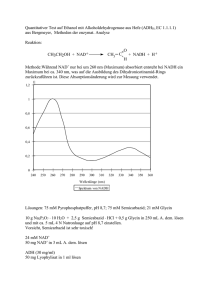
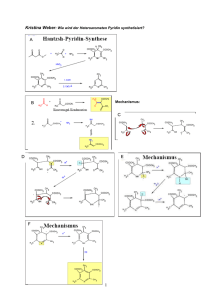
ZIP - FreiDok plus
Werbung

Funktionelle und strukturelle Charakterisierung von Varianten des respiratorischen Komplex I INAUGURALDISSERTATION zur Erlangung des Doktorgrades der Fakultät für Chemie und Pharmazie der Albert-Ludwigs-Universität Freiburg vorgelegt von Diplom-Chemikerin Klaudia Morina aus Freiburg Freiburg im Breisgau, 2014 Vorsitzender des Promotionsausschusses: Prof. Dr. Thorsten Koslowski Referent: Prof. Dr. Thorsten Friedrich Korreferent: Datum der mündlichen Prüfung: Prof. Dr. Oliver Einsle 02.04.2014 Teile der in dieser Arbeit vorgestellten Ergebnisse wurden in folgenden Artikeln veröffentlicht: Morina, K., Schulte, M., Hubrich, F., Dörner, K., Steimle, S., Stolpe, S., und Friedrich, T. (2011). Engineering the Respiratory Complex I to Energy-converting NADPH:Ubiquinone Oxidoreductase. J. Biol. Chem. 286, 34627–34634. Birrell, J.A., Morina, K., Bridges, H.R., Friedrich, T., und Hirst, J. (2013). Investigating the function of [2Fe-2S] cluster N1a, the off-pathway cluster in complex I, by manipulating its reduction potential. Biochem. J. 456, 139–146. per Nurijen dhe Salihin Inhaltsverzeichnis 1 Zusammenfassung ........................................................................................ 1 2 Einleitung ...................................................................................................... 4 3 2.1 Die aerobe Atmungskette von Escherichia coli ....................................................... 4 2.2 Die NADH:Ubichinon Oxidoreduktase ................................................................... 6 2.3 Elektronentransfer im Komplex I .......................................................................... 10 2.4 Die Fe/S-Zentren N1a und N7 ................................................................................ 13 2.5 Die NADH-Bindestelle des Komplex I ................................................................... 18 2.6 λ-Red vermittelte Rekombination .......................................................................... 23 2.7 Ziel der Arbeit .......................................................................................................... 26 Material und Methoden ............................................................................. 28 3.1 Chemikalien ............................................................................................................. 28 3.2 Stämme ..................................................................................................................... 31 3.2 Verwendete Vektoren .............................................................................................. 31 3.3 DNA-Oligonukleotide .............................................................................................. 35 3.4 Medien, Lösungen und DNA-modifizierende Enzyme ......................................... 38 3.4.1 Nährmedien ........................................................................................................ 38 3.4.2 Antibiotika-Lösungen ......................................................................................... 40 3.4.3 Lösungen und Puffer für die Molekularbiologie ................................................ 40 3.4.4 SDS-Polyacrylamid-Gelelektrophorese und Western Blot ................................ 40 3.4.5 Puffer und Lösungen für die Proteinbiochemie ................................................. 42 3.4.6 DNA-modifizierende Enzyme ............................................................................ 44 3.5 Mikrobiologische Methoden ................................................................................... 45 3.5.1 Bestimmung der optischen Dichte ..................................................................... 45 3.5.2 Vorkulturen ........................................................................................................ 45 3.5.3 Glycerin-Dauerkulturen ..................................................................................... 45 3.5.4 Herstellung elektrokompetenter Zellen .............................................................. 45 3.5.5 Bakterienanzucht ................................................................................................ 46 3.5.6 Überproduktion des E. coli Komplex I .............................................................. 46 3.5.7 Expressionstests ................................................................................................. 47 3.5.8 Überproduktion des A. aeolicus NuoEFHis ......................................................... 47 3.6 3.6.1 Plasmidpräparation ............................................................................................. 48 3.6.2 Reinigung von DNA........................................................................................... 49 3.6.3 DNA-Konzentrationsbestimmung ...................................................................... 49 3.6.4 Restriktionsanalysen und DpnI-Verdau ............................................................. 49 3.6.5 Polymerasekettenreaktion .................................................................................. 49 3.6.6 Agarose-Gelelektrophorese ................................................................................ 51 3.6.7 Klonierung .......................................................................................................... 52 3.6.8 Transformation durch Elektroporation ............................................................... 52 3.6.9 Sequenzierung von Plasmiden ........................................................................... 53 3.7 Proteinchemische Methoden ................................................................................... 53 3.7.1 Isolierung der E. coli Cytoplasmamembranen ................................................... 53 3.7.2 Isolierung des E. coli Komplex I ........................................................................ 53 3.7.3 Isolierung des A. aeolicus NuoEFHis .................................................................. 54 3.7.4 Bestimmung der Proteinkonzentration ............................................................... 56 3.7.5 Aktivitätsmessungen .......................................................................................... 57 3.7.6 SDS-Polyacrylamid-Gelelektrophorese ............................................................. 59 3.7.7 Western Blot-Analyse ........................................................................................ 60 3.7.8 Saccharosedichtegradienten-Zentrifugation ....................................................... 61 3.8 Kristallographische Methoden ............................................................................... 61 3.8.1 Kristallisation der A. aeolicus NuoEFHis-Varianten ........................................... 61 3.8.2 Herstellung von Keimkristallen ......................................................................... 62 3.9 4 Molekularbiologische Methoden ............................................................................ 48 EPR-Spektroskopie ................................................................................................. 62 Ergebnisse ................................................................................................... 64 4.1 Substratunterscheidung am Komplex I ................................................................. 64 4.1.1 Darstellung der Mutanten pBADnuo nuoFhis E183A/D/G/H/N und Q.............. 65 4.1.2 Komplex I-vermittelte Aktivitäten der Cytoplasmamembranen ........................ 67 4.1.3 Präparation des Komplex I und der Varianten E183A/D/G/H/N und QF .......... 69 4.1.4 4.2 Charakterisierung der Varianten E183A/D/G/H/N und QF ................................ 72 Strukturelle Untersuchung der Substratdiskriminierung ................................... 79 4.2.1 Optimierung der heterologen Expression von A. aeolicus nuoEF in E. coli...... 79 4.2.2 Darstellung der E185F-Varianten des A. aeolicus Subkomplexes NuoEF ......... 83 4.2.3 Kristallisation der Varianten E185XF................................................................. 86 4.2.4 Strukturen der Varianten E185DF und E185HF.................................................. 89 4.2.5 Struktur der Variante E185DF mit gebundenem NAD+ ..................................... 93 4.3 Die Position Tyr178F im E. coli Komplex I ........................................................... 98 4.3.1 Darstellung der Komplex I-Varianten Y178A/C/F und LF ................................ 99 4.3.2 Charakterisierung der Komplex I-Varianten Y178A/C/F und LF ...................... 99 4.4 Die Beteiligung von Trp221G am Elektronentransfer ........................................ 101 4.4.1 Darstellung der Doppelmutanten...................................................................... 103 4.4.2 Komplex I-vermittelte Aktivitäten der Cytoplasmamembranen ...................... 104 4.5 Der E. coli Stamm BW25113nptI Δnuo Δndh/pBADnuo nuoFhis ...................... 107 4.5.1 Oxidaseaktivitäten der Membranen.................................................................. 107 4.5.2 Überproduktion des E. coli Komplex I ............................................................ 111 4.6 Modulation des Mittenpunktspotentials von N1a............................................... 112 4.6.1 Darstellung von pBADnuo nuoFhis nuoE V96P, N142M und V96P/N142M.. 113 4.6.2 Präparation der Varianten V96PE, N142ME und V96P/N142ME .................... 114 4.7 Redox-induzierte Konformationsänderung des Glu95 auf NuoF ..................... 119 4.7.1 Darstellung der A. aeolicus NuoEFHis-Varianten G129DE und G129SE .......... 120 4.7.2 Strukturen der NuoEFHis-Varianten G129DE und G129SE .............................. 122 4.7.3 Darstellung der Komplex I-Varianten G135DE und G135SE ........................... 126 4.8 Die Oberflächen-Salzbrücke in NuoG ................................................................. 132 4.8.1 Darstellung der Mutante pBADnuo nuoFhis nuoG E617A............................... 132 4.8.2 Die Stabilität der Variante E617AG.................................................................. 133 4.9 Zuordnung des EPR-Signals N4 ........................................................................... 136 4.9.1 Darstellung der N4- und N5-Mutanten ............................................................ 138 4.9.2 Charakterisierung der N4- und N5-Varianten .................................................. 140 4.10 Mutationen der NADH-Bindestelle des A. aeolicus NuoEFHis ........................... 144 4.10.1 Darstellung der Mutanten ................................................................................. 145 5 Diskussion .................................................................................................. 146 5.1 Die durch Glu183F vermittelten Substratselektivität ......................................... 146 5.2 Die Position Tyr178F im E. coli Komplex I ......................................................... 148 5.3 Die Beteiligung von Trp221G am Elektronentransfer ........................................ 149 5.4 Überproduktion des E. coli Komplex I und des A. aeolicus NuoEFHis ............. 150 5.5 Modulation des Mittenpunktspotentials von N1a............................................... 152 5.6 Redox-induzierte Konformationsänderung des Glu95 auf NuoF ..................... 153 5.7 Oberflächen-Salzbrücke in NuoG ........................................................................ 156 5.8 Zuordnung des EPR-Signals N4 ........................................................................... 156 6 Literaturverzeichnis ................................................................................. 158 7 Anhang....................................................................................................... 173 7.1 H-Brücken Netzwerk um NAD+/NADH und FMN im A. aeolicus NuoEFHis... 173 7.2 Kristallisations-screens .......................................................................................... 178 7.3 Kristallographische Statistiken ............................................................................ 179 Zusammenfassung 1 Zusammenfassung Reaktive Sauerstoffspezies (reactive oxygen species; ROS) stehen im Verdacht an der Entstehung neurodegenerativer Erkrankungen, wie Morbus Parkinson und Morbus Alzheimer beteiligt zu sein. Zusätzlich wird ROS eine wesentliche Rolle beim Alterungsprozess zugeschrieben. Die NADH:Ubichinon Oxidoreduktase, auch respiratorischer Komplex I genannt, gilt als die bedeutendste Quelle von ROS im Mitochondrium. Der Enzymkomplex koppelt den Elektronentransfer von NADH auf Ubichinon mit der Translokation von Protonen über die Membran. Aufgrund seines einfacheren Aufbaus wird der bakterielle Komplex I als Modellsystem für den eukaryotischen Komplex angesehen. Der Escherichia coli Komplex I besteht aus 13 Untereinheiten, die mit NuoA bis NuoN bezeichnet werden und trägt neun Eisen-Schwefel (Fe/S)-Zentren sowie ein Flavinmononukleotid (FMN) als Kofaktoren. In dieser Arbeit wurden spezifische Fragen zum Mechanismus des Komplex I durch Charakterisierung ortsgerichteter Mutanten geklärt. Auswirkungen der Mutationen auf die enzymatische Aktivität wurden mit dem E. coli Komplex I untersucht. Die strukturellen Änderungen der Mutationen wurden, falls möglich, mit dem löslichen Subkomplex aus den Untereinheiten NuoE und F des Aquifex aeolicus Komplex I charakterisiert. NADPH ist ein schlechtes Substrat für den Komplex I, da der Rest Glu183F vermutlich die Bindung blockiert. Durch Mutagenese des Glu183F im E. coli Komplex I zu Ala, Asn, Asp, Gln, Gly und His wurde eine NADPH:Ubichinon Oxidoreduktase konstruiert. Dies wurde durch eine hohe katalytische Aktivität bei einer geringen ROS-Produktion gezeigt. Die katalytische Effizienz der Reaktion mit NADPH war bis zu 2400-fach erhöht, während die ROS-Produktion um mehr als 90% verringert wurde. Auch die Aktivität mit NADH war verbessert, allerdings auf Kosten einer erhöhten ROS-Produktion. Die Struktur der homologen NuoEF-Variante E185DF mit gebundenem NAD+ wurde bei einer Auflösung von 1.80 Å gelöst. Die Struktur zeigte eine veränderte Bindung des NAD+, wobei die AdenosinRibose um etwa 160° verkippt vorliegt, um sich an die neue Bindestelle anzupassen. Die veränderte Bindung erklärt sowohl die hohe Aktivität als auch die gesteigerte ROSProduktion bei der Umsetzung von NADH. Die Mutation des konservierten Tyr204 zu Cys der Untereinheit NDUFV1 wurde im Genom eines am Leigh-like-Syndrom erkrankten Patienten nachgewiesen. Dieses Tyrosin bildet eine H-Brücke mit einem Glutamat-Rest in der NADH-Bindetasche aus und stabilisiert damit die Bindung des NADHs. Die Auswirkung dieser Mutation auf den E. coli Komplex I wurde durch Mutagenese des homologen Tyr178F zu Ala, Cys, Leu und Phe untersucht. Die 1 Zusammenfassung katalytische Effizienz der Varianten war bis zu 80% verringert und die ROS-Produktion bis zu 13-fach erhöht. Der Verlust der H-Brücke verringert offensichtlich die Effizienz der Elektronenübertragung auf FMN. Zusätzlich erleichtert der geringe sterische Anspruch der eingeführten Aminosäuren die Diffusion von Sauerstoff in das aktive Zentrum. Beide Effekte führen zu einer erhöhten ROS-Produktion. Aufgrund des großen Abstands zwischen zwei Fe/S-Zentren wurde vermutet, dass das Trp221G am Elektronentransfer im Komplex I über einen hopping-Mechanismus beteiligt ist. Der Austausch von Trp221G gegen Leu verringerte die Komplex I-Aktivität in der Membran um etwa ein Drittel. Allerdings können strukturelle Ursachen für die geringe Aktivität nicht ausgeschlossen werden. Daher wurden in dieser Arbeit aliphatische Aminosäuren mit ähnlichen Abständen zu den Fe/S-Zentren wie das Leu221G gegen Trp ausgetauscht, mit der Absicht, Doppelmutanten mit erhöhter Aktivität zu erzeugen. Jedoch wurde für keine der sechs dargestellten Doppelmutanten eine erhöhte Komplex I-Aktivität gemessen. In dieser Arbeit wurde der E. coli Stamm BW25113nptI Δnuo Δndh/pBADnuo nuoFhis als Überproduktionsstamm für den Komplex I etabliert. In diesem Stamm sind die Gene, die für den Komplex I und die alternative NADH Dehydrogenase kodieren, deletiert. Daher kann bei Verwendung dieses Stamms zur Bestimmung der Komplex I-Aktivität in der Membran auf den Einsatz des kostspieligen Substrats d-NADH verzichtet werden. Um die heterologe Überproduktion des A. aeolicus Subkomplexes NuoEFHis zu erhöhen, wurde in dieser Arbeit der Vektor pETBlue-1nuoEFhis ohne nuoG kloniert. Da NuoG nur in Form von Einschlusskörpern produziert wurde, sollte der Stress der Zellen bei der Expression und Produktion vermindert werden. Mit Hilfe von Expressionstests wurde die Proteinausbeute mit diesem Plasmid etwa sechseinhalbfach erhöht. Das Fe/S-Zentrum N1a des bakteriellen Komplex I hat ein ungewöhnlich hohes Mittenpunktspotential im Vergleich zu seinem mitochondrialen Homolog. In einem Kooperations-Projekt mit der Gruppe von Dr. Judy Hirst (MRC, Cambridge) wurde das Mittenpunktspotential dieses Zentrums durch ortsgerichtete Mutagenese um bis zu 100 mV abgesenkt. Aufgrund dieses negativeren Potentials konnte das Zentrum N1a in der Variante V96P/N142ME nicht mit NADH reduziert werden. Dies hatte eine um 60% erhöhte H2O2Produktion zur Folge, was die funktionelle Bedeutung dieses Zentrums als Schutz vor einer erhöhten ROS-Produktion zeigt. Die Strukturen des NuoEFHis mit gebundenem NAD+ und NADH zeigten eine Konformationsänderung der Peptidbindung von Glu95F. In dem reduzierten NuoEFHis zeigt die Carbonyl-Gruppe des Glu95F vom FMN weg und erlaubt so die Bindung von NADH. Im 2 Zusammenfassung oxidierten NuoEFHis zeigt diese Carbonyl-Gruppe zum FMN hin und verdrängt dadurch das NAD+. Dieser mögliche „Produkt-Auswurf-Mechanismus“ wurde durch die Mutation des Gly129E zu Ser und Asp untersucht. Die neu eingeführten Reste sollten die Carbonyl-Gruppe des Glu95F durch H-Brücken in die Konformation des reduzierten Proteins zwingen. Die Struktur der Varianten G129DE und G129SE wurden bei 1.85 Å bzw. 2.44 Å Auflösung erhalten. Die Carbonyl-Gruppe war nur in der Variante G129DE in der Konformation des reduzierten Proteins fixiert. Die Auswirkungen der Mutationen auf die Aktivität des Komplex I wurde anhand der homologen Varianten G135DE und G135SE bestimmt. Die Komplex I-vermittelten Aktivitäten der Variante G135SE waren nur leicht verringert. Die NADH:Decyl-Ubichinon Oxidoreduktaseaktivität der Variante G135DE nahm während der Messung stetig ab, vermutlich weil das Produkt NAD+ nicht mehr effektiv aus der Bindetasche entfernt wird, was wiederum die Hypothese des „Auswurf-Mechanismus“ unterstützt. Die katalytische Untereinheit NarG der Nitratreduktase A ist sequenz- und strukturhomolog zur C-terminalen Domäne von NuoG. Die Deletion einer Salzbrücke an der Oberfläche von NarG führte zum Verlust der Kofaktoren dieser Untereinheit. NuoG besitzt ebenfalls eine Salzbrücke an der Proteinoberfläche in der Nähe eines Fe/S-Zentrums, das wichtig für die Stabilität des Komplex I ist. Der Austausch von Glu617G, einem Teil der Salzbrücke, gegen Ala hatte den Zerfall des Komplexes zur Folge, was vermutlich an dem Verlust des Fe/SZentrums liegt. Diese Daten unterstreichen die evolutionäre Verwandtschaft dieser Proteine. In den letzten Jahren wird die „klassische“ Zuordnung der EPR-Signale der Fe/S-Zentren des Komplex I zu den strukturellen definierten Fe/S-Zentren in Frage gestellt. Es wird vermutet, dass die EPR-Signale N4 und N5 den falschen strukturell definierten Fe/S-Zentren zugeordnet wurden. Um dies zu untersuchen, wurden die Aminosäuren, die die Zentren N4 und N5 koordinieren jeweils gegen Alanine ausgetauscht. Die meisten Mutanten zeigten keine Komplex I-vermittelte Aktivität in den Membranen, was auf den Verlust eines Zentrums hindeutet. Drei der Varianten mit einer Mutation im Motiv von N4 und eine mit einer Mutation um N5 konnten präpariert und EPR-spektroskopisch charakterisiert werden. Alle Präparationen zeigten das Signal, das dem Zentrum N4 zugeordnet wird. Es ist somit unwahrscheinlich, dass das EPR-Signal N4 von den strukturell definierten Fe/S-Zentren N4 oder N5 stammt. 3 Einleitung 2 Einleitung 2.1 Die aerobe Atmungskette von Escherichia coli Die bei katabolen Prozessen wie der Glykolyse, dem Citratzyklus und der β-Oxidation in Formen von reduziertem Nikotinamidadenindinukleotid (NADH) und Flavinen (FADH2 und FMNH2) anfallenden Reduktionsäquivalente werden in der aeroben Atmungskette genutzt, um Sauerstoff zu H2O zu reduzieren. Die Oxidoreduktasen der Atmungskette translozieren dabei Protonen über die innere Mitochondrienmembran der Eukaryoten bzw. über die Cytoplasmamembran der Prokaryoten. Der so erzeugte Protonengradient stellt die sogenannte protonenmotorische Kraft dar, die von der ATP-Synthase zur ATP-Produktion aus ADP und anorganischem Phosphat genutzt wird (Mitchell und Moyle, 1967). Dieser Prozess wird als Oxidative Phosphorylierung bezeichnet. Das gramnegative, fakultativ anaerobe Darmbakterium Escherichia coli besitzt mindestens 15 primäre Dehydrogenasen und zehn terminale Oxidasen oder Reduktasen (Unden und Bongaerts, 1997). Die aerobe Atmungskette von E. coli wird im Wesentlichen von der Succinat Dehydrogenase, der protonenpumpenden Typ I sowie der nicht energieumwandelnden Typ II NADH:Ubichinon Oxidoreduktase und den drei terminalen Ubichinol:O2 Oxidoreduktasen, der bd-I und -II sowie der bo3 Oxidase gebildet (Anraku und Gennis, 1987; Borisov et al., 2011a; Sousa et al., 2011). Abbildung 2.1 zeigt die schematische Darstellung der aeroben Atmungskette von E. coli. (d)-NADH (d)-NAD+ Succinat Fumarat NADH NAD+ Cytosol 2 H+/e− 2 H+/e− Q QH2 Q QH2 Q QH2 1 H+/e− ½O2 H2 O QH2 Q Periplasma Abbildung 2.1: Schematische Darstellung der aeroben Atmungskette von E. coli. Die Typ I NADH:Ubichinon Oxidoreduktase ist cyan (PDB-Eintrag: 4HEA; Baradaran et al., 2013), die Typ II NADH:Ubichinon Oxidoreduktase hellgrün (PDB-Eintrag: 4G6H; Feng et al., 2012), die Succinat Dehydrogenase grün (PDB-Eintrag: 1NEK; Yankovskaya et al., 2003) und die bo3 Oxidase blau (PDB-Eintrag: 1FFT; Abramson et al., 2000) dargestellt. Die Strukturen der beiden bd Oxidasen sind nicht bekannt, daher wird stellvertretend nur eine in violetten Blöcken dargestellt. Die katalysierten Reaktionen der einzelnen Enzyme sind durch Substrate und Produkte angedeutet. Die roten Pfeile zeigen die Protonentranslokationsreaktionen mit den jeweiligen Stöchiometrien. 4 Einleitung Die Typ I NADH:Ubichinon Oxidoreduktase (NDH-I) koppelt den Transfer von zwei Elektronen von NADH auf Ubichinon (Q) mit der Translokation von vier Protonen über die Membran (Friedrich, 1998, 2001; Yagi und Matsuno-Yagi, 2003). Prokaryoten und einfach aufgebaute Eukaryoten besitzen eine oder mehrere alternative NADH:Ubichinon Oxidoreduktasen (NDH-II) (Matsushita et al., 1987; Melo et al., 2004; Sousa et al., 2011). Die E. coli NDH-II trägt ein nicht kovalent gebundenes Flavinadenindinukleotid (FAD) und besteht aus einem einzelnen 47.2 kDa großen Peptid, das vom Gen ndh kodiert wird (Jaworowski et al., 1981). Es wurde gezeigt, dass die NDH-II als Dimer über jeweils eine C-terminale, amphipatische Helix an die cytoplasmatische Membran assoziiert ist (Villegas et al., 2011; Feng et al., 2012). Die NDH-I kann im Gegensatz zur NDH-II neben NADH auch das Derivat Desamino-(d-)NADH umsetzen (Matsushita et al., 1987). Die Succinat Dehydrogenase (Komplex II) ist in die Membran integriert und verbindet den Citratzyklus mit der Atmungskette. Der Enzymkomplex besteht aus vier Untereinheiten und trägt ein FAD, drei Eisen-Schwefel (Fe/S)-Zentren und ein Häm b als Kofaktoren. Komplex II überträgt zwei Elektronen von Succinat auf Ubichinon, transloziert dabei jedoch keine Protonen (Cecchini et al., 2002; Yankovskaya et al., 2003). Die zur Familie der Häm-Kupfer Oxidasen gehörende bo3 Oxidase besteht aus vier Untereinheiten und koppelt die Reduktion von molekularem Sauerstoff zu H2O mit der Translokation von Protonen über die Membran (Abramson et al., 2000; Anraku und Gennis, 1987). Alle Kofaktoren, ein Häm b, ein Häm o3 und ein CuB sind in der Untereinheit I gebunden (Abramson et al., 2000). Die bd-I und -II Oxidasen bestehen jeweils aus zwei Untereinheiten und tragen über eine skalare Reaktion zum Aufbau des Protonengradienten bei (Puustinen et al., 1991; Borisov et al., 2011a). Die Oxidasen sind homolog zueinander und tragen jeweils zwei Häm b und ein Häm d in der großen Untereinheit (Dassa et al., 1991; Borisov et al., 2011b). NAD+ ist essentiell für katabole Prozesse, da es das Hauptoxidationsmittel der Zelle darstellt. Eine wichtige Funktion der Atmungskette ist die Regeneration von NAD+. Unter aeroben Bedingungen nutzt E. coli dafür hauptsächlich die NDH-II und die bo3 Oxidase. Hierbei werden pro regeneriertem NAD+ insgesamt zwei H+ transloziert. Unter mikroaeroben Bedingungen werden vermehrt die NDH-I und die bd-I Oxidase produziert, was einer Gesamtausbeute von fünf translozierten H+ pro oxidiertem NADH entspricht (Calhoun et al., 1993; Unden und Bongaerts, 1997). Obwohl die ATP-Ausbeute pro oxidiertem NAD+ über NDH-I und bd-I Oxidase höher ist, steht unter aeroben Bedingungen die NAD+-Regeneration im Vordergrund (Melo et al., 2004). 5 Einleitung 2.2 Die NADH:Ubichinon Oxidoreduktase Die Typ I NADH:Ubichinon Oxidoreduktase (EC 1.6.5.3), auch bekannt als respiratorischer Komplex I, stellt bei Eukaryoten und den meisten Prokaryoten das erste und größte Enzym der Atmungskette dar (Weiss et al., 1991; Leif et al., 1995; Friedrich, 1998; Yagi und Matsuno-Yagi, 2003). Folgende Gleichung gibt die von Komplex I katalysierte Reaktion wieder. (2.1) Der etwa 1 MDa schwere mitochondriale Komplex I besteht aus 44 Untereinheiten und trägt acht Fe/S-Zentren und ein Flavinmononukleotid (FMN) (Carroll et al., 2006; Balsa et al., 2012). Er gilt als die Hauptquelle für reaktive Sauerstoffspezies (reactive oxygene species; ROS) im Mitochondrium (Walker, 1992; Murphy, 2009). Bei ROS handelt es sich um freie Radikale, wie z. B. O2•−, OH• sowie um reaktive sauerstoffhaltige Moleküle, wie H2O2 und O3. Diese Spezies sind einerseits ein essentielles Nebenprodukt des Sauerstoffmetabolismus, stellen andererseits als oxidativer Stress einen wichtigen pathophysiologischen Faktor dar (Devasagayam et al., 2004). Mutationen im Komplex I und die daraus möglicherweise resultierenden Dysfunktionen können zu einer erhöhten ROS-Produktion oder zu einer erniedrigten katalytischen Aktivität führen. Dies wiederum kann schwerwiegende neurodegenerative Störungen wie z. B. die Parkinson- und die Alzheimer-Krankheit zur Folge haben (Schapira, 1998; Dawson und Dawson, 2003; Lin und Beal, 2006; Pagniez-Mammeri et al., 2012). Die ROS-Produktion in Mitochondrien wird weiterhin mit dem Alterungsprozess in Zusammenhang gebracht (Balaban et al., 2005). Der bakterielle Komplex besteht in der Regel aus 14 Untereinheiten mit einer molekularen Masse von etwa 550 kDa (Friedrich, 1998). Er trägt bis zu zehn Fe/S-Zentren und ein FMN (Friedrich und Böttcher, 2004). Die Untereinheiten des bakteriellen Komplexes sind essentiell für die Katalyse und werden daher als Kernuntereinheiten bezeichnet. Homologe sämtlicher dieser Untereinheiten werden im mitochondrialen Komplex I gefunden. Darüber hinaus besitzt der bakterielle Komplex die gleichen Substratbindestellen, katalysiert die gleiche Reaktion und wird durch die selben Hemmstoffe blockiert wie der mitochondriale (Friedrich et al., 1994; Degli Esposti, 1998). Daher wird der bakterielle Komplex I als Modell für die eukaryotischen NADH:Ubichinon Oxidoreduktase angesehen (Friedrich und Scheide, 2000). 6 Einleitung In E. coli sind die nuo-Gene (NADH:Ubichinon Oxidoreduktase) in dem 15 kbp großen nuoOperon organisiert, wobei nuoC und nuoD fusioniert vorliegen. Der 535 kDa schwere Komplex wird entsprechend aus 13 Untereinheiten aufgebaut, die mit NuoA – N bezeichnet werden (Weidner et al., 1993, Braun et al., 1998; Friedrich, 1998). Erst 2013 wurde eine Kristallstruktur des Komplex I aus Thermus thermophilus bei einer Auflösung von 3.3 Å veröffentlicht (Baradaran et al., 2013). Dieser besteht aus 16 Untereinheiten Nqo1 – 16 (NADH:quinone oxidoreductase), die zusammen einen 536 kDa schweren Komplex bilden (Abb. 2.2). In dieser Arbeit wird der Übersicht wegen ausschließlich die E. coli Nomenklatur für die 14 Kernuntereinheiten verwendet. Der Komplex weist eine L-förmige Struktur auf, was schon vorher durch elektronenmikroskopische Aufnahmen des E. coli und Bos taurus Komplex I gezeigt wurde (Guénebaut et al., 1998; Grigorieff, 1999). Er besteht aus einen peripheren Arm, der in die wässrige Phase ragt und einem Membranarm, der in die Lipiddoppelschicht eingebettet ist. NADH NAD+ 140 Å Cytosol Q 30 Å QH2 Periplasma 180 Å Abbildung 2.2: Struktur des T. thermophilus Komplex I bei 3.3 Å Auflösung (PDB-Eintrag: 4HEA; Baradaran et al., 2013). Die Orientierung des Komplexes in der Membran ist gezeigt. Die Substratbindestellen sind durch schwarze Pfeile angedeutet. Der periphere Arm trägt neun Fe/S-Zentren und ein FMN, hier als Kalotten-Modelle dargestellt. Der Membranarm trägt keine Kofaktoren und ist für die Protonentranslokation verantwortlich. Die Dimensionen des Komplexes und der Membran sind angegeben. Der aus den Untereinheiten NuoB, NuoE – G, NuoI und den zwei löslichen Proteinen Nqo15 und Nqo16 bestehende periphere Arm trägt alle bekannten Kofaktoren des Elektronentransfers: ein FMN und neun Fe/S-Zentren. NuoE trägt das zweikernige Fe/SZentrum N1a, NuoF enthält die NADH-Bindestelle mit dem nicht-kovalent gebundenen FMN 7 Einleitung und das vierkernige Zentrum N3. Die größte Untereinheit NuoG trägt das zweikernige Fe/SZentrum N1b und die vierkernigen Zentren N4, N5 und N7 (Uhlmann und Friedrich, 2005). NuoI beherbergt die zwei vierkernigen Fe/S-Zentren N6a und N6b. Das distale vierkernige Zentrum N2 befindet sich auf NuoB. In der Literatur wird diskutiert, ob die Zuordnung der EPR-spektroskopisch detektierten Signale zu den strukturell definierten Zentren stimmig ist (Roessler et al., 2010). Es ist strittig, ob das EPR-Signal „N4“ nicht von den strukturell definierten Fe/S-Zentren N5 oder N6a erzeugt wird (Yakovlev et al., 2007; Nakamaru-Ogiso et al., 2008; Sinha et al., 2012). Der Übersicht wegen wird in dieser Arbeit die Nomenklatur nach Tomoko Ohnishi verwendet (Ohnishi, 1998; Sazanov und Hinchliffe, 2006). Die Untereinheiten Nqo15 und 16 sind nur in der Gattungen Thermus und Deinococcus konserviert, wobei Nqo16 keinen Einfluss auf die Aktivität hat, aber essentiell für die Kristallisation des gesamten Komplexes ist. Es wird daher davon ausgegangen, dass es sich um einen Assemblierungsfaktor handelt (Baradaran et al., 2013). Die Deletion des Gens, des zu Nqo15, das ein Mitglied der Frataxin-Proteinfamilie ist, homologen Proteins CyaY, hatte weder Einfluss auf die Assemblierung noch auf die Aktivität des E. coli Komplex I (Pohl et al., 2007a). Der Elektronentransfer findet im peripheren Arm statt. Die von NADH an FMN übertragenen Elektronen gelangen über die Kette aus Fe/S-Zentren zum Ubichinon, das zwischen beiden Armen gebunden wird. Die 30 Å lange, enge Ubichinon-Bindestelle wird von NuoB, NuoD und NuoH gebildet und ist vollständig vom Solvens abgeschirmt. Der schmale Eingang zur Bindestelle wird von hydrophoben Seitenketten gebildet, wobei geladene Reste das Innere des Bindungskanals auskleiden. Die Kopfgruppe des Ubichinons wird 15 Å oberhalb der Membran und in etwa 12 Å Abstand von N2 gebunden. Der periphere Arm wird hauptsächlich über hydrophobe Wechselwirkungen an den Membranarm gebunden. Die Kavität der Ubichinon-Bindestelle bildet dabei eine Ausnahme. Hier wechselwirken NuoB und NuoH über geladene Reste mit einander (Baradaran et al., 2013). Der Membranarm besteht aus den Untereinheiten NuoA, NuoH, NuoJ – N. Die Untereinheiten NuoL, NuoM sowie NuoN sind zueinander und zu Na+/H+-Antiportern homolog und bilden drei der vier Protonenkänale des Komplexes (Friedrich und Weiss, 1997). NuoL, NuoM und NuoN besitzen jeweils 14 transmembrane Helices (TM), von denen wiederum jeweils vier zwei zentrale Helix-Bündel bilden, die von den restlichen Helices flankiert werden. Jedes dieser Helix-Bündel bildet einen Halbkanal: einem N-terminalen aus TM4–8 und einem C-terminalen aus TM9 – 13. Die Halbkanäle TM4 – 8 sind im oxidierten Zustand der Präparation zum Cytoplasma hin geöffnet und enthalten ein zentrales Lysin auf TM7 (LysTM7) sowie ein Glutamat auf TM5. Die zum Periplasma geöffneten Halbkanäle TM9 –13 8 Einleitung enthalten ein zentrales Lysin (NuoL und NuoN) oder Glutamat (NuoM) auf TM12. Die zwei Halbkanäle sind über geladene Reste miteinander verbunden. NuoH trägt ebenfalls geladene Reste an analogen Positionen wie die Untereinheiten NuoL, M und N. Zusammen mit NuoJ und NuoK bildet NuoH den vierten Protonenkanal. Die zentralen geladenen Reste bilden eine Achse, die sich durch den gesamten Membranarm zieht (Abb. 2.3). Zusätzlich besitzt NuoL eine C-terminale, amphipatische Helix (HL), welche die Protonenkanäle miteinander verbindet. HL ist durch zwei transmembrane Helices in der Membran verankert, wobei die C-terminale Helix mit NuoJ, K und N über polare Reste interagiert (Efremov und Sazanov, 2011). Zudem besitzen die Protonenkanäle auf der periplasmatischen Seite jeweils zwei antiparallel ausgerichtete β-Stränge. Abbildung 2.3: Vorgeschlagener Kopplungsmechanismus des Komplex I. Der periphere Arm (grau, nicht maßstabsgetreu dargestellt) mit NADH (blau), FMN (orange) und den Fe/S-Zentren (orange) sowie der Membranarm mit den Protonenkanälen in verschiedenen Farben sind schematisch dargestellt. Die durch die Reduktion des Q initiierte Kaskade an konformativen Änderungen ist mit roten und die durch Reduktion von N2 hervorgerufenen Verschiebungen mit blauen Pfeilen dargestellt. Die zentrale Achse aus geladenen Resten ist durch rote bzw. blaue Punkte und die H+-Translokation durch schwarze Pfeilen gekennzeichnet. Die unterstützende Funktion der Helix HL und der β-Stränge ist mit Rechtecken und Pfeilen angedeutet (Baradaran et al., 2013). Folgender Redox-getriebener Kopplungsmechanismus von Elektronentransfer und Protonentranslokation wurde für Komplex I vorgeschlagen (Abb. 2.3; Baradaran et al., 2013): Nachdem ein Elektron von N2 auf Ubichinon übertragen wurde, initiiert das negativ geladene Q•− eine Kaskade an konformativen Änderungen, die über die geladene zentrale Achse an alle Protonenkanäle weitergeleitet werden. Die LysTM7 auf den zum Cytosol hin geöffneten 9 Einleitung Halbkanälen werden protoniert. Sobald das Q•− zu QH2 reduziert wird, schließen sich die Halbkanäle auf der cytosolischen Seite und die LysTM7 übertragen die Protonen auf die zentralen geladenen Reste der zum Periplasma hin geöffneten Halbkanäle. Diese geben die Protonen ins Periplasma ab und der Komplex I geht wieder in den oxidierten Zustand über. Die konformativen Änderungen werden durch die Helix HL auf der cytosolischen und die β-Stränge auf der periplasmatischen Seite unterstützt. Auch die Redoxreaktion von N2 verschiebt Helices in NuoB und NuoD und trägt damit zu den konformativen Änderungen im gesamten Komplex bei (Baradaran et al., 2013). 2.3 Elektronentransfer im Komplex I Der Elektronentransfer in biologischen Systemen wird generell durch einen Tunnel-Prozess beschrieben (Page et al., 1999). Nach Veröffentlichung der ersten Kristallstruktur des peripheren Arms des T. thermophilus Komplex I bei 3.3 Å waren die Abstände zwischen den Kofaktoren bekannt (Sazanov und Hinchliffe, 2006). Mit Hilfe der Mittenpunktspotentiale (Em, 7) wurde ein möglicher Elektronenweg ausformuliert (Moser et al., 2006; Abb. 2.4). NADH (Em, 7 = −340 mV) wird von NuoF gebunden und überträgt ein Hydridion und ein Proton auf das tief in der Substratbindetasche liegende FMN (Em, 7 = −320 mV; et al., 2008; Leif et al., 1995). Die Elektronen werden einzeln auf N3 (Em, 7 = −270 mV), das den Eintrittspunkt der Elektronentransferkette darstellt, über die isopotenziellen Fe/S-Zentren N1b, N4, N5, N6a, N6b (Em, 7 = −250 mV) auf N2 (Em, 7 = −220 mV) und schließlich auf Ubichinon (Em, 7 = +110 mV) übertragen (Leif et al., 1995; Ohnishi, 1998; Yano et al., 2003). Das Fe/S-Zentrum N7 ist über 20 Å von dem nächstgelegenen Zentrum N4 entfernt. Dies überschreitet die maximale Distanz von 14 Å für einen physiologisch relevanten Elektronentransfer (Page et al., 1999). Es wurde EPR-spektroskopisch gezeigt, dass N7 nicht mit NADH, aber chemisch mit Dithionit reduziert werden kann (Pohl et al., 2007b). Der durch Mutagenese des Bindemotivs bedingte Verlust des Zentrums hatte keinen Einfluss auf die Aktivität, sondern verminderte die Stabilität des Komplex I (Pohl et al., 2007b). Das zweikernige Fe/S-Zentrum N1a ist etwa 20 Å von N3, aber nur 12.3 Å vom FMN entfernt. Da N1a im Gegensatz zu N7 konserviert ist, ist es wahrscheinlich von funktioneller Bedeutung. Es wird angenommen, dass der Großteil der von Komplex I produzierten ROS am reduzierten FMN gebildet wird (Kussmaul und Hirst, 2006; Hirst et al., 2008). Es wird weiterhin vermutet, dass das gut vom Solvens abgeschirmte N1a als Zwischenspeicher für das 10 Einleitung zweite vom Flavosemichinonradikal übertragene Elektron dient, um die Lebensdauer des solvensexponierten Radikals so gering wie möglich zu halten (Sazanov, 2007). Aufgrund seines niedrigen Mittenpunktspotentials (Em, 7 = −330 mV; Leif et al., 1995) kann N1a erst von dem entstehenden Flavosemichinonradikal (E m, 7 = −390 mV; Sled et al., 1994) reduziert werden (Sazanov, 2007). Während des katalytischen turnovers bleiben einige Fe/S-Zentren der Elektronentransferkette teilweise und nur N2 vollständig reduziert (Krishnamoorthy und Hinkle, 1988; Kotlyar et al., 1990; Bridges et al., 2012). Erst wenn das distale Fe/S-Zentrum N2 durch Ubichinon oxidiert wird, rücken die anderen Elektronen nach und das Elektron von N1a kann über FMN und N3 in die Elektronentransferkette eingespeist werden (Sazanov, 2007). N7 N1b 10.9 (7.6) 14.2 (11.0) 24.2 (20.5) 13.0 (10.7) N4 16.2 (13.8) N3 13.5 (12.3) 12.2 (8.5) N5 N2 22.3 (19.4) N6a N1a N6b 16.9 (14.0) 14.0 (10.5) 12.2 (9.0) Abbildung 2.4: Anordnung der Kofaktoren im T. thermophilus Komplex I (PDB-Eintrag: 3IAM; Berrisford und Sazanov, 2009). FMN und NADH sind als Stabmodelle in Elementfarben und die neun Fe/SZentren sind als Kalotten-Modelle mit Eisen in Rot und Schwefel in Gelb dargestellt. Die blauen Pfeile markieren den möglichen Elektronenweg. Der grüne Pfeil deutet die mögliche reversible Elektronenübertragung auf N1a an. Alle Abstände sind in Ångström angegeben. Die Zahlen vor den Klammern geben die Mittelpunktsabstände und die Zahlen in den Klammern die Kante-zu-Kante Abstände zwischen den Kofaktoren wieder. Anhand der bekannten Abstände der Kofaktoren wurden die Elektronentransferraten durch den Komplex I berechnet (Moser et al., 2006). Hierfür wurde ein von der Marcus-Theorie abgeleitetes phänomenologisches Modell benutzt, bei dem nur die Abstände zwischen den einzelnen Kofaktoren berücksichtigt werden und das Protein als einheitliche Tunnelbarriere betrachtet wird (Page et al., 1999). Für alle Transferschritte, bis auf den zwischen N5 und N6a, wurden Elektronentransferraten (kET) von 106 – 107 s−1 berechnet. Der Kante-zu-Kante 11 Einleitung Abstand zwischen N5 und N6a beträgt 14 Å (Sazanov und Hinchliffe, 2006), was genau der Grenze des physiologisch relevanten Elektronentransfers entspricht (Page et al., 1999). Für diesen Schritt betrug die berechnete Transferrate 104 s−1. Obwohl der errechnete Wert die experimentellen Werte von 50 – 180 s−1 (Ragan und Racker, 1973; Stolpe und Friedrich, 2004; Sharpley et al., 2006) bei weitem überschreitet, wird er als Flaschenhals für die Gesamttransferrate von Komplex I angesehen (Moser et al., 2006). Die Gruppe von Prof. Thorsten Koslowski, Instititut für Physikalische Chemie der Universität Freiburg, veröffentlichte eine alternative Berechnung auf atomarer Ebene (Wittekindt et al., 2009). Bei dieser Beschreibung werden nicht nur die Abstände, sondern auch die chemische Umgebung der Redoxpartner berücksichtigt, was zu genaueren Werten führt. Es wurden vier mögliche Transfer-Mechanismen berücksichtigt: Tunneln (through space), E ek r nenüber ragung über σ-Bindungen des Proteins (through bond), indirekte Beteiligung e π-Systems der aromatischen Aminosäuren oder der Amidbindungen durch Erhöhung der elektronischen Amin Kopplung (superexchange) und direkte Beteiligung aromatischer uren, in em a zu über ragen e E ek r n in erme i r auf em π-System lokalisiert ist (hopping) (Wittekindt et al., 2009). Nur für den hopping-Mechanismus wurden Werte berechnet, die mit der physiologischen Transferrate von kET = 50 – 180 s−1 übereinstimmen (Wittekindt et al., 2009). Sequenzvergleiche zeigten, dass die von Wittekind et al. identifizierten aromatischen Aminosäuren entweder konserviert oder durch andere aromatische Aminosäuren ersetzt sind. Abbildung 2.5 gibt die Lage der aromatischen Seitenketten zwischen den Fe/S-Zentren wieder. In vorangegangenen Arbeiten unserer Gruppe wurden die in Abbildung 2.5 gezeigten aromatischen Aminosäuren gegen anderen aromatische Aminosäuren und Leucin ausgetauscht. Die Mutation der aromatischen Aminosäuren zu Leucin, die sich zwischen den Fe/S-Zentren N1b und N4 bzw. N6b und N2 befinden hatten keinen oder nur geringen Einfluss auf die Aktivität. Dies ist verständlich, da die Mutationen die Elektronentransferraten theoretisch nur unwesentlich beeinflussen sollten und dieser geringe Effekt mit den verwendeten Methoden nicht erfasst werden kann. Der Austausch von Phe515CD und Trp221G gegen Leucin, die beide zwischen N5 und N6a liegen, bedingte eine Abnahme der d-NADH-Oxidaseaktivität der E. coli Cytoplasmamembranen um mehr als 30% (Dörner, 2010). Obwohl Leucin von allen aliphatischen Aminosäuren die größte Seitenkette besitzt, müssen auch strukturelle Gründe für die erniedrigte Aktivität in Betracht gezogen werden. 12 Einleitung N1b 3 G F33 (F35 ) 3 G W247 (W221 ) N2 N4 N5 4 F328 CD (F515 ) N6b 4 H327 CD (H514 ) N6a 9 I F100 (F101 ) Abbildung 2.5: Position der am Elektronentransfer beteiligten aromatischen Aminosäuren in Komplex I nach Wittekind et al. 2009 (PDB-Eintrag: 3IAM; Berrisford und Sazanov, 2009). Die T. thermophilus und die E. coli Nomenklatur (in Klammern) der möglicherweise am Elektronentransfer beteiligten aromatischen Aminosäuren sind angegeben. Die Fe/S-Zentren sind als Kalotten-Modelle mit Eisen in Rot und Schwefel in Gelb dargestellt. Die berechneten hopping-Pfade sind mit blauen, gestrichelten und die through space-Pfade mit schwarzen, gestrichelten Linien angedeutet. 2.4 Die Fe/S-Zentren N1a und N7 Das Fe/S-Zentrum N1a wird von der Thioredoxin-ähnlichen Domäne von NuoE gebunden und fungiert vermutlich als Antioxidans (Sazanov und Hinchliffe, 2006). In vorangegangenen Arbeiten wurde gezeigt, dass N1a essentiell für die Stabilität des Komplex I ist (Dörner, 2010). Die N1a-koordinierenden Cysteine wurden gegen Alanin oder Serin ausgetauscht. Durch die Bestimmung der NADH-Oxidaseaktivität der Cytoplasmamembranen der Mutanten wurde gezeigt, dass die Varianten in verringertem Maß vorhanden sind. Zudem wurde durch Saccharosedichtegradienten-Zentrifugation gezeigt, dass einige der Varianten nach der Extraktion aus der Membran zerfielen. Die Varianten, die nicht zerfielen, zeigten eine deutlich verringerte NADH/Ferricyanid Oxidoreduktaseaktivität (Dörner, 2010). N1a kann nach Reduktion durch NADH EPR-spektroskopisch im E. coli Komplex I nachgewiesen werden (Leif et al., 1995; Uhlmann und Friedrich, 2005). Für den mitochondrialen Komplex I wurde das mit Dithionit reduzierte N1a nur in der einzeln produzierten Untereinheit NuoE oder in einem Subkomplex bestehend aus NuoE, NuoF und einer 10 kDa Untereinheit detektiert (Ragan et al., 1982; Barker et al., 2007). Im intakten mitochondrialen Komplex I ist N1a aufgrund seines niedrigen Mittenpunktspotentials von etwa −450 mV nicht mit NADH 13 Einleitung reduzierbar (Ohnishi et al., 1981). In Proben die mit dem sehr starken Reduktionsmittel Eu2+ behandelt wurden, war N1a EPR-spektroskopisch nicht nachweisbar. Daraus wurde geschlossen, dass das Potential von N1a niedriger als −1 V sein sollte (Reda et al., 2008). Allerdings kann es auch mechanistische Gründe geben, warum das Zentrum nicht reduziert wird. Anhand von Mutagenese-Studien an NuoE aus E. coli hat die Gruppe von Dr. Judy Hirst am MRC Cambridge, UK, versucht Reste zu identifizieren, die in der Lage sind das Mittenpunktspotential von N1a zu modulieren. In der zweiten Koordinationssphäre von N1a in Prokaryoten befinden sich die nicht konservierten Reste, Val96E und Asn142E. Im mitochondrialen Komplex I sind an diesen Positionen ein Prolin (Pos. 96) und ein Methionin (Pos. 142) konserviert (Abb. 2.6). Abbildung 2.7 gibt die Positionen der Aminosäuren in T. thermophilus NuoE wieder. 96 E. coli T. thermophilus A. aeolicus B. taurus Y. lipolytica VIRYCDSVVC HLQVCATLSC RIRVCVSIVC HIQVCTTTPC HLQICTTTPC 142 HINGYQGIQA KLAGAEELWD HLMGTNKLLK MLRNSDSILE QLCGSDGIME ----------- DKGPNMMIDE HTAPVIQVND SEAPVFMVND VNAPMVQIND VNAPMMAIND Abbildung 2.6: Ausschnitt des Sequenzvergleichs von NuoE. Die Aminosäuren Pro96 und Met142 sind in Eukaryoten konserviert, wie hier am Beispiel von Yarrowia lipolytica und Bos taurus NuoE gezeigt. Die E. coli Nummerierung ist angegeben. S96 V142 Abbildung 2.7: Die Thioredoxin-ähnliche Domäne von NuoE aus T. thermophilus (PDB-Eintrag: 3IAM; Berrisford und Sazanov, 2009). Die zu Val96E und Asn142E homologen Positionen sind gezeigt. Es wurde die Nummerierung nach E. coli verwendet. N1a ist als Stabmodell mit den koordinierenden Cysteinen dargestellt. 