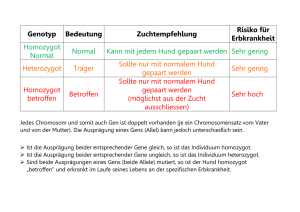
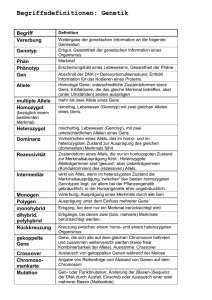
Studien - Ruhr-Universität Bochum
Werbung

Kandidatengenanalyse für generalisierte progressive Retina-Atrophie in dreißig Hunderassen Dissertation zur Erlangung des Grades eines Doktors der Naturwissenschaften der Fakultät für Biologie der Ruhr-Universität Bochum angefertigt in der Medizinischen Fakultät Abteilung für Humangenetik vorgelegt von Gabriele Dekomien aus Dortmund Bochum 2002 Die vorliegende Arbeit wurde in der Zeit von Januar 1998 bis Oktober 2002 in der Abteilung Humangentik der medizinischen Fakultät der Ruhr-Universität Bochum unter der Anleitung von Herrn Prof. Dr. Jörg T. Epplen angefertigt. 1. Gutachter: Herr Prof. Dr. Jörg T. Epplen 2. Gutachter: Herr Prof. Dr. Andreas Faissner Meiner Familie Inhalt I Inhalt INHALT ABKÜRZUNGSVERZEICHNIS I VI KAPITEL 1 1 EINLEITUNG 1 1.1 RETINA DER VERTEBRATEN 2 1.1.1 Organisation der Retina 2 1.1.2 Aufbau der Zapfen und Stäbchen 2 1.1.3 Retinales Pigmentepithel (RPE), Vitamin-A Zyklus 3 1.1.4 Phototransduktion 5 1.1.5 Wahrnehmungsunterschiede des Lichtes von Menschen und Hunden 6 1.2 PHÄNOTYPEN RETINALER ERKRANKUNGEN IM MENSCHEN UND IM HUND 7 1.2.1 Retinitis Pigmentosa 7 1.2.2 Generalisierte progressive Retina Atrophie Formen beim Hund 8 1.2.3 Natürliche Modellsysteme für retinale Erkrankungen 10 1.2.4 Mutierte Gene in natürlichen Tiermodellen 11 1.2.4.1 Photorezeptorspezifische Gene 11 1.2.4.1.1 cGMP Phosphodiesterase 11 1.2.4.1.2 RDS/Peripherin und ROM1 12 1.2.4.1.3 Phosducin 12 1.2.4.1.4 Myosin VIIA (USH1B) 12 1.2.4.1.5 RP Guanosin triphosphat GTPase Regulator (RPGR) 13 1.2.4.1.6 Rhodopsin 13 1.2.4.2 RPE spezifische Gene 14 1.2.4.2.1 Rezeptor Tyrosin Kinase (Mertk) 14 1.2.4.2.2 Retinales Pigmentepithel spezifisches Protein 65 (RPE65) 14 1.3 ZIELSETZUNG DER ARBEIT 15 Inhalt II KAPITEL 2 17 MATERIAL & METHODEN 17 2.1. MATERIAL 17 2.1.1 Chemikalien 17 2.1.2 Untersuchungsgruppe und DNA-Proben 17 2.1.3 Isolierung der DNA und Polymerase-Kettenreaktion (PCR) 20 2.1.3 DNA Sequenz-Analyse 20 2.1.4 Klonierung und Identifikation von Intron/Exon Grenzen 21 2.1.5 PCR-SSCP- und Mikrosatelliten-Analyse 22 2.2 BEDINGUNGEN FÜR DEN AUSSCHLUSS VON KANDIDATENGENEN IN DEN VERSCHIEDENEN HUNDERASSEN 22 2.2.1 Kopplungswahrscheinlichkeit einer gPRA-Mutation mit (Einzelnukleotidpolymorphismen, SNP) 22 2.2.2 Gründereffekt 23 2.3 DATENBANKEN / AUSWERTUNG / SOFTWARE 24 KAPITEL 3 25 PDE6A GEN 26 3.1 VORBEMERKUNG 26 3.2 RESULTATE UND DISKUSSION 26 3.2.1 Voraussetzungen für die Untersuchung des PDE6A Gens 26 3.2.2 Mutationsanalyse des PDE6A Gens 28 3.2.3 Schlussfolgerung 28 KAPITEL 4 31 PDE6B GEN 32 4.1 VORBEMERKUNG 32 4.2 RESULTATE UND DISKUSSION 33 4.2.1 Genomische Organisation des PDE6B Gens 33 4.2.2 Mutationsanalyse des PDE6B Gens 33 4.2.3 Schlussfolgerung 34 4.3 PCR-GESTÜTZTER GPRA TEST ZUR IDENTIFIZIERUNG DER 8-BP INSERTION IN SLOUGHIS 41 Inhalt III KAPITEL 5 43 SAG GEN 44 5.1 VORBEMERKUNG 44 5.2 RESULTATE UND DISKUSSION 44 5.2.1 Genomische Organisation des SAG Gens 44 5.2.2 Mutationsanalyse mittels PCR-SSCP 45 5.2.3 Schlussfolgerung 46 KAPITEL 6 50 PDC GEN 51 6.1 VORBEMERKUNG 51 6.2 RESULTATE UND DISKUSSION 51 6.2.1 Genomische Organisation des PDC Gens 51 6.2.2 Mutations-Analyse des PDC Gens 52 6.2.3 Schlussfolgerung 53 KAPITEL 7 58 RCV1 GEN 59 7.1 VORBEMERKUNG 59 7.2 RESULTATE UND DISKUSSION 59 7.2.1 Identifikation des RCV1 Gens 59 7.2.2 Mutationsanalyse des RCV1 Gens 60 7.2.3 Schlussfolgerungen 61 KAPITEL 8 65 PDE6D & PDE6G GENE 66 8.1 VORBEMERKUNG 66 8.2 RESULTATE UND DISKUSSION 67 8.2.1 Identifikation des Intron 1 des caninen PDE6D Gens 67 8.2.2 Mutationsanalyse des PDE6D Gens 67 8.2.3 Mutationsanalyse des PDE6G Gens 68 8.2.4 Schlussfolgerung 68 Inhalt IV KAPITEL 9 72 RPE65 GEN 72 9.1 VORBEMERKUNG 72 9.2 RESULTATE UND DISKUSSION 73 9.2.1 Genomische Organisation des RPE65 Gens 73 9.2.2 Mutationsanalyse des RPE65 Gens 74 9.2.3 Schlussfolgerung 74 KAPITEL 10 79 CRX GEN 79 10.1 VORBEMERKUNG 79 10.2 RESULTATE UND DISKUSSION 79 10.2.1 Bestimmung der Intronsequenzen des CRX Gens 79 10.2.2 Mutationsanalyse des CRX Gens 80 10.2.3 Schlussfolgerung 81 KAPITEL 11 84 GNGT1 GEN 84 11.1 VORBEMERKUNG 84 11.2 RESULTATE UND DISKUSSION 85 11.2.1 Identifizierung der Introns des GNGT1 Gens 85 11.1.2 Mutationsanalyse des GNGT1 Gens 85 11.1.3 Schlussfolgerung 86 KAPITEL 12 88 MITOCHONDRIALE GENE 88 12.1 VORBEMERKUNG 88 12.2 RESULTATE UND DISKUSSION 89 12.2.1 Mutationsanalyse der mitochondrialen Gene 89 12.2.2 Schlussfolgerung 89 Inhalt V KAPITEL 13 92 DISKUSSION 92 13.1 ZUR FORSCHUNGSSTRATEGIE DER DISSERTATION 92 13.1.1 Kandidatengenanalyse beim Menschen 92 13.1.2 Kurzer Überblick der Rassenentstehung 93 13.1.3 Kandidatengenanalyse beim Hund 94 13.1.4 PDE6B-Mutation in Sloughis 96 13.2 KANDIDATENGEN-AUSSCHLUSSVERFAHREN 99 13.2.1 Verteilung der Sequenzvariationen in den untersuchten Genen 99 13.2.2 Intragenische Marker 99 13. 3 AUSBLICK 101 KAPITEL 14 104 ZUSAMMENFASSUNG 104 SUMMARY 106 KAPITEL 15 107 LITERATUR 107 DANKSAGUNG 121 LEBENSLAUF 122 ANHANG 123 Abkürzungsverzeichnis VI Abkürzungsverzeichnis ABCR A. bidest. ad ar AS ATP BCM bp BSA CCCM cd cDNA CFA cGC-E cGMP Ci cM CNCG COD CORD CRALBP CRX CSNB CSRD d Da DOK DS DSK dd Del DMSO DNA E. coli EDTA EMBL eOD EOG erd ERG g GARP GCAP1 GDP GMP ATP-binding cassette, retina-specific bidestilliertes Wasser autosomal dominant vererbt Aland island eye disease Aminosäure Adenosintriphosphat Baylor College of Medicine Basenpaare Bovine-Serumalbumin carboxychemical cleavage of mismatches cone dystrophy complementary DNA canines Chromosom Guanylatzyklase E zyklisches Guanosin-Monophosphat Curie Centimorgan cyclic nucleotide-gated channel cone dystrophy cone-rod dystrophy cellular retinaldehyde binding protein cone rod homeobox protein kongenitale stationäre Nachtblindheit childhood-onset severe retinal dystrophy 2'-DesoxyriboDalton Dortmunder Ophthalmologen Kreis Doppelstrangbande Doppelstrangkontrolle 2', 3'-DidesoxyriboDeletion Dimethylsulfoxid Desoxyribonukleinsäure Escherischia coli Ethylendiamin-N,N,N',N'-tetraessigsäure-Dinatriumsalz European Molecular Biology Laboratory europäische Form der Oguchi-Erkrankung Elektrookulographie early retinal degeneration Elektroretinogramm Gramm glutamic acid rich protein Guanylatzyklase-aktivierendes Protein 1 Guanosin-Diphosphat Guanosin-Monophosphat Abkürzungsverzeichnis GNA(B,G)T gPRA GR GUCA GTP h HD Ins kb l J LCA LMBBS LHON M MEDLINE MERTK MERRF MG min mRNA MS NNaCl NARP NCBI ND nxg nm nt OD OMIM OLR p PAA PCR PDE (6A, 6B, 6D, 6G) PNR prcd q RAPD RetNet RCS rat rcd RCV rd RDS RFLP RHO RHOK RNA VII Transducin α- (β-, γ-)Untereinheit generalisierte Progressive Retina-Atrophie GTPase-Regulator Guanylatzyklase activator Guanosin-Triphosphat Stunde(n) Heteroduplex-Bande Insertion Kilobasenpaare Liter Joule Leber congenital amaurosis Laurence-Moon-Bardet-Biedl-Syndrom Lebersche heriditäre Optikus Atrophie Molar Medical Literature, Analysis, and Retrieval System Online Tyrosin Kinase Rezeptor Myoklone Epilepsie und ragged red fibers Molekularmasse Minute(n) messenger RNA Mikrosatellit AminoNatriumchlorid Neuropathie / Ataxie / RP National Center for Biotechnology Information Norrie-Erkrankung n-fache Gravitationsbeschleunigung Nanometer Nukleotid(e) Oguchi-Erkrankung Online Mendelian Inheritance in Man offener Leserahmen kurzer Arm eines Chromosoms Polyacrylamid polymerase chain reaction Phosphodiesterase (α- β- δ- γ- Untereinheiten) Photoreceptor-specific nuclear receptor progressive rod cone dysplasia langer Arm eines Chromosoms Random amplified polymorphic DNA Retinal Information Network Royal College of Surgeons rat rod cone dysplasia Recoverin rod dysplasia retinal degeneration slow Restriktionsfragmentlängenpolymorphismus Rhodopsin Rhodopsinkinase Ribonukleinsäure Abkürzungsverzeichnis ROM1 RP RPA RPE RPE65 RPGR rpm RT RT-PCR RVC s SDS SNP SMD SSCP T Tab. Taq TBE TE TIMP Tris tRNA TULP U Upm UTR UV V VNTR v/v W w/v XLPRA VIII rod outer segment membrane protein 1 Retinitis pigmentosa Retinitis punctata albescens retinal pigment epithelium retinal pigment epithelial protein 65 Retinitis pigmentosa-GTPase-Regulator Umdrehungen pro Minute Raumtemperatur Reverse Transkriptase-PCR Recoverin Sekunde(n) Natriumdodecylsulfat singel nucletide polymorphisms Sorsby-Makuladystrophy single strand conformation polymorphism Temperatur Tabelle Thermophilus aquaticus Tris-Borat-EDTA Tris-EDTA tissue inhibitor of metalloproteinases Trishydroxymethylaminomethan transfer RNA tubby-like protein unit Umdrehungen pro Minute nicht-translatierter Bereich ultraviolettes Licht Volt variable number of tandem repeats volume per volume Watt weight per volume X-linked progressive retinal atrophy Kapitel 1 Einleitung Kapitel 1 Einleitung Unter den vielen Krankheitsursachen, die in den Industrienationen Blindheit zur Folge haben, nehmen die vererbten Augenkrankheiten einen großen Anteil ein. Die größte Gruppe dieser genetisch und klinisch heterogenen Netzhauterkrankungen ist die Retinitis pigmentosa (RP). Bei dieser Erkrankung kommt es zur Degeneration der Photorezeptorzellen. Meist gehen zuerst die Stäbchen und daran anschließend auch die Zapfen unter. Weltweit ist einer von 5000 Individuen erkrankt, womit etwa 1,2 Millionen Menschen weltweit von RP betroffen sind (Weleber et al. 2001). In Deutschland liegt die Prävalenz zwischen 40000 und 60000. Aufgrund unterschiedlicher Vererbungsmuster trägt statistisch jeder 40. - 60. Mensch eine Anlage in sich, die im ungünstigsten Fall zu RP führen kann. Konsequenzen der unterschiedlichen Gendefekte führen zu einem weiten Spektrum von Phänotypen, vom frühen Krankheitsbeginn mit hoher Progressionsrate bis zur nichtprogressiver oder stationärer Nachtblindheit. Die Identifikation von Mutationen, die zu Störungen der Sehfähigkeit führen, ist ein entscheidender Weg, um die Funktionsweise der Gene und deren Proteinprodukte in der Retina zu verstehen. Dabei spielen Modellorganismen wie der Hund mit Gendefekten, die zu RP entsprechenden Augenerkrankungen führen, eine wichtige Rolle, da Experimente mit RPPatienten ausgeschlossen sind. Jede Hunderasse hat, ähnlich wie in menschlichen RP betroffenen Familien, ihre spezifische generalisierte progressive Retina-Atrophie- (gPRA)Mutation, so dass zwischen den von gPRA betroffenen Rassen vergleichbare Heterogenität wie im Menschen zu beobachten ist. Identifizierung der verschiedenen Gendefekte beim Hund hilft bei der Erforschung der Krankheitsursachen auf molekularer Ebene. Wenn der pathologische Effekt der Mutationen im Hund verstanden ist, könnten konventionelle oder Gentherapien eingeführt werden, die den Krankheitsbeginn verzögern oder sogar die Erkrankung heilen und die dann nach erfolgreichen Therapien beim Hund auch beim Menschen zum Einsatz kommen könnten. 1 Kapitel 1 Einleitung 1.1 Retina der Vertebraten 1.1.1 Organisation der Retina Die Retina entsteht aus einer Ausstülpung des Zwischenhirns und kleidet die hintere innere Oberfläche des Auges aus. Die neurale Retina umfasst drei Zellschichten. Die äußere Schicht besteht aus den Photorezeptorzellen, den Zapfen und Stäbchen. Sie stehen im engen Kontakt zum retinalen Pigment Epithel (RPE). Die Sinneszellen werden in der inneren nukleären Schicht synaptisch über Bipolarzellen mit den Ganglien in der inneren plexiformen Schicht verknüpft, deren Axone im Nervus opticus enden und ins Gehirn ziehen. Die synaptischen Querverbindungen innerhalb der Retina werden von den Horizontal- und amakrinen Zellen übernommen (Abbildung 1.1). In der Retina des Menschen wurden 100-120 Millionen Stäbchen aber nur ca. 6 Millionen Zapfen nachgewiesen. Zapfen findet man in der gesamten Retina, sie kommen aber konzentriert im gelben Fleck der Macula lutea vor (dem Bereich des scharfen Sehens mit dem Ort des schärfsten Sehens der Fovea centralis). Stäbchen sind gehäuft in der Peripherie der Retina und im Randbereich der Fovea centralis anzutreffen. Durch den Mangel an Stäbchen am gelben Fleck kann man in der Dämmerung keine Farbe wahrnehmen und nur unscharf sehen. An der Austrittsstelle des nervus opticus, dem blinden Fleck, gibt es keine Photorezeptoren (Schmidt 1998). 1.1.2 Aufbau der Zapfen und Stäbchen Das lichtempfindliche Außensegment der Stäbchen (Abbildung 1.2) enthält dichtgepackte, von der Plasmamembran getrennte Membranscheibchen, die "Disks", die zur Vergrößerung der sensorischen Oberfläche dienen. In der Diskmembran befindet sich das lichtempfindliche Pigment Rhodopsin in einer extrem hohen Dichte von ca. 30000 Moleküle pro µm2. Auch weitere Proteine der Phototransduktionskaskade sind hauptsächlich in den Disks der Rezeptoraußensegmente lokalisiert. Der Ort der Proteinsynthese und des Energiestoffwechsels ist das Innensegment mit seinem Kern und Mitochondrien. In der Synapse befinden sich glutamathaltige Vesikel, die den Neurotransmitter bei depolarisiertem Membranpotential ausschütten und damit das Signal auf die nachgeschalteten Zellen übertragen. Stäbchen zeigen eine sehr hohe Lichtempfindlichkeit; sie ermöglichen das Sehen bei sehr geringer Lichtintensität. Vom Rhodopsin der Stäbchen, die für das farblose Dämmerungssehen verantwortlich 2 Kapitel 1 Einleitung sind, wird das Licht des ganzen sichtbaren Spektrums absorbiert. Das Absorptionsmaximum der Stäbchen liegt bei 500 nm (Schmidt 1998). Bei den Zapfen (Abbildung 1) sind die Disks nicht von der Plasmamembran getrennt, sondern sie entstehen durch Einfaltung der Plasmamembran. Zapfen sind etwa 20-mal weniger lichtempfindlich als Stäbchen, reagieren aber schneller auf Lichtänderungen. Zapfen sind bei Tageslicht für die Wahrnehmung von Farben, Kontrasten und Bewegung verantwortlich. Beim Menschen gibt es drei verschiedene Zapfentypen mit unterschiedlichen Farbpigmenten die blauviolettes Licht, grünes Licht, gelbes Licht und rotes Licht ausreichend absorbieren (Stryer 1995). In Zapfen werden Isoformen der Phototransduktionkaskade wie Opsin, Transducin und die Phosphodiesterase verwendet, die auch in den Stäbchen vorkommen, allerdings mit einigen charakteristischen Unterschieden in den Aminosäuresequenzen (Peng et al. 1992). 1.1.3 Retinales Pigmentepithel (RPE), Vitamin-A Zyklus Direkt an der Retina liegt das retinale Pigmentepithel (RPE; Abbildung 1.2). Jede RPE-Zelle steht im engen Kontakt von ca. 45 Außensegementen der Photorezeptorzellen. Eine der wichtigen Funktionen des RPE ist die tägliche Phagozytose von ca. 10 % der Photorezeptoraußensegmente. Eine andere Funktion betrifft die Regeneration von Sehpigmenten im sog. Vitamin-A Zyklus. Die Sehpigmente in allen Vertebraten bestehen aus einem Apoprotein, dem Opsin und einem kovalent gebunden Chromatophor dem 11-cisRetinal, einem Aldehydderivat des Vitamin A. Photoaktivierung isomerisiert Retinal von 11cis in die all-trans Form. Die all-trans Form dissoziert dann vom Opsin und wird durch ein neues Molekül 11-cis Retinal ersetzt. In Stäbchen wird das freigesetzte all-trans Retinal Chromatophor in all-trans Retinol (dem entsprechenden Alkoholderivat des Vitamin A) umgesetzt und zum RPE weitergeleitet. Im RPE wird es zu einem Lipid verestert, chemisch zur 11-cis Konfiguration isomerisiert, von der Esterverbindung hydrolisiert am Ende zu einem Aldehyd oxidiert und anschließend den Photorezeptoren wieder zugeführt. Der Isomerisationszyklus in den Zapfen findet im Gegensatz zu den Stäbchen eher in der Retina als in dem RPE statt (Rando 1992; Rodieck 1998). 3 Kapitel 1 Einleitung (Nervus opticus) Abbildung 1.1 Schematische Darstellung eines Wirbeltierauges mit Struktur der Retina, PE = Pigment Epithelium (verändert nach http://webvision.med.utah.edu/). A. B. Abbildung 1.2 Schematische Darstellung und elektronenmikroskopische Aufnahmen von Stäbchen- und Zapfen-Photorezeptorzellen (verändert nach Eckert 2000). Die lichtempfindlichen Außensegmente enthalten dichtgepackte Membransysteme, die Disks, im Außensegment (AS) der Stäbchen (A) "freischwimmend", im Bereich des Ziliums wie in den Zapfen (B), eingefaltet. Im elektronenmikroskopischen Bild wird der enge Kontakt der Photorezeptorzellen mit dem retinalen Pigment Epithel (RPE) erkennbar (Pfeile). (IS): Innensegment. 4 Kapitel 1 Einleitung 1.1.4 Phototransduktion Bei Absorption eines Photons durch Rhodopsin wird das Lichtsignal zunächst in ein chemisches Signal umgewandelt. In der Erregungsphase der Phototransduktion (siehe Abbildung 13.1) wird ein Photon durch das Chromatophor des Rhodopsins, das Retinal, absorbiert. Eine lichtinduzierte Isomerisierung des Retinals aktiviert Rhodopsin (Rho*). Rho* seinerseits aktiviert das GTP-bindende Protein Transducin (T), das aus drei Untereinheiten (α, β und γ; GNAT; GNBT; GNGT) besteht. Bei Aktivierung dissoziiert die GTP-bindende αUntereinheit von dem βγ-Komplex. Dabei wird die Phosphodiesterase (PDE) durch Abtrennung der inhibitorischen γ-PDE-Untereinheiten aktiviert. PDE katalysiert die Hydrolyse des cGMP zu GMP. In der Plasmamembran der Außensegmente der Photorezeptorzellen sind cGMP-gesteuerte Kationenkanäle lokalisiert. Bei Belichtung fällt die cGMP-Konzentration. Dadurch werden die cGMP-gesteuerte Kationenkanäle (CNCG) geschlossen, wodurch dann der Na+- und Ca2+-Einstrom verringert wird. Daraus resultiert eine Hyperpolarisation der Membran, die zum Verschluss der synaptischen Ca2+-Kanäle führt und so die Freisetzung des Neurotransmitters Glutamat in den Synapsen der Rezeptoren stoppt, wodurch die Photorezeptorzelle inaktiviert wird. Die cGMP und Ca2+-Konzentration sind voneinander abhängig. Die cGMP-Synthese wird durch das Enzym Guanylylzyklase (GC) durch die regulatorischen Calcium-bindenden Proteine guanylyl cyclase activating protein 1 und 2 (GCAP) kontrolliert. GCAP aktiviert GC, aber nur bei niedriger Ca2+-Konzentration. Geringe intrazelluläre Ca2+Konzentration stimuliert eine negative Rückkopplungschleife, die die Zelle in ihren Anfangszustand versetzt (Palczewski & Saari 1997). Die Erholungsphase der Phototransduktion ist gekennzeichnet durch aktives Inaktivieren und Wiederaufbereitung der Transduktionskomponenten. Rhodopsin wird durch die kombinierte Aktion von der Rhodopsin Kinase (RHOK) und Arrestin (SAG) inaktiviert. RHOK phosphoryliert photoaktiviertes RHO und SAG bindet phosphoryliertes RHO und entfernt die Phosphatgruppe, wobei gleichzeitig all-trans Retinal freigegeben und 11-cis Retinal gebunden wird. Transducin hydrolysiert GTP zu GDP und Pi eine Reaktion, die durch ein Mitglied der G-Protein-Regulatorfamilie (RGS) beschleunigt wird. Durch RGS-9 wird die PDE in den inaktivierten Status zurückversetzt (Morimura et al. 1999). Hemmende Wirkungen der Sehkaskade wird u.a. durch Phosducin (PDC) erreicht. Es bindet sich an die β- und γ-Untereinheit des T und verhindert somit die erneute Anlagerung von T-α. Dadurch kann T-α kein GDP zu GTP umwandeln, der Kreislauf kann nicht mehr von neuem 5 Kapitel 1 Einleitung beginnen, was wiederum dem Phototransduktionsprozess entgegenwirkt. PDC wird bei geringem Licht von der Proteinphosphatase 2A (PP2A) dephosphoryliert (Lee et al. 1992). Ca2+-Ionen haben eine hemmende Wirkung auf SAG und Recoverin (RCV). Durch Absinken der intrazellulären Ca2+-Konzentration wird SAG und RCV freigesetzt. RCV aktiviert die GC, die aus GTP wieder cGMP herstellt, so dass der Phototransduktionsprozess erneut beginnen kann. Bei ansteigender Ca2+-Konzentration in der Zelle bindet sich Ca2+ an RCV, so dass dieses die GC nicht weiter aktivieren kann. Die Aktionen der GC führt zu einer Zunahme von offenen cGMP-gesteuerten Kationenkanälen (Yau et al. 1994). Calcium aktiviert die PDE bzw. hemmt die RHOK. Calcium hat somit auch Einfluss auf die Sensitivität der Photorezeptoren und die Adaptation an verschiedene Lichtverhältnisse (Yau et al. 1994). 1.1.5 Wahrnehmungsunterschiede des Lichtes von Menschen und Hunden Es gibt fundamentale Wahrnehmungsunterschiede zwischen Menschen und Hunden. Das visuelle System des Hundes ist gut auf Dämmerlicht abgestimmt. Was sich durch die Evolutionsgeschichte des Hundes als direkter Nachfahre des Wolfes erklärt. Der Wolf ist ein Dämmerungsjäger und ist dementsprechend an diese Lichtverhältnisse hervorragend angepasst. Das Optimum der Wahrnehmung erreicht der Mensch dagegen im hellem Licht (Miller at al. 1995). Menschen und Hunde zeigen eine unterschiedliche Stäbchen- und Zapfenverteilung in der Retina. Im Gegensatz zur Retina des Menschen wurden in der Hunderetina weniger Zapfen nachgewiesen. Mit einer der wesentlichen Gründe, weshalb Hunde sensitiver auf Licht reagieren und besser im Dämmerlicht sehen können. Ein weiterer Grund ist das in schillernden Farben schimmernde Tapetum lucidum. Es befindet sich im oberen Teil des Augenhintergrunds. Das angrenzende Tapetum nigrum ist dunkel pigmentiert und reduziert die Lichtstreuung, die von der Sonne ausgeht. Das Tapetum des Hundes ist ein effektiver Reflektor und reizt die Rezeptorzellen der Retina zweimal, einmal beim Eintritt und einmal beim Austritt des Auges. Dies führt zur zusätzlich Verbesserung der Wahrnehmung in der Dämmerung. Eine geringere Anzahl von Ganglionzellen, erkennbar durch die Anzahl der Nervenfasern die zum Nervus opticus führen, weist auf eine geringere Sehschärfe des Hundes als des Mensch hin. Der Nervus opticus beim Hund besteht aus ca. 167.000 Nervenfasern während der menschliche Nervus opticus ca. 1,2 Millionen Fasern umfasst. Die Fovea ist ein Bereich mit höchster Konzentration von Photorezeptoren und Ganglionzellen und somit der Bereich des schärfsten Sehens beim Menschen. Der Hund hat keine Fovea sondern einen sog. visuellen Streifen, ein ovaler Bereich, der direkt oberhalb des 6 Kapitel 1 Einleitung Nervus opticus, horizontal in der tapetalen Zone liegt (Peichl 1992). Er ist allerdings auch ein Areal mit der höchsten Konzentration von Photorezeptoren und Ganglionzellen und wird wie beim Menschen, somit zum Bereich des schärfsten Sehens. Die Lage in der tapetalen Zone führt zur Erhöhung der Wahrnehmung im Dämmerlicht, aber Lichtstreuung im hellem Licht verschlechtert die Sehschärfe. Die ovale Form ermöglicht dem Hund höchstwahrscheinlich sowohl den Horizont zu erfassen als auch peripheres Sehen (Peichl 1992). Geringere Dichte der Ganglionzellen im visuellen Streif sind in den meisten domestizierten Hunderassen im Vergleich zu Wölfen erkennbar. So kann davon ausgegangen werden, dass Wölfe durch umweltbedingte Notwendigkeiten, weit schärfer sehen als die meisten Rassehunde, die durch Domestikation diese Fähigkeit verloren haben (Peichl 1992). Hunde reagieren sehr gut auf Bewegung und Figurenunterschiede. Dies liegt an der höheren zeitlichen Bildauflösung im Hundeauge. Der Hund kann unter günstigen Lichtverhältnissen bis zu 80, der Mensch aber nur bis zu 60 Bilder pro Sekunde wahrnehmen (Miller & Murphy 1995). Auch das Raumempfinden bzw. das Tiefensehen ist unterschiedlich. Durch die seitliche Augenstellung beim Hund kann der Hund nur monokular sehen, also nur zweidimensional (Sherman & Wilson 1975), im Gegensatz zum Menschen der durch die frontale Augenstellung biokular sehen kann und so die Umwelt dreidimensional wahrnimmt. Der Hund ist im Vergleich zum Menschen, der bis zu 160 Farben unterscheiden kann, in seiner Farbenerkennung eingeschränkt. Dies liegt an der zum Menschen unterschiedlichen Art der Zapfen. Während der Menschen über drei unterschiedliche Zapfentypen verfügt, wurden beim Hund nur zwei deutlich von einander abzugrenzende Zapfenarten nachgewiesen. Der sensitive Spektralbereich dieser Zapfen liegt bei 429-435 nm (blau-violett) und bei über 555 nm (gelb-grün; Neitz et al. 1989; Jacobs 1983). Dieses Farbensehen entspricht nicht ganz dem Farbensehen eines „rot-grün-blinden“ Menschen, da die geringere Anzahl von Zapfen zu Farbspektrumverschiebungen führt. Allerdings können Hunde wegen der geringeren Menge an Zapfen Grautöne besser voneinander unterscheiden (Peichl 1992). 1.2 Phänotypen retinaler Erkrankungen im Menschen und im Hund 1.2.1 Retinitis Pigmentosa RP umfaßt eine Gruppe von genetischen Erkrankungen, deren charakteristische Eigenschaft progressive Photorezeptordegeneration ist. Charakteristische Merkmale dieser Augenerkrankung sind der frühe Verlust der peripheren Rezeptorzellen der Tunnelblick und Nachtblindheit. Im weiteren Verlauf der Erkrankung kommt es durch Degeneration der zentralen 7 Kapitel 1 Einleitung Photorezeptorzellen auch zum Ausfall des zentralen Gesichtsfeldes und führt im Endstadium zur Blindheit. Typische Krankheitszeichen sind periphere intraretinale knochenkörperchenartige Pigmentation, Verringerung der retinalen Blutgefässe und Veränderungen der Papille des Nervus opticus in der Retina. Die Diagnose RP wird durch verschiedene Untersuchungsmethoden oder Tests gesichert, unter anderem durch die Höhe der Amplituden im Elektroretinogramm (ERG), dem Test der Adaptation an die Dunkelheit und der Untersuchung des Gesichtsfelds und Störungen des Farbsinns. Die Erkrankung ist genetisch heterogen und folgt zusammengenommen allen bekannten Vererbungsmodi, dem autosomal dominanten (ad), dem autosomal rezessiven (ar), X-chromosomalen, maternalen und digenischen Erbgang (Dryja 1995; Danciger et al. 1995; Farrar et al. 1993; Kajiwara et al. 1994; Mansergh et al. 1997; Shastry 1997). Viele betroffene Patienten sind jedoch sog. Simplex-Fälle, in deren Familien keine weiteren Betroffen bekannt sind. Ihnen liegt meist eine autosomal rezessive oder X-chromosomale Vererbung zugrunde oder in ganz seltenen Fällen eine Neumutation. Die meisten RP-Patienten zeigen keine extraokulären Auffälligkeiten, aber die Erkrankung kann auch in Kombination mit Taubheit (UsherSyndrom), Polydaktylie oder mentaler Retardierung auftreten (Kellner 1997). 1.2.2 Generalisierte progressive Retina Atrophie Formen beim Hund Auch beim Hund sind eine große Anzahl von natürlich vorkommenden, erbliche Photorezeptorerkrankungen sowie Störungen des RPE bekannt. Der Krankheitsverlauf ist meist progressiv, in wenigen Fällen aber auch langsam fortschreitend, wobei hier entweder nur Stäbchen oder Zapfen betroffenen sind (Petersen-Jones 1998). Die progressiven Erkrankungen, wie z.B. die gPRA, umfassen eine heterogene Gruppe von genetisch differenzierbaren retinalen Erkrankungen mit sehr ähnlichen Phänotypen. In den meisten Hunderassen wird die gPRA ar vererbt (Clements et al. 1996). Nur bei sibirischen Huskys und Samoyede wurde zusätzlich ein X-gekoppelter Erbgang nachgewiesen (Acland et al. 1994; Zeiss et al. 2000). Kürzlich wurde bei englischen Mastiffs auch ein ad vererbte retinale Dysfunktion identifiziert, die durch eine Mutation im RHO verursacht ist (Kijas et al. 2002). Die ophthalmologischen Befunde und Wahrnehmungsdefizite, ausgelöst durch die Degeneration der Photorezeptorzellen, führen bei den betroffenen Hunden im Endstadium immer zur Blindheit und oft auch zum sekundären Katarakt. Der klinische Phänotyp der gPRA des Hundes ist homolog zur RP beim Menschen (siehe Abbildung 1.3). Die gPRA kann in zwei Klassen unterteilt werden, zum einen in Krankheiten mit Störungen der Entwicklung vor Zelldifferenzierung und zum anderen in Degenerationen der Photorezeptorzellen nach 8 Kapitel 1 Einleitung Zelldifferenzierung. Mehrere unterschiedliche Genloci für diese sog. Dysplasien, die durch einen fehlerhaften Aufbau von Zapfen oder Stäbchen während der Embryonalentwicklung charakterisiert sind, sind bekannt: rod dysplasia (rd; Aguirre 1978), rod cone dysplasia (rcd1; rcd2; rcd3; Ray et al. 1994; Wang et al. 1999; Petersen-Jones et al. 1999) und early retinal degeneration (erd; Acland & Aguirre 1987 und Acland et al. 1999). Die Dysplasien sind histologisch gut voneinander zu unterscheiden. Der Krankheitsbeginn ist schon im jungen Welpenalter und ist im Verlauf meist progressiv. Das Krankheitsbild der Photorezeptorzellendegeneration ist durch langsame Progression gekennzeichnet. Zuerst degenerieren zwar die Stäbchen relativ schnell. Die Zapfendegeneration erfolgt danach aber viel langsamer, sodass das Sehvermögen für einen längeren Zeitraum erhalten bleibt. Diese gPRA-Formen umfassen die progressive rod-cone degeneration (prcd; Aguirre & Acland 1988; Acland et al. 1998) und die XLPRA (Zeiss et al. 1999 und Zhang et al. 2001). Zu den nicht progressiven Augenerkrankungen zählen die cone degeneration (cd; Gropp et al. 1996), hier sind nur die Zapfen betroffen. Außerdem die congenitale stationäre Nachtblindheit (csnb), wobei ein defektes Protein des RPE zur Degeneration der Photorezeptorzellen führt. Mutationen in diesen Genloci wurden inzwischen in verschiedenen Hunderassen identifiziert (Tabelle 2.1). In mehr als 70 Hunderassen ist die gPRA noch nicht charakterisiert und die für die Erkrankung ursächlich Genloci sind nicht identifiziert. A. B. No No Abbildung 1.3 Retina eines gesunden und an gPRA-erkrankten Sloughis dargestellt mittels eines Ophthalmoskops. (A) gesunde Retina mit dem Nervus opticus (No), den miteintretenden retinalen Blutgefäßen und normaler Pigmentierung. (B) Retina eines gPRA-erkrankten Sloughis mit peripherer intraretinaler typischer Pigmentation und Verringerung der retinalen Blutgefässe und Veränderungen der Papille des Nervus opticus (Abbildungen von Dr. Brahm; DOK). 9 Kapitel 1 Einleitung 1.2.3 Natürliche Modellsysteme für retinale Erkrankungen Als natürliche Modellsysteme für RP und andere retinale Erkrankungen kommen eine Anzahl von Haustieren infrage, die der RP homologe Erkrankungen entwickeln (Tabelle 1.1). Der moderne domestizierte Hund ist eines der wichtigsten Modellsysteme für RP, da er eine Kombination von Charakteristiken zeigt, die für die Untersuchung von menschlichen genetischen Erkrankungen entscheidend sind. So ist der medizinische Überwachungsgrad des Hundes durchaus mit dem des Menschen vergleichbar. Diagnosen können durch erfahrene Tierärzte mit Hilfe moderner Geräte gestellt werden, sodass die klinischen, ophthalmologische und genetischen Daten zur Verfügung stehen. Der Vererbungsmodus ist für die meisten genetischen Erkrankungen durch die intensive Züchtung definiert. Meist folgen sie dem ar oder dem X-chromosomalen Erbgang. Dominant vererbte Erkrankungen spielen allerdings nur eine untergeordnete Rolle, da betroffene Hunde sofort nach dem Auftreten der Erkrankung von Züchtern frühzeitig aus der Zucht herausgenommen werden, sodass sich das krankheitsverursachende Allel nicht verbreiten kann. Ein weiterer Vorteil der Rassehunde für vergleichende genetische Studien, ist die Struktur der Hundepopulationen. Insgesamt bestehen aus mehr als 300 genetische Isolate (Rassen) sowie die heterogene Gruppe von Hundemischlingen. Der Genfluss zwischen den einzelnen Rassen wird durch die Registrierung der Eltern und Nachkommen, der Stammbaum-Barriere, beschränkt (Ostrander et al. 2000). Die nützlichsten Hundemodellsysteme sind jene mit natürlich vorkommenden genetischen Erkrankungen, die homolog zu genetischen Erkrankungen des Menschen sind. Mehr als 215 dieser genetischen Erkrankung, die beim Hund bekannt sind, zeigen klinische Symptome und entsprechende Laborbefunde die auch bei menschlichen Erkrankungen erkennbar sind. In 41 dieser Hunde wurde nachgewiesen, dass das gleiche Genprodukt, wie in der entsprechenden menschlichen Krankheit, defekt ist. Inzwischen wurde beim Hund insgesamt 21 genetische Erkrankungen auf der Genebene charakterisiert, die homolog zu genetischen Erkrankungen beim Menschen sind (Ostrander et al. 2000). Hierunter fallen u.a. die erblichen Retinaerkrankungen wie die gPRA und die CSNB. Für den ar Vererbungsmodus wurden in vier Genen krankheitsverursachende Mutationen in fünf verschiedenen Rassen und zwei Mutationen in einem Gen in zwei Rassen für den X-chromosomalen Erbgang identifiziert. Bisher nur in einer Rasse, den Englischen Mastiffs, wurde eine Mutation für den ad Vererbungsmodus im RHO Gen nachgewiesen (Kijas et al. 2002). Weitere wichtige Tiermodellsysteme sind die Maus und die Ratte. Hier spielen die kurzen Reproduktionszeiten und schnelle Verfügbarkeit von Zuchtlinen für die Forschung eine 10 Kapitel 1 Einleitung wichtige Rolle (Frederick et al 2000). Außerdem ist die Genomsequenz der Maus fast vollständig und die der Ratte größtenteils aufgeklärt. Allerdings haben Mäuse nur eine kurze Lebensdauer, sodass sie für altersbedingte Erkrankungen nicht kein gutes Modell darstellen. Zusätzlich sind blinde Mäuse schwer von ihren sehenden Geschwistern zu unterscheiden, da sie sich in ihrer vertrauten Umgebung nicht anders verhalten als sehende Mäuse. Deshalb ist der Verlust der Sehkraft nur durch hochentwickelte elektrophysikalische Methoden nachzuweisen. 