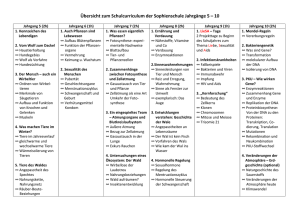
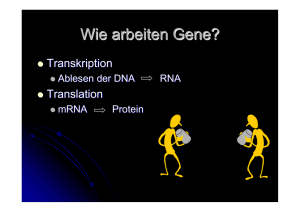
Prof. Dr. med. HP Bruch Die genomische Instabilität beei
Werbung

Aus der Klinik für Chirurgie der Universität zu Lübeck Direktor: Prof. Dr. med. H. P. Bruch Die genomische Instabilität beeinflusst das Transkriptom und Proteom von Subtypen des Endometriumkarzinoms. Inauguraldissertation zur Erlangung der Doktorwürde der Universität zu Lübeck - aus der Sektion Medizin - vorgelegt von Nana Kristin Bündgen aus Dortmund Lübeck 2012 1. Berichterstatter: Priv. Doz. Dr. med. Jens Habermann PhD 2. Berichterstatter: Priv. Doz. Dr. med. Dorothea Fischer Tag der mündlichen Prüfung: 20.11.2012 Zum Druck genehmigt. Lübeck, den 20.11.2012 - Promotionskommission der Sektion Medizin - Inhaltsverzeichnis Abkürzungsverzeichnis....................................................................................................... 1 1. Einleitung ......................................................................................................................... 2 1.1. Einführung und Ziele der Arbeit................................................................................... 2 1.2. Epidemiologie ............................................................................................................. 3 1.3 Anatomische Strukturen............................................................................................... 4 1.4. Histopathologie ........................................................................................................... 5 1.5. Ätiologie ...................................................................................................................... 6 1.6. Risikofaktoren ............................................................................................................. 6 1.6.1. Adipositas ............................................................................................................ 7 1.6.2. Menstruationszyklus............................................................................................. 8 1.6.3. Tamoxifen ............................................................................................................ 8 1.6.4. Hormonsubstitution .............................................................................................. 8 1.6.5. Ovulationshemmer ............................................................................................... 9 1.6.6. Nikotin .................................................................................................................. 9 1.6.7. Familie ................................................................................................................. 9 1.7. Präkanzerosen und Vorstufen des Endometriumkarzinoms ...................................... 10 1.7.1. Endometriumhyperplasien.................................................................................. 10 1.7.2. Endometriales Intraepitheliales Karzinom........................................................... 11 1.8. Diagnostik ................................................................................................................. 11 1.8.1. Vaginale Sonographie ........................................................................................ 12 1.8.2. Fraktionierte Abrasio und Hysteroskopie ............................................................ 12 1.9. Früherkennung und Screening .................................................................................. 13 1.10. Einteilung ................................................................................................................ 13 1.10.1. Endometrioides Adenokarzinom....................................................................... 16 1.10.2. Serös-papilläres Adenokarzinom...................................................................... 16 1.11. Prognosefaktoren.................................................................................................... 16 1.11.1. Tumorstadium .................................................................................................. 17 1.11.2. Differenzierung ................................................................................................. 17 1.11.3. Alter.................................................................................................................. 18 1.11.4. Histologie ......................................................................................................... 18 1.11.5. Hormonrezeptoren ........................................................................................... 18 1.11.6. Ploidie .............................................................................................................. 19 1.11.7. Protein 53 (TP53) ............................................................................................. 20 1.11.8. ERBB2 ............................................................................................................. 22 1.11.9. Mikrosatelliteninstabilität................................................................................... 22 1.12. Therapie.................................................................................................................. 23 1.12.1. Operation ......................................................................................................... 23 1.12.2. Radiotherapie................................................................................................... 24 1.12.3. Chemotherapie................................................................................................. 24 1.12.4. Hormontherapie................................................................................................ 24 2. Patienten ........................................................................................................................ 26 2.1. Klinische Daten ......................................................................................................... 26 3. Methodik......................................................................................................................... 27 3.1. Bild-DNA-Cytometrie................................................................................................. 27 3.2. Immunhistochemie .................................................................................................... 28 3.3. Comparative Genomische Hybridisierung ................................................................. 28 3.3.1. DNA-Extraktion aus Blut..................................................................................... 31 3.3.2. DNA und RNA Extraktion ................................................................................... 31 3.3.3. Metaphasen-Objekttäger .................................................................................... 32 3.3.4. Nick-Translation ................................................................................................. 33 3.3.5. DNA-Präzipitation............................................................................................... 34 3.3.6. Vorbehandlung der Objektträger ........................................................................ 34 3.3.7. Hybridisierung .................................................................................................... 34 3.3.8. Detektion............................................................................................................ 35 3.3.9. Aufnahme mittels CCD-Camera ......................................................................... 35 3.3.10. Metaphasen-Auswertung.................................................................................. 36 3.4. cDNA-Microarray....................................................................................................... 37 3.4.1. Herstellung der cDNA-Microarrays ..................................................................... 38 3.4.2. Scannen der Arrays............................................................................................ 40 3.4.3. Auswertung mit Gene Pix Pro............................................................................. 40 3.4.4. Microarray-Qualitäts-Bewertung und Datenanalyse (Zwei-Gruppen-KlassenVergleich):.................................................................................................................... 42 3.4.5. Signalweg-Analysen........................................................................................... 43 3.5. Protein-Analyse......................................................................................................... 43 4. Ergebnisse ..................................................................................................................... 44 4.1. Ploidie ....................................................................................................................... 44 4.2. CGH.......................................................................................................................... 45 4.2.1. Diploid endometrioide Endometriumkarzinome (EnD) ........................................ 45 4.2.2. Aneuploid endometrioide Endometriumkarzinome (EnA).................................... 45 4.2.3. Serös-papilläre Endometriumkarzinome (UPSC)................................................ 46 4.2.4. ANCA- und ANRA-Werte.................................................................................... 46 4.3. Genexpression.......................................................................................................... 48 4.3.1. Korrelation der Veränderungen im Gen- und Proteinexpressionsmuster ............ 49 4.3.2. Funktionale Einordnung der unterschiedlich exprimierten Gene und Proteine .... 50 5. Diskussion...................................................................................................................... 54 6. Zusammenfassung ........................................................................................................ 62 7. Literaturverzeichnis....................................................................................................... 63 8. Anhang ........................................................................................................................... 71 8.1. Protokolle.................................................................................................................. 71 8.1.1. Statische Bild-Cytometrie ................................................................................... 71 8.1.2. CGH ................................................................................................................... 72 8.1.3. cDNA-Microarray................................................................................................ 81 8.2. Tabellen und Abbildungen......................................................................................... 86 8.2.1. Klinische Daten mit Ergebnissen der Ploidie, Immunhistochemie und CGH. .......... 86 8.2.2. DEG Listen......................................................................................................... 89 8.2.3. Überblick der IPA-Analysen................................................................................ 96 8.2.4. Netzwerke basierend auf der IPA-Analyse ......................................................... 98 9. Danksagung ................................................................................................................. 100 10. Lebenslauf.................................................................................................................. 101 Abkürzungsverzeichnis ANCA Average number of copy number alterations; durchschnittliche Anzahl der Veränderung der Kopienzahl ANRA Average number of regional amplifications; durchschnittliche Anzahl der regionalen Amplifikationen CCD Charge coupled device CGH Comparative genomische Hybridisierung CV Coefficient of variation; Variationskoeffizient DEG Differentially expressed gene, unterschiedlich exprimiertes Gen DEP Differentially expressed protein; unterschiedlich exprimiertes Protein EIC endometriales intraepitheliales Karzinom EnA Aneuploid endometrioides Adenokarzinom EnD Diploid endometrioides Adenokarzinom EnCa Endometrial cancer; Endometriumkarzinom ERBB2 v-erb-b2 erythroblastic leukemia viral oncogene homolog 2 FIGO Fédération Internationale de Gynécologie et d’Obstétrique FISH Fluoreszens in situ Hybridisierung HNPCC hereditary non-polyposis colorectal cancer; heriditäres kolorektales Karzinom ohne Polyposis HRT hormone replacement therapy; Hormon-Ersatz-Therapie IPA Ingenuity pathways analysis KEGG Kyoto enciclopedia of genes and genomes MSI Mikrosatelliteninstabilität TP53 Protein 53 PCR „polymerase chain reaction“; Polymerasekettenreaktion SHBG Sex-hormone-binding-globulin; Sexualhormon bindendes Protein SSI Stem line scatter index; Stammlinien Verteilungs-Wert UPSC Uterine papillary serous carcinoma, serös papilläres Karzinom UPSC-A Aneuploid uterine papillary serous carcinoma; aneuploides serös papilläres Karzinom 1 1. Einleitung 1.1. Einführung und Ziele der Arbeit Das typische Karzinom der Gebärmutter geht von der Schleimhaut (Endometrium) aus und ist der häufigste maligne Tumor des weiblichen Genitaltraktes in Deutschland und der westlichen Welt [1,2]. Das klinische Verhalten dieser Tumorgruppe ist sehr indifferent und die Überlebensraten variieren stark. Obwohl die meisten Endometriumkarzinome in einem frühen Stadium entdeckt werden und mit einer relativen 5-Jahres-Überlebensrate von 72 85% [1] klinisch einen günstigen Verlauf zeigen, gibt es doch Untergruppen, die durch einen ausgesprochen aggressiven Verlauf mit frühzeitiger Metastasierung und geringen Überlebenszeiten gekennzeichnet sind. Es wurden bereits prognose-bestimmende Faktoren für Endometriumkarzinome identifiziert. Dazu zählen vor allem die Histologie, der Tumorgrad und die Metastasierung aber auch der Ploidiestatus. Insbesondere der Ploidiestatus hat sich beim Endometriumkarzinom, ebenso wie beim Mamma-, Prostata-, und Colonkarzinom, als ein signifikanter, unabhängiger Prognosefaktor herausgestellt [36]. Bei Endometriumkarzinom-Patientinnen mit einem diploiden Tumor konnte eine günstige 5-Jahres-Überlebensrate von 94% im Vergleich zu 83% bei aneuploiden Tumoren nachgewiesen werden [7]. Der Ploidiestatus kann auf chromosomalem Level mittels Comparativer Genomischer Hybridisierung (CGH) untersucht und dargestellt werden [8]. CGH-Studien an unterschiedlichen Tumorentitäten zeigten interessanterweise spezifische Muster von Chromosomen-Gewinnen und -Verlusten, die für die einzelnen soliden Tumoren charakteristisch waren [9]. So ließ sich bei Vaginalkarzinomen die häufigste chromosomale Veränderung als Zugewin von DNA auf dem Chromosomenarm 3q [10] detektieren, während bei Endometriumkarzinomen häufig 1q, 3q, 8q und 10q betroffen sind [11,12]. Die tumorspezifischen chromosomalen Alterationen führen zu einer Veränderung der Expression der auf den betroffenen Chromosomenabschnitten lokalisierten Gene. Diese Expressionsänderungen scheinen unabhängig vom Gewebeoder Zelltyp zu sein [13] und darüber hinaus zu einer irreversiblen Störung auf transkriptionaler Ebene in aneuploiden Zellen [14] zu führen. Beim Mammakarzinom haben Expressionsstudien bereits den Einzug der ArrayTechnologie in die klinische Anwendung ermöglicht und zu einer weiteren Individualisierung der Tumortherapie geführt. Spezielle kommerzielle Array-Chips wie der Mammaprint ® erlauben Onkologen, eine bessere Einschätzung des individuellen Rückfallrisikos und des möglichen Ansprechens auf eine Chemotherapie vorzunehmen. 2 Die Arrays basieren auf spezifischen Gensignaturen und werden an frischem Tumorgewebe angewandt. Für spezielle Tumorkonstellationen wurden Genexpressionstests für die Therapieentscheidung beim Mammakarzinom von den Fachgesellschaften bereits in die aktuellen Leitlinien aufgenommen [15]. Ziel dieser Arbeit war zu untersuchen, inwiefern sich Aneuploidie und genomische Instabilität auch auf das Transkriptom und Proteom von Endometriumkarzinomen unterschiedlichen Subtyps auswirken. Darüber hinaus soll gezeigt werden, durch welche spezifischen chromosomalen Veränderungen die Aneuploidie charakterisiert wird und inwieweit diese über das Transkriptom und Genom die sehr unterschiedliche Prognose der Endometrium-Subgruppen beeinflussen. Vor diesem Hintergrund war es weiter das Ziel dieser Studie, durch die verbesserte Charakterisierung von Endometriumkarzinomen Einblicke in die Onkogenese zu erlangen und Risikogene und -proteine zu identifizieren. Dafür wurden mittels Bild-Zytometrie, Comparativer genomischer Hybridisierung (CGH), cDNA Microarray-Technologie, 2dimensionaler Gelelektrophorese (2-DE) und Massenspektrometrie spezifische chromosomale Aberrationen und assoziierte Gen- und Proteinexpressionsmuster untersucht, die die besonders aggressiven Subtypen von den prognostisch günstigen unterscheiden. Die Analysen sollen hier mögliche neue Ansätze für eine verbesserte individualisierte Prognosebestimmung, Endometriumkarzinome aufzeigen. Diagnostik Untersucht wurden und drei Therapie der Subtypen des Endometiumkarzinoms, die sich im Ploidiestatus bzw. in der Histologie voneinander unterscheiden: endometriode diploide Adenokarzinome (EnD), endometriodie aneuploide Adenokarzinome (EnA) und aneuploide serös-papilläre Endometriumkarzinome (uterine papillary serous carcinoma; UPSC-A). 1.2. Epidemiologie Das Korpuskarzinom ist die häufigste Krebserkrankung des weiblichen Genitaltraktes in Deutschland mit einem Anteil von 5,1% an allen bösartigen Neubildungen der weiblichen Bevölkerung. Im Jahr 2008 erkrankten in Deutschland 11.280 Frauen an einem Endometriumkarzinom. Es stellte die viertgrößte Krebslokalisation bei Frauen nach der Brust mit 32,1%, dem Darm mit 13,5% und der Lunge mit 7,0% der Neuerkrankungen im Jahr 2008 dar. Das Endometriumkarzinom tritt klinisch meist frühzeitig durch unregelmäßige oder postmenopausale Blutungen auf und wird daher häufig in einem prognostisch günstigen Stadium diagnostiziert. Aufgrund der häufigen Diagnose in frühen Stadien und guter Prognose liegt der Anteil des Endometriumkarzinoms an den Krebssterbefällen in Deutschland mit 2,4% an elfter Stelle. Im Jahr 2008 starben in 3 Deutschland 2420 Frauen an einem Karzinom der Gebärmutter, welches mit einem mittleren Erkrankungsalter von 69 Jahren zu den Erkrankungen des höheren Lebensalters zählt [1]. Allerdings treten Endometriumkarzinome auch in bis zu 20% der Fälle prämenopausal und bis zu 5% sogar bei unter 45 jährigen Frauen auf [16]. In Anbetracht des demografischen Wandels in Deutschland könnte sich der Trend einer ansteigenden Prävalenz des Endometriumkarzinoms (von 1990 bis 2004 um 10 bis 20%) insbesondere in höheren Altersgruppen entsprechend fortsetzen [16,17]. In Deutschland erkrankt aktuell eine von 47 Frauen im Laufe ihres Lebens an einem Endometriumkarzinom. Im Vergleich zu anderen EU-Ländern liegt Deutschland im mittleren Bereich der Erkrankungshäufigkeiten. Die Mortalität durch das Endometriumkarzinom sinkt in den letzten Jahren in Deutschland deutlich [1]. Trotzdem hat dieses Karzinom in den letzten Jahren das Cervixkarzinom von Platz eins der häufigsten Genitalkarzinome abgelöst [1]. Die Inzidenz des Endometriumkarzinoms variiert weltweit und ist am höchsten bei weißen Frauen aus westlichen Bevölkerungsgruppen. Die höchsten Inzidenzen werden in Europa, Großbritannien, den Vereinigten Staaten, Kanada, Neuseeland und Australien registriert. Konstant niedrigere Inzidenzen findet man in Afrika, Zentral- und Südamerika und in Asien. Interessanterweise erkranken afro-amerikanische Frauen und Asiatinnen, die in den Vereinigten Staaten von Amerika leben, wesentlich häufiger an Endometriumkarzinomen als Frauen in ihren Ursprungsländern [18]. Epidemiologische Studien aus den USA wiesen nach, dass die Inzidenzraten für Endometriumkarzinome in der weißen Bevölkerung 60% höher waren als bei Afroamerikanerinnen. Im Gegensatz dazu war die Mortalität bei Afroamerikanerinnen im Vergleich zur weißen Bevölkerung um 30% höher. Unterschiedliche Studien zeigten, dass Afroamerikanerinnen häufiger an histologisch aggressiven Tumortypen erkranken wie dem serös-papillären Endometriumkarzinom [19]. 1.3 Anatomische Strukturen Die Gebärmutter (lat. Uterus) ist ein ca. 7-8 cm langes muskuläres Hohlorgan. Der Uterus liegt in einer Bauchfellduplikatur, dem ligamentum latum, im kleinen Becken der Frau. Die Gebärmutter grenzt ventral an die Harnblase und dorsal an das Rectum. Sie setzt sich zusammen aus Gebärmutterhals (cervix uteri) und Gebärmutterkörper (corpus uteri), der im Bereich des Fundus in die beiden Eileiter (tubae uterinae) übergeht. Die Gebärmutter wird von einem komplexen Bandapparat im kleinen Becken gehalten, bestehend aus dem seitlich verlaufenden ligamentum latum, dem ligamentum rotundum und dem ligamentum teres uteri. Die arterielle Versorgung erfolgt durch die arteria uterina aus der arteria iliaca interna, der venöse Abfluss erfolgt über mehrere Venenplexus in die venae iliacae 4 internae. Die Lymphe des Uterus drainiert in verschiedene Lymphknotenstationen. Der Lymphabfluss des Endometrium erfolgt über das Myometrium in Lymphknoten des Parametrium. Die Lymphe des Cervixbereiches fließt in die regionären Lymphknoten entlang der arteria iliaca interna, die des corpus uteri gelangt in die nodi lymphatici lumbales. Über das ligamentum teres uteri besteht ein weiterer Abflussweg in die oberflächlichen nodi lymphatici inguinales. Die sympatischen Nervenfasern aus dem unteren ganglion mesentericum ziehen in den plexuus uterovaginalis seitlich der cervix, in den auch die parasympatischen Nerven aus S3 und S4 einstrahlen [20]. Die Uteruswand besteht aus mehreren Schichten. Innen ist die Gebärmutter mit einer Schleimhaut, dem Endometrium, ausgekleidet, daran grenzt eine Muskelschicht, das Myometrium. Von außen ist der Uterus von einer Bindegewebsschicht umkleidet, in der Gefäße und Nerven verlaufen. Darüber legt sich das Perimetrium, eine peritoneale Schicht. Das Endometrium besteht zur Gebärmutterhöhle hin aus Epithel, darauf folgen nach außen das stratum basale und das stratum functionale. Das stratum functionale unterliegt bei der geschlechtsreifen Frau zyklischen Veränderungen und wird während der Menstruation abgestoßen. Das stratum basale setzt sich aus bindegewebigen Anteilen und den verzweigten Endabschnitten der glandulae uterinae zusammen, es beinhaltet die Basalarterien. Aus dem stratum basale wird nach der menstruellen Blutung das stratum functionale wieder aufgebaut. Die Schleimhaut der cervix uteri unterliegt im Gegensatz zum Endometrium des Uterus nur geringen zyklischen Schwankungen und wird während der Menstruation nicht abgestoßen. Auch der histologische Aufbau unterscheidet sich von dem des corpus uteri [20]. 1.4. Histopathologie Von den Karzinomen der Gebärmutter entstehen über 90% aus dem epithelialen Anteil (Endometrium), welcher den Uterus auskleidet. Diese werden als Endometriumkarzinome bezeichnet [21], wenngleich der Begriff Korpuskarzinom häufig synonym verwendet wird. Bokhman et. al. schlugen 1983 als Erste ein dualistisches Modell der Tumorgenese von Endometriumkarzinomen vor [22]: Typ I repräsentiert das häufige, in circa 80% der Fälle vorliegende, endometrioide Adenokarzinom und die sehr seltenen adenosquamösen Endometriumkarzinome. Unter den Typ II gruppieren sich histologisch seltene Formen wie das seröse-papilläre Karzinom (UPSC) als häufigster Vertreter und klarzellige Tumoren. Es hat sich gezeigt, dass sich Endometriumkarzinome der Gruppe I und II in der Pathogenese, dem klinischen Verlauf sowie in der Ätiologie wesentlich voneinander 5 unterscheiden (siehe Tabelle 1) [19,23-25]. Diese Unterteilung bietet seitdem die logische Grundlage für viele weitere klinische Studien und hat breite Akzeptanz gefunden [24]. Typ I Typ II Häufigkeit 80% 20% Histologie endometrioid (adenosquamös) serös papillär (klarzellig) Alter 55-60 Jahre >70 Jahre Vorstufe hyperplastisches Endometrium atrophes Endometrium, endometriales intraepitheliales Karzinom (EIC) Hormon ja nein selten häufig Ploidiestatus häufig diploid fast immer aneuploid Prognose günstig schlecht abhängig TP 53 Überexpression Tabelle 1. Vergleich klinischer Merkmale des Endometriumkarzinoms Typ I und Typ II. 1.5. Ätiologie Mit Hilfe neuer und etablierter Analyseverfahren wie der Microarraytechnik, PCRAnalysen und Immunoassays haben sich für Endometriumkarzinome zumindest zwei unterschiedliche Entstehungswege herauskristallisiert. Endometriumkarzinome des Typs I haben ihren Ursprung meist auf dem Boden einer Endometriumhyperplasie in Folge einer erhöhten Östrogenwirkung auf das Endometrium. Im Gegensatz dazu entstehen die selteneren serös-papillären Endometriumkarzinome des Typs II zumeist bei älteren Frauen aus atrophem Endometrium im Rahmen eines endometrialen intraepithelialen Karzinoms [19]. Es konnte gezeigt werden, dass in Ausnahmefällen ein UPSC allerdings auch aus einem Endometriumkarzinom des Typs I hervorgehen kann [26]. 1.6. Risikofaktoren Das Endometrium ist das Zielgewebe von Steroidhormonen. Sowohl die Epithelschicht als auch das Stromagewebe weisen zahlreiche Östrogen- und Progesteronrezeptoren auf. Für das endometrioide Adenokarzinom gilt eine anhaltende Östrogenexposition unter 6 relativem Gestagenmangel als gesicherter Risikofaktor. Auf das Endometrium haben Östrogene einen stimulierenden Effekt, indem sie die Proliferation fördern. Gestagene hingegen hemmen diesen Effekt [27]. So finden sich erhöhte Inzidenzen des Typs I Karzinoms auch bei Frauen, die aufgrund eines Granulosazelltumors der Eierstöcke einer erhöhten Östrogenproduktion ausgesetzt sind [28]. Bei Frauen, die an einem Endometriumkarzinom des Typs I erkrankt waren, fanden sich im Serum erhöhte Werte sowohl für Östrogene als auch für Androgene, wohingegen bei Patientinnen mit einem Endometriumkarzinom des Typs II die Serumwerte der Sexualhormone mit denen der gesunden Kontrollen übereinstimmten [29]. Auf dieser Grundlage lassen sich weitere Risikofaktoren erklären, die mit einer erhöhten Hormonexposition sowohl endogenen als auch exogenen Ursprungs einhergehen. 1.6.1. Adipositas Der Zusammenhang zwischen Adipositas und einem erhöhtem Risiko, an einem Korpuskarzinom zu erkranken, ist durch klinische Studien seit langem bekannt [30,31]. Das relative Risiko für übergewichtige Frauen, ein Gebärmutterkarzinom zu entwickeln, variiert je nach Studie zwischen Faktor zwei und zehn [18]. Biologisch lässt sich die Risikoerhöhung durch einen erhöhten Östrogenspiegel bei adiöpsen Frauen erklären. Dazu tragen unterschiedliche Mechanismen bei. Einerseits werden bei Adipositas vermehrt Östradiol und Östrogen in den Ovarien produziert. Dazu kommt eine Zunahme der Androgenproduktion in den Nebennieren und eine Verminderung der Hydroxilierung und somit Inaktivierung von Östrogen. Andererseits findet sich im vermehrten Fettgewebe auch eine erhöhte Aktivität des Enzyms Aromatase. Die Aromatase im peripheren Fettgewebe wandelt Androgene in Östrogene um, deren Blutspiegel sich somit weiter steigert [21,32]. Darüber hinaus ist bei übergewichtigen Frauen der Spiegel des Sex-hormone-bindingglobulin (SHBG) vermindert, so dass mehr freies, bioverfügbares Östrogen vorhanden ist [29]. Im Umkehrschluss fand man bei Patientinnen, die an einem serös-papillären Karzinom erkrankt waren im Gegensatz zu denjenigen mit einem endometrioiden Adenokarzinom erhöhte Spiegel an SHBG [19]. Auch Diabetes mellitus und arterieller Hypertonus wurden in Studien als eigenständige Risikofaktoren beschrieben. Die Studienergebnisse divergieren hier jedoch stark. So gibt es Hinweise, dass zum die erniedrigten SHBG-Spiegel bei Hyperinsulinämie mit Erhöhung des bioaktiven Östrogens einhergehen, die für Diabetes mellitus als einen nicht unabhängigen Risikofaktor sprechen. Allerdings könnten die erhöhten Spiegel an Insulinlike-growth-factor (IGF1), der zumindest in vitro als Promotor der Onkogenese bekannt ist, auch eine Rolle in der Karzinogenese des Endometriumkarzinoms spielen [18,33]. 7 1.6.2. Menstruationszyklus Der Einfluss der reproduktiven Zeitspanne auf die Entstehung von Endometriumkarzinomen wurde durch unterschiedliche Studien belegt. So zeigte sich sowohl eine höhere Inzidenz von Tumoren bei Frauen mit früher Menarche, Frauen mit später Menopause sowie insgesamt verlängerter reproduktiver Phase. Auch Frauen mit unregelmäßigem, anovulatorischen Zyklus erkrankten häufiger an einem Karzinom der Gebärmutter [18]. Als Erklärung wird zum einen die erhöhte kumulative Östrogenwirkung im Laufe des Lebens herbeigezogen, was auch die erhöhte Inzidenz bei Nullipara erklärt, zum anderen wird ein relativer Östrogenüberschuss als Erklärung angeführt, da bei Zunahme der anovulatorischen Zyklen unter Progesteronmangel die Östrogenmenge in Relation erhöht ist [21]. Auch bei Patientinnen mit polyzystischen Ovarien im Rahmen eines Stein-Leventhal-Syndroms sind die Östrogenspiegel als Folge des erhöhten luteinisierenden Hormons (LH) erhöht und führen zu einer vermehrten Inzidenz von Endometriumkarzinomen bei diesen Frauen [19]. 