14 Einleitung Der Austausch Val96E gegen Pro und Asn142E gegen Met verschob das Mittenpunktspotential des Zentrums in der einzeln produzierten Untereinheit NuoE jeweils um etwa −60 mV. In der Variante mit beiden Mutationen wurde das Mittenpunktspotential sogar um −100 mV verschoben (J. Hirst, persönliche Mitteilung). Erschwerend kommt hinzu, dass das Mittenpunktspotential von N1a von der chemischen Umgebung, genauer vom pH und der Ionenstärke abhängt. Daher bleibt zu untersuchen ob dieser an der einzelnen Untereinheit beobachtete Effekt der Mutationen auch bei dem Gesamtkomplex zu messen ist. Sequenzanalysen haben gezeigt, dass die N-terminale Domäne von NuoG, die N1b, N4 und N5 bindet, Ähnlichkeit zu Untereinheiten der [Fe/Fe]-Hydrogenasen und den NAD+-reduzierenden [Ni/Fe]-Hydrogenasen aufweist (Fearnley und Walker, 1992; Rothery et al., 2008). Die C-terminale Domäne, die das Zentrum N7 bindet, zeigt Struktur- und Sequenzähnlichkeit zu Enzymen der Molybdo-bis(Molybdopterin-Guanin-Dinukleotid) ([Mo(MGD)2])-Familie (Chow et al., 1991; Finel, 1998; Rothery et al., 2008). Da N7 nur im Komplex I einiger Bakterien wie z. B. E. coli, T. thermophilus und Aquifex aeolicus vorkommt, ist davon auszugehen, dass es sich bei diesem Zentrum um ein evolutionäres Relikt handelt. Es wird vermutet, dass der Vorläufer von NuoG ebenfalls einen [Mo(MGD)2]Kofaktor und ein weiteres Fe/S-Zentrum besaß, die im Laufe der Evolution verloren gingen. NuoG im A. aeolicus Komplex I besitzt ein weiteres Fe/S-Zentrum N8, das N7 mit der Elektronentransferkette verbinden könnte (Peng et al., 2010). Dies deutet darauf hin, dass der NuoG-Vorläufer als Elektroneneintritts-Modul für ein anderes Substrat als NADH fungierte. Im Genom von A. aeolicus, einem der evolutionär ältesten Eubakterien, sind die nuo-Gene nicht wie in anderen Bakterien in einem Operon organisiert, sondern sind auf drei verschiedenen Loci verteilt (Scheide et al., 2002). Die Tatsache, dass nuoEF und nuoG auf verschiedenen Loci organisiert sind, verstärkt die Vermutung der evolutionär späten Entstehung des NADH-Dehydrogenase-Moduls NuoEFG (Friedrich und Scheide, 2000). Ein typischer Vertreter der [Mo(MGD)2]-Familie ist die Chinol:Nitrat Oxidoreduktase (EC 1.7.5.1), die auch als Nitratreduktase A oder NarGHI bekannt ist. Unter anaeroben Bedingungen und in Anwesenheit von Nitrat nutzt E. coli NarGHI, um mit Hilfe von Menachinol (MQH2) Nitrat zu Nitrit zu reduzieren (Forget, 1974). NarGHI bildet mit der Formiat Dehydrogenase N (FdnGHI) eine anaerobe Atmungskette, die Elektronen von Formiat im Periplasma auf Nitrat im Cytosol transferiert. Beide Redoxreaktionen sind über den Menachinon (MQ)-Pool in der Membran gekoppelt (Abb. 2.8). Dabei werden Protonen über eine Redox-Schleife vom Cytosol ins Periplasma transloziert (Bertero et al., 2003). 15 Einleitung FdnGHI gehört auch zur [Mo(MGD)2]-Familie. Die Untereinheit FdnG hat ebenfalls Strukturund Sequenz-Ähnlichkeit zur C-terminalen Domäne von NuoG (Rothery et al., 2008). Periplasma Cytosol Abbildung 2.8: Schematische Darstellung der Nitratatmung in E. coli (Bertero et al., 2003). Der von FdnGHI und NarGHI katalysierte Elektronentransfer von Formiat (Periplasma) zu Nitrat (Cytosol) und der redox-gekoppelten Protonentransport über MQ/MQH2 in der Membran ist dargestellt. FS bezeichnen Fe/SZentren und bD und bP entsprechen Häm-Gruppen. Die Untereinheiten NarG, H und I bilden den 224 kDa schweren NarGHI-Komplex. Die Gene narGHJI sind in einem Operon organisiert. Das Gen narJ kodiert für das Chaperon NarJ, das essentiell für die Assemblierung des gesamten Enzyms und für die Reifung sowie Insertion der Kofaktoren ist (Blasco et al., 1998; Lanciano et al., 2007). Der Komplex trägt insgesamt acht Kofaktoren: ein [Mo(MGD)2], vier vierkernige und ein dreikerniges Fe/S-Zentrum sowie zwei Häm-Gruppen. Der Komplex ist über NarI, das die Häm-Gruppen bP und bD trägt, in der Membran verankert. NarH trägt das dreikernige Fe/S-Zentrum FS4 sowie die vierkernigen Zentren FS3, FS2 und FS1 und verbindet den Elektronentransfer von NarI zu NarG. Die zur C-terminalen Domäne von NuoG homologe, katalytische Untereinheit NarG trägt den [Mo(MGD)2]-Kofaktor und das vierkernige Zentrum FS0, das wie N5 in NuoG von drei Cys und einem His koordiniert wird. Abbildung 2.9 zeigt die Struktur des NarGHI-Komplexes bei einer Auflösung von 1.6 Å (Bertero et al., 2003). 16 Einleitung B) A) NarG NarH Cytosol NarI Periplasma Abbildung 2.9: Struktur des NarGHI Komplexes aus E. coli (PDB-Eintrag: 1Q16; Bertero et al., 2003). A) zeigt die Organisation der Untereinheiten und die Anordnung der Kofaktoren, die als Kalotten-Modelle dargestellt sind. B) zeigt die Koordination des Mo (cyan) und des Fe/S-Zentrums FS0 in NarG. Die zwei MGD (nach Elementfarben eingefärbt) und das FS0 (Fe in Rot, S in Gelb) sind als Stabmodelle abgebildet. In der Gruppe von Prof. Axel Magalon Universität Marseilles, Frankreich, wurde gezeigt, dass das Chaperon NarJ den [Mo(MGD)2]-Kofaktor und das FS0 in eine offene Form des löslichen NarGH-Intermediats einsetzt (Lanciano et al., 2007). NarG untergeht eine konformative Änderung und die geschlossene Form wird durch eine an der Oberfläche von NarG befindlichen Salzbrücke aus den Resten Arg107 und Glu793 stabilisiert. Es wurde gezeigt, dass nach Mutation einer der beiden Aminosäuren zu einem Alanin das Enzym noch produziert wird, aber weder den [Mo(MGD)2]-Kofaktor noch das Fe/S-Zentrum FS0 trägt. Diese Salzbrücke ist in der katalytischen Untereinheit von Enzymen der [Mo(MGD)2]Familie konserviert (Prof. Axel Magalon, persönliche Mitteilung). In NuoG ist diese Oberflächen-Salzbrücke ebenfalls vorhanden und wird im E. coli Komplex I von Arg280 und Glu617 gebildet. N7 in NuoG und FS0 in NarG sind in ähnlicher Position zu der jeweiligen Salzbrücke angeordnet (Abb. 2.10). Die Mutation der N7koordinierenden Cysteine bedingte den Zerfall des Komplex I (Pohl et al., 2007b). Die Auswirkung der Deletion der Salzbrücke auf die Assemblierung von Komplex I ist zu untersuchen. 17 Einleitung B) A) FS0 N7 Abbildung 2.10: Oberflächen-Salzbrücke in NarG A) und NuoG B). Die Strukturen sind nach ihren Sekundärstrukturelementen eingefärbt. A) zeigt NarG mit dem [Mo(MGD)2]-Kofaktor (rot) und dem Zentrum FS0 (PDB-Eintrag: 1Q16; Bertero et al., 2003). B) zeigt NuoG mit dem Zentrum N7 (PDB-Eintrag: 3IAM; Berrisford und Sazanov, 2009). Die Aminosäuren, die die Oberflächen-Salzbrücke bilden, sind farblich hervorgehoben. 2.5 Die NADH-Bindestelle des Komplex I In unserem Arbeitskreis wurden die Untereinheiten NuoE und NuoF des Komplex I aus dem hyperthermophilen Bakterium A. aeolicus als Subkomplex NuoEF in E. coli überproduziert, isoliert und kristallisiert et al., 2008). NuoF ist aus einer N-terminalen Domäne, die die glycinreiche Schleife trägt, einer ungewöhnlichen Rossmann-Faltung mit nur vier statt fünf β-Strängen, die von zwei α-Helices flankiert werden, einer Ubiquitin-ähnlichen und einer C-terminalen Domäne aus einem Bündel von vier α-Helices aufgebaut. Diese Helices enthalten die Cysteine 347F, 350F, 353F und 393F, die das vierkernige N3 tetraedrisch koordinieren. NuoE ist aus einer N-terminalen Domäne, die aus einem Bündel von vier αHelices besteht und mit NuoF wechselwirkt und einer Thioredoxin-ähnlichen Domäne, die das zweikernige N1a bindet, aufgebaut (Kohlstädt, 2009). N1a wird von den Cysteinen 86E, 91E, 127E und 131E quadratisch planar koordiniert. Abbildung 2.11 gibt die Quartärstruktur des Subkomplexes wieder. 18 Einleitung Abbildung 2.11: Struktur des A. aeolicus Subkomplexes NuoEF bei 2.0 Å. Das FMN ist in Elementfarben als Stabmodell dargestellt. Die Fe/S-Zentren N1a und N3 sind als Kalotten-Modelle mit Eisen in Rot und Schwefel in Gelb dargestellt. Die N-terminale (blau), die ungewöhnliche Rossmann-Faltung (grün), die Ubiquitin-ähnliche (beige) und die N3 koordinierende C-terminale Domäne (rot) von NuoF sind gezeigt. Die an NuoF bindende Domäne von NuoE ist in Grau und die N1a koordinierende Thioredoxin-ähnliche Domäne in Türkis dargestellt (Kohlstädt, 2009). Der Subkomplex wurde zudem mit NAD+ im oxidierten und NADH im reduzierten Zustand kristallisiert. Die Strukturen wurden jeweils bei 1.8 Å gelöst. Bei Anbindung des Nukleotids öffnet sich die Bindetasche, indem sich die glycinreiche Schleife in der Struktur des oxidierten Proteins um 0.4 bis 0.8 Å und in der des reduzierten Proteins um 0.6 bis 1.1 Å Struktur bewegt. Das Adenin des NAD+/H wird durch π-Wechselwirkungen mit Phe71F und Tyr205F stabilisiert. In beiden Strukturen bildet Lys76F Wasserstoff (H)-Brücken zum O3' der Adenosin-Ribose und O2P des FMNs aus. Das Glu185F bildet in beiden Strukturen mit seiner Seitenkette H-Brücken mit den Hydroxyl-Gruppen der Adenosin-Ribose und mit seinem Peptid-Stickstoff eine H-Brücke zum O2' des FMN aus. In der Struktur der NADPHabhängigen Glutathion Reduktase aus E. coli wechselwirken zwei Arginin-Reste und ein Lysin-Rest mit der zusätzlichen Phosphat-Gruppe des NADPHs (Scrutton et al., 1990; Mittl et al., 1994). Die Arginin-Reste befinden sich in ähnlicher Position wie Glu185F und Lys76F im Komplex I. Zudem bietet die NADH-Bindetasche etwas, aber nicht ausreichend Raum für die zusätzliche Phosphat-Gruppe des NADPH. Es ist zu vermuten, dass das Glu185F durch eine sterische Hinderung und/oder eine elektrostatische Abstoßung mit der zusätzlichen Phosphat-Gruppe die Anbindung des NADPHs erschwert, was die geringe Aktivität des Komplexes mit NADPH erklären würde (Stolpe, 2007). In der Struktur des reduzierten 19 Einleitung Proteins mit gebundenem NADH bildet die Seitenkette des Lys202F eine H-Brücke mit der Seitenkette von Glu184F und den Carbonyl-Gruppen von Arg200F und Ile392F. In der Struktur des oxidierten Proteins ist die Seitenkette des Lys202F verdreht. Dadurch verliert es die H-Brücken zu den Carbonyl-Gruppen und bildet eine neue mit der Seitenkette des Glu185F aus, was wiederum dessen H-Brücken zur Adenosin-Ribose schwächt (Abb. 2.12 A). Im Gegensatz dazu wurde für die Struktur des T. thermophilus Komplex I beschrieben, dass das Lys202F im reduzierten Zustand eine H-Brücke zum Pyrophosphat des NADHs ausbildet (Berrisford und Sazanov, 2009). A) B) E97 Y180 E97 Y180 F71 E184 E185 F71 K76 E185 K76 E184 F79 K202 Y205 F79 K202 Y205 Abbildung 2.12: Wechselwirkungen der Seitenketten von NuoF mit NAD+/H im Subkomplex NuoEF im oxidierten A) bzw. reduzierten B) Zustand. Oxidiertes NuoF (grau) mit gebundenem NAD + (grün) und reduziertes NuoF (hellblau) mit gebundenem NADH (magenta) sind dargestellt. Mit NAD + bzw. NADH wechselwirkenden Seitenketten sowie deren Nomenklatur sind gezeigt. H-Brücken wurden mit dem Programm UCSF Chimera nach Mills und Dean bestimmt (Mills und Dean, 1996; Pettersen et al., 2004) und sind durch gestrichelte Linien angedeutet. Das FMN ist in Gelb dargestellt. Ein sehr wichtiger Unterschied zwischen der Struktur des Proteins im oxidierten und im reduzierten Zustand ist die Umorientierung der Peptidbindung des Glu95F (Abb. 2.13). In der Struktur des reduzierten Proteins zeigt die Peptidbindung vom FMN weg. Dabei verdrängt sein Carbonyl-Sauerstoff ein H2O, das in der Struktur des oxidierten Proteins die gesamte Schleife über H-Brücken stabilisiert. In dieser Konformation ist genügend Platz für die Anbindung des NADH. In der Struktur des oxidierten Proteins zeigt die Carbonyl-Gruppe der Peptidbindung zum FMN hin. Aufgrund des sterischen Anspruchs der Carbonyl-Gruppe wird das NAD+ bei dieser Umorientierung aus der Bindetasche geschoben (Abb. 2.13). 20 Einleitung A) B) E95 E95 D94 Abbildung 2.13: Peptidrückgrate der NADH-Bindestelle. A) zeigt die überlagerten Strukturen des A. aeolicus NuoEF im oxidierten Zustand (grau) mit NAD+ (grün) und im reduzierten Zustand (hellblau) mit NADH (magenta). Das Rückgrat von Glu95F ist jeweils in der gleichen Farbe wie das gebundene Nukleotid dargestellt. Das in der reduzierten Struktur verdrängte H2O ist grün markiert. B) zeigt die überlagerten Strukturen des T. thermophilus (beige) und A. aeolicus (hellblau) NuoF jeweils im reduzierten Zustand mit gebundenem NADH (T. thermophilus: orange, A. aeolicus: magenta). Die Orientierung des Rückgrats an den Positionen Asp94F und Glu95F (Farbgebung der Nukleotide entsprechend) ist gezeigt. Die Umorientierung der Carbonyl-Gruppe scheint eine Art „Auswurf-Mechanismus“ für das Produkt NAD+ darzustellen. Aus der Struktur geht nicht hervor, ob die Änderung des RedoxZustands des FMN oder des N1a der Schalter für die Umorientierung des Rückgrats von Glu95F ist. Die Anbindung des Nukleotids kann als Schalter ausgeschlossen werden, da die Peptidbindung in der Struktur des oxidierten und reduzierten Proteins ohne gebundenes Nukleotid jeweils die entsprechende Orientierung einnimmt. In der Struktur des T. thermophilus Komplex I mit gebundenem NADH zeigt die Carbonyl-Gruppe des Glu95F ebenfalls vom FMN weg, jedoch weist die Peptidbindung des Asp94F eine andere Orientierung auf (Abb. 2.13 B, Berrisford und Sazanov, 2009). Aufgrund der geringen Auflösung von 3.1 Å ist es jedoch schwierig, die genaue Orientierung des Rückgrats in dieser Struktur anzugeben. In der A. aeolicus NuoEF Struktur ist die Seitenkette des Glu95F über H-Brücken mit den Amid-Stickstoffen von Arg103F und Asp104F und mit der Seitenkette des Tyr138F im oxidierten und reduzierten Zustand fest verankert. In der Struktur des reduzierten Proteins ist die Seitenkette des Glu95F zusätzlich über ein H2O-Molekül mit dem NN7 des NADH verbunden (Abb. 2.14). In beiden Strukturen ist der Amid-Stickstoff des Glu95F über ein H2O-Molekül mit dem N3 des FMNs verbrückt. Sehr wichtig scheint das von FMN, Asp94F und Tyr180F koordinierte Wasser zu sein (Abb. 2.14). In der Struktur des oxidierten Proteins bildet es zusätzlich eine H-Brücke zu der zum FMN hin orientierten CarbonylGruppe des Glu95F. In der Struktur des reduzierten Proteins bildet es zusätzlich zur H-Brücke 21 Einleitung zum O4- noch eine zum N5-Atom des FMNs (Abb. 2.14 B). In beiden Strukturen ist das Glu97F über ein H2O-Molekül mit dem O2' der Nikotinamid-Ribose verbrückt (Abb. 2.14). Das Glu97F verdreht sich in der Struktur des reduzierten Proteins, um mit Tyr180F eine HBrücke zu bilden. Mit seinem Amid-Stickstoff bildet es zusätzlich eine H-Brücke zum NO7 des Nikotinamids aus. Es wird vermutet, dass die H-Brücke zu Tyr180F die Anbindung des Nikotinamids in 3.0 Å Abstand zum Isoalloxazinring des FMNs stabilisiert, um den Hydridtransfer zu unterstützen. Die Mutation des Glu97F im E. coli Komplex I (Glu95F) zu Glutamin führte zu einer um etwa ein Drittel verminderten Aktivitäten (Euro et al., 2009). Die Mutation des Tyr180F zu Cys ist bei einem Patienten mit dem Leigh-like Syndrom verknüpft (Bénit et al., 2001; Pagniez-Mammeri et al., 2012). Zudem wird vermutet, dass das Tyr unter Ausbildung eines Tyrosyl-Radikals an der ROS-Produktion im Komplex I beteiligt sein könnte. A) B) E97 E97 E95 E95 Y180 Y180 D94 D94 Abbildung 2.14: H-Brücken-Netzwerk am aktiven Zentrum im oxidierten A) und reduzierten B) Subkomplex NuoEF. A) zeigt den oxidierten Subkomplex (grau) mit gebundenem NAD+ (grün) und B) den reduzierten (hellblau) mit gebundenem NADH (magenta). Die verbrückenden H2O sind dunkelblau markiert. H-Brücken sind durch gestrichelte Linien dargestellt. Eine komplette Auflistung aller von NAD+/NADH und FMN zu NuoF gebildeten H-Brücken in erster und zweiter Koordinationssphäre sind im Anhang 7.1 (Tab. 7.1 – 7.4) aufgeführt. 22 Einleitung 2.6 λ-Red vermittelte Rekombination Unter homologer Rekombination versteht man den Austausch von DNA-Abschnitten, die über homologe DNA-Sequenzen verfügen. Eukaryoten und Prokaryoten benutzen ihr Rekombinase-System als Reparaturmechanismus, um beschädigte chromosomale DNA über einen Austauschmechanismus zu reparieren. Das E. coli Rekombinase-System besteht hauptsächlich aus RecA und RecBCD (Clark und Sandler, 1994). Die Exonuklease-Aktivität des RecBCD-Komplexes erzeugt 3'-Überhänge, an die RecA bindet und die Rekombination vermittelt (Anderson und Kowalczykowski, 1997). Zur gezielten in vitro Manipulation von DNA ist das E. coli Rekombinations-System nicht geeignet, da es für den Austausch etwa 1 kbp lange, homologe Bereiche benötigt. Zudem verhindert die Exonuklease-Aktivität von RecBCD den Einbau linearer DNA. Das Rekombinase-System des λ-Phagen bietet die Möglichkeit, Gene durch lineare Einzelstrang (ss) und Doppelstrang (ds) DNA in einem E. coli-Wirt gezielt zu modifizieren (Murphy, 1998; Ellis et al., 2001). Das -Red System (recombinant defective) besteht aus den Proteinen Gam, Exo und Beta und benötigt für eine effiziente Rekombination etwa 36 – 50 nt lange homologe Bereiche (Datsenko und Wanner, 2000). λ-Gam inhibiert den RecBCD-Komplex des Wirts (Murphy, 1991). Die 5'-3' Exonuklease λ-Exo baut einen Strang des dsDNA-Fragments ab und lässt einen intakten ssDNA Strang zurück (Mosberg et al., 2010). λ-Beta bindet an die ssDNA, verhindert so den Abbau durch Nukleasen und vermittelt die Anlagerung an die komplementären Stränge (Court et al., 2002). Die lineare ssDNA wird hauptsächlich als Okazaki-Fragment an der Replikationsgabel auf dem Folgestrang eingebaut, wobei auch der Einbau am Leitstrang beobachtet wird (Mosberg et al., 2010). Die -Red vermittelte Rekombination kann unabhängig von der Größe der zu manipulierenden DNA eingesetzt werden und hat sich zu einer bewährten Methode zur Modifikation chromosomaler und episomaler DNA entwickelt (Jeong et al., 2013; Pohl et al., 2007b). Das in unserem Arbeitskreis zur Expression der E. coli nuo-Gene verwendete Plasmid pBADnuo nuoFhis ist aufgrund seiner Größe einer ortsgerichteten Mutagenese nicht zugänglich. Um Punktmutationen in einzelne Gene einzufügen muss zuerst eine ortsgerichtete Mutagenese und dann eine -Red vermittelte Rekombination angewendet werden (Abb. 2.15). Um die Effizienz der -Red vermittelten Rekombination zu steigern, wurde in dieser Arbeit das Suizid-System nptI-sacB verwendet (Ried und Collmer, 1987). 23 Einleitung 3. Ortsgerichte Mutagenese Subklon nuo pVO1100 5 – 7 kBp 5 – 7 kBp 1. PCR 4. PCR nptI-sacB-Kassette mit homologen Bereichen zum Zielgen mutiertes nuo-Zielgen 2. λ-Red vermittelte Rekombination und Selektion auf Kanamycin pBADnuo nuoFhis Subklon nuo pBADnuo nuoFhis::nptIsacB 21 kBp zur Darstellung des Expressionsvektor mit nptIsacB-Kassette im Zielgen 25 kBp 5. λ-Red vermittelte Rekombination und Selektion auf Saccharose pBADnuo nuoFhis 21 kBp Expressionsvektor mit Punktmutation im Zielgen Abbildung 2.15: Schematische Darstellung der Strategie zur Mutagenese des Komplex I- Expressionsplasmids. Zur Insertion der Selektions-Kassette in den Expressionsvektor pBADnuo nuoFhis wird zunächst mittels PCR die nptI-sacB-Kassette aus der Vorlage pVO1100 mit Oligonukleotiden, welche die für die Rekombination notwendigen homologen Bereiche tragen, amplifiziert. Im zweiten Schritt wird mittels λ-Red vermittelter Rekombination die nptI-sacB-Kassette in der Verktor insertiert und positive Klone auf kanamycinhaltigen Medium selektiert. Zur Insertion von Punktmutationen in den Expressionsvektor wird zunächst durch Ortsgerichtete Mutagenese an einem Subklon, der das Zielgen enthält, die gewünschte Mutation eingebracht. Im nächsten Schritt wird das mutierten Gen mittels PCR amplifiziert. Im letzten Schritt wird das mutierte Gen mit Hilfe der λ-Red vermittelten Rekombination in den Vektor pBADnuo nuoFhis eingebracht. Die homologen Bereiche sind grün und das Zielgen blau dargestellt. 24 Einleitung Zur Kontrolle wird die durch sacB vermittelte Empfindlichkeit gramnegativer Bakterien gegen Saccharose genutzt (Gay et al., 1985). Die durch nptI vermittelte Kanamycin-Resistenz wird zur Selektion auf den Einbau der Kassette verwendet. Zunächst wird die nptI-sacB-Kassette mittels Polymerasekettenreaktion (polymerase chain reaction, PCR) amplifiziert und durch -Red vermittelte Rekombination in das Zielgen insertiert. Die für die Rekombination benötigten homologen Bereiche werden durch die Oligonukleotide in die Sequenzen eingeführt. Zur Selektion positiver Klone wird die durch nptI vermittelte Kanamycin-Resistenz genutzt. Die gewünschten Punktmutationen werden durch ortsgerichtete Mutagenese in einen Subklon, der das Zielgen enthält, eingefügt. Es werden lineare Fragmente, welche die gewünschten Mutationen tragen, mittels PCR amplifiziert und durch -Red vermittelte Rekombination gegen die Selektions-Kassette im Plasmid pBABnuo nuoFhis::nptI-sacB ausgetauscht. Zur Selektion auf die gewünschten Klone wird die durch sacB vermittelte Empfindlichkeit gegen Saccharose genutzt. Um die Rekombination des PCR-Produkts mit genomischer DNA zu verhindern, wurde in dieser Arbeit der E. coli Stamm DH5αΔnuo/pKD46 verwendet (Pohl et al., 2007b). Der Vektor pKD46 trägt das red-Operon unter der Kontrolle eines ara-Promotors und ein Temperatur-sensitives Replikon, das die Replikation bei 37 °C verhindert. 25 Einleitung 2.7 Ziel der Arbeit NADPH wird von Komplex I nur sehr langsam umgesetzt. Strukturanalysen der NADHBindetasche haben gezeigt, dass vermutlich Glu183F aufgrund von sterischen und elektrostatischen Abstoßungen für die Substratspezifität von Komplex I verantwortlich ist. Um diese Annahme zu überprüfen, sollte Glu183F im E. coli Komplex I jeweils gegen Ala, Asn, Asp, Gln und Gly ausgetauscht werden. Zudem sollte, um eine elektrostatische Anziehung an diese Position einzubringen, das Glu gegen His ausgetauscht werden. Die strukturellen Effekte der Mutationen sollten durch die homologen Varianten des A. aeolicus NuoEFHis-Subkomplexes untersucht werden. Diese sollten präpariert und falls möglich kristallisiert werden. Auf dem Genom eines am Leigh-like-Syndrom erkrankten Patienten wurde die Mutation Tyr204Cys auf der Untereinheit NDUFV1 (homolog zu E. coli Tyr178CysF) nachgewiesen. Die Auswirkung der Mutation auf die Aktivität des Komplex I sollte durch Mutation der homologen Position im E. coli Komplex I gegen Phe, Leu, Ala und Cys untersucht werden. Die Varianten sollten produziert, präpariert und charakterisiert werden. Quantenmechanische Rechnungen deuten auf eine Beteiligung aromatischer Aminosäuren am Elektronentransfer im Komplex I hin. In vorangegangen Arbeiten wurde gezeigt, dass die Variante W221LG, mit der Mutation zwischen N5 und N6a, gegenüber dem Ausgangsstamm eine um 30% verminderte d-NADH-Oxidaseaktivität aufweist. Um strukturelle Gründe für die verminderte Aktivität ausschließen zu können, sollte versucht werden Doppelmutanten von der Mutante Trp221LeuG zu generieren, die die ursprüngliche Aktivität aufweisen. Hierfür sollten Aliphatische Aminosäuren an ähnlicher Position zwischen N5 und N6a gegen Trp ausgetauscht und die Aktivität der Doppelmutanten bestimmt werden. E. coli besitzt neben Komplex I noch die alternative NADH:Ubichinon Oxidoreduktase. Um die Komplex I-vermittelte NADH-Oxidaseaktivität in den Cytoplasmamembranen zu bestimmen, muss das kostspielige spezifische Substrat d-NADH verwendet werden. Um dies zu umgehen, sollte in dieser Arbeit der E. coli Stamm BW25113nptI Δnuo Δndh als Überproduzent für den E. coli Komplex I etabliert werden. In Kooperation mit Dr. Judy Hirst sollte untersucht werden, ob die Mutationen Val96ProE und Asn142MetE die in der einzeln produzierten Untereinheit NuoE das Mittenpunktspotential des Fe/S-Zentrums N1a drastisch erniedrigen, den gleichen Effekt im gesamten E. coli Komplex I haben. Die Varianten sollten dargestellt und die physiologischen Auswirkungen der Mutationen untersucht werden. 26 Einleitung Der Redox-induzierte Konformationswechsel der Peptidbindung von Glu95F wird als „Auswurf-Mechanismus“ für das Produkt NAD+ diskutiert. In dieser Arbeit sollte versucht werden, durch Mutationen H-Brücken einzubringen, die die Peptidbindung des Glu95F in einer Konformation fixieren. Zum strukturellen Nachweis der Fixierung sollte Gly129E in A. aeolicus NuoEF jeweils gegen Ser und Asp ausgetauscht werden. Die Varianten sollten präpariert und kristallisiert werden. Zur funktionellen Charakterisierung sollte die homologe Position Gly135E im E. coli Komplex I mutiert werden. In der katalytischen Untereinheit NarG der Nitratreduktase A führte die Deletion einer Oberflächen-Salzbrücke zum mangelnden Einbau oder zum Verlust der Kofaktoren. Die das Zentrum N7 bindende C-terminale Domäne von NuoG ist homolog zu NarG und weist eine Oberflächen-Salzbrücke zwischen Arg280G und Glu617G auf. In dieser Arbeit sollte Glu617G gegen Ala im E. coli Komplex I ausgetauscht und die Auswirkung auf die Stabilität des Komplexes untersucht werden. Die EPR-Signale des Komplex I wurden anhand biochemischer Charakterisierung den einzelnen Fe/S-Zentren zugeordnet. Allerdings wird diese Zuordnung seit der Veröffentlichung der Struktur des peripheren Arms scharf diskutiert. Es ist strittig, ob das EPR-Signal N4 von den strukturell definierten Fe/S-Zentren N4, N5 oder N6a generiert wird. In dieser Arbeit sollten die Liganden der strukturell definierten Zentren N4 und N5 jeweils gegen Alanine ausgetauscht werden. Anhand der Aktivitäten und der EPR-Spektren sollte versucht werden, die EPR-Signale den strukturell definierten Zentren zuzuordnen. In Zusammenarbeit mit Karoline Aierstock und Dr. Lothar Kussmaul, Boehringer Ingelheim, Biberach, sollten für enzymkinetische und strukturelle Untersuchungen sowie zur Bestimmung der ROS-Produktion die Positionen Glu95F, Glu97F und Tyr180F des A. aeolicus Subkomplexes NuoEFHis gegen verschiedene Aminosäuren ausgetauscht werden. 27 Material und Methoden 3 Material und Methoden 3.1 Chemikalien Tabelle 3.1 gibt alle in dieser Arbeit verwendeten Chemikalien und deren Hersteller wieder. Tabelle 3.1: Verwendete Chemikalien. Chemikalie Acrylamid-Bisacrylamid-Lösung (29:1, w/w) Hersteller Gerbu Adenosin-5'-diphosphoribose (ADP-Ribose) Sigma Aldrich Agar Agarose low EEO 9-Amino-3-chlor-7-methoxyacridin (ACMA) Ammoniumchlorid (NH4Cl) Applichem Applichem Sigma Aldrich Roth Ammoniumperoxodisulfat (APS) Merck Ammoniumsulfat ((NH4)2SO4) Grüssing Ampicillin, Natriumsalz Roth Amplex Red Antifoam 204 L-(+)-Arabinose Biobeads SM-2 1,3-Bis(tris(hydroxymethyl)-methylaminopropan) (BisTris) Borsäure Bromphenolblau Invitrogen Sigma Aldrich Roth BioRad Roth Applichem Sigma Aldrich Calciumchlorid (CaCl2) Fluka Carbonylcyanid-3-chlorphenylhydrazon (CCCP) Sigma Casein (säurehydrolysiert) Chloramphenicol Cobaltchlorid (CoCl2) Coomassie Brilliant Blue R-250 (CBB R-250) L-Cystein 3,3'-Diaminobenzidin 5-(Diethoxyphosphoryl)-5-methyl-1-pyrrolin-N-oxid (DEPMPO) 2,3-Dimethoxy-5-methyl-6-decyl-1,4-benzochinon (DecylUbichinon) Dithiothreitol (DTT) DNase I n-Dodecyl-β-D-maltopyranosid (DDM) Roth Roth Merck Applichem Applichem Sigma 28 Enzo Sigma Gerbu Applichem Applichem Material und Methoden Tabelle 3.1; Fortsetzung. E. coli polare Lipide (E. coli polar lipid extract) Eisen-Ammonium-Citrat (14.5 – 16% (w/w) Eisen) Essigsäure Ethanol abs. Ethidiumbromid-Lösung (1%ig) Ethylendiamintetraessigsäure, di-Natriumsalz (EDTA) Fenazaquin Glucose Glycerin (~86%) Glycin Hefeextrakt 2-(4-(2-Hydroxyethyl)-1-piperazinyl)-ethansulfonsäure, Natriumsalz (HEPES) Imidazol Isopropanol Isopropyl-β-D-thio-galactopyranosid (IPTG) Kaliumacetat Kaliumchlorid (KCl) Kaliumcyanid (KCN) Kaliumdihydrogenphosphat (KH2PO4) Kaliumhexacyanoferrat(III) (K3[Fe(CN)6]) di-Kaliumhydrogenphosphat (K2HPO4) Kaliumiodid (KI) Kalium-Natrium- Tartrat (Seignettesalz) Kanamycinsulfat Kupfersulfat Pentahydrat (CuSO4·5 H2O) Magnesiumchlorid (MgCl2) Magnesiumsulfat (MgSO4) Malonat D-Mannitol Meerrettichperoxidase (HRP) 2-Mercaptoethanol Methanol 2-Methylbutan Methylcyclohexan 2-N-Morpholino-ethansulfonsäure (MES) 3-N-Morpholino-propansulfonsäure (MOPS) Tri-Natriumcitrat Natriumchlorid (NaCl) Natriumdithionit (Na2S2O4) Avanti Polar Lipids Sigma VWR VWR Roth Sigma Aldrich Sigma Grüssing Roth Applichem Ohly Gerbu Applichem Sigma Aldrich Gerbu Roth Merck Merck Roth Applichem Merck Merck Grüssing Roth Grüssing VWR Grüssing Institutsbestände Applichem MP Biomedicals Roth Institutsbestände Merck Merck Applichem Applichem Roth VWR Merck 29 Material und Methoden Tabelle 3.1; Fortsetzung. Natriumdodecylsulfat (SDS) di-Natriumhydrogenphosphat (Na2HPO4) Natriumhydroxid (NaOH) tri-Natriumphosphat (Na3PO4) Natriumsulfat (Na2SO4) Nickelsulfat (NiSO4) Nicotinamid-Adenin-Dinukleotid, di-Natriumsalz, oxidierte Form (NAD+) Nicotinamid-Adenin-Dinukleotid, di-Natriumsalz, reduzierte Form (NADH) Nicotinamidadenin-Dinukleotid-2'-phosphat, Tetra-Natriumsalz Hydrat, reduzierte Form (NADPH) Nicotinamid-Hypoxanthin-Dinukleotid, Natriumsalz, reduzierte Form (d-NADH) desoxy-Nukleosidtriphosphatemix Pepton aus Casein (pankreatischer Verdau) Phenylmethylsulfonylfluorid (PMSF) Phosphorsäure (85%; w/w) Piericidin A Polyethylenglycol (PEG) 3350 Riboflavin Rinderserum Albumin (BSA) RNase A D-(+)-Saccharose Salzsäure (HCl) Succinat Superoxiddismutase (Rinderleber) N,N,N',N'-Tetramethyldiamin (TEMED) Thiaminhydrochlorid Trichloressigsäure (TCA) Tris-(hydroxymethyl)-aminomethan (Tris) N-[Tri(hydroxymethyl)-methyl]-glycin (Tricin) L-Tryptophan Tween 20 Valinomycin Wasserstoffperoxyd (H2O2) 30 Serva VWR VWR Grüssing Roth Aldrich Sigma Aldrich/ Roth Roth Sigma Aldrich Sigma Aldrich Thermo Scientific Applichem Applichem Merck Aus Institutsbeständen Sigma Aldrich Applichem Applichem Applichem Roth VWR Serva Sigma Aldrich Serva Merck Merck Applichem Roth Merck Acros Sigma Aldrich Sigma Aldrich Material und Methoden 3.2 Stämme Die in dieser Arbeit verwendeten Bakterienstämme sind in Tabelle 3.2 aufgeführt. Tabelle 3.2: Verwendete E. coli Stämme. E. coli Stamm DH5α DH5nuo BW25113 BW25113nuo BW25113ndh::nptI BW25113nptI nuo ndh Rosetta(DE3) Genotyp F- Φ80lacZ∆M15 Δ(lacZYAargF)U169 recA1 endA1 hsdR17 (rK- mK+) phoA supE44 λ- thi-1 gyrA96 relA1 DH5Δnuo lacIq rrnBT14 lacZWJ16 hsdR514 araBADAH33 rhaBADLD78 BW25113 nuo BW25113 ndh::FRTnptIFRT BW25113 nuo ndh; nptI F- ompT hsdSB (rB-mB-) gal dcm pRARE (camR) Referenz Invitrogen (Pohl, 2008) (Datsenko & Wanner, 2000) (Macedo Vranas, 2013) (Baba et al., 2006) T. Lenn (persönliche Mitteilung) Novagen 3.2 Verwendete Vektoren Die in dieser Arbeit verwendeten und dargestellten Vektoren sind in Tabelle 3.3 aufgeführt. Tabelle 3.3: Verwendete Vektoren. Vektor pCA24NnuoCD Genotyp pCA24NnuoCD T504W pCA24NnuoCD S519W pCA24NnuoE pCA24NnuoCD, nuoCD T504W pCA24NnuoCD, nuoCD S519W camR, ColE1 origin, lacI-PT5-lac his6-nuoE pCA24NnuoE V96P pCA24NnuoE V96P/N142M pCA24NnuoE G135D pCA24NnuoE G135S pCA24NnuoE N142M pCA24NnuoI pCA24NnuoE, nuoE V96P pCA24NnuoE, nuoE V96P/N142M pCA24NnuoE, nuoE G135D pCA24NnuoE, nuoE G135S pCA24NnuoE, nuoE N142M camR, ColE1 origin, lacI-PT5-lac his6-nuoI pCA24NnuoI L65W pCA24NnuoI, nuoI L65W camR, ColE1 origin, lacI-PT5-lac his6-nuoCDgfp Referenz (Kitagawa et al., 2005) (Dek vić, 2012) (Dek vić, 2012) (Kitagawa et al., 2005) diese Arbeit diese Arbeit diese Arbeit diese Arbeit diese Arbeit (Kitagawa et al., 2005) (Dek vić, 2012) 31 Material und Methoden Tabelle 3.3; Fortsetzung. pCA24NnuoF camR, ColE1 origin, lacI-PT5-lac his6-nuoF pCA24NnuoF E183A pCA24NnuoF E183D pCA24NnuoF E183G pCA24NnuoF E183H pCA24NnuoF E183N pCA24NnuoF E183Q pUC25nuoE´–G pCA24NnuoF, nuoF E183A pCA24NnuoF, nuoF E183D pCA24NnuoF, nuoF E183G pCA24NnuoF, nuoF E183H pCA24NnuoF, nuoF E183N pCA24NnuoF, nuoF E183Q ampR, pUC origin, lacZ´, ´nuoE-G nuoFcstrep-tagII pUC25nuoE´–G, nuoF Y178A pUC25nuoE´–G, nuoF Y178F pUC25nuoE´–G, nuoF Y178L pUC25nuoE´–G, nuoF Y178C pUC25nuoE´–G, nuoG H101A pUC25nuoE´–G, nuoG C105A pUC25nuoE´–G, nuoG V107W pUC25nuoE´–G, nuoG C108A pUC25nuoE´–G, nuoG C114A pUC25nuoE´–G, nuoG C153A pUC25nuoE´–G, nuoG C156A pUC25nuoE´–G, nuoG C159A pUC25nuoE´–G, nuoG C203A pUC25nuoE´–G , nuoG R219L pUC25nuoE´–G, nuoG R219W/W221L pUC25nuoE´–G nuoF Y178A pUC25nuoE´–G nuoF Y178F pUC25nuoE´–G nuoF Y178L pUC25nuoE´–G nuoF Y178C pUC25nuoE´–G H101A pUC25nuoE´–G C105A pUC25nuoE´–G V107W pUC25nuoE´–G C108A pUC25nuoE´–G C114A pUC25nuoE´–G C153A pUC25nuoE´–G C156A pUC25nuoE´–G C159A pUC25nuoE´–G C203A pUC25nuoE´–G R219L pUC25nuoE´–G R219W/W221L pUC25nuoE´–G R219W/W221R pUC25nuoE´–G W221L pUC25nuoE´–G E617A pBADnuo nuoFhis (Kitagawa et al., 2005) diese Arbeit diese Arbeit (Hubrich, 2010) (Hubrich, 2010) (Hubrich, 2010) (Hubrich, 2010) (Kohlstädt, 2009) diese Arbeit diese Arbeit diese Arbeit diese Arbeit (Burschel, 2013) (Burschel, 2013) (Dek vić, 2012) (Burschel, 2013) (Burschel, 2013) diese Arbeit diese Arbeit (Burschel, 2013) diese Arbeit (Dek vić, 2012) (Schäuble, 2013) pUC25nuoE´–G, nuoG R219W/W221R (Schäuble, 2013) pUC25nuoE´–G, nuoG W221L pUC25nuoE´–G, nuoG E617A camR, p15a origin, araC-ParaBAD nuoA-N, his6-nuoF pBADnuo nuoFhis, nuoCD:: nptI-sacR sacB, nuoG W221L (Dörner, 2010) diese Arbeit (Pohl et al., 2007c) pBADnuo nuoFhis nuoCD T504W nuoG W221L pBADnuo nuoFhis, nuoCD T504W, nuoG W221L (Dek vić, 2012) pBADnuo nuoFhis nuoCD S519W nuoG W221L pBADnuo nuoFhis, nuoCD S519W, nuoG W221L (Dek vić, 2012) pBADnuo nuoFhis nuoE::nptIsacB pBADnuo nuoFhis, nuoE::nptI-sacR sacB (Dörner, 2010) pBADnuo nuoFhis nuoE V96P pBADnuo nuoFhis, nuoE V96P diese Arbeit pBADnuo nuoFhis nuoE V96P/N142M pBADnuo nuoFhis, nuoE V96P/N142M diese Arbeit pBADnuo nuoFhis nuoCD::nptI-sacB nuoG W221L 32 (Dek vić, 2012) Material und Methoden Tabelle 3.3; Fortsetzung. pBADnuo nuoFhis, nuoE G135D diese Arbeit pBADnuo nuoFhis, nuoE G135S pBADnuo nuoFhis, nuoE N142M diese Arbeit diese Arbeit (Hubrich, 2010) pBADnuo nuoFhis Y178A pBADnuo nuoFhis Y178C pBADnuo nuoFhis Y178F pBADnuo nuoFhis Y178L pBADnuo nuoFhis E183A pBADnuo nuoFhis E183D pBADnuo nuoFhis E183H pBADnuo nuoFhis E183G pBADnuo nuoFhis E183N pBADnuo nuoFhis E183Q pBADnuo nuoFhis, hisnuoF::nptIsacR sacB pBADnuo nuoFhis, hisnuoF Y178A pBADnuo nuoFhis, hisnuoF Y178C pBADnuo nuoFhis, hisnuoF Y178F pBADnuo nuoFhis, hisnuoF Y178L pBADnuo nuoFhis, hisnuoF E183A pBADnuo nuoFhis, hisnuoF E183D pBADnuo nuoFhis, hisnuoF E183H pBADnuo nuoFhis, hisnuoF E183G pBADnuo nuoFhis, hisnuoF E183N pBADnuo nuoFhis, hisnuoF E183Q pBADnuo nuoFhis nuoGc::nptIsacB pBADnuo nuoFhis, nuoGcomplete:: nptI-sacR sacB (Dek vić, 2012) pBADnuo nuoFhis nuoG H101A pBADnuo nuoFhis, nuoG H101A (Burschel, 2013) pBADnuo nuoFhis nuoG C105A pBADnuo nuoFhis nuoG V107W W221L pBADnuo nuoFhis nuoG C108A pBADnuo nuoFhis nuoG C114A pBADnuo nuoFhis nuoG C153A pBADnuo nuoFhis nuoG C156A pBADnuo nuoFhis nuoG C159A pBADnuo nuoFhis nuoG C203A pBADnuo nuoFhis, nuoG C105A (Burschel, 2013) pBADnuo nuoFhis, nuoG V107W W221L (Dek vić, 2012) pBADnuo nuoFhis, nuoG C108A (Burschel, 2013) pBADnuo nuoFhis, nuoG C114A (Burschel, 2013) pBADnuo nuoFhis, nuoG C153A diese Arbeit pBADnuo nuoFhis, nuoG C156A diese Arbeit pBADnuo nuoFhis, nuoG C159A (Burschel, 2013) pBADnuo nuoFhis, nuoG C203A diese Arbeit pBADnuo nuoFhis nuoE G135D pBADnuo nuoFhis nuoE G135S pBADnuo nuoFhis nuoE N142M pBADnuo nuoFhis::nptI-sacB (Gnandt, 2012) (Gnandt, 2012) diese Arbeit (Gnandt, 2012) diese Arbeit diese Arbeit (Hubrich, 2010) diese Arbeit diese Arbeit (Hubrich, 2010) pBADnuo nuoFhis nuoG R219L pBADnuo nuoFhis nuoG R219W/W221L pBADnuo nuoFhis, nuoG R219L (Dek vić, 2012) pBADnuo nuoFhis, nuoG R219W/W221L (Schäuble, 2013) pBADnuo nuoFhis nuoG R219W/W221R pBADnuo nuoFhis, nuoG R219W/W221R (Schäuble, 2013) 33 Material und Methoden Tabelle 3.3; Fortsetzung. pBADnuo nuoFhis nuoG W221L pBADnuo nuoFhis nuoG E617A pBADnuo nuoFhis nuoG W221L nuoI2::nptI-sacB pVO1100 pBADnuo nuoFhis, nuoG W221L (Dörner, 2010) pBADnuo nuoFhis, nuoG E617A diese Arbeit pBADnuo nuoFhis, nuoG W221L, nuoI2:: nptI-sacR sacB (Dek vić, 2012) ampR, kanR, nptI-sacRsacB pKD46 ampR, R101 origin, araCParaBAD, exo, bet, gam pETBlue-1nuoEFhisG ampR, pUC origin, PT7lac, lacZ´, nuoEFaxa-his6 nuoG ampR, pUC origin, PT7lac, lacZ´, nuoEFaxa-his6 pETBlue-1nuoEFhis, nuoE G129D V. Spehr (persönliche Mitteilung) (Datsenko und Wanner, 2000) (Kohlstädt, 2009) pETBlue-1nuoEFhis pETBlue-1nuoEFhis nuoE G129D pETBlue-1nuoEFhis nuoE G129D pETBlue-1nuoEFhis E95D pETBlue-1nuoEFhis E95N pETBlue-1nuoEFhis E95Q pETBlue-1nuoEFhis E97D pETBlue-1nuoEFhis E97N pETBlue-1nuoEFhis E97Q pETBlue-1nuoEFhisY180A pETBlue-1nuoEFhisY180F pETBlue-1nuoEFhisY180L pETBlue-1nuoEFhisY180C pETBlue-1nuoEFhis E185A pETBlue-1nuoEFhis E185D pETBlue-1nuoEFhis E185G pETBlue-1nuoEFhis E185H pETBlue-1nuoEFhis E185N pETBlue-1nuoEFhis E185Q 34 diese Arbeit diese Arbeit pETBlue-1nuoEFhis, nuoE G129D diese Arbeit pETBlue-1nuoEFhis, nuoFhis E95D pETBlue-1nuoEFhis, nuoFhis E95N pETBlue-1nuoEFhis, nuoFhis E95Q pETBlue-1nuoEFhis, nuoFhis E97D pETBlue-1nuoEFhis, nuoFhis E97N pETBlue-1nuoEFhis, nuoFhis E97Q pETBlue-1nuoEFhis, nuoFhis Y180A pETBlue-1nuoEFhis, nuoFhis Y180F pETBlue-1nuoEFhis, nuoFhis Y180L pETBlue-1nuoEFhis, nuoFhis Y180C pETBlue-1 nuoEFhis, nuoFhis E185A pETBlue-1 nuoEFhis, nuoFhis E185D pETBlue-1 nuoEFhis, nuoFhis E185G pETBlue-1 nuoEFhis, nuoFhis E185H pETBlue-1 nuoEFhis, nuoFhis E185N pETBlue-1 nuoEFhis, nuoFhis E185Q (Engel, 2010) (Engel, 2010) (Engel, 2010) (Engel, 2010) (Engel, 2010) (Engel, 2010) (Engel, 2010) (Engel, 2010) (Engel, 2010) diese Arbeit (Burschel, 2011) diese Arbeit (Burschel, 2011) diese Arbeit diese Arbeit diese Arbeit Material und Methoden 3.3 DNA-Oligonukleotide Die in dieser Arbeit verwendeten DNA-Oligonukleotide sind in den Tabellen 3.4 bis 3.6 aufgeführt. Tabelle 3.4: Sequenzen der DNA-Oligonukleotide für die ortsgerichtete Mutagenese an den nuo-Genen von E. coli und A. aeolicus. Die für die Mutagenese der A. aeolicus nuo-Gene verwendete Oligonukleotide sind mit axa im Subskript gekennzeichnet. Die neu generierten Codons sind fett, die ausgetauschten Basen kursiv hervorgehoben. Zum Nachweis der Mutation wurde eine neue Restriktionsschnittstelle eingefügt. Diese ist durch Unterstrich gekennzeichnet und die ausgetauschten Basen kursiv hervorgehoben. Ausgetauschte Basen zur Vermeidung von Haarnadelstrukturen sind grün markiert. Die Sequenzen der für die ortsgerichtete Mutagenese notwendigen revers komplementären Oligonukleotide sind nicht aufgeführt. Sie werden im Text mit dem g eic en Namen un er En ung „rev“ erw n . Deletierte Schnittstellen sind mit * markiert. Oligonukleotid nuoCD T504W_fwd nuoCD S519W_fwd nuoE V96P_fwd nuoE G135S_fwd nuoE G135D_fwd nuoE N142M_fwd nuoF Y178A_fwd nuoF Y178C_fwd nuoF Y178F_fwd nuoF Y178L_fwd nuoF E183A_fwd nuoF E183D_fwd nuoF E183G_fwd nuoF E183H_fwd nuoF E183N_fwd nuoF E183Q_fwd Sequenz 5'-CCGAAAGAGCGCTGGCTGCAGCATATCG AAAC-3' 5'-CTTCCTGCAAGTGTGGTGGGGCCCGGTGA TGCC-3' 5'-GTGACAGCGTGCCGTGCCATATTAATGGT TATCAGG-3' 5'-CAACTTGCTGCTTAAGTAACTGTGATAAA G-3' 5'-CTTGCTGCCTGGACAACTGTGATAAAGGG CCCAACATGATGATC-3' 5'-GTGATAAAGGGCCCATGATGATGATCGA TG-3' 5'-GCAGGGCGCGCCATATGCGGGGAAG-3' 5'-GCAGGGCGCTGCATATGCGGGGAAG-3' 5'-GCAGGGCGCTTCATATGCGGGGAAG-3' 5'-GGGCAGGGCGATTAATCTGCGGGGAAG-3' 5'-CATCTGCGGGGAAGCGACAGCATTAATC AACTCC-3' 5'-CTGCGGGGAAGATACAGCATTAATCAACT CCCTGG-3' 5'-CATCTGCGGGGAAGGCACAGCATTAATC AACTCC-3' 5'-CTGCGGGGAACACACAGCATTAATCAACT CCCTGG-3' 5'-CTGCGGGGAAAACACAGCATTAATCAACT CCCTGG-3' 5'-CTGCGGGGAACAGACAGCATTAATCAACT CCCTGG-3' Schnittstelle PstI BanII VspI AflII ApaI BanII NdeI NdeI NdeI VspI VspI VspI VspI VspI VspI VspI 35 Material und Methoden Tabelle 3.4; Fortsetzung. nuoG H101A_fwd nuoG C105A_fwd nuoG V107W_fwd nuoG C108A_fwd nuoG C114A_fwd nuoG C153A_fwd nuoG C156A_fwd nuoG C159A_fwd nuoG C203A_fwd nuoG R219L_fwd nuoG R219W W221L_fwd nuoG R219W W221R_fwd nuoG E617A_fwd nuoI L65W_fwd nuoEaxaG129D_fwd nuoEaxaG129S_fwd nuoFaxa E95D_fwd nuoFaxa E95N_fwd nuoFaxa E95Q_fwd nuoFaxa E97D_fwd nuoFaxa E97N_fwd nuoFaxa E97Q_fwd nuoFaxa Y180A_fwd 36 5'-GTTGATGACCAACGCGCCGCACGACTG-3' 5'-CACCCGCACGACGCACCGGTATGTG-3' 5'-GCACGACTGTCCATGGTGTGAAGAGGG C-3' 5'-CACGACTGTCCAGTAGCTGAAGAGGGC-3' 5'-GAGGGCGGTAACGCGCATCTGCAGGATA TGAC-3' 5'-CTCTCACGAGATGAACCGCGCCATCGCCT GC-3' 5'-CTCTCACGAGATGAACCGCTGCATCGCCG CCTACCGCTG-3' 5'-CTGCTACCGCGCTGTGCGTTATTATAAAG ATTACG-3' 5'-CTGGTCGAAATTGCGCCAACCGGCGTATT TAC-3' 5'-CTCCGAGCGTTATAACCTGAAATGGGATA TGC-3' 5'-CTCCGAGCGTTATAACTGGAAACTGGATA TGCAGTTTGC-3' 5'-CTCCGAGCGTTATAACTGGAAACGTGATA TGCAGTTTGC-3' 5'-CTGCCAGCTTTGCTGCTAGCGACGGTACG GTGATC-3' 5'-CGTTGCGTAGCATGCAATTGGTGCGCGGT AGCC-3' 5'-CGTTCAGTGTCTGGATGCCTGCAGTGAAG CTCCCGTG-3' 5'-CGTTCAGTGTCTGAGCGCCTGCAGTGAAG CTCCCGTG-3' 5'-GCAACGCGGACGACTCAGAACCGGGTAC CTTTAAGGAC-3' 5'-GCAACGCGGACAACTCAGAACCGGGTAC CTTTAAGGAC-3' 5'-GCAACGCGGACCAGTCAGAACCGGGTAC CTTTAAGGAC-3' 5'-GGACGAGTCAGACCCGGGAACCTTTAAG G-3' 5'-GGACGAGTCAAACCCGGGAACCTTTAAG G-3' 5'-GGACGAGTCACAACCCGGGACCTTTAAG G-3' 5'-GGGGTGCGGGCGCCGCTATATGCGGTGA G-3' AgeI StyI CaiI PstI BauI BauI PsiI PsiI PsiI PsiI NheI PaeI PstI PstI KpnI KpnI KpnI SmaI SmaI SmaI EheI Material und Methoden Tabelle 3.4; Fortsetzung. nuoFaxa Y180C_fwd nuoFaxa Y180F_fwd nuoFaxa Y180L_fwd nuoFaxa E185A_fwd nuoFaxa E185D_fwd nuoFaxa E185G_fwd nuoFaxa E185H_fwd nuoFaxa E185N_fwd nuoFaxa E185Q_fwd 5'-GCGGGTGCCTGTATATGCGGTGAG-3' 5'-GGGGTGCGGGCGCCTTTATATTGCGGTGA G-3' 5'-GGGGTGCGGGGGCCCTCATATGCGGTGA G-3' 5'-GGTGCCTACATCTGTGGTGAGGCGACCG CTCTGATAG-3' 5'-GTGCCTACATCTGTGGTGAGGACACCGCT CTGATAG-3' 5'-GGTGCCTACATCTGTGGTGAGGGTACCGC TCTGATAG-3' 5'-GGTGCCTACATCTGTGGTGAGCATACCGC TCTGATAG-3' 5'-GTGCCTACATCTGTGGTGAGAACACCGCT CTGATAG-3' 5'-GTGCCTACATCTGTGGTGAGCAAACCGCT CTGATAG-3' NdeI* EheI ApaI NdeI* NdeI* NdeI* NdeI* NdeI* NdeI* Tabelle 3.5: Sequenzen der Oligonukleotide zur Amplifikation der nptI-sacB Selektionskassette. Die für die -Red vermittelte Rekombination notwendigen und zu den nuo-Genen homologen Bereiche sind fett gedruckt und die homologen Bereiche zur sacB-Kassette unterstrichen. Oligonukleotid Sequenz nuoD::nptI-sacB_fwd 5'-TCACCCGCTGACCACGCCGCCGCCGAAAGAGCGC ACGCTGCACGTTGTGTCTCAAAATCTCTGATG-3' nuoD::nptI-sacB_rev 5'-CTTTGGTCGCCTCAATCATCTGGAAAGATTCATTG GCAGGCATCACCGAATTCCCCGGGGGATCCG-3' nuoF::nptI-sacB_fwd 5'-CCGCTGACCTGGCGTCTGCGCGATGACAAACAGC CAGTGTGGCTGGACGGTACCGGATCCGTCGACCTG-3' nuoF::nptI-sacB_rev 5'-GCCAGGCTTTAAATTTCAGACCATCACGCATACCA CCGGCGTAATCTTCGGAATTCCCCGGGGGATCCG-3' nuoGbeg::nptI-sacB_fwd 5'-GAATACGAGGTCAACGGAGCGGACAACCTGCTGG AAGCTTCACGTTGTGTCTCAAAATCTCTGATG-3' nuoGend::npt-sacB_rev 5'-TCAACCGGCAGCGTGACCGTGTTGCCATCGTAAC TAAAGGAGACGCGGAATTCCCCGGGGGATCCG-3' nuoI2::nptI-sacB_fwd 5'-ACGCGAAACGCGAATGTACCCGGAAGAGCCGGTC TATCTGCACGTTGTGTCTCAAAATCTCTGATG-3' nuoI::nptI-sacB_rev 5'-GTATTCCCCCATTTCGAAATCCGGGGTTAACTGAA TCGCCGTGGTCGGAATTCCCCGGGGGATCCG-3' 37 Material und Methoden Tabelle 3.6: Sequenzen der DNA-Oligonukleotide zur Amplifikation linearer DNA-Fragmente. Die zur Amplifikation der A. aeolicus nuo-Gene verwendeten Oligonukleotide sind mit axa gekennzeichnet. Eingeführte Restriktionsschnittstellen sind mit Unterstrich hervorgehoben. Oligonukleotid nuoD_fwd nuoD_rev nuoE_fwd nuoE_rev nuoI_fwd nuoI_rev nuoF_fwd nuoF_rev nuoGbeg_fwd nuoGrec_rev axa_nuoE_fwd axa_nuoFhis_rev Sequenz 5'-TGGGTGGTGGCGTTTCTGAC-3' 5'-ATCGCCGCCGGAATTTGCTG-3' 5'-CACGAGAATCAACAACCACAAACCG-3' 5'-TTTATACCGCTCCAGCAGTTCAGG-3' 5'-TCTGGATGATCGGCCTGCAC-3' 5'-TCGCCCTTATCTTTGCCGTC-3' 5'-AACATTATCCGTACTCCCGAAACG-3' 5'-CAGCGCTCTTTCAGCAGGTTCG-3' 5'-CGATTAACGCTCAGTCTC-3' 5'-GTGCCTCCTTGAGATCCTC-3' 5'-TACCAGAATTCTAAGAAGGAGATATAGAT TATGTTTAAAACG-3' 5'-AGTTCGCTCAGCTTAATGATGATGATGATG ATGTCCGG-3' Schnittstelle EcoRI BlpI 3.4 Medien, Lösungen und DNA-modifizierende Enzyme 3.4.1 Nährmedien In Tabelle 3.7 sind alle verwendeten Nährmedien und deren Zusammensetzung aufgeführt. Tabelle 3.7: Zusammensetzung der Medien. Medium LB-Medium Zusammensetzung Pepton NaCl Hefeextrakt Konzentration 1% (w/v) 1% (w/v) 0.5% (w/v) SOC-Medium Pepton Hefeextrakt NaCl KCl MgSO4 MgCl2 Glucose 2% (w/v) 0.5% (w/v) 10 mM 2.5 mM 10 mM 10 mM 20 mM 38 Material und Methoden Tabelle 3.7; Fortsetzung. LB-Agar LB-Medium mit Agar 2% (w/v) YP/SUC-Agar Pepton Hefeextrakt Saccharose Agar 1% (w/v) 0.5% (w/v) 10% (w/v) 2% (w/v) Autoinduktionsmedium Pepton Hefe Na2HPO4 KH2PO4 NH4Cl Na2SO4 MgSO4 Riboflavin Eisenammonium-Citrat L-Cystein Mannitol Arabinose Glucose 1% (w/v) 0.5% (w/v) 25 mM 25 mM 50 mM 5 mM 2 mM 50 mg/L 30 mg/L 0.5 mM 0.5% (w/v) 0.2% (w/v) 0.05% (w/v) M9-Minimalmedium Na2HPO4 KH2PO4 NaCl NH4Cl CaCl2 MgSO4 Casein (säurehydrolysiert) L-Tryptophan Glycerin Thiaminhydrochlorid 25 mM 22 mM 8.5 mM 18.7 mM 0.1 mM 1 mM 0.1% (w/v) 5‰ (w/v) 25 mM 100 µg/mL NuoEFaxa-Medium Pepton NaCl Hefeextrakt Glucose Riboflavin Eisenammonium-Citrat L-Cystein 1% (w/v) 1% (w/v) 0.5% (w/v) 1% (w/v) 50 mg/L 30 mg/L 0.5 mM 39 Material und Methoden 3.4.2 Antibiotika-Lösungen In Tabelle 3.8 sind alle in dieser Arbeit verwendeten Antibiotika und die verwendeten Konzentrationen aufgeführt. Tabelle 3.8: Verwendete Antibiotika. Antibiotikum Ampicillin Stammlösung 50 mg/mL, in H2O Endkonzentration 100 μg/mL Chloramphenicol 34 mg/mL, in Ethanol 34 oder 170 μg/mL in f ü igen Me ien 20 μg/mL in fe en Me ien Kanamycin 50 mg/mL, in H2O 50 μg/mL 3.4.3 Lösungen und Puffer für die Molekularbiologie In Tabelle 3.9 sind die Zusammensetzungen der in dieser Arbeit verwendeten Puffer und Lösungen für die Plasmidpräparationen im analytischen Maßstab und für die AgaroseGelelektrophorese aufgeführt. Tabelle 3.9: Zusammensetzung der Puffer und Lösungen für die Molekularbiologie. Puffer P1 Zusammensetzung Tris/HCl, pH 8.0 EDTA RNase A Konzentration 50 mM 10 mM 100 µg/mL P2 NaOH SDS 200 mM 1% (w/v) P3 Kaliumacetat/Essigsäure, pH 5.5 3M TBE-Puffer Tris/Borsäure, pH 8.3 EDTA 89 mM 1 mM 3.4.4 SDS-Polyacrylamid-Gelelektrophorese und Western Blot Die für die SDS-Polyacrylamid-Gelelektrophorese (SDS-PAGE) und Western Blot-Analyse verwendeten Puffer und Lösungen sowie deren Zusammensetzung sind in den Tabellen 3.10 bis 3.12 aufgeführt. 