1.2.4 Mutierte Gene in natürlichen Tiermodellen 1.2.4.1 Photorezeptorspezifische Gene 1.2.4.1.1 cGMP Phosphodiesterase Die PDB ist ein Enzym der Sehkaskade, das die cGMP-Hydrolyse des internen Botenstoffes der Phototransduktion, dem zyklischen GMP (cGMP), in Zäpfchen nach Lichtaktivierung durchführt. Das Enzym ist aus fünf Untereinheiten zusammengesetzt, den zwei katalytischen α-, β- (PDE6A; PDE6B), zwei identischen hemmenden γ-Untereinheiten (PDE6G; Stryer 1996) und einer δ Untereinheit (PDE6D; Florio et al. 1996) mit noch nicht vollständig aufgeklärter Funktionsweise. Störungen in der PDE führen zu einem veränderten cGMP Metabolismus. Im an rcd1 erkrankten Irischen Setter konnte in der betroffenen Retina eine 10x höhere cGMP-Konzentration nachgewiesen werden (Aguirre et al. 1982). Im PDE6B Gen wurden Mutationen sowohl in der rd Maus als auch in zwei Hunderassen, dem Irischen Setter (Suber et al. 1993) und den Sloughis (im Rahmen dieser Arbeit; Dekomien et al. 2000) nachgewiesen. In der rd Maus wurde im Intron 1 die Sequenz eines Leukämievirus. In Exon 7 wurde ein Basenaustausch identifiziert, der zu einer Unsinn-Mutation führte (Pittler & Baehr 1991; Bowes et al. 1993). Im Irischen Setter ist eine Transition im Exon 21 im Kodon 806 des PDE6B Gens die Ursache für ein vorzeitiges Stoppkodon (Suber et al. 1993). Im gleichen Exon wurde im Rahmen dieser Arbeit in gPRA erkrankten Sloughis eine zum Stoppkodon führende Isertion von 8 bp nachgewiesen (Dekomien et al. 2000). Im PDE6A Gen ist eine 1 bp Deletion im Kodon 616 die Ursache für den rcd3 Defekt in Cardigan Welsh corgi (Petersen-Jones et al. 1999). Diese Mutationen verkürzen die Proteine, dadurch wird das Holoenzym PDE instabil und es verliert seine hydrolytische Funktion. In der Retina der betroffenen Tiere steigt der cGMP-Spiegel auf zelltoxische Werte an und löst so die Rezeptorzellendegeneration aus. 11 Kapitel 1 Einleitung Im Menschen mit ar RP wurden eine Reihe von Mutationen sowohl im PDE6B (Danciger et al. 1996; McLaughlin et al. 1993; Bayes et al. 1995) als auch im PDE6A (Huang et al. 1995) Gen identifiziert. 1.2.4.1.2 RDS/Peripherin und ROM1 RDS/Peripherin und das Membranprotein des Stäbchenausssegments (ROM1) sind Strukturproteine im Randbereich der Disks in den Außensegmenten der Zäpfchen. Sie bilden Heterotetramere in unterschiedlicher Zusammensetzung und sind wichtig für den Aufbau der Diskmembranen dieser Photorezeptorzellen (Bascom et al. 1995). Mutationen im RDS Gen führen, wie in dem retinal degeneration slow (rds) Mausmodell, zu retinalen Erkrankungen. Der Defekt in diesem rds Mausmodell ist eine Insertation von 10 bp eines Maus spezifischen genomischen repetitiven Elements in einem kodierenden Exon. Dies führt zu einem verkürzten funktionslosen Protein. Die daraus hervorgehende Photorezeptordegeneration entwickelt sich viel langsamer als in der rd Maus und die Mäuse werden erst relativ spät blind. Beim Menschen sind Mutationen nur im RDS Gen bekannt, die dem ad Vererbungsmodus folgen (Farrar et al. 1991; 1993; Kajiwara et al. 1991). Zusätzlich ist auch ein digenischer Erbgang in RP Patienten bekannt. Sie sind heterozygot für eine Mutation im RDS und zusätzlich auch im ROM1 Gen (Kajiwara et al. 1994). Diese Mutationen führen zur Außensegment-Desorganisation und anschließend zur Photorezeptordegeneration. 1.2.4.1.3 Phosducin Im PDC Gen wurde eine Fehlsinn-Mutation in homozygotem Zustand in erkrankten Zwergschnauzer entdeckt. Diese R82G Mutation führt höchstwahrscheinlich zur photoreceptor dysplasia (pd). Allerdings müssen Zwergschnauzer wohl noch eine weitere Mutation in einem anderen Gen aufweisen, da nicht in allen, sicher an gPRA erkrankten Hunden, diese Mutation nachzuweisen war (Zhang et al. 1998). Beim Menschen wurde noch keine RP relevante Mutation im PDC Gen nachgewiesen. 1.2.4.1.4 Myosin VIIA (USH1B) Das Usher Syndrom ist eine meist rezessive vererbte, aber genetisch heterogene Gruppe von Erkrankungen, die durch die Kombination von Taubheit und RP charakterisiert ist (Kloepfer & Laguaite 1966). Bei der sog. shaker-1 (sh1) Maus, die die typischen Merkmale des Usher Syndroms zeigt, wurde eine Mutation im homozygoten Zustand im USH1B Gen entdeckt. Die Funktion des Gens ist noch nicht vollständig aufgeklärt. Untersuchungen mit der sh1 Maus 12 Kapitel 1 Einleitung zeigen Expression eines mutierten Myosin VIIA Proteins, was höchstwahrscheinlich die Ursache für eine fehlerhafte Melanosomenverteilung innerhalb des RPE ist. Außerdem scheint sich die Hypothese zu bestätigen, dass Myosin IIA in der Außensegmentbiogenese der Retina von sh1 Mäusen für den intrazellulären Transport des Opsin entlang der Zilien zu den Außensegmenten wichtig ist (Liu et al. 1998). Es gibt allerdings auch sich widersprechende Aussagen über die Lokalisierung des USH1Bs in Primaten und Nagetieren, die die Hypothesen der Funktionsweise des Myosin IIAs aber wieder in Frage stellen (Liu et al. 1998; El-Amraoui et al. 1996). 1.2.4.1.5 RP Guanosin triphosphat GTPase Regulator (RPGR) Zwei X-Chromosomal gekoppelte gPRA Formen sind in Sibirischen Huskys und den Samojede bekannt, die homolog zu dem RP3 Typ im Menschen sind. Zwei unterschiedliche Mutationsorte beeinträchtigen die korrekte Bildung des gleichen Gens. Kopplungsstudien weisen nach, dass der XLPRA Lokus dem RP3-Lokus in den chromosomalen Abschnitt (Xp21) entspricht, wo das RPGR Gen lokalisiert wurde ( et al. 2000; et al. 2001; Optigen). Allerdings wurden nur wenige ursächliche Mutationen ( et al. 1997), unterschiedliche Spleißvarianten des Gens sowohl beim Menschen ( et al. 1999) als auch beim Hund identifiziert. Es ist noch nicht sicher nachgewiesen, welche der Varianten die Erkrankung auslöst. Die Funktion des RPGR Gens ist noch nicht vollständig aufgeklärt. Es zeigt aber große Homologie zu der Familie der RCC1 Familie der Guanin Nukleotid Austauschfaktoren. In XLPRA erkrankten Rüden degenerieren zuerst die Stäbchen. Dann schließt sich die Zapfendegeneration an und mündet in die komplette Retinaatrophie. In weiblichen Trägern des krankheitsverursachenden Allels ist ein stochastisches Muster der Photorezeptordegeneration zu erkennen, die der zufälligen Inaktivierung des X Chromosoms entspricht (Aguirre 2001). 1.2.4.1.6 Rhodopsin Erst kürzlich wurde eine dominant vererbte PRA Form in englischen Mastiffs identifiziert. Eine Punktmutation im RHO Gen führt zu einem Aminosäureaustausch von Thyrosin zu Arginin in AS-Position 4 des Proteins. Es gibt zwei unterschiedliche Krankheitsverläufe, die auf Mutationen im RHO Gen zurückzuführen sind. Ein Krankheitsbild mit hoher Progession, hier beginnt die Augenerkrankung sehr früh und führt durch gleichmäßige Degeneration der Zäpfchen zu einem schnellen Verlust der Sehkraft. Das andere Krankheitsbild ist durch 13 Kapitel 1 Einleitung Beeinträchtigung der Stäbchen nach starker Sonneneinstrahlung charakterisiert. Außerdem zeichnet sich diese Form nur durch lokale Degenerationen in der Retina und eine geringe Progressionsrate mit spätem Krankheitsbeginn aus. Diese zuletzt genannte Erkrankungsform wurde in der dominanten gPRA Form des Hundes nachgewiesen ( et al. 2001). Beim Menschen sind mehrere Mutationen bekannt die zur adRP, als auch zur arRP führen ( et al. 2001). 1.2.4.2 RPE spezifische Gene 1.2.4.2.1 Rezeptor Tyrosin Kinase (Mertk) Schon seit 1938 ist die Royal College of Surgeon (RCS) Ratte als Tiermodell für retinale Dystrophy bekannt (Bourne et al. 1938). Es wurden intensive zellphysiologische und molekulargenetische Studien mit den Zellschichten der Retina dieser Ratte durchgeführt, aber erst vor kurzem wurde eine ursächliche Mutation im Mertk Gen identifiziert (D’Cruz et al. 2000). Dieser genetische Defekt verhindert, dass im RPE die Stäbchenaußensegmente phagozytiert werden, ein entscheidender Prozess für die Funktion und den normalen Aufbau der Stäbchen. Daraus folgt, dass Mertk möglicherweise eine wichtige Rolle bei der RPEAdhäsion mit den Photorezeptoraußensegmenten spielt. Das homologe Gen beim Menschen wurde bei verschiedenen retinalen Dystrophien untersucht und es konnten drei verschiedene Mutationen in Patienten mit RP nachgewiesen werden (Gal et al. 2000). 1.2.4.2.2 Retinales Pigmentepithel spezifisches Protein 65 (RPE65) Das im RPE lokalisierte RPE65 gehört zu einer Gruppe von Proteinen, die in den Vitamin A Zyklus eingebunden sind. In diesem Zyklus finden eine Reihe von biochemischen Schritten statt, die das Chromatophor des RHO, das 11-cis Retinalaldehyd, für die Phototransduktionskaskade zur Verfügung stellt und später auch wieder aufbereitet (siehe Kapitel 1.1.3). Mutationen im RPE65 Gen führen zu einer RP mit sehr frühen Krankheitsbeginn und in einigen Fällen zum Leber (Amaurosis congenita) Syndrom (LCA; Morimura et al. 1998; Thompson et al. 2000). Als Hundemodellsystem steht für diese Erkrankung der Schwedische Briad zur Verfügung. Bei den erkrankten Hunden wurde eine 4 bp Deletion nach Kodon 153 des RPE65 Gens identifiziert, die zu einer Verschiebung des Leserasters und zu einer Verkürzung des Proteins führt (Aguirre et al. 1998; Veske et al. 1999). Bei diesen Hunde wurde erfolgreich eine Gentherapie per Gentransfer etabliert, die das Sehvermögen verbesserte (Acland et al. 2001). 14 Kapitel 1 Einleitung 1.3 Zielsetzung der Arbeit Im Rahmen des gPRA-Projekts am Lehrstuhl für Humangenetik wurden bereits eine Reihe von Genen in den vergangenen Jahren untersucht (Blümel 1996; Dekomien 1997; Dekomien et al. 1998; Klein et al. 1998; Janke 1998; Runte 1999; Runte et al. 2000). Meist basierten diese PCR-SSCP-Analysen jedoch lediglich auf cDNA-Sequenzen (GNAT1, GNGT1, PDE6A, PDE6B Dekomien 1997), so dass die intronischen Spleißkonsensussequenzen, die für die Prozessierung der mRNA notwendig sind, nicht untersucht werden konnten. Punktmutationen in den exonischen Primer-Bindestellen können auf diese Weise nicht entdeckt werden, die möglicherweise sogar ursächlich für die gPRA sind. In anderen Fällen konnte, obwohl die intronische Sequenzinformation vorhanden war, ein Gen innerhalb einer oder mehrerer Rassen trotz Untersuchung des Großteils der Sequenz nicht für die gPRA ausgeschlossen werden (CNCG1, Janke 1998; TIMP1, cGC-E, GNBT1, Gödde 2000). Es wurden auch Gene ausgewählt, die nach Literaturangaben Sequenzvarianten enthielten (PDC, PDE6D, RPE65 und SAG). Diese identifizierten Polymorphismen sind als intragenische Marker nützlich für die Einschätzung des Gens als gPRA-Ursache in den verschiedenen Rassen. Dem Forschungsprojekt zur Verfügung gestellte gPRA erkrankte Hunde können gezielt auf diese Variationen hin untersucht werden, um gegebenenfalls das jeweilige Gen dann z.B. bei nur homozygot identifizierten Sequenzvarianten, vollständig nach Mutationen zu durchsuchen. Dies bietet sich vor allem für erkrankte Hunde in größeren Stammbäumen an, da weiterführende Segregationsanalysen mit den Stammbauminformationen durchgeführt werden können. In Genen, bei denen schon intronische Sequenzen bekannt sind, aber noch keine intragenischen Marker für die Segregationsanalyse zur Verfügung standen, sollten die Introns komplett zur Identifizierung von Variationen untersucht werden (PDE6G). Die kleinen mitochondrialen Gene Cytochromb, ATPase 6 & 8 waren vollständig auf gPRA ursächliche Mutationen hin zu überprüften. In Fällen, in denen die intronischen Bereiche nicht bekannt oder veröffentlicht waren (PDE6A, PDE6B, RPE65, CRX, GNGT1) oder noch keine caninen Sequenzen (RCV1) identifiziert wurden, sollten diese entweder durch intron-überlappende PCR aus genomischer DNA oder über Klonierung aus einer genomischen Phagen-DNA-Bank ermittelt werden. Somit konnten diese Gene unter Einbeziehung der exonischen wie auch der intronischen Sequenzen mittels der PCR-SSCP-Analyse untersucht werden. Das Hauptziel dieser Arbeit war es, ursächliche Mutationen für gPRA in verschiedenen Rassen zu identifizieren und einen Gentest für die entsprechende Rassen zu etablieren. Allerdings war ein weiteres wichtiges Ziel durch identifizierte Sequenzvariationen 15 Kapitel 1 Einleitung intragenische Marker zu gewinnen, die dann für Gen-Ausschlüsse verwendet werden können und somit die ursächlichen Kandidatengene für die gPRA eingrenzen. Tabelle 1.1 Beispiele von erblichen Erkrankungen des RPE-Phororezeptorkomplexes in natürlichen Tiermodellen Erkrankung Symbol Ratte Retinale Dystrophie RCS Maus Retinale Degeneration Retinale degeneration slow Retinale Degeneration 7 nicht wahrnehmbare bKurve rd Huhn Retinale Degeneration Gen/ Lokalisation Referenzen Mertk D’Cruz et al. 2000 ARRP, CSNB PDE6B ADRP - Barnett & Curtis, 1985 ARRP - Narfström 1983 rcd1 ARPR PDE6B Suber et al. 1993 rcd rcd3 ARPR ARPR PDE6B PDE6B Dekomien et al. 2000 Petersen-Jones et al. 1999 erd ARPR HSA12p13-q13 Acland et al.1999 csnb CSNB Lebers congenitale Amourosis RPE65 Aguirre et al. 1998 Veske et al. 1999 prcd RP17 HSA 17q23 Acland et al. 1998 XLPRA RP3 HSA Xp21 Zeiss et al. 2000 cd Achromatopsia HSA 8q21-22 Sidjanin et al. 2002 rd Lebers congenitale Amaurosis GC1 Semple-Rowland et al. 1998 rds rd7 nob Katze Retinale rdy Dystrophie progressive rodprcd cone degeneration Hund rod cone dysplasia1 rod cone dysplasia rod cone dysplasia3 early retinal degeneration congenitale stationäre Nachtblindheit progressive rod cone degeneration X-gekoppelte progessive RetinaAtrophie Achromatopsia homologe Erkrankung beim Menschen Bowes et al 1990; Pittler & Baehr 1991; ADRP, Dystophiemuster & RDS/Peripherin Travis et al. 1989 Flavimaculatus Verstärktes S-Zapfen mPNR Akhmedov et al. 2000 Syndrom X-gekoppelte CSNB1 (CSNB1) Candille et al. 1999 16 Kapitel 2 Material & Methoden Kapitel 2 Material & Methoden 2.1. Material 2.1.1 Chemikalien Aceton (Merck); Acrylamid (Fluka); AG-801-x8 (Biorad); Agar (GIBCO BRL); Agarose (GIBCO BRL); Ammoniumperoxydisulfat = APS (Baker); Ampicillin (Sigma); Borsäure (Baker/MBI Fermentas); Bovin-Serumalbumin = BSA (Fluka); Bromphenolblau (Merck); Chloroform (Merck); Cycolheximid (Sigma); Desoxyribonukleotid-Tri-phosphate / dNTP (2‘Desoxy-Adenosintriphosphat / dATP, 2‘-Desoxy-Cytidin-triphosphat / dCTP, 2‘-DesoxyGuanosintriphosphat / dGTP, 2‘-Desoxy-Tymidintri-phosphat / dTTP; MBI Fermentas); DE52 Cellulose (Whatman); Dichlordimethylsilan (Merck); Dimethylsulfoxid = DMSO (Sigma); Ethanol (Riedel); Ethidiumbromid (Sigma); Ethylen-N,N,N‘,N‘-Tetraessigsäure- Dinatriumsalz = EDTA (Merck); Ethylen-polyethylenglycol = Nonidet P40 (Fluka); Ficoll 400 (Pharmacia), Formaldehyd (Baker); Formamid (Baker); Glycerin (Riedel); Harnstoff (Baker); Hefeextrakt (GIBCO BRL); Isoamylalkohol (Riedel); Isopropanol (Riedel); Magnesiumchlorid = Tetramethylethylendiamin MgCl2 = TEMED (Merck); (Riedel); Mineralöl (Sigma); N,N,N‘,N‘- N,N,N-Methylenbisacrylamid (Fluka); Natriumacetat = NaAc (Riedel); Natriumchlorid = NaCl (Riedel); Natriumcitrat (Riedel); Natriumdodecylsulfat = SDS (Biomol); Natriumhydroxid = NaOH (Riedel); Nonidet P40 = NP40 (Fluka); Phenol (Riedel); Polyethylenglykol (Sigma); Polyoxy-ethylen(20)- sorbitanmonolaurat = Tween 20 (Sigma); Polyvinyl (Sigma); Salzsäure = HCl (Baker); Sephadex G-50 (Pharmacia); Trishydroxymethylaminomethan = Tris (Biomol); Trypton (GIBCO BRL); Xylencyanol FF (Biorad); Xylol (Baker). 2.1.2 Untersuchungsgruppe und DNA-Proben Für die molekulargenetischen Untersuchungen standen Ende Juli 2002 EDTA-Blut von 30 Hundrassen mit insgesamt 953 Hunden mit 153 gPRA-erkrankter Individuen durch die Kooperation von verschiedenen Zuchtvereinen zur Verfügung [(Tab 2.1) Verband für das Deutsche Hundewesen (VDH); Nederlandse Vereiniging van Saarlooswolfhonden; Schweizer 17 Kapitel 2 Material & Methoden Kynologischen Gesellschaft (GKS)]. Durch die Beobachtungen der gPRA-Fälle in den Stammbäumen, gehen die Züchter in folgenden Rassen von einer ar Vererbung aus: Australian Cattle Dogs, Collies, Teckel, Engl. Cocker Spaniel, Entlebucher Sennenhund, Irish Setter, Labrador Retriever, Zwergpudel, Saarloos/Wolfshund, Salukis, Schapendoes, Sloughis und Tibet Terrrier. Die meisten Blutproben wurden von Hundehaltern, -züchtern und verschiedenen Tierärzten aus Deutschland zur Verfügung gestellt. Zusätzlich wurden aber auch Blutproben von Hunden aus verschieden anderen Ländern in die Untersuchung aufgenommen: Chesapeake Bay Retriever aus Großbritannien, Saarloos/Wolfshund und Schapendoes aus den Niederlanden, Sloughis aus der Schweiz, Schweden und der USA und Tibet Terrier aus der Schweiz. Ein erkrankter Hund ist ein Mischling aus den Rassen Vissler und Kouvac. Die jeweilige Diagnose wurde von erfahrenen, speziell geschulten Tierärzten des Dortmunder Ophthalmologen Kreises (DOK) aufgrund einer direkten oder indirekten Ophthalmoskopie gestellt, da sich die vorliegende Erkrankung in einer charakteristischen Veränderung des Augenhintergrundes manifestiert. 18 Kapitel 2 Material & Methoden Tabelle 2. 1 DNA-Proben bis Juli 2002 von verschiedenen Hunderassen im PRAForschungsprojekt Rasse (Abkürzung) Σ DNA-Proben erkrankte Hunde Afghanischer Windhund (AW) 7 1 Amerikanischer Cocker Spaniel (ACS) 2 1 Australian Cattle Dogs (AC) 22 2 Berger des Pyrénées (BDP) 48 1 Berner Sennenhund (BS) 1 1 Bologneser (Bo) 1 1 Chesapeake Bay Retriever (CBR) 4 1 Colli (Co) 3 2 Curly Coated Retriever (CCR) 1 1 Englischer Cocker Spaniel (ECS) 23 7 Entlebucher Sennenhund (ES) 31 25 Golden Retriever (GR) 47 4 Irish Setter (IRS) 3 2 Kuvasz/Viszla (KV) 1 1 145 5 Neufundländer (NF) 1 1 Polnischer Niederungshütehund (PON) 1 1 Riesenschnauzer (RS) 1 1 Rottweiler (Ro) 1 1 125 7 Saluki (Sal) 2 1 Schapendoes (SD) 14 5 Scottish Terrier (ScT) 1 1 213 5 Springer Spaniel (SP) 1 1 Teckel (rauhaar; Te) 94 20 Tibet Mastiff (TM) 2 2 Tibet Terrier (TT) 96 2 Yorkshire Terrier (YT) 2 1 Zwergpudel (ZP) 58 48 Labrador Retriever (LR) Saarloos/Wolfshund (Sa) Sloughi (Sl) Summe 30 Rassen 951 152 19 Kapitel 2 Material & Methoden 2.1.3 Isolierung der DNA und Polymerase-Kettenreaktion (PCR) Die genomische DNA wurde aus peripherem Blut nach dem Standardprotokoll (Miller et al. 1988) extrahiert. Die Sequenzen der verschiedenen Gene wurden in einem Thermozykler (Biometra, Göttingen, Deutschland) durch polymerase chain reaction (PCR) (Saiki et al. 1988) sowohl von genomischer DNA und von den in λ Phagen eingefügten GenomAbschnitten amplifiziert. PCRs wurden in 96-Loch Mikrotiterplatten (Thermowell Costar Corming, NY) durchgeführt. Der 10 µl Reaktionsansatz beinhalted 50 ng DNA, 100 mM Tris (pH8,3), 500 mM KCl, 1 U Taq Polymerase (Gencraft, Münster, Deutschland) je 0,2 mmol der NTPs, je 0,4 mM Primer (Metabion, Deutschland) und unterschiedliche MgCl2Konzentrationen (siehe Tabellen Kapitel 3-12). Dem Reaktionsansatz wurden für die single strand conformation polymorphismus- (SSCP)-Analysen 10mCi/ml [α-32P] dCTP zugefügt. Die Fragmente der genomischen Hunde-DNA aus dem λ Phagen wurden mit einem EinSchritt-PCR-Programm vermehrt. Für die Mutationsanalyse des Genoms wurden zwei verschiedene so g. "touchdown" PCR Programme in einem TG Thermocycler (Whatman Biometra, Göttingen, Deutschland) durchgeführt. Das meist angewandte Drei-SchrittProgramm beginnt mit einem ersten Denaturierungsschritt (3 min bei 95°C) gefolgt von zwei Zyklen von 6°C und 3°C unter der Primeranlagerungstemperatur (siehe Kapitel 3-12). Das selten angewandte Programm (Tabellen Kapitel 3-12) beginnt mit einer 10°C höheren Anlagerungstemperatur wobei nach 10 Zyklen mit -1°C die fragmentspezifische Hybridisierungstemperatur (siehe Tabellen Kapitel 3-12) erreicht wird. Danach folgen 25 Zyklen, die jeweils 30 s Denaturierung bei 95°C, 30 s spezifischer Primeranlagerungstemperatur der einzelnen Systeme (siehe Tabellen Kapitel 3-12) und 30 s Sequenzverlängerung bei 72°C umfassen. Die PCR wird mit einem Schritt von 5 min bei 72°C beendet. 2.1.3 DNA Sequenz-Analyse Die Sequenzreaktionen wurde nach der Didesoxyribonukleotid Kettenabbruchmethode nach Sanger et al. (1977) mit BDT (Perkin-Elmer. Norwalk, CT) entsprechend dem Standardprotokoll des Anbieters durchgeführt. Die Sequenzreaktionen wurden auf dem Trio-Thermoblock (Whatman Biometra Göttingen, Deutschland) durchgeführt. Auf ein Sequenziergel (7% (w/v) PAA [Acrylamid/Bisacrylamid 29:1], 8 M Harnstoff, 0,03% (w/v) APS, 4×10-4% (v/v) TEMED) wurden die Ethanol gereinigten Sequenzierprodukte in 4,5 µl Formamide Loading Dye (Amersham, Buckinghamshire, UK) nach Denaturierung bei 95°C aufgetragen und auf 20 Kapitel 2 Material & Methoden einem automatisierten DNA-Sequenziergerät (Applied Biosystems 373XL, Forster City, USA) mit dem Analysierungsprogramm ABIPrismTM ausgewertet. 2.1.4 Klonierung und Identifikation von Intron/Exon Grenzen Klone, die Anteile der Gene SAG, PDE6A, PDE6B, PDE6D, PDC, RVC enthielten, wurden von einer genomischen caninen λ-DNA Library (Lambda FIX-II Library; Wirt: E. coli XL1Blue MRA (P2), Stratagene, La Jolla, Ca, USA) nach dem Stratagene Standardprotokoll isoliert. Rekombinante λ-DNA wurde auf HybondTM-N Nylon Membranen (Amersham, Buckinghamshire, UK) fixiert und UV-vernetzt (1'70 J/cm2). Die DNA-Phagen-Bank wurde mit Sonden, PCR-Produkte von Exon-Sequenzen der zu identifizierenden Gene durchsucht. Diese Sonden wurden mit dem Megaprime Labelling System (Amersham, Buckinghamshire, UK) mit [α32P] dATP radioaktiv markiert. Die Hybridisierung wurde bei 65°C in 0,5 M Natrium Phosphat Puffer (pH 7,2 / 7% Natriumdodecylsulfat (Church & Gilbert 1984) durchgeführt. Nach der Hybridisierung wurden die Filter zweimal für 30 min in 2 x SSC / 1% SDS, einmal für 15 min in 0,2 x SSC / 1% SDS bei 65°C und 30 min mit 6 x SSC bei Raumtemperatur gewaschen. Die Filter wurden auf einer phosphoimager Matrix (STORM 860) exponiert und mit dem Programmen STORM Scanner Control und Image Quant (Molecular Dynamics) dargestellt und ausgewertet. Hybridisierte Klone wurden isoliert und die Plaques nach Sambrook et al. (1989) aufgereinigt. Die Größe, der in die Phagen einklonierten Hundegenom-Abschnitte, wurde durch PCR mit exonischen Primern bestimmt (PCR-Konditionen wie oben beschrieben mit 0,2 ng Phagen-DNA, 2 mM MgCl2, Anlagerungstemperatur 54°C). Die Exon-/Introngrenzen wurden durch Vergleiche der mRNA des Hundes und der genomischen Sequenz des Menschen mit dem Blast Search Programm (NCBI [http://www.ncbi.nlm.nih.gov/blast]) bestimmt. Introngrößen wurden durch überlappende PCRs mit den eingeschlossenen benachbarten exonischen Sequenzen geschätzt. Die PCRProdukte wurden mit dem Easy Pure extractions kit (Biozym, Deutschland) aus einem 1,5%igem Agarosegel isoliert und mit intronüberspannenden Primeren sequenziert (siehe Tabellen Kapitel 3-12). Die Sequenzreaktion von 2-3 Klonen wurde entsprechend der in Abschnitt 2.1.3 beschriebenen Protokoll durchgeführt. 21 Kapitel 2 Material & Methoden 2.1.5 PCR-SSCP- und Mikrosatelliten-Analyse Die Positionen der intronischen Primer für die Mutationsanalyse wurde nach der DNA Sequenzuntersuchung genomischer DNA und der Hundegenom-Anteile des λ Phagen der entsprechenden Gene (siehe Kapitel 3-12) bestimmt. PCR-Produkte wurden in Abhängigkeit von der Fragmentlänge (Jäckel et al. 1998) mit verschiedenen Restriktionsendonukleasen geschnitten. Bekannte Polymorphismen wurden, wenn möglich, nach der Restriktionsfragmentlängenpolymorphismus (RFLP) Methode untersucht (siehe Tabellen Kapitel 3-12). Für die PCR-SSCP-Analyse nach Spinardi et al. (1991) wurden 3 µl des amplifizierten Produktes mit 8 µl Ladepuffer (95% deionisiertes Formamid, 10 mM NaOH, 20 mM EDTA, 0.06% (w/v) Xylencyanol und 0.06% (w/v) Bromophenolblau) vermischt und bei 95°C 5 min denaturiert und 3 µl dieser anschließend eisgekühlten Proben mittels Elektrophorese in Polyacrylamid (PAA) Gelen (Acrylamid/Bisacrylamid 19:1) mit einem Base Ace sequencer (Stratagene, La Jolla, Ca, USA) bei 35-40 W für 4-7 h bei 4 °C in 1 x TBE Puffer analysiert. Die PCR-SSCP-Analyse wurde mit zwei verschiedenen Gelkonditionen (6% PAA, 10% Glycerin und 6% PAA, 5% Glycerin und 1 M Harnstoff) durchgeführt. Für die Mikrosatelliten-Analyse wurden 5 µl PCR-Produkt mit 5 µl Formamid-Ladepuffer bei 95°C denaturiert und auf einem hochdenaturierendem Gel (6% PAA, 8 M Harnstoff) in 1 x TBE bei 60 W bei Raumtemperatur aufgetrennt. Jedes Gel wurden auf GB002-Papier (Schleicher Schuell, Dassel, Deutschland) auf einem Gel Dryer Model 583 (Biorad) bei 80°C unter Vakuum getrocknet und durch Autoradiographie nach einer Expositionszeit von 1-10 Tagen analysiert. Ausgewählte DNA-Proben wurden entsprechend ihres, bei der PCR-SSCP-Analyse identifizierten Bandenmusters, aufgereinigt und wie im Abschnitt 2.1.3 beschrieben sequenziert. 2.2 Bedingungen für den Ausschluss von Kandidatengenen in den verschiedenen Hunderassen 2.2.1 Kopplungswahrscheinlichkeit einer gPRA-Mutation mit (Einzelnukleotidpolymorphismen, SNP) Bestimmte Allele von Polymorphismen können in einem Gen mit der gPRA-verursachenden Mutation gekoppelt sein und somit zur indirekten Gen-Analyse herangezogen werden 22 Kapitel 2 Material & Methoden (Bourgain et al. 2002). Je näher ein Polymorphismus in dem Bereich einer gPRA-Mutation liegt, desto niedriger ist seine Rekombinationswahrscheinlichkeit, die durch die MorganEinheit definiert ist. Eine Rekombinationshäufigkeit von 1% entspricht beim Säuger 1 cM oder ca. 1000 kb genomischer DNA. Die zu untersuchenden Gene sind mit ~1-20 kb nicht sehr groß. Somit kann von einer durchschnittlichen genetischen Distanz zweier Polymorphismen von maximal 0,001-0,020 cM ausgegangen werden. Rekombinationen sind bei dieser genetischen Distanz bei 1:100 000-5 000 Meiosen zu erwarten (Hudson & Kaplan 1985). Bis auf wenige Ausnahmen existieren die heutigen Hunderassen erst seit ~100-600 Jahren. Bei einem zweijährigen Fortpflanzungszyklus kann also mit maximal 300 Generationen gerechnet werden. Somit ist bei den wenigen zu erwartenden Meiosen in den meisten Hunderassen die Rekombinationswahrscheinlichkeit zwischen gPRA-Mutation und untersuchtem Polymorphismus sehr gering. 2.2.2 Gründereffekt Eine weitere wichtige Vorraussetzung für die Möglichkeit, single nucleotid polymorphism (SNP)s als indirekte Marker für den Ausschluss von Kandidatengenen in den einzelnen Rassen einzusetzen, ist die Annahme, dass in den meist recht kleinen Populationen von reinrassigen Hunden von einem Gründereffekt ausgegangen werden kann. Diese Hypothese wird dadurch bekräftigt, dass bisher meist nur rassenspezifische Mutationen nachgewiesen wurden. Bei einem Gründereffekt mit zusätzlicher Inzucht durch Phänotyperhaltung in der Rassenbildung wird, wie in menschlichen Isolaten, die Anreicherung von seltenen rezessiven Allelen gefördert (Kruglyak 1999). Polymorphismen sollten bei den oben vorrausgesetzten Bedingungen in gPRA erkrankten Hunden im homozygoten Zustand nachzuweisen sein, wenn Kopplung mit dem Erkrankungslokus vorliegt. Bei rezessiv vererbten Erkrankungen tritt die Erkrankung nämlich nur dann auf, wenn die Mutation homozygot vorliegt und so ein Ausfall des Proteins verursacht wird. Wenn in den untersuchten Genabschnitten bei erkrankten Hunden Heterozygotie nachzuweisen ist, gibt es keine Kopplung des SNPs zum gPRA-Allel auf beiden Chromosomen und das Gen kann damit als Ursache für die Erkrankung in dieser Rasse ausgeschlossen werden. Das Gen kann auch in den Rassen ausgeschlossen werden, wo der Polymorphismus sowohl in erkrankten wie auch in gesunden Hunden homozygot nachzuweisen ist. Auch hier ist die Wahrscheinlichkeit einer Kopplung mit einem gPRA-Allel ausgeschlossen, da ja ein Hund der homozygot für das gPRA-Allel ist, dieser Augenkrankheit erkranken würde. Daher ist, wenn von einem ar Erbgang ausgegangen wird, die indirekte DNA-Diagnostik hier sehr aussagekräftig. 23 Kapitel 2 Material & Methoden 2.3 Datenbanken / Auswertung / Software Zum Zwecke der Literaturrecherche, der DNA-Analyse und der Suche nach bereits veröffentlichten DNA-Sequenzen wurden verschiedene Dienste im WorldWideWeb in Anspruch genommen: Online Mendelian Inheritance in Man (OMIM): http://www3.ncbi.nlm.nih.gov/Omim/searchomim.html; PubMed (MEDLINE): http://www.ncbi.nlm.nih.gov/entrez/query.fcgi BCM Search Launcher: BLAST Search TESS und NNPP/Eukaryotic http://searchlauncher.bcm.tmc.edu/cgi-bin/seq-search/gene-search.pl ProDom NCBI-BLASTP2: http://prodes.toulouse.inra.fr/prodom/doc/prodom.html Fasta3-Search: http://www2.ebi.ac.uk/fasta3/?request Sequence Retrieval System: http://srs.ebi.ac.uk/ Blast-Search: http://www.ncbi.nlm.nih.gov/blast/blast.cgi?Jform=1 Webcutter 2.0: http://www.firstmarket.com/cutter/cut2.html RetNet-Retinal Information Network: http://www.sph.uth.tmc.edu/RetNet/home.htm Liste von Kandidatengenen für retinale Erkrankungen des Menschen http://eyegene.meei.harvard.edu/ . 