodell (rCr=1,256 Å) berechnet. Cr23C6 RG: F m 3 m – Nr. 225 kubisch, a= 10,6595(4)Å[172] Position x „ideal“[171] Ref. für x[172] 48h 0, x, x 0,1667 0,16991(2) Cr 2 32f x, x, x 0,3822 0,38199(2) Cr 3 8c ½, ½, ½ - - Cr 4 4a 0, 0, 0 - - C 24e x, 0, 0 0,274 Atom Wyckoff Cr 1 0,2751(3) Die Abbildung 4.1.1 zeigt die charakteristische Metallpolyederdarstellung. Diese Betrachtung der kubisch dichtesten Packung aus Kuboktaedern und Würfeln lässt die NaCl analoge Anordnung erkennen. Das Kuboktaeder erhält eine oktaedrische Koordination der Würfel und umgekehrt bilden die Würfel ein Oktaeder um die Kuboktaeder. Somit hat jedes Metallpolyeder sechs Nachbarn. Die Punktlagen 4a, 8c, 32f und 48h sind von Chromatomen besetzt. Der Kohlenstoff besetzt vollständig die Punktlage 24e, in der er eine quadratische antiprismatische Koordination erhält. Die Kristallstruktur der hochsymmetrischen Cr23C6-Struktur wird am besten über die Metallpolyeder beschrieben. Somit formen zwölf Metallatome (Cr4) auf der Position 48h das Kuboktaeder. Die Punktlage 4a bildet das Zentrum des Kuboktaeders, dieser Hohlraum ist groß genug um ein weiteres Metallatom unterzubringen (Cr1). Acht Metallatome (Cr3) auf der Position 32f gestalten einen Würfel, diese Würfellücke erwies sich als zu klein um weitere Atomen Einlass zu gewähren. Bei der Position 8c der Metallatome (Cr2) handelt es sich um ein vierfach überkapptes Friauf-Polyeder, diese Punktlage ist mit Abstand die interessanteste Lage der Struktur und wird in Abschnitt 4.1.3 genauer diskutiert. - 186 - τ-Boride - Literaturübersicht Abbildung 4.1.1: Kristallstruktur von Cr23C6, Polyederdarstellung der Metallatome: Die Kohlenstoffatome (24e) befinden sich zwischen den Flächen des Kuboktaeders und des Würfels. Bei der Koordination um das Zentrum des überkappten Friauf-Polyeders 8c [½,½,½] sind zwei Sorten Chromatome [4(Cr3) + 12(Cr4)] beteiligt. Diese Koordination um ein Atom konnte bereits schon in den so genannten Laves-Phasen beobachtet werden, bei denen die Koordinationszahl sechzehn beträgt. Das klassische Beispiel ist die intermetallische Verbindung MgCu2[173]. In dieser AB2-Verbindung ist jedes Cu-Atom von vier Kupfer- und zwölf Magnesiumatomen koordiniert. Abbildung 4.1.2 zeigt die Elementarzelle mit dem charakteristischen FriaufPolyeder. Die Kupferatome haben einen Abstand von 3,046(1)Å zueinander, während die Bindung der Kupfer-Magnesium-Abstand mit 2,916(1) Å etwas Abbildung 4.1.2: Kristallstruktur der kürzer ist. kubischen Laves-Phase MgCu2 - 187 - τ-Boride - Literaturübersicht 4.1.3 Sonderverhalten in der τ-Phase Die Untersuchungen an Einkristallen der τ-Boride im System Nickel-AluminiumBor[174,167] und Kobalt-Iridium-Bor[162] zeigte eine ungewöhnliche Substitution von Metallatomen gegen B4-Tetraeder. Das Ausgangsmodell für die Strukturverfeinerung der τPhase war die Cr23C6-Struktur. Die Strukturverfeinerung an den oben genannten Systemen zeigte eine deutliche Restelektronendichte auf einer neuen Position (32f; x=0, y=194, z=0,194). Zudem konnten bei den borreichen Ansätzen ein Anstieg der Gitterkonstanten mit beobachtet werden. Dieses zeigte eine Gitterkonstante von 10,486 Å für Ni20,5Al2,5B6 bis 10,617 Å für Ni20AlB14 im System Nickel-Aluminium-Bor. Auch das System Kobalt-Iridium-Bor präsentierte eine Aufweitung der Gitterkonstante, hier wurde auch die Beobachtung gemacht, dass ein höherer Borgehalt und einer höhere Restelektronendichte auf 32f vorlag (10,798 Å für Co15Ir8B6 bis 10,943 Å für eine Zusammensetzung von Co12,4Ir10,6B11,8). Bei der Untersuchung von borreichen Proben aus dem System bestehend aus Kobalt, Iridium und Bor zeigte sich auch ein erhöherer Einbau an Iridium in den untersuchten Kristallen, was sich auch deutlich in der Gitterkonstante widergespiegelt hat. Die Untersuchungen an borarmen und borreichen Einkristallen halfen einen chemisch plausiblen Substitutionsmechanismus aufzustellen[174]. Hier wird die Substitution der Metalllage M’ auf (8c) gegen B4-Tetraeder auf (32f) diskutiert, dieser ist in der Abbildung 4.1.3 dargestellt. Jedoch soll an dieser Stelle darauf hingewiesen werden, dass bei einer nicht vollständigen Besetzung der Punktlage 32f auch Bordreiecke wie auch Borpaare denkbar sind. Eine weitere Substitutionslage ist die Metalllage 4a. Diese bildet das Zentrum der Kuboktaeder. Hier kommt es je nach eingesetztem Metall zu einer Misch- oder Vollbesetzung durch eine Metallsorte. Substitutionsmuster M’ (8c) ↔ B4 (32f) Abbildung 4.1.3: Substitutionsmuster in der τ-Phase - 188 - τ-Boride - Literaturübersicht Aus Untersuchungen an Einkristallen konnten die zwei unterschiedlichen Substitutionsmechanismen erarbeitet werden, die eine Erklärung für die Aufweitung der Gitterkonstanten liefern. Die zwei oben diskutierten Systeme konnten mit Hilfe von Einkristalldaten zur Verifizierung des Mechanismus beitragen. Nur mit Hilfe der Einkristalluntersuchungen kann eine Aussage zum M / M’- bzw. M’ / B4-Austausch13 gemacht werden. Einige weitere Systeme zeigten zu höheren Bor/Metall-Verhältnis ebenfalls einen beachtlicher Anstieg der Gitterkonstanten. Diese Sonderfälle wurde in Tabelle 4.1.3.1 unter der Angabe des Homogenitätsbereichs zusammengefasst. Auch hier wurde bereits durch die Autoren schon auf den zusätzlichen Einbau von Bor hingewiesen. Tabelle 4.1.3.1: Systeme die auf borreiche τ-Boride hinweisen (* konnte nur aus einen überstöchiometrischen Borgehalt erhalten werden) τ-Phase Gitterkonstante [Å] y Ref. Ni23-xMgxB6+y 10,549 – 10,585 0–1 [133,191] Ni23-xZnxB5+y 10,498 – 10,555 0–3 [140,141,191] Ni23-xInxB6+y 10,581 – 10,609 0 – 1,5 [144,162,167] Ni23-xSnxB5+y 10,584 – 10,598 0–4 [146,191] Ni21Ca2B6+y 10,471 – 10,602 0 – 4,5 [134] Cr13Ir10B6+y* 11,195 9,3max. [137,162] Co23-xIrxB6+y 10,798 – 10,943 6,4max. [162,163] Ni23-xAlxB6+y 10,486 – 10,617 0-8 [167] Eine endgültige Aufklärung der Besetzungen der Metallatome und die Aussage über zusätzlichen Einbaus von Boratomen kann nur mit Hilfe der röntgenographischen Einkristallenuntersuchungen eindeutig dargelegt werden. 4.1.4 Das System Kobalt-Bor Ternäre Verbindungen stehen immer in Kristallisationskonkurrenz mit den binären Phasen. Somit ist ein Verständnis der binären Randsysteme unerlässlich. Der Homogenitätsbereich der τ-Phase befindet sich meist in der metallreichen Ecke, die als Hauptkomponente eingesetzt 13 B4-Tetraedern kommen nur bei einer Zusammensetzung von M20M’B6(B4) vor. Abweichungen vom Borgehalt können zur Ausbildung von Bordreieck oder Borpaar führen. - 189 - τ-Boride - Literaturübersicht wird. Hier zeigt sich der Bereich der Boride mit der Cr23C6-Struktur überwiegend sehr schmal und umgeben von den stabilen binären Phasen. In dieser Arbeit wurde überwiegend als Hauptkomponente das Übergangsmetall Kobalt eingesetzt. So soll die Abbildung 4.1.4 und die Tabelle 4.1.4.1 die drei binären Verbindungen aus dem Zweistoffsystem Kobalt-Bor vorstellen. Abbildung 4.1.4: Phasendiagramm des Systems Kobalt-Bor[175] Die Beschreibung der bekannten Phasen im binären System basieren auf den thermischen und röntgenographischen Untersuchungen von Schöbel und Stadelmaier[176]. Unterhalb 33,3 At.-% Bor gilt das Teilsystem das inkongruent. Hier sind die bei 1125°C inkongruent schmelzender Phase Co3B und die bei 1280°C kongruent schmelzender Phase Co2B erhältlich. Daneben existiert ein Teilsystem bestehend aus Kobalt und Co2B, das einfach eutektisch ist. CoB ist das einzige Borid, das im Bereich von 33,3 bis 100 At.-% Bor existiert. CoB schmilzt unzersetzt ab einer Temperatur von 1460°C. Die kobaltreichste Phase Co3B kristallisiert orthorhombisch im Cementit-Typ. Die Boratome habe hier eine einfach überkappte trigonale prismatische Koordination aus sechs Kobalt- und einem Boratom (CN=7) Abbildung 4.1.5. Das tetragonale Co2B leitet sich aus dem CuAl2-Typ ab. Hier sind die Boratome quadratisch antiprismatisch durch die Kobaltatome koordiniert (CN=8 Abbildung 4.1.7). CoB kristallisiert orthorhombisch im FeBTyp. Hier sind die Boratome zu Borketten verbunden, die von trigonalen Metallprismen umhüllt werden (Abbildung 4.1.6). - 190 - τ-Boride - Literaturübersicht Tabelle 4.1.4.1:Kristallographische Daten der binären Phasen im Kobalt-Bor-System Typ RG Z Gitterkonstante [Å] Volumen[Å3] Ref. Co3B Fe3C Pnma 4 a = 5,223; b = 6,629; c = 4,408 152,62 [177] Co2B CuAl2 I4/mcm 4 a = 5,015; b = 5,015; c = 4,220 106,13 [178] CoB FeB Pnma 4 a = 5,254; b = 3,043; c = 3,956 63,25 [178] Abbildung 4.1.5: Kristallstruktur von Co3B (Projektion auf [001]-Ebene) Abbildung 4.1.6: Kristallstruktur von CoB (Projektion auf [001]-Ebene) Abbildung 4.1.8: Angaben der Bindungen in den binären Phasen im Kobalt-Bor-System (*Der Abstand liegt hier bei über 3,0 Å) d(B-B) [Å] Abbildung 4.1.7: Kristallstruktur von Co2B (Projektion auf [001]-Ebene) d(Co-B) [Å] Ref. 1,9290 – [177] Co3B -* Co2B 2,1100 (x 2) 2,1436 (x 8) CoB 1,9179 (x 2) 2,3287 (x 8) 2,0563 – [178] [178] 2,1671 (x 7) Die Übergangsmetallboride besitzen mit mehr als 130 Strukturtypen eine enorm vielfaltige Strukturchemie[179]. Die Boratome zeigen meistens eine trigonal prismatische von Metallatomen koordinierte Umgebung. Seltener findet man auch oktaedrische und quadratisch antiprismatische Koordination. - 191 - Das System Kobalt-Iridium-Bor 4.2 Das System Kobalt-Iridium-Bor 4.2.1 Die Phasenbreite des ternären Systems Kobalt-Iridium-Bor Das ternäre Borid vom Chromcarbid Cr23C6-Typ im System Kobalt-Iridium-Bor zeigte in neuen Untersuchungen eine erstaunlich große Gitterkonstante von a = 10,9433(14)Å mit einer Zusammensetzung von Co12,35Ir10,65B11,76 [162] . In Arbeiten von Rogl et al. [163] wurde das τ- Borid mit einer Gitterkonstante von 10,85(5)Å bei einer ungefähren Zusammensetzung von Co~15Ir~8B6 angegeben. Einkristalluntersuchungen an den Komplexboriden zeigten eine ungeordnete Verteilung der Metallatome. Die bisherigen Untersuchungen ließen keinen Rückschluss auf die Phasenbreite der τ-Phase im gegebenen Dreistoffsystem zu. 4.2.2 Präparative Untersuchung im System Kobalt-Iridium-Bor Die präparativen Arbeiten in diesem System dienten dem Zweck, den Bereich der Primärekristallisation der borreichen und borarmen Seite der mit τ gekennzeichneten Phase abzusuchen. Somit stand die Züchtung von Einkristallen guter Qualität für eine Röntgenstrukturanalyse im Vordergrund. Hierzu wurden die Elemente in stöchiometrischen Verhältnissen zu Tabletten kalt verpresst und in Korundtiegel bei einer Temperatur von 1450°C unter Argon-Schutzgas umgesetzt. Die Zusammensetzungen der Proben wurden mehrfach variiert, um die Phasenbreite des ternären Borids mit der Cr23C6-Struktur zu erfassen. Jede Probe wurde einer pulverdiffraktometrischen Analyse auf die vorhandenen Phasen hin unterzogen. Aus den gewonnenen Daten konnte die Gitterkonstante des entstandenen τ-Borids verfeinert und eine Zuordnung der Nebenprodukte durchgeführt werden. In der Tabelle 4.2.2.1 ist die Übersicht, die neu erworbenen Erkenntnisse über Primärkristallisation, Nebenprodukte, Zusammensetzung sowie die verfeinerte Gitterkonstante zusammengefasst. Es konnte eine erstaunliche Phasenbreite festgestellt werden. Die Spanne reicht von dem borarmen Ansatz mit der Zusammensetzung von Co20Ir3B6 und einer Gitterkonstante von a = 10,656(8)Å bis zum borreichen und zugleich iridiumreichen Ansatz mit der Zusammensetzung von Co13,5Ir9,5B15 Å und a = 11,043(7) Å. Temperaturprogramm: 300°C/h RT 10°C/h 200°C/h 1450°C 1000°C (10h halten) (2h halten) RT - 192 - Das System Kobalt-Iridium-Bor Tabelle 4.2.2.1: Proben im System Kobalt-Iridium-Bor und ihre pulverdiffraktometrische Charakterisierung. Fettgedruckt sind die Hauptprodukte. Graue Unterlegung unterscheidet den Borgehalt. (*nicht Indiziert) Nr Zusammen- . setzung Gehalt [At.-%] Gitterkonstante a [Å] weitere kristalline Phasen Co Ir B von Co23-xIrxB6+y 1 Co20Ir3B6 69,0 10,3 20,7 10,626(8) Co, CoB,Co2B 2 Co17Ir6B6 58,6 20,7 20,7 - CoIr, Co, Ir, CoB 3 Co5Ir18B15 13,1 47,4 39,5 - CoIr, IrB1,15, Co 4 Co9Ir14B15 23,7 36,8 39,5 11,043(7) 5 Co13,5Ir9,5B15 35,5 25,0 39,5 10,9797(21) 6 Co19Ir4B15 50,0 10,5 39,5 - 7 Co16Ir7B15 42,1 18,4 39,5 - IrB1,1, Ir IrB1,1 Co2B, CoB Co7B3, IrB1,1, Ir, Co23-xIrxB6+y* Nur in drei von sieben Proben konnte das τ-Borid nachgewiesen werden. Jedoch trat in diesen Ansätzen die Bildung des ternären Borids sehr ausgeprägt in Erscheinung. Eine phasenreine Darstellung war nicht möglich. In der borarmen Probe konnte die τ-Phase als Hauptprodukt neben Co, CoB und Co2B beobachtet werden. Bei den borreichen Ansätzen wurde das binäre IrB1,1 als kristallines Nebenprodukt beobachtet, dieses trat jedoch nur in Spuren auf. 4.2.3 EDX-Untersuchung Geeignete Kristalle aus den borarmen (Co20Ir3B6 Schmelzzusammensetzung) und borreichen Ansatz (Co13,5Ir9,5B15 Schmelzzusammensetzung) wurden einer qualitativen Elementaranalyse mittels energiedispersiver Röntgenemissionsspektroskopie unterzogen. Tabelle 4.2.3.1: Gemitteltes Ergebnis der Metallverhältnisse aus unterschiedlichen Messstellen an den Einkristallen aus den Ansätzen Co20Ir3B6 und Co13,5Ir9,5B15. Der Atomsoll-Wert richtet sich nach der Einwaage. Borarmer Ansatz Co20Ir3B6 (a) Borreicher Ansatz Co13,5Ir9,5B15 (b) Element Atomist [%] σ [%] Atomsoll [%]* Element Atomist [%] σ [%] Atomsoll [%]* Co L 87,9 0,5 87,0 Co L 58,1 0,4 58,7 Ir L 12,1 0,5 13,0 Ir L 41,9 0,4 41,3 - 193 - Das System Kobalt-Iridium-Bor Aus der EDX-Untersuchung (Tabelle 4.2.3.1 a) der borarmen Probe konnte ein Metallverhältnis von 87 At.-% Kobalt zu 13 At.-% Iridium bestimmt werden. Dieses Ergebnis konnte mit der röntgenographischen Untersuchung des isolierten Einkristalls in Einklang gebracht werden (Tabelle 4.2.4.1 und Tabelle 4.2.4.2). Auch die borreiche Probe zeigte ein sehr gute Übereinstimmung der Ergebnisse aus der EDX-Untersuchung 59 At.-% Kobalt zu 41 At.-% Iridium (Tabelle 4.2.3.1 b) mit der Verfeinerung der Einkristalluntersuchung (Tabelle 4.2.4.4 und Tabelle 4.2.4.5) 4.2.4 Röntgenographische Untersuchung an Einkristallen Aus der borarmen und der borreichen Proben konnten nach einer mechanischen Zertrümmerung im Wolframcarbid-Mörser geeignete Einkristalle für eine Einkristalluntersuchung isoliert werden. Die Einkristalle der τ-Boride zeigen keine Neigung, als regelmäßige optische, einheitliche Einkristalle zu kristallisieren. Vielmehr besitzen die Einkristalle der τ-Phase den Charakter unregelmäßige, metallisch glänzende Polyeder zubilden. Diese unangenehme Eigenschaft macht gelegentlich Probleme bei der qualitativen Elementaranalyse der Elemente, jedoch hat dieses keinen Einfluss auf die Verfeinerung. 4.2.4.1 Untersuchungen an borarmen Einkristallen Der isolierte Einkristall aus der borarmen Probe zeigte eine Gitterkonstante von a = 10,608(3) Å in der kubischen Raumgruppe F m 3 m. Die Größe der Gitterkonstante war schon ein gutes Indiz dafür, dass es sich bei diesem Einkristall um einen borarmen Einkristall mit sehr geringem Iridiumgehalte handelt. Für die Strukturanalyse lag ein Datensatz mit 136 unabhängigen Reflexen vor. Als Startmodell für die Strukturverfeinerung wurden die Lageparameter von Co20Sn3B6 verwendet, wobei die Position 8c (¼, ¼, ¼) mit Iridium besetzt wurde. Anschließend wurde die 8c-Lage mit einer Mischbesetzung aus Iridium und Kobalt verfeinert. Alle Metalllagen wurden auf eventuelle Misch- oder Unterbesetzungen überprüft, jedoch konnte keine signifikante Abweichung festgestellt werden. - 194 - Das System Kobalt-Iridium-Bor Tabelle 4.2.4.1: Angaben zur Strukturanalyse von Co20Ir3B6. Summenformel Co20Ir3B6 Kristallsystem kubisch Raumgruppe F m3m a = 10,6082(12) Gitterkonstante [Å] 3 Volumen der Elementarzelle [Å ] 1193,8(5) Zahl der Formeleinheiten 4 3 röntgenogr. Dichte [g/cm ] 10,127 F(000) 3204,0 Kristallform, -farbe metallisch glänzender Polyeder STOE IPDS II (Mo-Kα-Strahlung; λ = 0,71073 Diffraktometer Å Graphitmonochromator 0° ≤ ω ≤ 180° Rotation ψ = 0°, 111; ∆ω = 2° Belichtungszeit [sec.] 300 Messtemperatur [K] Raumtemperatur [293(2)] 0,03 × 0,03 × 0,03 Kristallgröße [mm] -1 Absorptionskoeffizient [mm ] 60,16 Messbereich [°] 6°<2θ<65° -16 ≤ h ≤ 16, -16 ≤ k ≤ 16, Indexbereich -16 ≤ l ≤ 16 Zahl der gemessenen Reflexe 6317 Zahl der unabh. Reflexe mit I > 0 135 I > σ2(I): 83 freie Parameter 14 Korrekturen Lorentz, Polarisation Absorptionskorrektur numerical / equivalent method (XSHAPE[68]) Strukturlösung Isotypie Cr23C6[180] Strukturverfeinerung SHELXTL-97[199] w-1 = σ2F02 + (0,0000 * P)2 + 0,00 * P Wichtungsschema P = (F02 + 2 FC2) / 3 0,6727 Goodness of fit 0,00044(2) Extinktionskorrektur - 3 Restelektronendichte [e /Å ]: R-Werte für I > 0: Alle Reflexe +1,23 / -1,56 - 0,26 e-/Å3 R1(F) = 0,0210 wR2(I) = 0,0269 R1(F) = 0,0459 - 195 - Das System Kobalt-Iridium-Bor Tabelle 4.2.4.2: Ortskoordinaten, Besetzungsfaktoren (in [%]) und Auslenkungsparameter von Co20Ir3B6. Atom Lage x y z sof Ueq Co/Ir 1 48h 0 0,66915(7) 0,16912(5) 88(1)/12*# 0,0021(2) Co/Ir 2 0,11955(7) 0,61955(7) 0,38045(7) 88(2)/12*# 0,0017(2) Co 3 32f 4a 0 ½ 0 100* 0,0128(11) Co 4/Ir 4 8c ¼ ¾ ¼ 66(3)/34*# 0,0044(4) B1 24e 0 0,7201(12) ½ 100* 0,0084(2) * bei wechselweiser Freigabe der Besetzungsfaktoren keine signifikante Restelektronendichte vorhanden # Besetzungsfaktoren und Temperaturfaktoren gekoppelt und fixiert Atom U11 U22 U33 U23 U13 U12 Co/Ir 1 0,0047(2) 0,0047(2) 0,0047(2) 0,0012(3) 0,0012(3) 0,0012(3) Co/Ir 2 0,0048(3) 0,0048(3) 0,008(5) 0 0 0,005(3) Tabelle 4.2.4.3: Ausgewählte Abstände und Winkeln in der Co20Ir3B6-Struktur. Anzahl der Benachbarte Atome Abstand [Å] Co/Ir 2 - B1 2,112(8) x3 Co/Ir 4 2,3946(13) x1 Co/Ir 2 2,5391(15) x3 Co/Ir 1 2,6284(7) x4 B1 2,120(8) x2 Co/Ir 1 2,428(2) x1 Co 3 2,5378(12) x1 Co/Ir 1 2,5378(12) x4 Co/Ir 2 2,6284(7) x4 Co 3- Co/Ir 2 2,5377(11) x 12 Co/Ir 4 - Co/Ir 2 2,3946(13) x4 Co/Ir 1 2,9168(5) x 12 Co/Ir 2 2,112(8) x4 Co/Ir 1 2,120(8) x4 Co/Ir 1- B1- Bindungen - 196 - Das System Kobalt-Iridium-Bor 4.2.4.2 Strukturdiskussion Der borarme Ansatz lieferte ein ternäres Borid mit der Zusammensetzung von Co20Ir3B6 (Cr23C6-Typ) in der Raumgruppe Fm 3 m mit einer Gitterkonstante von a = 10,6082(12) Å. Die Verteilung der Metallatome ist an dem analysierten τ-Borid erwartungsgemäß von klassischer Natur. Die Kobaltatome besetzen die Metalllagen 48h, 32f, 4a sowie partiell 8c mit 80(6) %. Die Punktlage 8c wird als τ-Stabilisator bezeichnet (Kapitel 0 Sonderverhalten in der τ-Phase) und stellt ein besonderes Verhalten in der Struktur dar. Die Position 8c ist vollständig besetzt. Somit sind die restlichen 20(6)% der kristallographischen Punktlage mit dem Edelmetall Iridium aufgefüllt. Ein Vergleich der Radien der beteiligten Metallatome liefert eine Erklärung für den Anstieg der Gitterkonstanten (rCo = 1,25 Å und rIr = 1,36 Å[190]). Die Punktlage 48h bildet den Kuboktaeder, hier sind die Kobaltatome 2,5378(12) Å voneinander entfernt. Bei einer Polyederbetrachtung befinden sich die Würfel mit einer Kantenlänge von 2,5364(15) Å bestehen ebenfalls aus Kobaltatomen auf 32f zwischen den Kuboktaeder. Die Ecken der Polyeder nähern sich bis auf 2,6286(7) Å aneinander an. Zwischen den Grundfläche des Kuboktaeders und der des Würfels befinden sich Boratome im Abstand von dCo1-Co2: 2,145(8) Å x4 und dB1-Co2: 2,087(7) Å x 4. Die Hohlräume in den Kuboktaedern sind ausschließlich von Kobaltatomen besetzt und erhalten auf dieser Lage (4a) eine zwölffache Koordination von den Kobaltatomen der Lage 48h im Abstand von 2,5377(11) Å. Bei der 8c Lage (Co :80(6)%/ Ir 20(6)%) sind die Atome von Kobalt aus der Lage 48h 2,9163(6) Å zwölffach und von weiteren Kobaltatomen mit 2,3969(13) Å von 32f vierfach umgeben. - 197 - Das System Kobalt-Iridium-Bor 4.2.4.3 Untersuchungen an borreichen Einkristallen An einem untersuchten Einkristall aus einem borreichen Ansatz, mit der Schmelzzusammensetzung Co13,5Ir9,5B15 entnommen wurde, konnte eine Gitterkonstante von a = 10,9393(13) Å ermittelt werden. Ein Vergleich der Gitterkonstanten mit den Einkristallen aus dem borarmen Ansatz (oben: 4.2.4.1 Untersuchungen an borarmen Einkristallen) zeigt einen Anstieg der Gitterkonstanten um ca. 1,7%. Die Aufweitung der Gitterparameter resultiert zum einem aus der partiellen Substitution von Iridium gegen Kobalt auf den Metalllagen und zum zweiten durch den Einbau von zusätzlichem Bor auf einer 32f-Lage. Von den 6353 beobachteten Reflexen blieben nach der Mittelung in der Laueklasse m 3 m ein Datensatz mit 151 unabhängigen Reflexen. Als Startmodell wurden die Parameter des borreichen Ni-τ-Borids mit der Zusammensetzung Ni20AlB14[167] verwendet. Die anschließende Verfeinerung der 20 freien Parameter konvergierte nach der Mischbesetzung ausgewählter Metalllagen und die Einführung von anisotropen Temperaturfaktoren für ausgesuchte Metalllagen sowie die Freigabe der Borpositionen zu Zuverlässigkeitswerten von R1(F) = 0,0189 und wR2(I) = 0,0249. Eine wechselweise Freigabe der Besetzungsfaktoren aller Metallpositionen zeigte keine signifikante Abweichung von der vollständigen Besetzung der Metalle. Bei der Verfeinerung der Borlage mit dem Zentrum auf 32f zeigte sich eine deutliche Unterbesetzung. Hier wurde die Position entsprechend dem Ergebnis der EDX-Untersuchung mit B4-Tetraedern sowie mit Kobaltatomen besetzt. Alle Einzelheiten der Strukturverfeinerung sowie die Atomkoordinaten und Besetzungsfaktoren, ausgewählte Abstände und benachbarte Atome sind den folgenden Tabellen zu entnehmen. - 198 - Das System Kobalt-Iridium-Bor Tabelle 4.2.4.4: Angaben zur Strukturanalyse von Co12.3Ir8.9B10.5. Summenformel Co12.3Ir8.9B10.5 Kristallsystem kubisch Raumgruppe F m3m Gitterkonstante [Å] a = 10,9393(13) Volumen der Elementarzelle [Å3] 1309,1(3) Zahl der Formeleinheiten 4 röntgenogr. Dichte [g/cm3] 13,968 F(000) 4604 Kristallform, -farbe metallisch glänzender Polyeder Diffraktometer Rotation STOE IPDS II (Mo-Kα-Strahlung; λ=0,71073 Å Graphitmonochromator 0° ≤ ω ≤ 180° ψ = 0°, 111; ∆ω = 2° Belichtungszeit [sec.] 300 Messtemperatur [K] Raumtemperatur [293(2)] Kristallgröße [mm] 0,03 × 0,03 × 0,03 Absorptionskoeffizient [mm-1] 117,08 Messbereich [°] 2°<2θ<65° 0 ≤ h ≤ 9, Indexbereich 0 ≤ k ≤ 11, 1 ≤ l ≤ 16 Zahl der gemessenen Reflexe 6353 Zahl der unabh. Reflexe mit I > 0 151 I > σ2(I): 125 freie Parameter 20 Korrekturen Lorentz, Polarisation Absorptionskorrektur numerical / equivalent method (XSHAPE[68]) Strukturlösung Isotypie Cr23C6[180] Strukturverfeinerung SHELXTL-97[199] Wichtungsschema w-1 = σ2F02 + (0,0093 * P)2 + 0,00 * P P = (F02 + 2 FC2) / 3 Goodness of fit 0,778 Extinktionskorrektur 0,00016(1) Restelektronendichte [e-/Å3]: +2,12 / -1,93; R-Werte für I > 0: Alle Reflexe 0,35 e-/Å3 R1(F) = 0,0189 wR2(I) = 0,0249 R1(F) = 0,0294 - 199 - Das System Kobalt-Iridium-Bor Tabelle 4.2.4.5: Ortskoordinaten, Besetzungsfaktoren (in [%]) und Auslenkungsparameter von Co12.3Ir8.9B10.5. Atom Co 2/Ir 2 Co 1/Ir 1 Lage x 0,16603(5) 48h y z ½ sof Ueq 0,16603(5) 76,0/24,0#* 0,0081(2) #* 0,0099(1) 0,87603(3) 0,87603(3) 0,87603(3) 22,6/77,4 Co 3 32f 4a 0 0,5 0 94,6(8) 0,00513(12) Co 4 8c ¼ ¼ ¼ 3,3(4) 0,0051(8) B1 24e 0 0,275(2) ½ 95(5) #* 0,0123(24) B2 32 f 0,302(1) 0,1974(13) 0,19747(13) 56(6) #* 0,0123(24) # # * bei wechselweiser Freigabe der Besetzungsfaktoren keine signifikante Restelektronendichte vorhanden # Besetzungsfaktoren und Temperaturfaktoren gekoppelt und fixiert Atom U11 U22 U33 U23 U13 U12 Co 1/Ir 1 0,0099(2) 0,0099(2) 0,0099(2) 0,0010(1) 0,0010(1) 0,0010(1) Co 2/Ir 2 0,0080(2) 0,0080(2) 0,0080(2) 0 0 0 Tabelle 4.2.4.6: Ausgewählte Abstände in der Co12.3Ir8.9B10.5-Struktur. Anzahl der Benachbarte Atome Abstand [Å] Co/Ir 1 B1 2,216(9) x3 B2 2,260(2) x3 Co 4 2,3879(6) x1 Co/Ir 2 2,7071(5) x3 B1 2,170(9) x2 B2 2,214(18) x2 Co 3 2,5685(8)) x1 Co/Ir 2 2,5685(8) x4 Co 3- Co/Ir 2 2,5685(8) x 12 Co 4 - Co/Ir 1 2,3879(6) x 12 Co/Ir 2 3,0282(5) x4 Co/Ir 2 2,170(9) x4 Co/Ir 1 2,216(9) x4 B2 1,63(4) x3 Co/Ir 2 2,214(18) x3 Co/Ir 1 2,260(2) x3 Co/Ir 2 B1- B2- Bindungen - 200 - Das System Kobalt-Iridium-Bor 4.2.5 Strukturdiskussion Der borreiche Ansatz mit der Schmelzzusammensetzung von Co13,5Ir9,5B15 lieferte ein borreiches τ-Boride mit der Zusammensetzung Co12,3Ir8,9B10,5. Das ternäre Borid kristallisiert in der Raumgruppe Fm 3 m mit einer Gitterkonstante von a = 10,9393(13) Å. B B (B-B) 1,63(4) Å (B-B) 1,63(4) Å B B Abbildung 4.2.1: Struktur des Co12,3Ir8,9B10,5 τ-Borids (oben l.) sowie das isolierte B4-Tetraeder (oben r. ) mit einem Bor-Bor-Abstand von 1,63(4) Å. (Unten l. und r.) Fouriersynthese: Schnitt durch die Atomebene in den B2-Einheiten des B4-Tetraeders. Aus der EDX Untersuchung zeigte sich ein Iridiumanteil von 41,9(4) At.