40 Material und Methoden Tabelle 3.10: Zusammensetzung der Puffer und Gele für die SDS-PAGE nach Schägger und von Jagow, 1987. Puffer Zusammensetzung Konzentration Probenpuffer Tris/Acetat, pH 6.8 SDS Glycerin 2-Mercaptoethanol Bromphenolblau 0.2 M 16% (w/v) 48% (v/v) 8% (v/v) 0.04% (w/v) Anodenpuffer Tris/HCl, pH 8.9 0.2 M Kathodenpuffer Tris/HCl, pH 8.25 Tricine SDS 0.1 M 0.1 M 0.1% (w/v) Schägger-Gelpuffer Tris/HCl, pH 8.45 SDS 3M 0.3% (w/v) Färbelösung CBB R-250 Ethanol Essigsäure 0.5% (w/v) 50% (v/v) 10% (v/v) Entfärbelösung Ethanol Essigsäure 30% (v/v) 10% (v/v) Trenngel Acrylamid-Bisacrylamid-Lösung Schägger-Gelpuffer Glycerin TEMED APS 34% (v/v) 34% (v/v) 11% (v/v) 0.1% (v/v) 0.1% (w/v) Sammelgel Acrylamid-Bisacrylamid-Lösung Schägger-Gelpuffer TEMED APS 13% (v/v) 23% (v/v) 0.2% (v/v) 0.1% (w/v) Tabelle 3.11: Zusammensetzung der Puffer und Gele für die SDS-PAGE nach Laemmli, 1970. Puffer Probenpuffer Laufpuffer Zusammensetzung Tris/HCl, pH 6.8 SDS Glycerin Bromphenolblau EDTA DTT Tris/HCl, pH 8.3 Glycin SDS Konzentration 0.4 M 4% (w/v) 12% (w/v) 1% (w/v) 2.5 mM 3.5 mM 50 mM 390 mM 0.1% (w/v) 41 Material und Methoden Tabelle 3.11; Fortsetzung. Sammelgelpuffer Tris/Essigsäure, pH 6.8 1M Trenngelpuffer Tris/Essigsäure, pH 8.8 3M Sammelgel Acrylamid-Bisacrylamid-Lösung Sammelgelpuffer TEMED APS SDS Acrylamid-Bisacrylamid-Lösung Trenngelpuffer TEMED APS SDS 19% (v/v) 14% (v/v) 0.1% (v/v) 0.1% (w/v) 0.1% (w/v) 50% (v/v) 13% (v/v) 0.1% (v/v) 0.1% (w/v) 0.1% (w/v) Trenngel Tabelle 3.12: Puffer und Lösungen für den Western Blot nach Towbin et al., 1979. Puffer Blotpuffer Zusammensetzung Tris/HCl, pH 7.5 Glycin Methanol SDS Konzentration 25 mM 191 mM 20% (v/v) 0.05% (w/v) phoshate-buffered saline (PBS), pH 7.4 KH2PO4 Na2HPO4 NaCl KCl 1.5 mM 6.5 mM 137 mM 2.7 mM PBS-T PBS TWEEN 20 0.01% (v/v) Blockpuffer Magermilchpulver in PBS-T 3% (w/v) Na2HPO4-Lösung Na2HPO4, pH 7.4 25 mM Entwickler Na2HPO4, pH 7.4 NiSO4 CoCl2 3,3'-Diaminobenzidin H2O2 25 mM 1.1 mM 2.3 mM 2.8 mM 0.06% (v/v) 3.4.5 Puffer und Lösungen für die Proteinbiochemie In Tabelle 3.13 sind die Puffer und Lösungen für die Präparation von E. coli Cytoplasmamembranen, des Komplex I, die Saccharosedichtegradienten-Zentrifugation und die Aktivitätsbestimmungen sowie deren Zusammensetzung aufgeführt. 42 Material und Methoden Tabelle 3.13: Zusammensetzung der Puffer für die E. coli Cytoplasmamembran- und Komplex IPräparation, die Saccharosedichtegradienten-Zentrifugation und Aktivitätsbestimmungen. Puffer A-Puffer Zusammensetzung MES/NaOH, pH 6.0 NaCl Konzentration 50 mM 50 mM A*-Puffer MES/NaOH, pH 6.0 NaCl MgCl2 50 mM 50 mM 5 mM ADDM*-Puffer MES/NaOH, pH 6.0 NaCl MgCl2 DDM MES/NaOH, pH 6.0 NaCl MgCl2 DDM MES/NaOH, pH 6.3 NaCl Imidazol MgCl2 DDM 50 mM 50 mM 5 mM 0.1% (w/v) 50 mM 350 mM 5 mM 0.1% (w/v) 50 mM 500 mM 20 mM 5 mM 0.1% (w/v) Elutionspuffer MES/NaOH, pH 6.3 NaCl Imidazol MgCl2 DDM 50 mM 500 mM 500 mM 5 mM 0.1% (w/v) Saccharoselösung 5% Saccharose in A*-Puffer DDM 5% (w/v) 0.1% (w/v) Saccharoselösung 30% Saccharose in A*-Puffer DDM 30% (w/v) 0.1% (w/v) Biuret-Lösung NaOH Seignettesalz KI CuSO4 200 mM 0.5% (w/v) 0.5% (w/v) 0.3% (w/v) Rekonstitutionspuffer MES/NaOH, pH 6.0 NaCl DDM 5 mM 50 mM 0.1% (w/v) Lipidpuffer MES/NaOH, pH 6.0 NaCl DDM 5 mM 50 mM 2% (w/v) BDDM*-Puffer Bindepuffer 43 Material und Methoden Tabelle 3.13; Fortsetzung. ACMA-Messpuffer MES/NaOH, pH 6.0 KCl MgCl2 Valinomycin 5 mM 50 mM 2 mM 1 µM In Tabelle 3.14 sind die Zusammensetzungen der Puffer und Lösungen für die A. aeolicus NuoEFHis-Präparation aufgeführt. Tabelle 3.14: Zusammensetzung der Puffer für die A. aeolicus NuoEFHis-Präparation. Puffer Bindepuffer Zusammensetzung MOPS/NaOH, pH 7.0 NaCl Imidazol Konzentration 50 mM 200 mM 20 mM Elutionspuffer MOPS/NaOH, pH 7.0 NaCl Imidazol 50 mM 200 mM 500 mM A-Puffer MOPS/NaOH, pH 7.0 NaCl 50 mM 50 mM B-Puffer MOPS/NaOH, pH 7.0 NaCl 50 mM 350 mM 3.4.6 DNA-modifizierende Enzyme Alle in dieser Arbeit verwendeten Enzyme sind in den Tabellen 3.15 und 3.16 aufgeführt. Tabelle 3.15: Verwendete Restriktionsenzyme. Restriktionsenzyme Konzentration AflII, AgeI, ApaI, BamHI, BanII, BauI, BlpI, CaiI, DpnI, EcoRI, EheI, 10 U/µL KpnI, NdeI, NheI, PaeI, PsiI, PstI, SacI, SmaI, StyI, VspI Hersteller Thermo Scientific Tabelle 3.16: Verwendete DNA-Polymerasen und Ligasen. Polymerase Pfu-DNA-Polymerase Phusion-DNA-Polymerase Long PCR Enzyme Mix T4-DNA-Ligase 44 Konzentration 2.5 U/µL 2 U/µL 5 U/µL 5 U/µL Hersteller Thermo Scientific Finnzymes Thermo Scientific Thermo Scientific Material und Methoden 3.5 Mikrobiologische Methoden 3.5.1 Bestimmung der optischen Dichte Das Zellwachstum wurde anhand der optischen Dichte der Kulturen bei 600 nm (OD600) verfolgt (Ultrospec 1000 pro, UV/Visible Spectrophotometer, Amersham Pharmacia Biotech). Als Referenz diente das verwendete Medium. Es wurden Küvetten mit einem Lichtweg von 1 cm (Plastibrand 1.5 mL Halbmikro, Brand) verwendet. Ab einer OD600 von 0.4 wurde die Zellsuspension mit dem verwendeten Medium verdünnt. 3.5.2 Vorkulturen Für die Herstellung von Vorkulturen wurden 4, 50 bzw. 400 mL LB-Medium mit den entsprechenden Antibiotika versetzt und aus einer anderen Vorkultur oder aus einer GlycerinDauerkultur 1:1000 bis 1:5000 (v/v) angeimpft. Um Einzelkolonien zu kultivieren, wurden diese mit einem sterilen Zahnstocher von einer Platte gepickt und in LB-Medium überführt. Die Vorkulturen wurden unter Schütteln bei 30 bzw. 37 °C 16 – 18 h angezogen. 3.5.3 Glycerin-Dauerkulturen Zur Herstellung von Glycerin-Dauerkulturen wurden 150 µL 80%iges (v/v) steriles Glycerin in einem Eppendorfgefäß vorgelegt, mit 250 µL Vorkultur versetzt und sofort in flüssigem Stickstoff schockgefroren. Die Glycerin-Dauerkulturen wurden bei −80 °C gelagert. 3.5.4 Herstellung elektrokompetenter Zellen Zellen für die Elektroporation: Zur Herstellung elektrokompetenter Zellen für die Elektroporation wurde LB-Medium im Verhältnis 1:100 (v/v) mit einer E. coli Vorkultur angeimpft und unter Schütteln bei 37 °C inkubiert. Bei einer OD600 von 0.5 – 0.7 wurde die Zellsuspension 15 min auf Eis inkubiert. Die Zellen wurden zentrifugiert (4500×g, 10 min, 4 °C, Rotor A-4-44, Eppendorf Zentrifuge 5804 R) und der Überstand verworfen. Das Sediment wurde in dem Ausgangsvolumen sterilem, eiskaltem ddH2O resuspendiert, erneut 45 Material und Methoden zentrifugiert und der Überstand verworfen. Dieser Schritt wurde zweimal wiederholt. Das Sediment wurde im 1.5-fachen Volumen eiskaltem 15%igem (v/v) Glycerin gelöst. Die Zellen wurden bis zur Transformation auf Eis gestellt oder in 50 µL Aliquots in flüssigem Stickstoff schockgefroren und bei −80 °C gelagert. Ze en für ie λ-Red vermittelte Rekombination: Für ie λ-Red vermittelte Rekombination wurden 100 mL LB-Medium mit Ampicillin versetzt und im Verhältnis 1:100 (v/v) mit einer E. coli DH5αΔnuo/pKD46 Vorkultur angeimpft und bei 30 °C und 180 Upm (Certomat R, B. Braun Biotech International) inkubiert. Bei einer OD600 von 0.25 – 0.3 wurde die Expression des red-Operons durch Zugabe von 0.2% (w/v) L-(+)-Arabinose induziert. Nach exakt 20 min wurden die Zellen für 15 min auf Eis gestellt, zentrifugiert (4500×g, 10 min, 4 °C, Rotor A-444, Eppendorf Zentrifuge 5804 R) und der Überstand verworfen. Das Sediment wurde im Ausgangsvolumen sterilem eiskaltem 10%igem (v/v) Glycerin oder ddH2O resuspendiert, erneut zentrifugiert und der Überstand verworfen. Dieser Schritt wurde zweimal wiederholt. Anschließend wurde die OD600 der Zellsuspension auf 30 – 45 eingestellt. Die elektrokompetenten DH5αΔnuo/pKD46 Zellen wurden bis zu ihrer Verwendung auf Eis gelagert. 3.5.5 Bakterienanzucht Für die Anzucht von E. coli Stämmen ohne Plasmide wurde LB-Medium verwendet. 400 mL Medium wurden falls benötigt mit Antibiotika versetzt und 1:100 (v/v) aus einer 50 mL Vorkultur angeimpft. Die Kulturen wurden unter Schütteln bei 37 °C inkubiert. Beim Erreichen der stationären Phase wurde das Wachstum durch Inkubation auf Eis gestoppt und die Zellen durch Zentrifugation geerntet (11´900×g, 10 min, 4 °C, Rotor JLA 10´500, Avanti J-26 XP, Beckman Coulter). Das Zellsediment wurde in flüssigem Stickstoff schockgefroren und bei −80 °C gelagert. 3.5.6 Überproduktion des E. coli Komplex I 400 mL Maßstab: 400 mL Autoinduktionsmedium ohne Antibiotikum wurden 1:50 oder 1:25 (v/v) aus einer 100 oder 400 mL E. coli Vorkultur angeimpft. Die Kulturen wurden unter Schütteln bei 37 °C inkubiert. Kurz vor oder bei Erreichen der stationären Phase wurden die Zellen durch Zentrifugation geerntet (11´900×g, 10 min, 4 °C, Rotor JLA 10´500, Avanti J-26 46 Material und Methoden XP, Beckman Coulter). Das Zellsediment wurde in flüssigem Stickstoff schockgefroren und bei −80 °C gelagert. 10 L Maßstab: In einem Fermenter wurden 10 L Autoinduktionsmedium 1:25 (v/v) mit einer 400 mL E. coli Vorkultur angeimpft und bei 37 °C im Druckluftstrom aerob kultiviert. Die Zellsuspension wurde mit Hilfe eines KPG-Rührers durchmischt. Kurz vor oder bei Erreichen der stationären Phase wurden die Zellen durch Zentrifugation geerntet (11´900×g, 10 min, 4 °C, Rotor JLA 10´500, Avanti J-26 XP, Beckman Coulter). Das Zellsediment wurde in flüssigem Stickstoff schockgefroren und bei −80 °C gelagert. 3.5.7 Expressionstests Die Expressionsleistung einzelner Klone für die Überexpression von A. aeolicus nuoEFhis variiert stark. Geeignete Klone wurden über Expressionstests selektiert. Dafür wurden 50 mL LB-Medium wurden mit Chloramphenicol und Ampicillin versetzt und 1:50 (v/v) aus einer 4 mL Vorkultur angeimpft. Die Zellen wurden unter Schütteln bei 37 °C kultiviert. Bei einer OD600 von 0.6 – 0.7 wurde die Genexpression durch Zugabe von 0.7 – 1.0 mM Isopropyl-β-Dthio-galactopyranosid (IPTG) induziert. Nach 3 h Wachstum wurden die Zellen durch Zentrifugation geerntet (4500×g, 10 min, 4 °C, Rotor A-4-44, Eppendorf Zentrifuge 5804 R). Die Proteinproduktion der verschiedenen Stämme wurde optisch anhand der Färbung der Zellsedimente verglichen. Der dunkelste Klon wurde für die Zellzucht verwendet. 3.5.8 Überproduktion des A. aeolicus NuoEFHis 400 mL-Maßstab: 400 mL NuoEFaxa-Medium ohne Antibiotikum wurden 1:25 – 1:100 (v/v) aus einer 100 oder 400 mL E. coli Vorkultur angeimpft. Die Kulturen wurden unter Schütteln bei 37 °C inkubiert. Bei einer OD600 von 0.6 – 0.7 wurde durch Zugabe von 0.7 – 1.0 mM IPTG die Expression von nuoEFhis induziert. Beim Erreichen der stationären Phase oder spätestens 4 h nach Induktion wurden die Zellen durch Zentrifugation geerntet (11´900×g, 10 min, 4 °C, Rotor JLA 10´500, Avanti J-26 XP, Beckman Coulter). Das Zellsediment wurde in flüssigem Stickstoff schockgefroren und bei −80 °C gelagert. 47 Material und Methoden 10 L-Maßstab: In einem Fermenter wurden 10 L NuoEFaxa-Medium 1:25 (v/v) mit einer 400 mL E. coli Vorkultur angeimpft und bei 37 °C im Druckluftstrom aerob kultiviert. Die Zellsuspension wurde mit Hilfe eines KPG-Rührers gerührt. Bei einer OD600 von 0.6 – 1.0 wurde die Expression von nuoEFhis durch Zugabe von 0.7 – 1.0 mM IPTG induziert. Beim Erreichen der stationären Phase oder spätestens 4 h nach Induktion wurden die Zellen durch Zentrifugation geerntet (11´900×g, 10 min, 4 °C, Rotor JLA 10´500, Avanti J-26 XP, Beckman Coulter). Das Zellsediment wurde in flüssigem Stickstoff schockgefroren und bei −80 °C gelagert. 3.6 Molekularbiologische Methoden 3.6.1 Plasmidpräparation Analytischer Maßstab: Für Plasmidpräparationen im analytischen Maßstab wurden 1.5 – 2 mL einer Vorkultur zentrifugiert (2´300×g, 4 min, Raumtemperatur, Rotor F-45-36-8, Eppendorf 5415 D). Das Sediment wurde in 250 µL kaltem Puffer P1 suspendiert. Durch die Zugabe von 250 µL Puffer P2 wurden die Zellen lysiert. Nach exakt 4 min Inkubation bei Raumtemperatur wurde die Suspension mit 250 µL Puffer P3 (4 °C) neutralisiert, 15 min auf Eis inkubiert und anschließend zentrifugiert (20´800×g, 15 min, 4 °C, Rotor F45-30-11, Eppendorf 5417R). Der Überstand wurde vorsichtig in ein neues Eppendorfgefäß überführt un mi 750 µL I pr pan ver e z , 20 min bei −20 °C inkubiert und erneut zentrifugiert (20´800×g, 15 min, 4 °C, Rotor F45-30-11, Eppendorf 5417R). Die gefällte Plasmid-DNA wurde 1 – 2 h bei 37°C getrocknet und anschließend in 30 µL ddH2O (55 °C, 20 min, 700 Upm, Eppendorf Thermomixer compact) gelöst. Präparativer Maßstab: Bei Plasmidpräparationen im präparativen Maßstab wurde das Wizard Plus SV Minipreps DNA Purification System Kit (Promega) nach Herstellerangaben verwendet. Vor der Elution der gereinigten DNA wurde das Säulenmaterial 2 min bei 55 °C mit 50 µL ddH2O inkubiert. 48 Material und Methoden 3.6.2 Reinigung von DNA Zur Reinigung linearer DNA-Fragmente aus Lösungen oder aus einem Agarosegel wurde das Wizard SV Gel and PCR Clean-Up System (Promega) nach Angaben des Herstellers verwendet. Vor der Elution der gereinigten DNA wurde das Säulenmaterial 2 min bei 55 °C mit 25 – 50 µL ddH2O inkubiert. 3.6.3 DNA-Konzentrationsbestimmung Die Konzentration von DNA-Lösungen wurde in zehnfacher Verdünnung mit dem Quant-iT dsDNA-Assay am Qubit Fluorometer (Invitrogen) nach Angaben des Herstellers bestimmt. 3.6.4 Restriktionsanalysen und DpnI-Verdau Das Volumen der Restriktionsansätze betrug 20 µL. 0.1 – 1 µg DNA wurden mit 0.2 – 10 U Restriktionsenzym und 2 µL des entsprechenden Puffers versetzt. Die Lösung wurde 1 – 3 h bei 37 °C inkubiert. Für Restriktionsanalysen mit zwei Enzymen wurden die Protokolle von Thermo Scientific verwendet. Die Restriktions-Fragmente wurden durch Agarosegelelektrophorese aufgetrennt. Um nach der PCR die methylierte Templat-DNA abzubauen, wurde der gereinigte PCRAnsatz mit DpnI verdaut. Hierfür wurde das Volumen des Ansatzes bestimmt und mit der benötigten Menge Tango-Puffer und 10 U DpnI versetzt. Der Ansatz wurde 2 h bei 37 °C inkubiert und erneut gereinigt. 3.6.5 Polymerasekettenreaktion Die PCR wurde in 50 µL Ansätzen in 0.2 mL PCR-Gefäßen (Biozyme) in dem vom Hersteller mitgelieferten Puffer durchgeführt. Der Reaktionsansatz wurde vor Zugabe der Polymerase und bis zum Erreichen der initialen Denaturierungstemperatur auf Eis gekühlt. Die PCRProdukte wurden im Thermocycler (PTC-200, MJ Research) mit unterschiedlichen Temperaturprogrammen amplifiziert. Um den Erfolg der PCR zu überprüfen, wurden 2 – 10 µL des Ansatzes entnommen und mittels Agarose-Gelelektrophorese untersucht. Der restliche Ansatz wurde mit dem Wizard SV Gel and PCR Clean-Up System (Promega) 49 Material und Methoden gereinigt und mit DpnI verdaut. Zur Kontrolle wurden 2 – 4 µL der gereinigten Probe auf ein Agarosegel aufgetragen. Ortsgerichtete Mutagenese: Für die ortsgerichtete Mutagenese wurde die QuikChangeMethode (Stratagene) verwendet. Ein Ansatz enthielt 40 – 45 ng Vorlage-DNA, je 240 nM DNA-Oligonukleotide, 0.2 mM dNTP-Mix, 1×Pfu-Puffer + MgSO4 und 2.5 U Pfu-DNAPolymerase (Thermo Scientific). Tabelle 3.17 gibt das verwendete Temperaturprogramm wieder. Tabelle 3.17: Temperaturprogramm der PCR für die ortsgerichtete Mutagenese nach der QuikChangeMethode. Temperatur [°C] Zeit [s] Zyklen Initiale Denaturierung 95 120 1 Denaturierung 95 60 Anlagerung der Oligonukleotide 50 – 60 30 – 60 Elongation 72 60 – 120 pro 1 kbp 18 Amplifikation der nptI-sacB-Kassette: Um die nptI-sacB-Kassette durch -Red vermittelte Rekombination in das gewünschte nuo-Gen einzubringen, musste diese von homologen Bereichen des jeweiligen nuo-Gens flankiert sein. Es wurden Oligonukleotide verwendet, die über einen Bereich von etwa 36 – 47 bp homolog zu den jeweiligen nuo-Genen sind und 19 – 26 bp lange, homologe Bereiche zur nptI-sacB-Kassette enthalten (Tab. 3.5). Zur Amplifikation der Selektionskassette wurde eine kommerziell erhältliche Mischung aus Taq und einer anderen thermostabilen DNA-Polymerase mit Korrekturlesefunktion (Long PCR Enzym Mix, Thermo Scientific) verwendet. Der Ansatz enthielt 20 – 30 ng VorlageDNA, je 0.5 µM DNA-Oligonukleotide, 0.2 mM dNTP-Mix, 1×Long PCR Enzym MixPuffer + MgCl2 und 2.5 U Long PCR Enzym Mix (5 U/µL). Tabelle 3.18 zeigt das verwendete Temperaturprogramm. 50 Material und Methoden Tabelle 3.18: Temperaturprogramm zur Amplifikation der Selektionskassette. Temperatur [°C] Zeit [s] Zyklen Initiale Denaturierung 95 180 1 Denaturierung 95 30 Anlagerung der Oligonukleotide 58 30 Elongation 68 90 pro 1 kbp Abschließende Elongation 68 600 25 1 Amplifikation linearer DNA-Fragmente: Um lineare DNA-Fragmente zu amplifizieren, wurde folgender Ansatz verwendet: 20 – 40 ng Vorlage-DNA, je 2 µM DNA-Oligonukleotide, 200 µM dNTP-Mix, 1× Phusion HF-Puffer + MgCl2 (Finnzymes) und 1 U Phusion DNA Polymerase (2 U/µL, Finnzymes). Tabelle 3.19 gibt die Parameter des verwendeten Temperaturprogramms wieder. Tabelle 3.19: Temperaturprogramm zur Amplifikation linearer DNA-Fragmente. Temperatur [°C] Zeit [s] Zyklen Initiale Denaturierung 95 120 1 Denaturierung 95 20 Anlagerung der Oligonukleotide 55 – 60 30 Elongation 72 20 s pro 1 kbp Abschließende Elongation 72 300 25 1 3.6.6 Agarose-Gelelektrophorese In dieser Arbeit wurden ausschließlich 0.8%ige (w/v) Agarosegele in TBE-Puffer verwendet. Proben mit einem kleineren Volumen als 10 µL wurden mit ddH2O auf 10 µL aufgefüllt. Die Proben wurden mit 6×Probenauftragspuffer (Thermo Scientific) versetzt. Zur Bestimmung der Fragmentlängen wurden 500 ng λ-DNA/Eco130I (StyI) Marker 16 (Thermo Scientific) oder GeneRuler 1 kb DNA Ladder (Thermo Scientific) aufgetragen. Die Ansätze wurden elektrophoretisch bei 10 V/cm getrennt. Zur Visualisierung der DNA-Banden wurde den Gelen 200 ng/mL Ethidiumbromid zugesetzt. Auf einem Leuchtschirm wurde die Fluoreszenz des DNA/Ethidiumbromid-Komplexes bei 312 nm angeregt und fotografisch dokumentiert. 51 Material und Methoden 3.6.7 Klonierung Zu klonierende Gene wurde durch PCR amplifiziert. Das PCR-Produkt und das Plasmid, in welches das Gen eingebracht werden sollte, wurden mit zwei Restriktionsenzymen geschnitten und über ein Agarosegel gereinigt. Für die Ligation wurden linearisiertes Plasmid und Insert im molaren Verhältnis von 1:5 gemischt und mit ddH2O auf 17.5 µL aufgefüllt. Um Fehlanlagerungen der Überhänge zu minimieren wurde der Ansatz 5 min bei 45 °C und anschließend 5 min auf Eis inkubiert. Es wurden 2 µL T4-DNA Ligase-Puffer und 0.5 µL T4-DNA-Ligase (2.5 U) zugegeben und 45 min bei 16 °C inkubiert. Der gesamte Ligationsansatz wurde mit dem Wizard SV Gel and PCR Clean-Up System (Promega) gereinigt. E. coli DH5α Ze en wur en mi 1 µL des gereinigten Produkts transformiert und auf LB-Agar-Platten mit dem benötigten Antibiotikum ausgestrichen. 3.6.8 Transformation durch Elektroporation Für die Transformation von E. coli wurden 50 µL frisch hergestellte oder auf Eis aufgetaute elektrokompetente Zellen mit 10 – 500 ng DNA versetzt. Die Suspension wurde in einer zuvor auf Eis gekühlten Elektroporationsküvette (d = 0.1 cm, Eppendorf) für 5.0 – 6.0 ms einem Spannungspuls von 1700 V (Electroporator 2510, Eppendorf) ausgesetzt. Der Ansatz wurde sofort in 1 mL kaltem SOC-Medium aufgenommen, in ein 2.0 mL Eppendorfgefäß überführt und bei 37 °C (1 h, 750 Upm, Thermomixer compact, Eppendorf) inkubiert. Abhängig von der eingesetzten Menge an DNA wurde der gesamte oder 100 – 200 µL des Transformationsansatzes auf LB-Agar-Platten, die mit dem für die Selektion benötigten Antibiotikum versetzt waren, ausgestrichen. Die LB-Agar-Platten wurden für 16 h bei 37 °C oder für 20 – 24 h bei 30 °C inkubiert. Für die -Red vermittelte Rekombination wurden die elektrokompetenten Zellen frisch hergestellt. 50 µL der elektrokompetenten Zellen wurden in einem eiskaltem 1.5 mL Eppendorfgefäß vorgelegt und mit 50 ng Plasmid und 100 – 500 ng linearer DNA versetzt. Um die Transformationseffizienz zu erhöhen, wurde SOC-Medium bei Raumtemperatur verwendet. Wenn die nptI-sacB-Kassette eingefügt werden sollte, wurde der Ansatz auf LB-Agar-Platten mit Kanamycin ausgestrichen. Wenn die nptI-sacB-Kassette gegen lineare DNA mit eingefügten Mutationen ausgetauscht werden sollte, wurden die Zellen auf YP/SUC-Agar-Platten mit Chloramphenicol ausgestrichen. Die Agar-Platten wurden für 20 – 24 h bei 30 °C inkubiert. 52 Material und Methoden 3.6.9 Sequenzierung von Plasmiden Für die Sequenzierung wurden 20 µL DNA-Lösung (≥ 20 ng/µL) zur Firma GATC Biotech AG (Konstanz) geschickt. Um die Qualität der Sequenzierung von Plasmiden einer Länge von mehr als 21 kbp zu erhöhen, wurde eine höher konzentrierte Lösung (50 – 100 ng/µL) eingesendet. 3.7 Proteinchemische Methoden 3.7.1 Isolierung der E. coli Cytoplasmamembranen Alle Schritte wurden bei 4 °C oder auf Eis durchgeführt. Zwei bis fünf g Zellen (Feuchtgewicht) wurden im 6-fachen Volumen A-Puffer resuspendiert, mit 0.1 mM PMSF (0.1 M Stammlösung in Isopropanol) und einer Spatelspitze DNase I versetzt und in einem Teflon-in-Glas-Homogenisator homogenisiert. Die Zellen wurden in zwei Durchgänge mit einer French-Press Zelle (110 MPa, 17´000 psi, French Pressure Cell, SLM-Aminco) aufgeschlossen. Der Aufschluss wurde zentrifugiert (9500×g, 20 min, 4 °C, Rotor A8.24, RC-5, Sorvall Instruments) und das Sediment aus Zelltrümmern und nicht aufgeschlossenen Zellen verworfen. Zur Sedimentation der Membranen wurde der Überstand erneut zentrifugiert (252´000×g, 70 min, 4 °C, Rotor 70.1 Ti, L8-M Ultrafuge, Beckman). Die Membranen wurden mit A*-Puffer (1:1; w/v) und 0.1 mM PMSF versetzt und in einem Teflon-in-Glas-Homogenisator homogenisiert. Die NADH-Oxidaseaktivität, die NADH/Ferricyanid Oxidoreduktaseaktivität und der Proteingehalt der Suspension wurden bestimmt. Die Membranen wurden in flüssigem Stickstoff schockgefroren und bis zur Verwendung bei −80 °C gelagert. 3.7.2 Isolierung des E. coli Komplex I Alle Schritte wurden bei 4 °C oder auf Eis durchgeführt. 22 – 45 g E. coli Zellen (Feuchtgewicht) wurden im 6-fachen Volumen A-Puffer resuspendiert, mit 0.1 mM PMSF (0.1 M Stammlösung in Isopropanol) sowie einer Spatelspitze DNase I versetzt und in einem Labormixer (2 – 5 min, Einstellung: II; Braun MX 32) zerkleinert. Entstandener Schaum wurde durch Zugabe von wenig Antifoam 204 entfernt. Die Cytoplasmamembranen wurden 53 Material und Methoden wie beschrieben isoliert. Die Membransuspension wurde mit einer 20%igen (w/v) DDMStammlösung auf eine Endkonzentration von 3% (w/v) eingestellt. Der Ansatz wurde 15 min auf Eis inkubiert und alle 1 – 3 min mit einem Teflon-in-Glas-Homogenisator durchmischt. Der Detergenzextrakt wurde mit A*-Puffer auf ein Volumen von 32 – 96 mL verdünnt. Zur Abtrennung von nicht gelösten Bestandteilen wurde der Extrakt zentrifugiert (257´000×g, 4 °C, 15 min, Rotor 60 Ti, L8-M Ultrafuge, Beckman). Die NADH/Ferricyanid Oxidoreduktaseaktivität und der Proteingehalt des Überstands wurden bestimmt. Der Überstand wurde mit einer Flussgeschwindigkeit von 6 mL/min auf eine zuvor in A*DDMPuffer äquilibrierten 50 mL Anionenaustausch-Chromatographiesäule (26×95 mm, Fractogel EMD TMAE Hicap (M), Merck) aufgetragen (ÄKTA prime plus, Amersham Biosciences). Die Säule wurde mit 150 mM NaCl (33% B*DDM-Puffer) bei derselben Flussgeschwindigkeit gewaschen und gebundene Proteine in einem linearen Gradienten von 150 – 350 mM NaCl (33 – 100% B*DDM-Puffer) eluiert. Das Eluat wurde in 4 mL Fraktionen gesammelt und deren NADH/Ferricyanid Oxidoreduktaseativität bestimmt. Die Gipfelfraktionen der Aktivität wurden vereinigt und deren NADH/Ferricyanid Oxidoreduktaseaktivität und Proteingehalt bestimmt. Das Eluat wurde mit Elutionspuffer auf eine Imidazolkonzentration von 20 mM eingestellt. Die vereinigten Fraktionen der Anionenaustausch-Chromatographie wurden mit einer Flussgeschwindigkeit von 1 mL/min auf eine in Bindepuffer äquilibrierte 10 mL Affinitäts-Chromatographiesäule (16×50 mm, Ni2+-IDA, ProBond Resin, Invitrogen) aufgetragen (ÄKTA prime plus, Amersham Biosciences). Die Säule wurde mit Bindepuffer bei derselben Flussgeschwindigkeit gewaschen und gebundene Proteine in einem Stufengradienten von 140, 260, 380 und 500 mM Imidazol im Elutionspuffer eluiert. Das Eluat wurde in 2 mL Fraktionen gesammelt. Fraktionen, die signifikante NADH/Ferricyanid Oxidoreduktaseaktivität zeigten, wurden vereinigt und durch Ultrafiltration auf ein Volumen von 100 – 500 µL eingeengt (Amicon Ultra-15, MWCO: 100 kDa, Millipore, 3800×g, 4 °C, Rotor A-4-44, Eppendorf Centrifuge 5804 R). Das Konzentrat wurde auf A*-Puffer umgepuffert, indem es dreimal mit dem zehnfachen Volumen A*-Puffer verdünnt und wieder konzentriert wurde. Danach wurde die NADH/Ferricyanid sowie die NADH:Decyl-Ubichinon Oxidoreduktaseaktivität und der Proteingehalt bestimmt. Die Proteinlösung wurde aliquotiert, in f ü igem S ick ff c ckgefr ren un bei −80 °C gelagert. 3.7.3 Isolierung des A. aeolicus NuoEFHis Alle Schritte wurden bei 4 °C oder auf Eis durchgeführt. 5 – 10 g Zellen (Feuchtgewicht) wurden mit 4 – 6-fachen Volumen Bindepuffer, 0.1 mM PMSF (0.1 M in Isopropanol) und 54 Material und Methoden einer Spatelspitze DNase I versetzt und mit einem Teflon-in-Glas Homogenisator homogenisiert. Die Zellen wurden in zwei Durchgängen mit einer French-Press Zelle (110 MPa, 17´000 psi, French Pressure Cell, SLM-Aminco) aufgeschlossen. Durch Ultrazentrifugation (252´000×g, 4 °C, 70 min, Rotor 60 Ti, L8-M Ultrafuge, Beckman) wurde das Cytosol von nicht aufgeschlossenen Zellen, Membranen und Zelltrümmern abgetrennt. Von der cytosolischen Fraktion wurde die NADH/Ferricyanid Oxidoreduktaseaktivität sowie der Proteingehalt bestimmt. Das restliche Cytosol wurde mit einer Laufgeschwindigkeit von 1 – 1.5 mL/min auf eine in Bindepuffer äquilibrierte 10 mL Affinitäts-Chromatographiesäule (16×50 mm, Ni2+-IDA, ProBondTM Resin, Invitrogen) aufgetragen (ÄKTA prime plus, Amersham Biosciences). Es wurde mit Bindepuffer gewaschen bis die gemessene Absorbanz bei 280 nm wieder das Grundniveau erreicht hatte. Durch einen Waschschritt mit 68 – 116 mM Imidazol im Elutionspuffer (10 – 20% Elutionspuffer), wurden unspezifisch gebundene Proteine eluiert. NuoEFhis wurde durch einen 40 mL linearen Gradienten von 68 bzw. 116 mM auf 500 mM Imidazol im Elutionspuffer (10 bzw. 20 – 100% Elutionspuffer) eluiert und in 1.5 mL Fraktionen gesammelt. Die NADH/Ferricyanid Oxidoreduktaseaktivität der einzelnen Fraktionen wurde gemessen und Fraktionen mit signifikanter Aktivität vereinigt. Es wurde eine Probe zur Bestimmung der Gesamtaktiviät, der Proteinkonzentration und für die SDS-PAGE entnommen. Die vereinigten Fraktionen wurden durch Ultrafiltration (Amicon-Ultra 15, MWCO; 30 kDa, Millipore, 3800×g, 4 °C, Rotor A-4-44, Eppendorf Centrifuge 5804 R) auf weniger als 500 µL eingeengt und mit einer Flussgeschwindigkeit von 1 – 2 mL/min auf eine in A-Puffer äquilibrierte 120 mL Größenausschluss- Chromatographiesäule (Superdex 200 16/60) aufgetragen (ÄKTA prime plus, Amersham Biosciences). Das Eluat wurde in 2 mL Fraktionen gesammelt. Das Elutionsprofil zeigte zwei Absorptionsmaxima zwischen 70 – 74 mL und bei 80 mL Elutionsvolumen. Das erste Maximum entspricht der aktiven Form des Enzyms, was durch die Bestimmung der NADH/Ferricyanid Oxidoreduktaseaktivität überprüft wurde. Das zweite Maximum entspricht vermutlich einer falsch gefalteten Form, die eine deutlich geringere Aktivität aufweist. Für die folgenden Reinigungsschritte wurden die Fraktionen des ersten Gipfels verwendet. Fraktionen mit signifikanter Aktivität wurden vereinigt und eine Probe zur Bestimmung der Gesamtaktiviät, der Proteinkonzentration und für die SDS-PAGE entnommen. Die restliche Lösung wurde mit einer Flussgeschwindigkeit von 1.5 mL/min auf eine in A-Puffer äquilibrierte 5 mL Anionentausch-Chromatographiesäule (16×25 mm, Source 15Q, Amersham Pharmacia) aufgetragen (ÄKTA prime plus, Amersham Biosciences). Nach dem Auftrag wurde die Säule mit A-Puffer gewaschen bis die Absorbanz bei 280 nm wieder das Grundniveau erreicht hatte. Gebundene Proteine wurden mit einem 25 mL linearen 55 Material und Methoden Gradienten von 50 – 350 mM NaCl im Elutionspuffer (0 – 100% B-Puffer) eluiert. Es wurden 1 mL Fraktionen gesammelt und deren NADH/Ferricyanid Oxidoreduktaseaktivität bestimmt. Fraktionen mit signifikanter Aktivität wurden vereinigt und es wurde eine Probe zur Bestimmung der Gesamtaktiviät, der Proteinkonzentration und für die SDS-PAGE entnommen. Die vereinigten Fraktionen wurden durch Ultrafiltration auf weniger als 500 µL konzentriert. Die Probe wurde mit einer Flussgeschwindigkeit von 1.0 – 2.0 mL/min auf eine mit A-Puffer äquilibrierte 120 mL Größenausschluss-Chromatographiesäule (Superdex 200 16/60) aufgetragen (ÄKTA prime plus, Amersham Biosciences) und das Eluat in 2 mL Fraktionen gesammelt. Fraktionen mit signifikanter NADH/Ferricyanid Oxidoreduktaseaktivität wurden vereinigt. Das gereinigte Protein wurde auf eine Konzentration von 10 – 12 mg/mL eingestellt, zu je 65 µL aliquotiert, in flüssigem Stickstoff c ckgefr ren un bei −80 °C gelagert. 3.7.4 Bestimmung der Proteinkonzentration Biuret-Methode: Der Gesamtproteingehalt der Membranen und der Eluate der einzelnen Reinigungsschritte der Komplex I-Präparationen wurde nach der Biuret-Methode bestimmt. Hierfür wurden 10 – 20 µL Membransuspension, 10 – 20 µL Detergenzextrakt oder 20 – 40 µL Eluat der Anionenaustausch-Chromatographie mit 1 mL 6% (w/v) Trichloressigsäure versetzt, invertiert und anschließend zentrifugiert (16´100×g, 1 min, RT, Rotor F45-24-11, Eppendorf Zentrifuge 5415 D). Der Überstand wurde verworfen, das Sediment mit 1 mL Biuret-Reagenz versetzt und durch Schütteln bei 37 °C (1400 Upm, 30 min, Eppendorf Thermomixer compact) gelöst. Die Absorbanz bei 546 nm wurde gemessen (Halbmikro-Küvetten, d = 1 cm, Greiner, Ultraspec 1100 pro, Pharmacia Biotech). Durch Zugabe von wenigen Körnchen KCN wurden die Proben entfärbt und die Absorbanz bei 546 nm erneut bestimmt. Diese Absorbanz wurde als Hintergrundstreuung durch Lipide und Aggregate, abgezogen. Aus der Differenz wurde über den Vergleich mit einer mittels Rinderserumalbumin (BSA) erstellten Kalibrationsgeraden (0.1 – 1.0 mg BSA) die Proteinkonzentration der Probe bestimmt. Die Konzentration jeder Probe wurde dreifach bestimmt. Bradford-Methode: Der Proteingehalt der bei der Reinigung von NuoEFHis anfallenden Proben und der Präparation wurde nach der Bradford-Methode bestimmt. Dabei wurde 1 µL der jeweiligen Proteinlösung mit 19 µL ddH2O sowie 1 mL 1×Roti-Quant (Roth) Lösung versetzt, invertiert und 5 min bei Raumtemperatur inkubiert. Es wurde die Absorbanz bei 56 Material und Methoden 595 nm gemessen (Halbmikro-Küvetten, d = 1 cm, Greiner, Ultraspec 1100 pro, Pharmacia Biotech). Die Proteinkonzentration wurde mittels einer mit 1 – 7 µg BSA erstellten Kalibrationsgeraden bestimmt. Es wurden jeweils Doppelbestimmungen durchgeführt. UV-spektroskopische Methode: Die Konzentration des gereinigten Komplex I wurde spektroskopisch bestimmt (UV-Küvette micro, d = 1 cm, Plastibrand; TIDAS II, J&M). Die Absorbanz der Proteinlösung bei 280 nm wurde bestimmt und gegen den Wert des Puffers bei gleicher Wellenlänge korrigiert. Zur Korrektur der Lichtstreuung wurde von diesem Wert die korrigierte Absorbanz bei 310 nm abgezogen. Mit Hilfe des Lambert-Beerschen Gesetzes wurde die Proteinkonzentration bestimmt. Hierfür wurde der aus der Aminosäuresequenz abgeleitete Extinktionskoeffizient von ε280 = 763 mM−1 cm−1 (Gill und von Hippel, 1989) bei einer angenommenen molekularen Masse von Mr = 535 kDa verwendet. Die Konzentration des gereinigten Subkomplexes NuoEFHis wurde ebenfalls spektroskopisch bestimmt. Hierfür wurde der aus der Aminosäuresequenz abgeleitete Extinktionskoeffizient von ε280 = 80.7 mM−1 cm−1 (Gill und von Hippel, 1989) bei einer angenommenen molekularen Masse von Mr = 66 kDa verwendet. 3.7.5 Aktivitätsmessungen (d)-NADH/Ferricyanid Oxidoreduktaseaktivität: Die NADH/Ferricyanid Oxidoreduktaseaktivität wurde durch die zeitliche Abnahme der K3[Fe(CN)6]-Konzentration bei 410 nm spektroskopisch verfolgt. Zur Messung wurden 1 mL K3[Fe(CN)6]-Lösung (1 mM in A-Puffer) und 20 µL (d)-NADH (10 mM) in einer Küvette vorgelegt. Es wurden 1 – 5 µL Probe zugegeben, der Ansatz vermischt und die zeitliche Änderung der Absorbanz gemessen (Ultrospec 1000 UV/Visible Spectrophotometer, Pharmacia Biotech). Für die Aktivitätsberechnung mit Hilfe des Lambert-Beerschen-Gesetzes wurde ein molarer Ex ink i n k effizien v n ε410(K3[Fe(CN)6]) = 1 mM−1 cm−1 verwendet (Friedrich et al., 1989). (d)-NAD(P)H-Oxidaseaktivität: Zur Bestimmung der (d)-NAD(P)H-Oxidaseaktivität wurde die Abnahme der Sauerstoff-Konzentration mit Hilfe einer Clark-Sauerstoffelektrode (RE K11, Oxytec) gemessen. Zur Kalibrierung der Elektrode wurden 2 mL A-Puffer bei 30 °C mit einer Spatelspitze Natriumdithionit reduziert und das Signal aufgenommen. Zur Berechnung der Aktivität wurde von einer Sauerstoffkonzentration von 237 µM (Weiss, 1970) im Puffer 57 Material und Methoden ausgegangen. Für die Messung wurden 2 mL A-Puffer bei 30 °C vorgelegt und mit 5 µL Membransuspension versetzt. Die Reaktion wurde durch Zugabe von 5 µL 0.5 M (d)-NAD(P)H (Endkonzentration 1.25 mM) gestartet. Die Komplex I-vermittelte Aktivität der Probe wurde durch die Zugabe von 5 µL der spezifischen Hemmstoffe Piericidin A (10 mM in EtOH, Endkonzentration 25 µM) oder Fenazaquin (10 mM in Ethanol, Endkonzentration 50 µM) blockiert. Succinat-Oxidaseaktivität: Die Succinat-Oxidaseaktivität wurde ebenfalls als Sauerstoffverbrauch an einer Clark-Sauerstoffelektrode (RE K1-1, Oxytec) gemessen. Zur Kalibrierung wurden 2 mL PBS-Puffer mit einer Spatelspitze Natriumdithionit reduziert. Der gemessene Spannungsabfall entspricht einer Sauerstoffkonzentration von 237 µM (Weiss, 1970). Für die Messung wurden 2 mL PBS-Puffer bei 30 °C vorgelegt und mit 5 µL Membransuspension versetzt. Der Ansatz wurde 10 min unter Rühren inkubiert um eventuell gebundenes Oxalacetat aus dem aktiven Zentrum zu verdrängen (Maklashina und Cecchini, 1999). Die Reaktion wurde durch Zugabe von 5 µL 0.5 M Succinat (Endkonzentration 1.25 mM) gestartet. Die Succinat Dehydrogenase vermittelte Aktivität der Probe wurde durch die Zugabe von 20 µL des Hemmstoffes Malonat (1 M in ddH2O, Endkonzentration 10 mM) inhibiert. Um den Einfluss der Komplex I-spezifischen Inhibitoren Piericidin A und Fenzaquin auf die Aktivität der der restlichen Atmungskette in den verwendeten E. coli Membranen zu überprüfen, wurden der Reaktion jeweils 5 µL Piericidin A (10 mM in EtOH, Endkonzentration 25 µM) oder 10 µL Fenazaquin (10 mM in Ethanol, Endkonzentration 50 µM) zugesetzt. NADH:Decyl-Ubichinon Oxidoreduktaseaktivität: Zur Bestimmung der physiologischen Komplex I Aktivität wurde der Elektronentransfer von NAD(P)H auf das UbichinonAnalogon 2,3-Dimethoxy-5-methyl-6-decyl-1,4-benzochinon (Decyl-Ubichinon) verfolgt. Die Aktivität wurde über die zeitliche Abnahme der NAD(P)H-Konzentration bei 340 nm bestimmt. 5 µg Komplex I wurden 1:1 (w/w) mit E. coli polaren Lipiden (10 mg/mL) versetzt und für 30 min auf Eis inkubiert. Für die Messung wurden 981 µL A-Puffer vorgelegt und mit 10 µg der Komplex I/Lipid-Mischung und anschließend mit 3 µL Decyl-Ubichinon (20 mM in Ethanol, Endkonzentration 60 µM) versetzt. Es wurde 1 min bei Raumtemperatur inkubiert und die Messung durch Zugabe von 15 µL NADH (10 mM in H2O, Endkonzentration 150 µM) gestartet. Zur Berechnung der physiologischen Aktivität mittels des Lambert- 58 Material und Methoden Beerschen-Ge e ze wur e ein m arer Ex ink i n k effizien v n ε340(NADH) = 6.178 mM−1 cm−1 verwendet. Protonentranslokationsaktivität: Um die Protonentranslokation zu messen, wurde Komplex I zunächst in Liposomen rekonstituiert. Hierfür wurden 200 µg Enzym (5 mg/mL) in ADDM-Puffer mit 60 µL E. coli polaren Lipiden (20 mg/mL in Lipidpuffer) 1:6 (w/w) vermischt und mit 80 µL Rekonstitutionspuffer verdünnt. BioBeads SM-2 (Biorad) wurden mit Methanol und anschließend mit ddH2O aktiviert. Die Enzym/Lipid-Lösung wurde zum etwa 8-fachen Überschuss an BioBeads, bezogen auf die Detergenzbindekapazität von etwa 105 mg DDM/g BioBeads (Rigaud et al., 1998), gegeben und 3 h unter Rühren auf Eis inkubiert (Stolpe und Friedrich, 2004). Zur Abschätzung der in der Probe befindlichen DDMGesamtmenge, wurde die DDM-Konzentration in den Puffern und der Komplex I-Lösung berechnet (Böttcher et al., 2002). Die Proteoliposomen wurden vorsichtig mit einer Pipette von den BioBeads abgetrennt und zur Messung der Protonentranslokationsaktivität mittels Fluoreszenzlöschung von 9-amino-6-chlor-2-methoxyacridin (ACMA) eingesetzt. Alle Messungen wurden im Arbeitskreis von Prof. P. Gräber im Institut für Physikalische Chemie (Albert-Ludwigs-Universität, Freiburg) durchgeführt. 967 µL ACMA-Messpuffer wurden mit 5.1 µL Proteoliposomen (6.5 µg Komplex I), 5 µL Decyl-Ubichinon (10 mM in Ethanol, Endkonzentration 50 µM) und 8 µL ACMA-Lösung (25 µM in Ethanol, Endkonzentration 0.2 µM) versetzt und unter Rühren inkubiert bis die Fluoreszenzemission bei 480 nm (d = 1 cm, Makro-Küvetten Spezial PS, Ratiolab; LS 45 Luminescence Spectrometer, Perkin Elmer, Anregungswellenlänge 430 nm, Spaltbreite 10 nm) konstant blieb. Durch die Zugabe von 15 µL NAD(P)H (10 mM in ddH2O, Endkonzentration 150 µM) wurde die Reaktion gestartet. Die zeitliche Änderung der Fluoreszenzemission bei 480 nm wurde mit Hilfe eines des Programms FL WinLab (Perkin Elmer) aufgezeichnet. Zur Kontrolle wurde der durch die Reaktion aufgebaute Protonengradient durch die Zugabe von 10 µL CCCP (10 mM in Ethanol, Endkonzentration 100 μM) abgebaut oder die Reaktion durch die Zugabe von 5 µL Piericidin A (10 mM in Ethanol, Endkonzentration 50 µM) gestoppt. 3.7.6 SDS-Polyacrylamid-Gelelektrophorese Nach Schägger und von Jagow (1987): Um die Reinheit und Zusammensetzung von Cytoplasmamembranen und der Präparation des E. coli Komplex I zu untersuchen, wurde eine SDS-PAGE nach Schägger und von Jagow (1987) verwendet. Für die Auftrennung der 59 Material und Methoden Polypeptide wurde ein diskontinuierliches Gelsystem mit 3.9%igem Sammelgel und 10%igem Trenngel verwendet. Zur Probenvorbereitung wurden 70 – 300 µg gelöstes Protein oder Membranensuspension mit Schägger-Probenpuffer versetzt, für 30 min bei 37 °C unter Schütteln (750 Upm, Eppendorf Thermomixer compact) denaturiert und auf das Gel aufgetragen. Die Polypeptide wurden bei konstanten 45 mA (EPS 600, Pharmacia Biotech) aufgetrennt, bis die Farbstoffbande die untere Gelkante erreicht hatte. Die Proteinbanden wurden mit heißer Färbelösung gefärbt und durch anschließende Entfärbung des Hintergrunds mit Entfärbelösung sichtbar gemacht. Wenn Gele für den Western Blot verwendet wurden, wurden diese bis zum Transfer in Blotpuffer aufbewahrt. Nach Laemmli (1970): Um die Reinheit von A. aeolicus NuoEFHis-Proben zu untersuchen, wurde ein diskontinuierliches Gelsystem nach Laemmli (1970) aus einem 5%igem Sammelgel und einem 15%igem Trenngel verwendet. Es wurden 10 µg Protein mit ddH2O auf 10 µL aufgefüllt, mit 4 μL Pr benpuffer ver e z un bei 65 °C denaturiert (750 Upm, 10 min, Thermomixer compact, Eppendorf). Die Polypeptide wurden bei konstanten 35 mA (EPS 600, Pharmacia Biotech) für etwa eine Stunde elektrophoretisch aufgetrennt. Die Proteinbanden wurden wie oben beschrieben sichtbar gemacht. 3.7.7 Western Blot-Analyse Zur Untersuchung der Produktion einzelner E. coli Komplex I-Varianten wurden die durch SDS-PAGE aufgetrennten Proteinuntereinheiten auf eine zuvor in Methanol aktivierte PVDFMembran (Westran S, Whatman) transferiert. Es wurde eine mit Blotpuffer gefüllte Nass-Blot Apparatur bei 4 °C und ein über einen Zeitraum von 135 min konstanter Strom von 400 mA verwendet (Towbin et al., 1979). Um eine unspezifische Anbindung der primären Antikörper zu verhindern, wurde die Membran bei Raumtemperatur 1 h in Blockpuffer inkubiert. Danach wurde die Membran für 1 h oder über Nacht bei 4 °C mit dem primären Antikörper (Mouse Anti-His4, Qiagen, in PBS) inkubiert. Die Membran wurde zweimal mit PBS-T gewaschen (50 mL, 5 min) und 1 h mit dem sekundären Antikörper (Goat anti mouse IgG, HRP conjugated, Qiagen, in PBS) inkubiert. Die Membran wurde erneut zweimal mit PBS-T (50 mL, 5 min) und zweimal mit Na2HPO4-Lösung (50 mL, 5 min) gewaschen. Danach wurde die frisch angesetzte Färbelösung hinzugegeben. Sobald die Banden sichtbar waren, wurde die Membran in ein Wasserbad (RT) überführt, um die Färbereaktion zu stoppen. 60 Material und Methoden 3.7.8 Saccharosedichtegradienten-Zentrifugation Alle Arbeitsschritte wurden bei 4 °C oder auf Eis durchgeführt. E. coli Cytoplasmamembranen wurden isoliert, 1:1 (w/v) in A*-Puffer resuspendiert und 1:1000 (v/v) mit PMSF (0.1 M in Isopropanol) versetzt. 1.5 mL der Membranensuspension wurden mit 263 µL DDM-Stammlösung (20% (w/v), Endkonzentration 3%) versetzt und für 15 min auf Eis inkubiert. Alle 3 – 4 min wurde mit einem Teflon-in-Glas-Homogenisator durchmischt. Der Extrakt wurde zentrifugiert (150´000×g, 4 °C, 30 psi, 25 min, Rotor A-100, Airfuge, Beckmann). 400 µL des Extrakts wurden auf einen 12 mL Saccharosegradienten von 5 – 30% (w/v) Saccharose in A*DDM-Puffer mit 50 µM PMSF aufgetragen und zentrifugiert (222´000×g, 17 h, 4 °C, Rotor SW 41 Ti, Coulter-Optima LE-80K Ultracentrifuge, Beckman). Die Gradienten wurden zu 600 µL fraktioniert und die NADH/Ferricyanid Oxidoreduktaseaktivität der einzelnen Fraktionen bestimmt. 3.8 Kristallographische Methoden 3.8.1 Kristallisation der A. aeolicus NuoEFHis-Varianten Für Kristallisationsexperimente wurden die hanging drop und die sitting drop Methode in 24-Well-Kristallisationsplatten angewendet. Es wurden jeweils 2.5 µL Proteinlösung mit 5 –12 mg/mL mit 2.5 µL Reservoirlösung versetzt. Das Volumenverhältnis von Reservoirlösung zu Tropfengröße betrug immer 1:100. Die Kristallisationsansätze wurden bei 16 – 20 °C unter Lichtausschluss gelagert. Das Kristallwachstum wurde in regelmäßigen Abständen beobachtet, bevor die bis dahin gewachsenen Kristalle für röntgendiffraktometrische Messungen weiter präpariert wurden. Die Zusammensetzungen der einzelnen Reservoirs sind in der Tabelle 3.20 aufgeführt. Für die Visualisierung von Proteinstrukturen wurde das Programm UCSF Chimera verwendet (Petterson et al., 2004). 61 Material und Methoden Tabelle 3.20: Zusammensetzung der Reservoirs für die Kristallisation. Der pH-Wert für 0.1 M Tris/HCl ist angegeben. Alle Reservoirlösungen enthielten zusätzlich 0.2 M NaCl. 1 2 3 4 5 6 A pH 6.8, 0.6 M Na3Citrat pH 6.8, 0.7 M Na3Citrat pH 6.8, 0.8 M Na3Citrat pH 6.8, 0.9 M Na3Citrat pH 6.8, 1.0 M Na3Citrat pH 6.8, 1.05 M Na3Citrat B pH 6.9, 0.6 M Na3Citrat pH 6.9, 0.7 M Na3Citrat pH 6.9, 0.8 M Na3Citrat pH 6.9, 0.9 M Na3Citrat pH 6.9, 1.0 M Na3Citrat pH 6.9, 1.05 M Na3Citrat C pH 7.0, 0.6 M Na3Citrat pH 7.0, 0.7 M Na3Citrat pH 7.0, 0.8 M Na3Citrat pH 7.0, 0.9 M Na3Citrat pH 7.0, 1.0 M Na3Citrat pH 7.0, 1.05 M Na3Citrat D pH 7.1, 0.6 M Na3Citrat pH 7.1, 0.7 M Na3Citrat pH 7.1, 0.8 M Na3Citrat pH 7.1, 0.9 M Na3Citrat pH 7.1, 1.0 M Na3Citrat pH 7.1, 1.05 M Na3Citrat 3.8.2 Herstellung von Keimkristallen Um das Kristallwachstum zu initiieren oder hochwertigere Kristalle für die Röntgendiffraktometrie zu erhalten, wurden Keimkristalle (seeds) verwendet. Zu deren Darstellung wurden verwachsene Kristalle oder Kristallfragmente aus einem Kristallisationstropfen entnommen und in 10 µL Reservoirlösung mit einer Pipettenspitze grob zerkleinert. Diese wurden in einem 1.5 mL Reaktionsgefäß mit 50 µL eiskalter Reservoirlösung verdünnt. Seed-beads (Hampton Research) wurden zugegeben und durch zweiminütiges Rühren (Vortex-GENIE, Scientific Industries) die noch zu großen Kristalle zu Mikrokristallen zerkleinert. Um eine zu starke Erwärmung und damit ein Auflösen der Mikrokristalle zu vermeiden, wurde das Reaktionsgefäß alle 30 s auf Eis gestellt. Es wurden jeweils 10 µL der Lösung aliquotiert und Verdünnungen von 1:1000 und 1:10´000 hergestellt. Alle seed-Lö ungen wur en bei −80 °C gelagert. Bei der Kristallisation wurden 1.9 µL Reservoirlösung und 0.6 µL der 1:1000 oder 1:10´000 verdünnten seedstock Lösung mit 2.5 µL Proteinlösung in einem Tropfen vermischt. Anschließend wurde wie oben beschrieben weiterverfahren. 3.9 EPR-Spektroskopie Alle EPR-spektroskopischen Untersuchungen wurden in Zusammenarbeit mit Herrn Prof. Thorsten Friedrich oder Frau Dr. Katerina Dörner am Institut für Biochemie (Albert-Ludwigs62 Material und Methoden Universität, Freiburg) durchgeführt. Die EPR-Spektren wurden mit einem EMX 6/1 EPRSpektrometer (Bruker) ausgestattet mit einem ESR-9 Helium-Kryostaten (Oxford Instruments) aufgenommen. Zum Nachweis einzelner Fe/S-Zentren wurden 0.5 – 1.5 mg des isolierten Komplex I in 300 µL A-Puffer mit 1 – 3 µL NAD(P)H (1 M) und/oder mit einigen Körnchen Dithionit reduziert. Die Probe wurde durchmischt, luftblasenfrei in ein EPR-Röhrchen überführt und in einer Kältemischung (2-Methylbutan/Methylcyclohexan 5:1, v/v) bei ca. −120 °C eingefroren. Die Proben wurden in flüssigem Stickstoff gelagert. Während der NAD(P)H-Umsetzung durch Komplex I entstehende ROS wurden EPRspektroskopisch mit Hilfe des Radikalfängers DEPMPO bei Raumtemperatur detektiert (Vasquez-Vivar et al., 2000). Der 1 mL Reaktionsansatz (A-Puffer) enthielt 45 µg Komplex I rekonstituiert in E. coli polaren Lipiden, 1 mM NAD(P)H und 0.5 µL DEPMPO (Endkonzentration 100 mM). Die Reaktion wurde durch die Zugabe von 100 µM DecylUbichinon gestartet. Der Ansatz wurde 1 min bei Raumtemperatur inkubiert bevor 20 µL in einem Quarzröhrchen entnommen wurden. Zur Kontrolle wurden 100 U/mL Superoxiddismutase (SOD) bzw. 20 µM Piericidin A zugegeben, oder es wurde auf die Zugabe von Decyl-Ubichinon verzichtet. 