24 Kapitel 3 Animal Genetics 2000, 31 135-139 Short Communication Exclusion of the PDE6A gene for generalised progressive retinal atrophy in 11 breeds of dogs G Dekomien, J T Epplen Abstract The cyclic guanosine monophosphate specific phosphodiesterase (cGMP-specific PDE) is a key enzyme in the phototransduction cascade of the vertebrate retina. This enzyme consists of two catalytic α and β subunits, two identical inhibitory γ subunits as well as a δ subunit. Mutations in PDE6A and the PDE6B genes lead to autosomal recessive (ar) forms of retinitis pigmentosa (RP) in human and to the homologous disease in dogs, designated as generalised progressive retinal atrophy (gPRA). We investigated the PDE6A gene in 13 gPRA-affected dog breeds including healthy animals, obligate gPRA carriers and gPRA-affected dogs. In the coding region of PDE6A only a rare sequence variation (G103A; Asp35Asn) was found in exon 1 of 2 healthy Tibet Terriers and 1 affected Cocker Spaniel. Using single-stranded conformation polymorphism (SSCP) analyses we detected several sequence variations in 8 of the PDE6A introns in different investigated breeds. Most informative for excluding the PDE6A gene as a cause for gPRA were a polymorphic microsatellite ((GT)10CG(GT)2CG(GT)12) in intron 14 and four sequence variations in intron 18 for almost all breeds investigated. The sequence variations of PDE6A did not segregate together with gPRA in 11 breeds. Since diseased animals were heterozygous for the polymorphisms, the PDE6A gene is unlikely to harbour the critical mutation causing gPRA in the following breeds: Chesapeake Bay Retriever, Entlebucher Sennenhund, Labrador Retriever, Tibet Mastiff, Dachshund (long- and wire-haired), Tibetan Terrier, Miniature Poodle, Australian Cattle Dog, Cocker Spaniel, Saarloos/Wolfshound, Sloughi. 25 Kapitel 3 PDE6A Gen PDE6A Gen 3.1 Vorbemerkung Ein wichtiges Effektorenzym der Phototransduktionskaskade ist die cGMP PDE (zyklische Guanosin-Monophosphat spezifische Phosphodiesterase), ein heterotetramerisches Protein, das den Diskmembranen der Photorezeptorzellen angelagert ist. Die PDE hydrolysiert 3',5'cGMP zu 5'GMP. PDE6A und PDE6B sind katalytische Untereinheiten die sich in Stäbchen zu Heterodimeren verbinden. In Zapfen konnten dagegen auch PDE6A Homodimere nachgewiesen werden. Sowohl in dem PDE6A wie auch in dem PDE6B Gen wurden inzwischen zahlreiche Mutationen identifiziert, die zur RP in Menschen führen (Danciger et al. 1995; Huang et al. 1995; McLaughlin et al. 1993). In Cardigan Welsh Corgi wurde eine 1bp Deletion im Kodon 616 des PDE6A Gens identifiziert, die zu einer der klinischen Subtypen der gPRA, der Rod Cone Dysplasie (rcd) 3, führt (Petersen-Jones et al. 1999). Die chromosomale Lokalisation des caninen PDE6A Gens ist noch unbekannt. Das entsprechende menschliche Gen ist auf dem Chromosom 5q31.2-34 lokalisiert (Pittler et al. 1990). 3.2 Resultate und Diskussion 3.2.1 Voraussetzungen für die Untersuchung des PDE6A Gens Das PDE6A wurde als ein Kandidatengen für die gPRA in 13 verschiedenen Hunderassen mit insgesamt 231 Individuen und 59 eingeschlossenen an gPRA-erkrankten Hunden untersucht. Für die Mutationsanalyse wurde auf schon veröffentliche Exonsequenzen von Kylma et al. (1997) und Intronsequenzen von Petersen-Jones et al. (1999) zurückgegriffen. Für diese Kandidatengenuntersuchung zusätzlich identifizierte Intronsequenzen (Kapitel 2.1.4) wurden unter der EMBL-Accessions-Nr. AJ251206 veröffentlicht. Das PDE6A Gen besteht aus 22 Exons. Davon wurden Exons 1-4 und 10-22, die Introns 2, 11, 15, 16 und 18 komplett nach Sequenzvariationen durchsucht. Primer, PCR-Bedingungen Produktlänge und Restriktionsenzyme sind in Tabelle 3.1. aufgelistet. Die kleinen Exons 5-9 wurden nicht untersucht, da insgesamt 16 informative Variationen für die indirekte GenAnalyse des PDE6A Gens zur Verfügung standen. PCR-SSCP und Mikrosatelliten Analysen der genomischen DNA von gesunden und an gPRA erkrankten Hunden, deckten eine große Anzahl von Sequenzpolymorphismen auf. Die PCR-Produkte der eindeutig durch die Gelektrophorese ersichtliche PCR-SSCP oder Mikrosatelliten-Muster wurden sequenziert und 26 Kapitel 3 PDE6A Gen die dafür ursächlich Sequenzvariation identifiziert. Die Sequenzvariationen sind in der Tabelle 3.2 zusammengefasst. Tabelle 3.1 Primersequenzen und Bedingungen für die PCR-Systeme zur Amplifikation des PDE6A Gens PCR-Systeme Lokali& Primersation Bezeichnungen 1.1 1.2 2.-IF 3-IR 4-IF 4.2 10.1 10-IR 11-IF 12.2 13-IF 13.2 14.1 14.2 15-IF 15-IR 15-IF 16.2 16.1 17.2 18.1 19-IR 20-IF 20-IR 21-IF 21.2 22-IF 22UTR A Exon1 Exon1 Intron1 Intron3 Intron4 Exon4 Exon10 Intron10 Intron11 Intron12 Intron13 Exon13 Exon14 Exon14 Intron14 Intron15 Intron14 Exon16 Exon16 Exon17 Intron18 Intron19 Intron19 Intron20 Intron20 Exon21 Intron21 Exon 22 PrimersequenzA (5'→ →3') Länge der PCRProdukte (bp) PCR-Bedingungen [T-°C / MgCl2 -mM] Restriktionsenzyme für die SSCPAnalyse ATGGGTGAGGTGACAGCAG CTCTTCTGTGTTGACGACGT 488 59/1,0B Eco57I GATGAGCATTTCTGTGACTTTG CTGGCCACGCCGAGTCT 985 56/2,0 MseI TCTGTCTTGGAGTTATTGTGAA CTTCTGCTTGGTCATGTCTAA 212 57/2,0 - TCTTTGGCTCAATTCCTGGG CACCTGTCTCTGCCCTCAT 239 54/1,0 Sau3AI TCTAGACCTCAGATAGATAAA CTCCTGAGGAATGTGAAATTT 493 54/4,0C Sau3AI CAAAGTACACTCACACGTTA CACCAGCAAGGAGAACATG 233 54/1,0 - ACCGGAAAGCTGAAGCGATA TTCATCTGGTAGAGATTGTT 170 52/2,0 - AGGACTGGGTGAGGATGATA CACTCAAGCTTCTTGTGAGAA 241 54/2,0 HaeIII AGGACTGGGTGAGGATGATA TTGAAATACAGGGCGAGGTC 486 54/2,0 AluI AGCCTGAATATCTTTCAAAAC ATAACAATTTCCTTCCGTGTC 626 54/3,0 AluI/RsaI AGGGCCATGATGATGACCG GGTAAGATCACATGCACCTAT 501 57/2,0 MvaI AACAAGAATGCTTCATAGGT TCCTGAGGACCAAATGCCTT 326 54/2,0 GGTTGCCAAAGGAGGGTAA TGGCTGTCTGCTGCTTCTG 283 54/2,0 CCTTAGTTTGCAACTTGGTCTA CAAATCCGATTACTATTTTAC 222 53/3,0 HaeIII - PDE6A-Sequenzen von den EMBL-Accession-Nr. Z68340; AJ233678; AJ233686AJ233693 entnommen B 3% DMSO C 5% Formamid 27 Kapitel 3 PDE6A Gen 3.2.2 Mutationsanalyse des PDE6A Gens Im offenen Leserahmen (OLR) des Exon 1 wurde eine selten auftretende Substitution (G103A), die zu einem D35N Aminosäurewechsel führt, in zwei gesunden Tibet Terriern und einem erkrankten Cocker Spaniel gefunden. Alle durch die PCR-SSCP-Analyse identifizierten Sequenzvariationen sind in der Tabelle 3.3 aufgelistet. Intronische SNPs wurden in verschiedenen Rassen im Intron 2, 10, 18 und 20 nachgewiesen. Die für die Rassen spezifischen Deletionen des polymorphen Mikrosatelliten (GT)10CG(GT)2CG(GT)12 im Intron 14 werden in der Tabelle 3.3 deutlich gemacht. Im Intron 11 wurde eine C-Insertion in drei Rassen und eine 6 bp Deletion (AACAAT) im Intron 16 in zwei Rassen identifiziert. Die vier SNPs im Intron 4 traten immer gemeinsam auf und sind somit im Kopplungsungleichgewicht. Jede der gefunden Sequenzvariationen wurde sowohl in gesunden wie auch in gPRA erkrankten Hunden identifiziert. Im Auftreten und somit auch für die Interpretation der Polymorphismen im PDE6A Gen bestehten keine Unterschiede zwischen erkrankten und gesunden Hunden. Bis auf Collie und Yorkshire Terrier wurden die Polymorphismen in allen erkrankten Hunden der einzelnen Rassen im heterozygoten Zustand nachgewiesen. Somit können die Sequenzvariationen als indirekte Marker genutzt werden, um das PDE6A Gen als Ursache für die gPRA in 11 Rassen auszuschließen. Der Gründereffekt kann für die genetische Analyse herangezogen werden, da er eine wichtige Bedeutung für Rassenbildung hat. Hunde, die durch einen ar vererbtes Merkmal erkranken, sind nicht nur homozygot für die erkrankungsverursachende Mutation sondern sind auch homozygot für die eng gekoppelte nicht krankheitserregende Sequenzvariationen im gleichen Gen. Deshalb können die folgenden Fakten die auf die erkrankten Hunde zutreffen, einen Beweis für den Ausschluss des Kandidatengens für die retinale Erkrankung in einer bestimmten Rasse liefern: 1. Heterozygotie für jede Sequenzvariation oder 2. homozygote Muster für zwei verschiedene Allele des gleichen Polymorphismus in erkrankten Hunden der gleichen Rasse. 3.2.3 Schlussfolgerung Der erkrankte Collie ist für die Variationen im Intron 14 und 18, der Yorkshire Terrier für die Variationen im Intron 11, 14, 15 und 18 homozygot. Deshalb kann in diesen beiden Rassen aus den oben genannten Gründen das PDE6A Gen nicht als Ursache für die gPRA ausgeschlossen werden. gPRA in Collies ist durch ähnliche biochemische und histopathologische Veränderungen wie in Cardigan Welsh Corgis (Petersen-Jones et al. 1999) und Irish Setter (Suber et al. 1993) charakterisiert. Allerdings wurde die für die Cardigan Welsh Corgis 28 Kapitel 3 PDE6A Gen typische 1-bp Deletion im Kodon 616 in keinem der beiden obengenannten Rassen identifiziert und schließt sich somit als Ursache für die gPRA in diesen Rassen aus. Wang et al. (1999) schloss eine Mutation in dem PDE6A Gen als Ursache für die rcd2 in Collies aus. Weitere Untersuchungen sollten sich in Yorkshire Terrier anschließen, wenn weitere erkrankte Hunde zur Verfügung stehen. Allerdings kann das PDE6A Gen in 11 der 13 untersuchten Rassen als Ursache für die gPRA ausgeschlossen werden: Australian Cattle Dogs, Chesapeake Bay Retriever, Cocker Spaniel, Entlebucher Sennenhunde, Labrador Retriever, Saarloos/Wolfshunde, Sloughis, Teckel, Tibet Mastiffs, Tibet Terrier und Zwergpudel. 29 Kapitel 3 PDE6A Gen Tabelle 3. 2 Sequenzvariationen im PDE6A Gen in verschiedenen Hunderassen Gen / Lokalisation SequenzvariationA AminosäureAustausch ECSB, TT PDE6A Exon 1 G103A PDE6A Intron 2 IVS2+53C→T - AC, CBR, Te, TM, Sa PDE6A Intron 10 IVS10+51T→C - CBR PDE6A Intron 11 IVS11+82InsC - Te, TT, YT, ZP PDE6A Intron 14 MS:IVS14 -11(GT)11; -11Del(GT)32 -36 + -42C→A3 -63Del(GT)14 - Co, CBR2+4, ES, Sa, Sl1, Te1, TM2+4, YT, ZP1+3 PDE6A Intron 15 IVS15+36G→A - CBR, ES, Sl, TM, Sa, Te, YT, ZP PDE6A Intron 16 IVS16+41Del.6bp - Te, TM PDE6A Intron 18C IVS18+55C→T IVS18+65T→C IVS18+88A→C IVS18+121G→C - AC, Co, CBR, ECS, ES, Sl, YT, Sa, SH, Te, TM, TT, ZP PDE6A Intron 20 IVS20+29G→A IVS20+56A→C - ZP CBR, Sa A D35N Rasse - Position der Sequenzvariationen nach Kylma et al. 1997; Petersen-Jones et al. 1998; EMBL-Accessions-Nr. AJ251206. B Rassen mit heterozygoten Sequenzvariationsmustern in gPRA-erkrankten Hunden in fettgedruckten Buchstaben (Abkürzungen der Rassenamen siehe Tabelle 2.1). C Die Haplotypen im Intron 18 sind entweder C-T-A-G oder T-C-C-C in den entsprechenden Hunderassen. 30 Kapitel 4 Cytogenetics and Cell Genetics 2000, 90 261-267 Generalised progressive retinal atrophy of Sloughi dogs is due to an 8-bp insertion in exon 21 of the PDE6B gene Gabriele Dekomien, Maren Runte, Rene Gödde, Jörg Thomas Epplen Abstract We investigated the gene encoding the ß subunit of the cGMP phosphodiesterase (PDE6B) as a candidate for generalised progressive retinal atrophy (gPRA), an autosomal recessively transmitted eye disease in dogs. The PDE6B gene was isolated from a genomic library. Single strand conformation polymorphism (SSCP) analysis revealed 8 intronic variations in different subsets of the 14 dog breeds investigated. In addition, we have identified an 8-bp insertion after codon 816 in certain Sloughi dogs. Analysis of PRA-affected and obligatory carrier Sloughis showed that this mutation cosegregates with disease status in a large pedigree. All other exchanges identified are not located in functionally relevant parts of the gene, e.g. in the splice signal consensus sites. In most dog breeds (Labrador Retriever, Tibet Mastiff, Dachshund, Tibetan Terrier, Miniature Poodle, Australian Cattle Dog, Cocker Spaniel, Collie, Saarloos/Wolfshound, Chesapeake Bay Retriever and Yorkshire Terrier), the PDE6B gene was excluded as a candidate gene for gPRA because heterozygous allele constellations were detected in diseased animals. Therefore, the PDE6B sequence variations did not segregate together with the gPRA causing mutation(s). Direct and indirect DNA tests concerning gPRA can be offered now for a variety of different dog breeds. 31 Kapitel 4 PDE6B Gen PDE6B Gen 4.1 Vorbemerkung Die cGMP, ein Schlüsselenzym der Phototransduktionskaskade, ist in den Aussensegmenten der Photorezeptorzellen lokalisiert (Bennett & Clerk 1992). Die Phototransduktion beginnt mit der Absorption von Licht durch Rhodopsin. Photoaktiviertes Rhodopsin stimuliert die αUntereinheit des heterotrimerischen Guanin-Nukleotid-Binde-Proteins, das Transducin (Tαβγ). Dieses aktiviert die PDE unter Freigabe der inhibitorischen PDE γ-Untereinheiten von den katalytischen Komponenten der PDE, den PDE α- und PDE β-Untereinheiten. Aktiviertes PDE hydrolysiert cGMP; niedriger cGMP-Spiegel führt zur Schließung des Kationenkanals während der Hyperpolarisation der Zellmembran (Stryer 1996). Mutationen in verschiedenen Exons des Gen für die PDE β-Untereinheit (PDE6B) sind verantwortlich für ar RP im Menschen (McLaughlin et al.1993; Danciger et al. 1995). In zwei Tiermodellen führt ein verkürztes PDE6B Protein zum Photorezeptortod. Zum einen wird in der rd Maus die retinale Degeneration von einem integrierten murinen Leukemievirus im Intron 1 und von einer Unsinn-Mutation im Exon 7 des PDE6B Gens ausgelöst (Pittler & Baehr 1991; Bowes 1993). Zum anderen führt auch im Irishen Setter eine Unsinn-Mutation nach dem Kodon 806 zu einem Stoppkodon, die eine Verkürzung des Proteins verursacht (Suber et al. 1993). In Sloughis wird die gPRA ar vererbt mit einem frühen Krankheitsbeginn mit 1-11/2 Jahren, vergleichbar mit den Irishen Settern. Der Krankheitsverlauf ist progredient. Die betroffenen Hunde sind nach ca. 2-3 Jahren noch nicht vollständig erblindet. Einige deutsche und niederländische Züchter beobachteten schon in den letzten 15 Jahren "Blindheit" (vermutlich gPRA) bei bestimmten Sloughis. Jedoch liegen bis jetzt noch keine detaillierten elektrophysikalischen, histopathologischen und biochemikalischen Untersuchungen von der Retina gPRA-erkrankter Sloughis vor. Bei der Mutationsanalyse, die sich nach der Identifizierung von intronischen Sequenzen des PDE6B Gens von einer Hunde-Genombank anschloss, wurde eine 8-bp Insertion im Kodon 816 der PDE6B nachgewiesen, die zur gPRA in Sloughis führt, die aber von der in Irish Settern gefundenen PDE6B-Mutation abzugrenzen ist. 32 Kapitel 4 PDE6B Gen 4.2 Resultate und Diskussion 4.2.1 Genomische Organisation des PDE6B Gens Bis zur Veröffentlichung im Mai 2000 standen dem Projekt 349 Hunde mit insgesamt 75 gPRA erkrankte Hunde aus 14 verschiedenen Rassen zur Verfügung. Inzwischen wurde die Polymorphismusanalyse auf 6 weiteren Rassen ausgeweitet, da dieses Gen nach wie vor als potentielles Kandidatengen für die ar vererbte gPRA in weiteren Rassen gelten kann. Für die Identifizierung der Introns des PDE6B Gens wurden mehrere λ-Klone mit verschiedenen Abschnitten des PDE6B Gens aus der Lambda Fix Genombank isoliert. Diese Teilbereiche des PDE6B Gens wurden mit 4 radioaktiv markierte Sonden, PCR Produkte der Exons 1, 4, 10 und 21, durch Hybridisierung identifiziert, isoliert und charakterisiert. Das PDE6B Gen umfasst ∼ 15 kb, Promotor (EMBL Accessions-Nr X 92832) und 3' nichttranslatierter Bereich (UTR) eingeschlossen. Die Längen der 21 Introns wurden durch intronumspannende PCR Systemen festgelegt oder geschätzt. (Tabelle 4.1). Die Exon/Introngrenzen stimmen mit den Spleißkonsensussequenzen nach Sharp (1981) überein. Sehr große Introns wurden nicht vollständig sequenziert, aber die 5' und 3' Abschnitte, bis auf die 3' Region des Intron 3, charakterisiert (EMBL-Accession-Nr. AJ277998-AJ278009). Das canine PDE6B Gen ist ∼23 kb kürzer als das entsprechende Gegenstück des Menschen (Weber et al. 1991). Diese Differenz erklärt sich aus den kürzeren Introns 3, 10, 11, 12 und 21. Kleinere Unterschiede der Exongröße zwischen Hund und Mensch wurden nach der Sequenzierung der Intron-überspannenden PCR-Produkte festgestellt. Das canine Exon 14 umfasst 110 bp, das des Menschen 111 bp. Dieser Unterschied von einer Base findet sich im Exon 15 wieder und hat somit keinen Einfluss auf die Aminosäuresequenz. 4.2.2 Mutationsanalyse des PDE6B Gens Die Mutationsanalyse des PDE6B Gens wurde mittels der PCR-SSCP Methode für alle Exons bis auf die Exons 14, 15, 16 und 19 durchgeführt. Die letztgenannten 4 Exons ließen sich auf einem SSCP Gel nicht auswertbar darstellen, deshalb wurde die DNA ausgewählter erkrankter Hunde direkt sequenziert. PCR-Systeme wurden in der Tabelle aufgelistet. Im OLR wurden insgesamt 4 Sequenzvariationen identifziert. Wobei im Exon 1 der c267T→C Austausch ohne Aminosäurewechsel nur in Entlebuchern nachzuweisen ist, wobei der c352G→A Austausch einen Aminosäurewechsel von A117T verursacht, aber in erkrankten und gesunden Hunden dreier verschiedener Rassen erkennbar ist. Auch die Transition von 33 Kapitel 4 PDE6B Gen c2343T→C im Exon 20 bleibt ohne Konsequenzen für die Aminosäuresequenz und ist sowohl in gesunden als auch in kranken Hunden in 6 unterschiedlichen Rassen nachweisbar. Zahlreiche weitere Sequenzvariationen wurden im Promotorbereich und in den Introns 7, 8, 10 und 19 identifiziert (Tabelle 4.3). Im Exon 21 wurde nur in an gPRA erkrankten Sloughis eine Sequenzvariation nachgewiesen, die in Abbildung 3.1 zu sehen ist. In diesem SSCP-Gel ist die Bandenverschiebung des mutierten Allels deutlich im Vergleich mit dem Normalallel zu erkennen. Bei einem sicheren Träger eines mutierten Allels wird eine eindeutige Heteroduplexbildung ersichtlich. Jede dieser Bandenverschiebungen wurden durch Sequenzierung der PCR-Produkte untersucht und identifiziert. Es wurde eine 8-bp Insertion (TGAAGTCC) nach dem Kodon 816 in gPRA erkrankten und sicheren Trägern nachgewiesen (Abbildung 4.2). Dieser Einschub verursacht sofort ein Stoppkodon in der Position 817 wodurch das PDE6B Protein um 50 Aminosäuren verkürzt wird. Um diese Mutation als Ursache für die gPRA in Sloughis zu verifizieren wurden Blutproben von Mitgliedern der Familie der gPRA erkrankten Hunde und nicht verwandte Sloughis gesammelt. Die zwei gPRA-erkrankten Sloughis und ein blinder Sloughi, mit einer anderen Augenerkrankung, einem Glaukom, gehören zu einem gemeinsamen großen Stammbaum, wovon 88 Hunde untersucht wurden (Abbildung 4.3). 4.2.3 Schlussfolgerung Die 8-bp Insertion wurde homozygot in den beiden gPRA-erkrankten Hunden nachgewiesen. Der Sloughi mit der abweichenden Augenerkrankung zeigt ein normales Allel und ein mutiertes Allel. Sichere Träger waren immer heterozygot und keiner der klinisch gesunden Hunde war homozygot für die Mutation (Abbildung 4.3). Bis September 2002 wurden insgesamt 222 Hunde untersucht. Davon sind 5 Hunde homozygot und 68 Sloughis heterozygot für die Mutation. Weitere erkrankte Hunde mit dieser 8-bp Insertion wird es dank des direkten Gentests wohl nicht mehr geben, da inzwischen alle zuchtfähigen Hunde genetisch auf diese Mutation hin überprüft werden. So ist es möglich, identifizierte Träger mit homozygot gesunden Sloughis zu verpaaren, um den Genpool dieser kleinen Rasse nicht einzuschränken. Keine der anderen identifizierten Sequenzvariationen hat für das Protein funktionelle bzw. krankheitsauslösende Bedeutung. Zusätzlich wurden die Variationen heterozygot als auch homozygot in erkrankten und gesunden Hunden nachgewiesen (Tabelle 4.3) und können deshalb als indirekte Marker für den Ausschluss des PDE6B Gens als Ursache für die gPRA in diesen Rassen verwendet werden (Kapitel 2.3). Daher ist es möglich, das PDE6B Gen als 34 Kapitel 4 PDE6B Gen Ursache für die gPRA in den folgenden 17 Hunderassen auszuschließen: Australien Cattle Dogs, Berger des Pyrénées, Cheasapeak Bay Retrievern, Collies, Englische Cocker Spaniel, Entlebucher Sennenhunde, Golden Retriever, Irish Setter, Labrador Retriever, Rauhaarteckel, Riesenschnauzer, Saarloos/Wolfshunde, Schapendoes, Tibet Terrier, Tibet Matiff, Yorkshire Terrier und Zwergpudeln. In der Veröffentlichung Dekomien & Epplen (2000) konnte das PDE6B Gen für die Rassen, Entlebucher Sennenhund und Golden Retriever, aufgrund von homozygoten Sequenzvariationen in einigen erkrankten Hunden noch nicht als krankheitsverursachend ausgeschlosssen werden. Dem Forschungsprojekt wurden inzwischen weitere erkrankte Hunde dieser Rassen zur Verfügung gestellt. Nach gezielten Analysen der DNA dieser Hunde anhand der schon ermittelten Polymorphismen wurde dieses Gen als Krankheitsursache nachträglich auch in diesen Rassen ausgeschlossen. N T T A A N DSK DSK HE DS ES ES DS ES ES Abbildung 4.1 Repräsentative SSCP-Analyse des Exon 21 des caninen PDE6B Gens eines erkrankten, eines sicheren Trägers und eines ophthalmologisch als normal getesteten Sloughis. A: mutiertes Allel eines an gPRA erkrankten Hundes; T: Träger mit Heteroduplex; N: normales Allel; DS: Doppelstrang, ES: Einzelstrang, DSK. Doppelstrangkontrolle. Die Variationen die das Bandenmuster verursachten, wurden durch DNA Sequenzierung nachuntersucht. 35 Kapitel 4 PDE6B Gen a Sloughi: Normal N R K E W K A L A D E Y E A K K A L A D E Y E A * L K A L b Sloughi: Mutante N R K E W c Irish Setter: Mutante N R K E * Abbildung 4.2 Vergleiche von Sequenzen des PDE6B Gens von einem Sloughi mit dem Normalallel (a), dem mutierten Allel eines Sloughis (b), mit der Irish Setter spezifischen Mutation (c). In Sloughis wird eine 8 bp-Insertion gezeigt nach Kodon 816 (b), im Irishen Setter eine G→A Transition nach dem Kodon 806 (c). Die Mutationen, die Insertion und die Transition, sind mit Pfeilen markiert. 36 Kapitel 4 PDE6B Gen 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 Weibchen und Männchen, die für die Untersuchung nicht zur Verfügung standen. weibliche und männliche Hunde mit dem normalen Genotyp (Wildtyp). weibliche und männliche gPRA Träger. gPRA-erkrankte Männchen. Abbildung 4.3 Stammbaum mit insgesamt 88 untersuchten Hunden mit drei gPRA erkrankten Sloughis, zwei davon mit den typischen Krankheitszeichen. Der Sloughi mit der abweichenden Augenerkrankung wurde mit einem Pfeil markiert. 37 0 Kapitel 4 PDE6B Gen Tabelle 4.1 Exon 1-2 2-3 3-4 4-5 5-6 6-7 7-8 8-9 9-10 10-11 11-12 12-13 13-14 14-15 15-16 16-17 17-18 18-19 19-20 20-21 21-22 Primer für die PCR Amplifikation des caninen PDE6B Gens* Primersequenzen 5’ → 3’ GACAGACAGCCGGACACC GAAGAGACCCACGGCACA TGCCCCCACTTCAGCCC CGGCCGCCTAGATCCCATC CAAGCCTGCTGCCCGGGA TGAGGCAGCACTGCGGC GTGCTGCTGTGGTCGGC GTGCTCAGGGTGCGCCTT TCCTCCTGGCGCCGCG CCCCCGCGGGCCCTT CTCACCACCGCAGCGGT GAAGAGGACGGGGACACT CGGACACCCAGAGCCCT TTGCGGAACCACAGCGTCTT CCACCTCCACTGCGGA TCGGGCCGCAGGCGGAA TGACGGGCCGGGCCG CCTCCCCTCTGGGCAGAA ACTCGGGGTGGCCC GCCTCCAGGGTCCACCT ACGAGGGGGCGGCTCACT CTCTTGGGGCTCAGACCT GATGCTCCCAGGGGCACA GCACCGGCTCTCGGCCT CTGGGCCCGCGGTGCT TGCTTCCCGCGTCGCGAG CCCTGTGGGAGGGGGGTC TGTCGTCGCTGGGCCA CTCGCGACGCGGGAAGCA CGCGGCTCCTGGCTGCC GACCCGGGAGGGGGGCA GTCTGAGATCGACCAGCT CCTCGGTTTCTCTCCTAGAA CGTTCCTCTCCCTGGTCT GCCGTGGAGGGGTGGG AAGCGAGGCCCGAGGTTA TCTTTCCTCAATTTCTGACCAA TGGGGTTGCGGCAGGTCA CTCATCCCACGGACACTGT GGTCCGCGGGCGGCTAA CACCGGGCCGAGGAGGA AAGTTTCAAGATATCCAAAATTGT Exon Länge (bp) (1) 468 Intron Länge (bp) 1568 (2) 153 ~1000 (3) 90 ~700 (4) 141 85 (5) 75 ~740 (6) 65 ~800 (7) 68 85 (8) 48 ~900 (9) 150 121 (10) 144 521 (11) 66 368 (12) 147 545 (13) 108 85 (14) 110 102 (15) 88 ~500 (16) 101 179 (17) 108 469 (18) 64 345 (19) 75 ~900 (20) 84 ~1100 (21) 157 ~990 Exon-/Introngrenzen CAGAGgtgggct gccccagTGCCC AAGACgtgagtg gctgcagGTTTT GCCAGgtacctg AGAAGgtgaggc tgctcagGAATT GCCGTgtgagtc ttcacagGAAAT ATCCCgtaagtg cccgcagCCTCG GCTTTgtgagta cttccagATCTG TCCAGgtacgtc cctgcagGAAGG TGGAGgtaaagc tccgcagTCCCT TCCTGgtaccac cccctagCCAAC TCTTGgtcagaa cccacagAAGGA AGGAGgtggggg cccatagGTCCT TCACGgtgcgcg tccgcagACGGG ATGAAgtaggcg gccgcagATCCC AGGAGgtccgtg cccgcagACCCT TTCAAgtgggtg ggttcagGAAGA GTCATgtgagca cccgcagGGCCA GCAAGgtcagaa tcccaagGTTGC CTATTgtgagtg ccacaagCCGAT ACAAGgtgagca tgtccagGAGTT GAAAGgtctggg ctcgcagCGGGC PCR Bedingungen (T)/[MgCl2] 53°C/2mM 54°C/1mM 55°C/3mM 54°C/1mM 54°C/1mM 56°C/1mM 56°C/1mM 56°C/1mM 56°C/1mM 53°C/2mM 56°C/1mM 54°C/1mM 56°C/1mM 55°C/1mM 56°C/1mM 56°C/1mM 56°C/1mM 52°C/1mM 53°C/2mM 53°C/1mM 57°C/1mM * PCR Primer für die Identifikation der Exon-/Introngrenzen und für die Festlegung der Intronlängen der caninen genomischen PDE6B Klone (Exonsequenzen in Großbuchstaben; EMBL-Accession-Nr. AJ277998-AJ278009). 38 Kapitel 4 PDE6B Gen Table 4.2 Primersequenzen für die Mutationsanalyse der einzelnen Exons, Introns und des Promotors des PDE6B Gens Promoter/ PrimersequenzenA Exon 5’→ → 3’ Lokation (bp) PCRBedingungen [T-°C/ MgCl2-mM] PCRProdukt (bp) Restriktionenzyme für SSCP Analyse Pro CTAGCATTGAGGGAAGCCA TCAGCGTCTTCCCGAAGTA 1-19 329-347 50/2 346 HinfI 1 GACAGACAGCCGGACACC CTCTGTCACGTCCTGGAC 248-265 716-733 56/2 483 Sau3aI 2 TGCCCCCACTTCAGCCC GTCTTCGTCCTCGCTTGT 1298-1314 1433-1450 57/2 153 - 3 CAAGCTTGCTGCCCGGGA CGGCCGCCTAGATCCCAT 70-87 310-328 57/2B 259 MseI 4 GTGCTGCTGTGGTCGGC CTTCTCCTTGGTCATGTCC 1-17 123-141 54/4B 140 - 5 TCCTCCTGGCGCCGCG GTGCTCAGGGTGCGCCTT 153-169 314-331 61/1 183 - 6 CTCACCACCGCAGCGGT CCCCCGCCGGCCCTT 315-331 430-444 61/1 125 - 7 CGGACACCCAGAGCCCT GAAGAGGACGGGGACACT 77-93 259-280 61/1 198 - 8 CCACCTCCACTGCGGA TTGCGGAACCACAGCGTCTT 30-45 377-396 57/2 167 - 9 TGACGGGCCGGGCCG CGGGGCCACCCCGAGT 87-101 354-370 55/1 229 Sau3AI 10 TTCCGCCTGCGGCCCGA CCTCCCCTCTGGGCAGAA 301-315 582-599 56/2B 245 AvaII 11 CGAGGGGGCGGCTCACT GCCTCCAGGGTCCACCT 1117-1134 1253-1269 56/2 152 AluI 12 GATGCTGGGAGGGGCACA CTCTTGGGGCTCAGACCT 1428-1445 1655-1672 54/2 147 - 13 CTGGGCCCGCGGTGCT GCACCGGCTCTCGGCCT 27-43 230-246 63/1 219 - 17 CCTCGGTTTCTCTCCTAGAA GTCTGAGATCGACCAGCT 370-392 443-460 58/1 248 - 18 GCCGTGGAGGGGTGGGA CGTTCCTCTCCCTGGTCT 149-164 299-216 54/2 173 HphI 20 CTCATCCCACGGACACTGT TGGGGTTGCGGCAGGTCA 26-44 254-271 54/1 246 AvaII 21 CCCCCACTGCCTGGGAA GGTCCGCGGGCGGCTAA 124-140 362-379 65/1 282 AluI 22 CACCGGGCCGAGGAGGA AAGTTTCAAGATATCCAAAATT GT 235-251 458-481 54/3B 247 - A Sequenzen EMBL-Accession-Nr. AJ277998-AJ278009. B zusätzlich 5% DMSO. 39 Kapitel 4 PDE6B Gen Tabelle 4.3 Sequenzvariationen des PDE6B Gens in heterozygoten Mustern in gPRAerkrankten und gesunden HundenA. Aminosäure -Austausch RasseB Gen / Lokalisation Sequenzvariation PDE6B Promotor -45G→A -45C→A - TM TM PDE6B Exon 1 c267T→C c352G→A A89A A117T ES AC, BDP, ES, TT PDE6B Intron 7 IVS7+52G→A IVS7+63G→A - Co, GR, Ro, SD Sa, TM PDE6B Intron 8 IVS8+23A→G - BDP, ECS, ES, GR, YT PDE6B Intron 10 IVS10+18G→C IVS10+19G→C IVS10-19C→T IVS10-9A→C - TM, YT, ZP IRS, TM, TT, LR, ZP ECS, ES, LR, RS, TM, TT, ZP Co, GR, SD PDE6B Intron14 IVS14+37C→A - ES PDE6B Intron 19 IVS19-67G→A - Te, YT, ZP PDE6B Exon 20 C2349T F783F AC, ECS, CBR, ES, GR, TT PDE6B Exon 21 c2449-2456ins TGAAGTCC. K816 STOPP Sl A : Rassen mit gPRA-erkrankte Hunde in fettgedruckten Buchstaben (Abkürzungen siehe Tabelle 2.1). B : Position der Sequenzvariationen in der cDNA entspricht der: EMBL-Accession-Nr. L13262. 40 Kapitel 4 PDE6B Gen 4.3 PCR-gestützter gPRA Test zur Identifizierung der 8-bp Insertion in Sloughis Nach der Identifizierung der 8-bp Insertion im PDE6B Gen stellte sich die Frage nach einem einfachen und preiswerten Diagnostiksystem für den Nachweis der Mutation. Durch die Insertion ergab sich keine neue Restriktionsschnittstelle. Trotzdem war es möglich, die Insertion für die Mutationsdarstellung auszunutzen. Dafür wurden insgesamt vier verschiedene Primer (Tabelle 4.4) in einer PCR-Reaktion eingesetzt. So entstanden drei Primersysteme: 1. Bei einer Insertion wird ein 165 bp und ein Gesamtlängenfragment von 241 bp amplifiziert. 2. Bei einem normalen Allele entsteht bei der PCR-Reaktion ein 88 bp und ein 233 bp- Fragment. 3. Bei der DNA eines Mutations-Träger sind sowohl das 241 bp - 233 bp, 165 bp und das 88 bp-Fragment auf einem 2%igem Agarosegel erkennbar (siehe Abbildung 4.3). Diese Diagnostik kommt ohne Radioaktivität und Restriktionsenzyme aus und kann innerhalb eines Tages durchgeführt werden. Allerdings ist für eine optimale Darstellung der Insertion eine sichere Anlagerung der Primer zur Vermeidung einer Fehldiagnose Vorraussetzung. Deshalb sind Kontrollen, die DNA homozygot erkrankter und von gesicherten heterozygoten Hunden, unbedingt erforderlich. Um die Zuverlässigkeit dieser Methode zu überprüfen, wurde die gPRA Diagnostik sowohl per SSCP-Analyse (Primer in Tabelle 4.4) wie auch nach der PCR-Methode durchgeführt. Im Vergleich war die PCRMethode nicht immer eindeutig. Wohingegen sich die SSCP-Analyse als stabileres Diagnosemittel herausstellte. 41 Kapitel 4 PDE6B Gen Tabelle 4.4 Primersequenzen und Bedingungen für die Diagnostik zur Identifizierung der 8bp Insertion im Exon 21 des PDE6B Gens Untersuchungssystem PCR-Systeme & PrimerBezeichnungen PrimersequenzA (5'→ →3') Länge der PCRProdukte (bp) PCRBedingungen T/MgCl2 [(°C)/(mM)] PCR-SSCP 21 ins F 21ins E R CCCGTTTCCACGAAGAGATC CTTTCTTGGCTGTCGTCC 157 56/1,5 PCRAgarosegel 41 I 21 ins R 41 I 21+8 R 21-8 F 21 ins R CCCCCACTGCCTGGGAA CCTGCGGCCCAGACCTTT CCCCCACTGCCTGGGAA TCAGCTTGGACTTCAGGCC TGAGTACGAGGCCAAGCTG CCTGCGGCCCAGACCTTT 233 (+8) 57/1,5B A B 165 88 Sequenzen: EMBL-Accessions-Nr. AJ278008. -1 touchdown PCR-Programm. P N N M M T T LW 241 bp 233 bp 165 bp 88 bp Primer Abbildung 4.3 Repräsentative PCR-gestützte DNA Diagnostik des Exon 21 von PDE6B Gens auf einem 2%tigem Agarosegel. P: pUC19DNA/MspI Marker LW: Leerwertkontrolle (Abkürzungen siehe Abbildung 4.1). 42 Kapitel 5 BMC Genetics 2002, 3 12 Research article Screening of the arrestin gene in dogs afflicted with generalized progressive retinal atrophy Gabriele Dekomien, Jörg Thomas Epplen Abstract Background: Intronic DNA sequences of the canine arrestin (SAG) gene was screened to identify potential disease causing mutations in dogs with generalized progressive retinal atrophy (gPRA). The intronic sequences flanking each of the 16 Exons were obtained from clones of a canine genomic library. Results: Using polymerase chain reaction / single strand conformation polymorphism (PCRSSCP) and DNA sequence analyses we screened affected and unaffected dogs of 23 breeds with presumed autosomal recessively (ar) transmitted gPRA. In the coding region of the SAG gene 12 nucleotide exchanges were identified, 5 of which lead to amino acid substitutions (H14C; A111V; A113T; D259T; A379E). 7 other exonic substitutions represent silent polymorphisms (C132C; Q199Q; H225H; V247V; P264P; T288T and L293L). 16 additional sequence variations were observed in intronic regions of different dog breeds. Conclusion: In several breeds, these polymorphisms were found in homozygous state in unaffected and in heterozygous state in affected animals, respectively. Consequently these informative substitutions provide evidence to exclude mutations in the SAG gene as causing retinal degeneration in 14 of the 23 dog breeds with presumed ar transmitted gPRA. 43 Kapitel 5 SAG Gen SAG Gen 5.1 Vorbemerkung Arrestin (SAG) gehört zu einer Familie der inhibitorischen Proteine, die an Tyrosinphosphorylierten Rezeptoren binden, dadurch die Interaktion mit G-Proteinen blockieren und somit die Signalkette effektiv beenden. In der Phototransduktionskaskade arbeiten SAG und die Rhodopsin Kinase (RHOK) in der Wiederherstellungsphase des RHO zusammen. Die Abschaltung des aktiven RHO wird durch die Phosphorylierung mittels der RHOK eingeleitet. SAG vervollständigt die Deaktivierung durch die Bindung an das phosphorylierte RHO. SAG bleibt solange an RHO gebunden bis das all-trans-Retinal durch die Retinoldehydrogenase zu all-trans-Retinol reduziert ist. Dann dissoziiert SAG vom phosphorylierten RHO, welches dann von der Phosphatase 2A, somit wieder photolabil wird und so wieder bereit für die Signalkaskade ist (Wilson et al. 1993; Hofmann et al. 1995). Die Existenz von stabilen Komplexen zwischen RHO und dem Regulatorprotein SAG ausgelöst durch verschiedene Mutationen führt in Drosophila zu retinalen Degenerationen. Akkumulationen dieser Komplexe lösen den apoptotischen Zelltod aus, wobei gezeigt wird, dass retinale Degeneration abhängig von der Endozytose der Zellen ist (Alloway et al. 2000). Der Verlust der SAG-Funktion ist ursächlich für die Oguchi-Erkrankung in Japanern, eine Variante der kongenitalen stationären Nachtblindheit (Fuchs et al. 1995; Sippel et al. 1998). Allerdings kann die gleiche Mutation, die die Oguchi-Erkrankung auslöst, auch zur arRP in japanischen Familien führen (Nakazawa et al. 1998). Hier wird von der Bestimmung der Intronsequenzen des SAG Gens mit anschließender Mutationsanalyse in 23 verschiedenen Hunderassen berichtet. 