-% im untersuchten Einkristall. Bei der Strukturverfeinerung konnte Iridium auf die Metalllagen 48h und 32f partiell und statistisch verteilt werden. Somit konnte die Position 48h, welche das Kuboktaeder bildet mit 24% Iridium und 76% Kobalt vollständig besetzt werden (dCo2/Ir2Co2/Ir2: 2,70711(5) Å). Das Zentrum des Kuboktaeders ist vollständig durch Kobaltatome besetzt (12 x dCo1/Ir1-Co3: 2,5685(8) Å). Auch die Punktlage 32f zeigt eine Mischbesetzung durch beide Übergangsmetalle mit einem Verhältnis von 22,6% Kobalt und 77,4% Iridium. Diese Punktlage ist vollständig besetzt und bildet den Würfel mit einer Kantenlänge von 2,5686(8) Å. Zwischen den Flächen der beiden beschriebenen Polyeder befindet sich das - 201 - Das System Kobalt-Iridium-Bor Boratom mit einer achtfachen quadratisch antiprismatischen Koordination von dB2-Co/Ir2 : 2,214(18) Å (x4) und dB2-Co/Ir1 : 2,260(2) Å (x4). Das Zentrum des Friauf-Polyeders ist nahezu unbesetzt. Hier befände sich das Kobaltatom in einer sechzehnfachen Koordination bestehend aus den vier Dreiecksflächen des Kuboktaeders 2,3879(6) Å und den Ecken der Würfel 3,0282(6) Å. In der Abbildung 4.2.1 (unten) ist die Differenz-Fouriersynthese des Bortetraeders dargestellt. Gezeigt ist nur der Ausschnitt entlang der c-Achse, der die Kante des B4-Tetraeders in zwei unterschiedlichen Ebenen beschreibt. Die Bor – Bor Abstände betragen in den B4-Einheiten jeweils 1,63(4) Å. Das Zentrum des Bortetraeders liegt genau auf der kristallographischen Punktlage 8c. - 202 - Das System Kobalt-Aluminium-Bor 4.3 Das System Kobalt-Gallium-Bor Die Studie von Komplexboriden im System Kobalt-Gallium-Bor brachte zwei τ-Boride mit der Zusammensetzung von Co20Ga3B6 (a = 10,5020 Å)[181] und Co21Ga2B6 (a = 10,5190 Å)[182] zum Vorschein. Außer den τ-Boriden sind keine weiteren ternären Verbindungen in dem Dreistoffsystem bekannt. Abbildung 4.3.1: Projektion der Liquidusfläche bei 800°C im System Kobalt-Gallium-Bor Neben den ternären Verbindungen existieren zahlreiche binäre Verbindungen, die binären Kobaltboride wurden bereits diskutiert. Zudem gibt es mit CoGa3 eine intermetallische Verbindung in der galliumreichen Ecke. 4.3.1 Präparative Untersuchung im System Kobalt-Gallium-Bor Die präparativen Untersuchungen im System Kobalt-Gallium-Bor dienten der Überprüfung des Homogenitätsbereiches sowie der Prüfung auf die Anwesenheit von borreichen τ-Boriden. Somit wurde die Zusammensetzung der Elemente Gallium, Kobalt und Bor mehrmals variiert, um Ansätze von borarm zu borreich darzustellen. Alle Probe wurde aus den Elementen dargestellt, indem die Pulver Kobalt und Bor in den unten angegeben Verhältnissen gut homogenisiert und mit Hilfe einer Handtablettenpresse in Tabletten verdichtet wurden. Das Stückchen Gallium, das auf die benötigte Menge zugeschnitten wurde, wurde auf die - 203 - Das System Kobalt-Gallium-Bor Tabletten platziert. Die Proben wurden in Korundtiegel bei einer Temperatur von 1450°C für 10 Stunden unter Ar-Schutzgas zur Reaktion gebracht. Die Proben wurden sehr langsam abgekühlt, um eine möglichst gute Kristallisation zu ermöglichen Alle erhaltenen Proben wurden pulverdiffraktometrisch auf die gebildeten Phasen hin analysiert und die Gitterkonstanten τ-Boride des verfeinert. Für die Umsetzung wurde folgendes Temperaturprogramm verwendet. Temperaturprogramm: 300°C/h RT 15°C/h 150°C/h 1450°C 1000°C (10h halten) (2h halten) RT Tabelle 4.3.1.1: Proben im System Kobalt-Gallium-Bor und ihre pulverdiffraktometrische Charakterisierung. Fettgedruckt sind die Hauptprodukte. Graue Unterlegung unterscheidet den Borgehalt. Nr Zusammen- . setzung Co 1 Co22Ga1B6 75,9 3,4 20,7 10,5041(4) - 2 Co20Ga3B6 69,0 10,3 20,7 10,4976(11) >> Co 3 Co17Ga6B6 58,6 10,7 20,7 - Co2B, CoGa, 4 Co13Ga10B6 44,8 34,5 20,7 - CoGa, CoB 5 Co20Ga3B12 57,1 8,6 34,3 6 Co18Ga5B12 51,4 14,3 34,3 - CoGa, CoB, Co2B 7 Co13Ga10B12 37,1 28,6 34,3 - CoGa3, CoB 8 Co20Ga3B15 52,6 7,9 39,5 - Co2B, CoGa 9 Co13Ga10B15 34,2 26,3 39,5 - CoB, Ga, Gehalt [At.-%] Ga B Gitterkonstante a [Å] weitere kristalline von Co23-xGaxB6+y Phasen 10,4978(6) >>Co2B In Übereinstimmung mit den Ergebnissen von Stadelmaier konnte nur eine τ-Phase beobachtet werden, die kein Hinweis dafür liefert, dass der Einbau von zusätzlichem Bor stattgefunden hat. Bei den borreichen Ansätzen konnten nur binäre Phasen beobachtet werden. Der Ansatz mit der Schmelzzusammensetzung von Co22Ga1B6 zeigte als einziger eine phasenreine Darstellung des τ-Borids mit einer Gittekonstante von 10,5041(4) Å. Ein Vergleich der Gitterkonstante mit der Literatur zeigt, dass es sich hier um eine gallium- und borarme τ-Phase handeln muss. - 204 - Das System Kobalt-Gallium-Bor 4.3.2 Härtemessung an Einkristallen Aus dem phasenreinen Ansatz (Tabelle 4.3.1.1/Nr.1) wurden einige Einkristalle geeigneter Größe entnommen und für eine Härteprüfung nach Vickers und Knoop präpariert. Hierzu wurden zwei Einkristalle in Epoxidharz eingebettet, anschließend angeschliffen und poliert. Auf den polierten Flächen wurden von jedem Kristall drei Abdrücke entnommen. Dabei konnte eine mittlere Härte von 840 HV 0,2/10 und 826 HK 0,2/10 ermittelt werden. Die ermittelte Härte nach Vickers und Knoop liegt in der Größenordnung, wie sie bereits für andere borarme τ-Boride erhalten wurde. 4.3.3 Röntgenographische Untersuchung an Einkristallen Aus dem Ansatz der Schmelzzusammensetzung Co22Ga1B6 konnte ein geeigneter Einkristall isoliert und mit dem IPDS II vermessen. Aus den beobachteten 8687 Reflexen wurden nach der Mittelung in der Laueklasse m 3 m und den üblichen Korrekturen ein Datensatz von 139 unabhängigen Reflexen erhalten. Als Startmodell wurden für die Verfeinerung die Atomkoordinaten der Cr23C6-Struktur verwendet, wobei die Metalllagen mit Kobaltatomen und die Punktlage 24e mit Bor besetzt wurde. Nach einer Verfeinerung der Position 8c durch eine Mischbesetzung, bestehen aus Gallium und Kobalt konvergierte die Strukturverfeinerung zu Zuverlässigkeitswerten von R1(F) = 0,0173 und wR2(I) = 0,0207. Bei der Überprüfung der Besetzungsfaktoren durch Freigabe der drei Metalllagen (48h, 32f und 4a) konnte keine signifikante Abweichung von der geordneten, vollständigen Besetzung beobachtet werden. Dabei wurden die Temperatur- und Besetzungsfaktoren der Position 8c gekoppelt und fixiert. Die Freigabe der Borlage zeigt im Rahmen der Messgenauigkeit konstante Werte. Weitere Einzelheiten zur Strukturanalyse sind in folgenden Tabellen angegeben. - 205 - Das System Kobalt-Aluminium-Bor Tabelle 4.3.3.1: Angaben zur Strukturanalyse von Co22,13Ga0,87B6. Summenformel Co21,9Ga1,1B6 Kristallsystem kubisch Raumgruppe F m3m a = 10,4872(12) Gitterkonstante [Å] 3 Volumen der Elementarzelle [Å ] 1152,7(3) Zahl der Formeleinheiten 4 röntgenogr. Dichte [g/cm3] 8,233 F(000) 2648,0 Kristallform, -farbe metallisch glänzender unregelmäßiger Polyeder Diffraktometer Rotation STOE IPDS II (Mo-Kα-Strahlung; λ=0,71073 Å Graphitmonochromator 0° ≤ ω ≤ 180° ψ = 0°, 111; ∆ω = 2° Belichtungszeit [sec.] 300 Messtemperatur [K] Raumtemperatur [293(2)] Kristallgröße [mm] 0,04 × 0,04 × 0,04 Absorptionskoeffizient [mm-1] 32,79 Messbereich [°] 6,7° < 2θ < 70° -16 ≤ h ≤ 16, Indexbereich -16 ≤ k ≤ 16, -16 ≤ l ≤ 16 Zahl der gemessenen Reflexe 8687 Zahl der unabh. Reflexe mit I > 0 139 I > σ2(I): 90 freie Parameter 14 Korrekturen Lorentz, Polarisation Absorptionskorrektur numerical / equivalent method (XSHAPE[183]) Strukturlösung Isotypie Cr23C6[184] Strukturverfeinerung SHELXTL-97[199] Wichtungsschema w-1 = σ2F02 + (0,0000 * P)2 + 0,00 * P P = (F02 + 2 FC2) / 3 Goodness of fit 0,457 Extinktionskorrektur 0,00060(2) Restelektronendichte [e-/Å3]: +1,08 / -0,77; R-Werte für I > 0: Alle Reflexe 0,15 e-/Å3 R1(F) = 0,0173 wR2(I) = 0,0207 R1(F) = 0,0400 - 206 - Das System Kobalt-Gallium-Bor Tabelle 4.3.3.2: Ortskoordinaten, Besetzungsfaktoren (in [%]) und Auslenkungsparameter von Co21,9Ga1,1B6. Atom Lage x y z sof Ueq Co 1 48h 0,16935(5) 0,16935(5) 0 100* 0,00596(13) Co 2 0,61843(5) 0,61843(5) 0,61843(5) 100* 0,0066(2) Co3/Ga3 32f 8c ¼ ¼ ¼ 43,7/56,3(13)# 0,00703(34) Co4 4a 0 0 0 100* 0,0064(6) B1 24e 0,5 0,5 0,7232(8) 100* 0,0034(12) * bei wechselweiser Freigabe der Besetzungsfaktoren keine signifikante Restelektronendichte vorhanden # Besetzungsfaktoren und Temperaturfaktoren gekoppelt und fixiert Atom U11 U22 U33 U23 U13 U12 Co 1 0,0052(2) 0,0052(2) 0,0052(2) 0 0 0,0005(25) Co 2 0,0065(2) 0,0065(2) 0,0065(2) 0,0011(2) 0,0011(2) 0,0011(2) Co/Ga 3 0,0070(3) 0,0070(3) 0,0070(3) 0 0 0 Co 4 0,0064(6) 0,0064(6) 0,0064(6) 0 0 0 Tabelle 4.3.3.3: Ausgewählte Abstände in der Co21,9Ga1,1B6-Struktur. Anzahl der Benachbarte Atome Abstand [Å] Co 2 - B1 2,086(7) x3 Co/Ga 3 2,3969(13) x1 Co 2 2,5364(15) x3 Co 1 2,6286(7) x4 B1 2,1447(8) x2 Co 1 2,426(2) x1 Co 3 2,5377(11) x1 Co 1 2,5378(12) x4 Co 2 2,6286(7) x4 Co 3- Co 2 2,5377(11) x 12 Co/Ga 3 - Co 2 2,3969(13) x4 Co 1 2,9163(6) x8 Co 2 2,087(7) x4 Co 1 2,145(8) x4 Co 1- B1- Bindungen - 207 - Das System Kobalt-Gallium-Bor 4.3.4 Strukturdiskussion Co21,9Ga1,1B6 kristallisiert im Cr23C6-Typ in der Raumgruppe Fm 3 m mit einer Gitterkonstanten von a = 10,4872(12) Å. Co21,9Ga1,1B6 wurde zum ersten Mal in kristalliner Form dargestellt. Mit der Strukturanalyse an Einkristallen war es möglich, die exakte Lokalisierung des Galliums in der Struktur festzumachen. Position 8c ist durch eine Mischbesetzung gezeichnet, hier befinden sich Gallium- (44 %) und Kobaltatome (56 %) und zentrieren statistisch das vierfach überkappten Friauf-Polyeder. Die vier Dreiecksflächen der Kuboktaeder (geformt von Co 2) befinden sich im Abstand von 2,9163(6) Å. Weitere vier Ecken der Würfel (Co 1) sind an der Koordination am Zentrum des Friaufpolyeders (CN = 16) im Abstand von 2,3969(13) Å beteiligt. Die Würfel werden ausschließlich von Kobaltatomen auf der kristallographischen Lage 32f gebildet. Jeder dieser Würfel besitzt eine Kantenlänge von 2,5364(15) Å. Die Kuboktaeder bestehen aus zwölf Kobaltatomen auf der Position 48h. Jedes der Kuboktaeder besitzt ein weiteres Kobaltatom im Zentrum auf der Lage 4a. Die Kobaltatome auf dieser Position erfahren eine Zwölffachkoordination in einem Abstand von 2,5377(11) Å. Die Boratome besetzten die Punktlage 24e vollständig. Die quadratisch antiprismatische Koordination wird durch acht Kobaltatome verursacht. Hier sind je vier Kobaltatome aus dem Würfel im Abstand von 2,087(7) Å sowie vier weitere Kobaltatome aus der Grundfläche des Kuboktaeders (2,145(8) Å). - 208 - Das System Kobalt-Aluminium-Bor 4.4 Das System Kobalt-Aluminium-Bor Das ternäre System aus Kobalt-Aluminium-Bor wurde bereits sehr detailliert auf die Bildung der τ-Boride hin untersucht. Es konnte ein Homogenitätsbereich für die Existenz der borarmen τ-Phase erfasst und mit der Zusammensetzung Co23-xAlxB6 (x = 2,6-3,0) beschrieben werden[162,167]. Dabei wurden bei der Untersuchung immer wieder borreiche Verbindungen des Typs Co20-xAlxB6+y postuliert. Systematische Untersuchungen von Kuz’ma et al.[185] sowie der Beitrag von Stadelmaier[165] lieferten wertvolle Informationen und ergaben das ternäre Phasendiagramm, das in der Abbildung 4.4.1 und 4.1.2 dargestellt ist. Abbildung 4.4.1: Phasendiagramm im System Kobalt-Aluminium-Bor: Projektion der Liquidusfläche. Abbildung 4.4.2: Phasendiagramm im System Kobalt-Aluminium-Bor: isothermer Schnitt bei 800°C. Abbildung 4.4.2 zeigt einen isothermen Schnitt bei 800°C. Der Bereich für eine Kristallisation der mit τ bezeichneten Phase ist sehr schmal. Hier existiert ein Phasengleichgewicht von CoAl (β-Phase) und Co2B, sowie Co (α-Phase). Die τ-Boride sind die einzigen ternären Verbindungen die in dem Dreistoffsystem beobachtet wurden. Untersuchungen mit borreichen Einwaagen sollen nun zur Klärung führen, ob ein zusätzlicher Einbau von Bor in das τ- Borid möglich ist, wie es schon im System bestehend aus Nickel, Aluminium und Bor beobachtet wurde. - 209 - Das System Kobalt-Aluminium-Bor 4.4.1 Präparative Untersuchung im System Kobalt-Aluminium-Bor In nahezu stöchiometrischen Einwaagen wurden die Elemente Kobalt, Aluminium und Bor in einem Achatmörser innig verrieben und kalt verpresst. Die Umsetzung wurde bei einer Temperatur von 1250°C (10h) durchgeführt, unter sehr langsamen Abkühlraten von 15°C pro Stunde wurde eine gute Kristallisation gewährleistet. Eine pulverdiffraktometrische Charakterisierung aller Proben konnte nur in zwei Ansätzen die mit τ bezeichnete Phase eindeutig nachweisen. Zwar konnten Spuren der τ-Phase in jedem Ansatz beobachtet werden, doch dominierten die binären Phasen auf Kosten der ternären Verbindung. Eine phasenreine Darstellung der τ-Boride ist bei den gegebenen Bedingungen fehlgeschlagen. Temperaturprogramm: 300°C/h 15°C/h RT 150°C/h 1250°C 800°C (10h halten) (2h halten) RT Tabelle 4.4.1.1: Proben im System Kobalt-Aluminium-Bor und ihre pulverdiffraktometrische Charakterisierung. Fettgedruckt sind die Hauptprodukte. Graue Unterlegung unterscheidet den # Borgehalt. ( nicht Indiziert) Nr Zusammen- Gitterkonstante a [Å] weitere kristalline . setzung Co Al B von Co23-xAlxB6+y Phasen 2 Co20Al3B8 64,5 9,7 25,8 - 3 Co10AlB8 52,6 5,3 42,1 - 4 Co20Al3B12 57,1 8,6 34,3 - 5 Co12AlB12 48,0 4,0 48,0 - 6 Co20Al3B12 57,1 8,6 34,3 7 Co12Al10C14 33,3 27,8 38,9 8 Co20Al3C14 Gehalt [At.-%] 54,1 8,1 37,8 a = 10,5108(15); V=1161,2(3) * a = 10,439(7); V=1137,4(12) - Co2B, CoB >>Co20Al3B6#, Co2B, CoB >>Co20Al3B6#, Co2B, CoB >>Co20Al3B6, Co2B >, Co Co20Al3B6, Co2B AlCo (Co20-xAlxC6)* Co3AlC, Co20Al3B6#, Co - 210 - Das System Kobalt-Aluminium-Bor Die verfeinerten Gitterkonstanten liegen in guter Übereinstimmung mit der in der Literatur bekannten Phasen der borarmen τ-Boride. Durch die Versuchsserie mit einem deutlichen Überschuss an Bor, konnte deutlich gezeigt werden, dass es keine Anzeichen für eine borreiche τ-Phase im Dreistoffsystem Kobalt-Aluminium-Bor gibt. 4.4.2 Röntgenographische Untersuchung an Einkristallen Aus dem Ansatz der Schmelzzusammensetzung Co20Al3B12 konnten ein geeigneter Einkristall isoliert und mit dem IPDS II vermessen werden. Der metallisch glänzende, unregelmäßige Polyeder liefert 6512 Reflexe. Die erstaunlich kleine Gitterkonstante von a = 10,4817(12) Å deutete schon auf einen sehr geringen Anteil an Aluminium in der Probe hin. Nach den üblichen Korrekturen konnte ein Datensatz von 112 unabhängigen Reflexen erhalten werden. Die kristallographische Lage 8c wurde vollständig mit Aluminium besetzt. Die Position 4a wurde mit einer Mischbesetzung von Kobalt und Aluminium im Verhältnis 92% Co zu 8% Al verfeinert. Die restlichen Atomlagen wurden vollständig mit Kobaltatomen besetzt. Bei der Freigabe der Besetzungsfaktoren konnte keine signifikante Abweichung von der vollständigen Besetzung beobachtet werden. Nach einer Einführung von anisotropen Temperaturfaktoren konnten Zuverlässigkeitswert von R1(F) = 0,0152 und wR2(I) = 0,0169 erhalten werde. Die Restelektronendichte lag in einem plausiblen Bereich. Weitere Angaben und Einzelheiten der Strukturanalyse eines Einkristalls mit der Zusammensetzung Co20,1Al2,1B6 sind in den folgenden Tabellen aufgeführt. - 211 - Das System Kobalt-Aluminium-Bor Tabelle 4.4.2.1: Angaben zur Strukturanalyse von Co20,9Al2,1B6. Summenformel Co20,9Al2,1B6 Kristallsystem kubisch Raumgruppe Fm 3 m a = 10,4817(12) Gitterkonstante [Å] 3 Volumen der Elementarzelle [Å ] 1151,6(3) Zahl der Formeleinheiten 4 röntgenogr. Dichte [g/cm3] 7,810 F(000) 2487,9 Kristallform, -farbe metallisch glänzendes, unregelmäßige Polyeder Diffraktometer Rotation STOE IPDS II (Mo-Kα-Strahlung; λ=0,71073 Å Graphitmonochromator 0° ≤ ω ≤ 180° ψ = 0°, 111; ∆ω = 2° Belichtungszeit [sec.] 300 Messtemperatur [K] Raumtemperatur [293(2)] Kristallgröße [mm] 0,04 × 0,04 × 0,04 Absorptionskoeffizient [mm-1] 29,33 Messbereich [°] 6,7° < 2θ < 70° -16 ≤ h ≤ 16, Indexbereich -16 ≤ k ≤ 16, -16 ≤ l ≤ 16 Zahl der gemessenen Reflexe 6512 Zahl der unabh. Reflexe mit I > 0 112 I > σ2(I): 73 freie Parameter 15 Korrekturen Lorentz, Polarisation Absorptionskorrektur numerical / equivalent method (XSHAPE[186]) Strukturlösung Isotypie Cr23C6[187] Strukturverfeinerung SHELXTL-97[199] Wichtungsschema w-1 = σ2F02 + (0,0000 * P)2 + 0,00 * P P = (F02 + 2 FC2) / 3 Goodness of fit 0,440 Extinktionskorrektur 0,00101(3) Restelektronendichte [e-/Å3]: +0,62 / -0,87; R-Werte für I > 0: Alle Reflexe 0,14 e-/Å3 R1(F) = 0,0152 wR2(I) = 0,0169 R1(F) = 0,0311 - 212 - Das System Kobalt-Aluminium-Bor Tabelle 4.4.2.2: Ortskoordinaten, Besetzungsfaktoren (in [%]) und Auslenkungsparameter von Co20,9Al2,1B6. Atom Lage x y z sof Ueq Co 1 48h 0,16975(6) 0,16975(6) 0 100* 0,0065(2) Co 2 0,11723(5) 0,38277(5) 0,11723(5) 100* 0,0055(2) Al 3 32f 8c ¼ ¼ ¼ 100* 0,0037(9) Co/Al 4 4a 0 0 0 95(2)/5*# 0,0119(8) B1 24e 0,2732(9) 0 0 100* 0,0069(13) * bei wechselweiser Freigabe der Besetzungsfaktoren keine signifikante Restelektronendichte vorhanden # Besetzungsfaktoren und Temperaturfaktoren gekoppelt und fixiert Atom U11 U22 U33 U23 U13 U12 Co 1 0,0057(3) 0,0057(3) 0,0057(3) 0 0 0,0004(3) Co 2 0,0055(2) 0,0055(2) 0,0055(2) 0,003(2) 0,003(2) 0,003(2) Al 3 0,0039(9) 0,0039(9) 0,0039(9) 0 0 0 Co/Al 4 0,0119(8) 0,0119(8) 0,0119(8) 0 0 0 Tabelle 4.4.2.3: Ausgewählte Abstände in der Co20,9Al2,1B6-Struktur. Anzahl der Benachbarte Atome Abstand [Å] Co 2 - B1 2,084(6) x3 Al 3 2,4104(9) x1 Co 2 2,4576(10) x3 Co 1 2,6073(6) x4 B1 2,0834(5) x2 Co 1 2,379(2) x1 Al 3 2,5163(9) x1 Co 1 2,5163(9) x4 Co 2 2,6073(6) x4 Co/Al 4 - Co 2 2,5163(9) x 12 Al 3 - Co 2 2,4104(9) x4 Co 1 2,8778(5) x8 Co 2 2,083(6) x4 Co 1 2,084(5) x4 Co 1 - B1- Bindungen - 213 - Das System Kobalt-Aluminium-Bor 4.4.3 Strukturdiskussion Co20,9Al2,1B6 kristallisiert im Cr23C6-Typ in der Raumgruppe Fm 3 m mit einer Gitterkonstanten von a = 10,4872(12) Å. Die Gitterkonstante von 10,4872(12) Å liegt etwas über der des binären τ-Borids mit der Zusammensetzung von Co23B6[162]. Die Aufweitung der Zelle ist durch den zusätzlichen Einbau von Aluminium auf der Position 8c und 4a zu erklären. Das Zentrum des FriaufPolyeders ist vollständig durch Aluminium besetzt. Bei der Punktlage 4a konnte eine Mischbesetzung durch die beteiligen Metalle vernommen werden, somit befinden sich auf dieser Position 5(2) % Aluminium statistisch neben Kobalt verteilt. Der Kuboktaeder wird ausschließlich von Kobaltatomen geformt, die Kantenlängen des Kuboktaeders betragen 2,4576(10) Å. Das Zentrum des Kuboktaeder ist mit Kobalt und Aluminium statistisch Mischbesetzung dCo1-Co/Al4: 2,5163(9) Å. Die Würfel mit einer Kantenlänge von 2,379(2) Å bestehen ebenfalls nur aus Kobaltatomen. Der Platz zwischen dem Kuboktaeder und Würfel wird durch das Boratom vollständig besetzt. Das Boratom befindet sich fast im Zentrum des quadratischen Antiprismas und erfährt somit ein Koordination von acht Kobaltatomen (dCo1-B1 : 2,084(5) Å x 4) und (dCo2-B1 : 2,083(6) Å x 4). - 214 - 4.5 Das System Kobalt-Zinn-Bor 4.5.1 Der kobaltreiche Bereich des ternären Systems Kobalt-Zinn-Bor Die Abbildung 4.5.1 zeigt eine Schmelzflächenprojektion des Dreistoffsystem KobaltZinn-Bor. Dargestellt wurde des Phasendiagramm nach metallographischen Untersuchungen von Stadelmaier et al.[188]. Die einzelnen Felder sind nach den primär kristallisierenden Phasen bezeichnet und sind durch Kurven doppelter Sättigung der Schmelze gegeneinander begrenzt. Somit schmilzt das τ-Borid mit einem Maximum bei der Zusammensetzung Co70Sn7B23 (für die Struktur bezogene Formeleinheit Co21Sn2B6) kongruent und kristallisiert in einem recht großen Bereich im Phasendiagramm primär aus der Schmelze. Bei 800°C besteht ein Phasengleichgewicht mit den Verbindungen Co (α), Co2B, Co3Sn2 (γ) und CoSn. Einen Unterschied des vorliegenden Systems zu ähnlichen Borsystemen zeigt das Auftreten einer Mischungslücke in der Schmelze. (diese Fläche ist mit einem S im Phasendiagramm gekenn-zeichnet). Abbildung 4.5.1: Kobaltreicher Bereich im Phasendiagramm Kobalt-Zinn-Bor[188]. Projektion der Liquidusfläche bei 800°C. 4.5.2 Präparative Untersuchung im System Kobalt-Zinn-Bor Ziel der Synthese neben der Überprüfung des Homogenitätsbereiches der τ-Phase mit die Züchtung von Einkristallen, die geeignet sind für eine röntgenographische Untersuchung. Die Elemente wurden in entsprechenden nahezu stöchiometrischen Verhältnissen homogenisiert und kalt verpresst. Die Presslinge wurden in Korundtiegeln und in einer Argon-Schutzgasatmosphäre bei 1450°C bei einer Reaktionszeit von einer Stunde umgesetzt. Um eine gute Kristallisation zu gewährleisten wurde die Abkühlrate auf 15 K/h gewählt. - 215 - Das System Kobalt-Zinn-Bor Nach Erreichung der Temperatur von 900°C wurde mit 300 K/h auf Raumtemperatur gekühlt. Die Überprüfung der Phasenbreite erfolgte röntgenographisch am Pulver. Die Existenz der τPhase konnte nur für borarme Ansätze festgestellt werden. Eine phasenreine Darstellung der mit τ gekennzeichneten Verbindung konnte nicht erfolgen. Tabelle 4.5.2.1 fasst die gewonnenen Ergebnisse aus der pulverdiffraktometrischen Charakterisierung der Proben zusammen und gibt die weiteren kristallinen Phasen an, die neben dem τ-Borid beobachtet wurden. Temperaturprogramm: 300°C/h 15°C/h RT 1450°C 300°C/h 900°C (10h halten) RT (2h halten) Tabelle 4.5.2.1: Proben im System Kobalt-Zinn-Bor und ihre pulverdiffraktometrische Charakterisierung. Fettgedruckt sind die Hauptprodukte. Graue Unterlegung unterscheidet den Borgehalt. Nr Zusammen- . setzung Co 1 Co20Sn3B6 88,3 9,0 2,7 10,593(4) >>Co3Sn2 2 Co21Sn2B6 72,4 6,9 20,7 10,594(11) <CoSn2 3 Co22SnB6 90,6 4,9 4,5 10,597(19) >Co 4 Co13Sn10B6 50,1 45,6 4,3 - Co2B, CoSn, Sn 5 Co15Sn8B12 42,9 22,9 34,2 - Co2B, CoB, CoSn, Sn 6 Co20Sn3B12 57,1 8,6 34,3 - Co2B, CoSn 7 Co22Sn1B12 62,9 2,9 34,3 - Co2B, CoSn, Co 8 Co20Sn3,5B12 56,3 9,9 33,8 - Co2B, CoSn 9 Co13Sn10B12 37,1 28,6 34,3 - CoB, CoSn2 Gehalt [At.