63 Ergebnisse 4 Ergebnisse 4.1 Substratunterscheidung am Komplex I NADH und NADPH werden in verschiedenen metabolischen Prozessen verwendet. NADH wird bevorzugt in katabolen, NADPH hingegen in anabolen Prozessen genutzt. Die Struktur der NADH-Bindestelle des Komplex I zeigt, dass Glu183F Wasserstoffbrücken zu dem O2' und dem O3' der Adenosin-Ribose ausbildet. Sequenzanalysen von Dinukleotid-abhängigen Dehydrogenasen haben gezeigt, dass an der homologen Position ein Asp bei NAD- bzw. ein Asn oder ein Gly bei NADP-abhängigen Dehydrogenasen vorliegt (Lesk, 1995). Um zu untersuchen, ob das Glu183F für die Unterscheidung zwischen NADH und NADPH als Substrat verantwortlich ist, wurde es jeweils gegen Ala, Asp, Asn, Gln, Gly und His ausgetauscht. Florian Hubrich wurde im Rahmen seiner Zulassungsarbeit zu den Arbeiten zu Teilen dieses Kapitels angeleitet (Hubrich, 2010). Teile der hier gezeigten Ergebnisse wurden bereits veröffentlicht (Morina et al., 2011). Zur gleichen Zeit veröffentlichte die Gruppe von Isabelle Meynial-Salles an der Université de Toulouse, Frankreich, ähnliche Ergebnisse mit einem anderen methodischen Ansatz (Auriol et al., 2011). Hierbei wurde untersucht, wie sich E. coli an eine erhöhte intrazelluläre NADPH-Konzentration anpasst. Durch Inaktivierung des zweiten Enzyms der Glykolyse und des ersten des Entner-Doudoroff-Wegs war diese E. coli Mutante gezwungen Glucose über den Pentosephosphatweg, die Hauptproduktionsstätte von NADPH, zu verstoffwechseln. Nach bereits 20 Generationen wurden erhöhte Wachstumsraten beobachtet. Analysen der genomischen DNA zeigten eine einzelne Punktmutation; Glu183AlaF. Um die NADPH-Konzentration weiter zu erhöhen wurden die Gene der Phosphofruktokinase 1 und 2 inaktiviert, um anaplerotische Reaktionen der Glykolyse zu unterbinden. Nach mehreren Generationen wurden nur acht Klone erhalten, von denen fünf ebenfalls die Mutation Glu183AlaF und drei die Mutation Glu183GlyF trugen (Auriol et al., 2011). Zur Überproduktion der Glu183F-Varianten wurde hier der E. coli Stamm BW25113Δnuo und das Expressionsplasmid pBADnuo nuoFhis verwendet. Da das Expressionsplasmid pBADnuo nuoFhis mit etwa 21 kbp zu groß für eine ortsgerichtete Mutagenese ist, wurden Punktmutationen mit Hilfe der λ-Red vermittelten Rekombination eingeführt. Um die Effizienz der Rekombination zu erhöhen wurde das Gen nuoF auf pBADnuo nuoFhis durch Insertion der nptI-sacB-Selektionskassette inaktiviert. Die gewünschten Punktmutationen wurden mittels ortsgerichteter Mutagenese in den Subklon pCA24nuoF, der nur das Gen 64 Ergebnisse nuoF enthält, eingebracht. Von dem Subklon wurde ein lineares Fragment amplifiziert, das die gewünschte Mutation trug. Mit Hilfe der λ-Red vermittelten Rekombination wurde die Selektionskassette in dem Vektor pBADnuo nuoFhis::nptI-sacB gegen das mutierte lineare Fragment ausgetauscht. Die so dargestellten Mutanten wurden angezogen, die Komplex I-Varianten isoliert und biochemisch charakterisiert. 4.1.1 Darstellung der Mutanten pBADnuo nuoFhis E183A/D/G/H/N und Q Die nptI-sacB-Kassette wurde von dem Vektor pVO1100 mit den Oligonukleotiden nuoF::nptI-sacB_fwd und nuoF::nptI-sacB_rev (Tab. 3.5) amplifiziert. Die Oligonukleotide trugen die für die λ-Red vermittelte Rekombination benötigten homologen Bereiche zu nuoF. E ek r k mpe en e DH5αΔnuo/pKD46 wurden frisch hergestellt und mit 50 ng pBADnuo nuoFhis und 350 ng des PCR-Produkts nuoF::nptI-sacB kotransformiert. Der Ansatz wurde auf LB-Platten mit Kanamycin ausgestrichen und etwa 22 h bei 30 °C inkubiert. Es wurden einzelne Klone kultiviert, deren Plasmide isoliert und mittels Restriktion analysiert (Abb. 4.1). Länge [bp] 1 2 19´329→ 7743→ 4254→ 3472→ 2690→ Abbildung 4.1: Restriktionsanalyse des Vektors pBADnuo nuoFhis::nptI-sacB mit VspI. Spur 1 zeigt das Bandenmuster des λ-DNA/Eco130I(StyI) Marker16. Spur 2 zeigt das erwartete Bandenmuster mit Längen von 7238, 5907, 4545, 3508 und 3147 bp. Um die einzelnen Punktmutationen in den Expressionsvektor pBADnuo nuoFhis einzufügen, mussten diese zunächst durch ortsgerichtete Mutagenese in den Subklon pCA24nuoF eingebracht werden. Hierfür wurden die Oligonukleotid-Paare nuoF E183A_fwd/rev, nuoF E183D_fwd/rev, nuoF E183G_fwd/rev, nuoF E183H_fwd/rev, nuoF E183N_fwd/rev und nuoF E183Q_fwd/rev verwendet (Tab. 3.4). Die Oligonukleotide trugen neben den 65 Ergebnisse gewünschten Mutationen, auch stille Mutationen um eine neue Schnittstelle für VspI zu generieren. Für die PCR wurden 40 – 50 ng pCA24NnuoF als Vorlage und eine Anlagerungstemperatur von 60 °C für 60 s verwendet. Der Erfolg der PCR wurde durch Agarosegelelektrophorese überprüft. Das gereinigte PCR-Produkt wurde mit DpnI verdaut. 50 µL elektrokompetente E. coli DH5α wur en mi 1 µL des gereinigten Produktes transformiert und der gesamte Ansatz auf chloramphenicolhaltigen LB-Agar-Platten ausgestrichen. Es wurden einzelne Kolonien angezogen, die Plasmide der Kulturen isoliert und mittels Restriktion analysiert (Abb. 4.2). Der Erfolg der ortsgerichteten Mutagenese wurde durch Sequenzierung bestätigt. Länge [bp] 1 2 19´329→ 7743→ 4254→ 3472→ 1489→ 925→ 421→ Abbildung 4.2: Restriktionsanalyse des Vektors pCA24NnuoF E183A mit VspI. Die Agarosegele der anderen Mutanten sehen identisch aus und werden daher nicht gezeigt. Spur 1 zeigt das Bandenmuster des λ-DNA/Eco130I(StyI) Marker16. Spur 2 zeigt das erwartete Bandenmuster mit Längen von 3999, 1055 und 742 bp. Die erwartete Bande bei 59 bp ist nicht mehr im Agarosegel enthalten. Die Subklone pCA24NnuoF E183A/D/G/H/N und Q tragen genau wie der Expressionsvektor pBADnuo nuoFhis das Gen für die Chloramphenicol-Acetyltransferase, welche die Chloramphenicol-Resistenz vermittelt. Um die Effizienz der Rekombination zu erhöhen, wurden die Subklone zunächst mit CaiI geschnitten, das Fragment mit dem mutierten nuoF aus dem Agarosegel isoliert und als Vorlage zur Amplifikation der für die λ-Red vermittelte Rekombination benötigten linearen Fragmente verwendet. Für die PCR wurden die Oligonukleotide nuoF_fwd und nuoF_rev (Tab. 3.6) bei einer Anlagerungstemperatur von 60 °C verwendet. Der Erfolg der PCR wurde durch Agarose-Gelelektrophorese überprüft. Die Ansätze wurden gereinigt, mit DpnI verdaut und erneut gereinigt. Elektrokompetente DH5αΔnuo/pKD46 wurden frisch hergestellt und mit 50 ng pBADnuo nuoFhis::nptI-sacB und 300 – 400 ng der linearen Fragmente kotransformiert, auf YP/SUC-Agar-Platten mit 66 Ergebnisse Chloramphenicol ausgestrichen und 20 – 24 h bei 30 °C inkubiert. Es wurden einzelne Kolonien angezogen, die Plasmide der Klone isoliert und durch Restriktion analysiert (Abb. 4.3). Der Erfolg der λ-Red vermittelten Rekombination wurde durch Sequenzierung bestätigt. Länge [bp] 1 2 19´329→ 7743→ 6223→ 4254→ 3472→ 2690→ 925→ 421→ Abbildung 4.3: Restriktionsanalyse des Vektors pBADnuo nuoFhis E183A mit VspI. Die Agarosegele der anderen Mutanten sehen identisch aus und werden daher nicht gezeigt. Spur 1) zeigt das Bandenmuster des λ-DNA/Eco130I(StyI) Marker16. Spur 2) zeigt das erwartete Bandenmuster mit Längen von 7238, 5654, 4545, 3147 und 742 bp. 4.1.2 Komplex I-vermittelte Aktivitäten der Cytoplasmamembranen Für die Überproduktion der Komplex I Varianten wurde der E. coli Stamm BW25113Δnuo verwendet. Zellen dieses Stamms wurden mit den dargestellten Plasmiden transformiert. Zur Kontrolle wurden die Plasmide vor der Zellzucht aus dem Expressionsstamm isoliert und erneut über eine Restriktionsanalyse mit VspI charakterisiert (Daten nicht gezeigt). Die Mutanten BW25113Δnuo/pBADnuo nuoFhis E183A/D/G/H/N und Q wurden im 10 LMaßstab in Autoinduktionsmedium angezogen. Um den Einfluss der Mutationen auf das Zellwachstum zu untersuchen, wurde der Ausgangsstamm BW25113Δnuo/pBADnuo nuoFhis ebenfalls angezogen. Abbildung 4.4 zeigt die Wachstumskurven der einzelnen Mutanten und des Ausgangsstamms. Die Mutationen von E183F zu D, H, N und Q hatten keinen Einfluss auf das Zellwachstum, da alle Stämme eine OD600 von etwa 5.5 erreichten. Die entsprechenden Mutationen zu Ala und Gly beeinträchtigten das Wachstum stark. Die Stämme BW25113Δnuo/pBADnuo nuoFhis E183G und E183A erreichten etwa 2 h vor dem Ausgangsstamm die stationäre Phase bei einer OD600 von 2.5 bzw. 2.8, was weniger als der Hälfte der OD600 des Ausgangsstamms entspricht. 67 Ergebnisse A) B) Abbildung 4.4: Wachstumskurven der Stämme BW25113Δnuo/pBADnuo nuoFhis E183X. A) zeigt die Wachstumskurven der Stämme BW25113Δnuo/pBADnuo nuoFhis E183D (grün), E183H (rot), E183N (blau), E183Q (violett), und des Ausgangsstamms (schwarz). B) stellt die Wachstumskurven der Stämme BW25113Δnuo/pBADnuo nuoFhis E183A (rot), E183G (blau) und des Ausgangsstamms (schwarz) dar. Die Zellen wurden im 10 L-Maßstab in Autoinduktionsmedium angezogen. Die Cytoplasmamembranen der Mutanten sowie des Ausgangsstamms wurden präpariert und deren NAD(P)H-Oxidaseaktivitäten als auch NADH/Ferricyanid Oxidoreduktaseaktivitäten bestimmt. Die Membranen des Ausgangsstamms dienten als Referenz. Tabelle 4.1 gibt die Aktivitäten wieder. Tabelle 4.1: NADH/Ferricyanid Oxidoreduktaseaktivitäten und NAD(P)H-Oxidaseaktivitäten der Cytoplasmamembranen des Ausgangsstamms und der Glu183F-Mutanten. Für eine bessere Vergleichbarkeit wurden die Aktivitäten auf die des Ausgangsstamms normiert, wobei diese gleich 1.0 gesetzt wurde. BW25113Δnuo/ pBADnuo nuoFhis NADH/Ferricyanid Oxidoredukatseaktivität NADHOxidaseaktivität NADPHOxidaseaktivität Normierte Oxidaseaktivitäten NADH NADPH [µmol·min−1·mg−1] 68 unverändert 7.0 0.8 0.2 1.0 1.0 E183A 7.5 1.5 0.5 1.9 2.5 E183D 15.4 1.8 1.1 2.3 5.5 E183G 6.6 0.8 0.4 1.0 2.0 E183H 11.2 1.1 0.5 1.4 2.5 E183N 15.4 1.1 0.5 1.4 2.5 E183Q 7.9 0.8 0.5 1.0 2.5 Ergebnisse Die Werte sind höher als bei anderen Messungen (vgl. 4.4.2, 4.5.1 und 4.6.1). Untereinander sind die Werte vergleichbar, da die Membranen zeitgleich und unter identischen Bedingungen vermessen wurden. Der Grund für die erhöhte Aktivität ist nicht verstanden. Die Membranen der Mutanten weisen im Vergleich zum Ausgangsstamm eine deutlich erhöhte NADPHOxidaseaktivität auf. Die NADPH-Oxidaseaktivität der Cytoplasmamembranen der Mutante BW25113Δnuo/pBADnuo nuoFhis E183G ist doppelt so hoch, die der Mutanten BW25113Δnuo/pBADnuo nuoFhis E183A/H/N und Q zweieinhalbmal und die der Mutante BW25113Δnuo/pBADnuo nuoFhis E183D sogar mehr als fünfmal so hoch wie die des Ausgangsstamms. Die NADH-Oxidaseaktivitäten der Membranen der Mutanten BW25113Δnuo/pBADnuo nuoFhis E183G und Q sind im Vergleich zum Ausgangsstamm unverändert. Die der Mutanten BW25113Δnuo/pBADnuo nuoFhis E183H und N sind anderthalbfach erhöht und die Mutanten BW25113Δnuo/pBADnuo nuoFhis E183A und D zeigen eine doppelt so hohe Aktivität. Die NADH/Ferricyanid Oxidoreduktaseaktivitäten der Mutanten BW25113Δnuo/pBADnuo nuoFhis E183A/G und Q ähneln der des Ausgangsstamms. Bei den Mutanten BW25113Δnuo/pBADnuo nuoFhis E183D/H und N sind diese deutlich erhöht. Die Aktivitäten der Membranen geben einen Hinweis darauf, dass die eingeführten Mutationen die Umsetzung von NADPH begünstigen. 4.1.3 Präparation des Komplex I und der Varianten E183A/D/G/H/N und QF Um den Einfluss der Mutationen auf die Affinität zu NADH und NADPH zu untersuchen, wurden die Varianten nach etabliertem Protokoll isoliert. Da alle Varianten auf dieselbe Weise präpariert wurden, wird im diesem Kapitel die Reinigung der Variante E183DF exemplarisch gezeigt. Die übrigen Präparationen verliefen ähnlich. 31 g Zellen des E. coli S amm BW25113Δnuo/pBADnuo nuoFhis E183D wurden mit Hilfe einer Druckentspannungsanlage aufgeschlossen. Die Cytoplasmamembranen wurden präpariert und die Membranproteine mit 3% (w/v) DDM extrahiert. Die Variante wurde durch Anionenaustausch- und Affinitäts-Chromatographie gereinigt. Abbildung 4.5 A gibt das Elutionsprofil der Anionenaustausch-Chromatographie wieder. Die gebundenen Proteine wurden mit einem Gradienten von 150 bis 350 mM NaCl eluiert. Fraktionen mit NADH/Ferricyanid Oxidoreduktaseaktivität über 20 U/mL wurden vereinigt und über eine Ni2+-Affinitäts-Chromatographie weiter gereinigt. Gebundene Proteine wurden mit einem Stufengradienten von 140, 260, 380 und 500 mM Imidazol eluiert (Abb. 4.5 B). Die Variante 69 Ergebnisse eluierte bei 260 mM Imidazol. Fraktionen mit NADH/Ferricyanid Oxidoreduktaseaktivität über 15 U/mL wurden vereinigt. A) B) Abbildung 4.5: Elutionsprofile der Anionenaustausch-Chromatographie an Fractogel EMD TMAE A) und Affinitäts-Chromatographie an ProBond Ni2+-IDA B) der Präparation der Variante E183DF. Die Absorbanz bei 280 nm ist in Blau, die NaCl- bzw. die Imidazol-Konzentration in Grün und die artifizielle NADH/Ferricyanid Oxidoreduktaseaktivität in Rot dargestellt. Tabelle 4.2 gibt den Verlauf der Reinigung der Variante E183DF als Beispiel wieder. Die Ausbeuten der Präparationen aller Varianten und des ursprünglichen Komplex I sind in Tabelle 4.3 zusammengefasst. Tabelle 4.2: Präparation der Variante E183DF aus 31 g Zellen des Stamms BW25113Δnuo/pBADnuo nuoFhis E183D. Protein [mL] [mg] [µmol·min-1] [µmol·min-1·mg-1] [%] Membranen 18.0 1168 4722 4.0 100 Detergenzextrakt 60 786 3935 5.0 83 Fractogel EMD 44 119 1571 13.2 33 0.5 6.3 882 139.8 19 Präparationsschritt 2+ ProBond Ni -IDA 70 NADH/Ferricyanid Oxidoreduktaseaktivität total spezifisch Volumen Ausbeute Ergebnisse Tabelle 4.3: Ausbeute der Präparationen der Varianten E183A/D/G/H/N/QF und des ursprünglichen Komplex I. Variante Eingesetzte Zellen Protein NADH/Ferricyanid Oxidoreduktaseaktivität Ausbeute aus Gesamtaktivität Komplex I [g] 30.0 [mg] 6.0 [µmol·min−1·mg−1] 83.0 [%] 13 E183AF 31.0 2.1 80.0 8 F E183D 31.0 6.3 139.8 19 E183GF 25.0 4.5 64.8 9 F 20.0 6.1 66.2 26 F 23.0 2.0 60.0 5 F 22.0 2.9 81.3 7 E183H E183N E183Q Alle Varianten konnten in ausreichender Menge isoliert werden. Die Präparationstabelle der Variante E183DF zeigt, dass die spezifische NADH/Ferricyanid Oxidoreduktaseaktivität mit jedem Reinigungsschritt stieg. Diese lag für die meisten Präparationen zwischen 60 und 80 µmol·min−1·mg−1. Für die Variante E183DF betrug die Aktivität 140 µmol·min−1·mg−1 und lag damit über der des Komplex I mit 83 µmol·min−1·mg−1. Die Präparationen wurden mittels SDS-PAGE analysiert (Abb. 4.6). Das Acrylamidgel zeigt alle Banden der NuoUntereinheiten in den Präparationen. M 1 2 3 4 5 6 7 app. Mr [kDa] 85 → 70 → 50 → 40 → 30 → 25 → 20 → 15 → ← Nu G ← Nu CD ← Nu F ← NuoL/N ← NuoM ← NuoH ← NuoB } NuoI/J/E ← NuoA ← NuoK Abbildung 4.6: Polyacrylamidgel der Präparationen. Spur M zeigt die Banden des Proteinstandards PageRuler Unstained Protein Ladder (Thermo Scientific), Spur 1 die der Variante E183H F, Spur 2 die der Variante E183QF, Spur 3 die der Variante E183AF, Spur 4 die der Variante E183DF, Spur 5 die der Variante E183NF, Spur 6 die der Variante E183GF und Spur 7 die des unveränderten Komplex I. Es wurden jeweils 60 – 70 µg Protein auf das Gel aufgetragen. Die hydrophoben Untereinheiten NuoH, L, M und N werden nur schwach angefärbt. Die Banden sind den einzelnen Komplex I-Untereinheiten zugeordnet. 71 Ergebnisse 4.1.4 Charakterisierung der Varianten E183A/D/G/H/N und QF Um den Einfluss der eingeführten Mutation auf die Substratspezifität zu untersuchen, wurde die NAD(P)H:Decyl-Ubichinon Oxidoreduktaseaktivität der in polare Lipide rekonstituierten Varianten in Abhängigkeit von der Substratkonzentration bestimmt. Die Aktivitäten wurden für 5 – 200 µM NADH bzw. 5 – 1500 µM NADPH bei einer Decyl-Ubichinon-Konzentration von 60 µM bestimmt. Die Aktivitäten wurden nach Hanes (1932) gegen die Substratkonzentration aufgetragen. Nach linearer Regression wurde anhand der Steigung der Geraden und dem Schnittpunkt mit der Ordinate die Michaelis-Konstante Km und die maximale Umsatzgeschwindigkeit vmax bestimmt. Aus vmax wurde die Wechselzahl kcat berechnet und mit dieser wiederum die katalytische Effizienz (kcat/Km) bestimmt. In Tabelle 4.4 sind die kinetischen Parameter des Komplex I und der Varianten aufgeführt. Die kinetischen Parameter wurden in Zusammenarbeit mit Herrn Dipl.-Chem. Marius Schulte bestimmt. Tabelle 4.4: Kinetische Parameter des Komplex I und der Varianten E183A/D/G/H/N und QF mit NADH und NADPH als Substrat. Variante Km vmax kcat kcat/Km [µM] [µmol·min−1·mg−1] [s−1] [s−1·µM−1] NADH NADPH NADH NADPH NADH NADPH NADH NADPH Komplex I 13 ± 1 1870 ± 30 2.9 ± 0.2 0.37 ± 0.1 26 ± 2 3.0 ± 1 2.0 ± 0.3 0.0018 ± 0.001 E183AF 12 ± 1 8±1 3.9 ± 0.1 3.9 ± 0.2 35 ± 1 35 ± 2 2.9 ± 0.2 4.35 ± 0.005 E183DF 5.8 ± 1 390 ± 30 4.2 ± 0.2 3.8 ± 0.2 37 ± 2 34 ± 2 6.4 ± 0.4 0.09 ± 0.006 E183GF 11 ± 1 33 ± 2 2.1 ± 0.1 2.4 ± 0.1 19 ± 1 21 ± 1 1.7 ± 0.2 0.65 ± 0.004 E183HF 5.7 ± 1 25 ± 2 4.1 ± 0.2 1.1 ± 0.1 37 ± 2 10 ± 1 6.5 ± 0.3 0.40 ± 0.003 E183NF 14 ± 1 480 ± 50 1.2 ± 0.2 1.2 ± 0.3 11 ± 2 11 ± 3 0.8 ± 0.1 0.022 ± 0.006 E183QF 12 ± 2 45 ± 4 3.1 ± 0.2 1.6 ± 0.1 28 ± 2 14 ± 1 2.3 ± 0.5 0.32 ± 0.003 Die kinetischen Parameter der Variante E183QF mit NADH als Substrat sind im Vergleich zum Komplex I unverändert. Im Gegensatz dazu steigt die Affinität der Variante zu NADPH um das 40-fache und der vmax um das Vierfache. Die Affinität von E183NF zu NADH ist ebenfalls unverändert, aber vmax ist halbiert. Die Varianten E183DF und E183HF besitzen eine doppelt so hohe Affinität zu NADH; zudem ist vmax um etwa ein Drittel gesteigert. Alle Varianten zeigen eine deutlich erhöhte Reaktivität mit NADPH im Vergleich zum Komplex I. 72 Ergebnisse Die Affinität der Variante E183HF zu NADPH ist um das 75-fache, der vmax dreifach erhöht. Obwohl die Affinität der Variante E183DF zu NADPH nur fünffach erhöht ist, setzt diese NADPH 10-mal schneller um als der ursprüngliche Komplex I. Die Umsatzrate überschreitet sogar die des unveränderten Enzyms mit NADH als Substrat. Die Affinität von E183NF zu NADPH und der vmax sind im Vergleich zu Komplex I vierfach erhöht, wobei die Variante NADH und NADPH gleich schnell umsetzt. Da die katalytische Effizienz von Km und vmax abhängt, ist sie ein sinnvoller Parameter, um die Varianten untereinander zu vergleichen. Die katalytische Effizienz der Varianten E183DF und E183HF sind im Vergleich zu Komplex I mit NADH als Substrat dreifach erhöht. Die der Variante E183QF ist unverändert und die von E183NF ist mehr als halbiert. Die katalytische Effizienz aller Varianten mit NADPH als Substrat ist deutlich erhöht. Die der Variante E183HF ist 220-fach, die der Variante E183QF 180-fach, die der Variante E183DF 50-fach und die der Variante E183NF 12-fach erhöht. Die auch durch eine gerichtete Evolution erhaltenen Varianten E183AF und E183GF (Auriol et al., 2011) hatten den größten Effekt auf die Umsetzung von NADPH. Die Variante E183G F hat im Vergleich zu Komplex I eine unveränderte Affinität zu NADH, wobei vmax um ein Drittel erniedrigt ist. Mit NADPH als Substrat zeigt die Variante eine 6.5-fach erhöhte Umsatzgeschwindigkeit im Vergleich zum ursprünglichen Komplex. Die katalytische Effizienz mit NADH als Substrat ist etwas erniedrigt. Wegen der stark erhöhten Affinität dieser Variante zu NADPH ist die katalytische Effizienz 360-fach erhöht. Die kinetischen Parameter der Variante E183AF sind bemerkenswert. Diese Variante zeigt die gleiche Affinität zu NADH aber eine um ein Drittel erhöhte Umsatzgeschwindigkeit im Vergleich zum unveränderten Komplex I. Daraus ergibt sich eine anderthalbfach höhere katalytische Effizienz. Der Km der Variante zu NADPH ist sogar niedriger als zu NADH, wobei beide Substrate gleich schnell umsetzt werden. Die katalytische Effizienz für die Umsetzung von NADPH ist im Vergleich zu Komplex I 2400-fach gesteigert, wobei nur die Variante E183AF eine höhere katalytische Effizienz für Umsetzung von NADPH als für die Umsetzung von NADH aufweist. Um zu untersuchen, ob alle Fe/S-Zentren durch die Zugabe von NADPH reduziert werden, wurde die Variante E183HF EPR-spektroskopisch untersucht. 1.5 mg der Variante wurden mit 1.5 µL NADPH (1 M) reduziert und eingefroren. Es wurden Spektren bei 40 und 13 K aufgenommen. Die Proben wurden von Prof. Thorsten Friedrich vermessen. Abbildung 4.7 zeigt die EPR-Spektren des Komplex I und der Variante E183HF. 73 Ergebnisse A) B) a a b b c c Magnetische Flussdichte [mT] Abbildung 4.7: EPR-Spektren des Komplex I und der Variante E183HF. Die Spektren in A) wurden bei 40 K und 2 mW, die in B) bei 13 K und 5 mW aufgenommen. a zeigt die Spektren des mit NADH und b des mit NADPH reduzierten unveränderten Komplex I. c zeigt die Spektren der mit NADPH reduzierten Variante E183HF. Die Mikrowellenfrequenz betrug 9.462 GHz, die Modulationsamplitude 0.6 mT, die Zeitkonstante 0.164 s und die Aufnahmegeschwindigkeit 17.9 mT/min (Morina et al., 2011). In den EPR-Spektren der mit NADPH-reduzierten Variante E183HF sind die Signale aller Fe/S-Zentren enthalten. Im Vergleich zu den Spektren des mit NADH reduzierten Komplex I wurden alle Fe/S-Zentren der Variante E183HF in gleichem Maß von NADPH reduziert. Es wurde untersucht, ob die NAD(P)H:Decyl-Ubichinon Oxidoreduktaseaktivität der Variante E183HF mit der Translokation von Protonen gekoppelt ist. Die Variante wurde in Liposomen rekonstituiert und der durch die Redoxreaktion erzeugte pH-Gradient anhand der Fluoreszenzlöschung des pH-sensitiven Fluoreszenzfarbstoffs ACMA detektiert. Um zu überprüfen, ob die gemessene Fluoreszenzlöschung durch den von Komplex I aufgebauten Protonengradient erfolgte, wurde den Ansätzen jeweils der Komplex I-spezifische Inhibitor Piericidin A oder der Protonengradient-Entkoppler CCCP zugegeben. Alle Reaktionen wurden durch die Zugabe von 150 µM NAD(P)H gestartet (Abb. 4.8). Die Messungen wurden in Zusammenarbeit mit Herrn Dipl.-Chem. Stefan Steimle durchgeführt. 74 Ergebnisse A) B) C) D) Abbildung 4.8: Aufbau der Protonengradienten durch den Komplex I und die Variante E183HF. Die Proteine wurden in Liposomen rekonstituiert. Die pH-abhängige Fluoreszenslöschung von ACMA (ΔF) über die Zeit wurde detektiert. Die durch die Reaktion von Komplex I A) und der Variante E183HF C) mit NADH (schwarz) und NADPH (rot) aufgebauten Protonengradienten sind abgebildet. Zur Kontrolle wurden Komplex I B) und die Variante E183HF D), bevor die Reaktion mit NADH bzw. NAD(P)H gestartet wurde, mit Piericidin A (PicA) oder CCCP inkubiert (Morina et al., 2011). Die Reaktion von Komplex I mit NADH erzeugte einen Protonengradienten. Der Aufbau des Gradienten wurde durch die Zugabe des Inhibitors Piericidin A und durch die Zugabe von CCCP komplett unterbunden. Die Zugabe von NADPH zu Komplex I-Proteoliposomen erzeugte keinen Protonengradient, was vermutlich an der langsamen Umsetzung des NADPH durch Komplex I liegt (Tab. 4.4). Der Protonenrückstrom über die Membran lag vermutlich in der gleichen Größenordnung wie die Translokation durch Komplex I. Die Variante E183HF erzeugte durch die Umsetzung von NADH ebenfalls einen Protonengradienten. Im Gegensatz zum ursprünglichen Komplex I ist die Reaktion der Variante mit NADPH mit einer nachweisbaren Protonentranslokation gekoppelt. Die Inkubation mit Piericidin A und CCCP verhinderte die Ausbildung eines Protonengradienten. Der durch die Redoxreaktion der Variante mit NADH erzeugte Protonengradient nimmt schneller ab als der von dem ursprünglichen Komplex I erzeugte. Dies könnte durch eine unterschiedliche Qualität der Liposomen oder durch die schnellere Reaktion von E183HF mit NADH bedingt sein. Unterschiede in der durch NADH und NADPH induzierten Fluoreszenzlöschung könnten auf 75 Ergebnisse den unterschiedlichen Umsatzgeschwindigkeiten von E183HF mit den Substraten basieren (vgl. Tab. 4.4). Untersuchungen an Mitochondrien haben gezeigt, dass die Lipidperoxidation durch die Komplex I-vermittelte ROS-Produktion in Anwesenheit von NADPH erhöht ist (Glinn et al., 1997). Es wurde untersucht, ob die ROS-Produktion des bakteriellen Komplex I und der Variante E183HF mit NADPH ebenfalls erhöht ist. Für den qualitativen ROS-Nachweis wurde der Radikalfänger DEPMPO verwendet. Die mit Superoxid- und Hydroxyl-Radikalen gebildeten Addukte DEPMPO-OOH und DEPMPO-OH erzeugen charakteristische, EPRspektroskopisch detektierbare Signale (Vasquez-Vivar et al., 2000). Der Komplex I und die Variante E183HF wurden in E. coli polare Lipide rekonstituiert und mit DEPMPO (100 mM) und NAD(P)H (1 mM) versetzt. Die Reaktion wurde durch die Zugabe von Decyl-Ubichinon gestartet. SOD (100 U/mL) bzw. Piericidin A (20 µM) wurden zur Kontrolle zugegeben oder es wurde auf die Zugabe von Decyl-Ubichinon verzichtet. Die EPR-spektroskopischen Messungen wurden in Zusammenarbeit mit Frau Dr. Katerina Dörner durchgeführt. Abbildung 4.9 gibt die gemessenen Spektren wieder. Proben, die ausschließlich mit DEPMPO und dem zum Protein 1000-fachen molaren Überschuss Decyl-Ubichinon inkubiert wurden, erzeugten keine Signale. Die Zugabe von NADH zu der Komplex I-Probe lieferten kaum Signale, während bei der Variante kleine, aber deutlich sichtbare Signale nachweisbar waren. Die mit NADPH inkubierten Proben erzeugten beide das charakteristische EPR-Signal des DEPMPO-OOH Addukts, wobei das Signal mit der Komplex I-Probe deutlich intensiver war als mit der Variante E183HF. Die Signale wurden durch Zugabe von Piericidin A nicht, durch die Zugabe von SOD aber vollständig unterdrückt. Ohne Decyl-Ubichinon erzeugten beide Proben deutlich geringere Signale. 76 Ergebnisse A) a B) a b b c c d d e e f f Magnetische Flussdichte [mT] Magnetische Flussdichte [mT] Abbildung 4.9: EPR-spektroskopischer Nachweis der Superoxid-Produktion von Komplex I A) und der Variante E183HF B). Die Enzyme wurden in E. coli polare Lipide rekonstituiert, mit 100 mM DEPMPO und 100 µM Decyl-Ubichinon (a) sowie mit 1 mM NADH (b), mit 1 mM NADPH (c), mit 1 mM NADPH und 100 U/mL SOD (d), mit 1 mM NADPH ohne Decyl-Ubichinon (e) oder mit 1 mM NADPH und 20 µM Piericidin A (f) inkubiert. Die Spektren wurden bei Raumtemperatur aufgenommen. Die Mikrowellenfrequenz betrug 9.65 GHz, die Modulationsamplitude 0.1 mT, die Zeitkonstante 0.164 s und die Aufnahmegeschwindigkeit 5.4 mT/min (Morina et al., 2011). Um die ROS-Produktion von Komplex I und seinen Varianten zu quantifizieren, wurde ein von Herrn Dipl.-Chem. Marius Schulte (Schulte, 2014) für den E. coli Komplex I etablierter Amplex Red Assay verwendet. Amplex Red wird von der Meerrettich Peroxidase (HRP) in Anwesenheit von H2O2 zu Resorufin oxidiert. Die Bildung von Resorufin kann photometrisch bei 557 – 571 nm verfolgt werden. 5 µg Komplex I wurde in polaren Lipiden rekonstituiert und mit 60 µM Decyl-Ubichinon, 2 U/mL HRP sowie 10 µM Amplex Red inkubiert. Die Reaktion wurde durch Zugabe von 30 µM NAD(P)H gestartet. Die Abnahme der NADHKonzentration und die Zunahme der Resorufin-Konzentration wurden gleichzeitig aufgenommen. Somit konnte die prozentuale Zunahme von Resorufin und dadurch die Entstehung von H2O2 pro umgesetztem NADH berechnet werden (Tab. 4.5) Die Messungen wurden in Zusammenarbeit mit Herrn Dipl.-Chem. Marius Schulte durchgeführt. 77 Ergebnisse Tabelle 4.5: Prozentuale H2O2-Produktion pro umgesetztem NAD(P)H des Komplex I und der E183XFVarianten. Zur Bestimmung der H2O2-Produktion wurde das Amplex Red Assay verwendet. H2O2-Produktion pro umgesetztem NAD(P)H [%] Variante Komplex I + NADH + NADPH − NAD(P)H 0.8 ± 0.08 14.1 ± 0.4 0 F E183D 9.5 ± 0.9 24.4 ± 1.7 0 E183QF 2.1 ± 0.2 4.9 ± 0.3 0 F 2.2 ± 0.2 4.1 ± 0.2 0 F E183H 2.1 ± 0.2 3.6 ± 0.2 0 E183AF 1.6 ± 0.1 1.2 ± 0.1 0 E183GF 1.6 ± 0.1 1.3 ± 0.1 0 E183N Ohne die Zugabe von NAD(P)H konnte keine H2O2-Produktion beobachtet werden. Weniger als 1% der von NADH auf den Komplex I übertragenen Elektronen führen zur Produktion von ROS. Die ROS-Produktion pro umgesetztem NADPH ist 18-fach erhöht. Das ist wahrscheinlich durch die schlechte Bindung von NADPH bedingt, die zu einer sehr langsamen Umsetzung von 0.37 µmol·min−1·mg−1 führt. Alle Varianten zeigen mit NADH als Substrat mindestens eine doppelt so hohe ROS-Produktion; die der Variante E183DF ist sogar 12-fach erhöht. Die ROS-Produktion der Varianten, die durch gerichtete Evolution entstanden, ist nur in geringem Maß erhöht, beträgt aber dennoch das Doppelte der von Komplex I. Alle Varianten außer E183DF produzieren bei der Umsetzung von NADPH deutlich weniger ROS als der unveränderte Komplex I. Die ROS-Produktion der in dieser Arbeit erstmals beschriebenen Varianten beträgt mit NADPH als Substrat etwa ein Drittel von der des ursprünglichen Komplex I. Jedoch sind die Werte höher als bei der Umsetzung von NADH. Bei den von Auriol et al. (2011) beschriebenen Varianten E183AF und E183GF ist die ROS-Produktion bei der Umsetzung von NADPH im Vergleich zum ursprünglichen Komplex I um über 90% niedriger und ist sogar niedriger als der Wert mit NADH als Substrat. Die hohen ROS-Produktionsraten der Varianten mit NADPH als Substrat sind wahrscheinlich durch die hohen Km-Werte bedingt. Die Variante E183DF produziert aufgrund des hohen vmax von 3.8 µmol·min−1·mg−1 mit NADPH sehr viel mehr ROS als der ursprüngliche Komplex I mit NADH bei einem vmax von 2.9 µmol·min−1·mg−1. Das könnte durch die geringe Affinität zu NADPH (Km = 390 µM) erklärt werden. Da aber der Km der Variante E183NF mit 480 µM ungefähr in der gleichen Größenordnung liegt, die ROS-Produktion aber der von E183Q und HF ähnlich ist, kann die geringe Affinität zu NADPH nicht der einzige Grund für 78 Ergebnisse die erhöhte ROS-Produktion sein. Sehr interessant ist die Tatsache, dass alle Varianten trotz gleichem oder niedrigerem Km-Wert mit NADH eine deutlich erhöhte ROS-Produktion haben. Dies kann allein durch die kinetischen Daten nicht erklärt werden und erfordert daher weitergehende strukturelle Untersuchungen. 4.2 Strukturelle Untersuchung der Substratdiskriminierung In dieser Arbeit wurde mit enzymkinetischen Methoden gezeigt, dass das konservierte Glutamat 183 auf NuoF des E. coli Komplex I eine Schlüsselposition für die Unterscheidung zwischen NADH und NADPH als Substrat darstellt. Es wurden sechs Komplex I-Varianten dargestellt, die alle deutlich erhöhte Aktivitäten mit NADPH und teilweise auch mit NADH aufwiesen. Die Umsetzung von NADH war bei allen Varianten mit einer erhöhten ROSProduktion gekoppelt. Um die strukturellen Gründe für die veränderten Umsatzraten und die erhöhte ROS-Produktion der Varianten zu untersuchen, wurden die homologen Varianten des A. aeolicus Subkomplexes NuoEFHis erzeugt, produziert und kristallisiert. Sabrina Burschel wurde im Rahmen ihrer Bachlorarbeit und Sascha Jurkovic im Rahmen seiner Diplomarbeit zu Teilen der Arbeiten in diesem Kapitel angeleitet (Burschel, 2011; Jurkovic, 2013). Zunächst sollte die heterologe Expression der A. aeolicus Gene nuoEF in E. coli optimiert werden. In vorangegangen Arbeiten wurde für die Produktion von NuoEFHis der Expressionsvektor pETBlue-1nuoEFhisG verwendet , 200 et al., 2008). Bei der Präparation wurden die Untereinheiten NuoE und NuoF als löslicher Komplex NuoEF erhalten. NuoG lag in Form von Einschlusskörpern vor. Um den durch die Überproduktion möglicherweise bedingten Zellstress zu minimieren, sollte der Vektor pETBlue-1nuoEFhis ohne das Gen nuoG kloniert werden. 4.2.1 Optimierung der heterologen Expression von A. aeolicus nuoEF in E. coli Um die Expression der A. aeolicus Gene nuoEF zu optimieren, wurde der Vektor pETBlue-1nuoEFhis hergestellt. Dafür wurden die Gene nuoEFhis mit den Oligonukleotiden axa_nuoE_fwd und axa_nuoFhis_rev (Tab. 3.6) von dem Vektor pETBlue-1nuoEFhisG mittels PCR amplifiziert. Die Anlagerungstemperatur betrug 60 °C. Das Amplikon und der Vektor 79 Ergebnisse pETBlue-1 wurden mit BlpI und EcoRI geschnitten und ligiert. Die korrekte Insertion wurde durch Sequenzierung der Übergänge von Vektor zu Insert bestätigt. Der E. coli Stamm Rosetta(DE3) wurde mit dem Vektor pETBlue-1nuoEFhis transformiert und auf LB-AgarPlatten mit Chloramphenicol und Ampicillin ausgestrichen. Der Stamm wurde im 10 LMaßstab in NuoEFaxa-Medium angezogen (Abb. 4.10 A) und das Protein nach etabliertem Protokoll isoliert. Die erste NuoEFHis-Präparation aus 28 g Zellen dieses Stamms ergab 1 mg Protein. Dies entspricht 0.04 mg Protein pro g Zellen. Diese Ausbeute war deutlich geringer als die in vorangegangen Arbeiten beschrieben von 0.42 mg pro g Zellen (Ko et al., 2008). Bei einer weiteren Zellzucht aus derselben Glycerindauerkultur wurden sehr helle Zellen erhalten. Nach dem ersten chromatographischen Reinigungsschritt an Ni2+-IDA wurde in den entsprechenden Fraktionen nur eine farblose Lösung erhalten (Daten nicht gezeigt). Als Konsequenz daraus wurden Zellen des E. coli Stamms Rosetta(DE3) erneut mit dem Vektor pETBlue-1nuoEFhis transformiert. Es wurden zehn einzelne Kolonien im 4 mL-Maßstab angezogen. Je 50 mL LB-Medium wurden mit Chloramphenicol und Ampicillin versetzt und 1:50 (v/v) aus den 4 mL-Vorkulturen angeimpft. Die Expression der Gene wurde durch die Zugabe von 1 mM IPTG bei OD600 0.7 induziert. Die Zellen wurden für weitere 3 h angezogen und durch Zentrifugation geerntet. Nur vier der zehn Zellsedimente wiesen die für die Überproduktion von NuoEFHis charakteristische Braunfärbung auf. Der dunkelste Klon wurde für die Zellzucht ausgewählt und wieder im 10 L-Maßstab in NuoEFaxa-Medium kultiviert. Abbildung 4.10 B gibt die Wachstumskurve wieder. A) B) Abbildung 4.10: Wachstumskurven des Stamms Rosetta(DE3)/pETBlue-1nuoEFhis. A) zeigt die Wachstumskurve ohne vorangegangenen Expressionstest. B) zeigt die Wachstumskurve des Klons mit der nach optischen Kriterien, höchsten Expressionsleistung. Der Zeitpunkt der Induktion mit 1 mM IPTG ist mit einem Pfeil markiert. Es wurde im 10 L-Maßstab in NuoEFaxa-Medium angezogen. 80 Ergebnisse Der Klon, der durch Expressionstests ausgewählt worden war, zeigte nach der Induktion ein deutlich langsameres Wachstum und eine geringere Enddichte (OD600 = 2.7) als der nicht getestete Klon (OD600 = 6.2). Nach Induktion wurde kein exponentielles Wachstum des ausgewählten Klons beobachtet, was vermutlich auf den durch die erhöhte Expression und Produktion bedingten Stress zurückzuführen ist. Aus 10 L NuoEFaxa-Medium wurden 22 g Zellen erhalten, was einem Drittel der ersten Zellzucht entspricht. Da die Zellen eine sehr dunkle, braune Färbung aufwiesen, wurden nur 5 g für die Proteinpräparation verwendet. Die Zellen wurden in zwei Durchläufen mit Hilfe einer Druckentspannungsanlage aufgeschlossen. Das Cytosol wurde durch Ultrazentrifugation von unlöslichen Bestandteilen befreit. Das Protein wurde durch vier chromatographische Schritte gereinigt. Zuerst wurde das Cytosol auf eine Ni2+-Affinitäts-Chromatographiesäule aufgetragen. Gebundene Proteine wurden mit einem linearen Gradienten von 116 – 500 mM Imidazol eluiert. Fraktionen mit einer NADH/Ferricyanid Oxidoreduktaseaktivität von über 20 U/mL wurden vereinigt, durch Ultrafiltration auf Chromatographiesäule unter 500 µL (Superdex eingeengt 200 16/60) und auf eine aufgetragen. GrößenausschlussEs wurden zwei Absorptionsmaxima erhalten, wobei nur das erste bei etwa 72 mL Elutionsvolumen Aktivität aufwies. Der zweite Gipfel bei etwa 80 mL Elutionsvolumen wurde verworfen. Die Fraktionen des ersten Gipfels mit NADH/Ferricyanid Oxidoreduktaseaktivitäten über 40 U/mL wurden vereinigt und auf eine Ionenaustausch-Chromatographiesäule (Source 15Q) aufgetragen. Gebundene Proteine wurden mit einem linearen Gradienten von 50 – 350 mM NaCl eluiert. Erneut wurden die Fraktionen mit signifikanter Aktivität (≥ 30 U/mL) vereinigt, durch Ultrafiltration auf unter 500 µL eingeengt und auf eine GrößenausschlussChromatographiesäule aufgetragen. Es wurde nur ein Gipfel bei 72 mL Elutionsvolumen erhalten. Abbildung 4.11 gibt die Elutionsprofile der chromatographischen Reinigungsschritte wieder. Die Reinheit und die Zusammensetzung der Proben der einzelnen Reinigungsschritte wurde durch SDS-PAGE analysiert (Abb. 4.12). Das Polyacrylamidgel zeigt in jeder Spur NuoFHis bei etwa 50 kDa und NuoE bei etwa 16 kDa. Die Präparation weist nach Färbung keine Verunreinigungen auf. Die Präparation wurde unter den Standardbedingungen zur Kristallisation gebracht (Daten nicht gezeigt). Aus 5 g Zellen des Stamms Rosetta(DE3)/pETBlue-1nuoEFhis wurden 7.7 mg NuoEFHis isoliert, das entspricht 1.5 mg pro g Zellen. Durch Verwendung des Plasmids pETBlue-1nuoEFhis und eine Vorauswahl des Klons über Expressionstests wurde die Produktion von NuoEFHis mehr als verdreifacht. 81 Ergebnisse A) B) C) D) Abbildung 4.11: Präparation von NuoEFHis aus dem Stamm Rosetta(DE3)/pETBlue-1nuoEFhis. Gezeigt ist das Elutionsprofil der Affinitäts-Chromatographie an Ni2+-IDA A), das der ersten GrößenausschlussChromatographie an Superdex 200 B) das der Ionenaustausch-Chromatographie C) und das der zweiten Größenausschluss-Chromatographie an Superdex 200. Die Absorbanz bei 280 nm ist in Blau, die Imidazol- bzw. die NaCl-Konzentration in Grün und die NADH/Ferricyanid Oxidoreduktaseaktivität in Rot dargestellt. 1 2 3 4 5 M app. Mr [kDa] NuoFHis → ← 66.2 ← 45.0 ← 35.0 ← 25.0 NuoE → ← 18.4 ← 14.4 Abbildung 4.12: Polyacrylamidgel der Präparation von A. aeolicus NuoEFHis. Spur M zeigt die Banden des Proteinstandards Unstained Protein Molecular Weight Marker (Thermo Scientific), Spur 1 zeigt die Proteinzusammensetzung des Cytosols, Spur 2 die des Eluats der Affinitäts-Chromatographie, Spur 3 die der ersten Größenausschluss-Chromatographie, Spur 4 die der Ionenaustausch-Chromatographie und Spur 5 die des gereinigten Proteins. Es wurden jeweils 10 µg Protein bzw. 30 µg der cytosolischen Fraktion auf das Gel aufgetragen. Die Banden von NuoFHis und NuoE wurden zugeordnet. 82 Ergebnisse 4.2.2 Darstellung der E185F-Varianten des A. aeolicus Subkomplexes NuoEF Die Mutanten pETBlue-1nuoEFhis E185A/D/G/H/N und Q wurden durch ortsgerichtete Mutagenese mit den jeweiligen Oligonukleotid-Paaren (Tab. 3.4) generiert. Der Vektor pETBlue-1nuoEFhis diente als Vorlage. Es wurden je 40 – 50 ng Vorlage-DNA für die PCR verwendet. Die Anlagerungstemperatur betrug zwischen 50 und 60 °C. Die Ansätze wurden gereinigt, mit DpnI verdaut und erneut gereinigt. Zellen des E. coli Stamms DH5α wurden mit 1 µL der gereinigten PCR-Ansätze transformiert und der gesamte Ansatz auf LB-Agar-Platten mit Ampicillin ausgestrichen. Es wurden einzelne Klone angezogen, die Plasmide der Klone isoliert und mittels Restriktion analysiert. Die erhaltenen DNA-Fragmente wurden durch Agarose-Gelelektrophorese aufgetrennt und fotografisch dokumentiert (Abb. 4.13). Der Erfolg der ortsgerichteten Mutagenese wurde durch Sequenzierung bestätigt. Länge [bp] 1 2 19´329→ 7743→ 3472→ 2690→ 1882→ 1489→ 925→ Abbildung 4.13: Restriktionsanalyse des Vektors pETBlue-1nuoEFhis E185H mit VspI und NdeI. Die Restriktionsanalysen der anderen Mutanten sahen identisch aus und werden daher nicht gezeigt. Spur 1 zeigt das z Bandenmuster des λ-DNA/Eco130I(StyI) Markers16 und Spur 2 das erwartete Bandenmuster mit Längen von 2980 und 1742 bp. Zellen des Stamms Rosetta(DE3) wurden mit den mutierten Plasmiden transformiert und auf LB-Agar-Platten mit Chloramphenicol und Ampicillin ausgestrichen. Es wurden je zehn Klone angezogen und für Expressionstests verwendet. Die Ausbeute an positiven Klonen lag zwischen 30 und 80%. Die Zellen wurden im 10 L-Maßstab in NuoEFaxa-Medium angezogen (Abb. 4.14). 83 Ergebnisse A) B) Abbildung 4.14: Wachstumskurven der Stämme Rosetta(DE3)/pETBlue-1nuoEFhis E185X. A) zeigt die Wachstumskurven der Stämme E. coli Rosetta(DE3)/pETBlue-1nuoEFhis E185A (schwarz), E183D (rot), E183N (grün) und B) die der Stämme Rosetta(DE3)/pETBlue-1nuoEFhis E185G (schwarz), E185H (rot) und E183Q (grün). Die Pfeile markieren den Zeitpunkt der Induktion mit 0.7–1.0 mM IPTG. Es wurde im 10 L-Maßstab in NuoEFaxa-Medium angezogen. Aus den Zellzuchten wurden zwischen 16 und 30 g Zellen erhalten. Das unterschiedliche Zellwachstum war vermutlich durch die unterschiedliche Expressionsleistung der einzelnen Klone bedingt. Je dunkler die geernteten Zellen waren, desto geringer war die maximal erreichte Zelldichte. Die Varianten wurden nach dem für den NuoEFHis-Subkomplex etablierten Protokoll isoliert. Da die Präparationen sehr ähnlich waren, wird die der Variante E185AF exemplarisch gezeigt. Für die Präparation wurden 5 g Zellen des Stamms Rosetta(DE3)/pETblue-1nuoEFhis E185A aufgeschlossen und das Cytosol durch Ultrazentrifugation von Membranen und anderen unlöslichen Bestandteilen befreit. Die Variante wurde durch Affinitäts-Chromatographie, Größenausschluss-Chromatographie, Anionenaustausch-Chromatographie und erneuter Größenausschluss-Chromatographie bis zur elektrophoretischen Homogenität gereinigt (Abb. 4.15). Die Ausbeuten der einzelnen Präparationen sind in Tabelle 4.6 zusammengefasst. 84 Ergebnisse A) B) C) D) Abbildung 4.15: Präparation der Variante E185AF aus dem Stamm Rosetta(DE3)/pETBlue-1nuoEFhis E185A. A) zeigt das Elutionsprofil der Affinitäts-Chromatographie, B) das der ersten GrößenausschlussChromatographie, C) das der Anionenaustausch-Chromatographie und D) das der zweiten GrößenausschlussChromatographie. Die Absorbanz bei 280 nm ist in Blau, die Imidazol- bzw. die NaCl-Konzentration in Grün und die NADH/Ferricyanid Oxidoreduktaseaktivität in Rot dargestellt. Tabelle 4.6: Ausbeuten der Präparationen der Varianten E185XF. Für eine bessere Vergleichbarkeit wurden die NADH/Ferricyanid Oxidoreduktaseaktivitäten der Varianten auf die des ursprünglichen NuoEF His normiert, wobei diese gleich 100% gesetzt wurde. Variante Eingesetzte Zellen [g] Ausbeute Protein [mg] NADH/Ferricyanid Oxidoreduktaseaktivität [µmol·min−1·mg−1] Normierte Aktivität [%] NuoEFHis 5.0 7.7 48.4 100 F 5.0 6.8 26.7 55 F 5.1 9.1 26.0 54 F 5.4 10.3 18.3 38 F 10.0 12.3 22.0 45 F 5.2 10.8 20.7 43 F 5.0 8.2 17.5 36 E185A E185D E185G E185H E185N E185Q 85 Ergebnisse Die Ausbeuten der Präparationen lagen zwischen 1.2 und 2.1 mg Protein pro g Zellen. Im Gegensatz zu den homologen Varianten des E. coli Komplex I zeigen die NuoEFHis-Varianten eine deutlich verringerte NADH/Ferricyanid Oxidoreduktaseaktivität. Die Reinheit und Zusammensetzung der Präparationen wurde durch SDS-PAGE nach Laemmli überprüft (Abb. 4.16). 1 2 3 4 NuoFHis → 5 6 M app. Mr [kDa] ← 66.2 ← 45.0 ← 35.0 ← 25.0 ← 18.4 NuoE → ← 14.4 Abbildung 4.16: Polyacrylamidgel der Präparationen der NuoEFHis Varianten E185XF. Spur M zeigt die Banden des Proteinstandards Unstained Protein Molecular Weight Marker (Thermo Scientific), Spur 1 das Bandenmuster der Präparation der Variante E185AF, Spur 2 das von E185GF, Spur 3 das von E185NF, Spur 4 das von E185DF, Spur 5 das von E185HF und Spur 6 das von E185QF. Es wurden 5 – 10 µg Protein aufgetragen. Die Banden von NuoFHis und NuoE wurden zugeordnet. Das Polyacrylamidgel zeigt die korrekte Zusammensetzung der Präparationen ohne nennenswerte Kontaminationen. In jeder Präparation wurde NuoFHis bei etwa 50 kDa und NuoE bei etwa 16 kDa detektiert. 4.2.3 Kristallisation der Varianten E185XF Die Präparationen sollten unter den für den Subkomplex NuoEFHis etablierten Citratbedingungen et al., 2008) kristallisiert werden (Tab. 3.20). Für die Variante E185HF wurde die hanging drop, für die restlichen die sitting drop Methode verwendet. Nach zwei bis drei Tagen wurden in den Bedingungen 1 bis 5 klare, dunkelbraune Kristalle der Varianten E185D/H/N und QF beobachtet. Nach etwa neun Tagen wurde kein weiteres Kristallwachstum beobachtet. Die für die Röntgendiffraktometrie verwendeten Kristalle waren etwa 300 – 1000 µm groß und wiesen gleichmäßiges Wachstum mit scharfen Kanten auf (Abb. 4.17). 86 Ergebnisse A) B) C) D) Abbildung 4.17: Kristalle der Varianten E185D/H/N und QF, die in den Citratbedingungen erhalten wurden. A) zeigt einen Kristall der Variante E185DF in 0.1 M Tris/HCl (pH 6.9) und 0.7 M Na3Citrat, B) einen der Variante E185HF in 0.1 M Tris/HCl (pH 7.0) und 0.9 M Na3Citrat, C) einen der Variante E185QF in 0.1 M Tris/HCl (pH 7.1) und 0.6 M Na3Citrat und D) einen der Variante E185NF in Tris/HCl (pH 6.9) und 0.8 M Na3Citrat. Zudem enthielt jede Kristallisationsbedingung 0.2 M NaCl. Zahlreiche Proteinkristalle wurden auf ihr Beugungsverhalten getestet. Die Kristalle der Variante E185NF streuten maximal bis zu einer Auflösungsgrenze von 5 Å. Kristalle der Variante E185QF wiesen Beugung bis zu einer Auflösungsgrenze von etwa 2.5 Å auf. Allerdings konnte dieser Datensatz aufgrund einer mangelhaften Vollständigkeit der Reflexe nicht ausgewertet werden. Von Kristallen der Varianten E185D und HF wurden vollständige auswertbare Datensätze mit Auflösungsgrenzen von 2.4 Å bzw. 2.2 Å aufgenommen. Von den Präparationen der Variante E185AF konnten mit den Citratbedingungen nur Sphärolithe erhalten werden. Diese wurden verwendet, um Keimkristalle zu erzeugen. Die Kristallisationsversuche wurden mit Keimkristallen wiederholt, lieferten aber nur sehr kleine, nicht scharfkantige Kristalle in 0.1 M Tris/HCl (pH 6.8 – 7.0), 0.2 M NaCl und 0.9 M Na3Citrat (Daten nicht gezeigt). Die Variante E185GF kristallisierte nicht unter den Citratbedingungen. Um Kristallisationsbedingungen für die Variante E185AF zu verbessern und um Initialbedingungen für die Variante E185GF zu finden, wurde der Hampton Index HT screen verwendet. Für die Variante E185GF wurden keine geeigneten Kristallisationsbedingungen gefunden. In 1.5 M D/L-Äpfelsäure (pH 7.0) und 20% (w/v) PEG 3350 wurden nur Sphärolithe erhalten (Abb. 4.18 A). Für die Variante E185AF wurden in verschiedenen Puffern und bei verschiedenen pH-Werten mit (NH4)2SO4 als Präzipitant große, braune und scharfkantige Kristalle erhalten (Abb. 4.18 B). Kristallisationsbedingungen mit 0.49 M NaH2PO4 und 0.91 M K2HPO4 (pH 6.9) lieferten ebenfalls große, scharfkantige Kristalle. Von der Präparation der Variante E185GF wurden fine-screens um die Bedingung, in denen Sphärolithe erhalten wurden, angesetzt (Anhang 7.2; Tab. 7.5). Für die 87 Ergebnisse Kristallisationsversuche wurden Keimkristalle des Subkomplexes NuoEFHis verwendet. Unter keiner Bedingung konnten streufähige Kristalle erhalten werden. A) B) Abbildung 4.18: Kristallisationsversuche im Hampton Index screen. A) zeigt Sphärolithe der Variante E185GF in 1.5 M D/L-Äpfelsäure (pH 7.0) und 20% (w/v) PEG 3350. B) zeigt Kristalle der Variante E185AF in 0.1 M Tris/HCl (pH 8.5) mit (NH4)2SO4. Für die Variante E185AF wurde fine-screens für die (NH4)2SO4- und Phosphat-Bedingungen angesetzt. Die fine-screens um die Phosphat-Bedingungen (Anhang 7.2; Tab. 7.6) lieferten mit Keimkristallen zwar scharfkantige Kristalle (Abb. 4.19 A), diese zeigten aber kein Streuvermögen. Die (NH4)2SO4-Bedingungen (Tab. 4.7) lieferten mit Keimkristallen in einer 1:10´000 Verdünnung streufähige Kristalle in den Bedingungen 1-3 (Abb. 4.19 B und C). Tabelle 4.7: (NH4)2SO4-Bedingungen zur Kristallisation der Variante E185AF. 1 2 A 0.1 M MES/NaOH pH 6.0, 1.5 M (NH4)2SO4 0.1 M BisTris/HCl pH 6.5, 1.5 M (NH4)2SO4 0.1 M HEPES/HCl pH 7.0, 1.5 M (NH4)2SO4 0.1 M HEPES/HCl pH 7.5, 1.5 M (NH4)2SO4 0.1 M Tris/HCl 0.1 M Tris/HCl pH 8.0, pH 8.5, 1.5 M 1.5 M (NH4)2SO4 (NH4)2SO4 B 0.1 M MES/NaOH pH 6.0, 1.7 M (NH4)2SO4 0.1 M Bis-Tris/ HCl pH 6.5, 1.7 M (NH4)2SO4 0.1 M HEPES/HCl pH 7.0, 1.7 M (NH4)2SO4 0.1 M HEPES/HCl pH 7.5, 1.7 M (NH4)2SO4 0.1 M Tris/HCl 0.1 M Tris/HCl pH 8.0, pH 8.5, 1.7 M 1.7 M (NH4)2SO4 (NH4)2SO4 C 0.1 M MES/NaOH pH 6.0, 2.0 M (NH4)2SO4 0.1 M Bis-Tris/ HCl pH 6.5, 2.0 M (NH4)2SO4 0.1 M HEPES/HCl pH 7.0, 2.0 M (NH4)2SO4 0.1 M HEPES/HCl pH 7.5, 2.0 M (NH4)2SO4 0.1 M Tris/HCl 0.1 M Tris/HCl pH 8.0, pH 8.5, 2.0 M 2.0 M (NH4)2SO4 (NH4)2SO4 D 0.1 M MES/NaOH pH 6.0, 2.1 M (NH4)2SO4 0.1 M Bis-Tris/ HCl pH 6.5, 2.1 M (NH4)2SO4 0.1 M HEPES/HCl pH 7.0, 2.1 M (NH4)2SO4 0.1 M HEPES/HCl pH 7.5, 2.1 M (NH4)2SO4 0.1 M Tris/HCl 0.1 M Tris/HCl pH 8.0, pH 8.5, 2.1 M 2.1 M (NH4)2SO4 (NH4)2SO4 88 3 4 5 6 Ergebnisse Von den Kristallen der Variante E185AF wurde ein Datensatz mit einer Auflösungsgrenze von 1.8 Å am hauseigenen Diffraktometer aufgenommen. Allerdings konnten die Datensätze aufgrund fehlender Reflexe nicht ausgewertet werden. A) B) C) Abbildung 4.19: Kristalle der Variante E185AF. A) zeigt Kristalle in 0.3 M NaH2PO4 und 0.7 M K2HPO4, B) in 0.1 M Bis-Tris/HCl (pH 6.5) und 1.7 M (NH4)2SO4 und C) in 0.1 M HEPES/HCl (pH 7.0) und 1.5 M (NH4)2SO4. 