5.2 Resultate und Diskussion 5.2.1 Genomische Organisation des SAG Gens Verschiedene Anteile des SAG Gens, verteilt auf 7 λ-Klone, wurden nach der Hybridisierung einer caninen Genombank mit Sonden der Exons 2, 5 und 16 isoliert. Das SAG Gen umfasst 16 Exons, wobei sich im 5'UTR Exon 1 (156 bp) und Exon 2 (57 bp) und im 3'UTR das Exon 16 (137 bp) befinden. Die kodierende Region ist 1215 bp lang, wohingegen das gesamte Gen ∼35 kb groß ist. Die meisten Introns sind größer als 1,5 kb (Tabelle 5.1). Der Vergleich mit 44 Kapitel 5 SAG Gen dem menschlichen SAG Gen zeigt, dass das Intron 1 des Hundes im 5'UTR um 23 bp verschoben ist. Das bedeutet, dass Exon 1 des Hundes 23 bp kürzer und Exon 2 23 bp länger als der vergleichbare Bereich im SAG Gen des Menschen ist. Weiterhin ist Exon 15 in Hunden 6 bp länger und Exon 16 6 bp kürzer als die entsprechenden Exons beim Menschen. Das Protein besteht in beiden Spezies aus 405 Aminosäuren. Sie haben eine Ähnlichkeit von 89,8% zueinander. Die Größen der Introns sind im Vergleich vom Menschen zum Hund unterschiedlich, was zu unterschiedlichen Größen des Gens von ∼35 kb im Hund und ∼40 kb im Menschen führt. Die 5' und 3' Spleißstellen folgen der GT-AG Regel (Tabelle 5.1). Es wurden Canoindea-spezifische, tRNA short interspersed nucleotid elements (SINE; Bentolila et al. 1999) in den Introns 1, 3 und 14 identifiziert. Das menschliche SAG Gen ist auf dem Chromosom 2q37.1 lokalisiert. Auf der Basis der vergleichenden Studie von Chromosomenbereichen bei Mensch und Hund (Yang et al. 1999), kann davon ausgegangen werden, dass das Gen entweder auf CFA 28 oder 33, den homologen Chromosomen beim Hund, lokalisiert ist. 5.2.2 Mutationsanalyse mittels PCR-SSCP In 23 verschiedenen Hunderassen, eingeschlossen alle erkrankten und einige ausgewählte gesunde Hunde, wurden die Sequenzen des SAG Gens auf Mutationen, die ursächlich für die gPRA sein könnten durchsucht. Die 16 Exons und alle intronischen Signalsequenzen des Spleißapparates sowie die UTRs (d.h. die komplette cDNA und >3070 bp der Introns) wurden mittels der PCR-SSCP Methode analysiert. In der kodierenden Region des SAG Gens wurden 5 Protein-Polymorphismen (H14C, A111V, A113T, D259T und A379E) mit, und 7 Polymorphismen ohne Aminosäureaustausch (C132C, Q199Q, H225H, V247V, P264P, T288T und L293L; Tabelle 5.2) identifiziert. Zusätzlich wurden 13 Sequenzvariationen in 9 Introns in gPRA erkrankten und nicht erkrankten Hunden nachgewiesen (Tabelle 5.2). Einige der an gPRA erkrankten Hunde in 14 von den 23 untersuchten Rassen waren heterozygot für die oben erwähnten Polymorphismen (Tabelle 5.3). In 6 der 14 Rassen ist die Hauptursache für gPRA bereits aufgeklärt. Direkte Gentest werden inzwischen für den Irischen Setter und Sloughis angeboten (Suber et al. 1993; Dekomien & Epplen 2000). Indirekte Gentest werden für Australian Cattle Dogs, Englische Cocker Spaniel, Labrador Retriever und Zwergpudel bereitgestellt (patentiert durch OptiGen, USA). Diese Rassen wurden als Kontrollen mit in die Untersuchungen eingeschlossen. Eine zweite Mutation ist in den Irischen Settern möglich, da die klinische Symptomatik von der der Irischen Setter mit der bekannten Mutation abweicht. Der Erkrankungsbeginn ist bei diesem Hund ohne die Irish-Setter typische gPRA Mutation 45 Kapitel 5 SAG Gen erst nach 5 Jahren (und nicht früh). Auch bei den Zwergpudeln kann von einer zweiten Mutation innerhalb dieser Rasse ausgegangen werden, da die klinischen Merkmale bei erkrankten Hunden uneinheitlich sind. 5.2.3 Schlussfolgerung Keine der putativen Aminosäurewechsel, die bei der Mutationsanalyse identifiziert worden sind, ist für die Bindung des SAG mit dem aktivierten dephosphorylierten RHO wichtig (Fuchs et al. 1995; Sippel et al. 1998; Vishnivetskiy et al. 1999). Wie schon oben berichtet, führt die Deletion im Kodon 309 in Japanern zu der Oguchi-Erkrankung und zur arRP. Keine der Aminosäureänderungen, die in den untersuchten Rassen gefunden wurden, korrespondiert mit dieser Region. Allerdings können die hier identifizierten Sequenzvariationen unter bestimmten Bedingungen als intragenische Marker für die Segregations-Analyse mit der ar gPRA verwendet werden. Da die Ausschlusskriterien (Kapitel 2.2) für diese Rassen zutreffen, ist das SAG Gen in folgenden 16 Rassen nicht ursächlich für die gPRA: Australien Cattle Dogs, Berger des Pyrénées, Berner Sennenhund, Bologneser, Collies, Englische Cocker Spaniel, Golden Retriever, Irish Setter, Labrador Retriever, Neufundländer, Polnischer Niederungshütehund, Rauhaarteckel, Saarloos/Wolfshunde, Schapendoes, Sloughis und Zwergpudeln. In 6 dieser Rassen stand jeweils nur die DNA eines erkrankten Hundes für die Mutationsanalyse zur Verfügung (Tabelle 2.1). Deshalb muss berücksichtigt werden, dass der Ausschluss des SAG Gens von der gPRA-Diagnose abhängt, und eine falsche Diagnose zu einem unberechtigten Ausschluss des Gens für diese Rasse führen kann. In 6 Rassen (Afghanischer Windhund, Curly Coated Retriever, Springer Spaniel, Labrador Retriever, Rottweiler und Tibet Terrier wurden nur homozygote Sequenzvariationsmuster und in drei Rassen (Collies, Entlebucher Sennehund, und Riesenschnauzer) konnten keinerlei Sequenzvariationen nachgewiesen werden. Deshalb kann das SAG Gen für diese Rassen als Ursache für die gPRA nicht ausgeschlossen werden, da sich möglicherweise in den noch nicht untersuchten regulatorischen Regionen des Gens Mutationen verbergen können. 46 Kapitel 5 SAG Gen Tabelle 5.1 Primer zur Charakterisierung des SAG Gens Exon Primersequenzen (5'-3') Exon (#) und Länge (bp) Intron Länge (bp) 1-2 GGGCAACCCTGTCCAGG ACACCCTGGGGTCTGTGTC (1) 156 836 2-3 AGTGAAAGAAGCTACCAGGGA ACCCACAGGCTCTACTTGT (2) 132 ∼5500 3-4 AGGTGACCATCTACTTGGG AAAGTTCTTTCCTAGCACTAAGa (3) 61 ∼2600 4-5 AGATGGTATCGTGTTGGTGG ACCGTGAGCAGGAAGGG (4) 45 ∼1800 5-6 AGTGTATGTCTCTCTGACCTG ACCTTCCCCATGTCTTGCG (5) 194 ∼2000 6-7 CAAGTTTTACACACTGAGTGCa TCGCAGGCCACAGAGGAGAAGa (6) 60 ∼1300 7-8 GTGCTGTGGGGTCGACTTT CAGCACCCACAGCAGATTGa (7) 77 ∼1500 8-9 AGGAGGTCCGTGCGTTTTAC CTAATGCCTTTAATCTTCTTC (8) 136 905 9-10 CCTAGAAAGCCATGAGATTAAAa AAGCCAAGCATCCTACTTCC (9) 85 ∼1500 10-11 TGGAACAAGTGGCCAACGTTa ACATGGTGCTGCAAGG (10) 73 ∼2500 11-12 GGACTGATGGTGGCTTTATGa ACCCTGACACCGTGAGCTTC (11) 138 ∼3200 12-13 TGAGGGATGTGTTCATCTAGa TTCATAACACCTCTGAGCTACa (12) 78 1390 13-14 CTGGAGGTGAGCTCTCCCA GTGCTCAGGGACCACTAG (13) 24 ∼1400 14-15 AGTGAAGTGGCAACTGAGGT AAGCCGAAGACTGGGAGCa (14) 56 ∼3000 15-16 CCTGCTCACGATTCTCTTTC GTCCAGGTCGACCGACCT (15) 16 426 (16) 237 Spleißstellen 5' (gt) und 3' (ag) CACAAGgtacatg ttcccagCTTGCT AAGTCGgtaagtgg cctatagGTGACC CTGTGGgtaagttg ggtttagATGGTA AGAAAGgtaagaca cctccagTGTATGT CTCACGgtgggtg cccacagTTCCCTG GGGAAGgttagtg actgcagTGCTGTG CAAGAAgtaagagt tctgcagGAGCTCC AAAGAGgtgagcca ttttcagATCTATT CATTAGgtaggaac tctgcagTGGAACA AACACAgtgagtag cctacagAGAAAAA CACCATgtgagtcc tgagcagAATAAAG GTCAGGgtgagtgt ttcctagCTTTCTG CTCCAGgtaagcct tttccagTGAAGTG ACCCAGgtcagtcg cttacagCTACGGC GGAAAGgtgagccc tcttttagTTTTCA PCR Amplifikation des caninen SAG Gens: Primer zur Identifikation der Exon-/Introngrenzen und zur Bestimmung der Längen der Introns der caninen genomischen Sequenz in den SAG Klonen (Exonsequenzen in Großbuchstaben dargestellt), a Sequenzen des SAG Gens: EMBL-Accession-Nr. AJ426068-AJ426078. 47 Kapitel 5 SAG Gen Table 5.2 Primersequenzen für die Mutationsanalyse einzelner Exons/Introns des caninen SAG Gens PCRSystem Lokalisation Primersequenzen (5'-3')A PCR-Bedingungen [T-°C/ PCR Produkt (bp) Restriktionsenzyme für die SSCP Analyse 54/1.0 699 NlaIII/RsaI 50/1.0B 283 Tru1I 53/1.0 249 PvuIII; NlaIIIC 48/1.0B 224 NlaIII 52/1.0 189 AluI 56/1.0 343 MnlI 55/1.0 213 AluI 58/1.0 233 RsaIc 55/1.0 288 RsaI; PstIC 55/1.5 214 TruI1; StyIC 58/1.0 235 AluI 58/1.0 266 BsuRI 55/1.0 183 - 64/1.0 209 MvaI 50/2.0 229 AvaII 58/1.0 244 HphI 52/2.0B 347 AluI MgCl2-mM] UTR1F UTRI-1aR UTRI-1aF UTRI-1aR 1-IF 2-I2R 3-IF 3-IR 4-IF 4-IR 5-IF 5-IR 6-IF 6-IR 7-IF 7-IR 8-IF 8-IR 9-IF 9-IR 10-IF 10-IR 11-IF 11-IR 12-IF 12-IR 13-IF 13-IR 14-IF 14-IR 15-IF 15-IR 16-IF 16-ER Exon 1 Intron 1 Intron 1 Intron 1 Intron 1 Intron 2 Intron 2 Intron 3 Intron 3 Intron 4 Intron 4 Intron 5 Intron 5 Intron 6 Intron 6 Intron 7 Intron 7 Intron 8 Intron 8 Intron 9 Intron 9 Intron 10 Intron 10 Intron 11 Intron 11 Intron 12 Intron 12 Intron 13 Intron 13 Intron 14 Intron 14 Intron 15 Intron 15 Exon 16 GGGCAACCCTGTCCAGGT TCTATCATGACGGGACGCCT GAAAATGATATTTGCAAAGCAG TCTATCATGACGGGACGCCT CTAATGGGCACACAGCATCTC TTCTGTTAAGCCACTCACTTC TGTTTTTATCTAACACTGACTACTTC AAATAACAAAGTAGCAGCTGTC AACTGCAGATAAATATATGAAG AAAGTTCTTTCCTAGCACTAAG GGTTACCCCATGTTCACTTG GCTCCTGGTCACACTGCAAG CAAGTTTTACACACTGAGTGC ATTTTCCCCAGAGAAAAGGCTA CGGGAAGGGAGGTGCTGA TCGCAGGCCACAGAGGAGAAG ATCACAGCGTGAGTACGGGGAG CAGCACCCACAGCAGATTG CCTAGAAAGCCATGAGATTAA GACCAGACTGAGAAATTCTAG GGCACCATGCACATGCGTG AAGCCAAGCATCCTACTTCC GGACTGATGGTGGCTTTATG AGCAGACCAGCACCTCCTC TGAGGGATGTGTTCATCTAG GCCCATGGTGTTGGCTCTTG CATGCTTGGGACATGTCCAC TTCATAACACCTCTGAGCTAC CTCTGCAGCCACAGCCCTTC GTGCTCAGGGACCACTAG CCTGCTCACGATTCTCTTTC AAGCCGAAGACTGGGAGC GATCGGTCCCTTGTTGCA CACAGCTGAACAGACAAACTT A EMBL-Accession-Nr. AJ426068-AJ426078. B mit 5% Formamid. C für RFLP Analyse. 48 Kapitel 5 SAG Gen Tabelle 5.3 SAG Sequenzvariationen und Aminosäureveränderungen in verschiedenen Hunderassen Sequenzvariationen in Rasse(n)a Lokalisation Sequenzvariation* Aminosäurewechsel in gPRA-erkrankten und gesunden Hunden gPRA-erkrankte Hunde Intron 1 IVS1+393T→C - BDP, BS, GR, IRS, Rob, TT BDP, GR, Exon 2 UTR-5A→G - c255A→G H14C A111V A113T Intron 5 c526C→T c531G→A IVS5-30C→T Bo, BS, GRc, BDP, IRS, SDc, Sl, Bo, GR, SD TTb SI Sl c Sa Sa Sac Sa - ECS, Rob, Sac, ZP ECS, Sa, ZP Exon 6 c610C→T C132C GR GR Intron 7 IVS7+10C→T IVS7+52Ins.G IVS7-4Del.G - IRSb, GR, SPc IRS, GR, Te GR GR b c c b GR, IRS, SD, Te CCR GR, IRS, SD , Sl , SP , Te, c TT , ZP Exon 8 Intron 8 c811G→A IVS8+8A→G Q199Q - GR ECSc,GRc, LRc, Sa, Te Exon 9 c874T→C H225H Bo, BDP, CCRb, ECS, GRc, IRS, Bo, ECS, D, GR, NF, Rob, Sa, Slb, Te, TTbc, ZP IRS, NF, Sa, ZP Exon 10 c949G→T c983G→T c1000A→G V247V D259T P264P IRS, GR ACb AC, IRSb, GR, LR GR AC GR Intron 10 IVS10-18G→C IVS10-33C→T - GR, IRS, BDP GR, IRS GR GR Exon 11 c1063G→A c1076C→T T288T L293L BS, BDP, GR, IRSb, Sa BS, BDP, GRc, Sa, TT BS, GR, Sa BS, Sa Intron 11 IVS11-51Ins.TT - Sa, IRSb Sa b Sa Exon 5 c GR ECS, GR, Sa, Te Intron 13 IVS13+66C→G - Sa, IRS Intron 14 IVS14-45Del.A - PON PON Intron 15 IVS15+14C→T IVS15+86T→A IVS15-45Ins.C - Sa Sa PON, AWbc, SPb PON Exon 16 c1344C→A A379E IRSb, ECSb, GRc, PON, Sabc, TTb ECS, GR a Für Abkürzungen siehe Tabelle 2.1. b Homozygoter Zustand in gPRA erkrankten Hunden. c Homozygoter Zustand in gesunden Hunden. * cDNA: EMBL-Accession-Nr. X98460; genomische DNA: EMBL-Accession-Nr. AJ426068-AJ426078. 49 Kapitel 6 Molecular Vision 2002, 8 138-142 The canine Phosducin gene: characterization of the exon-intron structure and exclusion as a candidate gene for generalized progressive retinal atrophy in 11 dog breeds Gabriele Dekomien, Jörg Thomas Epplen Abstract Purpose: The exon-intron structure of the canine Phosducin (PDC) gene was identified and the gene evaluated as a candidate for generalized progressive retinal atrophy (gPRA) in 20 dog breeds. Methods: Intronic sequences of the PDC gene were analyzed after amplification using polymerase chain reaction (PCR) and following sequencing from clones isolated from a canine genomic library. Mutation screening was performed by PCR with single strand conformation polymorphism (SSCP) analysis. Conspicuous banding patterns were analyzed via sequence analyses to detect the underlying nucleotide variations. Results: No polymorphisms were identified after PCR-SSCP analysis within the entire coding region of the PDC gene. A 3 bp deletion in intron (intervening sequence (IVS) 3 (position -16 to -18) was observed in 9 breeds, a T→A transversion (position IVS3 -63) in 10 breeds and an A→T transversion (position IVS3 -64) in 2 dog breeds. Conclusions: PDC was excluded as a candidate gene for autosomal recessively (ar) transmitted gPRA in 11 of the 20 dog breeds investigated. 50 Kapitel 6 PDC Gen PDC Gen 6.1 Vorbemerkung Phosducin (PDC) hat eine hemmende Wirkung auf die Phototransduktionskaskade durch die Bindung an den βγ-Untereinheitenkomplex des Transducin (Tαβγ). Dadurch wird ein PDC(Tdβγ) Komplex gebildet der eine erneute Anlagerung von Tα verhindert. Nun kann Tα kein GDP zu GTP umwandeln was dem Phototransduktionsprozess entgegenwirkt (Lee et al. 1990). Die zyklische Adenosin 3'5' -monophosphatabhängige Proteinkinase (A-Kinase) katalysiert die Phosphorylierung des PDC. Phosphoryliertes PDC verliert die Bindeaffinität zum Transducin. Die Konsequenz daraus ist, dass Transducin licht-aktiviertes RHO nicht mehr hemmt (Lee et al. 1992; Schaad et al. 1991; Thulin et al. 1999). PDC ist hauptsächlich nur in den inneren Segmenten und der perinukleären Region der Stäbchenphotorezeptoren konzentriert (Bauer et al. 1992). Im PDC Gen wurde von Zhang et al. (1998) eine "Fehlsinn" (missense) Mutation identifiziert, die in einigen erkrankten Zwergschnauzern zur Photorezeptordysplasie (rd), einem Subtypen der gPRA führt. Die mRNA des caninen PDC Gens umfasst einen OLR von 735 nt, die für 245 Aminosäuren kodieren. Die PDC Nukleotidund Aminosäuresequenz sind in Säugetieren konserviert (Zhang et al. 1998). Hier wird über die Identifikation der intronischen Sequenzen und der anschließenden Mutationsanalyse des PDC Gens berichtet. 6.2 Resultate und Diskussion 6.2.1 Genomische Organisation des PDC Gens Vier verschiedene λ-Klone mit Teilbereichen des PDC Gens wurden mit radioaktiv markierten Sonden der Exons 2 (Position 59-143) und 4 (Position 296-667, EMBLAccession-Nr.CFY17697) identifiziert und charakterisiert. In keinem der Klone wurde das vollständige Intron 1 nachgewiesen. Der Vergleich mit der genomischen DNA des Menschen (EMBL-Accessions-Nr AL162722) zeigte eine Introngröße von 75 kb. Wenn im PDC Gen des Hundes eine entsprechende Größe des Intron 1 zu erwarten ist, kann das Intron niemals vollständig in einem λ-Klon einkloniert sei, da der Vektor nur Fragmente von 9-23 kb aufnehmen kann. Deshalb wurde der 3' Anteil des Introns 1 durch eine "nested" PCR mit den T7 Primer des λ Fix II Vektors und mit Hilfe eines exonischen Primers im Exon 2 51 Kapitel 6 PDC Gen amplifiziert. Durch eine s.g. Long-range PCR mit dem Elongase Enzym Mix (Gibco BRL, Karlsruhe, Deutschland) konnte unter Einhaltung der von der Firma vorgeschriebenen Anweisungen die ungefähre Größe von 14 kb und der 5'-Bereich des großen Introns bestimmt und anschließend sequenziert werden (EMBL-Accession-Nr. AJ429100). Die Sequenzdaten des caninen PDC Gens weisen auf 4 Exons hin. Exon 1 (69 bp) und 24 bp des Exon 2 umfassen den 5' UTR des PDC Gens. Die kodierende Region beinhaltet mit insgesamt 738 bp Exon 2 mit 85 bp, Exon 3 mit 152 bp und Exon 4 mit 525 bp, wobei zusätzlich 409 bp der 3'UTR zuzurechnen sind (siehe Abbildung 6. 1). Vergleiche des caninen mit dem humanen PDC Gen zeigen eine Verlängerung des OLRs im Exon 4 beim Menschen um 3 bp. Dies führt zu einer Proteinverlängerung von einer Aminosäure, sodass das humane PDC Protein aus 246 und das des Hundes aus 245 Aminosäuren besteht. Ansonsten besteht ein 90%ige Übereinstimmung der Aminosäuresequenz zwischen Mensch und Hund. Die Introns 2 und 3 umfassen beim Hund eine geringere Anzahl von Nukleotiden als beim Menschen (Intron 2: Mensch 2820 bp, Hund ∼1900 bp; Intron 3: human 1947 bp zu ∼1500 bp beim Hund). Die 5' und 3' Spleißstellen der caninen PDC-Introns sind in Übereinstimmung mit der GT-AG Regel (Tabelle 6.3). Das menschliche PDC Gen ist auf dem Chromosom 1q24.3-31.1 lokalisiert. Auf der Basis der vergleichenden Studie von Chromosomenbereichen von Mensch und Hund (Yang et al. 1999) kann davon ausgegangen werden, dass das Gen auf CFA 7, der homologen chromosomalen Region des Hundes, lokalisiert ist. 6.2.2 Mutations-Analyse des PDC Gens Die Polymorphismussuche des PDC Gens wurde in 20 verschieden Rassen durchgeführt. In die PCR-SSCP-Analyse wurden alle an gPRA erkrankten Hunde und ausgewählte normale und obligatorische gPRA-Träger eingeschlossen. Exons 2, 3 und 4, Teile der drei Introns sowie die UTRs wurden untersucht (genutzte Primer, Tabelle 6.1). PCR-Amplifikate von drei Exons wurden mit Restriktionsenzymen in direkt auswertbare Fragmentgrößen geschnitten (Exon 2 BsuRI, Exon 3 MnlI, Exon 4 Teil1 MboII und Teil 2 MnlI/RsaI). Im OLR des PDC Gens wurden keine Polymorphismen identifiziert. Jedoch wurden in den untersuchten Hunden von 11 Rassen Unterschiede in der Sequenz von Intron 3 identifiziert: eine TCT-Deletion in der Position -16 bis -18 (Abbildung 6.2 A) in Berger des Pyrénées, Collies, Englische Cocker Spaniel, Entlebucher Sennenhunde, Irish Setter, Labrador Retriever, Sloughis, Teckel, Tibet Mastiffs, Tibet Terrier und Zwergpudel sowie eine T→A Transversion in Position -63 in Berger des Pyrénées, Collie, Teckel, Englische Cocker Spaniel, Golden Retriever, Irish Setter, Labrador Retriever, Sloughis, Tibet Mastiffs, Tibet 52 Kapitel 6 PDC Gen Terrier und Zwergpudel sowie ein A→T Nukleotidaustausch in der Position -64 in Entlebucher Sennenhunde und Saarloos/Wolfshund (siehe Abbildung 6.1 & 6.2 B). Der Polymorphismus in der Position IVS3-64 wurde niemals zusammen mit der TCT-Deletion beobachtet. Die Allelfrequenzen der 3 Polymorphismen sind in Übereinstimmung mit dem Hardy Weinberg Gleichgewicht (Tabelle 6.2). 6.2.3 Schlussfolgerung Die R82G "Fehl-Sinn" (missense) Mutation, die im Exon 4 in Zwergschnauzern zur Photorezeptordysplasie (pd) führt (Zhang et al. 1998), konnte in keinem der hier untersuchten Hunde nachgewiesen werden. Auch die beiden veröffentlichten Polymorphismen im 3'UTR in Zwergpudeln und in Irischen Wolfshunden (Lin et al. 1998) wurden nicht in der zu untersuchenden Hundepopulation entdeckt. Die neuen Polymorphismen, die weiter oben spezifiziert wurden, sind in einigen der an gPRA erkrankten Hunde im heterozygoten Zustand nachweisbar. Somit können diese Polymorphismen als intragenische Marker für den Nachweis der fehlenden Kosegregation mit der gPRA genutzt werden. Durch Züchtung, kleine Populationsgrößen und gPRA Häufigkeiten wie in Kapitel 2.3.1 und 3.2.1 beschrieben, besteht bei einem ar Erbgang keine Kopplung zur gPRA-Mutation zu den untersuchten Polymorphismen. Deshalb kann nach Analyse dieser Sequenzvariationen das PDC Gen als Ursache für die gPRA für die folgenden 11 Hunderassen ausgeschlossen werden: Collies, Englische Cocker Spaniel, Entlebucher Sennenhunde, Golden Retriever, Irish Setter, Saarloos/Wolfshunde, Sloughis, Teckel, Tibet Mastiffs, Tibet Terrier und Zwergpudel. 53 Kapitel 6 PDC Gen Tabelle 6.1 Primersequenzen, PCR-Systeme und -Bedingungen zur Charakterisierung und Mutationsuntersuchung des caninen PDC Gens PCR-System PrimerBezeichnung 1a UTR Fa 2 (1) Rab 2 (1a)-IFb 2 (1) Rab 2 (1b)-2Fb 2 (1b)-IRb 2 (1b)-2Fab 3 (2) Ra 3 (2) Fa 4 (3)-1-Rab 3 (1b)-IFb 3 (2)-IRb 4 (3) Fa 4 (3)-1-Ra 4 (3)-1-IFb 4 (3)-1-Rab 4 (3)-2-Fab 4 (3)-2-Rab a Primersequenza (5'→ →3') Bindeposition E:Exon I:Intron AATTCGGCACGAAACAAGC CTGTATGTGTGGCCTGTC ACTCCTACCCTAGCATGCA CTGTATGTGTGGCCTGTC AGATTATATAAAATCAAATCC TTTATGCTGACCAATGGCAGT AGATTATATAAAATCAAATCC ACCTTTCTGCTGAATCTTTCTT AGGACCCAAAGGAGTAATAAA CCCACCTTTGTAGATGAGCA TCTGATGCAGAAGTTGATATCT TTTGCCAATGTAACCAATACTAT AGATGAGCATTCAAGAATATGA CCCACCTTTGTAGATGAGCA AAAGCCTGTTTCTCTATCT CCCACCTTTGTAGATGAGCA GCTGGGGACCGCTTTTCCTCAGA AAACAAGGAGTAAAAAAAGTCAAC E1 E2 I1 E2 E2 I2 E2 E3 E3 E4 I2 I3 E4 E4 I3 E4 E4 3'UTR E 4 Länge der PCRProdukte (bp) ∼14000 PCR-Konditionen T/MgCl2 [(°C)/(mM)] 59/1.5 191 54/1 169 51/1 ∼1900 54/2d ∼1500 54/2d 396 56/1 374 55/1 509 53/1c 512 56/1 PCR Primersequenzen von der caninen mRNA (EMBL-Accession-Nr. Y17697) für die Identifikation der Exon-/Introngrenzen und Intronsequenzen. b PCR-Primer für die Mutationsanalyse (Auf der Basis des caninen PDC Gens, EMBL-Accession-Nr. CFA417559-CFA17561). c Zusätzlich 5% Formamid. d 5% DMSO. 54 Kapitel 6 PDC Gen IVS3-64A→T&-63T→A IVS3-16-18DelTCT Abbildung 6.1 Genomische Organisation des PDC Gens. Weiße Kästen entsprechen dem OLR, die grauen Kästen von Exon 1 und 4 repräsentieren die 5' und 3'UTRs. Die Längen der abgebildeten Introns entspricht dem Anteil der veröffentlichen Sequenzen (EMBL-Accessions-Nr. AJ429100, CFA417559-417561). Die ungefähre Länge der Introns wurde durch überlappende PCRs inklusive der benachbarten Exons bestimmt. A. -70 -48 -26 -73 -51 -29 -5 -8 B. Abbildung 6.2 Sequenz-Chromatogramme der Polymorphismen im Intron 3 des PDC Gens. A: Die WildtypSequenz eines Dachshundes von -70 bis -48 sowie die Deletion (Pfeil) von -18 bis -16. B: Die entsprechende Teilsequenz eines Entlebucher Sennenhundes mit einer Substitution in Position -64. Da dieser Hund heterozygot für diesen Austausch ist, wird eine kombiniertes Signal (N) angezeigt. 55 Kapitel 6 PDC Gen Tabelle 6.2 Krankheitsbeginn, Altersverteilung von gesunden und gPRA-erkrankten Hunden bei DNA Isolierung und Allelfrequenzen des PDC Gens in den untersuchten Rassen Rasse Diagnose AC BDP Bo Co Te ECS ES GR IRS LR ZP NF PON Ro Sa ScT SD Sl TM TT a gPRA-erkrankt gesund gPRA-erkrankt gesund gPRA-erkrankt gPRA-erkrankt gesund gPRA-erkrankt gesund gPRA-erkrankt gesund gPRA-erkrankt gesund gPRA-erkrankt gesund gPRA-erkrankt gesund gPRA-erkrankt gesund gPRA-erkrankt gesund gPRA-erkrankt gPRA-erkrankt gPRA-erkrankt gPRA-erkrankt gesund gPRA-erkrankt gPRA-erkrankt gesund gPRA-erkrankt gesund gPRA-erkrankt gesund gPRA-erkrankt gesund (Laratta et al. 1988). Altersverteilungf (Jahr) IVS3 16-18 delTCT späta 10 2-4 5 1-10 9 4-8 8 1-13 6-13 3-11 6-14 5-13 1-7 5-10 3-6 0,6-7 3-13 8-12 3-13 5-12 1-12 3 9 3 7-9 2-10 6 2-6 3-6 2 0,1-12 4 4 7-8 2-10 17 28 50 50 50 14 29 20 50 38 50 34 11 mittelb spätb frühcd variabelb spätcd späte spätb frühcd/spätb spätcd spätcd mittelb spätb spätb spätb spätb frühd mittelb mittelb mittelcd b Allelfrequenz (%) gPRAKrankheitsbeginn IVS3 -63 T→ →A A 17 25 50 20 37 60 25 12 25 2 17 30 25 56 50 17 21 IVS3 -64 A→ →T T 59 29 19 27 - Bericht des Besitzers/Augenuntersuchungsbogen (DOK). c Klassifi- kation der verschiedenen Krankheitsbeginn-Arten der gPRA in den Übersichtsartikeln (Clements et al. 1996 und Gropp et al. 1997). (http://www.sheepdog.com/diseases/pra/clinical). e d Online-Information unter PRA Today (Spiess et al. 1994). f Zum Zeitpunkt der Blutabnahme für die DNA Analyse. 56 Kapitel 6 PDC Gen Tabelle 6.3 Vergleich der Exon-/Intronorganisation des PDC Gens von Hund und Mensch Exon # Exonlänge (bp) Hund Mensch 1 69 69 69 120404 AACCAAACTgtgagtctgcaggtgg ATCCAAACTgtgagtccacaagtag Hund Mensch 2 85 85 105 44942 tttgttcatactatagATTATATAA tttgttcctcctatagATTATATCA 189 44856 CACATACAGgtatgaaaatctagaa CACATACAGgcaagcaaagctagaa 226 42036 tttcttttattcagGACCCAAAG taatttcattccagGACCCAAAG 377 41885 AGCAGAAAGgtaagggaaaaatcaa AGCAGAAAGgtaagataatacaaaa 520 39937 cttttgtctattcccagATGAGCATT catttctttattctcagATGAGCATT Hund Mensch Hund Mensch 3 152 152 Hund Mensch Hund Mensch 4 934 918 Exon Nukleotidpositiona 5' (gt) und 3' Spleißstellen (ag) Intron Intron# länge (bp) 1 ∼15000 75390 2 ∼1900 2820 3 ∼1500 1947 Die Exon-/Introngrenzen folgen der gt-ag -Regel. Exonsequenzen sind in Großbuchstaben, Intronsequenzen in Kleinbuchstaben dargestellt a Nummern entsprechenden den Nukleotidpositionen der Exon-/Introngrenzen der PDC Gens (Hund: EMBL-Accession-Nr. AJ429100 und CFA417559 – CFA417561; Mensch: Rücksequenz, Klon des Chromosoms 1 RP13-317E13; EMBL-Accession-Nr. AL162722). 57 Kapitel 7 Molecular Vision 2002, 8 436-441 The canine Recoverin (RCV1) gene, a candidate for generalized progressive retinal atrophy Gabriele Dekomien, Joerg Thomas Epplen Abstract Purpose: We describe the cloning, sequence, and mutation analysis of the canine Recoverin (RCV1) gene, a candidate gene for generalized progressive retinal atrophy (PRA). Methods: The gene was isolated from a genomic lambda Fix II library using an exon 1 probe of the human RCV1 cDNA. Canine RCV1 sequences were identified by subcloning, polymerase chain reaction (PCR), and sequence analysis. Furthermore, selected DNA samples of 22 dog breeds (including all PRA-affected and several representative unaffected dogs from the pedigrees) were screened for mutations and polymorphisms using PCR-SSCP (single strand conformation polymorphism) and sequence analysis. Results: The canine RCV1 gene revealed 3 exons and an open reading frame of 606 bp, potentially coding for a protein of 202 amino acids. The deduced amino acid sequence of the canine RCV1 gene shares 89% identity with the homologous human, 94% with bovine, and 91% identity with the mouse genes. The protein sequence reveals two typical Ca2+-binding EF-hand motifs. In the ORF (open reading frame) of the RCV1 gene a C272A (exon 1) and a C4275A transversion (exon 3) were discovered. These exchanges result in amino acid substitutions (N3K and P202H), but they do not segregate with PRA in the breeds investigated. Additionally, two sequence variations were identified in the 5'-UTR, one in intron 1 and thirteen variations in intron 2 as well as one in the 3'-UTR. Conclusion: Using intragenic polymorphisms, we excluded the RCV1 gene as a candidate gene for autosomal recessively transmitted (ar) PRA in 16 dog breeds. In addition the RCV1 gene was excluded for presumedly autosomal dominant (ad) PRA in 8 out of the 22 dog breeds investigated. 58 Kapitel 7 RCV1 Gen RCV1 Gen 7.1 Vorbemerkung Recoverin (RCV) ist ein retinales Ca2+-Bindeprotein, das an der Erholungsphase der visuellen Erregungsphase und an der Adaption an verschiedene Lichtverhältnisse beteiligt ist. Die Ca2+gebundene RCV-Form verlängert die Lichtantwort wahrscheinlich durch die Blockade der Phosphorylierung des angeregtem Rhodopsin (siehe auch Abbildung 13.1). RCV umfaßt eine kovalent angelagerte Myristoyl- oder Acylgruppe am N-Terminus und hat mindestens 2 Ca2+Bindestellen sowie ein EF-hand Motiv. Der Name ist von den E und F Helices und der dazwischen liegenden Schleife abgeleitet (Tanaka et al. 1995). Beim Menschen liegt das Gen auf Chromosom 17p13.1, eine Region, in der sowohl Loci für ar wie auch ad Formen der RP nachgewiesen sind (RetNet; Goliath et al. 1995; Tarttelin et al. 1996). Als ein Mitglied der Phototransduktion wurde RCV1 als Kandidatengen für die ar RP in 42 spanischen Familien auf RP-ursächliche Mutationen hin untersucht und in 38 als Ursache der ar RP ausgeschlossen (Bayes et al. 1995). Zusätzlich wurde das Gen bei 596 Patienten mit RP, oder entsprechenden Erkrankungen auf Mutationen hin überprüft und als Ursache für diese Erkrankungen ausgeschlossen (Parminder et al. 1997). Um herauszufinden, ob das RCV1 Gen eine wichtige Rolle für die gPRA in den erkrankten Hunden spielt, wurde die gesamte Sequenz kloniert und mit PCR-SSCP in 22 Rassen untersucht. 7.2 Resultate und Diskussion 7.2.1 Identifikation des RCV1 Gens Um das canine RCV1 Gen zu identifizieren, wurde eine canine Genombank mit einer DNASonde des menschlichen Exon 1 durchsucht. Drei λ-Klone wurden durch die Hybidisierung entdeckt mit unterschiedlich langen und unvollständigen 5'- und 3'UTRs. Optimale Primerpositionen in Bereichen höchster Sequenzübereinstimmungen wurden durch Vergleiche der mRNAs von Maus (McGinnis et al. 1992; EMBL-Accession-Nr. X66196, Rind (Kutuzov et al. 1991; EMBL-Accession-Nr. AB001838) und Mensch (Murakami et al. 1992; EMBL-Accessions-Nr. AB001838) identifiziert (Tabelle 7.1). Teile des RCV1 Gen des Hundes wurde mit diesen Primer mittels PCR aus diesen Klonen amplifiziert. Nach der Sequenzierung des intronüberlappenden PCR-Produkte wurden intronische Primer hergestellt, um die vollständige Länge und Sequenzen der beiden Introns festzulegen (Sequenzen unter 59 Kapitel 7 RCV1 Gen EMBL-Accessions-Nr. AJ414401; Tabelle 7.1). Exon-/Introngrenzen wurden durch Vergleiche der menschlichen RCV1 mRNA mit dem genomischen Klon von Chromosom 17 (EMBL-Accession-Nr. AC005747) durch das Blast Search Programm bestimmt. Die Exon/Intronorganisation des Hundes stimmt im Vergleich mit den Grenzen des menschlichen Gens überein (Tabelle 7.2). Das canine RCV1 Gen ist 4523 bp lang davon verteilen sich auf das Exon 1 644 bp (5'UTR 263 bp, OLR 381 bp), Intron 1951 bp, Exon 2 112 bp, Intron 2 2456 bp und auf Exon 3 360 bp (OLR 116 bp, 3'UTR 244 bp). Das canine RCV1 Gen ist kürzer als das menschliche Gegenstück. Dieser Unterschied ist durch die kürzeren Introns bedingt. Zwei EF-hand Motive wurden mit dem ProDomNCBI-BlastP2-Programm in dem 202 Aminosäuren umfassenden OLR des RCV1 Gens identifiziert. Die größte Homologie (95%) mit den EF-hand Motivsequenzen wurde in der Amiosäureposition 26-95 und in der Position 143-191 der Proteinsequenz des Rindes beobachtet. Im Intron 1 wurde eine kurze Sequenzfrequenz (Nukleotid-Position 1103-1180), welche einem canine-spezifischen tRNA SINE entspricht, nachgewiesen (Bentolila et al. 1999). Das menschliche RCV1 Gen ist auf dem Chromosom 17p13.1 lokalisiert. Auf der Basis der vergleichenden Studie von Chromosomenbereichen von Mensch zum Hund (Yang et al. 1999) kann davon ausgegangen werden, dass das Gen auf CFA 5 der homologen chromosomalen Region des Hundes lokalisiert ist. 7.2.2 Mutationsanalyse des RCV1 Gens Nach der Isolierung und Charakterisation des caninen RCV1 Gens wurde das Gen nach der die Augenkrankheit auslösenden Mutation durchsucht. Für die PCR-SSCP-Analyse wurden Primer konstruiert, die die Exons flankieren, wobei die konservierten Spleißregionen und Anteile des Intron 2 mit berücksichtigt wurden (Tabelle 7.1). In dieser Studie standen insgesamt 805 Hunde inklusive 112 gPRA erkrankte Hunde aus 22 Rassen zur Verfügung. In vier Rassen (Australien Cattle dogs, Golden Retriever, English Cocker Spaniel, Rottweiler) wurden keine Mutationen in gPRA erkrankten Hunden identifiziert. Zwei SNPs wurden im OLR nachgewiesen. Die C272A Transversion verursacht einen Aminosäurewechsel (N3K) in Teckeln, Saarloos und Scottish Terriern. Die C4275A Variation resultiert in einen P202H Aminosäurewechsel im carboxyterminalen Bereich und zeigt sich homozygot bei Berger des Pyrénées und im heterozygoten Zustand in Bolognese, Teckel, Riesenschnauzer, Schapendoes und Sloughis. Weitere Sequenzvariationen wurden im 5'UTR-, Intron 1 und dem 3'UTR identifiziert. Intron 2 beherbergt 13 rassenspezifische Sequenzvariationen, davon sind vier SNPs (C2735T-C2843T) immer im Kopplungsungleichgewicht (Tabelle 7.3 und 7.4). Für 60 Kapitel 7 RCV1 Gen Irishe Setter und Sloughis (Suber et al. 1993; Dekomien & Epplen 2000) gibt es schon direkte und für Zwergpudel einen indirekten Gentest (patentiert durch Optigen). In Irish Settern und Zwergpudeln wird eine zweite gPRA-Mutation aufgrund der klinischen Symptome vermutet, und Sloughis wurden als Kontrollen in die Untersuchung mit einbezogen. 