1.6.3. Tamoxifen Tamoxifen ist ein nicht steroidales Antiöstrogen, welches seit vielen Jahren in der adjuvanten Therapie hormonsensibler Brustkarzinome eingesetzt wird. Seitdem wurden bei diesen Patienten vermehrt Fälle von Tamoxifen-assoziierten Endometriumkarzinomen beschrieben. Tamoxifen ist ein partieller Östrogenantagonist, der aber am Endometrium eine schwache östrogenähnliche Wirkung aufweist. Studien zeigten die Entwicklung verschiedener Endometriumveränderungen (Polypen, Hyperplasien, Karzinome und Sarkome) unter Tamoxifeneinnahme. Trotz des erhöhten Risikos, ein Zweitkarzinom zu entwickeln, haben Patientinnen mit hormonrezeptorpositivem Mammakarzinom unter Tamoxifentherapie einen deutlichen Überlebensvorteil [21]. Patientinnen unter LangzeitTamoxifengabe sollten regelmäßig auf Veränderungen des Endometriums hin untersucht werden und bei Auffälligkeiten im vaginalen Ultraschall frühzeitig einer weiterführenden Diagnostik zugeführt werden [15]. 1.6.4. Hormonsubstitution Unter reiner Östrogensubstitution ohne Gestagenkomponente steigt das Risiko, an einem Endometriumkarzinom zu erkranken bei postmenopausalen Frauen auf mehr als das Doppelte an. So folgte in den 70-er Jahren auf die zunehmende der Verschreibung von Hormon-Ersatz-Therapien bei klimakterischen Beschwerden ein starker Anstieg der Inzidenz von Endometriumkarzinomen in den USA. Mit dem Rückgang der Anwendung reiner Östrogensubstitution war auch der Peak in der Inzidenz in den späten 80-er Jahren 8 wieder rückläufig [18]. Einige Fall-Kontroll-Studien konnten sogar ein bis zu 12-fach erhöhtes Risiko bei alleiniger Östrogensubstitution zeigen, an einem Endometriumkarzinom zu erkranken [21]. Eine Metaanalyse unterschiedlicher FallKontroll- und Kohortenstudien zeigte eine Erhöhung des relativen Risikos für ein Endometriumkarzinom auf 2,3 zwischen Frauen, die nie Östrogen eingenommen haben und Frauen mit Hormonsubstitution. Aber auch die Dauer der Hormonersatz-Therapie mit Östrogenen lässt das relative Risiko weiter ansteigen [34]. Unter reiner Östrogensubstitution ließen sich vor allem Endometriumkarzinome des Typs I in frühen Stadien nachweisen, die mit einer guten Prognose einhergehen. Neuere Studien zeigten, dass sich unter Zusatz von einem Gestagen über mindestens 10 Tage pro Zyklus die Inzidenzsteigerung des Endometriumkarzinoms unter Östrogentherapie verhindern lässt [21]. 1.6.5. Ovulationshemmer Durch die kombinierte Einnahme von Östrogen mit einem Gestagen wird das Risiko des Endometriumkarzinoms um 50-60% reduziert. Dabei korreliert der Schutz vor der Entstehung eines Karzinoms mit der Dosis der Gestagenkomponente und ist umso höher, je länger der Einnahmezeitraum ist. Diese protektive Wirkung der oralen Kontrazeptiva kann vor allem bei Frauen mit anovulatorischen Zyklen genutzt werden, die einem erhöhten endogenen Östrogeneinfluss mit fehlender protektiver Gestagenkomponente ausgesetzt sind [35]. 1.6.6. Nikotin Interessanterweise hat sich gezeigt, dass Raucherinnen ein niedrigeres Risiko aufweisen, an einem Endometriumkarzinom zu erkranken als Nichtraucherinnen [32,36]. Auch dieser Faktor lässt sich auf eine erhöhte Östrogenexposition zurückführen. So kommen Raucherinnen eher in die Menopause und weisen erniedrigte Östrogenspiegel auf, da Nikotin den Metabolismus von Östrogen fördert und es zu einem schnelleren Abbau desselben kommt [36,37]. So fanden Cirisano et al. unter Raucherinnen wie erwartet erniedrigte Zahlen von endometrioiden Adenokarzinomen, im Gegensatz dazu aber erhöhte Zahlen von serös-papillären Karzinomen der Gebärmutter [36]. 1.6.7. Familie Der weitaus größte Anteil der Endometriumkarzinome tritt sporadisch auf. Ein kleiner Teil der Fälle ist, vor allem im Rahmen des autosomal-dominant vererbten Lynch-Syndroms, 9 genetisch bedingt. Das Lynch-Syndrom gehört zu der Gruppe von Erkrankungen mit Mikrosatelliteninstabilität (MSI), die durch Defekte in Mismatch-Repair-Genen hervorgerufen werden [38,39]. Die häufigsten Veränderungen treten in den Genen hMLH1 und hMLH2 auf, aber auch in anderen Genen dieser Gruppe wurden bereits Mutationen beschrieben (hMSH6, hPMS1, hPMS2) [40,41]. Das Syndrom ist definiert durch das zusätzliche Auftreten von Tumoren außerhalb des Kolons im Rahmen des familiären, nicht polypösen, kolorektalen Karzinoms (hereditary non-polyposis colorectal cancer/ HNPCC). Der häufigste Manifestationsort vor allem bei jüngeren Frauen mit einem mittleren Erkrankungsalter von 50 Jahren ist dabei das Endometrium [38,39]. Frauen mit HNPCC-Syndrom haben im Vergleich zur Normalpopulation ein etwa zehnfach erhöhtes Risiko, an einem Endometriumkarzinom zu erkranken [42]. Wir konnten HNPCCPatienten in unserem Kollektiv aufgrund des Erkrankungsalters und der Familien- und Eigenanamnese weitestgehend ausschließen. 1.7. Präkanzerosen und Vorstufen des Endometriumkarzinoms 1.7.1. Endometriumhyperplasien Endometriumkarzinome des Typs I entstehen zumeist auf dem Boden einer endometrialen Hyperplasie. Die Schleimhauthyperplasien entstehen durch eine unter Östrogen-Einfluss überschießende Proliferation, die nicht ausreichend von Gestagenen antagonisiert wird [35]. Die Hyperplasien des Endometriums werden anhand ihrer histologischen Besonderheiten und ihres Entartungsrisikos in vier Gruppen eingeteilt [43,44]. 1. Einfache Hyperplasie (zystisch ohne Atypie) Durch Proliferation von Endometriumdrüsen und Stromagewebe kommt es zu einer Verdickung des Endometriums. Dabei weisen Epithel und Stroma Charakteristika der Proliferationsphase auf. Wenn keine zellulären Atypien vorliegen, wird die Progression zu einem Endometriumkarzinom mit 1-3% angegeben. Die Einfache Hyperplasie ohne Atypie zählt nicht als obligate Präkanzerose. 2. Komplexe Hyperplasie (adenomatös ohne Atypien) Es liegt eine exzessive Drüsenproliferation vor mit zunehmend ausgedünntem Stroma und Bildung von intraluminalen Epithelpapillen. Es muss in 3-4% der Fälle mit einer Progression zum Karzinom gerechnet werden. 10 3. Einfache Hyperplasie mit Atypien Histologisch zeigen sich zytologische Atypien des Drüsenepithels, das ZellkernZytoplasma-Verhältnis steigt zugunsten der Zellkerne an, und es treten Kernpolymorphien auf. Das Karzinomrisiko liegt bei 5-10%. 4. Komplexe Hyperplasie mit Atypien Das Zellkern-Zytoplasma-Verhältnis und die Anzahl der Kernpolymorphien steigen weiter an. Das Progressionsrisiko zum Endometriumkarzinom liegt bei 29-30%. Die mikroskopische Entscheidung, ob eine atypische Hyperplasie vorliegt, oder ein reifes Endometriumkarzinom, kann sich als sehr schwierig erweisen. Als entscheidendes Kriterium für das Vorliegen eines Karzinoms gilt das Verschwinden des endometrialen Stroma [43]. 1.7.2. Endometriales Intraepitheliales Karzinom Das serös-papilläre Karzinom ist der häufigste Vertreter der Typ II Endometriumkarzinome und geht zumeist aus einem endometrialen intraepithelialen Karzinom (EIC) hervor. Diese Vorstufe eines invasiven Karzinoms entsteht als maligne Transformation im atroph veränderten Endometrium. Charakteristisch für das EIC ist der Ersatz der benignen oberflächlichen Zellen des Endometriums und der darunterliegenden Drüsen durch Zellen mit anaplastischen Kernen, die den Zellen serös-papillärer Karzinome gleichen. Definitionsgemäß überschreitet das EIC die endometriale Schicht nicht und ein invasives Wachstum in das darunterliegende Stroma fehlt. Die Aggressivität dieses intraepithelialen Karzinoms zeigt sich durch die gelegentlich beobachteten intraperitonealen Metastasen bei fehlendem mikroskopischen Nachweis einer Stromainvasion im Uterus [19,24]. Das EIC findet sich vor allem bei über 60 Jahre alten Frauen mit atrophem Endometrium. Mehrere Studien wiesen in endometrialen intraepithelialen Karzinomen mit einer deutlichen Immun-Positivität veränderte Genproliferate vonTP53, sowie Ki-67 nach [45,46]. 1.8. Diagnostik Das Endometriumkarzinom zeichnet sich durch seine meist frühzeitige klinische Diagnose in einem therapeutisch günstigen Tumorstadium aus. Das klassische Frühzeichen ist die perimenopausale und postmenopausale Blutung beziehungsweise die abnormale vaginale Blutung. Dazu gehören auch blutiger oder dunkler Fluor, die Diagnose einer 11 Hämato- oder Pyometra sowie Zwischenblutungen, Schmierblutungen, Hypermenorrhoen und Metrorrhagien. Bei 10% der Patientinnen, die sich mit dieser Symptomatik klinisch vorstellen, lässt sich im Verlauf ein Endometriumkarzinom diagnostizieren. Bei Patientinnen über 70 Jahren trifft dies sogar in 30% der Fälle zu, sofern die Blutung nicht durch eine exogene Hormonzufuhr ausgelöst wurde [47]. Das diagnostische Vorgehen lässt sich in invasive und nicht invasive Methoden unterteilen. Die invasive Diagnostik besteht aus der fraktionierten Ausschabung (Abrasio) nach Dilatation des Gebärmutterhalses, der Biopsie des Endometriums und der Hysteroskopie mit gezielter Biopsie. An nichtinvasiven Methoden lassen sich die Vaginalsonographie und die endometriale Zytologiegewinnung nennen, wobei letztere aufgrund ihrer Ungenauigkeit an Bedeutung verloren hat [21,35]. 1.8.1. Vaginale Sonographie In Deutschland hat sich als erstes diagnostisches Vorgehen bei symptomatischen Patientinnen oder Frauen mit zusätzlichen Risikofaktoren die vaginale Ultraschallsonographie weitgehend durchgesetzt. Dabei wird mit einem vaginalen Schallkopf der anteroposteriore Durchmesser des Endometriums gemessen. Im Allgemeinen wird bei postmenopausalen Frauen ohne Hormontherapie eine doppelte Endometriumbreite von kleiner 5 mm als unauffällig angesehen [48]. Problematisch ist die Beurteilung der Endometriumbreite prämenopausal, sowie bei Frauen unter Hormonersatztherapie (hormonal replacement therapy; HRT) oder Tamoxifengabe. Diese Medikamente führen zu einer Verbreiterung der endometrialen Schleimhaut. Auch durch neuere Studien konnte kein neuer Grenzwert der Endometriumbreite für Patientinnen unter einer solchen Therapie festgesetzt werden [49,50]. In den aktuellen Leitlinien wird eine alleinige diagnostische Messung der Endometriumbreite als nicht verwertbar angesehen [51]. Bei Frauen, die nach sechs Monaten HRT oder Tamoxifeneinnahme eine irreguläre Blutung zeigen, sollte eine histologische Abklärung des Endometriums durchgeführt werden [52]. Durch die Vaginalsonographie lassen sich auch morphologische Veränderungen des Endometriums beurteilen und geben wichtige Hinweise auf das Vorliegen von Karzinomen, Schleimhautpolypen oder Hyperplasien. Ein EIC lässt sich im Gegensatz zu den Präkanzerosen des Typs I Endometriumkarzinoms in der Regel sonografisch nicht detektieren [53]. 1.8.2. Fraktionierte Abrasio und Hysteroskopie Zur Diagnosesicherung gilt in Deutschland weiterhin die Hysteroskopie mit fraktionierter Abrasio der Korpus- und Zervixschleimhaut als Goldstandart [51]. Gerade für die 12 Identifizierung anderer Blutungsquellen wie intracavitäre Polypen und submuköse Myome gilt die Hysteroskopie als sichere Methode [54]. Auch kann mit Hilfe des Hysteroskops eine gezielte Probeentnahme durchgeführt werden und die Genauigkeit der Abrasio erhöht werden. Studien zeigten, dass bei alleiniger Dilatation und Kürettage bei bis zu 60% der Patientinnen weniger als die Hälfte der Schleimhautoberfläche kürettiert wurde und somit nur 50% des Endometriums der histologischen Untersuchung zugeführt werden konnten [55]. Allerdings zeigt eine neuere Studie, dass die zusätzliche Hysteroskopie die Sensitivität der Kürettage bei der Entdeckung von endometrialen Hyperplasien oder Karzinomen nicht erhöht [56]. 1.9. Früherkennung und Screening Ein generelles Screening mittels vaginalen Ultraschalls und Zytologiegewinnung wird bei asymptomatischen Frauen ohne Risikofaktoren als ineffektiv angesehen und in den Leitlinien der Deutschen Gesellschaft für Gynäkologie und Geburtshilfe, der Deutschen Krebsgesellschaft und der Arbeitsgemeinschaft für Gynäkologische Onkologie daher nicht empfohlen. (Adipositas, Früherkennungsuntersuchungen Diabetes mellitus, bei Frauen der Hochrisikogruppe polyzystisches-Ovar-Syndrom, bekannte Endometriumhyperplasie) werden in den Leitlinien als möglicherweise sinnvoll eingestuft. Eine Mortalitätssenkung konnte allerdings bisher nicht bewiesen werden [51]. Hohe Bedeutung kommt daher der Abklärung der abnormalen vaginalen Blutung zu. Insbesondere die postmenopausale Blutung hat eine hohe Signifikanz als frühes Zeichen eines Endometrialkarzinoms [48]. 1.10. Einteilung Bis 1988 wurden Endometriumkarzinome nach den Regeln der FIGO (Fédération Internationale de Gynécologie et d’Obstétrique) von 1971 anhand des klinischen Bildes eingeteilt. Aufgrund unterschiedlicher prospektiver Studien, die zeigten, dass vor allem Endometriumkarzinome der Stadien I und II im klinischen Staging im Vergleich zum chirurgischen in ihrer Ausdehnung unterschätzt wurden [57,58], änderte die FIGO ihre Empfehlung für das Staging von Endometriumkarzinomen. Seit 1988 empfiehlt das FIGO Komitee der Gynäkologischen Onkologie ein chirurgisches Staging; dieses dient vor allem der histologischen Verifizierung der Tumordifferenzierung und der Tumorausbreitung. Bei Patientinnen, die aufgrund eines ausgedehnten Tumorbefalls oder anderen Kontraindikationen keine primäre Operation erhalten, ist eine 13 klinische Klassifizierung noch gerechtfertigt. Das gleiche gilt in Ausnahmefällen für prämenopausale Patientinnen mit nicht abgeschlossener Familienplanung, bei denen bei Vorliegen von frühen Typ I Karzinomen fertlilitätserhaltend operiert werden kann. Die FIGO Einteilung wurde zuletzt am 01.01.2010 modifiziert [17]. Die folgende Tabelle 2 zeigt das Staging des Endometriumkarzinoms anhand der TNM und FIGO (alt/neu)– Klassifikation [17,21]. TNM FIGO Merkmal alt Merkmal neu T0 - Kein Hinweis auf Primärtumor Tis 0 Präinvasives Karzinom (carcinoma in Präinvasives Karzinom (carcinoma in situ) situ) T1 I Tumor auf das corpus uteri beschränkt Tumor auf das corpus uteri beschränkt T1a Ia Tumor auf das Endometrium Kein oder <1/2 des Myometrium beschränkt infiltriert Karzinom infiltriert <½ des > ½ des Myometrium infiltriert T1b Ib Myometrium T1c Ic Karzinom infiltriert >½ des Myometrium T2 II Invasion der cervix uteri T2a IIa Nur cervixepithel betroffen T2b IIb Stromainfiltration der cervix T3 III Ausdehnung des Tumors über den Ausdehnung des Tumors über den Uterus hinaus, aber auf das kleine Uterus hinaus, aber auf das kleine Becken begrenzt Becken begrenzt Uterusserosa, Adnexe; pos. Serosabefall u./o. Adnexbefall T3a IIa Invasion cervixstroma Peritonealzytologie T3b IIIb Vaginalbefall Vaginalbefall u./o. Parametriumbefall T3c IIIc Pelvine u./o. paraaortale Lymphknoten Pelvine u./o. paraaortale Lymphknoten T4 M1 IIIc1 Positive pelvine Lymphknoten IIIc2 Positive paraaortale Lymphknoten IVa IVb Infiltration Blasen- u./o. Infiltration Blasen- u./o. Rektumschleimhaut Rektumschleimhaut Fernmetastasen Fernmetastasen Tabelle 2. Einteilung des Endometriumkarzinoms nach FIGO und TNM Klassifikation. In der TNM Klassifikation werden zusätzlich Angaben zur Tumorstreuung in regionäre Lymphknoten (N) und Fernmetastasen (M) gegeben: 14 NX keine Angabe über regionäre Lymphknoten N0 keine regionären Lymphknotenmetastasen N1 regionäre Lymphknotenmetastasen MX keine Angaben zu Fernmetastasen M0 keine Fernmetastasen M1 Fernmetastasen Je nach Differenzierungsgrad des Tumorgewebes wird das Endometriumkarzinom in drei Grade eingeteilt, dabei entscheidet der am wenigsten differenzierte Tumoranteil über das Grading. Grad 1 Hochdifferenziertes Karzinom (< 5% des Tumors zeigt ein solides Tumorwachstum) Grad 2 Mäßig differenziertes Karzinom (6-50% des Tumors wächst solide) Grad 3 Undifferenziertes Karzinom (> 50% des Tumors zeigt ein solides Wachstumsverhalten) [59]. Karzinome des Endometriums treten fast ausschließlich in Form von Adenokarzinomen auf. Primäre Plattenepithelkarzinome der Gebärmutter sind eine Rarität. Bei der histologischen Begutachtung finden sich häufig Tumoranteile unterschiedlicher Differenzierung in einer Probe. Der quantitativ überwiegende Anteil entscheidet dabei über die morphologische Einteilung des Karzinomtyps. Bis zu 40% der Typ II Karzinome weisen Anteile von Typ I Karzinomen auf, allerdings ist in diesen Fällen der seröspapilläre beziehungsweise klarzellige Tumoranteil prognosebestimmend, selbst wenn nur fokal ein Typ II Anteil vorliegt [21,51]. Alle Endometriumkarzinome sollten nach mikroskopischer Beurteilung einem der folgenden histologischen Typen zugeordnet werden [21]: a) Endometrioides Karzinom (einschließlich Adenokarzinom, Adenoakanthom und Adenosquamöses Karzinom) b) Muzinöses Adenokarzinom c) Serös papilläres Adenokarzinom (UPSC) d) Klarzellkarzinom e) Adenosquamöses Karzinom f) Undifferenziertes Karzinom 15 1.10.1. Endometrioides Adenokarzinom Mit circa 80% ist das endometrioide Adenokarzinom der häufigste histologische Typ der Endometriumkarzinome und ist der typische Vertreter der Typ I Karzinome. Charakteristisch sind im histologischen Präparat relativ regelmäßig angeordnete Drüsen mit stark vermindertem Stromaanteil. Wichtig ist die Differenzierung zur atypischen Hyperplasie, der präkanzerösen Vorstufe, gekennzeichnet durch das Vorhandensein von Stromafibrosen und -nekrosen, sowie teilweise völliges Fehlen des Stroma zwischen den Drüsen mit Drüsenkonfluenz. Anhand des Verhältnisses zwischen soliden Tumoranteilen und Drüsen erfolgt das Grading (siehe oben). Stimmt das Ausmaß der Kernatypien mit dem Grad nicht überein, so erfolgt bei hoher mitotischer Aktivität die Einteilung in einen höheren Grad [21]. 1.10.2. Serös-papilläres Adenokarzinom Das serös-papilläre Adenokarzinom, auch als uterine papillery serous carcinoma (UPSC) bezeichnet, liegt in ca. 5% der Fälle vor und ist der häufigste Vertreter der Typ II Endometriumkarzinome. Charakteristisch ist ein papilläres Wachstum von mehr als 50% des Tumoranteiles. Unter dem Mikroskop fallen fibrös-papilläre Strukturen auf, die von zytoplasmaarmen Epithelzellen ausgekleidet sind. Zentral zeigen sich häufig abgelöste Zellhaufen mit Kernatypien und Nekrosen. Gelegentlich lassen sich Psammonkörperchen nachweisen. Insgesamt liegen ausgeprägte Atypien und zahlreiche Mitosen vor. Im Unterschied zum endometrioiden Adenokarzinom findet sich hier die Kombination aus einer gut differenzierten Gewebsarchitektur mit ausgeprägter Zellatypie, wohingegen sich beim endometrioiden Adenokarzinom Gewebsarchitektur und Zellkernatypie inkonkordant verhalten. Solide Tumoranteile zeigen sich selten [19,21,60]. Die Typ II Karzinome werden per definitionem als gering differenziert klassifiziert [17]. 1.11. Prognosefaktoren Im Allgemeinen hat das Endometriumkarzinom mit einer Fünf-Jahres-Überlebensrate von 75-83% über alle Stadien und histologische Typen hinweg betrachtet eine relativ gute Prognose [1]. Insbesondere die Typ I Karzinome werden häufig in einem frühen klinischen Stadium diagnostiziert, in ca. 80% der Fälle hat der Tumor zum Zeitpunkt der Diagnose den Uterus noch nicht überschritten. In diesen Fällen liegt die Überlebensrate bei bis zu 90-95% [61,62]. Im Gegensatz zum endometrioiden Endometriumkarzinom sind die allgemeinen klinischen Risikofaktoren wie die Myometriuminfiltration oder Gefäßeinbrüche durch den Tumor für 16 die Typ II Karzinome nicht spezifisch genug. So können Endometriumkarzinome des Typs II, insbesondere das serös-papilläre Karzinom, bereits eine extensive Metastasierung zeigen, wenn der primäre Tumor noch auf das Endometrium beschränkt ist [63]. Einige Studien wiesen bei 50-75% der serös-papillären Karzinome bereits im Stadium I eine Metastasierung außerhalb des Uterus nach [64]. 1.11.1. Tumorstadium Das Tumorstadium gilt weiterhin als das wichtigste Prognosekriterium [17] (siehe Tabelle 2). Mitbestimmend für das Tumorstadium ist die Infiltration des Myometriums durch das Karzinom, die auch maßgeblich die operative Therapie bestimmt. Seit der Einführung der FIGO-Kriterien von 1988 wird zwischen einer Infiltration nur der inneren Hälfte des Myometriums und einer Tumorausdehnung bis in die äußere Hälfte der Muskelschicht unterschieden. Eine Tumorinfiltration der äußeren Hälfte der Muskelschicht ist mit einer extrauterinen Ausbreitung und einer hohen Rezidivquote assoziiert. [65]. Die Myometriuminfiltration gilt als der wichtigste lndikator einer hämatogenen Streuung des Tumors. [66]. Auch Abeler et al. wiesen die myometriale Infiltration durch den Tumor als wichtiges Prognosekriterium aus. In einer groß angelegten Populationsstudie fanden sich 5- und 10-Jahres-Überlebensraten bei Patienten mit Karzinomen, die auf das Endometrium beschränkt waren, von 89% und 83%. Bei Patientinnen mit Infiltration der inneren Myometriumhälfte waren es nur 85% und 73%. Wenn auch die äußere Hälfte der Muskelschicht vom Tumor befallen war, sanken die Überlebensraten sogar auf 48% und 29% [67]. Ein Mitbefall des Zervixstroma, die Invasion in die Lymphgefäße und eine Ausbreitung des Tumors über die Uterusgrenzen hinaus gelten neben dem histologischen Typ als die wichtigsten Prädiktoren einer lymphogenen Metastasierung und peritonealen Ausbreitung mit dementsprechend verminderten Überlebensraten [68]. 1.11.2. Differenzierung Der Grad der Tumordifferenzierung von Endometriumkarzinomen korreliert direkt mit den Überlebensraten [67]. Je entdifferenzierter der Tumor, desto schlechter ist meist die Prognose. Für endometrioide Adenokarzinome zeigten sich 5-Jahres-Überlebensraten beim Stadium I Grad 1 von 92% und bei Grad 3 dagegen von 74%. Im Stadium II sank die 5-Jahres-Überlebensrate für Grad 1 Patientinnen auf 85,5%, für Grad 3 sogar auf 58,1% [21]. 17 1.11.3. Alter Das Alter bei Diagnosestellung gilt als ein wichtiger unabhängiger Prognosefaktor. Bei Patientinnen über 70 Jahre sinkt die Fünf-Jahres-Überlebensrate auf 60%. Mit zunehmendem Alter der Frau finden sich aber auch häufiger histologisch aggressivere Endometriumkarzinome vom Typ II [35,69]. So zeigte eine Studie, dass Frauen mit einem serös-papillären Endometrialkarzinom im Durchschnitt sechs Jahre älter waren als Patientinnen, die an einem Typ I Endometriumkarzinom erkrankten [32]. 1.11.4. Histologie Endometriumkarzinome des Typs II (seröse-papilläre Karzinome, Klarzellkarzinome, sowie undifferenzierte und squamöse Karzinome) gehen mit einer schlechten Prognose einher. Für das serös-papilläre Karzinom wurden die niedrigsten Überlebensraten ermittelt. Diese betragen für Tumoren des Stadiums 1 und 2 etwa 35-50% und für das Stadium 3 und 4 circa 0-15% [35,70]. Auch Tumorrezidive nach Therapie traten bei Patientinnnen mit serös-papillären Endometriumkarzinomen und Klarzellkarzinomen im Vergleich zu endometrioiden Adenokarzinomen signifikant häufiger auf [36]. 1.11.5. Hormonrezeptoren Der Steroidhormonrezeptorstatus hat sowohl für die Prognose als auch für die Therapieentscheidung Bedeutsamkeit. Östrogen und Progesteronrezeptoren lassen sich gewöhnlich in Endometriumhyperplasien und endometrioiden Adenokarzinomen nachweisen. Je höher die Differenzierung, desto höher ist zumeist der Gehalt an Steroidrezeptoren [71]. Serös-papilläre Karzinome und ihre Vorstufen zeigen im immmunhistochemischen Nachweis generell keine Hormonrezeporen [19]. Fukuda et al. zeigten, dass der Hormonrezeptorgehalt nicht nur mit dem Differenzierungsgrad korreliert, sondern auch mit dem Ploidiestatus, der extrauterinen Tumorausdehnung und einer höheren Überlebensrate [72,73]. Dabei kommt dem Progesteronrezeptor eine wesentlich höhere prognostische Bedeutsamkeit zu als dem Östrogenrezeptor [21]. Darüber hinaus ist der Nachweis von Progesteronrezeptoren auch für die Therapieentscheidung wichtig. So können gezielt Patientinnen ausgewählt werden, bei denen eine Gestagentherapie erfolgversprechend wäre. 18 1.11.6. Ploidie Der normale DNA-Gehalt einer Zelle ist diploid und entspricht 46 Chromosomen beziehungsweise 22 Chromosomenpaaren plus Geschlechtschromosomen. Chromosomeninstabilität kann zu strukturellen oder numerischen Veränderungen der Chromosomen führen und damit ein frühes Ereignis auf dem Weg der malignen Entartung einer Zelle mit potentieller Tumorentstehung darstellen. Der DNA-Gehalt einer Zelle spiegelt ihre genomische Stabilität wider und kann sowohl per Flow-Cytometrie als auch mit der statischen Bild-Cytometrie bestimmt werden. In den meisten Studien wurde die Flow-Cytometrie zur Bestimmung des Ploidiestatus angewandt. Mit dieser Methode kann eine große Anzahl von Zellkernen untersucht werden. Allerdings unterscheidet die Messung nicht zwischen Tumor- und normaler Zelle, und eine Zellselektion während der Messung ist nicht möglich. Die Bild-Cytometrie hingegen erlaubt die selektive Analyse von Tumorzellen unter dem Mikroskop. Somit ist die Bild-Cytometrie auch in der Lage, kleine aneuploide Tumorzellkolonien zu erkennen [74,75], so dass die Aneuploidierate in BildCytometrie-Studien gelegentlich höher ausfällt. Unterschiedliche Studien zeigten, dass die Ergebnisse der Flow-Cytometrie mit denen der Bild-Cytometrie aber zu circa 80% übereinstimmten [76]. Anhand einer großen Studie an Endometriumkarzinomen aller Grade und Histologien fanden Lundgren et al. eine Diploidierate von 61% und eine Aneuploidierate von 39% unter Verwendung der statischen Bild-Cytometrie. Diese Studie mit 376 Patientinnen wies die DNA-Ploidie neben dem Tumorstadium und dem histologischen Typ in der Multivariantenanalyse als eindeutigen Prognosefaktor aus [7]. Larson et al. differenzierten in einer Studie mit 208 Patientinnen mit Hilfe der FlowCytometrie 74% der Endometriumkarzinome als diploid und 26% als aneuploid. Die Gruppe kam zu dem Ergebnis, dass Patientinnen mit aneuploiden Tumoren ein signifikant höheres Risiko für Metastasen haben. Außerdem wiesen die Frauen mit aneuploiden Endometriumkarzinomen signifikant häufiger ein fortgeschrittenes Tumorstadium auf. Larson et al. bestimmten für aneuploide Tumoren eine 4,5 -fach höhere Prävalenz von Lymphknotenmetastasen im Becken und eine sogar 5,8-fach erhöhte Prävalenz von Metastasen entlang der Aorta. Weiterhin erlitt eine signifikant höhere Anzahl von Frauen mit aneuploiden Tumoren ein Rezidiv oder wies ein progressives Krankheitsgeschehen auf (22,2% versus 6,5%). Auch die Überlebensrate war unter den Patientinnen mit aneuploiden Endometriumkarzinomen signifikant niedriger: die 5-Jahres-Überlebensrate betrug bei ihnen 83% im Gegensatz zu 94% bei Frauen mit diploiden Tumoren [77]. Nur circa 15% der Endometriumkarzinome des Typs I weisen einen aneuploiden Chromosomensatz auf, wenngleich der Anteil an aneuploiden Tumoren mit zunehmendem Stadium steigt. Viele Studien haben gezeigt, dass Patientinnen mit diploiden Endometriumkarzinomen eine deutlich bessere Prognose haben, als diejenigen, 19 die an aneuploiden Tumoren erkrankt waren. Multivariantenanalysen wiesen den Ploidiestatus als unabhängigen Prognosefaktor aus [7,78-81]. Ein aneuploider Chromosomensatz findet sich zumeist in entdifferenzierten (Grad 3) endometrioiden Adenokarzinomen, Klarzellkarzinomen und serös-papillären Karzinomen, die dem Endometriumkarzinom des Typs II zugeordnet werden. Trotzdem zeigten einige Studien den Ploidiestatus als vom histologischen Typ unabhängigen Prognosefaktor [79,80,82,83]. Andere Studien konnten dies hingegen nicht belegen [84,85], allerdings unterschieden sich die Studien wesentlich in Bezug auf die Staging-Methode, die Anzahl der untersuchten Karzinome und die Einschlusskriterien. Im Gegensatz zu anderen wichtigen Prognosefaktoren, wie dem FIGO-Stadium und insbesondere der myometrialen Infiltration, konnte gezeigt werden, dass der Ploidiestatus auch schon im präoperativen Kürettagematerial mit hoher Sicherheit bestimmt werden kann [86,87]. So konnten Susini et al. in einer Sudie mit 50 Endometriumkarzinomen bereits im Kürettagematerial den Ploidiestatus mit einer Sensitivität und Spezifität von 100% voraussagen [87]. 