-%] Sn B Gitterkonstante a [Å] weitere kristalline von Co23-xSnxB6+y Phasen Bei der Zusammensetzung Co23-xSnxB6 konnte ein Homogenitätsbereich mit x=1-3 Anteilen Zinn festgestellt werden. Die borreichen Ansätze zeigten keinen Hinweis auf die Bildung der τ-Phase. Hier zeigte sich eine starke Dominanz der binären Verbindung Co2B neben geringen Anteilen der übrigen binären Phasen des Dreistoffsystems. Bei dem Ansatz Co20Sn3B6 zeigte das τ-Borid mit sehr geringen Anteilen von Co3Sn2 ein nahezu phasenreines Pulverdiffraktogramm. Das mit Hilfe monochromatischer Mo-Kα1-Strahlung erhaltene Pulverdiffraktogramm und die Zuordnung aller Reflexe ist in der Abbildung 4.5.2 dargestellt. Das erhaltene τ-Borid zeigt eine Gitterkonstante von 10,593(4) Å. Diese befindet sich in guter - 216 - Das System Kobalt-Zinn-Bor Übereinstimmung mit der verfeinerten Gitterkonstanten, die von Stadelmaier et al. am Pulver beobachtet wurde (a = 10,611 Å)[188]. 300 290 280 270 260 250 240 230 220 210 200 Lin (Counts) 190 180 170 160 150 140 130 120 110 100 90 80 70 60 50 40 30 20 10 0 4 10 20 30 Abbildung 4.5.2: Pulverdiffraktogramm der Probe mit der Zusammensetzung Co22Sn1B6. Die blau markierten Reflexe konnten als Co3Sn2 identifiziert werden. 4.5.3 EDX-Untersuchung Die energiedispersive Röntgenemmissionsspektroskopie (EDX) soll bei der genauen Bestimmung des Metallverhältnisses behilflich sein und die aus dem Röntgenbeugungsexperiment gewonnenen Daten ergänzen. Die Strukturverfeinerung zeigt ein Metallverhältnis von 93,2 At.-% Kobalt zu 6,8 At.-% Zinn. Die Einkristalle der τ-Boride zeigen keinerlei Neigung als definierte Polyeder zu kristallisieren, vielmehr sind es metallische Scherben mit geringer Symmetrie (Abbildung 4.5.3). Der zu untersuchte Einkristall wurde an mehreren Stellen vermessen. Die EDXUntersuchung zeigt eine gute Übereinstimmung mit den erhaltenen Daten aus der Strukturverfeinerung. Tabelle 4.5.3.1: Verhältnis der Metallatome in der Probe Co21Sn2B6. Element Gewichts. [%] Atom [%] σ [%] Co K 87,3 93,3 0,06 Sn L 12,7 6,7 0,06 - 217 - Das System Kobalt-Zinn-Bor Abbildung 4.5.3: REM-Aufnahme der Probe Co21Sn2B6 Abbildung 4.5.4: Messspektrum der Probe Co21Sn2B6. Detektiert wurde Kobalt und Zinn, das Verhältnis der Metallatome ist in der Tabelle 4.5.3.1 angegeben 4.5.4 Röntgenographische Untersuchung an Einkristallen Die Schmelzperle aus einem zinnarmen Ansatz der Zusammensetzung Co20Sn3B6 wurde durch mechanische Einwirkung zertrümmert. Aus den kleinen, metallisch glänzenden Bruchstücken wurde ein unregelmäßiger Splitter für eine Strukturanalyse isoliert. Als Startmodell für die Verfeinerung wurden die Atomkoordinaten von Cr23C6 verwendet. Die Metalllagen 48h, 32f, 4a, wurden vollständig mit Kobaltatomen besetzt. Bei der kristallographischen Position 8c wurden die Temperaturfaktoren gekoppelt und mit einer Mischbesetzung bestehend aus 79(6)% Zinn und 21(6)% Kobalt verfeinert. Die Nichtmetalllage wurde vollständig mit Bor besetzt. Bei einer wechselweisen Freigabe der Besetzungsfaktoren konnte keine signifikante Abweichung von der vollständigen Besetzung beobachtet werden. Nach Einführung anisotroper Temperaturfaktoren konverierte die Verfeinerung der 15 feien Parameter zu Zuverlässigkeitswerten von R1(F)=0,0147 und wR2(I)=0,0245. Es konnte kein Einbau von zusätzlichem Bor festgestellt werden. Bei dem analogen System mit Nickel als Hauptkomponente konnte ein höhere Borgehalt beobachtet werden (Ni20,9Sn1,9B7,1 a = 10,5958(3) Å[167]). - 218 - Tabelle 4.5.4.1: Angaben zur Strukturanalyse von Co21,43Sn1,57B6. Co21,43Sn1,57B6 Summenformel Kristallsystem kubisch Raumgruppe F m3m Gitterkonstante [Å] 10,5553(12) Volumen der Elementarzelle [Å3] 1176,01(2) Zahl der Formeleinheiten 4 3 röntgenogr. Dichte [g/cm ] 8,553 F(000) 2748,9 Kristallform, -farbe metallisch glänzende, unregelmäßige Splitter STOE IPDS II (Mo-Kα-Strahlung; λ=0,71073 Å Diffraktometer Graphitmonochromator 0° ≤ ω ≤ 180° Rotation ψ = 0°, 111; ∆ω = 2° Belichtungszeit [sec.] 300 Messtemperatur [K] Raumtemperatur [293(2)] 0,02 × 0,03 × 0,09 Kristallgröße [mm] -1 Absorptionskoeffizient [mm ] 32,51 Messbereich [°] 6,7° < 2θ < 70° -16 ≤ h ≤ 16, -16 ≤ k ≤ 16, Indexbereich -16 ≤ l ≤ 16 Zahl der gemessenen Reflexe 8623 Zahl der unabh. Reflexe mit I > 0 160 I > σ2(I): 123 freie Parameter 15 Korrekturen Lorentz, Polarisation Absorptionskorrektur numerical / equivalent method (XSHAPE[68]) Strukturlösung Isotypie Cr23C6[189] Strukturverfeinerung SHELXTL-97[199] w-1 = σ2F02 + (0,0109 * P)2 + 0,00 * P Wichtungsschema P = (F02 + 2 FC2) / 3 0,727 Goodness of fit 0,00168(7) Extinktionskorrektur - 3 Restelektronendichte [e /Å ]: R-Werte für I > 0: Alle Reflexe +0,67 / -0,72; 0,15 e-/Å3 R1(F) = 0,0147 wR2(I) = 0,0245 R1(F) = 0,0244 - 219 - Das System Kobalt-Zinn-Bor Tabelle 4.5.4.2: Ortskoordinaten, Besetzungsfaktoren (in [%]) und Auslenkungsparameter von Co22Sn2B6. Atom Lage x y z Sof Ueq Co 1 48h 0 0,16885(3) 0,16885(3) 100* 0,0055(2) Co 2 32f 4a 0,38403(4) 0,38403(4) 0,38403(4) 100* 0,0043(2) 0 0 0 100* Co 3 Co/Sn 4 8c B 24e ¼ ¼ ¼ 21(6)/79 0 ½ 0,22558(6) 100* 0,0049(4) #* 0,0043(7) 0,0051(9) * bei wechselweiser Freigabe der Besetzungsfaktoren keine signifikante Restelektronendichte vorhanden # Besetzungsfaktoren und Temperaturfaktoren gekoppelt und fixiert Atom U11 U22 U33 U23 U13 U12 Co 1 0,0055(2) 0,0055(2) 0,0055(2) 0,0019(1) 0,0019(1) 0,0019(1) Co 2 0,0054(2) 0,0037(2) 0,0037(2) 0,0001(1) 0 0 Co 3 0,0049(4) 0,0049(4) 0,0049(4) 0 0 0 Die folgende Tabelle zeigt benachbarte Atome, ihren charakteristischen Abstand und die Koordinationszahl. Tabelle 4.5.4.3: Ausgewählte Abstände in der Co21,43Sn1,57B6-Struktur. Anzahl der Benachbarte Atome Abstand [Å] Co 1 - B 2,082(4) x3 Co 1 2,4488(9) x3 Co 2 2,6398(5) x4 Co/Sn4 2,4499(5) x1 B 2,102(4) x2 Co 2 2,423(1) x1 Co 3 2,5203(6) x4 Co 2 2,5203(5) x4 Co 1 2,6398(5) x4 Co 3- Co 2 2,5203(6) x 12 Co/Sn 4 - Co 1 2,4499(8) x4 Co 2 2,9037(4) x8 Co 1 2,082(4) x4 Co 2 2,102(4) x4 Co 2 - B- Bindungen - 220 - Das System Kobalt-Zinn-Bor 4.5.5 Strukturdiskussion Co21,43Sn1,57B6 kristallisiert im Cr23C6-Typ in der Raumgruppe Fm 3 m mit einer Gitterkonstanten von a = 10,5553(12)Å. Dieses τ-Borid zeigt wieder mal das klassische Verhalten und bildet die Mischbesetzung auf 8c im Zentrum des Friauf-Polyeders. Mit einer sechzehnfachen Koordination aus Kobaltatomen. Die Position ist überwiegend mit Zinn (79(6) %) besetzt, dieses erklärt die Aufweitung der Gitterkonstante (Radienvergleich rCo = 1,25 Å vs. rSn = 1,41 Å)[190]. Alle weiteren Metallpositionen sind ausschließlich von Kobalt besetzt. Der Würfel besitzt eine Kantenlänge von 2,423(1) Å. Der Kuboktaeder sowie sein Zentrum zeigen eine vollständige Besetzung durch Kobaltatome (12 x dCo3-Co2: 2,5203(6) Å). Position 24e ist vollständig durch Bor in der achtfachen Koordinationssphäre fixiert (4 x d BCo1: 2,082(4) Å und 4 x dB-Co2: 2,102(4) Å). - 221 - 4.6 Das System Kobalt-Titan-Bor 4.6.1 Präparative Untersuchung im System Kobalt-Titan-Bor Bei der Überprüfung des Homogenitätsbereiches der τ Phase wurde das Metallverhältnis wie auch der Borgehalte verändert (Tabelle 4.6.1.1), die Synthese erfolgte aus den Elementen. Dazu wurden die kaltgepressten Proben bei einer Temperatur von 1450°C aufgeschmolzen und für zehn Stunden unter Argon-Schutzgasatmosphäre zur Reaktion gebracht. In der Untersuchung von Stadelmaiers et al.[191] im System Kobalt-Titan-Bor wurde der τ-Phase mir einer Gitterkonstantanten mir der Phasenbreite von a = 10.508 Å bis 10.552 Å berichtet. Dieses war ein Indiz für einen eventuell zusätzlichen Einbau von Bor. Die Untersuchung diente dem Ziel, Einkristalle mit borarmen und borreichen Gehalt zu erhalten und den Grund für die Gitteraufweitung zu klären. Tabelle 4.6.1.1: Proben im System Kobalt-Titan-Bor und ihre pulverdiffraktometrische Charakterisierung. Fettgedruckt sind die Hauptprodukte. Graue unterlegung unterscheidet den Borgehalt. (*nicht Indiziert). Nr. Zusammen- Gitterkonstante a [Å] weitere kristalline B von Co23-xTixB6+y Phasen Gehalt [At.-%] setzung Co Ti 1 Co22Ti1B6 75,9 3,4 20,7 10,532(5) Spuren Ti2Co 2 Co21Ti2B6 72,4 6,9 20,7 10,537(5) Spuren Ti2Co 3 Co20Ti3B6 69,0 10,3 20,7 10,529(6) > Co 4 Co17Ti 6B6 58,6 20,7 20,7 - 5 Co13Ti10B6 44,8 34,5 20,7 - 6 Co10Ti13B6 34,5 44,8 20,7 - 7 Co20Ti3B14 54,1 8,1 37,8 10,526(8) 8 Co10Ti13B14 27,1 35,1 37,8 - Co20-xTixB6*, Ti2Co, TiCo2, Ti, > Ti3Co5B2 Co3B, Ti und Spuren an Co20-xTixB6* CoTi und Co20-xTixB6* Co2B, Ti2B, TiB100, Co20-xTixB6 Co2B, Co, Co3B, TiCo - 222 - Das System Kobalt-Titan-Bor Alle Proben wurden pulverdiffraktometrisch auf die gebildeten Phasen hin analysiert. Eine Verfeinerung der Gitterkonstante wurde nur bei Proben durchgeführt, bei denen die τ-Phase dominierte. Temperaturprogramm: 300°C/h RT 10°C/h 300°C/h 1450°C 850°C (10h halten) (2h halten) RT Das Ergebnis der Untersuchung steht im Widerspruch zu den Werten, die von Stadelmaier et al. erhalten wurden. Der untersuchte Bereich zeigten die Bildung der τ-Phase mit einer Gitterkonstante von 10,526(8) Å – 10,537(5) Å. Dieser Homogenitätsbereich ist viel schmaler und gibt keinen Hinweis auf das Auftreten einer borreichen τ-Phase. Diese Gitterkonstanten der borreichen wie auch borarmen Proben besitzen ähnliche Größenordnungen (Tabelle 4.6.1.1). Somit konnte bei vier unterschiedlichen Ansätzen im Rahmen der Standardabweichung nahezu keine Zellenvergrößerung erfasst werden. Das nahezu phasenreine Ergebnis konnte bei den borarmen Ansätzen der Zusammensetzung Co22Ti1B6 und Co21Ti2B6 erhalten werden. Das Pulverdiffraktogramm der Probe Co21Ti2B6 ist in der Abbildung 4.6.1 dargestellt. Hier wurde eine Gitterkonstante von a = 10,527(7) Å in Fm 3 m ermittelt werden. 210 200 190 180 170 160 150 140 130 120 110 100 90 80 70 60 50 40 30 20 10 0 5 10 20 30 40 2-Theta - Scale Abbildung 4.6.1: Pulverdiffraktogramm der Probe mit der Zusammensetzung Co21Ti2B6 mit einer Gitterkonstanten von a = 10,527(7) Å. Die blau markierten Reflexe konnten als Spuren von Ti2Co identifiziert werden. - 223 - Das System Kobalt-Titan-Bor 4.6.2 Röntgenographische Untersuchung an Einkristallen Ein Einkristall aus einem Ansatz der Schmelzzusammensetzung von Co20Ti3B6 in Form eines metallisch glänzenden, unregelmäßigen Polyeders wurde vermessen. Bei der Verfeinerung der Metalllage 4a mit einer vollständigen Besetzung durch Titan und einer Mischbesetzung des Zentrums des Friauf-Polyeders mit 11(4) % Titan neben 89(4)% Kobalt konnten nach der Einführung von anisotropen Temperaturfaktoren Zuverlässigkeitswerte von R1(F) = 0,0141 und wR2(I) = 0,0248 erreicht werden. Die wechselweise Freigabe der Besetzungsfaktoren aller kristallographischen Lagen zeigte keine bemerkenswerten Restelektronendichten auf den Positionen. Alle Einzelheiten zu der verfeinerten Struktur von Co20,89Ti2,11B6 sind in den folgenden Tabellen angegeben. - 224 - Tabelle 4.6.2.1: Angaben zur Strukturanalyse von Co20,89Ti2,11B6. Summenformel Co20,89Ti2,11B6 Kristallsystem kubisch Raumgruppe F m3m a = 10,5161(12) Gitterkonstante [Å] 3 Volumen der Elementarzelle [Å ] 1162,96(2) Zahl der Formeleinheiten 4 3 röntgenogr. Dichte [g/cm ] 7,979 F(000) 2564,0 Kristallform, -farbe metallisch glänzender Splitter STOE IPDS II (Mo-Kα-Strahlung; λ=0,71073 Diffraktometer Å Graphitmonochromator 0° ≤ ω ≤ 180° Rotation ψ = 0°, 111; ∆ω = 2° Belichtungszeit [sec.] 300 Messtemperatur [K] Raumtemperatur [293(2)] 0,02 × 0,06 × 0,03 Kristallgröße [mm] -1 Absorptionskoeffizient [mm ] 30,18 Messbereich [°] 2°<2θ<70° -16 ≤ h ≤ 16, -16 ≤ k ≤ 16, Indexbereich -16 ≤ l ≤ 16 Zahl der gemessenen Reflexe 2378 Zahl der unabh. Reflexe mit I > 0 2368 I > σ2(I): 109 freie Parameter 15 Korrekturen Lorentz, Polarisation Absorptionskorrektur numerical / equivalent method (XSHAPE[68]) Strukturlösung Isotypie Cr23C6[189] Strukturverfeinerung SHELXTL-97[199] w-1 = σ2F02 + (0,0152 * P)2 + 0,0123 * P Wichtungsschema P = (F02 + 2 FC2) / 3 0,747 Goodness of fit 0,00056(4) Extinktionskorrektur - 3 Restelektronendichte [e /Å ]: R-Werte für I > 0: Alle Reflexe +0,45 / -0,67; 0,14 e-/Å3 R1(F) = 0,0141 wR2(I) = 0,0248 R1(F) = 0,0232 - 225 - Das System Kobalt-Titan-Bor Tabelle 4.6.2.2: Ortskoordinaten, Besetzungsfaktoren (in [%]) und Auslenkungsparameter von Co20,89Ti2,11B6. Atom Co 1 Lage x 0 48h Co 2 y z sof Ueq 0,32973(5) 0,17027(5) 100* 0,00749(20) * 0,00782(23) 32f Co 3/ Ti 2 4a 0,11721(5) 0,11721(5) 0,11721(5) 100 0 ½ 0 89(4)/11# 0,00059(20) Ti 1 8c ¼ ¼ ¼ 100* 0,00672(57) B1 24e 0 ½ 0,2730(12) 100* 0,0073(16) * bei wechselweiser Freigabe der Besetzungsfaktoren keine signifikante Restelektronendichte vorhanden # Besetzungsfaktoren und Temperaturfaktoren gekoppelt und fixiert Atom U11 U22 U33 U23 U13 U12 Co 1 0,00818(3) 0,0071(3) 0,0071(3) 0,0003(3) 0 0 Co 2 0,0078(2) 0,0078(2) 0,0078(2) 0,0003(3) 0,0003(3) 0,0003(3) Co 3 /Ti 2 0,006(2) 0,006(2) 0,006(2) 0 0 0 Ti 1 0,0067(6) 0,0067(6) 0,0067(6) 0 0 0 Tabelle 4.6.2.3: Ausgewählte Abstände in der Co20,89Ti2,11B6-Struktur. Benachbarte Atome Abstand [Å] Anzahl der Bindungen B1 2,091(6) x3 Co 2 2,4187(10) x3 Co 1 2,6125(6) x4 Ti 1 2,4187(11) x1 B1 2,091(6) x2 Co 1 2,3715(15) x1 Co 1 2,5323(8) x4 Co 3 / Ti 2 2,5323(8) x1 Co 2 2,6125(6) x4 Co 3 /Ti 2- Co 2 2,5323(8) x 12 Ti 1 - Co 1 2,4187(10) x4 Co 2 2,8840(4) x8 Co 1 2,091(6) x4 Co 2 2,091(5) x4 Co 2 - Co 1 - B1- - 226 - Das System Kobalt-Titan-Bor 4.6.3 Strukturdiskussion Co20,89Ti2,11B6 kristallisiert im Cr23C6-Typ in der Raumgruppe Fm 3 m mit einer Gitterkonstanten von a = 10,5161(12) Å. Die ermittelte Gitterkonstante liegt in einer sehr guten Übereinstimmung mit der Literatur. In Rahmen dieser Arbeit konnten zum ersten Mal Einkristalldaten von einem τ-Borid aus dem System Kobalt-Titan-Bor gesammelt werden. Die Strukturverfeinerung zeigt eine vollständige Besetzung der 8c Lage durch Titan. Die Lage 4a bildet das Zentrum des Kuboktaeders, dieser Position wird vollständig mit den Metallatomen Kobalt und Titan in einem Verhältnis von 89(4) zu 11(4) % gefüllt. Jedes Metall im Zentrum des Kuboktaeders wird im Abstand von 2,5323(8) Å zwölffach koordiniert. Das Kuboktaeder hat eine Kantenlänge von 2,4187(10) Å. Die Würfel werden von der Position 32f durch Kobaltatome geformt mit einer Kantenlänge von 2,3715(15) Å. Die Boratome besetzten die Position 24e vollständig in der üblichen quadratischen antiprismatischen Koordination (dCo1-B1 : 2,091(6)Å) und (dCo2-B1 : 2,091(6) Å). - 227 - 4.7 Das System Kobalt-Vanadium-Bor 4.7.1 Der kobaltreiche Bereich des ternären Systems Kobalt-Vanadium-Bor Der Bereich der τ-Phase des Kobalt-Vanadium-Bor Dreistoffsystems ist sehr schmal. In der Abbildung 4.7.1 ist die Projektion der Schmelzfläche dargestellt. Metallographische Untersuchungen wurden von Stadelmaier et al.192 durchgeführt. Das Ergebnis zeigte die primären Erstarrungen der kongruent schmelzenden, ternären Phase (mit τ gekennzeichnet) und die eutektische Phasenreaktionen. Abbildung 4.7.1:Projektion der Schmelzflächen im Dreistoffsystem Co-V-B. Abbildung 4.7.2:Isothermer Schnitt bei 800°C im Dreistoffsystem Co-V-B. Abbildung 4.7.2 zeigt einen isothermen Schnitt bei einer Temperatur von 800°C. Diese Darstellung bestätigt die Löslichkeit von Vanadium in Co2B und zeigt die Entstehung der τPhase. Der Borgehalt für das ternäre Borid mit der Cr23C6-Struktur scheint konstant innerhalb der experimentellen Fehlergrenzen bei einer idealen Zusammensetzung für Co23-xVxB6 von 20,7 At.-% B zu liegen. Den Untersuchungen zufolge ist ein Vanadiumgehalt von 6,5 bis 10,3 At.-% V möglich. Dieses lässt eine Gitterkonstantenvariation mit einem Minimum von amin = 10,476 Å bis zu einem Maximum von amax. = 10,497 Å zu. 4.7.2 Präparative Untersuchung im System Kobalt-Vanadium-Bor Mit einer gezielten Synthese sollten hier Einkristalle für weitere Untersuchungen gezüchtet werden. Die ausgewählten Einkristalle sollen zur Klärung der Frage nach der Mischbesetzung - 228 - Das System Kobalt-Vanadium-Bor in der Chromcarbidstruktur behilflich sein. Zudem soll eine Überprüfung des borreichen Bereiches der τ Phase erfolgen. Für die Synthese wurden die Elemente in unterschiedlichen stöchiometrischen Verhältnissen homogenisiert und kalt verpresst. Die Umsetzung erfolgte in Korundtiegeln bei einer Reaktionstemperatur von 1400°C unter Ar-Schutzgasatmosphäre. Nach einer Reaktionszeit von einer Stunde wurde mit einer Abkühlrate von 20K / h auf 850°C gekühlt. Unterhalb von 850°C sollte die Temperatur keinen Einfluss mehr auf die Kristallisation nehmen. Somit wurde hier eine Abkühlrate von 300K / h bis zur Raumtemperatur gewählt. Temperaturprogramm: 300°C/h RT 20°C/h 300°C/h 1400°C 850°C (10h halten) (2h halten) RT Tabelle 4.7.2.1: Proben im System Kobalt-Vanadium-Bor und ihre pulverdiffraktometrische Charakterisierung. Fettgedruckt sind die Hauptprodukte. Graue unterlegung unterscheidet den Borgehalt. (*nicht Indiziert). Nr. Zusammensetzung Gehalt [At.-%] Gitterkonstante a [Å] weitere kristalline Phasen Co V B von Co23-xVxB6+y 1 Co22V1B6 75,9 3,4 20,7 10,504(9) Co 2 Co21V2B6 72,4 6,9 20,7 10,476(6) Co 3 Co20V3B6 69,0 10,3 20,7 10,477(23) >> Co 4 Co17V6B6 58,6 20,7 20,7 - VB >> Co, Co20V3B6* 5 Co13V10B6 44,8 34,5 20,7 - VB >> Co, Co20V3B6* 6 Co10V13B6 34,5 44,8 20,7 - Co3V, VB, CoB, Co2B 7 Co20V3B14 54,1 8,1 37,8 - Co2B >>Co20V3B* 8 Co10V13B14 27,1 35,1 37,8 - 9 Co3V20B14 8,1 54,1 37,8 - Co3V, VB, CoB, CoV3 VB, >>Co20V3B* Die Überprüfung der vorhandenen Phasen und die Ermittlung der Gitterkonstanten wurden mit der pulverdiffraktometrischen Methode durchgeführt. Das Vorhandensein der τ Phase konnte nur für die bor- und vanadiumarmen Ansätze mit der Schmelzzusammensetzung von Co23-xVxB6 (x=1-3) nachgewiesen werden. - 229 - Das System Kobalt-Vanadium-Bor 4.7.3 Härtemessung an Einkristallen Aus dem Ansatz Nr. 2 mit der Schmelzzusammensetzung von Co22V1B6 wurden zwei Kristalle in Epoxidharz eingebettet und aufgearbeitet, so dass eine Härtemessung nach Vickers und Knoop möglich war. Abbildung 4.7.3: Vickers- und Knoopabdrücke auf der Oberfläche eines Einkristalls mit der Zusammensetzung von Co22,64V0,36B6. Die Härte wird als ein Mittelwert aus je drei Abdrücken ermittelt. Dazu wurden die hinterbleibenden Abdrücke auf der Oberfläche der Einkristalls nach Entfernung des Eindrinkkörpers vermessen. Somit konnte ein Mittelwert von 858 HV 0,2/10 und 853 HK 0,2/10 gebildet werden. Die ermittelte Härte erfüllt liegt in gleicher Größe wie schon zuvor an τ-Boriden beobachtet. 4.7.4 Röntgenographische Untersuchung an Einkristallen Aus der Probe der Zusammensetzung Co20V3B6 konnte unter einem Lichtmikroskop ein geeigneter Einkristall für eine röntgenographische Untersuchung mit dem IDPS II isoliert werden. Der metallisch glänzende, unregelmäßige Polyeder konnte in einem Winkelbereich von 180° in Schritten von zwei Grad mit einem Detektorabstand von 100 mm für fünf Minuten pro Aufnahme vermessen werden. Aus den gesammelten Daten konnten 135 unabhängige Reflexe nach einer Mittelung in der Laueklasse und den üblichen Korrekturen erhalten werden. Die Verfeinerung der 14 freien Parameter lieferte Zuverlässigkeitswerten von R1(F) = 0,0297 und wR2(I) = 0,0641. Die Strukturverfeinerung zeigt eine Mischbesetzung auf der Position 8c mit einem Anteil an 82(3)% Vanadium und 18(3)% Kobalt. Alle weiteren Metalllagen sind ausnahmslos mit Kobalt besetzt und zeigen im Rahmen der Standardabweichung keine signifikante Abweichung von der vollständigen Besetzung. Die Boratome konnten mit einer vollständigen Besetzung auf der kristallographischen Punktlage 24e lokalisiert werden. - 230 - Das System Kobalt-Vanadium-Bor Tabelle 4.7.4.1: Angaben zur Strukturanalyse von Co21V2B6. Summenformel Co21V2B6 Kristallsystem kubisch Raumgruppe F m3m 10,4756(12) Gitterkonstante [Å] 3 Volumen der Elementarzelle [Å ] 11496,57(2) Zahl der Formeleinheiten 4 3 röntgenogr. Dichte [g/cm ] 8,190 F(000) 2598,6 Kristallform, -farbe metallisch glänzende, unregelmäßige Polyeder STOE IPDS II (Mo-Kα-Strahlung; λ=0,71073 Å Diffraktometer Graphitmonochromator 0° ≤ ω ≤ 180° Rotation ψ = 0°, 111; ∆ω = 2° Belichtungszeit [sec.] 300 Messtemperatur [K] Raumtemperatur [293(2)] 0,05 × 0,05 × 0,05 Kristallgröße [mm] -1 Absorptionskoeffizient [mm ] 31,89 Messbereich [°] 2°<2θ<70° -16 ≤ h ≤ 16, -16 ≤ k ≤ 16, Indexbereich -16 ≤ l ≤ 16 Zahl der gemessenen Reflexe 6953 Zahl der unabh. Reflexe mit I > 0 5146 I > σ2(I): 135 freie Parameter 14 Korrekturen Lorentz, Polarisation Absorptionskorrektur numerical / equivalent method (XSHAPE[68]) Strukturlösung Isotypie Cr23C6[189] Strukturverfeinerung SHELXTL-97[199] w-1 = σ2F02 + (0,0356 * P)2 + 0,00 * P Wichtungsschema P = (F02 + 2 FC2) / 3 0,800 Goodness of fit 0,00102(28) Extinktionskorrektur - 3 Restelektronendichte [e /Å ]: R-Werte für I > 0: Alle Reflexe +0,96 / -0,94; 0,21 e-/Å3 R1(F) = 0,0296 wR2(I) = 0,0616 R1(F) = 0,0455 - 231 - Das System Kobalt-Vanadium-Bor Tabelle 4.7.4.2: Ortskoordinaten, Besetzungsfaktoren (in [%]) und Auslenkungsparameter von Co21V2B6. Atom Lage x y z sof Ueq Co 1 48h 0,17001(1) 0,17001(1) 0 100* 0,0217(13) Co 2 0,1193(1) 0,3807(1) 0,1193(1) 100* 0,0191(5) Co 3 32f 4a 0 0 0 100* 0,0175(5) V 8c ½ ½ ½ 100* 0,0148(9) B 24e 0,2697(17) 0 0 100* 0,009(3) * bei wechselweiser Freigabe der Besetzungsfaktoren keine signifikante Restelektronendichte vorhanden # Besetzungsfaktoren und Temperaturfaktoren gekoppelt und fixiert Atom U11 U22 U33 U23 U13 U12 Co 1 0,0190(6) 0,0190(6) 0,0190(6) 0,0 0,0 0,0005(6) Co 2 0,0175(5) 0,0175(5) 0,0175(5) 0,0011(5) 0,0011(5) 0,0011(5) Co 1 0,021(1) 0,021(1) 0,021(1) 0,0 0,0 0,0 Co 4/V 1 0,016(2) 0,016(2) 0,016(2) 0,0 0,0 0,0 Tabelle 4.7.4.3: Ausgewählte Abstände in der Co21V2B6-Struktur. Anzahl der Benachbarte Atome Abstand [Å] Co 2 - B1 2,119(9) x3 Co 2 2,491(2) x3 Co 3 2,5912(10) x4 V 2,372(2) x1 B1 2,062(9) x2 Co 2 2,372(2) x1 Co 1 2,5197(14) x1 Co 3 2,5197(14) x4 Co 2 2,5912(10) x4 Co 3- Co 3 2,5197(14) x 12 V- Co 3 2,372(2) x4 Co 2 2,8740(6) x8 Co 3 2,062(9) x4 Co 2 2,119(9) x4 Co 3 - B1- Bindungen - 232 - Das System Kobalt-Vanadium-Bor 4.7.5 Strukturdiskussion Co21V2B6 kristallisiert im Cr23C6-Typ in der Raumgruppe Fm 3 m mit einer Gitterkonstanten von a = 10, 4756(12) Å. Die sehr kleine Gitterkonstante liegt sehr nahe am binären Borid mit der Summenformel von Co23B6 (a = 10,4618(13) Å)[162]. In dieser Arbeit konnten zum ersten Mal Einkristalle des τ-Borids mit Vanadium isoliert und an Hand von Einkristalldaten verfeinert werden. Nur die Punktlage des τ-Stabilisators 8c zeigt eine Besetzung durch Vanadium mit 83(3) %, der Rest dieser Position wurde statistisch und vollständig mit Kobaltatomen aufgefühlt. Alle weiteren Metallpositionen sind vollständig mit Kobaltatomen besetzt. Die Punktlage 48h formt das Kuboktaeder. Die Metallatome auf dieser Lage liegen im Abstand von 2,5197(14) Å voneinander entfernt. Im Zentrum des Kuboktaeders befindet sich die Punktlage 4a, die von weiteren Kobaltatomen besetzt ist und in einer Distanz von 2,5197(14) Å zwölffach koordiniert wird. Die Eckpunkte der Dreiecksflächen liegen 2,372(2) Å voneinander entfernt. Der Würfel auf der Punktlage 32f, ebenfalls bestehend aus Kobaltatomen besitzt eine Kantenlänge von 2,491(2) Å. Bor ist wie üblich achtfach quadratisch antiprismatisch von Kobaltatomen umgeben (dCo1-B1 : 2,062(9)Å) und (dCo2-B1 : 2,119(19) Å). - 233 - 4.8 Das System Kobalt-Wolfram-Bor Das ternäre System bestehend aus Kobalt, Wolfram und Bor wurde bereits vom Ganglberger et al.