7.2.3 Schlussfolgerungen Die identifizierten intronischen Sequenzvariationen wurden in heterozygoten Zustandsformen in gPRA erkrankten Hunden verschiedener Rassen nachgewiesen. Durch die Kopplung der polymorphen Nukleotide der bestimmten RCV1 Allele in den gegebenen Rassen schließt sich Heterozygotie bei einer ar Vererbung in erkrankten Hunden aus. Der Irish Setter mit der bekannten Rassen-spezifischen PDE6B-Mutation (Suber et al. 1993) ist homozygot für die variablen Positionen in dem RCV1 Gen. Im Gegensatz dazu zeigt der Irish Setter mit der späten Form der gPRA (ohne die PDE6B-Mutation) heterozygote Zustandformen in den meisten Positionen. Aufgrund der für einen rezessiven Erbgang gegebenen Bedingungen (Kapitel 2.3; 3) kann das RCV1 Gen als Kandidatengen für die ar gPRA in den folgenden Rassen ausgeschlossen werden: Bologneser, Curly Coated Retriever, Collies, Entlebucher Sennenhunde, Irish Setter, Labrador Retriever, Neufundländer, Riesenschnauzer, Saarloos/Wolfshunde, Salukis, Scottish Terrier, Schapendoes, Sloughis, Tibet Terrier, Teckel und Zwergpudel. In ganz seltenen Fällen wird die PRA in Haustieren auch ad vererbt. Sowohl in Mastiffs und Abessinierkatzen wurde dominante Vererbung für retinale Augenerkrankungen nachgewiesen (Kijas et al. 2002; Curtis et al. 1987). Zusätzlich liegt das RCV1 Gen in einem chromosomalen Bereich, der sowohl für ad wie auch ar vererbte RP ursächlich sein könnte. Somit war zu überprüfen ob dominante Vererbung einer Mutation im RCV1 Gen als Ursache der gPRA in den zu untersuchenden Rassen möglich ist. Durch die Polymorphismusanalyse von 11 verschiedenen Rassen wurden insgesamt 12 verschiedene Haplotypen in erkrankten Hunden identifiziert (Tabelle 7.4). Da kein bestimmtes Allel mit der Erkrankung kosegregiert (Tabelle 7.4), kann auch eine mögliche dominante Vererbung der gPRA in 8 Rassen (Entlebucher Sennenhunde, Labrador Retriever, Saarloos/Wolfshunde, Schapendoes, Sloughis, Teckel, Tibet Terrier und Zwergpudel) ausgeschlossen werden. Der Polnische Niederungshütehund und der Berger des Pyrénées zeigen Seqzenzvariationen homozygot ohne Segregation mit der gPRA. In Australian Cattle Dogs, English Cocker Spaniel, Golden Retrievern und dem Rottweiler wurden keine Sequenzvariationen nachgewiesen. Deshalb ist 61 Kapitel 7 RCV1 Gen keine sichere Aussage für das RCV1 als Kandidatengen für die gPRA in diesen Rassen, möglich. Tabelle 7.1 PCR Systeme zur Charakterisierung und Mutationsanalyse des caninen RCV1 Gens PCR-System Primersequenz (5'→ →3')* H 1F H 1R I 1-331EF I 1-IR H 1F H 2R H 2F H 3R I 2F I 2R I/M 2 2F I/M 2 2R I/M 2Ht 2F I/M 2 2R M`5'UTR1F M/Ht Kl1ER H/M 1F M 1-IR M 2-IF M 2-IR M 3-IF M 3'UTR R CCCTGTCCAAGGAGATCCT CATGACGATCTCCAGCACTT TCTCGCTCTACGACGTGGA GACAGGGAACGGAGTGAAG CCCTGTCCAAGGAGATCCT CTTTCCAAAGTACTTCCAGATC TTCAAAATGATCACTCCCGAGG TCATCTTTTCCTTCACTTTTTGAG ACCGCCGAGCTCCCTGG ATGCGGAGACACTCCCACG TGAGAACTGGCCTTCGAGG CGTGACTGTGGCATTCACC GGGAGGCATTCCAGAACAG CGTGACTGTGGCATTCACC CTCCCTGAAGGCCAAGATG TTCGGAAGGAAACTGGTACC CCCTGTCCAAGGAGATCCT GAACCCCCTGGACCCAGGA GGAATCTGTATTTTCACCTCTG CTAGGAAGACCAATCTCACT GTGATGGGTTGTACCCTCAG GCATGTGCACGTGCTCACG Lokalisa-tion PCR-Fragment Größe (bp) Exon (E) Intron (I); (bp) E 1; 13 E 1; 381 E 1; 580 I 1; 1419 E 1; 13 E 2; 483 E 2; 388 E 3; 592 I 2; 2006 I 2; 3777 I 2; 2398 I 2; 3417 I 2 2658 I 2; 3417 UTR/E 1; 1 E 1; 374 E 1; 13 I 1, 709 I 1, 1483 I 2, 1790 I 2; 4019 3’UTR; 4358 Restriktionsenzym für SSCP Analyse PCR-Bedingungen [T-°C) / MgCl2-mM] 359 - 47/2** 837 - 54/2 1422 - 46/2** 2684 - 48/2** 1774 - 54/2** 1020: 208, 98, 160, 13, 188, 276, 77 760: 30, 160, 13, 188, 276, 77 372: 184, 132 56 424: 183, 71, 145, 65 308: 136, 172 340: 206, 134 DdeI 52/1 DdeI 54/1 PstI 55/1 NlaIII 54/1 DdeI 55/1 MboI 55/1 H: PCR-Primersequenzen von der humanen mRNA (EMBL-Accession-Nr. AB001838) abgeleitet für die Identifikation der Exon-/Introngrenzen und der caninen Gensequenzen. I: PCR-Primersequenzen des caninen RCV1 Gens EMBL-Accession-Nr. AJ414401 zur Amplifizierung des vollständigen Introns. M: PCR-Primer vom caninen RCV1 Gen für die Mutationsanalyse. Ht: Zusätzlicher PCR-Primer für die Haplotypanalyse * Nukleotidposition mit der die 5' Enden des Primers (fett gedruckt) hybridisieren. ** zusätzlich 5% DMSO. 62 Kapitel 7 RCV1 Gen Tabelle 7.2 Organisation der Exon-/Introngrenzen des caninen RCV1 Gens Exon # Exonlänge (bp) 1 2 644 112 3 360 Exon Nukleotidposition* 1 1596 1707 4163 Intron 5' -(gt) und 3' Spleißstellen (ag) # GATCGTCATGgtcagtctcg tgtgcctgcagGCTATTTTC AAAGATGATGgtgaattcc cccatgcacagATAAACTT Intronlänge (bp) 1 951 2 2456 Die Exon-/Introngrenzen folgen der gt-ag Regel. Exonsequenzen sind in Großbuchstaben, Intronsequenzen in Kleinbuchstaben dargestellt. * Die Nummern entsprechen den korrespondierende Nukleotidpositionen der Exon-/Introngrenzen des RCV1 Gens EMBL-Accession-Nr. AJ414401. Tabelle 7.3 Identifizierte Polymorphismen in heterozygoten Mustern in gPRA-erkrankten Hunden. Aufgelistete Hunderassen für den Ausschluss des RVC1 Gens als Ursache für ar gPRA. Lokalisation Sequenzvariation Aminosäure Rassen* wechsel Bo, ES, NF, RS, Sa, ScT, SD, Sl, Te, TT, ZP Bo, Te, ES, IRS, NF, ZP, ScT, Sl ScT, Sl, Te 5‘ UTR A127G - Exon 1 C229T C272A N3K Intron1 C1552T - Co, TT Intron 2 C2735T G2774A C2814T C2843T G2940C C2950T C2958A G3010A A3102G G3225A G3325A T3370C C3370G - Co, LR, NF, RS, SD, Sl, Te Co, LR, NF, RS, SD, Sl, Te Co, LR, NF, RS, SD, Sl, Te Co, LR, NF, RS, SD, Sl, Te NF, RS, SD, Sl, Te CCR, IRS, LR, NF, Sa, Sl, Te, TT, ZP ZP Sa, Te Bo, Co, LR, RS, Te, Sa, Sal, ScT, TT, ZP Sa NF LR, Sa, Te Bo, IRS, Sa, Sal, ScT, ZP Exon 3 3'UTR C4275A A4309G P202H - - Bo, RS, SD, Sl, Te Bo, ES, LR, Sa, Sal, SD, Sl, Te * Abkürzungen siehe Tabelle 2.1 (EMBL-Accession-Nr. AJ414401). 63 PON BDP ZP TT Te Sl SD Sa LR IRS ES Variation Rassen C C C o. T C T C C C C C C C C C C C C C C C or T C C C C C C C C C C C C C C C o. T C o. T C C A C229T G A A o.G G G G G A A A o. G A A A o. G A A A o. G A A G A o. G A o. G A A o. G A o. G A o. G G A A G A A A A A A o. G A o. G A A127G Intron 1 C C C C C C C C C C C C C C C C C C C C o. A C o. A C C C C C C o. A C C C C C C C C C C C C C C C C C C C C C C C C C C C C C C C C C C C C C C C C T T C or T C C C C C C C272A C1552T 5'UTR Exon 1 C C C C C o. T C T T C C o. T C C C C C C C o.r T C o. T C o. T C C o. T C o. T C C C C C o. T C C C C C C C C C C C G G G G G o. A G A A G G o. A G G G G G G G o. A G o. A G o. A G G o. A G o. A G G G G G o. A G G G G G G G G G G G C C C C C o. T C T T C C o. T C C C C C C C o. T C o. T C o.T C C o. T C o. T C C C C C o. T C C C C C C C C C C C C C C C C o.T C T T C C o. T C C C C C C C o. T C o. T C o. T C C o. T C o. T C C C C C o. T C C C C C C C C C C C C2735T G2774A C2814T C2843T C C C C C C C C C C C C C C o. G C C C o. G C o. G C o. G C C o. G C o. G C C G C C C C C C C C C C C C C C2940G C C C C C o. T C C T C C o. T C o. T C o. T C C o. T C C C C C o. T C C o. T C C C C T C o. T C T C C C C C C C C C C2950T C C C C C C C C C C C C C C C C C C C C C C C C C C C C C C C C C A C A C C C2958A Intron 2 G G G G A G G G G G G G o. A G G G G G G G G G G G G G or A G G G G G G G G G G G G G G3010A A A A A A G A A A A A o. G A A A o. G A G A o. G A A o. G A A o. G A o. G G A o. G A o. G A A A A A A A o. G A A A o. G A o. G A A A3102G G G G G G G G G G G G o. A G G G o. A G G G G G G G G G G G G G G G G G G G G G G G G G3225A C C C C C C C C C C C C C C C C o. A C o. A C o. A C o. A C o. A C C C o. A C C C C C C C C C C C C C C A T T T C o. G C T T T T o. C T o. C T T o. C C o. G T T T T T T T C T T o. C T T T T T T T T o. C T T C o. G C o. G T C C A G A A o. G A G A A A A A o. G A A o. G A o. G A A A o. G G A o. G A o. G A o. G A A A o. G A A o. G A A A A A A A G A o. G A o. G A o. G A A4309G Exon 3 3'UTR T3370C C4275A C3370G Table 7.4 Haplotypen des RVC1 Gens in gPRA-erkrankten Hunden von 11 Hunderassen (Abkürzungen siehe Tabelle 2.1) Kapitel 8 Manuskript am 22.07.02 bei Genetics Selection Evolution (GSE) eingereicht (in press) Analysis of PDE6D and PDE6G genes for gPRA mutations in dogs Gabriele Dekomien, Joerg T. Epplen Abstract The δ and γ subunits of the cGMP-phosphodiesterase (PDE6D, PDE6G) genes were screened in order to identify mutations causing generalised progressive retinal atrophy (gPRA) in dogs. In the PDE6D gene single nucleotide polymorphisms (SNPs) were observed in exon 4, in introns 2 and 3 and in the 3’ untranslated region (UTR) of different dog breeds. In the coding region of the PDE6G gene exclusively healthy Labrador Retrievers showed an A→G transition in exon 4 without amino acid exchange. SNPs were also observed in introns 1 and 2 in different dog breeds. The different SNPs were used as intragenic markers to investigate the involvement of both genes in gPRA. The informative substitutions allow us to exclude mutations in the PDE6D and PDE6G genes as causing retinal degeneration in 15 of the 22 dog breeds with presumed autosomal recessively (ar) transmitted gPRA. 65 Kapitel 8 PDE6D & PDE6G Gene PDE6D & PDE6G Gene 8.1 Vorbemerkung Die cGMP-Phosphodiesterase (PDE) der Stäbchen ist ein G-Protein-aktivierendes Effektorenzym, das den cGMP-Spiegel in den Photorezeptorzellen der Vertebraten reguliert (Deterre et al. 1988). Diese cGMP PDE besteht gewöhnlicherweise aus den katalytischen αund β-, zwei identischen γ-Untereinheiten (Stryer 1996) und einer zusätzlichen δUntereinheit. Verschiedene mRNAs wurden inzwischen von Mensch, Maus, Rind und Hund identifiziert (Florio et al. 1996; Li et al. 1998; Lorenz et al. 1998). Die exakte Funktion der δUntereinheit ist noch nicht bekannt. Allerdings konnte nachgewiesen werden, dass die δUntereinheit der PDE die Bindung des Holoenzyms an die Membran durch die Interaktion mit dem isoprenylierten C-Terminus verhindert. Sie hat somit eine mögliche regulatorische Funktion in der Sehkaskade (Florio et al. 1996). Der Verlust der γ-Untereinheiten zieht eine reduzierte hydrolytische Aktivität nach sich und führt dadurch zu einer Verringerung der PDE-Aktivität (Tsang et al. 1996). Defekte in den Untereinheiten der PDE, die zu retinalen Erkrankungen führen wurden schon in Kapitel 3 und 4 beschrieben. In einem Mausmodel für retinale Erkrankungen führt eine Mutation im homozygoten Zustand im PDE6G Gen zum Verlust der inhibitorischen Funktion dieser Untereinheiten. Dies ist ursächlich in den Mäusen für eine vollständige postnatale Degeneration der Retina innerhalb von drei Monaten (Tsang et al. 1996). Weder beim Menschen noch beim Hund wurden bisher Mutationen im PDE6D noch im PDE6G Gen identifiziert. Allerdings wurden Assoziationen mit ar RP mit den chromosomalen Regionen dieser beiden Gene nachgewiesen (PDE6D: 2q35-36; RP26; PDE6G: 17q25 RP17; RetNet). Auf der Basis der vergleichenden Studie von Chromosomenbereichen von Mensch zum Hund (Yang et al. 1999) kann davon ausgegangen werden, dass das canine PDE6D Gen auf CFA 33 und das PDE6G Gen auf CFA 9 der homologen chromosomalen Region des Hundes lokalisiert sind. Diese Gene wurden kürzlich als Ursache für die rod-cone dysplasia 2 (rcd2) in Collies ausgeschlossen (Wang et al. 1999). Allerdings könnten Mutationen in diesen Genen eine Ursache für die gPRA in anderen Rassen sein. Deshalb wurden diese Gene in 22 verschiedenen Hunderassen auf eine gPRA-ursächliche Mutation hin untersucht. 66 Kapitel 8 PDE6D & PDE6G Gene 8.2 Resultate und Diskussion 8.2.1 Identifikation des Intron 1 des caninen PDE6D Gens Genomische Sequenzen der PDE6D (Wang et al 1999; EMBL-Accession-Nr. AF113996) und PDE6G (Wang et al. 1999; EMBL-Accessions-Nr. CF49360) Gene standen für die Mutationsanalyse zur Verfügung. Es wurde davon ausgegangen, dass das canine PDE6D Gen vier Exons wie das von Ershova et al. (1997) beschriebene menschliche PDE6D Gen umfasst. Mittels PCR konnte das canine Exon 1 nicht amplifiziert werden. Deshalb wurde von einem zusätzlichen Intron 1 ausgegangen. Nach Computerrecherche zeigte sich, dass Lorenz et al. (1998) auch beim PDE6D Gen des Menschen fünf Exons nachweisen konnten. Um die Sequenz des Intron 1 zu identifizieren, wurde die canine Genombank mit einer radioaktiv markierten Sonde des PCR-amplifizierten Exon 2 durchsucht. Verschiedene Klone mit unterschiedlichen Anteilen des Intron 1 und des Exon 2 des PDE6D Gens wurden isoliert. In keinem dieser Klone wurde Exon 1 nachgewiesen. Der Vergleich mit der erst kürzlich veröffentlichen genomischen DNA vom Chromosom 2 des Menschen (EMBL-AccessionsNr. AC073476) zeigt eine Introngrösse von 41877 bp. Diese Basenanzahl überschreitet die klonierbare Fragmentgröße eines λ-Phagen. Die exakte Größe konnte auch mit einer long range PCR, wie in Kapitel 7 schon beschrieben, nicht bestimmt werden. Teile des Introns 1 wurden nach Subklonierung der genomischen PDE6D Fragmente des λ-Phagen und nach PCR-Analyse mit dem T7 Primer des Phagen und Primer des Exon 2 identifiziert und sequenziert (EMBL-Accession-Nr. AJ427396; 3' Spleißregion Intron1/Exon2: atatttgatcagAAATTGGATGAA). 8.2.2 Mutationsanalyse des PDE6D Gens Mittels der PCR-SSCP-Analyse wurden die gesamte kodierende Region, die 3' und 5' Spleißsequenzen bis auf 20 bp (Primersequenz) des Exon 1 und die Bereiche der 5' Spleißstellen des Intron 1 des PDE6D Gens untersucht (Primer und PCR-Bedingungen siehe Tabelle 8.1). In der kodierenden Region wurden keine Polymorphismen identifiziert. Um doch noch intragenische SNPs als Marker für den Ausschluss des PDE6D Gens als Ursache für die gPRA zu erhalten, wurden Intron 3 und der 3'UTR komplett analysiert. Dabei wurden Sequenzunterschiede im Vergleich zum veröffentlichten PDE6D Gen (Wang et al. 1999; EMBL-Accession-Nr. AF113996) in allen sequenzierten DNAs identifiziert (Intron 3 in fünf und im 3'UTR in einer Position; siehe EMBL-Accessions-Nr. AJ427396). Ein einzelner SNP 67 Kapitel 8 PDE6D & PDE6G Gene wurde im Intron 2 (IVS2 +67A→T) in Labrador Retrievern entdeckt. Im Intron 3 wurden zwei SNPs (IVS3+133C→T; IVS3+133T→C und drei im 3'UTR des Exon 5 (3'UTR+133C→T, 3'UTR+177T→C, 3'UTR+357C→T) in verschiedenen Rassen nachgewiesen (Tabelle 8.2). 8.2.3 Mutationsanalyse des PDE6G Gens Der gesamte OLR des PDE6G Gens und große Bereiche der Introns 1, 2, 3 sowie die UTRs wurden mittels der PCR-SSCP-Analyse untersucht (Primer-Systeme und PCR-Bedingungen Tabelle 8.1). Dabei wurde in dem OLR des PDE6G Gens eine Sequenzvariation (G2285A) ohne Aminosäureaustausch in gesunden Labrador Retrievern gefunden (Tabelle 8.2). Informative SNPs wurden in Intron 1 (G744A) in 14 und in Intron 2 (C1662T; G2285A) in 6 Rassen identifiziert (Tabelle 8.2). 8.2.4 Schlussfolgerung Das PDE6D Gen umfasst 5 Exons mit einem OLR von 150 Aminosäuren. Das PDE6G Gen codiert ein Protein von 87 Aminosäuren und besteht aus 4 Exons. Keiner der in den verschiedenen Rassen nachgewiesenen SNPs liegt in funktionell wichtigen Bereichen dieser kleinen Gene. Die identifizierten Sequenzvariationen wurden als heterozygote Allele sowohl in erkrankten wie auch in gesunden Hunden der verschiedenen Rassen nachgewiesen. Somit sind sie als indirekte Marker für den Ausschluß dieser Kandidatengene für die ar vererbte gPRA einsetztbar (siehe Kapitel 2.6). Deshalb kann sowohl das PDE6D Gen in 15 Rassen (Australian Cattle Dogs, Berner Sennenhunde, Bologneser, Collies, Englische Cocker Spaniel, Entlebucher Sennenhunde, Irish Setter, Labrador Retriever, Neufundländer, Rottweiler, Saarloos/Wolfshunde, Schapendoes, Sloughis, Teckel und Zwergpudel) und das PDE6G Gen in 15 Rassen (Berner Sennenhunde, Chesapeake Bay Retriever, Collies, Englische Cocker Spaniel, Entlebucher Sennenhunde, Golden Retriever, Irish Setter, Labrador Retriever, Rottweiler, Saarloos/Wolfshunde, Scottish Terrier, Schapendoes, Teckel, Tibet Terrier, Zwergpudel) als Ursache für die gPRA ausgeschlossen werden. In 8 der 22 untersuchten Rassen wurde kein Austausch identifiziert, somit stehen keine intragenischen Marker für den Ausschluß dieser Kandidatengene zur Verfügung. Da die Promotorbereiche der Gene noch nicht identifiziert und somit auch nicht untersucht werden konnte, ob in diesen regulatorischen Bereichen gPRA ursächliche Mutationen in den erkrankten Hunden dieser Rassen vorkommen, können die beiden Gene hier noch nicht sicher ausgeschlossen werden. 68 Kapitel 8 PDE6D & PDE6G Gene Das gilt für das PDE6D Gen für die Rassen Chesapeake Bay Retriever, Berger des Pyrénées, Golden Retriever, Polnische Niederungshütehunde, Scottish Terrier, Tibet Mastiffs, Tibet Terrier und für das PDE6G Gen für die Rasssen Australian Cattle Dogs, Berger des Pyrénées, Bologneser, Neufundländer, Polnischer Niederungshütehund, Sloughis, Tibet Mastiffs. Australian Cattle Dogs, Chesapeake Bay Retriever und Sloughis waren als Kontrollen mit in die Mutationsanalyse eingeschlossen. In diesen Rassen ist die ursächliche Mutation (Dekomien et al. 2000) oder die chromosomale Lokalisation (Acland et al. 1998) schon bekannt. Zusammen mit den Daten dieser Studie wurden somit die Untereinheiten der cGMPPDE bis auf die Promotorbereiche vollständig untersucht und für die meisten der zur Verfügung stehenden Rassen als Ursache für die ar gPRA ausgeschlossen (siehe auch Kapitel 3 und 4). 69 Kapitel 8 PDE6D & PDE6G Gene Tabelle 8.1 Primer und Bedingungen für PCR-Amplifikation des caninen PDE6D und PDE6G Gens für die Mutationsanalyse PCR Bedingungen [T-°C / MgCl2-mM] Länge der PCRProdukte (bp) Restrikionsenzyme für die SSCP Analyse CCGTCTGCGAGGCTCCGC ACAGTTTGAAGCCCTTCAGG 55/1 109 - 551 768 TCGTGGCACTTAGCAGATAG CACCAGGGACAGACAAGTC 54/1 218 TruI Exon 2 Intron 2 690 901 AGAAATTGGATGAACCTCCGG ACAACAACACATGCTGTG 59/1.5 213 XmnI 1-I2 F E2R Exon 3 Intron 3 1354 CTGTAGCTATCTCTGTGACT 1540 CCTTCTAGGCATTGCCCTTT 55/1 190 DdeI E2F E3R Exon 3 Exon 4 1412 AGCCCGTGTTCCCAAGAAAA 2318 ACGTTAGCACACTGGCGG 60/1 907 MnlI +XmbId E3F 3R Exon 4 Intron 4 2210 GAATGGTTCTTCGAGTTTGG 2458 ACAGAAGTCAGTAACCT 52/2 249 XbaI 4F 4R Intron 4 Exon 5 52/2e 4110 GGTTCTAAGTGGGTGCATGT 4783 CATTATGTAATAATAATATCAGTC 674 MboII/ HaeIIId UTR-A1 UTR-B1 5’UTR Intron 1 1 440 57/0.5e 440 AvaI UTR-A2 UTR-B2 Intron 1 Intron 1 496 ACCACCTGGGCTGGGGA 1283 CTGGAACCAGGAGACCCAGG 57/1e 887 PvuII 60/1,5 149 - Position (bp)a Primersequenzen (5'→ →3') 1F 1a R 5‘UTR Exon 1 1 109 1a-2 F E1-2 R Intron 1 Exon 2 1b F 1R Primer PDE6Db PDE6Gc GAGCACACCCGTGACCCT CCGGCTGCTCTGGCCCCT GGTGGTGCCTGGGCATCT ACCCTGCTCAAGGGCAA UTR-MA2 Intron 1 UTR-MB2 Intron 1 669 801 I-2A I-2B Intron 1 Intron 2 1196 CTTGCCTGACCCAGGTGGA 1611 CCCAATTCCTGGGTAGCC 53/2 416 HinfI /MseI I-3A I-3B Intron 2 Intron 3 1782 CCTGTGTCCCCGCATGCA 1934 CGGGAGAGTTGGGGGATC 58/1 153 - I-4A I-4B Intron 3 3’ÙTR 2181 CTCTGGGCGTGGACAACA 2385 GGCACCCGGAGCAGGGGA 58/1 205 - E-2A I-3B Intron 1 Intron 3 1398 TTCTCTGCCAACCCTGGCC 1934 CGGGAGAGTTGGGGGATC 58/2.5e 537 AvaII I-3A E-4B Intron 2 3’UTR 1782 CCTGTGTCCCCGCATGCA 2332 TGGGTCAGGCTCTGGCG 57/1.5 551 BsiHKAI Legende nächste Seite. 70 Kapitel 8 PDE6D & PDE6G Gene Legende zur Tabelle 8.1 a Nukleotidposition zu der das 5' Ende des Primers (fettgedruckte Buchstaben) hybridisiert. b Die veröffentlichten caninen PDE6D Sequenzen (EMBL-Accession-Nr. AJ427395 und AJ427396) dienten als Vorlage für die Positionierung der PCR-Primer. c Für die Mutationsanalyse wurden PCR-Primer basierend auf der Sequenz des caninen PDE6G Gens (Wang et al. 1999; EMBL-Accessions-Nr. CF49360) hergestellt. d Mutationsuntersuchung mit der RFLP-Analyse. e Zusätzlich 5% Formamid. Tabelle 8.2 PDE6D- und PDE6D-Sequenzvariationen und gPRA-erkrankte Hunde mit heterozygoten Allelen Lokation Sequenzvariation PDE6D Intron 2 IVS2+67A→T - LR PDE6D Intron 3 IVS3+269A→G - IVS3-45T→A - Bo, BS, Co, ES, LR, NF, Sa, SD, Ro, ZP BS, ES, LR, Ro 3'UTR+133C→T 3'UTR+177T→C - 3'UTR+357C→T - PDE6D Exon 5 Aminosäurewechsel Rassena Gen SD AC, BS, Co, ECS, ES, LR, Ro, Sl, ZP AC, Co, ECS, ES, IRS, LR, Ro, SD, Sl, Te, ZP PDE6G Intron 1 744G→A - BS, Co, ECS, ES, GR, IRS, LR, Ro, Sa, ScT, SD, Te, TT, ZP PDE6G Intron 2 1662C→T 1694G→A - CBR, ECS, ES, LR, Sa, SD CBR, ECS, ES, LR, Sa, SD PDE6G Exon 4 2285 G→A (L78L) a (LR)* Abkürzungen siehe Tabelle 2.1 (EMBL-Accession-Nr. PDE6D: AJ427396 und PDE6G DNA: CF49360). * Heterozygote Sequenzvariation in gesunden Labrador Retrievern. 71 Kapitel 9 RPE65 Gen Kapitel 9 RPE65 Gen 9.1 Vorbemerkung Das im retinalem Pigment Epithel (RPE) exprimierte RPE65 gehört zu einer Gruppe von Proteinen, die in den Vitamin A Zyklus eingebunden sind. In diesem Zyklus finden eine Reihe von biochemischen Schritten statt, die das Chromatophor des RHO, das 11-cis Retinalaldehyd, für die Phototransduktionskaskade zur Verfügung stellen und nach Isomerisation auch wieder aufbereiten. Mutationen im RPE65 Gen führen zur ar RP20 mit sehr frühem Krankheitsbeginn und in einigen Fällen zur LCA (Gu et al. 1997; Morimura et al. 1998; Thompson et al. 2000). Mäuse bei denen das RPE65 Gen ausgeschaltet wurde, zeigen desorganisierte Scheibchenmembranen in den Aussensegmenten der Photorezeptorzellen. Zusätzlich konnte mit diesen Mäusen die retinale Biochemie und Elektrophysiologie untersucht werden (Redmond et al. 1998). Bei schwedischen Briads mit congenitaler stationärer Nachtblindheit (csnb) wurde eine 4 bp Deletion nach dem Kodon 153 des RPE65 Gens identifiziert, die zu einem vorzeitigen Stoppkodon führt und somit eine Verkürzung des Proteins zur Folge hat (Aguirre et al. 1998; Veske et al. 1999). Bei der histologischen Untersuchung der Retina dieser Hunde wurden nur geringe Auffälligkeiten der Photorezeptorzellen, aber eine erhöhte Anzahl von lipidhaltigen Einschlüssen nachgewiesen (Wrigstad et al. 1992). Das Protein ist in Mammaliern auf Proteinebene hochkonserviert (Aguirre et al. 1998). Das menschliche RPE65 Gen ist in dem Bereich des Chromosom 1p31 lokalisiert. Auf der Basis der vergleichenden Studie von Chromosomenbereichen in Mensch und Hund (Yang et al. 1999) kann davon ausgegangen werden, dass das Gen auf CFA dem homologen Chromosom beim Hund lokalisiert ist. Mutationen im RPE65 Gen sind eine der Hauptursachen für die ar RP im Menschen (Morimura et al. 1998), deshalb wurde dieses Gen intensiv auf gPRA verursachenden Sequenzunterschiede in den 29 Hunderassen untersucht, da in diesen Rassen nach Züchterbeobachtungen die gPRA ar vererbt wird. 72 Kapitel 9 RPE65 Gen 9.2 Resultate und Diskussion 9.2.1 Genomische Organisation des RPE65 Gens Das canine RPE65 Gen umfasst 14 Exons. Sie sind zwischen 65 und 149 bp groß. Intronische Sequenzen des Gens wurden noch nicht veröffentlicht. Die Entscheidung über die Strategie der Introncharakterisierung beim RPE65 Gen wurde anhand der möglichen Introngrößen beim Hund getroffen. Deshalb wurden vergleichende Studien von der caninen mRNA (Aguirre et al. 1998; EMBL-Accessions-Nr. AF084537) mit der genomischen Sequenz des humanen Chromosom 1 (EMBL-Accessions-Nr. AL139413) durchgeführt. Diese Vergleiche dienten sowohl zur Identifizierung der Exon-/Introngrenzen als auch zur Bestimmung der Introngrößen des RPE65 Gens des Menschen. Die ermittelten Introngrößen im menschlichen RPE65 Gen liegen meist unter 2 kb. Deshalb konnte davon ausgegangen werden, dass die Introngrößen des RPE65 Gen des Hundes ähnlich, oder gemäß der Erfahrung mit schon identifizierter Introns in den RVC1 oder PDE6B Genen, eher kleiner sind. Somit wurden intronüberspannende PCRs mit genomischer DNA durchgeführt und dadurch fast alle Intronsequenzen bestimmt (PCR-Systeme und -Bedingungen Tabelle 9.1). Vergleiche der ermittelten RPE65-Sequenzen von Hund und Mensch zeigten, dass beim Hund die identifizierten Introns des RPE65 Gens bis auf Introns 1 und 6 überraschenderweise größer als beim Menschen sind (siehe Tabelle 9.2). Introns 2, 3 und 10 konnten wahrscheinlich aufgrund der Größe oder sequenzbedingter Konformationen nicht amplifiziert werden. Allerdings wurden insgesamt 21 Sequenzenvariationen mit Hilfe der PCR-SSCP-Methode, in fast allen Rassen nachgewiesen (Tabelle 9.3), sodass es nicht mehr notwendig war, diese Bereiche mit in die Untersuchungen einzuschließen. Die Vergleich der caninen intronischen Sequenzen mittels des Blast search Programms mit den in der Datenbank schon veröffentlichen Sequenzen, wies auf kurze Wiederholungselemente in den Introns 6 (Nukleotidposition 4-66; EMBL-Accessions-Nr. AJ506756) und 12 (Nukleotidposition 580-704; EMBL-AccessionsNr. AJ251207) hin, die canine-spezifischen tRNA SINE entsprechen (Bentolila et al. 1999). Ausserdem sind im Intron 11 zwei fragmentierte LINE Sequenzen (Nukleotidpositione 139224 und 312-359 EMBL-Accessions-Nr. AJ251207) entsprechend der veröffentlichen Sequenzen von Choi et al. (1999) lokalisiert. Vergleiche der 5' und 3' Spleißstellenumgebung des RPE65 Gens von Mensch und Hund zeigen die Einhaltung der GT-AG Regel und große Sequenzübereinstimmung in diesen Bereichen (Tabelle 9.2). 73 Kapitel 9 RPE65 Gen 9.2.2 Mutationsanalyse des RPE65 Gens 13 der 14 Exons des RPE65 Gens wurden mit den angrenzenden Intronbereichen mittels der PCR-SSCP-Analyse in 28 Rassen untersucht (Primer und PCR-Bedingungen Tabelle 9.1). Exon 3 sowie 20 bp (Primersequenz) der kodierenden Sequenz von Exons 4 und 10 waren in die Mutationsuntersuchung nicht eingeschlossen. Die DNA aller erkrankten Hunde und einige gesunde Hunde wurden auf gPRA ursächliche Mutationen hin überprüft. Die von Veske et al. (1999) nachgewiesenen SNPs im Exon 5 (T459C) wurden in 20 und im Exon 6 (G541A) in 8 verschiedenen Hunderassen identifiziert (Tabelle 9.3). Allerdings waren diese Substitutionen sowohl heterozygot wie auch homozygot in gesunden und erkrankten Hunden erkennbar, sodass diese Variationen nicht in allen Rassen als intragenische Marker genutzt werden konnten. Die von Veske et al. (1999) veröffentlichte Sequenzvariationen im Intron 11 wurde in keiner der untersuchten Rassen nachgewiesen. Dafür wurden in verschiedenen Rasssen spezifische Allelkonstellationen (C-A-A oder A-T-G) identifiziert, die immer in dieser Zusammensetzung bei der Sequenzierung zu beobachten waren. Zusätzlich wurde eine G→A Transversion in der Position 203 und eine T-Deletion in der Position 61 (Tabelle 9.3) in diesem Intron entdeckt. Im OLR wurden weitere SNPs in den Exons 9 (C900T) und 10 (A1026T) gefunden, die aber keinen Aminosäurewechsel zur Folge hatten. Deletionen wurden im Intron 1, 8 und 3'UTR, eine Insertion im Intron 6 und weitere SNPs im Intron 6, 7, 8 und 13 des RPE65 Gens nachgewiesen (siehe Tabelle 9.3). 9.2.3 Schlussfolgerung Nach PCR-SSCP-Analyse des gesamten Gens (bis auf 191 bp des OLRs) wurde keine ursächliche Mutation für die ar vererbte gPRA in den 29 Rassen identifiziert. Die 4-bp Deletion, die in schwedischen Briads zur CSNB führt (Aguirre et al. 1998; Veske et al. 1999), wurde in keiner dieser Rassen identifiziert. Eine Reihe von Sequenzvariationen in 4 Exons und 7 Introns wurden heterozygot in erkrankten Hunden von 27 Rassen nachgewiesen (Tabelle 9.3). So konnten diese Sequenzvariationen für das indirekte GenausschlussVerfahren verwendet werden. Da sich Heterozygotie bei einer angenommenen ar Vererbung der gPRA bei einer Kopplung mit der krankheitsverursachenden Mutation ausschließt, kommt das RPE65 Gen in den folgenden Rassen als Ursache für die gPRA nicht in Frage: Afghanische Windhunde, Australian Cattle Dogs, Amerikanische Cocker Spaniel, Berger des Pyrénées, Bologneser, Collies, Chesapeake Bay Retriever, Curly Coated Retriever, Englische Cocker Spaniel, Entlebucher Sennenhunde, Golden Retriever, Irish Setter, Kuvasz/Viszla, 74 Kapitel 9 RPE65 Gen Labrador Retriever, Neufundländer, Polnische Niederungshütehunde, Riesenschnauzer, Saarloos/Wolfshunde, Salukis, Scottish Terrier, Schapendoes, Sloughis, Springer Spaniel, Teckel, Tibet Mastiffs, Tibet Terrier, Yorkshire Terrier und Zwergpudel. Bei dem erkrankten Rottweiler wurden homozygote SNPs im Intron 7 und 8 gefunden. Zwar sind nur wenige bp des OLR nicht untersucht, aber die regulatorischen Elemente des Promotors konnten in die Untersuchung nicht eingeschlossen werden, da diese Sequenzen noch nicht identifiziert wurden. Deshalb kann das RPE65 Gen für Rottweiler als Ursache für die gPRA nicht ausgeschlossen werden. 75 Kapitel 9 RPE65 Gen Tabelle 9.1 Primer und Bedingungen für die PCR-Amplifikationen des RPE65 Gens PCR-System PrimerBezeichnung Bindestelle PrimersequenzA (5'→ →3') Länge der PCRProdukte (bp) RPE65UTR 1-F RPE65 I-1-R Exon1 Intron1 Exon 1 CGACCGTCTGTCCTGCCC GGGTATTTGCTTCAATCCATG 186 56/1,5B - Exon2 CTGGGAGACAATGTCCATC CGACGACAGCTCTTCCAC 1124 51/1,5C - RPE65 E4 F RPE65 E5 R Exon4 Exon5 AGGTTCATCCGCACCGATG ACCTGCTTAATTGTCTCCAG 357 54/1,5C AluI/HincII RPE65 I 4 F RPE65 I 5 R Intron4 Intron5 CACAGCTTGAAGGTTACTGGAC TCTATTTGGCCCTCATGAGC 286 54/1,5 HincII RPE65 E5 F RPE65 E6 R Exon5 Exon6 AGGTTTTTTTCTTACTTCCGA TGCTTGGAGTGGAGGGATC ∼2800 60/1,5 - RPE65 E-6-F RPE65 I 6 R Exon6 Intron6 AGGTTGATCTCTGCAACTAC CTGTGGAATACGACTTGGC 377 56/1,5 DdeI RPE65 E-6-F RPE65 E-7-R Exon6 Exon7 AGGTTGATCTCTGCAACTAC CTATGGACGTACGATGGC ∼2000 60/1,0 - RPE65 7EF RPE65 8ER Exon7 Exon8 AGACAAGGAAGATCCAATAAGC CCCATGGTTTCATTGGACTC 443 54/1,5C TruI RPE65 8EF RPE65 9ER Exon8 Exon9 TTTTGTGGAGACGCCAGTC CCTTTCCAGCAGCAGAGATC 369 60/2,5 BsiHKAI RPE65 E-9-F RPE65 E-10-R Exon9 Exon10 AGGTTTGGCTTCACATCGCTG TTGTCGATATTCAGAGGAAGC ∼2000 60/1,0 - RPE65 I-9 F RPE65 E-10-R Intron9 Exon10 ACACCCCAGAGCAAGCAGG TTGTCGATATTCAGAGGAAGC 229 59/1,5 EcoRI RPE65 E11-F RPE65 E12-R Exon11 Exon12 GCCGACACAGGCAAGAAC ACCCTGTCCGGAACGAAG 542 55/1,5 DdeI RPE65 E-12-F RPE65 I-13-R Exon 12 Intron 13 CCTCAAATCAACTATCAGAAGTC AACACACTAACATAGAGAACTC 580 58/1,0 - RPE65 I-12 F RPE65 I-13-R Intron 12 Intron 13 AAGAGAAAAGTAGTTTGAGTCAC AACACACTAACATAGAGAACTC 261 57/1,5C HphI RPE65 E13-F RPE65 E/I14R Exon 13 Exon 14 CTCTGCAAGCTGAACGTC CCATGTAATACACAGCAGGCTAA ∼1800 55/1,0 - RPE65 E 14-F RPE65 E 14-R Exon 14 Exon 14 AGGTGTAGTTCTGAGTGTG CCGTATACAGCAGGCTAAA 249 55/1,0 RsaI RPE65 UTR14F RPE65 UTR14R Exon 14 Exon 14 CAAAGTCAAGAAAAAGTGAGGT GCTTTGATGTTATGTAAGCTTTT 705 53/2,0 MseI RPE65 UTR14F Exon 14 RPE65 UTR214R Exon 14 CAAAGTCAAGAAAAAGTGAGGT CACTTTCATAATAGGAACAAGAA 277 55/1,5 MnlI RPE65UTR21F RPE65 E2-2R PCR-Bedingungen [T-°C / MgCl2 -mM] Restriktionsenzyme für die SSCPAnalyse A RPE65-Sequenzen vom Hund: EMBL-Accessions-Nr. AJ506753-AJ506759 und AJ251207. B 5% Formamid. C -1 touchdown PCR (siehe Kapitel 2.1.3). 