1.11.7. Protein 53 (TP53) Das TP53-Gen, lokalisiert auf dem kurzen Arm des Chromosoms 17 (17p13), gehört zu der Familie der Tumorsupressorgene. Lane und Levine entdeckten 1979 unabhängig voneinander erhöhte Konzentrationen von TP53 in Tumorzellen [88,89]. Aufgrund der Akkumulation von TP53 in Tumorzellen wurde es zunächst fälschlicherweise als Tumoronkogen bezeichnet. Das Protein TP53 hat in gesunden Zellen unterschiedliche Funktionen, vor allem wirkt es aber als Transkriptionsfaktor. Als sogenannter „Wächter des Genoms“ kontrolliert TP53 den Zellzyklus und verhindert eine unkontrollierte Replikation geschädigter DNA [90]. Kommt es in der DNA zu Schäden, akkumuliert TP53 durch posttranslationelle Stabilisierung in der betroffenen Zelle. TP53 kann als Transkriptionsfaktor durch die Induktion weiterer Moleküle den Zellzyklus stoppen und die Reparatur der geschädigten DNA ermöglichen. Bei irreparablen DNA-Schäden leitet TP53 über die Aktivierung von Bcl2-Genen eine Signalkaskade ein, die mit Hilfe von Caspasen zum programmierten Zelltod, der Apoptose, führt. So wird der Körper vor der Weitergabe von geschädigter DNA geschützt. Durch Mutationen im Genort auf Chromosom 17 kann es zur Produktion von inaktivem, fehlerhaften TP53 kommen. Die Zelle kann dann trotz geschädigter DNA in den Zellzyklus eintreten und sich vermehren. Dies ist ein kritischer Schritt in der Tumorgenese [91]. Der Wildtyp TP53 hat eine kurze Halbwertszeit und ist nur kurzfristig bei DNA-Schäden in der Zelle nachzuweisen. Im Gegensatz dazu findet man in vielen Tumoren mutiertes 20 TP53, welches aufgrund einer erhöhten Stabilität in der Zelle akkumuliert und daher im Gegensatz zum Wildtyp durch immunhistochemische Färbungen nachgewiesen werden kann [92]. Allerdings korrelierte in einer Studie von Soong et al. bei Endometriumkarzinomen der immunhistologische Nachweis einer TP53-Überexpression nur in circa 76% der Fälle mit einer echten Mutation [93]. Andere Studien wiesen bei circa 10-20% der Endometriumkarzinome eine Mutation des Tumorsuppressorgens TP53 nach, wobei höhere Mutationsraten bei serös-papillären und Klarzelltumoren auftraten [93,94]. Kounelis et al. zeigten in einer großen immunhistochemischen Studie, dass TP53 signifikant häufiger in serös-papillären Endometriumkarzinomen exprimiert war als in endometrioiden Adenokarzinomen. Darüber hinaus konnte die Gruppe nachweisen, dass bei endometrioiden Endometriumkarzinomen eine TP53-Überexpression signifikant assoziiert war mit einem höheren Tumor-Grad und -Stadium [95]. Berchuck et al. beschrieben Veränderungen im TP53-Gen bei etwa 10-15% der Endometriumkarzinome im frühen Stadium und in bis zu 50% der Karzinome im fortgeschrittenen FIGO-Stadium [96]. In immunhistologischen Färbungen wiesen Zheng et al. bei 29% der endometrioiden Adenokarzinome eine Überexpression von TP53 nach, bei serös-papillären Karzinomen lag die Überexpression mit 71% signifikant höher [97]. Auch Sherman et al. und Tashiro et al. fanden eine wesentlich erhöhte Rate von TP53 vor allem in serös-papillären Karzinomen (circa 90% der Fälle) [45,98]. Tashiro et al. wiesen bereits in den als Vorläufer der serös-papillären Karzinome angesehenen, endometrialen intraepithelialen Karzinomen in circa 78% der Fälle eine stark erhöhte Rate von TP53 nach [98]. Somit scheint, anders als bei Typ I Karzinomen, bei serös papillären Karzinomen die TP53Mutation ein früher Schritt in der Karzinogenese zu sein. Erhöhte TP53-Expressionsraten vor allem bei aggressiven histologischen Typen wie dem serös-papillären Endometriumkarzinomen scheinen einen Zusammenhang zwischen einem fortgeschrittenem Tumorstadium und verminderten Überlebensraten herzustellen. Uneinigkeit herrscht allerdings über die Signifikanz von TP53 als unabhängigen Prognosefaktor [7,99]. Mariani et al. sprachen der Überexpression von TP53 und dem Proliferationsmarker MIB-1 die größte Voraussagekraft für das Auftreten eines Rezidivs und einer kurzen Überlebenszeit zu [84]. In einer weiteren Studie wies die Gruppe TP53 als einzigen unabhängigen molekulargenetischen Marker für ein Rezidiv aus [100]. Pisani et al. fanden bei einer Studie Fünf-Jahresüberlebensraten von 12% bei Patientinnen mit erhöhter TP53-Expression im Vergleich zu 90% bei Patientinnen mit Endometriumkarzinomen, die keine TP53-Überexpression aufwiesen [101]. 21 1.11.8. ERBB2 ERBB (v-erb-b2 erythroblastic leukemia viral oncogene homolog 2) lokalisiert auf dem kurzen Arm des Chromosom 17 (17q11.2-q12) ist vor allem als wichtiger Biomarker bei Mammakarzinomen bekannt, der hier die Prognose und Therapie wesentlich mitbestimmt. Im Hinblick auf das Endometriumkarzinom gibt es einige Studien, die sich mit den Expressionsleveln von ERBB2 und deren Auswirkung auf die Prognose und das Überleben beschäftigen [100,101]. Die aktuellste und bisher größte Studie bezüglich der Expression von ERBB2 in Endometriumkarzinomen stellten Morrison et al. 2006 vor. In einer groß angelegten Untersuchung an Karzinomproben von 483 Patientinnen stellte die Gruppe mittels Tissue-Microarray die ERBB2-Expression in Endometriumkarzinomen unterschiedlicher Histologie und unterschiedlichen Tumorgrades und -stadiums dar. Dabei wurde die höchste Rate an ERBB2-Expression und -Amplifikation in serös-papillären Karzinomen gefunden (43% und 29%), während sich die niedrigsten Expressions- und Amplifikationsraten (3% und 1%) in den gut differenzierten endometrioiden Adenokarzinomen darstellen ließen. Es wurde eine erhöhte Expression von ERBB2 mit ansteigendem Tumorgrad nachgewiesen. Morrison et al. zeigten eine signifikante Korrelation zwischen ERBB2-Expression und den etablierten negativen Prognosefaktoren wie Differenzierungsgrad, Tumorstadium, nicht-endometrioider Histologie, positivem Lymphknotenstatus sowie der Invasionstiefe des Myometriums von mehr als 50%. In der Multivariantenanalyse stellte sich die ERBB2-Überexpression als signifikanter Faktor für das krankheitsfreie Überleben heraus. Obwohl sich eine ERBB2-Überexpression wesentlich häufiger in serös-papillären Karzinomen zeigte, war die negative Auswirkung auf die Überlebensraten auch in endometrioiden Adenokarzinomen nachzuweisen. Morisson et al. kamen zu dem Schluss, dass die ERBB2-Überexpression ein wichtiger Faktor in der Gruppe von Endometriumkarzinomen mit hohem Risiko ist, aber aufgrund der sehr viel häufigeren Gruppe von endometrioiden Adenokarzinomen (Grad I) ein generelles Screening auf ERBB2 nicht sinnvoll sei [102]. 1.11.9. Mikrosatelliteninstabilität In den letzten Jahren wurde auch einer Störung im DNA-mismatch-Reparatursystem als Tumorgenesefaktor vermehrt Beachtung geschenkt. Eine Inaktivierung dieses Reparatursystems führt zu einem DNA-Status, der sehr anfällig für Mutationen ist. Replikationsfehler entstehen dabei vor allem in repetetiven Sequenzen, zu denen auch die sogenannten Mikrosatelliten gehören [103]. Als Mikrosatelliten werden kurze, meist nicht kodierende DNA-Abschnitte bezeichnet, die aus einfachen, sich wiederholenden Nukleotidsequenzen bestehen. Mikrosatelliten lassen sich weit verteilt im Genom nachweisen [104]. Man nimmt an, dass die repetitiven 22 Sequenzen gehäuft zu Fehlern in der DNA-Replikation führen. In gesunden Zellen werden solche Replikationsfehler im Allgemeinen schnell durch spezielle DNA- Reparaturmechanismen korrigiert. In Tumorzellen aber führen Replikationsfehler bei Inaktivierung des Reparatursystems oft zu Veränderungen der Mikrosatelliten; dieses Ereignis wird als Mikrosatelliteninstabilität (MSI) bezeichnet. Findet man in Tumorzellen eine Mikrosatelliteninstabilität im Vergleich zu gesunden Zellen, so ist dies ein Hinweis auf Defekte im Mismatch-Repair-System. Defekte in diesem DNA-Reparatur-System können so über spontane Mutationen in Regulatorgenen, wie Tumorsupressorgenen, zur Entstehung von Karzinomen beitragen [105]. Die MSI wird als einer der Initialschritte in der Karzinogenese vor allem von Endometriumkarzinomen des Typs I angesehen [23]. MSI konnte in circa 20-54% der Endometriumkarzinome nachgewiesen werden [106]. Hierbei war die Häufigkeit in Typ I Karzinomen mit 33% der Fälle wesentlich häufiger als in nicht-endometrioiden Karzinomen mit 11% [107]. Bei MSI-positiven Endometriumkarzinomen wurde am häufigsten eine Inaktivierung des Mismatch-RepairGenes MLH1 durch Hypermethylierung der sonst unmethylierten CpG-Inseln in der Promoterregion gefunden. Dieses betrifft ca. 45-64% der sporadischen Endometriumkarzinome. Eine Methylierung des Mismatch-Repair-Genes MLH2 wurde hingegen nicht gefunden [11,106,108]. Neben der endometrioiden Histologie sind MSI positive Endometriumkarzinome typischerweise diploid, weisen keine Tp53-Mutationen auf und zeigen ein niedriges Tumorstadium mit guter Prognose [94]. Eine signifikante Assoziation fand sich zwischen dem Vorliegen einer MSI und Mutationen im PTEN-Gen. Diese häufig bei Endometriumkarzinomen vorliegende Genveränderung wurde bei bis zu 88% der MSI-Tumoren, aber nur zu 33% der mikrosatellit-stabilen Tumoren nachgewiesen [109]. 1.12. Therapie 1.12.1. Operation Die Operation gilt als Methode der Wahl bei der Behandlung des Endometriumkarzinoms. Im Falle eines Karzinoms des Stadium IIIb bis IVb ist eine Operation allerdings nur selten kurativ durchführbar. Trotzdem sollte eine möglichst komplette Resektion des Tumors erfolgen, um die Effizienz adjuvanter Therapien zu erhöhen. Für die Behandlung dieser Patientinnen und bei Vorliegen von Operationskontraindikationen kommt eine primäre oder sekundäre Strahlentherapie in Frage. Die operative Therapie umfasst die Entnahme einer Zytologie aus der Bauchhöhle, die Hysterektomie mit Adnexexstirpation beidseits sowie die pelvine und paraaortale 23 Lymphonodektomie bis zum Nierenstiel. Die Lymphonodektomie wird in den Stadien pT1a und G1/2 als fakultativ angesehen. Das weitere Vorgehen wird durch die Klassifikation des Tumors bestimmt und ist abhängig vom Tumorstadium, dem Grad der Differenzierung und der Histologie. Liegt ein seröses oder klarzelliges Karzinom vor, sollten zusätzlich peritoneale Biopsien entnommen werden, sowie eine Omentektomie ähnlich einer Operation von Ovarialkarzinomen erfolgen [35,51,62]. 1.12.2. Radiotherapie Eine adjuvante Strahlentherapie kann in Form der vaginalen Kontaktbestrahlung (Brachytherapie) und der perkutanen Strahlentherapie erfolgen. Obwohl die primäre Hysterektomie als Therapie der Wahl gilt, ist die alleinige Radiotherapie eine effektive Behandlung für Patientinnen, die aus medizinischer Sicht primär inoperabel sind. Obwohl in unterschiedlichen Studien die Senkung der Lokalrezidivrate durch eine adjuvante Radiotherapie deutlich belegt wurde, gibt es keine eindeutigen Zahlen aus prospektiven Studien, dass das Gesamtüberleben günstig beeinflusst wird. Bei Patientinnen mit hohem Risiko für ein Lokalrezidiv sollte daher eine adjuvante Strahlentherapie durchgeführt werden [17]. 1.12.3. Chemotherapie Die Frage, ob eine adjuvante systemische Chemotherapie, speziell bei Patientinnen mit einem hohen Rezidivrisiko, das Gesamt-Überleben verbessern kann, wird weiterhin kontrovers diskutiert. Studien, die einen Vorteil durch eine Chemotherapie mit Adriamycin und Cisplatin [110] bzw. Cyclophosphamid/Epirubicin/Cisplatin [111] im Vergleich zur Radiatio zeigten, werden aufgrund der Datenanalyse oder der geringen Fallzahl als nicht sicher eingeschätzt [17]. In den aktuellen Leitlinien wird eine Chemotherapie, ggf. in Kombination mit einer Strahlentherapie nur für höhergradige Stadien (T3, T4, oder positive Lymphknoten), sowie für Endometriumkarzinome des Typs II empfohlen [51]. 1.12.4. Hormontherapie Im Gegensatz zur Chemotherapie zeichnet sich die Gabe von Progesteron oder dem Antiöstrogen Tamoxifen durch ihre geringe Toxizität und patientenfreundliche Anwendung aus. Die Effektivität von Progesteron ist abhängig vom Rezeptorstatus und steigt, je höher der Differenzierungsgrad des Karzinoms ist. Die Response-Rate auf Progesteron lag in klinischen Studien bei 15,8% beziehungsweise bei 10% für Tamoxifen [112]. Eine 24 signifikante Erhöhung der Überlebensrate durch adjuvante Hormontherapie konnte in den Studien allerdings nicht gezeigt werden [62]. In den aktuellen Leitlinien wird eine adjuvante Progesteron (Gestagen) -Therapie beim Endometriumkarzinom aufgrund eines nicht ausreichend gesicherten Nutzens im Allgemeinen nicht empfohlen. Bei typischen Vorstufen des Typs I Karzinoms sowie bei frühen Stadien prognostisch günstiger endometrioider Endometriumkarzinome und dem Wunsch der Patientin nach einer fertilitätserhaltenden Therapie bei nicht abgeschlossener Familienplanung ist nach den aktuellen Leitlinien eine Therapie mit hochdosiertem Gestagen indiziert [51]. 25 2. Patienten Konform zu den Leitlinien der zuständigen Ethikkommission wurden Gewebeproben von Patientinnen gesammelt, die in den Jahren von 1997 bis 2003 am Karolinska Hospital aufgrund eines Endometriumkarzinoms operativ behandelt wurden. Bei allen Patientinnen wurde eine Hysterektomie mit beidseitiger Adnexektomie durchgeführt und ein operatives Staging nach FIGO-Kriterien vorgenommen. Keine der Patientinnen hatte präoperativ eine Chemotherapie oder Radiotherapie erhalten. Die histologische Beurteilung inklusive Grading erfolgte im Institut für Onkologie und Pathologie des Karolinska Hospitals. Die Histologie wurde von einem zweiten Pathologen anhand der HE-gefärbten Präparate des jeweiligen Tumors bestätigt. Vom Karzinomgewebe wurde jeweils eine etwa 1 x 1 cm große Probe entnommen und zunächst ein Abklatsch-Präparat für die Bild-Cytometrie gewonnen. Unmittelbar danach wurden die Proben schockgefroren und bis zur weiteren Analyse bei –80 C° aufbewahrt. 2.1. Klinische Daten Für die vorliegende Studie wurden insgesamt 33 Proben analysiert. Die histologische Beurteilung zeigte 25 endometrioide Adenokarzinome sowie 8 serös-papilläre Endometriumkarzinome. Das mittlere Erkrankungsalter der Patientinnen mit einem endometrioiden Adenokarzinom lag bei 66,9 (51 - 87) Jahren. Die Frauen mit einem serös-papillären Endometriumkarzinom waren bei Erstdiagnose im Mittel 80,3 (77 - 90) Jahre alt. Bei den Patientinnen mit einem endometrioiden Adenokarzinom wurde in 17 Fällen die Diagnose eines FIGO-Stadium 1a Tumors gestellt, in sechs Fällen lag ein Stadium 1b und in jeweils einem Fall ein Stadium 2 und 3a vor. Unter den Frauen, die an einem seröspapillären Endometriumkarzinom erkrankt waren, fand sich bei je einer Patientin ein FIGO 1a beziehungsweise 1b Tumor, bei vier Patientinnen lag bereits ein FIGO 3a und bei einer Frau ein FIGO 4b Stadium vor. Bei drei der 25 (12%) Patientinnen mit einem endometrioiden Tumor lagen bei Erstdiagnose Metastasen vor, demhingegen bei sechs von acht (75%) Frauen mit einem serös-papillären Endometriumkarzinom. Das Follow-up der Patientinnen betrug im Durchschnitt 58,5 (4 - 106) Monate (Tabelle 8.2.1). 26 3. Methodik 3.1. Bild-DNA-Cytometrie Der DNA-Gehalt der Tumorproben wurde mittels statischer Bild-Cytometrie bestimmt. Die daraus resultierenden Diagramme der DNA-Verteilung wurden klassifiziert und die Proben der diploiden bzw. aneuploiden Gruppe zugeordnet. Färbung, interne Standardisierung und Kriterien zur Zellauswahl wurden entsprechend der von Auer etablierten Methode durchgeführt [113]. Dazu wurden 8 µm dünne Schnitte des Tumors angefertigt, auf einem Objektträger platziert und nach Feulgen gefärbt. Diese Färbung wurde erstmals 1942 von Feulgen und Rossenbeck beschrieben und nach Auer modifiziert. Zunächst wurden durch eine saure Hydrolyse die Purinbasen der Desoxyribonucleinsäure entfernt und die Aldehydgruppen der DesoxyribopentoseMoleküle freigelegt. Das farblose Schiffs-Reagenz reagiert daraufhin mit den Aldehydgruppen und wird dabei in die farbige Form konvertiert und kovalent an die DNA gebunden. RNA und Proteine reagieren nicht mit dem Schiffs-Reagenz und müssen daher nicht entfernt werden. Im Einzelnen wurden die Schnittpräparate für zweimal zehn Minuten in Xylol deparaffiniert, in einer absteigenden Alkoholreihe rehydriert und in Formalin refixiert. Nach dem Abspülen erfolgte die saure Hydrolyse in 5M HCl für 60 Minuten bei Raumtemperatur. Anschließend wurden die Präparate für zwei Stunden bei Raumtemperatur in abgedunkeltes Schiffs-Reagenz gegeben. Auf die Waschschritte in Sulfidlösung folgten die Dehydratation in ansteigender Alkoholreihe, ein Xylenbad und die Eindeckung mit Entellan (Merck KG, Darmstadt, #1.07961.0100, Brechungsindex 1,4901,500). Die Präparate wurden auf einem Bildanalysegerät ausgewertet (Ahrens System ACAS, Bargteheide/Hamburg, Deutschland). Dieses setzt sich aus einem Zeiss Axioplan Mikroskop (Plan Objektiv 40/0,95), und einer Video-CCD-Kamera zusammen. Es wurden in jedem Fall mindestens 100 Epithelzell- beziehungsweise Tumorzellkerne gemessen. Zellkerne mit unscharfen Begrenzungen oder überlappenden Zellkernen wurden von der Auswertung ausgeschlossen. Als interner Standard wurden für jedes Präparat jeweils circa 20 Granulozyten oder Lymphozyten verwendet. Deren Medianwert wurde als diploider Referenzwert oder “2c-Wert” (2 DNA Kopien) festgelegt. Unter dem Mikroskop mit angeschlossener Kamera und Softwarezugang wurden mindestens 100 Interphasenzellkerne pro Probe aufgenommen und die Intensität der DNA-Färbung bestimmt. Der DNA-Gehalt der Tumorprobe wurde daraufhin in Form von 27 Histogrammen dargestellt. Als Kontrolle dienten nach Feulgen gefärbte normale, diploide Zellen. Alle DNA-Werte wurden in Relation zur korrespondierenden Kontrolle gesetzt, welche den Wert 2c, einem normalen diploiden DNA-Gehalt entsprechend, hatte. Die Proben wurden nach drei Gruppen klassifiziert: (i) diploide Tumoren mit einem einzelnen Peak in der normalen 2c Region, ohne Zellen, die den 5c Wert überschritten, (ii) aneuploide Tumoren, denen mindestens 5% der Zellen die Region 5c überschritten, mit einem Stammlinien-Verteilungs-Index (stemline scatter index; SSI) niedriger oder gleich 8,8, und schließlich die Gruppe (iii) der aneuploiden Tumoren mit einem SSI größer als 8,8. Das neue Ploidie-Klassifizierungssystem ist angelehnt an die Parameter, die von Kronenwett et al. etabliert wurden [114]: Der stemline scatter index ist definiert als Maß der klonalen Heterogenität in Tumorzell-Populationen. Der SSI wird berechnet aus der Summe des Prozentsatzes der Zellen mit DNA-Gehalt in der S-Phasen-Region (SPhase), dem Prozentsatz der Zellen mit einem DNA-Gehalt, der zweimal den Eigenwert plus 1c überschreitet (G2 exceeding rate) und dem Variations-Koeffizienten (coefficient of variation; CV) der zugehörigen Tumor-Stammlinie. 3.2. Immunhistochemie Die TP53-Expression in den Tumorproben wurde am Karolinska-Institute routinemäßig mittels immunhistochemischer Färbung bestimmt. Zur Detektion von akkumuliertem TP53-Protein wurde der murine monoklonale Antikörper DO-1 genutzt. Für detaillierte Protokolle wird an dieser Stelle auf die Arbeit von Lundgren et al. verwiesen [7]. Bei den endometrioiden Endometriumkarzinomen des Typs I zeigte sich weder bei den diploiden noch bei den aneuploiden Karzinomen eine p53-Überexpression in der Immunhistochemie. Dementgegen ließ sich bei den serös-papillären Karzinomen in sieben von acht Proben (87,5 %) eine TP53-Akkumulation nachweisen (Tabelle 8.2.1.). 3.3. Comparative Genomische Hybridisierung Die Technik der Comparativen Genomischen Hybridisierung (CGH) wurde erstmals 1992 von Kallionemi et al. beschrieben [8]. Die CGH ermöglicht die Darstellung von DNAVeränderungen des gesamten Genoms auf chromosomaler Ebene. In einer einzelnen Hybridisierung bietet die Methode einen Überblick über numerische Veränderungen von DNA-Sequenzen, wie Deletionen oder Amplifikationen [115]. Die CGH basiert auf einer zweifarbigen Fluoreszenz-in-situ-Hybridisierung (FISH). Hierbei wird die zu untersuchende DNA (Test-DNA, zum Beispiel aus Tumorgewebe) und DNA 28 eines gesunden Individuums (Kontroll-DNA) mit unterschiedlichen Markermolekülen (Biotin und Digoxiginin) markiert. Diese Markermoleküle werden dann mit Hilfe von gegen sie gerichteten fluoreszierenden Antikörpern lokalisiert. Die Hybridisierung von Test- und Kontroll-DNA erfolgt simultan auf Metaphasen-Chromosomen einer gesunden Kontrollperson und erlaubt somit die Generierung eines Karyogramms. Anhand des Karyogramms können dann numerische Veränderungen der Test-DNA im Vergleich zur Kontroll-DNA dargestellt werden. Die Methode wurde seit ihrer Einführung 1992 kontinuierlich weiterentwickelt und diente bereits der Charakterisierung spezifischer chromosomaler Veränderungen vieler unterschiedlicher Tumorentitäten [9]. In dieser Studie wurde für die CGH-Untersuchungen die Test-DNA aus den Tumorproben isoliert und als quantitative Kontrolle DNA aus Lymphozyten gesunder Spenderinnen gewonnen. Während der „Nick-Translation“ wurden unterschiedliche Haptene in die DNA eingebaut, Biotin-14-dATP in die Tumor-DNA und Digoxigenin-11-dUTP in die KontrollDNA. Die markierte Tumor- und Kontroll-DNA wurde zusammen auf speziell vorbereitete Metaphasenobjektträger hybridisiert. Um zu verhindern, dass fluoreszierende Antikörper unspezifisch an repetitive Sequenzen der perizentromerischen, heterochromatischen Regionen der Metaphasenchromosomen binden, wurden jeder Hybridisierung humane Cot-1-DNA zugesetzt. Würde die Blockierung der repetitiven Sequenzen unterlassen, käme es bei der späteren Auswertung zu großen Schwankungen in den Signalintensitäten und zur Artefaktbildung [115]. Die Metaphasenchromosomen wurden aus Lymphozyten gesunder Spender isoliert und vor der Hybridisierung denaturiert, um eine gute Bindung der Test-DNA zu erreichen. Dabei binden die markierten DNA-Sequenzen exakt an die korrespondierenden Sequenzen der denaturierten Metaphase-Chromosomen. Während der Detektion binden fluoreszierende Antikörper gegen Biotin beziehungsweise Digoxigenin an die auf die Methaphasen hybridisierte DNA. Für die Tumor-DNA wurden grün-fluoreszierende Antikörper (Anti-Biotin) und für die Kontroll-DNA rot-fluoreszierende Antikörper (AntiDigoxigenin) verwendet. Das Verhältnis der beiden Fluoreszensstoffe zueinander entlang der Chromosomen kann im Fluoreszensmikroskop sichtbar gemacht werden. Die Unterschiede in den Signalintensitäten der beiden Fluoreszensstoffe entlang der Metaphasen-Chromosomen reflektieren das Verhältnis von gebundener Tumor- und Kontroll-DNA zueinander. Konkurriert die gleiche Menge von Tumor- und Kontroll-DNA um die DNA-Sequenz des Metaphasenchromosoms, findet eine ausgeglichene Bindung von grün- (Tumor) und rot- (Kontroll) fluoreszierender DNA statt und die Mischfarbe erscheint gelb. Bei einem Zugewinn (gain) von DNA-Sequenzen im Tumor-Genom kommt es zur vermehrten Bindung von grün-fluoreszierender DNA und der betroffene Chromosomenabschnitt stellt sich grün dar. Ebenso kommt es bei einem relativen Verlust 29 (loss) von DNA-Sequenzen des Tumor-Genoms zu einer übermäßigen Bindung der rotfluoreszierenden Kontroll-DNA, und die entsprechende Chromosomenregion erscheint rot. Die mit der CCD-Kamera aufgenommenen Metaphasenbilder werden mit einem speziellen Computerprogramm analysiert. Die unterschiedlichen Signalintensitäten entlang der Chromosomen werden berechnet und graphisch dargestellt. Auch für das menschliche Auge primär nicht sichtbare Signalunterschiede der beiden Fluoreszensfarbstoffe zueinander können so ermittelt werden. Dadurch werden quantitative Unterschiede des Verhältnisses von Tumor- zur Kontroll-DNA für jede Region des Chromosoms detektiert. Verluste oder Gewinne von DNA-Sequenzen und auch ganze Trisomien können so ermittelt und quantifiziert werden. Profile der Signalunterschiede werden für jedes Chromosom erstellt. Durch die Analyse von fünf bis zehn Metaphasen derselben Probe und Mittelwertbildung können Artefakte klein gehalten werden. Es resultiert ein Karyogramm des analysierten Tumor-Genoms mit genauer Zuordnung numerischer Chromosomenaberrationen zu den betroffenen Chromosomenabschnitten. Die CGH hat sich als besonders hilfreich beim Nachweis chromosomaler Veränderungen von soliden Tumoren erwiesen. Eine relativ geringe Menge an reiner DNA (circa 0,5-2 µg) reicht bereits aus, um ein erfolgreiches CGH-Experiment durchzuführen. In einer einzelnen Hybridisierung zeigt die CGH die oft sehr komplexen genomischen Veränderungen von soliden Tumoren und ordnet sie spezifischen Chromosomenabschnitten zu. Sie lenkt das Interesse auf veränderte Regionen mit potentiellen Tumorsupressorgenen, beziehungsweise Onkogenen und bietet die Grundlage für weiterführende Untersuchungen. Ein weiterer Vorteil der CGH ist ihre Anwendbarkeit sowohl für frische, tiefgefrorene als auch in Paraffin gelagerte Proben. Die Darstellung genomischer Veränderungen beschränkt sich auf die Veränderung in der Anzahl von DNA-Sequenz-Kopien. Mutationen, die mit einer ausgeglichenen Anzahl von DNA-Kopien einhergehen, balancierte Translokationen, Inversionen und Punktmutationen, lassen sich durch die CGH nicht detektieren. Tumore, die aus sehr heterogenen Anteilen bestehen, können zu verfälschten CGH Ergebnissen führen, indem sich DNA-Verluste und -Gewinne in der Hybridisierungsreaktion ausgleichen und somit zu keiner Signalveränderung führen. Um Veränderungen in der DNA-Kopienzahl zuverlässig erkennen zu können, sollte der Anteil der reinen Tumorzellen an der Tumorprobe mindestens 70% betragen. Anderenfalls heben sich die Signalunterschiede zwischen Tumor- und Kontroll-DNA nicht eindeutig genug voneinander ab [115]. Der Tumorzellgehalt der Probe sollte daher vor Beginn der CGH histologisch bestätigt werden. Limitierend für die Zuverlässigkeit der CGH ist ihr Auflösungsvermögen. So lassen sich quantitative Veränderungen in der Signalintensität nur feststellen, wenn der veränderte 30 Chromosomenabschnitt zumindest zwei Megabasenpaare (Mbp) umfasst [115]. Ausgenommen sind Amplifikationen, die auch bei wesentlich kleineren Sequenzen (circa 50 kb) quantitativ nachweisbar sind [116]. Die Beurteilung der Regionen 1pter, 16, 19 und 22 bedarf einer vorsichtigen Interpretation, da diese Chromosomenbereiche aufgrund eines hohen Anteils repetitiver Sequenzen anfällig für Artefakte sind [115]. 3.3.1. DNA-Extraktion aus Blut Als Kontroll-DNA wurde mit Phenol aus Vollblut extrahierte DNA einer gesunden Spenderin eingesetzt. Dazu wurden 10 ml heparinisiertes Vollblut mit Lyse-Puffer versetzt, vorsichtig vermischt, auf Eis inkubiert und danach zentrifugiert. Der Überstand wurde verworfen und das Pellet in Lyse-Puffer unter leichtem Schütteln gelöst und erneut zentrifugiert. Das Pellet wurde zweimal mit SE-Puffer gewaschen und danach zur Entfernung der Proteine mit 40 µl Proteinase K und 20% SDS vermengt. Die Reaktion verlief über Nacht bei 37°C im Wasserbad. Nach Zugabe von Phenol wurde die Lösung 10 Minuten kräftig geschüttelt und anschließend zentrifugiert. Der Überstand mit der DNA wurde in ein neues Gefäß überführt und mit einer Mischung aus Phenol und Chloroform/Isoamylalkohol versetzt. Daraufhin wurde die Lösung 10 Minuten kräftig geschüttelt und erneut zentrifugiert. Der Überstand wurde in ein neues Gefäß transferiert und erneut mit Chloroform/Isoamylalkohol kräftig gemischt. Die DNA Präzipitation erfolgte mit 3M Sodiumacetat (pH 5,2), sowie Isopropanol und eisgekühltem Ethanol. Die DNA konnte dann vorsichtig mit einer Pipettenspitze in ein neues Gefäß überführt werden. Daraufhin wurde die DNA in 70% Ethanol gewaschen und dann in einem Eppendorfgefäß mit Ethanol bei 4°C und 14.000 rpm über Nacht zentrifugiert. Anschließend wurde das Pellet im Speed-Vac getrocknet, bevor die DNA in 0,5-1 ml sterilem Wasser über Nacht resuspendiert wurde. 3.3.2. DNA und RNA Extraktion Die bei –80 °C gelagerten Tumorproben wurden in TRIzol Reagenz (Invitrogen life technologies, Carlsbad, Canada) aufgetaut und mit Hilfe eines Mixers mechanisch zerkleinert. Durch Zugabe von Chloroform ergab sich nach circa 15 Minuten bei Raumtemperatur eine Phasentrennung, die durch Zentrifugation bei 8.000 rpm über 15 Minuten forciert wurde. Die obere klare Phase enthielt die RNA und wurde zur zusätzlichen Aufreinigung mit dem RNeasy Kit (Qiagen, Valencia, Canada) in ein neues Gefäß überführt. Die untere Phase mit DNA und Proteinen wurde für die DNA- und Protein-Extraktion aufbewahrt. 31 Während das Gefäß auf dem Vortex geschüttelt wurde, setzte man der RNA-Phase tropfenweise Ethanol zu. Die Flüssigkeit wurde dann in ein Gefäß mit Filtersäule überführt (RNeasy Kit). Durch mehrfaches kurzes Zentrifugieren bei 5.500 rpm wurden die RNASequenzen, welche länger als 200 Basenpaare lang waren, an die Membran der Säule gebunden. Die RNA wurde in zwei Zentrifugationsschritten mit Pufferlösungen des Rneasy Kits gereinigt. Die RNA-tragende Säule wurde nun in ein neues Röhrchen überführt. Durch viermalige Zugabe DEPC-behandelten Wassers und jeweils Zentrifugation bei 5.000 rpm wurde die RNA aus der Membran gelöst und gesammelt. Die RNA wurde dann durch Hinzufügen von 3 M Natrium-Acetat und 100% Ethanol bei 80°C prezipitiert. Durch 30-minütige Zentrifugation bei 8.000 rpm bildete sich dann ein RNA-Pellet, dass in 75% Ethanol gewaschen und erneut zentrifugiert wurde. Nachdem der Überstand vorsichtig entfernt worden war, wurde die RNA in RNAse freier Umgebung getrocknet und mit DEPC behandeltem Wasser resuspendiert. Die RNA wurde bei –80°C bis zur weiteren Verwendung gelagert. Bei der nun folgenden DNA-Extraktion war darauf zu achten, dass der RNA-enthaltende klare Überstand komplett entfernt war. Der Flüssigkeit wurde Ethanol beigefügt und das Gefäß nach kräftigem Schütteln zentrifugiert, bevor der Proteinüberstand entfernt werden konnte. Das DNA-Pellet wurde in 0,1 M Sodium-Citrat und Ethanol gewaschen. Nach erneutem Zentrifugieren wurde der Überstand verworfen. Ein weiterer Waschschritt mit 0,1 M Sodium-Citrat schloss sich an. Das DNA-Pellet konnte jetzt getrocknet werden, bevor es mit sterilem Wasser bei 37°C über Nacht resuspendiert wurde. Nach kurzer Zentrifugation wurde die DNA vorsichtig abpipettiert und konnte bei –30°C gelagert werden. 3.3.3. Metaphasen-Objekttäger Die Metaphasen-Objektträger wurden aus mitotischen Zellen aus Kurzzeit-Blutkulturen erstellt. Hierzu wurde zunächst 3-5 ml Vollblut von einer gesunden Spenderin gewonnen. Als Antikoagulanz wurde Sodiumheparin verwendet. In einer Zellkulturflasche wurden daraufhin 0,7 ml des heparinisierten Blutes mit Kulturmedium (RPMI 1640) gemischt und für 72 h bei 37°C in einem Zellkulturschrank inkubiert. Daraufhin wurden die Zellen mit Colcemid, einem Spindelgift, behandelt und inkubiert. Nach Zentrifugation konnte der Überstand verworfen werden, bis auf einen kleinen Rest, in dem das Pellet durch vorsichtiges Schütteln des Gefäßes wieder vom Boden gelöst wurde. Die Zellen wurden durch vorsichtiges Bewegen unter tropfenweiser Zugabe von vorgewärmten 0,0075 M KCl resuspendiert und im Wasserbad inkubiert. Durch die Beigabe von drei bis fünf Tropfen einer frischen Fixationslösung, bestehend aus Methanol und Acetatsäure im Verhältnis 32 3:1, wurde die Reaktion gestoppt und die Lösung erneut zentrifugiert. Der Überstand wurde bis auf einen kleinen Rest verworfen, mit dem das Pellet vorsichtig gelöst wurde. Die Zellen konnten nun nach Zugabe von Fixationslösung durch Auf-und Abpipettieren resuspendiert werden. Dann wurden innerhalb eines 37°C warmen Inkubators durch vorsichtiges Tropfen der Zelllösung auf Objektträger die Methaphasen-Objektträger hergestellt. Unter dem Mikroskop ließ sich die Qualität der Methaphasen beurteilen, bevor diese bis zur weiteren Verwendung kühl und trocken gelagert wurden. 