[113] auf die Phasen hin untersucht. Ganglberger et al. konnte als erster aus Pulverdaten die Struktur des τ-Borids mit einer Summenformel von Co21W2B6 bestimmen. Das Borid mit Cr23C6-Struktur kristallisiert in der Raumgruppe Fm 3 m mit einer Gitterkonstanten von a = 10,506 Å[156]. Es existiert nur ein kleiner Homogenitätsbereich von Co23-xWxB6 mit x = 1,4 -2 mit einer sehr schmalen Phasenbreite von 10,50 – 10,506. 4.8.1 Präparative Untersuchung im System Kobalt-Wolfram-Bor Die Synthese erfolgte aus den Elementen. Hier wurde Kobalt, Wolfram und Bor in entsprechenden Verhältnissen homogenisiert und in Tabletten gepresst. Es sollte zum einen die Phasenbreite des τ-Borids überprüft werden und zudem sollte ein zusätzlicher Einbau an Bor erfolgen. Des Weiteren sollte zum die synthese so modifiziert werden, dass Einkristalle für eine röntgenographische Untersuchung zugänglich werden. Das Aufschmelzen der Proben bei einer Temperatur von 1450°C in einer Argon-Atmosphäre und einer Haltezeit von 10 Stunden ermöglichte einen quantitativen Umsatz der Edukte. Die Schmelze wurde sehr langsam abgekühlt (10°C/h), dieses ermöglichte eine gute Kristallisation. Die pulverdiffraktometrische Charakterisierung aller Proben ermöglichte die Bestimmung der erhaltenen Produkte und die Verfeinerung der Gitterkonstanten des gewonnenen τ-Borids. In der Tabelle 4.8.1.1 sind die Proben mit ihrer Phasenanalyse zusammengefasst. Nur in drei Ansätze konnte die Bildung der τ-Phase beobachtet werden. In dem Ansatz mit der Schmelzzusammensetzung Co22WB6 wurde die ternäre Phase nahezu phasenrein dargestellt. In den borreichen Proben konnte kein Hinweis auf die Existenz von τ-Boriden verzeichnet werden. Viel mehr dominierte das ternäre Borid mit der Zusammensetzung von Co2WB2 neben weiten binären Kobaltboriden. Temperaturprogramm: 300°C/h RT 10°C/h 150°C/h 1450°C 950°C (10h halten) (2h halten) RT - 234 - Das System Kobalt- Wolfram-Bor Tabelle 4.8.1.1: Proben im System Kobalt-Wolfram-Bor und ihre pulverdiffraktometrische Charakterisierung. Fettgedruckt sind die Hauptprodukte. Graue Unterlegung unterscheidet den Borgehalt. Nr. Zusammensetzung Gitterkonstante a [Å] weitere kristalline B von Co23-xWxB6+y Phasen Gehalt [At.-%] Co W 1 Co22W1B6 75,9 3,4 20,7 10,503(6) W 2 Co21W2B6 72,1 6,9 20,7 - 3 Co20W3B6 69,0 10,3 20,7 - 4 Co17W6B6 58,6 20,7 20,7 - CoWB, W2B, Co 5 Co10W13B6 34,5 44,8 20,7 - CoWB, W, CoB 6 Co3W20B6 10,3 69,0 20,7 - W, Co2WB2 8 Co20W3B14 54,1 8,1 37,8 - Co2WB2, Co2B 9 Co10W13B14 27,1 35,1 37,8 - Co2WB2, CoWB, W CoWB, Co23-xWxB6* CoWB, W2B, Co23-xWxB6* Die Gitterkonstante der τ Phase konnte mit a = 10,503(6) Å bestimmt werden, in sehr guter Übereinstimmung mit den von Ganglberger et al. ermittelten Parameter. Unter einem Lichtmikroskop wurden metallisch glänzende unregelmäßige Polyeder für weite Untersuchungen isoliert werden. 4.8.2 Härtemessung an Einkristallen An dem Einkristall, der zuvor auf dem IPDS II vermessen wurde, konnte nach dem Einbetten in Epoxidharz und anschließendem Aufarbeiten der Oberflächen eine Härte nach Vickers und Knoop bestimmt werde. Abbildung 4.8.1: Vickersabdruck. d1=20,73µm, d2=21,13µm 854 HV Abbildung 4.8.2: Knoop-Abdruck: d = 57,93µm; 847 HK - 235 - Das System Kobalt- Wolfram-Bor Bei der Härtemessung nach Vickers konnte aus vier Abdrücken eine Härte von 865 HV 0,2/10 (8,5 GPa) ermittelt werden. Die Messung mit dem Knoopindenter lieferte ebenfalls konstante Werte in Höhe von 843 HK 0,2/10. Die ermittelten Härtewerte für das τ-Borid mit der Zusammensetzung von Co22,32W0,68B6 bewegen sich in der charakteristischen Größenordnung die für Verbindungen dieser Substanzklasse typisch sind. 4.8.3 Röntgenographische Untersuchung an Einkristallen Durch die röntgenographisch Untersuchung eines isolierten Einkristalls aus dem Ansatz mit der Schmelzzusammensetzung Co22WB6 konnte eine Gitterkonstante von a = 10,4911(12) Å bestimmt werden. In Rahmen dieser Arbeit konnte zum ersten Mal ein τ-Borid aus gegebenem Dreistoffsystem anhand von Einkristalldaten charakterisiert werden. Die Reflexintensitäten wurden auf dem IPDS II gesammelt, hierfür wurde ein Bereich von 0°≤ω≤ 180° mit ψ = 0°, 111; ∆ω = 2° und eine Belichtungszeit von fünf Minuten gewählt. Somit konnte nach einer Mittelung in der Lauenklasse m 3 m ein Datensatz mit 132 unabhängigen Reflexen erhalten werden. Eine Verfeinerung der 14 freien Parameter mit den Atomkoordinaten eines bekannten τ-Borids konvergierten nach einer vollständigen Besetzung der 8c Lage mit Wolfram 64(7) % und Kobalt 36(7) % bei Zuverlässigkeitswerten von R1(F) = 0,0425 und von wR2(I) = 0,0922. Weitere Einzelheiten der Strukturverfeinerung sowie die Besetzungsfaktoren der Metalllagen und Abstände wie auch die Koordinationssphäre der Atome sind den folgenden Tabellen zu entnehmen. - 236 - Tabelle 4.8.3.1: Angaben zur Strukturanalyse von Co22,32W0,68B6. Summenformel Co22,32W0,68B6 Kristallsystem kubisch Raumgruppe F m3m Gitterkonstante [Å] 10,4911(12) Volumen der Elementarzelle [Å3] 1154,68(20) Zahl der Formeleinheiten 4 röntgenogr. Dichte [g/cm3] 8,662 F(000) 2732,8 Kristallform, -farbe metallisch glänzender, unregelmäßiger Polyeder Diffraktometer Messtemperatur [K] Rotation STOE IPDS II (Mo-Kα-Strahlung; λ=0,71073 Å Graphitmonochromator Raumtemperatur [293(2)] 0° ≤ ω ≤ 180° ψ = 0°, 111; ∆ω = 2° Belichtungszeit [sec.] 300 Kristallgröße [mm] 0,025 × 0,025 × 0,025 Absorptionskoeffizient [mm-1] 37,80 Messbereich [°] 2°<2θ<70° -16 ≤ h ≤ 16, Indexbereich -16 ≤ k ≤ 16, -16 ≤ l ≤ 16 Zahl der gemessenen Reflexe 5206 Zahl der unabh. Reflexe mit I > 0 5195 I > σ2(I): 132 freie Parameter 14 Korrekturen Lorentz, Polarisation Absorptionskorrektur numerical / equivalent method (XSHAPE[68]) Strukturlösung Isotypie Cr23C6[189] Strukturverfeinerung SHELXTL-97[199] Wichtungsschema w-1 = σ2F02 + (0,0 * P)2 + 265,0 * P P = (F02 + 2 FC2) / 3 Goodness of fit 1,248 Extinktionskorrektur 0,00041(11) Restelektronendichte [e-/Å3]: +2,37 / -1,92; R-Werte für I > 0: Alle Reflexe 0,41 e-/Å3 R1(F) = 0,0425 wR2(I) = 0,0922 R1(F) = 0,0477 - 237 - Das System Kobalt- Wolfram-Bor Tabelle 4.8.3.2: Ortskoordinaten, Besetzungsfaktoren (in [%]) und Auslenkungsparameter von Co22,32W0,68B6. Atom Lage x y z sof Ueq Co 1 48h 0,16986(14) -0,16986(14) 0 100* 0,0104(6) Co 2 0,11877(14) -0,38123(14) -0,01187(1) 100* 0,0103(6) Co 3 32f 4a 0 0 0 100* 0,009(1) Co/W 1 8c ¼ ¼ ¾ 36/64(7)*# 0,0093(3) B1 24e 0,277(3) 0 0 100* 0,013(4) * bei wechselweiser Freigabe der Besetzungsfaktoren keine signifikante Restelektronendichte vorhanden # Besetzungsfaktoren und Temperaturfaktoren gekoppelt und fixiert Atom U11 U22 U33 U23 U13 U12 Co 2 0,0103(6) 0,0103(6) 0,0103(6) -0,0011(5) -0,0011(5) 0,0011(5) Co 1 0,0096(7) 0,0096(7) 0,0120(9) 0 0 -0,0010(6) Tabelle 4.8.3.3: Ausgewählte Abstände in der Co22,32W0,68B6-Struktur. Benachbarte Atome Abstand [Å] Anzahl der Bindungen B1 2,076(13) x3 Co/W 4 2,385(3) x1 Co 2 2,492(3) x3 Co 1 2,5994(13) x4 B1 2,104(13) x2 Co 1 2,3780(40) x1 Co 1 2,5202(20) x1 Co 3 2,5202(20) x4 Co 2 2,5994(13) x4 Co 2 - Co 1 2,5202(20) x 12 Co/W 4 - Co 2 2,385(3) x4 Co 2 2,8797(9) x8 Co 1 2,076(13) x4 Co 2 2,104(13) x4 Co 1 - Co 2 - B1- - 238 - Das System Kobalt- Wolfram-Bor 4.8.4 Strukturdiskussion Co22,32W0,68B6 kristallisiert im Cr23C6-Typ mit einer Gitterkonstanten von a = 10,4911(12) Å. Die Struktur des τ-Borids wurde bereits mehrfach diskutiert. Unterschied zu den zuvor diskutierten τ-Boriden gibt es kaum. Die Position 8c ist in disem Fall durch eine Mischbesetzung von Kobalt und Wolfram geprägt. Der geringe Anteil an Wolfram lässt die Gitterkonstante auf einen Wert von 10,4911(12) Å schrumpfen. Alle weiteren Metallpositionen sind mit Kobalt besetzt und bilden die charakteristischen Polyeder auf den zu erwarteten Positionen. So besitzen die Kanten der Würfel eine Länge von 2,492(3)Å. Im Kuboktaeder beträgt die Entfernung der Kobaltatome 2,3780(4) Å zueinander. Das Zentrum ist ebenfalls mit Kobaltatomen vollständig besetzt (dCo2-Co4 = 2,520(2) Å). Bor steht in der quadrarisch antiprismatischen Koordination zwischen den Flächen des Würfels und des Kuboktaeders (dCo1-B2 : 2,076(13) Å und dCo2-B3 : 2,104(13) Å). - 239 - Ternäre Boride 4.9 Das System Kobalt-Mangan-Bor Über ein ternäres Borid im System Kobalt-Mangan-Bor wurde bereits vom Stadelmaier et al. in den sechziger Jahren bereichtet. Hier wurden ein Homogenitätsbereich von a = 10,518 – 10,641 Å bei einer Zusammensetzung von Co23-xMnxB6 (x= 3 – 11,5) angegeben[169]. Alle Untersuchungen basierten auf Pulverfilmaufnahmen, somit gab es keinerlei Informationen zur Verteilung der Metalle in der Cr23C6-Struktur. Der enorme Homogenitätsbereich macht das System interessant für Studien an Einkristallen, um den Einfluss des τ-Stabilisators zu untersuchen. Zudem sollte überprüft werden, ob ein zusätzlicher Einbau von Bor möglich ist. 4.9.1 Präparative Untersuchung im System Kobalt-Mangan-Bor Das primäre Ziel der Synthese in diesem System war die Züchtung von Einkristallen. Dazu wurden die Elemente in stöchiometrischen Verhältnissen in Tabletten verpresst und in Korundtiegel unter Argon-Schutzgasatmosphäre bei einer Temperatur von 1450°C umgesetzt. Dabei wurde sehr langsam (10°C/h) bis auf eine Temperatur von 950°C abgekühlt. Eine röntgenographische Phasenanalyse aller Probe sollte den Homogenitätsbereich der mit τ bezeichneten Phase beschreiben und das Phasengleichgewicht darlegen. Temperaturprogramm: 300°C/h RT 10°C/h 300°C/h 1450°C 950°C (10h halten) (5h halten) RT Tabelle 4.9.1.1: Proben im System Kobalt-Mangan-Bor und ihre pulverdiffraktometrische Charakterisierung. Fettgedruckt sind die Hauptprodukte. Graue Unterlegung unterscheidet den Borgehalt. Nr. Zusammensetzung Gitterkonstante a [Å] weitere kristalline B von Co23-xMnxB6+y Phasen Gehalt [At.-%] Co Mn 1 Co22Mn1B6 75,9 3,4 20,7 10,564(4) > Co 2 Co21Mn2B6 72,1 6,9 20,7 10,543(6) > Co 3 Co20Mn3B6 69,0 10,3 20,7 10,551(4) > Co 4 Co17Mn6B6 58,6 20,7 20,7 10,597(7) - 5 Co10Mn13B6 34,5 44,8 20,7 - Co2B, Mn2B 6 Co3Mn20B6 10,3 69,0 20,7 - Mn2B, MnB 8 Co20Mn3B14 54,1 8,1 37,8 Co2B, Mn2B, B 9 Co10Mn13B14 27,1 35,1 37,8 MnB, Mn2B - 240 - Das System Kobalt- Mangan-Bor Bei den ersten vier Proben die sowohl borarm als auch manganarm sind, konnte die Existenz des τ-Borids beobachtet werden. Somit konnten die τ-Phasen mit Gitterkonstanten von a = 10,543(6) bis 10,597(7) Å verfeinert werden. Eine phasenreine Darstellung des ternären Borids war bei einer Schmelzzusammensetzung von Co17Mn6B6 gelungen. Bei den anderen Ansätzen konnte noch etwas elementares Kobalt beobachtet werden. Bei den Proben mit einem höheren Borgehalt wurde die Kristallisation der binären Phasen (Co2B, MnB und Mn2B) wahrgenommen. Es konnten keine Indizien für die Entstehung der ternären Phase mit Cr23C6 Struktur festgestellt werden. 4.9.2 Röntgenographische Untersuchung an Einkristallen Ein metallisch glänzender, unregelmäßiger Polyeder konnte aus der Probe mit der Schmelzzusammensetztung Co17Mn6B6 isoliert und auf dem IPDS II vermessen werden. Dieser Einkristall wurde in einem Winkelbereich von 180° in 2° Schritten vermessen. Aufgrund der Gitterkonstanten konnte davon ausgegangen werden, dass es sich hier um eine τ-Borid handelt. Somit wurde für die Verfeinerung mit unter verwendung der Atomokoordinaten von Cr23C6 begonnen. Nach Einführung einer Mischbesetzung auf den kristallographischen Lagen 32f und 4a und anisotroper Temperaturfaktoren für dir Metallpositionen konnten Zuverlässigkeitswerte von R1(F)=0,0337 und wR2(I)=0,0807 erhalten werden. Die Freigabe der Besetzungsfaktoren zeigte auf keine der vier kristallographischen Lagen eine signifikante Abweichung von der vollständigen Besetzung. Die Metallpositionen 32f und 4a wurden mit einer Mischbesetzung verfeinert. Somit lag die Restelektronendichte in einem plausiblen Rahmen von +0,97 und -0,96 e-/Å3. Weitere Einzelheiten zur Strukturverfeinerung, Besetzungsfaktoren sowie Temperaturfaktoren und Abstände sind den folgenden Tabellen zu entnehmen. - 241 - Das System Kobalt- Mangan-Bor Tabelle 4.9.2.1: Angaben zur Strukturanalyse von Co19,9Mn3,1B6. Summenformel Co19,9Mn3,1B6 Kristallsystem kubisch Raumgruppe F m3m 10,5657(12) Gitterkonstante [Å] 3 Volumen der Elementarzelle [Å ] 1179,2(5) Zahl der Formeleinheiten 4 3 röntgenogr. Dichte [g/cm ] 7,930 F(000) 2579,6 Kristallform, -farbe metallisch glänzender, unregelmäßiger Polyeder STOE IPDS II (Mo-Kα-Strahlung; λ=0,71073 Å Diffraktometer Graphitmonochromator Raumtemperatur [293(2)] Messtemperatur [K] 0° ≤ ω ≤ 180° Rotation ψ = 0°, 111; ∆ω = 2° 300 Belichtungszeit [sec.] 0,03 × 0,03 × 0,03 Kristallgröße [mm] -1 Absorptionskoeffizient [mm ] 30,26 Messbereich [°] 2°<2θ<70° -17 ≤ h ≤ 16, -17 ≤ k ≤ 15, Indexbereich -17 ≤ l ≤ 17 Zahl der gemessenen Reflexe 6279 Zahl der unabh. Reflexe mit I > 0 6250 I > σ2(I): 132 freie Parameter 14 Korrekturen Lorentz, Polarisation Absorptionskorrektur numerical / equivalent method (XSHAPE[68]) Strukturlösung Isotypie Cr23C6[189] Strukturverfeinerung SHELXTL-97[199] w-1 = σ2F02 + (0,0485 * P)2 + 0,0 * P Wichtungsschema P = (F02 + 2 FC2) / 3 1,015 Goodness of fit 0,00034(28) Extinktionskorrektur - 3 Restelektronendichte [e /Å ]: R-Werte für I > 0: Alle Reflexe +0,97 / -0,96; 0,27 e-/Å3 R1(F) = 0,0337 wR2(I) = 0,0807 R1(F) = 0,0508 - 242 - Das System Kobalt- Mangan-Bor Tabelle 4.9.2.2: Ortskoordinaten, Besetzungsfaktoren (in [%]) und Auslenkungsparameter von Co19,9Mn3,1B6. Atom Lage x y z Co 1 48h 0,17025(8) 0,17025(8) 0 Co/Mn 2 0,61782(9) 0,61782(9) Co/Mn 3 32f 4a ¼ Co 4 8c B 24e sof Ueq 100 0,0179(5) 0,61782(9) 89/11(3) 0,0178(5) ¼ ¼ 86/14(2) 0,0180(7) 0 0 0 100 0,0200(9) ½ ½ 0,72543 113(7) 0,022(3) * bei wechselweiser Freigabe der Besetzungsfaktoren keine signifikante Restelektronendichte vorhanden # Besetzungsfaktoren und Temperaturfaktoren gekoppelt und fixiert Atom U11 U22 U33 U23 U13 U12 Co 1 0,0175(5) 0,0175(5) 0,0187(7) 0 0 0,0005(4) Co/Mn 2 0,0178(5) 0,0178(5) 0,01788(5) 0,0011(3) 0,0011(3) 0,0011(3) Co/Mn 3 0,0202(9) 0,0202(9) 0,0202(9) 0 0 0 Co 4 0,0180(5) 0,0180(5) 0,0180(5) 0 0 0 Tabelle 4.9.2.3: Ausgewählte Abstände in der Co19,9Mn3,1B6-Struktur. Benachbarte Atome Abstand [Å] Anzahl der Bindungen B 2,096(9) x3 Co/Mn 3 2,419(2) x1 Co/Mn 2 2,489(2) x3 Co 1 2,6212(8) x4 B 2,110(9) x2 Co 1 2,383(2) x1 Co/Mn 3 2,5439(12) x1 Co 1 2,544(1) x4 Co/Mn 2 2,6212(8) x4 Co/Mn 3 - Co 1 2,544(1) x 12 Co 4 - Co/Mn 2 2,4190(16) x4 Co 1 2,8978(6) x8 Co/Mn 2 2,096(9) x4 Co 1 2,110(9) x4 Co/Mn 2 - Co 1 - B- - 243 - Das System Kobalt- Mangan-Bor 4.9.3 Strukturdiskussion Co19,9Mn3,1B6 kristallisiert in der Cr23C6-Typ in der Raumgruppe Fm 3 m mit einer Gitterkonstanten von a = 10,5657(12) Å. Bei diesem borarmen Kobalt τ-Borid sind die Positionen 48h und 8c ausschließlich mit Kobalt besetzt. Bei den zwei weiteren Positionen 32f (mit 89(3)% Co und 11(3)% Mn) und 4a (mit 86(2)% Co und 14(2)% Mn) konnte eine vollständige, statistische Mischbesetzung von Kobalt und Mangan beobachtet werden. Somit sind alle Kuboktaeder aus Kobaltatomen aufgebaut (dCo1-Co1: 2,5439(12)Å). Das Zentrum des Kuboktaeders ist mit den Atomen Kobalt und Mangan statistisch besetzt (dCo1Co/Mn3: 2,544(1)Å). Die Würfel werden ebenfalls durch die zwei Metallatome gestaltet und besitzen in der statistischen Verteilung einen Abstand von 2,489(2) Å zueinander. Die Punktlage 8c ist ausschließlich durch Kobaltatome besetzt. Hier wird das Zentrum des FrankKasper-Polyeders lediglich durch Kobalt (dCo4-Co/Mn 2: 2,491(2)Å) x 4 und dCo4-Co1: 2,8978(6)Å besetzt. Das Boratom (24e) ist wie üblich quadratisch antiprismatisch von den Grundflächen der Würfel (dB-Co/Mn 2: 2,096(9)Å) und der Kuboktaeder (dB-Co1: 2,110(9)Å) umgeben. - 244 - Ternäre Boride 4.10 Das System Kobalt-Molybdän-Bor Untersuchungen des Systems Kobalt-Molybdän-Bor zeigten die Existenz eines borarmen τBorids mit der Zusammensetzung von Co23-xMoxB6 (für x = 1,3 bis 2). Untersuchungen an Pulverproben zeigen eine Gitterkonstante, die sich in einer sehr schmalen Bereich bewegt (a = 10,500 – 10,504 Å)[156]. Alle gesammelten Informationen zu den τ-Boriden, bei denen der Einbau von Molybdän erfolgreich war, wurden anhand von Pulverdaten erhalten. Ziel dieser Untersuchung war, Einkristalle ausreichender Qualität für weitere Untersuchungen zu züchten, sowie auch die Überprüfung, ob ein Einbau von zusätzlichem Bor erfolgen kann. 4.10.1 Präparative Untersuchung im System Kobalt-Molybdän-Bor Für die Synthese wurden wie üblich die Elemente in stöchiometrischen Verhältnissen in einem Achatmörser homogenisiert und mit einer Handpresse verdichtet. Die Tabletten wurden in Korundtiegeln bei einer Temperatur von 1450°C aufgeschmolzen und nach einer Reaktionszeit von 10 Stunden langsam (15°C/h) auf ein Temperatur von 1050°C abgekühlt. Durch eine röntgenographische Phasenanalyse wurde Auskunft über die Produkte gesammelt, dabei konnte die Gitterkonstante des τ-Borids verfeinert werden. Die charakteristischen Reflexe für das ternäre Borid mit Cr23C6- Struktur konnten nur in den borarmen und molybdänarmen Ansätzen beobachtet werden. In einem Ansatz der Schmelzzusammensetzung Co22MoB6 wurde das τ-Borid als Hauptprodukt neben sehr wenig elementarem Kobalt beobachtet. Die Gitterkonstante konnte mit a = 10,524(6) Å bestimmt werden. Bei einer Erhöhung des Molybdänanteiles wurde die Bildung von MoCoB gefördert. Bei eine Molybdängehalt von mehr als x = 3 bezogen auf Co23-xMoxB6 konnte schließlich keine Hinweis für die Entstehung von einem τ-Borid vernommen werden. Bei den borreichen Boriden wurden zahlreiche Phasen beobachtet, jedoch konnte kein Indiz auf das Aufkommen einer Cr23C6-Struktur. Temperaturprogramm: 300°C/h RT 10°C/h 150°C/h 1450°C 950°C (10h halten) (2h halten) RT - 245 - Das System Kobalt- Molybdän-Bor Tabelle 4.10.1.1: Proben im System Kobalt-Molybdän-Bor und ihre pulverdiffraktometrische Charakterisierung. Fettgedruckt sind die Hauptprodukte. Graue Unterlegung unterscheidet den Borgehalt. Nr Zusammen- . setzung Gitterkonstante a [Å] weitere kristalline B von Co23-xMoxB6+y Phasen Gehalt [At.-%] Co Mo 1 Co22Mo1B6 75,9 3,4 20,7 10,524(6) >>Co 2 Co21Mo2B6 72,1 6,9 20,7 - 3 Co20Mo3B6 69,0 10,3 20,7 - 4 Co17Mo5B6 58,6 20,7 20,7 - MoCoB 5 Co10Mo13B6 34,5 44,8 20,7 - CoMo2B2, MoCoB 6 Co3Mo20B6 10,3 69,0 20,7 - Mo2B, Mo, CoB 8 Co20Mo3B14 54,1 8,1 37,8 - Co2B, CoMoB 9 Co10Mo13B14 27,1 35,1 37,8 - MoCoB, >>Co20Mo3B6* MoCoB, >>Co20Mo3B6*, Co CoMo2B2, CoB, Mo5Co2, Co 4.10.2 Röntgenographische Untersuchung an Einkristallen Ein Einkristall konnte aus dem nahezu phasenreinen Ansatz der Schmelzzusammensetzung Co22MoB6 unter einem Lichtmikroskop isoliert werden. Auch hier konnte nach der ermittelten Gitterkonstanten von einem τ-Borid ausgegangen werden, somit wurde die Mittelung in der Laueklasse m 3 m durchgeführt. Mit dem Datensatz von 135 unabhängigen Reflexen wurde ein Startmodell für die Verfeinerung mit den Lageparametern von Co23B6 gewählt, wobei die 8c Lage mit 32(6)% Molybdän und 68(6)% mit Kobalt besetzt wurde. Die Verfeinerung konvergierte nach Einführung von anisotropen Temperaturfaktoren schnell bei Zuverlässigkeitswerten von R1(F) = 0,0359 und von wR2(I) = 0,0569. Bei der Überprüfung der Besetzungsfaktoren konnte keine signifikante Abweichung von der vollständigen Besetzung festgestellt werden. Letztlich wurde bei 14 freien Parametern eine Zusammensetzung von Co22,34Mo0,66B6 erhalten. Weitere Einzelheiten zur Strukturverfeinerung sowie Besetzungsfaktoren und Abstände sind in den folgenden Tabellen zusammengefasst. - 246 - Ternäre Boride Tabelle 4.10.2.1: Angaben zur Strukturanalyse von Co22,34Mo0,66B6. Summenformel Co22,34Mo0,66B6 Kristallsystem kubisch Raumgruppe F m3m Gitterkonstante [Å] 10,4790(12) Volumen der Elementarzelle [Å3] 1150,5(8) Zahl der Formeleinheiten 4 röntgenogr. Dichte [g/cm3] 8,337 F(000) 2642,9 Kristallform, -farbe metallisch glänzender, unregelmäßige Polyeder Diffraktometer Rotation STOE IPDS II (Mo-Kα-Strahlung; λ=0,71073 Å Graphitmonochromator 0° ≤ ω ≤ 180° ψ = 0°, 111; ∆ω = 2° Belichtungszeit [sec.] 300 Messtemperatur [K] Raumtemperatur [293(2)] Kristallgröße [mm] 0,05 × 0,05 × 0,04 Absorptionskoeffizient [mm-1] 31,84 Messbereich [°] 4°<2θ<65° 0 ≤ h ≤ 9; Indexbereich 0 ≤ k ≤ 11; 2 ≤ l ≤ 15 Zahl der gemessenen Reflexe 4606 Zahl der unabh. Reflexe mit I > 0 135 I > σ2(I): 79 freie Parameter 14 Korrekturen Lorentz, Polarisation Absorptionskorrektur numerical / equivalent method (XSHAPE[68]) Strukturlösung Isotypie Cr23C6[193] Strukturverfeinerung SHELXTL-97[199] Wichtungsschema w-1 = σ2F02 + (0,0196 * P)2 + 0,00 * P P = (F02 + 2 FC2) / 3 Goodness of fit 0,707 Extinktionskorrektur 0,00091(15) Restelektronendichte [e-/Å3]: +1,86 / -1,71; R-Werte für I > 0: Alle Reflexe 0,38 e-/Å3 R1(F) = 0,0359 wR2(I) = 0,0569 R1(F) = 0,0788 - 247 - Das System Kobalt- Molybdän-Bor Tabelle 4.10.2.2: Ortskoordinaten, Besetzungsfaktoren (in [%]) und Auslenkungsparameter von Co22,34Mo0,66B6. Atom Lage x y z sof Ueq Co 1 48h 0,1698(1) 0,1698(1) 0 100* 0,0049(4) Co 2 0,6189(1) 0,6189(1) 0,6189(1) 100* 0,0063(6) Co 4/Mo 1 32f 8c ¼ ¼ ¼ 68(6) /32*# 0,0035(7) Co 3 4a 0 0 0 100* 0,0055(13) B1 24e 0,265(3) 0 0 100* 0,019(4) * bei wechselweiser Freigabe der Besetzungsfaktoren keine signifikante Restelektronendichte vorhanden # Besetzungsfaktoren und Temperaturfaktoren gekoppelt und fixiert Atom U11 U22 U33 U23 U13 U12 Co 1 0,0049(5) 0,0049(5) 0,0049(5) 0 0 0,003(6) Co 2 0,00628(6) 0,00628(6) 0,00628(6) 0,0014(5) 0,0014(5) 0,0014(5) Co 4 /Mo 1 0,0035(7) 0,0035(7) 0,0035(7) 0 0 0 Co 3 0,0055(1) 0,0055(1) 0,0055(1) 0 0 0 Tabelle 4.10.2.3: Ausgewählte Abstände in der Co22,34Mo0,66B6-Struktur. Anzahl der Benachbarte Atome Abstand [Å] Co 1- B 2,041(15) x2 Co 1 2,377(4) x1 Co 4 2,5165(18) x1 Co 1 2,5165(18) x4 Co 2 2,5957(12) x4 B 2,141(18) x3 Co 3/Mo 1 2,379(2) x1 Co 2 2,493(3) x3 Co 1 2,5957(12) x4 Co 2 2,379(2) x4 Co 1 2,8767(8) x8 Co 4- Co 2 2,5165(18) x 12 B- Co 1 2,041(15) x4 Co 2 2,141(18) x4 Co 2 - Co 3/Mo 1 - Bindungen - 248 - Das System Kobalt- Molybdän-Bor 4.10.3 Strukturdiskussion Co22,34Mo0,66B6 kristallisiert in der Cr23C6-Struktur in der Raumgruppe Fm 3 m mit einer Gitterkonstanten von a = 10, 4790(12) Å. Auch hier ist die Gitterkonstante sehr klein und erinnert an das binäre τ-Borid des Kobalts. Der Molybdänanteil in der Struktur ist so gering, dass er sich kaum in den Gitterkonstanten widerspiegelt. Ein Vergleich der Radien der beteiligten Metalle zeigt, dass der Radius von Molybdän (rMo = 1,362 Å)[190] nur um 8% größer als der von Kobalt (rCo = 1,253Å)[190] ist. Die Position des τ-Stabilisators (8c) ist überwiegend mit Kobaltatomen (68(6)%) besetzt, der Rest dieser kristallographischen Lage wird statistisch bis zu Vollständigkeit mit Molybdänatomen aufgefüllt. Bei dem Zentrum des Friauf-Polyeders kommt es wie schon öfters beschrieben zu einer sechzehnfachen Koordinationssphäre. Hier koordinieren die Dreiecksflächen der Kuboktaeder (dCo3/Mo1-Co1 = 2,8767(8) Å) und die Ecken der Würfel (dCo3/Mo1-Co2 = 2,379(2) Å) die Position 8c. Die Kuboktaeder auf der Lage 48h bestehen ausschließlich aus Kobaltatomen mit einem Abstand von 2,377(4) Å zueinander. Das Zentrum ist somit zwölffach im Abstand von 2,516(2) Å von Kobaltatomen umgeben. Die Würfel ebenfalls aufgebaut aus Kobaltatomen haben eine Kantenlänge von 2,493(3) Å. Die Borposition besitzt eine achtfach Koordination durch die Grundflächen der Würfel (dB-Co1 = 2,041(15) Å) und Kuboktaeder (dB-Co2 = 2,141(18) Å). - 249 - Ternäre Boride 4.11 Das System Kobalt-Magnesium-Bor Das Dreistoffsystem Kobalt-Magnesium-Bor wurde von Stadelmaier et al. auf die mögliche Anwesenheit der mit τ bezeichneten Phasen hin untersucht[110]. Aus den erhaltenen Pulverdaten konnte eine τ-Phase bestimmt werden, die eine Gitterkonstante von a = 10,541 Å zeigt, jedoch konnte diese weder einkristallin noch phasenrein dargestellt werden. 