76 Kapitel 9 RPE65 Gen Tabelle 9.2 Vergleichende Exon-/Intronorganisation des RPE65 Gens von Hund und Mensch Exon # Hund Mensch Hund Mensch Hund Mensch Hund Mensch Hund Mensch Hund Mensch Hund Mensch Hund Mensch Hund Mensch Hund Mensch Hund Mensch Hund Mensch Hund Mensch Hund Mensch Hund Mensch Hund Mensch Hund Mensch Hund Mensch Hund Mensch Hund Mensch Hund Mensch Hund Mensch Hund Mensch Hund Mensch Hund Mensch Hund Mensch A Exon-/IntrongrenzenA Länge (bp) 1 65 69 2 83 83 3 151 151 4 108 108 5 142 142 6 148 148 7 82 82 8 133 133 9 140 140 10 130 130 11 115 115 12 95 95 13 112 112 152/734 152/1023 14 TGTCCATCCAgtgagtatgc TATCCAGTAAgtatctctgg accttctcagAGTGGAGCAT accatttcagGGTTGAGCAT CACGTGACAGgt........ CATGTAACAGgttggtctcg ........agGCAGGATCCC ttcatcacagGCAGGATCCC ATCACAGAAGgt........ ACCACAGAAGgtaaagcagc ........agGTTCATCCGC tgtccttcagGTTCATCCGC TATTTTCCAGgttattgaaa TATTTTCCAGgttactgaac gcttctgtagGTTTTTTTCT gtttctacagGTTTTTTTCT AATTAAGCAGgtaggacgaa AATTAAGCAGgtgggacaca cctcccgcagGTTGATCTCT acttccgtagGTTGATCTTT CTCCAAGCAGgtcagtttac CTGCAAGCAGgtgagtttac tttatttcagACAAGGAAGA tttatttcagACAAGGAAGA ACGTCCATAGgtaacttgaa ACGTTCATAGgtaacttgaa ctctaaacagTTTTGGTTTG ttctgaacagTTTTGGTCTG AACCATGGGGgtaagtcggg ATGGGGGTAAgtcttagata atttttcaagGTTTGGCTTC atttttcaagGTTTGGCTTC GCTGGAAAGGgtaagagagg GCTGGAAAGGgtaagaaagg atttttgcagATTTGAATTC atttttgcagATTTGAGTTT TATCGACAAGgt....... TATTGACAAGgtaacctgct ........agGCCGACACAG tctctcctagGCTGACACAG CCTCGTCAAGgtgaggtgat CCTCGTCAAGgtgagatgat tattttgtagCCTTTGAGTT tatttcgtagCATTTGAGTT TCCGGACAGGgtaccgctcc TCCAGATAGGgtaattaatc tcacaagcagCTCTGCAAGC tcacaaacagCTCTGTAAGC GAAGATGATGgtaatgaaag GAAGATGATGgtaatgaaag tgattaacagGTGTAGTTCT tgataaacagGTGTAGTTCT Intron # Länge (bp) 1 925 1184 2 ? 1763 3 ? 1826 4 104 103 5 ∼2800 3530 6 ∼2000 1210 7 227 237 8 123 105 9 ∼1700 625 10 ? 6601 11 330 94 12 320 105 13 ∼1500 1137 RPE65-Sequenzen vom Hund: EMBL-Accessions-Nr. AJ506753-AJ506759 und AJ251207; Sequenzen vom Chromosom 1 des Menschen: EMBL-Accessions-Nr. AL139413. 77 Kapitel 9 RPE65 Gen Tabelle 9.3 Sequenzvariationen im RPE65 Gen in verschiedenen Hunderassen Gen / Lokalisation SequenzvariationA Aminosäure- RasseB Austausch - AC, AW, CCR, ECS, GR, LR, NF, Sal, TM, ZP T459C Y144Y AC, AW, BDP, ECS, CBR, Co, ES, GR, LR, RS, Sa, Sal, ScT, Sl, Te, TM, TT, YT, ZP Exon 6 G541A V172I ACS, IRS, Te, ECS, LR, TT, Sa, GR RPE65 Intron 6 IVS6+23T→C IVS6+31C→G IVS6+71-72Ins.GG IVS6+116G→C IVS6+164G→A RPE65 Intron 7 IVS7-69T→A IVS7-18C→A RPE65 Intron 8 IVS8+24T→C RPE65 Intron 1 IVS1 +76DelG RPE65 Exon 5 RPE65 - - - IVS8+109TIns/Del RPE65 Exon 9 C900T I291I RPE65 Exon 10 A1026T G333G RPE65 Intron 11C IVS11+110C→A IVS11+138A→T IVS11+143A→G IVS11+203G→A - AW, SP, RS RS, Sl ZP ZP SP, Sl BDP, ECS, ES, Ro, RS, Sa, Sl, Te, TT, ZP BDP, ECS, ES, Ro, RS, Sa, Sl, Te, TT, ZP AC ,BDP, Bo, ES, GR, IRS, SD, Te, PON, Sa, ZP AW, Bo, BDP, ECS, ES, IRS, GR, PON, Ro, RS, Sa, SD, Sp, Te, ZP BDP, ECS, Sa, Te SD, RS, TT, ZP AC, Co, CBR, ECS, ES, LR, Sa, ScT, Sl, Te, TM, TT, YT, ZP BDP, ECS, ES, IRS, NF, ScT, SD, Sa, Te, TM, TT, ZP AW, Te, TM, TT, ZP IVS11-61Del.T RPE65 Intron 13 IVS13+36 C→T - AW, BDP, ES, IRS, KV, ScT, Sp, Te, TM, NF, RS, Sa, TT, TM, ZP RPE65 Exon 14 c1856 Del.A - AC, ECS, CBR, ES, GR, LR, Sa, Sl, Te, TM, TT, YT, ZP A Position der Sequenzvariationen cDNA: EMBL-Accessions-Nr. AF084537; Introns: AJ506753-AJ506759 und AJ251207. B Rassen in fett gedruckten Buchstaben mit heterozygoten Sequenzvariationsmustern in gPRA-erkrankten Hunden (Abkürzungen der Rassenamen, siehe Tabelle 2.1). C Die Haplotypen im Intron 11 sind entweder C-A-A oder A-T-G in den entsprechenden Hunderassen. 78 Kapitel 10 CRX Gen Kapitel 10 CRX Gen 10.1 Vorbemerkung Das Cone-rod homeobox (CRX) Protein wird in den Photorezeptorzellen und Pinealocyten exprimiert. Es ist ein Transkriptionsfaktor, der in Interaktion mit anderen Faktoren z.B. dem neutral retinal leucin zipper (NRL) die Genexpression von mind. 10 Photorezeptorspezifischen Genen mit konsensualen CRX Bindeelementen (WSP- und Otx-SchwanzDomänen) reguliert (Furukawa et al. 1997; Mitton et al. 2000). In einem Tiermodell, Mäusen mit homozygoten Verlust des CRX Gens, erfolgte keine Entwicklung der Photorezeptoraußensegmente, und es werden keine Aktivitäten von Zapfen und Stäbchen nachgewiesen (Furukawa et al. 1999). Eine Reihe von dominanten, in seltenen Fällen auch rezessiven Mutationen im CRX Gen führen zur CORD2; LCA und RP des Menschen. Der Krankheitsbeginn und -verlauf variieren (Furukawa et al. 1997; Freund et al. 1997; 1998; Swain et al. 1997; Swaroop et al. 1999; Rivolta et al. 2001). Beim Hund wurde das Gen bereits für die canine cd in Alaskan Malamutes, rcd2 in Collies und zwei verschiedenen Formen der gPRA mit frühen Krankheitsbeginn in Amerikanischen Staffordshire Terrier und Tibet Terrier ausgeschlossen (Akhmedov et al. 2002). Das menschliche CRX Gen ist auf dem Chromosom 19q13.3 lokalisiert. Beim Hund wurde das Gen im Zentromer-Bereich CFA 1 nachgewiesen (Akhmedov et al. 2002). 10.2 Resultate und Diskussion 10.2.1 Bestimmung der Intronsequenzen des CRX Gens Die cDNA des CRX Gens (EMBL-Accessions-Nr. AF454668) und die intronischen Primersequenzen wurden von Akhmedov et al. (2002) veröffentlicht. Die intronischen Sequenzen für die Identifizierung der Restriktionsschnittstellen für PCR-SSCP-Analyse des Gens standen jedoch nicht zur Verfügung. Deshalb wurde mit Hilfe der schon veröffentlichen Primersequenzen zuerst Fragmente von Exon1/Intron1, Exon 2 mit den umgebenden Introns und wenige bp dem 3' Bereichs des Exon 3 amplifiziert und anschließend die Nukleotidfolge durch Sequenzierung bestimmt (Tabelle 10.1; EMBL-Accessions-Nr. AJ507727-507729). 79 Kapitel 10 CRX Gen Nach Recherchen in der Genbank mit den identifizierten Sequenzen mit dem Blast search Programm zeigte sich, dass das CRX Gen des Menschen wahrscheinlich mehr als 3 Exons umfasst. Weitere Nachforschungen mit dem Programm NCBI Ace View (http://www.ncbi.nih.gov/IEB/Research/Acembly/av.cgi?db=30&c=mRNA&a=fiche&l=CRX.a) wies auf 5 Exons hin, wobei Intron 1 im 5' UTR und das mögliche Intron 4 im 3'UTR lokalisiert ist. Diese Annahmen stützen sich auf die Daten der humanen mRNA (EMBLAccession-Nr. BC016664). Wie in Tabelle 10.2 dargestellt, folgen die Introns 1-3 der GTAG-Regel, wobei im Intron 4 diese Sequenzen von den Konsensussequenzen abweichen (GCGT). Exon-/Introngrenzen sind in Mammaliern, wie auch schon in den Kapiteln 3-8 aufgeführt, streng konserviert. Deshalb wurden die entsprechenden Sequenzbereiche des Hundes im Vergleich zu den menschlichen Exon-/Introngrenzen in Tabelle 10.2 aufgelistet. Hierbei sind große Übereinstimmungen in den Sequenzen des OLR erkennbar. Die Unterschiede sind in den UTRs erwartungsgemäß größer, da sie weniger dem Selektionsdruck unterworfen sind. Die identifizierten Introns des CRX Gen des Hundes sind um die Hälfte kleiner als die Introns im entsprechenden menschlichen Gen. Deshalb kann davon ausgegangen werden, dass die Größe von Intron 1 mit >12 kb beim Menschen beim Hund unterschritten wird. Deshalb wurde die Amplifikation des putativen Intron 1 des caninen CRX Gens aus genomischer DNA mit Primer UTR 1F (TGAGGACTCTGACTCCAACCAGGTCCTG) und Primer CRX(19)-I1 R (Tabelle 10.1) versucht. Dabei wurde ein DNA-Fragment von ca. 900 bp amplifiziert. Dieses zeigte nach Sequenzierung ein canin-spezifisches tRNA SINE (Bentolila et al. 1999), wobei in der Nukleotidfolge keine Sequenzen des putativen Exon 1 oder 2 des CRX Gens nachzuweisen waren. Es kann deshalb angenommen werden, dass es auch in der CRX-Sequenz des Hundes im 5'UTR ein Intron gibt, das von genomischer DNA mit den zur Verfügung stehenden Mitteln nicht zu amplifizieren ist. 10.2.2 Mutationsanalyse des CRX Gens Der gesamte OLR und die wichtigen Spleißsequenzen der bekannten Exon-/Introngrenzen wurden in die Mutationsanalyse von 30 Rassen einbezogen. Dabei wurden eine große Anzahl von Sequenzvarianten sowohl im OLR wie auch in den Introns identifiziert. Im OLR wurden insgesamt vier SNPs nachgewiesen. Im putativen Exon 3 wurde ein Austausch (c294G→C ) und drei Nukleotid-Austausche im Exon 4 (c720G→A; c832A→C; c857T→C) identifiziert. Die Positionen der SNPs in der Sequenz entsprechen der Nummerierung in der veröffentlichten mRNA des CRX Gens (Akhmedov et al. 2002; EMBL Accessions-Nr. AF454668). Keiner dieser SNPs führten zu einem Aminosäurewechsel (siehe Tabelle 10.3). 80 Kapitel 10 CRX Gen Im Intron 2 wurden 3 Sequenzvariationen identifiziert: ein seltener Austausch im IVS2+163T→C, der nur in vier Rassen und zwei sehr häufig auftretende SNPs (IVS2+200C→T; IVS2-220C→T), die in 14 bzw. 18 Rassen vorkamen. Ein weitere Sequenzvariante zeigte sich im Intron 3 in 15 Rassen (Tabelle 10.3). 10.2.3 Schlussfolgerung Das CRX Gen umfasst möglicherweise 4 bzw. 5 Exons. Das Protein besteht aus 299 Aminosäuren. Der Nukleotidaustausch im putativen Exon 3 liegt zwar in der Homeodomaine (Aminosäureposition 39-98) führt aber zu keinem Aminosäurewechsel. Keiner der weiteren exonischen SNPs befindet sich in den für die transkriptionale Aktivation wichtigen WSP und Otx-Schwanz-Domänen (Chen et al. 2002) des CRX Gens. Alle Sequenzvariationen im OLR oder in den Introns, wurden sowohl bei gesunden wie auch erkrankten Hunden identifiziert. Keine der Sequenzvariationen wurde nur in einer Rasse identifiziert, wobei dies bei einer Kombination von Gründereffekt und Zuchtverhalten in den Rassen zu erwarten wäre. Somit kann für den ar Erbgang der gPRA dieses Gen aufgrund der heterozygoten Variationen in den folgenden 16 Rassen ausgeschlossen werden: Amerikanische Cocker Spaniel, Collies, Englische Cocker Spaniel, Entlebucher Sennenhunde, Golden Retriever, Irish Setter, Kuvasz/Viszla, Neufundländer, Riesenschnauzer, Saarloos/Wolfshund, Saluki, Scottish Terrier, Schapendoes, Springer Spaniel, Teckel, Zwergpudel (Tabelle 10.3). In fünf Rassen (Afghanische Windhunde, Berger des Pyrénées, Bologneser, Curly Coated Retriever Polnische Niederungshütehunde) wurden nur SNPs im homozygoten Zustand beobachtet. Deshalb kann das Gen nicht sicher als Ursache für die gPRA in diesen Rassen ausgeschlossen werden. Es stehen noch Untersuchungen aus, die den Promotorbereich betreffen, sowie die möglichen Exon-/Introngrenzen von Intron 1 und 4. Es wäre somit möglich, dass Mutationen in regulatorischen Elementen und in den Spleißkonsensussequenzen, die die Funktion des Gens beeinträchtigen könnten, noch nicht identifiziert wurden. Weitere erkrankte Hunde dieser Rasse mit SNPs im homozygoten Zustand müssten in die Untersuchung noch eingebunden werden, damit eine ursächliche Mutation oder indirekte Marker für den Ausschluss identifiziert werden könnten. 81 Kapitel 10 CRX Gen Tabelle 10.1 Primer und Bedingungen für die PCR-Amplifikationen des CRX Gens PCRSystem Bindestelle PrimersequenzA (5'→ →3') E-1 F (19)-I1 R (16)-I1 F E-2R 2 E2 F (14)-I2 R 3 I-2F 3'UTR-R Exon 1 (2) Intron 1 (2) Intron 1 (2) Exon 2 (3) Exon 2 (3) Intron 2 (3) Intron 2 (3) 3'UTR AGATCATGATGGCGTATATG GGATGGCTCAACGGTTGAGCATC GCTCAACCGTTGAGCCATCCAG ACCTGAACCCTGGACTCAG CCATCTTAGGTGCCCCAAG TCCGTCTAGGTCTCATTGAAGG GGAACCATGGAGGATACTTCGGG TAAAATGGCAGCCAGCATCC A Länge der PCRProdukte (bp) PCR-Bedingungen [T-°C / MgCl2 mM] Restriktionsenzyme für die SSCPAnalyse 526 54/1,5 RsaI/MnlIB 527 56/1,5 BsuRI 481 53/2,5 BsuRIB 950 58/1,5D Eco130I Primersequenzen aus Akhmedov et al. (2002); B RFLP; D zusätzlich mit 5% Acetamid Tabelle 10.2 Vergleichende Exon-/Intronorganisation des CRX Gens von Hund und Mensch Exon-/IntrongrenzenA Exon # Hund Mensch Hund Mensch Hund Mensch Hund Mensch Hund Mensch Hund Mensch Hund Mensch Hund Mensch A Länge (bp) (1a) 1 66 (1b) 2 135 135 (2) 3 152 152 (3a) 4 (3b) 5 648/UTR453 648/UTR453 UTR 1844 785 IntronB # GTTTTTGGAA.......... CTTTCTGAAGgtgagcgtct ..........GCCCGCAGAC tctcttgcagGCCCCCTGAC TCCTATCCAAgtgagtacgg CCCTACCCAAgtgagtacag cccatcttagGTGCCCCAAG cccaccccagGCGCCCCCAG CAGGGTTCAGgtgcggtgtt CAGGGTTCAGgtggggtggt ttccccccagGTTTGGTTTA tatcccccagGTTTGGTTCA GTTTTAAGAA.......... TTTTTGTTTTgcagacacag ..........GCCCCACCAT cagagattgtGCCACTGTGC Länge (bp) (1a) 1 ? 12398 (1b) 2 789 1695 (2) 3 1059 2925 (3) 4 ? 2118 Canine CRX-Sequenzen nach EMBL-Accessions-Nr. AJ507727-AJ507729; CRX- Sequenzen des Menschen vom Chromosom 19, EMBL-Accessions-Nr. AC008745 (Exonsequenzen in Großbuchstaben); B Zahlen in der Klammer sind entsprechend der Anzahl der Exons nach Akhmedov et al. (2002). 82 Kapitel 10 CRX Gen Tabelle 10.3 Sequenzvariationen im CRX Gen in verschiedenen Hunderassen Gen / Lokalisation SequenzvariationA CRX IVS2+163T→C IVS2+200C→T Intron 2 (1b) Aminosäure RasseB -Austausch - IVS2-220C→T CRX Exon 3 (2) c294G→C CRX Intron 3 (2) IVS3+212G→A Exon 4 (3a) c720G→A c832A→C c857T→C CRX A P64P S206S G243G T254T CCR, Co, ES, Sa AC, BDP, Bo, Co, ECS, GR, IRS, KV, PON, SD, SP, Te, TT, ZP ACS, AW, BDP, ECS CCR, ES, IRS, GR, KV, LR, NF, Sa, Sal, ScT, SP, RS, TM, ZP ACS, ES, Sa, Te, TT, ZP ACS, ECS, IRS, GR, ES, KV, Sa, Sal, ScT, Sp, Te, TM, TT, RS, ZP IRS, SD, Te IRS, ES, SD, Te SD, Te Position der Sequenzvariationen in den Exons entsprechen der cDNA: EMBL-AccessionsNr. AF454668; der Introns: EMBL-Accessions-Nr. AJ507727-AJ507729. B Rassen in fett gedruckten Buchstaben mit heterozygoten Sequenzvariationsmustern in gPRA-erkrankten Hunden (Abkürzungen der Rassenamen siehe Tabelle 2.1). 83 Kapitel 11 GNGT1 Gen Kapitel 11 GNGT1 Gen 11.1 Vorbemerkung Transducin gehört zu einer Familie der heterotrimeren GTP-bindenen Proteine. Es bindet an das aktivierte RHO, wodurch sich die α-Untereinheit vom βγ-Komplex ablöst und die cGMPPDE aktiviert. Die Träger der GTPase-Funktion sind die α-Untereinheiten der G-Proteine. Die katalytischen Bereiche sind in der gesamten Fauna hoch konserviert. Deshalb wird davon ausgegangen, dass alle Isoformen (>20) durch Duplikationsereignisse aus einer Urform entstanden sind (Wilkie et al. 1992). Ansonsten zeigen die α-Untereinheiten eine große Vielfalt, wodurch die Spezifität des G-Proteins bestimmt wird. Die Unterschiede der verschiedenen β- und γ-Untereinheiten sind sehr viel geringer. Allerdings ist funktionsbedingt eine deutliche Abgrenzung der Struktur in Zapfen und Stäbchen zueinander erkennbar. Denn in beiden Photorezeptorzelltypen findet zwar die Phototransduktion statt, aber die Lichtreaktionen sind verschieden. Somit kann davon ausgegangen werden, dass das GProtein, das den Phototransduktionsprozess vermittelt, zusammen mit der α-Untereinheit zellspezifische Formen der β- und γ-Untereinheiten zeigt (Peng et al. 1992). Die γUntereinheiten ist die kleinste Untereinheit des Heterotrimers. Sie hat eine Prenylgruppe, über die der βγ-Komplex in der Membran verankert wird (Casey 1994), sowie eine Domäne für die βγ-Bindung (Neer 1995). Die cDNA des GNGT1 Gens (Kylma et al. 1997; EMBL-AccessionNr. Z75156) wurde schon mittels exonischer Primer durch eine PCR-SSCP-Analyse untersucht (Dekomien et al. 1998). Dabei führte eine A298G-Transition im 3'UTR in Teckeln und Zwergpudeln zum Ausschluss des Gens als Ursache für die gPRA in diesen beiden Rassen. Da dem Forschungsprojekt inzwischen 26 weitere Rassen für die Kandidatengenanalyse zur Verfügung standen, wurde für die umfassendere Untersuchung des Gens zuerst die intronischen Sequenzen des Gens bestimmt. 84 Kapitel 11 GNGT1 Gen 11.2 Resultate und Diskussion 11.2.1 Identifizierung der Introns des GNGT1 Gens Scherer et al. (1996) ermittelte eine partielle genomische DNA-Sequenz und Struktur des humanen GNGT1 Gens und lokalisierte es auf dem Chromosom 7q21.3. Das GNGT1 Gen des Hundes müßte aufgrund der Chromosomenvergleichsstudie zwischen Mensch und Caniiden von Yang et al. (1999) auf CFA 14 liegen. Das menschliche Gen umfasst drei Exons wobei Exon 1, und Teilbereiche des Exon 3 im UTR liegen. Die Exon-/Introngrenzen wurden über den Vergleich der cDNA des Hundes mit dem genomischen Klon des Chromosom 7 (EMBLAccession-Nr. NT_007933) identifiziert. Mit Intron-überspannenden Primern wurde das kleine Intron 1 mit der Standard-PCR-Methode (Kapitel 2.1.3) und das größere mit der Longrange PCR mit dem Elongase Enzym (Kapitel 6.2.1; Tabelle 11.1) von genomischer DNA amplifiziert und sequenziert. Vergleichende Studien des menschlichen und caninen GNGT Gens zeigten, wie in Tabelle 11.2 dargestellt, dass der 5'UTR des Menschen 77 bp größer (76 bp Exon 1, 1 bp Exon 2) und der 3'UTR um 19 bp kleiner ist. Intron 1 des Hundes ist 5 bp kleiner, Intron 2 ist ca. 200 bp größer. Erfahrungsgemäß sind die Introns beim Hund meist verkürzt. Vergleiche der Exon-/Introngrenzen von Hund zum Menschen zeigten eine hohe Übereinstimmung auf Nukleotidebene (Tabelle 11.2). Der OLR kodiert für 74 Aminosäuren und zeigt auf Proteinebene sogar eine Überstimmung von 100% zum humanen Protein. 11.1.2 Mutationsanalyse des GNGT1 Gens Bei der Sequenzierung des 5' Bereichs zur Identifizierung von Intron 2 wurden in der zufällig gewählten Sloughi DNA Sequenzvariationen im heterozygoten Zustand identifiziert. Deshalb wurden die Primer für die PCR-SSCP-Analyse so gewählt, dass diese Bereiche in die Untersuchung mit eingeschlossen waren (Tabelle 11.1). Diese SNPs wurden in neun (IVS2+336A→T) bzw. in acht Rassen (IVS2+272G→T) nachgewiesen (Tabelle 11.3). Bei der PCR-SSCP-Analyse des Fragments von Intron 2/Exon 3 wurden keine zusätzlich zu den schon veröffentlichten Sequenzvariation (Dekomien et al. 1998) identifiziert. Dieser SNP (c230A→G) wurde in 24 der 30 Hunderassen nicht vorgefunden. Neu identifizierte Sequenzen werden auf Besonderheiten (SINE, LINE) hin überprüft und mit schon veröffentlichten Sequenzen in der Datenbank mittels des Programms Blast search verglichen. Dabei wurde ein expressed sequence tag (EST) des Exon 3 des GNGT1 Gens (EMBL-Accession-Nr.AJ407851) mit Bereichen des Intron 2/Exon 3 identifiziert. Eine 85 Kapitel 11 GNGT1 Gen derartige EST-Sequenz enthält normalerweise nur exprimierte Bereiche. Die Intronsequenzen wurden in dieser veröffentlichten Sequenz nicht herausgespleißt, da in der AG-Spleißstelle ein G→A Austausch stattfand. Deshalb wurden die Exon-/Introngrenze des Exon 3 zusätzlich in erkrankten Hunden der Rassen, wo keine SNPs im heterozygoten Zustand nachzuweisen waren, direkt sequenziert. Dieser Austausch wurde bei der Untersuchung des PCR-Fragments in keinem der untersuchten Hunde nachgewiesen. 11.1.3 Schlussfolgerung In insgesamt 13 Rassen wurden Sequenzvariationen im Intron 2 und 3'UTR des GNGT 1 Gens nachgewiesen. Der gesamte kodierende Bereich ohne Promotorsequenzen wurde untersucht. In CBR, GR, Sl und TT wurden die entsprechenden Variationen sowohl homozygot wie auch heterozygot in gesunden Hunden identifiziert, was ein wichtiger Hinweis dafür ist, dass diese Austausche nicht ursächlich pathogen sind. Diese SNPs können in den Rassen, wo sie heterozygot bei erkrankten Tieren auftreten, als intragenische Marker für eine ar vererbte gPRA eingesetzt werden (Kapitel 2.2). So kann das GNGT1 Gen nun als Ursache für die gPRA in 11 Rassen ausgeschlossen werden: Bologneser, Berger des Pyrénées, Irish Setter, Labrador Retriever, Polnische Niederungshütehunde, Saarloos/Wolfshunde, Schapendoes, Sloughis, Teckel, Tibet Terrier und Zwergpudel. Tabelle 11.1 Primer und Bedingungen für die PCR-Amplifikationen des GNGT1 Gens PCRSysteme Bindestelle Primersequenza (5' - 3') UTR 1 F E 1/2 E 2-2 F E 2-2 R E 2-2 F I 1-2 R I 2F E 2-2 R I2F UTR 3 R UTR 5' Exon 2 Exon 2 Exon 3 Exon 2 Intron 2 Intron 2 Exon 3 Intron 2 3'UTR GACCATTCATAAGAAATCAGTTA CAGCATTCTTTCCAGGGT TGAGGACTTGACAGAAAAGG CTTATGAAATCACACAGCCTC TGAGGACTTGACAGAAAAGG CAATTATGGGAGGTTTTCAG TATGCATTGGTGGAAACCAG CTTATGAAATCACACAGCCTC TATGCATTGGTGGAAACCAG ATCCAAGCTATTCCAAGGAAC Länge der PCRProdukte (bp) 253 PCRBedingungen [T-°C)/MgCl2mM)] Restriktionsenzyme für die SSCPAnalyse 54/2,5 SspI 59/1,5b - 462 54/1,5 MnlI 250 54/1,5 - 280 54/1,5 BglII ∼4100 a Primersequenzen nach EMBL-Accession-Nr. AJ507014, AJ507015. b PCR amplifiziert mit dem Elongase Enzym-Mix (Kapitel 6.2.1). 86 Kapitel 11 GNGT1 Gen Tabelle 11.2 Vergleichende Exon-/Intron Organisation des GNGT1 Gens von Hund und Mensch Exon # Hund Mensch Hund Mensch Hund Mensch Hund Mensch A Exon-/Introngrenzen 61A 137B 2 106 107 OLR 129/UTR 159 ORL 129/UTR 140 3 # Länge (bp) 1 Intron AGTTCACTTAgtaagttaag AGTTTACTTTgtaagttaag attttttcagCAGGCAAAAG acatcttcagCAGGCAAAAA AAGAATGCTGgtaaactgct AAGAATGCTAgtaagttgct ttcttttaagGTCTCCAAAT tccccttaagGTTTCCAAAT Länge (bp) 1 86 91 2 ∼4100 3947 Länge des Exon 1 bestimmt nach EMBL- Accession-Nr. Z75156; Intronsequenzen des Hundes von EMBL- Accession-Nr AJ507014, AJ507015. B EMBL- Accession-Nr. NT_007933. Tabelle 11.3 Sequenzvariationen im GNGT1 Gen in verschiedenen Hunderassen (Abkürzungen der Rassenamen siehe Tabelle 2.1) Gen / Lokalisation Sequenzvariation GNGT1 Intron 2 IVS2+272G→T Amino- Rasse säure Austausch - Bo, BDP, IRS, PON, Sa, SD, Sl, Te, TT IVS2+336A→T GNGT1 Exon 3 c230A→G Bo, BDP, IRS, PON, SD, Sa, Sl, TT - CBR, GR, LR, Te, TT, ZP Sequenzposition im UTR entspricht der caninen cDNA (EMBL-Accession-Nr. Z75156). 87 Kapitel 12 Mitochondriale Gene Kapitel 12 Mitochondriale Gene 12.1 Vorbemerkung Mutationen in menschlichen mitochondrialen Genen führen zu Krankheiten mit vielen unterschiedlichen Symptomen unter anderem auch zur RP. Die ATPSyntase (oder Komplex V) ist ein Enzym der aeroben ATP Produktion. Sie ist in der inneren mitochondrialen Membran eukariotischer Zellen lokalisiert, zusammen mit vier Atmungskettenenzymen, die in die Elektronentransportkette mit eingebunden sind. Die ATPSyntase der Säuger ist aus einem rotierenden katalytischen F1-, einem transmembranen F0-Anteil und zwei Stielen zusammengesetzt, die die F1 und F0 Teile miteinander verbinden. Zwei Untereinheiten des F0Anteils der ATPSyntase sind die ATPase 6 und 8 die von der mitochondrialen DNA codiert werden (Leslie et al. 1999). Eine Mutation, die in der Position 8993 T→G der human mitochondrialen DNA im Gen der ATPase 6 ist, ist assoziert mit dem neurologischen "Muskelschwäche, Ataxie und RP" Syndrom (NARP; Holt et al. 1990; Tatuch et al. 1992). Eine Mutatione in der Position 9101 T→C führt zur "Leberschen hereditären Optikus Atropie" (LHON; Lamminen et al. 1995). Ein weiteres Mitglied der Atmungskettte ist das Cytochrom b ein Anteil des Komplexes III. Das Cytochrom b ist auch in der inneren mitochondrialen Membran lokalisiert und ist das zweite Enzym in der Elektronentransportkette der mitochondrialen oxidativen Phosphorylierung. Die G15257A Mutation führt zur LHON (Brown et al. 1992) und die A15579G Mutation zu multiplen Störungen wie Taubheit, mentale Retardardierung, RP, Katarakt, Wachstumsstörungen und Epilepsie (Wibrand et al. 2001). Das mitochondriale Genom besitzt ein eigenes Translationssystem, erkennbar am Vorhandensein von Genen für tRNA und rRNA. Störungen des Translations-Systems durch eine Mutation in der tRNASer führt zum Usher Syndrom (Taubheit und RP; Mansergh et al. 1999) und der tRNALys in der Position 8344 des mitochondrialen Genoms zur "Myoklonen Epilepsie und ragged red fibers" (MERRF; Shoffner 1990; Loksalisation der og Mutationen im mitochondrialem Genom siehe Abbildung 13.2). Bei den Saarloos aus Deutschland wurden zusätzlich zur gPRA Symptome wie Muskelschwäche, Taubheit und Epilepsie beobachtet. Deshalb wurden die mitochondrialen Gene Cytochrom b die ATPase 6 und 8 mit den angrenzenden tRNAs in die gPRAMutationsanalyse einbezogen. 88 Kapitel 12 Mitochondriale Gene 12.2 Resultate und Diskussion 12.2.1 Mutationsanalyse der mitochondrialen Gene Gesunde und kranke Saarloos/Wolfshunde mit den oben genannten multiplen Symptomen sowie gPRA erkrankte Hunde von 24 Rassen wurden in die Mutationsanalyse eingeschlossen. Sequenzen des gesamte canine mitochondriale Genom sind bekannt (Kim et al. 1998; EMBL Accession Nummer NC_002008). Bei der DNA-Isolierung aus Zellen erhält man ein Gemisch aus genomischer Kern- und ringförmiger mitochondrialen DNA, sodass die Amplifizierung von mitochondrialer DNA unproblematisch ist. Der OLR des ATPase 8 und 6 Gen sind überlappend, das Startkodon für das ATPase 6 Gen liegt vor dem Stoppkodon des ATPase 8 Gens. Insgesamt wurden drei Teilbereiche der mitochondrialen DNA mittels der PCR-SSCPAnalyse untersucht. Ein Fragment umfasst die tRNALys, das ATPase 8 und Teile des ATPase 6 Gens. Für das zweites Fragment wurde der Hauptanteil des ATPase 6 Gens Gens amplifiziert, mit den Sequenzbereichen des Rückwärtsprimers des 1. Teils und Teile des 5' Bereichs des Cytochorm-C-Oxidase (Primersequenzen siehe Tabelle 12.1). Das dritte untersuchte Fragment umfaßt das gesamte Cytochrom b Gen, eingerahmt durch die tRNAGlu und tRNATyr. Im ATPase 8 Gen wurden zwei SNPs und im ATPase 6 Gen sieben Variationen identifiziert. Ein Austausch wurde in der tRNAGlu und 10 Unterschiede in der Cytochrom b Sequenz nachgewiesen. Durch den 612 A→G Austausch geht eine Bsp143I-Schnittstelle verloren, so dass hier eine RLFP-Analyse des SNPs möglich war (siehe Tabelle 12.2). 12.2.2 Schlussfolgerung Es wird davon ausgegangen, dass die Häufigkeit für Nukleotidaustausche in mitochondrialer DNA 10-20mal höher ist als in Kern-DNA. Deshalb ist nicht ungewöhnlich, dass mehrere Unterschiede in diesen Sequenzbereichen in einzelnen Rassen nachzuweisen waren. Die identifizierten Sequenzvariationen wurden in Tabelle 12.2 aufgelistet. Im ATPase 6 Gen wurden nur zwei SNPs in mehreren Rassen nachgewiesen, der 48T→A Austausch in 10 Rassen und die 138G→A Variante in 6 verschiedenen Rassen. Die anderen Variationen wurden nur in gesunden und erkrankten Saarloos/Wolfshunden identifiziert. Sieben der identifizierten Variationen lösen keinen Aminosäurewechsel aus. Der 508C→G SNP im Polnischen Niederungshütehund führt zu einem konservativen L169V Aminosäureaustausch. Die SNPs in dem ATPase 8 Gen resultieren in einen Aminosäurewechsel: der 127T→C 89 Kapitel 12 Mitochondriale Gene Austausch wurde in vier Rassen und der 164A→C nur in einem der erkrankten Saarloos/Wolfshunde nachgewiesen. Im Cytochrom b Gen wurde nur ein konservativer Aminosäurewechsel bei den 10 identifizierten Variationen in zwei von sieben erkrankten Entlebucher Sennenhunden identifiziert. Acht SNPs sind rassenspezifisch, und zwei der identifizierten Variationen kommen in mehreren Rassen vor. Keine in diesen drei mitochondrialen Genen gefundenen Variationen kosegregieren in den verschiedenen Rassen mit der gPRA. Eventuell könnten diese SNPs, in Kombination mit weiteren Variationen in mitochondrialen Genen, für evolutionäre Studien eingesetzt werden. Tabelle 12.1 Primer und Bedingungen für PCR-Amplifikationen mitochondrialer Gene PCR-System PrimerBezeichnung tRNALysATPase8F tRNALysATPase8R MT-ATPase F MT-ATPase R ATPase6-2 F MT-ATPase R MT Cytb F MT Cytb R BindepositionA 7647 8261 7889 8772 8168 8772 14101 15454 Primersequenz (5'→ →3')B Länge der PCRProdukte (bp) PCR-Bedingungen [T-°C / MgCl2 -mM] Restriktionsenzyme für die SSCPAnalyse GCTTTATACCCATTGTTCTTG GAGAGTTGTGTTGTGGGCG 614 54/1,5 MnlI TTCAAATCACTACTACCCAG TAGGGCTATTGAGTTATAGTG 833 55/1,5 MnlI TGAGCTCTCATACTTATATCAC TAGGGCTATTGAGTTATAGTG 604 52/1,5 DdeI 1353 52/1,5 Bsp143I TATGTTATCATTATTCCTACATGG GAATAGTTTAAGAAGAATCTCAG A Nukleotidposition zu der das 5' Ende des Primers (fettgedruckte Buchstaben) hybridisiert. B Canine mitochondriale Sequenz nach Kim et al 1998; EMBL-Accession Nr. NC_002008). 90 Kapitel 12 Mitochondriale Gene Tabelle 12.2 Sequenzvariationen im ATPase 6, und 8, dem Cytochom b Gen und den angrenzenden tRNAs Gen / Lokalisation (bp)A SequenzvariationB Aminosäure- Rasse Austausch ATPase 8 7806 8009 127T→C 164A→C S43P N55T CCR, CS, TT, ZP Sa ATP-ase 6 79678647 48T→A G16G 138G→A 318T→C 360A→G 462G→A 508C→G 606A→G Q46Q I106I K120K M154M L169V L202L ACS, CCR, ES, ECS, IRS, Sa, SD, Te, TT, ZP Te, NF, ES, CS, ES, Sa Sa Sa Sa PON Sa - ACS I69I V118I Y155Y G204G P265P W326W L335L Q341Q E344E P346P Sal ES SD AC, ACS, GR, Sal, SD, Te, ZP LR ZP SD Sa AW, BDP, CS, PON, ScT, ZP tRNAGlu 1411114179 64A→T Cytochrom b 1418415333 207C→T 352G→A 465T→C 612A→G 795T→C 978A→G 1003T→C 1023A→G 1032A→G 1038T→A A Lokalisation in der vollständigen caninen Mitochondriensequenz nach EMBL-Accession Nr. NC_002008 B Die Nummerierung der Sequenzvariationen beginnt mit dem jeweiligen Start-Kodon des Gens 91 Kapitel 13 Diskussion Kapitel 13 Diskussion 13.1 Zur Forschungsstrategie der Dissertation Wichtige Kandidatengene für die gPRA beim Hund sind Gene, in denen Mutationen zu retinalen Erkrankung beim Menschen führen. Identifizierte Mutationen im homologen caninen Gen machen diese Hunde zu wichtigen natürlichen Modellsystemen für die weitere Erforschung der Funktionsweise dieser Gene und ihrer Produkte. Zusätzlich könnten Forschungsstrategien für die Gentherapie mittels dieses Gendefektes in diesen Hunden entwickelt werden. Daher wurde bei der Auswahl der Gene auf die umfangreichen Erkenntnisse aus der Erforschung der humanen RP zurückgegriffen. 13.1.1 Kandidatengenanalyse beim Menschen Die in dieser Arbeit angewendete Methode der Kandidatengenanalyse wurden beim Menschen schon mehrfach erfolgreich bei retinalen Erkrankungen angewendet (Dryja 1997). Der Zusammenhang zwischen einer Mutation in funktionell wichtigen Genen und einer vererbten retinalen Erkrankung konnte gerade dort hergestellt werden, wo nur kleine Stammbäume bzw. nur Simplexfälle für die Untersuchung zur Verfügung standen (Aguirre 2001). Gene werden als Kandidatengene für retinale Erkrankungen ausgewählt, wenn diese Gene speziell in den Photorezeptorzellen exprimiert werden. Dies sind Gene, die in den Phototransduktionsprozess, in den Auf- und Abbau und in biochemische Verläufe in den Photorezeptorzellen und angrenzenden Geweben eingebunden sind. Oft wurden Zusammenhänge auffälliger Expressionsmuster dieser Genprodukte in Tiermodellsystemen (siehe Kapitel 1.3.3) mit retinalen Erkrankungen aufgeschlüsselt (Aguirre 2001). Beim Menschen hat sich das positionsabhängige Kandidatengenverfahren für die Identifizierung von RP-Genen als sehr wertvoll erwiesen. Bei diesem Verfahren wird ein durch seine Funktion als Kandidatengen prädestiniertes Gen zusätzlich einer durch Kopplungsanalysen ermittelte chromosomale Teilregion zugeordnet. Zur Eingrenzung des Genlocus mit dem entsprechenden Krankheitsphänotyp wurden polymorphe DNA-Marker (SNP, RFLP- oder VNTR-Marker) eingesetzt (Strachan & Read 1996). In mehr als 108 kartierten mit RP assoziierten Loci wurden inzwischen RP-ursächliche Mutationen in 75 Kandidatengenen 92 Kapitel 13 Diskussion gefunden. 