3.3.4. Nick-Translation Die Nick-Translation ist eine Methode zur nicht-radioaktiven Markierung von DNA. Mit Hilfe der Enzyme Desoxyribonuklease I (Dnase I) und Escherichia coli DNA-Polymerase I werden die DNA-Stränge gekürzt und Lücken („nicks“) in die Stränge hydrolisiert. Mit Biotin (Tumor-DNA) und Digoxin (Kontroll-DNA) markierte DNA-Nucleotide werden durch die DNA Polymerase in die entstandenen Lücken eingebaut. Es entsteht mit Biotin markierte Tumor-DNA und mit Digoxin markierte Kontroll-DNA. Nach der Inkubation mit dem Enzym Dnase sollten die DNA-Moleküle idealerweise in einer Länge von 300-900 Basenpaaren vorliegen, um eine gute Hybridisierung auf die Metaphasen zu erlangen. Für jede Hybridisierung wurde eine Nick-Probe von 100 µl erstellt. Dazu wurden jeweils 2 µl DNA, 10 µl NT Puffer, 10 µl dNTP (enthält verschiedene DNA Nucleotide) und 10 µl ßMercaptoethanol gemischt. Bei der Tumor-DNA wurden 4 µl Biotin-16-dUTP und bei der Kontroll-DNA 4 µl Digoxin-11-dUTP hinzugefügt. Die Ansätze wurden mit destilliertem Wasser auf 100 µl aufgefüllt und kurz gemixt. Während die Proben auf Eis lagerten, wurden die Enzyme DNA-Polymerase und eine stark verdünnte Dnase-Mischung ergänzt. Die Reaktion lief bei 15°C im Wasserbad über 1,5 Stunden. Zur Feststellung der Länge der DNA-Fragmente wurden jeweils 5 µl der Probe auf einem Agarosegel zusammen mit dem Marker HAES III als Längenmaßstab aufgetragen. Die Proben liefen bei 100 Volt für etwa 30 Minuten. Das Gel wurde anschließend mit Ethidiumbromid gefärbt. Die Länge der DNA-Moleküle konnte unter UV-Licht beurteilt und fotografiert werden. Zu lange DNA-Proben wurden erneut mit DNase inkubiert. Die NickProben mit einer Moleküllänge von 300-900 Basenpaaren wurden mit EDTA versetzt und inkubiert, um die Dnase zu inaktivieren und die Reaktion zu stoppen. Die Nick-Proben wurden bei -20°C gelagert. 33 3.3.5. DNA-Präzipitation Vor der Hybridisierung der DNA auf die Metaphasenchromosomen wurden ca. 50-100 µl biotinmarkierte Tumor-DNA mit der gleichen Menge digoxinmarkierter Kontroll-DNA in einem Eppendorfgefäß vermischt. Zur Unterdrückung der DNA-Bindung an repetitive Sequenzen auf den Chromosomen wurde humane Cot 1 DNA hinzugesetzt. Nach Beigabe von Salmon Sperm wurden die Proben mit Natrium-Acetat und eisgekühltem Ethanol bei –80°C präzipitiert. Nach der Zentrifugation für 30 Minuten wurde der Überstand verworfen und die DNA im Speed-Vac getrocknet. Das DNA-Pellet wurde dann in deionisiertem Formamid gelöst und bei 37°C inkubiert. Danach wurde Master Mix hinzupipettiert und die DNA bei 80°C für fünf Minuten denaturiert, bevor die Probe bei 37°C zur Hybridisierung vorbereitet wurde. 3.3.6. Vorbehandlung der Objektträger Die Metaphasen-Objektträger wurden durch verschiedene Reinigungsschritte auf die nachfolgende Hybridisierung vorbereitet. Nach dem Waschen in SSC wurden die Objektträger mit einer Rnase enthaltenden Lösung für 45 Minuten bei 37°C inkubiert, um RNA-Reste zu entfernen und die Hybridisierung zu optimieren. Daraufhin wurden die Objektträger mehrfach in SSC gewaschen, bevor sie für 1,5 Minuten bei 37°C in einer Lösung aus Pepsin und 0,01 M HCl von Zytoplasmarückständen befreit wurden. Die Objektträger wurden nacheinander in folgenden Lösungen gewaschen: PBS, PBS/1MMgCl2 und 1%Formaldehyd/1XPBS/1 M MgCl2. Zum Abschluss wurden die Objektträger mit kaltem Ethanol ansteigender Konzentration dehydriert, bevor sie an der Luft trockneten. 3.3.7. Hybridisierung Die Objektträger wurden mit Formamid/SSC bedeckt. Danach wurden die Chromosomen für 1,5 Minuten bei 75°C denaturiert, um die DNA auf die Aufnahme der Nick-Probe vorzubereiten. Nach der Denaturierung wurden die Objektträger sofort in einer Ethanolreihe ansteigender Konzentration abgekühlt und dehydriert. Auf die getrockneten Objektträger konnte anschließend die DNA-Probe aufgetragen werden. Zum Schutz vor dem Austrocknen wurde die DNA mit einem Deckgläschen bedeckt und die Ränder mit Klebstoff abgedichtet. Die Hybridisierung erfolgte in einer Feuchtkammer bei 37°C für 48 Stunden. 34 3.3.8. Detektion Zur Detektion wurden spezifische fluoreszierende Antikörper an die Biotin- beziehungsweise Digoxin-markierte Tumor- und Kontroll-DNA gebunden. Dabei wurde das Fluochrom FITC (Emission von grünem Licht) an die Tumor-DNA und das Fluochrom TRITC (Emission von rotem Licht) an die Kontroll-DNA gebunden. Nach Anregung der Fluochrome mit kurzwelligem Licht kann dessen Intensität unter dem Mikroskop bestimmt werden, welche proportional zum Anteil der gebunden DNA ist. Zunächst mussten die auf den Objektträgern befindlichen Deckgläschen vorsichtig entfernt werden. Die Objektträger wurden daraufhin mehrfach in FA/SSC und 0,1xSSX gewaschen, bevor sie in 4xSSC/Tween20 getaucht wurden. Alle Waschvorgänge fanden bei 60°C statt. Durch die Waschungen wurden überschüssige, nichtgebundene Anteile der Nick-Proben entfernt, um die Reinheit der Metaphasen zu erhöhen. Um unspezifische Bindungen der Antikörper zu vermeiden, wurde daraufhin BSA-haltige Blockierungs-Lösung auf die Objektträger aufgetragen und diese bei 37°C in einer Feuchtkammer inkubiert. Danach wurden die Objektträger in 4xSSC/Tween20 getaucht, bevor die fluoreszierenden Antikörper nacheinander in vier Schritten mit jeweils einer Stunde Inkubationszeit bei 37°C in einer Feuchtkammer aufgetragen wurden. Nach jeder Inkubationsphase wurden die Slides erneut mit 4xSSC/Tween20 gewaschen, um überschüssige Fluoreszenzstoffe zu entfernen. Nach dem letzten Waschschritt wurden die auf dem Objektträger befindlichen Metaphasen mit 4,6-Diamino-2-phenylindol (DAPI) gefärbt, um die Auswertung der Karyogramme zu ermöglichen. Mit 2xSSC wurde überschüssiger Farbstoff entfernt, bevor zum Schutz vor dem Ausbleichen der Fluoreszenzstoffe Antifade (1,4-phenylene-diamine) aufgetragen wurde. Die Objektträger wurden mit einem Deckgläschen versehen und bei 4°C unter Lichtausschluss gelagert. 3.3.9. Aufnahme mittels CCD-Camera Zur Erstellung digitaler Bilder der hybridisierten Metaphasen wurde ein FluoreszenzMikroskop mit CCD-Camera genutzt. Zunächst wurde die Qualität der Hybridisierung unter dem 63x Öl-Immersions Objektiv beurteilt. Dabei wurde auf eine gleichmäßige Hybridisierung in allen drei Farbkanälen (DAPI, FITC und TRITC) Wert gelegt, sowie auf eine geringe Hintergrundfärbung und auf gleichmäßige, vollständige Metaphasen mit möglichst wenigen sich überlappenden Chromosomen. Es wurden mindestens zehn geeignete Metaphasen fotografiert. Mit Hilfe verschiedener Filter wurde jeweils ein Bild der DAPI Färbung, der grün fluoreszierenden Tumor-DNA und der rot fluoreszierenden Referenz-DNA aufgenommen. Durch individuelle Variation der Expositionszeiten konnte für jede Farbe und Hybridisierung ein Bild mit optimaler Qualität 35 für die nachfolgende computergestützte Auswertung aufgenommen werden. Die drei Bilder konnten so ausgerichtet werden, dass ein Einzelbild (Komposit) mit sich exakt überlagernden Chromosomen in den drei Farben entstand. Schon unter dem Mikroskop konnte man einen groben Überblick über Verluste und Gewinne an Tumor-DNA in der Probe erlangen. Im Komposit-Bild erschienen Verluste an DNA rot (Kontroll-DNA überwiegt) und Zugewinne in grün (Tumor-DNA überwiegt). Chromosomenabschnitte mit ausgeglichenen DNA-Anteilen erschienen durch ein ausgewogenes Verhältnis an grünem und rotem Farbstoff gelb. Die Metaphasen-Bilder wurden gespeichert, um sie später mittels softwaregestützt zu analysieren. 3.3.10. Metaphasen-Auswertung Um die CGH-Bilder mittels spezieller Software auswerten zu können, wurden zunächst die Tif-Dateien im Programm Leica CW 4000 Karyo V 1.0 (Leica Imaging Systems, Cambridge, UK) geöffnet. Anhand unterschiedlicher Parameter wie der Granularität oder der Homogenität der Metaphase konnte die Qualität der Aufnahme beurteilt werden. Im DAPI-Bild konnten dann die von der Software festgelegte Mittellinie der Chromosomen oder auch die markierten Zentromerregionen - wenn nötig - korrigiert werden, um Artefakte in der Auswertung zu vermeiden. Mit unterschiedlichen Werkzeugen der Software konnten die Chromosomen aufgerichtet und unvollständig gefärbte oder sich überlappende Chromosomen eliminiert werden. Anhand der DAPI-Aufnahme wurde das Karyogramm manuell erstellt. Nach mindestens zehn in dieser Weise analysierten Metaphasen wurde von der Software ein Gesamtprofil über die DNA-Verluste und Gewinne der Tumorprobe im Verhältnis zur Kontrolle entworfen. Dabei stellten sich Verluste als Abweichung der Kurve vom Mittelwert nach links und Gewinne als Abweichung der Kurve nach rechts dar. Zusätzlich wurden durch das Programm die Veränderungen im DNA-Gehalt als roter (loss) beziehungsweise grüner (gain) Balken an entsprechender Position des betroffenen Chromosoms abgebildet. Eine Amplifikation, dass heißt ein höhergradiger Zugewinn von bestimmten DNA-Sequenzen (gain), stellte sich durch ein entsprechend starkes Ausschlagen der Kurve und einen breiten Balken entlang der korrespondierenden Stelle des Chromosoms dar. Durch eine Gruppierung der Fälle in die zu untersuchenden Subgruppen EnD, EnA und UPSC-A ließen sich spezifische Gruppenprofile erstellen und charakteristische Muster numerischer Chromosomenveränderungen der Gruppen analysieren. 36 3.4. cDNA-Microarray Schena et al. veröffentlichten bereits 1995 die erste cDNA-Microarray Studie [117]. Mit der Entwicklung der Microarray-Technik steht eine Methode zur Verfügung, die es ermöglicht, in einer einzelnen Hybridisierung das Expressionsmuster von mehreren tausend Genen zu untersuchen. Hierzu ist nur eine geringe Menge von bis zu 20 µg reiner RNA nötig. Mit dieser Methode ergibt sich die Möglichkeit, genetische „Fingerabdrücke“ unterschiedlicher Gewebsproben zu identifizieren [118]. Die Technik basiert auf der quantitativen Zwei-Farben-Fluoreszenz-in-situ-Hybridisierung, die das Prinzip der spezifischen DNA-Basenpaarung nutzt (Adenosin bindet an Thymin und Guanin an Cytosin) [119]. Die Methodik der Arrays beruht auf der Bindung der zu analysierenden freien Nukleinsäuremoleküle („target“) an die Sonden („probe“), die als Nukleinsäuremoleküle auf einer festen Oberfläche immobilisiert wurden. Als Sonden dienen im Allgemeinen durch PCR gewonnene cDNA-Moleküle oder Oligonukleotide aus Datenbanken. Die Sonden werden als Spots hoher Moleküldichte und in großer Anzahl maschinell auf bestimmte Trägerstoffe aufgebracht, circa 100.000 Oligonukleotide beziehungsweise 10.000 PCR-Produkte pro cm². Durch die Auswahl der Sonden kann der Array einer spezifischen Fragestellung angepasst werden. Als Trägermaterial zeichnet sich Glas gegenüber anderen Trägerstoffen durch seine Stabilität, die glatte Oberfläche und geringe Hintergrundanfärbung als besonders geeignet aus [120,121]. Eine direkte Bindung der zu analysierenden RNA an die Sonden ist nicht möglich. Daher wird die Information der RNA durch das Enzym Reverse-Transkriptase unter Bereitstellung von freien Nukleinsäurebasen zunächst in cDNA-Einzelstränge umgeschrieben. Durch Nukleinsäurebasen wird vorherige Kopplung gleichzeitig die von Aminoallyl-Resten Bindungsstelle für die an die spätere Fluoreszinmarkierung in die cDNA-Einzelstränge eingebracht. Um am Ende der Reaktion eine quantitative Aussage über die in der Gewebsprobe vorhandene RNA treffen zu können, wird auch eine entsprechende Menge an Kontroll-RNA in cDNA umgeschrieben. Die Kontroll-RNA kann kommerziell erworben oder selbst extrahiert werden, zum Beispiel aus gesundem Gewebe. Die Referenz-RNA sollte in adäquater Menge die Gene vertreten, die auch als Sonden auf dem Array zu finden sind. Die Verwendung von kommerziell erwerblicher RNA erhöht die Vergleichbarkeit verschiedener Studien [122]. Die Markierung der Proben- und Kontroll-cDNA mit Fluoreszin gilt als die Methode der Wahl [123], aber auch eine radioaktive Darstellung ist möglich. Durch die Markierung mit zwei unterschiedlichen Farbstoffen für die Kontroll- und Tumor-cDNA kann eine Aussage über das Expressionsniveau der RNA getroffen werden [120]. Während der Inkubation in einer Feuchtkammer findet nach dem Prinzip der Basenpaarung eine kompetitive Hybridisierung der Target-cDNA an die Sonden auf dem Arrayslide statt. Unter diesen 37 Bedingungen ist die Menge der markierten cDNA, die an den Arrayspot bindet, proportional zu der ursprünglichen Menge der mRNA im Ausgangsmaterial [124] (siehe Abbildung 1). TumorDNA DNA Klone ReferenzDNA Laser 1 Laser 2 Reverse Transkriptase Reaktion Markierung mit Fluochromen Emission Amplifikation und Reinigung Druck auf Glas-Träger Computer Analyse Hybridisierung auf die Arrays Abbildung 1. cDNA-Microarray-Schema modifiziert nach Duggan et al. [125]. 3.4.1. Herstellung der cDNA-Microarrays Für die hier vorliegende Studie wurde die Methode der indirekten Markierung der cDNA mit amino-allyl-Resten, an die Fluorchrome gebunden werden können, angewandt. Die RNA wurde mit TRIzol (Invitrogen) extrahiert und mit Hilfe des Qiagen-RneasySäulen-Systems (Qiagen, Valencia, CA) gereinigt, um ein bestmögliches Hybridisierungsergebnis zu erreichen. Für die Hybridisierung wurden alle Arrays von einem Herstellungsvorgang (Printset) der NCI Array Core Facility verwendet. Diese Arrays enthalten 9.128 Spots mittels PCR amplifizierter cDNA, die auf einem Glasobjektträger mit poly-L-lysine-Oberfläche immobilisiert wurden. So umfasst ein Array die genetische Information von etwa 1/3 des menschlichen Genoms. Die Liste der hinterlegten Gene (Gene annotation file =GAL file; Hs-UniGEM2-v2px-32Bx18Cx18R.gal) ist auf der CoreFacility Website abrufbar (http://nciarray.nci.nih.gov). Zunächst wurde mit Hilfe des Enzyms Reverse-Transkriptase aus der vorliegenden Tumor- und Kontroll-RNA die jeweilige cDNA synthetisiert. Dabei wurden der Reversen 38 Transkriptase DNA-Bausteine zur Verfügung gestellt, an die ein amino-allyl-Rest gekoppelt war. Es wurden jeweils 20 µg der Tumor RNA und der Universal Human Reference RNA (Stratagene, La Jolla, Canada) verwendet. Beide Proben wurden mit einem Randome Hexamer Primer bei 70°C inkubiert. Nach dem Abkühlen auf ca. 42°C wurden der Reaktion eine Mischung aus einem Puffer, DTT, dem amino-allyl-dNTP-Mix und das Enzym Single-strang-II-Reverse-Transkriptase zugefügt. Die Reaktion erfolgte über Nacht bei 42°C. Die Reverse Transkriptase Reaktion wurde durch Zugabe von EDTA gestoppt. Zur Hydrolyse der cDNA wurde 1 M NaOH ergänzt und die Reaktion auf 65,5 °C erhitzt. Zur Neutralisierung wurde dann die gleiche Menge an 1 M HCl hinzugegeben. Daraufhin wurde die cDNA mit Hilfe von Quiagen PCR Zentrifugationssäulen gereinigt. Die cDNA wurde mit PB-Puffer an die Säulenmatrix gebunden und anschließend in mehrfachen Waschschritten mit Phosphat-Puffer gesäubert. Danach wurde die cDNA mit KPO4 aus der Säulenmatrix gelöst und im Speed-Vac getrocknet. Das Pellet wurde in CarbonatPuffer resuspendiert, die Tumor-Probe mit dem Fluoreszenz Cy5 und die Kontroll-cDNA mit Cy3 sorgfältig gemischt. Die Fluoreszenz-Kopplung an die amino-allyl-Reste der cDNA erfolgte während einer einstündigen Inkubationszeit unter Lichtausschluss. Danach wurde der Reaktion Natrium-Acetat zugesetzt und die cDNA wurde mit PB Puffer an die Säulenmatrix des QIAquick PCR Systems (Qiagen, Valencia, CA) gebunden. Durch Einsatz des PE-Puffers wurde die cDNA von überschüssigen Fluoreszenzstoffen gereinigt und anschließend mit einem Lösungs-Puffer aus der Säule in ein neues Gefäß ausgewaschen. Zur Überprüfung der Fluoreszenz-Inkorporation mittels Spectrophotometer (Nanodrop, Thermo Scientific, Wilmington, USA) wurde jeder Probe 1 µl entnommen, der Rest wurde im Speed-Vac getrocknet. Die Proben wurden in einem Hybridisierungs-Puffer resuspendiert und jeweils eine Cy5-markierte Tumor-Probe mit einer Cy3 markierten Kontroll-Probe sorgfältig vermischt. Dem Gemisch wurden Cot1DNA und PolyA-DNA hinzugesetzt, um die Bindung an repetitive Sequenzen auf den Arrays zu unterbinden. Die Proben wurden bei 95°C denaturiert und mit Hilfe eines Deckglases auf die vorbehandelten Array-Slides aufgetragen. Um eine gute Hybridisierung der cDNA auf die DNA-Spots der Array-Slides zu erreichen und die Hintergrundanfärbung zu minimieren, wurden die Slides vorhybridisiert. Dazu wurde ein Prehybridisierungspuffer mit bovinem Serumalbumin (BSA) genutzt. Die Hybridisierung lief in einer Feuchtkammer (Arraylt™ Hybridization Cassette, TeleChem Intl., Sunnyvale, CA, USA) im 42°C Wasserbad für 16-20 Stunden. Danach wurden die Arrays vorsichtig aus der Kammer entnommen und bei 42°C in verschiedenen 39 Waschlösungen gewaschen, um überschüssige, nicht gebundene cDNA zu entfernen. Der Waschvorgang führte zu einer deutlichen Reduktion der Hintergrundfärbung. 3.4.2. Scannen der Arrays Um eine computergestützte Auswertung der Arrays vorzunehmen, wurden die Slides mit einem Laserscanner gescannt. Verwendet wurde ein Axon Gene Pix 4000 Scanner (Axon Instruments, Union City, CA) mit dem Aufnahme- und Analyse-Programm Gene Pix Pro Version 4.0.1.17. Durch den Laserstrahl werden die an die Array-Spots gebundenen Fluroeszine Cy5 und Cy3 zur Emission von Strahlen angeregt. Die emittierten Strahlen werden registriert und auf dem Computerbildschirm in Form von Spots unterschiedlicher Intensität und Farbe abgebildet. Die Cy3 markierte Tumor-cDNA erscheint grün, die Cy5 markierte KontrollcDNA rot. Der Scanvorgang wurde mit einer Vorschau geringerer Auflösung begonnen, um die genauen Abmaße der Arrays zu bestimmen und die PMT-Werte anzupassen. Der Laser verwendet rotes und grünes Helium-Neon bei 633 nm für Cy5 (rot) und 543 nm für Cy3 (grün). Beide Kanäle wurden gescannt und für die spätere Auswertung gespeichert. Während des Scanvorgangs lassen sich Helligkeit, Kontrast und PMT-Werte überwachen und gegebenenfalls anpassen. Ziel war es, eine maximale Intensität der Spots bei minimaler Sättigung (weiße Spots) zu erreichen. Werden die gescannten Bilder der Kanäle Cy5 und Cy3 übereinander projiziert, so ergibt sich ein Kombinationsbild. Ein gelber Spot zeigt eine gleichmäßige Bindung der Tumor- und Kontroll-cDNA an die cDNA auf dem Array. Ein grüner Spot verdeutlicht ein Überwiegen der Kontroll-cDNA, ein roter Spot ein Überwiegen der Tumor-cDNA. Weiße Spots erscheinen bei Übersättigung mit Fluochromen, lassen sich nicht in die Auswertung einbeziehen und sollten daher in ihrer Anzahl gering gehalten werden (kleiner 1%). Nicht oder wenig intensiv gefärbte Spots lassen sich durch eine fehlende Hybridisierung sowohl der Tumor- als auch der KontrollcDNA erklären. Die Information der aufgedruckten cDNA-Spots liegt in diesem Fall weder im Genom des Tumors noch der Kontrolle vor. 3.4.3. Auswertung mit Gene Pix Pro Die Auswertung der cDNA-Microarrays erfolgte mit dem Computerprogramm Gene Pix Pro Version 4.0.1.17 von Axon Instruments. Mit diesem Programm können die Intensitäten der Cy3- und Cy5-Signale der gescannten Arrays gemessen werden. Es werden Signal- bzw. Expression-Ratios für jeden einzelnen Spot errechnet. Dazu wurden die nach dem Scanvorgang gespeicherten Dateien in Gene Pix Pro geöffnet und die zu 40 jedem Array korrespondierende Detektionsmaske aus der Gene Array Liste geladen (gal.file; http://nciarray.nci.nih.gov) (siehe Abbildung 2). Abbildung 2. Griding-Progress von Array Nr. 12 mit Gene Pix Pro. Der Annotation-File trägt die genaue Information, welche cDNA an welchem Spot zur Hybridisierung aufgetragen wurde. Somit dient der Annotation-File der Zuordnung der hybridisierten Spots zu den korrespondierenden cDNA-Klonen. Dieses ist eine wesentliche Information zur späteren Berechnung von RNA-Über- und -Unterexpression spezifischer Gene. Nachdem die Detektionsmaske angepasst ist, markiert das Programm selbstständig schlecht hybridisierte Spots, Artefakte, die zwar angefärbt, aber auf der ursprünglichen Array nicht aufgedruckt waren und Spots, die nicht gefunden wurden. Diese Markierungen müssen sorgfältig kontrolliert und gegebenenfalls korrigiert werden, um die Auswertung der Arrays zu optimieren [126]. Nach der manuellen Nachbearbeitung der Arrays wurden die Intensitätsratios berechnet und eine Ergebnisstabelle erstellt; diese wurde als Datei gespeichert. 41 3.4.4. Microarray-Qualitäts-Bewertung und Datenanalyse (Zwei-Gruppen-Klassen- Vergleich): Arrays, die unseren visuellen Qualitätskriterien aufgrund schlechter Hybridisierungsergebnisse oder hohen Hintergrundrauschens nicht entsprachen, wurden aus der Analyse ausgeschlossen. In die weitere Analyse konnten 13 diploide endometrioide Adenokarzinome, 9 aneuploide endometrioide Adenomkarzinome und 7 serös papilläre Karzinome eingeschlossen werden. Alle Werte, die die Kriterien der Qualitätskontrolle nicht erfüllten, wurden als fehlende Werte eingeordnet. Folgende Qualitätskriterien wurden zur Beurteilung im Gene-Pix-Ergebnis-File als Ausschlußkriterien verwendet: - Anzahl der gesättigten Pixel im Signal (F) größer als 20% - Spot-Diameter kleiner als 50 Pixel - Leerer Spot - Anzahl der Pixel kleiner als 40 - Mediane Test- und Referenz-Intensität kleiner als 100 - Weniger als 40% der Pixel stärker als der Hintergrund in einem von beiden Kanälen Die Intensitäts-Ratios (korreliert zum Hintergrund) wurden mit den medianen Intensitäten berechnet, welche mittels lokal-linearem Regressionsmodell (LOWESS; locally weighted scatterplot smoothing) für jede print-tip Gruppe normalisiert wurden. Die Fraktion der Messpunkte, die in der lokalen Regression (f) genutzt wurden, war 0.2, weitere Parameter wurden adjustiert wie von Cleveland empfohlen [127]. Der Wert f wurde mittels selfversus-self-Experiment bestimmt. Alle normalisierten Ratios eines Slides wurden logtransformiert (natural base). Über alle Proben hinweg konnten insgesamt 4.995 Gene identifiziert werden, die keine fehlenden Werte aufwiesen. Von diesen 4.995 wurden die unterschiedlich exprimierten Gene mittels Paarvergleich ermittelt: diploide endometrioide versus aneuploide endometriode Endometriumkarzinome, aneuploide endometriode Endometriumkarzinome versus aneuploide serös-papilläre Endometriumkarzinome und diploide endometrioide Endometriumkarzinome versus aneuploide serös-papilläre Karzinome. Um eine stabile Gen-Liste zu etablieren, wurden zwei statistische Tests genutzt. Nur Gene, die in beiden Analysen identifiziert wurden, fanden für die weiteren Auswertungen Verwendung. Zunächst wurde der Wilcoxon-Rangsummentest mit einem Permutationstest genutzt; wenn der p-Wert zwischen den Gruppen niedriger als 0,05 war, wurde der Wert zufällig in zwei Gruppen verteilt, der p-Wert generiert und 10.000 mal wiederhohlt. Alle Fälle, bei denen der p-Wert mit permutierten Werten unter 0,05 lag, wurden summiert und durch die totale Anzahl der Permutationen (10.000) dividiert. Dieser p-Wert bezeichnet die 42 Wahrscheinlichkeit, dass ein Gen eine kleinere oder gleiche Signifikanz per zufälliger Permutation hat als die ursprünliche Signifikanz, die vorher beschrieben war. Gene, die einen p-Wert kleiner 0,05 hatten, wurden als unterschiedlich exprimiert eingeordnet. Als zweiter Test wurde eine stufenweise (step-wise) Genselektions-Analyse angewandt [128,129]. Die zugrunde liegende Idee war, Gene einzeln zu einem Genset zu fügen, das zwei Klassen mittels Fishers linearem Diskriminations-Test am besten unterscheidet. Die step-wise Analyse wurde gestoppt, wenn der Wert, der die Entfernung zweier Ratios zwischen zwei Klassen beschreibt, kleiner als 0,0001 war. Abschließend wurde die Schnittmenge der signifikanten Gene aus beiden Test-Verfahren (Wilcoxon und stepwise-Analyse) bestimmt und nur diese für weitere Auswertungen verwendet. 3.4.5. Signalweg-Analysen Um die Einbindung der signifikant unterschiedlich exprimierten Gene in bekannten Signalwegen (Pathways) und Netzwerken zu ermitteln und darzustellen, wurde die Ingenuity Pathways Analysis (IPA) Software (v8.7, Ingenuity, Mountain View, CA) genutzt. IPA beschreibt Gruppen von Genen, die zusammen Netzwerke bilden. Diese Netzwerke zeigen an, wie bestimmte Gene sich vor dem Hintergrund von canonical Pathways beeinflussen können und miteinander in Beziehnung stehen. Die Pathways sind in der Kyoto Encyclopedia of Genes and Genomes (KEGG, www.genome.jp/kegg) hinterlegt. Nach Eingabe der Gene in die IPA-Software werden die generierten Netzwerke in einer Rangfolge angezeigt, in der die obersten „Top-Netzwerke“ einer geringeren Wahrscheinlichkeit unterliegen, nur durch puren Zufall generiert worden zu sein. Netzwerke mit einem Score von fünf und mehr Punkten wurden von uns als signifikant gewertet. 3.5. Protein-Analyse Nach der Isolierung der Proteine wurde der weitere experimentelle Teil der ProteinAnalyse, bestehend aus 2-dimensionaler Gelelektrophorese (2-DE), Massenspektometrie und IPA-Analyse im Rahmen einer anderen Dokorarbeit unserer Forschungsgruppe durchgeführt. Daher werden in dieser Arbeit erst im Rahmen des Ergebnissteils und der Diskussion auf die Proteindaten eingegangen, die im engen Zusammenhang mit den Array-Daten stehen. Für weitere Inormationen und Daten bezüglich der Proteinanalyse wird an dieser Stelle auf die Veröffentlichung unserer Arbeitsgruppe von Gemoll et al. [130] hingewiesen. 43 4. Ergebnisse 4.1. Ploidie Unter den endometrioiden Endometriumkarzinomen zeigten 16 Proben in der BildCytometrie eine eindeutige Stammlinie in der Region 2.0c und wurden somit als diploid klassifiziert. Neun endometrioide Endometriumkarzinome wurden anhand ihres cytometrischen Bildes der DNA-Verteilung als aneuploid klassifiziert. Alle acht seröspapillären Endometriumkarzinomproben wiesen in der Ploidiemessung ebenfalls typische aneuploide Charakteristika auf und wurden somit der aneuploiden Gruppe zugeschrieben. Der mittlere DNA-Stammlinien-Wert stieg von 2,23c in der EnD-Gruppe auf 2,98c in der EnA-Gruppe und schließlich bis auf 3,06c in der Gruppe der aneuploiden serös-papillären Endometriumkarzinome (UPSC-A; p < 0,0004). Die mittleren SSI-Werte waren 26,9% bei den diploiden endometrioiden, 45,4% bei den aneuploiden endometrioiden und 53,5% bei den serös-papillären Endometriumkarzinomen (p < 0,004). Der deutlich erhöhte SSI-Wert spiegelt die zunehmende genomische Instabilität bei aneuploiden im Vergleich zu diploiden Tumoren und insbesondere bei aneuploiden serös-papillären Karzinomen wider. Zum Zeitpunkt der Diagnose des Endometriumkarzinoms waren die Frauen mit endometrioiden Adenokarzinomen des diploiden Typs im Mittel 66,5 Jahre (51-87 Jahre), die Patientinnen mit einem endometrioiden Adenokarzinom des aneuploiden Typs im Mittel 67,7 Jahre (46-82 Jahre) alt. Die Diagnose eines aneuploiden serös-papillären Endometriumkarzinoms wurde bei deutlich älteren Patientinnen gestellt, die zum Zeitpunkt der Diagnosestellung im Durchschnitt 80,3 Jahre (66-90 Jahre) alt waren. In der Gruppe der endometrioiden Adenokarzinome fanden sich bei Patientinnen mit diploidem Tumor in nur 6,3% (1/16) der Fälle Metastasen, bei Frauen mit aneuploidem Tumor bereits in 22,2% (2/9) der Fälle. Dagegen waren unter den Patientinnen mit einem serös-papilläre Karzinom 75% (6/8) der Frauen von Metastasen betroffen. Von den endometrioiden Adenokarzinomen des diploiden Typs waren 62,5% (10/16) histologische Grad 1-Tumore, 31,3% (5/16) Grad 2 und 6,3% (1/16) stellten sich als Grad 3-Karzinome heraus. Anderes zeigte sich bei den aneuploiden Tumoren dieser Gruppe; hier waren jeweils 33,3% (3/9) der Karzinome histologisch Grad 1, Grad 2, oder Grad 3. 44 4.2. CGH Der ansteigende Grad der genomischen Instabilität von diploiden endometrioiden, über die aneuploiden endometrioiden bis hin zu den aneuploiden serös-papillären Endometriumkarzinomen war auch in der Auswertung der CGH-Ergebnisse durch eine insgesamt zunehmende Anzahl an Chromosomen-Alterationen zu beobachten (siehe Tabelle 3). 