4.11.1 Präparative Untersuchung im System Kobalt-Magnesium-Bor Bei der Darstellung der τ-Boride im System Kobalt, Magnesium und Bor wurden die Elemente in verschiedenen stöchiometrischen Verhältnissen eingewogen und zu Tabletten verpresst. Anschließend wurden die Tabletten in BN-Tiegeln überführt und in Tantalampullen verschlossen14. Diese wurden darauf bei einer Temperatur von 1450°C (erste Versuchsserie) und bei 1250°C (zweite Versuchsreiche) für 10 Stunden zur Reaktion gebracht. Die Abkühlung wurde in beiden Fällen sehr langsam (15°C/h) bis auf eine Temperatur von 850°C vorgenommen, um eine möglichst gute Kristallisation zu gewährleisten. Über eine röntgenographische Phasenanalyse konnten die Proben auf die entstandenen Phasen hin untersucht werden. Bei einer Schmelzzusammensetzung von Co13Mg10B6 konnte die Existenz der τ-Phase mit einer Gitterkonstanten von a = 10,5598(2) Å bei einer Reaktionstemperatur von 1250°C erhalten werden. Bei einer Reaktion im Temperaturbereich von 1450°C mit identischer Schmelzzusammensetzung konnte eine τ-Phase mit einer Gitterkonstanten von a = 10,577(6) Å neben Co2B, Co3B verfeinert werden. Bei der Reaktionstemperatur von 1250°C für zehn Stunden wurde das τ-Borid in höheren Anteilen erhalten. Temperaturprogramm I: 100°C/h RT 15°C/h 150°C/h 1250°C 850°C (10h halten) (2h halten) RT Temperaturprogramm II: 100°C/h RT 15°C/h 150°C/h 1450°C 850°C (10h halten) (2h halten) RT 14 Magnesium besitzt einen Siedepunkt von 1107°C. Somit ist es notwendig, entsprechende Reaktionsgefäße zu verwenden, damit sich das Magnesium nicht schon vor der Reaktion verflüchtigt und zudem sollte es auch nicht mit dem Reaktionstiegel in Wechselwirkung treten. Für Synthesen mit Magnesium haben sich Al2O3-Tiegeln als ungeeignet gezeigt. - 250 - Das System Kobalt- Magnesium-Bor Tabelle 4.11.1.1: Proben im System Kobalt-Magnesium-Bor und ihre pulverdiffraktometrische Charakterisierung. (I) bei 1250°C und (II) bei 1450°C beziehen sich jeweils auf die Temperaturführung Zusammen- Nr. setzung Gehalt [At.-%] Gitterkonstante a [Å] weitere kristalline Phasen Co Mg B von Co23-xMgxB6 1 Co20Mg3B6 (I) 69,0 10,3 20,7 - Co2B, CoB 2 Co13Mg10B6 (I) 44,8 34,5 20,7 10,5598(2) Co2B 3 Co20Mg3B6 (II) 69,0 10,3 20,7 - Co2B 4 Co13Mg10B6 (II) 44,8 34,5 20,7 10,577(6) Co2B ,Co3B Abbildung 4.11.1 zeigt das erhaltene Pulverdiffraktogramm der Probe mit der Schmelzzusammensetzung von Co13Mg10B6, die bei einer Reaktionstemperatur von 1250°C umgesetzt wurde. Die roten Reflexe konnte eindeutig der τ-Phase zugeordnet werden. Als Hauptprodukt bei dieser Untersuchung ist Co2B entstanden. Bei einer höheren Temperatur (1450°C) konnte das τ-Borid noch beobachtet werden, jedoch in einer geringeren Intensität. Vielmehr dominierten bei einer Temperaturerhöhung die binären Boride das Kobalts Co2B und Co3B. 3000 Lin (Counts) 2000 1000 0 6 10 20 30 40 50 2-Theta - Scale Abbildung 4.11.1: Pulverdiffraktogramm, der Probe mit der Zusammensetzung Co13Mg13B6 (rot) mit einer Gitterkonstanten von a = 10,5598(2) Å. Die blaumarkierten Reflexe konnten als Co2B identifiziert werden. Die grünmarkierten Reflexe konnten keiner bekannten Phase zu geordnet werden. Tabelle 4.11.1.2 gibt die gesammelten Pulverdaten der Co20-xMgxB6 Verbindung wieder. - 251 - Das System Kobalt- Magnesium-Bor Tabelle 4.11.1.2: Pulverdaten von Co20Mg3B6. No. h k l 2θ [obs] 2θ [calc.] Intobs. dobs. dcalc.. 1 4 0 0 15,420 15,431 29,5 2,6434 2,6416 2 4 2 0 17,254 17,265 31,7 2,3647 2,3628 3 4 2 2 18,919 18,928 40,0 2,1579 2,1569 4 5 1 1 20,094 20,088 100,0 2,0329 2,0335 5 4 4 0 21,906 21,890 31,6 1,8666 1,8679 6 5 3 1 22,902 22,906 43,4 1,7864 1,7861 7 6 0 0 23,250 23,235 35,1 1,7600 1,7611 8 5 3 3 25,367 25,429 66,3 1,6152 1,6114 9 6 2 2 25,672 25,728 35,2 1,5964 1,5930 10 8 2 0 32,160 32,135 27,8 1,2804 1,2814 11 6 6 0 33,140 33,093 35,5 1,2436 1,2453 12 7 5 1 33,828 33,796 32,1 1,2190 1,2201 13 8 4 0 34,919 34,939 42,6 1,1821 1,1814 14 8 4 4 38,412 38,398 37,4 1,0781 1,0784 15 10 0 0 39,273 39,222 30,0 1,0553 1,0567 16 8 2 45,311 45,364 37,5 0,9207 0,9197 8 Bei der Untersuchung aller Proben, unter einem Lichtmikroskop und dem vermessen auf dem IPDS II auf die Bildung von möglichen Einkristallen des τ-Borids, konnten nur Einkristalle der binären Kobalt Verbindung des Typs Co2B isoliert werden. Das τ-Borid mit Magnesium als τ-Stabilisator zeigt keine Anzeichen in einkristalliner Form aufzutreten. - 252 - Ternäre Boride 5 Ternäre Boride 5.1 Cr2ZrB6 Über das ternäre Borid mit der Zusammensetzung von Cr2ZrB6 wurde im Jahre 2001 als neue Variante des Y2ReB6-Typs[194] berichtet. Neben der Cr2ZrB6 Verbindung konnte auch noch Zr2CrB6 als ein weiterer Vertreter mit geordneter Verteilung der Metallatome erhalten werden. Beide Verbindungen konnten bei einer Temperatur von 1500°C gewonnen werden. Im Folgenden wird auf die Synthese und die Charakterisierung von Cr2ZrB6 eingegangen. Cr2ZrB6 konnte aus einer Aluminiumschmelze erhalten werden. 5.1.1 Präparative Untersuchungen Für die Züchtung von Einkristallen, wurden die Elemente Zirkonium, Chrom, Aluminium und Bor in verschiedenen Verhältnissen eingewogen und in einem Achatmörser innig verrieben. Das homogene Gemisch wurde in Tabletten mit Hilfe einer Handpresse überführt. Die Tabletten wurden in Korundtiegeln unter Argonatmosphäre bei einer Temperatur von 1500°C und 1600°C für je 10 Stunden umgesetzt. Um eine gute Kristallisation zu erreichen, wurde die Abkühlrate um 10°C pro Stunde bis auf eine Temperatur von 600°C gewählt. Für die Züchtung von Einkristallen wurden zwei unterschiedliche Strategien vorgenommen. Bei der ersten Strategie wurde als Hilfsmetall Aluminium eingesetzt. Die Umsetzung der Proben wurde, wie bereits von Etzkorn diskutiert, bei einer Temperatur von 1500°C durchgeführt (siehe Tabelle 5.1.1.1). Die zweite Versuchsreihe wurde mit einem deutlichen Überschuss an Aluminium vorgenommen (siehe Tabelle 5.1.1.2). Hier wurde somit das Hilfsmetall als Lösungsmittel eingesetzt damit, eine gute Aktivität der Reaktionspartner gegeben ist. Die detaillierten Temperaturprogramme sowie die Einwagen der Edukte mit pulverdiffraktometrischer Charakterisierung nach der Hydrolyse erhaltenen Produktpalette sind in folgenden Tabellen zu finden. Temperaturprogramm der ersten Strategie: 100°C/h RT 10°C/h 30°C/h 1500°C 600°C (10h halten) (10h halten) RT - 253 - Ternäre Boride Tabelle 5.1.1.1: Proben Charakterisierung. Nr. Zusammen- für die Synthese von Gehalt [At.-%] setzung Zr Cr Al Cr2ZrB6 und ihre pulverdiffraktometrische weitere kristalline Hauptprodukt Phasen B 1 ZrCr2B6 11,1 22,2 - 66,7 ZrB2 Al5BO9, ZrB, CrB2, AlB2 2 Zr2CrAlB6 20,0 10,0 10,0 60,0 ZrB2 CrB 3 ZrCrAlB6 11,1 11,1 11,1 66,7 ZrB2 ZrB 4 ZrCrAl2B6 10,0 10,0 20,0 60,0 ZrB2 Al5BO9, Cr2B, Al2Zr 5 Zr2CrAl2B6 18,2 9,1 18,2 54,5 ZrB2 CrB2, Cr5B3 6 ZrCrAl3B6 9,1 9,1 27,3 54,5 ZrB2 Al5BO9, CrB 7 ZrCrAl4B6 8,3 8,3 33,3 50,0 ZrB2 CrB2, 8 Zr2CrAl3B6 16,7 8,3 25,0 50,0 ZrB2 CrB2, AlZr Der abgekühlte Schmelzregulus wurde in verd. Salzsäure eingelegt. Der kristalline Rückstand wurde gewaschen und abgesaugt. Nach dem Trocknen wurde der Rückstand einer röntgenographischen Phasenanalyse unterzogen. Tabelle 5.1.1.1 zeigte das Ergebnis der Phasenanalyse. In keinem der acht Ansätze konnte auch nur ein Hinweis auf das ternäre Produkt des Y2ReB6-Typs[195] vernommen werden. Bei der zweiten Versuchsreihe wurde die Reaktionstemperatur um 100°C erhöht. Des Weiteren wurde die Einwaage so veränderte, dass ein deutlicher Überschuss an Aluminium vorlag. Eine genaue Übersicht der eingesetzten Verhältnisse ist in der Tabelle 5.1.1.2 zu finden. In dem Ansatz mit der Schmelzzusammensetzung von Zr3Cr1Al30B10 konnte eine ausgeprägte Bildung von Cr2ZrB6 neben ZrB2 beobachtet werden. Eine Verfeinerung der Gitterparameter zeigte in der Raumgruppe Pbam Gitterkonstanten von a = 3,297(3) Å, b = 8,703(3) Å und c = 10,998(2) Å. Diese liegen in sehr guter Übereinstimmung mit der Literatur. Bei der Reaktion der Zusammensetzung von Zr4Cr3Al30B10 wurden neben ZrB2 metallisch glänzende Nadeln mit der Zusammensetzung von Cr2AlB2 beobachtet, diese werden im Kapitel 5.2 diskutiert. Temperaturprogramm der zweiten Strategie: 100°C/h RT 5°C/h 30°C/h 1600°C 600°C (10h halten) (10h halten) RT - 254 - Ternäre Boride Tabelle 5.1.1.2: Proben Charakterisierung. Nr. 1 Zusammensetzung Zr4Cr1Al30B10 für die Synthese von Cr2ZrB6 Gehalt [At.-%] und ihre pulverdiffraktometrische Hauptprodukt Zr Cr Al B 8,9 2,2 66,7 22,2 ZrB2 weitere kristalline Phasen - Cr2ZrB6 2 Zr3Cr1Al30B10 6,8 2,3 68,2 22,7 (RG: Pbam; Z=4 < ZrB2 a = 3,297(3) Å; b = 8,703(3) Å; c = 10,998(2) Å) 3 Zr4Cr2Al30B10 8,7 2,2 65,2 21,7 ZrB2 Spuren Cr2AlB2 4 Zr4Cr3Al30B10 8,5 6,4 63,8 21,3 ZrB2 - 5.1.2 Röntgenographische Untersuchungen an Einkristallen Mit dem IPDS II wurde in einem Winkelbereich von 0°≤ω≤180° ° mit ∆ω=2 (ψ = 0°, 111°;) vermessen. Nach der Mittelung in der Laueklasse mmm und einer numerischen Absorptionskorrektur konnte ein Datensatz mit 619 unabhängigen Reflexen erhalten werden. Da aufgrund der Gitterkonstante schon auf Cr2ZrB6 bzw. Zr2CrB6 geschlossen werden konnte, wurden als Startmodell die Atomparameter von Y2ReB6 herangezogen. Die Verfeinerung der 56 freien Parameter konvertierte problemlos bei Zuverlässigkeitswerten von R1(F) = 0,0196 und wR2(I) = 0,0516. Die wechselweise Freigabe der Besetzungsfaktoren aller kristallographischen Lagen zeigte keine erwähnungswerten Abweichung von der vollständigen Besetzung. - 255 - Ternäre Boride Tabelle 5.1.2.1: Angaben zur Strukturanalyse von Cr2ZrB6. Summenformel Cr2ZrB6 Kristallsystem orthorhombisch Raumgruppe P bam a =8,7075(17); b = 10,9904(22); c = 3,2944(7) Gitterkonstante [Å] 3 Volumen der Elementarzelle [Å ] 315,27(11) Zahl der Formeleinheiten 4 3 röntgenogr. Dichte [g/cm ] 5,479 F(000) 472,0 Kristallform, -farbe metallisch glänzende Nadeln Diffraktometer Rotation STOE IPDS II (Mo-Kα-Strahlung; λ=0,71073 Å Graphitmonochromator 0° ≤ ω ≤ 180° ψ = 0°, 111; ∆ω = 2° Belichtungszeit [sec.] 360 Messtemperatur [K] Raumtemperatur [293(2)] Kristallgröße [mm] 0,05 × 0,05 × 0,04 Absorptionskoeffizient [mm-1] 9,73 Messbereich [°] 4°<2θ<65° 4 ≤ h ≤ 4; Indexbereich -12 ≤ k ≤ 12; -16 ≤ l ≤ 16 Zahl der gemessenen Reflexe 4384 Zahl der unabh. Reflexe mit I > 0 619 I > σ2(I): 511 freie Parameter 56 Korrekturen Lorentz, Polarisation Absorptionskorrektur numerical / equivalent method (XSHAPE[68]) Strukturlösung Isotypie Y2ReB6[196] Strukturverfeinerung SHELXTL-97[199] Wichtungsschema w-1 = σ2F02 + (0,0305 * P)2 + 0,75 * P P = (F02 + 2 FC2) / 3 Goodness of fit 1,098 Extinktionskorrektur 0,0130(11) Restelektronendichte [e-/Å3]: +1,03 / -1,03; R-Werte für I > 0: Alle Reflexe 0,20 e-/Å3 R1(F) = 0,0196 wR2(I) = 0,0516 R1(F) = 0,0243 - 256 - Ternäre Boride Die folgende Tabelle zeigt die Ortskoordinaten, Besetzungsfaktoren und Auslenkungsparameter aller Atome. Tabelle 5.1.2.2: Ortskoordinaten, Besetzungsfaktoren (in [%]) und Auslenkungsparameter von Cr2ZrB6. Atom Lage x y z sof Ueq Zr 1 4g 0.17730(4) 0.41399(3) 0 100* 0,0048(1) Cr 1 4g 0.05545(7) 0.12693(6) 0 100* 0,0085(2) Cr 2 4g 0.14268(7) 0.68266(5) 0 100* 0,0058(1) B1 4h 0.2969(5) 0.2624(4) 0,5 100* 0,0058(6) B2 4h 0.0201(5) 0.2863(4) 0,5 100* 0,0064(7) B3 4h 0.4033(5) 0.4740(3) 0,5 100* 0,0059(7) B4 4h 0.2570(5) 0.5801(4) 0,5 100* 0,0059(6) B5 4h 0.0541(5) 0.5658(4) 0,5 100* 0,0054(6) B6 4h 0.3716(5) 0.3202(4) 0,5 100* 0,0050(6) * bei wechselweiser Freigabe der Besetzungsfaktoren keine Abweichung von vollständiger Besetzung Atom U11 U22 U33 U23 U13 U12 Zr 1 0.00505(17) 0.0047(2) 0.0047(2) 0.0 0.0 0.00009(11) Cr 1 0.00783(27) 0.0091(3) 0.0087(3) 0.0 0.0 0.00043(19) Cr 2 0.0056(3) 0.0057(3) 0.0061(3) 0.0 0.0 0.00029(19) B1 0.0065(14) 0.0043(15) 0.0066(16) 0.0 0.0 0.00017(12) B2 0.0051(15) 0.0056(15) 0.0086(17) 0.0 0.0 0.0012(12) B3 0.0050(14) 0.0052(16) 0.0074(18) 0.0 0.0 0.0005(12) B4 0.0048(14) 0.0055(15) 0.0075(16) 0.0 0.0 0.00004(11) B5 0.0043(14) 0.00623(15) 0.0057(15) 0.0 0.0 0.0005(12) B6 0.0048(14) 0.0043(15) 0.0 0.0 0.0004(12) 0.0060(17) Die folgende Tabelle zeigt benachbarte Atome, ihren charakteristischen Abstand und die zugehörige Koordinationszahl. - 257 - Ternäre Boride Tabelle 5.1.2.3: Ausgewählte Abstände in der Cr2ZrB6-Struktur. Benachbarte Atome Zr 1- Cr 1 - Cr 2 - B 1- B2- Abstand [Å] Anzahl der Benachbarte Bindungen Atome B1 2,554(3) x2 B4 2,555(3) B2 B 3- Abstand [Å] Anzahl der Bindungen B6 1,714(6) x1 x2 B4 1,727(6) x1 2,560(3) x2 B1 1,778(8) x1 B6 2,576(3) x2 Cr 1 2,280(3) x2 B5 2,578(3) x2 Cr 1 2,387(3) x2 B5 2,612(3) x2 Zr 1 2,650(3) x2 B6 2,370(3) x2 B4 2,376(3) B3 B4 B4 1,727(6) x1 x2 B3 1,765(6) x1 2,380(3) x2 B1 1,774(6) x1 B3 2,387(3) x2 Cr 2 2,230(3) x2 B1 2,418(3) x2 Cr 1 2,376(3) x2 B2 2,424(3) x2 Zr 1 2,555(3) x2 B2 2,200(3) x2 B5 1,725(8) x1 B1 2,209(3) x2 B2 1,750(6) x1 B5 2,227(3) x2 B4 1,774(6) x1 B4 2,230(3) x2 Cr 2 2,227(3) x2 B6 2,339(3) x2 Zr 1 2,578(3) x2 Cr 2 2,699(1) x1 Zr 1 2,612(3) x2 Cr 2 2,712(1) x1 B2 1,681(6) x1 B3 1,714(6) x1 B6 1,745(6) x1 B3 1,725(6) x1 B4 1,765(6) x1 B1 1,744(6) x1 Cr 2 2,209(3) x2 Cr 2 2,239(3) x2 Cr 1 2,418(3) x2 Cr 1 2,367(3) x2 Zr 1 2,554(3) x2 Zr 1 2,576(3) x2 B1 1,681(6) x1 B6 1,744(6) x1 B5 1,750(6) x1 Cr 2 2,200(3) x2 Cr 1 2,424(3) x2 Zr 1 2,560(3) x2 B5 B6 - 258 - Ternäre Boride 5.1.3 Strukturdiskussion Cr2ZrB6 kristallisiert orthorhombisch in der Raumgruppe Pbam mit den Gitterkonstanten a = 8,7075(17) Å; b = 10,9904(22) Å; c = 3,2944(7) Å. Die bereits bekannte Cr2ZrB6 Verbindung kristallisiert im Y2ReB6-Typ. In der Zelle befinden sich vier Formeleinheiten. Das besondere Merkmal dieser Struktur ist, dass Cr2ZrB6 der erste Vertreter seiner Art, ohne den Einbau eines Seltenerdmetalls ist. Wie aus der Abbildung 5.1.1 zu entnehmen ist, liegen in dieser Struktur Borschichten vor, die deckungsgleich in der [001]-Richtung gestapelt sind. Die Boratome bilden Fünf-, Sechs- und Siebenringe aus entlang der a-b-Ebene. Alle prismatischen Lücken zwischen den Borlagen werden vollständig und geordnet mit den Atomen Zirkon und Chrom besetzt. Das größere Zirkon (rZr: 2,16 Å) erhält jeweils eine vierzehnfache Koordination und befindet sich in allen heptagonalen Prismen, die durch die Boratome gebildet werden. Abbildung 5.1.1: Kristallstruktur von Cr2ZrB6; Das Chromatom (rCr: 1,85Å) besetzt Polyderdarstellung der Metallatome alle Lücken der pentagonalen und hexagonalen Prismen. Somit entstehen alternierend Schichten bestehend aus einer Metalllage gefolgt von einer Borlage. Alle Zirkonatome werden von 14 Boratomen im Abstand von 2,554(3) bis 2,612(3) Å koordiniert. Die heptagonalen-Prismen sind verzerrt, was auf ihre Umgebeung zurückzuführen ist, denn an jeden heptagonalen-Prismen sind entlang der a-b-Ebene genau ein heptagonales-Prisma (besetzt mit einem Zirkonatom) (besetzt mit und je einem pentagonale-Prismen Chromatom) angeordnet. drei hexagonale-Prismen Chromatom) (besetzt sowie mit drei einem Abbildung 5.1.2: Koordinationssphäre des Zirkon (CN 14) - 259 - Ternäre Boride Es existieren zwei kristallographisch unterscheidbare Chromatome. Das Chromatom der Sorte 1 besetzt alle hexagonalen Prismen. Hier sind die Boratome mit 2,370(3) Å bis 2,424(3) Å um das Übergangsmetall angeordnet. Alle anderen Chromatome besetzen die Lücken der pentagonalen Prismen (dBx-Co2: 2,200(3) bis 2,339(3) Å). Abbildung 5.1.3: Koordinationssphäre des Chroms (Cr 1: CN 12 und Cr 2: CN, 10) - 260 - Ternäre Boride 5.2 Cr2AlB2 Bei der Untersuchung von ternären Boriden und der Synthese von Cr2ZrB6 aus einer Aluminiumschmelze, konnten metallisch glänzende Nadeln mit der Zusammensetzung von Cr2AlB2 aus einem Ansatz mit der Schmelzzusammensetzung von Cr2Zr4Al30B10 beobachtet werden. In der Literatur wurde die ternäre Verbindung mit der Zusammensetzung Cr2AlB2[197] anhand von Pulverdaten charakterisiert. Cr2AlB2 kristallisiert orthorhombisch in der Raumgruppe Cmmm und den Gitterkonstanten von a = 2,937 Å, b = 11,07 Å und c = 2,971 Å. 5.2.1 Präparative Untersuchungen Einkristalle konnten in Form von metallisch glänzenden Nadeln aus einem Ansatz erhalten werden, in dem die Elemente Chrom 0,036 g (0,392 mol), Zirkonium 0,25 g (2,72 mol), Aluminium 0,277 g (0,010 mol) und Bor 0,037 g (3,422 mmol) bei einer Temperatur von 1600°C miteinander umgesetzt wurden (in der Abbildung 5.2.1 wurde der untersuchte Einkristall dargestellt). Dazu wurden die eingewogenen Pulver in einem Achatmörser homogenisiert und mit einer Tablettenhandpresse zu Presslingen verarbeitet. Die Umsetzung erfolgte 24 Stunden in Korundtiegeln unter Argonschutzgas. Um eine gute Kristallisation zu gewährleisten, wurde mit 5°C pro Stunde bis auf eine Temperatur von 600°C abgekühlt. Weitere Details zur Temperaturbehandlung des Presslings sind unten zu entnehmen. 50 µm Abbildung 5.2.1: Einkristalle des Cr2AlB2 gewonnen aus eine Ansatz mit der Schmelzzusammensetzung von Cr2Zr8Al30B10. Temperaturprogramm: 100°C/h RT 5°C/h 30°C/h 1600°C 600°C (10h halten) (10h halten) RT - 261 - Ternäre Boride Der abgekühlte Schmelzregulus wurde mit konzentrierter konz. HNO3 behandelt, der erhaltene Rückstand wurde abgesaugt, gewaschen und über Nacht an der Luft getrocknet. Zurück blieben wenige gut ausgeprägte metallische Nadeln mit einer durchschnittlichen Länge von 150 µm und einer Dicke von 10 µm übrig. Diese wurden unter einem Lichtmikroskop erlesen und für weitere Untersuchungen, wie die röntgenographische Untersuchung an Einkristallen, auf dem IPDS II sowie ein EDX-Messungen weiter verwendet. 5.2.2 EDX-Untersuchungen Mit Hilfe der EDX-Untersuchung konnten die Elemente an den isolierten Einkristallen erfasst und eindeutig zugeordnet werden. Diese Zusatzinformation erwies sich als eine große Unterstützung bei der Strukturverfeinerung. Abbildung 5.2.2 und Tabelle 5.2.2.1 zeigen das Ergebnis der qualitativen Elementaranalyse. Abbildung 5.2.2: EDX-Messung an Einkristallen gewonnen aus der Schmelzzusammensetzung von Cr2Zr8Al30B10. Dargestellt ist: das Spektrum (links) und der gemessene Einkristall (rechts) sowie eine der fünf Messstellen. Tabelle 5.2.2.1 zeigt das Ergebnis aus der EDX-Untersuchung. Das dargestellte Resultat ist die Mittelung aus fünf Messstellen. Es konnten ausschließlich die Metalle Chrom, Aluminium und das Nichtmetall Bor15 detektiert werden. Die Messung der Elemente konnte die Strukturverfeinerung festigen und zeigte eine nahezu stöchiometrische Zusammensetzung an. 15 An dieser Stelle soll auch erwähnt werden, dass am untersuchten Einkristall an einigen Stellen geringe Mengen an Kohlenstoff festgestellt wurden konnten. - 262 - Ternäre Boride Tabelle 5.2.2.1: Verhältnis der Atome gemessen an Einkristallen Cr2AlB2. Aufgeführt sind die gewonnenen Mittelwerte aus fünf Messstellen. Element Gewichts [%] Atomist [%] σ [%] Atomsoll [%] Cr K 74,26 49,62 1,38 50,0 Al K 16,79 22,62 1,04 25 BK 8,95 27,76 0,34 25 5.2.3 Röntgenographische Untersuchungen an Einkristallen Mit dem IPDS II wurde der Einkristall in einem Winkelbereich von 180° in zwei Grad Schritten vermessen. Der Detektorabstand wurde auf 80 mm eingestellt, die Belichtungszeit pro Aufnahme betrug 5 Minuten. Es konnten 1357 Reflexe gesammelt werden. Eine Mittelung in der Laue-Klasse mmm und eine numerische Absorptionskorrektur ergaben einen Datensatz von 142 unabhängigen Reflexen. Ein Vergleich der erhaltenen Gitterkonstanten mit denen in der Literatur beschriebenen, wies auf die Cr2AlB2-Struktur (Fe2AlB2-Typ: a = 2,937 Å, b = 11,07 Å, c = 2,971 Å)[197] hin. Aufgrund der Ähnlichkeit der Zelle wurde in der Raumgruppe Cmmm mit der Strukturverfeinerung begonnen, die Verfeinerung konvergierte sehr schnell zum Fe2AlB2-Strukturtyp. Somit wurde für die Strukturverfeinerung das Startmodell mit den Atomkoordinaten aus den publizierten Pulverdaten von Chaban et al. verwendet. Die Verfeinerung der 14 freien Parameter lieferte nach der Einführung anisotroper Temperaturfaktoren Zuverlässigkeitswerte von R1(F) = 0,0248 und wR2(I) = 0,0421. Eine wechselweise Freigabe der Besetzungsfaktoren zeigte bei keiner der kristallographischen Lagen eine signifikante Abweichung von der vollständig geordneten Besetzung der Atomposition. Die durch die röntgenographische Strukturverfeinerung ermittelte Zusammensetzung konnte durch die EDX-Untersuchung bestätigt werden. Der Einbau des Zirkons konnte eindeutig ausgeschlossen werden. Alle Einzelheiten zur Strukturbestimmung sowie die Messparameter und ausgewählte Abstände mit Koordinationsumgebung können in den folgenden Tabellen entnommen werden. - 263 - Ternäre Boride Tabelle 5.2.3.1: Angaben zur Strukturanalyse von Cr2AlB2. Summenformel Cr2AlB2 Kristallsystem orthorhombisch Raumgruppe C mmm a = 2,9363(6); b = 11,0323(22); c = 2,9538(6) Gitterkonstante [Å] 3 Volumen der Elementarzelle [Å ] 95,68(3) Zahl der Formeleinheiten 2 3 röntgenogr. Dichte [g/cm ] 5,296 F(000) 142,0 Kristallform, -farbe metallisch glänzende Nadeln Diffraktometer Rotation STOE IPDS II (Mo-Kα-Strahlung; λ=0,71073 Å Graphitmonochromator 0° ≤ ω ≤ 180° ψ = 0°, 111; ∆ω = 2° Belichtungszeit [sec.] 300 Messtemperatur [K] Raumtemperatur [293(2)] Kristallgröße [mm] 0,02 × 0,02 × 0,15 Absorptionskoeffizient [mm-1] 11,29 Messbereich [°] 2°<2θ<70° 0 ≤ h ≤ 4, Indexbereich 0 ≤ k ≤ 17, 0≤l≤4 Zahl der gemessenen Reflexe 1357 Zahl der unabh. Reflexe mit I > 0 263 I > σ2(I): 142 freie Parameter 11 Korrekturen Lorentz, Polarisation Absorptionskorrektur numerical / equivalent method (XSHAPE[68]) Strukturlösung Fe2AlB2-Typ[198] Strukturverfeinerung SHELXTL-97[199] Wichtungsschema w-1 = σ2F02 + (0,0 * P)2 + 0,00 * P P = (F02 + 2 FC2) / 3 Goodness of fit 0,592 Extinktionskorrektur 0,028(9) Restelektronendichte [e-/Å3]: +1,21 / -1,15; R-Werte für I > 0: Alle Reflexe 0,23 e-/Å3 R1(F) = 0,0248 wR2(I) = 0,0421 R1(F) = 0,0405 - 264 - Ternäre Boride Tabelle 5.2.3.2: Ortskoordinaten, Besetzungsfaktoren (in [%]) und Auslenkungsparameter von Cr2AlB2. Alle Atome wurden anisotrop verfeinert. Atom Cr Al B Lage x 0 4i 2c 4j y z 0,14668(8) 0 sof Ueq 100* 0,0042(3) * 0,0052(5) 0,0053(9) 0 0,5 -0,5 100 0 0,2941 -0,5 100* * bei wechselweiser Freigabe der Besetzungsfaktoren keine signifikante Restelektronendichte vorhanden Atom U11 U22 U33 U23 U13 U12 Cr 0.0051(4) 0.0041(4) 0.0033(4) 0.0 0.0 0.0 Al 0.0080(11) 0.0033(9) 0.0044(10) 0.0 0.0 0.0 B 0.004(2) 0.007(2) 0.005(2) 0.0 0.0 0.0 Die folgende Tabelle zeigt benachbarte Atome und ihren charakteristischen Abstand in der typischen Koordination. Tabelle 5.2.3.3: Ausgewählte Abstände in der Cr2AlB2-Struktur. Anzahl der Benachbarte Atome Abstand [Å] Cr - B 2,183(2) x4 B 2,197(4) x2 Al 2,637(1) x4 Cr 2,712(1) x2 Cr 2,936(1) x2 B 2,271(6) x2 Cr 2,637(1) x8 Al 2,936(1) x2 B 1,761(4) x2 Cr 2,183(2) x4 Cr 2,197(4) x2 Al - B- Bindungen - 265 - Ternäre Boride 5.2.4 Strukturdiskussion Cr2AlB2 kristallisiert orthorhombisch in der Raumgruppe Cmmm mit den Gitterkonstanten a = 2,9363(6) Å; b = 11,0323(22) Å und c = 2,9538(6) Å. Cr2AlB2 ist ein Isotyp zu Fe2AlB2 und kristallisiert mit zwei Formeleinheiten pro Zelle. Die Betrachtung der Struktur in Blickrichtung entlang der c-Achse zeigt Bor-Bor-Zickzackketten mit einem charakteristischen Abstand von 1,761(4) Å. Ähnlich wie schon bei CrB (Abbildung 5.2.4: Abstand von B-B: 1,73 Å) zeigt die Cr2AlB2-Struktur die markanten Zickzackketten der Boratome entlang der Metallprismen. Trigonale Prismen aus Chromatomen sind sowohl über Rechtecks- (dCr-Cr: 2,936(1) Å) als auch Dreiecksflächen 2,712(1) Å) zu (dCr1-Cr1: Schichten verknüpft, welche entlang [010] übereinander gestapelt sind. Wie schon oft in ternären Verbindung beobachtet, liegen auch hier isolierte Metallschichten (dAl-Al: 2,936(1) Å) bestehend Aluminiumatomen vor. aus Somit befindet sich jedes AluminiumAbbildung 5.2.3: Kristallstruktur von Cr2AlB2: Dargestellt sind Bor-Bor-Ketten und die charakteristischen Metallpolyeder. atom in direkter Nachbarschaft zu acht Chromatomen, (dCr-Al: 2,936(1) Å), die einen Würfel ausbilden. Zudem sind je zwei Boratome (dB-Al: 2,637(1) Å) und je vier Aluminiumatomen (dB-Al: 2,271(6) Å) über die Fläche an der Koordination des AlCr8Würfels beteiligt. Die Koordination des Chromatoms besteht aus Teilen der Borkette (dCr-B: 2,183(2) Å und dCr-B: 2,197(4) Å) Abbildung 5.2.4: Kristallstruktur von der sowie aus vier Aluminiumatomen (dCr-Al: Verbindung CrB (Projektion auf die [100-Ebene]). 