4.2.4 Strukturen der Varianten E185DF und E185HF In Zusammenarbeit mit Prof. Oliver Einsle, Institut für Biochemie (Albert-LudwigsUniversität, Freiburg), wurde ein Datensatz von Kristallen der Variante E185HF am hauseigenen Diffraktometer aufgenommen. Als Kryoprotektant diente eine 25%ige Saccharose-Lösung in Tris/HCl-Puffer (pH 7.0). Der Kristall wurde am Diffraktometer langsam im Stickstoffstrom eingefroren und es wurde ein Datensatz mit einer Auflösungsgrenze von 2.20 Å aufgenommen. Die Struktur wurde von Prof. Einsle über das molecular replacement Verfahren mit Hilfe der Struktur des Subkomplexes NuoEFHis gelöst. Die Struktur der Variante E185DF wurde von Sascha Jurkovic im Rahmen seiner Diplomarbeit gelöst (Jurkovic, 2013). Beide Varianten kristallisierten wie NuoEFHis in der Raumgruppe P 21 21 21. Die Auflösungsgrenze der Kristalle der Variante E185DF betrug 2.40 Å. Die Quartär- und Tertiärstruktur der Varianten unterscheidet sich nicht von der des NuoEFHis-Subkomplexes (Abb. 4.20). Die kristallographischen Statistiken der von den Varianten E185D und HF erhaltenen Datensätze sind in Tabelle 7.7 (Anhang 7.3) zusammengefasst. 89 Ergebnisse A) B) C) Abbildung 4.20: Überlagerung der Strukturen der Varianten E185HF und E185DF mit NuoEFHis. A) zeigt die Struktur von E185HF (blau) bei 2.20 Å und B) die von E185DF (cyan) bei 2.40 Å. Die für die Bindung von NADH wichtigen Seitenketten sind gezeigt. C) zeigt die Überlagerung der Strukturen von NuoEFHis (rot) mit den Varianten. Die Wurzel aus der mittleren quadratischen Abweichung (root mean square deviation; r.m.s.d.) der Cα-Positionen bezogen auf NuoEFHis beträgt für E185HF 0.250 Å (416 Atom-Paare) und für E185DF 0.277 Å (417 Atom-Paare). Die Strukturen der Varianten unterscheiden sich kaum voneinander und von der des NuoEFHis. Obwohl beide Varianten im oxidierten Zustand kristallisiert wurden, weisen sie Merkmale der Struktur des unveränderten Subkomplexes im reduzierten als auch im oxidierten Zustand auf. Die Carbonyl-Gruppe des Peptidrückgrats von Glu95F ist in beiden Strukturen, wie in der Struktur des NuoEFHis im reduzierten Zustand orientiert und zeigt vom FMN weg, um mehr Platz für die Anbindung des NADH zu schaffen. Der B-Faktor des Peptid-Sauerstoffs von Glu95F beträgt für die Variante E185DF 40.3 Å2 (durchschnittlicher 90 Ergebnisse B-Faktor: 52.3 Å2) und für die Variante E185HF 30.5 Å2 (durchschnittlicher B-Faktor: 28.9 Å2). Somit kann die Orientierung der Carbonyl-Gruppe zwar genau angegeben werden, aber die hohen B-Faktoren deuten auf eine Flexibilität dieser Position hin. In beiden Varianten bildet die Seitenkette des Glu97F keine H-Brücke mit der Seitenkette des Tyr180F aus (Abb. 4.21 A und B). Dies stimmt mit der Struktur des NuoEFHis im oxidierten Zustand überein. Das Peptidrückgrat von Asp185F und His185F ist genau wie im NuoEFHis im oxidierten und reduzierten Zustand über zwei H-Brücken, eine vom Peptid-Stickstoff zum FMN und eine von der Carbonyl-Gruppe zum Peptid-Stickstoff des Ile189F, fest verankert (Abb. 4.21 C und D). A) B) E95 E97 Y180 E97 D94 Y180 D185 E95 D94 H185 D) C) N3 N3 E184 D185 I392 E184 H185 I392 I189 I189 K202 R200 K202 Abbildung 4.21: Strukturmerkmale der NuoEFHis-Varianten E185DF und E185HF. H-Brücken sind mit gestrichelten Linien dargestellt. A) zeigt das aktive Zentrum der Variante E185DF und B) das der Variante E185HF. Gezeigt ist die Orientierung des Peptidrückgrats von Glu95F in beiden Strukturen. Das strukturell konservierte H2O-Molekül (dunkelblau) ist in A) und B) wie in NuoEFHis im reduzierten Zustand koordiniert. C) zeigt die Orientierung der Seitenkette des Lys202F und Wechselwirkungen mit anderen Resten in der Variante E185DF und D) in der Variante E185HF. Die mit der Carbonyl-Gruppe von Asp185F bzw. His185F und dem Peptid-Stickstoff von Ile189F gebildeten H-Brücken sind angedeutet. 91 Ergebnisse Die Seitenkette des Lys202F in den Varianten E185DF und E185HF ist wie in dem reduzierten NuoEFHis verdreht (Abb. 4.21 C und D). In dem oxidierten NuoEFHis bilden die Seitenketten des Glu185F und des Lys202F eine H-Brücke aus (vgl. Abb. 2.12). Die um eine MethylenEinheit verkürzte Seitenkette des Asp185F kann keine H-Brücke mehr mit dem Lys202F ausbilden, daher verdreht sich dessen Seitenkette und bildet eine H-Brücke mit der CarbonylGruppe des Ile392F aus (Abb. 4.21 C). Allerdings beträgt der Mittelwert der B-Faktoren der einzelnen Seitenketten-Atome des Lys202F 45.7 Å2, was auf eine flexible Position hindeutet. Dennoch ist in der Elektronendichtekarte bei einer Konturstufe von 1 σ die Seitenkette von Lys202F teilweise und bei 2 σ vollständig definiert (Abb. 4.22 A). In der Variante E185HF stoßen sich die positiven Ladungen des His185F und Lys202F ab und bewirken dadurch das Verdrehen der Seitenkette des Lys202F (Abb. 4.21 D). Hier beträgt der Mittelwert der BFaktoren der einzelnen Seitenketten-Atome 33.4 Å2, was eine genauere Definition der Lage der Seitenkette erlaubt, die aber auch hier nicht vollständig fixiert ist. Abbildung 4.22 B zeigt die Elektronendichtekarte an dieser Position. A) B) R200 I392 I392 E184 E184 K202 K202 Abbildung 4.22: Ausschnitte der Elektronendichtekarten am Lys202 F. A) zeigt die 2 Fo−Fc Elektronendichte an den Seitenkette Lys202F, Glu184F und Ile392F der Variante E185DF bei einer Konturstufe von 1 σ (blau) und 2 σ (grün). B) zeigt die 2 Fo−Fc Elektronendichte an den gleichen Seitenketten und zusätzlich am Rückgrat von Arg200F bei einer Konturstufe von 1 σ (blau) und 2 σ (grün) der Variante E185HF. Durch die eingeführten Mutationen vergrößert sich die NADH-Bindetasche in dem Teil in dem die Adenosin-Ribose gebunden wird. Dies erklärt die erhöhte Affinität der E. coli Komplex I-Varianten zu NADPH. Das redox-induzierte Umklappen des Rückgrats von Glu95F wird als „Auswurf-Mechanismus‟ für das verbrauchte Substrat diskutiert. Die Orientierung der Carbonyl-Gruppe des Glu95F von der Bindetasche weg könnte in beiden Varianten auf eine nicht effiziente Freisetzung des NAD+ hindeuten, was wiederum im 92 Ergebnisse Kontrast zu den erhöhten Aktivitäten der E. coli Komplex I-Varianten steht. Zwar zeigen die B-Faktoren dieser Positionen, dass deren Konformation nicht rigide ist, aber die gut aufgelöste Elektronendichte an dieser Position verdeutlicht, dass die Varianten bevorzugt die Konformation des reduzierten NuoEFHis einnehmen. Die Strukturen der NuoEFHis-Varianten liefern keine Erklärung für die erhöhte Aktivität und ROS-Produktion der E. coli Komplex IVarianten. Da sonst keine konformativen Änderungen im Vergleich zu NuoEFHis beobachtet wurden, liegt die Erklärung vermutlich in der Substrat-Anbindung. 4.2.5 Struktur der Variante E185DF mit gebundenem NAD+ Um die Anbindung des Nukleotids strukturell zu charakterisieren, wurden die NuoEFHisVarianten E185XF mit NAD+ kokristallisiert. Die NAD+-Konzentration im Kristallisationstropfen betrug 10 mM. Die Varianten E185A/D/H und QF wurden mit NAD+ unter den gleichen Bedingungen kristallisiert wie ohne Nukleotid. Zur Kokristallisation der Variante E185NF mit NAD+ mussten die Standard-Citratbedingungen um die Bedingungen B2-4 und C2-4 (Tab. 3.20) verfeinert werden, da ansonsten nur Sphärolithe und verformte Kristalle ohne scharfe Kanten erhalten wurden. Es konnte nur von der Variante E185DF ein Datensatz aufgenommen werden, in dem die Elektronendichte des Nukleotids aufgelöst war. Die Struktur wurde von Sascha Jurkovic im Rahmen seiner Diplomarbeit gelöst (Jurkovic, 2013). Die Struktur der Variante ohne Nukleotid wurde für das molecular replacement verwendet. Die Auflösungsgrenze des Kristalls betrug 1.8 Å. Die Variante mit gebundenem NAD+ kristallisierte wie NuoEFHis in der Raumgruppe P 21 21 21. Die Statistiken des Datensatzes sind in der Tabelle 7.8 (Anhang 7.3) zusammengefasst. Aufgrund der geringen Affinität des Enzyms zu NAD+ sind die Bindestellen im Kristall nicht vollständig besetzt, wodurch elektronenarme Bereiche durch das allgemeine Signalrauschen überlagert wurden. Um die volle Elektronendichte des NAD+ zu erhalten, wurde mit dem Programm Coot (Emsley et al., 2010) die Elektronendichte des Proteins maskiert, so dass die Intensität der Elektronendichte des NAD+ herausgearbeitet wurde und das komplette Molekül dargestellt werden konnte (Abb. 4.23). 93 Ergebnisse Abbildung 4.23: Elektronendichte des NAD+ in der Variante E185DF. Die herausgearbeitete 2 Fo−Fc Elektronendichte des NAD+ ist bei einer Konturstufe von 2 σ gezeig . Am O2' der Adenosin-Ribose wurde in der Elektronendichtekarte eine für eine HydroxylGruppe zu ausgedehnte Elektronendichte beobachtet. Diese wurde der Sulfon-Gruppe eines MOPS-Moleküls zugeordnet, das als Puffer für die Präparation verwendet wurde (Abb. 4.24). Die Sulfon-Gruppe bindet an dieselbe Position, an welche die Phosphat-Gruppe des NADPH binden würde. Dies deutet darauf hin, dass diese Position für die Anbindung von negativgeladenen Molekülen oder Resten geeignet ist. Abbildung 4.24: Ausschnitt aus der Elektronendichtekarte am O2' des gebundenen NAD + in der Variante E185DF. Die für die Hydroxyl-Gruppe zu große 2 Fo−Fc Elektronendichte (Konturstufe 1.5 σ) am O2' der Adenosin-Ribose und das zugeordnete MOPS-Molekül (grün) sind gezeigt. Die Variante E185DF mit gebundenem NAD+ zeigt im Vergleich zu NuoEFHis ebenfalls keine Unterschiede in der Quartär- und Tertiärstruktur. Wie die Variante ohne Nukleotid weist die Nukleotid-gebundene Form dieselben Merkmale der Strukturen des reduzierten und 94 Ergebnisse oxidierten NuoEFHis auf. Beim Vergleich der drei Strukturen mit gebundenen Nukleotiden fallen interessante Unterschiede und Gemeinsamkeiten auf (Abb. 4.25). In der Variante E185DF wird der Adenin-Rest des NAD+ wie in den Strukturen des NuoEFHis über π-Wechselwirkungen mit den Seitenketten von Phe71F und Tyr204F stabilisiert (Abb. 4.25 A). Die Seitenkette des Phe79F trägt nicht zu der Stabilisierung bei. A) D185 S96 B) G68 F71 K76 E95 F79 E97 Y204 E185 Y180 E184 K202 C) Abbildung 4.25: Struktur der Variante E185DF mit gebundenem NAD+ A) und des oxidierten NuoEFHis mit gebundenem NAD+ B). C) zeigt die Struktur des reduzierten NuoEFHis mit gebundenem NADH. Die zur Stabilisierung der gebundenen Nukleotide beitragenden H-Brücken sind durch gestrichelte Linien gezeigt. Die Längen der H-Brücken sind in Ångström angegeben. Beteiligte H2O-Moleküle sind blau markiert. Das FMN ist gelb dargestellt. 95 Ergebnisse Da die Carbonyl-Gruppe der Peptidbindung von Glu95F in der Variante E185DF aus der Bindetasche herausgeklappt ist, liegt das NAD+ tiefer in der Bindetasche als in NuoEFHis. Seine Bindung in der Variante ähnelt daher eher der Bindung des NADH (Abb. 4.26 A). Wie das NADH im reduzierten NuoEFHis, bildet das NAD+ mit der Amid-Gruppe des Nikotinamidrings je eine H-Brücke zu den Amid-Stickstoffen des Glu97F und Ser96F aus. Zudem bilden die Hydroxyl-Gruppen der Nikotinamid-Ribose keine H-Brücken zur Carbonyl-Gruppe des Gly68F in der glycinreichen-Schleife (Abb. 4.25). In allen drei Strukturen bildet das Pyrophosphat des Nukleotids zwei H-Brücken mit den HydroxylGruppen des FMNs aus. Durch die kurze Seitenkette des Asp185F ist die Adenosin-Ribose des NAD+ gezwungen sich zu verkippen, um mit seinem O3' eine H-Brücke zum Asp185F auszubilden (Abb. 4.25 A). Dadurch verdreht sich das C5' der Adenosin-Ribose um ca. 160° (Abb. 4.26 B). Die zweite H-Brücke zur Adenosin-Ribose geht verloren, wodurch die Anbindung des NAD+ geschwächt wird. In dieser Konformation der Adenosin-Ribose vergrößert sich der Abstand zur Seitenkette des Lys76F, wodurch die H-Brücke um 0.5 Å länger ist, was eine schwächere Wechselwirkung impliziert (Abb. 4.25 A). A) B) E95 C5' Abbildung 4.26: Überlagerung der Strukturen der Variante E185DF (grün) mit gebundenem NAD+ (blau) und von NuoEFHis (hellblau) mit gebundenem NADH (magenta). A) zeigt die überlagerten Peptidrückgrate und die Konformation der Carbonyl-Gruppe der Peptidbindung von Glu95F in beiden Strukturen. Bis auf die verkippte Adenosin-Ribose des NAD+ unterscheidet sich die Anbindung der Nukleotide kaum. B) zeigt das um etwa 160° verdrehte C5' (rot) der Adenosin-Ribose. Da die Carbonyl-Gruppe der Peptidbindung von Glu95F der Variante E185DF dieselbe Konformation wie im reduzierten NuoEFHis einnimmt, scheint der postulierte „AuswurfMechanismus“ für NAD+ in dieser Variante nicht funktional zu sein. Jedoch führt die 96 Ergebnisse fehlende H-Brücke zum O2' der Adenosin-Ribose und die schwache H-Brücke zu Lys76F zu einer deutlich schwächeren Anbindung des NAD+, so dass dieses leichter aus der Bindetasche dissoziieren kann. Da die B-Faktoren des Carbonyl-Sauerstoffs zeigen, dass diese Position nicht rigide ist, könnte der eingeschränkte „Auswurf-Mechanismus“ und die schwächere Wechselwirkung der Adenosin-Ribose mit dem Enzym die erhöhte Aktivität der homologen E. coli Komplex I-Variante mit NADH erklären. Das durch die Mutation Glu185AspF induzierte Verkippen der Adenosin-Ribose des NAD+ schafft mehr Raum in der Substratbindestelle. Dies zeigen die Querschnitte der Oberflächendarstellung von E185DF mit gebundenem NAD+ und von NuoEFHis mit gebundenem NAD+ bzw. NADH. Durch die veränderte Konformation der Adenosin-Ribose wird der Boden und zum Teil auch das Innere der Bindestelle nicht mehr vollständig ausgefüllt (Abb. 4.27). A) B) C) Abbildung 4.27: Querschnitt durch die Oberflächendarstellung der Substrat-Bindestelle. Der Schnitt am Halbacetal-O4' der Adenosin-Ribose ist dargestellt. Gezeigt sind die Strukturen von NuoEFHis mit gebundenem NAD+ im oxidierten A) und gebundenem NADH im reduzierten B) Zustand. C) zeigt die Variante E185DF mit gebundenem NAD+. Die Oberfläche des Asp185F in der Variante bzw. die des Glu185F in NuoEFHis ist rot markiert. Das FMN ist in Gelb dargestellt. In die nicht mehr vollständig abgeschirmte NADH-Bindestelle der Variante könnte Sauerstoff eindiffundieren und vom reduzierten FMN reduziert werden. Dies würde die deutlich erhöhte ROS-Produktion der homologen E. coli Komplex I-Variante bei der Oxidation von NADH erklären. 97 Ergebnisse 4.3 Die Position Tyr178F im E. coli Komplex I Mutationen im mitochondrialen Komplex I und die damit einhergehende erniedrigte Aktivität und erhöhte ROS-Produktion führen zu zahlreichen neurodegenerativen Erkrankungen (Pagniez-Mammeri et al., 2012). Der Großteil der von Komplex I produzierten ROS entstehen am reduzierten FMN (Kussmaul und Hirst, 2006; Hirst et al., 2008). Strukturelle Analysen des A. aeolicus NuoEFHis-Subkomplexes zeigen, dass Glu97F (in E. coli Glu95F) bei der Bindung von NADH eine H-Brücke mit Tyr180F (in E. coli Tyr178F) bildet. Diese H-Brücke ist wichtig für die korrekte Bindung des NADH und gewährleistet den Hydridtransfer auf das FMN. Die Mutation des Glu95F zu Gln im E. coli Komplex I führte zur einer um etwa zwei Drittel verringerten NADH:Decyl-Ubichinon Oxidoreduktaseaktivität in der isolierten Variante (Euro et al., 2009). Zwar ist Gln immer noch in der Lage, eine H-Brücke mit der Seitenkette des Tyr178F auszubilden, jedoch ist diese aufgrund der geringeren Ladungsdichte im Vergleich zu Glu deutlich geschwächt. Glu95F und Tyr178F sind in Pro- und Eukaryoten konserviert, was ein weiteres Indiz für die funktionelle Bedeutung beider Reste ist (Abb. 4.28). E. coli T. thermophilus A. aeolicus P. denitrificans N. crassa Y. lipolytica B. taurus H. sapiens 90 100 180 190 YLLCN ADEMEPGTYK D--------VHTGAGRYIC GEETALINSL YLICN ADESEPGSFK D--------VHRGAGAYIC GEETALMNSL YFICN ADESEPGTFK D--------VARGAGAYIC GEETALIESL YLVIN ADESEPATCK D--------LHHGAGAYIC GEETALLESL YLVVN ADEGEPGTCK D--------LHRGAGAYVC GEETSLIESL YLVVN ADEGEPGTCK D--------IHRGMGAYVC GEETSLIESL YLVVN ADEGEPGTCK D--------VVRGAGAYIC GEETALIESI YLVVN ADEGEPGTCK D--------VVRGAGAYIC GEETALIESI *:: * *** **:: * * : * * *:* ****:*::*: Abbildung 4.28: Ausschnitt eines Sequenzvergleichs der Homologen von NuoF um die Positionen Glu95F und Tyr178F. Die Nummerierung nach der E. coli-Sequenz ist angegeben. Glu95F und Tyr178F sind gelb hervorgehoben. Die Doppelmutation Tyr178CysF/Cys180GlyF im menschlichen Genom führte bei einem Patienten zum Leigh-like Syndrom (Pagniez-Mammeri et al., 2012). Da beide Mutationen nahe der FMN-Bindestelle des Komplex I lokalisiert sind, haben die Autoren angenommen, dass durch die Mutationen die Anbindung des FMNs gestört wird, konnten dies aber anhand ihrer Untersuchungen experimentell nicht beweisen (Bénit et al., 2001). Es wird zudem vermutet, dass Tyr178F, unter Ausbildung eines Tyrosyl-Radikals an der Verringerung der ROS-Produktion beteiligt ist (Prof. Friedrich, persönliche Mitteilung). Die Auswirkungen der Mutation von Tyr178F zu Ala, Leu, Phe und Cys auf die Aktivität und die ROS-Produktion 98 Ergebnisse des E. coli Komplex I sollten in dieser Arbeit untersucht werden. Emmanuel Gnandt wurde im Rahmen seiner Diplomarbeit zu den Arbeiten in diesem Kapitel angeleitet (Gnandt, 2012). 4.3.1 Darstellung der Komplex I-Varianten Y178A/C/F und LF Die Mutationen Tyr178Ala/Cys/Phe und LeuF wurden mit Hilfe der λ-Red vermittelten Rekombination in das Expressionsplasmid pBADnuo nuoFhis eingebracht. Die ortsgerichtete Mutagenese wurde mit den Oligonukleotid-Paaren nuoF Y178A/C/F/L_fwd und rev (Tab. 3.4) in den Subklon pUC25nuoE´–G eingebracht. Die vier mutierten linearen DNAFragmente wurden mit dem Oligonukleotid-Paar nuoF_fwd und rev mittels PCR amplifiziert (Tab. 3.6). Für die λ-Red vermittelte Rekombination wurden 50 ng pBADnuo nuoFhis::nptIsacB und 150 – 350 ng der linearen DNA-Fragmente verwendet. Der Erfolg der Rekombination wurde durch Restriktionsanalyse und DNA-Sequenzierung bestätigt. Für die Überproduktion der Komplex I-Varianten wurde der E. coli Stamm BW25113Δnuo verwendet. Die Mutanten und der Ausgangsstamm wurden im 10 L-Maßstab in Autoinduktionsmedium angezogen. Die Zellausbeute lag zwischen 6 und 11 g pro L Medium. Die Varianten und der ursprüngliche Komplex I wurden wie oben beschrieben isoliert, wobei die Ausbeute zwischen 0.17 und 0.27 mg Protein pro g Zellen lag. 4.3.2 Charakterisierung der Komplex I-Varianten Y178A/C/F und LF Um den Einfluss der eingeführten Mutation auf die Aktivität zu untersuchen, wurden die Präparationen der Varianten und des Komplex I in polare Lipide rekonstituiert und die NADH:Decyl-Ubichinon Oxidoreduktaseaktivitäten in Abhängigkeit von der Substratkonzentration bestimmt. Die Aktivitäten wurden für 5 – 150 µM NADH bestimmt, wobei eine Decyl-Ubichinon-Konzentration von 60 µM verwendet wurde. Durch die Auftragung der gemessenen Aktivitäten nach Hanes und linearer Regression wurde über die Steigung der berechneten Ausgleichsgeraden und deren Ordinatenabschnitt die Michaelis-Konstante Km und die maximale Umsatzgeschwindigkeit vmax bestimmt (Hanes, 1932). Aus vmax wurde die Wechselzahl kcat berechnet und hieraus wiederum die katalytische Effizienz (kcat/Km). In Tabelle 4.8 sind die kinetischen Parameter des Komplex I und der Varianten aufgeführt. 99 Ergebnisse Tabelle 4.8: Kinetische Parameter des unveränderten Komplex I und der Varianten Y178A/C/F und LF. Km Variante Komplex I vmax −1 kcat −1 −1 kcat/Km [µM] [µmol·min ·mg ] [s ] [s−1·µM−1] 8.5 ± 0.5 3.1 ± 0.1 28 ± 1 3.3 ± 0.3 F 10 ± 1 1.1 ± 0.1 10 ± 1 1.0 ± 0.1 F 12 ± 1 1.1 ± 0.1 10 ± 1 0.8 ± 0.08 F Y178F 23 ± 2 2.3 ± 0.1 21 ± 1 0.9 ± 0.1 Y178LF 12 ± 2 0.8 ± 0.1 7±1 0.6 ± 0.1 Y178A Y178C Die Varianten Y178A/C und LF zeigen im Vergleich zu Komplex I eine leicht verringerte Affinität zu NADH, wobei die Km-Werte im Bereich der Fehler nahe beieinander liegen. Den größten Effekt auf die Substrat-Affinität zeigte die Variante Y178FF mit einem knapp dreifach erhöhten Km. Alle Varianten zeigen eine deutlich verringerte Umsatzgeschwindigkeit. Die der Variante Y178FF ist um etwa ein Viertel verlangsamt. Für die Varianten Y178AF und Y178CF beträgt vmax nur noch etwa ein Drittel des Komplex I. Die katalytische Effizienz der Varianten ist jeweils stark erniedrigt und beträgt 18 – 30% der des Komplex I. Die Auswirkung der einzelnen Mutationen auf die bei der Reaktion gebildeten ROS wurde mit dem Amplex Red Assay bestimmt. Der Ansatz enthielt 5 µg in polare Lipide rekonstituiertes Protein, 60 µM Decyl-Ubichinon, 2 U/mL HRP und 10 µM Amplex Red. Die Reaktion wurde durch Zugabe von 30 µM NADH gestartet. Die Abnahme der NADHKonzentration und die Zunahme der Resorufin-Konzentration wurden gemessen und aus den Steigungen die entstehende Menge H2O2 pro umgesetztem NADH berechnet (Tab. 4.9). Tabelle 4.9: H2O2-Produktion pro umgesetztem NADH und FMN-Gehalt des Komplex I und der Varianten Y178A/C/F und LF. Die H2O2-Produktion wurde mit dem Amplex Red Assay und der FMN-Gehalt wurde spektroskopisch bestimmt. Präparation Komplex I FMN-Gehalt − NADH + NADH [%] 0 1.1 ± 0.2 87 ± 3 F 0 8.1 ± 0.9 91 ± 5 F 0 5.4 ± 0.6 89 ± 1 F 0 13.0 ± 1.0 83 ± 4 F 0 6.5 ± 0.5 91 ± 5 Y178A Y178C Y178F Y178L 100 H2O2-Produktion pro umgesetztem NADH [%] Ergebnisse Für alle Varianten war die H2O2-Produktion im Vergleich zum Komplex I deutlich erhöht. Die Varianten Y178A/C und LF produzierten die fünf- bis achtfache Menge an H2O2 pro umgesetztem NADH im Vergleich zum Komplex I. Für die Variante Y178FF war die ROSProduktion sogar fast 13-fach erhöht. Der FMN-Gehalt der Präparationen wurde über Differenzspektroskopie bestimmt. Hierfür wurden 0.5 mg des Komplex I oder der Varianten mit Dithionit reduziert und der zeitliche Verlauf der Reoxidation durch Luftsauerstoff bei 450 nm (FMN) und 320 nm (Dithionit) verfolgt. Anhand des Differenzspektrums des vollständig reduzierten und reoxidierten Komplexes wurde mit Hilfe des Lambert-Beerschen Gesetzes die FMN-Konzentration bestimmt. Der Differenz-Extinktionskoeffizient (ε x−re ) für freies FMN (Δε450 = 9 mM−1·cm−1; Ghisla, 1980) wurde um den Beitrag der bei 450 nm absorbierenden Fe/SZentren (ε450 = 6 mM−1·cm−1; Scheide, 2000) erweitert. Somit wurde ε450 = 15 mM−1·cm−1 verwendet, um den prozentualen FMN-Gehalt des Komplex I und der Varianten zu bestimmen (Tab. 4.9). Keine Präparation enthält eine quantitative FMN-Besetzung. Da der FMN-Gehalt aller Präparationen im Rahmen der Fehlergrenzen mit dem des Komplex I identisch ist, kann der Vorschlag, dass die Mutation Tyr178CysF zu einem Verlust des FMNs führt, ausgeschlossen werden. 4.4 Die Beteiligung von Trp221G am Elektronentransfer Aromatische Aminosäuren wie Tryptophan und Tyrosin dienen Enzymen als Trittsteine für den Elektronentransfer (Gray und Winkler, 2003). Quantenmechanische Berechnungen haben die Beteiligung konservierter, aromatischer Aminosäuren am Elektronentransfer in Komplex I vorgeschlagen (Wittekindt et al., 2009). In vorangegangen Arbeiten unseres Arbeitskreises wurde gezeigt, dass die Mutationen Phe515LeuCD bzw. Trp221LeuG einen deutlichen Effekt auf die d-NADH Oxidaseaktivität der E. coli Cytoplasmamembranen der Mutanten hatten. Die Aktivität war um mehr als ein Drittel reduziert (Dörner, 2010). Um auszuschließen, dass die verringerten Aktivitäten strukturell bedingt sind, sollten in dieser Arbeit Doppelmutanten dargestellt werden, die möglicherweise die ursprüngliche Aktivität aufweisen. Da die Mutante 101 Ergebnisse Trp221LeuG die niedrigste Aktivität aufwies, wurde sie als Grundlage für die Konstruktion weiterer Doppelmutanten ausgewählt. Es wurden aliphatische Aminosäuren, die sich in einem ähnlichen Abstand zu N5 und N6a wie Leu221G befinden, identifiziert und gegen Tryptophan ausgetauscht (Abb. 4.29). N5 4 R245 G (R219 ) 3 T121 G (V107 ) 4 W247 G (W221 ) 4 L317 CD (T504 ) 4 9 N6a T332 CD (S519 ) L58 I (L65 ) Abbildung 4.29: Positionen der ausgewählten aliphatischen Aminosäuren im T. thermophilus Komplex I (PDB-Eintrag: 3IAM; Berrisford und Sazanov, 2009). Die T. thermophilus und die E. coli Nomenklatur (in Klammern) der Aminosäuren ist angegeben. Die Fe/S-Zentren sind als Kalotten-Modelle mit Eisen in Rot und Schwefel in Gelb dargestellt. Die Abstände zwischen den Fe/S-Zentren und den Cβ-Atome der Seitenketten sind in Ångström angegeben. Die quantenmechanischen Berechnungen der Gruppe von Prof. Koslowski zeigen, dass basische Aminosäuren in direkter Nachbarschaft zum Aromaten die Energie unbesetzter Orbitale senken, so dass diese leichter mit Elektronen besetzt werden können (Wittekindt et al., 2009). Um den Einfluss des zum Trp221G benachbarten Arg219G auf die Aktivität zu untersuchen, sollte dieser Rest ebenfalls gegen Leucin ausgetauscht werden. Zudem sollte eine Mutante dargestellt werden, in der Arg219G und Trp221G gegeneinander ersetzt sind, sowie eine in der Arg219G gegen Trp und Trp221G gegen Leu ausgetauscht ist. Doris Deković und Manuel Schäuble wurden im Rahmen ihrer Diplomarbeiten zu den Arbeiten zu Teilen dieses Kapitels angeleitet (Dek vić, 2012; Schäuble, 2013). 102 Ergebnisse 4.4.1 Darstellung der Doppelmutanten Das Expressionsplasmid pBADnuo nuoFhis nuoG W221L und der Subklon pUC25nuoE´–G W221L lagen bereits zu Beginn der Arbeit vor (Dörner, 2010). Für die Mutagenese von nuoCD und nuoI dienten die Vektoren pCA24NnuoCD und pCA24NnuoI als Subklone. Die Mutanten pCA24NnuoCD T504W, pCA24NnuoCD S519W und pCA24NnuoI L65W wurden mit den entsprechenden Oligonukleotid-Paaren (Tab. 3.4) durch ortsgerichtete Mutagenese dargestellt. Der Erfolg der ortsgerichteten Mutagenese wurde durch Restriktionsanalyse der aus einzelnen Kolonien isolierten Plasmide und DNA-Sequenzierung bestätigt. Die für die λRed vermittelte Rekombination notwendigen linearen DNA-Fragmente lin_nuoCD T504W, lin_nuoCD S519W und lin_nuoI L65W wurden mit den Oligonukleotid-Paaren nuoCD_fwd/rev bzw. nuoI_fwd/rev (Tab. 3.6) mittels PCR amplifiziert. Um die Mutationen in das Expressionsplasmid pBADnuo nuoFhis nuoG W221L einzufügen, musste zunächst die nptI-sacB-Selektionskassette in nuoCD und nuoI mit Hilfe der λ-Red vermittelten Rekombination eingebracht werden. Hierfür wurden die Selektions-Kassetten mit den Oligonukleotiden nuoD::nptI-sacB_fwd/rev und nuoI2::nptI-sacB_fwd und nuoI::nptIsacB_rev (Tab. 3.5), die jeweils homologe Bereiche zu den Zielgenen enthielten, amplifiziert. Elektrokompe en e Ze en e S amm DH5αΔnuo/pKD46 wurden hergestellt und jeweils mit 50 ng pBADnuo nuoFhis nuoG W221L und 200 ng nuoI::nptI-sacB bzw. 150 ng nuoCD::nptIsacB kotransformiert. Positive Klone wurden auf LB-Agar-Platten mit Kanamycin selektiert. Einzelne Klone wurden angezogen und die Plasmide der Kulturen isoliert. Der Erfolg der Rekombination wurde durch Restriktionsanalyse überprüft. Zur Darstellung der Mutanten pBADnuo nuoFhis nuoCD T504W nuoG W221L pBADnuo nuoFhis nuoCD S519W nuoG W221L und und pBADnuo nuoFhis nuoG W221L nuoI L65W wurden die Selektions-Kassetten gegen die linearen DNA-Fragmente ausgetauscht. Für die λ-Red vermittelte Rekombination wurden 50 ng der Vektoren mit Selektions-Kassette und jeweils 150 – 200 ng der linearen DNA-Fragmente verwendet. Positive Klone wurden auf YP/SUC-Agar-Platten mit Chloramphenicol selektiert. Der Erfolg der Rekombination wurde durch Restriktionsanalyse und DNA-Sequenzierung überprüft. Für eine effiziente Rekombination ist es wichtig, dass der Bereich in den die Mutation eingefügt werden soll, durch die nptI-sacB-Kassette ersetzt ist. NuoG ist die größte Untereinheit des Komplex I. Um die Rekombinations-Effizienz für nuoG zu erhöhen, wurden für die Insertion der nptI-sacB-Kassette homologe Bereiche am Anfang und am Ende des Gens ausgewählt. Die Selektionskassette nuoGc::nptI-sacB (c, complete) wurde mit den 103 Ergebnisse Oligonukleotiden nuoGbeg::nptI-sacB_fwd sowie nuoGend::nptI-sacB_rev von dem Vektor pVO1100 mi e PCR amp ifizier un mi Hi fe er λ-Red vermittelten Rekombination in pBADnuo nuoFhis eingebracht. Hierfür wurden 50 ng pBADnuo nuoFhis und 400 ng nuoGc::nptI-sacB verwendet. Positive Klone wurden auf LB-Agar-Platten mit Kanamycin selektiert. Einzelne Kolonien wurden angezogen und die Plasmide aus den Kulturen isoliert. Der Erfolg der Rekombination wurde durch Restriktionsanalyse überprüft. Der Subklon pUC25nuoE´–G diente als Vorlage-DNA für die ortsgerichtete Mutagenese an den Positionen Arg219G und Trp221G. Die Mutationen Arg219LeuG, Arg219Trp/Trp221ArgG und Arg219Trp/Trp221LeuG wurden mit den entsprechenden Oligonukleotid-Paaren (Tab. 3.4) eingefügt. Bei der Darstellung von pUC25nuoE´–G V107W/W221L mit dem Oligonukleotid-Paar nuoG V107W_fwd/rev diente der Subklon pUC25nuoE´–G W221L als Vorlage-DNA. Die linearen DNA-Fragmente wurden mit dem Oligonukleotid-Paar nuoGbeg_fwd/nuoGrec_rev Tab. 3.6) amp ifizier un mi Hi fe er λ-Red vermittelten Rekombination in den Expressionsvektor eingebracht. 4.4.2 Komplex I-vermittelte Aktivitäten der Cytoplasmamembranen Zur Überproduktion der Komplex I-Varianten wurden Zellen des E. coli Stamms BW25113Δnuo mit 10 ng der dargestellten Doppel- und Einzel-Mutanten sowie dem unveränderten Plasmid pBADnuo nuoFhis transformiert und auf LB-Agar-Platten mit Chloramphenicol ausgestrichen. Vor der Zellzucht wurden die Plasmide aus den Expressionsstämmen isoliert und zur Kontrolle erneut mittels Restriktion analysiert. Die Mutanten wurden im 400 mL-Maßstab in Autoinduktionsmedium unter Schütteln bei 37 °C kultiviert. Die maximalen OD600-Werte lagen zwischen 4.5 und 7.0, was einer Zellausbeute von ca. 4 – 7 g pro L Medium entsprach. Die Cytoplasmamembranen der Mutanten und des Ausgangsstamms wurden isoliert. Der Expressionsstamm BW25113Δnuo besitzt keinen genomisch kodierten Komplex I aber die alternative NDH-II. Um sicher zu gehen, dass nur die Komplex I-vermittelten Aktivitäten gemessen werden, wurde das NADH-Derivat d-NADH als Substrat für die Aktivitätsmessungen verwendet. Der Proteingehalt der Membranen wurde mit der Biuret-Methode bestimmt. Tabelle 4.10 gibt die gemessenen d-NADH Oxidase- und d-NADH/Ferricyanid Oxidoreduktaseaktivitäten wieder. Um die Menge an überproduziertem Komplex I in der Membran zu berücksichtigen, wurden die Oxidaseaktivitäten auf die d-NADH/Ferricyanid Oxidoreduktaseaktivitäten bezogen. 104 Ergebnisse Tabelle 4.10: Die d-NADH Oxidase- und d-NADH/Ferricyanid Oxidoreduktaseaktivitäten der dargestellten Mutanten und des ursprünglichen Stamms. BW25113Δnuo/ pBADnuo nuoFhis d-NADHOxidaseaktivität d-NADH/Ferricyanid Oxidoreduktaseaktivität Normierte d-NADHOxidaseaktivität [µmol min−1 mg−1] Verbleibende Aktivität [%] unverändert 0.25 ± 0.02 1.6 ± 0.2 0.25 ± 0.02 100 nuoG W221L 0.09 ± 0.02 1.4 ± 0.2 0.11 ± 0.01 40 ± 4 nuoG R219L 0.14 ± 0.01 1.3 ± 0.1 0.17 ± 0.01 68 ± 5 nuoG R219W/W221L 0.03 ± 0.01 1.4 ± 0.2 0.03 ± 0.01 12 ± 4 nuoG R219W/W221R 0.02 ± 0.01 1.2 ± 0.1 0.03 ± 0.01 12 ± 6 nuoG W221L/V107W 0.05 ± 0.01 1.5 ± 0.2 0.05 ± 0.01 20 ± 4 nuoG W221L nuoI L65W 0.06 ± 0.01 1.3 ± 0.1 0.07 ± 0.01 28 ± 5 nuoG W221L nuoCD T504W 0.04 ± 0.01 1.4 ± 0.2 0.05 ± 0.01 20 ± 5 nuoG W221L nuoCD S519W 0.04 ± 0.01 1.6 ± 0.2 0.04 ± 0.01 16 ± 4 Die Mutation Arg219LeuG verringerte die normierte d-NADH Oxidaseaktivität um ein Drittel. Dies deutet auf die Funktion dieser Aminosäure für den Elektronentransfer hin (Wittekindt et al., 2009). Keine der zusätzlich eingeführten Mutationen konnte den Effekt der Mutation Trp221LeuG auf die Aktivität ausgleichen. Die Aktivitäten der Doppelmutanten waren im Gegenteil deutlich verringert. Die Aktivität der Doppelmutante Arg219TrpG/Trp221ArgG betrug 12% der des ursprünglichen Stamms und etwa ein Drittel der der Mutante Trp221LeuG. Alle Mutanten wiesen eine signifikante d-NADH/Ferricyanid Oxidoreduktaseaktivität auf, die jedoch für einzelne Mutanten bis zu einem Viertel verringert war. Um zu untersuchen, ob die verringerten NADH-Oxidaseaktivitäten und NADH/Ferricyanid Oxidoreduktaseaktivitäten durch die Mutationen oder durch eine geringere Proteinproduktion verursacht werden, wurden die Proteine über einen Western Blot analysiert. Je 300 µg Membranproteine wurden durch SDS-PAGE aufgetrennt und auf eine PVDF-Membran transferiert. Zur Kontrolle wurden Membranen der Einzelmutanten und des ursprünglichen Stamms mit aufgetragen. Zum Nachweis wurden Mouse Anti-His4 primäre Antikörper gegen das N-terminal an NuoF fusionierte His6-Affinitätspeptid verwendet (Abb. 4.30). 105 Ergebnisse 1 2 3 4 5 M 6 7 8 9 70 55 35 25 15 10 Abbildung 4.30: Western Blot der Membranproteine verschiedener BW25113Δnuo/pBADnuo nuoFhisMutanten mit einem Mouse Anti-His4 Antikörper. Spur M zeigt die Banden des Proteinstandards PageRuler Plus Prestained Protein Ladder (Thermo Scientific). Die apparenten Molekülmassen sind in kDa angegeben. Spur 1 zeigt das Signal der Membranproteine der Mutante Trp221LeuG/Val107TrpG, Spur 2 das von Trp221LeuG/Thr504TrpCD, Spur 3 das von Trp221LeuG/Ser519TrpCD, Spur 4 das von Trp221LeuG/Leu65TrpI, Spur 5 das von Trp221LeuG, Spur 6 das von Trp221ArgG/Arg219TrpCD, Spur 7 das von Arg219LeuG, Spur 8 das des ursprünglich Stamms und Spur 9 das der Mutante Trp221LeuG/Arg219TrpG. Mittels Western Blot-Analyse wurde in den Membranen aller Mutanten eine Bande bei etwa 55 kDa nachgewiesen, die der katalytischen Untereinheit His6-NuoF (51.2 kDa) zugeordnet wurde. Anhand der Signale kann die Menge an Komplex I in der Membran zwar nicht exakt quantifiziert werden, dennoch lassen sich die ungefähren Intensitäten abschätzen. Die Bande für die Mutante Trp221LeuG hat die geringste Intensität. Die verringerte d-NADH/Ferricyanid Oxidoreduktaseaktivität der Membranen dieser Mutante ist durch eine geringere Produktion der Variante zu erklären. Auch die Intensität der Banden für die Doppelmutanten Trp221ArgG/Arg219TrpG und Trp221LeuG/Val107TrpG ist im Vergleich zum Ausgangsstamm geringer. Dies erklärt die um ein Viertel verringerte d-NADH/Ferricyanid Oxidoreduktaseaktivität der Membranen von Trp221ArgG/Arg219TrpG, aber nicht die kaum verringerte Aktivität von Trp221LeuG/Val107TrpG. Die verminderte d-NADH/Ferricyanid Oxidoreduktaseaktivität von Arg219LeuG lässt sich nicht durch den Western Blot erklären. Die Bandenintensitäten zeigen eindeutig, dass die doppelt mutierten Varianten in ungefähr gleichem Maß wie der ursprüngliche Komplex I produziert wurden. Dies lässt den Schluss zu, dass die deutlich verringerte d-NADH-Oxidaseaktivität der Doppelmutanten durch die neu eingeführten Mutationen verursacht wird. Da His6-NuoF die äußerste Untereinheit des peripheren Arms von Komplex I ist, zeigt die Western Blot-Analyse ebenfalls, dass die produzierten Varianten zumindest in der Membran stabil sind. 106 Ergebnisse 4.5 Der E. coli Stamm BW25113nptI Δnuo Δndh/pBADnuo nuoFhis Der E. coli Stamm BW25113Δnuo/pBADnuo nuoFhis wurde in vorangegangen Arbeiten als Überproduktionsstamm für den Komplex I etabliert (Dörner, 2010). Er besitzt keinen genomisch kodierten Komplex I, wodurch eine mögliche trans-Komplementation mit episomal kodierten nuo-Genen verhindert wird. Allerdings enthält dieser Stamm noch das Gen ndh, das für die nicht energiewandelnde NDH-II kodiert. Dies hat den Nachteil, dass das Komplex I-spezifische, sehr teure Substrat d-NADH verwendet werden muss, um die Aktivität des episomal kodierten Komplex I in der Membranen zu bestimmen. In dieser Arbeit sollte der Stamm BW25113nptI Δnuo Δndh/pBADnuo nuoFhis als Überproduzent für den E. coli Komplex I etabliert werden. Es wurde zudem vermutet, dass der Stamm ohne NDH-II mehr Komplex I assembliert, um ausreichend NAD+ für den metabolischen Bedarf zu regenerieren. Zunächst cytoplasmatischen sollte die NADH- Membranen der E. coli und Succinat-Oxidaseaktivität Stämme BW25113, von BW25113Δnuo, BW25113ndh::nptI und BW25113nptI Δnuo Δndh bestimmt werden. 4.5.1 Oxidaseaktivitäten der Membranen Die E. coli Stämme BW25113, BW25113Δnuo, BW25113ndh::nptI und BW25113nptI Δnuo Δndh wurden im 400 mL-Maßstab in LB-Medium angezogen und durch Zentrifugation geerntet. Der Stamm BW25113ndh::nptI wuchs ähnlich wie der Ausgangsstamm BW25113 (Abb. 4.31). Die NDH-II scheint somit kaum einen Einfluss auf das Wachstum von BW25113 zu haben. Für die Stämme BW25113 und BW25113ndh::nptI wurden 4 g Zellen pro L Medium gewonnen. Die BW25113-Stämme ohne genomisch kodierten Komplex I zeigten deutlich geringeres Wachstum. Auch hier ist klar zu erkennen, dass die Anwesenheit oder das Fehlen der NDH-II das Wachstum der Komplex IDeletionsstämme nicht beeinflusst. Für die nuo-Deletionsstämme wurden 1.6 g Zellen pro L Medium erhalten. 107 Ergebnisse Abbildung 4.31: Wachstumskurven von BW25113 und von NADH-Dehydrogenase Deletionsstämmen in LB-Medium. Die Wachstumskurve von BW25113 ist schwarz, die von BW25113Δnuo blau, die von BW25113ndh::nptI grün und die von BW25113nptI Δnuo Δndh rot dargestellt. Die cytoplasmatischen Membranen der Stämme wurden isoliert und die NADH- und Succinat-Oxidaseaktivität der Membranen bestimmt. Die Komplex I-vermittelten Aktivitäten der Membranen wurden mit 25 µM Piericidin A oder mit 50 µM Fenazaquin inhibiert. Zudem wurde der Effekt der Komplex I-spezifischen Inhibitoren Piericidin A und Fenazaquin auf die NDH-II untersucht. Tab. 4.11 listet die gemessenen NADH-Oxidaseaktivitäten und (d-)NADH/Ferricyanid Oxidoreduktaseaktivitäten der verschiedenen Deletionsstämme auf. Tabelle 4.11: (d)-NADH/Ferricyanid Oxidoreduktase- und NADH-Oxidaseaktivitäten der cytoplasmatischen Membranen der Stämme BW25113, BW25113Δnuo, BW25113ndh::nptI und BW25113nptI Δnuo Δndh. Alle Werte wurden dreifach bestimmt. Die Endkonzentration der eingesetzten Hemmstoffe betrug für Piericidin A 25 µM und für Fenazaquin 50 µM. E. coli Stamm NADH/Ferricyanid d-NADH/Ferricyanid OxidoreduktaseOxidoreduktaseaktivität aktivität [µmol·min−1·mg−1] NADH Oxidase Aktivität Inhibition durch Piericidin A Fenazaquin [%] BW25113 1.7 ± 0.1 1.2 ± 0.1 0.36 ± 0.02 82 ± 1 85 ± 1 BW25113 ndh::nptI 1.4 ± 0.1 1.0 ± 0.1 0.29 ± 0.02 90 ± 1 90 ± 1 BW25113 Δnuo BW25113 Δnuo Δndh 1.2 ± 0.1 0.6 ± 0.05 0.14 ± 0.02 50 ± 1 25 ± 1 0.6 ± 0.05 0.4 ± 0.05 0 - - 108 Ergebnisse Die NADH-Oxidaseaktivität von BW25113 ließ sich zu 82% mit Piericidin A bzw. zu 85% mit Fenazaquin inhibieren. Die nur von Komplex I vermittelte NADH-Oxidaseaktivität des Stamms BW25113ndh::nptI betrug ca. 80% der des Ausgangsstamms und wurde mit beiden Hemmstoffen zu etwa 90% inhibiert. Die von der NDH-II vermittelte NADH-Oxidaseaktivität betrug nur etwa 40% der des Ausgangsstamms, wurde aber erstaunlicherweise von den Komplex I-spezifischen Inhibitoren Piericidin A zu 50% und Fenazaquin zu 25% gehemmt. Diese Messungen zeigen, dass Komplex I den größten Beitrag zur NADH-Oxidaseaktivität des Stamms BW25113 liefert. Die Membranen des Stamms BW25113nptI Δnuo Δndh besaßen erwartungsgemäß keine NADH-Oxidaseaktivität. Die NADH/Ferricyanid Oxidoreduktaseaktivität der Membranen von BW25113 betrug 1.7 µmol·min−1·mg−1. Die d-NADH/Ferricyanid Oxidoreduktaseaktivität betrug etwa 70% von dieser. Die NADH/Ferricyanid Oxidoreduktaseaktivität von BW25113ndh::nptI war im Vergleich zu der des Ausgangsstamms leicht erniedrigt und die d-NADH/Ferricyanid Oxidoreduktaseaktivität betrug Oxi ebenfalls 70% re uk a eak ivi der NADH-vermittelten. Die d-NADH/Ferricyanid e Δnuo-Stamms betrug die Hälfte der des Ausgangsstamms, was erstaunlich ist, da d-NADH ein spezifisches Substrat für Komplex I sein sollte (Matsushita et al., 1987). Überraschenderweise zeigten ie Membranen e Δnuo Δndh-Stamms ebenfalls NADH und d-NADH/Ferricyanid Oxidoreduktaseaktivität. Diese betrugen etwa ein Drittel der des Ausgangsstamms. Da die Membranen des Stamms BW25113nptI Δnuo Δndh keine NADH-Oxidaseaktivität zeigen, kann ausgeschlossen werden, dass andere NADH oxidierende Oxidoreduktasen Elektronen in die Atmungskette von E. coli einspeisen. Damit trifft die von Bekker et al. diskutierte Beteiligung weiterer NADH-abhängiger Enzyme wie WrbA, QOR und YhdH an der aeroben Atmungskette nicht zu (Bekker et al., 2009). WrbA ist eine vermutlich monotopisch an der Membran assoziierte NAD(P)H:Chinon Oxidoreduktase und ist in der Lage, sowohl NADH als auch NADPH zu oxidieren. Die genaue physiologische Rolle dieses Enzyms ist nicht bekannt. Es wird vermutet, dass es als Schutz vor oxidativem Stress in der Zelle dient (Patridge und Ferry, 2006; Pyla et al., 2010). QOR ist eine lösliche NADPH:Chinon Oxidoreduktase und überträgt einzelne Elektronen von NADPH auf Chinone um Semichinone zu bilden (Pan et al., 2009; Thorn et al., 1995). Die YhdH ist eine Acryloyl-CoA Reduktase und dient als Schutz vor intrazellulär gebildetem Acryloyl-CoA (Todd et al., 2012). Aufgrund der für QOR und YhdH beschriebenen Reaktionen ist es unwahrscheinlich, dass diese Enzyme Elektronen von NADH in den Chinon-Pool der Atmungskette einspeisen. Für WrbA wurde eine artifizielle NAD(P)H/Ferricyanid 109 Ergebnisse Oxidoreduktaseaktivität nachgewiesen (Patridge und Ferry, 2006). Die NADH bzw. d-NADH/Ferricyanid Oxidoreduktaseaktivität der Membranen von BW25113nptI Δnuo Δndh könnte somit durch WrbA vermittelt sein. Die Succinat-abhängige Oxidaseaktivität der NADH Dehydrogenase-Deletionsstämme wurde bestimmt und zur Kontrolle mit Malonat (10 mM) gehemmt (Tab. 4.12). Um die inhibierende Wirkung der Komplex I-spezifischen Hemmstoffe Piericidin A (25 µM) und Fenazaquin (50 µM) auf die verbleibenden Enzyme der aeroben Atmungskette zu untersuchen, wurde deren Wirkung auf die Succinat-Oxidaseaktivität untersucht. Tabelle 4.12: Succinat-Oxidaseaktivität der cytoplasmatischen Membranen der Stämme BW25113, BW25113Δnuo, BW25113ndh::nptI und BW25113nptI Δnuo Δndh. Es wurden Doppelbestimmung durchgeführt. (n. b. = nicht bestimmt) Inhibition durch E. coli Stamm SuccinatOxidaseaktivität Malonat [10 mM] [µmol·min-1·mg-1] Piericidin A [25 µM] Fenazaquin [50 µM] [%] BW25113 0.15 ± 0.01 99 18 ± 1 24 ± 1 BW25113 ndh::nptI 0.15 ± 0.01 98 n. b. n. b. 0.12 ± 0.01 100 n. b. n. b. 0.14 ± 0.01 99 16 ± 1 22 ± 1 BW25113 Δnuo BW25113 Δnuo Δndh Die Succinat-Oxidaseaktivitäten aller vier Stämme sind im Rahmen der Fehler identisch. Somit kann ein reprimierender Effekt auf die Succinat Dehydrogenase durch die Deletion des Komplex I und der NDH-II ausgeschlossen werden. Die Succinat-Oxidaseaktivität wurde in den Membranen aller Stämme nahezu vollständig mit Malonat inhibiert. Die SuccinatOxidaseaktivitäten des Ausgangsstamms und des Δnuo Δndh-Stamms wurden durch Piericidin A und Fenazaquin zu ca. 17 bzw. ca. 23% inhibiert. Ob die Inhibitoren die Succinat Dehydrogenase oder die terminalen Oxidasen blockieren kann aus dieser Messung nicht gefolgert werden. 110 Ergebnisse 4.5.2 Überproduktion des E. coli Komplex I Zur Überproduktion des Komplex I wurde der Stamm BW25113nptI Δnuo Δndh/pBADnuo nuoFhis in verschiedenen Medien kultiviert. Die Zellen wurden in LB-Medium und in Autoinduktionsmedium mit 0.5% (w/v) D-Mannitol oder 0.5% (v/v) Glycerin angezogen (Abb. 4.32). Die Expression der episomal kodierten nuo-Gene der Zucht in LB-Medium wurde bei einer OD600 von 0.1 mit 0.2% (w/v) L-(+)-Arabinose induziert. Abbildung 4.32: Wachstumskurven des Stamms BW25113nptI Δnuo Δndh/pBADnuo nuoFhis in verschiedenen Medien im 400 mL-Maßstab. Das Wachstum in Autoinduktionsmedium mit 0.5% (w/v) D-Mannitol (schwarz), bzw. 0.5% (v/v) Glycerin (rot) und in LB-Medium (blau) ist gezeigt. Der Pfeil markiert den Zeitpunkt der Induktion mit 0.2% (w/v) L-(+)-Arabinose bei der Zellzucht in LB-Medium. Im Autoinduktionsmedium mit D-Mannitol als zusätzlicher Kohlenstoffquelle wurde die höchste OD600 erreicht. Aus dieser Zellzucht wurden aus 30 g Zellen 6.5 mg Komplex I, nach dem etablierten Protokoll, isoliert. Die Zucht wurde wiederholt, wobei die Zellen vor einer OD600 von 5.0 geerntet wurden. Aus 4.8 L Medium wurden 42 g Zellen erhalten. Aus der gesamten Zellzucht wurden 12.5 mg Komplex I isoliert. Das entspricht 0.3 mg Komplex I pro g Zellen. Die NADH:Decyl-Ubichinon Oxidoreduktaseaktivität der Präparation betrug 3.3 ± 0.1 µmol·min−1·mg−1. Im Vergleich zu der Präparation aus dem Stamm BW25113Δnuo/ pBADnuo nuoFhis konnte die Komplex I-Ausbeute nicht gesteigert werden. Aus diesem Stamm wurden 0.29 mg Komplex I pro g Zellen isoliert (Dörner, 2010). Bei dieser Präparation betrug vmax 2.9 ± 0.1 µmol·min−1·mg−1. Die leicht erhöhte Aktivität ist 111 Ergebnisse wahrscheinlich auf Proteinkonzentration geringere genauer Verunreinigung bestimmt wurde. zurückzuführen, Die Verwendung wodurch des die Stamms BW25113nptI Δnuo Δndh/pBADnuo nuoFhis bietet im Vergleich zu der von BW25113Δnuo/ pBADnuo nuoFhis deutliche Vorteile. Zum einen trägt die Doppeldeletionsmutante eine genomisch kodierte Kanamycin-Resistenz, was eine Selektion auf zwei verschiedenen Antibiotika erlaubt. Zum anderen kann bei der Messung der Oxidaseaktivitäten der Membranen auf das kostspielige Substrat d-NADH verzichtet werden, um spezifisch eine Komplex I-vermittelte Aktivität zu bestimmen. 