4.2.1. Diploid endometrioide Endometriumkarzinome (EnD) In der mit 16 Fällen grössten Gruppe der diploiden endometrioiden Adenokarzinome ergab die Auswertung der CGH-Profile insgesamt 15 Veränderungen des DNA-Gehaltes. Die Anzahl der Veränderungen pro Tumorprobe varierte bei den diploiden Karzinomen (DNA Histrogramm Typ I) von keiner bis zu vier Veränderungen pro Tumorprobe, wobei acht der Karzinome keine Abweichungen an DNA-Kopien mittels CGH zeigten. Der Tumor EnCaD7 zeigte mit drei Zugewinnen auf den Chromosomen 8 und 17 sowie dem Chromosomarm 18p und einem Verlust auf Chromosomarem 18q die meisten Veränderungen. Im Detail betrachtet war der lange Arm des Chromosoms 1 (1q) am häufigsten von einem Zugewinn an DNA-Kopien betroffen: Bei sechs von 16 (37,5%) diploid endometrioiden Karzinomen ließ sich ein Zugewinn auf 1q nachweisen. Wesentlich seltener lag mit 12,5% ein Zugewinn im Bereich des kurzen Arms des Chromosoms 16 (16p) vor. Hiervon waren zwei Karzinome betroffen. Alle weiteren Veränderungen traten jeweils nur einmal innerhalb der diploiden Karzinome auf: Zugewinne auf den Chromosomarmen 8p, 8q, 17p, 17q und 20q sowie Verluste auf 16q und 18q. 4.2.2. Aneuploid endometrioide Endometriumkarzinome (EnA) Die Gruppe der aneuploiden endometrioiden Adenokarzinome umfasste neun Tumorproben. Die Anzahl der Veränderungen pro Tumor variierte in dieser Gruppe zwischen 0 und 13. Bei den Tumorproben EnCaAe1, EnCaAe6 und EnCaAe8 ließen sich keine Veränderungen der DNA-Kopien per CGH nachweisen. Die meisten Abweichungen zeigte die Tumorprobe EnCaAe4 mit 13 betroffenen Chromosomenregionen einschließlich zweier Amplifikationen. Am häufigsten waren mit jeweils 33,3% der lange Arm des Chromosoms 10 (10q) sowie der lange Arm des Chromosoms 20 (20q) von einem Zugewinn an DNA betroffen. Diese Zugewinne ließen sich in jeweils drei Tumorproben nachweisen. Einen Zugewinn von DNA-Kopien auf den Chromosomenarmen 1q, 8q, 10p, 45 16p und 17q zeigten 22,2% der Proben. Ebenso häufig, in zwei von neun Tumorproben, trat im CGH-Profil ein Verlust von DNA-Kopien auf den Chromosomenarmen 9q, 16q, 17p, 19p, 19q und 22q auf. 4.2.3. Serös-papilläre Endometriumkarzinome (UPSC) Bei den acht aneuploid serös-papillären Tumoren ließen sich 79 Veränderungen in der Anzahl von DNA-Kopien per CGH darstellen. Die serös-papillären Karzinome wiesen zwischen 2 und 15 Veränderungen in der Zahl der DNA-Kopien auf, somit waren in dieser Gruppe bei jedem Tumor mindestens zwei Chromosomenbereiche von Veränderungen betroffen. Bei fünf der acht (62,5%) serös-papillären Karzinome lag ein Zugewinn an DNA-Kopien auf den Chromosomenarmen 2q, 8q, 17q oder 20p vor. Bei der Hälfte der Tumorproben ließ sich ein Zugewinn im Bereich des Chromosomenarms 20q nachweisen. Von einem Verlust an DNA-Kopien war in dieser Subgruppe der Arm 15q in 50% der Fälle am häufigsten betroffen. Ein häufiger Verlust zeigte sich in dieser Tumorgruppe mit 37,5% (3 von 8 Tumorproben) auch auf den Chromosomenarmen 4q, 8p und 17p. Die Hälfte der serös-papillären Endometriumkarzinome wiesen eine Amplifikation des Chromosomarms 8q auf. 4.2.4. ANCA- und ANRA-Werte Deutlich zeigte sich die zunehmende genomische Instabilität auch in der Berechnnung der ANCA- (average number of copy number alterations) und ANRA- (average number of regional amplification) Werte. Die ANCA- und ANRA-Werte errechnen sich aus der Summe der veränderten Chromosomen-Kopien (ANCA) bzw. der Summe der regionalen Amplifikationen (ANRA) pro Gruppe dividiert durch die Fallzahl der Gruppe. Im Detail stieg der ANCA-Wert der diploiden Tumore von 0,041 auf 0,145 bei den aneuploiden endometrioiden Tumoren und weiter auf 0,429 bei den aneuploiden seröspapillären Endometriumkarzinomen (p<0,001). Der gleiche Trend konnte für die Anzahl der Amplifikationen gezeigt werden. So stieg der ANRA-Wert von 0,003 bei den diploiden Endometriumkarzinomen auf 0,014 bei den aneuploiden endometrioiden Tumoren und weiter bis auf 0,065 bei den aneuploiden serös-papillären Endometriumkarzinomen (p<0,002). Die mittels CGH detektierten Veränderungen sind im Detail in Tabelle 3 dargestellt. 46 Chrom. EnD EnA UPSC-A n=16 n=9 n=8 gain: 6/16 (37,5%) gain: 2/9 (22.2%) gain: 2/8 (25%) Zugewinn insgesamt n=33 Verlust insgesamt Veränderungen insgesamt 1p 1q 2p 2q gain: 3/8 (37.5%) gain: 5/8 (62.5%) gain: 1/8 (12.5%); loss: 2/8 (25%) gain: 3/8 (37.5%) loss: 1/8 (12.5%) gain: 1/8 (12.5%); loss: 3/8 (37.5%) 3p 3q 4p 4q 10/33 (30,3%) 3/33 (9,1%) 5/33 (15,2%) 1/33 (3,0%) 3/33 (9,1%) 10/33 (30,3%) 3/33 (9,1%) 5/33 (15,2%) 2/33 (6,1%) 1/33 (3,0%) 3/33 (9,1%) 3/33 (9,1%) 1/33 (3,0%) 1/33 (3,0%) 3/33 (9,1%) 4/33 (12,1%) 1/33 (3,0%) 2/33 (6,1%) 1/33 (3,0%) 2/33 (6,1%) 2/33 (6,1%) 1/33 (3,0%) 2/33 (6,1%) 3/33 (9,1%) 3/33 (9,1%) 3/33 (9,1%) 1/33 (3,0%) 4/33 (12,1%) 3/33 (9,1%) 2/33 (6,1%) 3/33 (9,1%) 5/33 (15,2%) 5p 5q 6p 6q 7p gain: 1/9 (11.1%) 7q gain: 1/9 (11.1%) 8p 8q gain: 1/16 (6,3%) gain: 1/16 (6,3%) gain: 2/9 (22.2%) 9p gain: 1/9 (11.1%) 9q loss: 2/9 (22.2%) 10p gain: 2/9 (22.2%) 10q gain: 3/9 (33.3%) 11p 11q 12p 12q 13p 13q 16q gain: 2/16 (12,5%) loss: 1/16 (6,3%) 17p gain: 1/16 (6,3%) 17q gain: 1/16 (6,3%) 18p 18q 1/33 (3,0%) 3/33 (9,1%) 3/33 (9,1%) 4/33 (12,1%) 3/33 (9,1%) 5/33 (15,2%) 1/33 (3,0%) 2/33 (6,1%) 2/33 (6,1%) 6/33 (18,2%) 2/33 (6,1%) 2/33 (6,1%) 1/33 (3,0%) 2/33 (6,1%) 3/33 (9,1%) 1/33 (3,0%) gain: 1/8 (12.5%); loss: 4/8 (50%) 1/33 (3,0%) gain: 2/9 (22.2%) gain: 1/8 (12.5%) loss: 2/9 (22.2%) loss: 2/8 (25%) gain: 1/9 (11,1%); loss: 2/9 (22,2%) gain: 1/8 (12.5%); loss: 3/8 (37.5%) gain: 2/9 (22.2%) loss: 1/16 (6,3%) loss: 2/9 (22.2%) 19q 20p loss: 2/9 (22.2%) 21p 21q 22p 22q Xp 4/33 (12,1%) loss: 1/9 (11.1%) gain: 1/9 (11.1%) gain: 1/16 (6,3%) 2/33 (6,1%) 1/33 (3,0%) gain: 3/9 (33.3%) gain: 5/8 (62.5%); loss: 1/8 (12.5%) gain: 3/8 (37.5%) gain: 1/8 (12.5%); loss: 2/8 (25%) gain: 1/8 (12.5%); loss: 2/8 (25%) gain: 3/8 (37.5%) gain: 5/8 (62.5%) gain: 4/8 (50%) gain: 1/8 (12.5%) loss: 2/9 (22.2%) loss: 1/9 (11.1%) loss: 1/8 (12.5%) 8/33 (24,2%) 2/33 (6,1%) gain: 1/8 (12.5%) 19p 20q 8/33 (24,2%) 1/33 (3,0%) 14p 14q 15p 16p gain: 5/8 (62.5%) gain: 1/8 (12.5%); loss: 2/8 (25%) gain: 1/8 (12.5%); loss: 1/8 (12.5%) gain: 1/8 (12.5%) gain: 2/8 (25%); loss1/8 (12.5%) loss: 2/8 (25%) loss: 2/8 (25%) gain: 1/8 (12.5%) gain: 1/8 (12.5%); loss: 1/8 (12.5%) loss: 1/9 (11.1%) 15q gain: 1/8 (12.5%); loss: 1/8 (12.5%) gain: 2/8 (25%) gain: 1/8 (12.5%); loss: 2/8 (25%) gain: 2/8 (25%); loss1/8 (12.5%) gain: 2/8 (25%) gain: 1/8 (12.5%); loss: 3/8 (37.5%) 1/33 (3,0%) 5/33 (15,2%) 5/33 (15,2%) 6/33 (18,2%) 5/33 (15,2%) 5/33 (15,2%) 5/33 (15,2%) 3/33 (9,1%) 5/33 (15,2%) 8/33 (24,2%) 8/33 (24,2%) 4/33 (12,1%) 1/33 (3,0%) 9/33 (27,3%) 4/33 (12,1%) 1/33 (3,0%) 3/33 (9,1%) 4/33 (12,1%) 1/33 (3,0%) 3/33 (9,1%) 5/33 (15,2%) 4/33 (12,1%) 2/33 (6,1%) 5/33 (15,2%) 5/33 (15,2%) 5/33 (15,2%) 8/33 (24,2%) 8/33 (24,2%) 1/33 (3,0%) 1/33 (3,0%) 2/33 (6,1%) 2/33 (6,1%) 2/33 (6,1%) 2/33 (6,1%) 47 Chrom. EnD EnA UPSC-A n=16 n=9 loss: 1/9 (11.1%) n=8 loss: 1/8 (12.5%) Xq Zugewinn insgesamt n=33 Verlust insgesamt 2/33 (6,1%) Veränderungen insgesamt 2/33 (6,1%) Tabelle 3. CGH-Ergebnisse aufgeschlüsselt nach Chromosomenarm und Subgruppe. Chrom. = Chromosomenarm; Zugewinn insgesamt = DNA Zugewinn pro Chromosomenarm im gesamten Probenkollektiv; Verlust insgesamt = DNA Verlust pro Chromosomenarm im gesamten Probenkollektiv; Veränderungen insgesamt = DNA Zugewinne und Verluste pro Chromosomenarm im gesamten Probenkollektiv gain = Gewinn an DNA loss = Verlust an DNA Die CGH-Profile der einzelnen Fälle, sowie die Gruppen-Profile sind unter http://www.ncbi.nlm.nih.gov/sky/skyweb.cgi abrufbar. 4.3. Genexpression Um diejenigen Gene zu identifizieren, die in den drei verschiedenen Tumorgruppen unterschiedlich exprimiert waren und somit die Subgruppen charakterisierten, analysierten wir die Ergebnisse der cDNA-Microarrays mittels Wilcoxon-Test mit Permutation und einer step-wise Analyse [128]. In beiden Analysen signifikant unterschiedlich exprimiert waren im Vergleich der endometrioden diploiden mit den endometrioden aneuploiden Tumore 90 Gene (differentially expressed genes; DEGs)., im Vergleich der endometrioden diploiden mit den serös papillären Karzinomen 117 Gene und im Vergleich der endometrioiden aneuploiden mit den serös papillären schließlich noch 68 Gene. Nur die Gene, die in beiden Analysen als signifikant unterschiedlich detektiert wurden und sich im Gruppenvergleich als einzigartig herausstellten, waren Teil der weiteren Auswertungen. Zwischen den EnD-Proben und den EnA-Tumoren waren dies 54 Gene, im Gruppenvergleich EnD versus UPSC-A sogar 76 Gene und bei den EnA Tumoren im Vergleich mit den UPSC-A Tumoren noch 39 Gene. Der größte Unterschied ließ sich auf der Genexpressionsebene zwischen den diploid endometrioiden und aneuploid seröspapillären Tumoren, welche sich sowohl im Ploidiestatus, als auch in der Histologie voneinander unterscheiden, darstellen (76 DEGs) (siehe Tabelle 4). 48 Wilcoxon (p<0.05) EnD versus EnA 478 EnD versus UPSC-A 576 EnA versus UPSC-A 276 step-wise Analyse (hoch-/runterreguliert) EnD versus EnA 140/118 EnD versus UPSC-A 195/179 EnA versus UPSC-A 165/154 Signifikante Gene aus beiden Analysen _EnD versus EnA 90 EnD versus UPSC-A 117 EnA versus UPSC-A 68 Einzigartige Gene im Gruppenvergleich EnD versus EnA 54 EnD versus UPSC-A 76 EnA versus UPSC-A 39 Tabelle 4. Überblick der signifikant unterschiedlich exprimierten Gene. Insgesamt waren 114 der 275 (41,45%) DEGs auf Chromosomenregionen lokalisiert, bei denen auch Veränderungen mittels CGH gefunden wurden (siehe auch 8.2.2. DEG Listen). 4.3.1. Korrelation der Veränderungen im Gen- und Proteinexpressionsmuster Die mittels 2-dimensionaler Gelelektrophorese ermittelten unterschiedlich exprimierten Proteine (differentially expressed proteins; DEPs) aus der vorangehenden Studie unserer Arbeitsgruppe [130] wurden zu ihrem chromosomalen Ursprungsort der korrespondierenden Gene lokalisiert. Dabei zeigte sich, dass im Gruppenvergleich der EnD versus EnA 11 von 20 Proteinen (55%) und im Vergleich der EnD versus UPSC-A Karzinome 7 von 15 Proteinen (47%) zu Chromosomenregionen lokalisiert werden konnten, die in der CGH-Analyse auch von Veränderungen in der DNA-Kopien-Anzahl betroffen waren (vollständige DEP-Listen bei Gemoll et al. [130]). Fünf der 35 identifizierten Proteine waren als cDNA-Spot auf den verwendeten Microarrays vorhanden. Von den korrespondierenden Genen zeigten zwei, AKR7A2 und ANXA2, den gleichen Trend in der transkriptionalen und der translationalen Expression. Trotzdem erreichten beide Gene nicht den Signifikanzlevel in der Expressions-Analyse. Das Gen AKR7A2 war bei den endometrioden diploiden im Vergleich zu den serös-papillären Endometriumkarzinomen herunterreguliert, während ANXA2 bei den aneuploid endometrioden im Vergleich zu den diploid endometrioden und im Vergleich von den serös-papillären mit den diploid endometrioden Endometriumkarzinomen hochreguliert war. 49 4.3.2. Funktionale Einordnung der unterschiedlich exprimierten Gene und Proteine Mit Hilfe der IPA (Ingenuity Pathway Analysis) Software wurden die zwischen den EnD, EnA und UPSC-A unterschiedlich exprimierten Gene und Proteine in bekannte Netzwerke und Signalwege eingeordnet. Ein Überblick der IPA-Analysen findet sich tabellarisch unter 8.2.3. (Überblick der IPA-Analysen) und zwei schematische Netzwerke unter 8.2.4. (Netzwerke basierend auf der IPA-Analyse). Bei dem Vergleich von diploiden endometrioiden mit den aneuploiden endometrioiden Tumoren wurden 45 von 54 (83%) der unterschiedlich exprimierten Gene (DEG) von der IPA-Datenbank erkannt und drei Netzwerke generiert. Das Netzwerk „Lipid Metabolism, Small Molecule Biochemistry and Vitamin and Mineral Metabolism” wurde mit einem Punktwert von 48 am höchsten gelistet. Hier konnten 20 der 45 DEGs eingeordnet werden. Im Folgenden sind jeweils die Gene fettgedruckt, welche in der Gruppe der diploid endometrioiden Adenokarzinomen im Vergleich zu den aneuploid endometrioden Karzinomen hochreguliert waren: ADAM9, AKR1C4, BMP4, CYP1B1, EIF4A3, ELANE, HBB, IDO1, ISG20, ITGA3, ITPR2, ITPR3, LIF, LYN, PCOLCE, SATB1, SERPINF1, SOD2, TNFRSF12A, and TTK. NFkB, Jnk und ERK1/2 sind zentrale Knotenpunkte des Netzwerks und assoziiert mit Krankeitsbildern und Funktionen aus dem Kreis „Cancer, Hematological Disease, and Gastrointestinal Disease“ (p<0,00001 bis p<0,0161). An zweithöchster Stelle wurde das Netzwerk „Lipid Metabolism, Small Molecule Biochemistry and Genetic Disorder“ mit einem Punktwert von 30 gelistet. Dieses Netzwerk beinhaltete 14 der DEGs: ATPAF2, CYBRD1, DSC2, KIAA0020, LDB2, MAP7D3, MGST3, PRCP, PRMT3, REV3L, SCN5A, TAGLN2, TMSB10. Auf Rang drei (Punktwert 22) folgte ein Netzwerk assoziiert mit „Gene Expression, Nutritional Disease and Cellular Development“; hier konnten 11 der 45 unterschiedlich exprimierten Gene wiedergefunden werden: AKR1C1, APOD, CCDC50, CPSF6, FLRT2, HPGD, NAPSA, PAM, SORD, SPAG8, TSC22D1. Im Vergleich der aneuploid endometrioiden Endometriumkarzinome mit den aneuploid serös-papillären konnten 33 von 39 DEGs (84,6%) in der IPA-Datenbank detektiert werden. Drei sich überschneidende Netzwerke erreichten den Signifikanzlevel mit Punktwerten von 28 bis 22. Das am höchsten gelistete Netzwerk mit einem Punktwert von 28 war hier assoziiert mit „Cardiovascular System Development and Function, Cell Cycle, Lipid Metabolism“ und zeigte spezifische Verbindungen mit dem Cannonical Pathway „Genexpression“ (p<0,0245) und „Zelltod“ (p<0,0338). Folgende Gene waren involviert (fettgedruckt sind die Gene, welche bei den aneuploid endometrioden Karzinomen im Vergleich mit den aneuploid serös-papillären Karzinomen hochreguliert waren): ACOX1, ARF3, BIRC2, CPD, EAF1, EEF1D, MPP3, NDRG4, RABGAP1L, SAA1, SPOCK3, TXK. Interaktionen in diesem Netzwerk fanden sich via BIRC2, SAA und SAA1 mit dem am 50 zweithöchsten gelisteten Netzwerk (Punktwert 28), welches mit „Cell Death, Cellular Movement; Haematological System Development and Function assoziiert war (BIRC2, BIRC3, CPNE1, EFNB2, EPAS1, FHL2, KCNJ8, MAP3K5, PTPN13, SAA1, SALL2, TM7SF2). Über die Gene CTSH, ITPR und SAA1 bestand wiederum eine Verbindung zu dem am dritthöchsten gelisteten Netzwerk (Punktwert 22) assoziiert mit „Cellular Assembly and Organization, Cellular Function and Maintenance, and Cell Signaling“ (CA8, CIB1, CSTB, GALNT10, HOXB13, LYST, PLAGL2, RDH10, SAA1, TGIF2 (siehe 8.2.4.). Interessanterweise verbindet SAA1 auch das zweite und dritte Netzwerk miteinander. In diesen Netzwerken stellen IFNG, TGFB; MYC und NFkB zentrale Knotenpunkte dar. Von den 76 Genen, welche die EnD- von den UPSC-A-Proben unterscheiden, waren 67 (88,2%) in der IPA-Datenbank hinterlegt. Daraus resultierten fünf sich überlappende Netzwerke. Das führende erreichte einen Punktwert von 44 und vereinte folgende 20 der DEGs (fettgedruckt sind die Gene, die bei den EnD-Proben im Vergleich zu den UPSC-AProben hochreguliert waren): ADCYAP1R1, APOE, ATF3, CRYAB, CXCL5, DNM2, DYNC1LI2, GSN, IFI27, ISG15, ITPKB, KRT17, LPAR1, MTSS1, MX2, OAS1, PRKCA, RNASEH2A, SOCS3, TXNIP, mit einer Verbindung zum Pathway „Organism Injury and Abnormalities, Cardiac Necrosis/Cell Death, Cell Death“. Das zweitplatzierte Netzwerk in dieser Analyse erreichte einen Punktwert von 28 mit 14 abgebildeten DEGs (ACVR1B, CLUAP1, CYP7B1, DERL1, DLX4, FCHO1, GSN, HIBCH, KIF3C, KMO, MSLN, PLEC, SETX, SLC39A4) und zeigte eine Assoziation zu „Organ Morphology, Reproductive System Development and Function; Skeletal and Muscular Disorders“. Als drittes folgte ein Netzwerk mit 20 Punkten, welches 11 DEGs (CENPF, DDX39, DGKE, DNMT3B, KRR1, LUC7L3, MSX2, SLC38A1, TCF12, UBE2S, WNT7A) vereinte und assoziiert war mit „Cellular Development, Cellular Growth, Proliferation and Cancer“. Auf Rang vier stand ein Netzwerk mit einem Punktwert von 19. Hier waren ebenfalls 11 DEGs subsumiert (ARL4C, ASS1, CACNA1D, KRT2, LGALS3BP, LMO2, MLLT3, P2RY1, PAPSS1, RBBP7, SPOCK2) mit einer Verbindung zu den Pathways „Cardiovascular Disease, Haematological Disease, and Skeletal and Muscular Disorders“. Als fünftes wurde ein Netzwerk zu „Endocrine System Development and Function, Small Molecule Biochemistry, and Gene Expression“ mit einem Punktwert von 15 generiert. In diesem waren folgende neun der 67 DEGs abgebildet: ASAP2, CIRBP, PGR, PVRL3, SPTBN2, TACC3, TBCB, TMED10, TPBG. Alle Netzwerke in diesem Gruppenvergleich standen in Verbindung mit „Cancer“ (p<0,0196), „Genetic Disorder“ (p<0,0234), „Cellular Growth and Proliferation“ (p<0,0212) und „Cell-To-Cell Signalling and Interaction“ (p<0,0234). Weitere drei Netzwerke, die in 51 der IPA Analyse gefunden wurden, enthielten nur ein DEG und erreichten somit mit einem Punktwert <5 nicht den Signifikanzlevel. Ein Überblick über die unterschiedlich exprimierten Gene (DEGs) und Proteine (DEPs) unter Einbeziehung der Ploidie-Typen und der Histologie findet sich im Gruppenvergleich der Abbildung 3. 52 Abbildung 3: Beispiele der Ploidie-Typen und Anzahl der unterschiedlich exprimierten Gene (DEGs) und Proteine (DEPs). Die DNA-Histogramme zeigen den DNA Gehalt auf der x-Achse und die Gesamtzellzahl auf der y-Achse. 53 5. Diskussion In dieser Studie wurden ploidie-assozierte chromosomale Veränderungen dargestellt und korrespondierende Gene analysiert, die Endometriumkarzinome unterschiedlichen prognostischen Subtyps charakterisieren. Untersucht wurden Endometriumkarzinome, die sich im Ploidiestatus und/oder im histologischen Typ voneinander unterschieden, darunter 16 diploide endometrioide Endometriumkarzinome, neun aneuploide endometrioide Adenokarzinome, (Typ I Endometriumkarzinome) und acht aneuploide serös-papilläre Endometriumkarzinome (Typ II Endometriumkarzinome). Die Auswertung der Ploidie-Messung ergab einen signifikant ansteigenden SSI-Wert von den diploiden Karzinomen über die aneuploid endometrioiden bis hin zu den aneuploid serös-papillären Endometriumkarzinomen. Dieses reflektiert eine steigende genomische Instabilität und korreliert mit der klinischen Prognose der Subtypen. Interessanterweise zeigten alle Endometriumkarzinomproben bis auf eine diploide Probe SSI-Werte größer 8,8%. Der Grenzwert des SSI-Wertes von 8,8% wurde von Kronenwett et al. für Mammakarzinome etabliert. Dieser Grenzwert kann bei Brustkrebspatientinnen genomisch stabile Tumoren mit einem SSI-Wert bis 8,8% von genomisch instabilen Mammakarzinomen mit einem SSI-Wert größer 8,8% abgrenzen. Kronenwett et al. konnten zeigen, dass Frauen mit genomisch stabilen Karzinomen signifikant bessere Überlebensraten hatten im Vergleich zu Patientinnen mit genomisch instabilen Karzinomen [114,131]. Der SSI-Wert wurde bisher nicht für andere Tumorentitäten etabliert. In unserem Probenkollektiv der Endometriumkarzinome zeigten bis auf eine Probe alle einen SSI-Wert deutlich über 8,8%, so dass anzunehmen ist, dass der SSIGrenzwert von Mammakarzinomen nicht gleichermaßen für Endometriumkarzinome übernommen werden kann. Es bedarf weiterer Studien, um auch für Endometriumkarzinome möglicherweise einen prognostisch bedeutenden SSI-Grenzwert zu etablieren. Entsprechend den Ergebnissen anderer Studiengruppen waren unsere Patientinnen, die an einem aneuploiden serös-papillären Endometriumkarzinom erkrankten, im Mittel deutlich älter, als diejenigen mit der Diagnose eines diploid endometrioden und aneuploid endometrioden Tumors (80,3 Jahre, versus 66,5 und 67,7 Jahre) [32,35,37]. Patientinnen mit diploidem Tumor waren in nur 6,3% (1/16) der Fälle von Metastasen betroffen, diejenigen mit aneuploidem endometrioden Tumor bereits in 22,2% (2/9) der Fälle und die Patientinnen mit dem aggressiven Subtyp des serös-papillären Karzinoms sogar in 75% (6/8) der Fälle. 54 Der Trend der genomischen Instabilität auf Ebene der Ploidie-Messung spiegelte sich auch in den CGH-Ergebnissen durch einen signifikant ansteigenden ANCA- und ANRAWert wider. Bei den EnD-Tumoren waren die häufigsten Veränderungen, die mittels CGH detektiert werden konnten, ein Zugewinn auf den Chromosomenarmen 1q und auf 16p. Unter den EnA-Proben fanden sich am häufigsten Veränderungen der DNA-Kopien als Zugewinn von 10q und 20q. Aber auch andere Chromosomenbereiche waren in dieser Tumorgruppe öfter von Veränderungen betroffen wie Zugewinne auf 1q, 8q, 10p, 16p und 17q sowie Verluste auf 9q, 16q, 17p, 19p, 19q und 22q. In der Gruppe der UPSC-A-Karzinome fanden sich schließlich die meisten chromosomalen Abweichungen: Am häufigsten waren Zugewinne auf 2q, 8q, 17q, 20p und 20q, sowie Verluste auf 4q, 8p, 15q und 17p zu beobachten. Besonders deutlich ins Auge fielen hier die Amplifikationen der Region 8q in 50% der UPSC-A Proben. Über die Gruppen hinweg betrachtet waren die häufigsten per CGH ermittelten genomischen Inbalancen ein Zugewinn der Chromosomenarme 1q, 8q, 17q und 20q, sowie DNA-Verluste auf 15q, 16q und 17p. Ähnliche CGH-Muster bei Endometriumkarzinomen finden sich auch in der Literatur. Suzuki et al. wiesen in ihrer Studie an 15 Endometriumkarzinomen ebenfalls die häufigsten Zugewinne auf 1q und 8q nach. Betrachtet man nur die endometrioiden Adenokarzinome, so zeigten Suzuki et al. in 61% der 13 Fälle einen Zugewinn auf 1q sowie in 31 % der Fälle einen Zugwinn auf 8q [132]. In einer groß angelegten CGH-Studie untersuchten Suehiro et al. 51 primäre Edometriumkarzinome. Darunter fanden sich 46 endometrioide Adenokarzinome sowie vier adenosquamöse Karzinome und ein Adenoakanthom, aber keine serös-papillären Subtypen. Häufig gefundene Zugewinne zeigten sich auch hier auf Chromosom 1q21q41 (19,6%), 1q42-qter (17,6%), 8q22-23 (19,6%), 8q24-qter (27,5%) und 19p (17,6%). Verluste an DNA wurden besonders häufig auf Chromosom 4q31-qter (17,6%) detektiert. Dagegen beobachteten wir in unserer Studie einen Verlust auf 4q nur bei den seröspapillären Karzinomen, hier aber in 37,5% der Proben. Suehiro et al. wiesen eine signifikant höhere Anzahl an chromosomalen Alterationen bei Patientinnen, die am Endometriumkarzinom verstorben waren, im Vergleich zu den nach Therapie tumorfreien Frauen (12,6 versus 2,7, p<0,0001) nach. Die Anzahl der chromosomalen Veränderungen war außerdem signifikant höher in Tumoren der Stadien III und IV im Vergleich zu Karzinomen der Stadien I und II. Die Gruppe zeigte darüber hinaus in einer Multivariantenanalyse, dass Lymphknotenmetastasen, Zervixbefall und eine oder mehr DNA-Kopie-Verluste auf 9q32-q34, 11q23, oder Xq12-q24 signifikant mit dem Versterben an dem Endometriumkarzinom korrelierten. Das Auftreten von Lymphknotenmetastasen 55 war signifikant höher bei Karzinomen mit DNA-Zugewinnen auf 8q22-q23 und 8q24-qter [133]. Ein Zugewinn auf dem Chromosomenarm 8q konnte auch in unserer Studie vor allem bei den klinisch aggressiveren Subtypen gefunden werden. So zeigte sich bei EnA-Proben in 22,2% der Fälle ein Zugewinn auf 8q im Vergleich zu nur 6,3% in der klinisch günstigeren EnD-Gruppe. Bei den UPSC-A-Tumoren, welche klinisch die schlechteste Prognose aufweisen und am frühesten metastasierten, zeigten sich in unserer Analyse sogar in 62,5% der Fälle ein Zugewinn in dieser Region, darunter sogar vier Fälle (50%) einer Amplifikation von 8q. Die einzigen bisher veröffentlichten Daten, die bei der Auswertung der CGH-Profile Typ Iund Typ II-Endometriumkarzinome vergleichen, kommen von Micci et al. [134]. Die Gruppe untersuchte insgesamt 67 Endometriumkarzinome mittels CGH. Für die Gruppe der Typ I-Karzinome fand sich am häufigsten ein Gewinn an DNA-Kopien auf den Chromosomenarmen 1q und 8q sowie Verluste an DNA auf den Chromosomenarmen Xp, 9p, 9q, 17p, 19p und 19q. Bei den serös-papillären Karzinomen des Typs II stellte sich ein DNA-Gewinn auf 1q, 2p, 2q, 3q, 5p, 6p, 6q, 7p, 8q, 18q, 20p und 20q am häufigsten dar. Die meisten DNA-Verluste betrafen bei diesem histologischen Typ den Chromosomenarm 17p. Levan et al. analysierten eine Serie von 98 endometrioiden Adenokarzinomen mittels CGH. Veränderungen der DNA-Kopien wurden in 75% der Tumore gefunden. Die häufigsten Gewinne an DNA waren auf 1q (30%), 19p (26%), 19q (19%), 10p (14%) und 10q (13%). Die häufigsten DNA-Verluste stellten sich auf 4q, 16q und 18q in jeweils 8% der endometrioiden Endometriumkarzinome dar [135]. Insgesamt lässt sich sagen, dass beim Endometriumkarzinom bisher kein eindeutiges CGH-Profil etabliert wurde. Ein Grund dafür ist sicherlich auch die mangelnde Differenzierung zwischen den Subtypen durch einige Autoren und die oft geringe Fallzahl in den Studien, insbesondere hinsichtlich der klinisch selteneren aneuploiden endometrioden und serös-papillären Karzinome. Obwohl auch in unserer Studie die Fallzahl insbesondere bei den klinisch selteneren aneuploiden endometrioden und serös-papillären Karzinomen relativ klein war, so konnten wir insbesondere durch eine klare Trennung zwischen den Subtypen, eindeutige Unterschiede herausarbeiten. So zeigten die CGH-Ergebnisse einen signifikant ansteigenden ANCA- und ANRA-Wert von den EnD über die EnA bis zu den UPSC-A Karzinomen. Betrachtet man die CGH-Gruppenprofile im Vergleich (Abbildung 3), so kristallisieren sich im Vergleich der Subtypen eindeutig unterschiedliche Profile heraus. Die EnD-Gruppe zeigte am häufigsten chromosomale Veränderungen als Zugewinne auf 1q und 16p. Bei den EnA Endometriumkarzinomen waren hingegen 56 die Chromosomenarme 10q und 20q am häufigsten von DNA-Zugewinnen betroffen. Bei den UPSC-A Karzinomen schließlich waren Zugewinne auf 2q, 8q, 17q, 20p und 20q, sowie Verluste auf 4q, 8p, 15q und 17p am häufigsten zu beobachten. In dieser Subgruppe fielen auch besonders die Amplifikationen der Region 8q in 50% der Fälle ins Auge. Viele Chromosomenbereiche waren ausschließlich in der UPSC-A Gruppe von Veränderungen betroffen, insbesondere der Chromosomenarm 2q, der bei den serös-papillären Karzinomen in 62,5% der Proben einen DNA-Gewinn zeigte, war in den beiden anderen Subgruppen nicht von Veränderungen betroffen. Die Etablierung der Microarray-Technik hat in einigen Bereichen, insbesondere der Senologie, bereits Einzug in die Klinik gehalten. Im Vergleich zu älteren Verfahren steht mit der Array-Technik nun eine Methode zur Verfügung, die die zeitgleiche Darstellung des gesamten Transkriptoms möglich macht. Durch die Einführung der Microarrays ergaben sich neue Erkenntnisse in der Onkogenese verschiedener Tumorentitäten, die bereits zu Fortschritten und Verbesserungen im Tumormanagement führten. Die Etablierung von spezifischen Genexpressions-Profilen kann zu neuen Tumorklassifikationen führen, welche dem klinischen Verhalten von Tumorsubtypen besser gerecht wird [124]. Die Identifizierung von Expressionsmustern ermöglicht eine bessere Vorhersage des klinischen Verhaltens, der Prognose und der Therapieoptionen. Sie eröffnen neue Angriffspunkte für die onkologische Therapie und die frühzeitige Diagnose [136]. Zwischen den EnD-Proben und den EnA-Tumoren waren 54 Gene unterschiedlich exprimiert. Die aneuploid endometrioiden Tumoren unterschieden sich von den seröspapillären durch 39 DEGs. Der größte Unterschied auf Ebene der Genexpression zeigte sich mit 76 DEGs zwischen den diploid endometrioiden und den aneuploid serös- papillären Tumoren und bestätigt damit auch die Unterschiede im Ploidiestatus und in den CGH-Ergebnissen. Die zwischen den Subtypen unterschiedlich exprimierten Gene gehören zu verschiedenen Funktionskreisen. Sie sind in fundamentale biologische Prozesse involviert, deren Zusammenhang mit Tumorerkrankungen bekannt ist. Von den Genen ATF3, DNMT3B, LMo2 und TCF12 zum Beispiel weiß man, dass sie Vorgänge der DNA-Bindung, Transkription und Proliferation beeinflussen. Interessanterweise konnte bei aneuploiden Endometriumkarzinomen korreliernd dazu auch eine höhere Zellproliferationsrate im Vergleich zu diploiden Tumoren nachgewiesen werden [137]. Eine andere Gruppe der identifizierten Gene kodiert Enzyme wie Proteasen, Reduktasen und Transferasen. Viele von diesen Genen, wie aflatoxin B1 aldehyde reductase member 2 (AKR7A2), v-yes-1 Yamaguchi sarcoma viral related oncogene homolog (LYN), oder cytochrome b reductase 1 (CYP1B1) wurden bisher nicht in EndometriumkarzinomStudien gefunden und bieten somit neue interessante Ziele für weitere Studien zur 57 Verbesserung von Diagnose und Behandlung. AKR7A2 (lokalisiert auf Chromosomenabschnitt 1p36.13) ist ein Golgi-assoziiertes AKR7 Familienmitglied und wird in vielen unterschiedlichen Geweben exprimiert [138]. AKR7A2 ist in die Detoxifikation von Aldehyden und Ketonen des Phase I-Stoffwechsels involviert und erhöhte Proteinlevel sind auch im cerebralen Cortex von Alzheimer-Patienten gefunden worden [139]. Die Ergebnisse der Genexpression unterstützen an dieser Stelle die Daten unserer Proteomics-Studie, in der eine AKR7A2-Überexpression bei diploid endometrioiden im Vergleich zu aneuploid serös-papillären Endometriumkarzinomen gefunden werden konnte [130]. LYN, lokalisiert auf Chromosom 8q13, gehört zu den hochsignifikant unterschiedlich exprimierten Genen, war im Vergleich zu EnA-Karzinomen in den EnD überexprimiert und belegt eine enge Korrelation zwischen DNA- und RNAAlterationen. LYN ist ein Mitglied der Src-Kinasen, eine Familie der non-RezeptorThyrosinkinasen. Dem Gen gilt besonderes Interesse, weil es als Kinase ein möglicher Ansatzort für Medikamente ist und somit therapeutische Optionen bei Endometriumkarzinomen bieten könnte. Choi et al. identifizierten LYN bereits als mögliches therapeutisches Ziel insbesondere bei dem klinisch aggressiven basal-like Mammakarzinom [140]. Eine weitere Gruppe interessanter Gene ist in den Transport und die Bindung von Proteinen involviert. Die Annexine können in der Anwesenheit von Calcium-Ionen an Phospholipide binden und sind tyischerweise in der Lage, zu phosphorylieren und zu dephosphorylieren. Humane Annexine gehören zur Subfamilie A der Annexine und werden als A1-A11 und A13 klassifiziert. Eine Isoform, das Annexin A2 (ANXA2; lokalisiert auf Chromosom 15q22.2) konnte bereits in der Proteomics-Studie unserer Arbeitsgruppe detektiert [130] und die korrespondierende Überexpression auf Genebene mit dieser Studie bestätigt werden. Das Annexin A2 liegt im Cytoplasma als Monomer oder im Komplex mit einem Mitglied von S100A10 vor [141]. Es gibt Hinweise auf Wechselwirkungen zwischen Proteasen und extrazellulären Matrix-Proteinen über das Annexin A2. Hierbei scheint ANXA2 die