2,936(1) Å). Dargestellt sind die Bor-Bor-Zickzackketten. - 266 - κ-Carbide 6 κ-Carbide Bei der Reproduktion der Komplexcarbide vom Typ der h-Carbide des Wolframs W6Co6C[200], W3Co3C[200] sowie W4Co2C[200] in einer Aluminium/Kobalt-Schmelze bei 1500°C wurden metallisch glänzende Stäbchen mit einer hexagonalen Grundfläche beobachtet (Abbildung 5.2.1 und Abbildung 6.2.1). Die Einkristalluntersuchung des W-Al-Co-Carbids zeigte nach der Indizierung Gitterparameter von a = 7,9050(11) Å und c = 7,7869(16) Å in einem hexagonalen Kristallsystem mit der Raumgruppe P63/mmc. Ein Vergleich der Gitterparameter mit bekannten Carbiden zeigte eine Isotypie mit den bekannten Carbiden (W6Fe1,5Al5,5C3 und W6Mn1,5Al5,5C3), die als κ-Phasen[201] bezeichnet werden. Abbildung 5.2.1: Einkristalle des κ-Carbides W9Al3Co1C3. 6.1 Präparative Untersuchungen Die Wahl der Reaktionsbedingungen für die Züchtung der κ-Phasen aus einer Aluminium/Kobalt Schmelze erfolgte auf der Basis der Arbeit vom J. Etzkorn[202]. Ziel war die Züchtung des h-Carbids mit der Zusammensetzung W3Al2CoC ausgehend vom W3Co3CTyp. Die Substitution des Kobalts durch Aluminium konnte bereits bei der analogen Verbindung Ta3Al2CoC[203], abgeleitet aus Ta3Co3C[204] beobachtet werden. Die Einkristalle von W9Al3CoC3 wurden aus einem Ansatz mit 0,6105 g (3,321 mmol) Wolfram, 0,2323 g (8,941 mmol) Aluminium, 0,1384 g (2,348 mmol) Kobalt und 0,0190 g (1,582 mmol) Kohlenstoff in der Form von metallisch glänzenden hexagonalen Prismen - 267 - κ-Carbide erhalten. Die Pulver wurden homogenisiert und in eine Tablette gepresst. Diese wurde im Korundtiegel bei einer Reaktionstemperatur von 1500°C und einer Reaktionszeit von 30 Stunden unter Argonschutzgas umgesetzt. Die Probe wurde sehr langsam (4°/h) bis 900°C abgekühlt. Der Schmelzregulus wurde mit konzentrierter Salpetersäure behandelt. Der Rückstand wurde mit dest. Wasser gewaschen, abgesaugt und getrocknet. Temperaturprogramm: 150°C/h 4°C/h RT 30°C/h 1500°C 900°C (30h halten) (5h halten) RT 6.2 EDX- Untersuchung Der isolierte Einkristall der in der Abbildung 6.2.1 dargestellt ist, wurde an zehn unterschiedlichen Stellen mittels energiedispersiver Röntgenemissionspektroskopie untersucht. Das Ergebnis der EDX-Analyse bekräftigte dis Aussage der Strukturverfeinerung. Abbildung 6.2.1: REM-Aufnahme des κ-Carbids mit der Zusammensetzung W9Al3CoC3. In der Abbildung ist die dritte von zehn Messstellen markiert. Abbildung 6.2.2: Spektrum der W9Al3CoC3-Probe. Tabelle 5.2.4.1: Gemitteltes Ergebnis; Metallverhältnis aus zehn Messstellen an der Probe der Zusammensetzung W9Al3CoC3. Element Atomist [%] σ [%] Atomsoll [%] Al K 19,4 0,8 20 Co K 9,3 0,6 10 WM 71,3 1,5 70 - 268 - κ-Carbide Als weiteres Element wurde Kohlenstoff in der Probe detektiert werden. Im Rahmen der Standardabweichung konnte die Summenformel von W9Al3CoC3 durch die EDX bestätigt werden. 6.3 Härtemessung an Einkristallen Für die Härtemessung wurden drei der nadelförmigen Einkristalle ausgelesen und auf dem IPDS II eindeutig als W9Al3CoC3 identifiziert. Die ausgewählten Einkristalle wurden so präpariert, dass die Grundfläche und die Längsseite auf Härte geprüft werden konnte. Anschließend wurden die Kristalle in Epoxidharz mit dem Namen „Körapox“ kalt eingebettet. Nach dem Aushärten des Epoxidharzes wurden die Einkristalle an unterschiedlichen Stellen auf ihre Härte hin untersucht. Abbildung 6.3.1 zeigt zwei Vickers Härteeindrücke und einen Knoop Härteeindruck, die Vickerspyramide zeigt leichte Rissbildung an den Ecken der Pyramide (Abdruck rechts: Risse wurden mit rotem Kreis markiert). Vickers Abdruck 1614 HV 0,2/10 Knoop Abdruck 1329 HK 0,2/10 50 µm Abbildung 6.3.1: Vickers-(dØ: 15,15µm) und Knoopabdrücke (dlang: 46,08µm) auf der Oberfläche eines κ-Carbides mit der Zusammensetzung W9Al3CoC3 Einkristalls Die untersuchten Einkristalle zeigten reproduzierbare und konstante Härtewerte. Mit der Vickers Härteprüfung konnte eine mittlere Härte von 1615 HV 0,2/10 (~15,5 GPa) ermittelt werden. - 269 - κ-Carbide Anschließend an die Vickers Härteprüfung wurden alle Flächen der eingebetteten Einkristalle mit dem Knoop Eindringkörper auf anisotropes Verhalten untersucht, d. h. der rhombische Indenter wird richtungsorientiert in die Probe gedrückt. Die untersuchten Einkristalle zeigten einen kontinuierlichen Wert 1301 HK 0,2/ 10 unabhängig von der Prüfstelle. Untersuchte Prüfstellen: hexagonale Grundfläche entlang der sechszähligen Achse wie auch senkrecht zu dieser. Im Rahmen dieser Arbeit konnten Sinterkörper von Ta3Al2CoN und Ta3Al2CoC im Pulsplasmaofen synthetisiert werden. An diesen Proben war eine Härteprüfung nach Vickers wie auch nach Knoop möglich. Hier konnten eine Härte für Ta3Al2CoN von 13,3 GPa (ermittelt aus der Härteprüfung nach Vickers) und für Ta3Al2CoC ein Härtewert von 13 GPa erhalten werden. Die h-Carbids mit Wolfram als Hauptbestandteil zeigten eine wesentlich höhere Härte an. Wolfram ist als ein starker Carbidbildner bekannt, sein Einfluss zeigt sich oft in der Erhöhung der Zähigkeit, Wärmefestigkeit wie auch bei der Steigerung der Härte. Er ist deshalb ein wichtiger Bestandteil bei Schnell- und Warmarbeitsstählen. 6.4 Röntgenographische Untersuchung an Einkristallen Für eine vollständige Strukturbestimmung wurde auf dem IPDS II in einem Winkelbereich von von 180° in zwei Grad Schritten und einer Belichtungszeit von 5 Minuten pro Aufnahme vermessen. Bei der wechselweisen Freigabe der Besetzungsfaktoren aller Positionen, zeigte kein Atom eine signifikante Abweichung von der vollständigen Besetzung. Die Verfeinerung der 20 freien Parameter konvergierte nach der Einführung von anisotropen Temperaturfaktoren für alle Metalllagen bei Zuverlässigkeitswerten von R1(F) = 0,0290 und wR2(I) = 0,0605. - 270 - κ-Carbide Tabelle 5.2.4.1: Angaben zur Strukturanalyse von W9Al3CoC3. Summenformel W9Al3CoC3 Kristallsystem hexagonal Raumgruppe P 63/mmc – Nr. 194 a = 7,9051(11); c = 7,7869(16) Gitterkonstante [Å] 3 Volumen der Elementarzelle [Å ] 421,41(12) Zahl der Formeleinheiten 4 3 röntgenogr. Dichte [g/cm ] 14,426 F(000) 1536,0 Kristallform, -farbe metallisch glänzende Nadeln STOE IPDS II (Mo-Kα-Strahlung; λ=0,71073 Å Diffraktometer Graphitmonochromator Raumtemperatur [293(2)] Messtemperatur [K] 0° ≤ ω ≤ 180° Rotation ψ = 0°, 111; ∆ω = 2° Belichtungszeit [sec.] 300 Kristallgröße [mm] 0,03 × 0,03 × 0,13 Absorptionskoeffizient [mm-1] 124,42 Messbereich [°] 2°<2θ<65° -11 ≤ h ≤ 11, -11 ≤ k ≤ 11, Indexbereich -11 ≤ l ≤ 11 Zahl der gemessenen Reflexe 42641 Zahl der unabh. Reflexe mit I > 0 309 I > σ2(I): 212 freie Parameter 20 Korrekturen Lorentz, Polarisation Absorptionskorrektur numerical / equivalent method (XSHAPE[68]) Strukturlösung W9Co3C3 Strukturverfeinerung SHELXTL-97[199] w-1 = σ2F02 + (0,0342 * P)2 + 0,00 * P Wichtungsschema P = (F02 + 2 FC2) / 3 0,875 Goodness of fit 0,01004(6) Extinktionskorrektur - 3 Restelektronendichte [e /Å ]: R-Werte für I > 0: Alle Reflexe [200] +4,33 / -3,14; 0,67 e-/Å3 R1(F) = 0,0290 wR2(I) = 0,0605 R1(F) = 0,0453 - 271 - κ-Carbide Tabelle 5.2.4.2: Ortskoordinaten, Besetzungsfaktoren (in [%]) und Auslenkungsparameter von W9Al3CoC3. Atom Lage W1 12k W2 x y z 0,20447(6) 0,79553(6) 0,0690(1) Sof [%] Ueq 100* 0,0096(8) * 0,0093(3) 0,55274(8) 0,10549(16) 0,25 100 Co 6h 2a 0,0 0,0 0,0 100* 0,0142(14) Al 6h 0,2184(12) 1,1092(6) 0,25 100* 0,0105(16) C 6g 0,0 0,5 0,0 100* 0,026(11)* * bei wechselweiser Freigabe der Besetzungsfaktoren keine signifikante Restelektronendichte vorhanden Atom U11 U22 U33 U23 U13 U12 W1 0,0071(3) 0,0071(3) 0,01333(4) 0,0002(1) 0,00015(14) 0,0025(3) W2 0,0076(4) 0,0084(6) 0,0122(5) 0 0 0,002(3) Co 0,0123(2) 0,0123(18) 0,018(4) 0 0 0,0062(9) Al 0,002(6) 0,006(2) 0,022(5) 0 0 0,012(18) Die folgende Tabelle zeigt benachbarte Atome und ihren charakteristischen Abstand und die Koordinationszahl. Tabelle 5.2.4.3: Ausgewählte Abstände in der W9Al3CoC3-Struktur. Benachbarte Abstand Anzahl der Benachbarte [Å] Bindungen Atome C 2,1408(4) x2 Al 2,8056(4) x1 Al 2,8060(8) x2 W1 2,8189(17) x1 Co 2,8507(9) W2 W1 Atome W1- W2- Co - Abstand [Å] Anzahl der Bindungen Al 2,455(5) x6 W1 2,8507(9) x6 x Co 2,455(5) x2 x1 Al 2,590(1) x2 2,9706(9) x1 W2 2,658(7) x2 2,999(1) x1 W1 2,806(4) x2 W1 2,8060(8) x4 W2 2,0764(5) x2 W1 2,1408(4) x4 C 2,0754(5) x2 Al 2,658(7) x2 W2 2,702(2) x2 W1 2,9706(9) x4 W1 2,9940(9) x2 Al - C- - 272 - κ-Carbide 6.5 Strukturdiskussion Die Kristallstruktur des κ-Carbids mit der stöchiometrischen Zusammensetzung von W9Al3CoC3 kristallisiert in der hexagonalen Raumgruppe P63/mmc. In der Zelle befinden sich vier Formeleinheiten. Die Gitterkostanten betragen sind a = 7,9051(11) Å und c =7,7869(16) Å. Das Wirtgitter ist mit dem Mn3Al10-Typ verwandt. Teile des Gitters in dem W9Al3CoC3 Carbid bestehen aus [W6C]-Oktaedern, welche über Ecken miteinander verkettet sind. Somit besitzt jedes Kohlenstoffatom eine verzerrte oktaederische Koordination von Wolframatomen im Abstand von dW2-C: 2,0764(5) Å (x2) und dW1-C: 2,1408(4) Å (x4). Der Wolfram-Kohlenstoff Abstand Abbildung 6.5.1:Elementarzelle des κ-Carbids W9Al3CoC3. Blick entlang der c-Achse auf verkippte über Ecken verknüpfte [W6C]-Oktaeder. Aluminiumatome bilden hier liegt deutlich unter dem des WC (dW-C: 2,197 Å)[205]. über flächenverknüpfte Verkettung von [Al6Co]Oktaedern in Blickrichtung [001] den weiteren Aufbau der Zelle. Die Kobaltatome im Inneren des perfekten Al-Oktaeders (dCoAl: 2,455(5) Å) haben untereinander einen Abstand von 3,893(1) Å. Der Aluminiumoktaeder besitzt eine Kantenlänge von 2,590(1) Å. Abbildung 6.5.2: Elementarzelle des κ-Carbids W9Al3CoC3. Blick entlang der a-Achse auf verkippte über Ecken verknüpfte [W6C]-Oktaeder sowie die über die Flächenverknüpfte [W6Co]-Oktaeder entlang der c-Achse. - 273 - Ternäre Boride 7 Weiter Untersuchungen zur Härte Ein wichtiger Teil dieser Arbeit war, das Einrichten und Kalibrieren der Härtemessung nach Vickers und Knoop am Institut der Anorganischen und Analytischen Chemie in Freiburg. Hier bestand die Hauptaufgabe in der Inbetriebnahme des Härtetesters, Außerdem sollte eine Verfahren für die Präparation von Einkristallen für dir Mikrohärtemessung entwickelt werden Neben den vorgestellten Resultaten sollten im Rahmen dieser Arbeit weitere Verbindungen in einkristalliner Form auf ihre Härte hin untersucht werden. Alle untersuchten Einkristalle wurden zuvor auf dem IPDS II vermessen. Die Einkristalle wurden mit den zu prüfenden Flächen ausgerichtet und in Epoxidharz eingebettet. An dieser Stelle soll noch einmal auf die Schwierigkeit bei der Bestimmung der Mikrohärte an Einkristallen hingewiesen werden. Das größte Problem ergibt sich durch die Größe der zur Verfügung stehenden Fläche und durch die Reproduzierbarkeit der Messung, wenn ein Abdruck pro Kristall möglich ist. Eine weitere Unsicherheit bei der Messung ergibt sich, wenn eine Mindestdicke nicht gegeben ist. So kann es zum Einsinken des Prüfkörpers kommen. Die Messung der Mikrohärte an Einkristallen stellt eine große Herausforderung an die Härteprüfmethoden wie auch schon an die Präparation (Einbetten, Schleifen, Läppen und Polieren) dar. Im Folgenden werden Boride, Borcarbide und Bornidride auf ihre Härte hin untersucht und verglichen. 7.1 Boride Härtemessung an Mg1,1B12Si2 –Kristallen Bei dem Borid der Zusammensetzung von Mg1,1B12Si2 konnte nach der Messung von zwei Einkristallen mit jeweils 3 Abdrücken eine Härte von 1942 HV 0,2/10 und 1796 HK 0,2/10 ermittelt werden. Die Einkristalle der Probe zeigen Rissbildungen an den Ecken des Abdrucks beider Eindringkörper. Rissbildungen können bei Einkristallen mit großer Spröde oder schlechter Qualität auftreten. - 274 - Weitere Untersuchungen zur Härte Rissbildung Rissbildung Abbildung 7.1.1: Abbildung 7.1.2: d1=14,29µm, d2=12,29µm: 2038 HV Knoop-Abdruck. d1=39,31µm: 1877 HK Härtemessung an LiB12PC -Kristallen Von der Verbindung LiB12PC konnte nur ein geeigneter Einkristall für eine Härtemessung isoliert werden. Abbildung 7.1.3 präsentiert die zwei Prüfstellen, an diesen die Probe auf ihre Härte nach Vickers untersucht wurde. Eine Prüfung nach Knoop war durch die Einhaltung des Mindestabstandes von benachbarten Abdrücken nicht mehr realisierbar. Aus den zwei Prüfstellen konnten eine Härte von 2746 HV 0,2/10 (27 GPa) bestimmt werden. Beide Abdrücke waren riss- und verzerrungsfrei. Abbildung 7.1.3: d1=11,75µm, d2=11,76µm: 2682 HV Härtemessung an MgB7 -Kristallen Von den roten Einkristallen der Verbindung MgB7 wurden zwei Einkristalle auf ihre Härte geprüft. Es konnte eine Härte von 2125 HV 0,2/10 (20,5 GPa) nach 8 Abdrücken festgestellt werden. Die Knoophärte wurde aus vier Abdrücken ermittelt und kann mit einem Wert von 2004 HK 0,2/10 angegeben werden. Der Knoopabdruck war rissfrei und reproduzierbar, bei - 275 - Weitere Untersuchungen zur Härte der Vickersprüfung sind große Risse entstanden. Diese sind in der Abbildung 7.1.4 dargestellt. Rissbildung Abbildung 7.1.4: d1=13,95µm, d2=13,59µm 1997 HV Härtemessung an MgB12-Kristallen Eine Härte von 2360 HV 0,2/10 und 2345 HK 0,2/10 konnte an dem binären Borid mit der Zusammensetzung von MgB12 gemessen werden. Hier handelt es sich um eine sehr harte und spröde Probe. Die entstandenen Risse zerstörten den untersuchten Einkristall meist vollständig, da sich die Risse durch die gesamte Oberfläche erstreckten. Hier wurden fünf Einkristalle untersucht. Eine Härteprüfung nach Knoop konnte rissfrei und reproduzierbar durchgeführt werden. Rissbildung Abbildung 7.1.5: Vickersabdruck mit sehr starken Rissen Abbildung 7.1.6: Knoop:bdruck neben dem Vickers Abdruck Härtemessung an B12P2 -Kristallen Von der Verbindung B12P2 stand nur ein Einkristall ausreichender Größe für eine Härtemessung zur Verfügung. - 276 - Weitere Untersuchungen zur Härte Rissbildung Abbildung 7.1.7: Vickersabdruck: Abbildung 7.1.8: Knospabdruck: d1=11,47µm, d2=13,20µm: 2480 HV d1=36,87µm, 2133 HK Die Messung nach Vickers zeigte eine Härte von 2505 HV 0,2/10. Hier konnte, wie schon bei nahezu alle Einkristallen mit einer Härte über 2000 HV, eine Rissbildung beobachtet werden. Bei der Härteprüfung nach Knoop konnten rissfreie Abdrücke mit einer dazugehörigen Härte von 2134 HK 0,2/10 wahrgenommen werden. Härtemessung an AlB12-Kristallen Insgesamt konnten vier Einkristalle aus unterschiedlichen Ansätzen auf ihre Härte hin untersucht werden. Jeder Kristall wurde mit mindestens drei Abdrücken untersucht. Es konnte eine Vickershärte von 2445 HV 0,2/10 (24GPa) und eine Knoophärte von 2459 HK 0,2/10 ermittelt werden. Bei der Prüfung mit der Vickerspyramide konnten Risse beobachtet werden. Abbildung 7.1.9: Abbildung 7.1.10: Vickersabdruck: (dØ=12,27µm, 2513 HV) Knoopabdruck: (d = 34,61µm; 2424 HK) Härtemessung an Al2,7Cu2,2,B105-Kristallen - 277 - Weitere Untersuchungen zur Härte An zwei Einkristallen konnte die Härte nach Vickers und Knoop erfolgreich und reproduzierbar gemessen werden. Rissbildung Abbildung 7.1.11:Vickersabdruck: Abbildung 7.1.12:Knoopabdruck: d1=10,72µm, d2=11,54µm 3054 HV d = 34,42µm; 2449 HK Die untersuchen Einkristalle zeigten eine Vickershärteprüfung von 2909HV 0,2/10 (29 GPa). Somit sind die Al2,7Cu2,2,B105-Kristalle mit ihrer hohen Härte bei den härtesten Verbindungen mit dabei. Die Knoophärte lag bei 2467 HK 0,2/10. Bei allen Einkristallen der Zusammensetzung Al2,7Cu2,2,B105 konnten bei der Vickershärteprüfung Risse festgestellt werden. Härtemessung an Einkristallen der τ-Boride Die Härteprüfung an den τ-Boriden wurde bereits in den einzelnen Kapiteln besprochen. Hier erfolgt nur eine tabellarische Auflistung der untersuchten Systeme. Tabelle 5.2.4.1: Übersicht der gemessenen τ-Boride (details sind dem Kapitel 4 „τ-Boride“ zu entnehmen) Verbindung Vickershärte Knoophärte GPa [Vickers] Co21Mn2B6 805 HV 0,2/10 802 HK 0,2/10 7,9 Co22V1B6 858 HV 0,2/10 853 HK 0,2/10 8,4 Co22W1B6 865 HV 0,2/10 843 HK 0,2/10 8,5 Ni20AlB14 1071 HV 0,2/10 - 10,5 Ni20Ga3B6 912 HV 0,2/10 - 8,9 - 278 - Weitere Untersuchungen zur Härte 7.2 Boridcarbide Härtemessung an B4C -Kristallen Die Härteprüfung wurde an drei Einkristallen der Zusammensetzung B4C durchgeführt. Hier handelt es sich um eine schon länger bekannte Verbindung. An B4C wurde bereits vollständig auf die Härte untersucht und mit Werten in Höhe von 31 GPa (nach Vickers mit HV 1) und 29 GPa (nach Knoop mit 0,1 HK) dokumentiert[206]. Die Untersuchungen an den Einkristallen sollten nicht nur als Bezugspunkt sondern vielmehr als Kontrollpunkt für die eigenen Messungen an Hartstoffen dienen. Die Einkristalle wurden bei 1800°C aus einer CuSchmelze erhalten. Mit den Ergebnis von 3238 HV 0,2/10 (31 GPa) und 2787 HK 0,2/10 mit je vier Abdrücken an jedem der untersuchten Einkristalle, haben die gezüchteten Kristalle perfekte Übereinstimmung mit den in der Literatur vorhandenen Werten. Die Härteprüfung nach Vickers zeigt wieder einmal eine Rissbildung an den Ecken der Abdrücke der Vickerspyramide. Rissbildung Abbildung 7.2.1: Abbildung 7.2.2: Vickersabdruck: d1=10,31µm, d2=10,95µm Vickers-: (d1=10,87µm, d2=10,89µm) Knoopabdruck: d = 31,97µm Härtemessung an Mg3B50C8 -Kristallen Zwei Einkristalle der Verbindung Mg3B50C8 konnten nach Vickers und Knoop mit einer Laset von 1,961 N geprüft werden. Es wurde jeweils 3 Abdrücke genommen und gemittelt. Es konnte eine Vickershärte von 3286 HV 0,2/10 (32GPa) und eine Knoophärte von 3165 HK 0,2/10 ermittelt werden. Bei der Härteprüfung nach Vickers konnte eine starke Rissbildung an der Probe festgestellt werden. - 279 - Weitere Untersuchungen zur Härte Rissbildung Abbildung 7.2.3: Vickersabdruck: dØ=10,75µm, 3174 HV Die ermittelte Härte von 32 GPa ist erstaunlich. Somit sind die Einkristalle mit der Zusammensetzung von Mg3B50C8, gezüchtet aus einer Mg/Cu-Schmelze bei einer Temperatur von 1600°C, eine der härtesten Verbindungen überhaupt. Beim Ranking der Hartstoffe befindet sich Mg3B50C8 ganz weit vorne und zählt zu den härtesten Vebindungen. 7.3 Boridnitride Härtemessung an c-BN -Kristallen Bei der Probe des kubischen Bornitrids handelt es sich um die zweithärteste Verbindung, die in der Literatur geführt wird. Das kubische Bornitrid überzeugt mit einer Härte von 47 50 GPa (ermittelt nach Vickers). Messungen an den Einkristallen zeigten gute Übereinstimmung mit der Literatur. Es konnte ein Vickershärte von 5108 HV 0,2/10 (49 GPa) und 4612 HK 0,2/10 erhalten werde. Rissbildung Abbildung 7.3.1: Vickersabdruck d1=8,86µm, d2=8,48µm 5054 HV - 280 - Weitere Untersuchungen zur Härte Wie es zu erwarten war, zeigen auch die Einkristalle des c-BN einen enormes Risswachstum entlang der Oberfläche des Kristalls. Bei Verbindungen dieser Härte, wie am Beispiel von c-BN, erhält man oft eine hohe Härte auf Kosten der Zähigkeit. Härtemessung an MgB9N -Kristallen Nur ein Kristall erfüllte von der Größe her die Anforderungen für eine Härteprüfung nach Vickers und Knoop. Abbildung 7.3.2: Vickersabdruck d1=11,10µm, d2=11,92µm 2799 HV MgB9N Einkristalle sind nur als Nebenprodukt bei der Reaktion in einer Cu/Mg-Schmelze bei 1600°C nach 40 Stunden ausgefallen. Die Schwierigkeiten der Züchtung dieser Kristalle stellen somit auch ein großes Problem bei der Steuerung der Größe dar. Der einzige Einkristall der von der Qualität und Güte her geeignet war, wurde eingebettet und für die Mikrohärtemessung präpariert. Hier konnte nach dem Vickers Experiment aus 3 Abdrücken eine Härte von 2421 HV 0,2/10 ermittelt werden. Wie in der Abbildung 7.1.2 gut zu sehen ist, sind auch hier Risse während der Messung aufgetreten. Der KnoopEindringkörper lieferte rissfreie und ebenfalls konstante Werte. Aus der Messung von drei Abdrücken konnte eine Härte von 2390 HK 0,2/10 ermittelt werden. 