39% dieser Gene werden nur in der Retina exprimiert, 61% sowohl in der Retina wie auch in anderen Geweben (Rivolta et al. 2002; vollständige Liste unter: http://eyegene.meei.harvard.edu; RetNet; http://www.sph.uth.tmc. edu/RetNet/). Weitere RP verursachende Mutationen wurden in drei RPE spezifischen Genen sowie in fünf mitochondrialen Genen nachgewiesen. Die Identifizierung Retina- und RPE-spezifischer Gene wurde durch die Anwendung der seriellen Analyse der Genexpression (SAGE) Methode möglich. Hierbei werden Genexpressionsmuster in den spezifischen Geweben untersucht. Die Häufigkeit der Genprodukts ist ein mögliches Maß für die Wichtigkeit des Proteins (Funktion im entsprechendem Gewebe) wodurch es dann zu einem interessanten Kandidatengen für die Mutationsanalyse wird (Sharon et al. 2002). 13.1.2 Kurzer Überblick der Rassenentstehung Eine wichtige Vorraussetzung für das Gen-Ausschlussverfahren ist, wie in Kapitel 2.2 schon beschrieben, das Wissen um die Geschichte des domestizierten Hundes und die Entstehung der über 300 Hunderassen in der ganzen Welt. Diese Geschichte hat vor ca. 15000 Jahren begonnen, als der Mensch den Wolf in sein soziales Umfeld eingebunden hat. Gefangennahme, Auswahl und Vermischung mit der Wolfspopulation hat diese über 300 verschiedenen Hunderassen erst möglich gemacht. Auf molekularer Ebene ist dies erkennbar durch Untersuchungen mit mitochondrialer DNA, wo nur 0,2% Sequenzunterschiede bei vergleichenden Studien des grauen Wolfes mit dem domestizierten Hund nachgewiesen wurden (Wayne 1993), obwohl die Mutationsrate in den Mitochondrien sogar 10-20mal höher als in Kern-DNA ist. Diese Tatsache lässt vermuten, dass Eigenschaften des Hundes nur durch wenige (genregulatorische) Veränderungen des Genoms im Vergleich zu Wolf hervorgerufen werden. Verhaltens- und genetische Studien unterstützen diese Annahme (Scott & Fuller 1965). Dies gilt sowohl für die Veränderungen des Verhaltens, wie auch für die morphologischen Modifikationen, die höchstwahrscheinlich von Allelen entstehen, die in der Wolfspopulation eher selten, aber durch die Gefangennahme und Eingrenzung im Rahmen von Zuchtprogramme der Menschen nun in diesen Populationen gehäuft auftreten. Zur Etablierung bestimmter Merkmale einer Rasse wird oft eine Zuchtstrategie angewandt wodurch sich der Genpool durch Verpaarung eng Verwandter stark eingeengt. Genetische Erkrankungen sind in einer hohen Frequenz in diesen Populationen zu erwarten, da sich Rassen nur auf eine kleine Anzahl von Gründern aufbaut und die von Züchtern gewünschte schnelle Verbreitung und Vergrößerung der Population mittels Inzucht ihren Teil beiträgt (Ostrander et al. 2000). Ar Erkrankung sind hierbei naturgemäß das größte Problem. Da 93 Kapitel 13 Diskussion heterozygote Träger eines krankheitsverursachenden Allels keine Symptome zeigen, werden sie ohne DNA-Tests erst erkannt, wenn Nachkommen erkranken. 13.1.3 Kandidatengenanalyse beim Hund Molekulare Studien von erblichen retinalen Erkrankungen mit Hunden waren während dieser Studie noch auf Kandidatengenanalysen limitiert. Eine der Ursachen der Beschränkung auf diese Methode sind sehr geringe Mengen an Hundegenomsequenzen in den Genombanken, da das Hundegenomprojekt sich noch im Anfangsstadium befindet (Yang et al. 1999). Damit sind vergleichende Genominformationen zwischen Hund, Mensch und Maus noch nicht in ausreichenden Maße erhältlich. Dies verhindert beim Hund die positionsabhängige Klonierungsstrategie, die beim Menschen so erfolgreich für die Identifizierung von retinalen Erkrankungsloci in den letzten Jahren eingesetzt wurde (Strachan & Read 1996). Die auf Kandidatengene basierende Methode ist in Hunderassen mit nur wenigen sicher an gPRA erkrankten Hunden sinnvoll. Ausserdem zusätzlich dann anzuwenden, wenn keine informativen Stammbäume für die Kopplungsanalyse oder Genomsuche verfügbar sind (Aguirre 2001). Der erste Schritt für die Kandidatengenanalyse ist die Identifizierung von hundespezifischen Genen, hauptsächlich der cDNA Sequenzen, um die Mutationsuntersuchung in den codierenden Genregionen in erkrankten Hunden durchführen zu können. Eine Reihe von cDNAs und genomische Sequenzen von photorezeptorspezifischen Genen wurden inzwischen schon von verschiedenen Forschungsgruppen aus Deutschland, England, Finnland und USA in den Genbanken deponiert. Aufgabe dieser Dissertation war es, mittels Kandidatengenanalyse gPRA ursächliche Mutationen in den 30 zur Verfügung stehenden unterschiedlichen Rassen zu identifizieren. Dafür wurden intronische Sequenzen von schon veröffentlichten wichtigen Kandidatengenen identifiziert, von denen nur die cDNAs bekannt waren, um die Spleißkonsensus- und regulatorische Sequenzen in die Mutationsanalyse mittels der PCR-SSCP-Analyse einzuschließen. Zusätzlich wurde die noch unbekannte canine Sequenz vom RCV1 Gen isoliert und charakterisiert, das in einer Region lokalisiert ist, in der aufgrund von Assoziationsstudien Gene vermutet werden, die ursächlich für ad und ar RP beim Menschen sind. Insgesamt wurden 13 Gene untersucht (Kapitel 3-12). Neun dieser Gene exprimeren Proteine, die in den Außensegmenten der Stäbchen lokalisiert sind. Acht dieser Produkte sind direkt in den Phototransduktionsprozess (GNGT1, PDC, PDE6A, PDE6B, PDE6D, PDE6G, RCV, SAG1) und eines in die Photorezeptorentwicklung sowie als Transkriptionsfaktor in die Regulierung der Proteine des Lichtzyklus (CRX) eingebunden. Ein untersuchtes Gen (RPE65) 94 Kapitel 13 Diskussion liefert ein Produkt, das nur im RPE nachgewiesen wird. Es hat eine wichtige Funktion im Vitamin A Zyklus. Drei mitochondriale Gene codieren für Proteine, die in der inneren Membran der Mitochondrien lokalisiert sind und wichtige Funktionen in der Elektronentransportkette haben. Zahlreiche Mutationen in diesen Genen führen zu den unterschiedliche Ausprägungen der retinalen Erkrankungen beim Menschen (siehe Kapitel 3-12). Teilweise ist die normale Funktion dieser Proteinprodukte noch nicht vollständig verstanden, und für einige der Gene ist das Verständnis für die pathophysiologischen Konsequenzen von Mutationen noch sehr lückenhaft. Deshalb ist die Mutationsanalyse im Modellsystem Hund wichtig, da er in seiner Rassenvielfalt die unterschiedlichsten Mutationen aufweist, so dass er für eine ganze Reihe natürlicher Systeme für die Erforschung nicht funktionstüchtiger Proteine zur Verfügung steht. Beim Nachweis von Mutationen im Kandidatengen, können Hunde der Rasse mit diesem spezifischen Defekt unter anderem als Modellsystem für Gentherapie (wie bei den Briads; Acland et al. 2001) oder zur Identifizierung unbekannter Funktion des Gens (wie bei den RCS Ratten; D’Cruz et al. 2000) eingesetzt werden. Auch für die Zucht bietet ein Gentest große Vorteile gegenüber den klinischen Untersuchungsmethoden. Gerade für Rassen mit relativ spätem Krankheitsbeginn (4-5 Jahr), ist die Wahrscheinlichkeit groß, dass homozygot erkrankte Hunde das krankheitsverursachende Allel an ihre Nachkommen weitervererben. Erst wenn ein an gPRA-erkrankter Nachkomme erkannt wird, ist bei einem ar Erbgang sicher, dass die Elterntiere Träger des gPRA-Allels sind. Sie werden dann von der Zucht ausgeschlossen, haben aber möglicherweise schon mehrere Nachkommen gezeugt, die dann auch von der Zucht ausgenommen werden, um die Reduktion des gPRA-Allels in dieser Rasse zu erreichen. Als frühestmögliche diagnostische Mittel zur Verringerung des Anteils von gPRA-Anlageträgern steht die Elektroretinographie (ERG) zur Verfügung. Mit dieser Methode sind wenige spezialisierte Veterinärmediziner in der Lage homozygote phänotypisch noch unauffällige, Anlageträger anhand der gPRA charakteristischen Veränderungen im ERG zu diagnostizieren. Diese Methode ist sehr aufwendig und ist für eine routinemäßigen Untersuchung der gesamten Rasse nicht geeignet. Deshalb wäre es wünschenswert, über molekulare Diagnostik der gPRA auch in den anderen Rasse zu verfügen. Nur so können auch heterozygote Anlageträger zweifelsfrei nachgewiesen werden. So ist auch die genetische Variabilität und Vielfalt in kleinen Populationen durch die gezielte Verpaarung in der Zucht gegeben. Ansonsten könnten sich durch die Einengung des Genpools sich andere krankheitsverursachende Allele manifestieren, was für den Fortbestand der Rasse schädlich bzw. deletär sein könnte. 95 Kapitel 13 Diskussion 13.1.4 PDE6B-Mutation in Sloughis Besonders erfolgreich war die Mutationssuche in Sloughis. In dieser Rasse wurde eine 8-bp Insertion im Exon 21 des PDE6B Gens nachgewiesen, wodurch sofort ein Stoppkodon entstehrt. So entsteht ein verkürztes und instabiles Protein. Die Phosphodiesterase kann kein cGMP mehr spalten. Dies führt zur Überproduktion von cGMP in den Außensegmenten und die Phototransduktion wird blockiert (Farber 1995). Daduch wird die Degeneration der Photorezeptorzellen ausgelösts in Hunden mit homozygoten, krankheitsverursachenden Allelen des PDE6B Gens. Für die Sloughis wurde ein Gentest entwickelt und inzwischen der Genotyp von 223 Tieren bestimmt. 5 homozygot kranke Individuen, 68 heterozygote und 155 homozygote gesunde Hunde wurden nachgewiesen. Auf Betreiben des Zuchtverbandes hin wird die gesamte deutsche Sloughi-Population auf das Vorliegen der 8-bp Insertion untersucht. Wird bei einem Hund ein krankheitsverursachenden Allel identifiziert wird dieses Tier nur noch mit einem homozygot gesunden Hunden verpaart, damit gPRA-erkrankte Hunde in dieser Population nicht mehr vorkommen. In dieser Konstellation ist der Gentest zur Feststellung der Trägerschaft wichtig, da statistisch gesehen 50% der Nachkommen wiederum Träger des gPRA-Allels sind. Die Nachkommen von Eltern, die homozygot für das Wildtypallel sind, sind sicher homozygot für dieses Allel. Deshalb müssten die Nachkommen dieser Konstellation bei gesicherter Vaterschaft nicht mehr getestet werden. Der Krankheitsbeginn fällt bei den Sloughis mit ca. 2 Jahren in den Beginn der Geschlechtsreife. Deshalb kann durch den gPRA-Test in Sloughis ausgeschlossen werden, dass weitere gPRAErkrankungen, verursacht durch diese Insertion, in der Zucht auftreten. 96 Kapitel 13 Diskussion RPE 11-trans Retinol T-γ 11-trans Retnal RPE6 RPE65 PDE-αβδγ CRX CRX RCV SAG PDC Abbildung 13.1 Schematische Darstellung der Phototransduktionskaskade in den Photorezeptorzellen der Vertebraten (verändert nach Pugh et al. 1999). In die hier durchgeführte Kandidatengenanalyse eingebundene Genprodukte wurden mit einem Pfeil markiert. Abkürzungen: R* = aktiviertes RHO, G = Tαβγ, Rec = RCV, RK = RHOK, Arr = SAG, E = PDE, GC = cGC-E, P = PDC, cG = cGMP. A: Aktivierungsphase der Kaskade. Lichtaktivierung des RHO durch schnellen Kontakt mit T, Katalyse des GDP zu GTP, das so aktivierte T*. bindet an die PDE-γ-Untereinheiten, dadurch Aktivierung der αβ-PDE*. PDE katalysiert die Hydrolyse von cGMP, Der niedrige cGMP-Spiegel führt zur Schließung des Kanals und zur Blockade der Na+- und Ca2+-Einfuhr. Dies fördert die Hyperpolarisation der Zelle mit einem Membranpotential bei Belichtung von -70 mV. B: Inaktivierung von RHO*. Durch hohe Konzentration von Ca2+ im Dunkeln entsteht hoher Anteil an membrangebundenen RCV-Ca2+. So ist die Bildung eines Komplexes mit RHOK und Blockierung der Aktivität des RHOK möglich. Durch Lichtantwort Absinken des Ca2+Spiegels, dadurch gibt RCV Ca2+ frei und löst sich vom RHOK. Somit wird die Phospho- 97 Kapitel 13 Diskussion rylierung von RHO* möglich, dies löst die Bindung von SAG an das aktivierte RHO* aus und unterdrückt so die Aktivität des RHO*. C: Regulation der Kaskade durch PDC. Die Inaktivierung des Ts wird durch die Bindung des PDC an Tβγ ausgelöst. Dadurch kommt es zur Blockade der Wiederaufbereitung von TαGDP. Nach Phosphorylierung des PDC, Aufgabe der Tβγ-Bindung somit steht G-α für die Kaskade wieder zur Verfügung. Der Phosphorylierungszustand wird durch Ca2+ reguliert (genaue Beschreibung der Phototransduktionskaskade, Kapitel 1.1.4). LHON 15579 LHON 9101 NARP 8993 MERRF 8344 Abbildung 13.2 Schematische Darstellung der mitochondrialen DNA mit der Lokalisation der Gene der Enzymkomplexe. Abkürzungen: Komplex I: ND = Untereinheiten der NADH-UbiquinonOxireduktase; Komplex III: Cyt b = Cytochrom b ein Baustein der Succinat-UbiquinonOxidoreduktase. Komplex IV: CO = Bestandteile der Cytochrom c-Oxidase. Kompex V: ATPase = Untereinheiten der ATP-Synthase. Position der D-Loop beim Hund 15459 bp 16728 bp. Pfeile kennzeichnen die Positionen der Mutationen die zu den entsprechenden Erkrankungen beim Menschen führen (Kapitel 12.1). Diese gekennzeichneten Gene und angrenzende tRNAs wurden mittels der PCR-SSCP-Analyse untersucht. 98 Kapitel 13 Diskussion 13.2 Kandidatengen-Ausschlussverfahren 13.2.1 Verteilung der Sequenzvariationen in den untersuchten Genen Durch die Mutationsanalyse der 13 Gene wurde insgesamt 120 Sequenzvariationen gefunden. Davon wurden 1/4 im OLR und 3/4 im nicht-translatierten Bereichen identifiziert (Tabelle 13.1). Die Verteilung der Sequenzvariationen ist von Gen zu Gen mitunter sehr verschieden. Neben extrem konservierten Protein-codierenden Abschnitten wie in den PDE6D und GNGT Genen finden sich hochpolymorphe Bereiche wie im SAG Gen. Dies könnte für die funktionelle Bedeutung der entsprechenden Sequenzen der einzelnen Gene sprechen, oder alternativ ein Maß für das Alter der entsprechenden Gene in der Spezies/Rasse sein. Unter diesen Annahmen ist die größere Dichte der Sequenzvariationen in den nichttranslatierten Bereichen der Gene keine Überraschung. Aber in einigen Genen ist die Variabilität auf die gesamte Einheit des Gens ungleichmäßig verteilt. Die unterdurchschnittlich polymorphen Exons der PDE6B und der PDE6G Gene sind mit höchst variablen Introns verknüpft. Diese Ergebnisse ergänzen erste genomweite Befunde beim Menschen: Gewisse Regeln für die Verteilung / Dichte von Polymorphismen ergeben sich primär aus der gemeinsamen Genealogie sowie der Verteilung der genetischen Rekombinationseinheiten, weniger aus lokalen Mutationsraten (Reich et al. 2002). Daher muss die Menge informativer Marker im Rahmen von Kandidatengenanalysen für jedes Gen entsprechend angepasst werden. 13.2.2 Intragenische Marker Gerade für die ar vererbten Erkrankungen ist das Genauschlussverfahren über intragenische Marker möglich, wie in dieser Arbeit in mehreren Beispielen nachgewiesen. Auch wenn keine direkt gPRA-verursachende Mutation identifiziert wurde, ist durch diese Methode die Eingrenzung der Kandiatengene für die gPRA erreichbar (Bourgain et al. 2002). Bestimmte Ausprägungsformen polymorpher Allele können mit der gPRA-verursachenden Mutation gekoppelt sein und so für die indirekte Mutationsanalyse herangezogen werden. Je enger ein Polymorphismus mit der gPRA-Mutation gekoppelt ist, desto niedriger ist seine Rekombinationswahrscheinlichkeit (Hudson & Kaplan 1985). Deshalb sind Ausschlüsse bei erkrankten Hunden einer Rasse möglich, wenn Sequenzmuster für heterozygote Tiere auftreten. Erkrankte Hunde müssen bei einer ar vererbten Erkrankung homozygot für das entsprechende gPRA-Allel sein. Bei einem funktionstüchtigen Genprodukt ist die Versorgung der Zellen gewährleistet. Bei Kopplung und ohne Rekombination sind die Variationen im 99 Kapitel 13 Diskussion homozygoten Zustand in diesen relativ kleinen (∼5-30 kb) Genen nachzuweisen, da durch die geringe Größe der Gene von einer mittleren Distanz zweier Polymorphismen von maximal 0,005-0,050 cM ausgegangen werden kann. Dies entspricht einer Rekombinationsrate von 1:20000-3333 (Hudson & Kaplan 1985). Die verfügbaren Hunderassen, existieren erst seit ∼100-600 Jahren. Bei einem 2jährigen Fortpflanzungszyklus kann also mit maximal 300 Generationen gerechnet werden. Somit ist die Rekombinationswahrscheinlichkeit in diesen Rassen zwischen untersuchter Sequenzvariation und gPRA-Mutation sehr gering und die Wahrscheinlichkeit sehr hoch, dass zwei in einem Gen liegende Loci miteinander gekoppelt vererbt werden. Mit Hilfe der intragenischen Marker wurden 10 der untersuchten Gene der Kern-DNA in einer große Anzahl der 30 verschiedenen Rassen, wie in Kapitel 3-11 beschrieben, als Ursache für die gPRA ausgeschlossen (Tabelle 13.2). Das RPE65 Gen konnte bei insgesamt 28 Rassen ausgeschlossen werden, obwohl es mit 20 Sequenzvariationen nicht den höchsten Polymorphiegrad aufwies. Die meisten Sequenzvariationen (28) wurden im SAG Gen nachgewiesen. Diese führten zum Ausschluss von 16 Rassen, sowie auch in drei weiteren Genen, dem RCV1, dem PDE6B und dem CRX Gen. Allerdings wurden beim SAG Gen 19, im PDE6B Gen 15, und im CRX Gen nur 8 Variationen identifiziert. In den Genen PDE6D, PDE6G und GNGT konnte trotz geringer Anzahl von SNPs (PDE6D 6, PDE6G 4 und GNGT 3) diese Gene in 15 Rassen als gPRA-Ursache ausgeschlossen werden. Für zwei Gene (PDC und PDE6A) konnten die Marker nur in 11 Rassen für den indirekten Ausschluss eingesetzt werden, obwohl im PDE6A Gen sogar insgesamt 15 Variationen nachgewiesen wurde. Das PDC Gen zeigt mit insgesamt 3 Sequenzunterschieden die geringste Anzahl von Variationen (Tabellen 13.1 und 13.2). Da in 10 der 30 Rassen nur ein erkrankter Hund (Tabelle 2.1) für die Mutationsanalyse zur Verfügung stand, ist es wichtig, dass die gPRA in diesen Rassen eindeutig diagnostiziert ist. Bei einer Fehldiagnose ist dieses Tier kein "homozygoter Anlageträger", und somit ist das indirekte Ausschlussverfahren über intragenische SNPs nicht anwendbar. Weitere erkrankte Hunde müssten in die Untersuchungen eingeschlossen werden, um das Risiko einer Fehlinterpretation der Ergebnisse zu minimieren. Selbst eine Fehldiagnose in einem Hund bei mehreren erkrankten Hunden in einer Rasse könnte zum Ausschluss des Kandidatengens über die heterozygot identifizierten SNPs in diesem gPRAdiagnostizierten aber genotypisch gesunden Hund führen. Damit dieses Risiko so gering wie möglich gehalten wird, sollten die funktionell wichtigen Bereiche eines Gens möglichst vollständig untersucht werden. Zusätzlich sollten möglichst mehrere erfahrene und auf 100 Kapitel 13 Diskussion Augenkrankheiten spezialisierte Tierärzte nicht ganz eindeutige Befunde unabhängig voneinander überprüfen. 13. 3 Ausblick Die Manipulation in der caninen Genetik der letzten Jahrhunderte durch den Menschen ist für die heutige Forschung ein wichtiges Hilfsmittel für das Verständnis der Biologie dieser Vertreter der Mammalia. Durch die Rassenstrukturen wurden Barrieren aufgebaut, wodurch die Vermischung von Genpools verhindert wird. Dadurch gleicht jede Hunderasse für sich gesehen den menschlichen Isolaten in Finnland, Island, oder den Beduinenstämmen Israels und kann mit den gleichen statistischen Methoden untersucht werden. Deshalb ist der reinrassige Hund als Modellsystem wichtig für genetisch komplexe menschliche Erkrankungen. Weitere identifizierte Mutationen in der Phototransduktionskaskade oder Zyklen in angrenzenden Geweben im Hund, die zu homologen Augenerkrankungen im Menschen führen, können für weitere Fortschritte in der Gentherapie eingesetzt werden. Da sich Versuche der Gentherapie beim Menschen ausschließen, aber erfolgreich etablierte Therapien beim Hund dann beim Menschen angewendet werden können. So kann durch direkte oder indirekte Gentherapie der Krankheitsverlauf verzögert, gemildert oder wie bei den Schwedischen Briads (Acland et al. 2001) wieder rückgängig gemacht werden. Daher bleibt besonders zu hoffen, dass erste Ankündigungen zur Entwicklung des Genomprojekts für den Hund tatsächlich zeitnah in die Tat umgesetzt werden können. 101 Kapitel 13 Diskussion Tabelle 13.1 Häufigkeiten von Polymorphismen/Variationen, die in den Exons und Introns der untersuchten Gene identifiziert wurden Gen Anzahl der Exons Exons Introns/UTRs Σ Länge Sequenz- Σ Länge Sequenz- (bp) Variation (bp) Variation CRX 3 786 4 1542 4 GNGT1 3 224 - 1206 3 PDC 4 735 - 1279 3 PDE6A 22 2580 1 2557 14 PDE6B 22 2574 5 499 10 PDE6G 4 261 1 2072 3 PDE6D 4 450 - 1038 6 RCV1 3 600 2 1500 17 SAG 16 1257 12 2896 14 RPE65 14 1400 4 2253 16 ATPase 6 - 607 7 - - ATPase 8 - 600 2 - - Cytochrom b - 1380 10 - - 102 Kapitel 13 Diskussion Tabelle 13.2 Indirekte Ausschlüsse von Kandidatengenen als Ursache für PRA in verschiede- Sa Sal SD ScT Sl Sp Te TM TT YT ZP ATPase8 Cytb tRNALys AW ACS AC BDP BS Bo CBR ECS Co CCR ES GR IRS KV LR NF PON RS Ro U U U U U U U U U U U U U U U ATPase6 CRX RCV1 SAG RPE65 PDC GNGT1 PDE6D PDE6G Rasse* PDE6B Gene PDE6A nen Hunderassen U U l U U U U U U U U U U U U U U U U U U U U U U U U U U U U U U U U U U U U U U U U U U U U U U U U U U U U U U U U U U U ✤ U U U U U U U U U U U U U U U U U U U U: Untersucht, keine Mutation identifiziert; Ausschluss unmöglich, da mitochondriales Gen (wegen Heteroplasmie); *: Abkürzungen Tabelle 2.1; Rassen ausgeschlossen; Rassen noch nicht ausgeschlossen; ✤ gPRA-Mutation definiert (Dekomien et al. 2000). 103 Kapitel 14 Zusammenfassung Kapitel 14 Zusammenfassung 30 Hunderassen mit insgesamt 815 Tieren, von denen 115 an gPRA erkrankt sind, wurden für die Kandidatengenanalyse in das Forschungsprojekt eingebunden. Für die Mutationssuche mit genomischer DNA ist es erforderlich, für die vollständige Genanalyse intronische Sequenzen zu identifizieren, da in den meisten Fällen nur cDNAs von den zu untersuchenden Kandidatengenen zur Verfügung stehen. Aus einer Phagengenombank wurden zur vollständigen Untersuchung die Gene PDE6A, PDE6B, PDE6D, PDC, RCV1 und SAG identifiziert und charakterisiert. Die Intronsequenzen des PDE6D und PDC Gens waren bereits veröffentlicht. In diesen beiden Genen wurde ein noch nicht veröffentlichtes Intron im 5'UTR nachgewiesen. Insgesamt wurden im Rahmen dieser Arbeit 59801 bp canine DNASequenzen in der EMBL-Datenbank veröffentlicht, wovon 13454 exonische und 16842 intronische Nukleotide per PCR-SSCP-Analyse untersucht wurden. In 3 Genen (CRX, GNGT1, RPE65) ließen sich intronische Sequenzen direkt aus genomischer DNA durch intronüberspannende PCRs amplifizieren. Die Sequenzen des RCV1 Gens wurden vollständig aus der Hundegenombank isoliert, identifiziert und sequenziert. Zusätzlich wurden die bekannten mitochondrialen Gene die ATPase 8 und 6, das Cytochrom b und die angrenzenden tRNAs vollständig untersucht. In den mitochondrialen Genen wurden eine große Anzahl von meist rassenspezifischen Sequenzvariationen nachgewiesen. Es konnte keine Kosegregation mit der gPRA gezeigt werden. Eine gPRA-verursachende 8-bp Insertion im Exon 21 des PDE6B Gens wurde in Sloughis identifiziert. Die Kosegregation mit der gPRA wurde mittels Stammbaumanalysen nachgewiesen. Ein direkter Gentest wird seit September 2000 durchgeführt und bisher insgesamt 223 Sloughis untersucht. Zusätzlich wurden eine Reihe von Sequenzvariationen in diesem Gen als indirekte Marker für den Genauschluss eingesetzt. So konnte das PDE6B Gen für 17 Rassen als Ursache der gPRA ausgeschlossen werden. Dieses KandidatengenAusschlussverfahren mittels identifizierter Sequenzvariationen (SNPs, Deletionen, Insertionen) wurde bei den 10 Kern-exprimierten Genen angewendet. So führten Sequenzvariationen in den Genen GNGT, PDC und PDE6A in 11 Rassen, PDE6G und PDE6D in 15 Rassen, CRX, SAG und RCV1 in 16 Rassen und RPE65 in 28 Rassen zum Ausschluss des Kandidatengens als Ursache für die ar vererbte gPRA. Das RCV1 Gen konnte zusätzlich in 8 Rassen für die ad gPRA ausgeschlossen werden. Zusätzliche Studien dieser Art sollten diese 104 Kapitel 14 Zusammenfassung schwerwiegende Augenkrankheit in Hunden eliminieren. Auf der anderen Seite sind solche gründlich charakterisierten genetischen Modelle von ausserordentlichem Wert für die Erforschung gleichartiger Erkrankungen des Menschen. 105 Kapitel 14 Summary Summary 30 different dog breeds comprising 815 animals including 115 gPRA-affected dogs were studied by the candidate gene approach for gPRA mutations. Since in several instances only cDNAs existed for the candidate genes, it was necessary to identify intronic sequences for complete gene analysis, i.e. mutation search in genomic DNA. From a canine genomic phage library the introns of the genes PDE6A, PDE6B, PDE6D, PDC, RCV1 and SAG were identified and characterized. Intronic sequences of the PDE6D and PDC genes were published before. In these two genes novel introns were identified here. Altogether I have submitted 59801 bp of canine DNA sequences to the EMBL database wherefrom 13454 exonic and 16842 intronic nucleotides where investigated by PCR-SSCP analyses for the abovementioned animals. In 3 genes (CRX, GNGT, RPE65) the intronic sequences were amplified directly by means of intron-overlapping PCRs. The RCV1 gene was isolated from the canine genomic phage library, and completely characterized. Additionally, the sequences of the mitochondrial genes ATPase 6 and 8, Cytochrom b and the flanking tRNAs were thoroughly investigated. In the mitochondrial genes various, often breed-specific sequence variations were identified. But no co-segregation with the gPRA in these breeds was verified. A gPRA causing 8-bp insertion in exon 21 of the PDE6B gene was identified in Sloughis. Cosegregation with the gPRA was verified by pedigree analysis. A direct DNAtest for gPRA for Sloughis has been offered since September 2000. 223 Slougis were tested so far. Many additional sequence variations were identified in this gene. These variations were used as intragenic markers to exclude the PDE6B gene as cause of gPRA in 16 other dog breeds. Furthermore, sequence variations (SNPs, deletions, insertions) were used for excluding ar transmitted gPRA 10 other in nuclear genes. These intragenic markers lead to the exclusion in several breeds as cause for gPRA: RPE65 gene in 28 breeds; GNGT, PDC, PDE6A genes each in 11 breeds; PDE6D and PDE6G genes each in 15 breeds, the CRX and RCV1 gene seach in 16 breeds. Finally, for ad transmitted gPRA the RCV1 gene is excluded in 6 breeds. Additional studies of this kind should aid in eradicating this devastating eye disease in dogs. On the other hand such genetic models are thoroughly characterized of eminent value for similar maladies of mankind. 106 Kapitel 15 Literatur Kapitel 15 Literatur Acland GM, Aguirre GD. (1987) Retinal degenerations in the dog: IV. Early retinal degeneration (erd) in Norwegian elkhounds. Exp Eye Res 44:491-521. Acland GM, Blanton SH, Hershfield B, Aguirre GD. (1994) XLPRA A canine retinale degeneration inherited as an X-linked trait. Am J Med Genet 52:27-33. Acland GM, Ray K, Mellersh CS, Gu W, Langston AA, Rine J, Ostrander EA, Aguirre GD. (1998) Linkage analysis and comparative mapping of canine progressive rod-cone degeneration (prcd) establishes potential locus homology with retinitis pigmentosa (RP17) in humans. Proc Natl Acad Sci USA 95:3048-3053. Acland GM, Ray K, Mellersh CS, Langston AA, Rine J, Ostrander EA, Aguirre GD. (1999) A novel retinal degeneration locus identified by linkage and comparative mapping of canine early retinal degeneration. Genomics 59:134-142. Acland GM, Aguirre GD, Ray J, Zhang Q, Aleman TS, Cideciyan AV, Pearce-Kelling SE, Anand V, Zeng Y, Maguire AM, Jacobson SG, Hauswirth WW, Bennett J. (2001) Gene therapy restores vision in a canine model of childhood blindness. Nature Genet 28:92-95. Aguirre GD. (1978) Retinal degenerations in the dog. I. Rod dysplasia. Exp Eye Res 26:233253. Aguirre GD, Faber D, Lolley R, O`Brien P, Alligood J, Fletcher RT, Chader G. (1982) Retinal degenerations in the dog: III abnormal cyclic nucleotide metabolism in rod-cone dysplasia. Exp Eye Res 35:625-542. Aguirre GD, Acland GM. (1988) Variation in retinal degeneration phenotype inherited at the prcd locus. Exp Eye Res 46:663-687. Aguirre GD, Baldwin V, Pearce-Kelling S, Narfstrom K, Ray K, Acland GM. (1998) Congenital stationary night blindness in the dog: common mutation in the RPE65 gene indicates founder effect. Mol Vis 4:23. Aguirre GD. (2001) Genes and diseases in man and models. In: Concepts and challenges in retinal biology. A tribute to John E. Dowling. (Eds. S. Wu, H. Ripps, R. Miller and H. Kolb) Prog Brain Res 131:663-678. Akhmedov NB, Piriev NI, Chang B, Rapoport AL, Hawes NL, Nishina PM, Nusinowitz S, Heckenlively JR, Roderick TH, Kozak CA, Danciger M, Davisson MT, Farber DB. 107 Kapitel 15 Literatur (2000) A deletion in a photoreceptor-specific nuclear receptor mRNA causes retinal degeneration in the rd7 mouse. Proc Natl Acad Sci USA 9:5551-5556. Akhmedov NB, Baldwin VJ, Zangerl B, Kijas JW, Hunter L, Minoofar KD, Mellersh C, Ostrander EA, Acland GM, Farber DB, Aguirre GD. (2002) Cloning and characterization of the canine photoreceptor specific cone-rod homeobox (CRX) gene and evaluation as a candidate for early onset photoreceptor diseases in the dog. Mol Vis 8:79-84. Alloway PG, Howard L, Dolph PJ. (2000) The formation of stable rhodopsin-arrestin complexes induces apoptosis and photoreceptor cell degeneration. Neuron 28:129-138. Barnett KC, Curtis R. (1985) Autosomal dominant progressive retinal atrophy in Abyssinian cats. J Hered 76:168-170. Bascom RA, Liu L, Heckenlively JR, Stone EM, McInnes RR. (1995) Mutation analysis of the ROM1 gene in retinitis pigmentosa. Hum Mol Genet 4:1895-1902. Bauer PH, Muller S, Puzicha M, Pippig S, Obermaier B, Helmreich EJ, Lohse MJ. (1992) Phosducin is a protein kinase A-regulated G-protein regulator. Nature 358:73-76. Bayes M, Valverde D, Balcells S, Grinberg D, Vilageliu L, Benitez J, Ayuso C, Beneyto M, Baiget M, Gonzalez-Duarte R. (1995) Evidence against involvement of recoverin in autosomal recessive retinitis pigmentosa in 42 Spanish families. Hum Genet 96:89-94. Bayes M, Giordano M, Balcells S, Grinberg D, Vilageliu L, Martinez I, Ayuso C, Benitez J, Ramos-Arroyo MA, Chivelet P, et al (1995) Homozygous tandem duplication within the gene encoding the beta-subunit of rod phosphodiesterase as a cause for autosomal recessive retinitis pigmentosa. Hum Mutat 5:228-234. Bentolila S, Bach JM, Kessler Jl, Bordelais I, Cruaud C, Weissenbach J, Panthier JJ. (1999) Analysis of major repetitive DNA sequences in the dog (canis familiaris) genome. Mam Genome 10:699-705. Bennett N, Clerc A. (1992) cGMP phosphodiesterase dependent light-induced scattering changes in suspensions of retinal disc membranes. Biochemistry 31:1858-1866. Blümel B. (1996) Mutationen in Kandidatengenen für progressive Retina-Atrophie beim Hund. Diplomarbei Fakultät für Biologie Ruhr-Universität Bochum. Bourgain C, Genin E, Clerget-Darpoux F. (2002) Comparison of family based haplotype methods using intragenic SNPs in candidate genes. Eur J Hum Genet 10:313-319. Bourne MC, , Campbell DA, Tansley K. (1938) Hereditary degeneration of the rat retina. Br J Ophthalmol 22:613-623. 