Reorganisation der extrazellulären Matrix in physiologischen, aber auch pathologischen Prozessen wie der Tumorinvasion zu unterstützen [142]. Bei Kolorektalen- und Magenkarzinomen konnte bereits gezeigt werden, dass entsprechend unserer Studie an Endometriumkarzinomen [130] eine Überexpression von ANXA2 mit der Invasivität des Karzinoms und einer schlechten Prognose korreliert [143,144]. Darüberhinaus fanden wir im Vergleich der diploiden mit den aneuploiden endometrioiden Karzinomen das Gen Wnt-7a (wingless-type MMTV integration site family, member 7a) überexprimiert. Wnt-7a ist auf dem Chromosomenabschnitt 3p25 lokalisiert und wird mit verschiedenen Zellreaktionen in Verbindung gebracht, wie der Regulierung der 58 Zellidentität, der Differenzierung, sowie Apoptose und Onkogenese. Hohe Expressionsspiegel des Gens können im weiblichen Reproduktionstrakt nachgewiesen werden [145,146]. Es wird vermutet, dass eine Veränderung der normalen Wnt-7a Expression durch Diethylstilbestrol oder andere östrogene Bestandteile die uterine Cytoarchitektur stören und über diesen Mechanismus letzlich zu Neoplasien im weiblichen Genitaltrakt führen kann [147]. Im Vergleich der diploiden endometrioiden Karzinome mit den aneuploiden seröspapillären war der Progesteronrezeptor (PGR; Chromosom 11q22-q23) signifikant niedriger bei den serös-papillären Karzinomen exprimiert. Im Gesamtbild zeigte der Progesteronrezeptor hier sogar die höchste Ratio der unterschiedlichen Expression in unserer Studie. Hiermit stimmte auch das Muster der CGH-Auswertung überein: In 25% der Fälle trat ein DNA-Kopien-Verlust des gesamten Chromosoms 11 bei den UPSC-AProben auf, wohingegen bei den diploiden Karzinomen keine Veränderung des betreffenden Chromosoms mittels CGH nachgewiesen wurde. Der PGR gehört zu den Steroidhormon-Rezeptoren und vermittelt die natürliche Wirkung von Progesteron an der Zelle. Progesteron ist ein wichtiges Homon bei der weiblichen Reproduktion. In der Ätiologie des endometrioiden Endometriumkarzinoms spielen erhöhte, nicht durch Progesteron ausgeglichene Östrogenspiegel eine wesentliche Rolle [27]. Progesteron kontrolliert über den PGR die Zelldifferenzierung und Proliferation am Endometrium [148]. Kounelis et al. konnten basierend auf immunhistochemischen Untersuchungen nachweisen, dass eine Überexpression des Progesteronrezeptors bei endometrioiden Endometriumkarzinomen häufiger vorliegt, und die Überexpression auch signifikant mit einem prognostisch günstigen niedrigen Grading und frühem Tumorstadium assoziiert ist [95]. Unsere Studie bestätigt diese Daten auf Chromosomen- und Genexpressions-Ebene mit einer signifkant niedrigeren PGR-Expression bei den prognostisch ungünstigen seröspapillären Karzinomen im Vergleich zu den diploiden endometrioiden Tumoren. Auch auf chromosomaler Ebene konnten wir die niedrigen Expressionsraten von PGR beim UPSCA durch einen Verlust in der Chromosomenregion des PGR-Gens auf Chromosom 11 erklären. Durch Interaktion am Rezeptor moduliert Progesteron die östrogen-vermittelte Zell-Proliferation und kann somit dem mittels Östrogen stimuliertem Tumorzellwachstum entgegenwirken. Der PGR kann in zwei funktionell unterschiedlichen Isoformen vorliegen: PRA und PRB [148,149]. Decruze et al. konnten in einer Metaanalyse ein Ansprechen von Endometriumkarzinomen mit gutem bis mittlerem Differenzierungsgrad (G1-G2) auf eine primäre Progesteron-Therapie in 11-56% der Fälle zeigen. Die Ansprechrate auf die Therapie war abhängig vom positiven Rezeptorstatus. Darüber hinaus konnte eine Verbesserung der Therapieansprechrate von 8-17% auf 37-89% durch die Hormontherapie gezeigt werden [150]. Martin-Hirsch et al. zeigten in einer Metaanalyse 59 eine Reduktion der Sterberate an Endometriumkarzinomen und eine Verlängerung des krankheitsfreien Überlebens nach adjuvanter Progesteron-Therapie, allerdings mit einer erhöhten “nicht Endometriumkarzinom assoziierten” Sterberate [151]. Nach den aktuellen Leitlinien ist eine adjuvante Progesteron- (Gestagen)-Therapie beim Endometriumkarzinom aufgrund eines nicht ausreichend gesicherten Nutzens nicht indiziert. Bei typischen Vorstufen des endometrioden Endometriumkarzinoms sowie dessen prognostisch günstigen frühen Stadien und dem Wunsch nach einer fertilitätserhaltenden Therapie wird von den aktuellen Leitlinien allerdings eine Therapie mit hochdosiertem Gestagen empfohlen [51]. Unsere Daten zeigen, dass vor allem Patientinnen mit einem diploiden endometrioden Tumor von einer Progesterontherapie profitieren könnten. Die Ergebnisse können neue Ansätze bieten, um diejenigen Patientinnen besser zu selektieren, deren Tumor auf eine Hormontherapie ansprechen würde. Damit könnte gegebenenfalls mehr Frauen eine fertilitätserhaltende Therapie angeboten oder eine nebenwirkungsreiche adjuvante Chemotherapie erspart werden. Der Vergleich von aneuploiden endometrioiden mit den serös-papillären Endometriumkarzinomen ermittelte ein Netzwerk mit den unterschiedlich exprimierten Genen BIRC2 (baculoviral IAP repeat-containing 2; Chromosom 11q22), BIRC3 (baculoviral IAP repeat-containing 3; 11q22), MAP3K5 (mitogen-activated protein Kinase 5; Chromosom 6q22.33), und SAA1 (serum amyloid A1; Chromosom 11p15.1). Alle diese vier Gene stehen in Verbindung mit unterschiedlichen Krebsentitäten, aber eine direkte Verknüpfung zum Endometriumkarzinom wurde bisher nicht beschrieben. Bemerkenswerterweise waren alle Gene bei den UPSC-A-Karzinomen niedriger exprimiert. Diese Ergebnisse der Microarray-Analyse korrelieren auch in diesem Fall mit den CGH-Ergebnissen und entsprechen einem DNA-Kopien-Verlust auf Chromosom 6 und 11 in bis zu 25% der UPSC-A-Proben. Bei den EnA-Karzinomen stellte sich hingegen keine Änderung der DNA-Kopienzahl dieser Chromosomen dar. BIRC2 und BIRC3 gehören zu einer Familie von Proteinen, die über eine Bindung an TRAF1 und TRAF2 (tumor necrosis factor receptor-associated factors) den Vorgang der Apoptose verhindern können. Möglicherweise geschieht dies durch eine Störung, die zur Aktivierung der ICEähnlichen Protease führt. Die kodierten Proteine inhibieren diejenigen ApoptoseVorgänge, die durch Serum-Deprivation und Menadion (Vitamin K3), einem potenten Induktor von freien Radikalen, initiiert werden. Cheng et al. konnten zeigen, dass die Überexpression von BIRC2 möglicherweise eine Rolle bei der Brustkrebs-Karzinogense in Verbindung mit p53-Mutationen spielt [152]. Eine Überexpression von BIRC2 wurde 60 kürzlich auch mit Mammakarzinomen des Subtyps Luminal B in Verbindung gebracht [152,153]. MAP3K5 (auch bekannt als apoptosis signal-regulating kinase 1; ASK1) ist weitläufig als Schlüsselkomponente bei der Regulation von reaktiven Sauerstoffspezies (reactive oxygen species; ROS) anerkannt [154,155]. Es gibt Hinweise dafür, dass oxidativer Stress unter anderem in der Pathogenese von Prostatakarzinomen eine Rolle spielt [156]. Das Gen SAA1 schließlich kodiert das Apolipoprotein Serumamyloid A1, welches als Akut-Phase-Protein mit physiologischem Ursprungsort in der Leber in erhöhten Konzentrationen nach Traumata, bei Infektionen und Neoplasien zu finden ist. SAA1 besitzt Bindungstellen für unterschiedliche Moleküle und Stoffwechselprodukte [157]. Dem Serumamyloid A1 werden unterschiedliche Funktionen zugeschrieben, darunter die Beeinflussung von Zell-Adhäsion und -Migration, sowie Zell-Proliferation und Aggregation. Dem SAA1 kommt daher zum einen eine bedeutsame physiologische Stoffwechsel-Funktion zu, aber es bestehen auch deutlich Hinweise für seine Bedeutung in der Onkogenese [157]. Sung et al. konnten aktuell eine erhöhte Expression von SAA1 in Adenokarzinomen der Lunge nachweisen [158]. Kürzlich wurde auch eine deutliche Überexpression von SAA1 in Ovarialkarzinomen detektiert [159]. Dies ist besonders bedeutsam, da sich die seröspapillären Endometriumkarzinome, welche in unserer Studie ebenfalls SAA1 überexprimierten, im klinischen Verhalten Ovarialkarzinomen sehr ähneln. Die Prognose und selbst die operative und adjuvante Therapie der serös-papillären Endometriumkarzinome entspricht der von Ovarialkarzinomen. SAA1 bietet somit ein mögliches neues Target in der Diagnostik und Behandlung insbesondere der klinisch aggressiven serös-papillären Endometriumkarzinome. 61 6. Zusammenfassung In dieser Studie zur molekulargenetischen Charakterisierung von Subtypen des Endometriumkarzinoms mit unterschiedlicher klinischer Prognose konnten 16 diploid endometrioide, neun aneuploid endometriode und acht aneuploid serös papilläre Endometriumkarzinome mittels statischer Bildcytometrie, CGH, cDNA Microarrays und 2DE und Massenspektrometrie untersucht werden. Die Auswertung der Bildcytometrie ergab einen mittleren SSI-Wert von 26,9 bei den diploiden endometrioiden, 45,4 bei den aneuploiden endometrioiden und 53,5 bei den serös papillären Endometriumkarzinomen. Der deutlich erhöhte SSI-Wert spiegelte dabei die zunehmende genomische Instabilität der aneuploiden im Vergleich zu den diploiden Tumoren und insbesondere der aneuploiden serös-papillären Karzinomen wider (p < 0,004). Der ansteigende Grad der genomischen Instabilität zwischen den Subtypen des Endometriumkarzinoms bestätigte sich auch in der Auswertung der CGH-Ergebnisse durch eine insgesamt zunehmende Anzahl an Chromosomen-Alterationen. So stieg der ANCA-Wert von 0,041 bei den EnDKarzinomen über 0,145 bei den EnA- bis hin zu einem Maximum von 0,429 bei den UPSC-A-Karzinomen (p<0,001). Auf Ebene der Genexpression unterschieden sich die endometrioid diploiden von den endometrioid aneuploiden Endometriumkarzinomen durch 54 unterschiedlich exprimierte Gene und von den serös-papillären Karzinomen durch 76 unterschiedlich exprimierte Gene. Die endometrioid aneuploiden Endometriumkarzinome konnten von den serös-papillären Tumoren mittels Wilcoxon und stepwise Analyse in 39 unterschiedlich exprimierten Genen unterschieden werden. Von den 275 DEGs waren 114 (41,45%) auf Chromosomenregionen lokalisiert, bei denen auch Veränderungen auf chromosomaler Ebene mittels CGH dargestellt werden konnten. Insgesamt zeigte sich eine zunehmende genomische Instabilität der EnD- über die EnA- bis hin zu den UPSC-AKarzinomen, welche mit der klinischen Prognose korrelierte. Die Analysen belegten den Einfluss der Veränderungen auf chromosomaler Ebene auf die Gen- und darüber hinaus auch auf die Proteinexpression. Es konnten verschiedene Gene identifiziert werden, die durch ihr verändertes Expressionsniveau eine Unterscheidung der Subgruppen ermöglichen und in unterschiedlichen Netzwerken und Signalwegen miteinander kommunizieren. Diese Biomarker könnten sich für eine individualisierte und somit verbesserte Diagnostik und Therapie des Endometriumkarzinoms eignen, müssen aber in größeren Kohorten weiter validiert werden. Dennoch stellen diese Marker hoffnungsvolle Ziele dar, die im Rahmen patientenorientierter Medizin der Diagnostik und Therapie der klinisch sehr unterschiedlichen Subgruppen des Endometriumkarzinoms besser gerecht werden könnten. 62 7. Literaturverzeichnis 1. 2. 3. 4. 5. 6. 7. 8. 9. 10. 11. 12. 13. 14. 15. 16. 17. 18. 19. Husmann G, Kaatsch P, Katalinic A, et al. Krebs in Deutschland 2005/2006. Häufigkeiten und Trends. 7. Ausgabe. In. Berlin: Robert Koch-Institut (Hrsg) und die Gesellschaft der epidemiologischen Krebsregister in Deutschland e. V.; 2010 Jemal A, Siegel R, Xu J, Ward E. Cancer statistics, 2010. CA Cancer J Clin 2010;60:277-300 Gerling M, Meyer KF, Fuchs K, et al. High Frequency of Aneuploidy Defines Ulcerative Colitis-Associated Carcinomas: A Comparative Prognostic Study to Sporadic Colorectal Carcinomas. Annals of surgery 2010 Auer G, Eriksson E, Azavedo E, Caspersson T, Wallgren A. Prognostic significance of nuclear DNA content in mammary adenocarcinomas in humans. Cancer Res 1984;44:394-396 Lexander H, Palmberg C, Hellman U, et al. Correlation of protein expression, Gleason score and DNA ploidy in prostate cancer. Proteomics 2006;6:4370-4380 Salvesen HB, Akslen LA. Molecular pathogenesis and prognostic factors in endometrial carcinoma. Apmis 2002;110:673-689 Lundgren C, Auer G, Frankendal B, et al. Nuclear DNA content, proliferative activity, and p53 expression related to clinical and histopathologic features in endometrial carcinoma. Int J Gynecol Cancer 2002;12:110-118 Kallioniemi A, Kallioniemi OP, Sudar D, et al. Comparative genomic hybridization for molecular cytogenetic analysis of solid tumors. Science 1992;258:818-821 Ried T, Heselmeyer-Haddad K, Blegen H, Schrock E, Auer G. Genomic changes defining the genesis, progression, and malignancy potential in solid human tumors: a phenotype/genotype correlation. Genes Chromosomes Cancer 1999;25:195-204 Habermann JK, Hellman K, Freitag S, et al. A recurrent gain of chromosome arm 3q in primary squamous carcinoma of the vagina. Cancer genetics and cytogenetics 2004;148:7-13 Muresu R, Sini MC, Cossu A, et al. Chromosomal abnormalities and microsatellite instability in sporadic endometrial cancer. Eur J Cancer 2002;38:1802-1809 Muslumanoglu HM, Oner U, Ozalp S, et al. Genetic imbalances in endometrial hyperplasia and endometrioid carcinoma detected by comparative genomic hybridization. Eur J Obstet Gynecol Reprod Biol 2005;120:107-114 Upender MB, Habermann JK, McShane LM, et al. Chromosome transfer induced aneuploidy results in complex dysregulation of the cellular transcriptome in immortalized and cancer cells. Cancer Res 2004;64:6941-6949 Habermann JK, Paulsen U, Roblick UJ, et al. Stage-specific alterations of the genome, transcriptome, and proteome during colorectal carcinogenesis. Genes Chromosomes Cancer 2007;46:10-26 Scharl A. Aktuelle Empfehlungen zur Prävention, Diagnostik und Therapie primärer und fortgeschrittener Mammakarzinome. State of the Art 2011. deutsche Version 11.1.0 ed: Kommission Mamma der Arbeitsgemeinschaft Gynäkologische Onkologie e.V. in der Deutschen Gesellschaf für Gynäkologie und Geburtshilfe e.V. und Deutschen Krebsgesellschaft e.V.; 2011 Robert-Koch-Institut. Verbreitung von Krebserkrankungen in Deutschland. Entwicklung der Prävalenzen zwischen 1990 und 2010. Berlin: Robert-KochInstitut 2010 Denschlag D, Ulrich U, Emons G. The diagnosis and treatment of endometrial cancer: progress and controversies. Dtsch Arztebl Int 2011;108 Purdie DM, Green AC. Epidemiology of endometrial cancer. Best Pract Res Clin Obstet Gynaecol 2001;15:341-354 Sherman ME. Theories of endometrial carcinogenesis: a multidisciplinary approach. Mod Pathol 2000;13:295-308 63 20. 21. 22. 23. 24. 25. 26. 27. 28. 29. 30. 31. 32. 33. 34. 35. 36. 37. 38. 39. 40. 41. 42. Schiebler T, Schmidt W, Zilles K. Anatomie. Berlin, Heidelberg: Springer; 1999; 613-683 Baltzer J. Präneoplasien und Neoplasien des Endometriums. München, Jena: Bender, HG; 2001; 145-188 Bokhman JV. Two pathogenetic types of endometrial carcinoma. Gynecol Oncol 1983;15:10-17 Abal M, Planaguma J, Gil-Moreno A, et al. Molecular pathology of endometrial carcinoma: transcriptional signature in endometrioid tumors. Histol Histopathol 2006;21:197-204 Ellenson LH, Wu TC. Focus on endometrial and cervical cancer. Cancer Cell 2004;5:533-538 Lax SF, Kurman RJ. A dualistic model for endometrial carcinogenesis based on immunohistochemical and molecular genetic analyses. Verhandlungen der Deutschen Gesellschaft fur Pathologie 1997;81:228-232 Matias-Guiu X, Catasus L, Bussaglia E, et al. Molecular pathology of endometrial hyperplasia and carcinoma. Hum Pathol 2001;32:569-577 Key TJ, Pike MC. The dose-effect relationship between 'unopposed' oestrogens and endometrial mitotic rate: its central role in explaining and predicting endometrial cancer risk. Br J Cancer 1988;57:205-212 Rose PG. Endometrial carcinoma. N Engl J Med 1996;335:640-649 Davidson BJ, Gambone JC, Lagasse LD, et al. Free estradiol in postmenopausal women with and without endometrial cancer. J Clin Endocrinol Metab 1981;52:404-408 Ewertz M, Schou G, Boice JD, Jr. The joint effect of risk factors on endometrial cancer. Eur J Cancer Clin Oncol 1988;24:189-194 Kelsey JL, LiVolsi VA, Holford TR, et al. A case-control study of cancer of the endometrium. Am J Epidemiol 1982;116:333-342 Sherman ME, Sturgeon S, Brinton LA, et al. Risk factors and hormone levels in patients with serous and endometrioid uterine carcinomas. Mod Pathol 1997;10:963-968 Weiderpass E, Persson I, Adami HO, et al. Body size in different periods of life, diabetes mellitus, hypertension, and risk of postmenopausal endometrial cancer (Sweden). Cancer Causes Control 2000;11:185-192 Grady D, Gebretsadik T, Kerlikowske K, Ernster V, Petitti D. Hormone replacement therapy and endometrial cancer risk: a meta-analysis. Obstet Gynecol 1995;85:304-313 Kaufmann M, Costa S, Scharl A. Die Gynäkologie. Berlin: Springer; 2002; 2:355368 Cirisano FD, Jr., Robboy SJ, Dodge RK, et al. The outcome of stage I-II clinically and surgically staged papillary serous and clear cell endometrial cancers when compared with endometrioid carcinoma. Gynecol Oncol 2000;77:55-65 Austin H, Drews C, Partridge EE. A case-control study of endometrial cancer in relation to cigarette smoking, serum estrogen levels, and alcohol use. Am J Obstet Gynecol 1993;169:1086-1091 Berends MJ, Kleibeuker JH, de Vries EG, et al. The importance of family history in young patients with endometrial cancer. Eur J Obstet Gynecol Reprod Biol 1999;82:139-141 Lynch HT, Casey MJ, Shaw TG, Lynch JF. Hereditary Factors in Gynecologic Cancer. Oncologist 1998;3:319-338 Aaltonen LA, Salovaara R, Kristo P, et al. Incidence of hereditary nonpolyposis colorectal cancer and the feasibility of molecular screening for the disease. N Engl J Med 1998;338:1481-1487 Salovaara R, Loukola A, Kristo P, et al. Population-based molecular detection of hereditary nonpolyposis colorectal cancer. J Clin Oncol 2000;18:2193-2200 Watson P, Vasen HF, Mecklin JP, Jarvinen H, Lynch HT. The risk of endometrial cancer in hereditary nonpolyposis colorectal cancer. Am J Med 1994;96:516-520 64 43. 44. 45. 46. 47. 48. 49. 50. 51. 52. 53. 54. 55. 56. 57. 58. 59. 60. 61. 62. 63. Gerber B, Janni W, Friese K. Gynäkologische Onkologie. Ein Taschenbuch zu Epidemiologie, Ätiologie, Diagnostik, Therapie und Nachsorge. Stuttgart: Wissenschaftliche Verlagsgesellschaft mbH Stuttgart; 2006; 202-222 Lacey JV, Jr., Sherman ME, Rush BB, et al. Absolute risk of endometrial carcinoma during 20-year follow-up among women with endometrial hyperplasia. J Clin Oncol 2010;28:788-792 Sherman ME, Bur ME, Kurman RJ. p53 in endometrial cancer and its putative precursors: evidence for diverse pathways of tumorigenesis. Hum Pathol 1995;26:1268-1274 Zheng W, Khurana R, Farahmand S, et al. p53 immunostaining as a significant adjunct diagnostic method for uterine surface carcinoma: precursor of uterine papillary serous carcinoma. Am J Surg Pathol 1998;22:1463-1473 Creasman WT. Endometrial cancer: incidence, prognostic factors, diagnosis, and treatment. Semin Oncol 1997;24:S1-140-S141-150 Robertson G. Screening for endometrial cancer. Med J Aust 2003;178:657-659 Cheng WF, Lin HH, Torng PL, Huang SC. Comparison of endometrial changes among symptomatic tamoxifen-treated and nontreated premenopausal and postmenopausal breast cancer patients. Gynecol Oncol 1997;66:233-237 Karlsson B, Granberg S, Wikland M, et al. Transvaginal ultrasonography of the endometrium in women with postmenopausal bleeding--a Nordic multicenter study. Am J Obstet Gynecol 1995;172:1488-1494 Interdisziplinäre S2k-Leitlinie für die Diagnostik und Therapie des Endometriumkarzinoms. München, Wien, New York: Zuckschwerdt; 2008 Dietl J. [Actual aspects of endometrial carcinoma]. Zentralbl Gynakol 2002;124:356-361 Emons G. Endometriumkarzinom. Der Gynäkologe 2009;42 Townsend DE, Fields G, McCausland A, Kauffman K. Diagnostic and operative hysteroscopy in the management of persistent postmenopausal bleeding. Obstet Gynecol 1993;82:419-421 Leather AT, Savvas M, Studd JW. Endometrial histology and bleeding patterns after 8 years of continuous combined estrogen and progestogen therapy in postmenopausal women. Obstet Gynecol 1991;78:1008-1010 Ben-Yehuda OM, Kim YB, Leuchter RS. Does hysteroscopy improve upon the sensitivity of dilatation and curettage in the diagnosis of endometrial hyperplasia or carcinoma? Gynecol Oncol 1998;68:4-7 Boronow RC, Morrow CP, Creasman WT, et al. Surgical staging in endometrial cancer: clinical-pathologic findings of a prospective study. Obstet Gynecol 1984;63:825-832 Creasman WT, Morrow CP, Bundy BN, et al. Surgical pathologic spread patterns of endometrial cancer. A Gynecologic Oncology Group Study. Cancer 1987;60:2035-2041 Creasman WT, Odicino F, Maisonneuve P, et al. Carcinoma of the corpus uteri. J Epidemiol Biostat 2001;6:47-86 Hendrickson M, Ross J, Eifel P, Martinez A, Kempson R. Uterine papillary serous carcinoma: a highly malignant form of endometrial adenocarcinoma. Am J Surg Pathol 1982;6:93-108 Morrow CP, Bundy BN, Kurman RJ, et al. Relationship between surgicalpathological risk factors and outcome in clinical stage I and II carcinoma of the endometrium: a Gynecologic Oncology Group study. Gynecol Oncol 1991;40:5565 Benedet JL, Bender H, Jones H, 3rd, Ngan HY, Pecorelli S. FIGO staging classifications and clinical practice guidelines in the management of gynecologic cancers. FIGO Committee on Gynecologic Oncology. Int J Gynaecol Obstet 2000;70:209-262 Nguyen NP, Sallah S, Karlsson U, et al. Prognosis for papillary serous carcinoma of the endometrium after surgical staging. Int J Gynecol Cancer 2001;11:305-311 65 64. 65. 66. 67. 68. 69. 70. 71. 72. 73. 74. 75. 76. 77. 78. 79. 80. 81. 82. 83. 84. Sood BM, Jones J, Gupta S, et al. Patterns of failure after the multimodality treatment of uterine papillary serous carcinoma. Int J Radiat Oncol Biol Phys 2003;57:208-216 Shumsky AG, Brasher PM, Stuart GC, Nation JG. Risk-specific follow-up for endometrial carcinoma patients. Gynecol Oncol 1997;65:379-382 Mariani A, Webb MJ, Keeney GL, Calori G, Podratz KC. Hematogenous dissemination in corpus cancer. Gynecol Oncol 2001;80:233-238 Abeler VM, Kjorstad KE, Berle E. Carcinoma of the endometrium in Norway: a histopathological and prognostic survey of a total population. Int J Gynecol Cancer 1992;2:9-22 Mariani A, Webb MJ, Keeney GL, Aletti G, Podratz KC. Predictors of lymphatic failure in endometrial cancer. Gynecol Oncol 2002;84:437-442 Schmidt-Matthiesen H, Bastert G, Wallwiener D. Gynäkologische Onkologie Diagnostik, Therapie und Nachsorge – auf der Basis der AGO – Leitlinien. 7 ed; 2002 Nicklin JL, Copeland LJ. Endometrial papillary serous carcinoma: patterns of spread and treatment. Clin Obstet Gynecol 1996;39:686-695 Nyholm HC, Nielsen AL, Lyndrup J, Norup P, Thorpe SM. Biochemical and immunohistochemical estrogen and progesterone receptors in adenomatous hyperplasia and endometrial carcinoma: correlations with stage and other clinicopathologic features. Am J Obstet Gynecol 1992;167:1334-1342 Fukuda K, Mori M, Uchiyama M, et al. Prognostic significance of progesterone receptor immunohistochemistry in endometrial carcinoma. Gynecol Oncol 1998;69:220-225 Ehrlich CE, Young PC, Stehman FB, Sutton GP, Alford WM. Steroid receptors and clinical outcome in patients with adenocarcinoma of the endometrium. Am J Obstet Gynecol 1988;158:796-807 Kaern J, Iversen T, Trope C, Pettersen EO, Nesland JM. Flow cytometric DNA measurements in squamous cell carcinoma of the vulva: an important prognostic method. Int J Gynecol Cancer 1992;2:169-174 Bauer TW, Tubbs RR, Edinger MG, et al. A prospective comparison of DNA quantitation by image and flow cytometry. American journal of clinical pathology 1990;93:322-326 Lundgren C, Auer G, Frankendal B, Nilsson B, Nordstrom B. Prognostic factors in surgical stage I endometrial carcinoma. Acta Oncol 2004;43:49-56 Larson DM, Berg R, Shaw G, Krawisz BR. Prognostic significance of DNA ploidy in endometrial cancer. Gynecol Oncol 1999;74:356-360 Ikeda M, Watanabe Y, Nanjoh T, Noda K. Evaluation of DNA ploidy in endometrial cancer. Gynecol Oncol 1993;50:25-29 Fox H. Ploidy in gynaecological cancers. Histopathology 2005;46:121-129 Nordstrom B, Strang P, Lindgren A, Bergstrom R, Tribukait B. Carcinoma of the endometrium: do the nuclear grade and DNA ploidy provide more prognostic information than do the FIGO and WHO classifications? Int J Gynecol Pathol 1996;15:191-201 Mangili G, De Marzi P, Vigano R, et al. Identification of high risk patients with endometrial carcinoma. Prognostic assessment of endometrial cancer. Eur J Gynaecol Oncol 2002;23:216-220 Lukes AS, Kohler MF, Pieper CF, et al. Multivariable analysis of DNA ploidy, p53, and HER-2/neu as prognostic factors in endometrial cancer. Cancer 1994;73:2380-2385 Zaino RJ, Davis AT, Ohlsson-Wilhelm BM, Brunetto VL. DNA content is an independent prognostic indicator in endometrial adenocarcinoma. A Gynecologic Oncology Group study. Int J Gynecol Pathol 1998;17:312-319 Mariani A, Sebo TJ, Katzmann JA, et al. Pretreatment assessment of prognostic indicators in endometrial cancer. Am J Obstet Gynecol 2000;182:1535-1544 66 85. 86. 87. 88. 89. 90. 91. 92. 93. 94. 95. 96. 97. 98. 99. 100. 101. 102. 103. 104. 105. Konski AA, Myles JL, Sawyer T, et al. Flow cytometric DNA content analysis of paraffin block embedded endometrial carcinomas. Int J Radiat Oncol Biol Phys 1991;21:1033-1039 Podratz KC, Wilson TO, Gaffey TA, Cha SS, Katzmann JA. Deoxyribonucleic acid analysis facilitates the pretreatment identification of high-risk endometrial cancer patients. Am J Obstet Gynecol 1993;168:1206-1213; discussion 1213-1205 Susini T, Rapi S, Massi D, et al. Preoperative evaluation of tumor ploidy in endometrial carcinoma: An accurate tool to identify patients at risk for extrauterine disease and recurrence. Cancer 1999;86:1005-1012 Lane DP, Crawford LV. T antigen is bound to a host protein in SV40-transformed cells. Nature 1979;278:261-263 Linzer DI, Levine AJ. Characterization of a 54K dalton cellular SV40 tumor antigen present in SV40-transformed cells and uninfected embryonal carcinoma cells. Cell 1979;17:43-52 Lane DP. Cancer. p53, guardian of the genome. Nature 1992;358:15-16 Stiewe T. The p53 family in differentiation and tumorigenesis. Nature reviews 2007;7:165-168 Alkushi A, Lim P, Coldman A, et al. Interpretation of p53 immunoreactivity in endometrial carcinoma: establishing a clinically relevant cut-off level. Int J Gynecol Pathol 2004;23:129-137 Soong R, Robbins PD, Dix BR, et al. Concordance between p53 protein overexpression and gene mutation in a large series of common human carcinomas. Hum Pathol 1996;27:1050-1055 Lax SF, Kendall B, Tashiro H, Slebos RJ, Hedrick L. The frequency of p53, K-ras mutations, and microsatellite instability differs in uterine endometrioid and serous carcinoma: evidence of distinct molecular genetic pathways. Cancer 2000;88:814824 Kounelis S, Kapranos N, Kouri E, et al. Immunohistochemical profile of endometrial adenocarcinoma: a study of 61 cases and review of the literature. Mod Pathol 2000;13:379-388 Berchuck A, Kohler MF, Marks JR, et al. The p53 tumor suppressor gene frequently is altered in gynecologic cancers. Am J Obstet Gynecol 1994;170:246252 Zheng W, Cao P, Zheng M, Kramer EE, Godwin TA. p53 overexpression and bcl-2 persistence in endometrial carcinoma: comparison of papillary serous and endometrioid subtypes. Gynecol Oncol 1996;61:167-174 Tashiro H, Isacson C, Levine R, et al. p53 gene mutations are common in uterine serous carcinoma and occur early in their pathogenesis. Am J Pathol 1997;150:177-185 Williams JA, Jr., Wang ZR, Parrish RS, et al. Fluorescence in situ hybridization analysis of HER-2/neu, c-myc, and p53 in endometrial cancer. Exp Mol Pathol 1999;67:135-143 Mariani A, Sebo TJ, Webb MJ, et al. Molecular and histopathologic predictors of distant failure in endometrial cancer. Cancer Detect Prev 2003;27:434-441 Pisani AL, Barbuto DA, Chen D, et al. HER-2/neu, p53, and DNA analyses as prognosticators for survival in endometrial carcinoma. Obstet Gynecol 1995;85:729-734 Morrison C, Zanagnolo V, Ramirez N, et al. HER-2 is an independent prognostic factor in endometrial cancer: association with outcome in a large cohort of surgically staged patients. J Clin Oncol 2006;24:2376-2385 Atkin NB. Microsatellite instability. Cytogenet Cell Genet 2001;92:177-181 Eshleman JR, Markowitz SD. Microsatellite instability in inherited and sporadic neoplasms. Curr Opin Oncol 1995;7:83-89 Loeb LA. Microsatellite instability: marker of a mutator phenotype in cancer. Cancer Res 1994;54:5059-5063 67 106. 107. 108. 109. 110. 111. 112. 113. 114. 115. 116. 117. 118. 119. 120. 121. 122. 123. 124. 125. 126. 