4.6 Modulation des Mittenpunktspotentials von N1a Das zweikernige Fe/S-Zentrum N1a wurde im intakten mitochondrialen Komplex I nicht durch starke Reduktionsmittel reduziert. Daraus wurde gefolgert, dass es ein sehr negatives Mittenpunktspotential von etwa −1 V haben muss (Reda et al., 2008). In anderen Präparationen des mitochondrialen Komplexes wurde das Mittenpunktspotential von N1a zu etwa −450 mV bestimmt (Ohnishi et al., 1981). Im E. coli Komplex I hat N1a ein höheres Mittenpunktspotential von −330 mV (pH 7.0), wird somit durch NADH reduziert und ist EPR-spektroskopisch nachweisbar (Leif et al., 1995; Uhlmann und Friedrich, 2005). Der Grund für die unterschiedlichen Mittenpunktspotentiale von N1a in verschiedenen Organismen ist nicht bekannt. Durch Mutagenesestudien an NuoE aus E. coli identifizierte die Gruppe von Dr. Judy Hirst die Positionen Val96E und Asn142E in der zweiten Koordinationssphäre um N1a, deren Mutagenese das Mittenpunktspotential von N1a modulierte (Abb. 2.7). In Prokaryoten sind diese Positionen variabel, wohingegen in Eukaryoten an diesen Positionen ein Pro96 und ein Met142 konserviert sind. In den NuoEVarianten V96P und N142M war das Mittenpunktspotential von N1a jeweils um etwa 60 mV erniedrigt. Die Doppelmutation verschob das N1a Mittenpunktspotential um etwa −100 mV (Birrell et al., 2013). Ob dieser Effekt auf den gesamten E. coli Komplex I übertragbar ist, wurde in Zusammenarbeit mit Dr. James Birrell aus der Arbeitsgruppe von Dr. Judy Hirst untersucht. Es sollte geklärt werden, ob N1a unter physiologischen Bedingungen als Antioxidans im E. coli Komplex I wirkt. Die Arbeiten aus diesem Kapitel wurden publiziert (Birrell et al., 2013). 112 Ergebnisse 4.6.1 Darstellung von pBADnuo nuoFhis nuoE V96P, N142M und V96P/N142M Die Mutationen wurden mit den Oligonukleotid-Paaren nuoE V96P_fwd/rev und nuoE N142M_fwd/rev (Tab. 3.4) in den Subklon pCA24NnuoE eingeführt. Der Subklon pCA24NnuoE V96P und das Oligonukleotid-Paar nuoE N142M_fwd/rev wurden zur Konstruktion der Doppelmutation verwendet. Die mutierten Subklone wurden wie oben beschrieben dargestellt. Die Anlagerungstemperatur betrug 55 °C. Der Subklon und der Expressionsvektor vermitteln die gleiche Antibiotikum-Resistenz. Um die Rekombinationseffizienz zu erhöhen, wurden die mutierten Subklone mit BamHI geschnitten und das Fragment mit dem mutierten Gen aus einem Agarosegel isoliert. Diese Fragmente dienten als Vorlage zur Amplifikation der mutierten linearen DNA-Fragmente. Diese wurden mit dem Oligonukleotid-Paar nuoE_fwd/rev (Tab. 3.6) mittels PCR amplifiziert. Hier betrug die Anlagerungstemperatur 60 °C. Für die λ-Red vermittelte Rekombination wurden 50 ng pBADnuo nuoFhis nuoE::nptI-sacB (Dörner, 2010) und 100 – 450 ng der mutierten linearen DNA-Fragmente verwendet. Der Erfolg der Rekombination wurde durch Restriktionsanalyse und DNA-Sequenzierung überprüft. Um die von Komplex I und den Varianten V96PE, N142ME und V96P/N142ME vermittelten Oxidaseaktivitäten zu bestimmen, wurde der E. coli Stamm BW25113nptI Δnuo Δndh mit den dargestellten Plasmiden transformiert und im 400 mL-Maßstab in Autoinduktionsmedium angezogen. Die Membranen wurden wie oben beschrieben isoliert und ihre NADH-Oxidasesowie NADH/Ferricyanid Oxidoreduktaseaktivitäten bestimmt (Tab. 4.13). Tabelle 4.13: NADH/Ferricyanid Oxidoreduktaseaktivitäten und NADH-Oxidaseaktivitäten der Cytoplasmamembranen des unveränderten Stamms und der dargestellten Mutanten. BW25113nptI Δnuo Δndh/pBADnuo nuoFhis NADH/Ferricyanid Oxidoreduktaseaktivität Restaktivität NADH-Oxidase Aktivität Restaktivität [µmol·min−1·mg−1] [%] [µmol·min−1·mg−1] [%] unverändert 1.6 ± 0.1 100 0.25 ± 0.02 100 nuoE V96P 1.3 ± 0.1 81 ± 8 0.21 ± 0.02 84 ± 8 nuoE N142M 1.4 ± 0.1 88 ± 7 0.24 ± 0.02 96 ± 8 nuoE V96P/N142M 1.2 ± 0.1 75 ± 8 0.20 ± 0.02 80 ± 10 113 Ergebnisse Die NADH/Ferricyanid Oxidoreduktaseaktivität und NADH-Oxidaseaktivität der Membranen der Mutante Val96ProE und von Val96ProE/Asn142MetE sind geringer als die des Ausgangsstamms. Die Membranen der Mutante Asn142MetE zeigen keine veränderte Aktivität. 4.6.2 Präparation der Varianten V96PE, N142ME und V96P/N142ME Der E. coli Stamm BW25113Δnuo wurde zur Überproduktion der Varianten V96PE, N142ME und V96P/N142ME verwendet. Die Mutanten wurden im 10 L-Maßstab in Autoinduktionsmedium angezogen (Abb. 4.33). Die Mutationen hatten keinen Einfluss auf den Wachstumsverlauf. Die Zellen wurden kurz vor Erreichen der stationären Phase geerntet, um eine höhere Proteinausbeute zu erzielen. Aus 10 L Medium wurden 70 – 85 g Zellen erhalten. Abbildung 4.33: Wachstumskurven der NuoE-Mutanten und des Ausgangsstamms. Die Wachstumskurven von BW25113Δnuo/pBADnuo nuoFhis nuoE V96P (grün), N142M (rot), der Doppelmutante V96P/N142M (schwarz) und des Ausgangsstamms (blau) sind dargestellt. Die Zellen wurde im 10 L-Maßstab in Autoinduktionsmedium angezogen. Die Varianten wurden wie oben beschrieben isoliert. Für die Präparationen wurden jeweils 30 – 35 g Zellen eingesetzt. Die Elutionsprofile der chromatographischen Reinigung an Fractogel und Ni2+-IDA sind beispielhaft für die Variante V96P/N142ME gezeigt (Abb. 4.34). 114 Ergebnisse Die Präparationen der anderen Varianten verliefen ähnlich und werden daher nicht gezeigt. Tabelle 4.14 gibt die Ausbeuten der einzelnen Präparationen wieder. A) B) Abbildung 4.34: Elutionsprofile der Präparation der Variante V96/N142ME. A) zeigt das Elutionsprofil der Anionenaustausch-Chromatographie an Fractogel EMD TMAE Hicap (M) und B) das der AffinitätsChromatographie an ProBond Ni2+-IDA. Die Absorbanz bei 280 nm ist in Blau, die NaCl- bzw. ImidazolKonzentration in Grün und die artifizielle NADH/Ferricyanid Oxidoreduktaseaktivität in Rot dargestellt. Tabelle 4.14: Zusammenfassung der Präparationen der NuoE-Varianten und des ursprünglichen Komplex I. Präparation Komplex I E V96P N142M E V96P/N142M E eingesetzte Zellen Protein NADH/Ferricyanid Oxidoreduktaseaktivität Ausbeute der Gesamtaktivität [g] [mg] [µmol·min−1·mg−1] [%] 35.0 6.5 79.0 8 31.5 6.3 72.0 10 34.0 4.6 71.0 4 35.0 8.0 65.2 5 Wie in den Membranen zeigen auch die isolierten Varianten eine nur geringfügig verminderte spezifische NADH/Ferricyanid Oxidoreduktaseaktivität. Die Minderung der Aktivität ist jedoch nicht signifikant, da Schwankungen in diesen Größenordnungen auch für verschiede Präparationen des ursprünglichen Komplex I beobachtet wurden. Die Reinheit und Zusammensetzung der Präparationen wurde durch SDS-PAGE analysiert (Abb. 4.35). Das Polyacrylamidgel zeigt, dass die Präparationen der Varianten sämtliche Nuo-Untereinheiten enthalten. 115 Ergebnisse app. Mr [kDa] M 1 2 3 4 116.0 → ← Nu G 66.2 → 45.0 → ← Nu CD ← Nu F 35.0 → ← NuoL, NuoN ← NuoM 25.0 → ← NuoH ← NuoB NuoI/J/E 18.4 → ← NuoA 14.4 → } ← NuoK Abbildung 4.35: Polyacrylamidgel der Präparationen der NuoE-Varianten. Spur M zeigt die Banden des Proteinstandards Unstained Protein Molecular Weight Marker (Thermo Scientific). Spur 1 zeigt das Bandenmuster des ursprünglichen Komplex I, Spur 2 das der Variante V96PE, Spur 3 das der Variante N142ME und Spur 4 das der Variante V96P/N142ME. Von den Varianten wurden ca. 70 µg und vom unveränderten Komplex I ca. 60 µg aufgetragen. Die Banden wurden aufgrund ihrer molekularen Masse den entsprechenden Nuo-Untereinheiten zugeordnet. Das Mittenpunktspotential für N1a in den einzeln produzierten NuoE-Varianten wurde durch Cyclovoltammetrie an einem Proteinfilm bestimmt (Zu et al., 2002). Die Cyclovoltammetrie wurde in 20 mM Tris/HCl (pH 8.0) und 0.5 M NaCl durchgeführt. Das Mittenpunktspotential von N1a in den Varianten V96PE und N142ME war um −53 mV verschoben. In der Variante V96P/N142ME ist es um −100 mV abgesenkt. Da die Einzelmutationen den gleichen Effekt auf das Mittenpunktspotentiale der einzeln produzierten NuoE-Varianten hatten, wurden nur die Komplex I-Varianten V96PE und V96P/N142ME für weitere Untersuchungen verwendet. Der ursprüngliche Komplex I und die Varianten V96PE und V96P/N142ME wurden mit 5 mM NADH (−370 mV, pH 6.5) sowie 1 mM NADH und 1 mM NAD+ (−310 mV, pH 6.5) reduziert und EPR-spektroskopisch untersucht (Abb. 4.36). In der mit NADH (−370 mV, pH 6.5) reduzierten Variante V96PE war die Intensität des N1a-Signals um 40% im Vergleich zum ursprünglichen Komplex I verringert. In dem Spektrum des mit NADH/NAD+ (1:1) reduzierten ursprünglichen Komplex I sind die Signalintensitäten für N1a und N1b um 30% verringert. Das Spektrum der mit NADH/NAD+ (1:1) reduzierten Variante V96PE zeigt, dass das Signal für N1a sogar um 70% schwächer ist (Abb. 4.36). Dies deutet darauf hin, dass das Mittenpunktspotential von N1a der Variante niedriger ist als im Komplex I. Die Mutation ändert die Umgebung des Zentrums und verursacht so eine Verschiebung der gz- und gx116 Ergebnisse Werte von N1a. In den Spektren der unter diesen Bedingungen reduzierten Variante V96P/N142ME fehlt das Signal von N1a vollständig. Dies zeigt, dass das Mittenpunktspotential von N1a auch im intakten Komplex I deutlich erniedrigt ist. Ein Verlust des Zentrum kann ausgeschlossen werden, da in vorangegangen Arbeiten gezeigt wurde, dass N1a essentiell für die Assemblierung des Komplex I ist (Dörner, 2010). a) b) c) Magnetische Flussdichte [mT] Abbildung 4.36: EPR-Spektren des ursprünglichen Komplex I a) und der Varianten V96PE b) und V96P/N142ME c). Die Spektren der unter anoxischen Bedingungen mit NADH (−370 mV) reduzierten Proben sind schwarz abgebildet. Die grauen Spektren zeigen die mit NADH/NAD + (1:1, −310 mV) reduzierten Proben. In allen Spektren sind die Signale von N1b eindeutig zuzuordnen. Der g z- und der gx-Wert von N1a ist in der Variante V96PE durch die Mutation verschoben. In dem Spektrum der Variante V96P/N142ME ist kein Signal für N1a detektierbar. Die Spektren wurden bei 40 K und 1 mW aufgenommen und auf die Proteinkonzentration normalisiert. Die Mikrowellenfrequenz betrug 9.38 – 9.39 GHz, die Modulationsfrequenz 100 kHz, die Modulationsamplitude 1 mT, die Zeitkonstante 81.92 ms und die Konversionszeit 20.48 ms. Der für die Aufnahmen verwendete Puffer enthielt 50 mM MES/NaOH (pH 6.5), 50 mM NaCl, 0.1% DDM und 10% Glycerin (Birrell et al., 2013). In analoger Weise sollte das Mittenpunktspotential von N1a im Y. lipolytica Komplex I erhöht werden. Hierfür wurden die homologen Positionen von Pro103E (in E. coli Val96E) und Met149E (in E. coli Asn142E) gegen die entsprechenden E. coli NuoE-Reste ausgetauscht. Die 117 Ergebnisse Mutagenese des Y. lipolytica Komplex I wurde von Dr. James Birrell durchgeführt. In der einzeln produzierten Untereinheit NuoE von Y. lipolytica wurde das Mittenpunktspotential für N1a in den Varianten P103VE um etwa +65 mV und M149NE um etwa +80 mV verschoben. Für die Variante P103V/M149NE wurde das N1a Mittenpunktspotential sogar um 160 mV auf −270 mV erhöht. Allerdings konnte N1a in den intakten Y. lipolytica Komplex I-Varianten EPR-spektroskopisch nicht nachgewiesen werden. Für den B. taurus Komplex I wurde gezeigt, dass am FMN hauptsächlich Superoxidradikale (O2•−) gebildet werden, die zu H2O2 weiterreagieren. Dies spricht dafür, dass die Elektronen einzeln auf O2 übertragen werden (Kussmaul und Hirst, 2006). Der E. coli Komplex I produziert hingegen hauptsächlich H2O2 als Nebenprodukt der NADH-Oxidation (Esterházy et al., 2008; Schulte, 2014). Das Zentrum N1a im E. coli Komplex I wird als Zwischenspeicher für das zweite von NADH auf FMN übertragene Elektron diskutiert (Sazanov, 2007). Um einen möglichen Zusammenhang zwischen der Fähigkeit von N1a, Elektronen aufzunehmen und der Art der gebildeten ROS-Spezies zu untersuchen, wurden die FMN-vermittelten Reaktionen des E. coli Komplex I sowie der Komplex I-Varianten V96PE, N142ME und V96P/N142ME bestimmt (Abb. 4.37). Die mit der NADH Oxidation verbundene Reduktion von Ferricyanid (FeCN), Hexaminruthenium-(III) (HAR) und 3-Acetylpyridinadenindinucleotid (APAD+) und die dabei entstehenden ROS wurden bestimmt. Die H2O2-Produktion wurde mit dem Amplex Red Assay und die Superoxid- Relative Reaktionsraten [%] Produktion mit dem Dihydroethidium/DNA Assay bestimmt (Esterházy et al., 2008). Abbildung 4.37: Relative Reaktionsraten der Varianten V96PE (grau), V96P/N142ME (dunkelgrau) zum ursprünglichen Komplex I (hellgrau). Die NADH/Ferricyanid, NADH/HAR und NADH/APAD+ Oxidoreduktaseaktivität und die während der NADH Oxidation entstehenden ROS wurden bestimmt (Birrell et al., 2013). 118 Ergebnisse Die Daten zeigen, dass die Varianten V96PE und V96P/N142ME nicht mehr O2•− produzieren als der ursprüngliche Komplex I. Somit kann ausgeschlossen werden, dass durch die Reduktion des N1a im ursprünglichen E. coli Komplex I bevorzugt H2O2 statt O2•− gebildet wird (Birrell et al., 2013). Allerdings ist die H2O2-Produktion der Variante V96PE um 20% und die der Variante V96P/N142ME um 60% im Vergleich zum ursprünglichen Komplex, erhöht. Diese Messung zeigt die funktionelle Bedeutung der Reduzierbarkeit von N1a, da seine Reduktion wichtig für den Schutz von E. coli vor einer erhöhten H2O2-Produktion ist. 4.7 Redox-induzierte Konformationsänderung des Glu95 auf NuoF In unserem Arbeitskreis wurde der A. aeolicus Subkomplex NuoEFHis mit gebundenem NAD+ bzw. NADH kristallisiert et al., 2008). Strukturvergleiche zeigten, dass das Peptidrückgrat von Glu95F je nach Redox-Zustand eine unterschiedliche Konformation einnimmt. Im reduzierten Zustand zeigt die Carbonyl-Gruppe vom FMN weg und ermöglicht dadurch die Bindung des NADH. In dieser Konformation verdrängt die Carbonyl-Gruppe ein H2O-Molekül und ersetzt zwei der drei von diesem Wasser gebildeten H-Brücken (Abb. 4.38). Die Carbonyl-Gruppe bildet eine H-Brücke mit der Seitenkette von Arg135F und eine mit einem anderen strukturell konservierten H2O-Molekül aus (Abb. 4.38 B). Im oxidierten Zustand zeigt Carbonyl-Gruppe der Peptidbindung des Glu95F zum FMN hin. Es wird vermutet, dass dieses Umklappen als „Auswurf-Mechanismus“ für das NAD+ dient. In dieser Arbeit sollte die Konformationsänderung der Peptidbindung zwischen von Glu95F und Ser96F genauer untersucht werden. Die Peptidbindung sollte durch Einführen von H-Brücken in die Konformation des reduzierten Proteins gezwungen werden. Hierfür sollte das Gly129E im A. aeolicus Subkomplex jeweils gegen Serin und Aspartat ausgetauscht werden. Gly129E befindet sich auf einer Schleife in der ersten Koordinationssphäre von N1a (Abb. 4.38). Durch die Mutationen sollte versucht werden die beiden H2O-Moleküle zu verdrängen. Diese H2OMoleküle bilden H-Brücken sowohl mit der N1a koordinierende Proteinschleife als auch mit der, auf der sich das Glu95F befindet. Die NuoEFHis-Varianten sollten isoliert und kristallisiert werden. Um die physiologischen Auswirkungen zu untersuchen, sollten die gleichen Mutationen an der homologen Position (Gly135E) in den E. coli Komplex I eingeführt werden. 119 Ergebnisse B) A) C127 T100 E G129 F F F E97 C127 E G129 E95 T100 E F E97 E F E95 F F F Y138 Y138 F R135 R135 F Abbildung 4.38: Konformationsänderung der Peptidbindung des Glu95 F. H-Brücken sind als gestrichelte Linien angedeutet. A) zeigt die Konformation von Glu95F in der Struktur des oxidierten Proteins. Das in der Struktur des reduzierten Proteins verdrängte H2O ist grün dargestellt. Das zweite konservierte H2O (rot) bildet vier H-Brücken aus, von denen die zu Cys127E und den Carbonyl-Gruppen von Thr100F und Glu97E auch in der Struktur des reduzierten Proteins erhalten bleiben B). In der Konformation des reduzierten Proteins B) sind die zwei von der Carbonyl-Gruppe von Glu95F gebildeten H-Brücken zu Arg135F und dem rot dargestellten H2O gezeigt. Die dritte H-Brücke zu Tyr138F geht verloren. Die Position des Gly129E auf der N1a koordinierende Schleife ist rot dargestellt. 4.7.1 Darstellung der A. aeolicus NuoEFHis-Varianten G129DE und G129SE Die Mutanten pETBlue-1nuoEFhis nuoE G129D und G129S wurden durch ortsgerichtete Mutagenese an dem Vektor pETBlue-1nuoEFhis mit den entsprechenden OligonukleotidPaaren (Tab. 3.4) dargestellt. Der Erfolg der Mutagenese wurde durch Restriktionsanalysen und DNA-Sequenzierungen überprüft. Zur Überproduktion der Varianten wurde der E. coli Stamm Rosetta(DE3) mit diesen Plasmiden transformiert. Durch Expressionstests wurden geeignete Klone selektiert. Diese wurden in NuoEFaxa-Medium im 10 L- bzw. im 400 mLMaßstab angezogen. Die Zellausbeute lag bei 2 – 3 g Zellen pro L Medium. Die Varianten wurden wie beschrieben isoliert. Die Elutionsprofile der chromatographischen Reinigungsschritte der Varianten waren sehr ähnlich, daher werden nur die von G129DE beispielhaft gezeigt (Abb. 4.39). Tabelle 4.15 gibt den Präparationsverlauf wieder und Tabelle 4.16 zeigt die Zusammenfassung der Präparationen. Die Reinheit der Varianten wurde durch SDS-PAGE analysiert (Abb. 4.40). 120 Ergebnisse A) B) C) D) Abbildung 4.39: Elutionsprofile der Präparation der NuoEFHis-Variante G129DE. A) zeigt das Elutionsprofile der Affinitäts-Chromatographie, B) das der ersten Größenausschluss-Chromatographie, C) das der Anionenaustausch-Chromatographie und D) das der abschließenden Größenausschluss-Chromatographie. Die Absorbanz bei 280 nm ist in Blau, die Imidazol- bzw. die NaCl-Konzentration in Grün und die NADH/Ferricyanid Oxidoreduktaseaktivität in Rot dargestellt. Tabelle 4.15: Präparationsverlauf der Variante G129DE aus 5g Zellen des Stamms Rosetta(DE3)/pETBlue-1nuoEFhis nuoE G129D. Präparationsschritt Cytosol Probond Ni2+-IDA Superdex 200 16/60 SQ15 Superdex 200 16/60 Volumen Protein [mL] 62 21 10 5 1.2 [mg] 742 110 40 21 13.5 NADH/Ferricyanid Oxidoreduktaseaktivität total spezifisch -1 [µmol·min ] [µmol·min-1·mg-1] 2283 3.1 1105 10.0 808 20.2 735 35.0 616 45.6 Ausbeute [%] 100 48 35 32 27 121 Ergebnisse Tabelle 4.16: Zusammenfassung der Präparation von NuoEFHis und der Varianten G129DE und G129SE. Präparation eingesetzte Zellen [g] Ausbeute Protein [mg] NADH/Ferricyanid Oxidoreduktaseaktivität [µmol·min−1·mg−1] normierte Aktivität [%] NuoEFHis G129DE G129SE 5.0 5.0 14.6 7.7 13.5 22.0 48.4 45.6 46.2 100 45 44 app. Mr [kDa] 1 2 M ← 66.2 ← 45.0 NuoFHis → ← 35.0 ← 25.0 ← 18.4 NuoE → ← 14.4 Abbildung 4.40: Acrylamidgel der Präparationen der Varianten G129DE (1) und G129SE (2). Spur M zeigt die Banden des Proteinstandards Unstained Protein Molecular Weight Marker (Thermo Fisher Scientific). Die Banden sind den Nuo-Untereinheiten anhand ihrer molekularen Masse zugeordnet. Die Ausbeute der Präparation der Variante G129DE war im Vergleich zu der von NuoEFHis deutlich erhöht. Es wurden 2.7 mg Protein pro g eingesetzter Zellen isoliert. Da es sich bei nuoEFhis nuoE G129D um ein heterolog exprimiertes Gen handelt, ist die höhere Proteinausbeute auf eine bessere Expression zurückzuführen. Die spezifische NADH/Ferricyanid Oxidoreduktaseaktivität beider Varianten entsprach in etwa der von NuoEFHis. Dies ist ein starker Hinweis darauf, dass die NADH Oxidation in den Varianten nicht gestört ist. 4.7.2 Strukturen der NuoEFHis-Varianten G129DE und G129SE Die Varianten G129DE und G129SE wurden unter den etablierten Citratbedingungen (Tab. 3.20) kristallisiert et al., 2008). Für die Variante G129SE wurden nach zwei bis drei Tagen in den Bedingungen 1 – 4, eine Vielzahl von Kristallen beobachtet (Abb. 4.41 A). Die Variante G129DE kristallisierte nur sehr schlecht ohne Keimkristalle. Um die Flexibilität des Proteins einzuschränken, wurde die Variante mit NAD+ kokristallisiert. Mit Keimkristallen und NAD+ wurden nach 3 – 4 Tagen unter den Bedingungen 1 – 3, mehrere streufähige Kristalle erhalten (Abb. 4.41 B). 122 Ergebnisse A) B) Abbildung 4.41: Proteinkristalle der NuoEFHis Varianten G129SE und G129DE. A) zeigt einen Kristall der Variante G129SE in 50 mM Tris/HCl (pH 7.0), 0.8 M Na3Citrat und 0.2 M NaCl. B) zeigt Kristalle der Variante G129DE mit 10 mM NAD+ in 50 mM Tris/HCl (pH 6.8), 0.6 M Na3Citrat und 0.2 M NaCl. Von Kristallen beider Varianten wurden in Zusammenarbeit mit Herrn Prof. Oliver Einsle und Herrn Dipl.-Chem. Sascha Jurkovic Datensätze aufgenommen und die Strukturen gelöst. Die Struktur der Variante G129DE wurde bei einer Auflösungsgrenze von 1.85 Å gelöst. Die Variante kristallisierte in der Raumgruppe P 1 21 1. Die Variante G129SE kristallisierte wie NuoEFHis in der Raumgruppe P 21 21 21 und die Auflösungsgrenze betrug 2.44 Å. Die zugehörigen kristallographischen Statistiken sind im Anhang 7.3 in den Tabellen 7.9 und 7.10 dargestellt. Die asymmetrische Einheit setzt sich aus zwei Monomeren zusammen, die sich für G129DE ausschließlich in den äußeren, flexiblen Regionen und G129SE an der Position Glu95F unterscheiden (Abb. 4.42). A) B) Abbildung 4.42: Überlagerung der Monomere der asymmetrischen Einheit der Kristalle von G129DE und G129SE. A) zeigt die Monomere von G129SE. Der Unterschied in der Glu95F tragenden Proteinschleife ist mit einem roten Kreis markiert. B) zeigt die Monomere von G129DE mit gebundener ADP-Ribose. 123 Ergebnisse In der Elektronendichtekarte der Variante G129DE ist der Nikotinamidrest des NAD+ nicht aufgelöst. In beiden Monomeren der asymmetrischen Einheit ist die Esterbindung der ADPRibose zum Pyrophosphat verdreht. Dies ist ein Indiz dafür, dass das Nikotinamid durch Hydrolyse abgespalten wurde. Die Tertiär- und Quartärstrukturen der Varianten unterscheiden sich kaum von denen des NuoEFHis (Abb. 4.43). A) B) C) D) Abbildung 4.43: Überlagerung der Monomere der asymmetrischen Einheit der Varianten G129SE und G129DE mit NuoEFHis. A) und B) zeigen die Überlagerung der einzelnen Monomere von G129SE mit der Struktur des oxidierten NuoEFHis (grau). C) zeigt die Überlagerung eines Monomers von G129DE mit der Struktur des oxidierten NuoEFHis (grau) und D) die des selben Monomers mit der Struktur des reduzierten NuoEFHis (hellblau). Die Position von Glu95F ist mit einem roten Kreis markiert. Da die einzelnen Monomere der asymmetrischen Einheit der Variante G129SE sich an der Position des Glu95F unterscheiden, wurden diese einzeln mit der Struktur des oxidierten 124 Ergebnisse NuoEFHis überlagert. Es ist deutlich zu erkennen, dass eines der Monomere in der Konformation des oxidierten und das andere in der des reduzierten NuoEFHis vorliegt (Abb. 4.43 A und B). Dies deutet darauf hin, dass die Mutation Gly129SerE die Carbonyl-Gruppe des Glu95F zum Teil in die Konformation des reduzierten Proteins zwingt, aber nicht vollständig fixiert. Die Monomere der asymmetrischen Einheit der Variante G129DF zeigen keine großen Unterschiede. Daher wurde nur ein Monomer jeweils mit den Strukturen des NuoEFHis im oxidierten und reduzierten Zustand überlagert. Die Glu95F-tragende Proteinschleife befindet sich in derselben Position wie in der Struktur des NuoEFHis im reduzierten Zustand (Abb. 4.43 D). Der B-Faktor der Carbonyl-Gruppe des Glu95F beträgt 19.0 Å2 bei einem durchschnittlichen B-Faktor von 29.7 Å2. Der niedrige B-Faktor des Peptid-Sauerstoffs von Glu95F in der Variante G129DE weist auf eine geringe Flexibilität an dieser Position hin. In beiden Varianten wurden die verbrückenden H2O-Moleküle verdrängt (Abb. 4.44; vgl. Abb. 4.39). B) A) F T100 C) F T100 F E97 F T100 F E97 E D129 E E S129 S129 E95 F F E95 F Y138 R135 E95 F F F Y138 F F E97 Y138 F R135 R135 F Abbildung 4.44: Konformation des Peptidrückgrats von Glu95F in den Varianten G129DE A) und den Monomeren der asymmetrischen Einheit von G129SE B) und C). A) zeigt das Rückgrat von G129DE. Der Abstand zwischen der Seitenkette des Asp129E und der Carbonyl-Gruppe des Glu95F ist in Ångström angegeben. Ein Monomer der asymmetrischen Einheit von G129SE liegt in der Konformation des reduzierten (B) und eines in der des oxidierten NuoEFHis (C) vor. In B) ist die Länge der H-Brücke zwischen Ser129E und der Carbonyl-Gruppe von Glu95F in Ångström angegeben. In der Struktur der Variante G129DE muss beachtet werden, dass das Asp129E protoniert sein muss, um ein H-Brücke zur Carbonyl-Gruppe des Glu95F ausbilden zu können. Die Mutationen zeigten den gewünschten Effekt, allerdings in unterschiedlicher Ausprägung. Die Peptidbindung des Glu95F befindet in der Variante G129DE komplett und in G129SE zum Teil 125 Ergebnisse in der Konformation des reduzierten NuoEFHis. Zudem wurden beide verbrückenden H2OMoleküle durch die jeweils eingeführten Seitenketten verdrängt. Die unterschiedliche Orientierung der Carbonyl-Gruppe des Glu95F in den einzelnen Monomeren der asymmetrischen Einheit von G129SE weist auf eine eingeschränkte aber dennoch vorhandenen Flexibilität dieser Position hin. Der niedrige B-Faktor des Peptidrückgrats von Glu95F in der Variante G129DE hingegen deutet darauf hin, dass die Carbonyl-Gruppe in der Konformation des reduzierten NuoEFHis fixiert ist. 4.7.3 Darstellung der Komplex I-Varianten G135DE und G135SE Der Subklon pCA24NnuoE diente als Vorlage-DNA für die ortsgerichtete Mutagenese mit den Oligonukleotid-Paaren nuoE G135D_fwd/rev und nuoE G135S_fwd/rev (Tab. 3.4). Die Anlagerungstemperatur betrug 55 °C für 30 s. Die Plasmide einzelner Klone wurden isoliert und der Erfolg der Mutagenese durch Restriktionsanalyse und Sequenzierung untersucht. Um die Rekombinationseffizienz zu erhöhen, wurden die Subklone mit den Mutationen mit BamHI geschnitten und das Fragment mit der Mutation aus einem Agarosegel isoliert. Die linearen Fragmente wurden mittels PCR an den isolierten Fragmenten mit dem Oligonukleotid-Paar nuoE_fwd/rev (Tab. 3.6) amplifiziert. Hier betrug die Anlagerungstemperatur 60 °C für 30 sec. Zur λ-Red vermittelte Rekombination wurden 50 ng pBADnuo nuoFhis nuoE::nptI-sacB (Dörner, 2010) und 100 – 450 ng der linearen DNAFragmente eingesetzt. Die Mutationen wurden durch Restriktionsanalyse und DNASequenzierung überprüft. Zur Überproduktion der E. coli Komplex I-Varianten wurde der Stamm BW25113nptI Δnuo Δndh mit den Plasmiden pBADnuo nuoFhis nuoE G135D und G135S transformiert und auf LB-Agar-Platten mit Chloramphenicol und Kanamycin selektiert. Vor der Zellzucht wurden die Plasmide aus den Expressionsstämmen isoliert und zur Kontrolle mittels Restriktion analysiert. Die Mutanten wurden in Autoinduktionsmedium im 10 L- und 400 mL-Maßstab angezogen. Die Cytoplasmamembranen wurden wie beschrieben präpariert und deren Komplex I-vermittelte Oxidaseaktivität bestimmt (Tab. 4.17). 126 Ergebnisse Tabelle 4.17: NADH/Ferricyanid Oxidoreduktase- und NADH-Oxidaseaktivität der Membranen des Ausgangsstamms und der NuoE-Mutanten. BW25113nptI Δnuo Δndh/pBADnuo nuoFhis NADH/Ferricyanid Oxidoreduktaseaktivität Restaktivität NADH-Oxidase Aktivität Restaktivität [µmol·min−1·mg−1] [%] [µmol·min−1·mg−1] [%] unverändert 1.6 ± 0.1 100 0.25 ± 0.02 100 nuoE G135S 1.3 ± 0.1 81 ± 8 0.12 ± 0.02 48 ± 8 nuoE G135D 1.1 ± 0.1 69 ± 8 0.0 0 Die NADH/Ferricyanid Oxidoreduktaseaktivität der Membranen der Gly135SerE-Mutante waren um etwa ein Fünftel und die der Gly135AspE-Mutante um ein Drittel verringert. Jedoch liegen beide Werte im Rahmen ihrer Fehler nahe beieinander. Die NADH-Oxidaseaktivität der Gly135SerE-Mutante war im Vergleich zu der des Ausgangsstamms halbiert. Die Membranen des Stamms BW25113nptI Δnuo Δndh/pBADnuo nuoFhis nuoE G135D besaßen keine nachweisbare NADH-Oxidaseaktivität. Diese Messungen geben einen ersten Hinweis auf die mechanistische Relevanz der konformativen Flexibilität des Peptidrückgrats von Glu95F. Während die Menge des Enzyms in der Membran durch die Mutation nur unwesentlich beeinflusst wird, scheint die Mutation Gly135AspE die physiologische Aktivität vollständig zu blockieren. Aus den Membranen wurden die Varianten wie beschrieben isoliert. Die Elutionsprofile unterscheiden sich nicht von den anderer Präparationen und werden daher nicht gezeigt. Beispielhaft ist Verlauf der Präparation von G135DE in Tabelle 4.18 aufgeführt. Tabelle 4.19 gibt die Zusammenfassung der Präparationen wieder. Die Reinheit der Proben wurde durch SDS-PAGE analysiert (Abb. 4.45). Tabelle 4.18: Präparation der Variante G135DE aus 42 g Zellen des Stamms BW25113nptI Δndh Δnuo/pBADnuo nuoFhis nuoE G135D. NADH/Ferricyanid Oxidoreduktaseaktivität total spezifisch Volumen Protein [mL] [mg] [µmol·min-1] [µmol·min-1·mg-1] [%] Membranen 28.7 1828 2011 1.1 100 Detergenzextrakt 125.0 1409 1690 1.2 84 Fractogel EMD 60.0 358 1017 3.0 51 0.7 7.0 434 62.0 22 Präparationsschritt 2+ ProBond Ni -IDA Ausbeute 127 Ergebnisse Tabelle 4.19: Zusammenfassung der Präparationen des Komplex I und der Varianten aus den Stämmen BW25113nptI Δnuo Δndh/pBADnuo nuoFhis, nuoE G135D und nuoE G135S. Präparation Komplex I eingesetzte Zellen Protein NADH/Ferricyanid Oxidoreduktaseaktivität Ausbeute aus Gesamtaktivität [g] [mg] [µmol·min−1·mg−1] [%] 35.0 6.0 60.0 20 E G135D 42.0 7.0 62.0 22 G135SE 31.0 4.3 61.0 18 1 M 116.0 2 ← Nu G 66.2 ← Nu CD 45.0 ← Nu F ← NuoL ← NuoM 35.0 ← NuoB } NuoI/J/E 25.0 18.4 Abbildung 4.45: Polyacrylamidgel der Präparationen der Komplex I NuoE-Varianten. Spur M zeigt die Banden des Proteinstandards Unstained Protein Molecular Weight Marker (Thermo Scientific). Spur 1 zeigt die Banden der Präparation der Variante G135SE und Spur 2 die der Variante G129DE. Von den Varianten wurden etwa 50 µg aufgetragen. Die hydrophoben Banden waren aufgrund der geringen Anfärbung kaum zu erkennen. Die NADH/Ferricyanid Oxidoreduktaseaktivität beider Varianten ist unverändert zu der des ursprünglichen Komplex I. Die genaue Enzymkinetik ist der Dissertation von Herrn Dipl.Chem. Marius Schulte zu entnehmen. Die kinetischen Parameter der NADH:Decyl-Ubichinon Oxidoreduktaseaktivität der Varianten wurden in Zusammenarbeit mit Marius Schulte bestimmt. Für die Variante G135SE ergab sich ein vmax von 2.7 ± 0.2 µmol·min−1·mg−1 und ein Km von 13.6 ± 1.0 µM. Für die Variante G135DE konnten keine kinetischen Parameter bestimmt werden, da die Aktivität mit der Zeit abnahm (Schulte, 2014). Dies deutet auf eine inhibierende Wirkung des entstehenden NAD+ auf die Variante hin und unterstützt die Hypothese der Umorientierung des Peptidrückgrats von Glu95F als eine Art „Auswurf-Mechanismus“. Eine Inhibierung mit 128 Ergebnisse NAD+ konnte nicht gemessen werden, da der Zusatz von NAD+ zu einer Aktivierung führte. Das gleiche wurde auch für die Variante G135SE und den Komplex I beobachtet. Zur Kontrolle wurden die einzelnen Strukturelemente des NAD+ (AMP, ADP-Ribose, NMN, Nikotinamid) zu den Messungen gegeben. Diese hatten aber keinen Effekt auf die Aktivität. Durch Messungen unter anoxischen Bedingungen wurde ein mögliche Elektronenübertragung auf Sauerstoff ausgeschlossen. Daher ist die Aktivierung sehr wahrscheinlich auf Verunreinigungen des NAD+ mit Metallionen zurückzuführen (Schulte, 2014). Die NADH/HAR Oxidoreduktaseaktivität der Variante G135SE war im Vergleich zu der des Komplex I um 18%, die der Variante G135DE um 87% vermindert. Die NADH/Ferricyanid Oxidoreduktaseaktivität der Varianten ist im Vergleich zu der des Komplex I unverändert (Schulte, 2014). Der negativ geladene [Fe(CN)6]3−-Komplex erhält die Elektronen direkt vom reduzierten FMN, in dem er an die Position des NADH bindet. Dagegen bildet der positiv geladene [Ru(NH3)6]3+-Komplex einen ternären Komplex aus reduziertem FMN und gebundenem Nukleotid, um reduziert werden zu können. Hierfür bindet HAR vermutlich an das Pyrophosphat des NAD+(H) (Birrell et al., 2011; Birrell und Hirst, 2013). Da die NADH/Ferricyanid Oxidoreduktaseaktivität durch die eingeführten Mutationen nicht beeinflusst ist, ist die NADH Oxidation durch die Mutationen nicht beeinträchtigt. Es ist aber davon auszugehen, dass die Mutationen die Verweildauer des NAD+ im Komplex I erhöhen, da die NADH/HAR Oxidoreduktaseaktivität vor allem die der Variante G135DE stark verringert ist. Die Position von gebundenem NADH und NAD+ unterscheiden sich, so dass das über die Pyrophosphat-Gruppe gebundene Ru3+ sich nicht mehr in einer für einen schnellen Elektronentransfer benötigten Entfernung zum reduzierten FMN befindet. Um zu untersuchen ob die Fe/S-Zentren der Variante G135DE durch die Zugabe von NADH reduziert werden, wurde die Variante EPR-spektroskopisch untersucht. Abb. 4.46 zeigt die EPR-Spektren der mit NADH bzw. Dithionit reduzierten Variante G135DE. Die EPRspektroskopischen Untersuchungen wurden in Zusammenarbeit mit Prof. Thorsten Friedrich durchgeführt. Das Spektrum der mit Dithionit reduzierten Variante G135DE zeigt, dass alle detektierbaren Fe/S-Zentren stöchiometrisch vorhanden sind. Dem Spektrum der NADH reduzierten Variante ist zu entnehmen, dass die Fe/S-Zentren nicht vollständig reduziert sind. Dies ist wahrscheinlich auf die sehr geringe Aktivität der Variante zurückzuführen. 129 Ergebnisse N2 N7 N3 N4 N1a A) N1b N3 N4 N2 B) C) 300 320 340 360 380 Magnetische Flussdichte [mT] Abbildung 4.46: EPR-Spektren der Variante G135DE und des Komplex I bei 13 K. A) zeigt das Spektrum des mit Dithionit reduzierten Komplex I, B) das der mit NADH und c) das der mit Dithionit reduzierten Variante G135DE. Die Mikrowellenfrequenz betrug 9.36 GHz, die Modulationsamplitude 0.6 mT, die Zeitkonstante 0.164 s und die Aufnahmegeschwindigkeit 17.9 mT/min. Die ROS-Produktion der Variante G135DE wurde wie beschrieben mit dem Radikalfänger DEPMPO untersucht. Hierfür wurde die Variante und der Komplex I in E. coli polare Lipide rekonstituiert und mit 100 mM DEPMPO sowie 1 mM NADH versetzt. Die Reaktion wurde durch Zugabe von 100 µM Decyl-Ubichinon gestartet. Um zu untersuchen, ob die oben beschriebene Aktivierung der NADH:Decyl-Ubichinon Oxidoreduktaseaktivität durch NAD+ eine Erhöhung der ROS-Produktion zur Folge hat, wurde einem weiteren Ansatz der Variante 130 Ergebnisse G135DE NAD+ (2.7 mM) zugesetzt. Zur Kontrolle wurde der Fenton-Test mit FeSO4 (2 mM) und H2O2 (1%) eingesetzt. Abbildung 4.47 zeigt die gemessenen Spektren. A) B) C) D) 325 330 335 340 Magnetische Flussdichte [mT] Abbildung 4.47: EPR-spektroskopischer Nachweis der ROS-Produktion durch die Variante G135DE. A) zeigt die Signale des DEPMPO-OH Addukts, das durch die Hydroxylradikale der Fenton-Reaktion gebildet wurde. Die Variante G135DE und der ursprüngliche Komplex I wurden in E. coli polare Lipide rekonstituiert und mit 100 mM DEPMPO, 100 µM Decyl-Ubichinon sowie mit 1 mM NADH inkubiert. B) zeigt das Signal für Komplex I, C) das der Variante und D) das der Variante die zusätzlich mit 2.7 mM NAD+ inkubiert wurde. Für die Variante wurden keine erhöhte Radikal-Produktion detektiert. Die Spektren wurden bei Raumtemperatur aufgenommen. Die Mikrowellenfrequenz betrug 9.36 GHz, die Modulationsamplitude 0.1 mT, die Zeitkonstante 0.082 s und die Aufnahmegeschwindigkeit 5.4 mT/min. Mit der Variante G135DE konnte mit DEPMPO keine erhöhte Radikalproduktion nachgewiesen werden. Auch die Zugabe von NAD+ hatte keine ROS-Produktion zur Folge. Herr Dipl.-Chem. Marius Schulte konnte bei der quantitativen ROS-Bestimmung mit Hilfe 131 Ergebnisse des Amplex Red Assays ebenfalls keine erhöhte H2O2-Produktion für diese Variante beobachten. Die Variante produziert pro umgesetztem NADH genauso viel H2O2 wie der Komplex I (Schulte, 2014). 4.8 Die Oberflächen-Salzbrücke in NuoG Sequenz- und Strukturanalysen haben gezeigt, dass der C-Terminus von NuoG homolog zu der katalytischen Untereinheit NarG der Chinol:Nitrat Oxidoreduktase ist (Chow et al., 1991; Finel, 1998; Rothery et al., 2008). Es wurde gezeigt, dass die in NarG befindlichen Kofaktoren, das Fe/S-Zentrum FS0 und der [Mo(MGD)2]-Kofaktor, von dem Chaperon NarJ in die offene Form der Untereinheit eingebaut werden (Lanciano et al., 2007). Die geschlossene Form wird von einer solvensexponierten Salzbrücke aus einem Arg/Glu-Paar stabilisiert. Diese Salzbrücke ist in der katalytischen Untereinheit aller [Mo(MGD)2]-Kofaktor tragenden Enzyme konserviert. Die Mutation einer der beiden Reste zu einem Alanin führt dazu, dass NarG zwar produziert wird, aber weder das Fe/S-Zentrum noch den [Mo(MGD)2]Kofaktor trägt (persönliche Mitteilung Prof. Axel Magalon, CNRS, Marseilles). NuoG besitzt ebenfalls eine Oberflächen-Salzbrücke, die von Arg280G und Glu617G gebildet wird. Diese ist ähnlich zu N7 positioniert, wie die Salzbrücke in NarG zu FS0 (Abb. 2.10). In dieser Arbeit sollte der Effekt der Deletion dieser Oberflächen-Salzbrücke auf die Assemblierung des Komplex I untersucht werden. Hierfür wurde das Glu617G durch ortsgerichtete Mutagenese und λ-Red vermittelte Rekombination gegen Alanin ausgetauscht und die Assemblierung der Variante durch Saccharosedichtegradienten-Zentrifugation untersucht. 4.8.1 Darstellung der Mutante pBADnuo nuoFhis nuoG E617A Der Subklon pUC25nuoE´–G diente als Vorlage-DNA für die ortsgerichtete Mutagenese mit dem Oligonukleotid-Paar nuoG E617A_fwd/rev (Tab. 3.4). Der Subklon pUC25nuoE´– G E617A wurde wie oben beschrieben dargestellt. Die Anlagerungstemperatur bei der PCR betrug 55 °C. Der Erfolg der Mutagenese wurde durch Restriktionsanalyse und Sequenzierung überprüft. Das lineare DNA-Fragment lin_nuoG E617A wurde von dem Subklon mit Mutation mit dem Oligonukleotiden nuoGbeg_fwd und nuoGrec_rev (Tab. 3.6) bei 132 Ergebnisse einer Anlagerungstemperatur von 60 °C amplifiziert. Für die λ-Red vermittelte Rekombination wurden 50 ng des Plasmids pBADnuoFhis nuoGc::nptI-sacB und 200 ng lin_nuoG E617A verwendet. Es wurden einzelne Klone selektiert, die Plasmide isoliert und der Erfolg der Rekombination durch Restriktionsanalyse und Sequenzierung überprüft. 4.8.2 Die Stabilität der Variante E617AG Zur Überproduktion der Variante E617AG wurde der E. coli Stamm BW25113Δnuo verwendet. Die Zellen wurden in 10 L Autoinduktionsmedium angezogen und vor Erreichen der stationären Phase durch Zentrifugation geerntet (Abb. 4.48). Abbildung 4.48: Wachstumskurve der Mutante BW25113Δnuo/pBADnuo nuoFhis nuoG E617A in 10 L Autoinduktionsmedium. Aus 10 L Medium wurden 65 g Zellen erhalten. Die Membranen aus 5 g Zellen der Mutante und des Stamms BW25113Δnuo/pBADnuo nuoFhis wurden wie beschrieben isoliert. Aus 1.5 mL Membransuspension wurden die Proteine durch Zugabe von 3% (w/v) DDM gelöst. Der Extrakt wurde durch Zentrifugation (150´000×g, 30 Psi, 25 min, Rotor A-100, Airfuge, Beckmann) von unlöslichen Bestandteilen befreit. 400 µL des Extrakts wurden auf einen Saccharosegradienten (12 mL, 5 – 30% (w/v) Saccharose in A*DDM-Puffer mit 50 µM PMSF) aufgetragen und zentrifugiert. Die Gradienten wurden fraktioniert und die NADH/Ferricyanid Oxidoreduktaseaktivität der Fraktionen bestimmt (Abb. 4.49). 133 Ergebnisse Abbildung 4.49: NADH/Ferricyanid Oxidoreduktaseaktivität der Fraktionen des Saccharosedichtegradienten. Die Aktivitäten des Extrakts aus der Mutante BW25113Δnuo/pBADnuo nuoFhis nuoG E617A (rot) und des Ausgangsstamms BW25113Δnuo/pBADnuo nuoFhis (schwarz) sind dargestellt. Die Hauptaktivität der Ferricyanid-Redoxaktivität der Fraktionen des Saccharosegradienten, der mit dem Extrakt des Ausgangsstamms beladen war, lag in den für Komplex I typischen Fraktionen 12 – 16. Für die Mutante wurden die Aktivitätsmaxima in den für das NADHDehydrogenasefragment typischen Fraktionen 8 – 11 detektiert. In den Fraktionen 12 – 16 wurde ebenfalls Aktivität gemessen, diese war aber im Vergleich zum Ausgangsstamm deutlich verringert. Die Aktivitätsverteilung über den Saccharosegradienten zeigt, dass die Variante bei der Extraktion mit Detergenz zerfällt. Der Versuch, die Variante E617AE zu isolieren, schlug fehl. Während der Affinitätschromatographie eluierten bei 260 mM Imidazol keine Proteine. Bei 380 mM Imidazol wurde ein kleines Absorptionsmaximum erhalten. Eine SDS-PAGE zeigte zwar einige Banden, die möglicherweise Untereinheiten des Komplex I darstellen. Zudem wies das Polyacrylamidgel aber sehr starke Verunreinigungen auf (Daten nicht gezeigt). Um die Komplex I-vermittelte Aktivität in cytoplasmatischen Membranen zu bestimmen wurde der E. coli Stamm BW25113nptI Δnuo Δndh mit dem Plasmid transformiert und in Autoinduktionsmedium im 400 mL-Maßstab angezogen. Die Membranen wurden wie beschrieben isoliert und die Komplex I-vermittelte NADH/Ferricyanid Oxidoreduktase- und NADH-Oxidaseaktivität bestimmt (Tab. 4.20). 134 Ergebnisse Tabelle 4.20: NADH/Ferricyanid Oxidoreduktase- und NADH-Oxidaseaktivität der Membranen des Ausgangsstamms und der NuoG-Mutante. BW25113nptI Δnuo Δndh/pBADnuo nuoFhis NADH/Ferricyanid Oxidoreduktaseaktivität Restaktivität NADH Oxidase Aktivität Restaktivität [µmol·min−1·mg−1] [%] [µmol·min−1·mg−1] [%] unverändert 1.4 ± 0.1 100 0.21 ± 0.02 100 nuoG E617A 1.0 ± 0.1 71 ± 7 0.14 ± 0.02 67 ± 14 Die Aktivitäten der Membranen der Mutante waren um etwa ein Drittel geringer als die der Membranen des Ausgangsstamms. Um abzuschätzen, ob dies durch die Mutation oder eine geringere Proteinproduktion bedingt war, wurde der Gehalt der Membranen an NuoF über eine Western Blot-Analyse bestimmt. Hierfür wurden 100 µg der Membranproteine des Ausgangsstamms und der Mutante durch eine SDS-PAGE aufgetrennt und anschließend auf eine PVDF-Membran transferiert. Zur Detektion wurden Mouse Anti-His4 primäre Antikörper gegen das am N-Terminus von NuoF fusionierte His6-Affinitätspeptid verwendet (Abb. 4.50). 1 M 2 70 NuoFHis → 55 35 25 15 10 Abbildung 4.50: Western Blot der Membranproteine der Mutante BW25113nptI Δnuo Δndh/pBADnuo nuoFhis nuoG E617A (1) und des Ausgangsstamms (2). Spur M zeigt die Banden des Proteinstandards PageRuler Plus Prestained Protein Ladder (Thermo Scientific). Die apparenten Molekülmassen sind in kDa angegeben. In beiden Spuren ist eine Bande bei 55 kDa zu erkennen, deren Masse der His6-Affinitäspeptid fusionierten Untereinheit NuoF entspricht. Auf der PVDF-Membran war sowohl für die Membranen der Mutante als auch für die des Ausgangsstamms eine Bande bei etwa 55 kDa nachweisbar, die der Untereinheit His6-NuoF 135 Ergebnisse entspricht. Zwar kann die Produktion des Komplexes auf diese Weise nicht quantifiziert werden, aber es ist deutlich zu erkennen, dass die Bande von NuoF für den Ausgangsstamm deutlich intensiver als für die Mutante Glu617AlaE ist. Daher sind die verringerten Aktivitäten der Membranen der Mutante vermutlich auf eine geringere Proteinproduktion zurückzuführen. Zusammenfassend kann gesagt werden, dass die Variante E617AE in der Membran wohl noch stabil und aktiv ist, aber durch die Extraktion zerfällt. In vorangegangen Arbeiten wurde gezeigt, dass der Verlust von N7 zum Zerfall von Komplex I bei der Extraktion aus der Membran führt (Pohl et al., 2007b). Deshalb ist zu vermuten, dass durch die Deletion der Oberflächen-Salzbrücke N7 möglicherweise verloren geht. 4.9 Zuordnung des EPR-Signals N4 Der E. coli Komplex I trägt neun Fe/S-Zentren von denen nur N1a, N1b, N2, N3, N4 und N7 (Nomenklatur nach Ohnishi, 1998) EPR-spektroskopisch nachweisbar sind (Leif et al., 1995; Uhlmann und Friedrich, 2005; Pohl et al., 2007b). Die Zentren N5, N6a und N6b wurden im E. coli Komplex I bislang EPR-spektroskopisch nicht nachgewiesen. Die detektierbaren EPRSignale wurden durch die Charakterisierung von einzeln produzierten Untereinheiten und durch Mutagenese-Studien den jeweiligen Nuo-Untereinheiten zugeordnet (Ohnishi, 1998). Tabelle 4.21 gibt die im E. coli Komplex I durch EPR-Spektroskopie nachgewiesenen Fe/SZentren wieder. Tabelle 4.21: EPR-spektroskopisch detektierbare Fe/S-Zentren und die dazugehörigen Untereinheiten des E. coli Komplex I (Leif et al., 1995; Uhlmann und Friedrich, 2005; Pohl et al., 2007b). Feldposition Fe/SZentrum Art des Fe/SZentrums koordinierende Untereinheit gx gy N1a [2Fe-2S] NuoE 1.92 1.96 2.00 N1b [2Fe-2S] NuoG 1.94 1.94 2.03 N2 [4Fe-4S] NuoB 1.91 1.91 2.05 N3 [4Fe-4S] NuoF 1.88 1.92 2.04 N4 [4Fe-4S] NuoG 1.89 1.93 2.09 N7 [4Fe-4S] NuoG 1.89 1.95 2.05 gz Die Fe/S-Zentren N6a und N6b wurden über UV/Vis-Spektroskopie im HydrogenaseFragment bestehend aus NuoB, CD und I nachgewiesen (Rasmussen et al., 2001). Das 136 Ergebnisse Zentrum N5 wurde in einem anderen, biochemisch nicht scharf definierten Fragment des Komplex I nachgewiesen (Nakamaru-Ogiso et al., 2008). Mit der Veröffentlichung der Struktur des hydrophilen Arms des T. thermophilus Komplex I wurden die EPR-Signale den einzelnen strukturell definierten Fe/S-Zentren zugeordnet (Sazanov und Hinchliffe, 2006; Abb. 2.4). Seit einigen Jahren wird diese Zuordnung der EPRSignale diskutiert. 2007 veröffentlichte die Gruppe von Dr. Judy Hirst ein neues Modell zur Benennung der strukturell definierten Fe/S-Zentren (Yakovlev et al., 2007). Anhand von EPR-spektroskopischen Untersuchungen an der einzeln produzierten Untereinheit NuoG aus E. coli und durch den Vergleich mit den Spektren des gesamten Komplex I, wurde das Signal N4 einem der beiden auf NuoI lokalisierten Fe/S-Zentren N6a oder N6b zugeordnet. 