7.4 Diskussion der Härteergebnisse Gegenstand des Teils der vorliegenden Arbeit war die Untersuchung der Härte an Einkristallen. Die Schwierigkeit lag hier ganz klar auf der Präparation und Aufarbeitung der sehr kleinen Kristalle. Die im Arbeitskreis vom Prof. Dr. Hillebrecht dargestellten Stoffe gehören zu den härtesten Stoffen die bekannt sind. Die Härte geht meist Hand in Hand mit der Sprödigkeit. Folglich wurde bei der Härteprüfung nach Vickers nahezu allen Proben eine - 281 - Weitere Untersuchungen zur Härte Rissbildung, in extremen Fällen sogar eine Spaltung des Kristalls beobachtet. Die Härteprüfung nach Knoop konnte in den meisten Fällen einen rissfreien Abdruck hinterlassen. Somit wurden an so gut wie jedem untersuchten Einkristall beide Härtemessmethoden durchgeführt. Bevorzugt wurde die Methode nach Vickers, da diese heute in der Industrie einen hohen Stellenwert hat. Neben der in der Arbeit dargestellten Verbindungen wie Ti3Ga0,85Sn0,15C2, Ti3GeC2 Ta3Al2CoC, Ta3Al2CoN, W9Al3Co1C3 und den τ-Boride sollte auch die Härte von neuen Verbindungen aus dem Arbeitskreis Hillebrecht durchgeführt werden. Die Forschungsschwerpunkte liegen hier auf der Synthese von Boriden, Carbiden, Nitriden, Boridnitriden und Boridcarbiden. In Rahmen der Arbeit konnte bei der Verbindung Mg3B50C8 eine Härte von 3295 HV 0,2 ermittelt werden. Dementsprechend handelt es sich hier um einen der härtesten Werkstoffe der bekannt ist. Eine Übersicht der Ergebnisse aus der Messung nach Vickers, wurde in der Abbildung 7.2.1 dargestellt. - 282 - Weitere Untersuchungen zur Härte 3000 HV c-BN 5108 HV 0,2 B4C 3237 HV 0,2 Mg3B50C8 3295 HV 0,2 Al2,7Cu2,2B105 2909 HV 0,2 LiB12PC 2746 HV 0,2 AlB12 2625 HV 0,2 B12P2 2505 HV 0,2 MgB9N 2421 HV 0,2 MgB12 2360 HV 0,2 2000 HV MgB7 2125 HV 0,2 Mg1,1B12Si2 1942 HV 0,2 Li2B12Si2 2027 HV 0,2 Mg3B36Si9C 1755 HV 0,2 W9Al3CoC3 1614 HV 0,2 τ-Boride 800-900 HV 0,2 Ti3Ga0,85Sn0,15C2 308 HV 0,1 ═ 668 HV 0,1 ┴ Boride Boridcarbide Boridnitride Ti3GeC2 360 HV 0,2 ┴ Carbide Abbildung 7.4.1:Übersicht der Härteergebnisse von neuen Verbindungen aus dem Arbeitskreis Hillebrecht, eingeteilt nach Stoffklassen. - 283 - Experimenteller Teil 8 Experimenteller Teil 8.1 Allgemeine Angaben zu den Synthesen 8.1.1 Spezifikation der verwendeten Chemikalien In der Tabelle 8.1.1.1 sind sämtliche Chemikalien aufgelistet, die für die praktische Arbeit verwendet wurden. Bei unbekannter Reinheit der Chemikalie wurde röntgenographisch auf die Anwesenheit von Verunreinigungen geprüft. Tabelle 8.1.1.1: Spezifikation der für die Synthesen verwendeten Chemikalien. Chem. Hersteller / Symbol Herkunft Al ALFA Aesar Reinheit Nr. Substanz 1 Aluminium 2 3 Aluminiumnitrit AlN Ar Argon 4 Black carbon C 5 Bor B ABCR 99,5 6 Chrom Cr ALFA Aesar 99,95 Pulver 7 Eisen Fe MERK 99,9 Pulver 8 Gallium Germanium Ga (unbekannt) - Stücke Ge ABCR 99,999 Pulver 10 Indium In ABCR 99,99 11 Iridium Ir Degussa 100 Pulver 12 Kobalt Co ChemPur 99,5 Pulver 13 Kohlenstoff C ChemPur 99,9+ Pulver 14 Kohlenstoff „carban black“ C 99,9 Pulver 15 Kupfer Cu Merck 99,9 Pulver 16 Magnesium Mg ABCR - Pulver 9 [%] 99,8 Alfa Products SWF GmbH STREM CHEMICALS STREM CHEMICALS Form Pulver Pulver 99,999 Schutzgas 99,99 Pulver amorphes Pulver Shot Ø 1mm - 284 - Experimenteller Teil 17 Mangan Mn ALFA Aesar 99,9 Pulver 18 Molybdän Mo AVOCADO 99+ Pulver 19 Nickel Ni ALFA Aesar 99,9 Pulver 20 Niob Nb - Pulver 21 Palladium Pd Degussa 99,9 Pulver 22 Silizium Si Aldrich Chemical 99,0 Pulver 23 Tantal Ta (unbekannt) - Pulver 24 Titan Ti Merck 99,9 Pulver 25 Titan Ti ABCR 99,9 26 Titancarbid TiC Aldrich Chemical - Pulver 27 Titannitrit TiN ALFA Aesar - Pulver 28 Vanadium V ABCR technisches Pulver 29 Wolfrom W ALFA Aesar 99,95 Pulver 30 Zinn Sn (unbekannt) - Pulver 31 Zirkonium Zr ChemPur technisches Pulver 32 Dia-Pro 9 µm SiO2 Struers Dia-Pro 3 µm SiC Struers 34 OP-S 0,04 µm SiO2 Struers 35 Epoxidharz und Kömmerling Körapox 36 Salzsäure 33 Kristallhandel Kelpin Suspension zum Feinpolieren Suspension zum Feinpolieren Suspension zum Feinpolieren Einbett Glycidylneodecanoat Chemische Fabrik Zweikomponenten Harz HCl Merck ~ 37% tech. Die Homogenisierung der Pulver wurde in Achatmörser durchgeführt, anschließend wurde das homogene Gemisch mit einer Handpresse zu einer Tablette verarbeitet. Beim Arbeiten mit Gallium und Indium wurden die Metallstückchen jeweils auf die fertig gepresste Tablette platziert. Tiegel wurden ausschließlich aus gesintertem Korund (Al2O3) der Firma FRIATEC verwendet. Die Lagerung vom Borpulver erfolgte im evakuierten Exsikkator mit Orange Gel als Trockenmittel. - 285 - Experimenteller Teil 8.1.2 Synthesen in Hochtemperaturöfen Synthesen im „Gero“ – Ofen. Laborofen, Gero Hochtemperaturofen GmbH, HNeuhausen Die Hochtemperatursynthesen wurden in einem Rohrofen durchgeführt. In einem Rohrofen mit SiC- Heizrohr der Firma HERAEUS konnte eine Arbeitstemperatur von 1450°C – 1500°C erreicht werden. Zum Schutz der Schmelzen vor Oxidation durch Luft-Sauerstoff wurden die Proben in einem gasdichten Schutzrohr aus Korund (99,7% Al2O3) unter permanentem Argonstrom (50 ml/min; 99,999% Reinheit) in den Öfen platziert. Synthesen im „Astro“ – Ofen. Serial No. C072-9211, Thermal technology Inc., Astro Division, Santa Rosa, Kalifornien. Die Hochtemperatursynthesen wurden in einem Rohrofen durchgeführt. Mit einem Graphitheizelement konnte eine Arbeitstemperatur von 1650°C (max. 2000°C) erreicht werden. Zum Schutz der Schmelzen vor Oxidation an der Luft wurden die Proben unter permanentem Argonstrom (50 ml/min; 99,999% Reinheit) in den Öfen platziert. Synthesen im „Nabertherm“ – Ofen. Laborofen, Nabertherm Hochtemperaturofen GmbH, Bremen Mod. HT 08/17 Die Hochtemperatursynthesen wurden in einem Rohrofen durchgeführt. In einem Rohrofen mit SiC-Heizrohr der Firma HERAEUS konnte eine Arbeitstemperatur von 1450°C – 1650°C erreicht werden. Zum Schutz der Schmelzen vor Oxidation durch Luft-Sauerstoff wurden die Proben in einem gasdichten Schutzrohr aus Korund (99,7% Al2O3) unter permanentem Argonstrom (50 ml/min; 99,999% Reinheit) in den Öfen platziert. Synthese im Pulsplasma Ofen Dr. SINTER-LAB™ - SPARK PLASMA SINTERING SYSTEM SPS-515 S Die Synthesen wurden in einem Sinterplasmaofen im Labormaßstab, mit einer einachsigen vertikalen hydraulischen Presse, die Drücke bis zu 50 kN (die Einstellung des Sinterdruckes ist von dem Durchmesser der verwendeten Graphitmatrize abhängig) erzeugen kann. Für diesen Ofentyp sind Matrizen aus Graphit mit einem Reaktionsdurchmesser von 10 bis 20 Millimeter vorgesehen (Die Graphitmatrize wurde mit einer Graphitfolie vor jeder Reaktion ausgekleidet). Der leistungsstarke Gleichstrom-Generator arbeitet mit einer durchschnittlichen Stromstärke von 1500 A bei einer Spannung von bis zu 20 V. Dieses ermöglicht eine - 286 - Experimenteller Teil Arbeitstemperatur von bis zu 2000°C. Die Synthesetemperatur wurde unter der Betrachtung der Schmelzpunkte ausgewählt. Bei der Vakuumkammer handelt es sich um eine von außen mit Wasser gekühlte doppelschichtige Reaktionskammer aus Edelstahl, die sich auf 6,0 Pa (4,5·10-2 Torr) evakuieren lässt. Die Reaktionskammer besitzt zwei Fenster, das eine dient der Temperaturüberwachung durch das Pyrometer und das zweite Fenster bietet einen Einblick in das Reaktionsgeschehen für eine visuelle Überwachung. 8.1.3 Aufarbeitung der Proben Schmelzperlen: - Unter mechanischer Einwirkung wurde der Schmelzregulus in einem WolframcarbidMörser zertrümmert. Die Isolierung von geeigneten Einkristallen wurde unter einem Lichtmikroskop durchgeführt. - Bei einer Verwendung vom Hilfsmetall im Überschuss lagen die Produkte nach dem Erkalten der Schmelze in der Hilfsmetallmatrix eingebettet vor. Diese Matrix wurde in der Regel mit verd. (selten, aber auch mit halbkonz.) Salzsäure behandelt. Die Auflösung der Hilfsmetallmatrix konnte von einigen Minuten bis zur einer Woche dauern (je nach System). Starke Angriffe der Säure auf die gewonnenen Produkte konnte nicht beobachtet werden. Anschließend wurden die erhaltenen Produkte mit Wasser mehrmals gewaschen, abgesaugt und getrocknet. Die Isolierung von geeigneten Einkristallen wurde unter einem Lichtmikroskop durchgeführt. 8.2 Untersuchungsmethoden 8.2.1 Röntgenbeugung an Pulvern Die röntgenographischen Untersuchungen an Pulvern erfolgte mit einem Diffraktometer Stadi II / P der Firma STOE mit monochromatischer Cu-Kα1-Strahlung (Ge-Monochromator) und ausgerüstet mit einem gebogenen Imageplatedetektor (2theta = 145°) und Probenwechsler vorgenommen. Die Proben wurden auf Kunststoffstreifen präpariert und anschließend vermessen. Die Phasenanalyse erfolgten durch Anwendung der geräteeigenen Software WinXPOW und DIFFRACplus der Firma Bruker AXS. - 287 - Experimenteller Teil Die röntgenographischen Untersuchungen der Proben im System Kobalt- M` -Bor (M` = Mo, Ga, Ir, Al, Sn, Ti, V, W, Mn, Mg) wurde mit einem Diffraktometer Stadi II / P der Firma STOE mit monochromatischer Mo-Kα1-Strahlung (Ge-Monochromator) vorgenommen. Die Phasenanalyse erfolgte durch Anwendung der geräteeigenen Software WinXPOW und DIFFRACpius der Firma Bruker AXS. 8.2.2 Röntgenbeugung an Einkristallen Die röntgenographischen Untersuchungen der Einkristalle erfolgten an einem STOE IPDS II Diffraktometer mit Mo-Kα-Strahlung, image plate Detektor und Graphit-Monochromator unter Luft bei Raumtemperatur. Indizierung, Integration und numerische Absorptionskorrektur wurden mit Hilfe der geräteeigenen Software X-Area durchgeführt. 8.2.3 EDX – Untersuchungen an Einkristallen Die Einkristalle wurden einer qualitativen Elementaranalyse mittels energiedispersiver Röntgenemmissionsspektroskopie (EDX) unterzogen. (I) Hierzu stand ein Rasterelektronenmikroskop (Zeiss Digital Scanning Mikroskope DSM 960) mit integrierter Mikrosonde (Link Analytical Microprobe LEO 1525) zur Verfügung (Institut für Kristallographie, Universität Freiburg). Die emittierte Röntgenstrahlung wurde energiedispersiv mit einem Halbleiterdetektor gemessen. (II) Wie auch ein Rasterelektronenmikroskop (Zeiss Digital Scanning Microscope DSM 962) mit dem Oxford EDX-System (Software: Inca 300, Version 4.02, Fa. Oxford Instruments GmbH) mit Si(Li)-Detektor am Institut für Mikrosystemtechnik der Universität Freiburg. 8.2.4 Einbetten von Einkristallen Einkristalle, die zuvor einer röntgenographischen Untersuchung unterzogen wurden, wurden anschließend auf einem Träger aus Teflon ausgerichtet und in Epoxidharz (Körapox, - 288 - Experimenteller Teil Zweikomponenten Epoxidharz) eingebettet. Die Aushärtung erfolgte über zwei Tage. Der eingebettete Einkristall wurde anschließend geschliffen und poliert. 8.2.5 Härtemessung Die Härtemessung wurde mit dem Mikrohärtetester am Institut für Analytische und Anorganische Chemie der Universität Freiburg durchgeführt. Mit dem Mikrohärtetester MHT- 10 der Firma Anton Paar wurden Lasten zwischen 0,5 bis 2N mit einer Dauer von 10 Sekunden auf die Probe ausgeübt. Die Härteermittlung erfolgte durch Anwendung der geräteeigenen Software der Firma Anton Paar. - 289 - Zusammenfassung 9 Verwendete Programme Tabelle 8.2.5.1: Übersicht der verwendeten Programme Programm Verwendung Ref. X AREA Weiterverarbeitung von Einkristalldiffraktometerdaten, Verfeinerung von Main Menu Version 1.27 Strukturmodellen aus Einkristalldaten, Absorptionskorrektur. Diamond Version 3.0d Graphische Darstellung von Kristallstrukturen [207] DIFFRACplus EVA Auswertung von Pulverdiffraktogrammen (Fa. SIMENS) [208] STOE Win XPOW Auswertung von Pulverdiffraktogrammen (Fa. STOE) [209] XCH Version 5.03 Konvertieren von Pulverdiffraktogrammen [210] SHELXTL - 97 Strukturlösung, -verfeinerung anhand von Einkristalldaten (Patterson und [199] [68] Version 1.08 Direkte Methoden) Binary Phase Diagrams Binäre Phasendiagramme [116] - 290 - Zusammenfassung 10 Zusammenfassung und Ausblick Im ersten Teil dieser Arbeit wurden strukturchemische Untersuchungen an ternären Carbiden aus der Familie der sogenannten MAX-Phasen durchgeführt. Bei diesen MAXPhasen handelt es sich um ternäre Übergangsmetallcarbide der Zusammensetzung (MX)nMA mit in denen M ein frühes Übergangsmetall (z.B. Ti, V, Ta) und A ein Hauptgruppenelement (Ga, Al, Sn, Ge, In, Si) repräsentiert. Das Hauptinteresse lag auf der Optimierung der Synthesebedienungen für die Züchtung von Einkristallen ausreichender Qualität und Größe für weitere physikalische Untersuchungen wie die Härtemessung. So wurden Einkristalle der 211-, 312- und 413-Varianten, d.h. MAX-Phasen mit n = 1, 2 und 3, aus den unterschiedlichsten Systemen gezüchtet. Die MAX-Phasen sind durch ihren Charakter, metallische und keramische Eigenschaften zu vereinen, eine Stoffklasse für sich. Die physikalischen Eigenschaften der MAX-Phasen stellen somit ein abwechslungsreiches Einsatzfeld dar. Bei der einkristallinen Darstellung der MAX-Phasen wurde ausschließlich die Methode der Hilfsmetallbadtechnik verwendet, wobei das Schmelzmedium gleichzeitig als Reaktionsmedium fungierte und im deutlichen Überschuss eingesetzt wurde. Zudem wurden dem Reaktionsmedium weitere Hilfsmetalle zugesetzt, hier sollte auf die Substitution des Hauptgruppenmetalls und die Produktzusammensetzung eingegangen werden. Neben der Züchtung von Einkristallen sollten auch die Sinterkörper von MAX-Phasen dargestellt und charakterisiert werden. Diese Aufgabe beinhaltete die Inbetriebnahme des Pulsplasma Ofens „Dr. SINTER-LAB™ - SPARK PLASMA SINTERING SYSTEM SPS-515S“ am Institut. Im Rahmen dieser Arbeit gelang es mit Ti3Ga0,85Sn0,15C2 eine neue Verbindung aus der 312-Variante der MAX-Phasen aus einer Gallium/Aluminium Schmelze zu züchten. An dieser Verbindung konnten mit Hilfe von Röntgenbeugungsexperiment eine vollständige Strukturanalyse am Einkristall durchgeführt werden. Zudem konnten ausreichend große und von der Güte her optimale Einkristalle für eine Härtemessung dargestellt werden. Die Messung der Härte konnte das postuliert anisotrope Verhalten an den MAX-Phasen nun endlich bestätigen. Ti3Ga0,85Sn0,15C2 vervollständigt die Reihe der MAX-Phase, somit präsentiert das Dreistoffsystem Ti-Ga-C zum ersten Mal eine komplette Reihe aller Varianten. Bei den Untersuchungen in dem System Ti-(Al/Sn)-(C/N) konnten bei den Reaktionen in einer reinen Aluminiumschmelze nahezu ausschließlich die Bildung von Ti3AlC2-xNx (x = - 291 - Zusammenfassung 0,18) beobachtet werden. Bei der Zugabe von Zinn als Hilfsmetall konnte keine systematische Substitution von Zinn gegen Aluminium durchgeführt werde. Hier dominierte ein thermodynamisches Produkt mit einer ungefähren Zusammensetzung von Ti2Al0,5(2)Sn0,5(2)C/N. Erwähnenswert an dieser Stelle ist, dass bei der Zugabe von Zinn die 211-Variante deutlich in Präsenz trat, zudem konnte ein deutlicher Einbau von Stickstoff auf der Nichtmetalllage vernommen werden (welcher als Verunreinigung eingeschleppt wurde). Neben der Synthese der 211 und 312-Variante ist auch eine 413-Variante gelungen. Bei der Verbindung konnte ebenfalls eine Mischbesetzung auf der Hauptgruppenmetallposition durch Aluminium und Zinn sowie auf der Nichtmetalllage eine Besetzung durch Stickstoff und Kohlenstoff mit Hilfe der EDX und SXRD festgestellt werden. Somit konnten eine ungefähre Zusammensetzung von Ti4Al0,8(1)Sn0,2(1)N2,7(1)C0,3(1) diskutiert werden. Bei der Untersuchung der Kristallisationsbedingungen an MAX-Phasen wurden unterschiedliche Hilfsmetalle der Aluminiumschmelze zugesetzt. Neben den bereits erwähnten Zinn und Gallium wurden noch das Germanium und Indium zugegeben. So konnten Einkristalle der Zusammensetzung von Ti3GeC2 und Ti2Al0,85In0,15C aus der Schmelze gezüchtet werden. Neben den MAX-Phasen des Titans wurden auch die des Vanadiums betrachtet. Bei der Synthese von Vanadium MAX-Phasen aus einer Aluminiumschmelze mit dem Zusatz an Zinn als Hilfsmetall, konnte neben der 211-Phase mit der Zusammensetzung von V2Al0,92Sn0,08C auch eine 531-Phase mit der vermuteter Zusammensetzung von V5Al3C1-x (mit x bis 0,07) als Einkristalle in Form von metallisch glänzende Nadeln erhalten werden. Die Besonderheit dieser Struktur zeigt mögliche Kohlenstoff-Kohlenstoff Wechselwirkungen entlang der flächen-verknüpften [V6C]-Oktaeder. Als weiteres Hilfsmetall wurde das Gallium eingesetzt, hier war die Synthese einer V2GaC Phase ohne den zusätzlichen Einbau von Aluminium auf der Hauptgruppenmetallposition ermöglicht. Bei der Zugabe von Indium zur Aluminiumschmelze konnte eine Mischphase mit der Zusammensetzung von V2In0,63Al0,37C erhalten werden. Aus gleichem Ansatz wurde ein Einkristall mit deutlich kleinen Gitterkonstanten isoliert. Hier lag auf Grund der sehr kleinen Gitterkonstanten von a = 2,9190(4) Å; c = 12,989(3) Å die Vermutung auf eine MAX-Phase mit der Zusammensetzung von V2VC bzw. V3C vor. Neben den Hochtemperatursynthesen konnten die MAX-Phasen auch bei niedrigeren Temperaturen (im Allgemeinen ca. 200 bis 300°C) als Sinterkörper aus dem Pulsplasmaofen erhalten werden. Ti2AlC und Ti3AlC2 konnten nahezu phasenrein neben geringen Anteilen an - 292 - Zusammenfassung TiC erhalten werden. Bei der Herstellung von Ti2SnC-Sinterkörpern wurde als Hauptprodukt Ti8C5 erhalten Ti2SnC kristallisierte neben Ti5Sn3 und Graphit als Nebenprodukt. Die Nitride Ti2AlN und Ti4AlN3 konnten nebeneinander jedoch nicht phasenrein dargestellt werden, es konnten Spuren von TiN beobachtet werden. Ti2GeC konnte als Hauptprodukt neben zahlreichen anderen binären Phasen dargestellt werden. Ti3SiC2 konnte sehr ausgeprägt bei allerdings sehr hohen Temperaturen von 1600°C synthetisiert werden. Die MAX-Phasen mit Tantal als Hauptkomponente mit der Zusammensetzung von Ta2AlC und Ta4AlC3 wurden nebeneinander bei geringer Menge an TaC aus den Elementen dargestellt. Eine phasenreine Synthese war nicht möglich. Neben den MAX-Phasen konnte auch die h-Phase in Form von Carbiden und Nitriden aus dem SPS-Ofen erhalten werden. Die Sinterkörper von Ta3Al2CoC, Ta3Al2CoN und Nb3Al2CoC wurden in nahezu phasenreinem Zustand erhalten, nur geringe Anteile an TaC, TaN bzw. NbC0,84 wurden in der Probe beobachtet. Für Ta3Al2CoC und Ta3Al2CoN konnte ein Härtewert von 13,2 GPa mit Hilfe einer Mikrohärtemessung erhalten werden. Im zweiten Teil der vorliegenden Arbeit wurden strukturchemische Untersuchungen an einkristallinen ternären Boride mit der Cr23C6-Struktur durchgeführt. Hier wurden Kobalt τBoride mit der Zusammensetzung von M23-xM’xB6+y mit Kobalt als Hauptkomponente M untersucht. Als τ-Stabilisator (d.h. M’) wurden überwiegend Hauptgruppen- und Übergangsmetalle wie Iridium, Gallium, Aluminium, Zinn, Titan, Vanadium, Wolfram, Mangan und Molybdän so wie auch das Erdalkalimetall Magnesium eingesetzt. Dazu wurden Einkristalle aus der Schmelze gezüchtet und mit Hilfe von Röntgenbeugung (am Pulver und an Einkristall), energiedispersiver Röntgenemissionsspektroskopie sowie Härtemessung (an Einkristall) vollständig charakterisiert. In dieser Arbeit hat sich die Notwendigkeit der EDX-Untersuchung erneut bestätigt. Für eine eindeutige und ausnahmslose Charakterisierung der einzelnen Phasen ist es unentbehrlich, das genaue Verhältnis der Metalle zu wissen. Im System Kobalt-Iridium-Bor konnte die Substitution von Metallatomen gegen B4Tetraeder beobachtet werden. Der erstaunliche Austausch wurde erstmalig für ein Kobalt τBorid mit Hilfe von Einkristalluntersuchungen bestätigt. Hier konnten eine Phasenbreite von 20,7 bis 33,1 At.-% für das Bor gezeigt werden. Eine vollständige Besetzung konnte nicht bestätigt werden. Aus den röntgenographischen Untersuchungen am Einkristall konnte eine Zusammensetzung von Co12.3Ir8.9B10.5 mit einer Gitterkonstante von a = 10,9393(13) Å - 293 - Zusammenfassung aufgestellt werden. Das Tetraeder zeigte Bor-Bor-Abstände von 1,63(4) Å, diese sind etwas kürzer als die von Ade in dem borreichen τ-Boride mit der Zusammensetzung von Ni20AlB12 beobachtet (B-B: 1,68(2) Å). Für das τ-Boride aus dem System Kobalt-Aluminium-Bor konnte kein Hinweis für das Auftreten einer borreichen Verbindung vernommen werden (im Hinblick auf den denkbaren Einbau von B4-Einheiten sollte analog zu dem Ni-Al-B System das Co-Al-B System untersucht werden). Nur borarme τ-Boride konnten mit einer Mischbesetzung von Aluminium- und Kobaltatomen auf der Position 4a erfasst werden. Für die System Co23-xM’xB6 (M=Ga, Sn, Ti, V, W, Mn und Mo) konnte kein Anzeichen für eine Phasenbreite bezüglich des Borgehalts erfasst werden. Für die Systeme mit den Übergangsmetallen (Ti, V, X, Mn und Mo) wurden in dieser Arbeit zum ersten Mal Daten basierend auf Einkristalluntersuchungen gesammelt. Für die hier genannten Systeme wurde der Homogenitätsbereich bezüglich der Primärkristallisation auf Grundlagen von Schmelzdiagrammen analysiert. Bei der Untersuchung der Phasenbreite wurde auch das Metallverhältnis mehrfach variiert. Die gewonnenen Informationen aus den Einkristalluntersuchungen erlauben Rückschlüsse über das bevorzugte Einlagern von Metallen auf speziellen Positionen. Untersuchungen des Systems Co-Mg-B konnten das τBorid mit einer Gitterkonstanten von a = 10,5598(2) Å eindeutig neben wenig Co2B identifizieren, jedoch war eine einkristalline Darstellung nicht realisierbar. Mit der Reproduktion von Cr2ZrB6 konnte aus einer Aluminiumschmelze der erste Vertreter seiner Art ohne den Einbau eines Seltenerdmetalls dargestellt und vollständig charakterisiert werden. Als Nebenprodukt entstanden metallisch glänzende Nadeln von Cr2AlB2. Die Verbindung Cr2AlB2 konnte zum ersten Mal in Form von Einkristallen erhalten und vollständig crakterisiert werden. Mit der Verbindung W9Al3Co1C3 konnte ein neues κ-Carbid aus einer Aluminiumschmelze in einkristalliner Form dargestellt und charakterisiert werden. Das κ-Carbid ist ein schönes Beispiel für Wirtgitter-Systeme, hier besteht ein Teil der Struktur aus [W6C]-Oktaedern, welche über Ecken miteinander verkettet sind und über eine flächenverknüpfte Verkettung von [Al6Co]-Oktaedern. Abgesehen von den Synthesen der MAX-Phasen, τ-Boriden, κ-Carbide und h-Carbide in einkristalliner wie auch gesinterter Form sollte zur vollständigen Charakterisierung auch die - 294 - Zusammenfassung Messung der Mikrohärte durchgeführt werden. Hier standen die Härteprüfmethoden nach Vickers und Knoop zu Verfügung. In Rahmen dieser Arbeit wurde der Mikrohärtetester MHT-10 der Fa. Paar in Betrieb genommen. Somit wurden Einkristalle aus eigener Forschung wie auch Einkristalle aus dem Arbeitskreis Hillebrecht auf ihre Härte hin untersucht. Dabei konnten Hartstoffe wie Mg3B50C8 (3295 HV 0,2), Al2,7Cu2,2B105 (2909 HV 0,2), LiB12PC (2746 HV 0,2), AlB12 (2625 HV 0,2) und B12P2 (2505 HV 0,2) auf ihre Härte geprüft werden. Diese hier aufgelisteten Verbindungen zählen zu den härtesten Verbindungen, die je analysiert wurden. An weiteren Einkristallen der Boride, Boridcarbide und Boridnitride konnten Mikrohärtemessungen durchgeführt werden. Dabei konnte beobachtet werden, dass sich die Boride der Hauptgruppenmetalle in der Regel bei Härtewerte um 2000 HV bewegen. Bei den τ-Boride egal welcher Art konnte eine Härte in der Größenordnung von 800 - 900 HV 0,2 ermittelt werden. Im Laufe der Härteuntersuchung an Einkristallen gelang es erstmalig eine MAX-Phase mit der Zusammensetzung von Ti3Ga0,85Sn0,15C2 parallel (668 HV 0,1) und senkrecht (308 HV 0,1) zu basalen Schicht zu untersuchen. Erstmalig wurde in dieser Arbeit eine natürlich auftretende Anisotropie an den Einkristallen der MAX-Phase nachgewiesen. - 295 - Literaturverzeichnis 11 Literaturverzeichnis [1] F. Aldinger, H. J. Seifert, Z. Metallkd., 1993, 84, 2-10. [2] Stahlschlüssel-Taschenbuch, Wissenswertes über Stähle, Verlag Stahlschlüssel Wegst Gmbh, 18. Auflage, 1998. [3] H. H. Stadelmaier, Metal. Rich, Metal-Metalloid Phases, Developments in the Structural chemistry of Alloy Phases (Hesg.: B. C. Gissen), 1- Auflage. Plenum, New York, 1969, 141-180. [4] R. Telle, Chem. Unserer Zeit, 1988, 22, 93-99. [5] L. T. Jordan, R. K. Mathur, K. R. Brose, H. H. Stadelmaier, Z. Metallkd., 1981, 72, 782784. [6] K. H. J. Buschow, P. G. van Engen, R. Jongebreur, J. Magn. Magn. Master. 