127. MacDonald ND, Salvesen HB, Ryan A, et al. Frequency and prognostic impact of microsatellite instability in a large population-based study of endometrial carcinomas. Cancer Res 2000;60:1750-1752 Catasus L, Machin P, Matias-Guiu X, Prat J. Microsatellite instability in endometrial carcinomas: clinicopathologic correlations in a series of 42 cases. Hum Pathol 1998;29:1160-1164 Esteller M, Levine R, Baylin SB, Ellenson LH, Herman JG. MLH1 promoter hypermethylation is associated with the microsatellite instability phenotype in sporadic endometrial carcinomas. Oncogene 1998;17:2413-2417 Koul A, Willen R, Bendahl PO, Nilbert M, Borg A. Distinct sets of gene alterations in endometrial carcinoma implicate alternate modes of tumorigenesis. Cancer 2002;94:2369-2379 Randall ME, Filiaci VL, Muss H, et al. Randomized phase III trial of wholeabdominal irradiation versus doxorubicin and cisplatin chemotherapy in advanced endometrial carcinoma: a Gynecologic Oncology Group Study. J Clin Oncol 2006;24:36-44 Maggi R, Lissoni A, Spina F, et al. Adjuvant chemotherapy vs radiotherapy in highrisk endometrial carcinoma: results of a randomised trial. British journal of cancer 2006;95:266-271 Santin AD, Bellone S, O'Brien TJ, et al. Current treatment options for endometrial cancer. Expert Rev Anticancer Ther 2004;4:679-689 Auer GU, Caspersson TO, Wallgren AS. DNA content and survival in mammary carcinoma. Anal Quant Cytol 1980;2:161-165 Kronenwett U, Huwendiek S, Ostring C, et al. Improved grading of breast adenocarcinomas based on genomic instability. Cancer Res 2004;64:904-909 Kallioniemi OP, Kallioniemi A, Piper J, et al. Optimizing comparative genomic hybridization for analysis of DNA sequence copy number changes in solid tumors. Genes Chromosomes Cancer 1994;10:231-243 Ried T, Liyanage M, du Manoir S, et al. Tumor cytogenetics revisited: comparative genomic hybridization and spectral karyotyping. J Mol Med 1997;75:801-814 Schena M, Shalon D, Davis RW, Brown PO. Quantitative monitoring of gene expression patterns with a complementary DNA microarray. Science 1995;270:467-470 Smid-Koopman E, Blok LJ, Chadha-Ajwani S, et al. Gene expression profiles of human endometrial cancer samples using a cDNA-expression array technique: assessment of an analysis method. Br J Cancer 2000;83:246-251 Southern E, Mir K, Shchepinov M. Molecular interactions on microarrays. Nat Genet 1999;21:5-9 Maughan NJ, Lewis FA, Smith V. An introduction to arrays. J Pathol 2001;195:3-6 Clarke PA, te Poele R, Workman P. Gene expression microarray technologies in the development of new therapeutic agents. Eur J Cancer 2004;40:2560-2591 Macgregor PF, Squire JA. Application of microarrays to the analysis of gene expression in cancer. Clin Chem 2002;48:1170-1177 Lockhart DJ, Winzeler EA. Genomics, gene expression and DNA arrays. Nature 2000;405:827-836 Bertucci F, Houlgatte R, Nguyen C, et al. Gene expression profiling of cancer by use of DNA arrays: how far from the clinic? Lancet Oncol 2001;2:674-682 Duggan DJ, Bittner M, Chen Y, Meltzer P, Trent JM. Expression profiling using cDNA microarrays. Nat Genet 1999;21:10-14 Korn EL, Habermann JK, Upender MB, Ried T, McShane LM. Objective method of comparing DNA microarray image analysis systems. BioTechniques 2004;36:960967 Cleveland W. Robust Locally Weighted Fitting and Smoothing Scatterplots. . Journal of the American Statistical Association 1979;74:829-836 68 128. 129. 130. 131. 132. 133. 134. 135. 136. 137. 138. 139. 140. 141. 142. 143. 144. 145. 146. Xiong M, Li W, Zhao J, Jin L, Boerwinkle E. Feature (gene) selection in gene expression-based tumor classification. Molecular genetics and metabolism 2001;73:239-247 Kauraniemi P, Hautaniemi S, Autio R, et al. Effects of Herceptin treatment on global gene expression patterns in HER2-amplified and nonamplified breast cancer cell lines. Oncogene 2004;23:1010-1013 Gemoll T, Habermann JK, Lahmann J, et al. Protein profiling of genomic instability in endometrial cancer. Cell Mol Life Sci 2011 Kronenwett U, Ploner A, Zetterberg A, et al. Genomic instability and prognosis in breast carcinomas. Cancer Epidemiol Biomarkers Prev 2006;15:1630-1635 Suzuki A, Fukushige S, Nagase S, et al. Frequent gains on chromosome arms 1q and/or 8q in human endometrial cancer. Hum Genet 1997;100:629-636 Suehiro Y, Umayahara K, Ogata H, et al. Genetic aberrations detected by comparative genomic hybridization predict outcome in patients with endometrioid carcinoma. Genes Chromosomes Cancer 2000;29:75-82 Micci F, Teixeira MR, Haugom L, et al. Genomic aberrations in carcinomas of the uterine corpus. Genes Chromosomes Cancer 2004;40:229-246 Levan K, Partheen K, Osterberg L, Helou K, Horvath G. Chromosomal alterations in 98 endometrioid adenocarcinomas analyzed with comparative genomic hybridization. Cytogenetic and genome research 2006;115:16-22 Wulfkuhle J, Espina V, Liotta L, Petricoin E. Genomic and proteomic technologies for individualisation and improvement of cancer treatment. Eur J Cancer 2004;40:2623-2632 Nordstrom B, Strang P, Bergstrom R, Nilsson S, Tribukait B. A comparison of proliferation markers and their prognostic value for women with endometrial carcinoma. Ki-67, proliferating cell nuclear antigen, and flow cytometric S-phase fraction. Cancer 1996;78:1942-1951 O'Connor T, Ireland LS, Harrison DJ, Hayes JD. Major differences exist in the function and tissue-specific expression of human aflatoxin B1 aldehyde reductase and the principal human aldo-keto reductase AKR1 family members. The Biochemical journal 1999;343 Pt 2:487-504 Picklo MJ, Sr., Olson SJ, Hayes JD, Markesbery WR, Montine TJ. Elevation of AKR7A2 (succinic semialdehyde reductase) in neurodegenerative disease. Brain research 2001;916:229-238 Choi YL, Bocanegra M, Kwon MJ, et al. LYN is a mediator of epithelialmesenchymal transition and a target of dasatinib in breast cancer. Cancer Res 2010;70:2296-2306 Rodrigo Tapia JP, Pena Alonso E, Garcia-Pedrero JM, et al. [Annexin A2 expression in head and neck squamous cell carcinoma]. Acta otorrinolaringologica espanola 2007;58:257-262 Mai J, Waisman DM, Sloane BF. Cell surface complex of cathepsin B/annexin II tetramer in malignant progression. Biochimica et biophysica acta 2000;1477:215230 Emoto K, Sawada H, Yamada Y, et al. Annexin II overexpression is correlated with poor prognosis in human gastric carcinoma. Anticancer research 2001;21:13391345 Emoto K, Yamada Y, Sawada H, et al. Annexin II overexpression correlates with stromal tenascin-C overexpression: a prognostic marker in colorectal carcinoma. Cancer 2001;92:1419-1426 Inoue M, Isomura M, Ikegawa S, et al. Isolation and characterization of a human cDNA clone (GCN5L1) homologous to GCN5, a yeast transcription activator. Cytogenet Cell Genet 1996;73:134-136 Miller C, Sassoon DA. Wnt-7a maintains appropriate uterine patterning during the development of the mouse female reproductive tract. Development (Cambridge, England) 1998;125:3201-3211 69 147. 148. 149. 150. 151. 152. 153. 154. 155. 156. 157. 158. 159. Kitajewski J, Sassoon D. The emergence of molecular gynecology: homeobox and Wnt genes in the female reproductive tract. Bioessays 2000;22:902-910 Smid-Koopman E, Kuhne LC, Hanekamp EE, et al. Progesterone-induced inhibition of growth and differential regulation of gene expression in PRA- and/or PRB-expressing endometrial cancer cell lines. Journal of the Society for Gynecologic Investigation 2005;12:285-292 Gielen SC, Hanekamp EE, Hanifi-Moghaddam P, et al. Growth regulation and transcriptional activities of estrogen and progesterone in human endometrial cancer cells. Int J Gynecol Cancer 2006;16:110-120 Decruze SB, Green JA. Hormone therapy in advanced and recurrent endometrial cancer: a systematic review. Int J Gynecol Cancer 2007;17:964-978 Martin-Hirsch PL, Jarvis G, Kitchener H, Lilford R. Progestagens for endometrial cancer. Cochrane database of systematic reviews (Online) 2000:CD001040 Cheng L, Zhou Z, Flesken-Nikitin A, et al. Rb inactivation accelerates neoplastic growth and substitutes for recurrent amplification of cIAP1, cIAP2 and Yap1 in sporadic mammary carcinoma associated with p53 deficiency. Oncogene 2010;29:5700-5711 Frasor J, Weaver A, Pradhan M, et al. Positive cross-talk between estrogen receptor and NF-kappaB in breast cancer. Cancer Res 2009;69:8918-8925 Matsuzawa A, Ichijo H. Stress-responsive protein kinases in redox-regulated apoptosis signaling. Antioxidants & redox signaling 2005;7:472-481 Matsuzawa A, Saegusa K, Noguchi T, et al. ROS-dependent activation of the TRAF6-ASK1-p38 pathway is selectively required for TLR4-mediated innate immunity. Nature immunology 2005;6:587-592 Dakubo GD, Parr RL, Costello LC, Franklin RB, Thayer RE. Altered metabolism and mitochondrial genome in prostate cancer. Journal of clinical pathology 2006;59:10-16 Urieli-Shoval S, Linke RP, Matzner Y. Expression and function of serum amyloid A, a major acute-phase protein, in normal and disease states. Current opinion in hematology 2000;7:64-69 Sung HJ, Ahn JM, Yoon YH, et al. Identification and validation of SAA as a potential lung cancer biomarker and its involvement in metastatic pathogenesis of lung cancer. Journal of proteome research 2011;10:1383-1395 Urieli-Shoval S, Finci-Yeheskel Z, Dishon S, et al. Expression of serum amyloid a in human ovarian epithelial tumors: implication for a role in ovarian tumorigenesis. J Histochem Cytochem 2010;58:1015-1023 70 8. Anhang 8.1. Protokolle 8.1.1. Statische Bild-Cytometrie 8.1.1.1. Feulgen – Färbung A. Ansetzen des Schiffs-Reagenz 1. 150 ml 1 M HCL in eine Flasche (2l) mit dunklem Glas füllen, dazu 5 g Pararosanilin (Basic Fuchsin, Aldrich, 85,734-3) geben und die Flasche leicht schütteln, bis das Pulver gelöst erscheint, 2. dann eine Lösung aus 5 g Kaliumpyrosulfit (K2S2O5) und 850 ml Aqua dest. hinzufügen und die Flasche wiederum leicht schütteln. 3. Erscheint die Lösung nun rot gefärbt, die Flasche in Alufolie einwickeln und im Dunkeln über Nacht bei Raumtemperatur stehen lassen. 4. Alufolie entfernen, der angesetzten Lösung 3 g Aktivkohle hinzufügen, und die Flasche mindestens 4 min. gut schütteln, 5. die Lösung 2-mal filtrieren, zuletzt in eine Literflasche aus weißem Glas. 6. Erscheint die Lösung jetzt rosafarben transparent, wurde das Schiffs-Reagenz richtig angesetzt. Die Lösung kann in Alufolie eingewickelt bei 4 °C im Kühlschrank aufbewahrt werden. Diese Lösung ist bis zu 3 Tagen verwendbar. B. Paraffinschnitte: 1. Deparaffinierung: für 2-mal 5 min. im Xylolbad, 2. Rehydrierung der Schnitte in Ethanol absteigender Konzentration für jeweils 5 min. (100%,100%,95%,95%,70%). 3. Abspülen der Schnitte in Aqua dest. 4. Paraffinschnitte und luftgetrocknete Imprints refixieren über Nacht bei Raumtemperatur in 4% Formaldehyd. C. Feulgen-Färbung 1. Präparate vorsichtig in Leitungswasser spülen, bis die Küvetten nicht mehr nach Formaldehyd riechen. 2. Präparate in 5 M HCl 60 min. bei Raumtemperatur sauer hydrolysieren. 3. 3-mal vorsichtig in Aqua dest. spülen. 71 4. Inkubation im Schiffs-Reagenz für 120 min. im Dunkeln. 5. Vorsichtiges Spülen bis Spülflüssigkeit sich nicht mehr verfärbt, 6. für 3x10 min. in frischer Na2S2O5-Lösung waschen, 7. 5 min. unter fließendem Leitungswasser abwaschen, 8. Dehydratation der Präparate für je 5 min. in Ethanol aufsteigender Konzentration (70%, 95%, 95%, 100%, 100%). 9. Reinigung im Xylolbad und danach Eindeckeln der Präparate mit Entellan. 8.1.2. CGH 8.1.2.1. DNA-Extraktion aus Blut Lyse-Puffer 155 mM NH4Cl 8,29 g 10 mM KHCO3 1g 0,1 mM Na2EDTA 0,034 g oder 200 µl EDTA 0,5 M; 1 l Puffer mit 1 M HCl oder NaOH auf pH 7,4 justieren SE-Puffer 75 mM NaCl 4,39 g 25 mM Na2EDTA 8,41 g oder 50 ml EDTA 0,5 M; 1l TE-Puffer 10 mM Tris 1,21 g 1 mM Na2EDTA 0,34 g; 1l Puffer mit 1 M HCl auf pH 8,0 justieren 1. 10 ml Vollblut einem gesundem Spender entnehmen (EDTA Röhrchen, Heparin oder Citrat als Antikoagulanz), 2. 30 ml Lyse-Puffer hinzufügen und vorsichtig schütteln, 30 min. auf Eis inkubieren und dann 10 min. mit 1200 rpm zentrifugieren (4°C). 3. Überstand verwerfen, 10 ml Lyse-Puffer hinzufügen und das Zellpellet vorsichtig resuspendieren, erneut 10 min. bei 1200 rpm zentrifugieren (4°C). 4. Überstand verwerfen, das Pellet in 5 ml SE-Puffer resuspendieren und 10 min. bei 1200 rpm (4°C) zentrifugieren. 72 5. Überstand verwerfen, das Pellet in 5 ml SE-Puffer resuspendieren, 40 µl Proteinase K (10 mg/ml) und 250 µl 20% SDS hinzufügen, vorsichtig schütteln und bei 37°C über Nacht inkubieren. 6. 5 ml SE-Puffer und 10 ml Phenol hinzufügen, 10 min. schütteln, dann 5 min. bei 3000 rpm (10°C) zentrifugieren, 7. den Überstand in ein neues Gefäß überführen, 5 ml Phenol und 5 ml Chloroform/Isoamylalcohol (24:1) hinzufügen, die Lösung 10 min. schütteln, dann 5 min. bei 3000 rpm zentrifugieren (10 °C). 8. Den Überstand in ein neues Gefäß übertragen, 10 ml Chloroform/Isoamylalkohol (24:1) hinzugeben und 10 min. schütteln, dann 5 min bei 3000 rpm zentrifugieren (10 °C), 9. den Überstand wiederum in ein neues Gefäß füllen, 300 µl 3 M Sodiumacetat (pH 5,2) (1/10 des Volumens) und 10 ml Isopropanol (3-mal das Volumen) hinzugeben, die Lösung vorsichtig bewegen, bis die DNA präzipitiert, die DNA vorsichtig mit Hilfe einer Pipettenspitze in ein neues Gefäß übertragen. 10. DNA in 30 ml 70% Ethanol auf dem Shaker waschen (2 h), die DNA mit 1 ml 70 % Ethanol in ein Eppendorfgefäß überführen und bei 14000 rpm zentrifugieren (4°C), danach das Pellet im Speed Vac trocknen, DNA in 0,5-1 ml TE-Puffer oder sterilem Wasser über Nacht auf dem Shaker lösen (4°C), 11. DNA Konzentration messen. 8.1.2.2. DNA-und RNA-Extraktion aus tiefgefrorenem Material 1. Tiefgefrorene Probe in 6 ml Trizol auftauen und mittels Homogenizer zerkleinern, 2. 6 ml Trizol hinzufügen, 3. Gefäß 15 sek. schütteln, danach 5 min. bei Raumtemperatur ruhen lassen. 4. 2,4 ml Chloroform hinzufügen und die Lösung schütteln, bis sie homogen erscheint, 5. wenn die Phasentrennung deutlich sichtbar wird, (nach ca. 15 min. Ruhe bei Raumtemperatur) die Lösung bei 8000 rpm (4C°) 15 min. zentrifugieren. a) RNA-Extraktion: 1. Die obere klare Phase in ein 50 ml-Gefäß überführen, 2. unter ständigem Schütteln, tropfenweise 2 ml 100% Ethanol hinzufügen, 3. die Hälfte der Lösung auf eine Reinigungssäule (Quiagen) übertragen und bei 5500 rpm kurz anzentrifugieren (die Zentrifuge sollte während der nächsten Schritte langsam auf Raumtemperatur aufwärmen). 73 4. Den Überstand in ein neues Gefäß überführen und die zweite Hälfte auf die Säule geben, 5. die Säule bei 5500 rpm kurz anzentrifugieren, 6. den gesamten Überstand erneut auf die Säule geben und zentrifugieren. 7. Den Überstand verwerfen, die RNA befindet sich nun in der Säulenmatrix. 8. Säule zurück in das Gefäß setzen und ca. 5 ml RW1-Puffer (Quiagen) hinzufügen (zur Reduktion der Proteinverunreinigung). 9. Die Säule für 5 min. bei 5000 rpm zentrifugieren, 10. Flüssigkeit verwerfen und Schritte 8 und 9 wiederholen. 11. Ca. 5 ml RPE-Puffer (Quiagen) auf die Säule geben und für 5 min. bei 5000 rpm zentrifugieren. 12. Flüssigkeit verwerfen und Schritt 11. wiederholen mit einer Zentrifugationszeit von 10 min., 13. Säule in ein neues Gefäß setzen, 14. 500 µl DEPC-behandeltes Wasser auf die Säule geben und die Säule 1 min. bei Raumtemperatur stehen lassen. 15. Die Säule 15 min. bei 5000 rpm zentrifugieren; Flüssigkeit NICHT verwerfen, diese enthält die RNA!! 16. Schritte 14.-15. 3-mal wiederholen, beim 3. mal 10 min. zentifugieren. 17. Flüssigkeit sorgfältig in ein neues Gefäß überführen. 18. 1/10 des Volumens (ca. 200 µl) 3M Na Acetat (pH 5,5) hinzufügen, 19. das Doppelte des Volumens (ca. 4 ml) 100% Ethanol hinzufügen und mixen, 20. RNA prezipitieren lassen, bei –80 °C für 30 min., oder vorzugsweise bei –30 °C über Nacht. 21. RNA bei 8000 rpm 30 min. zentrifugieren (4°C), Überstand verwerfen, 22. RNA-Pellet mit 1000 µl Ethanol (75%) waschen und in ein Eppendorfgefäß überführen. 23. RNA bei 1400 rpm 15 min. zentrifugieren (4°C), Überstand vorsichtig entfernen, 24. RNA-Pellet im offenen Gefäß trocknen (nicht länger als 30 min. in RNAse freier Umgebung). 25. Pellet mit DEPC-behandeltem Wasser resuspendieren (10 min. Raumtemperatur, 10 min. bei 55 °C und 5 min. auf Eis). 26. Die RNA kann bei –80 C° aufbewahrt werden. b) DNA-Extraktion: 1. noch verbliebende RNA-Reste aus dem Gefäß entfernen, 74 bei 2. ca. 3,6 ml Ethanol (100%) hinzufügen (0,3 ml Ethanol pro 1 ml Trizol) und kräftig schütteln. 3. Die Lösung 5 min. bei Raumtemperatur stehen lassen, dann 15 min. bei 8000 rpm zentrifugieren (4 °C). 4. Der Überstand kann zur Proteinanalyse aufbewahrt werden. 5. DNA-Pellet in 12 ml 0,1 M Sodium Citrat/10% Ethanol waschen (1 ml pro 1 ml Trizol), 6. die Lösung gut mixen und 30 min. bei Raumtemperatur stehen lassen, zwischendurch mehrmals mixen. 7. 30 min. bei 8000 rpm zentrifugieren (4°C), 8. Überstand verwerfen 9. DNA-Pellet in 1000 µl 0,1 M Sodium Citrat/10% Ethanol lösen und in ein Eppendorfgefäß überführen, 10. mixen und 30 min. bei Raumtemperatur stehen lassen, zwischenzeitlich schütteln, 11. die Lösung 15 min. bei 1400 rpm zentrifugieren (4°C). 12. Überstand verwerfen, DNA-Pellet ca. 15 min. trocknen lassen, 13. DNA-Pellet in sterilem Wasser (ca. 300-500 µl) bei 37°C über Nacht resuspendieren, 14. die DNA 2 min. bei 1400 rpm zentrifugieren (4°C), 15. DNA abpipettieren und in ein neues Eppendorfgefäß überführen. 16. Die DNA kann bei –30 °C aufbewahrt werden. 8.1.2.3. Nick-Translation DNTP 100 mM dATP, dCTP und dGTP jeweils 5 µl 100 mM dTTP 1 µl steriles Wasser 984 µl vermischen und bei –20°C aufbewahren Dnase I Lösung Dnase I 10 mg NaCl, 1 M 1,5 ml Glycerol 5 ml mit sterilem Wasser auf 10 ml auffüllen, mischen und bei –20°C lagern 75 10x NT-Puffer Tris-HCl, 1 M, pH 8,0 500 µl Mg Cl2, 0,5 M 100 µl BSA, 10 mg/ml 50 µl vermischen und bei –20°C aufbewahren 1. Für jede DNA-Probe in einem Eppendorfgefäß ansetzen: 2 µg DNA, 10 µl 10xNT-Puffer, 10 µl dNTP, 10 µl 0,1 M b-Mercaptoethanol, 4 µl BIO-16-dUTP oder 4 µl DIG-11-dUTP (1 mM), x µl steriles Wasser (inklusive der Reagenzien aus Schritt drei sollten 100µl zusammenkommen). 2. Die Reagenzien mischen und die Gefäße auf Eis setzen. 3. 3-8 µl Dnase (1 mg/ml) 1:1000 und 3 µl Polymerase (Kronberg) hinzfügen, 4. DNA bei 15°C für 1,5-2 h inkubieren. 5. Gelelektrophorese: jeweils 5 µl der Probe mit dem DNA-Marker Lambda HAES III auf dem Gel laufen lassen, die ideale Länge der DNA nach der Nick Translation sind 500-900 bpm. 6. Falls die DNA zu lang ist, erneut Dnase hinzugeben und bei 15°C für 10-30 min. inkubieren und die Länge überprüfen, 7. die Nick-Translation stoppen mit jeweils 1 µl 0,5 M EDTA und Inkubation über 10 min. bei 65°C. 8. Die Proben können bei –20°C aufbewahrt werden. 8.1.2.4. Präparation von Metaphasen RPMI 1640 Full Medium RPMI Medium 1640 500 ml L-Glutamine-200 mM, 100x 5 ml Penicillin/Streptomycin 5000 U/ml/5000 µg/ml 10 ml oder Antibiotic-Antimycotic, 100x 5 ml Fetal Bovine Serum Qualified, hitzeinaktiviert 100 ml 76 Medium mit einem 0,22 µ Filter sterilisieren, das Medium ist dann für 2-3 Wochen bei 4°C haltbar. Hypotone Lösung KCI 5,6 g Destilliertes Wasser 1000 ml Fixationslösung Methylalkohol/eisgekühlte Acetatsäure 3:1 1. 3-5 ml Blut einem gesundem Spender entnehmen. 2. 0,7 ml davon in einer T25 Zellkulturflasche mit 10 ml RPMI 1640, sowie 0,2 ml Phythohämaglutinin mischen und über 72 h bei 37°C im Zellinkubator inkubieren, 3. 100 µl Colcemid hinzufügen, mischen und für 30 min. bei 37°C erneut inkubieren, 4. Überführung der Zellkultur in 15 ml Zentrifugationsgefäße und Zentrifugation bei 1200 rpm für 5 min., 5. bis auf ca. 0,5 ml oberhalb des Pellets den Überstand verwerfen. 6. Das Zellpellet im Überstand resuspendieren und ca. 2 ml 0,075 M KCl (37°C) tropfenweise hinzugeben, unterdessen das Gefäß vorsichtig schwenken, 7. weitere 8 ml KCl hinzugeben und sorgfältig mischen, 8. die Zellen 15 min. im Wasserbad (37°C) inkubieren. 9. Einige Tropfen der Fixationslösung hinzugeben und gut mischen. 10. Zellen erneut Zentrifugieren und Überstand verwerfen (s.h. Punkt 5). 11. Das Pellet in 10 ml Fixationslösung resuspendieren, dabei die ersten 2 ml unter vorsichtigem Schwenken tropfenweise hinzugeben, 12. nach 10 min. Inkubationszeit die Zellen erneut zentrifugieren und den Überstand entfernen (s.h. Punkt 5). 13. Den Fixationsprozess 2-3-mal wiederholen. 14. Nach der letzten Zentrifugation die Zellen in einer kleinen Menge Fixationslösung resuspendieren und auf gereinigte Objektträger tropfen (nicht verwendete Zellen bei –20°C aufbewahren). 8.1.2.5. Vorbehandlung der Methaphasen Pepsin Vorratslösung (100 mg/ml) 5 g Pepsin in 50 ml sterilem Wasser auflösen und bei –20°C aufbewahren. 77 1x PBS/MgCl2 1 M MgCl2 50 ml 1x PBS 950 ml Rnase-Lösung 20 mg/ml Rnase in sterilem Wasser auflösen und 15 min. kochen, auf Raumtemperatur abkühlen lassen und bei –20°C aufbewahren. 1% Formaldehyde/1x PBS/1 M MgCl2 Formaldehyde (37°C) 2,7 ml 1xPBS/1 M MgCl2 97,3 ml auf 37°C vorwärmen 0,01 M HCL 1 M HCL 1 ml dH2O 99 ml auf pH 2,0 adjustieren und auf 37°C vorwärmen 1. Objektträger in einem Gefäß mit 2xSSC 10 min. auf dem Rotomix waschen, 2. Rnase Vorratslösung mit 2xSSC verdünnen (1:200), 3. 100-200 µl Rnase auf den Objektträger auftragen und mit einem Deckglas abdecken. 4. Objektträger in einer Feuchtkammer bei 37°C inkubieren (45 min.). 5. Deckglas vorsichtig entfernen und den Objektträger 3-mal 5 min. in 2xSSC waschen, 6. 1,5 µl der Pepsinlösung mit 100 ml vorgewärmten 0,01 M HCL mischen, das Gefäß ins Wasserbad stellen (37°C). 7. Objektträger für 1,5 min. in dem Gefäß inkubieren, 8. Objektträger 2-mal 5 min. in 1xPBS auf dem Rotomix waschen, 9. Objektträger 1-mal 5 min. in 1xPBS/1 M MgCl2 waschen, 10. Objektträger in vorgewärmten (37°C) Formaldehyd/1xPBS/1 M MgCl2 10 min. inkubieren. 11. In 1xPBS 5 min. auf dem Rotomix waschen. 12. Methaphasen in einer Ethanolserie aufsteigender Konzentration (70%, 90%, 100%) dehydrieren, für jeweils 3 min. auf dem Rotomix, 13. Objektträger trocknen lassen. 78 8.1.2.6. DNA Präzipitation und Hybridisierung Master Mix Dextran sulfat, 50% 40 ml 20X SSC, pH 7,0 10 ml steriles Wasser 50 ml sorgfältig mischen und über Nacht auf einem Shaker belassen. 70% Formamid/2xSSC deionisiertes Formamid 70 µl 20x SSC 3 µl steriles Wasser 27 µl auf pH 7,5 justieren Folgende Substanzen in einem Eppendorfgefäß mischen: a) 1-2 µg Nick-DNA (Biotin markiert), b) 1-2 µg Nick-Kontroll-DNA (Digoxin markiert), c) 60 µl Human Cot-1 DNA (1 mg/ml), d) 10 µl salmon sperm DNA (10 mg/ml), e) 1/10 des Volumens an Na-Acetat hinzugeben, f) 2,5-3,0-mal des Volumens an Ethanol (100%) hinzugeben, 1. mischen und bei –20°C über Nacht, oder –80°C für 30 min. inkubieren. 2. Proben zentrifugieren (14000 rpm), bei 4°C 30 min., 3. Überstand verwerfen und das Pellet im Speed-Vac etwa 5 min. trocknen. 4. 6 µl deionisiertes Formamid (pH 7,5) hinzufügen und bei 37°C 30 min. auf dem Shaker inkubieren, währenddessen mehrmals schütteln. 5. 6 µl Master Mix zu der Reaktion dazugeben und mischen. 6. DNA bei 80°C über 5 min. denaturieren. 7. Proben bei 37°C für 1,5 h inkubieren. 8. Denaturierung der Metaphasenobjekträger mit 100 µl 70% Formamide/2XSSC welches mittels eines Deckglases aufgetragen wird, 9. Metaphasen bei 75°C für exakt 1,5 min. inkubieren, 10. Deckglas entfernen und den Objektträger sofort in eisgekühlten Ethanol (70%) tauchen, nach 3 min. zunächst in 90% Ethanol, dann 3 min. in 100% Ethanol tauchen, trocknen lassen. 11. Die DNA auf die Metaphasenobjektträger auftragen, mit einem Deckglas überdecken und mit Klebstoff abdichten. 79 12. Zur Hybridisierung die Objektträger für mindestens 48 h in einer Feuchtkammer bei 37°C inkubieren. 8.1.2.7. Detektion FA/SCC 20xSSC 30 ml dH2O 120 ml Formamid 150 ml Durch Zusatz von 1 M HCl auf pH 7,0-7,5 justieren und Lösung auf 37°C vorwärmen. 4xSSC/Tween 20 20xSSC 100 ml dH2O 400 ml Tween 20 0,5 ml Lösung auf 45°C vorwärmen. 0,1xSSC 20xSSC 2,5 ml dH2O 497,5 ml Lösung auf 60°C vorwärmen. Block-Lösung (3% BSA) Serum Albumin (Rind) 0,3 g 4xSSC/Tween 20 (37°C) 10 ml mixen, bis das Pulver bei 37°C gut gelöst ist. DAPI-Lösung (80 ng/ml DAPI in 2xSCC) DAPI (4',6-diamidino-2-phenylindole) 2 mg Steriles H2O 10 ml Arbeitslösung: DAPI-Lösung 40 µl 2xSSC 100 ml Antikörper: 1. Schicht avidin-FITC (1:200) und Maus-anti-DIG (1:100) 2. Schicht biotinylated anti-avidin (1:200) und Kaninchen-anti-Maus TRITC (1:200) 3. Schicht avidin-FITC (1:200) 4. Schicht Ziege-anti-Kaninchen TRITC (1:200) 80 1. Klebstoff und Deckglas von dem hybridisierten Objektträger entfernen, 2. Objektträger 3-mal 5 min. in FA/SCC auf dem Rotomix waschen, 3. Objektträger 3-mal 5 min. in 0,1xSCC auf dem Rotomix waschen, 4. Objektträger in 4xSSC/Tween 20 tauchen und feucht halten, 5. 100-140 µl Block-Lösung auf ein Deckglas geben und den Objektträger mit der hybridisierten Oberfläche vorsichtig auf das Deckglas drücken, Objektträger in einer Feuchtkammer bei 37°C 30 min. inkubieren. 6. Objektträger in 4xSSC/Tween 20 tauchen. 7. In Eppendorfgefäßen jeweils 200 µl 1% BSA-Lösung mit 1 µl des jeweiligen Antikörpers mischen, 8. ca. 100 µl der Antikörperlösung auf ein Deckglaß geben und Objekträger vorsichtig damit berühren, Objekträger in einer Feuchtkammer bei 37°C 60 min. inkubieren. 9. Objektträger in 4xSCC/Tween 20 3-mal 5 min. auf dem Rotomix waschen. 10. Weitere Antikörperschichten auftragen wie in Punkt 8. und 9. beschrieben, 11. Objektträger 7 min. in die DAPI-Lösung geben (unter Lichtausschluss), 12. Objektträger 10 min. in 2xSCC waschen (unter Lichtausschluss), 13. 30-35 µl Antifade mittels Deckglas auf den Objekträger aufbringen und die Objektträger bis zur Auswertung im Dunkeln aufbewahren. 8.1.3. cDNA-Microarray 8.1.3.1. Reverse Transkriptase Reaktion 1. Ca. 30 µg RNA mit DEPC behandeltem Wasser auf 16,4 µl auffüllen, 2. 3 µg/µl Random Hexamer hinzufügen, 3. die RNA bei 70 C° 10 min. inkubieren, dann auf ca. 42 °C abkühlen lassen, 4. 6 µl 5X Strand-Puffer hinzufügen, 5. 3 µl 0,1 M DTT hinzufügen, 6. 0,6 µl 50X aa-dNTP ergänzen, 7. 2 µl Reverse Transkriptase dazugeben, mixen und kurz zentrifugieren, 8. die Lösung 3 h bei 42 °C inkubieren. 8.1.3.2. Amino-allyl-Bindung 1. Zu jeder cDNA Probe jeweils 5 µl 500 mM EDTA (pH 8,0) hinzugeben, mixen und kurz zentrifugieren, 81 2. 10 µl 1 M NaOH hinzufügen, 3. Reaktion bei 65 °C 20 min. inkubieren, dann auf Raumtemperatur abkühlen lassen und kurz zentrifugieren. 4. Zur Neutralisation 10 µl 1 M HCl hinzufügen, mixen und kurz zentrifugieren. 8.1.3.3. Reinigung der Proben mit PCR-Säulen (Qiagen) 1. 300 µl PB-Puffer zur Reaktion hinzufügen und die Probe auf die Säule geben, 2. die Säule 1 min. bei 1000 rpm, dann 1 min. bei maximaler Geschwindigkeit zentrifugieren. 3. Überstand verwerfen und 740 µl Phosphat-Puffer auf die Säule geben. 4. 1 min. bei maximaler Geschwindigkeit zentrifugieren, 5. Überstand verwerfen und Waschschritte 3 und 4 wiederholen, 6. Eluat verwerfen und Säule für 1 min. bei maximaler Geschwindigkeit zentrifugieren. 7. Die Säule in ein neues Gefäß setzen, 30 µl Elutionspuffer hinzufügen und 1 min. inkubieren. 8. 1 min. bei maximaler Geschwindigkeit zentrifugieren. 9. Elution mit 30 µl Elutionspuffer wiederholen, 10. cDNA (Überstand) in ein neues Gefäß überführen und ca. 45 min. im Speed-Vac trocknen. 8.1.3.3. Cy dye Kopplung 1. Das cDNA-Pellet in 4,5 µl 0,1 M Carbonat-Puffer (pH 9,0) resuspendieren. 2. 4,5 µl NHS CY dyes hinzugeben (Cy5 für die Kontroll c-DNA; Cy3 für die Tumor cDNA), 3. Reaktion bei Raumtemperatur unter Lichtausschluss 1 h inkubieren. 8.1.3.4. Reinigung der Proben 1. 35 µl 100 mM Natrium Acetat (pH 5,2) zur Reaktion hinzufügen, 2. 250 µl PB Puffer dazugeben und dem QIAquick PCR Protokoll (Qiagen) folgen. 3. Probe auf die Säule auftragen und für 1 min. zentrifugieren, 4. Eluat verwerfen, 5. 0,75 ml PE Puffer hinzugeben und 1 min. zentrifugieren, 6. Eluat verwerfen und 1 min. mit maximaler Geschwindigkeit zentrifugieren 82 7. Säule in ein neues Gefäß setzen. 8. Probe zweimal mit je 30 µl Elutionspuffer auswaschen (je 1 min. Inkubationszeit und 1 min Zentrifugation). 9. 1 µl zur Analyse der Farbstoff-Inkorporation entnehmen. 10. Probe ca. 40 min. im Speed-Vac trocknen. 8.1.3.5. Farbstoff - Inkorporationsanalyse 1. 1 µl der Probe auf 1:200 verdünnen, 2. die Absorption bei 260 nm, 550 nm (Cy3) und 650 nm (Cy5) mittels Spectrometer bestimmen. 8.1.3.6. Prehybridisation 1. Objektträger in der Kürette mit Prehybridisationspuffer bei 42 °C für 45 min. inkubieren, 2. dann in DEPC H2O ca. 2 min. auf dem Rotomix bei Raumtemperatur waschen, 3 ca. 2 min. auf dem Rotomix bei Raumtemperatur in Isopropanol waschen, 4. Slides trocknen lassen und möglichst sofort hybridisieren. 5. Die Enden der Hybridisierungskammern mit 35 µl H2O befüllen und Objektträger hineinlegen. 8.1.3.7. Hybridisation 1. Proben jeweils in 12 µl 1xHybridisierungspuffer resuspendieren, 2. jeweils eine Cy3 und eine Cy5 markierte Probe miteinander gut mischen, 3. 2 µl COT1-DNA (10µg/µl) hinzufügen, 4. 2 µl Poly (A)-DNA (10µg/µl) hinzugeben, 5. Probe bei 95 °C 3 min. denaturieren. 6. Probe 1 min. bei maximaler Geschwindigkeit zentrifugieren. 7. Markierte Probe auf die prähybridisierte Microarrays auftragen und mit einem Deckglas bedecken. 8. Abgedichtete Hybridisierungskammer im Wasserbad bei 42°C inkubieren (16-20 h). 83 8.1.3.8. Waschen der Array 1. Array aus der Hybridisierungskammer nehmen und 2-mal 10 min. bei 42 °C im Waschpuffer (low-stringency) auf dem Rotomix waschen. 2. Vorsichtig das Deckglas während des Waschvorgangs lösen lassen, 3. Slide 2-mal 10 min. bei Raumtemperatur im Waschpuffer (high-stringency) auf dem Rotomix waschen. 4. Slide 2-mal 10 min. bei Raumtemperatur in 0,1SSC auf dem Rotomix waschen. 5. Arrays in der Zentrifuge bei 500 rpm 3 min. trocknen. 6. cDNA-arrays möglichst am gleichen Tag scannen, bis dahin unter Lichtausschluss aufbewahren. 