2010 wurde dieses Modell anhand von double electron-electron-resonance (DEER) Experimenten am B. taurus Komplex I spezifiziert und das Signal N4 dem strukturell definierten Fe/S-Zentrum N6a zugeordnet (Roessler et al., 2010). In der Gruppe von Prof. Tomoko Ohnishi wurden die koordinierenden Cysteine des strukturell definierten Zentrums N4 gegen Alanine ausgetauscht. Um NuoG einzeln löslich zu erhalten, wurde diese Untereinheit mit einem Maltose-Bindeprotein (MBP) fusioniert. Durch den Vergleich der EPR-Spektren des MBP-NuoG und der ΔN4-Variante wurde wiederum gezeigt, dass das EPR-Signal N4 auch von dem strukturell definierten N4 erzeugt wird (Ohnishi und Nakamaru-Ogiso, 2008). Zudem gelang es dieser Gruppe in einem Fragment des E. coli Komplex I bestehend aus NuoCDEFG das schnell relaxierende EPR-Signal N5 bei 3 K und 5 mW zu detektieren. Alle koordinierenden Aminosäuren des strukturell definierten Fe/S-Zentrums N5 wurden gegen Alanine ausgetauscht. In der ΔN5-Variante wurde das EPRSignal N5 nicht mehr detektiert (Nakamaru-Ogiso et al., 2008; Ohnishi und Nakamaru-Ogiso, 2008). Allerdings ordneten Yakovlev et al. (2007) dieses Signal dem strukturell definierten Zentrum N4 zu. EPR-Experimente mit dem E. coli Komplex I zur Lokalisierung der EPR-Signale N4 und N5 wurden entweder mit der einzeln produzierten Untereinheit NuoG oder mit Fragmenten des Komplexes durchgeführt. Da aber neben dem pH-Wert und der Ionenstärke die chemische Umgebung der Fe/S-Zentren einen entscheidenden Einfluss auf die Position der EPR-Signale hat und alle EPR-Signale der Fe/S-Zentren des E. coli Komplex I, in einem sehr engen spektralen Bereich auftreten und überlappen, sollte der Ursprung des EPR-Signals N4 mit dem gesamten Komplex untersucht werden. Die koordinierenden Aminosäuren der strukturell definierten Fe/S-Zentren N4 und N5 sollten einzeln gegen Alanine ausgetauscht werden. Sabrina Burschel wurde im Rahmen ihrer Masterarbeit zu den Arbeiten zu Teilen dieses 137 Ergebnisse Kapitels angeleitet (Burschel, 2013). N4 wird von Cys153G, Cys156G, Cys159G sowie Cys203G und N5 von His101G, Cys105G, Cys108G sowie Cys114G koordiniert (Abb. 4.51). C114 C159 C203 C108 N5 C156 C105 N4 C153 H101 Abbildung 4.51: Die Koordination der strukturell definierten Fe/S-Zentren N4 und N5 (PDB-Eintrag: 3IAM; Berrisford und Sazanov, 2009). Gezeigt ist NuoG aus T. thermophilus mit der Nummerierung nach E. coli. N4 und N5 sind als Stabmodelle mit Fe in Rot und S in Gelb dargestellt. 4.9.1 Darstellung der N4- und N5-Mutanten Die Subklone mit den Mutationen wurden durch ortsgerichtete Mutagenese an dem Subklon pUC25nuoE´–nuoG mit der entsprechenden Oligonukleotid-Paaren (Tab. 3.4) darstellt. Die für die λ-Red vermittelte Rekombination benötigten linearen Fragmente wurden mit den Oligonukleotiden nuoGbeg_fwd und nuoGrec_rev (Tab. 3.6) mittels PCR amplifiziert. Für die Rekombination wurden je 50 ng des Plasmids pBADnuo nuoFhis nuoGc::nptI-sacB und 200 – 500 ng der linearen Fragmente verwendet. Die Insertion der Mutationen wurde durch Restriktionsanalyse und DNA-Sequenzierung untersucht. Der Stamm E. coli BW25113nptI Δnuo Δndh wurde zur Überproduktion der Varianten verwendet. Die Mutanten wurden in 10 L Autoinduktionsmedium angezogen. Die Mutationen hatten keinen Einfluss auf das Wachstum der Mutanten (Daten nicht gezeigt). Um zu untersuchen, ob die Mutationen den Verlust der Fe/S-Zentren bedingen und somit die Elektronentransfer-Kette unterbrochen ist, wurden die Cytoplasmamembranen der Mutanten isoliert und deren NADH/Ferricyanid Oxidoreduktaseaktivität und NADH-Oxidaseaktivität bestimmt (Tab. 4.22). 138 Ergebnisse Tabelle 4.22: NADH/Ferricyanid Oxidoreduktaseaktivität und NADH-Oxidaseaktivität der cytoplasmatischen Membranen der N4- und N5-Mutanten. BW25113nptI ∆nuo ∆ndh/ pBADnuo nuoFhis Ligand des Fe/SZentrums unverändert NADH/Ferricyanid NADHOxidoreduktaseaktivität Oxidaseaktivität Restaktivität Oxidase [µmol·min−1·mg−1] [%] 2.7 ± 0.2 0.21 ± 0.02 100 nuoG C153A N4 1.5 ± 0.2 0 0 nuoG C156A N4 2.0 ± 0.2 0.16 ± 0.02 76 ± 10 nuoG C159A N4 1.7 ± 0.2 0.01 ± 0.01 5±5 nuoG C203A N4 2.0 ± 0.2 0 0 nuoG H101A N5 2.4 ± 0.2 0 0 nuoG C105A N5 2.2 ± 0.2 0 0 nuoG C108A N5 2.1 ± 0.2 0.01 ± 0.01 5±5 nuoG C114A N5 2.0 ± 0.2 0 0 Die NADH/Ferricyanid Oxidoreduktaseaktivitäten der N4- und N5-Mutanten sind im Vergleich zum Ausgangsstamm teilweise bis zu 50% verringert. Keine der N5-Mutanten wies signifikante NADH-Oxidaseaktivität auf. Mit Ausnahme der Membranen der Mutante BW25113nptI Δnuo Δndh/pBADnuo nuoFhis nuoG C156A besaßen auch die N4-Mutanten keine signifikante NADH-Oxidaseaktivität. Die Mutation Cys156AlaG führte demnach nicht zum Verlust des strukturell definierten Fe/S-Zentrums N4. Der Elektronentransfer von NADH auf Ubichinon wäre sonst aufgrund des zu großen Abstands zwischen den benachbarten Zentren N1b zu N5, unterbrochen (Page et al., 1999). Um zu untersuchen, ob die Varianten überhaupt produziert wurden oder ob der Verlust von N4 bzw. N5 zu einer strukturellen Instabilität oder einer unvollständigen Assemblierung führt, wurden die Membranproteine der Mutanten mittels Western Blot analysiert. Es wurden jeweils 250 µg Membranproteine mittels SDS-PAGE aufgetrennt und anschließend auf eine PVDF-Membran transferiert. Zur Detektion wurden Mouse Anti-His4 primäre Antikörper gegen das N-terminal an NuoF fusionierte His6-Affinitätspeptid verwendet (Abb. 4.52). 139 Ergebnisse 1 2 3 4 5 M 6 7 8 9 70 NuoFHis → 55 35 25 15 Abbildung 4.52: Western Blot der Membranproteine aus den Stämmen BW25113nptI ∆nuo ∆ndh/pBADnuo nuoFhis (Spur 1), nuoG C114A (Spur 2), C108A (Spur 3), C105A (Spur 4), H101A (Spur 5), C203A (Spur 6), C159A (Spur 7), C156A (Spur 8) und C153A (Spur 9). In Spur M ist der PageRuler Plus Prestained Protein Ladder (Thermo Scientific) aufgetragen. Die apparenten Molekülmassen sind gezeigt und NuoFHis mit einem Pfeil gekennzeichnet. Auf der PVDF-Membran war in allen Spuren eine Bande bei etwa 55 kDa detektierbar, die der Untereinheit His6-NuoF zugeordnet wurde. Es kann somit ausgeschlossen werden, dass die Varianten nicht produziert wurden. Sollte die Assemblierung nicht gestört sein, sind die Varianten zumindest in der Membran stabil. Die Bande auf der PVDF-Membran bei etwa 40 kDa ist wahrscheinlich auf eine unspezifische Bindung des primären Antikörpers zurückzuführen. Diese ist auch in der Spur des Ausgangsstamms zu beobachten, wodurch ihr Auftreten als Folge der Mutationen ausgeschlossen wird. 4.9.2 Charakterisierung der N4- und N5-Varianten Die Varianten und der Komplex I wurden wie beschrieben isoliert. Tabelle 4.23 gibt die Ausbeuten und Aktivitäten der Varianten wieder. Die Reinheit und Zusammensetzung der Untereinheiten wurde mit SDS-PAGE analysiert (Abb. 4.53). Für die Variante C156AG sind alle Nuo-Untereinheiten eindeutig zu erkennen. Für die Varianten C153AG, C159AG und H101AG sind trotz starker Verunreinigungen dennoch einige Nuo-Untereinheiten zuordenbar. Die anderen N5-Varianten C105AG, C108AG und C114AG sowie die N4-Variante C203AG (Daten nicht gezeigt) zerfielen während der Präparation. 140 Ergebnisse Tabelle 4.23: Zusammenfassung der Präparationen des Komplex I und der N4- und N5-Varianten. Variante eingesetzte Zellen Protein NADH/Ferricyanid Oxidoreduktaseaktivität Ausbeute aus Gesamtaktivität [g] [mg] [µmol·min−1·mg−1] [%] Komplex I 28.0 2.8 49.4 7 G C153A 38.0 1.2 11.5 1 C156AG 30.0 2.3 37.5 8 C159AG 34.0 0.5 16.6 1 G 45.0 1.0 14.6 1 G H101A 30.0 1.2 40.1 2 C105AG 36.0 0.5 20.1 1 C108AG 30.0 0.5 76.3 3 C114AG 36.0 1.2 21.1 5 C203A App. Mr [kDa] M 1 2 3 4 5 6 7 116.0 → ← Nu G 66.2 → ← Nu CD 45.0 → ← Nu F 35.0 → ← NuoL, N ← NuoM 25.0 → 18.4 → ← NuoB } NuoI/J/E 14.4 → ← NuoA ← NuoK Abbildung 4.53: Polyacrylamidgel der isolierten N4- und N5-Varianten. Spur M zeigt das Bandenmuster des Unstained Protein Molecular Weight Marker (Thermo Scientific). Spur 1 zeigt die Banden der Variante C153A G, Spur 2 die der Variante C159AG, Spur 3 die der Variante H101AG. Spur 4 zeigt das Bandenmuster der Variante C105AG, Spur 5 das der Variante C108AG, Spur 6 das der Variante C114AG und Spur 7 zeigt das Bandenmuster der Variante C156AG. Die Banden der Präparation sind anhand ihrer apparenten Molekülmassen den entsprechenden Untereinheiten zugeordnet. Es wurde die NADH:Decyl-Ubichinon Oxidoreduktaseaktivität der einzelnen Varianten bestimmt. Die maximale Umsatzrate betrug 3.3 ± 0.2 µmol·min−1·mg−1 für den Komplex I und 2.6 ± 0.2 µmol·min−1·mg−1 für die Variante C156AG. Das entspricht 79% der des 141 Ergebnisse Komplex I und steht im Einklang mit den Messungen in der Membran. Die anderen N4- und N5-Varianten wiesen keine messbare NADH:Decyl-Ubichinon Oxidoreduktaseaktivität auf. Die Präparationen der Varianten und des Komplex I wurden mit NADH und Dithionit reduziert und bei 40 K und 5 mW sowie 12 K und 10 mW im X-Band EPR-spektroskopisch untersucht (Abb. 4.54). Die Varianten C203AG, C105AG, C108AG und C114AG erzeugten keine oder keine zuordenbaren Signale, daher werden die Spektren nicht gezeigt. A) B) C) D) E) Magnetische Flussdichte [mT] Magnetische Flussdichte [mT] Abbildung 4.54: EPR-Spektren des Komplex I A) und der N4-Varianten C153AG B), C156AG C), C159AG D) und der N5-Variante H101AG E) bei 12 K und 10 mW. Zum besseren Vergleich ist das Spektrum des Komplex I zweimal abgebildet Die Mikrowellenfrequenz betrug 9.36 GHz, die Modulationsamplitude 0.6 mT, die Zeitkonstante 0.082 s und die Aufnahmegeschwindigkeit 71.5 mT/min. Das EPR-Signal N4 ist in den Varianten C153AG, C156AG, C159AG und H101AG eindeutig nachzuweisen. Somit kann ausgeschlossen werden, dass das Signal N4 von den strukturell definierten Fe/S-Zentren N4 oder N5 erzeugt wird. Die Aktivität der Varianten bestätigen, 142 Ergebnisse dass in den Varianten C153AG, C159AG und H101AG die strukturell definierten Zentren N4 bzw. N5 nicht mehr vorliegen. Das 40 K Spektrum der Variante C156AG zeigt, dass das Signal des zweikernigen Fe/S-Zentrums N1b nur noch sehr schwach detektierbar ist. Dieses ist sehr wahrscheinlich nicht mehr oder nur teilweise gebunden (Abb. 4.55). A) B) Magnetische Flussdichte [mT] Abbildung 4.55: EPR-Spektren des Komplex I A) und der N4-Variante C156AG B), bei 40 K und 5 mW. Die Mikrowellenfrequenz betrug 9.36 GHz, die Modulationsamplitude 0.6 mT, die Zeitkonstante 0.082 s und die Aufnahmegeschwindigkeit 71.5 mT/min. Durch Sequenz- und Strukturvergleiche wurde Glu36G in der T. thermophilus Struktur als potentieller Ligand für das strukturell definierte Fe/S-Zentrum N4 in der Variante C156AG identifiziert. Im E. coli Komplex I befindet sich an der homologen Position ein Histidin (Abb. 4.56 B). Der Abstand zwischen N3 und N5 beträgt 13.8 Å, somit sollte auch ohne N1b der Elektronentransfer im Komplex I theoretisch möglich (Page et al., 1999; Sazanov, 2007). In dieser Arbeit konnte dies anhand der NADH-Oxidaseaktivität und der NADH:DecylUbichinon Oxidoreduktaseaktivität der Variante C156AG, deren Gehalt an N1b maximal 10% der des Komplex I beträgt, bestätigt werden. Anhand der hier gezeigten Daten ist es 143 Ergebnisse auszuschließen, dass das EPR-Signal N4 von einem der strukturell definierten Zentren N4 oder N5 erzeugt wird. Dies lässt vermuten, dass das über DEER-Experimente identifizierte Fe/S-Zentrum N6a, das EPR-Signal N4 erzeugt (Roessler et al., 2010). A) N4 C156 N1b E36 B) T. thermophilus E. coli MVRVKVNDRIVEVPPGTSVMDAVFHAGYDVPLFCSEKHLSPIGACRMC MATIHVDGKEYEVNGADNLLEACLSLGLDIPYFCWHPALGSVGACRQC *. ::*:.: ** . .:::* : * *:* ** . *..:**** * Abbildung 4.56: Koordination der Fe/S-Zentren N4 und N1b in NuoG (PDB-Eintrag: 3IAM; Berrisford und Sazanov, 2009). A) zeigt die Fe/S-Zentren N1b und N4 als Kalottenmodelle. Die Abstände der Cβ-Atome von Glu36 und Cys156 zu dem zu koordinierenden Fe-Ion des N4 ist angegeben. Es wurde die E. coli Nummerierung verwendet. B) zeigt den zugehörigen Ausschnitt des Sequenzvergleichs zwischen T. thermophilus Nqo3 und E. coli NuoG. Die Position 36 ist gelb unterlegt. 4.10 Mutationen der NADH-Bindestelle des A. aeolicus NuoEFHis Strukturvergleiche der NADH-Bindestelle von A. aeolicus NuoEFHis im reduzierten und oxidierten Zustand haben gezeigt, dass einigen Aminosäuren bei der Bindung von NADH eine besondere Rolle zukommt. In Zusammenarbeit mit M. Sc. Biochemie Karoline Aierstock und Dr. Lothar Kussmaul (Boehringer Ingelheim, Biberach) sollten die Positionen Glu95F, Glu97F und Tyr180F des A. aeolicus Subkomplexes NuoEF gegen verschiedene Aminosäuren ausgetauscht werden. 144 Ergebnisse Für das Glu95F wurde gezeigt, dass seine Seitenkette über verschiedene H-Brücken im oxidierten und reduzierten Zustand fixiert ist. Das Glu95F sollte gegen Asn, Asp und Gln ausgetauscht werden. Das Tyr180F und das Glu97F bilden im reduzierten Zustand eine H-Brücke, die der Stabilisierung der NADH-Bindung dient. Die Mutation der Tyr180F zu Cys ist in einem Patienten mit dem Leigh-like-Syndrom assoziiert (Bénit et al., 2001). Die Mutation des Glu97F gegen Gln im E. coli Komplex I bewirkte einen drastischen Aktivitätsverlust (Euro et al., 2009). Das Tyr180F sollte gegen Ala, Cys, Leu und Phe und das Glu97F gegen Asn, Asp und Gln ausgetauscht werden. Therese Engel wurde im Rahmen ihrer Zulassungsarbeit bei den Arbeiten zu Teilen dieses Kapitels angeleitet (Engel, 2010). 4.10.1 Darstellung der Mutanten Das Plasmid pETBlue-1nuoEFhis diente als Templat für die ortsgerichtete Mutagenese. Die Mutanten pETBlue-1nuoEFhis E95D/N/Q, pETBlue-1nuoEFhis E97D/N/Q und pETBlue-1 nuoEFhis Y180A/C/F/L wurden mit den entsprechenden Oligonukleotid-Paaren (Tab. 3.4) dargestellt. Der Erfolg der ortsgerichteten Mutagenese wurde durch Restriktionsanalyse und Sequenzierung untersucht. Es wurden entweder die isolierten Plasmide oder GlycerinDauerkulturen an M. Sc. Biochemie Karoline Aierstock zur Überproduktion und Charakterisierung der Varianten versandt. Die Ergebnisse der strukturellen und kinetischen Charakterisierung sind bei Böhringer Ingelheim noch in Arbeit. Die dargestellten Mutanten sind in Tabelle 4.24 aufgelistet. Tabelle 4.24: Dargestellte nuoF-Mutanten des A. aeolicus NuoEFHis. Dargestellte Plasmide pETBlue-1nuoEFhis pETBlue-1nuoEFhis E95D pETBlue-1nuoEFhis E95N pETBlue-1nuoEFhis E95Q pETBlue-1nuoEFhis E97D pETBlue-1nuoEFhis E97N pETBlue-1nuoEFhis E97Q pETBlue-1nuoEFhis Y180A pETBlue-1nuoEFhis Y180C pETBlue-1nuoEFhis Y180F pETBlue-1nuoEFhis Y180L 145 Diskussion 5 Diskussion 5.1 Die durch Glu183F vermittelten Substratselektivität In dieser Arbeit wurde Glu183F als Schüsselposition für die Unterscheidung zwischen den Substraten NADH und NADPH durch Komplex I identifiziert. Um die sterischen und/oder elektrostatischen Ansprüche dieser Position zu senken, wurde Glu183F im E. coli Komplex I gegen Ala, Asp, Asn, Gly und Gln ausgetauscht. Zudem wurde durch den Austausch gegen das positiv geladene His, eine elektrostatische Anziehung zur Phosphat-Gruppe des NADPHs eingebracht. Daten die mit der Variante E183HF erzeugt wurden, zeigten, dass die Oxidation von NADPH mit der Translokation von Protonen gekoppelt ist (Abb. 4.8). EPRspektroskopische Analysen zeigten, dass alle Fe/S-Zentren der Variante auch mit NADPH reduziert werden (Abb. 4.7). Alle Varianten zeigten eine deutlich höhere NADPH:DecylUbichinon Oxidoreduktaseaktivität als der Komplex I (Tab. 4.4). Die katalytischen Effizienzen der Varianten E183NF und E183DF sind am wenigsten erhöht, dennoch betrugen sie das 12- bzw. 50-fache des Komplex I. Die Varianten E183HF, E183GF und E183AF zeigen eine sogar 220-, 360- bzw. das 2400-fach erhöhte katalytische Effizienz. Die konservative Mutation zu Glutamin erhöhte die katalytische Effizienz 180-fach. Die elektrostatische Abstoßung der Phosphatgruppe des NADPHs durch die Glu183F-Seitenkette hatte somit einen größeren Effekt auf die Substratunterscheidung als der sterische Anspruch. Die stark erhöhten Aktivitäten der Varianten E183AF und E183GF sind wahrscheinlich auf lokale strukturelle Änderungen zurückzuführen. Außer bei den Varianten E183NF und E183GF waren auch die NADH:Decyl-Ubichinon Oxidoreduktaseaktivitäten im Vergleich zum Komplex I erhöht. Bemerkenswerterweise waren die katalytischen Effizienzen mit NADH als Substrat für die Varianten E183DF und E183HF um mehr als das Doppelte erhöht. Die H2O2-Produktion bei der Oxidation von NADPH durch die Varianten war, mit Ausnahme von E183DF, im Vergleich zu Komplex I erniedrigt (Tab. 4.5). Das zeigt die durch die Mutationen bedingte Anpassung der NADH-Bindestelle an das Substrat NADPH. Die höhere katalytische Effizienz der Varianten bei der Umsetzung von NADH war immer mit einer höheren H2O2-Produktion gekoppelt. Für eine effiziente Elektronenübertragung von NADH auf Ubichinon ist also ein Glutamat an Position 183 erforderlich. Da die kinetischen Daten keine Erklärung für die erhöhten ROS-Produktionen lieferten, wurden die homologen Mutationen (E185A/D/G/H/N und QF) zur strukturellen Analyse in den A. aeolicus Subkomplex NuoEFHis eingebracht. Die Varianten E185H/D/N und QF 146 Diskussion kristallisierten unter denselben Bedingungen wie NuoEFHis. Für die Variante E185AF mussten neue Kristallisationsbedingungen gefunden werden. Die Variante E185GF konnte nicht zur Kristallisation gebracht werden. Die Kristallstrukturen der Varianten E185HF und E185DF wurden bei 2.20 Å bzw. 2.40 Å gelöst (Abb. 4.21). Anhand dieser Strukturen konnte keine Erklärung für die erhöhte Aktivität und H2O2-Produktion gefunden werden. Die Struktur der Variante E185DF wurde zusätzlich mit gebundenem NAD+ bei 1.80 Å gelöst. Aus dieser Struktur wurde die modifizierte Anbindung des Nukleotids ersichtlich (Abb. 4.27). Die Adenosin-Ribose war um etwa 160° verkippt, um mit der kürzeren Seitenkette des Asp185F eine H-Brücke zu bilden. Die zweite von Glu185F gebildete H-Brücke zur Adenosin-Ribose war in der Variante nicht mehr vorhanden, wodurch die Anbindung der Phosphat-Gruppe des NADPHs ermöglicht wurde. Zudem wurde die H-Brücke von Lys76F zum O3' der AdenosinRibose geschwächt. Der Verlust beider H-Brücken schwächt die Anbindung des NAD+, dadurch kann es leichter aus der Bindetasche gelöst werden, was die erhöhte Aktivität der E. coli Variante mit NADH erklärt. In allen drei Strukturen war interessanterweise das Peptidrückgrat des Glu95F in der Konformation des reduzierten Proteins. Die B-Faktoren zeigten jedoch, dass diese Position flexibel ist (Abb. 4.22). Da durch die Mutationen keine drastischen strukturellen Änderungen zu erkennen waren, ist die erhöhte ROS-Produktion der Varianten auf die veränderte Substratbindung zurückzuführen. Die Tatsache, dass alle Varianten eine unterschiedlich stark erhöhte ROSProduktion aufweisen, lässt darauf schließen, dass NADH in allen Varianten unterschiedlich gebunden wird. Der Querschnitt durch die Oberflächendarstellung von E185DF mit gebundenem NAD+ zeigt, dass der Boden und das Innere der NADH-Bindestelle nicht vollständig vom Nukleotid abgeschirmt werden (Abb. 4.28). Dadurch kann Sauerstoff in die Bindetasche diffundieren und am reduzierten FMN zu H2O2 reduziert werden, was die erhöhte ROS-Produktion der E. coli Komplex I-Varianten erklären würde. Um diese Hypothese zu untersuchen, könnten in weiterführenden Arbeiten Kristalle mit Xenon, das in Größe und Polarität molekularem Sauerstoff ähnelt, in einer Xenon-Druckkammer begast werden. Das Xenon diffundiert in die für Sauerstoff zugänglichen Bereiche und kann dann röntgendiffraktometrisch detektiert werden. 147 Diskussion 5.2 Die Position Tyr178F im E. coli Komplex I Die im Genom eines am Leigh-like-Syndrom erkrankten Patienten detektierte Mutation Tyr204Cys auf der Untereinheit NDUFV1 (Homolog von NuoF) wurde mit dem bakteriellen Komplex I als Modellsystem untersucht. Strukturelle Analysen mit dem A. aeolicus NuoEFHis im reduzierten und oxidierten Zustand zeigen, dass die homologe Position Tyr178F (Nomenklatur nach E. coli) durch eine H-Brücke mit Glu95F die Bindung des NADH stabilisiert. Der Verlust der für die H-Brücke notwendigen Hydroxyl-Gruppe des Tyr178F wurde durch den Austausch gegen Phe, Leu, Ala und Cys im E. coli Komplex I untersucht. Die Vermutung der Autoren, dass die Mutation Tyr178CysF zum Verlust des FMNs führt (Bénit et al., 2001), konnte durch die Bestimmung des FMN-Gehalts der Variante widerlegt werden (Tab. 4.9). In dieser Arbeit wurde gezeigt, dass das Tyr178F essentiell für die Aktivität des Komplex I ist (Tab. 4.8). Alle Varianten zeigten eine um mindestens 70% geringere katalytische Effizienz als der Komplex I. Die katalytische Effizienz der Variante Y178LF war sogar um mehr als 80% verringert. Für die E. coli Komplex I-Variante E95QF wurde bereits ein Aktivitätsverlust von 30% gezeigt (Euro et al., 2009). Die Varianten Y178A/C und LF produzierten etwa fünf- bis achtfach mehr H2O2 pro umgesetztem Substratmolekül als der Komplex I (Tab. 4.9). Die ROS-Produktion der Variante Y178FF war sogar fast 13-fach erhöht. Für die Variante E95QF wurde eine um das 25-fache erhöhte ROS-Produktion publiziert. Allerdings behaupten die Autoren, dass die Position Glu95F die ROS-Produktion im E. coli Komplex I kontrolliert (Knuuti et al., 2013). Diese Aussage wird aber mit den von unserem Arbeitskreis publizierten Daten widerlegt (Morina et al., 2011; Birrell et al., 2013). Durch den Verlust der sterisch anspruchsvollen Seitenkette und der Hydroxyl-Gruppe des Tyrosins kann die H-Brücke zu Glu95F nicht mehr gebildet werden. Dies führt zu einer ineffizienten Elektronenübertragung auf FMN. Die Seitenketten der neu eingeführten Reste haben alle einen geringeren Raumbedarf als Tyrosin, daher könnte mehr Sauerstoff in die Bindestelle diffundieren. Im Zusammenspiel mit dem Verlust der H-Brücke führt dies wahrscheinlich vermehrt zu Nebenreaktionen wie der Bildung von ROS. 148 Diskussion 5.3 Die Beteiligung von Trp221G am Elektronentransfer In vorangegangenen Arbeiten wurde die vorgeschlagene Beteiligung aromatischer Aminosäuren am Elektronentransfer in Komplex I untersucht (Dörner, 2010). Es wurde gezeigt, dass der Austausch Trp221LeuG die d-NADH-Oxidaseaktivität der Cytoplasmamembranen um etwa ein Drittel vermindert. Um strukturelle Gründe für die verminderte Aktivität möglicherweise ausschließen zu können, wurden in dieser Arbeit Doppelmutanten auf Basis der Mutante Trp221LeuG dargestellt. Mit Hilfe der Struktur des hydrophilen Arms des T. thermophilus Komplex I (Berrisford und Sazanov, 2009) wurden aliphatische Aminosäuren identifiziert, die sich in ähnlichem Abstand jeweils zu N5 und N6a befinden wie Leu221G. Diese wurden gegen Trp ausgetauscht, um im Idealfall eine Doppelmutante mit der Aktivität des Ausgangsstamms zu erhalten. Um die vorgeschlagene Beteiligung von Arg219G am Elektronentransfer zu untersuchen, wurde es gegen Leu ausgetauscht. Zudem wurde eine Mutante dargestellt, in der Trp221G und Arg219G gegeneinander ersetzt waren. Die Membranen der Doppelmutanten wiesen niedrigere d-NADH-Oxidaseaktivitäten auf als die der Mutante Trp221LeuG (Tab. 4.10). Die Doppelmutante Arg219Trp/Trp221ArgG wies 12% der Aktivität des Ausgangsstamms und nur etwa ein Drittel der Mutante Trp221LeuG auf. Die Mutation Arg219LeuG verringerte die d-NADH Oxidaseaktivität ebenfalls um ein Drittel und unterstreicht die funktionelle Bedeutung der positiven Ladung für den Elektronentransfer (Wittekindt et al., 2009). Eine Western Blot-Analyse zeigte, dass alle Varianten produziert wurden und in der Membran stabil vorlagen. Dies impliziert, dass die verringerten Aktivitäten auf die eingeführten Mutationen zurückzuführen sind. Die Tatsache, dass die Aktivität durch die neu eingeführten zweiten Mutationen nicht erhöht wurde, widerlegt nicht den postulierten hopping-Mechanismus. Da nicht nur die Distanz zu den aromatischen Seitenketten, sondern auch deren Orientierung zu den Fe/S-Zentren für den Elektronentransfer wichtig ist, kann ohne eine Struktur des E. coli Komplex I und der Varianten keine abschließende Aussage zum Mechanismus getroffen werden. Tryptophan und Tyrosin werden in einer Vielzahl von Enzymen für den Elektronentransfer genutzt (Gray und Winkler, 2003). Phenylalanin spielt für den Elektronentransfer eine geringere Rolle. Dennoch wurde gezeigt, dass Phe die zentrale Rolle bei der Elektronenübertragung zwischen einem high-potential Eisen-Schwefel Protein (high-potential iron-sulphur protein; HiPIP) und der tetrahäm Cytochrom c Untereinheit des Reaktionszentrums des Photosystems von Allochromatium vinosum spielt (Venturoli et al., 2004). 149 Diskussion Die aromatischen Aminosäuren Phe515CD und His514CD sind ebenfalls zwischen N5 und N6a lokalisiert. In vorangegangenen Arbeiten wurden beide Aminosäuren gegen Leu ausgetauscht. Die Aktivität der Mutante Phe515LeuCD war um etwa ein Drittel die der Mutante His514LeuCD um etwa 10% verringert (Dörner, 2010). Zudem wurde das Phe515CD von den Autoren als wahrscheinlichster Kandidat für den hopping-Mechanismus vorgeschlagen (Wittekindt et al., 2009). In weiterführenden Arbeiten könnten Doppelmutanten auf Basis der Mutante Phe515LeuCD entworfen werden. Zudem könnte eine Mutante dargestellt werden, in der die Positionen His514CD und Phe515CD gegeneinander ausgetauscht sind, um das Postulat des hoppings zu untersuchen. 5.4 Überproduktion des E. coli Komplex I und des A. aeolicus NuoEFHis In dieser Arbeit wurde der E. coli Stamm BW25113nptI Δnuo Δndh/pBADnuo nuoFhis als Überproduzent für den Komplex I etabliert. Zunächst wurden die NADH- und SuccinatOxidaseaktivitäten des Stammes BW25113 und seiner Abkömmlinge BW25113Δnuo, BW25113ndh::nptI und BW25113nptI Δnuo Δndh, sowie die Hemmbarkeit dieser Aktivitäten mit den Komplex I-spezifischen Inhibitoren Piericidin A und Fenazaquin bestimmt (Tab. 4.11). Die NADH-Oxidaseaktivität der Membranen des Ausgangsstamms ließ sich mit Piericidin A zu 82% und mit Fenazaquin zu 85% inhibieren. Die Aktivität der Membranen von BW25113ndh::nptI betrug etwa 80% der des Ausgangsstamms und war mit beiden Hemmstoffen zu 90% inhibierbar. Die NDH-II-vermittelte NADH-Oxidaseaktivität der Membranen des Stammes BW25113Δnuo betrug nur 40% der des Ausgangsstamms und war überraschenderweise mit Piericidin A zu 50% und mit Fenazaquin zu 25% inhibierbar. Der Stamm BW25113nptI Δnuo Δndh hatte erwartungsgemäß keine NADH-Oxidaseaktivität. Diese Messungen zeigten, dass die von Bekker et al. (2009) formulierte Beteiligung von weiteren NADH-verbrauchenden Enzymen in der aeroben Atmungskette von E. coli nicht zutrifft. Die Membranen des Stamms BW25113nptI Δnuo Δndh wiesen überraschenderweise noch ein Drittel der NADH bzw. d-NADH/Ferricyanid Oxidoreduktaseaktivität des Ausgangsstamms auf. Diese Aktivität könnte durch WrbA vermittelt sein, da für dieses membranassoziierte Enzym eine artifizielle NAD(P)H/Ferricyanid Oxidoreduktaseaktivität nachgewiesen wurde (Patridge und Ferry, 2006). 150 Diskussion Die Succinat-Oxidaseaktivität war für alle Stämme identisch und ließ sich nahezu vollständig durch Malonat hemmen (Tab. 4.12). Ein Effekt auf die restliche Atmungskette durch die Deletion von Komplex I und der NDH-II kann somit ausgeschlossen werden. Um die inhibierende Wirkung der Komplex I-spezifischen Hemmstoffe Piericidin A und Fenazaquin auf den Rest der Atmungskette zu untersuchen, wurde versucht auch die SuccinatOxidaseaktivität mit diesen Hemmstoffen zu inhibieren. Die Aktivitäten der Membranen des Ausgangsstamms und von BW25113nptI Δnuo Δndh ließen sich jeweils mit Piericidin A zu 17% und mit Fenazaquin zu 23% inhibieren. Allerdings geht aus den Experimenten nicht hervor, ob die Inhibitoren die Succinat Dehydrogenase oder die terminalen Oxidasen beeinflussen. Das Autoinduktionsmedium war für die Anzucht des Stamms BW25113nptI Δnuo Δndh/ pBADnuo nuoFhis am besten geeignet. Zwar konnte die Komplex I-Ausbeute im Vergleich zum Stamm BW25113Δnuo/pBADnuo nuoFhis nicht nennenswert erhöht werden, dennoch wies der isolierte Komplex eine um etwa 10% erhöhte spezifische Aktivität auf. Dies ist wahrscheinlich auf eine geringere Verunreinigung und die daraus resultierende genauere Konzentrationsbestimmung zurückzuführen. Zudem kann nun bei der Messung der NADHOxidaseaktivität auf das kostspielige, Komplex I-spezifische Substrat d-NADH verzichtet werden. Um die Expression der A. aeolicus Gene nuoEFhis zu erhöhen, wurde in dieser Arbeit das Plasmid pETBlue-1nuoEFhis ohne das Gen nuoG kloniert. Da NuoG bei der Präparation nur in Form von Einschlusskörpern erhalten wird, sollte durch die Verwendung des kleineren Vektors der Stress für die Zellen während der Expression und Produktion verringert werden. Erste Präparationsversuche lieferten sehr geringe Ausbeuten an NuoEFHis, zudem wurden Proteinspezies ohne Kofaktoren erhalten. Expressionstests zeigten, dass von den getesteten Klonen nur etwa 30 – 80% die durch den Subkomplex NuoEFHis vermittelte, charakteristische Braunfärbung aufwiesen. Die Ausbeute der Präparationen von NuoEFHis und seiner Varianten hing stark von den verwendeten Klonen ab. Es wurden 1.2 – 2.7 mg Protein pro g eingesetzter Zellen isoliert (Tab. 4.6 und Tab. 4.16). Zusammengefasst konnte durch die Verwendung des kleineren Plasmids und vorhergehenden Expressionstests die Proteinausbeute im Vergleich zum bisherigen Protokoll dt et al., 2008) um mehr als das Sechseinhalbfache gesteigert werden. 151 Diskussion 5.5 Modulation des Mittenpunktspotentials von N1a Das Fe/S-Zentrum N1a des E. coli Komplex I hat im Vergleich zu seinem mitochondrialen Homolog ein hohes Mittenpunktspotential von etwa −330 mV und kann deswegen durch NADH reduziert und EPR-spektroskopisch nachgewiesen werden (Leif et al., 1995; Reda et al., 2008). Der E. coli Komplex I produziert als Nebenreaktion am reduzierten FMN vornehmlich H2O2, wohingegen der mitochondriale Komplex Superoxid-Radikale bildet (Kussmaul und Hirst, 2006; Esterházy et al., 2008; Schulte, 2014). Die Fähigkeit von N1a Elektronen aufzunehmen wurde für die Bildung der unterschiedlichen ROS verantwortlich gemacht. Um dies zu untersuchen wurden in Zusammenarbeit mit der Arbeitsgruppe von Dr. Judy Hirst Komplex I Varianten dargestellt, in denen durch die Mutationen Val96ProE und Asn142MetE das Mittenpunktspotential von N1a um bis zu −100 mV verschoben war. Die NADH-Oxidaseaktivität der Membranen der Mutanten BW25113nptI Δnuo Δndh/ pBADnuo nuoFhis nuoE V96P und N142M waren im Rahmen der Messgenauigkeit nicht oder nur leicht verringert. Die Aktivität der Doppelmutante Val96Pro/Asn142MetE war um etwa ein Fünftel erniedrigt (Tab. 4.13). Die Varianten V96PE und V96P/N142ME wurden EPRspektroskopisch untersucht. In der Variante V96PE wurde N1a durch NADH nur zu 40% reduziert. Im EPR-Spektrum der Variante V96P/N142ME wurde N1a nicht mehr detektiert (Abb. 4.37). Die NADH/Ferricyanid, die NADH/HAR und die NADH/APAD+ Oxidoreduktaseaktivitäten sowie die H2O2- und Superoxid-Produktion der Varianten V96PE und V96P/N142ME wurden bestimmt (Abb. 4.38). Die NADH/Ferricyanid und NADH/HAR Aktivität der Varianten war im Vergleich zum Komplex I, um etwa 20 – 30% verringert. Die NADH/APAD+ Oxidoreduktaseaktivität beider Varianten betrug die Hälfte der des Komplex I. Die Superoxid-Produktion der Varianten war nicht erhöht. Somit kann ausgeschlossen werden, dass die Art der produzierten ROS vom Mittenpunktspotential von N1a abhängt (Birrell et al., 2013). Allerdings ist die H2O2-Produktion der Variante V96PE um etwa ein Fünftel und die der Variante V96P/N142ME um 60% erhöht. Dies ist ein Hinweis auf eine funktionelle Bedeutung des Zentrums N1a als Schutz vor erhöhter ROS-Produktion. Da für die Aktivitätsbestimmungen der Varianten nur artifizielle Elektronenakzeptoren verwendet wurden, könnten in weiterführenden Arbeiten die kinetischen Parameter der physiologischen NADH:Decyl-Ubichinon Oxidoreduktaseaktivität der Varianten bestimmt werden. Dies würde eine genauere Aussage zum Einfluss des N1a auf die Aktivität des Komplex I geben. Die H2O2-Produktion wurde ohne Elektronenakzeptor im Assay gemessen. Um einen physiologisch relevanten Wert für die ROS-Produktion zu erhalten, könnte in 152 Diskussion weiterführenden Arbeiten die H2O2-Bildung durch die Varianten während des katalytischen turnovers mit dem von Herrn Dipl.-Chem. Marius Schulte etablierten Amplex Red Assay bestimmt werden. 5.6 Redox-induzierte Konformationsänderung des Glu95 auf NuoF In unserem Arbeitskreis wurde der A. aeolicus Subkomplex NuoEFHis im oxidierten Zustand mit NAD+ bzw. im reduzierten Zustand mit NADH kristallisiert. Anhand des Vergleichs beider Strukturen wurde erkannt, dass die Peptidbindung zwischen Glu95F und Ser96F je nach Redox-Zustand in einer anderen Konformation vorliegt. Im reduzierten Zustand zeigt die Carbonyl-Gruppe des Glu95F vom FMN weg und ermöglicht so die Bindung des NADH. In dieser Orientierung verdrängt die Carbonyl-Gruppe ein H2O-Molekül, das nur in der oxidierten Konformation vorliegt und übernimmt teilweise die von diesem H2O-Molekül gebildeten H-Brücken (Abb. 4.39). Im oxidierten Zustand zeigt die Carbonyl-Gruppe zum FMN hin und verdrängt aufgrund seines sterischen Anspruchs das NAD+. In beiden Konformationen wird die Carbonyl-Gruppe durch H-Brücken zu verschiedenen H2O-Molekülen stabilisiert (Abb. 4.39 und Abb. 2.14). Dieser Konformationswechsel wird als „Auswurf-Mechanismus“ für das produzierte NAD+ interpretiert. Um die Bedeutung der Konformationsänderung für den Mechanismus zu untersuchen, wurde die Carbonyl-Gruppe des Glu95F durch das Einfügen neuer H-Brücken in der reduzierten Konformation fixiert. Hierfür wurde das Gly129E jeweils gegen Ser und Asp ausgetauscht. In einem Monomer der asymmetrischen Einheit der Variante G129SE zeigte die Carbonyl-Gruppe vom FMN weg, was der reduzierten Konformation entspricht. In dem anderen Monomer lag die CarbonylGruppe in der oxidierten Konformation vor. Dies deutet auf eine eingeschränkte Flexibilität hin, zeigt aber dennoch, dass die Peptidbindung immer noch flexibel ist (Abb. 4.44). Der niedrige B-Faktor der Carbonyl-Gruppe des Glu95F in der Variante G129DE deutet darauf hin, dass die Bindung in dieser Variante hingegen fixiert ist. Um die Auswirkungen der eingeschränkten Flexibilität der Peptidbindung auf die physiologische Aktivität zu untersuchen, wurden die homologen Mutationen (Gly135SerE und Gly135AspE) in den E. coli Komplex I eingeführt. Die NADH-Oxidaseaktivität der Cytoplasmamembranen der Variante G135SE war im Vergleich zum Ausgangsstamm in etwa halbiert. Die Membranen der Variante G135DE wiesen keine NADH-Oxidaseaktivität auf (Tab. 4.17). Die isolierten Varianten wurden in Zusammenarbeit mit Herrn Dipl.-Chem. Marius Schulte untersucht 153 Diskussion (Schulte, 2014). Die Mutation G135SE hatte nur einen geringen Einfluss auf die physiologische NADH:Decyl-Ubichinon Oxidoreduktaseaktivität. Die maximale Umsatzrate vmax betrug 2.7 ± 0.2 µmol·min−1·mg−1 und der Km lag bei 13.6 ± 1.0 µM. Das spricht dafür, dass die erniedrigte NADH-Oxidaseaktivität der Membranen dieser Mutante hauptsächlich auf eine geringere Produktion der Variante zurückzuführen ist. Die kinetischen Parameter der Variante G135DE konnte nicht aufgenommen werden, da die Aktivität im Laufe der Messung stetig abnahm. Dies deutet auf eine Inhibition der Reaktion durch das produzierte NAD+ hin. Diese eingeschränkte Aktivität zeigt die Bedeutung der konformativen Flexibilität der Glu95E-Peptidbindung und unterstützt die Hypothese des „Au wurf-Mec ani mu ‟. Die NADH/Ferricyanid Oxidoreduktaseaktivität der Varianten war im Vergleich zu Komplex I unverändert (Tab. 4.19). Die NADH/HAR Oxidoreduktaseaktivität der Variante G135SE war jedoch um 18% und die von G135DE um 87% im Vergleich zum Komplex I verringert (Schulte, 2014). HAR kann nur in einem ternären Komplex, bestehend aus HAR, reduziertem FMN und gebundenem NADH, reduziert werden (Birrell und Hirst, 2013). Daher wird vermutet, dass durch die Mutationen die Verweildauer des NAD+ in der Bindetasche verlängert wird. Die EPR-spektroskopische Untersuchung der Variante G135DE zeigte, dass alle Fe/S-Zentren durch NADH in einem geringeren Maß reduziert wurden als im Komplex I. Dies ist wahrscheinlich auf die langsamere Umsetzung von NADH und die Reoxidation der Fe/SZentren zurückzuführen. Das Spektrum der mit Dithionit reduzierten Probe zeigt, dass alle Fe/S-Zentren stöchiometrisch reduziert wurden. Für die Reaktion der Variante G135DE konnten mit dem Radikalfänger DEPMPO keine ROS detektiert werden. Dies stimmt mit den von Herrn Dipl.-Chem. Marius Schulte mit dem Amplex Red Assay bestimmten Werten überein (Schulte, 2014). In dieser Arbeit wurde durch Einführung von H-Brücken die Flexibilität der Peptidbindung zwischen Glu95F und Ser96F eingeschränkt. Durch die Bestimmung der NADHOxidoreduktaseaktivitäten mit verschiedenen, artifiziellen Elektronenakzeptoren konnte gezeigt wer en, a ie F exibi i er Pep i bin ung eine Ar „Auswurf-Mechanismus“ für das produzierte NAD+ darstellt. Allerdings ist der Auslöser dieser konformativen Änderung noch immer nicht identifiziert. Es könnte sich dabei um den Redox-Zustand des Zentrums N1a handeln, da die Carbonyl-Gruppe über ein H2O-Molekül mit der Proteinschleife, die N1a koordiniert, verbrückt ist. Für Flavodoxine wurde gezeigt, dass die Konformation einer Peptidbindung in der Nähe zum FMN von der Oxidationsstufe des Kofaktors abhängt (Ludwig et al., 1997). Bei diesen Proteinen zeigt in der reduzierten bzw. semichinoiden Form 154 Diskussion der Peptid-Sauerstoff zum FMN hin und bildet eine H-Brücke zu dem protoniertem N5-Atom. In der oxidierten Form zeigt der Peptid-Sauerstoff von dem FMN weg. Es wurde gezeigt, dass die Orientierung des Peptidrückgrats die Mittenpunktspotentiale der verschiedenen FMNOxidationsstufen moduliert (Ludwig et al., 1997). Für Flavodoxin II aus Azotobacter vinelandii wird angenommen, dass die Abwesenheit einer H-Brücke zum N5-Atom des FMNs die oxidierte Form destabilisiert und so die Elektronenaufnahme erleichtert (Alagaratnam et al., 2005). In Komplex I ist die von Glu95F und Ser96F gebildete Peptidbindung zu weit vom FMN entfernt, als dass der Peptid-Sauerstoff von Glu95F direkt mit dem FMN wechselwirken könnte. Allerdings befindet sich nahe am FMN ein H2O-Molekül, das in der oxidierten Struktur des A. aeolicus NuoEFHis von der Seitenkette des Asp94F, den PeptidSauerstoffatomen von Tyr180F und Glu95F sowie dem O4-Atom des FMN über H-Brücken gebunden wird (Abb. 2.14 A). In der Struktur des reduzierten Proteins zeigt der PeptidSauerstoff des Glu95F vom FMN weg, dadurch geht die H-Brücke zu diesem H2O-Molekül verloren. Jedoch bildet dieses nun eine neue H-Brücke zum protonierten N5-Atom des FMNs (Abb. 2.14 B). Diese H-Brücke könnte die semichinoide und vollreduzierte Form des FMNs stabilisieren, so wie es bereits für Flavodoxine beschrieben wurde. Die in Kooperation mit Dr. Judy Hirst durchgeführten Versuche mit der Variante V96P/N142ME zeigen, dass N1a in der Variante nicht von NADH reduziert wird. Dennoch beträgt die NADH/HAR Oxidoreduktaseaktivität noch ca. 70% des unveränderten Komplex I (Birrell et al., 2013). Wäre N1a der Schalter für den Konformationswechsel, müsste diese Aktivität stärker verringert sein. Es wird daher angenommen, dass die Änderungen des Redox-Zustands von FMN die Orientierung der Peptidbindung vorgibt. Um zu untersuchen, ob das strukturell konservierte H2O-Molekül das Mittenpunktspotential von FMN moduliert, könnte in weiterführenden Arbeiten das Asp94F in dem A. aeolicus Subkomplex NuoEFHis gegen verschiedene Aminosäuren ausgetauscht werden. Durch den Austausch gegen Asn könnte die H-Brücke zum Wasser geschwächt werden. Der Austausch gegen Glu könnte die Position des Wassers ändern, so dass dieses im reduzierten Protein eventuell keine H-Brücke mehr zum N5-Atom des FMN bildet. Die Mutation gegen Ala oder Gly könnte den Verlust dieses Wassers bedingen. Die Auswirkungen dieser Mutationen auf die physiologische Aktivität des E. coli Komplex I müssten untersucht werden. 155 Diskussion 5.7 Oberflächen-Salzbrücke in NuoG Das Fe/S-Zentrum N7 liegt in der C-terminalen Domäne von NuoG. Struktur- und Sequenzanalysen haben gezeigt, dass die Domäne homolog zur Untereinheit NarG der Nitratreduktase A ist (Chow et al., 1991; Finel, 1998; Rothery et al., 2008). Alle katalytischen Untereinheiten der [Mo(MGD)2]-Enzymfamilie tragen an der Protein-Oberfläche eine von einem Glu und einem Arg gebildete Salzbrücke. Eine von Arg280G und Glu617G gebildete Salzbrücke befindet sich ebenfalls auf der Oberfläche von NuoG. Für die Nitratreduktase A wurde gezeigt, dass eine Mutation die zum Verlust der Salzbrücke führt, in NarG die Assemblierung der Kofaktoren dieser Untereinheit blockiert. In dieser Arbeit wurde die Auswirkung der Mutation Glu617AlaG auf die Salzbrücke auf NuoG untersucht. Die Mutante Glu617AlaG wurde angezogen und die Membranen präpariert. Die NADH-Oxidaseaktivität der Membranen der Mutante war im Vergleich zu denen des unveränderten Stamms, um etwa ein Drittel verringert. Durch eine Western Blot-Analyse wurde gezeigt, dass die Variante in geringeren Mengen als der Komplex I produziert wurde. Daher ist die verringerte Aktivität auf eine geringere Konzentration der Variante in der Membran zurückzuführen. Durch Saccharosedichtegradienten-Zentrifugation wurde gezeigt, dass die Variante bei der Extraktion mit DDM aus der Membran zerfiel. In den Komplex I-typischen Fraktionen war für den Extrakt der Mutante kaum NADH/Ferricyanid Oxidoreduktaseaktivität nachzuweisen. Der Versuch die Variante zu isolieren schlug fehl. In vorangegangen Arbeiten wurde gezeigt, dass der Verlust des Zentrums N7 auf NuoG zum Zerfall des Komplexes bei der Extraktion aus der Membran führt (Pohl et al., 2007b). Es wird daher angenommen, dass die fehlende Salzbrücke ebenfalls zum Verlust des Zentrums führt. Dies unterstreicht die evolutionäre Verwandtschaft von NuoG zu NarG. Ob die Fe/S-Zentren in NuoG ähnlich assembliert werden wie das Zentrum FS0 und der [Mo(MGD)2]-Kofaktor in NarG bleibt zu untersuchen. 5.8 Zuordnung des EPR-Signals N4 In dieser Arbeit wurde versucht das EPR-Signal N4 den strukturell definierten Fe/S-Zentren zuzuordnen. Dafür wurden die koordinierenden Aminosäuren von N4 und N5 jeweils gegen Ala ausgetauscht. Anhand der NADH-Oxidaseaktivität der Membranen der Mutanten wurde gezeigt, dass den Mutanten ein Fe/S-Zentrum im Komplex I fehlt und dadurch der 156 Diskussion Elektronentransfer unterbrochen wurde. Nur die Variante C156AG wies in der Membran und nach der Präparation noch 80% der NADH:Decyl-Ubichinon Oxidoreduktaseaktivität des Komplex I auf (siehe unten). Beim Versuch die Varianten zu präparieren, zerfielen die N5-Varianten C105AG, C108AG und C114AG sowie die N4-Variante C203AG. Diese zerfallenen Komplexe zeigten keine EPR-spektroskopisch eindeutigen Signale. Die N4-Varianten C153AG, C156AG, C159AG und die N5-Variante H101AG konnten präpariert werden und die Präparationen zeigten Signale im EPR-Spektrum, die den Fe/S-Zentren des Komplex I entsprachen. In den Spektren der intakten Varianten war das EPR-Signal N4 eindeutig nachweisbar. Daher kann ausgeschlossen werden, dass das Signal N4 von den strukturell definierten Zentren N4 oder N5 erzeugt wird. Das lässt vermuten, dass das Signal N4 von dem strukturell definierten Fe/S-Zentrum N6a stammt, wie schon durch DEER-Experimente am mitochondrialen Komplex I vorgeschlagen wurde (Roessler et al., 2010). Diese Ergebnisse decken sich außerdem mit bereits veröffentlichten Experimenten, wobei das N6a koordinierende Cys63I im E. coli Komplex I gegen Ser ausgetauscht wurde. Im EPR-Spektrum dieser Variante konnte das Signal N4 nicht detektiert werden (Sinha et al., 2012). Interessanterweise zeigte das EPR-Spektrum der hier hergestellten Variante C156AG bei 40 K ein sehr schwaches Signal des Zentrums N1b. Durch Struktur- und Sequenzvergleiche wurde His36G als möglicher neuer Ligand des strukturell definierten Zentrums N4 identifiziert. Die durch das ligand swapping verursachten strukturellen Änderungen führten zu dem deutlichen Verlust von N1b. Die Koordination des strukturell definierten Zentrums N4 in der Variante müsste sich in einer Verschiebung der g-Werte des dazugehörigen EPR-Signals bemerkbar machen, dies ist aber nicht der Fall. Dies deutet ebenfalls darauf hin, dass das Signal N4 nicht von dem strukturell definierten Zentrum N4 stammt. Da der Abstand zwischen N3 und N5 unter 14 Å beträgt, ist ein Elektronentransfer im Komplex I auch ohne das Zentrum N1b theoretisch möglich (Page et al., 1999; Sazanov, 2007). Diese Arbeit liefert anhand der Aktivität der Variante C156AG einen experimentellen Hinweis darauf, dass dies wirklich möglich ist. Die Variante trägt eine ungerade Anzahl an Fe/S-Zentren. Da NADH zwei Elektronen auf das FMN überträgt, könnte dies bedeuten, dass das ein Elektron nun am FMN lokalisiert ist. Dadurch könnte die ROS-Produktion dieser Variante stark erhöht sein. In weiterführenden Arbeiten könnte für diese Variante die H2O2-Produktion pro umgesetztem NADH-Molekül mit dem Amplex Red Assay bestimmt werden. 157 Literaturverzeichnis 6 Literaturverzeichnis Abramson, J., Riistama, S., Larsson, G., Jasaitis, A., Svensson-Ek, M., Laakkonen, L., Puustinen, A., Iwata, S., und Wikström, M. (2000). The structure of the ubiquinol oxidase from Escherichia coli and its ubiquinone binding site. Nat. Struct. Biol. 7, 910–917. Alagaratnam, S., van Pouderoyen, G., Pijning, T., Dijkstra, B.W., Cavazzini, D., Rossi, G.L., Van Dongen, W.M.A.M., van Mierlo, C.P.M., van Berkel, W.J.H., und Canters, G.W. (2005). A crystallographic study of Cys69Ala flavodoxin II from Azotobacter vinelandii: structural determinants of redox potential. Protein Sci. 14, 2284–2295. Anderson, D.G., und Kowalczykowski, S.C. (1997). The translocating RecBCD enzyme stimulates recombination by directing RecA protein onto ssDNA in a χ-regulated manner. Cell 90, 77–86. Anraku, Y., und Gennis, R.B. (1987). The aerobic respiratory chain of Escherichia coli. Trends Biochem. Sci. 12, 262–266. Auriol, C., Bestel-Corre, G., Claude, J.