108 Kapitel 15 Literatur Bowes C, Li T, Danciger M, Baxter LC, Applebury ML, Farber DB. (1990) Retinal degeneration in the rd mouse is cause by a defect in the β subunit of rod cGMPphosphodiesterase. Nature 347:677-680. Bowes C, Li T, Frankel WN, Danciger M, Coffin JM, Applebury ML, Farber DB. (1993) Localization of a retroviral element within the rd gene coding for the β subunit of cGMPphosphodiesterase. Proc Natl Acad Sci USA 90:2955-2959. Brown MD, Voljavec AS, Lott MT, Torroni A, Yang CC, Wallace DC. (1992) Mitochondrial DNA complex I and III mutations associated with Leber's hereditary optic neuropathy. Genetics 130:163-173. Buraczynska M, Wu W, Fujita R, Buraczynska K, Phelps E, Andreasson S, Bennett J, Birch DG, Fishman GA, Hoffman DR, Inana G, Jacobson SG, Musarella MA, Sieving PA, Swaroop A. (1997) Spectrum of mutations in the RPGR gene that are identified in 20% of familie with X-linked retinitis pigmentosa. Am J Hum Genet 61:1287-1292. Candille SI, Pardue MT, McCall MA, Peachey NS, Gregg RG. (1999) Localization of the mouse nob (no b-wave) gene to the centromeric region of the X chromosome. Invest Ophthalmol Vis Sci 40:2748-2751. Casey PJ. (1994) Lipid modifications of G proteins. Curr Opin Cell Biol 6 219-225. Chen S, Wang QL, Xu S, Liu I, Li LY, Wang Y, Zack DJ. (2002) Functional analysis of cone-rod homeobox (CRX) mutations associated with retinal dystrophy. Hum Mol Genet 11:873-884. Choi Y, Ishiguro N, Shinagawa M, Kim CJ, Okamoto Y, Minami S, Ogihara K. (1999) Molecular structure of canine LINE-1 elements in canine transmissible venereal tumor. Anim Genet 30:51-53. Church GM, Gilbert W. (1984) Genomic sequencing. Proc Natl Acad Sci USA 81:1991-1995. Clements PJM, Sargan DR, Gould DJ, Petersen-Jones SM. (1996) Recent advances in understanding the spectrum of canine generalised progressive retinal atrophy. J Small Animal Pract 37:155-162. Curtis R, Barnett KC, Leon A. (1987) An early-onset retinal dystrophy with dominant inheritance in the Abyssinian cat. Clinical and pathological findings. Invest Ophthalmol Vis Sci 8:131-139. D’Cruz PM, Yasumura D, Weir J, Matthes MT, Abderrahim H, LaVail MM, Vollrath D. (2000) Mutationen of the receptor tyrosine kinase gene Mertk in the retinal dystrophic RCS rat. Hum Mol Genet 9:645-651. 109 Kapitel 15 Literatur Danciger M, Blaney J, Gao YQ, Zhao DY, Heckenlively JR, Jacobson SG, Farber DB. (1995) Mutations in the PDE6B in autosomal recessive retinitis pigmentosa. Genomics 30:1-7. Dekomien G. (1997) Mutationssuche in Kandidatengenen für progressive Retina-Atrophie beim Hund auf Genomebene. Diplomarbeit, Fakultät für Biologie, Ruhr-Universität Bochum. Dekomien G, Klein W, Epplen JT. (1998) Polymorphisms in the canine rod transducin gene and exclusion as cause for generalised progressive retinal atrophy (gPRA). J Exp Anim Sci 39:86-90. Dekomien G, Epplen JT. (2000) Exclusion of the PDE6A gene for generalised progressive retinal atrophy in 11 breeds of dogs. Anim Genet 31:135-139. Dekomien G, Runte M, Gödde R, Epplen JT. (2000) Generalised progressive retinal atrophy of Sloughi dogs is due to an 8-bp insertion in exon 21 of the PDE6B gene. Cytogenet Cell Genet 90:261-267. Dekomien G, Epplen JT. (2002) Screening of the arrestin gene in dogs afflicted with generalized progressive retinal atrophy. BMC Genet 1-8. Dekomien G, Epplen JT. (2002) The canine Phosducin gene: characterization of the exonintron structure and exclusion as a candidate gene for generalized progressive retinal atrophy in 11 dog breeds. Mol Vis 8:138-142. Deterre P, Bigay J, Forquet F. Robert , Chabre M. (1988) cGMP phosphodiesterase of retinal rods is regulated by two inhibitory subunits. Proc Natl Acad Sci USA 85:2424-2428. Dryja TP, Li T. (1995) Molecular genetics of retinitis pigmentosa. Hum Mol Genet 4:17391743. Dryja TP. (1997) Gene-based approach to human gene-phenotype correlations. Proc Natl Acad Sci USA 94:12117-12121. Eckert R. Tierphysiologie Thieme-Verlag 3. Aufl. 2000. El-Amraoui A, Sahly I, Picaud S, Sahel J, Abitbol M, Petit C. (1996) Human Usher 1B/mouse shaker-1: the retinal phenotype discrepancy explained by the presence/absence of myosin VIIA in the photoreceptor cells. Hum Mol Genet 5:1171-1178. Ershova G; Derre J; Chetelin S; Nancy V; Berger R; Kaplan J; Munnich A, de Gunzburg J. (1997) cDNA sequence, genomic organization and mapping of PDE6D, the human gene encoding the delta subunit of the cGMP phosphodiesterase of retinal rod cells to chromosome 2q36. Cytogenet. Cell Genet 79:139-141. Farber DB. (1995) From Mice to Men: The cyclic GMP Phosphodiesterase gene in vision and disease. Invest Ophthalmol Vis Sci 36:263-375. 110 Kapitel 15 Literatur Farrar GJ, Kenna P, Jordan SA, Kumar-Singh R, Humphries MM, Sharp EM, Sheils DM, Humphries P. (991) A three-base-pair deletion in the peripherin-RDS gene in one form of retinitis pigmentosa. Nature 354:478-480. Farrar GJ, Jordan SA, Kumar-Singh R, Inglehearn CF, Gal A, Greggory C. (1993) Extensive genetic heterogenety in autosomal dominant retinitis pigmentosa. In Retinal degeneration J. Hollyfield, RE Anderson, MM LaVail. eds. (New York Plenum Press), pp 63-77. Florio SK, Prusti RK, Beavo JA. (1996) Solubilzation of membran-bound rod phosphodiesterase by the rod phosphodiesterase recombinant delta subunit. J Biol Chem 271:24036-24047. Frederick J, Bronson JD, Baehr W. (2000) Animal models of inherited retinal diseases. Methods Enzymol 316:515-526. Freund CL, Gregory-Evans CY, Furukawa T, Papaioannou M, Looser J, Ploder L, Bellingham J, Ng D, Herbrick JA, Duncan A, Scherer SW, Tsui LC, LoutradisAnagnostou A, Jacobson SG, Cepko CL, Bhattacharya SS, McInnes RR. (1997) Cone-rod dystrophy due to mutations in a novel photoreceptor-specific homeobox gene (CRX) essential for maintenance of the photoreceptor. Cell 91:543-553. Freund CL, Wang Q-L, Chen S, Muskat, BL, Wiles CD, Sheffield VC, Jacobson SG, McInnes RR, Zack DJ, Stone EM. (1998) De novo mutations in the CRX homeobox gene associated with Leber congenital amaurosis. Nature Genet 18:311-312. Fuchs S, Nakazawa M, Maw M, Tamai M, Oguchi Y, Gal A. (1995) A homozygous 1-base pair deletion in the arrestin gene is a frequent cause of Oguchi disease in Japanese. Nat Genet 10:360-362. Furukawa T, Morrow EM Cepko CL. (1997) Crx, a novel otx-like homeobox gene, shows photoreceptor-specific expression and regulates photoreceptor differentiation. Cell 91:531-541. Furukawa T, Morrow EM, Li T, Davis FC, Cepko CL. (1999) Retinopathy and attenuated circadian entrainment in Crx-deficient mice. Nature Genet 23:466-470. Gal A, Li Y, Thompson DA, Weir J, Orth U, Jacobson S G, Apfelstedt-Sylla E, Vollrath D. (2000) Mutations in MERTK, the human orthologue of the RCS rat retinal dystrophy gene, cause retinitis pigmentosa. Nature Genet 26:270-271. Gödde R. (2000) Mutationssuche in sechs Kandidatengenen für die generalisierte Progressive Retinaatrophie. Diplomarbeit, Fakultät für Biologie Ruhr-Universität Bochum. 111 Kapitel 15 Literatur Goliath R, Shugart Y, Janssens P, Weissenbach J, Beighton P, Ramasar R., Greenberg J. (1995) Fine localization of the locus for autosomal dominant retinitis pigmentosa on chromosome 17p. Am Hum J Genet 57:962-965. Gropp KE, Szel A, Huang JC, Acland GM, Farber DB, Aguirre GD. (1996) Selective absence of cone outer segment beta 3-transducin immunoreactivity in hereditary cone degeneration (cd). Exp Eye Res 63:285-296. Gropp KE, Huang JC, Aguirre GD. (1997) Differential expression of photoreceptor-specific proteins during disease and degeneration in the progressive rod-cone degeneration (prcd) retina. Gene 30:231-239. Gu S, Thompson D A, Srikumari CS, Lorenz B, Finckh U, Nicoletti A, Murthy KR, Rathmann M, Kumaramanickavel G, Denton M, Gal A. (1997) Mutations in RPE65 cause autosomal recessive childhood-onset severe retinal dystrophy. Nature Genet 17:194-197. Hofmann KP, Jäger S, Ernst OP. (1995) Structure and function of activated rhodopsin. Isr J Chem 35:339-355. Holt IJ, Harding AE, Petty RK, Morgan-Hughes JA. (1990) A new mitochondrial disease associated with mitochondrial DNA heteroplasmy. Am J Hum Genet 46:428-433. Huang SH, Pittler SJ. Huang X. Oliveira L. Berson EL. Dryja TP. (1995) Autosomal recessive retinitis pigmentosa caused by mutations in the alpha subunit of rod cGMP phosphodiesterase Nature Genet 11:468-471. Hudson RR, Kaplan NL. (1985) Statistical properties of the number of recombination events in the history of a sample of DNA sequences. Genetics 111:147-164. Jäckel S, Epplen JT, Kauth M, Miterski B, Tschentscher F, Epplen C. (1998) Polymerase chain reaction-single strand conformation polymorphism or how to detect reliably and efficiently each sequence variation in many samples and many genes. Electrophoresis 19:3055-3061. Jacobs GH. (1983) Colour vision in animal. Endeavour 7:137-140. Janke B. (1998) Mutationssuche in drei Kandidatengenen für die progressive Retinaatrophie. Diplomarbeit, Fakultät für Biologie, Ruhr-Universität Bochum. Kajiwara K, Hahn LB, Mukai S, Travis GH, Berson EL, Dryja TP. (1991) Mutations in the human retinal degeneration slow gene in autosomal dominant retinitis pigmentosa. Nature 354:480-483. Kajiwara K, Berson EL, Dryja TP. (1994) Digenic retinitis pigmentosa due to mutations at the unlinked peripherin/RDS and ROM1 loci. Science 264:1604-1607. Kellner U. (1997) Hereditäre Netzhautdystrophien. Ophthalmologe 94:164-183. 112 Kapitel 15 Literatur Kijas JW, Cideciyan AV, Aleman TS, Pianta MJ, Pearce-Kelling SE, Miller BJ, Jacobson SG, Aguirre GD, Acland GM. (2002) Naturally occurring rhodpsin mutation in the dog causes retinal dysfunction and degeneration mimicking human dominant retinits pigmentosa. Proc Natl Acad Sci USA 99:6328-6333. Kim KS, Lee SE, Jeong HW, Ha JH. (1998) The complete nucleotide sequence of the domestic dog (Canis familiaris) mitochondrial genome. Mol Phylogenet Evol 10:210-220. Kirschner R, Rosenberg T, Schultz-Heienbrok R, Lenzner S, Feil S, Roepman R, Cremers FP, Ropers HH, Berger W. (1999) RPGR transcription studies in mouse and human tissues reveal a retina-specific isoform that is disrupted in a patient with X-linked retinitis pigmentosa. Hum Mol Genet 8:1571-1578. Klein W, Dekomien G, Holmes N, Epplen JT. (1998) Evaluation of ROM1 as a candidate gene in generalised progressive retinal atrophy in dogs. Animal Genetics 29:316-318. Kloepfer HW, Laguaite JK. (1966) The hereditary syndrome of congenital deafness and retinitis pigmentosa. (Usher's syndrome). Laryngoscope 76:850-862. Kruglyak L. (1999) Prospects for whole-genome linkage disequilibrium mapping of common disease genes. Nat Genet 22:139-144. Kutuzov MA, Shmukler BE, Suslov ON, Dergachev AE, Zargarov AA, Abdulaev NG. (1991) P26-calcium binding protein from bovine retinal photoreceptor cells. FEBS Letts 293:2124. Kylma T, Paulin L, Hurwitz MY, Hurwitz RL, Kommonen B. (1997) Cloning of the cDNA encoding rod cGMP-phosphodiesterase alpha- and betasubunit from the retinal degenerate labrador retriever dog Res Vet Sci 62:293-296. Lamminen T, Majander A, Juvonen V, Wikstrom M, Aula P, Nikoskelainen E, Savontous ML. (1995) A mitochondrial mutation at nt 9101 in the ATP synthase 6 gene associated with deficient oxidative phosphorylation in a family with Leber hereditary optic neuroretinopathy. Am J Hum Genet 56:1238-1240. Laratta LJ, Sims MH, Brooks DE. (1988) Progressive retinal degeneration in the Australien cattle dog. Proc Am Coll Vet Ophthal 19:9. Lee RH, Lieberman BS, Lolley RN. (1990) Retinal accumulation of the phosducin/T beta gamma and transducin complexes in developing normal mice and in mice and dogs with inherited retinal degeneration. Exp Eye Res 51:325-333. Lee RH, Ting TD, Lieberman BS, Tobias DE, Lolley DN, Ho YK. (1992) Regulation of retinal cGMP cascade by phosducin in bovine rod photoreceptor cells: Interaction of phosducin and transducin. J Biol Chem 267:25104-25112. 113 Kapitel 15 Literatur Leslie AG, Abrahams JP, Braig K, Lutter R, Menz RI, Orriss GL, van Raaij MJ, Walker JE. (1999) The structure of bovine mitochondrial F1-ATPase: an example of rotary catalysis. Biochem Soc Trans 27:37-42. Li N, Florio SK, Pettenati MJ. (1998) Characterization of human and mouse rod cGMPphosphodiesterase delta subunit (PDE6D) and chromosomal localization of the human gene. Genomics 49:76-82. Lin CT, Petersen-Jones SM, Sargan DR. (1998) Isolation and investigation of canine phosducin as a candidate for canine generalized progressive retinal atrophies. Invest Ophthalmol Vis Sci 40:1530-1544. Liu X, Ondek B, Williams DS. (1998) Mutant myosin VIIa causes defective melanosome distribution in the RPE of shaker-1 mice. Nature Genet 19:117-118. Lorenz B, Migliaccio C, Lichtner P, Meyer C, Strom, TM, D'Urso M, Becker J, Ciccodicola A, Meitinger T. (1998) Cloning and gene structure of the rod cGMP phosphodiesterase delta subunit gene (PDED) in man and mouse. Eur J Hum Genet 6:283-290. Mansergh FC, Millington-Ward S, Kennan A, Kiang AS, Humphries M, Farrar GJ, Humphries P, Kenna PF. (1999) Retinitis pigmentosa and progressive sensorineural hearing loss caused by a C12258A mutation in the mitochondrial MTTS2 gene. Am J Hum Genet 64:971-985. McGinnis J, Stepanik PL, Baehr W, Subbaraya I, Lerious V. (1992) Cloning and sequencing of the 23 kDa mouse photoreceptor cell-specific RT protein. FEBS Letts 302:172-176. McLaughlin ME, Ehrhard TL, Berson EL, Dryja TP. (1993) Recessive mutations in the gene encoding the beta-subunit of rod phosphodiesterase in patients with retinitis pigmentosa. Nature Genet 4:130-134. Miller PE, Murphy CJ. (1995) Vision in dogs. J Am Vet Assoc 207:1623-1634. Miller SA, Dykes DD, Polesky HF. (1988) A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res 16:1215. Mitton KP, Swain PK, Chen S, Xu S, Zack DJ, Swaroop A. (2000) The leucine zipper of NRL interacts with the CRX homeodomain. A possible mechanism of transcriptional synergy in rhodopsin regulation. J Biol Chem 275:29794-29799. Morimura H, Fishman GA, Grover SA, Fulton AB, Berson EL, Dryja TP. (1998) Mutations in the RPE65 gene in patients with autosomal recessive retinitis pigmentosa or leber congenital amaurosis. Proc Natl Acad Sci USA 95:3088-3093. 114 Kapitel 15 Literatur Morimura H, Saindelle-Ribeaudeau F, Berson El, Dryja TP. (1999) Mutations in RGR, encoding a light-sensetiv opsin homologue, in patients with retinits pigmentosa. Nat Genet 23:393. Murakami A, Yajima T, Inana G. (1992). Isolation of human retinal genes: recoverin cDNA and gene. Biochem Biophys Res Commun 187:234-244. Nakazawa M, Wada Y, and Tamai M. (1998) Arrestin gene mutations in autosomal recessive retinitis pigmentosa. Arch Ophthalmol 116:498-501. Narfström K, Nilsson SE. (1989) Morphological findings during retinal development and maturation in hereditary rod-cone degeneration in Abyssinian cats. Exp Eye Res 49:611628. Neer EJ. (1995) Heterotrimeric G proteins: organizers of transmembrane signals. Cell 80:249257. Neitz J, Geist T, Jacobs GH. (1989) Color vision in the dog. Vis Neurosci 3:119-125. Odom JV, Bromberg NM, Dawson WW. (1983) Canine visual acuity: retinal and cortical field potentials evoked by pattern stimulation. Am J Physiol 245:637-641. Ostrander EA, Galibert F, Patterson DF. (2000) Canine genetics come of age. Canine Genet 16:117-124. Palczewski K and Saari JC. (1997) Activation and inactivation steps in the visual pathway. Curr Opin Neurobiol 7:500-504. Parminder AH, Murakami A, Inana G, Berson EL, Dryja TP. (1997) Evaluation of the human gene encoding recoverin in patients with retinitis pigmentosa or an allied disease. Invest Ophthalmol Vis Sci 38:704-709. Peichl L. (1992) Topography of ganglion cells in the dog and wolf retina. J Comp Neurol 324:603-620. Peng YW, Robishaw JD, Levine MA, Yau KW. (1992) Retinal rods and cones have distinct G protein beta and gamma subunits. Proc Natl Acad Sci USA 89:10882-10886. Petersen-Jones SM. (1998) A Review of research to elucidate the cause of the generalized progressive retinal atrophies. Vet J 155:5-18. Petersen-Jones SM, Entz DD, Sargan DR. (1999) cGMP phosphodiesterase-α mutation causes progressive retinal atrophy in the cardigan welsh corgi dog. Invest Ophthalmol Vis Sci 40:1637-1644. Pittler SJ, Baehr W, Wasmuth JJ, McConnell DG, Champagne MS, Tuinen P, Ledbetter D, Davis LR. (1990) Molecular Characterisation of Human of Human and Bovine Rod 115 Kapitel 15 Literatur Photoreceptor cGMP Phosphodiesterase α-Subunit and Chromosomal Localisation of the Human Gene. Genomics 6:272-283. Pittler SJ, Baehr W. (1991) Identification of a nonsense mutation in the rod photoreceptor cGMP phosphodiesterase beta-subunit gene of the rd mouse. Proc Natl Acad Sci USA 88:8322-8326. Pugh EN, Nikonov S, Lamb TD. (1999) Molecular Mechanisms of Vertebrate Photoreceptor Light Adaptation. Curr Opin Neurobiol 9:410-418. Rando RR. (1992) Molecular mechanisms in visual pigment regeneration. Photochem Photobiol 56:1145-1156. Rattner A., Sun H, Nathans J. (1999) Molecular Genetics of human retinal disease. Ann Rev Genet 33:89-131. Ray K, Baldwin VJ, Acland GM, Blanton SH, Aguirre GD. (1994) Cosegregation of codon 807 mutation of the canine rod cGMP phosphodiesterase beta gene and rcd1. Invest Ophthalmol Vis Sci 35:4291-4299. Redmond TM, Yu S, Lee E, Bok D, Hamasaki D, Chen N, Goletz P, Ma JX, Crouch RK, Pfeifer K. (1998) Rpe65 is necessary for production of 11-cis-vitamin A in the retinal visual cycle. Nat Genet 20:344-351. Reich DE, Schaffner SF, Daly MJ, McVean G, Mullikin JC, Higgins JM, Richter DJ, Lander ES, Altshuler D. (2002) Human genome sequence variation and the influence of gene history, mutation and recombination. Nature Genet 32:135-142. Rivolta C, Peck NE, Fulton AB, Fishman GA, Berson EL, Dryja TP. (2001) Novel frameshift mutations in CRX associated with Leber congenital amaurosis. Hum Mutat 18:550-551 Rivolta C, Sharon D, DeAngelis MM, Dryja TP. (2002) Retinitis pigmentosa and allied diseases, genes and inheritance patterns. Hum Mol Genet 11:1219-1227. Rodieck RW. (1998) The first steps in seeing. Sunderland MA; Sinauer. 562 pp. Runte M. (1999) Klonierung des RDS/Peripherin-Gens und Mutationssuche in diesem und weiteren Kandidatengenen für die Progressive Retinaatrophie. Diplomarbeit, Fakultät für Biologie, Ruhr-Universität Bochum. Runte M, Dekomien G, Epplen JT. (2000) Evaluation of RDS/Peripherin and ROM1 as candidate genes in generalised progressive retinal atrophy and exclusion of digenic inheritance. Anim Genet 31:223-237. Saiki RK, Gelfand DH, Stoffel S, Scharf SJ, Higuchi R, Horn GT. (1988) Primer-directed enzymatic amplification of DNA with termostable DNA polymerase. Science 239:487491. 116 Kapitel 15 Literatur Sambrook J, Fritsch EF, Maniatis T. (1989) Molecular cloning: A laboratory Manual. 2nd ed. Cold Spring Harbor (NY); Harbor Press. Sanger F, Nicklen S, Coulson AR. (1977) DNA-sequencing with chain-termination inhibitors. Proc Natl Acad Sci USA 74:5463-5467. Schaad NC, Shinohara T, Abe T, Klein DC. (1991) Photoneural control of the synthesis and phosphorylation of pineal MEKA (phosducin) Endocrinology 129:3289-3298. Schmidt RF. (1998) Neuro- und Sinnesphysiologie. Springer-Verlag 3.Aufl. Scherer SW, Feinstein DS, Oliveira L, Tsui LC, Pittler SJ. (1996) Gene structure and chromosome localization to 7q21.3 of the human rod photorezeptor transducin gammasubunit gene (GNGT1). Genomics 35:241-243. Scott J, Fuller J. (1965) Genetics and the Social Behavior of the Dog. University of Chicago Press. Semple-Rowland SL, Lee NR, Van Hooser JP, Palczewski K, Baehr W. (1998) A null mutation in the photoreceptor guanylate cyclase gene causes the retinal degeneration chicken phenotype. Proc Natl Acad Sci USA 95:1271-1276. Sharon D, Blackshaw S, Cepko CL, Dryja TP. (2002) Profile of the genes expressed in the human peripheral retina, macula, and retinal pigment epithelium determined through serial analysis of gene expression (SAGE). Proc Natl Acad Sci USA 99:315-320. Sharp PA. (1998) Speculations on RNA splicing. Cell 23:643-646. Shastry BS. (1997) Signal transduction in the retina and inherited retinopathies. Cell Mol Life Sci 53:419-429. Sherman SM, Wilson JR. (1975) Behavioral and morphological evidence for binocular competion in the postnatal development of the dog’s visual system. J Comp Neurol 161:183-195. Shoffner JM, Lott MT, Lezza AM, Seibel P, Ballinger SW, Wallace DC. (1990) Myoclonic epilepsy and ragged-red fiber disease (MERRF) is associated with a mitochondrial DNA tRNA(Lys) mutation. Cell 61:931-937. Sidjanin DJ, Lowe JK, McElwee JL, Milne BS, Phippen TM, Sargan DR, Aguirre GD, Acland GM, Ostrander EA. (2002) Canine CNGB3 mutations establish cone degeneration as orthologous to the human achromatopsia locus ACHM3. Hum Mol Genet 11:18231833. Sippel KC, DeStefano JD, Berson EL, Dryja TP. (1998) Evaluation of the human arrestin gene in patients with retinitis pigmentosa and stationary night blindness. Invest Ophthalmol Vis Sci 39:665-670. 117 Kapitel 15 Literatur Sohocki MM, Daiger SP, Bowne SJ, Rodriquez JA, Northrup H. Heckenlively JR, Birch GD, Mintz-Hittner H, Ruitz SR, Lewis AR, Saperstein AD, Sullivan SL. (2001) Prevalence of Mutations Causing Retinitis Pigmentosa and Other Inherited Rethinopathies. Hum Mutat 17:42-51. Spiess BM. (1994) Inherited ocular diseases in the Entlebucher Mountain Dog. Schweiz Arch Tierheilk 136:105-110. Spinardi L, Mazars R, Theillet C. (1991) Protocols for an improved detection of point mutations by SSCP. Nucleic Acids Res 19:4009. Strachan T, Read AP. (1996) Molekulare Humangenetik Spektrum Akad. Verlag. Stryer L. (1995) Biochemistry. Freeman and Company, New York 4th ed. Stryer L. (1996) Vision: From photon to perception. Proc Natl Acad Sci USA 93:557-559. Suber ML, Pittler SJ, Qin N, Wright GC, Holcombe V, Lee RH, Craft CM, Lolley RN, Baehr W, Hurwitz RL. (1993) Irish setter dogs affected with rod/cone dysplasia contain a nonsense mutation in the rod cGMP Phosphodiesterase beta-subunit gene. Proc Natl Acad Sci USA 90:3968-3972. Swain PK, Chen S, Wang QL, Affatigato LM, Coats CL, Brady KD, Fishman GA, Jacobson SG, Swaroop A, Stone E, Sieving PA, Zack DJ. (1997) Mutations in the cone-rod homeobox gene are associated with the cone-rod dystrophy photoreceptor degeneration. Neuron 19:1329-1336. Swaroop A, Wang QL, Wu W, Cook J, Coats C, Xu S, Chen S, Zack DJ, Sieving PA. (1999) Leber congenital amaurosis caused by a homozygous mutation (R90W) in the homeodomain of the retinal transcription factor CRX: direct evidence for the involvement of CRX in the development of photoreceptor function. Hum Mol Genet 8:299-305. Tanaka T, Ames JB, Harvey TS, Stryer L, Ikura M. (1995) Sequestration of the membranetargeting myristoyl group of recoverin in the calcium-free state. Nature 376:444-447. Tao L, Pandey S, Simon MI, Fong HK. (1993) Structure of the bovine transducin gamma subunit gene and analysis of promoter function in transgenic mice. Exp Eye Res 56:497507. Tarttelin EE, Plant C, Weissenbach J, Bird AC, Bhattacharya SS, Inglehearn CF. (1996) A new family linked to the RP13 locus for autosomal dominant retinitis pigmentosa an distal 17p. J Med Genet 33:518-520. Tatuch Y, Christodoulou J, Feigenbaum A, Clarke JT, Wherret J, Smith C, Rudd N, PetrovaBenedict R, Robinson BH. (1992) Heteroplasmic mtDNA mutation (T-to-G) at 8993 can 118 Kapitel 15 Literatur cause Leigh disease when the percentage of abnormal mtDNA is high. Am J Hum Genet 50:852-858. Thompson DA, Gyürüs P, Fleischer L, Bingham EL, McHenry CL, Apfelstedt-Sylla E, Zrenner E, Lorenz B, Richards JE, Jacobson SG, Sieving PA, Gal A. (2000) Genetics and phenotypes of RPE65 mutations in inherited retinal degeneration. Invest Ophthalmol Vis Sci 41:4293-4299. Thulin CD, Howes K, Driscoll CD, Savage JR, Rand TA, Baehr W, Willardson BM. (1999) The immunolocalization and divergent roles of phosducin and phosducin-like protein in the retina. Mol Vis 5:40. Travis GH, Brennan MB, Danielson PE, Kozak CA, Sutcliffe JG. (1989) Identification of a photoreceptor-specific mRNA encoded by the gene responsible for retinal degeneration slow (rds). Nature 338:70-73 Tsang SH, Yamashita C, Tanabe T. (1996) Retinal degeneration in mice lacking the gamma subunit of the rod cGMP phosphodiesterase. Science 272:1026-1029. Veske A, Narfstrom K, Finckh U, Sargan DR, Nilsson SE, Gal A. (1997) Isolation of canine retinal arrestin cDNA and exclusion of three candidate genes for Swedish Briard retinal dystrophy. Curr Eye Res 16:270-274. Veske A, Nilsson SE, Narfstrom K, Gal A. (1999) Retinal dystrophy of Swedish briard/briard-beagle dogs is due to a 4-bp deletion in RPE65. Genomics 57:57-61. Vignal A, Milan D, SanCristobal M, Eggen A. (2002) A review on SNP and other types of molecular markers and their use in animal genetics. Genet Sci Evol 34:275-305. Vishnivetskiy SA, Paz CL, Schubert C, Hirsch JA, Sigler PB, Gurevich VV. (1999) How does arrestin respond to the phosphorylated state of rhodopsin? J Biol Chem 274:11451-11454. Wang W, Acland GM, Ray K, Aguirre GD. (1999) Evaluation of cGMP-phosphodiesterase (PDE) subunits for causal association with rod-cone dysplasia 2 (rcd2), a canine model of abnormal retinal cGMP metabolism. Exp Eye Res 69:445-453. Wang W, Zhang Q, Acland GM, Mellersh C, Ostrander EA, Ray K, Aguirre GD. (1999) Molecular characterization and mapping of canine cGMP-phosphodiesterase delta subunit (PDE6D). Gene 236:325-332. Watanabe Y, Kawasaki K, Miki N, Kuo CH. (1990) Isolation and analysis of the human gene encoding a retina-specific protein. Biochem Biophys Res Commun 170:951-956. Wayne RK. (1999) Molecular evolution of the dog. Trends Genet 9:218-224. Weber B, Riess O, Hutchinson G, Collins C, Lin BY, Kowbel D, Andrew S, Schappert K, Hayden MR. (1991) Genomic organization and complete sequence of the human gene 119 Kapitel 15 Literatur encoding the beta-subunit of the cGMP phosphodiesterase and its localisation to 4p 16.3. Nucleic Acids Res 19:6263-6268. Weleber and Gregory-Evens K. (2001) Retinits pigmentosa and allied disorders. In Ryan SJ. (ed.) Retina. Mosby, St. Louis pp 362-470. Wibrand F, Ravn K, Schwartz M, Rosenberg T, Horn N, Vissing J. (2001) Multisystem disorder associated with a missense mutation in the mitochondrial cytochrome b gene. Ann Neurol 50:540-543. Wilkie TM, Gilbert DJ, Olsen AS, Chen XN, Amatruda TT, Korenberg JR, Trask BJ, de Jong P, Reed RR, Simon MI, Jenkins NA, Copeland NG. (1992) Evolution of the mammalian G protein alpha subunit miltigene family. Nature Genet 1:85-91. Wilson CJ, Applebury ML. (1993) Arresting G-protein coupled receptor activity. Curr Biol 3:683-686. Wrigstad A, Nilsson SE, Narfstrom K. (1992) Ultrastructural changes of the retina and the retinal pigment epithelium in Briard dogs with hereditary congenital night blindness and partial day blindness. Exp Eye Res 55:805-818. Yamaki K, Tsuda M, Kikuchi T, Chen KH, Huang KP, Shinohara T. (1990) Structural organization of the human S-antigen gene: cDNA, amino acid, intron, exon, promoter, in vitro transcription, retina, and pineal gland. J Biol Chem, 265:20757-20762. Yamamoto S, Sippel KC, Berson EL, Dryja TP (1997) Defects in the rhodopsin kinase gene in the Oguchi form of stationary night blindness. Nat Genet 15:175-178. Yang F, O'Brien PC, Milne BS, Graphodatsky AS, Solanky N, Trifonov V, Rens W, Sargan D, Ferguson-Smith MA. (1999) A complete comparative chromosome map for the dog, red fox, and human and its integration with canine genetic maps. Genomics 62:189-202. Yau K-W. (1994) Photransduction mechanism in retinal rods and cones. Invest Ophthalmol Vis Sci 35:9-32. Zeiss CJ, Ray K, Acland GM, Aguirre GD. (2000) Mapping of X-linked progressive retinal atrophy (XLPRA), the canine homolog of retinitis pigmentosa 3 (RP3). Hum Mol Genet 9:531-537. Zhang Q, Acland GM, Parshall CJ, Haskell J, Ray K, Aguirre GD. (1998) Characterization of canine photoreceptor phosducin cDNA and identification of a sequence variant in dogs with photoreceptor dysplasia. Exp Eye Res 67:473-480. Zhang Q, Acland GM, Zangerl B, Johnson JL, Mao Z, Zeiss CJ, Ostrander EA, Aguirre GD. (2001) Fine mapping of canine XLPRA establishes homology of the human and canine RP3 intervals. Invest Ophthalmol Vis Sci 42:2466-2471. 120 Danksagung Danksagung Ich möchte mich bei allen Mitgliedern der Abteilung Humangentik der medizinischen Fakultät der Ruhr-Universität Bochum für die angenehme Arbeitsatmosphäre und Hilfsbereitschaft bedanken, die erheblich zum Gelingen dieser Arbeit beigetragen haben. Mein besonderer Dank gilt Herrn Prof. Dr. Jörg T. Epplen für das interessante Forschungsthema und seine uneingeschränkte Unterstützung und lehrreichen Diskussionen und sein stetes Interesse am Fortgang der Arbeit. Prof. Dr. Andreas Faissner danke ich vielmals für die bereitwillige Übernahme des Zweitgutachtens. Für die freundschaftliche Atmosphäre und vielfältige Unterstützung möchte ich mich noch einmal besonders bedanken bei Gudrun Rodepeter, Frauke Albustin, Dr. Erdmute Kunstmann, Dr. Wolfram Klein, Dr. Christof Hammans, Michaela Hagedorn, Natascha Dürig, Rene Gödde, Dr. Martin & Alexandra Gencik, Peter Jagiello und Gabi Vogel. Ich danke den Tierärzten des DOK für die gPRA-Diagnostik, den vielen Hundebesitzern und Vorsitzenden der Hundevereine, die meine Arbeit durch Blutproben und Stammbauminformationen massgeblich unterstützt haben. Zusätzlich danke ich der kynologischen Forschungsgesellschaft (GKF), die das Projekt finanziell förderte. Für die moralische Unterstützung und Organisation vieler familiärer Angelegenheiten, ohne die diese Arbeit gar nicht möglich gewesen wäre, danke ich ganz besonders meinem Ehemann Klaus Dekomien. 121 Lebenslauf Lebenslauf Gabriele Dekomien geb. Kastin Saturnstr. 31 44388 Dortmund Geburtsdatum: 29.09.1954 Geburtsort: Hamm Staatsangehörigkeit: deutsch 1961 - 1972 Volksschule/Hauptschule 1989 - 1992 Abendgymnasium der Stadt Dortmund Abschluss: Allgemeine Hochschulreife 1992 - 1997 Studium der Biologie an der Ruhr-Universität Bochum Diplomarbeit in der Abteilung für Humangentik der Medizinischen Fakultät Ruhr-Universität Bochum unter der Leitung von Herrn Prof. Dr. Jörg T. Epplen Titel: Mutationssuche in Kandidatengenen für progressive Retina-Atrophie beim Hund auf Genomebene 09.01.1998 Abschluss: Diplom-Biologin 01.1998-10.2002 Doktorarbeit unter der Leitung von Herrn Prof. Dr. Jörg T. Epplen in der Abteilung für Humangentik der Medizinischen Fakultät Ruhr-Universität Bochum 122 Anhang Anhang Teilergebnisse der Dissertation wurden veröffentlicht in: Animal Genetics 2000 31:135-139 Dekomien G, Epplen JT. Exclusion of the PDE6A gene for generalised progressive retinal atrophy in 11 breeds of dog. Cytogenetics and cell genetics 2000 90:261-267 Dekomien G, Runte M, Gödde R, Epplen JT. Generalized progressive retinal atrophy of Sloughi dogs is due to an 8-bp insertion in exon 21 of the PDE6B gene. Molecular vision 2002 8:138-142 Dekomien G, Epplen JT. The canine Phosducin gene: characterization of the exon-intron structure and exclusion as a candidate gene for generalized progressive retinal atrophy in 11 dog breeds. (BioMed Central) BMC Genetics 2002 3:12 Dekomien G, Epplen JT. Screening of the arrestin gene in dogs afflicted with generalized progressive retinal atrophy. Molecular vision 2002 8:436-441 Dekomien G, Epplen JT. The canine Recoverin (RCV1) gene: a candidate gene for generalized progressive retinal atrophy. Genetics Selection Evolution (GSE) (2003) in press Dekomien G, Epplen JT. Analysis of PDE6D and PDE6G genes for gPRA mutations in dogs 123