8.1.3.9. Reagenzien Prähybridisations-Puffer: - 74 ml DEPC-Wasser - 25 ml 20xSSC -1 ml 10% SDS -1 g BSA Hybridisierungs-Puffer: -240 µl DEPC-Wasser -250 µl 20xSSC -500 µl Formamid -10 µl 10% SDS Phosphat-Waschpuffer: -1 M KH2PO4 und 1 M K2HPO4 ansetzen, - 9,5 ml 1 M K2HPO4 und 0,5 ml 1M KH2PO4 zu 1M KPO4 (pH 8,5-8,7) mischen, - 0,5 ml 1 M KPO4, - 15,25 ml H2O, - 84,25 ml 95% EtOH dazugeben. Phosphat-Elutionspuffer: 1 M KPO4 (pH 8,5) auf 4 mM Phosphat-Eutionspuffer verdünnen: 84 - 0,4 ml 1 M KPO4 pH 8,5 - 99,6 ml DEPC-H2O Carbonat-Puffer: - 0,27 g Na2CO3, - 20 ml H2O, - pH mit 6M HCl auf 9,0 bringen, - mit H2O auf 25 ml auffüllen. 1. Posthybridisierungs-Waschlösung: - 186 ml H2O - 10 ml 20x SSC - 4 ml 10 % SDS 2. Posthybridisierungs-Waschlösung: - 195 ml H2O - 1 ml 20xSSC - 4 ml 10 % SDS 3. Posthybridisierungs-Waschlösung: - 199 ml H2O - 1 ml 20 x SSC 85 8.2. Tabellen und Abbildungen 8.2.1. Klinische Daten mit Ergebnissen der Ploidie, Immunhistochemie und CGH. Fall Histologie Ploidie SSI Alter bei Diagnose FIGO 1988 FIGO 2010 Grad Metastasen TP53 CGH Datenbank ANCA ANRA Beobachtung (Monate) EnCa D1 endometrioid diploid 35,0 83 1c 1b 1 keine nein 3191 0,043 0 74 EnCa D2 endometrioid diploid 31,7 59 1b 1a 1 keine nein 3192 0,087 0 42 EnCa D3 endometrioid diploid 45,3 60 1c 1b 1 keine nein 3193 0,13 0 94 EnCa D4 endometrioid diploid 16,1 52 1b 1a 1 keine nein 3194 0 0 92 EnCa D5 endometrioid diploid 6,8 87 1c 1b 2 keine nein 3195 0,043 0,043 15 EnCa D6* endometrioid diploid 13,2 67 1b 1a 2 keine nein 3196 0,043 0 109 EnCa D7* endometrioid diploid 17,6 78 3a 3a 2 ja nein 3197 0,174 0 4 EnCa D8* endometrioid diploid 13,7 78 1b 1a 1 keine nein 3198 0.043 0 104 EnCa D9 endometrioid diploid 17,3 59 1b 1a 1 keine nein 3193 0 0 94 EnCa D10 endometrioid diploid 12,4 55 1b 1a 1 keine nein 3200 0 0 94 EnCa D11 endometrioid diploid 14,5 81 1b 1a 1 keine nein 3201 0 0 86 EnCa D12 endometrioid diploid 46,5 72 1b 1a 1 keine nein 3202 0 0 75 Überleben (Monate) 42 15 4 86 Fall Histologie Ploidie SSI Alter bei Diagnose FIGO 1988 FIGO 2010 Grad Metastasen TP53 CGH Datenbank ANCA ANRA Beobachtung (Monate) Überleben (Monate) EnCa D13 endometrioid diploid 35,6 63 1c 1b 2 keine nein 3203 0 0 26 26 EnCa D14 endometrioid diploid 56,4 51 1b 1a 1 keine nein 3204 0 0 88 EnCa D15 endometrioid diploid 31,0 53 1b 1a 2 keine nein 3205 0 0 55 EnCa D16 endometrioid diploid 38,4 66 1b 1a 3 keine nein 3206 0,087 0 53 EnCa Ae1 endometrioid aneuploid 68,0 82 1c 1b 1 keine nein 3207 0,043 0 146 EnCa Ae2 endometrioid aneuploid 20,4 80 1b 1a 1 keine nein 3208 0,304 0 71 EnCa Ae3 endometrioid aneuploid 35,2 69 1b 1a 1 keine nein 3203 0,043 0 107 EnCa Ae4 endometrioid aneuploid 26,9 46 1b 1a 2 keine nein 3210 0,565 0,087 81 EnCa Ae5 endometrioid aneuploid 35,5 55 2a 2 3 ja nein 3211 0,087 0 73 EnCa Ae6 endometrioid aneuploid 30,7 66 1b 1a 2 keine nein 3212 0 0 63 EnCa Ae7 endometrioid aneuploid 71,0 79 1b 1a 2 keine nein 3213 0,13 0,043 55 EnCa Ae8 endometrioid aneuploid 59,6 60 1b 1a 3 keine nein 3214 0,043 0 51 EnCa Ae9 endometrioid aneuploid 61,7 72 1c 1b 3 ja nein 5521 0,087 0 42 EnCa Au1 serös-papillär aneuploid 70,9 77 3a 3a ja ja 3215 0,565 0,174 36 EnCa Au2 serös-papillär aneuploid 42,0 78 4b 4b ja nein 3216 0,609 0 51 36 87 Fall Histologie Ploidie SSI Alter bei Diagnose FIGO 1988 FIGO 2010 Metastasen TP53 CGH Datenbank ANCA ANRA Beobachtung (Monate) Überleben (Monate) EnCa Au3 serös-papillär aneuploid 28,5 66 1c 1b keine ja 3217 0,652 0,043 19 19 EnCa Au4 serös-papillär aneuploid 59,7 80 3a 3a ja ja 3218 0,565 0 27 27 EnCa Au5 serös-papillär aneuploid 48,6 90 3 3a ja ja 3219 0,13 0 9 9 EnCa Au6 serös-papillär aneuploid 27,5 88 3b 3b ja ja 3220 0,478 0,13 17 17 EnCa Au7* serös-papillär aneuploid 59,2 80 1a 1a keine ja 3359 0,348 0,13 7 7 EnCa Au8 serös-papillär aneuploid 91,3 83 3a 3a ja ja 5522 0,087 0,043 10 10 Grad EnCa D = EnD; endometrioid diploid EnCa A = EnA; endometrioid aneuploid EnCa Au = UPSC-A; serös papillär aneuploid SSI = stem line scatter index ANCA = average number of copy number alteration ANRA = average number of regional amplification * Array wurde aufgrund der nicht erfüllten Qualifikationskriterien aus der weiteren Analyse ausgeschlossen 88 8.2.2. DEG Listen 1. EnD versus EnA Spot Well-ID Klon Lokalisation Gen Ratios EnD vs. EnA Korrelation CGH 7413 167174 IncytePD:2418617 NaN NaN 0.31784 9252 169013 IncytePD:1854220 1q24 QSCN6 0.33034 8533 168294 IncytePD:1426031 NaN NaN 0.38965 3920 163681 IncytePD:1749102 8p12-p11 INDO 0.39094 550 160311 IncytePD:2132285 2p11.2 TMSB10 0.39303 2758 162519 IncytePD:566886 21q22.3 CSTB 0.40257 4457 164218 IncytePD:2171401 14q22.1 ERO1L 0.4064 504 160265 IncytePD:1739722 10p15.3-p15.2 PFKP 0.41218 x 5132 164893 IncytePD:496003 18q12.1 DSC2 0.41219 x 7796 167557 IncytePD:635178 6q25.3 SOD2 0.41415 74 159835 IncytePD:2070554 10p15-p14 AKR1C1 0.41872 x 1152 160913 IncytePD:1900114 7q11.23 CLDN4 0.43895 x 2391 162152 IncytePD:2131758 7q11.23 CLDN4 0.44753 x 1992 161753 IncytePD:5033671 10p15-p14 AKR1C4 0.45036 x 5562 165323 IncytePD:3139163 1q25.2-q25.3 PTGS2 0.46463 7200 166961 IncytePD:2174863 8q13 LYN 0.46587 x 82 159843 IncytePD:3085928 17q21.33 ITGA3 0.47301 x 2744 162505 IncytePD:1962141 12q12-q13 KRT7 0.4914 7389 167150 IncytePD:1922875 18q11.2 ABHD3 0.50664 7509 167270 IncytePD:1649959 12q12-q13 KRT7 0.5235 2610 162371 IncytePD:560466 NaN NaN 0.53333 5118 164879 IncytePD:86390 11p15.1-p14 SAA4 0.53614 269 160030 IncytePD:2790678 6p21 ITPR3 0.54984 3019 162780 IncytePD:417827 17q25.3 DDX48 0.55008 1122 160883 IncytePD:1845046 1q21-q22 EFNA1 0.55428 471 160232 IncytePD:468887 8p11.22 ADAM9 0.56878 8062 167823 IncytePD:2987878 22q12.2 LIF 0.57036 4772 164533 IncytePD:2636514 7q32.2 HIG2 0.57328 5760 165521 IncytePD:3199352 1q21-q25 TAGLN2 0.58252 1371 161132 IncytePD:2238363 15q26 ISG20 0.5981 9197 168958 IncytePD:3143579 22 NaN 0.61326 7887 167648 IncytePD:2840251 10q24 IFIT4 0.61517 x 4445 164206 IncytePD:1402615 16p13.3 TNFRSF12A 0.63732 x 7552 167313 IncytePD:2211625 14q11.2 PSME1 0.63985 9526 169287 IncytePD:2399253 19p13.3 ELA2 0.64316 8914 168675 IncytePD:3872557 11p15.5 HBB 0.66739 1657 161418 IncytePD:2680410 1q23 MGST3 0.74852 x x x 89 Spot Well-ID Klon Lokalisation Gen Ratios EnD vs. EnA Korrelation CGH 8795 168556 IncytePD:1805270 Xp11.23 USP11 1.3318 x 1246 161007 IncytePD:1334255 6q13-q21 TTK 1.425 15 159776 IncytePD:1890902 6q21 REV3L 1.4825 1185 160946 IncytePD:2736040 NaN NaN 1.5186 5999 165760 IncytePD:348143 9p24.2 XTP5 1.5554 8179 167940 IncytePD:2631284 Xq26.3 FLJ12649 1.5798 1225 160986 IncytePD:2169635 3p21 SCN5A 1.5823 1282 161043 IncytePD:1648517 17 ATPAF2 1.5991 959 160720 IncytePD:1685528 15q15.3 SORD 1.6201 9067 168828 IncytePD:1921725 18q21.1 TCF4 1.6429 6835 166596 IncytePD:1376090 NaN NaN 1.6441 6694 166455 IncytePD:3002566 12p11 ITPR2 1.6531 8241 168002 IncytePD:2378796 1p35.3-p34.1 TEKT2 1.6668 968 160729 IncytePD:1967983 NaN NaN 1.6689 2552 162313 IncytePD:197908 13q14 TSC22 1.6944 x 9964 169725 IncytePD:623523 15 NaN 1.7216 x 2669 162430 IncytePD:1985427 7p22 ETV1 1.7238 8593 168354 IncytePD:1672920 7q22 PCOLCE 1.7316 1878 161639 IncytePD:2862836 12q14.3 CPSF6 1.7557 4115 163876 IncytePD:1313183 2p22-p21 LTBP1 1.7772 2545 162306 IncytePD:5089227 15q12 SNRPN 1.7845 x 7238 166999 IncytePD:2175008 1q43 NID 1.7867 x 5854 165615 IncytePD:1731716 9p13.3 SPAG8 1.7925 1235 160996 IncytePD:1869110 11q14 PRCP 1.8058 9846 169607 IncytePD:1987738 Yp11.3 RPS4Y 1.8154 3174 162935 IncytePD:2499488 13q32.1 DZIP1 1.8428 9347 169108 IncytePD:2324195 3 C3orf6 1.844 6977 166738 IncytePD:1976279 5p13 DAB2 1.8889 867 160628 IncytePD:3543015 11p11.2-p11.1 SLC43A1 1.8896 5821 165582 IncytePD:1682642 5q14-q21 PAM 1.9083 3680 163441 IncytePD:1675571 8p11.23 FLJ14299 1.9431 x 6863 166624 IncytePD:1401817 9q13-q21 X123 1.9635 x 9228 168989 IncytePD:2989991 17p13.1 SERPINF1 1.9704 x 3045 162806 IncytePD:2262662 4p16 LDB2 1.9925 2674 162435 IncytePD:831941 1p13.2 MEP50 2.0122 9763 169524 IncytePD:1613615 14q22-q23 BMP4 2.1426 6238 165999 IncytePD:1909488 2q31.1 CYBRD1 2.1437 7384 167145 IncytePD:719318 2p21 CYP1B1 2.1613 600 160361 IncytePD:1740542 5p15.2 SEMA5A 2.1728 2294 162055 IncytePD:4741984 3p23 SATB1 2.2269 9940 169701 IncytePD:1999167 17q12-q21.1 IGFBP4 2.3449 5459 165220 IncytePD:551403 3q26.2-qter APOD 2.5479 x x x 90 Spot Well-ID Klon Lokalisation Gen Ratios EnD vs. EnA 8949 168710 IncytePD:15834 6q23 EPB41L2 2.6562 1979 161740 IncytePD:4094885 Xp11.4 NDP 2.6858 919 160680 IncytePD:3948420 3q26.2-qter APOD 2.7451 9749 169510 IncytePD:2654926 18q21.1 TCF4 2.8639 2845 162606 IncytePD:583146 11p15.1 PRMT3 2.9637 921 160682 IncytePD:4286401 14q24-q32 FLRT2 3.0483 7491 167252 IncytePD:1578941 4q34-q35 HPGD 3.0728 4173 163934 IncytePD:1525881 12q14.3 CPM 3.5486 1423 161184 IncytePD:2589487 19q13.33 NAP1 4.0899 9379 169140 IncytePD:588870 20q13.2-q13.3 EDN3 4.3388 7046 166807 IncytePD:1505977 4q32-q33 GRIA2 6.0075 Korrelation CGH x x 2. EnD versus UPSC-A Spot Well-ID Klone Map Gen Ratios EnD vs. UPSC-A Korrelation CGH 9947 169708 IncytePD:2506867 4p14 UCHL1 0.097803 880 160641 IncytePD:753651 10q23 RGR 0.13329 2973 162734 IncytePD:2622566 3p25 WNT7A 0.23921 2744 162505 IncytePD:1962141 12q12-q13 KRT7 0.26454 8239 168000 IncytePD:2842835 17q25 LGALS3BP 0.29259 2162 161923 IncytePD:4327691 NaN NaN 0.33472 4285 164046 IncytePD:2900277 14q32 IFI27 0.33617 316 160077 IncytePD:3016305 11q22.3-q23.1 CRYAB 0.33708 7509 167270 IncytePD:1649959 12q12-q13 KRT7 0.33898 4126 163887 IncytePD:1595081 9q32-q33.3 PTGS1 0.34934 7959 167720 IncytePD:1435811 12q13.11 SLC38A1 0.34944 5058 164819 IncytePD:1376121 11q13.3-q14.1 FOLR1 0.36771 2384 162145 IncytePD:1472042 8q24.3 LY6E 0.37422 x 4168 163929 IncytePD:1690670 21q22.3 MX2 0.377 x 2391 162152 IncytePD:2131758 7q11.23 CLDN4 0.38401 x 1152 160913 IncytePD:1900114 7q11.23 CLDN4 0.39355 x 649 160410 IncytePD:79797 17q21.33 DLX4 0.4029 x 855 160616 IncytePD:1652785 17q25.2 HUMPPA 0.41225 x 3659 163420 IncytePD:1444525 12q13.11 SLC38A1 0.41311 8597 168358 IncytePD:2655952 19p13.12 KIAA0290 0.44746 5895 165656 IncytePD:1981145 9q34.1 ASS 0.45126 x x x x 264 160025 IncytePD:1901073 7q35 AKR1B1 0.46078 x 2442 162203 IncytePD:1220385 17q25.3 SOCS3 0.4645 x 1169 160930 IncytePD:2211722 20q11.2 DNMT3B 0.46981 x 1395 161156 IncytePD:1701725 12q24.1 OAS1 0.48261 960 160721 IncytePD:1888594 1q32.3 ATF3 0.48685 91 Spot Well-ID Klone Map Gen Ratios EnD vs. UPSC-A Korrelation CGH 8724 168485 IncytePD:630458 8q24.3 SLC39A4 0.48869 x 6445 166206 IncytePD:2862971 1p36.33 G1P2 0.49863 7630 167391 IncytePD:734376 16p13.3 MSLN 0.5035 9093 168854 IncytePD:1985367 14q32.1 LGMN 0.50807 2617 162378 IncytePD:1965938 22 NaN 0.51664 2593 162354 IncytePD:5109040 NaN NaN 0.51806 1531 161292 IncytePD:2853091 8q21.3 CYP7B1 0.52476 9423 169184 IncytePD:485111 1q32-q41 CENPF 0.52549 1882 161643 IncytePD:3115514 2q37.2 ARL7 0.53209 1701 161462 IncytePD:1656490 6 NaN 0.53501 9068 168829 IncytePD:2926307 NaN NaN 0.5374 7525 167286 IncytePD:1923070 2p25 DDEF2 0.54146 409 160170 IncytePD:523 19p13.2 RNASEH2A 0.54632 6601 166362 IncytePD:2515389 19q13.2 APOE 0.56891 x 775 160536 IncytePD:3679736 17q12-q21 KRT17 0.57144 x 7887 167648 IncytePD:2840251 10q24 IFIT4 0.57542 x 3151 162912 IncytePD:2680168 3q13 PVRL3 0.58177 x 9502 169263 IncytePD:1674253 10pter-q25.3 SPOCK2 0.58195 x 4206 163967 IncytePD:1907232 8q24 PLEC1 0.59036 x 6078 165839 IncytePD:2135596 19p13.13 DDX39 0.59614 1085 160846 IncytePD:1594108 11q13 SPTBN2 0.60562 1724 161485 IncytePD:3935143 19p13.2 DNM2 0.60898 9930 169691 IncytePD:2057823 19q13.43 E2-EPF 0.60919 x 285 160046 IncytePD:1879888 8q24.13 MGC3067 0.62045 x 8335 168096 IncytePD:2496305 2p23 KIF3C 0.62885 x 2508 162269 IncytePD:508926 19q13.11-q13.12 CKAP1 0.63229 x 4108 163869 IncytePD:1871201 12q11-q13 KRT2A 0.63479 7552 167313 IncytePD:2211625 14q11.2 PSME1 0.64675 8692 168453 IncytePD:2056642 4p16.3 TACC3 0.64848 153 159914 IncytePD:1567992 12q13 ACVR1B 0.64848 6419 166180 IncytePD:1719657 8p22 MTSS1 0.6538 690 160451 IncytePD:2061241 20cen-q13.1 AHCY 0.66508 1122 160883 IncytePD:1845046 1q21-q22 EFNA1 0.69866 9592 169353 IncytePD:1666268 16q22.1 DNCLI2 1.4044 x 6553 166314 IncytePD:1383888 11p13 LMO2 1.4133 x 8058 167819 IncytePD:2918605 12p12.2 KIAA0528 1.4446 8773 168534 IncytePD:437868 12q21.1 HRB2 1.5008 1600 161361 IncytePD:2213276 9q34.3 KIAA0625 1.5426 8531 168292 IncytePD:998421 15q21 TCF12 1.5564 x 3680 163441 IncytePD:1675571 8p11.23 FLJ14299 1.5589 x 2939 162700 IncytePD:2360580 17q21 LUC7A 1.5628 4871 164632 IncytePD:1274957 15q21.3-q22.31 RAB11A 1.566 2530 162291 IncytePD:805942 9q32 EDG2 1.5998 2096 161857 IncytePD:3387976 13q12.13 NUPL1 1.6039 x x x x x x x 92 Spot Well-ID Klone Map Gen Ratios EnD vs. UPSC-A Korrelation CGH 5780 165541 IncytePD:238278 9q33 GSN 1.6245 9730 169491 IncytePD:340088 NaN NaN 1.6726 836 160597 IncytePD:1961218 3q25.2 P2RY1 1.6759 5625 165386 IncytePD:2912987 3p14.3 CACNA1D 1.6793 x 906 160667 IncytePD:1856587 15 NaN 1.705 x 9029 168790 IncytePD:11556 14q24.3 TMP21 1.7196 6845 166606 IncytePD:1525829 1q42-q44 KMO 1.7264 x 1104 160865 IncytePD:1890794 1q42.13 ITPKB 1.7295 x 752 160513 IncytePD:1998155 NaN NaN 1.7329 9868 169629 IncytePD:389707 9p22 MLLT3 1.7349 1956 161717 IncytePD:4225421 17q22 DGKE 1.7793 2408 162169 IncytePD:180164 19p13.3 CIRBP 1.7823 2128 161889 IncytePD:4309944 13q33.3 NaN 1.7834 959 160720 IncytePD:1685528 15q15.3 SORD 1.7915 x 7010 166771 IncytePD:1981939 16p13.3 KIAA0643 1.8006 x 5706 165467 IncytePD:1283532 6q14-q15 TPBG 1.8038 x 1540 161301 IncytePD:2634611 17q22-q23.2 PRKCA 1.8056 4115 163876 IncytePD:1313183 2p22-p21 LTBP1 1.8103 6784 166545 IncytePD:1403932 5q34-q35 MSX2 1.8292 2674 162435 IncytePD:831941 1p13.2 MEP50 1.8393 3000 162761 IncytePD:815861 6p21.2 CDKN1A 1.8394 908 160669 IncytePD:1864819 Xp22.22 RBBP7 1.8648 x 4565 164326 IncytePD:2888464 1q21.2 TXNIP 1.9465 x 9850 169611 IncytePD:1841989 4q24 PAPSS1 2.0115 x 6977 166738 IncytePD:1976279 5p13 DAB2 2.0472 8710 168471 IncytePD:3254666 2q32.3 HIBCH 2.0781 600 160361 IncytePD:1740542 5p15.2 SEMA5A 2.0842 7238 166999 IncytePD:2175008 1q43 NID 2.1095 6863 166624 IncytePD:1401817 9q13-q21 X123 2.1425 867 160628 IncytePD:3543015 11p11.2-p11.1 SLC43A1 2.1469 x 1979 161740 IncytePD:4094885 Xp11.4 NDP 2.189 x 9749 169510 IncytePD:2654926 18q21.1 TCF4 2.221 2107 161868 IncytePD:4401302 NaN NaN 2.2382 5734 165495 IncytePD:2498815 2q31 GAD1 2.2563 440 160201 IncytePD:698665 5q23.3 SLC12A2 2.2676 9940 169701 IncytePD:1999167 17q12-q21.1 IGFBP4 2.2844 6505 166266 IncytePD:4410949 4q12-q13 CXCL5 2.3334 x 8949 168710 IncytePD:15834 6q23 EPB41L2 2.4203 x 9220 168981 IncytePD:1636639 11p11.2 CREB3L1 2.6426 x 4067 163828 IncytePD:1737280 NaN NaN 3.2749 9379 169140 IncytePD:588870 20q13.2-q13.3 EDN3 3.3166 1699 161460 IncytePD:3214293 7p14 ADCYAP1R1 3.3822 1554 161315 IncytePD:2595612 11p15 MUC5AC 3.5522 4173 163934 IncytePD:1525881 12q14.3 CPM 3.5529 x x x x 93 Spot Well-ID Klone Map Gen Ratios EnD vs. UPSC-A Korrelation CGH 5610 165371 IncytePD:2242817 21q22.3 TFF3 3.6206 7046 166807 IncytePD:1505977 4q32-q33 GRIA2 4.8722 x 678 160439 IncytePD:685870 11q22-q23 PGR 12.2538 x 3. EnA versus UPSC-A Korrelation CGH Spot WellID Klon Map Gen Ratios_Ae_vs_Au 9947 169708 IncytePD:2506867 4p14 UCHL1 0.13753 9084 168845 IncytePD:1968446 NaN NaN 0.2665 5058 164819 IncytePD:1376121 11q13.3-q14.1 FOLR1 0.28208 4126 163887 IncytePD:1595081 9q32-q33.3 PTGS1 0.31468 x 264 160025 IncytePD:1901073 7q35 AKR1B1 0.37467 x 7852 167613 IncytePD:3078931 4q21.3 PTPN13 0.42708 4117 163878 IncytePD:1723401 9q32-q33.3 PTGS1 0.43031 x 3174 162935 IncytePD:2499488 13q32.1 DZIP1 0.45304 x 164 159925 IncytePD:2183703 NaN NaN 0.4822 7875 167636 IncytePD:2805762 14q11.1-q12 SALL2 0.49124 x 611 160372 IncytePD:460491 13q33 EFNB2 0.51122 x 1832 161593 IncytePD:2968067 4q32.3 SPOCK3 0.51859 2545 162306 IncytePD:5089227 15q12 SNRPN 0.52956 2384 162145 IncytePD:1472042 8q24.3 LY6E 0.53386 x 9093 168854 IncytePD:1985367 14q32.1 LGMN 0.54582 x 4331 164092 IncytePD:2479074 11q13 TM7SF2 0.55116 907 160668 IncytePD:1910461 20q11.23 CPNE1 0.55522 x 690 160451 IncytePD:2061241 20cen-q13.1 AHCY 0.56959 x 6009 165770 IncytePD:1416354 16q21-q22.1 NDRG4 0.5715 8795 168556 IncytePD:1805270 Xp11.23 USP11 0.60002 1531 161292 IncytePD:2853091 8q21.3 CYP7B1 0.60939 x 9966 169727 IncytePD:1001890 20q11.21 PLAGL2 0.61877 x 5994 165755 IncytePD:2190607 8q24.3 EEF1D 0.62906 x 1808 161569 IncytePD:2773217 17q12-q21 MPP3 0.63215 x 1844 161605 IncytePD:3068935 NaN NaN 0.63709 3523 163284 IncytePD:1405820 3 NaN 0.63718 x 7489 167250 IncytePD:1599532 20q11.2-q12 TGIF2 0.66356 x 3659 163420 IncytePD:1444525 12q13.11 SLC38A1 0.66807 2617 162378 IncytePD:1965938 22 NaN 0.68093 8122 167883 IncytePD:1798585 17q24-q25 ACOX1 0.68567 x 8804 168565 IncytePD:3186307 19 NaN 0.69092 x 8162 167923 IncytePD:1688468 12q13 ARF3 0.75279 94 Korrelation CGH Spot WellID Klon Map Gen Ratios_Ae_vs_Au 6027 165788 IncytePD:605831 12p11.23 KCNJ8 1.4061 2096 161857 IncytePD:3387976 13q12.13 NUPL1 1.4595 2013 161774 IncytePD:4626895 15q25.3-q26 CIB1 1.4642 x 8741 168502 IncytePD:1810777 11q22 BIRC2 1.4752 x 4871 164632 IncytePD:1274957 15q21.3-q22.31 RAB11A 1.5008 x 3000 162761 IncytePD:815861 6p21.2 CDKN1A 1.5513 2282 162043 IncytePD:1869139 NaN NaN 1.563 964 160725 IncytePD:1756503 1q24 HHL 1.59 9206 168967 IncytePD:1751692 6q23.3 KIAA1244 1.6348 x 7038 166799 IncytePD:1814912 6q22.33 MAP3K5 1.6483 x 2583 162344 IncytePD:2131914 2q12-q14 FHL2 1.6507 8704 168465 IncytePD:1964902 5q33.2 GALNT10 1.6625 8235 167996 IncytePD:2633207 2p21-p16 EPAS1 1.6809 7389 167150 IncytePD:1922875 18q11.2 ABHD3 1.6946 9515 169276 IncytePD:435572 1q42.1-q42.2 CHS1 1.7931 7921 167682 IncytePD:1731972 17p11.1-q11.2 CPD 1.8377 9252 169013 IncytePD:1854220 1q24 QSCN6 1.8934 9485 169246 IncytePD:377842 10q26.12 LOC118987 1.9608 2758 162519 IncytePD:566886 21q22.3 CSTB 1.9743 2128 161889 IncytePD:4309944 13q33.3 NaN 1.9776 5734 165495 IncytePD:2498815 2q31 GAD1 1.9944 7310 167071 IncytePD:1534482 4p12 TXK 2.0161 440 160201 IncytePD:698665 5q23.3 SLC12A2 2.121 5562 165323 IncytePD:3139163 1q25.2-q25.3 PTGS2 2.1722 5877 165638 IncytePD:1513214 11q22 BIRC3 2.3173 2246 162007 IncytePD:4714232 8q11-q12 CA8 2.3822 5118 164879 IncytePD:86390 11p15.1-p14 SAA4 2.4021 4820 164581 IncytePD:2506090 8q13.3 RDH10 2.6042 9220 168981 IncytePD:1636639 11p11.2 CREB3L1 2.6773 2610 162371 IncytePD:560466 NaN NaN 3.2671 7413 167174 IncytePD:2418617 NaN NaN 3.4535 9541 169302 IncytePD:446969 3p25.1 EAF1 3.5287 107 159868 IncytePD:1861743 17q21.2 HOXB13 3.7019 5610 165371 IncytePD:2242817 21q22.3 TFF3 4.0231 9372 169133 IncytePD:2631845 11p15.1 SAA1 5.1872 x 1554 161315 IncytePD:2595612 11p15 MUC5AC 6.4478 x x x x x x x x 95 8.2.3. Überblick der IPA-Analysen GruppenVergleich EnD vs. EnA Analyse Transcriptomics Proteomics § EnD vs. UPSC-A Transcriptomics Punktwert Top Erkrankungen p-Wert Anzahl der Moleküle Top Molekular und Zell Funktion p-Wert Anzahl der Moleküle 48 Cancer < 0.0161 23 Lipid Metabolism < 0.0184 6 Lipid Metabolism, Small Molecule Biochemistry, Genetic Disorder 30 Hematological Disease < 0.0132 12 Small Molecule Biochemistry < 0.0184 10 Gene Expression, Nutritional Disease, Cellular Development 22 Gastrointestinal Disease < 0.0105 9 Vitamin and Mineral Metabolism < 0.0175 12 25 Neurological Disease < 0.0106 7 Cellular Growth and Proliferation < 0.0382 7 Genetic Disorder < 0.0486 8 Cell Morphology < 0.0297 4 < 0.0346 6 Top Netzwerke Lipid Metabolism, Small Molecule Biochemistry, Vitamin and Mineral Metabolism Cellular Assembly and Organization, Nucleic Acid Metabolism, Small Molecule Biochemistry Organism Injury and Abnormalities, Cardiac Necrosis/Cell Death, Cell Death* Organ Morphology, Reproductive System Development and Function, Skeletal and Muscular Disorders # Cellular Development, Cellular Growth and Proliferation, Cancer Cardiovascular Disease, Hematological Disease, Skeletal and Muscular Disorders Proteomics § Endocrine System Development and Function, Small Molecule Biochemistry, Gene Expression Lipid Metabolism, Small Molecule Biochemistry, Cell *, # Morphology Cancer < 0.0394 7 Cellular Assembly and Organization 44 Cancer < 0.0196 31 Cellular Growth and Proliferation < 0.0212 28 28 Genetic Disorder < 0.0234 47 Cell-To-Cell Signaling and Interaction < 0.0234 11 20 Reproductive System Disease < 0.0196 12 Cell Death < 0.0234 26 Cancer < 0.0428 7 Amino Acid Metabolism < 0.0150 2 19 15 25 96 GruppenVergleich EnA vs. UPSC-A Analyse & Transcriptomics Top Netzwerke Cardiovascular System Development and Function, Cell Cycle, Lipid Metabolism Cell Death, Cellular Movement, Hematological System Development and Function Cellular Assembly and Organization, Cellular Function and Maintenance, Cell Signaling Top Erkrankungen p-Wert Anzahl der Moleküle Top Molekular und Zell Funktion p-Wert Anzahl der Moleküle Gastrointestinal Disease < 0.0214 4 Cell Morphology < 0.010 1 Inflammatory Disease < 0.0479 2 Cellular Assembly and Organization < 0.0125 2 28 Cardiovascular Disease < 0.0430 12 Gene Expression < 0.0245 7 28 Development Disorder < 0.0301 5 Cell Death < 0.0338 15 22 Connective Tissue Disorder < 0.0487 9 Cellular Movement < 0.0485 8 Punktwert * Überschneidung der markierten Netzwerke via platelet-derived growth factor beta polypeptide (simian sarcoma viral (v-sis) oncogene homolog) dimer (PDGFBB) # Überschneidung der marktierten Netzwerke via beta-Actin (ACTB) § Proteindaten nach Gemoll et al. [130] & Bei dem Vergleich EnA versus UPSC-A wurde nur ein DEP detektiert. Eine Netzwerkanalyse auf Proteinebene war somit nicht möglich. EnD = endometrioid diploid EnA = endometrioid aneuploid UPSC-A = serös-papillär aneuploid 97 8.2.4. Netzwerke basierend auf der IPA-Analyse A B Auf IPA basierende Netzwerk-Analyse der unterschiedlich exprimierten Gene. 98 Die rote Färbung markiert überexprimierte, die grüne Färbung unterexprimierte Gene in den betreffenden Gruppen. Blaue Pfeile und Umrundungen markieren zentrale Knotenpunkte des Netzwerks. A: endometrioid diploide versus endometriod aneuploide Endometriumkarzinome B: endometriod aneuploide versus serös- papilläre Endometriumkarzinome 99 9. Danksagung Für die großzügige Bereitstellung personeller und sachlicher Ressourcen aus der Klinik für Chirurgie der Universität zu Lübeck danke ich Herrn Prof. Dr. Hans-Peter Bruch. Entscheidenden Anteil am Gelingen dieser Arbeit trägt mein Doktorvater und Betreuer PD Dr. Dr. Jens K. Habermann, dem ich für seine fachliche Unterstützung sehr herzlich danke. Mein besonderer Dank gilt auch Prof. Dr. Thomas Ried und seinem Team, dem Ried-Lab am NIH in Bethesda, MD, USA, die mich während meiner Forschungsarbeit in ihrem Labor herzlich aufgenommen, mich unermüdlich die wichtigsten Techniken dieser Arbeit gelehrt und in jeglicher Art und Weise bei der Erstellung dieser Arbeit unterstützt haben. Gleiches gilt für Prof. Dr. Gerd Auer und sein Labor-Team des Karolinska-Instituts in Stockholm, insbesondere Prof. Britta Nordström und Dr. Caroline Lundgren, die mir wertvolle Einblicke in die Klinik und wichtige Unterstützung bei der Proben- und Datensammlung gewährt haben. Aus dem chirurgischen Forschungslabor in Lübeck möchte ich insbesondere Dr. Timo Gemoll herzlich danken, der mit seinem großen Engagement wesentlich zur Auswertung der wissenschaftlichen Daten für die Veröffentlichung beigetragen hat. Meinen lieben Eltern, Christa und Friedhelm Bündgen, danke ich sehr herzlich für die fortwährende Unterstützung und Ermutigung. Für die großzügige finanzielle Unterstützung, die die Durchführung meiner Forschungsarbeit in Bethesda und Stockholm ermöglicht hat, danke ich dem BoehringerIngelheim-Fonds und der Universität zu Lübeck. 100 10. Lebenslauf Name: Nana Kristin Bündgen Geburtstag/-ort: 17.07.1980 in Dortmund Schulausbildung 1990 - 1999 Bischöfliches Gymnasium St. Michael, Ahlen, Abitur Studium der Humanmedizin Okt. 1999 - Sept. 2001 Ernst Moritz Arndt Universität zu Greifswald Sept. 2001 Physikum Okt. 2001 Universität Schleswig Holstein, Campus Lübeck Aug. 2002 Erstes Staatsexamen März 2005 Zweites Staatsexamen Juni 2006 Drittes Staatsexamen 13. Juni 2006 Approbation Ärztliche Tätigkeit Juli 2006 - Jan. 2007 Assistenzärztin in der Frauenklinik des Städtischen Klinikums Lüneburg, Lehrkrankenhaus der Universität Göttingen, Prof. Dr. med. P. Dall März 2007 – Juni 2008 Assistenzärztin in der Universitäts-Frauenklinik Rostock, Prof. Dr. med. B. Gerber Juli 2008 – Sept. 2011 Assistenzärztin in der Frauenklinik des Universitätsklinikums Schleswig Holstein, Campus Lübeck, Prof. Dr. med. K. Diedrich 07. Sept. 2011 Annerkennung zur Fachärztin für Gynäkologie und Geburtshilfe, Ärztekammer SH, Bad Segeberg Seit Sept. 2011 Fachärztin und Funktionsoberärztin in der Frauenklinik des Universitätsklinikums Schleswig Holstein, Campus Lübeck, Prof. Dr. med. S. Becker (bis April 2012 Prof. Dr. med. K. Diedrich) Forschungsaufenthalte und Dissertation Okt. 2002 - April 2003 und Sept. - Okt. 2003 Forschungsarbeit im Labor von Prof. Dr. med. T. Ried, Section for Cancer Genomics, Genetics Branch, Center for Cancer Research, 101 National Cancer Institute, National Institutes of Health (NIH), Bethesda/Washington DC, USA Aug. 2003 Forschungsarbeit im Labor von Prof. Dr. med. G. Auer, Karolinska Institutet Stockholm, Schweden Okt. 2003 Übernahme beeinflusst des das Dissertationsthemas Transkriptom und „Die genomische Proteom von Instabilität Subtypen des Endometriumkarzinoms“. Stipendien 2002 Boehringer Ingelheim Fonds, Research Award 2003 Reisestipendium der Universität zu Lübeck Veröffentlichungen, die aus der vorliegenden Arbeit resultierten „Chromosomale Veränderungen korrelieren mit dem Ploidiestatus und Klinik des Endometriumkarzinoms“ Vortrag im Rahmen der Preisverleihung „Leuchtfeuer des Nordens“, Tagung der Norddeutschen Gesellschaft für Gynäkologie und Geburtshilfe (NGGG) 2008 in Hamburg “Chromosomal Aberrations Correlate with DNA Ploidy and the Clinical Course of Endometrial Carcinomas.” N.K. Buendgen, J.K. Habermann, C. Lundgren, J. Doering, B. Sundelin, R. Broll, H.-P. Bruch, B. Nordstroem, G. Auer, T. Ried Posterpräsentation beim NIH Research Festival 2003, National Institutes of Health, Bethesda/Washington DC, USA “Genomic instability influences the transcriptome and proteome in endometrial cancer subtypes” Habermann JK*, Buendgen NK*, Gemoll T*, Hautaniemi S, Lundgren C, Wangsa D, Doering J, Bruch HP, Nordstroem B, Roblick UJ, Jornvall H, Auer G, Ried T. Mol Cancer. 2011 Oct 31;10(1):132. (Epub ahead of print). * geteilte Erstautorenschaft. Artikel in Fachzeitschriften „Supportive Therapie mit Blut und Blutkomponenten Anämie und Thrombozytopenie“ N.K. Bündgen und B. Gerber Der Gynäkologe, Vol. 41, Nr. 8, 567-602 „Krebskrank und blutleer - Anämie und Thrombozytopenie gegensteuern“ N. Bündgen und B. Gerber CME, Springer 2009 102 „Polkörperdiagnostik für monogene Erkrankungen-Geburt von drei gesunden Kindern“ Griesinger, Georg; Bündgen, Nana; Salmen, Diana; Schwinger, Eberhard; Gillessen-Kaesbach, Gabriele; Diedrich, Klaus Dtsch Arztebl Int 2009; 106(33): 533-8 „Follikulogenese und In-vitro-Maturation aus humanem Ovargewebe - Aktueller Stand“ N. Bündgen, S. Lüke, K. Diedrich and G. Griesinger Gynäkologische Endokrinologie, Springer 2010, Volume 8, Number 3 „Präimplantationsdiagnostik und Präimplantationsscreening – Aktueller Stand“ N. Bündgen, G. Griesinger und K. Diedrich Der Gynäkologe, Springer 2011, Volume 44, Number 2 „Bericht vom 26. Jahrestreffen der Europäischen Gesellschaft für humane Reproduktion und Embryologie in Rom“ N. Bündgen, K. Diedrich und G. Griesinger Gynäkologische Endokrinologie, Springer 2010, Volume 8, Number 3 „Ovarfunktion und Fertilität“ N. Bündgen, A. Schultze-Mosgau, T. Cordes, K. Diedrich, G. Griesinger Der Gynäkologe, Springer 2011, Volume 44, Number 12 “Protein profiling of genomic instability in endometrial cancer.” Gemoll T, Habermann JK, Lahmann J, Szymczak S, Lundgren C, Bündgen NK, Jungbluth T, Nordström B, Becker S, Lomnytska MI, Bruch HP, Ziegler A, Hellman U, Auer G, Roblick UJ, Jörnvall H. Cell Mol Life Sci. 2012 Jan;69(2):325-33 “The gene expression signature of genomic instability in breast cancer is an independent predictor of clinical outcome.” Habermann JK, Doering J, Hautaniemi S, Roblick UJ, Bündgen NK, Nicorici D, Kronenwett U, Rathnagiriswaran S, Mettu RK, Ma Y, Krüger S, Bruch HP, Auer G, Guo NL, Ried T. Int J Cancer. 2009 Apr 1;124(7):1552-64 Poster-Vorstellungen „Aneurysma des interatrialen Septums (ASA), pränatale Diagnostik, Management und aktuelle Literatur“ Nana Bündgen, Andreas Schröer, Anja Dawson, David Hartge, Klaus Diedrich, Jan Weichert Kongress der Deutschen Gesellschaft für Perinatale Medizin (DGPM) in Berlin 2009 103 “Changes in AMH levels following ovarian cortex removal: implications for fertility preservation” N. Buendgen, S. Lueke, K. Diedrich, G. Griesinger 26. Jahrestagung der European Society of Human Reproduction and Endocrinologie (ESHRE) in Rom 2010 „Änderungen des AMH-Spiegels nach Kryokonservierung von Ovarkortex Implikationen für den Fertilitätserhalt“ N. Bündgen, S. Lüke, K. Diedrich, G. Griesinger Kongress der Deutschen Gesellschaft für Gynäkologie und Geburtshilfe (DGGG), in München 2010 “Initiation of ovarian stimulation independent of menstruation in a GnRH-antagonist protocol: a prospective, clinical, case-control, feasibility trial” N. Buendgen, T. Cordes, A. Schultze-Mosgau, K. Diedrich, G. Griesinger 27. Jahrestagung der ESHRE in Stockholm 2011 104