-B., Soucaille, P., und Meynial-Salles, I. (2011). Stress-induced evolution of Escherichia coli points to original concepts in respiratory cofactor selectivity. Proc. Natl. Acad. Sci. U. S. A. 108, 1278–1283. Baba, T., Ara, T., Hasegawa, M., Takai, Y., Okumura, Y., Baba, M., Datsenko, K.A., Tomita, M., Wanner, B.L., und Mori, H. (2006). Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol. Syst. Biol. 2, 2006. Balaban, R.S., Nemoto, S., und Finkel, T. (2005). Mitochondria, oxidants, and aging. Cell 120, 483–495. Balsa, E., Marco, R., Perales-Clemente, E., Szklarczyk, R., Calvo, E., Landázuri, M.O., und Enríquez, J.A. (2012). NDUFA4 is a subunit of complex IV of the mammalian electron transport chain. Cell Metab. 16, 378–386. Baradaran, R., Berrisford, J.M., Minhas, G.S., und Sazanov, L.A. (2013). Crystal structure of the entire respiratory complex I. Nature 494, 443–448. Barker, C.D., Reda, T., und Hirst, J. (2007). The flavoprotein subcomplex of complex I (NADH:ubiquinone oxidoreductase) from bovine heart mitochondria: insights into the 158 Literaturverzeichnis mechanisms of NADH oxidation and NAD+ reduction from protein film voltammetry. Biochemistry 46, 3454–3464. Bekker, M., de Vries, S., Ter Beek, A., Hellingwerf, K.J., und de Mattos, M.J.T. (2009). Respiration of Escherichia coli can be fully uncoupled via the nonelectrogenic terminal cytochrome bd-II oxidase. J. Bacteriol. 191, 5510–5517. Bénit, P., Chretien, D., Kadhom, N., de Lonlay-Debeney, P., Cormier-Daire, V., Cabral, A., Peudenier, S., Rustin, P., Munnich, A., und Rötig, A. (2001). Large-Scale Deletion and Point Mutations of the Nuclear NDUFV1 and NDUFS1 Genes in Mitochondrial Complex I Deficiency. Am. J. Hum. Genet. 68, 1344–1352. Berrisford, J.M., und Sazanov, L.A. (2009). Structural basis for the mechanism of respiratory complex I. J. Biol. Chem. 284, 29773–29783. Bertero, M.G., Rothery, R.A., Palak, M., Hou, C., Lim, D., Blasco, F., Weiner, J.H., und Strynadka, N.C.J. (2003). Insights into the respiratory electron transfer pathway from the structure of nitrate reductase A. Nat. Struct. Biol. 10, 681–687. Birrell, J.A., und Hirst, J. (2013). Investigation of NADH Binding, Hydride Transfer, and NAD+ Dissociation during NADH Oxidation by Mitochondrial Complex I Using Modified Nicotinamide Nucleotides. Biochemistry 52, 4048–4055. Birrell, J.A., King, M.S., und Hirst, J. (2011). A ternary mechanism for NADH oxidation by positively charged electron acceptors, catalyzed at the flavin site in respiratory complex I. FEBS Lett. 585, 2318–2322. Birrell, J.A., Morina, K., Bridges, H.R., Friedrich, T., und Hirst, J. (2013). Investigating the function of [2Fe-2S] cluster N1a, the off-pathway cluster in complex I, by manipulating its reduction potential. Biochem. J. 456, 139–146. Blasco, F., Dos Santos, J.P., Magalon, A., Frixon, C., Guigliarelli, B., Santini, C.L., und Giordano, G. (1998). NarJ is a specific chaperone required for molybdenum cofactor assembly in nitrate reductase A of Escherichia coli. Mol. Microbiol. 28, 435–447. Borisov, V.B., Murali, R., Verkhovskaya, M.L., Bloch, D.A., Han, H., Gennis, R.B., und Verkhovsky, M.I. (2011a). Aerobic respiratory chain of Escherichia coli is not allowed to work in fully uncoupled mode. Proc. Natl. Acad. Sci. U. S. A. 108, 17320–17324. 159 Literaturverzeichnis Borisov, V.B., Gennis, R.B., Hemp, J., und Verkhovsky, M.I. (2011b). The cytochrome bd respiratory oxygen reductases. Biochim. Biophys. Acta 1807, 1398–1413. Böttcher, B., Scheide, D., Hesterberg, M., Nagel-Steger, L., und Friedrich, T. (2002). A Novel, Enzymatically Active Conformation of the Escherichia coli NADH:Ubiquinone Oxidoreductase (Complex I). J. Biol. Chem. 277, 17970–17977. Braun, M., Bungert, S., und Friedrich, T. (1998). Characterization of the overproduced NADH dehydrogenase fragment of the NADH:ubiquinone oxidoreductase (complex I) from Escherichia coli. Biochemistry 37, 1861–1867. Bridges, H.R., Bill, E., und Hirst, J. (2012). Mössbauer spectroscopy on respiratory complex I: the iron-sulfur cluster ensemble in the NADH-reduced enzyme is partially oxidized. Biochemistry 51, 149–158. Burschel, S. (2011). Heterologous Production, Isolation and Crystallization of Aquifex aeolicus subcomplex NuoEF-variants E185GF and E185AF. Bachelorarbeit, Albert-LudwigsUniversität Freiburg. Burschel, S. (2013). Zuordnung des EPR-Signals N4 durch Mutagenesestudien am respiratorischen Komplex I aus Escherichia coli. Masterarbeit, Albert-Ludwigs-Universität Freiburg. Calhoun, M.W., Oden, K.L., Gennis, R.B., de Mattos, M.J., und Neijssel, O.M. (1993). Energetic efficiency of Escherichia coli: effects of mutations in components of the aerobic respiratory chain. J. Bacteriol. 175, 3020–3025. Carroll, J., Fearnley, I.M., Skehel, J.M., Shannon, R.J., Hirst, J., und Walker, J.E. (2006). Bovine complex I is a complex of 45 different subunits. J. Biol. Chem. 281, 32724–32727. Cecchini, G., Schröder, I., Gunsalus, R.P., und Maklashina, E. (2002). Succinate dehydrogenase and fumarate reductase from Escherichia coli. Biochim. Biophys. Acta 1553, 140–157. Chow, W., Ragan, I., und Robinson, B.H. (1991). Determination of the cDNA sequence for the human mitochondrial 75-kDa Fe-S protein of NADH-coenzyme Q reductase. Eur. J. Biochem. 201, 547–550. 160 Literaturverzeichnis Clark, A.J., und Sandler, S.J. (1994). Homologous genetic recombination: the pieces begin to fall into place. Crit. Rev. Microbiol. 20, 125–142. Court, D.L., Sawitzke, J.A., und Thomason, L.C. (2002). Genetic engineering using homologous recombination. Annu. Rev. Genet. 36, 361–388. Dassa, J., Fsihi, H., Marck, C., Dion, M., Kieffer-Bontemps, M., und Boquet, P.L. (1991). A new oxygen-regulated operon in Escherichia coli comprises the genes for a putative third cytochrome oxidase and for pH 2.5 acid phosphatase (appA). Mol. Gen. Genet. 229, 341–352. Datsenko, K.A., und Wanner, B.L. (2000). One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. U. S. A. 97, 6640–6645. Dawson, T.M., und Dawson, V.L. (2003). Molecular Pathways of Neurodegeneration in Parkin n’ Di ea e. Science 302, 819–822. Degli Esposti, M. (1998). Inhibitors of NADH-ubiquinone reductase: an overview. Biochim. Biophys. Acta 1364, 222–235. Deković, D. (2012). Role of aromatic amino acids in electron transfer in E. coli respiratory complex I. Diplomarbeit, Albert-Ludwigs-Universität Freiburg. Devasagayam, T.P.A., Tilak, J.C., Boloor, K.K., Sane, K.S., Ghaskadbi, S.S., und Lele, R.D. (2004). Free radicals and antioxidants in human health: current status and future prospects. J. Assoc. Physicians India 52, 794–804. Dörner, K.H. (2010). Der Elektronentransfer der Escherichia coli NADH:Ubichinon Oxidoreduktase sowie die ESR-spektroskopische Charakterisierung weiterer Metalloproteine. Dissertation, Albert-Ludwigs-Universität Freiburg. Efremov, R.G., und Sazanov, L.A. (2011). Structure of the membrane domain of respiratory complex I. Nature 476, 414–420. Ellis, H.M., Yu, D., DiTizio, T., und Court, D.L. (2001). High efficiency mutagenesis, repair, and engineering of chromosomal DNA using single-stranded oligonucleotides. Proc. Natl. Acad. Sci. U. S. A. 98, 6742–6746. Emsley, P., Lohkamp, B., Scott, W.G., und Cowtan, K. (2010). Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501. 161 Literaturverzeichnis Engel, T. (2010). Mutagenese der NADH-Bindestelle des Komplex I. Zulassungsarbeit, Albert-Ludwigs-Universität Freiburg. Esterházy, D., King, M.S., Yakovlev, G., und Hirst, J. (2008). Production of reactive oxygen species by complex I (NADH:ubiquinone oxidoreductase) from Escherichia coli and comparison to the enzyme from mitochondria. Biochemistry 47, 3964–3971. Euro, L., Belevich, G., Bloch, D.A., Verkhovsky, M.I., Wikström, M., und Verkhovskaya, M. (2009). The role of the invariant glutamate 95 in the catalytic site of Complex I from Escherichia coli. Biochim. Biophys. Acta 1787, 68–73. Evans, P. (2006). Scaling and assessment of data quality. Acta Cryst. D26, 72–82. Fearnley, I.M., und Walker, J.E. (1992). Conservation of sequences of subunits of mitochondrial complex I and their relationships with other proteins. Biochim. Biophys. Acta 1140, 105–134. Feng, Y., Li, W., Li, J., Wang, J., Ge, J., Xu, D., Liu, Y., Wu, K., Zeng, Q., Wu, J.-W., et al. (2012). Structural insight into the type-II mitochondrial NADH dehydrogenases. Nature 491, 478–482. Finel, M. (1998). Organization and evolution of structural elements within complex I. Biochim. Biophys. Acta 1364, 112–121. Forget, P. (1974). The bacterial nitrate reductases. Solubilization, purification and properties of the enzyme A of Escherichia coli K 12. Eur. J. Biochem. 42, 325–332. Friedrich, T. (1998). The NADH:ubiquinone oxidoreductase (complex I) from Escherichia coli. Biochim. Biophys. Acta 1364, 134–146. Friedrich, T. (2001). Complex I: a chimaera of a redox and conformation-driven proton pump? J. Bioenerg. Biomembr. 33, 169–177. Friedrich, T., und Böttcher, B. (2004). The gross structure of the respiratory complex I: a Lego System. Biochim. Biophys. Acta 1608, 1–9. Friedrich, T., und Scheide, D. (2000). The respiratory complex I of bacteria, archaea and eukarya and its module common with membrane-bound multisubunit hydrogenases. FEBS Lett. 479, 1–5. 162 Literaturverzeichnis Friedrich, T., und Weiss, H. (1997). Modular Evolution of the Respiratory NADH:Ubiquinone Oxidoreductase and the Origin of its Modules. J. Theor. Biol. 187, 529– 540. Friedrich, T., Hofhaus, G., Ise, W., Nehls, U., Schmitz, B., und Weiss, H. (1989). A small isoform of NADH:ubiquinone oxidoreductase (complex I) without mitochondrially encoded subunits is made in chloramphenicol-treated Neurospora crassa. Eur. J. Biochem. 180, 173– 180. Friedrich, T., van Heek, P., Leif, H., Ohnishi, T., Forche, E., Kunze, B., Jansen, R., Trowitzsch-Kienast, W., Höfle, G., und Reichenbach, H. (1994). Two binding sites of inhibitors in NADH:ubiquinone oxidoreductase (complex I). Relationship of one site with the ubiquinone-binding site of bacterial glucose:ubiquinone oxidoreductase. Eur. J. Biochem. 219, 691–698. Gay, P., Le Coq, D., Steinmetz, M., Berkelman, T., und Kado, C.I. (1985). Positive selection procedure for entrapment of insertion sequence elements in gram-negative bacteria. J. Bacteriol. 164, 918–921. Ghisla, S. (1980). Fluorescence and optical characteristics of reduced flavins and flavoproteins. Methods Enzymol. 66, 360–373. Gill, S.C., und von Hippel, P.H. (1989). Calculation of protein extinction coefficients from amino acid sequence data. Anal. Biochem. 182, 319–326. Glinn, M.A., Lee, C.P., und Ernster, L. (1997). Pro- and anti-oxidant activities of the mitochondrial respiratory chain: factors influencing NAD(P)H-induced lipid peroxidation. Biochim. Biophys. Acta 1318, 246–254. Gnandt, E. (2012). Herstellung und Charakterisierung von Tyrosin-Varianten des respiratorischen Komplex I. Diplomarbeit, Albert-Ludwigs-Universität Freiburg. Gray, H.B., und Winkler, J.R. (2003). Electron tunneling through proteins. Q. Rev. Biophys. 36, 341–372. Grigorieff, N. (1999). Structure of the respiratory NADH:ubiquinone oxidoreductase (complex I). Curr. Opin. Struct. Biol. 9, 476–483. 163 Literaturverzeichnis Guénebaut, V., Schlitt, A., Weiss, H., Leonard, K., und Friedrich, T. (1998). Consistent structure between bacterial and mitochondrial NADH:ubiquinone oxidoreductase (complex I). J. Mol. Biol. 276, 105–112. Hanes, C.S. (1932). Studies on plant amylases: The effect of starch concentration upon the velocity of hydrolysis by the amylase of germinated barley. Biochem. J. 26, 1406–1421. Hirst, J., King, M.S., und Pryde, K.R. (2008). The production of reactive oxygen species by complex I. Biochem. Soc. Trans. 36, 976–980. Hubrich, F. (2010). Mutagenese der Position E183 in der NADH-Bindestelle der NADH:Ubichinon-Oxidoreduktase. Zulassungsarbeit, Albert-Ludwigs-Universität Freiburg. Jaworowski, A., Mayo, G., Shaw, D.C., Campbell, H.D., und Young, I.G. (1981). Characterization of the respiratory NADH dehydrogenase of Escherichia coli and reconstitution of NADH oxidase in ndh mutant membrane vesicles. Biochemistry 20, 3621– 3628. Jeong, J., Cho, N., Jung, D., und Bang, D. (2013). Genome-scale genetic engineering in Escherichia coli. Biotechnol. Adv. 31, 804–810. Jurkovic, S.A. (2013). Strukturen der Aquifex aeolicus NuoEFHis-Varianten an der Position E185F mit und ohne gebundenem NAD+. Diplomarbeit, Albert-Ludwigs-Universität Freiburg. Kabsch, W. (2009). XDS. Acta Cryst. D66, 125–132. Kitagawa, M., Ara, T., Arifuzzaman, M., Ioka-Nakamichi, T., Inamoto, E., Toyonaga, H., und Mori, H. (2005). Complete set of ORF clones of Escherichia coli ASKA library (a complete set of E. coli K-12 ORF archive): unique resources for biological research. DNA Res. 12, 291– 299. Knuuti, J., Belevich, G., Sharma, V., Bloch, D.A., und Verkhovskaya, M. (2013). A single amino acid residue controls ROS production in the respiratory Complex I from Escherichia coli: ROS production by Complex I. Mol. Microbiol. 90, 1190–1200. Kohlstädt, M. (2009). Funktionelle und strukturelle Untersuchungen zur NADH - Bindung der bakteriellen NADH:Ubichinon Oxidoreduktase. Dissertation, Albert-Ludwigs-Universität Freiburg. 164 Literaturverzeichnis , M., Dörner, ., Laba zke, R., , C., Hielscher, R., Schiltz, E., Einsle, O., Hellwig, P., und Friedrich, T. (2008). Heterologous Production, Isolation, Characterization and Crystallization of a Soluble Fragment of the NADH:Ubiquinone Oxidoreductase (Complex I) from Aquifex aeolicus. Biochemistry 47, 13036–13045. Kotlyar, A.B., Sled, V.D., Burbaev, D.S., Moroz, I.A., und Vinogradov, A.D. (1990). Coupling site I and the rotenone-sensitive ubisemiquinone in tightly coupled submitochondrial particles. FEBS Lett. 264, 17–20. Krishnamoorthy, G., und Hinkle, P.C. (1988). Studies on the electron transfer pathway, topography of iron-sulfur centers, and site of coupling in NADH-Q oxidoreductase. J. Biol. Chem. 263, 17566–17575. Kussmaul, L., und Hirst, J. (2006). The mechanism of superoxide production by NADH:ubiquinone oxidoreductase (complex I) from bovine heart mitochondria. Proc. Natl. Acad. Sci. U. S. A. 103, 7607–7612. Laemmli, U.K. (1970). Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227, 680–685. Lanciano, P., Vergnes, A., Grimaldi, S., Guigliarelli, B., und Magalon, A. (2007). Biogenesis of a respiratory complex is orchestrated by a single accessory protein. J. Biol. Chem. 282, 17468–17474. Leif, H., Sled, V.D., Ohnishi, T., Weiss, H., und Friedrich, T. (1995). Isolation and characterization of the proton-translocating NADH:ubiquinone oxidoreductase from Escherichia coli. Eur. J. Biochem. 230, 538–548. Lesk, A.M. (1995). NAD-binding domains of dehydrogenases. Curr. Opin. Struct. Biol. 5, 775–783. Lin, M.T., und Beal, M.F. (2006). Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 443, 787–795. Ludwig, M.L., Pattridge, K.A., Metzger, A.L., Dixon, M.M., Eren, M., Feng, Y., und Swenson, R.P. (1997). Control of oxidation-reduction potentials in flavodoxin from Clostridium beijerinckii: the role of conformation changes. Biochemistry 36, 1259–1280. 165 Literaturverzeichnis Maklashina, E., und Cecchini, G. (1999). Comparison of catalytic activity and inhibitors of quinone reactions of succinate dehydrogenase (Succinate-ubiquinone oxidoreductase) and fumarate reductase (Menaquinol-fumarate oxidoreductase) from Escherichia coli. Arch. Biochem. Biophys. 369, 223–232. Matsushita, K., Ohnishi, T., und Kaback, H.R. (1987). NADH-ubiquinone oxidoreductases of the Escherichia coli aerobic respiratory chain. Biochemistry 26, 7732–7737. Melo, A.M.P., Bandeiras, T.M., und Teixeira, M. (2004). New Insights into Type II NAD(P)H:Quinone Oxidoreductases. Microbiol. Mol. Biol. Rev. 68, 603–616. Mills, J.E., und Dean, P.M. (1996). Three-dimensional hydrogen-bond geometry and probability information from a crystal survey. J. Comput. Aided Mol. Des. 10, 607–622. Mitchell, P., und Moyle, J. (1967). Chemiosmotic hypothesis of oxidative phosphorylation. Nature 213, 137–139. Mittl, P.R., Berry, A., Scrutton, N.S., Perham, R.N., und Schulz, G.E. (1994). Anatomy of an engineered NAD-binding site. Protein Sci. 3, 1504–1514. Morina, K., Schulte, M., Hubrich, F., Dörner, K., Steimle, S., Stolpe, S., und Friedrich, T. (2011). Engineering the Respiratory Complex I to Energy-converting NADPH:Ubiquinone Oxidoreductase. J. Biol. Chem. 286, 34627–34634. Mosberg, J.A., Lajoie, M.J., und Church, G.M. (2010). Lambda Red Recombineering in Escherichia coli Occurs Through a Fully Single-Stranded Intermediate. Genetics 186, 791– 799. Moser, C.C., Farid, T.A., Chobot, S.E., und Dutton, P.L. (2006). Electron tunneling chains of mitochondria. Biochim. Biophys. Acta 1757, 1096–1109. Murphy, K.C. (1991). λ Gam Protein Inhibits the Helicase and χ-Stimulated Recombination Activities of Escherichia coli RecBCD Enzyme. J. Bacteriol. 173, 5808–5821. Murphy, K.C. (1998). Use of Bacteriophage λ Recombination Functions to Promote Gene Replacement in Escherichia coli. J. Bacteriol. 180, 2063–2071. Murphy, M.P. (2009). How mitochondria produce reactive oxygen species. Biochem. J. 417, 1–13. 166 Literaturverzeichnis Nakamaru-Ogiso, E., Matsuno-Yagi, A., Yoshikawa, S., Yagi, T., und Ohnishi, T. (2008). Iron-sulfur cluster N5 is coordinated by an HXXXCXXCXXXXXC motif in the NuoG subunit of Escherichia coli NADH:quinone oxidoreductase (complex I). J. Biol. Chem. 283, 25979–25987. Ohnishi, T. (1998). Iron-sulfur clusters/semiquinones in complex I. Biochim. Biophys. Acta 1364, 186–206. Ohnishi, T., und Nakamaru-Ogi , E. 2008). Were ere any “mi a ignmen ” am ng ir n– sulfur clusters N4, N5 and N6b in NADH-quinone oxidoreductase (complex I)? Biochim. Biophys. Acta 1777, 703–710. Ohnishi, T., Blum, H., Galante, Y.M., und Hatefi, Y. (1981). Iron-sulfur N-1 clusters studied in NADH-ubiquinone oxidoreductase and in soluble NADH dehydrogenase. J. Biol. Chem. 256, 9216–9220. Page, C.C., Moser, C.C., Chen, X., und Dutton, P.L. (1999). Natural engineering principles of electron tunnelling in biological oxidation-reduction. Nature 402, 47–52. Pagniez-Mammeri, H., Loublier, S., Legrand, A., Bénit, P., Rustin, P., und Slama, A. (2012). Mitochondrial complex I deficiency of nuclear origin: I. Structural genes. Mol. Genet. Metab. 105, 163–172. Pan, X., Zhang, H., Gao, Y., Li, M., und Chang, W. (2009). Crystal structures of Pseudomonas syringae pv. tomato DC3000 quinone oxidoreductase and its complex with NADPH. Biochem. Biophys. Res. Commun. 390, 597–602. Patridge, E.V., und Ferry, J.G. (2006). WrbA from Escherichia coli and Archaeoglobus fulgidus is an NAD(P)H:quinone oxidoreductase. J. Bacteriol. 188, 3498–3506. Peng, G., Ermler, U., Clason, T., Bornemann, S., Hedderich, T., Ruiz, T., Meyer, B., Radermacher, M., Karas, M., und Michel, H. (2010). The structure of complex I from the hyperthermophilic eubacterium Aquifex aeolicus. Biochim. Biophys. Acta Bioenergetic 1797, 22. Pettersen, E.F., Goddard, T.D., Huang, C.C., Couch, G.S., Greenblatt, D.M., Meng, E.C., und Ferrin, T.E. (2004). UCSF Chimera – a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612. 167 Literaturverzeichnis Pohl, T. (2008). Lambda-Red vermittelte Mutagenese des nuo-Operons zur Herstellung von Protein-Varianten für die biophysikalische Charakterisierung der Escherichia coli NADH:Ubichinon Oxidoreduktase. Dissertation, Albert-Ludwigs-Universität, Freiburg. Pohl, T., Walter, J., Stolpe, S., Soufo, J.H.D., Grauman, P.L., und Friedrich, T. (2007a). Effects of the deletion of the Escherichia coli frataxin homologue CyaY on the respiratory NADH:ubiquinone oxidoreductase. BMC Biochem. 8, 13. Pohl, T., Bauer, T., Dörner, K., Stolpe, S., Sell, P., Zocher, G., und Friedrich, T. (2007b). Ir n−Su fur C u er N7 f e NADH:Ubiquin ne Oxi re uc a e C mp ex I) I E en ia for Stability but Not Involved in Electron Transfer. Biochemistry 46, 6588–6596. Pohl, T., Uhlmann, M., Kaufenstein, M., und Friedrich, T. (2007c). Lambda Red-mediated mutagenesis and efficient large scale affinity purification of the Escherichia coli NADH:ubiquinone oxidoreductase (complex I). Biochemistry 46, 10694–10702. Puustinen, A., Finel, M., Haltia, T., Gennis, R.B., und Wikström, M. (1991). Properties of the two terminal oxidases of Escherichia coli. Biochemistry 30, 3936–3942. Pyla, R., Kim, T.-J., Silva, J.L., und Jung, Y.-S. (2010). Proteome analysis of Azotobacter vinelandii ∆arrF mutant that overproduces poly-β-hydroxybutyrate polymer. Appl. Microbiol. Biotechnol. 88, 1343–1354. Ragan, C.I., und Racker, E. (1973). Resolution and reconstitution of the mitochondrial electron transport system. IV. The reconstitution of rotenone-sensitive reduced nicotinamide adenine dinucleotide-ubiquinone reductase from reduced nicotinamide adenine dinucleotide dehydrogenase and phospholipids. J. Biol. Chem. 248, 6876–6884. Ragan, C.I., Galante, Y.M., Hatefi, Y., und Ohnishi, T. (1982). Resolution of mitochondrial NADH dehydrogenase and isolation of two iron-sulfur proteins. Biochemistry 21, 590–594. Rasmussen, T., Scheide, D., Brors, B., Kintscher, L., Weiss, H., und Friedrich, T. (2001). Identification of two tetranuclear FeS clusters on the ferredoxin-type subunit of NADH:ubiquinone oxidoreductase (complex I). Biochemistry 40, 6124–6131. Reda, T., Barker, C.D., und Hirst, J. (2008). Reduction of the iron-sulfur clusters in mitochondrial NADH:ubiquinone oxidoreductase (complex I) by EuII-DTPA, a very low potential reductant. Biochemistry 47, 8885–8893. 168 Literaturverzeichnis Ried, J.L., und Collmer, A. (1987). An nptI-sacB-sacR cartridge for constructing directed, unmarked mutations in gram-negative bacteria by marker exchange-eviction mutagenesis. Gene 57, 239–246. Rigaud, J.-L., Lévy, D., Mosser, G., und Lambert, O. (1998). Detergent removal by non-polar polystyrene beads: application to membrane reconstitution and two-dimensional crystallization. Eur. Biophys. J. 27, 305–319. Roessler, M.M., King, M.S., Robinson, A.J., Armstrong, F.A., Harmer, J., und Hirst, J. (2010). Direct assignment of EPR spectra to structurally defined iron-sulfur clusters in complex I by double electron-electron resonance. Proc. Natl. Acad. Sci. U. S. A. 107, 1930– 1935. Rothery, R.A., Workun, G.J., und Weiner, J.H. (2008). The prokaryotic complex iron-sulfur molybdoenzyme family. Biochim. Biophys. Acta 1778, 1897–1929. Sazanov, L.A. (2007). Respiratory complex I: mechanistic and structural insights provided by the crystal structure of the hydrophilic domain. Biochemistry 46, 2275–2288. Sazanov, L.A., und Hinchliffe, P. (2006). Structure of the hydrophilic domain of respiratory complex I from Thermus thermophilus. Science 311, 1430–1436. Schägger, H., und von Jagow, G. (1987). Tricine-sodium dodecyl sulfate-polyacrylamide gel electrophoresis for the separation of proteins in the range from 1 to 100 kDa. Anal. Biochem. 166, 368–379. Schapira, A.H. (1998). Mitochondrial dysfunction in neurodegenerative disorders. Biochim. Biophys. Acta 1366, 225–233. Schäuble, M. (2013). Über die Beteiligung aromatischer Aminosäuren am Elektronentransfer in mp ex I, C arak eri ierung e S amm BW25113Δnuo ndh::nptI und Assemblierung der E617AG-Variante. Diplomarbeit, Albert-Ludwigs-Universität Freiburg. Scheide, D. (2000). Über den Mechanismus der NADH:Ubichinon Oxidoreduktase (Komplex I) aus Escherichia coli. Dissertation, Albert-Ludwigs-Universität, Freiburg. Scheide, D., Huber, R., und Friedrich, T. (2002). The proton-pumping NADH:ubiquinone oxidoreductase (complex I) of Aquifex aeolicus. FEBS Lett. 512, 80–84. 169 Literaturverzeichnis Schulte, M. (2014). Funktionelle Charakterisierung des Komplex I sowie strukturelle Untersuchungen an verwandten Enzymen. Dissertation, Albert-Ludwigs Universität Freiburg. Scrutton, N.S., Berry, A., und Perham, R.N. (1990). Redesign of the coenzyme specificity of a dehydrogenase by protein engineering. Nature 343, 38–43. Sharpley, M.S., Shannon, R.J., Draghi, F., und Hirst, J. (2006). Interactions between phospholipids and NADH:ubiquinone oxidoreductase (complex I) from bovine mitochondria. Biochemistry 45, 241–248. Sinha, P.K., Nakamaru-Ogiso, E., Torres-Bacete, J., Sato, M., Castro-Guerrero, N., Ohnishi, T., Matsuno-Yagi, A., und Yagi, T. (2012). Electron Transfer in Subunit NuoI (TYKY) of Escherichia coli NADH:Quinone Oxidoreductase (NDH-1). J. Biol. Chem. 287, 17363– 17373. Sled, V.D., Rudnitzky, N.I., Hatefi, Y., und Ohnishi, T. (1994). Thermodynamic analysis of flavin in mitochondrial NADH:ubiquinone oxidoreductase (complex I). Biochemistry 33, 10069–10075. Sousa, P.M.F., Silva, S.T.N., Hood, B.L., Charro, N., Carita, J.N., Vaz, F., Penque, D., Conrads, T.P., und Melo, A.M.P. (2011). Supramolecular organizations in the aerobic respiratory chain of Escherichia coli. Biochimie 93, 418–425. Stolpe, S. (2007). Über den Mechanismus der NADH:Ubichinon Oxidoreduktase aus Escherichia coli. Dissertation, Albert-Ludwigs-Universität Freiburg. Stolpe, S., und Friedrich, T. (2004). The Escherichia coli NADH:Ubiquinone Oxidoreductase (Complex I) Is a Primary Proton Pump but May Be Capable of Secondary Sodium Antiport. J. Biol. Chem. 279, 18377–18383. Thorn, J.M., Barton, J.D., Dixon, N.E., Ollis, D.L., und Edwards, K.J. (1995). Crystal structure of Escherichia coli QOR quinone oxidoreductase complexed with NADPH. J. Mol. Biol. 249, 785–799. Todd, J.D., Curson, A.R.J., Sullivan, M.J., Kirkwood, M., und Johnston, A.W.B. (2012). The Ruegeria pomeroyi acuI gene has a role in DMSP catabolism and resembles yhdH of E. coli and other bacteria in conferring resistance to acrylate. PloS One 7, e35947. 170 Literaturverzeichnis Towbin, H., Staehelin, T., und Gordon, J. (1979). Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc. Natl. Acad. Sci. U. S. A. 76, 4350–4354. Uhlmann, M., und Friedrich, T. (2005). EPR signals assigned to Fe/S cluster N1c of the Escherichia coli NADH:ubiquinone oxidoreductase (complex I) derive from cluster N1a. Biochemistry 44, 1653–1658. Unden, G., und Bongaerts, J. (1997). Alternative respiratory pathways of Escherichia coli: energetics and transcriptional regulation in response to electron acceptors. Biochim. Biophys. Acta 1320, 217–234. Vasquez-Vivar, J., Kalyanaraman, B., und Kennedy, M.C. (2000). Mitochondrial aconitase is a source of hydroxyl radical. An electron spin resonance investigation. J. Biol. Chem. 275, 14064–14069. Venturoli, G., Mamedov, M.D., Mansy, S.S., Musiani, F., Strocchi, M., Francia, F., Semenov, A.Y., Cowan, J.A., und Ciurli, S. (2004). Electron Transfer from HiPIP to the Photooxidized Tetraheme Cytochrome Subunit of Allochromatium vinosum Reaction Center: New Insights from Site-Directed Mutagenesis and Computational Studies. Biochemistry 43, 437–445. Villegas, J.M., Volentini, S.I., Rintoul, M.R., und Rapisarda, V.A. (2011). Amphipathic Cterminal region of Escherichia coli NADH dehydrogenase-2 mediates membrane localization. Arch. Biochem. Biophys. 505, 155–159. Walker, J.E. (1992). The NADH:ubiquinone oxidoreductase (complex I) of respiratory chains. Q. Rev. Biophys. 25, 253–324. Weidner, U., Geier, S., Ptock, A., Friedrich, T., Leif, H., und Weiss, H. (1993). The gene locus of the proton-translocating NADH:ubiquinone oxidoreductase in Escherichia coli. Organization of the 14 genes and relationship between the derived proteins and subunits of mitochondrial complex I. J. Mol. Biol. 233, 109–122. Weiss, R.F. (1970). The solubility of nitrogen, oxygen and argon in water and seawater. Deep-Sea Res. Oceanogr. Abstr. 17, 721–735. Weiss, H., Friedrich, T., Hofhaus, G., und Preis, D. (1991). The respiratory-chain NADH dehydrogenase (complex I) of mitochondria. Eur. J. Biochem. 197, 563–576. 171 Literaturverzeichnis Wittekindt, C., Schwarz, M., Friedrich, T., und Koslowski, T. (2009). Aromatic amino acids as stepping stones in charge transfer in respiratory complex I: an unusual mechanism deduced from atomistic theory and bioinformatics. J. Am. Chem. Soc. 131, 8134–8140. Yagi, T., und Matsuno-Yagi, A. (2003). The proton-translocating NADH-quinone oxidoreductase in the respiratory chain: the secret unlocked. Biochemistry 42, 2266–2274. Yakovlev, G., Reda, T., und Hirst, J. (2007). Reevaluating the relationship between EPR spectra and enzyme structure for the iron sulfur clusters in NADH:quinone oxidoreductase. Proc. Natl. Acad. Sci. U. S. A. 104, 12720–12725. Yankovskaya, V., Horsefield, R., Törnroth, S., Luna-Chavez, C., Miyoshi, H., Léger, C., Byrne, B., Cecchini, G., und Iwata, S. (2003). Architecture of succinate dehydrogenase and reactive oxygen species generation. Science 299, 700–704. Yano, T., Sklar, J., Nakamaru-Ogiso, E., Takahashi, Y., Yagi, T., und Ohnishi, T. (2003). Characterization of cluster N5 as a fast-relaxing [4Fe-4S] cluster in the Nqo3 subunit of the proton-translocating NADH-ubiquinone oxidoreductase from Paracoccus denitrificans. J. Biol. Chem. 278, 15514–15522. Zu, Y., Di Bernardo, S., Yagi, T., und Hirst, J. (2002). Redox Properties of the [2Fe-2S] Center in the 24 kDa (NQO2) Subunit of NADH:Ubiquinone Oxidoreductase (Complex I). Biochemistry 41, 10056–10069. 172 Anhang 7 Anhang 7.1 H-Brücken Netzwerk um NAD+/NADH und FMN im A. aeolicus NuoEFHis Die Tabellen 7.1 – 7.4 geben das H-Brücken Netzwerk in erster und zweiter + Koordinationssphäre von NAD bzw. NADH und FMN im A. aeolicus Subkomplex NuoEFHis wieder. Die H-Brücken wurden mit dem Programm UCSF Chimera nach den Kriterien von Mills und Dean (1996) bestimmt (Pettersen et al., 2004). H2O-Moleküle sind mit Nummern angeben. Konservierte H2O-Moleküle der Stuktur des oxidierten Proteins tragen in Klammern die Nummer des H2O-Moleküls der Struktur des reduzierten Proteins und umgekehrt. H-Brücken (HB) die nicht den exakten Kriterien nach Mills und Dean (1996) entsprechen sind grau und für die Aktivität wichtige Aminosäuren sind gelb markiert. Sterische Abstoßungen (SA) sind ebenfalls angegeben. Die Länge der HB bzw. der Abstand der SA sind angegeben. Die Abbildungen 7.1 und 7.2 geben die Strukturen von NAD+/H und FMN sowie deren Atomnummerierung wieder. Abbildung 7.1: Struktur des NAD+ bzw. des NADH. Abbildung 7.2: Struktur des FMN. 173 Anhang Tabelle 7.1: H-Brücken Netzwerk des NAD+ im A. aeolicus Subkomplex NuoEFHis im oxidierten Zustand. Erste Koordinationssphäre Zweite Koordinationssphäre Atom NAD+ AO3' InteraktionsPartner 1 Lys76F NZ InteraktionsPartner 1 Lys76F N Lys76F NZ Lys76F O Lys76F NZ InteraktionsPartner 2 Pro72F O Val218F O Ala8 0F N FMN O2P AO2' AO2' AO3' Glu185F OE1 Glu185F OE2 Glu185F OE1 HB 3.3 HB 2.6 HB 2.6 HB 2.4 Glu185F OE2 Glu185F O Glu185F N Glu185F OE1 HOH 483 (528) Lys202F NZ Ile189F N FMN O2' HOH 308 (103) HOH 308 (103) HB 2.9 HB 3.1 HB 3.0 HB 3.4 HB 2.8 AO2' HOH 483 (528) NO2' NO2' HOH 648 Gly68F O HB 2.8 HB 3.3 HOH 648 Gly68F N Glu97F OE1 HOH 129 HB 3.0 HB 2.7 NO3' Gly68F O HB 3.4 NO7 Glu95F O SA 2.9 NO7 HOH 545 HB 2.5 NO1 HOH 145 (534) HB 2.5 Glu95F O Glu95F N Glu95F OE2 Glu95F OE1 Glu95F OE1 Glu95F OE2 HOH 545 HOH 545 HOH 145 (534) HOH 483 (528) HOH 150 (6) HOH 79 (527) Asp103F N Arg104F N Tyr138F OH HOH 61 (526) HOH 61 (526) HOH 241 Glu184F OE2 Gly394F N HB 2.9 HB 3.1 HB 2.8 HB 3.2 HB 2.7 HB 2.7 HB 2.8 HB 2.9 HB 3.0 HB 2.9 NO2 NO2 FMN O2' FMN O3' HB 2.8 HB 2.6 AN3 HOH 483 (528) HB 2.5 Abstand [Å] HB 2.9 Abstand [Å] HB 3.2 HB 2.9 HB 3.2 HB 3.2 Tabelle 7.2: H-Brücken Netzwerk des FMN im A. aeolicus Subkomplex NuoEFHis im oxidierten Zustand. Erste Koordinationssphäre Atom FMN O1P InteraktionsPartner 1 Gly67F N HOH 12 (18) Zweite Koordinationssphäre Abstand[Å] HB 2.7 HB 2.7 O2P Lys76F NZ HB 3.2 O2P O2P HOH 134 (42) HOH 514 (26) HB 2.6 HB 2.7 O2P Asn219F ND2 HB 3.4 174 InteraktionsPartner 1 InteraktionsPartner 2 Abstand [Å] HOH 12 (18) HOH 12 (18) Ala69F N HOH 514 (26) HB 3.0 HB 2.8 Lys76F N Lys76F NZ Lys76F O Lys76F NZ HOH 134 (42) HOH 134 (42) HOH 514 (26) Asn219F OD1 Pro72F O Val218F O Ala80F N AO3' NAD+ Phe71F O Thr73F OG1 Phe71F N Trp77F NE1 HB 3.2 HB 2.9 HB 3.2 HB 2.9 HB 2.7 HB 3.2 HB 3.0 HB 3.0 Anhang Tabelle 7.2; Fortsetzung. O3P Asn219F ND2 HB 2.9 O3P Thr223F OG1 HB 2.6 O3P Asn220F N HB 2.9 O2 Asn220F ND2 HB 2.7 O2' NAD+ NO2 HB 2.8 O3' O3' NAD+ NO2 HOH 12 (18) HB 2.6 HB 2.9 O2' Glu185F N HB 3.4 O4 HOH 150 (6) HB 3.2 N3 HOH 79 (527) HB 3.2 Asn219F O Asn219F N Asn219F ND2 Thr223F OG1 Thr223F O Asn220F OD1 Asn220F ND2 Asn220F OD1 Asn220F O Asn92F N Ile90F O HOH 134 (42) Arg66F N Asn226F N Lys102F NZ Asp103F OD2 Glu222F N Ile224F N HB 2.8 HB 2.8 HB 3.2 HB 2.9 HB 3.1 HB 3.1 HB 2.9 HB 2.9 HB 3.2 HOH 12 (18) HOH 12 (18) Glu185F OE2 Glu185F O Glu185F OE1 Glu185F OE1 Glu185F OE2 Glu185F OE1 HOH 150 (6) HOH 150 (6) HOH 150 (6) HOH 79 (527) HOH 79 (527) HOH 79 (527) Ala69F N HOH 514 (26) Lys202F NZ Ile189F N HOH 308 (103) AO2' NAD+ AO2' NAD+ AO3' NAD+ Asp94F OD2 Glu95F O Tyr180F O Glu95F N Asn92F O Asp103F OD2 HB 3.0 HB 2.8 HB 2.9 HB 3.1 HB 3.4 HB 3.3 HB 2.6 HB 2.6 HB 2.4 HB 2.9 HB 2.8 HB 3.1 HB 2.7 HB 2.7 Tabelle 7.3: H-Brücken Netzwerk des NADH im A. aeolicus Subkomplex NuoEFHis im reduzierten Zustand. Erste Koordinationssphäre Zweite Koordinationssphäre AO3' InteraktionsPartner 1 Lys76F NZ Abstand [Å] HB 3.0 AO2' AO2' AO3' Glu185F OE1 Glu185F OE2 Glu185F OE1 HB 3.3 HB 2.5 HB 2.6 AO2' HOH 528 (483) HB 2.7 NO2' HOH 545 HB 2.7 NN7 HOH 526 (61) HB 2.9 NN7 Gly67 O HB 3.2 AN3 HOH 528 (483) HB 3.0 Atom NADH InteraktionsPartner 1 Lys76F N Lys76F NZ Lys76F O Lys76F NZ Glu185F OE2 Glu185F O Glu185F N Glu185F OE1 HOH 528 (483) HOH 528 (483) HOH 545 HOH 545 HOH 526 (61) HOH 526 (61) InteraktionsPartner 2 Pro72F O Val218F O Ala80F N FMN O2P HOH 540 Ile189F N FMN O2' HOH 103 (308) HOH 103 (308) AN3 Glu97F OE2 HOH 541 Glu95F OE2 Asp103F OD2 Abstand [Å] HB 3.3 HB 2.7 HB 3.4 HB 2.7 HB 2.7 HB 3.1 HB 3.0 HB 3.2 HB 2.9 HB 3.0 HB 2.9 HB 3.2 HB 2.7 HB 2.8 HOH 528 (483) HOH 528 (483) AO2' HOH 103 (308) HB 2.7 HB 2.9 175 Anhang Tabelle 7.3; Fortsetzung. NO7 Glu97F N HB 3.0 NO1 HOH 534 (145) HB 2.7 NO1 NO2 NO2 NO2 HOH 567 FMN O2' FMN O3' HOH 540 HB 2.7 HB 3.1 HB 2.7 HB 2.7 Glu97F O Glu97F O Glu97F OE2 Glu97F OE1 Glu97F OE2 Glu97F O Glu97F OE1 Glu97F OE2 HOH 534 (145) HOH 534 HOH 534 (145) HOH 567 Thr100F N Thr100F OG1 Tyr180F OH Ala296F N HOH 545 HOH 32 HOH 82 HOH 82 Gly394F N Glu184F OE2 HOH 514 HOH 261 HB 3.0 HB 3.2 HB 2.7 HB 2.9 HB 2.9 HB 3.1 HB 2.7 HB 3.3 HB 3.1 HB 2.7 HB 2.7 HB 2.7 HOH 540 Glu185F OE2 HB 2.7 Tabelle 7.4: H-Brücken Netzwerk des FMN im A. aeolicus Subkomplex NuoEFHis im reduzierten Zustand. Erste Koordinationssphäre Zweite Koordinationssphäre O1P O1P InteraktionsPartner 1 Gly67F N HOH 18 (12) O2P Lys76F NZ HB 2.7 O2P HOH 26 (514) HB 2.8 O2P HOH 42 (134) HB 2.7 O3P Asn219F ND2 HB 2.7 O3P HOH 42 (134) HB 3.5 O3P Thr223F OG1 HB 2.7 O3P Asn220F N HB 3.0 O2 Asn220F ND2 HB 2.7 O2' Glu185F N HB 3.0 Atom FMN 176 Abstand [Å] HB 2.8 HB 2.8 InteraktionsPartner 1 InteraktionsPartner 2 Abstand [Å] HOH 18 (12) HOH 18 (12) HOH 18 (12) Lys76F N Lys76F NZ Lys76F O Lys76F NZ HOH 26 (514) HOH 26 (514) HOH 42 (134) HOH 42 (134) HOH 42 (134) Asn219F OD1 Asn219F O Asn219F N Asn219F ND2 Thr223F OG1 Thr223F O Asn220F ND2 Asn220F ND2 Ala69F N HOH 26 (514) O3' Pro72F O Val218F O Ala80F N AO3' NAD+ Phe71F N HOH 18 (12) Thr73F OG1 Phe71F O Asn219F ND2 Trp77F NE1 Asn92F N Ile90F O HOH 42 (134) Arg66F N Asn226F N Asn92F O Asp103F OD2 HB 3.0 HB 2.7 HB 3.0 HB 3.3 HB 2.7 HB 3.4 HB 3.0 HB 2.9 HB 2.7 HB 2.9 HB 2.9 HB 2.9 HB 2.9 HB 2.8 HB 3.0 HB 2.9 HB 2.8 HB 3.2 HB 3.7 HB 2.8 Asn220F OD1 Asn220F O Glu185F O Glu185F OE1 Glu185F OE2 Glu185F OE1 Glu185F OE1 Glu185F OE2 Glu222F N Ile224F N Ile189F N AO2' NADH AO2' NADH AO3' NADH HOH 103 (308) HOH 540 HB 3.0 HB 3.1 HB 3.1 HB 3.3 HB 2.5 HB 2.6 HB 3.2 HB 2.7 Anhang Tabelle 7.3; Fortsetzung. O2' NADH NO2 HB 3.1 O3' O3' NADH NO2 HOH 18 (12) HB 2.7 HB 3.0 O4' Asn92F ND2 HB 2.9 O4 HOH 6 (150) HB 2.8 N3 HOH 527 (79) HB 2.9 N5 HOH 6 (150) HB 3.0 HOH 18 (12) HOH 18 (12) HOH 18 (12) Asn92F N Asn92F O Asn92F OD1 Asn92F ND2 Asn92F O Asn92F O HOH 6 (150) HOH 6 (150) HOH 6 (150) HOH 527 (79) HOH 527 (79) HOH 527 (79) Ala69F N HOH 26 (514) O1P Asn219F O Asn220F ND2 Asp94F N Gly183F O Val221F N HOH 527 (79) Asp94F OD2 Tyr180F O N5 Glu95F N Asn92F O Asp103F OD2 HB 3.0 HB 2.7 HB 2.8 HB 2.8 HB 3.7 HB 3.3 HB 2.8 HB 3.4 HB 2.8 HB 2.7 HB 2.9 HB 3.0 HB 3.1 HB 2.8 HB 2.6 177 Anhang 7.2 Kristallisations-screens Tabellen 7.5 und 7.6 gibt die fine-screens zur Kristallisation von E185AF und E185GF wieder. Tabelle 7.5: Fine-screens um Sphärolith-Bedingungen zur Kristallisation von E185GF. Alle Bedingungen waren auf pH 7.0 eingestellt. Alle Bedingungen enthielten PEG 3350 (PEG). 1 2 3 4 5 6 A 18% PEG 0.1 M Äpfelsäure 20% PEG 0.1 M Äpfelsäure 22% PEG 0.1 M Äpfelsäure 18% PEG 0.1 M Na3Citrat 20% PEG 0.1 M Na3Citrat 22% PEG 0.1 M Na3Citrat B 18% PEG 0.15 M Äpfelsäure 20% PEG 0.15 M Äpfelsäure 22% PEG 0.15 M Äpfelsäure 18% PEG 0.15 M Na3Citrat 20% PEG 0.15 M Na3Citrat 22% PEG 0.15 M Na3Citrat C 18% PEG 0.2 M Äpfelsäure 20% PEG 0.2 M Äpfelsäure 22% PEG 0.2 M Äpfelsäure 18% PEG 0.2 M Na3Citrat 20% PEG 0.2 M Na3Citrat 22% PEG 0.2 M Na3Citrat 18% PEG 5% Tacsimat 20% PEG 5% Tacsimat 22% PEG 5% Tacsimat 18% PEG 0.4 M Na3Citrat 20% PEG 0.4 M Na3Citrat 22% PEG 0.4 M Na3Citrat D Tabelle 7.6: Fine-screens um Phosphat-Bedingung zur Kristallisation von E185AF. A B C D 178 1 2 3 4 5 6 0.08 M NaH2PO4, 0.72 M K2HPO4 Σ 0.8M 0.16 M NaH2PO4, 0.64 M K2HPO4 0.2 M NaH2PO4, 0.8 M K2HPO4 0.24 M NaH2PO4, 0.96 M K2HPO4 0.3 M NaH2PO4, 1.2 M K2HPO4 0.24 M NaH2PO4, 0.56 M K2HPO4 0.3 M NaH2PO4, 0.7 M K2HPO4 0.36 M NaH2PO4, 0.84 M K2HPO4 0.45 M NaH2PO4, 1.05 M K2HPO4 0.32 M NaH2PO4, 0.48 M K2HPO4 0.4 M NaH2PO4, 0.6 M K2HPO4 0.48 M NaH2PO4, 0.72 M K2HPO4 0.6 M NaH2PO4, 0.9 M K2HPO4 0.4 M NaH2PO4, 0.4 M K2HPO4 0.5 M NaH2PO4, 0.5 M K2HPO4 0.6 M NaH2PO4, 0.6 M K2HPO4 0.75 M NaH2PO4, 0.75 M K2HPO4 0.48 M NaH2PO4, 0.32 M K2HPO4 0.6 M NaH2PO4, 0.4 M K2HPO4 0.72 M NaH2PO4, 0.48 M K2HPO4 0.9 M NaH2PO4, 0.6 M K2HPO4 0.1 M NaH2PO4, 0.9 M K2HPO4 Σ 1M 0.12 M NaH2PO4, 1.08 M K2HPO4 Σ 1.2 M 0.15 M NaH2PO4, 1.35 M K2HPO4 Σ 1.5 M Anhang 7.3 Kristallographische Statistiken Tabelle 7.7: Röntgendaten, Prozessierung und refinement Statistiken der Variante E185DF und E185HF. Die statistischen Werte wurden den, aus der Prozessierung resultierenden Proteindatenbank-Dateien (*.pdb) und den Protokolldateien der Programme Scala (Evans, 2006), XDS (Kabsch, 2009), PROCHECK (CCP4) entnommen. E185DF P 21 21 21 62.1; 115.8; 190.9; 90; 90; 90 E185HF P 21 21 21 63.3; 116.3; 190.6; 90; 90; 90 Auflösungsgrenze (Å) Auflösungsbereich (Å) Gesamte Vollständigkeit der Reflexe (%) 2.40 99.0 – 2.4 98.73 2.20 99.20 – 2.20 80.72 Anzahl einzigartiger Reflexe I/σ I) Rmerge (% ) 1 Rgesamt (%) 2 84765 1.98 22.8 26.12 55576 23.69 Rfrei (%) 28.43 31.23 0.0084 1.3459 0.020 2.218 In den bevorzugtesten Regionen (%) 97.4 88.9 In zusätzlich erlaubten Regionen (%) In großzügig erlaubten Regionen (%) In unerlaubten Regionen (%) 2.4 0.2 - 8.7 1.6 0.8 Raumgruppe Zellkonstanten a; b; c (Å) α β γ °) R.m.s. Abweichung der Idealwerte Bindungslängen (Å) Bindungswinkel (°) Φ, Ψ, Winkelverteilung fürAminosäurereste 3 1 Rmerge = 2 Rgesamt = Rfrei ist der Vergleichsprüfungs R-Faktor für einen Prüfsatz von 5% der einzigartigen Reflexe 3 Ramachandran Statistiken, die über PROCHECK definiert wurden 179 Anhang Tabelle 7.8: Röntgendaten, Prozessierung und refinement Statistiken der Variante E185DF mit gebundenem NAD+. Die statistischen Werte wurden den, aus der Prozessierung resultierenden Proteindatenbank-Dateien (*.pdb) und den Protokolldateien der Programme Scala (Evans, 2006), XDS (Kabsch, 2009), PROCHECK (CCP4) entnommen. Raumgruppe Ze k n an en a b c α β γ Å; °) P 21 21 21 62.3; 116.0; 190.4; 90; 90; 90 Auflösungsgrenze (Å) 1.8 Auflösungsbereich (Å) 99.0 – 1.8 Gesamte Vollständigkeit der Reflexe (%) 99.6 Anzahl einzigartiger Reflexe 128547 Multiplizität 2.0 I/σ I) 4.43 (0.69) 4 Rmerge (% ) 1 34.1 Rgesamt (%) 2 18.2 Rfrei (%) 22.5 R.m.s. Abweichung der Idealwerte Bindungslängen (Å) 0.01 Bindungswinkel (°) Φ, Ψ, Winkelverteilung fürAminosäurereste 1.04 3 In den bevorzugtesten Regionen (%) 91.8 In zusätzlich erlaubten Regionen (%) 7.8 In großzügig erlaubten Regionen (%) 0.4 In unerlaubten Regionen (%) - 1 Rmerge = 2 Rgesamt = Rfrei ist der Vergleichsprüfungs R-Faktor für einen Prüfsatz von 5% der einzigartigen Reflexe 3 Ramachandran Statistiken, die über PROCHECK definiert wurden 4 Die Zahl in Klammer beschreibt die höchste Auflösungsschale 180 Anhang Tabelle 7.9: Röntgendaten, Prozessierung und refinement Statistiken der Variante G129DE mit gebundener ADP-Ribose. Die statistischen Werte wurden den, aus der Prozessierung resultierenden Proteindatenbank-Dateien (*.pdb) und den Protokolldateien der Programme Scala (Evans, 2006), XDS (Kabsch, 2009), PROCHECK (CCP4) entnommen. Raumgruppe Ze k n an en a b c α β γ Å; °) P1211 63.38; 92.86; 124.51; 107.22; 90.03; 90.01 Auflösungsgrenze (Å) 1.85 Auflösungsbereich (Å) 38.97 – 1.85 Gesamte Vollständigkeit der Reflexe (%) 89.0 Anzahl einzigartiger Reflexe 118104 Multiplizität 2.33 I/σ I) 1.39 4 Rmerge (% ) 1 33.05 Rgesamt (%) 2 32.1 Rfrei (%) 36.9 R.m.s. Abweichung der Idealwerte Bindungslängen (Å) 0.012 Bindungswinkel (°) Φ, Ψ, Winkelverteilung fürAminosäurereste 1.24 3 In den bevorzugtesten Regionen (%) 91.4 In zusätzlich erlaubten Regionen (%) 8.2 In großzügig erlaubten Regionen (%) 0.4 In unerlaubten Regionen (%) - 1 Rmerge = 2 Rgesamt = Rfrei ist der Vergleichsprüfungs R-Faktor für einen Prüfsatz von 5% der einzigartigen Reflexe 3 Ramachandran Statistiken, die über PROCHECK definiert wurden 4 Die Zahl in Klammer beschreibt die höchste Auflösungsschale 181 Anhang Tabelle 7.10: Röntgendaten, Prozessierung und refinement Statistiken der Variante G129SE. Die statistischen Werte wurden den, aus der Prozessierung resultierenden Proteindatenbank-Dateien (*.pdb) und PROCHECK (CCP4) entnommen. Raumgruppe Zellkonstanten a; b; c (Å) α β γ °) Auflösungsgrenze (Å) P 21 21 21 62.8; 113.0; 188.1 90; 90; 90 2.44 Auflösungsbereich (Å) 52.71 – 2.44 Gesamte Vollständigkeit der Reflexe (%) 92.13 Anzahl einzigartiger Reflexe 44309 Rgesamt (%) 1 28.39 Rfrei (%) 34.07 R.m.s. Abweichung der Idealwerte Bindungslängen (Å) 0.011 Bindungswinkel (°) Φ, Ψ, Winkelverteilung für Aminosäurereste 2.742 2 In den bevorzugtesten Regionen (%) 88.5 In zusätzlich erlaubten Regionen (%) 9.5 In großzügig erlaubten Regionen (%) 1.1 In unerlaubten Regionen (%) 0.8 1 Rgesamt = Rfrei ist der Vergleichsprüfungs R-Faktor für einen Prüfsatz von 5% der einzigartigen Reflexe 2 Ramachandran Statistiken, die über PROCHECK definiert wurden 182 Danksagung Ich möchte mich hier bei meinem Doktorvater Prof. Dr. Thorsten Friedrich für die Bereitstellung der interessanten Themen und die vielen hilfreichen Anregungen und Diskussionen im Rahmen dieser Arbeit bedanken. Zudem bedanke ich mich herzlichst für sein stets offenes Ohr (und offene Tür!) und die sehr angenehme Arbeitsatmosphäre. Bei Prof. Dr. Oliver Einsle bedanke ich mich herzlich für die Übernahme des Korreferats, die große Hilfe bei der Proteinkristallographie und die Bereitstellung der Gerätschaften für die selbige. Dr. Stefan Gerhardt danke ich herzlich für die vielen Tipps und die Hilfe bei der Kristallographie. Meinen Kooperationspartnern Dr. Judy Hirst, Dr. James Birrell, Dr. Lothar Kussmaul und M. Sc. Biochemie Karoline Aierstock danke ich herzlich für die nette und fruchtbare Zusammenarbeit. Frau Dörte Thiel möchte ich für ihre tatkräftige Unterstützung im Labor und organisatorischen Arbeiten des Alltags danken. Unseren Damen im Büro: Frau Linda Williams, Frau Christiane Zähringer-Steffens und Frau Angelika Maßler gilt ein ganz lieber Dank für ihre Hilfe in allen bürokratischen Angelegenheiten. Ein besonderer Dank gilt Frau Helga Lay, die selbst über den Ruhestand hinaus eine unverzichtbare Hilfe beim Umzug des Labors, bei kleinen Fragen des Uni-Alltags und eine sehr nette Gesellschaft beim Kaffee trinken war. Christina Kress-Metzler danke ich für die sehr netten und hilfreichen Gespräche. Ich bedanke mich herzlichst bei den von mir betreuten Diplomanden, Master-Absolventen und Lehramtskandidaten Florian Hubrich, Therese Engel, Doris Kreuzer Deković, Sa c a Jurk vic, Emmanuel Gnandt, Manuel Schäuble, Ina Psarjow (du weißt) und Sabrina Burschel für ihre tatkräftige Unterstützung. Auch den von mir betreuten Bachelor-Absolventen und Praktikanten Sven Käfer, Uljana Botscharowa und Sabrina Burschel gilt ein großer Dank. Die Arbeit mit euch hat mir großen Spaß gemacht! Meinen Mitdoktoranden Katerina Dörner, Heiko Erhardt, Udo Glessner, Marta Vranas, Stefan Steimle, Doris Kreuzer Dek vić, Jo Hoeser und unseren "ganz jungen" Emmanuel Gnandt, Sascha Jurkovic, Ina Psarjow und natürlich Sabrina Burschel danke ich für die tolle, hilfsbereite Atmosphäre und die echt guten Partys!!! Die Mensa-Runde sei hier noch einmal besonders erwähnt..... ;-)!!! Ein ganz herzlicher Dank geht an meinen Mitstreiter seit der Diplomarbeit Marius Schulte. Unsere nächtlichen Labor-Sessions werden mir bestimmt fehlen! Mit Dir war es nur halb so schlimm!!! Meinen Mädels: Danke, dass es euch gibt!! Meiner gesamten Familie, vor allem meiner Mutter danke ich aus tiefstem Herzen für ihren Rückhalt und ihre Unterstützung in allen Lebenslagen. Meinen süßen Nichten dafür, dass sie mich die Welt mit ihren Augen sehen lassen. Ed danke ich einfach für alles!