1983, 38, 1-22. [7] B. W. MOTT, Die Mikrohärteprüfung. Übersetzt und herausgegeben von FRANK, K. F. Berliner Union, Stuttgart 1957. [8] DIN EN ISO 6506-1, Metallische Werkstoffe- Härteprüfung nach Brinell, Deutsche Fassung EN ISO 6506-1, 2005. [9] K. Nitsche, Werkstoffprüfung von Metallen, Band 1: Mechanische Prüfverfahren. J A Barth Verlag, Leipzig 1963, 200. [10] D. Tabor, The Hardness of Metals, Oxford University Press, 1951. [11] DIN EN ISO 6507-1, Metallische Werkstoffe- Härteprüfung nach Vickers, Deutsche Fassung EN ISO 6507-1, 2005. [12] B. W. Mott, Die Mikrohärteprüfung. Übersetzt und herausgegeben von Frank, K. F. Berliner Union, Stuttgart 1957. [13] DIN EN ISO 4516:2002, Mikrohärteprüfung nach Vickers und Knoop, für Metallische und andere anorganische Überzüge (Deutsche Fassung EN ISO 4516:2002). [14] F. Knoop, C. Peters, W. Emerson, A sensitivw pyramidal-diamondtool for indentation measurements, J. of Research, Mat. Bur. Dtds. 1939, 23, 39. [15] W. WEILER, Härteprüfung an Metallen und Kunststoffen, Expert Verlag, Sindelfingen 1985. [16] American Socienty for testing and Materials (ASTM), D 1474-68 Standard Test Methods for Indentation Hardness of Organic Coatings. Philadelphia, 1979. - 296 - Literaturverzeichnis [17] D. Dengel, E. Rossow, Untersuchungen über die Vergleichbarkeit der Vickers- und Knoop-Härte, HTM, 1971, 26, 21. [18] D. Dengel, E. Rossow, Vergleich der Vickers- und Knoop-Härte von Härtevergleichsplatten aus Stehl der Härte von ca. 100 bis 950 kp/mm2 in Prüfkraftberechen 0,2 bis 1 kp,VDI-Ber., 1972, 160, 73. [19] R. Riedel, Materials Harder than Diamond?, Adv. Mater. 1992, 4, No. 11, 175. [20] S. Otani; M.M. Korsukova; M.M. Korsukova; High-temperature hardness of ReB2 single crystals; J. of Alloys Com. Dez. 2008, Artikle in Press. [21] M. Hebbache, L. Stuparević and D. Živković, A new superhard material: Osmium diboride OsB2, Solid State Communications, 2006, 139, 227-231. [22] K. Inoue, U.S. Patent No. 3241956, 1962. [23] M. Tokita, Mechanism of Spark Plasma sintering an its application to ceramics, Nyn Sramikkasu, 1997, 10, 43-45. [24] http://www.scm-sps.com/e_htm/whatsps_e_htm/whatsps2_e.htm. [25] Modelling of the Field Assisted Sintering Technology (FAST) or Spark Plasma Sintering (SPS), K.U.Leuven, Department MTM, 1999. [26] http://www.scm-sps.com/e_htm/whatsps_e_htm/whatsps_e.htm. [27] W. Jeitschko, H. Nowotny, F. Benesovsky, Die H-Phasen Ti2InC, Zr2InC, Hf2InC, und Ti2InC, Monstsh. Chem. 1963, 94, 1201-1205. [28] Physical Review, 2005, B 72, 245. [29] Y. C, Zhou, X. H. Wang Sun, Electronic and structural properties of the layered ternary carbide Ti3AlC2; J. of Materials Chem., 2001, 11, 2335-2339. [30] H. Wolfsgruber, H. Nowotny, F. Benesovsky, Die Kristallstruktur von Ti3GeC2, Monatsh. Chem. 1967 98, 2403-2405. [31] S. Dubois, T. Cabioc’h, P. Chartier, V. Gauthier, M. Jaouen, A New Ternary Nanolaminate Carbice: Ti3SnC2, J. Am. Ceram. Soc., 2007, 90, 2642-2644. [32] C. J. Rawn, M. W. Barsoum, T. El-Raghy, A. Procipio, C. M. Hoffmann, C. R. Hubbard, Structure of Ti4AlN3 - a layered Mn+1AXn nitride, Mat. Research Bulletin, 2000, 35, 1785-1796. [33] J. C., Schuster, H. Nowotny, C. Vaccaro, The Ternary Systems: Cr-Al-C, V-Al-C, and Ti-Al-C and the Behavior of H-Phases (M2AlC), J. Solid State Chem. 1980, 32, 213219. [34] W. Jeitschko, H. Nowotny, Die Kristallstruktur von Ti3SiC2 - ein neuer KomplexcarbidTyp, Monatsh. Chem. 1967, 98, 329-337. - 297 - Literaturverzeichnis [35] M. Folchnandt, T. Schleid, CsYb3Se4: Ein gemischtvalenters Ytterbium (II, III)-Selenid mit Caesium Zeitschrift für Kristallographie, Supplement Issue, 1998, 15, 57-57. [36] W. Jeitschko, H. Nowotny, F. Benesovsky, Monstsh. Chem. 1963, 140, 672-676. [37] Y. C, Zhou, X. H. Wang Sun, Electronic and structural properties of the layered ternary carbide Ti3AlC2; J. of Materials Chem., 2001, 11, 2335-2339. [38] H. Wolfsgruber, H. Nowotny, F. Benesovsky, Die Kristallstruktur von Ti3GeC2, Monatsh. Chem. 1967 98, 2403-2405. [39] C. J. Rawn, M. W. Barsoum, T. El-Raghy, A. Procipio, C,. M. Hoffmann, C.R. Hubbard, Structure of Ti4AlN3 - a layered Mn+1AXn nitride, Mat. Research Bulletin, 2000, 35, 1785-1796. [40] A. Bouhemadou, R. Khenata; M. Chegaar, Structural and elastic properties of Zr2A X and Ti2AlX (X = C and N) under pressure effect, The European Physical Journal B. 2007, 56, 209-215. [41] C. Racault, F. Langlais and R. Naslain, Solid State Synthesis and Characterization of the Ternary Phase Ti3SiC2, J. Mater. Sci., 1994, 29, 3384. [42] X. Tong, T. Okano, T. Iseki, T. Yano, Synthesis and High Temerature Mechaanical Properties of Ti3SiC2/SiC Composites, J. Mater. Sci. 1995, 30, 3087. [43] T. Okano, T. Yano. T. Iseki. Synthesis and Mechanical Properties of Ti3SiC2, Ceramics, Mater. Letters., 1996, 27, 85. [44] M. W. Barsoum, T. El-Rayhy, L. Ogbuji, Oxidation of Ti3SiC2 in Air, J. Electrochem. Soc. 1997, 144, 2508. [45] J. J. Nickl, K. K. Schweitzer , P. Luxenberg, Gasphasenabschiedung im System Ti-C-Si, J. Less Common Metals, 1972, 26, 283. [46] R. Pampuch, L. Stobierski, M. Tymkiewicz, Solid Combustion Synthesis of Ti3SiC2, J. Eur. Ceram. Coc., 1989, 5, 283. [47] M. W, Barsoum, H.-I. Yoo, I. K. Polushina, V. Yu. Rud, Yu. V. Rud, T. El-Raghy, Electrical Conductivity, Thermopwer an Hall Effect of Ti3AlC2, Ti4 AlN3, and Ti3SiC2, Phys. Rev. B. 2000, 15, 10194. [48] N. Tzenov, M. W. Barsoum, Synthesis and Characterization of Ti3AlC1,8, J. Amer. Cer. Soc., 2000, 83, 825. [49] A. T. Procopio, M. W. Barsoum, T. El-Raghy, Characterization of Ti4Aln3 and Phase Equilibria in the Ti-Al-N System, Met. Mater. Trans. 2000, 31A, 373. [50] M. W. Barsoum, T. El-Raghy, A Progress Report on Ti3SiC2, Ti3GeC2 and the HPhases, M2AX, J. Mater. Synth. Process., 1997, 5, 197-216. - 298 - Literaturverzeichnis [51] M. W. Barsoum, d: Brodkin; T. El-Raghy, Layered Machinable Ceramics For High Temperature Applications, Scrip. Met. Et. Mater., 1997, 36, 535. [52] M. W. Barsoum, M. Ali, T. El-Raghy, Processing and Characterization of Ti2AlC, Ti2AlN and Ti2AlC0,5N0,5, Met. Mater. Trans., 2000, 31A, 1857-1865. [53] M. W. Barsoum, T. El-Raghy, C. J. Rawn, W. D. Porter, H. Wang, A. Payzant, C. Hubbard, Thermal Properties of Ti 3SiC2, J. Phys. Chem. Solids, 1999, 60, 429-439. [54] M. W. Barsoum, T. El-Raghy, W. D. Porter, H. Wang, J. C. Ho, S. Chakraborty, Thermal Properties of Nb2SnC, J. Appl. Phys, 2000, 88, 6316. [55] R. E. Taylor, S. Chakraborty, M. W. Barsoum, Synthesis and Characterization of Hf2PbC, Zr2PbC and M2SnC (M=Ti, Hf, Nb, and Zr), J. Europ. Cer. Soc., 2000, 20, 2619. [56] M. W. Barsoum, T. El-Raghy, C. Rawn, H. Wang, W. D. Poter, Electrical and Thermal Properties of Ti2AlC, 2000, 15, 241. [57] R. E. Taylor, Thermal Conductivity of TiC at High Temperatures, J. Amer. Cer. Soc., 1961, 44, 525. [58] W. Lengauer, S. Binder, K. Ainger, P. Ettmayer, A. Gillou, J. Debuigne, G. Groboth, Solid State Properties of Group IV b Carbonitrides, J. Alloys Compouds, 1995, 217, 137. [59] M. W. Barsoum et. al., The Mn+1AXn Phases: A New Class of Solids Prog. Solid State Chemistry, 2000, 28, 201-281. [60] H. Schnell: Wärmeaustauscher, Energieeinsparung durch Optimierung von Wärmeprozessen, 2. Ausgabe, Vulkan-Verlag Essen 1994. [61] E. Rudy, D. P. Harmon, and C. E. Brukl, AFML-TR-65-2, Part I, Vol. II 1965. [62] H. Bittner, H. Goretzki, Monatsh. Chem., 1962, 93, 1000-1004. [63] T. M. Gesing, W. Jeitschko., The Crystal Structure and Chemical Properties of U2Al3C4 and Structure Refinement of Al4C3, Z. Naturforsch. B, 1995, 50, 196-200. [64] J.-C. Schater, H. Isper, Z. Metallk., 1990, 81, 389-396. [65] C. McCullough, J. J. Valencia, C. G. Levi, R. Mehrabian, Acta Metall., 1989, 37, 13241336. [66] H. H. Hayes, Aluminium-Carben-Titanium, in Terrnary Alloys (Hersg.: G. Petzow, G. Effenberg) 1. Auflage VCH Verlagsgesellschaft, Weinheim. [67] W. Jeitschko, H. Nowotny, F. Benesovsky, Ti3AlC, ein Perowskit-Carbid, Monatshefte für Chemie, -108, 1977 und 1964, 95, 319-321. - 299 - Literaturverzeichnis [68] Weiterverarbeitung von Einkristalldiffraktometerdaten, Verfeinerung von Strukturmodellen aus Einkristalldaten, Absorptionskorrektur. [69] J. J. Nickl, K. K. Schweitzer, P. Luxenberg, Gasphasenabscheidung im System Ti-Si-C, J. Less-Common Met. 1972, 26, 335-353. [70] Phase equilibria of the quaternary system, Ti-Al-Sn-N at 900°C, J. Alloys Compd. 1997, 247, 198-201. [71] W. Jeitschko, H. Nowotny, F. Benesovsky, Die H-Phasen Ti2TlC, Ti2PbC, Nb2InC, Nb2SnC und Ta2GaC, Monatsh. Chem. 1964, 95, 431-435. [72] H. Lückeroth, Diploma Thesis, University of Bonn, Germany, 1998. [73 O. Ruff, B. Bergdahl, Z. Anorg. Allg. Chem., 1919, 106, 91-92 [74] S. Dubois, T. Cabioc, P. Chartier, V. Gauthier, Michel Jaouen, A New Ternary Nanolaminate Carbide: Ti3SnC2, J. Am. Ceram. Soc, 90, 2007, 2642-2644. [75] W. Jeitschko, H. Nowotny, F. Benesovsky, Kohlenstoffhaltige ternäre Verbindungen (H-Phase), Monatsh. Chem., 1963, 94, 672-676. [76] W. Jeitschko, H. Nowotny, F. Benesovsky, Ti2AlN, eine stickstoffhaltige H-Phase, Monatsh. Chem. 1963, 94, 1198-1200. [77] O. K. Belousov, I. I. Kornilov, Russ. Metall. 1978, 1,175-178. [78] E. Rudy, S. Windisch, C. E. Brukl. Planseeber. Pulvermetall. 1968, 16, 3-33. [79] H. Boller, E. Parthe, Monatsh. Chem., 1963, 94, 225-226. [80] M. Pötschke, K. Schubert, Z. Metallkd. 1962, 57, 474-488. [81] K. Schubert. H. g. Meissner, M. Pötschke, W. Rossteutscher, E. Stolz, Naturwissenschaften, 1962, 49, 57. [82] H. J. Wallbaum, Z. Metallkd., 1942, 34, 118-119. [83] Y. C. Zhou, X. H. Wang, Z. M. Sun, S. Q. Chen, Electronic and structural properties of the layered ternary carbide Ti3AlC2, J. Mater. Chem. 2001, 11, 2335-2339. [84] J. J. Nickl, K. K. Schweitzer, P. Luxenberg, Gasphasenabscheidung im System Ti-Si-C, J. Less-Common Met. 1972, 26, 335-353. [85] W. Jeitschko. H. Nowotny, F. Benesovsky., Die Kristallstruktur von Ti3InC, Ti3InN, Ti3TlC und Ti3TlN, Monatsh. Chem. 1964, 95, 436-438. [86] K. Anderko, Beitrag zu den binären Systemen des Titans mit Gallium, Indium und Germanium und des Zirkons mit Gallium und Indium, Z. Metallkd. 1958, 49, 165-172. [87] R. G. Johnson, R. J. Rrosen, The Indium Rich Side of the Indium-Titanium System, Trans., Metall. Soc. AIME, 1962, 224, 397-398. - 300 - Literaturverzeichnis [88] L. D. Gulay, J. C. Schuster, Investigation of the titanium-indium system, J. Alloys Compd., 2003, 360, 137-142. [89] W. Jeitschko, H. Nowotny, F. Benesovsky, Kohlenstoffhaltige ternäre Verbindungen (H-Phase), Monatsh. Chem., 1963, 94, 672-676. [90] H. Wolfsgruber, H. Nowotny, F. Benesovsky, Die Kristallstruktur von Ti3GeC2, Monatsh. Chem. 1967, 98, 2403-2405. [91 T. V. D'Yachkova, S. I. Alyamovskii, Y. G. Zainulin, The Possible Synthesis of Stoichiometric Vanadium Monocarbide at High Temperature and Pressure, Russ. J. Inorg. Chem., 1986, 31, 1258-1260. [92] V.S. Telegus, Y.B. Kuz'ma, The investigation of the transition metals of the fourth with carbon, Visn. L'viv. Derzh. Univ. (Ser. Khim.) 1971, 12-28. [93] B. Lönnberg, thermal expansion studies on the subcarbides of group V and VI transition metals, J. Less-Common Met., 1986, 120, 135-146. [94] K. Cenzual, L. M. Gelato, M. Penzo; E. Parthe, Anorganic sSructure Types with revised Spacegroups. I., Acta Crystallographica, 1991, 47, 433-439. [95] C. H. de Novion; R. Lorenzelli; P. Costa, Comptes Rendus Hebdomadaires des Seances de l'Academie des Sciences, Serie B 1966, 263, 775-778. [96] J. C. Schuster, H. Nowotny, C. Vaccaro, The Ternary System: Cr-Al-C, V-Al-C, and the Behavior of H-Phases (M2AlC), J. Solid State Chem., 1980, 32, 213-219. [97] T. Woelpl, W. Jeitschko, Crystal structures of VSn2, NbSn2 and CrSn2 with Mg2Cutype structure and NbSnSb with CuAl2-type structure, Journal of Alloys Compd. 1994, 210, 185-190. [98] N. E. Alekseevskii, I. I. Kornilov, N. M. Mateeva, Phase equilibria and superconductivity in the V3Al-V3Sn system, Doklady Akademii Nauk SSSR, 1970, 195(2), 417-420. [99] Y. B. Kuz'ma, O. N. Il'nitskaya, Russ. J. Inorg. Chem., 1988, 33, 1508-1510. [100] K. Girgis, W. Petter, G. Pupp, The Crystal Structure of V8Ga41, Acta Crystallogr. 1975, 31, 113-116. [101] H. D. Shashikala, P. V. Mohan Rao, K. S. N. Murthy, S. V. Suryanarayana, Thermal expansion of V3Ga, J. Phys. C: Solid State Phys., 1987, 20, 2063-2067, [102] J. M. Reddy, A. K. Storm, K. Knox, The crystal structure of V2Ga5, Z. Kristallogr. 1965, 121, 441-448. [103] Savitskii E.M., Krypyakevych P.I., Baron V.V., Efimov Y.V., Phase diagram of the Vanadium-Gallium System, Inorg. Mater., 1967, 3, 35-42. - 301 - Literaturverzeichnis [104] W. Jeitschko, H. Nowotny, F. Benesovsky, Kohlenstoffhaltige ternäre Verbindungen (H-Phase), Monatsh. Chem., 1963, 94, 672-676. [105] J. M. Léger, H. T. Hall, Pressure and Temperature formation of A3B compounds., Nb3Ge, Nb3Sn, Nb3Pb, V-In and V-Pb, J. Less-Common Met. 1974, 34, 17-24. [106] L. K. Borusevich, E. I. Gladyshevskii,E. I. Hladyshevskii, T. F. Fedorov, N. M. Popova, New representatives of the W3Fe3C structure type, hurnal Strukturnoi Khimii, 1965, 6, 313-314. [107] A. Westgren, G. Phragmèn, J. Iron Inst. 1928, 117, 383-400. [108] A. Westgren, Jernkontorets Ann. 1933, 117, 501-512. [109] A. Westgren, Natur,. 1933, 132, 480. [110] H. H. Stadelmaier, Z. Metallkd., 1961, 52, 758-762. [111] H. H. Stadelmaier, T. S. Yunn, Z. Metallkd. 1962, 53, 754-756. [112] H. H. Stadelmaier, R. A: Draughn, G. Hofer, Z. Metallkd, 1963, 54, 640-644. [113] E. Ganglberger, H. Nowotny, F. Benesovsky, Mh. Chemie, 1965, 96, 1144-1146. [114] E. Ganglberger, H. Nowotny, F. Benesovsky, Mh. Chemie, 1966, 97, 101-102. [115] Y. B. Kur’ma, Y. V. Voroshilov, E. E. Cherkashin, Inorg. Mater. 1965, 1, 1017. [116] Pearson’s Crystal Data CD, ASM International, 2007. [117] L. Y. Bezruk, E. T. Markovskii,G. E. Berlova, Inorg. Mater. 1971, 7, 50. [118] H. H. Stadelmaier, R. A. Gregg, Metall (Berlin) 1963, 17, 412-414. [119] V. Keimes, A. Mewis, Z. anorg. Allg. Chem. 1992, 618, 35-38. [120] M. Andersson-Söderg, Y. Andersson, J. Solid State Chem. 1990, 85, 315-317. [121] A. V. Gribanov, Y. D. Seropegin, J. Alloys Compd. 1994, 204, L9-L11. [122] H. Hirota, Y. Tawara, Y. Kamatsu, Jap. J. Appl. Phys. 1969, 8, 962. [123] K. H. J. Buschow, P. G. van Engen, R. Jongebreur, J. Magn. and Magn. Mater. 1983, 38, 1-22. [124] R. Teller, Chem. Unserer Zeit 1988, 22, 93-99. [125] E. Lugscheider. H. Reimann, R. Pankert Z. Metallkd. 1980, 71, 654-657. [126] D. Kotzott, Schwerpunktspraktikum, University of Freiburg, Germany, 2006. [127] E. W. Taylor, Correlation of Mohs’s scale of hardness with the Vickers’s hardness number. Nature, 1949, 163, 718-721. [128] L. Battrzzati, C. Antonine, M. Baricco, J. Alloys Comp. 1997, 247, 164-171. [129] U. Herold, U. Köstwe, Z. Metallkd. 1978, 58, 449-451. [130] Y. Khan, H. Wibbeke, Z. Metallkd.1991, 82, 703. [131] H. Y. Yakel, Acta Cryst. 1987, B43, 230-238. - 302 - Literaturverzeichnis [132] W. Jung, Z. Kristalloogr. 1980, 151, 113-120. [133] H. H. Stadelmaier, J. D. Schöbel, J. R. Sagmüller, Metall (Berlin) 1964, 18, 23-24. [134] M. L. Fiedler, H. H. Stadelmaier, I. K. Simonsen, Z. Metallkd. 1977, 68, 356-358. [135] R. Hawig, Y. Khan, H. Wibbeke, Z. Metallkd. 1991, 82, 646-649. [136] C. B. Finch, P. F. Becher, M. K. Ferber, V. J. Tennery, C. S. Yust, J. Cryst. Growth 1982, 58, 647-649. [137] P. Rogl, H. Nowotny, Mh. Chemie 1973, 104. [138] M. Ayel, R. Riviere, G. Monnier, C. R. 1967, 264C, 1756. [139] W. Steurer, P. Rogl, H. Nowotny, Mh. Chemie 1979, 110, 791-798. [140] H. H. Stadelmaier, R. A. Draughn, G. Hofer, Z. Metallkd. 1963. 54, 640-644. [141] H. H. Stadelmaier, , J. D. Schöbel, L. T. Jordan, Metall (Berlin) 1962, 16, 752-754. [142] H. H. Stadelmaier, A. C. Fraker, Metall (Berlin) 1962, 16, 212-214. [143] H. H. Stadelmaier, M.L. Fiedler, Z. Metallkd. 1969, 60,447-448. [144] J. D. Schöbel, H. H. Stadelmaier, Z. Metallkd. 1964, 55, 378-382. [145] H. H. Stadelmaier, F. M. Lee, Metall (Berlin) 1964, 67, 111-113. [146] H. H. Stadelmaier, M.L. Fiedler, Z. Metallkd. 1969, 53, 719-721. [147] G. Hofer, H. H. Stadelmaier, Metall (Berlin) 1964, 18, 963-964. [148] Y. B. Kur’ma, N. S. Bilonishko, Izv. Akad. Nauk SSSR. Materialy 1971, 7, 620. [149] E: Gangelberger; H. Nowotny, F. Benesovsky, Mh. Chemie 1965, 96, 1144-1146. [150] H. H. Stadelmaier, J. B. Ballance, Z. Metallkd. 1967. 58, 449-451. [151] V. Keimes, A. Mewis, Z. anorg. Allg. Chem. 1992, 618, 35-38. [152] A. V. Gribanov, Y. D. Seropegin, J. Alloys Compd. 1994, 204, L9 - L11. [153] M. Andersson-Söderberg, Y. Andersson, J. Solid State Chem. 1990, 85, 315-317. [154] N. F. Chaban, Y. B. Kuz’ma, Kotouska L. D. Dopov. Akad. Nauk URSR, Ser. A, 1980, 87-89. [155] E. Gangelgerger. H. Noweotny, F. Benesovsky. Mh. Chemie 1966, 97,101-102. [156] E. Gangelgerger. H. Noweotny, F. Benesovsky. Mh. Chemie 1965, 96, 1144-1146. [157] H. H. Stadelmaier, J. D. Schöbel, R. A. Jones, Metall, 1967, 21, 17. [158] J. D. Schöbel, H. H. Stadelmaier, Metall, 1969, 23, ??. [159] H. H. Stadelmaier, J. G. Avery, Z. Metallkd. 1965, 56, 508. [160] B. Kuz’ma, M. V. Chepiga, A. M. Plakhina, Izv. Akad. Nauk SSSR, Neorg. Materialy, 1966, 2, 1218. [161] Yu. V. Voroshilov, B. Kuz’ma, Izv. Akad. Nauk. SSSR, Neorg. Materialy, 1966, 2, 764. - 303 - Literaturverzeichnis [162] D. Kotzott, Diploma Thesis, University of Freiburg, Germany, 2006. [163] P. Rogl, H. Nowotny, Mh. Chemie 1973, 104. [164] L. T. Jorden, R. K. Mathur, K. R. Brose, H. H. Stadelmaier, Z. Metallkd. 1981, 72, 782-784. [165] H. H. Stadelmaier, R. A. Gregg, Metall 1962, 16, 407-408. [166] M. L. Fiedler, H. H. Stadelmaier, E. B. Snipes, Z. Metallkd. 1977, 68, 765-767. [167] M. Ade, Ph. D. Thesis, University of Freiburg, Germany, 1997. [168] J. B. Ballance, H. H. Stadelmaier, Z. Metallkd. 1976, 67, 729-731. [169] H. H. Stadelmaier, R. B. Fitts, Metall, 1962, 16, 773-774. [170] G. Hofer, H. H. Stadelmaier, Metall, 1965, 19, 1257-1258. [171] T. Hahn, International Tables for Cryatallography; 1983, Volume A, 678-681. [172] H. L. Yakel, Acta Cryst. 1987, B43, 230-238. [173] T. Ohba, Y. Kitano, Y. Komura, The Charge-Density Study of the Laves Phases, MgZn2 and MgCu2, Acta Crystallogr. 1984, 40, 1-5. [174] H. Hillebrecht, M. Ade, Al-Atome gegen B4-Tetraeder- eine überraschende Substitutionsmöglichkeit in den τ-Boriden Ni20Al3B6 und Ni20AlB14, Angew. Chem. 1998, 110, 981-983. [175] T. B. Massalski (Hrsg.), Binary Alloy Phase Diagrams, ASM, Metals Park, Ohio, 1986. [176] J. Schöbel, H. H. Stadelmaier, Das Zweistoffsystem Kobalt-Bor, Z. Metallkde. 1966, 57, 323-324. [177] P. Villars, L. D. Calvert, Pearson’s Handbool of Crystallographic Data for Intermetallic Phases, 1. Auflage., ASM, Metals Park, Ohio, 1985. [178] [179] T. Bjurstroem, Arkiv foer Kemi, Mineralogooch Geologo, A 1933, 11, 1-12. H. Nowotny, Kristallchemie von Komplexcarbiden und verwandten Verbindungen. Angew. Chem. 1972, 84, 973-983. [180] F. A. Cotton, Q. Rev. Chem. Soc. 1966, 20, 389; Angew. Chem. 1981, 93, 44-63. [181] M. L. Fiedler, H. H. Stadelmaier, E. B. Snipes, The Ternary System Cobalt-GalliumBoron, Z. Metallkd. 1977, 68, 765-767. [182] K. H. J. Buschow, P. G. Van Engen, R. Jongebreur, MAGNETO-OPTICAL PROPERTIES OF METALLIC FERROMAGNETIC MATERIALS, J. Magn. Magn. Mater. 1983, 38, 1-22. [183] Weiterverarbeitung von Einkristalldiffraktometerdaten, Verfeinerung von Strukturmodellen aus Einkristalldaten, Absorptionskorrektur. - 304 - Literaturverzeichnis [184] F. A. Cotton, Q. Rev. Chem. Soc. 1966, 20, 389; Angew. Chem. 1981, 93, 44-63. [185 ] N. F. Chaban, Y. B. Kuz’ma, Inorg. Mter. 1973, 9, 1886-1888. [186] Weiterverarbeitung von Einkristalldiffraktometerdaten, Verfeinerung von Strukturmodellen aus Einkristalldaten, Absorptionskorrektur. [187] [188] F. A. Cotton, Q. Rev. Chem. Soc. 1966, 20, 389; Angew. Chem. 1981, 93, 44-63. H. H. Stadelmaier, R. B. Fitts, Metallwissenschaft und Technik, 1962, 16, 773-774. [189] F. A. Cotton, Q. Rev. Chem. Soc. 1966, 20, 389; Angew. Chem. 1981, 93, 44-63. [190] UNI TERRA - Kindler & Gliech GbR Germany, Berlin, 1996 – 1999. [191] H. H. Stadelmaier, Metal-Rich Metal-Metalloid Phases, in Developments in the Structural Chemistry of Alloy Phases (Hrsg.: B. C. Giessen), 1. Aufl., Plenum, New York, 1969, 141-180. [192] H. H. Stadelmaier, J. G. Avery, Die Phasen mit der Borkoordinationszahl 8 im System Kobalt-Vanadium-Bor, Z. Metallkd., 1965, 56, 508-511. [193] F. A. Cotton, Q. Rev. Chem. Soc. 1966, 20, 389; Angew. Chem. 1981, 93, 44-63. [194] H. Hillebrecht, K. Gebhardt, 9. Jahrestagung der Deutschen Gesellschaft für Kristallographie (DGK), Bayreuth, 2001. [195] H. Hillebrecht, K. Gebhardt, 9. Jahrestagung der Deutschen Gesellschaft für Kristallographie (DGK), Bayreuth, 2001. [196] Yu. B. Kuz'ma, S. I. Svarichevskaya, Crystal structure of Y2ReB6 and its analogs, Kristallografiya, 1972, 17, 658-661. [197] N. F. Chaban, Yu. B. Kuz’ma, The triple System Cr-Al-B and Mn-Al-B, Izvestiya Akademi Nauk SSSR, Neorganicheskie Materialy, 1973, 9, 1908-1911. [198] [199] N. F. Chaban, Y. Kuz`ma, Inorg. Mater.,1973, 9, 1886-1888. Strukturlösung, -verfeinerung anhand von Einkristalldaten (Patterson und direkt Methode) [200] E. Lugscheider, H. Reimann, P. Roger, h-Carbide in den Systemen Co-W-C und FeW-C, Z. Metallkd. 1982, 73, 321-324. [201] E. Reiffenstein, H. Nowotny, F. Benesovsky, Neue Kappa-Phasen, Monatsh. Chem. 1966, 97, 499-505. [202] J. Etzkorn, Dissertation, Synthesen ternärer Carbide aus metallischen Schmelzen, 2001, 78-80. [203] J. Etzkorn, H. Hillebrecht, “Synthesis, Crystal Growth, and Structure of Ta3Al2CoC – An ordered quarternary cubic η-carbide and the first single crystal study of a ηcarbide”, J. solid State Chem., 2008, 181, 1342-1346. - 305 - Literaturverzeichnis [204] L. K. Borusevich, E. I. Gladyshevskii, T. F. Fedorov, N. M. popova, J. Struct. Chem., 1965, 6(2), 292-293. [205] A. D. Krawitz, D. G. Reichel, R. Hitterman, Thermal Expansion of Tungsten Carbide at Low Temperature, J. Am. Ceram. Soc. 1989, 72, 515-517. [206] http://www.esk.com/de/produkte-marken/produkte/duesen/strahlduesen.html. [207] Diamond Version 3.0d, Graphische Darstellung von Kristallstrukturen. [208] DIFFRACplus EVA, Auswertung von Pulverdiffraktogrammen (Fa. SIMENS). STOE Win XPOW (Version 1.08), Auswertung von Pulverdiffraktogrammen (Fa. STOE). [210] XCH Version 5.03, Konvertieren von Pulverdiffraktogrammen. - 306 -